






























































































































































































































































































































































































































































































Автор: Пилипенко А.Т. Пятницкий И.В.
Теги: аналитическая химия здравоохранение медицинские науки химия
ISBN: 5—7245—0751—X
Год: 1990




Текст
А Т. Пилипенко
И. В^ПятницкийАналитическая
химияКнига XДопущено Государственным
комитетом. СССР по народному
образованию в качестве учебного пособия
для студентов химических
и химико-технологических специальностей
высших учебных заведенийМОСКВА«ХИМИЯ»1990
ББК 543
П32
УДК 543(075.8)Рецензенты: кафедра аналитической химии МГУ
им. М. В. Ломоносова; кафедра аналитической химии МХТИ
им. Д. И. МенделееваПилипенко А. Т., Пятницкий И. В.П32 Аналитическая химия: В двух книгах: кн. 1 —
М.: Химия, 1990. 480 с.: ил.
ISBN 5—7245—0751—X (Кн. 1).
ISBN 5—7245—0507—XИзложены теоретические основы, а также цели и за¬
дачи аналитической химии, теория химических, физико-хв-
мических и физических методов анализа. Рассмотрены
принципы подготовки пробы к анализу, переведение ее в
раствор, определение состава материалов, препаратов, руд,
горных пород и анализ объектов окружающей среды. Даны
необходимые сведения о новых методах анализа и о ме¬
тодах определения структуры химических соединений. При¬
ведены методы обработки результатов.Предназначена для студентов химических факультетов
университетов. Будет полезна аспирантам и химнкам-ана-
литикам химических и геологических лабораторий, студен¬
там химико-технологических факультетов и вузов, а также
студентам нехимических естественных факультетов универ¬
ситетов и педагогических институтов.1707000000-079 ББК 543050(0))-90ISBN 5—7245—0751—X (Кн. 1).@ А. Т. Пилипенко,
ISBN 5—7245—0507—X И. В. Пятницкий, 1990
содержаниеПредисловие ЮВведение 12Раздел I. Общие проблемы аналитической хи¬
мии 15Глава 1. Методы аналитической химии .15§ 1. Задачи современной аиалитнчгской хим.ии 15§ 2. Химические методы анализа ..... 18§ 3. Физические методы анализа 29§ 4. Выбор метода анализа 34Глава 2. Химическое равновесие в гомогенной системе 37§ I. Закон действия масс 37§ 2. Примеры применения закона ;:енствия масс 45Глава 3. Правильность и статистическая обработкарезультатов анализа ......... 51§ J. Отбор пробы 52§ 2. Погрешности, возникающие на разных ста¬
диях анализа 53§ 3. Правильность анализа 57§ 4. Статистическая обработка результатов ана¬
лиза 60Глава 4. Концентрация растворов и вычисления в ко¬
личественном анализе ......... 74§ I. Общие понятия 74§ 2. Вычисления в титриметрнческок анализе . . 76§ 3. Вычисления н гравиметрическим анализе . 82Раздел II. Кислотно-основные реакции ... 85Глава 5. Современные представления о кислотах иоснованиях. Теория Бренстеда ..... 85§ I. Общие положения 85§ 2. Классификация неводных растворителей по
их дифференцирующему действию на силу
кислот 91Глава 6. Концентрация водородных ионов в водныхрастворах электролитов ........ 94§ 1. Концентрация водородных ионов води. Ион¬
ное произведение воды 941* 3
§ 2. Растворы сильных кислот и сильных осно¬
ваний 96§ 3. Растворы слабых кислот и слабых основа¬
ний 97§ 4. Растворы амфолитов 104§ 5. Буферные растворы 107Глава 7. Метод кислотно-основного титрования .113§ 1. Общие положения. Точка эквивалентности 113§ 2. Кривые титрования 119§ 3. Кислотно-основное титрование в неводныхрастворах . 127§ 4. Погрешности титрования 130Глава 8. Индикаторы . 138§ 1. Общая характеристика 138§ 2. Классификация индикаторов 142§ 3. Выбор индикатора при титровании кислота¬
ми и основаниями 154Раздел III. Реакции образования твердой фазы 158Глава 9. Реакции осаждения и растворения осадков 158§ 1. Равновесие между раствором и твердой фа¬
зой . . 158§ 2. Произведение растворимости и произведениеактивности 164§ 3. Растворимость осадков в воде и водныхрастворах электролитов 170§ 4. Влияние на растворимость реакций комплек¬
сообразования 175§ 5. Растворимость осадков в результате обра¬
зования комплексов с избытком осадителя 178
§ 6. Растворимость малорастворимых соединенийв кислотах 180§ 7. Механизм образования твердой фазы — тео¬
рия кристаллизации 185Глава 10. Соосаждение 189§ 1. Общая характеристика 189§ 2. Закономерности адсорбции на аморфныхосадках 192§ 3. Закономерности соосаждения на кристалли¬
ческих осадках 194§ 4. Методы устранения соосаждения ..... 197
§ 5. Использование соосаждения в аналитиче¬
ской химии 199Глава 11. Гравиметрический анализ .201§ 1. Основы метода 201§ 2. Некоторые органические осадители . . . 204
§ 3. Требования к осадкам и к гравиметриче¬
ском форме 207§ 4. Применение возникающих реактивов (оса¬
ждение из гомогенного раствора) .... 209
§ 5. Термогравиметрический анализ ..... 212Глава 12. Электрогравиметрический анализ .... 214§ 1. Общая характеристика ... 214§ 2. Значение напряжения при электролитиче¬
ском выделении металлов . 216§ 3. Физические условия осажден in металлов 222
§ 4. Химические условия осаждения металлов 226§ 5. Внутренний электролиз 231§ 6. Разделение металлов электролизом с при¬
менением ртутного катода . 233Раздел IV. Реакции образования комплексныхсоединений 235Глава 13. Сведения о комплексных соединениях . . 235§ 1. Общие положения .... 235§ 2. Диссоциация и устойчивость комплексов врастворах 239§ 3. Функции образования и закомплексованно¬
сти 245§ 4. Зависимость устойчивости комплексов в рас¬
творах от положения центрального атома в
периодической системе элементов .... 247
§ 5. Зависимость устойчивости комплексов врастворах от свойств лигандоЕ 255§ 6. Влияние концентрации компонентов, pH, ион¬
ной силы раствора, температуры на ком-плексообразование .... 261§ 7. Важнейшие комплексные соединения с не¬
органическими лигандами и применение их
в анализе 265Глава 14. Комплексные соединения в титриметрическоманализе . 269§ 1. Важнейшие неорганические и органическиетитранты 269§ 2. Комплексонометрия . 273§ 3. Металлохромные индикаторы и их выбор 287Раздел V. Абсорбционная спектроскопия ■ . . 292Глава 15. Фотометрический анализ 295§ 1. Общие положения .... 295§ 2. Типы соединений, применяемые в фотометри¬
ческом анализе 2965
§ 3. Свойства и природа электронных спектров 300
§ 4. Природа окраски, спектры поглощения со¬
единений, применяемых в фотометрическом
анализе. Направленный поиск веществ с
требуемыми оптическими свойствами . . • 307
§ 5. Контрастность фотометрических реагентов 312Глава 16. Поглощение электромагнитного излучения 315§ 1. Оптическая плотность (закон Бугера) . . . 315
§ 2. Молярный коэффициент поглощения . . . 319
§ 3. Характеристика светопоглощения .... 321
§ 4. Молекулярный коэффициент светопоглоще¬
ния, сила осциллятора 323§ 5. Причины отклонения от закона поглощенияэлектромагнитных колебаний 324Глава 17. Аппаратура и методы измерения, применяе¬
мые в фотометрическом анализе 327§ I. Методы измерения интенсивности окраски 327§ 2. Фотоэлектроколориметры 330§ 3. Спектрофотометры 338§ 4. Абсолютные методы определения веществ вотсутствие мешающих компонентов . . . 341§ 5. Фотометрическое титрование 343§ 6. Нефелометрический и турбидиметрическийметоды анализа 346§ 7. Дифференциальная спектрофотометрия . • 348
§ 8. Производная спектрофотометрия 350Глава 18. Люминесцентный анализ 352§ 1. Общие положения 352§ 2. Квантовый и энергетический выход и соот¬
ношение между ними 355§ 3. Тушение люминесценции 358§ 4. Качественный и количественный люминес¬
центный анализ 359§ 5. Флуориметры 360§ 6. Хемилюминесцентный анализ 363Раздел VI. Реакции окисления — восстановле¬
ния .369Глава 19. Окислительный потенциал и сопряженныереакции окисления — восстановления . . . 369§ 1. Характеристика реакций окисления — восста¬
новления 369§ 2. Окислительный потенциал 372§ 3. Сопряженные реакции окисления — восста¬
новления 387§ 4. Индикаторы окислительно-восстановительно¬
го титрования 390в
Глава 20. Титрование перманганатом (г ерманганато-метрия) 398§ 1. Общая характеристика. Приготовление исвойства раствора псрмлшанага .... 398
§ 2. Псрманганатомстричсское определение не¬
органических и органических воцеств . . . 402Глава 21. Иодометрия 411§ 1. Общая характеристика метода 411§ 2. Рабочие растворы иодомстрии 415§ 3. Примеры иодометрическнх ог^еделепий . . 420Глава 22. Другие методы окисления — восстановления.Методы предварительного восстановления 429§ 1. Броматометрия . .... 429§ 2. Хроматометрия 433§ 3. Методы предварительного вссстановления 434Глава 23. Скорость химических реакций н ее значениев аналитической химии . 440§ 1. Общая характеристика . . 440§ 2. Каталитические методы анализа .... 446Глава 24. Потенциометрический анализ 454§ 1. Общая характеристика потенциометрическо¬
го анализа 454§ 2. Металлические индикаторные электроды . . 460
§ 3. Иопселективные мембранные электроды . ■ 467
§ 4. Электроды сравнения ... 477Глава 25. Полярографический и кулономе'рический ме¬
тоды анализа ........... 481§ 1. Основы полярографического анализа . . .481
§ 2. Методы вольгамперометрии ..... 498§ 3. Полярография металлов . . 501§ 4. Полярография органических сс единений . . 509
§ 5. Кулонометрическин анализ . 511Раздел VII. Методы обнаружения и разделенияв аналитической химии 524Глава 26. Методы обнаружения и разделения посредст¬
вом осаждения 524§ 1. Характеристика аналитических реакций . . 524§ 2. Маскиропание 529§ 3. Специфичность, избирательное^ (селектив¬
ность) и специфические условия проведенияреакций 534§ 4. Методы обнаружения 5377
§ 5. Макро-, полумикро-, микро- и ультрамикро¬
анализ 541§ 6. Методы разделения 542§ 7. Другие методы разделения 562Глава 27. Разделение элементов методом экстракции 565§ 1. Общие положения . 565§ 2. Основные термины и количественные ха¬
рактеристики процесса экстракции .... 568
§ 3. Типы экстрагирующихся соединений . . . 572Глава 28. Хроматографический анализ 580§ 1. Общие положения 580§ 2. История развития хроматографии .... 582§ 3. Основы теории хроматографии 584§ 4. Жидкостная колоночная хроматография . . 595
§ 5. Твердожидкостная колоночная хроматогра¬
фия 599§ 6. Ионообменная хроматография 602§ 7. Гель-проникающая (молекулярно-ситовая)хроматография 609§ 8. Тонкослойная хроматография 610§ 9. Хроматография на бумаге 614§ 10. Газовая хроматография 617§ П. Измерение концентрации при помощи хро¬
матографических методов 627§ 12. Качественный анализ хроматографическимметодом 628§ 13. Области использования различных видовхроматографии 630Глава 29. Отбор пробы, подготовка образца к анализуи проведение анализа . 631§ 1. Отбор и подготовка пробы к анализу . . . 631
§ 2. Вода в пробе и методы ее определения . . 634§ 3. Выбор схемы и метода анализа 640§ 4. Разложение анализируемой пробы .... 642Раздел VIII. Физические методы анализа ■ . . 645
Глава 30. Спектральный анализ 645§ 1. Общие положения 645§ 2. Спектральные приборы 649§ 3. Источники возбуждения спектров . . . 658
§ 4. Качественный атомно-эмиссионный спек¬
тральный анализ 665§ 5. Количественный атомно-эмиссионный спек¬
тральный анализ 671§ 6. Пламенная фотометрия 693§ 7. Атомно-абсорбционный спектральный анализ 6978
Глава 31. Методы, основанные на взаимодействии ве¬
щества с магнитным полем 704§ 1. Краткие сведения о магнетизме 704§ 2. Метод статической магнитной восприимчиво¬
сти 707§ 3. Метод электронного парамагнитного резо¬
нанса (ЭПР) 713§ 4. Спектроскопия ядерного магнитного резо¬
нанса (ЯМР) ..... 727§ 5. Метод ядерной магнитной релаксации . . 738
§ 6. Ядернын квадрупольный резонанс (ЯКР) 742
§ 7. Ядерная \-резонансная спек- роскопия (ЯГР,Мессбауэровская спектроскопия) 745§ 8. Магнитная спектрополяримегрия 750§ 9. Масс-спектрометрИя 751Глава 32. Колебательная спектроскопиj 756§ 1. ИК-спектроскопня 756§ 2. Аппаратура и методы изуче-шя ИК-спектров 759§ 3. Применение ИК-спектров 766§ 4. Спектроскопия комбинации ного рассеяния 769Глава 33. Рентгенофлуоресцентный и другие физиче¬
ские методы 778§ 1. Рентгенофлуоресцентный ме,-од анализа . 778
§ 2. Радиоактивационный метод анализа . . 785§ 3. Фотоактивационный анализ 794§ 4. Рефрактометрия 795§ 5. Интерферометрия 798§ 6. Поляриметрия 800Раздел IX. Элементный, функциональный и фа¬
зовый методы анализа 805Глава 34. Анализ органических соединений .... 805§ 1. Общие сведения 805§ 2. Качественный элементный анализ органиче¬
ских соединений 806§ 3. Количественное определение отдельных эле¬
ментов 811§ 4. Автоматические анализаторы для элемент¬
ного анализа 815§ 5. Количественный анализ органических соеди¬
нений по функциональным группам .... 819Глава 35. Фазовый и вещественный методы анализа 824Список основной использованной литера¬
туры 827Предметный указатель 833
ПРЕДИСЛОВИЕВ предлагаемом пособии рассмотрены основные проб¬
лемы и методы современной аналитической химии.
Авторы стремились дать правильное представление о
всех важнейших методах анализа — химических, фи¬
зико-химических и физических, применяющихся для
обнаружения, разделения и количественного опреде¬
ления элементов. Уровень изложения рассчитан в ос¬
новном на студентов второго (третьего) курса; тем
не менее отдельные разделы, например, о фотометри¬
ческих методах анализа, хроматографии и др., могут
служить для более углубленного изучения предмета
и студентами старших курсов.При описании отдельных методов авторы стара¬
лись показать возможности их применения для ана¬
лиза веществ не только неорганической природы, но
также для исследования органических соединений;
элементному и функциональному анализу органиче¬
ских веществ посвящена отдельная глава.Проспект учебного пособия был разослан для зна¬
комства на кафедры аналитической химии многих
университетов страны. Авторы благодарны за сде¬
ланные по проспекту критические замечания и поже¬
лания и старались по возможности принять их во
внимание.Главы 1, 8, 10, 12, 27, §§ 1, 2, 3 гл. 33 написаны
И. В. Пятницким. Главы 9, 11, 13—18, гл. 26, 29, 35,
§§ 4—6 гл. 33 написаны А. Т. Пилипенко. Гл. 28 со¬
ставлена А. Т. Пилипенко совместно с С. В. Галушко,
гл. 30 — с О. П. Рябушко, гл. 31 и 32 — с С. В. Тра-
чевским, гл. 34 — с Н. С. Мирошниченко, §§ 3 и 4
гл. 15 — с Л. И. Савранским, § 6 гл. 18 —с Л. А. Пи¬
липенко.Содержание и текст всех разделов авторы обсу¬
ждали совместно.Авторы выражают глубокую благодарность
Г. С. Мацибуре, М. К. Искандерову, А. Л. Розен-
фельд, Л. П. Игнатенко и Т. Е. Гетьман, оказавшим
большую помощь в подготовке рукописи.Авторы признательны заведующему кафедрой
аналитической химии Московского государственного10
университета им. М. В. Ломоносова академику
И. П. Алимарину и сотрудникам кафедры, а также
заведующему кафедрой аналитической химии Москов¬
ского химико-технологического института им.
Д. И. Менделеева профессору О. М. Петрухину и со¬
трудникам кафедры, которые выполнили большой
труд по рецензированию рукописи и сделали ценные
замечания, способствующие улучшению рукописи.Авторы будут признательны за критические от¬
зывы о книге.
ВВЕДЕНИЕАналитическая химия представляет собой один из
разделов химии. Предмет химии — химические эле¬
менты и их соединения, она изучает процессы пре¬
вращения одних веществ в другие. Аналитическая хи¬
мия также занимается исследованием этих процес¬
сов, однако, в отличие от других разделов химии,
имеет своей главной задачей установление химиче¬
ского состава веществ. Поэтому аналитическую хи¬
мию определяют как науку, изучающую свойства и
процессы превращения веществ с целью установле¬
ния их химического состава. Химические и физиче¬
ские свойства веществ являются основой соответст¬
вующих методов анализа, поэтому нередко говорят
об аналитической химии как науке о методах уста¬
новления химического состава веществ. Приведенное
определение нуждается в расшифровке.Установить химический состав веществ означает
ответить на вопрос о том, какие элементы или (и)
их соединения и в каких именно количественных со¬
отношениях содержатся в анализируемом материале.
В зависимости от характера поставленной задачи
различают следующие виды анализа.1. Элементный анализ — установление наличия и
содержания отдельных элементов в данном веществе,
т. е. нахождение его элементного состава.2. Фазовый анализ — установление наличия и со¬
держания отдельных фаз исследуемого материала.
Так, углерод в стали может находиться в виде гра¬
фита и в форме карбидов — соединений железа (или
другого металла) с углеродом. Задача фазового ана¬
лиза — найти, сколько углерода содержится, в виде
графита и сколько в виде карбидов.3. Молекулярный анализ (вещественный анализ,
его иногда неправильно называют фазовым анали¬
зом)— установление наличия и содержания молекул
различных веществ (соединений) в материале. На¬
пример, в атмосфере определяют количество СО, С02,
N2, 02 и др.4. Функциональный анализ — установление нали¬
чия и содержания функциональных групп в молеку¬12
лах органических ‘ соединений, например амино-
(—NH2), нитро-(—N02), нитрозо- (—N0), гидрокси-
(—ОН), карбоксильных (—СООН) и других групп.В соответствии с задачами установления химиче¬
ского состава различают два вида анализа — качест¬
венный и количественный. Задача качественного ана¬
лиза — обнаружить, какие именно элементы или их
соединения входят в состав анализируемого мате¬
риала. Качественный анализ обычно предшествует
количественному; цель последнего — найти количест¬
венные соотношения между компонентами, найден¬
ными при качественном исследовании. Часто решает¬
ся и более узкая задача — определяют количество
только одного или нескольких (не всех) компонентов
пробы. Результаты анализа дают возможность уста¬
новить химические формулы синтетических и природ¬
ных соединений, оценить соответствие разнообразных
материалов требованиям производства.В зависимости от характера анализируемого ма¬
териала различают анализ неорганических и органи¬
ческих веществ. Выделение анализа органических ве¬
ществ в отдельный раздел аналитической химии свя¬
зано с некоторыми особенностями органических
соединений по сравнению с неорганическими. Часто
первый этап анализа состоит в переведении пробы
в раствор. При анализе неорганических материалов
растворителем чаще всего служит вода или водные
растворы кислот или щелочей. Полученный раствор
содержит катионы и анионы подлежащих определе¬
нию элементов. Для их обнаружения применяют реа¬
генты, которые взаимодействуют с определяемыми
ионами, как правило, очень быстро, причем в боль¬
шинстве случаев реакции доходят до конца. При
анализе органических соединений нередко необходимо
провести предварительную минерализацию пробы,
т. е. разрушить ее органическую часть прокаливанием
или обработкой концентрированными кислотами. Не¬
растворимые в воде органические соединения иногда
растворяют в органических растворителях; реакции
между органическими соединениями обычно проте¬
кают медленно и почти никогда не доходят до конца,
причем они могут протекать по нескольким направ¬
лениям с образованием разнообразных продуктов
реакции. В анализе применяют и некоторые другие13
специфические приемы, например определяют темпе¬
ратуру плавления или кипения вещества с целью про¬
верки чистоты препарата. В анализе сложных смесей
органических соединений главенствующая роль при¬
надлежит методу газовой хроматографии — его при¬
менение в анализе неорганических веществ ограни¬
чено. Эти и некоторые другие особенности оправды¬
вают деление анализа на две названные группы.Существуют физические и химические методы ана¬
лиза. Это деление несколько условно, между мето¬
дами обеих групп нет резкой границы. В обоих слу¬
чаях качественное обнаружение и количественное
определение составных частей анализируемого мате¬
риала основано на наблюдении и измерении какого-
либо физического свойства системы. Измеряют, на¬
пример, электропроводность, плотность, интенсивность
окраски, интенсивность радиоактивного излучения,
массу, объем, электрический потенциал и на этом
основании делают вывод о количестве данного эле¬
мента или его соединений. Однако при анализе физи¬
ческими методами наблюдение и измерение выпол¬
няют непосредственно с анализируемым материалом,
причем химические реакции либо совсем не проводят,
либо они играют вспомогательную роль. В химиче¬
ских методах пробу подвергают сначала действию
какого-либо реагента, т. е. проводят определенную
химическую реакцию, и только после этого наблю¬
дают и измеряют физическое свойство. В соответст¬
вии с этим в химических методах анализа главное
внимание уделяют правильному выполнению химиче¬
ской реакции, в то время как в физических методах
основной упор делается на соответствующее аппара¬
турное оформление измерения — определение физиче¬
ских свойств.
Раздел 1ОБЩИЕ ПРОБЛЕМЫ
АНАЛИТИЧЕСКОЙ ХИМИИГЛАВА 1МЕТОДЫ АНАЛИТИЧЕСКОЙ ХИМИИ§ 1. ЗАДАЧИ СОВРЕМЕННОЙ АНАЛИТИЧЕСКОЙ ХИМИИАналитическая химия тесно связана с различными
областями науки и производства. Химический анализ
применяют для контроля качества сырья, полуфабри¬
катов и готовой продукции. Каждая область науки и
производства ставит перед аналитической химией
свои специфические задачи. Так, в медицине большое
значение имеет качественное обнаружение и количест¬
венное определение отдельных элементов, которые
входят в состав тканей живых организмов и обуслов¬
ливают их нормальную физиологическую деятель¬
ность. Урожайность сельскохозяйственных культур
зависит в значительной степени от содержания в поч-
вах и в удобрениях многих микроэлементов. В связи
с этим возникла необходимость разработать методы
определения в удобрениях микроколичеств ряда эле¬
ментов (марганца, бора, железа, молибдена).Выплавляя чугун или сталь, металлург должен
знать содержание железа в руде, а также вредных
примесей — серы, фосфора, мышьяка и других эле¬
ментов в руде и готовом продукте. Эти сведения
дает аналитическая лаборатория завода. Полевые
анализы полезных ископаемых дают геологу возмож¬
ность оценить общие запасы элементов и сделать вы¬
вод об экономической целесообразности разработка
месторождения. Санитарная служба требует от хи-
мика-аналитика оценки качества питьевой воды; в пи¬
щевой промышленности необходимо контролировать
доброкачественность продуктов; для судебного экс¬
перта важно установить характер яда, вызвавшего
отравление человека и т. д.На основании этих и многих других примеров при¬
менения аналитической химии в различных областях15
науки и производства можно сформулировать не¬
сколько общих проблем. Одна из них — разработка
новых методов обнаружения и количественного опре¬
деления элементов, которые начали использовать в
широких масштабах сравнительно недавно. К таким
элементам принадлежат, например, ниобий, тантал,
цирконий, гафний, титан, молибден, вольфрам, редко¬
земельные элементы и др. Их используют в современ¬
ной технике в качестве компонентов жаропрочных
сплавов, конструкционных материалов в атомной
энергетике. Они входят в состав многих специальных
магниевых и других сплавов. Применение названных
элементов все время расширяется. Так, цирконий ь
прошлом не имел почти никакого практического зна
чения, хотя был открыт еще в конце XVIII в. В на¬
стоящее время цирконий, его сплавы и соединения
используют почти в 150 отраслях. Цирконий является
составной частью многих силикатных материалов, из
него изготавливают детали атомных реакторов, спла¬
вы алюминия, магния и др.Другая важная проблема — разработка методов
обнаружения и определения микроколичеств элемен¬
тов. Физические и химические свойства материалов
часто зависят от присутствия именно микрокомпонен¬
тов. Титан и хром долгое время считали хрупкими
металлами, которые нельзя ковать и прокатывать, од¬
нако недавно было установлено, что эти металлы в
очищенном состоянии пластичны и что их хрупкость
обусловлена незначительными примесями посторон¬
них элементов. Германий является одним из основ¬
ных материалов для изготовления полупроводниковых
приборов в радиотехнической промышленности, одна¬
ко он утрачивает свои полупроводниковые свойства,
если на десять миллионов атомов германия приходит¬
ся более одного атома фосфора, мышьяка или сурь¬
мы. Самая незначительная примесь гафния в метал¬
лическом цирконии делает последний непригодным
для использования в атомной промышленности. Ни¬
чтожные примеси титана, ванадия, висмута и некото¬
рых других металлов в сталях значительно изменяют
их механические и электрические свойства. Почти все
элементы периодической системы входят в очень не¬
больших количествах в состав тканей растений и жи¬
вых организмов, причем каждый элемент играет впол¬16
не определенную физиологическую роль. Требования
науки и техники к определению микроколичеств бес¬
прерывно возрастают. В 1947 году химики-аналитики
определяли примеси, составляющие тысячные доли
процента в основном материале; теперь необходимы
методы, которые были бы пригодны для определения
Ю-|00/о примесей, т. е. для нахождения одного атома
элемента в присутствии десятков миллиардов атомов
другого элемента.Разработка методов анализа органических веществ
является еще одной важной проблемой современной
аналитической химии. В последние годы возникло
много совершенно новых производств, вырабатываю¬
щих пластмассы, полимеры, элементоорганические
соединения, биологически активные и фармацевтиче¬
ские препараты, пестициды и др. Развилась промыш¬
ленность тяжелого органического синтеза, переработ¬
ки нефти, природного газа, угля. Для этих произ¬
водств необходимы надежные методы анализа сырья,
полуфабрикатов и готовой продукции.В связи с загрязнением атмосферы и водных бас¬
сейнов выбросами токсичных газообразных веществ
и промышленными стоками возникла необходимость
создания методов химического контроля степени очи¬
стки выпускаемых в реки и озера или в воздух отхо¬
дов производства. Нередко эти отходы содержат
очень сложные смеси самых разнообразных вредных
для здоровья человека неорганических и особенно
органических веществ: фтор- и хлорорганические
соединения, фенол и его производные,, формальдегид,
диоксид серы, сероводород, оксиды азота, оксид
углерода и др.Значительно повысились требования к точности и
чувствительности методов анализа. Все большее зна¬
чение приобретают экспрессные методы анализа; они
необходимы для оперативного контроля технологиче¬
ских процессов и позволяют идентифицировать мало¬
устойчивые промежуточные продукты химических,
биохимических и радиохимических превращений. В не¬
которых случаях нельзя обойтись без приемов ди¬
станционного анализа, когда.необходимо анализиро¬
вать высокоагрессивные, космические или подземные
объекты.17
§ 2. ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗАХимический анализ сложных материалов состоит в
большинстве случаев из следующих этапов.1. Отбор пробы для анализа. Лабораторная про¬
ба обычно составляет 10—50 г материала, который
должен быть отобран так, чтобы его средний состав
соответствовал среднему составу всей партии анали¬
зируемого объекта. Лабораторную пробу готовят по
специальным правилам, которые будут описаны ниже.2. Разложение пробы и переведение ее в раствор.
Пробу растворяют в воде или обрабатывают кисло¬
тами; при необходимости анализируемый материал
сплавляют с различными веществами, озоляют или
используют другие способы .уимиуестого воздействия
на него.Различные методы разложения описаны в гл. 29,§4.3. Проведение химической реакции. На определяе¬
мый компонент пробы X воздействуют реагентом R,
в результате чего образуется продукт реакции Р:x + R —о р. (1.1)Это важный этап анализа и его правильному выпол¬
нению должно быть уделено основное внимание.4. Измерение какого-либо физического параметра
продукта реакции, реагента или самого определяе¬
мого вещества. На основе этого измерения судят о
количестве или о содержании определяемого компо¬
нента в анализируемом материале.Классификация методов анализаВозможны три способа использования реакции (1.1)
В связи с этим все химические методы можно раз¬
делить на три группы.В первой группе методов измеряют количество
образовавшегося продукта реакции Р: устанавливают
массу продукта реакции путем взвешивания (грави¬
метрический анализ); измеряют интенсивность погло¬
щения света раствором Р, она при некоторых усло¬
виях пропорциональна массе этого вещества (фото¬
метрический анализ).18
Совершенно очевидно, что в обоих случаях к реак¬
ции (1.1) предъявляются одинаковые методические
требования. Следует прежде всего создать такие ус¬
ловия, при которых определяемое вещество полностью
превращается в продукт реакции Р, т. е. чтобы со¬
стояние равновесия реакции (1.1) было как можно
больше сдвинуто в правую сторону. Необходимо так¬
же, чтобы реагент R, вступая во взаимодействие с по¬
сторонними веществами, находящимися в растворе, не
давал продуктов реакции, физические свойства кото¬
рых были бы сходными с физическими свойствами Р.Эти два требования иллюстрируются такими при¬
мерами.Для определения железа гравиметрическим мето¬
дом ионы осаждаются раствором аммиака:Fe3+ + 3NH3 + 3H20 +=£ Fe(OH)3| + 3NH< . (1.2)Гидроксид железа затем переводят в оксид железа
РеаОз, который взвешивают.Определение будет правильным только тогда,
когда все железо из раствора полностью переведено
в осадок; кроме того, если в растворе нет других
ионов, образующих осадок с раствором аммиака, на¬
пример ионов алюминия, титана, висмута, сурьмы и
др. В то же время реагент NH3H20 может реагиро¬
вать с другими находящимися в растворе веществами,
если при этом не образуется другой твердой фазы.
Возможна, например, реакция NH3 с хлороводород¬
ной кислотой или с другими компонентами раствора:
HC1 + NH3 NH4CI. (1.3)Хлорид аммония хорошо растворим и поэтому не ме¬
шает определению.Для определения железа фотометрическим мето¬
дом его можно перевести в роданид железа, окрашен¬
ный в красный цвет:Fe3+ + 3SCN' +=± Fe(SCN),. (1.4)Затем измеряют интенсивность окрг!Ски роданида
железа.Как и при гравиметрическом определении, анализ
будет правильным только при полном превращении
железа в окрашенное соединение. Но и растворе дол¬
жны отсутствовать вещества, образующие с роданид-19
ионами окрашенные комплексы, например молибден:
МоО3* + 5SCN' + 2Н* :?=► Mo(SCN)3 + Н20. (1.5)Роданидный комплекс молибдена также красного
цвета.В то же время ионы цинка не мешают определе¬
нию, хотя они и реагируют с роданид-ионами:2п2+ + 4SCN" ц=± Zn(SCN)2', (1.6)но продукт реакции бесцветен и его присутствие не
отражается на результатах определения.Вторая группа методов основана на измерении ко¬
личества реагента, израсходованного на реакцию с
определяемым веществом X. Обычно измеряют объем
раствора реагента, концентрация которого точно из¬
вестна. Раствор реагента прибавляют до тех пор,
пока не будет достигнуто эквивалентное соотношение
между реагирующими веществами.Процесс прибавления титрованного раствора реа¬
гента R к раствору определяемого вещества X назы¬
вают титрованием. Отсюда общее название рассма¬
триваемой группы методов — титриметрические ме¬
тоды анализа.В титриметрических методах анализа необходимо
выполнение следующих требований: 1) взаимодейст¬
вие между X и R должно проходить стехиометриче-
ски, в точном соответствии с уравнением реакции;
2) реакция должна протекать быстро; 3) реагент не
должен вступать в реакцию с посторонними вещест¬
вами, находящимися в растворе; 4) необходимо рас¬
полагать способом установления точки эквивалент¬
ности, т. е. такого момента титрования, когда реагент
прибавлен в эквивалентном количестве.Применение титриметрических методов иллюстри¬
руется примером определения железа (II) титрова¬
нием перманганатом:5Fe2+ + MnO; + 8H+ = 5Fe’*+ Мп2+ + 4Н20. (1.7)При некоторых определенных условиях первые два
требования выполняются для реакции (1.7) доста¬
точно удовлетворительно. Третье требование реали¬
зовать трудно; определение становится невозможным,
если в растворе содержатся восстановители, напри¬
мер ионы Sn2+, I- и др. Четвертое требование реа¬20
лизуется вследствие того, что небольшой избыток
перманганата сверх эквивалентного количества окра¬
шивает раствор в розовый цвет, что указывает на
полное окисление железа.В третьей группе методов фиксируют изменения,
происходящие с самим определяемым веществом X в
процессе взаимодействия с реагентом R. К этой груп¬
пе относят, в частности, некоторые методы газового
анализа. Так, содержание С02 в сложной газовой сме¬
си можно определить, измерив его объем до и после
пропускания через раствор гидроксида натрия. По¬
следний поглощает С02 и уменьшение объема смеси
пропорционально содержанию С02 в ней:2NaOH + С02 = Na2C03 + Н2С. (1.8)Другая классификация методов химического ана¬
лиза основана на ином критерии, а именно на типе
химической реакции. Различают кислотно-основные
реакции, реакции образования комплексных соедине¬
ний и реакции окисления — восстановления.Такое разделение несколько условно. Нередко ре¬
акции различных типов сочетаются между собой, и
трудно четко определить принадлежность данной ре¬
акции к какому-либо определенному типу. Например,
реакция между металлической ртутью и HI —Hg + 2H+ + 4r = HgI24 +Н2, (1.9)это реакция окисления — восстановления, так как
здесь ртуть окисляется до Hg2+, а ионы Н+ восстана¬
вливаются до Н2. Однако реакция (1.9) сопрово¬
ждается образованием комплекса Hgl] , поэтому ее
можно причислить также к реакциям образования
комплексных соединений.Реакция между ионами алюминия и раствором
8-гидроксихинолина (НОх) выражается уравнениемА13++ ЗНОх = А10х3| + ЗН+. (1.10)Выделяется оксихинолинат алюминия. Осадок
АЮхз представляет собой комплексное соединение, по
этому признаку реакцию (1.10) следует отнести к ре¬
акциям комплексообразования. Однако при реакции
изменяется также кислотность раствора, поэтому с
равным правом реакция (1.10) может рассматри¬
ваться как принадлежащая к двум типам реакций.21
Однако приведенное выше деление реакций на три
типа удобно и облегчает рассмотрение и классифика¬
цию химических методов анализа.Необходимо также иметь в виду, что каждый тип
реакции может быть использован для одной из сле¬
дующих целей (или всех вместе): для отделения оп¬
ределяемого вещества (элемента, ионов или их соеди¬
нений) от других мешающих определению компонен¬
тов; для концентрирования определяемого вещества;
для качественного его обнаружения; для прямого ко-
личественного определения.Кислотно-основные реакции используют главным
образом для прямого количественного определения
сильных и слабых кислот и оснований или их солей.
На таких реакциях основан метод кислотно-основного
титрования (метод нейтрализации). В этом методе
применяют в качестве реагентов растворы сильных
кислот или сильных оснований — так называемые ра¬
бочие растворы. Возможны, например, такие опреде¬
ления:определение сильных кислот (или сильных основа¬
ний) титрованием растворами NaOH (или НС1)НС1 + NaOH = NaCl + Н20;определение слабых кислот (или слабых основа¬
ний)СНзСООН + NaOH = CH3COONa + Н20.В меньшей степени кислотно-основные реакции
применяют для качественного обнаружения отдельныхионов. Так, сульфит-ионы можно обнаружить, подкис-2 -ляя анализируемый раствор; присутствие SO3 легко
устанавливают по специфическому запаху. Прибавле¬
ние раствора гидроксида натрия к анализируемому
раствору, содержащему ионы аммония, вызывает вы¬
деление аммиака, который также обнаруживают но
запаху или какой-либо специфической реакцией.Реакции образования комплексных соединений.
Определяемые вещества действием реагентов перево¬
дят в комплексные ионы или соединения. Так, при
действии аммиака на ионы меди образуется комп¬
лексный аммиакат меди синего цвета. Ионы кобальта
реагируют с органическим реагентом 1-нитрозо-2-наф-
толом с образованием малорастворимого комплекс¬22
ного соединения и т. д. На реакциях комплексообра¬
зования основаны следующие методы разделения и
определения.Разделение посредством осаждения.
Многие комплексные соединения, и час ности те, ко¬
торые образуются при действии органических реаген¬
тов, нерастворимы в воде. Это дает возможность ис¬
пользовать комплексные соединения для разделения
ионов на отдельные группы. Так, нонь калия оса¬
ждают в виде комплексного соединения К3 [Со (Ы02)б],
отделяя их от ионов натрия, магния, бария, стронция,
кальция и ионов некоторых других металлов, которые
остаются в растворе. Осаждение никеля диметилглио-
кслмом позволяет отделить его от кобальта и железа.
Железо отделяют от алюминия, хрома, цинка, кад¬
мия, марганца и многих других элементов, осаждая
его органическим реагентом купфероном, с которым
железо образует нерастворимое комплзксное соеди¬
нение.Метод экстракции. Нерастворимые в воде
комплексные соединения нередко хорошо растворяют¬
ся в органических растворителях — хлороформе, бен¬
золе, диэтиловом эфире, амиловом или бутиловом
спиртах и др. На этом свойстве комплексных соедине¬
ний основано разделение элементов экстракцией. Эк¬
стракция— метод разделения, который заключается
в переведении соединения определяемое элемента из
водного раствора в слой органического растворителя,
не смешивающегося с водой. В органисеских раство¬
рителях часто растворяются также простые вещества,
например свободный иод, который ле’ко экстраги¬
руется из водного раствора хлороформом или бензо¬
лом. Однако чаще всего метод экстракции применяют
для переведения в органический растворитель именно
комплексных соединений.Железо экстрагируется диэтиловым эфиром в виде
комплексного соединения Н [FeCU] и отделяется та¬
ким способом от никеля и некоторых других элемен¬
тов. Существуют методы разделения элементов экс¬
тракцией дитизонатов, купферонатов, гидроксихино-
линатов, диэтилдитиокарбаминатов металлов, т. е.
комплексных соединений металлов с ргзличными ор¬
ганическими реагентами. Экстракцию используют и
как метод концентрирования: небольшие количества23
определяемых ионов, находящихся в водном растворе
в присутствии преобладающих количеств других
иоиов, сначала переводят действием определенных
реагентов в комплексные соединения, которые затем
экстрагируют небольшим количеством органического
растворителя.Фотометрический метод анализа. Из¬
меряют оптическую плотность растворов комплексных
соединений, образующихся при взаимодействии опре¬
деляемых ионов с неорганическими или органиче¬
скими реагентами. Так, для определения ионов же¬
леза к раствору прибавляют роданид калия или
аммония; оптическая плотность раствора образовав¬
шегося роданида железа пропорциональна количе¬
ству железа в растворе. Кремний, фосфор или мышь¬
як можно определить в виде гетерополикислот
Н4 [Si (Мо3Ою) 4], H3[P(Mo3Oio)4] или Н3 [As X
X (Мо3О10)4], окрашенных в желтый цвет.Еще чаще используют окрашенные комплексные
соединения с органическими реагентами. Так, алюми¬
ний определяют с ализарином, причем образуется
комплекс красного цвета. Кобальт образует окрашен¬
ный комплекс с нитрозо-Р-солью. Дифенилтиокарба-
зон (дитизон) реагирует с медью, золотом, серебром
и другими элементами, образуя окрашенные комп¬
лексные соединения, легко растворимые в различных
органических растворителях.Титриметрический анализ. Комплексоно-
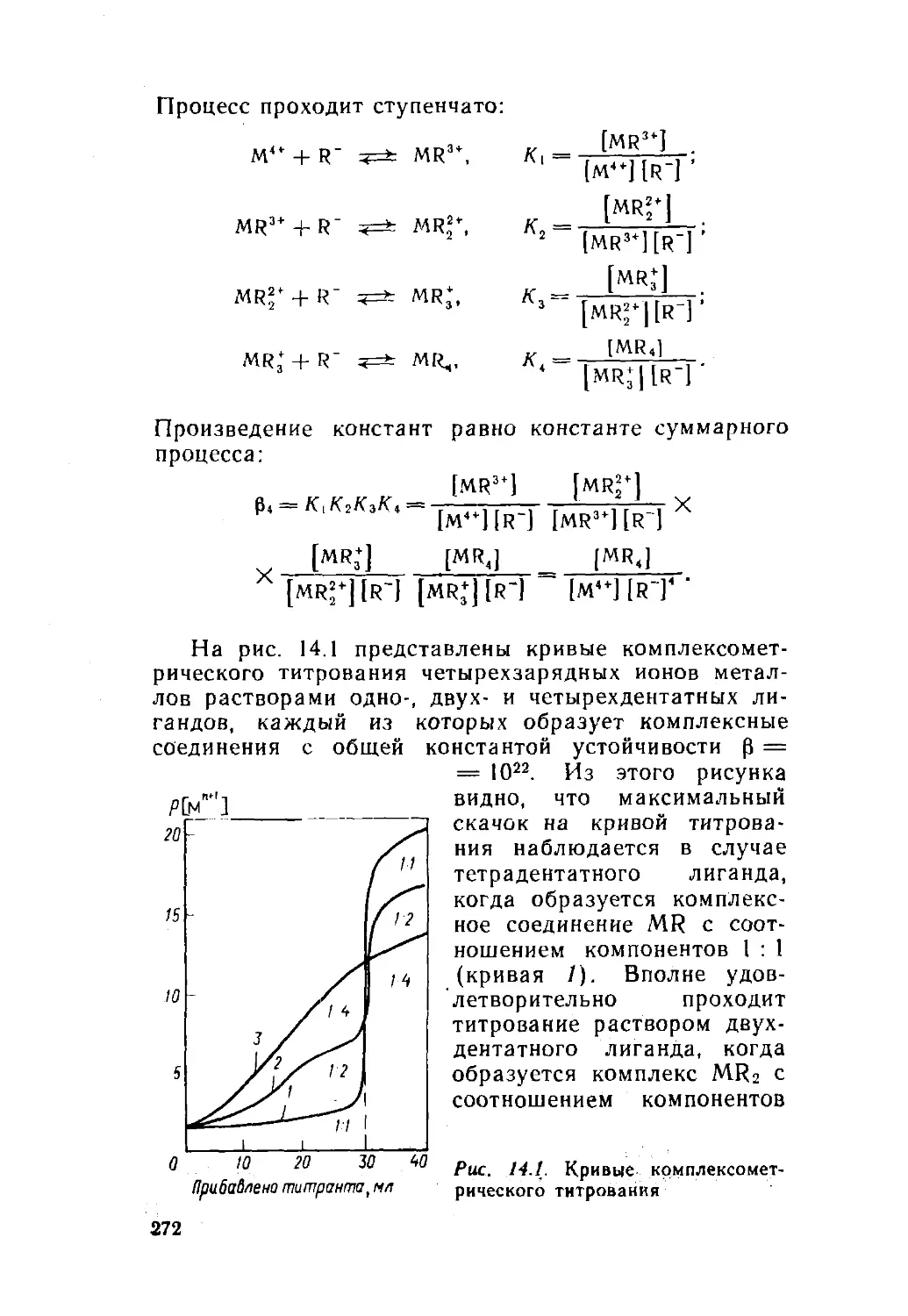
метрия — один из широко распространенных методов
анализа, основанный на применении комплексонов —
органических соединений, содержащих азот и карбо¬
ксильные группы. Титрование комплексонами различ¬
ного состава позволяет определять многие элементы:
цирконий, железо, висмут, кадмий, медь, цинк, маг¬
ний, кальций и др. Известны и другие титриметриче-
ские методы, в которых используют комплексные
соединения. Так, существует метод титрования фтори¬
дами— фторометрия, солями ртути (II) — меркуро-
метрия и др.Гравиметрический анализ. Многие нерас¬
творимые комплексные соединения используют для
гравиметрического определения различных элементов.
Так, алюминий можно осадить 8-гидроксихинолином,
осадок отфильтровать, высушить и взвесить. Известно24
определение никеля в виде диметилглиоксимата, ко¬
бальта— осаждением 1-нитрозо-2-наотолом и т. д.Реакции окисления — восстановления. На реак¬
циях этого типа основаны многие методы разделения,
концентрирования, идентификации и определения раз-
личных.элементов.Метод цементации. Цементацией называют
восстановление металлами ионов металлов, находя¬
щихся в растворе. Так, действием металлического
цинка на раствор соли меди можно зыделить из рас¬
твора всю медь в виде металла:Cu2t + Zn = Си + Zn2+.Цементацию металлическим цинком применяют
для восстановления ионов индия и отделения их от
ионов алюминия, которые остаются в растворе. На
металлической меди осаждают сурьму, которую мож¬
но таким способом отделить от ионоь олова и свинца.
Металлическим железом разделяют медь и кадмий
и т. д. Цементацию применяют также как метод кон¬
центрирования.Электролиз с ртутным катодом. При
электролизе раствора с ртутным катодом ионы мно¬
гих элементов восстанавливаются электрическим то¬
ком до металлов, которые растворяются в ртути, обра¬
зуя амальгаму. Ионы других металлов остаются при
этом в растворе. Метод применяют, например, для от¬
деления цинка от алюминия, железа от титана и во
многих других случаях.Методы идентификации. В качественном
анализе реакциями окисления — восстановления об¬
наруживают ионы марганца, xpoiv а, ртути, олова,
висмута и др. Так, для обнаружения ионов марган¬
ца (II) его окисляют бромом или хлором до марган¬
ца (VII); фиолетовая окраска образовавшегося
перманганата свидетельствует о присутствии ионов
марганца. Много других методов обнаружения ионов
также основано на реакциях окисления — восста¬
новления.Титриметрические методы. Широко рас¬
пространены методы титрования окислителями и вос¬
становителями — перманганатометрия, иодометрия,
хроматометрия, броматометрия, цериметрия и др.
В перманганатометрии, например определяемые25
ионы титруют раствором перманганата калия, в иодо-
метрии пользуются растворами иода и тиосульфата
натрия и т. д.Электрогравиметрический анализ.Через исследуемый раствор пропускают электриче¬
ский ток определенного напряжения. Находящиеся
в растворе ионы металлов восстанавливаются элект¬
рическим tokoiM до металлического состояния. Выде¬
лившийся металл взвешивают и по найденной массе
вычисляют содержание данного элемента в пробе.
Электрогравиметрический метод анализа применяют
для определения меди, кадмия, цинка, кобальта, ни¬
келя, свинца, серебра, золота и некоторых других
металлов.Каталитические методы анализа осно¬
ваны на измерении скорости химической реакции
(см. гл. 23).Полярографический метод анализа —
см. гл. 25.Кулонометрический метод анализа.
Количество вещества определяют по количеству элек¬
тричества, которое необходимо затратить для элек¬
трохимического превращения анализируемого веще¬
ства. Результаты анализа находят по закону Фарадея-.где М — количество определяемого элемента или иона; F—чис¬
ло Фарадея (96 500 кулонов); А — атомная масса элемента; п —
количество электронов, принимающих участие в электрохимиче¬
ском превращении данного элемента или иона; / — сила тока;
t — время электролиза, с; Q — количество электричества, К.Из приведенных примеров видно, что реакции ком¬
плексообразования применяют, в частности, для раз¬
деления элементов методами осаждения и экстракции,
когда происходит переход веществ из одной фазы в
другую. Существует классификация методов разделе¬
ния, основанная на использовании различных типов
фазовых равновесий.Фазовые равновесияФазой называют часть системы, отделенную от дру¬
гих ее частей определенной поверхностью раздела,
причем внутри каждой фазы вещество однородно в
физическом и химическом отношениях.26
Известны такие типы равновесий между фазами:
жидкость—газ (Ж—Г); твердое вещество — газ
(Т—Г); жидкость (1)—жидкость (2) (Ж1 — Ж2);
жидкость — твердое вещество (Ж—Т) Смесь двух
или нескольких газов не имеет повеэхности раздела
и представляет собой одну фазу.Равновесие Ж — Г или Т — Г. Выделение веществ
в газовую фазу часто используют в анализе. Методы,
основанные на таких равновесиях, немногочисленны,
но отличаются большей специфичностью по сравнению
с другими методами.Известны следующие методы, основанные на рав¬
новесии этих типов: выделение определяемых элемен¬
тов в виде летучих соединений с кислородом, напри¬
мер воды, диоксида углерода, серы в виде S02 или
S03); выделение элементов в виде летучих соедине¬
ний с галогенами, например отгонка AsCl3, СгС13,
GeCl4, SbCl3 и др.; выделение элементов в виде лету¬
чих соединений с водородом, например AsH3 и др.;
метод газовой хроматографии, в котором некоторые
неорганические вещества переводят в газообразное
состояние, например кремний, германий, мышьяк,
олово, бериллий определяют в виде летучих гидри¬
дов после их отделения от многих элементов, не об¬
разующих летучих соединений с водородом.Равновесие Ж1 — Ж2 наблюдается в методе экс¬
тракции, когда элементы переводят члще всего из вод¬
ной фазы в фазу органического растворителя. Обыч¬
но соединения экстрагируются в фоэме комплексов,
но известны и другие вещества, способные к такому
переходу.При электролизе с ртутным катодом также осу¬
ществляется этот тип равновесия.Равновесие Ж — Т характерно для процессов оса¬
ждения и процессов выделения на поверхности твер¬
дой фазы.На процессах осаждения — выделении определяе¬
мого вещества из водного раствора в твердую фазу —
основан один из наиболее старых и хорошо разрабо¬
танных методов анализа — гравиметрический анализ.
В гравиметрическом анализе к раствору определяе¬
мого вещества прибавляют раствор осадителя, выде¬
лившийся осадок после отделения от маточного рас¬
твора фильтрованием и высушивания взвешивают или27
сначала переводят в какое-либо другое соединение
(весовую форму) определенного состава. Один из
примеров — определение сульфат-ионов в виде суль¬
фата бария:H2S04 + ВаС12 = BaS04^ + 2HCI.Гравиметрические методы широко применяют для
определения многих веществ.В титриметрическом анализе часто используют
процессы осаждения — осадительное титрование. На¬
пример, ионы хлора титруют раствором нитрата се¬
ребра:NaCl + AgN03 = AgCl| + NaN03.Раствор AgNOa прибавляют до тех пор, пока не
будет достигнуто эквивалентное соотношение реаги¬
рующих веществ и практически весь хлорид не пре¬
вратится в осадок хлорида серебра.Перевод веществ из водной фазы в твердую ши¬
роко применяют для качественного обнаружения мно¬
гих ионов. Так, посредством раствора дихромата ка¬
лия легко обнаружить присутствие в растворе ионов
бария, так как при этом образуется характерный жел¬
тый осадок хромата бария. Образование желтого
осадка РЫг свидетельствует о присутствии в растворе
ионов свинца; выделение синего осадка KFe[Fe(CN)6]
при действии на исследуемый раствор K.4[Fe(CN)6] —
признак присутствия ионов железа (III) и т. д.На процессах осаждения основаны многие методы
разделения, которые широко применяют в качествен¬
ном и в количественном анализе. Так, действуя на ис¬
следуемый раствор раствором сульфида аммония мо¬
жно выделить в осадок нерастворимые в воде суль¬
фиды никеля, кобальта, марганца, цинка, железа и
отделить их таким способом от растворимых в данных
условиях солей бария, стронция, кальция и магния.Хроматографический анализ — один из методов
разделения, в котором используют способность осад¬
ков к адсорбции на их поверхности посторонних ио¬
нов (см. гл. 28).Образование твердой фазы (осадка) представляет
собой сложный процесс, который сопровождается по¬
бочными явлениями. Одно из них — соосаждение. Оно58
состоит в том, что осадок при образовании захваты¬
вает из раствора посторонние растворимые вещества.
Так, при осаждении сульфата бария может захваты¬
ваться из раствора перманганат калия, сульфат же¬
леза, хлорид кальция и другие вещества, хорошо рас¬
творимые в воде. При осаждении кремниевой кислоты
последняя захватывает из раствора соли железа,
алюминия, титана. Соосаждение используют как один
из методов концентрирования, т. е. извлечения из
большого объема раствора небольших количеств оп¬
ределяемых элементов, и для их отделения от основ¬
ного компонента пробы. Метод концентрирования
иногда называют также методом осуждения с кол¬
лектором.Подведем итог. К методам анализа относят:1) гравиметрический; 2) титриметрические (кислот¬
но-основной, осаждения, образовании комплексных
соединений, окисления — восстановления); 3) оптиче¬
ские (фотометрия, спектрофотометркя, турбидимет-
рия, нефелометрия и люминесценция] ; электрохими¬
ческие (потенциометрия, полярография, кулономет-
рия, электрогравиметрия); каталитические (кинети¬
ческие) .Нередко первые два метода называют химиче¬
скими (классическими) методами анализа, а осталь¬
ные— физико-химическими методами, хотя принци¬
пиальной разницы между теми и другими не су¬
ществует.К методам разделения относят: 1) осаждение,2) экстракцию, 3) хроматографию, 4) ионный обмен.К методам концентрирования относят: 1) осажде¬
ние, 2) экстракцию, 3) цементацию, 4| отгонку.§ 3. ФИЗИЧЕСКИЕ МЕТОДЫ АНАЛИЗАХарактерная особенность физических методов ана¬
лиза заключается в том, что в них непосредственно
измеряют какие-либо физические пара летры системы,
связанные с количеством определяемое элемента, без
предварительного проведения химической реакции.
Это не означает, однако, что при определении элемен¬
тов физическими методами химические реакции совер¬
шенно не происходят. Разложение пробы и подготов¬
ку вещества к анализу нередко проводят с помощью29
химических методов. Химические реакции могут про¬
ходить и без введения дополнительного раствора под
действием высокой температуры, электрического раз¬
ряда и т. д. Тем не менее перед заключительным оп¬
ределением нет необходимости прибегать к химиче¬
ским реакциям, и то время как в химических методах
анализа обязательной предварительной операцией яв¬
ляется действие некоторого химического реагента.Физические методы анализа включают три глав¬
ные группы методов: методы, основанные на взаимо¬
действии излучения с веществом или на измерении
излучения вещества; методы, основанные на измере¬
нии параметров электрических и магнитных свойств
вещества; методы, основанные на измерении плотно¬
сти или других параметров механических или моле¬
кулярных свойств вещества.Методы, основанные на взаимодействии излучения
с веществом или на измерении излучения веществаМетоды, основанные на энергетических переходах
внешних валентных электронов атомов включают
атомно-эмиссионные и атомно-абсорбционные ана¬
лизы.Атомно-эмиссионный анализ. 1. Фото¬
метрия пламени. Анализируемый раствор распыляют
в пламени газовой горелки. Под влиянием высокой
температуры пламени атомы переходят в возбужден¬
ное состояние. Внешние валентные электроны перехо¬
дят на более высокие, чаще всего соседние с основ¬
ным, энергетические уровни; обратный переход
электронов на основной энергетический уровень со¬
провождается излучением, длина волны которого за¬
висит от того, атомы какого элемента находились
в пламени. Интенсивность излучения при определен¬
ных условиях пропорциональна количеству атомов
элемента в пламени, а длина волны излучения харак¬
теризует качественный состав пробы. Метод фотомет¬
рии пламени чаще всего применяют для качествен¬
ного обнаружения и количественного определения
легко возбуждающихся щелочных и щелочноземель¬
ных металлов.2. Эмиссионный спектральный анализ. Пробу вво¬
дят в пламя вольтовой дуги или конденсированной30
искры. Под влиянием высокой тем тературы атомы
элементов переходят в возбужденное состояние. Тем¬
пература дуги или искры выше температуры пламени,
что приводит к возбуждению атомов большого числа
элементов; кроме того, электроны при возбуждении
переходят не только на ближайшие к основному, но
и на более отдаленные энергетические уровни. По¬
этому излучение представляет собой сложную смесь
световых колебаний самых разнообразных длин волн.
Это излучение — так называемый эмиссионный
спектр — разлагают на составные ^сти специаль¬
ными приборами (спектрографами) и фотографируют.
Сравнение положения и интенсивности отдельных ли¬
ний спектра с линиями соответствующего эталона с
точко известным содержанием отдельных элементов
дает возможность определить качественный и количе¬
ственный состав пробы.Атомно-абсорбционная спектромет¬
рия — метод атомной абсорбции. Он основан на из¬
мерении поглощения света определенной длины вол¬
ны, излучаемого специальным источником, невозбуж¬
денными атомами определяемого элемента. Источник
дает так называемое резонансное излучение, т. е. из¬
лучение, соответствующее переходу электронов на
наинизшую орбиталь с наименьшей энергией с бли¬
жайшей к ней орбитали с более высоким уровнем
энергии. Кванты света резонансной частоты перево¬
дят электроны атомов определяемого элемента в пла¬
мени в возбужденное состояние, т. е на ближайший
к основному более высокий энергетический уровень.
Уменьшение интенсивности света при прохождении
его через пламя пропорционально количеству невозбу¬
жденных атомов в нем. Поэтому предел обнаруже¬
ния в методе атомной абсорбции значительно ниже,
чем у двух предыдущих методов анализа.В методе атомной абсорбции применяют горючие
смеси с температурой до 3100 °С (известен и непла¬
менный вариант метода), что обеспечивает возмож¬
ность определения значительно большего количества
элементов, чем в методе фотометрии пламени. Атом¬
но-абсорбционный метод характеризуется также
высокой селективностью, определению данного эле¬
мента, как правило, не мешают многие другие эле¬
менты, содержащиеся в пробе.31
Методы, основанные на возбуждении глубинных
электронов атомов — рентгенофлуоресцентный и рент-
геноэмиссионный методы анализа. В более распро¬
страненном рентгенофлуоресцентном методе пробу
подвергают действию излучения рентгеновской труб¬
ки. Атомы пробы возбуждаются: внутренние элек¬
троны, находящиеся на ближайшей к ядру атома ор¬
битали, так называемые К-электроны, выбиваются из
атома. Их место занимают электроны с более отда¬
ленных от ядра орбиталей. Переход этих электронов
сопровождается возникновением вторичного рентге¬
новского излучения, длина волны которого связана
функциональной зависимостью с атомным номером
элемента. Измерение длины волны вторичного излу¬
чения дает возможность установить, какие именно
элементы входят в состав пробы; интенсивность же
вторичного излучения зависит от количества данного
элемента в пробе, т. е. ее измерение является осно¬
вой количественного рентгенофлуоресцентного метода
анализа.Методы, основанные на ядерных реакциях—ра-диоактивационный, или (его главная часть)—-ней¬
тронно-активационный метод анализа. Нейтронно-ак¬
тивационный метод возник после открытия атомной
энергии и создания действующих атомных реакторов.
Принцип метода заключается в следующем. Анализи¬
руемый материал подвергают действию нейтронного
излучения в атомном реакторе или посредством ней¬
тронного генератора. При взаимодействии нейтронов
с ядрами элементов происходят ядерные реакции и
образуются радиоактивные изотопы всех элементов,
входящих в состав пробы. Затем пробу переводят в
раствор и разделяют элементы химическими мето¬
дами. Завершающим этапом определения является
измерение интенсивности радиоактивного излучения
каждого элемента пробы.Параллельно анализируют эталонную пробу с точ¬
но известным содержанием отдельных элементов, ко¬
торое находят предварительно химическими методами
анализа. Эталон также облучают в реакторе потоком
нейтронов и обрабатывают химическими методами,
которые применяли при разделении элементов анали¬
зируемой пробы. Сравнивая интенсивность радиоак¬
тивного излучения отдельных фракций эталонной про¬32
бы и анализируемого материала, делают вывод о ко¬
личественном содержании элементов.Предел обнаружения элементов радиоактивацион-
ным методом очень низкий: удается определять эле¬
менты, содержание которых составляет 10~8—10-10 %.Методы, основанные на измерении паоаметров
электрических и магнитных свойств веществаКондуктометрический метод основан на измерении
электропроводности растворов или газов. Так, кон¬
центрацию серной кислоты в растворе определяют,
сравнивая электропроводность этого раствора с дан¬
ными таблицы, в которой содержатся значения элек¬
тропроводности растворов серной кис/оты различных
концентраций.Известен также термокондуктометрический метод
анализа газов. В газовую смесь поме цают нагретую
платиновую проволоку и измеряют ее электропро¬
водность. Последняя зависит от температуры прово¬
локи, которая в свою очередь изменяется с теплопро¬
водностью отдельных компонентов газе вой смеси. Так,
теплопроводность диоксида углерода очень сильно
отличается от теплопроводности азота или оксида
углерода. Поэтому изменение содержания СО2
в смеси с N2 и СО влияет на электропроводность
платиновой проволоки. На этом принципе основаны
различные конструкции термокондуктометрических
газоанализаторов.Потенциометрический метод. Следует различать
метод прямой потенциометрии и метод потенциомет¬
рического титрования. Во втором методе определяе¬
мое вещество титруют подходящим рабочим раство¬
ром, измеряя в процессе титрования потенциал какого-
либо электрода. Этот метод относится к химическим
методам анализа, так как он основан на проведении
химической реакции в растворе. В ме'оде же прямой
потенциометрии химическую реакцию не проводят: из¬
меряют потенциал электрода, погруженного в испы¬
туемый раствор, и затем по соответствующему урав¬
нению вычисляют концентрацию определяемых ионов.
Наиболее распространенный метод прямой потенцио¬
метрии— определение pH раствором посредством
стеклянного или какого-либо другого электрода.2 Аналитическая химия33
Термоэлектрический метод. Термоэлектродвижу¬
щая сила, возникающая при нагревании места сопри¬
косновения стали с другим металлом, сильно изме¬
няется в зависимости от содержания углерода и крем¬
ния в стали. На этом принципе основано действие раз¬
личных термоэлектрических карбометров.Масс-спектральный метод. Сложные газовые сме¬
си разделяют на составные части, подвергая их дей¬
ствию сильных электрических и магнитных полей.
Разделение происходит в соответствии с атомными
или молекулярными массами отдельных компонентов
смеси. Метод применяют при исследовании смесей
изотопов, смесей инертных газов или сложных смесей
органических веществ.Методы, основанные на измерении плотности
или других параметров механических
или молекулярных свойств веществДенситометрия — метод, основанный на измерении
плотности. Плотность растворов, газовых смесей,
сплавов зависит от концентрации определяемого ве¬
щества. Для анализа используют таблицы, составлен¬
ные на основании исследования зависимости плотно¬
сти от содержания в данном материале определяе¬
мого компонента. Плотность измеряют ареометрами,
пикнометрами, поплавками и другими приборами. Ме¬
тод применяют для определения концентрации раство¬
ров спиртов, кислот, солей, оснований и др.Другие методы. Для нахождения содержания
(в %) какой-либо составной части материала изме¬
ряют вязкость (анализ масел), поверхностное натя¬
жение (анализ растворов), скорость звука (анализ
газов). Для установления чистоты синтезированных
препаратов измеряют температуру плавления или ки¬
пения и др.§ 4. ВЫБОР МЕТОДА АНАЛИЗАВыбор наилучшего метода анализа диктуется мно¬
гими соображениями и представляет трудную задачу,
решение которой требует от химика высокой квали¬
фикации. Имеет значение химический состав анализи¬
руемого образца, оснащенность лаборатории необхо¬34
димой аппаратурой, наличие соответствующих реаген¬
тов и многое другое. Наиболее просто решается за¬
дача количественного определения одного элемента,
содержащегося в приготовленном растворе. Необхо¬
димо иметь хотя бы приблизительную информацию об
интервале концентраций этого элемента в растворе.
Если концентрация элемента очень мала, для анализа
пригодны почти все физические методы анализа, а из
химических — спектрсфотометрические, электрохими¬
ческие (полярография, кулономстрня), кинетические
и некоторые другие.Если определяемый элемент является основным
компонентом анализируемого материала, т. е. его со¬
держание велико, следует применять гравиметриче¬
ский или титриметрический методы. Существуют
также методы, которыми можно определять как ма¬
лые, так и большие количества, например рентгено¬
флуоресцентный метод.Выбор метода зависит также от того, какое коли¬
чество образцов необходимо проанализировать. Если
анализируют единичную пробу или небольшое число
проб, нет смысла применять физические методы, луч¬
ше воспользоваться, например, гравиметрическим или
спектрофотометрическим. В физических методах мно¬
го времени занимает предварительная калибровка ап¬
паратуры, построение градуировочных графиков, не¬
обходимо также иметь стандартные образцы для
сравнения и т. д. Поэтому применение этих методов
оправдано, если необходимо анализировать большую
серию проб приблизительно одинакового состава. Еди¬
ничные анализы лучше проводить химическими мето¬
дами.Так, большие количества алюминия следует опре¬
делять гравиметрическим или титриметрическим ме¬
тодом. Первый из них применяют, если необходима
высокая точность анализа и если длительность ана¬
лиза не имеет большого значения. В противном слу¬
чае лучше оттитровать алюминий комплексонометри-
чески — это быстрее, хотя точность анализа меньше.Если необходимо определить малые содержания
алюминия в большой серии однотипных образцов, ре¬
комендуется воспользоваться одним из физических
методов, однако выбор метода лимитирован наличием
в лаборатории соответствующей аппаратуры.2*35
Выбор метода осложняется, если анализируют ма¬
териал сложного состава, т. е. если он содержит
много сопутствующих элементов в различных коли¬
чественных соотношениях. Приходится учитывать хи¬
мическую природу сопутствующих элементов, их ко¬
личество и близость свойств с определяемым элемен¬
том. Так, микроколичества цинка легко определить
полярографически, однако большие количества меди
и кадмия мешают, потому что эти элементы восста¬
навливаются раньше цинка. Их необходимо предва¬
рительно отделить. Лучше применить экстракционно¬
фотометрический метод с дитизоном: в присутствии
тиосульфата натрия окрашенный комплекс цинка с
дитизоном экстрагируется тетрахлоридом углерода,
а медь и кадмий остаются в водном растворе в виде
тиосульфатных комплексов; окрашенный экстракт
фотометрируют.Гравиметрическое определение железа возможно
путем осаждения Fe(OH)3 аммиаком и последующего
взвешивания Ре2Оз, однако этот метод нельзя при¬
менить в присутствии, например, титана, который то¬
же образует нерастворимый гидроксид. В качестве
осадителя целесообразно выбрать 8-гидроксихинолин.
Он полностью осаждает железо уже при pH = 3, в то
время как титан остается в растворе.Имеет значение и требуемая точность анализа.
Высокая точность достигается обычно за счет боль¬
шого увеличения продолжительности анализа, обус¬
ловленного проведением многих дополнительных опе¬
раций. Так, кальций в силикатах нередко определяют
после осаждения в виде оксалата кальция; в присут¬
ствии магния последний в большей или меньшей сте¬
пени соосаждается с СаС204. Ь'сли высокая точность
не нужна, тогда соосаждением небольших количеств
магния можно пренебречь. Наоборот, при очень точ¬
ных определениях следует уменьшить соосаждение
магния, для чего осадок переосаждают, на это затра¬
чивается дополнительное время.Из приведенных примеров видно, что и в наибо¬
лее простых случаях, когда анализируемый материал
содержит всего два или три мешающих элемента, при
выборе метода анализа необходимо принимать во
внимание много различных обстоятельств. Но многие
реальные материалы имеют еще более сложный со¬36
став. Так, даже при сокращенном анализе силикатов
в них обычно определяют Si02, Ре2Оз, А120з, ТЮ2,
CaO, MgO, S03, К20, Na20. В некоторых специаль¬
ных случаях определяют еще Zr02, Cr203, V2Os, NiO,
СоО, BaO, SrO, BeO, CuO, F-, С1_, В2Оэ и др. В ме¬
таллической меди определяют примеси висмута, сурь¬
мы, мышьяка, железа, никеля, свинца, олова, серы
и др. Поэтому нельзя дать общего рецепта выбора ме¬
тода анализа: у исполнителя должна быть широкая
химическая эрудиция и твердое знание преимуществ
и ограничений доступных методов анализа.ГЛАВА 2ХИМИЧЕСКОЕ РАВНОВЕСИЕ
В ГОМОГЕННОЙ СИСТЕМЕ§ 1. ЗАКОН ДЕЙСТВИЯ МАССЗакон действия масс открыли в 1884 г. норвежские
ученые К. Гульдберг и П. Вааге. Это — фундамен¬
тальный закон химии. В аналитической химии закон
действия масс является теоретической основой многих
методов анализа. Он устанавливает количественные
соотношения между веществами, участвующими в об¬
ратимой химической реакции после достижения со¬
стояния равновесия. Обратимыми называют такие хи¬
мические реакции, которые идут не только в прямом,
но и в обратном направлении; равновесие устанавли¬
вается после того, как скорости прямой и обратной
реакций становятся одинаковыми.Рассмотрим вывод и формулировку закона дейст¬
вия масс на примере реакции между частицами А и
В, при взаимодействии которых образуются частицы
С и D:А + В С + D.Под термином «частицы» подразумеваются ионы,
молекулы или атомы элементов, находящиеся в рас¬
творе или в газовой фазе.Скорость реакции между А и В, т. е. количество
частиц, прореагировавших за единицу времени, зави¬37
сит от их химической природы, температуры и актив¬
ности. Для вывода закона действия масс учитывают
зависимость скорости реакции от активности частиц
А и В. Активность а определяется способностью ча¬
стиц в своем беспрерывном хаотическом движении
сталкиваться одна с другой, в результате чего проис¬
ходит химическое взаимодействие. Очевидно, чем
больше активность частиц, т. е. чем больше столкно¬
вений происходит за единицу времени, тем больше
скорость химической реакции. Отсюда следует, что
активность возрастает с увеличением общего количе^
ства частиц в системе, т. е. с ростом их концентрации.
Поэтому количественной мерой активности частиц
может при некоторых условиях служить их концент¬
рация:а = С.Это равенство справедливо только для очень раз¬
бавленных водных растворов или для идеальных газов.Таким образом, скорость реакции между А и В
пропорциональна произведению их концентраций:»,=МА][В]. (2.1)Через некоторе время в системе появляются ча¬
стицы С и D, которые, вследствие обратимости реак¬
ции, реагируют между собой с образованием частиц
А и В. Скорость обратной реакции пропорциональна
произведению концентраций С и D:v2 = k2 [С] [D], (2.2)Скорость реакции v1 из-за уменьшения концентра¬
ции частиц А и В со временем падает. В то же время
скорость обратной реакции v2 возрастает, так как кон¬
центрация С и D постепенно увеличивается. Уравне¬
ния (2.1) и (2.2) представляют собой математиче¬
скую формулировку закона действия масс: скорость
химической реакции прямо пропорциональна концен¬
трациям реагирующих веществ. В некоторый момент
времени скорости прямой и обратной реакции станут
одинаковыми:U|=r2 (2.3)и будет достигнуто состояние равновесия.38
Следовательно,MA][B] = Aa[C]|Dl. (2.4)Откуда= (2.5)[А] [В] k2Уравнение (2.5) представляет собой математиче¬
скую формулировку следствия из закона действия
масс: для обратимой химической реакции в состоянии
равновесия произведение концентраций продуктов ре¬
акции, деленное на произведение концентраций ис¬
ходных веществ, есть величина постоянная при по¬
стоянной температуре *; ее обозначают символом К
и называют константой равновесия.Состояние равновесия является динамическим, это
значит, что реакция между А и В и между С и D про¬
должается. Однако при равновесии сколько частиц
А и В реагирует в единицу времени с образованием С
и D, столько же частиц С и D взаимодействует за это
же время с образованием частиц А и В.В более общем случае, когда стехиометрические
коэффициенты отличаются от единицы, т. е. для реак¬
цииаА + йВ сС 4- dD (2.6)закон действия масс записывают следующим обра¬
зом:[C]c'[D]d■- —г = К. (2.7)[А] [В]Закон действия масс может быть выведен исходя
из термодинамических соображений. Протекание рас¬
смотренной выше реакции сопровождается не только
изменением концентрации реагирующих частиц, но
также изменением энергетических параметров систе¬
мы. Из термодинамики известно, что энергия Гиббса
G равна разности между энтальпией Н и произведе¬
нием абсолютной температуры Т на энтропию S:G = Н — TS. (2.8)* Это следствие обычно отождествляют с формулировкой :а-
мого закона действия масс.39
В случае растворенных р.еществ она зависит не
только от температуры и давления, но также от их
концентрации:G = О'0 + RT In С, (2.9)Химические реакции при постоянных давлении и
температуре протекают до тех пор, пока сумма сво¬
бодных энергий продуктов реакции не станет равной
сумме свободных энергий оставшихся непрореагиро¬
вавшими исходных веществ.Рассмотрим обратимую реакциюА + В C + D (2.10)в растворе.Система придет в состояние равновесия, когдаGA + GB = GC + GD, (2.11)где G с соответствующими индексами — энергия частиц А, В, С
и D.Подставляя в уравнение (2.11) вместо энергий их
значения из уравнения (2.9), получаемОд + RT In [А] + G°B + RT In [В] == G°C + RT In [С] + G°d + RT In [D].ОткудаRT In [A] + RT In [B] - RT In [C] - RT In [D] =ИЛИ y-iO i /•">() /"*0 о— oc + oD — о A — oBДО0 , [Cl ID]RT ~ | А] (В] ’ ^где AG° — разность между суммой энергий реагирующих веществ
А и В и суммой энергий продуктов реакции С и D.Имея в виду, что выражение в левой части уравне¬
ния (2.12) есть величина постоянная при постоянной
температуре, получаем1СПР1 .= * (2 13)[А][В] А’ ’где К—константа равновесия.Закон действия масс в своей концентрационной
форме [(2.5) или (2.7)] справедлив только для раз¬
бавленных водных растворов или для идеальных га¬40
зов. Рассмотрим причины отклонения от закона в вод¬
ных растворах электролитов. При увеличении концен¬
трации реагирующих частиц между ними начинают
проявляться силы межиониого или межмолекуляр-
ного взаимодействия, замедляющие их движение. По¬
этому при возрастании концентрации число столкно¬
вений, а следовательно и вероятность взаимодействия
между А и В увеличивается не прямо пропорцио¬
нально концентрации, а в меньшей степени, иными
словами, концентрация перестает быть количествен¬
ной мерой активности; последняя становится меньше
концентрации:а<С. (2.14)Аналогичное явление происходит в том случае, ко¬
гда в раствор, содержащий ионы реагирующих эле¬
ментов А и В вводят посторонний сильный электро¬
лит, ионы которого не вступают в химическое взаимо¬
действие с А и В. Этот важный для аналитической хи¬
мии случай необходимо рассмотреть более подробно.Предположим, что протекает реакция
А + В =«=fe С + D,где реагирующие частицы представлены ионамиFeCl2++ SCN~ *=fc FeSCN2++ СГ. (2.15)В очень разбавленных растворах, например при
миллимолярных концентрациях компонентов, межион-
ное взаимодействие между ними еще незаметно, оно
не оказывает тормозящего действия на движение ио¬
нов, и поэтому активности частиц а можно считать
численно равными их концентрации а = С.Введем в раствор сильный электролит, например
KN03, в большом избытке, ионы которого К+ и NO3
не реагируют ни с одним из ионов, обозначенных
в уравнении (2.15). Несмотря на это, скорость движе¬
ния реагирующих ионов, а следовательно, количество
столкновений между ними заметно уменьшится. При
той же самой концентрации активность ионов станет
меньше их концентрации. Эффект замедления вызвантем, что ионы К+ и NO3 вследствие сил электроста¬
тического притяжения образуют вокруг реагирующих
ионов оболочки из ионов противоположного знака;
образуется так называемая «ионная атмосфера»,41
которую можно схематически изобразить следующим
образом:N03NO34.N03К4'к4-киFeCl2+ ,SCN'МОаNO3NOaК"к.+ "t\'OiNO3. NO3к*к+-с->FeSCN+сгNOiNO3NOaк*Теперь уже активность ионов FeCl2+, SCN-,
FeSCN2+ и Cl- не равна их концентрации:а<С (2.16)и в этом случае в выражение закона действия масс
следует подставлять вместо концентрации реагирую¬
щих ионов их активности.Степень отклонения системы от идеальности вслед¬
ствие увеличения концентрации реагирующих ионов
или введения посторонних сильных электролитов ха¬
рактеризуется коэффициентом активности /. Это чис¬
ло (как правило, меньше единицы), на которое нужно
умножить концентрацию вещества (молекул или ио¬
нов), чтобы полученное произведение можно было
приравнять к активности:a = fC. (2.17)Понятие об активности и о коэффициентах актив¬
ности более глубоко и подробно рассмотрено в курсе
физической химии. Там же изложена теория Дебая и
Гюккеля, на основании которой выводятся уравнения
для вычисления коэффициентов активности с учетом
ионной силы растворов.Ионная сила растворов ц представляет собой по¬
ловину суммы произведений концентрации ионов С на
квадрат их заряда Z.clz2l + ctz?,+ ... +ctz}Ц = —~ ' (2',8)Связь коэффициентов активности ионов с ионной
силой выражается уравнениемlgt=—(2.19)
1 -(- Ва У М-42
где А и В — константы, зависящие от природы растворителя
(для воды при 20 °С /1= 0,5046, В = 0,3276), а — параметр
ионного размера, соответствующий диаметру гидратиропаиного
иона, А.В табл. 2.1 приведены значения а для некоторых
неорганических ионов.Таблица 2.1. Диаметры гидратированных, ионовНеорганические ионыaНеорганические ионыaRb+, Cs\ Tl+, Ag+2,5Sr2*, Ba2+, Cd2+, Hg2+,5К\ СГ, Вг“, Г, NO", NO",3,0S2-, Fe(CN)j'ОН', F", SCN", HS", СЮ'Li6сю;, ВгО", ю;, Мпо;,3,5Mg2+, Be2*8Hg2+, sosao|-. s2Oj_SeO2’, CIO;, PO]-,4,0Al3+, Fe3+, Cr3+, Sc3+,9Fe(CN)»-La3+, In3", H+Na+, НСО3", H2PO;, Pb2t,4,5Th4t, Zr4t, Ce‘l+, Sn4+Исо;2, Mno2-3 1 4Для ионов с значением а в пределах 2,5—4,0 А
произведение Ва близко к единице и уравнение (2.19)
можно записать в более простой форме:, t 0,522л/(х'“'-777Г ,г20)Уравнение (2.20) справедливо для растворов с
ионной силой в интервале 0 < р, < 0,1.Если ионная сила раствора находится в пределах0,1 < р. <1,0 — наиболее правильные результаты
дает вычисление по следующему уравнению:lg f = — 0,5Z2 ( -0,2ц\ (2.21)V 1 + Vm- 'Предположим, что равновесные концентрации
каждой реагирующей частицы в уравнении (2.15) не
превышают 0,001 М, а концентрация введенного элек¬
тролита KN03 равна 0,1 М. В этом случае при43
подсчете ионной силы раствора можно не принимать
во внимание концентрации реагирующих ионов, так
как они намного меньше концентрации KN03. По¬
этому^ = ———— = 0,1. (2.22)
Откуда для двухзарядных ионов FeCl24- и FeSCN2+[g / = - 0,5 • 4 = — 0,5i + Vo.iИf = 10~0S = 10“1 • ю°'5 = 0,3,
а для однозарядных частиц SCN- и С1~lg f = — 0,5 V°;U = — о,12И-л/0,1и/= Ю_0'12= ю-1 • Ю0,88 = 0,76.В выражение (2.5) закона действия масс для урав¬
нения реакции (2.15) следует подставить вместо рав¬
новесных концентраций соответствующих ионов ак¬
тивности, т. е. произведения концентраций на найден¬
ные коэффициенты активности а = fCu:асап т= КТ. (2.23)Константу равновесия К, выраженную через кон¬
центрации частиц в соответствии с уравнением (2.7),
называют концентрационной константой, ее значение
зависит не только от температуры, но также от ион¬
ной силы раствора. Величину Л7 в уравнении (2.23)
называют термодинамической константой равновесия,
и ее значение не зависит от ионной силы раствора.В аналитической химии чаще используют концен¬
трационные константы, однако для расчетов приме¬
няют термодинамические константы, приведенные в
таблицах, поскольку обычно можно принимать К =
= Л7.Закон действия масс указывает способы изменения
хода химической реакции. Так, если необходимо прак¬
тически полностью перевести вещество А в продукты
реакции С и D, следует увеличить концентрацию реа-44
гента В. Из уравнения (2.5) видно, что это повлечет
сдвиг состояния равновесия в правую сторону и
уменьшение равновесной концентрации А. Для дости¬
жения этой же цели можно вывести из сферы взаимо¬
действия один из продуктов реакции С или D, связав
его в малорастворимое, комплексное или малодиссо-
циирующее соединение или изменив степень его окис¬
ления.§ 2. ПРИМЕРЫ ПРИМЕНЕНИЯ ЗАКОНА ДЕЙСТВИЯ МАССОдин из способов количественного определения свин¬
ца состоит в осаждении PbSO^Pb2t + SC>r PbS04. (2.24)При стехиометрических соотношениях Pb?+ и SO4
некоторая часть свинца остается в растворе из-за за¬
метной растворимости осадка в воде. Для того чтобы
полностью перевести весь свинец в осадок, т. е. сдви¬
нуть равновесие вправо, вводят в раствор избыток
серной кислоты или сульфата натрия.Для отделения алюминия и железа от магния и
некоторых других двухвалентных металлов исполь- .
зуют явление гидролиза: :А13* + 3H20 :*=£ А1(ОН)3| + ЗН+. (2.25)Однако эта реакция идет вправо только в очень не¬
значительной степени. Перевести весь алюминий в
осадок можно, связав ионы водорода в недиссоцииро-
ванные молекулы уксусной кислоты. Для этого к рас- )
твору прибавляют ацетат натрия: jСНзСОО" + Н+ СНзСООН. (2.26) ■Концентрация ионов водорода резко уменьшается и
весь алюминий осаждается в виде гидроксида или
основного ацетата.Сульфид ртути с трудом растворяется в азотной
кислоте, но в присутствии хлороводородной кислоты
реакция протекает легко. Это вызвано тем, что ионы
хлора связывают ионы ртути в малодиссоциирован-
ный хлорид ртути:3HgS + 2NO3 + 6СГ + 8Н++=± 3HgCl2 + 3S + 2NO + 4Н20. (2.27)45
Гидроксид никеля практически нерастворим
в воде, его насыщенный раствор содержит очень
мало свободных ионов никеля:Ni(OH)a =f=* Ni2t + 20Н". (2.28)Однако гидроксид никеля легко растворяется в вод¬
ном растворе аммиака, который образует с ионами ни¬
келя комплексный ион — аммиакат никеляNi2+ + 6NH3 Ni(NH3)r, (2.29)вследствие чего концентрации продукта реакции —
ионов никеля—резко уменьшается и равновесие ре¬
акции (2.28) смещается вправо.Бромид серебра мало растворим в воде, концен¬
трация ионов брома в насыщенном растворе AgBr
ничтожна:AgBr Ag+ + Вг~. (2.30)Все ионы брома, связанные в AgBr, легко переве¬
сти в раствор, если обработать осадок сероводоро¬
дом:2Ag* + H2S = Ag2S + 2Н*. (2.31)При такой обработке ионы серебра образуют нерас¬
творимый сульфид серебра и выводятся из сферы ре¬
акции (2.30), концентрация ионов серебра сильно
уменьшается и равновесие реакции (2.30) полностью
смещается вправо.Константы равновесия отдельных процессов опре¬
деляются химической индивидуальностью реагирую¬
щих веществ. Есть реакции, которые идут практиче¬
ски полностью слева направо и характеризуются
большими константами равновесия. Так, реакция
между ионами серебра и аммиакомAg+ + 2NHj Ае(\'Н3)£ (2.32)идет практически до конца в сторону образования ам¬
миаката серебра, константа равновесия велика— 107.Многие другие реакции не доходят до конца, и
константы равновесия таких реакций невелики. На¬
пример, реакция между цианидным комплексом кад¬
мия и аммиакомCd(CN)2- + 4NH3 ^ Cd(NHs)r + 4CN~ (2.33)46
идет в очень незначительной степени, соответствую¬
щая константа равновесия очень мала.Константы равновесия отдельных типов химиче¬
ских реакций имеют разный смысл и названия. Так,
в кислотно-основных реакциях константа равновесия
представлена константами диссоциации кислот или
оснований, напримерСНзСООН СНзСОО' + н+,[СНзСОО-] [Н+][СНзСООН! к- (234)В реакциях комплексообразования константа рав¬
новесия— это константа устойчивости комплексного
соединения или иона:Ag+ + 2NH3 Ag(NH3)+. (2.35)Ее обозначают символом |5 и записывают следую¬
щим образом:[Ag(NH3)2+][Ag+Р2. (2.36)Константу равновесия, характеризующую малорас¬
творимое соединение, называют произведением рас¬
творимости. Так, для процессаAgBr Ag* + Вг- (2.37)эта константа произведения растворимости имеет вид
[Ag+][Br-] = /C = nP. (2.38)Для количественных расчетов необходимо знать
численные значения констант равновесия соответст¬
вующих реакций. Для многих реакций различных ти¬
пов — комплексообразования, окислительно-восстано¬
вительных, кислотно-основных — они найдены путем
экспериментального определения концентрации реаги¬
рующих частиц в равновесных системах или рассчи¬
таны теоретически, исходя из термодинамических па¬
раметров систем. Значения констант можно найти в
таблицах, например, в справочнике Ю. Ю. Лурье
«Справочник по аналитической химии» (М., Химия,
1989). Тем не менее эти таблицы не охватывают все¬
го многообразия химических реакций. Во многих слу¬
чаях для сложных химических процессов приходится47
прибегать к некоторым математическим выкладкам,
основанным на применении уже найденных ранее зна¬
чений констант более простых реакций.Рассмотрим в общем виде взаимодействие между
комплексом MR„ и лигандом R':MR„ + mR' =*=> т'т + пЯ. (2.39)*Константы равновесия реакций такого типа в таб¬
лицах обычно не приводятся, но их легко найти пу¬
тем несложных преобразований.Запишем выражение для константы равновесия
реакции (2.39):jMR^itRT[MRrt][R'p- (' )В таблицах можно найти численные значения кон¬
стант комплексообразования для комплексов MR„ и
MRm, т. е.йм ■ tMR«1 /ОДП ft [MR^] (042)Р «« [МЦКГ1 (] MR/n IMHR'r1' (' }
Легко видеть, что отношение констант 0 - /PMRт/ правно константе равновесия реакции (2.39), т. е.Р *MRК = -5—^- (2-43>PMR „Таким образом, константа равновесия сложной ре¬
акции с участием двух комплексных соединений равна
константе, характеризующей малодиссоциированный
продукт реакции, деленной на константу, характери¬
зующую малодиссоциированный исходный комплекс.
Это общее правило применимо и к другим типам хи¬
мических реакций.Оценим, например, численное значение константы
равновесия реакции (2.33):[С<ЦМНз)Г] [CN-]< pCd(NH3,l+ 10CSg _1Q-t0.5S[Cd(CN>;-] [NH3]4 Pcd(CN)?-Заряды частиц для простоты опущены.48
Константа равновесия очень мала, сдвинуть со¬
стояние равновесия реакции вправо можно, увеличив
концентрацию аммиака, но несложный расчет показы¬
вает, что влияние этого фактора крайне незначитель¬
но. Естественно, что в обратном направлении реак¬
ция будет протекать практически полностью.Более подробная информация о количественных
расчетах с использованием констант приведена в со¬
ответствующих разделах учебника.В настоящее время получили распространение так
называемые условные константы равновесия, предло¬
женные Шварценбахом и Рингбомом.Выясним физический смысл условных констант на
примере реакций комплексообразования металла М с
лигандом R — анионом слабой кислоты.Комплекс MR„ диссоциирует в соответствии с ура¬
внениемMR„ M + nR (2.44)и истинная константа устойчивости имеет следующий
вид:а [MRд] .{-IPmr„ ~ [M][R]n * (*В растворе со свободными ионами металла М и
лигандами R могут протекать различные побочные ре¬
акции. Так, ионы металла могут подвергаться гидро¬
лизу или связываться в комплексные ионы находя¬
щимися в растворе посторонними лигандами Y. Ли¬
ганд R, как анион слабой кислоты, склонен к прото¬
низации с образованием протонированных частиц RH,
RH2 и т. д. Вследствие этих процессов концентрация
свободных частиц М и R становится не равной общей
концентрации этих частиц, не связанных в комп¬
лекс MRn.В общем, можно записать, что[M'] = [M] + [MYJ + [MYa]+ .... (2.46)где М' — общая концентрация ионов металла, не связанных в
комплекс MR„; [М] — концентрация свободных ионов металла;
[MY], [MY2] и т. д. — концентрация комплексов металла с посто¬
ронним лигандом Y (например, с ионами ОН и т. д.).Вводя для этих комплексов константы
, [MY] , [MY,]Pmy [М] [Y] ’ Pmys [MJIY]1 ' '49
из уравнений (2.46) и (2.47) получаем[М'-=1 + РмУт + РмУ2т2+ =амт- (2-48)[М]Величину ам(У) называют коэффициентом побоч¬
ной реакции или a-коэффициентом для М, взаимодей¬
ствующего с Y.Аналогично для лиганда, который образует про-
тонированные частицы RH, RH2 и т. д., можно запи¬
сатьIRH] [RHa] qI f +1 ’ 2 r +iq Д* .J[R] [Н ] [R] [Н ]И-Ш. = 1 + /С, [Н+] + К2 [н+]2 + ... = ая (-н+). (2.50)Условная константа устойчивости имеет видя/ [MR,1 [MR„][M'][RT [M] [R]r‘aM(V)“R(H+) ‘ { }Связь между истинной и условной константами выра¬
жается уравнениемР' = 5 . (2.52)aM(Y)aR (н + )Условная константа р' отличается от истинной р
тем, что в знаменателе условной константы находятся
общие концентрации М' и R', не связанных в комп¬
лекс MR„, в то время как в знаменателе истинной
константы — концентрации свободных ионов М и R,
образующихся при диссоциации комплекса в соответ¬
ствии с уравнением (2.44).Условные константы устойчивости сохраняют по¬
стоянное значение только при строго конкретных ус¬
ловиях, например при постоянных значениях pH и
(или) постоянных концентрациях посторонних лиган¬
дов. Их следует применять в тех случаях, когда из¬
вестны значения а и их зависимость от pH или кон¬
центрации постороннего лиганда. Условные кон¬
станты можно использовать в расчетах точно так же,
как и обычные константы, однако результаты таких
расчетов относятся к конкретным частным условиям.Применение условных констант в аналитической
химии обусловлено тем, что химика-аналитика не¬50
редко не интересует концентрация всех присутствую¬
щих в растворе соединений; ему в большинстве слу¬
чаев важно знать полноту протекания данной реак¬
ции, характеризующуюся концентрацией образовав¬
шегося комплекса MR„. Например, при комплексоно-
метрическом титровании для химика неважно, в ка¬
ких именно формах находятся не связанные в комп¬
лекс этилендиаминтетрауксусная кислота или ионы
металла — более существенно оценить, сколько ионов
металла прореагировало с титрантом.Константы равновесия зависят от температуры.
Температура влияет по-разному на реакции, происхо¬
дящие с выделением или поглощением тепла. Равно¬
весие экзотермических реакций смещается с возра¬
станием температуры влево, следовательно, констан¬
ты равновесия уменьшаются. Эндотермические реак¬
ции, наоборот, протекают активнее с увеличением
температуры, и константы равновесия при этом воз¬
растают. Константы равновесия реакций в растворах
мало зависят от температуры, потому что реакции
протекают при температуре не выше 80—90°С. В ана¬
литической химии, где, как правило, имеют дело с
реакциями в растворах, обычно не принимают во вни¬
мание небольшие изменения констант равновесия с
изменением температуры. В реакциях между газами,
наоборот, изменение температуры является одним из
эффективных способов нарушения состояния равнове¬
сия реакций в нужном направлении. Очень сильно
смещает равновесие реакций в газовой фазе также
изменение давления, однако в растворах изменение
давления в небольших пределах не оказывает почти
никакого влияния на ход реакций, и этот фактор
можно не принимать во внимание.ГЛАВА 3ПРАВИЛЬНОСТЬ И СТАТИСТИЧЕСКАЯ
ОБРАБОТКА РЕЗУЛЬТАТОВ АНАЛИЗАЗадача количественного химического анализа — найти
содержание отдельных элементов в анализируемом ма¬
териале. Главное требование к анализу заключается
в том, чтобы полученные результаты отражали истин¬51
ное содержание элементов в пробе. Результаты ана¬
лиза будут удовлетворять этому требованию только
в том случае, если все операции анализа выполнены
правильно.Целесообразно выделить следующие три этапа в
любом аналитическом определении: 1) отбор пробы;2) проведение анализа; 3) статистическая обработка
результатов анализа.Рассмотрим каждый этап.§ 1. ОТБОР ПРОБЫПроба материала, поступающая в лабораторию (ла¬
бораторная проба) должна быть представительной,
т. е. состав пробы и всей партии объекта анализа
должны быть идентичными. На предприятие обычно
поступают для переработки большие партии сырья
или другого материала, например несдолько вагонов
или даже целый железнодорожный эшелон. Чаще все¬
го этот материал бывает неоднородным, т. е. в нем по¬
падаются куски разных размеров, причем содержа¬
ние отдельных элементов в них может быть разным.
Необходимо так отобрать пробу, чтобы она по своему
составу соответствовала составу всей партии мате¬
риала.Для каждого рода материала существуют спе¬
циальные правила отбора проб для анализа, регла¬
ментирующие порядок проведения отдельных опера¬
ций. Эти правила подробно рассматриваются в гл. 29.
Ниже, в виде примера, приведены правила отбора
проб твердых сыпучих материалов, например желез¬
ной руды, поступающей на металлургический завод.
На рис. 3.1 изображена железнодорожная платформа
с рудой (вид сверху). Цифрами показаны места от¬
бора проб материала, цифры в скобках — глубина от¬
бора (в метрах). Видно, что из платформы необхо¬
димо отобрать 20 от¬
дельных порций ма¬
териала. Железнодо¬
рожный состав мо¬
жет иметь 30—40Рис. 3.1. Отбор проб с
платформы52
Рис. 3.2. Сокращение пробыплатформ или вагонов;
существуют инструк¬
ции, предписывающие,
из каких именно ваго¬
нов следует брать пробы, а какие можно пропускать.Отобранного материала может быть очень много,
например около 100 кг или больше. Для получения
лабораторной пробы (около 0,5—1 кг) необходимо
провести сокращение с одновременным измельчением
более крупных кусков. Материал насыпают в виде ко¬
нуса и затем квартуют: конус превращают в плоскую
лепешку, которую делят на 4 части, как показано на
рис. 3.2. Материал двух противоположных секторов
отбрасывают, а двух оставшихся измельчают, снова
насыпают концом, превращают последний в лепешку
и операцию квартования и размельчения повторяют
до тех пор, пока проба не сократится до 1—2 кг, ко¬
торую передают в лабораторию для анализа.Лабораторную пробу снова сокращают и измель¬
чают описанным способом. Для анализа отвешивают
обычно около 1 г, при очень малом содержании опре¬
деляемых элементов навеску увеличивают до 5—10, а
иногда до 100 г.Отбор пробы — очень ответственная и важная под¬
готовительная операция анализа. Неправильно ото¬
бранная проба может совершенно исказить резуль¬
таты, в этом случае вообще бессмысленно выполнять
дальнейшие операции анализа.§ 2. ПОГРЕШНОСТИ, ВОЗНИКАЮЩИЕ НА РАЗНЫХ
СТАДИЯХ АНАЛИЗАХимический анализ сложных материалов обычно
включает много самых разнообразных операций.
Часть этих операций имеет чисто механический ха¬
рактер, другие сопряжены с разнообразными химиче¬
скими реакциями. Так, определение процентного со¬
держания кальция в известняке включает следующие
операции.1. Отвешивание определенного количества извест¬
няка на аналитических весах.53
2. Перенесение навески в стакан.3. Растворение навески в хлороводородной кис¬
лоте, при этом выделяется значительное количество
диоксида углерода.4. Нагревание раствора.5. Добавление раствора осадителя — оксалата ам¬
мония.6. Нейтрализация кислого раствора аммиаком до
щелочной реакции.7. Отстаивание выделившегося осадка оксалата
кальция.8. Отделение осадка от маточного раствора филь¬
трованием.9. Промывание осадка на фильтре.10. Высушивание фильтра с осадком в сушильном
шкафу.11. Перенесение осадка с фильтром во взвешен¬
ный тигель.12. Сжигание фильтра и прокаливание осадка при
температуре, при которой оксалат кальция превра¬
щается в весовую форму — в карбонат или в оксид
кальция.13. Охлаждение тигля с осадком в эксикаторе.14. Взвешивание охлажденного тигля с осадком.15. Вычисление результатов анализа.Правильные результаты можно получить толькопри очень тщательном и внимательном выполнении
всех перечисленных операций. Тем не менее даже в
этом случае неизбежны отдельные погрешности, т. е.
отклонения результата измерения от истинного зна¬
чения измеряемой величины. Так, ошибка взвешива¬
ния на аналитических весах обычно составляет
±0,0001 г. При перенесении навески в стакан воз¬
можны небольшие потери вещества. Растворение на¬
вески пробы в кислоте сопровождается выделением
СОг, вследствие чего отдельные частички раствора
могут быть вынесены вместе с газом из стакана.
Многократное промывание способствует растворению
части осадка и переходу его в фильтрат. Прокалива¬
ния при слишком низкой температуре недостаточно,
чтобы полностью перевести оксалат кальция в карбо¬
нат, а при слишком высокой температуре часть карбо¬
ната кальция превратится в оксид. Чрезмерно дли¬
тельное охлаждение прокаленного осадка в эксика¬54
торе (особенно, если весовая форма — оксид каль¬
ция) приводит к увеличению его муссы вследствие ги¬
гроскопичности и т. д.Часть отмеченных и некоторых других погрешно¬
стей, возникающих в процессе анализа, могут быть
одного знака и, складываясь, приводить к расхожде¬
нию между найденным и истинным содержанием оп¬
ределяемого элемента; возможна также частичная
или, как редкое исключение, почти полная компенса¬
ция погрешностей разного знака — положительных и
отрицательных. Как общее правило, найденный ре¬
зультат всегда в большей или меньшей степени
отличается от истинного.Анализ характера погрешностей приводит к
выводу, что они могут быть систематическими и слу¬
чайными.Систематические погрешности остаются постоян¬
ными или закономерно изменяющимися при повтор¬
ных измерениях одной и той же величины, их источник
может быть при внимательном отношении к работе
обнаружен и устранен. К систематическим погрешно¬
стям относят прежде всего индивидуальные погреш¬
ности исполнителя анализа. Например, переход ок¬
раски индикатора в точке эквивалентности может
быть зафиксирован неправильно, с отклонением от
точки эквивалентности в большую или меньшую сто¬
рону, что вызвано индивидуальными особенностями
восприятия цветов аналитиком, выполняющим титро¬
вание. Плохо подготовленный аналитик может вооб¬
ще неправильно выбрать индикатор. Так, правильное
титрование буры хлороводородной кислотой возможно
с метиловым оранжевым или метиловым красным,
замена этих индикаторов, например фенолфталеином,
приводит к грубой ошибке, хотя результаты парал¬
лельных титрований не отличаются один от другого.
Нередко индивидуальная погрешность вызвана неосо¬
знанным стремлением аналитика «подогнать» резуль¬
тат к полученному ранее или к среднему результату
и т. д.Другой род систематических погрешностей — ин¬
струментальные. К ним относят, например, погрешно¬
сти, связанные с' использованием неправильно отка¬
либрованных разновесов или с употреблением некаче¬
ственной мерной посуды. Так, если пипетка номи¬55
нальной вместимостью 25 мл в действительности отме¬
ривает объем раствора, равный 25,2 или 24,8 мл, па¬
раллельные определения будут хорошо сходиться ме¬
жду собой, однако результат аналич, г* равно ока¬
жется неверным.Наконец, возможны методические погрешности.
Оксид алюминия А1203 очень гигроскопичен; исполь¬
зование этого соединения в качестве весовой формы
чаще всего приводит к получению завышенных ре¬
зультатов. Погрешностей можно избежать только при
очень тщательном выполнении условий анализа. Взве¬
шивание сульфата свинца с целью гравиметрического
определения последнего обычно дает отрицательную
погрешность из-за значительной растворимости осадка
в воде. Иодометрическое определение двухвалентного
олова сопряжено обычно с отрицательной погреш¬
ностью из-за частичного окисления Sn2+ кислородом
воздуха в процессе титрования раствором иода.В рассмотренных примерах причина систематиче¬
ских погрешностей заключалась в химических свой¬
ствах используемых в анализе соединений. В других
случаях источником погрешностей является примесь
посторонних веществ в растворах и в реагентах. Так,
хлороводородная кислота нередко содержит примеси
железа (III). При фотометрическом определении по¬
следнего в виде роданидного комплекса необходи¬
мо пользоваться только химически чистой кислотой,
иначе железа будет найдено больше, чем его в дейст¬
вительности было в анализируемом материале.Как уже было сказано, систематическую погреш¬
ность всегда можно устранить или уменьшить, если
установлен ее источник.Существует, однако, другая группа погрешностей,
которые вызываются настолько разнообразными и не
поддающимися учету многочисленными причинами,
что их невозможно обнаружить и устранить. Это так
называемые случайные погрешности, т. е. такие, кото¬
рые изменяются случайным образом при повторных
измерениях одной и той же величины. Они приводят
к тому, что параллельные определения какого-либо
элемента, выполненные при совершенно одинаковых
условиях, никогда в точности не совпадают по ре¬
зультату; в качестве наиболее вероятного значения
обычно берут среднее значение из нескольких опре¬56
делений. Случайные погрешности составляют предмет
статистической обработки результатов. К такой обра¬
ботке необходимо приступать только после того, как
есть уверенность в правильности анализа.Кроме рассмотренных двух рядов погрешностей,
существуют еще так называемые промахи, т. е. явные
огрехи анализа, допущенные из-за небрежности или
заведомой некомпетентности аналитика. Приведенный
выше пример титрования буры хлороводородной кис¬
лотой в присутствии фенолфталеина относится к
числу таких промахов. Способ выявления промахов
описан ниже.§ 3. ПРАВИЛЬНОСТЬ АНАЛИЗАПравильность анализа характеризуется расхожде¬
нием между средним и истинным результатом. Пра¬
вильность анализа обеспечивается в том случае, если
удается полностью избежать систематических погреш¬
ностей. Правильность анализа нельзя установить по¬
средством статистической обработки результатов, за
исключением некоторых редких случаев. Необходимо
применять другие, экспериментальные, методы про¬
верки правильности. К ним относят следующие ме¬
тоды.1. Проведение анализа двумя или несколькими
независимыми методами. Например, свинец можно
определить гравиметрическим методом в форме суль¬
фата (PbS04) свинца. Иодометрическое определение
свинца основано на его осаждении в виде хромата
РЬСг04; далее осадок отфильтровывают, промывают,
растворяют в кислоте и прибавляют к полученному
раствору иодид калия:Ст2027~ + 6Г + 14Н+ = 2Сг3 + + 31, + 7Н20.Выделившийся иод титруют тиосульфатом натрия.При комплексонометрическом определении ионы
свинца титруют раствором этилендиаминтетрауксус-
ной кислоты. Наконец, свинец можно определить элек¬
тролитически, выделяя диоксид свинца(IV) на аноде
и взвешивая выделившийся осадок.Если все методы дают одинаковые результаты,
можно быть уверенным, что анализ выполнен пра¬
вильно. В дальнейшем для определения свинца можно57
уже с достаточной надежностью применять какой-
либо один метод из рассмотренных выше.2. Применение стандартных образцов. Стандарт¬
ные образцы — это эталоны химического состава ма¬
териалов. В Советском Союзе есть несколько органи¬
заций, специально занимающихся приготовлением и
аттестацией стандартных образцов. Так, образцы чер¬
ных металлов и сплавов — чугунов, сталей и др. — го¬
товит Всесоюзный институт стандартных образцов в
г. Свердловске. Сплавы цветных металлов, готовят и
аттестуют во Всесоюзном научно-исследовательском
и проектном институте вторичной цветной металлур¬
гии «Вниивторцветмет».Аттестация стандартных образцов — это установ¬
ление точного содержания отдельных элементов в них
посредством анализа несколькими самыми надеж¬
ными методами в нескольких наиболее ответственных
лабораториях страны. Полученные результаты тща¬
тельно сопоставляются и затем из этих данных вы¬
водят среднее содержание каждого элемента. Так,
аттестованное содержание элементов в алюминиевом
сплаве определенной марки равно (в %): Mg—1,56;
Si —0,90; Mn —0,65; Си —4,48; Zn — 0,28; Ni—
0,058 и т. д.Каждый стандартный образец снабжен официаль¬
ным документом — паспортом, в котором указано ат¬
тестованное содержание отдельных элементов. Чтобы
установить правильность анализа какого-либо мате¬
риала, из имеющегося в лаборатории набора стан¬
дартных образцов выбирают тот, который по своему
составу в наибольшей степени приближается к пред¬
полагаемому составу анализируемой пробы. Затем
проводят параллельно анализ пробы и стандартного
образца, устанавливая содержание отдельных элемен¬
тов в них. Считается, что использованный метод ана¬
лиза дает правильные результаты, если найденное
содержание отдельных элементов в стандартном об¬
разце соответствует паспортным данным, установлен¬
ным при аттестации этого образца.3. Метод добавок. Ведут несколько параллельных
определений, прибавляя к некоторым пробам точно
известные количества определяемого элемента. При
правильном анализе количество элемента, найденное
в пробах е добавками и без них, должно соответство¬58
вать количеству добавки. Например, определяя же¬
лезо в растворе, нашли, что проба без добавки содер¬
жала 0,2378 г Fe203, а проба с добавкой 0,1000 г
Fe203— 0,3375 г Fe203. Разность составляет 0,0997 г,
т. е. почти в точности равна введенному количеству
железа. Анализ можно считать правильным.При полном анализе какого-либо материала сум¬
ма всех компонентов должна быть равной 100 % или
только незначительно отличаться от этого значения.
Так, при анализе силиката определяют в нем сле¬
дующие компоненты: Si02, Fe203, ТЮ2, А1203, МпО,
CaO, MgO, Na20, КгО, Р2О5 и др. Если суммарное со¬
держание перечисленных составных частей значи¬
тельно отличается от. 100 % — анализ следует считать
неправильным. Вероятно, либо пропущена какая-то
составная часть (при сумме, меньшей 100%), либо
вкралась грубая погрешность (при сумме, значи¬
тельно большей 100 %).Результат, близкий к 100 %, дает некоторую уве¬
ренность в правильности анализа, хотя и в этом
случае возможны погрешности, связанные с компен¬
сацией положительных и отрицательных погрешно¬
стей. Так, если в осадке полуторных оксидов не опре¬
деляли титан, результат для алюминия окажется
завышенным, хотя сумма будет близка к 100 %■Изложенные методы, особенно два первых — ана¬
лиз различными независимыми методами и примене¬
ние стандартных образцов — дают основания для
достаточно надежного вывода о правильности ана¬
лиза. Тем не менее систематические погрешности все
же возможны. Важно, чтобы они не выходили за не¬
которые пределы.Рассмотрим зависимость результатов определения
от наличия систематических погрешностей. В одних
случаях результат может отличаться от истинного
всегда на одно и то же значение, т. е. погрешность
будет постоянной независимо от размера навески.
Так, определяя свинец, его осаждают в виде PbS04 и
промывают осадок водой. В результат вносится по¬
стоянная отрицательная систематическая погреш¬
ность, связанная с потерями за счет заметной раство¬
римости сульфата свинца. Эта погрешность по
абсолютному значению всегда будет одна и та же, не¬
зависимо от количества находящегося на фильтре59
осадка. Относительная же погрешность уменьшается
с возрастанием навески. Так, потеря из-за раствори¬
мости может составлять, например, 0,5 мг; при массе
PbS04 0,01 г она равна 5 %, а увеличение массы осад¬
ка до 0,1 г уменьшает относительную ошибку до 0,5 %.В других случаях систематическая погрешность
влияет на результаты анализа иначе. Увеличение на¬
вески анализируемого материала влечет за собой воз¬
растание только абсолютной погрешности, однако от¬
носительная погрешность остается при этом одной и
той же. Примером такого влияния может служить
иодометрическое определение меди в сплавах с незна¬
чительными примесями железа. Ионы меди реаги¬
руют с иодидом калия, выделяя эквивалентное коли¬
чество свободного иода:2Cu2v -f- 41' = 2CuI + Ь-Выделившийся иод титруют раствором тиосуль¬
фата натрия и по расходу последнего вычисляют со¬
держание меди в образце. Однако ионы железа реа¬
гируют с ионами иода тоже с выделением иода:2Fe3+4-2P = 2Fe2‘4-b.Поэтому в присутствии железа(III) получаются
повышенные результаты для меди, причем абсолют¬
ная погрешность возрастает с увеличением размера
навески. Однако относительная погрешность не зави¬
сит от количества взятого для анализа материала.
В самом деле, предположим, что навеске 0,1 г отве¬
чает абсолютная положительная погрешность 0,2 мг,
тогда при навеске 0,2 г она увеличится до 0,4 мг, од¬
нако в обоих случаях относительная погрешность ос¬
тается неизменной и составит0,0002-100 0,0004-100§ 4. СТАТИСТИЧЕСКАЯ ОБРАБОТКА РЕЗУЛЬТАТОВ
АНАЛИЗАПредмет статистической обработки результатов —
случайные погрешности анализа. Как уже было ска¬
зано, источники случайных погрешностей настолько
многообразны, что их нельзя заранее предусмотреть60
и устранить. Эти погрешности приводят к тому, что
результаты нескольких параллельных определений
практически никогда не совпадают; они рассеивают¬
ся вокруг некоторого среднего значения. Степень рас¬
сеивания результатов характеризует воспроизводи¬
мость анализа. Большой разброс результатов парал¬
лельных определений свидетельствует о плохой вос¬
производимости; наоборот, чем меньше отдельные ре¬
зультаты различаются, тем выше воспроизводимость.
При вдумчивом и внимательном отношении исполни¬
теля ко всем операциям анализа воспроизводимость
может быть значительно улучшена.Статистическая обработка результатов имеет две
основные задачи. Первая задача заключается в том,
чтобы представить результаты многих определений в
компактной форме или, иначе говоря, произвести
свертывание информации. Вторая задача — оценить
надежность полученных результатов, т. е. степень их
соответствия истинному содержанию определяемого
элемента в образце.Рассмотрим влияние случайных погрешностей на
результаты параллельных определений. Предполо¬
жим, что выполнено 16 параллельных анализов и что
имеется 4 источника погрешностей, которые могут
приводить к положительным или отрицательным
ошибкам одинакового значения. Положительные и от¬
рицательные погрешности могут компенсироваться,
поэтому в некоторых случаях будет наблюдаться от¬
сутствие погрешности; в других случаях, когда по¬
грешности имеют один знак, они складываются и
ошибки будут велики. Возможны и другие сочетания
погрешностей.В табл. 3.1 рассмотрены все возможные комбина¬
ции погрешностей для данного идеализированного
примера.Из данных таблицы следуют важные выводы. Вид¬
но, что чем меньше случайная погрешность, тем чаще
она появляется. Нулевая случайная погрешность по¬
является в 6 случаях из 16 определений. Чем больше
случайная погрешность, тем меньше частота ее по¬
явления. Так, погрешность в 2 единицы наблюдается4 раза из 16 определений, в 4 единицы — всего один
раз. Далее вероятность появления положительных и
отрицательных ошибок одинакова.61
Таблица 3.1. Возможные комбинации погрешностейдля идеализированного примера при проведении анализа№параллельныхопределенийКомбинациипогрешностейЗначениеслучайнойпогрешностиОтносительнаячастотапогрешности1+ + + +412- + + +3+ - + +4+ + - +245+ + + -6 + +7-|—| 8- + + -069+ +10+ - + -11- + - +12+ 13- + -2414 + -15 +16-41Данные таблицы представляют идеализированный
пример. Источников погрешностей может быть зна¬
чительно больше, чем четыре, а сами погрешности не
обязательно равны между собой. Поэтому при огра¬
ниченном числе наблюдений установленные зависимо¬
сти далеко не всегда проявляются так четко, как в
рассмотренном примере. Однако, когда число наблю¬
дений (измерений, определений) очень велико, слу¬
чайные ошибки распределяются по определенным за¬
конам: 1) нулевые или близкие к ним ошибки имеют
максимальную частоту появления; 2) вероятность по¬
явления отклонений разного знака (положительных и
отрицательных) одинакова; 3) с ростом ошибки веро¬
ятность ее появления экспоненциально уменьшается.Графическое изображение сформулированных за¬
кономерностей представляет собой кривую Гаусса или
кривую нормального распределения погрешностей
(рис. 3.3). Сама кривая является экспериментальной,
она построена по результатам очень большого числа
наблюдений. Кривую распределения можно описать
математическим уравнением. В это уравнение входит
в качестве одного из параметров так называемое«2
стандартное отклонение а или, что то же самое, сред¬
нее квадратичное отклонение:(х,-»)2п(3.1)где Xi, ц — измеренное н истинное значения измеряемой величи¬
ны соответственно; п — число наблюдений (измерений, опреде¬
лений).Стандартное отклонение представляет собой количе¬
ственную меру воспроизводимости, т. е. степени бли¬
зости отдельных результатов к среднему значению.Уравнение (3.1) справедливо для так называемой
генеральной совокупности наблюдений (определений),
т. е. когда число их очень велико.Квадрат стандартного отклонения называют дис¬
персией:В теории вероятности доказано, что кривую нор¬
мального распределения погрешностей (кривую Гаус¬
са) можно описать следующим уравнением:е-(х-а)У2 а-где (/—частота появления каждого значения; х— единичное из¬
мерение: (1 — истинное значение измеряемой величины (или сред¬
нее арифметическое из бесконечно большого числа измерений).Введем обозначениеЕсли принять р за условный нуль (р = 0) и отсчи¬
тывать отклонения в единицах стандартного отклоне-
нения (т. е. а = 1),тогда уравнение (3.3) преобразуется в еле- Iдующее:-Z41ного распределения ошибок -3-2-10123Площадь s, заключен-Рис. 3.3. Кривая нормаль-63
ная между кривой Гаусса и осью абсцисс, представ¬
ляет интегралs = —1=- [ e~zy2dZ. (3.6)V2 к JИзт 1Л1математики известно, что ^ е~гу2 dz = л/2л, по-— ООэтому интеграл (3.6) равен единице (или 100 %). Это
означает достоверность появления значения измере¬
ния, лежащего в области от —оо до +оо, т. е. ве¬
роятность такого события равна единице (100%).Интегрирование уравнения (3.6) в пределах от
1 — ст до 1 + ст дает
z= +1Lr- [ e~z’l2dZ = 0,683 или 68,3%. (3.7)
2я „ JV 2;Z=-IАналогично после интегрирования уравнения (3.6)
в пределах от 2 = —2 до Z = 2 и от Z = —3 до Z = 3
получаемZ-+ 2'=- ? e~zy2dZ = 0,955 или 95,5% (3.8)
2я _ JV2jiZ--2
Z= + 3s = —^=Г ^ e~z^2 dZ = 0,997 или 99,7%. (3.9)
V 2n Jv Z=-3Из уравнения (3.7) следует, что вероятность (так
называемая доверительная вероятность) появления
измерения, лежащего в области ±о, равна 68,3%,
т. е. в 68,3 случаях из 100 случайная погрешность лю¬
бого данного единичного измерения меньше (или рав¬
на) ±о. Аналогично из (3.8) и (3.9) выводим, что в
95,5 случаях и, соответственно, в 99,7 случаях из 100
случайные погрешности любого данного единичного
измерения меньше (или равны) ±2сг или ±3ст.Таким образом, доверительная вероятность — это
отношение площади под кривой нормального распре¬
деления для каждого значения Z к общей площади.
Иными словами, доверительная вероятность 68,3 %
означает, что Z — 1а и что, следовательно, в 68,3 слу-$4
чаях из 100 случайная погрешность любого данного
единичного определения (измерения, наблюдения) бу¬
дет меньше (или равна) 1 а. При доверительной ве¬
роятности 95,5 % Z = 2а и, следовательно, в 95,5 слу¬
чаях из 100 случайная погрешность любого данного
единичного определения будет меньше (или равна)
2а.Следовательно, доверительная вероятность опреде¬
ляется числом единиц стандартного отклонения, т. е.
величиной Z; в свою очередь, каждому значению до¬
верительной вероятности соответствует определенное
значение Z, т. е. Z зависит от заданной доверительной
вероятности. Некоторые другие значения доверитель¬
ной вероятности при разных значениях Z приведены
ниже:Р, % . 50 68 80 90 95 96 99,7 99,9
Z . . . ±0,67 ±1,00 ±1,29 ±1,64 ±1,96 ±2,00 ±3,00 ±3,29Все сказанное выше относится к генеральной со¬
вокупности наблюдений (измерений). Однако в дей¬
ствительности в условиях анализа никогда не бывает
слишком большого числа параллельных определений.
В обычных условиях проводят 4—5 параллельных
анализов. Кроме того, истинное значение измеряемой
величины |i также очень редко известно точно. По¬
этому вместо |ы берут среднее значение из нескольких
определений х:_=Xi+X2+Xi+X,+ ^ПДалее вычисляют стандартное отклонение так назы¬
ваемой малой выборки из генеральной совокупно¬
сти 5/ У (*/ — х)2S = Л/ _ , • (3.11)Стандартное отклонение — это количественная ха¬
рактеристика воспроизводимости. Оно может быть
равно нулю только в том случае, когда равна нулю
сумма (xi — х)2 = 0, т. е. если каждое единичное из¬
мерение равно среднему значению и отклонения от
среднего отсутствуют. Такая идеальная воспроизво¬
димость никогда не может быть реализована.Сопоставление стандартных отклонений при ана¬
лизе какого-либо образца двумя аналитиками или в3 Аналитическая химия65
двух различных лабораториях дает возможность оце¬
нить качество выполнения анализов и квалификацию
исполнителей.. Так, в двух различных лабораториях
были получены следующие результаты определения
оксида алюминия в некотором материале (в %):1-я лаборатория — 5,24; 5,37; 5,33; 5,38; 5,28х = = 5,32;52-я лаборатория — 5,26; 5,41; 5,49; 5,22; 5,48- 26’9 *47
х = —— = 5,37.5Средние результаты, полученные в обеих лабора¬
ториях, очень близки; это может свидетельствовать о
правильности анализа, т. е. об отсутствии системати¬
ческих ошибок. Тем не менее расчет стандартных от¬
клонений обеих лабораторий показывает следующее.
Стандартное отклонение первой лаборатории:s= д/64' 10~Ч + 25 ■ 10~4 + 1 • !(Г4 + 36- Ю~4 + 16 • 10~4 = n nfi
Стандартное отклонение второй лаборатории:S= V121-10 4+ 16 - 10 4 + 144 ■ 10 4 + 225 • 10 4 + 121 - 10 *4= 0,12.Во второй лаборатории стандартное отклонение в
два раза больше, чем в первой; вероятно, это являет¬
ся следствием более низкой квалификации или мень¬
шей внимательности исполнителей анализа.Однако расхождение может быть вызвано и слу¬
чайными ошибками, и, следовательно, тогда оба ряда
результатов принадлежат к одной генеральной сово¬
купности; иными словами, нет оснований считать, что
расхождение стандартных отклонений обеих лабора¬
торий вызвано систематическими погрешностями од¬
ной из них.Для решения этого вопроса привлекают /•'-крите-
рий (критерий Фишера). Вычисляют отношение двух
дисперсийF = (S2>S?)66
я сравнивают это отношение с табличными данными
для заданной доверительной вероятности {обычно
принимают Р — 0,95) и данного числа параллельных
проб.Ниже приведена выдержка из таблицы /^крите¬
риев для Р = 0,95. Здесь П\ и п2 — число параллель¬
ных результатов для > 5? соответственно:34567831,91,91,919,1519,319,3549.59,39,19,08,98,8557,06,71 6.4 |6,36,26,065,85,55,25,15,04,975,14,84,54,44,34,284,74,44,14,03,93,8В нашем случае F = 0,122/0,062 = 4, т. е. меньше
6,4 — табличного значения для п1 = п2 = 5. Поэтому
можно считать, что расхождение дисперсий вызвано
случайными погрешностями и оба ряда значений ха¬
рактеризуют одну и ту же генеральную совокупность.Иногда в серии параллельных определений один
из результатов представляется сомнительным, так как
довольно значительно отличается от всех других ре¬
зультатов. В этом случае необходимо решить, остав¬
лять ли этот результат для вычисления среднего или
отбросить как промах. С этой целью применяют так
называемый Q-критерий:где *i — сомнительный результат; хг — результат, который ближе
всего к х\\ R — размах варьирования, т. е. разность между пре¬
дельными значениями определяемой величины.Вычисленное значение Q сравнивают с таблич¬
ными значениями, найденными методами математиче¬
ской статистики. Сомнительный результат следует от¬
бросить, если Q > (?табл, в противном случае, его ос¬
тавляют для дальнейших операций.3*67
Табличные значения Q-критерия приведены ниже
для Р = 0,95 и различных п:п 3 4 5 6 7 8 9 10<3(0,95) . . 0,94 0,77 0,64 0,56 0,51 0,48 0,44 0,42Рассмотрим в качестве примера сомнительный ре¬
зультат параллельных определений содержания гра¬
фита в чугуне (в %): 2,86; 2,89; 2,90; 2,91; 2,99.Последний результат заметно отличается от дру¬
гих. В данном случаеВидно, что Q < (Этабл, поэтому результат 2,99 можно
оставить.Стандартное отклонение малой выборки S может
не совпадать со стандартным отклонением генераль¬
ной совокупности а, т. е. в общем случае 5 ф а. Тем
не менее опыт показывает, что достаточно хорошее
приближение 5 к а получается уже в том случае, если
количество измерений равно или больше двадцати.
Это дает возможность вычислить а из данных преды¬
дущих анализов аналогичного материала примерно с
одним и тем же содержанием данного элемента. Рас¬
смотрим последовательность вычислений.Так, при атомно-абсорбционном определении свин¬
ца в медном сплаве (образец № 1) были получены
следующие результаты (в %): 7,50; 7,75; 7,67; 7,63;
7,6]. Этих данных слишком мало для того, чтобы по¬
лучить хорошее приближение 5 к а. Однако такое
приближение можно реализовать, использовав архив¬
ные данные о результатах анализа аналогичных об¬
разцов, выполненных ранее, в другое время.
В табл. 3.2 приведены, наряду с данными анализа об¬
разца № 1, также результаты определения свинца
этим же методом в образцах №№ 2—5, которые ана¬
лизировались 2—3 недели тому назад. Приведены
также средние значения каждой серии измерений,
вычисленные по уравнению (3.10).Рассчитывают сумму квадратов отклонений от¬
дельных определений от среднего результата (х, — *);
порядок вычислений приведен только для образца
№ 1:(*у - х)2 = 0,13s + 0,122 + 0,04г + О2 + 0.022 = 0,0333.68
Таблица 3.2. Результаты анализа образцов сходного состава№образцаСодержание свинца, %Среднее значенне17,50;7,75;7,67;7,63;7,61*1 =38,165= 7,6324,57;4,65;4,63*2 =13,953= 4,653СОСП3,63;3,67;3,56*3 =14,394= 3,6046,00;5,70;5,75;5,81;5,76; 5,86•*4 =34,866= 5,8156,50;6,25;6,38;6,20;6,25*5 =31,585= 6,32Аналогичные данные, вычисленные для других об¬
разцов, приведены ниже: № 2— 0,0168; № 3 — 0,0123;
№ 4 — 0,0522; № 5 — 0,0602.Стандартное отклонение приведенной малой вы¬
борки S, близкое при данном числе параллельных оп¬
ределений к стандартному отклонению генеральной
совокупности а, можно вычислить далее по уравне¬
нию:^ а .. /У'. (ХИ ~ *|) + ^ (х21 ~ *2) /С (X3l ~ *з) ~1~ ■ ■ •V п — ш(3.12)где п — общее число определений (в нашем случае 23); m — чис¬
ло проб (в нашем случае 6).Применительно к рассматриваемому примеру. _ / 0,0333 + 0,0168 + 0,0123 + 0,0522 + 0,06025 — 1+3 — 1+4 — 1+6 — 1+Й — 1 =I-V( 0,1778
1Р= 0,1.Пользуясь найденным значением S=a = 0,l, мо¬
жно оценить надежность единичного и среднего зна¬
чения определения для каждого любого образца, на¬
пример для образца № 1.Под оценкой надежности результатов понимают
нахождение доверительных границ. Доверительные69
границы — это пределы области вокруг эксперимен¬
тально найденного единичного или среднего резуль¬
тата, внутри которой следует ожидать с заданной
степенью доверительной вероятности нахождения ис¬
тинного значения единичного или среднего результата.
Интервал, ограниченный этими пределами, называют
доверительным интервалом.Доверительные границы единичного определения
находят следующим образом.Из уравнения (3.4) следует, чтоДоверительные границы первого результата об¬
разца № 1 равныВ этом уравнении 0,l=5 = (j (см. выше).Обычно оценивают доверительные границы при
некоторой заданной доверительной вероятности, чаще
всего при 95%. Доверительная вероятность 95% со¬
ответствует Z— 1,96 (см. выше). ПоэтомуЭти границы обозначают, что в 95 случаях из 100
истинный результат единичного определения будет на¬
ходиться в пределах от 7,30 до 7,70 и что в 5 случаях
из 100 он может выходить за эти пределы. Иными
словами, вероятность того, что истинный результат
находится в границах 7,30—7,70, составляет 95 %,
т. е. нельзя дать 100 %-ной гарантии того, что пра¬
вильный результат находится в указанных пределах.Если имеется несколько параллельных определе¬
ний, как, например, при анализе образца № 1, можно
оценить надежность — доверительные границы — сред¬
него результата. Можно доказать, что доверительныйинтервал уменьшается в л!п раз для среднего из п
параллельных измерений, т. е. для образца № 1ц = х ± Za.(3.13)ц = 7,50 ± 0,1 Z,(3.14)ц = 7,50 ± (0,1 • 1,96)или доверительные раницы равны
7,30 < ц < 7,70.(3.15)70
Поэтому при доверительной вероятности 95 % дове¬
рительные границы равны7,63 ± 0,1или7,63 < ц < 7,73.Если задаются большей доверительной вероят¬
ностью, например равной 99,7%, тогда Z = 3 (см.
стр. 65) иВидно, что с чем большей вероятностью мы хотим
получить ответ, тем шире раздвигаются доверитель¬
ные границы.Очень часто архивные данные отсутствуют и нет
возможности получить хорошее приближение S к а.
В этих случаях необходимо пользоваться стандарт¬
ным отклонением малой выборки S. Но если S най¬
дено всего из нескольких параллельных определений,
тогда приведенное выше соответствие между Z и Р
нарушается. Определенному значению доверительной
вероятности Р будет теперь отвечать не величина Z,
а какая-то другая функция, большая, чем Z. Эту
функцию обозначают буквой t, она изменяется не
только в зависимости от доверительной вероятности,
но и от числа параллельных определений или от числа
степеней свободы (п— 1). Число t будет тем больше,
чем больше заданная доверительная вероятность и
чем меньше число степеней свободы. При большом
числе степеней свободы (большом числе параллель¬
ных определений) t стремится к Z и в конце концов
совпадает с Z, когда данную выборку можно считать
генеральной совокупностью.В случае малой выборки, характеризующейся
функцией t, речь идет не о нормальном распределе¬
нии, а о -^-распределении или распределении Стью-
дента. Численные значения t получают интегрирова¬
нием сложной функции, которую подробно рассматри¬
вают в курсе теории вероятности.7,63 ±0,13или7,50 < (i < 7,76.71
Таблица 3.3. Значения t при различной доверительной
вероятности аЧислостепенейсвободыКоэффициенты нормирован¬
ных отклонений при а (в %)ЧислостепенейсвободыКоэффициенты нормированных
отклонений при а (в %)959999 9959999.9112,763,763782,313,365,0424,303,9231,692,263,254,7833,185,8412,9102,233,174,5942,784,608,60112,203,114,4452,574,036,86122,183,064,3262,453,715,96132,163,014,2272,363,505,40142,142,984,14оо1,962,583,29Численные значения функции t называют коэффи¬
циентами нормированных отклонений. Они приведены
в табл. 3.3.При /-распределении доверительные границы нахо¬
дят из уравнений (3.16) и (3.17).Для единичного определения|x = *±<S. (3.16)Для среднего из нескольких определенийц = *±-^. (3.17)V пПри /-распределении доверительные границы при
одной и той же доверительной вероятности получают¬
ся более широкими, чем при нормальном распределе¬
нии.Рассмотрим в качестве примера результат анализа
образца № 1, предположив, что нет данных предыду¬
щих анализов и что поэтому нельзя вычислить стан¬
дартное отклонение генеральной совокупности.Из уравнения (3.11) получаемVT (х, - х)2 0,0333!— = — = 0,09.п — 1 4Из табл. 3.3 находим, что при а = 95 % для че¬
тырех степеней свободы (п = 5) t — 2,78. Поэтому
для единичного определения (см. уравнение 2.16)
ц = 7,50 ± ts = 7,50 ± 2,78 • 0,09 = 7,50 ± 0,25или7,25 < ц < 7,75.72
Доверительные границы среднего результата по
уравнению (3.17) равныц = х ± —-т=г = 7,63 ±V я2,78 • 0,09= 7.63 ± 0,11или7,52 < ц, < 7,73.В обоих рассмотренных случаях доверительные
границы получаются более широкими, чем при ис¬
пользовании стандартного отклонения генеральной со¬
вокупности о.Результаты статистической обработки можно ино¬
гда использовать для выявления систематической
погрешности новой методики анализа. Для этого не¬
обходимо располагать стандартным образцом с атте¬
стованным содержанием определяемого элемента, ко¬
торое принимается равным истинному содержанию ц.Выполняют несколько параллельных анализов
стандартного образца и находят среднее значение х.
Из уравнения (3.17) следуетЕсли среднее значение х отличается от истинного
значения р, только из-за допущенных случайных по¬
грешностей, тогда разность ц — х при заданной дове¬
рительной вероятности должна быть равна или мень-следует заключить, что кроме случайных погрешно¬
стей методика дает также систематическую погреш¬
ность, причину которой следует установить постанов¬
кой специального эксперимента.Например аттестованное содержание железа в
стандартном образце литейного алюминиевого сплава,
в соответствии с паспортными данными, ju. = ],39 %.
Проверяли новую фотометрическую методику опреде¬
ления железа с ацетилацетоном. В 6 параллельныхtsше tS/л/п, т. е.В противном случае, когда73
анализах были получены следующие результаты: 1^33;
1,27; 1,35; 1,36; 1,31; 1,26%. Среднее значение х =
= 1,31.Применяя уравнение (3.11), находим, что приРазность р. — х = 1,39 — 1,31 = 0,08.При доверительной вероятности 95 % и числе сте¬
пеней свободы п — 1=5 коэффициент нормирован¬
ных отклонений равен 2,57, откудапоэтому в методике есть невыявленная систематиче¬
ская погрешность, вывод о систематической погреш¬
ности справедлив в 95 случаях из 100, т. е. сущест¬
вует все же некоторая вероятность (5%) того, что
расхождения вызваны только случайными погрешно¬
стями.ГЛАВА 4КОНЦЕНТРАЦИЯ РАСТВОРОВ И ВЫЧИСЛЕНИЯ
В КОЛИЧЕСТВЕННОМ АНАЛИЗЕ§ 1. ОБЩИЕ ПОНЯТИЯРассмотрим некоторые термины и понятия, принятые
в Международной системе единиц (СИ) и употреб¬
ляющиеся при вычислениях в количественном ана¬
лизе.Моль — количество вещества, содержащее столь¬
ко определенных условных частиц, сколько атомов
содержится в 0,012 кг (или в 12 г) углерода— 12. Под
условными частицами подразумевают молекулы, ио¬
ны, электроны, доли молекул и ионов, например 1/5
часть молекулы KMnC>4, 1 /2 часть молекулы H2SO4,
1/6 часть иона СГ2О7" и т. д. Применяя терминл = 6■s/ п 6Разностьц — х = 0,08 > 0,04,74
«моль», необходимо всегда указывать, о каких имен¬
но условных частицах идет речь. Например, следует
говорить: 1 моль молекул серной кислоты, 0,1 моль
ионов железа и т. д. Количество вещества обо¬
значают буквой п и записывают следующим обра¬
зом: /z(H2S04) = 1 моль, rt(Fe2+)=0,l моль,
n(l/2H2S04) —0,1 моль и т. д.Слово «моль» после числа не склоняется.Молярная масса вещества х—масса
1 моль вещества, обозначают буквой М. Она равна
отношению массы т вещества к его количеству в мо¬
лях, т. е.Основной единицей молярной массы является кг-
•моль'1, а на практике чаще пользуются единицей
г-моль-1. Например, молярные массы атомов меди,
ионов водорода, молекул хлора, 1/2 молекул H2SO4
равны соответственно: М (Си) =63,54 г-моль-1;
М(Н+) = 1,074 г-моль-1; М(С12) = 70,916 г-моль-1;
M(1/2H2S04) =49,10 г-моль-1.Эквивалент в кислотно-основных реакциях —
условная частица, которая может присоединять, вы¬
свобождать или каким-либо иным способом взаимо¬
действовать с одним ионом водорода (или другого од¬
новалентного элемента).Эквивалент в реакциях окисления — восстановле¬
ния— условная частица, которая может присоединять
или высвобождать один электрон.Фактор эквивалентности f3KB — число,
обозначающее, какая доля условной частицы веще¬
ства х реагирует (эквивалентна) с одним ионом во¬
дорода в данной кислотно-основной реакции или с од¬
ним электроном в данной окислительно-восстанови-
тельной реакции. Например, фактор эквивалентности
перманганата калия в кислой среде/экв (КМПО4) = 1/5.Значения эквивалента и фактора эквивалентности
могут изменяться в зависимости от того, в какой
именно конкретной реакции участвует данное веще¬
ство.75
Молярная масса эквивалента* веще¬
ства х (Мэкв) — масса одного моля эквивалентов это¬
го вещества, равная произведению фактора эквива¬
лентности /экв на молярную массу вещества х:МЭкв = ^квМ (х) г ■ моль-'. (4.2)Рассмотрим способы выражения концентрации
растворов. Концентрацией раствора называют количе¬
ство вещества, содержащееся в единице объема рас¬
твора или растворителя. Различают следующие тер¬
мины.Молярная концентрация вещества х —
отношение количества вещества х (в молях) к объемуV раствора:= (4.3)Молярный раствор — раствор, в 1 л которого
содержится 1 моль вещества, например 1 моль моле¬
кул или 1 моль ионов или 0,1 моль эквивалентов
и т. д. Растворы, содержащие в 1 л 0,1 моль,0,01 моль,0,001 моль и т. д., называют соответственно деци-,
санти-, миллимолярными растворами. Концентрации
молярных растворов обозначают буквой М.§ 2. ВЫЧИСЛЕНИЯ В ТИТРИМЕТРИЧЕСКОМ АНАЛИЗЕДля вычислений в титриметрическом анализе сущест¬
венное значение имеет правильное определение экви¬
валентов и факторов эквивалентности реагирующих
веществ. Рассмотрим соответствующие примеры.Пример 1. Найти эквивалент и фактор эквивалентности
NaOH при титровании раствором хлороводородной кислоты. Ре¬
акция протекает по уравнениюNaOH + НС1 = NaCl + Н20.В этой реакции 1 моль молекул NaOH реагирует с 1 моль
молекул НС1. Каждая молекула НС1 содержит один ион водо¬
рода, поэтому эквивалентом NaOH является молекула NaOH,
а фактор эквивалентности равен единице.Молярные массы NaOH и НС1 равны соответственно
40 г-моль-1 и 36,6 г-моль-1, эти количества в данной реакции
эквивалентны.* Ранее молярную массу эквивалента называли грамм-экви¬
валентом.76
Пример 2. Найти эквивалент и фактор эквивалентности
Na2C03 в реакции с раствором НС1Na2C03 + НС1 = NaHC03 + NaOH.Здесь 1 моль молекул Na2C03 реагирует с 1 моль молекул
НС1 (или с 1 моль ионов Н+). Фактор эквивалентности, как и
в предыдущем случае, равен единице, а эквивалентом является
молекула Na2C03.Молярные массы Na2C03 и НС1 равны соответственно
106 г-моль-1 и 36,6 г-моль-1, эти количества в данной реакции
эквивалентны.Карбонат натрия может взаимодействовать с НС1 и по дру¬
гой реакции:Na2C03 + 2НС1 = Н2С03 + 2NaCl.С одним ионом водорода в данной реакции взаимодействует
условная частица 1/2 Na2C03, являющаяся в данном случае экви¬
валентом Na2C03, поэтому фактор эквивалентности равен 1/2.Из сопоставления двух последних реакций видно, что фактор
эквивалентности изменяется в зависимости от конкретной хими¬
ческой реакции, в которой участвует данное вещество.Пример 3. Найти эквивалент и фактор эквивалентности
Н3Р04 в реакции с. раствором NaOHН3РОч + 3NaOH = Na3P04 + 3H20.Один гидроксильный ион равноценен (эквивалентен) одному
иону водорода, поэтому фактор эквивалентности находим, ис¬
ходя из того, что три молекулы NaOH реагируют с одной моле¬
кулой Н3РОч. Следовательно, эквивалент Н3Р04 равен 1/3 части
молекулы Н3Р04, и фактор эквивалентности равен 1/3.Для определения фосфорной кислоты применяют другую ре¬
акцию:Н3Р04 + 2NaOH = Na2HP04 + 2Н20.Условная частица (эквивалент), реагирующая с двумя ОН-
(равноценными двум Н+)—это 1/2Н3Р04. Фактор эквивалент¬
ности равен, следовательно, 1/2.Молярные массы фосфорной кислоты и гидроксида натрия
равны соответственно 98 г-моль-1 и 80 г-моль-1, эти количества
в данной реакции эквивалентны.Пример 4. Найти эквивалент и фактор эквивалентности би¬
хромата калия в реакции:K2Cr2Or + 6KI + 7H3S04 = Cr2(S04)2 + 4K2S04 + 3I2 + 7НаО.Это реакция окисления — восстановления. Фактор эквива¬
лентности находят, принимая во внимание количество электро¬
нов, реагирующее с одним ионом Сг20^":Сг2°7' + 14Н* + = 2Сг3+ + 7Н2°-Эквивалент здесь — условная частица, присоединяющая один
электрон, т. е. 1/6К2Сг207, отсюда фактор эквивалентности ра¬
вен 1/6.77
Пример 5. Найти эквивалент и фактор эквивалентности
К2СГ2О7 в реакции:К2Сг207 + 2КОН = 2К2СЮ4 -f Н20.В отличие от предыдущего примера здесь не происходит
окисления или восстановления веществ; это кислотно-основная
реакция. Условшая частица (эквивалент) бихромата калия, реа¬
гирующая с одной молекулой КОН (равноценной с Н+)—это
1/2К2Сг207. Следовательно, фактор эквивалентности равен 1/2.Рассмотрим наиболее распространенный способ
вычислений в титриметрическом анализе. В первых
двух примерах факторы эквивалентности равны еди¬
нице, т. е. стехиометрические коэффициенты участни¬
ков реакции одинаковы. Одномолярные растворы со¬
держат по 1 моль молекул всех веществ, их количе¬
ства эквивалентны. Поэтому вещества, содержащиеся
в одномолярных растворах, реагируют между собой
при одинаковых объемах этих растворов. Аналогич¬
ный вывод справедлив для равных объемов 0,1 М,0,001 М и вообще для растворов веществ одинаковой
молярной концентрации. Растворы одинаковой моляр¬
ной концентрации, следовательно, реагируют между
собой в одинаковых объемах.В последующих примерах (3—5) факторы экви¬
валентности — дробные числа, меньшие единицы. Сте¬
хиометрические коэффициенты участвующих в реак¬
ции веществ различны: 1 моль молекул одного веще¬
ства (например NaOH) реагируют не с I моль моле¬
кул, а с 1 моль эквивалентов другого вещества (на¬
пример, Н3РО4). Поэтому для того, чтобы растворы
реагировали между собой в одинаковых объемах, не¬
обходимо выразить концентрацию другого вещества
в моль эквивалентах на литр.Рассмотрим зависимость между объемом раствора
и его молярной концентрацией. Чем концентрирован¬
нее раствор, тем меньший его объем необходимо
взять для того, чтобы он содержал такое же количе¬
ство растворенного вещества, какое содержится в не¬
котором объеме более разбавленного раствора. Так,
100 мл 0,1 М раствора по количеству вещества равно¬
ценны 10 мл 1 М раствора. Следовательно, между объ¬
емом раствора и его молярной концентрацией суще¬
ствует обратная пропорциональность. Определенный
объем раствора какой-либо молярной концентрации
всегда можно заменить большим или меньшим объ¬78
емом раствора другой молярной концентрации. Мате¬
матическое выражение этой зависимости имеет сле¬
дующий вид:IГ f '(4.4)V, С2V2 С, *где Vt и V? — соответственно объемы растворов с молярными
концентрациями С, и С2.Из приведенных рассуждений следует, что раство¬
ры разных молярных концентраций, выраженных в
моль эквивалентов на литр, реагируют между собой
в объемах, обратно пропорциональных их концен¬
трациям.При титриметрических определениях раствор оп¬
ределяемого вещества неизвестной молярной концен¬
трации Ci, взятый в объеме Vu титруют рабочим рас¬
твором известной молярной концентрации Сраб- р. Обо¬
значим объем рабочего раствора, израсходованного
на титрование, УРаб-р. Из сказанного можно заклю¬
чить, что между молярными концентрациями и объ¬
емами этих растворов существует соотношение= (4.5)'-'раб. р У IИЛИCiV] = Сраб. р^раб. р. (4.в)Из соотношения (4.6) можно найти неизвестную
концентрацию Сi определяемого вещества:Сраб. рУраб. рс,= рг . (4.7)Чаще бывает необходимо найти на основе данных
титрования массу вещества в граммах, содержащую¬
ся в некотором объеме раствора неизвестной концен¬
трации. Она равна произведению C\V\ на молярную
массу М миллиэквивалента вещества, т. е. на
/М/1000, где / — фактор эквивалентности. Тогда, учи¬
тывая уравнение (4.6), можно записать1^раб. рСраб. р/М1000(4.8)Следовательно, масса вещества в граммах В равна
произведению объема рабочего раствора Ураб- Р, из¬
расходованного на титрование, на его молярную кон¬79
центрацию Cpae p (выраженную в моль эквивалентах
на литр) и на 1/1000 часть произведения фактора эк¬
вивалентности /экв на молярную массу определяемого
вещества М.Нередко необходимо найти процентное содержа¬
ние вещества в анализируемом материале. На анали¬
тических весах берут определенную навеску пробы,
растворяют в воде или в другом растворителе и тит¬
руют полученный раствор рабочим раствором реа¬
гента. Процентное содержание определяемого веще¬
ства вычисляют из пропорцииg - 100 %Я-С%,где g — навеска пробы; В — масса вещества в граммах.Из этой пропорции получаем уравнение100В ^рзб. рСрзб. р/эквМс %= —= ^ • (4.9)Иногда применяют методы непрямого титрования,
один из них — метод титрования по остатку. К рас¬
твору определяемого вещества прибавляют отмерен¬
ное количество раствора реагента, взятого в опреде¬
ленном избытке. После этого титруют избыток реа¬
гента подходящим рабочим раствором. Результаты
титрования находят следующим образом.Предположим, что объем первого раствора, кото¬
рый израсходован на определение, равен V\, а его
молярная концентрация Сх. Объем и молярная кон¬
центрация второго рабочего раствора, который израс¬
ходован на взаимодействие с введенным избытком
первого, обозначим соответственно V2 и С2. Тогда
объем рабочего раствора Ураб.р (в мл), пошедшего на
реакцию с определяемым веществом, равен^раб. р = е,к, — с2к2. (4.10)Отсюда массу определяемого вещества вычисляем
по уравнению„ (С,К,~С21/2)/,квМ /J1Mв = iooo ' (4И1)Соответственно содержание (в %) вычисляем по урав¬
нениюС %=(CJV<~C£AL^. (4,2)80
На первых этапах развития титриметрического ана¬
лиза концентрацию рабочих растворов выражали ти¬
тром, т. е. массой вещества, содержащегося в 1 мл
раствора. Между титром и молярной концентрацией
раствора существует простая зависимость. В самом
деле, в 1 мл раствора молярной концентрации содер¬
жится /эквМ/1000 г вещества. Если молярная концен¬
трация раствора равна С, тогда, очевидно, в 1 мл рас¬
твора содержится7,==cJ9kbM. (413)1000 vВычислениями посредством титра удобно пользо¬
ваться в лабораториях, где выполняют много одно¬
типных анализов. В этом случае концентрацию
обозначают числом граммов определяемого вещества,
которое соответствует одному миллилитру рабочего
раствора. Так, при титровании карбоната натрия рас¬
твором хлороводородной кислоты можно заранее
вычислить, какой массе карбоната натрия соответст¬
вует 1 мл раствора хлороводородной кислоты. В этих
случаях говорят о титре хлороводородной кислоты по
карбонату натрия и обозначают Гна/Иагсоз- Для вы¬
числения массы вещества н этом случае, очевидно, не¬
обходимо помножить титр на объем рабочего рас¬
твора, израсходованного на титрование:В — ^раб. р^раб. р-Титр рабочего раствора по определяемому веществу
можно найти по уравнению (4.13), если вместо С под¬
ставить молярную концентрацию рабочего раствора,
а вместо /ЭквМ — произведение фактора эквивалент¬
ности на молярную массу определяемого вещества.
Следовательно,?экВМопр. враб. р/опр. р раб. р ЮОО ' 'Такой способ вычислений имеет определенные пре¬
имущества при использовании рабочего раствора для
определения только одного какого-либо вещества.
В других случаях целесообразнее пользоваться мо¬
лярными концентрациями растворов.Рассмотрим примеры вычислений по приведенным
уравнениям.81
Пример 6. Растворили в воде 1,46 г хлорида натрия и объем
раствора довели водой до 500 мл. Найти молярную концентра¬
цию этого раствора, имея в виду, что его используют в реакции
с раствором нитрата серебра.Реакция протекает в соответствии с уравнениемAgNOj + NaCl = AgCl + NaN03.Фактор эквивалентности равен единице и молярная масса хло¬
рида натрия, численно равная молярной массе его эквивалентна,
равна 58,46 г. Приготовленный раствор содержит 1,46 г хлорида
натрия в 500 мл, или 2,92 г в 1 л. Поэтому молярная концентра¬
ция раствора2 92С = J,, — 0,05 моль-л-'.5о,4оПример 7. Щавелевую кислоту в растворе определили по¬
средством титрования 0,1235 М раствором гидроксида натрия,
которого было израсходовано 13,50 мл. Реакция между Н2С2О4
и NaOH протекает по уравнениюН2С204 + 2NaOH = Na2C204 + 2Н20.Найти содержание щавелевой кислоты в растворе.Фактор эквивалентности щавелевой кислоты равен 1/2, мо¬
лярная масса Н2С204-2Н20 — 90 г моль-1. Вычисление проводим
по уравнению (4.10):О _т, п ^эквМН2С204-2Н20
аНгСгОА — kNaOHcNaOH fQOO = 13,5 • 0,1235 • 0,045 = 0,075 г.Пример 8. Растворили в воде 0,2 г технического гидрокарбо¬
ната натрия и раствор оттитровали 0,1 М раствором хлороводо¬
родной кислоты:NaHC03 + НС1 = Н2С03 + NaCl.На титрование израсходовали 20 мл НС1. Найти содержание
(в %) NaHC03 в техническом препарате.Для вычисления используем уравнение (4.9). Фактор эквива¬
лентности NaHC03 равен единице, а молярная масса —
84 г-моль-1. Поэтому^ ^HClCHCIMNaHC03 20,0-1,84 ^ ^10g — 10-0,2 / 3. ВЫЧИСЛЕНИЯ В ГРАВИМЕТРИЧЕСКОМ АНАЛИЗЕЗадача гравиметрического анализа обычно состоит в
определении содержания искомого элемента в пробе,
которое вычисляют на основании масс навески и гра¬82
виметрической (весовой) формы. Навеска g — это
масса вещества, взятого для анализа (в граммах);
гравиметрической формой называют соединение, кото¬
рое получают в процессе анализа из навески и массу
которого определяют на аналитических весах.Обозначим атомную (или мольную) массу опреде¬
ляемого элемента (или соединения) буквой А, моль¬
ную (или атомную) массу гравиметрической формы —
М, а количество атомов (моль) определяемого веще¬
ства в одной молекуле гравиметрической формы —
буквой п.Вычисление основано на следующих пропорциях.Один моль молекул гравиметрической формы со¬
держит п моль атомов (молекул) определяемого ве¬
щества. Если взвешиванием найдено, что масса гра¬
виметрической формы равна а, тогда массу х опре¬
деляемого вещества в а граммах гравиметрической
формы находим из пропорцииМ - А
а — хоткудапАа
* = -ЛГ*При вычислении содержания в процентах необхо¬
димо массу п выразить в процентах по отношению к
массе взятой навески. Поэтомуг - 100 %.х-в %откудаЮОяЛаВ = —Щ—Пример. Необходимо вычислить содержание (в %) магния в
сплаве с алюминием, исходя из следующих данных: навеска
сплава Q— 1,0135 г, гравиметрическая форма — MgjPjO?,
ее масса — 0,1325 г. Молярная масса М^РгСЬ равна 222,59, атом¬
ная масса магния — 24,32 и п — 2. Тогда24,32-2-0,1325- 100 _1,0135-222,59 ~ ,,)383
Для ускорения расчетов заранее вычисляют вели¬
чину пА/М, которую называют фактором или множи¬
телем пересчета.Удобно пользоваться таблицами факторов (мно¬
жителей). В них приведены факторы, необходимые
для вычисления результатов различных анализов. Та¬
кие таблицы помещены в справочниках*.Пример подобной таблицы приведен ниже:ОпределяютГГолучено (гравиметрическая
форма)МножительСаCaC2iO* ■ HjO0,2743СаCaSO*0,2944СаCaO0,7147СаОCaCaO+ ■ H200,3838MgMg2P 2O7! 0,2185MgOMg2P2070,3623В графе «Определяют» указан символ элемента
или формула вещества, содержание (в %) которого
определяют; в графе «Получено» — состав гравиме¬
трической формы; в графе «Множитель» — число,
представляющее собой отношение атомной или мо¬
лярной массы определяемого соединения к молярной
массе гравиметрической формы.* См., например, Ю. Ю. Лурье. Справочник по аналитиче¬
ской химии. М., Химия, 1989.
Раздел IIКИСЛОТНО-ОСНОВНЫЕ РЕАКЦИИГЛАВА 5СОВРЕМЕННЫЕ ПРЕДСТАВЛЕНИЯО КИСЛОТАХ И ОСНОВАНИЯХ.ТЕОРИЯ БРЕНСТЕДА§ 1. ОБЩИЕ ПОЛОЖЕНИЯВыдающийся литовский физик и химик Т. Гроттус
(Христиан Иоганн Дитрих) в 1805 г. впервые дал
правильное теоретическое обоснование процессов раз¬
ложения воды электрическим током и прохождения
тока через растворы электролитов, что заложило ос¬
новы теории электролитической диссоциации, основ¬
ные положения которой сформулированы шведским
химиком С. Аррениусом. Теория электролитической
диссоциации Аррениуса была разработана примени¬
тельно к водным растворам электролитов. По Арре¬
ниусу, кислоты — это электролиты, диссоциирующие в
водных растворах с образованием ионов водорода, ос¬
нования — электролиты, образующие в водных рас¬
творах ионы гидроксила:НА +=*= Н+ + А'; МОН ч=± М++ ОН\ (5.1)Аррениус считал, что в процессе диссоциации про¬
исходит также химическое взаимодействие растворен¬
ного вещества с растворителем. Но в литературе по¬
сле Аррениуса процесс диссоциации обычно описы¬
вали как чисто физический процесс, в котором не
участвует растворитель, рассматривая последний как
индифферентную среду. Известно, что Д. И. Менде¬
леев воздерживался от изложения теории электроли¬
тической диссоциации в «Основах химии», считая ос¬
новным ее недостатком игнорирование химического
взаимодействия между растворенным веществом и
растворителем. Д. И. Менделеев создал химическую
теорию растворов, придавая в ней большое значение
химическим процессам, проходящим в этих растворах.85
/Л. И. Менделеев писал, что он «...укрепил в себе
представление о природе растворов, сводящее их к
обычным случаям химического взаимодействия и к оп¬
ределенным, атомным соединениям, подобным — быть
может тождественным — с соединениями, содержа¬
щими кристаллизационную воду». По Менделееву,
растворы представляют жидкие диссоциационные си¬
стемы, образованные частицами растворителя, рас-
створенного тела и тех определенных нестойких, но
экзотермических соединений, которые между ними
происходят, одного или нескольких, смотря по при¬
роде составляющих начал.При экспериментальном исследовании свойств рас¬
творов веществ по их плотности Д. И. Менделеев
установил в растворах серной кислоты и спирта обра¬
зование гидратов различного состава, например
H2S04-H20, H2S04-2H20, H2S04-6H20, С2Н50Н-Н20,
С2Н50Н-ЗН20, С2Н50Н-12Н20 и др. 'Таким образом, в теории Аррениуса и многих ра¬
ботах, посвященных изучению растворов, мало вни¬
мания было уделено химическим процессам, проходя¬
щим при растворении веществ. Уже тогда было ясно,
что диссоциацию кислот в водных растворах нельзя
описывать уравнениемНА Н+ + А~.Свободный протон, размеры которого во много раз
меньше размеров других ионов, создает вокруг себя
электрическое поле большой интенсивности, в которое
втягиваются дипольные молекулы воды. Поэтому в
водных растворах нет свободных протонов, они, реаги¬
руя с молекулами воды, образуют ионы гидроксония
Н30+. Исходя из этих соображений, диссоциацию кис¬
лот более правильно записывать таким уравнением:HA + H20 н30+ + А~. (5.2)В водных растворах существуют и более сложные
частицы, например H(H20)^, H(H20)J и др.Изучение свойств неводных растворов кислот вы¬
явило и другие недостатки теории Аррениуса. Так,
растворы некоторых кислот в неводных растворите¬
лях, обладая кислотными свойствами, не содержат
ионов водорода. Например, раствор хлороводорода в86
бензоле не проводит электрический ток, но взаимодей¬
ствует с металлическим цинком, выделяя водород; он
характеризуется каталитической активностью и мо¬
жет быть нейтрализован основаниями. Оказалось, что
кислотные и основные свойства могут проявлять не
только ионы водорода и гидроксила, но и другие
ионы, например NHJ и NH2 в жидком аммиаке и т. д.Существует много различных теорий кислот и ос¬
нований. Одна группа теорий связывает кислотные
свойства с присутствием в соединении водорода. Наи¬
более важная из них — теория Бренстеда, которая
ниже рассматривается более подробно. Другая груп¬
па— это апротонные теории, не связывающие кислот¬
ные свойства с обязательным присутствием водорода
в составе соединения. Согласно этим теориям, носи¬
телем кислотных свойств может быть не только сво¬
бодный или сольватированный протон, но и любой дру¬
гой катион или частица. К числу таких теорий принад¬
лежат теории Усановича и Льюиса.Так, по Льюису, кислоты и основания — это веще¬
ства, являющиеся акцепторами и донорами протонов
соответственно. Атомы азота в аммиаке располагают
свободной парой электронов, поэтому аммиак высту¬
пает в качестве донора этой пары и является основа¬
нием. Бор в ВС13 имеет вакантные места для присо¬
единения пары электронов, поэтому BCI3 представ¬
ляет собой кислоту. Приведенное ниже уравнение
иллюстрирует взаимодействие между ВС13 и NH3:Усанович называет кислотами вещества, способ¬
ные выделять катионы, в том числе протоны, или
присоединять анионы, в том числе электроны. Основа¬
нием по Усановичу являются вещества, способные
присоединять катионы или выделять анионы.Теории Льюиса и Усановича не общепризнаны, не
имеют практического значения в аналитической хи¬
мии и здесь подробно не рассматриваются.Н CI
Н: N: + В : С1
Н С1н С1
Н: N: В : С1
Н С1.осно- ки-
вание слотасоль87
Теория Бренстеда устанавливает количественные
закономерности, характеризующие диссоциацию кис¬
лот и оснований в различных растворителях. Она ох¬
ватывает как водные, так и неводные растворы кис¬
лот и оснований и учитывает не только физические, но
и химические факторы, влияющие на диссоциацию
кислот.По Бренстеду, кислоты — это частицы, способны
отщеплять протоны, а основания — частицы, способ¬
ные их присоединять. Протон может выделяться толь¬
ко в том случае, если в системе есть основание, спо¬
собное этот протон присоединять. Кислотно-основное
взаимодействие поэтому представляет собой двойное
протолитическое равновесие, в котором принимают
участие две пары сопряженных кислот и оснований.
Например, диссоциация уксусной кислоты описы¬
вается следующим уравнением:СНзСООН + Н20 = СНзСОО' + н3о+.кислота I осно- основание I ки¬
вание II слота IIЗдесь одна сопряженная пара — это уксусная кис¬
лота и основание — анион этой кислоты, вторая па¬
ра — молекула воды и катион гидроксония.По Бренстеду, кислотами являются следующие со¬
единения или ионы.1. Нейтральные кислоты. Термин «нейтральные» не
означает, что вещество в растворе имеет нейтральную
реакцию. Нейтральность понимается как отсутствие
электрического заряда.2. Анионные кислоты HS03, НСОз, НРО4' и др.Катионные кислоты — ониевые катионы NHJ,CsHsNHj, лиониевые ионы растворителя Н30+ :?=±:Н20 + Н*, аквакомплексы металлов А1(Н20)б* 4=*
3=^ А1(Н20)50Н2* + Н+, комплексные ионы
Pt(NH3)5H204+ =р=* Pt(NH3)5OH3++ Н+.Основаниями являются следующие соединения
или ионы.1. Нейтральные основания Н20, NH3, C6H5NH2
и т. д.2. Анионные основания С1-, Вг_, 1~ СН3СОО-он-88
Многие частицы одновременно могут играть роль
оснований и кислот, т. е. для них характерна амфо-
терность, например:нсо; + н2о = н2со3 + он-осно* кисло- кислота I осно¬
вание Г та II ванне (IВ этом уравнении основание НСОз сопряжено с
кислотой Н2СОз- Частица НСОз является амфолитом,
так как способна и отщеплять, и присоединять про¬
тоны.Главная особенность теории Бренстеда — учет ро¬
ли растворителя в кислотно-основном равновесии.
Растворитель рассматривается не только как физиче¬
ская среда с определенным значением диэлектриче¬
ской проницаемости, но и как вещество, которое хи¬
мически реагирует с кислотой или основанием. При
взаимодействии кислоты НА с растворителем S про¬
текает реакцияНА + S ^ HS+ + А'. (5.3)Это взаимодействие является результатом двух со¬
пряженных реакций:реакции отщепления протонаНА ч=* Н+ + А' (5.4)и реакции присоединения протона растворителем,
выступающим в данном случае в роли основанияН* + S HS+. (5.5)Следовательно, суммарная константа двойного прото-
литического равновесия (5.3)[HS-] [а-]* HAS - -[НА] [S]определяется отношением двух константКна*has “тг*-. (57)HS+где„ _ [Hits] и у [Hi [A-] (5.8)HS+ [HS+] [НА] ‘89
Рис. 5.1. Зависимость рАГ хлор¬
ной кислоты от обратного зна¬
чения диэлектрической прони¬
цаемости неводных раствори¬
телей (карбоновых кислот)Это означает, что спо¬
собность кислот диссоци¬
ировать в данном раство¬
рителе зависит, с одной
и 01 0 2 03 1/D стороны, от способности
’ ' кислоты отщеплять про¬тон по уравнению (5,4), а с другой — от способности
растворителя присоединять этот протон в соответст¬
вии с уравнением (5.5).Некоторые наиболее важные выводы теории сфор¬
мулированы ниже.1. Для нейтральных (незаряженных) кислот типа
НА логарифм константы Кнas, взятый с обратным
знаком — pKhas, является линейной функцией обрат¬
ной величины диэлектрической проницаемости 1 /D:p^has == const + <5-9)Примером может служить изменение pKhas хлор¬
ной кислоты HCIO4 в растворителях с различными
значениями диэлектрической проницаемости. Если,
например, взять в качестве растворителей НСЮ4 без¬
водные карбоновые кислоты — на графике получается
линейная зависимость р/^нсю! от 1/D (рис. 5.1).2. Разность значений pKhas незаряженных (ней¬
тральных) кислот в двух разных растворителях дол¬
жна быть величиной постоянной:P*HAS2-P*HAS,=const- (5-Ю)Так, сила различных кислот в водной и бутаноль-
ной средах изменяется в одинаковой степени.
В табл. 5.1 в качестве примера приведены некоторые
данные, из которых видно, что разность АрК — вели¬
чина практически постоянная.Не во всех случаях теория Бренстеда правильно
объясняет изменение силы кислот при переходе от
одного растворителя к другому.Зависимости, выраженные уравнениями (5.9) и
(5.10), справедливы только в тех случаях, если срав-90
Таблица 5.1. Значения рК карбоновых кислот в воде
и в бутанолеКислотаVK в водерК в бутанолеДрКТрнхлоруксусная0,76,35,6Дихлоруксусная1,37.36,0Монохлоруксусная2,98,55,6л-Нитробензойная3,59,25,7Бензойная4,210,26,0Уксусная4,810,45,7нивают силу кислот в растворителях одинаковой хи¬
мической природы (см. табл. 5.1). Для растворите¬
лей и для кислот различной химической природы
имеет значение не только диэлектрическая проницае¬
мость, но и специфика химического взаимодействия,
которую пока не удается охарактеризовать какими-
либо количественными параметрами.Теория Бренстеда получила дальнейшее развитие
в работах Н. А. Измайлова, в которых подробно
рассмотрены причины отклонения от теории и разра¬
ботана более совершенная и общая теория элект¬
ролитов.Для аналитической химии большое значение имеет
дифференцирующее действие растворителей на силу
кислот и классификация неводных растворителей.§ 2. КЛАССИФИКАЦИЯ НЕВОДНЫХ РАСТВОРИТЕЛЕЙ
ПО ИХ ДИФФЕРЕНЦИРУЮЩЕМУ ДЕЙСТВИЮ
НА СИЛУ КИСЛОТВсе неводные растворители можно классифицировать
на основании их способности к взаимодействию с
протонами. По этому признаку растворители делятся
на апротонные и протолитические.Молекулы апротонных растворителей неспособны
к отщеплению протонов или к их присоединению, они
не ионизированы. Растворенные в апротонных рас¬
творителях кислоты и основания также неспособны к
диссоциации. Примером апротонных растворителей
служит бензол.Молекулы протолитических растворителей могут
присоединять или отщеплять протоны. Они в свою
очередь делятся на следующие группы.91
1. Амфипротные (амфотерные) растворители, мо¬
лекулы которых способны отщеплять и присоединять
протоны. К ним относятся, например, вода, метанол,
этанол, фенолы. Диссоциация воды проходит по урав¬
нениюН20 + н20 Ч=>: НаО+ + ОН". (5.11)В этом процессе проявляются амфотерные свойства
воды. Аналогично диссоциирует метанол:СН3ОН + СН3ОН СН30Н2" + СН/Г. (5.12)При диссоциации образуются катионы метоксония и
анионы СН30~.В амфипротных растворителях, которые прибли¬
зительно в одинаковой степени являются акцепто¬
рами и донорами протонов, диссоциируют и кислоты,
и основания.2. Протофильные (основные) растворители, моле¬
кулы которых очень легко присоединяют протоны.
В таких растворителях диссоциация кислот усили¬
вается. Примерами таких растворителей являются пи¬
ридин, гидразин.3. Протогенные (кислые) растворители, молекулы
которых мало склонны к присоединению протонов, но
легко их отщепляют. В таких растворителях легко
диссоциируют основания, диссоциация кислот незна¬
чительна. Примерами растворителей этого типа яв¬
ляются безводные карбоновые кислоты — уксусная,
масляная, муравьиная.Для аналитической химии важна классификация
растворителей по их способности дифференцировать
или нивелировать силу кислот и оснований. По этому
критерию все растворители делят на две группы.Нивелирующие растворители — это растворители,
в которых сохраняется соотношение в силе кислот или
оснований, характерное для водных растворов этих
электролитов. К нивелирующим растворителям отно¬
сят в первую очередь химические соединения, содер¬
жащие гидроксильные группы, например спирты, фе¬
нолы и др.Дифференцирующие растворители — это раствори¬
тели, в которых проявляется значительное различие
в силе кислот и оснований. В этих растворителях со¬
отношение в силе электролитов иное, чем в водных
растворах. Обычно такие растворители не обладают92
четко выраженным свойством донора или акцептора
протонов.В аналитической химии большее применение нахо¬
дят дифференцирующие растворители (альдегиды, ке-
тоны и нитрилы). Существует несколько типов диф¬
ференцирующего действия растворителей.Первый тип — изменение относительной силы кис¬
лот различной химической природы. По отношению к
кислотам дифференцирующее действие проявляют
практически все растворители. Наиболее яркие при¬
меры—изменение относительной силы бензойной
кислоты и 2,6-динитрофенола, хлороводородной кис¬
лоты и моно- или дихлоруксусной кислот. Бензойная
кислота и фенол (или производные фенола) являются
кислотами, но по своему химическому характеру это
соединения, принадлежащие к различным классам.
Кислотность растворов бензойной кислоты обуслов¬
лена наличием карбоксильной группы, а кислотность
растворов фенола — наличием оксигруппы. Хлорово¬
дородная и хлоруксусная кислота также сильно раз¬
личаются по своей химической природе: НС1 — мине¬
ральная кислота, хлоруксусные кислоты принадлежат
к классу органических карбоновых кислот. Дифферен¬
цирующее действие проявляется в следующем. В во¬
де бензойная кислота и 2,6-динитрофенол — кислоты
приблизительно одинаковой силы. Однако в этаноле
бензойная кислота становится почти в десять раз сла¬
бее 2,6-динитрофенола. Наибольшее дифференцирую¬
щее действие проявляет ацетон — в нем названные
кислоты различаются по силе примерно в 45 раз.
В воде хлороводородная кислота диссоциирует в
25 раз сильнее хлоруксусной кислоты. Однако в аце¬
тоне хлороводородная кислота становится сильнее
хлоруксусной в 9-105 раз.Второй тип дифференцирующего действия раство¬
рителей — изменение относительной силы минераль¬
ных кисло'г в протогенных растворителях. Так, в воде
приведенные ниже кислоты являются сильными кис¬
лотами и диссоциируют практически полностью. Од¬
нако в уксусной кислоте как растворителе они очень
сильно различаются по своим константам диссоциа¬
ции, а именно:Кислоты . . НСЮ4 HBr Н SO< НС1 HN03
p/C ... . 5,8 6,4 8,2 8,9 9,4S3
Диэлектрическая проницаемость уксусной кислоты
равна 6.1. Следовательно, диссоциация сильных кис¬
лот должна ослабляться при переходе от водных рас¬
творов к растворам в уксусной кислоте. Протогенные
свойства уксусной кислоты как растворителя также
должны обусловливать ослабление диссоциации кис¬
лот. Но из приведенных данных видно, что кроме ос¬
лабления диссоциации происходит также дифферен¬
циация силы кислот. В уксусной кислоте разность
значений р/С НСЮ4 и HNO3 составляет 3,6, т. е. эти
кислоты различаются по способности к диссоциации
приблизительно в 65 раз.Третий тип дифференцирующего действия раство¬
рителей — изменение относительной силы минераль¬
ных кислот в основных (протофильных) растворителях
с низкими значениями диэлектрической проницаемо¬
сти. Так, рК минеральных кислот в пиридине имеют
следующие значения:Кислоты . . HI HCIO4 НВг HNOa НС1 HFрК ... ■ 2,5 3,2 4,0 4,3 5,4 8,5В воде все эти кислоты, за исключением HF, силь¬
ные. Пиридин ослабляет силу кислот. Здесь прояв¬
ляется влияние диэлектрической проницаемости рас¬
творителя. Но сила кислот в пиридине ослабляется
меньше, чем в уксусной кислоте. Это объясняется раз¬
личием химической природы уксусной кислоты и
пиридина. Уксусная кислота — протогенный раствори¬
тель, мало склонный к присоединению протонов. Пи¬
ридин, наоборот, принадлежит к группе протофиль¬
ных растворителей, легко присоединяющих протоны,и, следовательно, усиливающих диссоциацию раство¬
ренных в нем кислот. Характерно, что сила кислот
уменьшается неодинаково: пиридин дифференцирует
силу кислот.ГЛАВА 6КОНЦЕНТРАЦИЯ ВОДОРОДНЫХ ионов
В ВОДНЫХ РАСТВОРАХ ЭЛЕКТРОЛИТОВ
§ 1. КОНЦЕНТРАЦИЯ ВОДОРОДНЫХ ИОНОВ ВОДЫ.
ИОННОЕ ПРОИЗВЕДЕНИЕ ВОДЫВода в очень незначительной степени диссоциирует с
образованием ионов гидроксония и гидроксила:Н20 + Н20 ^=±: НзО+ + ОН' (6.1)94
или в упрощенном видеН20 4=*: н* + ОН'.(6.2)По закону действия массИзмерения показали, что концентрация ионов во¬
дорода чистой воды, а следовательно, и концентра¬
ции ионов гидроксила равны[Н+] = [0Н']=И ■ 10'7М. (6.4)Концентрация воды в воде, т. е. количество молей
воды в 1 л равно1000Поэтому[Н20] = == 55,56 М. (6.5)1о*н2о = 1 5gja'"~= *'78 • 10~'6 (б6)Диссоциация воды очень незначительна, поэтому
концентрацию [Н20] можно считать практически по¬
стоянной, откуда/Hj0=[H+] [OH ] = 1 • 10 7 ■ 1 • 10 7 = 1 • Ю-и, (6.7)т. е. произведение концентрации ионов воды есть ве¬
личина постоянная при постоянной температуре. Ее
называют ионным произведением воды 1и2о-Диссоциация воды возрастает с температурой. Так,
при 99—100 °С ионное произведение воды равно
1-10-12, а концентрация водородных ионов равна
1 • 10® моль ионов в 1 л, т. е. увеличивается в десять
раз по сравнению с [Н+] при 20 °С.Кислотность водных растворов характеризуется
так: в нейтральных растворах [Н+] = [ОН-] =
= 10~7\ в кислых растворах [Н+] > [ОН-], [Н+] >
> 10-7; в щелочных растворах [Н+] < [ОН-],
[Н+] < ю-7.Кислотность растворов целесообразно характери¬
зовать не концентрацией водородных ионов, а водо¬
родным показателем:pH = — lg [Н+]. (6.8)95
[Н], 10“ to-' Ю'2 10'a 10-4 10-5 to 6 10'7 10-® 10-* 10-1° 10-tl 10-,2)0-’310-uMOfl'b
ИОНОЙ
В 1 RI I % v I i *__! » I % . I » « I « t —1-\ 1 I 1 vJpppf 111 1 1 irprrti 1 ii 1 111 "H* T 1^11pH 0 < 2 34 5 6 7 ( 9 10 11 12 13 U
I I I I I I I I I I IV 1 '-iпение кисло!* |Увеличение кислот- j Увеличение щелочности
ности 1рОН 14 ' 13 12 11 10 9 8 7 Б S « 3 2 1 0|_J I I I I I 1 I 1 1 1 1 L 110-14 10 ,а 10 12 10 "ю 1“10 9 ЮЧО-'Ю '10'5 10< 10-3 ю-2 10-1 10°[°н1. I.. I .. I.. I.. I.. I.. 1
моль рн Г 1 1Г71ГТПГТ^ИОНОВ !
BinсильнокислаяслабокислаяслабощелочнаясильнощелочнаянейтральнаясредаРис. 6.1. Связь между концентрациями водородных и гидроксиль¬
ных ионов, pH и рОНТогда водные растворы различной кислотности ха¬
рактеризуются следующими значениями pH: ней¬
тральные— pH = 7; кислые — pH < 7; щелочные —
pH > 7. Логарифмируя уравнение (6.7), получаемlg [н+] + lg [ОН-] = - 14 или pH + рОН = 14. (6.9)Связь между концентрацией водородных и гидро¬
ксильных ионов, pH и рОН представлена на рис. 6.1.§ 2. РАСТВОРЫ сильных кислот
И СИЛЬНЫХ ОСНОВАНИЙСильные кислоты и сильные основания диссоциируют
в водных растворах полностью. Следовательно, для
сильных кислот[н1 = Ск. (6.10)откудаpH = — lg [н+] = — lg Ск. (6.11)96
Так, для 0,01 М раствора хлороводородной кис¬
лоты находимрН= - lg ю 2 = 2,0.Для 0,03 М раствора этой же кислотыpH = — lg з • ю-2 = 2 - lg з = 1,5,а для 1 М раствора pH =0.Для сильных оснований[ОН-1==С0 (6.12)илиPOH = -1g[OH~l = -1gC0, (6.13)или, учитывая, чтоpH + рОН = 14,
рН= 14 — рОН = 14 + lgC0. (6.14)Так, для 0,01 М раствора NaOHрОН = - lg 10-2 = 2,0 (6.15)или pH =14 — 2=12.Одномолярный раствор гидроксида натрия имеет
pH = 14.§ 3. РАСТВОРЫ СЛАБЫХ КИСЛОТ
И СЛАБЫХ ОСНОВАНИЙВ соответствии с теорией Бренстеда (см. гл. 5, § 1)
кислотами являются вещества, способные отщеплять
протоны. У слабых кислот тенденция к отщеплению
протонов выражена незначительно, т. е. состояние
равновесия реакцииНА+Н20 ч=± НзО+ + А‘ (6.16)сдвинуто в левую сторону.К таким слабым кислотам принадлежат нейтраль¬
ные (не имеющие заряда) кислоты типа уксусной,
борной, синильной, а также катионные кислоты, на¬
пример катионы слабых оснований NH4, аквакомп¬
лексы металлов, например А1(Н20)в*> комплексные
ионы металлов [PtfNHaJsHjO44- и др.], катионы ор¬
ганических аминов (CSH5NH3, C5H5NH* и т. д.)4 Аналитическая химия97
Слабые основания-—это соединения, которые спо¬
собны быть акцепторами протонов:В + Н20 = ш-Г + ОН”. (6.17)В состав таких соединений не обязательно входят
гидроксильные ионы. К ним принадлежат NH3 и мно¬
гие его органические производные (амины — метил¬
амин CH3NH2, этиламин C2H5NH2, пиридин C2H6N,
2,2/-дипиридил), анионы слабых кислот (СНзСОО ,
В02 и др.), катионы [А1(ОН)(Н20)з' и др.]. К осно¬
ваниям присоединяются протоны воды.Из уравнений (6.16) и (6.17) видно, что в этих
равновесиях участвуют сопряженные пары кислот и
оснований. Так, сопряженным с кислотой НА является
основание А-, сопряженной с основанием В является
кислота ВН+. Легко установить связь между констан¬
тами сопряженных кислот и оснований.Для кислоты НА константа диссоциации (констан¬
та кислотности) запишется так:[Н+] [А ] (618)ак [НД]Для основания А- константа диссоциации основа¬
ния (константа основности) следует из уравненияА" + Н20 = НА + ОН" (6.19)[НА] [ОН’]Ко Рп—■ (620)Перемножая уравнения (6.18) и (6.20), получаемv. - Т ТГГ - [н*] [он-] - к„!0. <ы„т. е. произведение констант кислотности и основности
равно ионному произведению воды.Таким образом, если известно Кк, тогда из (6.21)
легко найти константу основности сопряженного осно¬
вания Ко (и наоборот).Уравнение, аналогичное уравнению (6.21), можно
получить и из уравнения (6.17).Рассмотрим вывод уравнения для вычисления pH
растворов слабых кислот на примере кислоты НА.98
Уравнение (6.10) в упрощенной записи имеет видНА я=^ Н+ + А". (6.22)Ионы водорода образуются не только вследствие
диссоциации кислоты, но также при диссоциации
воды. Однако для раствороЕ! не очень слабых кислот
можно не принимать во внимание диссоциацию воды
и считать, что[Н+] = [А']. (6.23)Концентрация недиссоциированных молекул кис¬
лоты НА равна[НА] = Ск — [Н+]. (6.24)Подставляя уравнения (6.23) и (6.24) в уравнение
(6.18), имеемfH+l'К к= -г- (6-м>Ск - [HiОткуда после преобразований приходи л к квадрат¬
ному уравнению[н+]2 + кк[н+]-л;кск = о. (6.26)Следовательно, [H+] = --^+д/^У+/СкСк. (6.27)В большинстве практически важных случаев урав¬
нения (6.25) и (6.27) можно упростить. Пусть, напри¬
мер, по условиям поставленной задачи при расчете
концентрации водородных ионов вполне допустима
погрешность, равная (или меньше) 5 % определяемой
величины, т. е. [Н+] ^ 0,05 Ск. Тогда, подставляя в
уравнение (6.25) [Н+] ^0,05СК, после преобразова¬
ний получим4^- > 400, (6.28)ЛКт. е. если общая концентрация кислоты по меньшей
мере в 400 раз больше константы диссоциации, можно
пренебречь в знаменателе уравнения (6.25) концен¬
трацией водородных ионов, и*К = -Ю. (6.29)^ КПри этом погрешность расчета не превысит 5 %•4* 99
Из уравнения (6.29)[н*] = У/СкСк(6.30)ирн = -рл:к -(6.31)Из уравнения (6.31) видно, что при изменении
концентрации кислоты в 10 раз pH изменяется на 0,5.
В соответствии с характером обозначения pH, при
увеличении концентрации кислоты pH уменьшается,
а при разбавлении раствора кислоты значение pH воз¬
растает.Пример 1. Вычислить pH 0,035 М раствора уксусной кис¬
лоты, Кк = 1,7-Ю-5.значительно больше 400. Отсюда следует, что при расчете по
упрощенному уравнению (6.31) погрешность будет меньше 5 %■
ПоэтомуПример 2. Вычислить pH 0,1 М раствора циановодородной
кислоты, p/Chcn = 9,2.Для 1 М раствора HCN в соответствии с уравнением (6.29),
pH равно 4,6. Для 0,1 М раствора pH должно быть больше на
0,5, т. е. pH = 5,1.Пример 3. Вычислить pH 0,01 М раствора фтороводородной
кислоты, p/Chf = 3,21, Khf = 6,2-10-4.т е. значительно меньше 400. [см уравнение (6.28)1. Поэтому
вычисление по упрощенной формуле даст ошибку, значительно
превышающую допустимые 5%. Расчет нужно вести по уравне¬
нию (6.27):рН = -рКк- -g-lgC*.но рАГк = - lg Лк = - lg 1,7 - 10“й = 5 - 0,23 = 4,77 и pH = XХ4.77 - 4- lg 3.5 • 10~2 = 2,39 - -1. (0,54 - 2.0) = 2,39 -+ ■ 1,46=
£ J. Л= 3,12.= -3,1-10 4 + 2,5-10 3 = 2,2 • 10-а
pH = — lg 2,2 ■ 10~3 = 3 — lg 2,2 = 3 - 0,34 = 2,66.100
В самом деле, если применить уравнение (6.3()
yP^HF - ~2pH — 'o'P^HF Т ^HF — 1,6 + 1 = 2,6[Н+] = 10 3- 10|),4 = 2,5 ■ 10 3,
т. е. погрешность расчета составит2,5 • 10_3 — 2,2 • 10~3 = 0,3 ■ 10_3
или в процентах по отношению к правильному результату0.3-ю-3-1.00 =13|6%-
2,2 ■ 10 3Уравнения (6.27) и (6.31) пригодны для вычисления pH рас¬
творов, содержащих слабые катионные кислоты NH*, CflH5NH3
и др.Пример 4. Вычислить pH О,! М раствора >лорида аммония;
по условиям задачи достаточно получить приближенные резуль¬
таты, р/Смн+=9,25.4В этом случае можно воспользоваться упрэщенным уравне¬
нием (6.31):pH = 9,2 - -- lg С „ = 5,1v 2 ' S! s nh4Для вычисления pH растворов многоосновных сла¬
бых кислот нужно рассмотреть вклад всех ступеней
диссоциации.Ступенчатая диссоциация, например угольной кис¬
лоты, выражается уравнениямиН2С03 ч=»: НС03 + Н\HCOj ч=± СО2" + Н+.Соответственно константы диссоциации равны
[н*] [нсо-1 _7
[н!со,1 -3-10 7^' =6'5)--[н + ] [СО*‘1
Кг = - Р- = 6-10 11 (р/С2== 10,2).[нсо-JТаким образом, вторая константа диссоциации в
4000 раз меньше первой, поэтому втор; я ступень дис¬
социации практически не имеет значения для pH
раствора слабой кислоты. Это правило имеет об¬
щий характер — соотношение между ступенчатыми, 101
константами диссоциации многоосновных кислот
обычно составляет Ki : К2 ■ Кз = 1 : Ю-5 : 10~10. Кро¬
ме того, необходимо иметь в виду, что в рассматри¬
ваемом примере концентрация ионов НСОз очень
мала, так как большая часть угольной кислоты нахо¬
дится в растворе в виде молекул Н2С03. Поэтому,
например, для 0,01 М раствора угольной кислоты
находим:pH = 1 рК, - 1 lgCK = 1 • 6,5 - 1 lg 10~2 = 3,25 + 1 = 4,25.Аналогичные зависимости справедливы и для дру¬
гих кислот.Таким образом, при вычислении pH растворов
слабых кислот необходимо принимать во внимание
только первую константу диссоциации.Вывод уравнения для вычисления pH слабых осно¬
ваний рассмотрим на примере уравнения (6.17):[ВН+] [ОН ] Ки„п
к° = [В] = <6-32>гдег [H+][BI ютмКк-~тТ- (6-33)Из уравнения (6.17) видно, что[ВН*] = [ОН-]. (6.34)При этом не принимается во внимание концентрация
гидроксильных ионов воды.Из уравнений (6.34) и (6.32) следует:[он-]2 кНа0[В] к к(6.35)Концентрация [В] = С0—[ОН-]. Однако для сла¬
бых оснований, у которых реакция (6.17) протекает
незначительно и, следовательно, С0 » [ОН-], можно
принять, что[ В] = Со. (6.36)Для более сильных оснований такое допущение не¬
правомерно, и дальнейшие выкладки нужно вести так,
как это показано в уравнениях (6.25) и (6.27) для
слабых кислот.102
Сочетая уравнения (6.35) и (6.36), после преобра¬
зований получаем: [ОН-] = V«rC°- ’ (6'37)откуда после логарифмированияpOH = 7-yp/?K-yIgC0 (6.38)или pH = 14 — рОН, поэтомуpH = 7 +1р*;к + 1 lgCo. (6.39)Из уравнения (6.39) видно, что pH сснований воз¬
растает с ослаблением диссоциации кислоты, сопря¬
женной с этим основанием: чем больше р/Ск, тем
выше pH. Из этого уравнения следует, что pH рас¬
твора уменьшается при разбавлении в 10 раз на 0,5.Пример 5. Вычислить pH 0,01 М водного р; створа пиридина
C2H5N, исходя из таких дачних:= jcsHsV]_[h ] = _ 10-Л = 5 23[С3Н3М1Г]Из уравнения (6.25) получим:pH = 7 + ~ ■ 5,23 + i- lg 10~2 = ;!,6.Пример 6. Определить pH 0,01 M растворд аммиака.
Применяя уравнение (6.25) голучаем:pH = 7 + у • 9,25 + ~ lg 10-2 = К ,64.Пример 7. Вычислить pH 0,1 М водного раствора цианида
калия.Принимая во внимание, что /(hcn = 7,2-10“ 0 или р/С = 9,14,
получаем:pH = 7 + ~ • 9,14 + 1 lg [CN-J = 11,07.Удобно запомнить формулу (6.25) для 1 М рас¬
твора основания, когда CQ = 1 и lgCo=0:pH = 7 -j—рКк- (6.40)Присоединение протонов из воды к многооснов-
ным основаниям типа А2-, А3- и т. д. происходит103
ступенчато, напримерА2' + Н20 ч=* НА' + ОН",HA~ + H20 Н2А + он-и т. д.Эти равновесия характеризуются следующими
константами:[НА-] [ОН'] [Н2А] [ОН ]
[АЧ И 01 [НА-] •Связь протона с основанием А2- обычно значи¬
тельно сильнее, чем с НА-, т. е. Ко, ^ К о,. Поэтому
вторую стадию реакции при расчете pH можно не
принимать во внимание.1' н*Из уравнения (6.21) следует, что/С0, = ~~jr.. к»В самом деле,„ „ [НА'] [ОН ] [Н+] [А2 ] ,,°‘ кг [А2'] [НА'] Н2°’откудаv Кн*°^н2о<<2Таким образом, при вычислении pH растворов мно¬
гоосновных оснований следует пользоваться послед¬
ней константой кислоты, сопряженной с данным осно¬
ванием, т. е.рн = 7 + — рКп + lg са,где рКл — последнее рК кислоты, сопряженной с основанием.
Пример 8. Вычислить pH 1 М раствора карбоната натрия.
По справочной таблице находим, что рК, = 6,5 и p/C2=10,2.
Поэтому pH = 7 + 1/2-10,2 = 12,1.Пример 9. Вычислить pH 0,1 М раствора Na3P04.Для фосфорной кислоты p/C, = 2,1, рл2 = 7,2 и рКз = 12,3.Следовательно, pH =7 ~ ■ 12,3 + ~ lg 10_ 1 = 12,65.§ 4. РАСТВОРЫ АМФОЛИТОВСреди упоминавшихся в § 3 кислот и оснований есть
и такие, которые имеют амфотерные свойства и мо¬
гут выступать и как доноры, и как акцепторы прото¬
нов. Так, среди кислот к амфолитам относят заря¬104
женные кислоты типа НСОз, Н2РО4 и др. К амфоли-
там — основаниям относится, например, аммиак.
Рассмотрим свойства амфолитов типа НА~.
Уравнение кислотной диссоциации имеет видНА" + Н20 == А2" + НзО*. (6.41)или в упрощенной формеНА' Н+ + А2“.1 (6.42)Анион НА- представляет собой сопряженное основа¬
ние кислоты, поэтому устанавливается и второе рав¬
новесие:НА'+Н20 ч=* Н2А + ОН". (6.43)В реакции (6.42) выделяются протоны, в реакции
(6.43) образуются ионы гидроксила. Если реакция(6.42) идет в большей степени, чем реакция (6.43),
концентрация Н+ превышает концентрацию ОН~ и
раствор амфолита будет иметь кислую реакцию. При
обратном соотношении реакция растрора будет ще¬
лочной.Если бы в растворе происходил только процесс(6.42) и не было бы взаимодействия НА- с водой,
тогда концентрация ионов водорода была бы равна
концентрации А2-:[Н-] = [А2-]. (6.44)Однако в растворе наряду с реакцией (6.42) идет
реакция (6.43). Гидроксильные ионы вступают в реак¬
цию с ионами водорода, вследствие чего [Н+] <
< [А2-] на концентрацию Н2А, которая численно
равна [ОН-]. Поэтому[Н1 = [А2 ]-[Н2А]. (6.45)Запишем выражения для первой и второй кон¬
стант диссоциации сопряженных кислот НА- и Н2А:^ [Н*] [НА-]*' “ [Н3А] (6'46)и„ _ [Hi [А2-]1 *105
Принимая концентрацию анионов НА- равной С0,
находим[Н2А1 = [Н;^ (6.48)A IИ[А2"] = 1нТ‘ (6'49)Подставляя значения [Н2А] и [А2-] из уравнений
(6.48) и (6.49) в уравнение (6.45), получаем:[Н*] = - 1н^с° . (6.50)[Н+] К,Откуда после обычных преобразований следует:[н+)= д/К,КцС о ^651JKi + С0Из этого уравнения видно, что концентрация во¬
дородных ионов амфолита в общем случае зависит от
его общей концентрации. Однако очень часто К\ зна¬
чительно меньше С0, поэтому в знаменателе можно
пренебречь К\ по сравнению с С0. Тогда[Н+] = VAV^J (6.52)и после логарифмирования и обычных преобразова¬
ний находим:рН= Р*' + Р*». (6.53)При вычислении pH растворов более высокозаряд¬
ных амфолитов этого типа, например НРО2", важно
правильно выбирать значения рК. Амфолит НРО^ под¬
вергается в растворе таким превращениям:НРО^ 4=* H* + POf (6.54)иНР042- + Н20 ч=^ Н2РО; + ОН\ (6.55)Анионы Р04" образуются при диссоциации фос¬
форной кислоты по третьей ступени, а анионы
НРО4' — по второй. Отсюда следует, что необходимоюв
учитывать только р/Сз и рК2, т. е.:рН = -Р^ + Р^1. (6.56)Аналогично для Н2РО4 найдем, что при вычисле¬
нии следует брать pKi и рК.2-рН = _Р*1±£*1.. (6.57)Пример 1. Вычислить pH раствора NaHCC3.По справочнику находим p/Ci = 6,5 и рКг = 10,2:рН-М + М_а,зБ.Пример 2. Вычислить pH раствора NaHSC3.Для сернистой кислоты p/Vi = 1,8 и рКг ■= 7,2. Поэтому:В этом случае реакция раствора кислая.§ 5. БУФЕРНЫЕ РАСТВОРЫБуферные растворы — это растворы, содержащие
смеси сопряженных слабых кислот и оснований. Так,
смесь растворов карбоната и гидрокарбоната натрия
представляет собой буферный раствоэ. Аналогичные
свойства имеет раствор, в котором находятся кислота
Н2РС>4 и сопряженное с ней основание НР04’ и т. д.Буферные растворы образуются 1ри титровании
слабых кислот или слабых оснований. Нередко их го¬
товят специально, если необходимо экспериментально
определить pH растворов фотометрическим методом
или провести химический экспериме1-т, связанный с
выделением или присоединением ионсв водорода, при
постоянном значении pH. Название «буферные» об¬
условлено тем, что такие растворы не изменяют за¬
метно pH при разбавлении или при добавлении неко¬
торых количеств растворов сильных кислот или силь¬
ных оснований. Постоянство pH буферных растворов
имеет значение в жизнедеятельности живых организ¬
мов или растений: кислотность крови или раститель¬
ных соков поддерживается постоянной из-за буфер¬
ного действия содержащихся в них составных ча¬
стей. Незначительное изменение pH три добавлении107
сильных кислот или оснований важно в химическои
технологии и в анализе: кислотность некоторой реак¬
ционной смеси остается постоянной при введении бу¬
ферного раствора независимо от выделения или по¬
глощения в реакции водородных или гидроксильных
ионов.Уравнение для вычисления pH буферных раство¬
ров можно вывести на примере сопряженных кислоты
НА и основания А- следующим образом.Слабая кислота буферного раствора диссоциирует
по схемеНА ч=£ Н+ + А'. (6.58)По закону действия масс[Hi [А']
[НА]*НА= ru»?-. (6.59)Для слабой кислоты можно принять, что концентра¬
ция недиссоциированных молекул равна их общей
концентрации:[НА]=Сна. (6.60)Введенная в буферную смесь соль МА диссоции¬
рует полностью. Концентрация образовавшегося осно¬
вания А значительно превышает концентрацию этого
же основания, возникающего вследствие диссоциации
слабой кислоты. Поэтому в первом приближении счи¬
тают, что.[ А~] = С0. (6.61)Подставляя значения [А~] и [НА] из уравнений
(6.61) и (6.60) в уравнение (6.59), получаем:[Hi С0ЛНА — . (6.62)НАоткуда[н+] = л:НА-^- (6.63)Ь0ИpH = pKHA + lg7^-. (6.64)НАИз уравнения (6.64) видна первая особенность бу¬
ферных растворов, а именно, pH не изменяется при
их разбавлении. В самом деле, при разбавлении из¬108
меняются абсолютные концентрации основания и кис¬
лоты, однако соотношение С0 : Ск остается постоян¬
ным. Следовательно, не изменяется также pH рас¬
твора. Изменяться pH может только фи очень боль¬
шом разбавлении раствора и если кислота буферной
смеси не очень слабая. В этих условиях [НА] не рав¬
няется уже общей концентрации кислоты Сна, а кон¬
центрация основания становится заметно больше об¬
щей концентрации С0.Буферные свойства имеют не толью смеси раство¬
ров одноосновных слабых кислот с сопряженными ос¬
нованиями, как было рассмотрено пыше, но также
смеси растворов многоосновных слабых кислот и ос¬
нований. Это же относится к смесям многоосновных
оснований с сопряженными кислотами. Например,
смесь растворов Na2C03 и ЫаНСОз гредставляет со¬
бой буферный раствор, так как СОз’ — основание и
НСОз — сопряженная кислота, и pH этого раствора
определяется уравнением:[соПPH = pK2+igJ—i-L. (6.65)[Hcu3]Здесь №гС03 и NaHC03 выступают соответствен¬
но в роли основания и кислоты. В гмеси растворов
Н2СОз и NaHC03 последний, наоборэт, является со¬
пряженным основанием по отношению к угольной
кислоте, и pH такого раствора равноГнсо-1рН=р*- + 'етайг (6-66)Необходимо обратить внимание на правильный
выбор р/С. В реакции Na2C03 с NaHC03 существует
равновесие между НСОз" и С03":НС03' ^=± Н* + СО*', (6.67)которое описывается второй константой диссоциации
угольной кислоты. Поэтому для вычисления pH необ¬
ходимо пользоваться их второй константой диссоциа¬
ции и, соответственно, рДг- В реакции Н2СО3 с
NaHC03 под знаком логарифма находятся концентра¬
ции НСОз и Н2СО3; следовательно, существует109
равновесиеН2СОа =?=fc Н+ + HCOj, (6.68)для которого можно записать первую константу дис¬
социации. Следовательно в уравнении (6.66) необхо¬
димо пользоваться р/СьКак уже было сказано, буферные растворы не ме¬
няют значение pH при добавлении некоторых коли¬
честв сильных кислот или оснований. Предположим,
что приготовлен 1 л буферного раствора, содержа¬
щего ацетат натрия и уксусную кислоту, причем кон¬
центрации обеих составных частей раствора равны
0,1 М. Для уксусной кислоты рд=4,8. Следователь¬
но, pH такого буферного раствора в соответствии с
уравнением (6.64) равно:pH = 4,8 + lg = 4,8.Введем в такой раствор 10 миллимоль сильной
хлороводородной кислоты. В результате концентра¬
ция слабой уксусной кислоты увеличитсяСНаСОСГ + Н+ ч=* СНзСООНи станет равной 0,1 + 0,01 — 0,11 М.Соответственно уменьшится концентрация основа¬
ния СН3СОО-: она станет равной 0,1—0,01 —
= 0,09 М. Теперь pH раствора равноpH = 4,8 + lg 5^- = 4,8 - 0,09 = 4,71.При введении вместо сильной кислоты такого же ко¬
личества основания последнее прореагирует со слабой
уксусной кислотойСНзСООН + ОН' СН3С00' + Н20,вследствие чего концентрация СН3СОО“ и СН3СООН
станет ранной 0,11 и 0,09 соответственно, а pH рас¬
твораpH = 4,8 + lg = 4,8 + 0,09 = 4,89.Расс мотрим влияние такого же количества силь¬
ной кислоты или сильного основания на pH незабуфе-ю
рированной чистой воды. Введение 10 миллимоль
НС1 или NaOH создает концентрацию водородных
или гидроксильных ионов, равную 0,01 М. В первом
случае pH станет равным 2, а во втором—12, т. е.
изменится на пять единиц pH по сравнению с pH чи¬
стой воды.Разумеется, если прибавить к буферному раствору
слишком много кислоты (или основания), все анионы
уксусной кислоты свяжутся в недиссоциированные мо¬
лекулы СН3СООН, и раствор утратит буферные свой¬
ства. Каждый буферный раствор характеризуется по¬
этому определенной буферной емкостью.Буферная емкость — это способность буферного
раствора не изменять заметно pH при добавлении
растворов сильной кислоты или сильного основания.
Мерой буферной емкости служит обычно количество
сильной кислоты или сильного основания, которое не¬
обходимо прибавить к раствору буферной смеси, что¬
бы pH этого раствора изменилось на единицу. Рас
смотрим, от каких факторов зависит буферная ем¬
кость раствора.Предположим, что исходные концентрации основа¬
ния и кислоты буферного раствора равны С0 и Ск, а
соотношение С0 : Ск = а, причемДля того чтобы pH буферного раствора измени¬
лось на единицу, необходимо добавить такое количе¬
ство раствора NaOH, чтобы соотношениедобавления раствора NaOH.Примем, что изменение общей концентрации С
вследствие прибавления раствора ЧаОН невелико,
т. е.Со 4- Ск — С.(6.69)(6.70)где С'0 и — концентрация компонентов буферной смеси после(6.73)(6.71)111
Принимая во внимание, что С'о = С — С', уравне¬
ние (6.72) запишем так:Со-СК
10а ’откуда после преобразований получаем:(673)Буферная емкость Б, т. е. количество (концентра¬
ция) раствора гидроксида натрия, прибавленного для
изменения pH на единицу, равна разности Ск — С'илиПредположим, что есть два буферных раствора с
pH = 5. Один из таких растворов готовят смешива¬
нием растворов ацетата натрия и уксусной кислоты*
причем С0 = Ск = 0,1, С = 0,2, а = 1, рК = 5. Вто¬
рой раствор представляет собой смесь растворов фта-
левой кислоты (НФ) и гидрофталата натрия, причем
Р/Снф = 3,0, Со = 0,1 и С, = 0,001, а = 100, С =
= 0,1001.В соответствии с уравнением (6.64)pH = рК + lg -jr- ■'-'КОба раствора имеют pH = 5.Однако эти растворы очень сильно различаются по
своей буферной емкости. По уравнению (6.74) для
первого раствораБ = 0,1 (10 -1 + 1) 0,2 = Q.CS2,а для второго_ 0,001 (10- 100+ I) — 0.1001 _ 1П—4
в- ш 9-10 .Следовательно, для того чтобы pH первого рас¬
твора увеличилось на единицу, необходимо прибавить
0,082 моль NaOH. При объеме буферного раствора
100 мл это соответствует 8,2 мл 1 М раствора NaOH.
Для того чтобы pH второго раствора также измени¬112
лось на единицу, необходимо прибавить всего 9Х
X Ю-4 моль NaOH, или при объеме раствора
100 мл - -0,09 мл 1 М раствора NaOH.Из этого примера видно, что буферная емкость
первого раствора приблизительно в стс раз больше
буферной емкости второго раствора.Отсюда следует, что буферная емкость зависит в
первую очередь от соотношения концентраций С0 : Ск.
Чем ближе к единице это соотношение, тем больше
буферная емкость. Максимальная буферная емкость
соответствует соотношению С0 : Ск = I.Буферная емкость зависит также oi абсолютных
концентраций С0 и Ск. Вычислим, какая будет ем¬
кость первого раствора при том же соотношении С0:
:СК = 1, но при абсолютных концентращях этих ком¬
понентов в десять раз больших, т. е. при С0 = Ск = 1.В соответствии с уравнением (6.74)или pH изменится на единицу после прибавления
0,82 моль NaOH. Следовательно, буферная емкость
возрастает с увеличением концентрации компонентов
буферной смеси.ГЛАВА 7МЕТОД КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ§ 1. ОБЩИЕ ПОЛОЖЕНИЯ. ТОЧКА ЭКВИВАЛЕНТНОСТИТитриметрические методы анализа основаны на из¬
мерении количества реагента R, израсходованного на
взаимодействие с определяемым вещестЕом X:X + R —► Р. (7:1)Реагент$эбычно применяют в виде рпствора точно
известной концентрации. Такой раствор называют ти¬
трованным или рабочим раствором. Ра(ючий раствор
постепенно прибавляют к раствору определяемого ве¬
щества до тех пор, пока не будет достигнуто эквива¬
лентное отношение реагирующих веществ. Измеряют
объем рабочего раствора, израсходованного на113
титрование, и вычисляют количество определяемого
вещества.В разделе о классификации методов химического
анализа указывалось, что в основе титриметрических
методов могут быть реакции самых различных типов:
кислотно-основные, окислительно-восстановительные,
реакции осаждения или образования комплексных со¬
единений. Тем не менее во всех этих случаях весьма
существенно правильно определить так называемую
точку эквивалентности, т. е. момент титрования, ког¬
да достигнуто эквивалентное отношение реагирующих
компонентов — определяемого вещества и реагента.Рассмотрим самый простой случай, когда соотно¬
шение реагирующих веществ в продукте реакции со¬
ставляет 1 : 1 (-в молях)X + R 4=fc XR. (7.2)Необходимо сразу же предостеречь от неправиль¬
ного понимания точки эквивалентности как такого мо¬
мента титрования, когда концентрация X равна нулю.
Все реакции, которые используют в титриметрическом
анализе, обратимы. Поэтому концентрация X в любой
момент титрования представляет собой некоторую ко¬
нечную величину, не равную нулю. Это относится и к
концентрации реагента R. Продукт реакции XR спо¬
собен в той или иной степени, в результате протекания
обратной реакции, давать снова исходные вещества X
и R. В точке эквивалентности, как и в любой другой
момент титрования, между реагирующими вещества¬
ми существует динамическое равновесие и реакция
продолжается в обоих направлениях с одинаковыми
скоростями.Правильное понимание точки эквивалентности со¬
стоит в следующем. Точка эквивалентности характе¬
ризуется тем, что концентрации реагирующих ве¬
ществ в ней становятся эквивалентными. Для реак¬
ции (7.2) это условие записывается так:[XJ-1RJ. . (7.3)В самом деле, в начале титрования в растворе есть
избыток вещества X; реагент R, который постепенно
прибавляют к раствору X, практически полностью рас¬
ходуется на образование продукта реакции XR. По¬
этому до точки эквивалентности соотношение между114
концентрациями реагирующих веществ характери¬
зуется неравенством[X] > [R], (7.4)После точки эквивалентности введен некоторый
избыток реагента, поэтому:[X] < [R], (7.5)Концентрации X и R становятся равными только в
точке эквивалентности.В более сложных случаях b молей вещества X
реагирует с а молями реагента R, т. е. реакция про¬
текает в соответствии с уравнениемйХ + aR XbRa (7.6)и условие точки эквивалентности соответствует равен¬
ствуa[X] = MRl. (7.7)Рассмотрим несколько примеров кислотно-основ-
ного титрования.Пример I. Титрование сильной хлороводородной кислоты
сильным основанием NaOH:НС1 + NaOH = NaCl + Н20или в ионном виде Н+ + ОН- = Н20.Условие точки эквивалентности записывается так:[Н-] = [ОН-].Ионы водорода и гидроксила образуются за счет диссоциа¬
ции молекул воды.Пример 2. Титрование слабой уксусной кислоты раствором
NaOH:СН3СООН + NaOH ч=>: CHsCOONa + Н20или в ионном виде:СН3СООН + ОН~ ^=4: СН3С00" + Н20.На первый взгляд может показаться, что в точке эквива¬
лентности не должно быть молекул уксусной кислоты и ионов
гидроксила, потому что они прореагировали между собой с об¬
разованием анионов CHjCOO- и молекул воды. За счет неко¬
торого сдвига состояния равновесия реакции влево создается
щелочная среда. В результате раствор содержит небольшое ко¬
личество молекул СИ3СООН и ионов ОН-. Поэтому условие
точки эквивалентности[CHjCOOH] = (ОН'1.
Пример 3. Карбонат натрия можно титровать раствором
хлороводородной кислоты. Реакция в первой стадии идет с об¬
разованием гидрокарбонат-иона:Na2C03 + HCl NaHC03 + NaClили в ионном видеСО='+Н+ =*=► НСО'.Условие этой первой точки эквивалентности выражается
уравнением:[со’"] = [н+].Во второй стадии реакции образуется угольная кислота:
NaHC03 + HCl ^=fc H2C03 + NaClили в ионном видеНС03- + Н+ ч=»= н2со3.Для второй точки эквивалентности записывают равенство[HCO-J = [н+].Пример 4. Фосфорную кислоту титруют обычно раствором
NaOH до образования Ыа2НР04:Н3РО* + 2NaOH =?=► Na2HPO* + 2НгО
или в ионном видеН3Р04 + 20Н' ч=>: НР0=- + 2Н20.Условие точки эквивалентности2[Н3Р04] = [ОН].Аналогичные соотношения справедливы также для
других типов реакций, например реакций осаждения,
комплексообразования, окисления — восстановления.Точку эквивалентности определяют эксперимен¬
тально посредством индикаторов. Индикаторы — это
вещества, которые претерпевают химические превра¬
щения, сопровождающиеся внешним эффектом, при
концентрациях реагента или определяемого вещества,
характерных для точки эквивалентности. Существуют
и физико-химические способы установления точки эк¬
вивалентности. Так, можно погрузить в титруемый
раствор металлический электрод и в процессе титро¬
вания измерять его потенциал; при концентрации ре¬
агента в точке эквивалентности потенциал электродаlie
приобретает некоторое вполне определенное значение.
При использовании любого способа несбходимо иметь
четкое представление об изменении концентрации реа¬
гирующих веществ в процессе титрования и особенно
вблизи точки эквивалентности. Это изменение обычно
выражают кривой титрования, передающей зависи¬
мость концентрации определяемого вешества от коли¬
чества прибавленного реагента.Рассмотрим характер изменения концентрации оп¬
ределяемого вещества в процессе титрования. Удобно
выражать это изменение графически — посредством
так называемой кривой титрования. Кривая титрова¬
ния обычно передает зависимость логаэифма концен¬
трации определяемого вещества, взято'о с обратным
знаком (—lgX), от объема прибавленного рабочего
раствора известной концентрации.Для реакции (7.2) кривая титроващя передает за¬
висимость —lgX от VR или от процентного отношения
прибавленного реагента к его эквивалентному коли¬
честву.Предположим, что константа, xaf актеризующая
устойчивость продукта реакции XR, разна[XR1- ю9[X][R]и что 100 мл 0,1 М раствора X титруют 0,1 М раство¬
ром R.Изменение концентрации X в процессе титрования,
вычисленное с учетом разбавления раствора и приве¬
денной константы, показано в табл. 7.1.Детали расчета данных, приведенных в табл. 7.1,
рассматриваются в последующих параграфах на кон¬
кретных примерах.•Таблица 7.1. Значения концентрации X при титровании
реагентом RПрибав¬
лено
0,1 М
раствора R,
мл1X1— lg 1X1Прибав¬
лено
0,1 М
раствора R,
мл1X1-Igixi01 • КГ11,0100,01,0- 10“55,0905,3- Ю~32,3100,1ю1оосч5,7995,0- Ю-43,3101,02,0- 10 76,799,95,0- 10~54,3110,02,1 ■ 10~87,7117
Кривая титрования, построенная по данным
табл. 7.1, показана на рис. 7.1. Из кривой (и табли¬
цы) видно, что по мере приближения к точке экви¬
валентности одинаковые изменения концентрации X
вызываются прибавлением все меньшего количества
реагента R. Иначе говоря, при равномерном прибав¬
лении рабочего раствора концентрация X изменяется
скачкообразно. Особенно большое изменение концен¬
трации X происходит вблизи точки эквивалентности.
'Так, при 99,9 мл введенного реагента она составляет5 -10-5 М, прибавление еще 0,1 мл R уменьшает кон¬
центрацию X в 5 раз, а при избытке реагента в 0,1 мл
снова падает еще в пять раз. Кривая вблизи точки
эквивалентности идет почти вертикально.Изменение -начальной концентрации X и концен¬
трации реагента R влияет на форму кривой титрова¬
ния. На рис. 7.1 пунктиром показан ход кривой в том
случае, если исходные концентрации X и R в десять
раз меньше, т. е. равны 0,01 М. Видно, что высота
скачка вблизи точки эквивалентности меньше, чем для
более концентрированных растворов, и кривая идет
уже не вертикально, а становится более пологой. Да¬
лее будет показано, что это отрицательно сказы¬
вается на правильном установлении точки эквива¬
лентности и на точности определения.Форма кривой титрования зависит также от проч¬
ности связи X и R в продукте реакции XR. Скачкооб¬
разное изменение концентрации определяемого веще¬
ства вблизи точки эквивалентности значительно боль¬
ше, если диссоциация продукта реакции очень мала
или практически отсутствует. На рис. 7.2 сопостав¬
лены две кривые титрования 0,1 М растворов X 0,1 М
растворами реагентов R и С. Первая кривая соответ¬
ствует образованию продукта реакции XR с констан¬
той устойчивости 109; вторая кривая соответствует
продукту реакции ХС с константой устойчивости в
10 000 раз большей, т. е. равной 1013. Из сравнения
кривых видно, что скачок концентрации вблизи точки
эквивалентности зависит от константы, характеризую¬
щей продукт реакции: он тем больше, чем больше
константа устойчивости, или, что то же самое, чем
больше прочность соединения, образующегося в ре¬
зультате взаимодействия. Большой скачок благо¬
приятно сказывается на точности титрования. По-118
IgW10(С-8Т.З2J1 1 .1 1 /6Т.З.42I ! 1 1О 20 40 60В.,м/i20 40 60 SO R,C,«/7Рис. 7.1. Кривая титрования вещества X веществом R. Сплошная
линия—0,1 MX и 0,1 Af R; пунктирная линия—0,01 MX и
0,01 М RРис. 7.2. Кривые титрования 0,1 М раствора X и 0,1 М раство¬
рами R (/) и С (2)этому реагент следует выбирать с таким расчетом,
чтобы он давал продукт реакции с как можно боль¬
шей энергией связи. Так, при титровании по методу
осаждения следует преимущественно использовать
реакции, в которых образуется наименее растворимое
соединение. В методе комплексообразования лучшие
результаты получаются, если образовавшееся комп¬
лексное соединение характеризуется высокой устойчи¬
востью. Аналогичные выводы справедливы для мето¬
дов кислотно-основного титрования и методов окисле¬
ния— восстановления.Отмстим, что в рассмотренных случаях концентра¬
ция X в точке эквивалентности соответствует точке
перегиба кривой титрования — эти точки располо¬
жены посредине скачка и показаны на рисунках.§ 2. КРИВЫЕ ТИТРОВАНИЯМетод кислотно-основного титрования пригоден для
определения сильных и слабых кислот и оснований.
В качестве рабочих растворов чаше всего используют
растворы хлороводородной кислоты и гидроксида на¬
трия. Другие сильные кислоты или основания, напри¬
мер HN03, H2S04, КОН применяют реже.Титрование сильной кислоты сильным основанием.
В качестве примера рассмотрим изменение pH при119
титровании 100 мл 0,1 М раствора сильной хлорово¬
дородной кислоты 0,1 М раствором гидроксида
натрия.В начале титрования, еще до прибавления раство¬
ра NaOH, имеем 0,1 М раствор НС1 с pH = 1. После
введения 90 мл 0,1 М раствора щелочи общий объем
составит 190 мл; в полученном растворе, который со¬
держит 10 мл 0,1 М раствора НС1, концентрация во¬
дородных ионов равна[H+] = iW' = 5'3'10_3’и pH = —lg 5,3 • 10~3 = 2,3.Аналогично вычисляем pH растворов после при¬
бавления последующих порций раствора щелочи.Уравнение реакции титрования имеет вид:Н+ + он- н20.Поэтому в точке эквивалентности[Н+] = [ОН-] = 10~7, pH = 7.После точки эквивалентности концентрация водо¬
родных ионов раствора определяется избытком рас¬
твора NaOH. Так, если избыток NaOH составляет
0,1 мл 0,1 М раствора, концентрация ионон ОН~
равна[OH'l = -W- = 5-1(r5’откуда[Н*] = —1 °— =2- 10-10 или pH = — lg 2 - 10~10 = 9,7.5- 10 5В табл. 7.2 приведены данные расчета.Графическое изображение данных таблицы пока¬
зано на рис. 7.3. Кривая характеризуется резким из¬
менением pH вблизи точки эквивалентности. Сущест¬
венно, что при pH = 4,3 и 9,7 соответственно в рас¬
творе остается всего 0,1 % неоттитрованной кислоты
или введен 0,1 % избытка NaOH. Поэтому даже
если индикатор или прибор зафиксирует точку экви¬
валентности не при pH — 7, а в пределах pH от 4,3 до
9,7 — ошибка титрования не превысит 0,1 %.Uo
Таблица 7.2. Дан^ше титрования сильной кием ты
сильным основаниемПрибав¬
лено
0,1 М
раствора
NaOH, млЭлектролит,
определяющий
pH раствораФормула для вычисления
pH раст юраpH00,1М НС1pH = — lg сНС1190,05,3- 10-3М НС1То же2,399,05,7- 10~4М НС1»3,399,95,0- 10“5Л1 НС14,3100,0—pH = рОН7,0100,15,0 • I0-4Af NaOHРон = — *8 CNaOH9,7110,05,0- 10-3М NaOHТо же11,7При титровании более разбавленного раствора кис¬
лоты более разбавленным раствором щелочи полу¬
чается аналогичная кривая, но изменение pH вблизи
точки эквивалентности менее резкое. На рисунке
пунктиром показана кривая титрования при исполь¬
зовании 0,01 М растворов кислоты и щелочи. Левая
ветвь кривой расположена выше, а правая — ниже со¬
ответствующих ветвей для 0,1 М растворов. Это об¬
стоятельство приводит к уменьшению точности титро¬
вания.Титрование сильным основанием слабой кислоты.Рассмотрим титрование 100 мл 0,1 М раствора уксус¬
ной кислоты 0,1 М раствором щелочи. В начале ти¬
трования в растворе находится 0,1 М уксусная кис¬
лота, pH которой равноРн = уР^ — у tecK = у'4,8 - уlg 10_1 = 2-9-При добавлении некоторых количеств щелочи об¬
разуется буферный раствор, содержащий уксусную
кислоту и ацетат-ионы. Для негоpH = рК + lgПусть, например, прибавлено 9 мл 0,1 М раствора
NaOH. Тогда образуется 9 мл 0,1 М раствора ацетата
натрия и останется 91 мл 0,1 М раствора уксусной121
pH121-
Т.Э-нсЮFrwWPJ26в//D 20 40 60 NaOH, мл0 20 40 60 NaOH, млРис. 7.3. Кривые титрования 0.1 М раствора HC1 0,1 M раство¬
ром fflaOH (сплошная) и 0,01 M раствора НС1 0,01 М раствором
NaOH (пунктирная линия)Рис. 7.4. Кривые титрования СНзСООН (сплошная) и НСЮ
(пунктирная) растворами гидроксида натриякислоты. В связи с разбавлением раствора, концен¬
трации основания и кислоты составят:Аналогично рассчитывают pH при других количе¬
ствах прибавленной щелочи.В точке эквивалентности в растворе находится
только ацетат натрия. Для этого раствораПосле точки эквивалентности в растворе окажется
избыток раствора гидроксида натрия, по которому
можно вычислить значение pH. Соответствующие дан¬
ные приведены в табл. 7.3.На рис. 7.4 показана кривая титрования, соответ¬
ствующая данным табл. 7.3. Отметим такие особен-иоткудаpH = 4,8 + lg 10_,) = 3,8.8,3 ■ 10 “pH = 7 + i- р/С + у lgC0 = 7 + 2,4 + y lg5- 10 2 = 8,75.125
ности кривой по сравнению с кривой т ггрования силь¬
ной кислоты: pH точки эквивалентности соответствует
слабощелочной реакции раствора, а ге нейтральной;
изменение pH вблизи точки эквивалентности значи¬
тельно менее резкое. Эти особенности сильно отра¬
жаются на правильности определена точки эквива¬
лентности и точности титрования.Если взять еще более слабую кислоту, например
НСЮ, с константой диссоциации 2,5-10-8 (р/С = 7,6),
кривая титрования, рассчитанная аналогичным спосо¬
бом, будет иметь вид, показанный на )ис. 7.4 пункти¬
ром. Левая ветвь кривой значительно смещена вверх
по сравнению с кривой для уксусной кислоты, а изме¬
нение pH возле точки эквивалентности очень неве¬
лико. Титрование таких слабых кислот с необходимой
точностью затруднено, а иногда, например в случае
еще более слабой борной кислоты, становится практи¬
чески невозможным.Многоосновные кислоты обычно являются слабыми
кислотами и диссоциируют ступенчатэ. В соответст¬
вии с этим на кривых наблюдается НесколькоТаблица 7.3. Данные титрования слабой уксусной кислоты
раствором гидроксида натрияПри¬бавлена0,1 мЭлектролит, определяющийФормула вычисленияpHраство¬раpH раствораpH раствораNaOH,мл00,Ш СНзСООНpH = lp^-i-lgCKса2,99,0Буферная смесь
СНзСООН + CH3COONapH = p/f-|- lg -ТГ-3,850,0То жеТо же4,891,0»щ *рН-7 + у,(С + ylgCo5,899,0»6,8100,0
(т. э)CH3COONa8,75100,1NaOHрон = gCNaOH9,7101,0То жеТо ке10,7110,0»11,7123
перегибов. Их расположение зависит от значений и
соотношения констант диссоциации, но изменение pH
вблизи каждой точки эквивалентности имеет менее
резкий характер, чем при титровании однооснрвных
кислот. Примером может служить титрование двухос¬
новной сернистой кислоты H2SO3. Первая стадия со¬
ответствует нейтрализации первого водородного иона:H2S03 + 0H~ *=* HSO'+HjO,а вторая — титрованию аниона HSO3:HSO' + ОН” so“_ + Н20,и на кривой титрования наблюдаются два перегиба.Вычисление pH растворов, образующихся при ти¬
тровании, в данном случае более сложно, чем обычно.
Сернистая кислота — это кислота средней силы, и при
расчете необходимо учитывать, что концентрация не-
диссоциированных молекул H2S03 меньше общей кон¬
центрации кислоты.Для вычисления pH буферных растворов, образую¬
щихся в процессе титрования, также необходимо при¬
менять более сложные уравнения. Поэтому точные
расчеты здесь не приводятся. На рис. 7.5 изображен
общий вид кривой титрования сернистой кислоты.
Видно, что скачки pH вблизи обеих точек эквива¬
лентности невелики, а кривые в этих областях накло¬
нены по отношению к оси ординат. Эти особенности
приводят к тому, что определение становится менее
точным, чем в случае одноосновных сильных или
даже слабых кислот.Титрование солей одноосновных или многооснов¬
ных слабых кислот сильной кислотой. Пример, иллю¬
стрирующий общий характер изменения pH в опреде¬
лениях подобного рода, — титрование раствора
карбоната натрия. Реакция проходит в две стадии.
Первая точка эквивалентности наступает после того,
как весь карбонат натрия прореагировал до гидро¬
карбоната.Na2C03 + НС1 = NaHCOs + NaCl.Следующая реакция:NaHCOa + НС1 = Н2СОэ + NaCI.Во второй точке эквивалентности раствор содер¬
жит угольную кислоту.124
Рис. 7.5. Кривая титрования 0,1 М H2SO3 0,1 М раствором NaOHРис. 7.6. Кривые титрования 0,1 М раствора Na2C03 0,1 М рас¬
твором НС1Рассмотрим изменение pH при титровании 100 мл0,1 М раствора Ыа2С0з 0,1 М раствором хлороводо¬
родной кислоты. До прибавления кислоты раствор со¬
держит только карбонат натрия, и pH равен:pH = 7 + i-p/C2 + ylgC0 = 7 + y 10,2 + -,-lg ю-1 = 11,6.После прибавления некоторого количества НС1 об¬
разуется буферный раствор, состоящий из карбоната
и гидрокарбоната натрия:Со fcoj-lp„_pK,+ lg-a-_10,2 + lei-^.Когда весь карбонат превратился в гидрокарбо-
нат-ион, pH равен:рН = Р*. + Р«2 .6,5+ 10,2 ,8 35Дальнейшее прибавление рабочего раствора кис¬
лоты приводит к появлению некоторого количества
свободной угольной кислоты и образованию буфер¬
ного раствора, содержащего НСОз и Н2С03. Следова¬
тельно,fHCO-l125
Таблица 7.4. Данные титрования карбоната натрия
хлороводородной кислотойПрибавлено0.1 М
раствора
НС1, млЭлектролит,
определяющий
pH раствораФормула для вычисления
pH раствораpH00,1 М Na2C03pH = 7 + — р/С2 + ~2 Cq11,69,0Буферная смесь
Na2C03 с NaHC03[col]pH-p/f2 + lg [НСО-]11,250.091.0100,0То же
»NaHC03То же»тт PKl + р/С2рИ- 210,29.28.3(1-я т. э)110,0Буферная смесь
NaHC03 с Н2С03[нсо-]pH - р/с, + lg -[Н4СОз57,5150.0190.0То жеТо же
»6.55.5200,0
(2-я, т. э)0,05 М И2С03рН = 1р/С, - у lgCK3,9201,05- 10-4 М НС1pH = — lg СНС|3,3210,05- 10-3 М НС1То же2,3Во второй точке эквивалентности, после реакции
всего гидрокарбоната, раствор содержит только сво¬
бодную угольную кислоту. ПоэтомуpH = yp/C1--*-IgCK.Данные соответствующих расчетов приведены в
табл. 7.4. Кривая титрования показана на рис. 7.6.
Из особенностей кривой отметим сравнительно малое
изменение pH и значительный наклон к оси ординат
вблизи первой точки эквивалентности. Участок кри¬
вой возле второй точки эквивалентности отличается
несколько большим скачком pH и меньшим наклоном,
однако обе эти характеристики выражены не так рез¬
ко, как, например, при титровании сильной кислоты.1Ж
§ 3. КИСЛОТНО-ОСНОВНОЕ ТИТРОВАНИЕ
В НЕВОДНЫХ РАСТВОРАХТитрованием кислот или оснований в неводных рас¬
творах можно повысить точность определения. Кроме
того, появляется возможность раздел >ного определе¬
ния смеси двух или нескольких кислот или оснований,
которые в водных растворах диссоциируют приблизи¬
тельно одинаково.Рассмотрим влияние неводных растворителей на
точность титрования. Точность определения кислот
(или оснований) зависит в первую очередь от их спо¬
собности к электролитической диссоциации. Сильные
кислоты титруются с очень незначительной погреш¬
ностью. Выше было показано, что погрешность опре¬
деления слабых кислот возрастает с уменьшением их
констант диссоциации. Так, погрешность определения
концентрации сильной хлороводородной кислоты в
водном растворе ничтожна, при титровании уксусной
кислоты (р7С = 4,8) она увеличивается до 0,1 %. а
слабую борную кислоту (р/С = 9,2) вообще нельзя оп¬
ределить с удовлетворительной точностью — ошибка
доходит до 10—12 %. При переходе от более сильных
кислот (СНзСООН) к более слабым (НВ02) возра¬
стают константы основности сопряжекных оснований
в соответствии с зависимостью*к*о = *нао- <7-8)Поэтому состояние равновесия реакцииА~ + Н20 :?=>: НА + ОН' (7.9)все более смещается в правую сторону, что приводит
к увеличению погрешности титрования и снижению
точности определения.Из уравнения (7.8) следует:1НДИ0Н1_^[А] КиВидно, что уменьшить константу основности Ко со¬
пряженного основания можно, если взять вместо воды
растворитель с меньшим ионным произведением, чем
у воды.Неводные растворители также способны к диссо¬
циации. Так, диссоциация метанола протекает по127
уравнениюСН3ОН + СН3ОН CH3OH2 + CH3CT.Уксусная кислота (безводная) как растворитель
диссоциирует следующим образом:СН3СООН + СН3СООН сн3соон2+ + СН3СОСГ.Приведенные процессы количественно характери¬
зуются ионными произведениями — константами авто-
протолиза. Константы автопротолиза некоторых рас¬
творителей приведены в табл. 7.5.Обычно при переходе от воды к неводному раство¬
рителю изменяется не только константа автопрото¬
лиза, но и константа диссоциации кислоты. Поэтому
в общем случае точность титрования не обязательно
должна увеличиваться. Важно не абсолютное значе¬
ние константы автопротолиза, но изменение соотно¬
шения Ks : Кна\ чем меньше это соотношение, тем в
меньшей степени проходит гидролиз (в воде) или
сольволиз (в неводном растворителе). Другими сло¬
вами, точность титрования увеличивается с возраста¬
нием разности pKs — рКна при переходе от одного
растворителя к другому.Сильные кислоты титруются и в водных растворах
достаточно точно, но в метаноле или этаноле точность
определения возрастает. Так, соотношение Ks : К на в
воде равно 1(Н\ а в этаноле Ks : Кнс\ = Ю~17 (раз¬
ность p/Cs — р/Снс! = 19,3 — 2,0=17,3 в этаноле иТаблица 7.5. Ионные произведения (константы
автопротолиза) некоторых растворителейРастворительФормульное выражение
ионного произведенияКонстантаавтопротолизаВода[н3о+][он_]1 ■ 10“14Муравьиная кислота[нсоон2+] [нсосг]5 ■ 1СГ7Уксусная кислота[сн3соон2+][сн3сосг]2,5- 10“13Метанол[СН3ОН2] [СН3О-]2,0- 10"17Этанол[с2н5он2+] [с2н6о ]8,0 • Ю~20ГидразинКН5+] Кнз]2,0 • 10“25128
Рис. 7.7. Кривые титрования хло- у
роводородной кислоты в воде (/), ”
этаноле (2), метилэтилкетоне (3)14.0 в воде). Поэтому точ¬
ность определения возра¬
стает — увеличивается ска¬
чок изменения pH вблизи
точки эквивалентности.Рис. 7.7 иллюстрирует элек¬
трометрическое титрованиехлороводородной кислоты в NaOH м/гводе (/), этаноле (2) и ме-
тилэтилкетоне (3).Значения рЛГ 2,6-динитрофенола в Еоде и этаноле
равны соответственно 4,0 и 8,2; рК воды и этанола —14.0 и 19,3. Разность p/Cs— р/Снд в воде составляет
14 — 4=10, в этаноле—19,3 — 8,2=11,1. Поэтому
титрование в этаноле дает меньшую ошибку, чем в
воде.Раздельное титрование смеси двух (или несколь¬
ких) кислот нельзя осуществить, если их константы
диссоциации одинаковы или близки. В этих случаях
нейтрализация обеих кислот происходит одновременно
и характеризуется одной общей кривей титрования,
на которой нельзя выделить двух скачков pH и двух
точек эквивалентности. Неводные растворители не¬
редко проявляют дифференцирующее действие: кис¬
лоты с одинаковыми константами дисссциации в воде
превращаются в неводных растворителях в кислоты
разной силы. Так, в воде разность р/( трихлоруксус-
ной и хлороводородной кислот Др/С =- р/Сг — р/Ci =
= 0,5, что недостаточно для раздельного определения,
обе кислоты титруются вместе. Однако в ацетоне эта
разность увеличивается и составляет уже 3,8, т. е.
хлороводородная кислота становится сильнее трихлор-
уксусной примерно в 100 раз. При титровании сна¬
чала нейтрализуется хлороводородная кислота, а за¬
тем трихлоруксусная, и кривая титрования имеет два
хорошо выраженных скачка pH.При переходе от водных к ацетоновым растворам
очень сильно увеличивается разность р/С пикриновой
и. салициловой кислот. В первом случае Др/С состав¬
ляет всего две единицы, во втором — равно шести,л■^1/23УууV.V5 Аналитическая химия129
т. е. раздельное титрование обеих кислот в ацетоно¬
вых растворах происходит со значительно большей
точностью, чем в водных.В неводных растворителях легко определять соли
слабых кислот, не поддающиеся определению с доста¬
точной точностью в водных растворах. Рассмотрим
титрование ацетата натрия:сн3сосг + н+ ензсоон.В водном растворе pH точки эквивалентности
определяется из уравнениярН = 1рк-1 igCK.Учитывая, что рК уксусной кислоты равно 4,8,
и принимая, что концентрация кислоты в точке экви¬
валентности равна 0,1 М, находим, что рНт. э = 2,9;
погрешность титрования доходит до 10%. Но уже
в 70 %-ном водном растворе ацетона диссоциация
уксусной кислоты значительно уменьшается, поэтому
pH точки эквивалентности увеличивается. В этих ус¬
ловиях можно титровать не только ацетат натрия, но
также соли почти всех слабых карбоновых кислот.§ 4. ПОГРЕШНОСТИ ТИТРОВАНИЯТочность определения зависит от многих факторов.
Один из них — точность отсчета объема рабочего рас¬
твора по бюретке, обычно применяемой для титрова¬
ния. Погрешность отсчета по шкале, разделенной на
десятые доли миллилитра, составляет, как правило,
не менее 0,02 мл. Оптимальный объем расходуемого
рабочего раствора примем равным 20 мл. В самом
деле, при значительно меньшем количестве израсхо¬
дованного на титрование раствора относительная по¬
грешность становится слишком большой. Затрата же
чрезмерно большого объема рабочего раствора, на¬
пример 100—150 мл, хотя и уменьшает относительную
погрешность, но неудобна по чисто практическим со¬
ображениям; нужно два или три раза наполнять бю¬
ретку, что приводит к дополнительным погрешностям
и увеличивает продолжительность анализа.Таким образом, при принятых условиях относи¬
тельная погрешность (Л) отсчета объема рабочего130
раствора по бюретке составляетПогрешность можно уменьшить, грименяя спе¬
циальные приспособления, например весовую бюрет¬
ку, когда измеряют не объем раствора, а его массу.
Однако такой способ работы сопряже! со значитель¬
ными экспериментальными трудностями и увеличи¬
вает продолжительность анализа. В опычных же ус¬
ловиях следует считать неизбежно? погрешность
0,1 %• Отсюда следует, что и при дру'их операциях,
связанных с определением, например i ри выборе ин¬
дикатора, нет необходимости стремиться обеспечить
погрешность меньшую, чем 0,1 %. В дьчьнейшем при
титриметрическнх определениях будем считать такую
погрешность допустимой.Погрешности, источником которых меняется непра¬
вильный выбор индикатора, можно ра (делить на две
группы.1. Индикатор выбран так, что ре:кое изменение
окраски наблюдается после введения некоторого из¬
бытка рабочего раствора по сравнению с эквивалент¬
ным количеством. В этом случае ра'.твор перетит-
рован.2. Индикатор выбран так, что переход окраски про¬
исходит до того, как достигнута точка эквивалентно¬
сти. В этом Случае раствор недотнтровлн.Погрешности первой группы легко найти по объ¬
ему избытка рабочего раствора, при£авленного для
изменения окраски индикатора. Способ вычисления
одинаков независимо от того, титруется сильная или
слабая кислота, сильное или слабое основание. На¬
пример, при титровании слабой уксусной кислоты
pH точки эквивалентности равно 8,9, необходим инди¬
катор с рТ* = 9 — лучше всего фенолфталеин. Пред¬
положим, что индикатор выбран неудачно — вместо
фенолфталеина взят тимолфталеин с рТ = 10. Инди¬
катор изменит цвет при pH = 10 и раствор, следова¬
тельно, будет перегитрован. Примем, что объем рас-* рТ — показатель титрования индикатора—значение pH,
при котором происходит наиболее резкое изменение окраски ин¬
дикатора.5*131
твора в конце определения составляет 50 мл. Для из¬
менения цвета индикатора необходимо прибавить
такой избыток 0,1 М раствора гидроксида натрия,
чтобы он был достаточен для создания pH = 10. По
соотношению между концентрацией и объемом на¬
ходим= V,M2, (7.11)где V, — объем раствора в конце титрования (50 мл); /И|[Н + ] =
= 10-10 или [ОН-] = 10-4, М2— молярность рабочего раствора
NaOH (0,1); — объем раствора щелочи, необходимый для из¬
менения окраски индикатора, мл.V,Mt 50 - 10-4 лпс
V2 - = i 0,05 мл.М2 10'"'Таким образом, для изменения цвета индикатора
необходимо прибавить избыток 0,1 М раствора NaOH,
равный 0,05 мл.Если принять, что исходное количество 0,1 М ук¬
сусной кислоты составляет 25 мл—относительная по¬
грешность титрования будет равна■ °>°5 ■ 100 _ 0 2 o/Q25Она больше допустимой погрешности отсчета объ
ема по бюретке. Нужно стремиться уменьшить по¬
грешность за счет выбора более подходящего инди¬
катора.Нели в рассматриваемом примере выбрать инди¬
катор с большим значением рТ, например с рТ= 11,
погрешность возрастет в десять раз и составит 2 %.Можно провести аналогичный расчет погрешности
первого рода при титровании, например, слабого осно¬
вания рабочим раствором хлороводородной кислоты.
Легко видеть, что если взят индикатор с рТ — 4 и
если для изменения его окраски необходимо перетит-
ровать раствор, введя некоторый избыток раствора
хлороводородной кислоты, погрешность также соста¬
вит 0,2 %, а при индикаторе с рТ = 3 она увеличи¬
вается до 2 %.Таким образом, для того чтобы погрешность опре¬
деления не превышала 0,2 %, следует выбирать инди¬
каторы с рТ = 10, применение индикаторов с рТ •< 4
или с рТ >> 10 приводит к слишком большим ошибкам.132
Погрешности второй группы можно определить как
отношение неоттнтрованного объема влцества к его
первоначальному объему, или, что то »е самое, отно¬
шение концентрации неоттитрованной части к перво¬
начальной концентрации вещества:^ [Неоттитрованиая часть] ■ 100 (7 121[Первоначальная концентрац1Я|Во многих случаях, когда погрешность не слишком
велика, принимают, что первоначальная концентрация
вещества равна концентрации в оттитрованной части:[Неоттитрованиая часть| • Н О[Оттитрованная часть](7.13)Рассмотрим, как влияет расхожденре между рТ и
pH точки эквивалентности на погре иность титро¬
вания.Выше было показано, что pH точ! и эквивалент¬
ности при титровании 0,1 М раствора уксусной кис¬
лотыСНзСООН + OFF СНг,СОО' -- н2оравно 8,9. Предположим, что взят индиьатор с рТ = 8,
в этом случае окраска изменится при pH = 8, т. е.
когда некоторая часть уксусной кислоты останется
неоттитрованной. ПоэтомуЛ [СН3СООН|[С На COO'(7.14)Отношение в правой части уравнення (7.14) нахо¬
дим из константы диссоциации кислоты,, [н*'1 [сн3соо | ,,1К|Д = [СНзСООН] ' (7Л5)Из уравнений (7.14) и (7.15) следуе' :[СНзСООЩ [н*|[CHiCooi кгде [11']-концентрация водородных ионов, нэи которой инди¬
катор изменяет окраску, т. е. [Н + ]= 10_s.Принимая во внимание, что К = 2-Ш^5, получим
1 п ^Д = =г^- = 5 • 10 4 или Д=Э,05%.2,0- 10133
Таким образом, уксусную кислоту можно опреде¬
лить с погрешностью 0,05%, если для титрования
взять индикатор с рТ = 8.Рассмотрим, насколько увеличится погрешность
определения, если взять, например, индикатор с рТ =
= 4,7. В этом случае изменение окраски произойдет
при pH = 4,7 или при [Н+]=2-10~5. Поэтомуt „МЛ, 2-10-=К 2-10 5Ответ показывает, что в момент изменения цвета
концентрации неоттитрованной и оттитрованной час¬
тей равны, т. е. половина кислоты еще не оттитро¬
вана. Погрешность составит 50 %.Из приведенного примера видна важность такого
выбора индикатора, при котором разность рНт.э—рТ
была бы наименьшей.При одной и той же разности между рНт. э и рТ
погрешность титрования возрастает с уменьшением
константы диссоциации титруемой кислоты. Покажем
это на примере определения трех кислот с разными
константами: уксусной (/С = 2-10-5, рК = 4,8), хлор¬
новатистой HCIO (К — 2,5-10~8, рК = 7,6), и борной
НВ02 (К = 6,3-10-'°, р/С — 9,2).При титровании уксусной кислоты pH точки экви¬
валентности равно 8,9. Аналогичный расчет по урав¬
нениюpH = 7 + i- vK + ~ IgCa (7.17)показывает, что для двух других кислот pH точки эк¬
вивалентности соответственно равно 10,3 и 11,1.Предположим, что во всех трех случаях взяты ин¬
дикаторы, показатели титрования которых меньше pH
точки эквивалентности на единицу, т. е. рТ равны со¬
ответственно 7,9; 9,3 и 10,1.Тогда погрешности титрования равны:для уксусной кислоты[СНзСООН] • 100 [н+] - 100 10-7 9 - 100 Л1П, .Д “ 1 .Q /0»[СН3СОО-] Хснзсоон 10134
для хлорноватистой кислоты[НСЮ] - 100 [н+] - 100 Ю-9Г1 - 100 _оп„;.Д — : i — 7 ,• — /0»[CIO-] А'нсю 10для борной кислоты[НВ02] - 100 [н+] - 100 10 "10Л - 100 10США = 7 \ — ZT) » А Э /о .[во-1 /Снп0а 10-9'2Полученные результаты объясняются изменением
характера кривых титрования при переходе от более
сильных кислот к более слабым. При таком переходе
все время уменьшается скачок pH и увеличивается
наклон участка кривой вблизи точки эквивалентности
по отношению к оси ординат, эти учас тки становятся
все более пологими. Рассмотренная ; акономерность
иллюстрируется приведенными в § 2 'л. 7 кривыми
титрования Вследствие возрастания наклона одина¬
ковые отклонения рТ индикаторов от pH точки экви¬
валентности обусловливают появление все больших
погрешностей определения.Из рассмотренного примера видно "акже, что при
обычных условиях определения — титрование 0,1 Af
растворов и не слишком большие отктонения рТ от
рНт., — кислоты с константами диссоциации, мень¬
шими 10-7 (например, хлорноватистую и борную)
нельзя уже определять с достаточно высокой точ¬
ностью: погрешность значительно превышает погреш¬
ность неправильного отсчета объема рабочего рас¬
твора по шкале бюретки. При титровании кислот
с /( < 10-7 в точке эквивалентности pH ^ 10; поэтому
данное правило можно сформулировать так: при опре¬
делении кислот, которые в точке эквивалентности
имеют pH ^ 10 погрешность титрована всегда будет
превышать 0,1 %•Аналогичные рассуждения приводит к выводу, что
при титровании слабых оснований следует соблюдать
условие: Ко ^ Ю-7, рНт. э ^ 4. Эти ебщие правила
можно сформулировать иначе: при титровании кислот
или оснований необходимо применят, только такие
индикаторы, показатели титрования которых нахо¬
дятся в пределах от 4 до 10.Так, титрование бората натрия идет в соответствии
с уравнениемВ02" + Hf HB02.135
Если принять, что концентрация кислоты в точке
эквивалентности равно 0,1 М, тогдарНт. 3 =-i-■ 9,2 - у lg io~* = 5,1,где 9,2 — р/( борной кислоты.Предположим, что взят индикатор с рТ = 6. Цвет
такого индикатора изменится еще до того, как весь,
борат натрия будет оттитрован. Поэтому погрешность
титрования выразится отношением концентрации не¬
оттитрованной (ВСЬ) и оттитрованной (НВ02) частей,
т. е.f ВО, 1 • 100 к„во, • юо 10-9-2 • 100
д = ^ = в- ,— = г— = о ,06 7о.[НВ02] Н+ 10“6Погрешность невелика, титрование с выбранным
индикатором вполне удовлетворительно.Особый случай — титрование многоосновных кис¬
лот или оснований до амфотерных соединений. Здесь
некоторый избыток рабочего раствора сверх эквива¬
лентного количества не приводит к появлению сво¬
бодной сильной кислоты или свободного сильного
основания. Этот избыток расходуется на дальнейшую
нейтрализацию НА- с образованием основания (тит¬
рование раствором щелочи) или слабой кислоты (тит¬
рование раствором сильной кислоты). Вычисление по¬
грешности титрования характеризуется некоторыми
особенностями.Рассмотрим в качестве примера титрование кар¬
боната натрия до гидрокарбоната:С02_ + Н+ ч=* НСО‘.В этом случае, учитывая, что p/Ci = 6,5, а рЛ'г =
= 10,2, pH точки эквивалентности равно 8,35.Если взять, например, индикатор с рТ = 9, тогда
раствор в конечной точке недотитрован и погрешность
равна[СО2'] • 100
А = - , 1 ,[НСО-]136
Из выражения второй константы диссоциации
угольной кислоты[н+][со*1К2[нСО.)][со| ] К2видно, что отношение -7 поэтому[НСО-] [Н-] *Кг ■ 100
[Н+] 10'Д=^77Т71-= = 6%-Погрешность очень большая.Вычислим погрешность при титровании с индика¬
тором, имеющим рТ = 8. В этом случае раствор будет
перетитрован. Однако избыток введеннэй кислоты не
останется в свободном состоянии, а грореагирует с
анионом НСОз:НСО" + Н+ =F=fc н.со.,Посколькук,[НСО-] ■
[Hi [нсо-][H2co3] ’[Н2С03]-100 [Н+]-100 1(Г8-Ю0[Н+] [НСО-] /с, ю-6’5Погрешность меньше, чем в предыдущем случае,
но все же еще достаточно велика. Ее можно умень¬
шить, более удачно выбрав индикатор. Однако в об¬
щем титрование до анионов НА- обычно не слишком
точно. В рассматриваемом случае луние титровать
до свободной угольной кислоты. Погрешность будет
меньше, что согласуется с общим характером кривой
титрования карбоната натрия, приведенной в § 2 гл. 7.Фосфорную кислоту обычно титруют только до
кислой соли Na2HP04, так как pH ра:твора ЫазРО*
слишком велико (для 0,1 М раствора pH = 11,6). Учи¬
тывая, что pKi, рКг и рК3 равны соответственно 2,0,7,0 и 11,0, pH раствора ЫагНР04 составляет 9,5.137
Применяя индикатор с рТ = 9, находим[н,ро;1-юо [н+] - юо юг9 - юо
А = -Ц Ц-. = = = I %.[НРО*] К2 Ю“7Если взять индикатор с рТ = 10, погрешность со¬
ставитГРОМ-ЮО /Со-100 10 _! 2 - 100д _ I 4 I J _ _ 1 0/[НРО^] [н+] ю~10В обоих случаях погрешность одинакова. Анало¬
гичный расчет показывает, что если титровать до
NaH2P04, получается приблизительно такая же по¬
грешность.ГЛАВА 8
ИНДИКАТОРЫ§ 1. ОБЩАЯ ХАРАКТЕРИСТИКАСуществуют различные методы установления точки
эквивалентности. Их можно разделить на две группы:
первая группа — индикаторные методы, вторая груп¬
па — физико-химические методы. Ко второй группе от¬
носят потенциометрический метод (наиболее распро¬
страненный), основанный на измерении в процессе
титрования потенциала погруженного в титруемый
раствор металлического электрода; амперометриче¬
ский метод, в котором измеряют силу тока, прохо¬
дящего через раствор; метод радиоактивных индикато¬
ров, в котором измеряемым свойством является ин¬
тенсивность радиоактивного излучения одного из
участников реакции и др.Индикаторы представляют собой химические соеди¬
нения, которые дают какой-либо внешний эффект при
концентрации реагирующих веществ, соответствующих
точке эквивалентности. Этот внешний эффект может
состоять в изменении, возникновении или исчезнове¬
нии окраски, образовании или растворении малорас¬
творимого соединения и др. Существенно при этом,
чтобы внешний эффект проявлялся только при такой138
концентрации определяемого вещества X или реаген¬
та R, которая соответствует точке эквивалентности.
Рассмотрим, например, титрование хлоридов раство¬
ром нитрата ртути. Определение основ* но на реакции2Cr + Hg2* HgCl2,в результате которой образуется очень лало диссоции¬
рованная хлорная ртуть. Для установления точки эк¬
вивалентности необходимо прибавить < раствору ве¬
щество, способное реагировать с иона ли ртути (или
хлора) и вызывать при этом какой-ннбудь внешний
эффект, например образование осадка. Можно попы¬
таться воспользоваться раствором сульфида натрия,
который реагирует с ионами ртути с образованием
черного осадка HgS. Однако опыт показывает, что
сульфид натрия непригоден в качестЕе индикатора:
первая же капля прибавляемого рабочего раствора
нитрата ртути приводит к осаждению Ь gS, что объяс¬
няется очень малой растворимостью осацка. Ионы рту¬
ти реагируют в первую очередь не с ионами хлора, а с
ионами S2~, и точка эквивалентности (>удет зафикси¬
рована неправильно. Ионы S2- в этом случае являют¬
ся чрезмерно чувствительным реагентом, который
фиксирует наличие ионов ртути прп значительно
меньшей их концентрации, чем в точке эквивалентно¬
сти. Для данного определения можнс использовать
раствор нитропруссида натрия Na2[Fe [CN)sNO], ко¬
торый образует осадок Hg[Fe(CN)5NO], но, в отли¬
чие от предыдущего примера, только госле того, как
все ионы хлора свяжутся в малодиссоц нированное со¬
единение HgCl2.Из приведенных примеров следует ьажный вывод:
индикатор должен реагировать с реагентом R или
с определяемым веществом X только гри такой кон¬
центрации одного из них, какая создаемся в точке эк¬
вивалентности.Рассмотрим титрование вещества X реагентом R:X + R =F=t XR. (8.1)Обозначим логарифм концентраци i реагента R
в точке эквивалентности, взятый с обратным знаком,
символом рЯэкв, т. е.IS RaKB = PRskb- (8-2)139
Логарифм концентрации R с обратным знаком обо¬
значим через рТ:- \g R = рТ. (8.3)Величину рТ называют показателем титрования
индикатора. Очевидно, условием правильного опреде¬
ления точки эквивалентности с помощью того или
иного индикатора является равенство этих двух ве¬
личин:рРэкп = рТ. (8.4)Аналогично, если индикатор реагирует с опреде¬
ляемым веществом X, наиболее благоприятным усло¬
вием точного определения будет возникновение внеш¬
него эффекта реакции прирХэкв~рТ- (8.5)Не всегда удается точно соблюсти эти условия.
Свойства продуктов реакции меняются в зависимости
от условий титрования. Поэтому точное выполнение
условий (8.4) или (8.5) означало бы необходимость
иметь для титрования каждого вещества отдельный
индикатор. Однако в практике нередко при определе¬
нии различных веществ применяют один и тот же
индикатор, допуская некоторые отклонения от ра¬
венств (8.4) или (8.5). Это приводит к тому, что ин¬
дикатор фиксирует конец титрования раньше или
позже, чем достигнута точка эквивалентности.Момент титрования, когда наблюдается внешний
эффект реакции между индикатором и рабочим рас¬
твором реагента R (или определяемым веществом X)
называют конечной точкой титрования. Расхождение
между конечной точкой титрования и точкой эквива¬
лентности, т. е. неравенство рТ и рХэкв (или pR3Kb)
является одной из причин возникновения погрешности
титрования. Важно правильно оценить влияние свойств
продукта реакции и реагента на точность титрования
и уметь определить допустимое расхождение рТ и
рХэкв (или pR,KB), которое не вносит еще чрезмерной
ошибки в определение.Рассмотрим влияние характера кривой титрования
и расхождения рТ и рХэкв на погрешность определе¬
ния. В качестве примеров возьмем кривые титрования
вещества X раствором реагента R при различных ис¬
ходных концентрациях компонентов (рис. 8.1). Кри-140
Рас. 8.1. Кривые титрова¬
ния вещества X веществом
R (см. подписи к рис. 7.1 и
7.2)вая 2 соответствует
титрованию 0,1 М рас¬
твора X 0,1 М раство¬
ром R, кривая / —0,01 М концентрациям
этих веществ. Величи¬
на рХэкв равна пяти и
отвечает средней точ¬
ке — точке перегиба
обеих кривых.Предположим, что
индикатор образует с
определяемым вещест¬
вом X окрашенное соединение, которог диссоциирует
на ионы при такой концентрации X, которая соот¬
ветствует рТ. В этот момент наблюдается внешний
эффект — изменение окраски — и фиксируется конеч¬
ная точка титрования. Из рисунка видно, что
рТ <С рХэкв, следовательно цвет изменится до того,
как будет достигнута точка эквивалентности, т. е.
когда раствор X несколько недотитрован. Определим
погрешность титрования как отношение количества
(концентрации) неоттитрованной части вещества X в
конечной точке титрования к ее общему количеству
(концентрации) в начальной точке титэования. В на¬
шем примере неоттитрованную часть (в случае 0,1 М
растворов) можно характеризовать отрезком I на оси
абсцисс, а общее количество веществ* X — отрезком
т. Из рисунка видно, что ошибка невелика; это обус¬
ловлено очень резким изменением концентрации X
вблизи точки эквивалентности. Поэтому даже доволь¬
но значительное расхождение между рХэкв и рТ —
в нашем примере оно составляет несколько меньше
двух единиц—мало отражается на тэчности титро¬
вания.Однако при 0,01 М концентрациям компонентов
(кривая /) погрешность при том ж; расхождении
рХэкв и рТ резко возрастает. Она выражается отрез¬
ком п на оси абсцисс. Причина возрастания погреш¬141
ности заключается в различном ходе обеих кривых.
При 0,1 М растворах участок кривой вблизи точки
эквивалентности почти вертикален, при уменьшении
концентрации реагирующих веществ в десять раз он
становится пологим, а скачок изменения концентра¬
ции — меньшим.Сравним кривые титрования вещества X двумя ре¬
агентами: реагентом R, который дает продукт реак¬
ции, заметно диссоциирующий на исходные вещества
X и R (кривая 2), и реагентом С, образующим веще¬
ство ХС, которое диссоциирует в значительно мень¬
шей степени (кривая 3). Видно, что кривые разли¬
чаются по высоте. Поэтому при одной и той же по¬
грешности определения разность рТ и рХ в первом
случае (вещество XR) может быть значительно боль¬
шей, чем во втором (вещество ХС).Из рассмотренных примеров следует, что погреш¬
ность определения увеличивается с возрастанием рас¬
хождения между рХ и рТ. При данном значении этого
расхождения погрешность возрастает с уменьшением
концентрации растворов реагирующих веществ, а так¬
же с уменьшением прочности связи между X и тит-
рантом.§ 2. КЛАССИФИКАЦИЯ ИНДИКАТОРОВВ основе классификации индикаторов могут быть раз¬
личные признаки.По технике применения различают внутренние и
внешние индикаторы. Внутренние индикаторы — веще¬
ства, которые вводят непосредственно в титруемый
раствор. Конечную точку титрования устанавливают
по изменению цвета индикатора, по появлению или
исчезновению осадка или по какому-либо другому
внешнему эффекту реакции. Внутренние индикаторы
широко распространены. Внешние индикаторы при¬
меняют в тех случаях, когда использование внутрен¬
них индикаторов невозможно. Например, ионы ни¬
келя можно титровать раствором диметилглиоксима.
Однако красный цвет осадка диметилглиоксимата
никеля мешает наблюдению эффекта реакции, вы¬
званного прибавлением индикатора. Поэтому в про¬
цессе титрования время от времени отбирают каплю
раствора, переносят ее на часовое стекло и прибав¬142
ляют реагент (тот же диметилглиоксим) на ионы ни¬
келя. Титрование ведут до отрицательной реакции на
определяемый ион. Применение внешних индикаторов
требует затраты дополнительного времени и высокой
квалификации исполнителя.По обратимости возникновения или исчезновения
внешнего эффекта реакции различают обратимые и
необратимые индикаторы. Обратимые индикаторы —
это соединения, способные существовать в двух (или
более) формах, причем переход одной формы в дру¬
гую обратим. К этому типу принадлежит большинство
известных индикаторов. Так, обратимые окрашенные
индикаторы легко изменяют окраску в зависимости от
избытка или недостатка введенного реагента или
определяемого вещества. Примером мэжет служить
метиловый оранжевый. В щелочной среде он окрашен
в желтый цвет, а в кислой — в красный. Переход
красной формы индикатора в желтую или наоборот
может происходить много раз в зависимости от изме¬
нения концентрации ионов водорода. Обратимые ин¬
дикаторы удобны тем, что при случа»ном введении
слишком большого избытка рабочего раствора опре¬
деление все же может быть правильньм. К перетит-
рованному раствору следует добавить ;ще некоторое
количество определяемого вещества (при этом вос¬
станавливается первоначальная окраска индикатора)
и осторожно дотитровать раствор.Необратимые индикаторы — это соединения, кото¬
рые разрушаются при введении избыто реагента и
цвет которых не восстанавливается от дополнитель¬
ного прибавления раствора определяе\ого вещества.
Тот же метиловый оранжевый может быть примером
необратимого индикатора в реакциях окнсления — вос¬
становления. Трехвалентную сурьму тигруют раство¬
ром бромата калия с метиловым оранжевым, до тех
пор, пока в растворе нет избытка окислителя, инди¬
катор окрашен в красный цвет. После точки эквива¬
лентности некоторое количество распора бромата
калия приводит к окислению индикатора, вследствие
чего раствор обесцвечивается. Естественно, что после
прибавления раствора трехвалентной сурьмы окраска
не восстанавливается, так как весь индикатор разру¬
шен. Необратимые индикаторы менее >добны и при¬
меняются редко.143
Индикаторы различают также по типу, химической
реакции, в которой их применяют. Индикаторы мето¬
дов кислотно-основного титрования — это окрашенные
органические соединения, существующие в двух фор¬
мах, в зависимости от pH раствора. Чаще всего обе
формы различаются по окраске, это так называемые
двухцветные индикаторы. Реже применяют одноцвет¬
ные индикаторы, в которых окрашена только одна
форма. Кислотно-основные индикаторы изменяют
окраску в зависимости от концентрации водородных
ионов раствора и в этом смысле являются специфиче¬
скими индикаторами на ионы водорода. Индикаторы
этой группы являются обратимыми.Для титрования мутных и окрашенных растворов
применяют люминесцентные и хемилюминесцентные
индикаторы. Использование люминесцентных индика¬
торов основано на применении веществ, которые при
освещении ультрафиолетовыми лучами изменяют ха¬
рактер свечения в зависимости от изменения свойств
среды (pH, концентрации ионов металлов или окис¬
лительно-восстановительного потенциала). Поэтому
люминесцентные индикаторы используют в методах
кислотно-основного титрования, комплексообразова¬
ния и окисления — восстановления. В табл. 8.1 приве¬
дены характеристики некоторых люминесцентных ин¬
дикаторов.Применение хемилюминесцентных индикаторов
основано на том, что в некоторых химических реак-Таблица 8.1. Люминесцентные индикаторыИндикаторpHпереходаЦветСалициловая кислота
Флуоресцеин
Днхлорфлуоресцеин
АкридинХромотроповая кислота
Магний-8-гидроксихинолинКумаровая кислота
Акридиновый оранжевый3.0-3,5
3,8-6,11.0-6,0
4 э —5 р6.0-7,07.0—7,2^.г-э.о8,4-10,4Бесцветный — темно-синий
Бесцветный — зеленый
То же3 еленыи — лиловыи
Бесцветный — синий
Бесцветный — золотисто¬
желтыйБесцветный — зеленый
Бесцветный — желто-зеле¬
ный144
циях выделяется энергия не в виде тгпла, а в виде
светового излучения. Эго явление называют хемилю¬
минесценцией. Например, в качестве хемилюминес-
центного индикатора можно применить люминол (гид-
разид аминофталевой кислоты). В щелочных раство¬
рах он окисляется пероксидом водорода в присутствии
некоторых катализаторов (например, солей меди, ко¬
бальта, железа и др.) по схеме:+ N2 + Н20 + ftv
COO-Реакция сопровождается свечением. В кислом рас¬
творе реакция не идет и свечение не наблюдается.
Однако если раствор кислоты, содержащий люминол,
пероксид водорода и следы ионов меди, титровать рас¬
твором гидроксида натрия, то после введения неко¬
торого избытка NaOH раствор начинает светиться,
что указывает на конечную точку титрования.Известно очень много индикаторов различного хи¬
мического строения. Их удобно разделить на несколь¬
ко групп. Ниже рассмотрены свойства типичных пред¬
ставителей основных групп индикаторов.1. Азоиндикаторы. Типичный представитель этого
ряда — натриевая соль 4-диметиламиноазобензол-4-
сульфокислоты или метиловый оранжевый:(CH3)2n——N=N—yZ/*—scb‘Na*-Метиловый оранжевый — оранжево-желтый поро¬
шок или кристаллические чешуйки, растворимые в
воде и практически нерастворимые в спирте. В вод¬
ных растворах находится в диссоциированной фор¬
ме— в виде аниона (I). Переход окраски связан с
присоединением к аниону (I) иона водорода и изме¬
нением строения молекулы.145
I II IIIЩелочная среда — Кислая среда — красная формажелтая формаН3С+ Н +К этой же группе относят много других индикато¬
ров аналогичного строения, например метиловый
красный •— 4-диметиламиноазобензол-2-карбоновая
кислота:(CH3)2N— N=N \ УНООС'2. Нитроиндикаторы. В кислой среде бесцветны,
в щелочной — окрашены в желтый цвет. Типичный
представитель — паранитрофенол. Возникновение жел¬
той окраски связано с изменением строения инди¬
катора:I и шКислая среда — Щелочная среда — желтая формабесцветная формаОН О О+ Н*0=N—ОН 0=N—O'146
Индикатор представляет собой бесцветные или
желтоватые кристаллы, легко раствор! мые в спирте,
хлороформе, диэтиловом эфире и умерс нно в холодной
воде. К этой группе индикаторов прингдлежат также
м-нитрофенол, 2,6-динитрофенол, 2,4 динитрофенол,
2,3-динитрофенол, 2,5-динитрофенол, 3,4-динитрофе¬
нол и др.3. Фталеины. Типичный представите.) ь — фенолфта¬
леин. В кислой среде он находится в б :сцветной фор¬
ме, в щелочной—красного цвета. Переход окраски
связан со структурными изменениями:Кислая среда — бесцветная Щелочная греда — краснаялактонная форма хиноидиая формаВ сильно щелочной среде образуется еще одна
бесцветная форма индикатора.Фенолфталеин — белый или слегка желтоватый
кристаллический порошок, нерастворимый в воде, рас¬
творяется в спирте или диэтиловом эфире, а также в
разбавленных растворах щелочей. Другие представите¬
ли этого ряда — а-нафтолфталеин, тимелфталеин и др.4. Сульфофталеины. Один из представителей — фе¬
ноловый красный. В кислой среде окрашен в желтый
цвет, в щелочной — в красный. Переход окраски об¬
условлен структурными изменениями:Кислая среда — Щелочная среда —желтая форма красная {юрмаИндикатор растворим в спирте и в растворе гидр¬
оксида натрия. Другие представители ряда — бром-НО+ 2Н*147
крезоловый пурпурный, бромтимоловый синий, бром-
феноловый синий и др.Из приведенных примеров видно, что во всех слу¬
чаях изменение цвета индикаторов происходит вслед¬
ствие структурных превращений их молекул. Эти пре¬
вращения являются предметом хромофорной теории
индикаторов, в которой рассмотрены цветность соеди¬
нений в зависимости от наличия в молекулах окрашен¬
ных веществ определенных групп атомов, так называе¬мых хромофорных групп I —N=N—, —N=0,и др. I, влияние системы сопряженных двой¬ных связей, наличие ауксохромных групп v—NH2,глощение света органическими соединениями.Очевидно, что изменение цвета всегда происходит
с участием ионов водорода, которые либо отщепляют¬
ся от молекул индикаторов, либо присоединяются
к ним. Изменение окраски, вызванное изменением
pH растворов, объясняет физико-химическая теория
индикаторов. С точки зрения этой теории индикато¬
ры— это окрашенные органические вещества кислот¬
ного, основного или амфотерного характера, которые
способны существовать в растворах в молекулярной
или ионной формах, имеющих различную окраску.
Так, в случае индикатора — слабой кислоты равнове¬
сие между двумя формами — молекулярной и ион¬
ной— можно записать следующим общим уравнением:Для индикаторов типа амфолитов, а также для ин¬
дикаторов— слабых оснований характерна аминная
группа — NH2. Такие индикаторы в общем виде мож¬
но изобразить формулой R — NH2, где R — органиче¬
ский радикал. Изменение окраски таких индикаторов
обусловлено нарушением равновесияи других факторов на по-Hlr.dН+ + Ind".(8 6)RNH2 + H+ =f=>rnh:.(8.7)148
Таблица 8.2. Значения рК и окраска молекуляр юй
и ионной форм некоторых индикаторовИнднкаторР кОк| »аскямолекулярнойформыионной формыМетиловый оранжевый4,0КраснаяЖелтаяБромфеноловый синий4,1ЖелтаяСине-фиолетоваяМетиловый красный5,2КраснаяЖелтаяПромкрезоловый6,4ЖелтаяКрасно-пурпурныйфиолетоваяНейтральный красный6,9Красна яЖелтаяФеноловый красный8,0ЖелтаяКраснаяФенолфталеин9,1Бесцветная»Тимолфталеин10,0*СиняяВажнейшей количественной характеристикой ин¬
дикаторов является константа диссоциации:К l[rlflnd |К ~ [Hind] (8 8)или в логарифмической форме- lg К = рК. (8.9)Значения рК важнейших индикатоэов и окраски
их молекулярной и ионной форм приведены в
табл. 8.2.Окраска индикаторов в молекулярной и ионной
формах может быть самой разнообразной. Суще¬
ственно, что константы диссоциации индикаторов
также различаются. В связи с этим как показано
далее, окраска индикаторов меняется при различных
значениях pH.Для титрнметрического анализа, а -акже для дру¬
гих применений индикаторов важны следующие две
их количественные характеристики: ^тервал pH из¬
менения окраски и показатель титрования. Рассмот¬
рим обе эти характеристики.Интервал pH изменения окраски индикаторов.
При постепенном изменении pH пере>од индикатора
из молекулярной в ионную форму, а, следовательно,
и изменение цвета, происходит такме постепенно.
В сильно кислой среде практически В':е кислотно-ос¬
новные индикаторы находятся в молекулярной форме,149
в сильно щелочной — в ионной, и наблюдатель вос¬
принимает окраску соответственно молекулярной или
ионной форм, несмотря на то, что в кислой среде в
равновесии с молекулярной формой находится неко¬
торое количество ионной формы, а в щелочной —на¬
ряду с ионной формой содержится некоторое неболь¬
шое количество молекулярной. Особенности восприя¬
тия цвета таковы, что окраска ионной формы наряду
с молекулярной (или молекулярной наряду с ионной)
становится заметной только при некотором опреде¬
ленном соотношении обеих форм. Опыт показывает,
что наблюдатель воспринимает изменение цвета на¬
чиная с того момента, когда в равновесии находится
наряду с 90 % одной формы 10 % другой. Проследим,
как изменяется соотношение между формами и, соот¬
ветственно, окраска в зависимости от pH раствора.Из уравнения (8.8) следует= (8.Ю)или после логарифмирования и преобразованийpH-pK + |2trSt (8||)Откуда,g ТВД =рН -рК- (8',2)Выражение в левой части уравнения (8.12) опре¬
деляет окраску индикатора. В табл. 8.3 приведены
значения соотношения концентраций ионной и молеку¬
лярной форм индикаторов и указана соответствую-Таблица 8.3. Соотношения концентраций молекулярной
и ионной форм и окраска индикатора метилового
оранжевогоpHfind ]В [НIп<11 рН Р*find 1
[Hind]Цвет1-31/1000Красный•2— 21/100)>3-I1/10Красно-оранжевый401/1Оранжевый5110/1Желто-оранжевыйь2100/1ЖелтыйУ31000/1»150
щая этим соотношениям окраска растгоров. В каче¬
стве примера взят индикатор метиловый оранжевый
с константой диссоциации К = Ю-4 (р/С = 4).Цвет одной части желтой ионной <|ормы индика¬
тора не заметен на фоне 1000 частей красной молеку¬
лярной формы и поэтому окраска воспринимается как
красная. Раствор имеет красный цвет также при со¬
отношении форм 1/100; при соотношении, равном 100
или 1000 раствор окрашен в желтый цв ;т. Только при
соотношении форм, характерном для промежуточных
значений pH наблюдается заметное изменение цве¬
та— от красно-оранжевого при pH = i, через оран¬
жевый при pH — 4 и до желто-оранжевого при
pH = 5.Интервал pH, в котором наблюдается изменение
цвета индикатора, называют интервалом перехода
окраски индикатора или просто интервалом перехода.
Из табл. 8.3 видно, что переход происходит в преде¬
лах pH от 3 до 5, т. е. начало и конед перехода от¬
мечаются при значениях pH, которые сличаются на
единицу н обе стороны от рК. В общем виде интервал
перехода окраски выражается уравненномpH = [>К ± 1. (8.13)Интервал перехода, описываемьн уравнением
(8.13), несколько условен. При других окрасках обеих
форм начало и конец перехода могут восприниматься
при иных соотношениях концентраций и в соответ¬
ствии с этим интервал перехода может (>ыть несколько
более узким или более широким, чем предусматривает
уравнение (8.13).Интервал перехода непосредственно связан с рК
индикатора. Поэтому изменение цвета различных ин¬
дикаторов происходит в неодинаковых пределах pH.
Это свойство индикаторов показано на рис. 8.2
в виде диаграмм. Диаграмма может служить основа¬
нием для приблизительной оценки pH растворов по¬
средством индикаторов. Так, если нейтральный крас¬
ный окрашен в растворе в оранжевый цвет — пере¬
ходный от красного к желтому—можно утверждать,
что pH раствора находится в пределах от 6 до 8. Если,
например, бромкрезоловый пурпурный окрашен в
желтый цвет—можно сделать вывод, что pH ^ 5,4.
Однако если в новой порции того же раствора будет151
обнаружено, что метиловый оранжевый также жел¬
того цвета, можно сделать вывод, что pH ^ 5. Окон¬
чательный вывод из этих двух опытов тот, что pH
исследуемого раствора находится в пределах 5,0—5,4.Из диаграммы также видно, что изменение цвета
одних индикаторов происходит в кислых растворах,
других — в нейтральных, третьих — в щелочных рас-,
творах. Индикаторы, изменяющие окраску в кислых
растворах, называют сильными индикаторами, а те,
которые изменяют цвет в щелочных растворах — сла¬
быми. Промежуточное положение занимают индика¬
торы средней силы, как, например, нейтральный крас¬
ный, цвет которого изменяется в пределах pH от 6 до 8.Показатель титрования рТ. Для установления точ¬
ки эквивалентности при титровании кислот оснований
или солей имеет большее значение показатель титро¬
вания индикатора. Наиболее резкое изменение цвета
наблюдается, как правило, при одинаковых концен¬
трациях обеих форм индикатора. Поэтому показатель
титрования во многих случаях равен или близок к
рК индикатора:рТ = р/С. (8.14)Отклонения возможны в связи с характерными
особенностями каждого индикатора — в зависимостиРис. 8.2. Изменение цвета наиболее распространенных кислотно¬
основных индикаторов в зависимости от pH раствора152
Таблица 8.4. Показатели титрования важнейших
индикаторовИндикаторр.'<ртМетиловый оранжевый3,64,0Метиловый красный5,25,0Лакмус7,07,0Фенолфталеинбольшая концентрация9,18,0малая концентрация9,19,0Тимолфталеин10,010,0от цвета молекулярной и ионной форм резкое из¬
менение окраски может наблюдаться \е при равен¬
стве концентраций форм, но при нескотько иных со¬
отношениях. Имеет значение также иьдивидуальный
фактор — различное восприятие цветов наблюдателя¬
ми. Поэтому, в отличие от р/С, показатель титрования
является субъективной характеристикой индикатора.Примерные значения рТ индикаторов приведены
в табл. 8.4.Необходимо иметь в виду, что рЛ индикаторов,
а следовательно, и интервалы перехода и показатели
титрования зависят от присутствия посторонних силь¬
ных электролитов, изменяющих ионнук силу раство¬
ров. Константы диссоциации индикаторов определены
обычно в отсутствие посторонних электролитов и со¬
ответствуют термодинамическим константам, т. е.
таким, которые выражены через актив iocth соответ¬
ствующих частиц:аII ]п<1fH4H+K„ld-[inrl(8.15)aHInd flllnd [Hind]где Kr — термодинамическая константа.В сильно разбавленных водных растворах индика¬
торов, какими обычно пользуются при титровании,
коэффициенты активности близки к единице и кон¬
центрационные константы диссоциации индикаторов
(Кк) близки к термодинамическим. Связь между эти¬
ми константами видна из уравнения (8. 6):,Т ^ Hind/<к = К'^H+^lnd(8.16)153
Введение сильного электролита в большой концен¬
трации, например, хлорида натрия, приводит к увели¬
чению ионной силы раствора и, следовательно, к
уменьшению коэффициентов активности заряженных
частиц Н+ и Ind-; в то же время коэффициент актив¬
ности незаряженной молекулярной формы индикатора
остается практически без изменения. Найдем, как из¬
менится концентрационная константа от введения0,1 М раствора хлорида натрия.Ионная сила раствора равнаC,ZT + C,ZH 0,1 ■ I2 4-0,1 ■ I2»*- 2 ~ * -«.I.Коэффициент активности частиц равен
lg / = —0,5—= -0,121 + Унили 10_| • IО0’84 = 0,76.Откуда, подставляя f в уравнение (8.16), полу¬
чаемь'К К! 7„тК =~Т~ 058 = 1'7КТаким образом, константа диссоциации индика¬
тора увеличивается от введения 0,1 М раствора хло¬
рида натрия примерно в два раза по сравнению со
значением константы в отсутствие NaCl. В -соответ¬
ствии с этим интервал перехода индикаторов-кислот
смещается в сторону более высоких концентраций во¬
дородных ионов, т. е. к более низким значениям pH,
а в случае индикаторов-оснований — в сторону высо¬
ких значений pH. Так, раствор фосфорной кислоты,
нейтрализованный по отношению к метиловому оран¬
жевому, снова восстанавливает красный цвет индика¬
тора при добавлении к раствору большого количе¬
ства хлорида натрия. Чтобы окраска соответствовала
нейтральной среде, надо добавить раствор гидроксида
натрия.§ 3. ВЫБОР ИНДИКАТОРА ПРИ ТИТРОВАНИИ
КИСЛОТАМИ И ОСНОВАНИЯМИОбщие требования к выбору индикаторов были рас¬
смотрены в § 1 данной главы. Для точного установле¬
ния точки эквивалентности в кислотно-основных мето¬154
дах необходимо выбрать индикатор < показателем
титрования, равным pH точки эквивалентности:РТ = рНт. э(8.17)В зависимости от свойств титруемого вещества
возможны отклонения от этого равенства.Титрование сильных оснований или сильных кис¬
лот. Сильные основания титруют сильными кисло¬
тами, а сильные кислоты — сильными основаниями.В обоих случаях признаком точки эквивалентно¬
сти является равенствоСледовательно, в обоих случаях, чтобы погреш¬
ность титрования была равна 0, необходимо выбрать
индикатор с рТ = 7.Степень допустимого отклонения р! от pH точки
эквивалентности оценивается на основании измене¬
ния pH в процессе титрования и вида кривой титро¬
вания, рассмотренных в § 2 гл. 7. В самом деле, из
данных табл. 7.2 и рис. 7.3 видно, что раствор хлоро¬
водородной кислоты, недотитрованный или перетит-
рованный на 0,1 %, имеет соответственно pH = 4,3
или 9,7. Следовательно, если при определении допу¬
стима ошибка 0,1 %, можно применять индикаторы
с показателями титрования, находящимися в преде¬
лах от 4,3 до 9,7. Практически сильнук: кислоту (или
сильное основание) можно титровать сз всеми инди¬
каторами, указанными в табл. 8.2. Н которые кор¬
рективы вносит присутствие в растворе растворенного
диоксида углерода.Титрование слабых кислот типа НА или ВН+. Для
титрования применяют рабочий раствор сильного
основания, чаще всего раствор гидро <сида натрия.
В результате образуются анионные или незаряжен¬
ные основания А- или В. Следовательно, pH точки
эквивалентности равноПри титровании 0,1 М раствора укс/сной кислоты
(рК = 4,8) в точке эквивалентности pH равно[Н + ] = [ОН‘]= 10~7 или pH = 7(8 18)pH = 7 + 2,4 - 0,5 = 8,9.(8.19)155
Поэтому для титрования необходимо взять инди¬
катор с соответствующим значением рТ. Из табл. 8.2
видно, что лучше всего подходит фенолфталеин.На основании кривой титрования, приведенной в
§ 2 гл. 7, можно сделать вывод, что допустимое от¬
клонение меньше, чем в случае титрования сильных
кислот.Титрование слабой двухосновной кислоты Н2А
можно в принципе осуществить с двумя различными
индикаторами. Можно титровать до одно- или двух¬
зарядного оснований НА- или А2~ в соответствии
с уравнениямиН2А + ОН~ ?=* НА" + НгО. (8.20)НА” -г ОН' ч=* А2 +Н20. (8.21)В первом случае образуются анионы НА-, во вто¬
ром— анионы А2~ и pH точек эквивалентности соот¬
ветственно равнырн = pKl + рК- . (8.22)pH = 7 + — р/Сг + ~2 Со- (8.23)Так, при титровании сернистой кислоты (рК\ =
= 2,0, р/С2 = 7,0) до оснований HSO3 и SO3 pH в
точках эквивалентности соответственно равно 4,5 и
10,0. Поэтому при титровании до HSO3 лучше всего
подходит метиловый оранжевый или метиловый крас¬
ный, а в случае титрования floSO:i—тимолфталеин.При титровании катионной кислоты — хлорида
анилиния СйНяМНзСГ реакция идет в соответствии
с уравнениемC.6H5NH^ + ОН" = C(.H5NH2 + Н20. (8.24)причем образуется незаряженное основание.п ^ о [С6Н,МН21[НЧ пс ,„-s ^ „сДля CGHr.NHлКк — —: ; = 2,5 10 и р/\ — 4,6.[c6h5nh;|Принимая, что концентрация анилина в точке эк¬
вивалентности равна 0,1 М, получаемpH = 7 + 2,3 - 0,5 = 8,8. (8.25)156
Лучше всего подходит по величине )Т индикатор
фенолфталеин.Титрование слабых оснований. В точке эквива¬
лентности образуется слабая незаряжешая или ка¬
тионная кислота НА или ВН+. ПоэтомуpH = -i-p/(--j«gCK. (8.26)При титровании 0,1 М раствора амниака (рК =
= 9,2)pH = 4,6 + 0,5 = 5,1.Лучший индикатор — метиловый красный.При титровании цианида калия реакция идет по
следующему уравнению:С1\Г + Н* = HCN. (8.27)Учитывая, что р/С = 9,2, и приним.я концентра¬
цию кислоты в точке эквивалентности равной 0,1 М,
получаемpH = 4,6 + 0,5 = 5,1Лучше всего подходит метиловый красный с
рТ = 5.Многоосновные основания, например А2-, титруют¬
ся в дес стадии. В первой стадии образуется НА-,
во второй — Н2А. Поэтому возможны два варианта
определения. Например, раствор карбоната натрия
титруют до аниона НСОз:СО^ +Н+ НСО;. (8.28)Учитывая, что p/Ci = 6,5 и р/(2 = 10,2, в первой
точке эквивалентностирН - 6,5 + 'О’2 =8,35.В этом случае необходимо пользозаться фенол¬
фталеином.При титровании до свободной кислот лСО*- + 2Н* 4=fc Н2СОа. (8.29)Если принять, что концентрация кислоты после
нейтрализации равна 0,1 М, тогдаpH = 3,25 + 0,5 = 3,75.Видно, что лучшим индикатором будет метиловый
оранжевый с рТ = 4,0.
Раздел IIIРЕАКЦИИ ОБРАЗОВАНИЯ
ТВЕРДОЙ ФАЗЫГЛАВА 9РЕАКЦИИ ОСАЖДЕНИЯ И РАСТВОРЕНИЯ
ОСАДКОВРеакции образования труднорастворимых соедине¬
ний— осадков — применяют в аналитической химии
для разделения ионов, а также для их обнаружения
в качественном анализе и для гравиметрического и
титриметрического осадительного определения в коли¬
чественном анализе. Процессы осаждения и растворе¬
ния соединений являются сложными физико-химиче¬
скими процессами и имеют большое значение не только
в химическом анализе, но и для разделения и выде¬
ления различных веществ в химической технологии.Способность к осаждению зависит от многих фак¬
торов: свойств катионов и анионов, входящих в состав
труднорастворимого соединения, концентрационных
условий, в которых проводят процесс осаждения, pH
раствора, температуры, ионной силы раствора, состава
и содержания других веществ в растворе, которые не
должны принимать прямого участия в образовании
осадка, однако могут соос.аждаться с ним или, наобо¬
рот, препятствовать осаждению. Все это необходимо
учитывать при проведении реакции осаждения.§ 1. РАВНОВЕСИЕ МЕЖДУ РАСТВОРОМ
И ТВЕРДОЙ ФАЗОЙРаствором называют гомогенную систему, состоящую
из двух или большего числа компонентов. При пере¬
ходе вещества в раствор происходит разрыв межмо-
лекулярных и ионных связей кристаллической решет¬
ки твердого вещества и переход его в раствор в виде
отдельных молекул или ионов, которые равномерно
распределяются среди молекул растворителя.Для разрушения кристаллической решетки веще¬
ства необходимо затратить большую энергию. Эта158
энергия освобождается в результате гидратации
(сольватации) ионов и молекул, т. е. химического
взаимодействия растворяемого вещества с водой (или
вообще с растворителем). Таким образ эм, раствори¬
мость вещества зависит от разности ве./ ичин энергии
гидратации (сольватации) и энергии кристаллической
решетки вещества.В зависимости от соотношения вели шн этих теп¬
ловых эффектов процесс растворения зещества мо¬
жет быть эндо- и экзотермическим.Если количество тепла, необходимое для разруше¬
ния кристаллической решетки 1 моль вещества, обо¬
значить Qp, количество тепла, выделяемого при взаи¬
модействии растворителя с частицами 1 моль веще¬
ства (сольватация) Qc, то теплота растворения 1 моль
веществаQpacT = Qc Qp-Теплота растворения различных веществ различна.
Так, при растворении в воде хлорида нагрия темпера¬
тура практически не изменяется, при растворении
нитрата калия или аммония температу )а резко сни¬
жается, а при растворении гидроксида калия или
серной кислоты температура раствора резко повы¬
шается.Энергию кристаллической решетки \ ожно рассчи¬
тать теоретически. Однако для теоретического расчета
энергии сольватации до сих пор нет надежных мето¬
дов. Поэтому в преобладающем большинстве случаев
нельзя заранее предвидеть, как будут растворяться
химические соединения различных типов в воде и дру¬
гих растворителях. Существуют лишь некоторые част¬
ные закономерности, которые связывают раствори¬
мость веществ с их составом. Известаа, например,
теоретическая закономерность, согласно которой для
ряда солей одного и того же аниона с разными ка¬
тионами (или наоборот) энергия ионной кристалличе¬
ской решетки максимальна, а поэтому растворимость
будет наименьшей в том случае, когда соль образо¬
вана ионами одинакового заряда и примерно одинако¬
вого размера. Так, растворимость сульоатов элемен¬
тов второй группы периодической системы увеличи¬
вается при переходе от бария к стронцию, кальцию
и магнию. Это объясняется тем, что ионы бария159
и сульфата по размерам больше всего подходят друг
к другу. В то время как катионы кальция и магния
намного меньше анионов SO^. Растворимость гидр¬
оксидов бария, стронция, кальция и магния, наоборот,
уменьшается от бария к магнию, потому что радиусы
катионов магния и анионов гидроксида практически
одинаковые, а катионы бария по размеру очень отли¬
чаются от небольших анионов гидроксила. Однако
имеются случаи, когда эта закономерность не отвечает
действительности, например для оксалатов и карбо¬
натов кальция, стронция, бария и др. Поэтому зави¬
симость между химическим составом соединений и их
растворимостью в воде до сих пор приходится описы¬
вать некоторыми эмпирическими правилами, имею¬
щими статистический характер.Эти правила следующие.1. Неорганические и органические кислоты в боль¬
шинстве случаев хорошо растворимы. Исключением
являются кремневая (НгЭЮз), оловянная (H2Sn03)
и сурьмяная (НБЬОз) кислоты.2. Все основания малорастворимы, за исключением
щелочей: LiOH, КОН, RbOH, CsOH, NaOH, Ва(ОН)2,
Sr(OH)2.3. Соли по растворимости разделяют на две боль¬
шие группы: соли сильных кислот, как правило, рас¬
творяющиеся хорошо, исключение представляют
сульфаты бария, стронция и свинца, хлориды, бро¬
миды и иодиды свинца, серебра и одновалентной рту¬
ти; соли слабых кислот, растворяющиеся плохо, за ис¬
ключением солей лития, натрия, калия, рубидия и це¬
зия, а также нитритов и ацетатов.Приведенные правила имеют приближенный ха¬
рактер, однако они помогают ориентироваться в рас¬
творимости соединений разных типов и дают возмож¬
ность предвидеть направление химических реакций,
которые происходят между ионами в растворе и в ре¬
зультате которых образуются малорастворимые со¬
единения. Приведем несколько примеров.Фосфат висмута — малорастворимая соль, так как
фосфорная кислота относится к слабым кислотам,
и преобладающее большинство солей этого типа труд¬
норастворимы.Если смешать растворы хлорида натрия и нитрата
кобальта, никакого химического взаимодействия не160
произойдет, потому что в растворе находятся только
анионы сильных кислот, а соли этих кислот, как пра¬
вило, хорошо растворяются.Если же смешать растворы сульфита натрия и хло¬
рида бария, то при этом должна произойти реакция,
так как в растворе присутствуют катионы натрия и
бария и анионы хлорида и сульфита. Ионы SO3 яв¬
ляются анионами слабой кислоты, а большинство со¬
лей слабых кислот, в том числе и соль бария, в воде
практически не растворяются. Уравнение реакции, ко¬
торая происходит, можно изобразить следующим об¬
разом:Ba2t + SO’- BaSCXj.Кроме химического состава, на растворимость со¬
единении в сильной мере влияют внешние условия
проведения реакции, в том числе концентрация реаги¬
рующих компонентов и температура.Растворимость большинства солей с повышением
температуры повышается. Так, растворимость суль¬
фата кобальта увеличивается почти в два раза с по¬
вышением температуры раствора от 15 до 100 °С. Ана¬
логичная зависимость характерна и для хлорида
серебра, сульфата бария и других солей. В качествен¬
ном анализе увеличение растворимости солей с повы¬
шением температуры иногда используют для обнару¬
жения и разделения некоторых ионов. Так, ионы
свинца отделяют от ионов серебра, переводя их в
хлориды, а затем нагревают раствор с осадком до
температуры кипения. При этом осадок РЬС12 прак¬
тически полностью растворяется, a AgCI остается в
твердой фазе. Зависимость растворимости солей от
концентрации реагирующих компонентов рассмотрена
ниже.Процесс растворения вещества всегда сопровож¬
дается диффузией, т. с. перемещением молекул из об¬
ластей более концентрированного раствора в области
с меньшей его концентрацией. Другими словами, ве¬
щество при растворении равномерно распределяется
по всей массе растворителя. Этот процесс происходит
до тех пор, пока концентрация данного вещества в
растворе не доходит до определенного значения, при6 Аналитическая химия161
котором наступает состояние равновесия:MR MRтвердая раствор
фазаТаким образом, растворение является двунаправ¬
ленным процессом: твердое вещество переходит в рас¬
твор, а растворенное вещество в свою очередь пере¬
ходит в твердую фазу. Следовательно, процесс
растворения — равновесный процесс. С увеличением
концентрации раствора замедляется установление
равновесия. Если количество вещества, переходящего
в раствор за единицу времени, равно количеству ве¬
щества, выделяющегося за то же время в твердую
фазу, то это значит, что произошло насыщение рас¬
твора. Раствор, отвечающий такому состоянию, назы¬
вают насыщенным раствором, концентрация насыщен¬
ного раствора при данной температуре является
величиной постоянной.С изменением температуры изменяется и концен¬
трация насыщенного раствора. При понижении тем¬
пературы раствор может в определенных условиях
некоторое время сохранять данную концентрацию ве¬
щества, т. е. концентрация раствора может оказаться
выше, чем в насыщенном растворе при данной темпе¬
ратуре. Такие растворы называют пересыщенными.
Насыщенные растворы являются стабильными систе¬
мами, т. е. они могут существовать при данной темпе¬
ратуре без изменения концентрации сколь угодно
долго. Пересыщенные же растворы являются неста¬
бильными системами. Достаточно перемешать такой
раствор или бросить самый маленький кристаллик
растворенного вещества (затравку), чтобы начала
выделяться твердая фаза. Этот процесс продолжается
до тех пор, пока концентрация вещества не достигнет
концентрации насыщенного раствора при данной тем¬
пературе. Растворы, содержащие меньше вещества,
чем необходимо для насыщения, называют ненасы¬
щенными. Очень многие вещества растворяются в
воде весьма слабо, или, как говорят, являются прак¬
тически нерастворимыми.В растворах электролитов непрерывно происходят
процессы ионизации и ассоциации. При этом поддер¬
живается равновесие, сохраняется постоянным состав162
раствора, но процесс электролитической диссоциации
не прекращается. Если же в раствор ввести некоторое
другое вещество, то его ионы могут вступить в реак¬
цию с первым веществом и образовать новое вещество,
которое не вводилось в раствор. Например, в отдельно
приготовленных растворах хлорида бария и сульфата
натрия устанавливается равновесие:в первом растворе: ВаС12 < * Ва2* + 2С1 ,
во втором растворе: Na2SC>4 2Na* + SO^'.Оба эти соединения представляют собой соли и отно¬
сятся к сильным электролитам, т. е. в разбавленных
растворах эти вещества находятся преимущественно
в виде ионов. Если слить эти два раствора, то ионы
SO4 встретятся не только с ионами натрия, но и с
ионами бария и вступят с ними в реакцию:SO^- + Ba2t +=± BaSO,.Эта реакция происходит достаточно быстро, так как
сульфат бария является малорастворимым соедине¬
нием и выпадает в осадок. В растворе останутся ка¬
тионы натрия и анионы хлора, но осадка не обра¬
зуется, потому что хлорид натрия хорошо растворим
в воде.Процесс осаждения происходит постепенно. Сна¬
чала образуются очень мелкие кристаллы — зароды¬
ши, которые постепенно вырастают в кристаллы боль¬
шого размера или группу кристаллов. Время с мо¬
мента смещения растворов до образования зароды¬
шей— мелких кристаллов называют индукционным пе¬
риодом. Продолжительность этого периода зависит от
индивидуальных свойств осадка. Так, в случае обра¬
зования хлорида серебра это время очень мало, в слу¬
чае образования сульфата бария этот период значи¬
тельно больше.Осаждение следует проводить так, чтобы образо¬
вывалось по возможности меньшее количество мелких
кристаллов (зародышей), тогда при постепенном при¬
бавлении осадителя будут нарастать имеющиеся
центры кристаллизации, т. е. будут расти крупные
кристаллы.6*163
§ 2. ПРОИЗВЕДЕНИЕ РАСТВОРИМОСТИ
И ПРОИЗВЕДЕНИЕ АКТИВНОСТИОсадки, используемые в химическом анализе, пред
ставляют собой соли, основания и кислоты. Чаще
всего используют малорастворимые соли. Соли неор¬
ганических кислот представляют собой, как правило,
сильные электролиты и в растворах практически пол¬
ностью диссоциируют на ионы.Таким образом, в насыщенном растворе неоргани¬
ческой малорастворимой соли MxRj, содержатся в
основном только отдельные ионы М"+ и Rm_, которые
находятся в равновесии с твердой фазой M.,RM:MxRj, ч=> + yRm~. (9.1)В соответствии с законом действия масс константа
равновесия этой реакции равна*--T-LRrr- м[MxRylКонстанта равновесия не зависит от абсолютного ко¬
личества твердой фазы и является величиной постоян¬
ной. Поэтому вместо равенства (9.2) можно записать[MB + ]*lRm~]l' = const = ПРМ 1; . (9.3)х уУравнение (9.3) показывает, что произведение кон¬
центрации ионов (точнее произведение активностей
ионов — см. ниже) в насыщенном растворе малорас¬
творимой соли при постоянных давлении и темпера¬
туре является величиной постоянной. Эту постоянную
величину называют произведением растворимости и
обозначают * ПР. Из уравнения (9.3) видно, что при
увеличении концентрации катионов осадка умень¬
шается концентрация анионов (и наоборот). Дело, ко¬
нечно, не в формальной математической зависимости,
а в физическом смысле явления. При возрастании1
концентрации ионов М"+ или Rm~ увеличивается ве¬
роятность встречи между ними. При этом скорость
осаждения увеличивается по сравнению со скоростью
реакции растворения осадка, и поэтому концентрация* Произведение растворимости обозначают по-разному: а од¬
них учебниках — Lp (от нем. Losliclvkeitsproduct); в других - Sp
(от англ. Solubility product). Мы пользуемся обозначением ПР —
сокращение русского термина «произведение растворимости».164
других разновидностей ионов в растворе уменьшается.
Произведение растворимости определенного осадка
обозначают через ПР с индексом, который соответ¬
ствует составу вещества, как это показано в уравне¬
нии (9.3).В реальных условиях анализа сравнительно редко
приходится иметь дело с насыщенными растворами
малорастворимых соединений, не содержащими ка¬
ких-либо посторонних ионов, которые способны взаи¬
модействовать с ионами осадка. Эти конкурирующие
реакции приводят к увеличению растворимости. В та¬
ких случаях для расчета растворимости удобно поль¬
зоваться так называемыми условными произведениями
растворимости. Условное произведение растворимо¬
сти — это произведение общих концентраций ионов
осадка М' и R' независимо от того, в какой форме —
свободной или связанной — эти ионы находятся в на¬
сыщенном растворе малорастворимой соли. Так, ка¬
тионы осадка могут вступать во взаимодействие с ка¬
ким-либо лигандом, образуя комплексы MR, MR2
и т. д. Тогда справедливо уравнение (заряды частиц
для простоты опущены)[M'1 = [M] + [MR1 + [MR2]+ .... (9.4)Константы устойчивости этих комплексов имеют
вид:[MRI „ _ [MR*] ,Q,.Pl [Ml [R] ' Рг [M] [R]2 " т- Д- ( • )Комбинируя уравнения (9.4) и (9.5), получаем
[М'|[Ml= I + P,[R1 + MRI2+ ■■■ =aM(R). <9-6>где [М| — концентрация свободных (не связанных в комплекс)
ионов металла в насыщенном растворе малорасткоримой соли;
a — так называемый коэффициент побочной реакции.Анионы осадка в свою очередь могут подвергаться
аналогичным или иным превращениям. Наиболее ча¬
стый случай — это протонизация анионов солей сла¬
бых кислот с образованием частиц HR, H2R и т. д.Тогда, по аналогии с (9.4) можно записать[R'] = [R] + [HR] + [H,R1+ .... (9.7)165
Вводя константы диссоциации образовавшихся
протонированных частиц,, [н+] [R] [Н*] [HR] ,оа)= [HR] ’ К[H2R]— " Т‘ Д' (9'8)и сочетая уравнения (9.7) и (9.8), находим[R'l , , [Hi [Hi _ (00)= 1 + —„ 1-тпг+ — aR(H)-' [R] ‘ КI 1 KiK,Запишем выражения для условного (ПР') и истин¬
ного ПР произведений растворимости осадкаПР' = [МП + Т (9.10)ПР = [мп + ]л [R'"-]^. (9.11)Подставляя в уравнение (9.10) значения [М'] и
[R'] из уравнений (9.3) и (9.9), находимПР' = [М"+Г№т-]%м(К,«Н(Н) (9-12)ИЛИПР' = ПР<Ч(Юа*(Н). (9.13)Уравнением (9.13) можно пользоваться для расче¬
тов растворимости осадков в реальных условиях ана¬
лиза, если заблаговременно из соответствующих кон¬
стант вычислить и свести и таблицы или графики
величины osm(R) и ccr(h) и их зависимости от концен¬
трации лиганда R и ионов водорода Н+. Необходимо
только помнить, что условные произведения раствори¬
мости не являются постоянными величинами и зави¬
сят от концентраций посторонних ионов, реагирующих
с ионами осадка.Правило постоянства произведения концентраций
следует из применения закона действия масс к насы¬
щенному раствору малорастворимого электролита.
Однако это правило имеет приближенный характер,
потому что равновесие между осадком и раствором
характеризуется более сложной зависимостью. Рас¬
смотрим случай, когда в насыщенном растворе мало¬
растворимой соли содержатся также ионы посторон¬
него сильного электролита. Тогда вокруг находящих¬
ся в растворе ионов осадка образуется ионная атмо¬
сфера — облака из ионов постороннего электролита
противоположного знака, которая замедляет передви¬
жение ионов и таким образом уменьшает вероятность166
встречи их между собою, а также с поверхностью
осадка.В системе (9.1) в результате такого действия ско¬
рость обратной реакции осаждения уменьшается. Ио¬
ны кристаллической решетки в это время продолжают
взаимодействовать с растворителем и переходят в рас¬
твор с прежней скоростью. Поэтому при введении в
насыщенный раствор труднорастворимой соли посто¬
роннего электролита состояние равновесия нарушает¬
ся, часть твердой фазы будет переходить в раствор и
растворимость осадка увеличится. Процесс растворе¬
ния твердой фазы проходит до тех пор, пока актив¬
ность ионов в растворе, т. е. их способность к взаим¬
ным столкновениям, не ст-анет такой же, как и до
введения в раствор постороннего электролита. После
этого снова установится динамическое равновесие
между осадком и ионами раствора. Таким образом,
из приведенного примера можно сделать вывод, что
постоянной величиной является не произведение кон¬
центрации ионов, а произведение их активности, по¬
этому правило произведения растворимости формули¬
руют следующим образом: в насыщенном растворе
малорастворимой соли произведение активностей
ионов при постоянной температуре и давлении яв¬
ляется величиной постоянной. Математически эту за¬
висимость записывают следующим образом:ах„.ау1П =ПА. (9.14)Mn+- Rm- \ IВеличину ПА называют произведением активно¬
сти; она, в отличие от произведения растворимости,
не зависит от концентрации посторонних ионов в рас¬
творе.Связь между произведением растворимости и про¬
изведением активности можно установить, исходя из
следующей зависимости:а = (С, (9.15)где а — активность; [—коэффициент активности; С — концен¬
трация ионов и растворе.Подставляя значение активности из уравнения
(9.15) в уравнение (9.14), имеемГмп+ [М" + ]* [Rm-]y = = ПАМЛ, (9.16)где fee — средний коэффициент активности ионов М'1+ и R"1-167
Для расчета коэффициентов активности можно
применить рассмотренное в § 1 гл. 2 уравнение (2.20),
если ионная сила раствора находится в пределах 0 <
<ц<0,1. Ионную силу вычисляют по уравнению
(2.18).Из уравнения (9.16) следует:ПА рпч« ^ = -7^ <917>/срТаким образом, из уравнения (9.17) можно вычис¬
лить произведение растворимости осадка и пользо¬
ваться им для дальнейших расчетов.Как было показано в § 1 гл. 2, коэффициенты ак¬
тивности приближаются к единице только в очень раз¬
бавленных растворах. Поэтому только в случае очень
разбавленных растворов произведение активности
практически равно произведению растворимости.
В присутствии же посторонних электролитов коэффи¬
циенты активности ионов, которые зависят от ионной
силы раствора, всегда меньше единицы. Отсюда мож¬
но заключить, что произведение растворимости, а так¬
же и растворимость малорастворимых соединений
увеличивается в растворах с повышением концентра¬
ции сильных электролитов.Из приведенных ниже примеров расчета раствори¬
мости осадков видно, что применение уравнения
(9.17) вместо приближенного уравнения (9.3) вносит
лишь небольшие поправки в результаты расчета и в
большинстве случаев увеличением растворимости
осадков под влиянием введения посторонних электро¬
литов можно пренебречь. Поэтому произведение рас¬
творимости является одной из основных характеристик
осадка. Пользуясь этой характеристикой, можно изме¬
нять растворимость осадка, рассчитывать оптималь¬
ные условия осаждения, предвидеть, какими реакция¬
ми осаждения лучше пользоваться для определения
тех или иных ионов.В рассмотренных выше примерах принималось, что
растворенная часть осадка MfR,, полностью диссоции¬
рована на ионы Мп+ и Rm~. Это чаще всего справед¬
ливо для неорганических соединений. Однако иногда,
особенно в соединениях ионов металлов с органиче¬
скими реагентами, в насыщенном растворе содержат¬168
ся не только ионы М"+ и Rm~, но также в большей
или меньшей концентрации недиссоциированные мо¬
лекулы M^Ry, т. е. процесс растворения соответствует
такому общему уравнению:M^Rу M*Rу ^ яМ" + + уЯт~■ (9.18)тв. ф растворИз неорганических соединений такой характер про¬
цесса свойствен, в частности, некоторым сульфидам
металлов.Общая концентрация металла в насыщенном рас¬
творе Р, в соответствии с уравнением (9.18), равнаP = x[Mn+] + je[MxRj,l. (9.19)Чтобы найти Р, необходимо знать, кроме произведе¬
ния растворимости осадка, также константу диссоциа¬
ции (устойчивости) соединения M*Ry:р = !МЛ] (9 20)Р [Мп + Г [Rm ]у ' }Из уравнения (9.20)[M,R„] = р [Мч + Г [Rm“ ]ы = pnPMj[R (9.21)Из уравнения (9.18) получаем, исходя из эквива¬
лентности концентраций ионов в насыщенном раство¬
ре, чтоУ [М"+] = х [Rm^] (9.22)и следовательно[R'"-] = -|-[Мп+]. (9.23)Подставляя в выражение произведения раствори¬
мости [М"+] * [Rm~]у значение [Rm~] из уравнения(9.23), находимх+и /ПР»[М" + ] = Л/ . (9.24)V (y/x)Jи из уравнений (9.19), (9.21) и (9.22) получаем
§ 3. РАСТВОРИМОСТЬ ОСАДКОВ В ВОДЕ
И ВОДНЫХ РАСТВОРАХ ЭЛЕКТРОЛИТОВРассмотренные выше уравнения пригодны для реше¬
ния практических задач. Наиболее простая задача —■
вычисление растворимости осадков в воде в отсут¬
ствие общих ионов или посторонних электролитов.
Под растворимостью обычно понимают концентрацию
ионов металла в насыщенном растворе малораство¬
римой соли.Для этой цели можно воспользоваться уравнениемВ наиболее простом случае, когда стехиометриче-
ские коэффициенты х и у равны единице, приведенное
уравнение превращается в следующее:Пример 1. Наптн концентрацию номов серебра в насыщенном
растворе хлорида серебра.Если ввести в раствор такое количество ионов хлора, чтобы
его концентрация стала равной 0,1 М, для вычисления раство¬
римости можно применить выражение для произведения раство¬
римости:В нашем случае х и у, а также п и т равны единице, и поэтомуВидно, что концентрация ионов С1~ увеличивается
в 10-' : 10-5 = 10 ООО р аз. В результате этого концен¬
трация ионов серебра уменьшится и 10 000 раз, т. е.
будет достигнуто более полное осаждение ионов се¬
ребра.(9.24):Р= [Mn+] = VfIPMR[Ag1 = ynPAgC1.Из справочника находят fIPAsci = 10-10, поэтомур = [Ag+] = V Ю"10 = 10~5А1[Мя + Г[«'"-]" = ПРм R .х У[M+][R-] = riPMRИ
Из этого примера следует, что при добавлении ре¬
актива, в котором содержатся ионы, одноименные с
ионами осадка, растворимость осадка уменьшается.
Этим широко пользуются в химическом анализе.Для достижения полного осаждения определяемых
ионов к исследуемому раствору прибавляют избыток
осадителя. При этом растворимость осадка умень¬
шается, количество определяемых ионов становится
незначительным и практически не влияет на резуль¬
таты анализа.Таким образом, казалось бы, что чем больше кон¬
центрация осадителя, тем полнее осаждение. Однако
иногда большой избыток одноименных ионов приводит
к нежелательным результатам, что объясняется по¬
сторонними процессами. Так, чтобы более полно от¬
делить ионы бария от ионов кальция с помощью суль¬
фата аммония, желательно прибавить большой избы¬
ток осадителя. В то же время при большом избытке
осадителя, кроме сульфата бария в осадок частично
переходит и сульфат кальция*. Другой пример: кад¬
мий можно отделить от цинка в виде сульфида, дей¬
ствуя сероводородом. Для более полного осаждения
CdS желательно увеличить концентрацию ионов суль¬
фида. Этого можно достичь уменьшением концентра¬
ции водородных ионов в растворе, однако тогда нач¬
нет выпадать и сульфид цинка. Таких примеров из¬
вестно много. В этих случаях, особенно когда разница
в растворимости соединений не очень велика, слиш¬
ком большой избыток реагента особенно отрицатель¬
но влияет на процесс разделения.Известны также случаи, когда с увеличением кон¬
центрации осадителя растворимость осадка увеличи¬
вается. Так, при осаждении сульфата свинца раство¬
ром серной кислоты осадок заметно растворяется в
избытке осадителя. Роданид серебра растворяется в
избытке SCN-. Растворение осадков в избытке осади¬
теля связано с образованием комплексных анионов
[Ag(SCN)2]_, которые легко растворяются в воде.Таким образом, избыток осадителя не всегда спо¬
собствует более полному осаждению, как это пока¬
зано на рис. 9.1. Поэтому осадителя необходимо* Часть кальция остается в водном растворе в виде ком¬
плекса.171
Рис. 9.1. Зависимость растворимости осадков от избытка осадн-
теляприбавлять столько, чтобы количество определяемых
ионов, остающихся в растворе, не имело практиче¬
ского значения.Приведем некоторые другие примеры вычисления
растворимости.При мер 2. Произведение растворимости сульфата кальция
равно 6,4- I0-5; однако в отличие от предыдущего примера, в
насыщенном растворе сульфата кальция содержатся в некотором
количестве также педиссоциированние молекулы CaS04, т. е со¬
стояние равновесия характеризуется уравнениемCaS04 = CaSO, = Ca2t + SO**.Tu. фаза РастиорИзвестно, что константа устойчивости CaS04 в растворе= 2- Ю2.[CaSO,[Са2 + ] [SOаствории
9.25):х + у /Р = , д/Для вычисления растворимости в данном случае необходимо
применить уравнение (9.25):4- *РПР.(у!х)уВ рассматриваемом случае х и у равны единице, поэтому
уравнение (9.25) превращается в следующее:Р = ^/ПРСай04 + рПРСа804,Р = VM- lO^5 + 2 ■ 102 - 6,4 ■ 10 5 = 2,08 ■ 10 2М.Таким образом, растворимость заметно отличается от топ,
которая была бы найдена без учета образования в насыщенном
растворе недиссоциированных молекул CaS04.172
Для вычисления растворимости в граммах необходимо най¬
денное значение умножить на молекулярную массу сульфата
кальция, равную 136. Находим, что в 1 л воды растворяется2,08- Ю-2- 136 = 2,8 г CaSO«2.Пример 3. Рассчитать растворимость хромата серебра
Ag2Cr04 в воде.ПРАв2СгО< = [АК+]2[СгОГ]=4-10-‘2.Исходим из общей формулы (9.24)Х+У/ПРМ R[мп+] =(.У/*)*В данном случае х = 2 и у = 1. Поэтому
[М"+] = ^2ПР^И[Ае+]=^2ПРА82Сг04 = ^2-4. 1(Г12 = 2. 1<Г4Л*.В самом деле, состояние равновесия в насыщенном растворе
этой соли характеризуется уравнениемAg2Cr04 ч=Ь 2Ag+ + CrOj'.тв. фазаИз уравнения видно, что концентрация иоиов серебра в рас¬
творе в два раза больше концентрации ионов хромата:[Ag+] = 2[CrOj~],Подставляя в уравнение произведения растворимости вместо
[Ag + | величину 2[CrOj~], имеем:ПРЛя2со,; = 2а [СЮ?’]2 [СЮ*'] = 4 [СЮ1У = 4 • 10“'2.Отсюда находим, что[CrOj] =-^l'i0“12= 1(Г4М и [Ag+] = 2- 10_17И.Таким образом, в отличие от примера с AgCl, для
уменьшения концентрации ионов ееребра в 100 раз
необходимо увеличить концентрацию ионов хромата
в 1/Ю-4 —10 000 раз. Это объясняется тем, что в
примере с AgCl мы имели дело е осадком, который
состоит из ионов одинакового заряда — бинарным
электролитом.173
В приведенных примерах не учтены коэффициенты
активности ионов, а расчеты проведены по прибли¬
женному правилу произведения растворимости. Ниже
приведены расчеты растворимости солей с учетом ион¬
ной силы раствора.Связь между произведением растворимости и про¬
изведением активности показана уравнением (9.17).Растворимость осадков в присутствии сильных по¬
сторонних электролитов вычисляют по этому уравне¬
нию. Коэффициенты активности ионов находят из ура¬
внений, связывающих эти коэффициенты активности с
ионной силой растворов (см. § 1 гл. 2).Рассмотрим примеры вычисления растворимости
осадков в присутствии сильных электролитов.Пример 4. ПАВа304 = (О-10. Найти растворимость BaS04
в 0,05 М растворе хлорида натрия.Ионная сила раствора0,05-12 +0,05 -I2
ц - - 0,05.Коэффициенты активности ионов бария и сульфат-ионов нахо¬
дили по уравнению0.5 • 22 лЛ ■ Ю~2lg / = — / - - = 0,36,откуда f = 0,45. Тогда1 + V5- 10 2ПР = ^ = -12 5 . ю-,°/2 0,452 ’а раствйрнмость
Р = [Ва2 + ] = [SO2 ] = ynPBaSo4 = V5 • I0-‘° = 2,2 ■ 10“sAf.Растворимость BaS04 в 0,05 растворе NaCI в 2,2 раза больше
растворимости в воде.Пример 5. .Значение nAAgci = 10-'°. Найти растворимость
AgCI в 0,05 М растворе сульфата магния.Ионная сила 0,05 М раствора сульфата магния0,057 • 22 + 0,05 • 22
ц = = 0,2.Коэффициент активности ионов серебра и хлорид-ионов находим
по уравнениюlg f = - 0,5 ( ■■-V'-L-- _ 0,2 • 0,2^ = - 0,13.' V 1 + Vo,2 )Откуда t = 0,74.174
ТогдаПР = ЦА =i£^. = 18.io-11 /2 0,742а растворимость Р= 1,8-10 10 = 1,4-10 5 М, т. е. в 1,4 раза
больше растворимости в воде.§ 4. ВЛИЯНИЕ НА РАСТВОРИМОСТЬ РЕАКЦИИ
КОМПЛЕКСООБРАЗОВАНИЯУвеличение растворимости малорастворимых соедине¬
ний часто связано с образованием комплексов. Во
многих случаях осаждаемые ионы способны взаимо¬
действовать с различными лигандами, в результате
чего они влияют на состояние равновесия между осад¬
ком и раствором. Так, ионы винной кислоты и других
оксикислот мешают осаждению железа в виде гидро¬
ксида. Растворимость сульфата свинца в присутствии
Pb(NOs)2 увеличивается в результате образования
комплексных катионов [Pb2S04]2+, [Pb3 (SO4) 2]2+
и др. При осаждении ионов серебра избытком хлоро¬
водородной кислоты или хлорида натрия часть осадка
хлорида серебра переходит в раствор в виде комп¬
лексных анионов [AgCl2]_ или [AgCl3]2~. Сдвиг со¬
стояния равновесия между осадком и раствором в сто¬
рону растворения осадка зависит от произведения
растворимости осадка, устойчивости комплекса, кон¬
центрации лиганда, кислотности раствора и других
условий.Образование комплексных соединений в процессе
осаждения во многих случаях нежелательно, так как
приводит к увеличению растворимости осадков.На реакциях комплексообразования основаны мно¬
гие процессы. Особенно широкое применение нашли
реакции комплексообразования в аналитической хи¬
мии для разделения элементов. Например, для разде¬
ления ионов меди и висмута к раствору солей обоих
металлов прибавляют избыток аммиака, при этом
медь образует растворимый аммиакат, а висмут оса¬
ждается в виде гидроксида. Железо можно отделить
от титана сероводородом в аммиачном растворе. Для
этого к раствору прибавляют винную кислоту, кото¬
рая в аммиачном растворе связывает (маскирует)
ионы титана в устойчивое растворимое комплексное175
соединение, а железо хотя и связывается винной кис¬
лотой, но сульфид железа очень плохо растворяется в
воде и поэтому при пропускании сероводорода вы¬
падает в осадок. Ионы меди и кадмия с цианидом
калия образуют комплексные соединения различной
устойчивости. При обработке такой смеси сероводоро¬
дом кадмий осаждается в виде сульфида, а медь
остается в растворе. Для разделения железа и„каль-
ция можно применить оксалат калия. Ионы С2О4" свя¬
зывают (маскируют) ионы железа настолько прочно,
что они не взаимодействуют даже с гексацианоферра-
том(П) калия. Кальций осаждается в виде СаСгО^
Хлорид серебра можно отделить от иодида, если об¬
работать смесь осадков водным раствором аммиака;
хлорид серебра, который является более растворимым
соединением по сравнению с иодидом, переходит в
раствор в виде аммиаката серебра.С помощью лигандов можно связать различные
ионы, которые мешают анализу, методом осаждения
или другими методами. По существу этот процесс со¬
ответствует разделению без фильтрования, промыва¬
ния и т. д., которые требуют много времени.Расчеты растворимости осадков при образовании
комплексов связаны с некоторыми затруднениями.
Обусловлено это тем, что для многих комплексных
соединений пока не установлены константы диссоциа¬
ции (константы нестойкости или устойчивости). Кроме
того, комплексные ионы, аналогично многоосновным
кислотам, образуются и диссоциируют ступенчато. Со¬
стояние равновесия между отдельными формами ком¬
плексных групп зависит от концентрации лиганда и
кислотности раствора.Рассмотрим несколько примеров расчета раствори¬
мости осадков при комплексообразовании.В общем виде взаимодействие между малораство¬
римым осадком М*Ау и лигандом R (заряд лиганда:
для простоты опущен) можно описать следующим
уравнением:М*А«, + pR = xtARp + уАт~, (9.26)откуда[MR Па'"-]"К = - - • (9.27)[RIP176
■Если умножить и разделить правую часть уравне¬
ния (9.27) на концентрацию М"+ в степени х, получим[MR,]* [Am-]y [мп+]хЛ'=- ~ i—.Тлх = прм А Р- (9.28)[RJP[M,1 + ] * Угде[Мч + 1 [R]pПоследующие расчеты основаны на использовании
уравнения (9.28).Пример 1. Рассчитать растворимость хлорида серебра в I М
растворе NH3 при условии, что после достижения равновесия
концентрация избытка аммиака fNH3] = 0,1 М.Растворимость хлорида серебра в растворе аммиака свя¬
зана с образованием аммиачного комплекса:AgCl + 2NH, ч=^ [Ag(NHs)s]Cl.Это соединение представляет собой сильный электролит и
диссоциирует на ионы fAg(NH3)2l+ и С! \ Таким образом, ком¬
плексным является катион fAg(NH3)2]+, который очень слабо
диссоциирует. Поэтому растворимость в этом случае связана с
взаимодействием ионов серебра с аммиаком по уравнению:Ag+ + 2NH3 ч=± [Ag(NH3hf.Константа устойчивости комплекса имеет вид:= 1,07 - 107.[Ag(NH,bl[Ag+] [NH3]2
Произведение растворимости хлорида серебраnpAgci = lAe+][cr]=i-io-,fl.Уравнение реакции между хлоридом серебра и аммиаком можно
записать следующим образом:AgCI+2NH3 +=± [Ag(NH3)2l+ + Cr.Константа равновесия этой реакции[Ag(NH^][cr]Л " INH;S]2Если это выражение умножить и поделить на концентрацию
ионов серебра fAg+] и подставить соответствующие значения, тог [Ag(NH,);|fcrl lAglА [NH3]2 [Ag+] AgC!P[Ag(NH3)2i;-177
Состояние равновесия реакции смещается вправо тем боль¬
ше, чем больше растворимость осадка и чем меньше константа
диссоциации комплекса, т. е. чем больше его устойчивость.При растворении осадка AgCl количество ионов С1~ и
[Ag(NH3)2]+ будет равно, т. е.[Cr] = [Ag(NH3)2r.Тогда можно записать:,, [MNH3)21[Ag(NH3)2-][NH3]2 AgCl lAg(NH3)2l +Отсюда[Ag(NH3)2+] = Vtf[NH3]2 = У 1,07 . |О-3 • 0,12 = 3,7 - 10~3M.Таким образом, растворимость AgCl, которая в воде равна
10~5 М, в 1 М растворе аммиака увеличивается до 3,7 ■ 10—3 М,
т. е. осадок практически полностью переходит в раствор.Пр имер 2. Рассчитать растворимость иодида серебра в усло¬
виях, приведенных для хлорида серебра.Произведение растворимости иодида серебраnPAgi = (Ag+i [Г] = 1,7 ■ 10~|в.Аналогично предыдущему находим:К =ПР К , ]+ = 1,7- 10~ 16 ■ 1,07 ■ 107 = 1,8 ■ 10~9.Agl [Ag(NH3)2]ОткудаfAg(NH3)2+] = ^/k[NH3]2 = -д/1,8 • 10“9 ■ 0,12 = 4,2 ■ 10“6М.Растворимость иодида серебра значительно возросла в при¬
сутствии 1 М раствора аммиака. Однако рассчитанная величина
показывает, что Agl, практически полностью остается в осадке.Приведенные расчеты показывают, что с помощью аммиака
как лиганда можно разделить ионы иодида и хлорида. Эти рас¬
четы обосновывают известный экспериментальный факт, что если
к смеси этих ионов и аммиака прибавить нитрат серебра, то
ноны хлора остаются в растворе, а ионы иодида переходят в
осадок в виде Agl.§ 5. РАСТВОРИМОСТЬ ОСАДКОВ В РЕЗУЛЬТАТЕ
ОБРАЗОВАНИЯ КОМПЛЕКСОВ С ИЗБЫТКОМ
ОСАДИТЕЛЯМногие труднорастворимые соединения растворяются
в избытке осадителя с образованием растворимых
комплексных соединений. Например, если к раство¬
рам солей цинка, кобальта, никеля и серебра приба¬
вить эквивалентное количество цианида калия, то об¬
разуются труднорастворимые цианиды этих металлов.178
Дальнейшее прибавление цианида калия приводит
к растворению осадков с образованием растворимых
цианидных комплексов. Аналогичные явления обра¬
зования комплексных соединении наблюдаются при
взаимодействии роданидов, иодидов и хлоридов многих
металлов с избытком осадителя.Известны многие случаи, когда прибавление из¬
бытка осадителя сначала приводит к снижению рас¬
творимости малорастворимого соединения, а затем
к ее увеличению. Поэтому в таких случаях необхо¬
димо иметь в виду, что обычный расчет растворимо¬
сти при избытке одноименных ионов приводит к оши¬
бочным результатам. Правило произведения раствори¬
мости основано на том, что с ростом концентрации
одноименных ионов увеличивается вероятность их
встречи с противоположно заряженными ионами, с ко¬
торыми образуется осадок. В связи с этим концентра¬
ция вещества и его растворимость уменьшается. При
комплексообразовании концентрация осаждаемых
ионов также уменьшается, но в результате этой реак¬
ции растворимость осадка растет. Вот почему правило
произведения растворимости нельзя применять меха¬
нически. Известны и такие осадки, растворимость ко¬
торых под действием избытка осадителя уменьшается.Таким образом, существует несколько типов осад¬
ков. Зависимость растворимости осадкон разных типов
от концентрации одноименных ионов характеризуется
кривыми (см. рис. 9.1), где на оси ординат отложены
значения растворимости осадка, а на оси абсцисс —
величины концентрации одноименных ионов (взятых
в избытке). Для соединений типа BaSC>4 избыток оса¬
дителя уменьшает растворимость осадков. Для соеди¬
нений типа Hgl2 избыток общих ионов (I" или Hg2+)
увеличивает растворимость осадка в результате обра¬
зования комплексных ионов:Hg[2 + 2l- (Hgui*-,осадок комплексв раствореHgt2 + Hg2+ 2Hgr.Кривые для осадков BaS04 и Hgl2 являются край¬
ними случаями. Большинство малорастворимых со¬
единений, которые применяют в анализе, по отноше¬
нию к избытку одноименных ионов занимают проме¬179
жуточное значение. Растворимость этих осадков при
добавлении одноименных ионов сначала уменьшается.
Если избыток осадителя окажется довольно большим,
начинает преобладать процесс комплексообразования
и растворимость осадка увеличивается. Примером та¬
кого типа соединений может быть хлорид серебра.
Растворимость хлорида серебра в растворах хлорида
натрия разной концентрации характеризуется такими
данными:КонцентрацияNaCl, М . . . О 0,0039 0,0092 0,036 0,35 0,9 2,9
Растворимость[AgCI] ■ 10—s, М 1,3 0,007 0,009 0,19 1,7 10 1000Приведенные данные показывают, что раствори¬
мость хлорида серебра сначала уменьшается, а при
увеличении концентрации хлорида натрия растет. Са¬
мая низкая растворимость AgCl наблюдается при кон¬
центрации хлорида натрия 0,0039 М. Затем раствори¬
мость растет и в 0,35 М растворе хлорида натрия
растворимость AgCl выше, чем в чистой воде, а с уве¬
личением концентрации хлорида натрия растет еще
больше.Из рассмотренных примеров можно сделать сле¬
дующие общие выводы: при осаждении не следует
применять очень большой избыток осадителя. Нельзя
рассчитывать растворимость, применяя механически
правило произведения растворимости. Применение
этого правила дает правильные результаты только
тогда, когда при реакции между ионами осадителя
и определяемого вещества образуется одно нераство¬
римое соединеиие.§ 6. РАСТВОРИМОСТЬ МАЛОРАСТВОРИМЫХ
СОЕДИНЕНИЙ В КИСЛОТАХМалорастворимые соли сильных кислот не раство¬
ряются в кислотах, однако соли слабых кислот, кото¬
рые плохо растворимы в поде, хорошо растворимы
в сильных кислотах.В общем виде уравнение реакции между малорас¬
творимой солью слабой одноосновной кислоты и иона¬
ми гидроксония имеет вид:МХАУ + уНзО* *МП + + уНА + yll2О. (9.29)180
Для этой реакции[Мп+Г(НА1!/К[НзОТЕсли умножить и разделить правую часть уравнения
на концентрацию анионов слабой кислоты в степени у,
тогда.. [м"+Пнл№"-Г[н3оГ[А"-Т ‘ (9-30)Так как [М"*]* [А"1 == ПР, а —г- является1 1 [н3о ] [А~]обратной величиной константы диссоциации слабойкислоты НА,, iM-trLHA!! , {9,з„К'нл [Н.О'1*Растворимость Р осадка в кислоте характеризуется
концентрацией свободных ионов М"^, которые пере¬
шли в раствор. Из уравнения (9.29) видно, что между
концентрациями ионои Мп+ и молекул НА существует
соотношениеУ [Mn + ] = JC [НА] (9.32)И[НА] = у/х [М'1 + !. (9.33)Подставляя в уравнение (9.30) вместо НА его зна¬
чение из (9.33), получаем[МП | ГУ (_у/х)У _ П{Ч-А,[f[j° 1 '4lAПоэтомуVПР«,А# [»Р*\У — . (9.35)К*нА (»/*■)“Из этого выражения видно, что растворимость
осадка в сильной мерс зависит от кислотности рас¬
твора, константы диссоциации кислоты и произведения
растворимости осадка. Кроме того, из формулы видно,
что концентрация водородных ионов по-разному влияет
на растворимость осадка. В тех случаях, когда вели¬
чина /Сил не намного меньше ПР осадка, осадки хо¬
рошо растворяются и кислотах (например, карбонаты,181
оксалаты и др.). Поэтому такие соединения осаждают
в нейтральных или слабощелочных растворах. Если
произведение растворимости намного меньше /(на, то
растворимость осадка очень мала даже в сильнокис¬
лых растворах (например, сульфиды катионов группы
сероводорода). Такие соединения осаждаются не толь¬
ко в щелочном и нейтральном, но и в очень кислых
растворах.Расчеты растворимости осадков в кислотах ослож¬
няются тем, что большинство слабых кислот являются
многоосновными (формула таких кислот НтА, а не
НА). В связи с этим следует учесть, что в зависимо¬
сти от концентрации Н30+ при растворении могут об¬
разовываться молекулы слабой кислоты или анионы
кислой соли (этой кислоты). Так, при малых кон¬
центрациях ионов гидроксония уравнение реакции
растворения оксалата бария имеет вид:Если кислотность раствора больше:ВаС204 + 2Н30+ =«=>■. Ва2* + Н2С204 + 2Н20.В большинстве практически важных случаев при
растворенни образуются молекулы слабой кислоты.
Если по уравнению реакции растворения образуется
двухосновная кислота, то общий вид уравнения (9.35)
будет следующий:Для двухосновной кислоты в знаменателе этой
формулы ставят величину, которая равна произведе¬
нию первой и второй ступени диссоциации этой кис-ВаС204 + Н3СГВа2* + нс2о; + н2о.(9.36)лоты, т. е. Кн2\К"ъь-
Первая ступеньн2а + Н20НэО+ + НА'; Ki[Н30+] [НА‘](9.37)[Н2А]Вторая ступень:НА- + н2о(9.38)182
Для приближенного расчета можно считать, что кис¬
лота диссоциируется по схеме:Н2А-Ь2Н20 ч=* 2НаО+ + А2'. (9.39)Тогда константа диссоциации будет иметь вид:= [Н»оТ[АЧ (9.40)[Н2А|Определив из уравнения (9.37) величину [Н2А], а
из уравнения (9.38) величину [А2-] и подставив их
в уравнение (9.40), имеем:[Н30 ] [А2 I к к (0 41)*НаА = [ЩА] ~К'К2- ( ’Величины констант диссоциации кислот и произведе¬
ния растворимости осадков для многих соединений
известны. * Пользуясь этими характеристиками,
можно с помощью формулы (9.35) рассчитать расгвэ-
римость осадков в зависимости от кислотности рас¬
твора.Рассмотрим несколько типичных примеров влия¬
ния кислотности на растворимость осадков.Пример 1. Рассчитать растворимость сульфида меди(II) н
0,1 М растворе кислоты.ПРС115 = 3 • 10“за; /\|Ь5 = /<-,К2 = I ■ 10 7 ■ I ■ 10 "13= 1 ■ 1<ГИ.Уравнение реакции растворения сульфида медн(П) в ионной
форме имеет вид:CuS + 2Н;,0+ :?=*: Cu2+ + II2S -f 2НоО.Для этого случая, в соответствии с уравнением (9.3G) и учиты¬
вая, что х = у — I, растворимость осадка составит:_ . /[H,o*]2npCuS /о.12-3- 10CuS V KHsS V 1 ■ ю-“--38— =1,7- 10 Af.Растворимость сульфида меди(II) в отсутствие кислоты равна:
pcus = Is2'] = = л/3-»0_:в = 1,7 ■ 1(Г ^* Константы .диссоциации кислот, произведения растворимо¬
сти и другие характеристики приведены в специальных справоч¬
никах и другой химической литературе.183
Таким образом, расчеты показывают, что в присутствии кис¬
лоты растворимостьо сульфида меди(П) значительно увеличи¬
вается.Пример 2. Рассчитать растворимость оксалата кальция в
0,1 М растворе хлороводородной кислоты.Произведение растворимости оксалата кальция ПРСаС2о4 =Растворимость осадка соответственно с этим уравнением рассчи¬
тывают, как и в предыдущем примере, по уравнению (9.30) с
учетом того, что х — у = 1:Эта величина показывает, что в условиях задачи кальций
в форме оксалата практически не осаждается.Пример 3. Рассчитать растворимость сульфида цинка в
0,01 М растворе хлороводородной кислоты, насыщенной серово¬
дородом. Произведение растворимости сульфида цинка nPzns =
= 7-10-27.Условия этой задачи отличаются от условий предыдущей
тем, что в растворе имеется избыток осадителя — сероводорода.
Если кислота увеличивает растворимость осадка, то избыток се¬
роводорода, наоборот, уменьшает ее.Концентрация сероводорода в насыщенном растворе состав¬
ляет 0,1 I и не равна равновесной, концентрации цинка. В этом
случае растворимость рассчитывается с помощью уравнения
(9.31). Запишем уравнение реакции растворения сульфида цинка
в кислоте:= 4 10 9.Константа диссоциации щавелевой кислоты*Н2С2О< = *1*2 = 6,5-10-2-6 1-10 5 = 4 10 б.Оксалат кальция растворяется в кислотах по реакцииСаС204 + 2Н30+ ц=£ Са2 + + Н2С204 + 2НгО.ZnS+2H3<y Zn2f -|- 2H20 + H2S.Из этого уравнения имеем:[Zn2t] [Н2S] riPZnS[НзО+]2 *H,S[H3o+]ИЛИ= 7 • 10 SM.^h2s[H2S]ю~‘ • 10-22184
§ 7. МЕХАНИЗМ ОБРАЗОВАНИЯ ТВЕРДОЙ ФАЗЫ -
ТЕОРИЯ КРИСТАЛЛИЗАЦИИОбразование твердой фазы — сложный процесс, со¬
стоящий из нескольких стадий.1. Дегидратация ионов, образующих осадок. Ионы
в растворе гидратированы, твердая фаза во многих
случаях не содержит воды, поэтому ионы перед обра¬
зованием кристалла должны утратить гидратную
воду.2. Образование первичных центров кристаллиза¬
ции. Два иона противоположного знака, объединяясь
в молекулу, не образуют еще кристаллической фор¬
мы. Образование первичного кристаллика проходит
через ряд промежуточных стадий. Так, при осаждении
сульфата бария из пересыщенных растворов происхо¬
дят такие процессы:Ва2+ + SO2" ч—Ba2 + S02' — образование ионной пары;Ba2 + S02~ + SO2” (или Ва2 + ) Ba(S04)2-[или Ba^SO,)2*];Ba(S04)22- + Ba2t (Ba2+S02)2;(Ba2tS02-)2 + SO2' (или Ba2+) *=k Ba2 (S04)J‘ [или Ba3(S04)2t];Ba2(so4)r + Ba2+ ^ (Ba2+soD3 и т- д-Это так называемый индукционный период.Таким образом, для образования первичного кри¬
сталлика в какой-либо точке раствора должны объ¬
единяться между собой по крайней мере по несколько
ионов противоположного знака. Только в этом случае
может образоваться первичный кристаллик (заро¬
дыш), например, кубической формы.3. Рост первичных центров кристаллизации за счет
осаждения на них все большего количества ионов, в
результате чего образуются кристаллы большего раз¬
мера, объединяющиеся между собой в более крупные
агрегаты, не способные, однако, еще выделиться из
раствора в виде осадка. Это — коллоидная стадия об¬
разования нерастворимого соединения.4. Образование в течение некоторого времени в
растворе настолько крупных кристаллов или их агре¬
гатов, что они не могут более удерживаться в рас¬
творе, и, наконец, происходит выделение твердой фа¬
зы (осадка).185
Рис. 9.2. Зависимость ско¬
ростей образования зароды¬
шей (/) и их роста (2) от
относительного пересыще¬
ния(Q-P)jPОбразовавшиеся
осадки могут отличать¬
ся по своим свойствам.
В одних случаях выде¬
ляются аморфныеосадки типа гидроксидов и др., других — кристал¬
лические типа сульфата бария и др. Характер осадка
зависит от соотношения скоростей двух процессов:
скорости образования зародышей — первичных цент¬
ров кристаллизации и,, и скорости роста размерЬв
зародышей v2. Значения у, и v2 определяются отно¬
сительным пересыщением, возникающим при добавле¬
нии раствора осадителя. Относительное пересыщение
определяется формулой (Q — Р)/Р. Здесь Q — кон¬
центрация растворенного вещества в пересыщенном
растворе в какой-либо момент времени, Р—раство¬
римость этого вещества при достижении состояния
равновесия между твердой фазой и раствором при
дайной температуре.Как показывает опыт, скорости названных процес¬
сов связаны с величиной относительного пересыщения
раствора следующими уравнениями:В большинстве случаев п « 4 и k2 > k\.Обычно при малых значениях относительного пе¬
ресыщения Vi > v\, при его возрастании скорости обо¬
их процессов становятся в некоторый момент времени
равными, и, наконец, скорость у, начинает преобла¬
дать, т. е. и2 становится меньше v\.Из рис. 9.2 видно, что при малых (Q — Р/Р) (ле¬
вая часть рисунка) преобладает скорость роста заро¬
дышей, а при больших значениях относительного пе¬
ресыщения преобладает (правая часть рисунка) ско¬
рость образования новых зародышей.Рассмотренные зависимости объясняют, почему в
одних случаях образуются аморфные, а в других —[|186
кристаллические осадки. При добавлении раствора
осадителя в первый момент времени образуется пере¬
сыщенный раствор. Если вещество очень мало рас¬
творимо (Р. мало), достигается большое относитель¬
ное пересыщение и скорость образования зародышей
значительно больше, чем скорость их роста (правая
часть рисунка). В этих условиях при добавлении по¬
следующих порций раствора осадителя образуется
большое количество новых центров кристаллизации.
Эти первичные зародыши слипаются в более крупные
агрегаты и выделяются из раствора в виде аморфного
осадка.Наоборот, если растворимость вещества не очень
мала, относительное пересыщение меньше, чем в пер¬
вом случае, и после прибавления раствора осадителя
образуется сравнительно мало зародышей, размеры
которых быстро увеличиваются. Из раствора выде¬
ляются отдельные кристаллы довольно значительных
размеров—образуется кристаллический осадок.Аморфные осадки состоят из множества слабо
связанных между собой очень мелких кристалликов.
Большинство таких осадков, как доказано рентгенов¬
ским методом, имеет кристаллическую структуру,
вследствие чего их называет также скрытокристалли¬
ческими. К аморфным осадкам относят, например,
очень мало растворимые гидроксиды и сульфиды ме¬
таллов, иногда и некоторые несколько более раство¬
римые вещества, например гидрофосфаты бария,
стронция, кальция и др.Характер осадка зависит и от условий осаждения.
Так, сульфат бария при осаждении из водных раство¬
ров осаждается в виде белых кристаллов. Если же его
осаждать из водно-спиртовой смеси с 30—60 % спир¬
та, который сильно понижает растворимость сульфата
бария, то образуется коллоидный раствор или аморф¬
ный осадок.Наиболее существенной характеристикой аморф¬
ных осадков является очень большая общая' поверх¬
ность. В связи с этим на поверхности аморфных осад¬
ков часто адсорбируются посторонние вещества. Кро¬
ме того, в связи с наличием большого количества
очень мелких агрегатов, которые слабо связаны ме¬
жду собой, при промывании аморфные осадки могут
образовывать коллоидные растворы.187
В виде кристаллических осадков осаждаются обыч¬
но не слишком мало растворимые соединения (по
сравнению, например, с гидроксидами металлов), как
сульфат бария, оксалат кальция, фосфат магния —
аммония и др. Время с момента смешения растворов
реагентов до появления зародышей называется индук¬
ционным периодом. Продолжительность этого пе¬
риода зависит от природы малорастворимого соеди¬
нения: он очень мал для хлорида серебра и аномально
велик для сульфата бария, фосфата магния — аммо¬
ния и других соединений.В процессе осаждения на поверхности осадка все¬
гда адсорбируются различные ионы. Адсорбируются
главным образом те ионы, которые находятся в из¬
бытке в растворе. Так, если осаждать ионы серебра
хлорид-ионами, то на поверхности осадка AgCl адсор¬
бируются главным образом ионы серебра, которые
имеются в избытке. Наоборот, при осаждении хло¬
рида прибавлением нитрата серебра на поверхности
адсорбируются главным образом ионы хлорида, так
как в этом случае они будут в избытке. Естественно,
что осадок будет адсорбировать и другие ионы, имею¬
щиеся в растворе, например ионы натрия или ни¬
трата, однако в первую очередь, как правило, адсор¬
бируются ионы, входящие в состав малорастворимого
соединения. Адсорбированные ионы кристаллической
решетки называют первично адсорбированными ио¬
нами. Вследствие адсорбции ионов поверхность осад¬
ка приобретает положительный или отрицательный
заряд в зависимости от того, какой ион, входящий в
состав осадка имеется в избытке. Под действием
этого заряда в зоны раствора, непосредственно при¬
мыкающие к частицам осадка, притягиваются проти¬
воположно заряженные ионы, которые называют про-
тивоионами. Эти противоионы удерживаются слабее
по сравнению с первично адсорбированными ионами.
Слой противоионов содержит также некоторое коли¬
чество других катионов и анионов. Адсорбированными
ионами на осадке будут преимущественно те ионы, ко¬
торые имеют наибольший заряд. Если же заряды ио¬
нов одинаковы, то в первую очередь адсорбируются те
ионы, которые образуют менее растворимые соедине¬
ния с первично адсорбированными ионами.188
ГЛАВА 10СООСАЖДЕНИЕ§ 1. ОБЩАЯ ХАРАКТЕРИСТИКАДавно было замечено, что при гравиметрическом оп¬
ределении элементов нередко возникают ошибки, вы¬
званные переходом в осадок присутствующих в рас¬
творе посторонних веществ. Это явление было на¬
звано соосаждением. Вначале предполагали, что со-
осаждение связано с несовершенством методик или
с некомпетентностью исполнителей анализа. Однако
подробное исследование выявило более глубокие при¬
чины. Оказалось, что соосаждение наблюдается при
самых точных методиках и при высокой квалифика¬
ции химиков-аналитиков, т. е. имеет общий характер.Приведем некоторые примеры.При осаждении сульфата бария осадок захваты¬
вает некоторое количество нитрата бария, хотя по¬
следнее соединение достаточно хорошо растворимо.Осадок оловянной кислоты, полученный из азотно¬
кислых растворов, всегда содержит небольшие коли¬
чества железа, меди и некоторых других элементов
несмотря на то, что нитраты этих элементов хорошо
растворимы.Железо отделяют от меди аммиаком. Медь обра¬
зует хорошо растворимый аммиачный комплекс, тем
не менее гидроксид железа содержит значительные
количества меди.Малорастворимая кремниевая кислота захваты¬
вает из раствора хлороводородной кислоты примеси
полуторных оксидов железа, алюминия, титана; между
тем эти элементы в кислых растворах не должны
были бы переходить в осадок.Подобных примеров можно привести очень цшого.Отметим следующие характерные особенности со-
осаждения.При соосаждении в осадок переходят вещества,
сами по себе в данных условиях хорошо растворимые.
К явлениям соосаждения относят также переход в
осадок сравнительно малорастворимых веществ, нахо¬
дящихся в растворе в таких ничтожных концентра-189
циях, при которых произведение концентраций соот¬
ветствующих соосаждающихся ионов значительно
меньше произведения растворимости. Иными словами,
в отсутствие основного осадка эти вещества должны
были бы оставаться в растворе.Например, ничтожные количества ионов стронция
в растворе при дсбавлении сульфат-ионов могут не
образовать твердой фазы вследствие того, что[ S г2+1 [so42'] < riPsrso,’ тем не менее они захваты¬
ваются осадком сульфата бария.Соосаждение следует отличать от совместного оса¬
ждения, когда оба вещества малорастворимы и при
данных условиях должны образовывать твердую фа¬
зу независимо от того, находятся ли они в растворе
вместе или порознь. Совместное осаждение сульфатов
бария и стронция не является соосаждением, если про¬
изведение концентрации обоих ионов на концентра¬
цию ионов сульфата превосходит соответствующие
значения ПР.Соосаждение происходит в процессе образования
осадка, но не после его образования. Так, осаждая
сульфат бария из раствора, содержащего ионы
MnOi, получают осадок розового или фиолетового
цвета из-за соосаждения этих ионов. Однако если
взять готовый препарат сульфата бария и взбалты¬
вать его с раствором перманганата калия — сооса¬
ждения не происходит, осадок остается неокрашен¬
ным.В преобладающем большинстве случаев осадок
нельзя освободить от соосажденных веществ промы¬
ванием водой или растворами каких-либо электро¬
литов.Явление соосаждения хорошо согласуется с меха¬
низмом образования твердой фазы, процесс кристал¬
лизации характеризуется определенным индукцион¬
ным периодом, т. е. происходит в течение конечного
отрезка времени, за которое успевает происходить за¬
хват посторонних примесей из раствора.Различают следующие виды соосаждения.1. Адсорбция посторонних веществ на поверхно¬
сти осадка — вид соосаждения, наиболее ярко выра¬
женный в случае аморфных осадков, отличающихся
сильно развитой общей поверхностью. Адсорбирован¬190
ные ионы удерживаются очень прочно и иногда не
удаляются при промывании осадка водой.2. Окклюзия — посторонние вещества находятся
внутри кристаллов осадка и более или менее равно¬
мерно распределяются по всему объему твердой фазы.
Окклюзия характерна в основном для кристалличе¬
ских осадков. Отличительная особенность окклюзии —
трудность извлечения поглощенных компонентов из
глубины кристаллов.3. Послеосаждение — наименее распространенный
вид соосаждения, отличается от предыдущих тем, что
переход примесей в осадок происходит не во время
формирования осадка, а после его выделения. По¬
этому иногда послеосаждение не рассматривают как
особый тип соосаждения. Так, при пропускании серо¬
водорода в кислый раствор, содержащий ионы меди
и цинка, сначала выделяется чистый осадок сульфида
меди; через некоторое время на поверхности этого
осадка адсорбируются молекулы сероводорода и кон¬
центрация последнего возрастает по сравнению с кон¬
центрацией в растворе. Это приводит к постепенному
осаждению цинка, несмотря на достаточно высокую
кислотность раствора.Какой-либо из рассмотренных видов соосаждения
не встречается в чистом виде в реальных условиях
анализа, обычно один или другой вид только преоб¬
ладает.Ниже обсуждены закономерности соосаждения на
аморфных (адсорбция) и кристаллических (окклю¬
зия) осадках. Различают микрокомпонент — веще¬
ство, находящееся в растворе в очень незначительной
концентрации и переходящее вследствие соосажде¬
ния в твердую фазу, и макрокомпонент — веще¬
ство, из которого состоит твердая фаза и количество
которого значительно превышает количество микро¬
компонента.Соосаждение с кристаллическими осадками, при
котором микрокомпонент распределяется по всему
объему твердой фазы, принимая участие в построении
кристаллической решетки микрокомпонента, назы¬
вают сокристаллизацией. Соосаждение микрокомпо¬
нента на поверхности твердой фазы макрокомпонента,
включая также внутреннюю поверхность, называют
адсорбцией.191
§ 2. ЗАКОНОМЕРНОСТИ АДСОРБЦИИ
НА АМОРФНЫХ ОСАДКАХАдсорбция вещества на поверхности осадка опреде¬
ляется многими факторами.1. Количество адсорбированного вещества зависит
от общей поверхности осадка: чем эта поверхность
больше, тем больше адсорбция.2. Количество адсорбированного вещества зависит
от его концентрации в растворе, возрастая не прямо
пропорционально этой концентрации. Зависимость
описывается изотермой адсорбции. Сначала увеличе¬
ние концентрации микрокомпонента в растворе приво¬
дит к увеличению количества адсорбированного веще¬
ства, после чего наступает насыщение: кривая пере¬
ходит в прямую, идущую параллельно оси абсцисс.3. Катионы адсорбируются поверхностью осадка
тем больше, чем меньше растворимость соединения,
образующегося из этих катионов и анионов осадка.
Справедлива и обратная зависимость: анионы адсор¬
бируются тем больше, чем меньше растворимость со¬
единения, образующегося из этих анионов и катионов
осадка.Эта зависимость называется правилом Панета —
Фаянса (она была установлена методом радиоактив¬
ных индикаторов). Данные, характеризующие эту за¬
висимость, приведены в табл. 10.1.4. Адсорбция ионов элементов-микрокомпонентов
подчиняется закону Гана. Ионы радиоактивного эле¬
мента адсорбируются на полярных кристаллах, если
поверхность кристалла имеет заряд, противополож-Таблица 10.1. Степень адсорбции радиоактивного
изотопа свинца в зависимости от растворимости
соответствующих солей свинцаАдсорбентРастворимость солей, образованных
катионами свинца и анионом
осадка, МСтепень
адсорбции, %AgBr2- КГ31,8Agl1 ■ Ю-32,1AgI035- КГ543,3Ag2C2048- I0-581,1Ag2CrO<2 • 10-797,9192
ный знаку заряда ионов. Закон Гана справедлив так¬
же для нерадиоактивных элементов, так как химиче¬
ские свойства радиоактивных и нерадиоактивных эле¬
ментов идентичны.Так, при осаждении ионов кальция в присутствии
ионов изотопа свинца ThB2+ избытком раствора сер¬
ной кислоты, когда поверхность осадка CaS04 заря¬
жена отрицательно, адсорбция ионов ThB2+ увеличи¬
вается по мере возрастания избытка прибавленной
серной кислоты. Наоборот, осаждение сульфат-ионов
избытком раствора хлорида кальция (при образова¬
нии осадка с положительно заряженной поверх¬
ностью) приводит к уменьшению адсорбции ионов
ThB2+ с увеличением избытка раствора хлорида каль¬
ция. Такая же зависимость наблюдается при адсорб¬
ции ионов ThB2+ на осадке Agl. Закон Гана характе¬
ризуется данными табл. 10.2.5. Высокозарядные ионы адсорбируются сильнее,
чем ионы с меньшим зарядом. Примером является
адсорбция катионов некоторых элементов на осадке
Agl. Эта адсорбция зависит также от концентрации
водородных ионов:[НзО + 1, м 0,0050,050,10Степень адсорбции, %
Ra2 + 7,9Незначительная0,0Ас3+ 75,525,17,0Th'+ 100,0100,050,56. Соосаждение зависит от свойств макрокомпо¬
нента, образующего осадок. Исследование соосажде-Таблица 10.2. Степень адсорбции ионов ThB2+ на осадках
CaSO, и AglОсадокОсадительИзбыток осадителя, %Степе нь
адсорбции, %CaSO*h2so4588,0»1092,2»10-кратный98,4СаС1г105,2»7-кратный1,7AglKI573,11076,9AgNOa104,5»1002,07 Аналитическая химия 193
ния микрокомпонентов на гидроксидах тяжелых ме¬
таллов показало, что степень соосаждения обуслов¬
лена кислотно-основными свойствами гидроксидов, ко¬
торые зависят от ионного потенциала катионов, обра¬
зующих эти гидроксиды. Так, соосаждение катионов
цезия или таллия возрастает в ряду гидроксидов та¬
ких металлов:Катион металла, образу¬
ющий гидроксид .... Mg2+<La3+<Th<+<Zr4+<Fe3 + <Ti4+
Ионный потенциал, Zjr 2,56 3,66 4,55 6,63 8,45 8,77На гидроксиде магния соосаждение цезия состав¬
ляет 5 % от взятого количества цезия, а на гидро¬
ксиде титана — 85 %.Для анионов наблюдается обратная зависимость.2 „Так, соосаждение ионов СгС>4 возрастает в ряду ги¬
дроксидов таких металлов:Катион металла, обра¬
зующий гидроксид . . . Ti4+ <Fe3+ <Zr4+ <Th4+ <Y3+
Ионный потенциал, Z/г 8,77 8,45 6,63 4,55 4,69Приведенные экспериментальные данные объяс¬
няют, почему по отношению к катионам микроком¬
понента адсорбционная способность больше у гидро¬
ксидов, образованных катионами с более высоким
значением ионного потенциала. Это обусловлено ка¬
чественным изменением внутренней структуры гидро¬
ксида, ослаблением связи водорода с кислородом, что
облегчает замещение иона водорода сорбирующимся
элементом.§ 3. ЗАКОНОМЕРНОСТИ СООСАЖДЕНИЯ
НА КРИСТАЛЛИЧЕСКИХ ОСАДКАХСоосаждающийся микрокомпонент распределен по
всему объему кристаллического осадка и входит в
кристаллическую решетку макрокомпонента. Такой
характер соосаждения часто обусловливается явле¬
нием изоморфизма.Изоморфизм был открыт Э. Митчерлихом в 1819 г.
Явление изоморфизма заключается в том, что анало¬
гичные по химическому составу вещества сходных по
химическим свойствам элементов способны кристал¬
лизоваться в одинаковых или близких формах. На¬194
пример, изоморфными являются кристаллы: ZnC03,
MgCOa, FeC03, MnC03, CdC03, CaC03. Ромбоэдриче¬
ские кристаллы их имеют угол а, соответственно рав¬
ный 103° 26', 103° 2 Г, 103° 04', 102° 50', 102° 30', 101 °55'.Изоморфные вещества способны поэтому к совме¬
стной кристаллизации и образуют фазы переменного
состава — твердые растворы (смешанные кристаллы).
Изоморфизм характерен для веществ с одинаковой
кристаллической структурой, имеющих одинаковое
количество атомов, аналогично связанных в мо¬
лекуле. Аналогия кристаллической структуры зависит
от числа частиц в соединении, близости их размеров
и поляризационных свойств, а также однотипности
химической связи. Так, NaCl и CuCl не изоморфны,
потому что при одинаковых размерах обоих катионов
их поляризационные свойства неодинаковы. По Гольд¬
шмидту, изоморфизм возможен в таких случаях: если
у обоих соединений сумма зарядов атомов, входящих
в их состав, и распределение зарядов одинаковы, на-
+2 +6 -2 +2 +8 -2
пример SrS04 и RaS04; если у обоих соединений сум¬
ма зарядов атомов, входящих в их состав, одинакова,+2 +6 -2но их распределение неодинаково, например SrS04 и+1 +7 -2КСЮ4; если сумма зарядов атомов, входящих в со¬
став обоих соединений, разная, но число атомов оди-+4-2 +2-1наковое, например ТЮ2 и MgF2.Изоморфное соосаждение . подчиняется закону
Хлопина: если два вещества являются изоморфными
и концентрация одного из них очень мала, распреде¬
ление микрокомпонента между кристаллической фа¬
зой и раствором при постоянной температуре харак¬
теризуется постоянной величиной и не зависит от ко¬
личественного соотношения фаз:где Хо и X — соответственно количества микрокомпонепта в си¬
стеме и кристаллах; Уд и К — количество макрокомпонента в си¬
стеме и в кристалле; D—коэффициент кристаллизации.Из уравнения (10.1) следует, что7*195
Рассмотрим случай, когда количество макроком¬
понента в системе остается постоянным, а изменяется
только количество микрокомпонента. Тогда правая
часть уравнения (10.2)—величина постоянная. Обо¬
значив ее буквой k, запишем уравнение (10.2) сле¬
дующим образом:X = kX0~kX. (10.3)Откуда<10-4)Следовательно, при изоморфном соосаждении количе¬
ство соосажденного вещества прямо пропорционально
общему количеству микрокомпонента Х0 в системе:
чем больше это количество, тем больше микроком¬
понента X переходит в твердую фазу макрокомпо¬
нента.Наиболее общая причина соосаждения — адсорб¬
ция посторонних примесей на поверхности растущих
кристаллов, т. е. внутренняя адсорбция. Вначале, в
первый момент осаждения, образуется некоторое ко¬
личество первичных центров кристаллизации. При
дальнейшем прибавлении раствора реагента продукты
реакции продолжают осаждаться на поверхности ра¬
нее образовавшихся кристаллов. Таким образом, в
процессе осаждения поверхность осадка непрерывно
обновляется, покрываясь новыми слоями вещества.
Каждый вновь образующийся слой поверхности ад¬
сорбирует примеси из раствора. При образовании
следующего слоя кристалла часть адсорбированных
примесей может быть вытеснена ионами, входящими
в основную решетку кристаллов. Однако в зависи¬
мости от порядка, скорости сливания растворов и
других условий часть адсорбированных примесей
остается внутри кристалла. Далее адсорбированные
примеси покрываются новыми слоями осаждающегося
вещества и таким образом остаются внутри кристал¬
ла и не могут быть удалены при промывании осадка.Внутренняя адсорбция подчиняется описанным в
§ 2 закономерностям. Для нее характерно явление
насыщения, описывающееся изотермой адсорбции.
Этим соосаждение в результате внутренней адсорб¬
ции отличается от изоморфного соосаждения, подчи¬
няющегося закону Хлопина.1%
§ 4. МЕТОДЫ УСТРАНЕНИЯ СООСАЖДЕНИЯВ гравиметрическом анализе соосаждение является
нежелательным. Его стараются уменьшить или уст¬
ранить. Методы устранения соосаждения зависят от
типа соосаждения. Так, в случае осаждения кристал¬
лических осадков основным видом загрязнения осад¬
ков является окклюзия. Захваченное вещество нахо¬
дится внутри кристаллов основного осадка. Эти
примеси практически нельзя отмыть. Их можно в зна¬
чительной мере уменьшить в процессе осаждения.
С этой целью рекомендуется вести осаждение в усло¬
виях, при которых растворимость труднорастворимой
соли достигает максимума. Кроме того, осадитель
прибавляют медленно, интенсивно перемешивая рас¬
твор. В этих условиях происходит медленный рост не¬
большого количества больших кристаллов. Еще лучше
проводить осаждение из гомогенного раствора, при¬
меняя возникающие реактивы (см. ниже). Поэтому
кристаллические осадки осаждают из горячих кис¬
лых растворов (условия максимального растворения
труднорастворимого соединения).Количество примесей, захваченных кристалличе¬
ским осадком, в значительной мере можно уменьшить
путем переосаждения осадка. Например, при осажде¬
нии ионов кальция оксалатом в присутствии ионов
магния осадок оксалата кальция в значительной мере
загрязнен магнием. Для уменьшения примеси магния
осадок растворяют в хлороводородной кислоте и за¬
тем раствор нагревают и постепенно нейтрализуют
гидроксидом аммония, при этом образуется осадок
оксалата кальция, содержащий значительно меньше
оксалата магния.Большое влияние на степень соосаждения оказы¬
вают также порядок сливания растворов и скорость
осаждения.Если кристаллический осадок образуется в среде,
содержащей избыток анионов осадка, то эти анноны
особенно сильно адсорбируются поверхностью осадка
и заряжают ее отрицательно. В этих условиях по¬
сторонние анионы не адсорбируются осадком и их со¬
осаждение резко уменьшается. В то же время при¬
сутствующие в растворе посторонние катионы будут197
сильнее соосаждаться, притягиваясь отрицательно за¬
ряженной поверхностью.Если поступать наоборот, т. е. получать осадок при
медленном приливании раствора электролита, содер¬
жащего анионы осадка, к раствору, содержащему ка¬
тионы осадка, последний формируется в среде, содер¬
жащей избыток катионов осадка, которые адсорби¬
руются и сообщают поверхности положительный за¬
ряд. При таком способе осаждения уменьшается со¬
осаждение посторонних катионов, но зато возрастает
возможность адсорбции анионов.Имеет значение и скорость осаждения. Так, при оса¬
ждении сульфат-ионов хлоридом бария соосаждаются
ионы хлора. Если раствор хлорида бария прибавить
сразу в большом избытке, то значительная часть осад¬
ка сульфата бария будет кристаллизоваться в среде,
содержащей избыток ионов бария. Такие условия спо¬
собствуют более сильному поглощению анионов. На¬
оборот, при медленном приливании раствора хлорида
бария большая часть кристаллов осадка растет в
среде, содержащей избыток сульфат-ионов, что сильно
уменьшает возможность соосаждения ионов хлора.В случае скрытокристаллических (аморфных)
осадков захваченное вещество находится главным об¬
разом на поверхности осадка, т. е. соосаждение обус¬
ловлено поверхностной адсорбцией. Такие примеси
также часто трудно отмыть водой. В этом случае хо¬
рошие результаты достигаются путем ионного обме¬
на между ионами, захваченными осадком, и ионами,
находящимися в растворе или в промывном растворе.
Естественно, что прибавлять для этого необходимо
такие электролиты, которые не мешают дальнейшему
гравиметрическому определению. Например, в про¬
мывные воды можно прибавить хлорид аммония. За¬
хваченные аммонийные соли при прокаливании осад¬
ка легко разлагаются и не мешают дальнейшему оп¬
ределению. Присутствие аммонийных солей в промыв¬
ной жидкости препятствует образованию коллоидных
растворов и проникновению осадка в фильтрат.В случае аморфных осадков также можно в зна¬
чительной мере уменьшить содержание примесей в
осадке в процессе осаждения. Для этого осаждение
проводят из концентрированных горячих растворов,
а затем быстро разбавляют горячей водой. При этом198
образовавшийся осадок имеет не очень большую по¬
верхность, а разбавление горячей водой приводит к
отмывке захваченных поверхностью осадка примесей.§ 5. ИСПОЛЬЗОВАНИЕ СООСАЖДЕНИЯ
В АНАЛИТИЧЕСКОЙ ХИМИИСоосаждение широко используют для концентриро¬
вания микрокомпонентов, находящихся в растворе в
очень малых количествах, которые нельзя определить
непосредственно в растворе каким-либо методом. Та¬
кое концентрирование микрокомпонентов дает воз¬
можность отделить микрокомпонент от основного ком¬
понента. При соосаждении применяют вещества, ко¬
торые захватывают из раствора микропримеси. Такие
вещества называют коллекторами. Они не должны
мешать дальнейшему определению микропримеси.
Коллекторы могут быть органические и неорганиче¬
ские. Преимущество органических и только некото¬
рых неорганических коллекторов состоит в том, что
их легко удалить сжиганием или прокаливанием.Приведем некоторые примеры.При радиоактивном распаде основного изотопа
урана 238U образуется изотоп тория 2goUX, причем на
1 г исходного урана приходится всего 10~" г 2goUX.
Разделить эти элементы можно следующим образом.
К раствору соли U02(N03)2 прибавляют раствор
FeCI3 и NH4OH. Образуются нерастворимые осадки
диураната аммония и гидроксида железа:2tJ02(N03)2 + 6NH3 + ЗН2<Э з=±: (NH4)2U207 + 4NH„N03;FeCI3 + 3NH„OH qpzfc Fe(OH)3 + 3NH„CI.Изотоп тория 2goUX превращается в Th(OH)4 и
адсорбируется осадком гидроксида железа. Затем до¬
бавляют избыток раствора (NH4)2C03, при этом
(NH4)2U207 растворяется вследствие образования хо¬
рошо растворимого двойного карбоната уранила
U02C03-2(NH4)2C03> а гидроксид тория остается в
осадке. Если бы гидроксида тория было много, он
также растворился бы в растворе карбоната аммония.
Однако его микроколичества настолько сильно адсор¬
бированы гидроксидом железа, что не переходят в
раствор при обработке карбонатом аммония.199
Для отделения нептуния от продуктов распада ис¬
пользуют натрийуранилацетатный метод. Нептуний в
степени окисления 6 осаждается с натрийуранилаце-
татом. Метод применяют для выделения индикатор¬
ных количеств солей нептуния из растворов солей об¬
лученного урана. Раствор соли облученного урана
при нагревании обрабатывают восстановителем (неп¬
туний восстанавливается до степени окисления 4), а
затем осаждают натрийураннлацетат. В осадок пере¬
ходит приблизительно 90—95 % солей урана. Из
фильтрата, содержащего остаток солей урана, соли
нептуния и других продуктов распада, осаждают диу-
ранат аммония, который растворяют в азотной кис¬
лоте, после чего окисляют нептуний броматом калия.
При повторном осаждении натрийуранилацетата
NaU02(CH3C00)3 соли нептуния соосаждаются, от¬
деляясь таким образом от продуктов распада.Для отделения плутония от урана массу урана с
примесями плутония растворяют в азотной кислоте,
причем образуются (J02 (NO-»)3 и PuOo(N03)2. Далее
плутоний восстанавливают до четырехвалентного, об¬
рабатывая SO2, и осаждают в виде PuF4, добавляя в
качестве коллектора смесь солей редкоземельных эле¬
ментов (РЗЭ). Чтобы отделить плутоний от фтори¬
дов РЗЭ, его снова окисляют до шестивалентного.Добывание радия основано на процессе соосажде-
ния. Урановые руды обрабатывают серной кислотой
и добавляют коллектор—соли бария. Радий сооса-
ждается вместе с сульфатом бария и таким способом
отделяется от урана. Сульфат бария переводят в кар¬
бонат, который растворяют в кислоте, после чего
разделяют радий и барий методом дробной кристал¬
лизации или каким-либо другим способом.Применяя в качестве коллектора хлорид индулина,
можно извлечь из 6 М раствора НС1 микроколичества
галлия, в то время как алюминий в концентрации в
109 раз большей, чем галлий, остается в растворе.Большое значение для концентрирования микро¬
компонентов имеют органические коллекторы — сооса-
дители. Органические соосадители—это малораство¬
римые в воде соединения, способные практически
полностью извлекать из очень разбавленных раство¬
ров микрокомпоненты. Основой такого извлечения яв¬
ляется соосаждение. Распределение микрокомпонента200
между осадком и раствором зависит в первую очередь
от степени гидрофобности соединения, в виде которого
соосаждается микрокомпонент.Коллоидно-химические соосадители состоят из двух
частей — основного красителя типа метилового фио¬
летового, кристаллического фиолетового, родамина и
других и коллоидного ингредиента, чаще всего тан-
нина. Эти два соединения, реагируя между собой, об¬
разуют малорастворимые соединения. Таннины — не
индивидуальные соединения. Это смесь веществ близ¬
кого строения, которые можно рассматривать как
эфиры глюкозы и галловой, дигалловой или тригалло-
вой кислот.ГЛАВА 11ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗРеакции осаждения применяют в следующих методах
анализа: гравиметрическом (весовом), осадительного
титрования с индикаторами, нефелометрическом,
турбидиметрическом, электрогравиметрическом, по¬
тенциометрического титрования, амперометрического
титрования, кондуктометрического титрования.В результате реакции, на которой основан метод,
должен образовываться практически нерастворимый
осадок.§ 1. ОСНОВЫ МЕТОДАСущность метода состоит в том, что к раствору опре¬
деляемого вещества прибавляют раствор осадителя и
выделяют определяемый компонент в виде труднора¬
створимого (практически нерастворимого) соедине¬
ния. Это означает, что после осаждения в исследуе¬
мом объеме раствора должно оставаться только та¬
кое количество данного соединения, которое нельзя
уже обнаружить при взвешивании на аналитических
весах. Чувствительность обычных аналитических ве¬
сов составляет ±0,0001 г. Поэтому практически нера¬
створимым соединением необходимо называть такое,
растворимость которого не превышает ±0,0001 г
в суммарном объеме раствора (1 л), остающегося
после осаждения, и в промывных водах. Допуская,
что средняя мольная масса осадка равна 100,201
растворимость практически нерастворимого соедине¬
ния должна составлять не более Р = 0,0001/100 =
= 10~б М или произведение растворимости бинарного
малорастворимого соединения равноПРМК =[Mn+][R'1 ] = [м”+]2 = (ю-6)2 = 10~12.Естественно, что в случае тринарного, тетрарного
и более сложного соединения произведение раствори¬
мости будет другим, но всегда растворимость мало¬
растворимого соединения не должна превышать 10~6 г
в 1 л раствора.После переведения определяемого вещества в оса¬
док его отделяют фильтрованием и отмывают от по¬
сторонних веществ, которые могут быть захвачены
поверхностью осадка. Затем осадок высушивают, про¬
каливают и после охлаждения взвешивают. Многие
осадки можно взвешивать после высушивания при
определенной температуре. Например, никель можно
осадить в виде диметилглиоксимата никеля и после
отделения от раствора его можно высушить и взвесить
в виде диметилглиоксимата. Можно также прокалить
осадок и взвесить образовавшийся оксид никеля. Взве¬
шиваемое соединение в гравиметрическом анализе на¬
зывают гравиметрической (весовой) формой.Таким образом, общая схема гравиметрического
анализа представляется в следующем виде:► MR
весовая
формаПо химическому составу весовая форма не всегда
совпадает с осадком. Так, при определении никеля
возможны два варианта:Ni2+ + 2H2Dm + 20Ыопреде- реактив
ляемый
ионвысушншшие ► Ni(HDm)2=P=fc Ni(HDm)2 - промлииание + 2НгОосадок > N12O3весоваяформам п+ + Rrt MRопре- осади- осадокделяе- тельмый (реак-ион трв)202
При определении ионов сульфата состав весовой
формы и осадка совпадают:SOj- + Ba2+ :<=fc BaSO, —> BaSO,опреде- реак- осадок весоваяляемый тив форма
ионОднако очень часто составы весовой формы и осадка
не совпадают. Например, при осаждении всех гидр¬
оксидов, магнийаммонийфосфата и многих других:Fe3+ + ЗОН Fe(OH)3 —> РегОзопреде- осади- осадок весоваяляемый тель формаионMg2+ + Na2HP04 + NH4OH MgNH4P04 + Н20 + 2Na+
опреде- осади- осадок Iпрокаливаниеляемый тель | ^ м„ р Qион евесоваяформаГравиметрический анализ является одним из наи¬
более старых и наиболее точных методов анализа.
Предел обнаружения гравиметрического метода огра¬
ничивается растворимостью осадка и чувствитель¬
ностью аналитических весов. Диапазон содержания
определяемых веществ колеблется в пределах от со¬
тых долей до десятков процентов.Основным недостатком гравиметрического метода
является длительность анализа. Однако в некоторых
случаях этот метод является незаменимым.Погрешности гравиметрического анализа зависят
главным образом от полноты осаждения и от чистоты
получаемого осадка. Кроме того, как и во всяком
другом методе анализа, могут быть случайные по¬
грешности.Погрешности могут быть вызваны комплексообра-
зованием, имеющем большое значение при растворе¬
нии осадков. Если образуется комплексное соедине¬
ние, то растворимость осадков значительно повыша¬
ется. Например, растворимость хлорида серебра203
в воде характеризуется произведением раствори¬
мости:nPAgCI = [Ag+] [Cl ] = 1.8 10-'°.Отсюда растворимость хлорида серебра в воде со¬
ставляетЕсли же на хлорид серебра подействовать раствором
аммиака, то растворимость резко повышается в ре¬
зультате реакции комплексообразования:Реакции комплексообразования могут изменять
потенциал системы. Так, аммиакат кобальта (II) на
воздухе быстро переходит в аммиакат кобальта (III),
так как последний значительно устойчивей.§ 2. НЕКОТОРЫЕ ОРГАНИЧЕСКИЕ ОСАДИТЕЛИпредставляет собой оранжево-бурые кристаллы или
порошок, мало растворимый в воде, легко раствори¬
мый в спирте (при нагревании), эфире, бензоле, ле¬
дяной уксусной кислоте и растворах щелочей. Темпе¬
ратура плавления а-нитрозо-|3-нафтола 109,0—109,5 °С.Реактив используют для открытия и гравиметри¬
ческого определения кобальта. Он образует с ионами
кобальта, железа (III), меди, циркония и другими
ионами малорастворимые соединения. При взаимо¬
действии с солями кобальта(II) образуется интенсив¬[Ag+] = У 1,78- 10 10 =1,3 10 5М.AgCl + 2NH3 [Ag(NH3)2]Cl.N=0а-Нитрозо-р-нафтол (или 1 -нитрозо-2-нафтол)204
но-красный осадок трехвалентного кобальта:N=04СоС12 + 12+ О-'гN=0N=0+ 8НС1 +2 Н20Кобальт в степени окисления 2 окисляется до ко¬
бальта в степени окисления 3 за счет кислорода воз¬
духа, хотя возможен процесс окисления и за счет
нитрозогруппы реактива.При гравиметрическом определении кобальта оса¬
док отфильтровывают, прокаливают до Со304 и взве¬
шивают.а-Диметилглиоксим (реактив Чугаева) Н3С—C=N—ОНпредставляет собой белый кристаллический порошок,
мало растворимый в воде, легко растворимый в спир¬
те, эфире и растворах щелочей.Реактив образует малорастворимые соединения с
ионами никеля, палладия и растворимый комплекс с
ионами железа (II). Строение диметилглиоксимата
никеля следующее:н3с—c=n—он/о—н • • • о~\./,сщN=C/ \/ \Т>*”Н—оЭто лучший реактив для обнаружения, отделения и
количественного определения никеля.
он8-Гидроксихинолин (оксин)представляет собой желтоватые кристаллы или кри¬
сталлический порошок с характерным запахом. Он
практически нерастворим в воде и эфире, хорошо ра¬
створим в спирте, ацетоне, хлороформе, бензоле, ук¬
сусной кислоте и в разбавленных растворах мине¬
ральных кислот и щелочей. 8-Гидроксихинолин имеет
амфотерные свойства:Н0х+Н30+ Н20х+ + Н20; /(' = 8-1(Г6,H0x + H20 *=fc НэО* + Ох"; К" = 1,4 • Ю~10.Температура плавления 74—76°С, температура
кипения 267°С. Для аналитических целей готовят
раствор реактива в уксусной кислоте. 8-Гидроксихи-
нолин взаимодействует с катионами большинства ме¬
таллов с образованием малорастворимых соединенийИзменяя pH раствора, 8-гидроксихинолином
можно осадить ионы одних металлов в присутствии
других. С применением этого реактива металлы опре¬
деляют гравиметрическим, титриметрическим и коло¬
риметрическим методами. При гравиметрическом
определении оксихинолинаты металлов высушивают
или прокаливают до оксидов и взвешивают.аммонийная соль N-нитрозофенилгидроксиламина
представляет собой белые или буроватые лепестки.
Реактив хорошо растворим в воде; при хранении раз¬
лагается и темнеет. Для аналитических целей исполь¬
зуют 6 %-ный водный раствор купферона. Баудиш
предложил эту соль в качестве специфического реак¬
тива на медь и железо, откуда и пошло название куп-Купферонч206
ферон. Однако позже было установлено, что купфе-
рон взаимодействует с большим количеством катио¬
нов многих других металлов с образованием малора¬
створимых соединений. Ионы металла замещают
ионы аммония и, кроме того, координационно связы¬
ваются с азотом или кислородом нитрозогруппы:N=0 или \ —N—N=0\=/ I . IО Мп+/я - О—fAn+ /пОсаждение купфероном в сильнокислом растворе
дает возможность отделить железо, ванадий, цирко¬
ний, титан, олово, ниобий, тантал от алюминия, бора,
бериллия, фосфора, марганца, никеля и урана. Куп-
феронаты осаждают при охлаждении, чтобы предуп¬
редить разложение купферона. Промывают купферо-
наты холодным раствором серной или хлороводород¬
ной кислоты с небольшим количеством купферона.
Гравиметрической формой являются оксиды ме¬
таллов.Разработаны также методы амперометрического
титрования с применением купферона.Разделение металлов в виде купферонатов прово¬
дят путем их осаждения при разной кислотности
раствора. Купферонаты металлов хорошо экстраги¬
руются неводными растворителями. Разделение ме¬
таллов методами экстракции имеет ряд преимуществ
перед методом осаждения. Поэтому для разделения
металлов лучше применять не осаждение, а экстрак¬
цию в виде купферонатов.Главным недостатком применения купферона
в анализе является то, что он легко разлагается (ос-
моляется). Поэтому купферонаты металлов не могут
быть весовой формой и их прокаливают до оксидов.
Разложение купферона ускоряется под действием
света, поэтому его хранят в склянках из темного
стекла.§ 3. ТРЕБОВАНИЯ К ОСАДКАМ
И К ГРАВИМЕТРИЧЕСКОЙ ФОРМЕВ основе гравиметрического анализа лежат реакции
осаждения. Однако для гравиметрического анализа
пригодны далеко не все реакции осаждения, хотя207
многие из них можно с успехом применять в качест¬
венном анализе. Так, ионы свинца легко обнаружить
посредством осаждения раствором иодида калия. Од¬
нако растворимость иодида свинца в холодной воде
слишком велика для того, чтобы можно было количе¬
ственно выделить свинец из раствора. Реакция осаж¬
дения синего гексацианоферрата(II) железа является
специфической реакцией определения железа. Это
очень чувствительная реакция, так как растворимость
осадка ничтожно мала. Однако его нельзя использо¬
вать в гравиметрическом анализе из-за непостоянства
состава: железо в форме гексацианоферрата (II)
осаждается вместе с ионами калия или натрия. Кро¬
ме того, осадок склонен к образованию коллоидного
раствора и отделение его часто представляет опреде¬
ленные трудности.Эти примеры показывают, что применяемые в гра¬
виметрическом анализе осадки должны соответство¬
вать определенным требованиям. Главные из них сле¬
дующие.1. Осадок должен быть практически нерастворим.
Это означает, что в растворе после осаждения и про¬
мывания осадка должно оставаться меньше вещест¬
ва, чем можно взвесить на аналитических весах.
С целью уменьшения растворимости осадка к рас¬
твору прибавляют избыток осадителя. Обычно счи¬
тается, что полуторный избыток против стехиометри-
чески необходимого достаточен для полного осажде¬
ния определяемого вещества. Иногда, если это необ¬
ходимо, прибавляют значительно большее количество
осадителя. Однако чрезмерное увеличение концентра¬
ции нецелесообразно, так как приводит к возраста¬
нию ионной силы раствора и, как следствие, к росту
растворимости осадка. Нередко увеличение концент¬
рации осадителя приводит к образованию раствори¬
мых комплексов. Растворимость солей слабых кислот
регулируют также изменением pH раствора.2. Состав гравиметрической формы (осадок после
высушивания и прокаливания) должен соответство¬
вать определенной формуле. Например, осадок гидр¬
оксида железа не соответствует формуле Fe(OH)3,
более правильно было бы писать Fe203-rcH20. Коли¬
чество воды в этом соединении непостоянно и зави¬
сит от многих факторов. После прокаливания гидр¬208
оксид железа переходит в соединение определенного
состава Fe203. Кроме воды, которую легко удалить
высушиванием или прокаливанием, скрытокристалли¬
ческие осадки во время их образования легко захва¬
тывают посторонние вещества из раствора. Чистые
осадки можно получить если придерживаться опре¬
деленных условий осаждения (температура, концент¬
рация или порядок сливания растворов).Для отделения осадка от раствора фильтрованием
размер зерна осадка должен превышать размеры пор
фильтра. Для получения крупных кристаллов осаж¬
дение проводят с определенной скоростью. Напри¬
мер, сульфат бария при быстром осаждении из нейт¬
ральных растворов выпадает в виде очень мелких
кристаллов, которые проходят даже через мелкопо¬
ристый фильтр. При медленном осаждении из кислого
и горячего раствора сульфат бария образуется в фор¬
ме более крупных кристаллов. Крупнокристалличе¬
ский осадок предпочтителен еще и потому, что он
имеет меньшую общую поверхность и поэтому посто¬
ронние вещества адсорбируются мало и их легче от¬
мыть при промывании осадка.3. Молекулярная масса гравиметрической формы
должна быть по возможности большей. Тогда относи¬
тельная погрешность определения в меньшей мере
влияет на результаты анализа. Например, фосфор
можно определить в виде М^гРгО? или Р2С)5-24МоОз.
Содержание фосфора во втором соединении в 16 раз
меньше, чем в первом. Абсолютная погрешность при
взвешивании на аналитических весах составляет
±0,0001 г. Поэтому относительная погрешность во
втором случае в 16 раз меньше.§ 4. ПРИМЕНЕНИЕ ВОЗНИКАЮЩИХ РЕАКТИВОВ
(ОСАЖДЕНИЕ ИЗ ГОМОГЕННОГО РАСТВОРА)Кристаллические осадки рекомендуется осаждать
медленно, прибавляя раствор осадителя по каплям и
тщательно перемешивая анализируемый раствор. Та¬
кой способ осаждения должен обеспечить равномер¬
ное распределение и малую концентрацию осадителя
во всем объеме раствора. Вследствие этого сте¬
пень пересыщения невелика и первичных центров209
кристаллизации образуется сравнительно немного.
Дальнейшее прибавление новых порций осадителя
приводит не столько к возникновению новых центров
кристаллизации, сколько к увеличению размеров уже
выделившихся центров и к образованию крупнокри¬
сталлических осадков. Такие осадки легко отделить
от маточного раствора фильтрованием. Кроме того,
общая поверхность крупнокристаллических осадков
невелика по сравнению с поверхностью мелкокри¬
сталлических (или аморфных) осадков, и адсорб¬
ция — соосаждение посторонних веществ — выра¬
жена слабо.Описанный способ осаждения не всегда приводит
к цели. Прибавление осадителя по каплям утоми¬
тельно и требует постоянного внимания со стороны
исполнителя анализа. Трудно устранить образование
местного избытка осадителя в местах падения ка¬
пель, что приводит к большой степени пересыщенно-
сти и к выделению слишком мелкокристаллического
осадка.Условия осаждения малорастворимых солей сла¬
бых кислот можно улучшить следующим образом.
В кислый анализируемый раствор вводят избыток
осадителя, причем из-за низкого значения pH осадок
не выделяется. Затем прибавляют какое-либо соеди¬
нение, подвергающееся в растворе медленному гидро¬
лизу и связывающее постепенно ионы водорода. При
этом pH раствора медленно увеличивается, возра¬
стает также концентрация аниона слабой кислоты,
входящего в состав осадителя. Последний распреде¬
ляется равномерно по всему объему раствора и его
концентрация в каждый момент времени невелика;
создаются именно те условия, которые благоприятст¬
вуют образованию компактных крупнокристалличе¬
ских осадков. Такой метод называют методом возни¬
кающих реактивов.Примером соединений, связывающих ионы водоро¬
да, могут служить уротропин, мочевина и др. Реак¬
ции идут следующим образом:<CH2)6N4 + 4Н* + 6Н20 4NHJ + 6HCHOилиШ2-СО—NH2 + 2Н+ + Н20 «=> 2NHt + C02.210
Рассмотренный метод можно применить для осаж¬
дения, например, оксалата кальция, хромата бария:Ca2t + H2C204 4=fc СаС204 + 2Н*.2Ва2++ Н2Сг207 + Н20 *=± 2ВаСгО, + 4Н*.В обоих случаях выделяющиеся ионы водорода
медленно связываются уротропином или мочевиной,
что приводит к постепенному сдвигу состояния равно¬
весия приведенных реакций в правую сторону.Применение возникающих реагентов также дает
возможность в значительной мере устранить соосаж¬
дение. Существуют реагенты, которые претерпевают
в растворе медленный гидролиз с выделением собст¬
венно осадителя. В зарубежной литературе процесс
осаждения с помощью возникающих реагентов назы¬
вают процессом осаждения из гомогенного раствора.
Это название не объясняет сути процесса и поэтому
является менее удачным.Возникающие реагенты были впервые применены
для получения сульфидов. Преимущество применения
органических возникающих реагентов для осаждения
сульфидов состоит и в том, что они менее токсичны
по сравнению с сероводородом. Для осаждения суль¬
фидов металлов применяют тиоацетат аммония, кото¬
рый при взаимодействии с солями тяжелых металлов
образует сульфиды и уксусную кислоту:МС12 + CH3COSNH* + Н20MS + СН3СООН + НС1 + NH4CI.Для осаждения сульфидов применяют также тио-
^NHацетамид СН3—С> , тиоформамид HN=CH—SH
^SHи много других соединений, содержащих сульфгид-
рильную группу.Особую ценность приобретают возникающие реа¬
генты в тех случаях, когда необходимо определять
микропримеси различных компонентов. Так, тиоацет-
амид был применен для определения микропримесей
кадмия, свинца, висмута и цинка при анализе жаро¬
стойких сплавов на основе никеля с молибденом или
вольфрамом, микропримесей молибдена в титановых211
сплавах и многих других металлов в различных мате¬
риалах.К числу возникающих реагентов принадлежат не¬
которые сложные эфиры — триэтилфосфат, диметил-
фосфат, метилоксалат, этилоксалат, диметилсульфат
и др. Триэтилфосфат применяют для осаждения фос¬
фатов циркония и гафния и для их фракционирован¬
ного выделения:(С2Н50)3Р=0 + ЗНгО = ЗС2Н5ОН + НзРОч.Посредством метил- или этилоксалата осаждают
кальций:СООСНзСООН + НгО = Н2С2Оч + СНзОН.Для определения стронция или бария используют
.диметилсульфат:(CH3hS04 + 2НгО = H2S04 + 2СН3ОН.Еще один вариант метода — синтез осадителя непос¬
редственно в анализируемом растворе. Так, осажде¬
ние кобальта 1-нитрозо-2-нафтолом можно выполнить
следующим образом.К кислому раствору 2-нафтола, содержащему
ионы кобальта, прибавляют нитрит натрия, выделяю¬
щий азотистую кислоту, которая реагирует с 2-на¬
фтолом:NОбразующийся 1-нитрозо-2-нафтол, концентрация
которого медленно возрастает, реагирует с ионами
кобальт^ с образованием осадка.S S. ТЕРМОГРАВИМЕТРИЧЕСКИП АНАЛИЗВ термогравиметрическом анализе наблюдают за из¬
менением массы вещества в процессе нагревания. Это
дает возможность судить о происходящих превраще¬
ниях вещества при нагревании и уцтановить состав
образующихся промежуточных продуктов. Различают
статический (изотермический) и динамический (поли-
термический) термографический анализ.212
В первом случае вещество нагревают до опреде¬
ленной температуры и, поддерживая температуру по¬
стоянной, наблюдают за изменением его массы. Ре¬
зультаты выражают в виде кривых зависимости
потери массы (в %) от времени (изотермия). Взвеши¬
вание проводят через определенные промежутки вре¬
мени до тех пор, пока масса вещества при заданной
температуре не станет постоянной. Полученные дан¬
ные пересчитывают на состав и наносят на диаграмму
состав — температура.В изотермическом термографическом анализе кри¬
вые изменения массы вещества в зависимости от вре¬
мени получают при постоянном давлении выделяе¬
мого веществом газа или пара (изобарные кривые),
например при исследовании гидратов в эксикаторе
над раствором серной кислоты с различным, заранее
заданным давлением пара. В этом случае опыты про¬
водят до установления постоянной массы вещества
при каждом заданном давлении. Затем полученные
данные пересчитывают на состав и наносят на график
давление—состав (тензиметрические кривые). Поль¬
зуясь этими кривыми, получают данные об образова¬
нии промежуточных соединений постоянного или пе¬
ременного состава.В случае политермического гравиметрического
анализа непрерывно регистрируют массу вещества в
процессе его нагревания и строят термографические
кривые. Эти кривые пересчитывают на содержание
летучего компонента в зависимости от температуры и
наносят на график. Такие кривые называют политер¬
мами. Политермы не отвечают равновесным данным,
так как нагревание в этом случае проводилось не до
постоянной массы. Однако с помощью этих кривых
можно установить число и состав промежуточных со¬
единений.Дериватный термографический метод также отно¬
сится к термогравиметрии. В этом случае записывают
производную-от термогравиметрической кривой, кото¬
рая показывает скорость изменения массы вещества
при его нагревании. Дериватную термогравиметрию
применяют обычно одновременно с политермическим
термогравиметрическим и дифференциальным терми¬
ческим анализом.213
Рис. 11.1. Термогравиметрн-
ческая кривая оксалата
кальцияТермогравиметриче¬
ский метод широко ис¬
пользуют для исследо¬
вания изменения соста¬
ва вещества при на¬
гревании, а значит н
для установления усло¬
вий высушивания или
прокаливания осадков. На рис. 11.1 приведена термо¬
гравиметрическая кривая для оксалата кальция. Из
этого рисунка видно, что температура прокаливания
оксалата кальция с целью переведения его в грави¬
метрическую форму СаО соответствует горизонталь¬
ному участку кривой 838—1025°С.ГЛАВА 12ЭЛЕКТРОГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ
§ I. ОБЩАЯ ХАРАКТЕРИСТИКАЭлектрогравиметрический анализ основан на элект¬
ролитическом выделении металлов и взвешивании
полученного на электроде осадка металлов. Первое
требование к осадкам в гравиметрическом анализе —
их практическая нерастворимость — хорошо выпол¬
няется в электроанализе, так как большинство ме¬
таллов не растворяется в воде. Однако электролити¬
ческое осаждение иногда бывает неполным вследствие
преждевременного прекращения электролиза. При
электролизе осаждение происходит только в момент
приближения определяемых ионов к поверхности
электрода, поэтому очень большое значение имеет
перемешивание раствора.Второе требование к осадку—его чистота, соот¬
ветствие состава осадка определенной формуле. Это
требование выполняется при электролитическом
осаждении значительно лучше, чем при обычных ме¬
тодах гравиметрического анализа.кСаС204Н20V.226 39б"|СаСОз478бзГ\773 \V, с»° ,838 102SТемпература, “С214
Основным физическим условием электролитиче¬
ского разделения металлов является определенное
напряжение, при котором осаждаются одни металлы
и не выделяются другие. Основным химическим усло¬
вием является присутствие определенных «буферных»
ионов, чаще всего ионов водорода, а также ионовNOa.Вследствие особого характера кристаллической
решетки металлов различные присутствующие в рас¬
творе соединения практически не захватываются при
электролитическом осаждении металлов. Здесь, сле¬
довательно, почти не наблюдается явление соосажде¬
ния. При очень крупнозернистых осадках иногда за¬
хватывается немного самого электролита, удерживаю¬
щегося между отдельными кристаллами осадка. В не¬
которых случаях наблюдается образование сплавов.Третье требование к осадку — получение его в оп¬
ределенной агрегатной форме, удобной для работы,
при электролизе обычно соблюдается хорошо. Осадок
при определенных физико-химических условиях полу¬
чается в форме плотного слоя на электроде, опера¬
ция фильтрования устраняется вовсе, а промывание
занимает очень мало времени.Электроанализ применяют главным образом для
очень точного определения некоторых металлов:
меди, никеля, цинка, кадмия, а также свинца путем
осаждения последнего на аноде в виде РЬ02.Некоторые металлы, например щелочные, доволь¬
но легко реагируют с водой и окисляются. Другие
осаждаются в обычных условиях электроанализа
медленно или неколичественно (например, висмут,
хром, железо). Поэтому применение электролитиче¬
ского осаждения в анализе ограничено. Кроме опре¬
деления названных и некоторых других металлов,
практическое значение имеет также определение ма¬
лых количеств некоторых металлов путем так назы¬
ваемого «внутреннего» электролиза.Большое значение для разделения ряда элементов
имеет электролитическое осаждение на ртутном като¬
де, причем осаждение облегчается образованием
амальгам. Так, адля определения примеси алюминия
в железных сплавах железо и многие другие металлы
осаждают из сернокислого раствора на ртутном ка¬
тоде, причем алюминий остается в растворе.Наконец,215
можно указать на применение анодного растворе¬
ния металлов. Например, для определения неметал¬
лических включений в стали и различных цветных
сплавах поступают следующим образом. Образец,
металла опускают в раствор соответствующего элек¬
тролита и включают ток, причем исследуемый металл
является анодом. Во время электролиза металл пе¬
реходит в раствор, а неметаллические примеси оста¬
ются в виде осадка. Этот метод имеет большое зна¬
чение для фазового анализа металлов.§ 2. ЗНАЧЕНИЕ НАПРЯЖЕНИЯ ПРИ
ЭЛЕКТРОЛИТИЧЕСКОМ ВЫДЕЛЕНИИ МЕТАЛЛОВПотенциал электрода. Поляризация и напряжение
разложения. Прохождение тока через раствор элек¬
тролита резко отличается от прохождения тока через
металл. Если к концам металлического стержня при¬
соединить провода от источника тока, то уже при са¬
мом небольшом приложенном напряжении через стер¬
жень будет идти поток электронов. Вещество метал¬
ла при этом не изменяется, часть тока затрачивается
только на некоторое нагревание проводника. Если же
провода от источника постоянного тока опустить в
раствор электролита, то электрический ток пойдет
только при некоторых определенных условиях. Про¬
хождение тока в этом случае связано с движением
ионов в растворе и с разрядом ионов на электродах
или с превращением атомов электрода в ионы. На
электродах начинаются электрохимические процессы,
которые приводят к изменению состава раствора и
электрода. Таким образом, два одинаковых электрода
становятся различными в результате прохождения
тока через раствор. Эти два проводника становятся
теперь различными полюсами гальванического эле¬
мента, возникающего внутри электролита; такое яв¬
ление, препятствующее прохождению тока через рас¬
твор, называют поляризацией.Так, при пропускании тока через раствор серной
кислоты на отрицательном полюсе (платиновый элек¬
трод) выделяется водород, так как электроны, посту¬
пающие на платиновый катод, восстанавливают ионы
водорода, превращая их в молекулы водорода:2Н+ + 2е~ н2. (12.1)216
На платиновом аноде, который отдает внешнему
источнику тока электроны, выделяется кислород:Н20 2Н* + |о2 + 2е‘. (12.2)Однако оба процесса обратимы. Поэтому выделив¬
шийся на катоде водород может снова восстанавли¬
ваться и переходить в раствор в виде ионов, отдавая
электроны платиновому проводнику. Эти электроны
по проводу поступают на другой электрод, содержа¬
щий кислород, и равновесие (12.2) смещается влево.Таким образом, при электролизе возникает галь¬
ванический элемент, ток которого направлен в сто¬
рону, обратную движению тока от внешнего источни¬
ка. Поэтому ток от внешнего источника будет идти
через электролит только в том случае, если приложен¬
ное напряжение будет достаточно для определенного
химического процесса, а именно для электролиза рас¬
твора или для образования ионов из материала элек¬
трода. Необходимое для этой цели напряжение назы¬
вают напряжением разложения, оно зависит прежде
всего от состава раствора.Пусть, например, в качестве электролита взяты
растворы сульфатов или нитратов серебра, меди,
свинца и цинка, причем каждый раствор содержит
1 М ионов металла в 1 л. При электролизе таких
растворов на платиновом аноде всегда идет один и
тот же процесс — выделение кислорода. На платино¬
вом катоде происходит восстановление ионов того или
иного металла. Из названных четырех ионов ионы
серебра восстанавливаются легче всего; поэтому для
электролиза раствора нитрата серебра достаточно
приложить сравнительно небольшое напряжение —
приблизительно 0,9 В*. Ионы меди восстанавливаются
труднее, чем ионы серебра, поэтому электролиз рас¬
твора сульфата меди будет идти только при значи¬
тельно большем напряжении, а именно около 1,4 В.
Ионы свинца и цинка восстанавливаются еще труднее,* Сверх указанного необходимо еще небольшое избыточное
напряжение для преодоления обычного («омического») сопротив¬
ления раствора, как всякого проводника. Кроме того, следует
иметь в виду, что указанное напряжение относится к раствору
нитрата серебра, содержащему в то же время водородные ионы
в 1 М концентрации.217
Рис. 12.1. Схема водородного электродаРис. 12.2. Схема для измерения потенциала металлического элек¬
трода/ — водородный электрод; 2— испытуемый электрод; Я — потенциометр;
4 —электролитический ключ; 5 —«пробки» из фильтровальной бумагии для электролитического разложения растворов
солей свинца и цинка необходимо приложить к элек¬
тродам еще большее напряжение (не менее 1,9 и2,5 В соответственно).Напряжение, необходимое для электролиза 1 М
растворов хорошо диссоциированных солей, можно
найти из ряда напряжений. Падение напряжения
в растворе характеризуется главным образом двумя
скачками потенциала — на аноде и на катоде. Для
измерения этих скачков потенциала в качестве стан¬
дартного («нулевого») электрода сравнения приме¬
няют водородный электрод. Этот электрод (рис. 12.1)
состоит из платиновой проволоки, помещенной в
стеклянную трубку. Платиновая проволока внизу по¬
крыта губчатой платиной. Часть губчатой платины
находится в атмосфере водорода, другая часть погру¬
жена в 1 М раствор серной кислоты. Для измерения
скачков потенциала между различными электродами
и растворами водородный электрод / соединяют с ис¬
пытуемым электродом 2 в гальванический элемент
(рис. 12.2). Напряжение испытуемого электрода из¬
меряют вольтметром, точнее — потенциометром 3.
Оба электрода (испытуемый и водородный) соеди¬
няют в гальванический элемент с помощью пористой218
перегородки или сифоном 4 (электролитический
ключ), наполненным каким-либо нейтральным элект¬
ролитом (КС1, NaN03) и снабженным «пробками» 5
из фильтровальной бумаги.С помощью описанной схемы возможно опреде¬
лить знак испытуемого электрода (положительный
или отрицательный) и численное значение его потен¬
циала по сравнению с потенциалом водородного
электрода. Потенциал водородного электрода принят
равным нулю, поэтому измеренное напряжение харак¬
теризует скачок потенциала между испытуемым ме¬
таллическим электродом и раствором соответствую¬
щей соли. В результате таких измерений составлен
ряд напряжений.На рис. 12.3 графически показано место потен¬
циалов некоторых электродов в ряду напряжений.
Численные значения электродных потенциалов отно¬
сятся к активности соответствующего иона в раство¬
ре, равной единице. Такие потенциалы называют
стандартными потенциалами. Ряд напряжений имеет
большое значение для всех электрохимических мето¬
дов анализа. В электроанализе ряд напряжений поз¬
воляет вычислить напряжение разложения электро¬
лита и дает возможность предвидеть условия разде¬
ления металлов.Пользуясь рядом напряжений, можно рассчитать
напряжение гальванического элемента, составленно¬
го из той или иной пары электродов. Так, гальваниче¬
ский элемент, составленный из водородного электро¬
да и подобного же по конструкции кислородного
электрода (при условии работы с губчатой платиной),
может дать напряжение 1,23 В (см. рис. 12.3). Кро¬
ме того, если в 1 М раствор H2S04 пропускать ток
через платиновые, покрытые губчатой платиной,
электроды, то На катоде выделяется водород, а на-0,5 0 0,5 1,0 1,51 I I I I I i I I I I I I I I I I 1 I I I I I I I I ' I I/ \ \ Ч \ \ t2ni+/ Pb2V 2HV Cu2V AgV Ог / 0, //Zn /РЬ /Н2 /Си /Ag /Н20 /Нг0Рис. 12.3. Ряд напряжений некоторых элементов219
аноде — кислород. Таким образом, в результате про¬
пускания тока образуется гальванический элемент, ко¬
торый может давать ток с напряжением 1,23 В, на¬
правленный против тока внешнего источника напря¬
жения.Очевидно, ток сможет идти через раствор только
в том случае, если приложенное напряжение будет не
менее 1,23 В.Если при пропускании тока в качестве анода взять
не губчатую платину, а обычную гладкую платину,
то выделение кислорода значительно затрудняется и
для электролиза необходимо большее напряжение, а
именно 1,7 В (см. рис. 12.3). Подобным же образом
из ряда напряжений можно найти значение напряже¬
ния разложения для растворов электролитов, если из¬
вестны процессы, идущие на катоде и на аноде. Так,
раствор, в 1 л которого содержится 1 моль ионов
серебра и 1 моль ионов водорода, при электролизе
образует гальванический элемент с кислородным и
серебряным электродами. Напряжение такого гальва¬
нического элемента, в соответствии с данными
рис. 12.3, будет составлять 1,7—0,8 = 0,9 В. .Это соот¬
ветствует приведенному выше напряжению разложе¬
ния раствора нитрата серебра. Аналогично рассчиты¬
вают из данных рис. 12.3 напряжение разложения
для раствора сульфата меди: 1,7 — 0,34 =1,36 В
и т. д.Из приведенных примеров видно, что напряжение
разложения представляет собой алгебраическую раз¬
ность потенциалов анода и катода и в общем случае
может быть вычислено без учета перенапряжения по
уравнению:£р — Еа /?к.где F.v — напряжение разложения, £а и Ек—соответственно ве¬
личины потенциалов анода и катода.Для выделения пузырьков водорода на чистом
цинковом катоде необходимо перенапряжение 0,70 В,
а для выделения на гладкой поверхности ртути 0,76 В.Перенапряжение зависит от ряда факторов. Имеет
значение прежде всего материал электрода: так пе¬
ренапряжение выделения водорода на ртутном катоде
значительно больше, чем на электродах из других ме¬
таллов. Перенапряжение, как правило, возрастает220
с увеличением плотности тока и зависит также os
температуры раствора.Перенапряжение необходимо иметь в виду при вы¬
числении напряжения разложения при выделении ме¬
таллов. Явление перенапряжения дает возможность
выделять ряд электроотрицательных металлов из вод¬
ных растворов их солей; если бы не было явления
перенапряжения, то при электролизе растворов солей
цинка или свинца вместо металлического цинка или
свинца должен был бы выделяться только водород
(см. рис. 12.3). Большое перенапряжение для выделе¬
ния водорода на ртути имеет значение в полярографи¬
ческом анализе, а также при использовании амальгам
металлов в качестве восстановителей.Влияние концентрации. Потенциал электрода за¬
висит не только от природы данных веществ, но и от
концентрации ионов в растворе. Выше был рассмот¬
рен пример электролиза 1 М раствора нитрата се¬
ребра; для выделения серебра из такого раствора не¬
обходимо приложить напряжение 0,9 В. Очевидно,
если концентрация серебра в растворе меньше 1 М,
восстановление будет идти труднее и придется при¬
ложить большее напряжение. Серебряный электрод
погружен в разбавленный (например 0,1 или 0,01 М)
раствор соли серебра, имеет по отношению к водород¬
ному электроду потенциал меньший, чем 0,9 В. Это
сооответствует увеличению напряжения разложения,
так как потенциал выделения металла сдвигается
влево, т. е. дальше от потенциала выделения кисло¬
рода (см. рис. 12.3).Зависимость потенциала металлического электрода
от концентрации ионов этого металла в растворе вы¬
ражается уравнением Нернста:£ = £•„+ -5L In с, (12.3)иггде Е — потенциал при данных температуре и концентрации;
Ео — стандартный потенциал (значение его может быть взято
из ряда напряжений); R — газовая постоянная (8,313 Дж); Т —
абсолютная температура; п — число электронов, принимающих
участие в электрохимической реакции; F — число Фарадея
(96 500 Кл); С — концентрация ионов металла и растворе.Уравнение (12.3) можно упростить. Для этого
вместо RT и F подставляют соответствующие
значения (Т принимают 293 К, а также учитывают221
коэффициент перехода от натуральных логарифмов
к десятичным (2,3026):Е = Еа + lg с. (12.4)Пусть, например, необходимо вычислить потенциал
серебряного электрода, погруженного в раствор, со¬
держащий 10_б М серебра в 1 л.По уравнению (12.4) находим:Е = Е0 + lg [Ag+] = 0,8 + 0,058 lg 10-6 == 0,8 — 0,058 ■ 6 = 0,45В.Полученный результат дает возможность вычис¬
лить напряжение разложения, необходимое для прак¬
тически полного выделения серебра, т. е. для того,
чтобы в растворе оставалось 10~6 М ионов серебра
в 1 л.Обычно применяют гладкий платиновый анод, на
котором выделяется кислород; раствор, во всяком
случае к концу электролиза, бывает кислым. Потен¬
циал кислородного электрода при этих условиях мож¬
но принять равным 1,7 В. Отсюда необходимое напря¬
жение разложения (Ер), очевидно, составляет:Ер — Еа — Ек = 1,7 - 0,45= 1,25В.§ 3. ФИЗИЧЕСКИЕ УСЛОВИЯ ОСАЖДЕНИЯ МЕТАЛЛОВВлияние напряжения. Напряжение влияет на все
характеристики осадка. Если к электродам приложить
недостаточное напряжение, металл не будет выде¬
ляться вообще или выделение его будет неполным
(см. § 1). Если приложить слишком большое напря¬
жение, то кроме интересующего нас металла, на элек¬
троде могут выделяться другие присутствующие в рас¬
творе металлы, т. е. получится загрязненный осадок.
Кроме того, при слишком большом напряжении не¬
редко образуется рыхлый, губчатый осадок металлов.Таким образом, электролитическое осаждение ме¬
таллов необходимо вести в определенном интервале
напряжения тока. В зависимости от химических ус¬
ловий (см. § 4) этот допустимый интервал может быть
более или менее узким. В большинстве случаев для222
Рис. 12.4. Схема прибо¬
ра для электроанализа:
/ — источник постоянного то-
ка; 2— вольтметр; $— рео¬
стат; 4— ползунок реостата;
5— амперметр; 6 — электро¬
лизерэлектролитического
осаждения выби¬
рают такие химиче¬
ские условия, при
которых нет необхо¬
димости строго ре¬
гулировать напря¬
жение. Тем не менее.X ’обычно во всех установках для проведения электро-
гравиметрического анализа предусмотрена возмож¬
ность регулировать напряжение.Наиболее простая схема для электроанализа по¬
казана на рис. 12.4. Движением ползунка 4 На рео¬
стате 3 можно изменять общее сопротивление цепи
и таким образом регулировать напряжение на клем¬
мах электролизера 6.Сила тока / в цепи зависит по закону Ома от на¬
пряжения Е, приложенного к электродам, напряжения
разложения Ер электролита в электролизере и от об¬
щего сопротивления цепи R:Е-Е рW~~(12.5)Обычно сопротивление цепи R состоит из двух
частей: сопротивления реостата и омического сопро¬
тивления раствора.Из рассмотренных в § 1 особенностей прохождения
тока через электролит, а также из приведенной фор¬
мулы видно, что ток будет идти в том случае, если
приложенное напряжение выше, чем напряжение раз¬
ложения, т. е. если Е > Ер. Избыток напряжения,
т. е. Е — Ер, распределяется между отдельными уча¬
стками цепи пропорционально их сопротивлению.Пусть, например, напряжение источника тока рав¬
но 6 В, напряжение разложения раствора в электроли¬
зере составляет 2 В, сопротивление реостата равно223
30 Ом и омическое сопротивление раствора 10 Ом. Из
общего напряжения 6 В на преодоление напряжения
гальванического элемента будет затрачено 2 В, обра¬
зующегося в электролизере, т. е. на разложение элек¬
тролита. Остаток 4 В распределится между клеммами
реостата и раствором. Отношение омического сопро¬
тивления реостата и раствора равно, согласно усло¬
вию, 30 : 10. Следовательно, избыток напряжения 4 В
распределится также в отношении 3: 1, т. е. ЗВ пой¬
дет на преодоление сопротивления реостата и 1 В на
преодоление омического сопротивления раствора.Вольтметр, включенный на клеммы электролизера,
покажет отклонение, соответствующее 3 В, из кото¬
рых 2 В соответствуют напряжению разложения и
1 В — преодолению омического сопротивления рас¬
твора.Из рассмотренного примера видно, что показания
вольтметра зависят не только от напряжения разло¬
жения, но также от омического сопротивления рас¬
твора. Омическое же сопротивление электролизера за¬
висит от расстояния между электродами, от темпера¬
туры и от концентрации электролита. Последние два
фактора обычно изменяются во время электролиза.
Таким образом, перед выполнением анализа TO4iio
рассчитать необходимое напряжение нельзя. Наконец,
напряжение разложения зависит также от того, осаж¬
дается ли металл из растворов простой, хорошо диссо¬
циированной соли или из раствора комплексной
соли.Все рассмотренные явления приводят к тому, что
при электрогравиметрическом анализе очень редко
применяют методы, в которых для осаждения металла
требуется точно определенное напряжение. Необходи¬
мый минимум напряжения можно рассчитать на осно¬
вании рассмотренного в предыдущем параграфе по¬
нятия о ряде напряжений с учетом уравнения Нернста.Если нужно выделить металл из такого раствора,
в котором нет других катионов, то, очевидно, целесо¬
образно повышать напряжение, так как это будет спо¬
собствовать более полному и быстрому выделению.
Верхний предел напряжения обусловливается в этом
случае только побочными причинами, как, например,
нагреванием раствора, выделением рыхлого осадка
металла и т. д.224
При необходимости разделения металлов иногда
приходится ограничивать напряжение. При этом, оче¬
видно, напряжение целесообразно повышать только до
тех пор, пока еще не будет превышено напряжение
разложения для раствора соли второго металла. На¬
пример, для разделения серебра и меди в растворе их
сульфатов следует применять напряжение не выше
1,4 В. Это значение соответствует напряжению разло¬
жения 1 М раствора сульфата меди, между тем как
в условиях анализа концентрация ионов меди обычно
значительно ниже. Следовательно, при напряжении
менее 1,4 В (например, при использовании железо-ни-
келевого аккумулятора) на электроде будет осаждать¬
ся только серебро.Легко видеть, что при количественном осаждении
серебра, т. е. когда его концентрация в растворе ста¬
нет равной 10_6 М, потенциал катода достигает зна¬
ченияЕх = 0,8 + 0,06 lg 10“6 = 0,44Ви, следовательно, уже при напряжении 1,7—0,44 =
= 1,26 В осаждение серебра должно быть практиче¬
ски полным.Влияние силы и плотности тока. Для полного
осаждения 1 моль эквивалента металла необходимо
пропустить через раствор по крайней мере 96 500 Кл
(число Фарадея), если даже при электролизе не будут
протекать побочные процессы.Для определения берут обычно 0,01—0,02 эквива¬
лента металла, например 0t3—0,6 г меди. При силе
тока 1 А для осаждения этого количества металла
достаточно 15—30 мин. В действительности прихо¬
дится пропускать значительно большее количество
электричества, чем рассчитанное по числу Фарадея,
так как на катоде протекают побочные процессы (вы¬
деление водорода, восстановление ионов N03' и т. п.).
Затрата количества электричества на побочные про¬
цессы часто уменьшается при перемешивании раство¬
ра, т. е. при быстром подводе ионов осуждаемого ме¬
талла к поверхности катода. Во всех случаях перед
окончанием электролиза необходимо проверить пол¬
ноту осаждения. Для этого платиновый катод погру¬
жают несколько глубже в раствор, пропускают элек¬
трический ток и наблюдают за выделением металла.8 Аналитическая химия225
Если свежая поверхность электрода не покрывается
выделяемым металлом, то это означает, что металл
выделился полностью и электролиз можно пре¬
кращать.Сила тока влияет на характер образующегося
осадка металла. Имеет значение не количество элек¬
тричества, а плотность тока на катоде, т. е. количе¬
ство ампер на единицу поверхности катода. При
очень малых плотностях тока металл иногда осаж¬
дается в виде крупных кристаллов, которые растут
отдельными ветвями. Такие ветви металла легко об¬
рываются, когда электрод вынимают из раствора. При
очень большой плотности тока может образоваться
рыхлый, губчатый осадок металла, так как происхо¬
дит быстрое осаждение металла и в слое раствора
вблизи электрода резко уменьшается концентрация
ионов металла. В результате нередко начинается вы¬
деление водорода и происходит ряд других явлений,
ведущих к разрыхлению осадка металла. Губчатый
осадок легко осыпается с электрода, сильно окисляет¬
ся при высушивании и поэтому очень неудобен в
работе.В некотором среднем интервале плотности тока
получается плотный осадок, хорошо пристающий к
поверхности электрода. Такой осадок легко промы¬
вается, не окисляется при высушивании и, следова¬
тельно, наиболее удобен в работе. Плотный осадок
получается обычно при плотности тока 1—3 А на
100 см2 поверхности электрода. В зависимости от хи¬
мических условий осаждения эти значения могут из¬
меняться, а иногда как, например, при осаждении се¬
ребра из раствора AgN03, очень трудно вообще полу¬
чить плотный осадок.§ 4. ХИМИЧЕСКИЕ УСЛОВИЯ ОСАЖДЕНИЯ МЕТАЛЛОВЗначение химических условий при электрогравимет-
рическом анализе частично отмечалось выше. Кислот¬
ность раствора, присутствие анионов азотной или сер¬
ной кислоты, введение анионов, образующих с метал¬
лами комплексы — все это имеет очень большое
значение при электролитическом выделении металлов.
Обычно химические условия еще в большей мере, чем226
физические, определяют полноту осаждения, чистоту
осадка и его внешние качества.Рассмотрим метод электролитического разделения
меди и цинка. Медь и цинк занимают различные ме¬
ста в ряду напряжений (см. рис. 12.3). Для разделе¬
ния таких металлов можно ограничиться определен¬
ными физическими условиями, а именно приложить
к электродам напряжение, достаточное для количе¬
ственного осаждения меди, но недостаточное для выде¬
ления цинка даже из концентрированных растворов
его солей. Для электролиза 1 М раствора сульфата
цинка необходимо напряжение £=1,7 — (—0,8) =
= 2,5 В. Если приложить меньшее напряжение, на¬
пример 1,7 В, цинк выделяться не будет. Полноту вы¬
деления меди в этих условиях можно вычислить из
уравнения Нернста. Напряжение разложения 1,7 В
при выделении на аноде кислорода в ряду напряжений
соответствует потенциалу на катоде, равному нулю,
т. е. потенциалу стандартного водородного электрода.
Подставляя это значение в уравнение Нернста *, на¬
ходим:0 = Е0 + lg [Cu2M = 0,34 + 0,03 lg [Сu2+].Отсюда рассчитываем концентрацию ионов меди, ко¬
торые могут остаться в растворе при данном катодном
потенциале:О Я4-lg[Cu^] =^_=11Доткуда[Си2 + ] = 10~11,1 = 5 • 10_|2Л1Следовательно, в этих условиях осаждение ионов
меди будет практически полным.Рассмотренный пример показывает, что разделение
меди и цинка вполне возможно. Одиако необходимост
ограничения напряжения тока вызывает большие не¬
удобства. В этом случае ток идет главным образом
только за счет переноса ионов меди, поэтому по мере
осаждения ионов меди сила тока уменьшается и осаж¬
дение замедляется. При работе в обычных условиях* Для упрощения подобных расчетов удо(5но принять, что
предлогарифмическнм коэффициент уравнения Нернста отнесен
к температуре 30 °С и в этом случае равен 0,06.8*227
по схеме включения, изображенной на рис. 12.4, не¬
обходимо тщательно следить за напряжением, по¬
скольку после осаждения меди (хотя бы в слое вбли¬
зи катода) напряжение на электродах будет повы¬
шаться, что приведет к осаждению цинка.Значительно удобнее для разделения меди и цинка
создавать определенные химические условия. Для
этого достаточно просто подкислить раствор азотной
кислотой. Ионы NO3 являются в данном случае
«буферными» ионами:no; + 10Н+ + 8е" = nh4+ + зн2о.После выделения меди напряжение на электродах по¬
вышается, однако это приводит к восстановлению
ионов NO3, а ионы цинка не восстанавливаются. Та¬
ким образом, нет необходимости во время электролиза
постоянно следить за напряжением тока.Условия разделения металлов, как видно из рас¬
смотренного примера, сильно зависят от кислотности
раствора. Большое значение имеет характер присут¬
ствующих анионов. Характер электролитических
осадков металлов обусловлен присутствием ионов, об¬
разующих комплексы с ионами металлов. Из раство¬
ров комплексных солей обычно получаются более
плотные осадки даже при больших плотностях тока.Осаждение из сернокислых растворов. При элек¬
тролизе сернокислых растворов на катоде разря¬
жаются главным образом определяемые ионы и осаж¬
даются в виде металла:М'1+ + пе~ = М.На аноде выделяется кислород:Н20 = 2е~ + 2Н+ у02.Следует иметь в виду, что кислотность раствора
в результате анодного процесса увеличивается. Это
иногда замедляет выделение металла на катоде. Так,
кадмий выделяется электролизом из слабого серно¬
кислого раствора, однако чрезмерное повышение кис¬
лотности препятствует его количественному осажде¬
нию. В этом случае необходимо частично нейтрализо¬
вать раствор, прибавив, например, ацетат натрия.228
Осаждение из азотнокислых растворов. При элек¬
тролизе азотнокислых растворов катодный и анодный
процессы в основном те же, что в предыдущем слу¬
чае. Подготовка раствора к электролизу должна быть
проведена так, чтобы обеспечить полное удаление
оксидов азота и азотистой кислоты. Обычно с азотно¬
кислым раствором приходится иметь дело после рас¬
творения металла или сплава в азотной кислоте; та¬
кой раствор всегда содержит оксиды азота, которые
следует удалить кипячением. Азотистая кислота яв¬
ляется более сильным окислителем, чем азотная, и
поэтому растворяет металлы, выделяющиеся при элек¬
тролизе на катоде. В ее присутствии осаждение метал¬
лов сильно замедляется или вообще не происходит.
Азотистую кислоту можно удалить, прибавив моче¬
вину:2HN02 + NHjCONHj = С02 + 2N2 + 3H20.Важным побочным катодным процессом при элек¬
тролизе из азотнокислых растворов является восста¬
новление анионов азотной кислоты до ионов аммо¬
ния:NG3 + ЮН* + 8е" = NH| + ЗН20.Нитрат-ионы восстанавливаются при более положи¬
тельных значениях потенциала, чем ионы водорода.
Поэтому водород не выделяется до тех пор, пока не
закончится восстановление NO3 до юнов аммонияи, следовательно, устраняется главная причина обра¬
зования губчатого осадка металла. Кроме того, ионы
NO3 играют роль «буферных» ионов.Осаждение в присутствии хлоридов. Электролиз
хлороводородной кислоты применяют очень редко.
При электролизе таких растворов на аноде выделяет¬
ся свободный хлор, который частично взаимодействует
с платиновым анодом и замедляет осаждение металла
на катоде. Металлы из растворов HCI выделяют в
присутствии какого-либо сильного восстановителя, на¬
пример гидроксиламина, который восстанавливает
выделяющийся хлор:2NH2OH + С12 = N2 + 2НС1 + Н20.Осаждение из растворов комплексных соединений.
Главным достоинством электролиза из растворов229
комплексных соединений является получение плотного
осадка металла. Так, серебро выделяется из азотно¬
кислого раствора в виде отдельных длинных кристал¬
лов, легко отделяющихся от катода. Напротив, из циа¬
нистого комплекса серебра получается равномерный
плотный осадок. Кроме того, применение комплексо-
образователей приводит к изменению потенциалов вы¬
деления отдельных металлов, что создает новые воз¬
можности для разделения.При электролизе чаще всего применяют такие
комплексообразователи как аммиак, цианид калия,
гидроксид натрия, щавелевая кислота и др.Электролиз аммиачных растворов применяют в
том случае, если определяемый ион образует раство¬
римый аммиакат, например, при определении никеля,
кобальта, меди и др. На катоде осаждается металл,
на аноде выделяется кислород.Электролиз щелочных растворов применяют глав¬
ным образом для определения цинка. Ионы цинка
с избытком едкого натра образуют цинкат:Zn2( + 40Н" = ZnO2- + 2Н20.Если в растворе присутствуют ионы NO3, цинк
осаждается только после того, как закончится восста¬
новление ионов NO3 до аммиака. Электролиз циани¬
стых растворов применяют для определения серебра,
кадмия и некоторых других элементов. На аноде раз¬
ряжаются, кроме гидроксильных ионов, также анионы
CN~ с выделением свободного дициана (CNb, кото¬
рый является ядовитым газом. Чтобы устранить его
выделение, необходимо прибавить к раствору немного
свободной щелочи, тогда происходит реакция:(CN)j + 2КОН = KCN + KCNO + Н20.При электролизе цианистых растворов, содержащих
кадмий и медь, в случае избытка цианида калия об¬
разуются комплексные соединения состава
K2[Cd(CN)4] и Кг[Си (CN)4].Комплексные ионы диссоциируют по схеме:[M(CN)4f-4 Mn+-MCN~.Реакция протекает в незначительной степени, и
концентрация свободных ионов кадмия и меди в рас¬230
творе очень мала. Поэтому напряжение разложения
для таких комплексных солей гораздо больше, чем
для простых солей. Более того, цианистый комплекс
меди диссоциирован значительно меньше, чем циани¬
стый комплекс кадмия. Поэтому при электролизе
сначала выделяется кадмий, несмотря на то, что в
обычных условиях (например, из раствора сульфатов)
осаждению кадмия предшествует осаждение меди.§ 5. ВНУТРЕННИЙ ЭЛЕКТРОЛИЗДля определения незначительных примесей посторон¬
них металлов в основном металле в некоторых слу¬
чаях применяют метод так называемого внутреннего
электролиза. Этот метод был предложен давно, но
простой бездиафрагменный вариант метода был раз¬
работан Ю. Ю. Лурье. Название «внутренний элект¬
ролиз» метод получил потому, что в нем не приме¬
няют внешний источник тока. Принцип метода ясен из
следующего примера.Пусть имеется раствор соли цинка, содержащий
незначительную примесь соли кадмия. Погрузим в
этот раствор цинковый и платиновый электроды, со¬
единенные между собой (рис. 12.5). Из ряда напря¬
жений металлов известно, что металлический цинк
вытесняет кадмий из его солей. Если опустить цинко¬
вую пластинку в раствор соли кадмия, часть атомов
цинка перейдет в раствор в виде положительных
ионов, а цинковый электрод приобретает отрицатель¬
ный заряд за счет освободившихся электронов. Элек¬
троны переходят также на платиновый электрод и на
нем начнут разряжаться ионы кадмия, находящиеся в
растворе. Кадмий может осаждаться также непосред¬
ственно на цинковой пластинке. Это явление назы¬
вают цементацией. Однако на платиновом электроде
разряд ионов кадмия значительно облегчается. Можно
подобрать такие условия (кислотность раствора, тем¬
пература, соответствующие концентрации определяе¬
мой примеси и основного компонента), при которых
кадмий будет осаждаться только на платиновом ка¬
тоде. Через некоторое время платиновый катод с осад¬
ком металлического кадмия вынимают из раствора,
отключают от цинкового анода и после нромынания
н высушивания взвешивают.231
Рис. 12.5. Схема прибора для внутреннего
электролиза:/ — платиновый катод; 2 — цинковый анодНа платиновом катоде выделяются
только те металлы, которые являются1 более электроположительными, чем
растворяющийся металл анода. По¬
этому метод внутреннего электролиза
применен для раздельного определе¬
ния примесей нескольких металлов,
если использовать аноды из различных металлов.В качестве примера рассмотрим определение меди
и кадмия в металлическом цинке. Если в раствор, со¬
держащий соль цинка и примеси солей меди и кад¬
мия, погрузить цинковый анод, соединенный с плати¬
новым катодом, то на катоде выделяются и медь, и
кадмий. Ьсли же взять железный анод, то на платине
выделится только медь, так как она более электропо¬
ложительна, чем железо. Ионы кадмия при этом
останутся в растворе, так как кадмий более электро¬
отрицателен, чем железо. После выделения меди по¬
средством железного анода можно выделить кадмий,
применив цинковый анод.Метод внутреннего электролиза в описанном выше
варианте пригоден только для определения очень не¬
больших количеств примесей. При большей их кон¬
центрации часто наблюдается цементация. Резуль¬
таты определения в этом случае, конечно, будут не¬
правильными.Ю. А. Чернихов предложил покрывать анод тон¬
кой пленкой из коллодия. Эта пленка предохраняет
анод от цементации, вследствие чего становится воз¬
можным определение значительно больших количеств
примесей. Однако электролиз при этом идет намного
медленнее.Преимущество метода внутреннего электролиза за¬
ключается в чрезвычайной простоте прибора, благо¬
даря чему его можно применять даже в недостаточно
оборудованных лабораториях. Известным ограниче¬
нием разделения элементов методом внутреннего
электролиза, как и других методов электролитиче¬
ского осаждения, является тот же ряд напряжений.
Очевидно, методом внутреннего электролиза можно!К232
определять примесь более электроположительного ме¬
талла в менее электроположительном, например медь
в кадмии, но не наоборот.Недостатком внутреннего электролиза является
невозможность его применения для определения боль¬
ших количеств компонентов и необходимость очень
строгого соблюдения условий определения (кислот¬
ность, температура, концентрация солей). При несо¬
блюдении этих условий результаты анализа могут
оказаться неправильными.Для успешного определения чрезвычайно важно,
чтобы между анодом и катодом был хороший кон¬
такт, так как нарушение контакта часто приводит
к цементации; по этой же причине необходимо приме¬
нять анод из чистого (лучше химически чистого) ме¬
талла с отполированной поверхностью.§ 6. РАЗДЕЛЕНИЕ МЕТАЛЛОВ ЭЛЕКТРОЛИЗОМ
С ПРИМЕНЕНИЕМ РТУТНОГО КАТОДАРтутный катод имеет следующие преимущества и осо¬
бенности по сравнению с платиновым катодом.]. Большинство металлов хорошо растворяется в
ртути с образованием амальгам или коллоидных рас¬
творов. В связи с этим электролитическое выделение
металлов значительно облегчается. Многие металлы
(железо, хром, молибден и др.), которые на твердом
катоде не выделяются вовсе или их электролитиче¬
ское осаждение сильно затруднено, на ртутном катоде
легко и количественно выделяются иг; раствора.2. Для разряда ионов водорода на зеркальной по¬
верхности ртути требуется значительно большее на¬
пряжение, чем для разряда на платине. Так, на пла¬
тиновых электродах водород выделяется (из раство¬
ров кислоты) при напряжении 1,7 В, а на ртутном
катоде это напряжение возрастает до 2,5 В и больше.
В связи с этим на ртутном катоде легко осаждаются
электроотрицательные металлы (цинк, кадмий, вис¬
мут и др.). Это осаждение происходит без выделения
водорода, которое в случае твердых электродов при¬
водит к получению губчатых осадков и затрудняет
выделение этих металлов.Практическое значение имеет применение ртутного
катода для отделения большего количества одного233
+ Рис. 12.6. Сосуд для электролиза с ртутнымI катодомили одновременно нескольких метал¬
лов, переходящих в амальгаму, от
примеси другого металла, остающе¬
гося в растворе.Электролиз с применением ртут¬
ного катода является прекрасным ме-II _ тодом отделения алюминия, титана,
циркония, магния, кальция, стронция,
itjo бария, бериллия, ванадия, фосфата,мышьяка и урана от железа, хрома,
цинка, никеля, кобальта, меди, олова,
молибдена, висмута и серебра, осаждающихся на
ртутном катоде. При этом осаждение ведут из серно¬
кислого раствора. В принципе можно осаждение про¬
водить также из раствора НС1, но при этом в элект¬
ролит необходимо прибавлять гидроксиламин. Схема
электролиза с ртутным катодом представлена на
рис. 12.6. В качестве анода обычно используют пла¬
тиновую проволоку. Электролиз проводят при силе
тока 5—6 А и напряжении 6—7 В. Конец электролиза
определяют капельной пробой на отделяемый эле¬
мент. Затем, не прерывая тока, сливают электролит
и промывают ртуть водой. Промывные воды присо¬
единяют к электролиту, перемешивают и определяют
интересующие компоненты,
Раздел IVРЕАКЦИИ ОБРАЗОВАНИЯ
КОМПЛЕКСНЫХ СОЕДИНЕНИИГЛАВА 13СВЕДЕНИЯ О КОМПЛЕКСНЫХ СОЕДИНЕНИЯХ
§ 1. ОБЩИЕ ПОЛОЖЕНИЯСреди трех типов химических реакций, лежащих в
основе химических и многих физико-химических ме¬
тодов анализа (кислотно-основные, окисления — вос¬
становления и комплексообразования), реакции ком-
плексообразования, по-видимому, имеют наиболее
широкое применение в аналитической химии. Реакции
комплексообразования имеют самостоятельное значе¬
ние для определения многих ионов граЕШметрическим,
титриметрическим, фотометрическим, люминесцент¬
ным и многими другими методами анализа. Кроме
того, комплексообразование широко применяют для
создания специфических условий определения тех или
других ионов.Таким образом, аналитическая химия широко ис¬
пользует достижения химии комплексных соедине¬
ний, которая в свою очередь широко исследует
комплексообразование с органическими лигандами.
Современная аналитическая химия невозможна без
развития химии комплексных соединений и учения об
органических реагентах.Комплексные соединения давно начали применять
в анализе, что было основано главным образом на
эмпирическом подборе цветных реакций без глубокого
выяснения химизма процессов, лежащих в основе
метода.В развитии химии комплексных соединений и их ис¬
пользования в анализе можно выделить три периода.Первый период до А. Вернера (до 1890 г.), когда
совершенно была неясна природа «молекулярных
комплексов». Однако этот период существовал почти
целое столетие. Еще в 1795 г. академик В. М. Север-
гин применил настой дубильных орешков для обнару-азб
жения и полуколичественного определения железа- в
сточных и природных водах. Для выявления и коло¬
риметрического определения ионов меди применяли
реакцию образования аммиаката. В 1844 г. был опуб¬
ликован метод определения фосфорной кислоты с по¬
мощью молибдата, т. е. фактически уже тогда приме¬
нялись гетерополикислоты для определения фосфора.
Для отделения железа(III) была применена экстрак¬
ция его хлоридного комплекса эфиром. Известна, на¬
конец, прекрасная работа М. А. Ильинского и
Г. Кнорре о применении а-нитрозо-Р-нафтола для вы¬
явления и определения кобальта.Второй период — это период расцвета теорииА. Вернера, который в 1891 г. объяснил образование
так называемых «молекулярных комплексов» из ста¬
бильных валентно насыщенных соединений. Этот пе¬
риод характеризуется интенсивным развитием работ
по синтезу и изучению свойств комплексных соеди¬
нений.Академик Н. С. Курнаков выполнил классические
исследования комплексов металлов с тиомочевиной и
гуанидином, проф. Л. А. Чугаев изучил комплексы
металлов с оксимами. Реактив Чугаева — диметил-
глиоксим является в настоящее время лучшим реак¬
тивом для определения никеля. Синтезированы и
исследованы комплексные соединения платиновых ме¬
таллов с тиомочевиной и другими лигандами. Акаде¬
миком И. И. Черняевым открыто явление трансвлия¬
ния. Г. Лей в 1904 г. исследовал гликолевые соедине¬
ния меди. Работы Л. А. Чугаева и Г. Лея положили
начало глубоким исследованиям внутрикомплексных
соединений. Это направление продолжает развиваться
и в настоящее время.Третий период — исследование комплексных соеди¬
нений в растворе и широкое применение их в фото¬
метрическом и других методах анализа. В этом на¬
правлении очень много сделано академиками
И. П. Алимариным, И. В. Тананаевым, академиком
АН УССР А. К. Бабко и их школами, Н. П. Комарем
с сотрудниками, а также многими другими коллекти¬
вами нашей страны. Очень много сделано по исследо¬
ванию комплексных соединений и применению их в
экстракционных методах разделения с последующим
определением различных ионов академиком Ю. А. Зо¬236
лотовым с сотрудниками. Проведение .этих работ дало
возможность выяснить и обосновать химизм процес¬
сов, применяемых в различных методах анализа, и
предложить много новых методов анализа.Особый интерес представили исследования
Г. Шварценбаха реакций металлов с аминополикарбо-
новыми кислотами — комплексонами — для титримет-
рического определения многих ионов. Сотрудниками
Всесоюзного научно-исследовательского института хи¬
мических реактивов и особо чистых химических ве¬
ществ профессорами Р. П. Ластовским, Н. М. Дятло¬
вой и В. Я. Темкиной синтезированы многие новые
комплексоны, которые применены в химической тех¬
нологии и других областях народного хозяйства.Большая заслуга в исследовании и применении
комплексных соединений в анализе принадлежит ака¬
демику И. В. Тананаеву. Его классические исследова¬
ния фторидных, ферроцианидных и других комплек¬
сов вошли в сокровищницу нашей отечественной и за¬
рубежной науки.Из комплексов с неорганическими лигандами боль¬
шое значение имеют гетерополикислоты, которые ши¬
роко применяют для определения фосфора, кремния,
мышьяка, ниобия и других ионов. Глубокие иссле¬
дования этих комплексов проведены академикамиВ. И. Спицыным, И. П. Алимариным и А. К. Бабко,
а также проф. А. И. Кокориным.После доклада А. К. Бабко на XX конгрессе
ИЮПАК в 1965 г. в Москве «О тройных комплексах
и применении их в анализе» исследования разноли¬
гандных комплексов развиваются во всех лаборато¬
риях мира. Открыты новые интересные реакции и на
их основе разработаны новые более специфические
методы определения различных ионов особенно близ¬
ких по свойствам элементов.Одновременно с развитием химии комплексных со¬
единений, в частности исследованием их в водных рас¬
творах, развивалось применение органических реа-к-
тивов в неорганическом анализе.Химия комплексных соединений, умение о примене¬
нии органических реагентов и фотометрический ана¬
лиз развивались одновременно и в сильной мере вза¬
имно влияли на развитие каждого из этих направле¬
ний.237
Известно много формулировок определения тер¬
мина «комплексное» или «координационное» соедине¬
ние, которые различаются только формально, но все
подчеркивают главное — сложный состав соединений
по сравнению с частицами, их составляющими. Так,
по А. А. Гринбергу, комплексные соединения — это
молекулярные соединения, при сочетании компонен¬
тов которых образуются положительно или отрица¬
тельно заряженные сложные ионы, способные к суще¬
ствованию как в кристаллах, так и в растворах. Это
определение имеет две характерные особенности:1) комплексные соединения — это молекулярные со¬
единения, т. е. такие, которые образовались за счет
сочетания двух или нескольких различных молекул,
например:Fe(CN)3 + 3KCN 3=* K3[Fe(CN)e];2) к комплексным соединениям относят такие соеди¬
нения, которые существуют не только в растворах, но
и в кристаллическом виде. Эти ограничения исклю¬
чают из комплексных соединений те, которые не отве¬
чают двум сформулированным требованиям.Другие авторы не связывают понятие комплекс¬
ного соединения с названными особенностями. Так,
Ф. Россоти и X. Россоти определяют «комплекс как
форму, образованную ассоциацией двух или более
простых форм, каждая из которых способна сущест¬
вовать независимо». По Ф. Коттону и Дж. Уилкин¬
сону, комплекс или координационное соединение об¬
разуется, если центральный ион М объединяется с
одним или несколькими лигандами R', И^ит. д. собра-
зоманием частицы THnaMR/R;, причем атом или ион
М, лиганды и образующиеся комплексы — все могут
нести электрический заряд. По К. Б. Яцимирскому, к
комплексным соединениям относятся соединения, об¬
разующие изолированные группы атомов (ионы или
молекулы), характеризующиеся наличием координа¬
ции, неполной диссоциацией по гетеролитическому ти¬
пу в растворе (или в вакууме) и сложностью состава
(несовпадение координационного числа и степени
окисления). По А. К. Бабко, комплекс — это всякая
сложная группа, которая существует в растворе как
одно целое, отличающееся по свойствам от компонен¬
тов, ее образующих. Это определение подчеркивает238
два наиболее существенных для аналитической химии
свойства комплекса: изменение свойств частиц вслед¬
ствие комплексообразования и способность существо¬
вания в растворах.Так, в аналитической химии качественное и коли¬
чественное определение элементов во многих случаях
основано именно на изменении свойств частиц вслед¬
ствие их связывания в комплекс, примем очень часто
взаимодействие, обусловливающее изменение этих
свойств, проходит именно в водных растворах. Ис¬
ходя из этих соображений, в дальнейшем будет при¬
нято определение комплекса по А. К. Бабко, хотя
имеются и другие определения, кроме рассмотренных.
В определении А. К. Бабко сказано еще об одной су
щественной особенности комплексов, а именно об их
малой склонности к диссоциации на составные части.
Комплекс — это группа, находящаяся в равновесии в
растворе с теми частицами, из которых она образова¬
лась. Диссоциация комплексной группы может изме¬
няться в очень широких пределах, однако нередко она
проходит в незначительной степени, причем именно
комплексы с малой склонностью к диссоциации наи¬
более важны для анализа.§ 2. ДИССОЦИАЦИЯ И УСТОЙЧИВОСТЬ КОМПЛЕКСОВ
В РАСТВОРАХВ преобладающем большинстве случаев образование
и диссоциация комплексов в растворах происходит
ступенчато. В зависимости от дентатности лигандов и
значения координационного числа центрального ато¬
ма последний способен присоединять несколько ли¬
гандов. Процесс образования комплексных (коорди¬
национных) соединений, т. е. процесс взаимодействия
гидратированных ионов с лигандами, характеризуется
константами равновесия (константами устойчивости).
Если ионы Мп+ взаимодействуют с монодентатным ли¬
гандом и при этом не прЬисходит образование нерас¬
творимых соединений, то состояние системы описы¬
вается следующими выражениями:Mn+ + R- ч=±: MR(n-1)+; =[Mre+][R ][MR(n-l) + ]Pi-[М»+] [R“]239
Образование комплекса с большим координационным
числом можно представить следующим образом:Если максимальное координационное число ионов
металла Мга+ по отношению к лиганду R равно N, та¬
ких уравнений будет также N. В зависимости от харак¬
тера лиганда координационное число может меняться.в котором координационное число кобальта равно 4.
При взаимодействии Со2+ с молекулами аммиака воз-ционное число кобальта равно 6. Аналогично при вза¬
имодействии А13+ с ионами С1- возможно образова¬
ние А1СЦ, т. е. максимальное координационное число
для ионов А13+ равно 4. При взаимодействии же ио¬
нов алюминия с ионами F- образуется ряд комплек¬
сов с координационным числом от 1 до 6: A1F2+,..., A1F6-, т. е. максимальное координационное число
ионов алюминия равно 6. Таким образом, координа¬
ционное число является не только свойством металла,
но также зависит от свойств лиганда. Между констан¬
тами устойчивости, или константами образования KiMR(n-l)+ + R- MR^_2)+; K2[MR(2n~2|+]илиMr(*-2)+ + r- ^ MR f3n~3) + , K3Mn+ + 2R~ MR^-2)+. p2[MR2<n -2,+]
[Mn+][R“]2 ’
[MR(3n_3,+l[MR'2ra-2,+j [R 1ИЛИИЛИТак, известен хлоридный комплекс кобальта СоС124-можно образование Co(NH3)2+, в котором координа-240
(Ки Кг, ... > Ki) и суммарной константой образования
(суммарной константой устойчивости p((0i, Р2, ■.., Pi)
существует следующая простая зависимость:= *1*2*3 ••• **ИЛИР,< = *,*2*3 ■■■ *к“П *-i =■ 1Иногда устойчивость комплексов характеризуют
константами диссоциации или константами нестойко¬
сти, т. е. обратными величинами констант устойчиво¬
сти:MR'-"* ,=* М"+ + R‘; K.-MlLfcj:+R-; «г-МГ^Ж1и т. д. Суммарная константа нестойкости комплекса
/(общ. равна произведению промежуточных констант
нестойкости:/С,А;2АГз ... Ki^Koem-Из этих уравнений видно, что чем меньше констан¬
та нестойкости комплекса, тем комплекс более про¬
чен и наоборот.Знание констант образования комплексов Ki поз¬
воляет характеризовать состояние комплексных ча¬
стиц в растворе в зависимости от концентрации ионов
металла и лигандов. В качестве примера приведем об¬
разование роданидных комплексов железа (III) в за¬
висимости от концентрации роданид-ионов (рис. 13.1).
Так, при содержании ионов железа(Щ), равном
10-3 М и избытке роданид-ионов порядка 5-10-2 М
состояние равновесия следующее: около 10 % общего
количества ионов железа остается свободным (гидра¬
тированные), около 25 % связывается в FeSCN2+, око¬
ло 30 % — в Fe(SCN)2 и часть образует комплексы
Fe(SCN)3, Fe(SCN); и др.241
Рис. 13.1. Образование же-
лезороданидных комплексовв зависимости от концен¬
трации роданид-иона:/ — свободные иоиы железа;
2—6 — роданидные комплексы с
координационным числом от 11,0 д° 6-lg [SCN ] /моль/л)Или другой пример:Cd2* + NH3=ё=* [Cd(NH3)]2t;К = 102'45;[Cd(NH3)]Jt + NH3*=* [Cd(NH3)2]2+;K= Ю2'10;[Cd(NH3)2]2+ + NHj4=fc [Cd(NH3)3]2‘;IIо[Cd(NH3)3]2+ + NH3[Cd(NH3),]2+;K= 10°'93; p4= 107'12.В случае образования цнанидных комплексов кадмия:Cd2+ + CN“^=± [Cd(CN)]+;K= Ю5-"8;[Cd(CN)] + CN~[Cd(CN)a];/С = 10s'2;[Cd(CN)2] -f- CIST 5i=fc [Cd(CN)3]~;К = Ю4’63;[Cd(CN)3r + CN'[Cd(CN)*]2-;tf=103’55; p4=10,e'*aТаким образом, если к раствору соли металла при¬
бавлять раствор лиганда, то сначала образуется про¬
стейший комплекс MR с координационным числом 1,
при дальнейшем прибавлении лиганда образуется
MR2, т. е. концентрация комплекса MR падает, а
MR2 — увеличивается, пока основной формой не ста¬
нет комплекс MRo. Последующее увеличение концен¬
трации лиганда R приводит к образованию комплекса
MR3 и т. д. до тех пор, пока не образуется MRn, где
п — максимальное координационное число для дан¬
ной системы.В аналитической химии, как правило, пользуются
концентрационными константами, т. е. константами,
выраженными через концентрации С соответствую¬
щих частиц. Однако значения концентрационных кон¬
стант зависят от ионной силы раствора.Связь между концентрационными и термодинами¬
ческими константами выражается следующим обра¬
зом.242
Активность частиц в растворе равна концентра¬
ции, умноженной на коэффициент активности, кото¬
рый обычно меньше единицы: а — fC. В случае комп¬
лекса MR3 : Ан = /м [М], Or = /r [R], аш!3 = fmr3 [MR3].
Тогдая [MR3] aMR3 Wr
[M] IR13 fMRjно так как pj = , где pj — термодинамическаяaMaRконстанта устойчивости, тойт Wft
РЗ — РЗ fIMR3ИЛИT д ?MRsmIrРз — Рз f ,3
1/Л1Константы устойчивости (нестойкости) являются
фундаментальными количественными характеристи¬
ками прочности связи между ионами металлов и ли¬
гандами в растворе подобно тому, как константы дис¬
социации кислот или произведения растворимости
труднорастворимых соединений характеризуют соот¬
ветственно прочность связи аниона кислоты с прото¬
нами или аниона кислоты с катионом металла в
осадке.Физический смысл констант нестойкости можно
выяснить, записав, например, выражение для общей
константы нестойкости комплекса:[М] [R]3AV[MR3Если принять, что общая концентрация комплекса и
равновесная концентрация лиганда )авны единице,
тогдаКн = [М],т. е. при указанных стандартных условиях константа
нестойкости численно равна концентрации свободных
ионов металла в растворе комплекса. Чем меньше
константа нестойкости, тем прочнее связь ионов ме¬
талла с лигандом R и тем меньше концентрация
ионов металла в растворе.243
Физический смысл константы устойчивости легко
понять из ее выражения:, IMRsl
Р [М] [R]3 ‘Допустим, что равновесные концентрации комплекса
и ионов (молекул) лиганда равны единице, тогдаРз =[М] •Эта величина будет тем большей, чем меньше концен¬
трация свободных ионов [М]. Таким образом, кон¬
станта устойчивости комплексного соединения в при¬
нятых «стандартных» условиях численно равна еди¬
нице, деленной на концентрацию свободных ионов
металла в растворе.При слишком больших константах устойчивости
(малых константах нестойкости) понятие свободных
ионов металла в растворе комплекса практически те¬
ряет смысл. Так, константа устойчивости тетраиодид-
ного комплекса ртути [Hgl4]2-. - N'!'![Hg2+][rПри [Г] = 1 и [HglM = l [Hg2+] = Ю-30 М.Число ионов металла в 1 М растворе равно
6,02-1023 (число Авогадро), тогда один свободный
ион металла содержится в 1,4-10® л воды, т. е. диссо¬
циация комплексов с константами устойчивости более
6-1023 в водных растворах практически не происходит.
Однако именно большим численным значением кон¬
станты устойчивости можно объяснить тот факт, что
металлическая ртуть, которая не растворяется в кис-
лотах-неокислителях (H2S04, НС1) с выделением во¬
дорода, легко растворяется в этих кислотах в присут¬
ствии KI или в иодистоводородной кислоте с выделе¬
нием водорода:Hg + 4HI =г=* H2[HgI4] + H2f.Отсюда можно сделать вывод, что металлическая
ртуть, залитая водой, дает больше свободных ионов
ртути, чем комплекс [Hgl^2-.244
Физический смысл ступенчатых констант нестой¬
кости можно выяснить, записав следующее выраже¬
ние:[MR11R][MR2]и, если [R] = К», то [MR] = |MR2]. Иными словами,
ступенчатая константа нестойкости комплекса чис¬
ленно равна такой равновесной концентрации лиган¬
да, при которой одна половина общего содержания
ионов металла находится в форме комплекса MR, а
другая в форме MR2i т. е. такой концентрации лиган¬
да, при которой одна половина общего количества
ионов металла наполовину перешла в другую форму,
присоединив (или утратив) один лиганд.Из термодинамики известно наиболее общее выра¬
жение, которое характеризует физический смысл кон¬
стант устойчивости:Л(?о = R1 In К ycTiгде Д00 — изменение свободной энергии (изобарно-изотермиче¬
ского потенциала) при образовании комплекса.Образование устойчивых комплексов связано со
значительным изменением свободной энергии; для ма¬
лопрочных комплексов это изменение невелико.Уравнение константы устойчивости (нестойкости^
показывает, что она является наиболее объективной
количественной характеристикой устойчивости комп¬
лексов в растворе или энергии химической связи ме¬
жду частицами, образующими комплекс.§ 3. ФУНКЦИИ ОБРАЗОВАНИЯ
И ЗАКОМПЛЕКСОВАННОСТИФункция образования п — это отношение концентра¬
ции связанного в комплексе лиганда ч общей концен¬
трации ионов металла; функция образования показы¬
вает среднее число лигандов, приходящееся на один
ион металла в комплексе:cR- [R1« = —гг •Функция образования может изменяться от нуля до
максимального значения координационного числа.
Так, если Сr= [R], то п = О, следовательно, в рас¬245
творе нет комплексообразования. При n = N, где N —
координационное число, в растворе находится только
один комплекс MR/v. Функция образования может
иметь дробное значение, если в растворе сосущест¬
вует несколько комплексов разного состава. Опре¬
делив экспериментально функции образования,
можно найти ступенчатые константы устойчивости
комплексов.Иногда пользуются функцией закомплексованно¬
сти Ф, которая представляет собой отношение общей
концентрации ионов металла к его равновесной кон¬
центрации:Функция закомплексованности может изменяться
от единицы до бесконечности. Если в растворе не про¬
исходит комплексообразование, тогда См — [М] и
Ф =1. Если ионы металла практически полностью
связаны в комплекс, т. е. концентрация свободных
ионов металла очень мала, следовательно Ф—voo.Обычно реакции комплексообразования проходят
быстро. Однако в некоторых случаях достижение со¬
стояния равновесного процесса комплексообразования
проходит довольно медленно, например при образо¬
вании гетерополикислот, при образовании большин¬
ства комплексов с платиновыми металлами и в дру¬
гих случаях. Константы же, как термодинамические,
так и концентрационные характеризуют строго равно¬
весные процессы при данной температуре. Таким об¬
разом, константы устойчивости (нестойкости) комп¬
лекса зависят только от природы вещества, от при¬
роды растворителя и всегда, для любого раствори¬
теля, от температуры.Иногда устанавливают «константы» при опреде¬
ленной температуре, но не достигая равновесия про¬
цесса, т. е. для определенного момента времени. Од¬
нако такие величины нельзя называть константами,
так как скорость процесса в сильной мере зависит от
изменения концентраций, посторонних веществ и дру¬
гих факторов. В то же время эти «константы» иногда
дают возможность с большим приближением предви¬
деть направление процесса и использовать данные
реакции в анализе.246
§ 4. ЗАВИСИМОСТЬ УСТОЯЧИВОСТИ КОМПЛЕКСОВ
В РАСТВОРАХ ОТ ПОЛОЖЕНИЯ ЦЕНТРАЛЬНОГО АТОМА
В ПЕРИОДИЧЕСКОЙ СИСТЕМЕ ЭЛЕМЕНТОВСвойства катионов металлов, образующих комплексы,
зависят от электронной конфигурации этих катионов,
а следовательно, и от положения соответствующих
элементов в периодической системе. Выделяют три
группы катионов.1. Катионы с электронной конфигурацией типа
инертного газа, у которых во внешней электронной
оболочке находится 2 или 8 электроноЕ;: Li+, Na+, К+,
Rb+, Cs+, Ве2+, Mg2+, Са2+, Sr2+, Ва2+, Al3+, Sc3+, Y3+,
La3+, TiIV, Zr'v, Hflv, а также ионы нисбил [NbO]3+ и
танталил {ТаО]3+.2. Катионы с недостроенной d-оболочкой: VIV, V1",
Сг111, Mn", Fe11, Со", Со111, Ni", Си11, а также катионы
платиновых металлов (рутения, родия, палладия, ос¬
мия, иридия и платины).3. Катионы, у которых во внешней электронной
оболочке находится 18 или 18 + 2 электронов: ZnM,
Cd", Hg", Gam, In'", Tl‘", GeIV, Sn'\ Asv, Sbv, ТГ,
Sn", Pb", As"1, Sb"1 и Bi1".Катионы первой группы образуют комплексы со
значительной долей ионной связи. Устойчивость оболо¬
чек типа инертных газов обусловливает малую поля¬
ризуемость и малую деформацию внешних электрон¬
ных оболочек при взаимодействии с различными ли¬
гандами. Поэтому катионы названного типа можно в
первом приближении рассматривать как жесткие ша¬
рики с положительным зарядом в центре, взаимодей¬
ствующие с лигандами в результате электростатиче¬
ского притяжения, силу которого определяют по за¬
кону Кулона:„ Nile2где N и и — заряды центрального иона н лигандов; с — заряд
электрона; г — расстояние между центрами положительно и от¬
рицательно заряженных частиц.Прочность комплексов, образованных такими ка¬
тионами, в значительной степени определяется заря¬
дом и радиусом этих частиц. Можно пользоваться и
объединенной величиной (т. е. отношением заряда к
радиусу Zjr), которую называют ионным потенциалом.247
Таблица 13.1. Устойчивость комплексов состава MR
в зависимости от радиусов катионов с электронной
конфигурацией типа инертного газа (приведены lgPi)ИоныРади¬усы,нм!gP( комплексов с лигандамиОНЭДТАОсиXиосии :СНзСНОНСОО"Ацетил-ацетонВе2+0,037,5_ Mg2+0,082,6—————Са2+0,111,310,960,778,180,82—Sr2+0,130,828,630,446,730,70—Ва!*0,140,647,630,416,410,55—Sc1+0,0812,323,1—_—8,0уз +0,11—18,1_——6,4La3+0,123,315,5———5,1Ионный потенциал характеризует напряженность
электрического поля данного иона.В табл. 13.1 приведены данные о зависимости
устойчивости комплексов простейшего состава MR от
заряда и радиуса катионов.Из данных таблицы видно, что комплексы тем
прочнее, чем меньше радиус катиона.Аналогичные зависимости наблюдаются для комп¬
лексов щелочноземельных элементов со многими дру¬
гими кислородсодержащими лигандами. В ряду скан¬
дий — иттрий — лантан изменение устойчивости также
связано с размерами катионов комплексообразо-
вателей.Из данных табл. 13.1 следует, что при равных ра¬
диусах катионов комплексообразователей устойчи¬
вость комплексов тем больше, чем больше заряд ка¬
тиона. Так, радиусы ионов кальция и иттрия одина¬
ковы, но устойчивость комплексов иттрия с ЭДТА
значительно выше, чем комплекса кальция с этим же
лигандом. Такие соотношения справедливы и для
комплексов ЭДТА со стронцием и лантаном, катионы
которых почти не различаются по размерам, но имеют
неодинаковые заряды.При разных зарядах и радиусах существует хоро¬
шая корреляция между значениями ионных потен¬
циалов и устойчивостью комплексов, образованных
катионами с одинаковой электронной конфигурацией,248
если даже они принадлежат к различным группам пе¬
риодической системы (табл. 13.2).Из данных правой части таблицы следует, что при
разных зарядах и радиусах ионов металлов устойчи¬
вость комплексов не всегда коррелируется с ионными
потенциалами. Прочность гидроксокомплексов увели¬
чивается в ряду бериллий > алюминий > торий, не¬
смотря на уменьшение ионного потенциала в этом
ряду. Заряды ионов и устойчивость лучше коррели-
руются, несмотря на различия в размерах ионов.Для катионов с недостроенной d-оболочкой харак¬
терно образование комплексов двух типов. Одни из
них, а именно двухзарядные катионы элементов чет¬
вертого периода, образуют обычные гак называемые
лабильные комплексы, у которых равновесие между
частицами в растворе устанавливается очень быстро,
как и у рассмотренных выше комплексов катионов с
оболочкой типа инертного газа. Трехзарядные катио¬
ны платиновых металлов, хрома и кобальта часто об¬
разуют стабильные комплексы. Стабильность в дан¬
ном случае — это не термодинамическая устойчивость,
а кинетическая инертность, вследствие чего находя¬
щиеся в растворе комплексы существуют в неравно¬
весном состоянии. Истинное равновесие устанавли¬
вается нередко очень медленно, в течение нескольких
суток или месяцев. Поэтому константы устойчивости
комплексов этой группы металлов определены только
для небольшого числа соединений, что затрудняет вы¬
яснение закономерностей устойчивости. В дальнейшем
будут рассмотрены только комплексы элементов чет¬
вертого периода, а именно комплексы катионов мар¬
ганца, железа, кобальта, никеля, меди и цинка.Таблица 13.2. Устойчивость комплексов МОН,
образованных катионами с различными зарядами
и радиусами, в зависимости от ионного потенциалаИоныРадиус,нмЗарядИонныйпотен¬циал,эВР.ИоныРадиус,нмЗарядИонныйпотен¬циал,эВе,1.Г0,0811,280,3Ве2+0,0325,907,0Са2+0,1121,891,5А13+0,0535,259,0уЗ +0,1132,827,0Thiv0,1143,6210,0249
Для катионов с недостроенной 18-электронной обо¬
лочкой в меньшей степени применимы простые элек¬
тростатические представления, основанные на законе
Кулона. Такие электронные оболочки при действии
электроотрицательных лигандов деформируются зна¬
чительно больше, чем 8-электронные оболочки катио¬
нов, и доля ковалентности химической связи металл —
лиганд сильно возрастает. Изменение устойчивости
комплексов элементов четвертого периода можно объ¬
яснить с позиций усовершенствованной электростати¬
ческой теории, которая принимает во внимание не
только чисто кулоновское взаимодействие между ча¬
стицами, но и форму орбиталей d-электронов. Речь
идет о теории кристаллического поля, созданной в
30-х годах этого столетия физиками Г. Бете и Ван-
Флеком и позже примененной химиками для объясне¬
ния спектров поглощения и магнитных свойств комп¬
лексов переходных металлов.Рассмотрим экспериментальные данные о констан¬
тах устойчивости комплексов переходных ^-элементов.В основном устойчивость комплексов изменяется
следующим образом:Mn2* < Fea* < Со2+ < Ni2+ < Cu2* > Zn2+.Это так называемый ряд устойчивости Ирвинга —
Вильямса. Приведенные в табл. 13.3 константы устой¬
чивости комплексов (lg Pi) подтверждают указанную
закономерность.Таблица 13.3. Усгойчипость комплексов MR (lg (3)
переходных элементов (ряд Ирвинга—Вильямса)•Лигандыleft( комплексом металловMn2 +Fe2tCo2 +n,2+Cu2*•3 2»2 nОН"43,03,94,44,66,54,4С О2'3,94,74,75,36,24,9NH,NH2CHaCOO'NH2CH2Clb\H2ЭДТАCHjCOCHjCOCH,0,83,42,713,54,21,44,314,52,15,25,916,15.42,85,97,718,56,14.2
8,410,618,98.32.45.5
5,716,65,1Ш
Из данных таблицы следует, что константы устой¬
чивости увеличиваются при переходе эт марганца к
железу, кобальту и никелю, достигают максимального
значения у комплексов меди и снова уменьшаются
при переходе от меди к цинку. Такие соотношения ха¬
рактерны и для комплексов этих металлов со мно¬
гими другими лигандами.Объяснить закономерности, выраженные рядом
Ирвинга — Вильямса, можно с позиций теории кри¬
сталлического поля. Эта электростатическая теория не
принимает во внимание особенности строения элек¬
тронных оболочек лигандов, а рассматривает послед¬
ние как точечные отрицательные заряды, располагаю¬
щиеся в определенной геометрической конфигурации
вокруг центрального катиона с положительным заря¬
дом. В отличие от упрощенной электростатической
теории, рассматривающей взаимодейс"вие между то¬
чечными частицами она учитывает энергетические из¬
менения в системе, связанные с формой орбиталей
^-электронов.После взаимодействия центрального иона с лиган¬
дами образовавшийся комплекс имеет меньший запас
энергии, чем отдельно взятые компоненты. В соответ¬
ствии с обычной электростатической теориейЭто уравнение не учитывает изменение энергии
электронов центрального иона под влиянием сил от¬
талкивания, возникающих между этими электронами
и отрицательно заряженными лигандами. В резуль¬
тате отталкивания энергия d-электронов увеличивает¬
ся. На рис. 13.2 в качестве примера горизонтальной
чертой А показан энергетический урсвень rf-электро-
нов свободного центрального иона до взаимодействия
с лигандами, а горизонтальной чертой Б — этот же
уровень после образования комплекса, например
[FeF6]3-.В этой схеме не учтена форма d-орбиталей. Пред¬
полагается, что заряд d-электронов в комплексе рав¬
номерно распределен на поверхности сферы, пред¬
ставляющей собой катион центрального атома. В дей¬
ствительности отдельные электроны центрального
атома в комплексе располагаются на различных251
Рис. 13.2. Энергетические уровни ^-элементов: Fe3+ (Л), FeFjj' (Б)
и после расщепления кристаллическим полем (В)энергетических уровнях. Орбитали d-электронов, или
конфигурации электронных облаков, имеют форму,
изображенную на рис. 13.3.Без внешних воздействий, например в свободном
ионе металла, все орбитали являются вырожденными,
т. е. они энергетически равноценны. Однако под влия¬
нием зарядов лигандов вырождение снимается, и элек¬
троны, занимающие различные d-орбитали, становят¬
ся в энергетическом отношении неравноценными. Од¬
ни из них занимают более высокие энергетические
уровни, другие — более низкие. Комплекс железа с
шестью ионами фтора имеет октаэдрическую конфи¬
гурацию. Шесть лигандов занимают места в верши¬
нах октаэдра, т. е. на осях координат х, у и z, вдоль
которых вытянуты электронные облака dA---y= и d?.
Энергия электронов, находящихся на этих орбиталях,
возрастает по сравнению с энергией, которую имели
бы эти электроны в комплексе, если бы их заряд был
распределен равномерно на поверхности сферы (уро¬
вень Б, см. рис. 13.3). Наоборот, энергия dKy, d2X и dzy
электронов уменьшается но сравнению с энергией, по¬
казанной на рис. 13.3, уровнем Б, так как их элек¬
тронные облака находятся в пространстве между ося¬
ми координат и испытывают меньшее отталкивание
под влиянием отрицательно заряженных лигандовУZУгzРис. 13.3. Конфигурация электронных облаков d-электронов
фторид-ионов, вследствие этого энергетический уро¬
вень Б (заряд электронов равномерно распределен на
поверхности сферы) расщепляется на два подуровня,
один из которых (более низкий) занимают электроны
с конфигурацией dxy, dxz и dzyу а другой (более высо¬
кий) — электроны с конфигурацией d,<a-y- и dx> (уро¬
вень В, см. рис. 13.2). Более высокий и более низкий
энергетические уровни принято обозначать символами
ее и Не¬
согласно теории химической связи, если разность
энергий уровней ее и t2g обозначить Л, то ее уровень
будет расположен на 0,6Д выше, а уровень t2g — на
0,4Л ниже, чем гипотетический уровень d-электронов,
расположенных на поверхности сферы (Б-уровень).
Появление уровней es и Hg под влиянием взаимодей¬
ствия с лигандами называют расщеплением кристал¬
лическим полем, а величину 0,4Д — энергией стабили¬
зации кристаллическим полем.В рассмотренном примере расщепление кристалли¬
ческим полем не приводит к какому-либо выигрышу
энергии. Выигрыш энергии при переходе трех элек¬
тронов на уровень t2g составляет 0,4Д-3=1,2Д. Од¬
нако проигрыш энергии вследствие перехода двух
электронов на уровень ее равен также 0,6Д-2 = 1,2Д,
т. е. эти противоположные эффекты компенсируются.
Так бывает не всегда. В табл. 13.4 сведены данные об
энергии стабилизации кристаллическим полем окта¬
эдрических комплексов катионов элементов четвер¬
того периода.На каждой d-орбитали может расположиться не
более двух электронов с противоположными спина¬
ми. Заполнение орбиталей электронами происходит
в соответствии с правилом Гунда: для электроновТаблица 13.4. Энергии стабилизации кристаллическим
полем комплексов некоторых переходных металловКатионИп2+fa2*Со2+Niz+Си2*Znz+Число Л-электронов
Эмргегмесмм уромнь *дЭмргетссшй урмаиь te,
Энергия стабиаизации, з85ЕШ6гггп7ЕШiЕШEISEE1.29ЕШ10ЕШГП7ГПlUltlflluluitlIHlMNlimImImi' 00.40,80.60253
энергетически выгоднее занимать свободные орби¬
тали, чем орбитали, занятые электронами. Однако
если отрицательно заряженные лиганды создают
сильное поле, последовательность размещения может
быть иной.Из данных табл. 13.3 следует, что энергия стаби¬
лизации максимальна у никеля и уменьшается в обе
стороны от него, т. е. комплексы никеля должны быть
прочнее, чем комплексы марганца, железа и кобальта,
а также меди и цинка. Однако наиболее прочные ком¬
плексы образует медь (см. табл. 13.3). Это объясняет¬
ся искажением октаэдрической структуры комплексов
меди. Выводы из данных этой таблицы основаны на
предположении, что все комплексы имеют строго ок¬
таэдрическую конфигурацию, т. е. лиганды располо¬
жены в вершинах октаэдра, совпадающих с электрон¬
ными орбиталями dx--Y} и dzкоторые вытянуты
вдоль осей координат.Для третьей группы катионов (во внешней элек¬
тронной оболочке находится 18 или 18 + 2 электро¬
нов) характерны иные зависимости. Большое число
электронов во внешней оболочке способствует их
сравнительно легкой деформируемости и поляризуе¬
мости. Жесткость электронной оболочки не так ве¬
лика, как у катионов первой группы. В комплексах
катионов третьей группы преобладает ковалентная
связь, осуществляемая парой электронов, находящих¬
ся в совместном владении катиона металла и лиганда.
Поэтому во многих случаях изменение устойчивости
комплексов катионов элементов одной и той же груп¬
пы периодической системы хорошо коррелирует со
способностью этих катионов к образованию ковалент¬
ной связи. С количественной стороны способность к
образованию ковалентных связей можно описать ко¬
валентной характеристикой, предложенной К. Б. Яци-
мирским для объяснения растворимости некоторых
малорастворимых соединений. Ковалентная характе¬
ристика представляет собой разность между энергией
ионизации атома и теплотой гидратации образующе¬
гося иона. Чем больше энергия ионизации, тем боль¬
ше энергии выделяется при обратном процессе — при¬
соединении к иону электронов, которые отдает лиганд
при образовании комплексного иона. С другой сто¬
роны, чем меньше теплота гидратации, тем меньше254
Таблица 13.5. Устойчивость комплексов MRи ковалентная характеристикаИоныг, нмК.кДж/мольlg Р, комплексов с лигандамиF~сгГЭДТАОН'Zn1*0,08565,7 —0,2-1,316,54,4Cd2+0,10666,2—1,32,316,52,3HgI+0,11955,3—5,312,921,810,3Ga3+0,06917,65,1—-20,310,6In3+0,08946,93,7—— •24,910,3энергии необходимо затратить на дегидратацию иона
и тем легче лиганду вытеснить вод> из внутренней
сферы иона металла и соединиться с him.Несмотря на преобладание 1%овалгнтной связи в
комплексах катионов металлов третьей группы имеют
значение также факторы, определяющие устойчивость
комплексов с преимущественно электровалентным ха¬
рактером связи, в частности заряды и радиусу частиц.
Поэтому ярко выраженной зависимости от какой-либо
одной из названных характеристик обычно не наблю¬
дается.В табл. 13.5 сопоставляются значения lg Pi некото¬
рых катионов третьей группы.Из данных табл. 13.5 следует, что устойчивость
комплексов цинка, кадмия и ртути с хлорид-, иодид-
ионами и ЭДТА, а также комплексов галлия и индия
с ЭДТА хорошо согласуется с величиной ковалентной
характеристики. Несмотря на увеличение радиусов
катионов устойчивость комплексов во:растает. Это по¬
казывает, что степень ковалентности связи имеет боль¬
шее значение, чем размеры ионов. Однако устойчи¬
вость гидроксокомплексов не подчиняется уже сфор¬
мулированному правилу. Устойчивость фторидных
комплексов галлия и индия также изменяется в об¬
ратной последовательности, т. е. определяется в боль¬
шей степени размерами ионов, чем ковалентной ха¬
рактеристикой.§ 5. ЗАВИСИМОСТЬ УСТОЙЧИВОСТИ КОМПЛЕКСОВ
В РАСТВОРАХ ОТ СВОЙСТВ ЛИГАНДОВУстойчивость комплексов в сильной мере зависит от
свойств лигандов, вступающих во взаимодействие с255
ионами металлов. И многих свойств лигандов можно
выделить основные, от которых в значительной сте¬
пени зависит прочность комплексов: 1) размеры ли¬
гандов и способность к образованию ковалентных свя¬
зей; 2) положение донорного атома лиганда в перио¬
дической системе; 3) способность к образованию
замкнутых циклов — хелатный эффект; 4) стерические
факторы.Донорными атомами в лигандах бывают чаще все¬
го атомыN о F
Р S С1
As Se Вг
Sb Те !Чаще других приходится иметь дело с галогенидными
комплексами и с комплексами лигандов, содержащих
донорно-активные атомы кислорода, серы и азота.Размеры лигандов и способность к образованию
ковалентных связей. Влияние этих факторов наибо¬
лее ярко проявляется у галогенидных комплексов ме¬
таллов, однако однозначной зависимости комплексов
от названных факторов не существует. Кроме того,
имеет значение то, с каким металлом образуется га-
логенидный комплекс.Для комплексов катионов металлов первой группы
(во внешней электронной оболочке находится 2 или
8 электронов) и для некоторых переходных металлов
(с недостроенным d-подуровнем) основным фактором
является размер лигандов. Фторидные комплексы
прочнее, чем хлоридные, а хлоридные прочнее бро-
мидных и иодидных. Так, бериллий, магний, алюми¬
ний, лантан, цирконий образуют прочные фторидные
комплексы (lg- Pi равны соответственно 4,3; 1,3; 6,1;
2,8; 8.8); устойчивость же комплексов названных эле¬
ментов с хлорид-, бромид- и иодид-ионами невелика
или они вообще не образуются. Из переходных ме¬
таллов такая же закономерность наблюдается, напри¬
мер, для железа и марганца; устойчивость фторидных,
хлоридных и бромидных комплексов этих металлов
характеризуется соответственно числами 5,3; 1,5 и
—0,3 (железо); а также 5,5 и 0,96 (марганец).Катионы металлов третьей группы с числом элек¬
тронов на внешней оболочке, равным 18 или 18 + 2,256
Таблица 13.6. Влияние ковалентности связи и размеров
лигандов на устойчивость комплексов MR
(приведены lgpi)lg Р,комплексов металловCd2+Hg2tBi3 +Zn2 +[n3tSn2*F"_ 3,7_сг1,35,32,5—0,251,41,1Вг‘1.79,14,3-0,61,10,7Г2,312,8—— 1,60,3 склонны образовывать комплексы с преимущественно
ковалентной связью. Поэтому здесь преобладающей
является ковалентная характеристика, а также спо¬
собность галогенид-ионов к потере электронов, наи¬
более ярко выраженная у иодид-ионов. В связи с
этим иодидные комплексы прочнее бромидных и хло-
ридных, а фторидные комплексы наименее прочны
или вообще не существуют. Известны случаи и обрат¬
ной последовательности (табл. 13.6).Из данных табл. 13.6 следует, что катионы кадмия,
ртути и висмута (элементов, в наибольшей степени
склонных к образованию ковалентных связей) дают
самые прочные иодидные комплексы. Однако для ка¬
тионов цинка, индия и олова в большей степени про¬
является зависимость от размеров лиггндов — в этой
группе прочнее всего фторидные комплексы, а наиме¬
нее устойчивы иодидные.Положение донорного атома в периодической си¬
стеме. Лиганды, содержащие электронодонорные ато¬
мы кислорода, серы и азота, чаще всего органические
реактивы, образующие хелатные или внутрикомплекс-
ные соединения. Однако можно указать на следующую
закономерность. Для катионов металлов с электрон¬
ной оболочкой типа инертного газа (2 лли 8 электро¬
нов) комплексы с кислородсодержащими лигандами
обычно более устойчивы, чем комплексы, содержа¬
щие лиганды с донорными атомами серы. Наоборот,
катионы металлов, на электронной оболочке которых
18 или 18 + 2 электронов, дают более устойчивые
комплексы с серосодержащими лигандами, чем9 Аналитическая химия257
Таблица 13.7. Устойчивость аммиачных и этилендиаминных
комплексов никеля (Mien)КомплексКомплексin P|Ni(HN3)2p5,0[Nien]2*7,5[Ni(NH3)*]2+7,9[Ni(en)2p13,9[Ni(NH3)6]2*8,6[Ni(en)3l2*18,3комплексы, образованные лигандами с донорными
атомами кислорода.Хелатный эффект. Хелаты металлов (комплексы
с замкнутыми циклами) устойчивее, чем комплексы с
аналогичными монодентатными лигандами. Это явле¬
ние получило название хелатного эффекта. Понятие
«хелатный эффект» было введено Т. Шварценбахом
в 1952 г. для того, чтобы отразить явление относи¬
тельно более высокой устойчивости хелатов металлов
по сравнению с аналогичными комплексами металлов
с монодентатными лигандами или с хелатообразую-
щими лигандами, но с меньшим числом хелатных цик¬
лов, содержащих те же донорные атомы. Так, ам¬
миачные комплексы металлов менее устойчивы, чем
комплексы этих металлов с этилендиамином, несмотря
на то, что координированные частицы содержат оди¬
наковое число атомов азота, присоединенных к ме¬
таллу. Хелатный эффект подтверждается данными
табл. 13.7.Две молекулы аммиака по числу донорных атомов
азота равноценны одной молекуле этилендиамина, но
устойчивость комплексов разная.Константы устойчивости комплексов связаны с из¬
менением свободной энергии реакций комплексообра¬
зования уравнениемдg0= - RT In /С.Из термодинамики известно следующее уравнение:
AG — АН — Т AS,где Н — энтальпия, или теплота, поглощенная при изобарическом
.процессе; С—энтропия.258
Сопоставляя оба уравнения, можно записать:RT In К = Q + Т AS,где Q — тепловой эффект реакции при постояь ном давлении.Комплекс тем более устойчив, чем больше тепло¬
вой эффект реакции и чем больше изменение
энтропии.По тепловому эффекту реакции присоединение
двух молекул аммиака приблизительно равноценно
присоединению одной молекулы этилендиамина. Од¬
нако из данных табл. 13.7 следует, что устойчивость
комплексов никеля с аммиаком и Э"илендиамином
различна; причина этого заключается в неодинаковом
изменении энтропии в обоих случаях. Действительно,
энтропия характеризует степень упорядоченности си¬
стемы, которая зависит от общего количества частиц
в этой системе. При координировании двух молекул
аммиака проходит реакция[Ni(H20)6]2+ + 2NH3 ч=± [Ni(NH3)2(H20)4]2*+ 2Н20,т. е. число частиц в системе не увеличивается.
Реакция[Ni(H20)6]z* + H2NCH2CH2NH2 :*=►:
ч=± [(H20)4^H2NCH2CH2NrH2)j2* -f 2НгОсопровождается увеличением числа частиц в системе,
что приводит к большему ее беспорядку и соответст¬
венно к большему увеличению энтрош-и, чем при об¬
разовании аммиачных комплексов никеля.Объяснить хелатный эффект (больнее возрастание
энтропийного члена при координировании одного би-
дентатного лиганда по сравнению с двумя моноден-
татными) можно иначе, основываясь на том, что эн¬
тропия является мерой вероятности состояния систе¬
мы, она возрастает при переходе системы из менее
вероятного состояния в более вероятюе. Замыкание
цикла при координировании бидентатного лиганда
происходит в две стадии. Сначала бидентатный ли¬
ганд присоединяется к иону металла госредством од¬
ного донорного атома. Более вероятно, что вместо
присоединения второго лиганда цикл будет замыкать¬
ся, что обусловлено присоединением к иону металла
второго донорного атома той же молекулы лиганда.9*259
Вероятность второй стадии больше, так как второй
донорный атом закрепленного одним концом лиганда
свободен меньше, чем незакрепленного, и находится
значительно ближе к иону металла, чем незакреплен¬
ные молекулы лигандов, находящиеся в растворе.
Большая вероятность процесса обусловливает боль¬
шее возрастание энтропии, а следовательно, и образо¬
вание более прочного комплекса.Устойчивость хелатов зависит также от числа ато¬
мов в цикле. Наиболее устойчивы пятичленные цик¬
лы; комплексы с меньшим или большим числом ато¬
мов в кольце характеризуются меньшей прочностью.
Например, у комплекса меди с этилендиамином
H2NCH2CH2NH2(en) в цикле находится пять атомов
(вместе с атомом металла), а с пропилендиамином
H2NCH2CH2CH2NH2(pn)—шесть. В соответствии с
этим устойчивость комплексов с этилендиамином вы¬
ше, чем комплексов с пропилендиамином. Влияние
числа атомов в цикле на устойчивость комплексов
видно из следующих данных:Комплексы . . [Cu(en)]2+ [Cu(en)2]2+ [Cu(pn)]2+ [Cu(pn)2]2+
IgP 10,7 20,0 10,0 17,2На устойчивость комплексов влияет также число
циклов: возрастание числа циклов приводит к увели¬
чению прочности. Приведем в качестве примера ком¬
плексы меди с этилендиамином H2NCH2CH2NH>(I),
диэтилентриамином H2NCH2CH2NHCH2CH2NH2(II) и
триэтилентетр аминомВо всех трех случаях медь координирует по од¬
ному лиганду, но этилендиамин присоединяется к ме¬
ди двумя атомами азота, образуя один пятичленный260H2NCH2CH2NHCH2CH2NHCH2CH2NH2(III):2 +H2Ov у.он2
''Си'H2rf 'nh2
\ /
Н2С— сн2
цикл; диэтилентриамин координируется тремя ато¬
мами азота, образуя два пятичленных цикла, а три-
этилентетрамин связан с медью четырьмя атомами
азота и замыкает три пятичленных цикла. В соответ¬
ствии с этим наиболее устойчив комплекс(III), а наи¬
менее— комплекс(1). Их прочность характеризуется
такими значениями lgp: 10,72(1); 15,9(11); 20,5(111).Стерические факторы. Введение в молекулу ли¬
ганда радикалов большого объема нередко затрудняет
координирование лиганда и уменьшает прочность
комплекса. Так, этилендиамин образует с металлами
более устойчивые комплексы, чем тетраметилэтилен-
диамин, (СНз^МСНгСНоЩСНз^, Метильные ради¬
калы создают стерические затруднения и ослабляют
связь атомов азота с ионами металла.'■■J у. '■§ 6. ВЛИЯНИЕ КОНЦЕНТРАЦИИ КОМПОНЕНТОВ, pH,
ИОННОЙ СИЛЫ РАСТВОРА, ТЕМПЕРАТУРЫ
НА КОМПЛЕКСООБРАЗОВАНИЕВлияние концентрации компонентов. Образование ком¬
плексов обычно проходит ступенчато, поэтому в рас¬
творе всегда присутствуют свободные ионы металла и
свободные лиганДы, а также все промежуточные ком¬
плексные соединения:М, MR, MR2, MR3 MR„.Процентное содержание каждой из форм зависит
от прочности (от константы устойчивости) комплекс¬
ного соединения, устойчивости промежуточных комп¬
лексов, а также от концентрационных условий. При
одной и той же устойчивости комплексов содержание
комплексов с большим координационным числом бу¬
дет тем больше, чем больше концентрация лиганда.
Это хорошо видно из выраженияA [MRnlРп [Ml [RIя ’При постоянстве р,г чем больше [R], тем меньше кон¬
центрация ионов металлов.Влияние pH, ионной силы и температуры. Влияние
концентрации водородных ионов на комплексообразо-
вание проявляется двояко. Это влияние зависит от со¬
става комплекса. В связи с этим слгдует отдельно261
рассмотреть комплексы, в состав которых входят
анионы сильных кислот, и комплексы, в состав кото¬
рых входят анионы слабых кислот.Комплексы, в состав которых входят
анионы сильных кислот. Повышение кислот¬
ности раствора (снижение pH) в этом случае практи¬
чески никакого влияния не оказывает, так как силь¬
ные кислоты в водных растворах практически пол¬
ностью диссоциированы:HR н* + R”и поэтому повышение концентрации водородных ионов
влияет только на изменение ионной силы раствора.
Повышение кислотности раствора в разумных преде¬
лах не влияет практически на процесс комплексооб¬
разования.Понижение кислотности (повышение pH раствора)
приведет к нежелательному процессу — гидролизу
комплексных ионов. Однако гидролиз последних про¬
ходит в меньшей мере по сравнению с простыми
ионами металлов. В самом деле, гидролиз простых
(гидратированных) ионов металла Мп+ (если не при¬
нимать во внимание ступенчатый характер процесса)
выражается уравнениемМган_ + rtHjO = М(ОН)„ + пН+. (13.1)тв. фазаКонстанта гидролиза равнаГн+1П*'-тЫг (13-2)или, если числитель и знаменатель уравнения (13.2)
умножить и разделить на концентрацию гидроксиль¬
ных ионов в степени п, получим[нТ [ОН'Г /7. 0
Кг= [мп+][онТ = ПРМ(ОН)^ ’ (133)где / — ионное произведение воды; ПР — произведение раствори¬
мости осадка.Из уравнения (13.3) видно, что при постоянной тем¬
пературе гидролиз будет протекать тем интенсивней,
чем меньше произведения растворимости (точнее ак¬
тивности) осадка.262
Однако в том случае, когда катионы металла Мп+
связаны в комплекс MR, получим иную зависимость.
Уравнение гидролиза следует записать так:и константа гидролиза, следовательно, равнагде Р — константа устойчивости комплекса MR.Учитывая, что 0 обычно больше единицы, заклю¬
чаем, что гидролиз комплекса MR проходит в мень¬
шей степени, чем гидролиз простых (гидратирован¬
ных) ионов металла.Физический смысл этого не трудно понять, так как
ясно, что образование комплексного соединения бло¬
кирует центральный ион от присоединения гидроксиль¬
ных ионов — от образования труднорастворимого ги¬
дроксида. Тем не менее повышение pH раствора все
же приведет к гидролизу и комплексных ионов. По¬
этому реакцию образования комплексов, в состав ко¬
торых входит анион сильной кислоты, лучше прово¬
дить в кислой среде.Комплексы, в состав которых входит
анион слабой кислоты. Рассмотрим, что про¬
изойдет, если комплексы образуются е, кислой среде.
В этом случае анионы слабой кислоты будут связы¬
ваться в слабодиссоциированные молекулы:и для обеспечения соответствующей концентрации ли¬
гандов R"- (для образования комплекса MR) необхо¬
димо повышать концентрацию кислоты H„R, что не
всегда возможно, так как это ограничивается раство¬
римостью слабой кислоты. Поэтому говышение кис¬
лотности раствора (снижение pH) пркводит к разру¬
шению комплексных соединений, в состав которых
входит анион слабой кислоты:MR + «Н20 = М(ОН)„ + Rn~ + пН*(13.4)тв. фазаПРМ(он,„Р 'R"- + гсН+ *=± H„R,(13.5)(13.6)MR + лН* ?=+ Mn+ + H„R.(13.7)263
Эффект этот будет тем сильней, чем меньше кон¬
станта диссоциации кислоты, анион которой входит в
состав комплекса. Разложение комплекса будет про¬
ходить тем в большей мере, чем больше концентра¬
ция водородных ионов (чем ниже pH раствора).Снижение концентрации водородных ионов (повы¬
шение pH раствора) приводит к двум процессам: ра¬
стет концентрация ионов-лигандов, что приводит к
увеличению концентрации комплекса, а также к обра¬
зованию комплексов с большим координационным
числом; увеличивается гидролиз комплексных ионов,
в результате чего образуются гидроксокомплексы' и
гидроксиды металлов:повышение pH i ► MOHRn_i, M(OH)2Rrt_2 М(ОН)пMR„ 1—► MR2, MR3 MR„Реакция гидролиза в этом случае имеет видMR + nHOH ч=* [М(ОН)„] + H„R. (13.8)ОтсюдаК — [HnR] (13 9)л г [MR] - (id.»)Если числитель и знаменатель уравнения (13.9) ум¬
ножить и разделить на концентрацию водородных ио¬
нов [Н+], концентрацию гидроксильных ионов [ОН-],
ионов металла [М+], ионов лиганда [R-] в соответст¬
вующих степенях, полупим:к = [HT[OHT|HnR][Mn1[Rn1 _ /^QpMR[H+]" [R"-] [MR] [M"+] [OH ]“ nPM(OH)nKHnR '(13.10)Таким образом, гидролиз комплексного соедине¬
ния, в состав которого входит анион слабой кислоты,
проходит тем в большей мере, чем слабее кислота
(чем меньше константа диссоциации), анион которой
входит в состав комплекса, и тем слабее, чем прочнее
комплекс (чем больше константа устойчивости ком¬
плекса), подвергающийся гидролизу.264
§ 7. важнейшие комплексные соединения
с неорганическими лигандами и применение
их в анализеКомплексы металлов с неорганическими лигандами
широко применяют как в качественном, так и в коли¬
чественном анализе. Среди таких комплексов имеют
большое значение аммиакаты, галогенидные и род-
анидные комплексы; реже находят применение пер-
оксидные, цианидные, фосфатные, сульфатные и
сульфитные комплексы.Аммиакаты. При взаимодействии ионов металлов
с аммиаком образуются аммиакаты: [Ag(NH3)2]+,
{Cu(NH3)4]2+, [Cd(NH3)4]2+, [Zn(NH3)4]2+,
[Ni(NH3)4]2+, [Co(NH3)4]2+, [Co(NH3)6] 3+ и др.
Очень малоустойчивый аммиакат образует хром (III)
[Cr(NH3}6]3+. Аммиакаты металлов применяют глав¬
ным образом для разделения ионов. Аммиакат меди
широко используют для обнаружения меди, так как
он окрашен в интенсивно синий цвет. Аммиакаты ни¬
келя и кобальта также окрашены в слабый зеленова¬
тый и темно-красный цвет. Аммиакат хрома образу¬
ется только при большом избытке аммиака и частич¬
но. Если же в растворе присутствуют ионы, которые
осаждаются при действии аммиака в виде гидрокси¬
дов, например железо(Ш), хром полностью осажда¬
ется в виде гидроксида, так что отделение хрома в
виде аммиаката невозможно. Другие ионы, образую¬
щие аммиакаты, широко применяют для их отделения
в виде аммиакатов.Галогенидные и роданидные комплексы. Галоге-
нидные и роданидные комплексы широко используют
для разделения элементов методом экстракции, |а
также в фотометрических методах для определения
ряда металлов. При взаимодействии галогенид- и ро¬
данид-ионов с ионами металлов обэазуются комп¬
лексные соединения, многие из которых характери¬
зуются определенными спектрами поглощения:
[Bib]- — желтого цвета, Fe(SCN)3 и Mo(SCN)5 —
красного, W(SCN)6 — желто-зеленого, [NbO(SCN)4]_
и [Bi(SCN)6]3_ — желтого, [СоС14]2- — голубого или
синего, [Co(SCN)4]2- — синего.Фторидные комплексы. Фториды кальция, свинца,
иттрия, лантана, редкоземельных элементов, строн¬265
ция, бария, алюминия, железа(III) мало раство¬
ряются в воде и часто применяются для отделения от
других ионов и для гравиметрического и титриметри-
ческого определения этих ионов. Однако необходимо
иметь в виду, что повышение концентрации фторид-
ионов часто приводит к образованию комплексов с
большим координационным числом, которые сравни¬
тельно хорошо растворяются в воде. Состав коорди¬
национно-насыщенных фторидных комплексов для
двух-, трех- и четырехзарядных катионов отвечает*2 0 +3 1- +4 2
формулам [MF4] [MF6] ‘ и [MF6] " соответственно.Фторидные комплексы применяют также для разде¬
ления ниобия и тантала.Относительная устойчивость простейших фторид¬
ных комплексов состава MF характеризуется рядом,
установленным А. К. Бабко и М. И. Штокало:
ZnIV > Thlv > La3 к > Nbv > Tav > Al3t > SnIV > Be2+ >
> Fe3t > B3+ > Ga3* > Tl3t > In3*.Для гравиметрического определения кальция при¬
меняют реакцию осаждения его в виде CaF2, свин¬
ца — в форме PbClF. Алюминий и железо отделяют
от титана путем осаждения их в виде Na3[AIFe] (по
данным И. В. Тананаева, состав этого осадка более
точно отвечает формуле HNaF-4AIF3) и Na3[FeF6].
Титан при этом остается в растворе в виде комплекса
Na2[TiF6]; при этом не осаждаются Be, Со, Ni, Мп,
Cd, Си, Nb и др. Разработаны методы определения
алюминия в феррониобии, ферротитане, ферроцирко¬
нии, ферровольфраме, феррохроме после отделения
его в виде llNaF-4AlF3.Фторидные комплексы бериллия и алюминия ис¬
пользуют для титриметрического их определения. Бе¬
риллий определяют по реакции:Ве2+ + 4F~ [BeF4]2'.В качестве индикатора используют смесь Fe3+ и Fe2+,
а точку эквивалентности устанавливают потенциомет-
рически, измеряя в процессе титрования потенциал
погруженного в раствор платинового электрода. Пос¬
ле связывания всех ионов бериллия во фторидный
комплекс образуются фторидные комплексы желе¬
за (III) и потенциал электрода резко уменьшается.МБ
Титрование алюминия проходит пс реакцииА13+ + 3F” A1F3.В качестве индикатора применяют салииилат железа
или метиловый оранжевый. Ионы алюминия в водных
растворах подвергаются гидролизу, вследствие этого
раствор имеет кислую реакцию и метиловый оранже¬
вый окрашен в красный цвет. После связывания всех
ионов алюминия во фторидный комплекс реакция
раствора становится нейтральной и икдикатор приоб¬
ретает желтую окраску.Для титриметрического определения кремниевой
кислоты промытый осадок ее обрабатывают фторидом
калия и прибавляют избыток титрованного раствора
хлороводородной кислоты:H2SiOa + 6KF + 4НС1 :?=*: K2[S iFGJ + IKCl + 3H20.Избыток хлороводородной кислоты титруют затем ра¬
бочим раствором гидроксида натрия.Фторид-ионы широко используют для маскирова¬
ния многих ионов. Так, при иодометрическом опреде¬
лении меди влияние ионов Fe3+, которые окисляют
иодид-ионы, устраняют фторидом. Образовавшийся
фторидный комплекс железа не окисляет иодид-ионы.Для отделения мышьяка, сурьмы, меди, свинца,
ртути, кадмия и других ионов от олова используют
осаждение их в виде сульфидов в присутствии фто-
рид-ионов, которые связывают олово. При фотомет¬
рическом определении кобальта в виде хлоридного
или роданидного комплексов вредное влияние желе¬
за (III) устраняют, связывая его в прочный фторид¬
ный комплекс.Широкое распространение получил экстракцион¬
ный метод отделения железа (III) в в^де H[FeCI4] от
многих других ионов, например от кальция, стронция,
бария, алюминия, редкоземельных и многих других
элементов. Тетрахлоридный комплекс железа экстра¬
гируют этилацетатом или диэтиловым эфиром.Одним из лучших методов определения хлоридов
является меркурометрический метод, основанный на
образовании устойчивого хлоридного комплекса2Cr + Hgst =f=* HgCI*
В качестве индикатора используют дифенилкар-
базонМН—NH—СвН5С1=С\n=N—СвН5образующий с ионами ртути интенсивно окрашенный
в синий цвет комплекс, или нитропруссид натрия
Na2[Fe(CN)5NO)], который с избытком ионов ртути
образует осадок Hg(Fe(CN)sNO].Пероксидные комплексы. Пероксид водорода обра¬
зует комплексы с титаном, ванадием, церием, нио¬
бием, танталом и др. Чаще всего пероксидные комп¬
лексы применяют для фотометрического определения
титана, ванадия, ниобия и тантала.Комплексы с другими неорганическими лигандами.
Устойчивые цианидные комплексы образуются с
ионами меди, кадмия, цинка, железа (III) и желе¬
за (II), кобальта, никеля и др. Однако в связи с боль¬
шой ядовитостью цианид мало применяют в анализе.
Его использование в анализе ограничивается маски¬
рованием посторонних ионов при определении неко¬
торых ионов другими методами, хотя в принципе воз¬
можно использование цианида в качестве титранта.Карбонатные комплексы малопрочные, их приме¬
няют главным образом для отделения урана от многих
элементов, так как карбонатные комплексы ура¬
на [ир2(С03)2]2_ или [и02(С0з)з]4- хорошо раство¬
римы в воде, в то время как карбонаты или гидрок¬
сиды многих других металлов в этих условиях плохо
растворимы в воде.Большое значение в аналитической химии имеют
фосфатные комплексы. Их широко применяют для
маскирования, например, железа(III) при фотомет¬
рическом определении титана в виде пероксидного
комплекса. При этом железо маскируют фосфорной
кислотой; оно образует растворимые в воде бесцвет¬
ные комплексы [FeHP04]+, [Fe(P04)2]3_ или
[Fe(P04)3]6-.Значительно большее значение в анализе имеют
комплексы фосфора с молибденом — молибдофосфор-
ные гетерополикислоты Нз [Р (МозОюЫ и восстанов¬
ленная форма (так называемые синие гетерополикис¬
лоты), которые широко используют д^я фотометриче¬
ского определения фосфора.Сульфатные комплексы применяют для косвенно¬
го фотометрического определения сульфата. При
этом готовят комплексы циркония с ализарином.
В присутствии сульфат-ионов образуется сульфатный
комплекс циркония и цирконализариновый лак раз¬
рушается, в результате чего растзор обесцвечи¬
вается.Сульфитные комплексы некоторых металлов обра¬
зуются только при очень большом избытке сульфит-
ионов и в анализе не нашли широкого применения. Не¬
которые из них экстрагируются неводными раствори¬
телями, что может быть применено для разделения
некоторых элементов.ГЛАВА 14КОМПЛЕКСНЫЕ СОЕДИНЕНИЯ
В ТИТРИМЕТРИЧЕСКОМ АНАЛИЗЕ§ 1. ВАЖНЕЙШИЕ НЕОРГАНИЧЕСКИЕ
И ОРГАНИЧЕСКИЕ ТИТРАНТЫТитриметрические методы, в основе которых лежат
реакции комплексообразования, известны уже более
150 лет. Такие методы называют комллексометриче-
скими, а по рекомендации Междунгродного союза
теоретической и прикладной химии (ИЮПАК) ком-
плексиметрическими. Первые работы, посвященные
комплексометрическому определению иодид-ионов
4KI + Hg<N03)2 =?=* K2[HgI,] + 2KN03относятся к 1834 г. В 1851 г. Либих списал комплек¬
сометрический метод определения цианидов раство¬
ром нитрата серебра:2KCN + AgN03 =<=>: K[Ag(CN)2]-h KN03.Однако неорганические титранты и в настоящее вре¬
мя находят ограниченное применение Наиболее ши¬
роко известен метод комплексометрического (мерку-
риметрического) титрования хлоридов, бромидов, ро-
данидов, цианидов и тиомочевины.2KCl+Hg(N03b HgCh + :'KN03.269
Органические титранты распространены значи¬
тельно шире. Как уже отмечалось выше, наибольшее
значение имеют комплексоны, впервые предложенные
Г. Шварценбахом, В последнее время применяют и
другие титранты, например тиолы, купферон, БФГА,
дитизон и др. Их преимущество по сравнению с ком-
плексонами в большей специфичности. Точку эквива¬
лентности при титровании органическими титрантами
устанавливают металлохромными индикаторами или
электрохимическими методами.Общие требования к реакциям в комплексометрии
такие же, как это было сформулировано в гл. 7: они
должны проходить быстро и практически до конца,
без каких-либо побочных процессов, связанных с за¬
тратой титранта.Селективность комплексометрических методов
обычно невелика и зависит от того, какие именно до-
норные атомы являются реакционными началами ти¬
транта. Так, при титровании иодидом калия селектив¬
ность достаточно высокая, потому что иодид-ионы об¬
разуют комплексы или осадки только с ионами ртути,
серебра, свинца, висмута. Аммиак и полиамины так¬
же более селективны по сравнению, например, с ком-
плексонами, так как они реагируют только с ионами
Со, Ni, Cu, Zn, Cd, Hg и Ag. Аммиак в качестве
титранта имеет некоторые недостатки, связанные
прежде всего с малой прочностью аммиакатов метал¬
лов. Применение полиаминов, например тетраэтилен-
пентамина имеет преимущество как по селективности
взаимодействия, так и по образованию прочных ком¬
плексных соединений.Большое значение в титриметрическом методе
анализа имеют комплексоны, среди которых в каче¬
стве титранта чаще всего применяют комплек¬
сен III — двунатриевую соль этилендиаминтетраук-
сусной кислоты (сокращенно ЭДТА или Na2H2Y).Комплексоны взаимодействуют с катионами очень
многих металлов, поэтому по селективности уступают
другим титр антам. В то же время высокая прочность
соединений и большая скорость взаимодействия поч-NaOOC—Н,СЧ;ноос—н2с/N—СН2—СН2—N./CH2-COONaГ^СНа—СООН
ти со всеми ионами металлов послужили причиной
широкого распространения комплексонов в титримет-
рическом анализе. Большим преимуществом комплек¬
сонов перед другими титрантами является то, что
практически со всеми катионами они дают комплексы
с соотношением 1 : 1<j)Na/С^0и ступенчатое комплексообразование отсутствует.Один из наиболее распространенных комплексо¬
нов— ЭДТА — представляет собой шестидентатный
лиганд, т. е. лиганд, молекула которого связана с ка¬
тионом металла посредством четырех атомов кисло¬
рода и двух атомов азота. Это имеет определенное
преимущество по сравнению с лигандами меньшей
дентатности. Рассмотрим, например, взаимодействие
ионов металла М с координационным числом, равным
четырем, с лигандами разной дентатности. Так, тет-
радентатный лиганд реагирует по схеме:МЛ++ Y4' MY(4-"b.Константа устойчивости образующегося комплекса[MY(4~n)~][Мл+] [Y4-] 'В случае монодентатного лиганда, например фторид-
ионов, можно записатьМ4+ + 4R" MR,.Ж
Процесс проходит ступенчато:=*: MR3", К,=М“ + R' :MR3* + R' ;MR** + R- 4=fc MR *MR* + R MR^, KtMR22\ K2*з =[MR3t]
(M4*][R"1 '_K1_.[MR3+] [R-J ’[mr:][MR’*][R-] '
[MR,][mr*| [r'1 •Произведение констант равно константе суммарного
процесса:[MR3+] fMR^l
= /С1/СгКэ/С4 = XX[m4+][r~] [mr3+][r ]
[MR*] [MR,] [MR,][mr2+] [r~] [mr+] [Rl [m4+] [R'T 'На рис. 14.1 представлены кривые комплексомет¬
рического титрования четырехзарядных ионов метал¬
лов растворами одно-, двух- и четырехдентатных ли¬
гандов, каждый из которых образует комплексные
соединения с общей константой устойчивости р == 1022. Из этого рисунка
видно, что максимальный
скачок на кривой титрова¬
ния наблюдается в случае
тетрадентатного лиганда,
когда образуется комплекс¬
ное соединение MR с соот¬
ношением компонентов 1 : 1
(кривая /). Вполне удов¬
летворительно проходит
титрование раствором двух-
дентатного лиганда, когда
образуется комплекс MR2 с
соотношением компонентовРис. 14.L Кривые комплексомет-
рического титрования272
1:2 (кривая 2) (Ki = Ю13, tf2=109; р= 1022).
В случае же титрования раствором однодентатного
лиганда с образованием комплекса (/(1 = Ю10,/с2 = I06, Кз = Ю4 и К* = Ю2; Р== Ь\-КгКз-К* =
= 1010 • 106 • 104 ■ 102 = I022) скачок в точке эквива¬
лентности практически не наблюдается (кривая 3) и
проведение определения по такому титрованию прак¬
тически невозможно.Для повышения селективности титранта можно
синтезировать полидентатные лиганды-титранты, ко¬
торые будут взаимодействовать с меньшим числом
ионов металлов. Так, более селективными йигандами-
титрантами по сравнению с комплексонами' являются
тиолы, например 2,3-димеркаптопропансульфонат
натрия (унитол) СН2—СН—СН2—S03Na, которыеI I
ОН онтакже с успехом применяют для титриметрического
анализа.§ 2. КОМПЛЕКСОНОМЕТРИЯПервые пробы применения ЭДТА (комплексона III,
Na2H2Y) в анализе были основаны на реакцииМ2 + -Ь H2Y2" MY2' + 2H+.Выделившиеся ионы водорода отти гровывали ще¬
лочью и таким косвенным методом определяли со¬
держание ионов металла. Затем бы/и предложены
прямые методы со специальными индикаторами —
металлохромными или металлометрическими. В на¬
чале титрования в растворе индикатор связан в ком¬
плекс с определяемым металлом. В конце титрования
концентрация ионов металлов уменьшается, потому
что комплексонат металла устойчивее комплекса ме¬
талла с индикатором. Поэтому в конце титрования
происходит реакция:MInd-i-H2Y2- ?=£ MY2" + HJnd.Цвет раствора в конце титрования изменяется от ок¬
раски комплекса с индикатором до дзета свободного273
индикатора. Таким образом, необходимым условием
прямого комплексонометрического титрования явля¬
ется неравенствогде К 2- — константа устойчивости комплекса металла с эта-
'MYленднамингетрауксусной кислотой; Км\пл — константа устойчиво¬
сти комплекса титруемого металла с индикатором.Известны следующие четыре способа проведения
комплексонометрического титрования: прямое титро¬
вание; обратное титрование; титрование по способу
вытеснения; косвенные способы.Прямое титрование — наиболее распространенный
прием. К анализируемому раствору прибавляют рас¬
твор титранта (например, ЭДТА — H4Y) до тех пор,
пока не будет достигнута точка эквивалентности, ко¬
торую устанавливают посредством индикаторов или
потенциометрически. По результатам титрования рас¬
считывают содержание определяемого элемента.Прямое титрование удобно применять, если есть
возможность подобрать соответствующий индикатор
и если взаимодействие между катионом металла и
ЭДТА происходит достаточно быстро.Точность титрования определяется изменением
концентрации ионов металла вблизи точки эквива¬
лентности. Зависимость этой величины от количества
прибавленного титранта удобно выражать графи¬
чески в виде кривой титрования, на оси ординат ко¬
торой откладывают —lgf-M] = р [М], а на оси абс¬
цисс — объем рабочего раствора, израсходованного
на титрование. Она дает наглядное представление о
ходе зависимости и может служить критерием выбора
оптимальных условий титрования.Рассмотрим способы вычисления концентрации
ионов металла и построения кривой титрования. Для
упрощения расчетов примем, что ионная сила раство¬
ра в процессе титрования не изменяется, а значение
pH поддерживается постоянным посредством введе¬
ния буферного раствора. В качестве примера ниже
приведены расчеты титрования соли магния раство¬
ром ЭДТА (H4Y).274
Концентрацию ионов магния можно найти из
уравненияMgs+ + Y4- :?=► MgY2*;[MgY2
'MgY2' [Mg2+][Y4-]‘Откуда[MgY2']2-= <141)Из уравнения (14.2) видно, что для расчета необхо¬
димо знать концентрации ионов Y4' и комплекса
MgY2-.Концентрация Y4- зависит от обще! концентрации
ЭДТА (не связанной в комплекс) Сщу и от pH рас¬
твора *: [нТ + лг,[н*]3 + /с,/с2[н+]2 + /с,а:2л:4[н+]+лг1лг2лГзА:-1= ^H4YaY(H)- . (1^-3)Для расчета кривых титрования многоосновных
кислот, а также для расчета концентрации свободных
ионов многоосновных кислот, какой является этилен-
диаминтетрауксусная кислота H4Y (p/(t = 1,996;
р/С2 = 2,672; р/Сз = 6,161; рА^ = 10,262), удобно кон¬
центрацию каждой анионной формы выразить через
величину ап в зависимости от pH раствора. Другими
словами, можно выразить концентрацию всех анион¬
ных форм многоосновных кислот как функцию от pH.
Так, в случае этилендиаминтетраук:усной кислоты
материальный баланс всех ее форм будет равен:Сн4^щ = [H«Y1 + tH’Y'l + lH*Y*'l + [W"] + [Y4]- (14‘4)* Уравнение (14.3) выведено, исходя и:> следующих сооб¬
ражений. Концентрация несвязанной в комплекс ЭДТА равна
=[Y4 ] + [HY3~] + fH2Y2] + [H3Y ] Ч [НД]. Путемподстановки вместо концентраций частиц в правой части этого
уравнения их значений из соответствующих констант диссоциа¬
ции легко прийти к уравнению (14.3).‘276
Тогда(,4'5)H,Y4 общfwcL. „«)H.Yo6uia,-p£L. „4.7,нл4 o6lll,„.8|Г у"-1Г[У 1 (14.9)СнЛобщСумма этих величин равна 1:Оо + О] + аг + оз + а* = 1.Величину а„ можно выразить через [Н30+] и со¬
ответствующие константы Ки Кг, Кз и К4- Тогда[HaV] = (1410)[Н30*1FTI ^1^2[Н<У]общ н J 11\[HsY 1_ [н^г ’ ( • }УС./Сг^Сз [H4Y]0<s[u1н3сг]|j-jy3-j __ 1 loom |2jf vi-l ^l^2^3^4 [^4^]о(5щ t t a iO\[Y ,! • (1413)Подставляя найденные значения [H4Y] в уравнения
(14.5) — (14.9), находим соответствующие значения а„:а0 = [н,о+Г [h3o+]4+Ki [н30+]3+^.л:2[Нз0+]2+К1л:2к3 [НзО^-ь/с.^Кз^’(14.14)а, = *,[н3оТ [НзО+)4+ЛГ,[НзО+]^+УС1/С2[НзО+]2+/С1/С2ЛГз[НзО+] Ч- ДС1/С2/СзЛГ4 ’(14.15)0,2 = л:гк3[н3оТ 1на0+]4Ч-/С,[н.,0+]34-/С,ЛГ21Нз0+]2+ /С,ЛГ2^Сз[НзО*] + /С,АГ2л:зАГ*’(14.16)276
а3 =
/С,К2Кз1НзО*][НэО+]4+ЯЛН^Р+Я./ГЛНзО*]8 + KlKsK3[haO+] + K,f<2K3Ki'(14.17)а4 = К<К2К3К4 _[н30+]Члг,[н30+]ЧА:1А:2(Нз0+]:г+А:1А:2Л'31н!0+] + /с,л:2А:эл:4'(14.18)На рис. 14.2 представлена зависимость состава
форм H4Y от pH раствора.Для построения кривой комплексонометрического
титрования ионов металлов с помощью H4Y в при¬
сутствии буферного раствора с определенным значе¬
нием pH удобно для расчета концентрации свободных
ионов Y4- пользоваться величиной а4:fY4iа4= -,L - (14.19)^4^общ— общая концентрация незакомглексованной эти-где Ц у4'общлендиаминтетрауксусной кислотыПодставляя значения произведения а4Сн2уобщ вме¬сто [Y4~] в выражение для константы устойчивости
комплексоната металла получим:-я,_1— а4^Му(4-я^ [м"4(14.20)■ «А,4 обшТак как а4 является постоянной величиной только
для данного значения pH, то произведение этой вели¬
чины на константу устойчивости комплекса является
условной константой, обозначаемой ч<-рез К[.
Однако этой величиной
удобно пользоваться
при построении кривых
комплексонометриче-
ского титрования.Рис. 14,2. Распределения
форм ЭДТА в зависимости
от pH раствора8 Ю П pH277
Таблица 14.1. Значения а<у«) для ЭДТА в растворах
с различными pHpHaY(H)pHaY(H)2,03,7-10"“8,05,4 • 10~33,02,5- 10"“9,05,2- 10-24,03,6-10-910,03,5 -10"15,03,5-10-711,08,5- 10“16,02,2 • 10-512,09,8- 10~‘7,04,8 -10-1Уравнением (14.18) неудобно пользоваться непос¬
редственно, так как в выражение ау<н> входит кон¬
центрация ионов водорода в четвертой и более низг
ких степенях. Лучше вычислить aY(m заранее для
разных pH и пользоваться готовыми значениями.
Значения ау(н> приведены в табл. 14.1.Преобразуем уравнение (14.1), подставив вместо
[Y4-] произведение Сщус^н):[MeY2i1g J . (14.21)[Mg2+] CHiYaY(H)ВыражениеK= [MgYlJ_ (14.22)tMe 1 cHtYназывают условной константой устойчивости, она от¬
личается от истинной константы (см. рис. 14.1) тем,
что в знаменателе стоит вместо [Y4-] общая кон¬
центрация свободного (не связанного в комплекс);
ЭДТА и, в соответствии с этим, «условная константа»
зависит от pH *. Тем не менее ею удобно пользовать¬
ся в сочетании с величинами ау<н).* В рассматриваемом примере для простоты учтен только
коэффициент конкурирующей реакции ау(Н), отражающий прото¬
низацию лиганда Y4-; в действительности, особенно при высоки»
pH, следует принимать во внимание также другой побочный)
процесс, а именно гидролиз катиона металла.m
Из уравнений (14.21) и (14.22) следуетА:' = /СаУ(Н) (14.23)и[Mg»«i - • (14.24)Пользуясь уравнениями (14.23) и (14.24) и данными
табл. 14.1, можно вычислить концентрацию свобод¬
ных ионов магния в любой момент титрования при
некотором определенном pH.Пример 1. Вычислить [Mg2+] при титровании 50 мл 0,01 М
раствора хлорида магния 0,01 М раствором ЭДТА в буферном
растворе с' pH = 10.Условная константа устойчивости равнаК' = Кау(Н) = 4,9- 108 • 3,5 • 10-1 = 1,72- 108,где 3,5-10-1 —ау(Н) при pH = 10.До точки эквивалентности в растворе содержатся свободные
и связанные в комплекс ионы магния, т. е. CMg = [Mg2+] +
+ [MgY2-.]. Концентрация MgY2- в свою очередь равна концен¬
трации прибавленной в раствор ЭДТА, так как последний при
избытке ионов магния практически полностью связывается в
комплекс MgY2-. Если, например, к 50 мл 0,01 М раствора соли
магния прибавлено 20 мл 0,01 М раствора ЭДТА, тогда[Mg2 + ] = 50,0-0,0. - 20,0- 0,0. = 4 29 [0-з миp[Mg2+] = 2,37.В точке эквивалентности, после прибавления 50 мл 0,01 М
раствора ЭДТА, общий объем раствора составит 100 мл и в нем
содержится комплекс MgY2-; последний частично диссоцииро¬
ван:MgY2' ч=* Mg2+ + Y4'.Анионы Y4- подвергаются протонизации с образованием ча¬
стиц, показанных в правой части уравнения. Поэтому [Mg2+] ф* [Y4-], но справедливо уравнение[Mgs + ] = CH4Y. (14.25)С другой стороны, [MgY2-] = 0,00500 — [Mg2+1 ж 0,00500 М.
Концентрацию Mg2+ в точке эквивалентности находим по урав¬
нению (14.22); К = 1,72-108.Лp[Mg2+]=5,27.Ш
p [Mg2*]р [Mg2 ]О 20 40 60 80
Прибавлено 0,01 М pacmSopa ЭДТА,ПрибаВлено 0,01 МpacmSopa ЗЦТД,
млРас. 14.3. Кривые титрования 0,01 М раствора ионов магния0,01 М раствором ЭДТА в буферных растворах с различными
значениями pHРис. !4.4. Кривые титрования некоторых металлов ЭДТА при
pH = 5После точки эквивалентности, например после прибавления
70 мл титранта- _ 50.0-0,0.00 _4(2<10-зМ(MBv2- 120=1,7.10- М.[Mg2+] = = *-44'10"8Иp[Mg2+] = 7,84.На рис. 14.3 представлены кривые титрования
ионов магния в буферных растворах с различными
значениями pH. Из этого рисунка видно, что замет¬
ное изменение p[Mg2+] достигается только при
pH > 8.На рис. 14.4 представлены кривые титрования
ионов различных металлов при pH = 5. Из этого ри¬
сунка видно, что ионы магния и кальция, прочность
комплексов которых невелика, не дают скачка изме¬
нения p[Mg2+], а ионы, образующие прочные комп-
лексонаты, могут быть оттитрованы удовлетворитель¬
но даже при этом значении pH.Кадмий титруют обычно в присутствии аммиака и
хлорида аммония, которые создают необходимое зна¬280
чение pH. В этих условиях аммиак образует аммиа¬
кат кадмия и выпадение гидроксида кадмия предот¬
вращается. Поэтому уравнение реакции титрования
кадмия раствором ЭДТА следует записать так:Cd (NH3)2* + HY3‘ CdY2'+ NH4++ 5NH3.В этом случае полнота протекания реакции, а сле¬
довательно, и высота скачка р [Мп+] зависит не толь¬
ко от pH раствора, а также и от концентрации ком¬
плексообразующего постороннего агента, в данном
случае аммиака.Влияние постороннего комплексообразователя учи¬
тывают подобно тому, как учитывали влияние pH на
концентрацию различных форм ЭДТА. Если обозна¬
чить чс'рез р7 отношение[м-+1 (1428|^•мгде См-г-общая концентрация всех форм, содержа¬
щих ионы титруемого металла, за исключением свя¬
занного в комплекс с ЭДТА, тогда для раствора, со¬
держащего ионы кадмия и аммиак, можно записать:см = [Cd2-] + [Cd(NH3)2*] + [Cd(NH3)2+] + [Cd(NH,)*s*] ++ [Cd(NH3)42t] + [Cd(NH3)j+] + [Cd(NH,)*+]. (14.27)Численное значение p' можно найти, зная концентра¬
цию аммиака и константы устойчивости образующих¬
ся комплексов. Например,„ [Cd(NH3)2+]Л 1 =отсюдазатем[Cd2+] [NH3J ’[Cd(NH3)2+] = /C,[Cd2+] [NH3J;[Cd(NH3)2t] = /C,/C2[Cda+] [NH3]2;
[Cd(NH3)3+] = [CdJ+] [NH3]3;[Cd(NH3)J+] = KMtKACd2+] [NH3]4;
[Cd(NH3)|+] = KiKiKzKbKACd2*] [NH3]5;
[Cd(NH3)r] = /С1 ifa/faA-4«‘s«"«[Cd2*J [NH3f.281
Подставляя эти величины в уравнение (14.26),
находимСм = [Cd2+] (1 + Ar,[NH3] + /f,/C2[NH3P + /С,/С2Л:з[МНз]3 ++ K^KzKiKtmh]* + KiKiKsKtKdNHrf + /С, /C2Ar3/C+/CsAre[NH3]®).Решая это уравнение совместно с (14.26) для случая
с кадмием, т. е. [М"+] = [Cd2+], находимо/ ! (14 28)Р _ I + K,[NH3] + /C,/(2[NH3]2 + /C.ATa/CsINTHa]3 + V 'Из уравнения (14.26) находим концентрацию
ионов кадмия [Мл+] = [Cd2+] = р'С„ и, подставляя
это значение в уравнение (14.20), получим:к' = а К [CdY2~]Lai • Cat p C»*(_u vM 4 общИЛИВеличина K^d^~ — новая условная константа,
применяемая только при единственном значении pH
и единственной концентрации аммиака и хлорида
аммония. Эту условную константу используют для
построения кривой титрования, как показано ниже,
в принятых условиях.Пример 2. Вычислить p[Cd2+] в растворах, содержащих 25 мл
0,00100 М Cd2+, к которому прибавлено 20,0; 25,0; 30 мл0,00100 М ЭДТА. Растворы кадмия и ЭДТА содержат 0,100 М
NH3 и 0,176 М NH4C1, при этом значение pH = 9,0. Константы
устойчивости аммиакатов кадмия: Ki =3,23-102; /Ci/С22,95-104;
KiKiK3 = 5,89-105; /С1К2/С3/С4 = 3,71 • 10°; KiKiK3KtK5 = 1,82-106;
К,К2К3К,К5К6 = 3,63-104.1. Расчет условной константы. Для расчета Р' принимаем
[NH3] = C'NH3 и подставляем в уравнение (14.28) значения
CNHj и ступенчатых констант устойчивости аммиакатов кадмия: 1 1 /. I п 6Р _ 1 + 32,3 + 2950+58900 + 371000+1820004-3630 — ’Подставляя в уравнение (14.29) значение ^cdy2- = 2,9 • 1016 и
а4 при pH « 9 (табл. 14.1) находим:K"dYs. = 5,2-.10^2- 1,6 - Ю~6-2,9- 10*6 = 2,41 • Ю9,ЭД2
2. Расчет [Cds+] после добавления 20 мл ЭДТА. Находим
концентрацию непрореагированного Cd2+ после прибавления
20 мл ЭДТА.25,0 • 0,00100 — 20,0 • 0,00100 -4 ,,Cd2+ _ 45,0 ’Пренебрегая диссоциацией различных комплексов кадмия,
подставляем в уравнение (14.26) общую концентрацию металла
^Cd2+ пРеДставляюи1Ую со^ой сумму равновесных концентрацийвсех комплексов кадмия, не содержащих ЭДТА, и рассчитываем
равновесную концентрацию ионов кадмия:[Cd2+] =Смр' = 1,11 ■ 10“4- 1,6- 10-6= 1,78- 10-ШМ,
p[Cd2+] = 9,78.3. Расчет fCd2+] после добавления 25 мл ЭДТА. В точке
эквивалентности концентрация CdY2- равна 5,00-Ю-1 М. Сумма
равновесных концентраций различных комплексов кадмия равна
сумме равновесных концентраций незакомплексованных форм
ЭДТА: См = Сну и [CdY2“] = 5,00-Ю"4 - См as 5,0‘ Ю-" М.Подставляя в уравнение (14.28) этот результат, получим.„9 5,00-Ю-4KcdY2-“2’41' 10 1См = V2,08 ■ 10~!3 = 4,4 • 10~7 М.Из уравнения (14.25) находим[Cd2+] = СМ ■ =4,4 ■ 10“7- 1,6- 10_б = 7,04 • 10~13 Af;
р[ Cd2+] = 12,18.4. Расчет [Cd2+] после добавления 30,0 мл ЭДТА. В этом
случае в растворе содержится избыток ЭДТА:= 30,0. 0,00100 - 25,0. 0,00100 ,Y 55[CdY2-] = -ZD’U = 4,54 ■ 10-4 М.25,0 ■ 0,00100
55После подстановки в уравнение (14.29) находимСм = 4,545-° д- =2,07- 10~~99,09- 10 5 ■ 2,41 ■ 109И[Cd2+] = См • р' = 2,07 - 10-9- 1,6- 10_6 = 3,31 ■ 10~15Л*,p[Cd21-]= 14,48.На рис. 14.5 представлены две теоретические кривые титро¬
вания кадмия раствором ЭДТА при р;Н == 9,00. Равновесна»Ш
Рис. 14.5. Теоретические кривые
титрования 0,00100 М раствора
ионов кадмия раствором ЭДТА
при pH = 9. Равновесные концен¬
трации аммиака 0,1000 М (кривая
1) и 0,0100 М (кривая 2)концентрация аммиака в одном
случае составляла 0,1000 М, в дру¬
гом—0,0100 М. Из этого рисун¬
ка видно, что аммиак вызывает
уменьшение скачка на кривой тит¬
рования. Поэтому концентрация
вспомогательного реагента не дол¬
жна превышать минимальной
концентрации, необходимой для предупреждения образования
оксида или гидроксида. Величина Р' не влияет на p[Cd2+]
после точки эквивалентности, но, с другой стороны, величина а
(см. рис. 14.4) и, следовательно, pH играют очень важную роль
в формировании этой области на кривой титрования.Таким образом, соединение металла с индикато¬
ром должно быть достаточно устойчивым, чтобы
можно было определить небольшие концентрации
ионов металла. Однако это соединение должно быть
менее устойчиво по сравнению с комплексонатом
этого же металла.Обратное титрование. Обратное титрование менее
удобно, однако к нему приходится прибегать, когда
для прямого титрования нельзя подобрать соответст¬
вующий индикатор, или когда катионы металла
очень медленно взаимодействуют с титрантом, или
когда при благоприятных значениях pH определяе¬
мый ион металла осаждается в виде гидроксокомп-
лексов. Во всех этих случаях к титруемому раствору
прибавляют избыток ЭДТА, раствор оставляют на
некоторое время, чтобы произошла реакция, или ус¬
коряют процесс взаимодействия, подогревая раствор.
Затем избыток ЭДТА титруют раствором второго
металла, для которого реакция с комплексоном соот¬
ветствует всем требованиям, предъявляемым к реак¬
циям комплексонометрического титрования.При обратном титровании комплекс определяемо¬
го металла с ЭДТА на определение точки эквивалент¬
ности практически не влияет, так как он образуется
медленно и также, как правило, медленно разлага¬
ется/ Кривая обратного титрования идентична кривойШ
прямого титрования, но противоположна по направ¬
лению.В качестве примера обратного комплексонометри¬
ческого титрования можно привести определение
хрома(III). Титрование избытка комплексона можно
проводить даже в кислой среде, применяя в качестве
титрантов растворы солей железа(III), висмута или
тория.Титрование по методу вытеснения. Вытеснительное
титрование применяют в тех же случаях, что и об¬
ратное. Вытеснительное титрование может до некото¬
рой степени повысить селективность определения
ряда ионов. К раствору определяемого металла при¬
бавляют избыток комплексоната металла определен¬
ной концентрации. Обычно применяют комплексонат
магния. При этом происходит реакция вытесненияМ"+ + MgY2' 3=Е Mg2+ + MY(n-4,+,после чего вытесненные ионы магния титруют комп-
лексоном. Если реакция между ионами Мп+ и комп-
лексонатом магния проходит медленно, необходимо
раствор оставить постоять или ускорить реакцию на¬
греванием.Селективность реакций в этом случае повышается
за счет того, что ионы металлов, которые образуют
менее прочные комплексы по сравнению с комплек¬
сом магния, практически не будут мешать опреде¬
лению.При выборе комплексоната металла для использо¬
вания в качестве рабочего раствора (в нашем случае
MgY2-) необходимо соблюдать условие ^MY(n-4)+ >АГМку2-' чтобы вытеснительная реакция проходила
достаточно полно.Обменная реакция протекает также при прямом
титровании, если для индикации точки эквивалентно¬
сти применяют металлохромный индикатор, который
образует комплекс с дополнительно введенным метал¬
лом. Концентрация этого металла должна быть
очень низкой, а устойчивость комплекса с индикато¬
ром должна быть намного меньше по сравнению
с устойчивостью комплекса данного металла с ЭДТА.
Кроме того, комплексонат этого металла должен
быть намного менее устойчив по сравнению с
комплексонатом определяемого металла, т. е. должны
соблюдаться условия:/CXY > ^Mlnd'Изменить направление вытеснительной реакции
можно также и в том случае, когда константа комп¬
лексоната определяемого металла меньше константы
комплексоната прибавляемого металла. Например,
при определении ионов бария, для которого нет хоро¬
ших металлиндикаторов, можно прибавить избыток
комплексоната цинка, который более устойчив по
сравнению с комплексонатом бария. Однако если
прибавить в раствор достаточное количество аммиа¬
ка, который в сильной мере будет связывать цинк
в комплекс, тогда произойдет реакцияВа2* + Zn Y2" + 4NH3 BaY2' + Zn(NH3)2\После чего аммиакат цинка титруют комплексоном
в присутствии эрнохрома черного Т.Косвенные способы титрования. Существует не¬
сколько вариантов косвенных методов. Определяемые
ионы можно осадить, а затем определить ионы ме¬
талла в осадке. Так, при определении натрия его
осаждают в виде натрий-цинк-уранилацетата:Na+ + Zn2+ + 3U02+ + 9СН3СОСГ NaZn(U02)3 (СН3ЭОО )«,затем в осадке определяют цинк титрованием комп¬
лексоном. Таким способом можно определить фос¬
фат-, пирофосфат-, молибдат-, вольфрамат- и другие
ионы, осаждая их соответственно в виде магний-ам-
моний-фосфата, пирофосфата цинка, молибдата и
вольфрамата кальция, а затем после переведения
осадка в раствор титровать комплексоном ионы маг¬
ния, цинка или кальция.Можно привести и другие примеры косвенного
комплексонометрического определения различных
ионов. Так, при взбалтывании растворов различных
солей с амальгамой цинка происходит обменная ре¬
акция, более электроположительные металлы перехо¬
дят в амальгаму, а ионы цинка переходят в раствор.
После определения содержания перешедшего в рас¬
твор цинка рассчитывают содержание определяемого
-металла;чт
$ з. метллохгомныЕ индикаторы и их выборИндикаторы комплексонометрии, так называемые
металлохромные индикаторы — это вещества, кото¬
рые при взаимодействии с ионами металлов образуют
окрашенные соединения, менее устойчивые по сравне¬
нию с их комплексонатами. Образующиеся соедине¬
ния отличаются по цвету от свободных ионов или
молекул индикатора. Рассмотрим свойства некоторых
наиболее распространенных металлохромных индика¬
торов.Мурексид—аммонийная соль пурпурной кислотыМурексид образует с солями кальция оранжево¬
красный пурпуреат кальция.Пурпурная кислота — трехосновная. Значения рК
следующие: pATi = 0, р/Сг = 9,2; р/Сз = 10,9. Водные
0,1 М растворы пурпуреата аммония в течение Г2—
15 дней полностью разлагаются. Скорость разложе¬
ния увеличивается с повышением pH. Из-за малой
устойчивости растворы мурексида готовят непосред¬
ственно перед анализом, растворяя навеску мурекси¬
да, смешанного для удобства взвешивания с хлори¬
дом натрия. Однако необходимо иметь в виду, что
ионы натрия также образуют комплексные соедине¬
ния с этилендиаминтетрауксусной кислотой. Хотя
устойчивость этих комплексов небольшая (pKNaY3- ~
= 1,66), при большей концентрации ионов натрия чет¬
кость интервала перехода индикатора ухудшается.Мурексид дает с ионами многих металлов комп¬
лексы, устойчивость которых увеличивается с повы¬
шением pH раствора.Состав комплексов металлов с мурексидом зави¬
сит от pH раствора. Так, в слабощелочной среде цинк
образует с мурексидом комплекс с соотношением
реагирующих компонентов 1 : 1, а в щелочной среде —
комплекс ZnR2- Мурексид чаще всего применяют при
титровании ионов кальция.HN—СО СО—NHHN—СО С NH287
Хромоген черный специальный ЕТ = 00 (эриохром
черный Т).Этот индикатор образует окрашенные комплекс¬
ные соединения с ионами многих металлов.Кислотный хром синий К(П) и кислотный хром
темно-синий(III) были предложены для комплексо¬
нометрического определения жесткости воды.Они содержат функциональные аналитические груп¬
пировки, аналогичные соответствующим группам
эриохромового черного Т, образуют с ионами кальция
и магния внутрикомплексные соединения оранжевого
цвета. Их применяют как индикаторы при определе¬
нии ионов этих металлов. Такого же цвета эти инди¬
каторы в кислой среде; в щелочной среде оба инди¬
катора окрашены в серовато-голубоватый цвет. Ин¬
дикаторы не специфичны — они дают окрашенные
соединения с ионами многих других металлов.o2nонон онS03NaIIОНОН ОНIII288
4-(2-Пиридйлазо) резорцин — ПАР,0НN=N'Применяют для титрования марганца, никеля,
свинца, цинка, кадмия, меди в среде ацетатного бу¬
ферного раствора. Комплексы окрашены в красный
цвет, а свободный индикатор — в желтый. Применяют0,05 или 0,2 %-ные водные растворы индикатора.Металлфталеин (фталеинкомплексон) имеет мно¬
го общего с фенолфталеином и в то же время яв¬
ляется сам комплексоном.Металлфталеин — шестиосновная кислота со сле¬
дующими значениями рК: pKi = 2,2; рК2 — 2,9 (дис¬
социация двух ацетатных групп); рКз = 7,0; рК* =
— 7,8 (кислотная диссоциация фенольных гидрокси-+лов); pKs = 11,4; рКи = 12 (диссоциация NH-rpynn).
При pH = 7—8 индикатор бесцветный, окрашивается
в бледно-розовый, при pH = 11 цвет становится ярко-
красным.Ксиленоловый оранжевыйНООС—Н,Сноос--Н,с/Ан,н2с/сн2—соон
.гI Ч'СН2—сооннон,оОсн36представляет собой трифенилметановый краситель,Ю Аналитическая химия289
который содержит иминодиацетатные группы, харак¬
терные для всех типичных комплексонов. При pH =
= 1—5 раствор индикатора желтого цвета, при pH =
= 7 — красно-фиолетового. Со многими металлами
при pH = 1—б он образует комплексы красного цве¬
та. Ксиленоловый оранжевый является одним из луч¬
ших реактивов для фотометрического определения
циркония, висмута, олова, индия, редкоземельных эле¬
ментов и др.Метилтимоловый синийпо спектрофотометрическим характеристикам превос¬
ходит ксиленоловый оранжевый. При образовании
комплексных соединений с металлами цвет изменяет¬
ся от слабо-серого до ярко-красного. Недостатком
индикатора является малая его устойчивость во вре¬
мени. Как правило, растворы готовят перед анализом.Рассмотрим более подробно процесс изменения
окраски металлохромных индикаторов в зависимости
от pH раствора и от их связывания в комплексы с ка¬
тионами металлов на примере эриохрома черного Т.Этот индикатор при возрастании pH переходит из
одной окрашенной формы в другую, в соответствии со
следующей схемой:красный
pH от 0 до 6, H2Ind290
no2синийpH от 7 до 10, Hind2-желтый
pH — 11. Ind3-Эриохром черный Т образует с катионами метал¬
лов, как правило, комплексы красного цвета в пре¬
делах pH = 7—10. Следовательно, в этом интервале
pH при титровании раствором ЭДТА вблизи или пос¬
ле точки эквивалентности происходит переход окрас¬
ки от красной (цвет комплекса Mind-) до синей
(цвет свободного индикатора в форме Hind2-). Ины¬
ми словами, сначала идет реакция Mrt+ -j- H2Y2* =?=ь
ч==* МУ(Л_Ч,+ + 2Н\ а затем реагируете окрашенным
комплексом:MInd“ + H2Y2' 4=fc MY2'+ HYJ-+ HIndJ- + H+.
Раздел VАБСОРБЦИОННАЯ СПЕКТРОСКОПИЯМетоды анализа, основанные на измерении умень¬
шения интенсивности (или мощности) потока элект¬
ромагнитного излучения составляют группу абсорб¬
ционных спектроскопических методов.Поглощая электромагнитные излучения, молекулы
и атомы вещества переходят в энергетически возбуж¬
денное состояние. Поглощенная атомами или молеку¬
лами избыточная энергия расходуется на повышение
их колебательной, вращательной или поступательной
энергии, а в некоторых случаях выделяется вторичное
излучение или проходят фотохимические процессы.Известно много различных видов электромагнит¬
ных излучений: у_лУчи. рентгеновское излучение,
ультрафиолетовое, видимое, инфракрасное, микровол¬
новое и радиочастотное (рис. 15.1). Согласно волно¬
вой теории все виды излучения представляют собой
колебания напряженности электрического и магнит¬
ных полей.В спектрофотометрии и колориметрии используют
избирательное поглощение молекулами вещества, по¬
лученного в результате предварительно проведенной
химической реакции с определяемым компонентом
(иногда без предварительного проведения реакции,
например при определении веществ по собственному
поглощению). Молекула поглощающего вещества при
поглощении энергии переходит из основного в воз¬
бужденное состояние, т. е. из состояния с минималь¬
ной энергией Е0 в состояние с большей энергией Е\.
В возбужденном состоянии молекулы или атомы, как
правило, находятся короткое время (10-9—10-8 с);
затем электроны самопроизвольно (спонтанно) пере¬
ходят на более низкий энергетический уровень или на
уровень основного состояния. Этот процесс сопровож¬
дается выделением энергии в виде тепла или электро¬
магнитного излучения, или одновременно того и дру¬
гого. Вызванные поглощением определенных квантов
электромагнитного излучения электронные переходы
характеризуются строго определенными полосами по-292
Тилы переходов,
вызванные
поглощением tO21Тип поглощаемого
излученияПереходы■внутреннихэлектроновПереходывнешнихЮ19Л17** tтри
1ниемУльтрафиолетовое
электронов Л t д|д-|| I .«акуумм^'"Молекулярное «•
колебание «10>3-льтафиолет.видим|11!|ТГ1Г'ИнфракрасноешинМолекулярное
вращение 1fl11Изменение л
спинового/1
состояния
лод действиеммагнитного поля10;Ml II I I I I III I
РадиоизлучениеL1UI III Ш11IQ-41010-10®'106 1-1010-2102104Увеличенная область
ультрафиолетового -
видимого излученияУльтрафиолетовое
ФиолётовоеЦеннее
Зеленое-Ю-2ЖелтоеОранжевоеКрасное•МО3• 5-104а-4 я. X■ к.- 2.5 10* jf
56 4о-1.66)0«|т- 0-10-5-125-10*Рис. 15.1. Электромагнитный спектр излученияглощения в электронных спектрах поглощающих мо¬
лекул. Необходимо отметить, что поглощение квантов
электромагнитного колебания происходит только в
том случае, если энергия поглощаемого кванта совпа¬
дает с разностью энергий А£ между квантованными
энергетическими уровнями в возбужденном Еt и ос¬
новном Ео состояниях поглощающей молекулы:AE = El—E0 — hy,где h — постоянная Планка, равная 6,625-Ю-34 Дж-с; v — ча¬
стота поглощаемого излучения, измеряемая в обратных секундах
(с-1), герцах (Гц).Частота определяется энергией поглощающего
кванта и равна отношению скорости распространения
излучения с (скорости световой волны в вакууме с =
= 3-1010 см/с) к длине волны А,Длину волны измеряют в микрометрах или микронах
(1 мкм = 1 мк = 1 • 10-6 м), нанометрах или милли¬
микронах (1 нм = 1 ммк =10 А = 1 X Ю-9 м).293
Рас. 15.2. Области, спектра,
используемые в фотометри¬
ческом анализеСпектршротометрический анализДлины волн электромагнитных излучений часто
характеризуют также волновым числом vВолновое число, а также частота пропорциональны
энергии, т. е. чем больше АЕ, тем больше волновое
число и частота. Длина волны, наоборот, обратно про¬
порциональна энергии, т. е. чем меньше АЕ, тем
больше длина волны. Волновое число измеряют в об¬
ратных сантиментах (см-1).В зависимости от типа поглощаемого излучения и
способа преобразования избыточной энергии вещест¬
вом различают следующие абсорбционные методы
анализа:1. Молекулярный абсорбционный анализ основан
на поглощении электромагнитных излучений молеку¬
лами или сложными ионами в ультрафиолетовой, ви¬
димой или инфракрасной областях спектра. К это»
группе методов относят спектрофотометрию, колори¬
метрию и ИК-спектроскопию.2. Нефелометрический и турбидиметрический ме¬
тоды анализа основаны на измерении рассеянного или
поглощенного света взвешенными частицами анали¬
зируемого вещества.3. Люминесцентный (флуориметрический) анализ
основан на измерении излучения (интенсивности или
суммы света), возникающего в результате выделения
избыточной энергии возбужденными молекулами ана¬
лизируемого вещества.4. Атомно-абсорбционный анализ основан на по¬
глощении световой энергии атомами определяемого
вещества (см. гл. 30).Между этими методами имеется существенное
различие, однако их часто объединяют в одну группу
спектроскопических или спектрохимических методов
анализа*294
В дальнейшем под фотометрическими методами
анализа будем подразумевать только колориметриче¬
ский и спектрофотометрический методы анализа
(рис. 15.2).ГЛАВА 15ФОТОМЕТРИЧЕСКИЙ АНАЛИЗ
§ 1. ОБЩИЕ ПОЛОЖЕНИЯФотометрическое определение состоит из двух частей:1) переведение определяемого компонента в погло¬
щающее электромагнитные колебания соединение;2) измерение интенсивности поглощения электромаг¬
нитных колебаний раствором полученного соединения.
Фотометрические методы разработаны для определе¬
ния практически всех элементов (рис. 15.3). Однако
не для всех ионов разработаны реакции получения со¬
единений, растворы которых поглощали бы электро¬
магнитные колебания в ультрафиолетовой, видимой и
ближней инфракрасной областях спектра. Поэтому
фотометрические методы делят на прямые и косвен¬
ные или, другими словами, методы фотометрического
определения основаны на реакциях трех типов:1. К + R ч—^ XR (прямой метод)опре- соединение,деляе- поглощаю-мы* ион шее электро¬магнитный
спектр2. MR + V MY (косвенный метод)соединенна, опре-поглошаю- деля¬
щее электро- емый
магнитный ипн
спектр3. X -f- R XR (косвенный метод)опре- нераство-деля- римве сое-емый дин«яиеимяШ
■й1. ■■■ 1 :IIin1У. r. v :■ ЫVIIm11Й 01. 'i■ ,(£2IILi D1Be а□ во ca к□o.адр[i; Me3111щ оMg 0a Ai□ Si"о pA Sд ClAi4IVк оCa □Sc □ti аv □Cf □Mn □F«DCftPN1 □V□ Си0 Zn□ Ga□ Ge□ As□ Se□ 0i5VIЙЬ оSr оY □Zr □Nb □Ho □Tc □RuDRhOPdPVII□ Afla Cd□ In□ SnP Sb□ Теа tXe«VIIIС* оBa оLl‘ □Hi □Та аW □Ha □Os а!( ПPmIX□ Аи□□ TIП Pb□ Bi□ PoAtRn7XFrRaAc** □Ku105□ Ce |o Pi |d Nd |о Рд|п Cm |о Eii |nGd |dTI) [п Оу |п Ho |d Ei |аТпг|о Vb |П la
**Акганоиды□ Pa□ uNpPuAmCm6kcrEsFmMdРис. 15.3. Прямые и косвенные методы определения ионов:□— прямые; Д— косвенные, основанные на разрушении соединений, погло¬
щающих электромагнитные колебания; О-косвенные, основанные на пред¬
варительном осаждении определенных ионов н последующем фотометриче¬
ском анализе осадкаВ последнем случае полученный осадок отделяют,
переводят в раствор и один из компонентов осадка
определяют фотометрическим методом. Таким обра¬
зом, косвенные методы основаны на разрушении со¬
единений, поглощающих электромагнитные колеба¬
ния, а также на реакциях осаждения с последующим
определением одного из компонентов осадка. Ясно,
что наилучшими являются прямые методы, косвенные
методы 3-го типа наихудщие, их используют только
в том случае, когда отсутствуют методы 1-го и 2-го
типов.Из рис. 15.3 видно, что косвенные методы приме¬
няют для определения щелочных металлов и большин¬
ства неметаллов, сульфат- и галогенид-ионов.£ 2. ТИПЫ СОЕДИНЕНИИ, ПРИМЕНЯЕМЫЕ
В ФОТОМЕТРИЧЕСКОМ АНАЛИЗЕСоединения, применяемые в фотометрическом ана¬
лизе, можно разделить на 8 групп.1. Однороднолигандные комплексы с неорганиче¬
скими лигандами. Эту группу следует разделить на286
четыре подгруппы: 1) роданидные и галогенидныс
комплексы (определение FeIU, Mo, W, Bi, Nb, Re, Со
и др.); 2) аммиака,ты (определение Си**); 3) комп¬
лексы металлов с пероксидом водорода (определение
Ti, V, Nb, Та, Се и др.); 4) гетерополикислоты (опре¬
деление Р, Si, As, Nb, V, Ge и др.).2. Однороднолигандные хелатные (внутрикомп-
лексные) соединения. Эту группу следует разделить
на пять подгрупп: 1) соединения металлов с поли фе¬
нолами и оксикислотами (определение Fe3+, Ti, Nb,
Та и др.); 2) соединения металлов с органическими
красител»Ш1 типа ализарина (определение AI, редко¬
земельных элементов, Zr, Hf и др.); 3) соединения
металлов с органическими реактивами, содержащими
аминный азот (определение Hg, Al, Mg, Со и др.);
4) соединения металлов с органическими реагентами,
содержащими нитро- и нитрозогруппы (определение
Со, К, FeIM и др.); 5) соединения металлов с органи¬
ческими реагентами, содержащими тионную и тиоль-
ную группы (определение Hg, Ag, F b, Cd, Cu, Bi, Sn,
Sb, As, Zn, Fe, Ni, Со и др.).3. Разнолигандные и разнометалльные комплексы.
К этой группе отнесен 21 тип соединений:1955 г. 1965 г. 1970 г. 1975 г... \НАяКц] 3. [mrX] 7. [МА,] [mrJ U. [MRJ [мв^]
2. (АИ)Л |MR^] 4. [MA,]^ 8. (AH^ [MRtfR^] 15. [M (HRJ^J R^R*К этой группе отнесены также ионные ассоциаты, ко¬
торые близки к ней, но отличаются по природе хими¬
ческой связи. Однако судя по сдвигу (хотя и неболь¬
шому) полос поглощения основных красителей и их
соединений с анионными комплексами или сложными
анионами и по другим свойствам, такое обобщение,
по-видимому, допустимо, тем более, что эти соедине¬
ния выделяются в отдельные подгруппы.б. Тройные6. MM'Rгетеро-лолн-кислотыЧмк*\К 1б-[мнлГ(н2*)+
10- ИЛ) К ,7- Iм w*] «АН>*
"■ ,8- [ммЧЛ]12. [мМ^Л,] R; 19. [MAxRj,] [m'R;Jis. [MR;R;Rz](AH)m го. [mr^x]21. [MRR'AJR*297
Разволигандные комплексы в последнее время наи¬
более широко применяют в фотометрическом анализе.4. Окрашенные соединения, получаемые при реак¬
циях окисления — восстановления (определение Мпт
Сг, Ni, As, Se, Те и др.).5. Малорастворимые соединения и соединения ад¬
сорбционного характера (определение NHt, Mg2*,
Na\ Sb3‘ и др.).6. Органические соединения, получаемые при ре¬
акциях синтеза с участием неорганических веществ
(определение NHi. NO^, NO^).7. Аква-ионы и другие простые соединения, обла¬
дающие собственным поглощением (определение Fe‘M,
Ni, Со, Си, Сг, оксидов азота и др. по собственному
поглощению).8. Кислотно-основные индикаторы (определе¬
ние pH).Однороднолигандные комплексы с неорганически¬
ми и органическими лигандами имеют большое значе¬
ние для фотометрического анализа. Однако особенно
важны для данного метода анализа, в том числе для
экстракционно-фотометрических определений, разно*
лигандные и разнометалльные комплексы, которые
дают возможность снизить предел обнаружения и по¬
высить селективность реакций. Соединения этого типа
применяли в фотометрическом анализе давно, но во
многих случаях была не ясна их природа. После вни¬
мательного исследования комплексных соединений в
растворах выяснилось, что образование разнолиганд-
ных комплексов является скорее правилом, а не ис¬
ключением, как это считалось ранее. Даже если в рас¬
творе находятся только два компонента, то и тогда
чаще всего нельзя говорить об однороднолигандных
соединениях, так как во многих случаях в состав ком¬
плексов входят молекулы растворителя, а также
гидроксил-ионы, которые образуются при гидролизе
или, наоборот, может присоединяться протояирован-
ный реагент.Отсюда вытекает новая задача: необходимо знать
состояние ионов металлов и реагента в условиях их
взаимодействия, которое в значительной мере влияет
ira реакционную способность этой пары. Выяснение
состояния взаимодействующих компонентов проли¬298
вает свет также и на природу продукта реакции. Ис¬
следование состояния ионов в растворе очень трудо¬
емкая, но и необходимая работа. Знание состояния
ионов в растворе нужно не только для выяснения хи¬
мизма аналитических реакций, но и для выяснения
химизма технологических процессов. Известно, что
применение маскирующих реагентов не всегда без¬
обидно, часто такой реагент может вступать в реак¬
цию с определяемым ионом. При этом образуется
разнолигандный комплекс, который может проявлять
совсем другие свойства.Применение разнолигандных комплексов во мно¬
гих случаях приводит к повышению селективности,
контрастности реакций, улучшению экстракционных и
других свойств. Приведем несколько примеров. Опре¬
деление малых количеств тантала в присутствии боль¬
ших количеств ниобия—очень трудная задача. Од¬
нако эта задача была успешно решена с применением
экстракционно-фотометрического метода определения
тантала в виде ионных ассоциатов гексафторидноп*
комплекса тантала с основными красителями. Анало¬
гичную трудность испытывали аналитики при опреде¬
лении малых количеств рения в присутствии больших
количеств молибдена. Только применение экстракции
с трифенилметановыми красителями дало возмож¬
ность определять очень малые количества рения в мо¬
либдене или молибденовых рудах с довольно низким
пределом обнаружения. Это же относится к определе¬
нию осмия в присутствии других платиновых метал¬
лов, определению бора и других элементов. Введение
второго реагента часто приводит к улучшению экс¬
тракционных свойств комплексов и снижению предела
обнаружения. Так, дитизонат никеля очень плохо экс¬
трагируется неводными растворителями. Для полной
его экстракции тетрахлоридом углерода требуется
примерно 24 ч. Если же ввести третий компонент —
1,10-фенантролин или 2,2/-дипиридил, то комплекс экс¬
трагируется очень быстро, а предел обнаружения ни¬
келя снижается в пять раз.Как было отмечено выше, фотометрические методы
определения анионов менее разработаны, чем катио¬
нов. На многие анионы нет цветных реакций, и в фо¬
тометрическом анализе для их определения приме¬
няют реакции разрушения окрашенных соединений.299
Между тем исследование разнолигандных комплексов
привело к разработке; прямых методов определения
фтора с ализаринкомплексоном, арсеназоЩ и дру¬
гими реактивами. Таким образов, одним из перспек¬
тивных направлений разработки фотометрических ме¬
тодов определения неметаллов, для которых нет цвет¬
ных реакций, является исследование окрашенных раз¬
нолигандных комплексов с участием этих ионов.Остановимся очень кратко на: образовании разно-
металльных комплексов. На рис. 15.4 представлены
кривые экстракции диэтиловым эфиром комплекса
железа с дюмогаллионом в присутствии других метал¬
лов. Как видно из рисунка, комплекс железа с люмо-
галлионом практически не экстрагируется, а в присут¬
ствии других металлов железо извлекается. Это ука¬
зывает на образование разнометалльных комплексов,
которые лучше экстрагируются. Объяснить экстрак¬
цию такого рода соединений труднее, чем рост экс¬
тракции разнолигандных комплексов. Приведем еще
один пример образования разнометалльно-разноли-
гандных комплексов. Галлий образует разнолиганд-
ный комплекс с пиридилазорезорцином и антипири¬
ном, а индий в этих условиях не образует подобных
комплексов. Однако если в растворе присутствует ин¬
дий, то образуется комплекс индия и галлия и моляр¬
ный коэффициент поглощения повышается в два раза.
Таким образом, можно сделать заключение о том, что
одним из важных направлений разработки более се¬
лективных фотометрических реакций и с меньшим пре¬
делом обнаружения является дальнейшее исследова¬
ние и применение разнолигандных комплексов.Необходимо иметь в виду, что образование разно¬
лигандных и разнометалльных комплексов может иг¬
рать и отрицательную роль при фотометрическом оп¬
ределении, поэтому при построении градуировочных
графиков в раствор сравнения следует вводить те же
компоненты, которые содержатся в исследуемом рас¬
творе.§ 3. СВОЙСТВА И ПРИРОДА ЭЛЕКТРОННЫХ СПЕКТРОВЭлектронными называют спектры, возникающие в ре¬
зультате поглощения (испускания) веществом кван¬
тов электромагнитного излучения, соответствующих300
Рис. 15.5. Общий вид простого
спектра поглощения и схема
расчета полуширины полосы
поглощенияф1lit1 3 5 pHРис. 15.4. Экстракция комплексных соединений железа(Ш) с лю-
могаллноном диэтиловым эфиром:;_Fe; J-Fe + Sc; Л-Fe + Y; 4-Fe + La: lFe*4]=2,2-IO—8 Af; (Sc). [La].
[Y]~=3,3-10“8 M (каждый); люмогаллиои — 7.5- I0-5Mчасти ультрафиолетовой, видимой и части инфра¬
красной областей спектра. Резкой границы между ве¬
личинами квантов света для области электронных
спектров нет, однако традиционно к электронным
спектрам относят область с длинами волн в интер¬
вале 2-102— 103 нм.В подавляющем большинстве случаев различные
окрашенные соединения, анализируемые фотометри¬
ческим методом, характеризуются довольно широкой
полосой поглощения. Спектром поглощения соедине¬
ния, поглощающего электромагнитные колебания, на¬
зывают более или менее сложную кривую зависимо¬
сти оптической плотности А или молярного коэффи¬
циента поглощения е от длины волны А. или частоты v.
Таким образом, спектр поглощения выражают в виде
кривой A=f(X), указывая толщину слоя и концен¬
трации истинную или формальную (рис. 15.5). Если
состав и состояние равновесия образования погло¬
щающего электромагнитные излучения соединения из¬
вестны, тогда спектр поглощения выражают как
функцию е = fCK).В фотометрическом анализе имеет значение ши¬
рина полосы поглощения. Очевидно, что чем шире по¬
лоса, тем труднее анализировать смесь нескольких301
Рис. 15.6. Спектр атомов, желез»соединений. При широких поло¬
сах поглощения реактива и ком¬
плекса более вероятно взаимное
наложение этих полос, что не¬
редко затрудняет анализ. Шири¬
ну полосы поглощения: устано¬
вить практически трудно, так
как по обе стороны от Х,Ма«с ин¬
тенсивность поглощения лишь
асимптотически приближается к
нулю. Поэтому пользуются ха¬
рактеристикой полуширины по¬
лосы поглощения.Полушириной ПОЛОСЫ П0ГЛ0‘
щения называют расстояние
Л'«енакс-^/2енакс (СМ. РИС. 15.5).
В большинстве случаев полуши¬
рина полосы поглощения про¬
стых молекул составляет 80'—
100 нм. Чем эта величина мень¬
ше, тем лучше. Возникновение
электронных спектров прежде
всего связано с перестройкой
(возбуждением) электронной
оболочки вещества, а не с движе¬
нием ядер, как при возникнове¬
нии колебательных или вращательных спектров. Од*
нако это замечание не следует понимать так, что
в веществе можно строго разделить движение элект¬
ронов и ядер. Особенности, и в частности, форму кон¬
туров полос в спектрах молекул можно понять только
с учетом движения и электронов, и ядер. Поэтому
точнее говорить не об «электронных» спектрах моле-
кул, а об электронно-колебательных (вибронных)
спектрах. Именно влиянием движения ядер объяс¬
няется различие вида электронных спектров молекул
и атомот. ¥ атомов’ спектры линейчатые (рис. 15.б);
тогда как молекулярные спектры характеризуются
полосат» разной ширины (рис. Г5.7). Б зависимости
от состава молекул и уел они# измерения спектров
(газовая или* конденсированная- фаза*, тип раствор»-302
•Рис. 15.7. Молекулярные спектры веществ:«—спектр состоит из одной полосы; б —спектр состоит из двух перекры¬
вающихся частично полостеля, температура) полосы в электронных спектр ах
могут быть гладкими или характеризоваться лучше
или хуже выраженной колебательной и вращательной
структурами (рис. 15.8).Спектры поглощения (испускания) веществ со¬
стоят из отдельных полос (линий). Одной из сущест¬
венных характеристик отдельных полос является есте¬
ственная ширина. Полоса, форма которой обусловле¬
на естественной шириной, не может быть разделена
на несколько отдельных полос при повышении разре¬
шающей способности спектрального прибора. Естест¬
венная ширина полосы обусловлена временем жизни
молекулы (атома, вещества) в определенном энерге¬
тическом состоянии. Как следует из соотношения не¬
определенностей Г айзенбергаДЕ М > ftуменьшение времени жизни частицы в определенном
состоянии (а значит и уменьшение неопределенностиРис. 15.8. Электронные спектры веществ:л — гдядкяй контур, колебательная структура не проявляется; б -на контуре
полосы вндны следы колебательной структуры; в -спектр поглощения паров
антраиеиа с четкой -колебятел&нвй структурой303
времени —А/) приводит к возрастанию неопределен¬
ности энергии. Как было показано выше, время жизни
свободных атомов в возбужденном состоянии 10~8 с,
тогда как время жизни молекул в возбужденном со¬
стоянии из-за большой вероятности безызлучательных
переходов составляет лишь 10-13 с Уменьшение на
5 порядков времени жизни приводит к возрастанию
на такую же величину естественной ширины спек¬
тральной полосы. Это объясняет почему атомные спек¬
тры— линейчатые, а молекулярные — полосатые.Разумеется, время жизни веществ в определенном
энергетическом состоянии зависит не только от
свойств отдельных атомов или молекул, но и от вза¬
имодействия между ними. Так, хорошо известно уши-
рение линий в атомных спектрах, вызываемое ростом
температуры исследуемого газа. Повышение темпе¬
ратуры вызывает учащение столкновений атомов и со¬
кращение времени их жизни в определенном энерге¬
тическом состоянии.Ширина полос резко увеличивается при переходе
к конденсированному состоянию вещества вследствие
сильного влияния соседних молекул кристалла или
жидкости.В соединениях редкоземельных элементов f-oрби-
тали атомов мало перекрываются с орбиталями ли-
гандов и локализованы на самих атомах (ионах)
РЗЭ. Поэтому для f~f* переходов сохраняются ли¬
нейчатые спектры, характерные для свободных ато¬
мов. Даже сильные комплексообразователи, такие как
оксикислоты, комплексоны и другие, очень мало
влияют на положение полос; более заметно влияние
комплексообразования на интенсивность полос редко-
земельных элементов.Формально, как и для любых молекулярных спект¬
ров, природу электронных спектров можно охаракте¬
ризовать переходами вещества в различные энергети¬
ческие состояния (рис. 15.9). Наиболее устойчивому
состоянию вещества соответствует наименьшая энер¬
гия (нулевой уровень на диаграмме рис. 15.9). Такое
состояние называют основным, все остальные состоя¬
ния называют возбужденными. Как показано на
рис. 15.9, вещество может поглощать кванты света
только определенной величины, например, hvi = Е\ —
— Е0 или h\2 = £2 — Е0, что соответствует поглоще-304
ii\
И 1i\hJj =)\hi, =-f,Рис. 15.9. Диаграмма энергети¬
ческих переходов вещества=Н=Ф£-н—свнв(донор)-W--н h(акцептор)Рис. 15.10. Схема переноса заряда в комплексе иода с бензолом
(пунктиром показаны уровни, между которыми происходит пере¬
нос заряда)нию излучения с определенными длинами волн Х| итак как k = c/hv.Поскольку каждое вещество характеризуется сво¬
ей системой энергетических уровней, то и спектры ве¬
ществ различаются как по числу полос, так и по их
положению в шкале длин волн.Таким образом, возможность рассчитать (предви¬
деть) положение полос в спектрах (Ямакс, см. рис. 15.7)
определяется умением рассчитать возможные энерге¬
тические состояния вещества.Другим важным свойством спектров является ин¬
тенсивность поглощения (испускания). Это свойство
в первую очередь обусловлено характером перестрой¬
ки электронной оболочки вещества, конкретнее —
электрического дипольного момента. Чем больше из¬
меняется дипольный момент вещества при возбужде¬
нии, тем интенсивнее полоса в спектре. Дипольные
моменты вещества в основном и возбужденном со¬
стояниях можно рассчитать, если известны волновые
функции вещества в этих состояниях.Существуют правила отбора (запрета), позволяю¬
щие заранее определить, какие из переходов не дол¬
жны проявляться или должны быть неинтенсивными
в спектрах.1. Запрещены переходы между энергетическими
состояниями, характеризующимися различным спи¬
ном (мультиплетностью). Это свойство особенно
важно при интерпретации спектров люминесценции,
так как возникновение долгоживущих возбужденных30S
состояний (медленно затухающей люминесценции,
фосфоресценции) как раз обусловлено таким за¬
претом.2. Запрет перехода по симметрии. Зная -симметрию
волновых функций основного и возбужденного состоя¬
ний, можно заранее знать, в каких случаях возбужде¬
ние не вызывает изменения дипольного момента ве¬
щества, т. е. полоса не должна проявляться в
спектре.Существуют и другие, менее общие, правила за¬
прета.Приведем классификацию электронных спектров»
основанную на указании типов уровней, между кото¬
рыми происходит переход. Типы уровней в свою оче¬
редь определяются составом и строением веществ.1. d* — d-, f* — /-спектры. Переходами между d-
или /-орбиталями обусловлена окраска соединений пе¬
реходных металлов. Эти переходы являются запрещен¬
ными по Лапорту и поэтому соответствующие полосы
в спектрах малоинтенсивны. Значения молярного ко¬
эффициента поглощения е находятся обычно в преде¬
лах J0—1000.2. я* — я-спектры возникают в молекулах с сопря¬
женными двойными связями. Интенсивность этих по¬
лос может варьировать в широких пределах и е мо¬
жет достигать значений 10s. Окраска большинства
молекул красителей обусловлена такими пере¬
ходами.3. п — я*-переходы возникают в молекулах с со¬
пряженными связями, которые содержат гетероатомы
с неподеленными парами электронов (/г-электроны).
Возбуждение электронов неподеленной пары на ва¬
кантный л*-уровень характеризует природу этих спек¬
тров. Такие переходы запрещены и поэтому соответ¬
ствующие полосы в спектрах малоинтенсивны.4. Полосы переноса заряда. Иногда структуру
энергетических уровней вещества можно приближенно
охарактеризовать группами уровней, относящимися к
различным составным компонентам (фрагментам) со¬
единения. Классическим примером соединения, окра¬
ска которого обусловлена переносом заряда, является
комплекс иода с бензолом. Систему уровней этого
■комплекса можно в хорошем приближении считать306
состоящей из групп уровней иода и бензола, лишь
слегка возмущенных взаимодействием (рис. 15.10).
Окраска обусловлена переносом электрона с верхнего
занятого уровня молекулы донора — бензола на сво¬
бодный антисвязывающий уровень молекулы акцеп¬
тора — иона. Поэтому говорят о переносе электрона
с бензола на иод и рассматривают этот переход как
перенос заряда. Полосы переноса заряда, как прави¬
ло, достаточно интенсивны, молярный коэффициент
поглощения имеет значения 103 — 104.§ 4. ПРИРОДА ОКРАСКИ,СПЕКТРЫ ПОГЛОЩЕНИЯ СОЕДИНЕНИИ,ПРИМЕНЯЕМЫХ В ФОТОМЕТРИЧЕСКОМ АНАЛИЗЕ.
НАПРАВЛЕННЫЙ понск веществ
с требуемыми оптическимиСВОЙСТВАМИВ фотометрическом анализе применяют вещества
практически всех известных классов. Основные требо¬
вания анализа — это достаточная интенсивность окра¬
ски, обеспечивающая надежное определение мнкро-
шмпонентов, т. е. низкий предел обнаружения веще¬
ства и контрастность реакции. Последнее свойство
определяется разностью положения полос поглощения
исходных веществ и продуктов реакции. Задача тео*
рии — предвидеть эти свойства. Природу спектров
лучше можно охарактеризовать, рассматривая отдель¬
ные классы соединений. Можно выделить следующие
классы соединений с точки зрения природы их
окраски.1. Координационные соединения металлов с кра¬
сителями, окраска обусловлена л* — я-переходамн
в молекуле красителя.Внутрисферные соединения: соединения металлов
с оксиантрахнно'нами, отаиаэокраситедями, красителя¬
ми трифенилметанового ряда (пирокатехиновый фио¬
летовый, бромпирогаллоловый красный и др.), триок-
сяфлуоронами и многими другим». Природа окраски
таких соединений наиболее полно изучена. Лиганды-
красители обычно являются слабыми кислотами.
Их. окраска изменяется при диссоциации. На¬
пример:307
ализаринО ОНH2R *=± HR* *=± R*
Xjiikci нм 430 535 565трноксифенилфлуоронH3R 3E=fc H2R‘ HR1'ЯМакс, нм 490 520 560Окраска комплексов металлов с такими реагентами
обычно определяется полосами поглощения с макси¬
мумами, расположенными между максимумами полос
поглощения нейтральной и одной из анионных форм
реагента (табл. 15.1). Это понятно, если принять, чтоТаблица 15.1. Положение максимумов полос поглощения
комплексов металлов с ализарином и фенилфлуорономСоединение илн ион*максСоединение или ион1лмаксАлизарин (HjR)430Фенилфлуорон (HaR)495HR'535H*R~520RJ-565HRS-560Комплексы ализарина сКомплексы фенилфлуоро-
на с металламиметалламиGeiv507АП"530Sniv510Y>4 и лан¬550Sbin530таноидыTaV530Zriv520Aim540GaHi530In545308
связь аниона реагента с металлом более ионная, чем
связь этого же аниона с ггротоном. Такое допущение
является вполне справедливым и позволяет прогнози¬
ровать контрастные возможности аналитических
реагентов такого типа. С этой точки зрения контраст¬
ность фотометрической реакции определяется раз¬
ностью положений максимумов полос двух кислотно¬
основных форм реагентов, одна из которых соответ¬
ствует форме свободного реагента в условиях
анализа, а другая — форме реагента в комплексе.Например, ализарин при pH = 5 находится в рас¬
творе в нейтральной форме с ?1Макс = 430 нм. В этих
же условиях алюминиевый комплекс ализарина имеет
полосу поглощения с ?1Макс = 530 нм, которая соответ¬
ствует дважды депротонированному иону (R2-) али¬
зарина. Контрастность реакции в этом случае боль¬
шая, АХ = 100 нм. Если же какой-либо металл
взаимодействует с ализарином в щелочной среде,
например Са, когда реагент находится в форме одно¬
зарядного аниона, Ямакс = 480 нм, то контрастность
реакции ухудшается, АХ = 50 нм. В сильнокислых
средах реакции также могут быть малоконтраст¬
ными. В этом случае, хотя реагент (например, ализа¬
рин) находится в нейтральной форме, часто обра¬
зуются протонированные комплексы, в которых ли¬
ганд находится в недиссоциированной форме. Так по¬
лучается, например, при взаимодействии ализарина с
Nbv в сильнокислых средах. Контрастность в этом
случае соответствует ДХ = 30 нм.Внешнесферные соединения (ионные ассоциаты):
соединения ацидокомплексов металлов с основными
красителями, ассоциаты катионных комплексов метал¬
лов с анионами кислотных красителей.Как отмечалось выше, к этому классу соединенийотносят ионные ассоциаты типа • mKp+ (R—ацидолиганд, часто галогенид-ион, Кр — основный
краситель) и комплексы типа МА™+ • mR~ (А —
обычно нейтральный лиганд, R- — анион кислотного
красителя). Существенной спектрофотометрической
характеристикой таких комплексов является очень ма¬
лая контрастность реакций. Спектры простой соли
красителя практически совпадают со спектрами ком¬
плексов (ассоциатов) (табл. 15.2). Поэтому такие309
Таблица 15.2. Ионные сиссоциаты. с осно$нь1ми
и кислотными красителямиЭлементКомплексный
ион металлаКрасительСостав2sАишAuci;Метиловый фиоле¬
товый (K+)Auci; • к*575Au(CN);Малахитовый зеле¬
ный (К+)Au(CN); ■ к*610TavTaF*"Кристаллический
фиолетовый (К+)TaF“' •2К+590Р, As, SiГПК7'То жеГМК7' -7К*595Bi1"Bii;Метиловый зеле¬
ный (К1)Bii;-K+660AgfAg(Phen)+Бромфенолоный
синий H2R[Ag Phen]+-1HR1'J410Fe“Fe(Phen)23 +То же(Fe(Phen;3]a+ • R2"600Примечание. ГПК — гег«рополикислотп: Phen — 1,10-фенантролйн.соединения используют обычно в экстракционно-фо¬
тометрических вариантах анализа, предполагающих
разделение комплекса и простой соли красителя. Из
сказанного ясно, что умение прогнозировать фотомет¬
рические характеристики соединений этого типа сво¬
дится к умению прогнозировать спектры сольватиро-
ванных ионов основных (кислотных) красителей.2. Координационные соединения металлов с неок¬
рашенными лигандами. Окраска обусловлена перено¬
сом заряда металл — лиганд. Такие соединения обра¬
зуют металлы с различными степенями окисления: Ti,
Nb, Mo, Fe, Си и др. Например, соединения TiIV и Nbv
с пероксидом водорода, соединения этих же металлов,
а также Movl, Fem с полифенолами, салициловой кис¬
лотой, роданидные комплексы Fe'n и Mov, комплексы
Си1 с дипиридилом и фенантролином и др.В табл. 15.3 и 15.4 приведены оптические характе¬
ристики ряда комплексов Fe11, Fem, Tilv с такими ли¬
гандами как амины, полифенолы, оксикислоты.Характерной особенностью этих соединений яв¬
ляется высокая контрастность реакций, так как избы¬
точные концентрации бесцветного реагента не мешают
определению металла в виде окрашенного комплекса.
При анализе этих соединений достигается более высо-310
Таблица /5.3. Положение полос переноса заряда,
в трисхеялтох Ft"Лю-аидV, эВЕя. Л2-Пир ид ил- N - м ет илал ьдими и
4,7-Дифенил-Г,Г0-фенантролин2J25-2.53 2,33—2.21 2,2'-Днпиридил2,37—a.is0,091,10-Фенантролин2,43— 1,880.092,4-Диамино-6-(пиридил-2) -1,3,5-2,72—0,75—триазин2,4-Диамино-6- (4-фенилпиридил-2) -2,64-1,571 Д5-трназинДи- (1^-метилкетимик)бутандион-2,32,1 8—3,430.11Бенэилдиимин1,99-3,340,129,ГО-Феи »ятрен хквоид тем от1,73—3,670,13кая селективность по сравнению с селективностью
определения соединений, окраска которых обусловле¬
на вяутрилигандными переходами и потому слабо за¬
висит от типа металла, особенно в ряду металлов с
одинаковой степенью окисления. В соединениях с пе¬
реносом заряда природа металла сказывается на
спектрах самым радикальным образом.В литературе есть примеры успешного прогнозиро¬
вания оптических свойств соединений с полосами пе¬
реноса заряда. Наиболее доступным является метод,Таблица 15.4. Положение полое переноса заряда
в комплексах Fe‘n и TilvЛигандFeIHТ»1*Ея- »ВV. эВЕ„. эВV. эВСалициловая кислота-9,802.34—9,873.022-Окси-З-нафтойная-9,162,10—9,232,61кислота1-Окси-2-нафтойная— 8.942.0—9,072,50кислота2-Оксм-2-нафтойньгй-9,272,29—9,271 2.48альдегидПирока-техи»-8,601,70-9.472,69Xромотролова я кислота—7,991,61—8,79%2Ьа- Нафтол-8,882,30(S-Нафтол—8.942,53311
основании# на использовании корреляционного урав¬
нения зависимости положения полос поглощения в
Спектрах комплексов одного металла с рядом одно¬
типных лигандов от рассчитанных значений энергии
одноэлектронных уровней лигандов. Например, для
расчета положения максимумов полос в спектрах Fe11
с а-дииминовыми лигандами можно пользоваться кор¬
реляционным уравнением:v = — 0,И£Я — 20,50Edn + 4,02,где v — энергия перехода, соответствующая максимуму полосы
поглощения (эВ); £п—энергия нижайшей свободной л-орбн-
тали лиганда, эВ; — энергия расщепления dn-уровней же¬
леза, эВ.Для расчета аналогичных оптических свойств со¬
единений Tilv с а-оксикислотами и О-диоксифенолами
получено уравнение:v = 0,42£„ — 1,36,где Ея — энергия верхнего занятого л-уровня (эВ), взятая со
знаком плюс.Соответствующие оптические характеристики при¬
ведены в табл. 15.3 и 15.4,Недостатком соединений такого типа с точки зре¬
ния фотометрии являете^ сравнительно малая интен¬
сивность окраски.3. Соединения переходных металлов с неокрашен¬
ными лигандами. Окраска обусловлена d— d- или
/-/-переходами. В аналитической практике исполь¬
зуют растворы простых солей переходных металлов,
если их концентрации достаточно высоки. Чаще дру¬
гих определяют Со11 по достаточно интенсивной окра¬
ске хлорида GoCli" или роданида Co(SCN)4~. а так¬
же редкоземельные элементы. Спектры соединений
РЗЭ характеризуются узкими полосами поглощения,
благодаря чему удается определять отдельные РЗЭ
при их совместном присутствии.§ 5. КОНТРАСТНОСТЬ ФОТОМЕТРИЧЕСКИХ РЕАГЕНТОВПри выборе органического реагента для фотометри¬
ческого определения тех или других ионов необходимо
учитывать многие факторы, один из которых — спек¬
тральная характеристика реагента и его комплекса с312
Рис. IS.lt. Спектр поглощения
реактива (1) и комплекса (2)определяемым ионом. Как
было показано выше, раз¬
мытость спектра поглоще¬
ния характеризуется полу¬
шириной полос поглощения,
чем меньше эта величина— '—л,«мтем легче выбрать длину
волны для спектрофотомет¬
рического определения одного компонента в присут¬
ствии других. С другой стороны, чем больше разница
в максимумах поглощения комплекса и реагента —
тем надежнее результат (рис. 15.11). Удовлетвори¬
тельным для фотометрического определения яв¬
ляется тот реагент, для которого эта разница равна
приблизительно 100 нм. Для неодинаковых реагентов
это расхождение является различным и зависит от
свойств реагента и растворителя. Известно, что пол¬
нее использовать контрастные возможности' реагента
типа ROH можно, например, путем подбора раствори¬
теля, способствующего отщеплению протона от гидро¬
ксильных групп, содержащихся в молекуле реагента.
Такими растворителями могут быть диметилформ-
амид и диметилсульфоксид, являющиеся достаточно
сильными протоно-акцепторными растворителями. На
рис. 15.12 представлены спектры водных (а) и диме-
тилсульфоксидных (б) растворов фенилфлуорона, со¬
держащих различные количества кислоты или щелочи.
Задача в этом случае состоит в том, чтобы насколько
возможно сдвинуть полосу поглощения ионной формы
реагента в длинноволновую область. Как видно, диа¬
пазон смещения положения максимума самой длинно¬
волновой полосы в водных растворах составляет
515 — 465 = 50 нм, тогда как в диметилсульфоксид-
ных растворах 600 — 480= 120 нм. Такое расширение
спектрального диапазона кислотно-основных форм фе¬
нилфлуорона свидетельствует о более полном исполь¬
зовании контрастных возможностей реагента (в дан¬
ном случае фенилфлуорона).Второй путь улучшения контрастности цветных ре¬
акций— введение подходящих заместителей в моле¬
кулу реагента. Так, в молекулу ализарина можно вве-313
еГ1АРас. 15.12. Спектры поглощения водных (а) и диметилсульфок-
сидных (б) растворов фенклфлуорона:/ — концентрированный раствор НС1; 2 — pH— 5.85; 3 — рН=7.30; 4 — рН*=8,75^
5 — кислая среда; 6 — В-10"-5 М раствор NaOH; 7-8-10 3 М раствор ОНсти нитрогруппу, не ухудшив его спектральную харак-
теристику. Если взять 3-нитро- и 4-нитроализарин, то
полосы поглощения их депротонированных форм сдви¬
нуты в более длинноволновую область по сравнению700 в00адо 500400 500Рис. 15.13. Спектры поглощения растворив. З-яитраализарина (о)
и 4-нитроализарина (б) в 50 % этаноле; титана(IV) с 3-«итро-
алнзаршшм (в), 4-нятроаляэарнном (г) и ализарином [д).
о « б: / — реагент в 5 М растворе НСЦ 2 — реагент в 5 М растворе КОН.
в {Рноог=2.«). г (рНввт=3,50) и 0 (рН0ПТ=Мв): /-ре*гент; 5-коя-
плекс с тятаном (IV)ш
Рис. IS.14. Зависимость экс¬
тракции хлороформом 3-н.итро-
алнзарина (/) и его комплекса
с цирконием (2) от pH рас¬
творас полосой ализарина. По¬
лосы поглощения их ком¬
плексов с металлами так¬
же сдвинуты в более
длинноволновую область, но несколько меньше
(рис. 15.13), вследствие чего контрастность цветной
реакции улучшается. Комплексы металлов с 3-нитро¬
ализарином экстрагируются хлороформом, что приво¬
дит к повышению селективности анализа (рис. 15.14).
Сочетание лучших спектральных характеристик 3-
нитроализарина и его комплексов с металлами, а так¬
же экстрагируемость этих комплексов указывает на
преимущество их применения в анализе по сравнению
с ализарином, комплексы которого не экстрагируются
неводными растворителями.ГЛАВА 16ПОГЛОЩЕНИЕ ЭЛЕКТРОМАГНИТНОГО
ИЗЛУЧЕНИЯ§ 1. ОПТИЧЕСКАЯ ПЛОТНОСТЬ (ЗАКОН БУГЕРА)Для количественного определения вещества фотоме¬
трическим методом после переведения определяемого
компонента в соединение, поглощающее электромаг¬
нитные излучения, необходимо определить ослабление
мощности (интенсивности) потока излучения при про¬
хождении его через поглощающую среду определен¬
ной толщины, т. е. необходимо количественно опреде¬
лить абсорбцию электромагнитного излучения полу¬
ченным раствором (газом или твердыми прозрачными
веществами).Рассмотрим поглощение электромагнитного излу¬
чения, проходящего через прозрачный слой газа, жид¬
кости или твердого тела. Проходя через такой слой,
часть электромагнитного излучения будет селективно315
: * Puc/ IMi Изменение ййтеи-'
снвяости- светового потока
при г црохрждениц через ок¬рашенный растворпоглощаться. Интесив-
ность электромагнит¬
ного излучения при
этом уменьшается. Та¬
ким образом, при про¬хождении монохроматического пучка электромаг¬
нитного излучения через слой прозрачного вещества
(газ, раствор или твердое тело), помещенного в кю¬
вету, часть этого излучения поглощается веществом,
небольшая часть излучения отражается, а часть про¬
ходит через раствор (рис. 16.1). Обозначим интенсив¬
ность падающего излучения через /0, прошедшего че¬
рез раствор — через /, поглощенного раствором — че¬
рез /п и отраженного через /0т, тогда интенсивность
падающего излучения равна сумме составляющих:Л> = / + /от + /п-Интенсивность отраженного излучения достаточно
мала по сравнению с интенсивностью излучения, по¬
глощенного веществом и прошедшего через раствор.
Кроме того, в фотометрическом анализе сравнивают
интенсивности излучения, прошедшего через исследуе¬
мый раствор и растворитель (или раствор сравнения),
в которых интенсивности отраженного излучения бу¬
дут равны. Поэтому интенсивностью отраженного из¬
лучения можно пренебречь.Разделим длину твердого тела, раствора или газа,
через которые проходит излучение на b единиц (см.
рис. 16.1). Когда излучение пройдет через первый уча¬
сток поглощающего вещества, его интенсивность ос¬
лабится в п раз и в конце первого участка будет
равнагде п — число, большее единицы, которое характеризует погло¬
щение данного вещества.Конец первого участка является в то же время на¬
чалом второго. Во второй участок раствора попадает/, =(16.1)П316
таким образом поток света с интенсивностью /i. При
прохождении через второй участок снова произойдет
ослабление излучения в такой же степени, т. е. в
п раз. Таким образом, в конце второго участка интен¬
сивность потока излучения равна/а= —. (16.2)
пПринимая во внимание уравнение (16.1), получим(,63)Аналогично найдем, что после прохождения третьего
участка интенсивность излучения, h /о
/s== —= -^итд-
Таким образом, когда поток излучения пройдет
через всю толщу b (т. е. согласно условию через Ь
участков), интенсивность выходящего потока равна1 = -~. (16.4)'я®Интенсивность поглощения раствора может быть оха¬
рактеризована отношением /о//: чем больше погло¬
щает раствор, тем меньше 1 по сравнению с /о и тем
больше отношение 1о/1■ Это отношение (ослабление
излучения) зависит также от толщины слоя раствора;
согласно уравнению (16.4) эту зависимость можно
выразить уравнением-!j-*=nb (16.5)или логарйфмируяlgA = 6ig„. (16.6)Величину lg(/0//) называют оптической плотностью А
раствора (газа или твердого тела):Л = 1е-у-. (16.7)Это же выражение называют также погашением или
экстинкцией раствора. Величина п в уравнениях(16.1) — (16.6) зависит от свойств поглощающего свет
вещества и для каждого данного вещества остается317
постоянной. Обозначим постоянную величину Ign че¬
рез е, тогда можно записатьA^eb~=ig-^-. (Ш)Ослабление интенсивности светового потока при
прохождении через раствор, газ или твердое тело, оче¬
видно, зависит от количества поглощающих свет цент¬
ров, встречающихся на пути потока излучения.Рассмотрим поглощение излучения раствором со¬
единения при условии, что с изменением концентрации
состав и структура этого соединения не меняются
tмногие органические красители, раствор хромата ка¬
лия, «забуферированный» боратом или карбонатом
натрия и т.д.). Концентрацию раствора обозначим че¬
рез С. Поместим немного раствора в цилиндр и будем
наблюдать поглощение излучения сверку в полном
слое. Если согласно условию при разбавлении рас¬
твора общее количество поглощающих свет центров
остается постоянным, общее светопоглощение также
не изменится. При разбавлении в п раз концентрация
раствора уменьшится в п раз, а толщина слоя во
столько же раз соответственно увеличится, поэтому
общее поглощение излучения не изменится. Следова¬
тельно, можно написатьА = еСЬ. (16.9)Таким образом, выражение оптической плотности
А будет иметь вид:A = lg ~ = йСЬ. (16.10)Эту зависимость называют законом Бугера, по имени
французского физика П. Бугера, открывшего его
в 1729 г.Закон Бугера справедлив для всех областей элек¬
тромагнитных излучений от рентгеновского излучения
до радиоволн. Если раствор, концентрация которого
равна Си при толщине слоя bi имеет такое же погло¬
щение излучения при определенной длине волны, как
и раствор того же вещества при большей толщине
слоя Ь2, то очевидно во втором растворе концентра¬
ция вещества С2 меньше, чем в первом растворе:А = BCibi = еС2Ь2 ... (16.1 Г) C,b, = €ib2 • • • (»6.Г2>»18
, Выражение (16.12.y- используют для расчета кон¬
центрации определяемого вещества в фотометриче¬
ском анализе. Этот закон всегда разделяют на две
части. Зависимость между оптической плотностью ве¬
щества А, толщиной слоя b [уравнение (16.8)] назы¬
вают законом Бугера, иногда Ламберта или Буге¬
ра — Ламберта. Другую часть — зависимость оптиче¬
ской плотности от концентрации (количества
поглощающих центров в единице объема) называют
законом Бера. Однако это неверно. Еще в 1924 г.
С. И. Вавилов писал: «...Трудно постигнуть основа¬
ния той упорной исторической несправедливости, с
которой ... законы, совершенно ясно и отчетливо фор¬
мулированные Бугером, соединяют с именами других
авторов (закон Бера, законы Ламберта и пр.)*.*
Частично эта несправедливость была исправлена: за¬
висимость поглощения излучения от толщины погло¬
щающего слоя теперь часто называют законом не
Ламберта, а Бугера. Однако зависимость ослабления
интенсивности излучения от числа частиц в погло¬
щающей среде и в настоящее время называют зако¬
ном Бера. Нелепость такого утверждения ясно пока¬
зана также Д. П. Щербовым **.§ 2. МОЛЯРНЫЙ КОЭФФИЦИЕНТ ПОГЛОЩЕНИЯЕсли в уравнении (16.10) концентрация С выражена
в молях поглощающего излучение соединения на 1 л*
а толщина слоя b — в сантиметрах, то е выражает
молярный коэффициент светопоглощения. Как видно
из уравнения (16.10), молярный -коэффициент погло¬
щения численно равен оптической плотности 1 М ра¬
створа при толщине слоя 1 см.Молярный коэффициент поглощения е характери¬
зует внутренние свойства вещества и не зависит от
объема раствора, толщины слоя и интенсивности ос¬
вещения. Поэтому величина е является наиболее важ¬
ной, общепризнанной и объективной характеристикой
возможной чувствительности фотометрического опре¬
деления. Значения е в области максимума для раз¬
личных поглощающих свет соединений сильно разли-* С. И. Вавилов. Успехи физ. п., 1924, т. 4, с. 215.** Д. П. Щербов. ЖАХ, 1971, т. 26, № 5, с. 1013.JW
чаются. Так, полосы поглощения «простых» ионов,
(аква-комплексор) меди, никеля и других в видимой'
части спектра характеризуются низкими значениями
е порядка 10—100. Окрашенные аммиакаты, пероксид-
ные и другие однороднолигандные комплексы имеют
значения е « 102—103. Наконец, многие комплексы с
органическими реактивами (ализаринаты, дитизона-
ты и т. п.) имеют очень высокие значения е — поряд¬
ка 104 и 105.Следует иметь в виду, что в некоторых случаях
расчет значения е может быть различным, в зависи¬
мости от принятых условий. Так, если хромофор (что
по гречески значит цветоноситель) представляет со¬
бой органический краситель, тогда е можно вычис¬
лить по отношению к металлу и по отношению к реак¬
тиву. Например, пусть ZrIV образует с некоторым ре¬
активом HR поглощающее свет соединение ZrR4.
При толщине слоя 1 см и концентрации циркония
1 • 10-5 М найдена оптическая плотность А, равная
1,2. Тогда из уравнения (16.10) получим:А 1,2 =1,2-10». (16.13)СЬ 10 5Этот результат характеризует предел обнаруже¬
ния метода определения циркония. Однако для объек¬
тивной оценки необходимо учитывать чувствитель¬
ность реактива HR, который применяют для опреде¬
ления не только циркония, но и элементов с меньшей
степенью окисления, образующих соединения MR3
или MR2. Иногда необходимо оценить сдвиг полосы
поглощения при переходе от HR к MR„. Если рассчи¬
тывают значение е исходя из концентрации окрашен¬
ного реактива — хромофора R, тоА ll2-rr =0,3- 105. (16.14)СЬ 4-10Аналогично интенсивность окраски раствора жел¬
той фосфорно-молибденовой кислоты можно отнести
к 1 моль фосфора, хотя окраска обусловлена измене¬
нием спектра поглощения молибдена. Если же рас¬
сматривать окрашенные соединения молибдена, то
значение е будет, разумеется, иным, так как с 1 моль
фосфора связано не менее 12 моль молибдена.320
Для смесей нескольких поглощающих свет соеди¬
нений, если они не взаимодействуют друг с другом,
соблюдается аддитивность оптической плотности:Лобщ = А, + A-i + Лз + ... + Ап (16.15)илиАайш. = (е 1С! + бгСо + бзСз + . . . + Zn.Cn) I- (16.16)Уравнение (16.16) применяют для расчетов в фо¬
тометрическом анализе смеси некоторых поглощаю¬
щих свет соединений, если они отличаются по спект¬
рам поглощения. Измерив оптическую плотность
смеси при нескольких длинах волн, можно составить
систему уравнений и решить их по отношению к кон¬
центрации Сь С2, С3, . ■ Сп.§ 3. ХАРАКТЕРИСТИКА СВЕТОПОГЛОЩЕНИЯВ фотометрическом анализе иногда удобно приме¬
нять обозначения еуСл (условное). Это целесообразно,
в частности, при косвенных методах фотометрическо¬
го анализа. Так, фотометрическое определение фтора
основано на ослаблении окраски, например, роданид-
ного комплекса железа. Наблюдаемое ослабление оп¬
тической плотности Л/4 можно пересчитать на извест¬
ную концентрацию фтора и выразить в виде еусл.
Разумеется, это не характеризует светопоглощение
какого-нибудь соединения фтора. Также целесообраз¬
но применять это обозначение при каталитических
методах и т. п.Зависимость оптической плотности или молярного
коэффициента поглощения от длины волны выража¬
ется более или менее сложной кривой. Для этой кри¬
вой принято название — спектр поглощения. Если на
оси абсцисс записывается длина волны (или частота),
то кривую всегда называют «спектр».Величины пропускания (% Т) или поглощения
(%/1) применяют в фотометрии редко, так как их за¬
висимость от концентрации более сложна по сравне¬
нию с зависимостью между А и С. Для иллюстрации
этого положения рассмотрим следующий пример.
Раствор какого-нибудь прочного окрашенного веще¬
ства (состав которого не изменяется при разбавле¬
нии) при концентрации Сi и толщине слоя bi погло¬
щает 50 % света. Интенсивность входящего светового] | Аналитическая химия321
потока /о можно принять равной 100 %; тогда интен¬
сивность светового потока, выходящего из раствора
/ь равна 50 %. Находим оптическую плотность рас¬
твораЛI = lg -у- = *{; 4^- = °.30. (16.17)Разбавим исходный раствор в 2 раза, т. е. до кон¬
центрации С2 = Ci/2, и будем измерять поглощение
света при той же толщине слоя Ьь Согласно закону
Бугера оптическая плотность раствора при этом
уменьшится в два раза:= ^ = 0,15. (16.18)Интенсивность светового потока, выходящего из вто¬
рого раствора, находим по формулеЛ2= lg ~у^-Откудаlg/2 = 1g/0 — Л2 = 2-0,15= 1,85 (16.19)или= 71 %.Таким образом, поглощение света в процентах со¬
ставляет для второго раствора 100 — 71= 29 %,
тогда как для раствора, вдвое более концентрирован¬
ного, оно равно 50 %•Теперь увеличим в два раза концентрацию пер¬
вого раствора (С:1 = 2С\) и измерим поглощение
света при той же толщине слоя b|. Если бы поглоще¬
ние света было прямо пропорционально концентра¬
ции, то этот раствор был бы совершенно непрозрач¬
ным, так как поглощение света составляло бы
50-2 =100%- В действительности же увеличится
в два раза не поглощение света в процентах, а опти¬
ческая плотность:А3 = 2А, = 2-0,3 = 0,6.Интенсивность выходящего светового потока най¬
дем из уравнения, аналогичного уравнению (16.19):lg h = lg /о — Л3 = 2 — 0,6 = 1,4322
Откуда/з = 25%,т. е. поглощение света в процентах составит
100 —25 = 75 %.§ 4. МОЛЕКУЛЯРНЫЙ КОЭФФИЦИЕНТ
СВЕТОПОГЛОЩЕНИЯ, СИЛА ОСЦИЛЛЯТОРАРазменость молярного коэффициента светопоглоще-
ния можно найти из уравнения (16.10), учитывая,
что А—число безразмерное, а размерность концент¬
рации С — моль/л:е = 74- . (16.20)Lb моль•смДля обсуждения некоторых физических аспектов
светопоглощении представляет интерес отнести эту
характеристику не к одному молю, а к одной моле¬
куле (или иону) поглощающего излучения соедине¬
ния. Расчет можно выполнить следующим образом.
Выше отмечалось, что численнос значение е отвечает
оптической плотности 1 М раствора при толщине
слоя 1 см. Предположим, что 1 л 1 М раствора налит
в кювету с толщиной слоя 1 см; очевидно, площадь
этой кюветы равна 1000 см2. Один моль вещества со¬
держит 6,02-1023 молекул (число Лвогадро). Таким
образом, на световой поток с поперечным сечением
1 см2 приходится п молекул:а = 6,02 - 1023: 1000 = 6.02- [О20. (16.21)И : уравнений (16.20) и (16.21) находим характе¬
ристику светопоглощении k отдельной молекулы ин¬
тенсивно окрашенного соединения, для которого мо¬
лярный коэффициент поглощения е ~ 10J:/<• = — = = >.(i • 10~'G- (10.122)п (3,02-1020 1Величину k называют молекулярным коэффициентом
светопоглощения и относят к одной молекуле (или
иону) поглощающего спет соединения. Размерность
k — см2; формально молекулярный коэффициент све¬
топоглощения отвечает непрозрачной площади
(в см2) молекулы. Молекула красителя имеет линей¬
ные размеры порядка 100 нм, т. е. площадь Q моле-11* 323
кулы составляет:Q == nr2 х 10 и см2.(16.23)Из уравнений (16.22) и (16.23) следует, что «не¬
прозрачная площадь» составляет около 1 % площади
молекулы. Очевидно, даже формально нельзя счи¬
тать, что молекула сложного красителя радиусом
100 нм может быть полностью непрозрачна. Поэтому
значение молярного коэффициента е = 105, принятое
при расчете по уравнению (16.22), является близким
к максимально возможному при столь упрощенной
схеме.Тот же вывод получается при использовании ста¬
тистических законов дл.я поглощения света, в которых
молярное и молекулярное поглощения характери¬
зуют вероятность поглощения квантов света моле¬
кулами окрашенного соединения. Вероятность погло¬
щения в общем пропорциональна интегральной пло¬щади ^ Evdv, ограниченной кривой спектра поглоще¬
ния; таким образом, в формуле (16.8) учитываются
молярные коэффициенты при всех частотах v.Молекулу красителя можно рассматривать как
электрический заряд, осциллирующий под действием
электромагнитного поля света. Вероятность поглоще¬
ния света определяется так называемой силой осцил¬
лятора /. Эта величина выражает отношение усред¬
ненной величины осциллирующего заряда в молекуле
к заряду одного электрона е:Величина / прямо пропорциональна площади под
кривой спектра поглощения, т. е.где т л с — масса и заряд электрона; с — скорость светгг. It4 —
молекулярным коэффициент поглощения свсга для чт еоты v.§ 5. ПРИЧИНЫ ОТКЛОНЕНИЯ ОТ ЗАКОНА ПОГЛОЩЕНИЯ
ЭЛЕКТРОМАГНИТНЫХ КОЛЕБАНИЙЗакон Бугера выведен для монохроматического излу¬
чения. Если же при измерении оптической плотности
пользуются светофильтром с достаточно широкой об-Осциллирующий заряд(16.24)Заряд электрона(16.25)324
ластью пропускания света, то наблюдается отклоне¬
ние от прямой пропорциональности между оптической
плотностью и концентрацией вещества. Кроме того,
закон Бугера справедлив только в том случае, если
с изменением концентрации вещества оно не претер¬
певает никаких химических изменений: не происходит
ассоциация молекул при высокой концентрации веще¬
ства, а также вещество не диссоциирует на ионы.Таким образом, причины отклонения от закона
Бугера могут быть физическими и химическими.Физические причины отклонения от закона Бугера.
Закон Бугера справедлив для разбавленных раство¬
ров, для концентраций веществ меньше 0,01 М. При
больших концентрациях частицы, поглощающие свет,
настолько близко расположены друг к другу, что
каждая частица влияет на распределение заряда со¬
седних частиц, что приводит к изменению способно¬
сти частиц поглощать свет данной длины волны.
В этом случае наблюдается отклонение от прямоли¬
нейной зависимости поглощение — концентрация.Погрешности измерения могут возникать также
из-за того, что при прохождении через границу воз¬
дух — стекло около 4 % светового потока отра¬
жается. Эта погрешность сводится к минимуму, если
сравнивают световые потоки, проходящие через ис¬
следуемый и стандартный (или холостой) растворы.
Тогда(ie.23)7р 1Поэтому, если /о считать интенсивностью светового
потока, прошедшего через кювету с холостым раство¬
ром, погрешность, связанная с отражением света при
прохождении через систему воздух — стекло и стек¬
ло— воздух будет сведена к допустимому минимуму.Значительное отклонение от закона Бугера свя¬
зано с немонохроматичностыо светового потока. Вы¬
делим на кривой светопоглощения раствора вещества
(рис. 16.2, а) два участка Л и Б. Зависимость оптиче¬
ской плотности от концентрации в случае измерения
ее при длине волны полосы поглощения А не будет
прямо пропорциональна, так как е в сильной мере
меняется внутри полосы участка А. Внутри полосы325
Рис. 16.2. Зависимость молярного коэффициента поглощения от
длины волны (а) и градуировочные графики при измерении оп¬
тической плотности при длине волны полос поглощения А и Б (б)Б г изменяется мало и зависимость оптической плот¬
ности от концентрации практически прямая.Лучшие результаты получают на спектрофотомет¬
рах, т. е. при измерении оптической плотности при
монохроматическом свете. Некоторая погрешность
связана также с показателем преломления раствора
(п). Для компенсации можно ввести поправку, под¬
ставляя в уравнение закона Бугера вместо е величину
гп/ (п2 + 2)2. Необходимо отметить, что при концен¬
трации веществ меньше 0,01 М, как правило, эта по¬
правка несущественна.Химические причины отклонений от закона Бу¬
гера. Кажущиеся отклонения от закона Бугера свя¬
заны с диссоциацией и ассоциацией химических со¬
единений, влиянием других веществ, присутствующих
в растворе, а также с другими химическими процес¬
сами, происходящими в растворах: гидролиз с обра¬
зованием гидроксокомплексов, гидроксидов, взаимо¬
действием с растворителем с образованием кислых
солей, изменением состава комплексных соединений
в связи со ступенчатым характером их образования
и др.Наиболее ярким примером может быть поведение
бихромата в растворе:Сг20*' + Н0Н +=*: 2НСг04 +=£ 2Н+ + 2Сг042'.Спектры поглощения бихромата и хромата сильно
различаются, поэтому погрешности определения мо¬326
гут достигать больших значений. Для устранения по¬
грешности определение хрома необходимо проводить
либо в сильно кислом растворе (определение СГ2О7")»
либо в достаточно щелочном растворе (определение
СгО^")- В обоих случаях наблюдается прямая зависи¬
мость между оптической плотностью и концентрацией
хрома в растворе.ГЛАВА 17АППАРАТУРА И МЕТОДЫ ИЗМЕРЕНИЯ,
ПРИМЕНЯЕМЫЕ В ФОТОМЕТРИЧЕСКОМ
АНАЛИЗЕ§ I. МЕТОДЫ ИЗМЕРЕНИЯ ИНТЕНСИВНОСТИ ОКРАСКИВ основе любого метода измерения интенсивности по¬
глощения электромагнитного излучения лежит опре¬
деление ослабления интенсивности этого излучения
после прохождения через исследуемый раствор. Для
этого обычно сравнивают два потока электромагнит¬
ного излучения: один, проходящий через исследуемый
раствор, а другой — через определенный стандартный
раствор или через растворитель.Если соединение определяемого компонента погло¬
щает электромагнитные излучения в видимой обла¬
сти спектра, то два световых потока можно сравни¬
вать визуально (именно с этого и началось развитие
фотометрических методов анализа) или посредством
фотоэлектрических приборов. Если наблюдение про¬
водит визуально, можно лишь твердо констатировать
наличие разницы в окраске, но оценить степень раз¬
личия ее с достаточной точностью практически невоз¬
можно. Поэтому при всех визуальных методах оба
световых потока должны быть одинаковыми. В соот¬
ветствии с законом Бугера этого можно достичь тре¬
мя путями: изменяя концентрацию раствора (методы
шкалы, разбавления и колориметрического титрова¬
ния— метод дублирования), изменяя толщину слоя
(применение колориметров) и изменяя интенсивность
светового потока.327
В связи с развитием и выпуском фотоэлектриче¬
ских приборов, визуальные методы утратили свое
значение. Однако при отсутствии фотоэлектрических
приборов (например, при работе в полевых условиях
различных экспедиций) эти методы можно применять
для фотометрического определения многих компо¬
нентов.Главным преимуществом фотоэлектрических мето¬
дов является облегчение условий работы аналитика
в связи с устранением утомляемости глаза. Особое
значение это обстоятельство имеет при массовых ана¬
лизах, для чего фотометрические методы нашли ши¬
рокое применение. Кроме того, применение фотоэле¬
ментов дает возможность автоматизировать контроль
производства. Наконец, большим преимуществом
фотоэлектрических методов является возможность
измерения оптической плотности растворов в ультра¬
фиолетовой и инфракрасной областях спектра, что
значительно расширило область применения фото¬
метрического анализа.Общий принцип всех систем фотоэлектрических
колориметров заключается в том, что поток электро¬
магнитного излучения, прошедший через кювету с
раствором или растворителем (раствором сравнения),
попадает на фотоэлемент, который превращает энер¬
гию излучения в электрическую. Согласно законам
фотоэффекта, сила возникающего фототока прямо
пропорциональна интенсивности электромагнитного
излучения, падающего на фотоэлемент. В связи с
этим отношение интенсивностей потоков электромаг¬
нитных излучений в математическом выражении зако¬
на Бугера может быть заменено отношением фотото¬
ков. Таким образом, при фотоэлектрическом опреде¬
лении оптической плотности растворов практически
измеряют не ослабление потоков электромагнитного
излучения, а значение фототоков, возникающих под
действием потока электромагнитных излучений.При работе с фотоэлементами следует иметь в
виду следующие обстоятельства, влияющие на точ¬
ность и воспроизводимость получаемых результатов.1. Для фотоэлемента, как и для глаза, хотя в зна¬
чительно меньшей мере характерно явление «утом¬
ляемости», т. е. при длительном освещении фотоэле¬
мента ярким светом сила фототока его уменьшается.328
Поэтому время от времени фотоэлементу необходимо
давать «отдохнуть» — прекратить на некоторое время
его облучать.2. Интегральная и спектральная чувствительность
фотоэлемента уменьшается со временем и после про¬
должительной работы его необходимо заменять.3. Чувствительность фотоэлементов по всей по¬
верхности бывает разная, что необходимо иметь
в виду для получения воспроизводимых результатов.4. С целью получения воспроизводимых результа¬
тов следует учитывать спектральные характеристики
испытуемых растворов и спектральную чувствитель¬
ность фотоэлемента.5. Для получения хорошей линейной зависимости
между интенсивностью падающего электромагнитного
излучения на фотоэлемент и силой фототока во внеш¬
ней цепи фотоэлемента необходимо, чтобы сопротив¬
ление гальванометра не превышало внутреннего со¬
противления фотоэлемента и было по возможности
меньше.Перед тем, как приступить к работе на фотомет¬
ре, фотоколориметре или спектрофотометре необхо¬
димо детально изучить инструкцию, прилагаемую
к прибору. Измерения на приборах проводят спустя
15—20 мин после включения. В течение этого вре¬
мени устанавливается режим накала лампы освети¬
теля.Чистота кювет имеет большое значение, поэтому
после работы они должны быть тщательно вымыты и
хранить их лучше всего в перевернутом виде на листе
фильтровальной бумаги, накрыв стеклянным колпа¬
ком. При работе брать кюветы необходимо за стенки,
через которые не проходит поток электромагнитного
излучения.Оптическую плотность стандартного и исследуе¬
мого растворов всегда измеряют по отношению к
раствору сравнения (нулевому раствору). В качестве
такого раствора используют растворитель, но лучше
брать раствор исследуемого объекта такой же кон¬
центрации, но без реактива, который применяют для
получения поглощающего поток электромагнитного
излучения. Если при данной длине волны применяе¬
мый реактив поглощает электромагнитные излучения,
тогда в качестве раствора сравнения применяют рас¬329
твор реактива в растворителе. Концентрация реакти¬
ва должна быть не выше его концентрации в анали¬
зируемой пробе.§ 2. ФОТОЭЛЕКТРОКОЛОРИМЕТРЫФотоэлектроколориметр ФЭК-М. Фотоэлектроколо¬
риметр ФЭК-М является двуплечим прибором с ком¬
пенсационной схемой, т. е. прибором с двумя фотоэле¬
ментами, включенными по принципу противотока.
Один из фотоэлементов находится в «контрольном»
световом потоке. Такая схема позволяет автоматиче¬
ски компенсировать колебания тока в цепи освети¬
теля. При помощи этого прибора измеряют оптиче¬
скую плотность или процент пропускания окрашен¬
ных растворов, а также мутность нефелометрических
взвесей. Для уравнивания двух световых потоков
в ФЭК-М применена щелевая диафрагма. Приемни¬
ками фототока служат селеновые фотоэлементы вен¬
тильного типа. В качестве нуль-инструмента приме¬
няют гальванометр, который вмонтирован под панель
прибора и, кроме того, может быть подключен сна¬
ружи к специально выведенным клеммам.Оптическая схема прибора показана на рис. 17.1.
Свет от лампы 1 при помощи конденсорных линз 2,2'
направляется на зеркала 4,4'\ перед попаданием све¬
тового потока на зеркало он проходит через тепло¬
вые фильтры 3,3', которые поглощают инфракрасные
лучи и предохраняют от нагревания раствор и фото¬
элементы. Световые потоки, отраженные от зеркал
4,4', проходят через светофильтры 5,5', линзы 6,6' и
попадают на кюветы 7,7'. Затем при помощи линз
8,8' и призм 9,9' световые потоки направляются на
фотоэлементы 10,10'. Перед фотоэлементом справа
установлена ножевая диафрагма 11, которая связана
с измерительными барабанами. Перед левым фото¬
элементом установлены два нейтральных клина:
большой плотности 12 для грубой наводки и малой
плотности 13 для тонкой наводки. Поэтому световой
поток справа перед фотоэлементом проходит через
ножевую диафрагму, а слева — через нейтральные
клинья. Фотоэлементы 10,10' подключены по диффе¬
ренциальной схеме к гальванометру 14.330
Рис. 17.1. Оптическая схема ФЭК-М:/—осветитель (лампа), 2, :?'-коиденсориые линзы; 3, 3' тепловые фильтры;
4, 4' — зеркала; 5, 5'— светофильтры; 6. 6' и S,3'-лнн_1ы; 7, 7' — кюветы; 9. 9' — по¬
воротные призмы; 10, /0'-- фотоэлементы; 11 — щелевая (ножевая) диафрагма;
12 —нейтральный клин большой плотности; 13 - нейтральный клин малой
плотности; }4— гальванометрПрибор питается от сети 220 В через ферро-
резонансный стабилизатор напряжения СТН-35М(220)
8 В, который обеспечивает достаточно постоянный
накал лампочки на 8 В. Если колебания напряжения
в сети не превышают 10—15 %, то точность стабили¬
зации составляет ±0,5 %•В приборе вмонтировано два набора с четырьмя
светофильтрами, которые передвигаются по кругу при
помощи переключателя. Номер на рукоятке переклю¬
чателя соответствует номеру светофильтра: 1 — нейт¬
ральный; 2 — зеленый; 3 — синий; 4 — красный.
Спектральная характеристика светофильтров приве¬
дена на рис. 17.2.Два измерительных барабана (левый и правый)
нанесены на одну ось, которая связана со щелевой
(ножевой) диафрагмой. Щелевая диафрагма пред¬
ставляет собой прямоугольник, который состоит из
двух отдельных пластинок с прямоугольными выре-331
Рис. 17.2. Спектральная характеристика светофильтров (2—4),
установленных в ФЭК-МРис. 17.3. Изображение нити лампы при правильной ее установкезами, передвигающихся навстречу друг другу. При
этом площадь прямоугольника может изменяться от
максимального значения до нуля. На обоих бараба¬
нах нанесены две шкалы: шкала оптической плотно¬
сти А (красная) и коэффициента светопропускания т
(черная). Зависимость между ними выражается
уравнением:Шкала первого барабана проградуирована так, что
максимальное светопропускание (100%) соответст¬
вует минимальному раскрытию щели, а 30 % — мак¬
симальному. Эта шкала коэффициентов пропускания
неравномерная и в области измерений 70—30 % дает
большую точность по сравнению со шкалой левого
барабана. Шкала левого барабана равномерная и
проградуирована так, что 100 % светопропускания
соответствует максимальному раскрытию щели, а
при полном закрытии щели светопропускание равно
нулю. Абсолютная погрешность по шкале светопро¬
пускания составляет ±1 %.В комплект прибора входят два набора прямо¬
угольных кювет. В каждом наборе имеется по две
кюветы со следующей рабочей длиной: 5,0; 3,0; 2,0;
1,0; 0,5; 0,3 и 0,1 см. Кюветы всегда должны быть чи¬
стыми и сухими.Для выполнения измерений включают стабилиза¬
тор и дают прибору принять постоянную температуру
в течение 15—20 мин, после чего наблюдается вполне
удовлетворительная стабилизация напряжения. Пе¬332
ред измерением проверяют правильность установки
осветителя. Для этого открывают шторки фотоэле¬
ментов и накладывают на окошки линз папиросную
бумагу. При правильной установке осветителя проек¬
ция нити лампы имеет вид, изображенный на
рис. 17.3. Если осветитель установлен неправильно,
то поворотом винтов добиваются правильной уста¬
новки лампы. Затем папиросную бумагу снимают.Оптическую плотность растворов измеряют при
помощи левого или правого барабана. Рекомендуется
всегда придерживаться одного приема измерения как
при построении градуировочного графика, так и при
измерении оптической плотности анализируемого
раствора.Измерения при помощи правого барабана прово¬
дят в тех случаях, когда оптическая плотность рас¬
твора не превышает 0,5 и требуется большая точ¬
ность измерений. На пути световых потоков устанав¬
ливают кюветы, наполненные растворителем, а
шкалу правого барабана устанавливают на нулевое
положение. Затем включают гальванометр на первую
чувствительность и при помощи нейтрального клина
(см. рис. 17.1) выводят стрелку гальванометра на ну¬
левое положение. После этого гальванометр переклю¬
чают на чувствительность «2» и при помощи нейтраль¬
ного клина /<? для тонкой наводки устанавливают
стрелку гальванометра на нуль. Гальванометр отклю¬
чают и на пути правого светового потока вместо
кюветы с растворителем устанавливают кювету с ис¬
следуемым раствором. Включают гальванометр на
первую чувствительность и, вращая измерительный
барабан, возвращают стрелку гальванометра в нуле¬
вое положение. Затем гальванометр переключают на
чувствительность «2» и уточняют измерение. Отсчеты
берут по шкале правого барабана.Измерения при помощи левого барабана можно
проводить при высоких и низких значениях оптиче¬
ской плотности. Точность измерений на левом бара¬
бане при оптической плотности менее 0,5 ниже по
сравнению с точностью измерений при помощи пра¬
вого барабана, но она вполне достаточна для прове¬
дения рядовых анализов. На пути левого светового
потока ставят кювету с растворителем, а на пути
правого светового потока — кювету с исследуемым333
раствором. Шкалу левого барабана уста 1авливают
на нулевое положение. Затем включают гальванометр
на первую чувствительность и при помощи серого
клина грубой наводки (см. рис. 17.1) возвращают
гальванометр в нулевое положение. После этого
гальванометр ставят на вторую чувствительность и
при помощи клина тонкой наводки устанавливают
гальванометр в нулевое положение. Гальванометр
отключают и на пути правого светового потока вме¬
сто кюветы с раствором ставят кювету с раствори¬
телем. Включают гальванометр на первую чувстви¬
тельность и измерительным барабаном возвращают
стрелку гальванометра в нулевое положение. Затем
переключают гальванометр на чувствительность «2»
и измерение уточняют. Отсчеты оптической плотности
или коэффициента пропускания берут по шкале лево¬
го барабана.Фотоэлектрический колориметр — нефелометр
ФЭК-56-2, Этот прибор, как и другие более новые
приборы этого типа — ФЭК-56, ФЭК-56М, имеет ряд
преимуществ перед ФЭК-М. Оптика прибора
ФЭК-56-2 позволяет работать в ближней ультрафио¬
летовой области спектра с применением ртутно-квар¬
цевой лампы СВД-120А.Оптическая схема прибора представлена на
рис. 17.4. Световой поток от источника света 1 прохо¬
дит через светофильтр 2, попадает на призму 3, ко¬
торая делит его на два равных потока (левый и пра¬
вый). Затем световые пучки проходят через линзы
4,4', попадают на зеркала 5,5' и после отражения
проходят через линзы 6,6' и далее попадают на кю¬
веты 7,7' с раствором или растворителем. Далее све¬
товые потоки попадают на линзы 8,8', в фокусе ко¬
торых помещены матовые зеркала 9,9', а за ними
сурьмяно-цсзисвые фотоэлементы Ф-4 10,10'. На пути
правого потока расположена щелевая диафрагма 11,
которая связана со шкалой барабана 12 и является
таким образом измерительной диафрагмой. Такая же
Диафрагма установлена на пути левого светового
потока; эта диафрагма служит для ослабления
светового потока, падающего на левый фото¬
элемент.Набор состоит из девяти светофильтров. Спект¬
ральная характеристика светофильтров приведена на334
Рис. 17.4. Оптическая схема ФЭК-56-2:/ — лампа накаливания; 2 — светофильтр; 3 — призма; 4, 4', 6, 6'. 8, 8'— линзы;
5, 5' —зеркала; 7, 7' — кюветы; 9, 3'--матовые стекла; 10, 10' — сурьмвно-цеаие-
вые фотоэлементы Ф-4; II, //' — щелевая диафрагма; 12, 12'—шкала барабанарис. 17.5. Эффективная длина волны светофильтров
следующая:Номер све¬
тофильтра . 12345678 9
Эффектив¬
ная длинаволны, нм . 315 364 400 440 490 540 582 610 Рабочая об¬
ласть про¬
пускания
630 нмМаксимумы пропускания светсфильтров прибли¬
зительно совпадают со спектральными линиями ртути
и поэтому их можно использовать при работе с ртут¬
ной лампой. Светофильтры устанавливают на пути
потока света рукояткой. Если наблюдается неста¬
бильность работы прибора (периодическое или бес¬
порядочное размыкание и смыкание сектора индика¬
торной лампы), устанавливают в световые потоки
еще добавочные нейтральные фильтры, которые335
t70ЮtiЛ,нмРис. 17.3. Спектральная характеристика светофильтров, установ¬
ленных в ФЭК-56-2уменьшают освещенность фотоэлементов. На измери¬
тельных барабанах нанесены две шкалы: черная —
процент пропускания и красная — оптическая плот¬
ность.В приборе вмонтирована усилительная лампа.
Нуль-инструментом служит индикаторная лампа. По¬
рядок работы на этих приборах такой же, как на
ФЭК-М, только для компенсации фототоков служат
не клинья, а диафрагмы, связанные с градуировоч¬
ными барабанами, которые установлены на отдельных
осях. Измерения начинают спустя 20 мин после вклю¬
чения при работе с лампой СП-98 и спустя 5—7 мин
при работе с ртутной лампой.Для проведения измерений при перекрытых свето¬
вых потоках устанавливают на пути левого потока
света кювету с растворителем, а на пути правого по¬
тока света — кювету с исследуемым раствором, и
правый барабан устанавливают на полное пропуска¬
ние (100%). Вращая левый измерительный барабан,
добиваются смыкания сектора индикаторной лампы.
Затем специальной рукояткой на пути правого све¬
тового потока ставят кювету с растворителем и вра¬
щением правого барабана снова добиваются смыка¬
ния сектора индикаторной лампы. Оптическую плот¬
ность отсчитывают по шкале правого барабана.Колориметр фотоэлектрический концентрацион¬
ный КФК-2 является однолучевым прибором и пред¬
назначен для измерения в отдельных участках диапа¬
зона длин волн 315—980 нм, выделяемых светофильт¬
рами, коэффициентов пропускания и оптической336
Таблица 17.1. Спектральные характеристики светофильтров,
установленных в фотоколориметре КФК-2№ свето¬
фильтраДлина волны,
соответствую¬
щая максимуму
пропускания,
нмПолу¬
ширина
полосы
пропуска¬
ния, нм№ свето¬
фильтраДлина волны,
соответствую¬
щая максимуму
пропускания,
нмПолу¬
ширина
полосы
пропуска¬
ния, нм1315±535 ±157590+1025+102364 ±525+108670 + 520 ±53400 ±545 ±109750 ±520±54440 ±1040 ±1510870 + 525±55490+1035 ±1011980 ±525 ±5в540 + 1025+10плотности растворов и твердых тел, а также опреде¬
ления концентрации веществ в растворах методом
построения градуировочных графиков. Спектральные
характеристики светофильтров приведены в табл. 17.1.Оптическая схема фотоколориметра КФК-2 пред¬
ставлена на рис. 17.6. Свет от малогабаритной лампы
(КГМ 6,3-15) 1 проходит через систему линз, тепло¬
защитный 2, нейтральный 3, выбранный цветной 4
светофильтры, кювету 5 с раствором сравнения или
с исследуемым раствором, попадает на пластину 6,
которая делит световой поток на два: 10 % света на¬
правляется на фотодиод (ФД-7К) 7 (при измерениях
в области спектра 590—980 нм) и 90 % — на фото¬
элемент (Ф-26) 8 (при измерениях в области спектра
315—540 нм). Регистрирующим устройством служит
микроамперметр типа М-907, оцифрованный в микро¬
амперах и имеющий шкалу 0—100 делений, соответ¬
ствующую шкале пропускания Т, или М-907-10 со
шкалой, оцифрованной в делениях пропускания и оп-Рис. 17.6. Оптическая схема фотоколориметра КФК-2:/ — источник света; 2--теплозащитный светофильтр; 3 — нейтральный свето¬
фильтр; 4 — цветной светофильтр; 5 —кювета с исследуемым раствором или
раствором сравнения; 6—пластина, которая делит световой поток на два
потока; 7— фотодиод; 8 — фотоэлемент337
тической плотности. На задней стенке крышки микро¬
амперметра имеются гнезда для подключения цифро¬
вого вольтметра с пределом измерения 0,1 В.Последовательность измерений приведена в инст¬
рукции к прибору.§ 3. СПЕКТРОФОТОМЕТРЫСпектрофотометры СФ-16, СФ-26. Эти однолуче¬
вые спектрофотометры предназначены для измерения
пропускания и оптической плотности растворов и
твердых тел в диапазоне 186—1100 нм.Оптическая схема прибора представлена на
рис. 17.7. Свет от источника / попадает на зеркаль¬
ный конденсор 2, затем на плоское зеркало 3, кото¬
рое отклоняет поток лучей на 90° и направляет его
в щель 4 (автоколлимационного монохроматора с уг¬
лом 30° при вершине), защищенную пластинкой 5.
Прошедший через щель свет попадает на дисперги¬
рующую призму 7, разлагающую его в спектр; дис¬
пергированный поток направляется на объектив 6,
который фокусирует лучи в щель 8. Призма соеди¬
нена с помощью специального механизма со шкалой
длин волн. Поворачивая призму вращением соответ¬
ствующей рукоятки на выходе монохроматора, полу¬
чают монохроматический поток света заданной длины
полны, который, после прохождения щели 8, кварце¬
вой линзы 9, фильтра 10, поглощающего рассеянныйРис. 17.7. Оптическая схема одмолучевого спектрофотометра:/—источник света; 2 — зеркальный конденсор; 3 — зеркало; 4—входная щель;
5J2— защитные пластины; б —зеркальный объектив; 7- кварцевая призма:
8 — выходная щель; 9 — кварцевая линза; /0-светофильтр; //--кювета с ис¬
следуемым или стандартным раствором; 13 — фотоэлемент338
свет, эталон (или образец) 11 и защитную пла¬
стину 12, попадает на фотоэлемент 13.В спектрофотометре СФ-26 после линзы 9 свет
проходит через эталон (или образец), линзу и с по¬
мощью поворотного зеркала собирается в один из
фотоэлементов: сурьмяно-цезиевый (для измерений
в области длин волн 186—650 нм) или кислородно¬
цезиевый (600—1100 нм). Источниками излучения
служат дейтериевая лампа (в области длин волн
186—350 нм) и лампа накаливания (320—1100 нм).Оптическую плотность (пропускание) измеряют
относительно эталона, пропускание которого прини¬
мают за 100%, а оптическую плотность — равной
нулю. Спектрофотометр СФ-26 может комплекто¬
ваться приставкой ПДО-5, позволяющей снимать
спектры диффузного отражения твердых об¬
разцов.Спектрофотометр СФ-46 представляет собой одно¬
лучевой прибор со встроенной микропроцессорной си¬
стемой и предназначен для измерения пропускания,
оптической плотности жидких и твердых веществ
в диапазоне длин волн 190—1100 нм. Диспергирую¬
щим элементом служит дифракционная решетка с пе¬
ременным шагом и криволинейным штрихом. Источ¬
ники и приемники излучения те же, что и в спектро¬
фотометре СФ-26.Спектрофотометры СФ-10, СФ-14, СФ-18. Эти
приборы представляют собой двухлучевыс спектрофо¬
тометры и предназначены для измерения пропуска¬
ния (оптической плотности) прозрачных и мутных
сред и коэффициентов диффузного отражения твер¬
дых и порошкообразных веществ в видимой области
спектра. Они состоят из осветителя, двойного приз¬
менного монохроматора, фотометра поляризационно¬
го типа, приемно-усилительной части и записываю¬
щего механизма.На рис. 17.8 представлена оптическая схема
СФ-14. Для измерения коэффициента пропускания
жидкостей имеется набор парных кювет с толщиной
слоя 5, 10, 20 и 50 мм.Твердые прозрачные образцы и кюветы с жид¬
костью помещают в специальные держатели, выпол¬
ненные в виде тисков, и зажимают винтом. Держа¬
тель с исследуемым раствором помещают в направ-339
Рис. 17.8. Оптическая схема спектрофотометра СФ-14:/ —источник света; 2 — конденсор; 3 — входная щель; 4—объектив коллима¬
тора; 5 — диспергирующая призма, разлагающая свет в спектр; б —объектив
первого монохроматора; 7 —зеркало; 8—нож; 9— выходная щель; 10 — линза;
//— двоякопреломляющая призма Ротона; 12 — диафрагма; 13— призма Вол¬
ластона; 14 — линза; 15 — полулннза; 16— барабан прерывателя; /7 —призма,
отклоняющая оба потока на 90°; /8— интегрирующий шар; 19 — фотоэлементляющую правой части (по ходу лучей) кюветного
отделения, а стандартный раствор — слева.На приборе СФ-14 можно проводить измерения
в двух диапазонах пропускания (0—100 % и 0—
10%) и в двух диапазонах оптической плотности
(0—2,5 и 0—1,0), что позволяет повысить чувстви¬
тельность и точность измерений.Запись одного спектра можно выполнить а тече¬
ние 2—4,5 мин.Оптическая схема спектрофотометров СФ-10 и
СФ-14 состоит из двух частей—спектральной (двой¬
ного монохроматора) и фотометрической.Нить лампы / (рис. 17.8) проектируется конден¬
сатором 2 через входную щель 3 в плоскости объек¬
тива 4 коллиматора. Входная щель расположена
в фокальной плоскости объектива. Выходящий из
него параллельный поток света проходит дисперги¬
рующую призму 5 и разлагается в спектр. Объек¬
тив 6 первого монохроматора дает спектральное изо¬
бражение входной щели в плоскости средней щели340
по линии А — А. Средняя щель двойного монохрома¬
тора, образованная зеркалом 7 и ножом 8, вырезает
участок спектра, который проходит во второй моно¬
хроматор и проектируется в плоскости выходной
щели 9. Выходя из монохроматора поток света про¬
ходит через линзу 10 и двоякопреломляющую приз¬
му Рошона 11. Первая дает изображение объектива
выходного коллиматора вблизи диафрагмы 12, вторая
разделяет это изображение на два, поляризованных
во взаимно перпендикулярных плоскостях: одно, сим¬
метричное оси, проходит через призму Волластона 13
и линзу 14, другое, смещенное, срезается диафраг¬
мой 12. Линза 14 дает изображение выходной щели
в плоскости полулинзы 15. Вследствие двойного луче¬
преломления призмы Волластона в плоскости полу-
линзы получаются два изображения выходной щели.
Пройдя полулинзу 15, установленную внутри бара¬
бана прерывателя 16, оба потока отклоняются на 90°
призмой 17, проходят через входные окна шара 18 и
попадают на окна, к которым прижимаются образец
и эталон (при измерении коэффициентов отражения)
или два эталона (при измерении коэффициентов про¬
пускания). Свет, отраженный от образца и эталона,
суммируется шаром и попадает на фотоэлемент 19,
расположенный за выходным окном шара. Фототок,
возникающий вследствие влияния суммарного свето¬
вого потока, передается через усилитель на кинема¬
тическую систему прибора, и значение оптической
плотности (пропускания) автоматически фиксируется
на бумажном бланке.§ 4. АБСОЛЮТНЫЕ МЕТОДЫ ОПРЕДЕЛЕНИЯ ВЕЩЕСТВ
В ОТСУТСТВИЕ МЕШАЮЩИХ КОМПОНЕНТОВМетод сравнения оптических плотностей стандарт¬
ного и исследуемого растворов. Отбирают пипеткой
аликвотную часть исследуемого раствора, проводят
химическую реакцию и получают раствор, который
поглощает энергию электромагнитных колебаний. Од¬
новременно готовят несколько растворов стандарт¬
ных образцов или стандартных растворов с близким
содержанием определяемого элемента. Измеряют оп¬
тическую плотность растворов исследуемого и стан¬
дартных. Оптическая плотность исследуемого рас-341
твора равнаAx = tKCxb*стандартного раствораА — е.С Ь .ст h ст стРазделив первое выражение на второе, получиме.С Ьк X XА е.С Ь 'СТ Л СТ СТУчитывая, что оптическую плотность измеряют при
одной и той же длине волны и в одной и той же кю¬
ветеАх С хАс т ^стоткудаТаким образом находят концентрацию вещества в
исследуемом растворе. Зная объем взятого для ана¬
лиза раствора, находят количество взятого для ана¬
лиза вещества.Определение по значению молярного коэффици¬
ента поглощения. Готовят ряд стандартных растворов
с применяемым реактивом и после выбора оптималь¬
ной длины волны определяют оптическую плотность
приготовленных, растворов. Зная концентрацию ве¬
щества и оптическую плотность, рассчитывают моляр¬
ный коэффициент поглощения:Ае* СЬ •Из полученных результатов находят среднее зна¬
чение £*. Затем готовят раствор исследуемого образца
с теми же реактивами и в тех же условиях и изме¬
ряют оптическую плотность приготовленного раствора.
Концентрацию вещества находят по формулеМетод градуировочного графика. Готовят серию
стандартных растворов с различным содержанием342
Рис. 17.9. Градуировочный график
зависимости аналитического сиг¬
нала А от концентрации Сопределяемого компонента
и измеряют оптическую
плотность в оптимальных
условиях при выбранных
длине волны и толщине
слоя (толщина кюветы).Для каждой точки готовят не менее 3 параллельных
растворов и берут средний результат полученного
значения оптической плотности. Необходимо, чтобы
выбранный интервал концентраций соответствовал
области возможных изменений концентраций анали¬
зируемых растворов. При выбранных длине волны и
толщине фотометрируемого слоя должна соблюдаться
зависимость А — ((С). Интервал значений оптической
плотности А, соответствующий интервалу стандартных
растворов, должен находиться в пределах максималь¬
ной воспроизводимости результатов измерений, т. е.
А « 0,14—1,9.По полученным результатам строят градуировоч¬
ный график А = f(С) (рис. 17.9). Затем отбирают
аликвотную часть исследуемого раствора, создают оп¬
ределенные условия и прибавляют реактив, аналогич¬
но приготовлению стандартных растворов, и измеряют
оптическую плотность раствора. По значению опти¬
ческой плотности раствора находят содержание оп¬
ределяемого компонента по градуировочному гра¬
фику.§ 5. ФОТОМЕТРИЧЕСКОЕ ТИТРОВАНИЕК фотометрическому методу анализа близко примы¬
кает фотометрическое титрование. При этом часто
используют те же реакции, что и в обычных фотомет¬
рических методах, и ту же аппаратуру. Однако о со¬
держании вещества судят не по интенсивности свето-
поглощения, а по количеству затраченного реактива.
Таким образом, фотометрическое титрование—это
разновидность титриметрического анализа, при кото¬
ром точку эквивалентности определяют с помощью
фотоэлемента.343
Далеко не все реакции, пригодные для обычного
фотометрического анализа, могут быть использованы
в фотометрическом титровании. Это прежде всего за¬
висит от прочности поглощающего свет комплекса.
В обычном фотометрическом анализе можно исполь¬
зовать и не.очень прочные комплексы, если применять
избыток реактива. При фотометрическом титровании
необходимо, чтобы уже при эквивалентном количестве
реактива было достигнуто практически полное связы¬
вание определяемого компонента. Поэтому, например,
железо можно титровать салицилатом натрия или кси-
леноловым оранжевым, но нельзя титровать родани¬
дом или хлоридом, которые образуют малопрочные
комплексы.Фотометрическое титрование применяют в следую¬
щих случаях: 1) если в результате титрования обра¬
зуется окрашенное соединение; 2) если цвет индика¬
тора изменяется постепенно; 3) при титровании
окрашенных растворов; 4) при титровании веществ,
поглощающих свет в ультрафиолетовой или ближней
инфракрасной области; 5) при титровании очень раз¬
бавленных растворов. Преимуществом фотометриче¬
ского титрования является также легкость автомати¬
зации его.Фотометрическое титрование проводят с примене¬
нием внутренних цветных индикаторов и без
индикаторов. В первом случае все закономерности,
выведенные для визуального наблюдения точки экви¬
валентности, сохраняются и для фотометрического
титрования. В начале титрования оптическая плот¬
ность раствора практически не изменяется. По мере
приближения к точке эквивалентности, когда индика¬
тор изменяет цвет, оптическая плотность раствора,
измеряемая при определенной длине волны, начинает
резко изменяться (уменьшаться или увеличиваться,
в зависимости от выбранной длины волны).При проведении фотометрического титрования в
отсутствие индикатора необходимо, чтобы титруемые
растворы или продукты реакции имели собственную
характерную полосу поглощения. На основании дан¬
ных титрования строят график в координатах оптиче¬
ская плотность (ось ординат)—объем израсходован¬
ного раствора (ось абсцисс). Точка пересечения двух
прямых (точка перегиба) соответствует точке эквива-344
I{M/IРис. 17.10. Кривые фотометриче¬
ского титрованияI/M/7лентности. Перпендикуляр, опущенный из этой точки
на ось абсцисс, показывает объем титрованного рас¬
твора, необходимого для достижения точки эквива¬
лентности.На рис. 17.10 представлены различные кривые фо¬
тометрического титрования: а) титруемый раствор не
поглощает свет, продукт реакции поглощает; 6) опре¬
деляемый компонент поглощает свет, титрант и про¬
дукт реакции не поглощают (при титровании в присут¬
ствии индикатора вид кривых может быть аналогич¬
ным); в) определяемый компонент и продукт реакции
не поглощают света, титрант поглощает; г) продукт
реакции не поглощает свет, определяемый компо¬
нент и титрант поглощают; (3) определяемый ком¬
понент, титрант и продукт реакции поглощают свет
(/ — определяемый компонент поглощает слабее, чем
поодукт реакции, 2— определяемый компонент и про¬
дукт реакции поглощают одинаково, а титрант погло¬
щает сильнее или слабее, 3— определяемый компо¬
нент поглощает сильнее, чем продукт реакции); е) про¬
дукт реакции и титрант поглощают свет, исследуемый
раствор не поглощает (1 — продукт реакции погло¬
щает сильнее, чем титрант, 2 — титрант поглощает345
сильнее, чем продукт реакции); ж) определяемый
компонент и продукт реакции поглощают свет, ти-
трант не поглощает (/ — продукт реакции поглощает
свет сильнее, чем определяемый компонент, 2— опре¬
деляемый компонент поглощает свет сильнее, чем
продукт реакции).Если в основе фотометрического титрования лежит
реакция разрушения окрашенного соединения, то кри¬
вая титрования имеет вид, показанный на рис. 17.10, б.
Пр имером такого титрования может быть определе¬
ние фторидов, сульфатов и других ионов.Как и другие методы титриметрического анализа,
фотометрическое титрование менее чувствительный
метод, чем обычные фотометрические методы. Фото¬
метрическое титрование, как правило, применяют для
определения больших количеств веществ (порядка
0,01—0,001 М).§ 6. НЕФЕЛОМЕТРИЧЕСКИИ И ТУРБИД ИМЕТРИЧ ЕСКИ Й
МЕТОДЫ АНАЛИЗАНефелометрический и турбидиметрический методы
анализа состоят в том, что определяемый компонент
переводят в малорастворимое соединение, которое на¬
ходится в виде взвеси, и измеряют интенсивность рас¬
сеянного света или ослабление светового потока этой
суспензией.Если содержание вещества находят по интенсив¬
ности рассеянного света (оис. 17.11, а), то такой ме¬
тод называют нефелометрическим. Метод определения
содержания вещества по ослаблению светового потока
суспензий (рис. 17.11,6) называется турбидиметриче-
ским. Таким образом, в случае нефелометрического и
турбидиметрического метода изменяется интенсив¬
ность светового потока, но спектральная характери¬
стика светового потока остается постоянной.В нефелометрическом и турбидиметрическом мето¬
дах анализа используют реакции осаждения. Основ¬
ные требования к реакциям, которые применяют в
этих методах, следующие: продукт реакции должен
быть практически нерастворимым; продукт реакции
должен находиться не в виде осадка, а в виде взвеси
(суспензии).346
Рис. 17.11. Схема хода
лучей при нефелометри-
ческом (а) и турбиди-
метрическом (б) методах
анализа ^ Мутный>Мутный■ ■ >раствор>раствор—№*1 UГлаз или
фотоэлементГяаэ ми
фотоэлементИнтенсивность рассеянного светового потока, в со¬
ответствии с законом Рэлея, зависит от многих фак¬
торов:I оNV-Tv(1 + cos2 Р),где Is — интенсивность рассеянного светового потока; Iо — ин¬
тенсивность первоначального светового потока; п* — коэффици¬
ент преломления частиц; п — коэффициент преломления среды;
N — количество частиц в данном объеме; V — объем шарообраз¬
ной частицы, рассеивающей свет; X — длина волны; — угол
между падающим и рассеянным световым потоком; г — расстоя¬
ние до наблюдателя.Большое значение имеет объем частиц, рассеиваю¬
щих свет. Поэтому условия приготовления взвесей ис¬
следуемой и стандартных должны быть одинаковыми.Для измерения интенсивности рассеянного света
пользуются специальными приборами — нефеломет¬
рами, которые по конструкции мало отличаются от
фотометров и фотоколориметров.С помощью нефелометрического и турбидиметри-
ческого методов анализа можно определять малые со¬
держания многих ионов, которые образуют малорас¬
творимые соединения. Так, сульфат-ионы определяют
в виде взвеси сульфата бария; хлорид-ионы опреде¬
ляют в виде взвеси хлорида серебра и т. д.Из приведенном выше формулы видно, что коли¬
чество частиц и объем их неодинаково влияют на рас¬
сеивание света. Между тем очень трудно добиться,
чтобы в стандартном и в исследуемом растворах по¬
лучались частицы одинакового размера. Кроме того,
влияет форма поверхности частиц. Мелкие кристаллы,
например кристаллы сульфата бария, могут прини¬
мать разнообразную форму, что сильно влияет на
рассеивание света. Таким образом, получение вос¬
производимых результатов затруднено. Поэтому в
настоящее время очень редко прибегают к нефело-347
метрическому методу, тем более, что разработаны
значительно более удобные и точные методы опреде¬
ления ионов с помощью других оптических или элект¬
рохимических методов анализа.§ 7. ДИФФЕРЕНЦИАЛЬНАЯ СПЕКТРОФОТОМЕТРИЯФотометрические методы были разработаны для оп¬
ределения очень малых количеств различных веществ.
Исходя из этого, в фотометрии допускались, особенно
при визуальных методах измерения интенсивности ок¬
раски, относительные погрешности 5—10%. Однако
с развитием приборостроения, переходом на измере¬
ния при монохроматическом излучении, выяснением
химизма процесса значительно уменьшились погреш¬
ности измерения в абсолютном методе фотометриче¬
ского анализа, когда оптическая плотность раствора
измеряется по отношению к оптической плотности
растворителя.Для определения больших количеств применяют
так называемый метод дифференциальной (сравни¬
тельной) фотометрии, в том числе и метод двусторон¬
ней дифференциальной спектрофотометрии.В дифференциальном методе получаемое значение
оитической плотности на спектрофотометре или фото¬
колориметре А представляет собой разницу между аб¬
солютными оптическими плотностями исследуемого
Анс и стандратного (нулевого) раствора А0 (метод од¬
носторонней дифференциальной спектрофотометрии)
или, наоборот, при меньшем содержании вещества в
исследуемом растворе по сравнению со стандартным
раствором (метод двусторонней фотометрии). Таким
образом, в дифференциальной фотометрии при
А не А оА = Лис А0 = бйСд = 6b (C{tc Cq);при С0 > Сис, т. е. Лис < А0А = А0 — Аис = еЬСо — ейСис = ?,Ь (С0 — Сис)где Сис, С0 — концентрации поглощающего свет соединения в ис¬
следуемом растворе и растворе сравнения.Сущность двусторонней дифференциальной фото¬
метрии заключается в том, что оптическая плотность348
-А0 -1,0 -0,5 0 0,5 1,0 1,5 -*4Рис. 17./2 Расчетные кривые точности двусторонней дифферен¬
циальной спектрофотометрии при различных значениях А0:1-0; 2— 0,25; 3 — 0,50; 4—1.0; .5—1,5; 6-2,0исследуемого раствора может быть как больше, так и
меньше оптической плотности раствора сравнения.В последнем случае измеряют оптическую плот¬
ность раствора сравнения по отношению к исследуе¬
мому раствору и значение оптической плотности бе¬
рут с отрицательным знаком. Этот вариант примерно
в два раза расширяет диапазон измеряемых концен¬
траций исследуемого раствора при небольшом увели¬
чении погрешности, как это показано на рис. 17.12.
На этом рисунке приведены расчетные кривые точно¬
сти определения вещества двусторонним дифферен¬
циальным методом при различной оптической плотно¬
сти раствора сравнения.Допустим, что интенсивность электромагнитного
излучения, прошедшего через раствор сравнения с
меньшим содержанием определяемого компонента,
равна /ср, а прошедшего через исследуемый раствор —
1х, тогда-еС b7<-Р = V 10 = 10,: (с<гс.х) ь ^ 1 о ЛСЙlx Iо- Ю”^Нели концентрация вещества в анализируемом
растворе будет меньше, чем в растворе сравнения, то¬
гда измеряют оптическую плотность раствора сравне¬
ния по отношению к анализируемому раствору.349
Относительная погрешность определения диффе¬
ренциальным методом зависит от отношения кон¬
центраций и, обычно, наименьшая, когда Сх ж С0,Q[ср/1X ^ 1 •Для экспериментального определения оптимальной
концентрации раствора сравнения С0 готовят несколь¬
ко стандартных растворов с такой разницей концен¬
траций АС, чтобы соответствующие им разности Д/4
были равны 0,3—0,4. Концентрации этих растворов
должны быть такими, чтобы соблюдался закон Бу¬
гера. Затем измеряют относительные оптические плот¬
ности каждого последующего раствора по отношению
к предыдущему и рассчитывают коэффициент:где Со — концентрация раствора, который используется в ка-
честве раствора сравнения в каждом i’-tom измерении.Раствор, для которого значение / наибольшее, и при¬
меняют в качестве раствора сравнения. Однако необ¬
ходимо всегда помнить, что удовлетворительные ре¬
зультаты будут получены, если через растворы про¬
ходит достаточное количество света и используе¬
мый прибор сравнительно легко устанавливается на
«нуль».Дифференциальную спектрофотометрию широко
применяет в практике, она дает возможность опреде¬
лять высокие содержания различных компонентов с
вполне удовлетворительней точностью. Сохраняя пре¬
имущества фотометрического метода, дифференциаль¬
ная спектрофотометрия может конкурировать по точ¬
ности с титриметрическим методом.§ 8. ПРОИЗВОДНАЯ СПЕКТРОФОТОМЕТРИЯПри дифференцировании кривой поглощения излуче¬
ния по длине волны или волновому числу изменяется
форма спектральной кривой (рис. 17.13). Из этого ри¬
сунка видно, что экстремумы первой производной
соответствуют точкам перегиба исходной полосы по¬
глощения, а нулевая точка — ее максимуму. Цент¬
ральный пик второй производной подобен контуру
исходной полосы, но значительно сужен. Таким обра¬
зом, даже в случае перекрывания спектров поглоще-350
А'А1/Ж/К' У\\/AV\f/'А\ .VРис. 17.13. Первая производная спектров определяемого (/) и
мешающего (2) компонентовРис. 17.14. Вторая производная спектров определяемого (/) и
мешающего (2) компонентовния после дифференцирования их производные спект¬
ров поглощения разделяются, что дает возможность
раздельно определять два и больше веществ.Линейность дифференцирования дает возможность
применять производную спектрофотометрию для ко¬
личественного спектрофотометрического определения.
При этом п-ная производная оптической плотностиdnA _ Cbdne
dxn ~ dxnЕсли дифференцироваиие провести во времени с
помощью аналоговых устройств и учесть, чтоdn vndne ( dx \=—-j—fj— = скорость сканирования I,то получимdnA Cbvndnz
din •Таким образом, при v — const производная про¬
порциональна концентрации.Так как правило аддитивности справедливо для
оптических плотностей, составляющих многокомпо¬
нентной системы, то оно справедливо и для их произ¬
водных. В таком случае при наличии компонентов351
Mi, спектры поглощения которых перекрываются, для
первой производной по длине волны можно записатьП. . „ , . X СМ 6 dEM
dA Cxb deM f^n■ т' dX dX dXЕсли в растворе два компонентаdA СMb d&M ' СMb deM.~dX = dX 1 dXПри совпадении области дифференцирования с
максимумом поглощения мешающего компонента
deM,/dX —> 0 и на оси X появляется точка нулевого
вклада х0 (см. рис. 17.13) мешающего компонента;
при этом фон подавляется.d2A *
Функция вторая производная также обла¬
дает двумя важными свойствами. На оси jc(X, v) по¬
являются точки нулевого вклада мешающих компо¬
нентов, лежащие между центральным пиком и пиком
мешающего компонента (рис. 17.14). При этом по¬
давляется фон, изменяющийся значительно медлен¬
нее, чем полоса поглощения.ГЛАВА 18ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗ
§ I. ОБЩИЕ ПОЛОЖЕНИЯЛюминесценцией называют свойство веществ излу¬
чать свет под воздействием различных возбуждаю¬
щих факторов. По определению С. И. Вавилова лю¬
минесценцией называют избыточное свечение тела
над тепловым (температурным) излучением того же
тела в данной спектральной области при данной тем¬
пературе и при условии, что это избыточное свечение
обладает длительностью 10~10 с и больше, т. е. пре¬
вышает период световых колебаний.Существует несколько систем классификации лю¬
минесценции. С. И. Вавилов и В. Л. Левшин разли¬
чают явления люминесценции двух типов: 1) свече-352
Рис. 18.1. Схема возникновения люминесценцииние дискретных (отдельных) центров; 2) рекомбина¬
ционные процессы свечения. В первом случае в про¬
цессе возникновения люминесценции принимает уча¬
стие только одна частица (центр свечения), которая
является как поглотителем энергии, так и излучате¬
лем световых волн. В рекомбинационных процессах
свечения поглощение энергии, как правило, осущест¬
вляется не теми частицами, которые излучают свето¬
вые волны.Если в основу классификации положен метод воз¬
буждения молекул или атомов ультрафиолетовым из¬
лучением (или коротковолновой видимой частью
спектра), то свечение называют фотолюминесценцией
или флуоресценцией, если возбуждение происходит
под действием катодных лучей — катодолюминесцен-
цией, под действием рентгеновских лучей — рентгено-
люминесцснцией, за счет энергии, возникающей при
механических деформациях вещества — триболюми
несценцией, за счет энергии нагревания вещества —
кайдолюминесценцией, за счет энергии химической
реакции — хемилюминесценцией, известны и другие
виды люминесценции.Процесс возбуждения и свечения в простейшем
виде представлен на рис. 18.1, а, где Н —нормальный
(невозбужденный) уровень основного состояния ато¬
ма с колебательными подуровнями 0, 1, 2, 3, 4; В —
уровень возбужденного состояния атомов с соответ-12 Аналитическая хнмня363
ствующими колебательными подуровнями 0, 1, 2, 3, 4;
М—метастабильное состояние. Вертикальные стрел¬
ки обозначают переход электронов при поглощении
или излучении световых квантов, причем длины стре¬
лок пропорциональны частотам или обратно пропор¬
циональны длинам волн. Переход с уровня Н0 на
уровни В0, В|, В2, В3 и В4 происходит при абсорбции
света, затем за время 10~9—10~8 с электроны пере¬
распределяются по колебательным подуровням и
излучаются кванты света в результате перехода элек¬
тронов с наиболее вероятного уровня В0 на подуров¬
ни Н0, Н1; Н2, Н3 и Н4. На рис. 18.1,6 показан меха¬
низм длительного самостоятельного свечения дис¬
кретных центров. Это происходит в том случае, когда
переход электрона с метастабильного уровня на уро¬
вень В становится невозможным и при этом спектр
излучения сдвигается в сторону длинных волн, так
как излучаемые кванты в данном случае меньше, чём
в случае а.Когда говорят о люминесцентном (или о флуорес¬
центном) методе анализа, под этим обычно пони¬
мают фотолюминесценцию. Различают обычно две
группы методов: анализ по непосредственному на¬
блюдению люминесцирующего материала и анализ,
основанный на переведении определяемого компонен¬
та в люминесцирующее соединение. Вторая группа
методов люминесцентного анализа близка к фотомет¬
рическому анализу. Известно немало случаев, когда
один и тот же реактив может быть применен для оп¬
ределения одного и того же элемента как фотометри¬
ческим, так и люминесцентным методом. В обоих
случаях необходимо перевести определяемый компо¬
нент в соединение, которое, возможно, более сильно
поглощает свет. При фотометрическом анализе изме¬
ряют непосредственно ослабление интенсивности
светового потока. Для люминесцентного же анализа
эту реакцию можно использовать только в том слу¬
чае, если значительная часть поглощенной энергии
выделяется не в виде тепла, а в виде света. Естест¬
венно, что это явление более редкое, поэтому в об¬
щем число люминесцентных методов меньше, чем фо¬
тометрических. В то же время люминесцентные
методы при некоторых условиях более чувствительны
по сравнению с фотометрическими.354
Рис. 18.2. Спектры поглощения (/)
и люминесценции (2)Для люминесценции харак¬
терно то, что часть энергии
возбуждения неизбежно теря¬
ется в виде тепла. Поэтому
энергия квантов света, выде¬
ляющегося при люминесцен¬
ции, будет меньше, чем энер¬
гия квантов возбуждающего света. Иначе говоря, дли¬
на волны люминесцентного свечения будет всегда
больше, чем длина волны возбуждающего света, за
исключением небольшого участка спектра, где полосы
возбуждения и люминесценции перекрываются. Эта
зависимость была установлена еще до квантовой тео¬
рии и известна как правило Стокса — Ломмеля:
спектр люминесценции всегда смещен в сторону более
длинных волн но сравнению со спектром поглощения
(рис. 18.2).Для многих веш.еств эти спектры зеркально сим¬
метричны (правило Левшина). Расстояние между
максимумами спектра поглощения и спектра люми¬
несценции называется стоксовым смещением. Чем
больше стоксово смещение, тем легче отделить воз¬
буждающий свет и таким образом устранить влияние
его («фон») на измерение люминесцентного свечения.
Правда, люминесценцию наблюдают в направлении,
перпендикулярном направлению потока возбуждаю¬
щего света. Однако и в этом случае возбуждающий
свет рассеивается поверхностью жидкости, стенками
кюветы, а также частицами пыли в растворе.§ 2. КВАНТОВЫЙ и энергетический выход
И СООТНОШЕНИЕ МЕЖДУ НИМИОчень важней закономерностью люминесценции яв¬
ляется связь между интенсивностями возбуждающего
электромагнитного колебания и света люминесцен¬
ции. Отношение излучаемой энергии люминесценции
Ел к энергии поглощенного кванта Ее называют энер¬
гетическим выходом люминесценции Вэ„:В,„ = (18.1)12*355
Отношение числа излучаемых квантов Мл к числу
поглощенных квантов М„ называют квантовым выхо¬
дом люминесценции Вкв:Энергетический и квантовый выходы характери¬
зуют эффективность преобразования энергии погло¬
щенного электромагнитного колебания в энергию
люминесценции.Учитывая, что энергия кванта Е — hv (где h —
постоянная Планка, v — частота волн электромагнит¬
ных колебаний), легко найти соотношение между
энергетическим и квантовым выходами:С. И. Вавилов установил зависимость энергетиче¬
ского выхода люминесценции от длины волны возбу¬
ждающего электромагнитного колебания. Эта зави¬
симость— закон С. И. Вавилова — формулируется
следующим образом: «При возбуждении люминесцен¬
ции коротковолновой частью спектра поглощения
величина энергетического выхода растет пропорцио¬
нально длине волны возбуждающего света; затем на
некотором спектральном интервале выход не изменяет
своей величины при увеличении длины волны возбу¬
ждающего света, после чего в области наложения
спектров поглощения и излучения происходит резкое
Падение выхода» (рис. 18.3). Таким образом, в опре¬
деленных областях спектра квантовый выход не за¬
висит от длины волны. Иначе говоря, спектр люми¬
несценции зависит от набора энергетических уровней
молекул и не зависит от того, какие именно кванты
света были израсходованы для возбуждения молеку-Вкп —(18.2)лы. Поэтому для возбужде¬
ния молекулы обычно при¬
меняют кванты ультрафио¬
летовой части спектра сческого выхода люминесценции
водно-аммыачмого раствора флуо
ресцеина от длины волны вс^буж ■
дающего снегаРис. 18.3. Зависимость энергети¬
ческого выхода люминесценцииО200£00356
большей энергией, что, разумеется, увеличивает до¬
лю энергии, превращающейся не в свет, а в тепло.
Однако для аналитических целей эти энергетические
«потери» не имеют значения.Квантовый выход характеризует предел обнару¬
жения вещества люминесцентным методом. Чем
больше квантовый вы; од, тем меньшие количества
вещества можно определить.Интенсивность люминесценции /л пропорциональ¬
на числу излучаемых ki антов Ыл:1л = -:Л/Л = -с BKB/V (18.4)где к — коэффициент пропорциональное] п.Количество поглощенных квантов N„ пропорцио¬
нально интенсивности поглощенного света:Л\, = *'(/0-0. (18.5)где !а. /-интенсивности падающего и прошедшего через рас¬
твор света соответственно; х’ — коэффициент пропорционально¬
сти.Согласно закону Бугера/ = /„• 10_ейс. (18.6)Подставляя в уравнение (18.5) значение / из ура¬
внения (18.6), получим‘V,; = х' (л, - /о ■ 10-е&с) = х-/-0 (1 - \(ГЕЬС). (18.7)После подстановки уравнения (18.7) в уравнение
(18.4) получим:/л = л:*'Вкв/,;(1 - Ю-£*с). - (18.8)При разложении 10“Eftc в ряд получим:= 1 - 2,3е.ЬС + ('2,3^Г— - i2’3f^-C-)- + . . . (18.9)При е.ЬС 10”2 значениями третьего и последую¬
щих членов можно пренебречь, так как эти величины
достаточно малы, поэтому в уравнении (18.9) можно
ограничиться первыми двумя членами. Если подста¬
вить в уравнение (18.8) результат разложении (18.9)
получим:1Л = 2,3.1х'ВКн/оЕ^С. (18.10)Обозначим все постоянные величины в уравнении
(18.10) через /С, получим:1Я = КС. (18.11.)357
Таким образом, при постоянстве квантового вы¬
хода, интенсивности возбуждающего света, постоян¬
ной толщине анализируемого слоя (толщина кюве¬
ты) и т. д. интенсивность люминесценции пропорцио¬
нальна концентрации. Необходимо иметь в виду, что
это справедливо для низких концентраций люмннес-
цирующих веществ. При увеличении концентрации
люминесцирующего вещества нарушится принятое
выше условие ebC ^ 10-2, в связи с чем нарушится и
линейность зависимости интенсивности люминесцен¬
ции от концентрации. Если концентрацию вещества
сильно повысить, то интенсивность люминесценции
может уменьшиться, т. е. может наблюдаться концен¬
трационное тушение люминесценции. Поэтому верх¬
ний предел концентрации растворов в люминесцент¬
ном анализе, как правило, не превышает 10_3 —
]0-4 М. Повышение температуры также может при¬
вести к тушению люминесценции (температурное ту¬
шение) .§ 3. ТУШЕНИЕ ЛЮМИНЕСЦЕНЦИИЯвление тушения люминесценции заключается в том,
что при увеличении концентрации разбавленных рас¬
творов вещества люминесценция возрастает сначала
пропорционально концентрации, а далее увеличение
интенсивности люминесценции «отстает» от увеличе¬
ния концентрации. Так, при увеличении концентра¬
ции флуоресцеина от 0,0003 до 0,003 М интенсивность
люминесценции возрастает почти в 10 раз. Однако,
например, в 1 %-ном растворе люминесценция флуо¬
ресцеина слабее, чем в очень разбавленных раство¬
рах. Такие явления были замечены давно для многих
веществ. Позже было показано, что аналогичный эф¬
фект резкого ослабления люминесценции флуорес¬
цеина и других веществ вызывается иногда добав¬
ками значительных количеств таких веществ, как, на¬
пример, иодид калия, которые в данных условиях не
реагируют с люминесцирующим веществом.Тушение люминесценции по С. И. Вавилову мо¬
жет быть двух родов. Тушение люминесценции, вы¬
зываемое процессами, связанными с их внутримоле¬
кулярными перегруппировками, которые могут про¬
исходить даже в тех случаях, когда молекулы нахо¬358
дятся и в невозбужденном состоянии, относят к пер¬
вому роду. В этом случае тушение не сопровождает¬
ся уменьшением длительности послесвечения. Туше¬
ние первого рода является причиной того, что при хи¬
мическом процессе люминесцирующее вещество пре¬
вращается в нелюминесцирующее. Таким образом,
тушение первого рода сопровождается изменением
внутримолекулярного взаимодействия составляющих
молекул и, как следствие, изменением спектров по¬
глощения и люминесценции.Тушение второго рода не сопровождается измене¬
нием спектров поглощения и люминесценции. Такой
процесс происходит вследствие воздействия на возбу¬
жденную молекулу внешних факторов, но не приво¬
дящих к образованию нового вещества.Наблюдается также концентрационное тушение,
которое может привести к получению ошибочных ре¬
зультатов в люминесцентном анализе. Причины ту¬
шения различны и далеко не всегда выяснена их фи-
зико-химическая природа.§ 4. КАЧЕСТВЕННЫЙ И КОЛИЧЕСТВЕННЫЙ
ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗМногие органические и неорганические вещества
характеризуются собственной люминесценцией. Так,
яркую люминесценцию проявляют соли редкоземель¬
ных элементов, особенно цериепой подгруппы: сама¬
рия, европия, гадолиния, тербия, диспрозия. Соб¬
ственной люминесценцией обладают таллий (Г), оло-
во(П), сурьма(Ш), свинец(П), висмут (III), ин¬
дий (III) и др. Люминесцируют многие органические
вещества, например вазелиновое масло (светло-сире-
невым цветом), парафин (светло-голубым), сосновая
смола (темно-зеленым с желтым оттенком), мине¬
ральное масло (светло-синим), канифоль (светло-си¬
ним), очищенный асфальт (темно-желтым или корич¬
невым).■ Для качественного анализа можно использовать
собственную люминесценцию, а также реакции обра¬
зования комплексных соединений неорганических ио¬
нов с органическими реагентами, в результате чего
появляется люминесценция. Так, многие катионы с
8-гидроксихннолином образуют соединения с харак¬
терной люминесценцией, бериллий с морином обра-359
зует комплекс, люминесцирующий ярко-зеленым цве¬
том, натрий-цинк-уранилацетат люминесцирует зеле¬
новато-желтым цветом, таких примеров можно при¬
вести много.В качественном анализе можно также использо¬
вать изменение цвета или тушение люминесценции
под действием обнаруживаемого вещества.При введении в некоторые нелюминесцирующие
кристаллы примесей других элементов (активаторов)
они проявляют характерную люминесценцию. Эти ве¬
щества называют кристаллофосфорами. По интен¬
сивности люминесценции кристаллофосфоров находят
элементы-примеси. Так, можно приготовить кристал-
лофосфоры на основе оксида кальция и определить
примесь селена и теллура по красной люминесцен¬
ции, таллия(1)—по желто-зеленой, висмута(Ш) —
по сине-фиолетовой с достаточно низким пределом
обнаружения.Для обнаружения вещества во многих случаях до¬
статочно визуально наблюдать люминесценцию. Од¬
нако если присутствует смесь люминесцирующих ве¬
ществ, то наблюдение люминесценции затрудняется;
в таких случаях применяют светофильтры для выде¬
ления люминесценции определенной длины волны или
проводят предварительное хроматографическое раз¬
деление.В количественном анализе используют зависи¬
мость интенсивности люминесценции от концентра¬
ции определяемого вещества. При использовании ре¬
акции образования люминесцирующего соединения
необходимо обратить внимание на наиболее полное
переведение определяемого компонента в соединение,
обладающее люминесценцией. В количественном ана¬
лизе большое значение имеет квантовый выход: чем
он больше, тем меньшие количества вещества можно
обнаружить.Интенсивность люминесценции можно измерять
визуально и с помощью специальных приборов —
флуориметров.§ 5. ФЛУОРИМЕТРЫЛюминесценция в анализируемом веществе возбу¬
ждается с помощью лучистого потока от источника
электромагнитных колебаний определенной длины360
Рис. 18.4. Оптическая схема прибора для измерения флуоресцен¬
ции растворов в «проходящем свете»:j — источник излучения', 2, 4—светофильтры; 3 —кювета с раствором, 5 фо¬
тоэлементволны, выделяемой оптической системой (монохрома¬
тором или светофильтрами). Приемник лучистой
энергии регистрирует свет флуоресценции.Задача оптической системы состоит в том, чтобы
минимально ослабить и практически полностью со¬
брать свет люминесценции на рабочую поверхность
приемника. Кроме того, на поверхность приемника не
должны попадать электромагнитные колебания ис¬
точника возбуждения, так как измерение интенсив¬
ности люминесценции в таком случае будет искаже¬
но. Для количественного измерения интенсивности
люминесценции обычно пользуются светофильтром,
установленным перед фотоэлементом, а для измере¬
ния спектрального распределения люминесценции
пользуются монохроматором.На рис. 18.4 представлена схема прибора для из¬
мерения люминесценции растворов «на просвет» или
в «проходящем свете». По такой схеме построены
приборы ФАС-1 и ФАС-2. Электромагнитные волны
источника излучения / проходят первичный свето¬
фильтр 2, выделяющий монохроматическое излуче¬
ние, которое попадает на кювету с раствором 3. Воз¬
никающий в растворе свет люминесценции проходит
через вторичный светофильтр 4 и попадает на фото¬
элемент 5. Интенсивность люминесценции регистри¬
руется гальванометром. Как видно из рис. 18.4, все
элементы схемы расположены на одной оси. Первич¬
ный светофильтр должен выделять электромагнитные
колебания с длиной волны возбуждения, а вторич¬
ный— с длиной волны только излучения. Эту схему
обычно применяют при возбуждении ультрафиолето¬
вым участком спектра с применением достаточно361
мГГг5 I/v, V д-'i jEI-ZРис. 18.5. Оптическая схема для измерения флуоресценции рас¬
творов под углом в 90°:/ — источник излучения; 2, 4 — светофильтры, 3 — кювета с раствором; 5 —фо¬тоэлементдлинной кюветы. При этом возбуждающий свет
практически полностью поглощается исследуемым
раствором.Приборы, построенные по схеме, изображенной
на рис. 18.4, имеют некоторые недостатки. В длин¬
ной кювете проявляется вторичное поглощение, что
приводит к искажению результатов. В таких случаях
люминесценция, возникшая в передней части кюветы,
ослабляется молекулами люминесцирующего веще¬
ства вследствие взаимного наложения спектров лю¬
минесценции и поглощения. При использовании уль¬
трафиолетового участка спектра для возбуждения
люминесценции наблюдаются ошибки из-за люминес¬
ценции материала кюветы.Лучшие результаты дает схема, изображенная на
рис. 18.5. Схему, изображенную на рис. 18.5, а, ис¬
пользуют в приборах ФАС-1 и ФАС-2, а на
рис. 18.5, б — в приборе ФО-1.Схемы, представленные на рис. 18.5, имеют пре¬
имущества по сравнению со схемой, изображенной на
рис. 18.4: 1) меньше сказывается люминесценция
материала кюветы; 2) требования, предъявляемые к
избирательности вторичных светофильтров, не очень
строгие, граница скрещивания светофильтров может
быть не очень четкой.362
Как видно из рис. 18.5, 6 возбуждающее излуче¬
ние, минуя стенки кюветы, падает непосредственно на
раствор сверху. Это дает возможность во многих слу¬
чаях использовать кюветы от фотоколориметров.§ 6. X ЕМ И ЛЮМИНЕСЦЕНТНЫЙ АНАЛИЗЯвление выделения света молекулами или атомами,
предварительно возбужденными за счет энергии хи¬
мической реакции, называют хемилюминесценцией.
Свободная энергия преобладающего большинства
экзотермических химических реакций выделяется в
виде тепла, однако в некоторых реакциях часть энер¬
гии выделяется в виде света.Явление хемилюминесценции известно давно. Оно
наблюдается во многих живых организмах (светляч¬
ки, гнилушки, свечение морских микроорганизмов и
др.). Свечение в них связано с ферментативным окис¬
лением веществ, вырабатываемых организмом, кис¬
лородом.Хемилюминесценция наблюдается лишь в том слу¬
чае, если в растворе происходят элементарные экзо¬
термические акты, энергия которых больше
170 кДж/моль.Схематически хемилюминесцентную реакцию мо¬
жно представить так:А + В —>■ Р* + Другие продукты реакции, Р* —у Р + hvОдновременно проходит и темновой (безызлуча-
тельный) процесс:Р* --> Р.Интенсивность хемилюминесценции / пропорциональ¬
на скорости v хемилюминесцентной реакции:где С — концентрация излучатели, моль; t — время, с.Коэффициент пропорциональности т) является
квантовым выходом хемилюминесцентной реакции.1 = rjuилиd £ (/iv)dC6,02 • 1023ti ,
dtdtж
где N„ — число люминесцирующих молекул; Л/0бщ — общее число
молекул излучателя.Максимально возможное значение г) равно 1;
квантовый выход некоторых биохимических реакций
близок к единице, однако при остальных хемилюми-
несцентных реакциях квантовый выход лишь изредка
достигает нескольких процентов.Механизм хемилюминесцентных реакций крайне
сложен и до конца не выяснен. Большое значение в
хемилюминесценции имеют процессы комплексообра¬
зования, каталитического разложения пероксида во¬
дорода, радикальные процессы.Хемилюминесценцию применяют для получения
кинетических характеристик реакций окисления —
восстановления; для изучения комплексных соедине¬
ний; при изучении свойств возбужденных молекул,
особенно в газообразных реакциях. На основе хеми¬
люминесцентных реакций созданы детекторы различ¬
ных радикалов и излучений. Используют хемилюми-
несцентные реакции в технологических схемах для
автоматического контроля производства, в биологии,
медицине и криминалистике.Основоположником применения хемилюминесцент¬
ных реакций в анализе является А. К. Бабко и его
ученики.В аналитической практике хемилюминесцентные
реакции используют: 1) для установления точки экви¬
валентности при титровании мутных или окрашенных
растворов (применение хемилюминесцентных индика¬
торов в методах нейтрализации, окисления — восста¬
новления, комплексообразования); 2) для определе¬
ния основных компонентов хемилюминисцентных ре¬
акций (хемилюминесцентного реактива, окислителя
или восстановителя), 3) для определения микроколи¬
честв ионов металлов, которые являются катализато¬
рами или ингибиторами хемилюминесцентных реак¬
ций; 4) для определения органических веществ, кото¬
рые являются ингибиторами хемилюминесцентных
реакций, по их окислению.Таким образом, хемилюминесцентный анализ яв¬
ляется одновременно разделом каталитических (ки¬
нетических) методов анализа с одной стороны, и лю¬
минесцентных методов — с другой. Однако в обычных
каталитических методах количество продукта реак¬364
ции устанавливают фотометрически или титриметри-
чески, в то время как хемилюминесцентный анализ
основан на измерении интенсивности или суммы вы¬
деленного света в химической реакции. ‘Интенсив¬
ность свечения или сумму выделенного света изме¬
ряют фотографическим методом или с помощью фо¬
тоумножителей или фотоэлементов.В хемилюминесцентном анализе нет необходимо¬
сти в применении источников возбуждения и поэтому
аппаратура метода более простая по сравнению с
той, которую применяют в люминесцентном анализе.
Меньшее влияние оказывает также фон. Многие ио¬
ны с незаполненными й?-уровнями гасят люминесцен¬
цию. В то же время эти ионы часто являются катали¬
заторами или активаторами (иногда и ингибитора¬
ми) хемилюминесцентных реакций и поэтому могут
быть определены этим методом.Экспериментальное изучение различных хемилю¬
минесцентных систем позволяет определить опти¬
мальные условия реакций, найти области, где суще¬
ствует прямолинейная зависимость между концентра¬
цией определяемого вещества и максимальной
интенсивностью свечения, или интенсивностью свече¬
ния через определенное время, или суммой выделив¬
шегося света за определенное время.Преимуществом хемилюминесцентного метода
анализа являются низкие пределы обнаружения
10-10— 10-4 г/мл при конечном объеме 2—5 мл., до¬
статочная точность определения, экспрессность, про¬
стота аппаратуры. Недостатком метода является ма¬
лая селективность реакций, однако варьирование ус¬
ловий определения и применение маскирующих аген-
тЬв часто позволяет устранить этот недостаток.При хемилюмннисцентных исследованиях необхо¬
димо применять высокочистые реактивы (хч, осч или
очищать реактивы перекристаллизацией, дистилля¬
цией или ионным обменом). Хранят растворы в поли¬
этиленовой, а лучше в кварцевой посуде. Вода для
приготовления растворов должна быть бидистиллиро-
ванной и, лучше, свежеперегнанной. Особое внимание
уделяется чистоте посуды и кювет (не должно на
стенках оставаться никаких капель).Наиболее распространенными и лучше изученны¬
ми в настоящее время являются следующие хемилю-305
минесцентные реактивы — люминол, люцигенин, си-
локсен.При реакции люминола (гидразид 3-аминофтале-
вой кислоты H2L) с пероксидом водорода в щелоч¬
ной среде (pH > 8,5) возникает голубое свечение;
реакцию схематически (на самом деле процесс идет
гораздо сложнее, со множеством промежуточных про¬
дуктов) можно представить так:nh2XоNH/NH+ <Э2 + 20Н"г у ^о-+N2+2H20+/iv.ЧгСвечение люминола впервые описал Альбрехт в
1928 г. Возникает свечение люминола не только при
действии Н2Ог, но и других окислителей: хлорной,
бромной, йодной воды, NaBi03 и др. Реакцию окис¬
ления люминола применяют для определения микро¬
количеств окислителей (Н202, I2, CIO- и др.), микро¬
количеств ионов металлов кобальта, меди, железа,
циркония, титана, платиновых металлов и других с
пределом обнаружения 10-9—10^8 г/мл. Ионы ме¬
таллов по-разному влияют на свечение. Ионы меди,
кобальта и других металлов усиливают свечение в
системе H2L—Н202, ионы циркония, титана и дру¬
гих— гасят свечение H2L—Си"—Н202.По реакции с люминолом определяют неметаллы
и органические вещества с пределом обнаружения
10-9—10-4 г/л: неорганические и органические суль¬
фиды, 8-гидроксихинолин, аминокислоты, аминофе-
нолы и др. Люминол применяют как индикатор в ти-
триметрии, например в комплексонометрии: к рас¬
твору соли цинка или кадмия добавляют избыток
титрованного раствора динатриевой соли этилендиа-
минтетрауксусной кислоты, который затем оттитро-
вывается раствором соли меди известной концентра¬
ции в присутствии люминола и Н202; сначала медь
связывается в прочный комплекс, а в точке эквива¬
лентности свободные ионы меди катализируют хеми-366
люминесцентную реакцию между H2L и Н2О2 и воз¬
никает свечение.Люцигенин (N.N'— диметилбиакридилнитрат, Lc)
описан в 1935 г. (Глей и Петч), излучает голубое све¬
чение под действием Н2О2 и восстановителей в при¬
сутствии кислорода при pH > 9. Схематически (не
более!) реакцию можно представить так:+ Н202 + 20Н'+ 2Н,0 + 2NO: + Av.Применяют люцигенин для определения ионов свин¬
ца, висмута, меди, кобальта и других ионов с пре¬
делом обнаружения 10~9—10-7 г/мл, различных вос¬
становителей и как индикатор в титриметрии.Силоксен (циклогексасилтриоксен) (1921 г., Каут¬
ский) — неорганическое соединение?\ /Н
| sr нH-Si/ "''Si'\н-Si. .s^1>Чо/ хноВ кислой среде (pH <С 5) при действии окислителей
наблюдается розовое свечение. Схематически реак¬367
цию, например с перманганатом, можно представить
так:(Si^Oj^ + MnO.'-l-fr —► 6/tSi02 • mHaO-f Мл2* + Av.Применяют силоксен как индикатор в методах охис-
ления — восстановления, для определения микроко¬
личеств церия, ванадия, золота, марганца и др.Сначала хемилюминесцентные реакции изучали
визуально. Затем для определения суммы света, вы¬
деляющегося при хемилюминесцентной реакции и
для практического применения в анализе был пред¬
ложен фотографический метод. Кюветы с плоским
прозрачным дном устанавливали над фотопластин¬
кой. Через заданное время или после окончания ви¬
димого свечения пластинку обрабатывали обычным
методом, высушивали и измеряли почернение пятен
и фона на микрофотометре.-На одной фотопластинке можно снять 12—24 пробы.
Объем раствора в кювете от 0,2 до 10 мл.Позже для изучения кинетических особенностей и
для применения в анализе были предложены фото¬
электрические установки с фотоумножителем и само¬
писцем или амперметром. На таких установках мож¬
но изучать распределение интенсивности свечения во
времени, определять максимальную интенсивность
свечения и сумму выделившегося света за определен¬
ное время т (интегральная площадь под описанной
самописцем кривой или сумма заряда на накопи¬
тельном конденсаторе).св 0
Раздел VIРЕАКЦИИОКИСЛЕНИЯ— ВОССТАНОВЛЕНИЯГ ЛАВ А 19ОКИСЛИТЕЛЬНЫЙ ПОТЕНЦИАЛ
И СОПРЯЖЕННЫЕ РЕАКЦИИ
ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯ§ I. ХАРАКТЕРИСТИКА РЕАКЦИЙ
ОКИСЛЕНИЯ — ВОССТАНОВЛЕНИЯРеакции окисления — восстановления — это реакции,
в которых реагирующие вещества присоединяют или
утрачивают электроны. Окислителем называется ча¬
стица (ион, молекула, элемент), которая присоеди¬
няет электроны и переходит при этом из более высо¬
кой в более низкую степень окисления. Восстанови¬
тель— это частица, которая утрачивает электроны и
переходит вследствие этого из более низкой в более
высокую степень окисления. Так, в реакции2Fe3t + Sn2> = 2Fe2+-f Sn,v (19.1)окислителем являются коны Fe3+, потому что пере¬
ход железа из более высокой в более низкую степень
окисления сопровождается присоединением одногоэлектрона:Fc3 + + е- =Fe2+. (192)Наоборот, ионы Sn2+ отдают электроны, в связи с
чем степень окисления возрастает:Sn2" = 2е~ + Snlv. (19.3)Таким образом, в реакциях окисления—восстановле¬
ния происходит перераспределение электронов между
реагирующими веществами.Окисление или восстановление может праиелодить
не только в том случае, если реагируют между собой
два вещества — окислитель и восстановитель. Источ¬
ником электронов может служить, например, гальва-369
нический элемент, на одном из полюсов которого про¬
исходит окисление или восстановление вещества.Реакции окисления — восстановления, по сравне¬
нию с рассмотренными выше другими типами реак¬
ций, являются наиболее сложными. Их сложность об¬
условлена тем, что они не всегда протекают в точ¬
ном соответствии с суммарным уравнением реакции.
В процессе окисления — восстановления часто обра¬
зуются нестойкие промежуточные соединения или ио¬
ны, элементы в которых имеют средние степени окис¬
ления участвующих в реакции веществ. Все эти про¬
межуточные соединения превращаются постепенно в
продукты реакции, однако их возникновение часто
обусловливает вовлечение в реакцию присутствую¬
щих в растворе веществ, непосредственно не реаги¬
рующих с окислителем или восстановителем. Такой
сложный ход процесса характерен в особенности для
реакций, в которых принимают участие два или бо¬
лее электронов. Они протекают через ряд одноэлек¬
тронных стадий.Так, реакцию между перманганат-ионами и иона¬
ми Fe2+ можно представить следующим уравнением:Mno; + 5Fe2* + 8Н* = Mn2t + 5Fe1+ + 4Н20. (19.4)Из уравнения видно, что взаимодействие может про¬
изойти только в том случае, если в определенный мо¬
мент времени в какой-либо точке пространства столк¬
нутся между собой один ион Мп04, пять ионов Fe2+
и восемь ионов Н+. Вероятность одновременного
столкновения четырнадцати ионов близка к нулю, и
поэтому реакция по описанному механизму должна
была бы протекать настолько медленно, что за изме¬
римый промежуток времени нельзя было бы обнару¬
жить в реакционной смеси продуктов реакции —
ионов Мп2+ и Fe3+. Между тем реакция идет практи¬
чески мгновенно, что свидетельствует о другом
механизме взаимодействия. В частности, было пока¬
зано экспериментально, что восстановление перман¬
ганат-ионов происходит ступенчато, причем в про¬
цессе восстановления образуются ионы, содержащие
марганец в степени окисления шесть, четыре и три.
В последующем механизм реакций подобного типа
будет рассмотрен более подробно.370
Реакции окисления—восстановления имеют очень
большое значение в аналитической химии, так как
многие методы анализа основаны на этих реакциях.
Приведем некоторые примеры.(.Реакции окисления—восстановления нередко
используют для идентификации ионов. Так, ионы
марганца(II) легко обнаружить по фиолетовой ок¬
раске раствора после их окисления до перманганат-
ионов. Иодид-ионы обнаруживают, окисляя их хло¬
ром до свободного иода, который образует с крахма¬
лом соединение синего цвета. Известно много других
подобных примеров.2. Анализируемый материал нередко переводят в
раствор действием азотной кислоты или ее смеси с
хлороводородной кислотой. Растворение сопрово¬
ждается окислением составных частей пробы. Так,
при анализе медных сплавов их растворяют в азот¬
ной кислоте, причем металлическая медь окисляется
до Си2+, а язот в азотной кислоте восстанавливается
до NO или N02.3. Цементация, т. е. вытеснение из раствора ио¬
нов более электроположительного металла более
электроотрицательным металлом, является широко
распространенным приемом концентрирования и раз
деления ионов перед их заключительным определе¬
нием. Например, металлическая медь легко восстана¬
вливает ионы ртути:Си + Hg2*" = Cu2t -f- fig.Ртуть обнаруживают затем по изменению цвета
медной пластинки.4. В титриметрическом методе анализа реакции
окисления — восстановления используют для количе¬
ственного определения многих веществ. Так, ионы
Fe2+ окисляются перманганатом до ионов Fe3+, что
дает возможность определить их содержание в рас¬
творе. В качестве окислителей применяют кроме пер¬
манганата калия также бихромат калия, ванадат на¬
трия, бромат калия и ряд других веществ. Известны
и методы титрования восстановителями, например
растворами SnCl2> TiCl3, СгС12 и др.5. Электрогравиметрический метод анализа осно¬
ван на взвешивании выделившегося на электроде
металла или его соединения. При электролизе содер¬371
жащиеся в растворе ионы восстанавливаются на элек¬
троде; в некоторых случаях выделение вещества на
электроде связано с его окислением. Так, ионы свин¬
ца окисляются на аноде и выделяются в виде осад¬
ка РЬ02 (см. гл. 12).6. В полярографическом методе анализа проводят
электролиз раствора, при котором определяемые ио¬
ны восстанавливаются или окисляются; вывод о ко¬
личестве вещества делается на основании измерения
силы тока, протекающего через раствор при электро¬
лизе.7. В кулонометрическом методе анализа нередко
определяемое вещество подвергается электрохимиче¬
скому превращению на электродах — окислению или
восстановлению—на что затрачивается определен¬
ное количество электричества. Измерение этого коли¬
чества дает возможность вычислить содержание дан¬
ного вещества в растворе.8. Каталитические методы анализа основаны пре¬
имущественно на реакциях окисления — восстановле¬
ния. Известно много медленных реакций этого типа,
которые можно ускорить, вводя в раствор катализа¬
торы— чаще всего ионы переходных металлов. Ско¬
рость реакции при некоторых условиях пропорцио¬
нальна концентрации катализатора, что используют
для количественного определения последнего.§ 2. ОКИСЛИТЕЛЬНЫЙ ПОТЕНЦИАЛНаправление реакций окисления — восстановления
зависит от соотношения энергии сродства высших
степеней окисления реагирующих частиц к электро¬
нам. Рассмотрим состояние равновесия в растворе,
содержащем частицы Ок], В|, Ок2 и В2, между кото¬
рыми может происходить реакция в соответствии с
уравнением.Ок, + В2 В, + Ок2. (19.5)Индексы 1 и 2 относятся к веществам 1 и 2 в
окисленной (Oki и Ок2) и восстановленной (В2 и Bi)
формах.Нели равновесие реакции (19.5) сдвинуто вправо,
т. е. если частица Oki окисляет частицу В2 с образо¬
ванием продуктов реакции Bi и Ок2 — уравнение372
(19.5) можно представить двумя следующими от¬
дельными уравнениями полуреакций:Ок, +й- = В, (19.6)иВ2 — е~ -{- Ок2. (19.7)Электроны при взаимодействии перераспреде¬
ляются: частица В2 отдает электроны, частица Ок( их
присоединяет. Поэтому реакция (19.5) будет проис¬
ходить слева направо только в том случае, если
сродство электрона к частице Ок, больше, чем его
сродство к частице Окг; при обратном соотношении
состояние равновесия должно быть сдвинуто в ле¬
вую сторону, т. е. окисленная форма вещества 2 бу¬
дет окислять восстановленную форму вещества 1 в
соответствии с уравнениями полуреакций:Ок2 е~ — В2 (19,8)иВ,=е-+Ок,. (19.9)В настоящее время не существует методов, поз¬
воляющих экспериментально определить абсолютное
значение энергии сродства электрона к находящимся
в растворе частицам и сопоставить эти значения ме¬
жду собой с тем, чтобы предвидеть направление ре¬
акций. Тем lie менее такое сравнение можно осуще¬
ствить, если условно принять энергию сродства элек¬
трона к высшей степени окисления какого-либо
элемента равной нулю. Для сравнения служит так
называемый водородный электрод, в котором принята
равной нулю энергия сродства электрона к ионам во¬
дорода:2Н* + 2<?- = Нг. (19.10)Водородный электрод представляет собой плати¬
новую пластинку, покрытую электролитически плати¬
новой чернью, которую насыщают газообразным во¬
дородом. Схема такого электрода, а также способ из¬
мерения энергии сродства электронов к окисленной
форме какого-либо элемента по отношению к водо¬
родному электроду показаны на рис. 19.1.Водородный электрод помещен в стеклянную
трубку с открытым нижним концом, присоединенную
к аппарату Кипа для получения газообразного373
Рис. 19.1. Схема для иэмере-
ния потенциала металлического
электрода:6Uп2 /--сосуд с исследуемым раствором;
2 — платиновый электрод; 3 — потен*
циометр; 4 — электролитическийключ; 5 — „пробки41 из фильтроваль¬
ной бумаги; 6— нодородный элект¬
род; 7 — сосуд с серной кислотой7водорода; электрод по¬
гружен в раствор серной
кислоты с активностью
ионов водорода, равной
единице (сосуд 7). В со¬суде I находится смесь окисленной и восстановленной
форм исследуемого вещества; активности обеих форм
и соотношение их активностей также равны единице.
В сосуд / погружен платиновый электрод 2. Раство¬
ры в сосудах 1 и 7 соединен^ специальным проводни¬
ком, обеспечивающим прохождение тока. Платиновый
и водородный электроды присоединены к потенцио¬
метру, измеряющему э. д. с. образованного обоими
электродами гальванического элемента. В сосуде 1
находятся ионы Fe3+ и Fe2h. В этом случае, как пока¬
зывает опыт, напряжение гальванического элемента
окажется равны 0,75 В; кроме того, электроны во
внешней цепи передвигаются по направлению от во¬
дородного электрода к платиновому электроду. Сле¬
довательно, в сосудах 7 и 1 происходят такие
процессы:т. е. энергия сродства электрона к ионам Fe3+ выше,
чем энергия сродства электрона к иону водорода. Ко¬
личественной характеристикой служит напряжение
гальванического элемента 0,75 В. Эта величина счи¬
тается положительной, если электроны во внешней
цепи двигаются от водородного к платиновому элек¬
троду.Заменим раствор в сосуде 1 раствором, содержа¬
щим смесь ионов Snlv и Sn2+ с такой же актив¬
ностью. Направление движения электронов останетсяН2=»2Н* + 2е~,
Fe3+ е~ = Fe2+,(19.11)(19.12)374
таким же, как в предыдущем случае, однако потен¬
циометр покажет напряжение 0,2 В.Наконец, если в сосуд / поместить раствор смеси
ионов Ti[V и Ti3+ с активностью каждого иона, рав¬
ной единице, электроны во внешней цепи будут дви¬
гаться в противоположном направлении, т. е. от пла¬
тинового к водородному электроду, а напряжение
гальванического элемента окажется равным 0,1 В.
В отличие от двух предыдущих случаев, электрохи¬
мические процессы в обоих сосудах выражаются ура¬
внениями:Ti^ = c_-f- Ti'v (19.13)И2H* + 2e- = H.j, (19.14)т. е. ионы Ti3+ удерживают электроны слабее ионов
водорода. В сосуде / идет процесс окисления тита-
на(III) до титана(IV), а в сосуде 7 — процесс восста¬
новления ионов водорода до свободного молекуляр¬
ного водорода. В этом случае напряжение гальвани¬
ческого элемента принято брать с противоположным
знаком: —0,1 В.Напряжение составленного описанным способом
гальванического элемента называют стандартным по¬
тенциалом. Он относится к системе из двух форм
элемента — окисленной и восстановленной, и являет¬
ся количественной характеристикой энергии сродства
электрона к высшей степени окисления элемента по
отношению к той же величине для ионов водорода,
которая условно принята равной нулю.Таким образом, стандартный потенциал — это
электродный потенциал полуреакции (относительно
стандартного водородного электрода) при условии,
что активности всех реагентов и продуктов реакции
равны единице. Стандартные потенциалы обозначают
символом £0. Значения стандартных потенциалов из¬
мерены непосредственно или вычислены из термоди¬
намических данных для многих окислительно-восста¬
новительных систем и их можно найти в таблицах.Следует подчеркнуть, что стандартные потенциа¬
лы относятся не к какому-либо элементу или иону,
а к системе из двух частиц, представляющих окис¬
ленную и восстановленную формы данного вещества.375
Так, для железа известны три значения стандартных
потенциалов, а именно:Fe3+ + e- = Fe2\ £' = 0,75 В;Fe3* + Зе' = Fe°, Е" = 0,00 В;Fe2+ + 2е" = Fe°, Б'" = -0,4 В.Для окислительно-восстановительных реакций с уча¬
стием хрома можно записать:Сг3+ + е“ = Сг2+, Е'0 = -0,4 В;СгО2" + Зе‘ + 8Н+ = Сгэ+ + 4Н20, Е" = 1,3 В.Значения стандартных потенциалов некоторых
окислительно-восстановительных систем приведены
на рис. 19.2. Пользуясь этими значениями, можно оп¬
ределить направление реакций окисления — восстано¬
вления, не прибегая к эксперименту. Окислителем в
данной реакции всегда будет выступать частица, пе¬
реход которой в низшую степень окисления характе¬
ризуется более высоким потенциалом; восстановите¬
лем является частица, переход окисленной формы ко¬
торой в восстановленную характеризуется более низ¬
ким потенциалом. Так, переход перманганат-ионов в
ионы Мп2+Мпо; + 8Н+ + 5е“ = Мп2+ + 4Н20характеризуется потенциалом £о=1,52 В, переход
Вг2 + 2е- = 2Вг-характеризуется потенциалом £'о = 1,08 В. В данном
случае £о> Ео, поэтому перманганат окисляет ио¬
ны брома до свободного брома. Реакция идет в соот¬
ветствии с уравнением2МпО; + 10Вг~ + 16Н+ = 2Мп2+ + 5Вг2 + 8Н20практически полностью слева направо.Значение стандартного потенциала определяется
не только природой данной окислительно-восстанови¬
тельной системы, но также активностью частиц, пред¬
ставляющих высшую и низшую степени окисления
элемента. Зависимость потенциала Ех от активности376
Рис. 19.2. Значение стан¬
дартных потенциалов не-
которых ' окислительно-
восстановительных си¬
стемвыражается урав-
нением Нернста:RTЕх = Еа Л — 1п-И .52
+ 1,36ОкnFав(19.15)где R — газовая постоян¬
ная; Т—абсолютная тем¬
пература; F — число Фа¬
радея; п — количество
электронов, принимаю¬
щих участие в переходе
окисленной формы в вос¬
становленную.Подставим в
уравнение (19.15)
вместо постоянных
величин их числен¬
ные значения, т. е.
£ = 8,31 Дж, F =
= 96 500 Кл, Т =
— 293 К, а также♦ 0.77+0,62+0,56+0,54+0.17+0,15-0,12МлО,- + <Н+ + 5е~ = Млг+ + 4НгОс/г * it = гаCr2of + 14H+ + 6е' = гсг'* + JM20
в1г + 2е=2Вг-. Fe3+ + e‘ = Fe2+- 2HgCl2 + 2e =Hg2Cl2 + 2C|-- As0i‘ + 2H+J 2e=As0| +Н20
■ I г + 2е=2ГСаг+ + e‘ = Cu+Sn4+ + 2e=Sn2+2N+ + 2e =H2CrOj + 4H20 + Зе' = Сг(0Н)з + 50H"
Cr3+ + е' = С|г+SnOf + H20 + 2e = SnO? + 2QH'преобразуем натуральные логарифмы в десятичные,
используя модуль перехода 2,3026. Тогда0,058Б* — Еа -|->g-(19.16)Обычно соотношение активностей близко к соот¬
ношению концентраций, если не происходит каких-
либо побочных процессов, нарушающих их. * Тогда
под знаком логарифма можно записать концентрации
соответствующих частиц:„ , 0,058 , [Ок]Ьх — ЕвЛ — lg -—я “ [В] <19'17>В таком виде уравнение Нернста удобно использо¬
вать для решения различных практических задач.* И случае побочных процессов правильнее применять тер¬
мин «формальный потенциал» вместо стандартного потенциала.377
Если в реакции окисления — восстановления при¬
нимают участие ионы водорода, необходимо записы¬
вать их концентрацию в соответствующей степени
в числителе выражения, стоящего под знаком лога¬
рифма. Так, для реакцииМпО; + 8Н* + 5е_ = Мп2* +• 4Н,0уравнение Нернста имеет вид:0,058 ГМпОгПнЧ8
— 'g-L- [м^г-- <1918>Таким образом, увеличение концентрации ионов
водорода приводит к возрастанию окислительного по¬
тенциала данной системы. Это — общий вывод для
всех реакций, протекающих с участием ионов водо¬
рода. В самом деле, ионы водорода участвуют в ре¬
акции в тех случаях, когда окисленная форма пред¬
ставляет собой сложный кислородсодержащий ион;
восстановленная же форма либо совсем не содержит
кислорода, либо содержит его меньше, чем окислен¬
ная форма. Поэтому в уравнениях, описывающих
переход элемента из высшей степени окисления в низ¬
шую, ионы водорода всегда будут находиться в ле¬
вой части уравнений. Это ясно из следующих при¬
меров:SO; + 2Н" + 2е = SOj" + Н20, As043'+2H++2e = As0^+H20,
СЮ*" + 8Н* + Зе = Сг3* + 4Н20, VO;+2H*+ е = V02t + Н20.Следовательно, уменьшение pH при восстановле¬
нии сложных кислородных ионов всегда способствует
возрастанию их окислительной способности.Нередко восстановленная форма представляет со¬
бой металл в свободном состоянии, например Zn, Ag
и т. д. В этих случаях концентрация восстановленной
формы — величина постоянная. Поэтому уравнение
Нернста записывается для таких систем так:ЕХ = Е0 + lg[Mn + ]. (19.19)Иногда окисленная форма представляет собой эле¬
мент в свободном состоянии, например для системыS -f- 2е~ = S2'..178
Легко видеть, что уравнение Нернста принимает тог¬
да следующий вид:ЕХ = Е o--^lg[S2l. (19.20)Определить направление реакции окисления — вос¬
становления можно в простейших случаях на основа¬
нии сопоставления стандартных потенциалов ве¬
ществ, участвующих в реакции, как это было пока¬
зано для систем Мп04/Мп2+ п Вг2/Вг . Известно, на¬
пример, что стандартные потенциалы приведенных
ниже реакций окисления — восстановления равны:
Fe3+ + е- = Fe2+, £0 = 0,75 В;Snlv + 2е' = Sri2*, £0 = 0,20 В.Потенциал полуреакции Fe3+ —>-Fe2f более поло¬
жителен, чем Sniv —>-Sn2+, поэтому ионы Fe3+ яв¬
ляются окислителями по отношению к ионам Sn!+ и
реакция между этими ионами2Fe3 + + Sn-2+ = 2Fe3 + + Snlvбудет проходить слева направо.Состояние равновесия наступит после того, как
потенциалы полуреакций обеих пар станут одинако¬
выми, т. е. £* = £*:Е'о + 0,058 lg = £'' + 0,029 lgЕсли в реакции принимают участие ионы водорода,
тогда при изменении pH раствора направление реак¬
ции может стать обратным. Например, для реакцииFe(CN)^ + VC)2+ + Н20= Fe(CN)*' + V02+ + 2Н +полуреакции и соответствующие им стандартные по¬
тенциалы следующие:[Fe(CN)G]3 +e' = [Fe(CN)0l4-, £' = 0,Э6 В;VO; + 2Н+ + е- = VO2* + Н30, £"=1,0 В.Из сопоставления стандартных потенциалов видно,
что при одномолярных концентрациях всех ионов
реакция будет проходить справа налево. Однако
увеличение pH раствора приводит к уменьшению по¬379
тенциала системы VO^/VO2*, в то время как потен¬
циал реакции [Fe(CN)e]3—/[Fe(CN)6]4— не изменяет¬
ся. Так, при pH = 10 и [VO.ll = [V02+JЕх = 1,0 + 0,058 lg- ( ю“|0)'-’ = —1,16 В.Поэтому при данных условиях окислителем в реак¬
ции является [Fe(CN)6]3_ и равновесие смещено
вправо.Металлическое серебро не окисляется ионами во¬
дорода, т. е. реакция2Ag + 2Н+ = 2Ag+ + Н2практически не проходит. Введение в систему ио-
дид-ионов, образующих с ионами серебра осадок Agl,
может способствовать протеканию этой реакции
вправо. Предположим, что в реакции2Ag + 2Hf+ 21 = 2AgI + Н2концентрации ионов водорода и иодид-ионов равны
единице. Уравнение Нернста для системы Agf/Ag
имеет вид:£* = 0,8 + 0,058 lg(Ag4где 0,8 — стандартиг.тп потенциал системы Ag'/Ag.При [I] = 1 из выражения произведения раствори¬
мости Agl находим:[Agl = -^=10“16 М.Если подставить найденную концентрацию в уравне¬
ние Нернста, тоЕк = 0,8 + 0,058 lg I0~ 10 = —0,13 В.Таким образом, введение' иодид-ионов приводит к
снижению потенциала Ag+/Ag, который становится
меньше, чем потенциал системы 2Н+/Н2 и, следова¬
тельно, рассматриваемая реакция должна проходить
слева направо.Последний пример иллюстрирует влияние реак¬
ций осаждения на ход реакций окисления — восста¬
новления: выведение одного из реагирующих компо¬
нентов из сферы реакции в виде нерастворимого со¬
единения может изменить направление реакции окис¬
ления — восстановления.380
Приведенные вычисления и выводы имеют качест¬
венный характер и не позволяют определить количе¬
ственное соотношение концентраций всех реагирую¬
щих веществ при установившемся равновесии.Более точные вычисления основаны на примене¬
нии уравнения, связывающего стандартные потенциа¬
лы с константой равновесия реакции окисления —
восстановления.Рассмотрим реакцию окисления — восстановле¬
ния, выражающуюся следующим уравнением:Юк,+аВ2 +=* 6В, + аОк2. (19.21)Константа равновесия этой реакции равна°к [Ок,] [В2]аРеакцию (19.21) можно представить в виде двух
полуреакций:60i<i пе~ ~ 6В] (19.23)ИаОк2 + пе~ = ав2. (19.24)Запишем для этих полуреакций уравнения Нерн¬
ста:£, + £0> + ^ig7^f; 09.25)п [В,]00,058 [Ок2)а „Е2 — Lc2+ п 'g (В2]а ' (19.26)В состоянии равновесия/^=£2, (19.27)поэтомуода JOKili= + ода* ,eJ25alf-. (19.28)01 п [В,]й 02 п [В2]аоткуда(£(Л— ^02) 1 [Ок2]“[В|]6 /10 1)01g [в2]а [Ок, )* (1' ;381
Выражение под знаком логарифма в уравнении
(19.29) равно константе равновесия, поэтому'» ~ (£д'о~5и’’ " ' (19-3°»Из уравнения (19.30) следует, что константа рав¬
новесия тем больше, чем значительнее различаются
стандартные потенциалы окислителя и восстанови¬
теля.Окислительный потенциал системы зависит не
только от pH раствора или от образования малорас¬
творимых соединений с одним из участников реак¬
ции, но также от связывания исходных веществ или
продуктов реакции в комплексные соединения. Так,
ионы железа (III) окисляют ионы иода, выделяя сво¬
бодный иод:2Fe3* + 2Г = 2Fea* + [,,так как окислительные потенциалы равны для систем
Fe3+/Fe2+ и 12/21“ 0,75 В и 0,53 В соответственно.Если, однако, прибавить к раствору фторид
натрия, образуются комплексные фториды железа
FeF2\ FeFj и другие, концентрация ионов Fe3+ рез¬
ко уменьшается и потенциал пары Fe3+/Fe2+ стано¬
вится меньше 0,53 В; поэтому железо во фторид-
ном растворе перестает выделять иод из иодида ка¬
лия.Ртуть не окисляется ионами водорода по реакции
Hg + 2H* = lVM-H2lтак как соотношение окислительных потенциалов сле¬
дующее.Hg2f+2e“ = Hg, £q = 0,85 В;2Н*" -|- 2е~ = Н „ £" = 0,0 В.Однако если прибавить иодид калия, концентрации
ионов ртути, находящихся в равновесии с ионами во¬
дорода в приведенной реакции, резко уменьшается
из-за связывания их иодид-ионами в прочный комп¬
лексный анион Hgl4 . В этом случае ртуть перехо¬
дит в значительном количестве в раствор в видеHgir.382
Сказанное подтверждается расчетом с использо¬
ванием уравнения (19.30). Пусть, например, необхо¬
димо вычислить константу равновесия и концентра¬
цию ионов Hgl2", образующихся в реакцииHg + 2Н+ + 4Г = Hgl*' + Н2.В этой реакции сочетаются реакции окисления и
комплексообразования. В отсутствие комплексообра¬
зующих веществ ртуть не растворяется в кислотах,
не являющихся окислителями. Однако в присутствии
иодид-ионов, связывающих ионы ртути в иодидный
комплекс, такая реакция может проходить.Предположим, что концентрации ионов водорода
и иодид-ионов поддерживаются во время реакции по¬
стоянными и равными 0,01 и 1 М соответственно.
Константа равновесия реакции представляет собой
сочетание двух констант, а именно константы, харак¬
теризующей процесс окисления ртути ионами водо¬
рода, и константы устойчивости иодидного комплекса
ртути. В соответствии с этим:jHgiy[н*ПггдеfHgiM1 s 4 1 . = ю30.[Hg2+][r]4Применив уравнение (19.30) и учитывая, что потен¬
циал реакции Hg2+ + 2£- = Hg равен 0,85 В, полу¬
чим:(0 - 0,85) ■ 2
g 0,058откудаMl[Hi2 [Г]410 29. 1030= 10.Так как [Н+] =0,01 и [I-] = 1, то в состоянии рав¬
новесия концентрация ионов [Hgl2] = 10~ЭМ, т. е.
металлическая ртуть окисляется ионами водорода и
переходит в значительном количестве в раствор ввиде Hgl/".Э8Э
Из уравнения (19.30) можно определить не толь¬
ко направление реакции окисления — восстановления
в данных условиях, но и вычислить равновесные кон¬
центрации всех реагирующих частиц в состоянии
равновесия.Пример 1. Вычислить концентрации реагирующих ионов
V02‘ + Fe2* + 2Н* VO2' + Fe3* + H20в состоянии равновесия при pH = 2,1 и при условии, что исход¬
ные концентрации VOj и Fe!* равны единице, a £oi = 1,0 В и
£02 = 0,75 В.В соответствии с уравнением (19.30), „ , [VO2*] [Fe1*] 1,0-0,75В °к ® [VO*] [Fe2*] [Н*]2 0,058откудаI VO-1 [Ре-Ч (H i*Концентрации продуктов реакции равны: fV02+] — fFe3 + |,
поэтому равновесные концентрации [VO*J = I — [VO2*] или
[V02] = I [F"с3 ] и [Fe2*] = 1 — [Fe3*]. Подставив эти далные
в уравнение и принимаяво внимание,что fHf] = IО-21, получим-.[ Ре3* ]°к 1— 1Ре3+]2 ’откуда[ Fe3+]2 = 1 — 2[Fe3 + ] + [Fe3 + ]2,[Fe3*] = [VO2*] =0,5 M, a [Fe2*] = [V02*| = 0,5 M.При заданном значении pH н указанных выше исходных
концентрациях ванадня(У) и железа(II) реакция будет проходить
слева направо на 50%.При мер 2. Определить направление реакцииМп02 + 2Ag-f 4Н* = 2Ag* + Мп2* + 2НгОпри pH = 4 и pH = 7, ес..пи1 изигетны стандартные потенциалы
реакцийМп02 + 4Н* + 2е~ = Мп2‘ + 2НгО, Е01 = 1,24 В;Ag* + ег = Ag, £„ = 0,8 8.В соответствии с уравнением (19.30)[Ag*]2[.Mn2+] (Е0, -£„,)• 2 (1,23-0,8)-2'g ''Ок — 'g r77+Ti = =14,7[Н*] 0,058 0,058или Кок -= 5-1014.384
Как показывает величина Кок, равновесие реакции при одно¬
молярной концентрации ионов водорода будет практически пол¬
ностью сдвинуто вправо.При pH = 4 [Ag+|2[Mn2+] = 5- 10'4- 10-,в = 5- [О-2.Принимая во внимание, что fAg+J = 2[Мп2+|, получим:[Ag+] = ^100-10~3 =0,46 моль/л,т. е. при данной кислотности значительная часть металлического
серебра будет окислена до ионов Ag+.При pH = 7 [Ag*l = • Ю~15 = 4,6 ■ 10“5 моль/л.т. е. диоксид марганца практически не окисляет металлическое
серебро.Из приведенных примеров видно, что направление
реакций окисления — восстановления, константы рав¬
новесия этих реакций, а также концентрации реаги¬
рующих веществ в состоянии равновесия всегда
можно найти, если известны стандартные потенциалы
и другие константы, характеризующие различные по¬
бочные процессы. К последним относятся, в частности,
процессы образования комплексных или малораство-
римых соединений и др. Однако в реальных условиях
анализа влияние этих побочных процессов удается
оценить далеко не всегда из-за отсутствия необходи¬
мых данных о константах протекающих реакций.
В этих случаях целесообразно пользоваться так на¬
зываемыми формальными или реальными потен¬
циалами.Формальный потенциал — это потенциал, относя¬
щийся к одномолярным концентрациям окисленной и
восстановленной форм вещества и к определенным
точно известным концентрациям остальных веществ,
содержащихся в растворе. Так, стандартный потен¬
циал пары Fe3+/Fe2+ при условии отсутствия побоч¬
ных процессов и при активности обеих частиц, равной
единице, равен 0,77 В. Однако потенциал этой же
пары в 1 М растворе хлорной кислоты, отнесенный
к I М концентрации (не активности) обеих частиц,
становится равным 0,73 В. Хлорная кислота не обра¬
зует каких-либо комплексных ионов с ионами железа;
изменение потенциала связано в данном случае с тем,
что высокая ионная сила раствора приводит к изме¬
нению коэффициентов активности частиц, причем для
ионов Fe3+- это изменение вследствие более высокого13 Аналитическая химия385
заряда (по сравнению с ионами Fe2+) происходит
в значительно большей степени, чем для ионов Fe24-.
В 1 М растворе хлороводородной или серной кислот
потенциалы становятся равными соответственно0,70 В и 0,68 В. Здесь кроме изменения ионной силы
раствора имеет значение также образование хлорид-
ных или сульфатных комплексов железа: ионы Fe3+
образуют более прочные комплексы, чем ионы Fe2+,
поэтому концентрация первых уменьшается в значи¬
тельно большей степени, чем вторых, что приводит
к уменьшению потенциала системы.Стандартный потенциал системы (Ag+ + ег = Ag)
равен 0,8 В. Однако в 1 М растворе хлороводородной
кислоты он снижается до 0,22 В, Это вызвано тем, что
концентрация ионов серебра из-за образования осад¬
ка AgCl снижается и определяется уже произведе¬
нием растворимости последнего. В данном случае зна¬
чение этого потенциала' может быть найдено простым
расчетом, подобно тому, как это было сделано ра¬
нее при оценке направления реакции между ионами
водорода и металлической ртутью в растворе иодово-
дородной кислоты.Применение формальных потенциалов, величины
которых для данных конкретных условий опреде¬
ляются экспериментально, дают возможность пра¬
вильно оценить состояние равновесия окислительно¬
восстановительной реакции и избежать ошибок, вы¬
званных отсутствием учета побочных процессов.Необходимо иметь в виду, что стандартные по¬
тенциалы характеризуют систему в состоянии устано¬
вившегося равновесия. Они определяют возможность
протекания данной окислительно-восстановительной
реакции, однако не дают никакой информации о ре¬
альной скорости достижения состояния равновесия.
Чем больше разность стандартных потенциалов окис¬
лителя и восстановителя, тем больше движущая сила
реакции и ее константа равновесия, которую можно
найти по уравнению (19.3), и тем в большей степени
реакция протекает слева направо. Так, стандартные
потенциалы полуреакцийН202 + 2Н+ + 2е~ = 2Н20иВг2 + 2е' = 2ВГ, 12 + 2е' = 2 Г386
равны соответственно 1,77, 1,08 и 0,53 В, т. е. перок¬
сид водорода является более сильным окислителем,
чем свободный бром, а бром — более сильным окисли¬
телем, чем иод.Константы равновесия реакцийН202 + 2Н+ + 21 = 2Н20 4- ЬВг2 + 2Г = :2ВГ + 12
равны, (1,77 - 0,53)-2 „00. „ _1П42,8.<g^OK = 42,8, Аок-Ю ,И'«к 0,058в — о,' 0,058»„ (1,08 - 0,53). 2 _ „ _ 19® Ок -n058i 19’ •Из сопоставления констант видно, что состояние
равновесия реакции между Н202 и I- сдвинуто вправо
в значительно большей степени, чем реакции между
Вг2 и I-. Однако первая реакция проходит крайне
медленно, и состояние равновесия достигается за до¬
вольно значительный промежуток времени. В то же
время бром окисляет ионы: иода практически мгно¬
венно.§ 3. СОПРЯЖЕННЫЕ РЕАКЦИ И
ОКИСЛЕНИЯ —ВОССТАНОВЛЕНИЯСопряженными реакциями окисления — восстановле¬
ния называют такие две реакции, из которых одна
протекает самопроизвольно, а вторая — только тогда,
когда в том же растворе происходит первая реакция.
По Н. А. Шилову, первую реакцию называют первич¬
ной, вторую — вторичной. Вещество, принимающее
участие в обеих реакциях, называют актором, веще¬
ство, которое принимает участие только в первичной
реакции — индуктором, вещество, участвующее только
во вторичной реакции — акцептором.|-—► А + В (индуктор) — первичная реакция
Актор —I'—»■ А + С (акцептор) — вторичная реакцияСопряженные реакции окисления — восстановле¬
ния отличаются от каталитических реакций:
после завершения реакции все три участвующих13*387
вещества — актор, индуктор и акцептор — превра¬
щаются в другие вещества.Известны многочисленные примеры сопряженных
реакций окисления — восстановления. Так, винная
кислота при определенных условиях восстанавливает
свободный хлор, однако не восстанавливает ионы
меди(II). При совместном присутствии всех трех ве¬
ществ восстанавливаются и хлор, и ионы меди. Сле¬
довательно, в этом примере имеем такую систему:■ *■ Н2С4Н406 + С12 (индуктор) — первичная реакцияАктор —1—»• H2C4H406 + Cu2f (акцептор) — вторичная реакцияХромовая кислота легко окисляет сернистую кис¬
лоту, однако не реагирует с винной кислотой. Вторая
реакция происходит только в том случае, если рас¬
твор содержит все три вещества. Таким образом:,—► Н2Сг04 + H2SG3 (индуктор) — первичная реакцияАктор ——► Н2СГО4+Н2С4Н4ОС (акцептор) —вторичная реакцияМожно подобрать такие условия, при которых би¬
хромат калия не выделяет иод из раствора иодида
калия. Однако если в растворе находятся ионы же¬
леза (II), то окисляется и железо, и иодид.Промежуточный продукт с неустойчивой степенью
окисления может образоваться за счет химического
превращения актора. При окислении железа хрома¬
том в условиях, когда последний не реагирует с иодид-
ионами (например, при pH = 5), на каждый ион Fe2+
в реакцию вовлекается два иона иода. Это дало осно¬
вание предположить, что механизм сопряженной ре¬
акции окисления иодид-ионов следующий:Н2Сг04 + Fe2*' —► Fev, Fev + 2Г —* Fe3t + I2>т. e. причиной сопряженного окисления иодид-ионов
является образование железа в степени окисле¬
ния (+5).Аналогичный механизм предложен для объясне¬
ния сопряженного окисления хлорид-ионов перманга¬
натом калия при титровании раствором последнего
ионов Fe2+. Таким образом, в обоих случаях промежу¬
точный продукт с неустойчивой степенью окисления
Fev образуется из индуктора (Fe2+).388
Промежуточный продукт с неустойчивой степенью
окисления может также образоваться за счет окисле¬
ния или восстановления индуктора и в других слу¬
чаях. Так, при исследовании системы из двух реакций,—»■ НВгОз + H2S03 (индуктор) — первичная реакцияАктор ——► HBr03-f-HAs02 (акцептор) — вторичная реакциянайдено, что при некоторых условиях вторичная ре¬
акция происходит только если в растворе находится
сернистая кислота, легко и быстро окисляющаяся
броматом. Однако вместо сернистой кислоты в каче¬
стве индуктора можно применить и другие восстано¬
вители, например формальдегид или бромоводород¬
ную кислоту. Поэтому был сделан вывод, что причи¬
ной данной сопряженной реакции не может быть
промежуточный продукт с нестойкой степенью окис¬
ления индуктора и что в процессе восстановления
образуется нестойкая кислота НВг02 за счет восста¬
новления актора, которая и вызывает протекание вто¬
ричной реакции.Наконец, в процессе реакции может образоваться
комплексное соединение из индуктора и акцептора,
примером чего является следующая система:Н2Сг04 -4- HAs02 — первичная реакции,Н2Сг04 + Н2С4Н406 — вторичная реакция.Хромовая кислота в определенных условиях окис¬
ляет винную кислоту только в присутствии мышьяко¬
вистой (или сурьмянистой) кислоты. Известно, что
винная кислота образует с мышьяковистой кислотой
комплексное соединение, причем связанная в комп¬
лекс винная кислота окисляется легче, чем свободная.Установлен также радикальный механизм сопря¬
женных реакций, обусловленных образованием
свободных радикалов. Например, ионы Fe2+ легко
окисляются пероксидом водорода, однако разложение
последнего при этом сильно ускоряется. Процесс
описывается следующими уравнениями:Н2Ог + 2Fe2+ = гре3* + 20Н~ — первичная реакция,Н202 + Н202 = 2Н20 + 02 — вторичная реакция.Первичная реакция проходит через такие стадии:Н202 + Fe2* = Fe3+ + ОН’ + . ОН, . ОН + Fe2+ = Fe3+ + ОН".389
Образующийся свободный радикал -ОН может во¬
влекать в реакцию пероксид водорода:НзОа + • ОН = • НО, + Н20, ■ Н02 + Н262 = Н20 + Ог + • ОНЦепная реакция разложения пероксида водорода
посредством радикала *ОН может проходить до тех
пор, пока последний не прореагирует с ионами Fe2+.§ 4. ИНДИКАТОРЫОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ТИТРОВАНИЯВ методах окисления — восстановления конечную точ¬
ку титрования можно фиксировать следующими спо¬
собами.1. По окраске избытка прибавленного рабочего
раствора.2. Введением в раствор веществ, специфически
реагирующих с веществом рабочего раствора или
с определяемым веществом с образованием окрашен¬
ных соединений.3. Введением в раствор веществ, разрушающихся
с исчезновением окраски от избытка рабочего рас¬
твора.4. Посредством окислительно-восстановительных
индикаторов, изменяющих окраску в зависимости от
изменения окислительно-восстановительного потен¬
циала системы.Установление точки эквивалентности по первому
способу характерно для метода перманганатометрии.
Раствор перманганата калия окрашен в фиолетово¬
красный цвет, поэтому после завершения титрования
лишняя капля КМп04 окрашивает оттитрованный
раствор в хорошо заметный розовый цвет. Метод не¬
применим при титровании окрашенных растворов,
а также при титровании окислителями или восстано¬
вителями, растворы которых бесцветны или окра¬
шены слабо.Второй способ применяют главным образом в ме¬
тоде иодометрии. В раствор вводят крахмал, который
образует с иодом соединение интенсивно синего цвета.
Появление (или исчезновение) синей окраски служит
признаком конца титрования.В качестве другого примера можно привести тит¬
рование ионов железа (III) раствором какого-либо390
восстановителя, например раствором TiCl3. Индикато¬
ром служит роданид калия или аммония, образующий
с ионами Fe3 h комплекс красного цвета. После вос¬
становления всего железа красная окраска исчезает.Третий способ — применение так называемых не¬
обратимых индикаторов, которые разрушаются (окис¬
ляются или восстанавливаются) избытком рабочего
раствора, вследствие чего окраска изменяется. Такой
прием применяют обычно в методе броматометрии.
В качестве индикатора можно брать, например, мети¬
ловый оранжевый или метиловый красный. При тит¬
ровании броматом калия выделяется свободный бром,
который окисляет сначала определяемое вещество,
а затем, после точки эквивалентности, и сам индика¬
тор. Необратимые индикаторы неудобны тем, что они
частично окисляются еще в процессе титрования,
вследствие чего окраска постепенно бледнеет и для
четкого перехода необходимо прибавлять новые пор¬
ции индикатора. Поэтому точка эквивалентности фик¬
сируется недостаточно резко, а первоначальная
окраска индикатора не восстанавливается при добав¬
лении к перетитрованному раствору некоторого коли¬
чества определяемого вещества.Четвертый способ — применение обратимых окис-
лительно-восстановительных индикаторов. Эти инди¬
каторы— преимущественно окрашенные органические
соединения, которые изменяют свой цвет в зависимо¬
сти от окислительно-восстановительного потенциала
титруемой системы. При введении избытка окислителя
образуется окисленная форма индикатора, а прибав¬
ление избытка восстановителя приводит к образова¬
нию его восстановленной формы. Процесс перехода
окисленной формы в восстановленную и обратно, со¬
провождающийся изменением окраски, можно повто¬
рить много раз без разрушения индикатора.Окислительно-восстановительные индикаторы при¬
меняют при титровании растворами бихромата калия,
сульфата церия, ванадата аммония и других, т. е.
в тех случаях, когда рабочий раствор окислителя бес¬
цветен или окрашен настолько слабо, что по его из¬
бытку нельзя с уверенностью установить точку экви¬
валентности. Кроме того, такие индикаторы можно
использовать при титровании окислителей восстано¬
вителями, например рабочими растворами TiCb,391
SnCl2, CrCl2, FeS04, H3As03, что значительно расши¬
ряет область применения титриметрических методов
анализа.Общая схема окисления — восстановления индика¬
тора может быть следующей:IndOK + пе~ + аН+ •—> IndB. (19.31)Откуда, применяя к этому выражению уравнение
Нернста, получаем:0,058 [lndOKl [Н+Г
ЕЯ-Е0 + ——’g ffHl (.9.32)Характерной особенностью индикаторов данного
класса является зависимость их потенциала от pH.
Из уравнения (19.32) видно, что при возрастании pH
потенциал индикаторной системы уменьшается.
С этим приходится считаться, когда в процессе тит¬
рования pH раствора заметно изменяется. Однако
чаще всего титрование окислителями или восстанови¬
телями ведут в присутствии достаточно большого из¬
бытка сильной кислоты, причем pH раствора остается
практически постоянным.Для дальнейшего рассмотрения можно принять,
что концентрация ионов водорода близка к 1 М.
Тогда уравнение (19.32) упрощается:0,058 IInd0l*w. + — '«Tisgp <19-33>Рассмотрим, как изменяется окраска индикатора при
титровании какого-либо восстановителя В, каким-
либо окислителем Ок2. Примем, что в уравнении
(19.31) /г = 2, что является наиболее частым случаем.В процессе титрования потенциал системы возра¬
стает, так как соотношение концентраций окисленной
и восстановленной форм определяемого вещества Ок2
и В] увеличивается. Пока этот потенциал имеет доста¬
точно низкое значение, намного меньше, чем стан¬
дартный потенциал индикаторной системы £о, прак¬
тически весь индикатор находится в восстановленной
форме In< 1 в и имеет окраску этой формы. По мере
увеличения потенциала системы Oki/Bj, вызванного
прибавлением рабочего раствора окислителя Ок2, из¬
меняется соотношение окисленной и восстановленной
форм индикатора. После точки эквивалентности,392
Таблица 19.1. Соотношения llndot]: [Inde] в процессе титрованияс*[IndOK] ех-е0ОкраскаЕхВ [lndB] 0.029[lnd„]0,605—51 : Ю5Восстановленной формы0,634-41 : Ю<То же0,663—31 : Ю3»0,692—21 : 100»0,721-11 : 10Восстановленной формы соттенком окисленной фор¬
мы0,75001 : 1Смешанная окраска окис¬
ленной и восстановленной
форм0,779110: 1Окисленной формы с от¬
тенком восстановленной
формы0,8082100: 1Окисленной формы0,83731000 : 1То жекогда введен некоторый избыток окислителя Окг, по¬
тенциал системы Ок2/Вг становится значительно
больше потенциала индикаторной системы Indcm/Inde
и практически весь индикатор будет находиться
в окисленной форме, окрашенной иначе, чем восста¬
новленная форма.В табл. 19.1 приведены данные, характеризующие
изменение соотношения концентраций IndoK и Inde
в процессе титрования для индикатора с £о = 0,75В.
Обычно изменение окраски восстановленной формы
индикатора начинает наблюдаться при соотношении
[IndoK] : [Inda]= 1 : 10, оттенок окраски восстанов¬
ленной формы еще заметен при соотношении [Indcm] :: [Inde] =10: 1. Наиболее резкое изменение окраски
соответствует соотношению, равному единице, т. е. ко¬
гда содержание каждой формы составляет 50 %•
Таким образом, из данных таблицы можно сде¬
лать вывод, что интервал изменения окраски индика¬
тора находится в пределах потенциалов, соответст¬
вующих уравнениюАЕХ = Е0± 0,029 (19.34)или в общем виде, если п Ф 2АЕХ^Е0±^-. (19.35)п393
Приведенные данные позволяют выбрать индика¬
тор с наиболее подходящим стандартным потенциа¬
лом для любого окислительно-вос'становительного тит¬
рования. Лучше всего, если наиболее резкое измене¬
ние цвета произойдет в точке эквивалентности; для
этого необходимо, чтобы стандартный потенциал ин¬
дикаторной системы был равен потенциалу точки эк¬
вивалентностиЕ0 = ЕТ..). (19.36)Рассмотрим титрование ионов двухвалентного
олова раствором соли трехвалентного железа:Sri2* + 2Fe3+ = SnIV + 2Fe2*\ (19.37)Потенциал точки эквивалентности равенОок аЕов
а + Ь(19.38)где £0ок и £ов — стандартные потенциалы систем Fe3+/Fe2+ и
Sn,v/Sn2+; а и в — стехиометрические коэффициенты при окисли¬
теле и восстановителе соответственно.Для рассматриваемого случая £0ок=0,77 В, £ов=
= 0,2 В, следовательно^^077 + 0^^0,38 В,т. е. для данного титрования наиболее подходит ин¬
дикатор со стандартным потенциалом £о = 0,38В.
Отклонения от этой величины приведут к погрешно¬
сти определения.Выясним, в какой степени Е0 индикаторной си¬
стемы может отклоняться от Ет.э, чтобы погрешность
титрования, обусловленная этим отклонением, не пре¬
вышала допустимых в титриметрическом анализе пре¬
делов. Ранее (см. гл. 7) было показано, что погреш¬
ность титриметрического анализа составляет прибли¬
зительно 0,1 %. Найдем значения потенциалов, при
которых ионы олова будут недотитрованы или пере-
титрованы на 0,1 %.Недотитрованный на 0,1 % раствор соответствует
соотношению [SnIV] : [Sn2+] = 1000: 1, откудаЕх = 0,2 + 0,029 lg 1000 = 0,29 В. (19.39)В перетитрованном на 0,1 % растворе содержатся
ионы Fe3+, причем соотношение [Fe3+] : [Fe2+] долж¬394
на быть равно 1 : 1000. ПоэтомуЕх = 0,75 + 0,058 lg 0,001 = 0,58 В.Для титрования с погрешностью, не превышающей
0,1 %, интервал изменения окраски индикатора дол¬
жен находиться в пределах 0,29—0,58 В, т. е. начало
изменения окраски индикатора может наблюдаться
при 0,29 В, а конец — при 0,58 В. Учитывая уравне¬
ние (19.34), приходим к заключению, что для данного
титрования можно использовать все индикаторы,
стандартные потенциалы которых находятся в преде¬
лах (0,29 + 0,03) — (0,58 — 0,03) или 0,32—0,55 В.При титровании ионов железа (II) раствором би¬
хромата калия реакция идет по уравнениюСг20*- + 6Fe2t + 14Н* == 2Cr3t + 6Fe3* + 7Н20.Учитывая, что £ „. = 13 В, а Е 2+ = 0,75 В,
для потенциала точки эквивалентности находим:Если раствор Fe2+ недотитрован на 0,1 %Ех = 0,75 + 0,058 lg 1000 = 0,92 В.Перетитрованный на 0,1 % раствор характери¬
зуется соотношением[СГ2О7"] : [Сг3+] = 1 : 1000, откудаЕх = 1,3 + ig icT6 = 1,24 В.Учитывая уравнение (19.34), находим, что для
данного определения пригодны все индикаторы, у ко¬
торых стандартные потенциалы находятся в пределах
0,95—1,21. В.Известно очень много обратимых окислительно¬
восстановительных индикаторов с различными значе¬
ниями стандартных потенциалов. Некоторые из них
приведены в табл. 19.2.Из таблицы видно, что для титрования ионов Sn2+
ионами Fe3+ наиболее подходят последние два инди¬
катора. Титрование железа бихроматом калия можно
осуществить с индикатором N-фенилантраниловой
кислотой или с ферроином.395
Таблица 19.2. Обратимые окислительно-восстановительные
индикаторыИндикаторИвет формЕ0. Вокисленнойвосстановленной5-Нитро-1,10-фенантро-Бледно-Красно-фиолето¬1,25линат железаголубойвыйN-ФенилантраниловаяКрасныйБесцветный1,08кислота1,10-Фенантролинат же¬Бледно-Красный1.11леза (ферроин)голубойЭриоглюцин АКрасныйЖелто-зеленый0,98Дифениламиносульфо-Красно¬Бесцветный0,85кислотафиолетовыйДифениламин -Фиолетовый»0,76Вариаминовый голубойСиний»0,60Метиленовый синий»»0,53Индиготетрасульфонатъъ0,36Механизм действия окислительно-восстановитель¬
ных индикаторов различен. Рассмотрим наиболее
важные индикаторы.При окислении дифениламина (I) сначала обра¬
зуется бесцветный дифенилбензидин(П):2 $ ^ NH ^ ^ >•\=/ WINH f у + 2Н+ + 2 .Эта реакция необратима. Далее дифенилбензидин
окисляется, образуя окрашенный дифенилбензидин
фиолетовый (III):*=± N=/^==/^=N--^^ + 2Hf + 2e-IIIПереход формы II в форму III уже обратим.396
Дифениламин мало растворим в воде, применяют
его раствор в концентрированной серной кислоте. При
титровании необходимо прибавлять только очень не¬
много дифениламина, иначе промежуточный про¬
дукт II может образовать с продуктом Ш трудно рас¬
творимое молекулярное соединение зеленого цвета
с довольно слабой интенсивностью окраски.Вместо дифениламина удобно применять дифенил¬
ам иносульфокислоту, которая хорошо растворима
в воде. Механизм окисления этого индикатора, также
как и N-фенилантраниловой кислоты, которые являют¬
ся производными дифениламина, аналогичен меха¬
низму окисления последнего.Широко применяют 1,10-фенантролянат железа
(ферроин). 1,10-Фенантролин имеет следующую фор¬
мулу:и образует с ионами железа комплексное соединение
за счет координационной связи с атомами азота; каж¬
дый ион железа координирует три молекулы фенан-
тролина (сокращенно Phen). Переходхорошо обратим и сопровождается изменением окрас¬
ки от бледно-голубой до красной. Нитропроизводное
ферроина имеет аналогичные свойства, он отличается
более высоким потенциалом.Окисление — восстановление вариаминового голу¬
бого происходит в соответствии с уравнениемЭтот переход происходит при pH = 2 и потенциале
около 0,6 В. С возрастанием pH потенциал изменения
окраски снижается.Существуют также неорганические обратимые
окислительно-восстановительные индикаторы. В каче¬Fe(Phen)f+ + е~ Fe^hen)^*ch>°43-n“0■=NHj + Н+ + 2е~.г397
стве примера можно привести кремнемолибденовую
кислоту H^SifMoaOiob], которая имеет слабую жел¬
тую окраску; при действии сильных восстановителей
образуется интенсивно окрашенная молибденовая
синь. Этот индикатор можно применять для опреде¬
ления железа по следующему методу.Трехвалентное железо сначала восстанавливают
раствором SnCb, который вводят в некотором из¬
бытке. Прибавляют в раствор кремнемолибденовую
кислоту, вследствие восстановления которой раствор
окрашивается в синий цвет. Постепенно прибавляют
раствор бихромата калия для окисления остатка ди¬
хлорида олова; признаком окончания окисления яв¬
ляется исчезновение синей окраски молибденовой
сини. Титруют двухвалентное железо раствором би¬
хромата калия в присутствии фенилантраниловой кис¬
лоты в качестве индикатора.ГЛАВА 20ТИТРОВАНИЕ ПЕРМАНГАНАТОМ
(ПЕРМАНГАНАТОМЕТРИЯ)§ 1. ОБЩАЯ ХАРАКТЕРИСТИКА.ПРИГОТОВЛЕНИЕ И СВОЙСТВА РАСТВОРА
ПЕРМАНГАНАТАПерманганат калия применяют для определения мно¬
гих неорганических и органических веществ. В кис¬
лых растворах он восстанавливается до ионов двух¬
валентного марганца:МпО; + 8Н+ + 5е' = Mn2t + 4Н20, £0 = 1,52 В. (20.1)В слабокислых, нейтральных и слабощелочных
растворах восстановление происходит до диоксида
марганца:МпО; + 4Н+ + Зе" = Мп02 + 2Н20, Е0 = 1,69 В. (20.2)Наконец, в сильнощелочных растворах реакция
происходит с образованием манганат-ионов:Мп04 +e' = MnOj*. £0 = 0,56 В. (20.3)398
В растворах, содержащих некоторые лиганды, ста¬
билизируется степень окисления +3. Например, в при¬
сутствии пирофосфатов (или фторидов) реакция вос¬
становления происходит следующим образом-МпО; + 8Н+ + 4е~ + ЗН^^- = Мп (Н2Р207)33' + 4Н20. (20.4)Последние две реакции используют редко. Чаще
всего титруют в кислом растворе, когда перманганат
восстанавливается до ионов Мп2+.Применяют прямые и косвенные методы титро¬
вания.Прямым титрованием, когда реакция происходит
в сильно кислом растворе по уравнению (20.1), опре¬
деляют многие неорганические ионы, находящиеся
в низшей или средней степени окисления, например
ионы Fe2+, As3+, Sb3+, Sn2+, H2O2, V02+, Mo3+, Mov,
U4+, Ti3+ и др.Многие другие ионы можно определять косвенно.
Их сначала осаждают в виде оксалатов, затем тит¬
руют перманганатом связанную с металлом щавеле¬
вую кислоту или избыток последней, введенный в рас¬
твор при осаждении. Так можно определять ионы
Са2+, Sr2+, Ва2+, Zn2+, Со2+, La3+, Th4+, Се3+, Ag+,
Pb2+ и др.Многие органические вещества также окисляются
перманганатом, например алифатические и аромати¬
ческие кислоты, оксикислоты, фенолы и др. Окисле¬
ние нередко идет до конечного продукта — диоксида
углерода. Однако реакции с органическими веще¬
ствами обычно протекают очень медленно, поэтому
прямое титрование применить трудно. В этих случаях
пользуются косвенным методом. К раствору опреде¬
ляемого вещества прибавляют щелочь и избыток пер¬
манганата, через некоторое время раствор подкисляют
и определяют каким-либо методом избыток не вошед¬
шего в реакцию перманганата.Реакцию (20.5) используют для определения ионов
двухвалентного марганца:2Мп04 + ЗМп2* + 2Н20 = 5Мп02 + 4Н+. (20.Б)Для подкисления раствора перед титрованием чаще
всего применяют серную кислоту. Хлороводородную
кислоту почти не используют, так как в ее399
присутствии возможно частичное окисление ионов
хлора перманганатом или различными промежуточ¬
ными оксидами, образующимися при основной реак¬
ции. Азотная кислота, особенно содержащая примеси
оксидов азота, вызывает ряд побочных процессов и мо¬
жет также окислять частично определяемое вещество.При титровании перманганатом редко применяют
специальные индикаторы, так как окраска перманга¬
ната обычно достаточна для установления точки экви¬
валентности. Однако необходимо иметь в виду, что
введенный избыток перманганата обычно разлагается
по реакции (20.5) и окраска постепенно исчезает.Из уравнения (20.1) видно, что эквивалент КМп04
равен одной пятой части молекулярной массы, т. е.
фактор эквивалентности /экв = 1 /5. Обычно поль¬
зуются растворами, содержащими в 1 л десятую долю
эквивалента, т. е. 158-1/5-1/10 = 3,16 г.Перманганат — очень сильный окислитель и по¬
этому способен окислять воду в соответствии с ре¬
акцией4МпО; + 2Н20 = 4Мп02 + 302 + 40Н~. (20.6)Эта реакция протекает крайне медленно и, как
правило, не вызывает каких-либо затруднений при
определении. Однако следы Мп02 ускоряют разложе¬
ние КМп04; разложение ионов перманганата уско¬
ряется также на свету, при введении в раствор кис¬
лот или оснований, при нагревании, а также при вве¬
дении ионов Мп2+. В последнем случае происходит
реакция (20.5), при которой образуется диоксид мар¬
ганца, еще более ускоряющий разложение.Диоксид марганца образуется также при восста¬
новлении перманганата следами органических ве¬
ществ, пыли и др. В связи с названными процессами
растворы перманганата калия постепенно изменяют
свою концентрацию.Для приготовления устойчивых во времени раство¬
ров необходимо устранить возможность загрязнения
перманганата диоксидом марганца. С этой целью для
приготовления раствора перманганата необходимо
брать свежеперегнанную дистиллированную воду, ко¬
торую перед приготовлением раствора следует пере¬
гнать еще раз, предварительно прибавив к ней КМп04
и КОН. При использовании воды, не перегнанной вто¬400
рично, приготовленный раствор следует выдержать
до тех пор, пока не закончится окисление органиче¬
ских веществ, а после этого в свободном от пыли по¬
мещении отделить фильтрованием через стеклянный
фильтр выпавший осадок диоксида марганца.Растворы перманганата калия защищают от по¬
падания пыли и хранят в темной бутыли, соединен¬
ной стеклянным сифоном с бюреткой.Разбавленные растворы перманганата, например
0,01 М, неустойчивы и их нельзя хранить продолжи¬
тельное время. Такие растворы готовят непосред¬
ственно перед анализом путем разбавления более
концентрированных растворов.Исходные вещества. Для установления концентра¬
ции раствора перманганата применяют обычно щаве¬
левую кислоту и оксалат натрия, а также триоксид
мышьяка, железоаммонийсульфат и другие вещества.
Основным недостатком щавелевой кислоты как ис¬
ходного вещества является постепенное выветривание
кристаллизационной воды, вследствие чего препарат
перед употреблением необходимо перекристаллизо-
вать. В этом отношении значительно лучшим исход¬
ным веществом является оксалат натрия; он не
содержит кристаллизационной воды, почти негигро¬
скопичен. Чистую соль легко получить перекристал¬
лизацией и высушиванием при 105—110°С. Перед
приготовлением раствора следы гигроскопической
влаги удаляют высушиванием при 105 °С. Растворы
оксалата натрия медленно разлагаются на свету и
частично разрушают стекло (образуется оксалат каль¬
ция). Поэтому запасные растворы оксалата натрия
готовить не следует.Установка концентрации перманганата калия. Ре¬
акция окисления щавелевой кислоты перманганатом
протекает по уравнениюCjOJ- = 2С02 + 2е'МпО; Ч- 8Н* + 5е' = Мп2* + 4Н205C20j- + 2MnO; + 16Н* = 10СО2 + 2Мп2+ + 8НгОили5Н2С204 + 2КМпО< + 3H2S04 == K2S04+2MnS04+ ЮС02+8Н20.
Однако это уравнение выражает только суммарный
результат реакции. В действительности процесс
окисления происходит значительно сложнее. Реакция
между перманганат-ионами и щавелевой кислотой
происходит очень медленно, но ускоряется в присут¬
ствии ионов двухвалентного марганца. Если их не
вводить специально в раствор, тогда необходимо
ускорить реакцию нагреванием до 70—80 °С. Однако
не следует нагревать раствор выше этой температуры,
так как это может привести к частичному разложе¬
нию щавелевой кислоты:н2с2о4 = Н20 + со2 + СО.Кроме того, при кипячении происходит частичное
разложение и самого перманганата с выделением
кислорода.При титровании к нагретому раствору щавелевой
кислоты прибавляют сначала несколько капель рас¬
твора КМп04 и ожидают, пока раствор обесцветится.
Восстановление следующих порций рабочего раствора
идет уже быстро. Конец титрования определяют по
появлению неисчезающей некоторое время розовой
окраски.§ 2. ПЕРМАНГАНАТОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ
НЕОРГАНИЧЕСКИХ И ОРГАНИЧЕСКИХ ВЕЩЕСТВОпределение железа в рудах. Железо в рудах обычно
содержится в виде оксидов: гематита Fe203, магне¬
тита Fe304, лимонита 3Fe203-3H20 и др. Разложение
пробы почти всегда заключается в обработке навески
концентрированной хлороводородной кислотой:Fe203 + 6НС1 = 2FeCl3 + 3H20.Чтобы ускорить растворение, приливают немного
раствора SnCI2. Дихлорид олова восстанавливает
трехвалентное железо, вследствие чего концентрация
Fe3+ уменьшается и равновесие реакции сдвигается,
вправо. Нерастворившийся силикатный остаток от¬
фильтровывают и, если он окрашен в темный цвет,
что свидетельствует о содержании железа, сплавляют
с содой. Плав растворяют в хлороводородной кислоте
и полученный раствор присоединяют к основному
раствору пробы.402
В полученном растворе хлоридов непосредственно
определяют железо, предварительно восстанавливая
его до двухвалентного. Восстановление можно прово¬
дить в цинковом, кадмиевом или висмутовом редук¬
торе; последним удобно пользоваться в присутствии
титана, который не восстанавливается висмутом, но
переходит в трехвалентное состояние при действии
цинка или кадмия.Менее точным, но весьма распространенным мето¬
дом восстановления, является восстановление железа
дихлоридом олова. Мешают определению ванадий,
молибден и вольфрам, которые иногда содержатся
в небольших количествах в железных рудах и также
восстанавливаются дихлоридом олова.Принцип метода заключается в следующем. К кис¬
лому раствору хлорида железа (III) приливают не¬
большой избыток раствора дихлорида олова:2FeCl3 + SnCl2 = 2FeCl2 -f SnCl4.Избыток SnCl2 необходимо перед титрованием
окислить, так как иначе перманганат будет расходо¬
ваться также на окисление дихлорида олова. Дихло¬
рид олова окисляют раствором хлорида ртути (су¬
лемы) :SnCl2 + 2HgCl2 = SnCl, + Hg2CI2.Выпадающий при этом осадок каломели очень
медленно реагирует с перманганатом. Подготовлен¬
ный таким образом холодный раствор хлорида же¬
леза титруют рабочим раствором перманганата до по¬
явления розовой окраски.При восстановлении железа(Ш) дихлоридом оло¬
ва необходимо обращать внимание на то, чтобы из¬
быток SnCl2 был очень мал — не более 2—3 капель.
При большом избытке идет восстановление сулемы и
каломели до металлической ртути:SnCl2 + HgCl2 = SnCU + Hg, SnCl2 + Hg2Cl2 = SnCl4+2Hg.Ртуть выделяется в мелкодисперсном виде и в та¬
ком состоянии легко окисляется перманганатом.Чтобы можно было установить тот момент, когда
прибавлено достаточное количество SnCb, раствор
FeCl3 перед восстановлением нагревают почти до ки¬
пения. Это приводит к значительному усилению403
интенсивности желтой окраски вследствие уменьше¬
ния диссоциации хлоридного комплекса железа. При¬
знаком полноты восстановления железа является ис¬
чезновение желтой окраски раствора. Затем раствор
охлаждают и разбавляют немного водой. В горячем
растворе возможно окисление хлороводородной кис-,
лоты перманганатом.Окисление железа(II) происходит в соответствии
со следующим уравнением:5Fe2+ + МпО; + 8Н+ = 5Fe3t + Mn2t + 4Н20или5FeCl2 + КМп04 + 8НС1 =5FeCl, + МпС12 + КС1 + 4НаО.Во время титрования в растворе появляется все
больше FeCI3, который в кислой среде в присутствии
хлорид-ионов окрашен в желтый цвет; в точке эк¬
вивалентности наблюдается переход окраски от жел¬
той к розовой. Этот переход воспринимается с трудом.
Для увеличения резкости перехода к раствору перед
титрованием прибавляют немного фосфорной кисло¬
ты, которая образует с ионами железа (III) бесцвет¬
ное комплексное соединение:Fe3+ + 2Н3РО, = H3[Fe(P04)2] + ЗН*.Титрование железа лучше всего проводить в сер¬
нокислом растворе, так как при этом не возникает
побочных реакций. Но железные руды растворяются
в хлороводородной кислоте легче, чем в серной кис¬
лоте, и поэтому удобнее пользоваться хлороводород¬
ной кислотой. Однако титрование в растворе хлорово¬
дородной кислоты осложняется сопряженной реак¬
цией окисления кислоты. Активные промежуточные
продукты, образующиеся во время реакции перманга¬
ната с железом(П), окисляют частично хлорид-ионы
до хлора, который выделяется из раствора в виде
газа. Вследствие этого перманганата расходуется
больше, чем необходимо для окисления железа(П).Причина сопряженного окисления—образование
промежуточного оксида железа(V); имеет значение
также образование ионов марганца(III), окислитель¬
ный потенциал пары Мп3+/Мп2+ снижается вслед¬
ствие связывания ионов Мп3+ в фосфатный комплекс.
Действие же Fev устраняется прибавлением к титруе-404
мому раствору достаточного количества сульфата
марганца. С увеличением концентрации Мп2+ пони¬
жается окислительный потенциал перманганата, окис¬
ление Fe2+ идет менее энергично и образование ак¬
тивного промежуточного оксида железа уменьшается.
Кроме того, по-видимому, железо в виде высшей ак¬
тивной степени окисления быстрее реагирует с ионом
Мп2+, чем с ионом CI-, образующиеся высшие валент¬
ные формы марганца, в отличие от хлора, не улету¬
чиваются и окисляют остающееся в растворе же¬
лезо (II).Защитная смесь, которую прибавляют перед тит¬
рованием к раствору, содержит смесь серной, фосфор¬
ной кислот и сульфата марганца; она устраняет
окраску ионов Fe3+ и предотвращает процесс сопря¬
женного окисления хлорида.Определение пероксида водорода. Торговый пре¬
парат пероксида водорода (пергидроль) содержит
около 30 % Н2Ог. Со временем пероксид водорода по¬
степенно разлагается с выделением кислорода:Н2О2 = НгО -f- 'ДЮг-Это разложение значительно замедляется в при¬
сутствии стабилизаторов — салициловой кислоты или
пирофосфата натрия. Однако при длительном хране¬
нии даже в присутствии стабилизаторов Н202 мед¬
ленно разлагается и ее содержание в растворе умень¬
шается. Для проверки качества реактива необходимо
определясь содержание Н202.Пероксид водорода обладает одновременно свой¬
ствами окислителя и восстановителя. Это объясняется
тем, что степень окисления кислорода в Н202 яв¬
ляется промежуточной между степенью его окисле¬
ния в воде и в 02. Когда пероксид водорода является
окислителем, ион 02‘ присоединяет два электрона:О2' + 2е- + 4Н* = 2Н20.Наоборот, действуя в качестве восстановителя, ион
ОГ отдает свои электроны с выделением молекуляр¬
ного кислорода:0^ = 2е+02НЛПН202 = 02 + 2Н* + 2е~.405
Для количественного определения пероксида во¬
дорода можно использовать и окислительные, и вос¬
становительные его свойства. Перманганатометриче¬
ский метод определения пероксида водорода состоит
в окислении его до свободного кислорода, т. е. здесь
используют свойства пероксида водорода как восста¬
новителя.Реакция протекает по уравнению2МпО; + 502“ + 16Н+ = 2Мп2* + 502 + 8НаОили2КМгЮ4 + 5Н202 + 3H2S04 = K2S04 + 2MnS04 + 502 + 8Н20.Титруют холодный раствор, точка эквивалентности
фиксируется по появлению неисчезающего розового
окрашивания избытка перманганата.Определение нитритов. Перманганат окисляет нит¬
риты в кислой среде до нитратов в соответствии
с уравнением5 NO“ + Н20 = N03 + 2Н+ + 2е‘2 Мпо; + 8Н+ + 5е~ = Мп2+ + 4Н202МпО; + 5N0“ + 6Н+ = 2Mn2* + 5N0' + ЗН20ИЛИ2КМп04 + 5KN02 + 3H2S04 = 2Mn S04 + K2S04+ 5KN03+ 3H20Однако непосредственное титрование кислого рас¬
твора нитрита перманганатом не дает правильных ре¬
зультатов. Это объясняется тем, что в кислом рас¬
творе нитриты разлагаются:2KNOa + H2S04 = K2S04 + N02 + NO + H20.Часть оксидов азота при этом выделяется из рас¬
твора в воздух и теряется для определения.Воспрепятствовать разложению нитритов можно
следующим образом. В колбу наливают некоторое ко¬
личество рабочего раствора перманганата калия и
разбавленную серную кислоту, затем прибавляют ней¬
тральный раствор определяемого нитрита с таким
расчетом, чтобы после его полного окисления в колбе
остался еще небольшой избыток перманганата. После
этого определяют количество не вошедшего в реак¬
цию КМПО4, например посредством титрованного рас¬
твора щавелевой кислоты. Однако при титровании406
перманганата щавелевой кислотой очень трудно уста¬
новить правильно точку эквивалентности: перманга¬
нат восстанавливается очень медленно, через ряд про¬
межуточных стадий, причем образуются окрашенные
в бурый цвет оксиды марганца различного состава.
Поэтому сразу приливают столько щавелевой кис¬
лоты, чтобы ее хватило на восстановление всего из¬
бытка КМп04 и, кроме того, остался еще некоторый
избыток Н2С204. Наконец, этот избыток оттитровы-
вают раствором перманганата калия обычным спосо¬
бом. Разность между общим количеством введенного
перманганата и количеством щавелевой кислоты соот¬
ветствует количеству определяемого нитрита.Для ускорения реакции окисления нитрита пер¬
манганатом смесь нагревают до 35—40 °С; реакцию
между щавелевой кислотой и перманганатом прово¬
дят как обычно при 70—80 °С.Определение марганца. Метод можно применять
для определения марганца в рудах. Руду растворяют
в концентрированной хлороводородной кислоте
Мп02 + 4НС1 = МпС12 + С12 4- 2Н20нагревая смесь до полного растворения навески и уда¬
ления хлора. При этом может остаться нераствори¬
мый силикатный остаток. Темный цвет остатка ука¬
зывает на наличие в нем некоторого количества мар¬
ганца. В этом случае остаток сплавляют с содой, плав
разлагают хлороводородной кислотой и присоединяют
полученный раствор к основному.Как уже было сказано, соли марганца (II) окис¬
ляются перманганатом в нейтральном или слабоще¬
лочном растворе до Мп02, причем перманганат вос¬
станавливается также до Мп023 Мп2 + + 2НгО = Мп02 + 4Н+ + 2ё~2 Мп04‘+ 4ЬГ + 3<Г == МгЮ2 + 2Н202МпО; + ЗМп2+ + 2Н20 = 5Мп02 + 4Н*или2КМп04 + 3MnS04 + 2Н20 = 5МпОг + K2S04 + 2H2S04.Диоксид марганца при титровании выделяется
в виде бурого осадка. Это вызывает некоторые за¬
труднения в определении конца титрования и требует
особой техники работы.407
Реакция между перманганатом и двухвалентным
марганцем идет уже в слабокислой среде. Из уравне¬
ния реакции видно, что для более полного сдвига рав¬
новесия вправо необходимо понизить концентрацию
водородных ионов. Однако в щелочной среде возни¬
кает побочная реакция, а именно: образуется гидр¬
оксид марганца (II), который легко окисляется кис¬
лородом воздухаМп2+ + 20Н~ + '/а02 = МпО(ОН)2.Таким образом, если к раствору прибавить ще¬
лочь— часть марганца (II) окислится не пермангана¬
том, а кислородом воздуха и результаты титрования
будут неправильными. Наиболее благоприятной сре¬
дой для титрования является раствор с pH = 5 или6. В этих условиях концентрация водородных ионов
понижена вполне достаточно для того, чтобы сдви¬
нуть равновесие реакции вправо, и в то же время зна¬
чение pH еще недостаточно для того, чтобы заметно
шла реакция окисления кислородом воздуха.Метод имеет другую особенность. Образующийся
при реакции осадок диоксида марганца имеет кислот¬
ный характер, поэтому Мп02 способен частично да¬
вать соли с некоторыми основаниями, или, во всяком
случае, заметно адсорбировать катионы различных
оснований. Если в растворе присутствуют главным
образом катионы двухвалентного марганца (в начале
титрования), то они в значительной степени связы¬
ваются диоксидом марганца и вместе с ним переходят
в осадок. Окисление перманганатом увлеченных
в осадок ионов Мп2+ идет очень медленно и конец
титрования получается нечетким. Для того чтобы уда¬
лить из осадка и перевести в раствор ионы марган¬
ца (II), необходимо ввести в раствор катионы других
металлов, которые будут вытеснять ионы марганца:Мп(Мп03) + М2+ ?=± М(Мп03) + Мп2*
осадокИЛИМп(НМпОз)2 + М2+ М(НМп03)2 + Мп2+.Из ряда катионов наилучшие результаты дают
ионы цинка. Поэтому при титровании в растворе
должны быть ионы цинка; в этом случае окраска408
вблизи точки эквивалентности изменяется более чет¬
ко и результаты получаются более правильные.Рассмотренные две особенности метода показы¬
вают, что для титрования марганца перманганатом
необходимо понизить концентрацию водородных ио¬
нов и ввести в раствор, ионы цинка. Удобнее всего
выполнить оба эти требования одновременно; для
этой цели к кислому раствору перед титрованием
прибавляют оксид цинка.Метод часто применяют для определения марган¬
ца в сплавах, рудах и шлаках, где обычно содержит¬
ся довольно много железа. Оксид цинка осаждает ио¬
ны железа(III), причем в растворе появляются ионы
цинка, необходимые для вытеснения адсорбирован¬
ных осадком ионов марганца:2Fe3+ + 3Zn(OH)2 = 2Fe(OH)3 + 3Zn2+.Титрование обычно ведут, не отфильтровывая
осадка гидроксида железа. Если титруют марганец
в отсутствие солей железа, а также в отсутствие сво¬
бодных кислот, то в раствор можно вводить лишь
очень мало оксида цинка (или даже вовсе не вво¬
дить его), так как титрование ведется в очень раз¬
бавленных растворах, но необходимо прибавить рас¬
творимую соль цинка, например ZnS04 или ZnCl2.При титровании выделяется бурый осадок Мп02,
который затрудняет наблюдение розовой окраски из¬
бытка перманганата в точке эквивалентности, кроме
того, осадок склонен переходить в коллоидное со¬
стояние. Поэтому раствор перед титрованием нагре¬
вают (для коагуляции осадка) и сильно разбавляют
горячей водой (чтобы облегчить установление точки
эквивалентности).Определение органических веществ. Некоторые ор¬
ганические вещества можно определять посредством
прямого титрования раствором перманганата калия.
К их числу относится, например, щавелевая кисло¬
та, служащая для установки концентрации раствора
перманганата. Титрование щавелевой кислоты мож¬
но также использовать для косвенного определения
ряда металлов, образующих нерастворимые оксала-
ты: кальция, лантана, тория, бария, стронция, церия,
свинца, серебра. Чаще всего этот прием применяют
для определения кальция.409
Ионы кальция осаждают раствором щавелевой
кислоты или оксалата аммония:С а С12 + (МН4)2С204 = СаС204 + 2NH4C1.Осадок оксалата кальция после фильтрования и
промывания растворяют в горячей разбавленной сер¬
ной кислоте, причем выделяется эквивалентное каль¬
цию количество щавелевой кислоты:СаС204 + H2SO, = CaSO, + H2C204.Образовавшуюся щавелевую кислоту титруют
раствором перманганата:2КМп04 + 5Н2С204 + 3H2S04 = K2S04+ 2MnS04+ 10С02+8Н20.Осадок оксалата кальция нельзя промывать окса-
латом аммония, так как избыток (NH4)2C204, смачи¬
вающий осадок и фильтр, также будет титроваться
перманганатом. Поэтому для получения правильного
результата осадок СаСг04 промывают холодной
водой.Можно применить и другой прием определения.
Осаждают кальций избытком титрованного раствора
щавелевой кислоты, разбавляют смесь до определен¬
ного объема и, не отделяя осадок, отбирают осто¬
рожно некоторую часть прозрачного раствора над
осадком пипеткой и титруют его перманганатом ка¬
лия. Количество кальция вычисляют по разности.Из других органических веществ перманганато¬
метрическое определение возможно для ряда окси-
кислот, например гликолевой, молочной, яблочной,
винной, лимонной; можно также титровать салици¬
ловую кислоту, метанол, фенол, пирокатехин, гидро¬
хинон, пирогаллол, нитрофенолы, пикриновую кисло¬
ту и некоторые другие вещества. Окисление происхо¬
дит до конечного продукта — диоксида углерода. Тем
не менее окисление происходит очень медленно, и по¬
этому применяют следующий прием. К анализируе¬
мому раствору прибавляют избыток раствора щелочи
и избыток раствора перманганата. Ожидают некото¬
рое время пока окисление закончится, затем раствор
подкисляют серной кислотой и определяют избыток
введенного перманганата каким-либо способом. Мо¬
жно, например, прибавить после подкисления избы-410
ток щавелевой кислоты, и ее остаток снова оттитро¬
вать перманганатом.Ниже приведены реакции окисления пермангана¬
том некоторых органических веществ:2MnO; + 5НС.ООН + 6Н+ = 2Мп2* + 5С02 + 8Н20,6МпО; + 5СН3ОН + 18Н+ = бМп2+ + 5СО, + 19Н20,28MnO; + 5С6Н6ОН + 84Н* = 28Мп2* + ЗОСОа + 57HsO.Муравьиную кислоту можно определить в присут¬
ствии других алифатических кислот с низкой молеку¬
лярной массой, например в присутствии уксусной,
пропионовой, масляной, капроновой кислот, которые
окисляются перманганатом очень медленно.В ряде случаев органическое вещество окисляет¬
ся не до конечного продукта — диоксида углерода, а
до промежуточных. Так, определение этанола осно¬
вано на реакции4МпО; + 5С2Н6ОН -f 12Н* = 4Mn2* + 5СН,СООН + 11Н20,т. е. окисление протекает только до уксусной кис¬
лоты.Многие органические вещества окисляются пер¬
манганатом до различных других промежуточных
продуктов, и стехиометрия реакции не может быть
установлена. Это относится, например, к титрованию
некоторых алифатических спиртов — пропанола, изо-
пропанола, бутанола и других, однако некоторые
многоатомные спирты с большой молекулярной мас¬
сой— глицерин, эритрит, маннит — окисляются пол¬
ностью до диоксида углерода.ГЛАВА 21ИОДОМЕТРИЯ§ 1. ОБЩАЯ ХАРАКТЕРИСТИКА МЕТОДАИодометрия — метод титриметрического анализа, ос¬
нованный на определении количества иода, которое
затрачивается на окисление восстановителей или вы¬
деляется при взаимодействии окислителя с раствором411
иодида калия. Основная реакция метода описывается
следующим уравнением:U + 2e~ «=>- 2Г. (21.1)Эта реакция обратима. В зависимости от условий
она может протекать в прямом или обратном направ¬
лениях. Иод представляет собой окислитель средней
силы: стандартный потенциал системы равен 0,54 В.
Поэтому сильные восстановители легко окисляются
свободным иодом. Примером может быть окисление
иодом ионов олова(II):Sn2t + h= Sn^ + 21'. (21.2)Другие неорганические ионы в низших степенях
окисления также можно определять посредством ти¬
трования раствором иода, например мышьяк(Ш), ва-
надий(1У), ртуть(1). Таким способом определяют
кислоты-восстановители, например сероводород, сер¬
нистую кислоту, тиосульфат и др. Иод окисляет
также многие органические вещества: соединения, со¬
держащие альдегидные группы, азот-, и серосодержа¬
щие соединения, оксисоединения и др.Сильные окислители способны, наоборот, выде¬
лять иод из растворов иодидов. По этому типу про¬
исходит взаимодействие между иодидом калия и
бихроматом или перманганатом калия, солями це-
рия(1У), ванадия(У), железа(Ш), меди(П), органи¬
ческими пероксидами и многими другими окислите¬
лями. Так, реакция между иодидом калия и ионами
железа(Ш)2Fe1+ + 2r = 2Fe2+ + I2 (21.3)является основой иодометрического определения же¬
леза. Таким образом, реакцию (21.1) можно исполь¬
зовать для определения и восстановителей, и окисли¬
телей. В первом случае применяют для титрования
рабочий раствор иода, во втором — рабочий раствор
тиосульфата натрия; последний реагирует с иодом в
соответствии с уравнением2Na2S203 +I2 = Na2S,Oe+ 2NaI. (21.4)Из уравнений (21.1) и (21.4) видно, что реакции
в системе происходят без участия ионов водорода.
Независимость двух основных реакций метода от кис¬412
лотности раствора представляет важную особенность
иодометрических определений. Окислительно-восста¬
новительный потенциал системы Ь/21- не зависит от
pH раствора, поэтому иодометрические определения
можно проводить в широком интервале кислотности
раствора (pH = 2—10). Это дает возможность под¬
бирать в каждом отдельном случае самые благопри¬
ятные для определения окислителей или восстанови¬
телей условия кислотности и применять иодометриче¬
ский метод для определения значительно более
широкого круга объектов, чем в других методах тит-
риметрического анализа. Иллюстрацией может быть
определение мышьяка. Реакция окисления мышья¬
ка (III) до мышьяка (V) проходит по уравнению:HAs02 + 2H20 H3AsO« + 2Н++ 2е~. (21.5)Из уравнения видно, что во время реакции выде¬
ляются ионы водорода, следовательно, мышьяк окис¬
ляется легче в щелочном или в слабо щелочном рас¬
творе. Применять для титрования мышьяка такие
окислители, как перманганат или бихромат калия, в
этом случае неудобно. Перманганат восстанавливает¬
ся до марганца (II) легче в сильно кислой среде; в
слабо щелочном растворе окислительный потенциал
перманганата снижается, вследствие чего реакция
между перманганатом и мышьяком происходит очень
медленно и не стехиометрически. Продуктами реак¬
ции являются соединения марганца(II) и марган¬
ца (IV), соотношение между количествами которых
обычно не постоянно и зависит от условий опреде¬
ления. В то же время иодометрическое определение
мышьяка проходит без всяких затруднений. Окисли¬
тельный потенциал системы Ь/21- не изменяется под
влиянием кислотности; pH раствора можно устанав¬
ливать только в зависимости от специфических усло¬
вий протекания реакции (21.5).Эта особенность иодометрического метода обус¬
ловливает его широкое применение при определении
самых разнообразных неорганических и органических
соединений.Другая особенность иодометрии заключается в
высокой точности установления точки эквивалентно¬
сти, что связано с наличием чувствительного специ¬
фического индикатора. В качестве индикатора приме¬413
няют раствор крахмала, который образует с наимень¬
шими количествами иода окрашенное в интенсивно
синий цвет адсорбционное соединение. Реакция отли¬
чается высокой чувствительностью — уже 0,00001 М
растворы иода окрашиваются в присутствии крах¬
мала в синий цвет. Точку эквивалентности можно
также установить по желтой окраске свободного ио¬
да; повысить чувствительность в этом случае можно
путем экстракции иода в слой органического раство¬
рителя (хлороформа, тетрахлорида углерода и др.).
Таким способом можно перевести наименьшие коли¬
чества иода из большого объема водного раствора
в небольшой объем органического растворителя и
значительно усилить интенсивность желтого окраши¬
вания.Существует две основных причины погрешностей
при иодометрических определениях. Первая обуслов¬
лена летучестью иода и его растворов. Поэтому все
работы, связанные с выделением иода, целесообразно
проводить в закрытых склянках или колбах. Вслед¬
ствие летучести иода и его малой растворимости в
воде для титрования применяют растворы иода в
концентрированном растворе иодида калия. При оп¬
ределении окислителей создают в растворе большой
избыток иодида калия, чтобы перевести выделяю¬
щийся при реакции твердый иод в раствор и умень¬
шить его летучесть. Вторая причина погрешностей —
окисление растворов иодида калия кислородом воз¬
духа. Реакция проходит по уравнению4Г + 4Н+ + 02 = 21* + 2Н20. (21.6)Скорость этой реакции зависит от ряда факторов.
В нейтральных растворах реакция происходит очень
медленно, «о высокая концентрация водородных
ионов способствует процессу окисления. Поэтому не
следует оставлять на долгое время на воздухе под¬
кисленные растворы иодида калия, которые предпола¬
гается использовать для иодометрических определе¬
ний. Скорость окисления возрастает также под влия¬
нием прямых солнечных лучей. Некоторые вещества,
например соли меди, оксиды азота, каталитически
ускоряют реакцию между иодидом калия и кис¬
лородом.414
§ 2. РАБОЧИЕ РАСТВОРЫ ИОДОМЕТРИИРаствор иода. Иод обычно содержит небольшое ко¬
личество воды и примеси хлора или брома. Для
очистки иода от примесей его растирают в ступке с
небольшим количеством иодида калия и оксида каль¬
ция. При этом примеси хлора или брома реагируют
с иодидом калия:Вг, + 2К1 = 2КВг + и. (21.7)Следовательно, вместо свободного брома (или
хлора) образуются нелетучие бромид и хлорид ка¬
лия. Вода реагирует с оксидом кальция:Н20 + СаО = Са(ОН)2. (21.8)Растертый в мелкий порошок иод переносят в
стакан и возгоняют, собирая возгон чистого иода на
дно кристаллизатора с холодной водой, который ста¬
вят на стакан. Очищенный таким способом иод со¬
храняют в эксикаторе в стакане с пришлифованной
крышкой.Из уравненияЬ + 2е' = 2Г (21.9)видно, что фактор эквивалентности /Эка равен 1/2.Иод плохо растворяется в воде, но его раствори¬
мость значительно возрастает в присутствии иодида
калия, что обусловлено образованием полииодида:12 + Г *=± I-. (21.10)Для растворения иода целесообразно брать 3—
4-кратный против эквивалентного количества избы¬
ток иодида калия. Такой избыток необходим, чтобы
увеличить растворимость иода; кроме того, летучесть
иода в растворах с высокой концентрацией иодида
калия значительно меньше по сравнению с лету¬
честью твердого иода, в результате чего практически
устраняется одна из основных причин погрешностей
при иодометрических определениях.Титрованные растворы иода необходимо хранить
в склянках из темного стекла, чтобы избежать окис¬
ления иодида калия кислородом воздуха с выделе¬
нием свободного иода.416
При иодометрических определениях обычнополь-
зуются 0,025 М растворами иода. Это обусловлено
тем, что, как уже было сказано, точность установле¬
ния точки эквивалентности посредством крахмала
достаточно высока; с другой стороны, иод является
дорогим препаратом, и поэтому нецелесообразно
применять растворы более высокой концент¬
рации.Раствор иода точной концентрации готовят сле¬
дующим образом: в стакан с пришлифованной крыш¬
кой помещают трехкратный по отношению к иоду из¬
быток иодида калия и растворяют его в воде. После
того как раствор примет комнатную температуру
(при растворении иодида калия в воде раствор охла¬
ждается) стакан взвешивают на аналитических ве¬
сах. Затем навеску иода переносят в стакан с раство¬
ром иодида калия, в котором иод быстро растворяет¬
ся, и снова взвешивают стакан с раствором. Точное
количество иода находят по разности двух взвешива¬
ний. Полученный раствор переносят количественно в
мерную колбу и разбавляют водой до метки.Раствор иода можно готовить и из неочищенного
иода, однако в этом случае нормальность раствора
необходимо устанавливать титрованием раствором
тиосульфата.Крахмал. Крахмал образует с раствором иоДа
адсорбционное соединение интенсивно синего цвета.
Чувствительность иод-крахмальной реакции зависит
от многих факторов: присутствия ионов иода, способа
приготовления и хранения раствора крахмала, тем¬
пературы и др. Молекулярный иод не дает с крахма¬
лом окрашенного соединения; для этого необходи¬
мо, чтобы в растворе были ионы иода. Поэтому все
иодометрические определения проводят в растворах,
содержащих избыток иодида калия. Чувствитель¬
ность иод-крахмальной реакции уменьшается с повы¬
шением температуры, поэтому лучше титровать холод¬
ные растворы. Чувствительность зависит также от
способа приготовления индикатора: правильно при¬
готовленные растворы крахмала окрашиваются в чи¬
сто синий цвет при добавлении одной капли 0,01 М
раствора иода. Неправильно приготовленные раство¬
ры крахмала или такие, которые сохранялись про¬
должительное время, нередко образуют с иодом со¬416
единение фиолетового цвета. Чувствительность реак¬
ции в этом случае значительно снижается.При иодометрическом определении окислителей
может возникнуть значительная погрешность вслед¬
ствие окисления крахмала иодом. Эта реакция идет
довольно медленно, однако при продолжительном
действии иода на крахмал часть иода расходуется
на эту побочную реакцию и результаты будут непра¬
вильными. Кроме того, в кислом растворе крахмал
постепенно гидролизуется, что также приводит к по¬
грешности. Поэтому иод титруют тиосульфатом, не
прибавляя в начале титрования крахмал; индикатор
прибавляют только в конце титрования, чтобы по
возможности уменьшить время контакта крахмала
с иодом.В качестве индикатора используют обычно
0,2 %-ный раствор крахмала: 1 г растворимого крах¬
мала взбалтывают с небольшим количеством воды и
полученную суспензию выливают в кипящую воду
(500 мл). Прозрачный охлажденный раствор приме¬
няют в качестве индикатора. Чтобы защитить раст¬
вор от проникновения микроорганизмов, которые де¬
лают крахмал непригодным для употребления, мож¬
но стабилизировать его, прибавив немного иодида
ртути.Тиосульфат натрия. Тиосульфат натрия после
окисления иодом превращается в тетратионат:2Na2S2On + [2 = Na2S406 + 2NaI. (21.11)Из уравнения видно, что эквивалент тиосульфата
натрия численно равен его молекулярной массе (фак¬
тор эквивалентности /эка = 1).Химическая формула тиосульфата натрия
Na2S203-5H20, однако кристаллизационная вода по¬
степенно выветривается и через некоторое время дей¬
ствительный состав соли перестает соответствовать ее
формуле. Концентрация растворов тиосульфата изме¬
няется при хранении под влиянием диоксида угле-:
рода воздуха:NajSjCb + СО,> + Н20 = NaHSOj + S + NaHCOj. (21.12)Чтобы предотвратить эту реакцию, тиосульфат
следует растворять в воде, из которой диоксид угле¬
рода устранен кипячением, а также защищать раст^
воры тиосульфата от проникновения СОг хлоркаль-(4 Аналитическая химий417
циевой трубкой, наполненной натронной известью.
Можно также ввести в раствор тиосульфата неболь¬
шое количество карбоната натрия. В этом случае СО2
реагирует в первую очередь с карбонатом натрия:Na2C03 + С02 + Н20 = 2NaHC03. (21.13)Другой характерной особенностью растворов тио¬
сульфата является способность окисляться кислоро¬
дом воздуха:2Na2S203 + 02 = S2 + 2Na2SO,. (21.14)Нестабильность растворов тиосульфата объясняет¬
ся, наконец, влиянием микроорганизмов, так назы¬
ваемых тиобактерий, которые попадают из атмо¬
сферы в раствор и поглощают из него серу, превра¬
щая ее в сульфат. Разложение раствора тиосульфата
тиобактериями сопровождается интенсивным выделе¬
нием серы и помутнением.Указанные обстоятельства делают нецелесообраз¬
ным приготовление точных растворов тиосульфата
непосредственно из навески; обычно готовят раствор
приблизительной концентрации, а точную нормаль
ность устанавливают по другим исходным веществам
Чаще всего применяют раствор бихромата калия
Препарат КгСг207 легко получить в чистом виде пе
рекристаллизацией из водного раствора и высушива
нием при 150—180°С; соль не содержит кристалли
зационной воды и не изменяет состав при хранении.
Водные растворы бихромата отличаются высокой ус¬
тойчивостью, их концентрация долгое время остается
неизменной.Навеску тиосульфата растворяют в дистиллиро¬
ванной воде, не содержащей С02, и выдерживают не¬
сколько дней в темной склянке. Для установки кон¬
центрации применяют раствор иода известной кон¬
центрации или раствор бихромата калия.Прямое титрование раствора тиосульфата натрия
раствором бихромата не рекомендуется из-за несте-
хиометричности реакции. Определение основано на
реакциях:С202,- + 14Н+ + 6е- = 2Сг3+ + 7НгО(21.15)2Г = 2е~ + 12418Сг20;- -1- 14Н+ + 6Г = 2Сг3+ + 3I2 + 7HsO
или в молекулярнбй формеKjCrjOj -f- 6KI + 7H2SO4 = Cr2(SO«)j + 31* + 4K2SO4. (21.16)Выделившийся иод титруют раствором тиосуль¬
фата натрия.В раствор вводят столько йодида калия, чтобы
его хватило для полного восстановления бихромата
калия и для растворения иода, выделяющегося в ре¬
зультате реакции. Обычно берут 3—4-кратный избы¬
ток KI по сравнению с эквивалентным количеством.Для проведения точного определения существен¬
ное значение имеет установление оптимальной кис¬
лотности раствора. Из уравнения (21.15) видно, что
восстановление бихромата происходит с участием
ионов водорода, причем увеличение концентрации по¬
следних повышает окислительный потенциал бихро¬
мата и облегчает восстановление хрома(VI) до хро-
ма(Ш). С другой стороны, с увеличением кислотно¬
сти, как уже было сказано, возрастает скорость
реакции окисления иодида калия кислородом воздуха.
Экспериментально установлено, что оптимальная
кислотность, при которой основная реакция между
К2СГ2О7 и KI проходит с достаточной скоростью, а
побочная реакция окисления иодида калия кислоро¬
дом воздуха за время определения почти не происхо¬
дит, соответствует прибавлению 15—20 мл раствора
серной кислоты (1:4). Необходимо при этом пом¬
нить, что концентрированная серная кислота легко
окисляет иодид калия:2H2S04 + 2KI = Is + K2SO4 + 2Н20 + S02. (21.17)Поэтому следует пользоваться только разбавленным
раствором серной кислоты.Препарат KI нередко содержит небольшую при¬
месь иодата калия КЮ3. Поэтому при подкислении
раствора иодида калия серной кислотой может выде¬
литься небольшое количество свободного иода:5K.I + KIOj + 3H2SO* = 3I2 + 3K2SO4 + 3H20. (21.18)Чтобы избежать связанной с этим погрешности опре¬
деления, к раствору бихромата калия не следует до¬
бавлять нейтральный раствор иодида калия; послед¬
ний сначала подкисляют серной кислотой и выделив¬
шийся иод восстанавливают, прибавляя по каплям14*419
0,01 М раствор тиосульфата натрия. Обесцвеченный
раствор иодида калия прибавляют затем к раствору
бихромата калия. (При титровании выделившегося иода раствором
тиосульфата натрия возможна побочная реакция раз¬
ложения тиосульфата:Na2S203 + H2S04 = Н20 + S02 + S + Na2S04, (21,1.9)Эта реакция протекает медленно, в то время «а«;
скорость реакции между иодом и тиосульфатом ве¬
лика. Однако выделение серы и диоксида серы все
же возможно, если раствор тиосульфата натрия при¬
бавляют чрезмерно большими порциями и плохо пе¬
ремешивают раствор.Ионы хрома, образующиеся при восстановлении
бихромата калия, окрашены в зеленый цвет. В точке
эквивалентности происходит переход окраски от си¬
ней (иод-крахмальная реакция) до зеленой. Этот пе?
реход в концентрированных растворах солей xpoMai
воспринимается плохо. Чтобы облегчить наблюдение
точки эквивалентности, целесообразно разбавить рас¬
твор перед титрованием дистиллированной водой.Крахмал необходимо вводить в раствор только
после того, как большая часть иода будет оттитро¬
вана и раствор окрасится в светло-желто-зеленую
окраску, иначе часть иода расходуется на окисление
крахмала.§ 3. ПРИМЕРЫ ИОДОМЕТРИЧЕСКИХ ОПРЕДЕЛЕНИЙОпределение меди. Иодометрический метод определе¬
ния меди основан на окислительном действии ионов
меди(II) по отношению к иодид-ионам. При взаимо¬
действии солей меди с иодидом калия медь(II) вос¬
станавливается до меди(1) с образованием нераство¬
римого осадка Cul и выделением свободного иода,
который титруют тиосульфатом:Си2* + 1" + е~ = Cul2Г = 2<Г +15(21.20)2Си2* + 41" = 2Си1 + 12
или в молекулярной форме2CuSO« + 4KI = 2CuI 2K2SO4 н 2.420(2121)
Из уравнения видно, что эквивалент меди в этой
реакции численно равен атомной массе.Йодометрический метод определения меди очень
точен. Этот метод применяют для определения меди
в цветных сплавах и в рудах. Преобладающее боль¬
шинство посторонних ионов, которые обычно содер¬
жатся в перечисленных материалах, не мешают оп¬
ределению меди, исключение составляют ионы, спо¬
собные окисляться иодом или окислять иодид-ионы.
Из окислителей чаще всего приходится иметь дело
с Солями железа(П1), оксидами азота и нитрит-иона¬
ми; Эти вещества способны окислять иодиды в соот¬
ветствий с уравнениями:2Fe3‘ + 2Г = Ij + 2Fe2*, (21.22)1 NOj+21' + 2Н* =1г+ N0 + Н£>. (2123): Влияние небольших количеств железа можно уст¬
ранить, связывая ионы Fe3* в комплексные иоНы.
Длй такого маскирования Применяют фториды или
пирофосфаты, напримерFo1+ + 6F_ = FeFj". (21.24)Вследствие прибавления фторид-ионов концентра¬
ция свободных ионов железа (III) в растворе резко
уменьшается и окислительный потенциал системы
F<i34 /1'е2+* понижается до значения, при котором окис¬
ление иодид-ионов становится невозможным. При
анализе материалов, содержащих слишком много же¬
леза, такой способ непригоден; железо необходимо
отделить от меди осаждением раствором гидроксида
аммония или каким-либо другим способом.Оксиды азота и азотистая кислота всегда обра¬
зуются при переведении пробы в раствор посред¬
ством обработки азотной кислотой. Даже самые не¬
значительные количества NO2 в растворе могут вы¬
звать интенсивное выделение иода. Это объясняется
тем, что при взаимодействии NO2 с I- обр!азуется
оксид азота NO, который легко окисляется кислоро¬
дом воздуха до диоксида:2N0 + 02 = 2N02.Последний снова реагирует с иодидом. Такой кру-
говрй, процесс происходит беспрерывно и может стать421
источником большой погрешности. Оксиды аэота мо¬
жно выделить иэ раствора кипячением; еще л у «иве
после растворения пробы полностью устранить азот¬
ную кислоту выпариванием раствора с серной кисло¬
той до появления тяжелых белых паров HjSO*.В очень кислых растворах мышьяк (V) н сурь¬
ма (V) также способны окислять иодиды. Выделе¬
ние иода в этом случае можно устранить, уменьшив
концентрацию водородных ионов раствора введением
буферной смеси.Необходимо отметить одну интересную особен¬
ность реакции между иодид-ионами и медью (II).
Стандартный потенциал системы Си^/Си^ равен0,17 В, а системы Ij/2I~ — 0,54 В. Сравнивая стан¬
дартные потенциалы, приходим к выводу, что иод
является более сильным окислителем, чем медь(II);
следовательно, реакция (21.20) должна проходить
справа налево, а не наоборот, как наблюдается в
действительности. Направление реакции обусловлено
в данном случае образованием малорастворимого
осадка иодида меди(1); концентрация ионов Си+ ста¬
новится вследствие этого очень незначительной, что
в свою очередь приводит к повышению окислитель¬
ного потенциала системы Cu2+/Cu+.Предположим, что в 0,1 М раствор соли меди(Н)
ввели для иодометрического определения 0,1 М рас¬
твор иодида калия. Из этих данных можно вычис¬
лить концентрацию ионов меди(1) в растворе в мо¬
мент образования осадка. Произведение растворимо¬
сти Cul равно 10-12:[Си+] [Г] = 1(Г12,откудаСледовательно, окислительный потенциал системы
Cu2+/Cu+ в начале титрования, в соответствии с ура¬
внением Нернста, равен:■^1 = 0,17 + 0,058 lg = -^ ‘[Си ] 10Ех = Е0 + 0,058 Ig= 0,17 + 0,058 lg = —^ = 0,75 В.Рассмотренный случай является одним из приме¬
ров изменения окислительного потенциала системы422
под влиянием образования в процессе реакции мало¬
растворимой соли. Приведенный расчет показывает,
что в результате этого окислительный потенциал си¬
стемы Cu*+/Cu+ увеличивается до 0,75 В, следова¬
тельно, ионы меди (II) проявляют относительно ио-
дид-нонов окислительные свойства и реакция(21.20) проходит слева направо.Точность иодометрического определения меди за¬
висит от pH раствора. Очень большая кислотность
недопустима, так как в этих условиях увеличивается
скорость реакции окисления иодид-ионов кислородом
воздуха. Наоборот, при слишком малой кислотности
замедляется основная реакция между медью(Н) и
иодидом. Экспериментально установлено, что опти¬
мальная кислотность находится в границах pH от 4£
до 5,5.Крахмал при титровании необходимо прибавлять
только после того, как основная часть иода будет от¬
титрована тиосульфатом и смесь окрасится в светло-
желтый цвет.Определение активного хлора в хлорной извести.
Хлорная известь представляет собой двойную каль¬
циевую соль хлороводородной и хлорноватистой кис¬
лот. Строение молекулы этой соли можно выразить
такой структурной формулой:Технический препарат содержит, кроме того, зна¬
чительную часть оксида кальция, хлорида кальция
и некоторых других веществ. Хлорную известь при¬
меняют для дезинфекции, отбеливания тканей, дега¬
зации отравляющих веществ и др. Качество препа¬
рата оценивается содержанием «активного хлора»,
т. е. хлора, который выделяется при взаимодействии
хлорной извести с хлороводородной кислотой:Иодометрическое определение «активного хлора»
в хлорной извести основано на следующих реакциях:чОС1CaCljO + 2НС1 = СаС12 + С12 + НаО. (21.25)CaCljO + 2HI = СаСЬ + 12 + НгО,
I2 2NaaS*Oj = Na^S^O^ -f- 2N>aL(21.26)(21.27)423
Навеску препарата взбалтывают с водой и сме¬
шивают эту суспензию с подкисленным раствором ио¬
дида калия, выделившийся иод титруют раствором
тиосульфата натрия. Из приведенных уравнений вид¬
но, что количество выделившегося иода эквивалентно
количес?ву хлора, образующегося при взаимодейст¬
вии хлорной извести с хлороводородной кислотой.
Поэтому по количеству выделившегося иода можно
вычислить процентное содержание активного хлора
в препарате.Определение кислот. Нейтральные растворы иодн-
да и иодата калия не реагируют между собой. Од¬
нако если к смеси растворов этих солей прилить
сильную кислоту, то Юз окисляет 1~ с выделением
свободного иода:Ю- + 5Г + 6Н+ = 312 + знгоилиКЮ3 + 5KI Ч-6НС1 = 6КС1 +312 + ЗН20. :Количество выделившегося иода эквивалентно ко¬
личеству прибавленной кислоты, на каждый эквива¬
лент кислоты приходится один эквивалент иода. Иод
титруют затем 0,1 М раствором тиосульфата нат¬
рия; по объему затраченного на титрование рабочего
раствора вычисляют количестве кислоты.Связью между окислительными потенциалами реа¬
гирующих, веществ и pH среды можно установить
следующим образом.Электронно-ионные уравнения реакций имеют вид:ю; + 6Н‘ + 5е' = 1° + 3H20, £q = 1,2 В;I- = 1° -4- е~, £" =0,53 В.Окислительный потенциал системы иодат-^иод
сильно зависит от pH раствора. Эта зависимость вы¬
ражается уравнением, , fiOi] [н-Г
^ = 4»0.0121rJ jbj— -Для упрощения расчета примем, что концентра¬
ции Юз, I и свободного иода приблизительно посто¬
янны и одинаковы (например, 0,1 М), тогда как кон¬
центрация водородных ионов изменяется в широких
пределах. Очевидно, что [Н+] -=^1 М (и при [Юз}-*-424
— [Г]) второй член уравнения становится р а видом
нулю и, таким образом, Е'х = Е'й= \,2 В: Следова¬
тельно, при 1 М концентрации кислоты окислитель¬
ный Потенциал системы иодат — иод (£' — 1,2 В) бу¬
дет значительно выше, чем потенциал системы иод—
иодид (0,53 В), и иодат будет выделять иод из ио¬
дида калия. По мере расхода кислоты концентрация
водородных ионов уменьшается, поэтому будет умень¬
шаться и потенциал иодата. Когда этот потенциал
(Ех) станет равным потенциалу системы иод — иодид
(0,53 Э), иод перестанет выделяться. Можно вычис¬
лить концентрацию водородных ионов, при которой
это произойдет:, [10:1 [Н*Г , , ,1в
е'х = 0,53 = 1,2 + 0,012 lg | jj-j = 1,2 + 0,012 tg [НЧ® == 1,2 -f- 0,012 ■ 6 lg [H+J.Отсюдаg 1 1 0,012-6. Таким образом, иодат перестанет выделять иод из
иодида калия при pH = —ig[H+] =9.Иодометрический метод пригоден для количест¬
венного определения сильных кислот — серной, хло¬
роводородной и азотной. В растворах слабых кислот
концентрация водородных ионов мала и реакция идет
медленно....... При практическом проведении определения необ¬
ходимо иметь в виду, что раствор должен содержать
избыток иодата и иодида калия по сравнению с пред¬
полагаемым количеством сильной кислоты. Избыток
иодида калия должен быть большим, чем избыток
КЮ3, так как некоторое количество KI необходимо
■ для растворения выделяющегося иода; обычно.доста¬
точно взять трехкратный избыток.В приведенных выше примерах иодометрический
метод использовали для определения окислителей.
Существует много методов, основанных на титрова¬
нии восстановителей раствором иода. Ниже приве¬
дены некоторые примеры.;■! Определение мыигеяка. Иодометри#ее*и*» метод
может быть применен для определения как. трехва-
лентного, так и пятивалентного мышьяка. Определе¬425
ние пятивалентного мышьяка основано на том, что в
сильно кислом растворе мышьяковая кислота восста¬
навливается иодидом калия с выделением эквива¬
лентного количества свободного иода, который затем
титруют раствором тиосульфата натрия. Чаще прихо¬
дится определять трехвалентный мышьяк. В слабо
щелочном растворе мышьяковистая кислота легко
окисляется свободным иодом до мышьяковой
кислоты.Подробно рассмотрим иодометрический метод оп¬
ределения арсенитов.Реакция между арсенитом и иодом идет по урав¬
нениюNaH2AsO., + I2 + Н20 = NaH2AsO< + 2HI. (21.28)В сильно щелочном растворе окисление проходит еще
легче, однако иод взаимодействует со щелочью:I2 + 2NaOH = Nal + NalO + Н20.После окисления мышьяковистой кислоты крах¬
мал не будет вызывать окраски, так как иод вступает
в реакцию с гидроксидом натрия. Поэтому для ней¬
трализации выделяющейся при реакции кислоты
(HI) удобнее применять гидрокарбонат натрия:NaHCOj + HI == Nal + H2COs.Для раствора гидрокарбоната натриярН=^1-+£^=6-5+10.-2=8,35.При взаимодействии HI с NaHC03 образуется буфер¬
ная смесь (Н2СОз + NaHC03), pH которой равен
приблизительно 7.Легко показать, что при pH = 7 реакция (21.28)
практически полностью идет слева направо. В самом
деле, стандартные потенциалы обеих систем равны:H2AsO; + 2Н+ + 2е~ = AsO' + 2НаО, Е'0 = 0,56 В;12 + 2е" = 21', £" = 0,54 В.В § 2 гл. 19 было показано, что логарифм константы
равновесия реакции окисления — восстановления ра¬
вен■ v (Еп — п
Ок о^58 •426
Для реакцииHAsOj + 2Н20 + U = H3AsO« + 2Н* + 2 Г (21.29)можно записать:и[HaAsCM [Hla [Г]а2,1 • 10“'.IHAs021 [12]Если концентрация водородных ионов равна 10-7,
тогдаИз этого выражения видно, что при pH = 7 со¬
стояние равновесия реакции (21.29) практически пол¬
ностью сдвинуто в правую сторону — иод количест¬
венно окисляет мышьяковистую кислоту до мышья¬
ковой.Определение органических веществ. В § 1 указы¬
валось, что многие органические вещества могут
быть также определены иодометрическим методом.
В преобладающем большинстве случаев речь идет
об использовании восстановительных свойств органи¬
ческих соединений, т. е. об их титровании растворами
иода. Некоторые органические соединения проявляют
окислительные свойства и могут быть определены на
основании их взаимодействия с раствором иодида ка¬
лия, сопровождающегося выделением свободного ио¬
да. К их числу относятся, например, органические
пероксиды.Ниже рассмотрены некоторые примеры титрова¬
ния органических восстановителей.Отличительной особенностью этих определений яв¬
ляется то, что раствор иода либо не реагирует с ор¬
ганическими восстановителями в кислых растворах,
либо реакции идут крайне медленно. Обычный при¬
ем заключается в том, что раствор иода прибавляют
к щелочному раствору органического соединения, вы¬
жидают некоторое время, затем подкисляют раствор
и титруют избыток не вошедшего в реакцию иода
раствором тиосульфата натрия. В щелочном растворе[H3AsOt] [Г]2IHAs02] [12]427
иод реагирует со щелочью с образованием гипоио-
дита:2NaOH + Ь = NalO + Nal + Н20.Поэтому окислителем в щелочном растворе высту¬
пает гипоиодит натрия. Однако стехиометрия реак¬
ции от этого не изменяется, так как после подкисле-
ния избыток гипоиодита восстанавливается иодидом
натрия и выделяется свободный иод.: Иодометрический метод применим для определе¬
ния соединений, содержащих альдегидные или кар¬
бонильные группы. Так, можно определять форм¬
альдегид:НСНО + Ь + 3NaOH = HCOONa + 2Nal + 2Н20;
глюкозу RCHO, где R—С5Нц05:RCHO + I2 + 3NaOH = RCOONa + 2NaI + 2Н20,причем образуются соли соответствующих карбоно¬
вых кислот: муравьиной — в первом случае и глюко-
новой—во втором.При окислении ацетона образуется йодоформ ,и
ацетат натрия:CHjCOCHj + 3I2 + 4NaOH = CHI3 + CH3COONa + 3NaT + 3H20'Рйй серосодержащих соединений также количествен¬
но окисляется иодом. Так, тиомочевина реагирует с
иодом в соответствии с уравнениемCS(MI2)2 + 4I2 + lONaOH — CO(NH2b + 8NaI + Na2S04 + 5H20.Некоторые оксисоединения реагируют с иодом,
причем последний замещ!ает водородные атомы в*ра^
дикале органического соединения. В качестве приме¬
ра приводим иодометрический метод определения ре¬
зорцина; он основан на следующей реакции:СбН«(ОН)2 + 31, = С6Н13(ОН)г + 3HI.К раствору резорцина прибавляют буферный рас-
твбр с pH = 5, затем раствор йода и избыток послед¬
него титруют раствором тиосульфата в присутствии
крахмала.428
ГЛАВА 22 tДРУГИЕ МЕТОДЫ ОКИСЛЕНИЯ —
ВОССТАНОВЛЕНИЯ. МЕТОДЫ
ПРЕДВАРИТЕЛЬНОГО ВОССТАНОВЛЕНИЯ§ I. БРОМАТОМЕТРИЯБромат калия в кислом растворе проявляет доста¬
точно сильные окислительные свойства. Электронно¬
ионную реакцию восстановления бромат-ионов можно
представить следующим уравнением:BrO" + 6Н* + 6е~ = Вг' + ЗН20, £0 = 1,46 В. (22.1)Однако в кислом растворе равновесие между
ионами бромата и бромида невозможно, так как они
взаимодействуют между собой с образованием сво¬
бодного брома:BrOj + 6Н+ + 5Вг~ = ЗВг2 4- ЗН20. (22.2)Таким образом, окислителем по существу является
свободный бром, потенциал которого значительно
ниже, чем потенциал бромата:2Вг' = 2е~ + Вг2, £„=1,07 В. (22.3)Эквивалент бромата калия, как это видно из урав¬
нений (22.1) или (22.2), равен одной шестой части
молекулярной массы.Бромат калия легко очистить от примесей перекри¬
сталлизацией из водного раствора. Перекристаллизо-
ванную соль высушивают в сушильном шкафу при
ЮО—150°С и сохраняют в закрытой склянке. Рабочий
раствор КВг03 можно готовить непосредственно из
навески высушенной соли. Иногда к рабочему рас¬
твору прибавляют при его приготовлении избыток
бромида калия; в этом случае при титровании таким
бромид-броматным раствором в кислой среде сразу
выделяется свободный бром [см. уравнение (22.2)],
который реагирует с находящимся в растворе восста¬
новителем.Рабочий раствор бромата калия обычно приме¬
няют в следующих случаях.1. Для прямого титрования неорганических ионов,
находящихся в низшей степени окисления. Известны429
методы определения трехвалентного мышьяка, перо¬
ксида водорода, олова (II), меди(1), таллия (I), же-
леза(П), урана(1У), ванадия(П) и др. Наиболее рас¬
пространен броматометрический метод титрования
сурьмы(Ш):КВгОз + 3SbCla + 6HCI = КВг + 3SbCl6 + ЗН20. (22.4)Титрование обычно проводят в растворе хлорово¬
дородной кислоты.2, Для прямого определения некоторых органиче¬
ских веществ. Примером может служить титрование
аскорбиновой кислоты (витамина С) в различных ме¬
дицинских препаратах.о=С С—ОН
i-OH °3 т [ I + Вго; + 5Вг~ + 6Н+ =НС —Iно—с—н
£н2он
о=с—Iс=оI Ос=о I= 3 I + 6НВг + ЗН20 (22.5)НС IНО—с—нIСН2ОНИзвестны и другие примеры. Так, тиомочевина ко¬
личественно окисляется броматом по реакции:3CS(NH2)2 + 4НВЮз + ЗН20 = 3CO(NH2)2 + 3H2SO« + 4HBr.(22.6)Продуктом реакции является мочевина.3. Для определения некоторых ароматических ор¬
ганических соединений и косвенного определения мно¬
гих неорганических ионов по реакциям замещения.Фенолы и его производные титруются раствором
бромид-бромата с образованием соответствующих430
бромзамещенных соединений. В случае фенола про¬
дуктом реакции является трйбромфенол:С6Н3ОН 4- ВгО- + 5Br~ 4- 6Н* = СвН2Вг3ОН 4- ЗНВг 4- ЗН,0.(22.7)Здесь происходит реакция бромирования фенолаАналогичные реакции бромирования лежат в осно¬
ве определения многих других органических соедине¬
ний ароматического ряда, например салициловой кис¬
лоты, крезолов, динитрофенолов, резорцина, 0-наф¬
тола, анилина, антипирина и др.Для косвенного определения металлов большое
значение имеет реакция бромата калия с 8-гидрокси-
хинолином. В кислой среде 8-гидроксихинолин реаги¬
рует с бромид-броматной смесью по уравнению:(22.8)ОН8-Гидроксихинолин образует с многими металлами
(алюминием, железом, титаном, медью, кадмием, цин¬
ком, кобальтом, никелем, магнием и др.) нераствори¬
мые осадки гидроксихинолинатов. Селективность
осаждения достигается регулированием pH раствора.
Так, полное осаждение гидроксихинолинатов алюми¬
ния, железа, титана, меди, магния, свинца достигается
при pH 4,4; 2,8; 4,8; 3,3; 8,2 и 8,4 соответственно. По¬
этому можно, например, осаждать алюминий в при¬
сутствии магния, железо — в присутствии свинца
и т. д. Другой путь повышения селективности — вве¬
дение маскирующих веществ. Описанные приемы по¬
зволяют определять один металл в присутствии ряда
других.Осадок гидроксихинолината металла отфильтро¬
вывают, промывают водой или другой промывной431
жидкостью и растворяют в хлороводородной кислоте.
Протекающие реакции описываются уравнениями:
осаждениеМ" + + «НОх = МОх„ + лН‘, (22.9)растворениеМОх„ + иН* = Мп + + пНОк, (22.10)где НОх —молекула гидроксихинолина.Полученный раствор гидроксихинолина, количе¬
ство которого эквивалентно количеству взятого ме¬
талла, титруют бромид-броматным раствором в соот¬
ветствии с уравнением (22.8). Видно, что, например,
с каждым ионом трехзарядного металла связано три
молекулы 8-гидроксихинолина, которые в свою оче¬
редь реагируют с четырьмя атомами брома. Поэтому
эквивалент определяемого металла равен одной две¬
надцатой части его ионной массы.Точку эквивалентности при броматометрическом
титровании устанавливают различными методами.
При определении сурьмы (также мышьяка и др.) не¬
редко применяют необратимые индикаторы, чаще
всего метиловый оранжевый; после введения неболь¬
шого избытка бромата выделяется свободный бром,
который окисляет индикатор, что сопровождается ис¬
чезновением красного окрашивания. Можно также
прибавить в конце титрования немного иодида калия
и раствор крахмала. Свободный бром реагирует с KI:Вг2 + 2К1 = Г2 + 2КВг (22.11)с выделением свободного иода, который образует
с крахмалом соединение интенсивно-синего цвета.
Этот метод применяют главным образом при титройа-
нии органических веществ. Иногда в титруемый рас¬
твор вводят некоторый избыток бромата калия и за¬
тем оттитровывают выделившийся иод раствором тио¬
сульфата натрия:I2 + 2Na2S2Oj = 2NaI + Na2S4Oe. (22.12)В точке эквивалентности синяя окраска исчезает.432
$ 2..ХР0МАТ0МЕТРИЯХроматометрия —метод титрования раствором бихро- ’
мата калия. Электронно-ионная реакция восстановле¬
ния К2СГ2О7 протекает в соответствии со следующим
уравнением:Сг2027' + 14Hf + 6е~ = 2Сг3* + 7НаО. (22.13)Стандартный потенциал этой системы £0=1,36В.Из уравнения видно, что эквивалент бихромата
калия равен 1/6 молекулярной массы (фактор экви¬
валентности равен 1/6).Стандартный потенциал бихромата калия ниже,
чем у перманганата, но все же достаточно высок. Это
позволяет титровать почти все те вещества, которые
определяют методом перманганатометрии. Однако
преимуществом К2СГ2О7 является то, что при титро¬
вании в растворе НС1 не наблюдается окисления хло¬
рид-ионов до свободного хлора, как в перманганато¬
метрии. Другое преимущество состоит в устойчивости
растворов бихромата — концентрация рабочего рас¬
твора при длительном хранении в закрытых сосудах
остается практически неизменной. Бихромат калия
легко очистить перекристаллизацией; полученный пре¬
парат высушивают при 130—150 °С. Рабочий раствор
нужной концентрации можно приготовить непосред¬
ственно из навески высушенного препарата.Бихромат калия восстанавливается до хрома(III);
ионы последнего окрашены в зеленый цвет. Поэтому
для установления конечной точки титрования необхо¬
димо применять специальные индикаторы.Самое большое распространение получил метод
хроматометрического определения железа(II)Сг20?- + 6Fe2+ + 14Н+ = 2Сг34 + 6Fe’* + 7Н20. (22.14)Титровать можно в растворах хлороводородной
кислоты, причем никаких побочных реакций не про¬
исходит.Некоторым недостатком по сравнению с пермац-
ганатометрическим определением является необходи¬
мость использования специальных индикаторов.
Обычно применяют обратимые окислительно-восста¬
новительные индикаторы — дифениламин, фенилан-
траниловую кислоту и др.433
Потенциал точки эквивалентности при титровании
железа равенЕт. экв = = 1,28 В.Из табл. 19.2 в гл. 19 видно, что лучше титровать
с N-фенилантраниловой кислотой, стандартный потен¬
циал которой (£о=1.08В) не очень сильно отли¬
чается от потенциала точки эквивалентности и, как
показано в § 4 гл. 19, находится в допустимых пре¬
делах. Однако можно взять и дифениламин с Ео =
= 0,76 В. В этом случае необходимо снизить потен¬
циал точки эквивалентности. Этого можно достичь
связыванием ионов Fe3+ в какое-либо комплексное
соединение — тогда концентрация свободных ионов
Fe34- становится очень небольшой и потенциал пары
Fe3+/Fe2+ значительно снижается. Удобнее всего при¬
менять для этой цели фосфорную кислоту, которая
образует с ионами железа(III) устойчивый фосфат¬
ный комплексFes* + Н3Р04 = FeHPO; + 2Ы, (22.15)тогда появление синего окрашивания окисленной фор¬
мы дифениламина происходит в точке эквивалентно¬
сти, после того как все железо окислится.§ 3. МЕТОДЫ ПРЕДВАРИТЕЛЬНОГО ВОССТАНОВЛЕНИЯМетодами окисления — восстановления можно опре¬
делять любые окислители или восстановители. Опи¬
сано применение для этой цели растворов перманга¬
ната, бихромата, бромата калия, иода. Используют
и другие окислители, например Ce(S04h, NH4VO3.
Известны и методы титрования окислителей восстано¬
вителями, например растворами FeS04, TiCl3 (тита-
нометрия), SnCl2, СгС12 (хромометрия). Эти методы
распространены мало, так как рабочие растворы
сильных восстановителей необходимо защищать от
окисления кислородом воздуха, сохраняя их и про¬
водя титрование в атмосфере инертного газа, что тре¬
бует применения специальных приспособлений. Кроме
того, для фиксации конечной точки титрования нужны
окислительно-восстановительные индикаторы, что не
всегда удобно или доступно.434
При титровании некоторыми окислителями нельзя
обходиться без индикаторов, например при использо¬
вании рабочих растворов сульфата церия, ванадата
аммония, бихрсмата калия. Поэтому наибольшее рас-- пространенне в практике получили методы титрования
окислителями, в частности растворами перманганата
и иода; в первом случае индикатором служит сам ра¬
бочий раствор, во втором — специфический по отно¬
шению к иоду индикатор — крахмал. Высокая устой¬
чивость растворов бнхромата калия и возможность
непосредственного использования его в качестве ис¬
ходного вещества нередко делают применение К2СГ2О7
более удобным по сравнению с другими окислите¬
лями. Кроме того, потенциал бихромата калия ниже,
чем у перманганата, вследствие чего титрование пер¬
вым проходит без побочных реакций, мешающих
определению. Бромат калия лучше всего применять
в качестве титра»та для определения ряда органиче¬
ских веществ, с которыми он реагирует стехиометри-
чески с образованием различных бромзамещенных
соединений.Несмотря на перечисленные достоинства, примене-
Hi с окислителей связано со следующими недостат¬
ками. Обычно предварительная подготовка пробы
к анализу состоит в переведении анализируемого ма¬
териала в раствор посредством обработки различными
кислотами; чаще всего применяют азотную кислоту
или ее смесь с хлороводородной или серной кислотой.
Так, медные сплавы растворяют в азотной кислоте,
причем содержащиеся в них элементы — железо,
олово и другие—превращаются в соединения выс¬
ших степеней окисления. При анализе различных чу-
гунов и сталей необходимо определять ванадий, мо¬
либден, вольфрам, титан и никоторые другие легирую¬
щие элементы, которые вследствие обработки пробы
окислительными агентами также содержатся в полу¬
ченном растворе в высших степенях окисления. Же¬
лезные руды содержат оксиды железа; растворяя их
в хлороводородной кислоте с добавками различных
окислителей, получают железо в степени окисления
+3 и т. д.Таким образом, титрование окислителями —
КМ1Ю4, К2СГ2О7 и другими — возможно только после
переведения определяемых элементов в низшие435
степени окисления. Восстановление необходимо прб-
вести количественно, для чего обычно вводят больший
или меньший избыток восстановителя. Перед титро¬
ванием этот избыток необходимо каким-либо спосо¬
бом устранить, иначе рабочий раствор окислителя бу¬
дет расходоваться не только на реакцию с определяе¬
мым элементом, но также на окисление восстанови¬
теля, и результат титрования окажется неправильным.Из многих восстановителей для предварительного
восстановления используют лишь некоторые.Восстановление газообразными веществами. При¬
меняют диоксид серы (сернистый газ, сернистую кис¬
лоту) и сероводород. Так, сернистая кислота восста¬
навливает железо до двухвалентного, ванадий —до
четырехвалентного в соответствии со следующими
уравнениями:2Fe3+ -f H2SOa -f НгО = 2Fe2+ + SO2' + 4H+ (22.16)или2V02 + H2S03 + 2H* = 2 VO2* + H2S04 + H20. (22.17)Сероводород реагирует с ионами Fe3+ следующим
образом:2Fe3+ + HjS = 2Fes+ + 2H+ + S. (22,)8)Избыток введенных H2S03 или H2S обычно уда¬
ляют кипячением раствора. Неудобство способа за¬
ключается в том, что трудно уловить момент, когда
восстановитель удален полностью, но окисление опре¬
деляемых элементов кислородом воздуха еще практи¬
чески не началось.Восстановление растворами восстановителей. Са¬
мый широко распространенный прием был описан
в гл. 20, посвященной перманганатометрическому
определению железа. Трехвалентное железо восста¬
навливают избытком раствора дихлорида олова:2FeCI3 + SnCl2 = 2FeCl2 + SnCI4. (22.19)Избыток SnCl2 окисляют раствором сулемы:SnC12 + 2HgCl2 = SnCl,+(Hg2Cl2. (22.20)Несмотря на широкое применение, с-нособ не очень
точен. Каломель медленно окисляется перманганатом,436
кроме того, возможно выделение свободной ртути:SnClj + HgCl2 = SnCI4 + Hg, (22.21)которая в мелкодисперсном виде также восстанавли¬
вает перманганат. Точное определение возможно
только при строгом соблюдении оптимальных условий
анализа.Для предварительного восстановления ванадия
применяют раствор FeSC>4.. VQj + Fe2* + 2Н* = V02t + Fe3+ + H20. (22.22)Избыток Fe2+ можно окислить персульфатом ам¬
мония, который в холодном растворе не взаимодей¬
ствует с V02+:■1 ^ : 2Feat 4- S,0'- = 2рё3+ + 2SOf\ • (22.23)Сильные i восстановители, как, например, СгС12,
легко окисляются кислородом воздуха. Поэтому при¬
менение СгСЬ для перевода многих катионов в вос¬
становленную форму удобно. Избыток СгСЬ легко
Окисляется при непродолжительном взбалтывании
раствора. Раствор хлорида хрома (II) получают вос¬
становлением бйхромата калия или хлорида хро¬
ма (III) металлическим цинком или амальгамой
цинка.: Восстановление твердыми восстановителями; Для
этой цели могут служить металлы с низкими значе¬
ниями окислительного потенциала: цинк (Е0=—0,76),
каДмий (£п = —0,4), свинец, висмут и некоторые дру¬
гие. Так, цйнк восстанавливает железо(Ш) до двух¬
валентного, ионы титана ^до титана(Ш) и т. д;Избыток восстановителя — твердого металла —
легко удаляется; достаточно восстановленный, рас¬
твор слить и промыть металл подходящей промывной
жидкостью.восстановление обычно ведут в кислом растворе.
В этом случае металл реагирует не только с восста¬
навливаемым веществом, но также с кислотой:Zn + 2HCI = ZnCI2 -f н2. (22.24)437
Неудобство метода состоит в том, что много ме¬
талла без пользы затрачивается на реакцию (22.24).
В связи с этим получили распространение методы
восстановления амальгамированными металлами. Ме¬
талл (цинк, кадмий) обрабатывают раствором
Hg2(N03)2; металлическая ртуть выделяется на по¬
верхности металла и образует тонкую пленку амаль¬
гамы. Такой амальгамированный металл восстанавли¬
вает так же, как и иеамальгамированный, однако
водород на поверхности амальгамы практически не
выделяется и кислоты расходуется очень мало.Большое практическое удобство представляют ре¬
дукторы. Редуктор — стеклянную трубку с краном
внизу — наполняют зернами металла, затем обраба¬
тывают металл раствором Hg2(N03)2 и промывают.
Анализируемый раствор медленно пропускают че¬
рез редуктор, выпуская восстановленный раствор
через кран.Редукторы с металлами можно' использовать для
проведения дифференциальных определений. Ниже
приведены данные о восстановительной способности
цинка и висмута по отношению к различным веще¬
ствам:Восстанавливаемые ионы . UO2* VOj MoO2* Fe3+ Ti4+
Продукты восстановленияZn U3+ V3+ Mo3+ Fe2t Ti3+Продукты восстановленияBi UO2* VO2* Mo03+ Fe2fАналогично цинку действует кадмий. Вместо вис¬
мута можно взять свинец или серебро.Из приведенных данных видно, что висмут дей¬
ствует слабее цинка. Основываясь на этом, можно
определять два или более элементов при их совмест¬
ном присутствии. Так, раствор, содержащий МоОГ и
Fe +, восстанавливают цинком и титруют сумму Мо®+
и Fe2+ перманганатом. В другой порции раствора бе¬
рут для восстановления висмут и снова титруют пер¬
манганатом. Разность количества перманганата, соот¬
ветствующего второму и первому титрованиям, соот¬
ветствует окислению молибдена от Мо3+ до Mov. Из438
Рис, 22.1. Амальгаматорэтих данных находят содержание мо¬
либдена в растворе, а затем и же¬
леза.Содержание железа (III) и тита¬
на (IV) можно найти следующим об¬
разом. Порцию раствора восстанавли¬
вают в цинковом редукторе и титруют
затем сумму Ti3+ и Fe2+; в другой
части раствора восстанавливают же¬
лезо в висмутовом (или в серебряном)
редукторе (титан не восстанавливает¬
ся) и титруют ионы железа перманга¬
натом. По разности находят содержа¬
ние титана.Нередко вместо редукторов с амальгамирован¬
ными металлами удобно пользоваться амальгамами
металлов: 2—3 г металла растворяют при нагревании
в 100 г ртути. Восстановление проводят путем взбал¬
тывания анализируемого раствора с амальгамой
в толстостенной склянке с притертой пробкой. При¬
меняют и специальные амальгаматоры, в которых бо¬
лее удобно отделять амальгаму от раствора. Нижнее
расширение амальгаматора (рис. 22.1) под краном
заполняют амальгамой, закрывают кран и остаток
амальгамы выливают через верхнее отверстие. Затем
наливают в амальгаматор подлежащий восстановле¬
нию раствор, закрывают прибор пробкой, открывают
кран, переворачивают амальгаматор и взбалтывают
раствор при закрытом кране. После восстановления
амальгаму снова выпускают в нижнее расширение.
Раствор можно затем перелить в колбу для титрова¬
ния или титровать непосредственно в амальгаматоре.Амальгамы восстанавливают катионы металлов до
таких же степеней, как и металлы в редукторах. От¬
личие заключается только в технике работы. При
пользовании амальгамами необходимо следить затем,
чтобы ртуть не разбрызгивалась. Пары ртути ток¬
сичны и отравляет атмосферу в лаборатории.
ГЛАВА 23СКОРОСТЬ ХИМИЧЕСКИХ РЕАКЦИИ
И ЕЕ ЗНАЧЕНИЕ В АНАЛИТИЧЕСКОЙ ХИМИИ§ 1. ОБЩАЯ ХАРАКТЕРИСТИКАСкорость химических реакций имеет большое значе¬
ние в аналитической химии. Измерение скорости ре¬
акций лежит в основе кинетических методов анализа.
Медленное протекание некоторых реакций затрудняет
аналйз. Иногда, наоборот, небольшая скорость хими¬
ческих процессов благоприятствует выполнению ана¬
лиза и играет положительную роль. Исследование
скорости реакций — предмет химической кинетики.Скорость реакций определяется количеством ве¬
ществ, прореагировавших за единицу времени. Рас¬
смотрим реакцию между веществаА и В:: ■ : - А + В == С + D.Ее скорость зависит от концентрации реагирую¬
щих компонентов:0 = 4f ==*|А1(В1, <23,)где х — количество вступившего в реакцию [А] (или fB}) за
время /; /( — константа скорости реакции, .В самом деле, при увеличении концентрации воз¬
растает общее количество частиц в единице объема,
число их столкновений между собой и, следовательно,
вероятность взаимодействия; поэтому за единицу вре¬
мени прореагирует больше вещества, чем при малых
концентрациях.■1 Физический-см-ывд нонста-нты екерости выясняется,
если принять концентрации [А] и [В] равными
1 моль/л. Тогда! 0 = if = (23.2)т. е. константа скорости равна количеству вещества,
прореагировавшего за единицу времени при [AJ =
= [В] == 1 моль..........Скорость реакций может изменяться в очень ши¬
роких пределах —от малых долей секунды до не¬
скольких часов или дней. Условно все реакции обычно,440
делят на быстрые и медленные. К быстрым реакциям
относят такие, в которых половина имеющегося коли¬
чества вещества реагирует за 10 с или за меньшее
время, все остальные реакции относят к медленным.Примером быстрых реакций могут служить мно¬
гие реакции между ионами противоположного знака
заряда, например реакции между водородными и
гидроксильными ионами, между окислителями и вос¬
становителями и т. д. Однако существуют и очень
медленные реакции. Так, исходя из значения стан¬
дартных потенциалов, следовало ожидать, что должна
происходить реакция между перманганатом и водой"
с выделением кислорода. Но эта реакция происходит
с бесконечно малой скоростью, в связи с чем водные
растворы перманганата калия вполне устойчивы и не
изменяют своей концентрации длительное время.Известны и другие реакции, характеризующиеся-
большими значениями констант равновесия, но про¬
текающие с очень малой скоростью. Наоборот, реак¬
ции с меньшими константами часто протекают быст¬
ро. Примером могут служить следующие реакции:Ij + AsO" .+ 2Н20 = 2 Г + AsOj + 4Н*. (23.3)H2Os + AsO~ = AsOj- + 2H*. (23.4)Реакция (23.3)—быстрая, а реакция (23.4) —
очень медленная. В то же время окислительный по¬
тенциал иода значительно меньше, чем у пероксида
водорода, и, следовательно, константа равновесия ре¬
акции (23.3) меньше, чем реакции (23.4). Значения
констант равновесия можно найти по уравнению: >.( Ь» (£fll £02) ^ /ЛЛ CV: g ^ = ' 0058~ ■ (23 5)И мея в виду, что £о,2/2|--=0 £2 В , Е oH202/H2(f= 1 77 В
•и Е . „з-/. = 0,56 В, для реакций (23 3) и (23.4)0asu4 /asu2получаем соответственно:ig Кз = 2,1 и 1ЙК«==41.7,т. е. в термодинамическом смысле реакция (23.4) зна¬
чительно более вероятна, чем реакция (233), и долж¬
на быть сдвинута в правую сторону в большей сте-
’ ленй, однако скорости обеих реакций находятся в об¬441
ратно пропорциональном отношении к значениям кон¬
стант равновесия.Пользуясь различными скоростями реакций, не¬
редко удается определять одно вещество в присут¬
ствии другого даже в том случае, если реагент взаи¬
модействует с обоими веществами. Рассмотрим две
реакции:2Ce(S04)2 + HAsOi + 2НгО = Ce2(S04)3 + H3AsO< + H2S04,(23.6)2Ce(S04)2 + 2FcS04 = Ce2(S04), + Fe„(S04)j. (23.7)Сульфат церия окисляет и мышьяк, и железо (II).
Однако реакция (23.6) идет с очень малой скоростью,
в то время как реакция (23.7) происходит мгновенно.
Таким способом удается оттитровать железо в присут¬
ствии мышьяка. Затем прибавляют катализатор OsCb,
который ускоряет реакцию (23.6), и оттитровывают
мышьяк.Другой пример — титрование иода тиосульфатом
в растворе, содержащем свободную кислоту. Тиосуль¬
фат взаимодействует и с иодом, и с кислотой:Правильные результаты определения иода по ре¬
акции (23.8) можно получить только потому, что ско¬
рости обеих реакций различаются во много раз. Реак¬
ция (23.9) протекает настолько медленно, что в про¬
цессе восстановления иода тиосульфат не успевает
разложиться в соответствии с уравнением (23.9).Скорость реакции можно регулировать различ¬
ными способами. Из уравнения (23.1) видно, что ско¬
рость зависит от концентрации реагирующих веществ;
следовательно, быстрая реакция замедляется при
уменьшении концентраций А и В, и, наоборот, она
возрастает, если концентрации соответствующих ве¬
ществ увеличиваются.Скорость реакций зависит от температуры. Зави¬
симость константы скорости реакции от температуры
определяется уравнением Аррениуса12 2Na2S203 *= 2NaI -f- N&2S4Oe,
2HCI + Na2S20j = H2SOj + S + 2NaCl.(23.8)(23.9)d In К E
dT = RT1 ’(23.10)где К — константа скорости; Е — энергия активации.
В случае простых реакций, идущих в одну стадию,
параметр Е показывает, какой минимальной энергией
(в расчете на 1 моль) должны обладать реагирую¬
щие частицы, чтобы они могли вступить в химическую
реакцию. Частицы, энергия которых больше или
равна Е, называют активными.В среднем повышение температуры на 10 °С при¬
водит к увеличению скорости реакций в растворе при¬
близительно в 2—3 раза. Этот прием часто исполь¬
зуют в анализе Так, реакция между щавелевой кис¬
лотой и перманганатом в холодных растворах идет
с очень малой скоростью, однако нагревание до 80—
90°С значительно ускоряет реакцию. Растворение ме¬
таллов или их солей идет значительно быстрее при
нагревании. При осаждении малорастворимых соеди¬
нений нагревание раствора способствует увеличению
скорости движения ионов в растворе и приводит
к быстрому росту центров кристаллизации и вслед¬
ствие этого к образованию крупнокристаллических
осадков. В кинетических и каталитических методах
анализа нередко необходимо в определенный момент
времени замедлить или вообще остановить реак¬
цию— охлаждение раствора является одним из мето¬
дов такого замедления.На скорость реакций влияет характер раствори¬
теля. Имеет значение диэлектрическая проницаемость
растворителя. Для наиболее распространенного в ана¬
литической химии случая, когда реагируют между со¬
бой ионы противоположного знака, скорость реакции
уменьшается с увеличением диэлектрической прони¬
цаемости. Большинство органических растворителей
имеет диэлектрические проницаемости меньше, чем
у воды, и поэтому скорость реакций в таких раство¬
рителях больше, чем в водных растворах. Однако та¬
кая корреляция наблюдается далеко не всегда. Она
справедлива только в пределах группы растворителей
одного гомологического ряда или для серии смесей
переменного состава, приготовленных из определен¬
ной пары растворителей.Скорость реакций зависит от ионной силы рас¬
твора. В наиболее простом случае, когда взаимодей¬
ствуют ионы А и В в растворе, зависимость лога¬
рифма константны скорости К от ионной силы рас-443
твора выражается следующим уравнением:lg* = lg *„•+ aZAZB ViT, (23.11)гдё! Ко — константа скорости реакции в среде, для которой коэф¬
фициенты активности исходных частиц А и В и активированного
комплекса между ними приняты равными единице; а — констан¬
та, .включающая величины диэлектрической проницаемости рас¬
творителя и абсолютной температуры; ZA, ZR — заряды частиц;
ц — ионная сила раствора.Из уравнения видно, что с увеличением ионной
силы, т. е. с введением в реакционную систему постог
ронних хорошо диссоциирующих солей, скорость ре¬
акции между ионами противоположного знака заряда
уменьшается. Объяснение заключается в том, что
ионы посторонних солей образуют вокруг реагирую-,
щих ионов ионную атмосферу из ионоп противополож¬
ного знака заряда, которая .препятствует непосред¬
ственному контакту между ионами и на разрушение
которой необходимо затратить определенное время.
Наоборот; если реагируют-между собой ионы с заря¬
дами •:одинакового; з^ака, скорость реакции должна'
возрастать. ;Изменение концентрации ионов водорода в реак-
ййяхчжисления — восстановления влияет не только на
окислительный потенциал системы, в соответствии
с уравнением Нернста, но также нередко на скорость
реакции. Так, окислительные потенциалы систем
fe3+/Fe2t и 12/21 — не изменяются в зависимости от
pH;, однако скорость реакции2Fe3* +2Г=2Ре2* f \г (23.12)сильнб,возрастает с увеличением концентрации вода-,
ро/шцх ирнов раствора. Аналогичное влияние на ско-,
рость процесса оказывает pH раствора в реакции: 2Cu2v + 41' —СиJ^-t- 12, (23.13)использующейся для иодометрического определения
меди. Видно, что ионы водорода не участвуют в сум-,
марном уравнении реакции и поэтому не влияют нд
окислительный потенциал реагирующих веществ. Од-.
на^Ь при слишком, малой кислот.нрсти раствора реак¬
ция (23.13) проходит очень медленно и ее скорость,
врзрартает с уменьшением pH раствора.Скорость реакций зависит от введения катализа-,
торов. Катализаторы могут ускорять или замедлять.
реакции, в последнем случае говорят об ингибирую¬
щем действии. Методы, основанные на каталитиче¬
ском действии вещества, представляют собой разно¬
видность кинетических методов, их называют катали¬
тическими.Рассмотрим принцип кинетических методов ана¬
лиза, основанных на измерении скорости некаталити¬
ческой реакции в зависимости от концентрации реа¬
гирующих веществ. Рассмотрим реакцию между
веществами А и В, которая идет по уравнению А + В =
== С-)-D, где А — определяемое вещество, а В — реа¬
гент. В начальный период взаимодействия, когда ко
личество прореагировавших веществ еще невелико
по сравнению с концентрациями А и В, можно запи¬
сатьV—%--KCACB. (23.Н)т. е. можно принять, что равновесные концентрации
А и В равны исходным концентрациям СА и Св.Преобразуем уравнение (23.14):dx = КСАСВ dt (23.15)и после интегрирования получим:л = КСАСВ1. (23.16)Таким образом, количество прореагировавшего ве¬
щества А пропорционально концентрации реагента Св
и времени протекания реакции. Если концентрация
реагента остается практически постоянной, тогда х
пропорционально концентрации определяемого веще¬
ства и времени протекания реакции.Практически можно поступать следующим обра¬
зом. Готовят серии растворов с переменной и извест¬
ной концентрацией Сд (при Св = const), измеряют че¬
рез определенный промежуток времени ((= const)
количество прореагировавшего вещества х и строят
градуировочный график, откладывая на оси орди¬
нат х, а на оси абсцисс — концентрацию С*
(рис. 23.1). По этому графику легко установить за¬
тем концентрацию Сд в анализируемом растворе, по¬
ставив аналогичный эксперимент с раствором, в кото¬
ром содержание С а «ензвестно.445
Рис. 23.1. Градуировочный график
для кинетического определения
веществаКинетический метод
можно применять для опре¬
деления двух компонентов
в смеси, в частности двух
органических веществ, на¬
пример двух органических
пероксидов. Определение
возможно только в том слу¬
чае, когда константы скорости реакций обоих веществ
с реагентами значительно различаются. Исследуют
скорость взаимодействия обоих пероксидов, напри¬
мер, с раствором иодида калия; о скорости реакции
судят по количеству выделившегося иода. График за¬
висимости концентрации иона (обнаруживают посред¬
ством крахмала) от времени протекания реакции со¬
стоит из двух участков с различным наклоном. Изме¬
рив тангенсы углов наклона <L\ и аг, можно найти
содержание каждого компонента, пользуясь тем, что
тангенсы углов наклона пропорциональны содержа¬
нию каждого вещества.Применяя специальные методы обработки резуль¬
татов, находят содержание двух компонентов смеси
и при близких скоростях обеих реакций.§ 2. каталитические методы анализаКаталитические методы анализа — вариант кинетиче¬
ских методов. Они основаны на измерении скорости
химической реакции, протекающей в растворе при
действии катализатора; нередко катализатор является
определяемым веществом. Известно много медленных
реакций, скорость которых в определенных условиях
увеличивается пропорционально концентрации введен¬
ного катализатора. Это дает возможность определить
количество катализатора по концентрации продуктов
реакции, образующихся за определенный промежуток
времени. Для таких определений пригодны медленные
реакции различных типов, однако наиболее распро¬
странены каталитические методы с использованием
реакций окисления — восстановления.446
Окислительно-восстановительные реакции катали¬
зируются преимущественно ионами переходных эле¬
ментов, характеризующихся большой склонностью
к образованию комплексных соединений и способ¬
ностью изменять степень окисления в растворе. Ука¬
занные две особенности имеют существенное значение
для объяснения каталитического действия. Рассмот¬
рим, например, медленную реакцию между окислите¬
лем Ок! и восстановителем В2:Ок, В2 В| + Ок2, (23Л7)где индексы 1 и 2 обозначают первое и второе веще¬
ство в окисленной и восстановленной формах соот-.
ветственно.Действие катализатора К обусловлено тем, что его
окисленная форма Кок быстро реагирует с восстано¬
вителем Вз, а образовавшаяся восстановленная фор¬
ма Кв также быстро переходит в окисленную форму
Кок при действии окислителя Окь Таким образом, ме¬
ханизм катализа можно схематически выразить сле¬
дующими уравнениями:КСк + В2 кв + Ок2 (быстрая реакция), (23.18)Кв+Ок1 К0к+В, (быстрая реакция). (23.19)Суммирование этих уравнений приводит к уравне¬
нию (23.17), причем после завершения цикла катали¬
затор снова находится в своей первоначальной форме.
Механизм катализа может быть также связан с обра¬
зованием нестойких промежуточных комплексных со¬
единений катализатора с веществами Ок, или В2. Эти
комплексы реагируют с основными участвующими
в реакции веществами Ок, и В2 значительно быстрее,
чем эти вещества взаимодействуют между собой.Выяснение механизма каталитических реакций яв¬
ляется сложной задачей и требует в каждом отдель¬
ном случае проведения специальных исследований.Главная особенность каталитических методов
определения — их высокая чувствительность. Это обус¬
ловлено тем, что одна и та же частица катализатора
действует многократно, вовлекая в реакцию большие
количества веществ Ок] и В2, и за короткий период
времени способствует образованию продуктов реак¬
ции, концентрация которых во много раз больше кон¬
центрации введенного катализатора. Так, каталитичсг
ским методом можно определить 1 -10—4 мкг (Ю-10 г)
кобальта, ванадия, рения, МО-6 мкг иода. Предел
обнаружения многих других элементов также очень
низкий.Иногда каталитические методы отличаются высо-
кой селективностью. Например, реакция между иона¬
ми Fe3+ и тиосульфатом2Fe3+ + 2SaO“' = 2FeJ* + S4Oj?"катализируется только ионами Cu2+, что дает возмож¬
ность количественно определять медь в присутствии
многих других элементов. Однако в большинстве слу¬
чаев перед определением необходимо мешающие эле¬
менты отделять или устранять их влияние каким-либо
другим способом.Для каталитического определения элементов чаще
всего применяют реакции окисления пероксидом во¬
дорода, анионами кислородных кислот, высокозаряд¬
ными катионами металлов. В табл. 23.1 приведены не¬
которые так называемые индикаторные реакции и
указаны элементы-катализаторы.Скорость реакции устанавливают, определяя кон¬
центрацию одного из продуктов реакции (или умень¬
шение концентрации исходных веществ) каким-либо
методом, чаще всего фотометрическим. Так, при ре¬
акции пероксида водорода с иодид-ионами добавляют
в раствор крахмал; выделяющийся иод образует с
крахмалом соединение интенсивно-синего цвета, из¬
меряя оптическую плотность которого, можно найти
количество иода. Из табл. 23.1 видно, что применяют
также реакции окислителя с каким-либо органиче¬
ским окрашенным реагентом. В этом случае за реак¬
цией можно следить по ослаблению окраски реаген¬
та, например метилового оранжевого или ализарина.Кроме ионов переходных металлов, высокая ката¬
литическая активность характерна для ферментов.
Ферменты — это вещества белкового происхождения,
имеющие в своем составе определенные функциональ¬
ные группы и катализирующие многие биохимические
процессы в живых организмах. Основное их преиму¬
щество— высокая специфичность действия. Фермент
обычно катализирует превращение только одного ве¬
щества— субстрата, что позволяет определить это со-44*
Таблица 23.1. Каталитические реакции, применяемые
для определения элементовИндикаторные реакцииКатализаторыОкислитель —н2о2 + s2oj-
н2о2 + г
н2о2 + nh2csnh,
н2о2 + н2о2Н202 + метиловый оранжевыйН202 + ализаринН,02 + люмииолН20, + гидрохинонН202 + п-фенилендиаминпероксид водородаП, Zr, Th, V, Nh, Та, Mo, W
Zr, Hr, Th, Та, Mo, W, Fe
Mo, W
Cu, Mn, Fe, Pd
Fe, Cu, Cr
Co
Co, Cu
CuCu, Fe, OsОкислители — анионы кислородных кислотСЮ’ + ГC.IO" + фенилендиамин
IOj + гидрохинон
TeQ^- + Sn2*МпО, + AsO~S2Og' + я-фенетидинV', Re, Ru, Os
V, Os
Mo
ReOsAgFe1* + S202-
Fe1" + SCN'
Ce4+ + As02
Се4" + СГОкислители — катионы металловCu
Г
Os, I'
Agединение в смеси близких ему по строению и свой¬
ствам других веществ. Выделено в чистом виде очень
много разнообразных ферментов. Одни из них — так
называемые оксиредуктазы — катализируют окисли¬
тельно-восстановительные реакции, другие — гидрола-
зы — способствуют разрыву связей С—О, С—N, С—С
с участием молекул воды в органических соединениях.
Наконец, третьи — изомеразы — катализируют геомет¬
рические или структурные изменения в молекуле суб¬
страта. Есть и другие разновидности ферментов.15 Аналитическая химия449
Для того чтобы обеспечить многократность исполь¬
зования ферментов, их подвергают иммобилизации.
Иммобилизацией называют перевод ферментов, обыч¬
но растворимых в воде, в водонерастворимое состоя¬
ние с сохранением их каталитической активности. Мо¬
лекулу фермента присоединяют за счет образования
ковалентной связи к какому-либо водонераствори¬
мому носителю — целлюлозе, стеклу, бумаге, силика¬
гелю, полистиролу и др. Можно также диспергиро¬
вать фермент в геле какого-либо вещества.Каталитические ферментативные методы анализа
используют преимущественно для определения мно¬
гих органических соединений, п частности таких, ко¬
торые содержатся в биологических объектах (в крови,
моче, тканях и др.). Таким способом можно опреде¬
лять мочевину, мочевую кислоту, аминокислоты и дру¬
гие органические кислоты, глюкозу и другие сахара,
антибиотики и т. д.Пределы обнаружения названных и многих других
органических веществ — субстратов ферментативны¬
ми реакциями находятся обычно в интервале 10~4—
ю-8 М.Определение каталитическим ферментативным ме¬
тодом обычно основано на измерении скорости изме¬
нения концентрации одного на участников реакции при
постоянной концентрации фермента. Например, опре¬
деление глюкозы основано на следующей реакции:Г люкоза 02 = Г люконовая кислота + Н2О2.Эта реакция катализируется ферментом глюкоз-
оксидазой. Скорость реакции определяют по количе¬
ству выделившегося пероксида водорода. Для иденти¬
фикации последнего можно использовать реакцию
окисления какого-либо бесцветного органического ве¬
щества, окисленная форма которого окрашена, напри¬
мер окисление о-толуидина. Тогда скорость увеличе¬
ния оптической плотности прямо пропорциональна ис¬
ходной концентрации глюкозы; необходимо только,
чтобы кислород и восстановленная форма красителя
находились в большом избытке по отношению к ко¬
личеству определяемой глюкозы.Для определения количества вещества фермента¬
тивными методами применяют и другие приемы. Так,
ионы многих металлов являются ингибиторами ката¬450
литического действия ряда ферментов; они снижают
их каталитическую активность в определенной инди¬
каторной реакции. Степень этого снижения при неко¬
торых условиях пропорциональна содержанию ионов
определяемого металла в анализируемом растворе.
Например, ионы ртути ингибируют каталитическую
активность фермента уреазы в реакции разложения
мочевиныУреазаNH2CONH2 + н20 ► 2NHt + С02.Скорость этой реакции уменьшается пропорцио¬
нально концентрации ионов ртути, и этим методом
можно определять ничтожные количества металлов.Пределы обнаружения ингибиторов очень низки.
Так, применяя различные ферментативные реакции,
можно определить 1-10~“ г/мл ртути, 10-12 г/мл
меди, 8 -10-12 г/мл цинка и т. д.Определение каталитическими методами старают¬
ся проводить в таких условиях, когда одно или оба
реагирующих вещества находятся в избытке и расход
их незначителен, вследствие чего можно принять, что
их концентрация в процессе определения остается
практически постоянной. Рассмотрим наиболее про¬
стой пример, когда стехиометрические коэффициенты
участвующих в реакции веществ равны единице.А + В = С + D,
тогда кинетическое уравнение имеет вид:~ = КСкСвСк,где х — концентрация одного из продуктов реакции; К — катали¬
тический коэффициент; Са, Св и С« — соответственно концен¬
трации веществ А, В и катализатора.Если концентрации СА и Св не изменяются в про¬
цессе определения, тогда после интегрирования урав¬
нения получим:л = /ССАСВСК<, (23 20)т. е. концентрация продукта реакции пропорциональ¬
на концентрации катализатора и времени протекания
реакции t.Такую же зависимость получим, если концентра¬
ция катализатора сохраняется постоянной, а изме¬
няется только концентрация Сд или Св.15*
Если можно считать постоянной концентрацию
только одного из участвующих в реакциях веществ,
например В (или катализатора), а концентрацию вто¬
рого— изменяющейся за период времени t, то кинети¬
ческое уравнение преобразуется в следующее:-~- = КСв(СА-х)Ск, (23.2!)где (Са — х) — текущая концентрация А в процессе реакции.Из уравнения (23.21) следует, что
dxС~-х = вСК dt- (23.22)и после интегрированияСАIn - - = KCHCKt. (23.23)А хВ данном случае пропорциональна концентрации
катализатора (или одного из веществ при постоянной
концентрации катализатора) и времени t не концен¬
трация продукта реакции х, а величина 1п[СА/(С'л —— х)], которую легко вычислить, если известно х.Существуют три метода определения концентрации
вещества: фиксированного времени t, фиксированной
концентрации х и тангенсов. Предположим, что кон¬
центрации веществ А и В заметно не изменяются.
Тогда следует применить уравнение (23.20).Практически поступают следующим образом. Если
исследуют зависимость скорости реакции от концен¬
трации катализатора, тогда готовят серию растворов
с постоянными концентрациями веществ СЛ и Св и
переменной концентрацией катализатора CVВ методе фиксированного времени измеряют че¬
рез определенный промежуток времени, например че¬
рез 10 мин, концентрацию продукта реакции х и на
основании полученных данных строят градуировоч¬
ный график (рис. 23.2, а). График выражается пря¬
мой линией, т. е. при фиксированном времени (/ =
= const) концентрация jc, как следует из уравнения(23.20), прямо'пропорциональна концентрации ката¬
лизатора.В методе фиксированной концентрации (*=const)
реакцию в каждом растворе проводят до тех пор,
пока концентрация продукта реакции к не станет во452
Рис. 23.2. Определение концентрации катализатора К по методам
фиксированного времени (а), фиксированной концентрации (б)
и по методу тангенсов (а, г)всех растворах одинаковой. Для этого необходимо
различное время t, и чем больше концентрация опре¬
деляемого катализатора, тем меньше времени потре¬
буется, чтобы концентрация * стала одинаковой во
всех растворах. На основании данных опыта строят
график, откладывая на оси ординат 1//, а на оси абс¬
цисс— Ск (рис. 23.2,6).Наиболее точные результаты получают в методе
тангенсов. В каждом растворе серии измеряют через
определенные промежутки времени концентрацию
продукта реакции х и на основании результатов экс¬
перимента получают график (рис. 23.2,в), наклоны
прямых которого тем больше, чем больше концентра¬
ция катализатора. По графику измеряют тангенс угла
наклона каждой прямой и изображают графически
зависимость tga от Ск (рис. 23.2,г).По одному из описанных методов легко найти кон¬
центрацию определяемого вещества в анализируемом
растворе.453
Если концентрация одного из участвующих в ре¬
акции веществ в процессе определения заметно изме¬
няется, то следует применять уравнение (23.23), т. е.
откладывать на оси ординат графика вместо х вели¬
чину 1п[Са/(Са — *)].ГЛАВА 24ПОТЕНЦИОМЕТРИЧЕСКИЙ АНАЛИЗ§ 1. ОБЩАЯ ХАРАКТЕРИСТИКА
ПОТЕНЦИОМЕТРИЧЕСКОГО АНАЛИЗАПотенциометрический анализ — метод определения
концентрации ионов, основанный на измерении элек¬
трохимического потенциала индикаторного электрода,
погруженного в исследуемый раствор. Потенциомет¬
рический метод был разработан еще в конце прош¬
лого столетия, после того как Нернст вывел уравне¬
ние, связывающее электродный потенциал с актив¬
ностью (концентрацией) компонентов обратимой
окислительно-восстановительной системы. В разбав¬
ленных растворах коэффициенты активности ионов
близки к единице, а активность близка к концентра¬
ции, поэтому можно пользоваться уравнениями Нерн-
ста в концентрационной форме, а именно:Е, = Е0 +^lgCM,1+, (24.1)или, если измерения проводят в растворах, содержа¬
щих ионы одного и того же металла в различных сте¬
пенях окисления:(21.2)В первом случае применяют электрод из вещества,
ионы которого содержатся в растворе. Во втором слу¬
чае применяют инертный металлический электрод,
чаще всего платиновый, потенциал которого зависит
от соотношения концентраций окисленной и восста¬
новленной форм ионов.Уравнения (24.1) и (24.2) лежат в основе потен¬
циометрических методов анализа. Существует два454
основных варианта использования этих уравнений в
аналитической химии. Первый вариант — это измере¬
ние потенциала электрода, погруженного в исследуе¬
мый раствор, и вычисление концентрации определяе¬
мых ионов непосредственно по уравнению (24.1).
Можно также пользоваться мембранными ионселек-
тивными электродами. Способ с применением мем¬
бранных электродов использовали раньше только для
определения активности ионов водорода, но в настоя¬
щее время он получил значительно более широкое
распространение в связи с конструированием мем¬
бранных электродов на ряд других ионов. Рассмот¬
ренный способ получил название прямой потенциомет-
рии. Этим способом устанавливают также состав и
константы устойчивости комплексных соединений,
применяемых в аналитической химии. В этих случаях
необходимо знать активность (концентрацию) свобод¬
ных ионов металла, образующихся при диссоциации
комплексного соединения. Ее можно найти, измеряя
потенциал металлического (или мембранного) элек¬
трода в растворе комплексного соединения и вычисляя
затем искомую концентрацию по уравнению (24.1).Второй вариант — это метод потенциометрического
титрования. Сущность метода заключается в том, что
в исследуемый раствор погружают индикаторный
электрод и титруют раствор, определяя в процессе
титрования потенциал электрода *. Концентрация
определяемых ионов изменяется в зависимости от
объема прибавленного рабочего раствора неравно¬
мерно: сначала это изменение невелико, затем стано¬
вится более заметным и, наконец, в точке эквивалент¬
ности достигает наибольшего значения. Из уравнения
(24.1) видно, что потенциал электрода зависит от кон¬
центрации ионов в растворе; поэтому он также изме¬
няется, причем максимальное изменение соответствует
эквивалентному отношению реагирующих веществ.
Зависимость потенциала от объема рабочего раствора
выражается кривой титрования, представленной на
рисунке 24.1. По кривой можно установить точку эк¬
вивалентности как точку перегиба кривой. Ордината* Его находят, измеряя э. д. с. гальванического элемента, со¬
стоящего из индикаторного электрода и электрода сравнения
(см. ниже).4S5
Рис. 24.1. Кривая потенциометрического титрованияРис. 24.2. Кривая титрования смеси хлорида и иодида калия
раствором нитрата серебраточки перегиба соответствует потенциалу электрода
в конце титрования, а абсцисса — объему рабочего
раствора, израсходованного на определение.Таким образом, потенциометрическое титрование
представляет собой обычный титриметрический метод
анализа; разница заключается в том, что точку экви
валентности определяют по характерному изменению
потенциала электрода в процессе титрования, а не
при помощи цветных индикаторов.Потенциометрическое титрование применяют глав¬
ным образом для определения сравнительно больших
количеств вещества. Его преимущества по сравнению
с обычным титрованием следующие.1. Можно титровать окрашенные растворы, когда
цветные индикаторы неприменимы; можно опреде¬
лить pH или общую кислотность в ваннах никелиро¬
вания или хромирования, когда неприменимы фото¬
метрический метод или титрование с индикатором.2. Легко определять, не прибегая к предваритель¬
ному разделению, несколько веществ в смеси. На кри¬
вой титрования получается неск@лько скачков потен¬
циала, по которым находят объем рабочего раствора,
израсходованного на взаимодействие с каждым ком¬
понентом сложного раствора.На рис. 24.2 приведена в качестве примера кривая
титрования смеси растворов хлорида и иодида калия
раствором нитрата серебра. На кривой наблюдается
два перегиба: первый соответствует осаждению всего
иодида (ПР = 8,3-10-17), второй — осаждению хло-456
Рис. 24.3. Кривая титрования сме-
си хромата, ванадата и желе-
за(Ш) раствором соли тита-
на(Ш)рида (ПР = 1,8-10-10). По
абсциссам перегиба нахо¬
дят объем рабочего раство¬
ра, израсходованного наСг2о* ++ vo®'1/титрование каждого аниона.Аналогично титруют смесь нескольких кислот, напри¬
мер хлороводородной и фосфорной, или нескольких
окислителей или восстановителей. Примером может
служить титрование хромата, ванадата и железа(III)
раствором соли титана(III) (рис. 24.3).Стандартные потенциалы полуреакций имеют сле¬
дующие значения:Сг20^ + бе' + 14Н+ = 2Сг3+ + 7Н20, Е[ = 1,36;В соответствии с этим хромат и ванадат восста¬
навливаются совместно соответственно до трех- и че¬
тырехвалентного состояния. Далее восстанавливается
трехвалентное железо; наконец, в следующей стадии
четырехвалентный ванадий восстанавливается до
трехвалентного. Все три этапа восстановления отме¬
чаются на кривой скачками потенциала электрода, по
которым можно определить содержание каждого эле¬
мента в смеси.Для потенциометрического анализа требуется спе¬
циальная аппаратура. Поэтому его имеет смысл при¬
менять только в тех случаях, когда нельзя решить
задачу посредством обычных титриметрических ме¬
тодов.Классификация потенциометрических методов
анализа такая же, как и обычных титриметрических
методов: в ее основе лежат различные типы химиче¬
ских реакций. Наиболее важные типы химических
реакций, используемые в потенциометрическом ти¬
тровании, следующие.V02+ +е' + 2Н+ = V02+ + Н20,
Fe3+ + е~ = Fe2\VQ2t + е~ + 2Н+ = V3+ + Н20,£'г = 1,08В;
£"' = 0,77В;
£'v = 0,34В.457
1. Методы осаждения и комплексообразования.
Широко распространено титрование раствором ни¬
трата серебра. Так можно определять анионы и их
смеси, образующие малорастворимые осадки с ио¬
нами серебра, например хлориды, бромиды, иодиды,
роданиды, фосфаты и др. Раствором хлорида натрия,
наоборот, можно титровать ионы серебра, висмута,
напримерBi(N03)3 + NaCI + Н20 = BiOCl + NaN03 + 2HN03.Гексацианоферрат(П) калия образует малораство¬
римые соли с катионами меди, цинка, кадмия, свинца,
серебра, что можно использовать для определения
этих катионов. Так, взаимодействие с солями цинка
происходит по уравнению3ZnCl2 + 2K4Fe(CN)e = K2Zn3[Fe(CN)6] + 6KCI.Можно указать также на титрование ионов ме¬
ди (I) раствором роданида калия, ионов молибдата—
раствором хлорида бария, ионов уранила — раство¬
ром фосфата натрия и др.На реакциях комплексообразования основано ти¬
трование катионов алюминия, бериллия, магния рас¬
твором фторида натрия. Реакция с алюминием про¬
текает в соответствии с уравнениемА1а* + 3F" = A1F3.Точку эквивалентности устанавливают по измене¬
нию потенциала платинового электрода в растворе,
содержащем помимо алюминия еще некоторое коли¬
чество солей железа(II) и (III). Можно также поль¬
зоваться мембранным ионселективным электродом на
ионы фтора.2. Методы окисления — восстановления. В данных
методах можно применять растворы многих окисли¬
телей и восстановителей. Часто пользуются раство¬
ром сульфата железа (И), которым титруют, напри¬
мер, хром и ванадий после их окисления персульфа¬
том аммония:К2Сг407 + 6FeS04 + 7H,SO* == K2S04 + Cr2(SOt), + 3Fe*(SO«)3 + 7H20,2HVO, + 2FeSOt + 3H2SO* = 2V0S04 + Fe2(S04)3 + 4H,0.458
Используют и раствор арсенита натрия для опре¬
деления хромата в присутствии ванадатов, так как
последние не восстанавливаются. Сильный восстано¬
витель— раствор соли титана(III)—можно приме¬
нять для определения железа и меди в смеси: сна¬
чала железо(III) превращается в двухвалентное, а
затем восстанавливается медь(II) до одновалентной.
Существуют и методы титрования другими сильными
восстановителями, например растворами солей хро¬
ма (II) или олова, хотя работа с такими растворами
сопряжена с необходимостью защиты их от действия
кислорода воздуха. Раствор хлорида олова(II) вос¬
станавливает молибден(VI) до молибдена (V) и ва¬
надий (V) до ванадия(III); так можно определить оба
элемента при их совместном присутствии.Гексацианоферрат(Ш) калия — слабый окисли¬
тель, стандартный потенциал полуреакцииFe(CN)a3-+<T = Fe(CN)‘-равен 0,36 В.Тем не менее в щелочных растворах им можно
окислить хром(Ш) до хрома(У1) и ванадий(1У) —
до ванадия (V). Известен потенциометрический метод
определения кобальта окислением до трехвалентного
состояния в аммиачном растворе:Со2" + Fe(CN)g' = Со3* + Fe(CN)J'.Раствор бихромата калия используют для титро¬
вания железа, олова и других восстановителей.3. Методы кислотно-основного титрования. Потен¬
циометрическое титрование используют для опреде¬
ления сильных и слабых кислот и их солей во всех
случаях, в частности, когда нельзя применить цвет¬
ные индикаторы.В потенциометрическом анализе важно правильно
выбрать подходящий индикаторный электрод для оп¬
ределения точки эквивалентности. К этим электро¬
дам предъявляют некоторые специальные требова¬
ния, рассмотренные в последующих параграфах.Существенным является также способ измерения
потенциала. Индикаторный электрод соединяют в
гальванический элемент с электродом сравнения
(стандартным электродом), имеющим известный и469
постоянный потенциал, и измеряют разность потен¬
циалов между этими электродами:Ех = Е инд Е ст.Способы измерения могут быть различны в зави¬
симости от цели анализа, требуемой точности и имею¬
щейся в лаборатории аппаратуры. В заводских и
контрольных лабораториях нередко применяют ме¬
тод некомпенсационного титрования, при котором
оба электрода присоединяют к вольтметру или к дру¬
гому измерительному прибору. Такой способ прост в
аппаратурном отношении, и им можно пользоваться
в работах, не требующих большой точности. Основ¬
ной недостаток этого метода заключается в том, что
в момент измерения разности потенциалов через рас¬
твор проходит электрический ток. Вследствие этого
электроды поляризуются, концентрация определяе¬
мых ионов вблизи поверхности электрода изменяется,
а продукты электролиза могут отлагаться на элек¬
тродах. Данные явления нередко нарушают соотно¬
шение между концентрацией ионов в растворе и по¬
тенциалом электрода и искажают результаты опре¬
деления.Более точен компенсационный метод, в котором
искомую разность потенциалов измеряют при отсут¬
ствии тока в цепи. Для этого пользуются специаль¬
ными приборами — потенциометрами, позволяющими
измерять потенциалы электродов с большой точ¬
ностью.§ 2. МЕТАЛЛИЧЕСКИЕ ИНДИКАТОРНЫЕ ЭЛЕКТРОДЫЭлектроды, применяемые для определения активно¬
сти (концентрации) ионов в растворах, называют ин¬
дикаторными электродами. Индикаторный электрод
соединяют с другим электродом в гальванический
элемент и измеряют разность потенциалов между
обоими электродами. Потенциал второго электрода
постоянен, и его значение известно; такие электроды
называют стандартными электродами, или электро¬
дами сравнения. Измерив разность потенциалов ме¬
жду индикаторным и стандартным электродами и
зная потенциал стандартного электрода, можно за¬
тем, если необходимо, найти потенциал индикатор¬
ного электрода.460
Требования к металлическим индикаторным элек¬
тродам различны и зависят от того, где их исполь¬
зуют, т. е. в прямой потендиометрии или при потен¬
циометрическом титровании. В обоих случаях инди¬
каторные электроды должны быть обратимыми, т. е.
их потенциал должен изменяться с изменением ак¬
тивности (концентрации) ионов металлов в растворе
в соответствии с уравнением Нернста. Дифференци¬
руя уравнение (24.1) по Ig С, получаемdEx 0,058
d lg С пОтсюда видно, что обратимый электрод должен из¬
менять потенциал при изменении концентрации ионов
в соответствии с этим уравнением. Свойство обрати¬
мости присуще не всем металлическим электродам.
Например, алюминиевый электрод необратим, так
как его поперхность покрывается оксидной пленкой;
нельзя использовать для потенциометрических изме¬
рений электроды из хрома, железа, молибдена, нио¬
бия, тантала, вольфрама и многих других элементов.Индикаторные электроды должны быть химически
устойчивы по отношению к веществам, находящимся
в растворе. Так, цинковый электрод непригоден в
кислых растворах, потому что металл будет раство¬
ряться в кислоте. Цинковым электродом нельзя вос¬
пользоваться, если в растворе содержатся катионы
более электроположительных металлов, например
меди, так как катионы меди будут вытесняться циН-
ком из раствора:Cu3+ + 2п = Си + Zn2+.Индикаторные электроды для потенциометриче¬
ского титрования должны удовлетворять еще такому
условию: потенциал должен устанавливаться быстро,
иначе титрование требует много времени. Сущест¬
вует сравнительно мало электродов, удовлетворяю¬
щих этому условию. Так, состояние равновесия ме¬
жду свинцовым, медным, висмутовым электродами и
растворами солей этих металлов устанавливается
очень медленно, и эти электроды редко применяют
для титрования.Медленное установление равновесия не препятст¬
вует применению металлических электродов в прямой461
потенциометрии. Поэтому здесь при соответствующей
постановке эксперимента можно пользоваться мед¬
ными, висмутовыми, свинцовыми, оловянными и
некоторыми другими электродами. Большинство на¬
званных электродов дает более четкие показания,
если растворить металл в ртути. Такие электроды на¬
зывают амальгамными, они имеют дополнительные
преимущества по сравнению с металлическими. Рас¬
творенный в ртути металл почти не реагирует с кис¬
лотами, так как водород на амальгаме выделяется
с большим перенапряжением по сравнению с чистым
металлом. Это позволяет применять цинковые, кад¬
миевые, свинцовые, висмутовые амальгамные элек¬
троды. Равновесие между амальгамным электродом
и раствором устанавливается быстрее, чем в случае
металлического электрода.Потенциал амальгамного электрода зависит не
только от активности ионов металла в растворе am,
но и от активности металла в амальгаме аАм:0,058 ам
Ех = Еа + —— lg—i(24.3)
и AMЕсли всегда применять амальгаму одной и той же
концентрации, то аш = К и уравнение (24.1) прини¬
мает вид:Ex = Eo + ^^r-1 <24-4)где_ 0,058 , ,Е0 - Е0 — ^ аА!Л = COnst.Поэтому при измерениях с амальгамными элек¬
тродами нельзя пользоваться табличными значения¬
ми стандартных потенциалов, а необходимо опреде¬
лять Ей экспериментально.Рассмотрим подробнее индикаторные электроды,
применяемые при титриметрическом анализе.Индикаторные электроды методов оваждения и
комплексообразования. Принцип этих методов со¬
стоит в переводе определяемых ионов в малораство¬
римые соединения или в связывании их в устойчивые
растворимые комплексные соединения. В обоих слу¬
чаях при титровании изменяется концентрация ионов
металла в растворе.462
Индикаторным электродом служит погруженная в
раствор металлическая проволока, реагирующая на
изменение концентрации одноименных ионов в рас¬
творе. Система металл — катион металла представ¬
ляет собой так называемый электрод первого рода.
Потенциал такого электрода определяется уравне¬
нием Нернста:£, = £o+^lgCM. (24.5)где Ех — потенциал в растворе с данной концентрацией ионов
металла в растворе; Еа — стандартный потенциал металла.В методах оааждения и комплексообразования
наибольшее распространение получил серебряный
электрод, который не имеет недостатков, присущих
большинству других металлических индикаторных
электродов. С серебряным электродом легко опреде¬
лять прежде всего ионы серебра, применяя для ти¬
трования, например, рабочие растворы хлорида на¬
трия, роданида калия, цианида калия и другие в со¬
ответствии с уравнениями:AgN03 + NaCl = AgCI + NaN03,AgNOj + 2KCN = KAg(CN)2 + KNO3.Известно много косвенных методов определения
основанных на применении электродов второго рода.
Металл, погруженный в раствор, содержащий мало
растворимую соль данного металла, является элек¬
тродом второго рода по отношению к аниону этой
соли. Так, серебряная проволока в растворе с осад¬
ком хлорида серебра представляет собой электрод
второго рода. Металл электрода, как и в случае элек¬
трода первого рода, изменяет потенциал в соответст¬
вии с изменением концентрации ионов серебра в рас¬
творе; однако концентрация ионов серебра в свою
очередь зависит от концентрации ионов хлора. По¬
этому в конечном счете потенциал такого электрода
изменяется с изменением концентрации ионов хлора.
Действительно£* = £<,+ 0,058 lg [Ag*J. (246).HO[Ag+] [R-] = Г1Р и [Ag+] = -jOE-,[R'lгде R~ — анион малорастворимого соединення серебра.463
ОткудаЕх = Е0 + 0,058 lg ПР - 0,058 lg [r-].(24.7)Первые два члена в правой части уравнения —
постоянные величины, тогдаНа уравнении (24.8) основаны различные косвен¬
ные определения с хлор-серебряным электродом.
К таким косвенным определениям относятся, напри¬
мер, титрование анионов хлора, брома, роданида и
других анионов, образующих малорастворимые соли
с ионами серебра.Известны также многочисленные примеры титро¬
вания с сернистосеребряным (аргентитовым) электро¬
дом. В качестве рабочего раствора можно применять,
например, раствор сульфида натрия, а определяемы¬
ми ионами могут быть катионы, образующие мало¬
растворимые сульфиды. Так, ионы цинка можно ти¬
тровать раствором сульфида натрия. В анализируе¬
мый раствор вводят небольшое количество твердого
сульфида серебра, создают щелочную среду и ти¬
труют раствором сульфида натрия. Пока в растворе
есть ионы цинка, концентрация сульфид-ионов опре¬
деляется в основном растворимостью осадка суль¬
фида цинка и изменяется мало. Концентрация суль-
фид-ионов определяет также концентрацию ионов се¬
ребра и потенциал серебряного электрода. Сильное
увеличение концентрации сульфид-ионов происходит
только после точки эквивалентности, когда весь цинк
осажден; точку эквивалентности фиксируют по рез¬
кому уменьшению потенциала серебряного элек¬
трода.В методах осаждения и комплексообразования
применяют также индифферентный платиновый элек¬
трод. Платиновый электрод наиболее широко исполь¬
зуют в методах окисления — восстановления; его по¬
тенциал является функцией соотношения концентра¬
ций окисленной и восстановленной форм:Ео + 0,058 lg ПР = К,
Я* =/С- 0,058 lg [R~].(24.8)(24.9)464
Некоторые косвенные определения методом оса¬
ждения и комплексообразования также основаны на
применении платинового электрода. В анализируе¬
мый раствор вводят окислительно-восстановительную
систему, т. е. раствор, содержащий ионы какого-либо
металла в двух степенях окисления. Далее в раствор
погружают платиновую проволоку и приступают к ти¬
трованию. Рабочий раствор подбирают так, чтобы он
реагировал с одним из ионов окислительно-восстано¬
вительной системы, но чтобы это взаимодействие про¬
исходило только после завершения основной реакции
между определяемым веществом и рабочим раство¬
ром. В результате этого взаимодействия нарушается
первоначальное соотношение концентраций окислен¬
ной и восстановленной форм, и в соответствии с этим
в точке эквивалентности изменяется потенциал пла¬
тинового электрода.Описанный метод применяют, например, при ти¬
тровании катионов алюминия раствором фторида на¬
трия:А13* + 6F" = AlFjj'.Индицирующей окислительно-восстановительной
системой служит раствор солей железа(II) и (III).
По окончании титрования алюминия избыточные
ионы фтора связывают ионы железа(III) во фторид-
ный комплекс:Fe3+ + F' = FeF2+.Это уменьшает соотношение [Fe3+] : [Fe2f], и в
связи с этим потенциал платинового электрода сни¬
жается.Последовательность взаимодействия ионов фтора с
ионами обоих металлов определяется их константа¬
ми устойчивости: для A1F|‘ и FeF|’ логарифмы этих
констант равны 20,7 и 16,1 соответственно.Примером применения индицирующих окислитель-
но-восстановительных систем в реакциях осаждения
может служить титрование катионов металлов рас¬
твором гексациаиоферрата (II) калия. Последний
образует с катионами многих металлов малораство¬
римые осадки; такие осадки образуют, например,
катионы меди, свинца, кадмия, цинка, галлия,
индия и др. Окислительно-восстановительную систему465
создают, прибавляя к рабочему раствору K4Fe(CN)6
небольшое количество раствора K-iFe(CN)s. Погру¬
женный в раствор платиновый электрод реагирует на
изменение соотношения концентраций окисленной н
восстановленной форм.[Fe(CN)»-JЕХ = Е0 + 0,058 lgРассмотрим пример определения цинка. Цинк реа¬
гирует с K4Fe(CN)6 по уравнению3ZnCl2 + 2K*Fe(CN)e — KzZnatFetCNM, + 6КС1.Малорастворимый осадок образуется также при
взаимодействии цинка с раствором K3Fe(CN)e
3ZnCl2 + 2KaFe(CN)e= Zn3[Fe(CN),]2 + 6KC1.Осадок KaZri3[Fe(CN)6b растворим меньше, чем
соединение Zn3[Fe(CN)6l2. Поэтому до точки экви¬
валентности концентрация анионов Fe(CN)^~ меньше,
чем концентрация анионов Fe(CN)|", и в соответ¬
ствии с установившимся соотношением концентраций
этих ионов потенциал платинового электрода прини¬
мает определенное значение. После точки эквива¬
лентности избыток введенного рабочего раствора
содержит соль K.4Fe(CN)6 в значительно большей
концентрации, чем соль K3Fe(CN)6; таким образом,
резко уменьшается отношение концентраций окислен¬
ной и восстановленной форм, и скачок потенциала
электрода указывает на точку эквивалентности.Индикаторные электроды в методах окисления—
восстановления. При окислительно-восстановительном
титровании изменяется соотношение концентраций
окисленной и восстановленной форм определяемого
иона в растворе. Так, при титровании ионов желе¬
за (II) перманганатом концентрация ионов Fe2+
уменьшается, а концентрация ионов Fe3+ возрастает.
После точки эквивалентности в растворе практически
отсутствуют ионы железа (II), но введенный избыток
перманганата создает окислительно-восстановитель¬
ную систему МпО^/Мп2*. Потенциал индикаторного
электрода должен фиксировать изменение соотноше¬
ния концентраций окисленной и восстановленной
форм. Для этого обычно применяют платиновый элек¬466
трод, потенциал которого в растворе определяется
уравнением (24.9).Применяют также электроды из других индиффе¬
рентных металлов, например из палладия, золота
и др. В отличие от индикаторных электродов методов
осаждения и комплексообразования здесь нет равно¬
весия между металлом электрода и ионами этого ме¬
талла в растворе. Индикаторный электрод служит
только проводником электронов, приобретая больший
или меньший потенциал в зависимости от интенсив¬
ности притяжения электронов окисленной формой,
что в свою очередь определяется природой находя¬
щихся в растворе ионов и их концентрацией.§ 3. ИОНСЕЛЕКТИВНЫЕ МЕМБРАННЫЕ ЭЛЕКТРОДЫИонселективные электроды — это электрохимические
полуэлементы, в которых разность потенциалов на
границе раздела фаз электродный материал — элек¬
тролит зависит от концентрации (точнее, от активно¬
сти) определяемого иона в растворе. Электродный
материал представляет собой твердую или жидкую
мембрану, в которую введено вещество, способное от¬
щеплять подлежащие определению ионы. Эти ионы
при соприкосновении с водой или с водным раство¬
ром электролита способны переходить в него. Ино¬
гда, наоборот, ионы из раствора проникают в мем¬
брану. В результате поверхность мембраны приобре¬
тает заряд, противоположный заряду перешедших в
раствор ионов, и на границе раздела фаз возникает
потенциал, значение которого зависит от активности
данных ионов в растворе. Если мембрана разделяет
два раствора с различной активностью, например од¬
нозарядных ионов, тогда потенциал определяется
уравнением Нернста:ПТ пг£ = £' + -^tn -Jjr, (24.10)где а' и а" — активности ионов в обоих растворах.Обычно в одном из растворов сохраняют актив¬
ность данных ионов постоянной; если, например, а"—
постоянная величина, тогдаЕ = К + In а'. (24.il)467
т. е. потенциал электрода зависит только от активно¬
сти а.К числу мембранных электродов относят прежде
всего давно известный стеклянный электрод, широко
применяющийся для определения активности ионов
водорода — измерения pH. В последние годы предло¬
жено много других мембранных электродов, посред¬
ством которых измеряют активность (концентрацию)
различных ионов и проводят потенциометрическое ти¬
трование. Известны, например, электроды для опре¬
деления ионов натрия, калия, кальция, магния, цин¬
ка, свинца, лантана, хлора, брома, иода, фтора, ни¬
трата, перхлората.Твердые мембранные электроды могут быть гомо¬
генными и гетерогенными. В гомогенных электродах
материалом для изготовления мембран служат мало¬
растворимые соединения, содержащие определяемые
ионы. Так, для определения ионов серебра или суль-
фид-ионов применяют мембрану из сульфида сереб¬
ра. Мембраны из фторидов редкоземельных элемен¬
тов, например из фторида лантана, пригодны для
определения ионов фтора. Гетерогенные мембраны
содержат какой-либо инертный связующий материал
(матрицу) для придания электроду необходимой ме¬
ханической прочности. В качестве такой инертной
матрицы используют парафин, каучук, полиэтилен,
агар-агар и другие материалы. Диспергированные в
них соли, например CaCr04, BaS04, Agl, Ag2S, ThF4t
обеспечивают возможность определения ионов, вхо¬
дящих в состав названных солей.Жидкие мембраны готовят из жидких или твер¬
дых ионитов или их растворов в подходящих органи¬
ческих растворителях, не смешивающихся с водой.
Так, ионы кальция можно определять посредством
электрода на основе кальциевой соли эфира фосфор¬
ной кислоты Активными группами электродов, имею¬
щих нитратную или перхлоратную функцию, т. е. чув¬
ствительных по отношению к ионам NO3 или CIO4,
являются нитраты фенантролинатов никеля
Ni(РЬеп)з(ЫОз)2 или перхлораты фенантролинатов
железа Fe(Phenb(C104)2 и т. д.Ионоселективные электроды очень чувствительны:
пределы обнаружения ионов посредством этих элек¬
тродов равны 10-5— 10-7 М (а иногда и до 10~19 М),468
а минимальное количество пробы, необходимое для
одного измерения, составляет 0,05—1 мл.Мембранные электроды при некоторых условиях
проявляют селективность по отношению к ионам оп¬
ределенного вида и пригодны для измерения их ак¬
тивности (концентрации) даже в растворах, содержа¬
щих посторонние ионы, которые не входят в состав
мембраны. Селективность мембраны в этом случае
зависит от двух основных факторов. Прежде всего —
это способность ионов мембраны обмениваться с по¬
сторонними ионами раствора. Пусть, например, в
мембране содержатся ионы М', а в растворе кроме
этих ионов находятся посторонние ионы М". Тогда
селективность мембраны по отношению к ионам М'
характеризуется степенью прохождения реакции об¬
мена:М" + М' М" -ь М'. (24.12)рас- мем- мем- рас¬
твор брана брана творСелективность тем меньше, чем больше состояние
равновесия реакции (24.12) сдвинуто вправо, т. е. чем
больше константа равновесия этой реакции.„ 1М"]м [М']р[М"]р [М'Ь(24.13)Селективность зависит также от соотношения по¬
движностей ионов М" и М' в мембране, уменьшаясь
с увеличением этого соотношения.Соотношение _ин. = (24.14)иМ'где i/M» и {7М, — подвижности ионов М" и М' в мембране, К —
константа равновесия реакции (24.13),называют коэффициентом селективности иона М' по
отношению к ионам М". Уравнение для потенциала
мембраны имеет вид:Е = Ео + ~ ъ Ы' + *М'/1й»вМ')- (24.15)Коэффициент селективности — количественная ме¬
ра относительной чувствительности электрода к двум469
ионам М' и М". Так, коэффициент селективности
стеклянного электрода, применяющегося для опреде¬
ления ионов натрия, по отношению к ионам калия
равен 3,6-10-4. Это значит, что раствор с концентра¬
цией ионов натрия 3,6-10-4 М будет давать такой же
эффект, что и раствор ионов калия с концентрацией1 М.Коэффициенты селективности ионселективного
электрода с жидкой мембраной для определения
ClOi по отношению к 1”, NO3, Вг”, F' и СГ равны
соответственно 1,2-1Q-2; 1,5-10-3; 5,6-10-4; 2,5-10~4 и
2,2-10~4. Из этих данных видно, что электрод являет¬
ся высокоселективным и может с успехом применять¬
ся для определения ионов CiOi в присутствии пере¬
численных анионов.Пределы концентрации ионов, в которых элек¬
тродная функция большинства ионселективных элек¬
тродов подчиняется уравнению Нернста, находятся
обычно в интервале от 1 М до 10-7 М.Мембранные электроды широко применяют для
измерения активности ионов в растворе методом пря¬
мой потенциометрии, например для определения
анионов CIO”, NOa, Г, Br”, Cl”, SCN”, катионных
и анионных поверхностно-активных веществ (ПАВ)
и др. Для аналитической химии представляет интерес
возможность определения ионов путем потенциоме¬
трического титрования по методам окисления — вос¬
становления, осаждения, комилексообразования.
В этом случае нет необходимости пересчитывать ак¬
тивность на концентрацию, так как точку эквивалент¬
ности при титровании устанавливают путем наблю¬
дения за изменением концентрации, а концентрация
и активность при постоянной ионной силе раствора
изменяются в одинаковой степени.Ниже описаны принципы действия и конструкция
стеклянного и некоторых других мембранных элек¬
тродов.Стеклянный электрод. Действие стеклянного элек¬
трода основано на возникновении разности потенциа¬
лов между тонкой стеклянной пленкой и водными
растворами с определенной концентрацией (актив¬
ностью) водородных ионов. Водородная функция
стеклянного электрода, т. е. зависимость разности ио-470
тенциалов от активности ионов водорода в растворе,
обусловлена их диффузией в стекло. К одному концу
открытой стеклянной трубки припаивают стеклянную
пленку из специального сорта стекла толщиной в не¬
сколько сотых миллиметра. В других конструкциях
выдувают на конце трубки шарик с тонкими стенка¬
ми. Обычно применяют легкоплавкое стекло, в состав
которого входит 72 % кремниевой кислоты, 6 % оксида
кальция и 22 % оксида натрия. Внутрь трубки нали¬
вают стандартный раствор кислоты, например 0,1 М
раствор хлороводородной кислоты, и погружают
туда какой-нибудь стандартный электрод, например
хлорсеребряный. Трубку с раствором кислоты и стан¬
дартным электродом погружают в исследуемый рас¬
твор. Последний соединяют электролитическим клю¬
чом со стандартным каломельным электродом и по¬
лучают цепь:Ag | AgCl | 0.ШНС11 Стекло | Н+ | КС\иас | Hg2Cl2 | Hg.Электродвижущая сила этой цепи связана с ак¬
тивностью ионов водорода в растворе уравнением:Ех = К + 0,058 lg ан*, (24.16)где К — постоянная, которая зависит от сорта стекла и устрой¬
ства электрода.Электропроводность стеклянной пленки очень ма¬
ла, ее сопротивление может составить несколько мил¬
лионов Ом. Поэтому проходящий в цепи ток перед
измерением необходимо усилить.Стеклянный электрод имеет ряд характерных осо¬
бенностей. Он пригоден для измерения pH в области
от 0 до 12—13. Электрод нечувствителен к различ¬
ным примесям в растворе (за исключением ионов
фтора), он не отравляется, им можно пользоваться
для измерения pH в растворах, содержащих сильные
окислители и восстановители, а также катионы раз¬
личных металлов. Равновесие между электродом и рас¬
твором устанавливается очень быстро. Недостатком
является хрупкость стеклянной пленки. Для защиты
от повреждений электрод помещают внутрь откры¬
той снизу стеклянной толстостенной трубки большого
диаметра. Такой электрод, прилагаемый к комплек¬
ту электронных рН-метров, показан на рис. 24.4.471
Рис. 24.4. Стеклянный электродЭлектрод нельзя погружать в кислые
фторидные растворы.Константа К в уравнении (24.16)
зависит от сорта стекла, поэтому
стеклянный электрод сначала калиб¬
руют по нескольким буферным рас¬
творам с определенной концентрацией
водородных ионов.Новый стеклянный электрод необ¬
ходимо перед применением выдержать
некоторое время в воде или в 0,01 М
растворе НС1. При этом ионы натрия,
содержащиеся в стекле, переходят в раствор, а их
место занимают ионы водорода из раствора. Возни¬
кающий на поверхности стекла гель имеет толщинуI —100 нм и определяет электрохимическое поведение
электрода.В щелочных растворах, содержащих ионы щелоч¬
ных металлов, стеклянный электрод приобретает на¬
триевую функцию, т. е. он реагирует на изменение
концентрации ионов натрия. Поэтому, измеряя pH в
щелочных растворах, необходимо вносить экспери¬
ментальные поправки на содержание этих ионов в
растворе.Специальный натрий-селективный электрод мож¬
но изготовить из другого сорта стекла, содержащегоII % Na20, 18% А1203 и 71 % Si02. Он избирательно
реагирует на ионы натрия в том случае, если значе¬
ние pH измеряемого раствора примерно на четыре по¬
рядка выше соответствующего значения pNa.Известны также стеклянные электроды для опре¬
деления ионов лития, калия, серебра состава: 15 %
Li20, 25% А1203, 60 % Si02 (литиевый электрод);
27 % Na20, 5 % А1203, 68 % Si02 (калиевый элек¬
трод); 28,8% Na20, 19,1 % А1203, 52,1 % Si02 (сереб¬
ряный электрод). Коэффициенты селективности та¬
ких электродов в присутствии ионов калия, натрия и
ионов водорода равны соответственно 10-3 (Li),
5-10-2 (К) и 10-5 (Ag). Однако коэффициент селек¬
тивности, например, литиевого электрода по отноше¬
нию к ионам натрия равен 0,3, т. е. эти ионы мешают
определению лития.472
Твердые мембранные электроды на основе суль¬
фида серебра. Спрессованная таблетка из сульфида
серебра может служить в качестве ионообменного
мембранного электрода для измерения активности
ионов серебра в растворе. Ионы серебра в решетке
Ag2S отличаются высокой подвижностью и при по¬
гружении такой таблетки в раствор электролита или
в воду способны переходить в него, образуя на обеих
сторонах поверхности мембраны двойной электриче¬
ский слой: прилегающий к поверхности слой раство¬
ра заряжен положительно за счет ионов серебра, а
сама поверхность имеет отрицательный заряд, обус¬
ловленный анионами серы. Если мембрана разделяет
два раствора с одинаковой активностью ионов сереб¬
ра, потенциалы на обеих сторонах ее поверхности
одинаковы. При различной активности ионов серебра
в обоих растворах возникает разность потенциалов,
определяющаяся уравнением Нернста:где а — активность ионов серебра в растворах, разделяемых
мембраной.Обычно электрод конструируют так, чтобы актив¬
ность ионов серебра в одном из растворов была по¬
стоянной. Тогда из уравнения (24.17) следует:т. е. электрод реагирует на изменение активности
ионов серебра в растворе.Мембрана из сульфида серебра пригодна также
для измерения активности сульфид-ионов. Произве¬
дение активности Ag2S равно:(24.17)(24.18)(24.19)Из уравнения (24.19) следует:(24.20)473
Подставляя значение aXg+ из уравнения (24.20) в
уравнение (24.18), получаем:RT ПА1/2
£ = £' + Jli lnii4— (24.21)Р п V2ИЛИ„ с, , RT . /=-г RT .Е = Е +—^-1пуПА ln as2-но так как второй член правой части уравнения(24.21)—величина постоянная, можно записатьЕ=Е" ~irlnas2- (24-22>т. е. потенциал изменяется в зависимости от вели¬
чины активности сульфид-ионов в растворе.Мембраны из сульфида серебра характеризуются
большой ионной проводимостью, они хорошо прово¬
дят ток по сравнению, например, с другими нераство¬
римыми соединениями серебра. Поэтому сульфид се¬
ребра был использован в качестве матрицы для из¬
готовления некоторых других электродов. Например
мембрана, содержащая наряду с Ag2S какой-либо га-
логенид серебра, представляет собой электрод на га-
логенид-ионы. Активность галогенида влияет в соот¬
ветствии с равновесиемAg++ НаГ = AgHalна активность свободных ионов серебра и на элек¬
тродный потенциал по уравнению:птЕ = Е' -р- In аНа|. (24.23)Матрица из Ag2S, в которую введены сульфиды
других металлов, например, сульфиды меди, свинца
или кадмия, работает как электроды на ионы назван¬
ных металлов. Так, изменение активности ионов кад¬
мия в растворе приводит к изменению активности
сульфид-ионов в соответствии с уравнением:nA('<j<;V-= „ • <24’24)S Cd2+474
В свою очередь это приводит к изменению актив¬
ности ионов серебраПодставляя в уравнение (24.25) вместо as2- его
значение из уравнения (24.24), получаемПервый множитель правой части уравнения (24.26) —
величина постоянная. Поэтому, подставляя значение
aAg* из уравнения (24.26) в уравнение (24.18) и объ¬
единяя постоянные величины, получим:Таким образом, мембрана Ag2S—CdS представляет
собой электрод, посредством которого можно изме¬
рять активность ионов кадмия.Мембранным ионселективный электрод с фтор ад-
ной функцией. В кристалле фторида лантана боль¬
шой подвижностью обладают ионы фтора. Поэтому
на поверхности мембраны из фторида лантана при
погружении ее в воду или в раствор электролита так¬
же образуется двойной электрический слой. Слой
раствора, прилегающий к поверхности мембраны, за¬
ряжен отрицательно, а поверхность мембраны — по¬
ложительно за счет находящихся там ионов лантана.
Мембрана функционирует как электрод для измере¬
ния активности ионов фтора.Жидкий мембранный электрод с кальциевой функ¬
цией. Ионы кальция играют большую роль в важных
физиологических процессах живых организмов. Про¬
блема измерения активности этих ионов в биологиче¬
ских жидкостях была решена после разработки
ионселективного жидкого мембранного электрода с
кальциевой функцией. Устройство одного из таких
электродов показано на рис. 24.5. Нижний конец от¬
крытой стеклянной трубки затянут целлюлозной плен¬
кой, проницаемой для всех ионов и служащей для удер¬
жания жидкой мембраны. Последняя представляет(24.25)(24.26)(24.27)475
5 ■7-ftЯ«с. 24.5. Ионселективный электрод
с кальциевой функцией:7— к потенциометру; 2— экранированный про¬
вод; 3 — полиэтиленовый корпус; 4 — сереб¬
ряная проволока; 5 —жидкий ионит; 6— ша¬
рик из плавленного сорбита, пропитанный
раствором хлорида кальция; 7—резиновое
кольцо; 5 —мембранасобой раствор кальциевой
соли додецилфосфорной кис¬
лоты в додецилфенилфосфо-
нате.Электрический контакт с
жидкой мембраной осуществ¬
ляется посредством серебря¬
ной проволоки с шариком из
плавленного сорбита, пропи¬
танного водным раствором
хлорида кальция. Электрод
погружают в исследуемый
раствор; его потенциал измеряют по отношению к
каломельному электроду сравнения посредством цепиAg | AgCl I CaCl, I Мембрана | И“^®уый | КС1 | Hg2CI2 | Hg.1,3 смвПотенциал такого мембранного электрода зависит
от активности ионов кальция в соответствии с урав¬
нениемЕ = К + -^\паСа-(24.28)Жидкие мембранные электроды — растворы ион¬
ных ассоциатов в органических растворителях. В ка¬
честве растворителей используют обычно различные
эфиры, например октиловый или дециловый эфиры
фосфорной кислоты, дибутилфосфат и др. Потенциал-
образующими ионами являются катионы или анио¬
ны ионных ассоциатов, т. е. электрод с катионоанион¬
ным ассоциатом чувствителен и к катионам, и к
анионам, входящим в состав ассоциата. Для умень¬
шения растворимости ассоциата в водной фазе, т. е.
для удержания его в основном в фазе органического
растворителя, обычно применяют ассоциаты потен-
циалобразующих ионов с противоионами большой
молекулярной массы, что обеспечивает достаточно476
большую гидрофобность ассоциата. В качестве при¬
мера мембраны с ионным ассоциатом для определе¬
ния анионов можно привести ассоциаты крупных
1,10-фенантролинатов никеля, железа (и некоторых
других металлов) NifPhen)!*, Fe(Phen)3* с анионами
многих кислот. Селективность таких анионочувстви-
тельных электродов определяется следующим рядом:сю: > scn" > г > bf; > no: > Br' > cr > f-,4 ч лкоторый отвечает ряду значений энергии гидратации
этих ионов.На основании этого ряда можно установить воз¬
можность определения одного из анионов в присут¬
ствии других. Так, определению, например, нитрат-
ионов мешают все анионы, стоящие в ряду влево от
него, и не мешают те, которые расположены вправо
от него.§ 4. ЭЛЕКТРОДЫ СРАВНЕНИЯЭлектроды сравнения служат эталонами, по отноше¬
нию к которым измеряют потенциал индикаторного
электрода. Поэтому любой индикаторный электрод
в принципе может служить также электродом сравне¬
ния, если создать условия, при которых потенциал та¬
кого электрода остается неизменным в процессе тит¬
рования. Для этого можно, например, поместить ин¬
дикаторный электрод в раствор, одинаковый по
составу с титруемым раствором, но отделенный от
последнего пористой перегородкой или электролитиче¬
ским ключом. Иногда можно даже помещать элек¬
трод сравнения непосредственно в титруемый раствор.
Так поступают в окислительно-восстановительных ме¬
тодах титрования. Индикаторным электродом служит
платиновый электрод, а электродом сравнения, напри¬
мер, вольфрамовый электрод. Поверхность последнего
быстро покрывается в растворе оксидной пленкой.
Такой электрод реагирует на изменение pH раствора,
но не на изменение соотношения концентраций окис¬
ленной и восстановленной форм определяемого эле¬
мента. Обычно титруемый раствор содержит большой
избыток свободной кислоты, и кислотность этого рас¬
твора практически не изменяется в процессе титрова¬
ния. В этих условиях вольфрамовый электрод выпол¬477
няет роль электрода сравнения, имея все время один
и тот же потенциал.Для потенциометрического титрования раствора
хлороводородной кислоты раствором гидроксида нат¬
рия в качестве электрода сравнения можно взять
хлорсеребряный электрод Ag|AgCl. При титровании
сильно изменяется только pH раствора, но не концен¬
трация ионов хлора. Поэтому концентрация ионов се¬
ребра и потенциал хлорсеребряного электрода оста¬
ются неизменными.Специально приготовляемые стандартные элек¬
троды сравнения имеют ряд преимуществ по сравне¬
нию с описанными. Величины потенциалов этих элек¬
тродов хорошо проверены и известны. Это позволяет
применять их в прямой потенциометрии, когда необ¬
ходимо определять абсолютное значение потенциала
индикаторного электрода, а не только изменение этой
величины в процессе титрования. Все эти электроды
являются электродами второго рода, содержащими
раствор электролита с анионом малорастворимой
соли в большой концентрации; часто применяют на¬
сыщенный раствор такой соли. Такое устройство
обеспечивает постоянство потенциала электрода даже
в том случае, если через него протекает ток.В самом деле, при пропускании тока металл элек¬
трода окисляется и переходит в раствор и виде ионов
или, наоборот, ионы металла восстанавливаются и от¬
лагаются на электроде в виде атомов. В обоих слу¬
чаях концентрация ионов металла в растворе изме¬
няется и потенциал электрода не остается постоян¬
ным. Введение электролита в большой концентрации
устраняет это нежелательное явление. Электрод вто¬
рого рода представляет собой систему, в котопой кон¬
центрация ионов металла определяется уравнением(м]=ТаТ (24'29)Предположим, что на электроде сравнения при
прохождении тока происходит процесс окисленияМ = М+ + е~.Но в присутствии избытка анионов А- образовав¬
шиеся катионы металла переходят в твердую фазу478
МА. Концентрация анионов А при этом изменяется
мало. Поэтому в соответствии с уравнением (24.29)
концентрация ионов металла, а следовательно, и по¬
тенциал электрода остаются неизменными.Каломельный электрод. Наиболее распространен
насыщенный каломельный электрод. Он удобен по¬
тому, что не нужно принимать мер предосторожности
против испарения воды. В электродах же с 0,1 Af или
1М растворами хлорида калия концентрация КС1 из¬
меняется от постепенного испарения воды. Потенциал
каломельного электрода определяется выражениемЕХ = Е0 - 0,058 lg[cr], (24.30)ш- щ jp'J-2-ljz.mi%ш.Рис. 24.6. Каломельные электроды сравнения:/ — ртуть; 2 —паста; 3 — раствор хлорида калия; 4 — электролитический ключРис. 24.7. Сочетание стандартного и индикаторного электродов:J — 4 — см. рис. 24.0; 5, 6—провода; 7—пробка; 8— широкая пробиркаКонцентрация ионов хлора в насыщенном рас¬
творе хлорида калия постоянна, следовательно, по¬
тенциал каломельного электрода также постоянен.Для приготовления каломельного электрода необ¬
ходимы чистая ртуть, каломель и хлорид калия. Тех¬
ника его приготовления подробно описана в руковод¬
ствах к практическим работам по аналитической хи¬
мии. Электролитические сосуды для каломельных
электродов бывают различной формы (рис. 24.6).Электрод «б» удобно применять для потенциомет¬
рических титрований. На рис. 24.7 представлено со¬
четание каломельного или другого стандартного и ин¬
дикаторного электродов в одном сосуде. Описанная
конструкция удобна для окислительно-восстанови¬479
тельных титрований; в этом случае индикаторный
электрод сделан из платиновой проволоки.Ртутносульфатный электрод. Электрод готовят
аналогично каломельному. Вместо каломели берут
сульфат ртути, а электролитом служит 1 М раствор
серной кислоты.Хлорсеребряный электрод. Чтобы приготовить та¬
кой электрод, серебряную проволоку погружают в на¬
сыщенный раствор хлорида калия, содержащий не¬
большое количество хлорида серебра. Лучше хлорид
серебра предварительно расплавить и серебряную
проволоку окунуть в расплавленную массу. После
охраждения на электроде получается равномерный
слой хлорида серебра, который хорошо проводит
электрический ток. Электрод помещают затем в насы¬
щенный раствор хлорида калия.Хлорсеребряные электроды входят в некоторые
комплексы измерительных приборов.Учебное пособиеПИЛИПЕНКО Анатолий Терентьевич
ПЯТНИЦКИЙ Игорь ВладимировичАналитическаяхимияКНИГА ПЕРВАЯРедактор В. Л. Абрамова
Художественный редактор Л. А. Леонтьева
Технические редакторы Н. Ю. Белякова,Ф. М. СкитинаКорректоры Н. А. Иванова, М. А. Ивлиева
ИБ № 2310Сдано в набор 23.08.89. Подписано в печать 24.01.90. Формат в4X1 Ов'/зi
Бумага типографская № 2. Гарнитура литературная. Печать высокая.
Уел. печ. л. 25,20. Уел. кр.-отт. 25,20. Уч.-изд. л. 24,48. Тираж 43 000 экэ.
Заказ 284. Цена 1 р. 10 к.Ордена «Знак Почета» издательство «Химия». 107076. Москва, Стро¬
мынка, 21, корп. 2Ленинградская типография № 2 головное предприятие ордена Трудового
Красного Знамени Ленинградского объединения «Техническая книга»
им. Евгении Соколовой Государственного комитета СССР по печати.
198052. г. Ленинград. Л-55. Измайловский проспект. 29.