














































































































Автор: Ярославцев А.Б.
Теги: физика химия термодинамика физическая химия химические вещества термохимия издательство научный мир
Год: 1995




Текст
А. Б. Ярославцев ОСНОВЫ ФИЗИЧЕСКОЙ ХИМИИ
А. Б. Ярославцев
ОСНОВЫ
ФИЗИЧЕСКОЙ ХИМИИ
НАУЧНЫЙ МИР
8
Введение
В XX веке был разработан ряд новых аппаратурных методов
исследования строения молекул и кристаллов (спектральные, масс-
спектральные, ЭПР, ЯМР), фотохимии и радиационной химии. По-
явились принципиально новые возможности определения ближай-
шего порядка в жидкостях и аморфных телах. Существенно возросла
чувствительность и разрешающая способность приборного парка.
Бурное развитие вычислительных методов и аппаратуры открыли
новые возможности в квантовой и статистической механике. В свою
очередь, развитие учения о строении вещества в совокупности с усо-
вершенствованием ранее известных и созданием новых методов ис-
следования привело к резкому увеличению работ в области химиче-
ской кинетики. И наконец, с середины XX века начала развиваться
термодинамика неравновесных процессов, являющаяся на настоя-
щий момент одной из наиболее перспективных отраслей физической
химии.
Итак, даже из этого краткого перечисления можно четко выделить
круг задач и явлений, изучаемых физической химией, но само опре-
деление предмета этой науки весьма расплывчато. Действительно,
химия изучает вещества и их превращения, физика - энергию и ее
превращение. Физической же химии надлежит описывать связь между
химическими и физическими явлениями.
Данное издание предназначено в первую очередь для студентов
младших курсов химических ВУЗов, которым знание физической
химии необходимо для углубленного понимания химических дисци-
плин на современном уровне. Исходя из этого в нем изложены лишь
основные представления этой науки. Причем, по возможности, автор
избегал использования высшей математики настолько, насколько
без нее вообще имеет смысл говорить об изучении физической хи-
мии. Тем не менее необходимо отметить, что этот материал может
быть использован лишь в качестве программы-минимума, знание ко-
торого, безусловно необходимо всем химикам. В издании частично
использовались задачи из Методических разработок к семинарским
занятиям по химии для студентов 1 курса Высшего колледжа наук о
материалах (М.: МГУ, 1993 г.), составленных автором совместно с
рядом сотрудников химического факультета МГУ.
Глава 1
ПЕРВЫЙ ЗАКОН ТЕРМОДИНАМИКИ
1.1. Основные понятия химической термодинамики
Термодинамика изучает количественные соотношения между
теплотой, работой и различными формами энергии, в том числе и
химической. Соответственно химическая термодинамика изучает
превращение энергии химических реакций в теплоту и работу.
Объектами ее изучения являются тепловые балансы физико-хими-
ческих процессов, фазовые и химические равновесия.
С точки зрения термодинамики нас ни в коей мере не интересует
внутреннее содержание системы и явления, происходящие на мик-
роскопическом уровне. Представляют интерес лишь макроскопи-
ческие свойства системы. Все положения термодинамики основаны
на ряде постулатов и рассматриваются исключительно для обоб-
щенных случаев, используя только общие законы природы. В этом
заключается как ее слабость, так и сила. Слабость состоит в том, что
наука эта крайне абстрактная, все ее положения, на первый взгляд,
далеки от реальных объектов. Поэтому для большинства химиков,
привыкших оперировать моделями и мыслить ассоциативно, вос-
приятие ее достаточно сложно. С другой стороны, опираясь лишь
на общие законы природы, термодинамика является наукой уни-
версальной, и все ее постулаты и законы представляют собой некие
непоколебимые твердыни. Именно поэтому Эйнштейн писал, что
термодинамика, по его мнению является единственной истинной на-
укой. В то же время следует заметить, что предпринимается масса
попыток модифицировать отдельные понятия термодинамики. Боль-
шинство из них касается попыток сужения границ применимости
Р го закона термодинамики и самого понятия энтропии, которое,
пенно, является наиболее сложным для восприятия.
про Д ТеРМОдинамической системой понимается некоторая часть
объект Нства со всеми включенными в нее компонентами, являющаяся
^^РаССМОтРе^- Непременным условием является наличие в
вшого числа частиц. Всякая термодинамическая система
10
Глава 1
должна быть ограничена некоторой воображаемой или реальной
границей, которую чаще называют поверхностью раздела. Через эту
поверхность может осуществляться обмен с окружающей средой ве-
ществом и различными формами энергии, такими как механическая
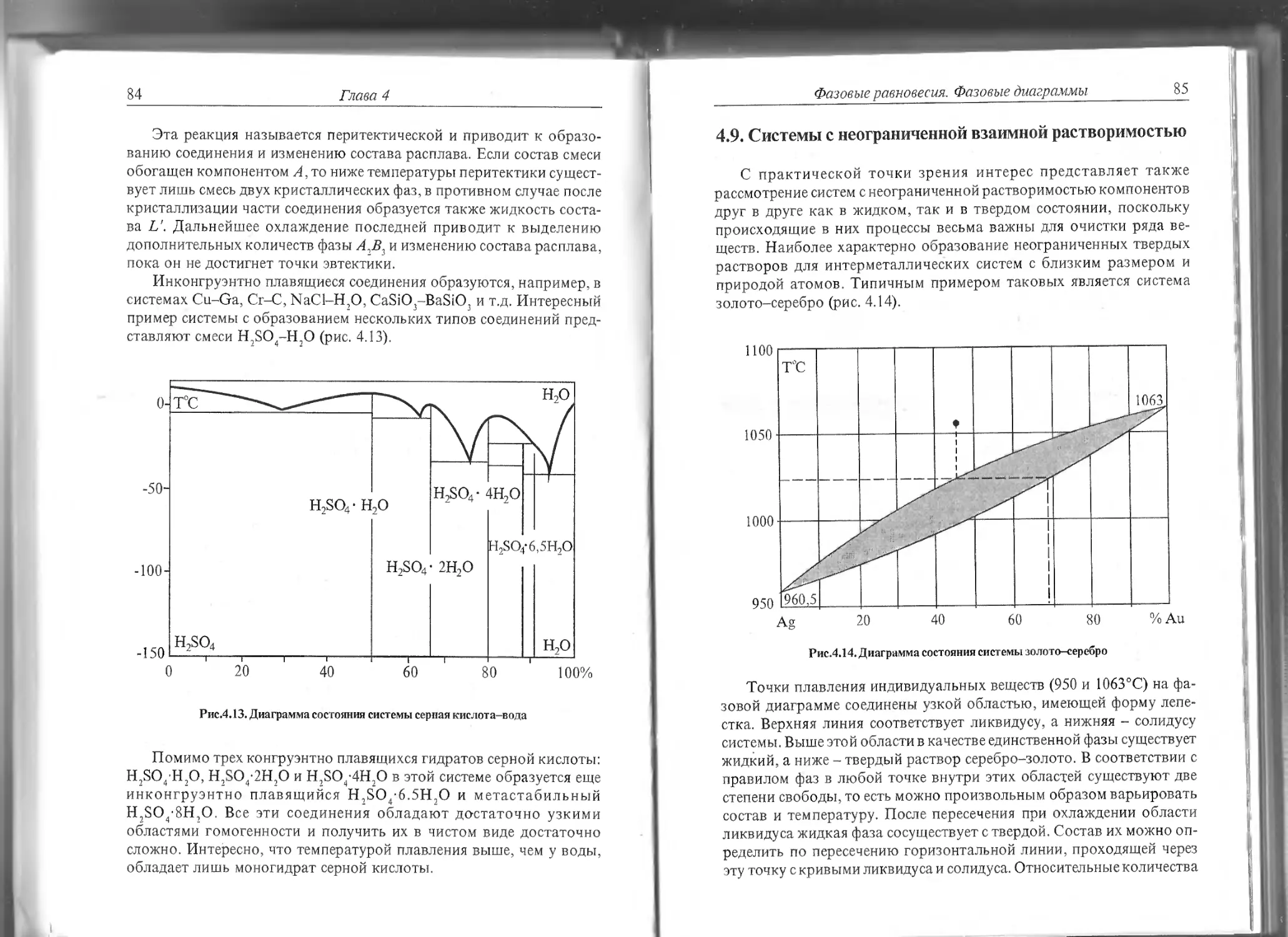
тепловая и т.д. Если все эти разновидности обмена разрешены, шо
система называется открытой. Типичными примерами являются
стакан с чаем, человеческий организм, который постоянно обме-
нивается с окружающим миром энергией и веществом, а также та-
кие глобальные геополитические образования, как отдельно взятая
страна. В случае, если обмен веществом запрещен, система называется
замкнутой или закрытой. Примерами могут служить запаянная ам-
пула, автоклав или воздушный шарик. Такая система может обме-
ниваться теплом с окружающей средой, а также совершать некоторую
работу. Если запрещен лишь обмен энергией, то система называется
адиабатической. Понятие это весьма абстрактное и встречается срав-
нительно редко. В некотором приближении адиабатические усло-
вия реализуются в термостатах, сосудах Дьюара и т.д. Наконец, если
все типы взаимодействия с окружающей средой запрещены, система
называется изолированной. Некоторым приближением ее является
закрытый термос.
Представляется целесообразным ввести также понятие окру-
жающей среды, которая служит неисчерпаемым резервуаром работы,
энергии или объема. Если рассматриваемый объект отдает или забирает
у окружающей среды некоторое количество тепла, температура ее не
меняется. Если воздушный шарик лопнет, это не приведет к изменению
объема окружающей среды. Последняя слишком велика для того,
чтобы реагировать на столь малые изменения. Окружающая среда
с точки зрения термодинамики это даже не то, что мы видим или
чувствуем. Понятие это существенно шире. Так, известно, например,
что средняя температура воздуха в крупных городах несколько по-
вышена за счет жизнедеятельности человека. Существует теория
глобального потепления климата на Земле в силу тех же причин.
Но все это практически не влияет на состояние Вселенной в целом.
Конечно Нерону во время поджога Рима удалось на какое-то
время повысить температуру в этом городе, однако явление это,
как показала история, оказалось сравнительно кратковременным.
Поэтому сомнительно, чтобы современные римляне могли хоть
немного согреться огнем того великого пожара. Хотя там и сейчас
достаточно тепло, никому не придет в голову связывать это с дей-
ствием тирана или повторять подобный эксперимент в условия
крайнего севера.
Первый закон термодинамики
И
Таким образом, окружающая среда есть вся Вселенная за исклю-
чеНием бесконечно малого на ее фоне изучаемого объекта.
Под термодинамическим состоянием системы понимается сово-
купность ее свойств. Под таковыми могут пониматься химический
состав, температура, давление, объем и т.д. То или иное изменение
состояния системы называется процессом. Хорошо известно, что
между некоторыми параметрами системы существует взаимосвязь.
Для идеального газа, например, она описывается уравнением Кла-
пейрона-Менделеева:
pV - (т!М) RT.
(1.D
Таким образом для описания системы оказывается достаточным
знание не всех, а лишь некоторого минимального набора параметров,
выбор которых по желанию исследователя можно варьировать исходя
из условий эксперимента. Свойства, выбираемые в качестве неза-
висимых переменных, называются параметрами состояния. Так, на-
пример, для моля газа в качестве таковых можно выбрать любое
из сочетаний: р и V, р и Т или V и Т.
Все параметры системы можно подразделить на два типа: эк-
стенсивные и интенсивные. Экстенсивные параметры определяются
количеством вещества в системе - это объем, масса, длина, площадь,
заряд и т.д. Все они могут быть измерены путем сопоставления с
некоторой стандартной величиной и подчиняются закону адди-
тивности, т.е. могут быть получены суммированием по всем частям
системы.
Интенсивные параметры (температура, давление, сила, напря-
женность поля и т.д.) не зависят от количества вещества и могут
быть измерены лишь опосредованно - через некоторую экстенсивную
величину. Например, для измерения температуры и давления можно
использовать высоту столбика жидкости в термометре или барометре.
По своему физическому смыслу все эти параметры являются аде-
кватными понятию силы в механике. Любые виды работы, совер-
шаемые термодинамической системой, могут быть охарактеризованы
произведением некоторого ее интенсивного параметра (У) на
изменение экстенсивного (X):
d^~YdX. (1.2)
вит^ЛЯ Работы по сжатию газа, например, это уравнение принимает
~ pdV
(1.3)
12
Глава 1
Уместно заметить, что удельные значения экстенсивных пара-
метров, такие как концентрация, удельный объем и т. д., в опре-
деленной степени также можно рассматривать как интенсивные
Для дальнейшего рассмотрения необходимо ввести играющее
значительную роль в термодинамике понятие равновесного состояния
системы, при котором ее свойства не меняются во времени при одно-
временном отсутствии потоков вещества и энергии. Следует особо
отметить, что для равновесной системы, находящейся в поле внешних
сил, интенсивные параметры могут меняться в объеме, как давление
в атмосфере Земли. Однако во всех своих дальнейших выкладках мы
будем оперировать такими типами систем, для которых этими изме-
нениями можно пренебречь.
Теперь можно сформулировать основное положение термоди-
намики, которое иногда называют ее нулевым законом. Если на
границе системы с окружающей средой поддерживаются постоянные
значения интенсивных параметров, то система рано или поздно придет
к состоянию равновесия. Особо подчеркнем, что для термодинамики
абсолютно несущественно, когда это произойдет, - главное, что это
неизбежно. Неважно, что все вокруг нас неравновесно _ цветет и бла-
гоухает, главное, что все это тленно.
Возникает вопрос, почему рассматриваемая система не может
перейти в иное стабильное состояние, например, с образованием
другой системы химических связей между атомами или перерас-
пределением энергии поступательного и колебательного движения
молекул. Но в этом случае одна из этих систем не будет являться
равновесной и в силу нулевого закона термодинамики неизбежно
перейдет в более устойчивое состояние.
1.2. Первый закон термодинамики
Хорошо известно, что химические реакции сопровождаются
выделением или поглощением энергии. При этом в ряде случаев
реакцию осуществляют именно для получения тепла, в других - вы-
деляющаяся теплота существенным образом влияет на протекание
процесса. Наконец, разогревание реакционной смеси во многих
случаях заставляет предъявлять особые требования к материалу
реактора. В связи с этим протекающие процессы часто представляют-
ся в виде термохимических уравнений. В этом случае в одной из частей
уравнения указывают количество теплоты, выделяемое в процессе
протекания реакции в данном направлении. В качестве единицы
Первый закон термодинамики
В
измерения тепловых эффектов как правило выбираются килокалория
(ккал) или килоджоуль (кДж). Для химиков, как правило, удобным
является отнесение этой теплоты к превращению одного моля ве-
щества. Поэтому они чаще пользуются размерностями типа ккал/моль
или кДж/моль. В мировой практике исторически сложилось так, что
первая из них в основном использовалась химиками и биологами, а
вторая - физиками. В течение длительного времени обе величины
успешно сосуществовали. Однако после принятия мировым сооб-
ществом системы СИ калории стали быстро вытесняться из научной
литературы, сохраняясь в основном в силу наличия старых спра-
вочников и неуступчивости биологов и медиков, которым чаще
приходится работать с обывателями, у которых понятие “калория”
напрямую связано с калорийными булочками (при всей своей ге-
ниальности Джоуль таковых изобрести, к сожалению, не пожелал).
На основании сказанного реакцию горения водорода можно пред-
ставить в виде:
2Н, + О, = 2Н,0 + 484 кДж , (1.4)
Н,+ 1/20, = Н,0 +242 кДж, (1.5)
2Н, + О, - 484 кДж = 2Н2О . (1.6)
Наиболее употребимым является представление уравнения в
форме (1.4) с указанием теплового эффекта в правой части и ми-
нимальными целочисленными коэффициентами. В соответствии с
законом сохранения энергии выделение тепла при протекании
процесса слева направо в точности соответствует поглощению его в
ходе обратного процесса.
Очевидно, что величина теплового эффекта определяется в первую
очередь различием энергии связей в исходных веществах и продуктах.
Каждое вещество, таким образом, характеризуется некоторым по-
тенциальным запасом энергии. Она включает все виды энергии дви-
жения и взаимодействия частиц, составляющих систему молекул,
атомов, их ядер, электронов и т.д. Величина эта получила название
внутренней энергии. Она зависит исключительно от этого состояния
термодинамических параметров системы, но не от путей достиже-
нг/я системой, являясь поэтому так называемой функцией состояния,
сплота процесса и произведенная им работа, напротив, опреде-
ляются путем его протекания и функцией состояния в общем случае
нс являются.
Таким образом мы пришли к формулировке первого закона
Рмодинамики: в ходе любого процесса приращение внутренней
14
Глава 1
энергии (MJ = U2-Ux) системы равно разности между количеством
сообщенной ей теплоты (О) и совершенной ею работой (А).
В качестве несколько упрощенной формулировки можно также
рассматривать положение, что совокупная энергия изолированной си-
стемы постоянна.
Проблема выбора знака для характеристики передачи энергии в
термодинамике решается весьма просто. Знак “+” выбирается в том
случае, если система приобретает энергию, то есть соответствует
увеличению внутренней энергии, сообщенной системе теплоте и
совершенной над ней работе, приводящей опять же к увеличению
внутренней энергии. Напротив, знаком обозначаются потери
системой внутренней энергии, теплоты и работа, совершаемая самой
системой. Таким образом, математическим выражением первого за-
кона термодинамики является соотношение
&U = Q-A. (1.7)
Можно найти и другие примеры функций состояния. Так, пройдя
от входной двери дома до квартиры, Вы совершили некоторую работу
по переносу своего тела на высоту, например, четвертого этажа (mgh).
Примерно на эту же величину изменилась ваша потенциальная энер-
гия, которую можно оценить, выпав из окошка на поршень с располо-
женным под ним газом. Путем повторения этого эксперимента можно
убедиться, что энергия эта практически не зависит от того, сколько
времени Вы при этом проговорили с соседями по подъезду, заходили
ли Вы к ним в гости и не спускались ли вновь за газетами к почтовому
ящику.
Таким образом, нетрудно убедиться, что внутренняя энергия не
является единственной функцией состояния. Другим ее примером
является энтальпия (Я), определяемая как
H=U + pV. (1.8)
В данном случае к внутренней энергии добавляется некоторая
величина, которая в первом приближении зависит лишь от количества
молей газа и его температуры. Действительно, из уравнения (1.1) для
идеального газа следует, что она равна (m!M)RT. Применение фун-
кции энтальпии целесообразно в случае совершения системой (или
над ней) работы по расширению (сжатию). Действительно, работа
по сжатию газа под действием некоторого постоянного внешнего дав-
ления (например, атмосферного, рис. 1.1) может быть представлена,
как
А =рМ' = р(Г2-Vl)=pV2-pV[ = \(pV)
(1.9)
Первый закон термодинамики
15
Рис.1.1. Работа по сжатию системы под действием постоянного внешнего давления
Учитывать величину работы расширения или сжатия газа при
постоянном давлении необходимо из-за того, что многие важнейшие
химические и физические процессы происходят с участием газовой
фазы именно при постоянном (атмосферном) давлении. Точно таким
же образом можно было бы учесть и энергию электромагнитного вза-
имодействия или энергию тяготения Земли. Однако эти состав-
ляющие оказываются существенными значительно реже. С другой
стороны, рыбам, наблюдающим преимущественно процессы, про-
текающие в практически несжимаемых конденсированных средах,
вряд ли понадобилось бы введение давления в качестве дополнитель-
ного параметра системы.
В случае, если в ходе процесса меняется и внутренняя энергия
системы:
А^+Д(рП =Д([/+/710 = АЯ (1.10)
При этом изменение энтальпии определяется исключительно
термодинамическими параметрами системы. Именно поэтому
энтальпия является мерой теплоты химических процессов, проис-
ходящих при постоянном давлении. Случай этот весьма распространен
’ по*алуй, наиболее часто встречается при описании различного
Р°Да физических и химических процессов. Очевидно, что в случае со-
ения объема работа сжатия равна нулю и
АИ=Дйг. (!!!)
16
Глава 1
Отсюда следует, что теплота процесса при постоянном объеме
является мерой изменения внутренней энергии.
Если давление, против которого совершается работа, изменяется
то процесс в целом можно разбить на ряд более мелких стадий, в ходе
которых изменением давления можно пренебречь, и тогда для каждой
из них
dA=pdV, (1.12)
и для процесса в целом (рис. 1.2)
A=$pdV. (1.13)
Рис. 1.2. Работа по сжатию системы против переменного внешнего давления
Работу системы при изотермическом расширении идеального газа
можно оценить из соотношения:
А =/pdV = nRT^dV/V = nRT\n(V,l V) = nRT\n(pJр2). (1.14)
Альтернативной термохимическим уравнениям формой записи,
учитывающей тепловой эффект реакции, является указание после
соответствующего процессу уравнения величин энтальпии или
изменения внутренней энергии. Например, уравнение горения
водорода может быть представлено в виде:
Первый закон термодинамики
. 17
2Н,+ 0 ~ 2Н.0 КН = -484 кДж/моль . (1-15)
Обратим внимание, что энтальпия процесса по знаку обратна
величине теплового эффекта. Обе эти величины меняют свои значения
с изменением температуры и давления. Поэтому имеет смысл
указывать их при некоторых стандартных условиях. В качестве
таковых обычно выбираются давление, равное одной атмосфере, и
температура 298 К, соответствующая 25°С. В этом случае энтальпию
принято записывать в форме КН°,98, где верхний индекс указывает на
стандартное состояние исходных веществ и продуктов, а нижний -
на температуру проведения эксперимента. Особо подчеркнем, что в
термодинамике температуру принято указывать в градусах Кельвина,
шкала которого берет отсчет от абсолютного нуля и имеет ту же
величину деления, что и шкала Цельсия. Пересчет температур, таким
образом, следует производить по формуле:
T,K = z°C + 273.15. (1.16)
Константа этого уравнения соответствует температуре плавления
льда.
Полезно уметь оценить соотношение между величинами ДЯи KU
различных реакций. Для этого заменим в соотношении (1.10) величину
pV ее эквивалентом для моля газа, взятым из уравнения Клапейрона-
Менделеева:
KH = K(U+pV) = KU + K(pV) = KU + KnrRT, (1.17)
где Кпг _ изменение числа молей газа в процессе реакции. Измене-
ние числа молекул, находящихся в конденсированной фазе, не учи-
тывается, поскольку они имеют при атмосферном давлении объем
меньше на три порядка. Так, для реакции (1.15) Д/?г = -1. Поэтому
при 298 К (KH-KU) = -0.00831 298 = 2.48 кДж/моль. С другой стороны,
при окислении глюкозы:
С6Н12О6(тв) + 60, (г) = 6СО, (г) + 6Н,0 (ж) (1.18)
количество молекул газа в обеих частях уравнения одинаково и
энтальпия реакции равна изменению внутренней энергии, хотя число
молекул твердой и жидкой фаз при этом различается в 6 раз.
1.3. Термохимия. Закон Гесса
пост СНовн°и задачей термохимии является определение теплот
мепо'* НИЯ Химических процессов. Из вышеизложенного следует, что
плоты химического процесса, протекающего при постоянном
18
Глава 1
объеме, служит изменение внутренней энергии. Для процессов
протекающих при постоянном давлении, такой характеристикой
является изменение энтальпии. Процессы, протекающие с выделе-
нием тепла (отрицательные значения ДЯ или ДУТ), называются
экзотермическими. Напротив, процессы, протекающие с поглощением
тепла (положительные значения или ДЯ), называются эндотер-
мическими.
В основе всех термохимических расчетов лежит закон Гесса
гласящий, что тепловой эффект химического процесса зависит только
от природы и состояния исходных веществ и продуктов, но не от пути
его осуществления, в том числе от выбора системы реакций и состояния
промежуточных продуктов. По сути своей это ни что иное как
несколько модифицированная формулировка закона сохранения
энергии. Этот закон помогает, например, осуществить определение
теплот процессов, измерить которые напрямую весьма сложно или
невозможно.
Классическим примером таких расчетов является определение
теплоты реакции образования монооксида углерода из графита и
газообразного кислорода.
С (графит) + 1/20, (г)= СО (г) ДЯ(. (1.19)
Очевидно, что невозможно подобрать такие условия, чтобы
остановить реакцию именно на этой стадии. С другой стороны, мож-
но легко оценить теплоты реакций полного сгорания графита и угар-
ного газа:
С (графит) + О, (г)= СО, (г) ДЯ, = -393.5 кДж/моль, (1.20)
и
СО (г) + 1/20, (г) = СО, (г) ДЯ3 =-110.5 кДж/моль. (1.21)
Процесс превращения, описываемый реакцией (1.20), можно
представить и как цепь последовательных превращений (1.19) и (1.21).
В таком случае в соответствии с законом Гесса можно записать
соотношение:
ДЯ,=ДЯ,+ДЯ3. (1.22)
Откуда ДЯ] = -393.5 + 1 10.5 = - 283 кДж/моль.
С помощью закона Гесса можно также рассчитать теплоты раз-
личных химических реакций. Естественно, трудно предположить, что
теплоты всех известных процессов сведены в виде табличных данных.
Можно себе представить, каков был бы объем этих таблиц и сколь-
ко времени занял бы поиск нужной величины. Но реально этого и
Первый закон термодинамики 19
к
не нужно. В соответствии с законом Гесса теплоту любого процесса
можно представить в виде разности между терлотами образования
продуктов и исходных веществ с соответствующими коэффициентами.
Так, в общем случае для реакции
а^А} + ... + аАп - Ь{В{ +... + ЬтВт (1.23)
ДЯ = ЪЬН. - ЕдН.. (1.24)
Это соотношение легко вывести из цикла, приведенного на
рисунке 1.3.
Рис.1.3. Определение теплоты химического процесса по теплотам образования
исходных веществ и продуктов реакции
Аналогичные соотношения можно предложить и для определения
теплот реакций через величины внутренних энергий. Поскольку
величины внутренней энергии и энтальпии некоторого соединения
можно измерить при исследовании процессов его образования из
Различных исходных веществ, а при этом искомые величины будут
существенно изменяться, необходимо определиться с точкой отсче-
та. В качестве таковой выбираются энтальпии простых веществ в
наиболее стабильных формах при стандартных условиях. Для
водорода это газ, для брома - жидкость, для углерода - графит,
разевание которого энергетически несколько более выгодно, чем
алмаза и т.д. Таким образом, энтальпия образования жидкой воды,
^пример, равна энтальпии реакции (1.15), оксида углерода - (1.19).
и3данН0 ЭТИ величины и приводятся в термодинамических справочных
20
Глава 1
Несколько реже, обычно для органических веществ, пользуются
теплотами сгорания, например по процессу (1.18). При этом учи-
тывается теплота сгорания до высших степеней окисления, например
СО,, SOr Если среди продуктов сгорания есть нестандартные
вещества: фториды, азот, НС1 и т. д., это оговаривается особо. Из
цикла, аналогичного приведенному на рисунке 1.3. можно заключить
что теплота реакции равна разности теплот сгорания исходных
веществ и продуктов. В качестве примера такого расчета можно
привести рассмотренную выше схему расчета теплоты образования
монооксида углерода.
1.4. Зависимость внутренней энергии и энтальпии
от температуры
Для того чтобы нагреть некоторую систему от температуры Т до
Т, , ей необходимо сообщить некоторое количество теплоты Q.
Отношение последней к разности температур называется тепло-
емкостью системы. Величина эта также зависит от температуры.
Истинная теплоемкость при данной температуре соответствует
бесконечно малому изменению последней:
CT = dQ!dT. (1.25)
Тогда при нагревании вещества от температуры 7\ до Т, будет
затрачена теплота
Q=$CrdT. (1.26)
Соответственно, средняя теплоемкость для этого интервала
температур может быть выражена соотношением:
Cp = eCTdT)/(T2-T). (1.27)
Следует различать теплоемкость при постоянном давлении и при
постоянном объеме. Если для твердых и жидких веществ разница
между ними относительно невелика, то для газов различие весьма
принципиально. Действительно, если при постоянном объеме под-
веденная теплота расходуется лишь на изменение внутренней энергии,
то при постоянном давлении совершается также некоторая работа
расширения. Поэтому поглощенная теплота будет соответствовать
изменению энтальпии вещества.
Cv = (dUldT)ln и С = (dHIdTjln , (1.28)
где п - количество вещества в молях. На графиках зависимости
внутренней энергии от температуры теплоемкость будет представлять
Первый закон термодинамики
21
собой касательную (рис. 1.4). Продифференцируем теперь по тем-
п натуре выражение для энтальпии (1.8) в случае идеального газа:
г = dHIdT = d(U+p V)!dT = d(U+RT)ldT = dUldT+R = Cv+R. (1.29)
p
Рис.1.4. Определение теплоемкости как производной по зависимое ги внутренней
энергии или энтальпии от температуры
Соотношение это удовлетворительно выполняется для сравни-
тельно разряженных (находящихся при низком давлении) газов, при
комнатной и более высоких температурах. Согласно молекулярно-
кинетической теории средняя кинетическая энергия молекулы газа
равна
Е-ЗПкТ или для моля U- Uo =3/2 RТ. (1.30)
Откуда следует, что для одноатомного идеального газа Су = 3/2R
и Ср = 5I2R. На каждую поступательную степень свободы приходится
вклад R/2. Точно такой же вклад должен приходиться на каждую
вращательную или колебательную степень свободы. Для N-атомной
молекулы (ЗД^ степеней свободы), таким образом, общая теплоемкость
Должна составлять Су - 3NR/2 или Ср = (3N+2)R/2. Однако эти со-
отношения хорошо выполняются лишь при высоких температурах.
Ри низких же, включая комнатную, вклад колебательных степеней
0 оды относительно мал и дополнительный вклад вносят лишь
Р щательные степени свободы, которых для двухатомной молекулы
’ а Для нелинейных молекул - 3.
22
Глава 1
В общем виде теплоемкость зависит от температуры более слож-
ным образом. Общий вид такой зависимости приведен на рисунке
1.5. При температурах, близких к О К, она меняется пропорционально
Т 3, далее - по более сложной зависимости, выражаемой обычно
эмпирическим полиномом типа
С-а +ЬТ+сТ2 +d/Tz +еТ3 + ... (1.31)
Значения коэффициентов уравнения (1.31) для большого коли-
чества веществ можно найти в термодинамических справочниках.
Точки разрыва на графике соответствуют фазовым переходам.
Таким образом, можно связать величины энтальпий вещества при
двух разных температурах:
Я(7’,)=Я(Г1) + /Ср</7’+гДЯфл. (1.32)
Последняя величина соответствует разности энтальпий фазовых
переходов исходных веществ и продуктов, происходящих в данном
интервале температур. В качестве таковых могут выступать как
переходы типа твердое тело-жидкость, жидкость-газ, так и ал-
лотропные превращения типа алмаз-графит, белый-красный фос-
фор. Очевидно, что кривая зависимости энтальпии вещества от
температуры будет аналогична приведенной на рисунке 1.5, однако
при нулевой температуре величина энтальпии не равна нулю.
Первый закон термодинамики
23
Аналогично можно представить и зависимость энтальпии неко-
торой реакции от температуры. Для этого также можно воспользо-
ваться законом Гесса и построить цикл типа:
исходные вещества при температуре Т\ - (нагревание до Г,) -»
- > исходные вещества при температуре Т. - (реакция) -»
— > продукты при температуре Г, - (охлаждение до Т\) —*
— > продукты при температуре Г,.
Откуда для реакции типа (1.23) легко получить:
т,
ьнт = дя(7,) + Me dT + S (ДЯфп)пр - S (ДЯфп)нс1 (1.33)
Таким образом, различие стандартных энтальпий некоторой
реакции при двух температурах равно разности теплот фазовых
переходов продуктов и исходных веществ плюс интегралу разности
теплоемкости продуктов и исходных веществ с соответствующими
коэффициентами в рассматриваемом интервале температур. Это
соотношение называется уравнением Кирхгофа.
реакции (а) и ее энтальпии (б) от температуры
Заметим, что в отличие от энтальпии индивидуального вещества
зависимость энтальпии химической реакции от температуры может
иметь экстремумы в точке, где теплоемкости продуктов и исходных
Веществ равны (рис. 1.6).
24
Глава 1
Задачи и вопросы:
1. Открытый металлический сосуд объемом 1 л опускают горло-
виной вниз в озеро на глубину 50 м. Какой объем будет занимать
воздух в конце процесса, если принять, что для него работает
приближение идеального газа. Оцените до какой температуры следует
охладить данный сосуд для достижения того же результата, если
исходная температура сосуда 10°С?
2. Голубой воздушный шарик с диаметром 20 см, заполненный
гелием, поднимается с поверхности земли, с температурой 20°С, имея
внутреннее давление, равное 1.05 атм. Перед завершением своей
карьеры он достиг вдвое большего размера, охладившись при этом
на 25°. Каково на этот момент было давление внутри шарика?
3. В откаченном контейнере, закрытом подвижным поршнем, при
300°С провели разложение 25 г хлорида аммония. Какую работу
произвела данная система?
4. Одинокий студент поставил на газовую плиту чайник с 1.5
литрами воды с температурой 10°С. Вернувшись с лекции любимого
преподавателя он обнаружил, что вода в чайнике иссякла. Оцените
величины АН и AU для данной системы, принимая теплоемкость воды
равной 75 Дж/мольК, а энтальпию испарения - 40.6 кДж/моль.
5. Укажите, какие из перечисленных процессов являются эндо-, а
какие экзотермическими? Таяние льда, конденсация водяного пара,
возгонка иода, изотермическое расширение газа, рост дерева, работа
холодильника.
6. Теплота испарения воды при 30°С равна 4407, а при 20°С -
4432 кДж/моль. Оцените среднее значение теплоемкости водяного па-
ра в этом интервале температур, если для жидкости она составляет
75.3 Дж/моль К?
7. Теплота сгорания 1 моля метана составляет -890.3 кДж/моль.
Оцените ДН° и AU0 образования метана.
8. Определите энтальпию гидратации серной кислоты до
моногидрата, если энтальпии следующих реакций составляют
(в кДж/моль):
Первый закон термодинамику
25
SO, = I/8S,(tb) + О, 296.9
So’(r) = SO,+1/20, 98.3
H,O(r) = Н,+1/20, 241.8
Н,О(г) = Н,О(ж) -44.0
SO,(г) + Н,О(ж) = Н,5О4(ж) -130.3
1/8S8(tb) + 2Н, +5/20,= Н,8О4Н,О(ж) -1128.0
9. Рассчитайте энтальпию фазового перехода графит-алмаз,
если энтальпия сгорания алмаза равна -395.4 кДж/моль, а графита -
-393.5 кДж/моль.
10. Составьте соотношение для расчета изменение энтальпии
реакции типа А + В = С, если в интересующем нас интервале
температур фазовые превращения исходных веществ и продук-
тов отсутствуют, а зависимость теплоемкости от температуры опи-
сываются формулой с = а + ЬТ + сГ + d/T.
Глава 2
ВТОРОЙ ЗАКОН ТЕРМОДИНАМИКИ
2.1. Работа и теплота обратимых процессов
Перед тем как перейти к рассмотрению второго закона термо-
динамики, введем понятие обратимого процесса, при котором в любой
фазе превращения все части рассматриваемой системы находятся в
равновесии друг с другом и с внешним окружением. Рассмотрим процесс
изотермического расширения некоторого газа от объема V до 1
Его можно представить себе протекающим при снятии грузиков
с поршня, перемещающегося в некотором сосуде без трения. Со-
вокупность грузов задает в нем давление р}. При этом система на-
ходится в равновесии, параметры которого связаны друг с другом
посредством уравнения Клапейрона-Менделеева (1.1). Снятие одного
из грузов приведет к снижению давления до р2 и расширению газа с
совершением работы против внешнего давления (рис. 2.1 я). Работа
эта по величине равна р,-ДЕ (заштрихованная площадь). Путем
последовательного осуществления такого рода операций мы
совершим полезную работу, равную всей заштрихованной площа-
ди. Вновь ставя гирьки на поршень, мы совершим большую работу
по сжатию газа, поскольку каждому изменению объема будет со-
ответствовать работа большего давления. Разность между совер-
шенной над системой работой сжатия и произведенной ею самой
работой расширения высвободится в виде тепла. Уменьшение веса
грузов приводит к снижению энергетических потерь (рис. 2.16). Пу-
тем последовательных приближений можно прийти к единственному
равновесному варианту, когда вес грузиков бесконечно мал. Это и
есть обратимый процесс. Он может быть осуществлен одним и только
одним путем, в ходе которого уравнение Клапейрона-Менделеева
выполняется в любой момент времени. Следовательно, высво-
бождаемая в ходе обратимого процесса работа является величиной
постоянной. Кроме того, этот процесс сопровождается постоянным
изменением внутренней энергии, поэтому как следует из первого
Второй закон термодинамики
27
закона термодинамики, постоянна и его теплота. Таким образом,
теплоту обратимого процесса можно рассматривать в качестве
функции состояния.
2.2. Второй закон термодинамики
Первый закон термодинамики дает представление лишь о
тепловом эффекте процесса, но не говорит о принципиальной
возможности его протекания. Казалось бы, самопроизвольно должны
протекать процессы, сопровождающиеся выделением тепла. Именно
такое мнение и было распространено в прошлом веке (У.Томсон,
*-Бертло). Однако, нетрудно убедиться, что существует великое
множество обратных примеров. Это, например, растворение нитра-
та аммония, а также такие в общем тривиальные процессы, как
Расширение газа, испарение жидкостей, возгонка твердых тел и,
наконец, существование большого числа обратимых процессов, со-
провождающихся отрицательным тепловым эффектом. Попробуем
януть на экзотермические реакции с другой стороны. В результате
ОсУЩествления внутренняя энергия системы понижается. С другой
28
Глава 2
стороны, эта же самая энергия рассеивается в окружающем про-
странстве. При этом его энергия увеличивается, то есть с этой точки
зрения процесс является противоестественным. Таким образом,
увеличив размер рассматриваемой системы мы пришли к абсурдно-
сти предположения Бертло и Томсона, поскольку нельзя отдать
предпочтение лишь одной части системы. В соответствии с первым
законом термодинамики совокупная энергия системы сохраняется и
никакого приращения энергии не происходит.
Для того чтобы выяснить принципиальную возможность про-
цессов различного рода и понять, какие из них могут протекать са-
мопроизвольно, вводится второй закон термодинамики. Он утверж-
дает, что существуют некоторые процессы, не противоречащие первом)
закону термодинамики, которые самопроизвольно протекать не могут.
К таковым следует отнести сжатие газа, передачу теплоты от хо-
лодного тела к горячему и некоторые другие. Такие процессы, само-
произвольное протекание которых невозможно, называются отри-
цательными. Второй закон термодинамики обычно представляется
в виде постулатов. Наиболее распространенной формой его пред-
ставления являются постулаты Клаузиуса и Томсона:
- теплота не может самопроизвольно переходить от холодного
тела к горячему;
- теплота более холодного из участвующих в процессе тел не может
служить источником работы.
Следует уточнить, что отрицательный процесс не может быть
лишь единственным результатом некоторого действия. Если таковой
наблюдается (как, например, в холодильнике, охлаждение которо-
го происходит за счет передачи тепла окружающей среде), то па-
раллельно с этим должен протекать и некоторый положительный
процесс (насильственная конденсация охлаждающей смеси в теп-
лообменнике, сопровождающаяся выделением тепла).
Для того чтобы охарактеризовать способность системы к само-
произвольным превращениям, вводится понятие энтропии. При
обратимом процессе, в ходе которого система проходит через не-
прерывный ряд равновесных состояний, изменение энтропии числен-
но равно отношению его теплоты к температуре протекания:
AS = S2-S, =Q/T. (2.1)
Это отношение не отражает физического смысла энтропии, но
фактически является математическим выражением второго закона
термодинамики. Выбор именно обратимого процесса представляет-
ся вполне естественным, поскольку лишь в этом случае его теплота
Второй закон термодинамики 29
приравнивается к изменению функции состояния - энтальпии или
внутренней энергии.
Если процесс является необратимым, то при его протекании
энтропия должна возрастать, то есть должно выполняться соотно-
шение
dS = dQIT-dl/T или \S<QtT, (2.2)
где - di поток энергии во внешнее пространство. Действительно, в
ходе таких превращений энтропия, как и любая функция состояния,
меняется на ту же величину, что и для равновесного процесса. С другой
стороны, произведенная системой работа оказывается меньше
максимально возможной, а выделяемая теплота - больше.
Таким образом, в изолированной системе самопроизвольно мо-
гут протекать лишь процессы, ход которых сопровождается по-
ложительным изменением энтропии. Следует обратить внимание, что
изменение энтропии при комнатной температуре в среднем на два с
половиной порядка меньше тепловых эффектов процесса и, поэтому,
измеряется не в кДж/моль К, а в Дж/моль К, иначе называемых просто
энтропийной единицей.
2.3. Статистическая природа второго закона
термодинамики
Для того чтобы понять, что же собой являет энтропия и в чем
состоит движущая сила самопроизвольных процессов, обратимся к
нашему жизненному опыту. Известно, что если привести в движение
бильярдный шар, то через некоторое время он неизбежно остановится.
При этом кинетическая энергия шара за счет силы трения постепенно
переходит в кинетическую энергию молекул газа, шара и бильярдно-
го стола. Это процесс вполне естественный. Направленное движе-
ние шара и составляющих его частиц постепенно переходит в бес-
порядочное движение молекул рассматриваемой системы. Для
осуществления обратного процесса в каждый конкретный момент
времени существенно большее число атомов воздуха и стола должно
Двигаться в ту же сторону, что и шар, подталкивая его в нужную
сторону (рис. 2.2). Даже чисто интуитивно легко догадаться, что такой
процесс невозможен с точки зрения вероятности.
Теперь рассмотрим закрытую систему, содержащую молекулы
скоторого газа. Разобьем эту систему на четыре равные части. Ве-
роятность того, что каждая из частей содержит соответственно h, k, I
Рис.2.2. Схема гипотетического процесса разгона бильярдного шара молекулами газа
и т молекул, пропорциональна
W= (h + к + I + т)\ I h\ к\ l\m\ (2.3)
Если таких молекул всего 4, то вероятность найти все молекулы в
одной части пропорциональна 4!/4! = 1, а равномерно распреде-
ленными - 4! = 24. Здесь и далее мы будем говорить о так называемой
термодинамической вероятности, характеризующей число микрососто-
яний, которым мы можем реализовать данное состояние системы.
В отличие от чаще используемого математического аналога сумма
термодинамических вероятностей всех состояний существенно
превышает единицу. Если молекул становится 8, то эти вероятности
составят соответственно 1 и 2520, для 16 молекул 1 и 63063000 и т. д.
То есть при увеличении числа молекул от четырех до восьми со-
отношение вероятностей нахождения их в одной ячейке и равномер-
но распределенными изменяется примерно в 100 раз, от восьми до
шестнадцати - еще в 25000 раз. Трудно даже представить, каково будет
соотношение этих величин для 1О23 молекул газа, заключенных в
наших легких! Именно поэтому отсутствует вероятность того, что
нормальный человек может задохнуться из-за случайного стечения
обстоятельств, в результате которого в его легкие не попало бы ни
одной молекулы кислорода. Точно так же при разрушении пере-
городки между двумя емкостями - заполненной газом и пустой -
молекулы газа устремляются в последнюю ввиду большей вероятности
равномерного распределения частиц в объеме.
Таким образом мы приходим к заключению, что система должна
стремиться к наиболее вероятному состоянию. Исходя из этого
представляется целесообразным отождествить энтропию с вероят-
ностью. Однако, как мы видели чуть выше, увеличение количества
вещества привело бы к огромному изменению энтропии, т.е. к
нарушению свойства аддитивности. Для тою чтобы избежать этой
Второй закон термодинамики
31
неприятности, Больцман предложил рассматривать энтропию как
величину, кратную логарифму термодинамической вероятности
состояния системы (W):
S = lc\nW, (2.4)
где к - постоянная Больцмана. При этом функция приобретает
свойство аддитивности. Действительно, увеличив в п раз количество
вещества, мы изменим совокупную термодинамическую вероятность
системы от РИдо Wn. При этом ее энтропия составит
S’-к In (Wn) = nk\nW = nS,
то есть также увеличится в п раз.
Только что рассмотренный пример наглядно характеризует
увеличение энтропии в процессе расширения газов. Действительно,
при увеличении объема газа в V,/V\ каждая его молекула получает
возможность находиться в объеме, в Г, /И, раз превышающем
исходный. Таким образом для расширения одного моля идеального
газа вероятность такого состояния возрастает в (V2/V\)N раз, где N-
число Авогадро. Тогда из уравнения (2.4) получим
AS = к In (ГДЕ/ = kN In (ГДЕ,). (2.5)
Величина постоянной Больцмана как раз и выбирается таким
образом, чтобы привести в соответствие полученную величину с
уравнением (2.1). Работа изотермического расширения идеального
газа равна «ЯTin (ИД Г,). Соответственно для расширения моля газа
эта величина равна ТТ1п (ИДИ,). Изменение кинетической энергии
молекул, а следовательно, внутренней энергии газа при неизменности
температуры равно нулю. Поэтому из первого закона термодинамики
Q - А = АТ1п (ИДИ,) и из уравнения (2.1) получаем:
&S=Q!T=R In (ИДИ,) (2.6)
Отсюда легко получается численное значение коэффициента к,
равное R/N - 8.314/6.02 • 1023 = 1.38-10'23 Дж/К. Аналогичным
описанному выше изменением энтропии будет сопровождаться и
самопроизвольно протекающий процесс смешения двух газов.
Естественно, что обратный процесс сжатия газа приведет к
понижению термодинамической вероятности данного состояния.
Следовательно, он не может быть самопроизвольным, а может про-
исходить лишь под воздействием некоторой внешней силы.
Из приведенного примера можно заключить, что энтропия явля-
ется мерой разупорядоченности системы, а собственно сам второй
закон термодинамики постулирует стремление любой системы к
порядку. Проявления этого мы постоянно видим в окружающей
32
Глава 2
нас действительности. Все мы периодически наводим порядок в своей
квартире. Однако через некоторое время непобедимая энтропии
сводит наши усилия на нет со скоростью, пропорциональной нашему
темпераменту. Поэтому через некоторое время приходится вновь
наводить порядок, совершая в буквальном смысле этого слова полез-
ную работу против сил природы, то есть осуществляя некий отрица-
тельный процесс за счет положительного. Однако, вследствие перво-
го закона термодинамики, пользы от этого в глобальном масштабе
мало. С неизбежностью все наши вещи должны в конце концов быть
разбросаны в самом низком месте квартиры (то есть на полу) и, вслед-
ствие второго закона термодинамики, перемешаны в замечательном
беспорядке.
Вернувшись к примеру с бильярдным шаром мы можем отметить
также, что энтропия должна способствовать преобразованию на-
правленного движения в хаотическое, т.е. сводить к беспорядку и
движение молекул системы. Представим процесс передачи тепла от
холодного тела горячему. При этом мы как бы концентрируем более
быстрые молекулы в одном теле, а обладающие меньшим запасом
кинетической энергии - в другом. Очевидно, что термодинамическая
вероятность такого состояния системы понижается и, следовательно,
процесс не является самопроизвольным. Напротив, самопроизволь-
ным будет процесс рассеивания энергии - передачи ее от горячего
тела к холодному. Обратный этому процесс невозможен в соответ-
ствии со вторым законом термодинамики.
С другой стороны, очевидно, что в природе происходят некоторые
процессы, сопровождающиеся уменьшением энтропии, например рост
и воспроизводство растений и животных. Сосуд, обмотанный мокрой
тряпкой, может заметно охладиться по сравнению с окружающей сре-
дой. Кажущееся противоречие на самом деле весьма легко разрешимо
- все это возможно лишь за счет увеличения энтропии окружающе-
го пространства. Действительно, жизнедеятельность растений проис-
ходит благодаря положительной работе солнечной энергии, а сосуд
остывает за счет процесса испарения воды (перехода ее из малого
объема в большой), что сопровождается громадным увеличением
энтропии.
2.4. Энтропия веществ и химических реакций
Каждому физическому телу, материалу или химическому соедине-
нию можно приписать некоторое значение энтропии. Наличие в
веществе примесей способствует повышению его энтропии. Напротив,
Второй закон термодинамики
33
очистка его невозможна без совершения некоторой работы. В процессе
охлаждения твердых тел каждый атом в кристалле, снижая амплитуду
своих колебаний и вероятность ее изменения, одновременно понижает
степень свободы, а следовательно, понижается и энтропия кристалла
в целом. В пределе в идеальном кристалле при абсолютном нуле все
атомы локализованы строго на присущих им позициях и принимают
участие лишь в одинаковых так называемых “нулевых” колебаниях
решетки. Согласно уравнению (2.4) термодинамическая вероятность
существования этого состояния равна единице и энтропия такого
кристалла равна нулю. Это так называемый постулат Планка, в соот-
ветствии с которым энтропия идеального кристалла при нуле градусов
Кельвина равна нулю. Строго говоря, постулат этот не является тако-
вым в чистом виде, так как следует из соотношения Больцмана (2.4).
В действительности бездефектных кристаллов не существует. Опре-
деляется это термодинамической выгодой наличия в них микро-
примесей. Кроме того, каждой температуре соответствует своя впол-
не определенная равновесная концентрация точечных дефектов,
понижающаяся с уменьшением температуры. Однако при глубоком
охлаждении перескоки атомов становятся заторможенными и кон-
центрация дефектов при этом сохраняется постоянной, превышая
равновесную. В то же время вклад таких дефектов в энтропию реаль-
ных кристаллов чистых веществ относительно мал и постулат Планка
оказывается для них достаточно хорошим приближением.
Нагревание кристалла приводит к увеличению амплитуды коле-
баний составляющих его частиц, появляется и реализуется возмож-
ность образования дефектных центров - вакантных узлов кристалли-
ческой решетки и/или нахождения части атомов в не присущих им
позициях. При этом, естественно, энтропия кристалла возрастает
прежде всего за счет распределения частиц по колебательным уровням
энергии. Рост ее происходит непрерывно (рис. 2.3) вплоть до точки
плавления. Здесь энтропия вещества увеличивается скачкообразно
ввиду дискретного увеличения степени разупорядоченности частиц.
Далее с увеличением температуры опять возрастает амплитуда ко-
лебаний, энтропия непрерывно повышается вплоть до перехода веще-
ства в газообразное состояние. После очередного скачка рост ее опять
происходит непрерывно.
Постулат Планка дает возможность рассчитать абсолютные зна-
чения энтропии для различных веществ. Проведем, например, расчет
мольной энтропии для этилена при постоянном давлении, исходя из
экспериментальных данных. Для этого необходимо знать теплоту
обратимого процесса его нагревания.
34
Глава 2
Рис.2.3. Зависимости теплоемкости (л), отношения теплоемкости к температуре (б)
и энтропии этилена (в) от температуры
Тогда S(T2)=S(T)+$dQo6p/T. (2.7)
Из предыдущее™ раздела следует, что при постоянном давлении
^20бр - dH = CdT. До 104 К этилен представляет собой кристалл,
величины теплоемкости которого сравнительно надежно измерены
Второй закон термодинамики
35
вЫШе 20 К. При более низких температурах величина ее эк-
страполируется с учетом закона кТ •’ (рис. 2.3а). Таким образом,
изменение энтропии при малом изменении температуры равно
(С / T)dT. Интегрируя эту величину до 104 К с начальным значением,
равным нулю, получаем:
Д5 - J (Ср! T)dT-= 52.3 Дж/моль К . (2.8)
При 104 К протекает процесс плавления этилена. Кристаллы его
формируются только за счет слабого ван-дер-ваальсова взаимодей-
ствия, и теплота плавления их сравнительно невелика - 3.36 кДж/моль.
Поскольку процесс этот протекает при постоянной температуре, эн-
тропия его составляет
д£ = ЬН!Т = 3360/104 = 32.3 Дж/моль К .
(2.9)
Далее следует процесс нагревания жидкости до 169 К, мало чем
отличающийся от аналогичного для твердого тела. Для него
Д5 = f (С/ T)dT = 33.2 Дж/моль К
При 169 К этилен кипит и переходит в газовую фазу. Прирост
энтропии в ходе этого испарения максимален и составляет
Д5 = AH IT = 13590 / 169 = 80.4 Дж/моль К .
Наконец, происходит нагревание газа при постоянном давлении:
AS = J (С / T)dT = 25.7 Дж/моль К .
Таким образом, величина стандартной энтропии этилена при
298 К должна равняться сумме всех приведенных величин и составля-
ет 223.9 Дж/моль К.
Следует отметить некоторые закономерности, характеризующие
величины энтропии различных веществ. Степень беспорядка жидко-
стей в ходе испарения меняется на сравнительно близкие величины.
Эта закономерность нашла отражение в эмпирическом правиле Тру-
тона: энтропия испарения при температуре кипения для слабо ассо-
циированных жидкостей составляет около 90 Дж/моль К. В случае
образования ассоциатов в газовой фазе энтропия кипения понижает-
я- Например, для уксусной кислоты, образующей пары, эя о значение
составляет 63 Дж/моль К. В случае же ассоциации жидкости с обра-
зованием пространственной сетки водородных связей эта величина
возрастает. Для воды, например, она составляет 109 Дж/моль К. В то
же время для подавляющего большинства жидкостей с помощью пра-
вила Трутона можно оценивать теплоту испарения по известной
температуре кипения. Теплоемкость газов, как правило, сравнитель-
иевелика. Поэтому во многих случаях для относительно узкого
" ’ ала температур ее принимают величиной постоянной. Тогда
36
Глава 2
соотношение 2.8 преобразуется в
AS = J (С IT)dT = Ср f (1 /T)dT= Ср In (Т2 /Т_). (2.10)
Изменение энтропии системы в ходе некоторого химического
процесса можно вычислить так же, как и в случае энтальпии, - по
разности энтропий продуктов и исходных веществ с соответств^.
ющими коэффициентами. Например для реакции
N, + О, = 2NO (2.U)
величина KS составляет 2-211.3 - 192.1 - 205.7 = 24.8 Дж/моль К. Для
реакции
2Н, + О, - 2Н2О(г) (2.12)
AS = 2'189.3 - 205.7 - 2-131.0 = -99.1 Дж/моль К. Если же продуктом
является жидкая вода,
2Н, + О2 = 2Н,О(ж) (2.13)
AS = 2-70.3 - 205.7 - 2-131.0 = -327.1 Дж/моль К.
Поскольку объем газа примерно на три порядка больше, нежели
объем конденсированных фаз, и энтропия газов существенно
превышает энтропию жидкостей и твердых тел, то энтропия
произвольной реакции в первую очередь определяется изменением
числа газообразных частиц в ходе процесса. Именно поэтому
изменение энтропии в ходе реакции, при которой число их не меняет-
ся (2.11), близко к нулю. Для реакции (2.12) число молекул 1аза
понижается на единицу, и ее энтропия характеризуется существенным
отрицательным значением, приближающимся к 100 Дж/моль К.
Наконец, для реакции, где их число меняется на три (2.13), измене-
ние энтропии значительно больше. Аналогичное заключение можно
сделать и для процессов с участием твердых и газообразных веществ,
например, процесс термического разложения карбоната кальция,
происходящий с выделением двуокиси углерода, сопровождается
существенным возрастанием энтропии.
Поскольку большинство газов при сравнительно невысоких
давлениях при комнатной и повышенных температурах близки к
идеальным, полезно уметь рассчитывать изменение энтропии
идеального газа в процессе нагревания. Пусть моль газа был нагрет
от температуры 1\ до Т, , расширившись при этом от объема Е, до Vr
Сообщенное ему количество теплоты на каждом малом участке ухо-
дит на увеличение внутренней энергии CvdT и совершение работы
расширения RTdV! V. Соответственно суммарное изменение энтропии
при этом составит:
Второй закон термодинамика
37
Д5= /С, dTIT + JA dV IV - Cv In (T,IT) + Д In (V, IГ,) . (2.14)
По сходным с ранее рассмотренными причинам энтропия
возрастает при растворении веществ. Так, энтропия растворения
хлорида натрия составляет:
S(Na+p.p) + S(C1 р.р) - S(NaCl) = 60.2+55.2-72.4 = 43 Дж/моль-К.
Теперь следует вернуться к определению энтропии неравновесного
процесса, например неравновесного расширения газа против
меньшего внешнего давления, сопровождавшегося нагреванием
системы. Так как энтропия является функцией состояния, то изменение
ее зависит лишь от начального и конечного состояний системы и будет
одинаково для всех путей перехода между ними, включая обрати-
мый. Тогда тот же процесс мы можем разбить на два равновесных -
равновесное расширение газа и нагревание его при постоянном
объеме. Суммарное изменение энтропии может быть оценено по
соотношению (2.14), Заметим, что в ходе неравновесных процес-
сов выделяемая теплота всегда больше, а совершаемая работа, со-
ответственно, меньше, чем для равновесных. Поэтому в их ходе уве-
личение энтропии больше, чем при равновесных процессах.
Пусть в результате некоторого процесса система отдает внешней
среде некоторое количество теплоты Q. Поскольку ресурсы последней
безграничны, то температура ее остается постоянной. Тогда внешняя
среда увеличивает свою энтропию на:
bS = Q!T. (2.15)
Эта величина положительна. Заметим, что совокупность вы-
бранного объекта с окружающей средой может рассматриваться в
качестве изолированной системы. Соответственно, энтропия от-
дельной ее части может понижаться за счет повышения для другой.
Именно так, например, и происходит работа холодильников, осу-
ществляющих передачу теплоты окружающей среде за счет нагрева-
ния последней. Подчеркнем еще раз, что положительное изменение
энтропии является признаком самопроизвольного протекания про-
месса лишь в изолированной системе.
Задачи и вопросы
1- Два металлических шара массой по 10 г двигались навстречу
дРУг другу со скоростью 20 м/с каждый. Определите изменение эн-
тР°пии данной системы после абсолютно неупругого столкновения.
38
Глава 2
2. Стальная пластина массой 10 кг с исходной температурой 99°С
была помещена в сосуд через который пропускали ток пара, полу-
ченного при кипении воды (Р = 1 атм). Определите каково изменение
энтропии пластины и системы в целом после окончания процесса
конденсации, если теплоемкость железа - 27 Дж/моль К, а теплота
испарения воды - 40.3 кДж/моль.
3. Серебряное кольцо массой 20г нагрели от температуры 930 до
1000°С. Определите энтропию этого процесса, если теплоемкость
твердого серебра - 295 Дж/кг К, жидкого - 305 Дж/кг К, температура
плавления серебра - 960°С, а удельная теплота плавления - 970 Дж/г.
4. В некотором сосуде емкостью 10 л, разделенном перегородкой
на две равные части находилось в одной — 4 г водорода и в другой -
32 г кислорода соответственно. Определите изменение энтропии
системы после изъятия перегородки.
5. Стандартные энтропии аммиака, хлористого водорода и
хлорида аммония составляют соответственно 186,193 и 96 Дж/моль К.
Рассчитайте стандартную энтропию процесса образования хлорида
аммония из приведенных веществ при комнатной температуре. Будет
ли данный процесс протекать самопроизвольно в изолированной
системе? Объясните полученный результат.
6. Принимая теплоемкость воды постоянной и равной
75 Дж/моль К, рассчитайте изменение энтропии в чашке с 90 мл чая с
исходной температурой 90°С после приливания в нее 10 мл холод-
ной воды (Т = 20°С).
7. Как изменится энтропия той же системы, если вместо холодной
воды использовать кубик льда той же массы при температуре 0°С?
8. При 298 К стандартная мольная энтропия аргона составляет
154.7 Дж/моль К. Рассчитайте энтропию его при 400 К.
9. Стандартная энтропия азота при 298 К составляет
199.9 Дж/моль К. Рассчитайте энтропию его при 400 К, если зави-
симость его теплоемкости от температуры задается соотношением:
Ср = 28.58 + 0.0037Т -50000,Т2, Дж/моль К.
10. Принимая теплоемкость твердого галлия равной 26 Дж/моль К,
а жидкого - 24 Дж/моль К, оцените изменение энтропии в процессе
кристаллизации переохлажденной жидкости при 10°С, если теплота
плавления его при 30°С составляет 5.59 кДж/моль.
Глава 3
Энергии Гиббса и Гельмгольца.
Химическое равновесие
3.1. Свободные энергии Гиббса и Гельмгольца
К сожалению, второй закон термодинамики, утверждающий,
что самопроизвольно должны протекать лишь процессы, сопро-
вождающиеся увеличением энтропии, выполняется только для
изолированных систем. Поэтому он не может адекватно исполь-
зоваться для произвольного случая. Действительно, процесс раз-
ложения карбоната кальция при любых приемлемых условиях
сопровождается увеличением энтропии, поскольку в результате его
образуется газообразный продукт. С другой стороны, может воз-
никнуть законное недоумение: а почему же в природе существуют
огромные количества меловых отложений, которые вовсе не спешат
увеличить свою энтропию. Дело тут вовсе не в кинетической затор-
моженности процесса разложения. Само равновесное давление
двуокиси углерода над СаСО3, как известно, является недостаточ-
ным для того, чтобы превысить аналогичную величину в атмосфере.
Если рассмотреть кусок мела в совокупности с окружающей средой,
что даст нам возможность говорить о такой большой системе в це-
лом как об изолированной, его поведение станет вполне понятным.
Помочь разрешить возникающие проблемы помогает объединение
первого и второго законов термодинамики.
Пусть внутри некоторой системы при постоянном давлении и
тепловом балансе с окружающей средой протекает процесс, сопро-
вождающийся изменением энтропии AS и энтальпии ДЯ. Тогда
теплота этого процесса, численно равная -ДН, будет рассеиваться в
окружающем пространстве, энтропия которого изменится на -AHIT.
ыберем в качестве новой термодинамической системы совокуп-
ность исходной системы со значительной частью окружающего
пространства (рис 3.1), в котором рассеивается выделившаяся тепло-
и компенсируется изменение объема. Новая система может рас-
сматриваться как изолированная, и изменение энтропии в ней будет
40
Глава 3
аддитивно складываться из энтропии исходной системы и окружаю-
щей среды:
(3.1)
Рис.3.1. Схема преобразования некоторой системы в изолированную
Критерием самопроизвольности протекания процесса в данной
изолированной системе будет выполнение неравенства:
Д^^ДХ-ДЯ/ТХ). (3.2)
или для удобства, домножив это соотношение на однозначно
положительную величину Т, получим.
ТД5-Д//>0 (3.3)
Аналогично, легко заключить, что для процессов, протекающих
при постоянном объеме, критерием самопроизвольного протекания
будет выполнение неравенства:
TkS-bU>0 (3.4)
Вид полученного соотношения наводит на мысль, что возмож-
ность самопроизвольного протекания процесса определяется про-
тивоборством двух тенденций: стремлением системы к минимальной
энергии (Д/7) и максимальному беспорядку (Т Д£). Это весьма близ-
ко к истине, хотя предпочтительнее расставить акценты несколько
по-другому. Изменение энтропии системы - это ее стремление к
увеличению беспорядка, а изменение энтальпии - аналогичное свой-
ство окружающей среды. Поэтому, если процесс протекает с малым
выделением тепла или при высоких температурах - преобладает
стремление системы к увеличению собственной энтропии, а при зна-
чительном тепловыделении и низких температурах - к увеличению
энтропии окружающей среды.
Энергии Гиббса и Гельмгольца. Химическое равновесие 41
К подобным результатам можно прийти и несколько другим
путем Вернемся к формулировке первого закона, свидетельствующей
о том, что изменение внутренней энергии системы в ходе процесса
равняется разности между сообщенной ей теплотой и совершенной
ею работой. Пусть в рассматриваемой системе совершается работа
расширения, которая сопровождает работу по осуществлению
химического процесса (Лхну) Если последняя величина оказывается
положительной, то химический процесс принципиально возможен.
В таком случае первый закон термодинамики можно переписать в
виде:
Д|7=е^ДГ-<„. (3.5)
Из второго закона термодинамики следует, что для обратимого
процесса теплоту можно представить в виде произведения изменения
энтропии на температуру. Тогда:
-Лхим=Д^-ТД5+рДЕ. (3.6)
Полученная величина характеризует ту часть внутренней энергии,
которую можно превратить в химическую работу при постоянных
давлении и температуре, что является самым ценным с точки зрения
большинства экспериментаторов. Она получила название свобод-
ной энергии Гиббса (или изобарно-изотермического потенциала) и
обозначается буквой G. Напротив, величина Т AS характеризуют
энергию связанную, которую невозможно перевести в работу нико-
им образом. Чем меньше величина свободной энергии Гиббса, тем
большую работу может совершить система и тем выше движущая сила
процесса. В общем случае эту функцию представляют как
G = U-TS+pV = H-TS (3.7)
или
AG = AH- TAS-SAT. (3.8)
Очевидно, что для процессов, происходящих при постоянной
температуре (что и предусматривает тепловое равновесие с окру-
жающей средой), последнее слагаемое равно нулю и
&G = AH-TAS. (3.9)
Критерием самопроизвольного протекания процесса будет являться
отрицательное значение изменения энергии Гиббса. Аналогично при
постоянном объеме пользуются свободной энергией Гельмгольца - F
'Из°хорно-изотермический потенциал):
F=U-TS. (3.10)
42
Глава 3
Если процесс протекает в конденсированной фазе или, в силу
некоторых причин, объем системы остается постоянным, изменения
свободной энергии Гиббса и Гельмгольца практически совпадают.
Важность использования этих функций при рассмотрении раз-
личного рода химических и физико-химических процессов подчер-
кивает наличие эндотермических реакций, которые сопровождаются
значительным увеличением энтропии. За счет этого величина сво-
бодной энергии Гиббса оказывается отрицательной, что и определяет
самопроизвольность таких процессов. Примером может служить ра-
створение ряда солей, сопровождающееся поглощением тепла.
Таким образом, при постоянных значениях температуры и давле-
ния процесс будет протекать в сторону понижения свободной энер-
гии Гиббса. При этом неизбежно должен наступить такой момент,
при котором ее величинадостигает минимального значения. При этом
производная изобарно-изотермического потенциала по степени пре-
вращения обращается в ноль, а любые колебания состава приводят к
его повышению, что будет являться движущей силой, возвращающей
систему в исходное состояние (рис 3.2). Эта ситуация аналогична та-
ковой в механике для шарика, попавшего в лунку, любое смещение
которого приводит к появлению сил, стремящихся вернуть его назад.
Как и в механике, это состояние называется положением равновесия.
Отметим, что принципиально возможны два случая, (а) при которых
равновесие наступает при частичном завершении реакции (например.
2NO, <-> N,O4) или (б) при полном превращении исходных веществ в
продукты (3Fe +20, -» Fe3O4).
Рис.3.2. Зависимость свободной энергии Гиббса от степени превращения для обратимых
(й) и необратимых (б) процессов
Энергии Гиббса и Гельмгольца. Химическое равновесие 43
Для необратимых процессов, как отмечалось ранее, Q > Т AS,
оТКуда AG = AU- Т AS + р АГ < 0, поскольку часть внутренней энер-
гии расходуется на выделение теплоты. Близкое к равновесному про-
текание процесса возможно, когда система совершает работу против
сил, уравновешивающих ее воздействие. Такой процесс может про-
текать. например, в ходе электролиза раствора при напряжении,
близком к равновесному. Если же приложенное напряжение выше
необходимого, это не приведет к дополнительному выделению ве-
щества, поскольку его количество определяется лишь перенесенным
зарядом. Избыточная энергия неизбежно превратится в теплоту.
Аналогичный пример для процесса расширения газа был рассмотрен
нами в предыдущем разделе.
Таблица 3.1
Условия самопроизвольного протекания процесса для различных систем
Условие достижения равновесия Условие самопроизвольного протекания процесса
Изолированная система Д5 = 0 максимум S Д5>0
V, Т- const ДР = 0 минимум ДР ДР<0
р,Т = const Д(7 = 0 минимум AG Д6 <0
В таблице 3.1 сопоставлены условия равновесия и самопроиз-
вольного протекания процесса в системах различного рода. Естест-
венно, что изобарно-изотермический и изохорно-изотермический
потенциалы используются значительно чаще, чем функция энтропии,
поскольку позволяют оперировать лишь процессами, происходящи-
ми непосредственно в исследуемой системе, и абстрагироваться от
внешней среды.
3.2. Расчет свободных энергий Гиббса и Гельмгольца
для химических процессов
Свободные энергии Гиббса и Гельмгольца являются функциями
состояния, то есть величины их определяются лишь начальным и ко-
Нецным состоянием системы. Это вполне естественно, поскольку они
представляют собой суммы одной функции состояния и произведения
44
Глава 3
другой на величину интенсивного параметра системы. Для ин-
дивидуальных веществ за стандартную энергию Гиббса принимает-
ся аналогичная величина для реакции их образования из простых
веществ, которую можно рассчитать, зная стандартные энтальпии и
энтропии всех участников процесса. Для простых веществ в стан-
дартном состоянии величины ДС и ДГ принимаются равными нулю.
Для облегчения термодинамических расчетов стандартные значения
этих параметров для различных химических соединений сведены в
таблицы, хотя большинство из них содержат лишь значения сво-
бодной энергии Гиббса, как наиболее употребимые, поскольку
большинство процессов протекает при постоянном давлении.
Расчет изменения AG и ДГдля различных химических процессов
можно провести двояким образом. Наиболее просто воспользоваться
способом, аналогичным применяемому для расчетов стандартных
изменений энтропии и энтальпии: вычитая из суммы стандартных
величин Д(7 и Д77 продуктов сумму аналогичных значений для ис-
ходных веществ с соответствующими коэффициентами. Так, напри-
мер, для реакции образования воды при 298 К и давлении, равном
1 атмосфере,
Д(7 = ДС(Н,О)-ДС(Н2)-1/2ДС(О2) = -237.2-0-0 = -237.2 кДж/моль.
По второму способу вначале из табличных данных вычисляют
ДУ/ и Д5* процесса, а, исходя из них, - величину ДС. Способ этот не-
сколько длиннее, но часто употребляется при отсутствии табличных
данных для свободных энергий Гиббса при нестандартной темпера-
туре. Кроме того, он используется и для термодинамических расчетов
по экспериментальным данным.
Для той же реакции синтеза воды из простых веществ
Д2/ = -285.8 кДж/моль,
\S = 70 - 0.5-205 -130.5 = -163 Дж/моль К,
ДС = -285800 - 298 (-163) = -237.2 кДж/моль.
Полученные результаты эквивалентны и свидетельствуют о
возможности протекания процесса синтеза воды из водорода и
кислорода. В то же время, мы знаем, что водород практически не
окисляется кислородом воздуха без нагревания. Понятно, что это
происходит не из-за того, что он “не хочет”, а из-за того, что “не мо-
жет”. То есть реакция, действительно, возможна, но у молекул по-
просту не хватает кинетической энергии для того, чтобы сблизиться
на расстояние, достаточное для эффективного взаимодействия. С
другой стороны, при бесконечно долгом сосуществовании молекулы
Энергии Гиббса и Гельмгольца. Химическое равновесие
45
одорода и кислорода в результате многочисленных флюктуаций
изредка будут получать достаточную энергию и, рано или поздно,
описанный процесс придет к успешному завершению, как это и пред-
сказывает термодинамика.
При вычислении величины свободной энергии Гиббса при
нестандартных условиях обычно предполагают, что величины из-
менения энтропии и энтальпии слабо зависят от температуры. Тогда
изменение Дб можно описать соотношением (3.9). Рассмотрим со-
четание величин Mi и Д5 с различными знаками (рис. 3.3, табл. 3.2).
Рис.3.3. Зависимость свободной энергии Гиббса химических реакций от температуры
Таблица 3.2
Возможность самопроизвольно! о протекания химических реакций и достижения
состояния равновесия для них
Возможность достижения равновесия Возможность протекания реакции
ЬН>0, &SX) возможно возможна при высоких Т
ЬНХ), &s<o невозможно невозможна при любых Т
&Н<0, &SX) невозможно возможна при любых Т
ЪН<0, &S<0 возможно возможна при низких Т
Очевидно, что реакции, характеризующиеся положительным
Значением ДЯ и отрицательным - ДД', невозможны ни при каких
Условиях, так как их энергия Гиббса всегда больше нуля. Напротив,
46
Глава 3
если величина АЯ отрицательна, a AS - положительна, реакция
должна протекать при любых условиях при отсутствии кинетических
затруднений. Если энтальпия и энтропия имеют одинаковый знак
то возможность самопроизвольного протекания реакции зависит от
температуры ее проведения. Причем, если обе эти величины отри-
цательны, протеканию прямого процесса способствует понижение
температуры, и наоборот - если они положительны.
Как видно из данных предыдущих разделов, энтальпия и энтропия
являются функциями температуры. И, поэтому, при пересчете их
величин из известных при некоторой температуре можно восполь-
зоваться разностью теплоемкостей продуктов реакции и исходных
веществ, взятых с соответствующими коэффициентами. Пусть в
простейшем случае она описывается зависимостью:
ЬСр=а + ЬТ (3.11)
Тогда АЯ, = АЯ, + f(o + b Т) dT = АЯ, + а\ dT + b\ TdT =
= АЯ, + а(Тг-Т,) + Ь/2(Г- Г), (3.12)
AS, = AS, + fra + ЬТ)/Т dT = AS, + «J dT/T + b\ dT =
= AS, 0(7,- T,) +flln(T,/T|). (3.13)
AG, = АЯ, + п(Т,- 71,) + b/2(T,2-Tf)-Т,[AS, + Ь(Т,-Т}) +
+ a\n(TJTJ] = АЯ, + а(Т,-Т)+Ь12(Т2-Т2) - T^S{ -
-bTAT.-T^-aTMTJT). (3 14)
При использовании более сложных форм зависимости тепло-
емкости от температуры получатся несколько более громоздкие со-
отношения.
Подчеркнем, что все перечисленные критерии свидетельствуют
лишь о принципиальной возможности осуществления процесса, а не о
реальном протекании его в заданных условиях.
А может ли протекать термодинамически запрещенный процесс?
Оказывается может, но не самопроизвольно, а под некоторым внеш-
ним воздействием, как, например, процесс фотосинтеза, идущий за
счет поглощения солнечной энергии. Возможны также процессы, про-
текающие с помощью сопряженных реакций, характеризующихся
отрицательными величинами AG. В качестве примера можно приве-
сти термодинамически запрещенный синтез сахарозы из глюкозы и
фруктозы в живых организмах (AG=21 кДж/моль). Он происходит с
помощью самопроизвольного гидролиза аденозинтрифосфорнои
Энергии Гиббса и Гельмгольца. Химическое равновесие
47
кислоты
ЛГФ + Н.О =ЛДФ + Н РО4 Д(7 =-50 кДж (3.15)
через последовательную цепочку превращений:
дТФ + глюкоза = глюкоза-1-фосфат + АДФ kG = -29 кДж (3.16)
Глюкозо-Гфосфат+фруктоза-сахароза+^ДГ)4 AG=0 кДж (3.17)
Каждая из этих реакций, по крайней мере, не запрещена, а
суммарный процесс может протекать самопроизвольно, хотя в
организме он еще и катализируется ферментами.
3.3. Химический потенциал
Ранее мы отмечали, что любой вид работы можно представить в
виде произведения некоторого интенсивного параметра на измене-
ние экстенсивного. Теперь зададимся вопросом: какие же факторы
определяют химическую работу системы? Представляется вполне есте-
ственным в качестве экстенсивного фактора выбрать количество
вещества, претерпевшего превращение в ходе реакции. Соответ-
ствующий же интенсивный параметр получил название химического
потенциала (ц). Таким образом, для реакции, протекающей при
постоянных давлении и температуре, можно записать:
<им = kG = kU-TkS+p ДГ = ц kn (3.18)
Если постоянными являются объем и температура:
-<им =AF = Д£7- Т kS = у kn. (3.19)
Продифференцировав выражение (3.18) по количеству /-го ком-
понента (/?.) получаем:
Ц^ДОЖ)^. (3.20)
Таким образом, химический потенциал некоторого компонента
системы характеризует изменение свободной энергии при добавлении
эгп°го компонента в систему при постоянных давлении, температуре и
количествах других веществ. При других условиях он может равнять-
ся м°льному изменению свободной энергии Гельмгольца, энтальпии
или внутренней энергии. Для индивидуального вещества химический
П^ен1^иал определяют как мольное изменение свободной энергии Гиб-
постоянных давлении и температуре.
в еРнемся к соотношению (3.18). Обратим внимание на то, что
ходе химического процесса происходит изменение количества
°льких веществ в соответствующих пропорциях. С другой
48
Глава 3
стороны, хорошо известно, что свободную энергию процесса можно
представить в виде:
AG = L(v.G;), (3.21)
где у,- стехиометрические коэффициенты реакции. Тогда при малом
изменении количества одного из веществ на dn., учитывая (3.20), мож-
но получить
dG.t = И, dn. (3.22)
или, поскольку изменения количества всех веществ можно связать с
dn. посредством стехиометрических коэффициентов:
dG =S (ц.д'и.) =dnkY (\Ldv), (3.23)
где vz - соотношение стехиометрических коэффициентов веществ / и
к. Продифференцировав выражение для свободной энергии Гиббса
по давлению, легко убедиться, что
dGIdp-V. (3.24)
Откуда для идеального газа можно найти следующую зависимость
свободной энергии Гиббса от давления:
G(p2) = G(p)+\Vdp = G(p) + j(nRT/p)dp = G(p})+nRT\n(p2/p}). (3.25)
Или для одного моля идеального газа:
Ц(?2) = Н(р,) + RT In (pjpj. (3.26)
Пользуясь этим выражением, можно описать химический потен-
циал газа, пользуясь стандартными величинами при давлении, рав-
ном одной атмосфере:
ц(р) = ц(1) + АТ1п(/э/1) =ц° + ЯЛпЛ (3.27)
где р - давление газа, выраженное в атмосферах. Крайне важно
обратить внимание на то, что давление, стоящее под знаком лога-
рифма, реально является безразмерной величиной, поскольку отнесе-
но к некоторому стандартному. Это дает возможность варьировать
набор стандартных величин и, как будет показано далее, использовать
вместо давления некоторые другие параметры системы.
Для реальных газов уравнение Клапейрона-Менделеева является
лишь некоторым приближением, хорошо работающим при низких
давлениях. Поэтому величина интеграла будет отлична от рассчи-
танной выше. Для сохранения формы уравнения (3.27) приходится
прибегать к замене давления некоторой другой величиной, назы-
ваемой летучестью (/). Таким образом, для реального газа получаем
уравнение:
ц(р) +ЯТ1п/ (3.28)
Энергии Гиббса и Гельмгольца. Химическое равновесие
49
Поскольку летучесть затруднительно измерить напрямую, ее все
же ‘“привязывают” к давлению соотношением:
f=yp, (3.29)
где у _ коэффициент летучести. Тогда уравнение (3.27) можно
переписать как
и(р) = ц° + 7?Т1п у + ATlnp. (3.30)
Величину коэффициента летучести можно получить интегри-
рованием экспериментальной зависимости объема от давления,
учитывая, что при низких давлениях летучесть приближается к дав-
лению и, соответственно, к нулю.
КТ]п(М\)=Мр. (3.31)
Аналогично за меру количества вещества можно выбрать и мо-
лярную концентрацию. В случае идеального газа из уравнения
Клапейрона-Менделеева имеем:
р = (m/M)RT/V = CRT (3.32)
Подстановка этого выражения в соотношение (3.26) приводит к:
ц(С2)=ц(С1) + ЯТ1п(С2/С1). (3.33)
Или, приняв за стандартную величину единичную концентрацию,
получаем:
Н(С) = ц° + ЯПпС. (3.34)
Заметим, что при этом будет отличаться от таковой при
единичном давлении на некоторую постоянную величину, равную
^7’1п(А7). Для того чтобы избежать связанных с этим осложнений,
для газов, как правило, в качестве стандартного принимают еди-
ничное давление, а для растворов - единичную концентрацию. В дан-
ном случае при переходе от идеальных растворов к реальным функцию
летучести выполняет активность, а коэффициента летучести - коэф-
фициент активности.
3-4. Равновесие в реакциях, протекающих в газовой фазе
Введем в некоторую систему, в которой поддерживаются по-
Ст°янные температура и давление, оксид азота и хлор в мольном
Со°тношении 2:1. При этом будет происходить образование хлорида
Нитрозила.
2NO + С12 <-> 2NOC1 . (3.35)
50
Глава 3
Однако эта реакция будет протекать далеко не до конца, достигая
равновесия при некоторой промежуточной степени превращения, что
особенно ярко проявляется при повышенных температурах, К тому
же состоянию можно прийти и исходя из эквивалентного количест-
ва чистого хлорида нитрозила. Очевидно, что прямая реакция
характеризуется отрицательной величиной изменения энтропии,
поскольку приводит к уменьшению числа частиц, находящихся в
газовой фазе. Однако, поскольку она все же протекает с определен-
ным выходом, величина А?/ для нее также должна быть отрицатель-
на. Значения этих величин составляют Д/7 = -77.4 кДж/моль и
AS =-118 Дж/моль К. Если при комнатной температуре степень
превращения достигает 99.98%, то при 800 К - всего 20%. Таким
образом, изменяя температуру процесса, мы изменяем и равновесные
концентрации компонентов. Положение равновесия можно сместить
и путем прибавления одного из участвующих в процессе веществ. Та-
кие реакции также называются обратимыми - здесь прослеживается
аналогия с обратимыми процессами, которые можно осуществить в
ту и другую сторону, изменив внешние условия. В то же время разница
между этими понятиями огромная, поскольку обратимая реакция не
обязательно должна протекать в равновесных условиях.
Зададимся вопросом, насколько велико число обратимых реакций.
С одной стороны, представляется, что число их не столь значительно.
При комнатной температуре большинство процессов протекают
практически до конца, если на их пути не возникает непреодолимых
барьеров, как в случае образования оксидной пленки при окислении
алюминия. С другой стороны, повышая температуру можно прийти
к таким условиям, в которых даже хлорид натрия будет разлагаться
на элементы. С этой точки зрения исключением, скорее, следует счи-
тать необратимые реакции.
Из ранее изложенного материала следует, что положение равно-
весия в системе реализуется при равенстве нулю изменения стан-
дартной энергии Гиббса. Так, для реакции
аА + ЬВ = сС +dD (3.36)
условие равновесия можно представить в виде равенства нулю ал-
гебраической суммы мольных значений свободных энергий Гиббса
или химических потенциалов:
ДО = - G„ = cGc + dGD - aG„ -bGB = 0. (3-37)
Выразим химические потенциалы реагентов через стандартные с
учетом давления всех компонентов с помощью соотношения (3.27):
И,- = Ц,° + АПп^..
Энергии Гиббса и Гельмгольца. Химическое равновесие
51
Тогда Дб = c[ic + ttyD - Z>gB =
= сцс° + Фо°' ЯКЛ V + RT(C [пРс + d }nPD ’ а 1пРл - ь 1п^) =
= д£0 + RT In [(рсУ (pD)d / (рАУ (рвУ]. (3.38)
Подученное выражение представляет одно из основных следствий
из законов химической термодинамики. Оно называется уравнением
изотермы химической реакции или изотермой Вант-Гоффа. Величина
в квадратных скобках есть ни что иное, как константа химического
равновесия для реакции между газами, выраженная в традиционной
форме. Поскольку при равновесии Дб = 0, то эта константа может
быть легко вычислена из соотношения
AG ° + RT In [(рсГ (Р„У I (Р„У (РВПр..„ = О
In [(/>,)'(Р„У /(РАУ (рП™ =\nKp = -^G’IRT (3.39)
ИЛИ
(рсУ (р0У/(рАУ (рвУ =К = ехр(-Д(?°/ RT) (3.40)
Таким образом, константу равновесия можно рассчитать из
величины энергии Гиббса рассматриваемого процесса. С помощью
соотношения (3.38) также можно вычислить энергию Гиббса в любой
точке химического процесса, определив таким образом направление
его самопроизвольного протекания и максимально возможную
химическую работу. Действительно, заменим в уравнении (3.38)
величину ДС °, выразив ее через константу равновесия.
ДС = RT {In [фсУ (р,УКрАУ (рвУ] - In Кр} (3.41)
Следуя этому уравнению, легко вычислить свободную энергию
химической реакции при любом соотношении компонентов, не
предпринимая дополнительных термодинамических расчетов. Особую
Ценность это имеет, если для части реагентов величины свободных
энергий неизвестны.
Константа равновесия реакций, происходящих в газовой фазе,
имеет размерность, выраженную в единицах давления в степени,
Равной разности числа молекул в ее правой и левой части (Д/?). Вопрос
заключается лишь в том, в каких же единицах выражено это давление.
Ри Дл, отличном от нуля, это существенно скажется на результатах
Расчетов равновесных концентраций, выполненных с использованием
п°лУченной константы. Для решения этого вопроса вернемся к
ИсХодному соотношению для расчета химического потенциала:
К =К° + ЯТ1пр..
52
Глава 3
Реально величина, стоящая под знаком логарифма, представая -
собой отношение двух давлений - текущего и стандартного, которое
в соотношении опущено, поскольку равно одной атмосфере. Таким
образом, проблема выбора решается однозначно, - для процес-
сов, протекающих в газовой фазе, термодинамическая константа
равновесия должна выражаться в атмосферах. При выборе иной раз-
мерности давления следует выбирать и другую величину коэффици-
ента R.
Используя полученные соотношения, легко заключить, например,
что при смешении всех исходных веществ и продуктов реакции (3.35)
с единичными парциальными давлениями при комнатной температу-
ре процесс будет протекать в прямом, а при 800 К - в обратном на-
правлении.
3.5. Равновесие в реакциях, протекающих в растворах
Для расчета констант равновесия реакций, протекающих в
растворе можно аналогичным образом воспользоваться зави-
симостью химического потенциала от концентрации (3.34) или от
активности:
=Ц(° + П1п С.,
ц. = ц.° + Я Tin а.. (3.42)
Нетрудно убедиться, что при этом будут получены уравнения,
аналогичные (3.40):
(СсУ (CDy / (САУ (СВУ = exp(-AG°/RT) = Кс, (3.43)
(асУ (aD)d / (аАУ (авУ = ехр(-ДС°/ RT) = Ка. (3.44)
Соотношение между этими величинами, в свою очередь, можно
получить, выразив активности компонентов реакции через коэффи-
циенты активности и концентрации:
«,=У,С. (3.45)
Откуда получаем:
к. = (acY (aDY I (аЛ)° (б„)‘ = (yQ/ (yQ/ / (yQ/ (yQ,‘ =
= [(Ccy (C„Y / (CJ« (CB)‘] [(yf)‘ (y,)' / (yj- (yB)‘j =
= *Д(ус)Чу0У/(ув)“(7г)Т 0.46)
Представляется естественным, что для термодинамических ра-
счетов следует пользоваться величиной константы равновесия,
Энергии Гиббса и Гельмгольца. Химическое равновесие
53
сраженной через активности. Ее можно определить, непосредственно
измеряя активности всех компонентов после установления равнове-
сия или пользуясь термодинамическими данными. Выражение (3.43)
является не вполне строгим С другой стороны, химикам чаще всего
приходится работать с растворами, для которых легко определяемы-
ми величинами являются именно концентрации. При отсутствии дан-
ных о коэффициентах активности реагентов можно получить лишь
концентрационную константу равновесия, которая может оказаться
вовсе не постоянной. При этом следует четко понимать, что указанная
связь ее с термодинамическими параметрами справедлива лишь для
весьма разбавленных растворов, для которых величина коэффициента
активности стремится к единице. Зачастую полагают, что отклонения
от единицы коэффициентов активности продуктов и исходных ве-
ществ взаимно компенсируют друг друга, то есть, что [(yc)f(yD)rf /
/ (ул)°(уб)Л] ® 1- В общем случае это неверно, хотя в ряде случаев
приводит к весьма правдоподобным результатам.
Зачастую исследователям приходится работать в четко ограни-
ченном узком интервале концентраций. При этом коэффициенты
активности реагентов можно считать величинами постоянными и для
текущих расчетов вполне можно пользоваться концентрационной
константой равновесия, хотя, не определив величину параметра
^c)f(yD)rf/(y^)a(yfi)/’], не следует задаваться целью определения термо-
динамических функций для процесса.
3.6. Равновесие в гетерогенных системах
При нахождении системы в состоянии равновесия должно вы-
полняться условие равенства всех интенсивных параметров во всех
присутствующих фазах. Это же с полным основанием можно сказать
0 величинах химических потенциалов всех участников реакции.
аким образом, химический потенциал ионов бария в насыщенном
Растворе BaSO4 должен равняться его потенциалу в равновесном
BaSO4« Ва2*(р-р) + SO4’- (р-р). (3.47)
н этом реальное различие концентраций указанных ионов в
Их Может достигать десяти и более порядков! Столь существенное
ба1Си71Н°Шение обусловлено тем, что в кристаллической фазе ионы
nvo, значительно прочнее связаны в решетке и величина ц.0 для них
0Казывается очень низкой.
54
Гпава 3
Рассмотрим с этой точки зрения процесс разложения карбоната
кальция:
СаСО3 = СаО + СОД . (3.48)
В положении равновесия изменение энергии Гиббса для этой
реакции должно быть равно нулю. Карбонат кальция и его оксид
не образуют между собой твердых растворов. Таким образом, кон-
центрации этих веществ в области протекания реакции, а следо-
вательно и их химические потенциалы, не будут меняться вплоть до
полного исчезновения одного из компонентов и будут равны соо-
тветствующим стандартным величинам. С другой стороны, зависи-
мость химического потенциала углекислого газа от его парциального
давления описывается следующим соотношением:
Н = К° +ЯТ1прС02. (3.49)
Откуда для равновесия при атмосферном давлении можно
получить:
- ЦС02 + ЦСа0 - ЦСаСОз - НС02 + RT1 1П£>СО2 + ^СаО° ‘ ^СаСОз° “
= АС0 + Т?Г1п^СО2 (3.50)
или
РСО2= exp (-AG0/ RT). (3.51)
Таким образом, константой равновесия данного процесса будет
являться парциальное давление двуокиси углерода. В общем случае
для получения константы равновесия гетерогенного процесса,
протекающего с участием газов, следует учитывать лишь парциаль-
ные давления последних. Аналогичное заключение можно сделать и
для реакций в растворах, сопровождающихся выделением или
растворением осадка.
Теперь представляется целесообразным рассмотреть причину
того, почему до конца протекает реакция сгорания железа на воздухе
3Fe + 20, = Fe3O4 , (3.52)
несмотря на существенно отрицательную величину ее энтропии.
Взаимная нерастворимость железа и его оксидов определяет
независимость их химических потенциалов от глубины протекания
процесса. С другой стороны, равновесное давление кислорода над
этой смесью в широком интервале температур крайне мало ввиду
значительного экзотермического эффекта (AG<<0). Таким образом,
парциальное давление кислорода воздуха в разумном диапазоне
температур существенно превышает равновесное, что делает эту
реакцию необратимой.
Энергии Гиббса и Гельмгольца. Химическое равновесие 55
3.7. Факторы, влияющие на константу равновесия.
Принцип Ле-Шателье-Брауна
Величина константы равновесия в существенной мере определяет
степень превращения исходных веществ и выход реакции в целом. В
свою очередь она зависит от условий осуществления реакции. Поэто-
му важно четко оговорить круг факторов, влияющих на величину
константы и положение равновесия. Естественно, что изменение
содержания одного из компонентов в смеси автоматически приво-
дит к нарушению равновесия. При этом, как следует из уравнения
изотермы химической реакции (3.41):
AG = RT {In [(рс)‘ (pD)u / (рЛУ (рв)1'] - In Кр}.
добавление продуктов приводит к увеличению величины AG и.
соответственно, смещению равновесия в сторону образования ис-
ходных веществ. При отсутствии кинетических затруднений процесс
будет протекать до тех пор, пока выражение в квадратных скобках
не окажется равным константе равновесия. И, напротив, добавление
одного из исходных веществ будет способствовать дополнительному
образованию продуктов. Это является проявлением так называемого
принципа подвижного равновесия Ле-Шателье-Брауна. если на систему,
находящуюся в состоянии подвижного равновесия, оказывается внеш-
нее воздействие, то положение равновесия смещается в сторону,
противодействующую этому воздействию. Обратим внимание, что
изменение концентрации одного из компонентов, участвующих в
процессе, не влияет на саму величину константы равновесия.
С другой стороны, при повышении общего давления в системе от
до р2, во столько же раз повысится и парциальное давление каждого
из компонентов. При этом величина в квадратных скобках выражения
(4.41) изменится в (p,lp})c+M' - (pjp^ раз, где А/? - изменение числа
молекул газа в ходе реакции. Естественно, что сместится и положение
равновесия, причем это смещение будет тем заметнее, чем больше
величина А/?. Опять же, в соответствии с принципом Ле-Шателье-
ьрауна при Ал > 0 повышение давления будет способствовать сме-
щению равновесия в сторону образования исходных веществ, то
есть снижению давления в системе. Так, при синтезе аммиака, со-
пР°вождающемся уменьшением числа частиц вдвое, повышение
Щего давления будет способствовать существенному росту выхода
Продукта.
Такое представление весьма наглядно, хотя и не вполне строго,
дольку, в общем случае, соотношение (3.41) справедливо при
56
Глава 3
постоянном давлении и включает константу равновесия при по ,и.
янных р и Т. Для того чтобы вывести соотношение между конста! . .)fi
равновесия и суммарным давлением в системе более строго, най лм
изменение энергии Гиббса для реакции (3.36) при изменении давления
от д, до р,. Из уравнения (3.38) следует, что:
AG = AG0 + /?7Чп [(pcy(pD)d / (^)Ч^У’]
При изменении суммарного давления от р{ до р парциальное
давление каждого из веществ также изменится отр.до (p.pjp,). Откуда:
AG(p.) - ДС(р,) =
= AG" + RT In Кр-AG = -
-ЛГ1п((р,гУ(р,0)'/(р,яНр,в)''] =
= RTIn [(pjp,)’(pjp,)‘ I (pjpytpjp,)"]. (3.53)
Таким образом, если в системе наблюдалось равновесие при
давлении р}, то при давлении ^ величина ДС равна RT In (р,/р,)д* =
= Ди RT In (pjp). Именно на эту величину и следует изменить сво-
бодную энергию Гиббса для достижения равновесия. Откуда:
Кр2-Кр1 -(р, //>,)-. (3.54)
Таким образом, полученное нами ранее на основании прибли-
женных расчетов соотношение оказывается верным.
Другим фактором, очевидно влияющим на константу равновесия,
является температура. В первом приближении зависимость константы
равновесия от температуры можно определить, считая величины эн-
тальпии и энтропии реакции не зависящими от температуры. Такое
приближение весьма эффективно работает в сравнительно узком тем-
пературном диапазоне, в котором, кроме того, отсутствуют фазовые
превращения исходных веществ и продуктов реакции. Воспользуемся
уравнением (3.39), выразив в нем свободную энергию Гиббса через
энтальпию и энтропию:
In А* = - &GIRT = -(&H- T&S)/RT = -\H/RT + &SIR. (3.55)
График зависимости логарифма константы равновесия от 1/Д
таким образом, в ограниченном температурном интервале должен
выражаться прямой линией, тангенс угла наклона которой к оси
абсцисс равен -&HIR, а отрезок, отсекаемый на оси ординат, - &S/R
(рис. 3.4). Эта закономерность обычно достаточно хорошо воспро-
изводится экспериментально. На основании этого химики, как при-
вило, и определяют термодинамические параметры различного рода
процессов.
Энергии Гиббса и Гельмгольца. Химическое равновесие
57
Из соотношения 3.54 следует, что для экзотермических процессов
увеличение температуры приводит к смещению равновесия в сторону
образования исходных веществ, что соответствует принципу Ле-
Шателье-Брауна.
До сих пор мы предполагали, что работаем в закрытой системе и
все продукты реакции непосредственно остаются в ней. Однако
реально многие процессы протекают в открытых системах и часть
продуктов может удаляться из сферы реакции. При этом система
постоянно выводится из состояния равновесия. Хорошо известно, к
примеру, что взаимодействие серной кислоты с нитратом натрия в
обычных условиях приводит к образованию смеси как минимум из
четырех веществ, включая серную и азотную кислоты, сульфат, нитрат
и бисульфат натрия. Константу равновесия этого процесса весьма
трудно представить в удобном виде, поскольку все участники реакции
в той или иной степени диссоциируют на ионы. Однако, умеренно
НагРевая систему под вакуумом, мы создаем условия, способствующие
Отг°нке азотной кислоты, и реакция протекает нацело:
H2SO4 + NaNO. = NaHSO4 + HNO3 Т (3.56)
Ярким примером осуществления необычных реакций в нерав-
ВИдеснь1х условиях является получение цезия путем взаимодейст-
ег° бихромата с металлическим цирконием при повышенных
58
Глава 3
температурах и пониженном давлении. При этом существенно бодее
активный цезий отгоняется в более холодную часть реактора ввиду
своей летучести. Таким образом, суммарный процесс описывается
уравнением:
2 Cs,Cr,O7 + Zr - Zr(Cr,O7), + 4 Cs Т . (3.57^
Следует заметить, что влияние, оказываемое на систему внешним
воздействием, например, отводом продуктов из зоны реакции или
наложеннием некоторой разности потенциалов, служит одним из
мощнейших факторов смещения равновесия. Путем приложения к
системе разности потенциалов можно изменить не только положение
равновесия, но и само направление протекания процесса, - так
например, получают фтор из термодинамически крайне устойчивых
солей плавиковой кислоты.
Таблица 3.3
Смещение равновесия некоторых реакций при изменении давления и температуры
Процесс АЯ Ап Смещение равновесия при росте
Т р
Hi + h = 2 HJ >0 = 0 не смещает
3H2+N2 = 2NH.? <0 <0 <— —>
N2O4 = 2 NO2 >0 >0 —> <—
В таблице 3.3 приведено влияние изменения давления и темпе-
ратуры на смещение положения равновесия некоторых химических
реакций с различными тепловыми эффектами и изменением числа
молекул газа. Данные примеры иллюстрируют выполнение принципа
Ле-Шателье-Брауна для химических процессов.
Задачи и вопросы
1. Для реакции синтеза аммиака стандартное значение свободной
энергии Гиббса равно -33.3 кДж/моль, а стандартная энтальпия -
-92.4 кДж/моль. Оцените максимальный выход его синтеза при 400 и
500°С и давлении 107 Па.
2. Пользуясь экспериментальными данными по зависимости
давления паров азотной кислоты от температуры определите стан-
дартную энтальпию ее испарения.
Энергии Гиббса и Гельмгольца Химическое равновесие
59
Vc 0 20 40 60 80 100
р, мм рт. Ст. 15 50 130 300 650 1300
3 Оцените возможность протекания при комнатной температуре
и возможные температуры достижения равновесия для следующих
реакций
H,S(r) + Cl,(г) = S(tb) +2 НС1(г)
H,Se(r) + Cl,(г) = 5е(тв) +2 НС1(г)
если
МГ °, кДж/моль
ДО0, кДж/моль
H,S(r) H,Se(r) НС1(г)
-21 '33 -92
-34 20 -95
4. Полагая, что в системе реализуются стандартные условия,
определите возможность самопроизвольного протекания реакции
Fe.O + СО = 3FeO + СО, и температуру достижения для нее состояния
равновесия если:
Fe3O4 СО FeO СО,
ДН°. кДж/моль -1118 -111 -265 -394
Д5°, Дж/моль К 146 198 61 214
5. Оцените температуру начала разложения нитрата серебра
атмосфере, содержащей 2 10'4 % диоксида азота если:
AgNO3 Ag NO,
кДж/моль -125 — 33' —
AS0-Дж/моль К 141 43 240 205
6. Определите величину давления в сосуде, в котором произошел
процесс испарения 0.5 г N,O( при комнатной температуре, если
константа равновесия его диссоциации при этом составляет 0.115 атм.
7- Степень диссоциации пентахлорида фосфора на трихлорид
®°сф°ра и хлор при 170°С и атмосферном давлении равна 20%, а при
С - 80%. Оцените значение энтальпии и энтропии его диссоциации
в этих условиях.
ПоЛ‘ ^Родолите максимальное количество энергии, которое можно
у ИТь из некоторой системы, состоящей из 5 молей веществ А и В
60
Глава 3
и одного моля АВ в 10 литрах раствора, если после установления
новесия в этой системе концентрация А достигла 0.1 моль/л.
Рав-
9. Определите равновесное давление углекислого газа над
карбонатом магния при 550°С, если стандартные энтальпии кар
боната, оксида магния и диоксида углерода составляют -1113, -607 и
-393.5 кДж/моль, а равновесное давление при 490°С - 78 атм.
Глава 4
ФАЗОВЫЕ РАВНОВЕСИЯ.
ФАЗОВЫЕ ДИАГРАММЫ
4.1. Правило фаз Гиббса
Практически все твердые и жидкие тела, включая вполне ста-
бильные при данных условиях, сосуществуют с газовой фазой. При
попадании их в замкнутую систему, включающую, например, жид-
кость и газ, между ними устанавливается равновесие, определяющее-
ся некоторыми, присущими данному веществу свойствами. Положе-
ние равновесия существенным образом зависит от температуры и
давления, которые, наряду с соотношением входящих в данную си-
стему веществ, являются параметрами системы. Если внутри системы
нет поверхности раздела, то она называется гомогенной. В качестве
гомогенных систем можно рассматривать различные газовые смеси,
водные растворы солей, твердые растворы золото-серебро и многие
другие. Система, в которой можно выделить части, различающиеся
по строению, свойствам и отделенные друг от друга границами разде-
ла, называется гетерогенной (рис. 4.1). К таковым относятся рас-
слаивающиеся смеси нескольких жидкостей типа вода-бензол, на-
сыщенные растворы, находящиеся в контакте с избытком твердого
вещества, жидкости, сосуществующие со своим паром, и др.
Рис.4.1. Примеры гомогенной и гетерогенной систем
62
Глава 4
Теперь можно дать более четкое определение термина “фаза”,
которым мы пользовались до сих пор, воспринимая его лишь из
соображений здравого смысла. Гомогенная часть гетерогенной
системы, отделенная от других поверхностью раздела и отличающаяся
от них по структуре и свойствам, называется фазой. Следует обратить
внимание на необходимость эквивалентности структуры и свойств
вещества в пределах одной фазы. Так, полиморфные модификации
одного и того же вещества, например ромбическая или моноклинная
сера, являются разными фазами, а частицы некоторого металла, пусть
даже разделенные поверхностями раздела, являются одной фазой.
Свойства твердых и жидких веществ, в отличие от газов, слабо зави-
сят от давления. На основании этого их зачастую объединяют под
названием конденсированных. Соответственно и системы, состоящие
лишь из твердой и жидкой фаз, а точнее системы, в которых газовая
фаза не оказывает существенного влияния, на фазовые и химические рав-
новесия, также называются конденсированными.
Каждая система состоит из набора веществ, которые могут либо
вступать во взаимодействие, либо, напротив, оставаться неизмен-
ными, слабо влияя на свойства друг друга. Все эти вещества приня-
то называть компонентами. Понятие числа компонентов играет важ-
ную роль в физической химии, особенно при рассмотрении фазовых
и химических равновесий. Можно заметить, что в нем заключена
некоторая неоднозначность. Так, если растворить хлорид аммония в
какой-либо жидкости, то с полным основанием можно считать его
одним компонентом. Если же рассмотреть процесс диссоциации NH4C1
да аммиак и хлористый водород, то компонентов будет уже три. Для
того чтобы обойти этот скользкий момент, вводят понятие числа неза-
висимых компонентов. Под таковыми понимается минимальное число
компонентов, из которых можно организовать данную систему. Оно
равно общему числу компонентов за вычетом количества связывающих
их уравнений.
Действительно, процесс диссоциации хлорида аммония описы-
вается уравнением:
NH4C1 <-> NH, + НС1 . (4.1)
Из того же уравнения ясно, что количества аммиака и хлористо-
го.водорода, образовавшиеся в процессе разложения, строго эк-
вивалентны друг другу. Таким образом, вычитая из трех компонентов
два уравнения (к счастью, аналогия с полутора землекопами, по-
лученными путем аналогичных преобразований Виктором Пере-
стукиным, здесь неуместна), получаем лишь один независимый
Фазовые равновесия. Фазовые диаграммы
63
компонент. Если подойти к тому же равновесию исходя из продук-
тов реакции, то их количества вовсе не обязательно должны быть
эквивалентными. В этом случае остается лишь одно уравнение, свя-
зывающее количества всех веществ в системе через константу рав-
новесия, а число независимых компонентов станет равно двум.
Основным вопросом, интересующим большинство исследо-
вателей, является выяснение соотношения между числом фаз, не-
зависимых компонентов и возможностью изменения параметров
системы без нарушения количества фаз. Для вывода такой законо-
мерности представим себе некоторую систему, состоящую из к ком-
понентов и содержащую f фаз. Как известно из предыдущих разделов,
условием равновесия фаз является равенство химических потенциалов
каждого компонента в каждой фазе. Таким образом, можно записать
следующую систему уравнений:
м/ = й/ = Ц/ =••••= и/
ц/ = ц/ = ц/ =....= ц/
...................................................... О
и/ = Ь2 = н, ’=•.-= Ц/
После того, как система пришла в состояние равновесия, величины
химических потенциалов могут меняться только при изменении
внешних условий, в качестве которых могут выступать температура
и давление. Нетрудно убедиться, что из каждой строки можно по-
лучить (/-7) независимых уравнений, а для всей системы - k(f-l)
таковых. С другой стороны, меняя, например, концентрацию одного
из компонентов газовой смеси, мы автоматически меняем и пар-
циальные давления (или концентрации) остальных. Но в целом при
данных температуре и давлении суммарная по всем компонентам
концентрация окажется величиной постоянной. Следовательно, в
каждой фазе имеем (/с-7) независимых концентраций, а для системы
в целом - flk-1) таковых. Кроме того, положение равновесия между
различными фазами можно изменить, поменяв хотя бы один из па-
раметров: температуру или давление. Поэтому общее число неза-
висимых переменных в системе составит f(k-l)+2. Если данное
состояние изменить невозможно не меняя числа фаз, то система дол-
жна иметь единственное решение и число независимых переменных
должно равняться количеству связывающих их уравнений, т.е.
f(k -1)+2 = к (/- 7) или У= к + 2 . (4.3)
С другой стороны, для большинства состояний многокомпо-
нентных систем возможны и вариации одного или двух параметров
без изменения количества компонентов. Так, в стакане с горячим чаем
(i I
Глава 4
легко можно растворить еще одну-две ложки сахара без образования
осадка. Максимальное число переменных, которые можно менять
независимо от других, не меняя при этом состояния системы, на-
зывается числом степеней свободы (с). Естественно, что при этом число
связывающих концентрации уравнений окажется меньше именно на
эту величину. Тогда можно получить следующее соотношение:
f + с = к + 2 . (4.4)
Это уравнение называется правилом фаз Гиббса, которое обычно
формулируют следующим образом: число степеней свободы равно-
весной термодинамической системы превышает разность между числом
независимых компонентов и количеством фаз на два.
Это правило формулируют различными способами, которые
определяются условиями проведения эксперимента. Так, для конден-
сированных систем, состоящих исключительно из жидких и твердых
фаз, например кристаллов некоторой соли, находящейся в равнове-
сии со своим насыщенным раствором, давление не оказывает су-
щественного воздействия на положение равновесия. При этом мы как
бы теряем одну из переменных и правило фаз следует видоизменить:
f+c-k + 1. (4.5)
С другой стороны, на состояние системы могут влиять и некоторые
другие факторы, например, электромагнитное поле. При этом, на-
против. число степеней свободы увеличится-
f+c = k + 2 + n, (4.6)
где п - число сторонних воздействующих на систему сил и полей.
4.2. Фазовые диаграммы однокомпонентных систем
Рассмотрим применение правила фаз Гиббса на примере од-
нокрмпонентной системы. В этом случае его можно представить в
виде:
f+c = 3. (4.7)
Минимальное число возможных фаз в системе равно единице. При
этом существуют две степени свободы. Это означает, что можно од-
новременно менять температуру и давление пара, жидкости или
твердого тела, оставаясь в области термодинамической стабильности
данной фазы. Строго говоря, это утверждение справедливо лишь для
систем, которые могут изменять свой объем. В противном случае, на-
пример, для газа, давление будет однозначно определяться уравнени-
ем Менделеева-Клапейрона.
Фазовые равновесия. Фазовые диаграммы
()5
Схема, отражающая области термодинамической стабильности
существующих в системе фаз и обозначающая границы раздела между
ними, называется фазовой диаграммой. Пример таковой для одно-
компонентной системы приведен на рисунке 4.2. Область, в которой
число и качественный состав фаз не меняется называется фазовым
полем. На приведенной диаграмме можно выделить три таких поля,
соответствующих областям стабильности газа, жидкости и твердого
тела. Для любого фазового поля диаграммы состояния однокомпо-
нентной системы справедливо утверждение о наличии двух степеней
свободы.
Рис.4.2. Фазовая диаграмма одиокомпонснтпой сие гемы (а) и кривая зависимости
температуры системы от времени ее охлаждения из точки “а” (6)
Охлаждая газ при постоянном давлении (с = 1, т.к. р = const)
(кривая “а-b” рис. 4.2), мы неизбежно достигнем некоторой
температуры, при которой начнется процесс конденсации пара в
жидкость или твердое тело (точка “Ь”). При этом параметры систе-
мы, включая и мольный объем газа, оказываются по-прежнему
связанными друг с другом. В точке конденсации получаем пар,
равновесный с данной жидкостью. Поскольку для индивидуальных
веществ давление насыщенного пара однозначно связано с тем-
пературой (или, что более привычно, - при постоянном давлении
66
Глава 4
жидкость кипит при одной температуре), процесс конденсации должен
протекать до тех пор, пока не исчезнет весь газ. Из правила фаз также
следует что f + с = 2 (т.к. р = const) и при наличии двух фаз с = 0. При
этом весь отвод тепла будет компенсироваться теплотой процесса
конденсации и температура системы сохранится постоянной столь
долго, пока она не перейдет в следующее фазовое поле (рис. 4.26).
Далее жидкость постепенно охлаждается (“b-с”, с - 1), затвер-
девает (точка “с” на кривой охлаждения соответствует следующей
платформе (“с-с' “) с постоянной температурой, с - 0), и, наконец,
следует охлаждение твердого тела (с = 1). Если давление в исследуемой
системе ниже соответствующего точке “о”, то процесс сублимации
газа в твердое тело будет происходить минуя жидкость.
Если же, напротив, постоянным будет объем системы (рис. 4.3),
то из уравнения Менделеева-Клапейрона можно заключить:
Рис.4.3. Охлаждение однокомпонентной системы при постоянном объеме
Все точки, через которые проходит процесс охлаждения, таким
образом, будут располагаться на прямой, проходящей через начало
координат. При некоторой температуре, соответствующей точке “Ь”
Фазовые равновесия. Фазовые диаграммы
67
начнется процесс конденсации. Это приведет к понижению давления
в системе. С уменьшением последнего понижается и температура
сосуществования газа и жидкости (продвижение по кривой “Ь-о”).
На этой кривой в равновесии находятся две фазы и с = 1, т.к.
понижение температуры оказывается однозначно связанным с по-
нижением давления. Такое равновесие называется моновариантным.
Продолжая процесс охлаждения по кривой “b-о”, мы обязательно
придем в точку “о”, в которой сосуществуют все три фазы - газовая,
жидкая и твердая. В соответствии с правилом фаз число степеней
свободы в ней равно нулю. Эта точка называется тройной, а соот-
ветствующее равновесие - нонвариантным (или инвариантным).
Кривые “О-о”, “о-Г и “o-s” соответствуют двухфазным рав-
новесиям и отражают соответственно зависимости температуры воз-
гонки твердого тела (сублимации), кипения жидкости и плавления
твердого тела от давления. Наибольший интерес представляет задача
определения угла их наклона. Его можно выразить в виде некоторой
функции термодинамических параметров. Действительно, условием
равновесия двух фаз является равенство их свободных энергий Гиббса.
Причем смещение по любой кривой, соответствующей двухфазному
равновесию, приводит к равному приращению свободных энергий в
обеих фазах. Продифференцировав Дб по температуре и давлению,
можно прийти к заключению, что полученные производные
соответственно равны -S и V. Тогда приращение dG для двух фаз ос и
Р можно выразить как
dG =-S dT+V dp, dG„ = -SdT + Vdp . (4.9)
a a a * fl fl fl *
При смещении по линии равновесия эти величины будут равны
друг другу, т. к. фазы находятся в равновесии. Тогда, приравнивая
правые части, получаем:
(5 -Sfi)dT = (Va-Vfl)dp или (S -S,)/(E-И,) = Д57Д V = dp/dT, (4.10)
где AS и ДИ- равновесные изменения энтропии и мольного объема
при фазовом переходе. Использование этого соотношения несколько
затрудняется тем, что данные об энтропии фазовых переходов
сравнительно мало распространены в справочной литературе. С
другой стороны, в условиях равновесия ДД' = ДЯ/Т. Подставляя это
соотношение в предыдущее уравнение, получаем:
dpldT= ШТ W. (4.11)
Полученное соотношение называется уравнением Клаузиуса-
Клапейрона. Для равновесных фазовых переходов оно описывает
взаимосвязь между температурой их протекания и давлением.
68
Глава 4
Очевидно, что все переходы из менее упорядоченной фазы в более
упорядоченную происходят с выделением теплоты. Кроме того,
конденсация газа в жидкость или твердое тело всегда связана с
уменьшением объема. Поэтому наклон кривых “О-о” и “о-Г всегда
положителен. Это означает, что равновесное давление пара над любой
конденсированной фазой всегда растет с ростом температуры. При
этом, поскольку удельный объем пара несравнимо больше, чем для
конденсированной фазы, последним можно пренебречь, а объем
газовой - выразить через RT/p. Подставив это значение в уравнение
Клаузиуса-Клапейрона и разделив переменные, получаем:
dplp = bHIR(dTIT2) (4.12)
или, после интегрирования:
In р = -AHIRT + С (4.13)
Константа интегрирования определяется по величине первого
слагаемого при единичном давлении. Поэтому графики зависимости
давления насыщенного пара некоторого вещества над твердой или
жидкой фазами от температуры элементарно строятся на основании
известных данных о теплотах сублимации и кипения. Исходя из тех
же данных, можно вычислить и положение тройной точки, поскольку
равновесию между твердой и жидкой фазами для нее соответствует
равенство давлений насыщенных паров над ними.
Для подавляющего большинства соединений процесс плавления
сопровождается увеличением объема. Поэтому температура плавления
для них растет с увеличением давления. Отклонений от этого правила
сравнительно немного. Наиболее известными их примерами являются
висмут, чугун и вода, для которых мольный объем жидкости меньше,
чем твердой фазы.
Наконец, зададимся вопросом: почему в точке “о” наклон кривой
“О-о” больше, нежели “о-Г? Исходя из того, что объем газа много
больше, нежели объем жидкости или твердого тела, легко заключить,
что изменения мольных объемов при кипении и возгонке в этой точке
практически равны. С другой стороны, поскольку энтропия жидкости
существенно выше, чем твердого тела, то величина Д5* для кипения
меньше. Отсюда и меньший наклон кривой кипения в точке “о”.
4.3. Диаграммы состояния воды и серы
Рассмотрим некоторые особенности диаграмм состояния одно-
компонентных систем на примере воды (рис. 4.4). Как уже отмечалось,
Фазовые равновесия. Фазовые диаграммы
69
мольный объем жидкости для нее меньше, чем у льда. Величины их
при 0°С составляют соответственно 18.018 и 19.652 мл. Теплота
плавления льда равна 6006 Дж/моль. Наклон графика зависимости
температуры плавления льда от давления должен составлять
-273.15(19.652-18.018)/6006 10й = -7.44.10'8 K/Па или -0.00754 К/атм.
Таким образом, изменение температуры плавления при увеличении
давления хоть и отрицательно, но крайне незначительно. Примерно
таким же порядком величины характеризуется наклон кривых
аналогичных зависимостей для других веществ. Давление паров воды
в тройной точке мало относительно атмосферного и составляет всего
0.00603 атм. Поэтому температуру, соответствующую этой точке
можно оценить как 0.00754 (1-0.00603) = 0.0075°С. Реальное значение
лишь на одну десятитысячную градуса больше. Это означает, что,
нагревая лед под вакуумом ниже указанного давления, можно перейти
к газовой фазе минуя жидкую. При давлениях выше 2000 атмосфер
термодинамически устойчивыми оказываются новые модификации
льда с плотностью, превышающей таковую для воды. Линии
плавления для таких форм льда имеют положительный наклон, как
для большинства других соединений (см. рис. 4.46).
Рис.4.4. Диаграммы состояния воды при низких (л) и высоких (б) давлениях
Мы уже обращали внимание на то, что стандартные химические
потенциалы ионов и молекул могут существенным образом от-
личаться в различных фазах. Для того чтобы иметь возможность
70
Глава 4
сопоставить их, отметим, что химические потенциалы различных
конденсированных фаз можно выразить через потенциал находя-
щегося в равновесии с ними пара:
ц = ц° +ЯПпр. (4.14)
Поскольку первое слагаемое во всех случаях одинаково, то можно
сделать вывод, что условием равновесия всех конденсированных фаз
является равенство давлений насыщенных паров над ними. Термо-
динамически стабильной при данных условиях будет являться та фаза,
которой соответствует меньшее давление равновесных паров. Ут-
верждение это весьма легко доказать. Действительно, при сосуще-
ствовании в замкнутой системе двух фаз с различным давлением паров
та фаза, для которой оно больше, неизбежно будет испаряться, а пары,
в свою очередь, будут конденсироваться в виде более устойчивой фазы.
При охлаждении воды может возникнуть и такая ситуация, при
которой отсутствие центров кристаллизации приведет к торможению
процесса образования твердой фазы. Для того чтобы наблюдать этот
эффект, надо пользоваться чистой водой, сосудом с гладкими стен-
ками и избегать любого рода механических воздействий. При этом
можно получить переохлажденную воду, давление паров над кото-
рой выше, чем надо льдом (пунктирная линия на рис. 4.4). Небольшие
механические воздействия (встряхивание, трение по стенке палочкой
или попадание затравки) могут привести к мгновенной кристал-
лизации части вещества. Такого рода явления встречаются иногда и
в очень чистых водоемах. Глубинные слои воды с максимальной плот-
ностью (при 4°С) изолируют верхний переохлажденный слой от
центров кристаллизации на дне. Образование переохлажденных
жидкостей наиболее характерно для вязких веществ типа глицерина.
Температура его кристаллизации 17°С, но закристаллизовать гли-
церин достаточно сложно. Высокая вязкость переохлажденной
жидкости приводит к тому, что она оказывается вполне устойчивой
кинетически (но не термодинамически). Дальнейшее охлаждение
может привести к образованию метастабильных стекол.
В качестве другого весьма интересного примера диаграмм
состояния однокомпонентных систем можно привести диаграмму
состояния серы (рис. 4.5). Главным ее отличием от таковой для воды
является то, что сера может образовывать кроме газовой и жидкой
еще и две твердые модификации - моноклинную и ромбическую. Из
правила фаз следует, что все четыре фазы для однокомпонентной
системы не могут сосуществовать одновременно. Поэтому на диа-
грамме состояния серы в идеальном случае м^ли бы существовать
Фазовые равновесия. Фазовые диаграммы
71
четыре тройные точки. Реально же их только три (см. рис. 4.5). Они
соответствуют сосуществованию паров серы с ромбической и мо-
ноклинной твердыми фазами при 95.5°С, с моноклинной и жидкой -
при 120°С и трех конденсированных фаз при 151°С и давлении
1288 атм.
Взаимные переходы двух кристаллических фаз иногда называют
также энантиотропными превращениями. Основной их характе-
ристикой является теплота фазового перехода, иногда называемая
скрытой теплотой (плавления, кипения или энантиотропного пре-
вращения). Это название отражает различное теплосодержание
разных форм вещества и наиболее часто применяется для объяснения
разности между теплотами реакций различных кристаллических
форм, например, между теплотами сгорания ромбической и моно-
клинной серы.
Процесс нагревания чаще всего неожиданно приводит к тому, что
ромбическая сера плавится, не переходя в моноклинную в некоторой
точке, расположенной на пересечении пунктирных линий рисунка 4.5.
При этом давление паров метастабильной в этих условиях ром-
бической серы выравнивается с давлением паров метастабильной же
72 Глава 4
жидкости, Образуется метастабильная система (та самая четвертая
тройная точка), которая отличается относительной устойчивостью.
С течением времени все существующие в этой области фазы неизбежно
должны перейти в моноклинную модификацию, характеризующую-
ся меньшим давлением насыщенных паров. Это проявление эмпи-
рического правила Оствальда, в соответствии с которым в случае
возможности реализации двух видов фазовых, переходов сначала об-
разуется более устойчивая, чем исходная, но метастабильная. фаза.
Другой пример проявления этого правила - образование при кон-
денсации из паров белого фосфора, хотя красный при этих условиях
более устойчив. Аналогично, при охлаждении жидкой серы до ком-
натной температуры зачастую кристаллизуется моноклинная моди-
фикация.
4.4. Краткие сведения о термическом анализе
Построение фазовых диаграмм и изучение фазовых равновесий
проводят чаще всего с привлечением термического анализа. Экспе-
риментальные методики строятся на подводе к веществу постоянного
количества тепла за единицу времени или на спонтанном его ох-
лаждении. Наблюдаемой величиной является либо температура
образца, либо электродвижущая сила, возникающая между вещест-
вом и стандартом и пропорциональная разнице их температур (по-
следний подход очень часто используется в широко распространен-
ных приборах, используемых для термического анализа, - дерива-
тографах). Методика интерпретации полученных данных для обоих
случаев одинакова. Результаты представляются в виде графика за-
висимости температуры вещества от времени. При отсутствии фазо-
вых превращений кривая охлаждения представляет собой плавную
линию, выгнутую в сторону начала координат. Это обусловлено по-
степенным понижением разности температур между образцом и ок-
ружающей средой, что снижает скорость отвода тепла (см. рис. 4.26).
Кривая нагрева идет с небольшим наклоном, однако при
протекании фазовых переходов происходит выделение или погло-
щение тепла, приводящие к резкому различию температур образца и
стандарта с появлением соответствующей разности потенциалов.
Таким образом, при протекании фазового перехода на графике будет
наблюдаться излом ввиду частичной или полной компенсации отво-
да тепла за счет теплоты фазового перехода. При -'том в моновариант-
ной системе температура продолжает снижаться с меньшим наклоном.
Фазовые равновесия. Фазовые диаграммы
73
например по мере конденсации паров и понижения их парциального
давления. В области нонвариантного равновесия превращение
происходит при одной температуре и не заканчивается до тех пор,
пока данный фазовый переход не завершится полностью. Размер
площадки будет тем больше, чем больше масса вещества и теплота
фазового перехода. Далее в обоих случаях охлаждение пойдет таким
же образом за исключением возможного изменения наклона за счет
изменения теплоемкости.
4.5. Термодинамика процесса смешения
В случае двухкомпонентных систем можно оперировать до-
полнительной степенью свободы, в качестве которой выступает
соотношение компонентов. Поэтому необходимо ввести новое
дополнительное измерение, обозначаемое “У”, соответствующее
мольному или весовому содержанию одного из компонентов. Оно
выражается в процентах или в мольных долях. Мольная доля
/-компонента смеси может быть представлена как
х. = п./Ъп., (4.15)
где п. - количество молей /-го компонента в смеси. Или для смеси из
двух компонентов:
Ха=Па/(Пи+П1)- (4.16)
Представляется очевидным, что газы всегда (а на самом деле почти
всегда) неограниченно растворяются друг в друге. Для жидкостей это
справедливо далеко не всегда. Для ряда из них, обладающих сходными
свойствами (спирт-вода), взаимная растворимость неограниченна,
для других (вода-бенол) она может быть очень мала. Таким же
образом обстоит дело и с растворимостью в жидкостях твердых
веществ. Еще хуже - с взаимной растворимостью двух твердых
соединений. Даже для пары металл-металл таковая, чаще всего, ока-
зывается существенно ограниченной.
Для того чтобы понять эти закономерности, рассмотрим процесс
смешения двух идеальных газов при постоянном давлении. Возьмем
п, и и,молей двух газов, занимающих объемы и К, соответственно.
Суммарный объем после смешения составит (Е) + К,), а число молей -
(/?/ + /12) = /7. Все перечисленные величины связаны друг с другом через
уравнение Менделеева-Клапейрона:
/7;/щ = V,l V2vix=n,l(n^n2) = + .
74
Глава 4
Представленный процесс можно рассматривать как сумму не-
зависимых процессов расширения каждого из газов до суммарного
объема. Тогда, используя известное соотношение для энтропии
расширения и учитывая аддитивность этой величины, получим:
AS = AS, + AS? = п,R In [(K, + K) / KJ + п2R In [(7,+ К,) / KJ =
= -R(n, +п2) {п , / (п, +/?,) In [ К, / (+ К,) + п2 / (/?, +п2) In [ К, / (К, + К,)] =
= -nR(x; lnxz + х,1пх,). (4.17)
Поскольку х; и х, < 1, полученная величина всегда является
положительной. Следовательно, для изолированной системы про-
цесс смешения газов самопроизволен. Что может этому помешать?
Естественно, только значительные затраты тепла, т.е. высокие
значения энтальпии смешения. Для идеального газа, молекулы
которого априори не взаимодействуют друг с другом, изменение
внутренней энергии при смешении равно нулю. Поскольку не
меняются и суммарный объем и давление, то и второе слагаемое
функции энтальпии (дЕ), а следовательно и вся величина энтальпии
смешения, равны нулю. Таким образом, процесс смешения идеальных
газов будет самопроизвольным при любых условиях. Взаимодействие
молекул любого газа между собой крайне мало. Поэтому полученный
вывод можно распространить на все газы вообще. С другой стороны,
если молекулы двух газов существенно различаются по природе, то
при сильном сжатии, когда свойства газа и жидкости сближаются,
процессы отталкивания между молекулами могут стать более суще-
ственными. Поэтому вблизи критической точки некоторые газовые
смеси могут расслаиваться.
Чем больше степень упорядочения вещества, тем прочнее в нем
межмолекулярные связи. При этом взаимодействие между одинако-
выми молекулами может существенно превысить таковое для разных.
Это, естественно, проявляется как “отталкивание” между молекулами,
и если энтальпия такового, отнесенная к температуре, превысит
выигрыш энтропии, смешение станет невозможным. Если же величина
АЯ положительна, но не столь велика, - взаимное растворение
происходит, хотя и с поглощением тепла, как в случае образования
водного раствора нитрата аммония.
4.6. Системы с простой эвтектикой
Очевидно, что фазовая диаграмма для двухкомпонентных смесей
будет трехмерной (оси р, Т, х~). Пользоваться такой диаграммой срав-
Фазовые равновесия. Фазовые диаграммы
75
нительно трудно, поэтому на практике всегда приходится опери-
ровать с одним из видов ее сечений или проекций. Начнем с наиболее
простого варианта - конденсированной системы, включающей лишь
твердые тела и жидкости. Давление сравнительно слабо влияет на
состояние такой системы, поэтому, как отмечалось ранее, для боль-
шинства случаев с хорошим приближением можно исключить его из
числа переменных. Тогда при наличии двух компонентов правило фаз
может быть записано как
c+f=2+l=3. (4.18)
Таким образом при равновесии одновременно могут сосущество-
вать максимально три фазы. В качестве независимых переменных при
этом выбираются состав и температура, причем из чисто практических
соображений удобным оказывается строить зависимость температуры
фазовых превращений от состава.
Рассмотрим конкретные типы фазовых диаграмм. Простейшим
случаем таковых является диаграмма системы с простой эвтектикой.
Для построения ее принимаются три предположения:
1) компоненты не образуют друг с другом химического соеди-
нения;
2) компоненты неограниченно растворяются друг в друге в жидкой
фазе;
3) компоненты абсолютно нерастворимы друг в друге в твердой
фазе.
Хорошим приближением для такого случая является диаграмма
плавкости системы свинец-олово (рис. 4.6). Как известно, чистые
компоненты плавятся соответственно при 327 и 232°С, а смесь их
образует более легкоплавкие системы. Это обусловлено тем, что
введение растворенного вещества в соответствии с уравнением (4.17)
повышает энтропию жидкости и понижает ее химический потенциал.
Поскольку мы оговорили взаимную нерастворимость компонентов в
твердой фазе, изменение состава не меняет химический потенциал
твердых компонентов. Поэтому выравнивание химических потен-
циалов жидкой и твердой фаз, которое является условием протека-
ния процесса плавления, достигается при более низких температурах.
При этом две кривые плавления неизбежно пересекутся в некоторой
точке, соответствующей минимальной температуре затвердевания.
Таковой для данной системы является смесь, содержащая 38.1 мас-
совых процентов свинца. Смесь этого состава называется эвтекти-
ческой. Общим правилом для эвтектических систем является большее
содержание в них более легкоплавкого компонента.
76
Глава 4
Рис.4.6. Диаграмма состояния системы олово-свинец и кривая охлаждения для
некоторого состава этой системы
Рассмотрим процесс затвердевания смеси, содержащий избыток
одного из компонентов (точка “а”), например свинца, относительно
эвтектического. В фазовом поле, соответствующем расплаву, в соот-
ветствии с правилом фаз система имеет две степени свободы (возможно
изменение состава и температуры). Поэтому можно произвольно ох-
лаждать ее без изменения состава. Однако при некоторой температуре
кривая охлаждения пересекается с кривой плавления, которая иначе
называется линией ликвидуса. В этой точке сосуществуют две фазы,
поэтому степень свободы остается лишь одна. После пересечения ли-
нии ликвидуса (точка “Ь”) часть тепла тратится на кристаллизацию
одного из компонентов и кривая охлаждения понижает свой наклон
(см. рис. 4.66). Состав выпадающих при данной температуре кристал-
лов, равновесных с раствором, можно найти по пересечению прямой,
проведенной из данной точки параллельно оси состава. Естественно,
что при отсутствии растворимости одного компонента в другом в
твердом виде кристаллизоваться будет чистый металл, в данном слу-
чае - свинец. Продолжая процесс охлаждения, приходим в некоторую
точку “х”, Состав равновесных расплава и твердой фазы находим
аналогично - по пересечению прямой, параллельной оси состава, с
Фазовые равновесия. Фазовые диаграммы 77
кривой ликвидуса и линией, соответствующей твердому компоненту.
Количество обеих фаз можно определить по правилу рычага - они
соотносятся между собой так же, как длины отрезков, соединяющих
три упомянутые точки (см. рис. 4.6), Мж/Мт = т/п.
Итак, в точке “х” сосуществуют две фазы, и в соответствии с
правилом фаз имеется одна степень свободы. Возникает противоречие
- находясь в области фазового поля, мы имеем столько же степеней
свободы, как и на его границе. Кроме того, казалось бы, мы можем
произвольно перемещаться в пределах поля в любом направлении,
сохраняя число фаз. Дело в том, что при изменении температуры в
соответствии с ранее сказанным однозначно меняется состав равно-
весной жидкости. В соответствии с определением фазы изменение
состава соответствует появлению новой фазы. То есть, равновесие
действительно будет моновариантным - сохранить состав жидкой
фазы можно лишь сохраняя температуру системы. В процессе охлаж-
дения мы реально движемся по границам этой двухфазной области, в
которой сосуществуют чистый свинец и расплав, состав которого
соответствует точке на кривой ликвидуса при данной температуре.
Таким образом моновариантность равновесия не вызывает сомнения.
Дальнейшее охлаждение приводит к совпадению состава раствора с
составом эвтектики (Е). Поэтому далее она кристаллизуется при
неизменной температуре. Эвтектика представляет собой смесь
мельчайших кристалликов олова и свинца. Таким образом, в точке
“с” сосуществуют три фазы: расплав, олово и свинец. Соответственно,
число степеней свободы оказывается равным нулю (точка “Е”
соответствует нонвариантному (инвариантному) равновесию). В
соответствии с этим температура системы не меняется вплоть до
полной кристаллизации всей эвтектики (рис. 4.6). Кривая, ниже
которой существуют только твердые фазы, называется кривой
солидуса. Для выбранного случая она вырождается в прямую,
параллельную оси состава. В любой ее точке равновесие будет ин-
вариантным, то есть последние фракции расплава всегда будут
затвердевать при одной и той же температуре.
В процессе кристаллизации образуется смесь из кристаллов
свинца, которые росли при охлаждении, и мельчайших кристалли-
ков эвтектики. Кристаллизация последней происходит за счет пер-
воначального образования мелкого кристаллика одного вещества (А),
вокруг которого образуется локальная область с пересыщенным
раствором другого, в результате чего вокруг образуются кристаллики
В и т.д. При протекании такого “колебательного процесса” получить
большие кристаллы одного из веществ оказывается невозможным
78
Глава 4
(рис. 4.7). После затвердевания всей эвтектической смеси тепло
расходуется только на охлаждение кристаллов твердых веществ и
наклон кривой охлаждения возрастает.
Рис.4.7. Схема кристаллизации
эвтектики двухкомпонентной
системы
Sn 20
40 60 80 %РЬ
Рис.4.8. Диаграмма состояния системы олово-свинец с учетом взаимной растворимости
компонентов
Фазовые равновесия. Фазовые диаграммы
79
На практике для всех систем характерна та или иная взаимная
растворимость веществ друг в друге (рис. 4.8). Причем для твердых
тел она растет с ростом температуры в соответствии с увеличением
вклада энтропии смешения. После достижения температуры эвтекти-
ки этот рост продолжается, но концентрация примеси в твердой фазе
падает ввиду увеличения количества равновесной с ней жидкой фазы
при относительно мало меняющемся коэффициенте распределения
“растворенного вещества” между ними. Полученная при дальнейшем
охлаждении расплава произвольного состава (кроме соответствую-
щего индивидуальному веществу с долей примеси меньше раство-
римости таковой) твердая масса (точка “е”) состоит из двух инди-
видуальных фаз, содержащих растворенную примесь второго ком-
понента. Соответственно, степень свободы в этом поле также одна.
Действительно, изменяя температуру, мы однозначно меняем и состав
сосуществующих твердых фаз. С другой стороны, при охлаждении
расплава из точки “а” до указанной температуры система приходит
в однофазную область (точка “с”). В ходе дальнейшего быстрого
охлаждения подвижность атомов в полученном твердом растворе
может оказаться недостаточной для его расслаивания. Таким образом
с помощью “закалки” можно получить пересыщенный раствор с
некоторыми особыми свойствами.
4.7. Системы с конгруэнтно плавящимися соединениями
В большом количестве бинарных систем оказывается возможным
образование химических соединений с различной устойчивостью.
Наиболее принципиальным для построения фазовых диаграмм
оказывается вопрос о соотношении температур плавления и разло-
жения соединения. Если разложение происходит при более высоких
температурах, то такое соединение называется конгруэнтно плавя-
щимся. Напротив, соединения, разлагающиеся уже в твердой фазе,
называются плавящимися инконгруэнтно. Примеров соединений
обоих типов чрезвычайно много. Кроме того, во многих системах
образуется по несколько соединений, часть из которых может пла-
виться конгруэнтно, а часть - инконгруэнтно.
Рассмотрим в качестве примера систему, содержащую хлориды
трехвалентного железа и одновалентной меди (рис. 4.9). В ней
образуется единственное соединение состава CuCl-FeCI,. Часто такие
соединения называют по мольному соотношению компонентов 1:1.
При этом фазовая диаграмма как бы разбивается на две - в левой ее
80
Глава 4
половине сосуществуют фазы CuCl и CuCl -FeCl,, а в правой - FeCl3 и
CuC!FeCl3. Температуры плавления индивидуальных веществ - 424
и 298°С, а соединения - 320°С. Последняя точка является локальным
максимумом на кривой ликвидуса, поскольку с обеих сторон от нее
температуры плавления понижаются примесями индивидуальных
хлоридов. В левой половине диаграммы образуется эвтектика с
температурой плавления 304, а в правой - 263°С. Области их
существования разделяются линией, соответствующей составу со-
единения и простирающейся от кривой ликвидуса до абсолютного
нуля либо до температуры перехода в другое соединение, которое
может быть более устойчивым при низких температурах. Степень
“закругленности” или приближения к плоскому максимума ха-
рактеризует устойчивость образующегося соединения.
Диаграмма такого рода может быть составлена из двух диаграмм
систем с простыми эвтектиками, объединенных общей осью (рис. 4.10).
Всю диаграмму можно разбить на четыре области, ограниченные
вертикальными линиями, проходящими через образующееся сое-
динение, а также через обе эвтектики. Для первой из них охлаждение
Фазовые равновесия. Фазовые диаграммы
81
Рис.4.10. Схема разбиения диаграммы состояния системы CuCl FeCl3na две с простыми
эвтектиками
жидкости ниже кривой ликвидуса будет приводить к выделению в
первую очередь кристаллов хлорида меди, для второй и третьей -
двойного хлорида и для четвертой - хлорного железа. После про-
хождения линий эвтектики в качестве твердых фаз будут образо-
вываться в двух первых областях - хлорид меди и двойной хлорид, а
в двух последних - хлорное железо и двойной хлорид.
Особое внимание следует уделить стехиометрическому составу
расплава. При его охлаждении образуется лишь одно вещество,
которое существовало и в жидкой фазе. Рассматриваемая система при
этом по сути является однокомпонентной и точке экстремума на линии
ликвидуса соответствует нонвариантное равновесие. Поэтому весь
расплав будет затвердевать при одной температуре. Следует заметить,
что реально практически все соединения могут допускать некоторые
отклонения от стехиометрического состава. По сути это ни что иное,
как проявление растворимости одного из исходных веществ в про-
дукте реакции. Так, например, хорошо известный оксид двухва-
лентного железа ввиду склонности его к окислению всегда содержит
некоторый избыток кислорода, т.е. имеет состав Fe( О. Часть позиций
82
Глава 4
Рис.4.11. Схема образования области
гомогенности для монооксида железа
атомов железа в кристаллической решетке этого соединения являются
вакантными. Причем величина х может варьировать в достаточно
широких пределах. Поэтому существованию “индивидуального”
соединения будет соответствовать не прямая, а область (рис. 4.11),
называемая областью гомогенности соединения. Особенностью
данного конкретного соединения является то, что область его гомо-
генности не включает стехиометрического состава.
4.8. Системы с инконгруэнтно плавящимися
соединениями
Целый ряд соединений, пусть даже весьма стабильных, разлагается
на исходные компоненты, не доходя до температуры плавления.
Получающиеся при этом диаграммы несколько сложнее. Пусть в
системе A-В образуется единственное соединение состава А2В3,
плавящееся инконгруэнтно. Тогда принципиально возможными
оказываются два варианта: разложение соединения может проис-
ходить ниже температуры одной или обеих эвтектик. В последнем
случае такая система мало отличается от системы с простой эвтек-
тикой и может быть получена из нее добавлением двух областей
сосуществования соединения с исходными компонентами ниже тем-
пературы эвтектики (рис. 4.12я). Для первого случая диаграмма
представлена на рисунке 4.126). На ней можно выделить две го-
ризонтальных прямых, соответствующих кристаллизации эвтектики
В+А,В3 и разложению соединения. Последняя называется линией
перитектики. Ниже нее правая часть диаграммы в целом аналогична
Фазовые равновесия. Фазовые диаграммы
83
Рис.4.12. Два варианта диаграмм состояния для систем с образованием инконгруэнтно
плавящегося соединения
разобранной ранее, а левая образована единственным полем,
соответствующим смеси твердых фаз А и АгВу Наибольший интерес
представляют превращения в ходе охлаждения расплавов, состав
которых находится над линией перитектики (см. рис. 4.126). Ох-
лаждение таковых ниже температуры линии, соответствующей
ликвидусу, приведет к кристаллизации вещества А и изменению
состава расплава в сторону обогащения компонентом В. Ситуация
эта вполне аналогична той, которая реализовалась бы для системы
A-В с простой эвтектикой. Однако при температуре перитектики
начнется кристаллизация вещества А,В3, причем этот процесс бу-
дет происходить при постоянной температуре, поскольку ввиду
сосуществования трех фаз состояние системы окажется инвариант-
ным. Действительно, на прямой перитектики будут сосуществовать
расплав, соединение и одна из фаз А или В. Выбор последней зависит
от того, левее или правее стехиометрического состава был выбран
исходный расплав. Для конденсированной двухкомпонентной си-
стемы число степеней свободы при температуре перитектики равно
нулю. При этом охлаждающаяся жидкость будет реагировать с уже
выделившимся веществом А по следующей реакции:
A+L^A,B2+L'. (4.19)
84
Глава 4
Эта реакция называется перитектической и приводит к образо-
ванию соединения и изменению состава расплава. Если состав смеси
обогащен компонентом Л, то ниже температуры перитектики сущест-
вует лишь смесь двух кристаллических фаз, в противном случае после
кристаллизации части соединения образуется также жидкость соста-
ва L'. Дальнейшее охлаждение последней приводит к выделению
дополнительных количеств фазы А,В^ и изменению состава расплава,
пока он не достигнет точки эвтектики.
Инконгруэнтно плавящиеся соединения образуются, например, в
системах Cu-Ga, Cr-C, NaCl-H,O, CaSiO,-BaSiO3 и т.д. Интересный
пример системы с образованием нескольких типов соединений пред-
ставляют смеси H,S04-H,0 (рис. 4.13).
Рис.4.13. Диаграмма состояния системы серная кислота-вода
Помимо трех конгруэнтно плавящихся гидратов серной кислоты:
H,SO4H,O, H,SO4-2H,O и H,SO4-4H2O в этой системе образуется еще
инконгруэнтно плавящийся H,SO4-6.5H2O и метастабильный
H,SO4-8H,O. Все эти соединения обладают достаточно узкими
областями гомогенности и получить их в чистом виде достаточно
сложно. Интересно, что температурой плавления выше, чем у воды,
обладает лишь моногидрат серной кислоты.
Фазовые равновесия. Фазовые диаграммы
85
4.9. Системы с неограниченной взаимной растворимостью
С практической точки зрения интерес представляет также
рассмотрение систем с неограниченной растворимостью компонентов
друг в друге как в жидком, так и в твердом состоянии, поскольку
происходящие в них процессы весьма важны для очистки ряда ве-
ществ. Наиболее характерно образование неограниченных твердых
растворов для интерметаллических систем с близким размером и
природой атомов. Типичным примером таковых является система
золото-серебро (рис. 4.14).
Точки плавления индивидуальных веществ (950 и 1063°С) на фа-
зовой диаграмме соединены узкой областью, имеющей форму лепе-
стка. Верхняя линия соответствует ликвидусу, а нижняя - солидусу
системы. Выше этой области в качестве единственной фазы существует
жидкий, а ниже - твердый раствор серебро-золото. В соответствии с
правилом фаз в любой точке внутри этих областей существуют две
степени свободы, то есть можно произвольным образом варьировать
состав и температуру. После пересечения при охлаждении области
ликвидуса жидкая фаза сосуществует с твердой. Состав их можно оп-
ределить по пересечению горизонтальной линии, проходящей через
эту точку с кривыми ликвидуса и солидуса. Относительные количества
86
Глава 4
фаз, как и ранее, определяются правилом рычага. Сосуществование
двух фаз означает, что система имеет одну степень свободы. Действи-
тельно, при изменении температуры меняется состав фаз. При прибли-
жении к линии солидуса растет содержание в системе твердой фазы.
Другим важным следствием сказанного является то, что твердая фаза
всегда оказывается обогащенной более высокоплавким компонентом
(в данном случае это золото), а жидкая - легкоплавким При низких
температурах диффузионные процессы в твердом теле оказываются
заторможенными и равновесие не успевает установиться. Поэтому в
конце процесса кристаллизации образуются накладывающиеся слои
сплава, все более обогащенного серебром (рис. 4.15).
Рис.4.15. Схема затвердевания расплава в
системе с неограниченной растворимостью
Рис.4.16. Диаграмма состояния системы марган-ч -медь
Фазовые равновесия. Фазовые диаграммы
87
Для ряда систем с неограниченной растворимостью компонентов
кривые ликвидуса и солидуса могут проходить через минимум или
максимум для некоторых промежуточных составов (причем точки
экстремума для этих кривых всегда является общим). В таком случае
в процессе кристаллизации жидкая фаза приближается к этому
составу, а твердая - обогащается одним из исходных компонентов. В
качестве примера можно рассматривать диаграмму системы марга-
нец-медь, приведенную на рисунке 4.16.
Задачи и вопросы
1. Каковы граничные точки на кривых кипения, сублимации и
плавления для диаграммы состояния однокомпонентной системы?
2. Почему кривые кипения и сублимации имеют больший наклон,
чем кривая плавления?
3. В закрытом литровом сосуде находятся 90 мл воды, и по 10 г
хлоридов натрия и кобальта. Определите число фаз, компонентов и
степеней свободы в системе. Что изменится, если вместо хлорида
кобальта в системе находилась питьевая сода? (Растворимости, г/100 г
раствора NaCl - 26.5, CoCi, - 35, NaHCO, - 8.76)
4. Известно, что при подъеме на каждые 300 м над уровнем моря
атмосферное давление понижается на 25 мм рт. ст, Полагая, что эн-
тальпия испарения воды (44.01 кДж/моль) не зависит от температу-
ры, рассчитайте температуру кипения воды на высоте 7000 м?
5. Для никеля известны следующие константы: Т = 1728К,
теплота плавления 310 Дж/г, с/та = 8.9 г/см3, rf. = 8.4 г/см3. Выразите в
виде уравнения график зависимости температуры плавления никеля
от давления.
6. При десяти градусах Цельсия в объеме 1 л находится 2 г воды
(р1ИС - 9 мм рт.ст.). Сколько фаз присутствует в системе? Какие процес-
сы будут происходить при охлаждении этой системы? Представьте
вид кривой охлаждения.
7. Нарисуйте график охлаждения расплава Ag-Au, содержащего
60% золота (см. рис. 4.14). Укажите состав жидкости и твердой фазы
88
Глава 4
в начале кристаллизации. Определите состав твердой и жидкой фаз
при 980°С.
8. Пользуясь приведенной ниже схемой фазовой диаграммы
диоксида углерода, опишите, какие процессы могут происходить при
быстром выпускании его из баллона с давлением 50 атм.
9. Опишите фазовые переходы и представьте кривые охлаждения
для точек на диаграмме воды при p=const, V=const.
10. Нарисуйте ТХ-диаграмму системы системы PbO-SiO,, по
известным температурам плавления составов и учитывая, что точки
3,5 и 7 соответствуют эвтектикам, а 4 и 6 - конгруэнтно плавящимся
соединениям. Определите состав соединений. Постройте кривую
охлаждения для расплава, содержащего 10 процентов SiO2.
1 2 3 4 5 6 7 8
SiCh, вес.% 0 6 8 13 16 22 29 100
t лпквнл, °C 890 800 710 740 716 760 726 1710
Фазовые равновесия. Фазовые диаграммы
89
И. Нарисуйте график кривых охлаждения растворов состава
H,SO4-1.2H,O, H2SO4-6H,O и H,SO46.5H,O (см. рис. 4.13). Какие
превращения протекают при этом в системе?
12. Для некоторой бинарной конденсированной системы получена
следующая кривая ликвидуса.
Достройте диаграмму состояния этой системы. Нарисуйте кривую
охлаждения расплава из точек 1 и 2. Укажите при охлаждении каких
расплавов можно получить чистые исходные вещества и соединение.
Глава 5
РАСТВОРЫ
5.1. Диаграммы состояния систем соль-вода
Водные растворы являются типичными представителями бинар-
ных систем, для которых характерны те же закономерности, что и
для ранее рассмотренных. Диаграмму состояния для них можно
разобрать на примере системы сульфат натрия-вода (рис. 5.1). Вид
ее, достаточно типичный для фазовых диаграмм вообще, оказывает-
ся несколько непривычным для растворов. Во-первых, вид кривой
растворимости, которую мы однозначно привыкли воспринимать как
зависимость концентрации насыщенного раствора от температуры,
на приведенном графике является зеркальным отображением таково-
го развернутым на 90 градусов. Во-вторых, зависимость растворимо-
сти от температуры при переходе через некоторую точку меняет не
только угол, но и знак наклона. В-третьих, представляется логичным,
что график этот должен исходить из 0°С, как это приводится в боль-
шинстве справочников. Однако нулевая температура соответствует
лишь точке плавления воды, а эвтектика расположена ниже по шкале
температуры.
Достаточно принципиальным является вопрос о зависимости
растворимости от температуры. Излом на графике обусловлен тем,
что десятиводный сульфат натрия является инконгруэнтно плавя-
щимся соединением (zZL7=32°C). Выше этой температуры он разла-
гается и величина растворимости обусловлена уже равновесием
раствора с безводным сульфатом натрия. Переход последнего в гидрат
сопряжен со значительным выделением тепла. Поэтому, несмотря на
то, что растворение безводного соединения в воде характеризуется
отрицательной энтальпией, для десятиводного эта величина положи-
тельна. Соотношение их можно проиллюстрировать следующим
циклом:
Na,SO4 + aq - Na,SO4- aq <0 (5.1)
Растворы
91
Рис.5 Л. Диаграмма состояния системы сульфат натрия-вода
Na,SO4 + 10Н,О = Na,SO4 ЮН,0 АЯ, <0 (5.2)
Na,SO4-ЮН,О + aq = Na,SO4* aq АЯ, (5.3)
АЯ, = АЯ, - АЯ, >0 (5.4)
В соответствии с принципом Ле-Шателье повышение температуры
смещает положение равновесия реакции (5.1) вправо, а (5.3) - вле-
во. Именно поэтому до тех пор, пока растворимость определяется
равновесием декагидрата с раствором, она растет, а после точки пе-
ритектики - падает с ростом температуры.
Рассмотрим процесс охлаждения системы, находящейся в точке
“В”. В этой точке сосуществуют безводный сульфат и раствор. По
мере охлаждения сульфат натрия растворяется, и после пересечения
кривой ликвидуса существует лишь единственная фаза - раствор.
Дальнейшее охлаждение приводит к пересечению со второй кривой
ликвидуса,'ниже которой будет происходить выделение новой фазы
- твердого декагидрата. Последний в каждой точке сосуществует с
насыщенным раствором, состав которого можно определить по
пересечению прямой, параллельной оси Ох, с кривой ликвидуса. По
мере охлаждения равновесный раствор обедняется сульфатом натрия
и, наконец, достигает состава, соответствующего эвтектическому.
92
Глава 5
Дальнейший отвод тепла сопровождается выпадением эвтектической
смеси кристаллов декагидрата и льда при постоянной температуре.
Результирующая кривая охлаждения приведена на рисунке 5.2. Точка,
соответствующая эвтектике для системы соль-вода, называется крио-
гидратной. Как правило, система при этом состоит преимущественно
из воды и имеет температуру плавления близкую к нулю. Однако для
некоторых соединений, интенсивно взаимодействующих с раствори-
телем, эти параметры отклоняются весьма значительно. Так крио-
гидрат фосфорной кислоты на 62.5% состоит из воды и имеет темпе-
ратуру кристаллизации -85°С. Криогидрат хлорида кальция плавится
при -55°С.
Рис.5.2. Кривые охлаждения
для фигуративных точек “А”,
“В” и “С “ (см. рис. 5.1)
При охлаждении состава, соответствующего точке “С”, до тем-
пературы перитектики безводное соединение будет растворяться с
параллельным образованием декагидрата. Охлаждение же из точ-
ки “D” при пересечении кривой ликвидуса будет сопровождаться
выделением кристаллов льда. Причем температура его кристал-
лизации будет понижаться с увеличением концентрации раствора.
Следует заметить, что растворимость неорганических солей во льду,
как правило, низка. Поэтому выделяющийся лед практически не
содержит примесей, и, таким образом оказывается возможным прово-
дить очистку воды от примесей вымораживанием.
Равновесие растворителя с растворенным веществом достигается,
как и для всех прочих случаев, при равенстве нулю изменения сво-
бодной энергии системы. Поскольку в твердой фазе концентрация
вещества постоянна, константой равновесия этого процесса являет-
ся непосредственно величина растворимости. Ее можно выразить
через термодинамические параметры процесса растворения:
Растворы
93
С = е .лаят = е ,siRe -ahirt (5.5)
Первый сомножитель является температурно независимым. Вклад
энтальпийного фактора полностью определяет зависимость раство-
римости соединения от температуры и может варьировать в доста-
точно широких пределах. При растворении некоторого вещества
затрачивается энергия, необходимая для разрушения его кристал-
лической решетки. С другой стороны, при этом высвобождается
энергия сольватации молекул или ионов. Их соотношением в основ-
ном и определяется энтальпия растворения. Для большинства веществ
она является величиной положительной, что соответствует росту ра-
створимости с ростом температуры. Известным примером таковых
является нитрат аммония. Для ряда веществ, среди которых преоб-
ладают безводные соли и низководные гидраты, она меньше нуля
(Na,SO4). Наконец, для некоторых соединений, например для хло-
рида натрия, энтальпия растворения близка к нулю. Растворимость
последних практически не зависит от температуры.
Причиной растворимости веществ с положительной энтальпией
растворения, как было показано в предыдущем разделе, является
существенный вклад энтропийного фактора. Действительно, при
растворении происходит разрушение упорядоченных кристаллов.
Кроме того, после этого молекулы растворителя перемешиваются с
молекулами или ионами растворенного вещества. Поэтому соб-
ственный вклад энтропийного фактора достаточно велик. Для его
компенсации необходимо, чтобы энергия кристаллической решет-
ки была очень большой, что характерно для соединений с полиза-
рядными катионами и анионами. Примерами могут служить суль-
фиды поливалентных металлов. Именно для таких соединений ока-
зывается характерной низкая растворимость.
Проиллюстрировать полученную зависимость удобно на приме-
ре галогенидов щелочных металлов (табл. 5.1). Легко показать, что
энергия кристаллической решетки для них будет расти с понижени-
ем суммы радиусов катиона и аниона, а также при приближении их
Таблица 5 1
Растворимость галогенидов щелочных металлов (г на 100 г воды)
НаГ 1 М+ Li Na К Rb Cs
F 0.3 4.1 95 300 565
CI 78 36 34 91 186
Вг 177 103 66 98 115
I 165 179 144 152 78
94
Глава 5
величин друг к другу. С другой стороны, энергия гидратации одно-
значно растет с понижением размера ионов. Поэтому растворимость
должна достигать минимальных значений для случая катионов и
анионов с близкими радиусами. Действительно, максимальной в
данном ряду соединений является растворимость иодида и бромида
лития, иодида натрия, фторида рубидия, фторида и хлорида цезия.
Минимальна же она для фторидов лития и натрия, а среди солей калия
и рубидия - у хлоридов.
5.2. Давление насыщенных паров над раствором
До сих пор мы оперировали только фазовыми диаграммами для
конденсированных систем. Однако достаточно важным оказывается
и равновесие, устанавливающееся между раствором и газом. Рас-
смотрим систему из двух взаимно неограниченно растворяющихся
жидкостей А и В. Каждая из них создает над раствором некоторое
давление насыщенных паров. Равновесие между жидкостью и паром
достигается при равенстве химических потенциалов обоих компо-
нентов. В частности, для компонента А идеального раствора можно
записать:
RT\npo = (ц°(ж) - ц°(г)) + RT 1пХд, (5.6)
где Ха - мольная доля компонента А в растворе. Для чистого раст-
ворителя, т.е. при X = 1, имеем
AT ln(p°ff) = (ц°(ж) - д°(г)) , (5.7)
откуда
RT\npa = ЛПпВД + АПпХ илиЛ = У ра°. (5.8)
Это выражение называется законом Рауля. Смысл его состоит в
том, что парциальное давление насыщенного пара растворителя и
растворенного вещества над раствором пропорционально их мольным
долям. Это соотношение может быть получено и при рассмотрении
кинетики испарения, скорость которого пропорциональна мольной
доле вещества на поверхности, а скорость конденсации - давлению
насыщенных паров. Аналогичное выражение можно получить и для
второго компонента.
Общее давление паров над раствором будет равно
Р = ХаР« + W = *Рай + (1 - W =Pb + W^°) (5.9)
Графическим выражением полученного соотношения является
прямая, соединяющая точки, соответствующие давлению паров
Растворы
95
чистых компонентов (рис. 5.3). Из этого соотношения при р° = О
можно получить и другую формулировку закона Рауля: относительное
понижение давления насыщенного пара растворителя равно мольной доле
растворенного вещества:
(р,’-р„Ур^^ь- (5.10)
Рис.5.3. Г рафическая
интерпретация закона
Рауля
Для вывода данного выражения использовалось предположение
о том, что раствор является идеальным, то есть силы взаимодействия
между молекулами растворителя и растворенного вещества равны
таковым для индивидуальных веществ. Энтальпия растворения в этом
случае равна нулю и каждый из компонентов ведет себя независимо
от других.
Строго говоря, критериям идеального не удовлетворяет ни один
реальный раствор. Исключение можно сделать лишь для случая, ког-
да оба вещества отличаются лишь изотопным составом. Предполо-
жим, что молекулы растворителя и растворенного вещества взаимо-
действуют друг с другом сильнее, чем в чистых веществах. Тогда каж-
дая частица растворенного вещества на поверхности связывает до-
полнительные молекулы растворителя, эффективно увеличивая свой
размер, уменьшая число частиц на поверхности и понижая тем самым
давление насыщенного пара (с точки зрения термодинамики это
означает понижение химического потенциала жидкости в растворе).
Примером может служить система эфир-хлороформ. Если же взаи-
модействие растворителя с веществом слабее, чем в индивидуальной
фазе (метиловый спирт-октан), ситуация меняется на обратную. Это
так называемые случаи отрицательного и положительного отклоне-
ния от закона Рауля. Графики давления пара над растворами типа
96
Глава 5
жидкость-жидкость для таких случаев приведены на рисунке 5.4. В
первом случае молекулы постороннего компонента как бы связывают,
а во втором - выталкивают молекулы растворителя. Такое поведение
однозначно определяется энтальпией растворения. Для первого типа
растворов она меньше, а для второго - больше нуля.
Рис.5.4. Давление насыщенных паров над системами эфир-хлороформ при 20° С (а)
и пиридин-вода при 80° С (б)
Для согласования поведения реальных растворов с законом Рауля
вводят понятие активности. Активность растворителя можно вы-
разить, например, через отношение давления его паров над данным
раствором к чистому растворителю. Активность вещества в раст-
воре представляет собой некоторую вспомогательную величину,
позволяющую оперировать с данным раствором как с идеальным,
ai=Pi!Pi> (5.И)
ri = ai!Xi = pi!Xip^ (5.12)
Величина коэффициента активности для разбавленных раство-
ров близка к единице. В целом она является функцией температуры,
давления и ионной силы раствора. Последняя представляет собой
полусумму произведений концентраций содержащихся в растворе
ионов на квадрат их заряда. Поэтому для растворов смеси веществ
коэффициент активности в первую очередь зависит не от концент-
рации данного компонента, а от обшей концентрации раствора.
Растворы
97
Коэффициент активности компонента характеризует степень от-
клонения его свойств в растворе от таковых для идеального раст-
вора. Так, над раствором, содержащим 0.042 мольных долей эти-
лового спирта, парциальное давление водяных паров составляет при
20°С 2.24-103 Па. Над чистой водой оно достигает 2.33-103 Па.
Следовательно, активность спирта составляет:
«п = (2.33 - 2.24)/2.33 = 0.039,
а коэффициент активности усп = 0.039/0.042 = 0.92.
Аналогично для воды аъ =1- 0.039 = 0.961 и yg = 0.961/0.958 =
= 1.003.
Этот пример весьма характерен для большинства водных ра-
створов. Коэффициенты активности для растворенных веществ
обычно несколько меньше единицы.
Многие растворы по свойствам близки к идеальным, и отклонения
от закона Рауля для них не столь велики. Примерами являются си-
стемы бензол-толуол, гексан-гептан, этиловый-пропиловый спирт.
Особенно хорошо закон Рауля соблюдается для разбавленных ра-
створов, для которых величина коэффициента активности близка к
единице.
5.3. Диаграммы кипения для бинарных систем
Рассмотрим теперь равновесие в двухкомпонентной системе
жидкость-газ. Пусть имеется 50%-ный раствор (рис. 5.5а, точка У).
Давление паров над ним можно найти по закону Рауля из приве-
денного графика. Из того же графика видно, что состав пара суще-
ственным образом отличается от состава жидкости. Газовая фаза
оказывается обогащенной легкокипящим компонентом (5).
Понятно, что полностью сконденсировать такой пар (состав Z) в
жидкость можно лишь при существенно большем давлении. Послед-
нее выравнивается с давлением насыщенных паров для исходной
жидкости лишь в случае чистых компонентов.
Таким образом, для любого состава пара можно получить две
различные температуры - начала и завершения процесса конденсации.
Теперь рассмотрим ТХ проекцию фазовой диаграммы для раствора
двух неограниченно растворимых друг в друге жидкостей (см. рис.
5.56). Проекция эта представляет собой узкий лепесток, соединяющий
земпературы кипения чистых компонентов. Пусть для жидкости А
температура кипения ниже. При нагревании раствора, находящегося
98
Глава 5
Рис.5.5. Состав жидкости и пара для бинарной смеси (а) и ТХ диаграмма для системы
азот-кислород(б)
в некоторой точке 1, происходит повышение давления насыщенного
пара. Когда оно достигнет величины, равной давлению в данной
системе, жидкость начнет кипеть. Температура кипения определяется
точкой пересечения вертикальной линии, проведенной из исходной
точки, с нижней линией графика (7,). При этом равновесный состав
пара можно найти, проведя горизонтальную линию из этой точки.
Нетрудно убедиться, что пар в таком случае оказывается обогащен-
ным низкокипящим компонентом В. Верхняя же линия соответствует
началу конденсации пара данного состава (Д). При этом жидкость
всегда обогащена относительно пара высококипящим компонентом.
Рис.5.6. Диаграммы кипения для систем хлористый водород-вода (я) и мстилаль-
сероуглсрод(б)
Для некоторых бинарных систем кривые кипения могут проходить
через максимум или минимум (рис. 5.6). Смесь, характеризующаяся
максимальной температурой кипения, называется азеотропной.
Растворы
99
Хорошо известна, например, азеотропная смесь воды и хлорида
водорода, содержащая 20% последнего. Температура ее кипения
108.6°С. При этом азеоторопному составу всегда соответствует ми-
нимальное давление насыщенных паров.
5.4. Температуры кипения растворов
Рассмотрим сечения диаграммы состояния для систем вода-соль
при постоянной концентрации последней. Как отмечалось ранее,
введение в растворитель некоторого вещества приводит к дополни-
тельному разупорядочению за счет разрушения кристаллической ре-
шетки и повышения термодинамической вероятности системы при
смешении. Следствием этого является увеличение энтропии жидкости
и понижение ее химического потенциала
р. = + АПпха = ца° + АПп(1-х). (5.13)
Таким образом, выравнивание его с химическим потенциалом
паров растворителя будет достигаться при более высоких темпера-
турах, что соответствует повышению температуры кипения с ростом
концентрации растворенного вещества. Поэтому на рТ-диаграмме
кривая зависимости давления насыщенных паров от температуры
несколько смещается вниз, хотя профиль ее остается практически
неизменным (рис. 5.7).
Рис.5.7. Схема изменения фазовой дна! раммы воды при введении в нее растворимых
примесей
100
Глава 5
В отличие от чистого растворителя, температура кипения раст-
вора при постоянном давлении не остается постоянной. Для двух-
компонентной системы правило фаз выглядит следующим образом:
с = 2+ 2-/=4-/. Поэтому даже при постоянном давлении равновесие
между двумя фазами остается моновариантным и при изменении
концентрации раствора температура кипения меняется. Действи-
тельно, по мере упаривания концентрация раствора возрастает, что
приводит к повышению температуры его кипения. Под температурой
кипения раствора понимается та температура, при которой давление
пара растворителя достигает одной атмосферы.
Из рисунка 5.7 видно, что фигуры дР1Т1 и ОАД , ограниченные
изобарными и изотермическими линиями и отрезками кривых зави-
симости давления паров от температуры, в первом приближении мож-
но считать подобными треугольниками. Тогда
АД /АД = кР; /ЬР,.
В соответствии с законом Рауля понижение давления пара пропор-
ционально концентрации соли. Таким образом,'
АД /АР, = Д /Д = АД /АД, (5.14)
т.е. повышение температуры кипения пропорционально мольной доле
растворенного вещества.
Мольные доли неудобны для практических расчетов. Поэтому
гораздо чаще пользуются понятием моляльной концентрации (коли-
чество молей вещества в 1 000 г растворителя). Между ними существует
достаточно простое соотношение:
Х-т!(т + 1000/ЛД), (5.15)
где т - моляльность раствора, а М - молекулярная масса раствори-
теля. Для невысоких концентраций т<<1000/М и т ~ 1000 XIM.
Поэтому полученное соотношение можно переписать в виде:
АТ = Дт, (5.16)
где Е - эбулиоскопическая постоянная - константа для данного
растворителя. В случае воды она равна 0.52°С. Действительно, для
всех эквимоляльных растворов неэлектролитов температура кипения
повышается одинаково. Так, 0.02 моляльные водные растворы таких
разных веществ, как мочевина, сахароза и глицерин, кипят при одной
и той же температуре - 100.01 °C.
Проведем оценку величины эбулиоскопической постоянной. Для
этого приравняем величины химических потенциалов растворителя
в газовой и жидкой фазах при температуре кипения:
цж(7Ч АД = цж°(Т+ АД + Р(Р+ АД 1п(1-х )= Иг(Т+ АД , (5.17)
Растворы
101
где Т- температура кипения чистого растворителя, а АТ- ее повы-
шение для раствора, содержащего мольную долю растворенного
вещества, равную xs. Преобразуем это выражение:
А 1п(1-х> [цг(/+ АТ) - цж°(Т+ ДТ)] /(Т+ АТ) =
= &G(T + &Т)/(Т + &Т). (5.18)
Полагая, что энтальпия и энтропия испарения не зависят от тем-
пературы, получаем:
7?1п(1-х) = АЯ/(Т+А7)-А5. (5.19)
Аналогично для чистого растворителя:
Я1п1 = 0 = ДЯ/Т-А5. (5.20)
Выразив величину энтропии кипения из последнего уравнения,
подставив ее в соотношение (5.19) и заменяя 1п(1-х$) на -xs (что
справедливо лишь для низких концентраций), получим:
Rxx = -AHI(T+ АТ) + АЯ/Т= А#Л77(Т+А7)Т. (5.21)
Для разбавленных растворов Т + &Т~ Тит ~ 1000 х/М.
Откуда
АТ= RT2mM/\W) ЛН. (5.22)
или Е~РТ2М/Ш0 кН. (5.23)
5.5. Температуры замерзания растворов
Поскольку твердые вещества малорастворимы во льду, кривая
зависимости температуры сублимации от давления для двухкомпо-
нентных систем остается практически неизменной. С другой стороны,
температура тройной точки понижается вместе с понижением дав-
ления насыщенных паров раствора. Следовательно и температура
кристаллизации также должна понижаться. Этот эффект мы наблю-
дали и при рассмотрении фазовых ТХ-диаграмм (рис. 5.8). На по-
следних, как и на рисунке 5.7, можно выделить подобные треугольники
ОХ,?, и ОХ,Т,. Из этого следует, что понижение температуры замер-
зания пропорционально мольной доле растворенного вещества. На
практике, однако, так же как и в предыдущем случае, пользуются
моляльными концентрациями. Таким образом, итоговое уравнение
можно представить в виде:
&Тм=Кт, (5.24)
где К- криоскопическая постоянная. Для получения этого результата
102
Глава 5
Рис.5.8. Фрагмент диаграммы состояния системы соль-вода
в количественном виде, включающем только термодинамические
параметры, можно полностью воспроизвести преобразования,
проведенные нами для получения эбулиоскопической постоянной. В
конечном выражении, которое строго аналогично (5.22), вместо
энтальпии кипения задействована энтальпия плавления. В случае
воды криоскопическая постоянная равна 1.86°С. Следовательно
температуры замерзания 0.02-моляльных растворов неэлектролитов
составят -0.037°С. Следует отметить два важных фактора. Во-первых,
под температурой замерзания раствора, как и в предыдущем случае,
подразумевается температура начала его замерзания. Поскольку при-
меси малорастворимы во льду, то вымораживается практически чи-
стый лед и концентрация раствора повышается, а вместе с этим по-
нижается и сама температура замерзания. Таким образом, даже при
постоянном давлении процесс замерзания не является инвариантным.
Во-вторых, по мере повышения концентрации растворов тангенс угла
наклона кривой замерзания на рисунке 5.8 меняется. Поэтому до-
статочно точные расчеты удается провести только для разбавленных
растворов. Так, на основании данных о температуре и составе системы
в криогидратной точке обычно удается лишь приблизительно судить
о величине криоскопической постоянной.
5.6. Осмотическое давление
Хорошо известно, что при различии химических потенциалов
какого-либо компонента в разных участках раствора между ними
будет протекать процесс диффузии вплоть до выравнивания
Растворы
103
концентраций во всех точках. Схожая картина наблюдается, если
внести в сосуд с чистым растворителем емкость с раствором, отделен-
ную от него полупроницаемой перегородкой (рис. 5.9).
Рис.5.9. Схема проявления
осмотического давления
Примером таковых могут являться пергамент, кожа и ряд других
природных тканей. Их отличительным свойством является способ-
ность пропускать через себя молекулы воды и не пропускать молекулы
и ионы растворенных веществ. При этом молекулы растворителя
преимущественно диффундируют в раствор до тех пор, пока уровень
в сосуде с раствором не поднимется на некоторую высоту. В конечном
итоге разница уровней столбов жидкости уравновешивает процесс
диффузии растворителя. Этот процесс можно предотвратить, напри-
мер, создав в сосуде с раствором некоторое избыточное давление.
Величину давления, которую надо приложить к раствору, чтобы
предотвратить диффузию в него через полупроницаемую перегородку
чистого растворителя, называют осмотическим давлением.
Рассмотрим термодинамику происходящих при этом явлений. При
условии равновесия в двух сосудах должно выполняться условие
равенства химических потенциалов растворителя (ца! = цо°). Про-
дифференцируем это уравнение по мольной доле растворителя (хф и
давлению (р) в сосуде с растворителем:
= (d^/dx)/>Tdxa + (dpPldp\rdp = . (5.25)
Поскольку давление и концентрация во втором сосуде не влияют
на первый, химический потенциал чистого растворителя остается
постоянным и = 0. Кроме того,
(d\La4 dp) = d'-GJ dxdp = (dValdx) = Va (5.26)
и (ф/1 dxa) = RT[<71 nxa/ dxa], (5.27)
104
Глава 5
откуда RTd Inxa + Vdp = 0 (5.28)
и dp = - RT/Vad\nxa. (5.29)
Интегрируя это уравнение от ха = 1, р = р0 до ха и р0 + п , где
р0 - внешнее давление, п - осмотическое давление, a Va - мольный
объем растворителя, получаем:
Ро + я -Ро = - [In ха - In (1)]
к =-RTIVJn ха. (5.30)
При малых концентрациях растворенного вещества
In ха = ln( 1 - xs) ~ -xs и х ~ Azs / Az(i Ха И = И _ра= V.
Откуда л = -RT/Va\n ха = xs RTI ха Va= xs RT/V = CRT, (5.31)
где Xs и Ха - число молей растворенного вещества и растворителя в
системе, а С - молярная концентрация. Это соотношение называ-
ется уравнением Вант-Гоффа и строго выполняется только для раз-
бавленных растворов (которые также подчиняются закону Рауля).
Для концентрированных растворов приходится пользоваться ак-
тивностью вместо концентрации.
Осмотическое давление, как следует из приведенного вывода,
является тем добавочным давлением, которое уравновешивает
понижение химического потенциала растворителя, определяемое
наличием в нем растворенного вещества. Следует заметить, что явление
осмоса играет значительную роль в жизнедеятельности живых
организмов. Оно обеспечивает приток воды внутрь клеток и, создавая
внутри них избыточное давление, обеспечивает упругость тканей.
Напротив, при консервировании продуктов концентрированные
растворы соли или сахара вытягивают воду из бактерий через ок-
ружающие их полупроницаемые перегородки. В результате этого
бактерии сжимаются и становятся недееспособными. Размножение их
подавляется и поэтому консервированные продукты практически не
портятся.
5.7. Определение молекулярных масс на основе
коллигативных свойств
Рассмотренные свойства (осмотическое давление, понижение тем-
пературы плавления и повышение температуры кипения растворов)
определяются только концентрациями растворенных веществ и (за
Растворы
105
единственным исключением) не зависят от их свойств. Действительно,
как бы прочно или, напротив, слабо не связывало растворенное ве-
щество молекулы воды, число его частиц остается строго неизменным.
Все свойства растворов, определяемые лишь количеством частиц ра-
створенного вещества в единице объема, объединяются под названием
коллигативных свойств. Вышесказанное позволяет использовать
эбулиоскопию, криоскопию, осмотическое давление и давление насы-
щенных паров растворителя для определения молекулярных масс
различных веществ.
Рассмотрим возможность такого измерения на примере эбулио-
скопии. Для решения поставленной задачи измеряют температуры
кипения некоторого заранее отмеренного количества растворителя
после последовательного добавления к нему навесок вещества с из-
вестной молекулярной массой. По графику зависимости температуры
кипения от концентрации вычисляют эбулиоскопическую постоянную
и, проделав аналогичную процедуру для исследуемого вещества, оп-
ределяют его молекулярный вес. Тангенсы угла наклона графиков
зависимости температуры кипения раствора от массы прибавленно-
го вещества обратно пропорциональны их молекулярным массам
(рис. 5.10).
Рис.5.10. График зависимости
температуры замерзания от
молярности раствора
Следует особо обратить внимание на то, что концентрации
растворов должны быть достаточно низкими, чтобы коэффициен-
ты активности приближались к единице. При этом возникает оп-
ределенное противоречие. С одной стороны, точность измерения
молекулярной массы в основном определяется точностью измерения
температуры кипения или плавления, Естественно, что чем больше
эти температуры отклоняются от таковых для чистого растворителя,
тем выше точность измерения АГ. С другой стороны, чем выше
концентрация вещества, тем больше коэффициент активности
отклоняется от единицы. Поэтому на практике приходится искать
разумный компромисс.
106
Глава 5
5.8. Сольватация растворенных веществ
Процесс растворения, как известно, не сводится к простому пе-
ремешиванию частиц растворяемого вещества с растворителем. При
этом протекают также и различные явления, обусловленные хими-
ческим взаимодействием. Так, например, хорошо известно, что при
растворении спирта в воде выделяется значительное количество теп-
ла, что обусловлено протеканием процессов взаимной сольватации.
Процесс взаимодействия растворенного вещества с водой называется
гидратацией, а в более широком смысле - с растворителем вообще -
сольватацией. Все растворители можно разделить на полярные и
неполярные в соответствии с полярностью или неполярностью обра-
зующих их молекул. При рассмотрении возможности растворения
вещества в гом или ином растворителе в первом приближении обычно
используют достаточно простой принцип: подобное растворяется в
подобном. В соответствии с этим неполярные растворители будут
лучше всего растворять неполярные вещества с небольшими моле-
кулами, образующие в твердом состоянии молекулярные кристаллы.
Основной тип взаимодействия между частицами в таких растворах -
слабые Ван-дер-Ваальсовы связи. Энергия их невелика и, как правило,
имеет порядок 10—20 кДж/моль. С другой стороны, для таких
соединений невелика и энергия кристаллической решетки, поэтому
АН растворения для них, как правило, слабо отличается от нуля.
Энергия же, высвобождающаяся при образовании кристаллов
полярных или ионных соединений, обычно существенно выше.
Поэтому они не склонны к растворению в неполярных растворителях.
Другим важным типом растворителей являются полярные
жидкости, в которых могут растворяться полярные и ионные ве-
щества. Среди полярных растворителей, в свою очередь, можно
выделить группу веществ, являющихся донорами и акцепторами
электронной пары. Типичными представителями первых являются,
например, ацетон, жидкий аммиак и т.д. Ко вторым относятся азот-
ная и хлорная кислоты. Следует заметить, что подавляющее боль-
шинство растворителей-доноров электронной пары’ может также
выступать и в качестве ее акцептора. Характерным примером явля-
ется вода, атом кислорода которой имеет две неподеленные электрон-
ные пары, а протоны могут образовывать водородные связи с ве-
ществами, содержащими электроотрицательные атомы. Энергия
сольватации в таких растворителях существенно выше, и поэтому они
могут растворять вещества с достаточно прочной кристаллической
решеткой.
Растворы
107
Рассмотрим процесс взаимодействия вещества с растворителем
на примере растворов ионных солей в воде (рис. 5.11). Процесс
растворения начинается с окружения ионов, находящихся на по-
верхности вещества, молекулами растворителя. При этом катионы
координируют вокруг себя отрицательные, а анионы - положитель-
ные концы диполей Н,0 Это облегчает отрыв ионов с поверхности
кристаллической решетки и их переход в раствор.
Рис. 5.11. Схема сольватации поверхности ионного кристалла в воде
В последнем катионы и анионы чаще всего размещают вокруг
себя строго заданное число молекул растворителя, определяемое
их координационными возможностями и размером молекул ра-
створителя. В случае воды для большинства катионов оно составляет
4-8. Так, например, ионы трехвалентного хрома всегда находятся в
окружении шести атомов кислорода воды, образуя устойчивую ча-
стицу [Cr(H,O)J1+, медленно обменивающуюся водой с окружающим
раствором. Чаще всего координационное число лимитируется раз-
мером катиона, поэтому чем больше последний, тем больше молекул
воды может содержать такой ассоциат. Так, в координационную
сферу крупных катионов циркония и гафния могут входить 8 или 9
молекул воды, а в случае бериллия - всего 4. С другой стороны,
прочность их удерживания зависит от поляризующей способности
катиона, определяющейся отношением заряда катиона к квадрату
его радиуса. Поэтому в ряду катионов щелочных металлов эффек-
тивная степень гидратации понижается от 5.5 у лития до 3 у натрия и
1 у цезия, хотя реальное число молекул воды, входящее в их первую
координационную сферу составляет 4, 6 и 8.
108
Глава 5
Процессам гидратации подвержены и анионы. В этом случае
молекулы воды координируют их посредством водородных связей,
например, типа НОН- -ОРО33 . Образовывать такие связи может
каждый атом кислорода аниона, а прочность их в первом прибли-
жении можно охарактеризовать плотностью отрицательного заряда
на анионе. Так, водородные связи молекул воды с перхлорат-ионом
значительно слабее, чем между собой (IIOH--OH,). Поэтому ион С1О(
сравнительно слабо подвержен гидратации.
Аналогичного типа процессы происходят и в других полярных
растворителях. Природа протекающих при этом явлений сохраняется
неизменной, хотя взаимодействие с различными растворителями
может иметь свои особенности. Так, ацетон, например, ввиду низкого
заряда на атомах водорода не склонен к образованию ими водород-
ных связей. Поэтому сольватация аниона в нем затруднена.
5.9. Электролитическая диссоциация
Итак, мы пришли к заключению, что в растворе ионные кри-
сталлы и некоторые полярные молекулы могут диссоциировать на
отдельные ионы. Это подтверждается, например, тем, что из водного
раствора, содержащего хлорид калия и сульфат натрия, при выпа-
ривании выделяются четыре продукта - хлориды и сульфаты натрия
и калия. Диссоциация приводит к существенному изменению кол-
лигативных свойств растворов, определяемых количеством частиц
растворенного вещества в единице объема. В соответствии с этим в
разбавленных эквимолекулярных растворах хлорида натрия, дис-
социирующего на две частицы, и мочевины осмотическое давление,
величины понижения давления насыщенных паров воды, изменения
температур замерзания и кипения различаются практически вдвое.
Для растворов хлористого кальция их значения возрастают еще в
полтора раза. Для приведения этих величин в соответствие с зако-
номерностями, характеризующими коллигативные свойства, вводят
понятие изотонического коэффициента (I), который показывает, на
сколько частиц распадается одна молекула растворенного вещества.
Изотонический коэффициент можно определить, например, из отно-
шения экспериментальной величины понижения давления водяного
пара к рассчитанному теоретически. Для разбавленных растворов
он близок к теоретическому и достигает для хлорида кальция - трех,
для хлорида алюминия - четырех, а для его сульфата - близок к пяти.
Растворы
109
В то же время для ряда веществ, примером которых может служить
уксусная кислота, он лишь немного превышает единицу.
Долю молекул, распавшихся в растворе на ионы, называют степенью
электролитической диссоциации (а). Рассмотрим взаимосвязь этой
величины с изотоническим коэффициентом. Пусть некоторый элек-
тролит распадается на п частиц. Тогда общая концентрация частиц
в растворе составит С + (д-1)осС. Величину изотонического коэф-
фициента можно получить, разделив это значение на исходную кон-
центрацию: i = 1 + (и-1)ос. Откуда
а = (/-1)/(«-1). (5.32)
Как будет показано несколько позже, эта величина не является
постоянной, однако, используя ее, все электролиты можно условно
разбить на сильные (со степенью диссоциации более 50% для раст-
воров средней концентрации (около 1М)), слабые (со степенью диссо-
циации менее 10%) и электролиты средней силы.
К сильным электролитам в водных растворах относится зна-
чительная часть неорганических кислот (НС1, HNO3, НС1О4, H,SO4
и т.д.), гидроксиды щелочных и щелочноземельных металлов и
практически все соли. К слабым электролитам относятся практически
все органические и некоторые неорганические кислоты (СИ3СООН,
НСО2Н, Н,СО,, H,SO3, НС1О и т.д.), гидроксиды большинства пе-
реходных элементов (за исключением AgOH и Т1ОН) и некоторые
соли, в основном элементов подгруппы цинка (HgCl,, Hg(CN),, Cdl,,
Znl,, Fe(CNS)3). Круг электролитов средней силы достаточно узок и
очерчен весьма условно. К ним можно отнести, например, такие ки-
слоты, как щавелевая, фосфорная и некоторые другие.
Степень диссоциации, как отмечалось ранее, не является посто-
янной - величина ее существенно зависит от целого ряда факторов.
Электролитическая диссоциация слабых электролитов является пол-
ноправным обратимым химическим процессом, который характе-
ризуется собственной константой равновесия. Рассмотрим взаимо-
связь различных характеристик электролитической диссоциации на
примере водного раствора уксусной кислоты. Диссоциация ее проте-
кает по схеме
СН3СООН +Н,0 Нзо+ +СН3СОО-. (5.33)
Поскольку концентрация воды, по крайней мере для разбавленных
растворов, есть величина постоянная, она не включается в константу
равновесия и последнюю можно выразить следующим соотношением:
К = [СН3СОО-][Н3О+] / [СН3СООН ]. (5.34)
но
Глава 5
Пусть суммарная концентрация раствора уксусной кислоты рав-
на С, а степень диссоциации - а. Тогда [СН3СОО'] = [Н3О+] = осС,
[СН^СООН] = (l-cx)C. На основании этого выражение для константы
диссоциации можно представить в виде:
К = Са2/(1-а). (5.35)
Для малых степеней диссоциации, когда (1-ос) близко к единице,
полученное выражение можно упростить:
к = с сГ или а =Vat /с. (5.36)
Это выражение иногда называется законом разведения Оствальда.
Смысл его заключается в том, что для слабых бинарных электролитов
степень диссоциации прямо пропорциональна квадратному корню из
константы диссоциации и обратно пропорциональна квадратному корню
из концентрации раствора. Таким образом, при разбавлении раствора
степень диссоциации слабых электролитов возрастает. Приведенные
в таблице 5.2 данные наглядно иллюстрируют правило разведения
Оствальда.
Таблица 5.2
Степень диссоциации уксусной кислоты в водных растворах различной
концентрации при 298 К
С(СНзСООН), м 1 0.1 0.01 0.001
а. % 0.42 1.3 4.1 12.4
Подойдем теперь к процессу диссоциации слабого электролита с
точки зрения термодинамики. Поскольку число частиц в ходе
диссоциации увеличивается, энтропия ее является величиной по-
ложительной. С другой стороны, константа диссоциации мала. Из
уравнения
RT\nK = -\H + TkS
можно заключить, что энтальпия диссоциации слабых электроли-
тов положительна и весьма велика. Следовательно, константа
диссоциации будет быстро возрастать с увеличением температуры.
Поэтому температура существенно влияет на степень гидролиза
(диссоциация слабых электролитов усиливается при нагревании).
Другим важным фактором является присутствие одноименных
ионов. Так, например, цинк при обычных условиях с заметной ско-
ростью реагирует с IM раствором уксусной кислоты
Zn + 2Н+ = Zn2+ +НД. (5.37)
Растворы
111
Внесение в реакционный сосуд ацетата натрия заметно замедляет
скорость этой реакции. Наблюдаемый эффект однозначно связан с
понижением концентрации ионов водорода. Действительно, в про-
цессе диссоциации кислоты образуется сравнительно небольшое коли-
чество ацетат-ионов. Поэтому с достаточной степенью достоверности
можно полагать, что концентрация их в растворе практически пол-
ностью определяется концентрацией в нем ацетата натрия. Тогда из
выражения для константы равновесия получаем:
№aC[NaCH3COO]/(l-a)C или а / (1-а) = К! [NaCH3COO]. (5.38)
Поскольку и без того малая степень диссоциации при этом пони-
зится, (1-ос) « 1 и это выражение можно переписать в виде:
ос =A/[NaCH,COO] . (5.38а)
Таким образом, степень диссоциации слабого бинарного элек-
тролита будет обратно пропорциональна концентрации в растворе
соли, содержащей одноименный ион. На оснований этого можно
построить график зависимости концентрации ионов водорода от
количества электролита с одноименным ионом (рис. 5.12). Для опи-
сания кислотности растворов, как известно, пользуются величиной
pH, которая равна отрицательному логарифму концентрации (ак-
тивности) ионов водорода в растворе. Из исходного уравнения
диссоциации можно получить следующее соотношение:
[Н3О+] = осС
и
p№-lgaC = -lg(^C/|NaCH3COO]) = lg [NaCH}COO] - IgAC. (5.39)
Рис.5.12. Зависимость pH
IM раствора уксусной
кислоты от концентрации
ацетата натрия
Аналогично, диссоциация подавляется присутствием в растворе
сильной кислоты, понижающей pH раствора.
112
Глава 5
5.10. Растворы сильных электролитов
Сильные электролиты при попадании в раствор диссоциируют
нацело или почти нацело. При этом понятие константы диссоциации
для них неприменимо. Действительно, величина знаменателя в вы-
ражении типа 5.3 в этом случае чаще всего не определена. Можно
полагать, что сильные электролиты диссоциированы нацело. При
разбавлении 0.1М раствора сильного бинарного электролита в 100
раз числитель возрастет практически в 104раз. В то же время нетрудно
убедиться, что pH 1М раствора соляной кислоты равно не нулю, а
0.1. Для объяснения этого явления вводится понятие кажущейся
степени диссоциации, характеризующей отношение активности ионов
сильных электролитов к их истинной концентрации. По мере
концентрирования раствора, как и для слабых электролитов, кажу-
щаяся степень диссоциации понижается, однако масштабы этих изме-
нений принципиально различаются.
Действительно, попадая в раствор, хлорид натрия полностью
диссоциирует на ионы. При этом трудно ожидать, что в растворе
существует заметная концентрация молекул NaCl. Однако за счет
кулоновского взаимодействия образуется некое подобие упорядо-
ченного расположения ионов, при котором катионы оказываются
преимущественно в окружении анионов и наоборот (рис. 5.13а)-
Рис.5Л3. Схема упорядочения ионов в растворе (л) и тормозящего дейст вия
“ионной шубы” (б)
Растворы
113
Характеристическое время образования и разрушения такой
“ионной шубы” имеет порядок 10’8с. Связь ее с ионом достаточно
эфемерна. Поэтому шуба оказывается как бы без застежек. И при
попадании раствора в электрическое поле, например, равновесие
нарушается. Катионы движутся к катоду, а анионы - к аноду. При
этом “ионная шуба сползает” и располагается с противоположной к
направлению движения иона стороны. Вследствие этого движение
иона тормозится за счет его электростатического взаимодействия с
противоположно заряженным окружением (см. рис. 5.136). По мере
увеличения концентрации раствора межатомные расстояния умень-
шаются. При этом кулоновское взаимодействие усиливается и ионы
проявляют большую склонность к ассоциации. Это выражается в том,
что гидратированные катион и анион получают возможность сов-
местного существования в растворе в течение некоторого времени.
Такие образования получили название ионных пар. Степень их
ассоциации растет с понижением радиуса и увеличением заряда ка-
тионов. Кроме того, степень ассоциации растет с уменьшением ди-
электрической проницаемости растворителя. Поэтому кажущаяся
степень диссоциации существенно понижается при переходе от водных
растворов к спиртовым.
На этом этапе целесообразно вернуться к понятию активности.
В случае сильных электролитов она играет роль вспомогательной
величины, которую можно использовать вместо концентрации ионов
для термодинамического описания системы. Основной причиной от-
клонения ее от концентрации являются межионные взаимодействия
в растворе. Возвращаясь к рассмотренному вначале примеру, можно
заключить, что активность ионов водорода в 1М растворе соляной
кислоты равна 0.79. И, поскольку реальная их концентрация равна
единице, коэффициент активности также равен 0.79. Коэффициент
активности безразмерен, а активность выражается в тех же единицах,
что и концентрация. Поэтому не следует удивляться, что ранее ак-
тивность использовалась в качестве эквивалента мольной доли, а в
настоящем случае (и наиболее часто) - в качестве эквивалента мо-
лярной концентрации.
Строго говоря, активности катиона и аниона в растворе могут
быть и неравны. Тем не менее обычно приписывают некую активность
всему электролиту в целом. При этом полагают, что искомая величина
равна среднему геометрическому из активностей анионов и катионов
с учетом их количества. В общем случае для соединения
а = (5.40)
114
Глава 5
Прочность парных взаимодействий в растворе определяется не
концентрацией данного электролита, а концентрацией и зарядом су-
ществующих в растворе частиц вообще. Действительно, при совмест-
ном растворении эквимолекулярных количеств хлорида натрия и
нитрата алюминия активность хлорид-ионов будет понижаться в пер-
вую очередь за счет взаимодействия с трехзарядными катионами алю-
миния. Поэтому коэффициент активности зависит от некоторой ад-
дитивной величины, называемой ионной силой раствора (7), которая
равна полусумме произведений концентраций всех существующих в
растворе ионов на квадрат их заряда. Разработана и теоретическая
модель для определения электрохимической активности ионов в ра-
створе, основанная на расчете работы удаления ионов друг от друга
в бесконечно разбавленном растворе. Для водного раствора, на-
пример, основанное на этой модели уравнение для определения ко-
эффициента активности может быть представлено в виде:
lg/=-0.51z2 (5.41)
Таким образом, для 0.1М раствора хлористого натрия
1 = 0.5(0.1 • 12 + 0.1-Р) = 0.1, lg/ = -0.51-Р(0.1)ш = -0.16;/= 0.69.
5.11. Произведение растворимости
До сих пор мы говорили о хорошо растворимых сильных элек-
тролитах. Однако многие ионные соли растворяются весьма слабо.
Это обусловлено тем, что энергия кристаллической решетки в них
значительно превышает энергию гидратации. Поэтому наиболее
часто среди малорастворимых встречаются соединения с полиза-
рядными катионом или анионом (например, сульфиды олова и
мышьяка) или с существенным вкладом в энергию связи в кристалле
ковалентной составляющей, которая не может компенсироваться при
сольватации (галогениды серебра и ртути). Процесс растворения
соединения состава XnYm в общем случае можно представить в виде:
А”л Ym (тв) <-> «AZffl+(p-p) + тУДр-р) . (5.42)
Поскольку при равновесии раствор сосуществует с твердой фазой,
активность которой равна единице, константа равновесия реакции
(5.42) выражается соотношением:
[А""+]'![УЛ-]"’ = e^GtRr = e-iH!RT e*SIR =ПР . (5.43)
Таким образом, величина [АУД/УД"’, называемая произведением
растворимости, является константой для данного вещества и зависит
Растворы.
115
только от температуры. Поскольку для малорастворимых веществ
ПР«\, а вклад энтропийного фактора ejM»l, то энтальпия
растворения является величиной положительной (АЯ >0) и произ-
ведение растворимости возрастает с ростом температуры.
Определим, например, растворимость хлорида одновалентной
меди (ПР = 10’6) в воде. Из условия эквивалентности концентраций
катиона и аниона и выражения для произведения растворимости
можно получить систему из двух уравнений:
[Си+] = [СР] и [Cu+][C1-] = Ю-6.
Откуда [Си+] = [СР] = 10’3.
Понизить растворимость данного соединения можно прилива-
нием раствора, содержащего одноименный ион. Так, в 1М растворе
хлорида натрия концентрация ионов хлора практически- полностью
определяется концентрацией последнего и приблизительно равна
единице. Тогда растворимость хлорида меди будет определяться кон-
центрацией в растворе ионов меди и составит
[Си+] = 10б/[СР] = ЮЛ
Повысить растворимость можно, связывая один из ионов, на-
пример за счет комплексообразования. Так, в 0.1М растворе циа-
нистого натрия ионы меди вступают в реакцию по схеме:
Cu+ + 4CN- = [Cu(CN)J-. (5.44)
Константа комплексообразования определяется соотношением:
[Cu+][CN-]4/ [[Cu(CN)J4 = 5-ЮЛ (5.45)
Из последнего соотношения получаем:
[Cu+] = 5-10'28[[Cu(CN)4]3‘] / IO'4 = 5-10-24[[Cu(CN)J3-].
Растворимость хлорида меди в 0.1М растворе цианистого натрия
можно определить из системы:
[Си+][СР] = 10-6 (из ПР),
[СР] = [[Cu(CN)4]3’] (из стехиометрии CuCl),
[Cu+] = 5-1 0-24[[Cu(CN)4]3’] (из 8.14).
Откуда: 5-10’24[СР]2 = Ю’6
и
[СР] = [[Cu(CN)4]3'] = (2-10|7)°-5= 4.5-108.
Полученная величина оказалась много больше исходной кон-
центрации цианистого натрия, что противоречит здравому смыслу.
116
Глава 5
Реально хлорид одновалентной меди будет растворяться до тех пор,
пока концентрация цианид-иона в растворе не будет удовлетворять
соотношению (5.45). При этом она окажется много меньше исходной
величины. Поэтому с хорошей степенью достоверности можно пола-
гать, что растворимость хлорида меди составит четверть от концен-
трации цианистого натрия, то есть 0.025М. Для строгого расчета не-
обходимо решить систему из пяти уравнений с пятью неизвестными.
Однако результат при этом окажется практически тем же. Таким
образом, связывание одновалентной меди в комплексное соединение
позволяет добиться увеличения растворимости ее хлорида. Как отме-
чалось ранее, добиться подобного эффекта можно и за счет нагревания
раствора, однако это приведет к не столь значительному повышению
растворимости.
5.12. Теория кислот и оснований
В соответствии с введенным в 1887 г. Аррениусом определением
кислотами называются вещества, которые в водном растворе диссо-
циируют с образованием ионов водорода, а основаниями - вещества,
диссоциирующие с образованием ионов гидроксила. В последующие го-
ды предложенная теория была несколько скорректирована. Действи-
тельно, протон, обладающий чрезвычайно малым радиусом порядка
10‘7 А, не способен к самостоятельному существованию в растворе.
Обладая огромной поляризующей способностью, он в любой момент
времени оказывается локализованным на каком-либо электроот-
рицательном атоме, имеющем неподеленную электронную пару.
Таким образом. в водных растворах кислот образуются ионы оксония
(Н3О+), в которых все три протона оказываются эквивалентными.
Более того, последние также склонны к образованию водородных
связей с тремя дополнительными молекулами воды, образуя ионы
Н9О4+, которые можно сопоставить с гидратированными ионами ме-
талла (M(H,O)/+ и [(Н3О)(Н,О)3+]). Аналогично и гидроксидные ио-
ны в растворах образуют частицы Н3О, с водородной связью более
прочной, чем между двумя молекулами воды.
Тем не менее даже с этими поправками теория Аррениуса не
позволила распространить определение кислот и оснований на об-
ширную область апротонных растворителей. Поэтому Бренстед пред-
ложил считать кислотами вещества, отдающие протон, а основания-
ми - принимающие его. В соответствии с этим определением в качестве
основания можно рассматривать аммиак, присоединяющий протон
Растворы
117
по реакции:
NH3 + Н+ = NH,+ . (5.46)
Соответственно ион аммония, напротив, является кислотой. В
свете этой теории каждой кислоте соответствует некоторое сопря-
женное основание:
А = Н+ + В . (5.47)
Еще более радикальное определение было сформулировано Льюи-
сом. Он предложил в качестве кислот рассматривать все вещест-
ва, которые при образовании ковалентной связи принимают пару
электронов. В таком случае основаниями будут являться вещества,
отдающие пару электронов при образовании такой связи. Таким
образом, к кислотам Льюиса следует отнести также различные ко-
ординационно ненасыщенные молекулы или ионы, способные до-
страивать свою координационную сферу за счет присоединения
лигандов. В первую очередь к таковым относятся соединения, об-
ладающие незавершенной восьмиэлектронной оболочкой (BF3),
оксиды и галогениды с ненасыщенными координационными обо-
лочками (PF5, SbF5, TiClJ и различные катионы, способные образо-
вывать комплексные соединения (Cr3+, Со3+, Ni2+). Соответственно
к основаниям относятся различного рода анионы (включая ОН") и
молекулы, способные выступать в комплексных соединениях в
качестве лигандов. Несмотря на существенное расширение понятий
о кислотах и основаниях, теория Льюиса имеет массу недостатков,
ограничивающих ее применение. Так, с ее помощью невозможно
предсказать силу кислот. Кроме того, классические кислоты как раз
и не укладываются в это определение, поскольку не способны давать
дополнительных ковалентных связей с соединениями, содержащими
электронные пары.
Обойти эти противоречия удалось существенно позднее Пирсону.
Он постулировал, что связь между кислотой и основанием не обяза-
тельно должна являться ковалентной, а может включать и другие
типы взаимодействия (в том числе и ионную связь). В соответствии с
этой теорией к реакциям нейтрализации относится даже процесс об-
разования кристаллической решетки ионами натрия и хлора. Наи-
более ценным в теории Пирсона является представление о наличии
жестких и мягких кислот и оснований. К жестким относятся частицы
с реакционноспособными центрами, обладающие мало деформируемой
электронной структурой. Это могут быть катионы с большим заря-
дом и малым радиусом, а также анионы элементов с высокой электо-
отрицатель'ностью (F, О, N) (или включающие эти элементы NH3,
118
Глава 5
С104", ОН ). Напротив, мягкие частицы имеют электронную струк-
туру с высокой поляризуемостью (катионы с cZ-электронной под-
кладкой, объемные анионы, лиганды с функциональными группами,
включающими атомы с низкой электроотрицательностью ит.д.). При
этом жесткие основания образуют наиболее прочные связи с жесткими
кислотами и наоборот. Так, например, такие жесткие кислоты, как
ионы В3+, А13+, Ti4+, предпочтительно образуют соли с жесткими ос-
нованиями (О2-, F ). С другой стороны, такие мягкие кислоты, как
Ag+, Т1+, напротив, более склонны к образованию солей с мягкими
основаниями, например, с иодид- или сульфид-ионами. В принципе
эта теория допускает и количественную трактовку жесткости.
5.16. Автопротолиз
Под протолитическими понимаются реакции переноса протона.
Рассмотрим некоторый растворитель состава АН (где А - некоторый,
не обязательно одноатомный анион), имеющий на протоне положи-
тельный заряд. Между двумя молекулами такого растворителя может
образоваться водородная связь. При этом основное взаимодействие
А-Н несколько ослабеет и окажется возможным перенос протона ме-
жду двумя молекулами, участвующими в образовании водородной
связи. Протекающие процессы могут быть описаны уравнениями:
А-Н + А-Н <-* А-Н—А-Н А—Н-А-Н <-> А’ + Н,А+. (5.48)
Вновь образовавшаяся избыточная связь А-Н, естественно, ока-
зывается слабее исходной. Поэтому равновесие реакции существенно
смещено влево и константа равновесия мала, однако некоторый
выигрыш в энтропии обеспечивает осуществление такого рода
процессов в любых не апротонных растворителях. Это явление по-
лучило название автопротолиза. Характерным примером такового
является равновесие, устанавливающееся в водном растворе:
Н,0 +Н,0 <-> Н3О+ + ОН’. (5.49)
Именно им обусловлена маленькая, но все же реально сущест-
вующая электропроводность чистой воды. Поскольку концентрация
воды - величина постоянная, константа равновесия может быть
выражена соотношением:
К = [Н3О+][ОН‘] . (5.50)
При 22°С эта константа равна 10'14. В соответствии с тем что этот
процесс характеризуется существенно положительными величина-
Растворы
119
ми А# и AS. константа автопротолиза увеличивается с ростом тем-
пературы. Это соотношение оказывается справедливым также для
любых растворов кислот, солей и оснований, поскольку концентра-
ция воды во всех этих системах меняется сравнительно слабо и близка
к 55.5 моль/л. Следует заметить, однако, что при растворении таких
веществ в воде меняется ионная сила раствора, что приводит к
величинам коэффициентов активности для катионов, отличным от
единицы. Поэтому для растворов с концентрацией более 0.01М вме-
сто концентраций следует пользоваться активностями. Исключи-
тельно из соображений удобства далее мы все же будем продолжать
пользоваться концентрациями, подразумевая, что все записанные
соотношения будут выполняться с точностью до коэффициента
активности.
5.14» Сила кислот и оснований
При введении в водный раствор аммиака его молекулы также
получают возможность образовывать водородные связи с молекула-
ми воды. Поскольку положительный заряд больше на протонах моле-
кул воды, а у аммиака ввиду меньшей электротрицательности азота
существенно более активна электронная пара, наиболее значимые
изменения в растворе будут определяться протеканием реакции:
H,O+NH3 <-+ [НО-Н—NH3 <-> HO-HNHJ <-> НО'+ NH/. (5.51)
В образующемся промежуточном комплексе с короткой водо-
родной связью перенос протона происходит существенно быстрей,
чем процесс распада.
Поэтому формы НО-Н—NH3 и НО—HNH3 с полным основанием
можно считать одним соединением. Таким образом, в растворе будет
увеличиваться концентрация гидроксид-ионов. Равновесие этого
процесса описываеться константой диссоциации основания:
К = [NH4+][HO-] / [NHJ = 2-ЮЛ (5.52)
Как отмечалось ранее, каждой кислоте соответствует сопряжен-
ное основание и наоборот. Так, водный раствор аммиака обладает
свойствами основания, однако в процессе диссоциации образуется
ион аммония, который в водном растворе является слабой кислотой:
NH/ ОН, <-> NH3 + Н3О\ (5.53)
A; = [NH3][H3O+]/[NHJ. (5.54)
120
Глава 5
Перемножая выражения для обеих констант, легко получить:
К К = [Н3О+][ОН-] = К = 10'14. (5.55)
Поэтому для таких сопряженных пар произведение констант
диссоциации всегда равно константе автопротолиза воды. Таким
образом, при 22°С в водных растворах сумма логарифмов констант
диссоциации сопряженных кислоты и основания всегда равна -14.
Отсюда легко рассчитать константу кислотной диссоциации иона
аммония, которая оказывается равной 5-10’8.
Из сказанного следует, что сильному основанию соответствует
слабая сопряженная кислота и наоборот. Так, хлорной кислоте со-
ответствует рКк = -10. Поэтому ее анион, имеющий рКо = 24, прак-
тически не обладает свойствами основания.
Зададимся вопросом: чем же определяется сила кислоты? При этом
ограничимся случаем, когда молекула кислоты электронейтральна.
В первую очередь, конечно, соотношением донорных свойств не-
поделенной электронной пары и склонности к диссоциации связи
Э-Н. Активность первой падает, а второй - возрастает при продви-
жении по периоду таблицы Менделеева слева направо. Во-первых,
это определяется ростом электроотрицательности связанного с про-
тоном атома, а во-вторых, тем, что донорная способность элек-
тронных пар тем выше, чем меньше их число. В том же ряду по-
нижается электронная плотность на связи Э-Н, что облегчает ее дис-
социацию. Именно этим обусловлен рост кислотных свойств в ряду
NH3-H,O-HF. При продвижении сверху вниз по подгруппе донор-
ные свойства электронной пары убывают в связи с уменьшением
“склонности к .^-гибридизации”. Последний термин постоянно
подвергается критике в научной литературе, несмотря на очевидное
удобство его использования. Иными словами, это явление можно
объяснить тем, что ввиду увеличения размера орбитали и понижения
разницы между энергией последней (р) и последующей (чаще всего d)
орбитали электронная плотность этой пары оказывается существенно
более делокализованной. С другой стороны, с ростом радиуса эле-
мента прочность связи Э-Н существенно убывает. Это объясняет
увеличение кислотных свойств в рядах HF-HCl-HBr-HI и H2O-H,S-
-H,Se-H,Te.
Существует и целый ряд кислородсодержащих кислот состава
Н ЭО . Сила их падает с понижением разности (m-ri) на несколько
порядков.
Это легко проследить, на примере кислородсодержащих кислот
хлора (таблица 5.3).
Растворы
121
Таблица 5.3
Величины рК для кислородсодержащих кислот хлора
Кислота НС1О4 НСЮз НС1О2 нею
Р« -10 0 2.0 7.2
Это обусловлено тем, что при образовании p-связи Э=О элек-
тронная плотность смещается с центрального атома гораздо сильнее,
чем в случае связи Э-ОН. С другой стороны, слева направо растет и
заряд центрального иона. Это способствует большему смещению
электронной плотности связи Э-0 с атома кислорода. В свою очередь,
кислород компенсирует эту потерю за счет смещения электронной
плотности со связи О-Н. Последняя существенно ослабевает и ста-
новится более склонной к диссоциации. Связь же Э-О, хотя бы исходя
из электростатических соображений, напротив, становится более
прочной, что понижает вероятность диссоциации связи Э-ОН по
основному механизму.
5.15. Гидролиз солей, образованных сильной кислотой
и слабым основанием
По определению Льюиса катион любого элемента склонен к
образованию координационной связи с электронными парами
лигандов и, следовательно, является кислотой. Действительно, в
водных растворах катионы координируют вокруг себя молекулы
воды. При этом, за счет образования координационных связей,
электронная плотность с последних смещается к металлу. Это в свою
очередь приводит к увеличению эффективного положительного
заряда на протонах воды и повышает вероятность кислотной
диссоциации по следующей схеме:
[(H2O)n.1MOHJ7+ + ОН2 [(Н2О)п ,МОН-Н—ОН2]" ++
[(Н2О)п.,МОН] *-)+ + Н3О+ . ' (5.56)
В принципе этот процесс может протекать до тех пор, пока не
образуются частицы нейтрально заряженного гидроксида, который
при высоких концентрациях будет коагулировать. Одновременно в
ходе реакции (5.56) образуются и гидратированные ионы водорода.
Суммарный процесс можно описать как взаимодействие соли с водой
с образованием кислоты и основания. Такие процессы называются
гидролизом.
122
Глава 5
Рассмотрим вначале случай гидролиза соли, образованной силь-
ной кислотой и слабым основанием. Примером таковой является
хлорид аммония. При его диссоциации образуются ионы NH4+ и СГ.
Последний, являясь основанием, сопряженным с сильной кислотой,
практически не склонен к гидролизу. Ион же аммония вступает с водой
в реакцию:
NH/ + ОН, <-> NH3 + Н3О+. (5.57)
Образующиеся ионы оксония и гидратированный аммиак по мере
увеличения их концентрации будут взаимодействовать друг с другом
с образованием исходных веществ. Константа равновесия этой ре-
акции, как отмечалось ранее, может быть выражена уравнением:
К = |NH3][H3O+] / [NH4+] • (5.58)
Очевидно, что это ни что иное, как константа диссоциации со-
пряженной с гидроксидом аммония кислоты. Гидролиз будет про-
текать до тех пор, пока концентрация ионов оксония не станет удов-
летворять выражению (5.58). Отношение концентрации гидролизо-
вавшихся продуктов к исходной (С) называется степенью гидролиза.
Обозначим ее |3. Из соображений стехиометрии
[NHJ = [Н3О+] = рС . (5.59)
Концентрация оставшихся ионов аммония составит (1-р)С.
Перепишем выражение для константы равновесия в виде:
А'=р2С/(1 -р). (5.60)
Поскольку величина р, как правило, мала, то (1-р) приближа-
ется к единице ups (К/С)]1г. Таким образом, степень гидролиза
увеличивается с понижением концентрации раствора. С другой
стороны, гидролиз всегда сопровождается увеличением числа частиц
и беспорядка в растворе, Поэтому для веществ с малой степенью
гидролиза, когда константа равновесия меньше единицы, он опре-
деленно является экзотермическим процессом и степень гидролиза
растет с увеличением температуры. Это наглядно проявляется, на-
пример, при кипячении растворов медного купороса. Из вполне
стабильного при обычных условиях раствора выделяется некоторое
количество основных солей, что сопровождается понижением pH.
Гидролиз можно подавить введением в раствор сильной кислоты.
P = |NH3]/[NH4+] =^/[Н,О+]. (5.61)
Поскольку основным поставщиком ионов оксония в раство-
ре будет кислота, то степень гидролиза будет изменяться обрат-
но пропорционально ее концентрации. К аналогичному эффекту
Растворы
123
приводит и добавление к исходному раствору другого продукта
гидролиза - аммиака.
Домножим числитель и знаменатель в выражении для константы
гидролиза (5.58) на концентрацию гидроксйд-ионов. При этом
получаем:
Кг = [NH3][H30+][OH-] / [ЫНДОН-] =
= К [NH J / [NH4+][OH-] = К /К . (5.62)
Таким образом константа гидролиза соли, образованной силь-
ной кислотой и слабым основанием, равна отношению константы
автопротолиза растворителя к константе диссоциации в нем соот-
ветствующего основания.
5.16. Гидролиз солей, образованных слабой кислотой
и сильным основанием
Другим важным случаем является гидролиз солей, образованных
слабой кислотой и сильным основанием. Типичным примером таких
солей является ацетат натрия. В растворе он диссоциирует на ионы
натрия и ацетат-ионы. Первые из них, как основание, сопряженное с
сильной кислотой, практически не подвержены гидролизу, а вторые
вступают во взаимодействие с водой по следующей реакции:
СН3СОО- + Н2О <-> СН3СООН + ОН- . (5.63)
При этом в растворе возникает избыток гироксид-ионов, что
определяет его щелочную реакцию. Образование щелочи будет
происходить до тех пор, пока концентрация гирдроксид-ионов и
ионов оксония в нем не достигнет равновесной с образующейся ук-
сусной кислотой, то есть пока не будут удовлетворяться обе кон-
станты равновесия - для диссоциации уксусной кислоты и автопро-
толиза воды. Константу равновесия гидролиза можно представить
в виде:
а; = [СН3СООН][ОН-] / [СН3СОО‘] . (5.64)
Домножим числитель и знаменатель этого уравнения на кон-
центрацию ионов оксония:
К, = [СН3СООН][ОН-][Н30+] / [СН3СОО-][Н30+] =
([ОН-][Н3О+]) • ([СН3СООН] / [СН3СОО-][Н3О+]) = А/А.. (5.65)
Таким образом в случае гидролиза соли, образованной сла-
бой кислотой и сильным основанием, константа гидролиза равна
124
Глава 5
отношению константы автопротолиза растворителя к константе
диссоциации сопряженной с анионом кислоты. Расчет равновесной
концентрации ионов, образующихся вследствие гидролиза, прово-
дится аналогично случаю соли, образованной сильной кислотой и
слабым основанием. Степень гидролиза таких солей можно увеличить
повышением температуры или разбавлением раствора, а также свя-
зыванием одного из продуктов реакции (гидроксид-ионов), то есть
добавлением сильной кислоты. Из этого, следует известная законо-
мерность, гласящая, что сильные кислоты вытесняют слабые из их
солей. Напротив, добавление щелочи или уксусной кислоты понижает
степень гидролиза.
Следует заметить, что даже если соль образована многоосновной
слабой кислотой и сильным основанием или сильной кислотой и
слабым многоосновным основанием, гидролиз обычно останавли-
вается на первой ступени. В качестве примера рассмотрим гидролиз
раствора некоторой натриевой соли слабой двухосновной кислоты
Н,А с константами диссоциации и К, и концентрацией С моль/л.
Гидролиз ее можно описать уравнениями:
А2’ + Н,0 <-> НА" + ОН', (5.66)
НА' + Н,0 <-* Н,А + ОН’ . (5.67)
Константы гидролиза по первой и второй ступени соответственно
равны:
К, = К/Кг = [НА-][ОН-] / [Л2'] (5.68)
и
Кг2 = KJK, = [Н,А][ОН‘] / [НА'] (5.69)
При достижении равновесия концентрации всех ионов в раство-
ре должны удовлетворять обеим константам. Пусть степень гидро-
лиза по первой ступени равна х, а по второй - рх, где р - доля ионов
НА", подвергшихся гидролизу по второй ступени. Тогда из уравне-
ний (5.68) и (5.69) следует, что [НА'] = С(х-рх), [ОН-] = С(х+рх),
[Н,А] = Срх, [А2']= С(1-х). Подставляя эти значения в выражения для
констант гидролиза получаем:
Ка 1К2 = С(х-рх)(х +рх) / (1 -х) (5.70)
и
KJK{ = С(х+рх)рх / (х-рх)
Разделив первое уравнение на второе, получаем:
(5-71)
Растворы
125
KJK, = (х-рх)2/рх(1-х) =х(\-рУ/р (1-х)= [х/(1-х)][(1-р)2/р)]. (5.72)
Отношение первой и второй констант диссоциации для подав-
ляющего большинства кислот составляет 10М06. Полагая, что оно
равно 105 и (1-р) s 1, имеем: х/(1-х) s 105р. Таким образом, напри-
мер, если р = 0.01, то х = 0.999. Это, в свою очередь, достижимо при
Кг = 10’17. Гидролиз по первой ступени при этом протекает практиче-
ски нацело (на 99.9%). Только при этом степень диссоциации по вто-
рой ступени достигнет 1%. Если же 1«х и 1«р, то (х/р) = KJK, =
= 105. Таким образом в большинстве случаев гидролизом по второй
ступени можно пренебречь. Это определяется тем, что выделяющиеся
на первой стадии гидроксид-ионы практически полностью подавляют
гидролиз по второй ступени. Заметной степени гидролиза по второй
ступени можно достичь лишь путем смещения равновесия для первой
за счет приливания кислоты. В пределе при этом происходит уже
упомянутая выше реакция замещения слабой кислоты сильной.
В то же время описанные условия иногда оказываются достижи-
мыми для солей слабых оснований полизарядных катионов. Примером
таковых могут служить соли циркония. При попадании в водный раст-
вор тетрахлорид циркония полностью гидролизуется по первым двум
ступеням:
ZrCl4 + Н,0 = ZrOCl, + 2НС1 . (5.73)
Если хлористый водород удаляется в газовую фазу, то в сущест-
венной мере может протекать и гидролиз по третьей ступени.
Следует заметить, что гидроксид-ионы в достаточно концентри-
рованных растворах солей полизарядных катионов, подверженных
гидролизу, могут выступать в качестве связующих мостиков между
ними. При этом происходит образование полиатомных кластеров.
Так, цирконил-ионы в растворе представляют собой кластеры, в
которых четыре катиона, расположенные в вершинах квадрата, сое-
динены друг с другом за счет пар гидроксид-ионов. Кроме того, каж-
дый атом достраивает свой координационный полиэдр четырьмя
молекулами воды:
8+
126
Глава 5
5.17. Гидролиз солей, образованных слабой кислотой
и слабым основанием
Для соли АХ, образованной слабой кислотой и слабым основанием
гидролиз протекает одновременно и по катиону, и по аниону:
X- + Н,0 <-» НХ + ОН’, (5.74)
А+ + 2Н,0 <-> АОН + Н3О+ , (5.75)
или
А+ + X- + Н,0 <-> АОН + НХ . (5.76)
При этом выделяющиеся гидроксид-ионы и ионы оксония взаи-
модействуют с образованием воды. Это приводит к смещению равно-
весия вправо. Поэтому гидролиз таких солей протекает в существенно
большей степени по сравнению с ранее рассмотренными случаями.
При этом концентрация образующихся кислоты и основания намного
превышает концентрации нейтрализующих друг друга ионов ОН' и
Н,О+
X- + А+ + Н,о <-> НХ + АОН . (5.77)
Поэтому можно полагать, что соблюдается соотношение: [НХ] =
= [АОН]. Пусть концентрация соли равна С, степень гидролиза - у?,
а константы диссоциации кислоты и основания К:. и Кп. Константа
гидролиза задается уравнением:
К = [НХ][АОН] / [Х-][А+]. (5.78)
Домножив числитель и знаменатель одновременно на ионное
произведение воды, получаем:
К = ([НХ]/[Х-][Н30+])([АОН]/[А+][ОН-])([Н30+][ОН-]) =К1КК, (5.79)
В связи с вышесказанным можно заключить, что [НХ] = [ЛОН] =
= ДС, а [Х-] = [А+] = (1 - /7)С. Тогда
а; = [/?с/(1 - д)С]2 = [z?/G -/0Г- (5-80)
Таким образом степень гидролиза соли, образованной слабой ки-
слотой и слабым основанием, не зависит от ее концентрации. Она
определяется соотношением:
/7/(1 - Д) = откуда р = (КУ'Ч[\ +(Х)!/2]. (5.81)
Обычно важно знать также и концентрацию ионов водорода в
растворе, характеризуемую величиной pH. Для ее определения ис-
пользуем выражение для константы диссоциации кислоты:
К = [Х-][Н3О+] / [НХ] = [Н30+]аС / (1 - ос)С = [Н,О+] Кт (5.82)
Растворы
127
[Н,0+] = K(KJKK^- = (К К/К )"\ (5.83)
Откуда pH = -1/2 lg(W^) = \/2(рКа + рК - рКд). (5.84)
То есть величина pH таких растворов определяется отношением
констант диссоциации кислоты и основания. Так, для ацетата ам-
мония, для которого константы диссоциации соответствующих ки-
слоты и основания близки по значению, реакция раствора близка к
нейтральной. Сульфид аммония, образованный основанием более
сильным, чем кислота, имеет слабощелочную реакцию.
Экстремальным случаем является гидролиз солей, при котором
хотя бы один из продуктов может удаляться из сферы реакции. Ши-
роко известным примером является гидролиз сульфида алюминия,
который сопровождается выделением осадка гидроксида алюминия
и сероводорода в газовую фазу. В этом случае гидролиз протекает
практически нацело:
A1,S3 + 6Н2О = 2А1(ОН)34 + 3H2St. (5.85)
Аналогично для тетрахлорида циркония:
ZrCl4 + Н,0 = ZrOCl2 + 2НС1Т . (5.86)
5.18. Буферные растворы
Если в одном и том же растворе присутствуют в сопоставимых
концентрациях слабая кислота НА и ее соль с сильным основанием,
например КА, то гидролиз соли, как было показано ранее, подав-
ляется. Аналогично подавляется и диссоциация кислоты. Поэтому с
полным основанием можно полагать, что в растворе соблюдаются
соотношения: [НА] = СПА, [Л‘] = Скд. Тогда из выражения для
константы диссоциации кислоты имеем:
К = [Н3ОЧ[А-] / [НА], [Н3О4] = КСНА/СКА (5.87)
pH =рК+р(С^1Скл). (5.88)
При равенстве концентраций кислоты и соли концентрация ио-
нов водорода должна оказаться равной константе диссоциации
кислоты. Добавим теперь к этому раствору некоторое количество
сильной кислоты, составляющее 10% от концентраций исходных ве-
ществ. При этом десятая часть соли КА перейдет в кислоту НА и pH
раствора изменится на р (1.1/0.9) = 0.087 единиц. Аналогичные из-
менения будут происходить при приливании щелочи. Поэтому если
использовать растворы соли и кислоты высокой концентрации, то
128
Глава 5
введение посторонних примесей пусть даже сильных кислот или
оснований будет слабо менять pH раствора.
Аналогичные процессы происходят при сливании растворов со-
ли сильной кислоты и слабого основания, например АО с соот-
ветствующим основанием АОН (pH = рКа -рКо + р(САС1 /Сдон)). Такие
растворы получили название буферных и широко применяются в
аналитической химии. Наиболее распространены ацетатный буфер,
состоящий из эквимолярных количеств ацетата натрия и уксусной
кислоты (pH = 4.76), и аммиачный буфер, состоящий из эквимоляр-
ных количеств аммиака и хлорида аммония (pH = 9.25).
Буферные растворы образуются и при смешении солей многоос-
новных кислот, например, моно- и дизамещенных натриевых солей
фосфорной кислоты. Именно этим обусловлено длительное отсутствие
изменения pH при титровании кислот щелочами на стадии нейтра-
лизации каждого из неэквивалентных протонов (рис. 5.14). Причем
значения pH для каждого плато соответствуют величине рК кислоты
по соответствующим ступеням (1.1,7.2 и 12.4). Однако возможность
проявления первого и последнего из них во многом определяется кон-
центраций исходной кислоты и щелочи, которой проводится тит-
рование. Так, если кислота имеет концентрацию ниже, чем 0.1М, pH
Рис.5.14. Кривая титрования 1М раствора фосфорной кислоты 1М щелочью
исходного раствора уже в начальный момент оказывается выше 1.1.
Аналогично, если титрование проводится 0.1М раствором щело-
чи, то при приливании двукратного ее избытка pH достигнет лишь
12.0-12.7 в зависимости от концентрации исходной кислоты. Поэто-
му последнее плато сольется с выходом раствора на pH окончания
титрования.
Растворы
129
Задачи и вопросы
1 .Во сколько раз различаются молекулярные массы двух веществ,
если их растворы одинаковой процентной концентрации в бензоле
(К - 5.1) и камфоре (К = 40.0) понижают температуру замерзания ра-
створителей на одну и ту же величину?
2. Раствор 0.1155г фосфора в 19.03 г бензола замерзает при 5.15°С.
Определите состав молекулы фосфора, если Тя бензола составляет
5.40°С, а К = 5.1.
3. Определите молекулярную массу белка, раствор 150 мг которого
в 100 мл воды при 25°С характеризуется осмотическим давлением
0.00985 ат.
4. Определите молекулярную массу некоторого неэлектролита, а
также температуру замерзания (К ~ 1,86) и давление паров воды
(р0 - 2338 Па) над раствором 3.2 г его в литре воды, если осмотическое
давление внутри раствора достигает 2420 гПа.
5. Рассчитайте степень диссоциации 0.5М раствора муравьиной
кислоты (Кд = 2-10’4).
6. Вычислите величину осмотического давления 0.1М раствора
иодной кислоты (A'JHJOf.) =3-10'2).
7. Вычислите pH 0.01М раствора аммиака (Кд =2-10‘5).
8. Определите концентрацию синильной кислоты (К = 7-10'|()) при
которой степень ее диссоциации равна 0.01%.
9. Вычислите pH насыщенного раствора и растворимость гидро-
ксида магния (ПР = 5-10'11 12).
10. Растворимость гидроксида некоторого двухвалентного метал-
ла равна 3-10'5 моль/л. Каково его произведение растворимости?
11. Растворимость сульфата кальция в воде равна 1.06 г/л. Како-
ва растворимость этого соединения в 0.1М растворе сульфата калия?
130
Глава 5
12. Как изменится степень гидролиза 0.1М раствора цианида
калия при добавлении к нему 0.01М серной кислоты на литр
(K(HCN)=710-10)?
13. Произведение растворимости некоторой соли АХ, образо-
ванной сильной кислотой и слабым основанием равно 10’4. Определи-
те pH насыщенного раствора этой соли, если константа диссоциации
основания АОН равна 10'4.
14. Определите концентрацию раствора щелочи, при взаимодей-
ствии которой с эквивалентным объемом 2М уксусной кислоты pH
раствора стал равен 5 (К((СН3СООН)=210‘5).
15. Рассчитайте pH буферного раствора, содержащего по 0.1 моль
карбоната и бикарбоната калия в одном литре (К3(Н,СО1)=4.810’11).
16. Как изменится pH 100 мл раствора, содержащего по 0.5 моль
ацетата натрия и уксусной кислоты при добавлении к нему 11.2 г
гидроксида кальция?
17. Оцените величину произведения растворимости сульфидов MS.
которые будут осаждаться из 0.1М раствора соли соответствующе-
го металла 0.1 М растворами а) сероводорода, б) сульфида натрия,
в) сульфида аммония (К (H,S): ^ = 10'7, А'3=10’3, K(NH4OH)=210‘5)?
18. Оцените величину произведения растворимости сульфидов MS,
которые будут растворяться в 1М уксусной кислоте, считая за начало
растворения достижение концентрации катиона в растворе Ю’3 М.
Глава 6
МЕТОДЫ ОЧИСТКИ ВЕЩЕСТВ
Прежде чем приступить к проблеме очистки веществ, целесооб-
разно выяснить принципиальную возможность получения абсолютно
чистого вещества. Все методы очистки основаны на различии чисто
физических или физико-химических свойств веществ или их произ-
водных. Для максимально эффективного разделения следует про-
водить его в условиях, близких к равновесным. Поэтому для всех про-
цессов очистки целесообразно использовать термодинамические кри-
терии применительно к равновесному состоянию. При этом любая
система должна стремиться к минимуму свободной энергии. Рассмот-
рим с этой точки зрения, термодинамику загрязнения вещества.
Внедрение в вещество некоторой посторонней примеси чаще все-
го приводит к повышению энтальпии. С весьма хорошей точностью
можно полагать, что проигрыш в энтальпии пропорционален числу
посторонних атомов, приходящемуся на моль вещества. С другой
стороны, введение примеси, безусловно, приводит к увеличению
беспорядка в системе, а следовательно, к росту ее энтропии. В первую
очередь возрастает вклад так называемой конфигурационной энтро-
пии, равный
£ = £1пЖ, (6.1)
где W - число возможных способов размещения атомов в узлах
кристаллической решетки. Если все N атомов эквивалентны (нераз-
личимы), то перемена их местами не меняет состояния системы.
Следовательно, для чистого вещества, не содержащего дефектов,
конфигурационная энтропия равна нулю. Если же появляется один
примесный атом, то число способов его размещения по N узлам
решетки равно N (для моля вещества 6.02-Ю23). Это достаточно
большая величина, и для ее компенсации при комнатной температу-
ре необходимо, чтобы мольное увеличение энтальпии для внедрения
примеси составило:
1.3 8-10'23 1п(6.02-1023) 6.02-1023-298/1000 = 136 кДж/моль .
132
Глава 6
Для реальных веществ всегда можно подобрать целый ряд
примесей, энтальпия внедрения которых существенно меньше это-
го значения. При внедрении следующего атома прирост конфигура-
ционной энтропии уже меньше:
Д5 = In W, - In W, = 1п[Я(Я-1)/2] - 1пЯ = 1п[(ЛЧ)/2]
(первый атом можно разместить по N позициям, второй - по (7V-1)
оставшимся, но поскольку эти два атома также неразличимы, каждое
состояние будет иметь эквивалентное с взаимной сменой мест атомов
примеси). И так далее с каждым последующим атомом примеси
выигрыш в энтропии существенно понижается, пока не достигнет нуля
при замещении половины атомов основного вещества. Таким образом
график зависимости энтальпии кристаллов от концентрации приме-
си в первом приближении можно представить в виде монотонно ра-
стущей прямой, а аналогичная кривая, соответствующая произве-
дению энтропии на температуру - вначале резко снижаясь, замедляет
свой ход и затем меняет его на обратный (рис. 6.1).
Рис.6.1. Изменение функций состояния некоторого вещества в зависимости
от количества введенной примеси
При равновесии должно соблюдаться соотношение:
ЛЯ-ТД5 = 0. (6.2)
Нетрудно убедиться, что эта разность также достигнет минимума
при некоторой концентрации примеси, что соответствует ее равновес-
ному содержанию.
Очевидно, что любое вещество, находящееся при температуре
выше абсолютного нуля, даже чисто из термодинамических сообра-
жений должно быть загрязнено теми или иными примесями. Кроме
Методы очистки веществ
133
того, для достижения каждой новой все более глубокой степени
очистки требуются все большие энергетические затраты. При этом
имеется целый ряд соединений с близкими свойствами, сопутствую-
щих друг другу, добиться разделения которых чрезвычайно трудно.
Наиболее ярким примером таковых являются соединения циркония
и гафния. Обладая одинаковым зарядом и, вследствие лантаноидно-
го сжатия, практически одинаковым размером атомов и ионов, эти
элементы чрезвычайно трудно поддаются разделению.
Все методы очистки веществ делятся на физические и химические.
К первым в основном относятся методы, позволяющие добиться
разделения веществ, находящихся в различных агрегатных состо-
яниях. Типичным примером является фильтрование. Для мелко-
дисперсных осадков, микрочастицы которых проходят через поры
любого фильтра или забивают их, применяется центрифугирование.
Две несмешивающиеся жидкости можно разделить декантацией или
с помощью делительной воронки. Но наибольший интерес пред-
ставляют физико-химические и химические методы разделения, по-
зволяющие достичь глубокой очистки веществ, находящихся в одном
и том же агрегатном состоянии. На них мы остановимся подробнее.
6.1. Перекристаллизация
Этот метод применяется для разделения двух (или более) твердых
веществ (чаще всего солей), находящихся в смеси друг с другом.
Наиболее просто такое разделение происходит, если один из
компонентов вовсе нерастворим (А). Тогда процесс растворения
134
Глава 6
проводят до полного перехода в раствор второго соединения, после
чего выделение обоих веществ не представляет труда. Возможно также
выбрать количество растворителя таким образом, чтобы в осадке
остался лишь один компонент (вернее - его часть). Однако наиболее
часто используется изменение растворимости веществ с температурой.
Этот метод в подавляющем большинстве случаев является более
эффективным. Пусть имеется Мг. слабозагрязненного вещества В,
растворимость которого увеличивается с температурой и при tI
составляет c)t а при Z, - с, г на 100 г растворителя. Тогда, необходимое
для насыщения раствора по В при температуре Г, количество ра-
створителя составит -100 М1с{. При температуре г, в нем останется
Mcjc} г В Выход после перекристаллизации составит:
М - Мсг/с} = М(\- (6.3)
г чистого В. Наибольшего выхода за один цикл можно добиться при
максимальной разнице температур растворения и кристаллизации.
Если растворимость с ростом температуры уменьшается, насыщение
проводят при более низкой температуре. Если растворимость слабо
меняется с температурой (как в случае поваренной соли), используют
частичное или полное упаривание раствора. Причем, если примеси
вовсе нерастворимы, упаривание можно проводить нацело после
отделения осадка.
Существенно более сложным является случай, когда оба компо-
нента растворимы. Пусть имеется смесь а г вещества А и b г вещества
В (Ь»а и величины эти заранее неизвестны), растворимости которых
при температурах /, и /, составляют соответственно т2 и пу пг г на
100 г растворителя и слабо влияют на растворимость партнера. Тогда
при Z] вся смесь растворится в 100Z?//?, г растворителя (если а > Ьт /п},
то часть А остается в осадке, что лишь облегчает задачу разделения,
- допустим, что выполняется обратное условие). При охлаждении,
как и в предыдущем случае, выделяется (b - bnjn) = Z>(1 - njn) г В и
{а - bmjn}) г А. Если а < hmjn{, то выделяется чистый компонент В
(не достигается концентрация насыщенного раствора А). Такая пе-
рекристаллизация является оптимальной, и при Ь»а это условие
выполняется практически всегда. В противном случаев осадке вновь
присутствуют два компонента, но при тг1п2 > alb (что также выпол-
няется с большой вероятностью при Ь»а} осадок обогащается
компонентом В. Таким образом доля примеси после перекристал-
лизации понижается. Полной очистки можно добиться либо путем
последовательного повторения таких операций, либо подобрав
температуру так, чтобы выполнялось условие а < bmjn}.
Методы очистки веществ
135
Если количества А и В сопоставимы, насыщение обычно проводят
в минимальном количестве растворителя. Количество его опре-
деляется тем веществом, у которого выше отношение его доли в смеси
к растворимости при температуре насыщения. Пусть а!тх > Ыпу Тогда
по завершении процесса раствор будет насыщенным по А и не
насыщенным по В. При охлаждении можно найти такую температу-
ру, при которой будет достигнуто соотношение а/т} = Ь/п2, что со-
ответствует образованию насыщенного раствора В. До насыщения
раствора по В будет выделяться чистый А. Однако, поскольку эн-
тальпии растворения двух веществ не равны, растворимости их при
дальнейшем охлаждении насыщенных растворов меняются неоди-
наково. Пусть при некоторой температуре 12 < 1Ь_ выполняется условие
mjn2 > mjn,. Тогда осадок в процессе дальнейшей кристаллизации
обогащается компонентом В, а раствор - компонентом А. Растворяя
осадок при более высокой температуре, можно выделить В, а при ра-
створении продукта упаривания раствора - А.
Таким образом, перекристаллизация в той или иной ее модифи-
кации позволяет провести очистку одного из компонентов. Вопрос
заключается лишь в выборе ее условий. Кроме того, следует учитывать
возможные фазовые превращения. Так, если необходимо получить
чистый декагидрат сульфата натрия (рис. 6.1), осаждение следует
проводить не выше 32°С. Напротив, безводный Na,SO4 надо выделять,
нагревая насыщенный при 33°С раствор или упаривая его.
До сих пор мы предполагали, что А и В не растворяются друг в
друге в твердом состоянии. В действительности же такого не бывает.
При наличии взаимной растворимости компонентов в процессе вы-
падения осадка А он будет содержать некоторое количество В.
Допустим, что мольная доля В в растворе при равновесии достигла
Аж, а в твердом А - Хт . Найдем соотношение между этими величи-
нами. При равновесии твердого и жидкого растворов с чистым
компонентом соблюдаются условия:
Нй1 = Нй(ж)° + ^7’1пАж’ и =ЦВ(Л?)П +ЯГ1ПХД (6.4)
где Хж' и Ат' - концентрации В в растворителе и твердом А, соот-
ветственно. Приравняв правые части, легко получить:
\Чцх)-^=КТ\П(Х^/Хж'У (6.5)
Изменив состав исходной смеси, Получаем новое равновесие, для
которого:
ЦВ(ж)’ + RTI1UV = цВ(т)" + RT 1пХт=. (6.6)
Объединяя эти два уравнения получаем:
136
Глава 6
RT\n(X^/X^)= RT\n(Xy2/X^). (6.7)
или
Х'/Хж1 = Х2/Х2 = К. (6.8)
Таким образом, отношение концентраций вещества В в двух
находящихся в равновесии фазах можно с хорошим приближением
считать постоянным. Полученная величина называется коэффи-
циентом распределения и показывает, что отношение концентраций
(активностей) компонента В в А и в растворе - величина постоянная.
Поскольку растворимость примеси в твердом теле обычно много
меньше, чем в жидкостях, ею весьма часто можно пренебречь. Тем не
менее иногда эти процессы следует учитывать. Так, концентрацию в
растворе радиоактивного стронция можно существенно понизить,
осаждая из него сульфат или карбонат кальция с меньшим произ-
ведением растворимости. Обладая в силу отмеченных причин высокой
склонностью к ионам стронция, полученные осадки будут весьма эф-
фективно извлекать их из водного раствора.
Другим осложняющим фактором могут являться отчетливо
выраженные сорбционные свойства некоторых осадков. Так, гид-
роксиды всех переходных элементов обычно содержат значительные
количества сорбированных анионов. Действительно, обладая
развитой поверхностью, содержащей большое количество способных
диссоциировать М-ОН групп (рис. 6.2), при контакте с раствором
они активно обменивают их на анионы по реакции:
(МОД.) - ОН + Х~ <-> (М ОД) - X + ОН' . (6.9)
Рис.6.2. Схема строения осадка
гидроксида поливалентного
вещества
Концентрация анионов в растворе чаще всего существенно выше,
чем гидроксильных групп, - вспомним, что гидроксиды обычно по-
лучают взаимодействием солей с щелочью или аммиаком. При этом
Методы очистки веществ
137
концентрация анионов меняется незначительно, а подавляющее
количество ионов ОН переходит в осадок. Величину pH по окончании
эксперимента предпочитают поддерживать на уровне не выше 7-9
во избежание растворения гидроксидов. Поэтому продукт может за-
грязняться весьма существенно, Удалить сорбированные анионы легче
всего, промывая осадок разбавленным раствором щелочи, смещающей
равновесие влево.
При значительном пересыщении скорость зародышеобразования
выше скорости роста кристаллов, поэтому осадок формируется из
мелких частиц, которые могут срастаться, образуя полости. Послед-
ние содержат исходный раствор вместе со всеми присущими ему
примесями. Предотвратить проявление этого эффекта, называемого
окклюзией, можно, проводя кристаллизацию в максимально при-
ближенных к равновесным условиях.
6.2. Кристаллизация из расплава
Вернемся к рассмотрению диаграммы состояния системы с
простой эвтектикой (см. рис. 4.6). При охлаждении некоторого
расплава, из него кристаллизуется в первую очередь тот компонент,
который находится в избытке по отношению к эвтектике. При этом
твердый продукт окажется загрязненным примесью второго ком-
понента за счет их взаимной растворимости в твердом состоянии.
При этом, как и в предыдущем случае, концентрация В в твердом А и
в расплаве будет характеризоваться определенным коэффициентом
распределения, приблизительно равным мольной доле В в насы-
щенном твердом растворе, поскольку мы полагаем, что растворимость
В в жидком А неограниченна. Таким методом, однако, невозможно
разделить эвтектическую смесь или получить в чистом виде компо-
нент, содержание которого в расплаве ниже, чем в эвтектике.
Перейдем к системам с неограниченной растворимостью А в В
как в жидком, так и в твердом состоянии. При охлаждении расплава
состава 1 (рис. 6.3) в первую очередь выделяются кристаллы состава
обогащенные высокоплавким компонентом В. Эта точка несколько
ниже кривой ликвидуса, поскольку первая фракция кристаллов уже
образовалась. После их отделения охладим далее жидкость состава
Л, (обогащенную А). При этом образуются кристаллы состава Х2 и
жидкость L, (еще более обогащенная А). Таким образом, по мере
охлаждения жидкость будет все более обогащаться А, приближаясь
по составу к чистому веществу. Однако конечного продукта будет
138
Глава 6
достаточно мало. Увеличить выход можно повторением операции.
Аналогично можно получить почти чистый В.
Рис.б.З. Охлаждение системы АВ с неограниченной взаимной растворимостью в жидком
и твердом состоянии
Рис.6.4. Схема процесса зонной плавки («), распределение примеси на различных
участках стержня в процессе первого прохождения печи (б, в) и после завершения
полного цикла разделения (г)
Процесс разделения сплава на компоненты можно осуществить
значительно быстрее и качественнее методом зонной плавки. Этот
способ основан на продвижении вдоль длинного стержня из
загрязненного материала (твердого раствора) печи с узкой зоной
нагрева (рис. 6.4д). При этом образующийся в этой зоне расплав
Методы очистки веществ
139
обогащается легкоплавким веществом (см. рис. 6.46) до тех пор, пока
не достигнет состава, равновесного с составом кристалла (после этого
состав стержня уже не меняется (см. рис. 6.4s)). В процессе плавки
стержень обогащается компонентом В. Проделав эту операцию
несколько раз, можно получить брусок, в области начала прохода
состав которого соответствует практически чистому В, а в конце - А
(см. рис. 6.4г).
Подобными методами не удается разделить сплавы, характеризу-
ющиеся экстремальной для данной системы температурой плавления
(если таковая существует), поскольку составы расплава и твердого
раствора для нее равны (см. рис. 4.16). По этой же причине нельзя
выделить из таких систем компонент, содержание которого меньше
соответствующего экстремуму.
6.3. Дистилляция
До сих пор мы разбирали примеры очистки твердых веществ. Если
же, напротив, стоит задача выделения из раствора жидкости, то
решить ее можно несколькими физико-химическими способами. Так,
из диаграммы 5.8 следует, что из разбавленных растворов часть
практически чистого растворителя можно выделить просто вымо-
раживанием до температуры, несколько выше эвтектической. Другим
возможным методом является очистка путем пропускания через
полупроницаемую перегородку с использованием избыточного
давления, превышающего осмотическое.
Однако оба эти метода характеризуются низкой производи-
тельностью. Существенно более часто применяется перегонка. С ее
помощью можно быстро и практически полностью отделить раст-
воритель от растворенных твердых веществ. Кроме того, этот метод
может применяться для разделения нескольких жидких веществ. Рас-
смотрим пару жидкостей, образующих идеальный раствор. ТХ-диа-
грамма этой системы изображена на рисунке 6.5.
При нагревании жидкости состава она начнет кипеть в точке,
соответствующей кривой ликвидуса. При этом равновесный пар будет
иметь состав X , обогащенный низкокипящим компонентом (В) При
конденсации выделяющегося пара образуется жидкость того же
состава Хп. Оставшаяся жидкость обогащается высококипящим
компонентом А (состав Хж). Следующая фракция кипит при несколько
большей температуре, но пар также обогащен компонентом В
относительно нового состава. Таким образом, в процессе перегонки
140
Глава б
Рис.6.5. Диаграмма состояния двухкомпонентной системы с неограниченной взаимной
растворимостью компонентов в жидком состоянии
Рис.6.6. Зависимость температуры перегонки
от времени: теоретическая равновесная
перегонка (л), перегонка с дифлегматором
(<Э, прибор для перегонки жидкостей (в)
мы движемся по кривой кипения жидкости, приходя в конечной точке
к практически чистому компоненту А. При этом температура кипения
жидкости непрерывно повышается от до ТА (рис. 6.6а). При тех же
температурах будет конденсироваться пар.
Методы очистки веществ
141
С другой стороны, в процессе реальной перегонки смеси двух
жидкостей выделяется вначале практически чистый легколетучий, а
затем - высококипящии компонент. Температура же конденсации
пара меняется также по иной схеме, в начале процесса практически
совпадая с Гв, а в его конце - с ТА (см. рис. 6.66). Переход между ними
соответствует сравнительно небольшой фракции жидкости. Для
повышения эффективности процесса перегонки на выходе из колбы
с раствором помещают вертикальную трубку, предшествующую
холодильнику, называемую дифлегматором (см. рис. 6.6<?), Для
обеспечения большего теплообмена с окружающей средой ее снаб-
жают специальными отростками или шариками, увеличивающими
площадь поверхности. При этом пары кипящей жидкости, попадая в
дифлегматор, конденсируются на его стенках, постепенно стекая в
колбу. Встречный поток пара, контактируя с жидкостью обогащается
более летучим компонентом, а жидкость по мере стекания - высоко-
кипящим. Таким образом, по мере подъема пара по дифлегматору он
как бы еще несколько раз приходит в равновесие с жидкостью, все
более обогащенной В. Поэтому на выходе дифлегматора приходят
существенно очищенные пары В. Напротив, в колбу стекает практи-
чески чистый А. Таким образом и стекающая в холодильник жидкость
конденсируется при температуре существенно ниже той, при которой
кипит раствор в колбе. Разделение осуществляется тем лучше, чем
большее число раз успевает установиться равновесие в дифлегмато-
ре. Поэтому для получения чистых продуктов следует увеличивать
его длину и медленнее подводить тепло к раствору. На практике при
разделении жидкостей на фракции обычно вполне достаточным
оказывается пользоваться температурой конденсации. При этом
принято говорить, что отобрали фракцию со следующей темпера-
турой кипения, хотя правильнее было бы говорить о температуре
конденсации. Но так как целью подобного эксперимента обычно
является получение достаточно чистых веществ, разница между этими
понятиями сравнительно мала. Такое разделение жидкостей называют
также фракционной перегонкой (при этом число фракций, как
правило, превышает две). В промышленности для разделения смеси
жидкостей на компоненты строят так называемые ректификационные
колонны, устроенные по подобному принципу.
С помощью перегонки нельзя очистить смеси азеотропного со-
става, так как в этом случае состав пара соответствует составу жид-
кости (см. рис. 5.6). Поэтому таким образом нельзя разделить на
компоненты 20%-ный раствор соляной кислоты или 4%-ный раствор
воды в спирте. Для их разделения применяются химические методы.
142
Глава 6
Рис.6.7. Прибор для перегонки иода
Возгонкой удается очистить и некоторые твердые вещества.
Например иод способен переходить в газовую фазу, минуя жидкую
(рис. 6.7).
Образующиеся пары конденсируют на некоторой холодной
поверхности, например на донышке колбы с водой. Для интенсифи-
кации процесса часто проводят возгонку при пониженном давлении.
Фазовые диаграммы веществ, разделяющихся подобным образом,
должны удовлетворять следующим требованиям:
- иметь давление тройной точки выше давления, при котором
осуществляется перегонка,
- парциальное давление паров возгоняемого вещества должно
быть не слишком низким и существенно превышать парциальные
давления паров примеси при данной температуре.
6.4. Экстракция
В качестве некоторого промежуточного между физико-химиче-
скими и химическими методами разделения можно рассматривать
разделение с помощью экстракции. Этот метод основан на различной
растворимости соединений в двух несмешивающихся жидкостях. Он
широко используется для разделения смесей, а также для концен-
трирования продуктов как в органическом, так и в неорганическом
синтезе (рис. 6.8).
Рис.6.8. Экстракция в делительной воронке
Методы очистки веществ
143
Концентрации двух находящихся в равновесии растворов одного
и того же вещества в двух различных растворителях, как было
показано выше (уравнение 6.8), характеризуются постоянным от-
ношением, определяемым концентрацией их насыщенных растворов.
Это отношение называется коэффициентом распределения и является
постоянным при данной температуре. Пусть объемы растворителей
равны VA и VB , а коэффициент распределения - К. Тогда после
экстракции соотношение концентраций экстрагируемого вещества в
обоих растворителях составит АТа/Кв . Так, при экстракции иода
10 мл сероуглерода из 100 мл водного раствора соотношение кон-
центраций его в двух фазах будет равно (60-1 )/1 = 59. При этом иод
концентрируется в 5.9 раз. Кроме того, при этом происходит и его
очистка. Водный раствор иода часто получают окислением KI хло-
ром. В таком случае в водном растворе присутствует значительное
количество хлорида калия, практически нерастворимого в сероуг-
лероде. Таким образом с помощью экстракции мы не только доби-
ваемся концентрирования иода, но и очищаем его раствор от примеси.
Экстракцию можно использовать в комплексе с химическими
методами. При этом дополнительный эффект разделения получается
чаще всего за счет различия констант устойчивости комплексов двух
ионов. Характерным примером является экстракционное разделе-
ние циркония и гафния. Как отмечалось ранее, четырехзарядные
катионы этих элементов характеризуются идентичными зарядами и
радиусами. Вследствие этого их разделение представляет огромную
сложность и коэффициент разделения оказывается достаточно низким
даже после десятикратной перекристаллизации солей. С другой сто-
роны, при приливании к азотнокислому раствору циркония, содер-
жащему природную примесь гафния, раствора трибутилфосфата
[(С1|Н9).Р=О] в некотором органическом растворителе достигается их
достаточно хорошее разделение. Ионы циркония, образуя комплекс
с трибутилфосфатом, образуют вокруг себя “гидрофобную шубу” и
практически полностью переходят в органическую фазу, в то время
как гафний, существенно менее склонный к образованию таких ком-
плексов, остается в растворе.
6.5. Химические методы разделения
Одними из наиболее эффективных способов очистки различных
веществ от примесей являются методы, базирующиеся на использо-
вании транспортных реакций. Транспортными реакциями называются
144
Глава 6
процессы переноса вещества между двумя зонами с различной
температурой с промежуточным образованием легколетучих веществ.
Так, при действии монооксида углерода на металлический никель
при 60-80°C образуется легколетучая жидкость состава Ni(CO)4,
называемая карбонилом никеля (рис. 6.9).
Рис.б.9. Схема установки для очистки никеля
При дальнейшем нагревании ее пары легко разлагаются, вновь
образуя металлический никель. Если зоны протекания этих процессов
разделены, то никель освобождается от всех примесей. Вещества,
образующие менее устойчивые карбонилы или не образующие их
вообще, остаются в исходной смеси. Вещества же, образующие более
устойчивые карбонилы, не разлагаются и остаются в газовой фазе.
Кроме того, этот метод успешно применяется для нанесения никеле-
вых покрытий на различные металлические изделия.
По сходной методике проводят очистку металлического циркония.
Практически все методы его получения основаны на взаимодействии
его оксида или галогенидов с различными активными металлами, в
качестве которых могут выступать натрий, калий, магний, кальций
и алюминий. Естественно, что полученный таким методом металл
оказывается существенно загрязненным примесями и представляет
собой в большинстве случаев пористую губку или порошок. Для
очистки последнего его совместно с небольшим количеством иода
загружают в кварцевый реактор, нагреваемый до 600°С. При этом
протекает процесс образования тетраиодида циркония:
Zr +21, = Zrl4 , (6.10)
который благодаря своей летучести переходит в газовую фазу. В тот
же сосуд помещают тонкую вольфрамовую или циркониевую нить,
нагреваемую за счет пропускания электрического тока до 1800°С
(рис. 6.10). При этом иодид циркония разлагается с образованием
высокочистого циркония. Последний может содержать в качестве
примесей некоторые количества титана и гафния, от которых по
возможности стараются избавиться заранее.
Методы очистки веществ
145
............................... :: \ ь - . Zr, 870К • • •. W. 2СЮИК
Рис.6.10. Схема установки
для глубокой очистки циркония
Транспортные реакции также часто используются для получения
высокочистых металлов, используемых в полупроводниковой технике,
или выращивания монокристаллов. Рассмотрим термодинамические
аспекты этого процесса. Пусть при некоторой температуре Т\ проис-
ходит взаимодействие веществ А и В с образованием продукта ABv.
Последний имеет возможность переходить в газовую фазу и разла-
гаться при более высокой температуре Т, > 1\. Условия протекания
этих реакций можно представить в виде двух неравенств:
ДЯ-Т,Д5<0, bH-T,AS>Q (6.11)
или
ДЯ/Т, < AS < ДЯ/Т,. ДЯ < О, Д5 < 0 . (6.12)
Осуществление транспортных реакций по той же схеме возможно
и при Т2 меньше Т}. В таком случае два последних условия следует
переписать в виде ДЯ > О, AS >0. Иными словами, термодинамическое
условие протекания транспортной реакции в общем случае можно
выразить в следующем виде: 1) энтальпия и энтропия реакции имеют
одинаковый знак (чаще отрицательный, так как соединения с более
высокой степенью окисления отличаются большей летучестью),
2) температура То - &HI&S должна находиться между Т, и Т}. Кроме
того, необходимо, чтобы уже при меньшей температуре давление
паров АВд. было достаточно высоким для осуществления его переноса
между холодной и горячей зонами. Выберем в качестве такого
критерия, например, давление 0.1 атм. Тогда
К =\п(р) =-2.3 <-(ДЯ -ТAS J/RT=AHJRT+ AS JR, (6.13)
равп ' исп 1 исп-’ исп исп 4 '
где ДЯ и AS - соответственно энтальпия и энтропия испарения
АВ .
.V
146
Глава 6
Можно выделить также целую группу сорбционных методов
очистки. Несколько ранее мы рассматривали сорбцию как явление,
мешающее очистке веществ с помощью перекристаллизации. Это же
явление может играть и положительную роль. Так, широко извест-
ным методом является очистка воды, газов и спиртов с помощью ак-
тивированного угля, Аналогично многие ионы растворенных в воде
солей могут сорбироваться на силикагеле или мелкодисперсном окси-
де алюминия. Образующиеся между ними связи отличаются срав-
нительно низкой энергией. Поэтому такая сорбция является обра-
тимой. Так молекулы ароматических соединений легко удаляются из
активированного угля при небольшом нагревании, а сорбированные
на оксиде алюминия анионы вымываются из него даже небольшим
током воды. При этом за счет различной прочности образующихся
связей и просто изменения размера ионов различается и время удер-
жания их в некоторой зоне сорбента. На этом может быть основан
метод разделения малых количеств химических соединений. Так, на
пластину с нанесенным оксидом алюминия (либо на колонку или
просто на бумагу) можно нанести небольшое количество раствора
смеси полифосфатов. При помещении нижней части пластины в воду
последняя медленно поднимается по ней за счет капилярных сил.
Одновременно поток воды постепенно перемещает анионы. Причем
скорость их перемещения существенно различается. В результате этого
каждому иону будет соответствовать своя зона на пластине (рис. 6.11).
Рис.6.11. Разделение ионов на
хроматографической пластине
После этого в зависимости от применяемой методики исследуемые
соли можно последовательно вымывать из пластины, отбирая
отдельные фракции жидкости, либо просто разрезать бумагу на
Методы очистки веществ
147
участки, содержащие только один сорт анионов с последующим
переводом их в раствор. Сорбционная емкость оксидных материалов
невелика. Поэтому сорбцию чаще применяют для очистки газов или
жидкостей, а описанная методика чаще всего используется для
аналитических целей.
Существенно большей прочностью связей и большей емкостью
отличаются ионообменные смолы на полимерной матрице, содер-
жащие большое количество функциональных групп. В зависимости
от природы последних различают катиониты, сорбирующие катио-
ны (функциональные группы -SO3H, -РО3Н, -СО,Н), и аниониты,
сорбирующие анионы (функциональные группы - амины, -NR3+). При
помещении такого ионита в раствор соли протекает процесс обмена,
например:
R-SO,H + К+ + Н2О R-SO3K + Н3О+. (6.14)
Константа равновесия этой обратимой реакции (коэффициент
распределения) может быть представлена в виде:
к = fl(H3o;)a(K\) / й(н+>(к;) , (6.15)
где нижние индексы “р” и “w” обозначают отнесение соответствующих
активностей катионов к фазам раствора и ионита соответственно.
Этот коэффициент может существенно меняться в зависимости от
свойств и состава смолы. Можно подбирать смолы, склонные к
сорбции различных ионов.
Наиболее часто применяется следующая методика разделения.
Через колонку, содержащую катионит в водородной форме, про-
пускают смесь солей двух металлов А+Х* и В+Х_. Независимо от
коэффициента распределения они вытесняют из колонки водород
(концентрация которого в растворе низка). Пусть соотношение кон-
центраций ионов в исходном растворе равно единице, а коэффициент
распределения равен 10. Тогда в верху колонки преимущественно
сорбируют ся катионы А+, а стекающий раствор обогащен В+. Причем
по мере приливания раствора в верхней части устанавливается
равновесие между фазой ионита и вновь поступающим раствором с
соотношением ионов А+ и В* 1:1. Тогда соотношение концентраций
А * и В+ в ионите в верхней части колонки рано или поздно оказывается
равным 10. На некотором участке, где ионы А+ иссякают, соотношение
их концентраций в растворе резко понижается. Но таким же образом
оно меняется и в равновесной с ним фазе катионита. Так, при
соотношении концентраций А+ и В+ в твердой фазе, равном 1:1,
равновесный раствор содержит в 10 раз меньше А+. При контакте с
последующим слоем его концентрация понизится еще в 10 раз. Таким
148
Глава б
же образом на каждом последующем участке равновесная кон-
центрация А+ будет понижаться и в узкой области упадет практически
до нуля. Поэтому далее идет слой, в котором сорбируется лишь В+.
Если суммарное количество А+ иВ+ меньше емкости колонки, в нижней
части существует некоторая переходная область от формы В+ к Н+ и
чистая водородная форма (рис. 6.12а).
Рис.6.12. Распределение катионов в колонке: при пропускании через нее раствора А+ и
В+ («), при вымывании их кислотой (б)
После этого колонку промывают раствором кислоты и смола вне
зависимости от коэффициентов распределения полностью переходит
в водородную форму. При этом вымываемый из верхней части раствор
содержит В+ и А+ в соотношении 10:1. Опускаясь по колонке он
вступает в равновесие с нижними слоями. При этом по указанным
ранее причинам концентрация А+ в колонке резко возрастает и, в
первую очередь, из нее уходит В+. Таким образом, фронты прак-
тически чистых А+ и В+ постепенно спускаются по колонке, разде-
ленные узкой полосой их смеси (см. рис. 6.126). В таком порядке
происходит и их вымывание. При этом количество вымывающегося
раствора со смесью неразделенных А+ и В+ тем больше, чем ко-
эффициент распределения ближе к единице. И, напротив, очистка
происходит тем полней, чем больше коэффициент распределения
отклоняется от единицы.
Методы очистки веществ
149
С этой точки зрения наилучшими перспективами обладают
неорганические ионообменные материалы, в которых величины ко-
эффициентов распределения даже для пары натрий-калий могут
достигать нескольких порядков. Это обусловлено тем, что в их
структуре уже в основном заложены параметры будущего коорди-
национного полиэдра катионов (рис. 6.13), которые более приемле-
мы лишь для некоторых из них.
Рис.6.13. Строение кислого фосфата циркония. В центре обозначены контуры
координационного полиэдра, в который может входить металл
В качестве примера можно привести строение одного из наиболее
известных неорганических ионообменных материалов - кислого
фосфата циркония. В нем четко просматриваются основные контуры
будущего октаэдра МО6 - в центре рисунка (рис. 6.13). Причем набор
межатомных расстояний оказывается существенно более подходящим
для крупных катионов щелочных металлов, что и предопределяет их
селективную сорбцию.
Задачи и вопросы
1. 60 г К,МпО4 при некоторой температуре обработали 500 мл
воды. Какое количество перманганата калия можно получить охладив
полученную смесь до 0°С если реакция протекает по уравнению:
ЗК,МпО4 + 2Н,0 = 2КМпО4 + МпО, + 4КОН, а растворимость
перманганата при 0°С равна 2.8%?
150
Глава 6
2. Какое количество воды и алюмокалиевых квасцов следует взять
для получения 20 г чистой соли, если насыщение вести при 60°С, а
кристаллизацию - при 0°С? (растворимость безводной соли при этих
температурах составляет 26.7 и 3.1% соответственно)
3. Смесь, содержащую 33.8 г КС1 и 3.38 г КСЮ, при 80°С раство-
рили в минимальном количестве воды и охладили до 0°С. Какова
оставшаяся после перекристаллизации примесь хлората?
Соединение \ температура о°с 80°С
KCI 22.2% 33.8%
КСЮз 3.2% 28.4%
4. Предложите на основании приведенной температурной
зависимости растворимости (%) бихромата и сульфата калия метод
очистки их смеси в массовом соотношении 1:1. Оцените чистоту
продуктов после первой и второй перекристаллизации.
В-во \ температура, °C 0 20 . 40 60 80 100
K2SO4 6.9 10.0 13.1 15.4 17.6 19.4
ЮСгзО? 4.4 11.1 20.6 31.2 41.1 50.5
5. В стакан, содержащий 30 г. водородной формы ионообменной
смолы с емкостью 5 мг-экв/г, прилили 100 мл 0.1М раствора АС1,
содержащего также 0.0IM ВС1, на литр. После установления
равновесия раствор слили, а к смоле прибавили 200 мл 2М раствора
НС1. Определите выход АС1, полученного при выпаривании обоих
растворов, если коэффициенты распределения ионов В2+, А+ и Н+ на
данной смоле соотносятся как 1000:1:1.
6. Предложите наиболее эффективный метод разделения за одну
перекристаллизацию смеси, состоящей на 50% по массе из А и на 50%
- из В, если известны следующие зависимости их растворимости (в г
на 100 г воды) от температуры:
t, °C 0 10 20 30 40 50 60 70 80 90 100
А 5 9 15 22 30 38 46 56 67 78 90
В 5 6 7 9 12 16 21 28 42 62 89
7. Брусок металла А, содержащего 10% примеси В подвергли
очистке с помощью зонной плавки с толщиной зоны расплава 4 мм.
Предполагая, что равновесие устанавливается дискретно с остываю-
щей зоной толщиной 2 мм нарисуйте профиль изменения концен-
трации примеси по длине бруска
Методы очистки веществ
151
а) после прохождения печью зоны в 6 мм ;
б) после прохождения печью зоны в 20 см;
в) после 100 прогонов по всему бруску
если кривые ликвидуса и солидуса для этой системы описаны в
таблице:
в, % Ликв. Солид. В, % Ликв. Солид. В, % Ликв. Солид.
0 800 800 27.5 734 678 55 609 605
2.5 797 787 30 725 670 60 600 600
5 793 774 32.5 714 661 65 611 602
7.5 789 761 35 703 653 70 630 610
10 785 750 37.5 692 645 75 648 622
12.5 780 738 40 680 638 80 665 635
15 774 726 42.5 666 632 85 680 648
17.5 768 715 45 655 626 90 690 665
20 760 705 47.5 638 620 95 696 681
22.5 752 696 50 625 615 100 700 700
25 743 686
Глава 7
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ
ПРОЦЕССЫ
Протекание многих химических процессов сопряжено с переносом
электрона между атомами или ионами. При этом последние меняют
степень окисления. Эта величина представляет собой некий фор-
мальный показатель, равный заряду данного атома в соединении, в
предположении, что оно состоит из ионов с целочисленным зарядом,
определяемым числом образуемых полярных связей с учетом их на-
правленности. Исходя из этого, степень окисления азота в гидро-
ксиламине равна -2+1= -1, а в молекуле N-^ - нулю. Следует учесть,
что этот показатель не имеет ничего общего с действительной элек-
тронной плотностью вокруг данного атома, которую можно опре-
делить, например, из данных по дифракции рентгеновских лучей. Так,
при изменении степени окисления хромай железа в оксидных системах
от +2 до +3 и +6 эффективный заряд атома в среднем понижается от
+ 1.8е до + 1.2е и +0.1 е. Это связано с тем, что ионы, имеющие высо-
кую положительную степень окисления, поляризуют электронные
оболочки окружающих их анионов. Это сопровождается сущест-
венным перераспределением электронной плотности. Тем не менее
понятие степени окисления является удобным для описания хими-
ческих соединений и их реакций. Поэтому оно так широко и прочно
вошло в научную литературу.
7.1. Электродвижущая сила
Рассмотрим пластинку некоторого металла, погруженную в
раствор его соли. В рассматриваемой системе протекают следующие
процессы:
М -+ М2+(р-р) + 2е (7.1)
и
Af2+(p-p) -> М - 2е.
(7.2)
Окислительно-восстановительные процессы
153
Первая реакция сопровождается переходом ионов металла в
раствор При этом принадлежавшие им ранее электроны остаются
на пластине. Вторая реакция приводит к прямо противоположному
результату. За счет преобладания одного из описываемых процессов
пластина принимает положительный или отрицательный заряд.
Пусть, например, преобладает первый процесс. Тогда заряд пластины
оказывается отрицательным и возникает некоторый дополнительный
электрический потенциал, притягивающий к ней катионы из раствора
и облегчающий их сорбцию. Напротив, прямая реакция тормозится.
Через некоторое время скорости прямой и обратной реакции
выравниваются и наступает равновесие, при котором распределение
зарядов между раствором и пластиной оказывается постоянным.
Такая пара металл/ион металла в растворе или в общем случае пара
соединений с различной степенью окисления одного и того же элемен-
та называется сопряженной окислительно-восстановительной парой.
Оба вещества (иона) могут находится в растворе, образуя пару типа
Fe3+/Fe2+.
Различные металлы при контакте с растворами солей могут при-
обретать существенно различающийся заряд. При этом у поверхности
металла в жидкости находится избыток ионов противоположного
заряда (противоионов), компенсирующий заряд пластины. Их кон-
центрация понижается при удалении от поверхности металла, быстро
доходя до нуля. Совокупность заряженных частиц, локализованных
на поверхности пластины и в растворе вблизи нее, образует так
называемый двойной электрический слой. Если соединить провод-
ником пару пластин различных металлов, находящихся в контакте с
растворами своих солей (рис. 7.1), то между ними будет протекать
электрический ток, обусловленный контактной разностью потенци-
алов. Так, для пары медь-цинк ионы последнего существенно более
активно переходят в раствор, оставляя электроны на металле. На
медной пластине будет осуществляться обратный процесс. Поэтому
цинковая пластина окажется катодом (заряд “-”), а медная - анодом
(заряд “+”). Соответственно избыток электронов будет перетекать с
цинка на положительно заряженную медь. Разность потенциалов
между ними (электродвижущая сила (Е)) достигает 1.1В. По мере
протекания тока заряды на пластинах понизятся, за счет чего возоб-
новится осаждение меди и растворение новых количеств цинка, ко-
торое может продолжаться вплоть до полного расходования одного
из компонентов. Однако, рассуждая таким образом, мы упустили из
поля зрения присутствующие в растворе анионы. Последние не будут
ни уходить из раствора, ни появляться в нем. Поэтому раствор в сосуде
154
Глава 7
Рис.7.1. Гальванический элемент на основе пары металлов медь-ципк
с солью цинка быстро приобретет положительный, а сосуд с солью
меди - отрицательный заряд. Это создаст разность потенциалов между
пластинами и раствором, мешающую протеканию описанных процес-
сов. Для того чтобы избежать этого явления, между сосудами помеща-
ют “солевой ключ'' (см. рис. 7.1), допускающий перемещение анионов.
Последний представляет собой стеклянную трубку, заполненную раст-
вором любого электролита, перекрытую пористой перегородкой для
предотвращения быстрой диффузии.
Таким образом, мы сконструировали гальванический элемент, под
которым понимается любое устройство, преобразующее энергию
химической реакции в электрическую. Рассмотренный нами элемент
является обратимым. Так, если к нему присоединить источник тока с
большей разностью потенциалов, приложенной в противоположном
направлении, то избыточный отрицательный заряд на цинке приве-
дет к тому, что, в отличие от самопроизвольного процесса, он начнет
сорбировать катионы из раствора. На меди будет протекать обратный
процесс. Если пропускание через систему тока в обратном направле-
нии приводит к протеканию тех же процессов в противоположном на-
правлении, электроды называются обратимыми. Экспериментальным
путем было установлено, что для выделения одного моля различных
металлов требуются кратные количества электричества, причем
Окислительно-восстановительные процессы
155
последние соотносятся как заряды катионов. Таким образом для
восстановления одного моля металла потребуется*расход электроэнер-
гии, равный nFКл, где п - заряд катиона, a F-число Фарадея, равное
96 487 Кл (в электрохимических расчетах его обычно округляют до
96 500).
Следует заметить, что далеко не все электроды являются обра-
тимыми. Так, если те же пластины поместить в раствор медного
купороса, то разность потенциалов между ними сохранится и в системе
будут протекать аналогичные ранее описанным процессы - раство-
рение цинка и выделение меди. Однако если подключить большую
разность потенциалов, направленную в противоположную сторону,
выделение меди начнется на цинковом электроде, а на медном - ее
растворение. Если же один электрод состоит из магния или алюминия,
то их окажется невозможным получить электролизом водного ра-
створа солей. Все это - типичные примеры необратимых электродов.
При работе гальванического элемента через проводник протека-
ет электрический ток. Максимальная работа, которую он может со-
вершить равна произведению электродвижущей силы гальваниче-
ского элемента на переносимый заряд. С другой стороны, она опре-
деляется изменением свободной энергии Гиббса данной системы.
Таким образом можно записать:
-&G = nFE. (7.3)
Из полученного соотношения следует, что электродвижущая сила
обратимого гальванического элемента может служить мерой изме-
нения свободной энергии или химического потенциала данной
системы. Предположим, что мы сконструировали гальванический
элемент на основе некоторого окислительно-восстановительного
процесса, который можно описать реакцией:
аА + ЬВ = сС +6D . (7.4)
Тогда его свободную энергию можно также представить в виде:
Д(7= фс+ а]хА- =
= Фс°/ф/- ац/- b^+RT\n([C]WI [«) =
--АТ1пА' + Л7Чп([Пда/[Л]°[5]л) --nFE, (7.5)
где К- константа равновесия. Естественно, что рассмотренный нами
гальванический элемент достаточно далек от равновесия, которое со-
блюдается лишь при достижении концентрациями всех участвующих
в реакции веществ некоторого предельного значения. Тем не менее
равновесия в такой системе можно достичь, прервав электрический ток
156
Глава 7
и поддерживая в системе некоторую разность потенциалов. Допустим,
что мы сделали это при концентрациях всех участвующих в описывае-
мом процессе веществ, равных единице. Тогда получаем некоторую
стандартную характеристику электродвижущей силы данной реакции,
называемую стандартной электродвижущей силой (Е
RT\nK = nFE °. (7.6)
Объединяя уравнения 7.5 и 7.6, получаем:
Е = Ей + RTI nF 1п([С]Ж/ [J]'W). (7.7)
Это соотношение, называемое уравнением Нернста, лежит в основе
всех электрохимических исследований и теорий. Для практических
расчетов, как правило, пользуются десятичными логарифмами.
Подставив численное значение отношения RTIF и перейдя от на-
турального логарифма к десятичному, уравнение Нернста .можно
представить в виде:
Е = Е0 + 0.059 / п lg([C] W/ [/]W). (7.8)
Из полученных выражений видно, что э.д.с. исследуемого элемента
будет в существенной степени зависеть от концентраций участвую-
щих в реакции веществ. Отсюда следует, что можно сконструировать
гальванический элемент, используя одинаковые электроды при
различной концентрации растворов. Действительно, поместив две
цинковые пластины в раствор цинка различной концентрации, можно
заметить, что между ними также возникнет разность потенциалов,
пропорциональная логарифму отношения концентрации растворов.
Это происходит за счет того, что в более концентрированном раство-
ре процесс сорбции происходит с большей скоростью, ввиду чего
пластина, опущенная в него, приобретает положительный заряд. Э.д.с.
такого концентрационного элемента можно выразить уравнением:
Е = + А77 2Aln([Zn2+]1 /[Zn2+]2), (7.9)
где п = 2 - число электронов, передаваемых одним ионом цинка.
7.2. Окислительно-восстановительный потенциал
Из соотношения (7.5) можно заключить, что электродвижущая
сила гальванических элементов является некоторой аддитивной
характеристикой окислительно-восстановительных процессов. Дей-
ствительно, для описанного процесса а молекул А, например, пе-
редают п электронов b молекулам В, переходя в С, а В при этом
превращается в D. Тогда, объединяя уравнения (7.5) и (7.6), получаем:
Окислительно-восстановительные процессы
157
EF-(aln) \кАъ+(Ып) ^.^-(dn) цс° -(din) jiD° +
+(RT!n) 1п([Лр/л[^]"л/ [QW'"),
(7.Ю)
где электродвижущая сила приведена к процессу передачи одного
электрона. Приводя вещество А во взаимодействие с другим
окислителем, можно получить аналогичное уравнение для электро-
движущей силы, половина составляющих которого останется не-
изменной. Таким образом, для каждой сопряженной окислительно-
восстановительной пары можно получить некоторую характеристи-
ку окислительно-восстановительной способности относительно пары
А/С. Для стандартизации этих величин следует определиться с точкой
отсчета. В качестве таковой был выбран стандартный водородный
потенциал -потенциал стандартного (или нормального) водородно-
го электрода (рис. 7.2). Последний представляет собой пластину,
покрытую платиновой чернью (мелкодисперсной платиной), на
которую подается ток водорода под давлением 1 атм. В контакте с
ней находится раствор серной кислоты с активностью ионов во-
дорода, равной единице. Потенциал водородною электрода принят
равным нулю неслучайно, поскольку протон является основной
действующей единицей в водных и многих других растворах. Стан-
дартный водородный электрод представляет собой половину галь-
ванического элемента. Объединяя с ним другие полуэлементы, со-
держащие сопряженные окислитель и восстановитель с единичными
активностями в растворе, можно определить стандартный потенциал
данной пары.
Рис.7.2. Стандартный
водородный электрод
158
Глава 7
Электродному потенциалу принято присваивать знак, соответ-
ствующий заряду этого электрода по отношению к стандартному
водородному. Так, сконструируем гальванический элемент, состоящий
из стандартного водородного и медного электродов. Поскольку медь
менее активна, чем водород, на ней будут сорбироваться катионы
меди из раствора, сообщая ей положительный (относительно водо-
родного электрода) потенциал. Электродвижущая сила такого эле-
мента при единичной активности меди в растворе составит 0.337 В.
Таким образом, потенциал стандартного медного электрода состав-
ляет +0.337 В. Если же активность меди в растворе не равна единице,
его можно найти по формуле:
£ = 0.337 + 0.0295 1g пСи2+ (7.11)
По отношению к большинству металлов, сосуществующих с ра-
створами своих солей, потенциал стандартного водородного элект-
рода является отрицательной величиной. Из свойства аддитивности
по уже приведенным значениям можно рассчитать и стандартный
потенциал цинкового электрода. Поскольку он заряжен отрицательно
относительно медного и разность потенциалов между ними составля-
ет 1.1 В, можно записать соотношение:
£Cu-£zn=l-10B,
откуда
£Zn = -1-10 + ECu = -Ы0 + 0.337 = -0.763 В .
Действительно, потенциал цинкового электрода, измеренный
относительно водородного, равен той же величине. В таком случае
возникает вопрос - зачем же пользоваться таким громоздким и до-
рогостоящим электродом, как водородный? Куда проще заменить его,
к примеру, на медный. Однако и этот электрод недостаточно хорош,
поскольку равновесие сорбция-десорбция меди весьма подвижно
и концентрация ее в растворе со временем существенно меняется.
Аналогичное заключение можно сделать практически для всех
электродов, образованных сопряженными парами металл/катион или
катион/катион. Представляется целесообразным использовать не
меняющиеся во времени, устойчивые электроды. Важнейшими из них
являются каломельный и хлорсеребряный. Они построены сходным
образом. В каломельном электроде паста из смеси металлической
ртути и каломели (Hg,Cl,) находится в контакте с насыщенным, деци-
или одномолярным раствором хлористого калия (рис. 7.3). Стан-
дартный потенциал пары Hg,2+/Hg равен +0.798 В. В то же время
концентрация (активность) ионов ртути задается, формулой:
Окислительно-восстановительные процессы
159
Рис.7.3. Каломельный электрод
[Hg22+]=77P(Hg2Cl2)/[Cr]\ (7.12)
В зависимости от концентрации раствора КС1 потенциал
каломельного электрода будет равен соответственно +0.245, +0.282
или +0.337 В. Потенциал полученного электрода определяется ак-
тивностью ртути и каломели (которые постоянны) и концентрацией
хлорида калия (которая в контакте с насыщенным раствором также
будет постоянной, а в других случаях меняется весьма слабо). Поэто-
му каломельный электрод значительно проще и стабильнее в обра-
щении, чем водородный. Аналогичным образом (но на основе пары
AgCl/Ag) устроен и хлорсеребряный электрод. Измеряя потенциал
некоторого электрода относительно описанных выше стандартных,
необходимо учитывать, что началом отсчета в данном случае будет
не нулевая величина.
Для большинства сопряженных окислительно-восстановительных
пар оба вещества находятся в растворе. В таком случае потенциал
этой системы измеряют с помощью инертного относительно данной
пары (например, платинового).электрода. Будучи опущенной в
раствор, содержащий, например, ионы двух- и трехвалентного железа,
платиновая пластина станет служить местом стока или источником
электронов, участвующих в реакции окисления (восстановления)
ионов железа. Так, например, в случае измерения потенциала данной
пары относительно каломельного электрода контакт между ними
осуществляется через платиновое днище электрода (см. рис. 7.3) (для
хлорсеребряного соответственно через серебро). При этом потенциал
данной пары будет задаваться уравнением:
Е = Е0 + RTI nF ln([Fe3+] I [Fe2+]), (7.13)
где E 0 - стандартный потенциал данной пары.
160
Глава 7
7.3. Направления окислительно-восстановительных
процессов
Из уравнения (7.3) следует, что свободная энергия химических
реакций связана с электродвижущей силой. Следовательно, с
помощью последней также можно определять направления окис-
лительно-восстановительных процессов. Рассмотрим реакцию (7.4).
Пусть в ходе ее а молекул А отдают п электронов, переходя в с молекул
С, в то же время В, принимая эти электроны, переходит в D. Разделим
левую и правую часть уравнения (7.10) на F и перепишем его в виде:
Е= 1/^(%°-ф/) + RT I nF\n([B\bl [£?') - 1/«Нснс°-ф/) -
-Wln([QW) =EbId-EcIa =-bGtnF. (7.14)
Поскольку величина nF всегда положительна, можно заключить,
что для самопроизвольного протекания реакции слева направо
должно соблюдаться соотношение:
^/„-^>0. (7-15)
Таким образом, условием протекания реакции будет положи-
тельная разность между потенциалом окислителя и восстановителя.
Например, можно заключить, что железо будет вытеснять медь из ее
солей (0.337 - (-0.440) - 0.777 В > 0), а серебро не будет (0.337 - 0.779 =
= -0.442 В<0). Следует заметить, что простое вычисление стандартной
разности потенциалов, как и в случае других термодинамических па-
раметров, не всегда позволяет с полной уверенностью предсказать
возможность взаимодействия в некоторой системе. Оказывается не-
обходимым учитывать еще и кинетические факторы. Так, к примеру,
алюминий и хром должны были бы интенсивно окисляться кисло-
родом воздуха и взаимодействовать с водой. В случае алюминия этого
не наблюдается ввиду образования на нем окисной пленки, а в случае
хрома еще и из-за значительного перенапряжения (это как бы экви-
валент энергии активации для электрохимических процессов), необ-
ходимого для его окисления.
Другим важным фактором является необходимость учитывать
концентрацию ионов в растворе. Так, стандартный потенциал серебра
выше, чем у одновалентной ртути. Поэтому серебро, на первый взгляд,
не должен было бы вытеснять ртуть, из 1М нитратного раствора.
Однако если бы ионы серебра и вовсе отсутствовали в растворе, их
окислительный потенциал был бы бесконечно мал. Поэтому реакция
взаимодействия между этими веществами протекает до некоторого
положения, определяемого условием равенства окислительно-
Окислительно-восстановительные процессы
161
восстановительных потенциалов:
Е = Е„ , 0.799+0.059 lg[Ag+] = 0.789 + 0.059/2 lg[Hg 2+],
откуда
[Ag+]2/ [Hg,2+] = exp(-0.01/0.0295) = 0.46 .
Поскольку для вытеснения одного иона Hg,2+ необходимо ра-
створение двух атомов серебра, [Ag+]2 / (l-l/2[Ag+]) - 0.46 откуда
[Ag+]2 + 0.23[Ag+] - 0.46 = 0 .
Решая это уравнение, можно прийти к заключению, что равно-
весная концентрация нитрата серебра в этом растворе должна со-
ставить 0.57 М! Таким образом, растворение все же будет протекать,
хотя концентрация серебра в растворе всегда будет ниже, чем ртути.
С другой стороны, в равновесии с раствором медной соли будет
находиться всего 3'10’8 М серебра.
В общем случае для реакции (7.4) константа равновесия может
быть вычислена из соотношения:
(Ш1 И№) = ехр[«(5м - Ес/л)/0.059]. (7.16)
Значительная часть элементов периодической системы может
существовать в нескольких степенях окисления. При этом в зави-
симости от партнера по окислительно-восстановительному процессу
и некоторых других условий (чаще всего величины pH раствора) они
могут отдавать или принимать различное количество электронов.
Предположим, что некоторый элемент может иметь шесть различных
степеней- окисления. Тогда для описания стандартных потенциалов
всех возможных переходов в системе потребуется 15 уравнений.
Поскольку, как будет показано позже, большинство таких процессов
имеют разные потенциалы в кислой и щелочной средах, это число
удвоится. Конечно, составление таких таблиц представляется до-
статочно громоздким. Поэтому чаще используют некоторые упро-
щенные методики представления стандартных потенциалов Окис-
лительно-восстановительных полуреакций, существенно облегчаю-
щие их систематизацию. Наиболее распространенной формой
является диаграмма Латимера, описывающая потенциалы превра-
щения между последовательными степенями окисления. Например,
для различных форм существования хлора в кислом растворе (или в
равновесии с ним) такая диаграмма может быть представлена в виде:
СГ <.?|-збв_ С12 нею НС1О2 СЮ/
162
Глава 7
Из приведенной диаграммы можно вычислить окислительно-
восстановительный потенциал любого из пятнадцати процессов.
Действительно, пусть необходимо вычислить потенциал перехода
некоторого элемента из состояния со степенью окисления Zx в Z . В
тоже время на диаграмме приведены только потенциалы переходов
Z] в Z, (Е12) и Z2 в Z3 (Е23). Тогда свободные энергии двух последних
превращений относительно стандартного водородного электрода
составят AG], = (Z2-Z1)JrZ12 и AG,3 =(Z3-Z2)/,Z23 , а для искомого
процесса - AG|2 = (Z\-Zx)FExr Тогда по закону Гесса AGt3=AG1l2+AG123,
откуда
(Z3-Z,)Z|3 = (Z3-Z2)Z23 + (Z2-Z,)Z12
или
Z|2 = [(Z3-Z2)£23 + (Z2-Z,)Z12]/(Z3-Zt). (7.17)
Вычислим, к примеру, потенциал перехода НС1О2 в С1,.
Е(НС1О2/С12) = [2Z(HC1O2/HC1O) + Z(HC10/Cl2)]/3 =
= (2-1.65 + 1.63)/3 = 1.64 В.
Аналогично можно рассчитать и любой другой потенциал.
Более наглядной является диаграмма Фроста. Для ее представления
строят график зависимости величины пЕ от степени окисления. За
начало отсчета принимается аналогичная величина для элемента.
Таким образом, по оси абсцисс откладывается степень окисления, а
по оси ординат величина пЕ для процесса перехода данной формы в
элемент. Примеры диаграмм такого рода для хлора в кислой и
щелочной средах приведены на рисунке 7.4 а,б. Для расчета по-
тенциала любого перехода следует найти разность между величинами
пЕ для соответствующих состояний и разделить на разность степеней
окисления. Кроме того, исходя из этой диаграммы можно предсказать
термодинамическую устойчивость к диспропорционированию любой
формы. Так, например, для реакции диспропорционирования неко-
торого соединения со степенью окисления Z,, одна из молекул
которого окисляется, переходя в степень окисления Z3, а другая
восстанавливается, переходя в Zx величина дС реакции диспропор-
ционирования составляет
AG = F {(ZXEX - Z2E.) + (Z3Z3 - Z2Z2)J = -F [2Z,Z, -Z,Z, -Z3E3].
Поэтому для самопроизвольного диспропорционирования ве-
личина, взятая в квадратные скобки, должна быть положительной.
Таким образом, если прямая, соединяющая точки, соответствующие
на диаграмме соединениям со степенями окисления Z} и Z3, проходит
Окислительно-восстановительные процессы
163
Рис.7.4. Диаграммы Фроста для хлора в кислом (<?) и щелочном (б) растворах
ниже, чем для Z,, диспропорционирование будет термодинамически
выгодно. Так, можно заключить, что хлор по отношению к дис-
пропорционированию в кислом растворе устойчив, а в щелочном -
нет. С другой стороны, НС1О2 с точки зрения термодинамики
неустойчив в любом растворе.
7.4. pH-зависимые процессы
В ряде случаев в элементарном окислительно-восстановитель-
ном процессе могут участвовать и ионы водорода. Чаще всего это
наблюдается, когда хотя бы один из участников сопряженной
окислительно-восстановительной пары является анионом кисло-
родсодержащей кислоты. Так, процесс восстановления перманганат-
ионов может быть представлен в виде:
МпО; + 8Н+ +5е = Мп2+ + 4Н,О. (7.18)
При этом величина окислительно-восстановительного потенци-
ала этой пары (МпО4' / Мп2+) определяется соотношением:
Е = Е0 + RT/ nF 1п([МпО4-][Н+]8 / [Мп2+]) = '
= 1.51 + 0.059/5 lg([MnO4-][H+]8 / [Мп2+]). (7.19)
164
Глава 7
Таким образом, при эквивалентных концентрациях ионов двух-
валентного марганца и перманганат-ионов потенциал системы может
быть представлен в виде:
£= 1.51 - 0.059 (8/5)рН. (7.20)
В координатах Е-pH это соотношение описывается прямой ли-
нией (рис. 7.5). Из представленного графика можно сделать заклю-
чение, что перманганат окисляет хлорид-ион лишь в сильнокислых
растворах, бромид - при pH > 5, а иодид - почти во всех средах.
Рис.7.5. Сопоставление окислительно-восстановительного потенциала Mn(VII)/Mn(II) с
парами XJX' (X = CI, Br, I) при различных значениях pH
В случае слабых кислот этот процесс усложняется тем, что выше
некоторой концентрации ионов водорода основной формой их суще-
ствования будет недиссоциированная кислота. При этом концен-
трация иона, участвующего в процессе, будет убывать и определяется
величиной потенциала и pH раствора. Таким образом, на графике
зависимости Е-pH такой системы будет наблюдаться излом в точке,
соответствующей рК кислоты (рис. 7.6).
Так, сопряженная пара НСЮ/СГ в кислом и щелочном растворе
характеризуется разными стандартными потенциалами:
НСЮ + Н+ + 2е = Cl' +Н2О + 1.50 В , (7.21)
СЮ’ + Н,0 + 2е = Cl’ + 2ОН- + 0.89 В . (7.22)
Окислительно-восстановительные процессы
165
Рис.7.6. Типичный вид зависимости окислительно-восстановительного потенциала
пары слабая кислота (ее анион) - восстановленная форма от pH раствора
Представим зависимость £-рН через уравнение Нернста при
единичных концентрациях кислоты (СЮ") и хлорид-аниона:
Янсю/Cl- = 1-50 + 0.0295 lg[H+] = 1.50 - 0.0295 pH, (7.23)
Яс1О7С1- = 0-89 + 0 0295 lg(l/[OH"]2) = 1.716 - 0.059 pH . (7.24)
Равенство этих потенциалов достигается при pH = 0.216/0.0295 =
= 7.32. Табличное значение рК для НСЮ - 7.30. Действительно, кон-
центрация аниона СЮ" в кислом растворе может быть рассчитана по
соотношению: [С10‘] = Кцсю ’ [НСЮ] / [Н+]. В кислом растворе дис-
социация кислоты подавлена. Поэтому [НСЮ] = [НСЮ] + [СЮ"] ~ 1.
Таким образом, уравнение Нернста в этом случае можно предста-
вить в виде:
£°НСЮ/С1- = £°СЮ-/С1- + 0.0295 lg([C10-] / [ОН"]2) =
Я°СЮ-/С1-" 0-0295 lg(KHClo/ [Н+][ОН‘]2) =
Я°СЮ-/С1- -0-0295 lg(KHC10[Н+] / КН2О2) =
= 0.89 - 0.0295(7.30-28) - 0.0295 lg[H+] = 1.50 + 0.0295 pH.
В общем случае уравнения двухэлектронного восстановления
одноосновных кислот НХОп для кислых и щелочных растворов можно
представить в виде:
166
Глава 7
HXOn + 2Н+ + le = НХО(п.0 + Н,0 , (7.25)
ХОп ’ + Н,0 + 2е = XO(n,0- + 20Н-. (7.26)
При этом их стандартные потенциалы должны отличаться на
0.059-14+0.02951g(AfHxon^HXO(n.l)) = 0-826 - 0.0295(р£НХОп ^нхо(п.п)-
Если одна из кислот является сильной, то даже в кислом растворе
анион является основной формой ее существования. Наиболее веро-
ятно, что таковой будет кислота, в которой X имеет высшую степень
окисления. Тогда разность стандартных потенциалов составит 0.826+
+ 0.0295рКцхо(п 1} и график зависимости £-рН в этом случае также
будет иметь излом при pH = рЛд1ХО(п ()- В этой точке тангенс угла на-
клона будет меняться от 0.0295 при низких на 0.059 при высоких pH.
7.5. Влияние на направления окислительно-
восстановительных процессов природы реагентов
На направление окислительно-восстановительных процессов
может влиять не только степень окисления элемента, но и природа
исходного реагента или продукта реакции. Так, металлическая ртуть
(•^Hg,2+/Hg ~ 0-789 В) вполне стабильна по отношению к нитрату меди
(Ecu2+/Cu ~ 0.337 В, ^cu2+/Cu+ = 0.153 В). В то же время хлорид меди
вполне способен ее окислить. Так, в 1М растворе хлорида меди концен-
трация ионов хлора равна 2М, поэтому концентрация находящегося
в равновесии раствора одновалентной меди составляет 27РсиС1/[С1-] =
= 10’б/2. Таким образом окислительный потенциал пары Cu2+/CuCl
составит 0.153 - 0.059 lg5-10‘7 = 0.525 В. С другой стороны, хлорид
одновалентной ртути также малорастворим (27PHg?ci, = 3-10'18).
Концентрация ионов Hg,2+ равновесная с раствором хлорида двухва-
лентной меди составляет 3-10‘18/4 = 7.5-10'19М, а окислительно-восста-
новительный потенциал пары Hg,Cl,/Hg - 0.789 -0.0295 lg(7.5-1O’l9)=
= 0.254 В. Из сопоставления вычисленных потенциалов видно, что в
хлоридном растворе двухвалентная медь с успехом окисляет ртуть до
каломели.
Неустойчивые степени окисления в ряде случаев могут стабилизи-
роваться за счет комплексообразования. Так, хорошо известно, что
платина не растворяется в азотной кислоте даже при нагревании, так
как даже потенциал пары Pt2+/Pt, равный 1.19 В, существенно пре-
вышает таковой для азотной кислоты (0.96 В). Для перевода платины
в раствор, как известно, применяют царскую водку, так как содержа-
щиеся в ней хлорид-ионы способны образовывать платинохлористо-
Окислительно-восстановительные процессы
167
водородную кислоту (H,PtCl6). Потенциал реакции
Pt + 6С1- = [PtCl62] + 4е' (7.27)
составляет всего 0.73 В.
Другим интересным примером изменения направления окис-
лительно-восстановительного процесса при комплексообразовании
является растворение металлического золота, имеющего потенциал
Au+/Au = 1.46 В, в цианидных растворах. Цианидный комплекс
[Au(CN)2]' имеет константу устойчивости 10-\ Для концентраций
аниона и комплекса в растворе, равных 1М, потенциал этой пары
составит
•£[Au(CN)2]Au = 1-46 - 0.059-28 = -0.192 В .
При этом металл оказывается вполне способным к окислению ки-
слородом воздуха в нейтральной среде (Е = 0.815 В).
Еще одним проявлением подобных закономерностей является
факт существования большою числа комплексных соединений трех-
валентного кобальта, несмотря на то, что потенциал пары Со37Со2+
равен 1.808 В (например, растворение гидроксида трехвалентного ко-
бальта в соляной кислоте сопровождается выделением хлора и пере-
ходом его в двухвалентный). Стабилизация комплексов трехвален-
тного кобальта обусловлена тем, что этот катион, имея электронную
конфигурацию d6 (рис. 7.7), проявляет высокую склонность к образо-
ванию прочных октаэдрических низкоспиновых комплексов. При
этом даже его аммиачный комплекс имеет рК ~ 35.2.
Рис.7.7. Схема заполнения L и е,
4? Я
орбиталей в комплексах двух- и
трехвалентного кобальта
В то же время появление одного лишнего электрона у двухва-
лентного иона приводит к тому, что все его комплексы оказываются
внешнесферными и существенно менее устойчивыми. Так, для
аммиачного комплекса Со2+ рКуст = 5.1. При равных концентраци-
ях комплексов двух- и трехвалентного кобальта в 1 М аммиачном
168
Глава 7
растворе потенциал пары [Co(NH3)6]3+/[Co(NH3)6]2+ составит 1.808 +
+0.059(5.1 - 35.2) = 0.03В. Поэтому в аммиачных, цианидных и многих
других растворах кобальт легко окисляется кислородом воздуха до
трехвалентного состояния.
7.6. Электролиз
Хорошо известно, что при подаче электрического тока достаточно
высокого напряжения на пару электродов, погруженных, например,
в раствор хлорида двухвалентного олова, на одном из электродов
(катоде) будет выделяться металлическое олово, а на втором (аноде)
-газообразный хлор. Рассмотрим условия протекания этого процесса.
При подаче напряжения на погруженные в раствор электроды к
отрицательно заряженному катоду движутся катионы Sn2+ и Н3О+.
Концентрация последнего в растворе составляет 10'7 М, поэтому его
потенциал ниже стандартного и составляет -0.059-7 = -0.413 В. Эта
величина заметно ниже, чем у олова (-0.136 В). К аноду же движутся
ионы СГ (£ci,/СГ = 1 -36 В) и гидроксид - анионы. Потенциал выделения
кислорода по реакции :
4ОН- = О,+ 2Н,О + 4е- (7.28)
при pH = 7 составляет 1.228 - 0.059-7 = 0.815 В. Однако при электроли-
зе невозможно образование молекулы кислорода в качестве едино-
временного акта-вначале должен образоваться атомарный кислород.
Для образования последнего необходимо приложить дополнительный
потенциал, примерно равный энергии химической связи в молекуле.
Вследствие этого для выделения кислорода необходима подача су-
щественно большего напряжения на электроды (перенапряжения). Вы-
сокое перенапряжение не позволяет протекать данному процессу в
описываемой системе. В итоге на электродах осуществляется выделе-
ние хлора и олова, а потенциал гальванического элемента, работаю-
щего за счет обратного процесса, достигает 0.815-(-0.136) = 0.951 В.
Таким образом, электролиз будет протекать при разности потенци-
алов на электродах выше этой величины. Кислород будет выделяться
также и при электролизе растворов фторидов, имеющих существенно
больший потенциал выделения, нежели кислород. Аналогично
выделение на аноде кислорода происходит и при электролизе солей
кислородсодержащих кислот.
Если потенциал пары раствор соли/металл выше 0.413 В,
электролиз будет приводить к выделению на катоде водорода.
Окислительно-восстановительные процессы 169
Поэтому такие активные металлы, как щелочные, щелочноземельные
и многие другие нельзя выделить электролизом растворов. Таким
образом, электролиз раствора сульфата натрия, например, будет
сводиться к выделению на катоде водорода и кислорода - на аноде.
Возникает вопрос: а нужен ли при этом сульфат натрия вообще?
Оказывается, функция электролита и в этом случае весьма важна -
он обеспечивает перенос тока через раствор, существенно ускоряя
процесс электролиза.
Рассмотрим теперь раствор некоторой медной соли, в который
опущены два медных электрода (рис. 7.8). При подаче даже незна-
чительного напряжения в такой системе будут обеспечиваться условия
для переноса меди с анода на катод. При этом все примеси, более
активные, чем медь, на аноде будут растворяться несколько быстрее,
но не будут выделяться на катоде, так как для их выделения нужен
больший потенциал, нежели для выделения меди. Выделению их
препятствует и более низкая концентрация. С другой стороны, менее
активные примеси оказываются не способными к ионизации и по мере
растворения анода выпадают на дно сосуда. Таким образом, на катод
должна переноситься практически чистая медь. Этот довольно эф-
фективный способ очистки широко применяется для малоактивных
170
Глава 7
металлов. Чистота продуктов электролиза существенно зависит от
подаваемого напряжения, поскольку с его увеличением возникает
возможность выделения примесей. С другой стороны, эффективность
процесса растет с ростом перенапряжения. Поэтому здесь, как и во
многих других случаях, приходится искать золотую середину. При
описании процессов электролиза мы полагали, что на электродах
должна присутствовать фаза чистого металла. Однако, как следует
из предыдущего раздела, внедрение в металлы примесей в небольших
количествах оказывается выгодным с точки зрения термодинамики.
Это существенно понижает потенциал их внедрения в материал
катода. Поэтому последний реально все же загрязняется малым ко-
личеством примесей.
Многие элементы чрезвычайно трудно или даже невозможно
получить с помощью химических реакций ввиду их высокой актив-
ности. В таком случае оказывается эффективным проведение элек-
тролиза расплавов их солей. Последние также являются хорошими
электролитами, однако, в отличие отводных растворов, при их элек-
тролизе отсутствуют конкурирующие процессы выделения на элек-
тродах водорода и кислорода, которые не позволяют выделять многие
элементы из растворов. Так, никаким другим способом нельзя полу-
чить фтор. Для его выделения используют близкую к эвтектической
смесь фторидов калия и водорода. Процесс сводится к выделению на
катоде водорода, а на аноде - фтора. В качестве материала для
контейнера и электродов используют никель, который быстро
пассивируется ввиду образования защитной пленки NiF,. Сходным
образом получают и алюминий при электролизе раствора его оксида
в расплавленном криолите (Na3AlF6). В данном случае на катоде
выделяется алюминий, а на аноде - кислород.
7.7. Электрохимические источники тока
Простейшим источником тока является уже разобранный нами
гальванический элемент. Недостатками его являются необходимость
присутствия жидкой фазы, достаточно малое выходное напряжение
и малая емкость. Некоторые из этих недостатков удалось скомпен-
сировать в широко используемых батарейках. Рабочими вещества-
ми в них являются цинк (отрицательный электрод) и влажная смесь
двуокиси марганца с хлоридом аммония, обеспечивающим кис-
лую реакцию (рис. 7.9). Положительным электродом является ин-
дифферентный в данных условиях углеродный стержень. Реакции на
Окислительно-восстановительные процессы
171
МпО2+НН4ОН
Рис.7.9. Схема устройства простейшего
источника тока
электродах могут быть представлены уравнениями:
Zn - 2е" = Zn2+ Е = -0.763 В , (7.29)
МпО, + 4NH/ + 1е~ = Мп2+ +Н2О + 4NH3 Е = +0.7 В. (7.30)
Потенциал последнего процесса ниже стандартного в кислой среде
(+1.228 В), существенно зависит от pH и несколько меняется в процессе
выделения аммиака, уходящего на образование комплексов с цинком
и марганцем. Поэтому величина электродного потенциала описан-
ного элемента не может быть охарактеризована с высокой точностью.
К сожалению, такие батарейки также являются необратимыми и их
не удается привести в исходное состояние пропусканием электри-
ческого тока в обратном направлении (при этом вместо цинка будет
выделяться водород). Кроме того, они характеризуется низким вы-
ходным напряжением.
Другой вариант попытки исправить положение - хорошо изве-
стный свинцовый аккумулятор (рис. 7.10). В нем протекают процессы
растворения свинца и его диоксида с образованием малорастворимо-
го сульфата (ПР = 1.6- 1О‘Е). Активность сульфат-ионов в концентриро-
ванном растворе серной кислоты имеет порядок 10’1 М. Тогда соответ-
ствующие электродные реакции имеют потенциалы:
Pb + SO/- - 2е' = PbSO4 - 0.126 - 0.0295-6.8 = - 0.327 В, (7.31)
PbO,+4H++SO42'+2e = PbSO4+2H,O +1.449+0.0295-6.8 = 1.65В. (7.32)
172
Глава 7
Рис.7,10. Схема устройства свинцового аккумулятора
Таким образом напряжение работы аккумулятора близко к 2 В и
существенно выше, чем для цинкового элемента. Кроме того, при-
веденные процессы являются обратимыми и аккумулятор можно пе-
резарядить. С другой стороны в нем опять же используется жидкий
электролит (да еще какой! Можно представить, какое количество
брюк автомобилистов бесследно исчезает в неравной борьбе с сер-
ной кислотой). Немногим лучше в этом отношении щелочные акку-
муляторы.
Весьма перспективным направлением является использование
твердых электролитов. Так, если разделить слои серебра и иода
перегородкой из иодида серебра (рис. 7.11), проводящей ионы Ag+,
последние будут диффундировать к иоду, вступая с ним в реакцию.
При этом электродные процессы могут быть описаны реакциями:
Ag-e’=Ag+, (7.33)
Ag++1/21, + е'= Agl, (7.34)
Несмотря на то что окислительный потенциал пары Ag+/Ag пре-
вышает таковой для 12/Г, образование термодинамически устойчивого
иодида приводит к тому, что электродвижущая сила, этого процес-
са превышает 1 В. Элемент с еще большим выходным напряжением
можно построить на основе пары литий-сера. Элементы такого типа
Окислительно-восстановительные процессы 17 3
Рис.7.11. Схема устройства источника тока на основе твердого электролита
не содержат жидкости, имеют высокую емкость и вполне обратимы.
Сравнительно малое их использование в настоящее время объясняет-
ся отсутствием твердых электролитов с высокой проводимостью по
ионам лития при комнатной температуре. Поэтому имеющиеся эле-
менты имеют сравнительно высокое внутреннее сопротивление и
могут использоваться лишь в низкоточных процессах, например в
батарейках для часов.
Рис.7 Л 2. Устройство топливного элемента на основе реакции окисления водорода
По аналогичной схеме построены топливные элементы. Кислород
и водород в них разделяются прослойкой материала с проводимостью
по ионам водорода (рис.7.12). Ионизируясь и оставляя электроны на
174
Глава 7
одном электроде, водород диффундирует к другому, вступая в реак-
цию с кислородом. Легко убедиться, что суммирование электродных
процессов приводит к тривиальной реакции окисления водорода
кислородом, которая, протекает с очень высоким к.п.д.
Н, - 2е" = 2Н+, (735)
2Н+ + 1/20, + 2е" = Н,0 . (736)
Препятствием для широкого использования топливных элементов
является низкая устойчивость или низкая электропроводность при
комнатной температуре таких твердых электролитов. Однако при
повышенных температурах они работают сравнительно хорошо.
Поэтому японские исследователи поставили своей задачей разра-
ботку на их основе двигателей для автомашин. Еще более ценным
представляется создание топливного элемента, основанного на
реакции окисления углерода. К сожалению, до сих пор не удалось
подобрать твердого электролита, способного эффективно перемещать
ионы углерода.
7.8. Коррозия
Весьма важным проявлением окислительно-восстановительных
процессов является коррозия, ежегодно приводящая к разрушению
примерно 10% от мирового производства металла. Для того чтобы
успешно бороться с этим явлением, необходимо представлять его
физико-химические основы. Коррозия протекает посредством
окисления металлических изделий за счет взаимодействия их с
окружающей средой:
М - пе" = Мп+ . (737)
Этот процесс называется катодным. Окислителем, как правило,
оказывается влажный воздух или растворенный в воде кислород. В
некоторых случаях в качестве агрессивной среды могут выступать
раствор кислоты или собственно вода. Окислительные потенциалы
некоторых наиболее типичных коррозийных сред приведены в
таблице 7.1. Перечисленные процессы называются анодными. Таким
образом, озон в кислой среде потенциально может окислить любой
металл, а в нейтральной - практически любой, за исключением золо-
та. Влажный воздух может окислить все металлы, находящиеся в ряду
напряжений левее серебра, и т.д. В то же время при отсутствии кис-
лорода окислительный потенциал воды оказывается примерно таким
же, как для процесса перехода железа в двухвалентное состояние.
Окислительно-восстановительные процессы
175
Таблица 7.1
Окислительные потенциалы некоторых наиболее типичных коррозийных сред
Окислительный процесс Е>\ В
Оз + 2Н+ + 2е- = Ог + НзО (кислая среда) (7.38) 2.07
Оз + НгО + 2е-= О2 + 2ОН- (нейтральная среда) (7.39) 1.66
Оз + 4Н+ + 4е- = 2НзО (кислая среда) (7.40) 1.23
Оз + 2НзО + 4е- = 4 ОН- (нейтральная среда) (7.41) 0.81
Оз + 2НзО + 4е- = 4 ОН- (щелочная среда) (7.42) 0.40
2Н+ + 2е- = Нз (кислая среда) (7.43) 0.0
2НгО + 2е- = Нз + 2ОН- (нейтральная среда) (7.44) -0.41
Однако, поскольку для осуществления коррозии необходимо обра-
зование пузырьков водорода, зарождение которых требует достаточно
высокого перенапряжения, железо не корродирует в отсутствии
кислорода.
Процессы коррозии могут активироваться или замедляться по
целому ряду причин. Так, например, сухой воздух обладает суще-
ственно более низкой окислительной активностью, поскольку протон,
принимающий участие практически во всех окислительных актах, в
данном случае отсутствует. Кроме того, давно было замечено, что
морская вода, а точнее присутствие в ней соли, существенно ускоряет
коррозию. Аналогичным образом действует и загрязнение металла
другим - менее активным.
Катодный и анодный процессы могут быть разнесены в про-
странстве ввиду проводимости металла. Так, если соединить медную
и цинковую пластину в растворе уксусной кислоты, то цинк за счет
большей активности будет постепенно растворяться (уравнение
(7.37)), а высвобождающиеся электроны - перетекать на менее
активную медь. На ней и осуществляется анодный процесс выделения
водорода (уравнение (7.43)). Таким образом, несмотря на то, что медь
не взаимодействует с водой, водород будет выделяться именно на ме-
ди. Если же опустить те же пластины в раствор хлорида натрия, со-
держащий фенолфталеин, то на меди будет протекать реакция (7.44)
и вблизи нее раствор будет окрашиваться в розовый цвет за счет
присутствия гидроксид-ионов.
Для защиты металла от коррозии его обычно покрывают раз-
личными изолирующими от агрессивных сред покрытиями, напри-
мер масляной краской. Все было бы хорошо, если бы изделие пос-
ле этого не использовали. При эксплуатации такое покрытие легко
повреждается и коррозия протекает по трещинам. Представляется,
что куда эффективнее оказалось бы покрытие металла другим - менее
176
Глава 7
активным, например, медью. При этом доступ окислителя к активному
металлу оказывается перекрытым, а медь, на которую стекают элек-
троны с более активного металла, не окисляется. Однако при повре-
ждении такого покрытия катионы с более активного металла, уже
имеющего некоторый положительный заряд, переходят в раствор еще
более интенсивно. При этом металл разрушается гораздо быстрее.
Существенно более эффективным является так называемая ка-
тодная защита - покрытие металла более активным, например железа
- цинком. Жесть с таким покрытием можно увидеть на крышах,
подоконниках и т.д. Даже при повреждении такого покрытия железо,
на которое стекает избыток электронов с цинка, не окисляется до тех
пор, пока не прокорродирует все покрытие. Кроме того, цинковые
болванки иногда присоединяют к подземным трубопроводам через
некоторое определенное расстояние. За счет проводимости металла
эффект оказывается почти идентичным. Того же результата можно
добиться и просто за счет подачи на изделие отрицательного заряда.
Наконец, всем известно, что существуют так называемые нержа-
веющие стали. Ингибитором коррозии в них является примесь хрома.
Хром, являясь более активным металлом по сравнению с железом, не
дает ему окисляться. Сам же хром практически не окисляется за счет
высокого перенапряжения для этого процесса.
Задачи и вопросы
1.Установлено, что следующие реакции в растворах протекают
слева направо:
Fe3+ + Ag +С1- = Fe2+ +AgCl
Fe + 2H+ = Fe2+ +H,
2AgCl +H2 = 2Ag +2H+ + 2CF
Покажите на основе этого, в каком направлении протекают сле-
дующие процессы:
Fe + 2AgCl = Fe2+ + 2Ag + 2C1’
3Fe2+ = 2Fe3+ + Fe
2.Основываясь на приведенных стандартных потенциалах полу-
реакций вычислите значения для неизвестных.
Е°(С1О47С1*)= 1.380 В, Е°(С1,/С1-)= 1.358 В, Е°(С1О47С1,) - ?
Окислительно-восстановительные процессы
177
£°(H,sos/H,S)=0.347 В, £°(S/h2S)=O. 142 В, £°(H,so3/S) - ?
Е°(Ю3-/Г)= 1.085 В, Г)=0.536 В, Е°(1О371,) - ?
З.Три железные пластины, две из которых были покрыты медью
и цинком а третья-окрашена, были оцарапаны таким образом, чтобы
открылся слой железа. Опишите скорость протекания коррозии железа
для всех трех случаев. Укажите продукты коррозии, если в качестве
агрессивной среды выступала а) вода, б) вода с растворенным кисло-
родом.
F°(Zn27Zn)= -0.763 В, E°(Cu27Cu) = +0.337 В, £°(Fe27Fe) = -0.44 В,
E°(Fei+/Fe) = -0.037 В, С°(О,,Н7Н,О)= +1.228 В.
4.Пользуясь следующими данными постройте диаграммы Фроста
для состояния серы в кислой и щелочной средах. Определите будет ли
диспропорционировать сера в этих условиях?
H,SO4: К2= МО’2; H,SO3: #=2-10-2, Л7,=6-106 * 8 * * *;
H,S: £=10Л /С2=10-13;
S +2е = S2'-0.480 В
H2SO3 + 4Н+ +4<? = S +ЗН2О+0.449 В
SO,2" + 4Н+ +2е = H,SO3 + Н,О+0.17 В
5.Составьте диаграмму Латимера для переходов различных
ионных форм железа при pH = 14, если при pH = 0 она выглядит
следующим образом: FeO42’ —И—> Fe3+ —1,77?-> Fe2+ —Fe,
а величины произведений растворимости для двух- и трехвалент-
ного железа составляют 810‘16 и 410-40. Укажите продукты восстанов-
ления шестивалентного железа избытком металлического в кислой и
щелочной средах.
6. Определите произведение растворимости двухвалентного
свинца и константу нестойкости аниона [РЬ(ОН)4]2', используя
величины стандартных потенциалов полуреакций:
£°(РЬО2/РЬ2+) = +1.455 В,
£°(РЬО2/ Pb(OH)2) = +0.385 В,
Е°(РЬО, / [Pb(OH)4]2-) = +0.21 В.
178
Глава 7
7. Считая активности всех участвующих в реакции веществ
равными единице, оцените pH при котором возможно протекание
реакции:
2МпО4' + 6Вг' + 4Н,0 = 2MnO, +3Br, + 8ОН',
если: МпО/ +2Н,0 +3е = MnO, +4ОН' Е° = 0.59 В,
1/2Вг,+е = Вг" Е° = 1.09В
8. При какой концентрации цианид-ионов в растворе произойдет
растворение осадка цианида серебра, если:
Ag+ + е = Ag Е° = 0.799В
AgCN + е = Ag +CN' Е°=-0.017В
Ag(CN)2‘ + е = Ag + 2CN" Е7 = -0.31В
(Считайте, условием растворения достижение единичной кон-
центрации комплексного иона в растворе).
9. Вычислите вторую константу диссоциации H,SeO3, если
К, = 2.510-3и:
SeO3 + ЗН2О + 4е = Se + 6ОН’ Е7 =-0.366В,
H,SeO3 +4Н+ + 4е = Se + ЗН,0 Е7 = 0.74В.
10. Пользуясь следующими справочными данными определите
константу устойчивости иона Co(CN)64‘:
Co(CN)63’ + е = Co(CN)64' = -0.83В
Со3+ +3е = Со Е° = 0.330В,
Со2+ + le = Со Е7 = -0.277В
Кп. [Co(CN)63-] = 10м.
11. При какой какой величине pH произойдет растворение
гидроксида серебра, если:
Ag+ + е = Ag £°= 0.799В
AgOH + е = Ag + ОН’ Е°=0.339В
(Считайте, условием растворения достижение 0.01М концент-
рации серебра в растворе).
Глава 8
КИНЕТИКА ХИМИЧЕСКИХ РЕАКЦИЙ
При изучении термодинамики химических процессов мы
задавались вопросом их принципиальной возможности, не прини-
мая во внимание реальные скорости их протекания. Исходя из этого,
например, можно заключить, что водород должен весьма успешно
взаимодействовать с фтором, хлором и кислородом, поскольку все
эти реакции имеют большие по модулю отрицательные значения
энергии Гиббса. Реально первая из них протекает самопроизвольно,
для активации второй достаточно сравнительно небольшой по
энергии и кратковременной световой экспозиции. Третья же пара
веществ даже на свету может сосуществовать весьма долго. Реакция
между кислородом и водородом осуществляется лишь при нагрева-
нии, подаче электрического разряда или при наличии в системе
катализатора. Аналогично лишь заторможенностью процессов окис-
ления органической материи объясняется возможность существова-
ния живых организмов и растений.
Таким образом, из термодинамических расчетов можно сделать
лишь заключение о том, что протекание реакции принципиально
невозможно, если ее энергия Гиббса имеет существенно положи-
тельное значение. В том же случае, если она оказывается близкой к
нулю или отрицательной, возможность осуществления реакции оп-
ределяется особенностями кинетики процесса. Химическая кинетика
изучает закономерности протекания, химических реакций во времени,
зависимость скорости реакции от различных факторов: температуры,
давления, концентрации реагентов, воздействия излучения, присутст-
вия катализатора и т.д. Как правило, конечной целью такого иссле-
дования является оптимизация химического процесса или выявление
методов его торможения. В свою очередь, для этого оказывается по-
лезным знание механизма, т. е. пути протекания реакции, или по край-
ней мере, наиболее медленной ее стадии.
Подводя итог сказанному, необходимо заметить, что термоди-
намика, несомненно, является более целостной и разработанной
180
Глава 8
областью знания. С другой стороны, химическая кинетика постоянно
совершенствуется, в ней совершаются новые открытия, разрабаты-
ваются новые подходы. И от этого она становится лишь более привле-
кательной для химиков. Однако для корректного описания кинетики
процесса крайне важно знать его термодинамические параметры.
8.1. Скорость химической реакции
Скорость химической реакции принято характеризовать
изменением концентрации одного из реагирующих веществ или про-
дуктов реакции в единицу времени. При этом в силу ряда причин, среди
которых чаще всего фигурирует изменение концентрации реагентов
или площади их поверхности, скорость химической реакции сущест-
венно меняется во времени. С этой точки зрения следует делать разли-
чие между средней скоростью реакции в течение некоторого времени
и ее истинной скоростью в данный момент. Математические выраже-
ния этих величин можно представить в виде:
w = kc/At и w-dddt. (8.1)
Очевидно, что найденные на основании приведенных соотноше-
ний для процесса
аА + ЬВ = сС + dD (8.2)
скорости, выраженные через концентрации различных веществ, будут
иметь различные значения, а выраженные через концентрации ис-
ходных веществ и продуктов - должны различаться и по знаку, хотя
сама по себескорость процесса-сутьвеличина положительная. Таким
образом, обычно либо конкретизируют, по концентрации какого
вещества определяют скорость, либо пользуются приведенной ско-
ростью:
w = -w,/ а = -wJ b =wJ с -wn! d, (8.3)
Л D С U v z
где числители дробей выражают скорость процесса по данному
веществу.
Для осуществления анализа кинетики химического процесса
строят графики зависимости концентрации одного из исходных ве-
ществ или продуктов от времени протекания процесса. Естественно,
что для наиболее корректного описания желательно проводить его
при постоянной температуре. При этом наиболее часто концентрацию
вещества фиксируют с помощью каких-либо физических или физико-
химических методов исследования (например по поглощению света
Кинетика химических реакций
181
определенной частоты, интенсивности сигнала спектров ЯМР, ЭПР,
изменению pH раствора и т.д.)- Достоинством таких методов является
отсутствие постороннего вмешательства в ход процесса или изменения
количества реагентов. В противном случае периодически проводят
пробоотбор таким образом, чтобы концентрации менялись прене-
брежимо мало. Типичные виды кинетических кривых приведены ри-
сунке 8.1. Обработкой такого рода данных получают кинетические
уравнения, связывающие скорость реакции с концентрацией реаги-
рующих веществ.
Рис.8 Л. Типичные формы представления кинетических кривых:
зависимое! ь концентрации веществ (а) или скорости реакции (б) от времени
Для протекания подавляющего большинства химических процес-
сов необходимым условием является соударение двух молекул. Из
этого автоматически следует, что скорость процесса должна быть
равна или, по крайней мере, пропорциональна частоте соударений.
Действительно, на первый взгляд представляется закономерным, что
для бимолекулярной реакции
А + В = АВ , (8.4)
протекающей в газовой фазе, каждому акту столкновения должно
соответствовать образование одной молекулы продукта. Число таких
соударений и, соответственно, скорость реакции можно легко под-
считать. Из молекулярно-кинетической теории следует, что средняя
кинетическая энергия молекулы должна быть равна ЗИкТ. Откуда
mv2 = ЗкТ или у = (ЗкТ/т)"1, (8.5)
где к - постоянная Больцмана, а у - среднеквадратичная скорость
молекулы. Средняя скорость молекулы может быть рассчитана по
уравнению
v= (8кТ/пт)'12. (8.6)
182
Глава 8
Для определения частоты столкновения молекул газов предпо-
ложим вначале, что одна из них является неподвижной, а остальные
движутся вдоль направления некоторой заданной оси со скоростью
v. Тогда за некоторый небольшой интервал времени dt с выбранной
частицей столкнутся все молекулы, находящиеся внутри цилиндра с
высотой vdt и сечением s, радиус которого из общих соображений
должен быть близок к сумме радиусов сталкивающихся молекул (для
близких по размеру молекул - к диаметру) (ст = nd2) (рис. 8.2).
Рис.8.2. Схема “трубы столкновений” для бимолекулярных реакций
Внутри данного цилиндра находятся с Na vdt молекул, где с - их
мольная концентрация в единице объема, a N-число Авогадро. Таким
образом, частоту столкновений можно определить как отношение
этой величины к временному интервалу, т. е.
v = cNerv (8.7)
Аналогичную "трубу столкновений” можно выделить для любого
направления. Поэтому нет необходимости вводить какие-либо по-
правки на размерность движения. Реально в движении находятся обе
молекулы, и средняя скорость их взаимного перемещения друг относи-
тельно друга составит vV2. Для нахождения полного числа соуда-
рений необходимо умножить данное выражение на полное число
молекул в единице объема, деленное на два (чтобы исключить дуб-
ирование при подсчете числа столкновений). Выразив среднюю
Кинетика химических реакций
183
скорость молекулы из уравнения (8.6), получим:
Z - N 2 с2' nd2 (№<= N2c2 d2 (bkTnlm)"2. (8.8)
Для случая столкновения двух различных молекул соотношение
несколько сложнее:
Z = N 2cAcBd 2ав , (8.9)
где dAB = гА + гв- сумма радиусов молекул, сА и св - их концентрации
в единице объема, ц = тАтв1 (тА + тв) - приведенная масса. Число
таких соударений очень велико: так, при комнатной температуре в
одном литре воздуха их происходит около 1033 в секунду. Соответст-
венно, скорость реакции, выраженная в молях, должна быть пропор-
циональна
w = NcAcBdAB2(SnkT/^12. (8.10)
Из полученного выражения непосредственно следует, что скорость
реакции, происходящей при столкновении двух молекул, будет про-
порциональна произведению их концентраций. Если необходимым
условием является столкновение трех частиц, то в результирующее
выражение будут входить три концентрации и т. д. Таким образом,
мы подошли к формулировке основного постулата химической кине-
тики, гласящего, что скорость химического процесса прямо пропорцио-
нальна произведению концентраций реагирующих веществ в степенях,
равных стехиометрическим коэффициентам его уравнения. Приоритет
в открытии этого факта приписывается Гульбергу и Вааге, хотя не-
сколько раньше в немного отличном виде его сформулировал Бекетов.
Таким образом, для рассмотренной выше реакции (8.2) скорость
может быть выражена в виде:
и> = А:[Л]0[5]\ (8.11)
Величина к называется константой скорости химической реак-
ции и численно равна ее скорости при единичных концентрациях
реагентов.
8.2. Порядок и молекулярность реакции
При рассмотрении кинетики химических реакций было замечено,
что часто коэффициенты а и Ъ уравнения (8.11) оказываются не рав-
ными стехиометрическим коэффициентам. Это связано с тем, что
большинство химических процессов являются сложными, т. е. проте-
кают через ряд стадий, называемых иначе элементарными (простыми)
реакциями. Последние определяют как реакции непосредственного
184
Глава 8
перехода реагентов в продукты без образования какого-либо рода
переходных состояний, отличающихся от продуктов. Так, реакция
взаимодействия перманганата калия с перекисью водорода в щелоч-
ной среде протекает через следующие элементарные стадии:
МпО; + НОО- = [ООМпОД + ОН . (8.12)
Образующийся пероксидный комплекс распадается далее на
весьма неустойчивый ион МпО/ с выделением кислорода:
[ООМпОД = МпО; + О2. (8.13)
Нестабильность этого иона обусловлена нетипичным для пятива-
лентного марганца низким координационным числом. Поэтому он
практически мгновенно достраивает координационный полиэдр за
счет присоединения молекулы воды:
МпО; + Н,0 = Н2МпО; . (8.14)
Продукт сопропорционирует с другим перманганат-ионом:
Н,МпО; + МпО; = 2НМпО; . (8.15)
После этого избыточные протоны нейтрализуют часть гидроксид-
ионов раствора:
НМпО; + ОН- = МпОД + Н,0 . (8.16)
Таким, образом, суммарный процесс задается уравнением:
2МпО; + Н,О, +2ОН- - 2МпОД + 2Н2О + О2. (8.17)
Весьма сложными могут оказаться простые на первый взгляд
процессы типа взаимодействия хлора с водородом или с угарным
газом. Инициирование последнего, например, происходит за счет
диссоциации молекулы хлора. Дальнейшее протекание процесса
определяется совокупностью четырех основных реакций:
С1 + СО = СОС1 •,
СОС1 + С1, = СОС1, + С1 •,
(8.18)
СОС1+С1 =СОС12,
СОС1 + СОС1 = СОС12 + со.
Возможно также несколько путей рекомбинации участвующих в
процессе радикалов, не приводящих к образованию основного про-
дукта. Это взаимодействие их на стенке сосуда или при столкновении
с третьей молекулой.
Таким образом, скорость процесса является функцией скорости
всех его элементарных актов. Для понимания взаимосвязи между ними
выберем некоторый модельный процесс взаимодействия, протекаю-
Кинетика химических реакций
185
щий в две стадии. Пусть среднее время протекания элементарных
актов составляет и Тогда время протекания суммарного процесса
равно (г2 + ?,). В силу ряда причин удобнее описывать их в терминах
частоты. Тогда, поскольку v = \h, для суммарного процесса
l/vf = 1/V] + 1/v, = (vj + v2) / V]V, . (8-19)
Очевидно, что при v1>>v2 \>c ~ v, , т. e. скорость протекания
процесса определяется наиболее медленной его стадией. Таким
образом, в первом приближении, коэффициенты уравнения (8.11)
должны относиться к наиболее медленной стадии процесса. Сумма
показателей степеней этого уравнения называется порядком реакции.
Следует различать порядок реакции как таковой с частным порядком
по отдельным реагентам. Первый из них зачастую является суммой
последних. При этом скорость образования продукта реакции на пер-
вых этапах может определяться одной стадией, а по мере протекания
процесса и убывания концентрации реагентов - другой. Поэтому
зачастую полные кинетические уравнения выглядят намного слож-
нее. Например, для синтеза бромистого водорода из простых веществ
кинетическое уравнение можно описать соотношением:
d [HBr] / dt = (^[HJfBrJ*5) / ([BrJ + K2[Br2]0). (8.20)
На начальных этапах скорость этого процесса будет пропорцио-
нальна произведению концентрации водорода на корень квадратный
из концентрации брома. По мере же протекания процесса частный
порядок его по брому будет постепенно возрастать от 0.5 до 1.5 при
[Вг,]« Л12[Вг2]0 . Понимание этого факта невозможно без знания меха-
низма протекания процесса, наиболее полное описание которого и
является привлекательным для любого кинетического исследования.
Само по себе понятие порядка реакции формально, т. к. оно лишь
косвенно связано с ее механизмом. Чаще всего можно предложить
одновременно несколько различных путей, предполагающих тот же
порядок процесса. С другой стороны, если данный механизм приводит
к порядку реакции, отличному от наблюдаемого, однозначно можно
заключить, что он не соответствует действительности.
Более конкретным понятием является молекулярность реакции,
определяющаяся числом частиц, которые должны одновременно
взаимодействовать в ходе одного элементарного акта. К одномолеку-
лярным относятся многие реакции термического разложения, изоме-
ризации молекул и некоторые другие. Примером таковых является
реакция разложения азометана:
CH3-N=N-CH3 = С2Н6 + N, . (8.21)
186
Глава 8
Однако на самом деле одномолекулярные процессы встречаются
не столь часто. Для того чтобы молекула распалась, она должна
обладать достаточным запасом энергии для разрыва химической
связи. Спрашивается, если она так богата энергией, то почему она не
распалась до сих пор? Действительно, валентные колебания, которые
фактически являются попытками молекулы разорвать одну из связей,
происходят с высокой частотой. Принципиально мономолекулярный
распад оказывается возможным для больших молекул с большим
числом колебательных степеней свободы. В таком случае избыточная
энергия исходно распределялась между этими колебаниями. Но
наиболее часто “избыточная” энергия сообщается молекуле именно
при соударении с другой аналогичной или отличной от нее частицей
либо со стенкой реакционного сосуда. Поэтому реально мономоле-
кулярные процессы сравнительно редки. Совершенно однозначно к
мономолекулярный реакциям можно отнести радиоактивный рас-
пад, скорость которого прямо пропорциональна концентрации радио-
активного вещества.
Наиболее распространенными являются бимолекулярные реак-
ции. Широкая распространенность их определяется, с одной стороны,
весьма высокой вероятностью двойных столкновений, а с другой -
легкостью перераспределения энергии между сталкивающимися ча-
стицами. Типичным примером таковых может служить процесс обра-
зования сложных эфиров типа:
СН3СООН + С,Н5ОН = СН3СООС2Н5 + Н2О . (8.22)
Другим, пожалуй наиболее часто встречающимся в монографиях,
примером служит разложение йодистого водорода:
2HI = Н2 + 12. (8.23)
Тримолекулярные реакции (если они вообще существуют) встре-
чаются весьма редко. Объяснением их низкой распространенности
является очевидно малая вероятность тройных столкновений. Наибо-
лее распространенными примерами таковых, приводимыми в боль-
шинстве учебных пособий служат процессы с участием оксида азота,
например:
2NO + O? = 2NO2. (8.24)
Однако, как будет показано ниже, эта реакция действительно
имеет третий порядок, но состоит исключительно из бимолекулярных
процессов.
Оказывается, что несоответствие молекулярности и порядка может
наблюдаться и для сравнительно простых реакций. Например, процесс
Кинетика химических реакций
187
гидролиза бромбутана
С4Н9Вг +Н2О = С4Н9ОН + НВг (8.25)
является бимолекулярным, но в силу огромного избытка воды и
практически постоянной концентрации последняя автоматически
включается в константу равновесия. Это так называемая реакция
псевдопервого порядка. Процессы псевдо (п-1) порядка, включаю-
щие молекулы растворителей, весьма распространены в химии.
Приведенные примеры однозначно показывают, что в случае
сложных процессов порядок реакции и ее молекулярность не сог-
ласуются друг с другом, причем порядок может быть как меньше, так
и больше молекулярности. И если они даже и совпадают, это не яв-
ляется свидетельством одностадийности процесса. Тем не менее поня-
тие порядка реакций весьма полезно для кинетических исследований
и определению его уделяется достаточно большое внимание.
8.3. Кинетические уравнения и методы определения
порядка реакции
Среди всего многообразия химических процессов встречаются
реакции разного порядка, некоторые из которых описываются очень
сложными кинетическими уравнениями. Не имея возможности охва-
тить все из них, остановимся на тех, скорость которых пропорцио-
нальна концентрации в некоторой степени, сделав акцент на случае,
когда она имеет целые значения.
Для реакций нулевого порядка скорость процесса не зависит от
концентрации, а следовательно, и от времени его протекания:
-dc/dt=k (8.26)
или после интегрирования,
c0-c = kt, (8.27)
где с - концентрация исходного вещества в момент времени t, cQ - та
же величина в начале процесса, а к - константа скорости. Таким
образом, концентрация исходного реагента будет линейно убывать,
а продукта-линейно возрастать во времени. Размерность константы
скорости при этом совпадает с размерностью собственно скорости
процесса (моль/л с). Для анализа кинетических данных часто ис-
пользуется понятие времени полупревращения (период полураспада).
Для реакций нулевого порядка оно равно
г„2=сД*., (8.28)
188
Глава 8
Для реакций первого порядка соответственно
- de / dt = kc. (8.29)
Разделяя переменные и интегрируя, легко получить:
f -de I с = kt или ln(c01 с) = kt. (8.30)
Константа скорости при этом имеет размерность с1, а время
полупревращения определяется соотношением:
= 1п2/£. (8.31)
В общем случае, для реакции «-порядка при равенстве исходных
концентраций реагентов, отнесенных к стехиометрическим коэффици-
ентам и п 1, имеем:
- de!dt=kcn, (8.32)
- del сп = (1 /(л-1)) 6/(1/^) =kdt
или 1/сл’1 - Мс*'1 - k(n-V)t = const t. (8.33)
Константа скорости при этом имеет размерность концентрации
в степени (п-1), деленной на время, а время полупревращения равно
т1/2 = (2^- 1)/^(и-1)с0"Л (8.34)
Способы определения порядка реакции отличаются завидным
разнообразием. Остановимся на некоторых наиболее простых и рас-
пространенных из них. Наиболее часто массив для кинетической
обработки эксперимента представляется в форме зависимости кон-
центрации одного из исходных веществ или продуктов реакции от
времени (см. рис. 8.1а). Первый наиболее простой метод основан на
тривиальной подстановке этих данных в различные кинетические
уравнения (табл. 8.1). Очевидно, что постоянные значения константы
получатся лишь для правильно выбранного уравнения. Несколько
более цивилизованный путь - построение графических зависимостей
в координатах спрямления, также сведенных в таблице 8.1. Примеры
построения таковых для некоторой реакции второго порядка при-
ведены на рисунке 8.3.
Таблиц 8.1
Параметры и соотношения для кинетических расчетов
Порядок реакции Кинетическое уравнение Координаты спрямления Период полупревращения Размерность к
0 -dddt = /< с - t cn/2k моль/л-с
1 -dddt = к с Inc - t 1п2 / к с 1
п -dddt = ксп \/c,hl - t (2й-'-1)/А-(л-1)с0"-' (л/мольр-'с1
Как видно из приведенных данных, результаты эксперимента
спрямляются лишь в координатах “1/с -1”. Однако можно заметить,
Кинетика химических реакций
189
О 5 10 15 20 25 30
что и в этом случае экспериментальные точки не совсем четко ложатся
на одну прямую. Разброс их может являться следствием ошибок в
определении концентрации, времени протекания процесса и коле-
баний температуры. Точность обработки существенно растет при
разумном увеличении числа экспериментальных точек. Критерием
истинности является минимизация суммы квадратов отклонений то-
чек расчетной кривой от экспериментальной.
Еще более привлекательный способ оценки порядка реакции
предложен Вант-Гоффом. Он основан на построении графических
зависимостей в логарифмических координатах. Действительно, про-
логарифмировав выражение для скорости реакции «-порядка в общем
виде, получим:
Inw = In k + п Inc (8.35)
Построив графическую зависимость в координатах “Inw - Inc”
или обсчитав эту зависимость в виде линейной регрессии (у = ах + Ь),
получаем порядок реакции в виде тангенса угла наклона графика и
логарифм константы скорости в виде свободного члена (рис. 8.4). Этот
метод, безусловно, представляется наиболее перспективным, но для
его использования необходимо иметь достаточно большое число экс-
190
Глава 8
периментальных точек со сравнительно малым шагом по изменению
концентрации, чтобы иметь возможность корректно отнести скорость
к конкретной величине концентрации.
Рис.8.4. Пример расчета кинетического эксперимента по методу Вант-Гоффа
Для остальных методов определения порядка реакции приходит-
ся проводить серию кинетических экспериментов. Например, если
мы задаем заведомо избыточные концентрации всех веществ, кро-
ме одного, то в результате кинетического эксперимента получается
порядок реакции именно по данному веществу, поскольку кон-
центрации всех остальных меняются слабо. Путем сложения порядков
реакции по всем компонентам можно получить суммарный порядок
процесса. Недостатком этого метода является то, что реально можно
получить порядок разных стадий процесса, не все из которых являют-
ся лимитирующими.
Другой способ основан на определении времени полупревра-
щения или, в общем случае, фиксированной степени превращения
для различных исходных концентраций. Далее методика обработки
экспериментальных данных проводится на основе кинетических
уравнений, приведенных в таблице 8.1.
Кинетика химических реакций
191
8.4. Обратимые реакции
Как было отмечено ранее, большинство химических процессов
являются в той или иной мере обратимыми. Поэтому для них на оп-
ределенном этапе эффект протекания прямой реакции начинает
существенно нейтрализоваться протеканием обратного процесса.
В таком случае на всех кинетических кривых будут возникать си-
стематические отклонения. Поэтому приведенные способы опреде-
ления порядка реакции хорошо работают лишь для сравнительно ма-
лых степеней превращения или в случае, когда они сопровождаются
значительным уменьшением энергии Гиббса, т. е. когда воздействие
обратного процесса сводится к минимуму. Именно поэтому ранее
рассмотренный метод определения порядка реакции Вант-Гоффа
иногда называют методом начальных скоростей Вант-Гоффа.
С другой стороны, из кинетических уравнений можно так же, как
и из термодинамических соображений, прийти к понятию о хими-
ческом равновесии. Действительно, равновесие наступает тогда и
только тогда, когда скорости прямой и обратной реакций сравняются.
Действительно, для процесса, описываемого уравнением (8.4), ско-
рости прямого и обратного процессов могут быть описаны уравне-
ниями:
мда и Чб=*о6 гада. (8.36)
Приравняв правые части этих уравнений, получаем:
^w = *o6 [C]W
или
= = (8.37)
Таким образом, константа равновесия химической реакции равна
отношению констант скоростей прямого и обратного процессов.
Следует заметить, что данный вывод для сложных реакций не являет-
ся строгим, хотя константа равновесия от этого, естественно, не пе-
рестает быть константой.
8.5. Теория активированного комплекса
Из соотношения (8.10) следует, что скорость бимолекулярной
газовой реакции при атмосферном давлении и комнатной температу-
ре должна составлять около 1010 моль/л-с. Очевидно, что скорость
192
Глава 8
протекания большинства процессов существенно меньше этой
величины. Более того, даже для сравнительно простых бимолеку-
лярных процессов константа скорости не пропорциональна квад-
ратному корню из температуры, а связана с ней экспоненциаль-
ной зависимостью. Для объяснения этих фактов было принято
предположение, что протекание реакции возможно только в том
случае, когда совокупная кинетическая энергия атомов А и В боль-
ше или равна некоторому пороговому значению, называемому энер-
гией активации. Действительно, энергия химической связи склады-
вается из двух составляющих: энергии притяжения и энергии оттал-
кивания. Первая преобладает на сравнительно больших расстояниях,
последняя - на более коротких. В результате при образовании
молекулы достигается некое оптимальное состояние с минимальной
энергией (рис. 8.5с). Для осуществления химической реакции
необходимо сближение двух частиц на расстояние, сопоставимое с
длиной химической связи между ними. При постепенном сближении
атомов в первую очередь начинают действовать силы отталкивания
между их электронными оболочками. И лишь после достижения
некоторой деформации последних, силы притяжения начинают
преобладать над силами отталкивания за счет формирования хими-
ческой связи (см. рис. 8.56). Легко заметить, что на некотором
расстоянии энергия системы достигает максимального значения,
которое и соответствует энергии активации данного процесса. Оче-
видно, что два атома или две молекулы могут вступить во взаимо-
действие с образованием новой химической связи только, если сумма
их кинетических энергий превышает энергию активации процесса.
Рис.8.5. Зависимость энергии молекулы от межатомного расстояния (а) и изменение
энергии пары частиц по мере их сближения (б)
Кинетика химических реакций
193
В ходе кинетических экспериментов, как правило, не ставится
задача исследования зависимости энергии системы от межатомного
расстояния и потому чаще эту зависимость представляют в несколь-
ко упрощенном виде (рис. 8.6), где по оси абсцисс откладывается
некоторая абстрактная величина, упрощенно называемая “обобщен-
ной координатой реакции”. Некоторую туманность ее физического
смысла с лихвой покрывают наглядность и простота представления.
Рис.8.б. Схема зависимости
энергии системы от “коорди-
наты реакции”
Преимущества изображения изменения энергии процесса в этих
координатах легко понять на примере рассмотрения следующего
процесса:
АВ + С = А + ВС. (8.38)
Перераспределение химических связей в данной системе про-
исходит через образование в промежуточном состоянии некоторого
активированного комплекса состава АВС. Очевидно, что сближение
атомов В и С сопровождается увеличением расстояния между А и В.
График зависимости энергии этой системы от упомянутых расстояний
будет трехмерным. Для построения такового удобно пользоваться
системой координат, в которой по осям абсцисс и ординат отложены
расстояния АВ и ВС, а энергия системы представляется в виде изо-
линий с одинаковыми значениями энергии (рис. 8.7).
Такой системой представления принято пользоваться для изо-
бражения высоты местности на географических картах. Естественно,
что реакция будет протекать по пути с минимальной энергией, обо-
значенному на рисунке пунктирной линией. Здесь уместно провести
аналогию с рекой, которая никогда не течет вверх по возвышенности,
194
Глава 8
Рис.8.7. Зависимость энергии системы АВС от межатомных расстояний
если рядом есть ложбинка. Крайнее верхнее состояние на приведенной
диаграмме соответствует исходным веществам (АВ + С), а крайнее
правое - продуктам реакции (ВС + А). Некоторая точка (*) в середине
пунктирной кривой соответствует максимальной энергии, т.е. акти-
вированному комплексу. Нетрудно убедиться, что такое представ-
ление энергетической диаграммы для процесса соответствует приве-
денному на рисунке 8.6 в существенно более простом виде. При этом
принятое на двумерной диаграмме обозначение “ход процесса”
представляется чуть ли не единственно возможным.
Энергии активации химических процессов различного рода
колеблются в весьма широких пределах - от единиц до нескольких
сотен кДж/моль. Эти величины определяются природой реагирующих
частиц и, в ряде случаев, состоянием взаимодействующих фаз и
Кинетика химических реакций
195
продуктов процесса. Средняя кинетическая энергия одноатомной
молекулы при комнатной температуре составляет около 4 кДж/моль,
а при 1000°С - около 15 кДж/моль. В случае, если эта величина соп-
оставима с энергией активации, значительная часть молекул имеет
энергию, достаточную для химического взаимодействия. Тогда
скорость протекания процесса определяется частотой столкновений
и может приблизиться к теоретической. Такие процессы принято на-
зывать дезактивационными. Они сравнительно мало распростране-
ны. Для большинства процессов Ет.» ЗПкТи энергию, достаточную
для успешного преодоления активационного барьера, имеет сравни-
тельно небольшое число частиц. Распределение их по энергиям, назы-
ваемое распределением Больцмана определяется соотношением:
W = exp(-£Kr/^7). (839)
Пример такого распределения для двух температур Т: > Т, при-
веден на рисунке 8.8.
Рис.8.8. Распределение частиц
по энергиям при различных
температурах
С повышением температуры максимум кривой этого распределе-
ния смещается в сторону высоких энергий и постепенно размывается.
Таким образом, доля частиц с энергией, превышающей искомую вели-
чину, резко возрастает.
С учетом вышесказанного, объединяя уравнения (8.10) и (8.39),
для константы скорости реакции нетрудно получить:
к = const (7)|/гехр(-£акт/ кТ) (8.40),
или, переходя от атомного уровня к привычному мольному:
к = const • (7)1'2 ехр(-£ кт/ Д7) . (8.41)
После логарифмирования и дифференцирования по Тполучаем:
d\nk/dT = (EaKi. + \I2RT) /RT-. (8.42)
196
Глава 8
Для большинства химических процессов Ейкт » \I2RTh поэтому
вполне можно пренебречь вторым слагаемым в числителе. При этом
получается широко известное уравнение Аррениуса:
dink/ dT = E.M(/ RT2 (8.43)
или
k = k^^(-EJRT). (8.44)
Предэкспоненциальный множитель для реакций второго порядка
имеет величину около 1010 - 10" л/моль с.
Из уравнения (8.44) можно найти и соотношение скоростей
процесса при двух температурах, различающихся на дТ:
Д1п/с = ln[(Kr+jZ) / wr] « MTEm /RT2 (8.45)
или
(H<r,jr)/wr = exP(A7’£tT/A7’!). (8.46)
Для сравнительно малых отношений Д77Г изменением знамена-
теля в первом приближении можно пренебречь, откуда следует, что
для равных изменений температуры должны наблюдаться близкие
изменения скорости процесса. Это соответствует широко известному
правилу Вант-Гоффа, гласящему, что при нагревании системы на 10°С
скорость реакции возрастает в 2-4 раза.
При сопоставлении теоретических соотношений для скорости
реакции с экспериментальными константами оказалось, что расчет-
ные величины предэкспоненциальн.ого множителя часто превыша-
ют экспериментальные. Для приведения их в соответствие вводят
некоторый дополнительный множитель Р, называемый стерическим
фактором. Для различных реакций его величины колеблются of 1 до
1O’S. Физический смысл его сводится к тому, что для продуктивного
взаимодействия молекул необходимо соблюдение некоторых до-
полнительных условий. Так, из четырех приведенных на рисунке 8.9
схем столкновений молекул йодистого водорода очевидно, что первое
не приведет к образованию новых продуктов, для второй и третьей
схем реакция принципиально возможна, но в качестве промежуточ-
ных продуктов должны фигурировать атомы водорода или иода,
обладающие высокой потенциальной энергией, и, следовательно,
энергия активации будет существенно завышена. Поэтому вероят-
ность их осуществления крайне низка. И только для последнего случая
может произойти успешное перераспределение связей. Фактически
смысл стерического множителя сводится к учету доли столкновений,
благоприятных с точки зрения геометрии. При образовании про-
межуточного состояния (см. рис. 8.9.4) с энергией, достаточной для
Кинетика химических реакций
197
Рис.8.9. Различные варианты
столкновений молекул йодистого
водорода
преобразования, есть два пути для дальнейшего его распада: на
исходные вещества и продукты реакции. В скрытом виде фактор
различных путей распада активированного комплекса также входит
в стерический множитель.
Правомерным и весьма продуктивным зачастую представляется
следующий подход. Скорость реакции можно представить в виде
произведения числа столкновений на некоторую дополнительную
величину:
w = kQ- ехр(-ДЯ / кГ) ехр(Д8а/ к) И][Б], (8.47)
где энтропийный вклад и представляет стерический фактор реакции.
Другим важным выводом, следующим из теории активированного
комплекса, служит то, что для простых реакций активированное
состояние является общим как для прямого, так и для обратного
процесса. Действительно, приведенное на рисунке 8.9.4 состоя-
ние должно реализоваться как при столкновении двух молекул йоди-
стого водорода, так и при столкновении молекул иода и водорода.
Аналогично, распад его приведет к одной из этих двух пар частиц.
Следствием этого является то, что разностью энергий активации пря-
мого и обратного процессов будет энтальпия химического процесса:
= ДН. (8.48)
Аналогичное выражение можно получить и для соотношения
энтропий этих процессов. Поэтому константа равновесия химической
реакции равна отношению констант скоростей прямого и обратного
процессов:
K-kJk^. (8.49)
198
Глава 8
8.6. Некоторые типы реагирующих частиц и механизмов
химических реакций.
Многие химические процессы могут протекать по различным
направлениям. В качестве примера можно рассмотреть реакции
нуклеофильного замещения. Простейший процесс такого типа,
протекающий через взаимодействие двух частиц с образованием про-
межуточного активированного комплекса, быстро распадающегося
на продукты, представляет собой реакцию второго порядка:
АВ + С = (А-В-С) = А + ВС. (8.50)
В качестве примера таких реакций можно рассматривать процесс
превращения бромпроизводных органических соединений в спирты
под действием щелочи:
RCH,Br + ОН- = RCH,OH + Вг . (8.51)
Этот механизм принято называть SN2 (substitution nucleofilic).
Альтернативным путем протекания процесса является изначальный
медленный распад АВ на. А и В с последующим быстрым взаимо-
действием В с С..Здесь скорость процесса лимитируется реакцией
диссоциации первого порядка. Такой механизм обозначается соответ-
ственно <$^1
АВ + С = А + В + С = А + ВС. (8.52)
Выбор между ними легко сделать на основании зависимости или
независимости скорости процесса от концентрации реагента С или
стереохимии продуктов. Так, при гидролизе вторичного бутилбро-
мида в качестве промежуточного комплекса образуется анион с ко-
ординационным числом атома углерода, равным пяти (рис. 8.10).
Рис.8.10. Строение переходного комплекса,
образующегося при гидролизе вторичного
бутилбромида
Образующийся комплекс крайне неустойчив и мгновенно рас-
падается с выбросом одного из лигандов (фактически образование
одной связи сопровождается параллельным разрывом другой).
Кинетика химических реакций
199
Поэтому скорость реакции в целом прямо пропорциональна кон-
центрациям обоих исходных веществ, а продукт состоит лишь из
одного стерического изомера. Напротив, атака дополнительным нук-
леофильным лигандом третичного углеродного атома стерически
затруднена. Поэтому на первой стадии гидролиза в этом случае
медленно образуется плоский третбутильный катион, который
мгновенно взаимодействует с электрофильным реагентом. Поэтому
скорость процесса определяется лишь концентрацией органическо-
го реагента, а продукт, в случае оптически активных веществ пред-
ставляет собой смесь двух изомеров в соотношении 1:1.
Большинство химических процессов протекает по существенно
более сложному механизму, однако суть их остается той же и сводит-
ся к про геканию массы последовательных, а иногда и параллельных
реакций, приводящих к образованию конечного продукта (или ряда
таковых). Причем в качестве реагирующих частиц на каждом эта-
пе могут выступать отдельные молекулы, ионы или радикалы. Для
выбора оптимального пути синтеза химику важно иметь представ-
ление о реакционной способности этих частиц.
Многие химические реакции начинаются с только что рас-
смотренного процесса диссоциации молекулы. Необходимым усло-
вием для его протекания является разрыв химической связи. Энергия
этого процесса может меняться в весьма широких пределах - от
нескольких кДж/моль до близкой к 1000 кДж для молекул КГ2 и СО
с кратной связью. Следует заметить, что энергия одинарных связей
существенно меньше и достигает 560-580 кДж/моль лишь для фто-
ридов водорода и лития. Энергия активации гомолитического распада
на атомы, как правило, оказывается равной энергии химической свя-
зи (действительно, для осуществления такового мы должны развести
взаимодействующие атомы на бесконечное расстояние).
Процесс диссоциации, как известно, может протекать и гетеро-
литически - с образованием пары противоположно заряженных
ионов. Для газовой фазы разность между энергиями гомолитиче-
ского и гетеролитического распадов равна разности энергии иониза-
ции атома, образующего катион, и сродства к электрону-для аниона.
Нетрудно убедиться, что процессы эти сближаются по энергии лишь
для экстремальных случаев - минимальной разностью между этими
величинами (около 14 кДж/моль) характеризуется хлорид цезия. Для
всех прочих молекул она существенно больше. В кристаллах ионы
стабилизируются лишь за счет больших значений энергии кристал-
лической решетки. Поэтому во всех случаях без исключения гомо-
литический распад в газовой фазе намного выгоднее. В растворах
200
Глава 8
ситуация меняется кардинально ввиду значительной энергии соль-
ватации ионов. Поэтому для них экзотермические реакции гетеро-
литической диссоциации происходят безактивационно (как в случае
галогеноводородных кислот), а для эндотермических реакций энер-
гия диссоциации равна тепловому эффекту процесса (например
автопротолиз воды):
НС1(г) = Н+(р-р) + СГ(р-р) ДН = -75 кДж/моль, Е = 0
Н2О(ж) = Н+(р-р) + ОН (р-р) дН = +56 кДж/моль,
Eq = 56кДж/моль .
Для сопоставления заметим, что для первой из приведенных
реакций в газовой фазе энергии гомо- и гетеролитического распада
составляют соответственно 431 и 1367 кДж/моль.
Большинство химических процессов представляют собой реак-
ции между двумя или более молекулами. Они, как правило, со-
провождаются значительными энергиями активации, которые ни в
коей мере не согласуются с тепловым эффектом процесса. Взаимосвязь
между этими величинами можно выявить лишь для однотипных
реакций типа диссоциации галогеноводородов. Процесс диссоциа-
ции их происходит по наиболее слабой связи. Так, в молекуле этана
энергия связи С-С - 337, а С-Н - 406 кДж/моль. Соответственно
константа скорости для его гомолитического распада на два радика-
ла СН3 примерно в 107 раз выше, чем на Н • и С,Н5 •.
Образование радикалов может существенно облегчаться при
взаимодействии двух молекул, обладающих взаимным сродством. Так,
энергия активации взаимодействия углеводородов с кислородом
R-H + О, = R • + НО, . (8.53)
в среднем на 200 кДж/моль ниже энергии активации соответствующего
гомолитического распада.
Существенно большей реакционной способностью отличают-
ся образующиеся в результате упомянутых процессов свободные
радикалы. Реакции взаимодействия между двумя радикалами или
молекулами и радикалами, как правило, безактивационны или
характеризуются низкими энергиями активации, поскольку взаи-
модействие между неспаренными электронами вполне компенсирует
межэлектронное отталкивание в ходе всего процесса. Реакции, осу-
ществляющиеся посредством взаимодействия пары радикалов, от-
носительно редки ввиду малой их концентрации. Безактивационные
же процессы столкновения радикалов с молекулами реагентов про-
исходят существенно чаще. Кроме того, при этом не нужно участие в
Кинетика химических реакций
201
процессе третьей частицы, необходимой для “сброса” избытка
энергии, выделяющегося при взаимодействии пары радикалов.
Низкая концентрация радикалов обусловлена нулевой энергией
активации и значительным тепловым эффектом реакции их реком-
бинации. Исключений из этого правила крайне мало, но они все же
встречаются среди веществ, для которых энергия рекомбинации со-
поставима с положительным вкладом в величину свободной энергии
энтропийного фактора. Примерами таких частиц являются оксиды
азота, хлора и некоторые органические радикалы, для которых ре-
комбинация, кроме того, может осложняться и стерическими пре-
пятствиями, например (С6Н5)3С •.
Заметим, что безактивационными могут являться и некоторые
реакции между молекулами, приводящие к образованию донорно-
акцепторной связи. При этом взаимодействие, как известно,
осуществляется между неподеленной электронной парой одной и
вакантной орбиталью другой молекулы:
H3N: + nBF3 = H3N:BF3. (8.54)
Следует заметить, что эти реакции также происходят между
активными электронными состояниями и, по сути, близки к реакциям
рекомбинации двух радикалов.
Подобны реакциям радикалов и реакции между ионами в
растворе, не связанные с окислительно-восстановительными про-
цессами. Экзотермические процессы для них безактивационны, а
энергия активации для эндотермических приближается к энталь-
пии процесса. Окислительно-восстановительные процессы осуще-
ствляются сравнительно легко, если они связаны с взаимодействием
ионов противоположного заряда или иона и нейтральной молекулы.
При этом лимитирующей чаще всего является стадия вхождения
лиганда в координационную сферу катиона с замещением ранее
расположенного там лиганда или искажением координационной
сферы. Окислительно-восстановительные процессы между двумя
катионами осуществляются более сложно - путем передачи электрона
через лиганд. Процесс этот в общем случае имеет квантовую природу.
Примером таких реакций является
ТР+ + Sn2+ = Т1+ + Sn4+ . (8.55)
8.7. Параллельные (конкурирующие) реакции
Параллельными называются реакции превращения исходного
вещества, которые могут протекать по двум и более независимым
направлениям:
202
Глава 8
А-> В и А-> С (8.56)
или
A+B->CvA+D-»F. (8.57)
Весьма известным примером таковых является процесс разложения
бертолетовой соли, который может происходить по двум схемам:
2КС1О3 = 2КС1 + 30, или 4КС1О3 = КС1 + ЗКСЮ4, протекающим с
большей или меньшей скоростью в зависимости от присутствия
катализатора или температуры проведения эксперимента. Другим
характерным примером может являться растворение смеси нескольких
оксидов или металлов в кислоте. Схема изменения концентраций
исходного вещества и продуктов в ходе типичного процесса такого
рода приведена на рисунке 8.11.
Рис. 8.11. Изменение концентраций
при протекании параллельных
процессов по реакциям (8.56 и 8.57)
Несколько реже встречаются так называемые сопряженные про-
цессы, осуществление одного из которых невозможно без одновре-
менного протекания второго. Так, иодистоводородная кислота при
обычных условиях не окисляется пероксидом водорода. Введение же
в систему ионов двухвалентного железа приводит к интенсивному
протеканию процесса окисления иодид-ионов, инициируемого
окислением двухвалентного железа. Как правило, осуществление
сопряженных реакций обусловлено тем, что в одной из них в качестве
реагента используется промежуточный или конечный продукт другой.
Во избежание загрязнения продукта химикам приходится
выбирать условия эксперимента таким образом, чтобы свести к ми-
нимуму скорость протекания нежелательной реакции. Чаще всего
этого добиваются подбором катализаторов, среды проведения
процесса или температурных условий. Пусть, например, реакции,
Кинетика химических реакций
203
описываемые схемой (8,56), являются процессами первого порядка и
при 500 К характеризуются соотношением скоростей 1:1 и энергиями
активации 50 и 80 кДж/моль соответственно. Тогда, повысив тем-
пературу системы до 700 К, мы получим соотношение скоростей 1:4,
а понизив до 300 К, - 25:1! Тем не менее очевидно, что получить
вещество С практически в чистом виде можно лишь при подборе
селективного катализатора.
С другой стороны, даже этому негативному с точки зрения
возможности получения чистого продукта явлению удается найти
замечательное применение. Метод конкурирующих реакций может
оказаться крайне полезным для изучения кинетики быстрых про-
цессов, которые практически невозможно исследовать обычными ме-
тодами. Действительно, пусть, например, продукт В по схеме (8.56)
образуется практически мгновенно, а скорость реакции образова-
ния С можно определить в ходе независимых экспериментов. Тогда,
проводя оба процесса одновременно и проанализировав концентра-
цию продуктов по завершении эксперимента, можно получить:
[5] / [Q = / к,. (8.58)
Аналогично для схемы (8.57) имеем:
[а/[^=/с,[^/ад. (8.59)
Меняя концентрации веществ В и D при меньшей концентрации
А, можно добиться сопоставимых скоростей осуществления обоих
процессов в заданных условиях. Таким образом, например, иссле-
дуется большинство реакций, протекающих с участием радикалов.
8.8. Последовательные реакции
Последовательными называются реакции, протекающие через
одну или более промежуточных стадий, причем в каждой из по-
следующих используется один из продуктов предыдущей. Простей-
шую схему таковых можно представить в виде:
А -> В -+ С. (8.60)
Из ранее сказанного следует, что скорость процесса в целом будет
определяться скоростью наиболее медленной стадии. Общий вид гра-
фиков зависимости концентраций исходного вещества и продуктов
реакции от времени представлен на рисунке 8.12.
Нетрудно убедиться, что изменение концентрации исходно-
го вещества меняется, как и при обычных процессах. Концентрация
204
Глава 8
Рис. 8.12. Зависимость концентрации веществ для процесса (8.60)
от времени протекания
промежуточного продукта, резко возрастая в начале реакции, до-
стигает максимального значения и затем медленно уменьшается. Со-
ответственно, скорость накопления конечного продукта С вначале
близка к нулю ввиду низкой концентрации В - это так называемый
индукционный период. По мере накопления В скорость образова-
ния продукта возрастает, достигая максимального значения при
максимальной концентрации интермедиата. При этом на интеграль-
ной кривой (рис. 8.12) наблюдается перегиб. Таким образом, на-
пример, протекает взаимодействие хлорида двухвалентной ртути с
парами натрия. В соответствии с логикой окислительно-восстанови-
тельных процессов на первом этапе происходит практически полное
восстановление ртути до одновалентного состояния, после чего она
восстанавливается до ртути металлической:
2HgCl, + 2Na = Hg,Cl2 + 2NaCl,
Hg2Cl2 + 2Na = 2Hg + 2NaCl. (8.61)
В случае галогенидов циркония, напротив, образующиеся на
промежуточном этапе низшие галогениды оказываются существен-
но менее стабильными и интенсивно диспропорционируют или вос-
станавливаются до металлического циркония, успешно конкурируя с
процессом восстановления его тетрахлорида:
ZrCl4 + Na -» ZrCI3 -* ZrCl, -> (ZrCl) -> Zr . (8.62)
Кинетика химических реакций
205
Исследование кинетики последовательных реакций в общем
виде достаточно сложно, поскольку получающаяся система диф-
ференциальных и балансовых уравнений не имеет строгого матема-
тического решения. В большинстве случаев достаточным оказывает-
ся использование некоторых приближений, позволяющих свести
систему к более простой. Нетрудно убедиться, что в случае, если
константа первой стадии процесса (8.60) много меньше второй, ве-
щество В расходуется практически сразу же в момент образования.
При этом скорость реакции в целом определяется константой скорости
первого процесса и график зависимости концентрации продукта от
времени имитирует аналогичный график для простого химического
процесса А -> С. При этом обнаружение даже следов промежуточно-
го продукта зачастую является достаточно сложной задачей. Решить
ее удается, лишь проводя процесс при пониженных температурах и
с помощью весьма чувствительных методов, например, с помощью
электронного парамагнитного резонанса в случае образования на
промежуточной ступени радикалов.
В зависимости от условий протекания такого процесса скорость
его в определенном временном интервале оказывается возможным
описать с помощью методов квазистационарных или квазиравно-
весных концентраций. Рассмотрим условия их применения на примере
ранее упомянутой реакции окисления монооксида азота кислоро-
дом. Собственно, отнесение ее к сложным процессам было сделано на
основании того, что формально она имеет отрицательную энергию
активации, что лишено физического смысла. Присутствие в реак-
ционной системе промежуточного вещества - неустойчивого димера
оксида азота (NO), - было подтверждено экспериментально. Итак,
на первом этапе осуществляется обратимая реакция образования
димера, сопровождающаяся незначительным тепловым эффектом
(Д/7 = -16 кДж/моль).
2NO-> (NO), , = k,[NO]2. (8.63)
Значительное понижение энтропии в ходе этого процесса
(AS = -100 кДж/моль) приводит к тому, что равновесие для него су-
щественно смещается влево (Д(9 = 20 кДж/моль) и обратный процесс:
(NO)2-^2NO, Г, = k,[(NO)2] (8.64)
характеризуется существенно большей константой скорости:
к} « к . На втором этапе протекает процесс окисления димера
кислородом:
(NO), + О, -> 2NO2, И2 = k2[(NO)J[02]. (8.65)
206
Глава 8
Однако скорость его сравнительно низка и для сопоставимых кон-
центраций исходных реагентов всегда соблюдается условие:
7, =M(NO)2] » К, = £,[(NO)2][O,]
или
к} » M°J • (8.66)
Это означает, что окисление димера весьма слабо влияет на
равновесие реакции (8.63). Собственно это и является основным
условием применимости метода квазиравновесных концентраций.
Приняв указанное допущение, мы получаем возможность прирав-
нять скорости процессов образования и разложения димера:
УЛ = У{> /c_,[(NO)J=/:1|NOF. (8.67)
Откуда [(NO),] = (£,/£,)[NO]2 . (8.68)
Подставляя полученное соотношение в уравнение для скорости
процесса (8.65), получим:
У, = U(NO)J[OJ = (кг кх / к ,)[NO]2[O,]. (8.69)
Полученное уравнение ничем не отличается от обычного кине-
тического уравнения для реакции третьего порядка, почему ее дли-
тельное время и принимали за тримолекулярную. Однако сложная
функция для константы скорости проявляется в замедлении скоро-
сти суммарного процесса с повышением температуры с формальной
энергией активации £ = -4.6 кДж/моль. Действительно, из соот-
ношения (8.69) легко получить:
£=£,+£,- Е_, = ДЯ + £,. (8.70)
Откуда, поскольку ДЯ = £, - Е, = -16 кДж/моль, легко найти
£,= 11 кДж/моль.
Для метода квазистационарных концентраций необходимым
условием является лишь низкая концентрация промежуточного про-
дукта, независимо от того, какая из реакций его превращения яв-
ляется превалирующей. В этом случае имеем:
d [NO,] I dt ~ -d [NO] / dt. (8.71)
Кроме того полагают, что поскольку концентрация димера крайне
мала, то скорость его образования должна с высокой точностью
равняться сумме скоростей реакций диссоциации и окисления:
£,[(NO),][O,J + к ,(NO)J = fc,[NO]2. (8.72)
Откуда [(NO),] = кх [NO]2/ (/с, [О,] + к (8.73)
d \Wydt = к,кх[NO]2[O,]/ (к, [О,] + к,). (8.74)
Кинетика химических реакций
207
Таким образом, при соблюдении условия кл >>£,[0,], результаты,
полученные с помощью обоих методов, будут эквивалентны. Однако,
в случае очень высоких парциальных давлений кислорода, порядок
реакции по этому веществу оказывается непостоянным и может стать
значительно меньше единицы. Такое явление, действительно, иногда
наблюдается при проведении процесса со значительным избытком
кислорода.
В общем, сравнительно небольшое допущение о пренебрежимо
малой концентрации промежуточного продукта позволяет перейти
от весьма сложного численного интегрирования, неизбежного при
строгом решении полученной системы дифференциальных и ба-
лансовых уравнений, к весьма простым соотношениям типа (8.69)
или (8.74). Следует отметить также, что метод квазиравновесных
концентраций использует несколько более грубое приближение и
применимость его ограничена.
Следует отметить, что с помощью подобного рассмотрения можно
объяснить и существование многочисленных реакций первого по-
рядка, представить существование которых, как отмечалось в разде-
ле 8.2, чисто теоретически весьма затруднительно. Весьма удачную
трактовку этого явления предложил Линдеман. В рамках его модели
на первой стадии происходит столкновение двух молекул исходного
вещества, в результате чего одна из них переходит в возбужденное
состояние:
Л + Л-> Л * + Л (/с2). (8.75)
После этого возбужденная молекула может перейти либо в про-
дукт по мономолекулярной реакции, либо в дезактивированное
состояние при столкновении с другой молекулой А\
А* + А-+А+А (/с 2), (8.76)
А*—> В (А;). (8.77)
Из условия квазистационарности процесса можно заключить:
*2[Л]- =/с,[Л*] . (8.78)
Откуда следует:
[Л*]=£2[Лр/(/с, + МЛ]). (8.79)
Суммарное кинетическое уравнение в таком случае может быть
выражено в виде:
d [Л] / dt = ЦА *] = кл кг [А]2 / (/с, + к, [Л]). (8.80)
Теперь, если предположить, что к{ « к [Л], то последнее
уравнение можно переписать в виде
208
Глава 8
d[B\l dt = kJ £,)И]. (8.81)
При низкой концентрации исходного вещества соотношение ki и
к2 [А] может измениться на обратное и реакция будет иметь второй
порядок, что в ряде случаев и было подтверждено экспериментально.
8.9. Цепные реакции
Перейдем теперь к рассмотрению некоторых весьма распростра-
ненных классов последовательных реакций. Цепными называются
процессы, осуществляющиеся путем последовательного чередования
реакций исходных веществ с образующимися в предыдущем процессе
радикалами. Наиболее часто скорость их определяется скоростью
инициирования свободных радикалов, поскольку превращение
последних происходит с низкой энергией активации. Очевидно, что
при этом происходит разрыв наименее прочной молекулы. Напри-
мер, для синтеза бромистого водорода из элементов реакция иници-
ирования следующая:
Вг, + X-» X + 2Вг (/<,). (8.82)
Необходимость участия в реакции второй частицы, в качестве
которой может выступать вторая молекула брома, стенка реакцион-
ного сосуда или квант какого-либо излучения определяется необ-
ходимостью привлечения дополнительного источника энергии для
образования из молекулы, пары радикалов.
Далее протекают процессы продолжения цепи:
Вг + Н, = HBr + Н • (кА, (8.83)
Н+Вг, = НВг + Вг(/<3). (8.84)
Рекомбинация радикалов происходит по одной из реакций:
Н + Вг + X = HBr + X (кА , (8.85)
H+H' + y = H, + Z (к5), (8.86)
Вг + Вг • + X = Br2 + X (кв). (8.87)
Поскольку атомы водорода обладают заметно большим избытком
энергии, полагают, что их время жизни и концентрация существенно
ниже, чем для радикалов брома. Поэтому преобладает процесс (8.86).
Кроме того, по мере накопления продукта возможно взаимодей-
ствие его с водородными радикалами с образованием молекулы
водорода и менее активного радикала - брома:
Кинетика химических реакций
209
Н + НВг = Н2 + Вг (£7). (8.88)
Используя принцип стационарных концентраций, можно сделать
заключение, что концентрации атомов водорода и брома в ходе
процесса близки к нулю. Поэтому скорости реакций их образования
и расходования будут равны. Объединяя все эти уравнения с учетом
сделанных допущений, можно получить ранее приведенное нами
кинетическое уравнение суммарного процесса.
Низкая концентрация радикалов приводит к тому, что их стол-
кновения происходят достаточно редко, хотя бы с вероятностной
точки зрения. Поэтому каждый образующийся в ходе таких реакций
радикал успевает поучаствовать в большом количестве процессов
продолжения цепи, прежде чем регенерирует. Число актов продол-
жения цепи, приходящееся на один радикал и усредненное по числу
генерированных радикалов, называется длиной цепи. Для ряда ре-
акций она может достигать 106!
Протекание некоторых процессов сопровождается разветвлением
цепи. Так, помимо достаточно обычных реакций продолжения цепи
для взаимодействия водорода с кислородом, встречаются следующие:
Н + О, = НО + О:, (8.89)
О:+Н2=Н +ОН . (8.90)
В обоих случаях из одной реакционноспособной частицы
образуются две, что приводит к разветвлению процесса, т. е. к
самопроизвольному размножению радикалов. Если в силу энергети-
ческих причин и достаточной концентрации реагентов все стадии
протекают со значительной скоростью, происходит взрыв.
Характерной особенностью цепных процессов является ярко
выраженная зависимость скорости от объема и даже формы реак-
ционного сосуда, стенки которого способствуют рекомбинации ра-
дикалов. Кроме того, скорость таких реакций может существенно
увеличиваться или уменьшается в присутствии примесей, способных
генерировать или нейтрализовать радикалы. Так, вместо воздействия
освещения реакция хлора с водородом может инициироваться пара-
ми натрия. Последние мгновенно реагируют с хлором с образованием
радикалов:
Na+Cl2 = NaCl + Cl- (8.91)
Напротив, оксиды азота ввиду наличия неспаренного электрона
в ряде случаев способны присоединять свободные радикалы, вызывая
их рекомбинацию:
Я +NO2=7?NO2. (8.92)
210
Глава 8
Аналогичную функцию могут выполнять и некоторые насыщен-
ные молекулы, которые, взаимодействуя с радикалами, образуют
новые - менее активные, не способные к продолжению цепи частицы.
Фактически роль их сводится к существенному понижению длины
цепи.
8.10. Катализ
В ходе рассмотрения кинетических закономерностей ряда про-
цессов мы уже обращались к понятию катализа. Под катализом под-
разумевается ускорение реакции под действием веществ, которые сами
по себе не претерпевают превращений после завершения процесса.
Для понимания сути этого явления вновь обратимся к диаграмме
изменения энергии в ходе химической реакции. Скорость таковой
определяется энергией образования промежуточного активирован-
ного комплекса:
АВ + С -> (ABC) ->А+ВС. (8.93;
Роль катализатора сводится к образованию промежуточного,
более реакционноспособного соединения катализатора с одним из
реагентов, который затем взаимодействует со вторым реагентом:
(КАВ)+С-+ А+КВ+С-» К+А+ВС. (8.94)
Из приведенных данных видно (рис. 8.13), что один процесс
сводится к альтернативному, представленному двумя процессами,
характеризующимися меньшими энергиями активации, а следо-
вательно, протекающими с большей скоростью. При этом реакция,
естественно, протекает по пути наименьшего сопротивления. В ре-
зультате этого ускоряется процесс в целом, а катализатор остается в
неизменном виде.
Весьма важно, что катализатор, как видно из приведенного
рисунка, понижает энергию активации и повышает скорость прямого
и обратного процессов. При этом изменение свободной энергии
реакции в целом сохраняет свое значение, а следовательно, неиз-
менным остается и положение равновесия. С другой стороны, для
конкурирующих необратимых процессов введение катализатора
может практически полностью сместить равновесие в сторону
образования одной группы продуктов, как это происходит при
добавлении следовых количеств оксида марганца к бертолетовой соли:
2КС1О3 Mn0;-> 2КС1 + ЗО2. (8.95)
Кинетика химических реакций
211
Рис.8.13. Сопоставление энергетических диаграмм для прямого и каталитического
процессов взаимодействия АВ с С
Следует различать гомогенный и гетерогенный катализ. Для
первого из них катализатор находится в том же агрегатном состоянии,
что и реагенты, как в случае катализа процесса окисления диокси-
да серы окислами азота, протекающего через образование нитро-
зилсерной кислоты:
2NO + O, = 2NO,( (8.96)
2SO, + 3NO2 + Н2О = 2NOHSO4 + NO , (8.97)
2NOHSO„ + H2O = 2H2SO4 + NO + NO, . (8.98)
Тот же процесс позже стали осуществлять на твердом ванадиевом
катализаторе (гетерогенный катализ). Здесь взаимодействие реагентов
протекает на границе раздела двух фаз. Оксиды ванадия, весьма легко
взаимодействуя с кислородом воздуха, окисляются, отдавая затем
присоединенный кислород сере;
VOv+^/20, = VOv+),, (8.99)
VOv+y + уН2О + _ySO, = VOv + yH,SO4. (8.100)
Доля активных центров на поверхности сравнительно невелика,
поэтому гетерогенные катализаторы склонны к отравлению, когда
они именно в силу своей повышенной реакционной способности
образуют устойчивые соединения с какой-либо примесью.
212
Глава 8
Весьма важную роль в органической химии играет кислотный и
основной катализ. В ходе первого из них протон переходит от кат-
ализатора, в качестве которого выступает некоторая кислота НХ, к
субстрату, переводя его в активированное состояние:
НХ + В = НВ+ + Х'. . (8.101)
Таким образом, например, осуществляются процессы гидролиза
сахарозы и сложных эфиров1
RCOOR* + Н+ = (RCO-HO+-R*) = RCO+-R<OH . (8.102)
Образующийся катион является крайне реакционноспособным и
мгновенно реагирует с молекулой воды, образуя кислоту и регенери-
руя протон:
RCO+-R*OH + Н,0 = RCOOH + Н++ R-OH. (8.103)
При основном катализе происходит обратный процесс - отщеп-
ление протона с образованием карбоаниона, подверженного элек-
трофильной атаке.
Весьма важную роль в ряде процессов играет так называемый
отрицательный катализ, или ингибирование, которое сводится к
замедлению процесса. В этом случае ингибитор как бы заманивает
реакцию в энергетическую западню, вынуждая активные частицы
переходить в некоторый термодинамически нестабильный продукт
по реакции с меньшей энергией активации (рис. 8.14).
Рис.8.14. Сопоставление энергетических диаграмм процесса взаимодействия некоторых
веществ в отсутствие и в присутствии ингибитора
Энергия активации перехода из этого состояния в конечный
продукт оказывается очень высокой и такой процесс оказывается
маловероятным. Поэтому промежуточному соединению не остается
ничего лучшего, как вновь перейти в исходное состояние.
Кинетика химических реакций
213
8.11. Особенности реакций гетерогенного катализа
Осуществление химических процессов с помощью реакций
гетерогенного катализа является одним из наиболее бурно разви-
вающихся направлений химии и химической промышленности.
Сущность происходящих при этом явлений сводится к значительному
ослаблению химической связи в молекулах, сорбированных на
поверхности твердых тел, и, соответственно, к повышению их ре-
акционной способности. Рассмотрим, например, процесс сорбции
молекул водорода на поверхности платины. При их сближении
начинает осуществляться взаимодействие разрыхляющих орбиталей
водорода с d-орбнталями металла. Следствием этого является
уширение зоны, соответствующей разрыхляющим орбиталям молекул
водорода, нижний край которой опускается ниже уровня Ферми
платины. Перенос части электронов металла на эти орбитали
приводит к образованию типичной донорно-акцепторной связи
платина-водород н ослаблению взаимодействия между атомами
последнего. При этом молекула водорода продолжает сближаться с
поверхностью платины, зона, соответствующая разрыхляющей
орбитали, уширяется, и энергия ее нижней границы понижается. В
завершении процесса происходит разрыв связи Н-Н и формирование
на поверхности типичных гидридных центров, соответствующих
энергии хемосорбции;
Н,(газ) = 2Н(адс). (8.104)
С точки зрения химии, основным положительным результатом
является то, что мы получили относительно слабосвязанные гид-
ридные атомы водорода, способные активно вступать в химические
реакции, минуя энергоемкую стадию разрыва молекул водорода на
атомы (радикалы) в газовой фазе. Напомним, что последняя и оп-
ределяет энергию активации многих химических процессов, проте-
кающих с участием молекул Н,.
Аналогичные явления могут протекать и при сорбции на
поверхности переходных металлов молекул с существенно более
прочной химической связью (О,, СО, N,). В литературе существуют
многочисленные экспериментальные данные, подтверждающие
возможность их диссоциации или ослабления связи на поверхности
некоторых переходных металлов. Процесс также начинается с пе-
реноса ^-электронов в зону, образуемую разрыхляющими р.орби-
талямн этих молекул. При этом кратная связь в них существенно
ослабляется и приближается по энергии к одинарной. Поэтому столь
214
Глава 8
активно используется гетерогенный катализ в ходе многих процессов,
протекающих с участием этих веществ (например каталитическое
дожигание выхлопных газов и промышленный синтез аммиака).
Эти исключительные свойства поверхности в течение длительного
времени используются в процессах гетерогенного катализа. Рассмот-
рим вначале основные особенности реакций такого типа в общем виде
на примере процесса
Л (г) +Б(г) = ЛЛ(г). (8.105)
Первым этапом является сорбция вещества А на активных центрах
поверхности сорбента, протекающая с ослаблением химической связи
внутри них
/4(г) = Л(адс). (8.106)
Далее возможны два пути продолжения процесса. В первом случае
осуществляется также и сорбция атомов В, которые, мигрируя по
поверхности катализатора, сближаются с атомами или молекулами
А, взаимодействуют с ними с образованием сорбированного продукта,
а последний десорбируется с поверхности вещества. Таким образом,
продолжение процесса можно представить в виде:
5 (г) = 5 (аде), (8.107)
А (аде) + 5(адс) = А5(адс), (8.108)
ЛВ(адс) =Л5(г). (8.109)
Такой механизм получил название реакции Лэнгмюра-Хиншел-
вуда. В зависимости от природы участников процесса атомы веществ
А и В могут сорбироваться совместно, занимая соседние центры. Этот
случай так называемой кооперативной сорбции является оптималь-
ным для осуществления процесса. При этом кинетическое уравнение
не имеет принципиальных отличий от полученных ранее.
В другом случае адсорбция А и В протекает раздельно с образо-
ванием “островков” с сорбированными атрмами А или В. При этом
частицы обоих веществ конкурируют за право занятия сорбционных
центров, вытесняя друг друга с поверхности сорбента. В последнем
случае степень занятости активных центров поверхности молекулами
каждого вещества можно выразить как
2, =-W(l+*Z, + W (8.110)
QB = KBPt /(1 JrKAPA + KBPB), (8.111)
где КА и Кв- некоторые константы, Поскольку десорбция протекает,
как правило, практически мгновенно, скорость суммарного процесса
Кинетика химических реакций 215
определяется взаимодействием сорбированных А и В. Выражение для
скорости суммарного процесса при этом может быть представлено в
виде:
d[AB]/dt = k QAQB (8.112)
или
d [AB]/dt = k KAPA KBPB /(1 + KAPA + KBPBy. (8.113)
Отличительной особенностью такого процесса является про-
хождение его скорости через максимум при фиксации концентрации
одного и увеличении парциального давления другого вещества, как
в случае окисления СО на платиновом или палладиевом катализаторе
(рис, 8.15).
Рис.8.15. Зависимость степени заселенности поверхности платинового катализатора
молекулами СО и О 2 и скорости образовании продукта от парциального давления СО
(рог=1.7-10‘< мбар)
Во втором случае сорбированные атомы А вступают во взаимо-
действие с В непосредственно из газовой фазы:
Л (аде) + 5(г) = Л.В(адс), (8.114)
Л5(адс) = А В(г). (8.115)
Этот тип получил название реакции Элея-Ридела. При этом, даже
если осуществляется конкуренция за сорбционные центры, скорость
216
Глава 8
суммарного процесса выражается в виде:
d[AB]ldi=k Qa Ps. (8.116)
или
d[AB]/dt=k КАРАРв/(\ +К4РА+КВРВ). (8,117)
Аналогичная зависимость скорости реакции от парциального
давления одного из веществ при фиксированном давлении второго,
увеличиваясь в области низких давлений, выходит на насыщение при
высоких.
Типичным примером реакции Лэнгмюра-Хиншелвуда является
процесс окисления угарного газа кислородом. Исследование его стало
особо актуальным в связи с обострением проблемы дожигаиия вых-
лопных газов автомобильных двигателей внутреннего сгорания. В
настоящее время удалось достигнуть высокой эффективности этого
процесса лишь при использовании катализаторов на основе пла-
тиновых металлов. Лимитирующей стадией является непосред-
ственное химическое взаимодействие СО и О, протекающее с энергией
активации 100-110 кДж/моль (рис. 8.16), что существенно меньше
энергии активации прямого взаимодействия в газовой фазе. Десорб-
ция двуокиси углерода с поверхности осуществляется сравнительно
легко с энергией активации около 20 кДж/моль. Суммарное тепло-
выделение в ходе процесса составляет 283 кДж/моль.
Рис.8.16. Изменение энергии системы в ходе каталитического дожигания СО
Еще одним примером подобного превращения является синтез
аммиака на катализаторе из магнетита (Fe3O4), допированного
оксидами алюминия. Скорость процесса определяется стадией
сорбции азота. Далее протекает поэтапное присоединение атомов
водорода.
Кинетика химических реакций
217
8.12. Общие сведения о кинетике твердофазных
процессов
Принципиальных отличий между кинетическими закономер-
ностями реакций в газовой фазе и в растворах немного. Основное из
них заключается в существенно более медленном перемещении частиц
в жидкости. Следовательно и сами столкновения становятся- реже.
Поэтому весьма важную роль для реакций в растворах могут играть
диффузионные процессы. С другой стороны, если молекулы уже
сблизились, то вероятность взаимодействия между ними существенно
выше, поскольку и время непосредственного контакта между
частицами оказывается значительно больше. Еще более глубоко
различие между реакциями, протекающими в растворе и в твердой
фазе.
Среди многообразия химических веществ, с которыми нам
приходится иметь дело в повседневной жизни, безусловно преобла-
дают твердые. С другой стороны, нетрудно убедиться, что боль-
шинству химиков свойственно проводить все интересующие их
процессы и синтезы в растворах или газовой фазе, к которым и от-
носятся все перечисленные ранее закономерности. Причина этого
парадокса заключается в глубоком различии кинетических зако-
номерностей протекания химических процессов в газовой фазе и
растворах, с одной стороны, и в твердом теле - с другой. Действи-
тельно, как было изложено ранее, скорость первых двух типов
процессов лимитируется скоростью образования активированного
комплекса или возбужденного состояния. В твердом же теле прин-
ципиально существуют две возможности: либо атомы реагентов уже
сблизились и тогда их взаимодействие практически неизбежно, либо
атомы реагентов разделены слоем других атомов или молекул
(например продукта реакции) и тогда, естественно, взаимодействие
невозможно. Следовательно, скорость твердофазной реакции опре-
деляться скоростью поставки реагентов к месту протекания реакции,
т.е. скоростью диффузии реагентов через слой продукта.
Действительно, при попытке осуществить твердофазное взаимо-
действие двух твердых веществ А и В с образованием твердого же
продукта АВ последний, очевидно, будет образовываться на границе
раздела фаз А и В (рис. 8.17). При этом скорость диффузии, соглас-
но закону Фика, будет пропорциональна площади поверхности раз-
дела и градиенту концентраций диффундирующего вещества в слое
продукта:
218
Глава 8
dnldt = -DS(dddX}, (8.118)
где X - толщина слоя продукта. Коэффициентом пропорциональности
в этом уравнении и будет коэффициент диффузии. Движущей же си-
лой процесса диффузии, как можно видеть из этого уравнения, являет-
ся градиент концентраций. Решение полученного соотношения выра-
жается в виде
Г=ЛГ (8.119)
или
X=(Dt)">. (8.120)
Рис.8.17. Схема распределении частиц исходных веществ и продуктов в реакционной
массе при использовании реагентов различной степени дисперсности (а) и (б)
Скорость диффузии в твердом теле весьма мала. Действительно,
давайте мысленно положим золотое кольцо Вашей бабушки в ла-
тунную сковородку. Налицо контакт двух фаз, поверхность раздела
и растворимость золота в меди и наоборот. Однако, сколь длитель-
ное время мы не проведем на кухне за этим занимательным экспе-
риментом, отгоняя назойливых домочадцев, кольцо не растворится
и не станет медным, а в сковородку вряд ли перейдет много атомов
золота. Лица, особенно любящие своих бабушек, могут усовершен-
ствовать эксперимент, учитывая, что коэффициент диффузии, соглас-
но уравнению Аррениуса
Z)=D0 ехр(-£/Л7), (8.121)
экспоненциально возрастает при увеличении температуры. Однако
и в этом случае эксперимент завершится исходом, благоприятным для
бабушки. Отсюда легко понять, что между скоростью большинства
твердофазных процессов и реакций, протекающих в жидкой и газовой
фазе, лежит бездонная пропасть.
Кинетика химических реакций
219
Тем не менее известен ряд твердофазных процессов, протекающих
с вполне удовлетворительной скоростью. Чаще всего они происходят
при высоких температурах, когда величина коэффициента диффузии
существенно повышается. Кроме того, не останавливаясь на сути
этого явления, отметим, что в природе существует конечное число
так называемых быстрых ионных проводников, отличающихся
аномально высокими коэффициентами диффузии по одному из со-
ставляющих их сортов ионов, иногда сопоставимыми с коэффициен-
тами диффузии их в растворах. Примерами таковых являются высо-
котемпературная модификация иодида серебра, £- алюминат натрия
и некоторые другие соединения.
Допустим, что нам необходимо все же осуществить некий твердо-
фазный синтез, например реакцию
ВаО + Fe2O3 = BaFe,O4. (8.122)
Для максимально быстрого осуществления процесса естественным
представляется нагревание смеси. Однако возможности этого мето-
да ограничены хотя бы максимальной температурой разогрева печи,
Куда более эффективным зачастую оказывается тривиальное меха-
ническое измельчение частиц. Соотношение скоростей протекающих
при этом процессов легко оценить с использованием уравнения (8.115).
Действительно, для завершения реакции между частицами, разли-
чающимися по размеру в 100 раз, время завершения процесса будет
различаться на 4 порядка. Понять закономерность этого в общем не
очевидного на первый взгляд явления можно из рисунка 8.17. Отсюда
следует вывод о том, что если Вам все же необходимо провести твер-
дофазный процесс, перед этим придется потрудиться физически, из-
мельчая реагенты. Весьма эффективным может оказаться соосаждение
мелкодисперсных или аморфных осадков исходных веществ (напри-
мер, гидроксидов двух- и трехвалентного металла). В этом случае
перемешивание реагентов обеспечивается уже на молекулярном
уровне, что существенно ускоряет процесс последующего взаимо-
действия.
Отметим еще одну закономерность твердофазных процессов.
Подавляющее большинство их происходит гетерогенно, т. е. форми-
рование продукта происходит с образованием новой фазы. Так, при
создании Л' молей продукта высвобождается энергия -N&H. Вместе
с тем при этом затрачивается энергия на образование новой повер-
хности раздела - ст5, где ст - коэффициент поверхностного натяже-
ния. Поскольку объем новой фазы пропорционален кубу, а поверх-
ность - квадрату линейного размера частиц, то для крупных частиц
220
Глава 8
будет превалировать первая, а для малых - вторая из перечисленных
величин. Итоговая зависимость энергии системы от линейного раз-
мера заданного числа частиц продукта приведена на рисунке 8.18.
Рис.8.18. Зависимость энергии твердофазной системы от размера фиксированного числа
зародышей продукта (а - линейный размер частиц)
Следовательно, можно выделить критический размер зародыша
новой фазы, соответствующий максимуму на приведенной кривой
зависимости А (7 от а. После достижения этого размера реакция будет
протекать с понижением энергии системы, т.е. самопроизвольно.
Однако для этого система также должна преодолеть некоторый энер-
гетический барьер. Причем в состав такого зародыша критического
размера входит порядка 104 частиц! Можно представить, как низка
вероятность концентрации значительной избыточной энергии на
стольких частицах! Именно поэтому подавляющему большинству
твердофазных процессов предшествует некоторый индукционный
период, в ходе которого скорость реакции крайне мала (стадия II
иа рис.8.19). Затем рост зародышей происходит одновременно с
увеличением нх числа и процесс вступает в так называемую стадию
разгона (III). В сумме на этих этапах происходит до 10% превраще-
ния исходных веществ в продукты. Затем скорость достигает мак-
симума и определяется лишь скоростью роста зародышей (IV), т.е.
Кинетика химических реакций
221
диффузионными процессами, и, наконец, падает по мере расходования
реагентов (V). Такой S-образньщ ход кинетической кривой является
визитной карточкой твердофазных процессов.
Хорошо известно, например, что диффузионными процессами
лимитируется скорость окисления многих металлов на воздухе. Так,
окисление алюминия заканчивается, едва начавшись, ввиду обра-
зования тонкой, но плотной оксидной пленки, характеризующейся
крайне низкими коэффициентами диффузии ионов алюминия и
кислорода. В пленке оксида никеля при высоких температурах ка-
тионы диффундируют с достаточной скоростью. Поэтому никель,
сравнительно хорошо окисляясь при нагревании, инертен при
обычных температурах. Оксидная же пленка на железе не отличается
сплошностью, поэтому диффузионные процессы весьма слабо тор-
мозят скорость коррозии. В результате ежегодные потери изделий из
этого металла достигают 10% от уровня производства.
Задачи и вопросы
1. Во сколько раз следует изменить общее давление в системе,
чтобы скорость окисления оксида азота до диоксида кислородом
воздуха возросла в 1 000 раз?
222
Глава 8
2. Некоторая реакция взаимодействия А с В проводилась при трех
значениях исходных концентраций, при которых были получены
следующие значения начальных скоростей превращения:
[А].М [В1.М >v, М/с
0.001 0.1 0.028
0.001 0.05 0.01
0,1 0.005 0 0032
Определите порядки реакции по реагентам и константу скорости
в данных условиях.
3. Период полураспада 235 U составляет 7-108 лет. За какое время
содержание его в образце понизится на 0.02%?
4. Определите константу скорости превращения некоторого веще-
ства, если в начальный момент времени скорость его составляла
1.18• 10 s М/с, а через 700 и 1690 с она уменьшилась соответственно в 2
и 4 раза.
6. Энергии активации реакций 1 и 2 равны 50 и 100 кДж/моль.
При комнатной температуре первая из них протекает в 100 раз бы-
стрее. Могут ли их скорости сравняться, и если да, то при какой темпе-
ратуре?
7. Вычислите Еа реакции, скорость которой при переходе от 25 к
35°С возросла в 3 раза.
8. Температурный коэффициент реакции при 300К равен 2.5.
Вычислите энергию ее активации.
9. В растворе, содержащем вещество А происходит некоторая
реакция. Определите порядок реакции по веществу А и константу
скорости, если содержание этого соединения следующим образом
убывает со временем: .
1 tc 1 ° 1 5 1 | 20 30 1
1 с, М 1 5 | 4,30 1 3.70 1 2.74 2 03 1 1.30 1
10. В растворе, содержащем вещества А и Б происходит некоторая
реакция. Определите порядок реакции по веществу А и константу
скорости, если содержание этого соединения следующим образом
убывает со временем:
Кинетика химических реакций
223
с, М
I О I 5 | 10 | 20 | 30 | 45 ~~I
| 2.5 | 2.41 I 2,33 | 2.16 I 2.00 I L74~|
11. В растворе, содержащем вещество А происходит некоторая
реакция. Определите порядок реакции по веществу А и константу
скорости, если содержание продукта взаимодействия следующим
образом изменяется со временем:
I t, с I 0 I 5 I 10 I 20 I 30 I 45 I
| с.М | 0 I 0.36 I 0.61 | 0.94 | 1,14 I 133 |
12. В растворе, содержащем вещества А и В в стехиометрических
количествах происходит некоторая реакция. Определите порядок
реакции по веществу А и константу скорости методом Вант-Гоффа,
если содержание этого соединения следующим образом убывает со
временем:
1 t, с | 0 | 5 1 1° 1 20 1 35 1 « 1
1 сТм Г 1 .5 | 1 08 I 0 89 I 0,69 | 0 59 I 0.49 |
13. Распад этана протекает с образованием метильных радикалов,
в то же время радикал С,Н5 распадается не на СН, и СН3. а на С,Н4
и Н. Какова причина этого явления?
14. Почему в растворах реакции между полизарядными ионами
часты, а в газах они отсутствуют?
15. Процесс образования АХ, протекает в две стадии:
А + Х«->АХ kj =105М'1с'1, к. = 1О5с-’
АХ + Х->АХ, к2 = 10гМ-’с-' [А]о = [Х]о = 10-'М.
Предложите методы корректного описания кинетики процесса,
сопоставьте полученные результаты и оцените скорость процесса в
момент уменьшения концентрации X вдвое.
16. Для процесса
А+В<->С к, =10-2М-1с-1, к =10* с1
А + С—>D + E к2=10*с-’ [А]о = [В]й = 2М.
Предложите схему, описывающую кинетику реакции.
Возможно ли ее упрощение?
224
Глава 8
17. Вещество А претерпевает превращение по реакции 3-го
порядка, а В - по реакции второго порядка. Начальная смесь содер-
жала компоненты в соотношении 2:1, а скорости этих процессов были
равны. Определите соотношение скоростей обратных реакций к
моменту остановки процесса, если концентрации А и В при этом
понизились соответственно в 2 и 4 раза.
18, При исследовании кинетики некоторого обратимого процесса
XY + Z = X + YZ с исходными концентрациями [ХУ] = [Z] = 2М;
[Л] = [YZ] ~ ОМ; при 400 и 450 К были зафиксированы начальные
скорости превращения 0.2 и 0.5 М/с соответственно. При достижении
равновесия концентрация вещества X достигла 1 и 1.2 М, соответст-
венно. Полагая, что прямой и обратный процессы являются односта-
дийными. определите энергию активации обратного процесса.
19. В системе с начальной концентрацией веществ А, В, С, D, F
равными соответственно 0.05, 0.5, 0, 5 и 0 М возможно протекание
параллельных процессов: А+В=С и A+D=F. Определите энергию
активации первого процесса, если для второго она составляет
30 кДж/моль, а после завершения процесса при различных темпера-
турах были зафиксированы следующие концентрации (М) продуктов:
В-во \ t, °C 0 50 100 150
С 0 043 0.031 0.019 0.011
F 0.007 0.019 0.031 0 039
20. Кинетическое исследование некоторого необратимого про-
цесса проводилось при помощи измерения концентраций одного из
исходных веществ с точностью 0.005 М. Пользуясь приведенными
экспериментальными данными, определите частный порядок по
данному веществу и энергию активации процесса.
t.c /Т,К 300 300 300 400 500
0 1.000 2.000 4.000 1.000 1.000
20 1.000 1.985 3 935 0.625 0 075
40 0.990 1.970 3.875 0 450 0.040
100 0 985 1.925 3.700 0.250 0.015
1200 0.805 1.350 2 040 0.025 0 000
3000 0.630 0.910 1 175 0.010 0.000
6000 0 450 0.585 0.690 0 005 0.000
10000 0.330 0 395 0.440 0.000 о.ооо
Кинетика химических реакций
225
21, При исследовании кинетики некоторого обратимого процесса
АВ + С <-* А + ВС с исходными концентрациями [АВ] = 4М; [С] = 2М;
[Л] = [5С] = О М; при 300 и 350 К были зафиксированы начальные
скорости превращения 0.1 и 0.33 М/с, соответственно. При достиже-
нии равновесия концентрация вещества С достигла 0.2 и 0.15 М, соот-
ветственно. Полагая, что прямой и обратный процессы являются одно-
стадийными, определите энергию активации обратного процесса.
22. Каталитический процесс образования вещества С происходит
по схеме:
А+К<->АК = Ю-’М-’с-1, ^ = 104с’
AK-A-+C-D + K fc^lOV
Предложите кинетическое уравнение для данного процесса. Как
изменится скорость реакции с увеличением температуры, если
известно, что дЯ1 >0 ?
23. Некоторая обратимая реакция А + В = С осуществлялась при
исходных концентрациях компонентов 1 моль/л, Скорость ее в
начальный момент времени составляла 0.5 моль/л с. Через 2 часа
концентрация продукта составила 1.5 моль/л. Какова при этом
скорость обратной реакции?
24. Для некоторой системы последовательных реакций А + В =
АВ, АВ + С - АС + В k} = к2 = 104 л/моль с. Оцените скорость
образования А С через 4 секунды после начала реакции, если исходные
концентрации А, В и С составляли 1 моль/л.
25. Для некоторой системы, в которой протекают конкурирующие
необратимые процессы А + В = АВ и А + С = АС при 25°С выход
продуктов составляет 1:5, При 60°С это соотношение составило 1:3.
Определите энергию активации реакции 2, если для первой она со-
ставляет 30 кДж/моль. Каково должно быть соотношение концен-
траций исходных веществ, для того, чтобы задача имела решение?
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
автопротолиз 118
адиабатическая система 10
адсорбция 214
азеотропная смесь 98
активированный комплекс 191-194, 197
активности коэффициент 49, 114
активность 49, 113
Аррениуса теория кислот 116
Аррениуса уравнение 196
бимолекулярные реакции 186
Больцмана распределение 195
Бренстеда теория кислот 116
буферный раствор 128
Вант-Гоффа изотерма 51
В ант-Гофф а метод 189-191
Вант-Гоффа правило 196
Вант-Гоффа уравнение 104
внутренняя энергия 13
возгонка 142
второй закон термодниамики 28
гальванический элемент 154
Гельмгольца энергия 41
Гесса закон 18
гетерогенная система 61
гетерогенный катализ 211, 213
Гиббса правило фаз 64
Гиббса энергия 41,44
гидратация 93, 106
гидролиз 121,123, 126
гидролиза константа 122, 123, 124, 126
гидролиза степень 122
гомогенная система 61
Предметный указатель
227
гомогенности область 82
гомогенный катализ 211
двойной электрический слой 153
диссоциации константа ПО
дистилляция 139
дифлегматор 140, 141
диффузии коэффициент 218
диффузия 217
длина цепи 209
закрытая система 10
замерзания температура 101, 102
замкнутая система 10
зонная плавка 138
изолированная система 10, 29
изотерма химической реакции 51
изотонический коэффициент 108
инвариантное равновесие 67
ингибирование 212
инициирование цепи 208
инконгруэнтно плавящиеся соединения 79
интенсивные параметры 11
ионная сила 114
ионные пары 113
ионный обмен 147
кажущаяся степень диссоциации 112
каломельный электрод 158
катализ 210
квазиравновесных концентраций метод 205, 206
квазнстационариых концентраций метод 205, 206
кинетика 179
кипения температура 100
Кирхгофа уравнение 23
Клаузиуса постулат 28
Клаузиуса-Клапейрона уравнение 67
коллигативные свойства 105
компонент 62
компонентов число 62
конгруэнтно плавящиеся соединения 79
конденсированная система 62
конденсированная фаза 62
конкурирующих реакций метод 203
230
Предметный указатель
цепные реакции 208
частный порядок реакции 185
эбулиоскопическая постоянная 100
эвтектика 75, 77
экзотермический процесс 18
экстенсивные параметры 11
экстракция 142
электродвижущая сила 153, 156
электродный потенциал 158
электроды необратимые 155
электроды обратимые 154
электролиз 168
электролитической диссоциации степень 109
электрохимический источник тока 170
элементарная реакция 183
Элея-Ридела реакции 215
эндотермический процесс 18
энергия активации 192
энтальпия 14, 15
энтропия 28, 29
Научное издание
Ярославцев
Андрей Борисович
ОСНОВЫ ФИЗИЧЕСКОЙ химии
«Научный мир»
119890. Москва, Знаменка, 11/11
ЛР№ 030671 от 09.12.95 г.
Подписано к печати
Формат 60x90/16
Гарнитура Таймс Нью Роман
Печать офсетная. Уел, печ. л. 14,5
Тираж 500 экз. Заказ
Издание отпечатано в типографии
«Рост-Т»