













































































































































































































































































































































Автор: Гершкович А.А. Кибирев В.К.
Теги: органическая химия химия биология монография органический синтез издательство наукова думка биоорганическая химия химический синтез пептиды
ISBN: 5-12-003103-Х
Год: 1992




АКАДЕМИЯ НАУК УКРАИНЫ
ИНСТИТУТ БИООРГАНИЧЕСКОЙ ХИМИИ И НЕФТЕХИМИИ
А. А. ГЕРШКОВИЧ
В. К. КИБИРЕВ
ХИМИЧЕСКИЙ
СИНТЕЗ
ПЕПТИДОВ
КИЕВ НАУКОВА ДУМКА 1992
УДК 547.964.4
Химический синтез пептидов / Гершкович А. А., Кибирев В. К.;
Отв. ред. Серебряный С. Б.; АН Украины. Ин-т биоорган, химии и нефте-
химии.— Киев : Наук, думка, 1992.— 360 с.— ISBN 5-12-003103-Х.
Монография посвящена современным методам химического синтеза
пептидов — регуляторов биохимических процессов в организме. Приво-
дятся методики синтеза важнейших реагентов для образования пептидной
связи и защиты функциональных групп аминокислот, описываются совре-
менные методы синтеза защищенных пептидов и способы их деблокирования.
Рассматриваются основные методы твердофазного синтеза пептидов, а так-
же способы конъюгации пептидов с белками. Осуществлена классификация
реагентов по принципу химического строения. Кратко рассмотрены меха-
низмы активации карбоксильной и аминогрупп и процессов, происходящих
при удалении защитных групп. В таблицах представлены характеристики
многочисленных производных аминокислот и полупродуктов для синтеза
пептидов.
Для научных работников химического и биологического профилей, а
также студентов, аспирантов и преподавателей вузов.
Ил. 10. Табл. 52. Библиогр.: 336—353 (537 назв.).
Ответственный редактор С. Б. Серебряный
Утверждено к печати ученым советом
Института биоорганической химии и нефтехимии АН Украины
Все права принадлежат издательству «Наукова думка». Любое использование
этого издания или его элементов/фрагментов, т. е. копирование, тиражирование,
распространение и т. д., возможно только при наличии предварительного пись-
менного соглашения с издателем.
Адрес издательства «Наукова думка»: Украина, 252601, Киев 4, ул. Терещен-
ковская, 3.
All rights reserved. No part of this issue may be reproduced by any mechanical,
photographic or electronic process or in the form of a phonographic recording, nor
may it be stored in aretrieval system, transmitted or otherwise copied for public or
private use without written permission of the Naukova Dumka Publishers.
Address of the Publishers: Ukraine 252601, Kiev 4, Tereshchenkivska St., 3.
1705000000-329 279 9„
Г 221-92
ISBN 5-12-003103-X
© А. А. Гершкович, В. К. Кибирев, 1992
СПИСОК СОКРАЩЕНИЙ
АК — аминокислота ного синтеза пептидов
БТФА — (трис)трифторацетат бора с использованием из- бытка смешанных ан-
ДМСО — диметил сульфоксид гидридов (REMA)
дцгк —N, N' - дици к л огекси л- ТГФ — тетрагидрофуран
карбодиимид (DCC) ЭЭДХ — М-этиЛоксикарбонил-2-
ДЦГМ — М,ЬГ-дициклогексил- мочевина этокси-1,2-дигидрохи- нолин (EEDQ)
ОБТ — 1-оксибензтриазол (НОВТ) ТФУ — трифтор уксусная кис- лота
ПСА-метод — метод последователь- ТА — тиоанизол
АМИНОКИСЛОТЫ
Ala — аланин Leu — лейцин
Arg — аргинин Lys — лизин
Asn — аспарагин Met — метионин
Asp — аспарагиновая кислота Phe — фенилаланин
Cys — цистеин Pro — пролин
Gin — глутамин Ser — серин
Glu — глутаминовая кислота Thr — треонин
Gly — глицин Trp — триптофан
His — гистидин Tyr — тирозин
He — изолейцин Vai — валин
Ас — ацетил
АсОН — уксусная кислота
Ас2О — уксусный ангидрид
АОС — /ире/и-амилоксикарбо- нильная защитная группа
В эс — трет-бутилоксикарбо- нильная защитная группа
ВиОН Ви* — бутанол — трет-бутил
Bz — бензоил
Bzl — бензил
BOP-CI — N,N'-6wc-(2-okco-3-ok- сазолидинил)фосфоро- диамидохлорид
(ВОС)2О — ди-/пре/п-бутилпиро- карбонат
3
DMAP — 4-диметиламинопири- NMM
дин Nps
Et8N — триэтиламин
ЕЮАс — этилацетат Nsp
EtOH — этанол
EEDQ — М-этилоксикарбонил-2- NCA
этокси-1,2-дигидрохи-
нолин OBu*
Fmoc — 9-флуоренилметилокси- OBzl
карбонильная группа OEt
Form — формильная группа OMe
MBS — метоксибензолсульфо- ON В
нильная группа
MTS — мезитилен-2-сульфо- ONp
нильная группа
MdS — 4-метокси-2,6-диметил- OPfp
сульфонильная группа
Mtb — 2,4,6-триметоксибен- Ph
золсульфонильная TFA
группа
Mb — 4-метокси-2,3,6-три- Tos
метилбензо л сульфо- Trt
нильная группа Z
Me — метил
MeOH — метанол
— N-метилморфолин
'— о-нитрофенилсульфе-
нил
— 2-нитро-4-сульфофени-
ловый эфир
— N-карбоксиангидрид
аминокислоты
— трет-бутиловый эфир
— бензиловый эфир
— этиловый эфир
— метиловый эфир
— п-нитробензиловый
эфир ,
— n-нитрофен иловый
эфир
— пентафторфениловый
эфир
— фенил
*— трифторацетильная
группа
— тозильная группа
— тритильная группа
— бензилоксикарбониль-
ная группа (КБЗ)
ВВЕДЕНИЕ
Известный чехословацкий специалист Й. Рудингер в 1963 го-
ду писал, что среди химиков-органиков широко бытует мне-
ние, якобы синтез пептидов является искусством или даже та-
инством со своей собственной символикой, языком и ритуалом,
нечто чуждое интересам тех, кто работает в других областях.
Для нас это высказывание представляет чисто исторический
интерес и сегодня ни у кого не вызывает сомнения, что хими-
ческий синтез веществ пептидно-белковой природы является
специальным разделом синтетической органической химии. Од-
нако практика показывает, что даже опытный химик-органик,
приступивший к освоению этой специальной области, испыты-
вает трудности, пока не приобретет определенные навыки в син-
тезе и очистке пептидов и не научится выбирать в большом ар-
сенале методов именно те, которые наиболее эффективны в каж-
дом отдельном случае, т. е. пока не овладеет искусством синтеза
пептидов.
За истекшие четверть века после высказывания Й. Ру-
дингера интерес к синтезу пептидов и белков не только не сни-
зился, а продолжает непрерывно расти. Для того чтобы понять
причину этого, необходимо перечислить задачи современной
биологии (а точнее — молекулярной биологии, биоорганичес-
кой химии, энзимологии, иммунологии, фармакологии и др.)
и биотехнологии, которые решаются с использованием синтети-
ческих пептидов.
1. Синтез отдельных фрагментов белков и природных пеп-
тидов позволяет устанавливать функционально важные участ-
ки, определяющие биологическую активность этих соединений:
«активные центры» ферментов, гормонов, нейропептидов; анти-
генные детерминанты антител; участки, ответственные за связы-
вание с рецепторами, мембранами, белками и нуклеиновыми
кислотами; «сигнальные пептиды», ответственные за транспорт
секреторных белков из клетки, и др.
5
2. Синтез модифицированных пептидов (укороченные или
удлиненные последовательности, замены одних аминокислот
другими, замена L-аминокислот на D-изомеры, циклизация лй-
нейных последовательностей, замена пептидной связи —
CONH — другими группировками, изменение порядка распо-
ложения аминокислот в цепи на обратный, использование ами-
нокислот небелковой природы или несуществующих в природе
аминокислот, и др.) позволяет изучать зависимость биологи-
ческой активности полипептидов от их аминокислотной после-
довательности и конформации в растворе, моделировать меха-
низмы их биологического действия и получать аналоги, более
активные, чем природные пептиды.
3. Синтез субстратов и ингибиторов протеолитических фер-
ментов — путь к изучению тонких механизмов ферментативно-
го катализа.
4. Получение набора модельных пептидов для ра работки
физических методов изучения конформации пептидов и белков:
ЯМР-, УФ-, ИК- и флюоресцентная спектроскопия, дисперсия
оптического вращения и круговой дихроизм (КД), рентгенсГ-
структурный анализ и др.
5. Как результат всех перечисленных выше подходов, дости-
жения пеПтидной химии находят самое широкое применение в
медицине и ветеринарии. «Мы считаем, что самые эффективные
лекарственные препараты (лекарства будущего) будут созданы
именно на основе изучения структуры и молекулярных меха-
низмов действия природных биорегуляторов»,— так оценива-
ют роль пептидных биорегуляторов в медицине и ветеринарий
исследователи из Риги (О. С. Папсуевич, Г. И. Чипенс,
С. В. Михайлова. Нейрогипофизарные гормоны. — Рига : Зи-
натне, 1986, с. 136). Что же могут предложить медицине хими-
ки, занимающиеся пептидами? Многое: гормоны, кинины, ней-
ропептиды, индукторы и репрессоры биохимических реакций,
иммуномодуляторы (регуляторы, активирующие или снижаю-
щие иммунитет); ингибиторы ферментов или субстраты, позво-
ляющие тестировать активность ферментов в организме; син-
тетические вакцины, полученйые путем конъюгации синтети-
ческих антигенных детерминант с белками-носителями;
заменители некоторых пищевых продуктов, например «сладкий
пептид» — аспартам, который в 200 раз слаще сахара, и
многое другое.
Даже очень короткий перечень задач современной пептид-
ной химии свидетельствует о ее огромной роли как в биоорга-
нической химии, так и в других областях биологии и медици-
ны. Следует отметить, что производство пептидов занимает
важнейшее место в продукции многих западных химических и
фармацевтических фирм. Каков арсенал современных методов
6
получения синтетических пептидов и белков? Исторически
сложились следующие основные направления.
1. Химический синтез пептидов, включает традиционный
синтез в растворе и твердофазный метод. Последний является
более эффективным и позволяет получать на автоматических
синтезаторах полипептиды, содержащие до 200 аминокислот.
Метод синтеза в растворе более удобен для синтеза пептидов
до 10 остатков, так как позволяет получать их в больших коли-
чествах и большей степени чистоты.
2. Ферментативный синтез пептидов. В этом методе специ-
альными способами используют способность протеолитических
ферментов работать в качестве синтетаз в водной или водно-ор-
ганической среде (некоторые ферменты, например папаин, рабо-
тают практически в неводной среде). Преимущества'данного
метода состоят в том, что можно использовать минимальное ко-
личество защитных групп и получать пептиды высокой степени
оптической чистоты (ввиду высокой стереоспецифичности фер-
ментов). Метод эффективен для синтеза небольших пептидов,
сшивки больших фрагментов и сочетания синтетических фраг-
ментов с белками (полусинтез), например для получения полу-
синтетического инсулина.
3. Метод генетической инженерии. Метод рекомбинантных
ДНК появился в начале 70-х годов и сразу принес впечатляю-
щие результаты. В основе метода лежит возможность внедре-
ния нужного гена (в котором записана информация о строении
белка) в клетку, которая наряду с собственными белками син-
тезирует и необходимый нам белок. Нужный ген может быть
выделен из природного источника или синтезирован. Метод ген-
ной инженерии позволил синтезировать такие важнейшие бел-
ки, как инсулин, гормон роста, интерферон и многие другие.
Он способствовал развитию «белковой инженерии», т. е. созда-
нию белков с заданными изменениями структуры, например
более активных ферментов и др. Используется для синтеза бел-
ков и больших полипептидов.
4. Бесклеточный синтез. Осуществляется в бесклеточной
системе, в которой имеются рибосомы, АТФ, набор всех тРНК
и аминоацил-тРНКсинтетаз, а также других необходимых ком-
понентов. Помещая в систему какую-нибудь мРНК, можно
получить любой белок на этой матрице. Метод широко использу-
ется в лабораторных исследованиях, однако лишь недавно
доведен до препаративного масштаба. Необходимую мРНК
синтезируют методом рекомбинантных ДНК (есть специальный
способ, которым синтезируется только мРНК, а белок не синте-
зируется). Большим преимуществом этого метода перед методом
генетической инженерии является возможность получения чи-
стого белка, тогда как в альтернативном методе получают смеси
7
белков, разделение которых иногда представляет большие труд-
ности. К неразрешенным проблемам данного метода следует
отнести невозможность получения в составе белков некоторых
аминокислот, которые в процессе биосинтеза белка в клетке
получаются в результате посттрансляционной модификации
(например, оксипролин и др.).
Из четырех указанных методов синтеза белковых молекул
каждый имеет свои преимущества и ограничения. Несомненным
преимуществом химического метода синтеза является возмож-
ность включать в состав пептидов и белков D-аминокислоты,
аминофосфо новые кислоты и вообще любые неприродные ами-
нокислоты, а также аминокислоты, обязанные своим появле-
нием в природных белках посттрансляционным модификациям.
В то же время метод рекомбинантных ДНК и бесклеточный син-
тез позволяют включать в состав белковых молекул (во всяком
случае, на данной стадии развития метода) только 20 «канони-
ческих» аминокислот, что ограничивает возможности модифика-
ций. Следует отметить, что с конца 80-х годов успешно разра-
батываются подходы для введения неприродных аминокислот
в белки методами генной инженерии [1], однако широкого при-
менения они пока не нашли. Таким образом, несмотря на впе-
чатляющие успехи нетрадиционных методов синтеза молекул
белково-пептидной природы, традиционный химический синтез
(а также ферментативный), по-видимому, еще долгое время бу-
дет занимать важнейшее место в арсенале методов пептидного
синтеза как в научных исследованиях, так и в промышленном
производстве.
Хотя непосредственно разработкой методов синтеза пепти-
дов и синтезом конкретных пептидов занимаются химики-орга-
ники, подавляющее их большинство относится к самому про-
цессу синтеза лишь как к способу решения различных биоло-
гических задач, и несмотря на то, что сама «кухня» (как химики
называют свою экспериментальную работу) отнимает очень
много времени, требует больших знаний, опыта и изобретатель-
ности, химик, посвятивший себя синтезу пептидов, должен обла-
дать широкими познаниями в биологии, биоорганической химии,
молекулярной биологии, генетике и других областях в зависимо-
сти от конкретных задач, которые решаются в его лаборатории.
Ввиду того, что наша книга посвящена чисто практическим
аспектам химического синтеза пептидов, мы рекомендуем чита-
телю для более глубокого ознакомления с вопросами, затрону-
тыми выше, обратиться к некоторым монографиям, учебникам,
обзорам и научно-популярным изданиям, указанным в списке
литературы [2—8].
Исходя из того, что белки являются гетерополимерами, по-
строенными из 20 аминокислот-мономеров, действительно,
8
теоретически их можно получить простой реакцией полимериза-
ции с образованием амидных (пептидных) связей. Однако таким
образом мы не сможем получить полипептид с заданной после-
довательностью, поскольку не сможем контролировать ни по-
рядка включения мономеров в полипептидную цепь, ни степени
полимеризации, т. е. длину цепи. Поэтому на практике обыч-
но используют химические эквиваленты фрагмента
—NH—CHR— СО— типа:
О О
II II
z—NH—CH—С—А или A—NH—СН—С—X,
R R
-I I
где Z-, X- и Y- — защитные группы для а-аминогруппы, а-
карбоксильной группы и боковых реакционноспособных групп;
А — активирующая группа для карбоксильной или аминогруп-
пы. Тогда схема дипептида будет выглядеть следующим об-
разом:
О О
II II
Z—NH—СН—С—А + H2N—СН—С—X-------
I I
Ri Ra
1 I
Y Y
д
или _>
о
II
Z—NH—CH—СООН + A—NH-CH-C-X--------
I I
Rj R2
Y V
-> Z—NH—CH—CONH—СН—СОХ
I I
Ri R 2
Y 4 Y
H2N—CHRx—CONH—CHR2—COOH
На приведенной схеме защищенный дипептид деблокирует-
ся на последней стадии синтеза и превращается в свободный
пептид. Защитные группы могут быть подобраны так, что уда-
ляются одновременно или поочередно разными способами. Ос-
новные методы классического синтеза в растворе следующие:
1) ступенчатый синтез с С-конца, при котором стартовая амино-
кислота защищена по карбоксильной группе, а аминогруппа
свободна; к ней присоединяют другую аминокислоту, у которой
9
аминогруппа защищена, а карбоксильная группа свободна; у
полученного защищенного дипептида деблокируют аминогруп-
пу и присоединяют к нему следующую N-защищенную амино-
кислоту ит. д.; на последней стадии синтеза удаляют N- и С-за-
щитные группы; 2) блочный метод синтеза, при котором внача-
ле синтезируют отдельные короткие пептиды (блоки), а затем,
деблокируя их по амино- или карбоксильной группе, соеди-
няют в более крупные фрагменты. Обычно в случае использова-
ния этих схем синтеза боковые защитные группы удаляются на
последней стадии синтеза.
Как известно, процесс образования амидной (пептидной)
связи термодинамически невыгоден и может проводиться лишь
в жестких условиях. Поэтому необходимы реагенты, которые
активируют карбоксильную группу (иногда аминогруппу) в
очень мягких условиях, что исключает побочные реакции и по-
терю оптической активности, обусловленной хиральностью
а-углеродного атома (т. е. не приводят к рацемизации). В настоя-
щее время имеется много методов, отвечающих самым строгим
требованиям пептидного синтеза [9—30].
Реагенты и условия проведения реакций синтеза биологи-
чески активных пептидов должны обеспечивать: 1) максималь-
ные скорости протекания реакции образования пептидной свя-
зи, высокий выход целевого продукта при минимальной рацеми-
зации; 2) минимум побочных реакций; 3) легкое удаление
промежуточных продуктов реакции; 4) большой выбор защит-
ных групп, позволяющий производить их селективное удаление;
5) относительную доступность и дешевизну реагентов; 6) неток-
сицность используемых реагентов.
Специфика пептидного синтеза заключается в том,<гго дале-
ко не всегда удается для очистки защищенных пептидов исполь-
зовать традиционный в органической химии метод кристалли-
зации, а свободные пептиды вообще, как правило, не очищают
этим методом. Поэтому защищенные пептиды в настоящее вре-
мя очищают от примесей хроматографическими методами на
силикагеле, оксиде алюминия и др. Для очистки свободных
пептидов используют весь арсенал методов, применяемых в хи-
мии белков — ионообменную хроматографию, гель-хромато;
графию (сефадексы, биогели), препаративный электрофорез на
бумаге и в геле, высокоэффективную жидкостную хроматогра-
фию (ЖХВД) на обращенной фазе и некоторые другие специа-
льные методы. ЖХВД в последние годы стала универсальным
методом очистки защищенных и свободных пептидов и белков.
Несмотря на то, что очистка пептидов зачастую является
большей проблемой, чем их синтез, следует помнить, что пра-
вильно выбранная схема синтеза и соблюдение всех необходи-
мых условий его проведения значительно облегчит и стадию
10
очистки. Это особенно важно в случае твердофазного синтеза,
в котором отсутствует очистка продуктов синтеза на промежу-
точных стадиях. Именно поэтому, несмотря на впечатляющие
успехи твердофазного синтеза (в сочетании с ЖХВД), многие
исследователи предпочитают синтезировать даже большие пеп-
тиды классическими методами в растворе, считая, что они га-
рантируют более высокую степень чистоты конечных про-
дуктов.
Еще одним «подводным камнем» пептидного синтеза явля-
ется возможность рацемизации в процессе различных химиче-
ских манипуляций, а определение оптической чистоты пептидов
является, как правило, делом непростым.
Ввиду того, что цель нашей, книги — ознакомление с хи-
мией процессов, имеющих место в пептидном синтезе (как в тео-
ретическом, так и практическом плане), мы не рассматриваем
методы очистки и отсылаем читателя к специальной литерату-
ре [31—35].
Глава i
МЕТОДЫ ОБРАЗОВАНИЯ
ПЕПТИДНОЙ СВЯЗИ
СПОСОБЫ АКТИВАЦИИ КАРБОКСИЛЬНОЙ ГРУППЫ
Образование пептидной связи представляет собой нуклео-
фильную азаку карбонильного атома углерода’одной аминокис-
лоты аминогруппой другой аминокислоты. Это достигается
превращением СООН-группы в такие производные, где есть за-
местители (атома или группы атомов), «оттягивающие» элект-
роны. Это осуществляется за счет индукционного эффекта
электроотрицательных заместителей или эффекта сопряжения
с л-электронной системой:
О о
II II
R—С—О -> СН2 -> С^.\ R—С—О—X
Высокая реакционная способность таких активированных эфи-
ров, как пирокатехиновые или 4-оксипиримидиновые объясня-
ется также внутримолекулярным катализом.
В табл. 1 приведены основные методы и реагенты активации
карбоксильной группы ЫН2-защищенных аминокислот и пеп-
тидов. Часть из них хорошо себя зарекомендовала и находит
широкое применение в синтезе пептидов, а некоторые предло-
жены недавно, но, на наш взгляд, весьма перспективны. Наи-
более широко применяются следующие методы образования
пептидной связи: 1) карбодиимидный; 2) азидный; 3) метод сме-
шанных ангидридов; 4) метод активированных эфиров; 5) кар-
боксиангидридный.
При использовании карбодиимида для конденсации пепти-
дов часто для подавления рацемизации добавляют «нуклеофиль-
ные добавки»—1-оксибензтриазол, N-оксисукцинимид, пента-
фторфенол и др. Кениг и Гейгер [61] тщательно исследовали вли-
яние нуклеофильных добавок на степень рацемизации пептидов*
полученных DCC-методом, на следующей модельной реакции:
TFA — L — Pro — L — Vai—ОН + НРго - ОВи* ->
TFA — L — Pro — L — Vai — L — Pro — ОВи',
12
Карбодиимидную конденсацию вели в течение 1 ч при О °C, а
затем 1 ч при 20 еС в различных растворителях; в качестве ос-
нования использовали N-этилморфолин. В табл. 2 представле-
ны данные, полученные этими авторами в диметилформамиде.
Поскольку в модельных системах создаются условия, в ко-
торых имеет место заведомо высокая рацемизация, то из доступ-
ных реагентов этот нежелательный процесс сильно снижает
1-оксибензтриазол.
В качестве добавок, снижающих рацемизацию, Якубка ссо-
авт. [66] испытал некоторые кислоты Льюиса: хлориды цинка,
олова, титана, сурьмы и алюминия.
Хорошим катализатором, повышающим выход при исполь-
зовании ДЦГК, оказался 4-диметиламинопиридин (ДМАП)
[67]. В работе [68] изучали возможность использования ДМАП
вместо 1-оксибензтриазола в карбодиимидном синтезе. Пока-
зано, что при активации N-ацилированных аминокислот с до-
бавкой ДМАП наступает полная рацемизация; при использо-
вании N-защитных групп уретанового типа выходы выше, чем
с 1-оксибензтриазолом, а степень рацемизации такая же. Более
подробно о нуклеофильном катализе в пептидной химии см. об-
зор [69].
Механизмы активации карбоксильной группы
Реагенты группы енольных эфиров
Карбодиимиды. Карбодиимиды — соединения об-
щей формулы (1), способны легко реагировать с карбоновыми
кислотами с образованием реакционноспособных О-ацилизо-
мочевин (3). Изучение механизма этой реакции показало, что,
по-видимому, вначале протон отрывается от гидроксила кис-
лоты и затем присоединяется к двойной связи карбодиимида:
R—С + н+ + Rx—N=C=N—Rt -> R—С + C+ ->
I
u u NH—R,
(1) (2)
О N—Rt
II II
—+• R—C—-O^"C
NH-R,
(3)
Образовавшийся карбкатион (2) подвергается нуклеофильной
атаке карбоксильным анионом и образуется О-ацилизомочеви-
на. В результате смещения электронов, как это показано в фор-.
13
Таблица 1. Реагенты и методы
Метод активации Формула реагента Название реагента
Активация с образованием
Карбодиимидный
метод
N ,Ы-Д ицикл огексил-
карбодиимид
С =
М-Циклогексил-Ь1-(2-
морфолинил-(4)-этил«
карбодиимид
CH3CtiH,SO3
14-Циклогексил-14-(я-
диметиламиноцикло-
гексил)-карбодиимид
Кетениминный
метод
C=C=N—
СН,
Дифен илкетен-п-толу-
имин
Использование про- изводных ацетиле- на СН=С—О—С2Н5 Этоксиацетилен
сн3.
О
СН3
4-Диметиламино-З-бу-
тин-2-он
Использование
производных циа-
намида
N==C—N<
И,Ь1'-Дизаме1ценныв _
цианамиды
14
активации карбоксильной группы
Формула промежуточного активиро-
ванного соединения
Примечание
промежуточных енольных эфиров
О N—
II II Х-
R—С-О-С
Наиболее широко используе- [361
мый реагент для синтеза пепти-
дов. Применяется для получе-
ния активированных эфиров
Растворим в воде. Использу- [37]
ется для синтеза пептидов и
модификации белков
£>_-С—О—С
Растворим в воде. Использу- [37]‘
ется для синтеза пептидов и
белков, модификации белков
Широкого применения не на- [38Г
шел
о
II
R—С—О—С=СН2
хос2нб
о
II
R—С-О-С—N (СН3)2
II z°
Н—С—c<f
хсн,
О NH
II II /Кг
R—С—О-С—N<
X
В настоящее время не исполь- [39J
зуется
Предложен недавно. Дает [40]
очень высокий выход пепти-
дов
Практического применения не [41]
нашли
О
15
Метод активации Формула реагента Название реагента
Изоксазолиевые
соли
Имидоил галоиды
N-Этил-5-фенил изок-
сазолин -3-сульфон ат
(реактив Вудворта)
Хлорангидридный
метод (с соляной
кислотой)
Азидный метод (с
азотистоводородной
кислотой)
РС1б; SO2C1
О
II
С—NHNH,
Метод смешанных
ангидридов с уголь-
ной кислотой
О
Активация через
Смешанные ангидри-
ды с неорганически-
ми кислотами:
Пятихлористый фос-
фор, тионил хлорис-
тый
Нитрат натрия в со-
ляной кислоте; трет*
бутилнитрат гидрази-
ды защищенных ами-
нокислот и пептидов
Алкиловые эфиры
хлоругольной кисло*
ты .
Использование
EEDQ
С2Н5О-С = О
М-Карбоэтокси-2-
этокси-1,2-дигидрохи-
нолин
Использование сме-
шанных ангидридов
на основе производ-
ных фосфорной кис-
лоты
/OPh
Cl—Р=О
\OPh
Дифен ил хлорофосфат
/OEt
Br—P=O
\OEt
Диэтилбромфосфат
16
Продолжение табл. 1
Формула промежуточного активирован-
ного соединения
Примечание
/—с=сн—с=о
NH-С2Н
Применяются реже, чем
ДЦГК
[42]
R—С—О—С—-Ri
II II
О N
Rs Rs
Предложен для синтеза пеп- [43]
тидов в водных растворах
образование ангидридов
,0 Практически не используется [44]
R—С", из-за наличия значительной
ХЛ рацемизации
/О
R—
4N,
z°
О
№-с<0
О о
II II
R-С—О—С—ОС2Н5
Чаще всего используется для [45]
конденсации пептидных фраг-
ментов. Не дает рацемизации
Широко применяются для [46]
синтеза пептидов
Популярный реагент, но ис- [47]
пользуется реже, чем ДЦГК
О
OPh
С-0—Р=0
Широкого применения не на- [46]
шел
xOPh
7 /ОС.Н,
R—С=0—Р=0
ЧОСгНв
Предложен недавно [48]
2 2 — 882
17
Метод активации Формула реагента Название реагента
Использование
В OP-хлорида
N,N' -Бис-(2-оксо-3-
оксазолидин)-фосфоро-
диамидохлорид ВОР-
Смешанные ангид-
риды с органиче-
скими кислотами
Пивалоилхлорид
R=(CH3)3—
Симметричные ан-
гидриды защищен-
ных аминокислот
«Рге-ппх»-метод
2 моль эквивалента
МН2-защищенной ами-
нокислоты и 1 моль
ДЦГК
N-карбоксиангид-
риды
H2N—CHR—-СООН,
СОС1а
Активация через образование
Карбонилдиимида-
зольный метод
М,ЬГ-Карбонилдии-
мидазол
Метод активированных эфиров
Алифатические Cl—СН2—Ch=N
эфиры (метод циан-
метиловых)
Хлор ацетонитрил
Тиофениловые эфи-
ры
Тиофенол или 4-нит-
ротиофенол, ДЦГК
°«N—4 */—SH
18
, Продолжение табл. 1
Формула промежуточного активирован-
ного соединения
Примечание
Новый реагент. Считается до- [49]
вольно перспективным
Используется редко (50)
Используется в основном для [52]
твердофазного синтеза пепти-
дов
HN—CHR
Ох=С/ С=О
Используется не очень часто [51]
в связи с возможностью про-
текания побочных реакций
активированных амидов
О
II
R—С—0->СНа—CsN
О
R—С—S—
Время от времени использу- [53]
ется для синтеза пептидов
Один из первых активирован- [54]
ных эфиров, используемых
для синтеза пептидов, В настоя-
щее время не применяется
Нельзя использовать при уда- [55]
лении защитных групп гидра-
тированием
2*
19
Метод активации Формула реагента Название реагента
Замещенные фени- ловые эфиры НО—X Карбодиимид и заме щенные фенолы: 4- нитро, 2-нитро-2,4,5- трихлор-, пентахлор и пентафтор-произ- водные и др.
Сложные эфиры с гетероароматиче- скими соединения- ми ОН 8-Оксихинолин, ДЦГК
2-Оксипиридин или
3-оксипиридин, ДЦГ
Сложные эфиры с
производными гид-
роксиламина
Ацетоксим, ДЦГК
N-Оксифтал имид,
ДЦГК
N-Оксисукцинимид,
ДЦГК .
1 -Оксибензтр иазол 9
ДЦГК
20
Продолжение табл. 1
Формула промежуточного активирован- ного соединения Примечание Литера- тура
Наряду с ДЦГК являются [12]
самыми распространенными ре-
агентами для синтеза пептидов
О
Высокореакционные соедине- [56]
ния. Широкого применения
не нашли
Реакционноспособные соеди- [57]
нения. Широкого применения
не нашли
Ч /
R—С—О—N=C<
СН3
сн8
Используется редко
[58]
В настоящее время не ис- [59]
пользуется
II
R-C-0
Широко используется для [60]
синтеза пептидов
О
Il
R—С—О—N N
Обычно в свободном виде не [61]
применяется. Не дает раце-
мизации
21
Метод активации Формула реагента Название реагента
Реагенты переэте-
рификации (вари-
ант метода активи-
рованных эфиров)
О-Трифторацетил-N-
оксисукцинимид
О С1^ /С1
о—С1
С1/ ХС1
О FK
II
СС18Ч-С—О—F
F/ \р
1-Р-Нафтилсульфони-
локсибензтриазол
0-Т р и х лорацетил-пен-
тахлорфенол
0-Т р ихлор ацетил -пен-
тафторфенол
Таблица 2. Степень
рацемизации модельного пептида
в присутствии нуклеофильных
добавок [61]
£§ ge
Нуклеофильная о 2 s cxtf
добавка f I епень Лии в ,
О СП о*
Без нуклеофиль-
ной добавки — 38,0
Пентахлорфенол 2 27,0
N-Оксисукцин- имид 2 17,2
1 -Оксибензтр иазол 1 3,4
3-Окси-4-оксо-3,4- дигидро-1,2,3-бенз- триазин 1 1,0
муле (3), происходит значи-
тельное снижение плотности
электронов на карбонильном
атоме углерода, что обуслов-
ливает значительную реакци-
онную способность О-ацилизо-
мочевины (3). В зависимости
от условий реакции и природы
нуклеофильного агента при
реакции ДЦГК с ЫН2-защи-
щенными аминокислотами мо-
гут образовываться симмет-
ричные ангидриды, пептид,
N-ацилмочевина и 5 (4Н)-окса-
золон. Два последних соедине-
ния особенно нежелательны,
так как приводят к загрязне-
нию целевого продукта побоч-
ными соединениями и повыше-
22
Продолжение табл. 1
Формула промежуточного активирован*
ного соединения
Примечание
Реагент Сакакибары [62]
Отмечена рацемизация при ~ [63]
его использовании в синтезе
пептидов
Рацемизация отсутствует [64]
Удобен для синтеза пептидов, [65]
рацемизации не дает
нию рацемизации. Имеется большое количество обзоров, в ко-
торых рассматриваются различные аспекты карбодиимидного
синтеза пептидов [9—14].
При взаимодействии промежуточного соединения (3) с ами-
нокомпонентом образуется пептид t4) и N, N'-замещенная мо-
чевина (5):.
О NRi
II II
R—С—О-С—NHR2
h,Jr,
I, ।
L HjN+r, nhr2J
(3)
о о
II II
R—C—NH—R3 + RiNH-C—NHR2.
(4) (5)
Из большого числа изученных карбодиимидов наиболее подхо-
дящим оказался N, N'-дициклогексилкарбодиимид (6), кото-
рый был введен в практику пептидного синтеза Шиэном и Хес-
23
COM [36] 2
O-N-c-N-<_>
(6)
Вторым продуктом реакции ДЦГК с аминокомпонентом явля-
ется дициклогексилмочевина, которая плохо растворяется в
органических растворителях и легко отделяется фильтрованием.
Синтез пептидов ДЦГК-методом идет с высокими выходами
(70—90 %), но может сопровождаться значительной рацеми-
зацией и нежелательными побочными реакциями. Так, реак-
ция ацилпептидов с эфирами аминокислот или пептидов в при-
сутствии ДЦГК сопровождается значительной рацемизацией,
а введение в полипептидную цепь остатков глутамина или ас-
парагина часто сопровождается образованием нитрилов. Ши-
рокое применение нашел карбодиимидный метод для синтеза
активированных эфиров защищенных аминокислот.
Введение в практику водорастворимых карбодиимидов (на-
пример, 1-циклогексил-3-(2-морфолинил-(4)-этил)карбодиими-
да) значительно упростило очистку конечных продуктов, так
как соответствующие мочевины - хорошо удаляются водными
промывками.
Карбодиимиды нашли широкое распространение как в клас-
сическом, так и твердофазном методах синтеза. Наряду с ДЦГК
в твердофазном методе используют N/N'-диизопропилкарболим-
мид [70], так как соответствующая диизопропилмочевина луч-
ше растворима. Были исследованы некоторые несимметричные
карбо диимиды для твердофазного синтеза, дающие хорошо
растворимые мочевины [71], например карбодиимид (7), имею-
щий строение:
Ph—CHa—СН2—СН2—N=C=N—СН~СН3
(7)
дает на модельном твердофазном пептиде такие же выходы, как
ДЦГК и диизопропилкарбодиимид, однако растворимость со-
ответствующей мочевины в метиленхлориде в 200 раз выше,
чем в случае ДЦГК, и в 100 раз выше, чем в случае диизопро-
пилкарбодиимида.
Производные цианамида. Активация карбо-
ксильной группы N-защищенных аминокислот при помощи циа-
намида во многом близка процессу, происходящему при ДЦГК-
методе. Реакция N, N'-диалкилцианамидов (8) с аминокомпо-
нентом приводит сначала к образованию активированного со-
единения (9), которое реагирует далее с аминокомпонентом и
24
дает пептид!
N О NH
III И II Н
R—СООН + С -> R-С-О-С
I I
/N\ ZN\
Ri R2 Ri R2
(8) (9)
0 0 p.
II II /R1
-> p—c—NH—R3 + H2N—C—N .
X
Метод широкого распространения не получил.
Кетенимины. Аналогично карбодиимидам реагируют
с карбоновыми кислотами непредельные соединения (10), так
называемые кетенимины:
X
c=c=n-r2.
r/
(10)
При взаимодействии кетениминов с ЫН2-защищенной амино-
кислотой образуется промежуточный продукт (11), который ре-
агирует с аминокомпонентом с образованием пептида:
R—СООН + C=C=N—R2 CH—C=N—R2
R/ (J '
I
O=C—R
(И)
0 R« °
II Rl\ II
-> R—C— NHR# + CH—C—NH—Rj,
r/
В качестве конденсирующего средства был испытан дифенил-
кетон-/г-толилимин [38] (12)
Ph\ =
C=C=N—/ СН8
Ph/ ~
(12)
Метод широкого распространения не получил.
П р о и з в о д.н ы е ацетилена. 1. Этоксиацепшлен.
В 1955 г. Аренс предложил использовать этот реагент для син-
теза пептидов [39]. При взаимодействии этоксиацетилена (13)
25
-г карбоксильным компонентом образуется эфир энола (14), ко-
торый далее ацилирует аминокомпонент по схеме:
R—/ + сн^с—OCaHs ->
''•ОН
о сна
II II
R—С—О—С
осан}
(14)
(13)
О СН3
II I
R—C—О—С—осан5
NHRi
О
~ 9н»-с-.2£1»^ R—CONH—Rf.
Соединение (13) использовано для синтеза большого числа раз-
личных пептидов. Его преимуществом является летучесть, ко-
торая позволяет легко освободиться от избытка реагента; кро-
ме того, отщепляющийся этилацетат также легко удаляется
отгонкой. Недостатком метода является необходимость примене-
ния большого количества этоксиацетилена, проведение реакции
при повышенной температуре и значительная рацемизация при
использовании хлоргидратов эфиров аминокислот. В настоящее
время реагенты типа (13) не применяются.
2. 4-Диметиламино-3-бутин-2-он. Это конденсирующее
средство было предложено недавно Гейсом [40]. При взаимодей-
ствии реагента (15) с карбоксильным компонентом образуется
енольный эфир (16), который затем перегруппировывается в бо-
лее стабильное соединение (17). Последнее способно ацилиро-
вать амино компонент с образованием пептида:
СН« /О СН3ч ,Н
CssC—С N—С=С -*
CH8Z ХСН3 СН/ к \С—СН8
I в
' R—(!) °
II
О
(15) (16)
СН’\ II I /° HN R
-> N—С—СН=С—О—С lAaN~K1-).
СН8/ 4R
(17)
О О
II II
-> СНз-С—СН2-С—N (СН8)а + R—CONH-Rj.
(18)
Реагент (15) принадлежит к большому классу реагентов —
«пуш-пулл» — ацетиленов, которые были предложены группой
Нойеншвандера [72] в 1978 г. Енольные эфиры (17) — устойчи-
26
вые кристаллические соединения. При синтезе пептидов ис-
пользуются два подхода: с выделением енольных эфиров или
без их выделения. В последнем случае реакцию ведут в апро-
тонном растворителе при —30 9С сначала с карбоксильньш
компонентом, а затем добавляют аминокомпонент. Выходы пеп-
тидов—91—99 %. Второй продукт реакции: N, N'-диметил-
ацетоацетамид (18) может быть легко удален из реакционной
смеси. Весьма вероятно, что 4-диметиламино-3-бутин-2-он най-
дет широкое применение в синтезе пептидов.
Соли изоксазолия. Для активации карбоксильной
группы N-защищенных аминокислот Вудвортом и соавт. [42]
предложены соли изоксазолия. При действии оснований на N-
алкилизоксазолиевые соли раскрывается гетероцикл и образу-
ются кетокетенимины (19). Последние реагируют с карбоксиль-
ным компонентом, превращаясь в енольные эфиры (20), которые
ацилируют NH2-rpynny аминокомпонента с образованием пеп-
тида:
/° V
R—С=^С—-R + Rr-C С +Ri=C ->
i iH 4°’ i' х°н
r/^C—NR
II
R4/OH Rx/O-C—Ri
-> C Rt -> С r CONH—R„,
II I II
c o—c=o c
r/^c/ r/xc-nh-r
II II
N—R О
(20)
Наиболее часто в синтезе пептидов используется «/(-реагент
Вудворта», т. е. М-этил-5-фенилизоксазолийсульфат (21) [42]:
—С=-СН
О СН
I
С2Н5
(21)
Наличие в молекуле отрицательно заряженной сульфогруппы
позволяет легко отделять продукты превращения соли (21) от
полностью защищенного пептида.
27
Синтезы пептидов с применением реагента Вудворта идут
с высокими выходами, при конденсации ацилпептидов наблю-
дается рацемизация.
И м и до и л г а л о г е н ид ы. Хегарти и сотр. [43] пред-
ложили новые реагенты для образования пептидной связи —
имидоил галогениды (22). По структуре продукты их взаимодей-
ствия с аминокислотами близки к виниловым эфирам (23):
Rl\
C=N—N
х/ XRS
(22)
Ri=Ph, С (СН3)3; R2 = CH3; R3=Ph=, 4-NO2—Ph; X=C1, Br.
Соединения (22) получают из замещенных гидразидов и РС1В.
При синтезе пептидов имеет место следующий ряд превраще-
ний:
Rix /Ra . /R2
C=N— N 22—» Rj—N 2*222—>
х/ XR3 XR3
О
II
R«—C—O\ ,RS
C=N-N r4_Q0NH-R6 +
r/
(23)
О p
II /K*
+ Ri—C—NH—N
XR»
Смешанные ангидриды
Смешанные ангидриды с неорганическими кислотами
Хлорангидридный метод. Хлорангидридный метод синтеза
пептидов является старейшим методом и в последние десятиле-
тия используется сравнительно редко. Однако он может быть
полезным для получения амидов и других производных амино-
кислот. Кроме того, в последние годы появилось несколько
удобных модификаций хлорангидридного метода, -что свиде-
тельствует о том, что он должен занимать надлежащее место в
арсенале методов пептидной химии. Особое место занимают
хлорангидриды хлоргидратов аминокислот, так как отпадает
необходимость деблокирования аминогруппы после введения их
в реакцию. Они нашли применение для получения сложных
28
эфиров и n-нитроанилидов аминокислот (подробнее см.
главу 4).
Наряду с классическими методами получения хлорангид-
ридов N-защищенных аминокислот (действием РС15, SOC12 и
других хлорангидридов неорганических кислот) предложены
более мягкие реагенты: несимметричные дихлоралкиловые эфи-
ры 173], системы тионилхлорид — пиридин [74] и тионилхло-
рид — ДМФА [751. При взаимодействии ДМФА с тионилхло-
ридом, РС15, фосгеном или оксалилхлоридом образуется N, N-
диметилхлорформамидий (24), который является очень мягким
хлорирующим агентом [75]:
Н„СХ ° гН8Сх /с11+
N-C-Н N=C 1C1~+SO2.
Н8б/ LHjC^ Хн 1
(24)
’ Реагент (24) может быть предварительно выделен и затем
добавлен в раствор защищенной аминокислоты или получен
in situ при низких температурах действием тионилхлорида на
защищенную аминокислоту в органическом раетворителе в при-
сутствии каталитических количеств ДМФА (или в растворе
ДМФА) [101.
Показано, что при действии на карбоновые кислоты стехио-
метрического количества смеси тионилхлорид — пиридин в
метиленхлориде при комнатной температуре они количествен-
но превращаются в хлорангидриды в течение 6 мин, а если ис-
пользовать дициклоаммонийные соли N-защищенных амино-
кислот, время реакции сокращается до 1 мин [74]. Последую-
щее добавление эфиров аминокислот или различных аминов
приводит к пептидам или амидам с очень высокими выходами.
Получены хлорангидриды защищенных аминокислот с раз-
личными N-защитными группами (карбобензокси, тозильной,
трифторацетильной, тритильной и др.), многие из которых по-
лучены в кристаллическом виде. Хлорангидриды карбобензок-
сиаминокислот склонны к циклизации с образованием N-карбо-
ксиангидридов, поэтому целесообразно не выделять их из рас-
твора.
Недавно Карпино с соавт. [76] получили 14 хлорангидридов
9-флуоренилметилоксикарбонил(Етос)-аминокислот в кри-
сталлическом виде, которые оказались стабильными при хра-
нении. Об использовании их для быстрого пептидного синтеза
в двухфазной системе будет рассказано в последнем разделе
данной главы.
Хлорангидриды N-защищенных аминокислот используют-
ся в синтезе в органических растворителях или в водно-органи-
ческой среде, содержащей избыток третичного амина (или
29
щелочи), для связывания образующегося хлористого водорода.
В случае N-защитной группы ацильного типа или использова-
ния хлорангидридов пептидов (за исключением С-концевого
остатка глицина) использование хлорангидридного метода при-
водит к полной рацемизации.
Азидный метод. Азидный метод был предложен Курциусом
в 1902 г. 177] и на протяжении 80 лет остается важнейшим спо-
собом конденсации при синтезе пептидов. Долгое время счита-
лось, что азидная конденсация фрагментов пептидов протекает
без рацемизации. Однако в 1970 г. было показано, что при кон-
денсации фрагментов в присутствии избытка триэтиламина ра-
цемизация идет; она особенно растет, если С-концевым остатком
является гистидин. При тщательном контроле условий реакции
и применении N-этилморфолина вместо триэтиламина азидный
метод дает минимальную рацемизацию.
Азиды (26) защищенных аминокислот и пептидов можно лег-
ко получить из соответствующих гидразидов (25). Синтез по-
следних соединений из метиловых, этиловых или бензиловых
эфиров N-защищенных аминокислот и пептидов также не пред-
ставляет большого труда. Ниже дана схема получения азида и
превращения последнего в пептид:
/° /°
NH2NH3.HgO HNOg
^ОМе ^NHNHj
(25)
/°
R—С R-CONH—Ri-HHN,.
XNS
(26)
На каждой из трех стадий! синтез гидразида, получение азида
и образование пептида — могут иметь место побочные реакции.
Поскольку защищенные гидразиды (25) являются весьма ста-
бильными соединениями, то на этапе их синтеза меньше всего
можно опасаться нежелательных процессов. Напротив, азиды
нестабильны и их вводят в конденсацию немедленно. При полу-
чении азидов могут протекать следующие нежелательные про-
цессы: образование амида вместо азида (26), нитрозирование
остатка триптофана и превращение метионина в метионинсуль-
фоксид (101. В водных растворах имеет место также перегруп-
пировка Курциуса, т. е. превращение азида в изоцианат!
о
R—С—N=N+<=N~ R—N«C=O + Ng.
Если получение азидов из гидразидов (25) вести в модификации
Хонзля и Рудингера 145] (т. е. в безводном органическом рас-
30
творителе действием mpem-бутилнитрата при избытке хлористо-
го водорода или трифторуксусной кислоты), то побочные реак-
ции значительно подавляются, а выход пептидов составляет
40—70 % и выше. В настоящее время азидную конденсацию
проводят почти исключительно этим способом.
Интересной разновидностью азидного метода является спо-
соб Хиршмана и сотр. [78, 79], заключающийся в использова-
нии азидной конденсации для синтеза активированных эфиров:
/° /° /°
п Bu^ONO n " 2НО^п D // H2N-Rt
1\—с —> К—С»------------->“ К—Ъ
4NHNHa Н XN3 XOSu
(26) (27)
-> R—CONH—Rt
К азиду (26) добавляют 2 моль-эквивалента N-оксисукцини-
мида и выдерживают 12 ч; сукцинимидный эфир (27) вводят в
реакцию с аминокомпонентом. Эта модификация позволяет по-
лучать более чистые пептиды. Выходы также повышаются.
Ряд усовершенствований разработан в синтезе гидразидов
пептидов. Поскольку скорость реакции гидразинолиза резко
падает при увеличении длины пептидов, было предложено сна-
чала синтезировать защищенные гидразиды аминокислот (28),
затем использовать их для наращивания пептида. В требуе-
мый момент производят отщепление защитной группы
R—с" + H2N—NH—Вос -> R—С
ХОН XNHNH—Вос
(28)
и образующийся гидразид превращают в соответствующий азид
для последующего образования пептидной связи:
о о о
R—С—NHNH—Вос — -> R—С—NHNH2 R-C-Ns.
В 1974 г. Шиори и Ямада предложили новый реагент, кото-
рый активирует карбоксильную группу, превращая ее в ани-
лазид — дифенилфосфоразид (29) [82J. Возможно, реакция про-
текает через образование промежуточного продукта (30), кото-
рый в результате внутримолекулярной перегруппировки дает
ацил азид (31):
О 0
R.-Z +N,J/oph,
\0- '•ОН.
(29)
31
О OPh
II I
-> Ri-C—O-P-OPh
n8/\o-'
(30)
Ri-CONH—Ra Ri-C
4Ng.
° /0Pb
R—С—O—P=O
xOPh
(32)
• +Nf
(31)
Однако продукт (30) может находиться в равновесии со смешан-
ным ангидридом (32), который атакуется азид-ионом и также
превращается в ацилазид (31). Авторы не исключают, что в
реакцию с аминокомпонентом может вступать не только соеди-
нение (31), но и промежуточные (30) и (32) [83].
Конденсация пептидов дифенилфосфоразидным методом
протекает без рацемизации, кроме того, при использовании
этого метода не нужно блокировать боковые функциональные
группы серина, треонина, тирозина и гистидина [83]. Реагент
используется как для ступенчатого синтеза пептидов, так и для
конденсации фрагментов [84]. Дифенилфосфоразид является
также хорошим реагентом для получения циклопептидов [851:
H2N-CH—CONH—CH—CONH—CH—CONH—CH—СООН +
Ri Ra Rg 1I4
О
|| yOPii
+ N3~P<0Ph
R3
N4 ,0
C'
R<—CH ^h-r2
I о
R!
Метод смешанных ангидридов с угольной кислотой. Один
из основных методов создания пептидной связи, вошедших в
практику синтеза пептидов под названием «метода смешанных
ангидридов», основан на использовании смешанных ангидри-
32
дов аминокислот с моноэфирами угольной кислоты (33). Он был
предложен в 1951 г. одновременно несколькими группами ис-
следователей: Вилайдом и Бернхардом [46], Буассона [801 и
Ваугханом [81]. Простота эксперимента, высокие скорости ре-
акции и чистота продуктов делают этот метод весьма ценным
способом получения пептидов.
При обработке триалкиламмониевых солей защищенных
аминокислот эфирами хлоругольной кислоты образуются сме-
шанные ангидриды (33), которые с высокими выходами реаги-
руют с аминокомпонентом, образуя соответствующие пептиды:
О
R—с/° + + Cl—С—OR1 R—С<^°
XO~NHR, >0
RiO—с/
(33)
г О’ о
I II
R—С—О—С—OR1
- h2n+r«
R—CONH—R2 + R1-0H + CO2.
Продуктами реакции наряду с пептидами являются спирт
и углекислота. При работе по методу смешанных ангидридов
возможны побочные реакции, например расщепление ангидри-
да (33) в нежелательном направлении, происходящее при ата-
ке нуклеофила по углеродному атому карбонильной группы
угольной кислоты. Степень этой побочной реакции зависит от
используемого растворителя, природы заместителя угольной
кислоты. Так, использование разветвленного изобутилхлор-
формиата вместо этилхлорформиата снижает образование по-
бочных продуктов. Метод смешанных ангидридов нельзя при-
менять для конденсации фрагментов. В этом случае рацемиза-
ция весьма значительна.
В последнее время на основе метода смешанных ангидридов
разработана методика быстрого синтеза пептидов — так назы-
ваемый ПСА-метод [86], т. е. метод последовательного синтеза
пептидов с использованием избытка смешанного ангидрида
(REMA).
Метод с использованием ЭЭДХ. Вместо классического ва-
рианта синтеза смешанных ангидридов в 1968 г. Беллеу и Да-
леком [47] был введен новый конденсирующий реагент N-эти-
локсикарбонил-2-этилокси-1,2-дигидрохинолин (ЭЭДХ), соеди-
нение (34), который в короткое время стало весьма популярным
при синтезе пептидов. При взаимодействии реагента (34) с кар-
3 2-882 33
боксильным
II II
+ R—С—О—С—ОС2Н5
H*N R‘-> R—С—NH—Rj + C2H5OH + CO2
(35)
компонентом образуется смешанный ангидрид (35), который
при взаимодействии с аминокомпонентом образует пептид. Дру-
гими продуктами реакции являются хинолин, этиловый спирт и
углекислый газ, которые легко могут быть удалены. Выход пеп-
тидов достаточно высок. При использовании ЭЭДХ для конден-
сации фрагментов имеет место рацемизация.
Вместо ЭЭДХ при синтезе пептидов аналогично можно ис-
пользовать реагент (36) — 1-изобутилоксикарбонил-2-изобу-
тилокси-1,2-дигидрохинолин (ИИДХ) [87]:
БОФ-хлорид [49]. В качестве хорошего конденсирующего
реагента для ацилирования спиртов и аминов карбоновыми кис-
лотами недавно был предложен М,М-бис(2-оксо-3-оксазолидин)-
фосфородиамидохлорид (БОФ-хлорид) — соединение (37).
При его взаимодействии с анионом кислоты образуется смешан-
ный ангидрид производных аминокислоты и замещенных фос-
форной кислоты. Это соединение способно ацилировать спирты
и амины с высоким выходом:
ОН
О
О О и
II II /X
R—С—О—Р-----N О
Соединение (37) нетоксично
34
Дифенилхлорфосфат. Для образования смешанных ангид-
ридов N-защищенных аминокислот и производных фосфорной
кислоты (соединение 39) Виланд и Бернхард [46] предложили
использовать дифенилхлорфосфат (38):
4OPh
(38)
О
R_c-o-p^oPh
xOPh
(39)
г- СГ ОРИ о
I I II
R—С—О-----Р=О -> R—CONH—Rj + PhO—Р—OPh,
Н—N+—Rx OPh OH
- H
В этой схеме дифенилхлорфосфат является конденсирующим
агентом.
Горецкая и сотр. [48] для тех же целей применили диэтил-
бромфосфат:
О
(С2Н5О)3 Р + Вг2 (C2H,O)2=P-Br RtC00~ -*
о о о
Rj—С—О—Р (OEt)2 CONH—R2 + HO—P—OEt.
J>Et
Выход пептидов составляет 85—92 %. В практике пептидного
синтеза реагент широкого применения пока не нашел.
Смешанные ангидриды с органическими кислотами
При взаимодействии солей защищенных аминокислот с
хлорангидридами карбоновых кислот образуются смешанные
ангидриды (40), ацилирующие аминокомпонент!
Г ° °” 1
R—С—О—С—С|
L J
R—С^ H.N_____R.
\О —R—CONH—R2 + Rj—С^ •
R,-C< ХОН
ХО
(40)
Главными побочными реакциями при использовании соедине-
ний (40) является реакция диспропорционирования и возмож-
3*
35
ность расщепления смешанного ангидрида из-за атаки амино-
компонента либо по карбонильному атому углерода аминокис-
лоты, либо углеродному атому карбоновой кислоты. Показа-
но, что если использовать для синтеза пептидов хлорангид-
риды карбоновых кислот с разветвленными радикалами, на-
пример хлорангидрид триметилуксусной (41) или изовале-
риановой (42) кислот, то эти побочные процессы незначительны
из-за стерических препятствий [50, 88, 89]:
СН3 О
| // Н3СХ ,0
Н8С-С-С )СН-СН,
I \Г1 Н3с/ - ХС!
СН3 С1
(41) (42)
Смешанные ангидриды N-защищенных аминокислот или пеп-
тидов с бензолсульфокислотой приводят к полной рацемизации.
Однако использование N-метилморфолина в качестве основания
вместо триэтиламина в десятки раз снижает рацемизацию. При-
менение смешанных ангидридов на основе соединений (41) и
(42) позволяет получать оптически чистые пептиды даже при
конденсации сегментов [90].
Симметричные ангидриды
Симметричные ангидриды N-защищенных аминокислот (43)
были получены Вейгандом с сотр. в 1957 г. [91]. Преимущест-
вом этих соединений перед смешанными ангидридами является
однозначность их расщепления при действии нуклеофильного
агента, а недостаток выражается в том, что в синтезируемый
пептид включается лишь половина ациламинокислоты:
2R—СООН
/°
R— С<^
>0
R-C/
(43)
h2n-r,
R-CONH-Ri + R—СООН.
Чаще всего симметричные ангидриды получают при действии
1 моль-эквивалента ДЦГК на 2 моль-эквивалента защищенной
аминокислоты; они также образуются, если в качестве конден-
сирующего средства использовать этоксиацетилен. Многие сим-
метричные ангидриды являются достаточно устойчивыми сое-
динениями; некоторые из них получены в кристаллическом ви-
де, но, как правило, соединения типа (43) из раствора не выде-
ляют. В последнее время симметричные ангидриды находят ши-
рокое применение в твердофазном методе синтеза пептидов;
здесь они являются, по-видимому, лучшими реагентами для
образования пептидной связи.
36
Бенойтон с сотр. показали, что симметричные ангидриды
можно использовать для синтеза в водно-органических смесях.
Выход пептидов составляет более 90 % [92].
N-Карбоксиангидриды
N-Карбоксиангидриды аминокислот (оксазолидин-2,5-дио-
ны), соединения (44), впервые получены Лейксом в 1906 г. [93].
Их легко можно синтезировать при действии фосгена на амино-
кислоту; обработка аминокислот тиофосгеном приводит к соот-
ветствующим тиоаналогам (45):
/О
R—СН—С"
। >
HaN—CHR—СООН
csci2
(44) (45)
N-Карбоксиангидриды образуются также из карбобензокси-
аминокислот при действии хлористого тионила за счет термиче-
ского разложения промежуточно образующихся хлорангид-
ридов КБЗ-аминокислот.
С теоретической точки зрения идеальными полупродукта-
ми для синтеза пептидов могли бы быть такие соединения, у
которых N-защитная группа может одновременно активировать
карбоксильную группу и удаляться при образовании пептид-
ной связи. N-Карбоксиангидриды до некоторой степени удов-
летворяют этому требованию, так как при действии нуклеофиль-
ных агентов имеют место превращения*
H2N—CHR—CONH—Rx
H2N—CHR—COORi
.H2N—CHR-COOH
При раскрытии цикла и отщеплении СО2 образуется новая нук-
леофильная NH2-rpynna; последняя способна реагировать с
другой молекулой N-карбоксиангидрида. В результате этого
образуются гомополиаминокислоты:
NH—CO-CHR—NHa
НС—С—Nu
И т. д.
—...—» полиаминокислота
37
Здесь Nu — нуклеофил-затравка (амин или вода). N-Карбр-
ксиангидриды являются лучшими реагентами для синтеза как
гомополиаминокислот, так и сополимеров аминокислот [94].
В 1966 г. Хиршман с сотр. [95] показали, что строго конт-
ролируемых условиях N-карбоксиангидриды можно использо-
вать для ступенчатого наращивания полипептидной цепи. Син-
тез ведут при pH 10,2 и 0—2 9С при мощном перемешивании.
Конденсация проходит за несколько минут. Таким образом
было получено несколько коротких пептидов с довольно высо-
ким выходом, которые затем использовали для синтеза рибо-
нуклеазы [51].
Аналогично N-карбоксиангидридам используют тиозоли-
дин-2,5-дионы (45). В этом случае побочные реакции протекают
в меньшей степени, так как образующиеся при аминолизе тио-
карбаминовые кислоты являются более устойчивыми соедине-
ниями. Позднее метод NCA был улучшен [96].
Для более широкого применения в практике пептидного
синтеза рекомендуют видоизмененный способ, при котором N-
карбоксиангидриды превращают в их Nps-производные [97].
Благодаря такой модификации метода устраняется опасность
нежелательной полимеризации NCA и появляется возможность
более контролируемого наращивания пептидной цепи:
Rj—CH—HRN—CHR—COOH v
Nps—N--С/
^O
-> Nps—NH—CH—CONH—CH—COOH _
E | | —Nps
Ri R
H2N—CH—CONH—CH—COOH
Ri R
r2_CH-Cc°
I >°
Nps—N--C4O
Nps—NH—CH—CONH—CH—CONH—CH—COOH.
Ra ,l!x 1!
Карбонилдиимидазольный метод
В 1958 г. Андерсон и Пол [53] предложили новый реагент —
N.N'-карбонилдиимидазол (46) для образования пептидной
связи. При взаимодействии с N-защищенными аминокислотами
соединение (46) дает активированный амид (47). Последний
38
легко реагирует с аминокомпонентом с образованием пептидов:
(47)
Реакция протекает быстро, почти без рацемизации. Реагент не-
редко применяют для синтеза пептидов. Тиоаналог соединения
(46), М,М'-тионилдиимидазол (48) был предложен для синтеза
пептидов Виландом [98]:
(48)
Метод активированных эфиров
В начале 50-х годов две группы исследователей предложили
принципиально новый способ активации карбоксильной груп-
пы, заключающийся в использовании сложных эфиров, содер-
жащих электроноакцепторные группы. Виланд [55] изучал воз-
можности тиофениловых эфиров защищенных аминокислот
(49), Швицер и сотр. [54] использовали цианметиловые эфиры
типа (50)
О о
Z—NH—CHR—С -> O->CH2CN Z—NH—CHR—С—S—
(49) (50) =
Во втором случае активация карбоксильной группы объясняет-
ся индукционным эффектом цианогруппы; реакционная способ-
ность тиофениловых эфиров растет из-за эффекта л-электронной
системы бензольного кольца. Естественно, что введение в аро-
матическое кольцо электроноакцепторных заместителей, таких
как —NO2, —CN, —CF3, Cl, F и др., должно усиливать этот
эффект. Эфиры типа (49) и (50) вскоре перестали применять из-
за сравнительно жестких условий их аминолиза, и им на смену
пришли активированные эфиры на основе замещенных фенолов.
Замещенные фениловые эфиры. n-Нитрофениловые эфиры
были предложены Бодански в 1955 г. [99] и быстро приобрели
большую популярность благодаря доступности, высокой реак-
ционной способности и устойчивости при хранении. С исполь-
зованием n-нитрофениловых эфиров удалось получить доста-
39
точно крупные пептиды путем ступенчатого наращивания поли-
пептидной цепи с С-конца [100].
Показано, что реакционная способность п-нитрофениловых
эфиров заметно возрастает при добавлении к реакционной сме-
си 1,2,4-триазола [101]. Очень хорошим катализатором явля-
ется 1-оксибензтриазол в ДМФА, но не в ТГФ [102].
В последние годы в связи с появлением более активных эфи-
ров n-нитрофениловые эфиры стали применять лишь для вве-
дения остатков глутамина и аспарагина, которые могут дегид-
рироваться при использовании карбодиимидного метода.
n-Нитрофениловые эфиры, как и большинство других активи-
рованных эфиров, получаются при взаимодействии защищенных
аминокислот и соответствующего фенола с добавкой ДЦГК:
/О .— zO /—ч
R~“C\ _ + н0~\ NO2 -> R-C-O-<f >-NO2
-* R—CONH—Rx + HO-^ ^-NO2.
O-
I +
r_C-NH2—
Из других эфиров этого типа широкое применение для синтеза
пептидов нашли 2,4,5-трихлорфениловые (51) [103], пентахлорфе-
ниловые (52) [104] и пентафторфениловые эфиры (53) [105, 106].
Высокая реакционная способность пентахлорфениловых
эфиров побудила Кишфалуди и сотр. [106] исследовать в каче-
стве активированных эфиров пентафторфениловые производные
типа (53) и испытать их пригодность для синтеза пептидов. Ока-
залось, что указанные эфиры — кристаллические соединения,
хорошо растворимые в органических растворителях; они актив-
нее других ариловых эфиров и не дают рацемизации при кон-
денсации фрагментов. На основе ПФФ-эфиров разработан быст-
рый метод синтеза пептидов. Вариантом метода ПФФ-эфиров
является применениетак называемого «комплекса F», т. е. комп-
лекса пентафторфенола с ДЦГК, который является весьма эф-
фективным реагентом для создания пептидной связи [106].
Замещенные гидроксиламина. Активированные эфиры на
базе замещенных гидроксиламина были впервые получены Неф-
кенсом и Тессером [59] в 1961 г., исходя из N-оксифталими-
40
да (54). Эти эфиры оказались весьма реакционноспособными
соединениями, но вскоре были вытеснены более доступными
N-оксисукцинимидными эфирами (55), которые предложил в
1963 г. Андерсон [60]:
Две карбонильные группы в гетероциклическом ядре зна-
чительно снижают электронную плотность на атоме углерода
СО-группы аминокислоты, что значительно облегчает атаку
нуклеофилами. Легкость пол у чени я N -оксису кци нимид ны х
эфиров в кристаллическом виде, отсутствие рацемизации пеп-
тидов при их использовании, их высокая реакционная способ-
ность и растворимость в воде продукта реакции — N-оксисук-
цинимида сделали активированные N-оксисукцинимидные эфи-
ры чрезвычайно популярными реагентами для создания пептид-
ной связи.
Из других активированных эфиров следует обратить внима-
ние на группу соединений, повышенная реакционная способ-
ность которых обусловлена внутримолекулярным катализом.
Это, в частности, производные 2-оксипиридина (56) и 8-оксихи-
нолина (57):
(56J (57)
Кинетические исследования а.минолиза 8-оксихинолиновых
эфиров позволили предложить следующий механизм реакции:
--► R-CONH-R1 +
41
Внутримолекулярный катализ проявляется в неполярных рас-
творителях: хлористом метилене, хлороформе и т. д., но резко
уменьшен в растворителях типа ДМФА.
Рассматриваемым эффектом объясняется достаточно высо-
кая активность пирокатехиновых эфиров (58) (1071, которые
могут быть легко получены из замещенных аминокислот и
о-фениленсульфита:
О
R—С<у + O-S( 1 || -> R-C-O-/ + SO2
НО7
(58)
Если в ароматическое ядро ввести несколько заместителей,
один из которых несет положительный или отрицательный за-
ряд, то можно создать активированные эфиры, растворимые в
воде или водно-органических смесях. В частности, такой под-
ход привел к созданию водорастворимых 2-нитро-4-сульфофе-
ниловых активированных эфиров (59) [108—ПО]. Соединения
(59) легко получают при взаимодействии N-защищенной амино-
кислоты с натриевой солью 2-нитро-4-сульфофенола в присутст-
вии ДЦГК в растворе ДМФА:
О
R-сУ + НО—SO3Na ДЦГК-> r_С-О—SO.Na
\0Н
o2n osn
(59)
Эфиры (59) можно использовать для создания пептидной связи
в водной среде [108—ПО].
Реакционная способность активированных эфиров опреде-
ляется кислотностью гидроксильной группы соответствующих
фенолов. Действительно, было показано, что существует вза-
имосвязь между константой диссоциации замещенных фенолов
и реакционной способностью соответствующих активирован-
ных эфиров. Помимо этого фактора на скорость ацилирования
аминокомпонента влияет также степень пространственного экра-
нирования за счет введения заместителя в орто-положение к
эфирной связи. Ковач и сотр. [111] изучали кинетику аминоли-
за активированных эфиров в модельной реакции
О
II
Z-Cys (S-Bzl)-C—OR + HValOMe -> Z—Cys (S—Bzl)—ValOMe,
используя пространственно затрудненные аминокислоты, и по-
казали, что реакционная способность эфиров определяется так-
же пространственным эффектом заместителя. Например, пен-
гафторфениловые и пентахлорфениловые эфиры имеют близкие
42
величины констант диссо-
циации ОН-группы; однако
скорость ацилирования
аминокомпонента хлор-за-
мещенными эфирами в 20
paj ниже, чем соответствую-
щими фторпроизводными
(табл. 3). Это объясняется
пространственным экрани-
рованием атомами хлора,
поскольку вандерваальсовы
радиусы хлора больше, чем
у атома фтора. Стерически-
ми причинами объясняется
также повышенная реакци-
онная способность 2,4,5-
три хлорфениловых эфиров
по сравнению с 2,4,6-три-
хлорариловыми производ-
ными, у которых два атома
хлора, находящиеся в орто-
положении, сильно затруд-
няют доступ аминокомпо-
нента к электрофильному
центру. Аналогичный эф-
фект проявляется у 2,6-ди-
нитрофениловых эфиров,
если сравнивать их реакци-
онную способность с соот-
ветствующими 2,4-динитро-
замещенными соединениями
(см. табл. 3). Следователь-
но, скорость ацилирования
аминокомпонента активиро-
ванными эфирами зависит
от двух факторов: 1) кон-
станты диссоциации заме-
щенных фенолов и 2) до-
ступности реакционного
центра атома углерода кар-
бонильной группы для нук-
леофильного агента.
Таблица 3. Сравнение
реакционной способности
активированных эфиров [111]
я k0 . 10*, — 1 1 моль 1 • с 1 Время ацил и- 1 рогания i:a । 90 %, мин ।
F\ zF
НО—40,4±9
F/
НО—^--NO2
no/
О^
НО—
О^
no2x
НО—
no/
С1Х /С1
НО-^-С1
с/ ЧС1
18,4±3
5,4±0,7
1,73±0,2
1,72±0,03
0,3±0,03
0,1 ±0,01
СЧ
но—<("-/— С1
с/
0,06±0,002
2,9
6,3
21
67
62
385
1068
1856
Активированные эфиры — чрезвычайно удобные реагенты
синтеза пептидов. Основным их преимуществом является то,
что эти соединения можно использовать непосредственно, т. е.
без предварительной активации карбоксильного компонента,
43
как это имеет место при использовании, например смешанных
ангидридов, азидного или карбодиимидного методов синтеза
пептидов. Кроме того, кристаллические активированные эфи-
ры удобны при хранении и дозировании в практике пептидного
синтеза. Активированные эфиры широко используются для на-
ращивания полипептидной цепи с С-конца. Применяя избыток
активированного эфира (30—50 %), можно достичь практичес-
ки количественного выхода желаемого продукта на каждой ста-
дии реакции. Например, ступенчатый синтез окситоцина, осу-
ществленный Бодански в 1959 г. на основе п-нитрофениловых
эфиров, позволил получить биологически активное соединение
с выходом 30 %, что явилось большим успехом пептидной хи-
мии. В 1967 г. этому же автору удалось синтезировать при по-
мощи активированных эфиров 27-членный пептид [100]. Избы-
ток активированных эфиров можно удалять после окончания
реакции добавлением М,М-диметилэтилендиамина (60), кото-
рый впервые применил Кишфалуди при синтезе ряда модель-
ных пептидов [112]. Соединение (60) ацилируется избытком не-
прореагировавшего эфира, в результате чего образуется соеди-
нение (61), способное легко растворяться в кислотах, и поэто-
му оно без труда удаляется при промывках растворами кислота
О
(60)
о
II /СН3
R—С—NH—СН.СН2—N<
ХСН3
(61)
|/сн’
хсн3
Практика пептидного синтеза показала, что при получении ди-
пептида целесообразно использовать избыток аминокомпонен-
та, а не активированного эфира, так как эфиры аминокислот
обычно дешевле, чем активированные эфиры. Избыток амино-
компонента можно удалить промыванием раствором кислоты.
Обнаружено, что реакции активированных эфиров ускоря-
ются добавлением триазола, имидазола или уксусной кислоты.
Поэтому при использовании активированных эфиров можно
применять аминокомпонент в виде ацетата, т. е. без добавления
третичного основания.
Активированные эфиры являются удобными соединениями
для синтеза пептидов с незащищенной карбоксильной группой
и незащищенным С-концевым аргинином.
Реагенты переэтерификации. В 1964 г. Сакакибара и сотр.
[62] предложили для синтеза n-нитрофениловых эфиров защи*
щенных аминокислот использовать п-нитрофенилтрифтораце-
44
тат (62):
О
CF3—С-О—NOa
052)
В последующие годы появилась целая серия подобных реа-
гентов на основе N-оксисукцинимида, N-оксифталимида, пен-
тахлор- и пентафторфенолов с трихлор-, трифторацетатными
группами и остатками сульфокислот. Указанные реагенты весь-
ма удобны для синтеза активированных эфиров, они устойчи-
вы при хранении, легко синтезируются и в отличие от ДЦГК
нетоксичны. Их можно использовать как для получения акти-
вированных эфиров, так и непосредственно для образования
пептидной связи без выделения соответствующего активирован-
ного соединения.
Исследование механизма этой реакции показало, что она,
по-видимому, идет не за счет переэтерификации, как думали
раньше, а через промежуточное образование смешанного ан-
гидрида. Сначала анион карбоксильного компонента атакует
атом углерода СО-группы реагента Сакакибары (62), что приво-
дит к промежуточному соединению (63):
О
/° II к
R-C<f +CF3-C-О-NO2
хо-
(62)
О=С
,0-С—CF3
O-^-NO,
:-NOa +
(63)
R—С—О—С—CF3
(64)
no2
-> О~
(65)
t После внутримолекулярной перегруппировки (63) образу*
ется смешанный ангидрид (64) и n-нитрофенол, который ацили-
руется смешанным ангидридом (64) с образованием активиро-
ванного эфира (65) и трифторацетата [10]. Указанный механизм
подтверждается также тем, что при ацилировании реактивом
Сакакибары аминокомпонента в виде трифторацетата кроме же-
лаемого пептида (66) образуется трифторацетильное производ-
ное аминокомпонента (67). Это легко понять, если предполо-
жить, что в результате реакции сначала образуется смешанный
ангидрид [10]:
О
R-С—О—^-NOg + HgN—CHRj—СООМе • CF3COO
ZO “ О
R-C< II
)О + HgN—CHRj—СООМе -> R—С—NH—CHRj—СООМе +
CF3-C<
ХО (66)
+ CFgCO—NH—CHR!—СООМе.
(67)
Подробнее о реагентах переэтерификации см. обзор [113].
МЕТОДИКИ СИНТЕЗА РЕАГЕНТОВ
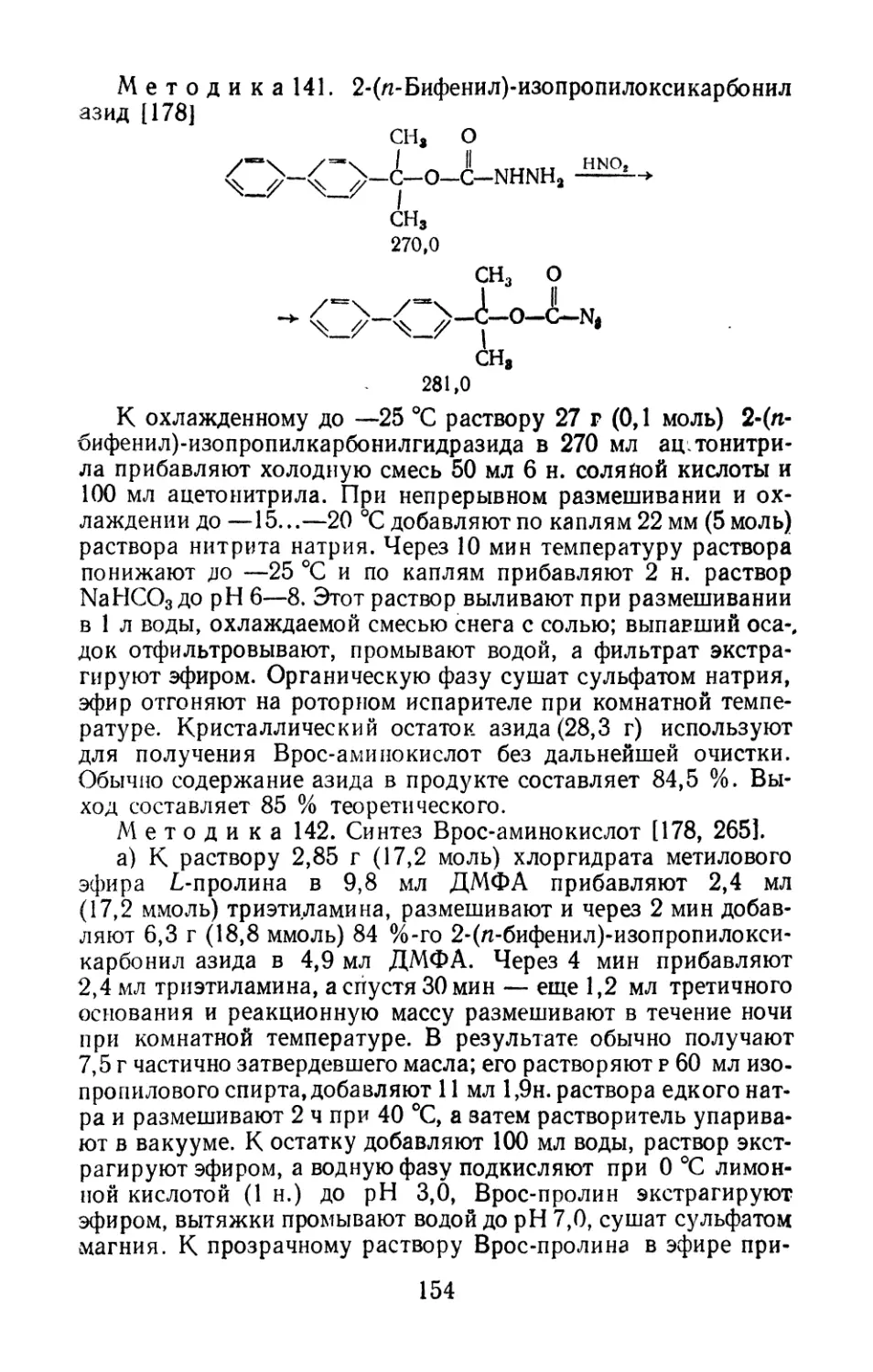
Карбодиимидный метод
Дициклогексилкарбодиимид
Методика 1. Дициклогексилмочевина [116]
О О
H2N—с— NHg + 2/ /—HN2 * х. /—NH—С—HN—/
60,1 1003 224,3 ~
Смесь 60 г (I моль) мочевины и 240 г (2,4 моль) циклогексил-
амина в 480 мл изоамилового спирта кипятят с обратным холо-
дильником 28 ч. После охлаждения раствора осадок отфильтро-
вывают, промывают эфиром и сушат на воздухе. Выход дицик-
логексилмочевины 200 г (89 %), т. пл. 234 °C. В н. амиловом
спирте выход продукта повышается до 94 %.
Методика -2. Дициклогексилкарбодиимид [116]
NH—С—NH—+ H8C-fy SO2C1 —
н X—/ Х=/
О 190,7
224,3
206 ~
46
Раствор 200 г (0,9 моль) дициклогексилмочевины и 300 г
(1,57 моль) n-толуолсульфохлорида в 600 мл сухого {пиридина
перемешивают в течение 1 ч при 70 °C и затем реакционную мас-
су выливают в колбу емкостью 3 л, содержащую 1,5 кг измель-
ченного льда. Продукт реакции извлекают эфиром. Получен-
ные органические вытяжки фильтруют, промывают водой до
pH 7,0, сушат сульфатом магния и эфир удаляют на роторном
испарителе. Остаток перегоняют с воздушным холодильником
при 11 мм рт. ст., собирая фракцию, кипящую в пределах 148—
152 °C. Выход дициклогексилкарбодиимида 152 г (82 %), т. пл.
35 °C. Продукт застывает в виде светлой воскообразной массы
и может длительно храниться в холодильнике. Д и ц и к л о»
гексил карбодиимид ядовит! Следует избегать
попадания его на кожу, так как он вызывает сильную аллерги-
ческую реакцию. Все работы с ДЦГК необходимо проводить
в вытяжном шкафу в резиновых перчатках.
Водорастворимый 1-этил-3 (3-диметиламинопропил) карбодиимид
Методика 3. 1-Этил-3(3-диметиламинопропил)мочевина
С2Н5—NCO + H2N—СН2СН2СН2—N (СН3)а ->
Н3С-СН2—NH—СО—NH—CH2CH2CH2-N (СН3)2.
К раствору 20,5 г (0,288 моль) технического этилизоциана-
та в 200 мл безводного эфира медленно по каплям добавляли
28,9 г (0,288 моль) М,М-диметил-1,3-диаминопропана в 100 мл
эфира. Температуру смеси поддерживали умеренной, охлаждая
на ледяной бане. После перемешивания при комнатной темпе-
ратуре в течение 2 ч эфир удаляли под уменьшенным давлением
и получали в остатке с количественным выходом неочищенную
мочевину в виде светло-желтого масла. Продукт можно исполь-
зовать без дальнейшей очистки для получения карбодиимида.
Методика 4. 1-Этил-3(3-диметиламинопропил)карбоди-
имид
Н3С—СН2—NH—СО—NH—СН2СН2СН2—N (СН3)2 +
ч- Н3С—SO2C1 Н8С—СН2—N=C—N-CH2CH2CH2N (СН8),.
К раствору 42 г (0,24 моль) неочищенной 1-этил-3(диметил-
аминопропил)мочевины в 750 мл метиленхлорида и 130 мл
триэтиламина’добавляли раствор' 91,3 г (0,48 моль) перекристал-
лизованного n-толуолсульфохлорида в 500 мл метиленхлорида.
Температура смеси поддерживалась 5 9С путем внешнего ох-
лаждения. После добавления всего п-толуолсульфохлорида
реакционную смесь оставляли нагреваться до комнатной тем-
пературы, а затем кипятили 3—4 ч. Затем реакционную смесь
перемешивали трижды с 200 мл 40 %-го водного раствора кар-
47
боната натрия, и после каждой промывки твердый осадок и вод-
ную фазу отделяли и тщательно промывали метиленхлоридом.
Комби тированные органические фазы концентрировали, и мас-
лянистый остаток экстрагировали несколькими порциями эфи-
ра. После отгонки эфира остаток перегоняли в вакууме и полу-
чили 19,3 г (51 %) 1-этил-3(3-диметиламинопропил)карбодии-
мида с т. кип. 53—54 °C (0,6 мм рт. ст.), п =* 1,4504.
Методика 5. Хлоргидрат 1-этил-3(3-диметиламино-
пропил)карбодиимида.
Смесь 1,71 г (0,011 моль) 1-этил-3(3-диметиламинопропил)~
карбодиимида и 1,07 г (0,0092 моль) хлоргидрата пиридина в
10 мл метиленхлорида перемешивали в течение 3 мин при ком-
натной температуре. При добавлении по каплям 100 мл безвод-
ного эфира выпадал кристаллический продукт, который
отфильтровывали и перекристаллизовывали из смеси метйлен-
хлорид — эфир. Выход 1,76 г (99,5 %), т. пл. 113,5—114,5 °C.
Когда загрузку увеличивали в 10—20 раз, выход продукта со-
ставлял 85—90 %, т. пл. 108—112,5 °C.
Методика 6. Синтез пептидов карбодиимидным ме-
тодом.
Обычно эквимолярные количества защищенной аминокис-
лоты, хлоргидрата, бромгидрата или бензолсульфоната амино-
компонента, третичного основания (триэтиламина, этилдиизо-
пропиламина или N-этилморфолина) и ДЦГК растворяют в под-
ходящем растворителе при 0 °C, перемешивают 1—2 ч и, нако-
нец, оставляют на 12 ч при комнатной температуре.
Лучшими растворителями для синтеза пептидов карбодии-
мидным методом являются хлористый метилен, диоксан, тетра-
гидрофуран и ацетонитрил. Если компоненты реакции не рас-
творяются в указанных растворителях, то используют ДА1ФА
или диметилсульфоксид; при этом, однако, следует помнить,
что в полярных растворителях наблюдается более высокая ра-
цемизация. Поскольку реакция с ДЦГК экзотермична, иног-
да рекомендуют начинать конденсацию не при 0 °C, а при болев
низкой температуре (—70 °C).
Выпавшую дициклогексилмочевину отфильтровывают и со-
бирают, так как ее можно в дальнейшем использовать для син-
теза ДЦГК. В качестве примера ниже приведен синтез пептида.
Методика 7. Метиловый эфир бензилоксикарбонилфе-
нилаланилсерина [114].
К раствору 9 г (30 ммоль) карбобензоксифенилаланина,
5 г (32 моль) хлоргидрата метилового эфира серина и
4,34 г (31 ммоль) триэтиламина в 100 мл хлористого метилена,
охлажденного до —10 °C, прибавляют 8,24 г (40 ммоль) ДЦГК
и размешивают 2 ч при этой температуре, азатем оставляют на
16 ч при 20 °C.
48
Выпавшую дициклогексилмочевину отделяют фильтровани-
ем, растворитель удаляют в вакууме, а оставшееся масло рас-
творяют в 200 мл этилацетата и последовательно промывают 1 н.
соляной кислотой, водой, 1 н. бикарбонатом натрия и 25 %-м
раствором NaCl. Органическую фазу сушат безводным сульфа-
том магния в течение I ч. Когда раствор становится прозрач-
ным, этилацетат упаривают на роторном испарителе. Остаток
растирают с петролейным эфиром до затвердения и кристал-
лизуют из смеси этилацетат — петролейный эфир. Выход 10,9 р
(91 %), т. пл. 125 ’С, [ct]^ = —5,7° (с = 1; ДМФА).
Избыток ДЦГК рекомендуют разлагать несколькими кап-
лями уксусной кислоты, что может, однако, привести к ацети-
лированию аминокомпонента. Лучше для этой цели использо-
вать щавелевую кислоту, что приводит к образованию дицикло-
гексил мочевины и выделению углекислого газа (частное сооб-
щение В. Ф. Позднева).
Синтез нуклеофильных добавок
Методика 8. 1-Оксибензтриазол £115]
ОН
NO, ^\/N\
| II + H,N-NHS • Н,0I II N
^/\С1 4/\N^
157,6 50,05 135,13
Смесь 31,52 г (0,2 моль) о-хлорнитробензола и 32 мл
(0,65 моль) гидразингидрата в 100 мл спирта кипятят с обрат-
ным холодильником в течение 9 ч. После охлаждения раствора
спирт упаривали в вакууме до консистенции сиропа. Этот про-
дукт|растворяют в воде, экстрагируют эфиром; водную фазу от-
деляют и подкисляют соляной кислотой до pH 1,0. Выпавший
осадок отфильтровывают и кристаллизуют из воды. Выход
16 г (60 %), т. пл. 157 °C. Продукт сначала плавится примерно
при 100 РС, затем затвердевает и снова плавится при 157 9С.
Методика 9. N-Оксисукцинимид (1181
100,1 69,49 115,09
Колбу вместимостью 1 л, в которой содержится смесь хоро-
шо высушенных компонентов реакции: 81,7г (0,82 моль) янтар-
ного ангидрида и 57,2 г (0,5 моль) солянокислого гидроксил-
4 2 — 882
49
мина, присоединяют к роторному испарителю, включают ваку-
ум водоструйного насоса, а колбу помещают в масляную баню»
нагретую до 125 °C. Температуру бани поднимают в течение 1 ч
до 160 °C и выдерживают еще 1 ч при указанной температуре.
После этого нагревание прекращают, но колбу оставляют в ба-
не до тех пор, пока температура не снизится до 125 °C. Образо-
вавшуюся жидкость выливают тонкой струйкой под вытяжкой
в стакан, содержащий 400 мл эфира. Необходимо хорошее пе-
ремешивание! Когда продукт затвердеет, эфир декантируют, а
остаток нагревают с 400 мл n-бутанола, раствор фильтруют и
быстро охлаждают до 0 °C. Образовавшиеся кристаллы от-
фильтровывают. Если осадок не образовался, то раствор упа-
ривают досуха на роторном испарителе, а остаток промывают
сухим н-бутанолом и эфиром. Полученный продукт плавится
при 89 °C. Для очистки кристаллическую массу помещают в
колбу объемом 1 л и несколько раз обрабатывают кипящим этил-
ацетатом, порциями по 0,5 л. Этилацетат собирают и охлаждают,
продукт выпадает в виде красивых бесцветных кристаллов. Вы-
ход 59 г (67 %), т. пл. 95—96 9С.
Примечание. Если при кристаллизации из я-бутанола
выпадает осадок, его также необходимо обрабатывать этилаце-
татом для удаления бутанола.
Ванг и соавт. [119] предложили новую методику синтеза
N-оксисукцинимида, которая проще в исполнении и лучше вос-
производима, чем вышеописанная методика.
Методика 10. N-Оксисукцинимид [119]
о н о
JI ch2-c-nh-oh U
С \ CH3ONa | п /°
HaN—ОН’НС1 + 1 О_-------I ° -----I N-OH4-H.O
СН2-С-ОН SZ
69,49 100,1 115,09
Свежеприготовленный раствор 6,7 г (0,291 моль) натрия в
100мл абсолютного метанола приливали к перемешиваемой сус-
пензии 22,2 г (0,291 моль) тонкоизмельченного хлоргидрата гид-
р жсиламина. Смесь кипятили 15 мин на паровой бане, охлажда-
ли 15 мин на бане со льдом и отфильтровывали осадск NaCl.
К фильтрату добавляли 29,1 г (0,291 моль) янтарного ангидрида
малыми порциями в течение нескольких минут при перемеши-
вании, а затем смесь кипятили 2 ч. После отгонки избытка рас-
творителя 1,2 л толуола (или ксилола) смешивали с вязкой
N-оксисукцинимидной кислотой и кипятили в чечение 4 ч. В про-
цессе кипячения использовали насадку Дина — Старка для
отделения смеси метанол — толуол — вода (около 200 мл). Го-
рячий растворитель был декантирован и при стоянии дал оса-
50
док технического N-оксисукцинимида с т. пл. 86—92 °C. Твер-
дый осадок был экстрагирован из реакционной колбы смесью
толуольного маточного раствора и метилэтилкетона (100 мл).
При концентрировании и охлаждении этого раствора получили
вторую порцию продукта. После перекристаллизации из эти-
лацетата получено 16,5 г (49 %) белого чешуйчатого вещества
с т. пл. 97—98 °C.
Методика 11. «Комплекс F» (комплекс ДЦГК с пента-
фторфенолом) [120]
В минимальном количестве гексана растворяют отдельно
каждый из следующих компонентов: 2,06 г (10 ммоль) дицикло-
гексилкарбодиимида и 5,6 г (30 ммоль) пентафторфенола. Полу-
ченные растворы соединяют и смесь выдерживают в течение
1 ч. Выпавший осадок отфильтровывают и кристаллизуют из
гексана. Выход 6,7 г (88 %), т. пл. 101,5—102,5 °C.
При синтезе пептидов прибавляют «комплекс F» к смеси
карбоксильного и аминокомпонента в эквимолярном количест-
ве; иногда используют небольшой избыток комплекса.
Синтез с использованием ЭЭДХ
Методика 12. 1-Этоксикарбонил-2-этокси-1,2-дигидро-
хинолин (ЭЭДХ) [121]
129,16
с2нло-с = о
247,29
108,53
Раствор 155 мл (1,0 моль) триэтиламина в 92 мл (2,0 моль)
абсолютного спирта прибавляют по каплям при —5 °C к смеси
97 мл (1,0 моль) этилхлорформиата и 130 г (1,0 моль) хинолина
в 300 мл сухого бензола. После перемешивания в течение 1 ч
4*
51
содержимое колбы промывают водой и водный слой экстраги-
руют несколько раз хлороформом. Соединенные вытяжки су-
шат сульфатом магния, хлороформ отгоняют в вакууме. После
добавления к образовавшемуся маслянистому остатку 20 мл
эрира вся масса затвердевает. Бесцветный продукт отфильтро-
вывают, промывают охлажденным эфиром и сушат. Выход
140 г (57 %), т. пл. 63,5—65 °C. После стояния маточного рас-
твора в холодильнике в течение ночи можно выделить еще 25 г
продукта. Общий выход 165 г (66 %).
Методика 13. Этиловый эфир бензилоксикарбонил-
аланилглицина [47].
К раствору 0,67 г (3 ммоль) бензилоксикарбонилаланина и
0,42 г (3 ммоль) этилового эфира глицина в 25 мл тетрагидро-
фурана прибавляют 0,74 г (3 ммоль) ЭЭДХ и смесь перемеши-
вают 3 ч при 25 °C. После удаления растворителя в вакууме
остаток кристаллизуют из смеси этилацетат — петролейный
эрир. Выход 0,85 г (90 %).
Карбонилдиимидазолидный метод
Метод и к а 14. N.N-Карбонилдиимидазол [531
68,08
98,9 162,15
+ 2НС1
2,2 л абсолютного бензола перегоняют над 10 г гидрида каль-
ция, собирая дистиллят в хорошо высушенную трехгорлую кол-
бу емкостью 3 л, которая содержит 112 г (1,65 моль) имидазо-
ла. Суспензию имидазола размешивают при нагревании до пол-
ного его растворения. В полученный раствор пропускают38,6 г
(0,4 моль) сухого фосгена. Выпадает в осадок солянокислый
имидазол. Для удаления избытка фосгена через раствор проду-
вают ток сухого азота; массу охлаждают до 30 °C и оставляют
до тех пор, пока жидкость не станет прозрачной (10—24 ч),
после чего колбу нагревают до 50 °C и жидкость фильтруют.
Бензол частично отгоняют на роторном испарителе, а выпав-
ший в осадок продукт отфильтровывают, сушат над фосфорным
ангидридом в эксикаторе. Выход карбонилдиимидазола 50,4 г
(77 %), т. пл. 113—117 °C.
Примечание. Реагент чрезвычайно гигроскопичен,
поэтому его хранят в эксикаторе над Р»О5.
Методика 15. Метиловый эфир бензилоксикарбонил-
глутаминилгистидина. 0,14 г (0,5 ммоль) карбобензоксиглута*
мина растворяют в 1,5 мл свежеперегнанного ДМФА и смеши
вают, исключая попадание влаги, с 0,097 г (0,6 ммоль) карбо’
52
нилдиимидазола. Как только прекратится выделение СО2,
добавляют раствор 0,085 г метилового эфира гистидина
(0,5 моль) в 1 мл ДМФА и смесь оставляют на ночь при 0 °C.
Образовавшийся дипептид осаждают из раствора большим ко-
личеством эфира, отфильтровывают и сушат. Выход 0,19 г
(88 %), т. пл. 170—173 °C; [а]?? = —32,2° (с = 1; 1 н. соля-
ная кислота).
Азидный метод
Методика 16. трет-Бутилнитрит [1231
СН9 СН,
СН8—С—ОН СН8—С—ONO
СН2 <Lh,
74,12 103,12
В стакан вместимостью 0,5 л, снабженный термометром,
мешалкой и капельной воронкой, помещают 74 г тр^т-бутилс-
вого спирта и прибавляют раствор 76 г нитрита натрия в 140 мл
воды. Смесь охлаждают до 0 °C и к ней при перемешивании при-
бавляют из капельной воронки 90 мл концентрированной со-
ляной кислоты так, чтобы температура раствора не поднималась
выше 5 °C. Раствор переносят затем в делительную воронку
вместимостью 1 л и встряхивают со 100 мл воды. Водный слой
отбрасывают, а органическую фазу промывают 5 %-м раство-
ром бикарбоната натрия (3 X 50 мл), затем водой (3 X 50 мл),
сушат хлористым кальцием и перегоняют, собирая фракцию,
кипящую при 62—64 °C. Выход 62 г (60 %), т. кип. 63 °C. трет-
Бутилнитрит представляет собой желтую подвижную жид-
кость, легко растворимую в органических растворителях.
Осторожно! Вдыхание паров mpem-бутилнитрита
вызывает сильную головную боль.
Методика 17. Гидразид бензилоксикарбонилфенил-
аланил-серина [114].
6,8 г (17 ммоль) метилового эфира бензилоксикарбонилфе-
нилаланил-серина растворяют в 40 мл метанола, прибавляют
2 мл (40 ммоль) гидразингидрата и раствор оставляют на 12 ч
при комнатной температуре. Образовавшиеся бесцветные кри-
сталлы отфильтровывают, промывают 20 мл метанола, 50 мл
смеси метанол — эфир (1:1) и затем 100 мл эфира. Выход
6,3 г (90 %), т. пл. 193 °C; [all? = —2,6° (с = 1, ДМФА).
Методика 18. Этиловый эфир карбобензоксифенилала-
нил-серил-триптофанил-глицил-аланина [1241
z—Phe-Ser—NHNH2 + Н—Trp—Gly—Ala—OEt -ON—->
Z—Phe—Ser—Trp—Gly—Ala—OEt.
53
К раствору 7,8 г (19,5 ммоль) гидразида бензилоксикарбо-
пилфенилаланил-серина в 20 мл ДМФА, охлажденному до
—20 °C, прибавляют раствор 80 ммоль хлористого водорода в
гетрагидрофуране и затем — 2,3 мл (20 ммоль) тре/п-бутил-
нитрита. Реакционную смесь размешивают 30 мин при —20 °C,
а затем прибавляют 11 мл (80 ммоль) триэтиламина (pH 8—9)
и раствор аминокомпонента, который получают гидрированием
9,8 г (20 ммоль) этилового эфира бензилоксикарбонилтрипто-
фанил-глицил-аланина.
Реакционную массу перемешивают при —10 °C в течение 1 ч
и затем оставляют на ночь в холодильнике. Соли отфильтровы-
вают,'растворитель отгоняют в вакууме, а остаток растирают
со 150 мл этилацетата. Этот раствор последовательно промыва-
ют 1 н. соляной кислотой, водой, 1 н. бикарбонатом натрия,
25 %-м раствором NaCl. Органическую фазу сушат сульфатом
магния, этилацетат отгоняют в вакууме, а остаток кристалли-
зуют из смеси спирт — вода. Выход 8,4 г (65 %), т. пл. 148—
151 °C; [а1д = —18,8° (с = 1; спирт).
Методика 19. /прет-Бутилоксикарбонилгидразид бен-
зилоксикарбонил глицина [1251
Z—Gly—ОН -I- H.N—NH—Вос Z—Gly—NH—NH—Вос.
К охлажденному до 0 °C раствору 2,9 р (10 ммоль) Z—
GlyOH в 100 мл этилацетата добавляют 2,9 г (10 ммоль) трет-
бутилоксикарбонилгидразида и 2,06 р (10 ммоль) ДЦГК.. Реак-
ционную массу размешивают 1 ч при 0 °C, а затем 2 ч при 20 °C.
После завершения реакции в раствор добавляют несколько
капель уксусной кислоты для того, чтобы разрушить избыток
ДЦГК, выдерживают 20 мин при 20 °C и выпавшую дицикло-
гексилмочевину отфильтровывают. Прозрачный раствор про-
мывают 1 н. бикарбонатом натрия, водой, 1 н. лимонной кисло-
той и снова водой. Органическую фазу сушат сульфатом магния
и этилацетат отгоняют досуха в вакууме. Остаток кристаллизу-
ют из смеси этилацетат — петролейный эфир. Выход 2,87 р
(89 %), т. пл. 81—82 °C.
Методика 20. Общий метод синтеза пептидов по Хирш-
ману [79]. >
К охлажденному до —40 °C раствору 2 ммоль гидразида
защищенного пептида в 15 мл ДМФА прибавляют при пере-
мешивании 10 н. раствор хлористого водорода в ТГФ до pH
2,5, а затем добавляют 2,1 ммоль трет-бутил нитрита. Реакци-
онную массу перемешивают 1 ч при —40 °C, после чего вносят
4 ммоль N-оксисукцинимида и добавляют триэтиламин до pH
6,0. Смесь выдерживают 24 ч при 5 °C и еще 48 ч при 20 °C.
Продукт выделяют обычным способом. Выход пептида 70—
«0 %.
54
Методика 21. Гидразид трет-бутилоксикарбонил-ф-
бензил)-аспартил-лейцил-е-бензилоксикарбонил-лизина [126]
1. дцгк
2 j-j эд_эдр]
Вос—Asp—Leu—Lys—ОН — новт—2—* ®ос—ASP—^eu—Lys—NHNH2.
i I II
OBz.l Z OBzl Z
2 г (2,86 ммоль) карбоксильного компонента растворяют в
7 мл ДМФА, охлаждают до О °C и прибавляют 0,108 г 3,4 (ммоль)
безводного гидразингидрата, затем прибавляют 0,963 г
(6,3 ммоль) 1-оксибензтриазола и 0,648 г (3,15 ммоль) ДЦГК и
перемешивают 17 чприО°С. После удаления выпавшей дицик-
логексилмочевины ДМФА отгоняют в вакууме, остаток раство-
ряют в 150 мл этилацетата и последовательно промывают водой,
1 н. раствором бикарбоната натрия, снова водой; сушат сульфа-
том натрия. После удаления растворителя остаток кристалли-
зуют из смеси метанол — эфир. Выход гидразида 1,46 г (72 %),
т. пл. 133—137 °C; [<x]d = —19,36° (с = 1, тетрагидрофуран).
Методика 22. Синтез дифенилфосфоразида [82]
О
PhO. ,,О |1 , OPh
)Р< + NaN. -> N3—Р< + NaCl
РИО/ ХС1 4OPh
268,64 65,01 275,20
Смесь 56,8 г (0,21 моль) дифенил хлорфосфата и 16,3 г
(0,25 моль) азида натрия в сухом ацетоне перемешивают при ком-
натной температуре 21 ч. Отфильтрованный раствор концентри-
руют на роторном испарителе и остаток перегоняют в ваку-
уме (0,15 мм рт. ст.) при 152—155 °C. Получено 53,3 г (92 %)
бесцветного масла.
М ето д и к а 23. Синтез этилового эфира карбобензокси-
глутамил-(гамма-тр#/п-бутил)-глицина [85].
К перемешиваемой смеси 8,44 г (25 ммоль) гамма-тр^т-бу-
тилового эфира карбобензоксиглутаминовой кислоты и хлор-
гидрата этилового эфира глицина (3,48 г, 25 ммоль) в 80 мл
ДМФА добавляют 4,08 г (25 ммоль) дифенилфосфоразида в
10 мл ДМФА при охлаждении льдом, затем вносят 5,06 г
(50 ммоль) триэтиламина в 10 мл ДМФА.Смесь перемешивают 4 ч
при охлаждении льдом, затем 15 ч при комнатной температуре.
Добавлена смесь этилацетат — бензол (4 : 1,1л), раствор про-
мывают обычным образом, высушивают сульфатом магния.
После удаления растворителя на роторном испарителе и крис-
таллизации остатка из смеси диэтиловый эфир — гексан полу-
чено 9,25 г (87 %) бесцветных кристаллов, т. пл. 67—68 °C;
[а]/? == —18,5Q (с == 1; 04, метанол).
55
Метод смешанных ангидридов
Методики синтеза пептидов хлорангидридным методом
Методика2 4. Синтез хлорангидридап-толуолсульфо-
нил-Ь-изолейцина [75].
Растворяли 2,9 г (10 ммоль) тозил-А-изолейцина в 15 мл су-
хого эфира, добавляли 2,2 г (10,6 ммоль) РС18 и содержимое кол-
бы встряхивали до растворения РС15. Раствор декантировали
с небольшого количества непрореагировавшего материала и
эфир удаляли в вакууме водоструйного насоса. Оставшуюся
хлорокись фосфора удаляли вакуумированием на масляном на-
сосе в течение 1 ч. Получали 3 г хлорангидрида в виде прозрач-
ного масла. Вещество пригодно для использования без дальней-
шей очистки.
Методика 25. Хлорангидрид трифторацетил-Е-фенил-
лаланина [127].
К суспензии 261 мг (1 ммоль) трифторацетил-1-фенилалани-
на в 5 мл сухого бензола добавляют 0,2 мл (2,7 ммоль) очищен-
ного тионилхлорида. Смесь кипятят с обратным холодильни-
ком с хлоркальциевой трубкой в течение 2,5 ч. Бензол и избы-
ток тионилхлорида удаляют в вакууме, остаток растворяют в
15 мл сухого бензола, упаривают в вакууме, опять растворяют
в 15 мл сухого бензола, горячий раствор фильтруют, охлажда-
ют и добавляют 10 мл петролейного эфира (т. кип. 30—60 °C).
Раствор выдерживают в течение ночи в холодильнике, отфильт-
ровывают осадок, промывают его эфиром и сушат в вакуум-
эксикаторе. После двух перекристаллизаций из смеси бензол —
петролейный эфир (2 i 1) получают продукт с т. пл. 109,5—
111,5 °C; [а]/> = +15,5° (с = 0,2, уксусная кислота). Добав-
лением небольших количеств ДМФА можно значительно со-
кратить время реакции.
Методика 26. Синтез метилового эфира тозил-8-бен-
зил-1-цистеинил-Ь-тирозил-Е-фенилаланииа [128].
2,75 г (4,5 ммоль) метилового эфира М.О-дикарбобензокси-
L-тирозил-Е-фенилаланина деблокировали бромистым водоро-
дом в уксусной кислоте, промывали сухим эфиром и сушили в
вакууме над Р2О6, Выход 1,5 г (80 %). Продукт растворяли в
10 мл воды и 10 мл этилацетата и добавляли к смеси раствор
0,5 г карбоната натрия в 18 мл воды. Смесь охлаждали до 0 °C
и вносили в нее раствор 1,3 г (3,5 ммоль) хлорангидрида тозил-
S-бензил-Е-цистеина в 7 мл этилацетата при сильном переме-
шивании. Продукт был выделен и? ограиической фавы. Выход
1,65 г (71 %),т.пл. 168—171РС; [а® «₽ — 23,1° (с = 2,4, АсОН).
Методика 27. Общий метод синтеза пептидов с исполь-
зованием катализа ДМФА [10].
56
К охлажденному раствору (—15...—30 °C) N-защищенной
аминокислоты в ДМФА прибавляют 1 экв. очищенного тионил-
хлорида и выдерживают в течение 30 мин. К полученному хло-
рангидриду прибавляют раствор хлоргидрата эфира амино-
кислоты или пептида и 3 экв. триэтиламина (или другого тре-
тичного амина) и оставляют на 18 ч при комнатной температу-
ре. Продукт выделяют обычным образом.
Методика 28. Синтез метилового эфира карбобензок-
си-£-фенилаланил-1-аланина [74].
К перемешиваемому раствору дициклогексиламмонийной
соли карбобензокси-£-фенилаланина (356 мг, 0,74 ммоль) и
пиридина (76 мг, 0,96 ммоль) в 1 мл СНаС12 прибавляют 106 мг
(0,89 ммоль) тионилхлорида при комнатной температуре в ат-
мосфере аргона. Смесь размешивают при этой температуре
1 мин, а затем добавляют 80 мг (0,57 мм зль) хлоргидрата метило-
вого эфира L-аланина и 151 мг (1,2 ммоль) 4-диметиламинопи-
ридина (D МАР) в 1 мл метиленхлорида. Смесь перемешивают
20 мин при комнатной температуре, затем выливают в 5 мл рас-
твора NaCl и продукт экстрагируют этилацетатом. Органиче-
скую фазу последовательно промывают 10 %-й лимонной кисло-
той, насыщенным раствором бикарбоната натрия, раствором
хлорида натрия и сушат над сульфатом натрия. После отгон-
ки растворителя получили 220 мг (99 %) дипептида. Перекрис-
таллизация смеси этилацетат — петролейный эфир дает 208 мг
(94 %) с т. пл. 126—127 °C, [а]2р = —22,9° (с = 1,25, этанол).
Этим способом получено 10 дипептидов с выходами 76—94 % .
Методика 2 9. Хлорангидрид 9-флуоренилметилокси-
карбонил-Л-валина. Общий метод получения хлорангидридов
9-флуоренилметилоксикарбонил-аминокислот [76].
Раствор 0,3 г (0,88 ммоль) Fmoc-L-валина в 5 мл СН2С12
обрабатывали 1,05 г (8,8 ммоль) тионилхлорида и кипятили
смесь 15 мин в атмосфере азота (в других случаях для заверше-
ния реакции требовалось 1—2 ч; в присутствии 0,1 экв. ДМФА
реакция завершается за 1 ч при 20 °C). Растворитель и избыток
тионилхлорида удаляли в вакууме и растворяли остаток в 1 мл
метиленхлорида. Добавлением 10 мл гексана выделяли 0,27 г ,
(85,3 %) чистого продукта с т. пл. 111—112 °C; [а]д = +5,5°
(с = 1, метиленхлорид). Этим методом получено 14 хлорангид-
ридов Fmoc-аминокислот о выходом 85—96 %.
Смешанные ангидриды
Методика 30. Метиловый эфир беязплоксикарбопил-
пролилаланина [129].
К раствору 12,4 г (50 ммоль) бензилоксикарбопилпролина
в 100 мл сухого хлороформа прибавляют при —12 СС 6,9 мл
57
(50 ммоль) триэтиламина и 6,55 мл (50 ммоль) изобутилхлорфор-
миата. Через 15 мин в реакционную массу вносят 7 г (50 ммоль)
хлс.ргидрата метилового эфира аланина в 50 мл ДМФА,
содержащего 6,9 мл (50 ммоль) триэтиламина. Смесь размеши-
вают 30 мин при —10 °C, затем 1 ч при 0 °C и оставляют на ночь
при комнатной температуре. Соли удаляют фильтрованием,
растворитель отгоняют в вакууме, а остаток растворяют в этил-
ацетате и промывают как обычно. Органическую фазу сушат
сульфатом магния, этил ацетат отгоняют досуха на роторном
испарителе, продукт растирают с петролейным эфиром до обра-
зования аморфной массы. После кристаллизации из смеси эти-
лацетат— петролейный эфир выход продукта 8,17 г (50 %),
т. пл. 72—75 °C.
Методика 31. Изобутиловый эфир хлоругольной кис-
лоты [81]
О
СН3 СН3 ||
>СН-СН2ОН + СОС12 -> )СН-СН2О-С—С1 + НС1
СН/ СН3/
74,12 98,92 136,58
Для синтеза используют установку, которая описана для
получения карбобензоксихлорида (см. главу 2). К 148,4 г
(8Ц5 моль) жидкого фосгена при охлаждении до —40 °C мед-
ленно при перемешивании прибавляют 74,1 г (8^1 моль) без-
водного изобутилового спирта. Убирают охлаждающую баню,
перемешивают еще 20 мин при 0 °C и оставляют содержимое
колбы на ночь при комнатной температуре. После продувки
сухого воздуха для удаления избытка фосгена и хлористого во-
дорода (см. главу 2) в течение 2 ч продукт перегоняют. Выход
изобутилхлорформиата 95—96 г (70 %), т. кип. 123—126 °C.
Внимание! При работе соблюдать все правила работы
с фосгеном, описанные в главе 2.
Ташнер и соавт. [88] показали, что использование для полу-
чения смешанных ангидридов изовалериановой и изобутиловой
кислот позволяет применять в качестве карбоксильного компо-
нента дипептиды практически без рацемизации.
Методика 32. трет-Бутиловый эфир бензилоксикар-
бонилфенилаланил-валил-фенилаланина [88]
Z—PheValOH Д- HPheOBi? Z—Phe—Val--PheOBuz.
Раствор 80 мг (0,2 ммоль) Z—Phe—Vai—ОН в 1 мл тетра-
гидрофурана смешивают при —15 °C с 22 мл (0,2 ммоль) N-ме-
тилморфолина. К раствору при размешивании прибавляют
0,21 ммоль хлорангидрида изовалериановой кислоты. Спустя
2 мин к охлажденному раствору прибавляют 46,4 мр
(0,21 ммоль) аминокомпонента в 0,8 мл ТГФ. Реакционную смесь
выдерживают 1 мин при —15 °C и 30 мин при комнатной темпе-
58
ратуре. Тетрагидрофуран отгоняют в вакууме, остаток раство-
ряют в 6 мл этилацетата; органическую фазу промывают (3 X
X 2 мл) последовательно 5 %-м бикарбонатом натрия, 2 мл
воды, 2 мл полупроцентной соляной кислоты, водой, сушат
сульфатом магния и растворитель отгоняют в вакууме. Выход
маслообразного продукта соответствует обычной методике.
Методика 33. Хлорангидрид триметилуксусной кис-
лоты (пивалоилхлорид) [130]
СН3 СН3 °
СН,—с!—СООН -Ь ЗОС12 -> СН,-С—С^ + SO, + НС!
I ' I ХС1
СН, СН,
102,8 118,90 120,57
В колбе емкостью 250 мл, снабженной обратным холодиль-
ником, нагреваем до кипения смесь 35 г (0,325 моль) триметил-
уксусной кислоты и 27 мл (0,325 моль) тионилхлорида. Нагре-
вание вели 5 ч, после чего реакционную смесь перегоняли на
колонке эффективностью пять теор. тарелок, собирая фракцию
104—106 °C. Выход пивалоилхлорида 30,2 г (78 %), «о =
= 1,4168.
Методика 34. Этиловый эфир тозилглицил-глици-
на (1311.
Раствор 2,29 г (10 ммоль) тозилглицина в 20 мл хлороформа,
содержащего 0,8 мл (10 ммоль) пиридина, обрабатывают при
—3 °C раствором 0,8 мл (10 ммоль) пивалоилхлорида в
20 мл хлороформа и выдерживают 5 мин при 0 °C. К смеси при
размешивании добавляют раствор 1 г (10 ммоль) этилового эфи-
ра глицина в 5 мл хлороформа, выдерживают 5 мин при 0 °C,
а затем 30 мин при комнатной температуре, после чего раство-
ритель отгоняют досуха в вакууме. Остаток последовательно
растирают 10 %-ной соляной кислотой, 5 %-м бикарбонатом
натрия и кристаллизуют из смеси бензол — петролейный эфир.
Выход 2,04 г, т. пл. 89—90 °C.
Тщательное исследование стадии активации карбоксильно-
го компонента было проведено Андерсоном с соавт. [132]. Для
сведения рацемизации к минимуму необходимо использовать
безводный ТГФ, N-метилморфолин, изобутилхлорформиат и
низкую температуру (—15 °C) реакции. Время активации не
должно превышать 1—2 мин.
В работе [133] исследовано влияние растворителей и основа-
ний на образование уретанов и рацемизацию. Показано, что
образование уретанов, а также степень рацемизации минималь-
ны при использовании N-метилпиперидина в метиленхлориде.
В работе [134] было показано, что в процессе синтеза элас-
тина при конденсации Вос—Vai—ОН с Н—Pro—OBzI мето-
59
дом смешанных ангидридов (изобутнлхлорформиат, N-метил»
морфолин) образуется 40—60 % уретана. Эту побочную реак-
цию удалось подавить добавлением 1-оксибензтриазола перед
введением аминокомпонента.
Интересная модификация метода предложена А. А. Мазуро-
вым с соавт. [1351: для избежания рацемизации используют
комплексы натриевых или калиевых солей защищенных ами-
нокислот с краун-эфирами, что дает возможность избежать при-
менения третичных аминов.
Синтез N, Ы-бис-(2-оксо-3-оксэзолидинил)
фосфодиамид хлорида
Методика 35. Оксазолидинон-2 [1361
С2Н6О. СН2—СН2
H2N-CH2CH2OH + )С=О -> I I 4-2.С2Н2ОН
C2H6OZ HN О
61,0 118,1 87,0
Смешивают 244 г (4 моль) 2-аминоэтанола и 472 г (4 моль)
перегнанного диэтилкарбоната и добавляют 0,5 г натрия, Ос-
торожно отгоняют около 6 моль (280 г) образующегося этанола
из смеси (экзотермическая реакция). Медленно прибавляют ос-
тавшееся масло к 2 л холодного (—40 °C) абсолютного эфира
(экзотермическая кристаллизация). Отфильтровывают образо-
вавшиеся кристаллы, промывают их несколько раз холодным
гексаном и сушат над Р2О6. Выход 313 г (90 %), т. пл.
87—89 °C.
Методика 36. М,М-(быс-2-Оксо-3-оксазолидинил)фос-
фодиамид хлорид (ВОР—С1) [136]
87,0 208,26
254,6
Добавляют 62,6 г (0,3 моль) РС15 порциями в течение 10 мин
к раствору 56,6 г (0,65 моль) оксазолидинона-2 в 300 мл ацето-
нитрила, поддерживая температуру реакционной смеси ниже
20 °C. Перемешивают 24 ч при комнатной температуре, охлаж-
дают до —5 °C и добавляют при энергетическом перемешивании
6 мл (0,3 моль) воды. Охлаждают до —40 °C и отделяют выпав-
ший осадок фильтрацией. Промывают кристаллы ацетонитри»
60
лом, холодной водой, снова ацетонитрилом, ацетоном и гекса-
ном сушат над Р2О5. Перекристаллизация из ацетонитрила. Вы-
ход ВОР—С1 27—42 г (35—55 %), т. пл. 195—198 °C (разл.).
Методика 37. Общий метод использования ВОР—С1
[136].
Прибавляют 1,1—1,2 экв. ВОР—С1 к раствору 1 экв. N-за-
щищенного аминокомпонента, затем, если потребуется 1 —
1,5 экв. 1-оксибензтриазола или имидазола и 1,2 экв. TEA (или
NMM) в метиленхлориде, ТГФ или ДМФА по выбору при О °C.
Перемешивают 5 мин, добавляют 1,1—1,2 экв. эфира аминоком-
понента (или его соли вместе с 1,1—1,2 экв TEA или NMM) и
по каплям в течение 0,5 ч (если количества превышают 4 ммоль)
вносят 1 экв. TEA или NMM. Перемешивают при комнатной
температуре до полного отсутствия аминокомпонента по ТСХ.
Отгоняют растворитель, растворяют в этилацетате, промыва-
ют обычным образом и выделяют полученный пептид. Выход
дипептидов 80—100 %. Примечания: 1) 1-оксибензтриа-
8ол или имидазол используют для эффективного подавления
рацемизации в случае активации дипептидов; 2) очень важно
использовать очищенный ВОР—С1; 3) растворять исходные
компоненты из расчета 5 мл/1 моль.
Методика 38. Специальная методика для производных
N-метил-аминокислот [136].
Добавляют 1,1—1,2 экв. ВОР—С1 к раствору 1 экв. карбок-
сильного компонента и 1,2 экв. NMM (или NEt3) в ТГФ или мети-
ленхлориде (из расчета 10 мл на 1 ммоль) ВОР—С1 при
—15 °C. Перемешивают 20 мин при этой температуре, отгоняют
растворитель наполовину и добавляют 1,1—1,2 экв. эфира ами-
нокомпонента (или соответствующей соли амина с 1,1—1,2 экв.
TEA или NMM) и 1 экв. NMM (или NEt3). Перемешивают при
0 °C необходимое время (контроль ТСХ) и выделяют продукт,
как в Приведенной выше методике. Выход дипептидов 80—
100 %.
Показано, что для эффективного подавления рацемизации
при конденсации пептидов с помощью ВОР—С1 нужно исполь-
зовать преактивацию карбоксильного компонента с добавкой
небольшого избытка 1-оксибензтриазола в ТГФ или ДМФА
(рацемизация 0—1,2 %) или имидазола в ТГФ (рацемизация
0,8 %) [137].
Методика 39. Общий метод синтеза симметричных
ангидридов N-защищенНых аминокислот [138].
К раствору 2 ммоль N-защищенной аминокислоты в 20 мл
хлористого метилена прибавляют при охлаждении лйДом
1 ммоль хлоргидрата М-этил-М'-(у-диметиламинопропил)-кар-
боднимида и раствор размешивают 2 ч при 0 °C. Хлористый
метилен отгоняют на роторном испарителе, к остатку добавля-
61
ют 25 мл этилацетата, и раствор последовательно промывают
охлажденным раствором лимонной кислоты (2 X 10 мл), 1 н.
раствором хлористого натрия, 1 н. бикарбонатом натрия и сно-
ва раствором NaCl. После охлаждения раствора до —5 °C вы-
падают кристаллы, их отфильтровывают и промывают петро-
лейным эфиром. Выход 51—86 %. В табл. 4 представлены фи-
зико-химические свойства синтезированных симметричных ан-
гидридов.
Таблица 4. Свойства симметричных ангидридов
Вос-аминокислот [138]
Аминокислота Выход, % T. пл., °C град (с «= 2, СНСЦ)
L-Аланин 75 95—97 — 16,9
р-Бензиловый эфир £-аспара- 86 Масло —4,0
гиновой кислоты
L-Валин 51 84—85 —4,0
у-Бензиловый эфир £-глута- 50 70—72 —27,0
миновой кислоты
Глицин 60 82—83
L-Изолейцин 55 68—69 —32,8
L-Лейцин 83 Масло — 16,6
L-Метионин 73 76—78 — 1,8
£-Пролин 80 Масло —60,8
о-Бензиловый эфир 54 Масло -3,5
L -сер ина
о-Бензиловый эфир L-тирози- 69 75—77 +8,1
на
L-Фенилаланин 76 95—97 + 19,6
Методика 40. Общий метод синтеза симметричных ан-
гидридов N-защищенных аминокислот без выделения [139].
Этот способ используют для синтеза пептидов твердофазным
методом. Смесь 2 ммоль Вос-аминокислоты и 1 ммоль ДЦГК
в минимальном количестве хлористого метилена выдерживают
20 мин при 0 °C, выпавшую в осадок дициклогексилмочевину
отфильтровывают, а раствор симметричного ангидрида исполь-
зуют для ацилирования амино компонента.
Методика41. Синтез пептидов в смеси ДМФА — во-
да [92].
К раствору 1 ммоль хлоргидрата, бромгидрата или тозила-
та эфира аминокислоты; или пептида в 5 мл смеси демитилфор-
мамид — вода (4 : 1) и 1 ммоль N-метилморфолина прибавляют
при перемешивании симметричный ангидрид, реакционную
смесь выдерживают в течение ночи при 23 °C. Если используют
избыток симметричного ангидрида, то спустя указанное время
62
прибавляют 1 ммоль 1-амино-З-диметиламинопропана'и разме-
шивание продолжают 1 ч. Затем прибавляют в колбу 100 мл
хлористого метилена и раствор последовательно промывают
водой (2 X 50 мл), 10 %-й лимонной кислотой, водой, насы-
щенным раствором бикарбоната натрия и, наконец, водой. Ра-
створ пептида в хлористом метилене сушат сульфатом магния
и растворитель отгоняют в вакууме. Защищенный пептид не
требует дополнительной очистки и может быть использован для
дальнейшего наращивания полипептидной цепи. Выход 90 %.
Карбоксиангидридный метод
Методика 42. Синтез N-карбоксиангидридов амино-
кислот с использованием фосгена [140]. N-карбоксиангидрид
глицина
101,0
H2N— СН2—СООН + СОС12
75,1
98,9
В сухую трехгорлую вместимость 2 л, снабженную трубкой
для подачи газа, обратным холодильником и мешалкой с си-
ликоновым затвором, помещают тонкоизмельченную суспен-
зию 8 г глицина в 375 мл сухого диоксана. При нагревании на
водяной бане до 40 °C в реакционную смесь медленно пропус-
кают ток фосгена в течение 4—5 ч; глицин при этом постепенно
растворяется. Для удаления избытка фосгена через раствор
продувают в течение 12 ч сухой воздух, после чего диоксан от-
гоняют на роторном испарителе при 40 °C. После добавления
небольшого количества сухого эфира остаток обычно кристал-
лизуется; кристаллы быстро отфильтровывают, промывают
сухим эфиром и хранят в эксикаторе над фосфорным ангидри-
дом. Выход 9,2 г (85 %).
Этим способом получают N-карбоксиангидриды других
аминокислот, однако время пропускания фосгена зависит от вре-
мени полного растворения аминокислоты.
Методика 43. Синтез N-карбоксиангидридов амино-
кислот с использованием хлористого тионила [93].
Раствор 5 г бензилоксикарбониламинокислоты в 25 мл све-
жеперегнанного хлористого тионила нагревают при 60 °C в
течение 30 мин, предохраняя реакционную смесь от попадания
влаги. Избыток тионилхлорида удаляют в вакууме, а остаток
растирают с сухим петролейным эфиром и затем кристаллизуют
из смеси этилацетат — петролейный эфир.
Методика 44. Синтез N-карбоксиландигдиров амино-
кислот через их серебряные соли [141]; N-карбоксиангидрид
63
серина
, NH----CHR
HaN—CHR—COO~Ag+ -I- COCL -> I I ~r AgCi
zCx zC\
0/ XOZ
Серебряная соль серина. Все операции должны проводить-
ся в темноте. К перемешиваемому раствору 105 г (1 моль)
L-серина в 2,5 л воды в течение 5 мин добавляют порциями 116 г
(1,1 моль) свежеприготовленной окиси серебра. Массу разме-
шивают 1 ч, а затем добавляют.50 г. активного угля и переме-
шивают еще 20 мин. Раствор фильтруют через целит, охлажда-
ют и прибавляют в течение 2 ч 750 мл метанола, затем в течение
ночи при размешивании и охлаждении до'О—5 °C прибавляют
по каплям 2 л метанола. Осадок отфильтровывают, примыва-.
ют метанолом и сушат в вакууме. Выход 126 г (40 %).
N-Карбоксиангидрнд серина. В приборе, защищенном от
света, пропускают рассчитанное количество фосгена через сус-
пензию 42,4 г серебрянной соли серина в 500 мл сухого диокса-
на, после чего смесь перемешивают 1 ч. Нерастворимый осадок
отфильтровывают, раствор лиофилизируют. Остаток растворя-
ют в минимальном объеме этилацетата и добавляют петролей-
ный эфир до помутнения раствора. Содержимое колбы переме-
шивают, охлаждая на ледяной бане. Когда жидкость станет
прозрачной, снова добавляют петролейный эфир до помутне-
ния и снова перемешивают. Выпавший в осадок продукт от-
фильтровывают, промывают петролейным эфиром и сушат в ва-
кууме. Выход 11 г (40 %), т. пл. 82 °C с разложением.
Методика4 5. Общий метод пептидов через N-карбок-
сиангидриды аминокислот [95] (табл. 5).
Аминокислоту (10 ммоль) растворяют в 10—50 мл натрий-
боратного буфера, pH 10. Сначала аминокислоту растворяют
в 0,45 моль борной кислоте и, используя 50 %-й раствор едко-
го натра, доводят pH до 10. С помощью pH-метра доводят pH
раствора до 10,2, раствор охлаждают до 0 °C и в качестве эмуль-
гатора добавляют каплю каприлового спирта. Смесь перено-
сят в миксер и включают мешалку. Карбоксиангидрйд соответ-
ствующей аминокислоты прибавляют весь сразу или порциями
в течение 15—30 с. Одновременно из автоматической бюретки
добавляют раствор едкого натра, поддерживая pH раствора
около 10,2. Обычно реакция заканчивается через 2 мин, и до-
бавление щелочи прекращают. При использовании NCA гли-
цина, аланина и пролина, которые лучше растворимы в воде,
чем NCA других аминокислот, синтез пептидов можно вести в
колбе с магнитной мешалкой. После окончания реакции рас-
твор подкисляют и пептид выделяют обычным способом.
Свойства N-карбоксиангидридов аминокислот указаны в
табл. 5.
64
Методика 46. Пролил-фенилаланин (951.
Раствор 1,65 г (10 ммоль) фенилаланина в 350 мл боратно-
го буфера охлаждают до 0 °C и доводят pH до 10,2. При интен-
сивном перемешивании в миксере к раствору добавляют 1,7 г
(12 ммоль) N-карбоксиангидрида пролина и размешивание про-
должают в течение 30 с; затем затвор подкисляют до pH 5 кон-
Таблица 5. Свойства N-карбоксиангидридов аминокислот
Название аминокислоты Вы- ход, % Растворитель для кристаллиза- ции Т. разл„ °C [«Id. град (с = = 1, аце- тон)
£-Аланин 92
£-Аспарагиновая кислота 25 — — —
P-Бензиловый эфир £-ас- — — 121 —
парагиновой кислоты
£-Аспарагин * 36 — — —
£-Валин 56,7 Гексан-этил аце- тат (9:1) 67—69 —44,4
Бензил-£-гистидин-НС1 — — 187—189
Глицин — — 100 —
у-Бензиловый эфир £-глу* — — 96—97 —•
тиминовой кислоты
£-Глутаминовая кислота 56,5 Этил ацетат гек- сан 83 —21,2
Бромгидрат £-гистидина * 50 — — —
£-Глутамин * 33 Метилэтил-кетон- гексан 270 —9,3
£-Лейцин —- — 76—77,5 —
е-Карбобензокси-£-лизин — — 100 —
£-Сер ин 42 — 82 —
е-трет-Б утилоксикарбо- 54,6 Ацетон-гексан 123 — 17,3
нил-£-лизин *
£-Пролин — — 45 —
о-Карбобензокси-£-тиро- — — 101 —
зин
£-Треонин * 49 Этилацетат-гек- сан 94—98 —62,7
• N-Карбоксиангидриды указанных аминокислот получены через серебряные
соли. *
центрированной серной кислотой. Кристаллы отфильтровыва-
ют, промывают водой и сушат. Выход 2,31 г (89 %), т. пл.
236—237,5 °C; [а]д =» —41,2° (с = 2,6; н. соляная кислота).
Методика 47. Метионил-тирозин [95J.
Синтез пептида ведут аналогично, но пептид выделяют ина-
че. После подкисления раствора до pH 5 его пропускают через
колонку с активным углем, на которой пептид адсорбируется.
Колонку сначала промывают 10 объемами воды, а продукт элю-
ируют смесью уксусной кислоты с 50 %-м ацетоном (1 s 3).
5 2-882
65
После удаления растворителя на роторном испарителе дипеп-
тид получают с выходом 80 %, т. пл. 259—265 °C.
В 1980 г. Кирхер и соавт. [96] опубликовали детальное ис-
следование по оптимизации условий проведения синтеза пеп-
тидов N-карбоксиангидридным методом. Условия реакции (для
минимализации побочных продуктов) представляют собой
компромисс значения pH, необходимого для повышения устой-
чивости карбамилата, с одной стороны, а с другой -г- для
уменьшения анионной полимеризации. Эти условия — pH 10,2
(0—3 °C) и добавление твердых N-карбоксиангидридов при ин-
тенсивном размешивании. Однако соблюдение этих условий не
гарантирует образования побочных продуктов (иногда в зна-
чительных количествах). Авторы работы [96] показали, что при
введении в реакцию больших избытков аминокислот (100—
200 %) в реакционной смеси содержится небольшое количество
простых примесей, которые легко разделить хроматографиче-
ски. Они получили 12 дипептидов при реакции N-карбоксиан-
гидридов со 100 %-м избытком аминокомпонента с хорошим вы-
ходом, и целевой продукт выделяли хроматографией на кизель-
геле.
Методика 48. Фенилаланил-глицин [96].
В реакционный сосуд добавляют 100 мл 0,5 н. калий-борат-
ного буфера, pH 10,2 и 0,75 г (10 ммоль) глицина. Раствор ох-
лаждают при перемешивании до 3 °C, доводят pH до 10,2 4 н.
КОН и после включения мешалки на полную мощность добав-
ляют 0,96 г (5 ммоль) N-карбоксиангидрида фенилаланина. Пу-
тем быстрого прибавления 4 н. КОН поддерживают pH 10,2.
Размешивают 0,5 мин на максимальных оборотах, а затем 1,5
мин — при уменьшенных оборотах, но так, чтобы температура
смеси не поднималась выше 4 °C- Затем реакционную смесь под-
кисляют 30мл полуконцентрированного раствора серной кис-
лоты до pH 3,0 в течение нескольких секунд интенсивного пе-
ремешивания и вслед за этим раствор нейтрализуют путем до-
бавления 4 н. КОН.
Полученный водный раствор упаривают досуха и пептидный
материал извлекают 300 мл горячего абсолютного метанола.
После удаления растворителя в вакууме остаток растворяют
в 30 мл смеси бутанол — уксусная кислота — вода (4:1:1)
и пропускают через колонку с кизельгелем (72 X 3 см), уравно-
вешенную этим же растворителем. Собирают фракции по 12 мл.
Фракции 40—70, содержащие нужный продукт, упаривают в
вакууме и оставшееся масло оставляют медленно кристаллизо-
ваться. Переосаждают продукт 200 мл эфира из минимального
количества метанола. Выход фенилаланил-глицина 1,05 г
(94,5 % из расчета на NCA), т. пл. 265—267 °C; [<x]z> — +101,5°
(с = 2, вода).
36
Метод синтеза с использованием
р -нитрофенилсульфенил N-карбоксиангидридов
аминокислот [97]
Методика 49. Синтез о-нитрофенилсульфенил-N-Kap-
боксиангидридов.
0,5 моля N-карбоксиангидрида аминокислоты растворяли в
300 мл ТГФ и охлаждали раствор до 0 °C на ледяной бане.
К раствору при перемешивании добавили 19 г (0,1 моль) кристал-
лического о-нитросульфенилхлорида. Затем при энергичном пе-
ремешивании добавили по каплям в раствор 14 мл триэтилами-
на. После окончания прибавления раствор перемешивали при
Таблица 6, о-Нитрофенилсульфенил-1М-карбоксиангидриды
аминокислот [97]
Аминокислота Выход. % T. пл., °C [a]p, град (ТГФ)
H-L-AlaOH 92 176—178 4-22,7 (c = 1)
H-L-Asp (OBzl) ОН 94 137—138 4-70,8 (c= 2)
H-L-ValOH 94 150—152 4-16,9 (c= 1)
H-GlyOH 94 168—170 —
H-L-Glu (OBzl) OH 94 масло 4-18,7 (c= 2,5)
H-L-HeOH 92 101 — 103 4-32,9 (c = 1)
H-L-LaH—OH 82 97—99 4-44,8 (c= 1)
H-L-Lys (Z) OH 94 121 — 123 4-23,5 (c = 2)
H-L-Phe—OH 92 151 — 153 4-31,2 (c= 2)
0 °C еще 15 мин. Выпавшие кристаллы хлоргидрата триэтилами-
на отфильтровывали, а фильтрат концентрировали на роторном
испарителе при 35 °C. Маслянистый остаток начинал кристал-
лизоваться при добавлении н. гексана. Кристаллы о-нитрофе-
нилсульфенил-М-карбоксиангидрида растворяли в небольшом
объеме этилацетата, а нерастворившийся материал удаляли
фильтрованием. После добавления к фильтрату гексана и ох-
лаждения его в холодильнике получалЪ бледно-желтые кри-
сталлы, которые перекристаллизовывали из этилацетата и полу-
чали чистый продукт, который отделяли фильтрацией и суши-
ли в эксикаторе над Р20б. Константы полученных соединений
указаны в табл. 6.
Методика 50. Общая методика синтеза пептидов с ис-
пользованием о-нитросульфенил-Ы-карбоксиангидридов ами-
нокислот [97].
0,1 моль хлоргидрата или n-толуол сульфоната эфира ами-
нокислоты растворяли в 200 мл ТГФ или ацетонитрила, до-
бавляли 14 мл (0,1 моль) триэтиламина и фильтровали соль.
Затем добавляли к этому раствору 0,105 моля о-нитросульфе-
нил-N-карбоксиангидрида аминокислоты и перемешивали 2 ч
67
при комнатной температуре. Упаривали растворитель на ро-
торном испарителе при 35 °C, оставшееся масло растворяли в
400 мл этилацетата и промывали раствор 5 %-й лимонной кис-
лотой, 50 %-м бикарбонатом натрия, водой и сушили над суль-
фатом натрия. Раствор концентрировали на роторном испарите-
ле при 40 °C и маслянистый остаток закристаллизовывали до-
бавлением 1 н. гексана. Перекристаллизацию производили из
этилацетата. Выход пептидов составил 86—98 %.
Использование N-карбоксиангидридов
для получения полиаминокислот
Изучение физико-химических свойств и пространственной
структуры синтетических полиаминокислот сыграло важную
роль в выяснении закономерностей и особенностей простран-
ственных структур белков, их иммуногенности и др. Полиамино-
кислоты подразделяют на гомополиаминокислоты (продукты
полимеризации одной аминокислоты) и гетерополиаминокис-
лоты (продукты сополимеризации двух или более аминокислот,
построенные по статистическим законам). Молекулярная мас-
са полиаминокислот колеблется от нескольких десятков до
100 000.
Наиболее эффективным способом получения полиаминокис-
лот является метод полимеризации NCA аминокислот с защи-
щенными боковыми реакционноспособными группами. Катали-
заторами полимеризации могут быть различные нуклеофилы,
которые раскрывают цикл:
R—СН—СО
I \о > НООС—NH—CHR-COOH —*
HN---СО
RCH—СО^
-> HaN—CHR—СООН ~СО ->
R-CH-CO
I >со
-► НООС—NH—CHR—CONH—CHR—СООН —“NrnCO ->
-> H2N—CHR—СО—[NH—СН—СО—j„ —NHCH—СООН-
I I
R R
Для получения степени полимеризации п > 100 используют
сильные основания: третичные амины, алкоголяты щелочных
металлов, гидроокиси щелочных металлов и другие, а для полу-
чения средней степени полимеризации (п —/20—100) применя-
68
ют воду, пиридин, аммиак, первичные и вторичные амины.
Ниже приводятся некоторые методики получения полиамино-
кислот. Мольное отношение NCA и инициатора обозначается
через A/I.
Методика 51. Поли-(0-ацетил)-£-тирозин [142].
Смешивают 7,9 г (31,6 ммоль) NCA О-ацетил-£-тирозина в
75 мл абсолютного диоксана с раствором 10,4 мг (0,6 ммоль)
(A/I — 50) аммиака в 10 мл абсолютного диоксана. Раствор
сразу мутнеет. Спустя 1 ч перемешивания при 20 сС массу на-
гревают в течение 40 ч при 100 °C, защищая от доступа влаги.
После охлаждения раствор смешивают с 31 мл 1 н. НС1 и 50 мл
воды, встряхивают в течение 3 ч, отфильтровывают выпавший
осадок (или центрифугируют), многократно промывают водой
и спиртом и сушат. Выход 5,7 г (88 %). Степень полимеризации
п = 18.
Методика 52. Поли-М-(2,4-динитрофенил)-Л-гистидин
[143].
К раствору 2,3 г (6 ммоль) NCA (Ы1'7г-2,4-динитрофенил)-£-
гистидина в 200 мл смеси абсолютных ДМФА-ацетон (1:1)
прибавляют 2,5 г сухого оксида серебра. Размешивают 30 мин,
фильтрат упаривают в глубоком вакууме при 25 °C, маслооб-
разный остаток растворяют 30 мл абсолютного диоксана и до-
бавляют 0,0084 мл (0,06 ммоль) триэтиламина (A/I = 100). Вы-
держивают 3 дня при 22 °C, осадок отфильтровывают, промы-
вают диоксаном, водой, диоксаном, эфиром и сушат в вакууме
над Р2О5. Выход 1,3 г. (72 %), степень полимеризации п = 110-
Методика 53. Поли-(№-карбобензокси)-Ь-лизина [1441-
При хорошем перемешивании сливают раствор 2,407 г
(7,8 ммоль) NCA №-карбобензокси-Ь-лизина в 240 мл абсолют-
ного диоксана с 0,095 мл 0,415ан. раствора метилата натрия
в метаноле (A/I == 200). Смесь оставляют на ночь, а затем
выливают в 1 л 0,5 %-го этанола, при этом выпадает липкий
материал. После декантации надосадочной жидкости поли-
конденсат растворяют в 200 мл хлороформа и выливают
этот раствор тонкой струйкой в 1 л гексана. Образовавшийся
осадок промывают гексаном и сушат в вакууме при 40 сС.
Выход 1,6 г (80 %). Степень полимеризации п = 5000.
Ниже приводится пример сополимеризации тирозина, ала
ни на' и глутаминовой кислоты.
Методика 54. Сополимер тирозин34-аланин81-(у-бен-
зил)глутаминовая кислота35 [145].
Растворяют в 300 мл абсолютного диоксана 6,21 г (30 ммоль)
NCA L-тирозина, 3,45 г (30 ммоль) NCA L-аланина и 7,8 г-
(30 ммоль) у-бензилового эфира L-глутаминовой кислоты. Пос-
ле добавления 0,025 мл триэтиламина (A/I = 500) раствор ос-
69
тавляют на 2 дня при 20 °C. После разбавления смеси водой
выпавший поликонденсат отфильтровывают и сушат. Выход
11,5 г. Общий состав! Туг i Ala ! Glu (OBzl) = 34 ! 31 i 35.
Метод активированных эфиров
Паранитрофениловые эфиры
Методика 55. п-Нитрофениловые эфиры N-защищен-
ных аминокислот [146].
К раствору N-защищенной аминокислоты (1 моль) в этил-
ацетате (для глутамина и аспарагина в качестве растворителя
используют ДМФА) прибавляют при 0 °C 1,1 моля п-нитрофе-
нола и 1,1 моля ДЦГК, перемешивают 1 ч при 0 °C и оставляют
на 20 ч при комнатной температуре. Выпавшую дициклогек-
силмочевину отфильтровывают, промывают этилацетатом, а
растворитель упаривают в вакууме. Остаток кристаллизуют
из спирта.
В табл. 7 представлены свойства n-нитрофениловых эфиров
N-защищенных аминокислот, полученных разными методами.
Таблица 7. Свойства /г-нитрофениловых эфиров аминокислот *
Аминокислота T. пл., °C [a]^, град Растворитель (c = I)
Z-L-AlaOH 79—79,5 —38,1 Этилацетат
Z-L-AsnOH 158—159,5 —31,5 ДМФА
Z-L-Asp (OBzl) ОН 76 —16,6 ДМФА
Z-L-Asp (OBi?) OH 86,5—87,5 + 1,5 Хлороформ
Z-L-Cys (Bzl) OH 93—94 —43 ДМФА
Z-L-GInOH 155—156 —24 ДМФА
Z-L-Gln (OBzl) OH 114—115 —32,5 Метанол
Z-L-Gln (OBt/) OH 60—61 —33,5 Метанол
Z-GIyOH 128 — —
Z-L-IleOH 60—62 — 15,5 ДМФА
Z-L-LeuOH 95 —33,5 ДМФА
Z-L-Ly’ (Boc) OH 88—91 — 14,8 Ацетон
Z-L-MetOH 96—97 —33,8 ДМФА
Z-L-PheOH 126—127 —24,7 ДМФА
Z-L-ProOH 94—96 —68 , ДМФА
Z-L-Ser (Bzl) OH 55—57 — 12 ДМФА
Z-L-ThrOH 93—95 —24 ДМФА
Z-L-TroOH 105 —4,5 ДМФА
Z-L-Tyr (OBzl) OH 148—150 —9,0 ДМФА
Z-LA'alOH 66—67 —25 ДМФА
Boc-L-AlaOH 83 —52,5 ДМФА
Boc-L-Arg (NO2) OH 126—128 —30 ДМФА
Boc-L-AsnOH 178—180 —36 ДМФА
Boc-L-Asp (OBzl) OH 104—106 - —36 ДМФА
70
Продолжение табл. 7
Аминокислота T. пл., °C [a]p град Раствори- тель (c = 1)
Boc-L-Cys (Bzl) ОН 95—96 —37,5 ДМФА
Boc-L-GlnOH 154—155 —35 ДМФА
Boc-L-Glu (OBzl) ОН 123 —33,5 ДМФА
Boc-GlyOH 70—71 — —
Boc-L-His (Вос) ОН 141—143 —22,8 Диоксан
Boc-L-HeOH 65—66,5 —32,5 ДМФА
Boc-L-LeuOH 94—95 —48 ДМФА
Boc-L-Lys (Z) OH 83—85 —23,6 ДМФА
Boc-L-MetOH 99—100 —48. ДМФА
Boc-L-PheOH 132 —21 ДМФА
Boc-L-Ser (Bze) OH Масло
Boc-L-TrpOH * 114—116 + 1,4 ДМФА
Boc-L-Туг (Bze) OH 145—146 —3,0 ДМФА
Boc-L-Val-OH 64—66 —38,4 Диоксан
♦ Примечание. Здесь и во всех последующих таблицах констант соединений
(для уменьшения количества ссылок в книге) не даются литературные ссылки на от-
дельные производные. Константы заимствованы из оригинальных работ, монографий
[9—12], а также обзорных таблиц Флэтчера и Джонса [28, 29].
Методика 56. п-Нитробензиловый эфир бензилокси-
карбонилглутаминил-нитроаргинина [1241
. 2ZGlnONp + НВг • ArgONB ♦ Z—Gin—Arg—ONB
I I
no2 no2
К раствору 10,8 г (27 ммоль) п-нитрофенилового эфира бен-
вилоксикарбонилглутамина в 70 мл ДМФА прибавляют 1,5 г
имидазола и 11,6 г (27 ммоль) бромгидрата п-нитробензилово-
го эфира нитроаргинина, растворенного в 40 мл ДМФА с 3,8 мл
(27 ммоль) триэтиламина. Раствор перемешивают 6 ч при ком-
натной температуре и затем оставляют на ночь. Диметилфор-
мамид удаляют в вакууме, а остаток растирают с петролей-
ным эфиром до образования амофного продукта, который за-
тем кристаллизуют из смеси ДМФА — вода (1 ! 10). Выход 11 г
(87 %), т. пл. 190—192 °C, [cc]d = —10,4° (с = 1; ДМФА).
Для более полного удаления л-нитрофенола продукт реак-
ции следует многократно растирать с раствором бикарбоната
натрия до исчезновения характерной желтой окраски л-нитро-
фенолята, а затем высушить и очистить кристаллизацией.
N-Оксисукцинимидные эфиры
Методика 57. N-Оксисукцинимидный эфир трет^у-
тилоксикарбонилаланина [1181.
К раствору 1,32 г (7 ммоль) /ире/и-бутилоксикарбонилаланина
и 0,805 г (7 ммоль) N-оксисукцинимида в 20 мл сухого тетра-
71
гидрофурана добавляют при О °C 1,59 г (7 ммоль) ДЦГК и
реакционную массу перемешивают 2 ч при О °C, а затем остав-
ляют на ночь в холодильнике. Образовавшийся осадок от-
фильтровывают, тетрагидрофуран отгоняют на роторном
испарителе, а остаток кристаллизуют из изопропилового
спирта. Выход 1,42 г (71 %), т. пл. 143—144 °C.
В табл. 8 представлены физико-химические характеристики
N-оксисукцинимидных эфиров N-защищенных аминокислот.
Таблица 8. Строение и свойства N-оксисукцинимидных эфиров
N-защищенных аминокислот [118]
Аминокислота Растворитель для кристаллизации Вы- ход, % Т. пл., °C (а]р» град (Сех 2; диоксан)
Z-L-AlaOH Изопропиловый спирт 65 123—123,5 —37,2
Zg-L-ArgOH Этилацетат-петролей- 80 85—86 —9,7
Z-GlyOH ный эфир Хлороформ-петролей- 86 113—114 —
Z-L-HeOH ный эфир Изопропиловый спирт 56 115,5—116 — 15,5
Z-L-LeuOH » в 51 116—117 —33,6
Z-L-PheOH » в 76 140—140,5 — 17,3
Z-L-ProOH в в 78 90 —54
Z-L-ValOH в в 53 116—117 —25,1
Z-L-MetOH в в 59 101—102 — 15,9
Boc-L-AlaOH » в 71 143—144 —49
Boc-L-CysOH в в 49 117—117,5 —54
S—Bzl Boc-GlyOH в » 62 168—170
Boc-L-HeOH в в 61 92—93 —25,5
Boc-L-LeuOH в в 48 116 —41,8
Boc-L-MetOH в в 59 128—129 -20,6
Boc-L-PheOH в в 81 152—153 — 19,0
Boc-L-ProOH » в 74 135—136 —55,3
Boc-L-TrpOH » в 37 153—154 —22,4
Boc-L-ValOH Хлороформ — метил- 74 128—129 —37,0
циклогексан
Синтезированные соединения можно хранить в течение не-
скольких месяцев в холодильнике без разложения.
Методика 58. Этиловый эфир бензилоксикарбонил-
фенилаланилтирозина [1181.
К раствору 1,98 г (5 ммоль) сукцинимидного эфира бензил-
оксикарбонилфенилаланина в 20 мл диоксана прибавляют рас-
твор 1,23 г (5 ммоль) хлоргидрата этилового эфира тирозина в
хлористом метилене, содержащем 0,69 мл (5 ммоль) триэтил-
амина. Реакционную смесь перемешивают 1 ч при 20 °C, затем
растворитель удаляют в вакууме, а остаток растворяют г этил-
ацетате и последовательно промывают 1 н. соляной кислотой,
72
водой, бикарбонатом натрия, снова водой. Органическую фазу
сушат сульфатом магния, этилацетат отгоняют в вакууме, а ос-
таток кристаллизуют из смеси спирт — вода. Выход 2,08. г
(85 %), т. пл. 156—158 °C; [а]Ь5 = —9,1° (с = 1, этилацетат).
Пентафторфениловые эфиры
Методика 59. Пентафторфениловые эфиры N-защи-
щенных аминокислот [105].
Раствор 5 ммоль /ярет-бутилоксикарбонил-аминокислоты и
5,5 ммоль пентафторфенола в 5 мл сухого этилацетата или сме-
си диоксан — ДМФА охлаждают до 0 °C и при размешивании
вносят 5,5 ммоль ДЦГК в 5 мл этилацетата. Реакционную смесь
перемешивают 1 ч при 0°С, дициклогексилмочевину отфильтро-
вывают, растворитель удаляют в вакууме, а остаток растирают
с гексаном и кристаллизуют из подходящего растворителя.
В табл. 9 приведены физико-химические характеристики
синтезированных пентафторфениловых эфиров N-защищенных
аминокислот.
Методика 60. Пептафторфениловый эфир бензилокси-
карбониласпарагина [105].
Раствор 1,33 г. (5 ммоль) бензилоксикарбониласпарагина
и 3,04 г (15 ммоль) пентафторфенола в смеси 3,5 мл ДМФА и
10,5 мл диоксана охлаждают до 0 °C и прибавляют 1,13 г
(5,5 ммоль) ДЦГК. Реакционную смесь размешивают 2 ч при
0 °C, затем дициклогексилмочевину отфильтровывают и рас-
творитель упаривают на роторном испарителе. К остатку до-
бавляют 8 мл эфира и растирают, образовавшиеся кристаллы
отфильтровывают, промывают охлажденным эфиром и сушат.
Выход 2,02 г (94 %), т. пл. 149—150 сС.
Методика 61. трет- Бут иловый эфир бензилокси-
карбониласпаргинилглицина [1051.
Раствор 0,43 г (1 ммоль) пентафторфенилового эфира бен-
вилоксикарбониласпарагина в 10 мл диоксана охлаждают до
10 °C и вносят 0,185 г (1,1 ммоль) хлоргидрата mptm-бутило-
вого эфира глицина. К суспензии прибавляют 0,154 мл
(1,1 ммоль) триэтиламина и перемешивают 0,5 ч при комнат-
ной температуре. Соли отфильтровывают, растворитель от-
гоняют в вакууме, а остаток растирают с зфиром. Осадок от-
фильтровывают, промывают эфиром, сушат. Выход 0,34 г
(90 %), т. пл. 150—151 °C.
Методика 62. Пентафторфениловые эфиры 9-флу-
оренилметилоксикарбонил аминокислот [1471.
К раствору 2 ммоль Fmoc-аминокислоты и 2 ммоль пента-
фторфенола в сухом диоксане, эти л ацетате или в смеси одного
из них с ДМФА (5—10 мл) добавляют при охлаждении до 0 °C и
73
Таблица 9. Строение и свойства пентафторфениловых эфиров
N-защищенных аминокислот [105, 106]
Формула аминокислоты Растворитель для кристал- лизации Вы- ход, % Т. пл., *С [ос]д, град (растворитель)
Z-Z,-AlaOH Гексан 84—85 —22,4 (этилацетат)
Z-L-ArgOH Спирт —• 98—99 —12,6 (этилацетат)
no2
Z-L-AspOH Bzl Спирт —- 95,5—98 —9,9 (этилацетат)
Z-Z,-AspOH 1 , Гексан — бен- зол — .76,5—77,5 —43,1 (хлороформ)
OBi?
Z-L-CysOH Этил ацетат — пентан — 85—86 —25,6 (этилацетат)
Z-L-GluOH 1 , Гексан — бен- зол — 85—87 —16,5 (этилацетат)
OBuz
Z-L-GluOH Спирт — вода — 116—118 + 10,9 (этилацетат)
Z-L-IleOH Z-L-LeuOH — масло масло —13,5 (этилацетат) —17,7 (этилацетат)
Z-L-LysOH Хлороформ — гексан — 105—107 +8,58 (этилацетат)
Z
Z-L-MetOH Спирт — 104—106 —18,7 (этилацетат)
Z-L-PheOH Спирт — 98—100 —26,7 (этилацетат)
Z-L-ProOH .— — Масло —55 (этил ацетат)
Z-L-SerOH Спирт — 142,5—143,5 —16,2 (этилацетат)
Z-L-TrpOH Хлороформ — гексан — 124—125 —26,1 (этилацетат
Z-A-ValOH Спирт — вода — 50—51,5 —12,3 (этилацетат)
Boc-L-AlaOH Этил ацетат — гексан 85 82—83 —31,2 (диоксан)
Boc-L-AsnOH Эфир — гексан 66 124—126 —16,7 (этилацетат)
Boc-L-AspOH — 98 масло —10,7 (этилацетат)
OBu'
Boc-L-CysOH 1 Этил ацетат — гексан 73 99—101 —42,0 (этилацетат)
Bzl
Boc-GlyOH Гексан 70 79—80 —
Boc-GlnOH Спирт 82 146—148 —25,8 (этилацетат) диоксан (1:1)
Boc-L-GluOH 1 , Диизопропи ловый эфир 72 90—91 —24,1 (этилацетат)
OBi?
Boc-L-HeOH Гексан 98 48—49 —16,9 (этилацетат)
Boc-L-LeuOH : Гексан 94 48—50 —3.0,2 (этилацетат)
74
Продолжение табл. 9
Формула аминокислоты Растворитель для кристал- лизации Вы- ход, % Т. пл., *С [а]д. град (растворитель)
Boc-L-LysOH 1 Z Этил ацетат — гексан 80 54—55 —17,1 (диоксан)
Boc-L-MetOH Эфир — гек- сан 89 80—82 —22,5 (этилацетат)
Boc-L-ProOH Гексан 88 61—52 —53,0 (диоксан)
Boc-L-PheOH Этил ацетат — гексан 97 111—112 —26,9 (диоксан)
Boc-L-ThrOH Этил ацетат — гексан 71 105—106 —32,6 (этилацетат)
Boc-L-TrpOH Этилацетат — гексан 88 109—111 —28,1 (диоксан)
Boc-L-SerOH Эти л ацетат — . гексан . 77 90—91 —29,8 (диоксан)
Boc-L-ValOH Гексан 78 62—64 —18,4 (диоксан)
• Все аминокислоты — 1 «изомеры.
• ♦ 1—2 %-е растворы.
перемешивании 2 ммоль ДЦГК; реакционную смесь размеши-
вают в течение 1 ч при О °C и затем 1 ч при комнатной темпе-
ратуре. Дициклогексилмочевину отфильтровывают, раствори-
тель отгоняют на роторном испарителе при 40 °C, а остаток ра-
стирают с гексаном. Затвердевший продукт отфильтровывают
и кристаллизуют из подходящего растворителя.
В табл. 10 приведены свойства синтезированных эфиров.
Для получения пентафторфениловых эфиров Fmoc-ами-
нокислот предложен 9-флуоренилметилпентафторфенилкарбо-
нат (68), который позволяет получать указанные соединения
в одну стадию [1481:
------ Fmoc ~ HN — CHR • • С — OPfP
75
Методика 63. 9-Флуоренилметилпентафторфенил кар-
бонат [148]
К раствору 25,9 г (0,1 моль) 9-флуоренилметил хлоркарбо-
ната и 18,4 г (0,1 моль) пентафторфенола в 200 мл диэтилового
Таблица 10. Строение и свойства пентафторфениловых эфиров
9-флуорофенилметилоксикарбонил аминокислот [147]
Формула аминокис- лоты Растворитель для кристаллизации Вы- ход, % Т. пл,, °C Га]р> град (с «в lj хлороформ)
Ala Этил ацетат — гексан 93 171—173 —22,7
Asn » 96 164—165 — 13,1 *
Asp Этилацетат — диизопро- 75 128—131 — 14,0 **,
1 п иловый эфир
OBzl
Cys i Этил ацетат — гексан 89 132—134 —31,0
1 Bzl
Gly Этилацетат 99 160—161 —
Gin » 97 151—153 — 19,8 ♦
Glu Этил ацетат — гексан 85 121—123 25,2
OBuz
He Гексан 78 96—98 — 13,4
Leu Этилацетат — гексан 96 114—116 —25,7
Lys i » 93 106—108 — 10,5
I Z
Met » 93 102—104 — 12,6
Pro Этилацетат 82 127—129 —59,2
Phe » 93 154—157 —20,3
Ser Этил ацетат — гексан 84 125—130 —21,3
Trp » 85 185—186 —42,1
Tyr’ 61 76—78 — 12,7
1 t OBu* __
Thr » 77 126—128 —33,0
Vai » 85 122—123 —21,9
♦ Оптическую активность измеряли в диоксане.
• ♦ Удельное вращение определяли в этилацетата.
76
эфира при О °C добавляли по каплям 14 мл (0,1 моль) триэтил-
амина. Смесь перемешивали 2 ч при 0 °C, затем трижды про-
мывали водой, сразу сушили над сульфатом натрия и обраба-
тывали активным углем. Растворитель удаляли в вакууме и
твердый остаток перекристаллизовывали из 120 мл н. гексана.
Выход продукта 29,6 г. Вторая порция была получена из ма-
точного раствора — 5 г. Общий выход 34,6 г (86 %), т. пл. 84—
86 °C.
Методика 64. Общий метод получения пентафторфенило-
вых эфиров 9-фл уоренилметилоксикарбонил аминокислот L 148].
I. 2,44 г (6,0 ммоль) 9-флуоренилметил пентафторфенил-
карбоната добавляли к перемешиваемой суспензии аминокис-
лоты или ее производного (6,6 моль) и раствор 1,09 г (13 ммоль)
NaHCO3 в смеси 30 мл воды и 15 мл ацетона при комнатной
температуре. Реакцию контролировали по ТХС. После пол-
ного прохождения реакции (в зависимости от растворимости
аминокислоты или ее производного от 4 до 24 ч) смесь подкис-
ляли до pH 3 концентрированной НС1, экстрагировали 3 раза
этилацетатом и сушили раствор над сульфатом натрия. (После
удаления этилацетата в вакууме Fmoc-ами но кислоты были вы-
делены с выходом 78—99 %).
II. Полученный этилацетатный раствор Fmoc-аминокисло-
ты и пентафторфенола охлаждали до 0 °C и добавляли к нему
1,36 г (6,6 ммоль) ДЦГК- После перемешивания смеси в тече-
ние 2 ч при этой температуре удаляли дициклогексилмочевину,
упаривали растворитель и выделяли продукты по описанной
методике. Выход 72—96 %. Пентафторфениловые эфиры Fmoc-
аминокислот являются лучшими реагентами для использова-
ния в твердофазном синтезе пептидов.
Предложен способ синтеза защищенных пентафторфенило-
вых эфиров аминокислот, заключающийся в обработке три-
ати л аммонийных солей защищенных аминокислот новым ре-
агентом — дипентафторфенилкарбонатом
F\ /F
О—/ F
0 / /-< 0 F\ zF
F\ /F
+ HO-<( ^-F + CO,.
f/xf
77
Методика 65. Синтез дипентафторфенилкарбоната
1149]
F F \ Fx /F
2НО-/~\—F 0—F
F/~\F f/ XF
К раствору 0,1 моль (18,4 г) пентафторфенола, 0,1 моль
(14 мл) триэтиламина в 150 мл эфира при —20 °C добавляют
при перемешивании раствор 0,05 моль (4,9 г) фосгена в 50 мл
эфира. Перемешивают 30 мин, повышая температуру до ком-
натной. Фильтруют, фильтрат упаривают в вакууме, остаток
кристаллизуют из гексана. Выход дипентафторфенилкарбона-
та количественный. Белый кристаллический порошок, не гиг-
роскопичен, т. пл. 49—50 °C.
Методика 66. Общий метод синтеза пентафторфениловых
эфиров защищенных аминокислот с использованием дипентаф-
торфенилкарбоната [ 150]
К раствору 10 ммоль защищенной аминокислоты в 20 мл
растворителя (преимущественно эти л ацетат а) добавляли 1,1 мл
(10 ммоль) N-метилморфолина, охлаждали до 0 °C, вносили 4 г
(10,5 ммоль) дипентафторфенилкарбоната и перемешивали
40 мин, контролируя процесс с помощью ТСХ. Если за это
время реакция не проходила полностью, вносили еще 0,1 ммоль
дипентафторфенилкарбоната и перемешивали 5—10 мин при
комнатной температуре. После окончания реакции выпавшие
кристаллы пентафторфенолята N-метилморфолина отфильтро-
вывали, фильтр упаривали в вакууме, растворяли в неболь-
шом (5—10 мл) количестве хлороформа, отделяли осадок соли
N-метилморфолина (если он образовывался) и хроматографи-
ровали на колонке (2,5 X 20 см) с силикагелем (элюент хлоро-
форм). Выход пентафторфениловых эфиров составляет 85—
98 %.
Модифицированные пентафторфениловые эфиры
защищенных аминокислот
Несмотря на широкое распространение пентафторфенило-
вых эфиров (Pfp) в пептидной химии, физико-химические ха-
рактеристики некоторых производных аминокислот неудов-
летворительны: Pfp-эфиры Вос-Gly, Вос-Leu, Вос-Не, Вос-
78
Таблица 11. Температуры плавления активированных эфиров
Вос-аминокислрт [152]
Производное аминокислоты Температура плавления, ®C
OPfp OTfp OTfc
Boc-Gly-OH 79—80 83—84 94—97
Boc-Ieu-OH 48—50 62—63 78—79
Boc-IPe-OH 48—49 68—69 77—78
Boc-Val-OH 62—64 66—69 76—78
Boc-Pro-OH 51—52 114—116 90—92
Вос-Ser (OBzl) OH 44—45 Масло 58—61
Vai, Вос-Рго и Вос-Ser (OBzl) имеют низкие температуры плав-
ления и вследствие этого плохо кристаллизуются. Недавно
американскими авторами [151] предложены 2,3,5,6-тетрафтор-
фениловые эфиры (Tfp) (68), а В. Н. Медведкин с соавт. [152]
предложили и-хлортетрафторфениловые эфиры (Tfc) (69), ко-
торые кристаллизуются лучше соответствующих пентафтор»
фениловых эфиров:
О F4 Т О F\
R—С—О—R—С—О—С1
f/~xf f/~xf
(68) (69)
В табл. 11 приведены характеристики некоторых активи-
рованных эфиров N-защищенных аминокислот.
По своей реакционной способности Tfc-эфиры аминокислот
практически не отличаются от Pfp-и Tfp-эфиров.
Реагенты переэтерификации
Методика 67. N-Оксисукцинимидный эфир трифтор-
уксусной кислоты [1531 (Реагент Сакакибары.)
218,0
115,1
К Ю г (87 ммоль) N-оксисукцинимида прибавляют 22,3 г
(103 ммоль) ангидрида трифторуксусной кислоты, смесь раз-
мешивают 20 мин при комнатной температуре, а затем избыток
ангидрида и трифторуксусную кислоту удаляют в вакууме при
79
температуре ниже 30 °C. Остаток переносят в эксикатор и хра-
нят там над фосфорным ангидридом. Выход 18,4 г (100 %),
т. пл. 69—70 °C. П р и м е ч а н и е. Реагент чрезвычайно гиг-
роскопичен1
Методика 68. Этиловый эфир бензилоксикарбонил-
глицил-фенил-аланил-глицина [1541.
К охлажденному до 0 °C раствору 0,8 г (25 ммоль) бензил-
оксикарбонилглицил-фенилаланина в 5 мл пиридина медленно
прибавляют 0,7 г (33 ммоль) N-оксисукцинимидного эфира
трифторуксусной кислоты в 2 мл пиридина. Реакционную
смесь размешивают 1 ч при 20 °C, а затем добавляют 0,02 мл
воды для разложения избытка реагента Сакакибары. При раз-
мешивании в колбу прибавляют 0,47 г (34 ммоль) хлоргидрата
этилового эфира глицина в 5 мл хлороформа, содержащего
0,47 мл (34 ммоль) пиридина, и выдерживают 3 ч при комнат-
ной температуре. После удаления растворителя в вакууме
водоструйного насоса остаток промывают этилацетатом. Вы-
ход 0,77 г (78,3 %).
Методи к‘а 69. Пентафторфениловый эфир трихлор-
уксусной кислоты [641
F\ /F 0 F\ /F
CC13—+ HO—/ S—F CC18—C—O—{ S—F
C1 /=< /=\
p/Z Хчр р/ \p
182,0 184,0 329,1
К раствору 39,8 г (0,22 моль) хлорангидрида трихлорук-
сусной кислоты и 40,1 г (0,22 моль) пентафторфенола в 150 мл
бензола прибавляют в течение 30 мин по каплям при переме-
шивании и охлаждении до 0 °C 17,8 мл (0,22 моль) пиридина,
а затем смесь размешивают еще 1 ч при 20 °C. Осадок соляно-
кислого пиридина отфильтровывают, бензол удаляют в ва-
кууме, а остаток кристаллизуют из небольшого количества
гексана и сушат. Выход кристаллического продукта 59,4 г
(82 %), т. пл. 43—45 °C.
Методика 70. Общий метод синтеза пептидов с ис-
пользованием пентафторфенилового эфира трихлоруксусной
кислоты [64].
Эквимолярные количества ациламинокислоты или N-за-
щищенного пептида, триэтиламина и пентафторфенилового эфи-
ра трихлоруксусной кислоты перемешивают 1—2 ч при 0—
20 °C в таких растворителях, как ДМФА, ацетонитрил, диок-
сан или хлористый метилен; затем добавляют аминокомпонент
(эфир аминокислоты или пептида) и оставляют на несколько
часов при комнатной температуре. Пептид выделяют обычным
способом. Выход продукта реакции 85—90 %.
80
Методика71. 1 -0- Нафтилсул ьфо нилоксибензтриазол [651
226,55 135,13 335.23
Раствор 22,2 г (165 ммоль) 1-оксибензтриазола в 60 мл
2,75 н. едкого натра (165 ммоль) добавляют по каплям при пе-
ремешивании и охлаждении до 0 °C к раствору р-нафталинсуль-
фохлорида 33,4 г (147 ммоль) в 600 мл ацетона. Спустя 1,5 ч
выпавший осадок отфильтровывают, промывают водой, сушат
в вакуум-эксикаторе и кристаллизуют из смеси ацетон — гек-
сан. Выход 40 г (82 %), т. пл. 139—140 °C.
Методика 72. n-Нитробензиловый эфир бензилокси-
карбонилглутаминиласпарагина [65]
/N\
Z—GlnOH + I II I /=< + н—AsnONB ->
о
Z—Gin—AsnONB
Раствор 0,28 г (1 ммоль) бензилоксикарбоннлглутамина и
0,348 г (1 ммоль) n-нитробензилового эфира аспарагина в 4 мл
ДМФА охлаждают до 0 °C и при размешивании добавляют
0,224 мл N-этилморфолина, а затем — 0,34 г (1 ммоль) 1-р-наф-
тилсульфонилоксибензтриазола. Реакционную смесь размеши-
вают ГО мин при 0 °C и затем оставляют на 10—12 ч при ком-
натной температуре. После удаления растворителя в вакууме
остаток промывают последовательно 10 %-ным бикарбонатом
натрия, водой, 2 н. соляной кислотой, снова водой, наконец,
спиртом. Продукт кристаллизуют из смеси ДМФА — вода.
Выход 0,46 г (90 %), т. пл. 220—222 °C; [a]2J = —10,6° (с =
= 2; ДМФА).
Синтез пептидов
с незащищенной карбоксильной группой
при помощи активированных эфиров
Использование n-нитрофениловых эфиров
N-защищенных аминокислот
Методика 73. Бензилоксикарбонилфенилаланил-со-то-
виларгинин [1551.
В 300 мл смеси диоксан — 1 н. NaOH (1 ! 1) суспендируют
25,8 г (78,5 ммоль) со-тозиларганина и 26,4 г (63 ммоль) п-нит-
6 2 — 882
81
рофенилового эфира бензилоксикарбонилфенилаланипа. При
размешивании в колбу добавляют из автоматической бюретки
смесь диоксана и 1 н. NaOH (1 • 1), поддерживая pH раствора
8,5. После размешивания реакционной массы в течение 24 ч
при комнатной температуре прибавление щелочи прекращают.
К полученному раствору добавляют этилацетат и водную фазу
подкисляют 1 н. серной кислотой. Продукт экстрагируют этил-
ацетатом, органическую фазу сушат сульфатом натрия, рас-
творитель отгоняют на роторном испарителе, а остаток расти-
рают с эфиром. Затвердевший пептид отфильтровывают и су-
шат. Выход 35,3 г (92 %); [а]^ = 4-1,9° (с = 1, метанол).
Методика 74. Бензилоксикарбонилпролил-аргинил-
пролин [1561
К раствору 0,22 г (8 ммоль) аргинил-пролина в 0,5 мл воды
прибавляют 0,39 г (8 ммоль) п-ни1рофенилового эфира карбо-
бензоксипролина в 0,5 мл диоксана. Реакционную смесь раз-
мешивают 2 ч при 45 °C, растворитель удаляют в вакууме, ос-
таток растворяют в 1 мл сухого ДМФА и оставляют на 12 ч при
40 °C. Трипептид осаждают затем сухим этилацетатом, от-
фильтровывают и сушат в вакуум-эксикаторе. Выход 3,2 г
(84 %), т. пл. 165—167 °C; [а]^5 = —34° (с = 0,3; вода).
Использование N-оксисукцинимидных эфиров
N-защищенных аминокислот
Методика 75. трет-Бутилоксикарбонил-у-бензил-
глутамил-глицин [157]
Раствор 0,434 г (1 ммоль) N-оксисукцинимидного эфира
у-бензилового эфира Вос-глутаминовой кислоты в 4 мл спирта
смешивают с раствором 0,075 г (1 ммоль) глицина и 0,168 г
(2 ммоль) бикарбоната натрия в 3 мл воды и оставляют па ночь
при комнатной температуре. Затем спирт удаляют в вакууме,
оставшийся водный раствор промывают два раза этилацета-
том и водный слой подкисляют лимонной кислотой. Образовав-
шийся осадок экстрагируют этилацетатом и последовательно
промывают 1 н. раствором лимонной кислоты, водой, сушат
сульфатом магния, и растворитель удаляют на роторном испа-
рителе. Остаток кристаллизуют из этилацетата. Выход 0,347 г
(88 %), т. пл. 114—115 °C; [а]^5 = —4,5° (с = 1, ДМФА).
Методика 76. трет-Бутилоксикарбонилфенилала-
нил-метионин [158]
0,6 г (4 ммоль) метионина растворяют в 7 мл (0,5 моль) ед-
кого натра, к раствору добавляют 0,34 г (4 ммоль) кристалли-
ческого бикарбоната натрия так, чтобы раствор имел pH 9,0.
В реакционную колбу вносят раствор 1,45 г (4 ммоль) N-окси-
сукцинимидного эфира трет-бутилоксикарбонилфенилаланина
в 5 мл ДМФА и затем добавляют еще 15 мл диметилформамида.
82
Реакционную массу перемешивают 18 ч при комнатной тем-
пературе, после чего растворитель удаляют в вакууме, а ос-
таток переносят в делительную воронку в смеси 50 мл этил-
ацетата и 50 мл 10 %-й лимонной кислоты. Органический слой
отделяют, промывают 20 мл насыщенного раствора NaCl, су-
шат сульфатом магния и этилацетат отгоняют на роторном ис-
парителе. Затвердевший остаток кристаллизуют из смеси
10 мл этилацетат и 30 мл петролейного эфира. Выход 1.2 г
(78 %), т. пл. 138—140 °C; [a]2J = —9,8е (с - 1; ДМФА).
Методика 77. N (а)-Тозил-валил-аргинин [159]
К раствору 3 ммоль N-оксисукципимидного эфира тознл-
валина в 5 мл диоксана прибавляют раствор 3 ммоль аргинина
в 2 мл воды, размешивают 3 ч при комнатной температуре и
затем оставляют на ночь. Растворитель упаривают в вакууме,
оставшийся маслообразный продукт растворяют в ДМФА и
дипептид осаждают этилацетатом. Бесцветный аморфный про-
дукт отфильтровывают, промывают этилацетатом и кристал-
лизуют из н-бутанола. Выход 0,65 г (66 %), т. пл. 158—160 Т,
[ос]2^ = —25,0° (с = 1; метанол).
Использование 2-нитро-4-сульфофениловых эфиров
Методика 78. Натриевая соль 2-нитро-4-сульфофепо
ла 1160]
НО—SOgNa + NaNO3 НО-Т~^—SO3Na • ЗН2О.
NOa
196,2 85,0 295,0
В трехгорлый реактор (1 л), снабженный мешалкой, ка-
пельной воронкой и термометром, вносят 132 г (0,67 моль)
натриевой соли 4-фенолсульфокислоты и 132 г (1,55 моль) нит-
рата натрия в 500 мл воды. Смесь нагревают, размешивая до
полного растворения натриевой соли фенолсульфокислоты.
Температуру раствора снижают затем до 35—40 °C так, чтобы
осадок еще не начал выпадать^ и прибавляют из капельной
воронки 73 мл концентрированной серной кислоты. Темпера-
тура реакционной! массы начинает медленно подниматься и
после достижения 60 °C колбу необходимо охлаждать на во-
дяной бане. При добавлении серной кислоты иногда начинает
выпадать осадок оранжевого цвета. После добавления кислоты
смесь размешивают 1 ч, а затем оставляют еще на 20 ч при ком-
натной температуре. Продукт, выпавший в осадок, отфильт-
ровывают, хорошо отжимают на фильтре и растворяют в го-
рячем насыщенном растворе хлористого натрия. При медлен-
ном охлаждении колбы выпадает осадок оранжевого цвета, его
6* ' 83
Таблица 12. 2-Нитро-4-сульфо-
фениловые эфиры некоторых
карбоновых кислот и аминокислот
[Ю9]
Формула исходной карбоновой кислоты или аминокислоты Выход Nsp эфира, % Хроматографи- ческая подвиж- ность i ри TCX (Rf) *
сн3 - соон 70 0,43
С0Н5 — соон 94 0,56
СН3 (СН2)7 — соон 75 0,58
^бН&х>сн — соон 66 0.57
Свн5/
Z-GlyOH 80 0,43
Z-GlnOH 80 0,49
Z-PheOH 68 0,52
Z-TrpOH 74 0,62
Tos-AlaOH 68 0,56
Boc-GluOH 85 0,66
OBzl
Boc-LysOH 65 0,69
Z
Boc-TrpOH 72 0,63
Boc-Gly-GiyOH 90 0,59
* Хроматографическую подвижность оп-
ределяли на пластинках «Силуфол» (Чехо-Сло<
вакия в системе бутанол — уксусная кисло-
та — вода (4:1: 1).
отфильтровывают, промы-
вают насыщенным раство-
ром NaC] и сушат. Выход
130 г (60 %).
Методика 79. Об-
щий метод получения Nsp-
эфиров N-защищенных ами-
нокислот [1091.
В 20 мл ДМФА раство-
ряют 10 ммоль N-защищен-
ной аминокислоты (или кар-
боновой кислоты) и 10 ммоль
натриевой соли 2-нитро-
4-сульфофенола, раствор
охлаждают до 0°Си вносят
10 ммоль ДЦГК. Реакци-
онную смесь перемешивают
1 ч при 0 °C, а затем остав-
ляют на ночь при комнат-
ной температуре. Дицикло-
гексилмочевину, выпавшую
в осадок, отфильтровыва-
ют, растворитель удаляют
в вакууме, а остаток раст-
воряют в 5 мл спирта. После
добавления к полученному
раствору эфира выдержива-
ют в течение 2 ч в холодиль-
нике, образовавшийся оса-
док отфильтровывают, про-
мывают эфиром и сушат.
После кристаллизации из
спирта получают продукт оранжевого цвета с выходом 50—
85 %. Характеристики соединений, полученных по этой мето-
дике, приведены в табл. 12.
Методика 80. трет-Бутилоксикарбонил-у-бензил-
глутамил-глицин [109]
К раствору 5,6 г (10 ммоль) Nsp-эфира у-бензилевого эфира
mpem-бутилоксикарбонилглутаминовой кислоты в 20 мл воды
прибавляют 1,5 г (20 ммоль) глицина и 20 ммоль бикарбоната
натрия. Реакционную смесь размешивают 4 ч при 0 °C и остав-
ляют на ночь при комнатной температуре. Раствор экстраги-
руют эфиром, затем подкисляют лимонной кислотой и пептид
экстрагируют этилацетатом. Органическую фазу промывают
водой до pH 7,0 и сушат сульфатом магния, этилацетат отго-
няют в вакууме, а остаток растирают с петролейным эфиром.
84
Продукт кристаллизуют из смеси ацетат — гексан. Выход 2,7 г
(70 %), т. пл. 114 °C, [сс]^5 - —2,2° (с = 1; ДМФА).
Методика 81. Тозилфенилаланил-лейцин [109]
К раствору 1,5 г (3 ммоль) Nsp-эфира тозилаланина в 5 мл
воды прибавляют при перемешивании 0,53 г (4 ммоль) лей-
цина в 0,84 г (10 ммоль) бикарбоната натрия. Реакционную
смесь размешивают 4 ч при комнатной температуре, а затем
подкисляют соляной кислотой до pH 3,0. Образовавшийся
осадок отфильтровывают, промывают водой и сушат. Выход
0,6 г (60 %), т. пл. 187—188 °C; [а]^5 =—29,5° (с = 1; спирт).
Методика 82. Карбобензокси-триглицил-глицин [109]
К раствору 3 г (6,1 ммоль) Nsp-эфира карбобензокси-
глицил-глицина в 30 мл воды прибавляют 1 г (7,6 ммоль) гли-
цил-глицина и 1,6 г (20 ммоль) бикарбоната натрия. Реакцион-
ную смесь перемешивают 2 ч при 0 °C, а затем выдерживают
в течение ночи при комнатной температуре. Раствор, окрашен-
ный в оранжевый цвет, подкисляют соляной кислотой до pH
3,0 и охлаждают. Образовавшийся осадок отфильтровывают
и кристаллизуют из воды. Выход 1,2 г (50 %), т. пл. 230—
231 °C.
Ускоренные методы синтеза пептидов
Методика 83. Метод последовательного синтеза пеп-
тидов с использованием избытков смешанных ангидридов
(REMA) [161].
Принцип метода заключается в использовании на каждой
стадии конденсации избытка ацилирующего агента и проведе-
ние наращивания полипептидной цепи без промежуточного
выделения и очистки пептида.
К охлажденному до —15 °C раствору N-защищенной ами-
нокислоты (1,5 экв.) и N-метилморфолина (1,5 экв.) в тетра-
гидрофуране или ДМФА добавляют 1,4 моль эквивалентов
изобутилхлорформиата, размешивают 2 мин и в один прием
к реакционной смеси добавляют 1 экв. аминокомпонента в
ДМФА. Размешивание продолжают 3 ч, затем температуру
поднимают до 0 °C и прибавляют раствор КНСО3 до начала
слабого выделения углекислого газа. Смесь энергично пере-
мешивают 30 мин, а затем пептид осаждают 4 объемами насы-
щенного раствора хлористого натрия. При синтезе больших
пептидов для этой цели используют воду. Защищенный пеп-
тид промывают водой и сушат в вакууме при комнатной тем-
пературе.
Высокий выход и чистота синтезируемого пептида позво-
ляют наращивать полипептидную цепь без выделения проме-
жуточного пептида. Полноту реакции ацилирования проверя-
85
ют ТСХ; после обработки пластинки флуорескамином интен-
сивность пятна сравнивают визуально с интенсивностью све-
чения стандартных количеств аминокомпонента и, если необ-
ходимо, ацилирование проводят повторно. После окончания
реакции и разложения избытка смешанного ангидрида не-
прореагировавшая N-защищенная аминокислота удаляется
водным раствором КНСО3, а защищенный пептид в органиче-
ском растворителе сушится, деблокируется и вводится в кон-
денсацию с новым карбоксильным компонентом.^
Этим методом был синтезирован 16-членный фрагмент га-
стрина 12—27 с общим выходом 31 % [161], а недавно этим
способом получен секретин свиньи (27 остатков аминокислот)
1162].
Методика 84. Ускоренный синтез пептидов с избыт-
ком пентафторфениловых эфиров [112].
Для полного ацилирования аминокомпонента применяют
двойной избыток пентафторфенилового эфира N-защищенной
аминокислоты, время образования пептидной связи 10—
30 мин, полноту реакции контролируют ТСХ. Удаление из-
бытка активированного эфира осуществляют добавлением
N,N диметилэтилендиамина в течение 5—10 мин с промывкой
органической фазы последовательно лимонной кислотой, во-
дой и 50 %-м раствором КНСО3. После высушивания органи-
ческий растворитель удаляют в вакууме, остаток обрабаты-
вают раствором НС] в диоксане в течение 10 мин. Хлоргидрат
аминокомпонента осаждают эфиром, промывают и суспенди-
руют в органическом растворителе, а затем третичным осно-
ванием доводят pH раствора до 7,5—3,5 и весь цикл операций
повторяют снова. Время одного цикла 65-—85 мин. Пептиды,
содержащие 9—10 аминокислот, можно синтезировать в те-
чение одного дня; выход 43—76 %.
Методика 85. Синтез пептидов методом «удержания
в растворе» [163].
Принцип метода заключается в том, что реакцию проводят
в гетерогенной среде, в тонкой эмульсии двух несмешива-
ющихся растворителей — воцы и дихлорэтана. В воде находят-
ся Вос-аминокислота, 1-оксибензтриазол и водорастворимый
карбодиимид; здесь следует в качестве аминокомпонента при-
менять бензиловые эфиры, которые распределяются между
органической фазой и водой. После окончания реакции все
непрореагировавшие продукты остаются в воде, а защищен-
ный пептид переходит в органическую фазу. После отщепле-
ния Вос-группы весь цикл превращений повторяют. Этим
способом был синтезирован ангиотензин II (8 остатков ами-
нокислот) и пептид дельта-сна (9 остатков аминокислот) с вы-
ходом 78 и 53 % соответственно.
86
Методика 86. Бензиловый эфир пропил-фенилала-
нина.
К раствору 0,222 г (0,3 ммоль) тозилата бензилового эфира
фенилаланина, 0,7 мл триэтиламина, 0,215 г (1 ммоль) Вос-
иролина и 0,135 г (1 ммоль) оксибензтриазола в 25 мл ди-
хлорэтана прибавляют при комнатной температуре раствор
0,192 г (1 ммоль) 1-этил-3-(3-диметил аминопропил) карбоди-
имида в 1 мл воды и смесь размешивают 2 ч при этой темпера-
туре. Реакционную смесь затем последовательно промывают
при 10 °C раствором 0,1 М НО (3x5 мл), водой (5 мл), рас-
твором 0,5 М бикарбоната натрия (5 мл) и снова водой (4 X
X 5 мл). Для отщепления Вос-группы к раствору пептида
в дихлорэтане прибавляют 8 мл (7,2 моль) раствора НО в
диоксане при температуре —10 °C и смесь выдерживают
30 мин при 0 °C. Затем к органическому растворителю добав-
ляют 18 мл (2 моль) бикарбоната натрия, размешивают,
водную фазу отделяют и раствор дипептида в дихлорэтане
последовательно промывают водой (2 X 4 мл), 0,5 моль би-
карбонатом натрия (4 мл) и снова водой (2x4 мл). Чистоту
полученного продукта контролируют ТСХ и аминокислот-
ным анализом.
М е т о д и к а 87. Использование хлорангидридов Fmoc-
аминокислот [76]
Хлорангидрид Fmoc-аминокислоты (11 —15 ммоль) кон-
денсируют с 10 ммолями соли эфира аминокислоты (этилового
или бензилового) в двухфазной системе хлороформ — вода
(вместо хлороформа можно брать метиленхлорид или дихлор-
этан) в присутствии NаНСО3 в течение 5—10 мин, водный
слой отделяют, органическую фазу промывают разбавленной
НС1, затем водой. Fmoc-группу удаляют большим избытком
4-(аминометил) пиперидина в течение 20 мин, промывают во-
дой, а аддукт дибензофульвена экстрагируют фосфатным бу-
фером pH 5,5. Время одного цикла 25—45 мин. Защищенный
лейцин-энкефалин был получен в течение 3—4 ч, а общий вы-
ход свободного пептида после гидрирования составил 50—
55 %.
СПОСОБЫ АКТИВАЦИИ АМИНОГРУППЫ
Основным способом образования пептидной связи являет-
ся активация карбоксильной группы, однако существует не-
сколько методов активации аминогруппы, которые в некото-
рых случаях предпочтительнее. Например, они дают хорошие
результаты при использовании в качестве аминокомпонептов
слабых оснований, в частности и-нитроаннлина, который ши-
роко применяется для синтеза пептидных хромогенных суб-
87
Механизмы активации аминогруппы
Изоцианатный метод. Впервые реакцию карбониламино-
кислот с защищенными аминокислотами применил в 1950 г.
Голдшмидт [166]. По-видимому, реакция протекает через
смешанный ангидрид карбоновой и карбаминовой кислот (69)
по схеме:
Z—-NH—R1—СООН 4- О—C=N—CHR2—СООХ ->
О О (70)
II II
Z—NH—CHR!—С— О—С—NH—CHR2—СОХ
Z—NH—-CHR1—CONH-— CHR2— COOX + СОа.
Реакцию обычно ведут в толуоле при НО °C до прекраще-
ния выделения двуокиси углерода или в пиридине при тем-
пературе от 20 до 50 °C. Эфиры карбониламинокислот получа-
ют действием фосгена на хлоргидраты эфиров аминокислот в
толуоле при 120 °C. Эфиры карбониламинокислот хорошо
перегоняются и хранятся без разложения. В случае исполь-
зования N-защищенных аминокислот рацемизации не на-
блюдалось, однако при использовании в качестве карбоксиль-
ного компонента защищенных пептидов возможна рацеми-
зация.
Интересная модификация метода предложена недавно Кун-
цем с соавт. [165]. Стабильная N-защитная 2,4-бис(метилтио)-
феноксикарбонильная группа (71) окислялась надмуравьи-
ной кислотой в 2,4-бис(метилсульфонил)феноксикарбониль-
ную группу (72), которая является сильным акцептором элек-
тронов и уменьшает электронную плотность на карбонильном
углеродном атоме. Под действием основания происходит эли-
минирование защитной группы с образованием изоцианата
73):
о 9
II
CH8S—' 4-0-C-NH-CHR1-CONH-CHR2-OBu^4iH7^co<5H^
\сн3
(71)
о
11 0
СНз-Ь-М 4 ||
II х=/ ,О—О—С—NH-CHRj—CONH—CHRa—ОВи'
О +
О" ХСН3 Ру
(72)
-> O=C=N—CHRX—CONH—CHR2—OBu‘.
(73)
90
По-видимому, механизм элиминирования включает отрыв
протона аминогруппы и образование аниона (74), который да-
лее путем разрыва связи С—О превращается в фенолят (75)
и изоцианат:
о> __ о’
r-^o^-c-n ~r'—^[r-o-c-n-r’*—*r--o-c = .\~r! ]—-
(74)
----R-O“+ O = C = N-R’.
(75)
Реакция проводится при комнатной температуре в метилен-
хлориде в присутствии пиридина. Была успешно использова-
на в синтезе циклотетрадепсипептидов [165]. Возможно, ука-
занная модификация сделает изоцианатный метод более по-
пулярным в пептидном синтезе.
Фосфоразный метод. Фосфоразный метод был предложен
для синтеза пептидов Голдшмидтом в 1950 году [166]. Пред-
полагается, что при взаимодействии двух молей эфира амино-
кислоты с одним молем треххлористого фосфора в присутствии
основания образуются реакционноспособные фосфоразо соеди-
нения (76), которые реагируют с карбоксильным компонентом
с образованием пептида:
РС13 4- 2H2N—CHRj—СООМе ..^Et3 >
R3
/N—CHRi—СООМе I
1 2Z—NH—СН—СООН
\nH--CHR,—СООМе
(76)
2Z-—NH—CHR2—CONH—CHR1—СООМеНРО2 + 3NEt3 • НС.
Для получения фосфоразосоединений в качестве раствори-
теля лучше всего использовать сухой пиридин, так как в этом
случае отпадает необходимость использования третичного
амина и можно использовать хлоргидраты эфиров аминокис-
лот. Реакцию проводят при комцатной температуре или крат-
ковременном нагреве. В случае конденсации с эфирами ами-
нокислот карбобензоксипептидов отмечалась рацемизация.
Фосфорноангидридный метод. В 1958 году Шрамм и Вис-
ман предложили метод синтеза пептидов, при котором взаи-
модействие эфиров аминокислот с фосфорным ангидридом
в диэтилфосфите при нагревании приводит к моноамиду мо-
ноэтилового эфира фосфорной кислоты (78) через предпола-
гаемый промежуточный этилметафосфат (77). Полученное
производное реагирует с защищенной аминокислотой, давая
91
пептид:
/ОН zo
с2н5о-р< с2н5О—
хос2н5 ^0
(77)
ОН
/ НО. /ОН
-> С2Н5О—Р—NHR R,-CONH-r2 + )р/
II с2н5о/ ^o
о
(78)
Смесь пятиоксида фосфора, карбоксильного и аминоком-
понента в диэтилфосфите нагревают в присутствии третичного
основания при 100° в течение 3 ч. В том случае, когда карбо-
ксильный компонент является защищенным пептидом, воз-
можна рацемизация.
Диэтилхлорфосфитный метод. Метод был предложен в
1951 г. Андерсоном с соавт. [168]. Некоторые амиды диэтил-
фосфорной кислоты (79), полученные из эфиров аминокислот,
являются устойчивыми соединениями и перегоняются без
разложения. При взаимодействии карбоксильного и амино-
компонентов и диэтилхлорфосфита при кипячении или при
комнатной температуре в бензоле, толуоле и других раствори-
телях образуются пептиды:
С2Н5О.
)Р—Cl + H2N—CHRi— СООМе
с2н5о/
санюк
\р__NH—CHRi—>
C2H5OZ
(79)
С2Н5О.
z—NH—CHR2—CONH—CHRi—COOMe + )P—OH.
c2H5o/
Ниже приводятся методики синтеза пептидов методами
активации аминогруппы.
Методики
Изоцианатный метод
Методика 88. Этиловый эфир N-карбонил-Л-лей-
цина [170].
В трехгорлую колбу, снабженную герметической мешал-
кой, трубкой для подвода газа и обратным холодильником,
вносят 17,5 г (0,095 моль) хлоргидрата этилового эфира-Л-лей-
цина (высушенного в вакууме над Р2О5 при 50°) и 50 мл аб-
солютного толуола. Колбу помещают на масляную баню и
пропускают через кипящий толуол при перемешивании ток
92
фосгена в течение 1,5 ч. Холодильник защищают от влаги
воздуха хлоркальциевой трубкой. Постепенно весь хлоргид-
рат переходит в раствор, и после его растворения толуол от-
гоняют с эффективной колонкой в вакууме при 45 °C, а оста-
ток фракционируют. Получают 16,9 г (91,5 %) этилового
N-карбонил-А-лейцина с т. к. 104,5° (15 мм рт. ст.); [а]^0 ==
— —22,4 (без растворителя). Полученное масло хранить без
доступа влаги!
Методика 89. Этиловый эфир карбобензокси-5-бен-
зил-Ь-цистеинилглицина [171].
К смеси 85 г (0,24 моль) Z-S-бензил-А-цистеина и 31,8 г
(0,24 моль) этилового эфира N-карбонилглицина добавляли
10 мл абсолютного пиридина, причем масса саморазогревает-
ся, вспенивается и выделяется углекислый газ. После завер-
шения бурной реакции массу нагревают на водяной бане в
течение 2 ч. Реакционная смесь застывает в серую массу, ко-
торую растворяют в 700 мл этилацетата. Полученный раствор
дважды промывают 100 мл 2 н. НС1, а затем 100 мл 10 %-го
раствора Na2CO3, сушат над сульфатом натрия и удаляют этил-
ацетат под уменьшенным давлением. Остаток перекристалли-
зовывают из смеси этилацетат — петролейный эфир. Выход
бесцветных игольчатых кристаллов 97,2 г (92 %), т. пл. 98—
99 °C; [а]^ = —26,8° (с = 6; уксусная кислота).
Фосфоразный метод
Методика 90. Этиловый эфир карбобензоксиглицил-
глицина [169].
Растворяют 5 г (34,5 м моль) хлоргидрата этилового эфира
глицина в 100 мл сухого пиридина при слабом нагревании и
охлаждают раствор до 0 °C. К затвердевшей массе при энер-
гичном перемешивании добавляют раствор 1,6 мл треххло-
ристого фосфора в 20 мл сухого пиридина, перемешивают
30 мин при 20 °C и затем прибавляют 7,5 г (36 ммоль) карбо-
бензоксиглицина. Реакционную смесь нагревают 3 ч на
кипящей водяной бане, упаривают в вакууме, к остатку до-
бавляют 20 мл воды и экстрагируют несколько раз этилацета-
том. Экстракты промывают 1 н. НО, водой, 10 %-м раствором
Na2COs и снова водой, сушат над сульфатом натрия, кипятят
для обесцвечивания с активным углем и упаривают до малого
объема. К остатку добавляют петролейный эфир, охлаждают
и отфильтровывают выпавший осадок. Выход этилового эфира
карбобензоксиглицил-глицина 9,1 г (96 %), т. пл. 82 °C.
Примечание: если реакционную смесь вместо нагре-
вания в течение 3 ч выдержать 60 ч при комнатной температу-
ре, выход продукта будет таким же.
93
Фосфорноангидридный метод
Методика 91. Этиловый эфир карбобензоксиглицил-
глицина [171]
Растворяют 1,4 г (0,01 моль) Р2О5при сильном перемеши-
вании (защита от влаги!) в 7 мл диэтилфосфита при 100 °C.
К бесцветному прозрачному раствору добавляют 6,5 мл
(0,035 моль) трибутиламина и 1,4 г (0,01 моль) хлоргидрата
этилового эфира глицина. Через 10 мин он полностью рас-
творяется. После этого вносят 2,1 г (0,01 моль) карбобензокси-
глицина и раствор размешивают в течение 3 ч при 100 °C, а
затем охлаждают до комнатной температуре. После того, как
раствор выливают в 60 мл насыщенного раствора бикарбо-
ната натрия, этиловый эфир карбобензоксиглицил-глицина
выпадает после охлаждения до —15 °C в течение 1 чл Допол-
нительной экстракцией маточника этилацетатом извлекают
дополнительное количество продукта. Объединенный продукт
кристаллизуют из смеси этанол — вода. Выход 2,45 г (83 %),
т. пл. 81 °C. П р и м е ч а н и е: вместо трибутиламина мож-
но использовать триэтиламин.
Глава 2
ЗАЩИТА ФУНКЦИОНАЛЬНЫХ
ГРУПП АМИНОКИСЛОТ.
ЗАЩИТА АМИНОГРУППЫ
Все аминокислоты содержат две реакционноспособные
группировки: карбоксильную и аминогруппы. При создании
пептидной связи необходимо временно блокировать одну из
этих групп с тем, чтобы реакция протекала в нужном направ-
лении
Z—HN—CHRi—СООН +
+ H2N—CHR2—COOX -> Z—HN—CHRi—CONH—СН—СООХ.
R2
В боковых радикалах некоторых аминокислот (лизина^
тирозина и др.) имеются реакционноспособные группы, кото-
рые во избежание побочных реакций также необходимо бло-
кировать.
Выбор защитных групп определяется схемой синтеза дан-
ного пептида, а также специфическими свойствами остатков
аминокислот, например устойчивостью в условиях удаления
защитных группировок. Предложено большое число разнооб-
разных защитных групп, которые отвечают требованиям, не-
обходимым для синтеза пептидов. Наиболее важными из этих
требований являются: 1) отсутствие рацемизации; 2) удале-
ние защитных групп без расщепления пептидной связи; 3) воз-
можность селективного удаления одних группировок при
сохранении других; 4) устойчивость блокирующих групп при
синтезе и хранении пептидов. Существенным моментом явля-
ется также доступность и простота исходных реагентов, а так-
же способность давать кристаллические производные амино-
кислот и пептидов.
Классификацию защитных групп можно проводить, взяв
за основу химическую природу блокирующей группы, способ
удаления защитной группы или какой-нибудь другой признак.
В табл. 14 суммированы свойства используемых в настоя-
щее время защитных групп для блокирования NH2-rpynn
аминокислот и пептидов.
95
Таблица 14. Защитные
Название Обозначе- ние Формула
Ароматические
Бензилоксикарбонильная Z, Cbz
(карбобензокси)
Защитные группы
О
II
/ СН2О—С—
«•Нитробензилоксикарбональ- Z (NO2)
ная
О
O2N-/_^-CH2O-C-
о-Нитробензилоксикарбониль- NBOC
ная
а,а-Диметил-3,5 диметокси- DDZ
бензилоксикарбонильная
n-Метоксибензил оксикарбо- Z (ОМе)
пильная
Изоникотинилексикарбональная Z-NOC
2-п-Дифенилизопропилокси- Врос
карбонильная
Ацетоксибензилоксикарбо- ABOG
нильная
О С
И II
R—СО—СН2—О—С—
96
группы для аминогрупп
Реагент для введения Способ удаления Примечание Лите- ратура
уретанового типа
о 1. НВг/АЮН Самая распро- (172]
II / \-СН2О-С-С1 2. Ha/Pd 3. HF страненная за- щитная груп- па. Устойчива в условиях уда- ления Вос, Аос,
О /—к II °aN-/ СН2О— С—С1 Те же Производные кристаллизу- ются лучше. Поглощает при 265 нм, что можно исполь- зовать для кон- троля [173]
О \ II /_^-СНаО-С-С1 УФ-облучени- ем при 320 нм Одна из не- многих групп, удаляемых фо- толизом [174]
xno2
СНаОх СН3 О 1 II 1. Тот же 2. 1—5% То же [175]
/ С—О—С—С1 СН3о/ (!н,
О II CHSO—СН2О-С—N8 , О t . ЛЛ и //X ^-СН2О-СО-^ NO2 1. 1 н. НС1 в ' диоксане 2. TFA 1. Zn/AcOH 2, H2/Pd В настоящее время приме- няется редко Очень перспек- тивная группа. Устойчива к действию НВг, НО, TFA, HF [176] [177]
СН3 О С >—С >—с—о-с-о-/ > 1 СН8 1. 80 % АсОН 2. НВг, НС1 : 3. TFA, HF Водн. NH3 Перспективная группа, селек- тивно удаляе- мая в присут- ствии Вос, Аос и др. [178]
О о II \ II R—СО—/ СН2—О—С—С1 Водн. NH3 В TFA более стабильна, чем соответствую- щая Z-rpynna [179]
X
7 2 — 882
97
Название
Обозначе-
ние
Формула
5-Вензоксазолилметиленокси- Bic
карбонильная
9-Флуоренилметилоксикарбо- Fmoa
пильная
Алифатические
mpe/n-Бутилоксикарбонильная Вос
СН3 О
1 К
СН8—С—О—С—
СН3
mpe/n-Амилоксикарбонильная Аос
Адамантилоксикарбонильная Adoc
Аллилоксикарбон ильная АОС
РДР-Трихлорэтоксикарбо- Теос
нильная
СН2=СН-СН2—О-С—
С1
С1—С—СН2О—С—
98
Продолжение табл. 14
Реагент для введения Способ удаления Примечание Лите- ратура
1. N (EEt)3 2. ДМФА (Na2SO8, 40 °C) Предложена недавно [180]
Пиперидин, морфолин и другие основа- ния Весьма пер- спективная группа, удаля- емая основани- ями в мягких условиях [1811
сна L НС1 в диок- Наряду с Z- [182]
сн,-с-о—с^° сане, ТГФ, МеОН группой явля- ется самой рас- пространенной
1 XN, 2. TFA
СН, 3. НВг/АсОН защитной груп-
ВосаО 4. Сульфокис- пой. Устойчива
лоты к каталитиче-
5. HF безвод- скому гидроге-
ный нолизу
6. Трифтор аце- тат бора
СН3 О Те же реаген- Используется [183]
1 II ты реже, чем Вос-
СН3СН2—с—О-С—С1 группа
СНэ Те же реаген- Adoc-производ- [184]
fl 1 0 ты ные лучше;растворимы:
чем соответствую- щие Вос- и
Аос-пептиды
^Н2=СН—СН2-~О~-С—Cl H2/Pd Представляют 1185]
II лишь теорети-
О ческий инте- рес. Практиче- ски не исполь- вуется
С1 2п/АсОН Представляет 1186]
Cl—С—СН2О—С—С1 интерес как группа, отщеп-
1 II ляемая восста-
С1 о новлением. Ши- рокого приме- нения не нашла
1
99
Название Обозначе» ние Формула
2-Фосфониэгоксикарбонильная Реос
с«н»\+ II
СН,—Р—СНаСНаО—с—
С.н/
Защитные группы
n-Метилфенилсульфонильная Тоз
Фталильная
Pht
Формильная
НСО
(Form)
Тр ифтор ацетил ьн а я
TFA
(CF3CO—)
О
II
CFe-C—
Дифен илфосфинамидная Dpp
100
Продолжение табл. 14
Реагент для введения Способ удаления Примечание Лите- ратура
О ОН** Новая перепек- [187] 5\+ || • тивная гидро- СН3—Р—СНЯСНЙО—С—С1 фильная за- с и / щитная груп- пе115 па. Предложе- на для синте- за пептидов в водном раство- ре ацильного типа u г сл rt Na/NHs Сыграла боль- [188] шую роль в {1891 пептидной хи- мии. В настоя- щее время при- меняется в ос- новном для за- щиты боковых групп амино- кислот 0 Гидразингид- В настоящее [190] || рат в спирте время не при- УСК меняется rv > ^/хс/ II о НСООН; 1. HCl/спирт Применяется [191] нтгп-1 1 /СН о 2. 15 % НЯОЯ редко из-за НСООН -г (СН8СО), О 3 Гидразин. склонности про- ацетат изводи ых к ра- цемизации (CF8CO)2 О; 1. NaOH Первая защит- [192] CF.-C-S-CH.CH. 2- NaBH« в аб- ная гРУппа’ 8 ц 88 солютном спир- удаляемая ос- q те кованиями. Ис- пользуется ред- ко из-за склон- ности произ- водных к ра- цемизации Ph. „О i. 80 % TFA Предложена не- 2. 0,4 М НС1 в давно. В отли- [193] Ph' ХС1 90 %-м три- чие от других фторэтаноле групп не наблю- 3. 2 экв. n-то- дается побочное луолсульфокис- алкилирование лоты триптофана
101
Название Обозиа- kчение Формула
о-Нитрофенилсульфенильная Nps
Защитные группы
Трифенилметильная (тритиль- Trt
пая)
Триметилсилильная TMS
5-Х лор-2-оксибензофенон кети-
минная
Защитные групп®
,Ph
Н
Vxq/
3,5-Ди-трг/и-бутил-4-оксо-1- PChd
фенил-2,5-циклогексадиениль-
ная
О
(СН8),СХ " /CtCH,),
II
/xPh
Тозиламинокарбонильная Тас
Защитные группы
О О
О
102
Продолжение табл, 14
Реагент для введения Способ удаления Примечание Лите- ратура
s~C1 1. Кислыми ре- агентами Широко ис- пользуемая за- [194]
^NOa 2. Тиольные щитная группа,
соединения которая легко и селективно удаляется в мягких усло- виях
алкильного типа
1. 50 % АсОН Сыграла боль- [195]
II 1 2. Ледяная шую роль в [196]
АсОН при ки- развитии син-
пячении тетических ме-
3. НС1/метанол тодов химии пептидов. В
0 настоящее вре- мя применяет-
ся редко
CNS Н2О, спирты Ввиду высокой лабильности [197]
CHg—Si—Ci применяется лишь в особых
сн3 случаях
арилиденового типа
О 80 % АсОН Широкого рас- [198]
II (80 °C, 20 мин, пространения
С1\^\/С\/Чх 1 II II 1 25 °C, 10 ч) не нашла
1 II II 1
(СНз)зС.
С (СН8)8
уреидного типа
О
У-Х II
СН3-< >-S-NCO
||
О
1. H2/Pd 2. 50 %-я ТФУ В C2H2CI2 Уникальная защитная груп- па, которая вводится элект- рохимическим путем; удаля- ется в мягких условиях [199]
1. Кипячение со спиртами, 1—3 ч 2. Нагревание со смесью CH3CN/H2O (95 : 5), 80 °C, 4 ч Очень интерес- ная группа, удаляемая ней- тральными аген- тами. Может найти широкое применение [200]
103
Таблица 15. Классификация защитных
Основные способы удаления
Кислотами Основаниями Восстанови
Сильными Слабыми Неорганиче- скими Органи- ческими H2/Pd Zr/AcOH
i-Noc—
Теос—
Z—, Z (NOJ— Trt— CF3CO— Pht- z—
Z (OMe)— Врос— ABOC—, Fmoc— Z (NOa)—
Peoc—
Boc— Pchd— Z (OMe)—
Aoc—, Adoc— Bic— i-Noc—
HCO— Ppp—, NpS— Pchd-
DDZ— AOC
Примечание. Подчеркнуты те защитные группы, для которых данный способ
ОСНОВНЫЕ СПОСОБЫ УДАЛЕНИЯ ЗАЩИТНЫХ ГРУПП
И МЕХАНИЗМЫ СООТВЕТСТВУЮЩИХ ПРОЦЕССОВ
В табл. 15 защитные группы классифицированы по спо-
собам их отщепления: кислотами и основаниями (в мягких
или жестких условиях), восстанавливающими агентами, ней-
тральными реагентами и фотолитическими методами. Рассмот-
рим кратко некоторые механизмы реакций, происходящих
при удалении указанных групп.
Защитные группы, удаляемые кислотами
В настоящее время главными методами отщепления за-
щитных групп является обработка кислотами или восстанав-
ливающими агентами. Большинство N-защитных групп урета-
нового, алкильного и арилиденового типов легко удаляются
действием таких кислот, как HBr, НС1, HF, трифторуксусная
кислота, уксусная кислота и т. д.
Защитные группы уретанового типа
Карбобензоксигруппа и ее производные отщепляются дей-
ствием сильных кислот. Предполагаемый механизм реакции
бимолекулярный [201]
RO—С—NH—СН —RO—С—NH—СН—COORj ->
II I II I
О Rj ОН Rj
104
групп по способам их удаления
Вспомогательные способы удаления
телями Нейтральными реагентами УФ-облу- ченйем
Na/NHa NaBH, R—SH R—ОН R—CN Н2О, Н8О
Tos— CFjCO— Nps— IMS— HCO— TMS— NBoc—
Jac— DDZ—
(Z-)
удаления группы является основным.
н—о+—н
, I
RO—С+—NH—CH-COOR, -> RO-C—NH—CH-COORj ->
II II
ОН, Rj ОН R1
ОН
. I
-» RO+—С—NH—СН—COOR, -> R—ОН + СО2 +
Н С^Н Rt
(80)
О
. II
+ h#n+—СН—С—or2.
Ri
В результате протонирования карбонильной группы обра-
зуется неустойчивое промежуточное соединение (80), которое
быстро распадается на соответствующий спирт, углекислый
газ и свободный аминокомпонент. Реакции способствует по-
вышенная электронная плотность в ароматическом ядре.
Как показали исследования Блага и Рудингера [201], ско-
рость отщепления замещенных КБЗ-групп возрастает в сле-
дующем ряду:
NO2<C1<F<H< СН8 < СвН6 < СН8О,
Действительно, КБЗ-группу обычно удаляют раствором
36 %-й НВг в ледяной уксусной кислоте, тогда как п-мето-
105
ксибензилоксикарбонильная группа отщепляется трифтор-
уксусной кислотой.
Для защиты N-конца аминокислот и пептидов наибольшее
распространение получила карбобензокси-группа (Z—,
Cbz—), предложенная Бергманом иЗервасом в 1932 г. [172];
она сыграла исключительную роль в развитии синтеза пепти-
дов. Этому способствовали легкость введения этой защитной
группы посредством КБЗ-хлорида, способность КБЗ-амино-
кислот и пептидов кристаллизоваться, простота ее удаления;
n-нитро- и n-метоксикарбобензокси-группы получили значи-
тельно меньшее распространение.
Защитные группы уретанового типа, содержащие алифа-
тический радикал, например Вос-группу и другие, удаляют-
ся кислотами в более мягких условиях: 1 н. НС1, трифторук-
сусной кислотой, муравьиной или уксусной кислотами при
комнатной температуре.
Вос-группа и родственные ей группировки реагируют с
кислотами по мономолекулярному механизму [202]:
RO—С+~NH—CH—COOR2 -> RO—С+—NH—CH—COOR2 ->
II II
ОН Ri ОН Ri
R+ + СО2 + h3n+-ch-coor2.
I I
олефин Rj
Образующийся катион R® может вызывать побочные ре-
акции. Так, при удалении Вос-группы при действии НС1 об-
разующийся т/2^т-бутилхлорид способен алкилировать ос-
татки триптофана или метионина [203]:
СН3 О СН8
1 11 ' НГ1 I 4_
СН3—С—О—С—NHR ЛЯ > СН8—С+ + СО2 + H2N—R
сн3 (2Н3
СН3 СН3 СН„
сн3-с+ -> с=сн2 ЛЯ. > СН3—С—С1
с!н3 сн3 сн3
Следует отметить, что при отщеплении адамантилоксикарбо-
нильной защитной группы образуется весьма стабильный
карбкатион, который не дает олефина.
По данным Эмбера и Изелина [204], по относительной
скорости отщепления некоторых алифатических групп уре-
106
танового типа их можно расположить в следующий ряд!
СН, СН, СН,
I I I
СН,—С—О-СО— < Ph—C-о—СО— < Ph—Ph—С—О—СО— <
I I I
СН, СН, СН,
1 700 2800
СН,
<СН,—/ С—О—СО—<
х==/ I
СН,
12 000
Дифенилизопропилоксикарбонильная группа (ВРОС) отщеп-
ляется почти в 3000 раз быстрее, чем Вос-группа; а, а-диме-
тил-3,5-диметилоксикарбонильная защитная группа занима-
ет промежуточное положение по скорости отщепления. DDZ-
защита отщепляется в 1400, а Врос-защита — в 3000 раз быст-
рее, чем Вос-группа. Из приведенных данных следует, что
скорость отщепления защитных групп уретанового типа
определяется стабильностью образующегося карбкатиона R+.
Введение заместителей, способствующих делокализации или
уменьшению положительного заряда, приводит к росту ско-
рости удаления защитной группы.
Среди N-защитных групп указанного типа наибольшее
распространение получила Вос-группа, которую в 1957 г.
предложили Мак-Кэй и Альбертсон [176] и независимо от них
Андерсон и Мак-Грегор [182]. Для введения Вос-группы
тре/и-бутилоксикарбонилхлорид использовать нельзя из-за
его неустойчивости. Поэтому было предложено несколько
других реагентов, среди которых наибольшую популярность
приобрели Вос-азид [205] и ди-тпрет-бутилпирокарбонат
[206]. Вос-группу можно селективно удалять в присутствии
карбобензоксигруппы.
Аналог Вос-группы mpe/n-амилоксикарбонильная защит-
ная группа получила меньшее распространение [183].
В 1968 г. Зибер иИзелин [178]предложили очень лабиль-
ную 2-(п-дифенил)-изопропилоксикарбонильную группу
для блокирования N-конца аминокислот и пептидов. Эта
группа нашла достаточно широкое применение в химии пеп-
тидов. Врос-группа в 3000 раз лабильнее Вос-группы, и поэ-
тому ее можно селективно удалять в присутствии Вос-груп-
пы. Для отщепления Врос-защиты используют смесь мура-
вьиной и уксусной кислот в течение 1,5 ч. Повышенная ла-
107
бильность Врос-группы позволяет удалять ее также в при-
сутствии тритильной группы.
Бирр и соавт. [175] ввели в химию пептидов чрезвычайно
лабильную а,а-диметил-3,5-диметоксибензилоксикарбони-
льную группу (DDZ), которую можно отщепить в присут-
ствии Вос-группы и mpem-бутиловых эфиров действием 1—
5 %-й ТФУ в хлористом метилене в течение 8—30 мин. Эта
группа нашла весьма широкое применение.
Защитные группы алкильного типа
Отщепление группировок алкильного типа под действием
кислот протекает, по-видимому, по механизму, включающему
протонирование атома азота аминогруппы и удаление алкиль-
ного остатка в виде карбониевого иона:
н4“ 4“
R1-NH—CH—COOR8 — -* Ri—NH—CH—COOR8 -> R+ +
r» A is
+ H,N—CH—COORj.
R.
Скорость деблокирования аминогруппы будет определять-
ся стабильностью'карбкатиона R+. Действительно, из приве-
денных ниже алкильных групп тритильная оказалась наи-
более лабильной [178]:
Ph л-СН8О—Ph n-CH8—Ph
I I I
Ph—С— > Н—С— > Н—С—
Ph л-СН8О—Ph n-CH8— Ph
На раннем этапе развития пептидного синтеза тритильная
группа находила применение и была достаточно распростра-
ненной защитной группой. Однако в настоящее время она
почти не используется, так как при синтезе тритильных про-
изводных аминокислот и пептидов имеют место стерические
препятствия, что снижает выход пептидов.
Защитные группы ацильного типа
Гидролитическое расщепление N-ацильных производных
аминокислот и пептидов идет, по-видимому, по механизму,
который аналогичен катализируемому кислотами гидролизу
108
амидов [207] i
о ОН он он
II н+ II | I +
R—С—NHa C-NHa + HaO я R—С—NHa R-C-NH,
+ 1 I
..OHj OH
он
14-
R—С—ОН + NH3 -> R—СООН + NH4 .
В пептидном синтезе нашли применение формильная и
нитрофенилсульфенильная группы. При использовании фор-
мильной группы для защиты N-конца аминокислот наблюда-
ется заметная рацемизация; поэтому ее используют главным
образом для блокирования функциональных групп боковых
радикалов аминокислот.
о-Нитрофенилсульфенильная группа (Nps) была введена
в практику пептидного синтеза Герделером и Хольстом [194]
в 1959 г. и впоследствии нашла широкое применение как
N-защитная группа, которая удаляется кислотами. Ее можно
отщеплять селективно в присутствии Вос-группы и mpem-бу-
тиловых эфиров. Nps-группа деблокируется также тиольны-
ми соединениями.
При удалении Nps-защиты в присутствии НС1 образуется
о-нитрофенилсульфенилхлорид; это следует учитывать при
синтезе пептидов, содержащих триптофан, так как послед-
ний может алкилироваться Nps — Cl.
Nps-аминокислоты довольно доступны. Они весьма легко
получаются обработкой аминокислот о-нитрофенилсульфе-
нилхлоридом в щелочной среде.
Защитную Nps-группу нельзя использовать в том случае,
если предполагается применение каталитического гидроге-
нолиза для деблокирования пептидов, так как отравляется
катализатор.
Из других ацильных групп недавно была предложена де-
финилфосфинамидная группа (Dpp), которая легко отщепля-
ется трифторуксусной кислотой, 0,4 М НС1 или п-толуол-
сульфокислотой в метаноле [193]. В отличие от Nps-группы
продукты, которые получаются при расщеплении Dpp-ами-
нокислот или пептидов при действии НС1, не алкилируют
остатки триптофана. В связи с этим можно думать, что Dpp-
защита найдет применение в синтезе триптофансодержащих
пептидов.
109
стратов протеолитических фер-
ментов [ 164]. Предложены так-
же оригинальные N-защитные
группы, которые после моди-
фикации превращают амино-
группу в изоцианатную (т. е.
активируют ее), что позволяет
использовать новые схемы
синтеза пептидов [165].
Следует отметить, что тер-
мин «активация» применяется
по отношению к аминогруппе
формально, по аналогии с кар-
боксильной группой. По это-
му поводу Шредер и Любке
[10] отмечают следующее:
«Аминогруппа в противопо-
ложность карбоксильной груп-
пе способна к образованию
амидной связи без дополни-
тельной активации благодаря наличию свободной пары элект-
ронов на атоме азота. Поэтому было высказано предположение,
Таблица 13.
Способ активации Формула реагента
Изоцианатный ме- СОС12 тод Фосфоразный ме- РС13 тод Фосфор ноангид- Р2О3, ридный метод /ОС2Н5 Н—Р' ^чос2нб Диэтилхлорфос- ,ОС2Н3 фитный метод С1—Р/ ХОС2Н5
* R — боковой радикал аминокислот)
что синтез пептидов на основе активирования аминогруппы в
действительности протекает через стадию образования промежу-
точного соединения с активированной карбоксильной группой».
Остановимся на этом несколько подробнее. Рассмотрим,
как происходит активация карбоксильной группы. Ранее мы
говорили о том, что карбоксильная группа является слабым
электрофилом для того, чтобы взаимодействовать с нуклеофи-
лом (аминогруппой) в мягких условиях. Поэтому все ранее
рассмотренные методы ее активирования направлены на пре-
вращение ее в сильный электрофил. В результате образования,
например, хлорангидрида, увеличивается электронная плот-
ность на карбонильном атоме углерода, в результате чего он
легко атакуется нуклеофилом:
О
ргч || _НС!
R-COOH ----R-C-Cl R-CONH-R'.
В случае «активации» аминогруппы, например путем пре-
вращения ее в изоцианат, аминокомпонент превращается из
нуклеофила в электрофил, а карбоксильная группа выступает
в роли нуклеофила (в виде карбаниона):
r~c/°”
R'— NH, R'— N=c+e=o-----> R-—NH—С—О—C—R ->
II II
О О
-> R—CONH—R' 4- co2.
88
Реагенты и методы активации аминогруппы*
Название реагента Формула активированного соедине- ния Примечание Лите- ратура
Фосген O=C=N—CHR—COOR' Эфиры карбонилами- нокислот не полимери- зуются и перегоняют- ся без разложения [166]
Треххлорис- тый фосфор ,NH—CHR—COOR' Р< .N—СН—R—COOR' В настоящее время применяется редко [166]
Фосфорный ангидрид в диэтилфос- фите О Н6С2О || )Р—NH—CHR—COOR' н5с2о/ То же [167]
Диэтил- хлорфосфит С2Н5ОЧ \p—NH—CHR—COOR' C2H6OZ » [168]
R' — алкил.
Таким образом, мы видим, что при «активации» аминогруп-
пы полностью изменяется механизм реакции. Возникает во-
прос, имеем ли мы право применять выражение «активация
аминогруппы», если ее активации не происходит? Рассмотрим,
насколько правильно выражение «активация карбоксильной
группы», которое возражений не вызывает. Если подойти к
проблеме строго, то следует отметить, что любые активиро-
ванные формы карбоксильной группы (сложные эфиры, ан-
гидриды, хлорангидриды и т. п.) являются не карбоксильной
группой, а карбонильными соединениями разных типов. Од-
нако ввиду того, что они реагируют с нуклеофилами по тако-
му же механизму, как и карбоксильная группа (электрофиль-
ный центр на карбонильном атоме углерода), мы принимаем
выражение «активированная карбоксильная группа». Нам
кажется, что несмотря на то, что выражение «активация ами-
ногруппы» является значительно менее строгим, его можно
использовать чисто формально, понимая, что оно отражает
только конечный результат реакции, а не ее механизм.
К этому следует добавить, что так как в результате акти-
вации аминогруппы происходит активация карбоксильной
группы, остаются те же проблемы, связанные с опасностью
рацемизации, что и в традиционных методах. Реагенты и ме-
тоды активации аминогруппы указаны в табл. 13.
89
Защитные группы, удаляемые основаниями
Защитные группы ацильного типа
Первой защитной группой, которая легко удаляется в
щелочной среде, была трифторацетильная группа, предло-
женная в 1952 г. Вейгандом и соавт. [192]. Сильный индук-
ционный эффект СР3-группы значительно снижает электрон-
ную плотность атома углерода карбонильной группы, что
облегчает атаку ионом гидроксила. Реакция, очевидно, идет
по механизму гидролиза амидов при катализе основаниями!
о
CF3—С—NH—CH—COOR2 —
Ri
+ H2N—CH—coor2.
ОН
CF3—С—NH—CH—COOR2
А-
Предложено также удалять TFA-группу боргидридом нат-
рия в абсолютном спирте [208].
TFA-аминокислоты легко синтезируются при действии
трифтор уксусного ангидрида или тиоэтилрвого эфира три-
фторуксусной кислоты на свободные аминокислоты.
В свое время для защиты а-аминогруппы пептидов при-
меняли фталильную группу, которую можно удалять гидра-
зингидратом [190]:
О
\н—CHR—СООН fr?!~Nt4H.o,
чу
о
NH
| + H2N—CHR-COOH.
NH
В настоящее время фтал ильная группа при синтезе пептидов
почти не используется.
110
Защитные группы уретанового типа
В 1973 г. Вюнш предложил новую интересную защитную
группу циано-тре/п-бутилоксикарбонильную, которая яв-
ляется модифицированной Вос-группой [209]:
сн3 о
I II
№С—СНа—С— О—С—
СН8
Эта группа удаляется водными растворами К2СО3 или три-
этиламина. Практического применения она, однако, не получила.
По-видимому, наиболее перспективной среди защитных
групп, лабильных в щелочной среде, является 9-флуоренил-
мётилоксикарбонильная группа (Fmoc), которую в 1972 г.
предложили Карпино и Хан [181]:
Fmoc-группа стабильна в кислой среде, но отщепляется осно-
ваниями: пиперидином, морфолином и др. В настоящее время
используется как в классическом, так и в твердофазном мето-
дах синтеза пептидов.
Механизм отщепления Fmoc-группы заключается в следую-
щем. В результате мощного электроноакцепторного эффекта
бензольных колец атом водорода, находящийся в 9-м положе-
нии, приобретает кислый характер и легко отрывается основа-
ниями. Это ведет к возникновению карбаниона (81):
Карбанион (81) быстро распадается, образуя свободный ами*
нокомпонент и дибензофульвен (82), который дает аддукт с ос-
нованиями.
Легкость деблокирования определяется в данном случае
стабильностью карбаниона типа (81), который образуется пу-
111
тем Р-элиминирования. По сходному механизму происходит
отщепление в щелочной среде защитной группы, предложен-
ной ЛеКорром с соавт. [179]. Она представляет собой модифи-
цированную карбобензокси-группу; в частности ЛеКорр и
другие исследовали n-ацилзамещенные производные типа:
О О
II II
Rj-C-O-/ \-СНа—О-С-
X
где R, = СН3—, C2HS—, (СН3)2СН—, (CH3)aCH-NH—,
(CH3)2N— и др., R2=H—, Cl—. Щелочной гидролиз произ-
водных аминокислот с указанными группами протекает следу-
ющим образом:
R1_C-O-/=>-CHa-O~C-NR2
А ^_/ о
(83)
О
+ О-—/СН2—О—A—NH—Ra
—- Ri-C-O- +
II
О
' элиминирование 1—6
(84) О~
-> О=/~\=СН2 + О=А—NH—R2 -со, } H2N-R2
_ZW) <85)
НО—/^-CHjOH
(87)
В результате нуклеофильной атаки основания ацетокси-груп-
па (83) образует n-оксизамещенное производное Z-защищен-
ной аминокислоты (84); последнее претерпевает элиминиро-
вание 1—6 и превращается в карбаминовое производное ами-
нокислоты (85) и хинон (86). После декарбоксилирования про-
дукта (85) образуется аминокислота (87). Для предотвращения
реакции между хиноном (86) и аминокомпонентом (87) в реак-
ционную смесь добавляют сульфит натрия, который восстанав-
ливает хинон, превращая его в n-оксибензиловый спирт.
При отщеплении групп типа (83) в качестве основания ис-
пользуют водный раствор аммиака с добавкой перекиси водо-
рода. При pH 9,5 анион НОО- является более сильным нук-
леофилом, чем гидроксильный ион.
Можно предположить, что рассмотренные группы окажутся
весьма перспективными для синтеза пептидов.
В 1978 г. Кэмп предложил оригинальную N-защитную груп-
пу — 5-бензизоксазолилметилоксикарбонильную группу (Bic)
112
1180]:
ch2-c-nh-r
NEt„ ДМФА
Элиминирование 1—6
(88)
I I + ’“ООО—NH—R -+
(90)
-> СОа + HaN—R
в апротонных растворителях
О
"O/X^ H
(89)
Устойчивое соединение (88
(ДМФА, ацетонитрил) при действии оснований легко изомери-
зуется в соединение (89). Например, при действии двух экви-
валентов триэтиламина в ДМФА при 25 °C изомеризация про-
текает за 30 мин; последующее выдерживание в течение 3 ч при
40 °C в водно-спиртовом растворе сульфита натрия (pH 7,0)
дает свободную аминокислоту; в этом случае бисульфит натрия
необходим для предотвращения взаимодействия хинона (90)
с аминокомпонентом.
Защитные группы,
отщепляемые каталитическим гидрированием
Для деблокирования некоторых защитных групп использу-
ют следующие восстановители: цинк в уксусной кислоте, нат-
рий в жидком аммиаке й боргидрид натрия. Однако наиболь-
шее распространение получил метод деблокирования путем
каталитического гидрогенолиза над палладием. Этим способом
удаляется Z-группа и ее производные, аллилоксикарбонильная
группа и расщепляются эфиры, используемые для блокирова-
ния карбоксильных и оксигрупп аминокислот.
Предполагается, что каталитическое отщепление Z-группы
протекает через стадию образования замещенной карбаминовой
кислоты с последующим выделением углекислого газа [1721:
О
СвН6— СНа—О—C-NH—CH—СООН СвНв—СН, +
О
II
4- НО—С—NH—CH—СООН -> СОа + HaN—СН—СООН
R R
В качестве катализатора при гидрировании используются:
палладиевая чернь, палладий на угл е или сульфате бария, ок-
8 2—882
113
сид палладия или оксид палладия на угле. Вместо молекуляр-
ного водорода для восстановления можно использовать такие
доноры водорода, как циклогексан [210, 211], циклогексадиен
[212], муравьиная кислота [213] иформиат аммония [214]. Эти
реагенты, предложенные в последние годы, позволяют вести
восстановление защитных групп быстрее и нередко дают более
чистые продукты.
Лоссе с соавт. [215] предложил в качестве катализатора для
гидрогенолиза использовать комплексную соль К3 [Со (CN)ftJ,
которая позволяет количественно удалять Z-группу и расщеп-
лять бензиловые эфиры. Этот катализатор дешевле палладие-
вого и может найти широкое применение.
Защитные группы,
удаляемые ультрафиолетовым облучением
Наиболее мягким методом удаления защитных групп явля-
ется по-видимому, фотолитическое расщепление. В 60-е годы
экспериментально было установлено, что введение некоторых
электронодонорных и электроноакцепторных заместителей
может приводить к гетеролитическому процессу ароматических
соединений при их облучении ультрафиолетовым светом. Сле-
дует отметить, что в возбужденном состоянии ориентирующее
влияние заместителей меняется по сравнению с их влиянием
в основном состоянии: электроноакцепторные группировки,
например NO2, оттягивают электроны от метаположения, а элект-
ронодонорные заместители «накачивают» электроны в метаполо-
жение. Следовательно, в фотореакциях метаположение бензоль-
ного кольца в нитробензоле является местом атаки нуклеофиль-
ными агентами, а в анизоле — местом электрофильной атаки:
В 1962 г. Барлтроп и Шоффилд [216] показали, что при
УФ-облучении Кбз-аминокислот последние расщепляются с
образованием свободной аминокислоты, бензилового спирта
и СО2. Снятие Кбз-защиты с Кбз-глицина происходит при облу-
чении светом с Лтах 254 нм, выход глицина 75 %.
О
“\-СН2-О-С—NH—СН2—СООН
,О
/_\-СН2ОН + СО2 + H2N-CH2-C<^oh
114
Поскольку введение NO2- или СН3О-групп изменяет чувстви-
тельность соединения к фотолитическому расщеплению, в даль-
нейшем было испытано несколько замещенных Кбз-групп в ка-
честве фотолабильных группировок.
Так, Чемберлен [217] предложил для этой цели 3,5-димет-
оксибензилоксикарбонильную группу (91), а Бирр с соавт.
[175] использовал а,а-диметил-3,5-диметоксибензилоксикар-
бонильную группу (92).
(91) (92)
В 1969 г. Пачорник, Амити Вудворд [218] изучали возможность
применения в химии пептидов таких фотолабильных групп,
как о-нитробензилоксикарбонильная (93), 6-нитровератрилок-
сикарбонильная (94) и 2,2'-динитродифенилоксикарбонильная
(95), используя УФ-свет с длиной волны 320 нм:
,NO2 О СНяО. О
/=\ II 11
/ СН2О—С— сн3о—/ \-CHgO—с—
XNOa
(93) (94)
/NOg О
z=< /=х II
NOa
(95)
В качестве промежуточных продуктов при фотолитическом
расщеплении образуются альдегиды. Для того чтобы они не
реагировали с аминогруппой, в реакционную смесь добавляют
либо минеральную кислоту, либо вещества, способные взаимо-
действовать с альдегидной группой (например, гидразин), ко-
торый превращает их в гидразоны.
МЕТОДИКИ СИНТЕЗА РЕАГЕНТОВ,
ИСПОЛЬЗУЕМЫХ ДЛЯ БЛОКИРОВАНИЯ АМИНОГРУПП
ПЕПТИДОВ И АМИНОКИСЛОТ
Карбобензоксигруппа
Карбобензоксихлорид
Гринштейн и Виниц [91 рекомендуют получать карбобен-
зоксихлорид по методу Фартинга, т. е. насыщением бензилово-
го спирта фосгеном при —10...—30 °C. Практика показывает,
8*
115
/7
Рис. 1. Схема прибора синтеза карбобензоксихлорида
что чистый карбобензоксихлорид можно получить лишь после
перегонки. Однако в некоторых руководствах отмечается, что
перегонка в вакууме может привести к взрыву, поэтому кар-
бобензоксихлорид лучше не перегонять. Применение неочищен-
ного препарата вызывает затруднения при кристаллизации
Кбз-аминокислот, которые получаются в этом случае в виде
масла.
Достаточно чистый карбобензоксихлорид без перегонки
удается получить при добавлении бензилового спирта к избыт-
ку жидкого фосгена при —30 °C. Для синтеза бензилоксикар-
бониламинокислот можно использовать также раствор Кбз-
хлорида в толуоле; получение этого раствора описано в «Синте-
зах органических препаратов» [219]. 4
Методика 92. Синтез карбобензоксихлорида с исполь-
з званием жидкого фосгена
О
СН2ОН + СОС12 — СН2О—(L-C1+HC1
84,0 109,0 156,5
Внимание! Всю работу с фосгеном необходимо прово-
дить в хорошо вентилируемом вытяжном шкафу; у каждого
сотрудника, работающего с фосгеном, должен быть наготове
противогаз.
Аппаратура для синтеза Кбз-хлорида (рис. 1) состоит из
двух блоков: установки для получения фосгена (I) и реактора
116
для синтеза карбобензоксихлорида (II). Трехгорлую колбу па
2 л (7), снабженную обратным холодильником (2) и капель-
ной воронкой (3) с хлоркальциевой трубкой, заполненной
СаС12, помещают в масляную баню (4). Синтез Кбз-хлорида
ведут в трехгорлой колбе емкостью 2 л (5), которую снабжают
низкотемпературным холодильником (6), мешалкой с ртутным
затвором (7) и хлоркальциевой трубкой (8). Колбу погружают
в баню, охлаждаемую до —70 °C ацетоном с сухим льдом; этой
же смесью наполняют и холодильник (6). Перед началом рабо-
ты на стенке колбы (5) делают отметку емкости 400 мл.
В колбу (7) помещают 200 г инфузорной земли или тефло-
новых стружек, 1 л сухого четыреххлористого углерода и на-
гревают на масляной бане до 80 °C. При этой температуре из
воронки (3) добавляют в течение 2—3 ч 200 мл 60 %-ного олеу-
ма с такой скоростью, чтобы СС14 оживленно кипел; выделяю-
щийся фосген конденсируется в колбе (5). После того, как собе-
рется необходимое количество жидкого фосгена до метки
400 мл (5,5 моль), подачу олеума прекращают, а нагрев масля-
ной бани выключают. Затем низкотемпературный холодиль-
ник (6) заменяют капельной воронкой на 100—200 мл, включа-
ют мешалку (7) на медленные обороты (для предотвращения
вскипания фосгена). Во избежание перегрева и вскипания фос-
гена перемешивание нельзя прекращать. Из воронки добавля-
ют в течение 1—2 ч по каплям 515 сухого бензилового спир-
та. Температуру охлаждающей бани поддерживают при —40. ..
—70 °C. После того, как весь бензиловый спирт будет добав-
лен, баню (9) убирают, а реакционную массу энергично разме-
шивают в течение 1—2 ч. По газоотводной трубке (8) удаляют
избыток фосгена и выделяющийся хлористый водород; эти газы
отводят в колбу с раствором щелочи, помещая газоотводную
трубку над поверхностью щелочного раствора.
Для полного удаления фосгена и хлористого водорода через
систему продувают воздух. Вместо капельной воронки встав-
ляют трубку (10), которую соединяют со склянкой Дрекселя,
содержащую концентрированную серную кислоту; последняя
необходима для поглощения влаги воздуха. Хлор* алытиевую
трубку (8) заменяют трубкой, которую присоединяют к ло-
вушке (13), заполненной 10 %-м раствором едкого натра, и
затем — к ловушке, содержащей спиртовой раствор аммиака.
Воздух просасывают через реактор (5) в течение 24 ч при помо-
щи водоструйного насоса.
Карбобензоксихлорид, полученный этим способом, доста-
точно чист и пригоден к употреблению; выход составляет
880 г (720 мл).
Для дегазации прибора содержимое колбы (1) разбавляют
серной кислотой и осторожно выливают под тягой в эмалирс-
117
ванное ведро с толченым льдом; содержимое нейтрализуют
щелочью и выливают в раковину в вытяжном шкафу.
Карбобензоксихлорид — бесцветная или слегка желтоватая
жидкость с резким запахом, раздражающим слизистую оболоч-
ку. Его следует хранить в холодильнике над сульфатом магния.
Для получения небольших количество фосгена можно
использовать реакцию взаимодействия четыреххлористого
углерода, формальдегида и хлористого алюминия [220].
М-етоди ка 93. Синтез фосгена [220]
СН2О + СС14 +• А1С13 -> СОС12.
Трехгорлую колбу (1 л) снабжают термометром, капельной
воронкой и обратным холодильником, верхняя часть которого
соединена с ловушкой, охлаждаемой сухим льдом и ацетоном.
В реактор загружают 15 г (0,5 моль) параформа, 100 р
(0,65 моль) сухого четыреххлористого углерода и 16,7 р
(0,125 моль) безводного хлористого алюминия. Реакционную
смесь нагревают на водяной бане до 35—40 °C и выдерживают
затем при этой температуре 2—2,5 ч, собирая жидкий фосген.
Когда приток фосгена уменьшится, содержимое колбы нагре-
вают до кипения и кипятят в течение 1 ч. Для прекращения
реакции баню удаляют, ловушку с фосгеном отсоединяют, а
охлажденную реакционную массу нейтрализуют добавлением
из-капельной воронки водного раствора аммиака. Выход фос-
гена 42 г (85 %).
Синтез бензилоксикарбониламинокислот
О
Сн2°—C-Cl + HgR—CHR—COO-
о
- II
-> СНаО-С—NH—CHR—СОО-.
Большинство Кбз-аминокислот получают действием кар-
бобензоксихлорида на водный раствор аминокислоты в ще-
лочной среде при низкой температуре.
Методика 94. Карбобензокси-/-триптофан [221]
К раствору 10,2 г (0,05 моль) L-триптофана в 50 мл 1 н. едко-
го натра прибавляют по каплям, размешивая и охлаждая до
0 9С, 8,4 г (0,05 моль) карбобензоксихлорида. Поддерживают
pH 9,0 добавлением (из другой капельной воронки) 50 мл 1 н.
NaOH. Через 1 ч после прекращения подачи карбобензоксихло-
риди содержимое колбы переносят в делительную воронку и
избыток карбобензоксихлорида экстрагируют 20 мл эфира. Вод-
ный раствор подкисляют 1 н. соляной кислотой до pH 3,0; при
118
этом карбобензокситриптофан выпадает в виде t масла. При
стоянии в холодильнике продукт кристаллизуется; его отделяют
фильтрованием, промывают на фильтре водой до нейтральной
реакции и сушат на воздухе. После кристализации из смеси
этил ацетат — петролейный эфир выход составляет 16,6 г
(85 %), т. пл. 126 °C.
Кб.з-производные аспарагина, глутамина, у-бензилов< го
эфира глутаминовой кислоты, а также р-бензилового эфира
аспарагиновой кислоты синтезируют иначе, так как для этих
аминокислот при высоких значениях pH может произойти
омыление эстерных групп и гидролиз амидов. Для синтеза
Кбз-производных тирозина, гистидина, лизина и аргинина, за-
щищенных лишь по а-аминогруппе, также необходимы спе-
циальные условия эксперимента. Ниже приведены соответст-
вующие методики.
Методика 95. Карбобензокси-£-глутамин [222].
К раствору 14,6 г (0,1 моль) L-глутамина в смеси 25 мл 4 н.
едкого натра и 100 мл 1 н. раствора бикарбоната натрия (pH 9,5)
одновременно прибавляют при охлаждении и перемешивании
в течение 1 ч 24,5 г (0,15 моль) карбобензоксихлорида в 40 мл
диоксана и 30 мл 4 н. NaOH; после этого смесь размешивают
еще 45 мин при 20 °C, затем 1 н. соляной кислотой доводят pH
до 7,0 и избыток Кбз-хлорида экстрагируют эфиром. Раствор
подкисляют до pH 1,0, выпавшие кристаллы отфильтровывают,
промывают водой до нейтральной реакции и сушат в эксикато-
ре. После кристаллизации из воды выход Кбз-глутамина 25 г
(86 %), т. пл. 133—137 °C.
Методика 96. у-Бензиловый эфир карбобензоксиглу-
таминовой кислоты [223].
23,7 г (0,1 моль) у-бенз^илового эфира L-глутаминовой кис-
лоты и 20 г (0,24 моль) бикарбоната натрия растворяют при
65 °C .в 1 л воды. К раствору при постоянном размешивании
добавляют по каплям 20 мл карбобензоксихлорида и продол-
жают перемешивание до тех пор, пока температура реакцион-
ной массы не снизится до 50 °C; на это требуется примерно
50 мин. Затем колбу охлаждают, раствор обрабатывают эфиром
(30 мл X 2), фильтруют и подкисляют до pH 2,0. Выпавшее
масло экстрагируют эфиром; эфирные вытяжки промывают
водой, содержащей 7 мл 1,4 н; раствора бикарбоната калия,
сушат сульфатом магния и эфир удаляют на роторном испари-
теле. Остаток растворяют в четыреххлористом углероде, упа-
ривают до объема 140 мл и оставляют на ночь в холодильнике.
Выпавшие бесцветные иглы отфильтровывают и сушат. Выход
19,8 г (54 %), т. пл. 76—78 °C.
Если продукт не удается закристаллизовать, то его можно
перевести в кристаллическую дициклогексиламмонийную соль.
119
Продукт реакции растворяют в эфире и добавляют небольшой
избыток дициклогексиламина. Через некоторое время выпадает
кристаллическая ДЦГА-соль у-бензилового эфира карбобен-
зоксиглутаминовой кислоты.
Аналогично [224] из 4,4 г (20 ммоль) р-бензилового эфира —
аспарагиновой кислоты и 4,1 г (24 ммоль) карбобензоксихлори-
да при 50 еС получили 5,3 г (75 %) карбобензоксипроизводного
с т. пл. 107—108 °C, [а]2* + 11,9 (с = 10, уксусная кислота).
Ввиду низкого выхода у-бензилового эфира карбобензокси-
глутаминовой кислоты нами предложен метод получения его
через триметилсилильное производное с выходом около 90 %.
Методика 97. у-Бензиловый эфир карбобензокси-L-
глутаминовой кислоты
H-Glu (OBzl) ОН gN^-MCI-> TMS-Glu (OBzl) OTMS
-> Z—Glu (OBzl) OTMS —Z—Glu (OBzl) OH.
К суспензии 10 г (41 ммоль) у-бензилового эфира L-глутамино-
вой кислоты в 60 мл безводного метиленхлорида добавляли
13 мл (100 ммоль) триметилхлорсилана и кипятили, защищая
от влаги, с обратным холодильником 1 ч. После охлаждения
раствора льдом вносят 7,6 мл (72 ммоль) NMM и 8 мл (55 ммоль)
карбобензосихлорида и перемешивают при этой температуре
1 ч, а затем еще 5 ч. После фильтрования и упарки раствора
остаток в колбе переносят в этилацетат, промывают 1 н. НС1 и
водой, сушат сульфатом натрия и отгоняют растворитель.
К раствору остатка добавляют гексан и растирают с ним, затем
растворяют масло в эфире и вносят небольшой избыток дицик-
логексиламина и оставляют на 1 ч при 0 °. Выпавшую дицикло-
гексиламмонийную соль отфильтровывают, хорошо промы-
вают эфиром и сушат. Выход 20,3 г (88 %), т. пл. 142—144 eG,
[а]25 = +7,0° (с = 1; метанол).
Методика 98. Карбобензокси-Ь-аргинин [225].
К охлажденному до 0° раствору 63 г (0,3 моль) хлоргидрата
L-аргинина в 300 мл 1 н. NaOH прибавляют в течение 1 ч при
перемешивании 55 мл карбобензоксихлорида и 165 мл 2 н. рас-
твора едкого натра, поддерживая pH 9—10. После добавления
всего карбобензоксихлорида размешивание продолжают еще
2 ч, а затем подкисляют до pH 7—7,5. Выпавший осадок
отфильтровывают, промывают 150 мл холодной воды и кристал-
лизуют из 400 мл кипящей воды. После отделения продукта
фильтрованием и высушивания кристаллы размешивают в те-
чение 1 ч со смесью 400 мл ацетона и 200 мл эфира, отфильтро-
вывают и сушат в вакууме при 50 еС. Выход бесцветных кри-
сталлов 82,7 г (89,5 %), т. пл. 184 еС; [<х]^5 = —9,3е (с == 1, 1 н.
соляная кислота).
120
В монографии [9] описан удобный метод синтеза карбобен-
зоксиаминокислот при комнатной температуре в присутствии
избытка бикарбоната натрия.
В качестве примера приводим очень удобную методику син-
теза карбобензокси-£-аргинина, которая дает возможность
получить очень чистый, прекрасно кристаллизующийся про-
дукт.
Методика 99. Карбобензокси-£-аргинин [226]
К суспензии 63 г бикарбоната натрия в 250 мл воды в колбе
емкостью 1 л прибавляют 42,2 г (20 ммоль) хлоргидрата £-арги-
нина и перемешивают при комнатной температуре до полного
растворения. Затем вносят 37,4 г (31 мл; 22 ммоль) карбобензок-
сихлорида пятью равными порциями в течение 30 мин и пере-
мешивают еще 1 ч при комнатной температуре. Смесь подщела-
чивают до pH 8,5 концентрированным раствором аммиака и
выдерживают 2 ч при 4 °C. Выпавший осадок отфильтровывают,
промывают холодной водой и кристаллизуют из воды, в которую
добавляют несколько капель аммиака. Выход белого кристал-
лического продукта 58,6 г (95 %), т. пл. 175 °C.
Физико-химические характеристика карбобензоксиамино-
кислот представлены в табл. 16.
Таблица 16. Свойства карбобензоксиаминокислот
Аминокислота Т. пл., °C 25 [а]р, град (растворитель)
/.-Аланин 87 —13,9 (с = 2; метанол)
/.-Аргинин 175 —9,2 (с== 2; 1 н. НС1)
<о-Нитро-/.-аргинин 134—136 —3,5 (<?«= 1; метанол)
(о-Дикарбобензокси-Д-арги- 138—139 + 16,8 (с = 1; СНС13)
нин £-Аспарагин 165 +7,6 (с~ 1; уксусная
L-Аспарагиновая кислота 116 кислота) +9,6 (с= 7; уксусная кислота) + 11,9 (с — 10; уксусная
P-Бензиловый эфир £-аспа- 107—107
рагиновой кислоты кислота)
ДЦГА-соль Р-бензилового 117—118 + 14,8 (с — 1; этиловый
эфира /.-аспарагиновой спирт)
кислоты
/.-Валин 59—60 + 1,5 (с — 5; этанол)
/.-Глутамин 139—141 —5,7 (с — 5; этанол)
/.-Глутаминовая кислота 120—121 —7,9 (с— 10; уксусная
у-Бензиловый эфир /.-глу- 75—76 кислота) —22,7 (с= 1; 4, 1 н.
таминовой кислоты КНСОз)
ДЦГА-соль у-бензилового 144—146 +7,9 (с= 2,9;, метанол)
эфира /.-глутаминовой кислоты Глицин 120 — '
121
Продолжение табл. 16
Аминокислота Т. пл., °C 25 [ос]р, град (растворитель)
/.-Гистидин 166—167 —25,0 (с =6; 6 н. НС1)
1\гш-Бензил-£-гистидин 214—215 (с +6,4 (с = 5; уксусная
разложением) кислота)
_\?/т-Карбобензокси-£-гисти- 90,5—92,0 +29,1 (с = 1; этилацетаТ
дин £-Изолейцин (с разложением) 44—46 +6,5 (с = 6; этанол)
А-Лейцин Масло —16,4 1,8; этанол)
ДЦГА-соль £-лейцина 151,5—152 —7,9 (с = 3; метанол)
Н8-Карбобензокси-£-лизин 278—280 ; 235 + 17,3 (с = 2; 2 н. НС!)
М,М-Дикарбобензокси-£- 88—90; 80 —5,0 (с = 2; метанол)
лизин £-Метионин 68—69 —31,6 (с « 1; метанол)
£-Оксипролин 106—107 —72,0 (с « 1; хлороформ)
L-Пр олин 76—77 —61,7 (с= 5; уксусная
£-Серин 121; 114—116 кислота) +5,6 (с= 6; уксусная
О-тре/н-Бутил-£-серин 85—87 кислота) +21,2 (с= 2,8; этанол)
£-Тирозин 101 + 11,1 (с = 3, уксусная
0-Бензил-£-тирозин 112—113 кислота)
О,Ы-Дикарбобензокси-£-ти/ розин £-Треонин 117 103—104 —5,0 (с = 10; уксусная кислота) —5,8 (с = 2; уксусная
ДЦГА-соль O-mpem-бутил- 146—147 кислота) +9,9 (с — 1; эталацетат)
£-треонина /.-Триптофан (126—126,5) 126 —
£-Фенилаланин 88—89 +5,1 (с — 2; этанол)
5-Бепзил-£-цистеин 135—136 —36,6 (с = 5; этанол)
Для получения Z-аминокислот предложен новый реагент —
дибензил-пирокарбонат (96) [227]. Синтез Z-аминокислот про-
водится в водно-щелочной среде или с добавкой диоксана. Вы-
ход производных 72—91 %. В отличие от карбобензоксихлори-
да новый реагент не дает примесей побочных продуктов — ди-
пептидов.
£ £—сн2о—с—о—с—он2с—£
II II
о о
(96)
Отмечаются следующие недостатки карбобензоксихлорида
[2281: 1) следы ионов металлов и хлорид-иона катализируют
разложение карбобензоксихлорида при хранении, что приводит
к загрязнению ^-аминокислот; 2) сложно получить карбо-
бензоксихлорид высокой степени чистоты; 3) синтез Z-амино-
122
Таблица 17. Некоторые производные для введения
карбобензоксигруппы
R Вы- ход, % Т. пл., °C Система для кристаллизации Литерату- ра
79—80
58—59
85—87
115—116
78—80
Масло
—о/хЧг
Бензол-петролейный [229]
эфир
То же [229]
Петролейный эфир [229]
Толуол [229]
Этилацетат-петролейпый [230]
эфир
[231]
кислот может сопровождаться образованием олигопептидов
через смешанные ангидриды— в зависимости от условий реак-
ции (pH, концентрации и температуры), в неочищенных пре-
паратах Z—SerOH и Z—ThrOH образуется до 20 % N-карбо-
бензоксипептидов. Для устранения этого недостатка предложе-
ны некоторые производные общей формулы (табл. 17):
О
СН2О—С—R
Методи к а 100. n-Нитрофенил-бензил карбонат [229]
О
/ \-СН2ОС-С1 + НО-/ >-no2 ->
156,5 "139,0
О
-* сн8о—<Lo—NO2
259,0
123
К раствору 6,95 (0,05 моль) /г-нитрофенола и 6,45 г
(0,05 моль) хинолина в 100 мл метиленхлорида добавляли по
каплям 8,5 г (0,05 моль) карбобензоксихлорида в течение
15 мин. Реакционную смесь выдерживали в течение ночи, про-
мывали 1 н. НС1, 0,5 н. NаНСО3 и водой и сушили сульфатом
натрия. После удаления растворителя в вакууме остаток кари-
сталлизовали из смеси бензол — петролейный эфир. Выход
11,2 г (83 %), т. пл. 79—80 °C.
Аналогично могут быть получены соединения, указанные
в табл. 17.
Удаление бензилоксикарбонильной группы
Методика 101. Получение насыщенного раствора^бро-
мистого водорода в ледяной уксусной кислоте.
Для получения газообразного бромистого водорода исполь-
зуют реакцию бромирования тетралина или нафталина.
В двугорлый реактор, снабженный обратным холодильником
и капельной воронкой, помещают избыток тетралина, а в ка-
пельную воронку — рассчитанное количество брома. Для по-
глощения паров брома, увлекаемых током НВг, газоотводную
трубку соединяют с двумя склянками Дрекселя, одна из кото-
рых заполняется тетралином, а другая — суспензией красного
фосфора в толуоле. Для предотвращения попадания влаги
из воздуха в колбу, содержащую уксусную кислоту, соединяют
с хлоркальциевой трубкой, наполненной плавленным СаС12.
Ток бромистого водорода регулируется скоростью подачи
брома. Сначала насыщение уксусной кислоты бромистым во-
дородом ведут при комнатной температуре, так как при охлаж-
дении уксусная кислота может затвердеть; затем колбу с уксус-
ной кислотой можно охладить, поместив ее в баню со льдом.
Если вместо тетралина используют нафталин, то целесооб-
разно добавить в реакционную колбу небольшое количество
бензола и при добавлении брома нагревать ее на водяной бане.
Степень насыщения уксусной кислоты бромистым водоро-
дом контролируют по привесу.
В табл. 18 представлены соответствующие данные из рас-
чета на 36 %-й раствор НВг в уксусной кислоте.
Свежеприготовленный раствор бромистого водорода в уксус-
ной кислоте почти бесцветен; при хранении, однако, он приоб-
ретает желтоватую окраску. Длительное хранение такого ра-
створа нежелательно.
Методика 102. Бромгидрат этилового эфира глицил-
L-аланина
В 15 мл безводной уксусной кислоты суспендируют 10 г
(0,033 моль) этилового эфира карбобензоксиглицил-£-аланина
124
и прибавляют равный объем
(15 мл) 36 %-го раствора
бромистого водорода в ле-
дяной уксусной кислоте.
Колбу закрывают пробкой,
снабженной U-образным
счетчиком пузырьков, ко-
торый заполняют серной
кислотой. Это предотвраща-
ет проникновение влаги из
воздуха и позволяет следить
за скоростью выделения
СО2. Обычно отщепление
Таблица 18. Количество НВч,
необходимое для насыщения
различных количеств уксусной
кислоты до 36 %
Исходная масса уксус- ной кислоты, взятой для насыщения, г Масса НВг, г Общая масса НВг в уксус- ной кислоте, г
50 28 78
100 56 156
150 84 234
250 140 390
300 168 468
500 280 780
карбобензоксигруппы про- ------------------------------
ходит за 30—40 мин. Спустя указанное время содержимое кол-
бы выливают тонкой струйкой в 200 мл сухого эфира; при этом
выпадает белый творожистый осадок бромистоводорбдной соли
дипептида. Надосадочную жидкость декантируют, осадок про-
мывают сухим эфиром, быстро отфильтровывают и хорошо про-
мывают эфиром. Бромгидраты пептидов часто расплываются на
воздухе, поэтому все операции промывания и декантации
следует вести так, что^ы над осадком оставался слой эфира;
после окончательной промывки продукт тотчас же помещают
в вакуум-эксикатор с сухим едким натром. Выход продукта
7 г (85 %).
Полученные бромгидраты пептидов без дополнительной
очистки можно вводить в пептидный синтез. Часто такие про-
дукты адсорбируют некоторое количество бромистого водорода,
который не удаляется даже в вакуум-эксикаторе. В связи с
этим рекомендуется переводить их в свободные основания сле-
дующим способом: к суспензии или раствору бромгидрата пеп-
тида сначала прибавляют рассчитанное количество третичного
ямина (триэтиламина, N-метилморфолина и т. д.), измеряют
pH раствора по индикаторной бумажке и, если необходимо, то
добавляют по каплям избыток соответствующего основания до
значения pH раствора 8—9.
Наряду с бензилоксикарбонильной группой бромистый во-
дород отщепляет и другие кислотолабильные защитные группы,
расщепляет /пре/п-бутиловые эфиры и частично бензиловые эфи-
ры пептидов. Следует также иметь в виду, что НВг/АсОН раз-
рушает остаток триптофана и разрывает пептидную связь меж-
ду глицином и пролином, которая, как известно, лабильна в
кислой среде.
При деблокировании гидрофобных пептидов их бромисто-
водородные соли могут сначала оставаться в растворе при вы-
ливании реакционной массы в эфир. Такое явление, в частности,
125
мы наблюдали при деблокировании Z-группы у п-Нитробепзи-
лового эфира карбобензоксифенилаланил-лейцина. Кристал-
лический осадок бромгидрата указанного пептида образовался
только при стоянии реакционной смеси в холодильнике в те-
чение 30 мин.
Отщепление Z-группы каталитическим гидрогенолизом
Карбобензокси-группа легко отщепляется при действии
водорода в присутствии таких катализаторов, как палладиевая
чернь, оксид палладия и палладий на угле с образованием
5 глекислого газа и толуола [232].
о
СН2О-С—NH—CHR— СООН
-> H2N—СН— R—СООН + СОа + <“>-сн3.
Восстановление ведут обычно при комнатной температуре при
атмосферном давлении в спиртовых, водно-спиртовых раство-
рах или смесях спирта, воды и кислоты. При необходимости
удаление Z-группы в присутствии других кислотолабильных
защитных групп гидрирование следует вести в отсутствии
кислоты. В процессе гидрирования одновременное карбобензо-
ксигруппой расщепляются бензиловые и /г-нитробензиловые
эфиры, а также деблокируется гуанидиновая группа со-нитро-
аргинина. Метиловые, этиловые и mpem-бутиловые эфиры
пептидов при этом не расщепляются; не происходит деблоки-
рование также mpem-бутилоксикарбонильной, тозильной и
некоторых других защитных групп. Это позволяет осуществ-
лять селективное деблокирование пептидов. Необходимо отме-
тить, что при гидрировании остаток триптофана не изменяется.
Ввиду того, что палладиевый катализатор легко отравляется
серосодержащими соединениями, гидрирование непригодно
для удаления Z-группы у цистеин- и метионинсодержащих пеп-
тидов, а также у пептидов, блокированных о-нитросульфофе-
нильной защитной группой. По этой же причине в приборе для
гидрирования следует избегать применения резиновых трубок
и пробок. Каталитическое гидрирование ведут на установке,
изображенной на рис. 2.
Палладиевая чернь пирофорна, поэтому катализатор хра-
нят под слоем воды. Более удобен в хранении оксид палладия:
его можно сохранять в сухом виде. При гидрировании оксид
палладия превращается в активный губчатый палладий. Пал-
ладиевую чернь используют чаще, чем оксид палладия, по-ви-
димому, из-за того, что ее легче получить.
126
Методика 103. Приго-
товление палладиевой черни
[232].
В колбу Кьельдаля вмести-
мостью 250 мл помещают 25 мл
«царской водки» и 6 г мелко
нарезанных кусков палладие-
вой фольги. Смесь выдержи-
вают 20 мин при комнатной
температуре, а затем осторож-
но нагревают в вытяжном
шкафу до полного растворения
металла. Полученный раствор
упаривают досуха, а затем
остаток растворяют в 15 мл
концентрированной соляной
кислоты и снова раствор упа-
ривают досуха; эту операцию
Рис. 2. Установка для каталитиче-
повторяют еще 1 раз, а затем ского восстановления
остаток растворяют в 40 мл 5 н.
соляной кислоты и фильтруют в колбу, содержащую 600 мл горя-
чей дистиллированной воды. К горячему раствору при взбалты-
вании добавляют по каплям 1,8 мл муравьиной кислоты, после
чего при энергичном встряхивании прибавляют порциями
около 100 мл 5 н. едкого кали до щелочной реакции на лакмус.
К полученному раствору при размешивании добавляют по
каплям муравьиную кислоту так, чтобы среда оставалась слабо
щелочной. Обильный осадок палладиевой черни отделяют де-
кантацией и многократно промывают дистиллированной водой.
Катализатор хранят в плотно* закрытом сосуде под слоем
воды, предохраняя его от контакта с серосодержащими мате-
риалами и реагентами (сероводородом, алкилмеркаптанами
и т. д.). Для проверки активности катализатора необходимо
внести небольшое количество губчатого палладия в пробирку,
промыть метанолом, а затем поместить на фильтровальную бума-
гу. Если катализатор активен, наблюдается возгорание и обуг-
ливание фильтра.
Методика 104. Получение оксида палладия на угле.
Очень хорошим катализатором является оксид палладия
на угле [233]. Его получают следующим способом:
Drl HNQ3 . njri ОН ПЛГнГ
Pd нс1 PdClg q PdO/C.
Для очистки 50 г активного угля заливают 150 мл 15 %-й со-
ляной кислоты и кипятят в течение 30 мин. Горячий раствор
фильтруют, промывают уголь горячей водой до pH 7,0 и су-
шат при 110 бС в течение 1 ч. Проба этого угля со смесью раз-
127
бавленных НО и HNO3 не должна давать реакции на железо с
железосинеродистым калием.
Для получения хлористого палладия 2,5 г палладия поме-
щают в фарфоровую чашку на кипящей водяной базе и при пере-
мешивании медленно добавляют около 60 мл царской водки до
полного растворения палладия. Раствор упаривают досуха, и
коричневый остаток растворяют в 300 мл дистиллированной во-
ды, слегка подкисляют НС1, фильтруют и разбавляют дистил-
лированной водой до объема 350 мл.
В стакан вместимостью 500 мл, снабженный механической
мешалкой, термометром и капельной воронкой и помещенный
па асбестовой сеуке, насыпают 50 г очищенного активного угля,
наливают 230 Мл дистиллированной воды, нагревают при
медленн м перемешивании до 80 °C и из капельной воронки мед-
ленно приливают полученный ранее раствор хлористого пал-
ладия. Затем при перемешивании при 80 °C по.каплям прили-
вают 10 %-й раствор NaOH до щелочной реакции раствора.
Раствор перемешивают и выдерживают при 80 °C еще 1 ч до
тех пор, пока из отфильтрованной пробы смеси, подщелочен-
ной едким кали, при кипячении с несколькими каплями 40 ,%*
го формалина не перестанет выделяться черный осадок пал-
ладия.
Уголь с адсорбированной на нем окисью палладия фильт-
руют и промывают теплой водой до исчезновения щелочной
реакции на лакмус. Осадок сушат при ПО °C и после измель-
чения в ступке хранят в банке с притертой пробкой.
Предложен очень эффективный катализатор гидрогенолиза:
палладий наЪолиэтилениминном носителе [234], который обла-
дает более высокой активностью, чем палладий на угле и пал-
ладиегап чернь. Корбобензокси-группа удаляется в присутст-
вии этого катализатора количественно за 20 мин.
Методика 105. Общий метод гидрогенолиза пептидов.
Навеску карбобензоксипептида (0,15 моль) суспендируют
или растворяют в 60 мл смеси метанола —- уксусная кислота —
вода (6:1: 1), прибавляют палладиевый катализатор и гидри-
руют 3—4 ч при комнатной температуре, контролируя ход
реакции деблокирования ТСХ, а также по прекращению выде-
ления СО2 (проба с гидроокисью бария). После окончания реак-
ции катализатор отфильтровывают, промывают водой, а рас-
творитель упаривают в вакууме при 40 °C. Остаток можно ис-
пользовать для синтеза пептидов без дополнительной очистки.
Методика 106. Восстановительное отщепление Z-груп-
пы циклогексеном [210, 211]
Z-HN-R + Q ™ Q| + HaN—R + СвН6-СН3,
128
Навеску карбобензоксиаминокислоты или Кбз-пептида рас-
творяют в этиловом спирте или метаноле, прибавляют требу-
емое количество циклогексена и палладия на угле. Используют
равное по массе пептида или полуторакратное количество ка-
тализатора. Смесь кипятят 15мин, затем катализатор отфиль-
тровывают, промывают водой, а растворитель упаривают в ва-
кууме. Выход продукта составляет 90—100 %.
Методика 107. Деблокирование гуанидиновой группы
нитроаргинина циклогексеном [211].
745 мг Вос—Phe—Arg (NO2)—ОН и 14 мл циклогексена
растворяют в 28 мл спирта. Для полного растворения пептида
прибавляют 3 мл уксусной кислоты. К раствору добавляют
75 мг палладиевой черни и реакционную смесь кипятят с обрат-
ным холодильником в течение 6 ч, контролируя степень отщеп-
ления нитрогруппы при помощи ТСХ. После окончания реак-
ции катализатор отфильтровывают, растворитель упаривают
в вакууме, а остаток переосаждают из смеси метанол — этила-
цетат. Выход ацетата пептида 88 %, т. пл. 175—180 °C.
Методика 108. Деблокирование пептидов циклогекса-
диеном [212].
Защищенный пептид (1 ммоль) растворяют в 4 мл абсолют-
ного этилового спирта в колбе, снабженной барботером. Реак-
ционную колбу помещают на водяную баню при 25 °C и проду-
вают током сухого азота. Затем прибавляют 10 %-й палладий
на угле (1 эквивалент на каждую удаляемую группу) и 0,94 мл
(10 ммоль) 1,4-дициклогексадиеня. Реакцию ведут по крайней
мере 2 ч, после чего катализатор отфильтровывают, промывают
ДМФА, уксусной кислотой и водой, растворитель упаривают в
вакууме, а остаток обрабатывают, как обычно. Выход около 90 %.
Этим способом можно деблокировать Z, Bzl, МО2-группы
у пептидов, содержащих остатки гистидина, аргинина, тирози-
на, метионина и других аминокислот.
Методика 109. Восстановительное деблокирование
Z-группы при помощи муравьиной кислоты [213].
Раствор 200 мг карбобензоксипептида в 2—10 мл 4,4 %-го
(по объему) раствора муравьиной кислоты (88 %-й в метаноле)
добавляют к 10 мл 4,4 %-й муравьиной кислоты в метаноле,
содержащей 200 мг палладиевого катализатора. Смесь разме-
шивают 10 мин при комнатной температуре в атмосфере азота,
затем катализатор отфилвтровывают, промывают 10 мл метано-
ла и 10 мл воды. Растворитель упаривают на роторном испа-
рителе и остаток кристаллизуют из подходящего растворителя.
Выход 90—97 %.
Следует отметить, что этим способом удалось гладко дебло-
кировать этиловый эфир карбобензокси-метионил-глицина,
что обычно не удается при традиционном методе отщепления
9 2-882 Д29
Z-группы. Гуанидиновая группа нитроаргинина отщепляется
муравьиной кислотой примерно за 5 ч. Для контроля оконча-
ния реакции удобно использовать ТСХ.
Методика ПО. Деблокирование пептидов формиатом
аммония [214].
К раствору защищенного пептида в метаноле или ДМФА’
добавляют от 0,1 до 0,5 частей по массе пептида 10 %-го палла-
дия на угле и 2—4 эквивалента формиата аммония. Реакцион-
ную смесь выдерживают в течение 5—10 мин и затем катализа-
тор отфильтровывают, растворитель удаляют в вакууме; а
остаток лиофилизируют или растворяют в этилацетате и затем
экстрагируют насыщенным раствором хлористого натрия.
В дальнейшем выделение пептид^ ведут, как обычно.
Время для удаления Z-группы в большинстве случаев со-
ставляет около 1 мин; нитрогруппа отщепляется примерно за
5—10 мин. Для деблокирования бензильных групп серийа и
треонина в смеси метанол — уксусная кислота (1:1) требуется
30 и 90 мин соответственно.
Методика 111. Восстановительный гидрогенолиз пеп-
тидов гидразингидратом [235].
Раствор защищенного пептида в метаноле или эталоне, со-
держащий палладиевую чернь (1/10 по массе пептида), пере-
мешивают при 50 °C и добавляют от 6 до 8 эквивалентов гидра-
зингидрата (99—100 %). После окончания гидрогенолиза
(Z—, NO2—, Туг (OBzl) — 0,5.. 1,5 ч; Ser (OBzl) — 8 ч) ката-
лизатор отфильтровывают, фильтрат упаривают в вакууме.
Для удаления избытка гидразингидрата продукт экстраги-
руют этилацетатом или хлороформом и промывают водой.
Выход 90—96 %.
Имеются сообщения отом, что каталитическое гидрирование
проходит не всегда гладко. При гидрировании нитроаргинин-
содержащих пептидов формиатом аммония или водородом над
палладием деблокирование проходит гладко, однако, если в их
состав входит остаток Тгр, деблокирование проходит на 20—
25 %. В кислой среде происходит гидрирование индольного
кольца Тгр [236].
При длительном деблокировании Тгр-содержащих пептидов
в муравьиной кислоте (до 5 ч) образуются производные 2,3-ди-
гидротриптофана [237].
Имеется сообщение [238], что в процессе деблокирования
пептида Вос—Asp (oBzl)—Ala—Phe—He—GlyOEt водородом
или формиатом аммония в присутствии палладия на угле обра-
зуется от 5 до 30 % побочного продукта — аминосукцинильно-
го производного. Эта побочная реакция полностью подавляется
добавлением нескольких эквивалентов уксусной или муравьи-
ной кислоты.
130
Удаление карбобензокси-группы
с использованием акцепторов
Предложен способ удаления карбобензокси-группы и за-
щитных групп бензильного типа трифторуксусной кислотой с
добавкой акцепторов, которые ускоряют реакцию и связывают
побочные продукты [2391. Реакция имеет место при протони-
ровании (жесткой кислотой) атома кислорода (жесткого осно-
вания) карбонильной группы и атаке атомов серы (мягкое осно-
вание) электрондефицитного бензильного углеродного атома
(мягкая кислота). Таким образом эта реакция включает коопе-
ративное действие мягкого нуклеофила и жесткого электрофи-
ла на субстрат («пуш-пулл механизм»), что благоприятствует
отщеплению карбобензокси-группы. Скорость реакции зависит
от природы акцептора и убывает в ряду: тиоанизол ) диме-
тил сульфид ) этандитиол ~ фенол ) анизол.
^~СН2-ОСО— NHR + CFSCOOH
~ t _
СН8—S— /
+ ” /—к +
-> СО2 + RNH8 . CF3COO- + " у—S—СН8 • CFSCOO’
Хотя тиоанизол оказался хорошим промотором реакции, он
является недостаточно хорошим акцептором бензильной груп-
пы в случае деблокирования О-бензил-тирозина, поэтому Та-
кейама с соавт. [240] предложили вносить дополнительно м-
крезол. Бодански и соавт. [241] предложили в качестве акцеп-
тора вместо смеси тиоанизола с ;и-крезолом использовать 4-
(мети л мер к апто)фенол.
Методика 112. Удаление Z-группы смесью трифторук-
сусная кислота — тиоанизол [239].
Деблокирование пептидов с удалением карбобензокси-
группы одновременно с О-бензильной группой терозина ведут
следующим образом. Навеску (0,1 ммоль) Z-аминокислоты или
пептида растворяют в 27 мл ТФУ, добавляют 5 ммоль тиоани-
зола и выдерживают реакционную смесь 3 ч при комнатной
температуре, затем растворитель удаляют в вакууме и вы-
деляют продукт обычным образом.
Удаление защитных групп триметилсилил-
трифторметилсульфонатом (тримех^лсилилтрифлатом)
Фуджи и соавт, [2421 показали, что триметилсилилтрйфлат
в смеси с тиоанизолом (1:1 моль) в ТФУ (1 М раствор) удаляет
многие защитные группы без заметных побочных реакций.
9*
131
Указанный реагент оказался более эффективным, чем трй-
фторметансульфокислота в ТФУ. Образующиеся в результате
реакции триметилсилильные производные гидролизуются во-
дой или фторидом аммония.
Ввиду того, что триметилсилилтрифлат используется в
основном для удаления боковых защитных групп на последней
стадии деблокирования защищенных пептидов, методики рабо-
ты с ним будут приведены в главах 4 и 5. В данном разделе мы
лишь отметим, что триметилсилилтрифлат гладко отщепляет
такие N-защитные группы, как Z, Вос, Trt, Врос и др.
Методика 113. Синтезтриметилсилилтрифлата [243].
О
CF3-SO3H + Si (СН3)4 CF8-S-O-Si (СН3)3 + СН4
А
Перемешивали 1,5 г (10 ммоль) трифторметансульфокисло-
ты и 1,1 г тетраметилсилана в атмосфере аргона при комнатной
температуре. Через 1 ч выделение метана прекращается, что
свидетельствует об окончании реакции. Вакуумная перегонка
дает 2,2 г (99 %) бесцветной жидкости ст. кип. 40 °C (11 мм. рт.
ст.) и 23—24 °C (3 мм рт. ст.); 4° « 1,225; 4°’5 - 1,3604.
mpem-Бутилоксикарбонильная группа
Основным методом введения Вос-группы является обработ-
ка аминокислот ди-трет-бутилпирокарбонатом [244—2461
или Вос-азидом [247, 2481.
Синтез реагентов для получения
Вос-аминокислот
Методика 114. Синтез /пре/п-бутилоксикарбонилазида
[247].
Вос-азид получают в три стадии: сначала готовят трет-бу-
тилэтилкарбонат (97), который превращают в /пре/п-бутилокси-
карбонилгидразид (98), а последний — в соответствующий
азид (99):
СН, О СН, О
I II II
а) СН,-С—ONa + Cl—С-ОС,Н, СН,—С—О—С—ОС,Н6;
сн, сн,
(97)
132
сн, о CH, о
6) CH,-C-O-COC2H6 + H2N—NH, -> СН,—С—О—(^—NHNH,;
СН, ? <4н8
(98)
СН, О
в) СН,-<СО—С—NHNH,
(!н,
СН, о
СНз-А-О-С-Н,.
<!н,
(99)
Методика 115. /ире/п-Бутилэтилкарбонат [246].
Круглодонный трехгорлый реактор (2 л), снабженный ме-
шалкой и эффективным холодильником с хлоркальциевой труб-
кой, закрепляют над плитой с закрытой спиралью. В реактор
помещают 250 мл абсолютного толуола и 69 г металлического
натрия, а затем включают мешалку и нагрев плиты. Когда
натрий расплавится, то мешалку запуск; ют на полные обороты.
Суспендирование натрия продолжают 20—30 мин. После по-
лучения тонкой суспензии натрия нагрев отключают и, про-
должая перемешивать, снижают температуру реакционной сме-
си до комнатной. Мешалку останавливают и толуол сливают, а
остаток его возможно полнее отсасывают при помощи пипетки
и водоструйного насоса.
Реактор помещают в баню, охлаждаемую сухим льдом и
ацетоном, и прибавляют ИЗО мл абсолютного третичного бу-
тилового спирта. Скорость реакции регулируют погружением
реактора в охлаждающую баню или удалением охлаждения.
Взаимодействие /ире/и-бутанола с натрием должно идти с такой
скоростью, чтобы обратный холодильник не «захлебывался».
После завершения реакции колбу подогревают некоторое вре-
мя, а затем оставляют затвердевшую массу на 24 ч при комнат-
ной температуре, защищая от попадания влаги и воздуха. Для
окончания реакции реактор нагревают на масляной бане при
125 еС в течение 5 ч и затем массу охлаждают.
Реактор снабжают капельной воронкой, термометром и
погружают в баню со льдом; при постоянном размешивании
добавляют в течение 1—1,5 ч по каплям 285 мл этилового эфира
хлоругольной кислоты (температура внутри колбы должна
быть не выше 24—28 °C). После того, как весьэтилхлорформиат
будет добавлен, смесь оставляют на ночь при комнатной темпе-
ратуре, а затем добавляют 600 мл воды, переносят в делитель-
ную воронку на 2 л и экстрагируют эфиром (200 мл X 3). Со-
единенные вытяжки сушат сульфатом магния (500 г) в течение
24 ч. Раствор фильтруют в двухлитровую круглодонную кол-
бу, снабженную дефлегматором, и эфир отгоняют. Остаток
133
нагревают на масляной бане, медленнс поднимая температуру
до 130 °C; при этом отгоняются примеси с температурой кипе-
ния до 83 °C. Остаток перегоняют в вакууме, собирая фракции
с температурой кипения 62—64 °C (38—40 мм рт. ст.). Выход
продукта 300—305 г (70 %).
Чистый трет-бутилэтилкарбонат — бесцветная или слегка
желтоватая жидкость с т. кип. 62 °C (35 мм рт. ст.).
Методика 116. /пое/п-Бутилоксикарбонилгидразид
[2481.
В круглодонную колбу на 2 л, снабженную обратным хо-
лодильником, через который пропущена мешалка, помещают
300—305 г mpem-бутилэтилкарбоната и 520 г (505 мл) гидра-
зипгидрата. Колбу устанавливают на плитку с закрытой спи-
ралью и кипятя! при непрерывном размешивании в течение
13 ч. После охлаждения до комнатной температуры содержи-
мое переносят в делительную воронку на 2 л и добавляют насы-
щенный раствор хлористого натрия (500 мл). Смесь экстраги-
руют затем эфиром (4 X 300 мл). Органическую фазу промывают
200 мл насыщенного раствора хлористого натрия и сушат суль-
фатом магния в течение ночи. Затем раствор фильтруют, эфир
удаляют на роторном испарителе. Остаток затвердевает. Вы-
ход бесцветного кристаллического продукта составляет 137—
145 г (52—56 %), т. пл. 38—40 °C, т. кип. 70—72 °C (3,5 мм рт.
ст.). Примечание. Если после отгонки растворителя
продукт не затвердевает, то его необходимо растворить в эфире,
высушить сульфатом магния и эфир отогнать.
Методика 117. /ирг/п-Бутилоксикарбонилазид [2471.
Раствор 100 г Вос-гидразида и 88 мл уксусной кислоты в
125 мл воды охлаждают льдом, и при сильном размешивании
добавляют в течение 50—60 мин при 10 °C 54,4 г нитрита на-
трия или его раствор в 80 мл воды. Затем реакционную смесь
перемешивают еще 30 мин и после этого добавляют 125 мл во-
ды. Содержимое колбы переносят в делительную воронку, отде-
ляют нижний водный слой, а верхний слой, окрашенный в
желтый цвет, промывают водой (3x50 мл) и 1 н. раствором
бикарбоната натрия (3X40 мл) и помещают в колбу, содер-
жащую 10 г сульфата магния. В таком виде Вос-азйд хранят
в холодильнике. Примечав и е. Вос-азид ядовит! Вдыха-
ние его паров вызывает головную боль. Продукт можно пере-
гонять при 73—76 °C (70 мм рт. ст.). Однако это нежелатель-
но, так как возможен взрыв. Вос-азид следует хранить в холо-
дильнике не более двух дней.
134
Синтез третп-бутилоксикарбониламинокислот
по Шнабелю
В 1967 г. Шнабель предложил получать Вос-аминокислоты
из Вос-азида и аминокислот в водно-диоксановом растворе при
строго контролируемых на pH-стате условиях pH среды [249].
До недавнего времени это был наиболее распространенный
способ получения Вос-аминокислот. В настоящее время по-
явились более современные методы введения Вос-группы, но
метод Шнабеля до сих пор распространен достаточно широко,
так как позволяет получать Вос-аминокислоты высокой
чистоты с отличным выходом. В табл. 19 представлены условия
синтеза Вос-аминокислот по Шнабелю.
Методика 118. /пре/и-Бутилоксикарбонил-Ь-изолей-
цин [2491
6,56г(50ммоль)£-изолейцина суспендируют в смеси 10 мл
диоксана и 10 мл воды, затем вносят на автотитраторе 7,8 г
Таблица 19. Условия синтеза Вос-аминокислот при помощи
Вос-азида
Аминокислота* Методика 117 Методика 121 Методика 122
Ж сх Время реакции, ч Выход, % Время реакции, ч Выход, % Время реакции, ч £ § со
Аланин 10,1 8 95 8 98
Аргинин 12,0 18 75 — — — —
(о-Нитроаргинин 9,8 24 93 — — 72 85
Аспарагиновая кислота 10,2 26 68 — — — —
Аспарагин 9,5 40 72 40 69 48 85
Валин 9,5 8 93 8 94 18 85
Глицин 10,0 2,5 90 2 91 4 92
Глутаминовая кислота — — — 12 87 — —
Гистидин — — — — 24 100
Глутамин 10,3 24 82 — — 48 55
Изолейцин 9,8 6,5 96 (45 °C) 18 82
Лейцин 9,75 9,0 97 — — 4 100
N®—Z-Л из ин 10,2 9 95 — —
Метионин 9,7 6 95 —- — 18 75
Пролин 8,6 0,5 96 —- 10 88
Серин 9,3 2.9 85 — 18 70
Треонин 9,5 30 88 18 75
Триптофан 9,8 9,5 84 — — 18 84
О-Бензил-тирозин 9,8 20 98 — 48 75
Тирозин 9,8 18 94 — — —
($-Бензил)цистеин 9,6 14 98 18 86
Фенилаланин 10,1 13,5 91 13 94 18 100
♦ Все аминокислоты L-ряда,
135
55 ммолей) Вос-азида и поддерживают pH 9,8 добавлением в
течение 6,5 ч 4 н. раствора едкого натра. Расходуется примерно
22 мл едкого натра. После прекращения подачи щелочи смесь
экстрагируют 50 мл эфира, водную фазу подкисляют лимонной
кислотой и продукт экстрагируют эфиром (2 X 50 мл) и этила-
цетатом (2 X 25 мл). Соединенные вытяжки промывают водой
(5x10 мл), сушат и растворитель удаляют на роторном испа-
рителе; оставшееся масло растирают с петролейным эфиром до
затвердевания продукта. Выход бесцветного Boc-L-изолейцина
11,1 г (96 %), т. пл. 66—68 °C [а]2о5 = +2,5° (с« 1; уксусная
кислота).
Методика 119. Стандартный метод выделения Na-Boc-
аминокислот [2491
После окончания реакции с Вос-азидом органический рас-
творитель (спирт, диоксан) отгоняют на роторном испарителе
при 40 °C, остаток в 1,5 раза разбавляют водой, раствор насы-
щают хлористым натрием и экстрагируют эфиром или петро-
лейным эфиром, а затем водный слой подкисляют избытком ли-
монной кислоты, небольшим избытком 1 н. серной кислоты или
раствором бисульфата натрия до pH 3—3,5 и Вос-аминокисло-
ту экстрагируют этилацетатом. Органический слой промывают
раствором хлористого натрия, сушат сульфатом магния и рас-
творитель упаривают. Остаток кристаллизуют из подходящего
растворителя.
Иногда Вос-аминокислоты выделяют в виде хорошо кристал-
лизующихся дициклогексиламмониевых солей (ДЦГА-солей).
Для использования в синтезе пептидов ДЦГА-соли необходимо
перевести в свободные Вос-аминокислоты.
Методика 120. Превращение ДЦГА-солей Вос-амино-
кислот в свободные Вос-аминокислоты
Навеску ДЦГА-соли Вос-аминокислоты суспендируют в
делительной воронке в этилацетате, используя 100 мл этила-
цетата на 20 г соли. К суспензии добавляют 1,2 эквивалента
1 н. раствора серной кислоты или 1 н. раствора бисульфата
натрия и встряхивают до тех пор, пока соль полностью не
растворится. Водный нижний слой отделяют и затем дважды
экстрагируют этилацетатом. Соединенные вытяжки промывают
водой и насыщенным раствором хлористого натрия, сушат без-
водным сульфатом магния и растворитель удаляют в вакууме.
Полученный продукт можно использовать для синтеза пеп-
тидов. Примечание. Если раствор Вос-аминокислоты
в этилацетате плохо отмыт от кислоты, то при его дальнейшем
промывании раствором хлористого натрия в осадок начинает
выпадать плохо растворимая соль хлоргидрата дициклогекси-
ламина.
136
При получении Вос-аминокислот по методу Шнабеля исполь-
зуют автотитратор. В более простых методиках pH раствора
поддерживают добавлением триэтиламина. При этом в качестве
растворителя прим няют водно-диоксановые растворы или
смесь воды с диметилформамидом. Рассмотрим две такие ме-
тодики.
Методика 121. Синтез Вос-аминокислот с триэтилами-
ном в водно-диоксановом растворе [2501.
10 ммоль аминокислоты растворяют в 15 мл воды, прибав-
ляют 4,2 мл триэтиламина и раствор 1,7 мл Вос-азида в 15 мл
диоксана. Часто при добавлении диоксана выпадают в осадок
соли. Реакционную смесь размешивают до тех пор, пока рас-
твор не станет прозрачным. Диоксан удаляют на роторном ис-
парителе, а водный раствор экстрагируют эфиром, охлаждают
и затем подкисляют охлажденным раствором соляной кислоты.
Выпавшее масло экстрагируют этилацетатом и обрабатывают,
как указано выше в методике 85. Время проведения реакции
и выход Вос-аминокислот даны в табл. 18.
Методика 122. Синтез Вос-аминокислот в смеси во-
да — ДМФА [251]
К раствору 10 ммоль аминокислоты в 5 мл воды и 4 мл
ДМФА прибавляют 1,7 мл (12 ммоль) Вос-азида и 3,1 мл
(22 ммоль) триэтиламина и размешивают от 4 до 72 ч при ком-
натной температуре. После этого к реакционной смеси добав-
ляют 25 мл воды, а избыток Вос-азида экстрагируют эфиром
(2 X 50 мл). Водный слой подкисляют до pH 2—3 раствором
бисульфата калия и Вос-аминокислоту извлекают этилацета-
том (4x20 мл). Вытяжки соединяют, промывают водой
(3 X 25 мл) и сушат сульфатом натрия. Органический рас-
творитель удаляют в вакууме и остаток кристаллизуют из под-
ходящего растворителя. В табл. 19 показан выход Вос-ами-
нокислот, синтезированных этим методом, и время проведения
реакции.
Синтез
т рет -бутилоксикарбониламинокислот
по Поздневу
В 1971 г. В. Ф. Позднее [2521 предложил для введения Вос-
группы использовать новый реагент: ди-/пре/п-бутилпирокар-
бонат [100].
СН8 СН8
сн,—d—о—с—о—с—о—А—сн,
Ан, А А Ан,
(100)
137
При взаимодействии этого соединения с аминокислотами
получаются Вос-аминокислоты. В серии работ [244—246,
254] Позднев с соавт. разработали бесфосгенный метод синтеза
ди-тре/п-бутилпирокарбоната и нашли условия получения
практически всех Вос-аминокислот, обычно используемых
для синтеза пептидов, Вос-аминокислоты получаются при
взаимодействии дикарбоната (100) с аминокислотами в вод-
но-органических смесях при температуре 20—45 9С в тече-
ние от 0,5 до 2 ч. Исследуя применимость ди-трет-бутилпи-
рокарбоната для синтеза Вос-аминокислот, Мородер и со-
авт. [2531 назвали его «идеальным реагентом» для. синтеза
пептидов.
Методика 123. Ди-тре/п-бутилпирокарбонат [244].
Синтез этого соединения включает три стадии! получение
/npem-бутилата натрия (101), превращение его в трет- бутил-
карбонат (102) и, наконец, синтез ди-тре/и-бутилпирокар-
боната (100):
СН8 СН, СН, О
1. СН,—С—ОН + Na -> СН,—ONa CH,-(L-О—(5— ONa
Сн, <5н3 <5н,
(101) (102)
74,12 23,0 96,11 140,11
СН, О О
2, СН,—<L—О—(2—ONa + С1,С—(5—С1
id,
(Ю2)
140,11 181,8
СН, О О
СН,-С—О—С—O-cLcCl,
<!н,
263,52
Boe ONa
сн8 о о сн8 о
-> CH,-i-O—i-O—(?—О-С—СН, + С1,С—С?—ONa
<5н, ii,
(100)
218,25
Методика 124. Синтез ди-тре/трбутилпирокарбоната
[244].
1. В термостойкую коническую колбу вместимостью 1 л
вносят 34,5 г (1,5 моль) металлического натрия и 230 г. сухо-
138
го толуола. Колбу закрывают обратным воздушным холодиль-
ником, снабженным хлоркальциевой трубкой, и нагревают
до тех пор, пока натрий не расплавится. Холодильник быстро
заменяют хорошо пригнанной корковой пробкой, оборачи-
вают колбу асбестовой тканью и интенсивно встряхивают в
течение нескольких минут. При этом образуется мелкая сус-
пензия натрия. Пробку заменяют хлоркальциевой трубкой,
асбестовую ткань удаляют и колбу оставляют медленно ох-
лаждаться до комнатной температуры.
Суспензию натрия быстро переносят в трехгорлый реак-
тор вместимостью 5 л, снабженный мешалкой с затвором из
силиконового масла, термометром и обратным воздушным
холодильником, закрытым счетчиком пузырьков. В колбу
прибавляют 2,1 л сухого толуола и при размешивании быстро
вносят 570 мл (6 молей) абсолютного mpem-бутилового спир-
та. Содержимое колбы нагревают при температуре 80—85 °C,
перемешивая до тех пор, пока весь натрий не растворится.
На это требуется примерно 4 ч. Затем реакционную массу
охлаждают до комнатной температуры. В результате реакции
образуется обильный белый осадок mpem-бутилата натрия.
Содержимое реактора можно оставить на ночь при комнатной
температуре, защитив от попадания влаги из воздуха.
2. Для осуществления второй стадии реакции гместо тер-
мометра в реактор вставляют барбатер и при интенсивном пе-
ремешивании пропускают в течение 3 ч углекислый газ. Для
высушивания СО2 предварительно пропускают через склян-
ку с концентрированной серной кислотой. По мере продува-
ния углекислого газа в колбе образуется прозрачный гель
mpem-бутилоксикарбоната натрия.
3. В реактор добавляют 170 мл неперегнанного сухого
ДМФА, реакционную смесь охлаждают до 8-—10 °C, и затем из ка-
пельной воронки добавляют в течение 1,5 ч при медленном пере-
мешивании раствор 80 мл (0,72 моль) хлорангидрида трихлор-
уксусной кислоты в 80 мл сухого толуола. Из-за высокой вяз-
кости раствора следует пользоваться более эффективной ло-
пастной мешалкой. После окончания подачи хлорангидрида
температуру реакционной смеси поднимают до 20 °C и интен-
сивно размешивают еще 2 ч при этой температуре. В резуль-
тате реакпии выпадает большое количество солей. Содержи-
мое реактора охлаждают до 15—17 °C, добавляют 1,3 л воды и
интенсивно размешивают, а затем после, отстаивания и рас-
слоения жидкости нижний водный слой удаляют при помощи
водоструйного насоса через трубку с оттянутым кончиком.
Органический растворитель снова промывают водой (по 1,3 л)
до pH 7,0 и наконец — 1 л насыщенного раствора хлорида
натрия. Толуольный раствор сушат 150 г безводногосул фага
139
натрия в колбе емкостью 3 л. После фильтрования толуол
отгоняют в вакууме на роторном испарителе при 50—60 °C,
а остаток перегоняют в вакууме, собирая фракцию, кипящую
при 72—76 °C (3—4 мм рт. ст.). При перегонке температур*
в холодильнике не должна быть ниже 20—25 °C. Выход ди-
/ире/и-бути л пирокарбоната 100—ПО г (64—70 %).
Ди-трет-бутилпирокарбонат — прозрачная вязкая жид-
кость с характерным запахом; при охлаждении затвердевает
Т. пл. 23—24 °C, т. кип. 75—78 °C (3—4 мм рт. ст.), 56—57 °C
(0,1 мм рт, ст.). Ди-трет-бутилпирокарбонат устойчив при
хранении.
Примечание. 1. При добавлении хлорангидрида
Трифторуксусной кислоты в реакционную массу может прои-
зойти закупорка трубки, погруженной в гель трет-бутил-
карбоната натрия. Чтобы этого не произошло, на кончик
капельной воронки надевают плотный тефлоновый шланг,
который при размешивании содержимого колбы будет вибри-
ровать, и это предохраняет его от закупоривания. 2. При пе-
регонке ди-трет-бутилпирокарбоната его не следует перегре-
вать, т. е. поднимать температуру выше 85 °C, так как может
произойти разложение продукта.
М е т о д и к ц 125. Общий способ получения Вос-амино-
кислот с использованием ди-трет-бутилпирокарбоната [254].
К раствору 10 ммоль аминокислоты в 10 мл 1 н. раствора
едкого натра добавляют раствор 0,5 г NaHCOs в 5 мл воды,
10 мл изопропилового спирта и 3 мл ди-трет-бутилпирокар-
боната. Смесь размешивают 2 ч при комнатной температуре,
а затем разбавляют водой до 50 мл и избыток реагента экстра-
гируют петролейным эфиром (2 X 20 мл). Водный раствор
подкисляют лимонной кислотой и экстрагируют этилацета-
том (2x15 мл). Соединенные вытяжки промывают насыщен-
ным раствором хлористого натрия, сушат сульфатом магния
и этилацетат удаляют на роторном испарителе. Остаток кри-
сталлизуют из подходящего растворителя или выделяют в ви-
де дициклогексиламмонийной соли.
Методика 126. (З-Бензиловый эфир mpem-бутилокси-
карбониласпарагиновой кислдты [246].
Раствор 2,23 г. (10 ммолей) Р-бензилового эфира А-аспа-
рагиновой кислоты в смеси 30 мл диоксан — вода (2:1) и 10 мл
1 н. NaOH размешивают и охлаждают до 0 °C, а затем при-
бавляют 2,4 г (11 ммоль) ди-трет-бутилпирокарбоната и
реакционную смесь размешивают 30 мин при комнатной тем-
пературе. Диоксан упаривают в вакууме, а водный раствор
охлаждают и прибавляют этилацетат, а затем подкисляют
1 н. раствором бисульфата калия до pH 2—3. Водную фазу
отделяют и промывают этилацетатом. Соединенные вытяжки
140
сушат сульфатом магния, этилацетат отгоняют на роторном
испарителе, а оставшийся продукт кристаллизуют из смеси
^тилацетат — петролейный эфир. Выход 8 г (83 %), т. пл.
100—101 9С.
Получить по этой методике у-бензиловый эфир Вос-глута-
миновой кислоты с приемлемым выходом не удается, однако
он хорошо получается в неводной среде [253].
Методика 127. у-Бензиловый эфир /преАп-бутил-
оксикарбонил-Ь-глутаминовой кислоты [253].
10 ммоль у-бензилового эфира L-глутаминовой кислоты и
10 ммоль триэтиламина или тетраметил гуанидина в 25 мл
ДМФА перемешивали с 2,4 г (11 ммоль)ди-тре/и-бутилпиро-
карбоната 30 мин при комнатной температуре. После удаления
ДМФА в вакууме остаток обрабатывали, как в предыдущей
методике. Выход 86 %, т. пл. 138—138,5 °C; (в виде дици-
клогексиламмонийной соли).
Таблица 20. Условия синтеза Вос-аминокислот при помощи
ди-тре/п-бутилпирокарбоната [253, 254, 246]
Воо-аминокислота Температура реакции, °C Растворитель Время реакции, ч Выход, %
L-Аланин 20 Изопропиловый спирт — вода (2 : 1) 2 84
£-Аргинин 20 То же ъ 1 96
£-Нитроаргинин 40 ДМФА — вода 2 85
P-Бензиловый эфир L- аспарагиновой кислоты 20 Диоксан — вода (2: D 0,5 83
£-Аспарагин 45—50 ДМФА — вода 3 77,5
Д-Валин 20 Диоксан — вода (2 : 1) 0,5 88
Глицин 20 Изопропиловый спирт — вода /о . п 2 95
у-Вензиловый эфир £• Глутаминовой кислоты 20 w * Диоксан — вода (2:1) 0,5 86
Ы*от-Вос-£-гистидин 20 Изопропиловый спирт — вода /о . 1\ 2 95
£-Лейцин 20 Vx . То же 2 96
Д-Пролин 20 » 2 95
4-Серин — ДЦГА-соль 40 » 1 85
£-Треонин — ДЦГА-соль 40 1 95
^-Триптофан 20 2 94
£-Тирозин ДЦГА-соль 20 2 84
L-Фенилаланин 20 » 2 86
8-Вос-£-цистеин 20 » 2 72
141
Таблица 21. Физико-химические характеристики Вос-аминокислот
Вос-аминокислота Т. пл., °C 25 а]р растворителя Растворитель для кристал* лизании
£-Аланин 83—84 —22,4 (с = 1; ук- сусная кислота) Эфир — петролей- ный эфир
£-Алапин • ДЦГА-соль 144—145 — —
£-Аргинин 181—182 —5,6 (с= 1; ук- сусная кислота) ТГФ
со-Нитро-£-аргинин 123—125 —5,9 (с = 2; ДМФА) —3,6 (с = 4; ДМФА) Этил ацетат
()-Тозил-£-аргинин 96—97
£-Аспарагин 174—176 —8,5 (с= 1 , ДМФА) Вода
£-Аспарагиновая кислота 114—116 —2,9 (с= 1; ДМФА) Этилаце- тат-петр о* лейный эфир
6-Бензиловый эфир £-ас- парагиновой кислоты 99—100 +8,2 (с = 2; ук- сусная кислота) То же
у-Бензиловый эфир £-ас- парагиновой кислоты • . ДЦГА-соль 130 + 17,1 (с= 1; - метанол)
£-Валин • ДЦГА-соль 140,5—141 0 (метанол) »
£-Валин 77—79 —6,3 (с ~ 1; ук- сусная кислота)
£-Глутаминовая кислота 110—112 —15,2 (с = 1; ук- сусная кислота)
у-Бензиловый эфир £- глутаминовой кислоты 65—66 — 13,4 (с = 1; . СНС13)
у-Бензиловый эфир £- глутаминовой кислоты • • ДЦГА-соль 138—139 + 11,9 (с= 1; МеОН)
L-Глутамин 116—118 —2,9 (с = 1; спирт) Этилацетат
Глицин 94—95 Этилаце- тат-*петро- лейный эфир
£-Гистидин 191—191,5 — 10,6 (с= 1; ДМФА)
М1>"-Бензил-£-гистидин 189—190 +23,2 (с = 1; МеОН)
+т-трет-Бутилоксикар- 157—159 + 17,6 (с= 2;
бонил-£-гистидин • ДЦГА- соль СНС13)
£-Изолейцин 66—68 +2,5 (с =. 1; ук- сусная кислота) Петролей- ный эфир
£-Изолейцин • ДЦГА-соль 127—128 +6 (с= 1,5; ДМФА)
£-Лейцин 86—87 —24 (с — 1; ук- сусная кислота) То же
142
Продолжение табл. 21
Вос- аминокислота Т. пл„ °C 26 [а]р растворителя Растворитель для кристал- лизации
£-Лизин 202—203 —10,7 (с = 0,85; уксусная кислота)
е-Карбобензокси-£-ли- зин • ДЦГА-соль 110—111 —9,3 (с= 1; ук- сусная кислота) Этилацетат
£-Метионин 47—49 —20 (с = 1,3; МеОН)
£-Метионин • ДЦГА-соль 134—135 + 16 (с== 1; ДМФА)
£-Фенилаланин 86—88 —4 (с = 1; ук- сусная кислота) Этилаце- тат-петро- л ейный эфир Этилаце- тат — пе- тролейный эфир
£-Серин 75—78 —4,6 (с — 1; ук- сусная кислота)
£-Серин ДЦГА-соль 142—144 + 13,3 (с — 3; МеОН)
О-Бензил-£-серин 60—62 +20,4 (с = 2; 80 % -ный спирт)
О-Бензил-£-се- рин • ДЦГА-соль 135,5—136 +24,3 (с= 2,5; МеОН)
£-Треонин 79—81 —9,2 (с = 0,2; уксусная кисло- Петрол ей- ный эфир
£-Треонин • ДЦГА-соль 154—155 та) + 11,4 (с= 1; МеОН)
О-Бензил-£-треонин 115—116 + 15,8 (с= 1,1; МеОН) —21,5 (с = 1; уксусная кисло- та)
£-Триптофан 135—137 Этилаце- тат — пе - тролейный эфир
£-Тирозин 136—138 +3,9 (с = 2; ук- сусная кислота)
£-О-Бензил-£-тирозин 107—109 +25 (с = 2; спирт) То же
£-Тирозин • ДЦГА-соль 205—208 +3,8 (с= 1; ук- сусная кислота)
О-трет-Бутиловый эфир 134—136 +24,4 (с =* 0,5;
£-тирозина • ДЦГА-соль >
£-Фенилаланин • ДЦГА- соль 210—212 +29,2
§-Бензил-£-цистеин 63—65 —41 (с=« 1, МеОН) Эфир — пе" тролейный эфир
8-Бензигидрил-£-цистеин 96—97 — 14,8 (с^ 2, МеОН)
143
Методика 128. /пре/п-Бутилоксикарбонил-со-нитро-
L-аргинин [2461
К раствору 1,5 г К2СО3 в 10 мл воды прибавляют 2,2 г
(10 ммоль) со-нитро-£-аргинина, затем добавляют 10 мл ДМФА
и 2,7 г ди-тре/и-бутилпирокарбоната и реакционную смесь
размешивают 2 ч при комнатной температуре. После оконча-
ния реакции избыток ди-/прет-бутилпирокарбоната экстра-
гируют эфиром, а водную фазу подкисляют лимонной кисло-
той до pH 3,0; продукт экстрагируют этилацетатом (один раз
30 мл и дважды по 15 мл). Органическую фазу промывают на-
сыщенным раствором хлористого натрия, сушат сульфатом
магния и этилацетат упаривают на роторном испарителе.
Остаток растирают с эфиром и образовавшийся аморфный
продукт отфильтровывают и сушат. Выход 2,7 г (85 %), т. пл.
119—120 °C; [а]д = 6,0° (с = 2; ДМФА).
Методика 129. N-Boc-S-ацетамидометил- L-цистеин
[246].
К раствору 2,28 г (10 ммоль) хлоргидрата S-ацетамидо-
метил-Ь-цистеина в 20 мл водного диоксана (1:1) и 15 мл 1 н.
NaOH добавили 2,4 г (11 ммоль) ди-трет-бутилпирокар-
боната и перемешивали 20 мин. После обработки по стандарт-
ной методике получили 2,74 г (94 %) кристаллического N-
Boc-S-ацетамидометилцистеина. После перекристаллизации
из смеси этил-ацетат-эфир его т. пл. ПО—112 °C, [а]д =»
= — 31,7°, (с = 0,3; Н2О).
Условия синтеза и выход Вос-аминокислот при использо-
вании ди-тр<?т-бутилпирокарбоната указаны в табл. 20.
Большую работу по оптимизации синтеза Вос-аминокислот
провели Позднев с соавт. [245]. Исследуя влияние раствори-
телей, оснований и температуры реакции, авторы нашли ус-
ловия получения Вос-аминокислот с очень высокими выходами.
В табл. 21 представлены характеристики /пре/п-бутилокси-
карбониламинокислот, которые были синтезированы различ-
ными методами.
Отщепление трет-бутилоксикарбонильной группы
Вос-группа легко отщепляется в кислой среде с образова-
нием изобутилена и углекислого газа. Чаще всего для дебло-
кирования Вос-пептидов или Вос-аминокислот используют
1—4 н. раствор НС1 в диоксане; при плохой растворимости
пептидов вместо диоксана применяют уксусную кислоту или
метанол. Вос-группа легко удаляется раствором бромистого
водорода в уксусной кислоте, а также безводным фтористым
водородом. Эта защитная группа отщепляется также обработ-
кой соответствующего производного пептида при помощи три-
144
фторуксусной кислоты в течение 30 мин. Для предотвращения
побочной реакции — алкилирования остатка триптофана —
в условиях" удаления Вос-группы применяют добавки 2 %
меркаптоэтанола или дитиотреитола. Рекомендуется отщеп-
лять Вос-группу у триптофансодержащих пептидов 98 %-ной
муравьиной кислотой в течение 3,5 ч [255].
В условиях удаления Вос-группы при комнатной темпе-
ратуре расщепляются mpem-бутиловые сложные эфиры, а так-
же некоторые кислотолабильные N-защитные группы, на-
пример такие, как о-нитрофенилсульфенильная, тритильная
и др. В этих условиях карбобензокси-группа не удаляется;
это позволяет селективно отщеплять Вос-группу при наличии
Z-группы.
Для селективного удаления Вос-группы в присутствии
mpem-бутиловых и n-метоксибензиловых эфиров было пред-
ложено обрабатывать Вос-производные и-толуолсульфокис-
лотой [256].
Методика 130. Отщепление Вос-группы п-толуол-
сульфокислотой [256].
К охлажденному до — 10...0 °C раствору Вос-пептида
(10 ммоль) в 50 мл эфира добавляют раствор 10 ммолей и-то-
луолсульфокислоты в 10 мл спирта в течение 30 мин, а затем
выдерживают 3 ч при комнатной температуре. Спустя это вре-
мя растворитель удаляют в вакууме, а остаток кристаллизуют
из этилацетата.
Для пептидов время деблокирования иногда приходится
увеличивать до 18—24 ч.
Методика 131. Отщепление Вос-группы муравьиной
кислотой [257].
В 50 мл 98—100 %-й муравьиной кислоты растворяют
5,77 г (11 ммоль) этилового эфира трет- бутилоксикарбонил-
нитро-аргинилтриптофанил-глицина и оставляют на 3,5 ч при
комнатной температуре. Муравьиную кислоту затем удаляют
в вакууме при 40—45 QC, а остаток растворяют в воде, рас-
твор промывают эфиром и водную фазу лиофилизируют. Вы-
ход продукта 4,76 г (91 %), т. пл. 107—111 ’С, [а]д = + 13 ±
± 0,5° (с — 2; метанол).
о-Нитрофенилсульфенильная группа (Nps)
о-Нитросульфенилхлорид
.Синтез о-нитросульфенилхлорида осуществляется в две
стадии: сначала получают 2,2'-динитродифенилдисульфид
(103), а затем его превращают в о-нитросульфенилхлорид
Ю 2-882
145
(104).
2 Ci + NasSa -> S—S—+ 2NaCl
4NOa ^NOj No/
(ЮЗ)
S—S—* 2 S—Cl
XNOa NO/ XNOa
(Ю4)
Методика 132. Синтез 2,2-дититродифенилсульфи-
да [258].
В круглодонной колбе на 250 мл, снабженной обратным хо-
лодильником, растворяют при нагревании на водяной бане
7,8 г (0,1 моль) сульфита натрия в 150 мл спирта; к полученно-
му раствору прибавляют 5,02 г (0,5 моль) тонкоизмельченной
серы и кипятят в течение 30—60 мин до полного растворения
серы.
Горячий раствор дисульфида натрия прибавляют неболь-
шими порциями к раствору 31,4 г (0,2 моль) о-нитро хлорбензо-
ла в 30 мл спирта, находящемуся в колбе на 350 мл, которая
снабжена обратным холодильником. Жидкость кипятят на во-
дяной бане в течение 3 ч; при этом выпадает в осадок кристал-
лический 2,2-динитродифенилдисульфид. Реакционную массу
охлаждают до 50—60 °C, осадок отфильтровывают и промывают
50 мл бензола, 20 мл спирта, 10 мл холодной воды, 20 мл спирта
и сушат при температуре, не превышающей 80 °C. Выход 21,6—
22,3 г (70—75 %), т. пл. 174—180 °C.
Синтез о-нитросульфенилхлорида хлорированием 2-2-дини-
тродифенилдисульфида описан в работе 1259]. В препаратив-
ном отношении более удобной, однако, является методика, ко-
торая была предложена Р. Э. Вегнером с соавт. [260], позволя-
ющая получать чистый о-нитросульфенилхлорид с более высо-
ким выходом.
Методика 133. о-Нитрофенилсульфенилхлорид [260].
Втрехгорлую колбу, снабженную обратным холодильни-
ком, счетчиком пузырьков, механической мешалкой и термомет-
ром, загружают 154,2 г (0,5 моль) 2,2-динитродифенилдисуль-
фида, 225 мл хлористого метилена и 47 мл (78 г; 0,575 моль) све-
жеперегнаиного хлористого сульфурила. Реакционную смесь
перемешивают 10 мин, затем повышают температуру до 40 СС
и выдерживают при слабом кипении растворителя до полного
растворения дисульфида, на что требуется примерно 2,5 ч. Кол-
бу охлаждают до 20 °C, содержимое переносят в коническую
колбу на 0,5 л и оставляют на ночь в холодильнике. Выпавший
кристаллический осадок о-нитросульфенилхлорида отфильтро-
146
вывают, маточник помеща-
ют в колбу с нисходящим
холодильником, отгоняют
100 мл растворителя, а
оставшуюся массу выдер-
живают в холодильнике 2 ч.
Выпавший осадок отфильт-
ровывают и соединяют с
первым продуктом, сушат в
вакуум-сушильном шкафу
при 40—50 °C в течение
30 мин в вакууме водо-
струйного насоса. Выход
159 г(84 %), т. пл. 74,5—
75 °C. После повторного
упаривания маточника до-
полнительно получают еще
25 г (13 %) продукта с
т. пл. 70—72 "С.
Методи ка 134. Об-
щий мелэд синтеза о-нитро-
фенилсульфениламинокис-
лот [261, 2621.
а) К раствору 0,02 моль
аминокислоты в смеси 10 мл
2 н. едкого натра и 25 мл
диоксана при энергичном
перемешивании добавляют
в течение 15 мин 0,022 моль
о-нитросульфенилхлорида
десятью равными порция-
ми, одновременно прибав-
ляя по каплям 12 мл 2 н.
едкого натра. Затем рас-
твор разбавляют 200 мл во-
Таблица 22. Условия синтеза
и выход NpS-аминокислот [261, 262]
Аминокислота Метод синтеза Выход, %
L-Аланин а 83
L-Аланин • ДЦГА-соль б 83
Р-Бензил-£-аспарагино- В 70
вая кислота • ДЦГА- соль
L-Аспарагин а 78
со-Карбобензокси-£-ар- а 90
гинин
£-Валин а 65
L-Валин • ДЦГА-соль б 64
Глицин • ДЦГА-соль б 60
L-Глутамин а 87
у-Бензил-£-глутамино- в 70
вая кислота * ДЦГА-
соль
L-Изолейцин а 70
L-Изолейцин • ДЦГА- б 85
соль
L-Лейцин • ДЦГА-соль б 90
N8' Карбобензокси-£- а 74
лизин-ДЦГА-соль
N8 -Карбобензокси-А-л и- б 70
зин • ДЦГА-соль
L-Метионин • ДЦГА- б 54
соль
L-Пролин • ДЦГА-соль б 45
£-Серин • ДЦГА-соль в 61
L-Треонин а 60
L-Треонин • ДЦГА-соль б 60
L-Тирозин • ДЦГА-соль б 62
L-Фенил аланин а 90
8-Бензил-£-цистеин • б 73
ДЦГА-соль
ды, отфильтровывают от небольшого количества образовавше-
гося 2,2'-динитродифенилдисульфида и подкисляют 1 н. серной
кислотой до pH 2—3. Выделившееся масло кристаллизуется при
растирании и охлаждении. Продукт отфильтровывают, промы-
вают водой, сушат и кристаллизуют из смеси этилацетат —
эфир или этилацетат — петролейный эфир.
б) Аминокислоту обрабатывают, как описано в пункте а).
После подкисления серной кислотой Nps-производное амино-
кислоты экстрагируют этилацетатом, эфиром или смесью этих
растворителей (1 ; 1). Органическую фазу промывают водой до
нейтральной реакции и сушат сульфатом магния. После добав-
10*
147
Рис. 3. Значение pH реакцион-
ной среды, которое поддержива-
ется в процессе ацилирования
аминокислот постепенным добав-
лением о-нитросульфенилхло-
рида
ления дициклогексиламина соот-
ветствующая ДЦГА-соль выпа-
дает в большинстве случаев в ви-
де игл.
в) В смеси 10 мл воды и 25 мл
диоксана растворяют (или сус-
пендируют) 20 ммоль аминокис-
лоты. При размешивании в тече-
ние 15 мин прибавляют десятью
равными порциями 22 моль о-нит-
росульфенилхлорида и одновре-
менно прибавляют по каплям
12 мл 2 н. раствора едкого натра.
Образовавшийся раствор подкис-
ляют разбавленной серной кис-
лотой и дальнейшую обработку
ведут, как описано в пункте б).
Условия синтеза и выход Nps-аминокислот, полученных
указанными способами приведен в табл. 21.
В работе Р. Э. Вегнера и соавт. [2601 показано, что .Nps-
аминокислоты можно получать с выходом 90—95 %, если аци-
лирование вести при pH 11,5—12 в начале реакции и затем
по мере добавления о-нитросульфенилхлорида постепенно сни-
жать значение pH среды до 8,0.
На рис. 3 изображен график, показывающий, какие значе-
ния pH необходимо поддерживать в реакционной среде в зави-
симости от количества использованного во время реакции
о-нитросульфенилхлорида. После добавления определенного
количества реагента pH реакции поддерживают в пределах
вацпрлхозанной части графика [260].
Мето д-и к а 135. о-Нитрофенилсульфенил-^-лейцин
1260]
К суспензии 13,1 г (0,1 моль) L-лейцина в 100 мл диоксана
добавляют раствор 2 н. едкого натра до pH 11,5 (около 50 мл).
К реакционной смеси при перемешивании добавляют со ско-
ростью примерно 2 мл в 1 мин раствор 20,9 г (0,11 моль) о-нит-
росульфенилхлорида в 40 мл диоксана и одновременно — рас-
твор 2 н. едкого натра так, чтобы pH среды соответствовало
меньшим значениям заштрихованной области графика, приве-
денного на рис. 3. После того, как весь о-нитросульфенилхло-
рид будет добавлен, прибавляют 1 л воды и содержимое колбы
фильтруют, а фильтрат подкисляют 1 М раствором серной кис-
лоты до pH 3,0. Выпавшее масло кристаллизуется после вы-
держивания 1—3 ч в холодильнике при 0—5 °C. Кристаллы
о фильтровывают, промывают водой и сушат на воздухе. Вы-
ход 25,5—26,9 г (90—95 %), т. пл. 103—107 °C.
148
Аналогично получены Nps-производные пролина с выходом
90—94 % и тирозина с выходом 94—98 %.
В табл. 23 представлены свойства о-нитрофенилсульфенил-
аминокислот.
о-Нитрсфенилсульфенильная группа легко удаляется в
условиях отщепления карбобензокси-, /пре/п-бутилоксикарбо-
нил-и mpem-амилоксикарбонильной защитных групп. Стандарт-
ный метод деблокирования Nps-производных заключается в об-
работке их двумя эквивалентами хлористого водорода в спирте
или эфире в течение 10 мин при комнатной температуре.
Другим способом отщепления Nps-группы является обра-
ботка Nps-аминокислот тиольными соединениями: тисацета-
мидом, тиогликолевой кислотой и другими тиосоединениями.
Следует иметь в виду, что при использовании Nps-группы в
качестве N-защитной группы пептидов исключается примене-
ние каталитического гидрогенолиза для деблокирования дру-
гих групп из-за отравления катализатора соединениями двух-
валентной серы. Для триптофансодержащих пептидов удале-
ние Nps-группы хлористым водородом не рекомендуется, так
как образующийся при этом о-нитросульфенилхлорид способен
алкилировать индольное кольцо триптофана.
Таблица 23. Физико-химические характеристики NpS-аминохислот
Аминокислота Т. пл., °C [а]д, град Растворитель для кристал- лизации
£-Алании 128—130 — 101,8 (с = 2; ДМФА) Спирт
£-Аланин • ДЦГА-соль 176—178 —56,5 (с = 2; МеОН)
£-Аргинин 164 —10,8 (с«= 2,5; ДМФА)
со-Нитро-£-аргинин X X ДЦГА-соль 150—151 —4,5 ДМФА)
<й-Карбобензокси-£-арги- нин 105 —-
£-Аспярагин 165—166 — 119,3 (с== 4; ДМФА) Метанол
Р-Бензил-£-аспарагиновая кислота • ДЦГА-соль 165—166 —38,0 (с=« 1; ДМФА) ДМФА
£-Валин 105 —127,8 (с - 2; ДМФА) ДМФА
Глицин 147 — ДМФА
Глицин • ДЦГА-соль 190—191 — ДМФА
£-Глутамин 165 —74,3 (с = 2; ДМФА) ДМФА
£-Глутаминовая кислота 132—133 —84,5 (с= 1; мета- нол) ДМФА
у-Бензиловый эфир £-глутаминовой кисло- 168 —34,0 (с = 3,5; хлороформ) ДМФА
149
Продолжение табл. 23
Аминокислота Т. пл., °C 95 |а]£. град Растворитель для кристал- лизации
ты • ДЦГА-соль
а-Бензиловый эфир-£- глутаминовой кисло* 154 —25,9 (с= 1; ДМФА) ДМФА
ты • ДЦГА-соль
£-Гистидин 185—188 —6,9 (с== 1; ДМФА) ДМФА
М,т-£-Гистидин 140—141 +39,9 (с= 1; ДМФА) ДМФА
£-Изолейцин 90—93 —102,2 (с== 2; ДМФА) ДМФА
£-Изолейцин • ДЦГА- соль 188—189 —53,8 (с = 2; метанол) Метанол
L-Лейцин 102—106 —99,7 (с = 2; ДМФА) в
£-Лейцин • ДЦГА-соль 182—183 —76,1 (с = 1; ДМФА) в
N8* Карбобензокси-£-ли- зин ДЦГА-соль 184—187 —29,1 (с = 0,7; ДМФА) Спирт
М8’Вос-£-лизин » ДЦГА- соль 194—195 —43,4 (с = 1,5; хло- роформ) в
£-Метионин • ДЦГА-соль £-Пролин 196—197 107—108 —34,4 (с = 0,7; метанол) Метанол
L-Пролин • ДЦГА-соль 151—154 —43,2 (с = 0,7; ДМФА) , Этилацетат
£-Серин • ДЦГА-соль 171 — 174 —89,0 (с== 1; ДМФА) —111,6 (с= 2; ДМФА) Спирт
£-Треонин 138—141 в
£-Т реон ин-ДЦГА-соль 181—182 —103,0 (с= 0,7; ДМФА) ’Спирт
£-Триптофан • ДЦГА-соль . 168—169 —20,4 (с = 2; ДМФА) ТГФ — эфир
£-Тирозин • ДЦГА-соль 173—175 +41,8 (с = 2; мета- нол) Спирт
О-Бензил-£-Тирозин 136—139 в
О-Бензил-£-тиро- зин • ДЦГА-соль 182—183 + 19,6 (с= 0,5; ДМФА) в
8-Бензил-£-цисте- ип . ДЦГА-соль 168—169 —43,5 (с = 0,7; ДМФА) в
£-Фенилаланин 134—135 —47,6 (с = 4; ДМФА) в
Деблокирование о-нитрофенилсульфенил-пептидов тиоль-
ными реагентами позволяет осуществить селективное удаление
Nps-группы, не затрагивая таких защитных групп, как Вос,
Врос и ОВи'.
В табл. 24 приведены условия отщепления Nsp-группы раз-
личными тиосоединениями.
150
Таблица 24. Условия отщепления NpS-группы различными
тиосоединениями *
Формула тиольного реагента Время реак- ции, мин Состав отщепляемой среды
zs CH8-C<f XNH, Na2S2O3 <5 <5 1,5 мл СН3ОН + 0,5 мл АсОН + + 100 мг (1,3 ммоль) реагента 1,6 мл СН8ОН + 0,2 мл АсОН + + 0,2 мл (1 моль) раствора реагента
H,N - C—NH, II S n <5 <3 1,5 мл СН8ОН + 0,5 мл АсОН + + 100 мг (1,3 ммоль) реагента ДМФА или СН2С1а+0,1 ммоля АсОН + 0,3—0,5 ммоль реагента
ri
* В реакцию вводили 0,1 ммоль Nps-аминокислоты [263, 264].
Методика 136. Удаление Nps-группы 2-меркаптопи-
ридином [264].
К раствору (или суспензии) Nps-аминокислоты или пептида
в метаноле, ДМФА или хлористом метилене (10 ммоль) прибав-
ляют 30—50 ммолей 2-меркаптопиридина и 10 ммоль ледяной
уксусной кислоты. Смесь выдерживают около 3 мин при 25 °C
и затем растворитель удаляют на роторном испарителе.
Продукт кристаллизуют из подходящего растворителя. Выход
почти количественный.
При использовании Nps-производных для твердофазного
синтеза пептидов время обработки Nps-пептидилполимера со-
ставляет 20—30 мин.
2-(/г-Дифенил)-изопропилоксикар6онильная
группа(Врос)
Эта группа предложена Заберем и Изелином в 1968г. [178,
265]. Врос-группу вводят реакцией 2-(и-дифенил)-изопропилфе-
нилкарбоната или 2-(и-дифенил)-изопропилоксикарбонилази-
да с аминокислотами или их эфирами в безводной среде:
Bpoc-N3 + H2N—СН—СООМе -> Bpoc-NH—СН-СООМв
R
Bpoc-N3 + H2N-CH—СОО~ • NR4 -> Bpoc-NH—СН-СООН
R l!
151
Методика 137. п-Ацетилдифенил [266]
О О
+ ^Н8 С С1-}“ AlClg —► —СНд
454,0 ~ 78,4 ""196,0 ""
К смеси 125 т дифенила, 40 г ацетилхлорида в 500 мл серо-
углерода при хорошем размешивании добавляют в течение 10
мин 90 г бе водного хлористого алюминия. Содержимое реак-
тора нагревают 2 ч на водянкой бане, а затем охлаждают и раз-
бавляют водой. Сероуглерод и избыток дифенила отгоняют с
водяным паром, а маслянистый остаток перегоняют с перегре-
тым водяным паром, нагревая колбу с продуктом на масляной
бане до 200 °C. n-Ацетилдифенил, полученный таким способом,
пригоден для синтеза различных препаратов. После кристал-
лизации из спирта выход 60—70 г (45—50 %), т. пл. 120,5 °C.
Методика 138. Диметил-п-бифенилкарбинол [267] "
О СН8
СН3—MgBr /’2\_/“\Д_ОН +
Д
196,0 119,2 242,0
+ MgBra
К раствору реактива Гриньяра, полученного из 48,6 р
(2 моль) магния и 190 г (2 моль) бромистого метила в 250 мл
абсолютного эфира прибавляют в течение 1 ч при размешива-,
нии 371 г (1,84 моль) n-ацетилдифенила в 1,5 л сухого бензола.*
После стояния реакционной смеси в течение ночи продукт кон-
денсации гидролизуют разбавленной серной кислотой. Орга-
нический слой отделяют, промывают водой, сушат сульфатом
магния и растворитель удаляют в вакууме. Выход продукта
330 г (85 %). После кристаллизации из смеси бензола и гекса-
на т. пл. 92—93 °C.
Методика 139. 2-(иБифенил)-изопропил-фенилкарбо-
нат [178]
сн8 о
О-О_<г~°н+С1Д-0-О
СН8
242,0 156,4
152
К охлажденному до —5 °C раствору 152 мл фенилхлоркар-
боната в 500 мл хлористого метилена добавляют при размеши-
вании смесь 212 г (1 моль) п-бифенил-диметилкарбинола, 1 л
хлористого метилена и 120 мл пиридина. При добавлении
реагентов образуется обильный осадок, который при размеши-
вании в течение ночи при 0 °C в основном растворяется. Реакци-
онную смесь при размешивании выливают на небольшое коли-
чество измельченного льда, разбавляют 1 л хлористого метиле-
на, затем органическую фазу отделяют, промывают 3 раза водой
и сушат сульфатом натрия. Хлористый метилен отгоняют в ва-
кууме. Затвердевший остаток растворяют в 1 л этилацетата
при нагревании до 60 °C, затем растворитель упаривают на ро-
торном испарителе до 600 мл и оставляют раствор кристалли-
зоваться при 0 °C. При этом получают первую фракцию кри-
сталлического продукта (около 260 г) с т. пл. 115—116 °C, а из
маточника выделяют после упаривания этилацетата до 100 мл
еще 31,5 г продукта с т. пл. 114—115 °C. Общий выход 192,5 г
(80 %).
Методика 140. 2-(п-Бифенил)-изопропилкарбонилгид-
разид [178]
332,1 50,06
270,0
Из 160 г (0,5 моль) n-бифенил-диметилкарбинола получают
карбонат, как описано в методике 139 и продукт реакции сус-
пендируют в 200 мл ДМФА, затем при небольшом охлаждении
добавляют 125 мл гидразингидрата и реакционную смесь раз-
мешивают 6 ч при комнатной температуре. После этого колбу
охлаждают льдом и разбавляют 1 л воды и оставляют на ночь в
холодильнике при 0 °C. Выпавший осадок отфильтровывают,
промывают холодным раствором 1 н. едкого натра, затем водой
до нейтральной реакции. Продукт (139 г) кристаллизуют из
смеси 200 мл четыреххлористого углерода и 40 мл петролейно-
го эфира. Выход 103 г (76 %), т. пл. 106—108 °C.
153
Методика 141. 2-(/г-Бифенил)-изопропилоксикарбонил
азид [178]
СН, О
сн3
270,0
281,0
К охлажденному до —25 °C раствору 27 г (0,1 моль) 2-(я-
бифенил)-изопропилкарбонилгидразида в 270 мл ацетонитри-
ла прибавляют холодную смесь 50 мл 6 н. соляйой кислоты и
100 мл ацетонитрила. При непрерывном размешивании и ох-
лаждении до —15...—20 °C добавляют по каплям 22 мм (5 моль)
раствора нитрита натрия. Через 10 мин температуру раствора
понижают до —25 °C и по каплям прибавляют 2 н. раствор
NaHCO3 до pH 6—8. Этот раствор выливают при размешивании
в 1 л воды, охлаждаемой смесью снега с солью; выпавший оса-,
док отфильтровывают, промывают водой, а фильтрат экстра-
гируют эфиром. Органическую фазу сушат сульфатом натрия,
эфир отгоняют на роторном испарителе при комнатной темпе-
ратуре. Кристаллический остаток азида (28,3 г) используют
для получения Врос-аминокислот без дальнейшей очистки.
Обычно содержание азида в продукте составляет 84,5 %. Вы-
ход составляет 85 % теоретического.
Методика 142. Синтез Врос-аминокислот [178, 265].
а) К раствору 2,85 г (17,2 моль) хлоргидрата метилового
эфира L-пролина в 9,8 мл ДМФА прибавляют 2,4 мл
(17,2 ммоль) триэтиламина, размешивают и через 2 мин добав-
ляют 6,3 г (18,8 ммоль) 84 %-го 2-(л-бифенил)-изопропилокси-
карбонил азида в 4,9 мл ДМФА. Через 4 мин прибавляют
2,4 мл триэтиламина, а спустя 30 мин — еще 1,2 мл третичного
основания и реакционную массу размешивают в течение ночи
при комнатной температуре. В результате обычно получают
7,5 г частично затвердевшего масла; его растворяют в 60 мл изо-
пропилового спирта, добавляют 11 мл 1,9н. раствора едкого нат-
ра и размешивают 2 ч при 40 °C, а затем растворитель упарива-
ют в вакууме. К остатку добавляют 100 мл воды, раствор экст-
рагируют эфиром, а водную фазу подкисляют при 0 °C лимон-
ной кислотой (1 н.) до pH 3,0, Врос-пролин экстрагируют
эфиром, вытяжки промывают водой до pH 7,0, сушат сульфатом
магния. К прозрачному раствору Врос-пролина в эфире при-
154
Таблица 25, Физико-химические характеристики Врос-аминокислот
Аминокислота Метод синтеза Выход, % T. пл., вС ГпИ2б [a]D (с « 1; МеОН) Система для кристаллиза- ции
£-Аланин В 71 139—140 —3,0 Этилацетат — петролейный эфир Эфир
£-Аспарагин в 43 176—178 —8,0
£-Валин • ДЦГА- а 49 178—180 —8,0 Изопропанол
соль
£-Валин • ДЦГА- б 68 178—180 -8,0 »
соль Глицин в 72 142—143 Этилацетат
Глицин • ДЦГА-соль а 73 192—193 — »
Глицин • ДЦГА-соль в 65 192—193 — »
£-Глутамин • ДЦГА- в 56 107—110 +7,0 Ацетон — пе-
соль тролейный эфир
у-/иретп-Бутиловый эфир L-глутаминовой в 64 174—175 + 15,0 Спирт
кислоты • ДЦГА-соль
£-Изолейцин в 68 225—240 —2,0 МеОН — вода
£-Лейцин а 61 227—230 — 12,0 Эфир
М8-Вос-£-Ли- а 70 Аморфный + ю,о Эфир — пет-
зин • ДЦГА-соль ролейный эфир
£-Лейцин в 70 227—230 — 12,0 Эфир
£-Метионин • ДЦГА- в 73 143—145 + 14,0 Этил ацетат —
соль петролейный эфир То же
£-Пролин • ДЦГА-соль а 80 173—175 —12,0
£-Серин • ДЦГА-соль в 72 152—152,5 +5,0 Спирт
О-трет-Бутил-£-се- в 69 180—181 +21,0 Этилацетат
рин • ДЦГА-соль £-Тирозин а 33 248—252 +39,0 Ацетон
£-Тирозин в 35 248—252 +39,0 »
£-Треонин • ДЦГА- в 63 168—170 + 1,0 Метанол —
соль этилацетат
О-трет-Бутил-£-трео- в 66 183—184 +7,0 Хлороформ-
нин • ЦГА-соль гексан
^Фенилала- нин • ДЦГА-соль в 59 116—119 +33,0 Этилацетат
бавляют 3,5 мл дициклогексиламина и оставляют кристалли-
воваться. Выход соли 7,35 г (80 %).
б) 1,17 г (10 ммоль) L-валина растворяют в 4,55 мл 2,2 н.
раствора бензилтриметиламмоний гидроксида («Тритона В») в
метаноле и органический растворитель упаривают в вакууме.
Для удаления следов воды остаток растворяют в 10 мл ДМФА и
ватем растворитель отгоняют на роторном испарителе; эту опе-
рацию повторяют дважды. Остаток растворяют в 5 мл ДМФА
155
и смешивают при 40 °C с 3,35 г (10 ммоль) 2-(п-бифенил)-изо-
пропилоксикарбонилазида и 2,75 мл (20 ммоль) триэтиламина,
а затем реакционную массу размешивают 1 ч при 40 °C. После
этого раствор разбавляют водой и экстрагируют эфиром. Вод-
ную фазу охлаждают до 0 °Си подкисляют 1 н. лимонной кис-
лотой до pH 2—3, а продукт экстрагируют эфиром. Органиче-
скую фазу промывают водой до нейтральной реакции и сушат
сульфатом магния. Прозрачный эфирный раствор смешивают
с 1,2 мл дициклогексиламина; при Этом сразу начинает кри-
сталлизоваться продукт реакции. Эфир немного упаривают в
вакууме, добавчяют равное количество петролейного эфира и
охлаждают до 0 °C. Выход ДЦ1 А-соли Bpoc-L-валина состав-
ляет 3,08 г (68 %).
в) В 93 мл 2,15 н. раствора «Тритона В» в метаноле раство-
ряют при 50 °C 26,2 г (0,2 мель) £-лейцина и метанол отгоняют
на роторном испарителе. Для удаления воды остаток раство-
ряют в 60 мл ДМФА и растворитель отгоняют в вакууме, остаток
снова растворяют в 60 мл ДМФА и его опять отгоняют на ротор-
ном испарителе. Наконец остаток растворяют в 80 мл ДМФА,
смешивают при 50 °C с 66,6 г (0,2 моль) 2>(п-бифенил)'изопро-
пилфенилкарбоната и реакционную массу размешивают
3 ч при 50 Г.
Для выделения продукта к смеси добавляют воду и эфир,
водный слой отделяют и экстрагируют эфиром, а затем подкис-
ляют при 0 °C 1 н. лимонной кислотой до pH 2—3 и продукт
экстрагируют эфирсм. Органическую фазу промывают водой
до pH 7,0, сушат сульфатом магния и эфир удаляют на ротор-
ном испарителе при 30 °C. Остаток растирают со смесью 30 мл
эфира и 100 мл петролейного эфира, кристаллический Bpoc-L-
лейцин отфильтровывают, сушат. Выход 52,1 г (70 %).
Физико-химические характеристики Врос-аминокислот
представлены в табл. 25.
Методика 143. Удаление Врос-группы [265].
К 7 мл смеси уксусная кислота — 83 %-й муравьиная кис-
лота—вода (7 : 1 : 2) прибавляют 739 мг (2 ммоль) Врос-£-лей-
цина и размешивают в течение 1,5 ч при 20 °C. Затем добавля-
ют 40 мл ацетона и 1 мл пиридина и выдерживают некоторое
время при 0 °C. Выпавший лейцин отфильтровывают. Выход
246 мг (94 %).
Трифенилметильная (тритильная) группа
Тритильная группа (Trt) нашла применение в химии пепти-
дов [9, 101 ввиду возможности ее селективного ацидолиза в
очень мягких условиях в присутствии Вос- и карбобензокси-
групп. Однако низкий выход тритиламинокислот и неудовлет-
ворительное ацилирование ими амиПокомпонента (по сравне-
156
нию с кислотолабильными Nps- и Врос-группами) привели к
тому, что в последние годы тритильная группа используется
редко.
Недавно были разработаны эффективные методы получения
тритиламинокислот [2681, а также получены устойчивые 1-ок-
сибензтриазоловые эфиры, позволяющие синтезировать дипеп-
тиды с выходом около 90 % [269].
Показано, что в течение 2—4 дней в растворах ДМФА или
ТГФ эти эфиры образуют равновесную смесь форм I и II [269]:
/\/N\ /\/NW
f | N II C—CHR—NH—Trt
^/\N/ ^/\N/ II
1 I
О—C—CHR—NH—Trt 0“
I II II
О
Однако ввиду того, что реакция образования пептидной связи
происходит значительно быстрее, чем эта перегруппировка,
фактически используются активированные эфиры в форме I.
По-видимому, эти последние достижения должны способство-
вать более широкому применению тритильной группы.
Методика 144. Общий метод синтеза тритиламинокис-
лот [268]
О
HSN [ —CHR—СОО“ Ме si— HN—CHR—С—О—SiMe
NEt3
О
-> Trt—NH—CHR—£—О—SiMe3 ---е-Н^ Trt—NH—CHR—СООН
К суспензии 109 ммоль аминокислоты в 1 л сухого хлоро-
форма добавляют 31,7 мл (250 ммоль) триметилхлорсилана и
35 мл (250 ммоль) триэтиламина (для Ala, Asn, GPy, Zle, Zeu,
Met, Phe, Pro, Vai) или 44,5 (350 ммоль) триметилхлорсилана
и 49 мл (350 ммоль) триэтиламина (для CyS, His, Zys, Orn, Asp,
Glu, Ser, Thr, Туг) и кипятят в течение 10 мин, защищая от
влаги. Охлаждают до комнатной температуры, вносят по кап-
лям раствор 27Д Г (ЮО ммоль) и 250 мл хлороформа (или 55,6 г
и (200 ммоль/ тритилхлорида, в случае Cys, His, Lys, Orn, кото-
рые получают в виде дитритильных производных, в 500 мл хло-
роформа) и кипятят реакционную смесь 2 ч. Охлаждают раствор,
разбавляют 50 мл метанола. Растворитель удаляют в вакууме,
остаток экстрагируют эфиром или этилацетатом и промывают
органическую фазу 10 %,-й.лимонной кислотой, затем экстра-
гируют раствором 0,5, g NaOH и промывают водную фазу
дважды эфиром. Пдсле"подкисления водной фазы ледяной
уксусной кислотой (pH 6—7) выделившееся масло экстраги-
руют этилацетатом, промывают органическую фазу 10 %-й
157
Таблица 26. Характеристики тритиламинокислот [268]
Аминокислота Вы- ход, % Температу- ра плавле- ния, °C [а]д, град
Аланин 1 86 157 —19 (с = 5; метанол) -
Аспарагин 72 174—175 — 6(с = 3; метанол)
Аспарагиновая кислота 2 80 163—164 —16,6 (с= 2; метанол)
Валин 1 90 160 +6,7 (с = 5; метанол)
Глутаминовая кислота 72 141 — 143 +4,6 (с = 2; метанол)
Глицин 82 179—180 —
Гистидин-(Ы1т-тритил) 84 179—199 +3,7 (с = 5; пиридин)
Гистидин 46 198—200 +23,7 (с= 3,3; пиридин)
Лейцин 1 95 154 +3 (с = 5; метанол)
Изолейцин 1 93 151—152 + 13 (с= 5; метанол)
Лизин-(№-тритил) 77 158—159 + 14,5 (с — 2; метанол)
Метионин х 87 152—153 +21,6 (с = 5; метанол)
Орнитин-(К^-тритил) 1 75 154—155 + 16,7 (с= 2; метанол)
Пролин 1 79 162—163 —60 (с= 5; хлороформ)
Серин 1 88 137—138 —29 (с = 1; метанол)
Треонин 81 165 —6,7 (с — 2; метанол)
Тирозин 3 89 151 — 153 + 14,5 (с= 2; метанол)
Фенилаланин 1 87 150 + 12 (с = 5; метанол)
Примечания: 1) соль с молекулой диэтиламина; 2) соль с двумя молекула-
ми диэтиламина; 3) триэтиламмонийная соль.
лимонной кислотой, водой, сушат над сульфатом натрия и уда-
ляют растворитель в вакууме. Тритиламинокислоты выделяют в
виде свободных кислот или диэтиламмонийных солей (табл. 26).
Диэтиламмониевые соли тритиламинокислот получают пу-
тем добавления диэтиламина к раствору тритиламинокислоты
в эфире или ацетоне с последующей кристаллизацией из горя-
чего ацетона. Для превращения их в свободные кислоты их
растворяют в строго эквимолярном количестве 0,2 н. раствора
NaOH, выдерживают в течение нескольких минут в вакууме
(для удаления большей части выделившегося диэтиламина) и
затгм подкисляют уксусной кислотой. Образовавшийся при ох-
лаждении осадок отфильтровывают, промывают водой и сушат
в вакууме над Р2О5.
Методи ка 145. N-Тритилгистидин [268].
К суспензии 7,8 г (50 ммоль) гистидина в 500 мл сухого хло-
роформа добавляют 20,2 мл (160 ммоль) триметилхлорсилава
и 30,7 мл (200 ммоль) триэтиламина и кипятят 2 ч с обратным
холодильником, защищая от влаги. Затем смесь охлаждают до
комнатной температуры, вносят по каплям раствор 13,9
(50 ммоль) тритилхлорида в 150 мл хлороформа и кипятят еще
2 ч. Охлажденную смесь промывают водой, сушат над сульфа-
том натрия и удаляют растворитель в вакууме. К маслообраз-
158
Таблица 27, Свойства L-оксибензтриазиловых эфиров
N-тритиламинокислот [269]
N-Тритил ами- нокислота Вы- ход, % Растворитель для кристал- лизации Т. пл., °C [а]д, град (с == 2, СНС13)
Trt-Аланин 98 Бензол-петролейный 145—147 + 52,7
Trt-Глицин 92 эфир Ацетон 137—140
Trt-Валин 95 —- Пена +9,3
Trt-Изолейцин 93 Петролейный эфир 87—90 +3,4
Trt-Лейцин 95 Ацетонитрил 135—136 —35,0
Trt-Метионин 97 Эфир — петролейный 142—143 +52,0
Trt-Пролин 98 эфир Пена — 109,7
Trt-Тирозин 94 — Пена — 10,8
Trt-Фенил ала- нин 92 — Пена —26,7
ному остатку добавляют 500 мл 95 %-го этанола, быстро на-
гревают до кипения, отфильтровывают нерастворившиеся при-
меси и охлаждают. Выпавшие иглообразные кристаллы сушат
в вакууме в течение 15 ч при 45 °C, при этом они превращают-
ся в аморфный порошок. Выход N-тритилгистидина 9,14 г
(46 %), т. пл. 198—200 °C, [alo = +23° (с = 3,3; пиридин).
Методи ка 146. Получение 1-о ксибензтриазоловых эфи-
ров тритиламинокислот [269].
Растворяют 10 ммоль N-тритиламинокислоты в 30 мл ТГФ
и охлаждают раствор на ледяной бане. Затем вносят 2,06 г
(10 ммоль) ДЦГК и 2 г (15 ммоль) L-оксибензтриазола. Реак-
ционную смесь перемешивают 30 мин при 0 °C и 2 ч при ком-
натной температуре, затем добавляют одну каплю уксусной
кислоты и несколько капель волы и перемешивают еще 15 мин,
растворитель удаляют в вакууме при комнатной температуре,
остаток переводят в эфир (50 мл) или этилацетат (в случае Leu
или Ala) и отфильтровывают дициклогексилмочевину. Раствор
промывают последовательно охлажденным раствором 5 % ли-
монной кислоты (1 X 20 мл), водой (1 х 20 мл), 1 н. NaOH
(2 X 20 мл) или 5 %-й NaHCO3 (в случае Gly и Туг) и водой
(4 X 20 мл). После сушки сульфатом натрия и отгонки раство-
рителя продукты получают в виде пены или кристаллов. Рас-
творение в небольшом объеме этилацетата и выдержка в холо-
дильнике в течение ночи приводят к хроматографически чи-
стым продуктам, которые фильтруют и после отгонки раствори-
теля сушат в вакууме до постоянной массы. Эти продукты мо-
гут быть превращены в аморфный пороше к или кристаллы с
помощью системы растворителей, указанные в табл. 27.
159
Методика 147. Общий метод синтеза эфиров N-тритил-
дипептидов [269]
Растворяют 5 ммоль 1-оксибензтриазилового эфира N-три-
тиламинокислоты в 6 мл ДМФА и охлаждают на ледяной бане.
Затем вносят 5,5 ммоль хлоргидрата или и-толуолсульфоната
алкилового эфира аминокислоты и 1,14 мл (10,5 ммоль) N-ме-
тилморфолина и перемешивают 15 мин при 0 °C и 1—2 ч при
комнатной температуре и затем разбавляют 30 мл раствора
NaCl. Экстрагируют раствор эфиром или этилацетатом, и ком-
бинированные органические слои (50 мл) промывают последо-
вательно 5 %-й лимонной кислотой (2 X 20 мл), водой (2 X
X 20 мл), 5 %-м раствором NaHCO3 (2 X 20 мл) и несколько
раз водой и раствором NaCl. После сушки сульфатом натрия,
удаления растворителя получают защищенные дипептиды с
выходом 80—89 %.
и-Толуолсульфонильная группа (TosJ
n-Толуолсульфонильная (тозильная) группа была предло“
жена в качестве защиты аминогрупп Э. Фишером еще в 1915 г.»
но долго не применялась. В последующие годы она сыгра-
большую роль при синтезе пептидов благодаря наличию неко-
торых положительных свойств: высокой стабильности в усло-
виях удаления других защитных групп, чрезвычайной доступ-
ности n-толуолсульфохлорида, используемого для введения
Tos-группы, а также в связи со способностью тозильных произ-
водных давать кристаллические продукты.
Тозиламинокислоты легко получаются при взаимодействии
n-толуолсульфохлорида с аминокислотами в щелочной среде:
СН3—/^^—SOaCl + H2N—CHR—COO”
-> СН8—SOa-NH-CHR—COO”.
В настоящее время эта защитная группа почти не исполь-
зуется для блокирования а-аминогрупп аминокислот и пепти-
дов из-за довольно жестких условий ее удаления (натрий в
жидком аммиаке) и появления большого количества новых бо-
лее лабильных защитных групп; она в основном используется
для блокирования некоторых реакционноспособных групп бо-
ковых радикалов аминокислот (см. главу 3).
Способы получения тозиламинокислот хорошо освещены в
монографии [9], поэтому ограничимся лишь одним примером
синтеза тозил-^-валина.
Методика 148. Тозил-Ь-валин [2701.
К раствору 5 г (0,04 моль) L-валина в 55 мл 1 н. едкого нат-
ра добавляют 11 г (0,06 моль) кристаллического п-толуолсуль-
160
фохлорида. Смесь интенсивно перемешивают при комнатной
температуре в течение 3 ч, затем избыток сульфохлорида от-
фильтровывают, а водную фазу экстрагируют эфиром и под-
кисляют 1 н. соляной кислотой до pH 2. Тозил-£-валин, вы-
павший в осадок, отфильтровывают, промывают водой, сушат
и кристаллизуют из смеси этилацетат — петролейный эфир.
Выход бесцветных пластинок 6,8 г (70 %), т. пл. 144 °C.
Трифторацетильная защитная группа (TFA)
Трифторацетильная группа является первой, нашедшей
практическое применение N-защитной группой, которая уда-
ляется в условиях мягкого щелочного гидролиза. Показано,
что при синтезе TFA-аминокислот прямым действием трифтор-
уксусного ангидрида на аминокислоты может наблюдаться
значительная рацемизация.
Вейганд и Гейгер показали,что ацилирование аминокислот
трифторуксусным ангидридом в безводной трифторуксусной
кислоте при низкой температуре позволяет получать соответст-
вующие TFA-производные в оптически чистом состоянии [192,
2711.
Методика 149. Трифторацетилирование аминокислот
[271].
Предварительно высушенную аминокислоту растворяют
(если необходимо, то при нагревании) в 10—15-кратном коли-
честве безводной трифторуксусной кислоты, снабдив колбу об-
ратным холодильником, закрытым хлоркальциевой трубкой с
Р2О5. Раствор охлаждают до —10 °C и в течение нескольких
минут добавляют при размешивании на магнитной мешалке
1,2 моль трифторуксусного ангидрида. Охлаждающую баню
убирают или заменяют водяной баней, нагретой до 10 °C, и раз-
мешивание продолжают еще полчаса, а затем избыток ангидри-
да и ТФУ удаляют в вакууме водоструйного насоса при темпе-
ратуре водяной бани не выше 30 °C; отгон собирают в колбу,
охлаждаемую смесью сухого льда с ацетоном. Для удаления
непрореагировавшей аминокислоты остаток растворяют в 20-
кратном количестве сухого эфира и отфильтровывают от не-
растворившегося материала. После отгонки эфира продукт ре-
акции кристаллизуют из подходящего растворителя.
Для реакции трифторацетилирования аминокислот удобен
прибор, изображенный на рис. 4. Раствор аминокислоты в
ТХУ помещают в колбу 7, установленную на магнитной мешал-
ке. Калиброванную трубку 2 заполняют требуемым количест-
вом трифторуксусного ангидрида. Сосуд 6 охлаждают смесью
сухого льда с ацетоном. После того, как раствор, содержащий-
ся в колбе 7, охладится до —10 °C, в правой части прибора co-
ll 2-882 161
Рис. 4. Схема прибора для трифторацетилирования аминокислот
здают небольшое разрежение за счет закрывания крана 3 и от-
крывания крана 4. После этого краны 4 и 5 закрывают, а кран
3 открывают и из трубки 2 отгоняют необходимое количество
трифторуксусного ангидрида, который конденсируется в кол-
бу 1. Затем кран опять закрывают и выдерживают реакцион-
ную смесь в течение получаса. После окончания реакции кра-
ны 4 и 5 открывают и растворитель из колбы 1 отгоняют в ва-
кууме в приемник 6.
Ниже приведены примеры синтеза некоторых трифтораце-
тиламинокислот.
Методика 150. N-Трифторацетил-Л-лейцин [271].
К раствору 0,262 г (2 ммоля) L-лейцина в 2,5 мл ТФУ при-
бавляют 0,35 мл (2,4 ммоля) ангидрида ТФУ при 0 °C и раствор
выдерживают, как было описано выше. После удаления рас-
творителя в вакууме остаток растворяют в водном растворе би-
карбоната натрия, встряхивают с эфиром и водную фазу под-
кисляют рассчитанным количеством разбавленной соляной кис-
лоты. Продукт экстрагируют эфиром, органическую фазу су-
шат сульфатом магния и эфир отгоняют в вакууме. Остаток —
густой бесцветный сироп — при хранении в течение 14 дней в
вакуум-эксикаторе над КОН кристаллизуется. После кристал-
лизации из бензола выход 0,291 г (64 %), т. пл. 71—73 °C.
Методика 151. N-Трифторацетил-Л-тирозин [271].
162
К раствору 1,81 г L-тирозина в 9 мл ТФУ, охлажденному
до О °C, прибавляют 5,4 мл абсолютного эфира, а затем —
50 %-й избыток трифторуксусного ангидрида и реакционную
смесь выдерживают 45 мин при 0 °C. После этого растворитель
отгоняют в вакууме, остаток продолжительное время кипятят
с водой и затем воду отгоняют на роторном испарителе. Остаток
растворяют в эфире, а тирозин, не вступивший в реакцию, от-
фильтровывают. Эфирный раствор слегка упаривают в вакууме
и затем смешивают с подогретым толуолом; при этом сразу же
начинается кристаллизация продукта, которая завершается
после удаления эфира в вакууме. Выход трифторацетил-тиро-
зина 2,3 г (95,5 %), т. пл. 192—193,5 °C.
Методика 152. Ангидрид трифторуксусной кисло-
ты [272]
,0
CF3—
6СГ3-СООН + Р2О5 -> >0 + 2Н3РО4,
ср.-с<0
114,02 141,98 210,04
В двугорлую круглодонную колбу вместимостью 0,5 л, снаб*
женную капельной воронкой и холодильником, помещают
200 г (1,4 моль) Р2О5; сразу же в колбу добавляют 80 г (0,7 моль)
ТФУ и смесь осторожно нагревают на масляной бане до 100 °C.
Образующийся ангидрид трифторуксусной кислоты отгоняют в
течение 1—1,5 ч. Полученный продукт еще раз перегоняют с
дефлегматором. Выход 70 г (90 %), т. кип. 39—40 °C.
Хорошим реагентом для синтеза TFA-аминокислот являет-
ся предложенный Шелленбергом и Кельвином тиоэтиловый
эфир трифторуксусной кислоты [2731. Он легко ацилирует ами-
нокислоты по Шотену—Бауману в водно-щелочной среде; вы-
ходы TFA-аминокислот при этом высокие (50—80 %).
Методика 153. Тиоэтиловый эфир трифторуксусной
кислоты [273]
О
II
(CF3COO)2 о + С2Н5— SH + CF3-C~SC2H5 + CF3COOH.
210,04 62,13 158,11 114,02
К охлажденному до 0 °C этилмеркаптану (31 г; 0,5 моль) до-
бавляют по каплям в течение 1 ч при размешивании и охлажде-
нии 130 г (0,62 моль) трифторуксусного ангидрида. Реакцион-
ную колбу снабжают обратным холодильником, защищенным
хлоркальциевой трубкой, оставляют на 1 ч при комнатной тем-
пературе, а затем нагревают 3 ч при 100 °C. Продукт промывают
холодным 5 %-м раствором КОН (2 X 500 мл) и затем — во-
дой, сушат сульфатом магния и перегоняют, отбирая фракцию,
кипящую при 90,5 °C. Выход 47,5 г (60 %).
163
Таблица 28. Физико-химические характеристики TFA-аминокислот
Аминокислота T. пл., °C Г0ЭД Растворитель
L-Аланин 66—68 —60,6 с — 2; вода
£-Аргинин 130—132 —3,5 с = 2; вода
Г-Аспарагин 154—165 — . —
А-Валин 86—88 — 15,2 с = 2; вода
L-Глутаминовая кис-
лота 192 —23,8 с = 2,5; вода
Глицин 117—119 —
Г-Лейцин 72—75 —39,5 с == 2; этанол
Л-Метионин 69—71 —22,7 с — 2; вода
L-Пролин 46—48 —60,3 с — 1; бензол
/.-Триптофан 162—163 — —
Л-Тирозйп 192,5—193,5 +45,0 с ~ 2; вода + 1 экв.
NaOH
S-Бензил-Г-цистеин 84-85 —84,5 с== 1,5; 99?/о-й спирт
Методика 154. Синтез TFA-аминокислот при помощи
тиоэтилового эфира трифторуксусной кислоты [2731
О
CFg-C-SCaH6 + H2N-CHR-COOH
о
II
CF3—С—NH-CHR—СООН + C2H3SH.
Внимание! Реакцию необходимо проводить в хорошо
вентилируемом вытяжном шкафу.
К раствору аминокислоты в 1 эквиваленте 1 н. едкого нат-
ра добавляют тиоэтиловый эфир трифторуксусной кислоты
(0,2 мл тиоэфира на 1 ммоль аминокислоты); молярное соотно-
шение (1,6 : 1). Колбу закрывают пришлифованным краном с
газоотводной трубкой, конец которой вставлен в отверстие
вытяжной шахты, и колбу встряхивают на механической мешалке
в течение 24 ч. Затем раствор подкисляют концентрированной
голяной кислотой при охлаждении на ледяной бане; выпав-
ший продукт отфильтровывают.
В табл. 28 представлены некоторые константы TFA-амино-
кислот. TFA-аминокислоты можно получать, используя в ка-
честве ацилирующего агента метиловый [274] или этиловый
эфир [275] трифторуксусной кислоты в присутствии органиче-
ских оснований.
Методика 155.Трифторацетилирование аминокислот и
пептидов этилтрифторацетатом [275].
В хорошо высушенную колбу вместимостью 25 мл, снаб-
женную тефлоновой мешалкой с каучуковым уплотнением, по-
164
мещают 10 ммоль аминокис-
лоты или пептида | (при ра-
боте с цистеином использу-
ют 5 ммоль аминокислоты);
воздух из колбы вытесняют
сухим аргоном и добавляют
5 мл абсолютного метанола.
При непрерывном разме-
шивании добавляют 2,4 мл
(10 ммоль) триэтиламина
(при ацилировании аспара-
гиновой кислоты использу-
ют 2,8 мл основания), а за-
тем — 1,5 мл (12,5 ммоль)
этилтрифторацетата. Реак-
ционную массу энергично
размешивают при комнат-
ной температуре до полного
Таблица 29. Время проведения
реакции и выход TFA-аминокислот
[275]
Аминокислота Время реак- ции, ч Выход, %
Д-Аланин 25 82
Д-Аспарагин Д-Аспарагиновая кисло? 45 77
та 14 75
Д-Валин 45 91
Д-Серин * 34 80
Д-Тирозин ** 17 85
Д-Триптофан 23 89
Д-Цистеин 15 83
Д-Фенилаланин 25 96
* В качестве растворителя использо-
вали ДМФА.
♦ ♦ Вместо триэтиламина применяли те-
траметилгуанидин.
растворения аминокислоты
(время проведения реакции
указано в табл. 26). После
этого прибавляют 5 мл ме-
танола, колбу охлаждают
до 10 °C и прибавляют 4 г (при синтезе TFA — AspOH — 6 г)
смолы Дауэкс-50 в Н+-форме. Полученную смесь размеши-
вают 10 мин, частицы смолы отфильтровывают, растворитель
удаляют в вакууме, а остаток кристалллизуют из подходяще-
го растворителя.
В табл. 29 указаны выход TFA-аминокислот и время прове-
дения реакции.
Удаление трифторацетильной защитной группы
щелочными реагентами
TFA-группа легко отщепляется при действии 0,2 и. раство-
ра едкого натра в течение 10 мин. Для этой цели можно исполь-
зовать также разбавленный водный раствор аммиака. Свобод-
ные пептиды удобно получать путем одновременного гидролиза
сложноэфирной и трифторацетильной группировкой щелочью;
недостатком TFA-группы явлется то, что ее практически не-
возможно избирательно удалить, не затрагивая сложноэфир-
ные группировки —ОСН3 и —ОС2Н8.
В 1970 г. Вейганд и Францендорфер предложили новый
способ деблокирования TFA-производных действием боргид-
гида натрия [2081.
165
Методика 156. Отщепление трифторацетильной группы
боргидридом натрия [208]
о О
TFA—NH-CHR,—С—NH—СН₽2—С—
о
II
-> H2N—CHRj—CONH—CHR3—С—O—Bu'.
К раствору или суспензии 1 ммоль бутилового эфира TFA-
иептида в 5 мл охлажденного этилового спирта прибавляют
4 ммоль хорошо измельченного боргидрида натрия (при работе
с пролинсодержащими пептидами используют 2 ммоль NaBHJ
и размешивают 1 ч при комнатной температуре (для пролин-
содержащих пептидов — 6 мин). За скоростью выделения во-
дорода следят, закрыв реакционный сосуд {/-образной трубкой
с парафиновым маслом в качестве запорной жидкости. По мере
протекания реакции суспензия постоянно растворяется. Для
разрушения избытка боргидрида натрия колбу охлаждают и
смешивают содержимое с равным объемом ацетона, размешива-
ют в течение 15 мин, затем растворитель удаляют в вакууме,
а к остатку добавляют воду для разрушения комплекса бора.
Водный раствор несколько раз встряхивают с этилацетатом,
органическую фазу отделяют, сушат сульфатом магния, рас-
творитель удаляют на роторном испарителе. Остаток кристал-
лизуют из подходящего растворителя. При работе с соединени-
ями, хорошо растворимыми в воде, для разложения комплекса
бора применяют насыщенный раствор К2СО3, а экстрагирова-
ние продукта ведут хлористым метиленом.
По указанной методике с выходом 93 % получен L-валнл-
Л-валин из 207 мг (0,56 ммоль) /npem-бутилового эфира TFA-Z,-
в лин-£-валина и 86 мг (2,25 ммоль) NaBH4 в 3,5 мл этилового
спирта. Выход 142 мг.
9-Флуоренилметилоксикарбонильная группа (Fmoc)
Наряду с трифторацетильной группой 9-флуоренилмето-
ксикарбонильная группа является одной из защитных группи-
ровок, которые устойчивы к действию кислот, но легко удаля-
ются основаниями в мягких условиях. Fmoc-группа была пред-
ложена Карпино и Ханом в 1970 г. [1811. Находит широкое
применение не только в классическом, но и в твердофазном
методе синтеза.
166
Синтез 9-флуоренилметилхлорформиата
Методика 157. 9-Флуоренилметанол [277]
н СН2ОН
196,25
В трехгорлый реактор вместимостью 5 л, снабженный меха-
нической мешалкой, обогревательным кожухом, двумя после-
довательно соединенными мощными обратными холодильника-
ми и капельной воронкой с противодавлением, вносят 166,6 г
(1 моль флуорена, а затем 1700 мл абсолютного этанола. Рас-
твор перемешивают и вносят в него 150rNaH (60 % дисперсия
в масле). Серая смесь бурно закипает, и этот процесс длится
15 мин. В капельную воронку заливают 185 мл этилформиата
и из нее сразу приливают в колбу 15 мл (во избежание бурного
вскипания смеси до начала реакции). Реакция начинается при-
близительно через 0,5 ч, что сопровождается бурным кипением
жидкости в реакторе. Оставшийся в воронке этилформиат при-
бавляют по каплям в течение 1,5 ч. Если реакция проходит
слишком бурно, колбу охлаждают ледяной водой. После до
бавления всего этилформиата прекращают охлаждение и через
3 ч, когда реакция замедляется, колбу осторожно нагревают и
греют при кипении в течение 12 ч.
После окончания реакции в колбу (после охлаждения ее в
ледяной бане) медленно при перемешивании вносят кусочки
льда (около 800 мл по объему замерзшей воды) до тех пор, по-
ка прекратится выделение газа. Затем заливают в реактор 2 л
холодной воды и перемешивают еще 5 мин. Разделяют водный
и органический слой в делительной воронке вместимостью 5 л
и затем водный слой экстрагируют дважды 750 мл петролей-
ного эфира (т. кип. 30—60 °C) и фильтруют через Ватман-410.
Водный фильтрат помещают в 5-литровую колбу, охлажда-
ют на водяной бане и подкисляют, добавляя по каплям 350 мл
ледяной уксусной кислоты до pH 5. Выделяется нерастворимое
масло. В эту смесь добавляют при перемешивании 1 л эфира, в
котором масло легко растворяется, эфирный слой отделяют, а
водный еще раз промывают эфиром (2 X 1,5 л). Эфирные экс-
тракты объединяют, промывают 1 л воды, 1 л 1 н. NaHCO3, су-
167
шат сульфатом магния и концентрируют на роторном испарите-
ле. Получают около 250 мл коричнево-красного масла.
Это масло (альдегид) растворяют в 700 мл метанола и пере-
носят в трехгорлый реактор, снабженный обратным холодиль-
ником, мешалкой и воронкой для сыпучих веществ. К раствору
добавляют еще 1 л метанола и охлаждают его на ледяной бане.
Вносят в реактор 60 г (1,6 моль) боргидрида натрия порциями
по 2—3 г, чтобы избежать сильного газовыделения. Если реак-
ция пойдет слишком бурно, охлаждают реактор льдом. Смесь
перемешивают 0,5 ч при охлаждении, затем 4 ч при комнат-
ной температуре, разбавляют 2,5 л воды (при этом выпадает
белый осадок), затем подкисляют 300 мл ледяной уксусной кис-
лоты до pH 5 и перемешивают еще 2 ч, контролируя величину
pH. Осадок удаляют фильтрацией, промывают его 2 л воды, а
все фильтраты объединяют и охлаждают на водяной бане, что
приводит к образованию второй порции кристаллов. Вещество
сушат в вакууме при 50 °C в течение двух дней. Выход 9-флуо-
ренилметанола 157,2 г (80 %); 1 порция — 144 г, т. пл. 93—
96 сС; 2 порция — 13,2 г, т. пл. 94,5—99,5 °C.
Примечание: окончание реакции восстановления
альдегида боргидридбм натрия контролируют по ТСХ на сили-
кагеле в системе метиленхлорид—этилацетат (1 1). Для
альдегида /?/ = 0,85; для продукта = 0,8.
Удобная методика синтеза 9-флуоренилметанола без примене-
ния амида натрия разработана и предоставлена нам И. Л. Ро-
дионовым из Филиала Института биоорганической химии
Российской АН (Пущино-на-Оке).
Методика! 58. 9-Флуоренилметанол.
Раствор этилата натрия, полученный из 11,5 г металличе-
ского натрия и 500 мл абсолютного этанола («продажный», без
дополнительной очистки), выпаривают на роторном испарите-
ле, после чего затвердевший этилат выдерживают в вакууме
водоструйного насоса при 30—40 °C в течение 15 мин и раство-
ряют в 135 мл сухого диметилсульфоксида («хч», дважды вы-
мораживают в холодильнике в течение ночи, каждый раз сли-
вая незатвердевшую часть и фракционируют в вакууме водо-
струйного насоса, отбрасывая первые 20 % дистиллята). По-
лученный раствор быстро переливают в трехгорлую колбу ем-
костью 1 л, снабженную магнитной мешалкой, капельной во-
ронкой с трубкой, уравновешивающей давление, термометром
и обратным холодильником с хлоркальциевой трубкой, добав-
ляют в один прием 66,5 г (0,4 моль) флуорена («ч»), интенсивно
перемешивают и через 20 мин (смесь приобретает интенсивную
коричнево-черную окраску) начинают прибавлять по каплям
этилформиат («ч», предварительно выдержанный несколько
суток над К2СО3, 15 г/100 мл) (48,6 мл; 0,6 моль) с такой скоро-
168
стью, чтобы температура в колбе не поднималась выше 40 °C
(около 1 ч). В процессе прибавления этилформиата наблюдает-
ся легкое вспенивание, и реакционная смесь меняет цвет на зе-
леновато-черный; по окончании прибавления этилформиата
продолжают перемешивание 3 ч при комнатной температуре,
после чего к гомогенной реакционной смеси добавляют 400 г
льда, перемешивают еще 0,5 ч и отделяют фильтрованием не-
большое количество не вступившего в реакцию флуорена. Оса-
док на фильтре отмывают 250 мл 5 %-го водного КОН, причем
промывные воды объединяют с основным фильтратом, а оса-
док отмывают до нейтральной реакции 2 %-й уксусной кисло-
той и водой, высушивают до постоянного веса (около 9 г, 15 %).
К интенсивно-коричневому основному фильтрату, собранному
совместно с щелочными промывными водами в 5-литровую кол-
бу Бунзена, интенсивно перемешиваемому на магнитной мешал-
ке, добавляют в один прием 7,6 г боргидрида натрия. Прибли-
зительно через 20 мин исходная окраска, обусловленная ено-
лятом 9-флуоренилальдегида, сменяется бледно-желтой; к
реакционной смеси прибавляют лед до суммарного объема 3—
3,5 л и по каплям раствор НС1 (1 : 10) до pH 4—4,5 с такой
скоростью, чтобы выделение водорода не протекало слишком
бурно (около 700 мл НС1 за 1,5—2 ч). Белый осадок 9-флуоре-
нилметанола отфильтровывают через 1 ч после достижения
указанного pH и промывают большим количеством дистилли-
рованной воды, а затем сушат в вакуум-эксикаторе над КОН.
По. у чают 56 г (82 %) продукта, не содержащего полимерных
прьмесей и пригодного для получения 9-флуоренилметилхлор-
формиата без дополнительной перекристаллизации.
Методика 159. 9-Флуоренилмстилхлорформиат [278,
279].
К охлажденному до 0 °C раствору 7,12 г фосгена в 75 мл хло-
ристого метилена добавляют по каплям при охлаждении и пере-
мешивании 12,8 г 9-флуоренилметанола. Размешивание и ох-
лаждение продолжают 1 ч, а затем оставляют на холоду еще на
4 ч. После этого растворитель и избыток фосгена удаляют в ва-
кууме; оставшееся масло через несколько часов кристаллизу-
ется. Выходнеочищенного продукта 16 г (95 %). После двух-
кратной кристаллизации из эфира получают 14,5 г (86 %) бес-
цветных кристаллов с т. пл. 61,5—63 °C.
Синтез 9-флуоренилметилоксикарбониламинокислот [279]
Мето дика 160. 9-Флуоренилметилоксикарбонил-£-трип-
тофан [279].
К раствору 1,58 г (7,8 ммоль) Л-триптофана в смеси 10 мл ди-
оксана и 20,5 мл 10 %-го раствора карбоната натрия медленно
169
Таблица 30. Свойства
9-флуоренилметоксикарбониламнно-
кислот
Аминокислота Т. пл., °C град (с=-- 1; ДМФА)
L-Аланин 143—114 — 18,0
L-Аспарагип 185—18(5 — 10,5
р-трет-бутиловый эфир Л-аспарагиновой 148—149 —20,0
кислоты Г-Валин 142--144 — 16,0
Глицин 174—175
/,-Глутамин 220—223 — 17,0
у-трет-Бутило- вый эфир L-глу- таминовой кисло- 76—80 — 14,5
ты
N 1т-Бензил-£- 142—143 — 11,5
гистидин L-Изолейцин 144—146 — 11,9
£-Лейцин 155—156 —24,0
+ -Вос-£-лизин 123—124 —11,5
£-Метионин 118—119 —28,0
£-Пролин 114—115 —33,0
0-Трет-бутил-£- 126—129 +25,0
серин 0-Трет-бутил+- 148—151 —27,0
тирозин 0-Трет-бутил-£- 129-132 + 15,5
треонин L-Триптофан 185—187 —26,0
L-Фенилаланин 180—183 —37,0
S-Трет-бутил-Г- цистеин 135—136 —23,0
добавляют при размешива-
нии и охлаждении до 0 °C
раствор 2 г (7,8 ммоль)
Эчрлуоренилметилхлорфор-
миата в 20 мл диоксана.
Смесь размешивают при ох-
лаждении 4 ч и при комнат-
ной температуре 8 ч, а за-
тем раствор выливают в
450 мл воды и встряхивают
с эфиром. Водный слой ох-
лаждают льдом и подкисля-
ют концентрированной со-
ляной кислотой по Конго
красному, и реакционную
смесь оставляют на ночь в
холодильнике. Выпавший
осадок отфильтровывают и
кристаллизуют из нитроме-
тана, а затем из смеси хло-
роформ — гексан. Получа-
ют 3 г (91 %) чистого Fmoc-
триптофана с т. пл. 185—
187 °C, [а]Ь5 = +6,4° (с =
= 1; этилацетат).
Методика 161. 9-
Флуоренилметилоксикарбо-
нил-глицин [279].
К раствору 0,57 г
(7,6 ммоль) глицина в
20,2 мл 10 %-го раствора
карбоната натрия добавля-
ют при размешивании и
охлаждении раствор 1,96г(7,7 ммоль)9-флуоренилметилхлор-
формиата в 20 мл диоксана. Смесь перемешивают 2 ч при ком-
натной температуре и затем выливают в 400 мл воды. Для уда-
ления небольшого количества образующегося 9-флуоренилме-
танола и высокоплавкого полимера дибензсфульвена водный
раствор дважды встряхивают с эфиром. Водный слой охлажда-
ют льдом и подкисляют концентрированной соляной кислотой
по Конго красному. Выпавший бесцветный осадок экстрагиру-
ют этилацетатом, органическую фазу промывают водой, сушат
сульфатом магния, и этилацетат отгоняют в вакууме. Выход
бесцветного кристаллического продукта 2 г (89 %), т. пл. 173—
176 °C. После кристаллизации из нитрометана получают 1,98 г
(88 %) Fmoc-глицина с т. пл. 174—175 °C.
170
В табл. 30 даны физико-химические характеристики Fmoc-
аминокислот, полученных указанными выше способами.
В последние годы обнаружено, что при ацилировании ами-
нокислот 9-флуоренилметилхлорформиатом происходит побоч-
ное образование Fmoc-дипептидов. Так, в различных препара-
тах Fmoc-глицина обнаружено от 3 до 9 % Fmoc-Gly—
Gly—ОН.
Недавно Зиглер и соавт. [281] провели тщательное исследо-
вание побочных реакций, происходящих в процессе получения
Fmoc-ами но кислот с использованием Fmoc-хлорида. Они пока-
зали, что при получении производных стандартными методами
большинство Fmoc-аминокислот загрязнены различными, но
заметными количествами Fmoc-олигопептидов. Причиной это-
го является образование относительно стабильных смешанных
ангидридов:
В работе [281] показано, что при этом может образовываться
до 20 % побочного продукта.
Оптимизирована процедура синтеза Fmoc-аминокислот с
использованием Fmoc-хлорида: оптимальные условия — 20—
25 %-й избыток аминокислоты по отношению к Fmoc-хлори-
ду; минимальный объем диоксана по отношению к водной
фазе (ок. 1 I 10); 4-кратный избыток Na2CO3 по отношению
к аминокислоте; энергичное перемешивание при комнатной
техмпературе. Авторы [281] рекомендуют избегать использова-
ние Fmoc-хлорида как общего метода синтеза Fmoc-амиио-
кислот. Исследованы некоторые карбонаты, среди которых
наиболее эффективным оказался 9-флуоренилметилсукцини-
мидоил карбонат:
Методика 162.
доил карбоната [281].
Синтез 9-флуоренилметилсукциними-
171
102,8 г (0,4 моль) Fmoc-хлорида и 58 г (0,5 моль) N-оксисук-
цинимида растворяли в 700 мл сухого* дважды перегнанного
диоксана в двухлитровой трехгорлой колбе при эффективном
перемешивании. Смесь охлаждали на ледяной бане и добавля-
ли 55,75 мл (0,4 моль) триэтиламина с такой скоростью, чтобы
температура не поднималась выше 10 °C. Реакция полностью
заканчивается через 4 ч, после чего осадок хлоргидрата триэ-
тиламина удаляли фильтрованием, хорошо промывали на
фильтре диоксаном, раствор концентрировали в вакууме и пе-
рекристаллизовывали остаток из петролейного эфира при 4 °C.
Выход 150 г (86 %), т. пл. 148—149 °C.
Продукт можно получать без предварительного выделения
Fnioc- хлорида.
Методика 163. Синтез 9-флуоренилметилсукциними-
доил карбоната [282].
Конденсируют около 400 мл фосгена при —78 сС (установ-
ку см. в методике 92). Добавляют к нему по частям 98 г
(6,5 моль) при этой температуре при перемешивании 9-флуоре-
нилметанола. Стенки колбы и воронки промывают приблизи-
тельно 40 мл толуола. Затем суспензию оставляют нагреваться
при перемешивании до комнатной температуры. После переме-
шивания в течение ночи остаток фосгена удаляют, а остаток в
колбе сушат путем добавления сухого диоксана и его отгонки.
Полученный технический Fmoc-хлорид в виде масла растворяем
в 1,25 л диоксана, и к полученному раствору добавляем 63,3 г
(0,55 моль) N-оксисукцинимида. После охлаждения в ледяной
бане при хорошем перемешивании добавляли к раствору 70 мл
(0,5 моль) триэтиламина с такой скоростью, чтобы температура
не поднималась выше 10—15°. После перемешивания в течение
4 ч при комнатной температуре удаляли осадок хлоргидрата
триэтиламина фильтрованием.
Фильтрат концентрировали на роторном испарителе до объе-
ма 250 мл и медленно выливали в 2,5 л эфира при перемешива-
нии. Выкристаллизовавшийся продукт отделяли фильтрацией,
промывали эфиром и сушили. Выход 115—121,5 г (85—90 %),
т. пл. 130—148 °C. Продукт может быть использован для по-
лучения Fmoc-аминокислот без дальнейшей очистки. (Для по-
лучения чистого продукта растворяют его в количестве 0,1 моль
в 60 мл хлороформа и кристаллизуют добавлением 15 мл эфира.)
Внимание’ При работе с жидким фосгеном соблюдать пра-
вила безопасности, указанные для получения карбобензокси-
хлорида (методика 92).
Методика 164. Общий метод получения Fmoc-амино-
кислот [282].
Растворяют 25 ммоль свободной аминокислоты в 25 мл воды
в присутствии 1 экв. триэтиламина. К смеси добавляют рас-
172
твор 24 ммоль Fmoc—OSu в 25 мл ацетонитрила (растворяется
при легком нагревании) одной порцией. В результате выделе-
ния N-оксисукцинимида pH раствора снижается и его нужно
поддерживать на уровне 8,5—9,0 путем добавления по каплям
триэтиламина. Через 15 мин величина pH становится постоян-
ной или изменяется очень медленно. После перемешивания
смеси еще 15 мин ее фильтруют, если необходимо, и концент-
рируют в вакууме. Остаток вносят в 100 мл 1,5 н. НС1 при пере-
мешивании. Если при этом продукт кристаллизуется, его отде-
ляют фильтрацией, промывают водой и сушат. Если продукт
выпадает в виде масла, его экстрагируют этилацетатом, промы-
вают водой и раствором NaCl и сушат сульфатом натрия. После
отгонки растворителя продукт перекристаллизовывают из под-
ходящего растворителя.
Модификации общего метода. При получении
Fmoc-LeuOH в воде с 1 экв. триэталамина процесс растворе-
ния происходит очень долго, поэтому его нужно растворять с
1 экв. NaOH.
При получении Fmoc-Asn количество растворителя было
удвоено для устранения проблемы растворимости. В случае
аминокислот с mpem-бутильными защитными группами в боко-
вых группах использовались 20 %-я лимонная кислота вместо
1,5 н. НС1. Выход Fmoc-аминокислот по этой методике 80—
100 %.
Недавно был предложен новый метод получения Fmoc-ами-
нокислот, свободных от примеси продуктов полимеризации
[283], основанный на обработке О, N-бис-триметилсилил-ами-
нокислот Fmoc-хлоридом:
H3N+—CHR—COO” -™S~CU
2NR3
О
[{CH3)3Si— HN-CHR—С—O-Si (СН3)3]
О
И 11 о
-> Fmoc-HN—CHR—С—OSi (СН3)8 —2—> Fmoe-HN-CHR—СООН.
Методи ка 165. Fmoc-производные Gly, Ala, Phe, Pro,
Met, Trp, Leu, He и Lys (S—Buz) [2831.
37,5 ммоль тонко измельченной аминокислоты помещали в
круглодонную колбу емкостью 250 мл, снабженную обратным
холодильником с хлоркальциевой трубкой. Добавляли 87,5 мл
сухого метиленхлорида и при сильном перемешивании вносили
сразу 9,52 мл (75 ммоль) триметилхлорсилана. Смесь кипятили
1 ч и охлаждали на ледяной бане. Затем добавляли 11,3 мл
(65 ммоль) диизопропиламина и 6,47 г (25 ммоль) Fmoc-хлори-
173
да. Раствор перемешивали при охлаждении 20 мин и затем на-
гревали в течение 1—1,5 ч до комнатной температуры. Раство-
ритель удаляли при пониженном давлении и распределяли ос-
таток между 200 мл эфира и 250 мл 2,5 %-го раствора бикарбо-
ната натрия. После отделения водной фазы ее промывали 2 X
X 50 мл эфира, а эфирные вытяжки экстрагировали водой 2х
X 25 мл. Объединенные водные слои подкисляли 1 н. НО до
pH 2 и экстрагировали 3 х 75 мл этилацетата. Этилацетатные
вытяжки сушили сульфатом натрия, фильтровали, удаляли
растворитель и перекристаллизовывали остаток из подходя-
щего растворителя (в большинстве случаев использовали смесь
этанол — вода).
Методика 166. Fmoc-производные Asp (OBuz), Glu
(OBiF), Ser (OBu9, Thr (OBu') и Lys (Ne — Boc) [283].
37,5 мм^ль тонко измельченной аминокислоты помещали в*
250 мл круглодонную колбу, снабженную обратным холодиль-
ником с хлор кальциевой трубкой, вносили 87,5 мл сухого ме-
тиленхлорида, и при сильном перемешивании добавляли 12,7 мл
(73 ммоль) диизопропилэтиламина. Затем вносили 9,5 мл
(75 ммоль) триметилхлорсилана и кипятили раствор в течение
1,5 ч. После охлаждения раствора на бане со льдом в него до-
бавляли 6,4 г (25 ммоль) Fmoc-хлорида в один прием, переме-
шивали 20 мин при охлаждении и оставляли нагреваться до
комнатной температуры 1—1,5 ч. Продукт выделяли аналогич-
но приведенной выше методике. Выход Fmoc-аминокислот по
этому методу 82—94 %.
Методика 167. Синтез Fmoc-Asn и Fmoc-Gln [283].
37,5 ммоль тонко измельченной аминокислоты суспендиро-
вали в 87,5 мл сухого диметилацетамида и сильно размешива-
ли. Добавляли 19,04 мл (150 ммоль) триметилхлорсилана, пе-
ремешивали 1 ч, охлаждали на бане со льдом, добавляли
19,78 мл (114 ммоль) диизопропилэтиламина и 6,47 г (25 ммоль)
Fmoc-хлорида. Перемешивали 20 мин при этой температуре и
нагревали до комнатной температуры в течение 1 —1,5 ч. Смесь
обрабатывали, как описано выше, исключая подкисление вод-
ных фаз. Выпавший белый осадок был отфильтрован, промыт
холодной водой и высушен в вакууме. Перекристаллизация из
смеси этанол — вода.
Методика 168. Синтез Fmoc-Tyr (OBuz) ОН [283].
6,52 г (27,5 ммоль) тонко измельченного O-mpem-бутило-
вого эфира тирозина суспендировали в 100 мл сухого диок-
сана, прибавляли при перемешивании 9,04 (52 ммоль) диизо-
пропиламина и 6,98 мл (55 ммоль) триметилхлорсилана и пе-
ремешивали при комнатной температуре 1 ч. Прибавляли к
раствору 200 мл метиленхлорида и охлаждали на бане со льдом.
После внесения 6,4 г (25 ммоль) Рмос-хлорида реакционную
174
смесь перемешивали 20 мин при охлаждении и еще 1,5 ч при
комнатной температуре. Раствор концентрировали при по-
ниженном давлении, обрабатывали, как описано в методике
167 (водный слой подкисляли 10 %-й лимонной кислотой),
экстрагировали 3 X 200 мл метиленхлорида, выделяли обыч-
ным образом и перекристаллизовывали из смеси этанол — вода.
Выход 11,1 г (85 %), т. пл. 150—151 °C, [aI2D5 = — 27,34 (с =
= 1; ДМФА).
Получениес помощью Fmoc—OPfp см. гл. 1 (методика 62).
Отщепление флуоренилметилоксикарбонильной
защитной группы
Fmoc-группа количественно отщепляется при комнатной
температуре аммиаком, пиперидином, морфолином или диэ-
тиламином. В препаративном отношении чаще всего исполь-
зуют следующие два метода: 1) Fmoc-пептид перемешивают
40 мин в пиперидине или 25 мин в морфолине при комнатной
температуре, затем выпавший осадок отфильтровывают, а
растворитель отгоняют; 2) Fmoc-пептид выдерживают 2 ч при
комнатной температуре в 10 %-м растворе диэтиламина в
ДМФА, после чего растворитель удаляют в вакууме.
Рассмотрим конкретный пример.
М е т о д и к а 169. Глицил-глицин [181].
Раствор 1,3 г (3,7 ммоль) Fmcc-глицил-глицина в 10 мл пи-
перидина размешивают 30 мин при комнатной температуре,
а затем разбавляют 200 мл воды. Пиперидии-дибензофульве-
новый аддукт, выпавший в осадок, отфильтровывают, фильт-
рат упаривают па роторном испарителе, а остаток кристалли-
зуют из смеси этиловый спирт — вода (10: 1). Выход глицил-
глицина 0,4 г (83 %).
Глава 3
ЗАЩИТА КАРБОКСИЛЬНОЙ
ГРУППЫ
В синтезе пептидов защита С-концевой карбоксильной груп-
пы аминокислот и пептидов имеет такое же значение, как и
блокирование аминогрупп. Несмотря на то, что при получении
некоторых пептидов с успехом используют солевую защиту
(см. главу 1), чаще всего, однако, в процессе наращивания
полипептидной цепочки С-конец пептидов блокируют. Защи-
та карбоксильной группы, как правило, способствует повыше-
нию растворимости пептидов в органических растворителях.
Наиболее простым способом блокирования СООН-группы
является превращение ее в сложноэфирную группировку.
О других способах защиты — превращении в бензилоксикар-
бонил- или /пре/п-бутилоксикарбонилгидразидную группи-
ровки — сказано в главе 1. Преимуществом сложноэфир-
ных производных является доступность сложных эфиров
аминокислот, относительная простота их расщепления и лег-
кость, с которой многие из них превращаются в амиды или
гидразиды — важнейшие продукты синтеза пептидов.
В практике пептидного синтеза для защиты карбоксиль-
ной группы аминокислот используют алифатические (мети-
ловые, этиловые или /npem-бутиловые), алкилароматические
(бензиловые, п-нитробензиловые> 4-пиколиновые), ' аромати-
ческие (фениловые) эфиры.
Для расщепления указанных производных используют
ряд методов: щелочной или кислотный гидролиз, катали-
тическое восстановление над палладиевым катализатором,
восстановление цинком в уксусной кислоте, фотолиз и другие
способы, суммированные в табл. 31 и 32. Большинство С-за-
щитных групп удаляют в одну стадию, но некоторые из них
целесообразно сначала модифицировать в более лабильные
соединения, которые расщепляются в мягких условиях.
Примером может служить р-метилтиоэтильная группа (105),
которая легко гидролизуется в щелочной среде после предва-
рительной обработки перекисью водорода или иодистым ме-
176
тилом [284].
О
II
R—С—О—СН2СНа—S—СН8
(105)
О О
->• R—С—О—СНаСН2—S—СН,
II
О
о
'{jpj R—С—О—CHjCHg—S^*—-CH3
CH3
В некоторых случаях С-защитную группу модифицируют так
чтобы она превратилась в активированную группировку для,
последующего наращивания пептидной цепи. Примеры; трет-
бутилоксикарбонилгидразидная группа или предложенная
недавно Ю. В. Митиным [285] п-диметиламинофенильная груп-
па (106); последнюю после алкилирования превращают в
активированный 4-(М-триметил)аминофениловый эфир (107),
который используют для наращивания полипептидной цепи;
О
Z—NH—CHR—С—О-/ ^>—NMe, CH,r ,
(106)“
О
(107)
Для блокирования карбоксильной группы Пасс и соавт. [286]
предложили весьма интересную 5-бром-7-нитроиндольную
защиту. После облучения соединения (108) ультрафиолето-
вым светом с длиной волны 420 нм происходит активация ука-
занной группировки и она становится способной легко и без
рацемизации ацилировать аминокомпонент;
Выбор той или иной блокирующей группы диктуется ске-
мой синтеза пептидов. Для селективного удаления защиты в
присутствии других блокирующих группировок необходимо
12 2-882
177
Таблица 31. Защитные группы для блокирова
Название группы Обозначение группы Формула
Метиловые эфиры —ОМе
О
II
R—С—ОСН3
Этиловые эфиры —OEt
Бензиловые эфиры —OBzl
О
II
R-C'-OC2H5
о
II
R—С—О—СН2—
л-Нитробензиловые —ONB
эфиры
О
R-C—O-CH2-<f >-no2
/ире/п-Бутиловые эфиры —OBt?
О сн8
II I
R—С—О—С--СН3
х
и-Метоксибенз иловые
эфиры
-OBzl (ОМе)
II
R—С—О—СН2—-ОСН3
О
4-Пиколиновые эфиры —OPic
Трихлорзтиловые эфи- —ОТС1
ры
R—С—ОСН2 —СС13
178
ния карбоксильной группы аминокислот и пептидов
Реагент для введения Способ удаления Примечание Лите- ратура
СН8ОН + НС1; 1 Ht щелочь Получили широкое рас- [287]
СН3ОН + SOCla пространение; легко по- лучаются. Удаляются водно-спиртовым рас- твором NaOH. При взаи- [288|
С2Н,ОН + НС1; To же модействии гидразингид- [287,
С2Н»ОН + SOC13 ратом легко превраща- ются в гидразиды 288)
PhCH9OH 4- PhSO3H; H2/Pd, ОН—, Используются весьма [2891
PhCHaBr + NEt3 Na/NH3 (HF безводный) широко. Селективно удаляются в присутст- вии Вос-группы и OBtA эфиров и других кис- лотолабильных групп
4 — NO2 — PhCH2OH + + PhSO3H; 4 — NO2 — PhCH2Br + H2/Pd, Na2S2O4 Более устойчивы к дей- ствию НВг/АсОН, чем бензиловые эфиры. [290J
+ NEt3 Можно селективно уда- лять в присутствии бен- зильной защиты [291]
CH#4 1 h. НС1/ТГФ, Нашли очень широкое
>C=CH2+HaSO4 TFA (HBr/AcOH, распространение. Селек-
CH3/ HF безводный) тивно удаляются в при- сутствии Z-, -OBzl, -ONb и других групп. Устойчивы в щелочной среде и условиях ката- литического гидрогено- лиза
CH8O-/2)>-CHaOH» H® TFA, (H2/Pd) Применяются ограни- ченно. Селективно уда- ляются в присутствии Z-, -OBzl, -ONB и дру- гих групп [292]
CH2OH + H+ H2/Pd (OH"), Устойчивы к действию НВг/АЮН и HF без- водного [2931
CH2Br, NR3
CCI3OH 4- H® СС1ЭОН, дцгк Zn/AcOH Возможно селективное удаление в присутствии других защитных групп [294]
12*
179
Название группы Обозначение группы Формула
Фениловые эфиры —OPh
О
Бензгидриловые эфиры —OBzh
/Ph
R—С—О—СН
II Хрь
О
Триметилсилиловые —OTMS
эфиры
R—С—О—SiMe8
2-Триметилсилилэтило- —OTMSe
вые эфиры
R—С—ОСН аСН2—Si Ме8
2-Метилтиоэтиловые ОМТе
эфиры
II
R—С—О—СНаСН2—SMe3
9-Флуоренилметиловые —-OFmoc
эфиры
2-(1-Адамантил)-про- —OAdpoc
иан-2-иловые эфиры
2-(Трифторметил)-6-хро- —ОТсгот
монилметиловые эфиры
180
Продолжение табл. 31
Реагент для введения Способ удаления Примечание Лите- ратура
C,H6O-S-OC,H5 II о ОН~ Применяются редко Н2Оа + NaOH ' (pH 10,5) [293, 296]
(Ph)2CHOH, Н® (Ph)2CHBr, NEt3 TFA Применяются редко H2/Pd НС1/АсОН; (ОН—) [297]
Me3SlCl, NEL
CHj-C^N—SiMe3
OSiMe,
HO-CH2CHa—SiMe,
+ ДЦГК
нао ROH Очень лабильны. Используются в специ- альных случаях [298]
Устойчивы к гидроли- зу и каталитическому гидрированию. Расщеп- ляются NaOH диокса- не. По-видимому, перс- пективны, так как мо- гут 'расщепляться в присутствии других эфиров [299]
[284]
CH8—S—CH2CH,OH+
•4-PhSO3H _
CH3—S—CH2CHaCl +
+NEt3
H20a в щелочной
среде или СНЭ1
в щелочной среде
+ ДСС + ДМАР.
NHEta или пи-
перидин в СН2С12
Предложены недавно.
Возможно их селектив- [300]
ное удаление в присут- [301]
ствии кислотолабйль-
ных групп
CH3
C—Adam, H®
II
CH2
TFA, НС1 Предложены недавно [302]
C3H7NHaj
NHaNHa . HjO
Устойчивы к действию [303]
кислых реагентов. Се-
лективно удаляются в
присутствии Вос-,
-OBt? и Fmoc-rpynn
181
Название группы
Обозначение
группы
Формула
Пентаметилбензиловые
эфиры
О Нзсч >СН3
и >«<
С-ОСН2-/^-- сн3
Фенациловые эфиры —- OPhao
о-И итробензгидрил овыа
эфиры
5-Б ром-7-нитроин до- BNI
лильная группа
[H8N-CH—С—О—-Со (NH8)J (BFJi
Комплекс с пентаамино-
кобальтом
Таблица 32. Способы удаления ^защитных групп
Метод удаления Символ удаляемой группы
Кислотами: в жестких условиях OBur, OBzl (ОНе), OBzh, (-OAdpoc) в мягких условиях OAdpoc Основаниями: в жестких условиях ОМе, OEt, OPh, (OBzl), (ONB), (OPic), (OTce), (OBzh), (OTMSe) в мягких условиях OMTe, OFmoc, OTcrom
82
Окончание табл. 31
Реагент для введения Способ удаления Примечание Лите- ратура
TFA [304]
СН,Х /СН,
Н8С—/‘=\—СН2Вг,
СН3/ хсн3
_ NEtg
СО'—СН2Вг,
NEt,
Ph\
сн—он, н+
Ph/
1ю, '
Zn/AcOH, H2/Pd, [305]
PhS—, hv [ЗОв]
R—COOH+SOC1,+
NaBH4, NaSH
Уникальная защитная [286]
группировка: при об-
лучении УФ-светом
(420 нм) превращается
в активированную груп-
пу
Принципиально но- [307]
вая нековалентная С-
защитная группа. Ус-
тойчива в условиях от-
щепления Вос-группы
Окончание табл. 32
Метод удаления Символ удаляемой группы
Каталитическое гидрирование OBzl, ONB. OPia, (OBzl (ОМе)], (OFmoc), (ОТегош)
Восстановление цинком в уксус- ной кислоте OPhac, OTcl, (OPlc)
Обработка NK^F— OTMSe, (OBzl)
Гидролиз или алкоголиз OTMS
Обработка при помощи NaaSjO* ONB
Обработка боргидридом натрия Комплекс e пецтааминокобальтом OBzh (NO2), 6N1
Облучение УФ-светом
183
иметь достаточно большой набор различных С-защитных групп,
удаляемых разными методами. В настоящее время число Су-
концевых защитных групп, отщепляемых в мягких условиях,
намного меньше набора группировок, используемых для бло-
кирования N-конца аминокислот и пептидов. Это объясняется
особенностями химического строения аминогрупп, а также
тем, что для избежания рацемизации наращивание полипеп-
тидной цепи обычно ведут с С-конца, то есть в условиях по-
вторяющегося деблокирования аминогруппы пептидов при
сохранении до определенного момента карбоксильной группы
в защищенном состоянии. Часто защитную карбоксильную
группу удаляют вместе с другими блокирующими группа-
ми на заключительной стадии синтеза пептидов.
В последние годы были предложены весьма лабильные С-
защитные группы, которые раньше нашли применение для
конструирования лабильных N-защитных групп уретанового
типа. Речь идет, например, о 2-триметилсилилэтиловых эфи-
рах, 9-флуоренилметиловых эфирах и 2-(1-адамантил)-пропан-
2-иловых эфирах. Возможно, что эти соединения в дальнейшем
найдут применение в синтезе пептидов.
В табл. 30 представлены формулы С-защитных групп, ис-
пользуемых в синтетических целях, а в табл. 31 эти группы
классифицированы по способам их удаления.
ОСНОВНЫЕ СПОСОБЫ ПОЛУЧЕНИЯ СЛОЖИ О ЭФ ИРНЫХ
С-ЗАЩИТНЫХ ГРУПП
Этерификация спиртами
в присутствии кислых катализаторов
Наиболее распространенным методом синтеза метиловых,
этиловых, бензиловых, n-нитробензиловых и других эфиров
аминокислот является этерификация аминокислот соответ-
ствующими спиртами в присутствии кислых катализаторов,
хлористого водорода, серной кислоты, п-толуолсульфокис-
лоты, бензолсульфокислоты*
г +он ОН п
II I
R—С-ОН R—С—ОН
L + J
О
II н+
R-C—он —
Г °н -1
R—С—ОН
-R1-Ан _
-нон г
О
' R—С=О+Н] нон ||
R— C-ORj + H.O,
LRi-O
184
Согласно общепринятому механизму реакции на первой ста-
дии протонируется карбоксильная группа; это повышает
чувствительность к нуклеофильному реагенту, т. е. к соот-
ветствующему спирту. Так как на последней стадии образо-
вания сложного эфира отщепляется молекула воды, то рав-
новесие можно сместить вправо, в сторону образования слож-
ного эфира путем удаления воды из сферы реакции, например
путем азеотропной отгонки; для этой цели обычно используют
большой избыток спирта.
Метиловые и этиловые эфиры аминокислот чаще всего по-
лучают по методу Фишера пропусканием безводного хлорис
того водорода через раствор аминокислоты в абсолютном
метаноле или этаноле. Более удобным является, однако, спо-
соб, согласно которому этерификацию осуществляют в при-
сутствии хлористого тионила [288] по схеме:
О
Z—NH—СН—СООН + СаНбОН Z~NH—CH-C-OCBH5 + HCL
I —20 G ,
R R
Предполагается, что реакция идет через образование проме-
жуточного эфира хлорсульфоновой кислоты
О
II
C8H6OH + SOCla C2HbO-S-Cl + HCI.
Синтез бензиловых, n-нитробензиловых, п-метоксибензи-
ловых сложных эфиров аминокислот проводят путем нагре-
вания суспензии аминокислот в бензоле или четыреххлорис-
том углероде с соответствующими спиртами в присутствии
сульфокислот, осуществляя одновременно азеотропную от-
гонку образующейся в процессе реакции воды.
Этерификация солей защищенных аминокислот
алкилгалогенидами
Взаимодействие солей защищенных аминокислот с алкил-
галогенидами наряду с описанным выше методом является об-
щим способом получения большинства эфиров защищенных
аминокислот:
Z—NH—СН—СООН Z—NH—СН—COOR. 4- HN+Et3 • X-.
| NEt3 । 11 3
R R
При использовании алкилхлоридов или алкилбромидов
для синтеза эфиров в качестве катализатора применяют иодид
натрия. Защищенные аминокислоты вводят в виде триэтилам-
монийных, натриевых или серебряных солей. Метод нашел
185
применение для получения n-нитробензиловых, п-метокси^
бензиловых, 4-пиколиновых, бензгидрильных и других эфиров
защищенных аминокислот.
Этерификация аминокислот эфирами
п-толуолсульфокислоты
Недавно был предложен метод мягкой этерификации ами-
нокислот (например, триптофана), лабильных в кислой среде.
В нем применяют сложные эфиры n-толуолсульфокислоты и
соответствующего спирта [308J; он оказался весьма удобным,
так как не требует предварительного получения алкилгало-
генидов и блокирования аминогруппы аминокислот!
R- ОН + H,c-/2^-SO,Cl
-> HsC-/_\-SO,R
To»H,NCHRtGOpH
Tos • H,N—CHRj—COOR.
Этерификация аминокислот алкенами
трет-Бутиловые эфиры и 2-(1-адамантил)-пропан-2-иловые
эфиры аминокислот получают взаимодействием аминокислот
и алкенов в присутствии кислых катализаторов:
О СН,
R-COOH + Н,С=С< R-С—О—С—СН,.
СН’ I
СН,
Как катализатор чаще всего используют серную кислоту»
mpem-бутиловые эфиры аминокислот можно синтезировать
также гидрогенолизом трет-бутиловых эфиров карбобензокси-
аминокислот (109): '
О О
II f H./Pd ' II t
Z—NH-CHR—С—OBu' HaN—CHR—C-OBiA
(109)
В. Ф. Позднее предложил новый метод синтеза соединений
типа (109) с весьма высоким выходом, заключающимся во
взаимодействии mpem-бутанола, защищенных аминокислот и
ди-/прет-бутилпирокарбоната [309]:
О
Z—NH-CHR—СООН ?°9.*^уН9?ц<-> Z—NH—CHR—С—ОВи',
(109)
Предполагается, что реакция идет через образование про-
межуточного симметричного ангидрида карбобензоксиамино-
кислоты.
186
Этерификация защищенных аминокислот
в присутствии карбодиимидов
В отдельных случаях для получения фенильных эфиров
аминокислот используют дициклогексилкарбодиимид [295].
Этот же реагент применяют для синтеза метиловых и этиловых
эфиров аминокислот в присутствии таких соединений, как
1-оксибензтриазол [310] или 4-диметиламинопиридин; для
этих же целей используют водорастворимый карбодиимид и
4-диметиламинопиридин. По-видимому, реакция идет через
образование 1-оксибензтриазиловых эфиров (ПО) или ацилпи-
ридиниевых солей (111), которые далее способны ацилировать
Спирты без рацемизации:
Другие методы получения сложноэфирной защиты карбо-
ксильных групп аминокислот — переэтерификация, метод
смешанных ангидридов, карбонилдиимидазольный, метод
диазоалканов — применяются довольно редко.
СВОЙСТВА ЗАЩИТНЫХ ГРУПП
И СПОСОБЫ ИХ УДАЛЕНИЯ
В главе 2 рассматривались свойства многих N-защитных
групп и способы их отщепления. Ввиду того, что защитные
группы уретановогб типа являются сложными эфирами кар-
баминовой кислоты (112), можно было бы ожидать, что они
способны расщепляться в условиях деблокирования сложно-
эфирных группировок (113).
о о
II И -OR, /°
R->0—С— NH-Rr* /С—NH—Ri R—С—ORj -----— -> R—C<f
H0/ |-CO, 0H
I
H 2N—Rj
(112) (113)
Эта аналогия, однако, не всегда правомочна. Так, Э. Фишер
в свое время пытался полностью деблокировать этиловый эфир
187
N-этилоксикарбонилглицина щелочным гидролизом, но проис-
ходило лишь деблокирование карбоксильной группы;
С2Н6О—С—NH—СН2—С—ОС2Н5 -22^-» С»Н6О—С—NH—CHj-COO-.
II II II
о о о
Устойчивость большинства защитных групп уретанового
типа при щелочном гидролизе метиловых, этиловых, бензи-
ловых и других эфиров широко используется в настоящее
время в химии пептидов для получения защищенных пепти-
Та блица 33. Условия деблокирования амино- и карбоксильных
групп пептидов
N-защитная группа С-защитная сложно- уретанового типа эфирная группа Условия удаления защитных групп
Бензилоксикарбониль- Бензиловый эфир ная трет-Бутилоксикарбо- /ире/и-Бутиловый эфир нильная Трихлорэтоксикарбо- Трихлорэтиловый эфир нильная п -Метоксибензилокси- п-Метоксибензиловый карбонильная эфир 1 -(1 -Адамантил)-1 -метил- 2-( 1 -Адамантил)-пропан- этоксикарбонильная 2-иловый эфир 2-Триметилсилилэтилок- 2-Триметилсилилэтило- сикарбонильная вый эфир 9-Флуоренилметилокси- 9-Флуоренилметиловый карбонильная эфир Каталитический гид’ рогенолиз над Pd ТФУ, 1 н. НС1 в ди- оксане Циук в уксусной кис- лоте Трифторуксусная кислота То же Т етр абутил аммоний фторид Диэтиламин, пипе- ридин
дов со свободной карбоксильной группой. В противополож-
ность щелочному гидролизу деблокирование пептидов кисло-
тами или каталитическим гидрогенолизом часто сопровож-
дается одновременным удалением как защитных групп урета-
нового типа, так и сложноэфирных группировок, это также
используется в синтетических целях.
В табл. 32 представлены некоторые N-защитные группы
уретанового типа и С-защитные сложноэфирные группировки,
которые удаляются в одинаковых условиях отщепления.
Данные, приведенные в табл. 39, свидетельствуют о том,
что для конструирования новых лабильных С-защитных слож-
ноэфирных групп можно использовать те же спирты, которые
применяются для создания лабильных N-защитных групп
уретанового типа. Следует, однако, подчеркнуть, что скорость
расщепления родственных С- и N-защитных * групп может
заметно различаться. Это часто используют для селективного
удаления тех или иных защитных группировок. Например,
Вос-группу удается селективно отщепить при наличии трет-
188
бутиловой сложноэфирной группировки (см. главу 2). Точно
так же при строго контролируемых условиях каталитического
гидрогенолиза удается избирательно деблокировать N-конец
пептида, защищенный карбобензоксигруппой при наличии
на С-конце соединения сложноэфирной бензиловой группиров-
ки, так как Кбз-группа при каталитическом отщеплении уда-
ляется быстрее, чем происходит расщепление бензиловых
эфиров. Таким образом при конструировании новых С-защит-
ных групп по аналогии с известными аналогичными группами
уретанового типа следует иметь в виду, что условия их дебло-
кирования могут различаться весьма значительно. Условия
деблокирования защитных групп боковых цепей тирозина, се-
рина, треонина и гистидина также сильно отличаются от ус-
ловий, при которых осуществляется удаление соответствую-
щих группировок с N- или С-конца пептидов (более подрбб-
но см. в главе 4).
Защитные группы, удаляемые основаниями
Среди С-концевых защитных групп, удаляемых основания-
ми, прежде всего следует рассмотреть метиловые и этиловые
эфиры пептидов, препаративным способом расщепления ко-
торых является щелочной гидролиз. Эти эфиры, предложен-
ные Курциусом и Гебелем в 1883 г. [287], сыграли большую
роль в химии пептидов, что прежде всего связано с легкостью
их получения, простотой расщепления, а также устойчиво-
стью при ацидолизе других защитных группировок. При об-
обработке эфиров пептидов раствором аммиака в спирте они
превращаются в соответствующие амиды; взаимодействие
эфиров пептидов с гидразингидратом дает гидразиды:
О
о Н2О R_c_NHNH,
R—С—ОСН, — ' О
Омыление метиловых и этиловых эфиров проводят при
комнатной температуре в водно-спиртовых растворах едкого
натра или в смеси раствора NaOH с такими органическими
растворителями, как ацетон, метанол, диоксан. Предпола-
гается, что реакция идет по следующему механизму:
О 0-0 0
II он- I II II
R—С—ОСН3 ~R—С—ОСН, R—c+0CHf R-С + СН,ОН
(!>н dn d-
189
В указанных условиях расщепляются также бензиловые, п-
нитробензиловые, бензгидриловые, 4-пи кол иновые, • 2-три-
хлорэтиловые и другие сложные эфиры пептидов, хотя щелоч-
ное омыление не является основным способом их расщепле-
ния. Фениловые эфиры (114) гидролизуются в жестких усло-
виях при pH 11. Кеннер с соавт. [295] разработали мягкий
способ расщепления этих соединений — омыление в водно-
ацетоновой среде щелочью и перекисью водорода в течение
20 мин; рацемизация при этом незначительна.
О О
II II
R-C—OPh + ООН” -> R—С—ООН + Ph О™
(114) (115) Н2О (116) (117)
О
II
R—С— О' + ООН"
(118)
Образующийся в щелочной среде перекисный анион (115)
является сильным нуклеофилом и расщепляет фениловый
эфир с образованием перкарбоновой кислоты (116) и фенолят-
аниона (117). Соединение (116) легко разлагается с образо-
ванием аниона кислоты (118), перекисного аниона и фенола.
Фениловые эфиры были успешно использованы группой Кен-
нера для синтеза последовательности 1 —129 лизоцима [312].
Некоторые С-защитные группы омыляются щелочами в
очень мягких условиях. Так, 2-п-(толуолсульфо)этильная
группа, предложенная Миллером с соавт. в 1968 г. [313], рас-
щепляется водным раствором карбоната натрия за 2 ч. или 1 н.
едким натром за 3 мин. Эта группировка относится к защит-
ным группам, представляющим собой 0-замещенные этило-
вые эфиры. В щелочной среде идет отрыв протона от Р-угле-
родного атома, который активирован электроотрицательным
заместителем. В результате Р-элиминирования образующийся
карбкатион (119) высвобождает свободную карбоксильную
группу:
О
II /-\
R—C-OCH2CH2-SO2—s
он----н+ R-COO-+CH2=CH-S02-^2/-CH»
б
R—С—О—СН2 —СН —SO2— — сн8
(119)
190
ПсГаналогичному механизму идет расщепление 9-флуоренил-
метиловых эфиров, введенных в практику пептидного синтеза
недавно Боданским с соавт. [300] и Кесслером с соавт. [ЗОН.
Соединения (133) обычно получают переэтерификацией о-нит-
рофениловых эфиров в присутствии 4-диметиламинопириди-
на [301].
R-eo-ONp
ЯСС, ДМАР
--------
R-COOH
9-Флуоренилметиловые эфиры устойчивы в кислой среде,
но частично расщепляются при гидрировании; удаление С-
защитной группы этих соединений проводят диэтиламином
или пиперидином в ДМФА или хлористом метилене в течение
1 —2 ч. Механизм расщепления аналогичен механизму удале-
ния Fmoc-группы (см. главу 2):
Очень интересной С-защитной группой пептидов является
предложенная Кемпом с соавт. в 1981 г. [303] 2-(трифторме-
тил)-6-хромонилметильная (Terom) (122). Ее вводят в амино-
кислоты, действуя на соответствующие цезиевые соли 2-(три-
фторметил)-6-бромметилхромоном:
Эта группа довольно легко отщепляется пропиламином или
разбавленным спиртовым раствором гидразина и устойчива
к действию эфиров аминокислот и кислых реагентов. Она
инертна также к вторичным аминам, поэтому ее можно селек-
тивно удалять в присутствии Fmoc-группы. Легкость разры-
ва хромонового кольца при действии первичных аминов
191
способствует реакции отщепления данной группы:
(122) (123)
Промежуточное соединение (121) подвергается реакции
элиминирования с образованием свободной кислоты и Хино-
на (122), который действием избытка пропиламина превра-
щается в соединение (123), Тсгот-группа устойчива в усло-
виях удаления Вос-, Fmoc- и других защитных групп.
Используя Tcrom-группу, авторы успешно синтезировали
Met-энкефалин [303].
Защитные группы, удаляемые кислотами
Как известно, метиловые, этиловые, бензиловые и дру-
гие сложноэфирные группы расщепляются при действии силь-
ных кислот. Однако жесткие условия реакции не позволяют
использовать этот способ деблокирования в практике пептид-
ного синтеза. Первой С-защитной кислотолабильной группой,
которая нашла весьма широкое применение в синтезе пепти-
дов, является mpem-бутильная группа, предложенная Реш-
ке в 1963 году [291]. трет-Бутиловые эфиры устойчивы в ще-
лочной среде и при гидрогенолизе, но чрезвычайно лабильны
в трифторуксусной кислоте и других сильных кислотах. Лег-
кость кислотного расщепления этих эфиров можно объяснить
образованием стабильного mpem-бутильного катиона:
о сн. он сн3 он сн.
II I н+, || 11+
R—С—О—С—СНа 7LZ1R—с—О—С— CHS?±R—с=о+сн8—С -и
<!н, <к (!н3
192
бутиловые эфиры устойчивы в условиях отщепления
тритильной, Врос-, Adpoc-rpynn; их можно расщеплять в
присутствии большинства защитных групп. При удалении
таких групп, как Вос-, Аос-, Adoc- mpem-бутильная слож-
ноэфирная группировка также отщепляется.
Устойчивые катионы образуются и при расщеплении слож-
ных эфиров аминокислот со вторичными спиртами. Это обстоя-
тельство позволило синтезировать безгидриловые эфиры (126),
лабильные в кислой среде [2971.
О Ph
н+ И /
HaN—CHR—СООН + (Ph)a СНОН —— -> H2N—CHR—С—О—-СН
он v
xPh
(126)
При введении в n-положение метаксильной электронодо-
норной группы устойчивость карбокатиона типа (127) возрас-
тает.
*-^-^-с+н2
(127)
Это облегчает ацидолиз n-метоксибензиловых эфиров по сра-
внению с бензиловыми. Эта группа была описана Вейгандом
и Хайгером в 1962 г. и применялась для синтеза пептидов [292].
Еще более кислотолабильную группу — пентаметилбензиль-
ную — предложил Стюарт в 1968 г. [304]. Она удаляется три-
фторуксусной кислотой при 20 °C в течение 2 мин:
О СНЗК ,СН3
II 3\_/
Z-NH—СН—С—О-СН2—/ S—СН.. ..Т£Л-»
I /“<
R СН/ ХСН3
О СН3\ ZCH,
-^Z-HN—СН—С—ОН+ н8с—сн2он
СН3/ XCHS
Недавно Ф. К. Мутулис и соавт. [302] исследовали весьма
лабильную 2-(адамантил)-пропан-2-иловую защитную груп-
пу (128), которую получали по схеме!
R-COOH
13 2-882
193
Эта группа удаляется в условиях, аналогичных отщеплению
Adpoc-защиты, т. е. может деблокироваться в присутствии
Вос-группы и mpcm-бутиловых эфиров.
Защитные группы, удаляемые восстановлением
Каталитическое гидрирование. Возможность удаления
бензильных и n-нитробензильных защитных групп гидриро-
ванием над палладиевым катализатором способствовала их
широкому применению для синтеза пептидов. Одним из про-
дуктов гидрогенолиза n-нитробензиловых эфиров является
ацетат п-толуидина (115), от которого можно легко осв’обо-
диться в вакууме:
О
R_C_OCHs-^2^-NOs R—СООН + H3C-<^>-NH2 •
. АсОН. ~
Поскольку при каталитическом гидрировании отщепляют-
ся многие другие защитные группы (см. табл. 14), данный спо--
соб деблокирования С-концевой группы пептидов весьма удо-
бен для одновременного удаления всех защитных групп на
последней стадии синтеза.
Кроме газообразного водорода для каталитического гидро-
генолиза нашли применение различные реагенты — доноры
водорода: циклогексен, циклогексадиен, муравьиная кислота,
формиат аммония и другие соединения.
Восстановление цинком в уксусной кислоте. Ряд защит-
ных групп удается «снять» действием цинка в уксусной кис-
лоте. К таким соединениям относятся 4-пиколиновые эфиры,
2-трихлорэтиловые эфиры, фенациловые эфиры. Предпола-
гается, что атом цинка выступает как источник электронов,
вызывая согласованное элиминирование блокирующих групп:
О С! **4 О
R_C-(PcH2-XCCb —R-C-OH + СН3-СС13 .
Восстановительное расщепление w-нитробензиловых эфи-
ров. Этот интересный способ селективного удаления п-нитро-
бензиловой группы был предложен в 1982 г. [314]. При дей-
ствии избытка дитионата натрия на n-нитробензиловые эфи-
ры аминокислот идет избирательное расщепление этих эфи-
ров:
0 °
r-c-och2-^^>—no2 - Na^£4-> r-c-o~+
+ HOCH2—nh2.
194
В этих условиях стабильны карбобензоксигруппа, Вос-груп-
па, бензильная, нитрогруппа, блокирующая гуанидиновую
функцию аргинина, метиловые и этиловые эфиры пептидов.
Метод может оказаться полезным, так как он является прак-
тически единственным способом удаления п-нитробензольной
группы в присутствии бензильной защиты.
Другие пособы удаления защитных групп
Кроме рассмотренных общих способов удаления С-защит-
ных групп, существует ряд специальных методов отщепления
редко используемых защитных группировок. Среди мало при-
меняющихся, но широко известных групп, блокирующих С-
конец пептидов, следует прежде всего назвать триметилси-
лильную группу, предложенную Биркофером и Риттером в
1960 г. [298]. Эти авторы показали, что при действии гекса-
метилсилазана на аминокислоты образуются О, N-триметил-
силиламинокислоты (129):
H2N—СН—СООН + Me3Si—NH—SiMe3
I
R
О
II
-> r—CH—С—О—SiMe3H-NH3.
HN—SiMe3
(129)
Триметилсилильная группа легко гидролизуется при дей-
ствии воды и спиртов:
О
II
Me3Si —NH—-СН— С—О—Si Меа
| 2RtOH | Н2О
H2N— СН—СООН + 2Me3SiOR1 H2N— СН—СООН +
R R
+ Me3Si—О—SiMe3
Крайняя лабильность ТМС-группы очень затрудняет работу
с ней. О, N—ТМС-аминокислоты получаются действием
триметилхлорсилана на натриевые соли аминокислот [315]:
О
H2N— CHR—COONa + 2Me3SiCl -> Me3Si—NH—CHR—C—OSiMe3.
Как оказалось, благодаря достаточно высокой прочности
связи Si—О можно избирательно отщеплять N-защитную
13:
195
группу и получать О—ТМС-эфиры аминокислот в свободном
виде (130) или в виде их хлоргидратов (131):
О
о > НС1 • H2N-CHR—C-OTMS + Me3SiCl
II (130)
TMS-NH-CH-C-OTMS- 0
R H2N—CHR—C—OTMS +
+ Me3SiOSiMe3
(131)
Позднее Биркофер и соавт. [316] показали, что ТМС-груп-
па не препятствует атаке аминогруппы ацилирующими аген-
тами. Это обстоятельство авторы использовали для синтеза
дипептидов методом смешанных ангидридов:
0
Z-NH-CHRi—С< || _со
г u п г/® + Me3Si—NH—CHR2—С—OSiMe3 -c2H6O-SiMea~*
о
II н20
Z—NH-CH-CONH—СН—С—OSiMe3 —>
Ri R3
^Z—NH—CH—CONH—СН—СООН.
Ri R2
Применение O,N — ТМС-аминокислот в качестве аминоком-
понентов очень удобно, так как в отличие от аминокислот
указанные производные хорошо растворимы в растворителях,
применяемых для синтеза пептидов, а С-защитная ТМС-группа
легко удаляется при промывании продукта реакции раство-
рами кислот. Е. П. Крысин и соавт. [317] показали, что при
этом вместо смешанных ангидридов могут использоваться
N-оксисукцинимидные эфиры. В этом случае бнс(О,П-три-
метилсилил)-аминокислоты получают действием 6hc(O,N-
триметилсилил)ацетамида (146) на суспензию J аминокислот
в сухом хлористом метилене:
О—SiMe3
HjN—CHR—СООН + 2СН3—C=N—SiMe3 —
(146)
О
О
Me3Sl—HN—СН—С—О—SiMe, + 2СН3—С—NH-SiMe
196
При этом отпадает необходимость очищать триметилсилили-
рованные аминокислоты перегонкой в вакууме. В обзоре
С. М. Андреева с соавт. [318] детально рассмотрено использо-
вание ТМС-группы в пептидном синтезе.
Перспективной, по нашему мнению, является 2-триметил-
силилэтильная группа, введенная в практику пептидного
синтеза Зибером с соавт. [299] в 1977г. Соответствующие
эфиры получают по схеме:
О
R—СООН + НО—СН2СН2—SiMe3 £ —R-C—ОСН2СН2—SiMe3.
TMSE-группа удаляется тетраметил- или тетрабутиламмоний
фторидом в ДМФА при 20 °C в течение 10—60 мин:
° + _ °
R—С-О-СН2СН2—SiMe3 -» R—С—0“ + СН2 == СН2 +
+ Me3Si—F.
Она также медленно расщепляется хлористым водородом в
органических растворителях и едким натром при деблокиро-
вании метиловых эфиров.
В 1982 г. Изайд с соавт. [307] предложили оригинальную
нековалентную С-защитную группу. Блокирование карбок-
сильной группы достигается здесь .за счет комплексообразо-
вания с пентааминокобальтом, который получают по схеме:
l(NH3)B СООНа]3+ + H2N-CHR-COO” ->
О
II +з
[HoN-CHR-C—ОСо (NH3)61+ ,
(АКСо)
Ступенчатый синтез пептидов осуществляли по схеме:
Boc-(AK)2-0->~^-x + [АК1-Со] ]Вос-АКа—AKi-Co]
[Н—АК2—AKj—Со] Е**^*-» [Вос-АК3—АКа—
-АКх-Со] "J&f» Вос-АКз-АКх-АКх.
Новая защитная группа устойчива в условиях удаления
Вос-группы и отщепляется действием боргидрида натрия или
гидросульфидом натрия.
Из других экзотических способов защиты и последующего
удаления С-защитных групп следует отметить фотолабильную
о-нитробензгидрильную группу, которая отщепляется при
освещении УФ-светом в бензоле или четыреххлористом угле-
197
роде [306]:
О
R—СООН + \ С—ч7~/
\оа
ПРЕПАРАТИВНАЯ ЧАСТЬ
Метиловые и этиловые эфиры аминокислот
Методика 170. Этерификация аминокислот в при-
сутствии хлористого водорода [287].
В суспензию 10 г аминокислоты в 150 мл абсолютного спир-
та, находящуюся в колбе, снабженной обратным холодиль-
ником с хлоркальциевой трубкой, пропускают без внешнего
охлаждения ток сухого хлористого водорода. Реакционная
смесь при этом разогревается; когда вся. аминокислота рас-
творится, колбу помещают в баню со льдом и при температуре
0—5 °C продолжают пропускать хлористый водород до насы-
щения. Реакционную смесь выдерживают 3—4 ч при комнат-
ной температуре. Если выпадает осадок, то колбу оставляют на
ночь в холодильнике, затем осадок отфильтровывают, промыва-
ют холодным спиртом, эфиром и сушат в вакуум-эксикаторе над
едким кали. Если продукт растворим и никакого осадка не
образуется, то спирт удаляют на роторном испарителе, а ос-
таток растирают с эфиром, аморфный продукт отфильтровы-
вают и выдерживают в вакуум-эксикаторе над едким кали.
Хлоргидраты эфиров аминокислот обычно хорошо крис-
талл изуютря из смеси соответствующего спирта и эфира. При
этерификации глутаминовой и аспарагиновой кислот, а та^-
же при получении сложных эфиров лизина пропускание хло-
ристого водорода ведут при кипячении спиртового раствора
аминокислоты в течение 1—3 ч и последующим выдержива-
нием смеси в течение 2 ч при комнатной температуре (табл. 34).
Методика 171. Этерификация аминокислот действием
хлористого тионила в спирте [288]
H2N-CHR~COOH + СН3ОН + SOC12
О
НС1 . H2N—CHR—СООН + СН3О—S—С1 ->
о
II
-> НС1 . H2N—CHR—С—ОСН3 + so2.
198
Таблица 34. Свойства хлоргидратов метиловых эфиров аминокислот
Аминокислота Температура плавления, °C [ос]д, град
L-Аланин 110—111 +5,8 (с — 3; метанол) L-Аргинин, 2 НС! 196 +21,7 (с = 2,5; МеОН) <о-Нитро-Ь-аргинин 159—161 +17,5 (с = 3; метанол) L-Аспарагин а, (З-Диметиловый эфир 116—117 — L-аспарагиновой кислоты р-Метиловый эфир Г-аспара- 191—193 +21,4 (с~ 1; этанолвода, гиновой кислоты 1 : 3) L-Валин 175 +16,0 (с ~ 2; вода) L-Глутамин 145—147 L-Гистидин. 2 НС1 199—202 +10,2 (с~ 2; вода) М1т-Бензил-£-гистидин • 111—115 — . 2 НС! у-Мет иловый эфир L-глута- 167 — миновой кислоты Глицин 175 — L-Изолейцин 100—101 +26,6 (с = 2; вода) £-Лейцип 150—151 —13,4 (с = 5; вода) L-Лизин, 2НС1 212 — №-Карбобепзокси-£-лизин- 117 +16,7 (с = 2; вода) И8-Бос-£-лизин 158—159 +19,0 (с™ 1; метанол) /.-Метионин 150 -j-26,8 (с = 5; вода) L-Оксипполин 162—164 разл. — Г-Пролип 71 ' —34,0 (с ~ 1; вода) £-Серин 165—166 +0,9 (с — 6; вода) (0-Бензил)-£-серин * 169 —2,7 (с == 3; вода) L-Тирозин 190 +74,3 (с = 3; пиридин) (0-Бснзнл)-£-тирозин 18'1 +11,5 (с = 4; метанол) L-Треонин (основание) 63—65 —21,0 (с = 1; 1 н. НС1) £-Триптофан 216 +17,7 (с — 2; метанол) £-Фенилаланин 71 —4,6 (с — 5; вода) £-Цистеин 140 —2,9 (с == 10; МеОН) (5-Бензил)-£-цистеин 150 —13,9 (с =2,9; вода)
В коническую колбу, снабженную магнитной мешалкой
и пробкой с хлоркальциевой трубкой, помещают 30 мл абсо-
лютного спирта и охлаждают его до 20 °C смесью сухого
льда и ацетона; затем по каплям добавляют 1,45 мл (20 ммоль)
перегнанного тионилхлорида и вносят 10 ммоль аминокисло-
ты. Суспензию перемешивают 2 ч при —5 °C, затем темпера-
туру реакционной смеси медленно поднимают до 40 °C и смесь
выдерживают при этой температуре еще 2 ч. По мере этери-
фикации аминокислоты осадок растворяется. Растворитель
удаляют на роторном испарителе; эту операцию повторяют
дважды, каждый раз добавляя 30 мд спирта, наконец, оста-
ток растирают с сухим эфиром. Этот метод получения эфиров
аминокислот очень удобен и непродолжителен. Он особенно
199
Таблица 35. Свойства хлоргидратов этиловых эфиров аминокислот
Аминокислота Температура плавления, °C [а]^, град
L-Аланин 77—78 +8,1 (с = 1; вода)
а, 0-Диметиловый эфир L-аспарагиновой кислоты 109—110 +8,1 (с= 1; вода)
P-Этиловый эфир L-acna- рагиновой кислоты 199—200 —
L-Валин 102—104 +6,7 (с= 2; вода)
Глицин 144 —
у-Этиловый эфир L-глута- 134—135
миновой кислоты (основа-
ние)
а, у-Диэтиловый эфир L- 116—118
глутаминовой кислоты (ос- нование) L-Лейцин 134 + 18,4 (с — 5; этиловый спирт) + 11,7 (с= 2,5; этанол)
L-Лизин 143,5—144,5
1Ч8‘Карбобензокси-£-лизин 115,5—116,5 + 12,2 (с = 2; 0,1 н. НС1)
L-Метионин 81—82 + 18,7 (с= 2,2; этанол)
L-Оксипролин 147—148
L-Серин 130—131 —4,8 (с== 2; вода)
L-Тирозин 166 —
L-Триптофан 225—226
L-Фенилаланин 154—155 —7,6 (с= 3; вода)
L-Цистеин 115 —.
S-Бензил-£-цистеин 165—166 —4,2 (с = 5; метанол)
пригоден для этерификации тех аминокислот, которые неус-
тойчивы в кислой среде, например триптофана и метионина.
Свойства хлоргидратов этиловых эфиров аминокислот пред-
ставлены в табл. 35.
Методика 172. Хлоргидрат метилового эфира трип-
тофана [319].
К 300 мл абсолютного метанола, охлажденного до —10 °C,
прибавляют по каплям 22 мл (31 ммоль) свежеперегнанного
тионилхлорида и затем при размешивании быстро вносят 30 р
(14,7 ммоль) L-триптофана. Аминокислота при непрерывном
размешивании в течение 2 ч при— 10 °C постепенно рас-
творяется. Когда осадок растворится, смесь выдерживают 48 ч
при комнатной температуре и затем выливают при размеши-
вании в 1 л сухого эфира. Хлоргидрат метилового эфира трип-
тофана медленно кристаллизуется. Продукт отфильтровывают,
промывают эфиром. Выход 35,1 г (94 %), т. пл. 216 °C.
Омыление метиловых и этиловых эфиров пептидов
Метиловые и этиловые эфиры пептидов обычно омыляют
водным раствором едкого натра в смеси с органическими рас-
творителями- метанолом, этиловым спиртом, диоксаном или
200
ацетоном. Процесс ведут при комнатной температуре в тече-
ние 1—2 ч; иногда время реакции может достигать 20 ч. Рас-
щепление сложных эфиров аминокислот и пептидов в кислой
среде, например, соляной кислотой в ацетоне в течение 1 <»
применяется редко.
Методика 173. тр^т-бутилоксикарбонил-фенилала-
нил-серил-триптофанил-глицин [320].
К раствору 3,13 г (5 ммоль) этилового эфира трет-буп-
локсикарбонил-фенилаланил-серил-триптофанил-глицина в
30 мл метанола прибавляют 3 мл 2 н. раствора едкого натра
(1, 2 эквивалента) и перемешивают при комнатной темпера-
туре в течение 1,5 ч. Затем растворитель упаривают при по-
ниженном давлении, а остаток разбавляют охлажденной во-
дой, подкисляют~лимонной кислотой до pH 3,0 и продукт
экстрагируют этилацетатом. Органическую фазу промывают
водой, сушат сульфатом магния, этилацетат упаривают до-
суха в вакууме. Остаток растворяют в этилацетате и продукт
осаждают петролейным эфиром. Выход аморфного пептида
2,8г (98 %), т. пл. 125—130°C; 1а]р = + 39,2° (с = 1; ДМФА).
Ниже представлено описание этерификации аминокислот
в мягких условиях. Преимуществом указанной методики яв-
ляется то, что при этом не наблюдается разрушения трипто-
фана и других кислотолабильных остатков аминокислот [248].
Методика 174. n-Толуолсульфонат этилового эфира
триптофана [309].
К раствору 2 г (10 ммоль) L-триптофана и 1,7 г (10 ммоль)
n-толуолсульфокислоты в 20 мл спирта добавляют при ком-,
натной температуре 2,3 г (12 ммоль) я-толуолсульфохлорида;
смесь затем нагревают до кипения и кипятят 1,5 ч. После это-
го растворитель удаляют в вакууме, а остаток растирают с
эфиром и продукт кристаллизуют из смеси этанол — эфир.
Выход 3,6 г (90 %), т. пл. 143 °C; [а]р =+ 16,6° (с = 2; эти-
ловый спирт).
Методика 175. Хлоргидрат бензилового эфира L-
триптофана [309].
Триптофан (10 ммоль), п-толуолсульфокислоту (10 ммоль)
и n-толуолсульфохлорид (12 ммоль) добавляют к 20 мл бен-
зилового спирта, и смесь нагревают в течение 1,5 ч при 80 °C.
Колбу затем охлаждают до комнатной температуры, содер-
жимое разбавляют 200 мл хлороформа и промывают 1 и. рас-
твором бикарбоната натрия. Хлороформ упаривают в вакуу-
ме до объема около 100 мл, добавляют 2 мл 7,5 н. НС1 в диок-
сане. Выпавший бесцветный осадок отфильтровывают из смеси
метанол — эфир. Выход 2,8 г (85 %); т. пл. 215 °C; [а]р ==
=а +5,4° (с = 2, метанол).
201
Рис. 5. Прибор для синтеза бензило-
вых эфиров аминокислот
Бензиловые эфиры аминокислот
Методика 176. п-Толуол-
сульфонат бензилового эфира
глицина [226].
В круглодонную колбу на
500 мл, снабженную насадкой
Дина — Старка и обратным холо-
дильником (рис. 5), помещают
18,8 г (0,25 ммоль) глицина,
48,5 г (255 ммоль) п-толуолсуль-
фокислоты, 100 мл бензилового
спирта и 50 ил бензола. Смесь
нагревают до кипения и кипя-
тят, осуществляя азеотропную
отгонку воды; когда соберут око-
ло 9 мл воды, реакционную смесь
охлаждают и добавляют к ней
0,5 л эфира, выдерживают 2 ч при
0 °C и осадок п-толуолсульфоната
бензилового эфира глицина от-
фильтровывают, промывают эфи-
ром и кристаллизуют из смеси
метанол —- эфир. Выход 50 г (90 %), т. пл. 132—134 °C.
В табл. 36 представлены свойства /г-толуолсульфонатов
бензиловых эфиров аминокислот.
Таблица 36. Свойства я-толуолсульфонатов бензиловых эфиров
аминокислот
Аминокислота Т. пл., °C 9 5 falp, град
Г-Аланин 116—118 —66,0 (с = 4; метанол)
о)-Нитро-£-аргинии
а, Р-Дибензиловый эфир Г-аспарагиновой кислоты 158—160 4-1,0 (с = 1; метанол)
Г-Валин 158—160 4-1,2 (с = 2; метанол)
Глицин 132—134 —
у-Бензиловый эфир Г-глу- 144—145 4-7,6 (с = 2; метанол)
таминовой кислоты
£-Изолейцин 153-154. —ОД (с — 2; метанол)
Z.-Лейцин 158,5—160 —1,7 (с — 2; метанол)
Г-Лизин (дитолизат) 147—149 —2,8 (с = 2; вода)
L -Оксипролин 107—109 —21,8 (с= 2; вода)
Г-Тирозин 179—180,5 —12,2 (с= 2; метанол)
L-Фенилаланин 170,5-171,5 —7,2 (с — 2; метанол)
(S-Бензил )-£-цистеин 162—163 —20,9 (с— 1; метанол)
202
Получение ^-бензилового эфира аспарагиновой кислоты и
^-бензилового эфира глутаминовой кислоты описано в главе 4.
В синтезе пептидов большое распространение получили
n-нитробензиловые эфиры аминокислот, которые имеют ряд
преимуществ перед соответствующими бензиловыми эфира-
ми: они устойчивы при обработке бромистым водородом в ле-
дяной уксусной кислоте, кроме того, n-нитробензиловые эфи-
ры аминокислот хорошо кристаллизуются.
Для расщепления их используют гидрогенолиз или щелоч-
ное омыление.
п -Нитробензиловые эфиры аминокислот
Методика 177. n-Нитробензиловые эфиры аминокис-
лот [260]
H,N—CHR—СООН 4- O2N—S—СН2ОН >
о
-> PhSO3H . H2N—CHR—С—ОСН2—NO2 + Н2О.
Суспензию 1,65 г (0,01 ммоль) L-фенилаланина, 2 г
(0,011 моль) бензолсульфокислоты и 2,1 г (0,014 моль) п-нитро-
бензилового спирта в 130 мл сухого хлороформа кипятят с об-
ратным холодильником в течение 15—20 ч. Конденсирующую-
ся жидкость перед возвращением в реакционную колбу про-
пускают через колонку с силикагелем или безводным сульфатом
кальция.
Спустя указанное время растворитель декантируют и ос-
таток растворяют в 95 %-ном ацетоне. При медленном добав-
лении к полученному раствору безводного эфира выпадают
кристаллы, которые очищают кристаллизацией из горячей
воды после обработки активным углем. Продукт отфильтро-
вывают и сушат в эксикаторе над Р2О5. Выход 2,93 г (65 %),
<г. пл. 190—191 °C; [ccJd + 11,0° (с = 4; ДМФА).
Время этерификации различных аминокислот обычно со-
ставляет от 1 ч до 2 дней. Выходы n-нитробензиловых эфиров
60—90 %.
Бромгидраты п-нитробензиловых эфиров аминокислот по-
лучают в два этапа: синтезируют n-нитробензиловые эфиры
карбобензоксиаминокислот, которые затем обрабатывают
бромистым водородом в уксусной кислоте:
а) Z—NH—CHR—COO- + С1СН2—^>— NOS ->
О
II Л-Х
Z-NH-CHR-C-OCH2-^ %-^NOi
203
б)
О
Z_NH—CHR—С—ОСН2—NO2 нвг/АсОн
о”
Таблица 37. Свойства
бензолсульфатов тг-нитробензиловых
эфиров аминокислот
Ам инокислота Т. пл., °C 95 град
L-Аланин 158—160 +7,1 (с~ «= 7,5; пи- ридин)
/.-Аспарагино- вая кислота (Ди-и-нитробен- зиловый эфир) 162—163 —8,4 (с = = 1; пи- ридин)
L-Валин 155 + 15,1 (с== == 1; пи- ридин
Глицин 191—192 —
L-Гистидин 92—95 —4,95 (с= — 3; пи- ридин)
L-Лейцин 213—215 + 16,7 = 1; пи- ридин)
№-Тозил-£-ли- зин 170—172 + 3,2 (с = = 4; ДМФА)
L-Сер ин 157—158 — 14,4 (с= = 1; пи- ридин) + 15,2 (с= = 1; пи- ридин)
L-Тирозин 218—219
/.-Фенилаланин 190—191 + 11,0 (с== = 4; ДМФА)
S-Бензил-/.- цистеин 170—171 —20,5 = 1;
ДМФА)
Методика 178. п-Нитробензилхлорид [322]
С1СН2~\1/ ++' С1СН2-<?>"№2
В трехгорлую колбу,снабженную мешалкой, термометром
и капельной воронкой, помещают 617 мл (14,8 моль) концент-
рированной азотной кис-
лоты (d = 1,51). Колбу ох-
лаждают смесью сухого
льда и ацетона до — 15 °C,
и при перемешивании до-
бавляют по каплям 160 мл
(2,8 моль) ледяной уксус-
ной кислоты, поддерживая
температуру — 15 °C.
К полученной нитрирую-
щей смеси прибавляют
46 мл (0,4 моль) бензилхло-
рида, размешивают при
—15 °C в течение 30 мин и за-
тем смесь выливают на 1,5 кг
размельченного льда с та-
кой скоростью, чтобы тем-
пература не превышала 0 °C.
Выпавший желтоватый оса-
док отделяют фильтровани-
ем, промывают 100 мл ле-
дяной воды, кристаллизуют
из спирта. Получают 61,7 г
(90 %) п-нитробензилхлори-
да в виде бесцветного кри-
сталлического соединения с
т. пл. 72,6 °C.
Методика 179. Син-
тез n-нитробензиловых эфи-
ров карбобензоксиамино-
кислот [323].
Смесь 20 ммоль кар-
бобензо ксиаминокислоты,
30 ммоль /г-нитробензилхло-
рида (или бромида) и
204
30 ммоль триэтиламина в этилацетате кипятят с обратным хо-
лодильником в течение 12 ч. Полученный раствор охлаждают,
фильтруют и затем последовательно промывают водой, 1 н.
соляной кислоты, раствором хлористого натрия и сушат сульфа-
том магния. Этилацетат упаривают под уменьшенным давлени-
ем и остаток кристаллизуют из смеси этилацетат — гексан.
Если карбобензоксиаминокислота нерастворима в этил-
ацетате, то в качестве растворителя применяют диметилфор-
мамид.
Методика 180. n-Нитробензиловый эфир карбобен-
зокси-со-нитро-Л-аргинина [3241.
Смесь 15,5 г (44 моль) карбобензоксиаргинина, 9,3 г (71 ммоль)
триэтиламина, 14,3 г (71 ммоль) п-нитробензилбромида или
соответствующее количество n-нитробензилхлорида и 0,5 г
йодистого натрия в 70 мл сухого ацетона, кипятят в течение
7 ч в колбе с обратным холодильником. Соли отфильтровы-
вают, ацетон удаляют на роторном испарителе, остаток рас-
творяют в 80 мл этилацетата и 20 мл воды и последовательно
промывают 1 н. соляной кислотой, 1 н. бикарбонатом натрия,
снова водой, сушат сульфатом магния и, наконец, раствори-
тель отгоняют в вакууме. Остаток растворяют в минимальном
количестве этилацетата и выдерживают в течение ночи в хо-
лодильнике. Выпавшие кристаллы отфильтровывают, промы-.
вают холодным этилацетатом и сушат на воздухе. Выход
15 г (70 %), т. пл. 118 °C; [а]% = — 9,0° (с = 1; ДМФА).
В табл. 37 даны физико-химические характеристики п-нит-
робензиловых эфиров аминокислот.
трет -Бутиловые эфиры аминокислот
Методика 181. Синтез изобутилена [265]
СН3
I н+ Н8СХ
Н3С—С—ОН ------>С = СН2 + Н2О.
L„ в,с/
Сг13
Изобутилен можно легко получить путем дегидратации
трг/п-бутилового спирта действием кислот: серной, щавеле-
вой, бензолсульфокислоты или в присутствии сульфокатионов.
Небольшие количества изобутилена можно получить по
следующим методикам.
1. В колбе с обратным холодильником нагревают до кипе-
ния избыток /npem-бутилового спирта с небольшим коли-
чеством щавелевой кислоты. Изобутилен отводят через об-
ратный холодильник в приемник, охлаждаемый смесью сухого
льда и ацетона.
205
2. В двугорлый реакционный сосуд, снабженный обрат-
ным холодильником и капельной воронкой, помещают 50 г
кристаллической щавелевой кислоты, предварительно высу-
шенной при 100—105 °C. Реактор нагревают на водяной бане
до 60 °C, и затем по каплям прибавляют 100 г (127 мл) очищен-
ного mpem-бутилового спирта. Образующийся изобутилен
отводят через обратный холодильник в конденсатор, охлаж-
даемый сухим льдом и ацетоном. Температуру в бане посте-
пенно повышают до 100 °C. Выход изобутилена 75 г (количест-
венный).
Синтез трет -бутиловых эфиров
аминокислот
В качестве реакционного сосуда для получения трет-6у-
тиловых эфиров аминокислот удобно использовать бутылку
из-под шампанского. После закрывания ее пробкой и закреп-
ления пробки проволокой бутылку оборачивают полотенцем,
так как при достижении комнатной температуры внутреннее
давление может разорвать ее.
После проведения реакции бутылку перед удалениехМ проб-
ки необходимо осторожно охладить в бане с сухим льдом и
ацетоном.
Методика 182. mpe/n-Бутиловый эфир (З-бензиласпа-
рагиновой кислоты [291].
К охлажденному раствору 3 г (13 ммоль) Р-бензилового
эфира аспарагиновой кислоты в смеси 25 мл диоксана и 2,5 мл
концентрированной серной кислоты в толстостенном стеклян-
ном или металлическом сосуде добавляют 25 мл жидкого изо-
бутилена, сосуд закрывают и встряхивают при комнатной
температуре в течение 4 ч. Затем содержимое охлаждают до
— 40 °C и быстро выливают в холодную смесь 200 мл эфира и
125 мл 1 н. едкого натра; эфирный слой отделяют, а водный
экстрагируют эфиром. Эфирный раствор сушат сульфатом
натрия, растворитель удаляют на роторном испарителе до
объема 5 мл и прибавляют 25 мл эфира. При пропускании су-
хого хлористого водорода через полученный раствор обра-
зуется кристаллический осадок хлоргидрата т/?ет-бутилово-
го эфира (3-бензил-аспарагиновой кислоты. После кристалли-
зации из этилацетата выход продукта 3 г (73 %), т. пл. 115—
117 °C.
Другие характеристики соединения приведены в
табл. 38. Все mpem-бутиловые эфиры, формулы которых
указаны в этой таблице, синтезированы по описанной методи-
ке; исключение — трет-бутиловый эфир тирозина.
Методика 183. тргт-Бутиловый эфир L-тирозина [291]
206
Таблица 38. Свойства хлоргидратов трет-бучиловых эфиров
аминокислот
Аминокислота Выход, % Т. пл.. °C Т. кип. оснований, °C [а]р, град (0= 1; спирт)
Р-Бензиловый эфир 73 115—117 — +23,3
L-аспарагиновой кислоты
а-Бензиловый эфир 60 110—112 — —2,6
L-аспарагиновой кислоты
L-Валин 65 147—149 63 (1,25 мм рт. ст.)
а-Бензиловый эфир 67 124—126 — + 13,8
L-глутаминовой кислоты
Т-Бензиловый эфир 62 107—108 — + 16,4
L-глутаминовой кислоты
Глицин 136 30 (2 мм рт. ст.) —
L-Изолейцин 60 158—160 52 (0,45 мм рт. ст.) +30,9
L-Лейцин 62 166—167 45 (0,15 мм рт. ст.) + 12,4
№-Тозил^-лизин 72 136—138 — + 14,8
Ne-Kap6o6eH3OKCH-L- 147—149 — + 12,2
лизин
N® Boc-L-лизин * 139—140 — + 12,2
L-Пролин 27 110—112 57 (1,5 мм рт. ст.) —30,5
0-/лре/п-бутил4-се- рин *** 170—170,5 70—71 (0,9 мм рт. ст.) —6 **
L-Тирозин 45 143—145 —— +24
L-Фенилаланин 75 Разлаг. 107 (0,25 мм рт. ст.) +44,2
♦ В метаноле.
♦ * В диметилформ амиде.
• ♦ ♦ Получен по ме тодике [326].
В толстостенном стеклянном или металлическом сосуде
растворяют 3 г L-тирозина в смеси 25 мл диоксана и 6 г п-то-
луолсульфокислоты или 2,5 мл концентрированной серной
кислоты, раствор охлаждают до — 40 °C, прибавляют 25 мл
жидкого изобутилена, реактор закрывают и встряхивают 20 ч
при комнатной температуре. Затем бутылку открывают пос-
ле предварительного охлаждения и раствор выливают в хо-
лодную смесь 100 мл этил ацетата, 100 мл воды и 5 мл 5 н.
едкого натра, поддерживая pH 9,1. Продукт дважды экстраги-
руют этилацетатом, органическую фазу сушат сульфатом нат-
рия и после удаления растворителя в вакууме получают крис-
таллический осадок. Выход 1,8 г (45 %); т. пл. 140—143 °C.
Хлоргидрат mpem-бутилового эфира глицина по этому
способу получить не удалось.
207
Следует отметить, что mpem-бутиловые эфиры аминокис-
лот в вакууме перегоняются без разложения (см. табл. 38).
В работе Андерсена и Каллахана [327] изложен двухста-
дийный метод синтеза трет-бутиловых эфиров аминокислот,
заключающийся в обработке изобутиленом карбобензокси-
аминокислоты и последующем деблокировании аминогруппы
каталитическим гидрогенолизом над палладиевым катализа-
тором. Ю. А. Давыдович и соавт. [269] для синтеза mpem-бу-
тилового эфира глицина использовали в двухстадийном ме-
тоде трифторацетилглицин вместо КБЗ-производного.
Методика 184. трет-Бутиловый эфир глицина [328]
снзх
СН. QH-
TFA-GlyOH ----трр---»TFA—-GlyOBu*-----> Н—GlyOBu' .
. а) трет-Бутиловый эфир трифторацетилглицина. В толг
стостенный стеклянный сосуд помещают 140 г трифторацетил-
глицина в 200 мл диоксана и 10 мл концентрированной сер-
ной кислоты. Полученную смесь охлаждают до —30 °C, и
прибавляют 400 мл жидкого изобутилена. Сосуд закрывают
и встряхивают 12 ч при комнатной температуре до образова-
ния прозрачного раствора. Затем сосуд охлаждают, вскры-
вают, прибавляют 300 мл 1 н. едкого натра и продукт экстра-
гируют эфиром. Органическую фазу отделяют и сушат суль-
фатом магния, эфир упаривают в вакууме. Получают 162,9 г
(90 %) продукта с температурой кипения 86 °C (3 мм рт. ст.).
б) трет-Бутиловый эфир глицина.К 25,5 г mpem-бутило-
вого эфира N-трифтора цетил глицина прибавляют 600 мл 0,2 н.
едкого натра и перемешивают массу 10 мин при комнатной
температуре. Образовавшийся продукт экстрагируют эфи-
ром, органический слой сушат сульфатом магния, эфир упа-
ривают, а остаток перегоняют в вакууме. Выход 12,3 г (80 %)f
т. кип. 54 °C (12 мм рт. ст.).
Новый способ синтеза трет-бутиловых эфиров N-защи-
щенных аминокислот предложили Дхаон с соавт. [311]. Он
заключается в этерификации N-защищенных аминокислот
mpem-бутанолом посредством водорастворимого карбодиими-
да в присутствии катализатора — 4-диметиламинопиридина.
Методика 185. mpem-Бутиловые эфиры КБЗ-амино-
кислот.
Раствор 2,2 ммоль карбобензоксиаминокислоты, 0,2—
1,0 ммоль 4-диметиламинопиридина и 2,5 мл mpem-бутилового
спирта в 8 мл хлористого метилена охлаждают до 0 °C, и при
перемешивании добавляют 2,4 ммоль хлоргидрата 1-этил-З-
(3-диметиламинопропил) карбо диимида. Размешивание про-
должают 2 ч при 0 °C и затем, в течение ночи, — при комнат-
ной температуре. После удаления органического растворителя
208
в вакууме остаток растворяют в 25 мл этилацетата и 5 мл во-
ды. Органический слой отделяют и промывают насыщенным
раствором бикарбоната натрия (2 X 15 мл), водой (2 X 15 мл)
и сушат сульфатом натрия. Этилацетат удаляют на роторном
испарителе и продукт очищают на колонке с силикагелем-60
230—400 меш в смеси гексанацетон (9:1). Выходы эфиров
хорошие: Z—Ala—OBuz — 76 %; Z—ValOBu'— 79 %; Z—
Gly—ОВи? — 78 % и Z—Pro—OBuz — 88 %.
Триметилсилильные эфиры
аминокислот
Методика 186. N-Триметилсилилацетамид [329]
О
/Р CISiMe. И
CH3-C<f ....'• -» снз—С—NH—Si (СН3)3 + NEt3 . HCI.
NH2
59,0 108,6 131,1
К кипящей смеси 78 г (1,32 моль) сухого ацетамида, 108,6 г
(1,08 моль) сухого триэтиламина и 400 мл бензола, высушен-
ного над натрием, прибавляют при перемешивании в течение
1—1,5 ч 108,6 г (1,0 моль) триметилхлорсилана (через обрат-
ный холодильник), а затем еще нагревают с обратным холо-
дильником в течение 1 ч. Реакционную смесь охлаждают до
комнатной температуры, фильтруют и промывают бензолом
осадок солянокислого триэтиламина. Полученный коричне-
вый фильтрат упаривают в вакууме (12 мм рт. ст.), перего-
няют N-триметилсилилацетамид, который кипит при 84 °C
(13 мм рт, ст.). Выход продукта ИЗ г (90 %).
Примечание: все описанные выше операции необ-
ходимо производить при полном исключении попадания
влаги.
Методика 187. бш>(О,М-Триметилсилил)-ацетамид [330]
/О
СНз-С<
xnh2
59,0
ClSiMe3
Net3
сн3—C=N—-SiMe3
C)—-SiMe3
108,6 203,44
К смеси 295 г (5 моль) ацетамида и 2,7 л триэтиламина в
колбе на 12 л, снабженной мешалкой, обратным холодиль-
ником и капельной воронкой, добавляют при перемешивании
1460 г (1745 мл; 133 моль) триметилхлорсилана. Реакцион-
ную массу защищают от влаги воздуха при помощи трубки,
наполненной дриеритом.
14 2-882
209
После окончания прибавления всего триметилхлорсилана
смесь слабо кипятят от 8 до 15 ч. При сильном кипячении проис-
ходит сублимация хлоргидрата триэтиламина, и это ведет к
закупированию холодильника. Реакционную массу охлаж-
дают, фильтруют в токе сухого азота, осадок промывают на
фильтре сухим триэтиламином (500 мл), и фильтрат концент-
рируют в вакууме, нагревая жидкость не выше 100 °C. Оста-
ток (темная жидкость) фракционируют с дефлегматором в ва-
кууме. Выход основного продукта 813,6 г (80 %), т. кип. 71 —
73 °C (35 мм рт. ст.).
Побочный продукт — (М-триметилсилил)ацетамид — ки-
пит при 105—107 °C (35 мм рт. ст.) и застывает при комнатной
температуре в твердую массу.
Методика 188. Синтез /прет-бутилоксикарбонил-L-
валил-глицина [317].
Смесь 0,4 г (5,3 ммоль) глицина, 3 г (14,8 ммоль) бис-
(О,М-триметилсилил)ацетамила и 3 мл сухого хлористого мети-
лена интенсивно размешивают в плотно закрытой колбе при
комнатной температуре до полного растворения глицина. Од-
новременно проводят активацию карбоксильного компонента.
Для этого к раствору 1,1 г (5 ммоль) Boc-L-валина и 0,8 мл
триэтиламина в 5 мл сухого хлористого метилена, охлажден-
ному до —15 °C, добавляют 0,6 г (5,5 ммоль) этилхлорфор-
миата и смесь размешивают при указанной температуре
15 мин.
К полученному смешанному ангидриду прибавляют рас-
твор бис-(триметилсилил)глицина, и реакционную массу раз-
мешивают при —15 °C 90 мин. Затем растворитель удаляют
в вакууме, остаток растворяют в этилацетате, промывают вод-
ным раствором лимонной кислоты и потом водой. При этом
карбоксильная группа дипептида полностью деблокирует-
ся. Органический растворитель сушат сульфатом магния, и
эти л ацетат упаривают на роторном испарителе. Это дает
1,35 г (98 %) хроматографически чистого Boc-L-валил-гли-
цина.
Синтез можно вести также в безводном диметилформами-
де. Если для получения дипептида использовать N-оксисук-
цинимидный эфир Boc-L-валина, то реакцию ведут в течение
48 ч при комнатной температуре.
Амиды аминокислот и пептидов
Ввиду большого сходства в химическом отношении амид-
ной и пептидной связей амидирование карбоксильной групт
пы не может использоваться для ее временной защиты. Од-
210
нако амиды могут рассматриваться как С-защитные группы
в том случае, когда синтезируется пептид, С-концевая амино-
кислота которого является а-амидом, например природные
пептидные регуляторы окситоцин, вазопрессин и др.
В этом случае полипептид может быть получен в виде слож-
ного эфира с последующим его аммонолизом или использова-
нием амида С-концевой аминокислоты.
Амиды аминокислот обычно получают аммонолизом их
N-защищенных метиловых, этиловых или бензиловых эфиров
аммиаком в метаноле или этаноле. Однако для получения не-
замещенных и замещенных амидов используются методы, при-
меняемые для образования пептидной связи (хлорангидрид-
ный, смешанных ангидридов, карбодиимидный, фосфоразо-
метод, активированных эфиров и др.). Хорошие результаты
получены при использовании для этих целей ди-трет-бутил-
пирокарбоната [331].
Следует отметить, что N-замещенные амиды защищенных
аминокислот и пептидов имеют важное значение как субстра-
ты и ингибиторы ферментов. Широкое практическое приме-
нение.нашли так называемые хромогенные субстраты, напри-
мер n-нитроанилиды, Р-нафтиламиды и др., которые позво-
ляют определять активность протеолитических ферментов в
организме (см. обзор [332]).
Ниже приводятся некоторые методики получения амидов
аминокислот и пептидов.
Методика 189. Амид А-метионина [333].
Растворяли 20 г (0,1 моль) хлоргидрата метилового эфи-
ра L-метионина в 200 мл метанола, раствор насыщали аммиа-
ком при 0 °C и оставляли на 5 дней при 20 °C. Растворитель
упаривали досуха, а остаток растворяли в воде и обрабаты-
вали Амберлитом IRA-410 (ОН“-форма). Фильтрат после
ионообменной смолы упаривали, к остатку добавляли этанол
и бензол и опять упаривали досуха. Остаток дважды пере-
кристаллизовывали из смеси этилацетат — петролейный
эфир. Выход9,2 г (62 %), т. пл. 40—45 °C; [сс]^ = + 28° (с = 2;
95 % АсОН); [al2D5 — 28° (с = 2; ДМФА).
Удобная методика получения амидов N-защищенных амино-
кислот без использования сухого аммиака была предложена в
работе [334];
Методика 190. Синтез амидов /тгрет-бутилоксикар-
бонил- и бензилоксикарбониламинокислот [334].
К перемешиваемому раствору Вос- или Z-аминокислоты
(0,2 моль) в 200 мл ТГФ, охлажденному до — 45°, добавляли
22,2 мл (0,2 моль) N-метилморфолина и 26,3 мл (0,2 моль) изо-
бутилхлорформиата. Через 10 мин охлаждали реакционную
смесь до — 55 °C и добавляли 50 мл (0,7 моль) концентрирован-
14* 2П
кого аммиака. Реакционную смесь снова охлаждали до — 55 °C
и вновь добавляли такое же количество концентрированного
аммиака. Смесь перемешивали 5 ч при —15 °C. Способ вы-
деления амидов зависит от их растворимости.
Выделение Вос- Gly - NH2. Упаривали раствори-
тель, добавляли 800 мл этилацетата и отфильтровывали хлор-
гидрат N-метилфорфолина. Фильтрат промывали 100 мл КНСО3
(раствор КНСО3 представляет смесь равных объемов насы-
щенного раствора КНСО3 и насыщенного раствора NAC1),
100 мл 2 н. НС1 (2 н. раствор НС1, насыщенный NaCl) и
сушили органическую фазу безводным сульфатом натрия.
Этилацетат упаривали, и остаток кристаллизовали из сме-
си этилацетат — петролейный эфир. Выход 83 % т. пл. 98—
09, 5 °C;
Выделение Вос- PheNH2, В о с - V а 1 NH2, Z-
G 1 у NH2, Z-PehNH2. 400 мл насыщенного раствора
NaCl добавляли к реакционной смеси (в случае Z-Phe NHa
добавляли некоторое количество ТГФ, необходимое для рас-
творения осадка). Органический слой отделяли, промывали
40 мл КНСО3 и 2 X 40 мл 2 н. НС1 и отгоняли ТГФ. Остаток
растворяли в подходящем растворителе (если необходимо,
отфильтровывали NaCl) и перекристаллизовывали. В случае
Z-ValNHa добавляли 400 мл насыщенного раствора NaCl к
реакционной смеси, осадок амида отфильтровывали и крис-
таллизовывали.
Получены: Boc-PheNH2 (93 %), т. пл. 149,5—152 °C (ме-
танол— вода); Boc-ValNH2 (95 %), т. пл. 160,5—162°С (мета-
нол—вода); Z-GlyNH2 (86%), т. пл. 139—141° (хлороформ);
Z-PheNH2 (94 %), т. пл. 165—167°; Z-ValNH2 (98 %), т. пл.
211—212,5° (метанол).
Методика 191. Амид карбобензоксиглицина [335].
Добавляли хлорангидрид, полученный из 4,2 г (20 ммоль)
карбобензоксиглицина к 50 мл безводного эфира, предвари-
тельно насыщенного аммиаком. Выпадает белый осадок. Про-
пускали через смесь аммиак в течение 15 мин, оставляли на
ночь и отфильтровывали осадок. Этот материал экстрагирова-
ли 40 мл этил ацетата при кипячении, а остаток после экстрак-
ции экстрагировали в течение 7 ч этилацетатом в непрерывно
действующем экстракторе. Экстракты были объединены,
нагреты до кипения и фильтровались горячими. Фильтрат
упаривали до тех пор, пока при охлаждении не начиналась
кристаллизация продукта.
Дополнительное количество продукта было получено путем
добавления к фильтрату гексана. Выход 2,4 г (58 %), т. пл.
133—136 °C.
Характеристики амидов аминокислот указаны в табл. -39..
212
Таблица 39. Характеристики амидов аминокислот [22]
Аминокислота Т, пл., °с [а$. град Концентрация с, % (растворитель)
Аланин 71—72 +6 5,2 (вода)
Аланин • НС1 225 +11 1,24 (метанол)
Аргинин • 2НС1 • 2Н2О — + 13,8 1,0 (вода)
Аспарагин • НС1 214—215 —15,8 1,0 (вода)
Аспарагиновая кислота 212 + 15,5 1,5 (0,1 н. НС1)
Гистидин • 2НС1 260—261 +22,0 1,0 (вода)
Глицин 67—68 — —
Глицин • НС1 200—203 — —
Глицин • НВг 199—203 — —
Глицин • АсОН
Глутамин 98-100 — —
Глутамин • 2НВг 193—196 +20,1 1,0 (вода)
Глутаминовая кислота' 186—187 +21,2 6,5 (вода)
Глутаминовая кислота X 216—217 + 17,3 6,2 (вода)
X НВг
Изолейцин • НС1 246 +21,2 1,0 (вода)
Лейцин • НС1 244 + 10,1 2,0 (вода)
Лейцин • НВг 235—238 +8,7 5,0 (вода)
Лейцин • АсОН 125—126 +9,3 5,0 (вода)
Лизин • 2НВг 250—260 + 18,8 1,0 (вода)
Метионин 50—51 +22,5 2,0 (1 и. НС1)
Валин • НС1 268 +28,3 1,0 (вода)
Валин • НВг 242 + 22,4 2,0 (вода)
Валин • CF3COOH 124—125 —
Пролин 99
Пролин • НС1 182 —70,7 2,0 (этанол)
Серин 103 +24,6 3,0 (АсОН)
Серин • НС1 187—188 + 13,2 1,1 (вода)
Серин • НВг 131—132 + 11,0 1,0 (вода)
Треонин • НС1 230—233 +33,1 1,5 (метанол),
Триптофан 167—170 —7,9 2,0 (этанол)
Триптофан • НС1 257—260 +24,0 1,6 (вода)
Тирозин 153—154 + 19,2 4,1 (вода)
Тирозин • НС1 • 2Н2О • 238—239 +28,8 0,74 (80 % этанол)
Фенилаланин 93—94 +20,7 1,8 (вода)
Фенилаланин • АсОН 119—120 — —
Цистеин ♦ НС1 190—191 — —
Цистеин «НВг 178—180 +8,2 1,8 (вода)
213
Методика 192. Амид П-тритилглицил-(е-карбобенз-
окси)-£-лизил-Л-пролил-Л-валина [336].
Через раствор 15,9 г (20 ммоль) метилового эфира N-тритил-
глицил-(8-карбобензокси)-£-лизил-А-пролил-А-валина в 500 мл
безводного метанола пропускали аммиак при 0 °C. После
выдержки 10 ч при 20 °C раствор упаривали досуха и остаток
затирали с эфиром. После перекристаллизации из смеси ме-
танол—эфир получили 9,5 г (61 %) продукта, т. пл. ПО °C
(разл.); [аЙ = — 39,0 ± 0,5° (с = 2,6, метанол).
Ниже приводятся методики получения п-нитроанилидов
некоторых производных аминокислот для синтеза хромоген-
ных субстратов.
Методика 193. л-Нитранилид со-нитроаргинина [337J.
К раствору 20 ммоль со-нитро-А-аргинина и 20 ммоль л-
нитранилина в 20 мл абсолютного пиридина при охлаждении
вносили раствор 24 ммоля ДЦГК в 10 мл пиридина, выдер-
живали 24 ч, и получали л-нитроанилид со-нитроаргинина в
количестве 8 г (84,6%), т. пл. 186—191 °C, [а]р = — 2(с = 2;
эти л ацетат).
По аналогичной методике из хлоргидратов карбобензокси-
и Вос-аргинина получены л-нитроанилид карбобензоксиарги-
нина и Вос-аргинина (в виде хлоргидратов) с выходом соот-
ветственно 81 и 90 %.
Методика 194. л-Нитроанилид Ь1а-карбобензокси-
№-Вос-£-лизина.
1. Пара-нитрофенилизоцианат [338]
нс1 • h2n—no2 o2n—nco + знс!.
В 200 мл диоксана, содержащего 20 г л-нитроанилина, вво-
дят при комнатной температуре сухой НС1 до насыщения, а
затем пропускают в течение 1 ч при 50 °C фосген до образова-
ния прозрачного раствора. После удаления фосгена в течение
30 мин током сухого азота (в поглотительный сосуд с раство-
ром NaOH) раствор упаривают досуха при 40 °C, а остаток
кипятят с 120 мл сухого СС14 в течение 2,5 ч, фильтруют, упа-
ривают досуха и перекристаллизовывают из СС14. Выход 21,8 р
(92,4 %) т. пл. 56 °C.
2. л-Нитроанилид №-карбобензокси-№ЧЗос-£-лизина [339]
OeN-^r"4-NCO
Вос — Lys (Z) ОН --~&Et-----* Вос — ^У3 (2) — PNA.
/ а
7,6 г (20 ммоль) измельченного и высушенного Ы^-карбобенз-
окси-Ма-Вос-£-лизина растворяли в 50 мл свежеперегнанно-
214
го гексаметиленфосфотриамида при 20 °C, добавляли 20 ммоль
триэтиламина и 4,92 г (30 ммоль) м-нитрофенилизоцианата,
и выдерживали 24 ч при комнатной температуре. Выливали
раствор в 0,5 л 20 раствора NaHCO3, выпавший осадок про-
мывали на фильтре раствором NaHCO3, 0,5 н. НО, водой и
сушили. Продукт из осадка извлекали метанолом и перекрис-
таллизовывали из метанола. Выход 6,7 г (67 %), [а]р —
= — 8,5° (с = 1; метанол).
Методика 195. Хлоргидрат n-нитроанилина Nq-to-
зил-Л-аргинина [340]
Раствор 0,23 мл (2,5 ммоль) РС13 в 5 мл безводного пири-
дина, охлажденный до —20 °C, добавляли к раствору 0,7 г
(5 ммоль) n-нитроанилина при —20 °C. Выдерживали смесь
30 мин при комнатной температуре, вносили в нее раствор
1,85 г (5 ммоль) хлоргидрата тозил-£-аргинина в 10 мл гекса-
метиленфосфотриамида и оставляли на 3 дня при комнатной
температуре. Растворитель удаляли в вакууме, а оставшее-
ся масло растворяли в 15 мл изопропилового спирта и добав-
ляли 125 мл 1 М НО, пока раствор не начинал мутнеть. Вно-
сили в раствор затравку кристаллического хлоргидрата п-
нитроанилида Ма-тозил-£-аргинина и выдерживали 12 ч при
0 °C. Выпавшие кристаллы отфильтровывали и перекристал-
лизовывали из 65 мл 20 %-й уксусной кислоты. Охлаждали
в течение 12 ч при 0 °C, фильтровали и промывали неболь-
шим количеством холодной воды. Выход 0,9 г (37 %), т. пл.
238—242 °C; Nd = — 17,4° (с = 0,4; 50 %-я уксусная ки-
слота).
М е т о д и к а 196. п-Нитроанилид-£-фенилаланина [341].
1. Хлоргидрат хлорангидрида L-фенилаланина. Добавляли
21 г (0,1 моль) РС15 к 280 мл сухого СС14 при перемешивании.
Через 15 мин прибавляли 15 г (0,09 моля) L-фенилаланина,
оставляли перемешиваться 18 ч. Осадок отфильтровывали,
промывали сухим СС14, петролейным эфиром и сушили в
вакуум-эксикаторе над Р2О5. Выход 16,3 г (99%).
2. п-Ншпроанилид L-фенилаланина. Растворяли 14,9 г
(0,108 моля) n-нитроанилина в 100 мл сухого диоксана, со-
держащего 12 мл пиридина. К этой смеси при перемешивании
для защиты от влаги в течение 30 мин прибавляли 16,3 г
(0,074 моля) хлоргидрата хлорангидрида L-фенилаланина, и
оставляли смесь на 3 ч. Прибавляли 0,5 л воды, подкисляли
конц. НС1 до pH 2. Отфильтровывали желтый осадок и от-
брасывали. Маточный раствор доводили до pH 9 насыщенным
раствором карбоната натрия и оставляли на 24 ч в холодиль-
нике. Выпавший осадок отфильтровывали, промывали водой
и сушили. После перекристаллизации из этанола выход
15,4 г (71 %), т. пл. 160—161 °C.
215
Таблица 40. Примеры многофункциональных защитных групп
Название Обо- значе- ние Формула Дополнительные функции Примечания Лите- ратура
2-Фосфониоэтоксикар- Реос
бонильная
О
PhX »
Ph-)p+- СН.СН2—О—С—NHR
Ph/
Положительный за-
ряд. Способствует
растворимости в воде
Применяется для син- [187]
теза пептидов в водном
растворе
4-Триметиламмоний-
хлоридобензилокси-
*— карбонильная
О То же
4- /—к II
(Н3С) N— " у-СН2О—С—NHR
То же [343]
4-Сульфобензильная
О
R—C-OCH2—>-807
в X ______ / о
Отрицательный заряд
1. Хорошая раствори- [344]
мость защищенных пеп-
тидов в полярных раст-
ворителях позволяет
использовать для очи-
стки ионообменную
хроматографию
2. Деблокированные по
аминогруппе С-защи-
щенные пептиды обра-
зуют цвиттер-ионы, ко-
торые можно кристал-
лизовать из воды
217
Т р иэтиленг л ико льиз-
опропилоксикарбо-
нильная
4-Пиколильная
Тос
СН3 (ОСН2СН2)3 О—СН2СН2- сн3 1 ’ Повышенная гидро- фобность по сравне- Может использоваться [345] для повышения раство-
—С—-О— 1 нию с Вос-группой римости защищенных пептидов
0 II сн3 -
—C—NHR
4-Фенилазобензилок-
сикарбонильная
4-Диметиламиноазо-
бензольная
Полимерная бензиль-
ная группа
\_/-N= 0 =n-^)-ch2o-c—nhr Окрашенная группа Позволяет вести спект- рофотометрический кон- троль в процессе синте- за и очистки пептидов [346]
Pic о II R—C—OCH2— В кислой среде при- Используется как обретает положитель- «якорная» группа в пре- ний заряд цессе очистки ионооб- менной хроматографией 1298]
о R—C— OCH2—^-N= =N— NMe2 1. Окрашенная труп- Используется как ок- па рашенная «якорная» 2. В кислой среде группа в процессе очи- приобретает положи- стки пептидов тельный заряд 1347]
О ВОС—NH—CH—C—OHC2—у R Сополимер стирола и Используется в твердо- дивинилбе'нзола. Де- фазном синтезе пепти- —Рлает продукты пеп- дов с С-конца. Отделя- тидного синтеза нера- ется от реагентов створимыми фильтрацией [348]
218
Продолженис табл, -hl
Название Обо- значе- ние Формула Дополнительные функции Примечание Л ите- ратура
Полимерная карбо- бензокси-группа О Z-X 11 Р—" ^—СН2О—С—NH--CH- R —СООН То же Предложена для твер- дофазного синтеза пеп- тидов с N-конца. Прак- тического применения не нашла ввиду воз- можности рацемизации при этой схеме синтеза [349]
Полимерный алкило- вый эфир о II Z__NH—CH—с—ОСН2СН2 х R X (ОСЩОЩ),, Полиэтиленгликоль мол. массы 20 тыс. дальтон. Хорошо рас творим в метилен- хлориде и воде Используется в жидко- фазном синтезе пепти- дов в метиленхлориде Пептидил-полимер отде ляется от реагентов пу тем ультрафильтрации через мембрану в вод- ном растворе [350]
4-Диметиламинофе- ниловый эфир ° _ /СН3 R—С—О—N = хсн3 ;сн31 ? _ +/СНэ R—С—О—/ S—N—СНЯ = хсн3 Устойчивая С- защит- ная группа, которая после алкилирования СН31 приобретает за- ряд и превращается в активированный эфир Пример сложной фупк ции «защита — актива- ция» - [351]
Таблица 41, «Днуступенчатые» защитные группы
Название Обо- значе- ние Формула Модифицированная форма Модифици- рующий реагент Лите- ратура
2-(Метилтио)этильная R-C—OCH2CH2-S—сн3 II о + R—С—ОСН2СН2—S—СН3 • I"" II 1 о сн3 СН31 [284]
2-(Метилтио)этоксика р- бонильная Mtc О II Н3С—S—СН2СН2О—-С—-NHR о Н3С—S—СН2СН2О—с—NHR II II О О Н2О2 [352]
2-Бромэтильная О II R—С—ОСН2СН2В₽ О II + R—С—ОСН2СНа NEt3 • Вг~ Net3 [353]
1,3-Дитиан-2-ил)ме- — тильная <£> Dim О II /S-ч. R—С—ОСН2—/ 2 О Ьч /О II /s-\ R-C-OCH2-/ 2 0^0 Н2О2 [354]
2-Бромэтоксикарбониль- ная В ДОС о II ВгСН2СН2О—С—NHR о + II (Ph)3 Р— СН2СН2О—С— NHR Вг- Р (РЬ)з [355]
2-(Дифенилфосфонио)- этильная ДРРЕ О II /Ph R—С—ОСН2СН2—Р< xPh 0 Ph Il +zHn R—С—ОСН2СН2—Р—СН3 . 1“ XPh СН31 [356]
2-(4-Пиридил)этоксикар- бонильная 4— Руос /=\ О z^ch2ch2°~-c~nhr И-О-СИ ° - с-NHR 1~ СН31 [357]
Методика 197. n-Нитроанилид Boc-L-пролина [331L
Раствор 1,3 г (6 ммоль) Boc-L-пролина, 0,4 мл пиридина,
0,9 г (6,5 ммоль) n-нитроанилина и 1,5 мл (6,8 ммоль) ди-mpem-
бутил-пирокарбоната в 10 мл бензола перемешивали 20 ч, до-
бавляли 0,5 мл ди-трет-бутил-пирокарбоната и перемешива-
ли еще 2 ч, причем образовывался небольшой желтый осадок.
Раствор разбавляли этилацетатом до объема 50 мл, фильтро-
вали, и после стандартной обработки остаток кристаллизо-
вали из эфира. Выход 1,8 г (89 %); т. пл. 198—199 °C, [а]о =
= — 54,5° (с = 1; ДМФА).
Методика 198. 4-Метилкумарил-7-амид №-карбо-
бензокси-Ма-Вос-А-лизина [331].
К раствору 1,3 ммоль №-карбобензокси-№е-Вос-Л-лгзина>
0,1 мл пиридина и 0,35 мл (1,6 ммоль) ди-трет-бутил-пи-
рокарбоната в 5 мл тетрагидрофурана добавляли 290 мг
(1,6 моль) 4-метил-7-аминокумарина и перемешивали 20 ч.
Разбавляли 30 мл этилацетата, а затем после стандартной об-
работки кристаллизовали остаток из смеси бензола с эфиром.
Выход 0,3г (43 %) т. пл. 138-139° С; [a$ = — 9,1° (с = 1;
ДМФА).
СЛОЖНЫЕ ЗАЩИТНЫЕ ГРУППЫ
Среди многочисленных защитных групп химиками были
предложены группы, которые характеризуются более слож-
ным строением и назначением. Эти группы представляют не
только практический интерес, но являются наглядным при-
мером творческого мышления химиков. По-видимому, наи-
больший интерес представляют два типа защитных групп —
многофункциональные и «двуступенчатые».
Многофункциональные защитные
группы
Общей особенностью защитных групп является то, что
они являются временными группами, т. е. выполняют времен-
ные защитные функции и затем удаляются. Это позволяет
«навязывать» им некоторые дополнительные временные функ-
ции: изменение растворимости, молекулярной массы, заряда,
спектральных свойств, замену защитной функции на активиру-
ющую и другие, что ведет к упрощению синтеза и очистки
пептидов.
Частным случаем использования многофункциональных
защитных групп является твердофазный синтез пептидов
(см. главу 5).
220
Более подробно этот вопрос рассмотрен в обзоре [342], а
некоторые примеры многофункциональных групп приведены
в табл. 40.
«Двуступенчатые» защитные группы
Разработкой этих групп в последние годы эффективнэ за-
нимался немецкий ученый X. Кунц, который и дал им на-
звание «двуступенчатые». Эти группы устойчивы в процессе
пептидного синтеза и удаляются в мягких щелочных усло-
виях после химической модификации реакцией [3-элимиииро-
вания. Цель модификации — создание электрофильного цент-
ра на Р-углеродном атоме путем введения электроноакце-
пторной группировки, что способствует атаке его нуклеофи-
лом. Примеры «двуступенчатых» защитных групп приведены
в табл. 41.
Глава 4
ЗАЩИТА БОКОВЫХ
ФУНКЦИОНАЛЬНЫХ ГРУПП
В процессе образования пептидной связи необходимо за
щищать реакционноспособные боковые группы некоторых
аминокислот. К ним относятся: е-аминогруппа лизина, б-
аминогруппа орнитина, гуанидиновая группа аргинина, кар-
боксильные группы аспарагиновой и глутаминовой кислот,
гидроксильные и тиольные группы серина, треонина, тиро-
зина и цистеина, имидазольное кольцо гистидина. В некото-
рых случаях осуществляют блокирование боковых групп ме-
тионина, аспарагина и глутамина. Недавно предложены за-
щитные группы для индольного кольца триптофана.
В зависимости от природы конденсирующих реагентов и
тактики пептидного синтеза боковые группы указанных ами-
нокислот иногда не блокируют, однако следует помнить, что
защита реакционноспособных группировок аминокислот по-
зволяет избежать побочных реакций и способствует раствори-
мости пептидов в органических растворителях. Хорошим
примером синтеза весьма крупного пептида с использованием
тактики полной защиты является получение а-бунгаротокси-
на [358].
К боковым защитным группам предъявляют те же требо-
вания, что и для N- и С-защитных групп (см. главы 2 и 3). Ту
или иную защитную группу конкретной аминокислоты вы-
бирают таким образом, чтобы обеспечить возможность селек-
тивного удаления других группировок. Весьма привлекате-
лен такой план синтеза пептидов, когда боковые защитные
группы удаляются в конце синтеза одновременно с N- и С-
защитными группировками. Например, защитные группы
бензильного типа можно одновременно элиминировать ката-
литическим гидрогенолизом,, если в составе получаемого пеп-
тида нет серосодержащих аминокислот. Сложные и простые
znpe/n-бутиловые эфиры аминокислот в сочетании с Вос-груп-
пой можно одновременно отщеплять обработкой НС1 в диок-
сане или трифторуксусной кислотой.
222
Некоторые защитные группировки в процессе синтеза и
отщепления могут давать нежелательные продукты. В послед-
ние годы эти побочные реакции тщательно исследуются (см.,
например, обзор [359]); продолжается также интенсивный
поиск новых защитных групп, не имеющих нежелательных
процессов.
В табл. 42 приведены реагенты, используемые для защи-
ты боковых групп трифункциональных аминокислот, и усло-
вия их отщепления.
ЗАЩИТА БОКОВЫХ ФУНКЦИОНАЛЬНЫХ ГРУПП
ДИКАРБОНОВЫХ АМИНОКИСЛОТ
Для защиты р- и у-карбоксильных групп аспарагиновой
и глутаминовой кислот их превращают в сложные эфиры.
Часто для этой цели используют бензиловые и mpezn-бутило-
вые эфиры, трет-Бутиловые эфиры получаются по общей ме-
тодике (глава 3) из соответствующих бензиловых эфиров ас-
парагиновой или глутаминовой аминокислот с последующим
удалением бензильной группы каталитическим гидрированием
по схеме
О
H2N—СН—С—О—СН2—
сн2—соон
о
-> H2N—СН— С—О-СН2—-> H2N—СН—соон
СН2—С—OBi? СНа—С—ОВ1Л
о А
Если соответствующие аминокислоты являются С-концевыми,
то при синтезе пептида могут использоваться соответствующие
а-бензиловые эфиры с трет-бутилыюй защитой боковых кар-
боксильных групп. Если аспарагиновая или глутаминовая
кислота локализована в N-концевой части пептида, то их трет-
бутиловые эфиры переводят сначала в N-карбобензокси-, или
в N-Boc-, или другие подобные производные, а затем исполь-
зуют в синтезе. Следует подчеркнуть, что на С-конце синте-
зируемых олигопептидов удобно использовать дибензиловые
эфиры аспарагиновой и глутаминовой кислот, а на N-конце —
соответствующие N-ацильные производные [3- и у-бензиловых
эфиров (см. главу 2).
Для синтеза бензиловых эфиров боковых карбоксильных
групп также проводят избирательный гидролиз дибензиловых
223
Таблица 42. Защитные группы для
Аминокис- лота Защитная группа Обозначе- ние Формула защитной группы
Аминодикарбо
Глутамино- Бензиловые —OBzl
вая и аспа- эфиры
ратиновая
ислоты
п-Нитробензило- —ONB
вые эфиры
п-Хлорбензило-
вые эфиры
R—С—ОСН2—2
О
R—С—ОСН2—NO2
О
r_C—OCH2—С1
трет-Бу тило- —ОВи*
вые эфиры
Циклогексило- —ОсНех
вые эфиры
Диаминокар
Лизин (ор- Бензилоксикар- Z*-, Cbz-
нитин) * бонильная
R—NH—С— ОСН2—
II
трет-Бут ило к- Вос*-
сикарбрнильная
2,6-Дихлорбен- (С1)2—Z-
зилоксикарбо-
нильная
О сн3
li I
R—NH—-С—О—С—СН3
сн3
О С1
II >-ч
R—NH—С—ОН2С—
с/
4-Бром-бензил- (4-Br) Z-
оксикарбониль-
ная
Трифтор ацет иль- TFA*-
ная
О
II
R—NH~C~OH2C—^-Вг
О
II
r_NH-C-CF8
224
боковых функций аминокислот
Реагент для введения защитной группы Способ удаления Примежание Лите- ратура
новые кислоты
СН2ОН, н+
H2/Pd, ОН~„ Очень распростри-, [360];
(НВг/TFA), HF йена
О2Н-^~^-CHgOH, н+ H2/Pd, он— Применяется ред- ко [360]
ci—СН4Вг H2/Pd, ОН— Используются в (т/ф) * синтезе, бо- лее устойчивы, чем OBzl [362]
СН, >с=сн„ н+ сн/ TFA, HCI, CHsSO3H и др. Распространенная группа [361]
НО—+ ДМАР + Используются в (т/ф) синтезе, бо- лее устойчивы, чем OBzl [363]
+ ДЦГК
боновые кислоты
О \7/_СН.ОС-С1, он- H./Pd, , HF. CH8SO8H, HBr/AcOH Самая распрос-, траненная группа [364]
СН >с=сна, н+ сн/ н+ То же [365]
/С1 о снао—С-С1, ОН- [Ht/Pd, HF Используется в т/ф синтезе [366]
ХС1
о » То же [367]
Br-/ СН2О—C-G1.OH-
О II CF3—С—S—СН2СН3 OH—, NaBH, Устойчива в кис- лой среде [368, 273]
15 2 — 882
225
Аминокис- лота Защитная группа Обозначе- ние Формула защитной группы
Формильная НСО- (Form)- О 11 NH—С—H
Тозильная Tos*- R—NH—SO2—CH3
4-Метокси-2,3, 6-триметил бен- зол сульфон иль- ная MtS NH II —NH—C—NH— 0 CH3k /СН. . —s z 7—och3 II 0 CH3Z
Аргинин о-Нитрогруппа NO2- —NH—C—NH—NOa и
II NH
со, (о'-Дибензил- оксикарбониль- ная z- .NH—Z —NH—Cz ^N—Z
Тозильная Tos- /NH—Tos —NH-CZ ^NH
4-Метоксибен- золсульфон иль- ная MbS NH О II н —NH—C—NH—S—7—OCH3 11 x— /
II — о
Мезитилен-2- сульфой ильная MtS 0 CH3 II / X —NH—C— NH—S—" CH3 II II xrz NH о j,Hs
со, о/-бис-Ада- Adoc мантилоксикар- бонильная N Adoc —NH— ^NH—Adoc
4-Метокси-2,6- MdS-
диметилбензол-
сульфон ильная
NH О С*1’
II II / X
__NH-C—NH-S-Z >-ОСН8
и Y=z
° СН3
226
Продолжение табл. 42
Реагент для ведения защитной группы Способ удаления Примечание Лите- ратура
НСООН, Ас2О н+ [369]
Н3С—{ S—SO2C1, он—
СНах zCHe
Н3СО—ЗО2С1, он-
хсн,
Na/NH3, HF
CH3SO3H/TFA
CF3SO3H/TFA
HF
[370]
Устойчива к TFA [371]
NaNO3 + H2SO4
О
CH2O—(®—Cl, OH-
H2/Pd, HF Используется ча- [372] сто
H2/Pd, НВг/АсОН, HF [226]
H3C—SO2C1, OH-
H3CO-<^ SO2C), OH-
Na/NHg, HF Широко исполь- зуется в т/ф синтезе [373]
CH3SO3H, CF3SO3H, HF Перспективная группа [374]
H3CO—SO3C1, OH-
~Чн,
TFA, HC1
To же
[375]
Легко удаляется, [376]
более гидрофобна,
чем Вос
CF3SO3H,
CH3SO3H/TA
[377]
15*
227
Аминокис- лота Защитная группа Обозначе- ние Фурмула защитно! группы
Пентаметилбен- Pme-
зол сульфон иль-
ная
Тистидин Бензильная Bzl
Тозильная Тоз-
2,4-Динитрофе- Dnp-
нильная
трет-Бутил ок- Вос-
сикарбонильная
Фенацильная
трет-Бутокси-
метил ьная
Тритильная Trt
Nx N-C(Ph)3
Адамантилокси- Adoc-
карбонильная
228
Продолжение табл. 42
Реагент для введения защитной группы Способ удаления Примечание Лите- ратура
сн,х /СН8 TFA/TA, HF Устойчива к TFA [378]
Нзс~\ SO2C1, ОН-*
сн3/ чсн,
—СНаС1> он—
H2/Pd, HF
Распространенная [379]
группа
HaC-^ Ч—SO2C1, он**
НОВТ, Ас2О/Ру Удаляется в очень [380]
мягких условиях
R—SH
Удобна при т/ф [381]
синтезе
'2
ВосаО, NEtg TFA, НС1 1251]
СН2Вг, ОН
НВг/т'РА,
Zn/AcOH, УФ
стойчива к [382]
IBr/AcOH
(СН3)3СО — СН2С1, он— Н+
Ph TFA, НС1
Ph—С—Cl, ОН—
Ph
TFA, НС1
Удаляется в кис- [383]
лой среде
Устойчива к ще-
лочам и H2/Pd
[384]
Используется ред- [184]
ко
229
Аминокис- лота Защитная группа Обозначе- ние Формула защитной группы
Г идроксилсодержащие
RO—СН,—
С,\
RO—СН,—
01/
Тирозин Бензильная Bzl-
2,6-Дихлорбен- (2,6) СР
зильная Bzl
З-Бромбензиль- (З-Вг) Bzl-
ная
о-Нитробензиль- ONB-
ная
Бензилоксикар- Z-
бонильная
/&
R—О—СН,—
R—О-СН,—
NO,/
RO—Z
трет-Б ути лок- Вос-
сикарбонильная
mpem-Бутильная -ОВи*
RO—Вос
RO—С (СН#)а
Диметилфосфо- ДМРТ-
нотиольная
р/сн*
11ХСН,
S
Серин, Бензильная Bzl-
треонин
mpem-Бутильная -OBiU
RO—С(СН8)8
Цистеин Бензильная Bzl
Ацетамидомети- льная Acm-
Бензамидоме- Ваш-
тильная
Серосодержащие
R—S—Н,С—
о
II
R—S—Н,С—NH—С—СН,
о
R—S—Н,С—NH—С—'
230
Продолжение табл. 42
Реагент для введения защитной группы Способ удаления Примечание Лите- ратура
аминокислоты
СН,СЦОН" H,/Pd, HF Используется ши- [385] роко
/С1 СНаС1, ОН- ЧС1 HF Более устойчива [386] к TFA, испольау- ется в т/ф синтеза
Вг\ TFA/TA То же [367]
СН„С1, ОН"
СН.С1, ОН" 4NO, УФ, он— Фотолабильная [387] группа
О CH,O(L-C1, ОН" Шк/АсОН, ОН- Используется ред- [388] ко
Boc-Ns или ВосвО, NEts То же [389]
СН, 4. х.с=сн2, н+ сн/ TFA, НС1, HF Используется ши- [390] роко
S СН8 II >Р—Cl, NEt, сн. он— [391]
СН,Вг. NaH g|/Pd, НВг/TFA То же [392]
СН,.' )С=СН„ н+ сн,/ TFA, HC1 > » [393]
аминокислоты
СН8С1, ОН" Na/NH„ HF Используется ред- [394] ко
о и Hg*+ Используется ши- [395]
СН,—С—NH—СН2ОН, Н+ роко
о II Hg2+ То же [396]
/ С—NH—СН,ОН, Н+
231
бензилового спирта. Продолжая размешивание, добавляют
небольшими порциями 15,1 г (0,1 моль) L-глутаминовой кис-
лоты. Полученный раствор выдерживают 20 ч при комнатной
температуре, после чего эфир удаляют на роторном испари-
теле, а к остатку добавляют 200 мл этилового спирта и 50 мл
пиридина. Продукт реакции начинает тотчас же кристалли-
зоваться. Смесь выдерживают 10 ч При 0 °C, выпавшие крис-
таллы отфильтровывают, промывают эфиром и сушат. Для
очистки продукта его растворяют в 190 мл кипящей воды,
раствор фильтруют и оставляют на 6 ч при 0 °C. Образовав-
шиеся кристаллы отфильтровывают, промывают вфдойисушат.
Выход 18 г (76 %), бесцветные пластинки с т. пл. 189 °C [а]д =
+ 19,6° (с = 5; уксусная кислота).
P-Бензиловый эфир L-аспарагиновой кислоты получают
с выходом 40—45 % по аналогичной методике [4101, т. пл.
218—220 °C, [а]2о = + 28,1° (с = Г, 1 н. соляная кислота).
Методика 200. у-Бензиловый эфир L-глутаминовой
кислоты [411].
К раствору 10 г (20 ммоль) n-толуолсульфоната дибен-
зилового эфира L-глутаминовой кислоты в 140 мл этилового
спирта прибавляют раствор 20 г (80 ммоль) сульфата меди в
350 мл воды, pH раствора доводят, добавляя 1 М NaOH, до
8,0 и реакционную смесь выдерживают 1 ч при 32 °C, после
чего pH раствора снижают 3 М соляной кислотой до 3,0. Об-
разовавшийся медный комплекс у-бензилового эфира глута-
миновой кислоты отфильтровывают, последовательно промы-
вают водой, спиртом и, наконец, эфиром. Полученный комп-
лекс добавляют к раствору 7,8 г (21 ммоль) динатриевой соли
этилендиаминтетрауксусной кислоты (Трилон Б) в 100 мл во-
ды, кипятят и горячим фильтруют. Пр-и стоянии и охлажде-
нии'раствора выпадает осадок у-бензилового эфира глутами-
новой кислоты. Выход продукта 3,5 г (74 %), т. пл. 169—
170 °C; [а]о = + 19,3° (с = 5; уксусная кислота).
р-Бензиловый эфир L-аспарагиновой кислоты синтезируют
аналогично. Выход 67 %.
Хеесвик и соавт. [413] предложили удобный метод полу-
чения различных замещенных бензиловых эфиров глутами-
новой и аспарагиновой кислот путем алкилирования солей
их медных комплексов:
О\\ Hj
х:-(сн2)п n о о
/ \ / ч / о
-° I Си I
(СНа)п—X
на о-
236
X Cu2+
H—Glu
-OH-J-HN=C^
N(CHS),
N(CH3),
dmf/h2o
N(CH3)a
X 2HN=C+
R—CHaX i
XN(CH8)2
% H2
ЧС-(СНа)„ч/\
» RCHt-O/ V /C\ zl4
О О N (CHa)n
Hg
H3N—CH—coo-
ЭДТА t
O—CHa—R
(CHa)n-C—OCHjR 4- ЭДТА • Cu
В табл. 43 указаны характеристики некоторых «-замещен-
ных бензиловых у- и 0-эфиров этих аминокислот.
Методика 201. Медная соль медного комплекса глу-
таминовой кислоты 1413].
Раствор 41,2 г (206 ммоль) ацетата меди (II) в 750 мл воды
приливали по каплям в течение 1 ч к перемешиваемому рас-
твору 29,4 г (200 ммоль) L-глутаминовой кислоты, растворен-
ной в 750 мл воды, при 70 °C. Смесь затем выдерживали
2 дня при комнатной температуре для полного осаждения
комплекса. Продукт отделяли фильтрацией, промывали водой,
спиртом, эфиром и сушили в вакууме (10 мм рт. ст.) при 50 °C.
Выход тетрагидрата медной соли медного комплекса L-глу-
таминовой кислоты — 45 г (92 %).
Методика 202. Медная соль медного комплекса аспа-
рагиновой кислоты [413].
Получена аналогично из 26,6 г (200 ммоль) L-аспарагино-
вой кислоты в 1100 мл воды. Выход октагидрата 52,1 г (98 %).
Методика 203. Тетраметилгуанидиновая соль мед-
ного комплекса глутаминовой кислоты [413].
Добавляли медленно 1,4 мл (11,2 ммоль) N,N,N',N'-TeTpa-
метилгуанидина (ТМГ) в течение 30 мин при перемешивании
к смеси 1,37 г (2,8 ммоль) медного комплекса, полученного
237
Таблица 43. Замещенные у-эфиры глутаминовой и f-эфиры аспарагиновой кислот [413]
Эфир Алкилирующий реагент Выход, % * Т. пл., вС [а] р, град • • (растворитель)
А Б
у-Эфиры глутами- новой кислоты СН2Вг 93 73 174—176° (Н2О) +19,9 (АсОН)
С1—СН2С1 93 79 175—175,5 (АсОН/Н2О) +18,3 (АсОН)
Н3СО—^2/—СН2С1 67 52 173—174 (Н2О) +17,5 (АсОН)
ьо со оо O2N-<fСН2Вг 77 60 171—172 (АсОН/Н2О) 19,0 (94 % АсОН)
Н3с—СН2Вг 91 81 180—181 (НгО) +18,2 (АсОН)
₽-Эфиры аспараги- СН2Вг 85 65 219—219,5 (Н2О) +28,1 (1 н. НС1)
новой кислоты С1—СН2С1 78 69 211—213 АсОН/Н2О +7,6 (АсОН)
Н8СО—СН2С1 59 44 205 Н2О +7,0 (АсОН)
0,N—CH2Bf 90 86 189— 190АсОН/НгО +10,2 (АсОН)
* а — выход алкилированных медных комплексов; Б — выход свободных эфиров из расчета на исходные аминокислоты.
♦ ♦ Концентрация С = 1.
по приведенной выше методике, и 0,83 г (5,6 ммоль) L-
глутаминовой кислоты в смеси 5 мл ДМФА и 0,8 мл воды.
После полного растворения комплекса (около 2 ч) добавляли
еще 4 мл ДМФА. Полученный голубой раствор использовали
непосредственно для получения у-эфиров глутаминовой кис-
лоты. Для алкилирования /г-нитробензилбромидом раствор
разбавляли 15 мл смеси ДМФА — вода (10: 1).
Методика 204. Тетраметилгуанидиновая соль мед-
ного комплекса аспарагиновой кислоты [413].
Получали аналогично из 1,49 г (2,8 ммоль) комплекса,
0,75 г (5,6 ммоль) А-аспа ратиновой кислоты и 1,4 мл (11,2 ммоль)
ТМГ в смеси 5 мл ДМФА и 0,6 мл воды, и разбавляли 4 мл
ДМФА.
Методика 205. Общий "метод получения у- и р-алки-
ловых эфиров медных комплексов глутаминовой и аспараги-
новой кислот [411].
К перемешиваемому раствору полученных комплексов до-
бавляли 11,8 ммоль алкилгалоида в один прием. Нестабиль-
ный n-метоксибензилбромид вносили в течение 30 мин. Смесь
перемешивали 24 ч при комнатной температуре или при 40 °C
(для n-хлорбензилхлорида, 1-нафтилбензилхлорида и и-ме-
тилбензилхлорида). Полученный осадок или полученную мас-
су перемешивали с ацетоном, пока не образовывался твердый
осадок. Его суспендировали в 25—50 мл воды и перемешивали
до получения тон ко дисперсного осадка, который отфильтро-
вывали, промывали водой и сушили до постоянного веса в
вакууме над Р3О5.
Методика 206. Получение свободных эфиров [413].
Влажный осадок (или полностью высушенный) переноси-
ли в пересыщенный раствор дииатриевой соли этилепди-
аминтетрауксусной кислоты (предварительно полученной пу-
тем добавления порциями 1,6 г ЭДТА к перемешиваемому
раствору 0,9 г бикарбоната натрия в 11 мл воды). Смесь встря-
хивали до полного разложения комплекса (0,5—1 ч), разбав-
ляли 15 мл воды и 5 мл ацетона, охлаждали до 10°, отфильт-
ровывали, выделившиеся кристаллы промывали холодной
водой и сушили в вакууме. '
Среди замещенных эфиров глутаминовой и аспарагиновой
кислот большое значение приобрели циклогексиловые эфиры,
ввиду их большей устойчивости в кислой среде, чем у бензи-
ловых эфиров [363].
Методика 207. Общий метод получения у- и (J-цикло-
гексиловых эфиров глутаминовой и аспарагиновой кислот
[363].
К раствору 1 экв. у- или р-бензилового эфира Вос-глута-
миновой или Вос-аспарагиновой кислоты в метилен хлориде
239
при О °C добавляли 1 экв. хлоргидрата 1-этил-3-(3-диметилами-
нопропил)карбодиимида, 3 экв. циклогексилового спирта и
10 мольных % 4-диметиламинопиридина и перемешивали в
течение 1 ч. После стандартной обработки (1 и. бисульфат нат-
рия, вода, бикарбонат натрия, вода) раствор, сушили над
сульфатом натрия, фильтровали и упаривали этилацетат.
Остаток растворяли в этаноле и гидрировали над 5 % палла-
дием на сульфате бария. Выход: Р-циклогексилового эфира
L-аспарагиновой кислоты 85 %, т. пл. 93—95 С°; у-циклоге-
ксилового эфира глутаминовой кислоты (дициклогексиламмо-
нийно’1 соли) 82 %, т. пл. 135—136 °C.
Р- и Бутиловые эфиры соответственно аспараги-
новой и глутаминовой кислот получаются по методике [361].
fi-mpem-Бутиловый эфир L-аспарагиновой кислоты; т. пл.
189—190 °C, [ccId = + 8,5° (с = 1; 90 %-я уксусная кисло-
та); у-трет-бутиловый эфир L-глутаминовой кислоты* т. пл.
168—169 ’С; [а]д = + 8,15° (с = 2; вода).
ЗАЩИТА БОКОВЫХ ФУНКЦИОНАЛЬНЫХ ГРУПП
ДИАМИНОКАРБОНОВЫХ КИСЛОТ
Защита е-аминогруппы лизина
Для защиты 8-аминогруппы лизина чаще всего применяют
карбобензокси- или Вос-группу, значительно реже исполь-
зуют формильную и трифторацетильную группы. Для прида-
ния большей устойчивости защитной группе е-аминогруппы
лизина при твердофазном методе синтеза пептидов используют
замещенную карбобензоксигруппу! например, такую как 2-
хлоп- или 2,4-дихлоркарбобензоксигруппу [366]. Эти произ-
водные оказались более устойчивыми, чем незамещенная Кбз-
группа соответственно в 60 и 90 раз при удалении Вос-группы
трифторуксусной кислотой.
Методика 208. №-Карбобензокси-£-лизин [414].
Навеску 1,8 г (10 ммоль) монохлоргидрата лизина раство-
ряют в 15 мл воды, добавляют избыток карбоната меди, смесь
кипятят в течение 30 мин, затем фильтруют и добавляют 5 мл
2 н. раствора едкого натра. Реакционная смесь окрашивает-
ся в интенсивный голубой цве-т. Раствор охлаждают и при пе-
ремешивании добавляют десятью порциями в течение 30 мин
2 мл карбобензоксихлорида и 10 мл 2 н. едкого натра. В ре-
зультате реакции выпадает медный комплекс 8-карбобенз-
оксилизина. Его отфильтровывают, промывают водой, затем
спиртом и суспендируют в 200 мл воды, в суспензию при раз-
мешивании пропускают ток сероводорода до полного разло-
жения медного комплекса. Реакционную массу нагревают
240
до кипения и фильтруют в горячем состоянии; при охлажде-
нии кристаллизуется е-карбобензоксилизин в виде длинных
бесцветных игл. Оставляют на ночь в холодильнике и отфиль-
тровывают осадок. Дополнительно количество продукта мож-
но получить после отгонки растворителя до малого объема и
последующей кристаллизации. Общий выход 2,3 г (80 %),
кристаллизация из воды дает продукт ст. пл. 235 °C; [а] д =
= 4- 14,4° (с = 1,6; разбавленная соляная кислота).
Методика 209. е-/пре/п-Бутилоксикарбонил-£-ли-
зин [365].
Раствор 10 г (55 ммоль) хлоргидрата L-лизина в 80 мл во-
ды и 10 г основного карбоната меди нагревают 30 мин до ки-
пения. Темно-голубой раствор отфильтровывают от избытка
карбоната меди, охлаждают, смешивают с 3 г оксида магния.
При энергичном перемешивании к охлажденному раствору
прибавляют по каплям 11,5 г (80 ммоль) трг/п-бутилкарбо-
нилазида в 150 мл метанола. Реакционную массу размеши-
вают в течение ночи при 45 °C. При этом выпадает медный комп-
лекс 8-Вос-лизина в виде светло-голубого осадка. Для удале-
ния избытка окиси магния смесь осторожно подкисляют при
размешивании и охлаждении 50 мл 2 «н. уксусной кислоты.
Размешивание продолжают в течение 2 ч при 2 °C, а-затем комп-
лекс отфильтровывают, промывают водой, метанолом и су-
шат. Выход 7,1 г (30 %), т. пл. 230—240 °C.
Медный комплекс (14 г) хорошо растирают в ступке и за-
тем суспендируют в 300 мл воды. При охлаждении льдом до-
бавляют 50 мл 2 н. раствора аммиака и при интенсивном раз-
мешивании пропускают в течение 3 ч сероводород. Для удале-
ния избытка сероводорода колбу охлаждают льдом, добав-
ляют 75 мл 2 н. уксусной кислоты и продувают через раствор
ток воздуха до тех пор, пока не исчезнет запах сероводорода.
После фильтрования раствора через угольные фильтры вы-
падают кристаллы 8-Вос-Л‘Лизина, их отфильтровывают, про-
мывают холодной водой и сушат. Выход 11 г (88 %), т. пл.
220—225 °C; [а]о = + 17,Г (я == 1; уксусная кислота).
Методика 210. М,Й'-Дикарбобензокси-А-лизин [414].
К раствору 125 г (684 ммоль) хлоргидрата L-лизина в 342 мл
воды и 684 мл 2 н. едкого натра, охлажденному до — 10 °C,
добавляют при интенсивном размешивании в течение 75 мин
852 мл 4 н. едкого натра и 360 мл карбобензоксихлорида (пред-
варительно охлажденных до 0 °C) с такой скоростью, чтобы
pH раствора не снижался ниже 10. Перемешивание продол-
жают еще 10 мин, а затем раствор при охлаждении подкис-
ляют до pH 1,5 1 л 2 н. соляной кислоты. Продукт дважды
экстрагируют эфиром, органическую фазу промывают 1 н. со-
ляной кислотой и водой, а затем обрабатывают 1 н. раствором
16 2-882 241
аммиака сначала 2 л, а затем дважды по 0,5 л. Аммиач-
ные растворы промывают эфиром и затем подкисляют при 0 °C
при помощи 700 мл 4 н. соляной кислоты в присутствии 500 г
измельченного льда и 2 л эфира. Органический слой отделяют,
а водную фазу экстрагируют 400 мл эфира. Эфирные вытяж-
ки соединяют и экстрагируют 1 н. раствором аммиака (1 раз
порцией по 2 л и 2 раза по 400 мл). Рствор продукта в аммиа-
ке дважды промывают эфиром и затем подкисляют при 0 °C и
хорошем перемешивании 750 мл 4 н. НС1 в присутствии 2 л.
эфира. Органический слой отделяют, водный раствор экстра-
гируют эфиром (2 X 200 мл). Органический слой отделяют и
сушат сульфатом магния, эфир удаляют на роторном испари-
теле. Полученное масло (243 г) растворяют в толуоле, а затем
растворитель удаляют на роторном испарителе в вакууме;
эту операцию повторяют дважды. Остаток после отгонки то-
луола кристаллизуют из смеси 350 мл эфира и 1000 мл пет-
ролейного эфира. После фильтрования, промывания петро-
лейным эфиром и высушивания получают 223 г (79 %) дикар-
бобензокси-Д-лизина с т. пл. 88—90 РС; [а]р — 5Q = 2;
метанол).
Поскольку при твердофазном методе синтеза пептидов ис-
пользуют многократную обработку пептидилполимера три-
фторуксусной кислотой, для устранен и-1 побочных реакций,
связанных с частичным отщеплением t-карбобензоксигруппы
лизина, была предложена и нашла широкое применение в
90 раз более устойчивая к ацидолизу защитная 2,4-дихлор-
бензилоксикарбонильная группа. Ниже приведены прописи
получения №-(2,4-дихлорбензилоксикарбонил)-£-лизина [366].
Методика 211. 2,4-Дихлорбензиловый спирт [366].
52 г (0,272 моль) 2,4-дихлорбензойной кислоты (предвари-
тельно очищенной кристаллизацией из смеси хлороформ —
бензол (1:1 по объему) с т. пл. 160,2—161 РС растворяют в
250 мл сухого тетрагидрофурана; полученный раствор при-
бавляют в течение 20 мин при перемешивании в токе азота к
раствору 9,5 г (0,25 моль; 1,2 экв) алюмогидрида лития в
750 мл сухого ТГФ. Смесь кипятят до тех пор, пока вся 2,4-
дихлорбензойная кислота не прореагирует. Обычно на это
требуется около 20 ч. Затем реакционную массу охлаждают
и осторожно подкисляют,6 н. соляной кислотой и экстраги-
руют этилацетатом. Органическую фазу промывают водой,
затем 0,1 М бикарбонатом натрия, сушат и растворитель уда-
ляют на роторном испарителе. Кристаллизация из гексана
дает 32,7 г (68 %) 2,4-дихлорбензилового спирта с т. пл. 57,2—
58,4 °C.
Методика 212» №-(2,4-ДихлорбензиЛЪксикарбонил^
L-лизин [366].
242
35,4 г (0,2 моль) 2,4-дихлорбензилового спирта в 1,25 М
растворе фосгена в бензоле выдерживают.при температуре
20 °C в течение 37 ч в плотно закрытой склянке, после чего рас-
творитель выпаривают на роторном испарителе в вакууме во-
доструйного насоса, поместив перед ним ловушку, охлажден-
ную сухим льдом с ацетоном Следы фосгена удаляют двукрат-
ным добавлением и последующей отгонкой в вакууме сухого
диоксана (50 мл). Оставшуюся прозрачную жидкость (52 г)
растворяют в 120 мл сухого диоксана; это г раствор 2,4-дихлор-
бензилхлорформиата используют в дальнейшем для ацили-
рования медного комплекса лизина.
К раствору хлоргидрата L-лизина (44,1 г, 0,24 моль) в
500 мл горячей воды добавляют небольшими порциями 30,9 г
(0,13 моля) порошкообразного основного карбоната меди;
реакционную смесь нагревают до кипения и затем фильтруют
в колбу Бунзена на 2 л, а осадок на фильтре промывают 20 мл
горячей воды. К полученному голубому раствору добавляют
120 мл 2 М раствора бикарбоната калия и 300 мл диоксана и
раствор охлаждают до 0 °C, погружая колбу в баню с ледяной
водой. Продолжая охлаждение, в колбу вносят каждые 10 мин
порцию раствора 2,4-дихлорбензилхлорформиата (10 мл) и
4 М раствора КОН (4,2 мл). Добавление этих реактивов зани-
мает примерно 2 ч, а затем реакционную массу размешивают
в течение 10 ч при 0 °C. Образовавшийся осадок отделяют
фильтрованием и тщательно промывают водой, этиловым спир-
том и эфиром. Выход медного комплекса 65,1 г (85 %).
Полученный продукт добавляют в течение 30 мин неболь-
шими порциями к суспензии ЭДТА (30 г; 0,1 моль) в 0,33 М
соляной кислоте (1,2 л), смесь размешивают в течение 1,5 ч,
а затем осадок отсасывают. Продукт суспендируют в 1 л сме-
си этиловый спирт — вода (7 < 3 по объему), кипятят в течение
некоторого времени и затем фильтруют. При охлаждении рас-
твора выпадают бесцветные призмы №-(2,4-дихлорбензилокси-
карбонил)-Л-лизина (25,L г). Упариванием маточника и по-
следующим охлаждением можно получить дополнительно еще
27,6 г продукта. Общий выход составляет76 %.
/прет-Бутилоксикарбонил-№-(2,4-дихлорбензилоксикар-
бонил)-Ь-лизин в виде вязкого бесцветного масла получают
с выходом 97 % обычными методами (см. главу 2).
Методика 213. №-Трифторацетил-Л-лизин [273, 368].
К раствору 1,83 г (10 ммоль) хлоргидрата L-лизина в 10 мл
1 н. NaOH добавляли 2 мл тиоэтилового эфира трифторуксус-
ной кислоты (см. методику 153). Гетерогенную смесь встря-
хивали 6 ч. Постепенно выпадало значительное количество
осадка. Реакционную смесь охлаждали до 0 °C и. отфильтро-
вывали осадок. Выход 1,81 г (75 %). Технический продукт
16*
«243
растворяли в 10 мл горячей воды и добавляли 15 мл горячего
этанола. При охлаждении выпали белые кристаллы. Выход
1,25 г (69 %); [а]2д — -ф 21,7 (с = 3; раствор монохлоруксус-
ной кислоты).
№‘-Вос-№-трифторацетиллизин плавится при 103 °C [368].
Методика 214. Общий метод выделения е-ацил-лизи-
на и 6-ацилорнитина из их медных комплексов [415].
Метод А. (для соединений, умеренно растворимых в воде).
Растворяли 0,005 моль медного комплекса при кипячении в
100 мл 0,1 раствора ЭДТА, охлаждали до 0 °C и отделяли вы-
павший осадок.
Метод Б (для соединений, плохо растворимых в воде). К
раствору 0,005 моль медного комплекса в 50 мл 2 н. НС1 до-
бавляли 100 мл 0,1 н. раствора ЭДТА и нейтрализовали 2 н.
NaOH. Отделяли выпавший осадок. Выход различных заме-
щений (Z-, Tos-, Z (NO)2-, Рогш-)лизина — 60—85 %, №-
карбобензоксиорнитина — 86 %.
Недавно Тейлор и соавт. [416] предложили использовать
для разложения медных комплексов аминокислот тиоацет-
амид. Авторы показали, что при обработке медных комплексов
№-карбобензокси L-орнитина и №-карбобензокси-Ь-лизина
тиоацетамидом в щелочной среде образуются соответствую-
щие карбобензоксиаминокислоты с хорошими выходами, а
сульфид меди легко удаляется.
Методика 215. №-карбобензокси-L-орнитин [416].
5,4 г (9 ммоль) медного комплекса Жкарбобензокси-Л-
орнитина суспендировали в 50 мл воды и добавляли к суспен-
зии 1 г (13,3 ммоль) тиоацетамида. Далее добавляли 2 н. рас-
твор NaOH до pH 8,0 и смесь перемешивали 26 ч при комнат-
ной температуре. Подкисляли раствор 2 н. соляной кислотой
до pH 1,6 и удаляли сульфид меди фильтрацией, его промы-
вали на фильтре разбавленной соляной кислотой, а затем во-
дой. Для осаждения №-карбобензокси-Ь-орнитина фильтрат
подщелачивали до pH 8,0 2 н. NaOH, отделяли осадок фильт-
рованием, промывали его водой и продукт реакции очищали
переосаждением его раствора в соляной кислоте, добавляя
р створ едкого натра. Выход 4,8 г (87,5 %), т. пл. 253 °C;
[а] о = + 18 °C (с — 1; водный ацетон 1 ! 1 + 2 экв. НС1).
Методика 216. №-карбобензокси-Ь-лиЗин [416].
500 мг (0,8 ммоль) медного комплекса №-карбобензокси-Ь-
лизина обрабатывали по описанной выше методике. После
кристаллизации из смеси этанол — вода выход №-кар-
бобензокси-Л-лизина составил 200 мг (88 %), т. пл. 255 °C;
[а]2о° -» + 1,5°С (с = 1,5; 2 н. НС1).
244
Защита гуанидиновой группы аргинина
Несмотря на то, что в последнее время для синтеза аргинин-
содержащих пептидов широко используют аргинин со свобод-
ной гуанидиновой функцией, т. е. аргинин в протонирован-
ной форме, во многих случаях удобнее работать с блокирован-
ной гуанидиновой группой. Наиболее распространенными
защитными группировками ее являются нитро-, дикарбобенз-
окси- и тозильная группы.
Нигрогруппу вводят при обработке аргинина смесью олеу-
ма и азотной кислоты [417], однако лучшие результаты полу-
чаются при нитровании аргинина нитратом аммония в серной
кислоте [418]:
H2N—СН—соон
(СН2)з
NH
NH4NO, I
с
С
HaN— СН— СООН
(СН2)8
lijH
HN NH-NO,
Тозильную группу и родственную ей п-метоксибензолсуль-
фонильную группу вводят, используя соответствующие суль-
фохлориды, в условиях реакции Шоттен — Баумана.
Весьма удобным производным для введения N-концевого
аргинина в пептиды является трикарбобензоксиаргинин, а так-
же Ы1-бензилоксикарбонил-Ы2, Ы3-ди(адамантил-1 -оксикар-
бонил)-аргинин. Ниже даны примеры синтезов этих произ-
водных аргинина.
Методика 217. со-Нитро-Ь-аргинин [418].
К 200 мл концентрированной серной кислоты при комнат-
ной температуре добавляют при перемешивании небольшими
порциями 100 г (0,43 моль) хлоргидрата аргинина. Выделяю-
щийся хлористый водород отсасывают водоструйным насосом
до тех пор, пока не прекратится выделение пузырьков газа.
К образовавшемуся прозрачному раствору медленно добавляют
при перемешивании 50 г NaNO3 и через 15 мин опять удаляют
выделяющийся газ водоструйным насосом. После прекраще-
ния выделения пузырьков газа реакционную массу выливают
при размешивании на мелко измельченный лед. Полученный
раствор фильтруют и доводят pH раствора до 6,8 водным ам-
миаком. Смесь выдерживают несколько часов при 0 °C. Обра-
зовавшийся осадок отфильтровывают, промывают холодной
водой и кристаллизуют из воды. Из-за небольшого различия
в растворимости нитроаргинина в горячей и холодной воде
245
кристаллизацию удобно проводить небольшими порциями,
используя маточник для очистки следующих порций нитро-
аргинина. Выход продукта 90 г (86,5 %), т. пл. 255 °C с раз-
ложением; [а]^ == + 24,1 (с = 1,9; 2 н. НС1).
Методика 218. со-Тозил-Л-аргинин [373].
12,5 г (40ммоль) карбобензоксиаргинина^ растворяют в
смеси 40 мл 4 М едкого натра и 320 мл ацетона, охлаждают до
0 9С и небольшими порциями добавляют при перемешивании
19 г n-толуолсульфохлорида в 60 мл ацетона. Реакционную
массу размешивают 2 ч при 0 °C, подкисляют до pH 5,0 соля-
ной кислотой. Ацетон отгоняют на роторном испарителе, а
остаток обрабатывают эфиром (2 X 30 мл) для удаления из-
бытка n-толуолсульфохлорида. Продукт экстрагируют из
водного раствора 150 мл этилацетата. Соединенные орга-
нические вытяжки промывают водой, а N-карбобензокси-со-
тозиларгинин экстрагируют 5 %-м раствором бикарбоната
натрия. Раствор при размешивании осторожно подкисляют
до pH 3,0, продукт экстрагируют 150 мл этилацетата. Орга-
ническую фазу промывают 2 н. НО, водой и сушат сульфатом
магния, а этилацетат отгоняют в вакууме. Остаток в виде
густого сиропа затвердевает при выдерживании над Р2О5.
Переосаждение из этилацетата петролейным эфиром при
охлаждении до — 70 °C дает 13,5 аморфного продукта (75 %),
т. пл. 75—85 °C; [а]р = — 1,3° (с == 2; метанол).
Полученный N-Кбз-со-тозиларгинин гидрируют над пал-
ладиевой чернью в смеси метанол — уксусная кислота —
вода (6:1*1). После удаления катализатора растворитель от-
гоняют, остаток растворяют в минимальном количестве ки-
пящей воды и оставляют кристаллизоваться при 0 °C. Обра-
зовавшийся продукт отфильтровывают и сушат. Выход (80 %),
т. пл. 146—150 °C; [а]о = — 6,1Q (с — 1; метанол).
Методика 219. №^-Карбобензокси-со-метоксибензол-
сульфонил-£-аргинин. ДЦГА-соль [374].
55,5 г (180ммоль) карбобензокси-Ь-аргинина растворяют
при комнатной температуре в смеси 180 мл 4 н. едкого натра
и 1300 мл ацетона. Раствор охлаждают до 0 °C и при интен-
сивном перемешивании к нему добавляют по каплям в течение
30 мин раствор 74,4 г (0,36 моля) п-метоксифенилсульфохло-
рида в 300 мл ацетона. Перемешивание продолжают 2 ч при
0 °C и затем еще 2 ч при комнатной температуре, после чего
реакционную смесь подкисляют раствором лимонной кисло-
ты. Растворитель удаляют при пониженном давлении и тем-
пературе 40 °C. Оставшееся масло растворяют в этилацетате
и дважды промывают водой, а продукт экстрагируют
5 %-м раствором бикарбоната натрия (400 X 3), водный слой
246
подкисляют осторожно лимонной кислотой, и выпавшее мас-
ло экстрагируют этилацетатом. Органический слой дважды
промывают раствором лимонной кислоты, затем водой и су-
шат сульфатом магния. После удаления растворителя полу-
чают 86 г продукта в виде масла. Его растворяют в 600 мл этил-
ацетата, к полученному раствору добавляют 36 мл (0, 18 моль)
дициклогексиламина и смесь оставляют на ночь в холодиль-
нике. Выпавшие кристаллы ДЦГА-соли отфильтровывают и
кристаллизуют из ацетонитрила. Выход 77 г (61,1 %), т. пл.
110—112 °C; [a]2D + 5,1° (с = 1; метанол).
Методика 220. со-п-Метоксифенилсульфонил-L-ap-
гинин [314].
2 г (4,2 ммоль) Ма-Кбз-со-п-метоксибензолсульфонил-Л-
аргинина в виде масла растворяют в 20 мл метанола и гидри-
руют в течение 3 ч в присутствии 0,2 г палладиевой черни. Ка-
тализатор удаляют фильтрованием, метанол упаривают в
вакууме, а остаток кристаллизуют изводы. Выход 0,9 г (63 %),
т. пл. 144—146 °C; [а]^ = — 6,1° (с = 0,7; метанол).
Это соединение авторы работы [374] использовали для
синтеза пептидов с С-концевым аргинином. При этом N-за-
щищенная аминокислота в виде п-нитрофенилового эфира со-
четалась с триэтиламмонийной солью (о-п-метоксифенилсуль-
фониларгинина в ДМФА.
Для синтеза пептидов с С-концевым аргинином весьма удоб-
но использовать также n-нитробензиловый эфир со-и-метокси-
фенилсульфониларгинина, который получается обработкой
соответствующего Кбз-производного 25 %-м раствором бро-
мистого водорода в уксусной кислоте.
Методика 221. n-Нитробензиловый эфир о-п-мето-
ксифенилсульфонилкарбобензокси-£-аргинина [374].
К суспензии 7 г (0,01 моль) ДЦГА-соли Кбз-со-п-метокси-
фенилсульфонил-Ь-аргинина в 200 мл этилацетата в делитель-
ной воронке прибавляют 60 мл 0,2 н. серной кислоты, охлаж-
денной до 0 °C. Воронку встряхивают до тех пор, пока весь
продукт не растворится. Органический слой дважды промы-
вают водой и сушат сульфатом натрия. Растворитель удаляют
на роторном испарителе, а остаток растворяют в 30 мл диме-
тилформамида; к полученному раствору добавляют 1,68 г
(0,012 моль) триэтиламина и 2,5 г (0,012 моль) п-нитробензил-
бромида. Смесь перемешивают 2 ч при 80 °C, а затем выливают
в 300 мл воды. Выпавшее масло растворяют в эти л ацетате,
органическую фазу промывают 0,1 н. соляной кислотой, а за-
тем — 5 %-м раствором бикарбоната натрия и сушат сульфа-
том натрия. После удаления этилацетата в вакууме получен-
ное масло растирают с петролейным эфиром. Выход аморф-
247х
ного продукта 6,2 г (100 %), т. пл. 90—ПО °C; [а]д == — 3,4°
(с = 0,59, ДМФА).
Методика 222. Трикарбобензокси-£-аргинин [419].
В течение 1 ч интенсивно перемешивают при 0—3 °C 43,5 г
(14,3 ммоль) карбобензоксиаргинина (см. методику 97) в 280 мл
2 н. NaOH, 100 мл диоксана с 98 мл (70 ммоль) карбобензокси-
хлорида и 353 мл 2 н. NaOH (добавление карбобензоксихлори-
да и щелочи производят попеременно примерно 10 порциями).
Спустя 1 ч перемешивания при 0—3° отфильтровывали вы-
павший осадок натриевой соли трикарбобензоксиаргинина и
промывали ее 300 мл холодного 5 %-го раствора карбоната
натрия. Хорошо отжатый на фильтре продукт растирали с эфи-
ром, а затем снова переносили на фильтр, промывали эфиром
и сушили. Затем сухую натриевую соль растворяли в абсолют-
ном этаноле (иногда нужно отфильтровывать нерастворив-
шуюся часть) и охлаждали полученный раствор в холодиль-
нике. Выпавший осадок отфильтровывали и сушили в вакуум-
эксикаторе.
Суспензию полученной натриевой соли (63 г) в 1,5 л этил-
ацетата смешивали при охлаждении льдом с 350 мл 2 %-й хо-
лодной серной кислоты. В полученном растворе отделяли вод-
ную фазу, а органическую промывали 2 %-й серной кисло-
той, водой и сушили над сульфатом натрия. После удаления
в вакууме этилацетата остаток растирали с петролинейным
эфиром и дважды кристаллизовали из этилацетата. Выход
25,5 г (32 %),т. пл. 137—138 °C; [а]д = + 16,8 (с = 1; хлоро-
форм).
По-видимому, лучшим является метод, предложенный
Смитвиком и Шуманом [420], который заключается в предвари-
тельном триметилсилилировании аргинина с последующим его
бензилоксикарбонилированием. Общий выход конечного про-
дукта 52 %.
H2N—СН—СООН
(Ана),
NH
J,
HN^NHa
О С1ч ХС1
CeHe-CHjO-C-O-^^Cl
с/ ХС1
СНз-С-NH-SiMe,
Z—NH—СН—СООН
(Ан,)»
NH
Z—XNH—Z
Методика 223. Бензилпентахлорфенилкарбонат[229].
К раствору 13,32 г пентахлорфенола (0,05 моль) и 6,45 г
(0,05 моль) хинолина в 100 мл метиленхлорида добавляют
8,5 г (0,05 моль) карбобензоксихлорида в течение 15 мин. Реак-
ционную смесь выдерживают в течение ночи, промывают 1 н.
НС1 (2 X 50мл), 0,5 н. NaHCO3 (4 X 50мл), водой (2 Х50 мл),
248
сушат сульфатом натрия и удаляют в вакууме растворитель.
Продукт кристаллизуют из толуола. Выход 16,0 г (78 %),т. пл.
115—116 °C.
Методика 224. Трикарбобензокси-£-аргинин [420].
1. Литиевая соль трикарбобензокси-Ь-аргинина. К 210 мл
сухого ДМФА добавляют 20,88 г (0,12 моль) L-аргинина и
160 г (0,4 моль) бензилпентахлорфенилкарбоната. Получен-
ную суспензию нагревают при перемешивании до 60 °C,
вносят 78,6 г (0,6 моль) N-триметилсилилацетамида и пере-
мешивают 60 ч при 60 °C. После прибавления 20 мл воды в
реакционную смесь ДМФА удаляют в вакууме. Остаток ра-
створяют в абсолютном спирте и этот раствор добавляют к го-
рячему насыщенному раствору 50 г ацетата лития в спирте.
Полученную смесь медленно охлаждают до комнатной темпе-
ратуры и затем выдерживают ночь при 4 °C. Образовашийся
осадок отфильтровывают, обрабатывают горячим этилацета-
том и затем перекристаллизовывают из минимального коли-
чества метанола. Выпавший осадок отделяют фильтрацией и
сушат в вакууме. Выход 39 г (56 %), т. пл. 153—155 °C. Ана-
литический образец, полученный кристаллизацией из смеси
метанол — ацетон, плавится в интервале 156—157 °C; [а]я ==
= + 10,6° (с = 1,5; метанол).
2. Трикарбобензокси-Ь-аргинин. Литиевую соль трикар-
бобензокси-£-аргинина (10 г; 0,017 моль) суспендировали в
этилацетате. Полученную суспензию нейтрализовали добав-
лением 2 %-й серной кислоты. Этилацетатный слой отделяли,
сушили сульфатом магния и удаляли в вакууме. Остаток пе-
рекристаллизовывали из этилацетата и сушили в вакууме.
Выход 9,1 г (92 %). Т. пл. 138—139 °C; [alb5 = + 15,1° (с =
== 1,5; хлороформ).
По этой методике был получен п-метоксибензилоксикар-
бонил-М.М'-дибензилоксикарбониларгинин с выходами: на
первой стадии 63 % (т. пл. 209—210 °C); [а]д = 4- 9,9° (с =
= (1; метанол); на второй стадии 91 % (т. пл. 139—141 РС);
laic = + 16,6я (с =з 1,5; хлороформ).
Примечание. Синтез N-триметилсилилацетамида
описан в главе 3.
Методика 225. 1-Хлорформилокси-адамантан [376].
К раствору 30 г фосг'*:: ;1! в 100 мл сухого бензола добав-
ляют по каплям в течение 1 ч при перемешивании и охлажде-
нии до 4 °C раствор 8 г 1-оксиадамантана и 7 г пиридина в
200 мл бензола. После того, как выпадет бесцветный осадок,
добавляют еще примерно 100 мл бензола. Реакционную смесь
* Установка для получения жидкого фосгена описана в главе 2,
249
выдерживают 1 ч при комнатной температуре, затем выпав-
шие соли отфильтровывают, а раствор промывают ледяной
водой. Органический слой сушат сульфатом натрия, раство-
ритель упаривают при 20 °C до одной пятой первоначального
объема и оставляют в холодильнике. Полагают, что выход
продукта количественный. Можно выделить 1-хлорформил-
оксиадамантан в твердом виде с т. пл. 46—47 °C после упари-
вания раствора досуха и кристаллизации осадка из петро-
лейного эфира при —20 ₽С.
Методика 226. К-Бензилоксикарбонил-№,№-ди-
(адамантил-1-оксикарбонил)- L-аргинин [376]
К раствору 20,7 г (67 ммоль) Кбз-аргинина в 40 мл диок-
сана и 134 мл 2 н. раствора NaOH при непрерывном размеши-
вании и охлаждении до 6—8 °C добавляют в течение 1 ч по
каплям одновременно раствор 57,4 г (268 ммоль) свежепри-
готовленного 1-хлорформилоксиадамантана в 50 мл диоксана
и 200 мл 2 н. раствора едкого натра. Спустя 3 ч непрерывного
размешивания при 5—8 °C массу центрифугируют и осадок
растирают с эфиром, отсасывают и промывают эфиром. Рас-
творитель удаляют на роторном испарителе, а остаток расти-
рают с петролейным эфиром, отсасывают и промывают пет-
ролейныМ^ эфиром, потом суспендируют в воде и подкисляют
0,5 М лимонной кислотой до pH 2—3. Выпавший продукт из-
влекают эфиром, органический слой сушат сульфатом натрия,
эфир удаляют на роторном испарителе. Остаток в виде пены
кристаллизуют из метанол — вода. Выход 39,2—41,1 г (88—
92 %), т. пл. 120—122 °C с разложением; [сс]д = + 20,8° (с ==
= 1; хлороформ). Ниже приведен пример использования не-
защищенного по гуанидиновой группе N-бензилоксикарбонил-
аргинина в качестве N-концевой аминокислоты.
Методика 227. Дибромгидрат метилового эфира кар-
бобензокси-Ь-аргинил-А-аргинина [421].
Суспензию 154 г (0,50 моль) карбобензи-£-аргинина и 193 г
(0,55 моль) дибромгидрата метилового эфира. L-аргинина в
1500 мл сухого пиридина перемешивают 20 мин при температу-
ре 0 °C, а затем вносят 124 г (0,60 моль) дициклогексил кар-
бодиимида. Реакционную смесь выдерживают 16 ч при 20 °C,
образовавшуюся дициклогексилмочевину отфильтровывают,
а растворитель упаривают. Остаток растирают с эфиром, за-
тем продукт суспендируют в ацетоне (1л). После фильтрова-
ния ацетон удаляют в вакууме и получают 259 г (81 %) дипеп-
тида, т. пл. 115—120 °C; [а]^ =» —10о (с == 2; метанол).
250
№-Мезитилен-2-сульфонил-£ -аргинин [375]
Методика 228. 2,4,6-Триметилбензолсульфохлорид
ZCH3 /СН,
Н3С-^~> + C1SO3H -> H3C-^~^>-SO2Cl + НС1.
'СН, хсн,
120,2 116,5 218,7
24 г (0,2 моль) чистого мезитилена добавили при непре-
рывном перемешивании к 42 г (0,33 моль) хлорсульфоновой
кислоты, охлаждаемой смесью соли со льдом, с такой ско-
ростью, чтобы температура реакционной смеси не превышала
0 QC. После того, как весь мезитилен был добавлен, смесь ос-
тавили нагреваться до комнатной температуры и осторожно
вылили на лед. Из полученного фиолетового раствора мези-
тиленсульфохлорид выделялся в виде красноватого осадка.
После промывки водой осадок растворяли в эфире, эфирный
раствор сушили сульфатом натрия, фильтровали и упаривали
до конца. Остаток — слегка фиолетового оттенка — пере-
кристаллизовывали из легкого петролейного эфира и полу-
чали 20 г (46JJ6) бесцветных призм с т. пл. 56 °C.
Методика 229. №-Мезитилен-2-сульфонил-Ь-арги-
нин [375].
1. При — 10° к 49,7 г (0,15 моль) п-метоксикарбобензокси-
аргинина в смеси 600 мл (0,6 моль) 4 н. NaOH и 750 мл ацето-
на прибавляли по каплям 65,6 г (0,3 моль) мезитиленсульфо-
хпорида в ацетоне. Смесь перемешивали 30 мин при 0 °C и 2 ч
при 20 °C, отгоняли в вакууме ацетон, остаток в воде промы-
вали этилацетатом, подкисляли лимонной кислотой, экстра-
гировали этилацетатом. Органическую фазу промывали 0,2 н.
НС1, водой, сушили сульфатом натрия и упаривали раство-
ритель. Остаток растворяли в ацетонитриле, добавляли 14,9 г
(0,15 моль) циклогексиламина и выделяли выпавшую ЦГА-
сольп-метоксикарбобензокси-№-мезитиленсульфонил-£-арги-
нина. Выход 56,7 г (61 %), т. пл. 125—128 °C (метанол — аце-
тонитрил); [а]’о = + 5,5° (с = 0,7; метанол).
Аналогично получают ЦГА-соли Boc-Mts-аргинина (вы-
ход 68 %, т. пл. 122—125°С;[а]р = +15,1 (с = 0,5; метанол))
HCbz-NG-Mts-aprHHHHa (выход62 %,т. пл. 113—114°С; [а]д
= +5,5Q (с = 1,1; метанол)).
2. К 10,5 г (17 ммоль) ЦГА-соли п-метокси-№-М1з-арги-
нина в 100 мл метанола прибавляют 17 мл 1 н. НО, раствори-
тель удаляют, остаток растворяют в этилацетате, промывают
10 % лимонной кислотой и раствором NaCl, упаривают, оста-
251
ток обрабатывают 25 мл ТФУ в присутствии 5 мл анизола
60 мин при 0 °C. Продукт осаждают эфиром, растворяют в во-
де, доводят 5 %-м раствором аммиака pH до 6 и выпавший
осадок №-мезитилен-2-сульфониларгинина перекристаллизо-
вывают из воды. Выход 5,3 г (88 %), т. пл. 158—161 °C; (ala =
= — 5,2° (с == 0,4; метанол). Аналогично продукт получают
обработкой как указано выше Вос-№-М1з-аргинина или гид-
рированием СЬг-№-М1з-аргинина в метаноле над Pd (выход
99//о).
Удаление боковых защитных групп аргинина
В работе [375] показано, что при обработке 10 мкмоль
№-М1з-аргинина 50 экв. метансульфокислоты (или трифтор-
метансульфокислоты) в присутствии 5 экв. анизола Mts-rpyn-
па удаляется на 98,2 % при 20 °C в течение 1 ч, а смесью
ТФМСК—ТФУ (1:1) —на 100 %.
В работе [423] Mts-группу удаляли раствором 1 — 1,5 М
метансульфокислоты и 1 М тиоанизола в ТФУ в течение 1,5 —
2 ч при 0 °C.
Нитрогуанидиновая группа обычно деблокируется при
каталитическом гидрогенолизе; аналогично отщепляется кар-
бобензоксигруппа. Указанные защитные группировки и так-
же тозильная группа гладко отщепляются безводным фторис-
тым водородом; эту методику используют в основном при
твердофазном методе синтеза пептидов [16]. Метоксифенил-
сульфонильная группа легко удаляется метансульфокислотой
или тристрифторацетатом бора [424].
Методика 230. Деблокирование гуанидиновой rpvnnbi
(о-нитроаргинина.
со-Нитрогруппу нитроаргинина отщепляют при катали-
тическом гидрировании над палладием (см. главу 2). Для этих
целей можно также использовать другие восстановители, на-
пример двухлористое олово [418] или цинк в трифторуксусной
кислоте [425].
Удаление нитрогруппы гидрированием над палладиевой
чернью продолжается обычно 12—30 ч; иногда для этого
требуется повышенная температура. Для удаления нитро-
группы весьма удобны методы восстановления без примене-
ния водорода (см. главу 2). Например, при обработке нитро-
аргинина муравьиной кислотой или формиатом аммония в
присутствии палладия защитная группа отщепляется в пер-
вом случае за 5 ч, а во втором — за 10 мин. Практика нашей
работы показала, что нитрогруппа легко удаляется циклогек-
сеном, в частности при этом легко деблокируются те нитро-
252
аргининсодержащие пептиды, которые плохо деблокируются
каталитическим гидрированием.
Методика 231. Деблокирование нитрогуанидиновои
группы циклогексеном.
К суспензии 745 мг (1,05 ммоль) Вос-фенилаланил-нитро-
аргинил-триптофанил-глицина в 14 мл циклогексена добав-
ляют 28 мл этилового спирта и 3 мл уксусной кислоты. При
непрерывном размешивании в колбу вносят 75 мг свежепри-
готовленной палладиевой черни и смесь кипятят до полного
деблокирования пептида, что контролируют при помощи ТСХ;
обычно на это требуется около 6 ч. Катализатор отфильтро-
вывают, растворитель удаляют на роторном испарителе при
температуре не выше 40 °C, а остаток промывают водой для
удаления ацетата аммония. Продукт сушат в вакуум-эксика-
торе и после растворения его в метаноле осаждают этилаце-
татом. Выход Вос-фенилаланиларгинил-триптофанил-глици-
на 670 мг (88 %), т. пл. 175—180 °C.
Методика удаления нитрогруппы безводным фтористым
водородом описана в монографии [16].
Из других защитных групп гуанидиновой функции аргини-
на рассмотрим удаление со-и-метоксифенилсульфонильной
(MBS-группы). Как известно, MBS-группа устойчива к дейст-
вию бромистого водорода в уксусной кислоте, но легко отщеп-
ляется безводным фтористым водородом, трис(трифторацета-
том) бора и метансульфокислотой.
Методика 232. Удаление MBS-группы метансуль-
фокислотой [374].
34,4 г (0,1 ммоль) co-MBS-аргинина обрабатывают при
комнатной температуре 0,5 мл метансульфокислоты, содер-
жащей 0,025 мл анизола, в течение 40 мин, затем к смеси до-
бавляют 10 мл эфира и выпавшее масло многократно расти-
рают с эфиром.
Защита имидазольного кольца гистидина
Несмотря на то, что имидазольное кольцо гистидина об-
ладает основными свойствами, ряд пептидов, содержащих
гистидин, был синтезирован без защиты этого ядра. Хотя при
этом не было отмечено побочных реакций, использование сво-
бодного гистидина вызывает некоторые затруднения и прежде
всего — это невозможность промывания продукта реакции
разбавленной кислотой, поскольку гистидинсодержащие
пептиды растворяются в кислых растворах. Применение N,
М*'”-дикарбобензоксигистидина устраняет указанное затруд.
нение. Однако данное соединение обладает одним существен,
ным недостатком: оно крайне неустойчиво при хранении.
253
В связи с этим реагент не нашел широкого применения при син-
тезе пептидов. Для блокирования имидазольного кольца гис-
тидина в настоящее время успешно используют Ы/т-бензил-
гистидин, который был предложен еще ДюВиньо и Беренсом
[379]. Бензильную группу можно удалить натрием в жидком
аммиаке или при каталитическом гидрировании в течение 72 ч.
Гидрирование над палладием в присутствии циклогексена
позволяет отщепить бензильную группу за 3 ч (см. главу 2)
Для введения гистидина в N-концевую часть молекулы пеп-
тида используют ди-трет-бутилоксикарбопилгистидин [289].
В последние годы в качестве весьма удобной защитной груп-
пы имидальзолыюго кольца гистидина находи? применение
тозильная группа [291]. Ее можно легко удалить 1-оксибен-
ззтоиазолом или ангидридами карбоновых кислот в пириди-
не [380].
Методика 233. Ы^-Бензил-Л-гистидин [379]
н2п-сн-соон
(Jh2
h2n-ch-cooh
/ \ PhCHoC)
Nx NH ----------2—<
В круглодонную трехгорлую колбу вместимостью 500 мл,
снабженную мешалкой с ртутным затвором и содержащую
200 мл безводного жидкого аммиака, добавляют 20 г монохлор-
гидрата Г-гистидина, охлаждая колбу до ~70 °C смесью сухо-
го льда и трихлорэтилена. При размешивании к реакцион-
ной массе добавляют небольшими порциями 9 г натрия до по-
явления устойчивой синей окраски раствора, после чего смесь
обесцвечивают добавлением небольшого количества гистидина.
К светло-коричневой реакционной смеси, содержащей бесцвет-
ный осадок, при интенсивном перемешивании медленно добав-
ляют по каплям 12 мл хлористого бензила. Перемешивают
еще 30 мин и оставляют при комнатной температуре для испа-
рения большей части аммиака. Следы аммиака удаляют в
вакууме. Остаток растворяют в 100 мл воды, охлажденной
до 0 °C; полученный раствор тотчас же промывают эфиром, а
затем через водный слой просасывают воздух при помощи во-
доструйного насоса до исчезновения запаха эфира. Раствор
фильтруют и при размешивании медленно добавляют 2 н. сер-
ную кислоту до начала выпадения осадка (pH 8—8,5). Смесь
оставляют на 3 ч при 0 °C, выпавший осадок фильтруют, про-
мывают и кристаллизуют из 70 %-го спирта. Выход бензил,
гистидина 13,4 г (57 %), т. пл. 248—249 °C; [odif =* +20,5е
(с « 2; вода с 1 эквивалентом НС1).
254
Ы^-Тоэнл- L -гистидин [380]
Z —NH —СН —СООН Z —NH —СН —СООН HBrHsN-CH~COOH
СН2 <гН2 <*Нг
У’х То» —CI ,/=\i т НВг/АсОН к/5==\,
NH-------•— Nx >N~Tos ---------------► Ы N-Tos
он-
Методика 234. Карбобензокси-М""-тозил-гисти-
дин [380].
К раствору 10 г (35 ммоль) Кбз-гистидина и 7 г Na2CO8 в
100 мл воды при энергичном перемешивании и охлаждении
до —10 °C добавляют порциями 9 г (47 ммоль) п-толуолсуль-
фохлорида в течение 30 мин. Реакционную смесь размешивают
потом 4 ч при комнатной температуре, а затем промывают
эфиром (2 X 50 мл) для удаления избытка п-толуолсульфо-
хлорида. Водную фазу отделяют, подкисляют 1 н. соляной
кислотой до pH 2,0, и масло экстрагируют этилацетатом (3 X
X 100 мл). Соединенные вытяжки сушат сульфатом магния,
и органический растворитель удаляют в вакууме. Остаток
растворяют в 100 мл этилацетата, добавляют 5,5 г дицикло-
гексиламина и оставляют кристаллизоваться. Образовавший-
ся осадок ДЦГА-соли отфильтровывают и продукт кристал-
лизуют из смеси метанол — этилацетат. Выход 15,4 г (70,5 %),
т. пл. 151—152 °C (разл.); [a]2J = +19,1° (с = 1; ДМФА).
Методика 235. Н<т-Тозил-£-гистидин [380].
Суспензию 10 г (16 ммоль) ДЦГА-соли Кбз-М^-тозил-Л-
гистидина в 1 н. серной кислоте энергично встряхивают
в делительной воронке с этилацетатом. После растворе-
ния осадка органический слой промывают водой до pH 7,0 и
сушат сульфатом магния. Эти л ацетат удаляют в вакууме водо-
струйного насоса, а остаток обрабатывают в течение 1 ч 20 мл
4 н. раствора бромистого водорода в ледяной уксусной кислоте
при комнатной температуре. Продукт осаждают добавлением
200 мл сухого эфира, его быстро отфильтровывают, промывают
сухим эфиром. После выдерживания в вакуум-эксикаторе над
. Таблица 44. Строение и свойства N-защищенных производных
тозилгистидина [380]
Формула соединения т. пл.» °C г„,25 [a]D» град Растворитель
Вос-His (Tos) ОН 160—162 4-26,5 с == 1; метанол
Аос-His (Tos) ОН 109—111 +ю,о с = 1; пиридин
Nps-His (Tos) ОН 141—142 +39,9 с = 1; ДМФА
Врос-His (Tos) ОН 123—125 + 15,1 с == 1; метанол
255
едким натром бромгидрат тозилгистидина растворяют в 100 мл
метано за и тозилгистидин осаждают, добавляя 6 мл сухого
пиридина для связывания НВг. Осадок отфильтровывают и
тщательно промывают спиртом. Выход 3,5 г (51,7 %), т. пл.
140—145 °C.
Из тозилгистидина обычными методами могут быть полу-
чены его N-защищенные производные. В табл. 44 приведены
формулы некоторых из них и их физико-химические характе-
ристики.
Удаление тозильной группы Н 1т -тозилгистидина
Защитная группа №т-тозилгистидина устойчива к катали-
тическому гидрированию, действию трифторуксусной кисло-
ты или бромистого водорода. Ее можно удалить 1 н. раство-
ром NaOH (1 ч, 20 °C), безводным фтористым водородом (30 мин,
0 °C), 1-оксибензтриазолом (1 ч, 20 °C), натрием в жидком
аммиаке и, наконец, ангидридами карбоновых кислот в пири-
дине.
Методика 236. Удаление защитной группы Ы1Ш-тозил-
гистидилпептидов.
1. К раствору 1 моль защищенного пептида в третрагидро-
фуране или диоксане добавляют 2 моль 1-оксибензтриазола
в тех же растворителях и выдерживают при комнатной тем-
пературе в течение 1 ч. Выделение пептида ведут обычными
методами [380].
2. К смеси 25 мл уксусного ангидрида и 0,5 мл пиридина
добавляют 2,6 ммоль защищенного пептида, и раствор переме-
шивают 3,5 ч при комнатной температуре. Выделяют пептид,
как обычно [380].
Методика 237. Синтез трет-бутилоксикарбонил-^'п-
динитрофени л гистидина [381]
он- ______ \NOa
Вос-His—OMe —-—> Boc-His-OH Вос—His—ОН.
I
Dnp
2,7 г (10 ммоль) метилового эфира mpem-бутилоксикарбо-
нил-£-гистидина растворяют в 20 мл спирта и добавляют 10 мл
1 н. NaOH. Через 45 мин раствор нейтрализовали при 0 °C
1 н. раствором НС1 и отогнали спирт на роторном испарителе.
К полученному раствору добавляют раствор 2,3 г NaHCO3 в
10 мл воды, а затем медленно прибавляют раствор 1,4 мл 2,4-
динитрофторбензола в 15 мл метанола в течение I ч. Реакци-
онную смесь оставляют на 10 ч при комнатной температуре.
Метанол отгоняют на роторном испарителе, а водную фазу
256
промывают эфиром (3 раза по 20 мл) , подкисляют до pH 3,5 1 н.
HCI при 0 °C и экстрагируют три раза по 20 мл этилацетата.
Экстракты соединяют, промывают водой, сушат 'над сульфа-
том магния и отгоняют этилацетат в вакууме. Маслянистый
продукт трижды кристаллизуют из смеси этанол—петролейный
эфир (т. кип. 30—60 °C). Выход 2,6 г (62 %), т. пл. 94 °C;
lain = +55,3° (с=1; этилацетат).
Удобными для использования в пептидном синтезе оказа-
лись производные №',Мм,-ди-трет-бутилоксикарбонил-£-гис-
тидина. N"”-Boc-rpynny удается селективно удалить обра-
боткой ди-Вос-производного метанольным раствором аммиака
при 20 °C в течение 24 ч [427].
Методика 238. Получение кристаллических производ-
ных Na,N ‘т-ди-/прет-бутил-оксикарбонил-£-гистидина [428].
К раствору 10 ммоль L-гистидина и 1,5 г карбоната калия
в 10 мл воды добавляют 10 мл изопропилового спирта и
23 ммоль ди-трет-бутил-пирокарбоната. Смесь выдерживают
1,5 ч при комнатной температуре, разбавляют водой до 50—
70 мл и экстрагируют 15—20 мл петролейного эфира. Водный
раствор подкисляют 2—6 г кристаллической лимонной кис-
лоты и экстрагируют продукт этилацетатом (20 + 2х 10 мл),
экстракт промывают водой, раствором NaCl, упаривают в ва-
кууме и получают 4 г ди-Вос-гистидина. Его растворяют в
15—30 мл сухого эфира, охлаждают до 0—5°, прибавляют
1,2 мл диэтиламина и выдерживают в холодильнике 1 ч. Оса.
док отделяют, промывают эфиром и сушат над серной кисло-
той. Выход диэтиламмонийной соли 84,3 %,т. пл. 115—116°С;
[<x]d = +46,0° (с= 1; метанол).
Аналогично получают циклогексиламмонийную соль. Вы-
ход 88 %, т. пл. 140—142 °C; [а]о — 38,8°(с — 1; метанол).
Описано получение пентахлор- и пентафторфениловых эфи-
ров ди-Вос-гистидина [429].
Перспективной защитной группой для гистидина является
М^-фенацильная группа [382]. Она устойчива в кислой и
щелочной среде и удаляется цинком в уксусной кислоте
(10 мин), гидрированием над палладием в 80 %-й уксусной
кислоте (2 ч) и обработкой бромистым водородом в трифторук-
сусной кислоте (1,5 ч).
17 2-882
Синтез N irn -фенацил-А- гистидина [382]
( Л )
AgOAc
80%АсОН
С
NaOH II
----- Вос -NH-CH-C-OH
Методика 239. Метиловый эфир Ма-/7гре/тг-бутилокси-
к арбонил-^^-фенацил-Л-гистидина [382].
1. Растворяют 6 г (12 ммоль) метилового эфира Na-Boc-
N^-Trt-L-гистидина в 10 мл эфира и добавляют 2,39 г
(12 ммоль) фенацилбромида при перемешивании. Через 48 ч
отделяют осадок, промывают его эфиром (4 х 20 мл) и сушат.
Выход бромида 8,1 г (95 %); т. пл. 177—179 °C; [alp = —9,2
(с = 1; метанол).
2. 7,1 г (10 ммоль) полученной соли растворяют в 20 мл
80 %-й уксусной кислоты и добавляют 1,67 г (10 ммоль) ацета-
та серебра при интенсивном перемешивании. Отфильтровыва-
ют осадок бромида серебра и перемешивают раствор 48 ч при
комнатной температуре. Растворитель отгоняют в вакууме и
к остатку добавляют насыщенный водный раствор (100 мл)
бикарбоната натрия. Смесь экстрагируют эфиром (2 х 50 мл),
органическую фазу сушат и оставляют стоять 48 ч при комнат-
ной температуре. При этом выпадают кристаллы метилового
эфира №с-Вос-М(лг)-фенацил-Л-гистидина. Выход3,25 г (84 %);
т. пл. 139—141 °C; [а]р = —9,3 (с = 1; метанол).
Методика 240. На-Вос-Ц^-фенацил-Л-гистидин [382).
Растворяют 3 г (7,8 ммоль) полученного по приведенной
выше методике продукта в 3 мл метанола, добавляют 7,8 мл
258
1 н. NaOH и смесь перемешивают 1 ч при комнатной темпера-
туре. Разбавляют водой, упаривают метанол и подкисляют
1 н. НС1 до pH 3,8. Затем насыщают водную фазу NaCl и экст-
рагируют ее метиленхлоридом (6 х 50 мл). После сушки над
сульфатом натрия, упарки метиленхлорида и обработки ос-
татка эфиром получают желтый осадок, который переосажда-
ют из смеси метанол — эфир. Выход 2,57 г (85 %); т. пл. 181 —
183J°C;[a]p = +9,4 (с = 1; метанол).
В работе [382] описано также получение карбобензокси-
производного ^£)-фенацил-£-гистидина; т. пл. 205—208 сС;
[allo = +10,9° (с = 0,5; пиридин); +2,8° (с = 0,6; метанол).
Методика 241. Удаление фенацильной группы цинком
в уксусной кислоте [382].
- Растворяют 0,083 моля N^-фенацил-гистидинсодержа-
щего пептида в 50 %-й водной уксусной кислоте
(3 мл) и добавляют 100 г (1,5 моль) цинковой пыли порциями
при перемешивании в течение 5 мин. Пропускают через рас-
твор сероводород и отфильтровывают сульфид цинка. Фильт-
рат упаривают досуха, остаток растворяют в 0,01 М триметил-
аммоний-ацетатном буфере (2 мл, pH 5,0), добавляют 0,5 мл
0,27 М раствора динатриевой соли ЭДТА и пропускают через
колонку (0,9 х 30 см) с КМ-целлюлозой Watman СМ-32.
Защита гидроксильной группы тирозина
Из-за высокой реакционной способности оксигруппы ти-
розина ее, как правило, защищают при синтезе тирозинсодер-
жащих пептидов. Для этой цели чаще всего применяют бен-
зильную защиту, которая в бромистом водороде более устой-
чива, чем Кбз-группа. Бензильную защиту можно легко
удалить каталитическим гидрогенолизом. Обычно применяют
N-защищенные производные бензилтирозина, а также его мети-
ловые, этиловые и бензиловые эфиры. Помимо бензильной
защиты для блокирования гидроксила тирозина нашла приме-
нение также /пр£7и-бутильная группа [390], а в твердофазном
методе синтеза пептидов для тех же целей предложены 3-бром-
бензил- и 2,6-дихлорбензильные производные [367, 386], ко-
торые в 50 раз более устойчивы, чем О-бензилтирозин при об-
работке пептидов трифторуксусной кислотой. Бензильная и
замещенные бензильные группы отщепляются при обработке
безводным фтористым водородом в течение 1 ч.
Для получения пептидов с N-концевым остатком тирози-
на удобно применять Ы,О-дикарбобензокситирозины. После
завершения синтеза обе Кбз-группы можно легко удалить ка-
талитическим гидрированием.
17*
259
При твердофазном методе синтеза пептидов широкое при-
менение нашел О-2,6-дихлорбензил-Л-тирозин, который ока-
зался более устойчивым, чем О-бензилтирозин в условиях мно-
гократной обработки пептидил-полимера трифторуксусной
кислотой. После завершения синтеза защитная группа легко
удаляется безводным фтористым водородом в течение 1 ч
при О °C.
/Методика 242. О- Бензил -А-тирозин [385]
~ H2N-CH-COO“
I
сн2
он
Си -I- 2CGH5CH2Br
h2n—СН-СОО—
2H3N<-CH--COOH
СН2
I
овн
НС1
СВ;!
К раствору 72,4 г (0,4 моль) L-тирозина в 200 мл 2 н. едко-
го натра прибавляют водный раствор 49,9 г CuSO4 ♦ 5Н2О и
нагревают некоторое время на водяной бане. После охлаж-
дения выпавший медный комплекс растворяют, добавляя
1500 мл метанола и 200 мл 2 н. едкого натра, после чего за один
прием в реакционную колбу добавляют 50 мл бензилбромида.
Смесь размешивают 1 ч при комнатной температуре. Выпав-
ший медный комплекс О-бензил-А-тирозина отфильтровыва-
ют, промывают на фильтре смесью метанол — вода (1 : 3,5),
затем сушат до постоянного веса при 60 °C. Выход продукта
88—96 г (72—79 %).
Полученный комплекс многократно растирают с 1 н. НС1,
отсасывают и последовательно промывают на фильтре водой,
разбавленным аммиаком и смесью ацетон — эфир. После
кристаллизации из 80 %-й уксусной кислоты получают 66—
72 г (60—65 %) бесцветных кристаллов с т. пл. 223 °C; [а]р —
= —9,9° (с = 1; 80 %-я уксусная кислота).
Методика 243. П,О-Дикарбобензокси-£-тирозин [430J.
К раствору 11,5 г (0,06 моль) L-тирозина в 60 мл 2 н. едко-
го натра поочередно добавляют небольшими порциями в те-
чение 1 ч при 0 °C 25 г (21 мл) карбобензоксихлорида и 40 мл
4 н. едкого натра. Реакционную смесь интенсивно перемеши-
260
вают и скорость подачи реагентов регулируют таким образом,
чтобы кислотность среды поддерживалась при pH 9—11. Пе-
ремешивание продолжают до тех пор, пока температура рас-
твора не достигнет 20 °C, после чего раствор экстрагируют эфи-
ром (2 х 100 мл), водный слой отделяют и подкисляют 4 н.
HCI до pH 3, выпавший продукт экстрагируют эфиром (2 х
X 100 мл), органическую фазу промывают водой, сушат суль-
фатом магния, растворитель упаривают на роторном испарите-
ле, а остаток дважды кристаллизуют из четыреххлористого
углерода. Выход 25,3 г (89 %), т. пл. 117 °C; [а]д = —5,0°
(с = 1; 10 %-я уксусная кислота).
Методика 244. 0-(2,6-Дихлорбензил)-£-тирозин [386].
К раствору 3,6 г (20 ммоль) L-тирозина в 40 мл-(40 ммоль)
1 М едкого натра добавляют при перемешивании 17 г (10 ммоль)
СиС12 • 2Н2О и 20 мл метанола. Зеленовато-голубой осадок
вскоре растворяется при перемешивании. К реакционной мас-
се добавляют раствор 4,5 г (18,8 ммоль) 2,6-дихлорбензилбро-
мида в 25 мл метанола; она приобретает темно-голубую ок-
раску. Смесь размешивают 13 ч при комнатной температуре,
а затем фильтруют. Осадок на фильтре последовательно про-
мывают смесью вода — метанол (1 : 1 по объему), эфиром (2 х
х50 мл), затем растворяют в горячей смеси 50 мл уксусной
кислоты, 17 мл 1 М НС1 и 2,92 г (10 ммоль) ЭДТА. Горячий
голубой раствор фильтруют, удаляя нерастворившуюся ЭДТА(
разбавляют 400 мл холодной воды. Образовавшуюся желеоб.
разную массу отделяют на фильтре, хорошо промывают во-
дой и сушат в вакууме. Выход 2,64 г (41 %), т. пл. 211—212 °С;
Wd = —10,6° (с = 2; 80 %-я уксусная кислота).
тре/п-Бутилоксикарбонил-О-(2,6-дихлорбензил)-Ь-тирозин
может быть получен любым из указанных ранее способов
введения Вос-группы (см. главу II). Его т. пл. 108—ПО °C;
[а]д = +21,0° (с = 2; этиловый спирт) [431].
Удобным соединением для введения тирозина является
П,О-ди-тре/п-бутилоксикарбонил-£-тирозин, синтез которо-
го приводим ниже.
Методика 245. Ди-шре/тг-бутилоксикарбонил-Л-тиро-
зин [432].
К суспензии 10 ммоль L-тирозина в 25 мл воды приливают
раствор 27 ммоль ди-/пре/п-бутилпирокарбоната в 15 мл изо-
пропилового спирта и при энергичном перемешивании добав-
ляют по каплям 8 М КОН до pH 12. Перемешивают реак-
ционную смесь при комнатной температуре, поддерживая
pH 11,5—12 (лучше контролировать pH-метром) до исчезнове-
ния пятна N-Вос-тирозина на ТСХ (силуфол; бензол : ацетон :
:уксусная_кислота — 100 : 50 : 1). Затем реакционную смесь
261
разбавляют водой (75 мл), экстрагируют эфиром (20 мл), промы-
вают эфирный экстракт 1 н. NaOH и объединенный водный
экстракт подкисляют до pH 2,5—3,0 и экстрагируют этил-
ацетатом (30 + 2 х 15 мл). Этилацетатный экстракт промы-
вают водой, сушат и упаривают в вакууме. Остаток растира-
ют с петролейным эфиром. Выход 3,6—3,8 г (94—99 %); т. пл.
95—97°С;[а]о = +29,3 (с = 1; диоксан).
Широкое распространение в синтезе пептидов получили
различные производные О-/прет-бутил-£-тирозина. Ниже при-
водится его синтез. Синтез О-тргт-бутил-Л-тирозина [390].
Методика 246. Этиловый эфир И-карбобензокси-Д-ти-
розина [390].
Вносят 131 г (0,626 моль) этилового эфира тирозина и
97,4 мл карбобензоксихлорида в раствор 202 г десятиводного
карбоната натрия в 385 мл воды и перемешивают при комнат-
ной температуре 2 ч. Полученный продукт отделяют, промы-
вают водой в растворе этилацетата, сушат и отгоняют раство-
ритель. После перекристаллизации из смеси этилацетат-пет-
ролейный эфир получают 205,7 г (95,7 %) кристаллического
продукта с т. пл. 88—91 °C.
Методика 247. N-Карбобензокси-А-тирозин [390].
80 г (0,233 моль) этилового эфира карбобензокси-£-тиро-
зина в растворе 500 мл смеси диоксан — вода (4:1) обраба-
тывают 2 ч 223 мл 2 н. едкого натра (0,466 моль). Отгоняют в
вакууме диоксан, раствор разбавляют водой, охлаждают и
подкисляют 5 н. серной кислотой. Выпавшее бесцветное масло
быстро закристаллизовывается. Кристаллы отделяют, рас-
творяют в растворе бикарбоната калия и опять осаждают 2 н.
серной кислотой. После сушки в вакууме над Р2О5 получают
67 г (91,2 %) продукта с т. пл. 92—95 °C.
Методика 248. n-Нитробензиловый эфир N-карбобен-
зокси-Д-тирозина [390].
Кипятят 62 г (около 200 ммоль)1 Ы-карбобензокси-Д-тиро-
зина, 42 мл (300 ммоль) триэтиламина и 64,8 мл (300 ммоль)
/г-нитробензилбромида в 300 мл этилацетата 8 ч. Разбавляют
реакционную смесь метиленхлоридом и продукт выделяют
обычным образом. Кристаллизуется в виде призм из смеси
этилацетат-бензол-петролейный эфир с т. пл. 117—119 °C;
[а]р — —11,16° ± 0,5 {с = 1; метанол).
Методика 249. n-Нитробензиловый эфир N-карбобен-
зокси-(О-трет-бутил)-А-тирозина [390].
Получают в автоклаве обычным образом (см. главу 3) из
40 г описанного выше эфира, 400 мл изобутилена и 3 мл кон-
центрированной серной кислоты в растворе 240 мл метилен-
хлорида. Перекристаллизовывают из смеси этилацетат-бензил-
петролейный эфир. Выход 35,9 г (80 %); т. пл. 73,5—74,5 °C.
262
Методика 250. О-трет-бутл-Ь-трозт (390).
Гидрируют 20 г описанного выше продукта над палладием
в смеси метанол-уксусная кислота — вода (6 : 1 : 1). После
окончания гидрирования катализатор отфильтровывают, от-
гоняют в вакууме растворитель и перекристаллизовывают ос-
таток из смеси вода — метанол. Выход 8,3 г (85 %); т. пл.
248—249,5 °C (с разл.); [а]2/? = —25,77 ± 0,5 (с = 1; вода).
Защита гидроксильной группы
серина и треонина
Довольно часто остатки серина и треонина вводят в расту-
щую полипептидную цепь без защиты гидроксильной группы
этих аминокислот. Однако возможность побочных реакций
привела к пониманию того, что лучше использовать при син-
тезе пептидов производные серина и треонина с блокированной
ЙН-группой. Наиболее широко блокирование гидроксйла се-
рина и треонина достигается за счет превращения его в соот-
ветствующие бензиловые или mpem-бутиловые эфиры.
Синтез О-бензил-Ь-серина до недавнего времени был со-
пряжен с рядом трудностей. Окава [433] впервые получил
О,Л-(0-бензил)-серин по следующей схеме:
СН2=СН—СООСН3 СН2Вг—СНВг—СООСН3 CHzON*-»-
СвН5—СНг-ОСН2—СНВг—соосн, ->
-> С„Н5—СН2—ОСН2—СНВг—СООН
-> С6Н5—СН2—ОСН2—СН—СООН.
I
nh2
Оптически чистый О-бензил-Л-серин был выделен при расщеп-
лении Р,£-изомера N-ацетил-О-бензил — серина ферментом
такадиэстеразой. Вюнш и Ферст разработали способ разделе-
ния рацемического М-формил-(О-бензил)-О,£-серина на оп-
тические антиподы через соли с бруцином или хинином [434].
По-видимому, более удобным методом разделения рацемиче-
ского продукта является преимущественный гидролиз одно-
го из изомеров, как это было осуществлено Чи-Хуэй Вонгом
и соавт. [435] при обработке метилового эфира М-Вос-(О-бен-
зил)-Р,Ь-серина папаином:
Boc-D,Z.-Ser—ОМе —fa™ - Boc-L-Ser-OH + Boc-D-SerOMe
OBzl J)Bzl J>Bzl
-+• Boc-£>-Ser (OBzl) OH-
(81 %)
В лабораторных условиях более пригоден метод синтеза Вос-
(О-бензил)-Г-серина, предложенный Сугано и Мийоши в
1976 г. [392].
263
Удаление О-бензильной защитной группы осуществляется
довольно легко безводным фтористым водородом или же ката-
литическим гидрогенолизом в присутствии палладия.
Методика 251. трет-Бутилоксикарбонил-(О-бензил)-
L-серин [392]
Boc-L-SerOH 'Тип Boc-L-SerOH
OBzl
К раствору 2,05 г (10 ммоль) Boc-L-серина в 50 мл диме-
тилформамида добавляют при 0 °C 820 мл (22 ммоль) 65 %-го
гидрида натрия; при этом выделяется водород. Когда водо-
род перестанет выделяться, к раствору прибавляют 1,88 г
(11 ммоль) свежеперегнанного бензилбромида и реакционную
массу размешивают 5 ч при 25—30 °C. Растворитель удаляют
в вакууме на роторном испарителе при температуре 40 °C,
остаток растворяют в 50 мл воды и экстрагируют эфиром (2 *х
X 20 мл). Водную фазу отделяют и подкисляют 3 н. НС1 до
pH 3,5, а продукт экстрагируют этилацетатом (5 х 20 мл).
Комбинированные органические вытяжки промывают водой,
сушат сульфатом магния. После удаления этилацетата в ва-
кууме получают бесцветное масло; его растворяют в 30 мл эфи-
ра и добавляют 0,9 г дициклогексиламина. Кристаллы от-
фильтровывают и кристаллизуют из этилацетата. Выход 2,2 г
(47 %), т. пл. 159—160 °C; [a]2D5 + 29,0° (с = 1; метанол).
Методика 252. /ире/и-Бутиловый эфир (О-тре/п-бутил)-
L-серина [393].
Суспензию 2,1 г (20 ммоль) L-серина в 20 мл диоксана сме-
шивают при 0 "°C с 1,5 мл концентрированной серной кислоты
и 5,4 мл изобутилена и встряхивают в течение трех дней при
комнатной температуре. Затем реакционную массу нейтрали-
зуют триэтиламином, растворитель упаривают на роторном
испарителе, остаток растворяют в этилацетате и Промывают
насыщенным раствором бикарбоната натрия, сушат сульфатом
натрия и растворитель отгоняют на роторном испарителе.
Остаток перегоняют в вакууме и получают продукт, кипящий
при 70—71 °C (0,9 мм рт. ст.) в виде масла с выходом 83 %;
Ыя = —2,9° (с = 1; метанол). Условия удаления Q-tnpem-
бутильной группы: HBr/TFA или HC1/TFA 30 мин, TFA — 6 ч.
Защита меркаптогруппы цистеина
Несмотря на значительное количество защитных групп,
предложенных для блокирования SH-группы цистеина, поиск
новых реагентов продолжается, так как каждая из используе-
мых групп имеет ряд недостатков. Наиболее широко применя-
264
емой защитной группой является S-бензильная группировка^
предложенная еще в 1935 г. Зиффердом и ДюВиньо 1394]. Эта
защитная группа гладко удаляется натрием в жидкрм аммиаке
или безводным фтористым водородом при комнатной темпе-
ратуре. В еще более мягких условиях отщепляется безводным
фтористым водородом n-метоксибензильная группа, введенная
в пептидный синтез Сакакибара с сотр. [398]; ее целесообразно
использовать в твердофазном методе синтеза пептидов. Из но-
вых защитных групп перспективными являются, по-видимо-
му, ацетамидометильная и бензаминометильная группы, они
легко удаляются ионами ртути в кислой среде.
Методика 253. 8-Бензил-/.-цистеин [436]
НС1. h2n-ch-cooh h2n-ch-cooh
CH2SH
CH2S—Bzl
Раствор 157,5 г (1 моль) хлоргидрата L-цистеина в 2 л ох-
лажденного раствора едкого натра смешивают с 256,5 г
(1,5 моль) бензилбромида. Смесь встряхивают несколько ча-
сов до образования гомогенного раствора, после чего подкис-
ляют уксусной кислотой до pH 5,0. Образующийся осадок от-
фильтровывают, хорошо промывают водой и, наконец, сушат
в вакуум-эксикаторе над оксидом фосфора. Выход 169 г (80 %)„
т. пл. 216—218 °C; [a]2D5 = +23,5° (с= 1, 1 н. NaOH).
В условиях деблокирования пептидов фтористым водоро-
дом n-метоксибензильная группа удаляется в цистеинсодер-
жащих пептидах более гладко, чем бензильная, поэтому она
нашла применение в твердофазном методе синтеза [14].
Методика 254. Хлоргидрат S-4-метоксибензил-Л-ци-
стеина [437].
* Растворяли 78 г (0,5 моль) хлоргидрата L-цистеина в 0,5 л
2 н. NaOH и 600 мл этанола, добавляли 103 г (0,55 моль) 4-
метоксибензилбромида при перемешивании. Через несколько
минут начинается экзотермическая реакция и выпадает оса-
док. Перемешивая еще 45 мин, доводим до pH 6—7, охлажда-
ем смесь до 0 ± 5° и отфильтровываем осадок. Промываем его
на фильтре водой, спиртом и эфиром. Выход 63 г (52 %), т. пл.
215—223 °C (из воды).
Boc-S-4-метокси-Л-цистеин плавится при 90—92 °C; [а]^° =
== —22,5 (с = 1; этанол).
Синтез
$-ацетамидометил-Ьцистеина [395]
Методика 255. N-Оксиметилацетамид [438]
О
CH3CONH2 h БСНО СН3—С—NH—СН2ОН,
265
В стакан емкостью 250 мл помещали 120 г (2 моль) 98,3 %
ацетамида и 62 г (2 моль) 96,8 % параформа, приливали 5 мл
50 % раствора КОН. и нагревали при перемешивании стек-
лянной палочкой до 60—65° (при этом реакционная смесь раз-
жижается). Выдерживали еще 30—40 мин до растворения кру-
пинок параформа. Выход технического продукта в виде густо-
го светло-желтого масла 170—175 г (95 %).
Методика 256. Хлоргидрат S-ацетамидометил-Л-цис-
теина [395]
Смесь 127 г (1,43 моль) N-оксимети л ацетамида и 228 г
(1,3 моль) гидрдта хлоргидрата L-цистеина, растворенного в
350 мл воды,-охлаждали на ледяной бане и добавляли в нее
50 мл концентрированной НС1 (pH 5). Колбу плотно закрыва-
ли и оставляли на 1—2 дня при комнатной температуре в ат-
мосфере азота. Когда ТСХ (н. бутанол-уксусная кислота —
вода — 10:2’3) показывала, что реакция прошла полнос-
тью (Rf — 0,19), добавляли абсолютный этанол и упаривали
несколько раз для полного удаления воды. Остаток после упа-
ривания растворяли в минимальном количестве метанола при
комнатной температуре и добавляли эфир до помутнения.
Смесь выдерживали несколько дней в холодильнике. Отфильт-
ровывали белый кристаллический осадок, промывали его эфи-
ром и сушили в вакууме. Выход 158 г (52 %); т. пл. 159—
163 °C (разл.); [ац>5 = —30,7° (с = 1; вода).
Методика 257. трет-Бутилоксикарбонил-5-ацетамидо*
метил-£-цистеин [245].
К раствору 2,28 г (10 ммоль) хлоргидрата S-ацетамидометил-
L-цистеина в 20 мл водного диоксана (1 ’ 1) и 15 мл 1 н. NaOH
добавляли 2,4 г (311 ммоль) ди-/пре/п-бутилпирокарбоната и
перемешивали 20 мин. После стандартной обработки получа-
ли 2,74 г (94 %) кристаллического продукта. После перекрис-
таллизации из этилацетата с эфиром т. пл. 110—112 °C; [ocJd =
= —31,7° (с = 1; вода).
S-Бензамидометилцистеин [396]
Методика 258. N-Оксиметилбензамид [439]
• 1СОН
\=/ \nh2 XNH-СН2ОН.
В колбу вместимостью 100 мл помещают 21 г (0,17 моль)
бензамида, прибавляют 0,6 г карбоната калия в 21 мл воды
и встряхивают при 50 °C, затем добавляют 12,3 мл формалина
(а =- 1,095) и продолжают встряхивание при температуре не
выше 50 °C до полного растворения бензамида. После этого
реакционную смесь оставляют на 24 ч при комнатной темпе-
ратуре. Бесцветные кристаллы отфильтровывают, промывают
холодной водой (2x5 мл), сушат и кристаллизуют из хло-
роформа (75 мл). Выход N-оксимети л бензамида 22 г (85 %),
т. пл. 100—104 °C.
М е т о д и к а 259. Хлоргидрат S-бензамидометил-Л-цис-
теина [396].
К смеси 45,6 г (0,3 моль) хлоргидрата Л-цистеина, 39,3 г
(0,3 моль) N-оксиметилбензамида и 200 мл воды, охлажден-
ной до 0°, добавляли при перемешивании 200 мл концентри-
рованной НО. Через 15 мин из раствора при комнатной тем-
пературе начинает выпадать осадок и вся масса быстро загус-
тевает. После охлаждения массы в течение нескольких часов
в холодильнике продукт отфильтровывают и перекристалли-
зовывают из смеси метанол — эфир. Выход 66,1 г (87 %);
т. пл. 168—168,5 °C (разл.); [а]р = —27,8 (с = 1; вода).
Методика 260. трет-Бутилоксикарбонил-Б-бензами-
дометил-Ь-цистеин [245].
К раствору 14,5 г (50 ммоль) хлоргидрата S-бензамидоме-
тил-Ь-цистеина в 200 мл ДМФА добавляли 7 мл триэтиламина,
перемешивали 15 мин, осадок отфильтровывали, к фильтрату
добавляли 3,5 мл триэтиламина и 15 мл (65 ммоль) ди-mpm-
бутилпирокарбоната и перемешивали еще 1 ч. После стан-
дартной обработки получили вязкое масло, легко кристалли-
зующееся растиранием с гексаном. Кристаллический продукт
отделяли и перекристаллизовывали из смеси этилацетат —
петролейный эфир. Выход 16,5 г (96 %); т. пл. 138—139 °C;
[а]д = —27,5 (с = 0,12; метанол).
Методика 261. Удаление S-бензамидометильной груп-
пы [440]
О
II 1 Но^4
Ph—С—NH—CH2—S-CH2-CH—СООН - ->H2N-CH-CQQH 4-
nh2 ch2-sh
о
II
+ HgS + Ph—C—NH—CH2OH.
37 мг (0,1 ммоль) трифторацетата S-бензамидометил-Л-цис-
теина растворяют при нагревании в 5 мл смеси метанол — вода
(1:1). К прозрачному раствору добавляют при комнатной
температуре 32 мг (0,1 ммоль) ацетата ртути и смесь переме-
шивают в течение 1 ч. Затем через раствор продувают в тече-
ние 10 мин ток сероводорода и образующийся осадок сульфида
ртути удаляют фильтрованием. Деблокированный продукт
выделяют обычным методом.
267
Bam-группа устойчива к действию щелочи, трифторуксус-
ной кислоты, гидразин-гидрата и других аналогичных реа-
гентов.
S-Тритильная защитная группа цистеина может селектив-
но удаляться действием ионов Hg2+ и Ag+ или иодом (с од-
новременным окислением в цистин) что позволило ей найти
широкое применение.
Методика 262. S-Тритил-Л-цистеин [4Q0].
Смесь 1,58 г (0,01 моль) хлоргидрата L-цистеина, 2,6 г
(0,01 моль) трифенилметанола в 10 мл ледяной уксусной кис-
лоты и 1,4 мл (10 % избыток) эфира трифторида бора нагре-
вали 30 мин на паровой бане, выдерживали температуру 20 °C
в течение 45 мин и переносили в стакан с 15 мл этанола. Обра-
батывали раствор 5 мл воды и 3 г безводного ацетата натрия.
Добавление 40 мл воды приводило к образованию вязкой мас-
сы, которая затвердевала при обработке холодной водой. Пос-
ле последовательных промывок водой, ацетоном и эфиром
продукт сушили в вакууме над Р2Об и NaOH. Выход 3,08 г
(85 %); т. пл. 181—182 °C (с разл.). Перекристаллизация из
смеси ДМФА — вода дает т. пл. 183,5 °C (с разл.);, [а]д =
= +114 ± 2 (с = 0,832; 0,04 н. НС1 в этаноле).
Методика 263. Ы-тре/и-Бутилоксикарбонил-З-тритил-
L-цистеин [245].
К раствору 0,73 г (2 ммоль) 5-тритил-£-цистеина в 4 мл
1 н. NaOH и 4 мл диоксана добавляют раствор 0,48 г (2,2 ммоль)
ди-/ире/и-бутилпирокарбоната в 2 мл mpem-бутилового спир-
та и смесь перемешивают 20 мин при 25 °C. После обработки
по стандартной методике получают 0,82 г продукта (88 %),
т. пл. 65—70 °C. ДЦГА-соль с т. пл. 210 °C; [а]2В = +25,8
(с = 0,11; этанол).
Перспективной является также S-трети-бутильная группа,
которая селективно удаляется о-нитрофенилсульфенилхло-
ридом, а также восстановительными агентами — тиолами и
боргидридом натрия. Кроме того, она гладко отщепляется три-
метилсилил-трифторметансульфонатом [242].
Методика 264. Хлоргидрат 5-трет-бутил-£-цистеи-
на [397].
Раствор 175,6 г (1 моль) хлоргидрата L-цистеина (гидрата)
в смеси 450 мл 2 н. НС1 и 123 мл (97 г; 1,3 моль) mpem-бута-
нола кипятят с эффективным холодильником 10—12 ч. Выде-
ляется некоторое количество изобутилена. Раствор концент-
рируют под уменьшенным давлением, отфильтровывают крис-
таллический продукт и промывают его сухим ацетоном. Вы-
ход 209 г (90 %), т. пл. 198—200 °C. Перекристаллизация из
4 н. НС1 дает т. пл. 204 °C; [a]2D° = +6,35 (с = 2,5; 1 н. НС1).
268
Методика 265. Ы-трет-бутилоксикарбонил-З-трт-
бутил-/.-цистеин [397].
К раствору 6,39 г (30 ммоль) хлоргидрата 5-тр<?гп-бутил-
L-цистеина в 50 мл воды добавляют 21 мл (150 ммоль) три-
этиламина и раствор 5,23 г (35,5 моль) Вос-азида в 20 мл диокса-
на при перемешивании. Через 48 ч диоксан отгоняют, остаток
экстрагируют эфиром, водный слой подкисляют бисульфатом
калия и выпавшее масло экстрагируют эфиром. После стан-
дартной обработки продукт осаждают петролейным эфиром из
концентрированного эфирного раствора. Выход 7,29 г (78 %);
т. пл. 89—90 °C; [а]д = —3 (с — 2; ацетон).
Недавно Фуджии с соавт. [441] предложили для деблоки-
рования и одновременного окисления тиольных групп цисте-
ина трифторацетат таллия (III) в трифторуксусной кислоте.
Реагент расщепляет в течение 60 мин такие S-защитные груп-
пы, как /npem-бутильную, адамантильную, ацетамидометиль-
ную, бензильную, тритильную и 4-метоксибензильную.
ЗАЩИТА АМИДНЫХ ГРУПП ГЛУТАМИНА
И АСПАРАГИНА
Как известно, введение в полипептидную цепь остатков
глутамина и аспарагина карбодиимидным методом сопровож-
дается иногда побочной реакцией дегидратации амидной груп-
пы. Однако если при этом добавить эквимолекулярное коли-
чество 1-оксибензтриазола или же использовать активирован-
ные эфиры глутамина или аспарагина, то указанный нежела-
тельный процесс не идет. Другим подходом для подавления
этой побочной реакции является использование в синтезе ас-
парагин- или глутаминсодержащих пептидов их производ-
ных с блокированными амидными группами. Следует подчерк-
нуть, что при этом улучшается растворимость пептидов в ор-
ганических растворителях, что было продемонстрировано, на-
пример, при синтезе бунгаротоксина [358].
4,4'-Диметоксибензгидрильная
группа (Mbh)
N-Вензилоксикарбонил (4, 4'-диметокси)-
бензгидрил -£ -глутамин [405]
ОСНз
СООН О ifY
I II /\/
СН—СН2—СНа—С—NH—сн
I
ZNH
269
Методика 266. 4,4'-Диметоксибензофенон [442]
4 ^-ОСН8
AiCI3
cs2
170,59 108,14
О
СН3О-^-ОСН3'
~ 242,27
107 г (0,63 моль) хлорангидрида анисовой кислоты смеши-
вают в 750 мл сероуглерода с 72,3 мл (0,68 моль) анизола и
90 г (0,68 моль) безводного хлористого алюминия. Смесь сра-
зу бурно закипает. Начинает выпадать хорошо кристаллизу-
ющийся желтый осадок и происходит слабое выделение хлорис-
того водорода. Реакционную смесь выдерживают ночь и на
следующий день кипятят еще 3 ч, а затем разлагают смесью со-
ляной кислоты со льдом и после стояния в течение нескольких
часов отфильтровывают осадок, а маточный раствор выбрасы-
вают. Осадок перекристаллизовывают из пропилового спирта.
Выход 135 г (88 %), т. пл. 143—144 °C.
Примечания. 1. Во время работы прибор должен
быть хорошо защищен от влаги. 2. Работа с сероуглеродом со-
пряжена с опасностью. Сероуглерод является самым легковос-
пламеняющимся из всех растворителей, а также сильным ядом
для нервной и кровеносной систем.
Методика 267. 4,4'-Диметоксибензгидриловый спирт
[405]
сн3о-/“^~с-^^-осн3 —аВ--4 >
II
о
242,27
-> сн3о-<^ Ч—СН-<^ ^-ОСН3
=/ i.H =
244,29
В раствор 96 г (0,5 моль) 4,4'-диметоксибензофенона в 1,5 л
96 %-ного этанола при нагревании вносят 8 г NaBH4, затем
кипятят 2—3 ч с обратным холодильником, фильтруют и вы-
ливают фильтрат в 4 л воды. Осадок отфильтровывают, еще
влажным растворяют в этйлацетате, сушат сульфатом натрия
и растворитель удаляют в вакууме. Остаток затирают с пет-
ролейным эфиром и отфильтровывают кристаллы.-Выход 76,8 г
(75 %), т. пл. 72—73 °C.
270
Методика 268. Ы-бензилоксикарбинол(4,4'-диметокси-
бензгидр и л) -L-гл утами и [405]
Z-GlnOH + СН3О-^“Ч—сн—S— ОСН3 -> Z-Gln(Mbh)—ОН.
\=/ ।
ОН
К раствору 28 г (0,1 моль) карбобензокси-Г-глугамина и
24 г (0,Х моль) 4,4-диметоксибензгидрилового спирта в 250 мл
уксусной кислоты добавляют при комнатной температуре 0,5 л
концентрированной серной кислоты, выдерживают раствор в
течение ночи при комнатной температуре и выливают в 750 мл
воды. Выпадает масло, которое вскоре закристаллизовыва-
ется. Осадок отфильтровывают и переводят в этилацетат, ор-
ганический слой после промывки водой сушат сульфатом нат-
рия и удаляют этилацетат в вакууме. Остаток растирают с эфи-
ром и отделяют кристаллы фильтрацией. После перекристал-
лизации их из смеси ТГФ — петролейный эфир выход соста-
вил 45,8 г (90 %), т. пл. 117—120 °C; [а]о = —6,75° (с = 2;
ДМФА).
Аналогично из 27 г (0,1 моль) карбобензокси-Л-аспарагина
в 300 мл уксусной кислоты получают 47,5 г (96 %) N-бензи-
локсикарбонил (4,4'-диметоксибензгидрил)-£-аспарагина. Т.
пл. 176—180 °C; [а]д = +2,43° (с = 2; Д/МФА).
Условия удаления Mbh-группы
4,4'-Диметоксибензгидрильная группа легко удаляется
трифторуксусной кислотой с анизолом при нагревании или
комнатной температуре, устойчива при каталитическом гидро-
генолизе и щелочной обработке. Приводим условия удаления
Mbh-группы [405]:
кипящая трифторуксусная кислота 15 мин
кипящая трифторуксусная кислота/анизол (10: 1) 5мин
трифторуксусная кислота/анизол (10: 1) при 22°С 2—3 ч
муравьиная кислота/анизол при 80 °C 1ч
Как видно из приведенных данных, в присутствии Mbh-
группы можно селективно удалять карбобензоксигруппу и
бензиловые эфиры гидрогенолизом, а также в мягких услови-
ях такие кислотолабильные группы, как Врос, Nps, Trt и неко-
торые другие. В то же время при удалении Mbh-группы одно-
временно удаляются Вос- и Аос-группы и расщепляются mpetn-
бутиловые эфиры.
Методика 269. 4,4'-Диметоксибензгидрил)-Д-глута-
мин [405]
z—L—Gin (Mbh) ОН H-L—Gin (Mbh)OH.
Гидрируют 5,2 г (10 моль) М-бензилоксикарбонил(4,4'-ди-
метокси)-Г-глутамина в растворе уксусная кислота — мета-
271
нол (1 ! 1) до прекращения выделения СО2. После фильтрации
раствор упаривают в вакууме, остаток растирают с эфиром и
образовавшийся осадок отфильтровывают. Выход 2,81 г
(76 %), т. пл. 205—208 °C; [а]д = +7,5° (с = 2: СН3СООН).
4,4'-Диметоксибензгидрил-7,-аспарагин получают-анало-
гично. После перекристаллизации из воды выход 59 %, т. пл.
226—230 °C; [а]д = +9,75° (с = 2; СН3СООН).
Методика 270. Метиловый эфир бензилоксикарбо-
нил(4,4'-диметоксибензгидрил)-£-аспарагил- (О-трет - бутил)-
А-тирозина [4051
Z- (Mbh)-Asn-Tyr-OMe TF~~* HAsn-Tyr-OMe.
OBu'
Раствор 1,45 г (2,02 ммоль) Z-Asn (Mbh)-Tyr (OBu*) ОН в
10 мл трифторуксусной кислоты и 1 мл анизола выдерживают
2 ч при температуре 20 °C. Упаривают растворитель в ва-
кууме при температуре бани 20 °C, остаток обрабатывают эфи-
ром, отфильтровывают осадок и промывают его эфиром. Вы-
ход 846 мг (94 %). Т. пл. 196—199 °C; [а® = +7,75° (с = 1;
ДМФА).
Методика 271. (О-/пре/п-бутил)-£-тирозил-(4,4'-диме-
токсибензгидрил)-£-глутамин [405]
Н —Tyr-GlnOH —H-Tyr-Gln-OH
। J
OBuz Mbh
2 р (О-трет-бутил)-£-тирозил-(4,4'-диметоксибензгид-
рил)-£-глутамина растворяют в 2 мл анизола и 20 мл трифтор-
уксусной кислоты и полученный раствор кипятят в течение
5 мин с обратным холодильником. Затем растворитель отго-
няют в вакууме, остаток растворяют в воде, водный слой экс-
трагируют эфиром, а затем встряхивают с ионообменником
«Леватит IR-45» (в ацетатной форме) до достижения pH 4,0.
После удаления смолы фильтрованием воду упаривают в ва-
кууме. Выход 1 г (95 %).
Защита индольного ядра триптофана
Хорошо известно, что индольное ядро триптофана в кис-
лой среде весьма склонно к окислению. Для того, чтобы избе-
жать этого, при удалении защитных групп триптофансодер-
жащих пептидов трифторуксусной кислотой добавляют в ре-
акционную смесь 2 % меркаптоэтанола или дитиотреитола,
а при обработке указанных пептидов безводным фтористым
водородом используют для тех же целей анизол. Разрушение
272
триптофана практически не идет при деблокировании катали-
тическим дегидрогенолизом или при обработке триптофансо-
держащих пептидов концентрированной муравьиной кисло-
той. Сузуки с соавт. [441] показали, что удаление Вос-группы
у триптофансодержащих пептидов можно проводить 2 М рас-
твором сульфокислот, например n-толуолсульфокислотой, ме-
та нсульфокислотой, этансульфокислотой в диоксане (или в
уксусной кислоте) в присутствии 2 % анизола и 2 % меркап-
тоэтанола при комнатной температуре в течение 20 мин без
разложения триптофана.
Согласно работам М. Оно с соавт. [407], удаление Вос-
группы триптофансодержащих пептидов можно осуществлять
0,1 н. НС1 в муравьиной кислоте. В этих условиях формирует-
ся индольное ядро триптофана, что защищает его от дальней-
ших нежелательных превращений.
Методика 272. №"-Формил-£-триптофан [407]
HCI
98% НСООН
HCI - h2n-ch-cooh
н2с
268’7 сно
Через раствор 7 г L-триптофана в 100 мл муравьиной кис-
лоты (98—100 %) при комнатной температуре пропускают
ток сухого хлористого водорода. В определенные промежутки
времени из реакционного сосуда отбирают аликвоту, разбав-
ляют ее водой и записывают УФ-спектр. Когда поглощение при
298 нм достигнет максимального значения (на что требуется
около 3 ч), растворитель удаляют в вакууме при слабом нагре-
вании. К сиропообразному продукту добавляют эфир, и фор-
милтриптофан кристаллизуется; его отфильтровывают и про-
мывают. Выход 9,2 г (100 %), т. пл. 218—220 °C; [а]р =
= —4,7° (с = 1,9; вода).
Примечание. №л-формил-£-триптофан не флуорес-
цирует в ультрафиолете и не дает фиолетовой окраски с реак-
тивом Эрлиха.
Методика 273. №-Вос-№"-формил-£-триптофан [407]
К раствору 2,68 г (10 ммоль) хлоргидрата №п-формил-£-
триптофана в 60 мл ДМФА добавляют 4,2 г (30 ммоль) три-
этиламина, а затем — 3 мл Вос-азида. Смесь перемешивают
при комнатной температуре в течение 2 дней. Вначале обра-
зуется гелеобразная масса, которая по мере протекания реак-
ции постепенно растворяется. Реакционную смесь выливают
в 300 мл 0,5 М раствора лимонной кислоты и выпавшее масло
экстрагируют 150 мл этилацетата. Органический слой про-
18 2 — 882
273
мывают 4 раза водой, сушат сульфатом натрия, растворитель
упаривают в вакууме при температуре 40 °C досуха. Сиропо-
образный остаток после растирания с петролейный эфиром
кристаллизуется; продукт отфильтровывают, промывают пе-
тролейным эфиром и сушат. Выход 2,9 г (89 %), т. пл. 102—
104 °C; [а]д = -|-22,Зо (с = 1,6; этилацетат).
Методи ка 274. Удаление Ы‘п-формильной группы
триптофана [407].
['[‘"-формильная группа триптофансодержащих пептидов
легко отщепляется при обработке соответствующих соедине-
ний 0,1 М раствором пиперидина в воде или 0,1 М водным три-
этиламином в течение 10 мин при температуре 0 °C с последу-
ющей лиофилизацией.
Синтез На-т/>ет-бутилоксикарбонил-
-2,4,6-триметоксибензол-сульфонил-
£-триптофана [408]
Методика 275. 2,4,6-Триметоксибензолсульфохлорид
[408]
/ОСНз /ОСН,
СН3О—+ C1SO3H -+ СН3О—SO3H ►
ЧОСН3 • хосн3
168,2 116,5 248,3
/ОСН,
-> СН3О-^~SO3C1
XOCH3
266,8
К раствору 18,7 г (0,11 моль) 1,3,5-триметоксибензола в
30 мл сухого четыреххлористого углерода добавляли по кап-
лям 8,64 мл (0,3 моль) хлорсульфоновой кислоты. Смесь вы-
держивали 30 мин при комнатной температуре и затем охлаж-
дали. В процессе добавления выпадали кристаллы 2,4,6-три-
метоксибензолсульфокислоты. Их отделяли и вносили в 100 мл
ледяной воды, содержащей 80 г Na2CO3. Осадок отделяли
фильтрацией и сушили при 100 °C над Р2О5. Полученную на-
триевую соль вводили в реакцию с 28 мл (0,3 моль) РОС13 при
100 °C в течение 2 ч, и затем реакционную смесь выливали в
ледяную воду. Раствор экстрагировали хлороформом (3 X
X 100 мл), объединенный органический слой промывали водой
и сушили сульфатом натрия. После отгонки хлороформа ос-
таток обрабатывали эфиром и получали кристаллы, которые
перекристаллизовывали из смеси хлороформ — эфир. Выход
10,4 г (35,5 %); т. пл. 133—134 °C.
274
Методика 276. №е-трет-Бутилоксикарбонил-№п-2,4,6-
триметоксибензолсульфонил-£-трииптофан [408].
К раствору 789 г (2 ммоль) бензилового эфира Boc-L-трип-
тофана и 6,4 мг (0,02 ммоль) цетилтриметиламмоний хлорида
в 10 мл метиленхлорида добавляют по каплям 810 мг (3 ммоль)
2,4,6-триметоксибензолсульфохлорида в 3 мл метиленхлори-
да присутствии 200 мг (5 ммоль) тонко измельчённого NaOH
и интенсивно перемешивают смесь при комнатной температуре
30 мин. Добавляют 10 мл 1 н. НС1 при охлаждении. Отделя-
ют органический слой, сушат над сульфатом натрия и уда-
ляют растворитель при уменьшенном давлении. Остаток рас-
творяют в 10 мл этанола, к полученному раствору добавляют
по каплям при охлаждении 2,2 мл 1 н. NaOH и перемешивают
1 ч при комнатной температуре. Отгоняют этанол, к оставше-
муся водному раствору добавляют 20 мл воды и экстрагиру-
ют его 20 мл эфира. Водный слой отделяют и подкисляют при
охлаждении 1 н. НС1 до pH 3. Продукт экстрагируют этил-
ацетатом, органическую фазу промывают водой и сушат суль-
фатом натрия. Упаривают растворитель и остаток обрабаты-
вают петролейный эфиром, что приводит к образованию твер-
дого остатка. Выход 976 мг (91,2 %); т. пл. 82—84 °C; [а]р =
= —15,4 (с = 0,5; ДМФА).
Описанная Mtb-группа устойчива к действию ТФУ и уда-
ляется фтористым водородом или метансульфокислотой без
разрушения остатков триптофана.
Защита боковой группы метионина
В большинстве случаев синтез метионинсодержащих пеп-
тидов проводят без специальной защиты тиоэфирной группы
метионина. Однако замечено,' что при деблокировании Кбз-
группы метионинсодержащих пептидов бромистым водоро-
дом в уксусной кислоте может происходить расщепление тио-
эфирной связи с образованием S-бензилгомоцистеина. Для
устранения Этой побочной реакции предложено использовать
вместо метионина в пептидном синтезе метионинсульфоксид.
Производные метионинсульфоксида легко получаются при
окислении метионина (или соответствующих его производных)
перекисью водорода в соляной или уксусной кислоте.
Методика 277. L- Метионин-d, /-сульфоксид [403]
HaN—СН—СООН HaN—СН—СООН
(<[Н2)а “ХЕЗн’* (9На)’
S—СН, о S—сн,
149,2 165,2
18* 275
Суспензию 1,49 г (10 ммоль) L-метионина обрабатывают в
30 мл уксусной кислоты при 10 °C 1,33 мл (12 ммоль) 30 %-го
Н2О2 (9 н. раствором) при встряхивании, постепенно доводя
температуру раствора до комнатной. Через 20 мин исходный
материал растворяется, а через 60 мин реакция заканчивается
(йодометрическая проба на присутствие Н2О2). Раствор упа-
ривают в вакууме при температуре не выше 40° С и остаток
перекристаллизовывают из смеси вода — ацетон. Выход 1,59 г
(96 %); [а)д = 4-37,2° (с = 2,1; 1 н. НС1).
Методика 278. N-бензилоксикарбонил-Л-метионин-
d,/-сульфоксид [403].
К раствору 5 г (30 ммоль) L-метионин-сульфоксида в 7,5 мл
4 н. NaOH добавляют при перемешивании и охлаждении до
5 °C по каплям одновременно 6 г (35 ммоль) карбобензокси-
хлорида и 7,5 мл, 4 н. NaOH, поддерживая pH 8—9 (в течение
около 15 мин). Затем перемешивали еще 30 мин при 5 °C и
30 мин при комнатной температуре, экстрагиваровали эфиром
и подкисляли концентрированной НС1 при 0 °C до pH 2. Вы-
делившееся масло экстрагировали этилацетатом, промывали
этилацетатный раствор водой до pH 7, сушили сульфатом магния
и удаляли растворитель в вакууме. Остаток кристаллизовали из
смеси этилацетат — эфир (0°С). Выход J,44 г (83 %), т. пл. 93—
96 °G. Перекристаллизация из смеси ацетон — эфир дает про-
дукт е т. пл. 104—106 °C; [а]р = —16,0° (с = 4,1; этанол).
Удаление защитной группы метионина
Метионин-сульфоксид легко восстанавливается в метионин
восстанавливающими агентами: тиогликолевой кислотой,
меркаптоэтанолом, водородом над палладиевым кристаллиза-
тором, никелем Рэнея, натрием в аммиаке и др. Лучше всего
проводить восстановление тиогликолевой кислотой.
Методика 279. Пролил-тирозил-(е-тозил)-лизил-
метионин [403].
Суспензию 354 мг (0,5 ммоль) H-L-Pro-L-Tyr-L-Lys(Tos)-L-
Met(O)—ОН в 5 мл воды обрабатывают 0,3 мл. (5 ммоль) све-
жеперегнанной тиогликолевой кислоты в атмосфере азота при
встряхивании в течение 20 ч при 50 °C (вначале прозрачный
раствор постепенно выделяет продукт реакции в виде масла).
После удаления растворителя в вакууме маслянистый осадок
многократно затирают с эфиром и получившийся аморфный
порошок растворяют в небольшом количестве метанола и до-
бавляют в него раствор аммиака в метаноле (2,5 н. раствор)
до слабощелочной реакции. Через 30 мин выпавшие кристал-
лы отфильтровывают, растирают с горячим метанолом, фильт-
руют и сушат. Выход 299 мг (86 %).
276
Удаление защитных групп
безводным фтористым водородом
В 1965 г. Сакакибара и Шимониши [4431 показали, что для
деблокирования защищенных пептидов может быть успешно
использован безводный фтористый водород. Для предотвра-
щения полимеризации бензилфторидов добавляют анизол.
В этих условиях гладко отщепляются следующие защитные
группы: Кбз-группа, mpem-бутилоксикарбонильная, нитро-
группа аргинина, тозильная, mpem-бутильная, о-нитрофе-
нилсульфенильная группа, защитные группировки бензиль-
ного типа, динитрофенильная группа ДНФ-гистидина и другие
группы. При этом метиловые, этиловые и п-нитробензиловые
эфиры не гидролизуются. Ленард и Робинсон [444] в 1967 г.
впервые применили безводный фтористый водород для дебло-
кирования и отщепления отсмолы пептидов, синтезированных
твердофазным методом.
Обычно отщепление защитных групп безводным фтористым
водородом завершается при О °C за 60 мин. При твердофазном
методе синтеза пептидов этот способ деблокирования стал ос-
новным. Установка для удаления защитных групп и отщепле-
ния пептидов от смолы описана в монографии Стюарта и
Янг [16].
Как известно, HF является одной из сильнейших кислот и
мощным катализатором ионных реакций; применение его может
привести к различным побочным реакциям, о которых в по-
следнее время накопилось довольно много экспериментальных
данных. В обзорной работе Вольпиной и соавт. [445] освеще-
ны результаты исследования разнообразных побочных про-
цессов, протекающих при использовании безводного фторис-
того водорода. Рассмотрим некоторые из них.
N О-Ацильная миграция
Пептиды, содержащие остатки серина и треонина, в при-
сутствии HF претерпевают N -> О-ацильную миграцию:
О н н О
|| |+ I НО O-CH-R1 II
R—С—N—С—Rl -> ;С< + I -> R—С-О-СН—R1
| | Rz XN—СН—R» + |
Н СН н/\м HaN—СН—R’
НО/Ч<
Изучали эту реакцию при условиях, при которых она идет с
высоким выходом (12—24 ч, температура 20—30 °C); в обыч-
ных условиях деблокирования пептидов HF она незна-
чительна. Кроме того, оказалось, что обработка пептидов
277
раствором бикарбоната натрия количественно приводит к об-
ратной 0 —► N миграции без сколько-нибудь ваметной раце-
мизации.
Алкилирование тирозина
При отщеплении О-бензильной группы тирозина в резуль-
тате реакции внутримолекулярного алкилирования образу-
ется устойчивый к действию HF остаток 3-бензилтирозина1
—NH—СН—СО— —NH—СН—СО—
Ан» СН,
I I
I JI I J
V
OBrl OH
Для устранения этого побочного процесса предложено бло-
кировать тирозин более устойчивыми защитными группами,
такими как О-(2,6-дихлор)бензильная или О-цикло гексиль-
ная группа.
Реакция транспептидации производных
аспарагиновой кислоты
Бензиловые эфиры аспарагиновой кислоты могут подвер-
гаться транспептидации с образованием смеси а- и 0-пептидов!
О О
н,сн/С\
е- ! j
А
О
II
Н2с/ XNHR*
R— HN-CH^c/OH
О
Для устранения этой побочной реакции предложено добав-
лять фенолы или 1-оксибензтриазол; использование вместо
бензиловых эфиров соответствующих znpe/n-бутиловых или
циклогексиловых производных также предотвращает ука-
занный нежелательный процесс.
278
Алкилирование боковой цепи триптофана
Отщепление Вос-группы при помощи безводной HF сопро-
вождается mpem-бутилированием боковой цепи триптофана.
Для защиты индольного ядра от этой побочной реакции необ-
ходимо добавлять тиоанизол, диметилсульфид и другие подоб-
ного типа реагенты.
Наиболее надежным способом предотвращения указанного
алкилирования является применение в синтезе №’”-формил-
триптофана, который неспособен mpem-бутилироваться; кро-
ме того, он устойчив в условиях окислительного разрушения
триптофана.
Нежелательные процессы протекают также при отщепле-
нии S-бензильной группы цистеина; в частности, имеет место
неполное деблокирование защищенной сульфгидрильной
группы.
Следует иметь в виду, что степень протекания указанных
побочных реакций зависит от природы синтезируемого пеп-
тида. Однако во всех случаях использования безводного HF
такие процессы могут иметь место, особенно при синтезе белков.
Другие эффективные реагенты
для удаления защитных групп
Ввиду того, что работа с безводным фтористым водородом
требует специального оборудования и повышенной осторож-
ности, в последние годы вместо него применяют другие дебло-
кирующие реагенты как в традиционном, так и в твёрдофазном
методах синтеза пептидов.
Трис(трифторацетат) бора (BTFA)
Для удаления защитных групп пептидов Плесе и Бауэр в
1973 г. [424] предложили вместо безводного фтористого водо-
рода использовать трис(трифторацетат) бора, применение ко-
торого удобнее и безопаснее, чем HF; кроме того, при этом не
нужна особая аппаратура.
Реактив получают непосредственно перед использованием
для удаления защитных групп по следующей схеме;
ВВг3 4-3CFSCOOH В (OOCCF,) + ЗНВг.
Раствор BTFA в трифторуксусной кислоте при температуре
О °C отщепляет подобно безводному фтористому водороду Z-,
Bog-, Tos-, (Me)Z-, -ОВи*, -NO2, а также группы бензильного
типа и многие другие. Этот способ удаления защитных групп
успешно примёняли для получения ряда биологически актив-
ных пептидов.
279
Методика 280. Трис(трифторацетат) бора [424].
Эквивалентные количества трехбромистого бора [446] и
трифторуксусной кислоты смешивают в хлористом метилене
при температуре 0 °C; при этом образуется осадок. Реакци-
онную смесь упаривают в вакууме при 20 °C досуха, а остаток
растворяют в ТФУ и полученный раствор используют непо-
средственно для отщепления защитных групп.
Методика 281. Общая методика отщепления защит-
ных групп при помощи BTFA [424].
Навеску защищенного пептида растворяют в трифюрук-
сусной кислоте и смешивают при температуре 0 °C по крайней
мере с тремя эквивалентами трис(трифторацетата) бора в триф-
торуксусной кислоте на каждую отщепляемую группу. Для
удаления нитро-, тозильной или /?-метоксибензильной групп
используют от 5 до 6 экв. реагента. Спустя 1 ч растворитель
упаривают при 20 °C, а остаток обрабатывают обычным спо-
собом.
Соединения бора удаляют при помощи многократного до-
бавления и последующего упаривания в вакууме метанола;
от них можно освободиться также при хроматографической
очистке пептида.
Трифторметансульфокислота (TFMSAj
Трифторметансульфокислота как реагент для удаления
кислотолабильных групп была предложена Яджима с соавт.
[447] в 1974 г. Несколько позднее они показали [4481, что 1 М
трифторметансульфокислота деблокирует в присутствии тио-
анизола и л<-крезола в ТФУ при 0 °C в течение 60 мин защит-
ные группы бензильного типа, Mts-защиту гуанидиновой груп-
пы аргинина и др. Эта система хорошо зарекомендовала себя
в пептидной химии.
Методика 282. Общий метод деблокирования защи-
щенного пептида трифторметансульфокислотой [449].
200 мг (29 мкмоль) защищенного пептида (44 аминокислот-
ных остатка) обработали 7 мл 1 М TFMSA — тиоанизол в ТФУ
в присутствии 60 экв. ж-крезола на ледяной бане в течение
60 мин. Затем смесь разбавляли сухим эфиром, осадок отде-
ляли центрифугированием и сушили в вакуум-эксикаторе
над КОН перед дальнейшей очисткой.
Метансульфокислота (MSA)
Метансульфокислота является эффективным, но более до-
ступным реагентом, чем TFMSA. Впервые ее применили для
этой цели Яджима с соавт. [449] в 1975 г. В табл. 45 приведены
280
данные из этой работы по
эффекти в но сти д ебл о пиро-
вания. Использовали
100 экв. MSA на удаляемую
ващитную группу.
Как видно из табл. 45,
нитро-, тозильная группы
аргинина и бензильная
группа тирозина удаляют-
ся неэффективно.
Ниспен с соавт. [4511
предложили использовать
смесь MSA — ТФУ с до-
бавкой тианизола и ^-кре-
зола в твердофазном синте-
зе для удаления некоторых
защитных групп и отщеп-
ления пептида от полимера.
По их данным, эта смесь
оказалась эффективнее, чем
TFMSA-трифторуксусная
кислота.
В 1989 г. В. В. Самуков
и соавт. [423] предложили
использовать раствор 1,0—
Таблица 45. Эффективность
удаления защитных групп MSA
[449]
Защищенные аминокис- Выход свобод-
лоты ной аминокис-
лоты, %
о-Нитрофенилсульфе- 100
нил-£-ванил
глре/п-Бутилоксикарбо- 96,5
нил-Л-серин
4—Метоксикарбобензок- 94,0
си-глицин
Карбобензокси-£-трипто- 100
фан
у-Бензиловый эфир 99,6
L-глутаминовой кисло-
ты
о-Бензнл-Ь-серин 97,7
о-Бензил-£-треонин 100
о-Бензил-£-тирозин 30,2
(о-Нитро-Ь-аргинин 58,5
(д-Тозил-А-аргинин 49,2
Трикарбобензокси-L-ap- 100
гинин
5-4-Метоксибензил-Л-ци- 94,6
стеин
1,5 М метансульфокислоты, 1 М тиоанизола в трифторуксус-
ной кислоте для удаления защитных групп бензильного типа и
Mts-группы. Они показали, что после деблокирования смесь
содержит меньше примесей, чем в случае использования си-
стемы TFMSA — ТФУ.
Методика 283. Общий метод деблокирования пепти-
дов метансульфокислотой в трифторуксусной кислоте [4231.
Пептид растворяют при 0 °C в безводной ТФУ, добавляют
10 % (по объему) тиоанизола и 5 % л/-крезола, затем вносят
MSA до концентрации 1—1,5 моль/л. Общий объем смеси
определяют из расчета 5—10 экв. MSA на деблокируемую
группу. Смесь выдерживают 2 ч при 0 °C, разбавляют 20 объ-
емами сухого эфира, отделяют осадок центрифугированием,
промывают эфиром и сушат в вакууме.
Триметилсилил трифторметансульфонат
(триметилсилил трифлат, TMSOT/)
Этот реагент был предложен недавно Фуджии с соавт. [242]
и, по-видимому, является очень перспективным, поскольку
удаляет большое количество защитных групп. Эффективность
281
Таблица 46. Удаление различных защитных гру в трифторуксусной кислоте [242 пп IM TMSOTf/TA
Защищенные аминокислоты Выход свободных аминокислот, %
Время обработки, мин
10 30 60
Ма-4-Метоксикарбобензокси-М8-карбобензокси-£-ли- 93,6 98,7 — ЗИН ЬМ-Метоксикарбобензокси-о-бензил-£-серин 90,6 91,7 — М-Трет-Бутилоксикарбонил-о-бензил-£-треонин 98,0 — — М-4-Метоксикарбобензокси-у-бензил-£-глутаминовая 100,0 — — кислота М-4-Метоксикарбобензокси-[3-бензил-£-аспарагиновая 99,2 — — кислота М«/прет-Бутилоксикарбонил-|3-Циклогептил-£-аспара- 95,3 100,0 — гиновая кислота М-тре/п-Бутилоксикарбонил-о-бензил-Л-тирозин 97,8 — — М»/пре/п-Бутилоксикарбонил-о-(2,6-дихлор)-бен- 98,1 — — зил-Ь-тирозин М-/пре/п-Бутилоксикарбонил-Ь^'п-тозил-Ь-гистидин 94,5 — — Ь1-/пре/п-Бутилоксикарбонпл-М4,п-бензилоксиметил- 88,9 — — L-гистидин N-mpem-Бутил оксикарбо нил-Nuz-Me3HTii лен-2-сульфо- 100,0 — — нил-Л-триптофан ]^а-4-Метоксикарбобензокси-^°-мезитилин-2-сульфо- 97,6 — — нил-А-аргинин №&-4-Метоксикарбобензокси-№)-4-метоксибензолсуль- 75^5 93,5 94,2 фонил-Л-аргинин №с-Карбобензокси-№с-тозил-Ь-аргинин 31,3 62,1 85,8 Ь]а-Карбобензокси-М®-нитро-£-аргинин 1 8,6 11,0 14,7 S-4-Метоксибензил-А-цистеин 95,4 Г^-трет-Бутилоксикарбонил-З-тре/п-бутил-Д-цистеин 79,5 87,3 96,8 Sr Адамантил-L-цистеин 100,0 — — 8-Бензил-£-цистеин 0 — — М-/пре/п-Бутилоксикарбонил-5-ацетамидометил-£-ци- 0 — — стеин М-4-Метоксикарбобензокси-Ь-метионинсульфоксид 17,8 27,6 44,2 То же (вместо тиоанизола — диметилселенид) 88,8 90,0 90,6
удаления защитных групп смесью TMSOTf — ТФУ — ТА при
О °C при использовании 30 экв. реагента на каждую удаляе-
мую группу приведена в табл. 46.
Как видно из табл. 46, TMSOTj в трифторуксусной кислоте
в присутствии тиоанизола удаляет практически все защитные
группы, кроме нитрогруппы аргинина, S-бензильной и S-аце-
282
тамидометильной групп цистеина. Очень важно, что реагент
полностью удаляет S-mpem-бутильную группу цистеина. В ра-
боте [242] приведена предполагаемая схема деблокирования:
СН,
(CH,),—Si—O,S—CF, СН,—^i—СН, + CF,SOf
R_CH,—О—<^н2—Ч -> R—СН,—i СН,—Ч
+ Х=/ I Н,о —. + I
ч | R—СН,ОН + Ч—S—СН,
S—СН, z
Авторы [2421 считают, что так как TMSOT; не является кис-
лотой Бренстеда, по-видимому, в растворе ТФУ она играет
роль сверхжесткой кислоты, а тиоанизол — жесткого основа-
ния. (Получение реагента см. в главе 2, (методика ИЗ).)
Общая стратегия
применения защитных групп
В начале главы 2 уже упоминалось о том, что большое чис-
ло защитных групп, предложенных на сегодняшний день,
вызвано необходимостью их селективного удаления в процес-
се синтеза. Однако поиск ведется и в направлении улучшения
характеристики защитных групп, используются более мягкие
условия удаления, подавление побочных реакций, их устой-
чивость при очистке и хранении пептидов и др.
Именно большой выбор группировок защитных групп по-
зволяет выбрать наиболее гибкую тактику синтеза каждого
конкретного полипептида.
Хотя набор существующих защитных групп позволил в
течение последних 20 лет достичь впечатляющих результатов
(синтезированы пептиды, состоящие из 100 и более аминокис-
лотных остатков), тем не менее проблема защитных групп ос-
тается актуальной. В статье, посвященной этой проблеме [452],
Янг отмечает: «Я имею право утверждать, что трудности в син-
тезе в настоящее время больше обусловлены несовершенством
защитных групп — частичное отщепление в процессе синтеза
Или неполное удаление в конце синтеза, чем недостатком кон-
денсирующих агентов (хотя они также нуждаются в усовер-
шенствовании)».
Каким же требованиям должны удовлетворять «совершен-
ные» защитные группы? В 1977 г. Бэрени и Мэррифилд [453]
предложили концепцию «ортогональной системы» в пептидном
синтезе. Ортогональная система определяется как набор пол-
ностью независимых классов защитных групп. В этой сис-
теме каждый класс защитных групп может быть удален в
283
Рис. 6. Схема селективного удаления защитных групп ортогональной сис-
темы. А,ВиС — триклассазащитныхгруппдляа-амино-, а-карбоксиЛьной
и боковых функциональных групп
npi еутствии в?ех других классов. Идеальной общей стратегией
пептидного синтеза, по мнению авторов указанной работы, бы-
ло бы использование по крайней мере трех классов защитных
групп, удаление которых осуществлялось бы реакциями в
различными механизмами. Схематически это изображено на
рис. 6.
В качестве примера ортогональной системы авторы работы
[453] предлагают следующие три класса защитных групп: ди-
тиосукцинильную (Dts), удаляемую тиолизом; о-нитробен-
зильную (ONB), удаляемую УФ-облучением; /пре/п-бутиль-
ную (Ви'), удаляемую ацидолизом,— для блокирования
боковых функциональных групп (рис. 7). Можно легко постро-
ить подобные «триады» ортогональной системы и для других
защитных групп.
По-видимому, ортогональная система защитных групп мо-
жет находить применение в отдельных специальных случаях.
Нам кажется, что для большинства схем пептидного синтеза
она «избыточна», поскольку чаще всего нет необходимости се-
лективного удаления трех классов защитных групп. В процес-
се синтеза пептидов обычно чаще всего приходится удалять
N-защитные группы (ступенчатый синтез в растворе, твердо-
фазный и жидкофазный синтезы), реже — защиту карбоксиль-
ной группы (блочный синтез пептидов). Защитные группиров-
ки боковых функциональных групп обычно удаляют на по-
следней стадии синтеза, поэтому Хиршман [14] называет их
284
W//////6
Bi?
7ZZZZZZZZX 0N8
JDtS ttzzzzzzzzzzzzzza'
Df SONB
Рис. 7. Пример использования некоторых классов защитных групп орто-
гональиой системы в синтезе пептидов
«стабильными» группами в отличие от указанных выше «вре-
менных» защитных групп.
Проблема удаления защитных групп детально рассмотрена
в обзоре Вольпиной и соавт. [445], где системы защитных груп-
пировок подразделяются по способам их удаления на конеч-
ной стадии синтеза на четыре группы:
1. Каталитическое гидрирование над палладием. Это очень
мягкий способ деблокирования. Удаляются все защитные
группы бензильного типа (и некоторые другие). Использова-
ние метода не подходит для пептидов, содержащих цистеин и
метионин.
2. Обработка трифторуксусной кислотой. Один из самых
мягких способов деблокирования. При его использовании вы-
бирают для защиты боковых функций аминокислот защитные
группы /пде/п-бутильного типа (Вос, Аос, Adoc, OBu*). В ка-
честве N-защитной группы можно использовать Кбз-группу
(в случае отсутствия Cys и Met), а также более кислотолабиль-
ные группы (Врос, Ddz и др.) или щелочнолабильные группы
(Fmoc и др.).
3. Обработка безводным фтористым водородом при О °C в
течение 1 ч. Этим путем удаляется большинство защитных
групп. Может вызывать побочные реакции (см. главу 4). Яв-
ляется стандартным методом деблокирования и отщепления
пептидов от полимера в твердофазном синтезе.
285
4. Обработка натрием в жидком аммиаке. Удаляются мно-
гие защитные группы. Метод может приводить к глубокой де-
струкции пептидов, поэтому в настоящее время применяется
редко.
Таким образом, наиболее распространенными схемами син-
теза пептидов в настоящее время являются такие, где исполь-
зуют: 1) боковые защитные группы бензильного типа в соче-
тании с кислотолабильными N-защитными группами и 2) бо-
ковые защитные группы кислотолабилыюго типа с N-защит-
ными группами бензильного или щелочнолабильного типа
(например, Fmoc-группы).
Следует остановиться на еще одной важной проблеме, об-
суждавшейся в работе [445],— проблеме защитных групп при
синтезе крупных полипептидов и белков. В уже упоминавшейся
выше работе [452] отмечается, что с ростом длины полипеп-
тидной цепи возрастают требования как к стабильности за-
щитных групп, так и к полноте их отщепления. Известно,
что неполное удаление Вос-группы при твердофазном синте-
зе приводит к появлению ошибочных последовательностей.
Другой проблемой, возникшей при синтезе крупных пептидов,
является неустойчивость некоторых защитных групп при мно-
гократном деблокировании а-аминогруппы кислыми реаген-
тами. Например, при обработке пептидов в условиях твердо-
фазного синтеза трифторуксусной кислотой в хлористом ме-
тилене (33—50%-й раствор) частично отщепляются некоторые
защитные группы бензильного типа (Lys(Z), Tyr(Bzl), Туг (Z),
Cys(MBzl)). Для устранения этого используют другие защит-
ные группы этих аминокислот (Lys(3ClZ), Lys(2,6Cl2Z),
Lys (TFA), Tyr(BzCl2) HCys(4-CH3Bzl)). Кроме того, перспек-
тивным является использование вместо кислотолабильных
N-защитных групп Fmoc-группы, которая снимается основа-
ниями в очень мягких условиях.
При планировании синтеза пептидов важное значение име-
ет правильный выбор системы защитных групп. Здесь получи-
ли распространение две стратегии: минимальной защиты; мак-
симальной защиты.
Хиршман и Вебер [14] указывают на ряд преимуществ так-
тики минимальной защиты: отсутствие некоторых побочных
реакций защитных групп, ухудшение растворимости крупных,
полностью защищенных пептидов в полярных растворителях,
возможность промежуточной очистки пептидов с минимальгым
числом защитных групп в водно-органических средах. Вместе
с тем они признают определенные недостатки тактики мини-
мальной защиты — возможность побочных реакций некото-
рых незащищенных функциональных групп, что ограничива-
ет выбор конденсирующих агентов. Они считают, что необхо-
286
димо защищать только такие сильные нуклеофильные группы,
как 8-аминогруппа лизина и тиольная группа цистеина.
По мнению авторов обзора [445], тактика максимальной
защиты боковых функций аминокислот (особенно при синте-
зе крупных пептидов и белков) хотя и усложняет синтез, но
обладает рядом ценных преимуществ — улучшение раство-
римости пептидов и ограничение возможности побочных реак-
ций. Синтез а-бунгаротоксина [358] и ряда других крупных
пептидов является хорошим подтверждением этого.
Глава 5
ТВЕРДОФАЗНЫЙ МЕТОД СИНТЕЗА
ПЕПТИДОВ
Материал предыдущих глав свидетельствует о том, что
классический метод синтеза пептидов в растворе является весь-
ма трудоемким и связан со значительными затратами времени
и усилий. В отличие от него твердофазный метод синтеза пеп-
тидов является быстрым, эффективным, удобным, позволяю-
щим получать пептиды не только в аналитическом, но и в пре-
паративных количествах.
ОБЩИЕ ПРИНЦИПЫ ТВЕРДОФАЗНОГО МЕТОДА
СИНТЕЗА ПЕПТИДОВ
Твердофазный метод является особым вариантом ступен-
чатого химического способа получения пептидов, при кото-
ром растущая полипептидная цепь ковалентно «прикрепляет-
ся» к нерастворимому полимерному носителю так, что все эта-
пы процедуры можно провести в одном и том же реакторе, а
избыток реагентов и растворителей, а также продуктов реак-
ции можно легко удалить простым фильтрованием и промы-
ванием смолы без обычных потерь, связанных с очисткой при
синтезе в растворе. По сравнению с классическими методами
твердофазный вариант характеризуется следующими преи-
муществами: увеличивается скорость рабочих операций; все
манипуляции становятся более простыми и удобными. Из-за
однотипности операций возможна автоматизация всего про-
цесса; кроме того, снижаются механические потери материала
при различных стадиях синтеза и достигается высокий выход
конечного продукта.
Недостатком метода является образование микрогетеро-
генного продукта, возникающего из-за неполноты протекания
реакций, а также в результате частичного разрушения поли-
пептида в ходе химических манипуляций. Для подавления
этих нежелательных процессов и достижения количественно-
го выхода используют большие избытки реагентов, а иногда
стадии повторяют дважды.
Детальная схема синтеза пептидов, предложенная Мер-
288
рифильдом [16, 348, 454—4571, представлена ниже:
1. Функционализация полимера
2. «Прикрепление» первой Вос-
_р*
С1— СН2—ОСН3 =|Z SnCl4
Cl—СН2—Р
аминокислоты
Boc-NH—СН—СООН
EtsN
I
R, _ 4
Boc-NH—СН—COO—СН2—^-Р
।
R(
1. ТФУ/СН2С12
2. Et3N _ ..
3. Удаление Иа-защитной H2N—СН— СОО— СН2—р
группы I
Ki
4. Конденсация со следу- Boc-NH—СН—( ООН Et3N
ющей защищенной ами- | ДЦГК
нокислотой Й2 __
Boc-NH—СН—СО—NH—СН—СОО—X—Р
R» Ri 1
5. Последовательное наращивание :
полипептидной цепи |
Boc-NH—СН—СО— ..•
I
Rn
—NH—СН—СО—NH-CH-COO—
R2 Ri
6, Отщепление свободного пептида от смолы и его
деблокирование
HoN—СН—СО- ... — NH-CH— СО—NH—СН-СООН
I I I
Rn Ra Rr
7. Очистка пептида
*Р — сополимер стирола и дивинилбензола
Метод включает следующие этапы: стадию «функционализа-
ции» полимера, т. е. получение смолы с реакционносгособной
группировкой X; стадию ковалентного присоединения пер-
вой (обычно С-концевой). защищенной аминокислоты; стадию
селективного удаления Na-защитной группы присоединенной
аминокислоты; взаимодействие со следующей защищенной
аминокислотой; стадии наращивания полипептидной цепи,
затем удаление синтезированного пептида со смолы, его пол-
ное деблокирование и, наконец, стадию очистки пептида.
В качестве строительных блоков используют по Мерри-
фильду защищенные аминокислоты, ступенчато присоединя-
емые в направлении отС-конца к N-концу растущего пептида.
19 2-882
289
Принципиально закрепление пептида можно осуществлять по
N-концевой аминокислоте или за счет боковой цепи аминокис-
лоты, находящейся в середине пептида. Наращивание цепи
можно вести от N-конца к С-концу или в обоих направлениях,
а вместо защищенных аминокислот использовать фрагменты
защищенных пептидов [458]. Однако при такой стратегии час-
то возникают дополнительные трудности.
Важнейшим условием успешного применения твердофаз-
ного метода является использование подходящего полимерно-
го носителя. В наиболее распространенном варианте метода
в качестве полимера используют предложенный Меррифиль-
дом гранулированный поперечно сшитый (1—2 %) хлорме-
тилированный полистирол с размерами частиц 20—80 мкм,
который обладает достаточной химической и механической
прочностью и хорошо набухает в органических растворителях.
При этом диаметр частиц увеличивается в 5—6 раз, что прев-
ращает носитель в хорошо сольватированный материал, в ко-
торый способны легко диффундировать используемые реаген-
ты и растворители. Кроме хлорметилированной смолы для
твердофазного метода были предложены материалы на основе
полиметилметакрилата, полисахаридов, фенольных смол, по-
лиакриламидов (см. соответствующие ссылки в обзорах и мо-
нографиях [16, 455—458]), но лишь полиакриламиды получили
широкое распространение [460, 461].
Для улучшения механических свойств смолы (что особен-
но важно в методе непрерывного проточного синтеза пептидов
на колонке [458, 4621) созданы материалы, состоящие из твер-
дой инертной основы (например, кизельгура, кремниевого
стекла с контролируемым размером пор), покрытой полисти-
ролом, полиамидом или полиэтиленгликолем [457—459].
Другой важной модификацией смолы является введение
между полимером и пришиваемой С-концевой аминокислотой
пептида специальной «ручки», т. е. особой-бифункциональной
группиэовки, на одном конце которой содержится группа, по-
зволяющая мягко отщеплять пептид от носителя, а на дру-
гом — группа, способная взаимодействовать с функционализи-
рованной смолой. В качестве «ручек» чаще всего используют
фенилацетамидометильную (РАМ) — или п-алкоксибензиль-
ную группировку [464, 465].
Этап ступенчатого наращивания полипептидной цепи вклю-
чает повторяющиеся циклы деблокирования и сочетания со
следующей защищенной аминокислотой. Для пептидообразо-
вания использовались практически все наиболее употреби-
тельные реагенты и методы [455], но самое широкое примене-
ние нашел дициклогексилкарбодиимид (иногда диизопропил-
карбодиимид) [466] и методы активированных эфиров
290
N-защищенных аминокислот [467—469] или их симметрич-
ные ангидриды (см. предыдущие разделы) [470].
В качестве «временных» Ж-защитных групп в современных
вариантах твердофазного метода используют главным обра-
зом mpem-бутилоксикарбонильнуюили флуоренил метилокси-
карбонильную защиту, которые могут быть количественно уда-
лены соответственно ацидолизом или обработкой смолы ос-
нованиями. Нередко находят применение временные защитные
группы, удаляемые за счет других химических процессов; на-
пример, дитиасукциноильная группа отщепляется тиола-
ми [471].
Постоянными защитными группировками боковых цепей
аминокислот являются производные простых и сложных эфи-
ров на основе бензилового, mpem-бутилового, а также цикло-
пентилового или циклогексилового спиртов [16, 455—458].
Хорошие результаты дает стратегия синтеза с использова-
нием принципа ортогональности [471], когда отщепление вре-
менных и постоянных защитных групп, а также удаление
пептида со смолы осуществляется при действии трех совер-
шенно различных химических реакций.
Ключевым подходом твердофазного метода является стрем-
ление довести выход реакций до 100 %. Поэтому разработаны
разнообразные количественные и качественные методы чтобы
удостовериться в этом. Наиболее распространенным являет-
ся нингидриновый тест Кайзера (см. методику 295). Другой
подход заключается в непрерывной спектрофотометрической
регистрации изменений концентрации продуктов реакции. Дан-
ный способ нашел применение при Fmoc-стратегии синтеза
пептидов на колонке [458, 469]. Ряд авторов предпочитает не
тестировать свободные аминогруппы, а повторять стадию кон-
денсации дважды или же блокировать свободные аминогруппы
за счет реакции ацетилирования.
Для отщепления пептида от смолы можно использовать
разнообразные методы [16, 455, 4561: обработку кислотами,
например жидким фтористым водородом, раствором бромис-
того водорода в трифторуксусной кислоте, трифторуксусной
кислотой, трифторметансульфокислотой и другими реаген-
тами; взаимодействие с основаниями, аммиаком, гидразином,
а также гидрирование в присутствии ацетата палладия или
фотолиз [455, 458]. В зависимости от используемой смолы и
методов отщепления образующийся пептид можно получить в
виде кислоты, амида, эфира или гидразида [4571. При этом
могут быть сконструированы материалы, позволяющие осу-
ществлять отщепления несколькими путями. Например, у со-
единения (132) отщепление пептида от смолы можно достичь
при помощи жидкого фтористого водорода (путь А) или за
19*
291
Таблица 47. Некоторые полимерные матрицы
к с Название смолы Структура функциональной группы полимера
С1— СН2—Р
1. Хлорметилированный
полистирол; «смола
Меррифильда»
2. Оксиметил ированпый
полистирол
3. 4-(Оксиметил)-фени-
лацетильный поли-
мер; «РАМ-смол а»
X—СН2—{ S—CH2—СО—NH—
4. Алкоксибензильный
полимер; «смола Ван-
га*
5. 2-Метокси-4-алкокси-
бензильный полимер;
«смола Сасрин» *
6. Дифенилметильный
полимер
X = —ОН, —Вг
но—сн2—Ч—о—сн,—р
7. Бензгидриламино по-
лимер; «БАГ-смола»
CI
Y = — Н, —СН,, — ОСН3
h2n—сн—Ч—р
—СН—" Р
8. Оксиэтилтиометиль-
ная смола
НО-СН2—CH2—S—СН2—Р
I
о
о о
/—ч
НО—СН2—СН2—S—НС,—" Р
292
для твердофазного метода синтеза пептидов
Методы отщепления и форма, в которой отщепляется пептид (приведено в скоб- Примечание «я £Х. Р «
ках) Л и 7 тур
1. HF, НВг/ТФУ (свободный пептид) Самая распространная смола; [348] обычно используют полимер с
2. Щелочной гидролиз (свобод- ный пептид) 3. Переэтерификация (эфир пеп- тида) 4. Аммонолиз (амид пептида) 1—2 % поперечной сшивки
То же То же [472, 473]
> > Используется весьма часто. [464] Связь смолы с аминокислотой в 100 раз более стабильна к ТФУ, чем у смолы Меррифильда
ТФУ в СН2С12 (свободный пеп- тид) Используется при работе с Врос- [474] или Fmoc-группами
0,5—1 % ТФУ (свободный пеп- тид) Используется при работе с [475] Fmoc-защитой. Расщепляется в очень мягких условиях
ТФУ в СН2С12 (свободный пеп- тид) Карбинольная смола. Примени- [476] ется при работе с Врос- и Fmoc- группами. Используется редко
ТФУ, в СН2С12 (амид пептида)
Используется при работе с Врос- [477]
защищенными аминокислотами
а) С1-^\-С
Ч/ ^ООН
б) 4 н. NaOH в смеси диоксан /
метанол (1 : 30 : 9) (свободный
пептид)
Смола расщепляется в два этапа: [478]
сначала активируется относите-
льно стабильная связь (за счет
окисления серы в сульфопроиз-
водное), а затем осуществляется *
расщепление под действием нук-
леофила
293
гт J3 Название смолы Структура функциональной группы полимера
9. 4-Оксифенилтиоме- тильная смола О но-^Хсн2-^ Ч—р II О
10. Полиамидная смола
на кизельгуре, (смо- ла Шеппарда) Таблица 48. Сведения о некоторых
Фирма, модель синтезатора, страна Основные характеристики аппарата
«Эпплайд биосистемз», США, модель 430 А Экономичный, высокоэффективный, пол- ностью автоматический синтезатор, уп- равляемый микропроцессором. Три ре- актора (по 40 мл) позволяют последова- тельно вести синтез трех пептидов. Раз- мешивание смолы вихревое
«Био^ёрч», США, модели 9500 и 9600 Автоматический синтезатор, управляе- мый ЭВМ. Щадящее перемешивание смо- лы током азота
«Миллиджен», США, серия 9000 Простой и удобный проточный синтеза- тор, содержащий три колонки; может ра- ботать в автоматическом или полуавтома- тическом режиме
ЛКБ, Швеция, «Биолинкс 4170» Полностью автоматизированный проточ- ный синтезатор, снабженный 3 колонка- ми, что дает возможность получать одно- временно аналоги пептидов; непрерыв- ный контроль за ходом реакции;
294
Продолжение табл. 47
Методы отщепления и форма, в
которой отщепляется пептид
(приведено в скобках)
Примечание
а) 112О2/СН3СООН
б) Нуклеофил: NaOH или КОН
(свободный пептид), спирты в
присутствии щрет-аминов (эфи-
ры пептидов), аммиак (амиды
пептидов), гидразин (гидразиды
пептидов)
То же
[479|
HF
Используется для Fmoc-страте- [458,
гии при синтезе пептидов на ко- 460,
лонках 462]
синтезаторах пептидов
Реагенты и методы Примечание
Смола бензгидриламино- или РАМ-по-
листирол;
^-Защитные группы: Вос- или Fmoc-
группа;
методы конденсации: симметричные ан-
гидриды или НОВТ-активированные эфи-
ры. Продолжительность 1 цикла около
1 ч.
Смола полистирольная или полиамид-
ная;
На-3ащитные группы: Вос- или Fmoc-
группа;
методы конденсации: диизопропилкар-
бодиимид, НОВТ-активированные эфи-
ры. Продолжительность цикла 1,5 ч
Смола: полиамидная; Na-защитная
группа — Етос-; методы конденсации:
пентафторфениловые или дигидрооксо-
бензотриазиновые активированные эфи-
ры, ди изопропил карбоди имид, реагент
Кастро
Смола полиамидная; Ыа-защитная груп-
па — Fmoc-; метод конденсации — акти-
вированные эфиры
Одна загрузка позволяет осущест-
вить до 50 циклов конденсации и
получить 0,5 ммоль пептида на
1 г смолы
Одна загрузка позволяет осуще-
ствить от 12 до 35 циклов конден-
сации; используется от 100 мг до
20 г смолы, что дает от 0,5 до
10 ммоль пептида на 1 г смолы
Позволяет получать от 0,1 до
5 ммоль пептида на колонку
Одна загрузка реагентов рассчи-
тана на 20 циклов. Позволяет по-
лучить 0,25 ммоль пептида на ко-
лонку
295
Фирма, модель синтезатора, страна
Основные характеристики аппарата
«Биолинке 4175»
«Бэкман инструменте», США,
система 990
Неавтоматический малогабаритный и де-
шевый проточный синтезатор, снабжен-
ный двумя колонками
Удобный, надежный и многофункцио-
нальный синтезатор, позволяющий ра-
ботать в автоматическом или ручном
режиме
счет облучения при 350 нм (путь В), а в соединении (133) соот-
ветственно — фтористым водородом или различными нуклео-
филами [458, 466]
А В
r-o-ch2—СН2—СО—О—СН—СО—Р
СНЯ (132)
Рас. 8. Простейший ручной прибор — качалка для твердофазного синюза
296
Продолжение табл. 48.
Реагенты в методы j
!
Примечание
То же Позволяет получать 0,25 ммоль
пептида на колонку
Смола: полистирольная или полиамид- Используется загрузка от 100 *.:г
ная; Ма-защитные группы: Вос- или до 20 г смолы
Fmoc-; методы конденсации: диизопро-
пилкарбодиимид, активированные эфи-
ры, симметричные ангидриды
А В
О—СО—СН2—Р.
А
I II
W (133)
В табл. 47 представлены сведения о некоторых смолах, ис-
пользуемых для твердофазного синтеза пептидов.
Сейчас созданы и выпускаются
рядом фирм специальные аппара-
ты, которые по специальной про
грамме могут вести наращивание
полипептидной цепи в автоматиче-
ском, полуавтоматическом и руч-
ном режиме. Эти синтезаторы пеп
тидов состоят из систем реактора и
контроля. Система реактора вклю-
чает реакционный сосуд, набор раз
нообразных вентелей для подачи
реагентов и растворителей, а также
резервуары для этих растворов.
Порядок добавления реагентов и
время нахождения их в реакторе,
а также все управление этой систе
мой может быть запрограммирова
но. К настоящему времени выпус
каются высокоэффективные, пол
ностью автоматизированные синте-
заторы, управляемые микропроцес
Рис. 9. Ручной прибор для твердофазной
синтеза УДПС-1. Выпускается ФИБХ
им. М. М. Шемякина совместно с НПС
«Бноприбор» Российской АН
297
Рис. iO. Полуавтоматический прибор для твердофазного синтеза У ДПС-2
сором или ЭВМ. В табл. 48 представлены некоторые сведения
о синтезаторах, а на рис. 8—11 изображено несколько подоб-
ных аппаратов.
МЕТОДЫ СИНТЕЗА РЕАГЕНТОВ
Получение полимерных носителей
Методика 284. Хлорметилирование полистирольной
смолы [16, 454].
Для набухания смолы в трехгорлой круглодонной колбе
размешивают в течение 2 ч при комнатной температуре 50 г
гранулированного полимера в 200 мл хлороформа, а затем
смесь охлаждают до 0 °C. Из капельной воронки при переме-
шивании добавляют охлажденный раствор 3,8 мл безводного
хлорного олова в 100 мл хлорметилированного эфира. Пере-
298
Рис 11. Автоматический пептидный синтезатор «Эпплэйд Байосистсм»,
модель 430 А (США)
мешивание при 0 °C продолжают еще 30 мин, затем смолу от-
фильтровывают на воронке Бюхнера с пористым стеклянным
фильтром и промывают следующими растворителями (по 1 л
каждого): смесью диоксана с водой (3:1), диоксана с 3 н. НО
(3:1), каждый раз давая возможность растворителям хорошо
проникнуть в гранулы полимера. Смолу затем тщательно про-
мывают диоксином, водой, метанолом и сушат в течение ночи в
вакуум-эксикаторе над хлористым кальцием.
Для определения степени хлорметилирования 200 мг смо-
лы нагревают в ампуле с 3 мл пиридина в течение 2 ч при
100 °C. Смесь количественно переносят в колбу Эрленмейера
емкостью 125 мл, содержащую 30 мл 50 %-й уксусной кис-
лоты, добавляют 5 мл концентрированной азотной кисло-
ты. Содержание хлорид-аниона в смеси количественно опре-
деляют модифицированным методом Фольгарда.
Методика 285. Полиамидная смола на кизельгуре [458].
Свежеперегнанные М,Ы-диметилакриламид (33,3 г;
336 ммоль) и метиловый эфир акрилоилсаркозина (2,83 г;
18 ммоль) прибавляют к раствору бис-акрилоилэтилендиами-
на (3,90 г; 23,2 ммоль) в смеси диметилформамид — вода
299
(3,12 : 2; 137 мл), прибавляют также 10 %-й водный раствор
персульфата аммония, смесь немедленно вливают в кизельгур
(диаметр частиц 335—500 мкм; 100 г) и перемешивают стек-
лянной палочкой. Реакционную массу помещают в эксикатор
и вакуумируют в течение 2 мин на водоструйном насосе. При
этом происходит сильная дегазация смеси. Эксикатор выдер-
живают под вакуумом в течение 15 мин, а затем в него осто-
рожно вводят ток азота и реакционную смесь выдерживают
2,5 ч при комнатной температуре. После этого смолу тщатель-
но промывают водой на фильтре с пористым стеклом и про-
пускают через сито с диаметром отверстий 700 мкм, смолу про-
мывают водой при декантации (4 раза), отфильтровывают, про-
мывают ацетоном, эфиром и сушат при высоком вакууме над
Р2Об. Выход 125,5 г.
Полимер на кизельгуре (2,5 г; 0,27 ммоль/г саркозина) ос-
торожно встряхивают с избытком этилендиамина в течение
12 ч, а затем смолу промывают диметилформамидом, перено-
сят в стеклянную колонку диаметром 15 мм и промывают ди-
метилформамидом со скоростью 3,3 мл в мин до отрицательной
реакции на нингидрин (обычно около 30 мин).
В качестве внутреннего стандарта используют норлейцин.
Для этого симметричный ангидрид флуоренилметилоксикар-
бонил-норлейцина (0,475 ммоль) в ДМФА вводят в колонку
и осуществляют ацилирование в течение 40 мин; за его ходом
следят спектрофотометрически. После промывания колонки
диметилформамидом и отщепления Fmoc-группы сразу же
добавляют активированный 2,4,5-трихлорфениловый эфир п-
оксиметилфеноксиуксусной кислоты (0,184 г; 0,5 ммоль) и 1-
оксибензтриазол (0,076 г; 0,5 ммоль) в 2 мл ДМФА. Реакцию
ацилирования ведут в течение 40 мин. После промывания смо-
лы диметилформамидом колонка готова к работе.
Методика 286. Присоединение Вос-аминокислоты к
хлорметилированной смоле в присутствии триэтиламина [ 161.
К суспензии хлорметилированной смолы (1,0 ммоль хлора)
в абсолютном спирте (2—5 мл на 1 г смолы) добавляют Вос-
аминокислоту, являющуюся в пептиде С-концевой (1 ммоль),
и триэтиламин (0,9 ммоль; из расчета 0,14 мл на 1 ммоль; плот-
ность 0,723), и смесь осторожно кипятят с обратным холо-
дильником (снабженным трубкой с хлористым кальцием) на
масляной бане, нагретой до 90 °C в течение от 24 ч (для боль-
шинства Вос-аминокислот) до 65 ч (для Вос-нитроаргинина).
Реакцию присоединения С-концевой Вос-аминокислоты
можно проводить в течение 5 ч при 40 °C, если в качестве ка-
тализатора использовать иодистый калий [480].
Вос-аспарагин этерифицируют в этилацетате, чтобы исклю-
чить алкоголиз амида.
300
После окончания реакции полимер отфильтровывают на
воронке со стеклянным пористым фильтром и трижды про-
мывают каждым из следующих растворителей: спиртом, водой,
метанолом и хлористым метиленом. Полимер, суспендирован-
ный в хлористом метилене, переносят в делительную воронку,
перемешивают с этим растворителем (20 мл на 1 г смолы) и
выдерживают до тех пор, пока основная масса полимера не
всплывает на поверхность. Растворитель с мелкими частица-
ми смолы сливают и выбрасывают, а операцию флотации повто-
ряют не менее 3 раз, после чего полимер переносят на фильтр,
растворитель удаляют с помощью водоструйного насоса и
продукт сушат в течение ночи в вакуум-эксикаторе.
Методика 287. Вос-фенилаланил-полимер. Присо-
единение Вос-аминокислоты с использованием гидрооки и
тетраметиламмония [481].
К раствору Вос-фенилаланина (2,69 г; 10,1 ммоль) в 10 мл
метанола добавляют 15,5 мл 0,62 н. гидроокиси тетраметилам-
мония. Последнее соединение получают путем растворения эк-
вимолярных количеств хлорида тетраметиламмония и едко-
го кали в абсолютном спирте, удаления КС1 фильтрованием и
упариванием спирта на роторном испарителе. Метанол уда-
ляют в вакууме, а к маслообразному остатку добавляют диок-
сан и снова упаривают; операцию повторяют дважды. Оста-
ток растворяют в метаноле и снова упаривают. После высу-
шивания в вакуум-эксикаторе над фосфорным ангидридом в
течение 1,5 ч соль (3,5 г) растворяют в диметилформамиде
(50 мл), добавляют хлорметилированный полимер (5,02 г) и
размешивают в течение 14 ч при комнатной температуре. Смо-
лу отфильтровывают и промывают сначала диметилформами-
дохМ (200 мл), а затем по 100мл каждым из следующих раст-
ворителей: метанолом, водой, снова метанолом. Продукт
сушат в вакуум-эксикаторе над фосфорным ангидридом
в течение ночи. Выход 5,6 г. Содержание фенилаланина
0,43 ммоль/г.
Методика 288. Получение цезиевых солей Вос-ами-
нокислот (4821.
К раствору 2,0 г Вос-аминокислоты в 15 мл спирта добав-
ляют 3—5 мл воды. На pH-стате значение pH раствора дово-
дят до 7,0 добавлением водного раствора гидроокиси цезия.
Затем нейтральный раствор упаривают на роторном испари-
теле до малого объема. Для удаления следов воды добавляют
несколько раз бензол и упаривают его на роторном испарите-
ле. Продукт сушат над фосфорным ангидридом и использу-
ют без дополнительной очистки.
Методика 289. Получение калиевых солей Вос-ами-
нокислот [483].
301
В смеси 6 мл спирта и 4 мл воды растворяют 1,0 г Вос-ами-
нокислоты и добавляют 1 экв. 1 н. раствора едкого кали. Смесь
подвергают азеотропной отгонке с толуолом, продукт сушат
в вакуум-эксикаторе над фосфорным ангидридом.
Методика 290. Присоединение цезиевых солей Вос-
аминокислот к смоле Меррифильда [4821.
В сосуд с завинчивающейся крышкой помещают 1 ммоль
сухой цезиевой соли Вос-аминокислоты, 1 мэкв. смолы Мер-
рифильда в диметилформамиде (6—8 мл на 1 г смолы). Смесь
размешивают на магнитной мешалке при 50 °C на водяной ба-
не в течение ночи. Затем полимер отфильтровывают, после-
довательно промывают диметилформамидом, смесью ДМФА —
вода (9 : 2), снова диметилформамидом, спиртом и, наконец,
сушат.
Методика 291. Присоединение калиевых и цезие-
вых солей Вос-аминокислот в условиях межфазного катали-
за [484].
К суспензии полимера (200 мг) в 1,5 мл диметилформамида
добавляют соответствующее количество калиевой или цезие-
вой соли Вос-аминокислоты (на 1 моль активного хлора при-
бавляют 1,2 моль соли), затемтетрабутиламмоний бро-
мида (1,2 моль) и встряхивают в течение 48 ч при комнатной
температуре или при 50 °C (для Вос-производных таких ами-
нокислот, как аспарагин, нитроаргинин, ацетамидометилцис-
теин). Полимер отфильтровывают и трижды промывают
(3 мл) следующими растворителями: диметилформамидом, сме-
сью ДМФА — вода (9 : 1), снова диметилформамидом и, на-
конец, спиртом (5 мл). Смолу сушат в вакууме.
Методика 292. Присоединение калиевых солей Вос-
аминокислот в присутствии 18-краун-эфира [483].
К раствору 1 экв. калиевой соли Вос-аминокислоты и 1 экв.
18-краун-6-эфира в диметилформамиде добавляют пятикрат-
ное количество хлорметилированного полимера и смесь вы-
держивают в течение 18 ч при комнатной температуре. Даль-
нейшую обработку проводят как обычно. Выход продукта
достаточно высокий.
Наргщизание полипептидной цепи
На рис. 8 изображен ручной прибор, используемый при
твердофазном синтезе пептидов [16]. Размер качающегося ре-
акционного сосуда зависит от количества загружаемой смо-
лы. Самый маленький сосуд предназначен для использова-
ния не более 1 г носителя, средний — для 1,5—4,5 г полиме-
ра, а большой — для 10 г смолы. Полимер с закрепленной на
нем С-концевой Вос-аминокислотой помещают в реакционный
302
Таблица 49. Программа синтеза пептида
Этап синтеза Используемый реагент или растворитель Объем раство- ра, мл Время, реак- ции , мин
Промывка Диоксан, 3 раза »0 5 Деблокирование Раствор 4 н. НС1 в диоксане 10 30 Промывка Диоксан, 3 раза 10 5 Промывка Хлороформ, 3 раза 10 5 Нейтрализация Раствор триэтиламина в хлоро- 10 10 форме (1:9) Промывка Хлороформ, 3 раза 10 5 Промывка Хлористый метилен, 3 раза 10 5 Введение карбоксиль- Раствор Вос-аминокислоты 6 5 ного компонента Конденсация Раствор ДЦГК в СН2С12 (50%-й) 0,15 120 Промывка Хлористый метилен, 3 раза 10 5
сосуд и выдерживают в растворителе для набухания смолы,
а затем осуществляют определенные манипуляции, задавае-
мые программой синтеза конкретного пептида (методики 293—
296). Реактивы и растворы вводят через боковой патрубок, а
их удаление происходит за счет фильтрования через пористый
стеклянный фильтр, являющийся дном реакционного сосуда.
На боковой отвод помещают во время синтеза трубку с хло-
ристым кальцием.
Ниже представлено несколько программ последователь-
ности операций, используемых при различных вариантах твер-
дофазного метода.
Методика 293.Синтез Leu-Asp-Ala-Gly-Arq-полимера [ 161.
OBzl
Вос-нитроаргинин-полимер (0,5 г; 0,284 ммоль/г смолы;
общее количество аминокислоты 0,142 ммоль) загружают в
малый реакционный сосуд, добавляют 10 мл диоксана и встря-
хивают в течение 5 мин. Используют 2,5 экв. Вос-аминокислог
и ДЦГК, применяя 50 %-й раствор карбодиимида в хлористом
метилене (1 г ДЦГК растворяют в 2 мл СН2С12).
Последовательно выполняют операции, представленные в
табл. 49.
После окончания синтеза пептидил-полимер помещают на
взвешенную воронку с пористым фильтром и промывают ук-
сусной кислотой и спиртом, а затем сушат в вакуум-эксикато-
ре над едким кали. Аликвотную часть пептидил-полимера
отбирают на гидролиз и определение аминокислотного состава.
Методика 294. Синтез рибонуклеазы [485].
Вос-валил-полимер (2,0 г; 0,21 ммоль/г смолы; общее коли-
чество аминокислоты 0,42 ммоль) помещают в реакционный
303
Таблица 50. Программа синтеза рибонуклеазы А (124 аминокислоты)
Этап синтеза Реагент или растворитель Объем раство- ра, мл Время, мин
Промывка Хлористый метилен, 2 раза Раствор ТФУ в СНаС1а (50%-й) 25 2
Смачивание смолы 25 2
Деблокирование Раствор ТФУ в СН2С12 (50%-й) 25 21
Промывка Хлористый метилен, 5 раз 25 2
Промывка Хлороформ, 3 раза 25 2
Нейтрализация Раствор триэтиламина в хлоро- форма (10 %-ный) 25 7
Промывка Хлороформ, 3 раза Хлористый метилен, 3 раза Раствор (3 экв.) Вос-аминокисло- ты в СН2С12 7 мл + 5 мл СН2С12 для смывки реагента со стенок 25 2
Промывка 25 2
Добавление кар- боксильного компонента 12 7
Конденсация Раствор (3 экв.) ДЦГК в 7 мл СН2С12 + 5 мл СН2С12 для смы- вок 12 300
Промывка Хлористый метилен, 3 раза 25 2
Промывка Этиловый спирт, 3 раза 25 2
Примечания: 1. При введении Вос-ш-нитроаргинина и Вос-гистидина эти
соединения растворяют в смеси 4,8 мл ДМФА и 2,2 мл СН2С12. 2. Вос-глутамин и
Вос-аспарагин вводят в виде п-нитрофениловых эфиров (4 экв.) в ДМФА в течение
10 ч. После деблокирования Вос-группы вместо СН2С12 промывание смолы ведут ди-
метилформамидом; стадию конденсации с ДЦГК исключают. 3. Ход синтеза контро-
лируют, отбирая 10 мг смолы после введения 6—10 аминокислотных остатков, гид-
ролизуя пробу и определяя аминокислотный состав. 4. После завершения синтеза
рибонуклеазу отщепляли жидким фтористым водородом или раствором HF в три-
фторуксусной кислоте.
сосуд, добавляют 25 мл хлористого метилена и встряхивают
2 мин. Используют следующие аминокислоты: Р-бензил-ас-
парагиновую кислоту, у-бензил-глутаминовую кислоту, S-
бензил-цистеин, О-бензил-серин, О-бензил-треонин, О-бензил-
тирозин, е-бензилоксикарбонил-лизин, нитроаргинин, метио-
нинсульфоксид. Гистидин применяли незащищенный (табл. 50).
Методика 295. Синтез пептида на РАМ-смоле [487].
На I г аминоацил-полимера использовали 10 мл раствори-
теля (см. табл. 51).
Методика 296. Синтез пентадекапептида Lys-Leu-
Ser-Val-Ala-Thr-Lys-Gly-Pro-Leu-Thr-Val-Ser-Asp-Gly- на ко-
лонке [458].
Полидиметилакриламидный гель на кизельгуре, функцио-
нализированный метоксикарбонильными группами, был прев-
ращен в аминоформу при реакции в течение 15 ч с избытком
этилендиамина. Смолу помещали в стеклянную колонку диа-
метром 15 мм и промывали со скоростью 3,3 мл/мин диметил-
формамидом до отрицательной реакции на нингидрин, что
занимает около 30 мин.
304
Таблица 51. Твердофазный синтез пептида на РАМ-смоле
№ п/п Этап синтеза Реагент или растворитель л ь ©. S CQ S
1 Промывка Хлористый метилен, 3 раза Раствор ТФУ в СН2С12(1 : 1) 1
2 Деблокирование 30
3 Промывка Хлористый метилен, 4 раза 1
4 Нейтрализация Диизпропилэтиламин в СН2С12 • 1Q1 5
5 Промывка Хлористый метилен, 3 раза 1
6 Добавление карбокси- Раствор Вос-аминокислоты в льного компонента СН2С12 (3 экв.) 5
7 Конденсация * Раствор ДЦГК в СН2С12 (3 экв.) 60
8 Промывка Хлористый метилен, 6 раз 2
Примечания: 1. В случае необходимости операцию 7 повторяют.
2. Для введения Вос-аспарагина и Вос-глутамина использовали добавление экви-
валентного количества 1-оксибензтриазола в ДМФА.
3. Пептид от смолы отцепляли в смеси HF-анизол (9 : 1) при О °C в течение
30 мин.
Использовали следующие Fmoc-аминокислоты: 8-Вос-ли-
зин, О-тргт-бутил-серин, O-mpem-бутил-треонин, $-трет-
бутил-аспарагиновая кислота. Лизин вводили через /г-нитро-
фениловый эфир в присутствии 0,5 моль 1-оксибензотриазола.
детальные аминокислоты «пришивались» по методу сим-
метричных ангидридов. Синтез этих ангидридов осуществля-
ли из Fmoc-аминокислот (1 ммоль) в СН2С12 (5 мл) с несколь-
кими каплями ДМФА и ДЦГК (0,475 ммоль) в 1 мл СН2С12 в
течение 10 мин. Мочевину удаляли фильтрованием, а раство-
ритель упаривали при пониженном давлении. Остаток рас-
творяли в 2 мл ДМФА и помещали на колонку; за реакцией
ацилирования следили фотометрически. Поскольку симмет-
ричный ангидрид Fmoc-глицина выпадает в осадок, то после
упаривания хлористого метилена его вместе с мочевиной раз-
мешивают в 3 мл ДМФА и раствор фильтруют прямо в колон-
ку (см. табл. 52).
Присоединение внутреннего стандарта — норлейцина —
и «ручки» — остатка оксиметилфеноксиуксусной кислоты1—
описано в методике 285.
Учитывая, что побочной реакцией при Fmoc-стратегии яв-
ляется потеря дипептидов из-за образования дикетопипера-
зинов (причем пиперидин является катализатором этого неже-
лательного’ процесса), японские исследователи предложили
использовать для удаления Fmoc-группы раствор (0,05—0,1 М)
тетрабутиламмоний фторида в диметилформамиде в течение
2 мин при комнатной температуре [486]. Метод апробирован
при синтезе тетрапептида Leu-Ala-Gly-Val.
20 2-882 305
Таблица 52. Синтез пептида на колонке
Этап синтеза Процедура синтеза Время,- мин
Ацилирование 1. Нанесение на колонку предварительно полученного симметричного ангидрида F-тос-аминокислоты (4—6 экв.) 2. Рециркуляция этого раствора 20 3. Пауза, отбор пробы смолы на анализ 4. Рециркуляция ДМФА с Fmoc-аминокис- лотой Промывка 5. Пропускание ДМФА 6,5 6. Рециркуляция ДМФА 0,3 7. Пропускание ДМФА 5 Отщепление Fmoc-8. Пропускание через колонки 20 %-го 10 группы раствора пиперидина в ДМФА Промывка 9. Элюирование колонки ДМФА 8 10. Пауза, отбор пробы на анализ 11. Пропускание ДМФА 12
При использовании Fmoc-аминокислот часто применяли сим-
метричные ангидриды (табл. 52). Несмотря на то, что симмет-
ричные ангидриды Вос-аминокислот широко используются в
твердофазном синтезе, они оказались менее эффективными в
случае применения Fmoc-производных (по-видимому, из-за
стерических препятствий). Поэтому кроме пентафторфени-
ловых эфиров в случае применения Fmoc-аминокислот эффек-
тивно используются 1-оксибензтриазиловые эфиры. Эти эфи-
ры имеют ряд преимуществ перед симметричными ангидри-
дами — меньший расход защищенных аминокислот, большая
устойчивость в процессе синтеза и более высокий выход.
Ввиду того, что 1-оксибензтриазиловые эфиры Вос-амино-
кислот также хорошо зарекомендовали себя в пептидном син-
тезе, их довольно часто применяют вместо симметричных
ангидридов. Обычно используют 2—Зэкв. 1-оксибензтриази-
лового эфира на 1 моль аминокомпонента, при этом ацили-
руется полностью аминокомпоненты в течение 1 —1,5 ч.
Для получения 1-оксибензтриазилового эфира растворяют
1 моль Вос- или Fmoc-аминокислоты в ДМФА, добавляют
1 моль 1-оксибензтриазола и 1 моль ДЦГК и выдерживают
40 мин в холодильнике. Отфильтровываютдициклогексилмо-
чевину и вносят раствор 1-оксибензтриазилового эфира в ре.
акционный сосуд. Все операции в случае использования Вос.
и Fmoc-аминокислот проводят в ДМФА, кроме удаления Вос.
группы трифторуксусной кислотой в метиленхлориде и по.
следующей нейтрализации триэтиламином g метиленхлориде.
Использование Вос-№т-тозилгистидина нежелательно, так
как 1-оксибензтриазол снимает защитную тозильную группу.
306
Основным фактором, влияющим на гомогенность конечно-
го продукта твердофазного синтеза, является полнота ацили-
рования на стадии образования пептидной связи, поэтому осо-
бое значение приобретает контроль полноты реакции на этой
стадии. Ниже приводятся основные методы контроля.
Методы контроля полноты реакции
пептидообразования
Методика 297. Нингидриновый тест Кайзера [488].
Готовят следующие растворы: 1) 500 мг нингидрина в 10 мл
этилового спирта; 2) 80 г фенола в 20 мл этанола; 3) 2 мл
0,001 М раствора KCN разбавляют пиридином до 100 мл.
Аликвоту смолы (10—20 мг) помещают в пробирку (раз-
мером 12 X 75 мм) и добавляют по капле каждого из указан-
ных выше растворов. Пробирку нагревают в течение 2—5 мин
на кипящей водяной бане, периодически встряхивая ее. Если
гранулы смолы остаются бесцветными, а раствор — желтым,
это считают отрицательным тестом. Окрашивание частичек
смолы в голубой цвет, а раствора — в слабый зеленый оцени-
вают как слабую положительную реакцию. Темно-синий цвет
как гранул, так и раствора свидетельствует о положительной
реакции. Если N-концевой аминокислотой является |3-бен-
зил-аспарагиновая кислота или пролин, то при положитель-
ном тесте развивается коричневая или красно-коричневая ок-
раска.
Метод позволяет определить 5 мкмоль свободных амино-
групп на 1 грамм смолы. Положительная окраска развивается
даже если прореагирует 99 % свободных групп пептидил-по-
лимера.
Методика 298. Количественный нингидриновый тест
[489].
Готовят следующие реагенты. Раствор 1 : В 10 мл абсо-
лютного этилового спирта растворяют при нагревании 40 г
фенола (ч. д. а.), добавляют 4 г смешанной смолы Амберлит
МВ-3, перемешивают 45 мин и ионообменник отфильтровы-
вают.
Растворяют 65 мг KCN в 100 мл воды. Затем 2 мл этого рас-
твора разбавляют до 100 мл пиридином свежеперегнанным над
нингидрином, добавляют 4 г амберлита МВ-3, размешивают и
смолу удаляют. К фильтрату прибавляют предыдущий рас-
твор фенола.
Раствор 2: В 50 мл абсолютного этилового спирта растворя-
ют 2,5 г нингидрина, плотно закрывают и склянку хранят в тем-
ноте над азотом. Пробу анализируемой смолы промывают два
раза 5 %-м раствором диизопропилэтиламина в хлористом
20* 307
метилене, трижды — хлористым метиленом и сушат в ваку-
уме при комнатной температуре.
Процедура определения свободных
аминогрупп при их низком содержании
(д о 0,1 м к м о л ь). К навеске (2—5 мг) пептидил-полимера в
пробирке (размером 10 X 75 мм) прибавляют 100 мкл рас-
твора 1 (6 капель) и 25 мкл раствора 2 (2 капли). В пробирку
сравнения добавляют лишь эти растворы. Пробирки вы-
держивают в нагревательном блоке при 100 °C в течение 10 мин,
а затем переносят в холодную воду. К смеси прибавляют 1 мл
60 %-го этилового спирта, перемешивают и отбирают раствор
пипеткой Пастера, содержащей тампон со стеклянной ватой.
После двукратного ополаскивания пробирок 0,20 мл раствора
хлористого тетраэтиламмония в хлористом метилене (0,5 М)
содержимое разбавляют 60 %-м этиловым спиртом до 2 мл и
измеряют поглощение света при 570 нм против контрольного
раствора.
Процедура определения свободных
аминогрупп при их высоком содержании
(0,1—2 мкмоль). Навеску пептидил-полимера (2—5 мг)
обрабатывают так, как описано выше, но добавляют не 1 мл,
а 2 мл 60 %-го этилового спирта и пробирки ополаскивают
раствором тетраэтиламмоний хлорида по 0,5 мл, а содержимое
разбавляют 60 %-м этиловым спиртом до 25 мл.
Если навеска смолы превышает 5 мг, то добавляемый объ-
ем смеси растворов 1 и 2 увеличивают на 20 мкл на каждый мил-
лиграмм полимера.
Количество свободных аминогрупп (Q) вычисляют по фор-
муле
Q = ‘X . юв,
е570 * W
где А -поглощение света при 570 нм; V — объем пробы, мл;
в' — коэффициент экстинкции, равный 1,5 X 104 М • см-1;
W — навеска, мг.
Чувствительность метода — 0,3 мкмоль аминогрупп на
1 г смолы. Это позволяет определять свободные аминогруппы
при блокировании их на 99,9 %.
При особо точных измерениях необходимо каждый раз
определять коэффициентэкстинкции для разных аминокислот
растущего полипептида.
Методика 299. Хлораниловый тест [490].
К пептидил-полимеру (около 1 мг), находящемуся в про-
бирке (размером 7 X 50 мм), добавляют по 200 мкл следую-
щих реагентов: ацетальдегида (при анализе первичных ами-
нов) или ацетона, если определяют вторичную аминогруппу
308
(например, пролина), и 1 %-й раствор 2,3,5,6-тетрахлор-1,4-
бензохинона (хлоранила). Смесь размешивают и спустя 45—
€0 с наблюдают возникающую окраску. Черный или темно-
синий цвет полимера свидетельствует о наличии очень боль-
шого количества непрореагировавших аминогрупп, синий —
много свободных аминогрупп, зеленый — имеется менее 0,5 %
аминогрупп, желтый — все аминогруппы проацилированы.
Чувствительность метода 2—5 мкмоль пролина на 1 г смо-
лы или 5—8 мкмоль других аминокислот.
Для успешного осуществления теста следует помнить, что
нельзя использовать для его проведения полиэтиленовые про-
бирки. Аминогруппа пептидил-полимера должна быть в неио-
низированной форме. Следует тщательно отмывать азотсодер-
жащие соединения, например триэтиламин. Появление бурой
окраски часто свидетельствует о присутствии в смоле адсор-
бированных третичных аминов.
Методика 300. Флуорометрический метод определе-
ния свободных аминогрупп [491].
Аликвотную часть пептидил-полимера (5—10 мг) переносят
на фильтр со стеклянным пористым дном и промывают смолу
следующими растворителями (по 3 раза): хлористым метиле-
ном, этиловым спиртом, снова хлористым метиленом и, нако-
нец, хлороформом. Смолу обрабатывают 10 %-м раствором три-
этиламина в хлороформе и промывают (1 мл X 4) растворите-
лями: хлороформом, этиловым спиртом, снова хлороформом.
Вакуум отсоединяют, смолу смачивают несколькими капля-
ми 10 %-го раствора триэтиламина в хлороформе и добавляют
1 мл раствора флуорескамина в хлороформе (10 мг/мл), кото-
рый можно хранить при 4 °C в течение месяца. После выдер-
живания пробы при комнатной температуре в течение 10 мин
растворитель отсасывают, а смолу промывают хлороформом,
метанолом и снова хлороформом. При облучении воронки со
смолой длинноволновым ультрафиолетовым светом (366 нм)
сравнивают флуоресценцию пробы со стандартом. В качестве
последнего используют исходную хлорметилированную смолу,
обработанную в той же последовательности, что и проба. По
мере протекания реакции пептидообразования интенсивность
флуоресценции пробы все больше приближается к флуоресцен-
ции стандарта. Указанным методом положительная реакция
наблюдается при блокировании свободных аминогрупп на
99,9 %.
Следует помнить, что флуорофорсодержащий полимер мо-
жно хранить при комнатной температуре неопределенно дол-
го. Это позволяет сравнивать между собой результаты прове-
дения реакции на разных стадиях наращивания полипептид-
ной цепочки. При повышенной температуре флуоресценция
309
5
Рис. 12. Принцип работы «свеллографа» в синтезаторах типа У ДПС:
а — упрощенная схема «свеллографа»: /— регистрирующее устройство; 2—перо самопис-
ца; 3 — стеклянная трубка; 4 — подвижный поршень; 5 — груз для прижимания поршня
к полимеру; 6 — устройство в нижней части реактора; Р — полимер; б — «свеллограм-
ма» процесса твердофазного синтеза с использованием Вос-аминокислот: / — промывка
ДМФА; 2 — деблокирование 50 % ТФУ/ метиленхлорид; 3 — нейтрализация 10 % ра-
створом триэтиламина в ТГФ; 4 — промывка метиленхлоридом; 5 — промывка 50 %
раствором /ире/и-бутанола в ТГФ; ПС — образование пептидной связи; ПС' — повтор-
Iая операция
резко снижается из-за образования у-лактона. Необходимо
подчеркнуть, что перед добавлением к пробе раствора флуорес-
камина смолу следует обязательно смачивать раствором три-
этиламина для предотвращения изомеризации флуорофор-по-
лимера в нефлуоресцирующий у-лактон.
Методика 301. Пикриновый тест [492].
Аликвоту смолы (примерно 20 мг) оставляют набухать в
хлористом метилене (0,5 мл; это же количество используют и
для других растворителей) в течение 5 мин, нейтрализуют при
помощи б %-го раствора диизопропилэтиламина в хлорис-
том метилене (2 раза по 3 мин), промывают хлористым мети-
леном (3 X 2 мин), а затем обрабатывают 0,1 М раствором пик-
риновой кислоты в хлористом метилене (2x3 мин). Избыток
пикриновой кислоты вымывают хлористым метиленом (5 X
X 2 мин). Пикриновую кислоту, сорбированную на смоле в
результате взаимодействия со свободными аминогруппами,
элюируют диизопропилэтиламином или с помощью 0,1 М
раствора гидрохлорида пиридина в хлористом метилене (2 X
X 3 мин). Полимер промывают хлористым метиленом (3 X
X 2 мин) и разбавляют 95 %-м этиловым спиртом до 10 мл.
Измерение поглощения света при 358 нм позволяет вычислить
количество свободных аминогрупп, используя коэффициент
молярной экстинкции пикриновой кислоты 14 500.
В приборах УДПС-1 и У ДПС-2 (рис. 9 и 10), разработан-
ных в ФИБХ им. академика М. М. Шемякина совместно с НПО
310
«Биоприбор» Российской АН, используется новый принцип
контроля за ходом реакций в твердофазном синтезе. В проточ-
ном реакторе (рис. 12) установлен специальный подвижный
поршень — датчик, связанный с регистрирующим устройством.
Поршень прижимается к полимеру и передвигается по верти-
кали при изменении объема смолы. Изменение объема смолы
происходит в процессе промывок, образования пептидной свя-
зи, деблокирования, нейтрализации, смены растворителя и
т. п. В процессе образования пептидной связи происходит на-
бухание смолы, что приводит к увеличению ее объема. Пре-
кращение увеличения объема свидетельствует о прекращении
реакции (полнота реакции определяется одним из описанных
выше методов). Таким образом легко установить оптимальное
время всех стадий твердофазного синтеза.
Для индикации полноты образования пептидной связи в
автоматических синтезаторах используются активированные
З-гидрокси-4-оксодигидробензтриазиновые эфиры М-защи-
щенных аминокислот:
В процессе ацилирования аминогрупп выделяющийся 3-гид-
роксид-4-оксобензтриазин в присутствии неацилированных
аминогрупп окрашивается в желтый цвет, придавая полимеру
желтый оттенок. Если ацилирование прошло полностью, на-
чальный белый цвет полимера восстанавливается.
Отщепление пептида от смолы
Методика 302. Применение безводного фтористого
водорода [16, 443]
Внимание! Всю работу со фтористым водородом необ-
ходимо проводить в хорошо вентилируемом вытяжном шкафу,
так как HF может вызвать серьезные ожоги, а вдыхание его
смертельно. При работе необходимо иметь под рукой 20 %-й
раствор глюконата кальция в глицерине. При попадании фто-
ристого водорода на кожу пораженное место обильно промы-
вают водой, а затем наносят раствор глюконата кальция.
Отщепление пептида от смолы удобно осуществлять на
установке, изготовленной из тефлона (рис. 13), которая предус-
матривает создание в ней вакуума. Она должна также выдер-
живать внутреннее давление до 2 атмосфер (196,13 кПа).
В реакционный сосуд 2 загружают сухой пептидил-поли-
мер, помещают магнитный перемешивающий стержень и при-
311
Рис. 13. Схема прибора для отщепления пептида безводным HF
бавляют анизол не менее 50 экв. на 1 экв. пептида. В предохра-
нительный резервуар 3, также снабженный магнитом, поме-
щают несколько граммов фторида кобальта. После этого ре-
зервуар и ловушку 1 погружают в сосуд Дьюара, наполненный
жидким азотом. Систему соединяют с водоструйным насосом
и создают вакуум, после чего закрывают все краны. Медлен-
но открывают вентиль баллона с HF и собирают конденсиру-
ющийся газ в резервуар 3 в количестве, достаточном для
нескольких отщеплений. Вентиль баллона закрывают, а резер-
вуару позволяют нагреться до комнатной температуры, по-
гружая его в сосуд с водой при температуре 20—25 °C. Пере-
мешивание жидкого HF проводят со скоростью, обеспечива-
ющей удовлетворительное суспендирование фторида кобаль-
та. Затем охлаждают реакционный сосуд 2 так, чтобы HF в
резервуаре 3 закипел и началась его перегонка в реакционный
сосуд 2. После того, как в сосуде 2 соберется достаточное ко-
личество фтористого водорода, все краны закрывают, под ре-
акционный сосуд подставляют баню с температурой воды 20—
25 °C и осуществляют перемешивание в течение от 30 мин до
1 ч при 0 °C
После окончания реакции, не прекращая перемешивания,
осторожно открывают кран между сосудом 2 и ловуш-
кой 1 и фтористый водород перегоняют в ловушку. Затем от-
крывают кран, соединяющий установку с вакуумом, и из
реакционного сосуда 2 в течение 1 ч удаляют анизол. Сухой
порошок суспендируют в этиловом спирте и переносят из реак-
ционного сосуда в сосуд для промывания. Вещество промывают
этилацетатом, сушат в вакууме и пептид элюируют раство-
ром уксусной кислоты, отфильтровывают от смолы и лиофи-
лизируют. Если пептид плохо растворяется в воде, то экстрак-
цию осуществляют пиридином или уксусной кислотой.
Фтористый водород удаляют из ловушки при помощи водо-
струйного насоса.
312
Методика 303. Раствор НВг в трифторуксусной кис-
лоте [16].
Внимание! Работу следует проводить в хорошо вен-
тилируемом вытяжном шкафу, исключая попадание раствора
НВг в трифторуксусной кислоте на кожу. Следует также из-
бегать вдыхание паров указанных соединений.
Пептидил-полимер суспендируют в трифторуксусной кис-
лоте (1 г полимера в 10 мл ТФУ) в сосуде для отщепления
(рис. 14). Прибавляют, если требуется, анизол или метилэтил-
сульфид (50 моль на 1 моль чувствительной аминокислоты),
а затем через суспензию пропускают медленный ток безводно-
го НВг в течение 1,5 ч. Если пептид содержит тирозин или
триптофан, то НВг предварительно пропускают через про-
мывную склянку, содержащую раствор 2 г анизола или резор-
цина в трифторуксусной кислоте.
После окончания реакции открывают кран на дне сосуда
для отщепления и раствор пептида отсасывают в круглодон-
ную колбу с помощью водоструйного насоса. Полимер промы-
вают 3 раза трифторуксусной кислотой (по 10 мл на 1 г смо-
лы), а затем растворитель удаляют на роторном испарителе.
Для избавления от избытка НВг продукт растворяют в смеси
уксусная кислота — вода (3 : 1) или метанол — вода (1 .* 1) и
упаривают. Если при отщеплении использовали анизол, то пеп-
тид тщательно экстрагируют эфиром, затем сушат в вакууме.
Методика 304. Отщепление пептида от бензгидрила-
миновой смолы при помощи трифторметансульфокислоты
14931.
21 2-882
313
Пептидил-полимер (8,0 г) суспендируют в 40 мл трифтор-
уксусной кислоты и 13,2 мл тиоанизола, а^затем встряхивают
в течение 5 мин в атмосфере сухого азота, добавляют 4,8 мл
трифторметансульфокислоты, и смесь встряхивают в течение
6 ч при комнатной температуре. После этого смолу отфильтро-
вывают, промывают трифторуксусной кислотой. Фильтрат упа-
ривают при пониженном давлении, а остаток растирают с хло-
ристым метиленом (50 мл X 2) и эфиром (50 мл X 3).
Сообщалось, что трифторметансульфокислота вызывает
превращение а-производных аспарагиновой кислоты в р-про-
изводные [494]. Действительно, в отщепленном пептиде обна-
ружено около 1 % Р-производного аспарагиновой кислоты.
Методика 305. Отщепление пептида посредством ме-
та нсульфокислоты [451 ]
К суспензии пептидил-полимера в трифторуксусной кисло-
те (6,5—9 мл на 1 г смолы), содержащей 1 % анизола, при-
бавляют метансульфокислоту (20 экв. на каждую функцио-
нальную группу: Z, OBzl, Bzl, Mbs и эфирную связь пептида
со смолой) и смесь выдерживают от 2 до 2,5 ч при комнатной
температуре. При наличии в пептиде остатка тирозина вмес-
то анизола используют тиоанизол (20 экв. на остаток тирози-
на). Затем смолу отфильтровывают, растворитель удаляют в
вакууме, а пептид очищают на колонке с силикагелем в сис-
теме н-бутанол — пиридин — уксусная кислота — вода (8:31
: 1 : 4 по объему).
Методика 306. Отщепление аммиаком [161.
Безводный метанол насыщают при 0 °C сухим аммиаком.
В раствор загружают пептидил-полимер (1 г смолы на 10 мл
раствора), в колбу помещают магнитный стержень, колбу за-
крывают и ее содержимое перемешивают на магнитной мешалке
в течение 2 дней при комнатной температуре. Затем полимер
отфильтровывают, промывают метанолом, растворитель упа-
ривают в вакууме, а остаток очищают подходящим ме-
тодом.
Методика 307. Отщепление гидразином [16]
Пептидил-полимер суспендируют в очищенном диметилфор-
мамиде (1 г смолы на 5 мл ДМФА), к.суспензии добавляют без-
водный гидразин (30 экв. на 1 экв. пептида). Смесь размеши-
вают 2 дня при комнатной температуре, после чего полимер от-
фильтровывают и промывают диметилформамидом. Растворитель
упаривают в вакууме, а остаток очищают подходящим ме-
тодом.
Методика 308. Отщепление пептида путем переэтери-
фикации [16]
Пептидил-полимер суспендируют в безводном метаноле
(1 г смолы в 40 мл метилового спирта). К суспензии добавляют
314
триэтиламин (50 ммоль на 1 ммоль пептида) и смесь перемеши-
вают при комнатной температуре в течение 20 ч. Смолу затем
отфильтровывают, растворитель упаривают, а остаток очи-
щают подходящим способом.
Этиловый эфир пептида можно получить при перемешива-
нии пептидил-полимера в 10 %-м растворе триэтиламина в эти-
ловом спирте при температуре 45 °C в течение 90 ч.
Методика 309. Трансэтерификация в бензиловом
спирте в присутствии KCN [497].
В н и м а н и е! Всю работу необходимо проводить в хо-
рошо вентилируемом вытяжном шкафу/
Пептидил-полимер (200 мг) размешивают в 5 мл бензило-
вого спирта, содержащем KCN (10—50 мг), при температуре
25—50 °C в течение 24 ч в закрытом сосуде. Затем смолу от-
фильтровывают, промывают теплым диметилформамидом. Вы-
деление пептида осуществляют тем или иным подходящим
методом.
Методика 310. Отщепление пептида от смолы гидри-
рованием [498]
Пептидил-полимер (3,5 ммоль) прибавляют к раствору
2,37 г (10,5 ммоль) ацетата палладия в 55 мл диметилформами-
да при 40 °C. Для набухания смолы суспензию встряхивают в
течение 15 мин, затем при помощи водорода создают неболь-
шое избыточное давление и смесь встряхивают в течение 24 ч
при 40 °C. После окончания реакции смолу отфильтровывают,
растворитель удаляют в вакууме, а остаток очищают на колон-
ке с Сефадексом LH-20, используя систему хлороформ — мета-
нол— уксусная кислота — вода (7 з 3 : 2 I 4).
Методика 311..Отщепление амида пептида от 3-нит-
ро-4-амино-метил-бензоил-амидной смолы фотолизом [499]
Пептидил-полимер суспендируют в метаноле в атмосфере
азота и колбу освещают светом при 350 нм. Для того, чтобы
«отсечь» излучение света с длиной волны ниже 320 нм, исполь-
зуют 40 %-й раствор сульфата меди. После окончания реак-
ции смолу отфильтровывают, метанол упаривают, а пептид
очищают подходящим способом.
Методика 312. Отщепление пептида от смолы с по-
мощью диметиламиноэтанола [4961.
Пептидил-полимер (1,3 ммоль) встряхивают в течение 24 ч
при комнатной температуре в смеси диметилформамида и ди-
метиламиноэтанола (1 : 1). Затем раствор удаляют фильтро-
ванием, смолу промывают диметилформамидом (30 мл), поли-
мер снова суспендируют в 40 мл смеси ДМФА —ДМАЭ(1 : 1}
и процедуру повторяют как описано выше. Фильтрат и про-
мывную жидкость объединяют, растворитель удаляют в ва-
кууме при температуре 20 °C, остаток — маслообразный про-
21
315
дукт желтого цвета — растирают с эфиром и получают по-
рошкообразное соединение. Выход составляет 85 %. Обра-
зовавшийся диметиламиноэтиловый эфир пептида (0,5 ммоль)
выдерживают при комнатной температуре в смеси 5 мл ДМФА
и 5 мл воды в присутствии каталитических количеств имида-
зола (0,05 ммоль). За реакцией гидролиза следят с помощью
ТСХ. Обычно через 20 ч реакция завершается, растворитель
удаляют в вакууме при 20 °C. К остатку добавляют 10 мл во-
ды и снова упаривают. Продукт кристаллизуют из спирта.
Выход 79 %.
Глава 6
КОНЪЮГАЦИЯ ПЕПТИДОВ
С ВЫСОКОМОЛЕКУЛЯРНЫМИ НОСИТЕЛЯМИ
Конъюгаты пептидов с высокомолекулярными носителями
(белками или синтетическими полимерами) широко исполь-
зуются в современных биохимических исследованиях. Они
могут быть моделями биополимеров: вирусных и бактериаль-
ных белковых антигенов, белковых .гормонов, ферментов и
др. С другой стороны, конъюгаты пептидов с нерастворимыми
полимерами (синтетическими полимерами, полисахаридами,
полимерами на основе соединений кремния) нашли примене-
ние в качестве аффинных сорбентов для выделения чистых
биологических активных молекул: антигенов и антител, ре-
цепторных и мембранных белков, ферментов.
Большинство реагентов, используемых для конъюгации
пептидов, можно применять для указанных выше целей конъю-
гации. Наиболее широкое применение белково-пептидные конъ-
югаты нашли в иммунологии [5001 для изучения антигенной
структуры антител и создания синтетических вакцин против
вирусных и микробных инфекций.
Для создания конъюгатов синтетических пептидов с бел-
ками-носителями используют реакционноспособные боковые
радикалы аминокислот, входящих в состав белков: 8-амино-
группу лизина, карбоксильную группу глутаминовой и аспа-
рагиновой кислот, сульфгидрильную группу цистеина, фе-
нольную группу тирозина и имидазольное ядро гистидина.
В синтетических пептидах обычно используют только кон-
цевую амино- или карбоксильные группы, так как использо-
вание боковых радикалов входящих в их состав аминокислот
может повлиять на иммуногенность канъюгируемого пептида.
РЕАГЕНТЫ, ИСПОЛЬЗУЕМЫЕ ДЛЯ КОНЪЮГАЦИИ
На практике чаще всего используют наиболее доступные
реагенты — водорастворимые карбодиимиды щ бифункцио-
нальные сшивающие реагенты, хотя они обладают рядом не-
достатков.
317
Водорастворимые карбодиимиды. Карбодиимиды могут ре-
агировать с карбоксильными группами белков и пептидов, об-
разуя промежуточные О-ацилизомочевины (134), которые ре-
агируют с аминогруппами (а также с сульфгидрильными и
оксигруппами) или с другой карбоксильной группой, давая
ангидрид (135), также обладающий высокой реакционной спо-
собностью:
R—N=C=N—Ri + Prot—СООН
О
|| Pep-NH,
Prot—С—О—С ---------------- Prot—CONH—Pep
(134) j XNH—Rj
o; О
II || Pep—NH,
Prot—C—O—C—Prot --------* Prot—CONH—Pep,
(135)
где Pep"*— пептид, Prot — белок.
Чаще всего используют 1-этил-3 (3-диметиламинопропил)
карбодиимид гидрохлорид и 1-циклогексил-3-(2-морфолино-
4-этил)карбодиимид (см. главу 1).
К недостаткам карбодиимидов как сшивающих реагентов
следует отнести, во-первых, неоднозначность пришивки пеп-
тидов с С- или N-конца к белку (этого можно избежать, ис-
пользуя С- или N-защищенные пептиды), во-вторых, образо-
вание межмолекулярных сшивок белковых молекул, что не
всегда желательно.
Бифункциональные сшивающие реагенты. Бифункцио-
нальные реагенты разделяют на две основные группы: гомо-
бифункциональные реагенты, содержащие две одинаковые
реакционные группы и реагирующие с одинаковыми группа-
ми в белках и пептидах, и гетерофункциональные реагенты,
которые имеют разные реакционные группы, ито позволяет
проводить их реакцию с белком и пептидом последовательно.
Кроме того, обе указанные группы реагентов подразделяют
на расщепляемые, которые позволяют легкоотделить конъю-
гированный пептид от белка-носителя, и нерасщепляемые.
В большинстве случаев нет необходимости в использовании
расщепляемых бифункциональных реагентов. В основном для
конъюгации пептидов используют те же бифункциональные
реагенты, что и для сшивки белковых молекул как внутри-
молекулярных, так и межмолекулярных [501].
318
Гомобифунациональные сшивающие реагенты
Глутаровый альдегид. Глутаровый альдегид реагирует
главным образом с аминогруппами в белках и пептидах, обра-
зуя основания Шиффа [502]:
Prot—NHa СНО Prot—NH=C
+ (СН2)8 -> (СН2)3
Pep—NH2 СНО Pep—NH=C
Частично он реагирует также с боковыми группами цисте-
ина, тирозина и гистидина. К недостаткам реагента можно от-
нести склонность к полимеризации в процессе хранения, к
достоинствам — хорошую растворимость в воде.
Диимидоэфиры. Хлоргидраты диимидоэфиров дикарбоно-
вых кислот хорошо растворимы в воде и реагируют в мягких
условиях (pH 7—10) преимущественно с аминогруппами [502]:
ci-.h2n+ ^n+h2.ci-
^С-(СН2)П-С
H3COZ хосн3
где п = 2—8.
Реакция с боковыми цепями цистеина, тирозина, гистиди-
на и аргинина очень незначительна. Образующиеся в резуль-
тате реакции амидины несут положительный заряд, то есть
сохраняют общий заряд белка после конъюгации, а также не
гидролизуются в процессе кислого гидролиза для аминокис-
лотного анализа. К недостаткам имидоэфиров следует отнести
высокую скорость гидролиза, сравнимую со скоростью амино-
лиза.
В качестве расщепляемого аналога можно привести ди-
хлоргидрат диметил-3,3'-дитио-бис-пропионимидата [503]:
Prot—NH2 C1“*H2n+ /N+H2.C1-
+ ^C-(CH2)2-S-S-(CH2)a-C\ ->
Pep—NH2 H3COZ XOCH3
(ДТВР)
Prot— NH—C—(CH2)2—S—S—(CH2)2—C—NH—Pep
II II
+nh2 • Cl~ | R_SH +nh2 . Cl~
n+h2.2C1-* n+h2 • Cl~
II II
Prot—NH—C—CH2CH2—SH + Pep—NH—C—CH2CH2—SH
Активированные эфиры дикарбоновых кислот. Для повы-
шения устойчивости к гидролизу были предложены дисукци-
нимидные эфиры дикарбоновых кислот [504], например дисук-
319
цинимидный эфир пробковой кислоты (DSS):
о о
II II
•— Prof - NH - С- (СН2)6 - С - NH - Pep + 2HOS11
В качестве расщепляемого реагента используют дисукци-
нимидный эфир дитио-б/лс-пропионовой кислоты (DSP) [505]:
О
- О-С-CH2CH2S - s - СН2СН2-
Дисукцинимидные эфиры дикарбоновых кислот устойчивы к
щелочному гидролизу, но нерастворимы в воде и вносятся в вод-
ный раствор при конъюгации в органических растворителях.
Водорастворимый аналог описанного выше реагента —
быс-(Ь[-оксисульфосукцинимидный) эфир пробковой кислоты —
описал Старое [506]:
Им также получен [506] расщепляемый 3,3'-дитио-бмс(М-окси-
сульфосукцинимидил пропионат):
О о
II II
-O-C-CH2CH2-S-S-CH2CH2-C-O-N
Водорастворимые бис-(2 питрю-4-сульфофениловые) эфирь
адипиновой и азелаиновой кислот описаны в работе [5071:
О О
NaO3S—О—С— (СН2)Л—С—О—SO3Na,
XNO2 OjN/
где п равно 4 и 7.
320
Диизоцианаты. Различные алифатические и ароматиче-
ские диизоцианаты реагируют преимущественно с аминогруп-
пами и используются для конъюгации пептидов. Наиболее ши-
роко используется толуил-2,4-диизоцианат [508]:
ZN=C=O
Prot—NH2 z—<
+ O=C=N-<^ ^-CH3
Pep—NHa
О
I’
Prot—NH—C—NH—
HN—C—NH—Pep
Изоцианаты являются высокореакционными устойчивы-
ми соединениями, но вносятся в органических растворителях,
так как нерастворимы в воде.
Гетеробифункциональные сшивающие реагенты
Идеальным реагентом для конъюгации пептидов можно
было бы считать такую систему из двух реакционных центров
А и В, которые реагировали бы только друг с другом, но нес
реакционноспособными группами в пептидах и белках:
Pep + А Рер — А
4—>- Рер—А—В—Prot.
Prot 4~ В -> Prot — В
Этим требованиям в общем удовлетворяет пара: сульфгид-
рильная группа — малеимидная группа, которая реагирует
по схеме:
В качестве гетерофункциональных реагентов, реагирующих
по этой схеме, использовались: сукцинимидил-4-(п-малеими-
дофенил)бутират (136) [509], малеимидобензоил-М-оксисукци-
нимидный эфир (137) [509], сукцинимидил 4-(Ы-малеимидоме-
тил) циклогексан-1-карбоксилат (138) [510], а также 2-нитро-
4-сульфофениловый эфир N-малеимидо-б-аминокапроновой
321
Эти реагенты ацилируют белки по аминогруппам в мягких
условиях, а затем реагируют с пептидами, в которые вводят
остаток цистеина (с N- или С-конца). Следует отметить, что ма-
леимидная группа может частично реагировать по аминогруп-
пам, поэтому ацилирование проводят при pH 8 (при этих зна-
чениях pH реакционная способность малеимидной группы по-
нижена), а затем снижают pH до 6,2 и вводят цистеинсодер-
жащий пептид. В этих условиях аминогруппы протонируются
и возрастает скорость реакции с тиольными группами.
Кроме малеимидной группы для^реакции с тиольными груп-
пами в пептидах используются и другие реагенты. Бернатович
с соавт. [512] предложили N-оксисукцинимидный эфир бром-
уксусной кислоты;
и 11
N-O-C-CH2Br—► Prot — NH —С — СН213г + Pep-SH—*
О
II
---- Pep-S-CH,~C-NH-Prot
М-Сукцинимидил-3-(2-пиридилдитио)пропионат(5РБР) [513]
вводится в белки, а затем в мягких условиях реагирует с
322
тиольными группами цистеинсодержащих пептидов:
а О
О
iN s- S- СН2СН2- С-О- N
о
II Pep-SH
S — S — CH2CH2—С —NH — Prot —
О
II
---► Pep—S — S — CH2CH2—С —NH — Prot +
Н
Иногда реакцию конъюгации проводят в обратном поряд-
ке — сначала вводят бифункциональные реагенты в пептиды,
а затем проводят их реакцию с тиольными группами белка,
нативными или специально введенными. Для введения тиоль-
ных групп в белки применяли следующие эффективные реаген-
ты: 2-иминотиолан хлоргидрат (140) [514], И-оксисукинимид-
ный эфир S-ацетилтиогликолевой кислоты (141), [515] и ан-
гидрид S-ацетилмеркаптоянтарной кислоты (142) [516]:
н2с—сн2 N+H2C1-
Нас/ \c=n+h„ • С1~ + H2N—Prot -+ HS—(CH2)3—С—NH—Prot;
х S z
(140)
О О
II II
Ac—S—СН2—C-OSu + H2N—Prot -> Ac—S—CH2—C—NH—Prot;
SATA
(141)
О
II . О
Af__c_/\ II
| 04- H2N—Prot -> Ac—S—CH—C—NH—Prot.
'll СН,—COOH
О
(142)
При модификации белков реагентом (142) кроме тиольной
группы дополнительно вводится карбоксильная группа, что
улучшает растворимость модифицированных белков-носите-
лей в водно-щелочной среде. S-Ацетильная группа легко уда-
ляется гидроксиламином.
Для взаимодействия синтетических пептидов с нативными
или введенными SH-группами белков удобно использовать N-
323
хлорацетил-пептиды [517]
О О
II II
Prot—SH + Cl—СН2—С—NH—Pep -> Prot— S—СН2—С—NH— Рер.
Другой подход заключается во введении в синтетические
пептиды 5-(3-нитро-2-пиридинсульфенил)цистеина посредст-
вом Вос(Ыруз)-цистеина [516, 518, 519]. S-Защитная Npys-
группа устойчива в условиях кислой обработки в процессе
твердофазного синтеза и реагирует с тиольными группами бел-
ка в мягких условиях:
H-S
I
Prot
Pep —S
Prot — S
Синтетические пептиды, содержащие S-Npys-цистеин, удобно
хранить без опасности окисления, что обычно имеет место
при работе с пептидами, содержащими незащищенный цис-
теин.
В работе [518] использовали N-оксисукцинимидный эфир
Вос(Мруз)цистеина как для введения его в пептиды, так и для
введения SH-групп в белки; при этом модифицированные бел-
ки могли реагировать с пептидами, содержащими свободные
SH-группы (схема А) или после обработки дитиотреитолом —
с Npys-цистеинилсодержащими пептидами (схема В):
(A) Pep-SH
О
II
C-NH-Prot
Р<р — (S—Npys) CvsОН
Pep — S S
Вос —NH
324
Другие методы конъюгации пептидов с белками
Среди эффективных методов конъюгации пептидов следует
отметить превращение белков (или других полимеров) в ак-
тивированные по карбоксильным группам соединения.
С. М. Андреев и соавт. [520] разработали метод активации сук-
цинированного бычьего сывороточного альбумина (BSA) и же-
латина, а также полиакриловой кислоты и сополимера винил-
пирролидона с малеиновым ангидридом.
При сукцинировании BSA или желатина в молекулу вмес-
то е-аминогрупп лизина посредством обработки ее янтарным
ангидридом вводили дополнительные карбоксильные группы,
которые превращали в N-оксисукцинимидные эфиры при по-
мощи трифторацетоксисукцинимида. Конъюгацию активиро-
ванных белков (или других полимеров) проводили в органи-
ческом растворителе
Prof —NH2----Prot-NH — С — СН2СН2 —СООН--------------
----4>rot- NH — С — СН2СН2 — С — NH — Pep .
Pep-NH2
ДМФА
В работе [521] вместо N-оксисукцинимидных эфиров полу-
чали активированные о-нитрофениловые эфиры белков и синте-
тических полимеров.
Следует отметить, что обычно белки конъюгируют с пепти-
дами в водных или водно-органических растворах, однако бел-
ки, активируемые по описанному выше способу, становятся
растворимыми в ДМФА, что позволяет вести реакцию конъю-
гации в неводной среде.
Конъюгацию активированных по карбоксильной группе
полностью защищенных пептидов обычно не проводят из-за
плохой растворимости этих соединений в воде и необходимос-
ти последующего деблокирования конъюгированных пепти-
дов. Поэтому определенный интерес представляет способ конъ-
югации пептидов, предложенный в работе [522]. Синтез пеп-
тидов осуществляется твердофазным методом и после удале-
325
ния концевой Вос-группы пептид сукцинируют. Образовав-
шуюся карбоксильную группу превращают в активированные
эфиры (оксисукцинимидные, пентахлорфениловые или 2-нит-
ро-4-сульфофениловые), которые не расщепляются при отщеп-
лении пептида от смолы посредством мягкой обработки фто-
ристым водородом. Полученные таким способом активирован-
ные пептиды реагируют с белками по аминогруппам в водно-
щелочной среде:
янтарный
Вос—HN—Pep—Р — —> H2N—Pep—Р
ангидрид
О
НООС—(СН2)2—NH—Pep—Р Н.°—SuO—С— (СН2)2— NH—
ДЦГК
-Pep—Р SuO—С—(СН3)2—Рер—СООН,
где Р — полимер.
Естественно, что метод непригоден, если в состав пептида
входит остаток лизина.
ГЕТЕРОБИФУНКЦИОНАЛЬНЫЕ ФОТОРЕАКТИВНЫЕ РЕАГЕНТЫ
Эти реагенты используются в биохимических исследова-
ниях для введения так называемых фотоаффинных меток для
изучения рецепторов пептидных гормонов, активных центров
ферментов и антител, участков связывания белков с нуклеи-
новыми кислотами и др. Для этих целей используются глав-
ным образом арилазиды, которые под действием облучения с
длиной волны 300—400 нм превращаются в очень реакционно-
способные нитрены:
R—N3 R—N +H—С-----> R—NH—С—
-N, •• I *|
Ввиду того, что время жизни нитренов составляет всего
10“~4 с, они успевают прореагировать с близкорасположенны-
ми С—Н, С=С- или С—N-связями, образуя новые ковалент-
ные связи. Поэтому фотореактивные реагенты удобно исполь-
зовать для специфического связывания пептидных лигандов с
участками белков, к которым они имеют большое сродство [523].
Среди широко используемых фотореактивных реагентов
следует отметить N-оксисукцинимидил 4-азидобензоат (143)
[524], хлоргидрат метил-4-азидобензимидат (144) [525], я-ази.
дофенацил бромид (145) [526], Ы-сукцинимидил-6-(4'-азидо.
326
2'-нитрофениламино)гексаноат (146) [5271:
Обычно фотореактивные реагенты вначале вводят в моле-
кулу белка, а затем облучают в присутствии пептида (или на-
оборот) :
Pro;—Ч-c—OSu ->• Prot—NH—С—Ч-N3 ->
II II
— Prot-NH-C-/—NH—Pep.
~rPeP II 4=/
о
Следует отметить, что описано огромное количество фото-
аффинных меток для различных биомолекул и благодаря их
использованию получено много ценной информации. Напри-
мер, введение в синтетический пептид 4-азидофенилаланина
позволяет непосредственно метить им специфические связы-
вающие участки в белках [528]:
Pep—Phe—
hv
"Prot
Pep—Phe—
Prot—NH
Для аффинной метки белков тирозинсодержащими пептида-
ми был предложен удобный бифункциональный реагент — 4-
азидоанилин (160) [529], который после диазоторирования реа-
гирует с остатками тирозина и гистидина в пептиде. После
облучения в присутствии белка и реакции азидной группы с
ним пептид может быть отделен, а метка, в которую вводится
радиоактивный иод или тритий, остается в участке связывания
327
эелка с пептидным лигандом:
-> Рер—Туг ~ + Prot—NH— NH,
J\ Ч1*
V4nh2
он
Ниже приводятся методики синтеза реагентов для конъю-
гации пептидов и примеры получения пептидно-белковых конъ-
югатов.
Методика 313. Конъюгация при помощи водораство-
римого карбо диимида [530].
Синтетический пептид и кроличий сывороточный альбу-
мин в молярном соотношении 1 ! 2 растворяли в 2 мл воды и
добавляли к раствору 200 мг 1-этил-3-(3-диметил-аминопро-
пил)карбодиимида, растворенного в 0,5 мл дистиллированной
воды. Реакцию вели при pH 7 при комнатной температуре 24 ч.
Избыток карбодиимида удаляли диализом реакционной сме-
си против дистиллированной воды в течение 24 ч при охлаж-
дении. По данным аминокислотного анализа, соотношение бе-
лок ; пептид в конъюгате составляло 7:1.
Для получения конъюгатов, содержащих большое коли-
чество пептида, в реакцию вводят большой молярный избыток
пептида по отношению к белку.
Методика 314. Конъюгация при помощи глутарово-
го диальдегида [531].
328
Добавляли пептид в 30-кратном молярном избытке к
0,15 мкмоль бычьего сывороточного альбумина (BSA) в 1 мл
0,1 М натрий-фосфатного буфера (pH 7,5). К полученному рас-
твэру добавляли по каплям в течение 0,5 ч при комнатной тем-
пературе при перемешивании 300 мкл 21 мМ раствора глута-
рового альдегида. Через 0,5 ч перемешивания при комнатной
температуре конъюгат очищали на колонке с Сефадексом G-15.
Методика 315. Синтез 1-окси-3-сульфо-2,5-пирроли-
диндиона(Ы-оксисульфосукцинимида) [506]
К раствору 1,54 г (12,8 ммоль) N-оксималеимида в абсо-
лютном спирте в токе азота добавляли водный раствор Na2S2O6.
Реакционную смесь перемешивали 2 ч при комнатной тем-
пературе в токе азота, а затем реакционную смесь упаривали
на роторном испарителе в вакууме водоструйного насоса при
40 °C. Остаток — желтое масло — растворяли в воде и лиофи-
лизировали. Светло-желтый твердый остаток растирали с эфи-
ром, и полученный не совсем белый порошок отфильтровывали
и сушили. Выход 2,66 г (96 %), т. пл. выше 300 РС, Rf — 0,32
(н. бутанол — уксусная кислота — вода, 5 ! 2 • 3).
Методика 316. Динатриевая соль бис-(М-оксисуль-
фосукцинимидного)эфира пробковой кислоты [506]
Растворяли 0,44 г (2 ммоль) натриевой соли N-оксисульфо-
сукцинимида, 0,174 г (1 ммоль) пробковой кислоты и 0,41 р
(2 ммоль) ДЦГК в 5 мл диметилформамида и перемешивали при
комнатной температуре в течение ночи, затем 2 ч при 0 9С.
Отфильтровывали дициклогексилмочевину и продукт осаж-
дали 20-кратным количеством этилацетата. Твердый осадок
отфильтровывали и сушили.
22 2 — 882
329
Методика 317. Динатриевая соль бш>(2-нитро-4-суль»
фофенилового) эфира адипиновой кислоты [507]
О N°a
СООН С-О-^“^-SO3Na
(СН,)4 + 2HO-^~^-SO3Na —+ ((Ь,)4
СООН С—О—SO3Na
О
К раствору 1,46 г (10 ммоль) адипиновой кислоты и 6 р
(20 ммоль) тригидрата натриевой соли 2-нитро-4-сульфофено-
ла в 50 мл ДМФА при 0 °C и перемешивании прибавляли 4,2 г
(20 ммоль) ДЦГК, выдерживали 2 ч при этой температуре и
оставляли на ночь при 20 °C. Отфильтровывали дициклогек-
силмочевину, удаляли в вакууме ДМФА и остаток растирали о
эфиром. После перекристаллизации из спирта получали 5 р
(85 %) желтого порошка с т. пл. 305—307 °C (разл.).
Аналогично получали динатриевую соль бис-(2-нитро-4-
сульфофенилового) эфира азелаиновой кислоты с выходом
60 %, т. пл. 300—302 °C (разл.)
Методика 318. Общий метод конъюгации пептидов
при помощи водорастворимых эфиров дикарбоновых кислот.
Растворяли 1,5 мг BSA в 0,5 мл 0,1 М растворе бикарбона-
та натрия (pH 8,5), добавляли к нему 0,5 мг пептида и 0,5 мр
сшивающих реагентов, описанных в приведенных выше мето-
диках. Выдерживали реакционную смесь ночь при комнат-
ной температуре, разлагали непрореагировавшие активные
группы водным аммиаком и отделяли конъюгат на колонке
с сефадексом G-25 (или диализовали). Оптимальные условия
конъюгации (время и молярное отношение пептид : белок) в
каждом отдельном случае нужно подбирать экспериментально.
Методика 319. Синтез малеимидо-6-аминокапроновой
кислоты [511]
О
I /О
СН—С—NH—(СНа)4-^СООН СН—се
Jh-cooh ’ <41>-(сн.)._соон
• Кипятили раствор 20 г (0,09 моль) N-малеиламинокапро-
новой кислоты в 500 мл уксусной кислоты в течение 24 ч. Уда-
ляли уксусную кислоту в вакууме и оставшееся масло рас-
творяли в 100 мл хлороформа. Незациклизовавшаяся N-мале-
иламинокапроновая кислота выпадала в осадок при стоянии
в течение ночи при комнатной температуре. После фильтрации
330
хлороформенный раствор упаривали до малого объема и очи-
щали на колонке с силикагелем (объем неподвижной фазы
600 мл) в смеси хлороформ: уксусная кислота (95:5). Рас-
твор, содержащий чистые фракции, упаривали и получали 9 г
продукта, который перекристаллизовывали из 30 мл горячего
хлороформа.
Методика 320. Натриевая соль 2-нитро-4-сульфофе-
нилового эфира малеимидо-6-аминокапроновой кислоты [511]
СН—с<^
8 /
сн-Ц
о
N—(СН2)5—СООН + HO-^“)>-SO3Na
0 NO2
ДЦГК (
,0 о
-> СН-С^ II Z—.
|| >N-(CH2)5-C-O-X у-SO3Na
СН—С<С
0 NO3
К раствору 2,42 г (100 ммоль) 2-нитро-4-сульфофенола (нат-
риевой соли) в 10 мл ДМФА прибавляли 2,1 г (100 ммоль) ма-
леимидо-6-аминокапроновой кислоты и раствор 2,06 г
(100 ммоль) ДЦГК в 4 мл ДМФА. Раствор выдерживали 6 ч
при комнатной температуре, отделяли дициклогексилмочеви-
ну и разбавляли 200 мл сухого эфира. Выпавшее желтое масло
растирали с сухим эфиром до получения твердого осадка. Вы-
ход технического продукта (85—90 % чистоты) составил 3,7 г.
Он пригоден для кэтьогации, хорошо растворим в воде.
Мегодика 321. N-Оксисукцинимидный эфир N-ма-
леимидо-6-аминокапроновой кислоты [532].
Реагент получали обычным методом, используя эквимо-
лярные количества N-малеимидокапроновой кислоты, N-окси-
сукцинимида и ДЦГК в диоксане. Выход сукцинимидного
эфира 87 % (перекристаллизация из смеси хлороформ — изо-
пропиловый спирт), т. пл. 62—65 °C (разл.); /?х — 0,73 (ме-
танол— хлороформ (1 : 1)), Rf2 = 0,43 (хлороформ — уксусная
кислота (95 : 5)). Реагент нерастворим в воде. Его прибавляют
к водному раствору белка в ДМФА, спирте и др.
Методика 322. Конъюгация пептидов при помощи
2-нитро-сульфофенилового эфира N-малеимидо-б-капроновой
кислоты [511].
Белок (BSA) растворяли в 0,1 М фосфатном буфере (pH 7,4)
до концентрации 11 мг/мл. К 2 мл белкового раствора, содер-
жащего 22 мг (0,33 мкмоль) BSA (или 19 мк/экв остатков ли-
зина), добавляли 11 мг (25 мкмоль) реагента, то есть 1,2 моляр-
ный избыток по отношению к аминогруппам лизина. Смесь вы-
22=
331
держивали 30 мин, а избыток непрореагировавшего реагента
отделяли на колонке с Сефадексом G-25 (2,5 X 18 см), урав-
новешенной 0,1 М фосфатным буфером с pH 6,0. В раствор,
собранный с колонки, добавляли 22 Мг (13 мкмоль) цистеин-
содержащего пептида, выдерживали несколько часов при ком-
натной температуре и диализовали против дистиллированной
воды. В этих условиях около 50 % малеимидных групп проре-
агировали с тиольными группами пептида.
Аналогично используют соответствующий N-оксисукцини-
мидный эфир N-малеимидо-б-аминокапроновой кислоты (а
также аналогичные реагенты; SMPB, MBS и SMEC, описан-
ные в вводной части данной главы). Так как они нерастворимы
в воде, их прибавляют к раствору белка в ДМФА. Обычно аци-
лирование белка ведут при pH 7,4 в течение 30 мин, затем сни-
жают его до 6,0, диализуют и вносят в раствор цистеинсодер-
жащий пептид.
СИНТЕЗ N-BOC-
[5-(3-НИТРО-2-ПИРИДИЛСУЛЬФЕНИЛ)] ЦИСТЕИНА
Методика 323. 5мс-(3-нитро-2-пиридил)дисульфид
[533]
67 г (0,42 моль) 2-хлор-З-нитропиридина и 38,05 г (0,5 моль)
тиомочевины кипятили в колбе о обратным холодильником в
760 мл спирта в течение 7 ч. К смеси полученной тиурониевой
соли с 32 г карбоната натрия в 950 мл воды добавляли при
0 °C раствор 48,5 г едкого натра в 950 мл воды и выдержи-
вали 1 ч при комнатной температуре. Подкисляли соляной
332
кислотой до pH 3, отфильтровывали осадок З-нитро-2-тиопи-
ридина, промывали его водой и растворяли в растворе 48,5 г
едкого натра в 950 мл воды. Для окисления его в дисульфид
добавляли раствор K3Fe (CN)e в 1 л воды при 20 РС и отфильт-
ровывали выпавший осадок. Получили 48 г (74 %) бис-(3-
нитро-2-пиридил)дисульфида с т. пл. 249—250 °C (разл.).
Методика 324. З-Нитро-2-пиридил сульфенилхлорид-
(533, 534]
310,30
190,59
В суспензию 50 г (16 ммоль) бнс-(3-нитро-2-пиридил)дисуль-
фида в 1 л метиленхлорида при перемешивании пропускаем
хлор, вначале 0,5 ч при 0 °C, а затем 1 ч при 20 °C. Раствор
фильтруем и растворитель отгоняем в вакууме. Выход 30,8 г
(100 %), т. пл.. 217—222 °C (разл.).
Методика 325. Еос-(8-Нруз)-£-цистеин [535].
К 1,2 г (10 ммоль) L-цистеина в 100 мл 90%-й муравьиной
кислоты прибавляли 2,1 г (11 ммоль) З-нитро-2-пиридил суль-
фенилхлорида в течение 1 ч при перемешивании при 20 °C.
Отфильтровывали нерастворившийся материал и осаждали чис-
тый хлоргидрат S-Npys-L-цистеина добавлением 200 мл эфи-
ра. После переосаждения продукта эфиром из метанола его
сушили в вакууме над Р2О5. Полугидрат гидрохлорида S-
Npys-Л-цистеина получен в количестве 2,23 г (81,2 %), т. пл.
188—189 °C, [а]д = +140,5° (с = 1; метанол).
Вос-производное можно получить любым из способов, опи-
санных в главе 2 (в работе [535] для этой цели использовали
не очень распространенный трет-бутил-5-(4,6-диметилпири-
мидин-2-ил)тиокарбонат). Продукт кристаллизовали из смеси
этилацетат — петролейный эфир, т. пл. 153—155 °C. S-Npys-
группа стабильна в трифторуксусной кислоте при 25 °C и К
HF при 0 °C, то есть удобна для синтеза пептидов твердофаз-
ным методом.
Методика 326. N-Оксисукцинимидный эфир Boc-[S-
(3-нитро-2-пиридилсульфенил)]-Л-цистеина [518].
К 100 мг (0,27 ммоль) Вос-(8-1Мру5)-£-цистеина и 34 мг
(0,29 ммоль) N-оксисукцинимида в смеси 0,5 мл ДМФА и
0,5 мл метиленхлорида, охлажденного до 0— 4 °C, добавили
55 мг (0,27 ммоль) ДЦГК при перемешивании. Через 3 ч
(20 °C) большую часть растворителя удаляли в вакууме и
к остатку добавили 1 мл эфира. Смесь охлаждали 0,5 ч
333
сухим льдом и отфильтровывали дициклогексилмочевину.
Фильтрат упаривали на роторном испарителе, а оставшее-
ся в колбе желтое масло сушили в вакууме до постоянного
веса. Продукт получили с количественным выходом (130 мг)
в виде густого сиропа, который постепенно затвердевал. Может
быть использован для реакций модификации без дальнейшей
очистки. Rf = 0,53 (10 % метанол — хлороформ).
Методика 327. Общий метод конъюгации (Npys)-mic-
теинил-пептидов с белками.
1. К раствору 7,5 мг гемоцианина из моллюска (предвари-
тельно восстановленного дитиотреитолом и диализованного)
добавляли 5—10 мг лиофилизированного пептида при pH 5,0
и оставляли на ночь. Затем подщелачивали раствор 1 н. NaOH
до pH 7,0 и выдерживали еще 3 ч. Проводили диализ конъюга-
та против 10 миллимолярного карбоната аммония [519].
2. Готовили три раствора: 1) раствор 4,5 мг BSA (модифи-
цированного ангидридом S-ацетилмеркаптоянтарной кисло-
ты) в 1 мл 0,1 М натрий-фосфатного буфера; 2) раствор
1,2 мкмоль (Ыруз)-цистеинил— пептида в 1 мл этого буфера;
3) раствор 100 мг солянокислого гидроксиламина в 4 мл буфе-
ра (для деацетилирования тиольных групп белка). К переме-
шиваемой смеси растворов 1 и 2 добавляли 0,2 мл раствора 3
в токе азота. Появляется желтая окраска З-нитро-2-тиопири-
дона. Через 3 ч раствор очищают от низкомолекулярных ком-
понентов на сефадексе G-50 [517].
Аналогично конъюгируют цистеинсодержащие пептиды с
белками, модифицированными SPDP [536].
Методика 328. Конъюгация пептидов с белками с ис-
пользованием Ы-сукцинимидил-3-(2-пиридилтио)пропионата
(SPDP) [536]
100-молярный избыток пептида по отношению к гемоциа-
нину из моллюска или BSA, модифицированным SPDP, в аце-
татном буфере (pH 4,6) перемешивали 3 ч при комнатной тем-
пературе, затем диализовали.
Методика 329.. N-Оксисукцинимидный эфир S-ацетил-
тиогликолевой кислоты [5153.
S-Ацетилтиогликолевую кислоту получали кипячением тио-
гликолевой кислоты с уксусным ангидридом; т. кип. 158—
159 °C (17 мм. рт. ст.), 115—118 °C (2,5 мм. рт. ст.).
2,68 г (0,02 моль) S-ацетилтиогликолевой кислоты раство-
ряли в сухом метиленхлориде (5 мл), добавляли^ к раствору
N-оксисукцинимида (2,3 г; 0,02 моль) в 30 мл метиленхлорида
и перемешивали 10 мин при 20 °C. К реакционной смеси до-
бавляли 4,15 г (0,02 моль) ДЦГК в 2 мл метиленхлорида и пе-
ремешивали 18 ч при комнатной температуре. Отфильтровыва-
л и дициклогексилмочевину и фильтрат упаривали при пони-
334
женном давлении, затем перемешивали остаток с 50 мл метилен-
хлорида и удаляли фильтрацией остаток дициклогексилмочеви’
ны. Фильтрат упаривали и остаток в виде желтого масла закрш "
таллизовывали растиранием с эфиром. После перекристалли-
зации из изопропанола получили 3,16 г (68 %) N-оксисукци-
нимидного эфира S-ацетилтиогликолевой кислоты ст. пл.
85—86°.
Для ацетилирования белка реагент вносили в водный рас-
твор белка (pH 7,5) в небольшом количестве ДМФА (соотноше-
ние объемов ДМФА — водный раствор — 1 : 50).
Деацетилирование белка, модифицированного N-оксисук-
цинимидным эфиром S-тиогликолевой кислоты, производи-
ли солянокислым гидроксилами ном в динатрий-фосфатном
буфере (pH 7,5) в присутствии ЭДТА. В течение 1 ч освобож-
далось более 90 % свободных SH-rpynn.
Таким же образом можно вводить SH-группу в пептиды,
модифицируя его по концевой аминогруппе.
АКТИВАЦИЯ КАРБОКСИЛСОДЕРЖАЩИХ ПОЛИМЕРОВ
Методика 330. N-Оксисукцинимидный эфир сукци-
нилированного BSA [520].
К суспензии 0,52 г сукцинилированного BSA в 10 мл су-
хого ДМФА прибавляли 1 мл пиридина и 1,06 г О-трифтор-
ацетил-М-оксисукцинимида (см. главу 1). Реакционную массу
выдерживали 20 мин при 20 °C до полного растворения осад-
ка и при интенсивном перемешивании выливали в 250 мл изо-
пропанола. Осадок отделяли центрифугированием (4000 об/мин),
промывали изопропанолом, эфиром и высушивали в вакууме.
Выход 0,55 г. Содержание активированных карбоксильных
групп — 0,79 ммоль/г модифицированного белка.
Методика 331. Общий метод иммобилизации пепти-
дов на сукцинилированных полимерах [537].
К раствору 100 мг сукцинированного желатина в 1 мл
ДМФА прибавляли 20—50 мкмоль пептида и 50—100 мкмоль
N-метилморфолина, через 24 ч к полученному вязкому раствору
для удаления избытка активированных групп прибавляли
50-кратный избыток водного аммиака. Через 1 ч разбавляли
50 мл воды, диафильтровали на мембране РМ 10 против воды
не менее 3 раз. Содержание пептида определяли с помощью
аминокислотного анализа.
СПИСОК ЛИТЕРАТУРЫ
1. Noven С. J.t Anthony‘Cahill S, J., Griffith Mt Cti Schultz Pt 0» //
Science.— 1989.— 244, N 4901.— P. 182—188.
2. Овчинников Ю. А. Биоорганическая химия.— M. : Просвещение,
1987.— 815 с.
3. Шульц Г., Ширмер Р. Принципы структурной организации белков,—
М. : Мир, 1982.— 354 с.
4. Чипенс Г. И., Полевая Л. К.> Веретенникова Н. И,, Крикис А. Ю.
Структура и функции низкомолекулярных пептидов.—Рига : Зинатне.
1980.— 328 с.
5. Никифорович Г. Почти природные лекарства.— М. : Молодая гвардия,
1986.— 207 с.
6. Samanen J.И Bioactive Polym. Syst. / Ed. C. G. Gebelin,— New York;
London, 1985.— P. 279—344.
7. Сассон А. Биотехнология: свершения и надежды.— М. : Мир, 1978.—
411 с.
8. Kullmann W. Enzymatic peptide synthesis,— Boca Raton : CRC press,
1987.— 350 p.
9. Гринштейн Дж.» Виниц M, Химия аминокислот и пептидов.— М.
Мир, 1967.— 821 с.
10. Шредер Э.» Любке К» Пептиды.— М. : Мир, 1967,— Т. 1.— 496 с,
11. Шредер Э., Любке К. Пептиды.— М. : Мир, 1969.— Т. 2.— 723 с.
12. Пептиды. Основные методы образования пептидных связей / Под ред.
Э. Гросса, И. Майенхофера.— М. : Мир, 1983.— 421 с.
13. Химия биологически активных природных соединений / Под ред.
Н. А. Преображенского, Р. П. Евстегнеевой,— М. : Химия, 1970.—
512 с.
14. Химия полипептидов / Под ред. П, Катсояниса.— М. : Химия, 1977.—
462 с.
15. Защитные группы в органической химии / Под ред. Дж. Макоми.—
М. : Мир, 1976.— 391 с.
16. Стюарт Дж.» Янг Дж. Твердофазный синтез пептидов.— М. : Мир,
1971.— 176 с.
17. Митин Ю. В.» Запевалова Н. П.П Успехи химии,— 1977,— 46, Кг 5,—
С. 852—877.
18. Буров С. В., Смирнова М. /7.//Там же.— 1982,— 51, № 9,— С, 1567—
1578.
19. Биополимеры / Под ред. Ю. Иманиши,— М. : Мир, 1988.— 544 с,
20. Гершкович А. А.» Кибирев В. К. Синтез пептидов. Реагенты и методы,—
Киев : Наук, думка, 1987.— 263 с.
21. Finn F. М.» Hoffmann К.И The proteins / Eds Е, Neurath, R. L. Hill.—
New York : Acad, press, 1976.— Vol. 2.— P. 105—253.
336
22. Synthese von Peptiden // Methoden der Organishen Chemie (herausgegeben
von E. Muller). Bd 15/1.— Georg Theime Verlag Stutgart, 1974.—
Teil 1,— S. 1 — 1005; Teil 2.—S. 1—806.
23. Bodanszky M. Peptide Chemistry. A practical textbook.— Berlin etc.:
Springer Verlag, 1988. — 200 p.
24. Bodanszky M., Bodanszky A. The practice of peptide synthesis.— Berlin
etc. : Springer-Verlag, 1984.— 284 p.
25. The peptides. Major methods of peptide bond formation / Eds. E. Gross,
J. Meienhofer.— New York etc. : Acad, press, 1979.— Vol. 1.— 435 p.
26. The peptides. Special methods of peptides synthesis.— New York : Acad,
press, 1980.— Vol. 2.— 596 p.
27. The peptides. Protextion of functional groups in peptide synthesis.—
New York : Acad, press, 1981.— Vol. 3.— 379 p.
28. Fletcher G. A., Jones J. H.U Int. J. Peptide and Protein Res.— 1972 —
4, N 6.— P. 347—371.
29. Fletcher G. A., Jones J. H.U Ibid.— 1975.— 7, N 2.— P. 91—102.
30. Фрайдфелдер Д. Физическая биохимия.— M. : Мир, 1980.— 582 с.
31. Препаративная жидкостная хроматография / Под ред. Б. Бидлингмей-
ера.— М. : Мир, 1990,— 358 с.
32, CRC Handbook of HPLC for separation of amino acids, peptides, and
proteins / Ed. W. S. Hancock.— Boca Raton (Florida) : CRC press,
1986.— Vol. 1.— 489 p.; Vol. 2.— 522 p.
33. Дэвени T., Гергей Я. Аминокислоты, пептиды и белки.— М. : Мир,
1976.- 364 с.
34. Остерман Л. А. Хроматография белков и нуклеиновых кислот.— М. :
Наука, 1985.— 535 с.
35. Лурье А. А. Хроматографические материалы : Справочник.— М. : Хи-
мия, 1978.— 438 с.
36. Sheehan J. С., Hess G, Р,Н J. Amer. Chem. Soc.— 1955.— 77, N 4.—
P. 1067—1068.
37. Sheehan J. C.t Hlavka J. J.U}. Org. Chem.— 1956.— 21, N 4.—
P. 439—441.
38. Stevens C. L.t Munk M. E.// J, Amer. Chem. Soc.— 1958.— 80, N 15.—
• P. 4069—4071.
39. Arens J. F.H Rec. Trav. Chim.— 1955.— 74, N 6,— P. 769—770.
40. Gais H. J.// Angew. Chem. Int. Ed.— 197.— 17, N 8.— P. 597—598.
41. LosseG., Weddige H.U Liebigs Ann. Chem.— I960.— 636.— S. 144—149,
42. Woodwart R. B.fOlofsonR. A. } Mayer H.// J. Amer. Chem. Soc.— 1961.—
83, N 4.— P. 1010—1012.
43. Woodwart R. B., Olofson R. A., Mayer H.U Ibid.— 1961.— 83, N 4.—
P. 1010—1012.
44. Bergmann M., Zervas L., Fruton J. .S./l J. Biol. Chem.— 1936.— 115,
N 3.-P. 593—611. -
45. Honzl JRudinger J.// Coll. Czech, Chem. Commun.— 1961.— 26,
N 9.— P. 2333—2344.
46. Wieland T., Berhard H, // Liebigs Ann. Chem.—1951.—572. N 3.—.
S. 190— 194.
47, Belleau В , Malek G. // J. Amer. Chem. See.—1968,— 90, N 6. —
P. 1651 — 1652.
48. Gorecka A., Leplawy M.t Zabrocky J.> Zwierrak A.// Synthesis.— 1978.—
N 6.— P. 474—476.
49. Diago-Meseguer JPalomo-Coll A. L.} Fernandez-Lisarbe J.R.tZugara-
Bilbao A.11 Synthesis.— 1980, N 7.— P. 547—551.
50. Leplawy M. T.t Jones D. S., Kenner G. W., Sheppard R. C.H Tetrahed-
ron.— I960.— 11, N 1.— P. 39—51.
51. Dankewalier R. G., Veber D. F., Holly F. W., Hirschmonn R.H J. Amer.
Chem. Soc.— 1969.— 91, N 2.— P. 502—503.
337
52. SchdsslerH., Zahn H.U Chem. Ber.— 1962.— 95, N 5.— S. 1076—1080,
53. Paul R.> Anderson G. W.H J. Amer. Chem. Soc.— I960.— 82, N 17.—
P. 4596—4600.
54. Schwyzer R.f Fsurer Iselin B.. KasiHJI Helv. Chim.Acta.— 1955.—
38, N 1.—P. 80—83.
55. Wieland T.t Schafer W.> Bockelmann ВЛ Liebigs Ann. Chem.— 1951,—
573, N 2.— S. 99—104.
56. Jakubke H.-D., Voigt A.//Chem. Ber.— 1966.— 99, N 8.—S. 2419—
2429.
57. Taschner E., Rzeszotarska B.. Lubiewska Lj! Liebigs Ann, Chem.—
1965.— 690.-S. 177—181.
58. Losse G.> Barth A.. Schatz /(.//Liebigs Ann. Chem.— 1964.- 678,—
S. 185—190.
59. NefkensG. H. L..TesserG. I.// J .Amer.Chem. Soc.— 1961.— 83, N 5.—
P. 1263.
60. Anderson Q. W., Zimmerman J. E.t Callahan F. M./l Ibid.— 1963,—
85, N 19.—P. 3039.
61. Konig W.t Geiger R.//Chem. Ber.— 1970.— 103. N 3.—S. 788—798.
62. Sakakibara S.t Jnukai N./1 Bull. Chem. Soc. Jap.— 1965.— 38, N 1.—
P. 1979—1984.
63. Fujino M.> Hatanaka Ch.H Chem. Pharm. Bull.— 1968.— 16, N 5.—
P. 929—932.
64. Гудков A. T., Шехватова T. B.H Журн. общ. химии.— 1978.— 48,
№ 9.— С. 2146.
65. Devadas В., Pandey R., Mathur К JI Ind. J. Chem.— 1979.— B16,
N 11.— P. 1026—1027.
66. Jakubke H. D., KJebenCh., Berger E.f Neubert К JI Tetrahedron Lett.—
1978.— N 17.— P. 1497—1500.
67. Wang S. S.,Tam J. P., Wang B. S. HMerrifield R. B.H Int. J. Peptide
Protein Res.— 1981.— 18, N 5.—P. 459—467.
68. Garnet J .-P., Jaequier R., Verducci J.// Tetrahedron.- 1984.— 40,
N 11.—P. 1995—2001.
69. Bodanszky M.ll J. Protein Chem.— 1985.— 4, N 2.— P. 69—86.
70. Orlowska A.t Holodowiez E.t Drabarek SJI Polish J. Chem.— 1982.—
8.- P. 1067—1070.
71. Tartar A., Gesquiere J.-C.H J. Org. Chem.— 1979.— 44, N 26.—
P. 5000—5002.
72. Neuenschwander H.t Fahrni H.-Р.. Lienhard U.// Helv. chim. acta.—
1978.— 61, N 7.—S. 2437—2451.
73. Poduska K-> Gross H.U Chem. Ber.— 1961.— 94, N 2.—S. 527—537.
74. Matsuda F., Itoh S., Hattori H.t et al.ll Tetrahedron.— 1985.— 41,
N 18,— P. 3625—3634.
75. Zarr.<l M., Arnold f.H Tetrahedron Lett.— 1960.— N 14.— P. 12.
76. CarpinoL. A.,CohenlB. J ..Stephens K. E.et al.// J .Org. Chem.— 1986.—
51, N 19.—P. 3732—3734.
77. Curtius Th.II Chem. Ber.— 1902.— 35.— P. 3226—3228.
78. Pat. 1918549 Germany, IC2 C07C, A 61 K. Peptide synthesis by N—hyd-
roxysuccinimide esters./ Joshua H., Hirschmann R. E., Paleveda W. J.—
30.10.69. Chem. Abstr.— 1970.- 72, P. 32215.
79. Коломийцева Л. А., Кравцов В. Ф.> Швачкин Ю. ПЛ Журн. общ. хи-
мии.— 1976.— 46, № 5.—С. 1176—1181.
80. Boissonnas R. АЛ Helv.. Chim. Acta.— 1951.— 34, N 104.—P. 874—879.
81. Vaughan J. R^Osato R. L.ll J; Amer. Chem. Soc.— 1952.- 74, N S.-
Р. 676—678.
82. Shiori T., Yamada ShJI Chem. Pharm. Bull.— 1974.— 22, N 4.—
P. 849—854.
83. Shiori Yamada ShJI Ibid.— P. 855—858.
338
84. Shiori T.t Yamada ShJ/ Chem. Pharm. Bull.— 1974.— 22, N 4.—
P. 859—863.
85. Hamada Y.t Rishi S.t Shiori T., Yamada Sh.// Ibid.— 1977.— 25,
N 2.— P. 224—230.
8G. Tilak M. Ad/ Tetrahedron Lett.— 1970.— N 11.— P. 849—854.
87. Kiso Y., Rai Y., Yajima Н.1/ Chem. Pharm. Bull.— 1973.— 21, N 11.—
P. 2507—2510.
88. Taschner E.t Smulokowski M., Lubiewska-Nakonieczna L./l Liebigs
Ann.— 1970.— 739 — S. 228—230.
89. Унковский В. И., Васильева Г, А., Евстигнеева Р. П., Кулиш М. А.И
Журн. общ. химии.— 1983.— 53, № 1.— С. 216—221.
90. Sokolowska Т., Kupryszewski G., Taschner Ed / Roczniki Chem.— 1958.—
32.— Р. 813—819.
91. Weygand G., KHnke P.t Eigen 7.//Chem. Ber.— 1957.— 90, N 6.—
S. 1896—1905.
92. Benoiton N. L., Chen F. M. F.H FEBS Lett.— 1981.— 125, N 1.—
P. 104—106.
93. Leuchs //.//Chem. Ber.— 1906.— 39.—S. 857—861.
94. Katchalski E.U Adv. Protein Chem.— 1951.— 6.— P. 123—185.
95. Hirshmann R., Strachan R. G., Schwam H. et aidI J. Org. Chem.—
1967.— 32, N 11 —P. 3415—3425.
96. Kircher K-> Berndt H.t ZahnH.II Liebigs Ann. Chem.— 1980.— N 2.—
S. 275—284.
97. Katakai Rd/ J. Org. Chem.— 1975.— 40, N 19.—P. 2697—2702.
98. Wieland Th.t VogeZer K.//Angew. Chem.— 1961.— 73, N 12.—S. 435.
99. Bodanszky M./l Nature.— 1955.— 175, N 4459.— P. 685—686.
100. Bodanszky M.t Ondetty M. A., Levine S. D., Williams N. I.// J. Amer.
Chem. Soc.— 1967.— 89, N 5.— P. 6753—6757.
101. Beyerman H. C., Van den Brink W. M., Weygand et al.11 Reel. Trav.
Chim.—1965.—84, N 2.—P. 213—231.
102. Kbnig W., Geiger R. Chemistry and biology of peptides / Ed. J. Mein-
hofer.— Michigan, 1972.— 450 p.
103. Pless J., Boissonnas R. A. И Helv. chim. acta.— 1963.— 46, N 176.—
S. 1609—1625.
104. Kupryszewski G., FormelaM.H Roczniki Chem.— 1963.— 37.— P. 161—
165.
105. Kisfaludy L.f Roberts J. E.> Johnson R. H. et aid I J. Org. Chem.—
1970.— 35, N 10.— P. 3563—3564.
106. Kisfaludy L., Low M.t Nyeki 0. et aid! Lieb. Ann. Chem.— 1973.—
№9 —S. 1421 — 1429.
107. Jones J. H.t Young G. Т.Н J. Chem. Soc. (C).— 1968.— P. 436—441.
108. Гершкович А. А., Серебряный C. Ed/ IV Всесоюзный симпозиум по
химии белков и пептидов (Минск, 5—8 сент. 1977. г.) : Тез. докл.
Минск : 1977, С. 188.
109. Гершкович А. А., Серебряный С. Б.11 Биоорган, химия.— 1979.— 5,
N 8.— С. 1125—1132.
НО. Klausner Y, S., Meiri Т. Н., Schneider Ed/ Proc. 5th Amer. Pept.
Symp./ Eds. M. Goodman, J. Meienhofer.— New York: J. Wiley and
sons.— 1977. P. 536—538.
111. Kovacs J., Mayers G. L.f Johnson R. H, etal.H J. Org. Chem.— 1970.—
35,— P. 1810—1815.
112. Kisfaludy L., Schon J.t Szirtes 7\ e/aZ.//Tetrahedron Lett.— 1974.—
N 19.—P. 1785—1786.
113. Гершкович А. А., Серебряный С. Б.Н Биоорган, химия.— 1985.—
11, Яг 7.—С. 869—894.
114. Boissonnas R. A., Guttmann St., Jaquenoud P.-A.H Helv. chim. acta.—
I960.— 43, N 6.—P, 1349—1358.
339
115. Nietzky R., Braunschweyg Ed/ Chem, Ber.— 1894.— 27.—S. 3381—
3384.
116. Альбертсон H. Ф.П Органические реакции.— M. : Мир, 1965.—
Т. 12.—С. 174—379.
117, Sheehan J. С., Cruickshank Р A., Boshart G, L,H J. Org. Chem,—-
1961.— 26, N 7,— P. 2525—2528.
118. Anderson G. W.t Zimmerman J. E., Callahan F. M,// J. Amer. Chem.
Soc.— 1964.— 86, N 9.— P. 1839—1842.
119. Wang K. T.t Brattesani D. N.. Weinstein В.П J. Heterocycl, Chem.—
1966.— 3, N 1.— P. 98.
120, Kovacs J .t Kisfaludy L.t Ceprini M. Q.// J. Amer. Chem. Soc.— 1967.—
89, N 1.— P. 183—184.
121. Физер Л., Физер M. Реагенты для органического синтеза.— М. : Мир,
1970.— Т. 5.— 236 с.
122. Kappeler Н.П Helv. chim. acta.— 1961.- 44, N 2.—P. 476—491.
123. Col C. S.f Doumani Th. Fd/ J. Amer. Chem. Soc.— 1948,— 70, N 4—
P. 1516—1519.
124. Гершкович А. А., Кибирев В. К.» Серебряный С. Б. и др.И Химия
природ, соединений.— 1976.— 4.— С. 507—517.
125. Suzuky К., Abiko Т., Endo N. et aid! Chem. Pharm. Bull.— 1973.—
21, N 12.—P. 2627—2633.
126. Wang S. S.f Kulesha I. D.t Winter D. P. et al.// Int. J. Peptide and
Protein Res.— 1978.— 11.— P. 297—300.
127. Schallenberg E. E., Calvin Md/ J. Amer. Chem. Soc.— 1955.— 77,
N 10.— P. 2779—2783.
128. Zaoral M.t Flegel M./l Collect. Czech. Chem. Commun.— 1972.— 37,
N 5.— P. 1539—1545.
129. Никифорова H. В., Радина Л. Б.П Журн. общ. химии.— 1979.—
48, № 8.— С. 1878—1883.
130. Корнилов Н. Ю., Юрченко А. Г., Туров А. В. и др.И Реактивы и особо
чистые вещества.— М. : НИИТЭхим, 1975.— Вып. 1 (27).— С. 72—
76.
131. Zaoral Md/ Coll. Czech. Chem, Commun.— 1962.— 27, N 5.—
P. 1273—1277.
132. Anderson G. W., Zimmerman J. E., Callahan F. М.П J. Amer. Chem.
Soc.— 1967.— 89, N 19.—P. 5012—5017.
133. Sole N., Torres J. L.t Garcia A. J. M. et aid/ Tetrahedron.— 1986.—
42, N 1.—P. 193—198.
134. Prasad K. U., Jgbai M. A., UrryD. W.// Int. J. Peptide and Protein
Res.— 1985.— 25, N 4.—P. 408—413.
135. Мазуров А. А., Кабанов В. M., Андронати С. А./7 Докл.АНСССР.—
1989.— 306, № 2 — С. 364—366.
136. Van der Auwera С., Anteunis М. J. О.// Bull. Soc. Chim. Belg.—
1986.— 95, N 3.— P. 203—205.
137. Van der Auwera C., Van Damme S., Anteunis M. J. О.// Int. J. Peptide
and Protein Res.— 1987.— 29, N 4.— P. 464—471.
138. Chen F. M. F., Kuroda K-> Benoiton N. L.ll Synthesis.— 1978.—
N 12.— P. 928—929.
139. Noble R. L.t Yamashiro D., Li Ch. Hd/ Int. J. Peptide and Protein
Res.— 1977.— 10.— P. 385—393.
140. Farthing A. Cd/ J. Chem. Soc.— 1950.— P. 3213—3217.
141. Hirschmann R., Schwan H., Strachan R. G. et aid/ J. Amer. Chem.
Soc.— 1971.— 93, N 11.—P. 2746—2754.
142. Schldgl K.> Wessely F.t Wawersich Ed/ Monatsch. ftir Chem.— 1953,—
84, N 2.— S. 705.
143. Fridkin M.f Shaltiel S.//Arch. Biochem. and Biophys.— 1971.—
147, N 2,— P, 767—771,
340
144. Fasman G. D.t Idelson M., BloutE. R.H J. Amer. Chem. Soc.— 1961.—
83, N 3.— P. 709—712.
145. Ramachandran J., Berger A., Kalchalski E.U Biopolymers.— 1971.—
10, N 10.— P. 1829—1851.
146. Bodanszky M., du Vtgneaud V.H J. Amer. Chem. Soc.— 1959.— 81,
N 21.— P. 5688—5691.
147. Kisfaludy L., Schon J.И Synthesis.— 1983.— N 4.— P. 325—327.
148. Schon J., Kisfaludy L.ll Ibid.— 1985.— N 4.— P. 303—305.
149. A. C. 724501 СССР, МКИ2 C07 C 103/52. Способ получения пентафтор-
фениловых эфиров аминокислот / В. Н. Медведкин.— Опубл. 30.03.80.
Бюл. N 12.
150. Медведкин В. Н., Митин Ю. В., Клименко Л. В. и др.II Биоорган,
химия, 1989.— 15, № 4.— С. 460—464.
151. Hui К. Y., Holleran Е. ЛК, Kovacs J.ll Int. J. Peptide and Protein-
Res.— 1988.— 31, N 3.—P. 205—211.
152. Медведкин В. H., Клименко Л. В., Митин Ю. В. и др.II Биоорган,
химия.— 1989.— 15, № 7.— С. 983—984.
153. А. с. 747854 СССР, МКИ2 С 07 207/40. Способ получения О-трифтор-
ацетил-ЬГ-оксисукцинимида / С. М. Андреев, Л. А. Павлова, С. В. Ро-
гожин.— Опубл. 15.07.80. Бюл. № 12.
154. Sakakibara S., Inukai N.I! Bull. Chem. Soc. Jap,— 1966.— 39, N 7.—
P. 1567 — 1572.
155. Guttmann St., Pless J., Boissonnas R. A.H Helv. chim. acta.— 1962.—
45, N 9.— P. 170—177.
156. Равдель Г. A.t МонановаH. H.t КритН.А.П Химия природ, соедине-
ний.— 1975.— 1.— С. 47—56.
157. Suzuky К., Sasaki Г.//Chem. Pharm. Bull.— 1973.— 21, N 12.—
P. 2634—2638.
158. Bower J. D., Guest К. P-> Morgan B. A.H J, Chem, Soc. Perkin Trans I.—
1976.— N 23.— P. 2488—2492.
159. Романова В. П., Серебряный С. B.H Биоорган, химия.— 1982.— 8,
№ 10.—С. 1302—1306.
160. Ананьин Л., Сластенина Е.Н Реактивы и препараты лабораторного
назначения.— Л.; М. : ГОНТИ, 1938.— Вып. 1,— С. 283—293.
161. Van Zon A., Beyerman Н. С.1/ Helv. chim. acta.— 1973.— 56.—
Р. 1729—1740.
162. Beyerman Н. C.t Kranenburg P., Schaaper W. M. M., Vaskamp D.H
Int. J. Peptide and Protein Res.— 1981.-18, N 3.—P. 276—283.
163. Nozaki S., Muramatsu Id! Bull. Chem. Soc. Jap.— 1982.— 55, N 7.—
P. 2165—2168.
164. Гершкович А. А., Кибирев В. Kdl Биоорган, химия.— 1988.— 14,
N 11.—С. 1461—1488.
165. KunzH., Lasowski Н.-J.H Angew. Chem.— 1986.—98, N 2.— S. 170—
171.
166. Goldschmidt S./l Ibid.— 1950.— 62, N 22.— S. 538.
137, Erlanger B. F., Kokowsky N.H J. Org. Chem.— 1961.— 26, N 7.—
p 2534_______2536.
168. Anderson G. W., Welcher A. D., Young R. W.H J. Amer. Chem. Soc.—•
1951.— 73, N 2.—P. 501—502.
169. Goldschmidt S., Lautenschlager H.U Liebigs Ann. Chem.— 1953,—
580.- S. 68—82.
170. Goldschmidt S., Wick M.ll Ibid.— 1952.— 575.- S. 217—231.
171. Schram G.t Wissmann H.U Chem Ber.— 1958.— 91, N 5.— S, 1073—
1082.
172. Bergmann M., Zervas L.ll Ibid.— 1932.— 65.,— S. 1192—1201.
173. Carpenter F. H.,GishD.T.H J. Amer. Chem. Soc,— 1952,—74, N 15,—
P, 3818—3821.
341
174. P at chornik A. t Amit В. t Woodward R. B.H Ibid.— 1970.— 92, N 21.—
P. 6333—6335.
175. Birr Ch., Lochinger W., Stohnke G. et al.H Lieb. Ann. Chem.— 1972.—
763.— S. 162—172.
176. McKay F. C.H J. Amer. Chem Soc.— 1957.— 79, N 17.— P. 4686—
4690.
177. Veber D. F., Paleveda W. J.t Ir., Lee Y. C., Hirschmann R.// J. Org.
Chem.— 1977.— 42, N 20.— P. 3286—3288.
178. Sieber P.f Iselin B.H Helv. chim.. acta.— 1968.— 51.— P. 622—632.
179. Le Corre G.> Guibe-Jampel E.9 Wakselman H.H Tetrahedron.— 1978.—
34, N 20.— P. 3105—3112.
180. Kemp D. S.f Hoyng Ch. F.H Tetrahedron. Lett.— 1975.— N 52.—
P. 4625-4628.
181. Carpino L. A., Han G. YJI J. Org. Chem.— 1972.— 37, N 22.—
P. 3404—3409.
182. Anderson G. W., McGregor А. C.H J. Amer. Chem. Soc.— 1957.— 79,
N 23.—P. 6180—6183.
183. Hoida I., Shitnonishi Y., Sakakibara S.H Bull. Chem. Soc. Jap.—
1967.— 40, N 10.—P. 2415—2418.
184. Haas W. L., Krumkalns E. V., Gerson КЛ J. Amer. Chem. Soc.—
1966.— 88, N 9.— P. 1988—1992.
185. Stevens С. M., Watanabe R.H Ibid.— 1950.— 72, N 2.— P. 725—727,
186. Carson J. F.H Synthesis.— 1981.— N 4.— P. 268—270.
187. Kunz II.// Andew. Chem.— 1978.— 90, N L— P. 63—64.
188. Fisher E.// Chem. Ber.— 1915.— 48.— S. 93—102.
189. Schonheimer R.H Z. Physiol. Chem.— 1920.— 154, N 5.— P. 203—224.
190. Kidd D. A.,t King F. E.H Nature.— 1948.— 162.— P. 776.
191. Hillmann A.t Hillmann GJ/ Z. Naturforsch. — 1951.— 66.— P. 340—
341.
192. Weygand F., Csendes E.H Angew. Chem.— 1952.— 64, N 5.— P. 136.
193. Kenner G. W., Moore G. A., Ramage R.H Tetrahedron Lett,— 1976.—
N 40.- P. 3623—3626.
194. Goerdeler J., Holst АЛ Angew. Chem.— 1959.— 71.— P. 775.
195. Helferich B., Moog L., JUnger A.H Chem. Ber.— 1925. — 58.—
S. 872—886.
196. Zervas L.. Theodoropoulos D, M.H J. Amer, Chem. Soc.— 1956,— 78,
N 7.— P. 1359—1363.
197. Birhofer L., Ritter A.H Liebigs Ann.— 1958.— 612.— S, 22—23,
198. Hope A.t Halpern ВЛ Tetrahedron Lett.— 1972.— N 22.— P. 2261 —
2264.
199. Khalifa M. H.t Jung G.f Reiker A.//Angew. Chern.— 1980.— 92,
jq g______§ 739____740.
200. Kunzl H., Studer R. 0Л Helv. chim. acta.— 1975.— 58, N 17—18.—
S. 139-146.
201, Blaha К.» Rudinger J.H Coll. Czech. Chem. Commun,— 1965.— 30,
N 2.— P. 585—598.
202. Hirschmann R.H Intra-Sci. Chem. Rep.— 1971.— 5, N 3.— P. 203—
220.
203, Wunsch E.t Jueger E., Kisfaludy L., Low M.H Angew. Chem. 1977.—
89, N 5.—S. 330—331.
204. Sieber P., Iselin B.H Helv..Chim. acta.— 1968.— 51. — S. 614—622.
205. Carpino L. АЛ J. Amer. Chem. Soc.— 1957,— 79, N 16,— P. 4427—
4431.
206. Позднее В. Ф.Н Химия природ, соединений.— 1974. — 6.— С. 764—
767.
207, Кери Ф,, Сандберг Р. Углубленный курс органической химии,—
М. ; Химия, 1981,— Т. 1.— 518 с.
342
208. Weygand F., Frauendrfer E.U Chem. Ber.— 1970.— 103, N 8.
S. 2437—2449.
209. Wunsch E., Sprangenberg 7?.//Ibid.— 1971 — 104, N 8.—S. 2427—
2429.
210. Jackson A. E., Jonstone R. A. W.H Synthesis.— 1976. — N 10.—
P. 685—687.
211. Ananthramaiah G. M., Sivanandaiah К. М.1/ J. Chem. Soc. Perkin-
Trans.— 1977.— 1, N 5.— P. 490—491.
212. Felix A. M., Heimer E. P., LambrosT. J. etal.H J. Org. Chem.— 1978.—
43, N 21.— P. 4194—4196.
213. Amin В. E., Anantharamaiah G. M., Roger G. P.t Means G. ЕЛ Ibid.—
1979.— 44, N 19.— P. 3442—3444.
214. AnwerM.K., SpatolaA. F./l Synthesis.— 1980.— N 11.— P. 929—932.
215. Losse G., Stiehl H. U.//Z. Chem.— 1981.—21, N 5.—S. 188.
216. Barltrop J. A., Schofield P.H Tetrahedron Lett.— 1962.— N 16.—
P. 697—699.
217. Chamberlin J. W.H J. Org. Chem.— 1966.— 31, N 5.— P. 1658—1660.
218. Patchornik A., Atrilt B., Woodward R. B.H J. Amer. Chem. Soc.—
1970.— 92, N 21.—P. 6333—6335.
219. Картер Г., Франк, Р.» Джонстон ХЛ Синтез органических препара-
тов.— М. : Иностр, лит., 1952.— Т. 3.— С. 100—103.
220. Хейфец М. В., Лопырев В. А,, Айзенштадт И. Я.//Журн. прикл.
химии.— 1968.— 41, № 6.— С. 1380.
221. Smith Е. L.H J. Biol. Chem.— 1948.— 175.— Р. 39—47,
222. Katsoyannis Р. G.,Gish D. T.t Hess G. P., du Vigneaud V.H J. Amer.
Chem. Soc.— 1958.- 80, N IL— P. 2558—2562.
223. TritschG. L., Woolly D. W.H Ibid.— I960.—82, N 11.— P. 2787—2793.
224. Benoiton L.//Can. J. Chem.— 1962.—40, N 3.—P. 570—571.
225. Boissonnas R. A., Guttmann St., Huguenin R. L. et al JI Helv. chim.
acta.— 1958.—41.—S. 1867—1882.
226. Zervas L., Winitz M., Greenstein J. PJI J. Org. Chem.— 1957,— 22,
N IL—P. 1515—1521.
227. Sennyey G., Barcelo G., Senet J.-P.H Tetrahedron Lett.— 1986,— 27,
N 44.— P. 5375—5376.
228. Rotn mi S., Moroder L., Bovermann G., Wunsch ЕЛ Synthesis.—1985.—
N 8.— P. 738—742.
229. Wolman Y., Ladkany D.t Frankel M./l J, Chem. Soc. (C).— 1967.—
N 8.— P. 689—690.
230. Frankel M., Ladkany D., Gllon C„ Wolman YЛ Tetrahedron Lett.—
1966.— N 39.— P. 4765—4768.
231. Kim S., Lee J. I., Yi K. Y.H Bull. Chem. Soc. Jap.— 1985.— 58,—
эд 12______P. 3570___3575
232. Wlllstatter R., Wal d'schmidt-Leitz. ЕЛ Chem. Ber.— 1921.— 54.—
S. 113—138.
233. Препаративная органическая химия.— M.; Л. : Химия, 1964.— 623 с.
234. Coleman D. R., Royer G. РЛ J. Org. Chem.— 1980.— 45, N IL—
P. 2268—2269.
235. Anwer M. К», Khan S. A., Sivanandaiah К. МЛ Synthetic.—
1978.—N 10.—P. 751—752.
236. Куликов С. В., Соколова H. Ю., Самарцев M. А.//Журн. орган, хи-
мии.— 1985.— 21, № 8.— С. 1779—1787.
237. Kikugawa Y.t Kashimura M.// Chem. and Pharm. Bull.— 1982,—
30, N 9.— P. 3386—3388.
238. Persco G., Forino R., Galantino M. et al.// Int. J. Peptide and Protein
Res.— 1986.— 27, N L— P. 51—60.
239. Kiso Y., Ukawa K-, Akita ТЛ J. Chem. Soc. Chem. Communs,— 1980,—
N 3,— P. 101—102.
343
240. Takeyama M., Koyama К., Inoue К» st al JI Chem. and Pharm. Bull.—
1980.— 28, N 6.— P. 1873—1883.
241. Bodanszky M., Bodanszky A 11 Int. J. Peptide and Protein Res.—
. 1984.— 23, N 2.—P. 287—291.
242. Fuiii A., Otaka A., Ikemura 0 et al J! J. Chem, Soc. Chem. Communs,—
1987.— N 4.— P. 274—275.
243. Dcmuth M., Mikhail GJ I Synthesis.— 1982.— N 10.— P. 827.
244. Позднее В. Ф., Смирнова Е. А., Подгорнова Н. Н, и др.//Журн.
орг. химии.— 1979.— 15, № 1.—С. 106—109.
245. Позднее В. Ф., Подгорнова Н. Н.> Зенцова Н. К. и дрЛ Химия природ,
соединений.— 1979.— № 4.— С. 543—548.
246. Позднее В. ФЛ Биоорган, химия.— 1977.— 3, № 12.— С. 1605.
247. Carpino L. АЛ J. Amer. Chem. Soc.— I960.— 82, N 11.— P. 2725—
2731.
248. Carpino L. At, Giza C. A,f Carpino В. АЛ Ibid.— 1959.— 81, N 4.—
Pe 955______957
249. Schnabel ЕЛ Lieb. Ann. Chem.— 1967.— 702.— S. 188—192.
250. Gronka Z., Lammek В.//Synthesis.— 1974.— N 9.— P. 661—662.
251. Shybir K., Sivanandaiah К. М.П Indian J. Chem. B.— 1977.— 15,
N 1.— P. 80—81.
252. Позднее В. ФЛ Химия природ, соединений.— 1971.— № 3<— С. 384—
385.
253. Moroder L., Hallett A., Wilnsch E. et alJ! Hoppe-Seyler’s Z. Physiol.
Chem.— 1976.— 357, N 11.—S. 1651—1653.
254. Позднее В, ФЛ Химия природ, соединений.— 1974.— № 6.— С. 764—
767.
255. Halpern В., Nitecki D. ЕЛ Tetrahedron Lett.— 1967.— N 31.—
p 3031_______3033.
256. Goodacre J., Pousford I. R., Stirling I.// Ibid.— 1975.— N 42.—
P. 3609—3612.
257. Inouyl K., Tanaka A., Otsuka H.UBvM. Chem. Soc. Jap. — 1970.—
43, N 4.— P. 1163—1172.
258. Беркенгейм A. M. Практикум по синтетическим лекарственным и ду-
шистым веществам и фотореактивам.— М. : Госхимиздат, 1942.—
С. 199.
259. Синтез органических препаратов.— М. : Иностр, лит., 1949. — Т. 2.—
С. 560—562.
260. Вегнер Р. Э., Полевая Л. К., Восекална И. Л. и др Л Изв, АН ЛатССР,
1979.—№ 1.—С. 103—106.
261. Zervas L., Borovas D., Gazis ЕЛ J. Amer. Chem. Soc.— 1963,— 85,
N 22.— P. 3660—3666.
262. Zervas L., Hamalidis СЛ1 Ibid.— 1965.— 87, N 1.— P. 99—104.
263. Kessler W., Iselin B.H Helv. chim. acta.— 1966.— 49.— S. 1330—1344.
264. Tun-Kyi AJ/W\d.— 1978.— 61, N 3.—S. 1086—1090.
265. Sieber P., Iselin B.// Ibid.— 1969.— 52.—S. 1525—1531.
266. Gull H. C., Turner E. E.//Chem. Soc.— 1929.— P. 491—500.
267. Mowry D. T., Dazzi J., Renoll M., Shortridge R. W.H J. Amer. Chem.
Soc.— 1948,— 70, N 10.— P. 1916—1917.
268. Mamos P., Sanida C., Barlos КЛ Liebigs Ann. Chem.— 1988.—
№ 11.— S. 1083—1084.
269. Barlos K., Papaioannou D., Theodor opoulos D.//Int. J. Peptide and
Protein Kes.— 1984.— 23.— P. 300—305.
270. Harris J. I., Work T. SJI Biochem. J.— 1950.— 46.— P. 582—589.
271. Weygand F., Geiger RJ/Chem. Ber.— 1956.— 89, N 3,—P. 647—
652.
272. Кошелева Г. H., Налецкая Г. НЛ Методы получения химических реак-
тивов и препаратов.— М. : ИРЕА, 1966,— G. 46—49.
344
273» Shallenberg E. E.t Calvin МЛ J* Amer. Chem. Soc.-— 1965,-77,
N 10.— P. 2779—2783.
274. Steglich W., Hinze S.H Synthesis.— 1976.—N 6.—P. 399—401.
275. Curphty T. J.H J. Org. Chem.— 1979.- 44, N 15.—P. 2885—2887.
276, Chang C’D., Felix A. M., Jimenez M. H., Meienhofer J.// hit. J. Peptide
and Protein Res.— 1980.— 15, N 5,— P. 485—494.
277. Carpino L. A.H J. Org.Chem.— 1980.- 45, N 21P. 4250—4252.
278. Carpino L. A., Han G. Y.// ibid.— 1973.— 38, N 24.— P 4218.
279. Carpino L.A., Han G. Y.// Ibid.- 1972.— 37, N 22.— P. 3404—3409.
280. Tessier M., Albericio F., Redroso E. et al.H Int. J. Peptide and Protein
Res.- 1983.- 22.- P. 125-128.
281. Sigler G. F., Fuller W. D., Chaiurvedl N. C. et a/.//Biopolymers.—
1983.- 22.- P. 2157—2162.
282. Ten Kortenaar P. B. W., van Dijk B.G., Peelers J. AL el al.H Int. J. Pep-
tide and Protein Res.— 1986.— 27, N 4.— P. 398—400.
283. Bolin D._ R., Sytwa 1.4.» Humiec F., Meienhofer J.// Ibid.—
1989.— 34, N 5.— P. 353—359.
284. Amaral M. J. S. А.» BarettG. С.» Rydon H. N., Willett J. E.H J. Chem.
Soc (C).— 1966.— P. 807—813.
285. Митин Ю. В., Надеждина Л. Б./l Журн. орган, химия.— 1968.— 4,
М 7.—С. 1181 —1184.
286. Pass Sh., Amit В., Patchornik A.H У. Amer. Chem. Soc.— 198L— 103,
N 25.— P. 7674—7675.
287. Curtins T., Goebel F.H J. Prakt, chem,- 1888.— 37,- P, 150,
288. Brenner М.» Huber W.H Helv. chim. acta.— 1953.— 36.— S 1109—
• 1115.
289. Bergmann М.» ZervasL., Salzmann L.H Chem. Ber.— 1933.— 66, M 8.—
S. 1288—1290.
290. Schwyzer R.» Sieber P.H Helv. chim. acta.— 1959.— 42.— S. 972—977,
291. Roeske R.H J. Org. Chem.— 1963.— 28, N 3.—P. 1251—1253.
292. Weygand F.» Hunger О Chem. Ber.— 1962,- 95, N 1.— P. 1—6.
293. Camble R., Garner R., Young G. Т.Н Nature.— 1967.— 217, N 5125.—
P. 247—248.
294. Mariner В.» Kim Y. C,9 Navarre J.-M.H Can. J. Chem.— 1973.—
51, N 9.— P. 208—214.
295. Kenner G. W.H Angew. Chem.— 1959.— 71.- P. 741—742.
296. Galpin I. J.» Hardy P. M., Kenner G. W. et al.H Tetrahedron,— 1979.—
35, N 21.— P. 2577—2582.
297. Hiskey R.G., Adams J.B.H J. Amer. Chem. Soc.— 1965.— 85, N 17.—
P. 3969______3973.
298. Birkofer L.» Ritter A.H Chem. Ber.— I960.— 93, N 2.—S. 424-^27.
299. Sieber Р.» Andreatta R. Н.» Eisler B. et ah// Proc. 5th Amer. PepL
Symp./ Eds. M. Goodman, J. Meienhofer. J. Wiley and Sons: New
York.— 1977.— P. 540—542.
300. Bednaker M. A., Bodanszky M.H Int. J. Peptide and Protein Res,—
1983.—21, N 2.—P. 196—201.
301. Kessler Н.» Siegmeir R.H Tetrahedron Lett.— 1983.— 24, N 3.—
P. 281—282.
302. My ту лис Ф. К., Мутуле И. Э.» Полис Я. Ю. и др Л VI Всесоюз .сим-
поз. по химии белков и пептидов (Рига, 20—25 ноября 1983 г.) : Тез,
докл. Рига : 1983.—С. 230—231.
303. KempD.S., HansonQЛ J. Org. Chem.— 1981.— 46, N 24.— P. 4971—
4975.
304. Stewart F. H. C.H Austral. J. Chem.— 1968.— 21, N 11.— P. 2831—
2834.
305. Stelakatos G. C., Paganou А.» Zeruas L.H J. Chem, Soc. (C).— 1966,—
N 13.—P. 1191 — 1199.
23 2~882
345
306. Barltrop J. A., Plant P. J., Schofield P.H Ibid.— P. 822—823.
307. Isied S. S., Vassilian A., Lion J. M., H J- Amer, Soc.— 1982.— 104,
N 14.— P. 3910—3916.
308. Aral JMuramatsu I.// J. Org. Chem.— 1983.— 48, N 1.— P. 121—123 .
309. Позднее В. Ф.Н VI Всесоюз. симпоз. по химии белков и пептидов (Рига,
20—25 ноября 1983 г.) : Тез. докл. Рига : 1983.— С. 284—285.
310. Weber U.HZ. Naturforsch.— 1976.—31b, N 8.—S. 1157—1158.
311. Dhaon M. K., Olsen R. K., Ramasamy КЛ J. Org.? Chem,— 1982.—
47, N 10.—P. 1962—1965.
312. Gal pin I, J., Hancock F. E.t HanadaB. K.etalJI Tetrahedron.— 1979.—
35, N 23.— P. 2771—2778.
313. Miller A. W., Stirling C. J, МЛ J. Chem. Soc. (C).— 1968.— P. 2612—
2617.
314. Guibe-Jympel E., Wakselman M.H Synth, Commun.— 1982,— 12,
N 3._______p, 219__223.
315. Ruhlmann КЛ J. Prakt. Chem.— 1959.— 9.— S. 86—91.
316. Birkofer L., Ritter A., Neuhausen P.HLkb. Ann.— 1962.— 659.—
S. 190—199.
317, Крысий В. П., Карельский В. Н., Антонов А. А., Глинка Э. ДЛ
Химия природ, соединений.— 1978.— № 4.— С. 482—485.
318. Андреев С. M.t Козюков В. П., Миронова Н. В Л Обзорная информа-
ция.— М. : НИИТЭХИМ, 1980.— С. 21 (Сер. Элементоорганические
соединения и их применение).
319. Guttmann S., Boissonnas R, А.И Helv. chim. acta.— 1958.— 41.—
S 1852________1867
320. Suzuki K>> Sasaki Y.H Chem. Pharm. Bull.— 1973.— 21, N 12.—
P. 2634—2638.
321. Shields J. E., McGregor W. H., Carpenter F. H.U J. Org. Chem.— 1961.—
26, N 5.— P. 1491 — 1494.
322. A. c. 422724 СССР, МКН2 C 07 C/12. Способ получения n-ннтробен-
зилхлорида / С. Я. Микста, Г, И. Чипенс, М. Б. Звайгзне.— Опубл,
15.03.74. Бюл. N 13.
323. Schwarz Н., Arakawa КЛ J. Amer. Chem. Soc.— 1959.— 81, N 21.—
P. 5691—5695.
324. Гершкович, А. А., Кибирев В. К.» Серебряный С. Б. и др.Н Химия
природ, соединений.— 1976.— № 4.— С. 507—517.
325. Болотов Б. А., Комаров В. А., Низовкина Т. В. Практические работы
по органическому катализу.— Л. : Изд-во ЛГУ, 1959.— 116 с.
326. Schroder ЕЛ Lieb. Ann.— 1963.— 670.—Р. 127—136.
327, Anderson С. W., Callahan F. M.H J. Amer. Chem. Soc,— 1960.—
82, N 13.— P. 3359—3363.
328. Давидович Ю. А., Галкин О. M., Рогожин Ct ВЛИзв. АН СССР.
Сер. хим.— 1977.— № 6.— C. 1444.
329. Birkofer L., Ritter A., Dickopp Я.//Chem Ber.— 1963.— 96, N 6.—
S. 1473—1474.
330. Klebe J. F.t Finkbeiner H.t White D. МЛ J. Amer. Chem. Soc,—
1966.- 88, N 14.—P. 3390—3395.
331. Позднее В. Ф.,Н Биоорган, химия.— 1985.— 11, № 5.— С. 583—589.
332. Гершкович А. А., Кибирев В. КЛ Там же.— 1988.— 14, № 11.—
С. 1461—1488.
333. Sandrin Е., Boissonnas R. АЛ Helv. chim. acta.— 1963.— 46, N 5.—
S. 1637—1669.
334. Rzeszotarska B., Makowski MKubica Z. Synthesis of t-butoxycarbonyl
amino acid amides // Org. Prep, and Proced. Int.— 1987. 16, N 2,
P. 136—139.
335. Fox S. W„ Wax H./lJ. Amer. Chem. Soc.— 1951, - 73, N 6.—
P. 2936—2937,
346
336. Boissonnas R. A., Guttmann St., Huguentn R. L., et al JI Helv. chim.
acta.— 1958.— 41, N 6.—S. 1867—1882.
337, Заявка 60—61553 Япония, МКИ*С07 С 129/12. Получение л-нитроа-
нилида аргинина / Куроива К., Накацуяма X., Нагасава Т. Опубл,
24.07.85. Рефер. журн. «Химия» — 1986, № 12, реф. 1 гО16П.
338. Nishi /V., Tokura S., Noguehl J.// Bull. Chem. Soc. Jap.— 1970.— 43,
N 9. — P. 2900—2907.
339. Пат. 700060 СССР МКИ* С 07 С 103/52, А61 К37/02. Способ получения
пептидов или их солей / Б. Т. Экенстам, Л. Э. Аурелл, К. И. Клаэсон
и др.— Опубл. 25.11.79. Бюл. N 13.
340. Kasafirek Е., Chavko М., Bartik М.Н Collect. Czech. Chem. Commun.—
1971.— 36, N 12.— P. 4070—4074.
341. Erlanger B. F., Edel F., Cooper A. GJ/ Arch. Biochem.— 1966.— 115,
N 1.- P. 206—210.
342. Гершкович, A. A.H Биоорган, химия,— 1991,— 17.— № 7.— G. 870—
875.
343. Zhang YWang X., Li I., Zhang P.H Чжунго Кэсюэ. Sin, Sin. В,—
1986.- 29, N 10.—P. 1009—1017.
344. Habbuch A., Bindewanol R., Fohles 1., Zahn Я.//Angew. Chem,—
1980.- 92, N 5.— P. 394—395.
345. Anriger H., Mutter H., Bayer E./l Angew. Chem.— 1979.— 91, N 9,—
P. 747—748.
346. Schwyrer R., Sieber P., Zatako A.//Helv. chim. acta.— 1958,— 41f
N 2.— P. 491-498.
347. Wieland Th., Racky IF.//Chimia.—1968.— 22, N 4.—P. 375—377,
348. Merrifield R. B.H J. Amer. Chem. Soc.— 1963.— 85, N 14.— P. 2149—
2154.
349. Letsinger R. L., Kernel M. 1 JI Ibid.— 1963.—85, N 14,— P* 3045—
3046.
350. Mutter M.. Uhmann R., Bayer E.H Liebigs Ann. Chem.— 1975.—
N 5.- S. 901-915.
351, Митин Ю. В., Надеждина Л. Б.//Журн. орган, химии.— 1968.—
4, № 7.— С. 1181 — 1184..
352. Kunz Я.//Chem. Вег.— 1976.— 109, N 11.—S. 3693—3706.
353. Kunz Н., Bucholz М.Н Ibid.— 1979.— 112, N 6.—S. 2145—2157.
354. Кши И., Waldmann H.H Angew. Chem.— 1983.— 95, N 1.—S. 47—
48
355. Kunz H.H Liebigs Ann. Chem.- 1976.- N 9.— S. 1674—1679.
356. Chantreux D., Garnet J.-P., Jaequier R., Verducci J.// Tetrahedron.—
1984.— 40, N 16.—P. 3087—3094.
357. Kuntz H., Birmbach S.H Tetrahedron Lett.— 1984.— 25, N 33.—
P. 3567—3570.
358. Вольпина О. M.t Дешко T. H., Михалева И. И., Иванов В. ТЛ Био-
ерган. химия.— 1980.— 6, № 8.— С. 1155—1162.
359. Bodanszky М., Martinez JЛ Synthesis.— 1981.— N 5.— Р. 333—356,
360. Schnruder Е., Kleiger ЕЛ Lieb. Ann.— 1964.— 673.— S. 196—207.
361. Klieger E., Gibian H.H Lieb. Ann.— 1962.— 655.—S. 195—210.
362. Przstidge R. L., Harding D. R. K., Hancock W. S.H J. Org. Chem,—
1976.- 41, N 15.— P. 2579—2583.
363. Tan J. P., Wong T.-W., Riemcn M. W. et al.H Tetrahedron Lett.—
1979.— N 42.— P. 4033—4036.
3b4. Neuberger A., Sanger FJI Biochem. J.— 1943.— 37.— P. 515—518.
365. Schwyzer R., Rittel W.// Helv. chim. acta.— 1961.— 44.— P. 159—169.
366. Fru.kson B. W., Merrifield R. B.H J. Amer. Chem. Soc.— 1973.— 95,
N 11.-- P 3757-3763.,
367. Yamashiro D., Nable R. L., Li С. НЛ Peptide / Ed, Nesvadba.—
Amsterdam : North-Holland, 1971,—P, 197, s'
23*
347
368. Anfinsen С. В., OntjesD., Ohno M. et al.// Proc. Nat. Acad. ScL USA,—
1967.— 58.—P. 1806—1811.
369, Hoffmann K., Stutz E., Spuhbr G. et al.H J. Amer. Chem. Soc.— 1960 —
82, N 14.— P. 3727—3732.
370. Roeske R. W., Stewar F. H. C., Stedman R. J., du Vigneaud VJl Ibid. —
1956.— 78, N 22.— P. 5883—5887.
371. Fakuda T., Wakimasu M., Kabayashi Sh., Fujino МЛ Chem. and Pharm.
Bull.— 1982.— 30, N 8.—P. 2825—2835.
372. Bergmann M., Zervas L., Rinke НЛ Hoppe-Seyler’s Z. Physiol.
Chem.— 1934.— 224, N 1/2.—S. 40—44.
373. Guttmann S., Pless J., Boissonnas R. A.// Helv. chim. acta.— 1962.—
45.—S. 170—177.
374. Nishimura 0., Fujino МЛ Chem. and Pharm. Bull.— 1976.— 24,
N 7.— P. 1568—1575.
375. Yajima H,, Takeyama M., Kanaki J.//Ibid.— 1978. — 26, N 12.—
P 3752________3757
376. Jager G., Geiger RJt Chem. Ber.— 1970.— 103, N 3.— S. 1727—
1747.
377. Fujino M., Nishimura 0., Wakimasu M.t Kitada CJ/ J. Chem. Soc,
Chem. Commun.— 1980.— N 14.— 668—669.
378. FujinoM., Wakimasu M., KitadaCJ/ Chem. and Pharm. Bull.— 1981.—
29, N 10.— P. 2825—2831.
379. Du Vigneaud V., Behreus O. КЛ J. Biol. Chem.— 1937.— 117.—
P. 27—36.
380. Fujii T., Sakakibara SJ/ BM. Chem. Soc. Jap.— 1974.— 47, N 12.—
P. 3146—1351.
381. Chillemi F.t Merrifield R. B./I Biochemistry.— 1969.— 8, N 11.—
P. 4344—4346.
382. Brown T., Jones J. H.H J. Chem. Soc. Chem. Commun.— 1981.—
P. 648—649.
383. Colombo R., Colombo F., Jones J. НЛ Ibid.— 1984.— N 7.— P. 292—
293.
384. Stelakatos G. C., Theodoropoulos D. M., Zervas LJ/ J. Amer. Chern,
Soc.— 1959.— 81, N 11.—P. 2884—2887.
385. Wiinsch E., Fries G., Zwick A.// Chem Ber.— 1958.—91, N 3.—
S. 542—547.
386. Erickson B. W., Merrifield R. В JI J. Amer. Chem. Soc.— 19'3.-- 95,
N 11.— P. 3750—3756.
387. Amit B., Hazum E., Fridkin M., Patchornik A J/ Int. J. Peptide and
Protein Res.— 1977.— 9, N 2.— P. 91—96.
388. Overell B. G., Petrov VJI J. Chem. Soc.— 1955.— P. 232—236.
389. Позднее В. ФЛ Химия природ, соединений,— 1982.—Кй 1.— С. 129—
130.
390. Wiinsch Е., Jentsch JЛ Chem Ber.— 1964.— 97, N 9.— S. 2490-2496,
391. Ueki M., Inazu ТЛ Bull. Chem. Soc. Jap.— 1982.— 55, N I.—
P. 204—207.
392. Sugano H., Miyoshi M.//J. Org. Chem.— 1976.— 41, N 13.—
P 2352_______2353.
393. Schroder f.//Lieb. Ann. Chem.— 1963.— 670.—S. 127—136.
394. Sifferd R. H., du Vigneaud VJ! J. Biol. Chem.— 1935.— 108, N 3.—
P. 753—761.
395. Veber D. F., Milkowskl J. D., Varga S. L. et alJ/ J. Amer. Chem.
Soc.— 1972.— 94, N 15.—P. 5456—5461.
396. Папсуевич О. С., Арш Г, Ill., Миксча С. ЯЛУКуръ. общ. химии,—
1975.- 45, № 6.—С. 1384—1388.
397. Pasturzak I. I., Chimiak АЛ J, Org, Chem»— 1981,— 46, N 9,—
P. 1868—1874.
348
398. Sakakibara S., Nabuhara Y., Simonishl Y., Ktyoi R.ll Bull. Chem.
Soc. Jap.— 1964.— 37, N 3 — P. 433—434.
399. Berse C., Boucher R.t Piche L.H J. Org. Chem.— 1957.— 22, N 7.—
P. 805—808.
400. Hiskey R. G.f Adams J. B.H Ibid.— 1965.— 30, N 4,— P 1340.
401. Zervas L., Phoiaki I Л J. Amer. Chem. Soc.— 1Г62.— 84, N 20.—
P. 38$7—3897.
402. Otaka A., Marimoto H., Fujii N. et al.II Chem. ad Pl.afm. Bull.—
1989,- 37, N 2.— P. 526—528.
403. Ariely S,, Fridkin M., Patchornik A.// Biopolymers.— 1969.— 7,
N 3.— P. 417-422.
404. Iselin B.H Helv. chim. acta.— 1961.— 44, N 8.— S. 61-78.
405. Konig W.f Geiger R.ll Chem. Ber.— 1970,— 103, N 7 — S. 2041—
2051.
406. Shimonishi Y., Sakakibara S., Akabori S.ll Bull. Chem. Soc. Jap.—
1962.- 35, N 12.—P. 1966—1970.
407, Ghno M., Tsukamoto S., Makisumi S., IzumiyaH Ibid.— 1972.—45,
N 6.— P. 2852—2855.
408. Fukuda T.> Wakimasa M., Kobayashi Sh., Fujino M.11 Chem. and
Pharm. Bull.— 1982.— 30, N 8.— P. 2825—2835.
409. Franzen H., Grehn L., Ragnarsson U./7 J. Chem. Soc. Chem. Commun,—
1984.—N 24.—P. 1699—1700.
410. Sergheraert C.H'J. Organomettalic Chem.— 1982.— 240, N 2.—
P. 163—168,
411, Prestidge R. L., Harding D. R. K., Battersby J. E., Hancock IF. S.ll
J. Org. Chem.— 1975.- 40, N 22.— P. 3287—3288.
412. Benoiton L.H Can. J. Chem — 1962.— 40, N 3.— P. 570-572.
413, Heeswijik IF. A. R., Eenink M. J. D.,Feijen J.//Synthesis.— 1982.—
№ 9,— p. 744—747.
414, Boissonnas R. A.f Gutimann St., Huguenin R. L. et al.II Helv. chim.
acta.— 1958.— 41.— P. 1867-1882.
415. Ledger R., Stewart F. H. C./l Australian J. Chem.— 1965.— 18, N 6.—
p 933________дзз
416. Tailor U. F., DyckesD. F., Cox 1. R.l/ Int. J. Peptide and Protein Res.—
1982.— 19, N 2.— P. 158—161.
417. Gibian H.t Schroder Е.1/J. Lieb. Ann. Chem.— 1961.- 642.—
S. 145—162.
418, Hayakawa T., Fujiwara Y., Noguchi I.// Bull. Chem. Soc. Jap.— 1967.—
40, N 5.— P. 1205—1208.
419. Wunsch E., Wend Aber ger G.//Chem. Ber.— 1967.— 100, N 1.—
S. 160—172.
420, Smithwlck E. L.. Jr., Shuman R, ТЛ J. Org. Chem.— 1974.— 39,
N 23.— P. 3441—3442.
421, Boissonnas R. A . Gutimann St., Jaquenoud P.-А. et al.// Helv. chim.
acta.— 1961.— 44, N 16.—P. 123—130.
422. Demeny L.H Rect. trav. chim.— 1931.— 50.— P. 50—59.
423. Самукое В. В., Калашников В. В., Швалье А. Ф., Офицеров В. И.И
Журн. общ. химии.— 1989.— 59, № 3.— С. 703—711.
424. Pless Bauer W.H Angew. Chem.— 1973.— 85, N 1.—S. 142.
425. Turan A., Patihy A., Bajusr S.ll Acta Chim.— 1975.— 85.— P, 327—
332.
426. Van der Eijk I., Nolte R. I., Zwikker 1. W.H J. Org. Chem.— 1980*—
45, N 3.— P. 547—548.
427. Schnabel E., Stoltefuss J., Offe H. AKlanke ЕЛ Liebigs Ann. Chem.—
1971.__ 743.__ S. 57—68.
428. Позднее В. Ф.Н Химия природ, соединений.— 1980.— № 3,—
С. 379—383.
349
429. А. с. 979341 СССР, МК1Р С 07 Д 233/64, С 07 С 103/52. Активирован-
ные ьфи, ы Na, Н1т.ди-/ир#п-бутилоксикарбонилгистидина
В. Ф. По днев.— Опубл. 12.07.82. Бюл. № 45.
430. Katehcdski Е., Sela МЛ J. Аглет* Chem. Soc.— 1953.— 75, N 21*—
Р. 5284—5289.
431. Yamashiro D., Li С. НЛ J, Amer. Chem. Soc,— 1973*— 95, N 4,—
P. 1310—1315.
432* Позднее В. ФЛ Химия природ, соединений.— 1982.— Ns 1.— С. 129—
130.
433. Okawa A.//Bull. Chem. Soc. Jap.— 1956.— 29, N 5.— P. 486—488*
434. Wiinsch E., Furst G.H Hoppe-Seyler’s Z. Physiol. Chem,— 1962.—
329, N 1/2.—S. 109—112.
435. Chi-Hueu Wong, Meug-Fei Ho, Kung-Tsung Wang 11 J. Org. Chem.—
1978.— 43, N 18.— P. 3604.
436* Greenstein J, P., Winitz M. Chemistry of amino acids,— New York;
London : John Willey and Sons, 1961.— Vol. 3.— 1920 p,
437. Birr ChJlJ, Liebigs Ann. Chem.— 1973.—N 10.—S. 1652—1662.
4 38. Виденина P. Ф., Дыханов H. НЛ Реактивы и особо чистые вещества.—
М. : НИИТЭХИМ, 1976.— Вып. 4.—G. 6—10.
439. Современные методы биохимии / Под ред4 Ореховича.— М, : Медици-
на, 1968.— 157 с.
440. Chakaravary Р. К., Olsen R. КЛ J. Org. Chem,— 1978,— 43, N 6,—
Р. 1270—1271.
441. Fujii N.. Otaka A., Funakoshi S, et al.H J. Chem. Soc. Chem, Commun.—
1987.— N 3.— P. 163—164.
442. Bergmann E., Hervey J.// Chem. Ber,— 1929*— 62, N 2»— S. 893—
916.
443. Sakakibara S., Shimoriishi V.//Bull. Chem. Soc. Jap.— 1965.— 38,
N 8.— P. 1412—1413.
444. Lenard J., Robinson А. ВЛ J. Amer. Chem. Soc.— 1967.— 89, N 1.—
P. 181—182..
445. Вольпина О. M., Михалева Ht И., Иванов В. ТЛ Биоорг. химия,—
1982.- 8, № 1.— С. 5—49.
446. Руководство по препаративной неорганической химии / Под ред.
Г. Бауэра.— М. : Иностр, лит., 1956.— 371 G.
447. Yajima Н., Fujii N., Ogawa Н.> Rawatani Н.П J. Chem. Soc. Chem,
Commun.— 1974.— N 3.— P. 107—108.
448. Yajima H.> Fujii N.H J. Amer. Chem. Soc.— 1981.— 103.— P. 5867*
449. Fujii N., Shimokura M., Nomizu M. et a/.//Chem. Pharm. Bull.—,
1984.— 32, N 2.— P. 520—529.
450. Yajima H., KisoYOgawaH. et al.H Chem. and Pharm. Bull.— 1975.—
23, N 5.- P. 1164—1166.
451. Nispen J. W., Polderdijk J. P., Jansen W.P. A., Greven Н.МЛ Rec*
trav. chim. Pays-Bas.— 1981.— 100, N 11.— P. 435—436.
452. Young G. ТЛ Biopolymers.— 1981.— 20, N 10.—P. 1805—1809.
453. Barany G., Merrifield R. B.H J. Amer* Chem. Soc.— 1977,— 99,
N 22.- P. 7363—7365.
454. Merrifield R. ВЛ Biochemistry.— 1964.— 3, N 9.— P. 1385—1390.
455. Merrifield R. В.// Edv. Enzymology / Ed. F* F. Nord.— 1969.— 32*—
P. 221—296.
456, Pettit G, R. Synthetic peptides.— New York : Acad. Press., 1975*—
Vol. 3.— 438 p.
457. Merrifield R. B.//Science.— 1986.— 232, N 4748.— P. 341—347.
458. Dryland A., Sheppard R. C.H J, Chem. Soc. Perkin Trans I.— 1986,—
N 1.— P. 125—137.
459. Barany G., Merrifield R. B.H The Peptides / Eds E. Gross, J. Meien-
hofer,— New York : Acad* press, 1979.— Vol, 2,— P, 1—284,
350
460. Atherton E., Clive D. L. J., Sheppard R. C.H J. Amer. Chem. Soc.—
1975.— 97, N 22.— P. 6584—6585.
461. Smith C. W., Skala G., Walter Rj/ J. Peptide and Protein Res.—
1980.— 16, N 2.— P. 365.
462. Atherton E., Brown E.> Sheppard R. C.H J. Chem. Soc. Chem. Commun.—
1981.- N 21.— P. 1151 — 1152.
463. Lukas T. J., Prystowsky M. B., Erickson B. W.H Proc. Natl. Acad.
Sei. USA — 1981.— 78.— N 5.— P. 2791—2795.
464. Mitchell A. R., Erickson B. W., Ryabtsev M. N. et al.H J. Amer.
Chem. Soc.- 1976.— 98, N 23.— P. 7357—7362.
465. Mitchell A. R., Kent S. B., Engelhardt M., Merrifield R. B.H J. Org.
Chem.- 1978.— 43, N 13.— P. 2845—2852.
466. Sarsntakis D., Teichman J., Lien E. L.t Fenichel R. L.H Biochem.
Biophys. Res. Commun.— 1976.— 73— N 2.— P. 336—342.
467. Bodanszky M., Bednarek M. A.H J. Prot. Chem.— 1989.—8, N 4.—
P. 461—469.
468. Atherton E., Sheppard R. C.H J. Chem. Soc., Chem. Commun.—
1985.— N 3.- P. 165—166.
469. Atturton E., Cameron L., Meldal M., Sheppard R.C.H Ibid.— 1986.—
N 24.— P. 1763-1765.
470. Hag/nmaier H., Frank N .H Hoppe-Seylers Z. Physiol. Chem.— 1972.—
353.—N. 12.— P. 1973—1976.
471. Barany G., Merrifield R. B.H J. Amer. Chem. Soc.— 1977.— 99.—
N 22.— P. 7363—7365.
472. Bodanszky M., Sheehan J.// Chem Ind.— 1966.— P. 1597—1598.
473. Gisin B. F., Merrifield R. B.H J. Amer. Chem. Soc.— 1972.— 94,
N 17.- P. 6165—6170.
474. Wang S.-S.H Ibid.— 1973.— 95, N 4.—P. 1328—1333.
475. Mergler M., Tanner R., Gosteli J.t Grogg P.H Tetrahedron Lett.—
1988.-29, N 32.—P. 4005—4008.
476. Pictta J. M., Marshall G. R.H J. Chem. Soc. (D).— 1970.— N 7.—
P. 650—651.
477. Southard G. L., Brooke G. S.t Pettee J. M.H Tetrahedron.— 1971.—
27, N 13.—P. 2701—2703.
478. Tesser G. I., Buis J. T. W. A. R. M., Wolters E. T. M.,
Bothe-HelmsE. G. A. M.H Tetrahedron.— 1976.— 32, N 9.— P. 1069—
1072.
479. Tesser G. T., Ellenbrock B. W. J.// Peptides 1966 / Eds. Beierman H. C.,
Linde van den W., Massen W.— North-Holland : Amsterdam.— 1967,
144 p.
480. Даванков В. A., Рогожин С. В., Коршак В. В., Цюрупа М. П.Н Изв.
АН СССР. Сер. хим.— 1967.—: №7.— С. 1612—1614.
481. Yamashiro D., Li С. H.ll J. Amer. Chem. Soc,— 1973.— 95, N 4,—
P. 1310—1315.
482. Gisin B. F.H Helv. Chim. acta.— 1973.— 56, N 5.—P. 1476—1482.
483. Roeske R. W., Gesellchen P. D.H Tetrahedron Lett.— 1976.— N 38.—
P. 3369—3372.
484. Куликов С. В., Соколова H. Ю., Леонова E. Б., Самаруева H. A.H
Биоорган, химия.— 1989.— 15, № 3.— С. 348—353.
485. Gutte В., Merrifield R. B.H J. Biol. Chem.— 1971.— 246, N 6.—
P. 1922.— 1941.
486. Ueki M., Amemiya M.H Tetrahedron Lett.— 1987.— 28.— N 52.—
P. 6617—6620.
487. Barany 6.//Int. J. Peptide and Protein Res.— 1987.— 30, N 6.—
P. 705—739.
488. Kaizer E.t Colescott R. L.t BossingerC. D., Cook P. I JI Anal, Bio-
chem.— 1970.— 34, N 2.— P. 595—598.
351
489. Sarin V. К*, Kent S. В., Tam J. P., Merrifield R. В.// Ibid.— 1981.—
117, N 1,—P. 147—157.
490. Christensen Т.П Acta Chem. Scand.— 1979,— B33, N 10.— P. 763—766.
491. Felix A. M., Jimenez M. //.//Anal. Biochem.— 1973.— 52, N 2.—
-P. 377—381.
492. Gisin B. F.H Anal. Chim. Acta.— 1972,— 58.— P. 248—249.
493. Heavner G. A., Doyle D. L., Riexinger D.// Tetrahedron Lett.— 1985.—
26, N 38.- P. 4583-4586.
494. Yajima H„ Fujii N.H Biopolymers.— 1981.— 20,— P. 4583—4586.
495. Nispen J. IF., Polderdijk J. P.t Jansen W. P> A., Greven H. M./l Rec,
trav. chim. Pays-Bas.— 1981.— 100, N 11.—P. 435—446.
496. Barton M. A., Lemieux R. U.f Savoie J. У JI J. Amer. Chem. Soc.—
1973.— 95.— N. 14.— P. 4501—4506.
497. Moore G. J., Kwok Y. CJI Can. J. Biochem.— 1980.— 58, N 8.—
P. 641—643.
498. Schlatter J. M.t Mazur R. H.U Tetrahedron Lett.— 1977.— N 33.—
P. 2851—2852.
499. Rich D. M., Gurwara S. КЛ Ibid.— 1975.— N 5.— P. 301—304.
500. Петров P. В., Хаитов P. M.H Успехи соврем, биологии.— 1979.—
88, № 6.—С. 307—321.
501. Han К.-К., Richard C., Delacourte A.H Int. J. Biochem,— 1984.—
16, N 2.— P. 129—145.
502. Pfaff E.ll EMBO J.— 1982.— N 1.— P. 869—874.
503. Wang K., Richards F. M.H J. Biol. Chem.— 1975.— 250, N 4.—
P. 6622—6626.
504. Hill M. Bechet J. J., D'Albis A.H FEBS Lett.— 1979,- 102.—
P. 282—286.
505. Lomant A., FairbanksG.il J. Mol. Biol.— 1976.— 104, N 3,— P. 243—
261.
506. Staros J. VJI Biochemistry.— 1982.— 21, N 17.—P. 3950—3955.
507. Гершкович А. А., Радавский Ю.Л., Партешко А. В.t Гончаренко В. С.
// Биоорг. химия.— 1989.— 15, № 8.— С. 1056—1059.
508. Methods in Immunology and Immunochemistry I Eds C. A. Williams,
M. W, Chase.—New York; London, 1967.—Vol. 1.— P. 152—153.
509. Kitagawa T., Aikawa Т.Н J. Biochem.— 1976.— 79, N 5.— P. 233—
233.
510. Yoshitake S., Imagava M., Ishikawa I.H Anal. Lett.— 1982.— 15,
N 2.—P. 147—160.
511. Aidwin L., Nitecki D. E.//Anal. Biochem.— 1987.— 164, N 5.—
P. 494—501.
512. Bernatowicz M. S.> Matsueda G. R.ll Ibid.— 1986,— 155, N 1,—
P. 95—102.
513. Carlsson J., Drevin H., Axen RJI Biochem, Jt— 1978.— 173, N 1,—
p 723________737.
b]A. 'Traut R- R>, Bollen A., San T. T. et al.H Biochemistry.— 1973.—
12, N 12.— P. 3266—3273.
515. Duncan R. J. S„ Weston P. D., Wrigglesworth R.H Anal. Biochem.—
1983.— 132, N 1.— P. 68—69.
516. Drijfhout J. W„ Perdijk E. W.t Weiger W. J., Bloemhoff W.H Int.
J. Peptide and Protein Res,— 1988.— 32, N 3.— P. 161—166.
517. Lindner W.. Robey F. A.//Ibid.— 1987.— 30, N 6.—P. 794—800.
518, bernatowicz M. Matsueda G. R.ll Biochem. Biophys. Res. Com-
muns.— 1985.— 132, N 3.—P. 1046—1050.
559 A lb eric io F., Andrea D., Giralt E. et olJl Int. J. Peptide and Protein
Res.— 1989.— 34, N 2.— P. 124—128.
520. Андреев Q. M., Сидорова M< B.> Ракова О\ А. и dp JI Биоорг, химия,—
1987,— 13, № 5,— С. 696—700.
352
621. Цветков Д, Е,, Андреев С. М., Фонина Л. А. идр.Н Тамже.— 1989.—
15, № 2.— С. 155—156.
522. Bhatnagar Р. К., Nitecki D. Е,, Raubitschek АЛ Peptides. Synthesis —
structure — function: Proc. 7th Amer. Pept. Symp./ Eds D. H. Rich.,
E. Gross.— Rockford IL: Piere Chemical Company, 1981.— P. 97—100.
523. Peters K-> Richards F< M./I Ann. Rev. Biochem.— 1977.— 46, N 6.—
p 523________551,
524. Yip C. C.t Yeung C. W.> Moule M.H], Biol. Chem.— 1978.— 253,
N 8.— P. 1743—1745.
525. Sutoh K., Matsuzaki FM Biochemistry.— 1980.— 19, N 11.—
P 3878_______3882
526. NgoT .T .,YamC. FLenhoff H. M., Ivy J M I. Biol. Chem.— 1981.—
256, N 12.—P. 11313—11318.
527. Lotnani A. J., Fairbanks GM J. Mol. Biol.— 1976.— 104, N 3.—
P. 243—261.
528. Fahrenholz F., Thierauch K.-HM Int. J. Peptide and Protein Res.—
1980.— 15, N 5.— P. 323—330.
529. Escher E. H. F., Robert H.> Guillemette GM Helv. chim. acta.— 1979.—
62, N 4.—S. 1217—1222.
530. Sagar A. J,t Mahalakshmi Y, V.» Nagaraj R., Pandit A.- W.ll Int.
J. Peptide and Protein Res.— 1989.— 33, N 6.— P. 452—456.
531. Strecker H.-J. Brussow H., Sare K.> Werchau НЛ J. Cell. Biochem.—
1986.— 31, N 3.—P. 277—287.
532. Keller O., Rudinger J.// Helv. chim. acta.— 1975.— 58, N 2.—
S. 531-541.
533. Matsueda R., Walter RM Int. J. Peptide and Protein Res.— 1980. —
16, N 5.—P. 392—401.
534. Matsueda R., Aiba КЛ Chem. Lett.— 1978.—N 9.—P. 951—952.
535. Bernatowicz M. S., Matsueda R,f Matsueda G. RM Int. J. Peptide and
Protein Res.— 1986.— 28, N 2.—P. 107—112.
536. Kennedy R. C., Henkel R, D., Pauletti D. et al.// Science.— 1986.—
231, N 4745.— P. 1556—1559.
537. Цветков Д. E., Ильина А. В., Андреев С. M. и дрЛ Биоорган, хи-
мия,— 1990,— 16, № 2,—G, 166—178.
ПРИЛОЖЕНИЕ 1
Основные реагенты для синтеза пептидов
Реагент Молеку- лярная масса Т. п„ ®С Т. кип., °C dz, г/см Количество 10 ммоль ре- агента , г (мл)
Дициклогексил карбодиимид 206,32 35 149—152/11 мм, рт. ст. — 2,06
1-Этил-З (3-ди- метиламинопро- пил) карбоди- имид, хлоргид-’ рат 188,56 113—115 1,89
N-Этиоксикар- 247,29 бонил-2-это- кси-1,2- ди гид- рохинол (EEPQ) 63,5—65 2,47
Изобутилхлор- формиат 136,58 —- 128—129 1,045 1,37(1,3)
Пивалоилхло- рид 120,57 — 105—108 0,98 1,21 (1,25)
Пентафтор- фенол 184,03 35—36 142—143 — 1,84
п-Нитрофенол 139,11 112—114 —. — 1,39
N-Оксисукци- нимид 115,09 95—96 — — 1,15
1-Оксибенз- триазол . Н2О 153,14 155—158 — — 1,53
Карбояилди- имидазол 162,15 115,5—116 — — 1,62
Дифенилфосфо- разид 275,20 — 152—155/0,15 мм. рт. ст. 1,273 2,75 (2,16)
трет-Бутил- нитрит 103,12 — 61—63 0,868 1,03(1,2)
Гидразингидрат 50,06 118—122 1,030 0,5 (0,48)
Карбобензо- ксихлорид 170,6 — 178—180 1,203 1,7(1,42)
Ди-трет-бутил- пирокарбонат - 218,25 22—24 (21—22) 75—78/3 мм. рт. ст. 0,94 2,18(2,3)
трет -Бути л- карбонил гидразид 132,16 4 39—40 70—72/3,5 мм. рт. ст. (63—65/0,1 мм, рт. ст. 1,32
о-Нитрофенил- сульфенил- хлорид 189,62 70—72 — — 1,89
Тритилхлорид 278,78 109—111 —— — 2,79
9-Флуоренилме- тилоксикарбо- нил хлорид 258,71 61,5—63 — 2,59
9-Флуоренил- метилсукцини- мидилкарбонат 337,33 149—150 3,37
854
Продолжение прил. 1
Реагент Молеку- лярная масса Т. п„ °C Т. кип., °C ^8, г/см Количество 10 ммоль ре- агента, Г (МЛ)
п-Толуолсуль- фохлорид 190,65 66—69 — — 1,9
Трифторуксус- ный ангидрид 210,04 —— 39—40 1,506 2,1 (1,39)
п-Нитробензил- хлорид 171,58 70—73 — — 1,72
Фенанилбромид 199,05 49—50 —> — 1,99
Дифенилмети- ловый спирт 184,24 65—67 — — 1,84
Триметилхлор- сплан 108,64 — 56—57 0,857 1,09 (1,27)
Трифторуксус- ная кислота 114,03 — 71—73 1,49 1,14(0,77)
Метансульфо- кислота 96,10 17-19 120—122/0,1 мм. рт. ст. 1,483 0,96 (0,65)
Трифторметан- сульфокислота 150,08 — — 1,708 1,5(0,88)
Триэтнламин 101,10 —- 89,4 0,728 1,01 (1,39)
N-Метилмор- фолин 101,15 — 113—116 1,434 1,01 (0,71)
Диизопрэпил- этиламии 129,25 — 123—126 0,755 1,29(1,7)
Дициклогекси- ламин 181,32 — 119—120/10 мм. рт. ст. 0,913 1,81 (2,0)
Циклогексила- 99,18 —-• 134—135 0,866. 0,99 (1,14)
мин
ПРИЛОЖЕНИЕ 2
Таблица молекулярних масс отдельных аминокислотных звеньев
полипептидной цепи — HN—CHR—СО— и защитных групп
для расчета молекулярных масс полипептидов *
Остатки аминокис- лот и защитные группы Сокра- щенное обозна- чение Молеку- лярная масса Остатки аминокислот и защитные группы Сокра- щенное обозна- чение Молеку- 1 лярная масса
1. Остатки амино- Глутаминил GLN, Q 128,1
кислот Глутамил GLU, Е 129,1
(—Ml—CHR- Глицил GLY, G 57,1
СО—) Гистидил HIS, Н 137,2
Аланил ALA, А 71,1 Гидроксипролил HYP, 113,2
Аргинил ARG, R 156,2 Изолейцил ILE, I 113,2
Аспарагинил ASN, N 114,1 Лейпил LEU, L 113,2
Аспартил ASP, D 115,1 Лизил LYS, К 128,2
Валил VAL, V 99,1 Метионил MET, M 131,2
355
Продолжение прил. 2
Остатки аминокислот и защитные группы Сокращенное обозначение Молекуляр- ная масса Остатки аминокислот и защитные группы Сокращенное обозначение Молекуляр- ная масса
Орнитил ORN, 114,2 mpem-Бутилок- Вос 100,1
Пироглутамил Пролил pGLU, PRO, Р 111,1 97,1 сикарбонильная о-Нитрофенил- Nps 153,2
Серил Тирозил SER, S TYR. Y 87,1 163,2 сульфенильная Тритильная Trt 242,3
Треонил .THR, T 101,1 Трифтораце- TFA 96,0
Триптофанил Фенилаланил TRP, W PHE, F 186,2 147,2 тильная 9-Флуоренилме- Fmoc 222,2
Цистеинил 2. С-Защитные группы (—С—R1) II О Амид CYS, C —NHa 103,2 17,0 токсикарбониль- ная /г-Толуолсуль- фонильная Дифенилизопро- пилоксикарбо- нильная Ацетильная Tos Врос Ac 154,2 238,3 42,0
Метиловый OMe 32,0 Формильная Form 28,0
эфир Этиловый эфир OEt 46,0 Дансильная 2,4-Дпнитро- Dns Dnp 233,3 166,1
Бензиловый эфир OBzl 108,1 фенильная Бензоильная Bz 104,1
п-Нитробензи- ONB 152,1 Сукцинильная Sue 100,1
ловый эфир трет-Бутило- вый эфир п-Нитрофенило- OBi? ONp 73,1 138,1 Глутарильная 4. Другие замес- тители Аминогруппа GPut NHS 114,1 15,0
вый эфир Пентафторфени- OPfp 183,0 Бром Хлор Bp Cl 78,9 34,5
ловый эфир N-Оксисукцини- OSu 114,1 Фтор Оксигруппа F OH 18,0 16,1
мидный эфир Гидразид —NHNHt 32,0 Нитрогруппа . Метильная NOS Me 45,0 14,0
БОК-гидразид п-Нитроанилид Boc-NHNH- pNA • 131,1 138,1 группа Бензильная Bzl 90,1
f-Нафтиламид 4-Метилкума- NA MCA 143,2 175,2 группа Метоксигруппа OMe 30,1
рил-7-амид 3. N-Защитные группы (R’-NH-) Карбобенэокси Z 134,1 п-Нитробен- зильная группа Сульфогруппа Ацетамидоме- тильная группа NB SOfH A cm 135,1 80,1 71,0
356
Продолжение прил, 2
Остатки аминокислот
и защитные группы
Остатки аминокислот
и защитные группы
В 5
§5
< в
Бензамидоме- Ват 133,1 Мезити- Mts 212,3
тильная группа лен-2-сульфо-
Триметоксибен- TMBS 230,3 группа
зосульфогруппа
* Для вычисления молекулярной массы пептидов нужно: 1) суммировать молеку
лярные массы аминокислотных остатков (поз. 1); 2) прибавить молекулярную массу
С-концевых заместителей (прибавляют 18, если имеется свободная карбоксильная груп-
па, 17 — в случае амида, 32 — в случае метилового эфира и т. п.), при этом молеку-
лярная масса заместителя увеличена на 1, что учитывает добавления атома водо-
рода к концевой аминогруппе; 3) прибавить молекулярные массы N-концевых замес-
тителей (поз. 3) и заместителей в боковых радикалах (поз. 4), а также других замес
жителей (поэ. 4), молекулярная масса которых уменьшена на 1, что учитывает за-
мещение ими атома водорода.
ПРИЛОЖЕНИЕ 3
Распространенные системы растворителей для тонкослойной
хроматографии защищенных аминокислот и пептидов на силикагеле
(456]
Система растворителей Соотношение в объемных частям
н. Бутанол — уксусная кислота — вода * 4:1:1 4:1:2; 10:30:35
н. Бутанол — пиридин — уксусная кисло- та — вода * Этилацетат — пиридин — уксусная кисло- та — вода * 15:10:3:12; 15:12:3:10 5:5:1:3; 60:20:6:11
Пиридин — уксусная кислота — вода Хлороформ — метанол * Хлороформ — метанол — уксусная кислота 10:6:3; 4:1:1 1:1; 2:1; 4:1; 5:1. 5:4:1; 7:1:2; 82:15:3
Хлороформ — метанол —- аммиак Изонропанол — уксусная кислота — вод а /пг/п-Бутанол — уксусная кислота — вода 12:9:4; 50:47:3 7:1:2: 10:1:1 10:1:1; 4:1:1; 4:1:2 10:3:9; 10:1:3
Изопропанол — аммиак — вода Гептан — трет-бутанол — уксусная кисло- та Изопропанол — аммиак — вода н. Бутанол — уксусная кислота — этила- цетат — вода Метиленхлорид — метанол ** 7:1:2 3:1:1; 5:1:1 7:1:2 1:1:1:1 9:1
mpem-Бутанол — метилэтилкетон — вода — 4:3:2:1
аммиак Этилацетат — хлороформ — метанол 5:3:2; 6:3:1
Примечание: • наиболее распространенные системы, ♦* система удобна для
аналитического и препаративного разделения аргининсодержащил пептидов.
ОГЛАВЛЕНИЕ
Список сокращений 3
Введение ..................................... ♦ 5
Глава 1. МЕТОДЫ ОБРАЗОВАНИЯ ПЕПТИДНОЙ СВЯЗИ 12
Способы активации карбоксильной группы 12
Механизмы активации карбоксильной группы 13
Смешанные ангидриды ....................................... 28
Метод активированных эфиров 39
Методики синтеза реагентов 46
Карбодиимидный метод...................................... 46
Синтез нуклеофильных добавок .............................. 49
Синтез с использованием ЭЭДХ............................... 51
Карбонилдиимидазолидный метод ...........................' 52
Азидный метод.............................................. 53
Метод смешанных ангидридов ................................ 56
Смешанные ангидриды ......................... , 57
Карбоксиангидридный метод ...............'................. 63
Метод активированных эфиров................................ 70
Модифицированные пентафторфениловые эфиры защищенных ами-
нокислот ................................................ 78
Синтез пептидов с незащищенной карбоксильной группой при по-
мощи активированных эфиров................................. 81
Ускоренные методы синтеза пептидов......................... 85
Способы активации аминогруппы............................... 87
Механизмы активации аминогруппы............................ 90
Методики.........«......................................... 92
Г л а в а 2. ЗАЩИТА ФУНКЦИОНАЛЬНЫХ ГРУПП АМИНО-
КИСЛОТ. ЗАЩИТА АМИНОГРУППЫ ............................. 95
Основные способы удаления защитных групп и механизмы соответ-
ствующих процессов .................................. 104
Защитные группы, удаляемые кислотами ............... . 104
Защитные группы уретанового типа...................... 104
Защитные группы алкильного типа . 158
Защитные группы ацильного типа........................ 108
Защитные группы, удаляемые основаниями................. НО
Защитные группы ацильного типа^. ............ НО
Защитные группы уретанового типа................... Н1
Защитные группы, ощепляемые каталитическим гидрированием ИЗ
Защитные группы, удаляемые ультрафиолетовым облучением 114
358
Методики синтеза реагентов, используемых для блокирования
аминогрупп пептидов и аминокислот........................... 11г
Карбобензоксигруппа...............1 15
/иргт-Бутплоксикарбонильная группа.......................... 13?
р-Нитрофеиищульфенильная группа (Nps)....................... 145
2-(п-Дифенил)-изопропилоксикарбонильная группа (Врос) (пред-
ложена Забером и Изелином в 1968 г. [178, 265])............. 151
Трифенилметильная (третильная) группа....................... 156
n-Толуолсульфонильная группа (Tos)................., * , • 160
Трифторацетильная защитная группа (TFA)..................... 161
9-Флуоренилметилоксикарбонильная группа (Fmos) ............. 166
Гл а в а 3. ЗАЩИТА КАРБОКСИЛЬНОЙ ГРУППЫ .................... 176
Основные способы получения сложноэфирных С-защитных групп 184
Этерификация спиртами в присутствии кислых катализаторов 184
Этерификация солей защищенных аминокислот алкилгалогенида-
ми.......................................................... 185
Этерификация аминокислот эфирами ц-толуолсульфокислоты 186
Этерификация аминокислот алкенами ......................... 186
Этерификация защищенных аминокислот в присутствии карбоди-
имидов ..................................................... 187
Свойства защитных групп и способы их удаления .......... » 187
Защитные группы, удаляемые основаниями....................♦ 189
Защитные группы, удаляемые кислотами......................» 192
Защитные группы, удаляемые восстановлением ................. 194
Другие способы удаления защитных групп . ............... . . 195
Препаративная часть ........................................ 19&
Метиловые и этиловые эфиры аминокислот..................... 19,8
Омыление метиловых и этиловых эфиров пептидов............... 200
Бензиловые эфиры аминокислот........................ . . . . 202
/1-Нитробензиловые эфиры аминокислот ....................... 203
тргт-Бутиловые эфиры аминокислот............................ 205
Синтез трет-бутиловых эфиров аминокислот.................... 206
Триметилсили тьные эфиры аминокислот........................ 209
Амиды аминокислот и пептидов............................... 210
Сложные защитные группы..................................... 220
Многофункциональные защитные группы...................... . . 220
«Двуступенчатые» защитные группы .......................... 221
Г л а в а 4. ЗАЩИТА БОКОВЫХ ФУНКЦИОНАЛЬНЫХ ГРУПП 222
Защита боковых функциональных групп дикарбоновых аминокис-
лот ........................................................ 223
Защита боковых функциональных групп диаминокарбоновых ки-
слот ....................................................... 240
Защита е-аминогруппы лизина............................♦ . . 240
Защита гуанидиновой группы аргинина ........................ 245
Защита имидазольного кольца гистидина . .................... 253
Защита гидроксильной группы тирозина ....................... 259
Запита гидроксильной группы серина и треонина ....... 263
Защита меркаптогруппы цистеина.............................. 264
Защита амидных групп глутамина и аспарагина................. 269
Защита индольного ядра триптофана........................... 272
Защита боковой группы метионина......................... , 275
Удаление защитных групп безводным фтористым водородом 277
Доугие эффективные реагенты для удаления защитных групп 279
Общая стратегия применения защитных групп................... 283
359
Г лава 5. ТВЕРДОФАЗНЫЙ МЕТОД СИНТЕЗА ПЕПТИДОВ 288
Общие принципы твердофазного метода синтеза пептидов , • . 288
Методы синтеза реагентов................................ 298
Получение полимерных носителей .•..•.»•••»»»•. 298
Наращивание полипептидной цепи .
Методы контроля полноты реакции пептидообразования ..... 307
Отщепление пептида от смолы.............................. 311
Г л а в а б. КОНЪЮГАЦИЯ ПЕПТИДОВ С ВЫСОКОМОЛЕКУ-
ЛЯРНЫМИ НОСИТЕЛЯМИ..................................... 317
Реагенты, используемые для конъюгации ...•.••••••• 317
Гомобифункциональные сшивающие реагенты 319
Гетеробифункциональные сшивающие реагенты ........ 321
Другие методы конъюгации пептидов с белками . ....... 325
Гетеробифункциональные фотореактивные реагенты ....... 326
Синтез М-Вос-[$-(3-нитро-2-пиридилсульфенил)] цистеина 332
Активация карбоксилсодержащих полимеров •..•••••> 335
Список литературы ...................................... 336
Приложение 1 354
Приложение 2 ....... • 355
Приложение 3...............................................357
Научное издание
ГЕРШКОВИЧ Александр Абрамович
КИБИРЕВ Владимир Константинович
ХИМИЧЕСКИЙ
СИНТЕЗ
ПЕПТИДОВ
Оформление художника С. В. Назарова
Сдано в набор 16.03.92. Подп. в печ. 29.09.92. Формат 84Х108/32. Бум. тип. № 1.
Лит. гари. Выс. печ. Усл. печ. л. 18,9. Усл. кр.-отт. 18,9. Уч.-изд. л. 20,3,
Зак. 2—882.
Издательство <Наукова думка». 252601, Киев, ул. Терещенковская, 3.
Отпечатано с матриц Головного предприятия республиканского производственного
эбъединения «Полиграфкнига». 252057, Киев, ул. Довженко, 3, в Жовковской го-
родской типографии 292310 Жовква, Львовской обл., ул. Василианская, 8.
Зак. 2204.