

























































































































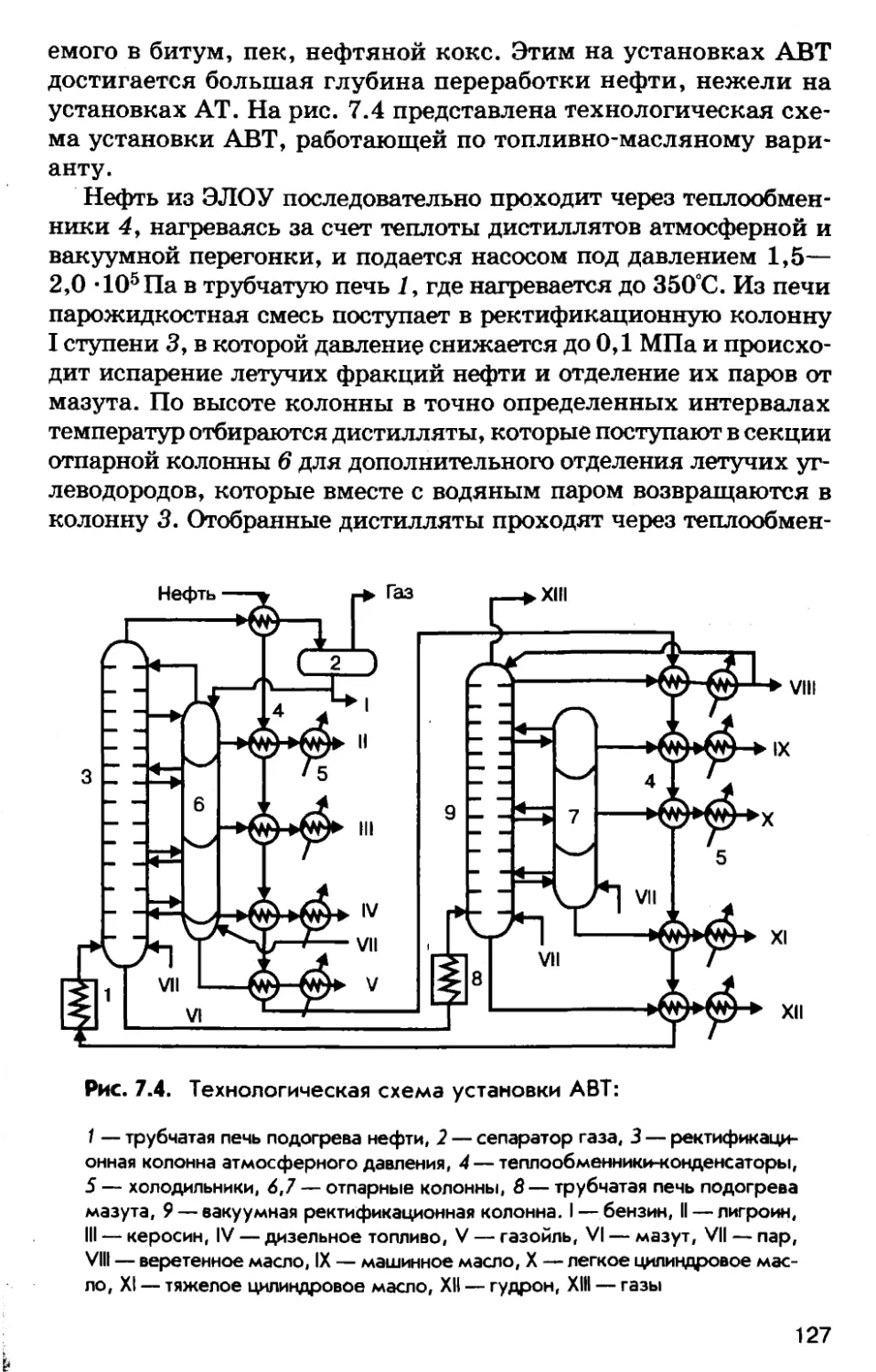



























































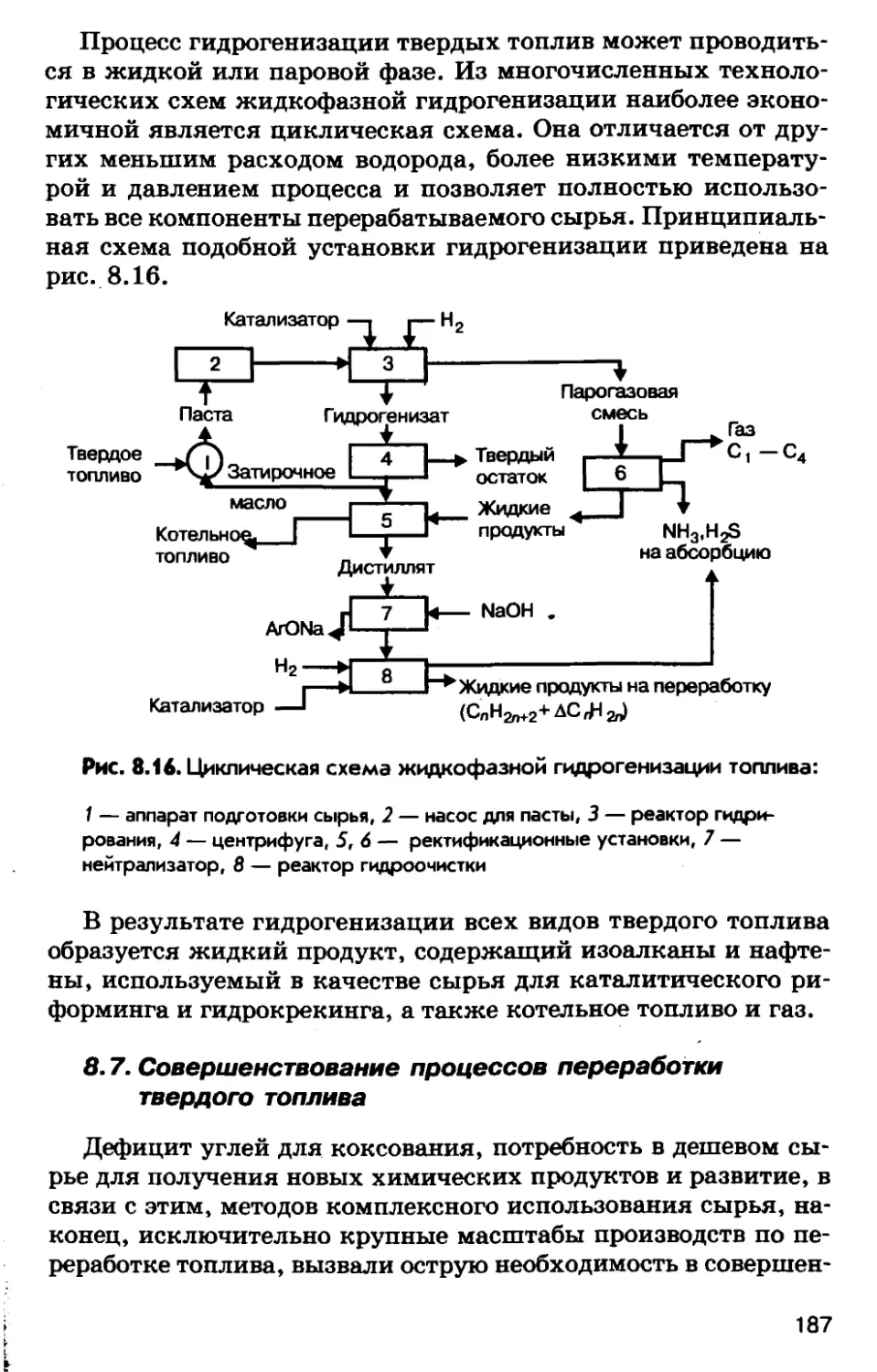

































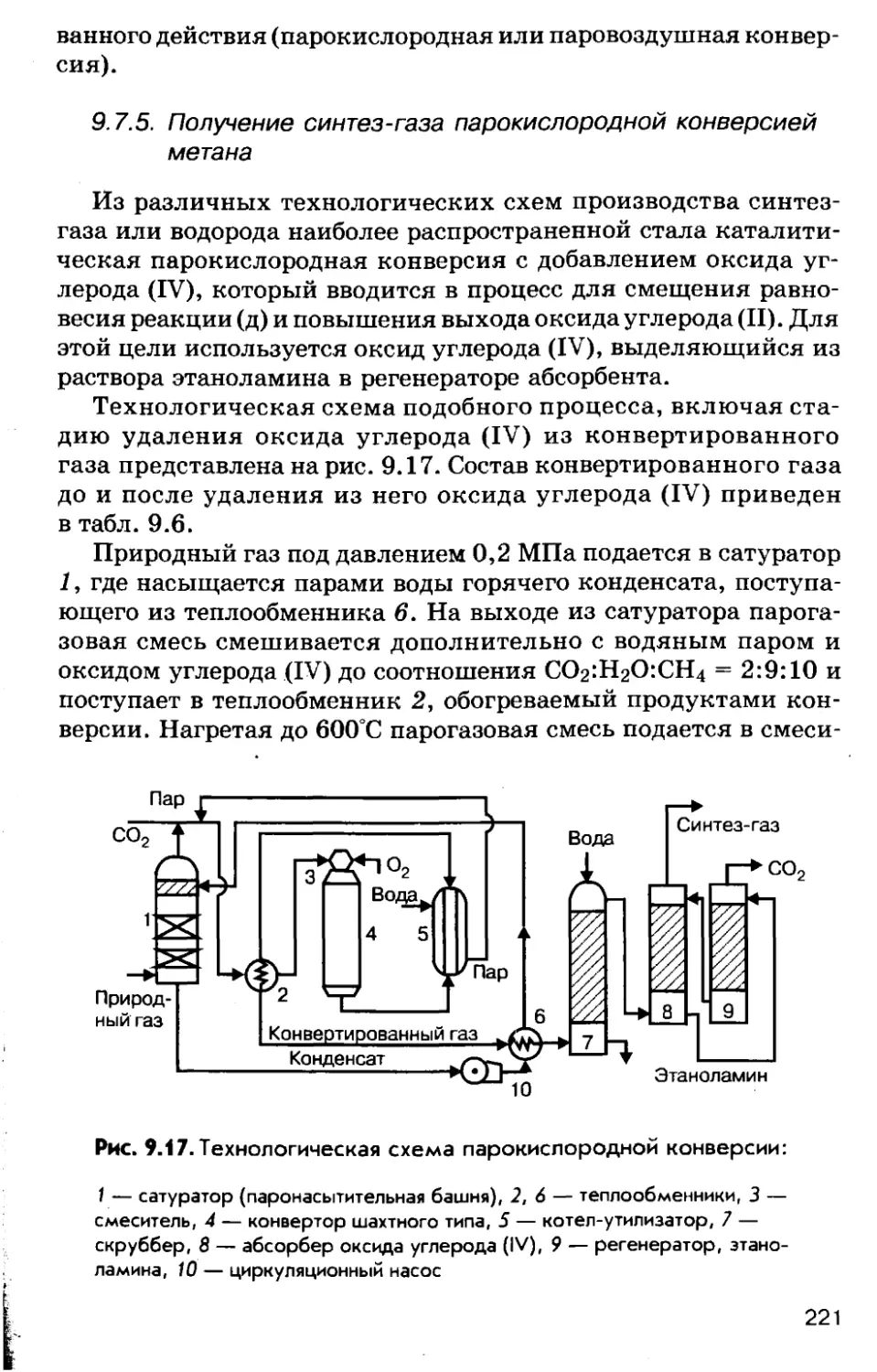
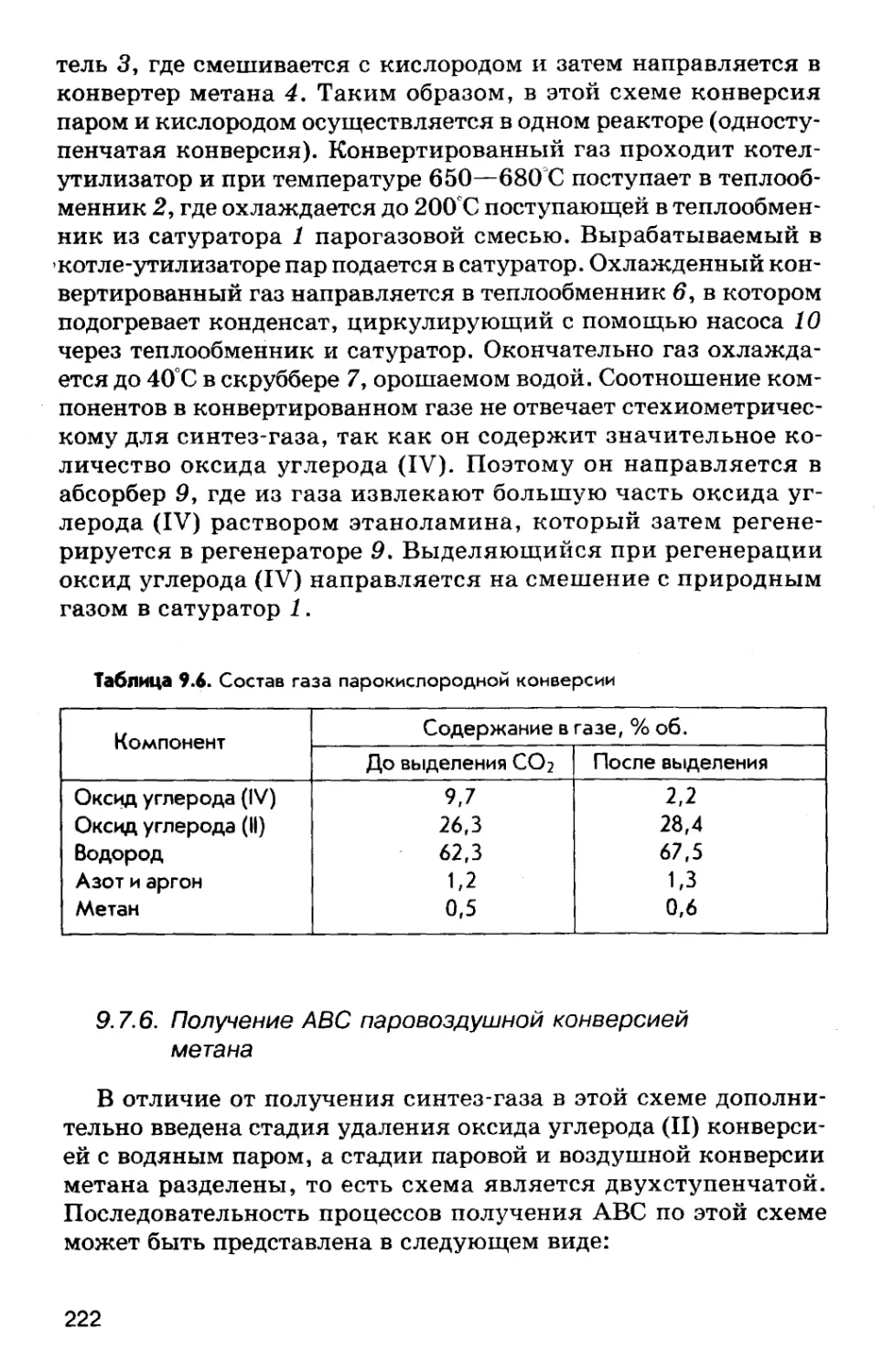










































































































































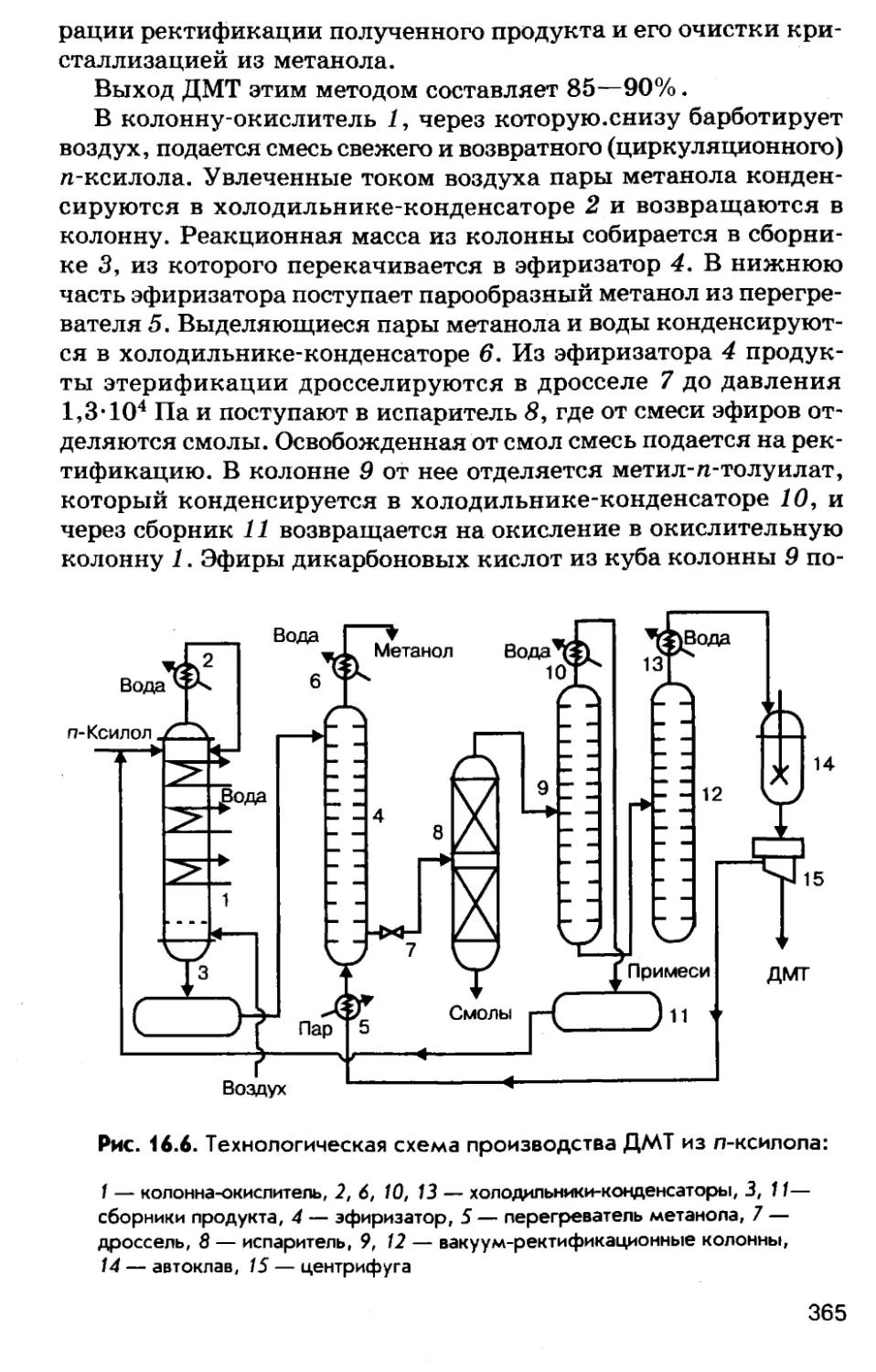





















































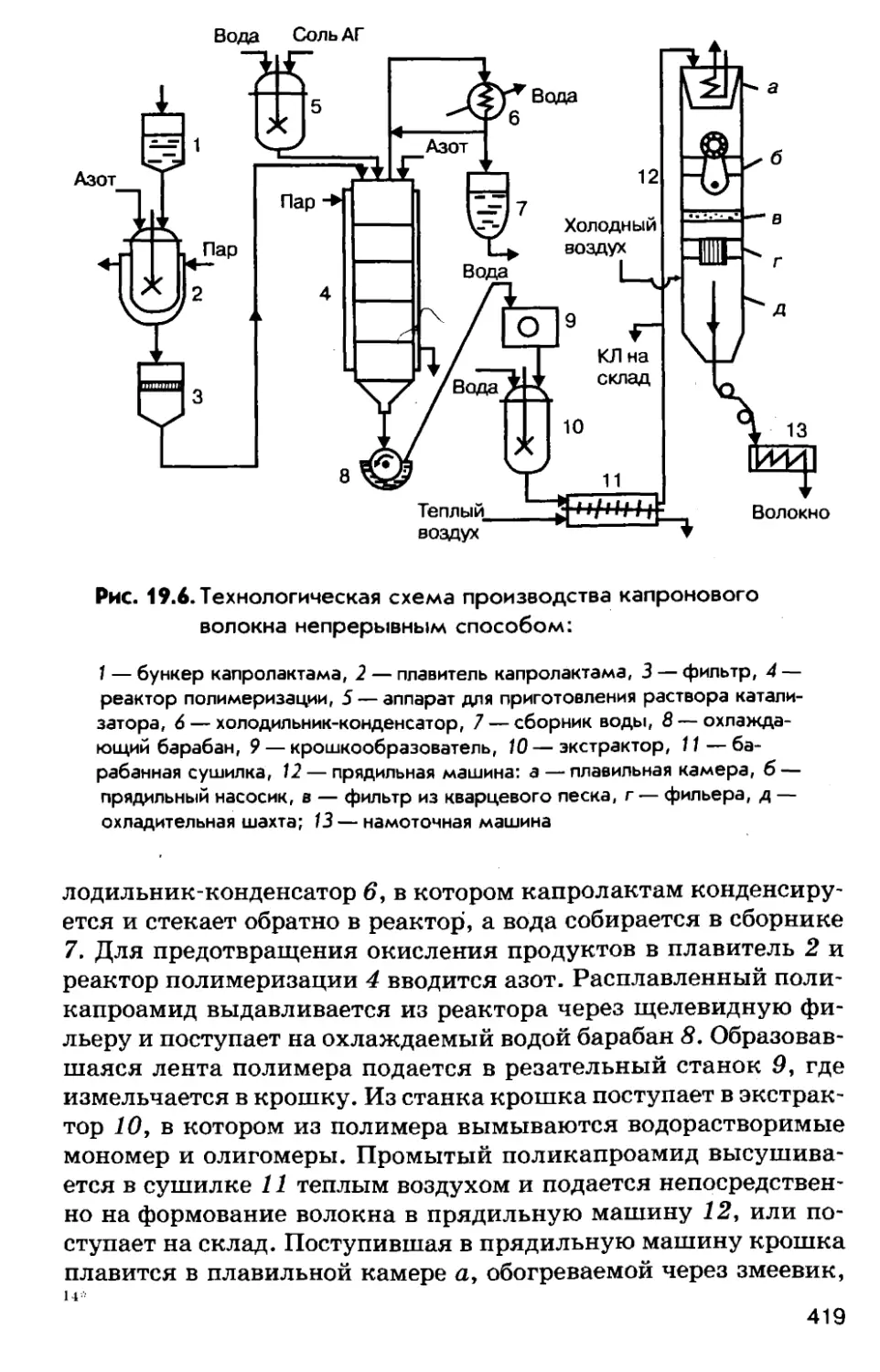





























Автор: Соколов Р.С.
Теги: химическая технология химические производства учебное пособие для студентов
ISBN: 5-691-00355-0
Год: 2000




Текст
УЧЕБНОЕ ПОСОБИЕ ДЛЯ ВУЗОВRC. СоколовХИМИЧЕСКАЯ
ТЕХНОЛОГИЯВ двух томах
Том 2Металлургические процессы
Переработка химического топлива
Производство органических веществ
и полимерных материаловРекомендовано
Министерством образования Российской Федерации
в качестве учебного пособия для студентов
высших учебных заведенийМоскваПШЛЮПЛГМЫЯмаялткяъсюШ
ББК 35я73
С59Рецензенты;
кафедра химии и химической технологии МПГУ
(зав. кафедрой профессор ЭЖ. Нифантг>ев);
доктор педагогических наук,
профессор ПЛ. Оржековский (МОПУ);
доктор химических наук, профессор MJC. Грачев(МПГУ)Соколов Р.С.С59 Химическая технология: Учеб. пособие для студ. высш.
учеб. заведений: В 2 т. — М.: Гуманит. изд. центр BJIA-
ДОС, 2000. — Т. 2: Металлургические процессы. Перера¬
ботка химического топлива. Производство органических
веществ и полимерных материалов. — 448 с.ISBN 5-691-00355-0.ISBN 5-691-00357-7(11).Книга является учебным пособием по курсу «Химическая тех¬
нология» для студентов химических и биолого-химических факуль¬
тетов педагогических вузов. Во втором томе рассмотрены металлур¬
гические процессы: производство черных и цветных металлов, про¬
изводство и обработка стали. Даны сведения о производстве и пере¬
работке нефти, каменных углей и газообразных топлив. Значитель¬
ное внимание уделено технологии производства и применения орга¬
нических веществ, а также полимерных материалов.ББК 35я73© Соколов Р.С., 1999
© «Гуманитарный издательский
центр ВЛАДОС», 1999
ISBN 5-691-00355-0 © Серийное оформление.ISBN 5-691-00357-7(11) Художник Токарев Ю.В., 1999
Часть IМЕТАЛЛУРГИЧЕСКИЕ ПРОЦЕССЫ
Глава IМЕТАЛЛЫ1ш1ш Свойства и классификация металловМеталлы, наряду с древесиной и керамикой, относятся к
числу наиболее распространенных «традиционных* конструк¬
ционных материалов и йзвестны человечеству с глубокой древ¬
ности. Производство металлов по масштабам соизмеримо с про¬
изводством таких промышленных продуктов как цемент, цел¬
люлоза, полимерные материалы. Так, для сравнения, в 1980—
1987 гг. мировое производство составило (млн. тонн в год): чу¬
гуна 509; стали 737; алюминия (без СССР) 12,6; меди (без СССР)
7,65; цемента 1051; бумаги 150,7; пластических масс 93,5. В
Российской Федерации в 1992 году в общем цромышленном про¬
изводстве страны доля черной металлургии составляла 8,6% и
доля цветной металлургии 9,1%. “Значение металлов как важнейших материалов современной
техники и, как следствие, возрастающая роль их в народном
хозяйстве, несмотря на внедрение полимерных материалов и
композитов, обусловлены рядом их специфических качеств. К
таким качествам относятся:— способность к взаимному растворению и образование мно¬
гочисленных сплавов разнообразного состава, что позволяет в
широких пределах изменять в заданном направлении физико¬
механические и физико-химические свойства металлических
материалов;— комплекс ценных механических, физических и химичес¬
ких свойств, в том числе тепловых (высокие теплопроводность
и коэффициент термического расширения, низкая теплоем¬
кость), электрических и магнитных (низкое удельное сопротив¬
ление, способность к термоэлектронной эмиссии, ферро- и па¬
рамагнетизм), механических (упругость, пластичность, проч¬
ность), химических (окисляемость);— возможность фазовых превращений при изменении тем¬
пературы и существование в нескольких полиморфных моди¬
фикациях с различными структурой и свойствами;— способность деформироваться в холодном и горячем состо¬
янии без нарушения сплошности материала.3
Существенную роль имеет также широкое распространение
металлов в литосфере и гидросфере Земли. Металлы составля¬
ют 86% известных химических элементов. К металлам отно¬
сятся: s-элементы, кроме водорода и гелия, все d-элементы, все
/-элементы, часть p-элементов (алюминий, галлий, индий, тал¬
лий).Значение того или иного металла в народном хозяйстве стра¬
ны принято оценивать долей его производства в общем производ¬
стве металлов или в производстве железа и его сплавов. Удель¬
ный вес различных металлов существенно меняется со време¬
нем. Появление новых отраслей техники (ракетостроение, атом¬
ная энергетика, электроника и др.) вызывает потребность в ма¬
териалах с новыми свойствами и стимулирует развитие новых
направлений в металлургии. Так уже после 1945 года промыш¬
ленное значение приобрели такие металлы как титан, молибден,
цирконий, ниобий. В настоящее время в цветной металлургии
производятся более 30 металлов, являющихся редкими элемен¬
тами, и сотни их сплавов. Поэтому доля производства различных
металлов со временем меняется. Например, за последние годы
существенно возросла доля производства алюминия, но практи¬
чески не изменилась доля производства меди.Научно обоснованной классификации металлов не суще¬
ствует. В основу классификации положен промышленный
принцип, учитывающий сложившуюся структуру металлур¬
гической промышленности, распространение в природе и свой¬
ства металлов. На рис. 1.1 представлена промышленная клас¬
сификация металлов.В соответствии с промышленной классификацией металлы
делятся на черные, к которым относятся железо и его сплавы,
марганец и хром, производство которых связано с производ¬
ством чугуна и стали, и цветные. Термин «цветные металлы*
достаточно условен, так как из всех металлов этой группы толь¬
ко золото и медь имеют ярко выраженную окраску. Из цветных
металлов основные тяжелые металлы получили название из-за
больших («тяжелых») масштабов производства и потребления.
Малые тяжелые металлы являются природными спутниками
основных тяжелых металлов, их получают попутно и в мень¬
ших количествах.1.2. Металлические рудыСырье в производстве металлов — металлические руды. За
исключением небольшого числа (платина, золото, серебро, иног-4
МЕТАЛЛЫ —ЧЕРНЫЕ: железо и его сплавы, марганец, хром+> Основные тяжелые: медь, свинец, никель,
цинк, олово
+> Малые тяжелые: висмут, мышьяк, сурьма,
ртуть, кадмий, кобальт
+Легкие: алюминий, магний, титан, натрий,
калий, барий, кальций, стронций
ЦВЕТНЫЕ: - ►! » Благородные: золото, серебро, платина,платиноидыРедкие■Тугоплавкие: вольфрам, мо¬
либден, тантал, ванадий,
ниобий, цирконий
Легкие: литий, бериллий,
рубидий, цезий
Рассеянные: галлий, индий,
таллий, германий. гафний,
рений, селен, теллур
Редкоземельные: скандий,
иттрий, лантан, лантанойды
Радиоактивные: радий, уран,
торий, актиний, трансураныРис. 1.1. Промышленная классификация металловда ртуть и медь) металлы находятся в природе в виде химичес¬
ких соединений, входящих в состав металлических руд.Металлической рудой называется горная порода, содержа¬
щая в своем составе один или несколько металлов в таких со¬
единениях, количествах и концентрациях, при которых воз¬
можно и целесообразно их извлечение при современном уровне
обогатительной и металлургической техники.Понятие «руда» имеет геологический, технический и эконо¬
мический смысл. Так, например, с появлением ртутного и циа^
нидного методов извлечения золота ранее неиспользуемые ста¬
рые отвалы приобрели промышленное значение и стали рас¬
сматриваться как золотоносные руды. Металлические руды
классифицируются по следующим признакам:1. По качеству и количеству металла руды делят на промыш¬
ленные и непромышленные. К промышленным относят те руды,
в которых содержание металла превышает его рентабельный
минимум^ то есть то минимальное содержание основного метал¬
ла, которое определяет возможность и целесообразность метал¬
лургической переработки данной руды. По мере развития про¬
изводства рентабельный минимум снижается. В конце XIX сто¬
летия к промышленным рудам относили горные породы с содер¬
жанием меди выше 1,5%. В настоящее время эта величина сни-5
до 0,4% • Снижению рентабельного минимума способствуй
tep ш кт вованиюбо гатителной и металлургической тех -
и повышение комплексности использования металл ичес-
Тжах руд.J 2. П о числу содержащихся в руде металлов их делят на мо¬
нометаллические (простые) и полиметаллические (комплекс¬
ные). К полиметаллическим относится большинство руд цвет¬
ных металлов (медные, медно-никелевые и свинцово-медно-
цинковые руды), содержащие до 10—15 различных металлов.
Полиметалличность большинства руд делает экономически не¬
обходимым их комплексное использование, то есть организа¬
цию безотходных или малоотходных производств.3. По содержанию металла руды подразделяют на богатые,
средние и бедные. Руды цветных металлов, как правило, отно¬
сятся к очень бедным, однако сопутствующие основному метал¬
лу в них другие элементы по ценности могут значительно пре¬
восходить основной компонент руды (табл. 1.1).Таблица 1.1. Ценностная структура медно-цинковой рудыКомпонент рудыСодержание,%Ценность,%Медь2,526,0Цинк2,515,6Сера40,041,7Золото2'10-410,4Серебро3-10"36,34. По форме нахождения металла руды делятся на:— самородные у содержащие металлы в свободном состоянии
(Me);— окисленные, в которых металлы присутствуют в форме
различных кислородных соединений (оксидов МеОл, гидрокси¬
дов Ме(ОН)д, солей многоосновных кислородных кислот
МеМепОп, МеЭОп);— сульфидные у содержащие сульфиды (MeS) и полисульфи¬
ды (MeSn) металлов;— галогенидные, в которых содержатся соли галогенводород-
яых кислот (МеГл)., Использование металлов в народном хозяйстве зависит не
только от их специфических свойств, но и от разведанных за¬
пасов и доступности руд и возможности промышленного выде¬
ления металлов из их соединений в этих рудах. В земной коре
содержание металлов весьма неравномерно. Наиболее распрос¬
транены элементы, имеющие нечетные и малые номера в пери¬
одической таблице. В табл. 1.2 приведено содержание метал¬
лов в земной коре в порядке его убывания.Доступность металлических руд как сырья для производства
металлов зависит от их состава (бедные, богатые), географичес¬
кого положения региона их нахождения и распределения в зем¬
ной коре.Руды или образуют в земле естественные скопления (место¬
рождения), или содержатся в очень небольших концентрациях
в виде изоморфных примесей в основных минералах (рассеян¬
ные металлы).Таблица 1.2. Распространение металлов в земной кореМеталлСодержание, %МеталлСодержание, %Алюминий7/50Вольфрам7-10“3Железо4,70МолибденМО"3Кальций3,40Свинец8-10“4Натрий2,64Олово6-10“4Калий2,40Уран5-10”4Магнии1,94Селен8-1Q-JТитан0,58Платина2'10“5Цинк0,02Серебро4-1 O'6Никель0,018Золото5-10”7Медь0,01РенийМ0“7Месторождением металлов называется скопление металли¬
ческих руд, пригодное для их промышленного использования.
Естественно, что концентрация металлов в месторождениях
значительно выше их средней концентрации в земной коре.
Богатство месторождения оценивают его металлоносностью (М),
представляющей отношениекларк месторождения” кларк планеты*Для металлических руд различают также:— запасы, то есть выявленные количества руды, для кото¬
рых известны метод и стоимость извлечения из них металла, и— потенциальные ресурсы, то есть количества руды, распо¬
ложение и метод добычи которых неизвестен.В табл. 1.3 приведено содержание важнейших металлов в
земной коре и их рудах.7
fjtwn l.3. Содержание металлов в земной коре и рудахМеталлСодержание металла, тКларк металла,
г/тв земной корев рудахАлюминий1,5-Ю165-10®81000Железо8,8* 1015МО1146000Цинк2,4-10138,2-10780Медь1,8-10131,8-10870Основную массу металлов извлекают из руд, содержащихся
в земной коре (литосфере). Потенциальным источником метал¬
лов можно считать и воды Мирового океана, содержащие до
3,5% растворенных солей, а также залежи металлсодержащих
конкреций на дне океана.1.3. Металлургические процессыВ основе производства металлов лежат металлургические
процессы, то есть технологические процессы извлечения метал¬
лов из руд и отходов производства. В общем случае металлур¬
гический процесс включает три последовательных стадии:— подготовка руды — превращение ее в состояние, обеспе¬
чивающее извлечение из руды металла;— восстановление химического соединения, в виде которо¬
го металл содержится в руде, до свободного металла;— вторичная обработка полученного металла.Подготовка руды состоит из ряда механических и физико¬
химических операций, содержание которых зависит от состава
руды и формы химического соединения металла в ней. К таким
операциям относят измельчение или укрупнение, классифика¬
цию и обогащение руды, а также превращение содержащего ме¬
талл соединения в форму, пригодную для восстановления. Не¬
обходимость последней операции связана с тем, что восстанов¬
лению подвергаются преимущественно оксиды, реже галогени-
ды металлов, поэтому все остальные соединения (сульфиды,
гидроксиды) должны быть переведены в них. Это достигается
воздействием на обогащенную руду высокой температуры или
соответствующих реагентов: ^ МеОТемпература, реагент
В соответствии с методом технологические процессы подго¬
товки руды подразделяются на пирометпаллургические и гидро¬
металлургические.Пирометаллургические процессы проводятся при высоких
температурах с полным или частичным расплавлением руды,
К ним относят:— обжиг — процесс, проводимый при высокой (500—1200°С)
температуре в твердой фазе с целью изменения химического
состава руды. Условия обжига зависят от состава руды и назна¬
чения процесса. Руды, содержащие сульфиды металдов, под¬
вергаются окислительному обжигу до их оксидов; руды для
последующего магнитного обогащения подвергаются восстано¬
вительному обжигу;— восстановительная плавка — процесс восстановления
оксидов металлов при температурах, обеспечивающих полное
расплавление руды;— дистилляция — процесс испарения перерабатываемого
вещества с целью разделения его компонентов на основе их раз¬
личной летучести.Гидрометаллургические процессы проводятся в водных сре¬
дах при температурах до 300°С на границе раздела твердбЬ и
жидкой фаз.Наиболее распространенным гидрометаллургическим про¬
цессом является выщелачивание — процесс перевода в жидкую
фазу (раствор) извлекаемых из руды соединений металлов при
воздействии на нее растворителей. Выщелачивание может быть
физическим процессом (растворитель вода) или химическим
процессом (растворитель — реагент, взаимодействующий с из¬
влекаемым компонентом).Восстановление химического соединения рассматривается в
разделе 1.4.Вторичная обработка восстановленного металла проводит¬
ся для его очистки, а также с целью перестройки кристалли¬
ческой структуры металла, изменения его состава и свойств.
К операциям вторичной обработки относятся рчистка метал¬
ла методами дистилляции, электролиза, электрошлакового пе¬
реплава и зонной плавки; получение сплавов, закалка, отжиг,
отпуск, цементирование и др. Некоторые из них рассматрива¬
ются ниже.На рис. 1.2 представлена общая схема производства метал¬
лов из руд (металлургического процесса).9
[OIT11т► МеОя ► м<В- IГ^НГ► МеГтт—IIРис. 1 .2. Общая схема производства металлов из руд:I — подготовка руды, II — восстановление, III — вторичная обработка, В —
восстановитель, [О] — окисление, Т — термическая обработка1,4. Физико-химические основы восстановления
металлов из рудПроцессы восстановления металлов из руд различаются по
природе восстановителя и по условиям восстановления. В ка¬
честве восстановителей применяют химические вещества (во¬
дород, оксид углерода (II), углерод, металлы) или электричес¬
кий ток, а процесс восстановления можно проводить в раство¬
ре, в расплаве или в твердой фазе. В зависимости от этого раз¬
личают следующие методы восстановления:1. Гидрометаллургическое восстановление — восстановле¬
ние химическими восстановителями из водных растворов, на¬
пример:2. Пирометаллургическое восстановление — восстановле¬
ние химическими восстановителями при высокой температуре
из расплавов или твердой фазы, например:3. Электрогидрометаллургическое восстановление— вбс-
становление электрическим током из водных растворов, напри¬
мер:4. Электропирометаллургическое восстановление — вос¬
становление электрическим током при высокой температуре из
расплавов, например:C11SO4 + Zn = Си + ZnSC>4FeO + СО = Fe + С02CuS04 + 2ё = Си + S04‘2А120з + 6ё = 2А1 + ЗО"210
Оба случая восстановления электрическим током представ¬
ляют собой процессы электролиза в водных растворах или рас¬
плавах электролитов, при которых восстанавливаемый металл
выделяется на катоде.При восстановлении металлов из их соединений необходимо
учитывать как принципиальную осуществимость этого процес¬
са, так и полноту его протекания, от которой зависит экономич¬
ность процесса.Принципиальная возможность процесса восстановления оп¬
ределяется условием AG<0, где AG — изобарно-изотермический
потенциал равный:' AG =АН - TAS (1.1)Скорость самопроизвольно протекающей реакции восстанов¬
ления тем выпде, чем больше по абсолютному значению вели¬
чина АG. Из 1.1 следует, что соблюдению неравенства AG<0 спо¬
собствует уменьшение энтальпии (ДН<0), увеличение энтропии
(AS>0) и проведение процесса цри максимально осуществимой
в данных условиях температуре Т. Использование температур?
ного фактора позволяет в ряде случаев применять для восста¬
новления слабые восстановители, которые не «работают* при
обычных температурах. Наиболее распространенный процесс восстановления окси¬
дов металлов описывается общим уравнением:МеО + В^Мё + ВО (а)где: В — восстановитель, ВО — продукт окисления восстанови¬
теля.Изобарно-изотермический потенциал этого процесса равен:AG = AGbo — (АСмеО + ЛСв) < 0 (1*2)Очевидно, что для восстановителей — простых веществ (ме¬
таллы, углерод, водород) AGb = 0 и уравнение 1.2 превращается
в уравнение:AG = AGbo~ ДСтлео (1.2а)Рассмотрим конкретные случаи восстановления оксидов ме¬
таллов различными восстановителями.1. Восстановление водородом.МехОу + уН2 *=* хЫе + Z/H2O (б)AG = yAGn2o - А(?мехоу < О2.Восстановление углеродом.МехОу +1/С хМе + уСО (в)AG = yAGco ~ А&мехоу < О11
3. Восстановление оксидом углерода (II).Ме*0у -1- уСО JtMe + г/СОг (г)AG = yAGco2 ~ (AGuexoy + i/AGco)<04. Восстановление металлами (металлотермия).Частным и наиболее распространенным случаем металлотер¬
мии является восстановление алюминием — алюминотермия:ЗМе*Оу + 2уА1 «=± ЗхМе + г/А^Оз (д)AG = z/AGai2o3 _ 3AGMexo1/ < ОИз 1.1 следует, что при достаточно низких температурах вос¬
становления, критерием возможности протекания процесса ме¬
таллотермии:MejO + Мв2 ^ Ме20 + Мехявляется условие ЬН.2<ЬЛ\% где AHi и Ai/2 — энтальпии образо¬
вания оксидов восстанавливаемого и восстанавливающего ме¬
таллов, соответственно. В табл.1.4 приведены энтальпии обра¬
зования некоторых распространенных металлов в расчете на г.а-
том кислорода в них. Из табл. следует, что методом алюмино¬
термии могут быть, например, получены из их оксидов такие
металлы как титан, марганец, хром, железо, никель, медь эн¬
тальпия образования оксидов которых алгебраически больше,
чем энтальпия образования оксида алюминия. Наоборот, метод
алюминотермии непригоден для восстановления бериллия и
магния.Таблица 1.4. Энтальпия образования оксидов металловМехО уДН°298. КДЖМе„0 уAH°298. кДжВеО-610,3v2o5-311,8МдО-601,1SnO-285,9АЬОз-555,9Ре2Оз-273,8ТЮ2-455,6FeO-266,3МпО-384,6NiO-244,1Сг2Оз-375,8CuO-155,19 У5. Восстановление электрическим током в расплаве.Ме*0 у± 2 уё хМе + 1/2уС>2 (е)В этом случае-AG = nFV<0 (1.3)где: V = Ek~Ea — разность потенциалов разряда ионов на като¬
де и аноде, то есть фактическое напряжение электролиза12
(15.2.1). Так как теоретически возможно неограниченное по¬
вышение напряжения , то и AG-*°°, то перед методом электро¬
литического восстановления металлов открываются неограни¬
ченные возможности.6. Термическая диссоциация галогенидов.Особым случаем восстановления металлов из их соединений
можно считать термическую диссоциацию галогенидов метал¬
лов или реакцию внутримолекулярного окисления-восстанов¬
ления, протекающую по уравнению:МехГу ^ хМе + у/2 Гг, (ж)ДЛЯ КОТОРОЙ AG = —ДСгМеЛ1 (1*4)УЗдесь мерой термической стабильности галогенида и мерой
легкости его распада при нагревании является значение изобар¬
но-изотермического потенциала галогенида Ме*Гу, зависящее
от природы галогена. В табл. 1.5 приведены значения потенци¬
ала для галогенидов циркония.Полнота протекания процесса восстановления металла из его
оксида зависит, очевидно, от сдвига вправо равновесия «а*. Ус¬
ловием этого является создание гетерогенной системы, что обес¬
печивает: — ———... — распределение компонентов по различным фазам систе¬
мы, например:Ме(ж)«—►Восстановитель (г) илиМе(ж)«—^Восстановитель (т),— удаление продукта окисления восстановителя (вода, ок¬
сиды углерода) в виде газа;— отделение жидкого расплавленного металла от твердого
оксида металла с более высокой.температурой плавления.Таблица 1.5. Изобарно-изотермический потенциал галогени¬дов цирконияГ алогенидZrCI4 ZrBr4 ZrJ4AG°298# кДж/мольПадение термической стабильностиПовышение температуры диссоциации-890 -725 -481 : ' —► :—: Таким образом, в основе любого металлургического процес¬
са восстановления металла из его соединений лежит принцип
перевода обрабатываемого сырья в гетерогенную систему, состо¬
ящую из двух, трех и более фаз, отличающихся друг от друга
составом и физическими свойствами. При этом одна из фаз обо-13
гкщается извлекаемым металлом, в то время как другие фазы
им обедняются и, наоборот, обогащаются примесями и побоч¬
ными продуктами восстановления.Контрольные вопросы1. На каких свойствах металлов основано широкое использование их?2. Приведите промышленную классификацию металлов.3. Какие виды природного ископаемого сырья относятся к металли¬
ческим рудам?4. Что такое металлоносность рудного месторождения?5. Приведите общую схему производства металлов из руд.6. Укажите особенности гидрометаллургического, гдроэлектрометал-
лургического, пирометаллургического и электропирометаллурги-
ческого методов восстановления металлов из их соединений.7. Чем определяются принципиальная возможность и полнота про¬
текания процесса восстановления металлов из руд?
Глава IIПРОИЗВОДСТВО АЛЮМИНИЯV2.1. Свойства и применение алюминияАлюминий относится к числу важнейших легких цветных
металлов. По масштабам производства и потребления он зани¬
мает второе место среди всех металлов (после железа) и первое
место среди цветных металлов. Поэтому в цветной металлур¬
гии производство этого металла выделено в отдельную специа¬
лизированную подотрасль 4Алюминиевая промышленность»
включающую добычу сырья для алюминиевой промышленное-
ти, производство алюминия, глинозема и фтористых солей.Алюминий — твердый серебристо-серый металл. Легко под¬
дается ковке, прокатке, волочению и резанию. Пластичность
алюминия возрастает с повышением его чистоты. Плотность
алюминия 2,7 т/м3, температура плавления 660,2°С, темпера¬
тура кипения 2520°С. В расплавленном состоянии жидкотекуч
и легко поддается литью. ___Алюминий имеет высокие тепло- и электропроводность, ко¬
торые зависят от его чистоты. Для алюминия высокой чистоты
электропроводность составляет 65% от электропроводности
меди.Алюминий химически активен, легко окисляется кислоро¬
дом воздуха, образуя прочную поверхностную пленку оксида
AI2O3, что обусловливает его высокую коррозионную стойкость.В мелко раздробленном состоянии при нагревании на воздухе
воспламеняется и сгорает. Алюминий реагирует с серой и гало¬
генами. При нагревании образует с углеродом карбид AI4C3 и с
азотом нитрид A1N. Как амфотерный металл алюминий раство¬
ряется в сильных кислотах и щелочах. Нормальный электро- ,
дный потенциал алюминия равен 1,66 В при рН<7 и 3,25 В при
рН>7.Вследствие комплекса ценных свойств (малая плотность,
пластичность, высокие тепло- и электропроводность4, неток-
сичность, немагнитность, коррозионная стойкость в атмос¬
фере), а также недефицитности сырья и относительно низ¬
кой стоимости алюминий в чистом виде и в сплавах широ¬
ко применяется в различных отраслях техники и народном
хозяйстве. В табл. 2.1 приведены данные об использовании
алюминия.15
*2.1 .Использование алюминия в народном хозяйствуДЮГ|1г |Г| 1—-■ ОтрасльОсновные направления
использованияПотребле- I
ние, %ТранспортАвиаконструкции двигатели f
тру бы, корпуса судов,
отделка вагонов18 —21СтроительствоАнгары, конструкционные детали
зданий, рамы, хранилища хими¬
ческой продукции24—30ЭлектротехническаяпромышленностьКабели, шинопроводы, конденса¬
торы, выпрямители12—14Машино-, приборо¬
строениеМоторы, блоки цилиндров, насо¬
сы, картеры, кино-фотоаппарату¬
ра, контрольно-измерительная
аппаратура5—7Тара и упаковочные
материалыФольга для пищевых продуктов,
емкости для консервирования14—17Предметы домаш¬
него обиходаПосуда, столовые приборы7—10Прочие потребителидо 10Алюминий образует с кремнием, медью, магнием, цинком,
марганцем и другими металлами два типа сплавов — деформи¬
руемые и литейные. Из деформируемых сплавов наиболее рас¬
пространены дуралюмины — сплавы алюминия с медью, мар¬
ганцем и магнием. Они применяются для изготовления мето¬
дами прокатки и штамповки изделий различного профиля (ли¬
сты, стержни, панели, трубы, проволока, емкости и др.).Из литейных сплавов, называемых силуминами, содержа¬
щих кремний, изготавливаются фасонные отливки различной
конфигурации.Алюминий высокой степени чистоты используют в ядерной
энергетике, полупроводниковой электронике, радиолокации,
для изготовления отражающих поверхностей рефлекторов и
зеркал. В металлургической промышленности алюминий при¬
меняется в качестве восстановителя при получении ряда метал¬
лов (алюминотермия), раскисления стали, для сварки отдель¬
ных деталей.Широкое использование алюминия в различных областях
народного хозяйства вызвало интенсивное развитие его произ¬
водства в мире, которое уже в 80-х годах составило более 20 млн.
тонн. В РФ в настоящее время алюминий стал источником ва¬16
лютных поступлений. В 1993 году экспорт алюминия из РФ
составил около 900 тыс. тонн. В то же время потребление алю¬
миния внутри страны сокращается и составляет всего 3,0 кг/год
на человека, тогда как в США оно равно 18 кг/год.2.2. Краткая история развития производства алюминияI лВпервые металлический алюминий был получен в 1825 году Г. Эр¬
стедом химическим методом восстановлением хлорида алюминия
амальгамой калия. В 1856 году этот метод был усовершенствован и
алюминий стали получать восстановлением двойной соли AlCl3*NaCl
металлическим натрием. Н.Н. Бекетов в 1865 году предложил метод
получения алюминия восстановлением криолита NaaAlFe магнием.
Производство алюминия химическим методом просуществовала до
1890 года и з% 35 лет его использования было получено всего около
200 тонн алюминия.В 1886 году, основываясь на работах А.Сент-Клера Девиля
(1856 г.), Н. Эру во Франции и Ч. Холл в США разработали ме¬
тод производства алюминия электролизом расплава глинозема
в криолите, который до настоящего времени является един¬
ственным методом промышленного производства алюминия.
После внедрения этого метода мировое производство алюминия
быстро росло и с 5,7 тыс. тонн в 1900 году достигло почти 2и млн.
тонн в 1980 году (без СССР).Промышленное производство алюминия в нашей стране было
организовано в 30-х годах XX столетия после строительства пер¬
вых крупных электростанций. Теоретической основой произ¬
водства явились исследования отечественных ученых, выпол¬
ненные в конце XIX — начале XX вв. П.П.Федотьев изучил и раз¬
работал теоретические основы электролиза системы « глинозем-
криолит», в том числе растворимость алюминия в электролите,
анодный эффект и другие условия процесса. В 1882—1892 гг.
К.И. Байер разработал «мокрый» метод получения глинозема вы¬
щелачиванием руд, а в 1895 году Д.Н. Пеняков предложил метод
производства глинозема из бокситов спеканием с сульфатом на¬
трия в присутствии угля. А.И.Кузнецов и Е.И. Жуковский разра¬
ботали в 1915 году способ получения глинозема методом восстано¬
вительной плавки низкосортных алюминиевых руд.На основе этих исследований в 1930 году в Ленинграде был пу¬
щен опытный завод, а в 1932 году введен в строй первый в стране
Волховский алюминиевый завод на базе Волховской ГЭС. В после¬
дующие годы отечественная алюминиевая промышленность раз¬
вивалась быстрыми темпами. В1933 году был пущен Днепровский
алюминиевый завод, на базе Днепрогэса, введены в эксплуатацию
Новокузнецкий (1943) и Богословский (1945) заводы.
В послевоенное время наряду с восстановлением Волховско¬
го и Днепровского заводов были построены новые алюминие¬
вые заводы: Канакерский (1950), Кандалакшский (1951), Но-
водвоицкий (1954), Сумгаитский (1955), а позднее заводы на
базе электроэнергии ГЭС на Волге и реках Сибири: Волгоградс¬
кий (1959), Иркутский (1962), Красноярский (1964), Братский
(1966) и Таджикский (1975). Одновременно вводились в строй
и предприятия по производству глинозема: Пикалевский
(1959), Ачинский (1970), Павлодарский (1964), Кировобадский
(1966) и Николаевский (1980) комбинаты.2.3. Сырье для производства алюминияАлюминий входит в состав многих минералов, однако в ка¬
честве алюминиевых руд используются только бокситы, нефе¬
лины, алуниты и каолины. Они различаются составом и кон¬
центрацией оксида алюминия. Важнейшей алюминиевой рудой
являются бокситы, содержащие гидратированный оксид алю¬
миния А1203-пН20. В зависимости от степени гидратации алю¬
миниевый компонент в бокситах может находиться в форме ди¬
аспора А1203-Н20 (или иначе НА102) или в форме гидроаргели-
та А120з*ЗН20 (или А1(ОН)з). Помимо оксида алюминия в со¬
став бокситов входит оксид кремния (от 0,5 до 20%) и различ¬
ные соединения железа, кальция и магния. Основная характе¬
ристика бокситов, от которой зависит выбор метода их перера¬
ботки — кремневый модуль — отношение содержания в них ок¬
сида алюминия к содержанию оксида кремния А120з/вЮ2. Для
бокситов, используемых в качестве сырья в алюминиевой про¬
мышленности, модуль должен быть не ниже 2,6; для бокситов
среднего качества он составляет 5—7, чему соответствует содер¬
жание оксида алюминия 46—48%.Нефелины представляют собой сложную тройную соль соста¬
ва (Na,K)20*Al203'2Si02 и входят как составная часть в апати-
то-нефелиновую руду, содержащую кроме нефелина апатит
' ЗСаз(Р04)2'СаГ2. Для производства алюминия используют не¬
фелиновый концентрат с содержанием оксида алюминия 20—
30%. Его получают наряду с апатитовым концентратом пере¬
работкой апатито-нефелиновой руды (рис. 2.1).Алуниты представляют двойную основную сернокислую
соль алюминия и калия состава K2S04*A12(S04)3*4A1(0H). Содер¬
жание оксида алюминия в алунитах не превышает 20%.В РФ крупные разведанные месторождения бокситов нахо¬
дятся в Ленинградской (Тихвинское), Свердловской (Северо-18
АлюминиевыеРУДЫ■ ► ДиаспорБОКСИТЫ30—60%А1203 —► Гидроаргвлит. Нефелины30% А1203 (концентрат)^ Алуниты
20% А1203_w Каолины
до40%А1203Рис. 2.1. Основные алюминиевые рудыуральское) и Челябинской (Южноуральское) областях. Основ¬
ные месторождения нефелинов расположены на Кольском по¬
луострове (Хибинское) и в Кемеровской области.2.4. Общая схема производства алюминияТехнология получения металлического алюминия из руд до¬
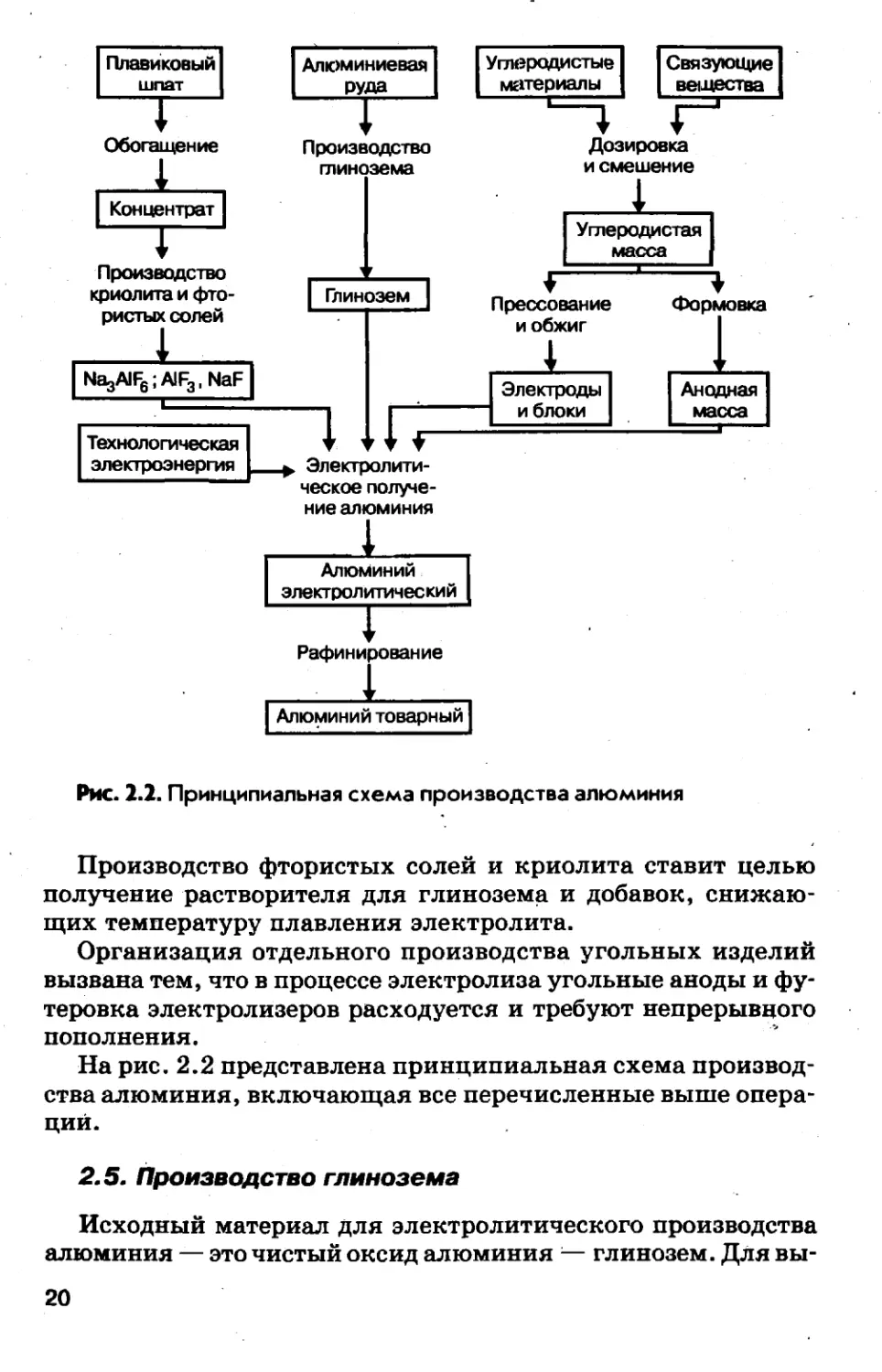
статочно сложна и состоит из четырех производств, свяэаппых
между собой технологической цепочкой и производимыми про¬
дуктами. Она включает:— производство глинозема (оксида алюминия);— производство фтористых солей и криолита;— производство угольных изделий (электродов и блоков фу¬
теровки);— производство электролитического алюминия.Основными производствами, составляющими технологичес¬
кую цепочку Руда—^Глинозем—»Алюминий, является производ¬
ства глинозема и алюминия. Территориально они обычно раз¬
делены. Вследствие высокой энергоемкости процесса электро¬
литического восстановления алюминия алюминиевые заводы
располагаются в районах с дешёвой электроэнергией ГЭС. Про¬
изводства глинозема, наоборот, базируются в местах добычи
алюминиевых руд с тем, чтобы сократить расходы на перевоз¬
ку сырья. Примером производства с полным циклом (от руды
до рафинированного металлического алюминия) являются Вол¬
ховский и Каменец-Уральский заводы. На других предприяти¬
ях этой отрасли осуществляется только часть технологической
цепочки: производство глинозема (Ачинск, Бокситогорск) или
выплавка алюминия (Кандалакша, Волгоград, Новокузнецк,
Братск, Красноярск).19
Рис. 2.2. Принципиальная схема производства алюминияПроизводство фтористых солей и криолита ставит целью
получение растворителя для глинозема и добавок, снижаю¬
щих температуру плавления электролита.Организация отдельного производства угольных изделий
вызвана тем, что в процессе электролиза угольные аноды и фу¬
теровка электролизеров расходуется и требуют непрерывного
пополнения.На рис. 2.2 представлена принципиальная схема производ¬
ства алюминия, включающая все перечисленные выше опера¬
ций.2.5. Производство глиноземаИсходный материал для электролитического производства
алюминия — это чистый оксид алюминия — глинозем. Для вы-20
деления глинозема из алюминиевых руд его переводят в раство¬
римую соль (алюминат натрия), которую отделяют от осталь¬
ных компонентов руды, осаждают из ее раствора гидроксид алю¬
миния и кальцинацией последнего получают глинозем.Метод выделения глинозема из руды зависит от ее состава.
Эти методы подразделяются на химико-термические (пироме-
таллургические), кислотные и щелочные (гидрометаллургичес¬
кие). К пирометаллургическим методам относится метод спе¬
кания; к гидрометаллургическим методам — щелочной метод
Байера.2.5.1. Производство глинозема методом БайераМетод Байера (мокрый метод, метод выщелачивания) явля¬
ется наиболее распространенным методом производства глино¬
зема. В основе метода лежит обратимый процесс взаимодей¬
ствия гидратированного оксида алюминия с водным раствором
гидроксида натрия с образованием алюмината натрия. Метод
применяется для выделения глинозема из бокситов с малым
(менее 5%) содержанием оксида кремния. При большем содер¬
жании последнего метод становится экономически невыгодным
вследствие высокого расхода дорогой щелочи на взаимодействие
с оксидом кремния.Процесс выделения глинозема по методу Байера состоит из
следующих операций.1 .Дробление боксита и мокрый размол его в среде оборотно¬
го щелочного раствора с образованием пульпы.2. Выщелачивание оксида алюминия оборотным раствором
гидроксида натрия по реакциям:HA102+Na0H+H20 ^ NaA102 +2Н20 NaAl(OH)4 (диаспор)
Al(OH)3+NaOH ^ NaA102 +2Н20 ^ NaAl(OH)4 (гидроаргелит)уОдновременно протекает реакция образования силиката на¬
трия, на что расходуется часть реакционной щелочи:Si02 + 2NaOH ^ Na2Si03 + Н20Выщелачивание представляет гетерогенный процесс насы¬
щения водного щелочного раствора оксидом алюминия, ско¬
рость которого зависит от дисперсности твердой фазы (бокси¬
та), концентрации раствора гидроксида натрия и температуры.
Режим процесса выщелачивания определяется степенью гид¬
ратации оксида алюминия в боксите: диаспор выщелачивают
при 240°С и давлении 3 МПа; гидроаргелит — при 100°С и дав¬
лении ОД МПа. Степень извлечения оксида алюминия (X) до¬21
стигает 0,92% за три часа и в даль¬
нейшем практически не изменяет¬
ся; концентрация гидроксида на¬
трия в щелочном растворе при
этом падает (рис. 2.3).3. Разбавление-самоочищение
при добавлении к пульпе воды с об¬
разованием нерастворимого гидра¬
тированного алюмината натрия:2NaAl(OH)4 + 2Na2Si03 *
=Na20-Al203.2Si02*2H20 + 4NaOHВ результате этой реакции часть
алюминиевого компонента теряет¬
ся, при этом тем больше, чем выше содержание оксида крем¬
ния в боксите. Осадок алюмосиликата, окрашенный оксидом
железа (III) в красно-бурый цвет, получил название красного
шлама.4. Фильтрование раствора алюмината натрия, отделение
и промывка красного шлама.5. Декомпозиция раствора алюмината натрия («выкручи¬
вание») при понижении температуры и интенсивном переме¬
шивании пульпы:NaAl(OH)4 ^ А1(ОН)3 + NaOHДекомпозиция — это самопроизвольно протекающий про¬
цесс гидролиза алюмината натрия. Он ускоряется введением
кристаллического гидроксида алюминия («затравки»), что од¬
новременно способствует образованию крупных кристаллов гид¬
роксида алюминия за счет создания в системе центров кристал¬
лизации.6. Сгущение пульпы с последующим отделением гидроксида
алюминия на вакуум-фильтре и классификация полученного
продукта с выделением основной фракции.7. Упаривание маточного раствора до образования оборот¬
ного щелока и его подкрепление гидроксидом натрия.8. Каустифйкация образовавшегося карбоната натрия гид¬
роксидом кальция и возвращение образовавшегося белого шла¬
ма в технологический процесс:Na2C03 + Са(ОН)2 = 2NaOH + СаС039. Кальцинация (обезвоживание) гидроксида алюминия при
1200°С:2А1(ОН)3 = А1203 + ЗД20Рис. 2.3. Изменение степени
извлечения 1 и концентрации
2 щелочи во времени:1 — степень извлечения АЬОз,2 — концентрация NaOH22
г Полученный по методу Байера глинозем представляет смееь
а-Дюдифйкации (корунд) и у-модйфикации оксида алюминия.
Технический продукт представляет белое кристаллическое ве¬
щество и выпускается нескольких марок, различающихся чис¬
тотой. Наиболее вредными примесями в глиноземе являются
оксид кремния, оксид железа (П1) и оксид титана (IV). Кроме
этого, в глиноземе регламентируется содержание оксидов ка¬
лия и натрия и оксида фосфора (V).К техническому глинозему предъявляется ряд требований по
физическим свойствам: влажности, плотности, насыпной мас¬
се, гранулометрическому составу и др. От этих свойств зависит
поведение продукта при транспортировке, загрузке в электро¬
лизеры и само проведение процесса электролиза.На рис. 2.4 представлена принцициальная, а на рис. 2.5тех-
нологическая схемы производства глинозема по методу Байе¬
ра. На принципиальной схеме номерами указаны химические
• 'операции, соответствующие реакциям, указанным выше.Из принципиальной схемы процесса следует, что в методе
выщелачивания Байера осуществляется замкнутый технологи¬
ческий цикл по щелочи. Щелочь, затраченная на выщелачива¬
ние оксида алюминия из боксита, регенерируется на стадиях
декомпозиции и каустификации и возвращается в процесс на
обработку новых порций боксита. Таким образом, в методе Бай¬
ера реализуется принцип организации малоотходного производ¬
ства. ^Технологический процесс производства глинозема по мето¬
ду Байера организуется следующим образом (рис. 2.5). Бокси¬
товая пульпа из смесителя 1 подается в подогреватель 2, обо¬
греваемый паром из сепаратора 5. Из подогревателя пульпа по¬
ступает в батарею греющих автоклавов 3 и затем в батарею ре¬
акционных автоклавов 4, где протекает процесс выщелачива¬
ния, откуда направляется в сепаратор 5. В сепараторе давление
снижается от 3 МПа до атмосферного, вследствие чего пульпа
вскипает и образовавшийся пар направляется в подогреватель
2. После этого пульпа, coctfоящая из щелочного раствора алю¬
мината натрия и красного шлама, разбавляется в разбавителе 6
и поступает в сгуститель пульпы 7 и, далее, для отделения крас¬
ного шлама на фильтр 9. Отделившийся шлам промывается во¬
дой в промывателе 8, а раствор алюмината натрия поступает в
декомпозер 10, где перемешивается барботирующим воздухом.
Из декомпозера гидратная пульпа, состоящая из кристаллов
гидроксида алюминия и маточного раствора, направляется в се¬
паратор кристаллов 11, где кристаллы отделяются от маточ-23
ИзвестьОборотныйрастворКаустификация 8Отделение
- карбоната
натрияВыпаривание <4-Боксит |Дробление
+ •Мокрый
" размолВыщелачивание 23 РазбавлениеСгущение Фильтрация _
раствораДекомпозицияСгущение, про¬
мывка и фильт¬
рацияКрасныйшлам~~г~- Промывнаяводакк1ывка►iКрасный
шлам в отвалЗатравкаГидроксидалюминияКальцинацияГлиноземРис. 2.4. Принципиапьная схема производства глинозема
по методу Байераного раствора и, пройдя бункер 12, поступают в трубчатую
печь кальцинации 139 после чего охлаждаются в трубчатом
холодильнике 14. Отделенный маточный раствор соединяет¬
ся с раствором из промывателя шлама 8 и направляется на
упаривание.Основными аппаратами в технологической схеме являются
реакционный автоклав и декомпозер.Реакционный автоклав для выщелачивания боксита пред¬
ставляет вертикально расположенный сварной сосуд диаметром
до 2,5 м и высотой 14—18 м, снабженный штуцером в верхней
крышке для подачи пульпы и трубой для разгрузки содержи¬
мого, доходящей до дна аппарата. Автоклав обогревается ост¬
рым паром, подаваемым в пульпу или с помощью обогреваемых
паром змеевиков. Время пребывания пульпы в автоклаве состав-24
ВодаГлиноземНа упариваниеРис. 2.5. Технологическая схема производства глиноземаиз боксита (содержащего диаспор) по методу Байера:1 — смеситель пульпы, 2 — подогреватель пульпы, 3 — греющие автокла¬
вы (два), 4 — реакционные автоклавы (6), 5 — сепаратор пара и жидкости,
6 — разбавитель, 7 — сгуститель шлама, 8 — промыватель шлама, 9 —
вакуум-фильтр, 10 — декомпозер, 1 / — сепаратор глинозема, 12 — бун¬
кер, 13 — трубчатая печь, 14 — трубчатый холодильникляет 2—3 часа, что обеспечивает степень разложения сырья до0,9 долей ед.Декомпозеры для операции выкручивания могут быть с ме¬
ханическим и пневматическим перемешиванием пульпы. Наи¬
более совершенный декомпозер с воздушным перемешиванием
представляет собой стальной бак с коническим дном диамет¬
ром Эми высотой до 35 м. Для циркуляции вводимых в пульпу
кристаллов затравки в декомпозер встроен аэролифт (воздуш¬
ный подъемник), состоящий из двух концентрических труб, в
который подается сжатый воздух, образующий воздушно-пуль-
повую смесь, поднимающуюся по внешней трубе в верхнюю
часть декомпозера.25
Расходные коэффициенты на 1 тонну глинозема, произвол
димого по методу Байера.Боксит 2,0^—2,5 т Пар 7—9 тГидроксид 0,07—0,09 т Вода 150 манатрияИзвестняк 0,12 т Электроэнергия 300 кВт-ч2.5.2. Производство глинозема методом спеканияВ основе метода спекания лежит процесс образования алю¬
минатов натрия (и калия в случае нефелинов) в результате вза¬
имодействия при высокой температуре оксида алюминия руды
с карбонатами металлов, с последующим выщелачиванием алю¬
минатов водой и разложением их оксидом углерода (IV). При¬
рода карбоната зависит от содержания в руде натриевого ком¬
понента: для спекания бокситов используют смесь карбонатов
натрия и кальция, а для спекания нефелинов, содержащих в
своем составе оксиды натрия и калия, только оксид кальция.
Карбонат кальция при спекании бокситов связывает присут¬
ствующий в них оксид кремния и позволяет существенно сни¬
зить расход дорогого карбоната натрия... Процесс производства глинозема методом спекания универ¬
сален и пригоден для переработки всех видов алюминиевого
сырья. На практике его применяют для нефелинов и бокситов с
высоким (более 5%) содержанием оксида кремния! Процесс
спекания состоит из следующих операций.1. Измельчение алюминиевой руды и известняка, мокрый
размол шихты в содовом растворе и корректировка состава пуль¬
пы.2. Спекание пульпы при 1300°С по реакциям:
в случае боксита2НА102 + Na2C03 = 2NaA102 + С02 = Н20или 2А1(ОН)з + Na2CC>3 = 2NaA102 + С02 + ЗН20
и образования дикальцийсиликатаSi02 + 2СаС03 = 2Ca0Si02 + 2С02,
в случае нёфелина(Na,K)2OAl203-2Si02 + 4СаС03 == 2 (Na,K)A102 + 4С02 + 2(2Ca0Si02)Процесс спекания протекает в твердой фазе и заключается в
проникновении частиц одного реагента в кристаллическую ре¬
шетку другого. Поэтому, скорость спекания определяется ско-26
ростью межкристаллической диффузии, которая возрастает при
повышении температуры. Расплавление легкоплавких компо¬
нентов шихты и образование жидкой фазы ускоряет процесс
спекания. В отходящих газах спекания содержится до 10—
12% оксида углерода (IV).3 .Дробление образовавшегося опека и выщелачивание из него
водой алюминатов натрия и калия.4. Обескремниваниераствора — удаление из раствора алю¬
минатов непрореагировавшего оксида алюминия и примеси
оксида кремния в виде белого шлама, возвращаемого в про¬
цесс.5. Карбонизация раствора алюминатов действием оксида уг¬
лерода (IV) и осаждение гидроксида алюминия по реакции (в
случае боксита):2NaA102 + С02 + ЗН20 = ЗА1(ОН)3 + Na2C03При карбонизации раствора, полученного обработкой нефе¬
лина, в качестве побочного продукта образуется смесь карбона¬
тов натрия и калия, получившая название «содопродукт*:2(Na,K)A102 + С02 + ЗН20 = 2А1(ОН)3 + (Na,K)2CQ3Обычно, содопродукт состоит на 85% из карбоната натрия и
на 15% из карбоната калия. Содержащий карбонаты маточный
раствор упаривается и возвращается на операцию мокрого из¬
мельчения, или из него извлекается твердый содопродукт.6. Отделение гидроксида алюминия от маточного раствора и
его промывка водой.7. Кальцинация гидроксида алюминия при 1200°С:2А1(ОН)3 = А1203 + ЗН20К глинозему, полученному методом спекания, предъявля¬
ются те же требования в отношении чистоты и физико-хими¬
ческих характеристик, что и к глинозему, полученному мето¬
дом Байера.Шлам, полученный при очистке раст&ора алюминатов, пос¬
ле добавления известняка перерабатывается на портландце¬
мент.В расчете на 1 т оксида алюминия в руде образуется около 1
тонны содопродукта и 7—8 тонн цемента.На рис. 2.6 представлена принципиальная и на рис. 2.7 тех¬
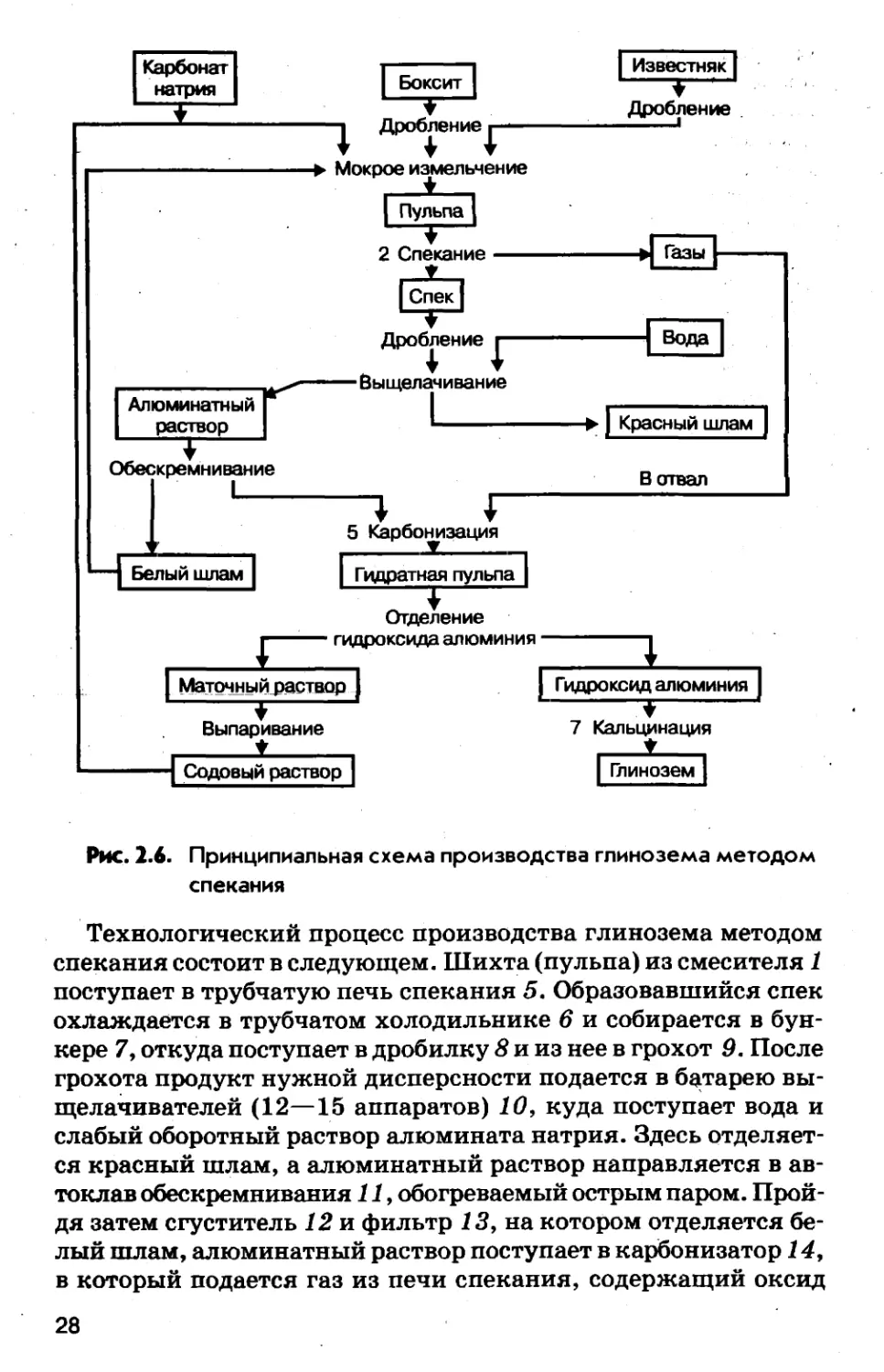
нологическая схемы производства глинозема из боксита мето¬
дом спекания. На принципиальной схеме указаны номера хи¬
мических операций, соответствующие указанным выше./27
Рис. 2.6. Принципиальная схема производства глинозема методом
спеканияТехнологический процесс производства глинозема методом
спекания состоит в следующем. Шихта (пульпа) из смесителя 1
поступает в трубчатую печь спекания 5. Образовавшийся спек
охлаждается в трубчатом холодильнике 6 и собирается в бун¬
кере 7, откуда поступает в дробилку 8 и из нее в грохот 9. После
грохота продукт нужной дисперсности подается в батарею вы¬
щелачивателей (12—15 аппаратов) 10, куда поступает вода и
слабый оборотный раствор алюмината натрия. Здесь отделяет¬
ся красный шлам, а алюминатный раствор направляется в ав¬
токлав обескремнивания 11, обогреваемый острым паром. Прой¬
дя затем сгуститель 12 и фильтр 13, на котором отделяется бе¬
лый шлам, алюминатный раствор поступает в карбонизатор 14,
в который подается газ из печи спекания, содержащий оксид28
углерода (IV). Выпавший гидроксид алюминия отделяется и
после промывки поступает на кальцинацию, а раствор карбо¬
натов после подкрепления направляется на приготовление пуль¬
пы (шихты). Отходящие из печи спекания газы проходят цик¬
лон 4 у электрофильтр 3 и скруббер-осадитель 2, где освобожда¬
ются от пыли спека, возвращаемой в процесс, и выбрасывают¬
ся в атмосферу.Основными аппаратами технологической схемы являются
печь спекания, выщелачиватель и карбонизатор. Вращающая¬
ся трубчатая печь имеет диаметр 3—5 м и длину 50—185 м и
обогревается топочными газами. В рабочей зоне печи поддер¬
живается температура 1200—1300°С. Отходящие из печи топоч¬
ные газы содержат до 12% оксида углерода (IV) и используют¬
ся для карбонизации алюминатного раствора.1 — смеситель шихты, 2 — скруббер-осадитель пыли, 3 — электро¬
фильтр, 4 — циклон, 5 — трубчатая печь спекания, 6 — трубчатый холо¬
дильник, 7 — бункер спека, 8 — дробилка, 9 — грохот, 10 — выщелачива¬
тель, 11 — автоклав обескремнивания, 12 — сгуститель, 13 — фильтр
белого шлама, 14 — карбонизаторИзвесть | БокситГ12007Газы 10% С02
* Глинозем на кальцинациюРис. 2.7. Технологическая схема производства глинозема
из нефелина методом спекания:29
Для выщелачивания спека применяют аппараты (выщела¬
чиватели) различной конструкции: диффузоры, перколляторы
и трубчатые выщелачиватели. Наиболее распространенные из
них — диффузоры представляют цилиндрические аппараты с
коническим верхом высотой 5 м и диаметром 1,5 м, снабжен¬
ные системой трубопроводов, по которой подается выщелачи¬
вающая жидкость. Раствор алюминатов выходит из верхней ча¬
сти аппарата, а красный шлам выгружается через нижнюю
крышку аппарата. Диффузоры в технологической схеме объ¬
единяются в батареи по 10—12 аппаратов, работающих по прин¬
ципу противотока.Карбонизатор представляет цилиндр с коническим дном ди¬
аметром Ими общей высотой 16 м, емкостью около 600 м3. То¬
почные газы поступают в коническую часть аппарата через бар-
ботажное устройство и перемешивают пульпу, которая аэролиф¬
том поднимается в верхнюю часть карбонизатора.2.6. Электролитическое производство алюминияОксид алюминия (III) при обычной и умеренно высоких тем¬
пературах не восстанавливается до металлического алюминия
такими химическими восстановителями как водород, углерод
и большинство металлов. В табл. 2.2 приведены значения изо¬
барно-изотермического потенциала оксидов алюминия, водоро¬
да, углерода и таких активных металлов как натрий и магний.Таблица 2.2. Изобарно-изотермический потенциал оксидовМеО*А1203н2ожсо2Na20MgOAG°29a, кДж/ моль-1583-238-395-378-569Расчеты по этим данным для таких восстановителей как во¬
дород, углерод и натрий показывают, что при нормальных тем¬
пературе и давлении изобарно-изотермический потенциал про¬
цесса восстановления, оксида алюминия по уравнениюAI2O3 + В = 2А1 + ВО3составляет, соответственно, +869, +1981 и +449 кДж/моль, то
есть АСг процесса существенно больше нуля. Повышение темпе¬
ратуры восстановления, даже в том случае, если это практичес¬
ки ооуществимо, например, для углерода, приводит к образо¬
ванию выше 2000‘С, когда достигается условие AG<0, карбида
алюминия:2А1203 + 9С = AI4C3 + 6СО30
Таким образом, единственным промышленным методом по¬
лучения металлического алюминия из его оксида является элек¬
тролиз его расплава.Температура плавления чистого оксида алюминия в его а-
модификации, устойчивой выше 900°С, равна 2053°С. Электро¬
лиз его расплава связан с весьма высоким расходом электроэнер¬
гии на расплавление и поддержание высокой температуры ван¬
ны и приводит к низкому выходу по энергии. Поэтому в произ¬
водстве алюминия применяют не чистый оксид алюминия, а
систему, состоящую из оксида алюминия и криолита Na3AlFe,
то есть криолито-глиноземный расплав.Криолит плавится при 1100°С. Он
образует с оксидом алюминия при t°C , /содержании последнего около 15%
мае. эвтектику с температурой плав- 1100
ления 938°С. Дальнейшее увеличе¬
ние содержания глинозема в рас-
плаве приводит к резкому повыше- 5,00
нию температуры плавления систе-мы (рис. 2.8). 15 •Для снижения температуры Рис 2 8 д^мграмма ebtfrrth
плавления электролита, увеличе- ния системы «кри0лит-ок-
ния его электропроводности, улуч- сид алюминИя»
шения смачиваемости им анода в
расплав вводятся добавки фторидовалюминия, магния, лития и кальция. Промышленный элект¬
ролит имеет состав: Na^AlFe 75—-90%, А120з 1—10%, AIF3 5—
12%, CaF2 2—4%, MgF2 2—5%. Электролит подобного состава
не содержит ионов с потенциалом разряда меньшим, чем потен¬
циал разряда ионов алюминия, и имеет температуру плавления
950—970°С.В результате диссоциации основных компонентов системыА1203 ^ А1+3 + AIO3-3и NaeAlFe ^ 3Na++ A1F6-3а также добавок фтористых солей, расплавленный электролит
представляет сложную многокомпонентную систему, содержа¬
щую ионы:А1+3, Na+, Са+2, Mg+2, Li+, А103‘3, A1F6"3, F“.Последовательность разрядов ионов электролита определя¬
ется значением их потенциалов разряда в соответствии с пра¬
вилом разряда ионов. В табл. 2.3 приведены потенциалы раз¬
ряда ионов оксида алюминия и криолита на алюминиевом ка-31с.*
тоде (расплавленный алюминий) и угольном аноде» используе¬
мых в промышленных электролизерах, и уравнения первичных
процессов электролизаТаблица 2.3. Первичные процессы при электролизеЭлектродПотенциал разряда, ВПервичная реакцияКатод+2,71Na++1,66А1+3 + Зё = Д1Анодболее -0,5AlF,f3-0,472АЮз~3 - 6ё = А1203 + 1 #502Следовательно, при электролизе криолито-глиноземного рас¬
плава разряжаются ионы А1+3 и АЮ3"3. Теоретическое напряже¬
ние разложения на этих электродах А1 — С равно 1,66 - (-0,47) =
=2,13 В.Напряжения разложения добавок фтористых солей AIF3,
NaF, MgF2 LiF и CaF2, рассчитанные по термодинамическим
данным для температуры 1300°С, равны, соответственно: 3,97;
4,37; 4,61; 5,11; 5,16 В. Поэтому при электролизе эти примеси
не подвергаются электрохимическому разложению.Вторичные процессы при электролизе криолито-глинозем¬
ного расплава протекают как в анодном, так и в катодном про¬
странствах.В анодном пространстве при температуре электролиза про¬
исходит непрерывное окисление угольных анодов с образова¬
нием смеси оксида углерода (II) и оксида углерода (IV):С + О — СО и с + о2 = со2Адсорбируясь на поверхности анодов пузырьки газа меня¬
ют его природу и повышают потенциал разряда ионов. Поэто¬
му практическое напряжение разложения на этих электродах
А1—С*С02 значительно выше теоретического и составляет
4,3—4,5 В.Суммируя реакции первичных и вторичных процессов, по¬
лучаем суммарные уравнения реакций электролиза:А1203 + ЗС = 2А1 + ЗСО2А1203 + ЗС = 4А1 + ЗС02Основным аппаратом в процессе электролитического полу¬
чения алюминия является электролизер или алюминиевая ван¬
на (рис. 2.9).Электролизер состоит из катодного и анодного устройств.
Катодное устройство представляет металлических кожух пря-32
Рис. 2.9. Схема электролизера: а — ванна с самообжигающимися
анодами, б — блок с предварительно обожженным
анодом1 — кожух, 2 — огнеупорная футеровка, 3— футеровка из угольных плит,4 — глинозем, 5 — слой расплавленного электролита, & — гарнисаж, 7 —
спой алюминии, 8—рабочж бяок обожженного анода, 9—токолодводы,10 — наращтаемый блок, 11 — кожух анодной массы, 12 — анодная массамоугольной формы с огнеупорной изоляцией, футерованный
изнутри угольными плитами и блоками. Нижние (подовые) бло¬
ки являются одновременно токоподводами для расплавленно¬
го алюминия, играющего роль катода. Электролизер снабжен
системой газоулавливания и дожигания оксида углерода (II),
устройством для непрерывной подачи глинозема и системой
откачивания металлического алюминия.Анодное устройство состоит из угольных анодов, частично (до0,5 м) погруженных в расплавленный электролит, и запрессо¬
ванных в них токоподводов. Так как материал анодов участвует
во вторичных процессах электролиза, то они непрерывно обго¬
рают, что приводит к изменению расстояния между электрода¬
ми и нарушает ^эежим процесса. Для устранения этого явления
применяют непрерывные аноды различной конструкции. В со¬
временных электролизерах используют непрерывные аноды
двух типов; самообжигающиеся и предварительно обожженные.Самообжигающийся анод состоит из алюминиевого кожуха,
в который помещена брикетированная анодная масса с запрес¬
сованными в нее токоподводами. При обгорании анод опуска¬
ется в электролизер с помощью специальных направляющих
по заданной программе. При этом анодная масса постепенно пе¬
ремещается в зону все более высоких температур, спекается и
превращается в твердое монолитное углеродистое вещество —2 Хкническм технологи*. Том 233
угольный анод. По мере сгорания анода алюминиевый кожух
наращивается сверху и заполняется анодной массой. Для ано¬
дов этого типа характерны высокое электрическое сопротивле*
ние, повышенный расход энергии, нестабильность плотности
тока и высокая загазованность вследствие выделения летучих
веществ из анодной массы.В современных электролизерах высокой мощности применя¬
ют предварительно обожженные аноды, которые состоят из бло¬
ков, наращиваемых сверху по мере их обгорания. Токоподводы
впрессованы сбоку в готовые блоки.Плотность криолита, алюминия и глинозема в твердом состо¬
янии равны, соответственно: 2,95; 2,70; 3,90 т/м3. При темпера¬
туре электролиза плотность расплавленйого алюминия состав¬
ляет 2,3 т/м3, а электролита около 2,0 т/м3. Вследствие разно¬
сти плотностей жидкий алюминий отделяется от криолито-гли-
ноземного расплава и собирается на дне ванны. В процессе элек¬
тролиза в результате охлаждения ванны наружным воздухом на
поверхности расплава образуется твердый слой электролита
(гарнисаж), который утепляет ванну и снижает расход энергии.
Для извлечения из ванны расплавленного алюминия использу¬
ют вакуумные ковши или сифоны, засасывающая труба которых
вводится в жидкий алюминий через слой гарнисажа.Глинозем непрерывно подается в электролизер с помощью
пневматического штокового устройства, позволяющего проби¬
вать корку гарнисажа и дозировать глинозем.Система газоулавливания электролизера предназначена для
сбора выделяющихся при электролизе газов и удаления их в
газоочистную систему. В ваннах с самообжигающимися анода¬
ми для улавливания газов применяются специальные колоко¬
ла, обеспечивающие возможность дожигания летучих продук¬
тов коксования анодной массы и оксида углерода (II). В ваннах
с непрерывными предварительно обожженными анодами при¬
меняют, как правило, газоизоляцию всего электролизера, что
исключает подсос в него воздуха извне.Современные электролизеры для производства алюминия с
непрерывными предварительно обожженными анодами имеют
характеристики:Сила тока 50—150 кА
Расход электроэнергии 13,8—15,0 МВт-ч/т
Рабочее напряжение 4,2—4,5 В
Выход по току 0,9 долей ед.Выход по энергии 0,3 долей ед.34
Производительность подобных электролизеров составляет от0,5 до 1,2 тонны алюминия в сутки и может быть рассчитана по
формуле:П = О.ЗЗЛВДО-6 (2.1)где: П — производительность электролизера, т/су^;J — сила тока, А;т — время электролиза, ч;В, — выход по току, долей ед.2.7. Очистка и рафинирование алюминия\Примеси значительно ухудшают механические, электричес¬
кие и литейные свойства алюминия и снижают его коррозиоы-
ную стойкость. Для очистки от механических примесей и раство¬
ренных газов алюминий, выкачанный из ванны, хлорируют не¬
посредственно в вакуум-ковшах. При этом хлорируются водород
и некоторые металлы, а образовавшиеся хлориды и механичес¬
кие примеси, всплывают на поверхность металла и удаляются:
{А1 + Mg + Са} — {MgCl2 + СаС12 + А1С13} + А1После хлорирования алюминий выдерживают в электричес-
ких печах для удаления остатков примесей и усреднения соста¬
ва, после чего отливают в слитки. После такой очистки получа¬
ют алюминий марки А85, который содержит не менее 99,85%
металла. Для получения алюминия
высокой и особой чистоты его под¬
вергают дополнительному рафини¬
рованию. В промышленности при¬
меняются два метода рафинирова¬
ния: электролитический и с помо¬
щью субсоединений алюминия.В основе электролитического
трехслойного метода рафинирования
лежит процесс анодного окисления и
последующего катодного восстанов¬
ления алюминия. Анодом (нижний
слой) электролизера является рафи¬
нируемый алюминий, содержащий Рис. 2.10. Схема электроли-
для увеличения плотности до 40% тического рафинирования
меди, катодом (верхний слой) —очи- алюминия:
щенный алюминий. Между катодом 1 _ слой рафинируемого
и анодом располагается расплавлен¬
ный электролит, состоящий из сме¬
си хлоридов бария и натрия и фто-р^ 2,35 т/м3р2=2,7т/м3р3= 3,2 т/м3алюминии, содержащего
медь, 2 — слои электролита,
3 — слой чистого алюминия2*35
ридов алюминия и натрия (рис. 2.10). В процессе рафинирования,
так как Ер<Ещ,имесей> алюминий растворяется на аноде:А1-Зё = А1+3,ионы его, вследствие разности плотностей слоев загрязненного
алюминия и электролита, проходят через слой последнего и вос¬
станавливаются на катоде:А1+3 + Зё = А1В то же время примеси с большим потенциалом остаются в
слое рафинируемого металла и накапливаются в слое электро¬
лита. По мере накопления примесей анодный спав и электро¬
лит периодически заменяют. Энергоемкость процесса электро¬
литического рафинирования составляет около 18 МВт-ч на тон¬
ну металла. Полученный этим методом рафинирования алюми¬
ний имеет чистоту 99,99%.Рафинирование с помощью субсоединений основано на воз¬
гонке легколетучих субсоединений одновалентного алюминия,
образующихся при высокотемпературной обработке рафиниру¬
емого алюминия хлоридом алюминия (III). Примеси при этом
не перегоняются и остаются в остатке от рафинирования. При
охлаждении продуктов перегонки до 700°С субсоединения раз¬
лагаются на алюминий и хлорид алюминия, который возвра¬
щается в процесс;книге г*~п{А1 +П}+А1С13 ► {А1С1+П} А 700°СI—► AlCl ►A1+AJC13Чистота алюминия, полученного через субсоединения равна
99,9995%.Алюминий сверхвысокой чистоты (99,9999%) может быть
получен методом зонной плавки.Расходные коэффициенты на 1 тонну алюминия составля¬
ют:Глинозем 2,0 т Криолит 0,1тАнодная масса 0,7 Электроэнергия 18МВт*ч2.8. Производства криолита и угольных изделийПроизводства криолита и разнообразных угольных изделий
для электролизеров являются сопутствующими, но необходи¬
мыми элементами производства алюминия электролитическим
методом.36
Криолит — двойная соль натрия и алюминия и фтористово¬
дородной кислоты 3NaF-AlF3 (или NaaAlFe) может быть полу¬
чен через стадию кислотного разложения плавикового пшата
(фторида кальция) или из отходов суперфосфатного производ¬
ства. Кислотный способ производства криолита состоит из сле¬
дующих стадий:1. Разложение фторида кальция серной кислотой при 200°СCaF2 + H2S04 - 2HF + CaS042. Получение фторалюминиевой кислоты6HF + А1(ОН)3 = H3AIF6 + ЗН203. Нейтрализация фторалюминиевой кислоты карбонатом
натрия2НзАШ6 + 3Na2C03 = 2 Na^AlFe + 3C02 + ЗН20Выпавший осадок криолита отделяют от раствора, фильтруют,
промывают на вакуум-фильтре и сушат в трубчатой сушилке.Более экономично производство криолита из отходов супер¬
фосфатного производства. В этом производстве на 1 тонну вы¬
рабатываемого суперфосфата выделяется около 6 кг фтора в
виде тетрафторсилана, при улавливании которого образуется ра-
створ, содержащий до 12% гексафторкремневой кислоты. Ее
перерабатывают на криолит по схеме:H2Si*6 + 2А1(ОН)3 = 2A1F3 + Si02 + 4Н20
H-jSiFe + 3Na2C03 « 6NaF + Si02 + 3C02 + H203NaF + AIF3 - NasAlF6Производство угольных материалов связано с тем, что они
используются для изготовления анодов и элементов футеровки
электролизеров. Эти детали работают при весьма жестких усло¬
виях и должны удовлетворять определенным требованиям по
термостойкости, механической прочности, электропроводности
и стойкости к расплавленным солям. Углеродистые материалы
делят на футеровочные блоки, обожженные аноды и анодные
массы для самообжигающихся анодов. Их изготавливают из
твердых углесодержащих материалов, составляющих их осно¬
ву (каменноугольный и нефтяной кокс, антрацит), и связующих
веществ, коксующихся при обжиге (каменноугольный пек, ка¬
менноугольная смола). Принципиальные схемы изготовления
углеродных материалов различны и зависят от природы сырья.Себестоимость электролитического алюминия. Производ¬
ство алюминия относится к числу материало- и энергоемких ме¬
таллургических производств. Поэтому в структуре себестоимо¬37
сти алюминия затраты на сырье й энергию являются основны¬
ми. В табл. 2.4 приведена структура себестоимости алюминияи, для сравнения, структура себестоимости продукции цветной
металлургии в целом.Таблица 2.4. Структура себестоимости алюминияСтатья затратДоля затрат f%АлюминийПродукций цветной
металлургииСырье м основные материалы64,053,6В том чиоге: глинозем49,5—фтористые соли5,6 анодыв,9 1Электроэнергия24,17,9Топливо— .3,8Заработная плата3,414,5Амортизация и прочие расходы9,112,3Контрольные вопросы1. Какие горные породы используются в качестве алюминиевых руд?2. Как влияет природа сырья на выбор метода производства глинозе¬
ма?3. Из каких операций состоит процесс получения глинозема методом
выщелачивания? Приведите принципиальную схему процесса.4. Почему метод спекания в производстве глинозема может приме¬
няться для всех видов алюминиевого сырья?5. Почему электролиз расцлава глинозема является единственным
промышленным методом производства алюминия?6. Для чего электролизу подвергают не чистый глинозем, а его ра¬
створ в криолите?7. Что такое «самообжигающиеся» аноды, используемые в электро¬
лизере?8. На чем основан метод электролитического рафинирования алюми¬
ния?
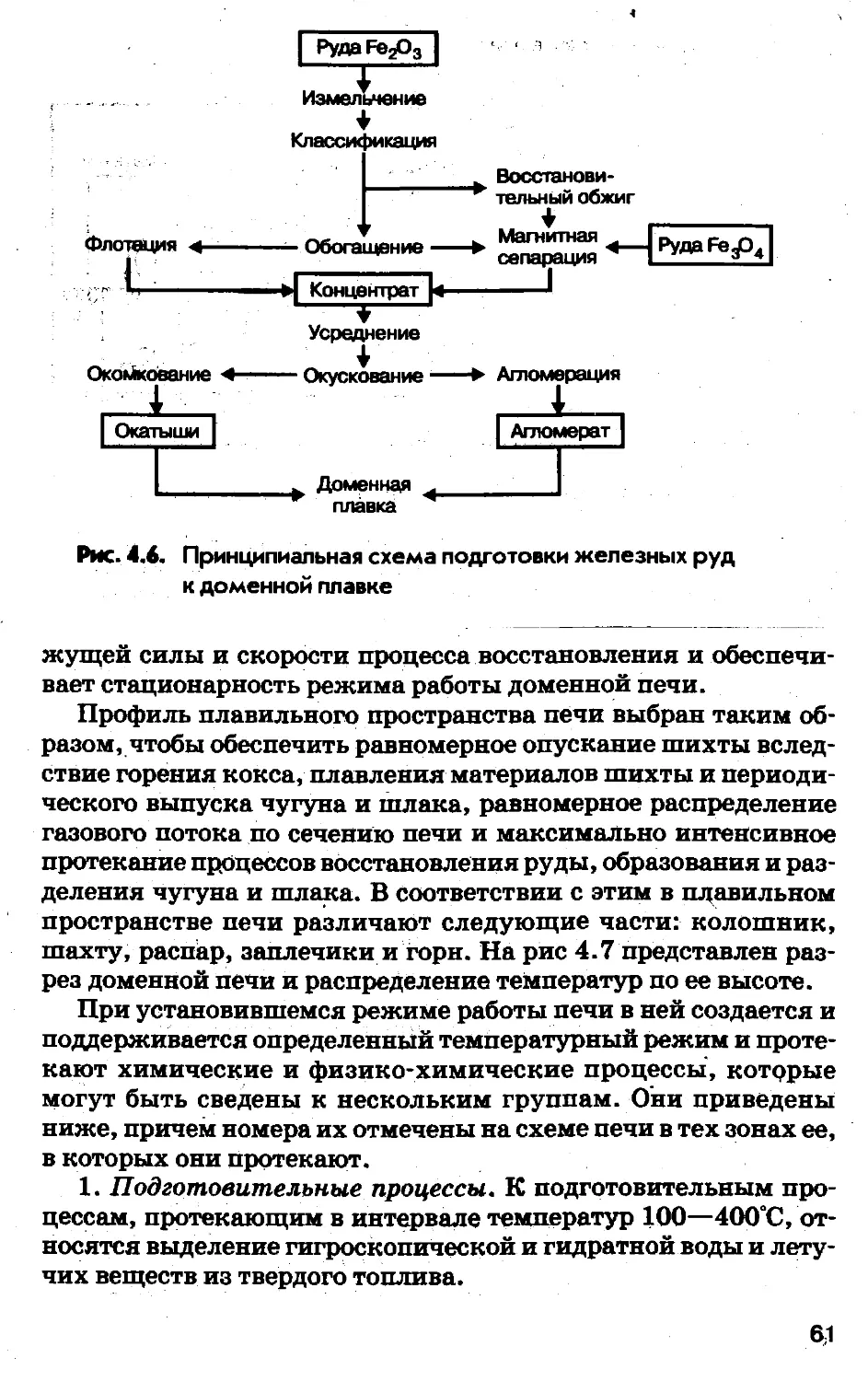
Глава IIIЧЕРНЫЕ МЕТАЛЛЫВ соответствии с промышленной классификацией металлов
(глава I) к черным металлам относятся железо и его сплавы с
углеродом — железоуглеродистые сплавы.3.1. Свойства железа и его сплавовЖелезо — один из самых распространенных элементов в при¬
роде. Его содержание в земной коре составляет 4,2%. Как со¬
ставная часть железо входит почти во все горные породы.Железо представляет собой твердый светло-серебристый ме¬
талл, пластичный, легко поддающийся ковке, прокатке, штам¬
пованию и волочению. Временное сопротивление на разрыв со¬
ставляет 170—210 МПа. Плотность железа 7,87 т/м3, темпера¬
тура плавления 1539°С, температура кипения 3200°С. Многие
свойства железа существенно зависят от его чистоты.В отсутствии влаги чистый металл химически стоек, не реа¬
гирует с кислородом, серой, галогенами, однако в высокодис¬
персном состоянии пирофорен. Техническое железо и его спла
вы корродируют в атмосфере паров воды, оксида углерода (IV)
и кислорода с образованием пористого слоя гидратированного
оксида железа (II) ГеОпЩО. Не взаимодействует с щелочами.
С углеродом при высоких температурах образует растворимый
в металле карбид железа FegC (цементит) с содержанием угле-
рода6,67% и температурой плавления 1550°С,атакже два типа
твердых растворов. Железо так же образует многочисленные
сплавы с другими металлами.Железо полиморфно и в зависимости от температуры суще¬
ствует в четырех аллотропических формах (модификациях),
связанных обратимыми переходами:768°С 910°С 1400°С 1539°Са-железо +*■ (3-железо «=* у-желебо «=* 8-железо расплавМодификации железа различаются типом кристаллической
решетки, удельным объемом, теплоемкостью, магнитными и
механическими свойствами и растворимостью в них углерода
(табл. 3.1).В основе полиморфных превращений железа лежит процесс
кристаллизации; они протекают через стадию образования и
роста зародышей новой фазы с последующим формированием
вокруг них кристаллов иной структуры. Ввиду узости темпера¬
турных интервалов переходов a-железа в p-железо (142°С) и39
5-железа в жидкое состояние (139°С) ими можно пренебречь и
рассматривать только два перехода:910'С 1539“Са-железо у-железо з=* жидкий металл,на основании которых и строить упрощенную диаграмму состо¬
яния.Таблица 3.1. Аллотропические формы железаВид кристалли¬
ческой решеткиМагнитныесвойстваРастворимость
углерода,% мае.Жидкоесостояние——■■Ч 6-FeОЦКНе магнитно—\y-FeГЦКНе магнитно2,14 при 11474:0-Fe \ОЦКНе магнитно-—a-Fe \ОЦКМагнитно0,025 при 723°СОЦК — кубическая объемноцентрироаанная решетка, ГЦК — кубическая
грвмцентрироаанная решетка.В зависимости от того, в какую кристаллическую решетку
внедряется углерод при его растворении в железе, различают
два типа твердых растворов — феррит и аустенит.Феррит это твердый раствор внедрения углерода в а-железо
(ОЦК). По свойствам он близок к чистому железу, пластичен,
имеет малую прочность, обладает магнитными свойствами.Аустенит (по имени Р.Остена) представляет собой твердый
раствор внедрения углерода в у-железо (ГЦК). Он устойчив при
температурах выше 72ТС, тверд, но хрупок, не обладает маг¬
нитными свойствами.Изменения фазового состава и структуры железоуглеродистых
сплавов, то есть системы «железо—углерод» в зависимости от тем¬
пературы при различном содержании компонентов в ней представ¬
лены на упрощенной (не учитывающей существование р-и S-форм
Железа) диаграмме состояния этой системы (рис. 3.1). Буквенные40
обозначения узловых (критических) точек на диаграмме всюду
стандартизованы и приняты во всех странах.Обозначение фаз: Ф — феррит, А — аустенит, Ц) — цементит первич¬
ный, Ц} — цементит вторичный, П = Ф + Ц — перлит, J1 = А + Ц< —
ледебурит, Д, = П + Ui — ледебурит превращенный, Р — расплавЛевая часть диаграммы (до точки Е) описывает превращения,
происходящие в сталях, то есть в сплавах с содержанием угле¬
рода до 2,14%. Правая часть — превращения, происходящие в
чугун ах — сплавах с содержанием углерода от 2,14 до 6,67%.
Так как цементит (карбид железа FeaC) представляет собой как
химическое соединение самостоятельный компонент системы,
диаграмма состояния ограничивается этим содержанием угле¬
рода. К тому же, сплавы, содержащие более 6,67% углерода,
практического значения не имеют. Таким образом, в диаграм¬
ме левая ордината характеризует чистое железо в а-модифика¬
ции до точки G и в у-модификации в интервале точек G и А.
Правая ордината соответствует цементиту.Линии ACD и AECF на диаграмме характеризуют фазовые
превращения в системе. Выше линии ACD (линии ликвидуса)
все сплавы находятся в жидком состоянии, ниже линии AFCF
(линия солидуса) — в твердом состоянии. Между линиями лик¬
видуса и солидуса находятся двухфазные области: при охлаж-41
детаи расплава ниже линии АС из него кристаллизуется &уоте-
нит, а ниже линии CD — цементит, называемый первичные* так
как выделяется из расплава, В точке С затвердевает эвтектичес¬
кая смесь аустенита и первичного цементита — ледебурит (по
имени исследователя Ледебура) с содержанием углерода 4,3%.Твердая фаза с содержанием углерода менее 2,14%, соответ¬
ствующая сталям, описывается областью диаграммы AGSE й
представляет однородный твердый раствор аустенит. Из диаг¬
раммы следует, что температура плавления сталей (линия AJE)
зависит от их состава, то есть содержания углерода.Твердая фаза в области, лежащей между линиями EGF и PSK
с содержанием углерода более 2,14%, соответствующая белым
чугунам, имеет различный состав. Доэвтектические чугуны
(2,14—4,3% углерода) состоят из аустенита и ледебурита, эв¬
тектические (4,3%) из ледебурита и заэвтектические (4,3—
6,67%) из цементита и ледебурита. При этом, в отличие от ста¬
лей, температура плавления чугунов (линия EGF) постоянна и
не зависит от содержания в них углерода.При дальнейшем понижении температуры в системе проте¬
кают превращения в твердой фазе, связанные с переходом а-
модификации железа в у-модификацию и изменением раство¬
римости углерода в железе. Это приводит к распаду аустенита
и выделяющийся избыточный углерод образует с железом це¬
ментит, который в отличие от первичного называется вторич¬
ным. Для сталей это превращение начинается при температу¬
рах, отвечающих линии GSE и продолжается до линии PSK
(723°С). При этом, в сталях, содержащих менее 0,83% углеро¬
да, выделяется феррит, а в сталях с большим содержанием уг¬
лерода — вторичный цементит. В точке S аустенит распадает¬
ся с образованием эвтектойдной смеси феррита и цементита —
перлита (от Perl — жемчуг). Это может быть представлено в
следующем виде:В точке S (0,83% углерода)Феррит —*Аустенит —1_^ Цементит _|—► 08ВЛИТВ области с содержанием углерода менее 0,83%
ФерритАустенитДигтоиит —Цементитг» Феррит 1 •-►Феррит —| _1+ Аустенит !_► ^ _|ПерлитВ области с содержанием углерода более 0,83%i—fr Цементит
Аустенит —I I—► Феррит
► Аустенит 1Аустенит 1 -► Перлит~ Цементит42
При охлаждении сплавов, содержащих в структуре ледебу¬
рит (чугуны) ниже линии РК, не сущзэтвделций при низких
температурах аустенит в составе последнего превращается в
перлит. Ледебурит, в котором аустенит превратился в перлит и
состоящий, следовательно, из перлита и цементита, получил
название ледебурита превращенного.Таким образом ,при температурах ниже 723‘С (линия РК)
железоуглеродные сплавы в зависимости от содержания угле¬
рода имеют следующий фазовый состав:—доэвтектоидные стали (менее 0,83% С) — перлит и феррит;— эвтектоидные стали (0,83% С)— перлит;— заэвтектоидные стали (более 0,83% С) — перлит и цемен¬
тит;— доэвтектические чугуны (менее 4,3% С) — перлит, леде- ■»■■■
бурит превращенный и цементит вторичный;— эвтектические чугуны (4,3% С) — ледебурит превращен¬
ный;— заэвтектические чугуны (более 4,3% С) — ледебурит пре¬
вращенный и цементит первичный.Диаграмма состояния «железо—углерод* позволяет проана¬
лизировать сущность ттреврятттений, происходящих в железо-
углеродных сплавах при нагреве и охлаждении и, исходя из это¬
го, выбрать соответствующие режимы термической обработки
сталей и чугунов с целью придания им определенных свойств и
структуры.3.2. Классификация черных металловВ основу классификации черных металлов положен их хи¬
мический состав. В общем случае черные металлы — это слож¬
ные системы, которые помимо железа и углерода содержат раз¬
нообразные примеси (серу, азот, фосфор, кремний и др.), вно¬
симые в металл из исходного сырья в процессе производства, а
также металлы целенаправленно добавляемые с целью прида-
. ния сплаву определенных свойств.По содержанию углерода черные металлы делят на две основ¬
ные группы: стали,(менее 2,14% С) и чугуны (более 2,14% С).
Так как это количественное различие связано с особенностями
модификаций железа и структурой металла (см. 3.1), подобная
классификация является естественной классификацией.Стали делят на углеродистые, в которых другие металлы со¬
держатся в незначительных количествах и не влияют на их
свойства и легированные, в состав которых введены такие ме-43
таллы, как хром, никель, вольфрам, ванадий, молибден, и др.
для ттрид&ния им определенных свойств. По содержанию угле-
рода углеродистые стали делят на низко-, средне- и вгисокоугле-
родистые.Чугуны делят на белые (передельные), серые (литейные) и
модифицированные. Белые чутуны содержат углерод в форме
карбида железа ГезС (цементита) и образуются при кристалли¬
зации расплавов. В серых чугунах углерод находится частично
в виде графитовых включений различной конфигурации, вы¬
деляющихся из жидкой или твердой фазы при медленном ох¬
лаждении (графитизация). Модифицированные чугуны содер¬
жат добавки, улучшающие распределение графита и структу¬
ру чугунов (кремний, магний, алюминий).Особую группу черных металлов составляют ферросплавы —
малоуглеродистые сплавы железа с высокой концентрацией
других элементов: ферромарганец, ферросилиций, феррохром
и т.п.На рис. 3.2 представлена классификация черных металлов
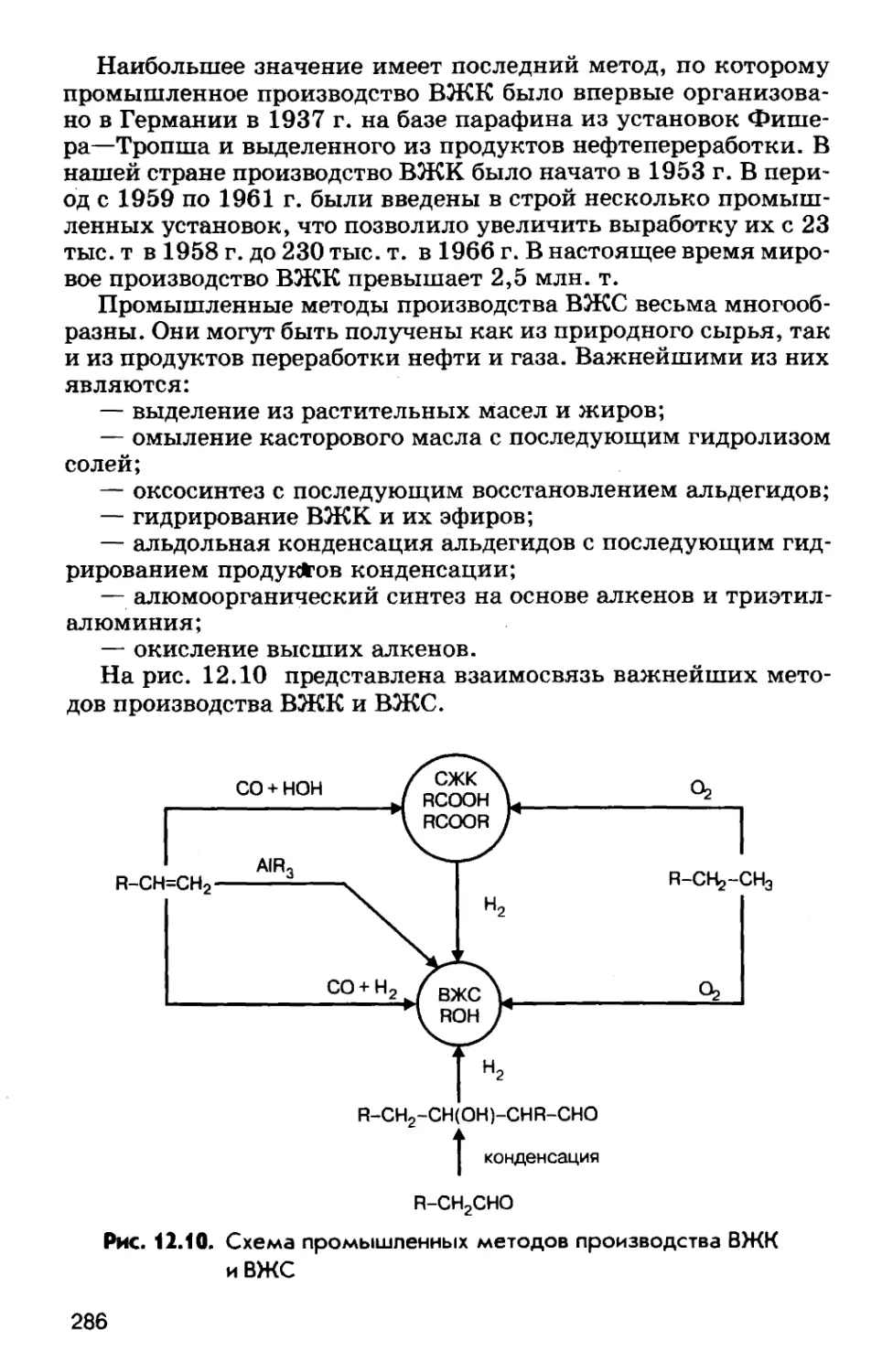
по химическому составу.Низкоугперодис-
• тые С < 0,25%Углеродистые х*. Сроднеугперодис-
{Fe + С} I стые С = 0,25—0,6%СталиВьюокоутеродис-
тые С > 0,6%2,14%!-► Хромистые-► НикелевыеЖелезо-
утперо-.
дистые
сплавы^ Легированные -► Ванадиевые
{Fe + C + Me}^ Содержащиенесколько леги¬
рующих метал*
ловФерро¬сплавыБелые
2,2—3,5%^ Чугуны Серые
2,14% ~ “*2,5—4,0%44
i3.3. Масштабы производства и области применения
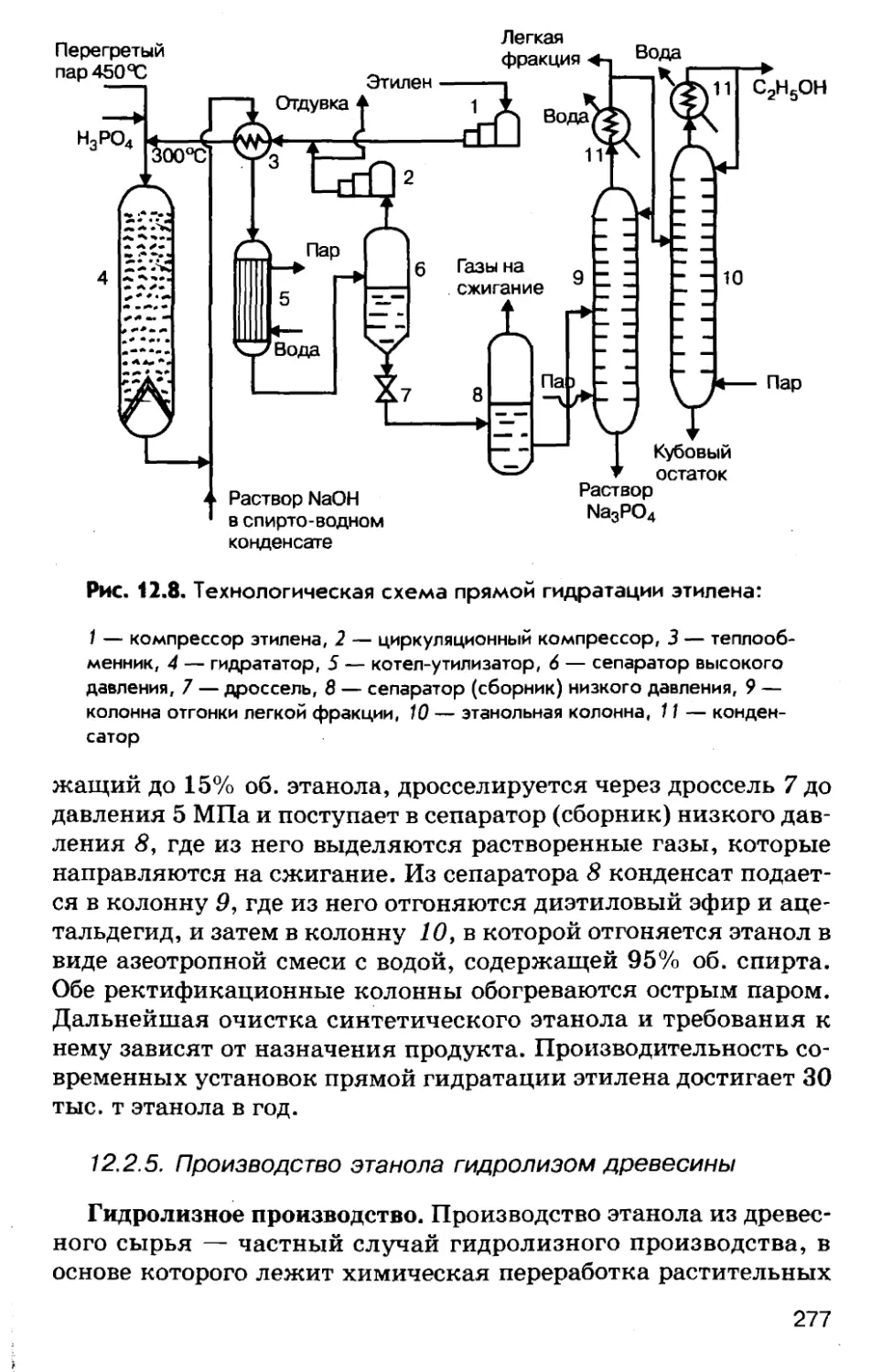
черных металловЧерные металлы относятся к числу важнейших конструк¬
ционных материалов, являются основой современной индуст¬
рии ц, в определенной степени, мерилом уровня ее развития. В
табл. 3.2 приведены данные о производстве на душу населения
черных металлов и, для сравнения, некоторых полимерных ма¬
териалов в СССР и развитых странах по данным 1987 года.Таблица 3.2. Производство материалов на душу населенияСтранаМатериал, кг/челЧугунСтальПластическиемассыХимическиеволокнаСССР40457516,05,4США180336104,516,1Япония59881282,013,9Широкое использование черных металлов обусловлено бо¬
гатой сырьевой базой, относительной простотой производства
и их высокими механическими свойствами. Различные желе¬
зоуглеродные сплавы составляют около 92% от всех произво¬
димых металлов. Подчеркивая это значение железа П.П. Ано¬
сов писал:«Природа как бы предвидела необходимость железа для челове¬
ческого рода, распространив металл сей в количестве, несравненно
большем против всех прочих металлов: нет ни одной страны, в коей
не было бы признаков железа, и нет ни одного состояния людей, ко¬
торые не имели бы в нем надобности».Мировое производство черных металлов составило (1987 год):
чугуна 509 млн. т, стали 737 млн. т. По прогнозу ЮНЕСКО миро¬
вое производство стали должно достигнуть к 2000 году 1200 млн. т
или 500—700 кг на душу населения Земли. Производство черныхметаллов в РФ (млн. т) представлено в таблице 3.3.Таблица 3.3. Производство черных металлов в РФ (млн.т)ПродуктГод1992199419961998Чугун46,136,537,234,8Сталь67,048,349,343,845
Из табл* 3.3 следует, что аа последние годы происходит сни¬
жение отношения железная руда/готовый металл. Этот процесс
стал следствием интенсификации производства чугуна и стали
и повышения степени регенерации металла.Область применения черных металлов как конструкционных
материалов чрезвычайно многообразна и практически неогра¬
ничен. Серые чугуны используют как материалы для произ¬
водства фасонных отливок. Они хорошо обрабатываются реза¬
нием, имеют повышенную сопротивляемость износу, вследствие
включений графита хорошо работают в условиях трения. Ис¬
пользуются в станкостроении и автостроении (станины, корпус¬
ные детали, зубчатые колеса, гильзы, блоки цилиндров, порш¬
невые кольца и др. детали), для изготовления товаров массово¬
го потребления (ванны, раковины, отопительные батареи, по¬
суда и др.). Из модифицированных высокопрочных чугунов
изготавливают детали прокатных станов, коленчатые валы и
детали двигателей автомобилей.Белые чугуны хрупки, плохо обрабатывается резанием. Их
используют для переделки в сталь и производства методом от¬
жига ковкого чугуна. Ковкий чугун хорошо воспринимает пе¬
ременные и ударные нагрузки, занимая промежуточное место
между серым чугуном и сталью. Он используется для изготов¬
ления деталей высокой прочности (цепи, тормозные колодки,
ступицы маховиков, крюки и т.п.).Область использования сталей значительно шире. По назна¬
чению стали подразделяются на конструкционные, предназна¬
ченные для изготовления деталей машин, конструкций и при¬
боров, инструментальные — для изготовления режущего,
штамповрго и измерительного инструмента и стали специаль¬
ного назначения — коррозионностойкие (нержавеющие), изно¬
состойкие, жаропрочные и т.п.Наиболее распространенные углеродистые конструкционные
стали используют для производства кровельного и котельного
железа, жести, стального листа, мягкой проволоки, деталей,
изготавливаемых методом проката (балки, рельсы, трубы) или
литья. Легированные конструкционные стали с содержанием
легирующих элементов от 2,5 до10% применяют в производ¬
стве шарикоподшипников, деталей транспортных машин
(валы, поршни, шестерни), химической аппаратуры, корпусов
летательных аппаратов, газовых турбин, реактивных и ракет¬
ных двигателей.46
Инструментальные стали могут быть высокоуглеродистыми
или легированными. Они характеризуются высокой твердо¬
стью, достаточной вязкостью и большой износо- и теплостой¬
костью. Из них изготавливают резцы, фрезы, штампы, рабочие
детали измерительных инструментов.Ферросплавы применяют для раскисления чугуна и стали
(ферромарганец, ферросилиций) или их легирования (ферро¬
ванадий, феррохром, ферровольфрам и др.). Ферросплавы
имеют более низкую чем соответствующие элементы, темпе¬
ратуру плавления, что облегчает их введение в жидкие сталь
и чугун.К черным металлам относят и магнитные материалы, при¬
меняемые для изготовления сердечников трансформаторов,
электрических машин, измерительной аппаратуры и т.п. Осо¬
бую группу веществ, близких к ним по свойствам, составляют
ферриты — соединения оксида железа (Ш) с оксидами других
металлов, широко используемые в приборостроении.Для обозначения чугунов и сталей принята определенная
система обозначений. Серые чугуны маркируются буквами СЧ
с указанием пределов прочности при растяжении и при изгибе,
например, СЧ12-28. Ковкий чугун обозначается буквами КЧ с
указанием предела прочности при растяжении и относительного
удлинения, например КЧ50-4.Углеродистые стали в зависимости от назначения подразде¬
ляют на группы Л, Б и В, а внутри каждой группы разделены
на категории, указанные в марке стали, например: СТЗ — сталь
группы А третьей категории, БСТ2 — сталь группы Б второй
категории и т.д.Легированные стали маркируют набором цифр и букв. Буква
обозначает легирующий элемент (В — вольфрам, Н — никель,
X — хром, М — молибден, К — кобальт, Г — марганец, С —
кремний, Ю — алюминий, Т — титан), цифра перед буквой —
содержание углерода в сотых долях процента, цифра после бук¬
вы —содержание легирующего элемента, превышающее 1% в
целых процентах. Например, сталь марки 30X13 содержит 0,3%
углерода и 13% хрома, сталь 20ХН2Т — 0,2% углерода, 2% ни¬
келя, а также хром и титан в количествах менее 1%.3.4. Краткая история развития производства черных
металловЖелезо и его сплавы известны человечеству в течение нескольких
тысячелетий. С ними неразрывно связано создание и совершенствова¬
ние разнообразных орудий труда и развитие материальной культуры,47
что.нашло отражение в исторической терминологии в выражении «же*
лезный век». \До ХШ столетия единственным методом получения железа был сы¬
родутный процесс, при котором руда восстанавливалась в горнах дре¬
весным углем с принудительной подачей воздуха ручными мехами. Из-
за низких температур при этом получался мягкий пластичный металл,
содержащий значительное количество шлака. Для его удаления полу¬
ченную губчатую массу — крицу обжимали молотом, получая свароч¬
ное железо. Процесс был мало производителен (до 60 кг в сутки с горна),
требовал большого расхода угля (до 16 кг на кг железа) при весьма низ¬
кой степени извлечения железа из руды (не более 0,4 долей ед.)<По мере увеличения высоты горна и интенсификации дутья (меха¬
нические мехи) температура плавки возрастала, что приводило к на¬
углероживанию железа и образованию наряду с мягким металлом
жидкого чугуна. Вначале чугун из-за хрупкости рассматривали как
отход производства, затем его стали использоваться как литейный
материал, а с XIV столетия его начали перерабатывать в специальных
печах — кричных горнах в железо. В связи с этим сыродутный горн
постепенно трансформировался в шахтную печь высотой до 8 метров —
домницу, предназначенную уже для производства исключительно
чугуна. Это был прототип современной доменной печи. Подобный двух¬
ступенчатый метод переработки железных руд оказался более совер¬
шенным — возросла производительность печи, снизился расход угля,
увеличился выход чугуна.В дальнейшем развитие техники выплавки чугуна (доменный про¬
цесс) и получения стали (сталеплавильное производство) шло парал¬
лельно в направлении совершенствования технологии, увеличения
мощности агрегатов, внедрения новых материалов и снижения стоимо¬
сти выплавляемого металла.В 1735 году в качестве топлива в доменных печах был предложен
вместо древесного угля каменноугольный кокс и с XIX века началось
его интенсивное внедрение в доменное производство, что способство¬
вало развитию черной металлургии в степных безлесных районах. В
18$8 году был выдан патент на применение в доменных печах для ду¬
тья подогретого воздуха. Это позволило за счет повышения темпера¬
туры в горне сократить расход топлива и увеличить производитель¬
ность печи. В1832 году в конструкцию доменной печи был введен зак¬
рытый колошник, что обеспечило возможность улавливания домен¬
ного газа и его использование в качестве топлива для подогрева дутья,
одновременно улучшив экологию. Дальнейшее совершенствование
доменного процесса заключалось в применении обогащенного кисло¬
родом воздушного дутья, повышении давления дутья, использовании
газообразного и жидкого топлива для снижения расхода кокса в связи
с дефицитом коксующихся углей.Одновременно совершенствовался и способ производства стали.
Кричное мягкое железо не могло удовлетворить всех потребностей. Его
переработка в сталь (науглероживание) вплоть до середины XIX века
осуществлялась или в твердой фазе или в тиглях (тигельный метод). В
1784 году для переделки чугуна в железо был предложен метод пуд-48
лингования в пламенных отражательных печах, в которых тонка от¬
делялась от ванны с расплавленным чугуном. Примеси удалялись из
чугуна под воздействием окислительной атмосферы в печи. Получен;
ную крицу подвергали обжиму под молотом.В 1856 году Г. Бессемер предложил высокопроизводительный ме¬
тод переделки чугуна в литую сталь продувкой его воздухом в конвер¬
торе — конвертерный метод. Модификация метода, отличавшаяся
материалом футеровки конвертера была разработана в 1878 году С.То**
масом. В1864 году П. Мартен предложил способ получения литой ста¬
ли в регенеративных пламенных печах, использовав для этой цели
изобретенную Ф. Сименсом печь для варки стекломассы. Тем самым
была решена и проблема переработки стального лома, то есть регене¬
рации металла.С начала XX столетия для выплавки стали, главным образом efe вы¬
сококачественных легированных марок, стали использоваться элект¬
рические дуговые и индукционные печи.Решающий шаг в совершенствовании сталелитейного производства
был сделан в 50-х годах XX века разработкой кислородно-конвертер¬
ного метода выплавки стали, который позволил использовать в каче¬
стве сырья не только чугун, но и стальной лом и руду, существенно
повысить производительность и улучшить качество стали. В настоя¬
щее время этот метод, наряду с электроплавильным, преобладает.Дальнейшее совершенствование металлургии черных металлов шло
по пути разработки и промышленно го внедрення метода прямого вос-
становлёния железа, минуя доменный процесс, и создания непрерыв¬
ных металлургических комплексов переработки чугуна в сталь.Теоретические основы металлургии чугуна и стали создавались на
протяжении нескольких столетий. К числу первых исследований в
этой области следует отнести фундаментальный труд Г. Агриколы (Ба¬
уэра) «12 книг о металлах» (1556 г.), обобщивший многовековой опыт
извлечения металлов из руд, работу М.В. Ломоносова «Первые осно¬
вания металлургии или рудных дел» (1763 г.), созданную им на осно¬
ве изучения движения воздуха и газов в пламенных металлургичес¬
ких печах, разработку П.П. Аносовым теоретических основ получе¬
ния высококачественной литой стали (1828 г.), метода прямого полу¬
чения железа из руд и принципов микроскопического метода изуче¬
ния структуры сталей.В период 1868—1876 гг. Д.К. Чернов проводит цикл исследова¬
ний по установлению взаимосвязей между тепловой обработкой
стали, ее структурой и свойствами и создает теорию кристаллиза¬
ции стали. В ряде работ он формулирует основы современного ме¬
талловедения, теорию термической обработки стали, устанавливает
значения критических точек в диаграмме состояния «железо—уг¬
лерод». В 1891 году выходит 'в свет его курс «Сталелитейное
дело» — первый в России труд по металловедению. В последующие
годы исследования Ф. Осмонда, Р. Остена, А.А. Байкова, Н.Т. Гуд-
цова и П.Геренса позволили уточнить диаграмму состояния «же¬
лезо—углерод» и вместе с работами М.А. Павлова, Н.С. Курнако-
ва и И.П. Бардина создать теоретический фундамент доменного и49j
сталеплавильного производств и существенно усовершенствовать
их технологию/Промышленное производство чугуна, и стали в России возникло в
начале ХУШ века с постройкой доменных печей в Тульской губернии
и металлургических заводов на Урале. В 1699 году был построен Не¬
вьянский завод, после чего начинается бурное развитие отечественной
черной металлургии, затормозившееся к XIX веку в связи с истоще¬
нием запасов древесного угля.В конце XIX века производство черных металлов развивается на
lore России. В1870 году построен Сулинский и позже Юзовский заво¬
ды и на бдое руд Кривого Рога и донецких углей создана мощная ме¬
таллургическая база. В1870 году на Сормовском заводе (Н.Новгород)
вступают в строй первые в стране мартеновские печи, а на заводах До*
нецкого бассейна — конвертеры. В 1910 году в России установлена
первая дуговая электропечь, а в 1917 году под Москвой построен элек¬
трометаллургический завод.В 30-е и 40-е годы в стране были построены Магнитогорский и Куз¬
нецкий металлургические комбинаты, Запорожский и Криворожский
заводы, реконструированы Днепропетровский, Макеевский, Нижне¬
днепровский и Таганрогский заводы, строятся заводы высококаче¬
ственных сталей «Электросталь» и «Днепроспецсталь». В послевоен¬
ные годы в стране продолжается рост производства черных металлов,
строятся Новолипецкий, Западно-Сибирский, Череповецкий и другие
заводы. В металлургическом производстве начинают применяться кис¬
лородные конвертеры емкостью 350 т, 900-тонные мартеновские печи,
двухванные сталеплавильные агрегаты, доменные печи с полезным
объемом до 5000 м3. Одновременно развивается металлургия специ¬
альных сталей и сплавов, в производство внедряются методы электро-
шлакового (ЭШП), вакуумного индукционного (ВИП), вакуумно-ду-
гового (ВДП), электронно-лучевого (ЭЛП) и плазменно-дугового (ПДТТ)
переплава стали.3.5, Железные рудыИсходное сырье для производства черных металлов — желез¬
ные руды. Вследствие высокого сродства к кислороду железо
находится в природе в виде оксидов различной степени окисле¬
ния. Минеральные породы, содержащие подобные оксиды в
смеси с пустой породой называются железными рудами.Пустая порода руды состоит из оксидов кремния, алюминия,
кальция и магния, образующих разнообразные силикаты и алю¬
мосиликаты. Кроме пустой породы в железных рудах содержат¬
ся в виде оксидов такие металлы как марганец, хром, никель,
молибден, вольфрам, ванадий.По содержанию железа руды делятся на бедные (менее 30%),
средние (30—50%) и богатые (более 50%).50
По форме соединений железа руды делятся на следующие
разновидности:1. Магнетитовая руда (магнитный железняк). Содержит
железо в виде магнетита РезС>4 (78,4% Fe), который можно рас¬
сматривать как состоящий из оксйдов железа II и П1 FeOFe^a.
Степень окисления магнетита, то есть соотношение в нем окси¬
дов железа, характеризуется отношением Feo6m:FeO. Для чис¬
того магнетита оно {>авно 2. В рудах, за счет окисления оксида
железа (II), оно повышается до 7 и выше. Подобные руды назы¬
ваются мар шитом. В наиболее распространенных рудах содер¬
жание железа составляет 50—60%, обычно они содержат серу
и фосфор и трудно восстанавливаются.2. Гематитповаяруда (красный железняк). Содержит желе¬
зо в виде безводного оксида Fe20g (70,0% Fe), образовавшегося
в результате выветривания и окисления магнитного железня¬
ка. Содержит 50—70% железа с небольшим количеством серы
и фосфора. Легко восстанавливается.3. Бурый железняк. Содержит железо в виде гидратирован¬
ного оксида железа (III): лимонита 2Fe203*3H20 и гетита
ГегОз'НгО и образуется при выветривании и окислении других
железных руд. Наиболее распространенный лимонит содержит
значительную примесь оксидов алюминия и кремния при со¬
держании железа 30—40%. Легко восстанавливается.4. Сидерит (шпатовый железняк). Содержит железо в виде
его карбоната FeCC>3 без каких либо вредных примесей. Содер¬
жание железа не превышает 30—40%.Основными месторождениями железных руд на территории
РФ являются Курская магнитная аномалия (42 млн. т), содер¬
жащая мартито-гематитовые железняки и железистые кварци¬
ты; Качканарское, Магнотогорское и Гусевское месторождения
на Урале (15 млрд. т), содержащие магнетитовые руды; место¬
рождения Горной Шории и Абаканское в Зап. Сибири, содер¬
жащие магнетиты; Ангаро-Питское и Нижне-Туруханское ме¬
сторождения в Вост. Сибири; Оленегорское месторождение в
Мурманской области и Костамукшское месторождение в Каре¬
лии.3.6. Общая схема производства черных металловИз всех черных металлов наибольшее промышленное значе¬
ние имеет сталь. Она может быть получена из железной руды
переделкой чугуна, прямым восстановлением руды в твердой
фазе и гидрометаллургическим методом через хлориды желе¬51
за. В настоящее время в промышленности используется прак¬
тически только первый метод. Мировое производство стали пря*
мым восстановлением не превышает 2—3% от общего ее про¬
изводства. Гидрометаллургический метод находится в стадии
проработки. Помимо стали к основным целевым продуктам чер¬
ной металлургии относятся серый (литейный) чугун и метал-’
лические порошки. На рис. 3.3 представлена общая схема пе¬
реработки железной руды в целевые продукты.Рис. 3.3. Общая схема переработки железной рудыСовременное производство черных металлов представляет
собой сложное комбинированное производство, состоящее из
основных и вспомогательных цехов и заводов. Оно включает:— шахты по добыче руды и каменного угля;— горнообогатительные комбинаты по измельчению, обога¬
щению и окускованию руд и концентратов;— коксохимические цехи и заводы, обеспечивающие подго¬
товку углей, их коксование и улавливание продуктов коксова¬
ния;— энергетические цехи для получения кислорода, сжатого
воздуха для дутья и очистки газов металлургических произ¬
водств;— доменные цехи для выплавки передельного и литейного
чугунов;— заводы для производства ферросплавов;52
‘—сталеплавильные цехи (конвертерные, мартеновские,
электросталеплавильные) для производства стали;— прокатные цехи.Заводы с полным металлургическим циклом «руда — чу¬
гун — сталь — прокат» вследствие высокой материалоемкости
производства (6 т сырья Н£ 1 т продукции) размещены в райо¬
нах месторождений руды или угля; предприятия по выплавке
ферросплавов и специальных сталей — в районах, обеспечен¬
ных дешевой электроэнергией. В РФ предприятия черной ме¬
таллургии сосредоточены в трех основных регионах: Уральском
(Н.Тагил, Магнитогорск, Челябинск, Новотроицк), Сибирском
(Новокузнецк, Новосибирск, Красноярск) и Центральном
(Тула, Череповец, Липецк, Москва, Нижний Новгород, Ст. Ос¬
кол).Контрольные вопросы1. В каких модификациях может существовать металлическое же¬
лезо?2. В чем различие свойств а- и у-модификаций железа и какое значе¬
ние это имеет для производства?3. Чем отличаются стали от чугунов?4. Какие стали называются легированными ?5. Какие превращения происходят в системе «железо—углерод» при
затвердеваний расплава и последующем охлаждении?6. Перечислите основные виды промышленных железных руд.
Глава IVПРОИЗВОДСТВО ЧУГУНАПроизводство чугуна — это первая стадия двухступенчатого
процесса переработки железных руд в сталь, который в настоя¬
щее время преобладает. Чугун выплавляют из железорудного
сырья в доменных печах, в которых за счет сгорания топлива
создаются высокие температуры, обеспечивающие процессы
восстановления оксидов железа руды, образования жидкого
чугуна и отделения пустой породы в виде шлака. Подобный
процесс получил название доменного процесса или доменной
плавки.4.1. Сырье доменной плавкиВ качестве сырья в доменном процессе используют специаль¬
но подготовленные железные руды (агломерат, окатыши), твер¬
дое, жидкое и газообразное топливо, флюсы, марганцевые руды
и воздух. Смесь твердых компонентов сырья, загружаемого в
доменную печь, называется шихтой. Виды железных руд, при¬
меняемых для выплавки чугуна, рассмотрены в главе III.1. Топливо обеспечивает создание в печи высоких темпера¬
тур, ттрпбуодттмтлу для ттрптекяния реакций восстановления ок¬
сидов железа, образование оксида углерода (II) и водорода* йв-
ляющихся газообразными восстановителями, диффузию угле¬
рода в восстановленное железо и образование чугуна. В каче¬
стве топлива используется преимущественно каменноугольный
кокс и, для снижения его расхода, добавки газообразного (при¬
родный и коксовый газы), жидкого (мазут) и аэрозольного
(угольная пыль) топлив. Доменный кокс должен обладать вы¬
сокой прочностью, сопротивлением к истиранию, не спекаться
в условиях доменного процесса и содержать минимальные ко¬
личества золы, серы и фосфора. Так, например, повышение со¬
держания серы в коксе на 1 % увеличивает расход кокса на 10%
и снижает производительность печи на 20%. Обычно, в метал¬
лургическом коксе содержится: золы 8—12%, серы 0,5—2,0%
и фосфора до 0,5%.2. Флюсы вводят в шихту для образования с пустой поро¬
дой руды и золой кокса, содержащих тугоплавкие оксиды
кремния, алюминия и кальция, легкоплавкого жидкотекуче¬
го и легко отделяемого от чугуна шлака. В качестве флюсов
используют не содержащие серы и фосфора карбонат кальция
и доломит СаСОз’МяСОз. В современном процессе флюсы вво-54
дят не непосредственно в доменную печь, а в шихту при ее
подготовке, образуя так называемый офлюсованный железо¬
рудный материал.3. Марганцевые руды, содержащие оксиды марганца МпОг
и Мп20з или карбонат марганца МпСОз, используются при вып¬
лавке ферромарганца и высокомарганцовистых (с содержани¬
ем около1%) сортов литейного и передельного чугунов.4. Воздух, обогащенный кислородом, обеспечивает горение
топлива и образование газообразных восстановителей.5. Железные руды перед доменной плавкой проходят специ¬
альную подготовку. Цель подготовки — повышение содержа¬
ния железа в железорудных материалах, обеспечение необхо¬
димых дисперсности и газопроницаемости их и, как следствие,
увеличение производительности доменной печи, снижение рас¬
хода кокса и флюсов. Основными операциями подготовки яв¬
ляются обогащенные руды и окускование рудной мелочи. Пе¬
ред этим сырье подвергают обычным операциям дробления, тон¬
кого измельчения и классификации (грохочения).Метод обогащения зависит от со¬
става руды, гидрофобности пустой з
породы и формы нахождение желе:
за в руде. В зависимости от этого для
обогащения используют промывку
(отмывание пустой породы), флота¬
цию, гравитационную и магнитную
сепарацию. При этом оптимальную
степень обогащения выбирают из
технико-экономических соображе¬
ний. По мере повышения содержа¬
ния железа в концентрате возраста- Рис. 4.1 . Определение
ют затраты на обогащение (кривая оптимальной степени обога-
CD на рис. 4.1), но сокращаются зат- щения:
раты на доменный процесс (кривая з — затраты и себестоимость,
АВ). Очевидно, что оптимальная и — содержание железа в
степень обогащения соответствует шихте
точке пересечения этих кривых(точка К), чему отвечает минимум на кривой себестоимости
чугуна (кривая EF).Наиболее распространенный метод магнитной сепарации,
которым получают до 90% всего железорудного концентрата,
основан на различной магнитной проницаемости минералов
руды и пригоден для обогащения магнитных железняков. Маг¬
нитная сепарация может быть сухой, когда руду загружают на55
^ь(у) ошйрррр\•ио иРис. 4.2. Схема магнитного ленточного сепаратора:1 — снимающая лента, 2 — питающая лента, 3 — бункер шихты, 4 — бун¬
кер магнитного концентрата, 5 — бункер пустой породы, 6 — магнитыбарабаны магнитных сепараторов, и мокрой, когда рудную
, пульпу подают в ванну под вращающийся барабан с электро¬
магнитом, извлекающим из пульпы ферромагнитный минерал.
На рис. 4.2 представлен магнитный сепаратор для сухой сепа¬
рации ленточного типа. Производительность магнитных сепа¬
раторов составляет более 400 т/ч при степени обогащения до0,75 долей ед.Для обогащения методом магнитной сепарации немагнитных
бурых и красных железняков их предварительно подвергают
магнетизирующему (восстановительному) обжигу при 600—
800°С в восстановительной атмосфере, образующейся при непол¬
ном сгорании топлива:ЗГегОз + СО 2Fe304 + СО2ЗРегОз + Н2 ^ 2Fe3C>4 + Н2ОМагнетизирующий обжиг приводит к повышению содержа¬
ния железа в концентрате и по существу является химическим
обогащением.Полученные в результате обогащения концентраты усредня¬
ются по составу, после чего поступают на операцию окускова-
ния.4.2. Окускование рудной мелочиПолученный обогащением рудный концентрат мелкодиспер¬
сен и не может непосредственно использоваться для доменйой
плавки. Поэтому он подвергается операции окускования. О кус-56
кованием называется процесс превращения мелкого железоруд¬
ного материала в кусковой материал необходимых размеров,
которые обеспечивают стабильный и высокопроизводительный
процесс доменной плавки. В черной Металлургии используют
два способа окускования: агломерацию и окомкование (окаты¬
вание).1. Агломерацией называется способ окускования рудного
концентрата путем спекания до частиц нужного размера за счет
сжигания твердого топлива в слое концентрата или подвода теп¬
ла извне, с одновременным удалением из него серы, мышьяка
и пустой породы (оксида кремния). В металлургическую прак¬
тику метод агломерации был введен в 1911 году и стал широко
применяться с 30-х годов XX столетия.Агломерацию осуществляют путем просасывания воздуха
через слой шихты, состоящей из рудного концентрата (40—
50% ), известняка (15—20%), возврата агломерата (20—30%),
коксовой мелочи или коксика (4—6%) и воды (6—9%), при тем¬
пературе около 1400°С. Процесс агломерации протекает через
три последовательных стадии:— подготовительная, начинающаяся после зажигания ших¬
ты (нагрев шихты, испарение влаги, воспламенение топлива);— стадия сгорания, сопровождающаяся опусканием зоны го¬
рения вниз (сгорание топлива с образованием оксида углерода
(II), образование жидкой фазы, частичное восстановление ок¬
сидов железа);— стадия охлаждения образовавшегося агломерата холод¬
ным воздухом.На рис. 4.3 представлены эти стадии процесса агломерации.ХолодныйвоздухIIIЗона горенияIIРис. 4.3. Схема агломерационного процесса:а — подготовительная стадия, 6 — стадия сгорания, a — стадия охлажде¬
ния57
При агломерации в шихте протекают процессы:— неполного окисления топлива и восстановления оксидов
железа2С + О2 ~ 2СО
ЗГегОз + СО = 2Fe304 + СО2Fe304 + СО = 3FeO + С02— термической диссоциации известняка и образования си¬
ликатов (шлак)СаСОз = СаО + СО22СаО + Si02 = 2Ca0Si022FeO + Si02 = 2Fe0Si02— диссоциации дисульфида и обжига сульфида железа2FeS2 = 2FeS + S22FeS + 3,502 = Fe203 + 2S02В результате агломерации из концентрата удаляется до 95%
серы и до 20% мышьяка, значительное количество оксида крем¬
ния и повышается до 65% содержание железа. Применение для
доменной плавки подобного офлюсованного (полученного с до¬
бавкой флюсов) агломерата:— исключает процесс диссоциации флюсов, вводимых в домен¬
ную печь, сокращая за счет этого на 10—12% расход топлива;— улучшает восстанавливаемость рудного сырья вследствие
образования силикатов и освобождения железа из его соединений;— уменьшает объем шихты, загружаемой в доменную печь;— улучшает условия шлакообразования и отделения шлака
от металла. В результате использования при доменной плавке
офлюсованного агломерата производительность доменной печи
возрастает на 10—12%.На рис 4.4 представлена технологическая схема агломера¬
ции.Процесс спекания шихты происходит на агломерационных
машинах. Наиболее распространены машины непрерывного
действия ленточного типа с поверхностью спекания до 800 м2.
Машина АКМ-800 этого типа имеет длину 102 м, ширину 8 м,
что при скорости движения ленты 12 м/мин. дает производи¬
тельность 30000 т/сут.2. Окомкованием (окатыванием) называется способ Окус-
кования тонко измельченного рудного концентрата путем пре¬
вращения его в комки (окатыши) с последующим упрочняющимI58
Рис. 4.4. Технологическая схема агломерации концентрата:1 — бункера, 2 — транспортер, 3 — барабанный смеситель, 4 — агломе¬
рационная машина, 5 — барабанный окомкователь, 6 — отсасывающий
вентилятор, 7 — зажигательный горн, 8 — слой шихты, 9 — грохотобжигом. Производство окатышей в промышленных масшта-
бах было начато в США в 1955 году, а в нашей стране в 1964
году. Окатыши используют как сырье доменной плавки (до 20%
состава железорудной части шихты) и в качестве полупродук¬
та во внедоменном производстве стали.Окомкование концентрата осуществляется в грануляторах
барабанного или тарельчатого типа. При этом в шихту добав¬
ляется около 1,5% мелкодисперсной глины (бентонита) в ка¬
честве связующего материала и известняк, если изготавлива¬
ются офлюсованные окатыши. Производительность грануля-
торов составляет 125^150 т/сут. при диаметре окатышей 6—
7 мм. Для обжига окатышей применяются ленточные обжи¬
говые машины с площадью спекания до 500 м2 и производи¬
тельностью 2500—9000 т/сут. При обжиге сырые окатыши
последовательно проходят зоны сушки при 250—400°С, обжига
при 1200—1300°С, где обогреваются продуктами сгорания газа
или мазута, просасываемыми через решетку и охлаждения хо¬
лодным воздухом (рис. 4.5).Целью обжига окатышей является упрочнение их до такого
состояния, при котором они могут выдерживать без значитель¬
ных разрушений транспортировку, перегрузки и процесс домен¬
ной плавки. При этом, в отличие от агломерации, при обжиге
окатышей не происходит спекания материалов шихты. Обыч¬
но окомкованию подвергают концентраты магнетита, поэтому59
Рис. 4.5. Технологическая схема изготовления окатышей:1 — бункера компонентов шихты, 2 — сборный транспортер, 3 — смеси¬
тельный барабан, 4 — бункер бентонита, 5 — тарельчатый гранулятор,6 — обжиговая машина, 7 — вентиляторы, в — грохот, 9 — транспортер
шихтыпри обжиге происходит частичное окисление его до оксида же¬
леза (III):4Fe3C>4 + О2 = 6Fe2C>3,
который образует в офлюсованных окатышах легкоплавкие
ферриты кальция CaOFeaOe и 2Ca0-Fe203, выполняющие роль
шлаковой связки. Содержание железа в окатышах составляет
обычно 80—90%. В болыпинствеслучаев для доменной плавки
используют смесь агломерата и окатышей в отношении 2:1.На рис. 4.6 представлена общая принципиальная схема под¬
готовки железорудного сырья к доменному процессу, включая
операции обогащения, агломерации и окомкования.4.3. Теоретические основы доменного процессаПроцесс доменной плавки происходит в плавильных агрега¬
тах непрерывного действия шахтного типа — доменных печах,
представляющих реакторы идеального вытеснения РИВ-Н. В
них непрерывно движутся навстречу друг другу два материаль¬
ных потока: сверху вниз твердая шихта и снизу вверх газооб¬
разные восстановители, образующиеся в результате горения
топлива и взаимодействия продуктов горения с компонентами
шихты. Подобный режим противотока создает постоянство дви-60
Флотация <4-
1_Оки&ование -4-r—i—| ОкатышиPyflaFejOa I" IИзмельчениеКлассификация’ Обогащение ‘Восстанови-
тельный обжиг+ - Магнитная^—|рудаРёр4-►I Концентрат U-+ Усреднение— Окускование —сепарацияАгломерация
| Агломерат |, Доменная .
плавкаРис. 4.6. Принципиальная схема подготовки железных руд
к доменной плавкежущей силы и скорости процесса восстановления и обеспечи¬
вает стационарность режима работы доменной печи.Профиль плавильного пространства печи выбран таким об¬
разом, чтобы обеспечить равномерное опускание шихты вслед¬
ствие горения кокса, плавления материалов шихты и периоди¬
ческого выпуска чугуна и шлака, равномерное распределение
газового потока по сечению печи и максимально интенсивное
протекание процессов восстановления руды, образования и раз¬
деления чугуна и шлака. В соответствии с этим в плавильном
пространстве печи различают следующие части: колошник,
шахту, распар, заплечики и горн. На рис 4.7 представлен раз¬
рез доменной печи и распределение температур по ее высоте.При установившемся режиме работы печи в ней создается и
поддерживается определенный температурный режим и проте¬
кают химические и физико-химические процессы, котррые
могут быть сведены к нескольким группам. Они приведены
ниже, причем номера их отмечены на схеме печи в тех зонах ее,
в которых они протекают.1. Подготовительные процессы. К подготовительным про¬
цессам, протекающим в интервале температур 100—400°С, от¬
носятся выделение гигроскопической и гидратной воды и лету¬
чих веществ из твердого топлива.61
2. Образование газообразных вос¬
становителей. Газообразные восста¬
новители образуются в результате
полного сгорания углерода кокса и
природного газа при температурах
1800—1900°С и последующего вос¬
становления продуктов окисления
при контакте с раскаленным коксом:
С + О2 = СО2 и СН4 + 2С>2 = 2НгО;
СО2 + С = 2СО и Н2О + С = СО + Н2Суммируя, соответственно, полу¬
чаем уравнения образования газооб¬
разных восстановителей:2С + 02 = 2СО -АЯ
СН4 + 0,502 = СО + 2Н2 -АН3. Восстановление оксидов желе¬
за. Восстановление оксидов железа
начинается при температуре выше
570°С и в соответствии с теорией
А. А. Байкова протекает ступенчато
от высших оксидов к низшим, то
есть:Fe203 * Гез04 FeO FeЗаканчивается восстановление при температуре 1200°С.При доменной плавке прямое восстановление оксидов желе¬
за углеродом кокса играет незначительную роль и возможно
лишь при высоких температурах. В доменной печи преоблада¬
ет процесс косвенного восстановления их оксидом углерода (II)
и водородом, описываемый уравнениями:3Fe203 + СО = 2Fe304 + С02 - АН АН = 53,74 кДж (а)Fe304 + СО +£ 3FeO + С02 + АН АН = 36,68 кДж (б)FeO + СО ^ Fe + С02 - АН АН = 16,06 кДж (в)3Fe203 + Н2 +£ 2Fe304 + Н20 - АН АД = 12,89 кДж (г)Fe304 + Н2 ^ 3FeO + Н20 + АН АН = 77,54 кДж (д)FeO + Н2 Fe + Н20 + АН А# = 24,79кДж (е)Из этих реакций — (а), (в) и (г) протекают с выделением теп¬
ла, реакции (б), (д) и (е) — с поглощением тепла. Расчет изобар-
но-изотермических потенциалов реакций (а), (б) и (в) по значе¬
ниям AG оксидов (табл. 4.1) дает для них значения, соответ-Рис. 4.7. Профиль доменной
печи62
ственно, -66, -54 и -6 кДж, что подтверждает справедливость
теории ступенчатого восстановления оксидов железа.Таблица 4.1. Изобарно-изотермические потенциалы оксидовОксидFeOFe203РезОдСОСОзМпОSiOjAG°298кДж/моль-252-744-1020-137-395-364V)001Каждой из приведенных выше реакций соответствует рав¬
новесный состав газовой фазы, характеризуемый отношением
СО/СОг и зависящий от природы оксида железа (его термичес¬
кой стабильности) и температуры. На рис. 4.8 представлены
кривые равновесного состава газовой фазы над твердой фазой в
функции температуры для реакций (а), (б) и (в). Так как реак¬
ция (а) практически необратима, кривая для нее приближает¬
ся к оси абсцисс; следовательно, даже небольшой концентра¬
ции оксида углерода (II) в газовой фазе достаточно для восста¬
новления РегОз до FeaO*. Реакция (в) является экзотермичес¬
кой, поэтому равновесная концентрация СО для нее возрастает
при повышении температуры, в то время как для эндотерми¬
ческой реакции (б) она падает. 7 ‘
Из рис. 4.8 следует, что если оксиды железа находятся в рав¬
новесии с газовой фазой, содержащей СО и СОг, то в поле диаг¬
раммы ниже кривой 1 устойчив оксид Те%Ог в поле между кри-Рис. 4.8. Равновесный состав газовой фазы реакций восстановле¬
ния оксидов железа оксидом углерода (II):1, 2, 3 — кривые равновесия для реакций а, б, в, соответственно, 4 —
кривая равновесного состава для реакции СОг + С = 2СО, 5 — кривая,
отвечающая составу реального газа в печи63
вышк 1 и 2 оксид Fe$C>4, в поле между кривыми 2 и 3 оксид FeO
и в поле вьппе кривой 3 металлическое железо. Если в системе
находится в избытке углерод, то состав газа будет определять¬
ся кривой 4. По условиям термодинамики восстановление ок¬
сида FesO* в оксид FeO может начаться при температуре, отве¬
чающей точке А, а оксида FeO в железо — в точке Б. Фактичес¬
кий состав газовой фазы в печи таков (кривая 5), что только при
высоких температурах достигаются равновесные концентрации
СО, отвечающие реакции СОг+ С = 2СО + АН. При низких тем¬
пературах фактическое содержание СО выше равновесного и по
отношению к железу в печи всегда поддерживается восстано¬
вительная атмосфера (27—30% СО в газе).Суммируя реакции а, б ив, получаем уравнение восстанов¬
ления оксида железа (П1) до металлического железа в виде:Fe203 + ЗСО ^ 2Fe + ЗС02 -АН АН = 25,58 кДж,из которого следует, что процесс в целом является экзотерми¬
ческим.Скорость косвенного восстановления оксидов железа оксидом
углерода (П) пропорциональна концентрации СО в газовой фазе:U = KMF( Ссо-Ссо*) (4.1)где: Ссо — концентрация СО в газовой фазе действительная,
Ссо* — равновесная концентрация СО над твердой фазой,
и тем выше, чем более система удалена от состоя¬
ния равновесия.Из приведенных выше реакций образования газообразных
восстановителей (стр. 62) следует, что этому способствует уве¬
личение концентрации кислорода в подаваемом в печь воздухе
и повышение давления, что и используется на практике.При температуре выше 900°С происходит интенсивное вос¬
становление оксида углерода (IV) на поверхности раскаленного
кокса и процесс может быть описан уравнениями:FeO + СО = Fe + С02 -АН и С + С02 = 2 СО +ДНСуммируя их, получаем:FeO + С = Fe + СО + АН АН = 153 кДж,то есть процесс восстановления оксида железа FeO можно рас¬
сматривать как протекающий в твердой фазе, как прямое вос¬
становление. Этой реакции на рис. 4.8 отвечает заштрихован¬
ная часть поля, следовательно, доля прямого восстановления
растет с увеличением температуры доменного процесса.4. Науглероживание железа и получение чугуна. Науглеро¬
живание губчатого металлического железа происходит при его64
контакте с доменными газами, содержащими оксид углерода
(II) и раскаленным коксом. Процесс науглероживания начина¬
ется в твердой фазе при температуре около 600°С на поверхнос¬
ти свежевосстановленного железа, катализирующего реакцию
распада оксида углерода (II). При этом образуется высокоактив¬
ный сажистый углерод, реагирующий с железом с образовани¬
ем карбида железа:2СО = С02 + С и 3Fe + С = Fe3Cили 3Fe + 2СО — Fe^C + СО2 - АН АН = 180,5 кДж.При переходе металла в жидкое состояние науглероживание
становится более интенсивным и протекает уже непосредствен¬
но за счет взаимодействия железа с раскаленным коксом с пос¬
ледующим растворением карбида железа в железе. По мере на¬
углероживания температура плавления железа понижается с
1539°С (чистое железо) до 1135°С (сплав, содержащий 4,3% уг¬
лерода).5. Восстановление примесей- В состав металлизированных
материалов шихты (агломерат, окатыши) входят помимо окси¬
дов железа оксиды различных элементов,. По возрастанию срод-
ства к кислороду и/термодинамической прочности их оксидов,
они располагаются в ряд: Си, As, Ni, Р, Zn, Mn, V, Сг^ Si, Ti, Al,
Mg, Ca. Степень восстановления этих элементов в доменной печи
соответствует их положению в этом ряду. Медь, мышьяк, фос¬
фор подобно железу почти полностью восстанавливаются и пе¬
реходят в чугун; цинк, хотя и восстанавливается, но возгоняет¬
ся; ванадий и хром восстанавливаются на 70—90%. Алюминий,
кальций и магний при доменной плавке не восстанавливаются.Важнейшие элементы в шихте — марганец и кремний, со¬
держащиеся в виде различных силикатов в составе агломера¬
та, золы, кокса и оксидов марганца в составе марганцевых руд.
Кремний и марганец (из его низшего оксида) восстанавливают¬
ся только по реакциям прямого восстановления при темпера¬
туре 1150—1300°С.Si02 + 2С ^ Si + 2СО +АН АН = 635,1 кДжМпО + С ^ Mn + СО +АН АН = 288,3 кДжВ отличие от оксида марганца (И) высшие оксиды его легко
восстанавливаются в газовой фазе оксидом углерода (II) уже при
200—500°С в последовательности, аналогичной последователь¬
ности восстановления оксидов железа:М11О2 * МП2О3 *■ М113О4 *■ МпО3 Химическая технология. Том 265
Расчет изобарно-изотермического потенциала реакций вос¬
становления кремния и марганца по значениям AG соответству¬
ющих оксидов (табл 4.1) показывает, что они составляют, соот¬
ветственно, +61 и -31 кДж, а для реакции восстановления же¬
леза из FeO +109 кДж. Поэтому, прямому восстановлению под¬
вергаются, в первую очередь, кремний и марганец.6. Образование шлака. При доменной плавке одновременно
с образованием чугуна образуется шлак в виде различных си¬
ликатов за счет взаимодействия невосстановленных основных
(CaO, MgO, МпО и FeO) и кислотных (А120з и Si02) оксидов. По
мере перемещения в зону высоких температур (выше 1400вС)
шлак обогащается оксидами кальция, магния, алюминия и
кремния за счет восстановления оксидов железа и марганца и
становится жидкотекучим. Состав доменного шлака: 85—95%
CaO, Si02, А1203; 2—10% MgO; 0,2—0,6% Fe0;0,3—3,0% МпО;
1,5—2,5% серы в виде CaS.7. Реакции с участием серы и фосфора. Сера и фосфор вно¬
сятся в доменную печь с материалами шихты: сера в виде орга¬
нических соединений, сульфидов и дисульфидов железа и дру¬
гих металлов, а также сульфатов с коксом и агломератом, фос¬
фор — в виде тетракальцийфосфата с пустой породой и флюса¬
ми. Оба элемента ухудшают качество как чугуна, так и выплав¬
ляемой из него стали, поэтому содержание их в металле долж¬
но быть ограничено.Восстановление фосфора в присутствии оксида кремния ших¬
ты протекает при температуре выше 1100°С по реакциям:4Са0Р205 + 4Si02 = 4Ca0Si02 + Р205Р205 + 5С = 2Р + 5СОВосстановленный фосфор растворяется в железе и полностью
переходит в чугун. Поэтому получить металл с низким содержа¬
нием фосфора можно только на основе низкофосфористой шихты.Сера из шихты в процессе плавки частично, на 10—20%, пе¬
реходит в газовую фазу в виде оксида серы (IV), сероводорода и
др. соединений, но большая часть ее остается в шихте в виде
сульфидов железа FeS, марганца MnS и кальция CaS. Из них
сульфиды железа и марганца хорошо растворимы в металле, а
сульфид кальция — в шлаке. Поэтому для удаления серы из чу¬
гуна необходимо перевести сульфиды железа и марганца в суль¬
фид кальция, который не растворяется в чугуне, (рис. 4.9).Для этого в печи должны быть созданы условия получения жид¬
кого хорошо нагретого шлака основного характера с высоким со¬
держанием оксида кальция. При взаимодействии такого шлака с66
сульфидами железа и марганца про¬
текают обменные реакции:FeS + СаО = FeO + CaSи MnS + СаО = MnO + CaSОбразовавшиеся оксиды метал¬
лов восстанавливаются углеродом
кокса до свободных металлов, по¬
этому процесс обессеривания чугу¬
на может быть выражен суммар¬
ным уравнением, например, для сульфида железа:FeS + СаО 4- С = Fe + СО + CaS (в шлак)4.4. Технологическая схема доменного производстваОрганизация технологического процесса выплавки чугуна из
руды должна предусматривать:—. непрерывность процесса плавки при периодичности опе¬
раций загрузки материалов шихты, выпуска чугуна и шлака;— осуществление противотока реагентов (шихты и газообраз¬
ных компонентов сырья);— использование теплоты продуктов процесса;— герметизацию оборудования и возможность непрерывно¬
го отбора доменного газа.В соответствии с этими задачами технологическая схема до¬
менного производства (рис. 4.10) включает:— рудный двор и бункерную эстакаду для хранения, дози¬
ровки и загрузки в скипы (тележки) материалов шихты;— доменную печь;— систему подогрева и подачи воздушного дутья;— устройство для уборки и транспортировки жидких про¬
дуктов плавки (чугуна и шлака);— систему очистки доменного газа.Агломерат и кокс загружаются на рудном дворе в бункера 1
и 2, из которых они поступают в скипы подъемника 3. Скипы с
материалами шихты в определенной последовательности напри¬
мер, АККАК (где А — скип с агломератом, К — скип с коксом),
подаются в загрузочное устройство (засыпной аппарат) 5 домен¬
ной печи 4. В нижнюю часть печи через фурмы с помощью по-
здуходувки из каупера (воздухонагревателя) 9 подается воздух,
нагретый до 1200—1300°С. Образующиеся в процессе плавки
жидкие чугун и шлак периодически выпускаются из нижней
части печи через специальные отверстия — летки. Выпускае-
з*67CaSШлакЖелезо«-V-FeS•4+4-MriSJРис. 4.9. Схема передачи
серы из шихты в шлак
Рис. 4.10. Технологическая схема доменного производства:1 — бункер агломерата, 2 — бункер кокса, 3 — скиповый подъемник, 4 —
доменная печь, 5 — засыпной аппарат доменной печи, 6 — пылеулови¬
тель, 7 — промывной скруббер, 8 — мокрый электрофильтр, 9 — каупер,
работающий на подогрев дутья, 10 — каупер, работающий на разогрев
насадки, 11 — дымовая трубамый чугун собирается в ковши емкостью 90—140 т, или в мик¬
серы, футерованные огнеупорным материалом емкостью до
420 т, установленные на железнодорожных платформах. Шлак
отводится по желобу в шлаковозы и направляется на грануля¬
цию. Доменный газ, выходящий из колошника печи, очищает¬
ся от пыли в пылеуловителе б, скруббере 7, орошаемом водой,
и окончательно в мокром электрофильтре 8. Очищенный и ох¬
лажденный до 40°С газ направляется в каупер 10, где сжигает¬
ся в токе воздуха и разогревает насадку. Кауперы работают, та¬
ким образом, попеременно: один на подогрев воздушного дутья,
другой — на разогрев насадки, что обеспечивает бесперебой¬
ность подачи подогретого воздуха в доменную печь.Основными аппаратами доменного производства являются
доменная печь и каупер.Доменная печь имеет форму башни из огнеупорного кирпи¬
ча, заключенную снаружи в металлический сварной кожух.
Изнутри печь имеет огнеупорную футеровку. Для уменьшения
выгорания кладки она снабжена специальными холодильника¬
ми. Масса печи достигает 30 000 тонн, поэтому она устанавли¬
вается на мощном фундаменте. Размеры печи зависят от ее по¬
лезного объема, который определяется ее полезной высотой (от68
верхнего уровня шихты до чугунной летки). Размеры осталь¬
ных Элементов печи (рис. 4.7) зависят от ее полезного объема.
Так, ядя наиболее мощных современных печей полезным объ¬
емом 5000 м3 полезная высота равна 32,2 м, высота шахты
19,$ м, диаметр и высота горна 14,9 и 4,5 м, соответственно.
Материалы шихты подаются в колошник через загрузочное ус¬
тройство, обеспечивающее равномерное распределение их по
сечению печи и герметичность в момент загрузки. В горн печи
через равномерно расположенные по его окружности отверстия
(от 24 до 40) с помощью фурм подается из общего кольцевого
трубопровода подогретый воздух (воздушное дутье) под давле¬
нием от 0,1—0,35 МПа. Для выпуска чугуна и шлака в горне
печи имеются отверстия — летки (от 10 до 18), забиваемые ог¬
неупорной глиной.Каупер представляет регенератор периодического действия,
в котором используется теплота сгорания доменного или при¬
родного газа. Он выполнен в виде металлического цилиндра
высотой до 50 м и диаметром 6—9 м общим объемом до
4000 м3, выложенного внутри огнеупорным материалом. Внут¬
ренне пространство каупера разделено на две части: камеру
сгорания и камеру с насадкой из огнеупорного кирпича, спаб-
женной сквозными каналами. В камере сгорания сжигается
доменный газ, к которому для увеличения теплоты сгорания
добавляется природный газ, и продукты горения обогревают
насадку во второй камере. По достижении 1200—1300°С ды¬
моход перекрывается и через нагретую насадку пропускается
холодный воздух, а обогревающий газ переключается на дру¬
гой каупер.4.5. Технико-экономические показатели работы печиДоменная печь представляет весьма мощный и высокопро¬
изводительный агрегат, в котором расходуется огромное коли¬
чество шихтовых материалов воздушного и газового дутья. Так,
печь полезным объемом 5000 м3 потребляет в сутки 23000 т
шихты, 18000 т воздушного дутья, 1700 т природного газа и
выдает 12000 т чугуна, 4000 т шлака и 27000 т доменного (ко¬
лошникового) газа. Поэтому вопросы экономики доменного
производства имеют первостепенное значение.Основные показатели работы доменной печи — это произво¬
дительность в единицу времени и расход кокса на единицу мас¬
сы выплавляемого чугуна. Для оценки производительности пе¬
чей различного объема применяют относительный показа-69
тедь — коэффициент использования полезного объема печи
(КИПО), определяемый по формуле:полКИПО = м3сут/Т (4.2)1TV чуггде: Vn0J1— полезный объем печи, м3,/Пчуг — среднесуточная (за месяц, квартал, год) выплавка
чугуна, т/сут.Чем ниже КИПО, тем более производительно работает печь.
За последние 50 лет в нашей стране КИПО улучшился более чем
вдвое, снизившись с 1,19 в 1940 году до 0,55 (а для лучших пе¬
чей до 0,43) в 1989 году. Помимо КИПО работа доменной печи
может оцениваться также съемом чугуна с 1 м3 полезного объ¬
ема печи в сутки, то есть:1С = т/м3-сут. (4-3)КИПОДругим важным показателем работы печи, характеризую¬
щим экономичность ее работы, является удельный расход кок¬
са на плавку (УРК):т кУРК = — т/т (4.4)чуггде: /пк — масса кокса, т/сут,л*чуг — масса выплавляемого чугуна, т/сут.Величина УРК не только влияет на производительность
печи, но и показывает эффективность использования хими¬
ческой и тепловой энергии топлива в плавильном простран¬
стве печи. УРК отражает уровень техники и технологии до¬
менной плавки и степень подготовки сырья к ней. За 40 лет
УРК в стране снизился с 0,93 до 0,54, достигая в лучших пе¬
чах 0,40 т/т. УРК может быть существенно снижен за счет
использования при плавке природного газа и жидкого топли¬
ва. Так, если УРК в печах, работающих только на коксе, ра¬
вен 0,8, то для печей с добавкой газа он снижается до 0,55, а
при введении мазута до 0,4.Себестоимость чугуна складывается из затрат на офлюсо¬
ванную металлическую шихту (агломерат, окатыши), на тех¬
нологическое топливо и расходов по переделу, включая обще¬
заводские расходы. Первая и вторая группы затрат не зависят
от масштабов производства и определяются стоимостью и ка¬
чеством материалов шихты и уровнем технологии. В среднем
они составляют до 90% себестоимости чугуна. Расходы по пе¬
ределу составляют около 10% и являются затратами на пере¬
работку шихты в чугун. Их величин^, зависит от масштабов
производства, уровня техники и технологии, стоимости энер¬
гии, зарплаты и т.п. Среди этих затрат основное место зани¬
мают энергетические, связанные с использованием в домен¬
ном процессе пара, воды, электроэнергии, дутья, сжатого воз¬
духа и др. В табл. 4.2 представлена структура себестоимости
чугуна.Таблица 4.2. Структура себестоимости чугунаСтатьяДоля в себестоимости, %Металлическое сырье30—40Кокс40—50Природный газ2—4Расходы по переделу13—18Заработная плата0,3—0,74. б. Продукты доменного
производстваДоменный процесс организуется для производства целевого
продукта чугуна. Однако, помимо чугуна, конечными продук¬
тами доменной плавки являются также шлак и доменный газ.Из всего выплавляемого чугуна до 90% его составляет чугун
передельный, используемый для производства стали и пере¬
плавки в литейный чугун и только 10—15% составляет серый
литейный чугун.Доменный шлак — побочный продукт плавки. Выход его
зависит от состава шихты и составляет 0,3—0*6 т на тонну чу¬
гуна. Шлак используют в качестве сырья в производстве цемен¬
та, вяжущих веществ, шлаковой пемзы и ваты, ситаллов, ма¬
териалов для дорожного покрытия. В эти продукты перераба¬
тывается до 75% годового количества шлака. гДоменный (колошниковый) газ содержит до 60% азота, 10—
18% оксида углерода (IV), 24—32% оксида углерода (И) и не¬
значительные количества метана и водорода. Теплота сгорания
доменного газа составляет около 4000 кДж/м3. Он использует¬
ся в качестве топлива для обогрева кауперов, в прокатных це¬
хах для нагрева стальных слитков и др.71
4.7. Интенсификация доменного процессаПод интенсификацией понимают методы и приемы ускорю-
ния протекающих в доменной печи процессов с целью повыше¬
ния ее производительности и улучшения показателей работы.
К ним относят:1. Применение сырья улучшенного качества, в том числе,
офлюсованных железорудных материалов с повышенным со¬
держанием железа и малосернистого кокса. Так, повышение
содержания железа в агломерате на 1% увеличивает произво¬
дительность печи на 2,5% при одновременном снижении УРК
на 2%; уменьшение массы известняка на 0,1 т/т чугуна при за¬
мене обычного агломерата на офлюсованный, снижает УРК на
3%, а понижение содержания серы в коксе на 1%, соответствен-
но, на 2,5%.2. Повышение температуры воздушного дутья увеличива¬
ет поступление в печь физического тепла, что позволяет сни¬
зить расход кокса и, соответственно, вносимой с ним золы, и
расход флюсов на ее связывание. Так, при повышении темпе¬
ратуры дутья от 1000 до 1200"С УРК снижается на 5%.3. Обогащение воздушного дутья кислородом увеличивает
скорость горения топлива и последующих процессов образова¬
ния восстановителен^ восстановления оксидов железа, умень¬
шает количество тепла, выносимого из печи с балластным азо¬
том воздуха. Так, при повышении содержания кислорода в ду¬
тье с 21 до 30% об. УРК снижается на 9%, а производительность
печи увеличивается на 10%. При более высоком содержании
кислорода в печи развиваются излишне высокие температуры,
что может вызвать подвисание шихты и замедление плавки.4. Повышение давление газов в печи увеличивает количество
воздуха, подаваемого в печь и форсирует ее работу. При этом,
за счет лучшего использования газообразных восстановителей
снижается расход топлива и вынос пыли с газом. Так, примене¬
ние давления на колошнике снижает УРК на 3—7% и повыша¬
ет производительность печи на 4—15%.5. Использование природного газа. Стоимость кокса состав¬
ляет до 50% себестоимости выплавляемого чугуна, к тому же
запасы коксующихся углей ограничены. Замена части кокса
природным газом обогащает газовую фазу восстановителями, в
том числе, водородом, снижает температуру в горне при исполь¬
зовании обогащенного кислородом дутья,повышает удельный
вес процессов косвенного восстановления оксидов железа и
уменьшает количество образующейся золы и, следовательно,72
шлака. Применение природного газа повышает производитель¬
ность печи на 15—20%.( 1 . -Контрольные вопросы1. Назовите основные компоненты сырья в доменном процессе и ук-
жите их назначение.2. Какие руды могут подвергаться обогащению методом магнитной
сепарации?3. Что такое агломерация и окомкование рудного сырья?4. Какие процессы и на каком уровне протекают в доменной печи?5. Чем объясняется последовательность восстановления оксидов же¬
леза в доменном процессе?6. За счет каких процессов образуется доменный шлак при выплавке
чугуна?7. С какой целью подогревается воздух перед подачей в доменную
печь?8. Перечислите технико-экономические показатели работы доменной
печи и укажите пути их улучшения.9. Для чего в доменную печь вводят природный газ или жидкое топ¬
ливо?
4Глава VПРОИЗВОДСТВО И ОБРАБОТКА СТАЛИ5.1. Сталелитейное производствоПроизводство стали — это второе звено в производственном
металлургическом цикле: руда—>чугун—►сталь—► изделие. Основ¬
ными способами выплавки стали в настоящее время являются
кислородно-конвертерный (более 60% от всей массы выплав¬
ляемой в мире стали), электросталеплавильный (около 25%) и
мартеновский (около 20%) способы. Для улучшения качества
стали или получения металла с особыми свойствами, выплав¬
ленная одним из этих методов сталь подвергается вторичной об¬
работке: рафинированию после выпуска из сталеплавильного
агрегата (ковшовая металлургия) или переплаву уже затвер¬
девших слитков (переплавные процессы). В связи с потребнос¬
тями новых отраслей техники роль вторичной обработки стали
непрерывно возрастает.Особое место в черной металлургии занимают внедоменные
процессы, используемые для производства сырья для выплав¬
ки стали (металлизированных окатышей) и железных порош¬
ков для порошковой металлургии. Несмотря на малый удель¬
ный вес этих процессов (не более 2%) роль их в техническом
прогрессе весьма велика.Из трех основных методов производства стали доля марте¬
новского непрерывно падает, что объясняется его неконкурен-
тноспособностью с кислородно-конвертерным и электросталеп¬
лавильным методами, обеспечивающими, наряду с получени¬
ем высококачественных сортов стали, высокую экономичность
производства. Так, по данным Кузбасского металлургического
комбината за 1990 год переход с мартеновских печей на кисло¬
родные конвертеры дал годовую экономию металла 7*105т,
энергии (топлива) 6*105 т и рабочей силы 200 человек. В 1987
году во всех индустриально-развитых странах Европы сталь
выплавлялась исключительно в кислородных конвертерах и
электрических печах. В США доля мартеновской стали состав¬
ляла всего около 4%.В нашей стране последняя мартеновская печь была пущена
в 1970 году; в последующие годы происходила только реконст¬
рукция действующих печей путем переделки их на двухванные
печи. Тем не менее, доля мартеновской стали, выплавляемой в
РФ, достаточно велика и значительно превышает долю стали,
получаемой кислородно-конвертерным и электросталеплавиль-74
ным методами. В табл. 5.1 представлено производство стали
этими методами в РФ.'«Таблица 5.1. Производство стали в РФ,%/МетодГод199219941996Кислородно-конверторный23,121,125,3Электросталеплавильный10,46,56.1Мартеновский66,572,468,6Основную долю себестоимости стали и стальных изделий
(прокат) составляют затраты на материалы и энергию. В РФ 92%
всех конструкций изготавливаются с использованием металлов,
главным образом, черных. Продукция металлургической отрас¬
ли производства составляет около 70% всего ВНП. Поэтому, ма-
териало- и энергоемкость сталеплавильного производства име¬
ет большое экономическое значение. В табл. 5.2 приведены дан¬
ные по энерго- и материалоемкости сталелитейного производ¬
ства в различных странах по состоянию на 1992 год.Таблица 5.2. Материало- и энергоемкость производства сталиСтранаМатериало- и энергоемкость
сталелитейного производства
в РФ в% к СШАЭнергоемкость
стального проката,
тУТ/т прокатаРФ1521,70Япония901,27ФРГ1021,31США1001,40Процесс превращения металлического сырья в сталь заклю¬
чается в уменьшении содержания в нем углерода, кремния и
марганца и полном удалении таких примесей как сера и фос¬
фор (II). Это достигается окислением этих компонентов до сб-
единений, образующих газообразную или жидкую, отделяющу¬
юся от металла, фазу (шлак):' В газовую фазу{Fe + С + Si + Mn + n}Fe—► Fe*CO -I- co2 + S02FeO + MnO + P20§ + CaS
« 1 -
. В жидкии шлак75
Окислителями в процессе выплавки стали могут быть:— кислород газовой фазы;— кислород, растворенный в металле;— кислород в составе оксида железа, растворенного в шлаке;— оксиды железа в металлической шихте (руда, стальной
лом).Так как в этих случаях металл и шлак являются растворите¬
лями, то для выражения концентрации растворенных в них
компонентов приняты обозначения в виде квадратных скобок
для раствора в металле, например, [С], [О] и круглых скобок для
раствора в шлаке, например, (Мп). Так, запись[Mn] + (FeO) = Mn + Feозначает, что реакция протекает между растворенным в метал¬
ле (железе), марганцем и растворенным в шлаке оксидом желе¬
за (II).При окислении углерода и примесей часть металлического
железа окисляется до оксида FeO (угар металла). Для умень¬
шения потерь металла его регенерируют, то есть восстанавли¬
вают до железа, В соответствии с этим в процессе выплавки ста¬
ли выделяют два последовательно протекающих периода —
окислительный и восстановительный, что может быть пред¬
ставлено схемой:окисление
► {Fe + FeO + СО + ПО}восстановление 1Выплавка стали требует больших затрат тепловой энергии.
Источниками теплоты являются:— в кислородно-конвертерном методе тепловой эффект окис¬
ления компонентов шихты (AH^Q);— в электросталеплавильном методе энергия электрическо¬
го тока (£^Q);— в мартеновском методе теплота сгорания топлива (Qr^Q)*5.2. Выплавка стали в кислородном конвертереКислородно-конвертерный метод выплавки стали — это один
из вариантов конвертерного метода, предложенного в 1856 году
Г.Бессемером. В настоящее время он полностью вытеснил как
бессемеровский (в конвертере с кислой футеровкой), так и то-
масовский (в конвертере с основной футеровкой) конвертерные
процессы с воздушным дутьем. К преимуществам кислородно¬{Fe + С+ П}76
конвертерного метода по сравнению с мартеновским и электро¬
сталеплавильным методами относятся:— более высокая производительность сталеплавильного аг¬
регата, достигающая 500 т/ч (производительность других ме¬
тодов не превышает 100 т/ч);— более низкие капитальные затраты вследствие простоты
устройства конвертера;— меньшие расходы по переделу сырья в сталь, в том числе
отсутствие затрат на топливо;—■ простота автоматизации и управления процессом плавки.В то же время применение для продувки чистого (не менее
99,5%) кислорода вместо воздуха позволяет по сравнению с дру¬
гими конвертерными методами:— получать сталь, не содержащую азота;— за счет избытка тепла, сверх необходимого для разогрева
металла до температуры выпуска, использовать шихту, содер¬
жащую до 25% твердого сырья (стальной лом, руда), что суще¬
ственно снижает себестоимость выплавляемой стали.5.2.1. Теоретические основы процессаКислородно-конвертерный процесс — один из видов переде¬
ла чугуна в сталь без использования топлива путем продувки
чугуна в конвертере током кислорода. При этом окислительный
и восстановительный периоды плавки разделены не только по
времени, но и в пространстве: первый протекает в конвертере,
второй — после выпуска стали в ковше.Л. Окислительный период плавки совпадает по времени с
операцией продувки конвертера кислородом. При подаче кис¬
лорода под давлением 0,9—1,4 МПа в ванне под воздействием
кислородных струй и потоков выделяющихся пузырьков газо¬
образного оксида углерода (II) создается микрогетерогенная
система «металл—шлак» с интенсивным массо- и теплообме¬
ном. Это ускоряет реакции окисления компонентов металли¬
ческой шихты, вследствие чего выделяющееся тепло не рассе¬
ивается в окружающую среду, а кумулируется в системе, обес¬
печивая интенсивность процессов нагрева металла и расплав¬
ление твердых составляющих шихты.В процессе продувки окисляются углерод, кремний, большая
часть марганца, фосфор и незначительная часть железа. При
этом лишь незначительная часть их окисляется непосредствен¬
но газообразным кислородом; основная часть компонентов окис¬
ляется кислородом, растворенным в металле [О] и «кислоро¬77
дом», растворенным в шлаке (FeO). Ниже рассмотрены реак¬
ции окисления каждого из компонентов шихты.1. Окисление железа. Вследствие высокой (приближающей¬
ся к 100%) концентрации железа в шихте оно окисляется не¬
посредственно газообразным кислородом на поверхности метал¬
ла в зоне его контакта с кислородной струей:2Fe + 02 = 2Fe0-Atf (а)Образующийся оксид железа FeO растворяется частично в
металле:FeO—{Fe + [О]} (б)и частично в шлаке:FeO—{Ш + (FeO)} (в)В незначительной степени железо может окисляться в массе
металла за счет высших оксидов железа, содержащихся в руде
и стальном ломе:Fe + ГегОз = 3FeO — АН2. Окисление углерода является основной реакцией при вып¬
лавке стали, так как снижает его содержание до предела, за
которым чугун превращается в сталь. Угдерод окисляется пре-
мущественно до оксида углерода (II); окисление до оксида уг¬
лерода (IV) незначительно (не более 10—15%) и возможно лишь
при малой концентрации его в чугуне. Окисление начинается с
момента подачи кислорода в конвертер и происходит главным
образом за счет кислорода, растворенного в металле:[С] + [0] = С0-Д н (г)и в шлаке: [С] + (FeO) = GO + АН (д)Суммируя, соответственно, реакции (а), (б), (г) и (а), (в), (г), по¬
лучаем уравнение итоговой реакции окисления углерода, отража¬
ющее начальное и конечное состояние системы в конвертере:С + 0,502 = СО3. Окисление марганца и кремния. Эти реакции, как и окис¬
ление углерода, начинаются с момента подачи кислорода и про¬
исходят за счет кислорода, растворенного в металле:[Мп] + [О] = (МпО) - АН[Si] + 2[0] = (Si02) - АНи в меньшей степени за счет кислорода, растворенного в шла¬
ке, на границе раздела фаз «шлак—металл»:[Мп] + (FeO) = (МпО) + Fe - АН78
[Si] + 2(FeO) = (Si02) + 2Fe - АНОбразовавшиеся при окислении оксиды марганца и кремния
переходят в шлак. Таким образом, для окислительного перио¬
да плавки в кислородном конвертере характерны как прямое
окисление железа в зоне его контакта с кислородной струей
(«первичная реакционная зона»), так и окисление остальных
компонентов за счет вторичных реакций на границе с первич¬
ной реакционной зоной и во всем объеме системы.Последовательность окисления элементов при кислородно¬
конвертерной плавке с достаточной точностью может быть оха¬
рактеризована значением энтальпии образования их оксидов
(табл. 5.3).Таблица 5.3. Энтальпия образования оксидовОксидSi02МпОСОFeOАН298°. КДЖ-861-386-111-267Последовательность окисления —— ► ВремяОт последовательности окисления элементов зависит изме¬
нение во времени состава ванны в конвертере (рис. 5.1). Из него
следует, что почти весь крёмний и большая часть марганца вы¬
горают (окисляются) в первые минуты продувки. Окисление
кремния заканчивается через 3—5 мин, причем в результате
связывания образовавшегося оксида кремния с-известью шла¬
ка реакция становится необратимой и металл полностью обеск-
ремнивается.Окисление марганца наиболее
интенсивно происходит в начале
продувки и за 3—5 мин. окисляет¬
ся до 70% марганца, содержащего¬
ся в чугуне. В дальнейшем окисле- *. ние марганца определяется равно¬
весием реакции:[Mn] + (FeO)^(MnO) + Fe - АНВысокая скорость окисления
марганца и относительно низкая
температура в начале плавки огра¬
ничивает скорость окисления угле- ^ис *т*а Изменение состава
рода. В дальнейшем, при снижении металла от времени продувки79
содержания марганца и кремния скорость окисления углерода
возрастает и падает только при его выгорании.4. Реакции дефосфоризации и десульфуризации. Удаление из
металла фосфора и серы необходимо потому, что фосфор увели¬
чивает х ладо ломкость, а сера красноломкость выплавляемой
стали. Фосфор растворяется в железе в значительных количе¬
ствах и переходит в него из чугуна и железного лома. При прю-
дувке конвертера фосфор окисляется уже в начале процесса и
переходит в шлак:2[Р] + 5(FeO) + З(СаО) = (ЗСа0Р205) + 5Fe - АНРазвитию этой реакции способствуют относительно невысо¬
кая температура, окислительная среда и высокая основность
шлака. Конечное содержание фосфора в выплавляемой стали
зависит от содержания его в исходном чугуне и, обычно* состав¬
ляет 0,002—0,004%.Сера неогранйченно растворяется в жидком и ограниченно в
твердом железе и поступает в него из чугуна и неочищенного от
смазочных масел стального лома. Десульфуризация металла
происходит в течение всей продувки за счет перехода ее из ме¬
талла в шлак по реакции:[FeS] + (СаО) = (CaS) + (FeO) - АЯ,чему способствуют высокая температура, интенсивное переме¬
шивание фаз и низкое содержание в шлаке оксида железа FeO
при его высокой основности. Так как в конвертере эти условия
полностью не соблюдаются, степень десульфуризации не пре¬
вышает 40% . Поэтому, содержание серы в выплавляемой ста¬
ли определяется главным образом ее содержанием в чугуне, ко¬
торое не должно быть выше 0,06% при выплавке углеродистых
и 0,035% при выплавке качественных сталей.5. Образование шлака. Шлак в процессе плавки образуется
в результате взаимодействия основных, кислотных и амфотер-
ных оксидов различных элементов в составе металлической
шихты:МеО + ЭОл = МеО-ЭОл,где: Me — металл,Э — элемент, образующий кислотный или амфотерный
оксид.Основным источником шлакообразования становится посту¬
пающий в конвертер оксид кальция СаО и продукты окисления
компонентов чугуна (SK>2, MnO, FeO, Р2О5), а также оксиды из
растворяющейся футеровки конвертера (СаО, MgO). В твердых
шлаках эти оксиды присутствуют в связанном состоянии в виде
силикатов и фосфатов, например, ЗСаОРгОб или в виде свобод¬
ных оксидов. От содержания последних зависит основность
(СаО) и окислительная способность (FeO) шлака. В жидком со¬
стоянии вследствие диссоциации шлаки представляют распла¬
вы, содержащие катионы Fe+2, Mn+2, Са+2, Mg+2 и анионов
02“2, S"2, Si04"4, РО4 3, AIO3-3, Fe02"2 и др.В процессе плавки шлак выполняет несколько функций:— связывает оксиды, образующиеся при окислении компо¬
нентов металлической шихты;— служит переносчиком кислорода из газовой фазы к жид¬
кому металлу;— является теплопередающей фазой (при электроплавке);— защищает металл от насыщения газами из газовой фазы.Вследствие этого состав, вязкость, количество и скоростьобразования шлака при плавке оказывают существенное влия¬
ние на качество и выход стали и стойкость футеровки конвер¬
тера.К важнейшим свойствам сталеплавильных шлаков относят¬
ся:— основность (кислотность), зависящая от отношения моль-
ных долей (или процентного содержания) оксидов СаО/ЗЮг, и— окислительная способность, характеризуемая содержани¬
ем в шлаке оксидов железа, главным образом, оксида FeO.Все реакции окисления компонентов чугуна, протекающие
в конвертере экзотермические. При этом количество выделяю¬
щегося тепла существенно зависит от состава металлической
шихты. В некоторых случаях такие ее компоненты как крем¬
ний и фосфор могут быть основным «топливом» при конвертер¬
ной плавке. Однако особое значение для температурного режи¬
ма плавки, процесса шлакообразования и создания микрогете-
рогенной системы имеет окисление углерода, при котором об¬
разуются газообразные продукты.Вследствие экзотермичности реакций окисления и их высо¬
кой скорости окислительный период плавки в кислородном
конверторе протекает в автотермичном режиме и не требует
притока тепла извне. При этом обеспечивается нагрев стали,
выпускаемой из конвертера, до 1600—1650°С, что значительно
выше температуры заливаемого чугуна (1250—1400°С).Б. Восстановительный период плавки при кислородно-кон¬
верторной выплавке стали пространственно отделен от окисли¬
тельного и протекает после выпуска стали из конвертера в ков¬
ше. Одновременно с восстановлением оксида железа FeO в вос¬81
становительный период происходит процесс перевода раство¬
ренного в стали кислорода в нерастворимые в металле соединен
ния и отделение их от стали. Поэтому, восстановительный пе¬
риод плавки называется также операцией раскисления стали.
Удаление кислорода из стали устраняет возможность реакции
между растворенным в ней углеродом и кислородом при ее
медленном охлаждении и образования так называемой «ки¬
пящей » стали за счет выделения из нее пузырьков оксида уг¬
лерода (II).В качестве реагентов для раскисления, восстанавливающих
оксид железа FeO и связывающих растворенный в стали кисло¬
род, используют так называемые раскислители, к которым от¬
носятся элементы с большим сродством к кислороду, чем желе¬
зо. Обычно для этой цели применяют марганец и кремний в виде
соответствующих ферросплавов, алюминий и сплавы некото¬
рых редкоземельных металлов. При этом протекают реакции:Мп + [О] = (MnO); Si + 2[0] = (Si02) иMn + (FeO) = Fe + (MnO); Si + 2(FeO) = 2Fe + (Si02)Образовавшиеся оксиды раскисляющих элементов перехо¬
дят в шлак.Обычно процесс раскисления стали совмещается с введени¬
ем в нее легирующих добавок в виде ферросплавов, содержа¬
щих соответствующие легирующие элементы.5.2.2. Аппаратура и технология плавкиСталеплавильный агрегат в кислородно-конвертерном мето¬
де выплавки стали включает собственно конвертер, систему по¬
дачи кислорода и систему отвода и
очистки конвертерных газов.Устройство конвертетера.
Кислородный конвертер является
реактором^ периодического дей¬
ствия РИС-П. Это поворачиваю¬
щийся на цапфах в вертикальной
цлоскости стальной сосуд груше¬
видной формы, имеющий концент¬
рически расположенную горловину
для заливки чугуна, завалки твер¬
дых компонентов шихты и отвода
газа. Через горловину вводится в
те конвертер фурма для подачи кисло-82
рода и происходит слив жидкой стали и шлака. Изнутри кон¬
вертер футерован огнеупорным материалом. Конфигурацию
внутренней полости конвертера (рис. 5.2) выбирают таким об-
разом, чтобы исключить выброс жидкого металла при продув¬
ке и обеспечить стабильный режим работы.Основные размеры конвертера — высота рабочего простран¬
ства Ну диаметр D, диаметр горловины d и глубина ванны жид¬
кого металла в спокойном состоянии I (рис. 5.2) — зависят от
емкости (мощности) конвертера, которая рассчитывается по
массе жидкой стали. В РФ ГОСТом установлен ряд типовых ем¬
костей кислородных конвертеров от 50 до 400 тонн.Важнейшими характеристиками конвертера являются:— отношение высоты к диаметру H/D, которое уменьшает¬
ся с увеличением емкости конвертера, и— удельный объем, то есть объем конвертера, приходящий¬
ся на 1 т перерабатываемого чугуна УУз который для различ¬
ных конвертеров изменяется незначительно.Футеровка конвертера состоит из трех слоев: примыкающе¬
го к кожуху арматурного слоя, внутреннего рабочего слоя и со¬
единяющего их промежуточного слоя. Арматурный слой из
магнезитового кирпича служит дда снижения теплопотерь и
защиты кожуха конвертера при прогаре рабочего слоя. Он не
требует замены в течение нескольких лет. Рабочий слой изго¬
тавливается из безобжиговых огнеупоров на основе каменно¬
угольной смолы или пека имеет толщину от 0,4 до 0,7 м и вы¬
держивает от 500 до 800 плавок. Так как он подвергается хими¬
ческому воздействию шлака, размывающему действию потоков
металла и шлака и ударам при загрузке шихты, то изнашива¬
ется во время работы и требует периодической замены. В пос¬
леднее время для восстановления рабочего слоя используется
метод горячего ремонта (торкретирование футеровки) путем
вдувания в конвертер смеси из магнезитового шлака и коксо¬
вой пыли. Торкретирование позволяет не останавливать кон¬
вертор на длительное время для ремонта.Система подачи кислорода. По принципу подачи кислорода
конвертеры делятся на три типа: с верхней продувкой, с дон¬
ной продувкой и с комбинированной продувкой. Наиболее рас¬
пространены конвертеры первого типа. В них кислород подает¬
ся под давлением 0,9—1,5 МПа через охлаждаемую водой фур¬
му, вводимую на время продувки в конвертер через его горло¬
вину.В конвертерах с донной продувкой, используемых с середи¬
ны 60-х годов, кислород подается через систему фурм (от 7 до 2283
в зависимости от емкости конвертера), установленных в днище*
конвертера. Для охлаждения фурм в их кольцевое пространство
поступает жидкое или газообразное топливо, термическая дис¬
социация которого, протекающая с поглощением тепла, компен¬
сирует избыточное тепло, выделяющееся при продувке в зоне,
прилегающей к фурмам. При донной продувке улучшается пе¬
ремешивание ванны, повышается скорость выгорания углерода,
увеличивается степень дефосфоризации и десульфуризации й
уменьшаются потери металла со шлаком. В результате сокраща¬
ется время плавки и увеличивается выход стали. Однако, дон¬
ная продувка требует сокращения содержания стального лома в
шихте, так как часть тепла при продувке расходуется на диссо-
цацию угловодородов охлаждающего фурма топлива.В последнее время получают распространение конвертеры с
комбинированной продувкой, в которых через верхнюю фурму
подается большая часть кислорода, а через донные фурмы или
пористые огнеупорные элементы днища вдувается остальная
часть кислорода или смесь его с инертным газом. В таких кон¬
вертерах сочетаются преимущества реакторов первого и второ¬
го типов.Расход кислорода на продувку зависит от емкости конверте¬
ра и достигает 2000 м3/мин. при интенсивности подачи от 2,5
до SjOmV^mhh. Так как от интенсивности подачи кислорода
зависит время продувки, то для его сокращения выгодно уве¬
личивать ее до предела, за которым возможен выброс металла
*и шлака.Система отвода и очистки конвертерных газов. В процес¬
се продувки образуется большое количество конвертерных га¬
зов, нагретых до 1450—1650°С. При интенсивности выхода га¬
зов 5—14 м^/т металла, объем их для 350-тонного конвертера
достигает 5000 м3. Конвертерные газы состоят главным обра¬
зом из продуктов окисления углерода и содержат около 85% ок¬
сида углерода (II), 10% оксида углерода (IV) и 5% азота, а так¬
же значительное количество (до 250 г/м3) мелкодисперсных ча¬
стиц оксида железа (III) — бурый дым.Система отвода и очистки конвертерных газов включает ко¬
тел-утилизатор, в котором используется теплосодержание га¬
зов, мокрые скрубберы и электрофильтры для удаления пыли.
Очищенный газ собирается в газгольдерах или выбрасывается
в атмосферу через дожигающее оксид углерода (II) устройство.Шихтовые материалы. Шихта для кислородно-конвертер¬
ной выплавки стали состоит из металлической части (жидкий
чугун, стальной лом или скрап), неметаллической части (из¬84
весть, плавиковый шпат, охладители) и легирующие добавки.-
Чугун является основным материалом и доля его в металличес¬
кой части шихты может составлять от 100 до 70%, доля метал¬
лического лома не более 25—30%. В кислородном конвертере
можно перерабатывать любой передельный чугун с содержани¬
ем углерода 3,9—4,3%. Однако для получения высоких ТЭП и
стал и надлежащего качества к составу чугуна предъявляют оп¬
ределенные требования по содержанию фосфора (не более 0,2—0,3%), серы (не более 0,04—0,06%) и марганца (не более 0,7—
1,1%). Металлический лом должен быть малогабаритным, с
размерами кусков не более 0,3x0,3x1,0 м3, и содержать мини¬
мальное количество вредных примесей и ржавчины. Помимо
лома используют также металлизированные окатыши и брике¬
тированное губчатое железо.Известь, используемая в качестве шлакообразующего мате¬
риала, должна содержать не менее 90% оксида кальция и огра¬
ниченное количество кремния (не более 3%) и серы (не более0,2%). Для разжижения шлака используется добавка плавико¬
вого шпата CaF2.При переработке чугунов с повышенным содержанием крем¬
ния во избежание подъема температуры плавки сверх оптималь-
ной, в состав шихты вводят охладители в виде железной руды,
боксита и агломерата.Технологический процесс. Процесс выплавки стали в кисло¬
родном конвертере состоит из нескольких операций, которым
соответствует определенное положение конвертера относитель¬
но его оси.1. Конвертер в наклонном положении:— завалка металлолома с помощью завалочной машины;— заливка жидкого чугуна из ковша с помощью заливочно¬
го крана;2. Конвертер в вертикальном положении:— введение фурмы и продувка кислородом с одновременной
загрузкой части шлакообразующих материалов;загрузка остальной части шлакообразующих материалов
до окончания продувки и выведение фурмы из конвертера, ана¬
лиз стали.3. Конвертер в горизонтальном положении:— выпуск стали через летку в ковш.4. Конвертер в положении горловиной вниз:— слив шлака через горловину в шлаковый ковш.Продувку ведут до достижения в металле заданного содер¬
жания углерода, определяемого результатами анализа стали,
объемом израсходованного кислорода и временем продувки.85
Для раскисления стали в ковш вводят в определенной пос¬
ледовательности раскислители. Правильный режим раскисла
ния позволяет снизить угар металла.Технико-экономические показатели. Годовая производи¬
тельность кислородного конвертора рассчитывается по форму¬
ле:1440/7ШП= л (5.1)хгде: т — емкость конвертера по массе жидкой стали, т
1440 — число минут в сутках,
п — число рабочих суток в году,
т — длительность плавки, мин.,
j\ — выход годной стали от шихты, долей ед.Для современных конвертеров длительность плавки колеб¬
лется от 25 до 55 мин. в зависимости от их емкости. Из этого
времени на продувку падает 35—50%. Часовая производитель¬
ность конвертеров большой емкости достигает 400—500 т или
18—20 т с 1 т емкости конвертера.Себестоимость конвертерной стали определяется, главным
образом, стоимостью металлической шихты и расходами на пе¬
редел (кислород, огнеупоры, электроэнергия, заработная плата).
На выплавку 1 тонны конвертерной стали расходуется, в сред¬
нем, 45—57 м3 кислорода и 2,0—5,0 кг огнеупорных материа¬
лов. Структура себестоимости кислородно-конвертерной стали:Стоимость металлической шихты 80—86%Стоимость вспомогательных материалов 1,3—3,5%Расходы по переделу 10—19%.5.3. Выплавка стали в электрических печахВыплавка стали в электрических печах основана на исполь¬
зовании для нагрева, расплавления и поддержания металла в
расплавленном состоянии электрической энергии, трансформи¬
руемой в теплоту. В отличие от кислородно-конвертерного ме¬
тода при электроплавке выделение тепла не связано с исполь¬
зованием окислителей. Поэтому, плавку в электрических пе¬
чах можно вести в любой атмосфере — окислительной, восста¬
новительной, нейтральной (инертный газ) и в широком диапа¬
зоне давлений — в вакууме, при атмосферном или повышенном
давлениях.К основным достоинствам электросталеплавильного метода
относятся:86
гг быстрый нагрев металла, что позволяет вводить болыпЬе
количество легирующих добавок;— применение безокислительных шлаков и как следствие,
малый угар легирующих элементов;“ точное и плавное регулирование температуры и состава
металла;— высокая степень раскисления металла;— возможность получения сталей с низким содержанием
серы и фосфора.Однако, несмотря на эти достоинства электроплавки, высо¬
кое потребление электроэнергии обусловило использование ее
преимущественно для производства легированных и высокока¬
чественных (с низким содержанием серы, фосфора, кислорода
и других вредных примесей) сталей, в том числе, инструмен¬
тальных, жаростойких, шарикоподшипниковых и т. п. В пос¬
леднее время, в связи с внедрением в металлургическое произ¬
водство электропечей большой мощности (до 400 т), электро¬
плавка стала применяться й для получения рядовых углероди¬
стых сталей по упрощенной технологии с их последующим пе¬
реплавом.5.3.1. Электрические печи ^Для выплавки стали применяются электрические печи пе¬
риодического действия реакторы РИС-П. По принципу гене¬
рирования теплоты они делятся на дуговые и индукционные.В дуговых печах тепло выделяется в результате гсфения элек¬
трической дуги. По расположению дуги относительно металли¬
ческой шихты дуговые печи подразделяются на печи косвенно¬
го нагрева, печи прямого нагрева и печи комбинированного на¬
грева. Наиболее распространены печи прямого нагрева, в кото¬
рых электрические дуги горят непосредственно ме^сду каждым
из электродов и металлом.В индукционных печах тепло выделяется за счет возникно¬
вения в толще шихты индукционных токов. В основе действия
индукционных печей лежит трансформаторный принцип пере¬
дачи энергии от первичной цепи к вторичной. При этом первич¬
ной обмоткой — индуктором является соленоид, а вторичной —
металлическая шихта.Дуговые печи. Для электроплавки применяются главным
образом трехфазные дуговые печи переменного тока с тремя
электродами и непроводящим подом. В соответствии с ГОСТ
7206-63 подобные печи выпускаются емкостью от 0,5 до 400 т.87
Рис. 5.3. Дуговая печь;1 — свод, 2 — эйектроды, 3 —
желоб, 4 — под, 5 — рабочее
окно, 6 — шлак, 7 — металлПечь (рис. 5.3) состоит из металли¬
ческого сварного корпуса, укреп¬
ленного в люльке, позволяющей на¬
клонять печь, и поворотного свода,
через который в плавильное про¬
странство печи вводятся электроды.
Корпус печи изнутри футерован ог¬
неупорным материалом и имеет вы¬
пускное отверстие (желоб) для
слива металла и рабочее окно, через
которое загружают шлакообразую¬
щие материалы, руду и ферроспла¬
вы и скачивают шлак. Печь снабже¬
на механизмом наклона в сторону
сливного отверстия и в сторону рабочего окна, механизмом
подъема и отворота свода при загрузке печи и механизмом по¬
ворота кожуха для ускорения плавления шихты. Печи после¬
дних конструкций имеют дополнительное устройство для элек¬
тромагнитного перемешивания расплавленного металла, что
ускоряет процесс плавки.Электроды печи должны обладать высокой электропроводи¬
мостью, выдерживать высокие температуры и иметь достаточ¬
ную механическую прочность. Этим требованиям удовлетворя¬
ют исключительно изделия на основе углерода: угольные и гра-
фитированные электроды, получаемые обжигом малозольных
углеродны^ материалов. Для уменьшения расхода материала
верхнюю чйсть электрода изготавливают из стали и охлаждают
водой. Электроды укреплены в специальных зажимах и в про¬
цессе плавки могут перемещаться в вертикальном направлений
в соответствии с заданной программой, что обеспечивает посто¬
янство длины дуги.Рабочее напряжение, подаваемое на электроды, колеблется
в зависимости от емкости печи от 110 до 800 В. Для уменьше¬
ния потерь энергии электрический ток подается к печам под
напряжением 6—10 кВ через понижающий трансформатор. В
табл. 5.4 приведены характеристики наиболее распространен¬
ных дуговых печей ДСП-100 и ДСП-200.Расход энергии на плавку в дуговых печах составляет 500—
800 кВт*ч на тонну выплавляемой стали. Расход электродной
массы 5—9 кг/т стали.Индукционные печи. Электроплавильные индукционные
печи имеют емкость от 0,01 до 12 т и питаются током от различ¬
ных источников. Для питания малых печей применяют лампо¬88
вые генераторы с частотой 50—1000 кГц, печей средней и боль¬
шой мощности — машинные генераторы с частотой 500—10000
кГц, наиболее крупные печи питаются непосредственно от сети
током промышленной частоты 50—60 Гц.Таблица 5.4. Характеристики дуговых печейХарактеристикаТип п<вчидсп-tooДСП-200Емкость, тДиаметр кожуха внутренний, м
Диаметр электрода, м
Ход электрода при плавке, м
Диаметр ванны, м
Глубина ванны, мМощность трансформатора, MB'А
Вторичное напряжение, В1007,20,5553,65,301,1032160—4802009,20,6105,06,851,4845160—600Индукционная печь (рис. 5.4)состоит из огнеупорного тиг-
ля с сливным носком, помещенного в индуктор в виде солено¬
ида из медной трубки, охлаждаемой водой. Печь заключена в
металлический кожух, закрываемый сверху сводом. Для сли¬
ва металла печь может наклоняться в сторону сливного нос¬
ка. Процесс плавки в индукционных печах протекает весьма
быстро. В качестве металлической шихты в них используется
металлический лом известного состава, который точно рассчи¬
тан по содержанию углерода, серы, фосфора и легирующих
элементов.Так как в индукцион¬
ных печах отсутствуют электроды,
выплавляемая в них сталь не заг¬
рязняется углеродом и продуктами
их обжига, угар легирующих эле¬
ментов весьма мал. Поэтому индук¬
ционные печи применяют для вып¬
лавки только высококачественных
сталей и сплавов сложного хими¬
ческого состава. Расход энергиипри плавке в индукционных печах Рис- 5.4. Индукционная печь:составляет 500—700 кВт*ч на тон- 1 — кожух, 2 — тигель, 3 —
ну стали. подовая плита, 4 —соленоид»5 — сливной носок, 6 — токо-
подводы89
5.3.2. Технологический процессТехнологические процессы выплавки стали в электрических
печах весьма разнообразны. Конкретный режим плавки зави¬
сит от природы и состава металлической шихты, типа печи и
материала ее футеровки. Наиболее распространена плавка в
дуговых печах, в частности:— плавка с полным окислением примесей;— переплав стальных отходов без окисления;— плавка на жидкой шихте (дуплекс-процесс) й др.Ниже рассмотрен один из подобных процессов — типоваяплавка в дуговых электропечах на углеродистой шихте с окис¬
лением, применяемая для выплавки большинства типов сталей.
Процесс включает следующие операции:— заправка печи,— нагрузка шихты в печь,— плавление шихты,— окислительный период плавки,— восстановительный период плавки,— выпуск металла из печи.Заправка печи. Цель этой операции — исправление изно¬
шенных и поврежденных в процессе плавки участков внутрен¬
ней футеровки пёчи. Д ля этого впечь забрасывается магнези¬
товый порошок или смесь порошка со связующим в виде камен¬
ноугольного пека.Загрузка шихты. Основным сырьем для электроплавки яв¬
ляется стальной лом, содержание которого в металлической
шихте составляет 90^100%. Для повышения содержания уг¬
лерода в шихту вводят до 10% чугуна. В качестве сырья для
плавки в электропечах используют также губчатое железо, со¬
держащее 85—93% металла, и металлизированные окатыши,
содержащие не менее 90% металла. Шихта загружается в печь
порциями с помощью бадей и плотно укладывается, что обес¬
печивает ее проводимость и устойчивое горение дуги.Плавление шихты. По окончании загрузки шихты элект¬
роды опускают до касания с шихтой и включается ток. Под воз¬
действием высокой температуры шихта под электродами пла¬
вится и расплавленный металл стекает вниз, образуя в шихте
«колодцы*, в которые постепенно по программе опускаются
электроды, пока не достигнут нижнего положения. Затем, по
мере накопления жидкого металла, электроды вновь поднима¬
ют. В период плавления происходит частичное окисление ком¬
понентов шихты кислородом воздуха и оксидами железа в руде90
и окалине и формируется шлак. В период плавления полнос¬
тью окисляется кремний, до 50% марганца и частично углерод
и железо. Время плавления шихты составляет от 1 до 3 часов.А. Окислительный перибд плавки. В этот период в печи
протекают процессы окисления углерода до заданного содер¬
жания, уменьшение содержания в металле фосфора, азота и
водорода и нагрев металла до температуры близкой к темпе¬
ратуре выпуска. В качестве окислителей используются окси¬
ды железа, содержащиеся в руде и агломерате в составе ших¬
ты, или газообразный кислород, подаваемый под давлением в
печь. В конце окислительного периода из печи скачивают об¬
разовавшийся шлак.Б. Восстановительнщй период плавки. В этот период в печи
происходит раскисление металла, удаление серы и состав ста¬
ли доводится до заданного. Для этого в печь подаются раскис-
лители (ферромарганец, ферросилиций, алюминий) и шлако¬
образующие компоненты. Одновременно в печь вводят легиру¬
ющие добавки.Выпуск металла. По достижении металлом температуры на
120— 130°С выше температуры ликвидуса металл выпускается
из печи в ковш.5.3.3. Технико-экономические показателиГодовая производительность дуговой электрической печи
рассчитывается по формуле:24 mnП= т) (5.2)хгде: m — емкость печи по жидкой стали, т,
п — число рабочих суток в году,24 — число часов в сутках,
т — длительность плавки, ч,т\ — выход годной стали от жидкой стали, дол. ед.Значительные простои электрических печей связаны с необ¬
ходимостью ремонта ее футеровки и составляют от 5 до 10%
всего времени. Продолжительность плавки в электропечах за¬
висит от емкости печи, состава выплавляемой, стали, техноло¬
гии плавки, метода раскисления стали (в печи или в ковше) и
мощности трансформатора тока. Для печей емкостью 100 т она
равна 4—6 часам. Применение кислорода в окислительный пе¬
риод плавки сокращает время плавки на 10—20% и, соответ¬
ственно, увеличивает производительность печи.91
I5.4. Выплавка стали в мартеновских печахДействующие современные мартеновские печи — это круп¬
ные сталеплавильные агрегаты сложной конструкции с боль¬
шим количеством различных дополнительных устройств- Стро¬
ительство их связано с крупными капитальными затратами. По¬
этому одновременный отказ от мартеновского способа производ¬
ства стали и переход к кислородно-конвертерному и электро¬
сталеплавильному способам экономически нецелесообразен.
Этим объясняется высокая доля мартеновской стали, выплав¬
ляемой до настоящего времени в нашей стране.5.4.1, Особенности мартеновского способа плавкиМартеновский процесс выплавки стали ведут на поду пла¬
менной отражательной печи, снабженной регенераторами теп¬
ла отходящих газов для подогрева воздуха и топлива, подавае¬
мых в печь. В зависимости от состава металлической печи раз¬
личают две разновидности процесса:1. Скрап-процесс, в котором основным компонентом шихты
является стальной лом (скрап) с добавкой 25—40% чугуна, об¬
легчающего расплавление лома, являющегося источником уг¬
лерода* Скрап-процесс используется в цехах металлургических
и машиностроительных заводов, в которых нет доменного про¬
изводства.2. Скрап-рудный процесс, в котором основным компонентом
шихты является жидкий чугун с добавкой 45—25% скрапа и
железной руды для окисления примесей в чугуне. Этот процесс
применяется на заводах, имеющих собственное доменное про¬
изводство.При выплавке стали в мартеновских печах протекают про¬
цессы окисления углерода и примесей в шихте и образования
шлаков аналогичные тем, которые идут в кислородном конвер¬
тере. Однако мартеновский процесс имеет ряд существенных
особенностей. К ним относятся:— использование в качестве источника тепла реакций сго¬
рания непосредственно в печи газообразного и жидкого топли¬
ва (природный газ, мазут, смесь коксового и доменного газов);— поступление тепла от горящего факела топлива к ванне
сверху и отвод тепла снизу, вследствие чего температура шла¬
ка превышает температуру металла;— окислительный характер газовой фазы, состоящей во все
периоды плавки из оксида углерода (IV),кислорода, паров воды
и азота;92
— макрогетерогенность системы «металл—шлак», в которой
металл находится под слоем шлака. Вследствие этого все добав¬
ки, кислород и тепло поступают в металл через шлак. Поэтому
изменение состава,консистенции и температуры шлака суще¬
ственно влияют на состав и качество выплавляемой стали;; -^участие пода печи в протекающих в ней процессах шла¬
кообразования вследствие длительности процесса плавки.5:4*2. Двухванные печи •Недостатки мартеновского способа выплавки стали (боль¬
шие капитальные затраты, низкая по сравнению с кислородно¬
конвертерным способом производительность, затраты на топ¬
ливо , сложность обслуживания регенераторов вследствие раз¬
рушения их насадки) не могут быть полностью компенсирова¬
ны такими методами интенсификации процесса как повыше¬
ние давления и обогащение кислородом воздушного дутья и
предварительная карбюрация топлива. Это вызвало необходи¬
мость изменения уже не технологии, а конструкции мартенов¬
ских печей — создания двухванных сталеплавильных агрега¬
тов (рис. 5.5). В основу их действия положен принцип работы
кислородного конвертера — окисление углерода и примесей
продувкой шихты кислородом. При этом в двухванных печах
для нагрева шихты используют часть выделяющегося тепла в
виде теплосодержания отходящих газов и теплового эффекта
дожигания оксида углерода (II).Конструктивно двухванные
печи состоят из двух ванн, рабочее
пространство которых спарено
между собой общим сводом. Каж¬
дая ванна имеет головку, в которой
размещены фурмы для подачи кис¬
лорода или топливно-кислородной
смеси и фурмы для вдувания твер- Рис Двухванная печь:дых материалов. Ванны соединены * — топливно-кислородные
вертикальным каналом с шлакови- Фурмы* 2 ~ Фур*ы для твеР-Т(§ « дых материалов, 3 — шлакови¬ком. Когда в одной ванне протека- ки 4 _ ПОДЬ1 ааннют процессы, требующие затратытепла (завалка, прогрев и началоплавления металлической шихты), в другой ванне идет про¬
дувка металла, то есть процессы с выделением тепла (интен¬
сивное плавление, рафинирование). Так, на рис. 5.5 в левой
ванне идет прбдувка жидкого металла кислородом, а в правой93
загруженная шихта нагревается газами, отходящими из ле¬
вой ванны.Режим плавки в двухванных печах требует очень четкой
синхронизации работы обеих ванн: к концу продувки металла
в левой ванне должна быть закончена завалка шихты в правой
ванне. При содержании в шихте более 65% жидкого чугуна
двухванные печи могут работать без подачи топлива. При боль¬
шем содержании металлического лома в шихте в ванне сжига¬
ется топливо, подающееся с помощью газокислородных горе¬
лок.Целесообразность внедрения в сталеплавильное производ¬
ство двухванных печей связана, в первую очередь, с тем, что в
них без значительных капитальных затрат могут быть переде¬
ланы существующие мартеновские печи. Это позволяет увели¬
чить производство стали в рамках существующих мартеновс¬
ких печей с использованием их коммуникаций и вспомогатель¬
ного оборудования.Технико-экономические показатели, Производительность
двухванных печей рассчитывается по формуле:тиП=— (5.3)тгде: тП — масса стали за одну плавку, т,
т — время плавки, ч,П — производительность печи, т/ч.Для двухванных печей время плавки составляет около 4 ча¬
сов, то есть каждые два часа из одной из ванн выпускается сталь.Важным ТЭП работы печи является съем стали или масса
стали, выплавляемая в сутки и отнесенная к 1м2 пода ванны
печи:где: т
S5.5. Вторичная обработка сталиДля многих изделий авиационной, приборостроительной,
радиоэлектронной и космических отраслей промышленности,
для конструкций, работающих при низкой температуре или в
широком интервале температур, требуются стали с ничтожноЛ mС=— (5.4)масса стали, выплавляемой одной ванной в сутки, т,
площадь пода одной ванны, м2.94
малым содержанием газов и неметаллических включений, од¬
нородные по свойствам, не содержащие микропор. Сталь, вып¬
лавленная в конвертере, электрической и мартеновской печи
удовлетворить всем этим требованиям не может. Поэтому, для
получения стали ответственного назначения ее подвергают вто¬
ричной обработке. При этом, помимо основной цели — удале¬
ния из металла примесей и улучшения его структуры, повы¬
шается производительность основного сталеплавильного агре¬
гата (конвертера, печи) ,так как часть технологических опера¬
ций выносится из его рабочего пространства в ковш, агрегат
переплава и т. п. К вторичной обработке стали относятся вне-
печное рафинирование и переплавные процессы.5.5.1. Внепечное рафинирование сталиВ основе метода внепечного рафинирования лежит процесс
удаление примесей из жидкой стали путем обработки ее раз¬
личными рафинирующими средами. Исходя из природы рафи¬
нирующих сред технологические схемы внепечного рафиниро¬
вания можно свести к следующим типам:— обработка разреженным гяаом (вакуумирование); ^ — обработка нейтральным газом (продувка);— обработка жидкими шлаками (шлаковое рафинирование).Вакуумирование стали. Вакуумная обработка жидкой ста¬
ли может производиться в ковше или в струе металла при его
переливе из ковша в ковш. В первом, наиболее распространен¬
ном на практике методе, ковш с жидкой сталью помещается в
вакуумную камеру и выдерживается в ней при разряжении от
13 до 2 кПа. При этом цз металла выделяются газы (водород,
оксиды углерода, азот) и снижается содержание в металле уг¬
лерода и оксидов за счет смещения вправо равновесий[С] + [О] СО и (МеО) + [С] Me + СОПродувка инертным газом. Метод продувки основан на том
же эффекте, что и вакуумирование. При продувке жидкого ме¬
талла аргоном масса металла пронизывается пузырьками газа,
каждый из которых подобен миниатюрной вакуум-камере, так
как парциальное давление газов, содержащихся в металле, в
этих пузырьках равно нулю. Кроме этого, интенсивный барбо-
таж при продувке приводит к усреднению состава металла и
переходу в шлак из металла неметаллических включений.Шлаковое рафинирование. При шлаковом рафинировании
стали в ковш заливают жидкий синтетический шлак, состоя¬
щий из 45% оксида алюминия и 55% оксида кальция, и в него.95
с высоты 3—4 м выпускается струя металла из сталеплавиль?
ного агрегата. За счет интенсивного перемешивания и эмуль¬
гирования фаз резко (в 104—105 раз) возрастает поверхность их
контакта и увеличивается скорость взаимодействия металла и
шлака. В результате достигается эффективное рафинирование
стали и ее дополнительная десульфуризация, раскисление и
удаление неметаллических включений.Переплавные процессы, В отличие от внепечного рафини¬
рования, при котором обрабатывается жидкая сталь, переплав¬
ные процессы представляют различные способы переплава уже
готовых слитков или стальных заготовок с целью удаления из
них примесей. В промышленных масштабах используют сле¬
дующие виды переплава: вакуумно-дуговой, электронно-лу¬
чевой, плазменно-дуговой, электрошлаковый. Принципи¬
альные схемы этих переплавных процессов приведены на
рис. .5.6 (а—г).Вакуумно дуговой переплав (ВДП) осуществляется в ваку¬
умных печах (рис. 5.6а) в которых очищаемый металл, игра¬
ющий роль электрода, плавится под воздействием электричес¬
кой дуги, возникающей между ним и формируемым слитком
чистого металла, находящимся в охлаждаемой водой излож¬
нице (кристаллизаторе). Для устойчивости дуги переплав ве¬
дут на'шэтоянном токе ’/при этом электрод является катодом,
а слиток чистого металла — анодом. В процессе переплава в
печи поддерживается разряжение, за счет чего капли плавя¬
щегося металла дегазируются. Охлаждение расплавленного
металла в кристаллизаторе ведется с такой скоростью, чтобы
обеспечить направленный характер ее — сверху вниз. Вслед¬
ствие этого из металла удаляются твердые включения, кон¬
центрирующиеся в верхней части слитка (метод направлен¬
ной кристаллизации).Электроннолучевой переплав (ЭЛП) проводится в элект¬
ронно-лучевых печах (рис. 5.66). В них нагрев и плавление
металла происходят под воздействием тепла, выделяющегося
при резком торможении электронов, поток которых, выходя¬
щий из электронной пушки, направлен на металл. При нагре¬
ве до высокой температуры в глубоком (1,3-10”2—1,3*10"3Па)
вакууме катод пушки испускает электроны, которые форми¬
руются в направленный поток с помощью фокусирующих и
отклоняющих устройств при приложении высокого (до 40 кВ)
напряжения между анодом и катодом пушки. Для обеспече¬
ния равномерного нагрева обычно используются несколько
пушек.96
~©—ок вакуум-
насосуВакуумная дуговая печь: 1 —
электрод (очищаемый металл),2 — слиток (очищенный металл),3 — жидкий металл, 4 — кристал¬
лизатор, 5 — вакуумная камера,6 — токоподвод56даlil»1Сгиу\Vi£Ф-Электронно-лучевая печь: 1 —
электрод (очйщаемый металл), 2 —
слиток (очищенный металл), 3 —
жидкий металл, 4 — кристаллизатор,5 — вакуумная камера, 6 — фокуси¬
рующее устройство, 7 — электрон¬
ная пушкаПлазменно-дуговая печь: 1 —
плазмотрон, 2 — подовый элект¬
род, 3 — жидкий металл, 4 —
устройство для перемешивания
металла, 5 — выпускное отверстиеЭлектрошлаковый переплав: 1 —
электрод (очищаемый металл), 2 —
слиток (очищенный металл), 3 —
жидкий металл, 4 — кристаллиза¬
тор, 5— герметизированная
камера, 6 — шлаковая ванна, 7 —
источник питанияРис. 5.6. Схемы устройств для ВДП, ЭЛП, ПДП и ЭШП4 Химическая технология. Том 297
tПлазменно-дуговой переплав (ПДП) осуществляется в плаз¬
менных дуговых печах (рис. 5.6в), конструктивно близких к
обычным дуговым электрическим печам. Однако в них нагрев
и расплавление шихты происходит при помощи плазменной
дуги, возникающей между катодом плазмотрона и металлом,
находящимся в контакте с охлаждаемым водой анодом. Источ¬
ником тепла в плазменно-дуговых печах является низкотемпе¬
ратурная плазма с температурой порядка 3* 104*С. Современные
плазменные печи достигают емкости 30 т.Электрошлаковый переплав (ЭШП) — это наиболее простой,
экономичный и не требующий дорогостоящего оборудования
способ переплава стали. Поэтому он широко используется в ста-
леплавлении. Источником тепла при ЭШП служит шлако¬
вая ванна, наполненная жидким электропроводным синтети¬
ческим шлаком, состоящим из 70% фторида кальция и 30% ок¬
сида алюминия. Электрический ток подводится к электроду
(неочищенный металл) и поддону, находящемуся в контакте со
слитком из очищенного металла. При прохождении тока шлак
нагревается до 2000°С и погружаемый в него электрод плавит¬
ся. Капли жидкого металла проходят через шлак и охлажда¬
ются в кристаллизаторе, образуя слиток. В результате контак¬
та со шлаком и последующей медленной направленной снизу
вверх кристаллизации из металла удаляются примеси. Мето¬
дом ЭШП в настоящее время получают стальные слитки мас¬
сой до 300 т (рис. 5,бг).5.6. Разливка сталиЗаключительная операция сталеплавильного производ¬
ства — выпуск стали в сталеразливочный ковш и ее разлив¬
ка. От правильного проведения процесса разливки зависят
ТЭП производства, оцениваемого по выходу готовых слитков.
Существуют два принципиально различных методов разлив¬
ки стали:— разливка в изложницы,— непрерывное литье заготовок.Первый метод разливки до настоящего времени преоблада¬
ет, хотя имеет ряд существенных недостатков, главным из ко¬
торых является высокий дополнительный расход тепла на ра¬
зогрев слитков перед прокатом.Метод непрерывного литья заключается в том, что жидкую
сталь из сталеразливочного ковша непрерывно подают через
промежуточный ковш (демпферная емкость) в охлаждаемый98
водой кристаллизатор. Образую¬
щийся в кристаллизаторе слиток
вытягивается из него с помощью
валков и поступает в зону вторично¬
го охлаждения, где орошается водой
из форсунок до полного затвердева¬
ния. Затвердевший слиток без оста¬
новки его движения режется на за¬
готовки заданной длины с помощью 5.7. МНРС радиальногогазорезки или гидравлических нож- типаниц. Скорость разливки (вытягива- 1 — разливочный ковш, 2 —ния слитка) зависит от сечения за- промежуточный ковш, 3 —- Л л 1Л кристаллизатор, 4 — зона вто-готовки и колеблется от 0,4 до 10 м ги w ричного охлаждения, 5 —в минуту. Методом непрерывного метаппа, 6 — устрой-литья можно получать слитки (за- ство для резки, 7 — отводящий
готовки) различного сечения: квад- рольганг,
ратного (блюмы 500 х 500 мм) пря¬
моугольного (слябы шириной до 2500 мм), профильные заготов¬
ки для труб, балок, рельсов, то есть формовать изделие в про¬
цессе разливки металла. Непрерывное литье осуществляется в
агрегатах, получивших название машин дли иеирерьшной раз-
ливки стали (МНРС). По расположению струи металла МНРС
делятся на машины вертикального типа, машины с изгибом
слитка, машины криволинейного (радиального) типа и маши¬
ны горизонтального типа. Наиболее распротранены МНРС ра¬
диального типа (рис. 5.7), сочетающие преимущества МНРС
вертикального типа с относительно небольшой высотой конст¬
рукции (10—12 м против 35—40 м).К достоинствам метода непрерывного литья относятся:— упрощение и сокращение с 140 до 6 часов технологичес¬
кого цикла;— снижение на 90% расхода тепла;— сокращение в три раза потерь металла и увеличение вы¬
хода готовой продукции; /— высокая степень автоматизации и механизации процессаи, как следствие, повышение производительности установки.Вследствие этого метод непрерывной разливки стали интен¬
сивно внедряется в производство, составляя в настоящее время
в индустриально развитых странах свыше 90% всего объема
разлитой стали. В РФ доля непрерывной разливки составляет
около 25% от общего количества.4*99
5.7. Экономика сталелитейного производстваВ табл. 5,5 приведена структура себестоимости стали, вып¬
лавляемой в кислородных конвертерах, электрических и мар¬
теновских печах без учета стоимости вторичных процессов пе¬
реплава. Из табл. 5.5 следует, что основной статьей в себестои¬
мости стали являются затраты на сырье и основные материа¬
лы. Они примерно одинаковы для мартеновского и кислород-
но-конвертерного методов производства и существенно меньше
для электросталеплавильного, что связано с составом исполь¬
зуемой шихты.Таблица S.5. Структура себестоимости стали,%Статья затратМетод выплавки сталиКислородно¬конвер¬торныйМарте¬новскийЭлектроста-леплавиль-,ныйСырье и основные мате¬87,483,866,7риалы за вычетом отхо¬дов и бракаДобавочные материалы2,43,28,1Расходы по переделу,9,812,524,3в том числе, топливо0,12,20,2Технологическая элект¬— 5.1роэнергияЗаработная плата0,60,82,0Общезаводские расходы0.40,50.9Повышенные затраты на добавочные материалы в электро¬
сталеплавильном методе связаны с расходом угоольных элект¬
родов. Расходы по переделу и на технологическую энергию так¬
же максимальные в этом методе, что объясняется принципи¬
альной особенностью метода. В то же время, затраты на техно¬
логическое топливо присущи только мартеновскому методу
выплавки.Снижение себестоимости стали может быть достигнуто:— увеличением отношения стальной лом/чугун за счет вне¬
дрения более эффективной технологии (за последенее время это
отношение возросло с 0,33 до 0,38);
— уменьшением угара металла за счет перевода операций
раскисления и легирования из сталеплавильного агрегата в раз¬
ливочный ковш.5.8. Непрерывные сталеплавильные процессыВ современном производстве черных металлов периодичес¬
кие процессы "(все методы выплавки стали и переплава) сочета¬
ются с процессами непрерывными (доменный процесс и непре¬
рывная разливка стали). Координация работы агрегатов пери¬
одического и непрерывного действия вызывает определенные
затруднения, приводит к неоправданным материальным и эко¬
номическим потерям и препятствует созданию единой техно¬
логической цепочки. В связи с этим перед сталеплавлением
встает задача организации полностью непрерывного металлур¬
гического цикла,что возможно лишь при замене сталеплавиль¬
ного агрегата периодического действия (печь, конвертер) агре¬
гатом непрерывного действия, как это показано на схеме:Доменная Сталепла- мнрспечь L/ Г вильная печь Т1 II I[ Агрегат не- [
прерывного “ “
действияПервые варианты сталеплавильных агрегатов непрерывно¬
го действия (САНД) появились еще в Д898 году в России. В
последующем были предложены их разнообразные конструк¬
ции. В зависимости от принципа, положенного в основу их дей¬
ствия, они делятся на две группы. К первой группе относятся
огрегаты, в которых плавление и технологические операции по
превращению чугуна в сталь протекают в одном и том же рабо¬
чем пространстве. Такие САНД относительно просты в устрой¬
стве, но технологически несовершенны. Ко второй группе от¬
носятся агрегаты, в которых отдельные стадии процесса проте¬
кают последовательно в отдельных реакционных зонах с опти¬
мальным для данной стадии режимом. Такие САНД являются
аппаратами, в которых металл перемещается из одной емко¬
сти в другую, или перетекает из одной части рабочего про¬
странства аппарата в другую, причем в каждой емкости или
части аппарата протекает одна или несколько технологичес¬
ких операций. Такие САНД наиболее распространены и де¬
лятся на три типа.Прокатный,станСтальноеизделие101
1. Агрегаты струйного типа, в которых струя жидкого чу¬
гуна последовательно обрабатывается тонкоизмельченными
флюсами и кислородом. При этом капельки металла и шлака
падают в приемный ковш, в котором металл отстаивается от
шлака и поступает в МНРС:ЧугунIСаОгСтруя-► Металл
Шлакна МНРС2 .Агрегаты желобчатого типа (проточные печи), в которых
чугун перетекает по желобу через ряд емкостей, в каждой из
которых осуществяется одна из реакций окисления примесей.3. Агрегаты последовательного окисления, в которых про¬
цесс переделки чугуна в сталь разделен на последовательные
операции, осуществляемые в системе отдельных аппаратов. В
каждом из этих аппаратов последовательно протекают реакции
окисления серы, кремния и марганца и перехода оксидов в
шлак, реакции удаления фосфора и обезуглероживания метал¬
ла, процессы вакуумирования и легирования образующейся ста¬
ли, как это представлено на рис. 5.8.Рис. 5.8. Схема САНД последовательного окисления:1 — печь для плавки шихты, 2 — миксеры, 3 — дозатор, 4 — аппарат для
удаления серы, 5 — трехсекционныи аппарат для удаления марганца,
кремния и фосфора, 6 — аппарат обезуглероживания, 7 — вакуум-ап-
парат, б — аппарат для легирования, 9 — разливочный ковшВ настоящее время все конструкции САНД отрабатываются
в полупромышленных масштабах и пока не конкурентноспо¬
собны с кислородно-конвертерным и электросталеплавильным
методами производства стали.102
5.9. Прямое получение железа«Я полагаю, что придет со временем
пора искать способ прямого получения
железа и стали, минуя чугун».Д.И. МенделеевПроцессами прямого получения железа (внедоменное произ¬
водство стали, безкоксовая металлургия) называются способы
получения губчатого железа, металлизированного сырья, ли¬
того железа или стали непосредственно из железорудного сы¬
рья, минуя доменный процесс. Причинами, вызвавшими появ¬
ление этого нового направления в черной металлургии, явля¬
ются:— рост дефицита коксующихся углей;— потребность в железных порошках вследствие интенсив¬
ного внедрения методов порошковой металлургии;— развитие производства высококачественных сталей ис¬
пользующего металлизированное сырье (губчатое железо, кри¬
цы, окатыши). Существующие методы прямого получения железа подраз¬
деляются:1. По физическому состоянию получаемого продукта, и со¬
ответственно, по температуре процесса на:— получение губчатого железа и металлизированных окаты¬
шей при температурах ниже температуры плавления пустой
породы (твердый продукт);— получение крицы, то есть слипшейся массы губчатого
железа при температуре плавления пустой породы с образова¬
нием шлака (тестообразный продукт);'— получение жидкой стали при температурах выше темпе¬
ратуры плавления железа (жидкий продукт).2. По природе используемого восстановителя на:— использование твердых восстановителей (коксик, низко¬
сортное твердое топливо);— использование газообразных восстановителей (оксид уг¬
лерода (П), водород, смесь этих газов).3. По состоянию слоя обрабатываемого сырья и, соответствен¬
но, по конструкции применимого оборудования на:— восстановление в плотном неподвижном слое (ретортах и
тиглях);— восстановление в плотном подвижном слое (в шахтных
печах);103
— восстановление во взвешенном слое (в колоннах, цикло¬
нах);— восстановление в кипящем слое (в колоннах, трубчатых
печах).Из этих методов наибольшее распространение получили про¬
цессы получения губчатого железа и металлизированных ока¬
тышей из высококачественных руд и рудных концентратов вос¬
становлением в шахтных печах газообразными восстановите*
лями.В зависимости от степени металлизации (Хм) и содержания
железа, получаемые металлизированные продукты подразделя¬
ются на:— шихту для выплавки стали с содержанием железа 0,9—0,93 мае. дол. и Хм = 0,85—0,90 и— губчатое железо для производства железного порошка с
содержанием железа 0,98—0,99 мае. долей и Хм = 0,98—0,995.Одним из подобных процессов можно назвать разработанный
в США процесс «Мидреке» («Мидлен Росс»), по которому рабо¬
тают промышленные установки в США, Канаде, ФРГ, странах
Южной Америки. В РФ этот процесс используется на Старо-
Оскольском электрометаллургическом комбинате для получе¬
ния металлизированных окатышей, переплавляемых в сталь.
Комбинат работает на железистых кварцитах КМА, которые
непосредственно в карьере перерабатываются в концентрат,
который после добавки к нему оксида кальция транспортиру¬
ется в виде пульпы по трубопроводу на расстояние 26 км до ком¬
бината. На рис. 5.9 представлена принципиальная и на рис. 5.10
технологическая схемы этого производства.Сырые окатыши из бункера 3 поступают в верхнюю часть
двухзонной шахтной печи 2, в среднюю часть которой из кон¬
вертора метана 1 подается конвертированный газ, полученный№► ПульпаТранспортировкаО ком кование ►Металлизированные^ nVQTUIIIUокатыши► Электроплавка ■ ц СтальРис. 5.9. Принципиальная схема процесса «Мидрекс»104
IМеталлизированный продуктРис. 5.10. Технологическая схема процесса «Мидрекс»:1 — конвертер метана, 2 — шахтная печь, 3 — бункер окатышей, 4—
бункер металлизированных окатышей, 5— охлаждающие газ скрубберы,6 — грохот, 7 — электрическая печьпо реакции неполного окисления СН4 + СО2 *= 2СО + 2Н2 и со¬
держащий 65% водорода и 35% оксида углерода (II). В печи
протекают реакцйи восстановления оксидов железа в той же
последовательности, как и в доменном процессе:
Fe^—^304—►FeoA.Fe. Отходящий газ поступает в скруббер 5
для охлаждения и очистки. Часть (1/3) очищенного газа сме¬
шивается с метаном и подается в конвертер для конвертирова¬
ния, а остальной газ направляется в топку конвертера для его
обогрева. Восстановленные (металлизированные) окатыши
опускаются из восстановительной зоны печи А в конусную зону
охлаждения Б и при температуре 40—50°С выгружаются на гро¬
хот 6. После отсева мелочи окатыши с грохота поступают в бун¬
кер 4 емкостью 5000 т, в котором хранятся в атмосфере инерт¬
ного газа во избежание вторичного окисления. Из бункера 4
металлизированные окатыши направляются в электрическую
печь для выплавки стали 7. Охлаждение окатышей в зоне печи
Б происходит газом, который отсасывается из средней части
печи и охлаждается в скруббере 5.Основной аппарат в процессе «Мидрекс* — это шахтная печь
объемом 300 м3. Рабочее пространство печи разделено на две105
зоны: зону восстановления А при 900°С и зону охлаждения Б,
где восстановленный продукт охлаждается до 40—50°С. Время
пребывания окатышей в печи составляет 8—12 часов, из них
4—6 часов в восстановительной зоне. Производительность печи
составляет 400000 т металлизированных окатышей в год. На
производство 1 т окатышей расходуется 400 м3 природного газа
(метана) и 150 кВт-ч электроэнергии. Процесс полностью авто¬
матизирован. На комбинате установлены и действуют три печи
первой очереди общей мощностью свыше 700000 т в год.Контрольные вопросы1. Перечислите основные способы производства стали.2. В чем заключается сущность процесса переделки чугуна в сталь?3. Укажите преимущества кислородно-конвертерного метода выплав¬
ки стали по сравнению с другими конвертерными методами и мар¬
теновским методом.4. Какие реакции и где протекают в окислительном и восстановитель¬
ном периодах при выплавке стали в конвертере?5. Чем объясняется последовательность окисления примесей при
конвертерной плавке?6. Какие сорта сталей преимущественно выплавляются в электропе¬
чах?7. Перечислите разновидности промышленных электроплавильных
печей и укажите их особенности.8. Чем мартеновский метод выплавки стали принципиально отлича¬
ется от конвертерного?9. Что такое двухванные мартеновские печи?10. Какие виды переплавных процессов испльзуются в производстве
стали?11. В чем преимущества непрерывной разливки стали перед разлив¬
кой ее в изложницы?12. Какие процессы называются процессами прямого восстановления
железа?
Часть IIПЕРЕРАБОТКА ХИМИЧЕСКОГО ТОПЛИВА
ГлаваМХИМИЧЕСКОЕ ТОПЛИВО6. У. Определение, классификация и состав топливТопливом называется одно- или многокомпонентная систе¬
ма, представляющая в результате протекающих в ней процес¬
сов источник энергии. Поэтому тоилива называют также энер¬
гоносителями. По природе процессов, протекающих в топливах
при их использовании, они делятся на ядерные и химические.В ядерных топливах энергия выделяется в результате деле¬
ния ядер тяжелых элементов, процесса воспроизводства ядер-
ного топлива и управляемого термоядерного синтеза между яд¬
рами легких элементов.В химических топливах энергия выделяется в результате
протекающих в них экзотермических реакций окисления—вос-
становления.Химические топлива подразделяются:1. По происхождению на: природные (ископаемые угли,
нефть, природный и попутный газы, торф, горючие сланцы),
искусственные или синтетические (кокс, моторное топливо,
технологические газы).2. По агрегатному состоянию на: твердые, жидкие, газооб¬
разные.Первые два вида топлив называются также конденсирован¬
ными.3. По составу: на унитарные (однокомпонентные), в которых
горючее и окислитель находятся в одной фазе (например, бал-
листитный порох), и многокомпонентные, в которых горючее
и окислитель составляют различные фазы. Многокомпонентные
топлива, в свою очередь, делятся на совмещенные, представля¬
ющие единую систему из горючего и окислителя (например,
твердое ракетное топлива из твердых органического горючего и
минерального окислителя), и раздельные, в которых горючее и
окислитель являются отдельными веществами, взаимодейству¬
ющими в момент их использования как источник энергии.Энергетика химической промышленности и других отраслей
народного хозяйства базируется, в основном, на химическом107
топливе раздельного типа. Так как в этом случае окислителем
является только кислород воздуха, понятие «топливо»относят ис¬
ключительно к его горючему компоненту. Таким образом, под
химическим топливом понимают природные или искусственные
горючие органические вещества, используемые в качестве источ¬
ника энергии или сырья для химической промышленности. В за¬
висимости от назначения химическое топливо делится на:— энергетическое топливо, используемое для выработки теп¬
ловой и электрической энергии на ТЭЦ и в котельных, и— технологическое топливо, используемое непосредственно
в различных агрегатах химического производства, в том числе,
в промышленных печах и для проведения технологических
процессов (коксование, выплавка металлов, обжиг, сушка, рек¬
тификация и др.).Для сравнения различных видов топлива и суммарного уче¬
та его запасов и расхода используется такая единица как услов¬
ное топливо (УТ). УТ называется топливо, имеющее низшую
теплоту сгорания 29260 кДж/кг. 1 кВт энергии эквивалентен0,123 г УТ.Общие запасы химического топлива на Планете оценивают¬
ся величиной 12800 млрд. т условного топлива. По запасам энер¬
гии химическое топливо расположено в конце ряда потенциаль¬
ных энергоносителей:ядерная энергия — энергия солнца — энергия приливов —
каменный уголь — горючие сланцы — торф — нефть — энергия
ветра — природный газ — энергия рек.В табл. 6.1 приведены мировые запасы основных видов топ¬
лива.Таблица 6.1. Запасы химического ископаемого топливаВид топливаРазведанные запасыкВт-чУТ,тИскопаемые угли1139,4-101511,2-1012Нефть и конденсат4,16-10157,4-Ю11Природный газ1,9-10156,3-Ю11Доля химического топлива в мировом энергетическом балан¬
се составляет около 90%. При этом соотношение между твер¬
дым, жидким и газообразным видами топлива меняется. В на¬
стоящее время около 70% мирового энергопотребления покры¬
вается за счет использования нефти и газа. Это объясняется их
преимуществами перед ископаемыми углями (меньшая себе¬108
стоимость добычи, транспортабельность, простота использова¬
ния), а также все возрастающим применением их в качестве
сырья для химической промышленности, масштабы которого
сопоставимы с их потреблением в качестве источников энергии.
Однако ограниченность запасов нефти и газа вновь выдвигает в
настоящее время в качестве основного вида химического топ¬
лива ископаемые угли. Так например, в современном топлив¬
ном балансе США доля угля составляет 80%, газа 14% и нефти
всего 6%.В табл. 6.2 представлена добыча основных видов ископаемо¬
го топлива в РФ и время исчерпания его ресурсов.Таблица 6.2. Добыча топлива в РФВид топливаПроизводство по годамВремя ис¬
черпания, лет1991199219931994Каменный уголь, млн. т3543373002502000Нефть, млн. т45639435732736Природный газ, млрд. м3643647638620—Эффективность использования химического топлива в каче¬
стве источника энергии зависит от условий сжигания и состава
топлива. Все природные химические топлива состоят из горю¬
чей массы, минеральных веществ и воды (так называемое рабо¬
чее топливо). После удаления влаги (W) получают обезвожен¬
ное топливо (сухое топливо). Горючая часть топлива включает
вещества, содержащие углерод и водород (органическая масса)
и окисляемые соединения серы (органические и неорганичес¬
кие сульфиды). Минеральные вещества топлива представляют
различные соли металлов (карбонаты, силикаты, сульфаты и
др.), образующие при сжигании топлива золу (А). Для различ¬
ных состояний топлива приняты буквенные обозначения, пред¬
ставленные на схеме (рис. 6.1). Для соответствующих состоя-Рабочее топливо «р»Сухое топливо «с» Влага (W)Горючая масса «г » Зола (А)
IОрганическая Горючаямасса «о» сераРис. 6.1. Состав химического топлива109
ний топлива эти обозначения указываются в виде показателей
при его энергетических характеристиках и составе, например:
QP, цг и т. д.Энергетические характеристики и другие свойства топлив
зависят от элементного состава их горючей массы, содержа¬
ния золы и влаги. С учетом этого состав топлива в различном
состоянии может быть представлен в следующем виде:
рабочее топливо (С, Н, О, N, S + А + W),
сухое топливо (С, Н, О, N, S 4- А),
горючая масса (С, Н, О, N, S),
органическая масса (С, Н, О, N).Основным элементом всех химических топлив является ак¬
тивный углерод, то есть углерод в степени окисления равной
или меньшей +3. Он входит в состав биогенных ископаемых
(90%) и живых организмов (10%).Для использования в качестве химического сырья твердого
топлива большое значение имеет также содержание в нем лету¬
чих веществ, то есть продуктов, которые удаляются из топлива
при нагревании его до определенной температуры без доступа
воздуха.В табл. 6.3 приведены состав и теплота сгорания различных
видов химического топлива.Таблица 6.3. Средний состав химических топливТопливоСостав
органической
части,%Содержание, %Высшаятеплотасгора¬ния,кДж/кгСНN+OSWАЛетучиеДревесина50644 30^400,4до 7019000Торф596350,4254,5до 7024000Бурый уголь755252—3до 504,045—5526000Каменный уголь825132—63—56,08—5034000Антрацит95231—21—1,56,0до 834000Нефть87130,30,1—5—оГо—40000Природный газ7525——— —40Q006.2. Энергетические характеристики топливК энергетическим характеристикам, от которых зависит эф¬
фективность химического топлива как энергоносителя относят¬
ся теплота сгорания, жаропроизводительность и энергоемкость
(для моторных топлив).110
Теплота сгорания (устаревший термин — теплотворная спо¬
собность) является основной технологической характеристикой
топлив, которая используется для оценки как конденсирован¬
ных так и газообразного топлив.1. Теплотой сгорания называется количество тепла, выде¬
ляемое при полном сгорании единицы массы или. объема топ¬
лива в атмосфере кислорода и охлаждении продуктов сгорания
до начальной температуры процесса.Под термином полное сгорание понимают окисление углеро¬
да до оксида углерода (IV), водорода — до воды и азота — до
молекулярного азота. В зависимости от температуры, до кото¬
рой охлаждаются продукты горения различают высшую и низ¬
шую теплоты сгорания топлива.Высшей теплотой сгорания QB называется количество теп¬
ла, выделяемое при полном сгорании 1 кг или 1 м3 топлива и
охлаждении продуктов сгорания до 298 К. Следовательно, выс¬
шая теплота сгорания учитывает теплоту конденсации паров
образовавшейся воды и может быть определена эксперимен¬
тально калориметрическим методом.Низшей теплотой сгорания QH называется количество теп¬
ла, выделяемое при полном сгорании 1 кг или 1 м3 топлива в
условиях сгорания, то есть без учета теплоты конденсации
воды. Очевидно, что высшая теплота сгорания больше низшей
теплоты сгорания.В отличие от теплоты горения топлива, которая зависит от
условий его сжигания, теплота сгорания является константой,
зависящей от природы и элементного состава топлива.Для конденсированных топлив, низшая теплота сгорания
рабочего топлива может быть с достаточной точностью рассчи¬
тана исходя из его элементного состава по формуле Д.И. Мен¬
делеева:пкДж/кг, (6.1)1где: цр — массовая доля элемента и влаги в рабочем топливе,
а — постоянный коэффициент для данного элемента и
влаги.Значения коэффициента «а» для различных элементов
(кДж):ас = 33940; ан = 103075; aw = -2515; a(0-s) = -10895.Из этих значений следует, что максимальный тепловой вклад
вносит водород. Поэтому, от отношения водород/углерод в топ-111
ливе существенно зависит теплота его сгорания. Положитель¬
ный тепловой вклад серы объясняется выделением тепла при
окислении «горючей» серы. Наоборот, отрицательный вклад
влаги и кислорода, входящего в состав образующейся воды,
объясняется затратой тепла на ее испарение.Высшая и низшая теплоты сгорания связаны зависимостью:Qhp=Qbp-2515(hP+9hP) (6.2)где: цР и 9^ — содержание в рабочем топливе влаги и водоро¬
да, соответственно.В формуле 6.2 вычитаемая из Qp сумма представляет общеесодержание конденсирующейся воды, так как при сгорании 1
массовой части водорода образуется 9 мае. частей воды.В большинстве случаев элементный состав приводится не для
рабочего топлива, а для его горючей массы. Для пересчета со¬
держания элемента в горючей части топлива на рабочее топли¬
ва используется формула:Цэр = Цэг [1 — (Hwp + Цар) (6.3)где: цэр и цэг — содержание элемента в рабочем топливе и вгорючей части его,Hwp и |хар — содержание влаги и золы в рабочем топли¬
ве.Теплота сгорания газообразного топлива рассчитывается по
формуле:пQp=S^-con кДж/нм3 (6.4)1где: qp — теплота сгорания компонента топлива, кДж/нм3,
со — объемная доля компонента в топливе.По формуле 6.4 может быть рассчитана как высшая, так и
низшая теплоты сгорания топлива в зависимости от того, как
выражены теплоты сгорания его компонентов.2. Жаропроизводительностью топлива называется макси¬
мальная температура горения (Тмакс), развиваемая при полном
сгорании топлива без избытка воздуха в условиях, когда вся
выделяющаяся теплота расходуется на нагрев продуктов сго¬
рания. При подсчете жаропроизводительности начальная тем¬
пература топлива и воздуха принимается равной нулю. Жаро-
производительность топлива пропорциональна его теплоте сго¬112
рания и обратно пропорциональна расходу тепла на нагрев про¬
дуктов сгорания ДО Тмакс.Жаропроизводительность положена в основу энергетической
классификации химических топлив. В соответствии с ней топ¬
лива делятся на:— топлива высокой жаропроизводительности, для которых
акс.^ЗОСГК (природный, нефтяные и некоторые технологичес¬
кие газы, каменный уголь, кокс, антрацит, древесный уголь) и— топлива пониженной жаропроизводительности, для кото¬
рых Тмакс.<2300°К (дрова, торф, бурый уголь, горючие сланцы,
доменный газ).3. Энергоемкостью топлива называется количество потен¬
циальной энергии, заключающееся в единице объема жидкого
моторного топлива, кДж/литр. Характеристика имеет ограни¬
ченное применение и используется для оценки исключительно
моторных топлив.Контрольные вопросы1. Какие системы называются топливами?2. Приведите классификацию химических топлив по происхожде¬
нию, агрегатному состоянию, составу.3. Какие топлива называются унитарными?4. От чего зависит эффективность использования топлива как источ¬
ника энергии?5. Что такое рабочее топливо? условное топливо?6. Что такое высшая и низшая теплоты сгорания топлива?
Глава VIIПЕРЕРАБОТКА ЖИДКОГО ТОПЛИВАК жидким химическим топливам относятся нефть и продук¬
ты ее переработки (нефтепродукты), а также продукты гидри¬
рования твердого топлива. В настоящее время практическое
значение имеют только нефтепродукты, для производства ко¬
торых сырьем является нефть.7.1. Нефть, ее происхождение и составНефтью называется жидкое ископаемое топливо, распрост¬
раненное в осадочной оболочке литосферы Земли. Свое назва¬
ние нефть получила от персидского слова «нафта» — вытекаю¬
щая, просачивающаяся.В настоящее время общепринята теория органического (био¬
генного) происхождения нефти, согласно которой она образо¬
валась в результате воздействия бактериального и геологичес¬
ких факторов на останки низших животных и растительных
организмов, обитавших в толще воды (планктон) и на дне водо¬
емов (бентос). В верхних слоях осадочных пород этот захоро¬
ненный органический материал подвергался воздействию кис¬
лорода и бактерий и разлагался с образованием газов (оксид
углерода, азот, аммиак, метан и др.) и растворимых в воде жид¬
ких продуктов.В дальнейшем, по мере погружения на глубину 1,5—3 км в
толщу осадочных пород, органические вещества нерастворимо¬
го остатка разложения подвергались в течение миллионов лет
уже в восстановительной атмосфере действию высоких (120—
200° С) температур и давлений (10—30 МПа) и каталитическо¬
му воздействию окружающих пород (алюмосиликаты глин). На
этой стадии в результате термических и термохимических про¬
цессов липиды органического вещества остатка (жиры, масла,
воска) превращались в смесь углеводородов, составляющих
нефть.Большинство нефтей представляют маслянистые жидкости
от темно-коричневого до темно-бурого цвета, который зависит
от содержания в них окрашенных смолистых веществ. Плот¬
ность нефтей составляет 0,82—0,90 т/м3, температура затвер¬
девания лежит в пределах от -20°С до +20°С. Вязкость нефтей
значительно выше вязкости воды. Элементный состав нефтей
колеблется в очень незначительных пределах: углерод 84—114
87%, водород 12—14%, сера ОД—5%, кислород и азот (в сум¬
ме) до 1,0%.В нефти различают углеводородную часть, неуглеводород¬
ную часть и минеральные примеси. Углеводородная часть не¬
фти представляет собой раствор газообразных и твердых угле¬
водородов в смеси жидких углеводородов различной природы
и сложности. В низкомолекулярной части нефти, перегоняю¬
щейся до 350 С, содержатся вещества с молекулярной массой
не более 250—300, а именно: алканы, моно-, би- и трицикли-
ческие нафтены, моно- и бициклические ароматические угле¬
водороды, углеводороды смешанного строения. В состав вы¬
сокомолекулярной части нефти, перегоняющейся выше
350С, входят вещества с молекулярной массой от 300 до
1000 — высокомолекулярные алканы, моно- и полицикли-
ческие нафтены с боковыми цепями, ароматические угле¬
водороды с боковыми цепями, конденсированные много¬
ядерные соединения и полициклические углеводороды сме¬
шанного строения.В зависимости от того, углеводороды какого класса преобла¬
дают в составе нефти, они подразделяются на парафиновые,
парафино-нафтеновые, нафтеновые, парафино-нафтено-арома-
тические, нафтено-ароматические, ароматические. Наиболее
распространены нефти так называемого смешанного основания,
в которых нельзя выделить определенный класс углеводородов.
В соответствии с технологической классификацией нефти под¬
разделяются на группы по выходу фракций, выкипающих до
350° С, по потенциальному содержанию масел, по содержанию
парафина и др.В неуглеводородную часть нефти входят разнообразные кис¬
лородные (фенолы, нафтеновые кислоты, гетероциклы), азоти¬
стые (производные пиридина и хинолина, амины) и сернистые
(тиофен, тиоспирты и тиоэфиры) соединения. По содержанию
серы нефти делятся на:— малосернистые (с содержанием до 0,5%),— сернистые (с содержанием от 0,5 до 2,0%) и— высокосернистые (с содержанием выше 2,0%).Основная масса всех этих соединений концентрируется ввысокомолекулярной части нефти.-"^Минеральные примеси в нефти составляют различные соли,
перешедшие в нее из пластовых вод, механические примеси
песка и глины и эмульгированная вода. В нефтях в весьма ма¬
лых количествах содержатся такие элементы как ванадий, ни¬
кель, железо, титан, германий и др.115
В природе нефть находится в виде нефтяных залежей, так
зываемых ловушек, образовавшихся в результате движения
фти и газа по пористым пластам породы под воздействием
авитационного и тектонического факторов. При достаточно
пылом объеме этих залежей они называются нефтяными ме-
юрождениями. В большинстве случаев нефтяные залежи рас-
ложены на глубине от 900 до 2300 м.Мировые разведанные запасы нефти оцениваются в 90—» млрд. т, прогнозируемые ресурсы составляют 250—0 млрд. т. Распределение нефтяных месторождений по Пла¬
те неравномерно. Наиболее крупные из них сосредоточены в
[удовской Аравии, Кувейте, Ираке, Венесуэле, Алжире, Ира-
, Ливии и США. В РФ Наиболее крупными и известными ме-
орождениями являются: Самотлорское, Усть-Балыкское,
фгутское и Северо-Советское в Западной Сибири, Ромашкин-
ое, Арланское, Туймазинское в Волго-Уральском нефтегазо¬
юном районе, Усинское в Республике Коми. На Западно-Си-
срский регион приходится 71% добычи нефти, на Волго-
эальский регион — 23%. На территории РФ действуют пять
новных магистральных нефтепроводов: Волго-Уральский, За-
1Дно-Сибирский, Северо-Кавказский, Тимано-Печерский,
1льневосточный, общей протяженностью 70 тыс, км с пропус-
юй способностью до 80 млн. тонн нефти в год. Развитая сеть
‘фтепроводов обеспечивает работу многочисленных нефтепе-
>гонных заводов, расположенных по всей территории РФ:
нгарск, Волгоград, Грозный, Кириши, Комсомольск-на-Аму-
S Краснодар, Москва, Нижний Новгород, Омск, Пермь, Ря-
LHb, Саратов, Сызрань, Туапсе, Ухта, Хабаровск, Ярославль.В зависимости от условий залегания и давления в нефтенос-
ш пласте методы извлечения нефти из пробуренных скважин
длятся на фонтанный, компрессорный и глубинно-насосный,
ри высоком давлении нефть поступает из недр земли под соб-
’венным давлением и через запорную аппаратуру направля¬
йся в сборные емкости (фонтанный метод). При малом давле-
ии нефть извлекают методом газлифта путем накачивания в
ольцевое пространство между трубами природного газа под
явлением до 5 МПа. В скважине газ смешивается с нефтью,
меныпает ее вязкость и «транспортирует» ее на поверхность
сомпрессионный метод). При глубоком залегании нефти и низ¬
ом давлении в пластах нефть извлекают с помощью поршне-
эго насоса, опущенного в скважину, и приводимого в движе-
ии балансирным станком-качалкой, который обеспечивает
эзвратно-поступательное движение плунжера насоса.16
При современном уровне техники и технологии добычи и*
фтяных пластов извлекается лишь около 50% содержащей-
[ в них нефти. Увеличение нефтеотдачи пластов до 80—90%
эжет быть достигнуто тепловым воздействием на пласты (за-
1чивание в скважину горячей воды, прогрев пласта сжигани-
I нефти), введением в скважину ПАВ, гидравлическим раз¬
овом пласта и другими интенсифицирующими извлечение
>фти из недр методами.7.2. НефтепродуктыВ настоящее время вся извлекаемая из недр нефть подверга-
ся переработке с целью получения из нее разнообразных неф-
епродуктову которые используют как в качестве целевых про¬
ектов, так и в качестве сырья для дальнейшей переработки.
ге нефтепродукты можно разделить на следующие группы.1. Моторные топлива, в том числе:— карбюраторное для поршневых двигателей с зажигани-I от электрической искры (автомобильные и тракторные бен-
ены);— дизельное для поршневых дизельных двигателей с воспла-
энением от сжатия (дизельное топливо).2. Котельные топлива для топок паровых котлов, генера-
фных установок, металлургических печей (мазут, гудрон).3. Реактивное топливо для авиационных реактивных и га-
►турбинных двигателей (авиокеросины).4. Смазочные масла для смазки трущихся деталей машин с
глъю уменьшения трения и отвода тепла (моторное, индуст-
1альное, турбинное, компрессионное, цилиндровое масла).5. Консистентные смазки для уменьшения трения между
сталями, защиты от коррозии, герметизации соединений, со-
‘ржащие загустители (мыла, церезин, силикаты).6. Продукты, используемые для нефтехимического синте-
i (мазут, широкая фракция и др.).Нефтепродукты, используемые в качестве топлив и смазоч-
лх материалов должны удовлетворять определенным требо-
1ниям. Так, основными эксплуатационными характеристика-
^1^ефтяных смазочных масел являются вязкость, вязкостно-
>мпературные свойства, маслянистость, подвижность при низ-IX температурах, химическая стабильность, защитные свой-
•ва. К аналогичным характеристикам топлив для двигателей
1утреннего сгорания относятся детонационная стойкость,
ракционный состав, химическая стабильность, антикоррози-117
онные свойства, а для дизельных топлив также вязкость, тем¬
пература застывания и коксуемость. Важнейшей характерис¬
тикой моторных топлив является их устойчивость к детона¬
ции — детонационная стойкость.Детонацией называется особый ненормальный режим сго¬
рания топлива в двигателе, при котором часть топливной сме¬
си, находящаяся перед фронтом пламени, воспламеняется мгно¬
венно, в результате чего скорость распространения пламени до¬
стигает 1500—2500 м/с. Это приводит к резкому скачкообраз¬
ному возрастанию давления в цилиндре и возникновению удар¬
ной детонационной волны. На режиме детонации мощность дви¬
гателя падает, расход топлива увеличивается и ускоряется из¬
нос деталей.Мерой детонационной стойкости для карбюраторных двига¬телей является октановое, а для дизельных двигателей цетано-
вое числа. В основе их определения лежит принцип сравнения
испытуемого топлива со смесями эталонных топлив.Октановым числом (04) называется условная единица из¬
мерения детонационной стойкости, численно равная содержа¬
нию в объемных процентах изооктана (2,2,4-триметилпента-
на) в смеси с н-гептаном, которая детонирует при той же сте¬
пени сжатия в цилиндре карбюраторного двигателя, что и топ¬
ливо.При этом октановое число изооктана СНз-С(СНз)2-СН2~
СН(СНз)-СНз принимается равным 100, а «-гептана СН3-
(СН2)б“СНз равным 0.Октановое число зависит от класса, молекулярной массы и
строения углеводорода, как это видно из ниже приведенных
данных.Октановое число повышается с увеличением молярной мае-и при переходе от алканов к алкенам, нафтенам и ароматичес¬
ким углеводородам с одинаковым числом углеродных атомов:СНз-С(СНз)2-СН2-СН2-СНз
СН3-С(СНз)2-СН(СНз)-СНз89104СбН14
26 63C6Hi2 цикло-С6Н12 СбНб
77 106118
Цетановым числом (ЦТ) называется условная единица из¬
мерения детонационной стойкости, численно равная содержа¬
нию в объемных процентах цетана (гексадекана) в смеси с а-
метилнафталином, которая детонирует при той же степени сжа¬
тия в цилиндре дизеля, что и топливо.При этом цетановое число цетана С16Н34 принимается рав¬
ным 100, а а-метилнафталина а—С10Н7—СН3 равным нулю.7.3. Краткая история развития переработки нефтичНефть и природный газ известны человечеству с глуббкой древнос¬
ти. Описание источников нефти содержится в трудах Геродота (V век
до н. э.), Плутарха и Плиния Старшего (I век до н. э). Гиппократ (IV—
V века до н. э.) рекомендовал лекарства, содержащие в своем составе
нефть. Нефть применялась в качестве топлива, как средство освеще¬
ния, в военном деле («греческий огонь»).В средние века нефть добывали из специально вырытых колодцев.
Уже в XIII веке в районе Баку функционировали нефтяные источни¬
ки. В последующем вместо колодцев стали использовать скважины,
что позволило извлекать нефть из более глубоких слоев. Первые сква¬
жины бурили ударным способом с помощью металлического долота, а
нефть после окончания фонтанирования извлекали специальными
ведрами (желонками). Недостаток в так называемых светлых нефтях,
используемых для освещения, вызвал потребность в разработке мето¬
дов переработки нефтей, сначала для повышения выхода осветитель¬
ного масла (фотогена). В России подобный метод был освоен в XVI—
XVII веках, после того, как в царствование Б.Годунова (XVI век) в Мос¬
кву из Ухты была завезена ♦горячая вода густа» (нефть).Интерес к процессам перегонки нефти для получения различных
ценных продуктов появился в первой половине XIX века. В 1821—23
гг. в Моздоке братьями Дубиниными была построена первая кубовая
установка для перегонки нефти, на которой из нее выделялось до 40%
фотогена (керосина). Легкая часть — бензин при этом методе терялась,
а мазут использовали для смазки колес. В 1837 году в Баку инжене¬
ром Воскобойниковым был сооружен первый нефтеперегонный завод.
Подобное производство керосина из нефти в Англии было организова¬
но в 1848 году и в США в 1860 году.Бурное развитие нефтеперерабатывающей промышленности начи¬
нается с 60-х годов XIX века. В 1869 году в Баку существовало уже 23
нефтеперегонных завода, а к 1876 году число их возросло до 123. В
этот период основным целевым продуктом переработки являлся осве¬
тительный керосин, выход которого составлял около 25%. Бензино¬
вая фракция (всего около 0,5%) и мазут промышленного применения
не находили. С 1876 года после изобретения В.Г. Шуховым форсунки
для сжигания жидкого топлива, мазут стал широко использоваться в
топках паровых котлов. К этому же времени было налажено произ¬
водство из мазута смазочных масел.119
Значительный вклад в разработку химии и технологии нефтепе¬
реработки внесли А.А. Летний, В.Г. Шухов, Л. Г. Гурвич и другие
исследователи. А.А. Летним был открыт процесс пиролиза нефтяного
сырья и выделены из продуктов нефтепереработки ароматические уг¬
леводороды. Работы Л.Г. Гурвича легли в основу разработки процес¬
сов очистки нефтепродуктов. В 1890 году В.Г. Шухов и Гаврилов за¬
патентовали трубчатую нефтеперегонную установку непрерывного
действия, которая стала прообразом современных установок АТ и АВТ.
В этом методе предусматривалась и возможность проведения процес¬
са с расщеплением углеводородов нефти (крекинга).Одновременно с развитием методов переработки нефти осваивались
новые районы ее добычи в Грозном, Фергане, на реке Эмбе. В 1913
году добыча нефти в России достигла 9 млн. т, из которых 82% добы¬
валось в Баку.Коренной переворот в методах переработки нефти происходит пос¬
ле изобретения двигателя внутренного сгорания. В связи с этим, бен¬
зин, не находивший ранее промышленного применения, становится с
начала XX века одним из важнейших нефтепродуктов.В России предприятия по переработке нефти вначале были сосре¬
доточены на Кавказе: к 1917 году в Баку действовало 53 и в Грозном 6
заводов. С 1928 года в нефтеперерабатывающей промышленности
страны вводят методы термического крекинга и строят новые уста¬
новки в Баку, Грозном, Ярославле, Батуми и Туапсе — всего 18 мощ¬
ных АВТ и 23 установки крекинга. К 1937 году число установок воз¬
растает, соответственно, до 46 и 73. Повышается мощность АВТ, дос¬
тигающая 1 млн.т в год, возрастает глубина крекирования и выход
светлых продуктов. Одновременно расширяется география нефтепе¬
рерабатывающей промышленности: строят заводы в Уфе, Саратове,
Одессе Хабаровске, Москве. Интенсивно вводят в строй нефте-газо-
проводы, создавая единую систему снабжения.В послевоенный период вводят в строй новые нефтеперерабатыва¬
ющие заводы в Уфе, Самаре, Омске, осваивается производство бензи¬
нов с высоким октановым числом, в практику нефтепереработки вне¬
дряют методы газофракционирования, алкилирования, селективной
очистки масел и др. В 1950 году вступает в строй первая установка
каталитического крекинга, в 1958 году внедряется процесс каталити¬
ческого риформинга. Широкое применение получают методы гидро¬
очистки, карбамидной депарафинизации нефтей, что позволило пере¬
рабатывать высокосернистые нефти. Потребности цветной металлур¬
гии в электродном коксе вызвали развитие процесса коксования тя¬
желых остатков, в частности замену малопроизводительных кубовых
установок на установки непрерывного коксования.Начиная с 1965 года в стране развиваются мощности вторичных
процессов нефтепереработки, увеличивается производство моторных
топлив. Так в 1970 году доля высокооктановых бензинов составляла
уже 50,7% и продолжает возрастать. В последующие годы в нефтепе¬
рерабатывающей промышленности внедряются новые высокопроиз¬
водительные процессы, комбинированные технологические установ¬
ки (ЭЛОУ-АВТ), переработка нефти приближается к районам потреб¬
ления нефтепродуктов.120
Возрастающая потребность в моторных топливах с высоким окта¬
новым числом для двигателей со степенью сжатия 9—10, потребовала
значительного углубления переработки нефти с целью более эффек¬
тивного ее использования и модернизации действующих нефтепере¬
рабатывающих заводов. Это было достигнуто за счет интенсивного
внедрения в нефтепереработку новых термических и каталитических
процессов, позволивших в 1,5—1,8 раза увеличить выход светлых про¬
дуктов. В результате к 1989 году глубина переработки нефти, которая
оценивается количеством целевых нефтепродуктов, отбираемых из
нефти при ее переработке:тцел.прод.Глубина=нефтиГде. ЯХцел.прод. ^ТХнефти (^мазута ^таза ^потерь) ДОСТИГЛа 80 /о ,превысив этот показатель в США.Вследствие кризисных явлений в народном хозяйстве РФ в
целом, и в нефтеперерабатывающей промышленности в част¬
ности, в настоящее время глубина переработки снизилась до
64% и значительно уступает таковой в США (90%). Так, из од¬
ной тонны нефти производится бензина, керосина и дизельно¬
го топлива в США 700 кг, в РФ — 400 кг, а доля мазута составля¬
ет, соответственно, 80 и 400 кг.7.4. Общая схема переработки нефтиВ общем случае переработка нефти на нефтепродукты вклю¬
чает ее подготовку и процессы первичной и вторичной перера¬
ботки.Подготовка извлеченной из недр нефти ставит целью удале¬
ние из нее механических примесей, растворенных солей и воды
и стабилизацию по составу. Эти операции проводят как непос¬
редственно на нефтяных промыслах, так и на нефтеперераба¬
тывающих заводах.Первичная переработка нефти (первичные процессы) зак¬
лючается в разделении ее на отдельные фракции (дистилляты),
каждая из которых представляет смесь углеводородов. Первич¬
ная переработка является физическим процессом и не затраги¬
вает химической природы и строения содержащихся в нефти
соединений. Важнейшим из первичных процессов является
прямая гонка нефти.Вторичная нефтепереработка (вторичные процессы) пред¬
ставляет собой разнообразные процессы переработки нефтепро¬
дуктов, полученных методом прямой гонки. Эти процессы со-121
провождаются деструктивными превращениями содержащих¬
ся в нефтепродуктах углеводородов и изменением их природы,
то есть являются химическими процессами.Вторичные процессы нефтепереработки весьма многообраз¬
ны. Они подразделяются:а) По назначению на:— процессы, проводимые с целью повышения выхода легко-
кипящих фракций за счет высококипящих (крекинг);— процессы, проводимые с целью изменения углеводородно¬
го состава сырья (риформинг);— процессы синтеза индивидуальных углеводородов (алки-
лирование);— процессы удаления из нефтепродуктов примесей (гидро¬
очистка).б) По условиям протекания на:— термические процессы, протекающие под воздействием
высоких температур и давлений;— каталитические процессы, протекающие под воздействи¬
ем высоких температур в присутствии катализаторов.в) По состоянию перерабатываемого сырья на:— процессы в жидкой фазе;— процессы в паровой фазе.Важнейшими из вторичных процессов является термичес¬
кий и каталитический крекинг, риформинг, алкилирование,
коксование и гидроочистка нефтепродуктов. На рис. 7.1 пред¬
ставлена общая схема переработки нефти и нефтепродуктов.Рис. 7.1. Общая схема переработки нефти122
7.5. Подготовка нефти к переработкеИзвлеченная из скважин сырая нефть содержит попутные
газы (50—100м3/т), пластовую воду (200—300 кг/т) и раство¬
ренные в воде минеральные соли (10—15 кг/т), которые отри¬
цательно сказываются на транспортировке, хранении и после¬
дующей переработке ее. Поэтому, подготовка нефти к перера¬
ботке обязательно включает следующие операции:— удаление попутных (растворенных в нефти) газов или ста¬
билизация нефти;— обессоливание нефти;— обезвоживание (дегидратация) нефти.На крупных месторождениях нефти эти операции объедине¬
ны в единую систему, включающую сбор, транспортировку и
обработку нефти, газа и воды. На рис. 7.2 представлена подоб¬
ная система.Сырая нефть из скважин 1 под собственным давлением на¬
правляется к групповым замерным установкам (ГЗУ) 2, в кото¬
рых нефтяной газ отделяется от жидкости и замеряются коли¬
чества этих продуктов. Затем газ вновь смешивается с нефтью
и водой и полученная смесь подается по коллектору (длиной до
8 км) 3 в дожимную насосную станцию 4, где газ отделяется от
нефти. Газ поступает на газоперерабатывающий завод (ГПЗ) 5,
а частично дегазированная нефть направляется на установку
подготовки нефти (УПН) 6. На УПН проводятся операции окон¬
чательной дегазации, обессоливания и обезвоживания нефти.
Газ далее направляется на ГПЗ, а вода — на установку очистки7. Очищенная вода закачивается насосами 8 в нефтяной пласт
через нагнетательные скважины 9. Обессоленная и обезвожен¬
ная нефть из УПН поступает в герметизированные резервуары—►ы-,.111 ►6 —►10—►1113Рис. 7.2. Схема сбора нефти, газа и воды на нефтяных промыслах:I — скважины, 2 — групповая замерная установка, 3 — коллектор, 4 —
дожимная насосная станция, 5 — газоперерабатывающий завод, 6 —
установка подготовки нефти, 7 — установка очистки воды, 8 — насосы,9 — нагнетательные скважины, 10 — герметизированные резервуары,II — установка «Рубин», 12 — товарные резервуары, 13 — магистраль¬
ный нефтепровод123
10, из которых насосами перекачивается в установку «Рубин»11 для определения качества и количества нефти. При удовлет¬
ворительном результате нефть подается в товарные резервуары12 и из них в магистральный нефтепровод 13, транспортирую¬
щий нефть на нефтеперерабатывающие заводы. При неудовлет¬
ворительном качестве подготовки нефти она возвращается из
установки «Рубин» в У ПН.В настоящее время разрабатываются методы магистральной
транспортировки газонасыщенных нефтей, то есть доставки
потребителю нефти и газа по одному трубопроводу. Это позво¬
ляет уменьшить расход энергии на перекачку продукта за счет
снижения его вязкости и более полно утилизировать попутные
нефтяные газы.Стабилизация нефти. Сырая нефть содержит значительное
количество растворенных в ней легких углеводородов Ci—С4.
При транспортировке и хранении нефти они могут выделять¬
ся, вследствие чего состав нефти будет меняться. Чтобы избе¬
жать потери газа и вместе с ним легких бензиновых фракций и
предотвратить загрязнение атмосферы, эти продукты должны
быть извлечены из нефти до ее переработки. Подобный процесс
выделения легких углеводородов из нефти в виде попутного газа
называется стабилизацией нефти. В зависимости от условий
стабилизацию нефти осуществляют методом сепарации непос¬
редственно в районе ее добычи на замерных установках, дожим-
ных станциях и У ПН (рис. 7.2), или на газоперерабатывающих
заводах (рис. 7.3).В первом случае попутный газ отделяют от нефти многосту¬
пенчатой сепарацией в сепараторах-газоотделителях (траппах),
в которых последовательно снижаются давление и скорЪсть по¬
тока нефти. В результате происходит десорбция газов, совмест¬
но с которыми удаляются и затем конденсируются летучие жид¬
кие углеводороды, образуя «газовый конденсат». При сепара-
ционном методе стабилизации в нефти остается до 2% углево¬
дородов состава Ci—С4.Обессоливание и обезвоживание нефти. Удаление из нефти
солей и воды происходит на промысловых установках подготов¬
ки нефти и непосредственно на нефтеперерабатывающих заво¬
дах (НПЗ).В обоих случаях процессы обессоливания и обезвоживания
нефти связаны с необходимостью разрушения эмульсий, кото¬
рые образует с нефтью вода. При этом, на промыслах разруша¬
ются эмульсии естественного происхождения, образовавшиеся
в процессе добычи нефти, а на заводе — искусственные эмуль¬124
сии, полученные при многократной промывке нефти водой для
удаления из нее солей. После обработки содержание воды и хло¬
ридов металлов в нефти снижается на первой стадии до 0,5—
1,0% и 100—1800 мг/л соответственно, и на второй стадии до0,05—0,1% и 3—5 мг/л.Для разрушения нефтяных эмульсий используются механи¬
ческие (отстаивание), термические (нагревание), химические и
электрические методы. При химическом методе обезвоживания
нагретую нефтяную эмульсию обрабатывают деэмульгаторами.
В качестве последних используются различные неиногенные
ПАВ типа защитных коллоидов: оксиэтилированные жирные
кислоты, метил- и карбоксиметилцеллюлоза, лигносульфоно-
вые кислоты и др. Наиболее эффективное удаление солей и воды
достигается при электротермохимическом методе обессолива-
ния, в котором сочетаются термохимическое отстаивание и раз¬
рушение эмульсии в электрическом поле.Установки электротермохимического удаления солей и
воды иди электрообессоливающие установки (ЭЛОУ) исполь¬
зуются как на промыслах, так и на нефтеперегонных заводах.
В этом методе разрушение нефтяной эмульсии происходит в ап¬
паратах — электродегидрататорах под воздействием перемен¬
ного тока напряжением 30—45 кВ, что вызывает передвижение
и слипание капель воды, содержащих соли, и ее отделение от
нефти. На рис. 7.3 представлена принципиальная схема ЭЛОУ.Нефть из сырьевого резервуара 1 с добавками деэмульгатора
и слабого щелочного или содового раствора проходит через теп¬
лообменник 2, подогревается в подогревателе 3 и поступает вРис. 7.3. Принципиальная схема ЭЛОУ:I — резервуар нефти, 2 — теплообменник, 3 — подогреватель, 4 —
смеситель, 5 — электродегидрататор I ступени, 6 — электродегидрататорII ступени, 7 — холодильник, 8— сборник обессоленной нефти, 9 —
нефтеотделитель125
смеситель 4, в котором к нефти добавляется вода. Образовав¬
шаяся эмульсия последовательно проходит электродегидрата-
торы 5 и б, в которых от нефти отделяется основная масса воды
и растворенных в ней солей, вследствие чего содержание их сни¬
жается в 8—10 раз. Обессоленная нефть проходит теплообмен¬
ник 2 и после охлаждения в холодильнике 7 поступает в сбор¬
нике 8. Отделившаяся в электродегидрататорах вода отстаива¬
ется в нефтеотделителе 9 и направляется на очистку, а отделив¬
шаяся нефть присоединяется к нефти, подаваемой в ЭЛОУ.Обессоливание и обезвоживание нефти увеличивает сроки
межремонтной работы установок гонки нефти и снижает рас¬
ход тепла, а также уменьшает расход реагентов и катализато¬
ров в процессах вторичной переработки нефтепродуктов.7-6. Первичная перегонка нефтиПервичная перегонка нефти (прямая гонка) — процесс пере¬
работки нефти, основанный на разделении смеси составляющих
ее углеводородов методом фракционной разгонки (ректифика¬
ции) на отдельные дистилляты (фракции) с определенными
интервалами температур кипения. Прямой гонке подвергается
вся добываемая нефть. В соответствии с назначением получае¬
мых дистиллятов различают три варианта прямой гонки:— топливный процесс (получение различных видов топлив);— топливно-масляный процесс (получение топлив и масел);— нефтехимический процесс (получение сырья для химичес¬
кого производства).Процесс прямой гонки проводится в установках трубчатого
типа (название — по названию трубчатых печей), которые вклю-
чайт трубчатые печи различного типа, ректификационные и
отпарные колонны, теплообменники и холодильники. В зави¬
симости от глубины переработки нефти установки прямой гон¬
ки делятся на:— одноступенчатые, работающие при атмосферном давле¬
нии (АТ) и— двухступенчатые (атмосферно-вакуумные АВТ), в кото¬
рых одна ступень работает при атмосферном давлении, а дру¬
гая при остаточном давлении 5—8 кПа.Продуктами прямой гонки на установках АТ являются мо¬
торные топлива (бензин, авиационный керосин), дизельное топ¬
ливо и значительное количество остатка — мазута. На установ¬
ках АВТ на второй ступени подвергается разгонке мазут с обра¬
зованием смазочных масел и остатка — гудрона, перерабатыва¬126
емого в битум, пек, нефтяной кокс. Этим на установках АВТ
достигается большая глубина переработки нефти, нежели на
установках АТ. На рис. 7.4 представлена технологическая схе¬
ма установки АВТ, работающей по топливно-масляному вари¬
анту.Нефть из ЭЛОУ последовательно проходит через теплообмен¬
ники 4, нагреваясь за счет теплоты дистиллятов атмосферной и
вакуумной перегонки, и подается насосом под давлением 1,5—2,0 -105 Па в трубчатую печь i, где нагревается до 350°С. Из печи
парожидкостная смесь поступает в ректификационную колонну
I ступени 3, в которой давление снижается до 0,1 МПа и происхо¬
дит испарение летучих фракций нефти и отделение их паров от
мазута. По высоте колонны в точно определенных интервалах
температур отбираются дистилляты, которые поступают в секции
отпарной колонны 6 для дополнительного отделения летучих уг¬
леводородов, которые вместе с водяным паром возвращаются в
колонну 3. Отобранные дистилляты проходят через теплообмен-► VIIIРис. 7.4. Технологическая схема установки АВТ:1 — трубчатая печь подогрева нефти, 2 — сепаратор газа, 3 — ректификаци¬
онная колонна атмосферного давления, 4 — теплообменники-конденсаторы,
5 — холодильники, 6,7 — отпарные колонны, 8 — трубчатая печь подогрева
мазута, 9— вакуумная ректификационная колонна. I — бензин, II — лигроин,III — керосин, IV — дизельное топливо, V — газойль, VI — мазут, VII — пар,
VIII — веретенное масло, IX — машинное масло, X — легкое цилиндровое мас¬
ло, XI — тяжелое цилиндровое масло, XII — гудрон, XIII — газы127Ь
ники 4 и после охлаждения в холодильниках 5 отводятся как то¬
варные продукты из установки. Бензиновый дистиллят через теп¬
лообменник 4 поступает в сепаратор газа 2 и после отделения газа
выводится как товарный продукт, а частично подается на ороше¬
ние колонны. Образующийся в количестве до 55% мазут из ниж¬
ней части колонны 3 подается в печь 8 и оттуда в колонну II сту¬
пени 9, работающую при остаточном давлении 0,005—0,008 МПа,
где разделяется на дистилляты. В нижнюю часть колонн 3 и 9
подается острый пар, что снижает температуру кипения и спо¬
собствует более полному отделению легких фракций.Аппаратура, применяемая при прямой гонке нефти и дру¬
гих процессах нефтепереработки должна обеспечивать нагре¬
вание сырья до высоких температур, при которых процесс про¬
текает с достаточной скоростью и достаточно четкое разделение
получаемых продуктов. Основными аппаратами в этих процес¬
сах нефтепереработки являются трубчатые печи и ректифика¬
ционные колонны.В трубчатых печах нефть и мазут проходят по трубам, распо¬
ложенным внутри печи и нагреваются за счет теплоты сгорания
жидкого или газообразного топлива. Печь состоит из двух камер:
радиационной, где размещаются горелки и радиантные трубы,
воспринимающие теплоту излучение и конвекционной, в кото¬
рой расположены трубы, обогреваемые дымовыми газами, вы¬
ходящими из камеры радиации. Конструкции трубчатых печей
весьма разнообразны. Они различаются способом передачи теп¬
ла (радиантные, конвекционные, радиантно-конвекционные),
способом сжигания топлива (с пламенным и беспламенным го¬
рением), расположением труб змеевика. Экономически наибо¬
лее эффективным являются печи беспламенного типа с излуча¬
ющими стенками. Производительность трубчатых печей устано¬
вок АВТ составляет от 100 до 1000 т/ч при коэффициенте полез¬
ного действия (коэффициенте использования теплоты) до 80%.Из многочисленных конструкций ректификационных ко¬
лонн в установках прямой гонки используются, главным обра¬
зом, барботажные колпачковые колонны тарельчатого типа.
Они содержат от 30 до 60 тарелок прямоточного действия с под¬
вижными клапанами, что обеспечивает динамический режим
работы колонны и постоянство скорости паров ректифицируе¬
мого продукта.Состав и выход продуктов прямой гонки зависит от типа про¬
цесса и состава перегоняемой нефти. В табл. 7.1 приведен вы¬
ход дистиллятов прямой гонки нефти по топливно-масляному
варианту процесса.128
Таблица 7.1. Состав продуктов прямой гонки1 -1ПродуктыИнтервал температур
кипения, °СВыход,% мае.Первая ступень АВТБензиндо 17014,5Лигроин160—2007,5Керосин200—30018,0Дизельное топливо300—3505,0Мазут (остаток)выше 35055,0Вторая ступень АВТ (перегонка мазута)Веретенное масло230—25010—12Машинное масло260—3055Легкое цилиндровое масло315—3253Тяжелое цилиндровое масло350—3707Гудрон (остаток)выше 37027—30На современных нефтеперерабатывающих заводах исполь¬
зуются комбинированные установки ЭЛОУ—АВТ, в которых со¬
вмещены процессы обессоливания и прямой гонки нефти. Мощ¬
ность их достигает 6 млн. т перерабатываемой нефти в год. При¬
менение комбинированных установок значительно улучшает
технико-экономические показатели процесса переработки не¬
фти. Расходные коэффициенты для таких установок составля¬
ют: пар 49 кг, вода 4,8 м3, электроэнергия 37,5-103 кДж, жид¬
кое топливо 33,4 т на 1 т перерабатываемой нефти.7.7. Крекинг нефтепродуктов7.7.1. Виды крекинг-процессаКрекингом называется вторичный процесс переработки неф¬
тепродуктов, проводимый с целью повышения общего выхода
бензина. Применение вторичных процессов в нефтепереработ¬
ке позволяет увеличить на 30—35% выход светлых продуктов
(моторных топлив), повысить их антидетонационные свойства
и термическую стабильность, а также расширить диапазон про¬
изводимого переработкой нефти химического сырья.Крекинг нефтепродуктов может быть термическим и ката¬
литическим. Термический крекинг проводится при температу¬
рах от 420 до 550°С и давлениях до 5 МПа. В настоящее время
термический крекинг используется для получения ограничен¬
ного числа продуктов: котельного топлива из гудрона (висбре-5 Химическая технология. Том 2129
кинг), высокоароматизированного сырья, сырья для техничес¬
кого углерода (сажи), а-олефинов для производства моющих ве¬
ществ. Для получения светлых нефтепродуктов, в том числе
Зензина, используется преимущественно метод каталитическо¬
го крекинга. Это объясняется тем, что каталитические процес¬
сы нефтепереработки по сравнению с термическими имеют ряд
преимуществ. К ним относятся:— высокая скорость превращений углеводородов и, как след¬
ствие, более мягкие условия процесса и меньшие энергозатра¬
ты;— увеличенный выход товарных продуктов, в том числе,
высокого качества (октановое число, стабильность);— возможность проведения процесса в заданном направле¬
нии и получение продуктов определенного состава;— использование сырья с высоким содержанием серы вслед¬
ствие гидрирования сернистых соединений и выведения их в
газовую фазу.7.7.2. Термохимические превращения углеводородовПри высоких температурах углеводороды нефтяного сырья
подвергаются разнообразным превращениям. Это первичные
реакции деструкции, приводящие к образованию продуктов с
меньшей молекулярной массой и вторичные реакции изомери¬
зации и конденсации, в результате которых образуются продук¬
ты с той же или большей молекулярной массой.Тип этих реакций и, следовательно, скорость, глубина и пос¬
ледовательность превращений зависят от стабильности углево¬
дородов различных классов в условиях крекинга. Мерой ста¬
бильности с достаточной степенью точности может служить ве¬
личина изобарно-изотермического потенциала образования уг¬
леводородов AG°o6i который является сильной функцией тем¬
пературы. В табл. 7.2 приведены значения AG°06 углеводородов
различных классов с одинаковым числом атомов углерода и уг¬
леводородов одного класса (алканов) с различным числом ато¬
мов углерода.Из табл. 7.2 можно сделать следующие выводы:1. При низкой температуре (298К) углеводороды различных
классов, но с одинаковым числом углеродных атомов в молеку¬
ле, по уменьшению их стабильности располагаются в ряд (ряд
термической устойчивости):Cnii2n+2>^CnH.2n > СлН2п >130
Таблица 7.2. Значения AG°o6> кДж/моль углеродаУглеводородФормулаТ емпература, °К2988001200ексанС6Н14-0,294иклогексанс6н1231,8317,9554,9ексен-1С6Н128 7,61внзолс6н6129,9221,2300,5ЛетанСН4-50,8-2,341,0Этанс2нА-32,966,6151,61ропанС3Н8-23,5127,4255,4>утанс4н10-17,1185,0355,12. Термодинамическая устойчивость углеводородов всех
тассов понижается с ростом температуры, но в различной сте¬
гни, поэтому, при высокой температуре (температуре крекин-l) положение углеводородов в ряду термической устойчивос-I меняется (рис. 7.5):СпН2п-б ^ СПН2П > АСпН2п ^ СпИ2п+23. Термическая устойчивость углеводородов одного класса
1дает с увеличением их молекулярной массы (числа атомов
\дерода) (рис. 7.6).Таким образом, при температуре крекинга в первую очередь
*струкции подвергаются алканы и нафтены преимуществен-
з с высокой молекулярной массой, а наиболее устойчивыми4С. 7.5. Зависимость AG06 Рис. 7.6. Зависимость AG0e алка-леводородов различных нов от числа углеродных атомов вlaccoB от температуры молекуле лс131
являются ароматические углеводороды и алкены. В результате
в продуктах крекинга накапливаются ароматические углеводо¬
роды и низшие алкены, которые затем вступают во вторичные
реакции полимеризации.Реакции превращения углеводородов нефтяного сырья при
крекинге могут быть сведены к следующим типам.1. Термическая деструкция алканов по схеме:СдН2д+2 * CmH2m+2срн2рICgH2<j+2 + Схн2хгде: n = m+p;m = q + xПри этом, в соответствии с рядом термической устойчивос¬
ти, из продуктов реакции деструктируются далее в первую оче¬
редь алканы. Для низших алканов помимо реакции деструк¬
ции по связи С-С, энергия которой равна 315—370 кДж/моль,
становится возможной и реакция дегидрирования с разрывом
связи С-Н, энергия которой составляет 380—410 кДж/моль и
становится соизмеримой с первой. Поэтому в газе крекинга все¬
гда содержится водород.2. Превращения нафтенов, в том числе реакции:
дегидрирования^0^-С^Н2га+1 + ЗН2,
деалкилированияO-CJWi Ос,н2р+1 + ст н2т,гидрирования с разрывом цикла0-С„Н2л+1 + н2 - СНз-СН2-СН-СН2-СН2-СНзСлН2л-Ь13. Превращения алкенов, в том числе реакции:
деструкции с образованием низших алкенов, алканов и ал-кадиеновСпН2л *2Сд/2Нд И СлН2п * СтЙ2т+2 ^р^2р-2^изомеризации R-CH=CH-CH3 — R-C=CH2,СН3полимеризации CnH2n^C2raH4„4. Синтез и превращения ароматических углеводородов по
реакциям конденсации алкенов и алкадиенов, напримерС2н4 + С4Н6 - <о) + 2Н2,132
деалкилирования<0>-С,Н2,+1- <Q> + СпН-2пконденсации с алкадиенами<0> + С4Нб-+ 2Н2Из этих реакций реакции деструкции алканов и ал кенов, де¬
алкилирования и превращения ароматических углеводородов
протекают по радикально-цепному механизму, а реакции терми¬
ческого распада нафтенов по молекулярному механизму.Скорость реакций первичной деструкции алканов и высших
алкенов, а также скорость реакции деалкилирования прибли¬
женно описывается уравнением реакции первого порядка:где: кср — усредненная константа скорости,X — степень превращения сырья,
т — время.При углублении процесса крекинга константа скорости
уменьшается вследствие тормозящего действия продуктов де¬
струкции и в уравнение 7.1 вводятся эмпирические поправ¬
ки. Глубина превращения крекируемого сырья и выход целе¬
вого продукта — бензина зависят от температуры, времени
пребывания сырья в зоне высоких температур и давления.С ростом температуры выход бензина сначала увеличивает¬
ся вследствие ускорения деструкции нестабильных тяжелых
углеводородов, а затем падает в результате разложения образо¬
вавшихся легких углеводородов до газообразных продуктов
(рис. 7.7 а).При увеличении времени контактирования выход бензина сна¬
чала также возрастает, а затем снижается вследствие тех же при¬
чин (рис. 7.7 б). Влияние давления при достаточно высокой и по¬
стоянной температуре на выход бензина аналогично влиянию тем¬
пературы. Поэтому, для повышения выхода бензина процесс кре¬
кинга ведут при умеренно повышенном давлении, а для увеличе¬
ния выхода газа — при пониженном давлении (рис. 7.7 в).Таким образом, максимальный выход бензина при крекин¬
ге достигается при некоторых опитимальных значениях пара¬
метров процесса.(7.1)133
а 6 вРис. 7.7. Зависимость выхода бензина при крекинге от температу¬
ры (а), времени контактирования (6) и давления (в)7.7.3. Каталитический крекинг нефтепродуктовКрекинг нефтяного сырья в присутствии катализаторов (ка¬
талитический крекинг) имеет ряд особенностей, которые обус¬
ловили широкое использование его в нефтеперерабатывающей
промышленности для производства моторных топлив. К этим
особенностям относятся:— высокая скорость процесса, в 500—4000 раз превышаю¬
щая скорость процесса термического крекинга;— увеличенный выход бензинов с большим содержанием
изоалканов и малым содержанием алкенов, характеризующих¬
ся высоким октановым числом и стабильностью при хранении;— большой выход газообразных продуктов, содержащих уг¬
леводороды Ci—С4, являющихся сырьем для органического
синтеза.К катализаторам, используемым в каталитическом крекин¬
ге, предъявляются следующие требования:— селективность;— высокая активность при температуре крекинга,— стабильность активности;— устойчивость к истиранию, действию высоких температур
и водяного пара.Мерой активности катализатора при крекинге является «ин¬
декс активности», определяемый экспериментально на лабо¬
раторных установках. Индекс активности равен выходу бензи¬
на, перегоняющегося до 200°С при крекинге эталонного сырья
в стандартных условиях.Стабильностью катализатора называется его способность со¬
хранять свою активность во время эксплуатации. Катализато¬134
ры каталитического крекинга достаточно быстро дезактивиру¬
ются вследствие отложения на поверхности зерен кокса и нуж¬
даются в регенерации.В каталитическом крекинге в качестве катализаторов ранее
применялись природные глины с содержанием оксида алюми¬
ния до 25% и индексом активности около 35. В настоящее вре¬
мя все установки каталитического крекинга работают на син¬
тетических алюмосиликатных катализаторах, содержащих в
своем составе цеолиты с индексом активности около 50:
rtNa20-mAl2037?SiC>2-<7H20. Селективность катализатора может
быть повышена введением в его состав оксида рения.Носителем активности подобных катализаторов является
гидратированный алюмосиликат HA102eSi02, сохраняющий
активность до 700 С. Все реакции, протекающие на поверхнос¬
ти алюмосиликатного катализатора, имеют цепной характер.
Последовательность реакций крекинга углеводородов различ¬
ных классов определяется скоростью адсорбции их на зернах
катализатора, так как при температуре крекинга процесс идет
в диффузионной области и лимитируется скоростью диффузии
молекул сырья к поверхности катализатора. При этом арома¬
тические углеводороды деалкилируются с образованием алке-
нов и простейших ароматических углеводородов, нафтены де¬
гидрируются, деалкилируются и расщепляются с разрывом
цикла. Алкены, образовавшиеся при крекинге, деструктируют-
ся, изомеризуются и гидрируются с образованием циклических
и ароматических углеводородов.Важнейшим направлением превращений при каталитичес¬
ком крекинге являются реакции алканов, которые подвергают¬
ся реакциям деструкции и изомеризации. Последовательность
реакций алканов на алюмосиликатном катализаторе может
быть представлена в следующем виде.1. Протонирование катализатора:HA102-Si02^HAlSi04^H® + AlSi04e2. Дегидрирование алкана до алкена под воздействием тер¬
мического фактора:R-CH2-CH2-CH3^R-CH=CH-CH3 + Н23. Образование вторичного карбкатиона:R-CH=CH-CH3 + h^r-^h-ch2-ch34. Превращение вторичного карбкатиона по двум схемам:
крекинг до алкена:R-GH-CH2^CH3^R-CH=CH2 + СНз
изомеризация в стабильный третичный карбкатион через не¬
стабильный первичный карбкатион:с алкеном R-C-CH3 + R-CH=CH2 ^ R-C=CH2 + R-ffl-CH3СНз СНзс образованием конечных продуктов — изоалкана и изоалкена
и вторичного карбкатиона генерирующего цепь. *Распад молекул алканов может происходить в различных
участках углеродной цепи, однако вероятность распада с обра¬
зованием метана, этана и этилена незначительна. Поэтому, в
газе каталитического крекинга содержатся преимущественно
углеводороды С3—С4. Таким образом, при каталитическом кре¬
кинге образуются преимущественно алканы и алкены изостро¬
ения и ароматические углеводороды и крекинг-бензин имеет
высокое октановое число.Вследствие низкой энергии активации реакций на алюмоси-
ликатных катализаторах скорость их незначительно зависит от
температуры. Она определяется, главным образом, активнос¬
тью катализатора. Давление влияет на скорость реакций поли¬
конденсации и коксообразования и практически не оказывает
влияния на скорость распада углеводородов, протекающих на
поверхности катализатора.Характерной особенностью процесса каталитического кре¬
кинга является перераспределение (диспропорционирование)
водорода. Это явление связано с тем, что в системе протекают
одновременно как реакции дегидрирования с образованием ал-
кенов, полимеризующихся на поверхности катализатора до
кокса, так и реакции гидрирования и образования насыщенных
соединений. Таким образом, в процессе крекинга одни молеку¬
лы обедняются водородом, а другие им насыщаются:0 © 0R-ffl-CH2^CH3)^R-CH-CH2^ R-C-CH3СНз5. Превращение третичного карбкатиона по реакциям:0* ИЗО-СлН2л+2СлН2л+2Конденсированные► Н 2 + АгН* системы► Кокс136
Перераспределение водорода в процессе каталитического кре¬
кинга вызывает отложение кокса на поверхности катализатора
и потерю его активности. Вследствие этого появляется необхо¬
димость в непрерывной регенерации катализатора, что достига¬
ется выжиганием кокса в токе воздуха. Поэтому, работа катали¬
затора при крекинге складывается из двух последовательных
стадий: рабочего процесса в реакторе и восстановления активно¬
сти в регенераторе (регенерация), как показано на рис. 7.8.Сырье|-> Продукты крекингаРабота
катализатора
в реактореВоздух |_^С02tДезактиви-
► рованный *
катализатор-Регенерация
катализатора
в регенератореРис. 7.8. Схема действия катализатора при крекингеСуществующие установки каталитического крекинга делят¬
ся на три типа:— периодического действия со стационарным слоем катали¬
затора;— непрерывного действия с движущимся слоем катализатора;— непрерывного действия с кипящим слоем микросферичес-
кого или пылевидного катализатора. Установки этого типа наи¬
более распространены.Сырьем для каталитического крекинга служат нефтепродук¬
ты, выкипающие в интервале 200—500°С. К ним относятся:— широкая фракция прямой гонки мазута;— соляровая фракция термического крекинга;— газойль коксования нефтяных остатков.Сырье должно удовлетворять определенным требованиям по
содержанию смол, сернистых и азотистых соединений. Поэто¬
му, перед крекингом его подвергают гидроочистке.Основными параметрами процесса каталитического крекинга
являются температура, время контактирования паров сырья с ка¬
тализатором и кратность циркуляции катализатора. Современ¬
ные промышленные процессы каталитического крекинга исполь¬
зуют непрерывно циркулирующий поток катализатора. Отноше¬
ние массы катализатора к массе сырья, подаваемых в реактор, на¬
зывается кратностью циркуляции катализатора (кг/кг):N = mK/mc (7.2)где: тк — масса катализатора, подаваемая в реактор, кг/ч;
тс — масса сырья, подаваемая в реактор, кг/ч.137
От кратности циркуляции катализатора зависит время пре¬
бывания его в зоне реакции и степень его закоксованности, а
также количество теплоты, вносимой с катализатором в реак¬
тор как теплоносителем. С увеличением кратности циркуляции
возрастает активность катализатора, повышается выход бензи¬
на и газа, но увеличиваются размеры регенератора и расход
энергии на транспортировку катализатора в установке. Опти¬
мальные значения параметров каталитического крекинга: тем¬
пература 480—490° С, давление 0,1—0,2 МПа, объемная ско¬
рость сырья 1,5—3,0 ч"1, кратность циркуляции катализатора
2,5—7,0 кг/кг.На рис.7.9 представлена технологическая схема установ¬
ки каталитического крекинга с кипящим слоем катализатора1—А/1—М. Крекируемое сырье через теплообменники 1 пода¬
ется в печь 2. Нагретое сырье смешивается с рециркулятом (ча¬
стью тяжелой фракции) и по катализаторопроводу поступает в
реактор крекинга 3. В нижнюю отпарную зону реактора вводит¬
ся водяной пар для отдувки катализатора. Пары продуктов ре¬
акции и водяной пар при температуре 450°С из верхней части
реактора 3 поступают в нижнюю часть ректификационной ко¬
лонны 4. Пары бензина и водяной пар отбираются с верхней
части колонны, проходят холодильник-конденсатор 5 и посту¬
пают в сепаратор 6, в котором разделяются на водяной слой, бен¬
зиновый слой и газ. Газ компрессируется и подается на газо-
фракционирование, а бензин поступает на ректификацию.
Часть бензина отбирается на орошение колонны.Дизельное топливо и тяжелая фракция проходят через сек¬
ции отпарной колонны 7, охлаждаются в теплообменниках 1 и
холодильниках 8 и отводятся как товарные продукты. Часть
тяжелой фракции в виде рециркулята смешивается с сырьем и
подается в реактор 3, а часть направляется на орошение ниж¬
ней части колонны 4. Смесь тяжелых жидких продуктов кре¬
кинга и катализаторной пыли из низа колонны 4 поступает в
шламоотделитель 9, из которого шлам возвращается в реактор3, а богатый ароматическими углеводородами декантат отводит¬
ся с установки.Дезактивированный в процессе работы катализатор из ки¬
пящего слоя реактора опускается в его отпарную зону и ката-
лизаторопроводом отводится в узел смешения с воздухом 10. Из
него за счет воздушного потока катализатор переносится в ре¬
генератор 12, в котором создается кипящий слой. Основная
часть воздуха для выжигания катализатора подается непосред¬
ственно в регенератор. Газы, образовавшиеся в результате вы-138
возддекантат► вода
сырье' ное топ-
8 ливо^ дизель-газбензинтяжелаяфракцияРис. 7.9. Технологическая схема установки крекинга 1—А/1—М:1 — теплообменники, 2 — трубчатая печь, 3 — реактор «КС», 4— ректи¬
фикационная колонна, 5—холодильник-конденсатор, 6 — газоотдели-
тель, 7 — отпарная колонна, 8 — холодильники, 9 — шламоотделитель,10— узел смешения, 11 — регенератор катализатора «КС», 12 — котел-
утилизатор, 13—электрофильтржигания кокса, проходят котел-утилизатор 12, электрофильтр13 для улавливания катализаторной пыли и выбрасываются в
атмосферу. Регенерированный катализатор из нижней части
регенератора 11 поступает в катализаторопровод и вместе с сы¬
рьем и рециркулятом возвращается в реактор 3.Основными аппаратами установки каталитического крекин¬
га являются реактор кипящего слоя, и регенератор катализато¬
ра кипящего слоя. Реактор крекинга «КС» представляет цилин¬
дрический стальной аппарат диаметром 4 м и высотой 40 м с вер¬
хним штуцером для ввода паров сырья и нижним — для вывода
отработанного катализатора. Внутренний объем реактора разде¬
лен на три зоны: реакционную, отпарную и отстойную. В отпар-
ную зону подается водяной пар для отделения адсорбированных
на катализаторе углеводородов. Реакционная зона реактора за¬
полнена кипящим слоем катализатора, который создается пара¬
ми сырья высотой 5—6 м и плотностью 400 кг/м3. Производи¬
тельность реакторов составляет 800 т/сутки.Регенератор катализатора «КС» выполнен в виде стального
цилиндрического аппарата диаметром 12 м и высотой 30 м, фу¬
терованного изнутри огнеупорным кирпичом. Регенератор139
внутри разбит на зоны, в каждой из которых размещены уст¬
ройства для подвода воздуха, вывода газов регенерации и змее*
вики для отвода реакционного тепла. Кипящий слой в регене¬
раторе создается током воздуха. Температура выжигания кок¬
са в регенераторе составляет 650—720С при расходе 12—15 кг
воздуха на кг кокса. Производительность регенератора харак¬
теризуется массой кокса, выжигаемого в единицу времени с
единицы реакционного объема. Для установок с микросфери-
ческим катализатором она составляет 12—14 кг/ч-м3.Продуктами каталитического крекинга являются кре¬
кинг-бензин, легкий газойль (дизельное топливо), тяжелый
газойль (широкая фракция) и крекинг-газ. В табл. 7.3 пред¬
ставлен выход и состав продуктов каталитического крекин¬
га.Таблица 7.3. Выход и состав продуктов каталитического
крекингаПродуктВыход,% массовых
от сырьяСостав отбираемой
фракцииТемпература,°СКрекинг -газ10—20Углеводороды Сз—С5
80%, из них изостро¬
ения до 40%Крекинг-бензин30—55изоалкены 25 %, изо-
алканы до 55%, арома¬
тические углеводороды
20—30%до 195Дизельное топливо25—30Ароматические углево¬
дороды 40—80%195—350Широкая фракция5—20Конденсированные уг¬
леводороды 40—60%350Выход кокса при каталитическом крекинге составляет 4—
8%.Расходные коэффициенты для установки крекинга «КС»
(на 1 т сырья): топливо жидкое 6,7 кг, топливо газообразное
9,5 кг, электроэнергия 3,2'105кДж, катализатор 1,9 кг, водя¬
ной пар (потребляемый) 270 кг, водяной пар (вырабатывае¬
мый) 685 кг.140
7.7.4. Гидрокрекинг нефтепродуктовОсобую разновидность крекинг-процессов представляет гид¬
рокрекинг. Он относится к так называемым гидрогенизацион-
ным процессам нефтепереработки и проводится в среде водоро¬
да при высоких температуре и давлении, в присутствии бифун¬
кциональных катализаторов, катализирующих одновременно
реакции расщепления, изомеризации и гидрирования углево¬
дородов.Подобные сложные контактные системы содержат гидриру¬
ющий компонент — металл (кобальт, никель, молибден, пла¬
тина, вольфрам) и деструктирующий и изомеризующий ком¬
понент — алюмосиликаты или цеолиты. Применяются также
оксиды и сульфиды металлов на алюмосиликатах.Важнейшая особенность гидрокрекинга заключается в том,
что в нем, наряду с реакциями распада тяжелых углеводородов
сырья, свойственными крекинг-процессу, протекают реакции
гидрирования образовавшихся продуктов распада. Основными
реакциями при гидрокрекинге являются:1. Деструкция высокомолекулярных алканов и алкенов и
дегидрирование продуктов деструкции:СпН2и+2 ~* СтН2т+2 + СрН2рСрН2р + Н2 СрН.2Р+2 и окончательно,СпН2л+2 + Н2 —^ СтЩт-2 СрН2р+22. Гидрирование алкенов сырья, что в условиях гидрокрекин¬
га термодинамически более вероятно, чем их полимеризация и
циклизация:СлН2л + Н2 —* С„Н2л+23. Изомеризация алкановн- С„Н2л+2^изо- С„Н2п+24. Распад, дециклизация (гидрогенолиз) и деалкилирование
нафтенов, например:Q + н2 - с5н12^ ^-СдНгд+х + Н2 —*• <( У + С„Н2п+25. Деалкилирование и гидрирование ароматических углево¬
дородов:<0>-r + h2- <3-r<^0^-СпН2п-1 + Н2 <Р> + Cnii2n+2141
По убыванию реакционной способности в условиях гидро¬
крекинга углеводороды могут быть расположены в следующий
ряд:конденсированные полициклические наф- алканы, алкилбен-ароматические угле- > тены, ароматические > золы, нафтеныводороды с числом углеводороды с чис- с числом цикловциклов более 3 лом циклов 1—3 1—3Таким образом, при гидрокрекинге, в отличие от катали¬
тического крекинга, легче всего вступают в превращения аро¬
матические полициклические соединения и образуются с вы¬
соким выходом легкие насыщенные углеводороды, в том чис¬
ле изостроения. При этом, одновременно с реакциями углево¬
дородной части сырья, происходит гидрирование и удаление
неуглеводородных соединений — гидроочистка нефтепродук¬
тов (см. 7.9).В целом применение гидрокрекинга позволяет повысить глу¬
бину переработки нефти и получить бензин высокого качества,
не содержащий сернистых соединений.Сырьем для гидрокрекинга служат тяжелые нефтяные ди¬
стилляты (газойли прямой гонки и каталитического крекин¬
га), мазут, гудрон. В зависимости от вида сырья гидрокрекинг
проводится в одну или две ступени, которые различаются ре¬
жимом работы. Основными параметрами процесса гидрокре¬
кинга, от которых зависит выход и состав продуктов, являют¬
ся температура, давление водорода, объемная скорость сырья,
соотношение между объемами циркулирующего водородсодер¬
жащего газа и сырья (кратность циркуляции) и содержание
водорода в этом газе. Например, для установки одноступенча¬
того гидрокрекинга JI-16-1 с алюмо-кобальт-молибденовым ка¬
тализатором принят следующий режим: температура 400—
410°С, давление 5 МПа, объемная скорость 1,0 ч~х, кратность
циркуляции водорода 600 м3/м3, содержание водорода в цир¬
кулирующем газе 75% об.Процесс гидрокрекинга используется для производства ав¬
томобильных бензинов, реактивного и дизельного топлива, сы¬
рья для нефтехимического синтеза и, в частности, для получе¬
ния бензина с высоким содержанием изоалканов для добавки к
бензину риформинга с целью снижения в нем содержания аро¬
матических углеводородов (рис. 7.10).142
Рис. 7.10. Схема корректировки состава бензина риформинга7.8. Каталитический риформинг нефтепродуктовРиформингом называется вторичный процесс переработки
нефтепродуктов, проводимый с целью получения индивидуаль¬
ных ароматических углеводородов, водорода или бензина с по¬
вышенным содержанием ароматических углеводородов. Про¬
цесс риформинга проводится в присутствии катализаторов (ка¬
талитический риформинг).7.8.1. Физико-химические основы процессаВ условиях каталитического риформинга превращениям на
катализаторе подвергаются углеводороды всех классов. Важ¬
нейшими реакциями при этом являются следующие.1. Дегидроциклизация и изомеризация алканов:C6H13-R «=* <0>-R + 4Н2Н-СлН2л+2 ИЗО-С„Н2л+22. Дегидрирование шестичленных и изомеризация с расши¬
рением цикла и дегидрирование пятичленных нафтенов:O-R - <0>-R + ЗН2
[3-CH2-R - <0>-R + ЗН23. Циклодегидрирование алкеновC6Hn-R <0>-R + ЗН24. Деалкилирование и дегидроконденсация ароматических
углеводородов:<0>-С„Н2п+1 - <о> + с„н2„2 <о>=00+20+21,2Реакции конденсации приводят к образованию кокса, отла¬
гающегося на поверхности катализатора и дезактивирующего143
его. Чтобы уменьшить отложение кокса процесс риформинга
проводят в атмосфере водорода. Однако повышение давления
водорода смещает равновесие реакций дегидрирования и дегид¬
роциклизации влево. Поэтому, оптимальное парциальное дав*
ление водорода в процессе риформинга определяется совмест¬
ным влиянием обоих факторов. Очевидно, что интенсивность
отложения кокса на катализаторе зависит от давления водоро¬
да: она незначительна при высоком давлении и весьма суще¬
ственна при низком. Так как высокая закоксованность катали¬
затора вызывает необходимость его регенерации, то в зависи¬
мости от давления, процесс риформинга может проводиться в
двух технологических вариантах:— без регенерации катализатора и— с регенерацией катализатора (ультраформинг).Выбор катализатора риформинга определяется механизмом
реакций, протекающих на нем. Реакции гидрирования и дегид¬
рирования протекают по окислительно-восстановительному
механизму и катализируются металлами, реакции изомериза¬
ции и гидрокрекинга протекают по ионному механизму и ката¬
лизируются кислотами. Поэтому, в каталитическом крекинге
используются бифункциональные катализаторы состава {Me +
+AI2O3}, где: Me = молибден, платина, рений, AI2O3 — катали¬
затор изомеризации, промотируемый фторидами или хлорида¬
ми металлов, являющийся одновременно носителем.В соответствии с природой катализатора различают следую¬
щие разновидности процесса риформинга:— платформинг (катализатор — платина),— рениформинг (катализатор — рений),— риформинг на молибденовом катализаторе.Вследствие низкой активности молибденовых катализаторовони в настоящее время в промышленности не используются. Вы¬
сокой активностью и селективностью обладают полиметалличес¬
кие катализаторы, содержащие платину, кадмий и рений, напри¬
мер, катализатор, КР-104, стабильно работающие без регенерации
до одного года и обеспечивающие выход бензина с 04 до 90. Все
катализаторы на основе платины чувствительны к каталитичес¬
ким ядам, к числу которых относятся соединения серы, азота и
некоторых металлов. Поэтому сырье, перед подачей на операцию
риформинга, подвергается гидроочистке и сушке.Превращения углеводородов при риформинге описываются
уравнением реакции 1 порядкаU = кср(а-х) (7.3)где: кср — усредненная константа скорости.144
Тепловой эффект процесса зависит от удельного веса в нем
эндотермических реакций ароматизации (АН\) и, следователь¬
но, от содержания в сырье нафтенов, и экзотермических реак¬
ций гидрокрекинга (АЯ^)- Соотношение это таково, что суммар¬
ный тепловой эффект риформинга АН = АНi - АН2 < 0, Рифор-
минг на платиновом катализаторе (платформинг) характеризу¬
ется следующими параметрами процесса:— температура 470—520°С,— давление водородсодержащего газа 2—8 МПа,— объемная скорость сырья 1—2 ч-1,— кратность циркуляции водородсодержащего газа 1300—
1800 м3/м3. Понижение температуры приводит к увеличению
выхода бензина и уменьшению содержания в них ароматичес¬
ких углеводородов. Повышение давления снижает скорость об¬
разования газа и кокса, но уменьшает выход ароматических уг¬
леводородов. Снижение объемной скорости сырья влияет анало¬
гично повышению температуры, однако при меньших скоростях
возрастает объем аппаратуры и падает экономичность процесса.7.8.2. Технология каталитического риформингаВ зависимости от цели процесса существует две разновидно¬
сти каталитического риформинга:— ароматизация — получение ароматических индивидуаль¬
ных углеводородов и— облагораживание бензина — получение бензина с высо¬
ким содержанием ароматических углеводородов и высоким ОЧ.Эти процессы различаются природой сырья, технологичес¬
ким режимом и составом получаемых продуктов. В табл. 7.4
приведены основные данные по этим процессам платформинга.Таблица 7.4. Характеристика процессов платформингаХарактеристикаВариант платформингаОблагораживаниеАроматизацияЦель процесса
СырьеТемпература, °С
Давление, МПа
Продукты процессаПрименение продуктовПовышение октаново¬
го числа бензина
Широкая фракция бен¬
зина прямой гонки
480—520
3—4Катализат 85%,
газ 15%Автобензин, газ для
гидрокрекингаСинтез индивидуальных
углеводородов
Узкие фракции бензина
прямой гонки
480—520
2Бензол, толуол,
ксилолыСырье для органическо¬
го синтеза145
Рис. 7.11. Схема платформинга для облагораживания бензинаУстановки каталитического риформинга состоят из трех бло¬
ков:— блока предварительной гидроочистки сырья;— блока платформинга очищенного сырья (гидрогената);— блока стабилизации бензина (катализата) в случае обла¬
гораживания бензина или блока выделения углеводородов в
случае ароматизации.Установки платформинга по режиму работы делятся на:— установки со стационарным слоем катализатора и— установки с движущимся слоем катализатора.На рис. 7.11 представлена принципиальная схема платфор¬
минга для получения облагороженного бензина.Технологическая схема установки со стационарным слоем
катализатора АП-64 производительностью один миллион тонн
в год бензина АИ-95 приведена на рис.7.12.Исходное сырье, пройдя теплообменник 2, смешивается с
циркулирующим газом гидроочистки и избыточным водород¬
содержащим газом риформинга и нагревается в первой секции
печи 2. Образовавшаяся газосырьевая смесь поступает в реак¬
тор гидроочистки 3, где очищается от соединений серы, азота и
кислорода. Очищенная парогазовая смесь охлаждается в теп¬
лообменнике 1 и холодильнике 4 и постуцает в сепаратор гид¬
роочистки высокого давления 5, где разделяется на циркуля¬
ционный гай и жидкий гидр(огенизат (очищенный бензин). Газ,
содержащий водород и сероводород, подается в абсорбер 6, где146
Рис. 7.12. Технологическая схема облагораживания бензина:1, 8 — теплообменники, 2 — печь двухсекционная, 3 — реактор гидроочи¬
стки, 4,10, 14 — холодильники, 5 — сепаратор гидроочистки, 6 — этанола-
минный абсорбер, 7 — отпарная колонна, 9 — реактор платформинга,11 — сепаратор платформинга высокого давления, 12 — сепаратор
платформинга низкого давления, 13 — колонна стабилизацииочищается от сероводорода раствором этаноламина, после чего
в виде циркуляционного газа смешивается с сырьем, поступа¬
ющим на гидроочистку. Гидрогенизат из сепаратора 5 поступа¬
ет в отпарную колонну 7, где из него удаляют остатки сероводо¬
рода, водяные пары и газообразные углеводороды. Стабильный
гидрогенизат выводится из нижней части колонны, проходит
теплообменник 8, смешивается с водородсодержащим газом
риформинга и, пройдя вторую секцию печи 2, поступает в ба¬
тарею из трех реакторов платформинга 9. Из последнего реак¬
тора батареи газопродуктовая смесь проходит теплообменник
8 и холодильник 10 и охлажденная до 30°С поступает в сепара¬
тор высокого давления 11 для отделения циркуляционного газа
от жидкого катализата. Циркуляционный газ возвращается в
систему платформинга и гидроочистки, а нестабильный ката-
лизат (бензин) поступает в сепаратор низкого давления 12. Из
сепаратора катализат направляется в колонну стабилизации 13,
где из него отделяются легколетучие продукты, направляемые147
на сжижение. Стабильный бензин отбирается из нижней части
колонны и пройдя холодильник 14 поступает на фракциониро¬
вание.Технологический процесс платформинга , проводимый с це¬
лью получения индивидуальных ароматических углеводородов
(ароматизация) не отличается принципиально по аппаратуре и
условиям от процесса облагораживания бензина, но имеет ряд
особенностей:1. Сырье (бензин прямой гонки) предварительно разгоняет¬
ся на узкие фракции, каждая из которых ароматизируется от¬
дельно. Отбирают фракции с интервалами температур кипения:
головная до 60С, бензольная 62—85°С, толуольная 85—115°С и
ксилольная 115—150°С.2. Полученные ароматические углеводороды выделяются из
ароматизированных фракций экстракцией этиленгликолем или
диэтиленгликолем, в которых не растворяются алканы и нафте-
ны.3. Смесь ксилолов разделяется сверхчеткой ректификацией
на колонне с 320 тарелками, а пара- и метаизомеры — кристал¬
лизацией.На рис. 7.13 представлена принципиальная схема аромати¬
зации.Рис. 7.13. Схема платформинга для ароматизации
148
Выход ароматических углеводородов при ароматизации со¬
ставляет от массы бензина: бензол 25%, толуол 30%, ксилолы
20—30%, газ, содержащий водород, метан, этан и пропан до
20%.7.9. Очистка нефтепродуктовПолученные в результате прямой гонки и различных вторич¬
ных процессов нефтепродукты содержат компоненты, отрица¬
тельно сказывающиеся на их эксплуатационных свойствах. В
светлых нефтепродуктах (бензин, керосин, дизельное топливо)
содержатся алкены и алкадиены, органические соединения
серы (тиоспирты тиоэфиры), нефтяные кислоты, высшие ами¬
ны и азотсодержащие гетероциклы. Помимо этих примесей, в
дизельном топливе присутствуют высшие алканы с температу¬
рой затвердевания -ЮС и выше, которые кристаллизуются при
низких температурах. В нефтяных маслах, полученных разгон¬
кой мазута, могут содержаться также смолы и полицикличес-
кие ароматические углеводороды с боковыми цепями.Многие из этих соединений вызывают нестабильность
свойств нефтепродуктов при хранении и транспортировке, кор-
ррозию аппаратуры, образование нагара и токсичных продук¬
тов сгорания. Для их удаления используют методы депарафи-
низации и очистки нефтепродуктов.Депарафинизацией называется процесс выделения из неф¬
тепродуктов твердых углеводородов, выпадающих в виде крис¬
таллов при охлаждении. Наиболее распространенным методом
депарафинизации стал метод с использованием селективных
растворителей, основанный на различной растворимости угле¬
водородов. В качестве растворителей используются ацетонто-
луольная или метилэтилкетон-толуольная смеси и спиртовой
раствор карбамида. При карбамидной депарафинизации карба¬
мид образует с алканами нормального строения с числом угле¬
родных атомов более шести и циклическими углеводородами с
длинными алифатическими радикалами кристаллические ком¬
плексыRH + CO(NH2)2 ^ RH-CO(NH2)2 -аЯПолнота извлечения алканов возрастает с понижением тем¬
пературы (-АН), увеличением времени обработки нефтепродук¬
тов растворителем и снижением вязкости системы. Для этого
используют растворители, хорошо растворяющие как алканы,
так и карбамид. Образовавшиеся кристаллические комплексы
отделяют от нефтепродуктов отстаиванием, фильтрованием или149
центрифугированием. Для очистки нефтепродуктов от приме¬
сей используются методы адсорбции, абсорбции и гидрирова¬
ния.При адсорбционной очистке в качестве адсорбентов исполь¬
зуют естественные глины, синтетические алюмосиликаты, ак¬
тивированный уголь. Для повышения адсорбционной активно¬
сти поглотители предварительно активируют обработкой кис¬
лотами и прокаливанием и диспергируют до размеров частиц
около 0,1 мм.При абсорбционной очистке используют такие селективные
растворители как фенол, фурфурол, смесь фенола с пропаном,
жидкий оксид серы (IV), серная кислота, гидроксид натрия. Так
например, при щелочной абсорбционной очистке протекают
реакции:H2S + 2NaOH = Na2S + 2Н20,RSH + NaOH = RSNa + H20,RCOOH + NaOH = RCOONa + H20с образованием соединений, растворимых в воде и удаляемых
при очистке.При кислотной очистке из нефтепродуктов удаляются, глав¬
ным образом, алкены, ароматические углеводороды и некото¬
рые соединения серы:R-CH=CH2 + H2S04 = R-CH(0S03H)-CH3,СбН6 + H2S04 = C6H50S03H + H20,H2S + H2S04 = S + S02 + 2H20,2RSH + H2S04 = R-S-S-R + S02 + 2H20Технологическая схема абсорбционной очистки нефтепро¬
дуктов включает операции экстракции, разделения образую¬
щихся фаз, непрерывной регенерации растворителя и его обез¬
воживания.Заключительной операцией очистки нефтепродуктов, кото¬
рую проходят почти все нефтяные топлива прямой гонки, кре¬
кинга и риформинга, является гидроочистка. Ее используют
также для облагораживания смазочных масел. Гидроочистка
представляет одну из разновидностей гидрогенизационного
процесса и протекает в условиях, близких к условиям гидро¬
крекинга и на тех же катализаторах.В процессе гидроочистки из нефтепродуктов удаляются со¬
единения серы, азота, кислорода и некоторых металлов и гид¬
рируются ненасыщенные углеводороды:150
RSH + H2 = RH + H2S
R2S + 2H2= 2RH + H2SRNH2 + H2 = RH + NH3
ROH + H2 = RH + H20
R-CH=CH2 + H2 = r-ch2-ch3Образовавшиеся продукты гидрирования отделяются от неф¬
тепродуктов путем поглощения их сорбентами (этаноламин,
раствор гидроксида натрия)7.10. Коксование нефтяных остатковКоксованием называется термохимический процесс превра¬
щения тяжелых остатков нефтепереработки (гудрон, асфальт,
крекинг-остаток) в нефтяной кокс и светлые нефтепродукты
(бензин, газойль). Коксование позволяет не только получать
беззольный электродный кокс, но и увеличить выход светлых
нефтепродуктов за счет расщепления высококипящих углево¬
дородов коксуемых остатков и тем самым повысить глубину
переработки тяжелого нефтяного сырья.Коксования нефтяных остатков может проводиться в уста¬
новках различного типа:— в горизонтальных кубах периодического действия;— в необогреваемых коксовых камерах полунепрерывного
действия;— в реакторах кипящего слоя непрерывного действия.При коксовании в кипящем слое нагретое сырье контакти¬
рует в реакторе «КС» с подвижным, нагретым до более высокой
температуры, чем сырье, инертным теплоносителем и коксует¬
ся на поверхности частиц этого теплоносителя. В современных
установках этого типа (рис. 7.14) теплоносителем является гра¬
нулированный кокс с размерами частиц до 0,3 мм, который со¬
здает в реакторе кипящий слой.В этом кипящем слое одновременно протекают три процесса:— собственно коксование, сопровождающееся образованием
продуктов разложения и уплотнения;— прокаливание кокса и удаление из него летучих веществ;— вторичные реакции продуктов коксования в паровой фазе.Жидкое сырье подается в реактор 1, в котором поступающимснизу водяным паром создается кипящий слой кокса. Парога¬
зовая смесь продуктов коксования поступает в парциальный
конденсатор реактора 2, где разделяется на газ, бензиновый151
a,дымовыегазы4~Тводапарж3нагретый, ка-=- z.кокс-Ч ^ ^газ^ бензин на
t d * ректификациюгазойльсырье6 h=-dвоздухпарРис. 7.14. Схема коксования нефтяных остатков:1 — реактор кипящего слоя, 2 — парциальный конденсатор, 3 — коксонаг-
реватель, 4 — котел-утилизатор, 5 — коксовый холодильник, 6 — бункер
коксадистиллят, направляемый на ректификацию, и газойль. Кок¬
совый теплоноситель из реактора подается в коксонагреватель3, где частично сжигается в токе воздуха. Нагретый кокс ох¬
лаждается в коксовом холодильнике 5 и поступает в бункер
кокса 6, а частично возвращается в реактор 1. Дымовые газы из
коксонагревателя 3 проходят котел-утилизатор 4 и выбрасыва¬
ются в атмосферу.Выход продуктов коксования (сырье — гудрон) составляет:
кокс 14%, газойль 63%, бензин 12%, газ 10%.Контрольные вопросы1. На какие классы подразделяются нефти по составу?2. Из чего состоят углеводородная, неуглеводородная и минеральная
части нефти?3. Какие вещества называются нефтепродуктами? Перечислите ос¬
новные из них.4. Приведите общую схему переработки нефти.5. Для чего проводятся операции стабилизации, обессоливания и
обезвоживания добытой нефти?6. Какие процессы нефтепереработки называются первичными и вто¬
ричными?7. Что является целью прямой гонки нефти?8. Какие превращения претерпевают углеводороды различных клас¬
сов при крекинге нефтепродуктов?9. Что характеризует стабильность углеводородов в процессе крекин¬
га и как она зависит от температуры и сложности молекулы угле¬
водорода?152
10. В чем преимущества каталитического крекинга перед термичес¬
ким?11. Что такое гидрокрекинг и где он используется в нефтепереработ¬
ке?12. Что такое риформинг нефтепродуктов?13. Чем отличается облагораживание бензина (повышение октаново¬
го числа) от ароматизации (синтез индивидуальных углеводоро¬
дов)?14. С какой целью и как проводится очистка нефтепродуктов?
Глава VIIIПЕРЕРАБОТКА ТВЕРДОГО ТОПЛИВА8. 1. Виды и происхождение твердых топливТвердые топлива, используемые как источник энергии и сы¬
рье для химического производства, подразделяются на топлива
естественного происхождения — природные и топлива искусст¬
венные — синтетические. К природным топливам относятся
торф, бурые и каменные угли, антрацит, горючие сланцы. Они на¬
зываются также ископаемыми твердыми топливами. Искусствен¬
ными топливами являются каменноугольный, торфяной и нефтя¬
ной кокс, полученные пирогенетической переработкой различных
видов природного топлива, а также брикеты и угольная пыль —
продукты механической переработки твердого топлива.Ископаемым твердым топливом (твердым горючим иско¬
паемым) называются естественные твердые горючие вещества
органического происхождения, образовавшиеся из остатков
отмерших растений и планктонов в результате бактериального
воздействия.- В земной коре твердые горючие ископаемые на¬
ходятся в виде углеродистых осадочных пород, образующих
месторождения или бассейны. Все ископаемые твердые топли¬
ва по материалу, из которого они образовались, делятся на сап-
ропелиты и гумолиты.Сапропелиты возникли в результате восстановительного
разложения остатков сапропеля — илистых отложений, обра¬
зовавшихся на дне водных бассейнов из планктона и низших
растений. К сапропелитам относятся горючие битуминозные
сланцы и некоторые другие ископаемые.Гумолиты возникли в результате окислительного разложе¬
ния остатков высших растений. Они подразделяются на:— гумиты, состоящие в основном из гумусовых веществ и— линтобиолиты, образовавшиеся из стойких структурных
элементов низших растений (споры, пыльца и т. п.).Основные виды ископаемых твердых топлив (торф, бурые и
каменные угли, антрацит) относятся к гумитам.В настоящее время общепринята биогенная теория проис¬
хождения ископаемых твердых топлив, согласно которой они
образуются в результате синтеза из продуктов разложения рас¬
тительных остатков за счет жизнедеятельности микроорганиз¬
мов. Процесс формирования твердого топлива класса гумитов
может быть представлен в такой последовательности. Он начи¬
нается с биогенного распада растительных остатков в результа¬154
те протекания энзиматических реакций деструкции, гидроли¬
за, окисления—восстановления. Эти реакции приводят к обра¬
зованию продуктов, которые частично используются микроор¬
ганизмами для питания и размножения, но главным образом
вступают во вторичные процессы полимеризации и поликонден¬
сации. При этом образуются новые вещества различного соста¬
ва и структуры, в том числе гуминовые высокомолекулярные
кислоты, содержащие конденсированные ароматические соеди¬
нения, составляющие основу гумитов. Эти процессы сопровож¬
даются отщеплением оксида углерода (IV), воды и простейших
углеводородов, в результате чего масса «углефицируется» —
обогащается углеродом и обедняется водородом и кислородом.
Они продолжаются до завершения образования буроугольной
залежи, до покрытия органических отложений минеральными
осадочными породами. Дальнейшие превращения бурых углей
в каменные угли происходят под воздействием уже чисто абио¬
генных факторов: высокой температуры, давления, влияния
окружающих залежь минералов.Глубина превращения исходных биогенных материалов в
результате углеобразования в твердые топлива характеризу¬
ется так называемой степенью углефикации (метаморфизма)
их, под которой понимают среднее содержание углерода в топ¬
ливе (в мас.% или дол.). По возрастанию степени углефика¬
ции твердые гумитовые топлива образуют генетический ряд:торф —► бурые угли —► каменные угли —► антрацитСтепень углефикации их приведена в табл 8.1.Таблица 8.1. Степень углефикации ископаемых твердых
топливТопливоТорфБурыеуглиКаменныеуглиАнтрацитСтепень углефикации,
% мае.58—6267—7576—9293—96Твердые топлива составляют основную массу известных ис¬
копаемых топлив на Планете. Их суммарные запасы на несколь¬
ко порядков превосходят запасы жидкого (нефть) и газообраз¬
ного топлива. В табл. 8.2 приведены общегеологические запа¬
сы (ОГЗ) и условно доступные запасы (УДЗ) твердого ископае¬
мого топлива в мире и на территории СНГ.155
Таблица 8.2. Запасы твердого ископаемого топлива,
млрд. т УТТопливоНа территории СНГМировыеОГЗУДЗОГЗУДЗИскопаемые угли61721617112102880Торф60159826Горючие сланцы5013114288-2. Каменные угли8.2.1. Строение и свойства каменных углейКаменные угли различной природы являются наиболее рас¬
пространенным видом твердого ископаемого топлива. Это нео¬
днородные твердые вещества черного или черно-серого цвета,
включающие четыре типа макроинградиентов, различающих
по блеску, внешнему виду и составу: блестящий (витрен), по-
лублестящий (к л арен), матовый (дюрен) и волнистый (фюзен).
Соотношение этих инградиентов, составляющих органическую
массу каменных углей, характеризует их структуру, химичес¬
кий и минералогический состав и обусловливает их многооб¬
разие и различие свойств.В состав органической части каменных углей входят биту¬
мы, гуминовые кислоты и остаточный уголь. Молекулярная
структура органической части угля представляет собой жест¬
кий трехмерный полимер нерегулярного строения, содержащий
подвижную фазу в виде разнообразных мономолекулярных со¬
единений. Обе фазы построены из отдельных фрагментов, вклю¬
чающих ароматические, в том числе многоядерные и гидриро¬
ванные системы с алифатическими заместителями и азотсодер¬
жащие гетероциклы, соединенные мостиковыми связями С-С,
С-О-С, C-S-C и C-NH-C. Степень конденсированности фраг¬
ментов (п) зависит от степени углефикации каменного угля.
Так, при степени углефикации 78% п = 2, при степени 90% п =4, для антрацита п = 12. В составе каменных углей установлено
также наличие различных функциональных групп: гидро¬
ксильной (спиртовые и фенольные), карбонильной, карбоксиль¬
ной и серусодержащих групп -SR- и -SH.Важнейшими характеристиками каменных углей, от кото¬
рых зависят возможность и эффективность их использования,
являются зольность, влажность, сернистость, выход летучих156
веществ и механические свойства, а для углей, применяемых в
качестве сырья для термохимической переработки, также спе-
каемость и коксуемость.1. Зольность. £}олой называется негорючая часть угля, со¬
стоящая из минеральных веществ, содержащихся в топливе. В
состав золы входят оксиды алюминия, кремния, железа (III),
кальция и магния. Высокая зольность снижает теплоту сгора¬
ния угля и ухудшает качество получаемого кокса. Зольность
каменных углей колеблется от 3 до 30% и может быть снижена
их обогащением. Угли, используемые для коксования, долж¬
ны иметь зольность не выше 7—7,5% .2. Влажность. Общая влажность угля складывается из
внешней, образующей капли или пленку на поверхности, и
внутренней (пирогенетической), выделяемой в процессе кок¬
сования. Влага, являясь балластом, удорожает транспортиров¬
ку угля, затрудняет подготовку его к коксованию, хранение и
дозировку, а также повышает расход тепла на коксование и
увеличивает время коксования. Влажность углей, используе¬
мых для термохимической переработки, не должна превышать
7%.3. Сернистость. Сера в каменных углях находится в виде
колчеданной, сульфатной и органической. Общее содержание
серы в углях колеблется от 0,4 до 8% . Так как в процессе кок¬
сования большая часть серы остается в коксе и может при вып¬
лавке чугуна переходить в металл, вызывая его краснолом¬
кость, уголь необходимо десульфировать обогащением.4. Выход летучих веществ. Летучими веществами камен¬
ного угля называются парообразные и газообразные вещества,
выделяющиеся из угля при нагревании его без доступа возду¬
ха при определенной фиксированной температуре. Выход ле¬
тучих веществ зависит от условий образования, химического
состава и степени углефикации угля, а также от температу¬
ры, скорости нагревания и выдержки при заданной темпера¬
туре. С увеличением степени углефикации выход летучих ве¬
ществ уменьшается. Так, для торфа он составляет около 70% ,
для бурых углей 65—45%, каменных углей 45-10% , для ант¬
рацита менее 10%. Методика выхода летучих веществ стан¬
дартизирована. Он определяется нагреванием навески угля
при 850°С и выдерживании при этой температуре в течение
семи минут.5. Коксуемость. Это свойство углей рассматривается в8.5.2.157
8.2.2. Классификация каменных углейВ основу технологической классификации каменных углей
положены выход летучих веществ и толщина образующегося
при нагревании пластического слоя. В РФ для каждого уголь¬
ного бассейна принята своя классификация (стандарт), так как
при одинаковых классификационных параметрах по спекаемо-
сти угли различного происхождения должны давать кокс оди¬
накового качества. В табл. 8.3 приведена технологическая клас¬
сификация углей одного из бассейнов, по которой они делятся
на 7 марок (классов).Таблица 8.3. Технологическая классификация углейМарка угляВыход летучих
веществ, %Толщина
пластического
слоя, ммНаименованиеОбозначениеДлиннопламенныйД42—Г азовыйГ356—15ЖирныйЖ35—2713—20КоксовыйК27—1814—20Отощенный спекающ.ОС22—146—13Тощийт17—19—АнтрацитА9—8.2.3. Запасы каменных углей и их использованиеПо материалам IX Международной энергетической конфе¬
ренции (1974 год) большая часть энергетических ресурсов твер¬
дого топлива сосредоточена на территории РФ, США, КНР и
Канады, на долю которых приходится более 90% мировых за¬
пасов ископаемых углей.В РФ значительная часть запасов (около 30%) приходится
на долю газовых углей. Удельный вес остальных марок, в том
числе коксующихся углей, не превышает 25%. Распределение
ископаемых углей по территории страны неравномерно: более
90% геологических запасов их приходится на восточные райо¬
ны, в том числе 60% на Сибирь и 30% на Дальний Восток. В РФ
представлены ископаемые угли всех степеней углефикации,
при этом основную массу составляют каменные угли. Они пре¬
обладают в Европейской части страны, в то время как на Урале
расположены запасы бурых углей. В Сибири запасы каменных
углей в четыре раза превосходят запасы бурых. Из каменных158
углей около 950 млрд. т, или более 10% , приходится на коксу¬
ющиеся угли, основные запасы которых сосредоточены в Куз¬
нецком, Печерском и Южно-Якутском угольных бассейнах. Ос¬
новная масса ресурсов углей сосредоточена в нескольких круп¬
ных бассейнах: Ленском (1650 млрд. т), Тунгусском (2300 млрд.
т), Канско-Ачинском (638 млрд. т), Кузнецком (637 млрд. т). К
другим достаточно мощным месторождениям углей в стране
следует отнести Печерский, Подмосковный и некоторые дру¬
гие бассейны. Основная масса каменных углей в РФ добывает¬
ся в настоящее время в Новокузнецком (37%), Канско-Ачинс-
ком (16 %) и Восточно-Сибирском (12%) месторождениях.Угольные ресурсы различаются глубиной залегания пластов,
степенью углефикации, географическим распространением.Методы добычи каменных углей зависят от глубины их за¬
легания. В РФ до 50% всех угольных запасов расположены на
глубине до 300 м, около 30% — на глубине 300—600 м и толь¬
ко около 10% на глубине от 600 до 1800 м. Поэтому в РФ полу¬
чил широкое распространение способ открытой добычи угля,
составляющий в настоящее время 57% от общего объема угле¬
добычи. Открытая добыча угля, в 2—4 раза более дешевая, чем
шахтный метод, практикуется в Канско-Ачинском, Южно-
Якутском и частично в Кузнецком, Иркутском и даже в Под¬
московном бассейнах, хотя это связано с необходимостью ре¬
шать дорогостоящие задачи рекультивации земли.В настоящее время около 60% добываемых ископаемых уг¬
лей используется для выработки тепловой (технологический
пар, горячая вода) и электрической энергии и до 30% для про¬
изводства металлургического кокса. Остальное количество
угля потребляется коммунально-бытовым хозяйством и мел¬
кими потребителями. В связи с совершенствованием домен¬
ного процесса (снижение УРК за счет использования газооб¬
разного и жидкого топлива), повышением степени регенера¬
ции черных металлов и развитием метода внедоменного про¬
изводства стали, доля каменного угля, используемого для про¬
изводства кокса в РФ как и во всем мире непрерывно снижа¬
ется.8.2.4. Ископаемые угли как химическое сырьеЗначительная часть ископаемых углей (8.2.1) подвергается
высокотемпературной (пирогенетической) переработке, то есть
является химическим сырьем. Цель такой переработки — по¬
лучение из угля ценных вторичных продуктов, используемых159
в качестве топлива и полупродуктов основного органического
синтеза.Все методы переработки ископаемых углей основаны на ге¬
терогенных, в большинстве случаев некаталитических, процес¬
сах, протекающих в многофазной системе при высоких темпе¬
ратурах. В этих условиях материал угля претерпевает глубо¬
кие изменения, приводящие к образованию новых твердых,
жидких и газообразных продуктов. По назначению и условиям
процессы пирогенетической переработки твердого топлива под¬
разделяются на три типа: пиролиз, газификация и гидрирова¬
ние.Пиролизом или сухой перегонкой называется процесс нагре¬
вания твердого топлива без доступа воздуха с целью получения
из него твердых, жидких и газообразных продуктов различно-
го назначения. В зависимости от условий процесса и природы
вторичных продуктов различают низкотемпературный пиро¬
лиз или полукоксование и высокотемпературный пиролиз или
коксование. По масштабам производства, объему и разнообра¬
зию производимой продукции процесс коксования занимает
первое место среди всех процессов переработки твердого топли¬
ва.Полукоксование проводят при 500—580°С с целью получе¬
ния искусственного жидкого и газообразного топлива транспор¬
табельного и более ценного, чем исходное твердое топливо. Про¬
дукты полукоксования — горючий газ, используемый в каче¬
стве топлива с высокой теплотой сгорания и сырья для органи¬
ческого синтеза, смола, служащая источником получения мо¬
торных топлив, растворителей и мономеров и полукокс, исполь¬
зуемый как местное топливо и добавка к шихте для коксова¬
ния. Сырьем для полукоксования служит низкосортные камен¬
ные угли с высоким содержанием золы, бурые угли и горючие
сланцы.Процессы гидрирования и газификации ставят целью полу¬
чение из твердого топлива, соответственно, жидких продуктов,
используемых в качестве моторного топлива, и горючих газов.
Внедрение этих методов переработки повышает значение твер¬
дых топлив и каменных углей в частности в топливном балансе
страны.8.3. Коксование каменного угляКоксованием называется разновидность сухой перегонки
(пиролиза) каменного угля, проводимая при 900—1200°С с це¬160
лью получения кокса, горючих газов и сырья для химической
промышленности.8.3.1. Общая схема коксохимического производстваСовременное коксохимическое предприятие — это крупно¬
масштабное комплексное производство, в котором утилизиру¬
ются и перерабатываются все компоненты коксуемого сырья.
Существует два типа коксохимических предприятий:— заводы с полным циклом коксохимического производства,
размещаемые отдельно от металлургических предприятий и— коксохимические цеха (производства), входящие в состав
металлургических комбинатов, и размещаемые на одной пло¬
щадке с ними.Металлургический кокс составляет важнейший компонент
сырья в доменном процессе и транспортировка его экономичес¬
ки невыгодна. Кроме того, коксохимические заводы часто коо¬
перируют с производствами аммиака и азотной кислоты, основ¬
ного органического синтеза, красителей, взрывчатых веществ
и ракетных топлив, пластических масс, в которых в качестве
сырья используются продукты коксохимии.В соответствии с назначением все цехи коксохимического
завода подразделяются на основные и вспомогательные. К ос¬
новным производственным цехам относятся:1) Углеподготовительный цех, где осуществляется прием,
хранение и подготовка углей к коксованию. Готовая продукция
цеха — угольная шихта.2) Коксовый цех, в котором происходит основной процесс —
переработка угольной шихты с получением целевого продукта
кокса и летучих химических продуктов — прямого коксового
газа (ПКГ) — коксование.3) Цех улавливания, в котором происходит охлаждение пря¬
мого коксового газа и выделение из него химических продук¬
тов: сырого бензола (СБ), каменноугольной смолы (КУС) и со¬
единений аммиака.4) Перерабатывающие цехи (коксовый, смолоперегонный,
ректификации и другие), в которых химические продукты, по¬
ступающие из цеха улавливания, подвергаются дальнейшей пе¬
реработке. Готовой продукцией этих цехов являются индиви¬
дуальные ароматические углеводороды, нафталин, фталевый
ангидрид, фенолы и пиридиновые основания, пек, пековый кокс
и другие.К вспомогательным цехам относятся: железнодорожный, ре¬
монтный, энергетический, хозяйственный, ОТК, ЦЗЛ и другие.6 Химическая технология. Том 2161
углеводороды (NH4)2S04Рис. 8.1. Общая схема коксохимического производстваНа рис. 8.1 представлена общая схема коксохимического
производства.8.3.2. Сырье коксохимического производстваОсновной продукт коксохимического производства — искус¬
ственное твердое топливо, кокс, выход которого составляет до
75% от массы коксуемого сырья. Кокс необходим в черной и
цветной металлургии (металлургический кокс), литейном про¬
изводстве и химической промышленности. Около 80% произ¬
водимого в стране кокса используется в доменном производстве,
поэтому к металлургическому коксу предъявляются определен¬
ные требования по прочности, однородности гранулометричес¬
кого состава, зольности, содержанию серы и др. Обеспечить эти
требования можно только при использовании сырья с опреде¬
ленными свойствами. Важнейшим из этих свойств является
спекаемостпь — способность угля при нагревании без доступа
воздуха образовывать из разрозненных зерен твердый остаток
в виде прочных кусков. Этим свойством обладают угли марок
«Г», «Ж», «К» и «ОС». Однако из этих марок углей образовы¬
вать металлургический кокс способны только угли марки «кок¬
совые».162
Коксуемость углей зависит от их петрографического соста¬
ва, степени углефикации, выхода летучих веществ, температур¬
ного интервала перехода в пластическое состояние, степени вяз¬
кости в этом состоянии, динамики газовыделения, а также тех¬
нологии подготовки угольной шихты и режима коксования.Ограниченные запасы коксующихся углей привели к необ¬
ходимости использовать в качестве сырья коксохимического
производства смеси углей различных марок, взятых в опреде¬
ленном соотношении. Состав подобной шихты должен обеспе¬
чивать образование кокса с заданными техническими характе¬
ристиками, необходимую полноту спекания при коксовании,
надлежащий выход газа и химических продуктов коксования.Состав шихты рассчитывается на основании свойств ее ком¬
понентов по результатам технического анализа и других испы¬
таний их по правилу аддитивности:Пш = ПаЦа + Пв|1в + ... + ПпДл (8.1)где: Пш — рассчитываемый показатель качества шихты,Па, Пв ... Пп — показатель качества компонентов шихты,
ца> Цв Цл — массовая доля компонентов в шихте.Технологический процесс составления угольной шихты (уг-
леподготовка) осуществляется в специальном углеподготови¬
тельном цехе и включает следующие операции:— прием и разгрузка углей,— складирование, усреднение состава и хранение углей,— обогащение углей,— дозирование компонентов шихты,— измельчение шихты (или, ранее, ее компонентов),— составление шихты (шихтовка).Углеподготовительный цех — важнейший цех коксохими¬
ческого завода. От ритмичности его работы зависит нормаль¬
ный режим работы всего предприятия. Цех располагается па¬
раллельно блоку коксовых печей на расстоянии, обеспечиваю¬
щем транспортировку угля конвеерной лентой или автомати¬
зированным железно-дорожным транспортом.Усреднение состава угольной шихты ставит целью вырав¬
нивание качества углей внутри каждой группы их и проводит¬
ся на складе в процессе разгрузки и укладки штабелей. Усред¬
ненными считаются угли, у которых все показатели качества
разовых проб соответствуют среднему показателю за все время
отбора проб.Обогащение углей для понижения содержания в них мине¬
ральных примесей проводится методами отсадки, сепарации ифлотации. Отсадкой называется процесс разделения смеси ком-
в*163
понентов по их плотности в турбулентном водном потоке, ко¬
леблющемся за счет пульсирующего тока воздуха в вертикаль¬
ном направлении с определенной амплитудой и частотой. Этим
методом обогащается до 50% углей.Обогащение методом сепарации основано на разделении
компонентов угля по плотности в тяжелых средах, в которых
более легкий уголь всплывает. В качестве тяжелых сред ис¬
пользуются стойкие минеральные суспензии пирита, барита
и магнезита.Методом флотации в настоящее время обогащается около
15% углей. В большинстве случаев для этого используются фло¬
тационные машины механического типа, в которых в качестве
реагентов-собирателей применяются керосин, камфарное мас¬
ло, флотореагент АФ-2 .Флотированный уголь подвергается за¬
тем обезвоживанию и сушке в барабанных сушилках или «КС*.Дозирование компонентов имеет большое значение для пос¬
ледующего составления угольной шихты заданного состава. Для
этой цели угли шихты из бункеров с помощью дозаторов раз¬
личной конструкции поступают на транспортер, которым по¬
даются на окончательное измельчение. В качестве дозирующих
устройств используются качающиеся, ленточные и тарельчатые
питатели производительностью до 200 т в час.Измельчение коксуемого сырья проводится для повышения
однородности шихты, что способствует улучшению качества
кокса. Так как насыпная масса шихты зависит от ее измельче¬
ния, что в свою очередь, определяет экономические показатели
работы углеподготовительного и коксового цехов, то для шихт
различного состава выбирают некоторую оптимальную степень
измельчения. При этом, для обеспечения возможно более вы¬
сокой плотности загрузки, выдерживают определенное соотно¬
шение частиц различного размера в шихте. Для измельчения
углей используют дробилки различного типа: молотковые, ро¬
торные, ударного действия, инерционно-роторные и другие.
Окончательное измельчение сырья для коксования может про¬
водиться по двум схемам: по схеме ДШ, при которой измельча¬
ется вся масса шихты, и по более совершенной дифференциро¬
ванной схеме ДК, учитывающей различную твердость измель¬
чаемого материала, при которой каждый компонент шихты из¬
мельчается отдельно. Эти схемы представлены на рис. 8.2.Шихтовка или смешение компонентов - это заключитель¬
ная операция приготовления угольной шихты для коксования.
Шихтовка осуществляется в смесительных машинах различной
конструкции: дезинтеграторных, валковых, тарельчатых и в164
жОСшшУБРис. 8.2. Схемы измельчения коксового сырья:
а — обычная ДШ, б — дифференцированная ДКГ, Ж, К, ОС — марки углей для шихты, Д — дробление, С — смешение,УБ — угольная башня (склад измельченного сырья)машинах барабанного типа производительностью до 1200 т
шихты в час.О1ОВода8.3.3. Физико-химические основы процесса коксованияКоксование — это сложный двухфазный эндотермический
процесс, в котором протекают термофизические превращения
коксуемого сырья и химические реакции с участием компонен¬
тов его органической части. Коксование проводят в коксовых
печах, являющихся реакторами пе¬
риодического действия с косвенным
нагревом, в которых теплота пере¬
дается к коксуемой угольной ших¬
те через стенку реактора. Поэтому
термофизические процессы при
коксовании включает:— теплопередачу от стенки к ма¬
териалу шихты,— диффузию продуктов пироли¬
за (паров воды и летучих веществ)
через слой шихты,— удаление этих продуктов из
шихты (рис. 8.3).«к-%ФГ"-*ЛетучиевеществаРис. 8.3. Схема термофизи¬
ческих процессов при коксо¬
вании шихты:/ — стенка, 2 — шихта165
При установившемся режиме процесса коксования количе¬
ство теплоты, передаваемое за единицу времени, определится
из уравнения:Q = KTFAt (8.2)где: Кт — коэффициент теплопередачи, кДж/м2-град* ч,F — поверхность теплопередачи, м2,At = tT - tm — разность температур обогревающих стен¬
ку камеры печи газов tr и нагреваемой шихты (тем¬
пературы коксования)Коэффициент теплопередачи рассчитывается по формуле: 1 т 1/а1+51/А.14- 52/А-2+1/а2 ^ ^где: ах и а 2 — коэффициенты теплопередачи, соответственно,
от греющих газов к стенке печи и от стенки к ших¬
те, кДж/м2-град* ч,81 — толщина стенки, м,82 = Ь/2 — половина толщины загрузки шихты, м,и \2 — соответственно, коэффициенты теплопроводнос¬
ти стенки и шихты, кДж/м2трад-ч
Для увеличения теплового потока и, как следствие, интен¬
сивности обогрева печи, необходимо повышать коэффициент
теплопередачи Кг и поверхность обогрева F. Так как коэффи¬
циент теплопередачи угольной шихты весьма мал, то из фор¬
мулы 8.2 следует, что для увеличения коэффициента теплопе¬
редачи Кт помимо повышения ai иаг, необходимо уменьшать
толщину слоя угольной шихты. Поэтому ширина камеры печи
достаточно жестко регламентирована и составляет, обычно,
0,40—0,41 м. Из этого следует, что поверхность теплопередачи
F определяется двумя другими размерами камеры печи — дли¬
ной и высотой.Коксовая печь — реактор периодического действия, поэто¬
му температура угольной шихты в ней изменяется во времени.
Следовательно, изменяется и движущая сила процесса, то есть
разность температур между греющими газами и угольной ших¬
той At = tr - tm . Непосредственно после загрузки шихты fш мала
и разность At велика. Поэтому в холодную шихту поступает в
единицу времени большее количество теплоты и уголь у стенок
камеры начинает коксоваться, в то время как вследствие низ¬
кой теплопроводности шихты средние слои остаются холодны¬
ми. По мере прогрева шихты ее температура возрастает и дви¬
жущая сила процесса At падает при одновременном повышении
температуры по сечению камеры.166
Распределение температур в шихте по ширине камеры для
различного времени коксования выражается так называемыми
изохроническими кривыми коксовой печи (кривые постоянно¬
го времени). На рис. 8.4 представлены изохронические кривые
печи (а) и состояние коксуемого материала в ней (б).С течением времени (тх^хз) температура по сечению камеры
выравнивается, слой образующегося кокса перемещается к цен¬
тру камеры и по достижении определенного времени угольная
шихта полностью закоксовывается.Химические превращения при коксовании могут быть све¬
дены к реакциям двух типов: первичным и вторичным.К первичным реакциям, протекающим в шихте при ее на¬
гревании , относятся:— реакции деструкции сложных молекул,— реакции фенолизации,— реакции карбонизации органической части угля,— реакции отщепления атомов водорода, гидроксильных,
карбоксильной и метоксильной ОСНз групп.В процессе первичных превращений из угольной шихты вы¬
деляются первичный газ и пары первичной смолы и образуется
кокс. К вторичным реакциям, которые протекают при контак¬
те выделившихся первичного газа и первичной смолы с нагре¬
той стенкой печи, относятся:— реакции крекинга алканов5 4 3 2 Лx3>t2>x1Рис. 8.4. Изохронические кривые коксовой печи (а) и состояние
коксуемой шихты в печи (6):1 — стенка камеры, 2 — кокс, 3 — полукокс, 4 — уголь в пластическом
состоянии, 5 — угольная шихта167
СяН2я+2 ~* CmH2m+2 + СрН2р,— реакции полимеризации алкенов:ЗСпН2л ЛС„Н2„,— реакции дегидрогенизации нафтенов:ЛС„Н2„ - СяН2п-6 + ЗН2,— реакции конденсации ароматических углеводородов, на¬
пример:— реакции образования карбенов с последующим превраще¬
нием их в полукокс и кокс.Продуктом вторичных превращений является сложная смесь
газообразных и парообразных при температуре коксования ве¬
ществ различной природы — прямой коксовый газ (ПКГ). На
рис. 8.5 представлена схема химических превращений при кок¬
совании.Рис. 8.5. Схема химических превращений при коксованииПоследовательность процессов, протекающих в шихте при
повышении температуры в печи может быть представлена в сле¬
дующем виде:250°С Отщепление НгО, СО, СО2, Н2300°С Начало выделения КУС, выделение пирогенетичес-
кой воды350—500°С Пластификация угольной шихты
500—550°С Разложение органической части угля с выделе¬
нием первичного газа и паров первичной смолы, спекание
твердого остатка с образованием полукокса600—700°С Разложение полукокса и полное выделение ле¬
тучих веществ700°С Упрочнение твердой массы и образование кокса.2СбНб СюНв + С2Н4,КаменноугольнаяшихтаКоксПервичный газ168
8.3.4. Технологический процесс коксованияПроцесс коксования осуществляется в коксовых печах — ре¬
акторах периодического действия. Современная коксовая печь
представляет сложное теплотехническое сооружение, состоя¬
щее из:— камеры для загрузки угольной шихты,— обогревательного простенка, в котором расположены 28—
32 отопительных каналов (вертикалов), где происходит горение
нагретого газообразного топлива для обогрева стенок камеры,— системы газораспределительных и воздухоподводящих
каналов для подачи газа и воздуха для отопления печи,— регенераторов для подогрева газообразного топлива и возду¬
ха, подаваемых в печь и для отвода продуктов горения топлива,— системы отвода летучих продуктов коксования.Для снижения тепловых потерь на излучение, удобства экс¬
плуатации и повышения производительности труда коксовые
печи объединяют в батареи, состоящие из п камер и n + 1 про¬
стенков. Число печей в батарее определяется конкретными ус¬
ловиями производства, главным образом, возможностью раци¬
онального использования машин общего назначения и равно
обычно 68—78.Камера коксовой печи по конфигурации представляет паралле¬
лепипед, размеры которого зависят от ряда факторов. Ширина ка¬
меры определяется толщиной слоя коксуемой шихты (8.3.3), вы¬
соту и длину выбирают исходя из обеспечения равномерности обо¬
грева камеры, качества шихты, размеров территории цеха и др.
Камеры современных печей имеют длину 14—16 м и высоту 4,3—7,0 м. На рис. 8.6 приведена схема коксовой печи.Угольная шихтасмесь (ПКГ)Парогазовая5Парогазовая
смесь <ПКГ)_Горячие газы, обогревающие камеруКоксРис. 8.6. Схема коксовой печи:1 — бункера для загрузки шихты, 2 — стояк для отвода летучих продук¬
тов, 3 — передняя дверца, 4 — задняя дверца, 5 — коксовыталкиватель169
В верхнем перекрытии камеры есть загрузочные отверстия
для подачи шихты и отверстия для отвода летучих продуктов
коксования (прямого коксового газа), которые через газоотвод
поступают в газосборник, откуда направляются в цех улавли¬
вания. С торцов камера закрывается дверями, которые снима¬
ются по окончании коксования для выдачи готового кокса из
камеры с помощью коксовыталкивателя.Конструкция коксовой камеры полностью обеспечивает ее
герметичность и исключает подсос наружного воздуха и отопи¬
тельных газов. Каждая печь имеет по два регенератора, распо¬
ложенных под камерой. Газообразное топливо подается в каж¬
дый простенок батареи через распределительный газопровод.
Батарея коксовых печей обслуживается единым комплексом
механизмов для загрузки угольной шихты и выгрузки готово¬
го кокса, в котором входят углезагрузочный вагон, коксовытал¬
киватель, разравнивающая шихту штанга, машина для съема
дверей камеры и коксотушильный вагон.В коксохимическом производстве применяются печи, разли¬
чающиеся конструктивными особенностями (расположение
камер, способ подвода газа и воздуха, способ утилизации теп¬
лоты продуктов горения топлива и др.) и технологическим ре¬
жимом (последовательность подъема температуры, состав обо¬
гревающего газа и др.). Однако все они могут быть сведены к
двум типам: печи с перекидными каналами (ПК) и печи с ре¬
циркуляцией продуктов горения (ПВР).В печах ПВР для улучшения равномерности обогрева по дли¬
не и высоте камер в вертикалах, осуществляется рециркуляция
продуктов горения путем подачи части их в пламя горящего
газа, что замедляет процесс его горения и удлиняет факел пла¬
мени. Печи этого типа являются наиболее распространенными.
В табл. 8.4 приведены характеристики печей ПВР, наиболее
распространенных в РФ.Таблица 8.4. Характеристики коксовых печей типа ПВРV„m3Размеры,мтШ1 т*к, 4Пк, т/годW, мм/чЬh/21,60,4074,314,0823,31672927,332,30,4105,516,0023,514730 41,60,4107,016,0030,614100032,0Vu — полезный объем камеры, b — ширина камеры, h — вы¬
сота камеры, I — длина камеры, тш — масса загружаемой в170
камеру шихты с влажностью 0,085 мае. дол., тк — время кок¬
сования, Пк — производительность по коксу с влажностью0,06 мае. дол., W — скорость коксования.Процесс коксования состоит из следующих стадий1. Загрузка шихты в камеру печи и разравнивание шихты
штангой (планиром). Во избежания задымления атмосферы в
камере в период загрузки шихты создается разряжение путем
инжекции пара, газа или аммиачной воды или с помощью сис¬
темы отсоса газа из камеры.2. Коксование. Производительность коксовой печи опреде¬
ляется так называемым периодом коксования — временем от
окончания загрузки камеры до выдачи кокса, в течение кото¬
рого в шихте происходят все изменения, приводящие к обра¬
зованию кокса и ПКГ. Период коксования хк зависит от ши¬
рины камеры, то есть толщины слоя шихты, толщины клад¬
ки и материала огнеупоров стенового канала, свойств уголь¬
ной шихты и температуры в вертикалах печи. С достаточной
степенью точности период коксования определяется по фор¬
муле:Ы“-*тт«=мГ <8-4->где: а — коэффициент температуропроводности, м2/ч, а значе¬
ние величин МшА приведены для формул 8.2 и 8.3.Приняв для расчета практические значения b = 0,4 м, tm =
=1100°С, tr = 1400°С, получим тк = 13 часов, что согласуется с
реальным режимом процесса коксования.Период коксования с добавкой времени на операции загруз¬
ки шихты и выгрузки кокса (9—10 минут) называется време¬
нем оборота или оборотом печи. Оборот печи сокращается при
повышении температуры в вертикалах, уменьшением толщи¬
ны стенового кирпича и снижении влажности шихты, а также
при улучшении организации работ по обслуживанию коксовой
батареи.Газообразным топливом для обогрева коксовых печей слу¬
жит обратный коксовый газ, доменный газ, их смеси и, зна¬
чительно реже, смесь доменного и природного газов. Темпе¬
ратура продуктов сгорания топлива, следовательно, темпера¬
тура газов, обогревающих стенки камеры *г, определяется как
отношение количества поступающего тепла к средней тепло¬
емкости:171
QhP + Qt 9дис Qoctr= (8.5)где: QHP — низшая теплота сгорания газообразного топливакДж/м3, равная для ОКГ 18500, для доменного
газа 3800,QT — теплосодержание вводимых топлива и воздуха,
кДж/м3,Ядис — теплота диссоциации продуктов горения, кДж/м3,
Qoc — потери теплоты в окружающую среду, кДж/м3,
Сг — средняя теплоемкость продуктов горения, кДж/м^К.Удельный расход тепла на коксование может быть рассчи¬
тан как:V Qp(8.6)тгде: q — расход теплоты, кДж/кг, коксуемого угля (шихты),
VT — расход газообразного топлива, м3/ч,
т — масса коксуемого угля, кг/ч,QHP — низшая теплота сгорания топлива, кДж/м3.Технологический режим работы коксовых печей во все вре¬
мя коксования регулируется автоматически. При этом парамет¬
ры процесса: температура в вертикалах, разряжение в регене¬
раторах и коэффициент избытка воздуха, подаваемого в печь,
постоянно поддерживается на среднем заданном уровне.3. Выгрузка кокса (выдача коксового «пирога» ) с помощью
коксовыталкивателя в тушильный вагон. Режим загрузки ших¬
ты в печи коксовой батареи и выгрузки кокса из них подчиня¬
ется определенным правилам. Эти операции должны проводить¬
ся в строгой последовательности, которая называется серийно¬
стью выдачи кокса. Серийность выдачи обеспечивает сохран¬
ность кладки печей и одинаковые температурные условия в
простенках по длине батареи.Расчет показывает, что при периоде коксования 13—16 ча¬
сов и числе печей в коксовой батарее 68—78, выдача кокса сле¬
дует каждые 10—12 минут. Поэтому, коксовую батарею в це¬
лом можно рассматривать как реактор непрерывного действия
РИВ-Н, хотя каждая отдельная печь в ней работает периоди¬
чески.Производительность коксовой батареи при установившемся
режиме работы и постоянном качестве угольной шихты зави¬
сит от периода коксования и рассчитывается по формуле:172
П=-пУп'5обtl-w)(8.7)где: П — производительность батареи, т/ч,Vu — полезный объем камеры, м3,5 — насыпная масса (плотность) шихты, в пересчете на
сухую шихту, т/м3,Хоб ~ время оборота печи, ч
W — влажность кокса, мае. дол.4. Тушение кокса. Кокс, выгружаемый из печи в коксоту¬
шильный вагон, имеет температуру 950—1100°С. Чтобы предот¬
вратить его горение на воздухе и обеспечить возможность транс¬
портировки до склада и хранение, кокс должен быть охлажден
до температуры 250—100°С, при которой исключается его са¬
мовозгорание. Для этого раскаленный кокс интенсивно охлаж¬
дают (тушат) мокрым или сухим методом.При мокром тушении вагон с коксом интенсивно орошается
в тушильной камере водой. Расход воды на тушение составляет
4—5 м3/т кокса. Недостаток мокрого метода тушения — значи¬
тельная потеря тепла, так как все тепло кокса, поглощаемое
водой, идет на ее испарение и не утилизируется. С парами воды
теряется до 50% тепла, затраченного на коксование.При сухом тушении раскаленный кокс охлаждается цирку¬
лирующими инертными газами, теплосодержание которых ис¬
пользуется затем в котле-утилизаторе (рис. 8.7). В качестве
инертных газов используются топочные газы (С02 + N2), обра¬
зующиеся при пуске установки тушения в результате продув¬
ки воздухом первой порции раскаленного кокса. Преимуще¬
ствами сухого тушения является:Нагретые газыРис. 8.7. Схема сухого тушения:1 - тушильная камера, 2 — вагон с коксом, 3 — котел-утилизатор173
— отсутствие выбросов пара и сточных вод,— получение кокса с минимальной влажностью,— утилизация теплоты кокса и выработка технологическо¬
го пара.5. Сортировка кокса. Кокс после тушения сортируется по
классам крупности на грохотах различной конструкции. Для
доменного производства применяется кокс класса более 40 мм,
в цветной металлургии кокс класса 10—25 мм, для производ¬
ства карбида кальция кокс класса 25—40 мм. Коксовая мелочь
используется в процессе агломерации железных руд.8.4. Улавливание и разделение летучих продуктов
коксования8.4.1. Состав и выход летучих продуктовЛетучие продукты, выделяющиеся при коксовании и обра¬
зующие прямой коксовый газ (ПКГ) составляют до 15% от мас¬
сы коксуемой шихты, или около 300 нм3 на тонну шихты. В
состав ПКГ входят пирогенетическая вода, смесь высококипя-
щих многоядерных и гетероциклических соединений — камен¬
ноугольная смола (КУС), ароматические углеводороды ряда бен¬
зола, нафталин, аммиак, соединения циана, сернистые соеди¬
нения и, образующие после их отделения обратный коксовый
газ (ОКГ), водород, метан, оксиды углерода (II) и (IV) и газооб¬
разные углеводороды различной природы. В ПКГ содержатся
также в незначительных количествах сероуглерод CS2, серок -
сид углерода COS, тиофен C4H4S и его гомологи, пиридин C5H5N
и пиридиновые основания.В табл. 8.5 приведено содержание основных компонентов в
ПКГ.Таблица 8.5. Основные компоненты ПКГВеществоСодержание, г/м3Пары воды (пирогенетической и влаги шихты)250—450Каменноугольная смола (пары)80—150Ароматические углеводороды30—40Аммиак8—13Нафталиндо 10Сероводород6—40Цианистый водород0,5—2,5174
В цехе улавливания и разделения из ПКГ извлекаются ос¬
новные компоненты не в виде индивидуальных химических
соединений, а в виде их смесей: каменноугольной смолы (КУС)
и сырого бензола (СБ). Все соединения аммиака и свободный
аммиак перерабатываются при этом в сульфат аммония:Q H2S04 АбсорбентПКГ I i— | ►окгКУС (NH4)2S04 сбВыход продуктов коксования зависит от степени углефика-
ции, насыпной плотности, выхода летучих и влажности уголь¬
ной шихты, конструкции печей, режима коксования (темпера¬
туры) и других факторов. В частности выход КУС и СБ выше
для углей с большим выходом летучих веществ, то есть марок
«Г» и «Ж», как это показано на рис. 8.8. Этим, помимо каче¬
ства кокса, объясняется использование при составлении уголь¬
ной шихты углей именно этих марок.8.4.2. Основные процессы и принципиальная схема
разделения ПКГЛетучие продукты коксования, составляющие ПКГ, имеют
различные физические и химические свойства, что использу¬
ются для их разделения. В технологии улавливания и разделе¬
ния ПКГ используются:— процессы теплообмена (охлаждение и конденсация паров),— процессы разделения фаз (от¬
стаивание и осветление),— процессы массопередачи (аб¬
сорбция и десорбция, хемосорбция
реагентами, реагирующими с кис¬
лыми и основными продуктами),— процессы ректификации и
фракционной конденсации.Большинство этих процессов в
коксохимическом производстве яв¬
ляется непрерывными, что повы¬
шает производительность аппарату¬
ры , улучшает качество выделяемых
из ПКГ продуктов и позволяет авто¬
матизировать технологические про¬
цессы .Выход, VoРис. 8.8. Выход продуктов
коксования (% к сухой шихте)
для различных углей:/ — ОКГ, 2 — КУС, 3 — СБ175
ПКГ из коксовых камер при температуре 650—670°С посту¬
пает в газосборники коксовой батареи, где усредняется по со¬
ставу и охлаждается впрыскиванием холодной надсмольной
воды до 85—90°С. После этого газ направляется в цех улавлива¬
ния и разделения, в котором после дополнительного охлажде¬
ния до 25—35°С из него выделяются КУС, СБ и соединения ам¬
миака. Последовательность этих операций представлена на схе¬
мах (рис. 8.9 а, б, в, г).-ОпкгtхолоднаяНСВ-► Газ-280°Счасть КУС* НСВРис. 8.9 а. Охлаждение (I стадия)На этой стадии конденсируется часть КУС и собирающаяся
над ней надсмольная вода НСВ (отсюда—ее название). Увлечен¬
ная током газа каменноугольная пыль в смеси с КУС оседает в
виде фусов.Газ-2-ОГаз-330°IГаз-4 (NH3)► КУС <4-Туман КУС*НСВ (NH3)Рис. 8.9 6. Охлаждение (II стадия) и выделение КУСНа этой стадии в холодильниках, орошаемых НСВ конден¬
сируется основная масса КУС, к которой добавляется КУС, осе¬
дающая из ее тумана в электрофильтрах, и НСВ, содержащая
до 30% аммиака в виде его солей. Остальной аммиак (до 70%)
остается в газе.Рис. 8.9 в. Выделение аммиака176
Аммиак после охлаждения ПКГ содержится в свободном со¬
стоянии в газе-4 и в виде растворенных солей аммония в НСВ,
образовавшихся при взаимодействии аммиака с сероводородом,
оксидом углерода (IV), хлористым водородом, цианистым во¬
дородом и другими кислыми компонентами ПКГ. Эти соли по
отношению к нагреванию делятся на две группы:— стойкие при высокой температуре NH4CN, NH4CI.(NH4)2S04 и— разлагающиеся при нагревании (NH)4S, (NH4)2C03,Если технологией не предусмотрено производство из ПКГиндивидуальных сульфида аммония, цианистого водорода и
роданистых солей, то все соединения аммиака, содержащиеся
в ПКГ, переводятся в стабильный, легко выделяемый сульфат
аммония. Для этого нестойкие соли аммония, содержащиеся в
НСВ разлагают нагреванием острым паром, например:(NH4)2C03 = 2NH3 + С02 + Н20 ,
а стойкие обработкой гидроксидом кальция, например:2NH4C1 + Са(ОН)2 = 2NH3 + СаС12 + 2Н20Выделившийся свободный аммиак соединяется с содержа¬
щим аммиак газом-4 и поступает в сульфатное отделение цеха,
где поглощается серной кислотой с образованием сульфата и
бисульфата аммония:2NH3 + H2S04 = (NH4)2S04; NH3 + H2S04 = NH4HS04rОКГГаз-5-► Газ-6H t'10H8 ■“ПМРастворСБвПМ I*СБРис» 8.9 г. Выделение сырого бензолаСБ, представляющий смесь ароматических углеводородов с
температурой кипения до 180°С, извлекается из ПКГ абсорбци¬
ей высококипящими растворителями с температурой кипения
более высокой, чем температура кипения СБ с последующей
отгонкой последнего. В качестве подобных сорбентов (ПМ) ис¬
пользуются соляровое масло (tK = 300—350‘С) или фракция КУС
(tK = 230—300°С). Предварительно из газа охлаждением выде¬
ляют остатки нафталина.Конечными продуктами на стадии улавливания и разделе¬
ния ПКГ становятся каменноугольная смола, сырой бензол,177
сульфат аммония и обратный коксовый газ. Выход этих про¬
дуктов от массы коксуемой шихты (в расчете на сухую шихту)
представлен в табл. 8.6.Таблица 8.6. Выход продуктов коксованияПродуктВыход, мае. долейКокс0,77—0,73Каменноугольная смола0,03—0,04Сырой бензол0.01—0,012Сульфат аммония0,01—0,013Обратный коксовый газ0,15—0,188.4.3. Технологическая схема улавливания и разделения ПКГТехнологическая схема улавливания и разделения прямого
коксового газа представлена на рис. 8.10.Отсасываемый газодувками из коксовых камер, ПКГ охлаж¬
дается в газосборнике 1, орошаемом холодной НСВ, и поступа¬
ет в сепаратор 2, в котором из газа конденсируются КУС, НСВ и
выделяются твердые частицы—фусы. Образовавшаяся смесь
этих продуктов разделяется в отстойнике-осветлителе 3. Газ,
пройдя сепаратор, охлаждается до 25—30°С в трубчатом холо¬
дильнике 4, орошаемом НСВ, где из него конденсируются ос¬пкгокг■Ч/*ФусыСа(ОН)2 (NHJgSO,СБ+ПМСБРис. 8.10. Технологическая схема улавливания и разделения ПКГ:1 — газосборник, 2 — сепаратор, 3 — отстойник-осветлитель, 4 —
трубчатый холодильник, 5 — электрофильтр, 6 — аммиачная колонна, 7 —
турбогазодувка, 8 — подогреватель газа, 9 — сатуратор, 10 — водяной
холодильник, 11 — абсорбер178
татки КУС и НСВ, которые поступают, соответственно, в отстой¬
ник 3 и сепаратор 2. НСВ из отстойника подается в аммиачную
колонну 6, в которую вводится раствор гидроксида кальция и
подается острый пар для разложения аммонийных солей.Газ после холодильника 4 освобождается от тумана КУС в
электрофильтре 5 и соединяется с током газообразного аммиа¬
ка из аммиачной колонны. Общий поток газа подается турбога-
зодувкой 7 через подогреватель 8 в сатуратор 9, барботирует
через раствор серной кислоты. Выпавшие в сатураторе кристал¬
лы сульфата аммония, отделяются, а газ, после охлаждения в
водяном холодильнике прямого смешения 10, направляется в
абсорбер с насадкой 11, который орошается циркулирующим
поглотительным маслом. В абсорбере из газа извлекается СБ и
раствор его в поглотительном масле (ПМ) направляется на рек¬
тификацию. СБ отгоняется из раствора, а регенерированное ПМ
возвращается на абсорбцию. В холодильнике 10 из газа выде¬
ляется твердый нафталин, который экстрагируется из водной
суспензии горячей КУС, подаваемой в нижнюю часть холодиль¬
ника. Из абсорбера 11 выходит обратный коксовый газ (ОКГ).8.5. Переработка продуктов коксования8.5.1. Переработка сырого бензолаСырой бензол — это сложная смесь соединений, основными
компонентами которой являются бензол и его гомологи. Кроме
них в СБ содержатся в небольших количествах непредельные и
сернистые соединения. В табл. 8.7 приведен средний состав
сырого бензола.Таблица 8.7. Состав сырого бензолаКласс соединенийВажнейшие представителиСодержа¬
ние, % мае.Ароматические угле¬
водородыНепредельные соеди¬
ненияСернистые соединения
Алканы и циклоалканыБензол, толуол, ксилолы,
этилбензол, триметилбензол
Стирол, циклопентен, цикло-
гексен, инден, кумарон
Сероводород, сероуглерод,
тиофен, метилтиофен, диме-
тилтиофенЦиклопентан, метилциклопен-
тан, циклогексан, гексан, гептан80—95
5—15
0,2—2,00,3—2,0179
Цель переработки СБ—получение индивидуальных арома¬
тических углеводородов путем разделения смеси составляющих
СБ веществ на отдельные компоненты. Для этого используют
методы ректификации и раздельной кристаллизации.Процесс переработки СБ включает следующие операции:— отделение легкокипящей сероуглеродной фракции и по¬
лучение бензольно-толуольно-ксилольной фракции (ВТК),— очистка фракции ВТК,— ректификация очищенной фракции ВТК и получение ин¬
дивидуальных ароматических углеводородов.Важнейшая операция при переработке СБ — это очистка
фракции ВТК. Она проводится для удаления из фракции непре¬
дельных и сернистых соединений, которые могут вызвать не¬
стабильность полученных продуктов и отравление катализато¬
ра, если они используются в качестве сырья для последующей
переработки. Для очистки фракции БТК используют сернокис¬
лотный метод и метод каталитической гидроочистки.При сернокислотной очистке протекают процессы полиме¬
ризации непредельных соединений с образованием смол, отде¬
ляемых от фракции при ректификации:н+7ПСлН2л * (СлН2/г)/п»реакции образования растворимых в воде эфиров серной кис¬
лоты:R-CH=CH2 + H2S04 — R-CH-CH3bso3Hи реакции сульфирования теофена и его гомологов до сульфо¬
кислот, также растворимых в воде:Qs + H2SO4 — Qs + н2оso3hПри использовании серной кислоты концентрацией 92—94%
сульфирование ароматических углеводородов практически не
происходит.При гидроочистке с использованием алюмо-молибден-ко-
бальтового катализатора протекают реакции гидрирования не¬
предельных углеводородов:С6Н5-СН=СН2 + Н2 - С6Н5-СН2-СН3,
азотсодержащих и кислородсодержащих соединений:CH3CN + ЗН2 — С2Н6 + NH3;180
C5H5N + 5H2 — C5H12 + NH3C6H5OH 4 H2 —*■ C6H6 + h2o
и сернистых соединений:CS2 + 4H2 — 2H2S + CH4;R-SH + H2 -» H2S + RHQS + 4H2 — H2S + C4H10Ректификация фракции ВТК, а также первоначальная отгон¬
ка головной сероуглеродной фракции от СБ проводятся в колон¬
нах барботажного типа с колпачковыми или ситчатыми тарел¬
ками. При отгонке сероуглеродной фракции колонна обогрева¬
ется глухим паром, при ректификации фракции ВТК в колонну
для понижения температуры кипения вводится острый пар.
Продуктами ректификации СБ являются бензол, толуол и техни¬
ческий ксилол, состоящий из изомеров диметилбензола и этил бен¬
зол а. Для разделения изомеров ксилола используют метод сверх-
четкой ректификации в колоннах с 320 тарелками, а для отделе¬
ния метаизомера от параизомера с температурами кипения 139,1
и 138,4°С, соответственно,—метод кристаллизации.На рис. 8.11 представлена принципиальная схема переработ¬
ки сырого бензола.СБh2so4-► БТК-фракцияCS,■■ '| ►ПримесиNaOH+Кислыйдистиллят-► Очищенная ■
БТК-фракцияNa2S04в^6^6С6Н5-СН3о, п, М-С6Н4(СН3)2См-изомеро-С6Н4(СН3)2
м, п-С6Н4(СН3)2
£_ п- изомерРис. 8.11. Принципиальная схема переработки СБ8.5.2. Переработка каменноугольной смолыКаменноугольная смола (КУС) представляет сложную смесь
веществ, в состав которой входит несколько сот органических
соединений (идентифицировано около 500 соединений, состав¬181
ляющих 60% массы смолы) различных классов. Их можно под¬
разделить на:— нейтральные соединения, к которым относятся конден¬
сированные ароматические углеводороды (нафталин, антрацен,
фенантрен, хризен и др.) и гетероциклические соединения (ти-
офен, тионафтен, индол, карбазол и др.)>— соединения кислой природы, главным образом одно- и
многоатомные фенолы (фенол, крезолы, ксиленолы и др.),— соединения основной природы, к которым относятся пи¬
ридин и его гомологи (пиридиновые основания) и хинолин.В табл. 8.8 представлено содержание основных компонентов
в обезвоженной КУС.Таблица 8.8. Основные компоненты КУСВеществоСодерж.
% мае.ВеществоСодерж.
% мае.НафталинГомологи нафталинаФлуоренФенантренАнтраценКарбазолОксибензолКрезолы8—12
3,7
1,2-2,0
4—5
1,0—1,3
1,2-1,5
0(2—0,5
0f6—1,2ФлуорантенПиренХризенДифенилХинолин и гомологи
Аценафтен
Дифениленоксид
Пиридин и гомологи2.2—3,31.2-2,1
2,00,2—0,4
2,0—3,0
1,4-1,8
0,6—0,8
0,5—1,5Остальную часть КУС (50—60%) составляют высокомолеку¬
лярные вещества неопределенного состава.Процесс переработки КУС состоит из следующих операций:— обезвоживание, обессоливание и обеззоливание смолы,— ректификация с отбором фракций смолы,— переработка фракций и их очистка от фенолов и пириди¬
новых оснований,— получение чистых индивидуальных соединений.Ректификация КУС осуществляется в трубчатых установкахс использованием принципов однократного испарения и фрак¬
ционной конденсации. Температурные интервалы и выход
фракций при ректификации КУС приведены в табл. 8.9.Схема ректификации КУС представлена на рис. 8.12. Про¬
изводительность современных смолоразгонных установок со¬
ставляет 105—2'105 т смолы в год.182
Таблица 8.9. Выход фракций при разгонке КУСНазвание фракцииИнтервал
отбора, °СВыход,
% от КУСЛегкаяФенольнаяНафталиноваяПоглотительнаяАнтраценоваяПек170
170—210
210—230
230—270
270—360
3600,5—1,0
2—4
9—12
7—10
19—25
55—60Фракции, полученные разгонкой КУС подвергаются даль¬
нейшей переработке с целью выделения из них индивидуаль¬
ных веществ. На рис. 8.13 представлена общая схема подобной
переработки и получаемые продукты.Для выделения из фракций КУС индивидуальных соедине¬
ний используются методы кристаллизации, ректификации и
возгонки, а для выделения фенолов и пиридиновых основа¬
ний — обработка фракций растворами щелочей и серной кис¬
лотой с образованием соответствующих фенолятов и солей.Оставшийся после выделения из ПКГ всех химических про¬
дуктов (пирогенетическая вода, КУС, СБ, соединения аммиа¬
ка) обратный коксовый газ имеет средний состав: (в об. процен¬
тах): водород 54—59, метан 23—28, другие низшие алканы 2—3, оксид углерода (II) 5—7, оксид углерода (IV) 1,5—2,5, а так¬
же около 4—5% азота и кислорода, попавших в газ в результа¬
те подсоса воздуха в печь при коксовании.Легкая фракция _5> 10 Па2-1СГ ПаКУСч-Э:110'400<1 \105ПаПарыКУС341—*\ПекПарФенольнаяНафталиноваяПоглотительнаяАнтраценоваяФракции КУСРис. 8.12. Схема ректификации каменноугольной смолы:1 — трубчатая печь, 2 — испаритель первой ступени, 3 — испаритель
второй ступени, 4 — ректификационная колонна183
ФлуоратенПиренДобавкакСБШпалопропиточное ^
масло, Легкая
фракцияСажевоепроизводствоАнтраценовоемасло+Антраценовая
фракцияАнтраценКарбазолСыройантрацен+Дифенил о кс ид ФенантренПоглотительная
фракцияФенольная Нафталиновая
фракция фракция/ IКрезолы I Фенол ^* ТехническийКсиленолы нафталинМетил-нафта¬линИндолФлуоренФенолыАценафтенХинолиновыел?Хинолиноснования\ХинальдинИзохиналинРис. 8.13. Общая схема переработки фракций КУСПродукты коксования широко применяются в различных об¬
ластях народного хозяйства. Потребление их составляет (в %):— в химической промышленности 30— в цветной металлургии 30— в сельском хозяйстве 23— в строительстве и транспорте 12.На основе химических продуктов переработки ПКГ в РФ
выпускается свыше 200 наименований продукции, в том чис¬
ле, полимерные материалы, минеральные удобрения, химичес¬
кие средства защиты растений, растворители и др. Остающие¬
ся при переработке фракций КУС масла используются в произ¬
водстве руберойда, асфальта, для пропитки деревянных конст¬
рукций. На основе пека получают угольные электроды и анод¬
ные массы.8.6. Гидрирование твердого топливаГидрированием (гидрогенизацией ) твердого топлива называ¬
ется процесс превращения органической части топлива в жид¬
кие продукты, обогащенные водородом и используемые как
жидкое топливо.184
Проблема гидрирования твердого топлива возникла в связи
с возросшим потреблением нефти и необходимостью эффектив¬
но использовать низкокалорийные и высокозольные ископае¬
мые угли, представляющие сложности при их сжигании. В про¬
мышленном масштабе гидрирование твердого топлива впервые
было организовано в 30-х годах XX века в Германии и получи¬
ло развитие в связи с необходимостью использовать для произ¬
водства моторных топлив тяжелых смолистых нефтей с высо¬
ким содержанием серы. В настоящее время в различных стра¬
нах работают установки деструктивной дегидрогенизации топ¬
лив производительностью от 200 до 1600 т/сутки.Гидрирование твердого топлива представляет деструктив¬
ный каталитический процесс, протекающий при температуре
400—560°С под давлением водорода 20—70 МПа. В этих усло¬
виях происходит разрыв межмолекулярных и межатомных (ва¬
лентных) связей в органической массе топлива и протекают ре¬
акции:— деструкции и деполимеризации высокомолекулярных
структур угля{С}л + пН2 —* СЛН2п— гидрирования образовавшихся алкеновСпН.2п + н2 -* слн2п+2— деструкции высших алканов с последующим гидрирова¬
нием алкенов и образованием алканов меньшей молекулярной
массы^пН2л+2 СщН2т+2 “Ь СрН2р и срн2р + н2 * срн2р+2— гидрирования конденсированных ароматических систем
с последующим разрывом цикла и деалкилированием§0 - §fsfi§i+2c*^2Нб— раскрытия пятичленных циклов с образованием изоалка-
новQ-R + Н2 - СН3-СН-СН2-СН2-СН3Rи другие.Так как процесс гидрогенизации протекает в избытке водо¬
рода, то реакции полимеризации и поликонденсации первич¬
ных продуктов деструкции подавляются и при достаточно вы¬185
соком отношении водород/углерод
продукты уплотнения почти не об¬
разуются. На рис. 8.14 представле¬
на зависимость выхода продуктов
гидрогенизации в зависимости от
удельного расхода водорода.Одновременно с гидрированием
углеродных соединений протекают
реакции гидрирования соединений,
содержащих серу, кислород и азот
по реакциям, аналогичным реакци¬
ям гидроочистки нефтепродуктов
(глава VII).Процесс гидрогенизации являет¬
ся каталитическим. В качестве ка¬
тализаторов используют контакт¬
ные массы на основе соединений
молибдена, никеля или железа с
различными активаторами, например:Мо03 + NiS + СаО + ВаО + А1203катализатор активатор носительИзменением параметров процесса (температура, давление,
время контактирования) и состава катализатора процесс гид¬
рогенизации может быть направлен в сторону получения про¬
дуктов заданного состава. Выход
жидких и газообразных продуктов
гидрирования твердого топлива
существенно зависит от содержа¬
ния в нем летучих веществ, то есть
от степени его углефикации (рис.
8.15).Из рисунка следует, что угли с
Содержание углерода в угле, высокой степенью углефикацииУдельный расход Н2, кг/кг, топливаРис. 8.14. Выход продуктов
гидрогенизации каменного
угля в зависимости от расхо¬
да водорода:1 — газ, 2 — бензин, 3 — сред¬
нее масло, 4 — тяжелое мас¬
ло, 5 — асфальт, 6 — остаточ¬
ный угольмае. дол.Рис. 8.15. Зависимость выхо¬
да жидких и газообразных
продуктов от углефикации
сырья:1 — нерастворимый остаток,2 — пек, 3 — пирогенетическая
вода, 4 — жидкие продукты и
газ(антрацит, тощие угли) не могут
быть использованы в качестве сы¬
рья для гидрогенизации. Из топлив
для этой цели пригодны бурые угли
или каменные угли с отношением
водород/углерод не ниже 0,06 и со¬
держанием золы не более 0,13 мае.
дол. например, угли Канско-Ачин-
ского бассейна.186
Процесс гидрогенизации твердых топлив может проводить¬
ся в жидкой или паровой фазе. Из многочисленных техноло¬
гических схем жидкофазной гидрогенизации наиболее эконо¬
мичной является циклическая схема. Она отличается от дру¬
гих меньшим расходом водорода, более низкими температу¬
рой и давлением процесса и позволяет полностью использо¬
вать все компоненты перерабатываемого сырья. Принципиаль¬
ная схема подобной установки гидрогенизации приведена на
рис. 8.16.КатализаторtПастаН-Твердоетопливо3"ГГидрогенизатЗатирочноемаслоКотельно^ 1
топливо4Т5тДистиллятТвердыйостатокЖидкиепродуктыNH3,H2S
на абсорбциюКатализатор]<— NaOH .Жидкие продукты на переработку^ 2л)Рис. 8.16. Циклическая схема жидкофазной гидрогенизации топлива:1 — аппарат подготовки сырья, 2 — насос для пасты, 3 — реактор гидри¬
рования, 4 — центрифуга, 5, 6 — ректификационные установки, 7 —
нейтрализатор, 8 — реактор гидроочисткиВ результате гидрогенизации всех видов твердого топлива
образуется жидкий продукт, содержащий изоалканы и нафте-
ны, используемый в качестве сырья для каталитического ри¬
форминга и гидрокрекинга, а также котельное топливо и газ.8.7. Совершенствование процессов переработки
твердого топливаДефицит углей для коксования, потребность в дешевом сы¬
рье для получения новых химических продуктов и развитие, в
связи с этим, методов комплексного использования сырья, на¬
конец, исключительно крупные масштабы производств по пе¬
реработке топлива, вызвали острую необходимость в совершен¬
ствовании коксохимического и других производств по перера¬
ботке твердого топлива. Здесь можно выделить четыре основ¬
ных направления.1. Интенсификация процесса коксования и сокращение вре¬
мени его за счет:— снижения влажности коксуемого сырья,— повышения теплопроводности материалов печи,— увеличения размеров и полезного объема коксовых камер,— автоматизации управления процессом.2. Создание новых технологических процессов коксования
и переработки продуктов, в том числе:— введение непрерывных процессов коксования,— использование брикетированных угольных шихт из мел¬
кого угля,— организация формованного металлургического кокса,— проектирование энерготехнологических схем использова¬
ния каменных углей с использованием энергии МГД-генерато-
ров (рис. 8.17).3. Повышение комплексности переработки углей и других
видов твердого топлива для утилизации всех их компонентов и
получения продуктов многоцелевого назначения. В качестве
примера подобного производства приведена комплексная хими¬
ческая переработка торфа (рис. 8.18).4. Получение новых продуктов, в том числе:— извлечение германия из над смольной воды (рис. 8.19).— производство чистых рад анидов аммония и натрия, циа¬
нистого водорода,— производство коллоидной серы, пирена и др.Рабочеетопливо► Сушка-Рециркуляция тяже¬
лого пиролизатаt IТермичес- Высокоско- Конденса-► кая пере-1 ► ростной пи—► ция и пе¬
ра ботка ролиз реработкаУксуснаякислота идругие ге-тероатом- ►ные сое¬диненияСжиганиетвердогоТранспорти¬
руемое твер¬ТеплотаТвердыйПРТОТЛ^►^остаткаUU1 d 1 UKдое топливоЭлектро¬энергияВысоко¬
калорий¬
ный газ—г~Жидкийгаз~Т~IФенолыПиридин
и другие
гетеро¬
циклыРис. 8.17. Энерготехнологическая схема использования каменного
угля188
H2S04
ГТорф|—► ГидролизNaOH -тНейтра¬лизацияINaOHИнверсия,
нейтрали¬
зацияДрожжи СолиОсахарен-
ный торфБиосинтезМелассаКормовыедрожжиМинеральныеудобрениячалОстатокПереработка <+
NaOHТорфоще¬лочныереагентыI ОстатокIУпаривание ~ОПереработка*ОКорм для
скотаАктивиро¬
ванный
угольНаполни¬
тели для
пластмассРис. 8.18. Схема комплексной химической переработки торфаНадсмольнаяводаТаннин
(дубильный экстракт)Отходы -4-Н20Комплекс
германия и таннина II45%-ный конденсат
оксида германия +-
(IV) Ge02iЖидкий GeCI4
Ионнообменная очистка
i^ GeCI4
Ge02ОкислительHCIРис. 8.19. Схема выделения оксида германия из надсмольной воды189
Контрольные вопросы1. Что такое степень углефикации твердого топлива?2. Перечислите основные методы химической переработки каменных
углей.3. Что такое коксуемость углей? выход летучих?4. Какой процесс называется гидрированием твердого топлива?5. Приведите общую схему коксохимического производства.6. Какие требования предъявляются к углям для коксования?7. Что такое шихтовка каменных углей и для чего она проводится?8. Чем определяется профиль камеры коксовой печи?9. Что такое изохронические кривые коксовой печи?10. Какое топливо используется для обогрева коксовых печей?11. В чем преимущество сухого гашения кокса перед мокрым?12. Какие продукты выделяются из прямого коксового газа при улав¬
ливании?13. Приведите в виде схемы последовательность операций при разде¬
лении прямого коксового газа.14. В виде какой соли и почему извлекаются из прямого коксового газа
соединения аммиака?15. Какие соединения входят в состав «сырого бензола»?16. На какие фракции ректифицируется каменноугольная смола и
какие основные продукты выделяются из этих фракций?
Глава IXПРОИЗВОДСТВО И ПЕРЕРАБОТКА ГАЗООБРАЗНОГО
ТОПЛИВА9.1. Классификация и состав газообразных топливГазообразным топливом называется топливо, находящееся
в состоянии газа при температуре и давлении его эксплуатации.
Таким образом, газообразное топливо может храниться и транс¬
портироваться в жидком (сжиженный газ) и в твердом (напри¬
мер, гидриды водорода) состоянии.По происхождению газообразное топливо подразделяется на
природное (ископаемое топливо) и синтетическое. К газообраз¬
ному топливу относятся также отходы некоторых металлурги¬
ческих (доменный газ) и химических производств.К природному газообразному топливу относятся различные
природные горючие газы, представляющие естественные сме¬
си углеводородов различного состава и строения:— собственно природные газы, месторождения которых не
связаны с месторождениями нефти;— попутные газы, которые растворены в нефти или находят¬
ся над скоплениями ее в виде «газовой шапки», то есть газы,
месторождения которых генетически связаны с месторождени¬
ями нефти;— газы газоконденсатных месторождений, обогащенные
жидкими легкокипящими углеводородами, которые отделяют¬
ся от газа при снижении давления в виде жидкой фазы—кон¬
денсата.К синтетическому газообразному топливу относятся разно¬
образные горючие газы, полученные при переработке твердого
и жидкого топлива: газы нефтепереработки, генераторные газы,
обратный коксовый газ и др.Классификация газообразных топлив представлена на рис.19.1.Состав газообразного топлива зависит от его природы, про¬
исхождения и способа получения. Природные газы состоят пре¬
имущественно из метана с незначительным содержанием дру¬
гих низших алканов, оксида углерода и азота. В попутных га¬
зах содержится значительное количество алканов от этана до
пентана и выше, при относительно низком содержании мета¬
на. Газы газоконденсатных месторождений по составу занима¬
ют промежуточное место. Содержание конденсата в них колеб-191
Г"ПриродноеГАЗООБРАЗНОЕ ТОПЛИВО
1 Отходы другихПопутныенефтяныегазыГазыконденсатныхместорожденийСинтетическоеПриродныегазынефтепереработки► термокрекингакаталитического
крекингакаталитического
риформинга► пиролиза► гидрокрекингапереработки
твердого
топливагазыпиролизагенераторныегазыполукоксовыи ► воздушный► водянойподземной
газификацииРис. 9.1. Классификация газообразного топливалется от 10 до 350 г/м3. Во всех углеводородных газах природ¬
ного происхождения содержатся в различных количествах азот,
оксид углерода (IV), сероводород, аргон и гелий.В состав газов нефтепереработки входят помимо алканов
низшие алкены и водород. Горючие газы, получаемые перера¬
боткой твердого топлива, существенно различаются по соста¬
ву, однако общим для них является высокое содержание водо¬
рода, азота и оксида углерода (IV), что связано с особенностями
их получения.Средний состав газообразного топлива различного происхож¬
дения приведен в табл. 9.1, 9.2 и 9.3.Таблица 9.1. Состав газов нефтепереработки (% об.)КомпонентМетод переработкиТерми¬ческийкрекингПиролизкеросинаКатали¬
тичес¬
кий кре¬
кингКатали¬
тичес¬
кий ри-
формингГидро¬крекинг123456Метан16,0 141,43,413,06,9Этан17,08,32,821,014,0Пропан21,51,010,732,044,7Бутан19,55,431,028,034,4192
Продолжение таблицы123456Этилен2,522,04,5——Пропилен9,014,523,8——Бутилены14,311,723,7——Водород0,20,60,16,0—Таблица 9.2. Состав газов природного происхождения
(% об.)Вид газаСН4с2нйС3н8ОXиС5н12и вышеСС>2, N2,
Н2, НеПриродный98,90,30,20,1 0,5Попутный30,87,521,520,419,8 Г азоконденсат-
ных месторож¬
дений84,74,61,60,81,96,4Таблица 9.3. Состав газов переработки твердого топлива
(% об.)Компонент газаВид газаПолукок¬сованияОбратныйкоксовыйСланцевыйПодземнойгазификацииВодородМетанАлкеныСероводородОксид углерода (II)Оксид углерода (IV)КислородАзот17—3055—652—7
0,2—0,35—84—100,1—33—852—60
22—301.5—3
0,2—0,35.5—8
1#7—2,8
0,3—1,24—822—28
15—18
5,2—5,5
0,2—0,3
9—12
14—17
0,5—10
23—3113—9
1,5-2,5
0,2
0,5—1,7
3—19
10—29
0,2—0,6
48—569.2. Сырьевые источники природного газообразного
топливаВсе запасы газообразного топлива на Планете по степени их
разведанности делятся на три категории:— промышленные (разведанные) запасы, на основании ко¬
торых составляются проекты разработки месторождений и про¬
екты газопроводов;7 Химическая технология- Том 2193
— запасы, установленные на основании благоприятных гео-
лого-физических данных и являющиеся основанием для орга¬
низации разведочных работ на конкретных земельных площа¬
дях;— прогнозируемые запасы, устанавливаемые на основе ана¬
лиза общих геологических критериев нефтегазоносности.Разведанные запасы природного газообразного топлива на
Земле превышают 60 трлн. м3, а прогнозируемые запасы оце¬
ниваются в 200 трлн. м3. Крупнейшие газовые месторождения
находятся в Алжире, США, Иране, Нидерландах. В РФ откры¬
то около 500 газовых, газоконденсатных и газонефтяных мес¬
торождений с запасами топлива в них свыше 1 трлн. м3. Важ¬
нейшими из них являются Уренгойское, Заполярное, Медве¬
жье, Ямбургское газовые, Оренбургское, Вуктыльское (Коми
АССР) и Астраханское газоконденсатные месторождения. Все
разведанные запасы газа в стране оцениваются в 31 трлн. м3.Месторождения газообразного топлива в РФ распределены
неравномерно как по территории, так и по глубине залегания.
Из общего количества разведанных запасов 75% приходится на
районы Сибири и Дальнего Востока, причем основные запасы
газа падают на месторождения Тюменской области. Глубина за¬
легания газовых месторождений колеблется от 0,1 до 5 км; при
этом на глубинах 1—3 км сосредоточено около 85% всего газа.Попутный нефтяной газ выделяется из нефти при ее добыче
и стабилизации (глава XVII). Природный газ и газ газоконден¬
сатных месторождений находятся в газовых залежах под дав¬
лением 5—10 МПа, которое создается напором пластовых вод и
давлением горных пород. Поэтому эти газы извлекают через
сеть скважин фонтанным способом, при котором газ поднима¬
ется на поверхность за счет пластового давления. Так как при
свободном истечении газа энергия пласта расходуется нераци¬
онально и возможно разрушение скважины, расход газа огра¬
ничивают, устанавливая на выходе скважины штуцер, с помо¬
щью которого регулируют количество отбираемого из скважи¬
ны газа.Современный газовый промысел представляет сложный
технический комплекс в состав которого входят эксплуатаци¬
онные скважины, газосборные сети с установками по сбору
газа, компрессорные, холодильные и газораспределительные
станции.Сбор газа на промысле организуется по кольцевой или луче¬
вой схемам. На рис. 9.2 представлена наиболее распространен¬
ная кольцевая схема сбора газа.194
1Рис. 9.2. Кольцевая схема сбора газа на промысле:1 — газовые скважины, 2 — промысловый коллектор, 3 — сборный пункт
газа, 4 — перемычка на кольцевом коллекторе, 5 — регулировочные
штуцерыЧисло скважин, подключаемых к пунктам сбора газа, зави¬
сит от размера месторождения, конфигурации газовой залежи,
системы расположения скважин и параметров извлекаемого
газа (состав, давление, температура).Важнейшей характеристикой работы газового промысла яв¬
ляется коэффициент извлечения газа при эксплуатации или
газоотдача месторождения. В отличие от коэффициента извле¬
чения твердых ископаемых (50—60%) и нефтеотдачи (30—
40%) газоотдача значительно выше и составляет в среднем 85% ,
достигая в отдельных случаях 92% и более. Это объясняется
малой вязкостью и высокой упругостью газа по сравнению с
нефтью и низким коэффициентом сорбции газа горными поро¬
дами. При указанной газоотдаче время эксплуатации газового
месторождения составляет 15—20 лет.Масштабы добычи природного газообразного топлива непре¬
рывно возрастают. С начала разработки газовых месторождений
из недр добыто 24,4 трлн. м3 газа. Если в 1970 году мировая до¬
быча газа составляла 1,03 трлн. м3, то в 1987 году она возросла
до 1,77 трлн. м3. ВРФв 1993 году было добыто 638 млрд. м3 газа,
что составило 30% энергетического баланса страны.7*195
Добытый газ перед транспортировкой очищают от механи¬
ческих примесей и капель жидкости в сепараторах различного
типа, после чего для предотвращения коррозии газопроводов
подвергают осушке и очистке от сернистых соединений.Для осушки газа используют абсорбционный или адсорбци¬
онный методы. При абсорбционной осушке газ промывают в
скрубберах диэтиленгликолем, при адсорбционной—влагу уда¬
ляют при пропускании газа через колонны, наполненные си¬
ликагелем или синтетическим цеолитом.Для удаления сернистых соединений применяют мокрые
методы, основанные на поглощении соединений серы раство¬
рами слабо щелочных реагентов, обычно, карбонатовК2С03 + H2S KHS + КНСОзили моноэтаноламина2HOC2H4NH2 + H2S «=* (HOC2H4NH3)2S-2,с последующей регенерацией абсорбента нагреванием раствора
соли.Газы газоконденсатных месторождений помимо общей очис¬
тки подвергаются низкотемпературной сепарации. При этом, за
счет резкого снижения давления, пары воды и жидких углево¬
дородов в газе конденсируются и отделяются от газа, после чего
конденсат разделяется на воду и углеводородный слой.Для транспортировки газообразного природного топлива ис¬
пользуются газопроводы диаметром до 1 м, по которым газ под
давлением 7 МПа перемещается со скоростью до 500 метров в
минуту. Для компенсации падения давления по длине газопро¬
вода, через каждые 80—100 км установлены специальные ком¬
прессионные станции, оборудованные газоперекачивающими
агрегатами. Длина газопроводов в РФ составляет 65000 км. Важ¬
нейшими и наиболее протяженными из них являются газопро¬
воды: Саратов—Москва (843 км), Ставрополь—Москва (1254
км), кольцевой газопровод Московской области (1000 км), Крас¬
нодар—Серпухов (1773 км), Саратов—Череповец (1118 км), Сер¬
пухов—Санкт-Петербург (803 км) и другие.9.3. Использование газообразного топлива9.3.1. Подготовка газообразного топливаГазообразное топливо используется в качестве источника
энергии и сырья для химической промышленности. Из общего
потребления газообразного топлива в стране 55% его перераба¬
тывают в промышленности, 26% сжигается в ТЭЦ, 15% расхо-196
дуется на бытовые нужды и 4% потребляется другими отрасля¬
ми народного хозяйства. В настоящее время с применением га¬
зообразного топлива производится до 90% стали и чугуна, 65%
цемента и 85% всех минеральных удобрений.По сравнению с твердым газообразное топливо имеет ряд
преимуществ. К ним относятся:— образование гомогенных систем с воздухом и, как след¬
ствие, малый избыток воздуха и минимальные потери теплоты
с продуктами горения;— легкость воспламенения;— возможность предварительного нагрева и получение вы¬
сокой температуры пламени;— отсутствие золы и шлака при горении и простота конст¬
рукции топки;— отсутствие дыма и малое содержание оксида серы (IV) в
продуктах горения, что улучшает экологическую обстановку в
зоне ТЭЦ;— удобство и дешевизна транспортировки.Энергетическая ценность газообразного топлива зависит отего природы и состава. Так, низшая теплота сгорания его ко¬
леблется от 3600—3800 кДж/м3 (генераторный воздушный газ
и газы подземной газификации угля) до 16500—46000 кДж/м3
(обратный коксовый газ и попутный нефтяной газ).При использовании газообразного топлива в качестве хими¬
ческого сырья его предварительно разделяют на индивидуаль¬
ные компоненты или пригодные для дальнейшей переработки
фракции. Для этого используют следующие методы.1. Низкотемпературная конденсация, при которой газ в ре¬
зультате охлаждения превращается в двухфазную систему, ме¬
ханически затем разделяемую на жидкость и газ. В качестве
охлаждающих агентов используются вода, жидкий аммиак и
сжиженные этан и пропан. В некоторых случаях конденсация
сочетается со сжатием газа, что способствует сжижению тяже-
локипящих компонентов разделяемого газа.2. Абсорбция, при которой отдельные компоненты газа из¬
влекаются из него при охлаждении жидкими углеводородами
с последующей десорбцией полученных растворов в отпарной
колонне-десорбере. Для уменьшения потерь абсорбента в виде
паров с газом применяют двухступенчатую абсорбцию: в каче¬
стве основного абсорбента используется бензин, а выходящий
после первой ступени абсорбции газ дополнительно промыва¬
ется тяжело кипящим газойлем, который извлекает из газа уне¬
сенный им бензин.197
3. Низкотемпературная ректификация, при которой пред¬
варительно охлажденный газ в смеси с образовавшимся при
этом конденсатом разделяется под давлением в ректификаци¬
онной колонне. Обычно ректификация завершает процесс раз¬
деления газообразного топлива и применяется для получения
индивидуальных углеводородов высокой чистоты. В этом слу¬
чае на ректификацию подается только конденсат, выделенный
из газа конденсационно-компрессионным методом.Для химической переработки выделенных из газа углеводо¬
родов используются, практически, все основные реакции орга¬
нического и нефтехимического синтеза: пиролиз, конверсия,
окисление, гидрирование и дегидрирование, гидратация, алки-
лирование, реакции введения функциональных групп — суль¬
фирование, нитрование, хлорирование, карбонилирование и др.
Наряду с процессами разделения они позволяют получать на ос¬
нове газообразного топлива водород, оксид углерода (II), синтез-
газ, азотоводородную смесь, ацетилен, алкадиены, цианистый
водород, разнообразные кислородсодержащие соединения,
хлор, нитропроизводные и многое другое. В свою очередь эти
полупрЬдукты являются сырьем в производстве многочислен¬
ных целевых продуктов для различных отраслей народного хо¬
зяйства: высококачественного топлива, пластических масс, эла¬
стомеров, химических волокон, растворителей, фармацевтичес¬
ких препаратов, стройматериалов и др., как это показано ниже.9.3.2. Изделия и материалы, производимые на основе
переработки газообразного топливаХимическая промышленность: формальдегид, пластмассы,
волокна, синтетические каучуки, красители, лаки, этанол, ам¬
миак, растворители, пластификаторы, ПАВ, сажа.Машиностроение и металлургическая промышленность:
детали и части аппаратуры, смазочные масла, формы для ли¬
тья, резка металла, антикоррозионные покрытия, универсаль¬
ные клеи.Горная промышленность: ткани для фильтров, транспортер¬
ные ленты, взрывчатые вещества, ПАВ.Пищевая промышленность: пищевые кислоты, консервиру¬
ющие средства, упаковочные материалы, сети и канаты.Деревообрабатывающая промышленность: антисептики,
клеи для фанеры и древесностружечных плит, лаки и краски.Медицинская промышленность: хлороформ, формалин, на¬
шатырный спирт, уротропин, душистые вещества.198
Строительная промышленность: органическое стекло, кро¬
вельные материалы, антисептики, строительные плиты, лино¬
леум, трубы, облицовочный материал, звуко- и теплоизоляци¬
онные материалы.Топливно энергетическая промышленность: антифризы, га¬
зообразное топливо, сажа, высокооктановое жидкое топливо,
изоляционные материалы, добавки к моторному топливу.Текстильная промышленность: химические волокна,
эмульгаторы, искусственная кожа, клеи, моющие средства, кра¬
сители.Сельское хозяйство: пестициды, стимуляторы роста, протра¬
вители семян, азотные удобрения, пленки.Культура и быт: кино- и фотопленки, фотореактивы, пред¬
меты быта, игрушки.9.4. Переработка нефтяных газовК нефтяным газам относятся попутный нефтяной газ и газы
нефтепереработки — крекинг-газ и газ риформинга.9.4.1. Переработка попутного нефтяного газаПроцесс переработки попутного нефтяного газа заключает¬
ся в разделении его на узкие фракции и выделении из них ин¬
дивидуальных углеводородов в соответствии с их назначением.
Для этого используют метод низкотемпературной компресси¬
онной ректификации.Так как попутный газ относится к так называемым «жир¬
ным» газам, то есть содержит помимо алканов Ci—С4 значитель¬
ное количество (до 50 г/м3 и более) паров пентана и высших уг¬
леводородов, его перед ректификацией подвергают операции
отбензинивания—удаляют из газа высшие алканы в виде кон¬
денсата. Этот конденсат (газовый бензин) используется как до¬
бавка к бензинам, а также служит сырьем для выделения из него
пентановой и изопентановой фракций. На рис. 9.3 представлена
схема подобного разделения попутного нефтяного газа.Полученные разделением попутного газа фракции исполь¬
зуются:— этановая — сырье для пиролиза, хладоагент;— пропановая — сырье для пиролиза, хладоагент, бытовой
сжиженный газ индивидуального газоснабжения;— бутановая — сырье для производства синтетического кау¬
чука, и пиролиза, компонент бытового сжиженного газа, добав¬
ка к автобензинам;199
{ПопутныйнефтянойгазГазовый I
бензин ГI Фракция Ii с* ГДобавка
к бензинуi II Сжиженный 1 foul
1 газ ] -[с2нб[J Фракция I I Фракция L
[ изо-С5 J [ С4 jI Фракция I
[ изо-С4 [' гJ Фракция 11 Сз гРис. 9.3. Разделение попутного нефтяного газа— изобутановая — сырье для производства изопренового ка¬
учука и бутилкаучука, реагент алкилирования;— пентановая — сырье для пиролиза и изомеризации;— изопентановая — сырье для производства изопренового
каучука, компонент высокооктанового бензина.Использование попутного нефтяного газа имеет большое эко¬
номическое значение. Тем не менее, на большинстве нефтепро¬
мыслов его сжигают во избежание отравления атмосферы. В
1988 году только в Западной Сибири было сожжено в факелах
попутного газа на 16 млрд. руб. (в ценах того времени). Одной
из причин этого является излишняя централизация процессов
стабилизации нефти и сосредоточение их на немногих крупных
газобензинных заводах. Гораздо более эффективным является
установка небольших газобензинных станций на отдельных
скважинах. Такой способ практикуется на зарубежных нефте¬
промыслах.9.4.2. Переработка крекинг-газаВ отличие от попутного нефтяного газа газы крекинга содер¬
жат значительное количество (до 40% об.) алкенов от этилена
до бутиленов. Разделение крекинг-газа на фракции совмеща¬
ется с процессом стабилизации крекинг-бензина, то есть про¬
цессом извлечения из него растворенных газообразных углево¬
дородов. Подобная переработка крекинг-газа и крекинг-бензи¬
на осуществляется на газофракционирующих установках
(ГФУ) конденсационно-компрессионного или абсорбционного
типа. На рис. 9.4 представлена принципиальная схема этого
процесса, а на рис. 9.5 приведена технологическая схема ГФУ200
Нестабильный ]
крекинг-бензинГКрекинг-газСтабилизиро¬
ванный бензин ► На складН2 + CH4+C^-lg
+ сзИбРис. 9.4. Принципиальная схема работы ГФУРис. 9.5. Технологическая схема стабилизации крекинг-бензина
на ГФУ абсорционно-ректификационного типа:1 — компрессор, 2,3 — насосы, 4,5, 6 — водяные холодильники, 7,8,9 —
кипятильники, 10, 11, 12 — ректификационные колонны (фракционирую¬
щие абсорберы)абсорбционно-ректификационного типа, работающей под дав¬
лением 1,4 МПа.Сжатый в компрессоре 1 крекинг-газ смешивается под дав¬
лением 1,4 МПа с нестабильным бензином, подаваемым насо¬
сом 2, охлаждается в холодильнике 4 и подается в фракциони¬
рующий абсорбер (абсорбер-десорбер) 10, орошаемый стабиль¬
ным бензином. В верхней части абсорбера из газа бензином из¬
влекаются углеводороды Сз—С4, а в нижней, которая обогрева¬
ется бензином, циркулирующим через кипятильник 7, проис¬
ходит десорбция их из раствора и регенерация абсорбента. В
результате этого из верхней части абсорбера выходит «сухой»201
газ, содержащий водород и углеводороды Ci—С2, а из нижней,
вместе с регенерированным абсорбентом, выводятся углеводо¬
роды С3—С4. Насыщенный ими бензин проходит последователь¬
но ректификационные колонны 11 и 12, работающие в анало¬
гичном режиме. С верха колонны 11 отбирается фракция газа
С3, а с верха колонны 12 — фракция С4. Для полноты отгонки
углеводородов из бензина часть их конденсируется в холодиль¬
никах 5 и б и направляется на орошение колонн. В отдельных
случаях схема ГФУ может быть дополнена специальной колон¬
ной для отбора изопентановой фракции. Стабильный бензин
выводится с низа колонны 12 и поступает в хранилища.Продукты стабилизации бензина на ГФУ используются:— пропан-пропиленовая фракция—в качестве сырья для
производства фенола, ацетона, бутанолов, синтетических мою¬
щих веществ;— бутан-бутиленовая фракция—как сырье в процессах ал-
килирования и полимеризации, в производстве СК, присадок к
маслам, для производства метилэтилкетона, метилбутанола.9.4.3. Алкилирование изобутанаПроцесс алкилирования проводится с целью получения сме¬
си жидких изоалканов, добавляемых к бензинам для повыше¬
ния их октанового числа. В основе алкилирования лежит реак¬
ция присоединения алкенов к алканам:С„Н2л+2 + СпН2п —> CmH2m+2 АНПодбирая соответствующие углеводороды можно получить
в одну стадию изоалканы различного строения. На практике
методом алкилирования получают разнообразные смеси алка¬
нов — алкилат. В промышленности наиболее распространен
процесс алкилирования изобутана н.бути ленами, а также ал¬
килирования бутанбутиленовой фракции из ГФУ, содержащей
как изобутан, так и изобутилен.Реакция алкилирования протекает в присутствии кислотных
катализаторов по цепному электрофильному механизму через
стадию образования третичного карбкатиона. В случае алкили¬
рования изобутана изобутиленом до 2,2,4-триметилпентана
(изооктана) в присутствии серной кислоты эта реакция может
быть представлена в следующем виде:H2S04 ^ Н+ + HS04
сн3- с = сн2 + н+ — СН3-С+-СН3СНз СН3202
СН3-С+-СН3 + СН2=С-СН3 — (СНз)3С-СН2-С+-СНз
СН3 СН3 СН3(СНз)зС-СН2Ч:+^15+Ш- С(СНз)з (СНз)зС-СН2-СН-СНз +I IСНз СНз+ (СНз)зС+Максимальный выход алкилата и высокое октановое число
алкилированного бензина достигаются при концентрации сер¬
ной кислоты 0,9—0,94 мае. дол., температуре 5—10°С и избыт¬
ке изобутана, что подавляет реакцию полимеризации изобути¬
лена. На рис. 9.6 представлена технологическая схема алкили-
рования изобутана бутиленами.Жидкий изобутан из ресивера 1, жидкий бутилен и свежая
и оборотная серная кислота поступают в реактор 4. За счет эк-
зотермичности реакции алкилирования часть изобутана испа¬
ряется и его пары возвращаются в ресивер 1, откуда после сжа¬
тия в компрессоре 2 поступают в холодильник 3, где конденси¬
руются. Пройдя затем дроссель 11 изобутан испаряется и ох¬
лажденный за счет дросселирования подается в ресивер. Смесь,
содержащая избыток изобутана, образовавшегося изоктана иРис. 9.6. Технологическая схема алкилирования изобутана:1 — ресивер, 2 — компрессор, 3,10 — холодильники, 4 — реактор
алкилирования, 5, 7 — сепараторы, 6 — нейтрализатор, 8 — теплообмен¬
ник, 9 — ректификационная колонна (дебутанизатор), 11 — дроссельный
вентиль203
другие углеводороды, из реактора 4 поступает в сепаратор 5 для
отделения серной кислоты, которая в виде оборотной кислоты
смешивается со свежей и возвращается в реактор. Углеводород¬
ный слой из сепаратора нейтрализуется водным раствором гид¬
роксида натрия и поступает в сепаратор 7, где разделяется на
два слоя. Нейтрализованная смесь углеводородов через тепло¬
обменник 8, обогреваемый алкил атом, подается в ректифика¬
ционную колонну-дебутанизатор 9, где из нее отгоняется избы¬
ток изобутана, возвращаемого через ресивер 1 в процесс. Товар¬
ный алкилат отбирается с низа колонны, охлаждается в тепло¬
обменнике 8 и поступает на склад. Свежий изобутан вводится в
верхнюю часть колонны 9.Основным аппаратом в технологической схеме алкилирова-
ния является реактор алкилирования (алкилатор). Из различ¬
ных их типов наиболее эффективны каскадные самоохлаждаю-
щиеся реакторы. Они состоят из нескольких секций, каждая из
которых снабжена мешалкой. Алкилируемая смесь подается в
каждую секцию, а испаряющиеся углеводороды отбираются из
реактора и после конденсации возвращаются в него. Тепловой
режим в реакторе поддерживается за счет испарения части реа¬
гирующих компонентов, что регулируется их количеством. От¬
работанная серная кислота выводится из отстойных зон.9.5. Переработка обратного коксового газаОбратный коксовый газ (ОКГ) представляет собой сложную
смесь газообразных веществ, основной компонент которой—
водород. Его содержание достигает 60% по объему (глава XVIII).
Поэтому переработка ОКГ с целью выделения из него водорода
является одним из наиболее доступных промышленных спосо¬
бов его производства.9.5.1. Водород в промышленностиВодород широко распространен в природе. Он входит в со¬
став воды, некоторых горных пород, ископаемого топлива, всех
растительных и животных организмов. Содержание водорода в
земной коре (литосфере и гидросфере) составляет около 1 % мае.,
в атмосфере в свободном состоянии водород присутствует в нич¬
тожных количествах (10~4% об.). Основными промышленны¬
ми источниками водорода являются вода, природные углеводо¬
родные газы, обратный коксовый газ, генераторные газы. По¬
мимо этого, водород — побочный продукт ряда производств:
синтеза ацетилена, электролитического получения щелочей.204
Масштабы производства водорода непрерывно возрастают.
Если в 1977 году мировое производство его составляло около
30 млн. тонн, то в 1985 году оно достигло уже 45—50 млн. т.
Это объясняется все возрастающим использованием водорода
в различных областях техники и народного хозяйства: хими¬
ческой промышленности, металлургии, ракетной технике,
энергетике.В химической промышленности водород применяют для про¬
изводства азотоводородной смеси (синтез аммиака), синтетичес¬
кой соляной кислоты, синтез-газа и разнообразных продуктов
на его основе (метанол и др.)? в процессах ароматизации, ри¬
форминга, гидрокрекинга, гидрогенизации углей, жидких топ¬
лив и жиров, гидроочистки нефтепродуктов и в разнообразных
процессах восстановления в органическом синтезе.В металлургии водород используют для восстановления ме¬
таллов из их соединений, резки и сварки металлов.В ракетной технике водород нашел применение как высоко¬
эффективное жидкое топливо, используемое в паре с кислоро¬
дом для последних ступеней ракет.Особое значение приобрели изотопы водорода дейтерий и
тритий как компоненты заряда водородного оружия и перс¬
пективное термоядерное топливо (управляемый термоядерный
синтез).Наконец, проблемы энергетики выдвинули в настоящее вре¬
мя задачу использования водорода в качестве топлива будуще¬
го (глава II).Из всех методов получения водорода промышленное значе¬
ние приобрели следующие:— извлечение из обратного коксового газа,— Конверсия природного газа,— газификация твердого топлива,— конверсия газа полукоксования,— электролиз воды.Электролитическое производство водорода из водных щелоч¬
ных растворов позволяет получать газ высокой чистоты (более
99,9% об.), но весьма энергоемко. Расход электроэнергии в нем
составляет 5,5 кВт-ч/м3 водорода, причем до 90% себестоимос¬
ти составляет энергия. Это ограничивает масштабы промыш¬
ленного производства электролитического водорода и он исполь¬
зуется в ограниченных целях, главным образом, в ракетной тех¬
нике. Для снижения расхода энергии, помимо классической
схемы электролиза, предложены методы высокотемпературно¬
го электролиза водяного пара с использованием оксидных элек¬205I:
тродов и комбинированный метод, в основе которого лежат раз¬
личные термохимические циклы. В этих процессах расход элек¬
троэнергии составляет около 3,0 кВт*ч/м3, что значительно
ниже, чем при электролизе водных щелочных растворов.Производство водорода конверсией углеводородных газов
будет рассмотрено ниже.9.5.2. Выделение водорода из обратного коксового газаПромышленный метод производства водорода из ОКГ осно¬
ван на ступенчатой (фракционированной) конденсации газа при
глубоком охлаждении его до температуры ниже температур
конденсации всех компонентов кроме водорода. Следователь¬
но, температура охлаждения ОКГ должна удовлетворять сле¬
дующему условию:t конд. примесей > t охлаждения > t конд. водородаВ табл. 9.4 приведен состав очищенного сухого ОКГ и темпе¬
ратуры конденсации составляющих его веществ.Таблица 9.4. Состав ОКГ и температуры конденсации
веществВеществоСодержание,% об.Температура
конденсации, °СЭтилен и пропилен/ 2 3-47,0Пропилен1-103,8Метан23—28-161,5КислородоТсоо“183,0Оксид углерода5—7-191,5Азот3—5-195,8Водород57—61-252,8Из табл. 9.4 следует, что указанному выше условию удовлет¬
воряет режим последовательного охлаждения ОКГ до темпера¬
туры ниже -190 195°С.В основе процесса фракционированной конденсации лежит
принцип использования холода последующих выделенных га¬
зообразных фракций газа для конденсации предыдущих фрак¬
ций с более высокими температурами конденсации. Схема по¬
добной конденсации приведена на рис. 9.7.Переработка ОКГ может производиться как с целью получе¬
ния водорода, так и азотоводородной смеси. В общем случае этот
процесс состоит из следующих стадий:206
ГазФ2VК1t3 X-ФЗФЗФ1Ф2"ГК2I?Газ■>Рис. 9,7, Принципиальная схема фракционированной конденсации:1,2 — теплообменники, охлаждаемые, соответственно, фракциями Ф1 и
Ф2, 3 — дроссели. Ф1, Ф2, ФЗ — отбираемые газообразные фракции с
понижающейся температурой конденсации, К1, К2— конденсаты фракций
Ф1, Ф2 соответственно— очистка ОКГ,— фракционированная конденсация очищенного газа,— корректировка состава продуктов разделения газа.1. Очистка. В этом процессе происходит удаление из ОКГ
высококипящих примесей, оксидов азота и сероводорода. Та-
кие вещества как вода, бензол, нафталин, оксид углерода (IV)
при низких температурах могут кристаллизоваться на стенках
аппаратуры, ухудшая теплообмен. Оксиды азота способны об¬
разовывать взрывоопасные смеси. Удаление из газа сероводо¬
рода, помимо предотвращения коррозии аппаратуры, вызвано
также целесообразностью его последующего использования для
производства элементарной серы и серной кислоты, так как в
ОКГ переходит до 30% серы, содержащейся в коксуемой уголь¬
ной шихте.Для очистки ОКГ от сероводорода используются так называ¬
емые мокрые методы, из которых наиболее распространенным
является мышьяково-содовый метод очистки, позволяющий
переработать извлекаемый из газа сероводород в элементарную
серу. Этот метод включает операции:1) Приготовление поглотительного раствора оксисульфомы-
шьяковистого натрия:4Na2C03 + А1203 + 2Н20 = Na4As205 + 4NaHC03
Na4As205 + 5H2S = Na4As2S5 + 5H20
Na4As2Ss + 02 = Na4As202S5207
2) Абсорбция сероводорода из ОКГ поглотительным раство¬
ром:Na4As2C>2S5 + H2S = Na4As2SeO + Н2О3) Регенерация раствора и выделение элементарной серы:2Na4As2S60 + О2 = 2Na4As202S5 + 2SТяжелые углеводороды удаляют из ОКГ абсорбцией погло¬
тительным маслом (ПМ), а непредельные углеводороды—ката¬
литическим гидрированием. На рис. 9.8 приведена принципи¬
альная схема очистки ОКГ.As-раствор ПМ
ОКГ ► Газ-2 —I 1S * H2S ^6^61^10^8КтjNeOH Н2^Газ-3 ► Газ-4 ► Газ-51 1С02, H^S с2н2, с2н4—► с2н6Рис. 9.8. Принципиальная схема очистки ОКГ2. Фракционирование. Очищенный ОКГ сжимается до дав¬
ления 1,8—2 МПа и поступает на разделение. На рис. 9.9 пред¬
ставлена последовательность и температурный режим выделе¬
ния фракций ОКГ при его фракционированной конденсации.При охлаждении ОКГ из него последовательно конденсиру¬
ются фракции: при -110°С пропиленовая, при -145°С этилено¬
вая, при -180°С метановая, содержащая до 85% метана. Затем
газ переходит через трубки испарителя жидкого азота и при
-190°С из него конденсируется оксидная фракция, состоящая
из 70—75% азота и 16—23% оксида углерода (II). Оставшийся
газ, содержащий около 15% азота и 85% водорода отмывается
от примеси оксида углерода (II) в колонне, орошаемой жидким■{ Газ-5 )■-80°С ^ f Н2, N2,CO-110°С „Н2, N3, со|СН4 С2— С зсн4 с2н41\г1гk^6^6Сзн6и2оN2)K(н2 М21-1
[СО СН4Г-180°Сн2 '-190яс'н2 'м2►^ 4м2соь 1соIСН4 (N2+CO)Рис. 9.9. Схема разделения ОКГСО-145°СТс2н4N2 ггы1 1-Т-НABC (I)'2 (II)N,208
азотом, и поступает на завершающую стадию. Для получения
ABC (вариант I) газ обогащается газообразным азотом до содер¬
жания 25% об., а для получения водорода (вариант II) подается
на операцию конденсации азота. Разделением ОКГ может быть
получен водород чистотой до 97% об.Смешением трех первых фракций получают так называемый
«богатый» газ, содержащий 65% метана, около 17% оксида уг¬
лерода (II) и 3% водорода, который возвращается на коксохи¬
мическое производство. При переработке ОКГ на установке про¬
изводительностью 30700 м3/ч образуется 24800 м3/ч ABC,
11000 м3/ч богатого газа и 1300 м3/ч этиленовой фракции, по¬
ступающей на дальнейшую технологическую переработку.9-6. Газификация твердого топливаГазификацией твердого топлива (ГТТ) называется процесс
превращения органической части топлива в горючие газы пу¬
тем воздействия на него окислителей. ГТТ представляет одно
из направлений совершенствования переработки экологически
«грязного» топлива, в процессе горения которого выделяются
зола, оксиды азота и серы. Метод ГТТ известен с 1670 года и в
настоящее время приобрел значение как источник получения
беззольного газообразного топлива и различных технологичес¬
ких газов для химической промышленности. Он стал универ¬
сальным процессом переработки топлива, так как позволяет
перерабатывать любые виды твердого топлива, получать газы
заданного состава, использовать процесс в установках различ¬
ной мощности—от автотранспорта до крупных стационарных
агрегатов. Реакторы, в которых осуществляется процесс ГТТ
называются газогенераторами; поэтому газы, полученные ГТТ
получили название генераторных газов.9.6.1. Физико-химические основы процесса ГТТГазификация твердого топлива представляет негетерогенный
некаталитический процесс. Он включает последовательные ста¬
дии диффузии газообразного окислителя, массопередачи и хи¬
мических реакций неполного окисления. В качестве окислите¬
лей при ГТТ используются воздух (воздушное дутье), кислород
(кислородное дутье), водяной пар (паровое дутье), а также их
смеси (паровоздушное и парокислородное дутье). Природа про¬
текающих при этом реакций, а, следовательно, состав соответ¬
ствующего генераторного газа, зависят от типа окислителя.209
При кислородном дутье:С + 02 ^ С02 -АН АН = 395 кДж (а)2С + 02^2С0-ЛЯ ДЯ=218кДж (б)При паровом дутье:С + Н20 СО + Н2 + АН АН = 136 кДж (в)С + 2Н20 <=* С02 + 2Н2 + АН АН = 89,8 кДж (г)а также вторичная реакция:СО + Н20 «=* С02 + Н2 - АН АН = 41 кДж (д)При парокислородном дутье: реакции «б» и «в», то есть:2С + 02 «=* 2СО и С + Н20 ^ СО + Н2Воздушное и паровоздушное дутье описывается теми же ре¬
акциями, что и в случае кислородного и парокислородного ду¬
тья, но в продуктах газификации содержится азот.Помимо основных реакций (а—д) при газификации протека¬
ют побочные реакции, влияющие на состав генераторного газа.
Важнейшей из них является реакция диспропорционированияС02 + С <=* 2СО,а при газификации под давлением реакция образования метана.Реакции газификации обратимы и протекают с увеличени¬
ем объема или при постоянном объеме, большинство из них яв¬
ляются эндотермическими. В условиях работы газогенераторов
(нормальное или относительно невысокое давление, температу¬
ра 900—1100С, избыток окислителя) равновесие их смещено в
сторону образования конечных продуктов. Поэтому режим га¬
зификации определяется, главным образом, кинетикой диффу¬
зионной и химической стадий процесса. Так как твердая фаза
(топливо) принимает непосредственное участие в реакции и
количество ее изменяется во времени, скорость окисления уг¬
лерода, например, водяным паром (реакция в) выражается урав¬
нением:dm г ЬРноО(9.1)dx 1+КрН2где: U — скорость процесса, выраженная в единицах массы
углерода, реагирующей в единицу времени,
k — константа скорости реакции,К — константа равновесия абсорбции-десорбции водоро¬
да в твердой фазе,Рн2о и Рн2—парциальные давления паров воды и водорода.210
Из 9.1 следует, что скорость реакции (в) тормозится десорб¬
цией водорода с поверхности угля, значит процесс газифика¬
ции протекает в переходной области (глава VI) и может быть
интенсифицирован факторами, ускоряющими как химическую
реакцию (повышение температуры и давления), так и диффу¬
зию (увеличение скорости дутья и использование реакторов,
конструкция которых обеспечивает максимальное развитие
поверхности контакта фаз и их перемешивание).9.6.2. Технологические схемы ПТВо всех случаях экономически и технологически целесооб¬
разно использовать для газификации низкосортное твердое топ¬
ливо—торф, бурые угли, сланцы, полукокс, отходы лесоразра¬
боток и др. Таким топливом являются, например, угли Канско-
Ачинского бассейна, которые даже при низкой зольности и ма¬
лом содержании серы, не могут эффективно использоваться как
твердое топливо из-за низкой теплоты сгорания.Технологическая схема и режим процесса ГТТ зависят от со¬
става генераторного газа и назначения газогенераторной уста¬
новки. В настоящее время в мире эксплуатируются сотни про¬
мышленных стационарных газогенераторных установок, кото¬
рые конструктивно классифицируются по следующим призна¬
кам:а) по состоянию топлива в реак¬
торе:— с топливом в стационарном
слое,— с топливом в кипящем слое,— с топливом во взвешенном со¬
стоянии;б) по принципу подвода тепла в
реактор:— автотермические с использо¬
ванием теплоты сгорания части га¬
зифицируемого топлива,— автотермические с использо¬
ванием внешнего тепла, в том чис¬
ле, энергии атомных реакторов.На рис. 9.10 представлен один из
наиболее распространенных видов
газогенераторов—реактор шахтно¬
го типа для проведения процесса вРис. 9.10. Г азогенератор:I — загрузочная коробка, 2 —
конусный затвор, 3 — шахта,4 — колосниковая решетка, 5 —
чаша. I — зона газификации,II — зона сухой перегонки, III —
зона сушки топлива211
стационарном слое топлива. Газогенератор выполнен в виде
цилиндра высотой 4,5 м и диаметром 3,5 м, футерованного
изнутри огнеупорным кирпичом. Нижняя часть шахты по¬
гружена во вращающуюся чашу, наполненную водой для со¬
здания гидравлического затвора. В чаше укреплена колос¬
никовая решетка, через которую в реактор подается дутье.
Твердое топливо периодически поступает в реактор через заг¬
рузочную коробку при опущенном конусе затвора. При под¬
нятии конуса топливо попадает в шахту. Образующаяся зола
проходит через колосниковую решетку и гасится водой в
чаше.Максимальной интенсивностью обладают газогенераторы
кипящего слоя, в которых используется тонкодисперсное топ¬
ливо. На рис. 9.11 представлена технологическая схема произ¬
водства водяного газа газификацией в кипящем слое.Рис. 9.11. Технологическая схема ГТТ в кипящем слое:1 — бункер топлива, 2 — газогенератор «КС», 3 — котел-утилизатор, 4 — цик¬
лон, 5 — сборник пыли, 6 — конденсатор-холодильник, 7 — каплеуловительКипящий слой измельченного топлива в подобной установ¬
ке создается за счет подачи в газогенератор водяного пара. Про¬
изводительность установки достигает 105 м3 генераторного газа
в час.Состав полученных генераторных газов зависит от природы
окислителя (вида дутья), типа газифицируемого топлива и ре¬
жима процесса. В зависимости от назначения генераторные
газы делятся на:— энергетические (генераторные),— технологические (синтез—газ),— восстановительные (для металлургии) и— отопительные (бытовые).В табл. 9.5 приведен средний состав газов, полученных на
различном дутье.212
Таблица 9.5. Состав генераторных газовДутьеНазвание газаСостав газа, % об.НизшаятеплотасгораниякДж/м3СОн2со2СН4N,ВоздухВоздушный305,04,01,0604950Водяной парВодяной36508,0—6,010000Воздух и вод.
парПаровоз¬душный30164,02,0486300Кислород
и вод.парПарокисло¬родный66311,01,01,09850Из данных таблицы следует, что наиболее богаты ценными
компонентами—оксидом углерода (II) и водородом водяной и
парокислородный генераторные газы.9.6.3. Особые виды газификации твердого топливаОдним из вариантов процесса газификации твердого топли¬
ва является метод подземной безшахтной газификации камен¬
ных углей (ПГУ), идея которого была выдвинута в 1888 году
Д.И. Менделеевым. В этом методе газификация протекает не¬
посредственно в угольном пласте, который является подземным
газогенератором, без извлечения топлива на поверхность. Этот
метод исключает трудоемкие горные работы и сохраняет от
вскрытия земельные участки.При ПГУ с поверхности земли к угольному пласту бурят сква¬
жины, отстоящие друг от друга на расстоянии 25—30 м. Затем
забои этих скважин соединяют по угольному пласту каналом
газификации. Одна часть скважин предназначается для дутья
(дутьевые скважины), другая—для отвода образующегося газа
(газоотводящие скважины). В результате этого под землей об¬
разуется газогенератор, состоящий из системы дутьевых и га¬
зоотводящих скважин, соединенных реакционным каналом
(рис. 9.12).Станции ПГУ работают на каменном и буром углях при глу¬
бине скважин до 500 м и по мощности эквивалентны добыче
угля 100—400 тыс. тонн в год. Основным недостатком метода
ПГУ, ограничивающим его промышленное использование, яв¬
ляется низкое содержание в газе водорода и оксида углерода (II)
и малая теплота сгорания (3000—4400 кДж/м3).Прогрессивным направлением в ГТТ стали процессы плаз¬
мохимической переработки углей. Среди других процессов с213
Рис. 9.12. Схема подземной газификации угля (ПГУ):1 — угольный пласт, 2 — газоотводящие скважины, 3 — дутьевые скважи¬
ны, 4 — огневой (реакционный) канал, 5 — скруббер для очистки газа, 6 —воздуходувкаиспользованием плазмы они занимают особое место, так как
представляют крупнотоннажные промышленные производства.
Плазмохимические процессы в угольной промышленности ис¬
пользуются для получения из каменного угля синтез-газа, вос¬
становительных газов, ацетилена и других продуктов.Преимуществами плазмохимического метода газификации
углей являются:— высокая селективность метода, позволяющая получать с
высоким выходом газы заданного состава;— возможность переработки различных видов твердого топ¬
лива;— высокая производительность и малые габариты аппара¬
туры;— отсутствие выбросов золы, оксидов серы и азота и, следо¬
вательно, экологическая чистота технологии.Для газификации твердого топлива в плазме используются
плазмотроны различной конструкции. Они включают:— генератор плазмы,— смеситель твердой (топливо) и газовой (плазма) фаз,— плазмохимический реактор дугового или плазмоструйно-
го типа с раздельной или совместной подачей угольного порош¬
ка и плазменной струи,— закаливающее устройство для быстрого охлаждения вы¬
ходящего газа,— систему сбора и разделения продуктов реакции. Принци¬
пиальная схема плазмохимической газификации каменного
угля представлена на рис. 9.13.В смеситель 2 подается угольный порошок и плазменная
струя из генератора плазмы 1. Выбор плазмообразующего газа214
оПлазмообра- Угольный Охлаждающийзующий газ порошок агентРис. 9.13. Принципиальная схема плазмохимической газификации:1 — генератор плазмы, 2 — смеситель твердого топлива и плазмы, 3 —плазмохимический реактор, 4 — закалочное устройствозависит от заданного состава получаемого генераторного газа.
В соответствии с этим плазмохимическую газификацию прово¬
дят в плазме водорода, водяного пара, воздуха, в плазме паро¬
воздушной смеси. Из смесителя 2 двухфазная система «уголь—
плазма» поступает в реактор 3. В реакторе последовательно про¬
текают:— нагрев угольного порошка до температуры разложения,— деструкция угля с образованием кокса и выделением ле¬
тучих веществ,— превращение летучих продуктов в газовой фазе плазмы,— нагрев кокса до температуры реакции,— взаимодействие кокса с газообразными продуктами раз¬
ложения и плазмообразующим газом.Образовавшийся генераторный газ из реактора 3 при темпе¬
ратуре 1800°К поступает в закалочное устройство 4У где охлаж¬
дается со скоростью не менее 107 К/с до температуры 500°К
впрыскиванием сначала бензина, а затем воды.9.7. Конверсия углеводородных газов*9.7.1. Конверсионные процессы в промышленностиКонверсией называется технологический процесс переработ¬
ки газообразного топлива с целью изменения его состава. Наи¬
более распространенными видами этого процесса являются кон¬
версия углеводородных газов и конверсия оксида углерода (II),
проводимая для удаления его из продуктов конверсии углево¬
дородного сырья. Сырьем для конверсии являются: природный
газ (метан), попутный нефтяной газ, газы нефтепереработки.215
По своему содержанию процесс конверсии — это неполное
окисление углеводородов, одним из продуктов которого явля¬
ется водород:CnH2n+2 JTlA * (Л “Ь 1)Н2 “Ь С^А^где: А — окислитель, С„Ат — продукт окисления.В качестве окислителей в конверсионных процессах исполь¬
зуются: кислород, воздух, водяной пар, оксид углерода (IV),
оксиды металлов. В соответствии с природой окислителя раз¬
личают три основных вида конверсии:— паровая конверсия (окислитель — водяной пар),— неполное окисление (окислитель — кислород или воздух),— окислительный пиролиз.Часто для получения конвертированного газа определенно¬
го состава и обеспечения автотермичности в одном процессе ис¬
пользуют различные окислители (комбинированная схема кон¬
версии).Конверсию углеводородных газов проводят для получения
технологических газов (синтез-газ, ABC), используемых в про¬
изводстве метанола, аммиака, высших спиртов, синтетическо¬
го бензина, водорода и других продуктов органического и неор¬
ганического синтеза; восстановительного газа для прямого по¬
лучения железа, ацетилена. Производство ацетилена методом
конверсии метана (окислительный пиролиз) рассмотрено в гла¬
ве XXI. Процесс конверсии газообразного топлива осуществля¬
ется в реакторах различного типа—конвертерах, а полученный
методом конверсии газ называют конвертированным газом.9.7.2. Физико-химические основы конверсионных процессовКонверсия углеводородных газов газообразными окислите¬
лями может проводиться в присутствии катализаторов или без
них (высокотемпературная конверсия), при атмосферном или
повышенном давлении. Наиболее распространены процессы
каталитической конверсии в присутствии гетерогенных ката¬
лизаторов.1. Конверсия с водяным паром. Реакция окисления метана и
его гомологов водяным паром может быть представлена урав¬
нениями:СН4 + н20 ^ СО + ЗН2 +ЛЯ АЯ = 206 кДжCrtH2n+2 + ftH20 ^ пСО + (2п + 1)Н2Состояние равновесия этой системы зависит от температу¬
ры, давления и состава парогазовой смеси. Равновесная степень216
превращения возрастает с повыше- СН4, %об.
нием температуры и увеличением
отношения Н20:СН4. Так как реак¬
ция протекает с увеличением объ¬
ема, то повышение давления влия¬
ет положительно только на скорость
реакции конверсии.На рис. 9.14 представлена зави¬
симость содержания остаточного
СН4 в конвертированном газе от дав- Рис. 9.14. Зависимость
ления для различных температур содержания СН4 в газе от
при соотношении Н20:СН4 в исход- давления и температуры
ной смеси 2:1. Из рис. 9.14 следует,что если при атмосферном давлении достаточно полная конвер¬
сия метана может быть достигнута уже при 700—750°С, то при
давлении 3 МПа—только при температурах выше 1000°С. Тем
не менее, для увеличения скорости реакции и уменьшения объ¬
ема аппаратуры и газопроводов, паровую конверсию метана про¬
водят под давлением, используя для этого естественное давле¬
ние природного газа, поступающего на установку конверсии 1—4 МПа. Скорость паровой конверсии при повышенном давлении
может быть рассчитана по формуле:v_dPcH4 _kp снdx р н(9.2)где: k — константа скорости конверсии, с-1,
т — время конверсии, с,Рсн4 и рщ—парциальные давления метана и водорода.Так как скорость установления равновесия в системе при
указанных выше температурах мала, а повышение температу¬
ры выше 1300 С приводит к крекингу метана, конверсию ведут
в присутствии никелевого катализатора на носителе AI2O3. Это
обеспечивает быстрое достижение высокой равновесной степе¬
ни превращения метана при температуре около 700 С и высо¬
ких объемных скоростях парогазовой смеси, подаваемой в ре¬
актор.2. Конверсия с кислородом. Процесс неполного окисления
метана кислородом (и воздухом) описывается реакциями окис¬
ления части метана до оксида углерода (IV):СН4 + 202 ^ С02 + 2Н20 -АН АН = 891 кДж (а)217
и последующего взаимодействия метана с продуктами окисле¬
ния:СН4 + С02 ^ 2СО + 2Н2 + ЛН АЯ = 248 кДж (б)
СН4 + Н20 СО + ЗН2 + АН АН = 206 кДж (в)Так как все эти реакции обратимы и протекают с увеличени¬
ем объема, а процесс конверсии кислородом, описываемый урав¬
нением суммарной реакции:СН4 + 0,502 ♦=* СО + 2Н2 - АН АН = 35 кДж (г)идет с выделением тепла, кислородная (и воздушная) конвер¬
сия подчиняется тем же закономерностям, что и паровая кон¬
версия. Ее проводят при температуре около 900С, в избытке
окислителя, под давлением и на аналогичном по составу нике¬
левом катализаторе.3. Конверсия оксида углерода (II). Конверсия оксида угле¬
рода (II) проводится только с водяным паром и представляет
обратимую экзотермическую реакцию:СО + Н20 С02 + Н2 — АН АН = 41 кДж (д)В отличие от реакций конверсии
метана эта реакция протекает без
изменения объема, поэтому повы¬
шение давления не влияет на состо¬
яние системы, но ускоряет реак¬
цию. Равновесная степень превра-тает с увеличением отношения
Рис. 9.15. Зависимость Н20:С0 и снижается при повыше-равновесной степени превра- нии температуры (рис. 9.15). На
щения СО от состава парога- практике отношение Н20:С0 под-
зовой смеси и температуры держивается около 4:1.Скорость конверсии, проводи¬
мой при повышенном давлении,Т\<Т2<Т звыражается формулой:dPcoU=dxРн, 'о(9.3)где: k —*Рсоконстанта скорости реакции,равновесное парциальное давление оксида углеро¬
да (II),Рсо, Ря2 и Рщо —парциальные давления оксида углерода
(II), водорода и паров воды, соответственно.218
Процесс конверсии оксида углерода (II) ускоряется введени¬
ем катализаторов. В современных установках конверсии при¬
меняются железохромовый и цинкхромомедный катализаторы.
Железохромовый промотированный оксидами калия и кальция
катализатор состава {РегОз+С^Оз+КгО+СаО} является высоко¬
температурным катализатором и обеспечивает необходимую
скорость конверсии только при 450—500°С; при этом в конвер¬
тированном газе остается до 4% оксида углерода (II). Этот ката¬
лизатор используется на первой ступени конверсии. Цинкхром-
медный катализатор состава {ZnO+Cr2C>3+CuO} работает при
200—300°С и позволяет довести содержание оксида углерода (II)
в газе до 0,2%. Этот катализатор используется на второй ступе¬
ни конверсии.Конверсия оксида углерода (II) проводится не как самостоятель¬
ный процесс, а для удаления его из конвертированного газа при
получении водорода и ABC. Одновременно при этом в конвертиро¬
ванном газе повышается содержание водорода (реакция д).9.7.3. Общая схема конверсии углеводородных газовТехнологическую схему конверсии выбирают исходя из на¬
значения и состава конвертированного газа. При этом учиты¬
вается как качественный состав газа (наличие азота, оксида уг¬
лерода (II) и т.п., так и соотношение компонентов (например,
азота и водорода для синтеза в ABC).На рис. 9.16 представлены наиболее распространенные виды
конверсии метана, состав конвертированного газа и назначение
процесса.Неполное окисление
+паровая конверсия02; Н20водородОкислительныйпиролизО.С2Н2н2соацетилен
водород
синтез-газНеполное окисление+ воздух; Н20паровая конверсия *+конверсия СОШ-► ABCРис. 9.16. Основные виды конверсионных процессов219
При выборе технологической схемы конверсии учитывают
также возможность организации автотермического процесса в
целом и полноту использования углеводородного сырья.В настоящее время в промышленности используют только
процессы конверсии метана и оксида углерода (II) при повышен¬
ном давлении. К их преимуществам относятся:— уменьшение расхода энергии на сжатие конвертирован¬
ного газа, объем которого существенно больше объема исход¬
ных газов;— уменьшение размеров аппаратуры и коммуникаций;— более полное использование теплоты влажных газов за
счет повышения температуры их конденсации.Технологическая схема конверсии углеводородных газов,
независимо от типа процесса, включает операции: компрессия
газа и окислителя, очистка газа от сернистых соединений, соб¬
ственно конверсия и очистка конвертированного газа.9.7.4. Очистка газа для конверсииПриродный газ, используемый в качестве сырья для конвер¬
сии, содержит механические примеси и масла, дезактивирую¬
щие поверхность катализатора, и сернистые соединения, отрав¬
ляющие катализатор. К таким соединениям серы относятся:
сероводород, сульфидооксид углерода, сероуглерод, тиофен,
органические сульфиды, меркаптаны и др. Для удаления соеди¬
нений серы газ подвергают двухстадийной очистке.На первой стадии сернистые соединения гидрируют на алю-
моникельмолибденовом катализаторе с образованием сероводо¬
рода:CS2 + 4Н2 = 2H2S + СН4
COS + Н2 = H2S + СОC4H4S + 4Н2 = H2S + С4Н10На второй стадии образовавшийся и содержавшийся в газе
сероводород поглощают адсорбентом на основе оксида цинка:H2S + ZnO = ZnS + Н20При более высоком содержании сернистых соединений для
очистки природного газа применяется адсорбционный метод с
использованием синтетических цеолитов состава Na20*Al203*
•Si02, которые затем регенерируют пропусканием азота или очи¬
щенного природного газа при 300—400 С.Очищенный газ поступает на стадию конверсии для получе¬
ния водорода, синтез-газа или ABC, на установках комбиниро¬220
ванного действия (парокислородная или паровоздушная конвер¬
сия).9.7.5. Получение синтез-газа парокислородной конверсией
метанаИз различных технологических схем производства синтез-
газа или водорода наиболее распространенной стала каталити¬
ческая парокислородная конверсия с добавлением оксида уг¬
лерода (IV), который вводится в процесс для смещения равно¬
весия реакции (д) и повышения выхода оксида углерода (II). Для
этой цели используется оксид углерода (IV), выделяющийся из
раствора этаноламина в регенераторе абсорбента.Технологическая схема подобного процесса, включая ста¬
дию удаления оксида углерода (IV) из конвертированного
газа представлена на рис. 9.17. Состав конвертированного газа
до и после удаления из него оксида углерода (IV) приведен
в табл. 9.6.Природный газ под давлением 0,2 МПа подается в сатуратор1, где насыщается парами воды горячего конденсата, поступа¬
ющего из теплообменника 6. На выходе из сатуратора парога¬
зовая смесь смешивается дополнительно с водяным паром и
оксидом углерода (IV) до соотношения СС^^ОгСЩ = 2:9:10 и
поступает в теплообменник 2, обогреваемый продуктами кон¬
версии. Нагретая до 600°С парогазовая смесь подается в смеси-СО,Пар рПрирод¬
ный газ-К§)гВода>|
4 5ПарКонвертированный газВодалV/.КонденсатСинтез-газ
СО,810ЭтаноламинРис. 9.17. Технологическая схема парокислородной конверсии:1 — сатуратор (паронасытительная башня), 2,6 — теплообменники, 3 —
смеситель, 4 — конвертор шахтного типа, 5 — котел-утилизатор, 7 —
скруббер, 8 — абсорбер оксида углерода (IV), 9 — регенератор, этано¬
ламина, 10 — циркуляционный насос221
тель 3, где смешивается с кислородом и затем направляется в
конвертер метана 4. Таким образом, в этой схеме конверсия
паром и кислородом осуществляется в одном реакторе (односту¬
пенчатая конверсия). Конвертированный газ проходит котел-
утилизатор и при температуре 650—680 С поступает в теплооб¬
менник 2, где охлаждается до 200 С поступающей в теплообмен'
ник из сатуратора 1 парогазовой смесью. Вырабатываемый в
котле-утилизаторе пар подается в сатуратор. Охлажденный кон¬
вертированный газ направляется в теплообменник б, в котором
подогревает конденсат, циркулирующий с помощью насоса 10
через теплообменник и сатуратор. Окончательно газ охлажда¬
ется до 40°С в скруббере 7, орошаемом водой. Соотношение ком¬
понентов в конвертированном газе не отвечает стехиометричес-
кому для синтез-газа, так как он содержит значительное ко¬
личество оксида углерода (IV). Поэтому он направляется в
абсорбер 9, где из газа извлекают большую часть оксида уг¬
лерода (IV) раствором этаноламина, который затем регене¬
рируется в регенераторе 9. Выделяющийся при регенерации
оксид углерода (IV) направляется на смешение с природным
газом в сатуратор 1.Таблица 9.6. Состав газа парокислородной конверсииКомпонентСодержание в газе, % об.До выделения СОгПосле выделенияОксид углерода (IV)9,72,2Оксид углерода (II)26,328,4Водород62,367,5Азот и аргон1,21,3Метан0,50,69.7.6. Получение ABC паровоздушной конверсией
метанаВ отличие от получения синтез-газа в этой схеме дополни¬
тельно введена стадия удаления оксида углерода (II) конверси¬
ей с водяным паром, а стадии паровой и воздушной конверсии
метана разделены, то есть схема является двухступенчатой.
Последовательность процессов получения ABC по этой схеме
может быть представлена в следующем виде:222
НоОiwВоздух1н2о1сн4сосо* ► -н2н2n2ЭтаноламинiCO.H2n2ICO,{Я!ЛНа рис. 9.18 приведена технологическая схема этого процес¬
са, которая включает также стадию очистки природного газа
от соединений серы (см. 9.7.4).Природный газ, сжатый в компрессоре до давления 4 МПа,
проходит подогреватель 1, обогреваемый дымовыми газами кон¬
вертора метана 6, и поступает в систему очистки газа от серни¬
стых соединений. Эта система состоит из реактора каталитичес¬
кого гидрирования 2 и адсорбера сероводорода 3 (см. 9.7.4).
Очищенный от соединений серы природный газ поступает в са¬
туратор (паронасытительную башню) 4> в которой смешивает¬
ся с водяным паром в отношении НгОсгаз = 4:1. Образовавшая¬
ся парогазовая смесь подогревается до 380 С в теплообменнике5 и подается в трубчатый конвертер паровой конверсии метана
(I ступень) 6. Трубки конвертера обогреваются за счет сжига¬
ния в нем метана, обеспечивая температуру 800°С. После пер¬
вой ступени конверсии газ, содержащий до 10% об. остаточно¬
го метана и водяной пар в отношении 0,8:1, направляется в
шахтный конвертер воздушной конверсии метана (И ступень)
7, в который вводится подогретый воздух. При этом в конвер¬
тере за счет экзотермичности реакции (г) поддерживается авто-
термический режим работы. Из реактора 7 конвертированный
газ при температуре 900—1000°С поступает в котел-утилизаторS, в котором вырабатывается пар высоких параметров (10 МПа,
480°С). Затем охлажденный до 380°С газ последовательно про¬
ходит конвертер оксида углерода (II) I ступени 9, котел-утили¬
затор 10, в котором дополнительно охлаждается до 25°С, и кон¬
вертер оксида углерода (II) II ступени 11. Выходящий из кон¬
вертера газ содержит всего 0,5% объема оксида углерода (II).
Для удаления из газа образовавшегося в результате конверсии
оксида углерода (IV) газ проходит через орошаемый водой скруб¬
бер 12у в котором охлаждается до 30°С и направляется на этано-
ламинную очистку в скруббер 13 и регенератор 14 (см. 9.7.7).223
Природ¬
ный газ■2) Г'к*,тег1 г-т%VZnSЛшК„ВоздухВода
охлаж¬
денная f вода
Газ ABC ВодаС°2±. *1413ЭАРис. 9.18. Технологическая схема паровоздушной конверсии
метана:1 — подогреватель природного газа, 2 — реактор гидрирования сернис¬
тых соединений, 3 — адсорбер сероводорода, 4 — сатуратор (паронасы-
тительная башня), 5 — теплообменник, 6 — трубчатый реактор конверсии
метана, 7 — шахтный реактор конверсии метана, 8, 10 — котлы утилиза¬
торы, 9 — конвертор оксида углерода I ступени, 11 — конвертер оксида
углерода II ступени, 12 — охладительный скруббер, 13 — абсорбер окси¬
да углерода (IV), 14 — регенератор раствора этанол-аминаВ процессе конверсии состав газа от стадии к стадии меняет-
ся. В табл. 9.7 приведен состав конвертированного газа паро¬
воздушной конверсии метана на различных стадиях процесса.Таблица 9.7. Состав конвертированного газаКомпонентСодержание в газе,% об.После конвер¬
тора метана
II ступениПосле конвер¬
тора со
II ступениПосле очистки
этаноламиномВодород57,661,774—75Оксид углерода (II)11,20,50,7Оксид углерода (IV)8,417,40,1Азот и аргон22,520,124—25Метан0,30,30,7224
Основными аппаратами в конверсионных процессах явля¬
ются реакторы-конвертеры трубчатого и шахтного типа. Труб¬
чатые конвертеры выполнены в форме прямоточной трубча¬
той печи, состоящей из камеры радиации и камеры конвек¬
ции, соединенных дымоходом. В камере радиации размеще¬
ны трубы, заполненные катализатором, общим объемом око¬
ло 20 м3, и инжекторные горелки факельного типа. В конвек¬
ционную камеру встроены подогреватели газа и пароперегре¬
ватель.Конвертер шахтного типа — это цилиндрический вертикаль¬
но расположенный аппарат, диаметром около 4 м и высотой 18
м. В верхней части конвертера находится смесительная каме¬
ра, а в нижней конусной размещены шары из глинозема, на ко¬
торых расположен слой катализатора. Внутри конвертер футе¬
рован жаропрочным бетоном.Конвертеры оксида углерода (II) выполнены в виде реактора
радиального типа. Катализатор в них размещен в перфориро¬
ванных корзинах, расположенными соосно между центральной
трубой и наружной обечайкой. Между обечайкой и корпусом
реактора находится кольцевой канал, по которому вводят сы¬
рье и отводят продукты конверсии.9.7.7. Очистка конвертированного газаКонвертированный газ, полученный паровоздушной конвер¬
сией метана, содержит до 20% объема оксида углерода (IV). Для
превращения подобного газа в ABC оксид углерода из него нуж¬
но удалить. Кроме того, оксиды углерода являются каталити¬
ческими ядами в синтезе аммиака.Газ парокислородной конверсии метана для производства
синтез-газа также содержит излишнее количество оксида угле-
рода (IV), который должен быть удален из него. Поэтому зак¬
лючительной стадией процесса конверсии природного газа в
обоих случаях является очистка конвертированного газа от ок¬
сида углерода (IV). Методы очистки от других примесей, так
называемая тонкая очистка газа, были рассмотрены в главе XI.
Наиболее распространенный метод удаления оксида углерода
(IV) из конвертированного газа — этаноламинная очистка. В
ее основе лежит хемосорбция оксида углерода 20% -ным раство¬
ром моноэтаноламина (МЭА). Образующиеся при этом карбо¬
нат и бикарбонат МЭА нестойки и при нагревании выше 100*С
диссоциируют с выделением оксида углерода‘(IV) и регенери¬
руют раствор МЭА:8 Химическая технология. Том 2225
2CH2OH-CH2NH2 + C02 + H20 ^ (CH2OH-CH2NH3)2C(V2
CH2OH-CH2NH2 + C02 + H20 ^ (CH2OH-CH2NH3)+ HCO3-Растворы МЭА обладают высокой поглощающей способнос¬
тью даже ири малых парциальных давлениях оксида углерода
(IV), что позволяет вести абсорбцию при атмосферном давлении.
В реальных условиях процесс поглощения проводят под давле¬
нием 1—3 МПа, а процесс регенерации МЭА при 120—130°С и
давлении 0,25—0,3 МПа. В этих условиях очищенный конвер¬
тированный газ содержит ОД—0,01% об. оксида углерода (IV).Расход тепла на регенерацию раствора МЭА рассчитывается
как:где: Qp, QH, (?д, Qu и QT — теплоты регенерации раствора, нагре¬
ва раствора до температуры регенерации, десорбции оксида уг¬
лерода (IV), испарения воды при отгонке оксида углерода и теп-
лопотери в окружающую среду, соответственно.На рис. 9.19 приведена технологическая схема этаноламин-
ной очистки конвертированного газа.Конвертированный газ при температуре 25°С подается под
давлением 2,8 МПа в абсорбер 1, орошаемый на двух уровнях
раствором МЭА. В нижней секции абсорбера газ очищается до
содержания оксида углерода (IV) 5—7%, в верхней—более пол¬
но. Карбонизированный раствор, выходящий из абсорбера, раз¬
ветвляется на три потока, которые направляются в регенера-Рис. 9.19. Технологическая схема моноэтаноламинной очистки:1 — абсорбер, 2, 3, 9 — воздушные холодильники, 4,5,6 — теплообмен¬
ники, 7 — регенератор, 8 — кипятильник, 10 — сепаратор конденсата игазаQP = QH + Фд + Qn + Qt(9.4)ный газ ►Кубовый остаток226
тор 7. Один поток поступает в верхнюю часть аппарата, другой
через теплообменник 4 — в среднюю часть, третий поток, до¬
полнительно нагреваемый в теплообменниках 6 и 5,—несколь¬
ко ниже второго потока. Окончательная десорбция оксида уг¬
лерода (IV) из раствора происходит в кипятильнике S, обогре¬
ваемом конвертированным газом. Регенерированный раствор
МЭА из регенератора 7 возвращается на абсорбцию в абсорбер 1
двумя потоками. Первый поток груборегенерированный прохо¬
дит через теплообменники 5 и 4 и воздушный холодильник 3 и
поступает на орошение нижней секции аппарата. Второй поток
глубокогенерированный, пройдя теплообменник 6 и воздушный
холодильник 2, поступает на орошение верхней секции аппа¬
рата. Парогазовая смесь из регенератора 7 охлаждается в воз¬
душном холодильнике 9, где конденсируется водяной пар, и
поступает на разделение в сепаратор 10. Из сепаратора оксид
углерода (IV) направляется потребителям, а конденсат возвра¬
щается в регенератор.9.7.8. Расчет состава конвертированного газаВ табл. 9.6 и 9.7 приведены составы технологических газов
парокислородной (синтез-газ) и паровоздушной (ABC) конвер¬
сии, полученных в реальных технологических процессах и оп¬
ределенные аналитически. Однако состав газов конверсии мо¬
жет быть рассчитан теоретически, если известен удельный вес
(доля) каждого вида конверсии в данном технологическом про¬
цессе.Основные виды конверсии описываются уравнениями:
полная паровая конверсияСН4 + Н20 = СО + ЗН2 * (а)полная кислородная конверсияСН4 + 0,502 = СО + 2Н2 (б)полная воздушная конверсияСН4 + 0,502 + 0,5-3,76N2 = СО + 2Н2 + 0,5-3,76N2 (в)полная конверсия оксида углерода (И)СО + Н20 = С02 + Н2 (г)Здесь коэффициент 3,76 представляет отношение содержа¬
ния N2/02 в воздухе для воздушной конверсии.Используя эти уравнения и задавшись долей каждой реак¬
ции в комбинированном процессе конверсии можно рассчитать
состав газа конверсии, исходя из следующих соображений.8-227
Пусть доля реакции «а» в конверсионном процессе состав¬
ляет х (дол.ед.) и реакций «б» и «в» соответственно 1-л: (дол.
ед.). Тогда, в том случае, если конверсия протекает полностью
и степень превращения равна единице, уравнения парокисло¬
родной и паровоздушной с удалением оксида углерода (II) па¬
ровой конверсией могут быть записаны в следующем виде:
Парокислородная конверсия:
х СН4 + *Н20 = *СО + 3*Н2
(1-х) СН4 + 0,5(1-*)02 = (1-*)СО + 2(1-*)Н2СН4 + *Н20 + 0,5(1-*)02 = СО + (*+2)Н2Паровоздушная конверсия с удалением СО:СН4 + *Н20 + 0,5(1-*)02 + 0,5(1-*)*3,76N2 == СО + (*+2)Н2 +0,5(1-*)-3,76 N2СО + Н20 = С02 + н2СН4 + (*+1)Н20 + 0,5(1-*)02 + 0,5(1-*)*3,76N2 == (*+3)Н2 + С02 + 0,5(1-*)*3,76N2Если принять объем метана равным 1 нм3, то объем конвер¬
тированного газа паровоздушной конверсии после удаления СО
составит1 + (*+3) + 0,5(1-*) *3,76 = 5,88 - 0,88* нм3и состав его (об. дол.): водород (х + 3):(5,88 - 0,88*)оксид углерода (IV) 1:(5,88 - 0,88*)
азот 0,5(1-х)‘3,76:(5,88 - 0,88*)Пример. Рассчитать состав газа парокислородной конверсии для
получения синтез-газа, при условии, что доля реакции паровой кон¬
версии х = 0,5 дол. ед.0,5СН4 + 0,5Н20 = 0,5СС) + 1,5Н20,5СН4 + 0,2503 - 0,5СО + Н2СН4 + 0,5Н20 + 0,2502 = СО + 2,5Н2Объем газа после конверсии 1 + 2,5 = 3,5 нм3
Состав газа: водород 2,5:3,5 = 0,714 об. дол.оксид углерода (II) 1:3,5 = 0,286 об. дол.228
9.8. Производство кислорода и азота разделением
воздухаЧистый кислород или обогащенный кислородом воздух ис¬
пользуются в процессах конверсии углеводородных газов, в
металлургии, для окисления в органическом синтезе, в каче¬
стве окислителя в ракетной технике, в медицине. Жидкий азот
применяется для тонкой очистки водорода от оксида углерода
(II) и метана, получения ABC стехиометрического состава, в
качестве хладоагента.Источником получения кислорода и азота, а также большин¬
ства инертных газов (кроме гелия) является атмосферный воз¬
дух, запасы которого практически неисчерпаемы и составляют5,1 '1012 т. Состав воздуха, за исключением оксида углерода (IV)
и паров воды, постоянен. Воздух содержит (по объему): азота
79,09%, кислорода 20,95%, аргона 0,93% , а также незначитель¬
ные количества неона, криптона, ксенона, гелия (1,6*10_3—
8-10“6%) и водорода (5-10_5%). Содержание оксида углерода (IV)
изменяется в зависимости от близости к населенным пунктам
и промышленным предприятиям и составляет, в среднем,0,03%. Содержание водяного пара в воздухе зависит от време¬
ни года, погодных условий, близости к водоемам и колеблется
от 0,1 до 2,8%.В промышленности разделение воздуха с целью получения
кислорода, азота и аргона осуществляется путем сжижения его
с последующей низкотемпературной ректификацией. Изучает¬
ся также возможность разделения воздуха методом абсорбции на
цеолитах и диффузионным методом, основанном на различной
скорости диффузии газов через полупроницаемые мембраны.9.8.1. Теоретические основы процессаВ основе метода получения кислорода и азота лежит процесс
глубокого охлаждения и конденсации предварительно сжатого
воздуха при теплообмене его с охлажденным, за счет расшире¬
ния (дросселирования), воздухом с последующей ректификаци¬
ей жидкого воздуха:Охлажденный расширенный воздух
Сжатый воздух ТК0НА Азот <4 Жидкий Кислородвоздух229
Таким образом, в процессе разделения воздуха на кислород
и азот можно выделить две стадии: стадию глубокого охлажде¬
ния и стадию ректификации, каждая из которых подчиняется
своим закономерностям,1. Глубокое охлаждение воздуха. Для получения глубокого
холода может быть использовано изоэнтальпическое (эффект
Джоуля—Томсона) или изоэнтропическое (с совершением
внешней работы) расширение газа.Если при постоянной температуре Т сжать реальный газ от
начального давления Рн до давления Рк, а затем снизить его дав¬
ление до первоначального Рн путем расширения (дросселиро¬
вания) через устройство, создающее сопротивление (вентиль,
диафрагма), без совершения внешней работы и теплообмена с
окружающей средой, то конечная температура газа Г понизит¬
ся вследствие затраты внутренней энергии его на преодоление
сил межмолекулярного взаимодействия. Очевидно, что расши¬
рение идеального газа в этих условиях будет происходить без
изменения внутренней энергии и его температура при расши¬
рении останется постоянной.Поэтому, чем больше отклонение реального газа от идеаль¬
ного состояния, то есть чем больше разность АР = Рк - Рн, тем
значительнее понижение его температуры при расширении
AT =Ti~T2 (так называемый дроссель эффект газа). В табл. 9.8
приведены значения дроссель-эффекта воздуха при его расши¬
рении до атмосферного давления (Рн) при различных давлени¬
ях сжатия Рк для двух начальных температур воздуха (Т\).Таблица 9.8. Значения дроссель-эффекта воздуха (градусов)т,° сНачальное давление, МПа5101520+3010,320,027,334,0-5021,943,861,272,7Из данных табл. 9.8 следует, что использовать для сжиже¬
ния воздуха только дросселирование нецелесообразно, так как
понижение температуры при этом невелико. Расчеты, напри¬
мер, показывают, что для конденсации воздух должен быть
сжат до Pfe = 45 ГПа, что технически неосуществимо. Поэтому
на практике принцип дросселирования всегда сочетается с теп¬
лообменом—охлаждением сжатого воздуха. Однако даже в
этом случае понижение температуры АТ составляет всего 0,1—230
0,3 градуса на атмосферу (0,1 МПа) и холодильный эффект про¬
цесса невелик.Значительно более эффективным является расширение
предварительно сжатого в изотермических условиях воздуха с
совершением внешней работы. В этом случае расширение про¬
текает в адиабатических условиях, без теплообмена с окружа¬
ющей средой в поршневой или турбинной машине (поршневом
или турбодетандере). При таком процессе разность АТ и холо¬
дильный эффект, создаваемый детандером, в несколько раз
выше, чем при дросселировании. Применение детандера не ис¬
ключает того, что часть сжатого воздуха дросселируется. Тог¬
да суммарное понижение температуры определяется как:АТ = АТдет + АГдрос (9.5)где: ДТдех — понижение температуры за счет расширения воз¬
духа в детандере,А Т дрос — дроссель-эффект.2. Ректификация жидкого воздуха. Разделение воздуха со¬
провождается уменьшением энтропии и поэтому требует затра¬
ты внешней работы. Для равновесного обратимого процесса она
равна:-А = АН — ToAS (9.6)где: А — внешняя работа на разделение компонентов воздуха,
АН— изменение энтальпии системы, АН = AU + pAV,AS — уменьшение энтропии системы, AS = Q/T.Внешняя работа в подобном процессе при получении кисло¬
рода чистотой 99% составляет 0,074 кВт -ч/нм3 газа. В реаль¬
ном необратимом процессе разделения воздуха вследствие по¬
терь холода в окружающую среду и гидравлического сопротив¬
ления аппаратуры расход энергии
на разделение воздуха значительно
выше и составляет не менее 0,5 кВт-
■ч/нм3 газа.При этом расход энергии тем
выше, чем больше чистота выделя¬
емых азота и кислорода. Существен¬
ное значение для расхода энергии
на разделение воздуха имеют поте¬
ри холода в процессе. С уменьшени¬
ем потерь расход энергии снижает¬
ся. Это может быть достигнуто ин¬
тенсификацией охлаждения за счет
применения более эффективныхА, кВт ч/нм3Рис. 9.20. Зависимость
удельного расхода энергии
от мощности установки и
чистоты получаемого газа
(кислорода)231
теплообменников и регенераторов и снижением потерь холода
в окружающую среду путем увеличения мощности установок.
На рис. 9.20 представлена зависимость расхода энергии А от
мощности установки П при производстве кислорода чистотой
99,5% и 95%.9.8.2. Технологический процесс разделения воздухаПроизводство азота и кислорода из воздуха состоит из трех
стадий: очистки и осушки воздуха, сжижения воздуха и ректи¬
фикации жидкого воздуха.1. Очистка и осушка воздуха.Воздух, особенно в промышленных районах, содержит пыль,
влагу, оксид углерода (IV) и ацетилен. Эти загрязнения могут
вызвать повышенный износ турбокомпрессоров, забивку аппа¬
ратуры, что ухудшает теплопередачу и увеличивает гидравли¬
ческое сопротивление, и даже привести к взрывам в случае об¬
разования твердого ацетилена.Для очистки воздуха от пыли перед компрессором устанав¬
ливают самоочищающиеся масляные фильтры. Оксид углеро¬
да (IV) удаляют из воздуха абсорбцией раствором едкого натра,
а ацетилен—адсорбцией силикагелем.Осушка воздуха осуществляется вымораживанием при ох¬
лаждении воздуха после сжатия или адсорбцией на синтетичес¬
ких цеолитах. При адсорбционной осушке одновременно с вла¬
гой из воздуха поглощаются оксид углерода (IV) и ацетилен.
Этим методом достигается достаточно тонкая очистка воздуха.
Адсорбция проводится при температуре не выше 10°С.2. Сжижение воздуха глубоким охлаждением.Очищенный воздух подается на установку сжижения. Кон¬
структивно эти установки весьма разнообразны и классифици¬
руются:а) по мощности—от 7 до 24*103 нм3 кислорода в час;б) по назначению и агрегатному состоянию вырабатываемых
газов—для получения газообразных азота и кислорода, для по¬
лучения жидкого кислорода, для получения инертных газов идр.;в) по чистоте вырабатываемых газов—выпускающие 95%-
ный технологический кислород, 99,5%-ный технический кис¬
лород, 99,998%-ный кислород для специальных целей и т.д.;г) по методу расширения сжатого воздуха—с дроссель-эффек-
том и с совершением внешней работы в поршневых и турбоде¬
тандерах.232
Поршневые детандеры работают при давлении сжатия Рк =
= 20 МПа и применяются в установках относительно небольшой
мощности, так как требуют большого расхода энергии. По срав¬
нению с поршневыми турбодетандеры (предложены в 1938 году
П.Л.Капицей) гораздо более экономичны, так как работают в
интервале низких давлений (Рн = 1,3-105 Па, Рк = 6-105 Па) и
имеют высокий коэффициент полезного действия, достигаю¬
щий 85%. Турбодетандеры применяются в установках большой
мощности.Для сжатия воздуха в установках малой и средней мощнос¬
ти используются поршневые компрессоры производительнос¬
тью 25—40-103 нм3/ч газа, в установках большой мощности—
турбокомпрессоры производительностью 95*103 нм3/ч газа,
имеющие шесть ступеней сжатия.На рис. 9.21 представлена схема агрегата глубокого охлаж¬
дения низкого давления с турбодетандером и турбокомпрессо¬
ром.Очищенный воздух сжимается турбокомпрессором 1 от дав¬
ления, близкого к атмосферному (Рн = 0,13 МПа) до давления
Рк = 0,6 МПа, проходит водяной хо¬
лодильник 2, охлаждается в тепло¬
обменнике 3 испаренным холодным
воздухом и разделяется на два пото¬
ка. Большая часть воздуха (90% об.)
расширяется в турбодетандере 4 до
давления 0,13 МПа, охлаждаясь
при этом до температуры, близкой
к температуре конденсации, и отда¬
ет свой холод последовательно в
конденсаторе 5 и теплообменнике 3.Меньшая часть воздуха (10% об.)
охлаждается и конденсируется в
конденсаторе 5. Затем жидкий воз¬
дух дросселируется в дросселе 6 с
понижением температуры, при
этом часть жидкого воздуха испаря¬
ется. Жидкая фаза и газ разделяют¬
ся в сепараторе-сборнике 7. Сжи¬
женный воздух выводится из сепа¬
ратора и поступает на ректифика¬
цию, а холодный газообразный сме¬
шивается с основным потоком из
турбодетандера 4 и отдает свой хо¬Рис. 9.21. Схема агрегата
глубокого охлаждения
воздуха:1 — турбокомпрессор, 2— во¬
дяной холодильник, 3 — тепло¬
обменник, 4 — турбодетандер,5 — конденсатор, 6 — дроссель,
7 — сепаратор233
лод в конденсаторе 5 и теплообменнике 3, поступающему на
сжижение сжатому воздуху из холодильника 2.3. Ректификация жидкого воздуха.Для разделения воздуха на составные компоненты применя¬
ют аппараты однократной и двукратной ректификации. При
однократной ректификации нельзя получить азот с содержани¬
ем кислорода менее 7% об. Поэтому для более полного разделе¬
ния воздуха используют аппараты двукратной ректификации,
позволяющие получить чистый азот с содержанием N2 не ме¬
нее 99,998% об. Схема аппарата двукратной ректификации
приведена на рис. 9.22. Аппарат состоит из верхней колонны 1,
нижней колонны 2 и испарителя 3. Нижняя колонна служит
для предварительного разделения воздуха на азот и воздушно¬
кислородную смесь. В ней поддерживается давление около 0,6
МПа. В верхней колонне происходит окончательное разделениегазовой смеси на азот и кислород.
Испаритель-конденсатор 3, распо¬
ложенный между колоннами, слу¬
жит для верхней колонны испари¬
телем кислорода, а для нижней —
конденсатором азота. Так как при
атмосферном давлении, которое
поддерживается в верхней колонне,
температуры кипения кислорода и
азота равны, соответственно,
-182,8ГС и -195,6ГС, то азот явля¬
ется низкокипящим, а кислород—
высококипящим компонентами.
Поэтому при ректификации жидко¬
го воздуха азот отбирается с верх¬
ней, а кислород с нижней части вер¬
хней ректификационной колонны.
Жидкий воздух вводится в аппарат
в нижнюю кубовую часть.При работе аппарата сжатый до
давления 12—20 МПа воздух, ох¬
лажденный предварительно до
-125°С, поступает в змеевик в ниж¬
ней части колонны, где охлаждает¬
ся до -160 С кипящей воздушно¬
кислородной смесью. Затем воздух дросселируется и подается
на средние полки нижней колонны. Пары, обогащенные азотом,
поднимаются вверх и конденсируются в испарителе-конденса¬Рис. 9.22. Аппарат двукрат¬
ной ректификации воздуха:1 — верхняя колонна, 2 — ниж¬
няя колонна, 3 — испаритель-
конденсатор, 4 — карман, 5 —
дроссели, 6 — куб234
торе, а жидкость, обогащенная кислородом стекает вниз. В кар¬
манах нижней колонны собирается жидкость с содержанием
азота до 99,5%. Отсюда часть жидкого азота стекает вниз, а
часть подается на орошение верхней колонны. Воздушно-кис¬
лородная смесь из нижней колонны, содержащая до 40% объ¬
ема кислорода, подается в среднюю часть верхней колонны. Сте¬
кая вниз, жидкость обогащается кислородом, а поднимающие¬
ся пары азота отмываются от кислорода жидким азотом, стека¬
ющим сверху.Наиболее распространена в промышленности стационарная
установка по разделению воздуха АКт-16 (А—азот чистый, со¬
держащий не менее 99,998% объема N2, Кт — кислород техно¬
логический, содержащий 92—95% объема 02, производитель¬
ность по азоту — 16-103 нм3/ч).Расход электроэнергии составляет: на 1 м3 кислорода 0,53
кВт-ч, азота 0,09 кВт-ч.Контрольные вопросы1. Какое химическое топливо называется газообразным?2. Чем отличаются природный газ от попутного нефтяного газа?3. В чем преимущества газообразного топлива перед твердым при
сжигании?4. Как перерабатывается попутный нефтяной газ?5. На чем основано выделение водорода из обратного коксового газа?6. Что такое газификация твердого топлива?7. Какие конверсионные процессы используются в промышленнос¬
ти?8. Какой вид конверсии применяется для получения синтез-газа из
метана?9. Из каких последовательных стадий состоит процесс конверсии ме¬
тана для производства азотоводородной смеси?
Часть IIIПРОИЗВОДСТВО ОРГАНИЧЕСКИХ ВЕЩЕСТВ
Глава XОСНОВНОЙ ОРГАНИЧЕСКИЙ СИНТЕЗ10.1. Продукты основного органического синтезаОсновным или тяжелым органическим синтезом (00С) на¬
зывается совокупность производств органических веществ от¬
носительно простого строения, вырабатываемых в очень боль¬
ших количествах и используемых как в качестве целевых про¬
дуктов, так и полупродуктов в других отраслях органической
технологии.К целевым продуктам ООС относятся синтетическое жидкое
топливо, смазочные масла, растворители и экстрагенты, моно¬
меры, пластификаторы полимерных материалов, пестициды,
средства защиты растений и другие. В качестве полупродуктов
ООС используются, как правило, простейшие представители
гомологических рядов соответствующих соединений: углеводо¬
родов (этилен, пропилен, бензол), галогензамещенных (дихлор¬
этан, винилхлорид), спиртов (метанол, этанол), альдегидов и
кетонов (ацетальдегид, ацетон), органических кислот (уксусная
кислота) и т.д.В отличие от ООС тонкий органический синтез (ТОС) объ¬
единяет производства органических веществ сложного строе¬
ния, вырабатываемые в относительно небольших количествах
и используемые преимущественно в качестве целевых конеч¬
ных продуктов. К ним относятся красители, фото- и кинореак¬
тивы, фармацевтические препараты, взрывчатые вещества, пар¬
фюмерные средства и т.п.Принятый в органической технологии термин «основной»
органический синтез указывает на то, что он объединяет про¬
изводства продуктов, являющихся основой всей остальной про¬
мышленной органической химии. При этом, в большинстве слу¬
чаев, ООС отождествляется с нефтехимическим синтезом. Это
связано с изменением структуры сырьевого баланса производств
органической технологии в последние десятилетия и переори¬
ентацией их на нефтяное сырье.“Продукция ООС исчисляется многими сотнями тысяч и мил¬
лионами тонн в год. Так, например, мировое производство двад¬
цати продуктов этой отрасли превышает 1 миллион тонн в год,236
а производство более чем ста продуктов достигает 200 тыс. т в
год каждого.Масштабы производства продуктов основного органическо¬
го и нефтехимического синтеза иллюстрирует табл. 10.1. В ней
приведены данные о мировом производстве некоторых целевых
продуктов и полупродуктов основного органического синтеза за
период 1975—1985 годов.Таблица 10.1. Мировое производство продуктов ООС(млн. т/год)ВеществоКоличествоВеществоКоличествоАкрилонитрил2,5Изопрен1,0Ацетальдегид2,0Окись этилена4,8Ацетон 13,0Пропилен15,4Бутадиен5,0Стирол8,0Винилацетат2,7Уксусная кислота3,5Винил хлорид15,6Фенол v3,0Высшие кислоты2,5Этилен30,5Высшие спирты3,0Этиленгликоль12,0Дихлорэтан19,5Этанол2,510.2. Сырье и процессы основного органического
синтезаПроизводство продуктов ООС базируется в настоящее время
на ископаемом органическом сырье: нефти, природном газе,
каменном угле и сланцах. В результате разнообразных хими¬
ческих и физико-химических процессов превращения этого
сырья (крекинг, пиролиз, риформинг, ректификация, конвер¬
сия, коксование и полукоксование), получают исходные веще¬
ства для ООС, которые могут быть объединены в пять групп:— алканы (от метана до парафинов С15—С40).— алкены (от этилена до пентенов),— ароматические углеводороды (бензол, толуол, ксилолы,
нафталин),— ацетилен,— оксид углерода (II) и синтез-газ.Ниже рассмотрены наиболее важные производства ООС на
основе:— синтез-газа (метанол и формальдегид),— алканов (высшие кислоты и спирты, бутадиен и изопрен),— алкенов (этанол, изопропанол),237
— ацетилена (ацетальдегид, уксусная кислота и ее ангидрид),— ароматических углеводородов (этилбензол и стирол, фе¬
нол),— нафтенов (капролактам).При этом для удобства, они объединены в соответствующие
классы органических соединений.Выбор конкретного процесса переработки сырья в продукты
ООС зависит от природы синтезируемого соединения, структу¬
ры и доступности сырья, экономической целесообразности про¬
изводства. В процессах ООС используются почти все реакции
препаративной органической химии, многие из которых идут в
присутствии катализаторов, при воздействии высоких темпе¬
ратур и давлений, в условиях фото- и радиационного облуче¬
ний.Процессы ООС протекают, как правило, в кинетической об¬
ласти. Поэтому, общая скорость процесса определяется скорос¬
тью химической реакции и описывается уравнением:U = -3—= й-аДС (10.1)ахгде: ДС — движущая сила процесса, равная произведению дей¬
ствующих концентраций реагирующих веществ в со¬
ответствии с кинетическим уравнением, определя¬
ющим порядок реакции,
k — константа скорости или коэффициент массопереда-
чи,а — параметр, зависящий от гомогенности или гетеро¬
генности процесса (см. также том I, часть I).В процессах ООС в системе обычно протекают несколько пос¬
ледовательных или параллельных реакций. При последователь¬
ных реакциях скорость одной из них определяет скорость все¬
го процесса. В этом случае константа общей скорости процесса
является сложной функцией констант скоростей отдельных
реакций:k = f(k\, h2, k3... kn), (10.2)которая в большой степени зависит от температуры, давления,
концентрации реагирующих веществ и других параметров про¬
цесса. При параллельных реакциях существенное значение
имеет селективность процесса по целевому продукту, которая
может быть обеспечена использованием селективно работаю¬
щих катализаторов, влияющих не только на характер зависи¬
мости (10.2), но и на порядок реакции.238
Технологий ООС присущи некоторые особенности, которые
отличают ее процессы от процессов общей химической техно¬
логии. К ним относятся: ^1. Многовариантность производственных процессов, обус¬
ловленная с одной стороны тем, что один и тот же продукт мо¬
жет быть получен из различных видов сырья (например, аце-
тальдегид из ацетилена и этилена) или различными методами
(например, стирол из этилбензола каталитическим дегидриро¬
ванием и через гидроперекись), а так же с тем, что одно и то же
сырье может быть использовано для производства различных
продуктов (например, из этилена могут быть получены этанол,
винилацетат, уксусная кислота и другие продукты).2. Многостадийность технологии как следствие сложнос¬
ти взаимосвязей исходного вещества с готовым продуктом:Исходное —► Полупродукт-1 —* Полупродукт-2 —► Готовыйсырье продуктПри этом, на определенных стадиях полупродукты могут
использоваться как для получения из них готовых целевых
продуктов, так и сами являться таковыми (как этиленгликоль
в схеме, приведенной ниже):Газы нефте—► Этилен —► Окись этилена —> Этилен—> Лавсанпереработки гликоль1Растворитель3. Многомаршрутность прохождения промежуточных про¬
дуктов, связанная с многочисленностью и многообразием про¬
цессов и аппаратов ООС и большое количество возможных тех¬
нологических схем получения одного и того же продукта. Так
например, для производства винилацетата из ацетилена и ук¬
сусной кислотыС2Н2 + СНзСООН — СН2=СН-ОСОСН3предложено в настоящее время свыше 30 технологических схем,
различающихся конструкцией реактора, методом конденсации
газообразных продуктов синтеза и условиями ректификации
реакционной смеси.4. Кооперирование и комбинирование различных технологи¬
ческих процессов, установок и производств в целом, взаимосвя¬
занных единой технологией, для более полного использования
сырья и эгнергии, утилизации отходов, сокращения капиталь¬
ных затрат на строительство. Например, при органиации подоб-239
ного производства на одном комбинате могут производиться на
основе этилена ацетальдегид, уксусная кислота, винилацетат,
поливинилбутираль и другие продукты.5. Высокая степень автоматизации на всех уровнях про¬
изводства, обеспечивающая точное соблюдение технологичес¬
ких параметров и, следовательно, высокое качество продукции.6. Использование совмещенных процессов, в которых объеди¬
нены несколько реакционных процессов или реакционные и
массообменные процессы. Примером первых являются сложные
химические процессы с совмещением экзо- и эндотермических
реакций (например, термоокислительный крекинг метана в про¬
изводстве ацетилена), примером вторых — реакционно-ректи-дкационные и реакционно-экстракционные процессы.При размещении производств органического синтеза все ста¬
дии их стремятся организовать на одной территории, в непос¬
редственной близости друг от друга, и только завершающая ста¬
дия получения конечного продукта может быть расположена в
районе потребления.10.3. Краткий исторический очерк развития ООСПроизводство органических веществ, используемых для нужд че¬
ловека, зародилось в древности и в дальнейшем развивалось вместе с
развитием теоретической органической химии. Первоначально оно ба¬
зировалось на процессах переработки растительного или животного
сырья, путем выделения из него ценных продуктов (жиры, масла, са¬
хар) или расщепления содержащихся в сырье веществ на более про¬
стые (спирт, уксусная кислота, глицерин, мыла).Промышленный органический синтез или производство более
сложных веществ из ограниченного набора простых органических со¬
единений появился в середине XIX века на базе успехов синтетичес¬
кой органической химии. «Едва ли можно говорить о промышленной
органической химии до 1855 года. Некоторое число природных про¬
дуктов подвергалось очистке или переработке другими способами, но
лишь немногие впервые созданные соединения изготавливались в мас¬
штабах больших, чем в лаборатории» (Ф.С. Тейлор «История промыш¬
ленной химии»).Первоначально органическая химия могла решать прямые техно¬
логические задачи только в отдельных редких случаях, как например,
разработка К.С. Кирхгофом способа осахаривания крахмала,синтез
У.Г. Перкиным красителя мовеина и некоторые другие. Создание и
развитие структурной теории органической химии имело решающее
значение для развития и промышленного органического синтеза. Тео¬
ретическая органическая химия с ее мощным теоретическим аппара¬
том и возможностью предсказывать неизвестные соединения, обога¬
щенная в XX веке новыми синтетическими методами, нашла множе¬
ство практических приложений. На ее основе выросла промышленная240
органическая химия в виде основного и тонкого органического синте¬
за, наложившая отпечаток на всю историю человечества XX столетия,
как это предсказывал А.М Бутлеров, анализируя значение синтеза
ализарина из антрацена каменноугольной смолы: «Это отразится, без
сомнения, на состоянии промышленности целых стран и на приложе¬
нии труда тысяч рук (А.М. Бутлеров речь «О практическом значении
научных химических работ» 1871 год).Успехи органического синтеза вызвали поворот химической про¬
мышленности к органической химии. Перед ней была поставлена за¬
дача целенаправленных' поисков новых органических соединений и
разработки новых методов получения уже известных веществ, имею¬
щих практическое значение и производимых в промышленных масш¬
табах. При этом от процессов, направленных на замену природных
продуктов (красителей, лекарственных и душистых веществ) синте¬
тическими, промышленность органического синтеза переходит к про¬
цессам производства новых, не встречающихся в природе соединении
и структурных аналогов уже известных.В последующие годы в своем развитии ранее единая промышлен¬
ность органического синтеза разделяется на ряд отраслей, которые и
составляют основной органический и нефтехимический синтез и тон¬
кий органический синтез.В процессе развития промышленности органического синтеза из¬
менялась и ее сырьевая база. Место каменного угля все более и более
занимало нефтяное сырье и природный газ. В процессе этого перехода
и появился термин «нефтехимический синтез». В нашей стране осно¬
вы промышленности ООС закладываются в 30-е годы XX века, а как
самостоятельная отрасль она оформляется только в 40-е годы. До это¬
го времени ассортимент и объем, вырабатывавшихся на основе коксг
и пищевого сырья, органических продуктов был незначителен. Так, i1913 году было произведено всего 200 т сырого бензола, 112 т сырогс
фенола, 90 т ацетона, 56 т нафталина и 1000 т уксусной кислоты. Е1914 году из 5754 коксовых печей только на 98 печах было организо
вано полное улавливание химических продуктов коксования, а основ
ными продуктами нефтепереработки являлись керосин и мазут.Развитие сырьевой базы ООС в нашей стране начинается с восста
новления и пуска коксохимических заводов Донбасса с полным улав
ливанием летучих продуктов коксования, что позволило организован
промышленное производство ряда органических продуктов: бензола
толуола, ксилолов, фенола, нитробензола и др. и к 1932 году отказать
ся от их импорта. В последующие годы выпуск этих и других продук
тов на базе коксохимического сырья продолжает возрастать. В 194«:
году начинаются исследования по промышленной химической пере
работке нефтяных газов (синтез толуола деалкилированием ксилол*
и др.). Ввод в строй промышленных установок каталитического ри
форминга нефтепродуктов позволил изменить структуру сырья дл*
производства ароматических углеводородов и уменьшить долю кок
сохимии в их сырьевом балансе. Так с 1963 по 1970 год доля аромати
ческих углеводородов, получаемых из нефтяного сырья, возрастает
бензола с 1 до 37%, толуола с 48,7 до 76,5%, ксилолов с 74,2 до 93,6%24
В табл. 10.2 приведена динамика роста производства продуктов ООС в
нашей стране с 1970 по 1987 год.Таблица 10.2. Производство продуктов ООС (в тыс. т)ПродуктГод1970197519801987Ацетилен228294311—Метанол1004147719003333Пропилен302580824—Уксусная кислота146190239—Фенол347413496521Этилен799136617732971Этанол синт. (миллионов
декалитров)909592,4Одновременно с увеличением общего объема продукции промышлен¬
ность ООС, начиная с 1960 года, по темпам роста начинает значительно
опережать химическую и нефтехимическую промышленность в целом.10.4. Значение и перспективы развития ООССовременная промышленность ООС решает две основные за¬
дачи: крупномасштабное производство полупродуктов для дру¬
гих отраслей промышленности и получение целевых продук¬
тов общего назначения. Поэтому, в настоящее время нет ни од¬
ной отрасли народного хозяйства, в которой не использовались
бы продукты ООС и их переработки (рис. 10.1).Каменный угольГазАроматическиесоединениятСинтез-газГ1АцетиленI[ Алкены] [^АлканыОсновной органический (нефтехимический) синтезi Т111Промежу¬Раствори¬ПАВ иСинтети¬Пести¬Моно¬Прочиеточныетели, экс¬моющиеческоецидымерыпрод.продуктытрагентысредстватопливоШДругие отрасли
химической
промышленностиНародное хозяйство
и потреблениеПолимеры, мате¬
риалы и изделия
из нихРис. 10.1. Место промышленности ООС в народном хозяйстве242
Основные тенденции развития промышленности ООС, на¬
правленного на повышение экономической эффективности про¬
изводства и качества продукции, связаны с экономией сырья,
энергии и снижением капитальных затрат. Важнейшими из них
являются следующие.1. Создание новых технологических процессов, базирующих¬
ся на более доступном и дешевом сырье. Например, переход от
дорогого ацетилена к ароматическим углеводородам, алкенам,
алканам и, наконец, синтез-газу в соответствии со шкалой их
ценности:С2Н2 > СпНйп-в > СДН2„ > С„Н2„+2 > (СО +Н2)2. Переход к прямым методам синтеза, исключающим по¬
требление неорганических реагентов. Например, замена серно¬
кислотной гидратации этилена прямой каталитической в про¬
изводстве этанола.3. Повышение селективности процессов за счет оптимизации
параметров, выбора аппаратуры и подбора высокоселективных
катализаторов.4. Сокращение числа стадий производства. Например, заме¬
на двухстадийного процесса синтеза ацетальдегида из этилена
через этанол (I) одностадийным окислительным процессом:С2Н4Н2° » С2Н5ОН (I)-Н2^ ► СНзСНО (II)5. Увеличение единичной мощности аппаратов и снижение
за счет этого капитальных затрат. Например, в производстве
метанола до 300—500 тыс. т/год.6. Экономия энергии и повышение коэффициента полезного
действия агрегатов, в частности за счет использования вторич¬
ных энергоресурсов и внедрения энерготехнологических схем.Контрольные вопросы1. Производства каких продуктов относятся к основному органичес¬
кому синтезу?2. Что является сырьем для производства основного органического
синтеза?3. Перечислите основные особенности технологии органического син¬
теза.243
Глава XIПРОИЗВОДСТВО АЦЕТИЛЕНА11.1. Технологические свойства и применение
ацетиленаАцетилен (этин) С2Н2 —это бесцветный газ, обладающий в чи¬
стом виде слабым эфирным запахом, с температурой кипения -
83,8°С, температурой плавления -80,8°С (при 0Д7 МПа) и плот¬
ностью 1,09 кг/м3. Критическая температура ацетилена 35,5°С.При нагревании до 500С и при сжатии до давлений выше 2-105
Па ацетилен, даже в отсутствии кислорода, разлагается со взры¬
вом. Разложение инициируется искрой и трением. Взрывоопас¬
ность ацетилена возрастает в контакте с металлами, способными
образовывать ацетилениды, например, с медью. Это необходимо
учитывать при выборе материала аппаратуры. С воздухом ацети¬
лен образует взрывчатые смеси с пределами воспламенения 2,3 и
80,7% объема. При этом взрывоопасность смесей снижается при
разбавлении их инертными газами (азот, метан) или парами.Ацетилен значительно лучше, чем другие газообразные уг¬
леводороды, растворим в воде. При температуре 15°С и давле¬
нии 105 Па в одном объеме воды растворяется 1,15 объемов. В
других растворителях растворимость ацетилена составляет: в
ацетоне 25, этаноле 6, бензоле 4, уксусной кислоте 6 объемов.
Растворимость в ацетоне возрастает с повышением давления и
при 1,25 МПа составляет уже 300 объемов в одном объеме. Ра¬
створимость ацетилена в различных растворителях имеет боль¬
шое значение для его выделения из смесей с другими газами, а
также при хранении в баллонах в виде раствора в ацетоне.Ацетилен является эндотермическим соединением с энталь¬
пией образования +227,4 кДж/моль. Поэтому, при сгорании его
в кислороде выделяется большое количество тепла и развива¬
ется высокая температура, достигающая 3150°С. Это обуслови¬
ло использование ацетилена для сварки и резки металлов, на
что расходуется до 30% всего его производства. Вследствие вы¬
сокой взрывоопасности ацетилен хранится и транспортирует¬
ся в баллонах, заполненных древесным углем, или в растворе в
ацетоне под давлением 1,5—2,5 МПа.Основная масса ацетилена используется в качестве сырья в
различных производствах основного органического синтеза для
получения многих важных в техническом отношении продук¬
тов. Это объясняется высокой реакционной способностью аце¬
тилена, в молекуле которого содержатся два активных фрагмен-244
та: тройная связь -С=С- и подвижный «ацетиленовый» атом
водорода =С-Н. В соответствии с этим реакции ацетилена мо¬
гут быть сведены к двум основным типам:— реакции винилирования, то есть реакции введения ви-
нильной группы -СН2=СН- в соединения, обладающие подвиж¬
ным атомом водорода, и— реакции с участием «ацетиленового» атома водорода.Методом винилирования получают винилхлорид, акрило-нитрил, ацетальдегид, винилацетат, алкилвиниловые эфиры,
винилацетилен и другие соединения, перерабатываемые на вто¬
ричные целевые продукты.По реакциям второго типа могут быть получены гомологи
ацетилена,разнообразные продукты присоединения альдегидов
и кетонов, например, бутиндиол, используемые в производстве
бутандиола и бутадиена.На рис. 11.1 представлены важнейшие направления перера¬
ботки С2Н2.с2н2гидрохлорирование^ СН2=СН-С1 —> СН2=СС12НОННС1гидроцианировани^ CH2=CH-CN ►
HCNалоксилирование CH2=CH-OR
ROH
ацетилированиеROHCH2=CH-COOHch2=ch-coorсн2=сн-ососн3СНзСООНдимеризацияСН=СНэтилинированиеСН20С1Н01 f—► сн2=с-сн=сн2н-сн=с-сн=сн2сн2=сн-сн=сн2носн2-с=с-сн2он *сн=с-сн2онНОНСН3СООС2Н5СНзСООН(СН3С0)20сн2=сн-сн=сн2хлорирование^ СНС12-СНС12СЬСНС1=СС12СС12=СС12Рис. 11.1. Основные направления переработки ацетилена245
Наиболее крупнотоннажными продуктами химической пе¬
реработки ацетилена стали винилхлорид (31% по данным
1975 г. в США), акрилаты (26%) и винилацетат (18% ).На долю
хлорорганических растворителей и других продуктов приходит¬
ся около 25% производимого ацетилена.11.2. Промышленные методы производства ацетиленаТак как ацетилен не содержится в природных продуктах,
например в нефти или природном газе, особое значение приоб¬
ретают не связанные с нефтехимией синтетические методы его
промышленного производства.Ацетилен был открыт Э. Дэви в 1836 г., синтезирован из угля
и водорода М. Бертло в 1862 г. и впервые получен разложением
карбида кальция водой Ф. Вёлером в том же 1862 г. После от¬
крытия А. Муассаном метода синтеза карбида кальция из угля
и извести, карбидный метод производства ацетилена стал од¬
ним из основных промышленных методов, сохранившим свое
значение до настоящего времени:Основными недостатками карбидного метода получения аце¬
тилена являются высокая энергоемкость на стадии производ¬
ства карбида кальция (более 3000 кВт*ч/т карбида), многоста-
дийность процесса и высокие капитальные затраты. К достоин¬
ствам метода следует отнести высокую концентрацию получае¬
мого ацетилена и возможность использования дешевых камен¬
ных углей.Начиная с 40-х — 50-х годов XX столетия приобретает про¬
мышленное значение метод производства ацетилена пиролизомнизкомолекулярного углеводородного сырья, основанный на его
высокотемпературной деструкции по схеме:Процесс получения ацетилена из углеводородного сырья про¬
текает в одну стадию, менее энергоемок, требует меньших ка¬
питальных затрат и, в целом, на 20% экономичнее карбидного
процесса. Однако в этом методе ацетилен разбавлен водородом,
а это требует более сложной системы его выделения из синтез-
газа и очистки.В нашей стране до 60-х годов ацетилен производился исклю¬
чительно из карбида кальция. В 1958—59 гг. было организова¬кокс (С)СпН2„+2 - п/2 С2Н2 + (п/2 + 1)Н2246
но производство ацетилена методом пиролиза на Лисичанском
и Саратовском химических комбинатах. С 1970 года более 50%
ацетилена производится из углеводородного сырья.Ацетилен является важным многотоннажным продуктом
основного органического синтеза. Мировое производство его (без
РФ) в 1970 г. составило 1,795 млн. тонн, в том числе из углево¬
дородного сырья 0,657 млн. тонн.11.3. Производство ацетилена из карбида кальция11.3.1. Физико-химические основы карбидного методаПроцесс производства ацетилена карбидным методом скла¬
дывается из двух последовательных стадий: получение карби¬
да кальция и его разложение водой (гидратация).Реакция образования карбида кальция представляет необра¬
тимую эндотермическую реакцию, протекающую в форме «ра¬
створения» углеродистого материала в расплаве смеси оксида
кальция и образовавшегося карбида кальция:СаОрасп. + ЗС - СаС2расп. + СО + АН АН = 462,3°С (а)Реакция протекает с поглощением большого количества теп¬
ла и начинается при температуре 1700—1800 С. При повыше¬
нии температуры выше 2200 С карбид кальция испаряется и
разлагается по уравнению:СаС2 = Са + 2С (б)поэтому температуру процесса поддерживают в пределах 1900—
1950С.В качестве углеродистых материалов для синтеза использу¬
ются кокс или антрацит. Для снижения содержания в ацетиле¬
не вредных примесей к сырью предъявляются жесткие требо¬
вания по чистоте. Так, известняк должен содержать не менее
97% карбоната кальция, а углеродистые материалы — не бо¬
лее 6—8% летучих веществ и минимальные количества серы и
фосфора. Соотношение оксида кальция и углеродистого мате¬
риала зависит от заданного «литража». Литражом карбида
кальция называется объем ацетилена в литрах, приведенный к
20°С и ОД МПа, полученный при полном разложении 1 кг кар¬
бида кальция водой. Теоретический литраж 100%-го СаС2 ра¬
вен 377,73 л. С увеличением количества углерода в шихте лит¬
раж карбида кальция повышается, но выход его падает. Обыч¬
но применяется шихта с содержанием углерода 40—50% . При
этом литраж колеблется в пределах 230—300 л. При образова¬247
нии карбида кальция на 1 т его выделяется 400 м3 газа состава:
80—90% СО, 6—15% Н2, 0,5—3% С02, 2—7% N2, 1% СН4.Реакция гидратации карбида кальция с образованием аце¬
тилена представляет экзотермическую необратимую гетероген¬
ную реакцию взаимодействия карбида кальция с водой:СаС2 + 2Н20 = С2Н2 + Са(ОН)2 -АН АН = 129,9 кДж (в)Скорость реакции гидратации существенно зависит от дис¬
персности карбида кальция, интенсивности перемешивания и
температурного режима процесса. Для обеспечения необходи¬
мой скорости процесса разложения используют карбид кальция
в виде кусков размером 50—80 мм, интенсивно перемешивают
реакционную массу для удаления с поверхности слоя гидрокси¬
да кальция и обеспечивают отвод тепла, чтобы исключить по¬
лимеризацию образующегося ацетилена, поддерживая темпе¬
ратуру 100—400°С.11.3.2. Технологическая схема производства ацетилена
из карбида кальцияВ зависимости от условий, в которых проводится процесс
гидратации карбида кальция, различают два способа производ¬
ства ацетилена.1. Мокрый способ по принципу «карбид в воду», при кото¬
ром карбид кальция подается в реактор, содержащий большой
объем (до 10 м3 на 1 т карбида) воды, а реакционное тепло отво¬
дится за счет нагревания этой воды до температуры 50—60°С. В
этом случае гидроксид кальция получается в виде суспензии,
содержащей до 70% воды, что затрудняет его транспортировку
и последующее использование.2. Сухой способ по принципу «вода на карбид», при котором
вода подается в реактор, содержащий карбид кальция, только
в таком количестве, чтобы обеспечить полное протекание реак¬
ции гидратации (в), а реакционное тепло отводится за счет ис¬
парения этой воды. В этом случае гидроксид кальция получа¬
ется в виде твердого порошкообразного продукта, содержащего
не более 5% воды (пушонка), который направляется на рецир¬
куляцию в виде возвратной извести для регенерации в оксид
кальция, или используется для производства строительных
материалов. Так как возвратная известь составляет до 2/3 все¬
го известкового сырья, то за счет этого сухой способ производ¬
ства ацетилена значительно более экономичен и получил ши¬
рокое распространение.Газ, полученный по карбидному методу, достаточно концен¬
трирован (до 99,5% ацетилена), но содержит большое количе¬248
ство твердых частиц (пыли) и примеси аммиака, сероводорода
и фосфина. Для их удаления полученный ацетилен промывает¬
ся водой и слабым раствором гипохлорита натрия с добавкой
активного хлора для окисления примесей.Производительность установок для получения ацетилена из
карбида кальция составляет от 500 м3/ч ацетилена для генера¬
торов мокрого типа и до 2000 м3/ч для генераторов сухого типа.
Средний расход электроэнергии в карбидном методе равен
104 кВт*ч на тонну ацетилена.Современное производство ацетилена по карбидному методу
является комбинированным производством. В нем объединены
производства оксида кальция обжигом известняка, получения
карбида кальция и его гидратации, а также регенерация воз¬
вратной извести и использование оксида углерода (II) для обо¬
грева обжиговых печей и машин кальцинации гидроксида каль¬
ция. Технологическая схема подобного производства ацетиле¬
на по сухому способу представлена на рис. 11.2.СаСОШламРис. 11.2. Технологическая схема производства ацетилена:1 — обжиговая печь, 2 — бункер оксида кальция, 3 — бункер кокса, 4, 5—грохоты, 6 — загрузочный бункер обычной шихты, 7 — загрузочный бун¬
кер мелкой шихты, 8 — полый электрод печи, 9 — карбидная печь, 10 —
дробилка, 11 — сухой генератор, 12 — очистной скруббер, 13 — установ¬ка сухой очистки газа, 14 — машина для кальцинирования возвратнойизвести, 15 — отстойник известкового шлама
Обожженная известь (оксид кальция) из обжиговой печи 1
подается в бункер 2, где смешивается с оксидом кальция, по¬
ступающим из машины кальцинации 14. На грохоте 4 известь
разделяется на две фракции. Крупная фракция поступает в заг¬
рузочный бункер 6, в котором смешивается с крупной фракци¬
ей кокса с грохота 5. Мелкая фракция, пройдя грохот 4, посту¬
пает в загрузочный бункер 7, в котором смешивается с мелкой
фракцией кокса, прошедшей грохот 5. Обычная шихта, образо¬
вавшаяся смешением крупных фракций извести и кокса, пода¬
ется в карбидную печь 9 непосредственно, а мелкая шихта по¬
током газа-носителя, выходящего из карбидной печи, через
полый электрод 8. Карбид кальция после выпуска из печи и зат¬
вердевания измельчается в дробилке 10 и направляется в гене¬
ратор 11. Выходящий из генератора ацетилен с парами воды
направляется в скруббер 12, орошаемый водой, где охлаждает¬
ся до температуры 20—25 С и освобождается от пыли, после чего
подается на дальнейшую очистку от примесей аммиака, серо¬
водорода и фосфина. Твердый гидроксид кальция (пушонка)
выгружается из нижней части генератора на транспортер и по-
дается на кальцинацию в машину 14. Образовавшаяся в скруб¬
бере 12 взвесь гидроксида кальция в воде (известковое молоко)
поступает в отстойник 15, из которого водный слой после от¬
стаивания шлама добавляется к воде, орошающей скруббер.
Выходящий из карбидной печи 9 газ очищается от пыли в уста¬
новке 13 и используется как топливо в печи обжига известняка1 ив машине кальцинации 14.Основными аппаратами в производстве ацетилена по кар¬
бидному способу являются карбидная печь и генератор ацети¬
лена.Карбидная печь (рис. 11.3) пред¬
ставляет закрытую электротерми¬
ческую печь с вращающейся или
колеблющейся ванной, что способ¬
ствует разрыхлению шихты и уско¬
ряет ее плавление. Печь изнутри
футерована огнеупорным матери¬
алом и снабжена устройством для
отвода газа и леткой для выпуска
жидкого карбида кальция. Печь
имеет три электрода из самообжи-
гающейся углеродистой массы,
снабженных центральным кана¬Рис. 11.3. Карбидная печь:1 — корпус, 2 — футеровка,3 — покрытие печи, 4 — элект¬
род, 5 — летка, 6 — трубопро¬
вод для отвода газа, 7 — рельс250
лом, через который в печь вводится 25% от общей массы ших¬
ты в пылевидном состоянии. С помощью газодувки в печи
поддерживается небольшое избыточное давление, что предот¬
вращает подсос воздуха в печь извне. Производительность
печи мощностью 6-104 кВт, составляет 150 тыс. т карбида в
год, что соответствует 48 тыс. т
ацетилена при литраже 300 л.Генератор ацетилена сухого
типа (рис. 11.4) выполнен в виде
цилиндра с конической нижней ча¬
стью, снабженного полками и греб¬
ками, насаженными на общий вал.Карбид кальция из бункера со шлю¬
зовым затвором, обеспечивающим
герметичность аппарата, поступаетна верхнюю полку генератора. В Л А г* * * Рис. 11.4. Генератор:
верхнюю часть генератора с помо-- 1 — корпус, 2 — полки, 3 —Щью разбрызгивающего устройства скребкИ14 _ га3оотвод, 5 -подается с заданной скоростью вода, мешалка
Смоченный карбид кальция переме¬
щается гребками по спирали сверху вниз, пересыпаясь через
отверстия в полках, расположенных попеременно в центре и на
периферии, что обеспечивает полноту гидратации карбида.
Производительность сухих генераторов составляет 3500 м3/ч
газа или 30 тыс. т ацетилена в год.Таким образом, современное производство ацетилена по кар¬
бидному методу представляет комбинированное производство,
в котором объединены цехи по получению оксида кальция из
известняка, карбида кальция из шихты и ацетилена его гидра¬
тацией. Это позволяет рационально использовать сырье и энер¬
гию, стоимость которых достигает 80% себестоимости ацети¬
лена.Расходные коэффициенты в производстве ацетилена по сухому ме¬
тоду в расчете на литраж 300 л составляют на 1 т карбида кальция:Известь 950 кг.Кокс 550 кг.Электродная масса 10—15 кг.Электроэнергия 3150 кВт-ч.СаС251
11 Л. Производство ацетилена из углеводородного
сырья11.4.1. Физико-химические основы процесса высокотемпе¬
ратурного пиролиза алкановВысокотемпературный пиролиз алканов, используемый в
производстве ацетилена из углеводородного сырья, представля¬
ет эндотермическую обратимую реакцию их деструкции, про¬
текающую по радикально-цепному механизму:2СН4 ► 2СН3 ► 2СН2 ► 2-СН. -2Н ц -2Н . |j -2НСН3-СН3 сн2=сн2 сн-сни описываемую уравнениями:
в случае метана2СН4 ^ С2Н2 + ЗН2 + АНх АН1 = 376 кДж (а)в случае этанаС2Н6 С2Н2 + 2Н2 + АН2 ЛЯ2 = 311 кДж. (б)Равновесная степень превращения и выход ацетилена уве¬
личиваются при повышении температуры и понижении давле¬
ния (табл. 11.1 и рис. 11.5).Таблица 11.1. Выход ацетилена при различной температуре
и давлении (%)Давление, Па105104Температура, °С7271,54,8212799,8799,99Равновесие реакций (а) и (б) заметно смещается вправо уже
при температуре 1000—1300 С и выше 1500 С метан практи¬
чески полностью превращается в ацетилен с достаточной для
практических целей скоростью. Однако, при этой температуре
ускоряется побочная реакция термического распада образовав¬
шегося ацетилена:С2Н2 = 2С + Н2 + ДНз Л#з = 227,4 кДж. (в)Из уравнений реакций (а) и (в) следует, что реакция образо¬
вания ацетилена может рассматриваться как промежуточная
стадия цикла превращений:252
2С + 4Н2(г)- 2СН4 «=* С2Н2 + ЗН2Чтобы остановить процесс на
этой стадии и предотвратить распад
целевого продукта — ацетилена,
следует ограничивать степень кон¬
версии метана, уменьшая время
пребывания углеводорода в зоне ре¬
акции, снижая давление и приме¬
няя «закалку» продуктов реакции
путем быстрого охлаждения их до
температуры, при которой реакция
(в) практически не идет. Этим тре¬
бованиям удовлетворяет время кон¬
тактирования 0,01—0,001 с и сте- Рис. 11.5. Влияние температу-
пень конверсии метана не выше 0,5. ры на равновесную степеньРеакция пиролиза углеводоро- превращения при пиролизе
дов протекает при высокой темпера- метана и этана
туре. По способу подвода тепла креакционной системе методы пиролиза делятся четыре типа.1. Регенеративный пиролиз, при котором сырье нагревает¬
ся за счет контакта с предварительно разогретой насадкой печи
(регенератора);2. Гомогенный пиролиз, при котором сырье вводится в поток
горячего топочного газа, полученного сжиганием части сырья
или другого топлива;3. Электрокрекинг, при котором сырье нагревается в пла¬
мени электрической дуги;4. Окислительный пиролиз, при котором источником теп¬
ла является тепловой эффект сгорания части сырья. В этом
методе в одном аппарате совмещаются экзотермическая реак¬
ция горения углеводородов и эндотермическая реакция их пи¬
ролиза.В данной главе рассматриваются наиболее экономичный и
распространенный метод производства ацетилена окислитель¬
ным пиролизом и метод электрокрекинга.11.4.2. Производство ацетилена окислительным пиролизом
метанаПри окислительном пиролизе деструкция метана происхо¬
дит за счет тепла, выделяющегося при сжигании части его в
кислороде. Подвод тепла и пиролиз метана протекают непос¬
редственно в факеле горения, что способствует теплообмену253
между источником тепла и газом. При недостатке кислорода и
высокой температуре сгорание метана протекает, в основном,
по реакции:СН4 + 02 = СО + Н2 + Н20 -АН АН = 272 кДж (д)Скорость сгорания метана весьма высока, поэтому образова¬
ние ацетилена по реакции (а) начинается лишь в зоне, в кото¬
рой практически отсутствует кислород. В этой же зоне проис¬
ходит конверсия оксида углерода (II) по реакции водяного газа:СО + Н20 ^ С02 + Н2Для того, чтобы процесс окислительного пиролиза протекал
в автотермическом режиме, необходимо обеспечить оптималь¬
ное соотношение количества метана, сгорающего с выделением
тепла по реакции (д) и количества его, подвергающегося эндо¬
термической реакции пиролиза по реакции (а). Для этого уста¬
навливают, соотношение начальных объемов метана и кислоро¬
да в газовой смеси 1 : 0,65, что также лежит за пределами взры-
ваемости метан-кислородных смесей. В этих условиях при ус¬
тановившемся режиме процесса на горение (реакция д) рас¬
ходуется 55% метана, на образование ацетилена (реакция а)
23—25% и на образование сажи (реакция в) около 4%. Ско¬
рость подвода газа должна быть выше скорости распростра¬
нения пламени, чтобы оно не распространялось в обратном
направлении.При оптимальных условиях процесса, то есть применении
нагретого до 400—600С 98%-ного кислорода, температуре пи¬
ролиза 1450—1500 С и времени контактирования 0,004—0,006
с, степень конверсии метана в ацетилен достигает 0,3 при об¬
щей степени превращения метана 0,9 и кислорода 0,99. Газ про¬
цесса окислительного пиролиза метана имеет состав (% об.):
С2Н2 —8,0; С2Н4 — 0,5; С02 — 4,0; СО — 26,5; Н2 — 54,0; N2 —
3,0; СН4 — 4,0. Кроме того, в газе содержится 0,2—0,3% гомо¬
логов ацетилена, следы ароматических соединений и 1—3 г/м3
сажи и смолы.Для выделения и очистки ацетилена используется его боль¬
шая, чем у других компонентов пирогаза, растворимость в не¬
которых растворителях. Для этой цели в качестве сорбентовиспользуются метанол и ацетон при температуре
N-СНз -70 С , или, чаще, диметилформамид (CH^NCHOгq и N-метил пирролидон при обычной температу¬ре. В процессе очистки пирогаз сначала освобож¬
дается от сажи и смолы, затем от ароматических соединений и
гомологов ацетилена (форабсорбция), после чего из него извлека-254
ЗакалкаН20н2оДМФАIН2, СО, С02, СИ4-^синтез-газСИСажаIАгНгомологиtДМФАРис. 11,6. Принципиальная схема окислительного пиролиза метанают абсорбентом ацетилен, который затем очищают методом сту¬
пенчатой десорбции.На рис. 11.6 представлена принципиальная, а на рис. 11.7
технологическая схемы производства ацетилена окислитель¬
ным пиролизом метана.ВодаВодаНа десорбцию II ступениРис. 11.7. Технологическая схема производства ацетилена окисли¬
тельным пиролизом метана:1 — трубчатая печь (подогреватель) метана, 2 — трубчатая печь (подогре¬
ватель) кислорода, 3 — реактор, 4 — полый водяной скруббер, 5 — мок¬
рый электрофильтр, 6 — холодильник непосредственного смешения, 7 —
форабсорбер, 8 — газгольдер, 9 — отстойник сажи и смолы, 10 — ком¬
прессор, 11 — абсорбер, 12 — скруббер, 13 — дроссель, 14 — десор-
бер, 15 — скруббер255
Метан и кислород подогревают до 600 С в трубчатых печах 1
и 2У обогреваемых газом, соответственно, и поступают в реак¬
тор 3. Из реактора пирогаз с температурой после «закалки» во¬
дой 80°С проходит полый, орошаемый водой, скруббер 4 и мок¬
рый электрофильтр 5, в которых из газа осаждаются сажа и
смола. Затем пирогаз охлаждается водой в холодильнике непос¬
редственного смешения б, промывается в форабсорбере 7 не¬
большим количеством диметилформамида (ДМФА) и посту¬
пает в газгольдер 8. Вода, стекающая из реактора 3, скруб¬
бера 4 и электрофильтра 5, содержащая сажу, поступает в
отстойник 9, из которого водный слой возвращается в реак¬
тор для закалки, а собранная сажа с примесью смолы направ¬
ляется на сжигание. Газ из газгольдера 8 сжимается в комп¬
рессоре 10 до давления 1 МПа и подается в абсорбер 11, где
из него ДМФА извлекается ацетилен. Непоглощенный газ,
состоящий из водорода, метана и оксидов углерода, посту¬
пает в скруббер 12, орошаемый водой, в котором из газа улав¬
ливается унесенный газом ДМФА. Оставшийся газ использу¬
ют как топливо или в качестве синтез-газа. Раствор ацетилена
в ДМФА из абсорбера 11 проходит дроссель 13, где давление
снижается до 0,15 МПа, и поступает в десорбер 14. Десорбиро¬
ванный из раствора ацетилен про¬
мывается в скруббере 15 водой и вы¬
водится с установки. Основным ап¬
паратом в производстве ацетилена
окислительным пиролизом метана
является реактор.Реактор окислительного пиро¬
лиза (рис. 11.8) состоит из корпуса
прямоугольного сечения размером1,5x1,8 м2, футерованного огне¬
упорным материалом, внутри кото¬
рого размещены смесительная ка¬
мера, диффузор и горелочная кера¬
мическая плита с 780 отверстиями.
В горелочной плите расположены
две форсунки для подачи в реактор
«закалочной» воды. Метан и кисло-Рис. 11.8. Реактор пиролиза: Р°Д смешиваются в смесительнойкамере, проходят диффузор и посту-/ — корпус. 2 — смесительнаякамера, 3 - камера горения, пают чеРез отверстия горелочной
4 — диффузор, 5 — горелоч- плиты в камеру горения. Образовав-
ная плита, 6 — форсунки воды ШИЙСЯ ПИрОГаЗ ОТВОДИТСЯ ИЗ НИЖ-256
ней части реактора, в которой осаждается и большая часть сажи
и смолы.Расходные коэффициенты в производстве ацетилена окисли¬
тельным пиролизом метана составляют на 1 т ацетилена:— природный газ 1000 м3— кислород (98% -ный) 3600 м3— электроэнергия 1570 кВт-ч.11.4.3. Производство ацетилена электрокрекингом метанаПроцесс электрокрекинга заключается в быстром пропуска¬
нии метана через зону высоких температур, создаваемых элек¬
трической дугой. Реактором в этом методе служит электроду-
говая печь, в которой при пропускании постоянного тока на¬
пряжением 7000—8000 В создается дуга с температурой около
2000°С. Электродуговая печь вертикального типа (рис. 11.9)
состоит из верхней цилиндрической реакционной камеры диа¬
метром 1 м и высотой 0,4 м и трубы диаметром 0,1 м и длиной
1,0 м. На камере установлен медный катод в виде гильзы, а на
верхней части трубы — анод. Катодная гильза и анодная труба
снабжены рубашками водяного охлаждения. Метан под давле¬
нием подается тангенциально в ка¬
меру, за счет чего поток газа приоб¬
ретает вихревую скорость около 100
м/с и направляется от периферии к
трубе. При этом он как бы втягивает
электрическую дугу в кольцевое про¬
странство анода, где при температу¬
ре 1600°С и происходит пиролиз ме¬
тана. Продукты пиролиза проходят
со скоростью 600—1000 м/с через ох¬
лаждаемую водой анодную трубу, ох¬
лаждаясь при этом до 600°С и посту¬
пают в закалочное устройство. В нем
за счет впрыскивания воды пирогаз
быстро охлаждается до 150С. Мощ¬
ность электрической печи по метану
составляет 2800 м3/ч, что соответ¬
ствует производительности по ацети- ^ ^ Элект опечь-лену 15 т/сут. Степень конверсии ме-0С к 1 — катод, 2 — реакционная,55 э г ." " ’ л камера, 3 — анод, 4 —при расходе электроэнергии 10 заКалочное устройство, 5 -кВт-ч/кг ацетилена. водяная рубашка9 Химическая технология. Том 2257
Газ электрокрекинга метана имеет состав (% объемных):
С2Н2 — 13—14; С2Н4 — 1; СН4 — 30—35; Н2 — 50—55. Из
103 м3 метана при электрокрекинге образуется 300 кг ацетиле-
на, 26 кг этилена, 1170 м3 водорода и свыше 20 кг высококаче¬
ственной сажи. Себестоимость ацетилена, полученного методом
электрокрекинга составляет около 35% себестоимости карбид¬
ного ацетилена.Контрольные вопросы1. Охарактеризуйте ацетилен как химическое сырье для ООС.2. Из каких стадий состоит производство ацетилена по карбидному
методу?3. Что такое литраж карбида кальция?4. Какими методами может быть получен ацетилен из углеводород¬
ного сырья?5. Какие реакции лежат в основе окислительного пиролиза метана?6. Для чего нужна «закалка» продуктов реакции при синтезе ацети¬
лена?7. Что является источником энергии при производстве ацетилена ме¬
тодом электрокрекинга метана?
Глава XII
ПРОИЗВОДСТВО СПИРТОВ12.1. Производство метанола12.1.1. Технологические свойства метанолаМетанол (метиловый спирт) СН3ОН представляет бесцвет¬
ную легкоподвижную жидкость с температурой кипения
64,65°С, температурой кристаллизации -97,9°С и плотностью0,792 т/м3. Критическая температура метанола равна 239,65°С.
Метанол смешивается во всех отношениях с водой, спиртами,
бензолом, ацетоном и другими органическими растворителями,
образуя с некоторыми из них азеотропные смеси. Не растворим
в алифатических углеводородах. В водных растворах образует
эвтектику, содержащую 93,3% мол. метанола. Хорошо раство¬
ряет многие газы, в том числе оксиды углерода, ацетилен, эти¬
лен и метан, вследствие чего используется в технике для абсор¬
бции примесей из технологических газов. В твердом состоянии
существует в двух кристаллических формах, переходящих одна
в другую при -115,75°С. Пары сухого метанола образуют с воз¬
духом взрывчатые смеси с пределами взрываемости: нижний
6,0% объемных и верхний 34,7% объемн. Метанол токсичен,
вызывает отравление через органы дыхания, кожу и при при¬
еме внутрь, действуя на нервную и сосудистую системы. ПДК
составляет 5 мг/м3. Прием внутрь 5—10 мл приводит к тя¬
желому отравлению, доза 30 мл и более может быть смертель¬
ной.Метанол впервые был обнаружен Р. Бойлем в 1661 году в
продуктах сухой перегонки древесины (отсюда название ме¬
танола — древесный спирт). В чистом виде выделен в 1834
году Ж. Дюма и Э. Пелиго, установившими его формулу, син¬
тезирован омылением хлористого метила М. Бертло в 1857
году. Промышленное производство метанола синтезом из во¬
дорода и оксида углерода (II) впервые было осуществлено в
1923 году и с тех пор непрерывно совершенствуется. В на¬
шей стране производство метанола впервые организовано в1934 году в объеме 30 т в сутки на Новомосковском хими¬
ческом комбинате из водяного газа, получаемого газифи¬
кацией кокса.9*259
12.1.2. Синтезы на основе синтез-газа
или оксида углерода (II)Современное производство метанола представляет один из
примеров промышленного органического синтеза на основе син¬
тез-газа (СО + Н2) или оксида углерода (II). Из многочисленных
каталитических превращений этого типа можно выделить сле¬
дующие наиболее важные направления.1. Синтез углеводородов (Ф. Фишер и Г. Тропш, 1928—1935 гг.) с целью получения моторного топлива (синтина):СОпСО + (2п + 1)Н2 —> С„Н2п+2 + лН20 илиFe2nCO + (п + 1)Н2 * С„Н2л+2 + пС022. Оксосинтез или гйдроформилирование (О. Релен 1938 г.)
с целью получения первичных спиртов и кислот на их основе,
например:СН2=СН2 + СО + Н2 — СН3-СН2-СНОН2сн3-сн2-сн2онс2сн3-сн2-соона также высших спиртов линейного строения:Н2к-сн=сн2 + со + н2 к-сн2-сн2-сно R-CH2-CH2 -сн2он,
где R = Cg—Cie3. Синтез карбоновых кислот и их производных (В. Реппе
1949 г.) из алкенов, ацетилена и спиртов, например:сн2=сн2 + со + н2о — сн3-сн2-соон
СН-СН + СО + ROH — ch2=ch-coorROH + СО — RCOOH4. Синтез первичных спиртов (Патар 1924 г.)
пСО + (тН-1)Н2 —► C„H2n+iOHИз этих процессов наибольшее промышленное значение по¬
лучил синтез метанола. Сырьём для этого синтеза служит син¬
тез-газ, получаемый конверсией природного газа, газификаци¬
ей углей, переработкой нефти и нефтепродуктов, а также обра¬
зующийся как отход других производств. Структура сырьевого
баланса в производстве метанола по данным 1980 года представ¬
лена в табл. 12.1.260
Таблица 12.1. Структура сырья в производстве метанола, %СырьеВ миреВ нашей странеПриродный газ73,870,7Нефть и нефтепродукты24,44,0Отходы других производ.—17,4Каменный уголь1,87,912.1.3. Физико-химические основы синтеза метанолаРеакция синтеза метанола из синтез-газа представляет гете-
рогенно-каталитическую обратимую экзотермическую реак¬
цию, протекающую по уравнению:СО + 2Н2 ^ СН3ОН -AHi A# 1 = 90,7 кДж (а)Тепловой эффект реакции возрастает с повышением темпе¬
ратуры и давления и для условий синтеза составляет 110,8 кДж.В отличие от синтеза аммиака (том 1) при синтезе метанола
параллельно с основной реакцией протекают побочные реакции:СО + ЗН2 ^ СН4 + Н20 - АЯ2 Aif2 = 209 кДж (б)
2СО + 2Н2 «=* СН4 + С02 - ДНз Aif3 = 252 кДж (в)СО + Н2 ^ СН20 - АН4 АЯ4 = 8,4 кДж (г)а также продукционная реакция образования метанола из со¬
держащегося в синтез-газе оксида углерода (IV):С02 + ЗН2 СН3ОН + Н20 -Aif5 АЯ5 = 49,5 кДж (д)Кроме этого, образовавшийся метанол может подвергаться
вторичным превращениям по реакциям:2СН30Н ^ СН3-О-СН3 + Н20
СН3ОН + пСО + 2лН2 ^ СН3(СН2)л ОНСН3ОН + Н2 ^ СН4 + Н20Реакции (а—д) протекают с выделением тепла и уменьше¬
нием объема, но различаются величиной теплового эффекта и
степенью контракции. Поэтому, хотя для всех этих реакций
степень превращения возрастает с увеличением давления и по¬
нижением температуры, в наибольшей степени повышение дав¬
ления влияет на равновесие основной реакции синтеза (а), для
которой степень контракции максимальна и составляет 3:1. В
то же время, понижение температуры ниже некоторого преде¬
ла нецелесообразно, так как при низких температурах скорость261?1г
Т.процесса синтеза настолько мала,
что не существует катализатора,
который в этих условиях мог бы су¬
щественно ускорить достижение
высокой степени превращения сы¬
рья. На рис. 12.1 показано влияние
давления на равновесный выход7’з>72>7’1Рис. 12.1. Влияние давления на
выход метанола при различ¬
ной температуре процесса... метанола при различных темпера-патурах синтеза.Вследствие противоречивого
влияния температуры на скорость
процесса и равновесную степеньпревращения выход метанола за один проход реакционной сме¬
си через реактор не превышает 20%, что делает необходимой
организацию циркуляционной технологической схемы синте¬
за.Температура процесса зависит главным образом от активно¬
сти применяемого катализатора и варьируется в пределах от 250
до 420°С. В соответствии с температурным режимом работы ка¬
тализаторы синтеза метанола подразделяются на высокотемпе¬
ратурные и низкотемпературные. Высокотемпературные ката¬
лизаторы, получаемые методом соосаждения оксидов цинка и
хрома, например, катализатор СМС-4 состава 2,5 ZnOZnCr2C>4,
термостойки, мало чувствительны к каталитическим ядам, при¬
чем отравляются обратимо, имеют высокую селективность, но
активны только при высоких температурах (370—420°С) и давле¬
ниях (20—35 МПа). Низкотемпературные катализаторы, напри¬
мер, цинк-медь-алюминиевый состава ZnOCuOA^Og или цинк-
медь-хромовый состава ZnOCuOC^Oa, менее термостойки, нео¬
братимо отравляются каталитическими ядами, но проявляют
высокую активность при относительно низких температурах
(250—300°С) и давлениях (5—10 МПа), что более экономично.Оба типа катализаторов проявляют свою активность и селек¬
тивность в узком интервале температур 20—30°С. Исходя из тем¬
пературного режима работы катализаторов выбирается давле¬
ние синтеза, которое тем больше, чем выше температура синте¬
за.Состав исходной газовой смеси оказывает существенное вли¬
яние как на степень превращения оксидов углерода, так и на
равновесную концентрацию метанола в продуктах синтеза. С
увеличением объемного отношения Нг:СО в синтез-газе степень
превращения оксидов углерода возрастает, причем оксида уг¬
лерода (IV) более интенсивно (рис. 12.2). Из рисунка также еле-262
CCHgOHРис. 12.2. Зависимость степени Рис. 12.3. Зависимость равновес-
превращения СО и СО2 от ной концентрации метанола отсостава исходной смеси (% об.) состава исходной смеси (% об.)дует, что оптимальный состав газовой смеси отвечает отноше¬
нию Н2:СО = 5:1. Равновесная концентрация метанола в про¬
дуктах реакции проходит через максимум, который отвечает
стехиометрическому отношению Н2 : СО в исходной газовой
смеси (рис. 12.3).Синтез метанола на окисных катализаторах приведенного
выше состава проходит через те же стадии, что и синтез аммиа¬
ка на железном катализаторе (том 1) и лимитируется наиболее
медленной стадией хемосорбции молекул водорода на поверхно¬
сти катализатора. Он может быть представлен в следующем виден2 + [Г *чн2ГСО + 2[Н2]х ^ СН3ОН + [ ]СО + 2Н2 5=* СН3ОНгде: [ ] — свободная поверхность катализатора.Скорость образования метанола является функцией многих
переменных:uJCc»,o3 ,tT,p) (12.1)dxгде: к — константа скорости реакции синтеза метанола;Ск — концентрация компонентов исходной газовой
смеси,
х — время контакта,Т — температура,Р — давление.263
Образующиеся при синтезе побочные продукты оказывают
существенное влияние на стадию хемосорбции и на кинетику
образования метанола в целом. Поэтому, для реакции синтеза
метанола предложено большое количество различных кинети¬
ческих уравнений, выведенных на основе выдвинутых их авто¬
рами предположений о механизме реакции. Независимо от это¬
го, время контактирования для реальных условий процесса син¬
теза может быть рассчитано по формуле:273 3600 Р
Т= *°ек^ (12.2)где: Р — давление, 1 МПа
Т — температура, КW — объемная скорость газа при нормальных усло¬
виях, с-1.12.1.4. Общая схема производства метанолаМногочисленные технологические схемы производства ме¬
танола включают три обязательных стадии:— очистка синтез-газа от сернистых соединений, карбони¬
лов железа и частиц компрессорного масла,— собственно синтез,— очистка и ректификация метанола-сырца.В остальном технологические схемы различаются аппаратур¬
ным оформлением и параметрами процесса. Все они могут быть
разделены на три группы.1. Синтез при высоком давлении проводится на цинк-хро-
мовых катализаторах при температуре 370—420‘С и давлении
20—35 МПа. В настоящее время этот процесс устарел и вытес¬
няется синтезом при низком давлении.2. Синтез при низком давлении проводится на цинк-медь-
алюминиевых или цинк-медь-хромовых катализаторах при
температуре 250—300С и давлении 5—10 МПа. Использование
в этом методе низкотемпературных катализаторов, активных
при более низких давлениях, позволяет снизить энергозатраты
на сжатие газа и уменьшить степень рециркуляции непрореа¬
гировавшего сырья, то есть увеличить степень его конверсии.
Однако, в этом методе требуется особо тонкая очистка исходно¬
го газа от соединений, отравляющих катализатор.3. Синтез в трехфазной системе «газ—жидкость—твер¬
дый катализатор», проводимый в суспензии из тонкодиспер¬
сного катализатора и инертной жидкости, через которую бар-264
ботируется синтез-газ. Этим процесс отличается от первых двух,
которые проводятся в двухфазной системе «газ—твердый ка¬
тализатор». В трехфазной системе может быть обеспечено бо¬
лее благоприятное состояние равновесия системы, что позволя¬
ет повысить равновесную концентрацию метанола в реакцион¬
ной смеси до 15% вместо 5% при использовании двухфазных
систем, доведя степень конверсии оксида углерода (II) до 35%
вместо 15% и ещё более уменьшить рециркуляцию газа и энер¬
гозатраты.Основным аппаратом в синтезе метанола служит реактор —
контактный аппарат, конструкция которого зависит, главным
образом, от способа отвода тепла и принципа осуществления
процесса синтеза. В современных технологических схемах ис¬
пользуются реакторы трех типов:— трубчатые реакторы, в которых катализатор размещен
в трубах, через которые проходит реакционная масса, охлаж¬
даемая водным конденсатом, кипящим в межтрубном простран¬
стве;— адиабатические реакторы с несколькими слоями ката¬
лизатора, в которых съем тепла и регулирование температуры
обеспечивается подачей холодного газа между слоями катали¬
затора;— реакторы для синтеза в трехфазной системе, в которых
тепло отводится за счет циркуляции жидкости через котел-ути-
лизатор или с помощью встроенных в реактор теплообменни¬
ков.12.1.5. Технологические схемы производства метанолаВследствие большого объема производства и весьма крупных
капитальных затрат в производстве метанола сейчас использу¬
ют все три типа технологических процессов. На рис. 12.4 пред¬
ставлена технологическая схема производства метанола при
низком давлении на цинк-медь-алюминиевом катализаторе из
синтез-газа состава: Н2 — 67%, СО — 22%, С02 — 9% объем¬
ных, полученного конверсией метана, производительностью
400 тыс. т в год.Очищенный от сернистых соединений синтез-газ сжимается
в компрессоре 1 до давления 5—9 МПа, охлаждается в холодиль¬
нике 3 и поступает в сепаратор 4 для отделения сконденсиро¬
вавшейся воды. Пройдя сепаратор, синтез-газ смешивается с
циркуляционным газом, который поджимается до рабочего дав¬
ления в компрессоре 2. Газовая смесь проходит через адсорбер265
Циркуляционный газРис. 12.4. Технологическая схема производства метанола
при низком давлении:1 — турбокомпрессор, 2 — циркуляционный компрессор, 3, 7 —холодиль¬
ники, 4 — сепаратор, 5 — адсорбер, 6 — реактор адиабатического дей¬
ствия, 8 — теплообменник, 9 — котел-утилизатор, 10 — сепаратор, 11 —
дроссель, 12 — сборник метанола-сырца, 13, 14 — ректификационные
колонны5, где очищается от пентакарбонила железа, образовавшегося
при взаимодействии оксида углерода (II) с материалом аппара¬
туры, и разделяется на два потока. Один поток подогревают в
теплообменнике 8 и подают в верхнюю часть реактора 6, а дру¬
гой поток вводят в реактор между слоями катализатора для от¬
вода тепла и регулирования температуры процесса. Пройдя ре¬
актор, реакционная смесь при температуре около 300°С также
делится на два потока. Один поток поступает в теплообменникS, где подогревает исходный синтез-газ, другой поток проходит
через котел-утилизатор 9> вырабатывающий пар высокого дав¬
ления. Затем потоки объединяются, охлаждаются в холодиль¬
нике 7 и поступают в сепаратор высокого давления 10, в кото¬
ром от циркуляционного газа отделяется спиртовой конденсат.
Циркуляционный газ дожимается в компрессоре 2 и возвраща¬
ется на синтез. Конденсат метанола-сырца дросселируется в
дросселе 11 до давления близкого к атмосферному и через сбор¬
ник 12 поступает на ректификацию. В ректификационной ко¬
лонне 13 от метанола отгоняются газы и диметиловый эфир,266
которые направляются на сжигание. В ректификационной ко¬
лонне 14 метанол отгоняется от тяжелокипящих высших спир¬
тов, которые также сжигаются. Полученный товарный метанол
с выходом 95% имеет чистоту 99,95% .На рис. 12.5 приведена технологическая схема производства
метанола по трехфазному методу на медь-цинковом катализа¬
торе из синтез-газа, полученного газификацией каменного угля,
производительностью 650 тыс. т в год.Очищенный от соединений серы синтез-газ сжимается в ком¬
прессоре 1 до давления 3—10 МПа, подогревается в теплообмен¬
нике 5 продуктами синтеза до 200— 280°С, смешивается с цир¬
куляционным газом и поступает в нижнюю часть реактора 4/
Образовавшаяся в реакторе парогазовая смесь, содержащая до
15% метанола, выходит из верхней части реактора, охлажда¬
ется последовательно в теплообменниках 5 и б и через холодиль¬
ник-конденсатор 7 поступает в сепаратор 8> в котором от жид¬
кости отделяется циркуляционный газ. Жидкая фаза разде¬
ляется в сепараторе на два слоя: углеводородный и метаноль-
ный. Жидкие углеводороды перекачиваются насосом 9 6 реак-Рис. 12.5. Технологическая схема производства метанола
в трехфазной системе:1 — компрессор, 2 — циркуляционный компрессор, 3, 9 — насосы, 4 —
реактор кипящего слоя, 5, 6 — теплообменники, 7 — холодильник-
конденсатор, 8 — сепаратор, 10 — котел-утилизатор267
тор, соединяясь с потоком углеводородов, проходящих через
котел-утилизатор 10. Таким образом жидкая углеводородная
фаза циркулирует через реактор снизу вверх, поддерживая ре¬
жим кипящего слоя тонкодисперсного катализатора в нем, и
одновременно обеспечивая отвод реакционного тепла. Метанол-
сырец из сепаратора 8 поступает на ректификацию или исполь¬
зуется непосредственно как топливо или добавка к топливу.Разработанный в 70-х годах трехфазный синтез метанола ис¬
пользуется в основном, для производства энергетического про¬
дукта. В качестве жидкой фазы в нем применяются стабильные
в условиях синтеза и не смешивающиеся с метанолом углеводо¬
родные фракции нефти, минеральные масла, полиалкилбензо-
лы. К указанным выше преимуществам трехфазного синтеза
метанола следует добавить простоту конструкции реактора, воз¬
можность замены катализатора в ходе процесса, более эффектив¬
ное использование теплового эффекта реакции. Вследствие это¬
го установки трехфазного синтеза более экономичны по сравне¬
нию с традиционными двухфазными как высокого так и низко¬
го давления. В табл. 12.2 приведены показатели работы устано¬
вок трех- и двухфазного процесса одинаковой производительно¬
сти 1800 т/сут.Таблица 12.2. Показатели работы установок синтеза
метанолаПоказательТип установкиТрехфазнаяДвухфазнаяДавление, МПа7,6510,3Объемная скорость газа, ч-140006000Отношение циркуляционного газак исходному синтез-газу1:15:1Концентрация метанола на выходе, % мол.14,55,0Мощность, потребляемая аппаратурой, кВт9574855Термический коэффициент полезногодействия,%97,986,3Относителные капитальные затраты0,771,0012.1.6. Применение метанола и перспективы развития
производстваИнтенсивное развитие производства метанола (например, в
нашей стране оно выросло с 1970 года по 1985 год более, чем в268
четыре раза) вызвано многообразием и непрерывным расшире¬
нием сфер его использования.Метанол — сырье для многих производств органического
синтеза. Основное количество его расходуется на получение
формальдегида. Он служит промежуточным продуктом в син¬
тезе сложных эфиров органических и неорганических веществ
(диметилтерефталата, метилметакрилата, диметил сульфата),
пентаэритрита. Его применяют в качестве метилирующего сред¬
ства для получения метиламинов и диметиланилина, карбофо¬
са, хлорофоса и других продуктов. Метанол используют также
в качестве растворителя и экстрагента, в энергетических целях
как компонент моторных топлив и для синтеза метил-трет-бу-
тилового эфира — высокооктановой добавки к топливу. В пос¬
леднее время наметились новые перспективные направления
использования метанола, такие как производство уксусной кис¬
лоты, очистка сточных вод, производство синтетического про¬
теина, конверсия в углеводороды с целью получения топлива.
В табл. 12.3 представлена структура потребления метанола по
основным направлениям в нашей стране и в Западной Европе
(данные 1985 года).Таблица 12.3. Структура потребления метанола, %Область примененияНаша странаЗападная ЕвропаПроизводство формальдегида34,642,4Производство СК12,6—Производство диметилтерефталата1,84,3Производство уксусной кислоты2,06,0Компонент моторного топлива1,06,3Процессы метилирования4,710,7Прочие направления использования43,330,3Возросшая потребность в метаноле вызвала разработку но¬
вых перспективных методов его производства. Помимо описан¬
ного выше трехфазного синтеза к ним относятся:— синтез метанола прямым окислением метана воздухом на
цинк-никель-кадмиевом катализаторе, позволяющий исполь¬
зовать в качестве сырья природный газ непосредственно из сква¬
жин;— совместное производство из синтез-газа метанола и спир¬
тов С2—С4 в виде так называемой «спиртовой композиции»,
используемой как добавка к моторному топливу;— совместное производство метанола и аммиака на основе
конвертированного газа по малоотходным энерготехнологичес¬269
ким схемам, обеспечивающим рациональное и комплексное
использование сырья.Несмотря на то, что доля метанола используемого на произ¬
водство моторного топлива в настоящее время еще невелика (см.
табл. 12.4), использование его для топливно-энергетических
целей стало весьма перспективным. Это обусловлено возмож¬
ностью получения метанола из любого углеродсодержащего сы¬
рья и неограниченными запасами его, что позволяет использо¬
вать метанол в качестве полупродукта в производстве синтети¬
ческого моторного топлива.Так, например, по технологической схеме «Мобиль» осуще¬
ствляется следующий цикл: уголь —* газификация —* метанол
—► синтетический бензин.Процесс протекает в две стадии: дегидратация метанола до
диметилового эфира и, далее, до алкена:2СН3ОН - СН3ОСН3 + Н20 — СН2=СН2 + 2Н20и последующие превращения алкенов в парафины, циклопара¬
фины и ароматические углеводороды. В качестве катализато¬
ров используются синтетические цеолиты.12.2. Производство этанола12.2.1. Технологические свойства и применение этанола**Этанол (мети л карбинол, этиловый спирт) — бесцветная под¬
вижная жидкость с жгучим вкусом и характерным запахом.
Температура кипения этанола 78,4°С, температура плавления
—114,15°С, плотность 0,794 т/м3. Этанол смешивается во всех
отношениях в водой, спиртами, глицерином, диэтиловым эфи¬
ром и другими органическими растворителями. С некоторыми
из них (водой, бензолом, этилацетатом, хлороформом) он обра¬
зует азеотропные смеси различного состава. Азеотропная смесь
с водой, содержащая 95,6% об. этанола, кипит при постоянной
температуре 78,ГС. Поэтому, для получения безводного («аб¬
солютного») этанола в промышленности используют специаль¬
ные методы его обезвоживания, например, абсолютирование
бензолом. Этанол образует алкоголяты с солями кальция и маг¬
ния, например: СаС12*4С2Н50Н и MgCl2-6C2H50H.Температура самовоспламенения этанола составляет 422,8 С.
С воздухом пары его образуют взрывчатые смеси с температу¬
рой вспышки 13,0°С в достаточно узких пределах взрываемос-
ти: нижний 3,28% об. и верхний 18,95% об. Этанол обладает
наркотическим действием, ПДК этанола равна 1000 мг/м3. Дли¬270
тельное воздействие этанола на организм вызывает тяжелые
органические заболевания нервной системы, пищеварительно¬
го тракта и печени, сердечно-сосудистой системы.Этанол является одним из наиболее важных и крупномасш¬
табных продуктов основного органического синтеза (мировое
производство в 1980 году составило более 2,5 млн. т). Он исполь¬
зуется в качестве растворителя в различных отраслях промыш¬
ленности (лакокрасочной, фармацевтической, в производстве
взрывчатых веществ, кино-, фото-, бытовой химии), антисеп¬
тика, сырья для производства синтетического каучука, кормо¬
вых дрожжей, ацетальдегида и уксусной кислоты, хлорофор¬
ма, диэтилового эфира, этилацетата, моно- и диэтиламинов и
других органических продуктов; компонента ракетных топлив
и антифризов. Значительная часть производимого этанола рас¬
ходуется на приготовление спиртных напитков, в парфюмер¬
ной промышленности. В табл. 12.4 представлена структура по¬
требления этанола (США, 1970 год).Таблица 12.4. Структура потребления этанола, %Область потребления этанола%Производство ацетальдегида50,5Органический синтез20,0Использование в качестве растворителя10,3Парфюмерная промышленность7,6Для пищевых и медицинских целей4,4Для производства антисептиков3,9Прочие области потребления3,312.2.2. Промышленные способы производства этанолаПромышленные способы производства этанола, а вместе с тем
и структура потребляемого для этой цели сырья, непрерывно
изменялись. На смену ректификации вина (отсюда название
этанола — винный спирт) пришли методы, основанные на хи¬
мической переработке сырья. В настоящее время все промыш¬
ленные методы производства этанола и, соответственно, сорта
производимого продукта делятся на четыре группы:— гидратация этилена (синтетический этанол — I),— гидролиз древесины (гидролизный этанол — II),— осахаривание крахмала (ферментативный или пищевой
этанол — III),271
ПирогазсернокислотнаяСн гидратация2 4прямая► С2Н5ОНц Отходы _
древесиныЗерно —^III КрахмалосахариваниеЦеллюлозагидролиз p-D-глюкозаa-D-глюкоза^ брожениеL'6M1ZJ6 Картофель^ Сульфитные
щелокаРис. 12.6. Промышленные способы производства этанола— переработка сульфитных щелоков (сульфитный этанолНа рис. 12.6 представлена взаимосвязь промышленных ме¬
тодов производства этанола.Выход этанола существенно зависит от вида сырья и состав¬
ляет (в л на 1 т сырья): для этилена 740, картофеля 93—117,
зерна 185—361, древесины 160—200, сульфитных щелоков
90—110 (в расчете на 1 т древесины). При использовании в ка¬
честве сырья древесины и сульфитных щелоков помимо этано¬
ла образуются дрожжи, фурфурол, лигнин и лигниносульфо-
наты и гипс. Во всех вариантах биохимического метода произ¬
водства этанола выделяется оксид углерода (IV).12.2.3. Краткий исторический очеркЭтанол известен человеку с глубокой древности. Первые упоми¬
нания о нем относятся к VIII в. В XI—XII вв. этанол получают ректи¬
фикацией виноградного вина. С XII в. этанол применяли в медицине,
а с 1600 г. в химических экспериментах для экстракции органичес¬
ких веществ. В 1682 г. И. Бехер впервые описывает метод получения
водного этанола из картофеля, а в 1748 г. опубликовано сообщение
Шведской академии наук о промышленном использовании этого ме¬
тода. Безводный этанол впервые получает в России в 1796 г. Т.Е. JIo-
виц. В 1798 г. А. Арганд применяет метод ректификации для перегон¬
ки этанола и с 1820 г. его используют в промышленном масштабе. В
1783 г. А. Лавуазье устанавливает элементный состав этанола и пы¬
тается объяснить механизм спиртового брожения, окончательно вы¬
ясненный в XIX в. В 60-х гг. XIX в. Д.И. Менделеев детально исследу¬IV).производства272
ет сиотемы «этанол—вода», используя полученные данные для разра¬
ботки гидратной теории.В нашей стране этанол до 1934 г. получали исключительно из пи¬
щевого сырья. В 1934 г. было освоено производство гидролизного эта¬
нола и в 1935 г. построен первый гидролизный завод в Ленинграде. На
основе работ по сернокислотной гидратации этилена в 1936 г. был пу¬
щен первый завод в Баку. После освоения метода прямой гидратации
этилена в 1952 г. в Сумгаите, в период с 1953 по 1958 г. были введены
в строй аналогичные заводы в Саратове, Уфе, Грозном и Самаре. В ре¬
зультате к 1960 г. доля синтетического этанола в общем объеме его
производства достигла 25%. С 1964 г. в стране было полностью прекра¬
щено использование пищевого этанола для технических целей. В на-
стоящее время перевод производства бутадиена с этанольного на угле¬
водородное сырье высвобождает значительное количество синтетичес¬
кого этанола. Его предполагается использовать как сырье для эко¬
логически чистого производства кормовых дрожжей. В то же вре¬
мя, в связи с переориентацией гидролизных заводов также на про¬
изводство кормовых дрожжей, производство гидролизного этано¬
ла прекращается.12.2.4. Производство этанола прямой
гидратацией этиленаПроизводство этанола прямой гидратацией этилена представ¬
ляет собой частный и наиболее важный случай получения спир¬
тов гидратацией алкенов. Присоединение воды к алкенам мо¬
жет быть осуществлено двумя методами:— через алкилсульфаты (сернокислотная гидратация) и— взаимодействием с водой в присутствии катализаторов
(прямая гидратация).Процесс сернокислотной гидратации связан с необходимо¬
стью упаривать большой объем (до 4 т на 1 т этанола) образую¬
щейся разбавленной до 40—50% серной кислоты, что эконо¬
мически нецелесообразно. Поэтому, в настоящее время серно¬
кислотная гидратация сохранила свое значение только для
получения изопропанола и изобутанолов, производимых в от¬
носительно небольших количествах. Для производства этано¬
ла используется исключительно метод прямой гидратации.Условия процесса абсорбции алкенов серной кислотой при
сернокислотной гидратации зависят от реакционной способно¬
сти алкена и выбираются так, чтобы свести к минимуму побоч¬
ные реакции его полимеризации. В табл. 12.5 приведен режим
абсорбции алкенов различного строения серной кислотой при
сернокислотной гидратации.Г;к273
Таблица 12.5. Условия процесса абсорбции алкеновАлкенКонцентрация
H2S04, мае. допейДавление,МПаТемпература,°ССН2=СН20,962,570СН2=СН-СН30,700,870СН2=СН-СН2-СНз0,800,445СН2=С(СН3)20,650,430Физико-химические основы процесса. Присоединение воды
по двойной связи (гидратация) может проводиться в жидкой или
паровой фазе. В промышленном масштабе используется преиму¬
щественно второй вариант.Гидратация в паровой фазе этилена представляет гетероген¬
но-каталитическую обратимую экзотермическую реакцию, про¬
текающую по уравнению:С2н4 (г) + н20 (П) ^ С2Н5ОН (П) - АН АН = 45,6 кДж (а)Реакция гидратации катализируется кислыми и нейтраль¬
ными катализаторами, из которых наиболее распространена
фосфорная кислота на носителе кизельгуре или силикагеле. В
присутствии фосфорной кислоты протекает электрофильное
присоединение воды к этилену по схеме:Н3РО4 ^ Н+ + Н2Р04-СН2=СН2 + Н+ сн3-сн2+СН3-СН2+ + Н-О-Н CH3-CH2-d< д 5=s СН3-СН2-ОН + н+Свободная фосфорная кислота находится в жидком состоя¬
нии в виде пленки на поверхности зерен катализатора. Таким
образом, при формально твердом катализаторе, катализ проте¬
кает фактически в жидкой фазе. Вследствие этого активность
катализатора зависит от концентрации кислоты и, следователь¬
но, от парциального давления паров воды в системе и темпера¬
туры. При концентрации кислоты ниже 83% массовых, актив¬
ность контактной массы резко снижается,поэтому вопреки тре¬
бованиям термодинамики, процесс гидратации нельзя вести в
избытке водяного пара, так как это уменьшает концентрацию
кислоты. На практике мольное отношение Н2О : С2Н4 составля¬
ет 0,6:1. Время работы фосфорного катализатора достигает 500
часов, после чего активность катализатора падает за счет уноса
части кислоты током газообразных продуктов. Во избежание
этого в систему в процессе работы непрерывно вводится свежая
фосфорная кислота.274
Реакция гидратации протекает с выделением тепла и умень¬
шением объема. Константа равновесия этой реакции выража¬ется уравнением:1 gKD=2093-6,304+^103 625004V/(12.3)Из уравнения следует, что рав¬
новесная степень превращения эти¬
лена возрастает с понижением тем¬
пературы и повышением давления
(рис. 12.7). Однако применение вы¬
сокого давления повышает затра¬
ты на аппаратуру и энергию, а при
низких температурах скорость ре¬
акции весьма мала. Оптимальными
условиями процесса являются: тем¬
пература 260-300С, давление 7—8
МПа, объемная скорость парогазо- Рис. 12.7. Влияние температу-
вой смеси 1800—2500 ч*1. ры и давления на степеньСодержание в этилене инертных превращения этилена в эта-
примесей отрицательно влияет как нол при реакции гидратации,
на скорость реакции, так и на рав- Ъ> Т2> Т\
новесную степень превращения.Поэтому газ, поступающий на гидратацию, должен содержать
не менее 85% об. этилена. В этих условиях степень превраще¬
ния этилена за один проход составляет не более 6%, поэтому
процесс гидратации строится по циркуляционной схеме. При
этом общий выход этанола по этилену достигает 95% .При гидратации этилена, наряду с основной реакцией (а)
протекают побочные реакции дегидратации этанола до диэти-
лового эфира:2С2Н5ОН ^ (С2Н5)20 + Н20, (б)и этилена:(в)с2н5он сн2=сн2 + н2о,дегидрирования до ацетальдегида:С2Н5ОН ^ СНзСНО + Н2, (г)а также реакции образования олигомеров различного соста¬
ва.Селективность процесса гидратации по этанолу возрастает
при увеличении отношения Н20:С2Н4 и уменьшении темпера¬
туры и снижается с приближением системы к состоянию рав-275
новесия. Поэтому степень конверсии этилена невыгодно дово¬
дить до величины, близкой к равновесной.Технологическая схема прямой гидратации этилена. Сырь¬
ем для производства этанола методом прямой гидратации слу¬
жит этилен, выделяемый из пирогаза, полученного пиролизом
низкооктанового бензина, газов нефтепереработки и попутно¬
го газа, из этиленовой фракции обратного коксового газа, а так¬
же полученного пиролизом этана.В технологической схеме предусмотрены приготовление па¬
рогазовой смеси этилена и водяного пара, пополнение потерь
катализатора, нейтрализация уносимой с потоком газа фосфор¬
ной кислоты, отдувка циркуляционного газа, теплообмен с ис¬
пользованием теплоты реакции гидратации.Парогазовую смесь готовят совместным нагреванием паров
воды и этилена в теплообменниках и трубчатой печи или сме¬
шением этилена с перегретым паром высокого давления. В про¬
мышленности используются обе схемы, однако вторая, реали¬
зованная в нашей стране, экономически более целесообразна
при наличии ТЭЦ вблизи производства этанола.Технологическая схема производства этанола прямой гидра¬
тацией этилена с использованием пара высокого давления пред¬
ставлена на рис. 12.8.Свежий этилен сжимается в компрессоре 1 до 8 МПа и сме¬
шивается с циркуляционным газом, предварительно поджатым
в компрессоре 2. Полученная смесь нагревается в теплообмен¬
нике 3 продуктами реакции до 300°С и смешивается с перегре¬
тым до 450°С паром, подаваемым под давлением 8 МПа. Обра¬
зовавшаяся парогазовая смесь поступает в реактор — гидрата-
тор 4. Гидрататор представляет стальной цилиндр диаметром
1,5 м и высотой 10 м, футерованный изнутри медью и напол¬
ненный катализатором в виде носителя, пропитанного 60% -ной
фосфорной кислотой, насыпанным на перфорированный конус
в нижней части гидрататора. Перед поступлением в гидрататор
в парогазовую смесь вводится фосфорная кислота для компен¬
сации ее потерь. Продукты реакции, состоящие из паров этано¬
ла, воды и непрореагировавшего этилена, обрабатываются ра¬
створом гидроксида натрия для нейтрализации унесенной по¬
током фосфорной кислоты, охлаждаются в тепплообменнике 3
и поступают в котел-утилизатор 5, вырабатывающий пар низ¬
кого давления, направляемый затем в пароперегреватель. Из
котла-утилизатора продукты поступают в сепаратор высокого
давления 6, в котором от них отделяется циркуляционный газ,
поступающий в компрессор 2. Спирто-водный конденсат, содер-276
Рис. 12.8. Технологическая схема прямой гидратации этилена:1 — компрессор этилена, 2 — циркуляционный компрессор, 3 — теплооб¬
менник, 4 — гидрататор, 5 — котел-утилизатор, 6 — сепаратор высокого
давления, 7 — дроссель, в — сепаратор (сборник) низкого давления, 9 —
колонна отгонки легкой фракции, 10 — этанольная колонна, 11 — конден¬
саторжащий до 15% об. этанола, дросселируется через дроссель 7 до
давления 5 МПа и поступает в сепаратор (сборник) низкого дав¬
ления 8, где из него выделяются растворенные газы, которые
направляются на сжигание. Из сепаратора 8 конденсат подает¬
ся в колонну 9, где из него отгоняются диэтиловый эфир и аце-
тальдегид, и затем в колонну 10, в которой отгоняется этанол в
виде азеотропной смеси с водой, содержащей 95% об. спирта.
Обе ректификационные колонны обогреваются острым паром.
Дальнейшая очистка синтетического этанола и требования к
нему зависят от назначения продукта. Производительность со¬
временных установок прямой гидратации этилена достигает 30
тыс. т этанола в год.12.2.5. Производство этанола гидролизом древесиныГидролизное производство. Производство этанола из древес¬
ного сырья — частный случай гидролизного производства, в
основе которого лежит химическая переработка растительных277
материалов путем превращения содержащихся в них полиса¬
харидов в моносахариды. При этом непищевое растительное
сырье (отходы древесины, подсолнечная лузга, кукурузные ко¬
черыжки и др.) может быть превращено в пищевые, кормовые
и технические продукты. Наиболее распространенным сырьем
в гидролизном производстве, в том числе, для получения эта¬
нола, является древесина. Это сложная структурированная си¬
стема, состоящая из целлюлозы, гемицеллюлозы, лигнина, а
также незначительных количеств смол, дубильных и красящих
веществ. Элементный состав органической части древесины
практически постоянен: углерод 49—51%, водород 6,1—6,9%,
кислород 43—45%, азот 1% . Содержание целлюлозы, лигнина
и гемицеллюлоз зависит от природы древесины. В сухой древе¬
сине хвойных пород, используемой для производства этанола,
содержится 52—58% целлюлозы, 28—29% лигнина и около
20% гемицеллюлоз различной степени полимеризации, состо¬
ящих из пентозанов (СбНвО^л и гексозанов (CeHioOs)^.В результате гидролиза полисахаридов образуются водные
растворы моносахаридов — гидролизаты. Из них кристаллиза¬
цией получают пищевую глюкозу и техническую ксилозу; гид¬
рированием — ксилит и сорбит; дегидратацией — фурфурол;
окислением — органические кислоты; микробиологической
переработкой — этанол, бутанол, ацетон, кормовые дрожжи,
антибиотики.Из лигнина, остающегося после отделения гидролизата, тер¬
мической обработкой получают активированный уголь, уксус¬
ную кислоту и фенол; химической переработкой — активиро¬
ванный лигнин и щавелевую кислоту; прессованием — строи¬
тельные материалы.Таким образом, гидролизное производство — это пример ма¬
лоотходного производства, в котором используются все компо¬
ненты сырья.В зависимости от назначения целевых продуктов гидролиз¬
ное производство организуется по той или иной технологичес¬
кой схеме. Так, если в качестве сырья используются древесные
отходы, то из 1 т абсолютно сухой древесины получается 220
кг кормовых дрожжей, или 35 кг дрожжей и 175 л этанола, или
110 кг дрожжей и 80 кг фурфурола.Физико-химические основы производства. Получение гид¬
ролизного этанола складывается из двух последовательных ста¬
дий: гидролиза древесины и брожения гидролизата.Гидролиз древесины представляет каталитический процесс
взаимодействия полисахаридов (целлюлозы, пентозанов и гек-278
созанов гемицеллюлоз) с водой с образованием соответствую¬
щих моносахаридов — ксилозы, a-D-глюкозы и т.д., например:н+(СэНвО^л + 71Н2О —> ЛС5Н10О5Н+(СбНю05)л 4- 71Н2О —* яСбН120бПроцесс гидролиза протекает в несколько стадий и катали¬
зируется кислотами и кислыми солями, причем каталитичес¬
кое действие кислот тем сильнее, чем выше степень их диссо¬
циации. Скорость гидролиза возрастает с увеличением концен¬
трации кислоты и температуры и зависит от природы гидроли¬
зуемого полисахарида. Полисахариды делятся на легко и труд¬
ногидролизуемые, располагаясь в ряд по скорости гидролиза:галактан > ксилан > маннан > целлюлоза.Условия процесса гидролиза и качество получаемого гидро¬
лизата существенно зависят от природы катализатора. Катализ
в присутствии разбавленной серной кислоты концентрацией
0,4—0,7% об. проводится при температуре 120—190°С, и дав¬
лении 6—12 МПа. В результате образуется гидролизат, содер¬
жащий фурфурол, органические кислоты и другие примеси. Это
объясняется тем, что параллельно с гидролизом протекают ре¬
акции разложения образовавшихся моносахаридов; скорость
реакций с повышением температуры также возрастает. Поэто¬
му, несмотря на то, что константа скорости реакции гидролиза
больше константы скорости реакций разложения, выход гек-
соз в этом случае не превышает 70% при степени гидролиза око¬
ло 0,9. Гидролиз в присутствии концентрированных кислот, та¬
ких как 70—80%-ная серная кислоты или 31—41%-ная соля¬
ная кислота, проводится при температуре не выше 60 С и ат¬
мосферном давлении. Он дает более чистый гидролизат с выхо¬
дом моносахаридов до 95%.Брожение или ферментация — процесс разложения углево¬
дов под воздействием микроорганизмов или выделенных из них
ферментов. В производстве этанола методом гидролиза (также,
как и в методах осахаривания крахмала и из сульфитных ще¬
локов) используют один из видов брожения — спиртовое бро¬
жение, вызываемое ферментом зимазой, содержащемся в дрож¬
жевых клетках. Из моносахаридов брожению подвергаются
только гексозы. Процесс спиртового брожения a-D-глюкозы,
являющейся структурной единицей глюкозы, происходит без
доступа кислорода (анаэробное брожение) и состоит из ряда ста-279
дий с участием фосфорорганических соединений, играющих
роль переносчиков фосфора. В результате сложных превраще¬
ний из глюкозы образуется этанол и оксид углерода (IV), что
может быть выражено суммарным уравнением:С6Н1206 = 2С2Н5ОН + 2С02Параметры процесса брожения выбирают исходя из опти¬
мальных условий жизнедеятельности дрожжевых клеток и по¬
давления развития их спутников — кислотообразующих бак¬
терий молочнокислого и уксуснокислого брожения. Оптималь¬
ные температуры размножения дрожжевых клеток и развития
бактерий практически совпадают. Чтобы подавить развитие
бактерий повышают кислотность среды, вводя в гидролизат сер¬
ную или молочную кислоты. При рН<4,2 дрожжевые клетки
интенсивно растут, а бактерии не размножаются. Поэтому в
производстве процесс брожения проводят при температуре 27—
30*С, атмосферном давлении и в слабо кислой среде (pH = 3,8—
4,0).Технологическая схема производства. В процессе производ¬
ства гидролизного этанола стадии гидролиза древесины и сбра¬
живания образующегося гидролизата объединены в единую тех¬
нологическую схему. В нашей стране распространен метод гид¬
ролиза древесины разбавленной серной кислотой, для которо¬
го в качестве сырья используют отходы хвойной древесины с
высоким содержанием гексозанов.По этой схеме производство этанола представляет полунеп¬
рерывный перколяционный процесс, в основе которого лежит
принцип непрерывной фильтрации кислоты через периодичес¬
ки загружаемое в реактор сырье с непрерывным отбором обра¬
зующегося гидролизата в течение нескольких часов. Подобная
схема производства этанола приведена на рис. 12.9.Древесное сырье в виде опилок или измельченной щепы заг¬
ружается в гидролиз-аппарат 1, представляющий цилиндричес¬
кий стальной сосуд.футерованный изнутри кислотоупорным
материалом. Затем в аппарат через специальное оросительное
устройство подается нагретая до 180—190 С серная кислота
концентрацией 0,5% массовых. Вода для разбавления кисло¬
ты подогревается в подогревателе 2. В гидролиз-аппарат пода¬
ется пар под давлением 1—1,2 МПа. Образующийся гидроли¬
зат непрерывно выводится из нижней части аппарата через
фильтрующее устройство в виде перфорированных медных тру¬
бок и направляется в испаритель 4. Вследствие снижения дав¬
ления гидролизат вскипает и пары, содержащие фурфурол (тем-280
ВодаСырьем '-ИаР
h2so4~ 4КормовыеВодаV68-Г"Вода iГипсС2Н5ОНРис. 12.9, Технологическая схема производства этанола гидроли¬
зом древесины:1 — гидролиз-аппарат, 2 — подогреватель воды, 3 — сборник лигнина, 4—
испаритель гидролизата, 5 —донденсатор, 6 — нейтрализатор, 7 — от¬
стойник шлама, 8 — холодильник, 9 — сборник гидролизата, 10 — бро¬
дильный чан, /1 — сепаратор дрожжей, 12 — газгольдер оксида углеро¬
да, 13 — сборник бражки, 14 — бражная колонна, 15 — спиртовая колон¬
на, 16 — метанольная колонна, 17 — конденсатор этанолапература кипения 161,7°С при атмосферном давлении) посту¬
пают в конденсатор 5. Цикл непрерывной работы гидролиз-ап¬
парата от загрузки до выгрузки составляет несколько часов,
после чего оставшийся в аппарате лигнин передавливается в
сборник 3. Затем в аппарат загружается новая порция сырья.
Гидролизат после отделения фурфурола поступает из испари¬
теля 4 в нейтрализатор 6, куда вводится раствор гидроксида
кальция для нейтрализации серной кислоты, и из него в отстой¬
ник гипса 7. После охлаждения до 30°С в холодильнике 8 гид¬
ролизат перекачивается через сборник 9 в бродильный чан 10,
где происходит процесс сбраживания и размножения дрожже¬
вых клеток. Продукт сбраживания (бражка) подается в сепара¬
тор 11, где от бражки отделяются дрожжи. Часть дрожжей воз¬
вращается в бродильный чан, а остальные выводятся из систе¬
мы в качестве кормовых дрожжей. Оксид углерода (IV), выде¬
ляющийся из бродильного чана, собирается в газгольдере 12 и
в дальнейшем перерабатывается на сухой лед. Осветленная281
бражка, содержащая около 1,5% об. этанола, поступает в сбор¬
ник 13 и оттуда подается в систему ректификации, состоящую
из трех колонн. В бражной колонне 14 от бражки отделяется
барда, после чего жидкость с содержанием этанола 15—20% об.
поступает сначала в спиртовую колонну 25, а затем в метаноль-
ную колонну, в которой этанол концентрируется и освобожда¬
ется от примеси метанола.Из 1 т абсолютно сухой хвойной древесины получается 150—
180 кг этанола и до 120 кг оксида углерода (IV).12.3. Производство высших жирных (синтетических)
спиртов и кислот12.3.1. Технологические свойства и применениеВысшие жирные кислоты (ВЖК) — техническое название
смесей карбоновых кислот алифатического ряда природного
или синтетического происхождения с числом углеродных ато¬
мов в молекуле от 6 до 24.Природные ВЖК содержатся в маслах и жирах в виде их гли¬
церидов в восках в виде эфиров высокомолекулярных спиртов.
Они имеют нормальное строение и четное число атомов углеро¬
да в цепи.Синтетические ВЖК — это смеси насыщенных монокарбоно-
вых кислот нормального и изостроения с четным и нечетным
числом углеродных атомов в цепи. В дальнейшем под термином
«ВЖК» понимаются исключительно синтетические кислота
(СЖК). Технический продукт под таким названием выпускает¬
ся в виде фракций различного состава в зависимости от назначе¬
ния. Свойства фракций, в том числе температуры их плавления,
зависят от молярной массы входящих в них кислот. Так, фрак¬
ции С'5—Сб, С7—Сд, Сд—Сю и Сю—С13 представляют маслянис¬
тые жидкости с температурами кипения от 200 до 284°С; фрак¬
ции Сю—Ci6 и С13—Сю — мазеподобные продукты с температу¬
рами кипения от 270С до 215°С (при 2 кПа), фракция С17—С20 —
твердое вещество с температурой размягчения 60—75°С. Все
ВЖК имеют цвет от белого до светло-желтого, они легче воды
(плотность 0,86—0,92 т/м3) и не растворяются в ней. Растворя¬
ются в щелочах с образованием соответствующих солей (мыла),
которые разлагаются минеральными кислотами и легко гидро¬
лизуются. ВЖК умеренно токсичны, оказывают раздражающее
действие на кожу и слизистые оболочки. ПДК паров суммы кис¬
лот (в пересчете на уксусную кислоту) равна 5 мг/м3.282
Высшие жирные спирты (ВЖС) — техническое название
смесей одноатомных насыщенных спиртов алифатического ряда
с числом углеродных атомов в молекуле от б до 20. ВЖС полу¬
чают методами органического синтеза, почему называются так¬
же синтетическими жирными спиртами (СЖС). В дальнейшем,
как и в случае кислот, под термином «ВЖС» понимаются СЖС.
Физические свойства ВЖС зависят от их молярной массы, ВЖС
с числом атомов углерода в цепи от 6 до 11 представляют жид¬
кости с температурами кипения 157—286°С, с большим числом
углеродных атомов — твердые легкоплавкие вещества светло-
желтого цвета с температурами плавления от -5 до 65°С. Все
ВЖС легче воды (плотность 0,6—0,7 т/м3). Растворимы в эта¬
ноле и диэтиловом эфире. Растворимость в воде падает с увели¬
чением молярной массы и спирты, начиная с Cs в воде практи¬
чески нерастворимы. ВЖС огнеопасны. Взрывоопасность паров
ВЖС в смеси с воздухом увеличивается с уменьшением моляр¬
ной массы. ПДК для ВЖС равна 10 мг/м3.Возникновение и интенсивное развитие производства ВЖК и
ВЖС было связано с необходимостью замены пищевого сырья,
идущего на технические цели (растительные масла и животные
жиры), непищевым сырьем. Достаточно сказать, что еще в 1970
году расход растительного масла на технические цели в нашей
стране составлял более 27% к его общей выработке.В настоящее время области применения ВЖК и ВЖС весьма
многообразны. ВЖК используют в производстве пластичных
смазок, синтетических масел и пластификаторов, синтетичес¬
ких спиртов, латексных изделий и лакокрасочных материалов,
мыла и синтетических моющих средств, эмульгаторов полиме¬
ризации, в качестве присадок к дизельному топливу, флотореа-
гентов в производстве минеральных удобрений.ВЖС применяются в качестве флотореагентов, растворите¬
лей синтетических смол, селективных экстрагентов некоторых
солей, депрессоров испарения воды из водоемов. Основное ко¬
личество производимых ВЖС перерабатывается в другие про¬
дукты целевого назначения:— пластификаторы полимерных материалов (фталаты, ади-
паты и сложные эфиры других кислот);— компоненты синтетических смазочных масел и консистен¬
тных смазок (адипаты, себацинаты);— фотокинореагенты;— депрессорные присадки к топливам и моторным маслам
(полимерные эфиры метакриловой и других непредельных кис¬
лот);283
— флотореагенты (ксантогенаты, дитиофосфаты, натрийал-
килсульфонаты);— вспомогательные вещества, улучшающие технологию и
качество химических волокон.Особое значение ВЖК и ВЖС приобрели в связи с внедрени¬
ем в технику и народное хозяйство поверхностно-активных ве¬
ществ.Поверхностно-активными веществами (ПАВ) называются
вещества, концентрирующиеся на поверхности раздела фаз ге¬
терогенной системы и снижающие поверхностное (межфазное)
натяжение. Молекулы ПАВ содержат гидрофильные атомные
группы, обеспечивающие растворимость ПАВ в воде, и гидро¬
фобные, чаще всего углеводородные атомные группы, которые
при достаточно большой молярной массе способствуют раство¬
рению ПАВ в неполярных растворителях. С помощью ПАВ мож¬
но влиять на энергетическое состояние и структуру межфазной
поверхности системы и этим регулировать свойства гетероген¬
ных и микрогетерогенных систем.Подобные свойства ПАВ обусловили широкое использование
их в качестве синтетических моющих веществ, флотореагентов,
диспергаторов, пеногасящих материалов, в строительном и до¬
рожном деле (пенобетоны, смачивающие гравий композиции),
в нефтедобыче и сельском хозяйстве (фиксация пестицидов в
почве). Особое значение ПАВ приобрели в качестве действую¬
щей основы синтетических моющих средств.Синтетическими моющими средствами (СМС) называют
композиции из синтетических моющих веществ (СМВ) и раз¬
нообразных добавок, изменяющих pH среды, улучшающих кол¬
лоидную структуру раствора, вызывающих эффект отбелива¬
ния. СМВ представляют ПАВ различной природы. Их делят на
ионогенные, в которых содержатся атомные группы, диссоци¬
ирующие в водных растворах и неионогенные, в которых по¬
добных групп нет. Ионогенные СМВ подразделяются на анио¬
ноактивные и катионоактивные.К анионоактивным СМВ относятся наиболее распространен¬
ные промышленные моющие вещества, в том числе мыла, по¬
лученные на основе синтетических ВЖК.Катионоактивные СМВ распространены значительно мень¬
ше и чаще всего представляют соли аминов и четвертичных
аммониевых оснований, например [К-СбН4-СН2~К(СНз)2]+СГКак и мыла на основе природных ВЖК эти СМВ, получен¬
ные из синтетических ВЖК, обладают рядом недостатков:— щелочная реакция, вызывающая разрушение тканей;284
— образование нерастворимых солей с ионами кальция и
магния, снижающее моющее действие;— эффективность лишь при значительных концентрациях.Эти недостатки отсутствуют у анионоактивных веществ типанатриевых солей органических сульфокислот и кислых эфиров
серной кислоты. К таким СМВ относятся алкиларилсульфонаты
(сульфонолы) R-CetLi-SC^ONa, алкилсульфонаты R-S020Na и
алкилсульфаты R-0S020Na, в которых R=Ci2—С\%.В последнее время стали использоваться и неионогенные
СМВ, синтезируемые из оксида этилена и веществ с активны¬
ми атомами водорода (карбоновые кислоты, спирты, амины),
например, продукты полиоксиэтилирования высших жирных
спиртов:ROH + пСН2-СН2 — R-(OCH2-CH2)„-OHхоВ подобных СМВ гидрофильные свойства обеспечены за счет
присутствия в молекуле оксиэтильной цепи.Для производства ПАВ и СМВ в частности, используют син¬
тетические жирные спирты состава Сю—С2о и фракции синте¬
тических жирных кислот Сю—Сю и Ci7—С2о-В табл. 12.6 представлена структура потребления ВЖК.Таблица 12.6. Структура потребления ВЖК, %Область потребления%Производство мыла и моющих средств61,2Производство высших жирных кислот8,6Производство резино-технических изделий3,9Производство синтетических каучуков4,6Парфюмерная и кожевенная промышленность2,3Лакокрасочная промышленность2,4Прочие отрасли17,012.3.2. Сырье и способы производства ВЖК и ВЖССинтетические ВЖК могут быть получены:— гидрокарбоксилированием алкеновr-ch=ch2 + со + нон — r-ch2-ch2-cooh— гидрокарбалкоксилированием алкенов с последующим
омылением полученных эфиров кислотНОНR-CH=CH2+CO+R‘OH — r-ch2-ch2-coor‘ —► r-ch2-ch2-cooh— окислением высших алканов.285
Наибольшее значение имеет последний метод, по которому
промышленное производство ВЖК было впервые организова¬
но в Германии в 1937 г. на базе парафина из установок Фише¬
ра—Тропша и выделенного из продуктов нефтепереработки. В
нашей стране производство ВЖК было начато в 1953 г. В пери-
од с 1959 по 1961 г. были введены в строй несколько промыш¬
ленных установок, что позволило увеличить выработку их с 23
тыс. т в 1958 г. до 230 тыс. т. в 1966 г. В настоящее время миро¬
вое производство ВЖК превышает 2,5 млн. т.Промышленные методы производства ВЖС весьма многооб¬
разны. Они могут быть получены как из природного сырья, так
и из продуктов переработки нефти и газа. Важнейшими из них
являются:— выделение из растительных масел и жиров;— омыление касторового масла с последующим гидролизом
солей;— оксосинтез с последующим восстановлением альдегидов;— гидрирование ВЖК и их эфиров;— альдольная конденсация альдегидов с последующим гид¬
рированием продуктов конденсации;— алюмоорганический синтез на основе алкенов и триэтил-
алюминия;— окисление высших алкенов.На рис. 12.10 представлена взаимосвязь важнейших мето¬
дов производства ВЖК и ВЖС.r-ch2choРис. 12.10. Схема промышленных методов производства ВЖК
и ВЖС286
Из схемы следует, что окисление алканов является общим
методом получения как ВЖК, так и ВЖС, опирающимся на
одну сырьевую базу. Ниже рассматривается промышленное
производство ВЖК и ВЖС этим методом.Процесс окисления алканов протекает в жидкой фазе кис¬
лородсодержащим окислительным газом по цепному механиз¬
му через образование и превращения радикалов R*, R-C*,
R-£-0- и R-(^COO. ОО ОМолекулярный кислород присоединяется к молекуле угле¬
водорода с образованием гидропероксида, который вступает за¬
тем во вторичные превращения. При окислении алканов изост¬
роения кислород присоединяется к третичному углеродному
атому, у алканов нормального строения — к метиленовой груп¬
пе. Присоединения кислорода к метильной группе практичес¬
ки не происходит. Механизм радикального окисления алканов
может быть представлен в следующем виде.1. Образование и развитие цепи.R-CH2-CH2-R' + 02 — R-CH-CH2-R + НО 2R-CH-CH2-R + 02 — R-CH-CH2-RI0-0*r-ch-ch2-r' + RH — r-ch-ch2-r' + R".I I0-0- O-OH2. Вторичные процессы распада гидропероксида с образова¬
нием конечного продукта — спирта (без разрыва углеродной
цепи).r-ch-ch2-r‘ -> r-ch-ch2-r' + он
о-он о-r-ch-ch2-r' + r"h —► r-ch-ch2-r' + R *
о- он3. Вторичные процессы распада гидропероксида через кетон
и кетогидропероксид, разлагающийся с образованием конечных
продуктов — кислоты и альдегида (с разрывом углеродной
цепи).r-ch-ch2-r' — r-c-ch2-r' + н2о
о-он оR-C-CH2-R' + 02 — R-C-CH-R' — R-COOH + R-CHOи м Iо о о-он287г
Направление вторичного процесса распада гидропероксида
зависит от условий окисления: концентрации кислорода в окис¬
лительном газе, скорости газового потока (времени пребывания
продуктов в зоне окисления), наличия или отсутствия катали¬
затора. В зависимости от этого, процесс окисления алканов идет
или без разрыва углеродной цепи в сторону образования ВЖС,
или с разрывом цепи в сторону образования ВЖК.В табл. 12.7 представлен режим окисления алканов для по¬
лучения высших жирных кислот и высших жирных спиртов.Таблица 12.7. Условия окисления алкановПродуктСоставокисляемогосырьяКатализаторСодержание
О2 в газе, %Темпера-
тура, °СВЖКПарафинС18—С 44МпОг-ЗНгО21 и более110ВЖСПарафинС ш—С20Нет3—517012.3.3. Производство высших жирных кислотВысшие жирные кислоты получают окислением парафина
состава Cis—С44 в жидкой фазе в присутствии катализатора на
основе соединений марганца. В качестве окисляющего газа ис¬
пользуется воздух, обогащенный кислородом до содержания
более 21% об. Катализатор в виде водного раствора перманга¬
ната калия вводится непосредственно в нагретый парафин. Пос¬
ле испарения воды катализатор восстанавливается до
МпС>2’2Н20, который распределяется в объеме парафина. По¬
добное дисперсное состояние катализатора позволяет обеспе¬
чить высокую скорость процесса окисления при температуре
110—120°С и атмосферном давлении. Чтобы избежать развития
реакций глубокого окисления процесс продолжают не более
15—20 часов и прекращают при достижении глубины окисле¬
ния парафина 30—35% .Продуктами окисления парафина являются фракции ВЖК,
выход и состав которых зависит от назначения процесса и его
условий. Так, при окислении парафина С30 выход ВЖК состав¬
ляет (%):фракция Ci—С4 —5—10%; фракция Сю—С^ — 25—28%;фракция С5—Сб — 3—5%; фракция C17—C20 — 15—20%;фракция C7—С9 — 8—10%; фракция выше С20 — 20—25%.288
Суммарный выход всех фракций ВЖК составляет в среднем
80%.На рис. 12.11 представлена принципиальная схема производ¬
ства ВЖК окислением парафина.Этилацетат1Г Раствор
J RCOOH[ Ci—с4IРаствор
RCOOH
в этил-
ацетате1+ * “ + +Cl С2 С3 с,
Низшие кислотыВоздух-[парафин V
if Продукты
-4 окисления -I Парафин .г 1 ,I Высшие
A RCOOH
[ Парафин ,J Мыла 1
1 RCOONa ГJ-Высшие
RCOOHi iV Ii}КатализаторВодаNaOHH2S04iNa2SQ4ФРАКЦИИ ВЖКРис. 12.11. Принципиальная схема производства ВЖКТехнологическая схема производства ВЖК приведена на рис.
12.12.Свежий парафин и возвратный парафин в отношении 1:2 и
раствор перманганата калия поступают в смеситель 2. Образо¬
вавшаяся смесь подается в окислительную колонну 3, вы¬
полненную из алюминия или специальной стали, и снабжен¬
ную выносным холодильником для отвода реакционного теп¬
ла. В нижнюю часть колонны через распределительное устрой¬
ство подается подогретый до 120*С воздух, который барботиру-
ет через смесь. Выходящий из верха колонны газ промывается
в скруббере 4 водой для извлечения из него низших кислот, очи¬
щается от примесей дожиганием в печи 5 и выбрасывается в
атмосферу. Оксидат из окислительной колонны охлаждается
до 80°С в холодильнике 6 и поступает в отстойник 7, где из него
отделяется катализаторный шлам, из которого затем регенери¬
руется катализатор. Из отстойника оксидат перекачивается в
промывную колонну S, орошаемую водой, в которой он от-10 Химическая технология. Том 2289
Вода КМп04 — Вода В атмосферуВоздухФ1г/л ф.ф2Ф.17КИСЛОТВозвратный парафин С^— С4Na2S04Рис. 12.12. Технологическая схема производства ВЖК:1 — аппарат для приготовления катализатора, 2 — смеситель, 3 — окис¬
лительная колонна, 4 — скруббер, 5 — печь дожигания, 6 —холодильник,7 — отстойник катализаторного шлама, 8, 16 — промывные колонны,9, 10 — омылители, 11 — теплообменник, 12 — автоклав, 13 — печь, 14 —
дроссель, 15 — гидролиз-аппарат, 17 — колонна ректификациимывается от водорастворимых кислот Ci—С4. Раствор кислот
из нижней части колонны подается на абсорбцию этилацетатом
(на схеме не указана) и на дальнейшее разделение. Освобожден¬
ный от водорастворимых кислот оксидат поступает в омылите¬
ли 9 и 10. В омылителе 9, орошаемом водным раствором карбо¬
ната натрия, нейтрализуются высшие кислоты:2R-COOH + Na2C03 = 2RCOONa + С02 + Н20,а в омылителе 10, орошаемом щелочью, происходит гидролиз
сложных эфиров лактонов (побочных продуктов окисления):RCOOR' + NaOH = RCOONa + ROHОмыление завершается в теплообменнике 11 и автоклаве 12
при температуре 180°С под давлением 2 МПа. Из автоклава 12
непрореагировавший парафин возвращается в смеситель 2, а
раствор солей ВЖК (мыло) поступает в трубчатую печь 13, где
нагревается до 320°С, после чего дросселируется в дросселе 14
до атмосферного давления. При этом от мыла отделяется нео-
мыленная органическая фаза, которая через теплообменник 11,
обогревающий оксидат из омылителя 10, возвращается на окис-290
ление или выделение из нее спиртов. Расплавленное мыло раз¬
бавляется водой до концентрации 40% и обрабатывается сер¬
ной кислотой в гидролиз-аппарате 15. Выделившиеся свобод¬
ные ВЖК отмываются в промывной колонне 16 от сульфата
натрия и подаются на ректификацию в колонну 2 7. В процессе
ректификации из смеси ВЖК выделяют соответствующие фрак¬
ции, указанные выше. Расходный коэффициент по парафину
составляет 1,3 т/т ВЖК, при выходе на израсходованное сырье
76—82%.12.3.4. Производство высших жирных спиртовВысшие жирные спирты получают аналогично ВЖК окис¬
лением парафина в жидкой фазе, но в иных условиях. Сырье
окисляется при температуре 165—170 С азотокислородной сме¬
сью, содержащей 3—5% кислорода, без катализатора. Чтобы
избежать дальнейшего окисления образующихся спиртов, про¬
цесс ведется в присутствии борной кислоты, дающей со спирта¬
ми триалкилбораты (ЕО)зВ. Они легко выводятся из сферы ре¬
акции. В результате цепь окислительных превращений преры¬
вается, обеспечивая селективность процесса. Борную кислоту
в количестве 5% от массы парафина вводят в виде суспензии в
парафине. Так как в этом случае процесс окисления протекает
без разрыва углеродной цепи, то для получения спиртов с дос¬
таточно высокой молярной массой используют так называемые
«мягкие» парафины Сю—С2о- Оксидат имеет состав: ВЖС —
67%, ВЖК — 11,5%, низкомолекулярные продукты окисле¬
ния — 12%, кубовый остаток 11,5% .На рис. 12.13 представлена принципиальная, а на рис. 12.14
технологическая схемы производства ВЖС окислением пара¬
фина.Суспензия борной кислоты в свежем парафине и возвратный
парафин смешиваются в отношении 1:1,2 в смесителе 1 и по¬
ступают в окислительную колонну 2, в нижнюю часть которой
через распределительное устройство подается окислительный
газ (смесь кислорода и азота). Отходящие из колонны газы про¬
ходят холодильник-конденсатор 3, в котором конденсируется
уносимый газом парафин, возвращаемый в колонну, и
выбрасываются в атмосферу. Оксидат из колонны 2 поступает
в отстойник 4, в котором осаждается избыток борной кислоты
и перекачивается через нутч-фильтр 5 для отделения остатка
кислоты в вакуумную дистилляционную колонну 6. В ней от
триалкилборатов отгоняется непрореагировавший парафин,
10*291
н3во3н3во3
I RCOONa -*■♦ -Гпарафин!- -<—" IГБораты (RO)oB 1
\ Парафин,3 уГ Бораты (RO)3B 1 «4-
1 RCOOH JRCOOH-ЧRCOOH
ROH
*Г RCOOH 1
1 ROH JАзотокислородныйгазВодаNaOH-Г ВЫСШИЕ ROH V, I . 'I + + *ФРАКЦИИ ВЖС
Рис. 12.13. Принципиальная схема производства ВЖСОтходящиегазыПарафинН3ВО3NaOHк вакуум-
насосуКубовыйостатокNa3B03Рис. 12.14. Технологическая схема производства ВЖС:1 — смеситель, 2 — окислительная колонна, 3, 11 — холодильники-
конденсаторы, 4 — отстойник, 5 — нутч-фильтр, 6, 10 — дистилляционные
колонны, 7 — нейтрализатор, 8 — сепаратор, 9 — гидролизер, 12 —
сборник ВЖСкоторый после нейтрализации содержащихся в нем карбоновых
кислот в нейтрализаторе 7 и отделения раствора их солей в се-
параторе S, возвращается в процесс. Триалкилбораты выводят-292
ся из нижней части колонны 6 и подаются в гидролизер 9, оро¬
шаемый водой. Из гидоолизера образовавшаяся смесь сырых
ВЖС, состоящая преимущественно из вторичных изомеров,
поступает в вакуумную дистилляционную колонну 10, из кото¬
рой целевые фракции ВЖС собираются в сборнике 12.Контрольные вопросы1. Перечислите основные промышленные синтезы на основе синтез-
газа и оксида углерода (II).2. Какими свойствами обладает метанол?3. За счет чего при синтезе метанола из синтез-газа достигается
необходимая селективность процесса?4. Какие технологические схемы используются в производстве мета¬
нола?5. Перечислите важнейшие области использования метанола.6. Из каких видов сырья может быть получен в промышленных мас¬
штабах этанол?7. Объясните преимущества метода прямой гидратации этилена пе¬
ред методом сернокислотной гидратации в производстве синтети¬
ческого этанола.8. Какие катализаторы используются при производстве этанола пря¬
мой гидратацией этилена в паровой фазе?9. Что такое гидролизное производство? Почему оно является мало¬
отходным?10. Из каких стадий состоит гидролизное производство этанола и чем
катализируется каждая стадия?11. Какие соединения относят к высшим синтетическим жирным кис¬
лотам (ВЖК) и спиртам (ВЖС)?12. Укажите основные промышленные методы производства ВЖК и
ВЖС.13. Что общего в химизме получения ВЖК и ВЖС окислением алка-
нов?14. Каким образом в производстве ВЖС прерывают процесс окисле¬
ния, не допуская деструкции молекулы алкана?15. Что такое синтетические моющие средства и какова их связь сDWP DUCITO
Глава XIII
ПРОИЗВОДСТВО АЛЬДЕГИДОВ13.1. Производство формальдегида и формалина13.1.1. Технологические свойства и применение
формальдегидаФормальдегид (метаналь, муравьиный альдегид) НСНО —
бесцветный газ с острым раздражающим запахом, с температу¬
рой кипения -19,2°С, температурой плавления -118°С и плот¬
ностью (в жидком состоянии при -20°С) 0,815 т/м3. С воздухом
образует взрывчатые смеси с пределами воспламеняемости 5,5
и 34,7% объемн. Формальдегид хорошо растворим в воде, спир¬
тах, ограниченно растворим в бензоле, эфире, хлороформе, не
растворим в алифатических углеводородах. Легко полимеризу-
ется, особенно при нагревании и в присутствии полярных при¬
месей, образуя твердый полимер линейного строения (пара¬
форм) с оксиметиленовыми звеньями:гсНСНО + Н20 <=* Н-(-0-СН2-)„-0Н,где: а = 8—100.Процесс полимеризации обратим, поэтому параформ легко
деполимеризуется под воздействием щелочных и кислотных
реагентов, что используется на практике для хранения и
транспортировки формальдегида. Токсичен, ПДК составляет0,05 мг/м3.Товарный продукт выпускается обычно в виде 37% -ного вод¬
ного раствора (формалин), в котором формальдегид содержит¬
ся в форме гидрата НСНОН2О и низкомолекулярных полиме¬
ров — полиоксиметиленгликолей. Для предотвращения более
глубокой полимеризации формальдегида и выпадения осадка,
который может отлагаться в аппаратуре, в формалин добавля¬
ется 6—15% объема метанола.Формальдегид вырабатывается в очень больших масштабах
(мировое производство в 1980 году составило свыше 2 млн. т) и
широко используется в различных областях органического син¬
теза, а также в качестве дезинфицирующего и дезинсекцион¬
ного средства. В больших количествах формальдегид применя¬
ют для производства феноло-, карбамидо- и меламидоформаль-
дегидных полимеров, в качестве полупродукта в синтезах изоп¬
рена, пентаэритрита, гексаметилентетрамина (уротропина). Он294
используется в многочисленных синтезах, лежащих в основе про¬
изводства химических волокон, многоатомных спиртов, краси¬
телей, взрывчатых веществ, фармацевтических препаратов.Формальдегид может быть получен окислением метана и его
гомологов или из метанола. При окислении метана в газовой
фазе воздухом или кислородом при атмосферном давлении пос¬
ледовательно протекают реакции:СН4 + 0,502 = СНзОН -АН (а)СН3ОН + 0,502 = НСНО + Н20 - ЛЯ (б)Реакция (б) селективно ускоряется катализаторами на осно¬
ве соединений меди и серебра. Однако достаточная для промыш¬
ленного использования селективность процесса по формальде¬
гиду может быть достигнута только при очень малой степени
окисления метана и недостатке кислорода, то есть при весьма
большой кратности циркуляции метанола. В противном случае
образовавшийся формальдегид подвергается дальнейшему
окислению:НСНО + 0,502 = НСООН и (в)НСООН + 0,502 = С02 + Н20 (г)Вследствие этого и, следовательно, малого выхода формаль¬
дегида технологический процесс прямого окисления метана
становится экономически невыгодным. Основная масса фор¬
мальдегида производится поэтому из метанола по двум мето¬
дам: окислительным дегидрированием и окислением.13.1.2. Производство формальдегида окислительным
дегидрированием метанолаОкислительное дегидрирование метанола представляет гете¬
рогенно-каталитический процесс, протекадющий в газовой фазе
на твердом катализаторе. В этом процессе совмещены экзотер¬
мическая реакция окисления метанола:СНзОН + 0,502 = НСНО + Н20 - А#1 AHi = 156,3 кДж (д)
и эндотермическая реакция его дегидрирования:СНзОН «=* НСНО + Н2 - ЛЯ2 АЯ2 = 85,3 кДж . (е)При соотношении реакций (д) и (е) равным 0,55 : 0,45 тепло¬
вой эффект процесса достаточен для возмещения потерь тепла
системы в окружающую среду и для нагревания исходных про¬
дуктов до нужной температуры. Если это отношение соблюда¬
ется, а в исходной паровоздушной смеси содержится около 45%
об. метанола, что лежит за верхним пределом взрываемости ее295
(34,7%), процесс можно проводить в реакторах адиабатическо¬
го типа, не имеющих поверхностей теплообмена.В качестве катализаторов процесса окислительного дегидри¬
рования используют медь (в виде сетки или стружки) и сереб¬
ро, нанесенное на пемзу. Одновременно с основными реакция¬
ми (д, е) протекают побочные реакции глубокого окисления (в,
г), а также реакции дегидрирования и гидрирования, приводя¬
щие к образованию смеси продуктов:СН3ОН — НСНО — СО; СН3ОН + Н2 — СН4 + Н20,для подавления которых в метанол вводится до 10% воды. Во из¬
бежание глубокого окисления метанола процесс окислительного
дегидрирования проводится при недостатке кислорода. В то же
время реакция дегидрирования (г) инициируется кислородом, что
позволяет уменьшить удельный вес побочных реакций. Процесс
окислительного дегидрирования проводится при температуре
500—600°С и времени контактирования около 0,02 с. В этих ус¬
ловиях выход формальдегида в расчете на пропущенное сырье
составляет 80—85% при степени контактирования 0,85—0,90.На рис. 13.1 представлена технологическая схема произ¬
водства формальдегида окислительным дегидрированием ме¬
танола.ВодаОтходя-
jr f щие
1—1 газы
128 ФормалинРис. 13.1. Технологическая схема окислительного дегидрирования
метанола:/ — напорный бак метанола, 2 — испаритель, 3 — брызгоуловитель, 4 —
перегреватель, 5 — реактор, 6 — холодильник реактора, 7 — абсорбер,8 — холодильник, 9 — сборник формалина, 10 — санитарная башня, 11 —
вакуум-компрессор, 12 — водоотделитель296
Метанол, содержащий 10% воды, из напорного бака 1 посту¬
пает в испаритель 2, обогреваемый горячей водой или паром из
холодильника реактора 6. В испаритель подается также очи¬
щенный от пыли воздух, барботирующий через слой метанола.
Образовавшаяся паровоздушная смесь освобождается от брызг
в брызгоуловителе 3 и через перегреватель 4> обогреваемый так¬
же горячей водой из холодильника реактора 6, подается в реак¬
тор 5, в верхней части которого находится катализатор. Про¬
дукты реакции быстро охлаждаются для предотвращения рас-
пада формальдегида в подконтактном холодильнике 6 и направ¬
ляются в абсорбер 7, орошаемый водой. Образовавшийся в аб¬
сорбере 37% -ный раствор формальдегида (формалин), содержа¬
щий для стабилизации 7—12% метанола, охлаждается в
холодильнике 8 и поступает в сборник формалина 9. Непогло¬
щенные газы проходят санитарную башню 10 и вакуум-комп¬
рессором 11 подаются в водоотделитель 12, после чего выбра¬
сываются в атмосферу.13.1.3. Производство формальдегида окислением метанолаВ этом новом, относительно недавно внедренном в практику
методе, метанол окисляется в избытке воздуха при температу¬
ре 350—430 С и атмосферном давлении на окисном железо-мо-
либденовом катализаторе состава МоОз*Гв2(Мо04)з. Этот ката¬
лизатор имеет высокую активность и малочувствителен к ката¬
литическим ядам.Технологический процесс прямого окисления отличается от
ранее описанного процесса окислительного дегидрирования
высокой степенью конверсии метанола (0,99), селективностью
по формальдегиду, достигающей 96% и высокой экзотермич-
ностью. Поэтому для окисления метанола в нем используют
трубчатые реакторы с интенсивным охлаждением циркулирую¬
щей в межтрубном пространстве водой или другими хладоаген-
тами. К достоинствам метода относятся также низкие расход¬
ные коэффициенты по сырью и энергии. На рис. 13.2 представ¬
лена технологическая схема производства формальдегида пря¬
мым окислением метанола.Метанол испаряется в теплообменнике 1, обогреваемом ре¬
акционной смесью, смешивается с воздухом, нагнетаемым тур¬
бокомпрессором 2 и через теплообменник 3 подается в реактор4. Съем тепла и тепловой режим в реакторе обеспечивается хла-
доагентом, циркулирующим через котел-утилизатор 5. Реакци¬
онная смесь, выходящая из реактора 4, охлаждается в теплооб-297
Циркуляционный газ На дожиганиеРис. 13.2. Технологическая схема окисления метанола:1,3 — теплообменники, 2 — турбокомпрессор, 4 — реактор, 5 — котел-
утилизатор, 6 — абсорбер, 7 — выносные теплообменники, 8 — сборник
формалинаменниках 3 и 1 и поступает в абсорбер 6, орошаемый водой. Теп¬
ло абсорбции отводится и утилизируется в выносных теплооб¬
менниках 7, подогревающих обессоленную воду, подаваемую на
абсорбцию и питающую котел-утилизатор 5, вырабатывающий
технологический пар. Образующийся формалин выводится из
нижней части абсорбера и поступает в сборник 8. Часть отходя¬
щих из верхней части абсорбера 6 газов смешивается с возду¬
хом перед входом его в реактор для снижения взрывоопасности
смеси воздуха с парами метанола, а остальное количество их
направляется в печь для дожигания (на схеме не указана) и
выбрасывается в атмосферу. Производство формальдегида по
этой схеме работает по замкнутому циклу и в нем отсутствуют
отходы, сточные воды и вредные газовые выбросы.Производство формальдегида из метанола-сырца. Рассмот¬
ренные выше схемы производства формальдегида дегидриро¬
ванием и окислением метанола предусматривают использова¬
ние преимущественно пемзосеребрянных катализаторов, весь¬
ма чувствительных к контактным ядам. Поэтому в них исполь¬
зуют метанол-ректификат, тщательно очищаемый от соедине¬
ний железа, хлора, серы и некоторых органических соединений
(олефинов, альдегидов и др.). Необходимость подобной очист¬
ки увеличивает капитальные затраты и значительно (на 15—298
20%) повышает себестоимость сырья и удорожает производство
формальдегида.Исходя из этого была предложена технологическая схема
производства формальдегида непосредственно из метанола-сыр¬
ца, в которой совмещены стадии каталитической очистки сы¬
рья и получения формальдегида. Подобная технология, пред¬
ложенная в нашей стране в 1978—79 гг., позволяет, не меняя
принципиально технологической схемы процесса, не только
использовать вместо метанола-ректификата сырец, но и утили¬
зировать содержащиеся в последнем побочные продукты, сни¬
зить расход пара на ректификацию и, в целом, повысить тех¬
нико-экономические показатели производства без снижения
качества конечного целевого продукта.13.2. Производство ацетальдегида13.2.1. Технологические свойства и применение
ацетальдегидаАцетальдегид (этаналь, уксусный альдегид) СН3СНО пред¬
ставляет бесцветную легкокипящую жидкость с резким удуш¬
ливым запахом, с температурой кипения 20,2°С, температурой
плавления -123,5°С и плотностью 0,783 т/м3. Критическая тем¬
пература ацетальдегида 188°С, температура самовоспламенения
156°С. С воздухом ацетальдегид образует взрывчатые смеси с
пределами воспламеняемости при 400°С 3,97 и 57,0% об. Смеси
с кислородом воспламеняются при более низкой температуре —
около 140°С. Токсичен, ПДК составляет 5 мг/м3.Ацетальдегид смешивается во всех отношениях с водой, эта¬
нолом, диэтиловым эфиром и другими органическими раство¬
рителями, с некоторыми образует азеотропные смеси. Под воз¬
действием минеральных кислот полимеризуется с образовани¬
ем жидкого циклического тримера — паральдегида с темпера¬
турой кипения 124,4°С и температурой плавления 12,б°С.I 1ЗСН3СНО 5=ь СН3СН-0-СН(СН3)-0-СН(СН3)-0и кристаллического тетрамера — метальдегида:4СН3СНО ^ (СН3СНО)4,которые при нагревании с серной кислотой деполимеризуются до
исходного ацетальдегида. На этом основано использование во
многих случаях паральдегида вместо мономерного ацетальдеги¬
да, так как он более удобен при хранении и транспортировке.299
Ацетальдегид - это один из важнейших многотоннажных
продуктов переработки ацетилена и этилена и применяется в
широких масштабах в промышленности органического синте¬
за. Важнейшие направления использования ацетальдегида:— окисление в уксусную кислоту и уксусный ангидрид;— получение циангидрина с последующей переработкой его
в акрилонитрил, эфиры акриловой кислоты, молочную кис¬
лоту;— альдольная конденсация и переработка альдоля в бутан-
диол-1,3 и бутадиен-1,3, н-бутанол, кротоновый альдегид;— конденсация с аммиаком с образованием гомологов пири¬
дина и винилпиридинов;— конденсация с формальдегидом до пентаэритрита.В настоящее время на производство уксусной кислоты и ее
ангидрида, этилацетата и 2-этилгексанола расходуется в мире
95%, а в нашей стране 75% всего производимого ацетальдегида.13.2.2. Промышленные методы производства ацетальдегидаСырьем для производства ацетальдегида служат ацетилен и
этилен. Из ацетилена ацетальдегид получают:— прямой гидратацией в жидкой фазе на ртутном катализа¬
торе или в паровой фазе на твердом кадмиевом катализаторе;— через виниловые эфиры низших насыщенных спиртов.На основе этилена ацетальдегид может быть получен:— через этанол, каталитическим дегидрированием или окис¬
лительным дегидрированием его;— прямым окислением на твердом палладиевом катализа¬
торе.Взаимосвязь промышленных методов получения ацетальде¬
гида представлена на рис. 13.3.ROHНОНСН-СНro-ch=ch2НОН + Н g+2
НОН + Cd+2сн2=сн2НзСНОРис. 13.3. Промышленные методы получения ацетальдегида300
С момента открытия М.Г. Кучеровым реакции прямой гидратации
ацетилена в жидкой фазе (1881 г.), она в течение более 60 лет была
единственным промышленным методом производства ацетальдегида.
Недостатки гидратации в присутствии ртутного катализатора — ток¬
сичность и сложность регенерации раствора, значительные потери
ртути, агрессивная сернокислая среда — привели к разработке нового
метода гидратации ацетилена на менее токсичном кадмиевом катали¬
заторе в паровой фазе. Одновременно приобрел промышленное значе¬
ние метод производства ацетальдегида из ацетилена через бутилвини-
ловый эфир. В настоящее время основная масса ацетальдегида почти
во всех странах производится на базе альтернативного более дешевого
сырья — этилена, окислением его воздухом или кислородом в присут¬
ствии палладиевого катализатора. Эта реакция, впервые описанная в
1894 г., была использована для промышленных целей в 1959 г. Сей¬
час мировое производство ацетальдегида этим методом превышает 2
млн. т в год, а в ряде стран он является единственным (Япония).В нашей стране промышленное производство ацетальдегида было
освоено в 1948 г. по методу Кучерова и из этилена через этанол. До
настоящего времени значительное количество ацетальдегида все еще
производится прямой гидратацией ацетилена на ртутном катализато¬
ре. В 1963—1968 гг. на основе исследований ВНИИСКа процесса па¬
рофазной гидратации ацетилена и разработки кадмийкальцийфосфат-
ных катализаторов (Ю.А. Горин и С.М. Момозон) были построены и
введены в эксплуатацию крупномасштабные производства ацетальде¬
гида по этому методу на Лисичанском и Невинномысском химичес¬
ких комбинатах. В 1970 г. на Омском заводе синтетического каучука
было организовано более экономичное производство ацетальдегида
прямым окислением этилена воздухом по двухстадийной схеме мощ¬
ностью 270 тыс. т в год. В дальнейшем, в связи с недостатками этой
схемы, была разработана и внедрена новая технология производства
ацетальдегида по одностадийной схеме — кислородный метод окисле¬
ния. Предполагается, что в дальнейшем производство ацетальдегида
будет полностью переведено на этиленовую базу.В настоящей главе рассматриваются технологические процессы
производства ацетальдегида из ацетилена гидратацией в паровой фазе,
через бутилвиниловый эфир и из этилена прямым окислением в одну
стадию.13.2.3. Производство ацетальдегида гидратацией
ацетилена в паровой фазеГидратация ацетилена в паровой фазе представляет гетеро¬
геннокаталитическую экзотермическую реакцию, протекаю*
щую по уравнению:С2Н2г + н20п = СНзСНО - АН АН = 162,8 кДж (а)Реакция катализируется оксидами и солями различных ме¬
таллов, в том числе фосфатами, ванадатами, хроматами и воль-301
фраматами. Наибольшей активностью и стабильностью обла¬
дает кислый кадмийкальцийфосфатный катализатор состава
С<ШР04'Саз(Р04)2. Он активен при температуре 350—400С, не
токсичен, однако быстро теряет активность и требует регенера¬
ции через каждые 100 часов работы.Реакция гидратации протекает через образование промежу¬
точного комплекса ацетилена с ионом кадмия, который затем
разлагается с образованием ацетальдегида: Cd+2С\ ОН“СН=СН + Cd+2 3=s CHV;H «=* Cd+-CH=CH-OHн+Cd+-CH=CH-OH Cd+-CH2-CHO ^ СН3-СНО + Cd+2Одновременно с основной реакцией гидратации (а) протека¬
ют побочные реакции дезактивации катализатора за счет его
восстановления:2СсГ 2 + СН3СНО + Н20 ^ 2Cd+ + СН3СООН + 2Н+ (б)
кротоновой конденсации:2СН3СНО ^ СНз-СН-СН-СНО + н20 (в)и реакции последующего смолообразования.При парофазной гидратации удаление ацетальдегида по мере
его образования невозможно, а накопление его в реакционной
зоне увеличивает вероятность побочных реакций, особенно кро¬
тоновой конденсации (в). Для подавления побочных реакций
необходимо устранение местных перегревов, при которых раз¬
вивается реакция (в), имеющая более высокую энергию актива¬
ции, применение избытка водяного пара, способствующего вы¬
воду ацетальдегида из сферы реакции, и проведение процесса
при невысокой степени конверсии ацетилена.Поэтому, процесс парофазной гидратации ацетилена прово¬
дят при объемном отношении водяного пара к ацетилену (7—
10): 1 и степени конверсии не выше 0,5. Образующийся ацеталь-
дегид сорбируется из реакционной смеси водой. В этих услови¬
ях выход ацетальдегида чистотой до 99,5% мае. достигает 90%.
В качестве побочных продуктов образуется 0,5—1,0% кротоно¬
вого альдегида, 0,5—1,0% уксусной кислоты и 0,3% ацетона.На рис. 13.4 представлена технологическая схема производ¬
ства ацетальдегида гидратацией ацетилена в паровой фазе.Ацетилен и водяной пар смешиваются в смесителе 1 и реак¬
ционная смесь, подогретая в подогревателе 2 до 400°С, подается
сверху в гидрататор колонного типа 3, содержащий катализа¬
тор. Температурный режим в гидрататоре поддерживается вво-302
Отходя¬щиегазыВоздух
Циркулирующая водаФормалинВода gРис. 13.4. Технологическая схема гидратации ацетилена в паровой
фазе:1 — смеситель ацетилена и водяного пара, 2 — подогреватель реакцион¬
ной смеси, 3 — гидрататор, 4 — котел-утилизатор, 5 — холодильник-кон¬
денсатор, 6 — сборник, 7 — промывной скруббер, 8 — подогреватель,9 — ректификационная колоннадом воды в межполочное пространство. Продукты реакции про¬
ходят котел-утилизатор 4, вырабатывающий технологический
пар, и поступают в холодильник-конденсатор 5, где конденси¬
руются вода, ацетальдегид и кротоновый альдегид. Конденсат
собирается в сборнике б, а несконденсировавшаяся парогазо¬
вая смесь подается в промывной скруббер 7, орошаемый водой,
где из нее извлекается ацетальдегид. Непоглощенный ацетилен
возвращается в смеситель 1 в виде циркуляционного газа, а
часть его выводится на очистку (отдувка). Раствор ацетальде-
гида из скруббера соединяется в сборнике б с конденсатом и в
виде 10%-ного водного раствора подается в колонну 9 предва¬
рительной ректификации через подогреватель 8. Отбираемая из
колонны верхняя фракция, содержащая ацетальдегид, посту¬
пает на окончательную ректификацию и очистку.13.2.4. Производство ацетальдегида из ацетилена
через бутилвиниловый эфирПроизводство ацетальдегида из ацетилена этим методом
представляет двухстадийный процесс винилирования н-бутано-
ла ацетиленом с образованием винилбутилового эфира (ВБЭ):С2Н2 + С4Н9ОН «=* СН2=СН-0-С4Н9 (г)и последующего гидролиза ВБЭ:303
CH2=CH-0-C4H9 + н20 «=* СНзСНО + С4Н9ОН (д)Таким образом, в этом процессе бутанол находится в рецикле
и добавляется только для пополнения производственных потерь,
а на получение ацетальдегида расходуется только ацетилен.Реакция винилирования (г) протекает в присутствии гидро¬
ксида калия при температуре 400—440°С. Конверсия ацетиле¬
на составляет 0,6—0,8 при расходном коэффициенте по ацети¬
лену 0,39—0,5 т/т ВБЭ. Реакционная смесь, содержащая 75—
80% ВБЭ, около 20% непрореагировавшего бутанола, воду и
легкую фракцию, перед гидролизом разделяется методами рек¬
тификации или экстракции. На гидролиз направляется фрак¬
ция, содержащая 99,5% вини лбу тилового эфира.Реакция гидролиза ВБЭ (д) протекает в парожидкостной сре¬
де при температуре, близкой к температуре кипения смеси
«ВБЭ—вода», в присутствии катионнообменного катализатора
КУ-2ФПП, который обеспечивает степень конверсии ВБЭ, близ¬
кую к единице. В результате гидролиза образуется система
«ацетальдегид—вода—бутанол», из которой ректификацией
выделяется целевой продукт — ацетальдегид, а бутанол возвра¬
щается в процесс.На рис. 13.5 представлена технологическая схема производ¬
ства ацетальдегида из ацетилена через бути л виниловый эфир.Ацет-Винилбутиловый эфир альдегидРис. 13.5. Технологическая схема производства ацетальдегида
из ацетилена через ВБЭ:1 — реактор винилирования, 2 — подогреватель, 3 — колонна отгонки
ВБЭ, 4 — холодильник-конденсатор, 5 — колонна отгонки бутанола, 6 —
гидролизатор, 7 — холодильник-конденсатор ацетальдегида304
Бутанол и ацетилен вводятся противотоком в секционный
кожухотрубный реактор винилирования 1. Продукты винили¬
рования подогреваются в подогревателе 2 и подаются в колон¬
ну 3, где из них отгоняется ВБЭ, поступающий в холодильник-
конденсатор 4 и затем в гидролизатор 6, куда вводится вода. Об¬
разовавшийся в результате гидролиза ацетальдегид кон¬
денсируется в холодильнике-конденсаторе 7 и направляется на
ректификацию, а бутанол соединяется с продуктом, отогнан¬
ным в колонне 5, и в виде рецикла возвращается в реактор ви¬
нилирования 1.13.2.5. Производство ацетальдегида окислением этиленаОкисление этилена до ацетальдегида представляет гомоген¬
но-каталитическую реакцию, протекающую в среде жидкого
катализаторного раствора хлорида палладия с высокой степе¬
нью селективности. Реакция описывается общим уравнениемС2Н4 + 0,502 = СН3СНО - АН АН = 221,5 кДж (е)В процессе окисления этилена хлорид палладия восстанав¬
ливается до металлического палладия, теряя свою активность,
и должен восстанавливаться за счет окисления кислородом:PdCl2 + С2Н4 + Н20 = СНзСНО + Pd + НС1 (ж)Pd + 2НС1 + 0,502 = PdCl2 + Н20 (з)Суммирование реакций (ж) и (з) дает реакцию (е).Так как скорость регенерирования палладаевого катализа¬
тора (реакция «з») значительно меньше скорости его восстанов¬
ления (реакция «ж»), активность катализатора в ходе процесса
падает очень быстро. Для увеличения скорости реакции (з) в
катализаторный раствор вводятся промоторы — соли меди или
железа в солянокислой среде, играющие роль переносчиков
кислорода. Окисляя металлический палладий, они восстанав¬
ливаются, например:Pd + 2CuCl2 - PdCl2 + Cu2Cl2 (и)Образовавшиеся соли меди или железа низшей степени окис¬
ления легко окисляются кислородом, переходя в исходное со¬
стояние высшей степени окисления:Cu2Cl2 + 0,502 + 2НС1 = 2СиС12 + Н20 (к)Процесс окисления протекает через стадию образования
промежуточного я-комплекса палладия с этиленом, который
под воздействием воды распадается по нескольким направле¬
ниям.305
Скорость суммарной реакции окисления, селективность про¬
цесса, выход ацетальдегида и побочных продуктов зависят от
условий процесса: состава катализаторного раствора и его кис¬
лотности, давления, температуры, соотоншения этилена и окис¬
ляющего агента (кислорода или воздуха). Так при избытке хло¬
рида меди (II) в катализаторном растворе, реакция (и) протека¬
ет количественно, поэтому содержание хлорида палладия (II) в
нем может поддерживаться минимальным. На практике со¬
отношение Cu:Pd поддерживается не менее,чем 15:1, что со¬
ответствует катализаторному раствору, содержащему 0,3—0,5% PdCb и 10—25% СиС12. В подобном растворе процесс
окисления протекает с достаточно высокой скоростью при
температуре 100—130°С. При этом, для поддержания реак¬
ционной массы в жидком состоянии его проводят под давле¬
нием около 1 МПа.Для возможно более полного окисления этилена и соблюде¬
ния безопасности процесса мольное отношение этилена и кис¬
лорода поддерживается в пределах (2,5^4): 1. При соблюдении
этих условий выход ацетальдегида достигает 95% при степени
конверсии этилена 0,5. В качестве побочных продуктов образу¬
ются уксусная кислота, кротоновый альдегид, муравьиная кис¬
лота и различные хлорпроизводные.Технологический процесс производства ацетальдегида из
этилена может быть одностадийным или двухстадийным. В од¬
ностадийном процессе окисление этилена с восстановлением
хлорида палладия и регенерация потерявшего активность ка¬
тализаторного раствора совмещены в одном аппарате — реак¬
торе. Образующийся ацетальдегид испаряется за счет теплоты
реакции и извлекается из реакционной смеси конденсацией и
промывкой водой. В качестве окислительного газа использует¬
ся кислород, почему метод получил название кислородного. В
двухстадийном процессе окисление этилена и регенерация ка¬
талитического раствора разделены и осуществляются в двух
отдельных аппаратах — реакторе и регенераторе, а в качестве
окислительного газа используется воздух.На рис. 13.6 представлена технологическая схема производ¬
ства ацетальдегида одностадийным окислением этилена.Свежий этилен смешивается с циркуляционным газом в сме¬
сителе 1 и подается в нижнюю часть реактора 2, выполненного
в виде барботажной колонны и заполненного катализаторным
раствором. В нижнюю часть реактора на высоту около одного
метра вводится кислород. Реакционная парогазовая смесь вы¬
водится из верхней части реактора и поступает в холодильник-306
Рис. 13.6. Технологическая схема одностадийного окисления
этилена:1 — смеситель, 2 — реактор, 3 — холодильник-конденсатор, 4 — сепара¬
тор, 5 — водяной скруббер, 6 — сборник конденсата, 7 — отгонная ко¬
лонна, 8 — обратный конденсатор, 9 — сборник Кубового продукта, 10 —
ректификационная колоннаконденсатор 3, в котором конденсируется вода, содержащая
компоненты катализатора. Конденсат отделяется в сепараторе
4 ив виде катализаторного раствора возвращается в реактор 2,
где он регенерируется поступающим в реактор кислородом.
Несконденсировавшаяся парогазовая смесь из сепаратора 4 по¬
дается в скруббер 5, орошаемый водой, где из нее извлекаются
ацетальдегид и другие продукты. Непоглощенный этилен воз¬
вращается в смеситель 1, а часть его выводится для очистки
(отдувка). Конденсат из сборника 6 направляется в отгонную
колонну 7, снабженную обратным холодильником 8. В колон¬
ну подается острый пар для отгонки избытка этилена и кисло¬
рода, которые возвращаются в смеситель 1. Сконденсировав¬
шийся кубовый продукт собирается в сборнике 9, из которого
подается в ректификационную колонну 10 для выделения аце¬
тальдегида.13.2.6. Технико-экономические показатели производства
ацетальдегида различными методамиШирокое использование ацетальдегида в органическом син¬
тезе и большие масштабы производства делают актуальным воп¬
рос о экономической эффективности различных методов его про¬
мышленного получения. В табл. 13.1 представлены относитель¬307
ные ТЭП производства ацетальдегида из ацетилена и этилена.
ТЭП двухстадийного процесса из этилена приняты за 100%.Таблица 13.1. ТЭП производства ацетальдегида, %Т ехнико-экономические
показателиОкисление этиленаГидратация ацетиленаДвухста¬дийныйпроцессОдноста¬дийныйпроцессВ жидкой
фазеВ паровой
фазеЕдиничная мощность агрегата100267686850—22Удельные капитальные затраты100504572—203Производительность труда1001785880РК по сырью100100251272РК по энергии1003878414Себестоимость ацетальдегида10082293286—320Из таблицы следует, что себестоимость ацетальдегида, про¬
изводимого из этилена, почти вдвое ниже себестоимости про¬
дукта, получаемого гидратацией ацетилена. При этом наиболее
экономичным является производство ацетальдегида из этиле¬
на одностадийным методом. Поэтому, основной тенденцией раз¬
вития производства ацетальдегида является переход его с аце¬
тиленового на этиленовое сырье.Контрольные вопросы1. В каких областях народного хозяйства используется формальде¬
гид?2. Почему формальдегид получают не окислением метана, а более
сложным путем через метанол?3. В чем отличие процессов производства формальдегида из метано¬
ла окислением его и окислительным дегидрированием?4. Перечислите основные направления использования ацетальдеги¬
да.5. Приведите схему промышленных методов производства ацеталь¬
дегида из различного вида сырья.6. Какими методами ацетальдегид может быть получен из ацетиле¬
на?7. В чем преимущества производства ацетальдегида из этилена по
сравнению с производством из ацетилена?
Глава XIVПРОИЗВОДСТВО УКСУСНОЙ кислоты
И АНГИДРИДА14.1. Технологические свойства и применениеУксусная кислота (этановая кислота) представляет бесцвет¬
ную жидкость с резким запахом, с температурой кипения
118,ГС, температурой плавления 16,75°С
и плотностью 1,05 т/м3. Безводная, так ^ ***
называемая «ледяная» уксусная кислота СН3С ССН3образует за счет водородных связей димер \ q_ц
циклического строения.Критическая температура составляет 321,6°С. Уксусная
кислота смешивается во всех отношениях с этанолом, диэти-
ловым эфиром, бензолом и другими органическими раствори¬
телями и с водой. Растворяет некоторые неорганические и
органические вещества, например, серу, фосфор, ацетаты цел¬
люлозы. С воздухом уксусная кислота образует взрывчатые
смеси с пределами воспламенения от 3,3 до 22,0% об. Темпе¬
ратура вспышки равна 34°С, температура самовоспламенения
354С.Уксусная кислота слабая. Константа ее диссоциации
1,75*10-5. Образует многочисленные растворимые в воде соли
(ацетаты) и этерифицируется спиртами с получением слож¬
ных эфиров. Уксусная кислота обладает высокой коррозион¬
ной активностью по отношению ко многим металлам, особен¬
но в парах и при температуре кипения, что необходимо учи¬
тывать при выборе материалов для аппаратуры. В ледяной
кислоте стойки как на холоду, так и при температуре кипе¬
ния, алюминий, кремнистый и хромистый чугуны, некоторые
сорта нержавеющей стали, но разрушается медь. Техничес¬
кая уксусная кислота обладает большей коррозионной актив¬
ностью, которая усиливается в контакте с воздухом. Из неме¬
таллических материалов стойки по отношению к уксусной
кислоте специальные сорта керамики и эмали, кислотоупор¬
ные цементы и бетоны и некоторые виды полимерных мате¬
риалов (полихлорвиниловые и фенолальдегидные пластмас¬
сы). Ингибитор коррозии в растворах уксусной кислоты —
перманганат калия.В парах уксусная кислота обладает раздражающим действи¬
ем на дыхательные пути, ПДК для нее составляет 5 мг/м3.309
Уксусный ангидрид (СНзС0)20 представляет бесцветную под¬
вижную жидкость с резким запахом, с температурой кипения
139,9°С, температурой плавления -73, ГС и плотностью 1,08 т/м3.
Растворим в этаноле, диэтиловом эфире, бензоле, хлороформе,
уксусной кислоте, холодной воде. В горячей воде гидролизует¬
ся до уксусной кислоты. Это необходимо учитывать при его про-
изводстве. Температура вспышки равна 40°С, температура са¬
мовоспламенения 389°С. Раздражает дыхательные пути и вы¬
зывает ожоги кожи. ПДК составляет 5*10~4%.Уксусная кислота и уксусный ангидрид находят широкое и
разнообразное применение во многих отраслях промышленно¬
сти, главным образом в органическом синтезе. Уксусная кис¬
лота используется:— как ацетилирующий агент в производстве различных аце¬
татов;— для получения уксусного ангидрида и ацетилхлорида;— в производстве красителей и фармацевтических препара¬
тов;— для получения ацетона;— в производстве ацетилцеллюлозы и винилацетата (наря¬
ду с уксусным ангидридом);— для получения монохлоруксусной кислоты — промежу¬
точного продукта в производстве инсектофунгицидов и моющих
средств;— в качестве растворителя и коагулянта латексов;— в пищевой и текстильной промышленности.Основную массу уксусной кислоты потребляют производстваацетилцеллюлозы и винилацетата.14.2. История и промышленные методы производства
уксусной кислоты и уксусного ангидридаУксусная кислота — первая из органических кислот, которая ста¬
ла известна человеку. Впервые она была получена И.Глаубером в
1648 г. и в концентрированном виде путем вымораживания ее водных
растворов и разложением ацетата кальция серной кислотой Г.Шталем
в 1666—1667 гг. Элементный состав уксусной кислоты был установ¬
лен Я.Берцелиусом в 1814 г. До начала XIX века уксусную кислоту
производили исключительно из природного сырья: пирогенетической
обработкой древесины и окислительным уксуснокислым брожением
пищевого этанола. В настоящее время производство уксусной кисло¬
ты из лесохимического сырья имеет второстепенное значение, хотя
масштабы его измеряются сотнями тысяч тонн. В этом методе уксус¬
ную кислоту выделяют из сконденсированной части парообразных
продуктов термической обработки древесины (жижки), получаемой310
в процессе углежжения. Выход кислоты составляет около 20 кг на
1 м3 древесины.Биохимический метод производства уксусной кислоты использу¬
ют только для производства натурального пищевого уксуса.Появление синтетических методов производства уксусной кисло¬
ты связано с разработкой и промышленной реализацией реакции по¬
лучения ацетальдегида по Кучерову. В1910—1911 гг. патентуется спо¬
соб производства уксусной кислоты окислением ацетальдегида, а в
годы первой мировой войны в Германии и Канаде по этому методу было
организовано промышленное производство. С некоторыми технологи¬
ческими изменениями этот метод сохранил свое значение и в течение
более пятидесяти лет является одним из основных.Кроме метода окисления ацетальдегида уксусная кислота в
промышленных масштабах производится следующими метода¬
ми.1) Из ацетона пиролизом его до кетена с последующей гид¬
ратацией последнего (с 40-х годов):СНз-С-СНз — сн2=с=о + СН4оСН2=С=0 + Н20 — СНзСООН2) Из метанола карбонилированием его на кобальтовом ка¬
тализаторе (с 1964 года):СН3ОН + СО — СНзСООН3) Из алканов окислением их в жидкой и паровой фазе:— пропанбутановых фракций (с 1952 года),— бензиновых фракций прямой перегонки (с 1966 года);4) Из продуктов окисления парафина при синтезе ВЖК.Следует также учесть в балансе производимой уксусной кис¬
лоты кислоту, рекуперированную из производства ацетата цел¬
люлозы, получаемого этерификацией целлюлозы уксусным
ангидридом:Цел-ОН + (СН3С0)20 - Цел-ОСОСН3 + СН3СООНУксусный ангидрид в промышленности производится:— совместно с уксусной кислотой окислением ацетальдеги¬
да;— разложением этилидендиацетата;— дегидратацией уксусной кислоты через кетен:СНзСООН — сн2=с=о + Н20СНзСООН + СН2=С=0 — (СН3С0)20Мировое производство уксусной кислоты составляет в насто¬
ящее время свыше 3,5 млн. т в год, в нашей стране в 1980 г.311
было произведено 250 тыс. т. Основная масса уксусной кисло¬
ты производится из ацетальдегида, окислением бутановой и бен¬
зиновой фракций.Первая установка по производству синтетической уксусной кисло¬
ты каталитическим окислением ацетальдегида была пущена на Чер-
нореченском химическом заводе в 1932 г., а в 1948 г. было организо¬
вано ее промышленное производство. К 60-м годам уксусная кислота
производилась также пиролизом ацетона через кетен, окислением уз¬
ких фракций бензина, а также выделением из продуктов окисления
твердого парафина. В результате развития синтетических методов
производства уксусной кислоты удельный вес их вырос с 50% в 1963 г.
до 70% в 1965 г. и до 90% в 1970 г. За эти же годы общий объем произ¬
водства уксусной кислоты в стране вырос в три раза.В 1963 г. были введены в строй новые предприятия по совместному
производству уксусной кислоты и уксусного ангидрида каталитичес¬
ким окислением ацетальдегида в жидкой фазе и к 1965 г. производ¬
ство уксусной кислоты этим методом составляло уже 17% от общего
объема ее производства в стране. В эти же годы было освоено в про¬
мышленном масштабе производство уксусной кислоты карбонилиро-
ванием метанола.14.3. Производство уксусной кислоты окислением
ацетальдегида14.3.1. Физико-химические основы процессаОкисление ацетальдегида молекулярным кислородом пред¬
ставляет гомогенную каталитическую реакцию, протекающую
в жидкой фазе и выражаемую общим уравнением:СН3СНО + 0,502 = СНзСООН - АЯ (а)Реакция протекает по цепному механизму через стадию об¬
разования надуксусной кислоты (НУК):СНз-СНО + 02 — СНз-С-ООН, (б)Окоторая, являясь сильным окислителем, окисляет ацетальде¬
гид до уксусного ангидрида:СНз-СНО + СНз-С-ООН - (СН3С0)20 + Н20 (в)ОУксусный ангидрид при достаточном количестве воды гид¬
ролизуется до уксусной кислоты:(СН3С0)20 + Н20 — 2СН3СНО (г)Таким образом, в системе всегда сосуществуют уксусная кис¬
лота, уксусный ангидрид и вода. Очевидно, что, остановив про¬312
цесс на стадии реакции (в), можно получить в качестве конеч¬
ного продукта не уксусную кислоту, а уксусный ангидрид. Из
этого вытекают два технологических процесса окисления аце¬
тальдегида: получение индивидуальной уксусной кислоты и
получение совместно уксусной кислоты и уксусного ангидри¬
да. Эти процессы различаются природой катализатора, темпе¬
ратурой, составом окислительного газа, методом удаления воды.При получении уксусной кислоты в качестве катализатора
используется раствор ацетата марганца Мп(СНзСОО)2, ускоря¬
ющий реакции (б) и (в), а в качестве окислительного газа при¬
меняется чистый кислород. Чтобы избежать накопления легко
разлагающейся со взрывом НУК, процесс проводят в растворе
циркулирующей уксусной кислоты при температуре не выше
65—70С.При совместном получении уксусной кислоты и уксусного
ангидрида в качестве катализатора для ускорения реакции (в)
используется смесь растворимых ацетатов кобальта
Со(СНзСОО)2 и меди Си(СНзСОО)2 в отношении 1:(1+3). Меха¬
низм действия этого катализатора может быть представлен в
следующем виде:СНз-С-ООН + Со+2 — СНз-С-О + ОН- + Со+3 (д)
ё ОСНз-СНО + Со+3 — СНз-С + Н+ + Со++ (е)ОСНз-С-0 + СНз-С — (СН3С0)20 (ж)О оПри этом лимитирующей процесс реакцией является реак¬
ция (е). Повышение концентрации радикалов СН3СО, опреде¬
ляющих выход уксусного ангидрида, способствует разбавление
окислительного газа до содержания кислорода 7—9% об. и про¬
ведение процесса в диффузионной области при температуре не
выше 50—60°С. Чтобы снизить вероятность реакции гидролиза
образовавшегося уксусного ангидрида (реакция г) из системы
необходимо удалять воду. Этого достигают введением добавок,
образующих с водой легко удаляемые азеотропные смеси, на¬
пример, этилацетата или диизопропилового эфира. При соблю¬
дении этих условий продукт, образующийся при совместном
получении уксусной кислоты и уксусного ангидрида, содержит
их в отношении (3+5):(7+5).В обоих методах производства в качестве побочных продук¬
тов образуются ацетон, эти л ид ен диацетат, формальдегид, ме-313
тилацетат, муравьиная кислота и оксид углерода (IV), которые
удаляются при ректификации сырого продукта.14.3.2. Технологическая схема производства уксусной
кислотыТехнологический процесс производства уксусной кислоты
окислением ацетальдегида состоит из трех последовательных
стадий:— окисление ацетальдегида,—^выделение непрореагировавшего ацетальдегида из паро-
газа,— выделение уксусной кислоты из реакционной смеси и ее
очистка.На рис. 14.1 представлена технологическая схема производ¬
ства уксусной кислоты из ацетальдегида на марганцевом ката¬
лизаторе.Растворы катализатора и ацетальдегида в циркуляционной
уксусной кислоте подаются из смесителей 1 и 2 в нижнюю часть
окислительной колонны — реактора барботажного типа 3. Тем¬
пературный режим в колонне поддерживается с помощью раз¬
мещенных в ней охлаждающих змеевиков, по которым цирку¬
лирует вода. По всей высоте в колонну через несколько труб
подается под давлением 4-105 Па кислород, который барботи-Рис. 14.1. Технологическая схема производства уксусной кислоты:1 — смеситель катализаторного раствора, 2 — смеситель раствора
ацетальдегида, 3 — окислительная колонна (реактор), 4 — брызгоулови-
тель, 5 — рассольный конденсатор, 6 — сепаратор жидкости и газа, 7 —
ректификационная колонна314
рует через жидкость, заполняющую колонну. Парогазовая
смесь, содержащая продукты окисления, выводится из колон¬
ны 3 через брызгоуловитель 4 и поступает в конденсатор 5, ох¬
лаждаемый рассолом, и из него в сепаратор 6. Из сепаратора
конденсат, состоящий из уксусной кислоты и ацетальдегида,
возвращается в окислительную колонну, а несконденсировав-
шиеся газы промываются водой и выпускаются в атмосферу.
Для предотвращения возможности взрыва НУК парогазовая
смесь, выходящая из колонны, разбавляется азотом, который
подается в брызгоуловитель 4. Жидкая уксусная кислота, вы1
ходящая из брызгоуловителя колонны 3, делится на два пото¬
ка. Меньший из них (циркуляционная кислота) направляется
в смесители 1 и 2 для приготовления растворов катализатора и
ацетальдегида, а больший поступает на ректификацию в ко¬
лонну 7 для получения товарного продукта. Из нижней части
колонны 7 выводится в виде кубового остатка раствор катали¬
затора, поступающий на регенерацию.Основной аппарат технологической схемы — окислительная
колонна. Она представляет цилиндр с расширенной верхней
частью, играющей роль брызгоуловителя, высотой 12 и диамет¬
ром 1 м. Колонна изготовлена из алюминия или хромоникеле¬
вой стали, мало подверженных коррозии в уксуснокислой сре¬
де. Внутри колонна имеет полки, между которыми размещены
змеевиковые холодильники для отвода реакционного тепла и
несколько труб для подачи кислорода.Товарным продуктом в этом методе является уксусная кис¬
лота концентрацией после двухкратной ректификации 97,5—
98,5% мае. Выход уксусной кислоты составляет 92% при сте¬
пени превращения ацетальдегида 0,98.14.4. Технологическая схема совместного производства
уксусной кислоты и уксусного ангидридаМетод совместного производства уксусной кислоты и ук¬
сусного ангидрида окислением ацетальдегида является наи¬
более экономичным. В промышленности применяют два ва¬
рианта этого производства. В первом варианте процесс прово¬
дится в присутствии этилацетата и вода удаляется из системы
в виде азеотропной смеси с этилацетатом. Во втором варианте
процесс ведется без постороннего растворителя, а в реактор
подают большой объем газа, что обеспечивает сильную турбо-
лизацию жидкой фазы и способствует удалению воды в виде
пара.315
На рис. 14.2 представлена технологическая схема совмест¬
ного производства уксусной кислоты и уксусного ангидрида с
азеотропной отгонкой воды.Свежий воздух смешивается в смесителе 1 с циркуляцион¬
ным газом и полученная смесь, содержащая 7—9% кислорода,
25—30% ацетальдегида, 1% уксусной кислоты (по объему) и
азот, подается в нижнюю часть реактора 2, заполненного ката-
лизаторным раствором. Тепловой режим в реакторе обеспечи¬
вается системой змеевиков, охлаждаемых водой. Выходящая
из реактора парогазовая смесь охлаждается в холодильнике 3 и
поступает в сатуратор 4, куда вводится ацетальдегид. За счет
испарения жидкого ацетальдегида температура в сатураторе
понижается и из парогазовой смеси конденсируются оставшие¬
ся продукты. Циркуляционная парогазовая смесь из верхней
части сатуратора 4 возвращается в смеситель 1, а частично вы¬
водится в скруббер 5, орошаемый уксусной кислотой, для из¬
влечения из нее паров ацетальдегида. Конденсаты из сатурато¬
ра и скруббера собираются в сборнике 6 в виде сырого продук¬
та, содержащего (в % объемных): 60 уксусного ангидрида, 30
уксусной кислоты, 10 воды и примесь этилидендиацетата, аце-Рис. 14.2. Технологическая схема совместного производства
уксусной кислоты и уксусного ангидрида:1 — смеситель воздуха и циркуляционного газа, 2 — реактор, 3 — холо¬
дильник, 4 — сатуратор, 5 — скруббер, 6 — сборник конденсата, 7 — ко¬
лонна азеотропной осушки, 8 — дефлегматор, 9 — сепаратор, 10 —ко¬
лонна отгонки этилацетата316
тальдегида и формальдегида. Из сборника 6 сырой продукт по¬
дается в колонну азеотропной осушки 7, в которую вводится
эти л ацетат. Отгоняемая смесь паров воды и этилацетата прохо¬
дит дефлегматор 8 и поступает в сепаратор 9У где от этил ацета¬
та отделяется вода. Этилацетат возвращается в колонну 7, из
которой смесь уксусной кислоты, уксусного ангидрида и эти¬
лацетата подается в колонну 10, где от нее отгоняется этилаце¬
тат, возвращаемый, после добавки свежего для пополнения про¬
изводственных потерь, в колонну азеотропной осушки 7. Товар¬
ный продукт в виде смеси уксусной кислоты и уксусного ангид¬
рида, содержащий 0,5—0,8 тонн ангидрида на 1 тонну кисло¬
ты, выводится из нижней части колонны и направляется на рек¬
тификацию.Расходные коэффициенты на 1 т суммарного продукта составля¬
ют:Ацетальдегид, кг
Этилацетат, кг
Катализатор, кг
Электроэнергия, кВт*ч
Сжатый воздух, м3
Оборотная вода, м3Контрольные вопросы1. Перечислите важнейшие направления использования уксусной
кислоты и уксусного ангидрида.2. Из каких видов сырья может быть получена уксусная кислота и
уксусный ангидрид?3. В чем преимущества метода совместного производства уксусной
кислоты и уксусного ангидрида?4. Каким путем в этом методе обеспечивается азеотропная отгонка
воды?87060,012230440725
Глава XVПРОИЗВОДСТВО МОНОМЕРОВ.
ПОЛИМЕРИЗАЦИОННЫЕ МОНОМЕРЫВ этой и последующих главах рассмотрено производство не¬
которых продуктов органического синтеза, которые использу¬
ются в качестве мономеров для получения полимерных мате¬
риалов. Производство этих соединений занимает одно из самых
важных мест в органическом и нефтехимическом синтезе, обес¬
печивая сырьем промышленность пластических масс, химичес¬
ких волокон, эластомеров (каучуков), синтетических лаков и
клеев и пленочных материалов.Мономеры производят в весьма больших количествах. Для
наиболее распространенных представителей их (этилен, винил-
хлорид и др.) объем мирового производства составляет милли¬
оны и десятки миллионов в год.Производство некоторых мономеров было уже рассмотрено
в этой книге. Производство других рассматривается ниже.15.1 ш МономерыМономерами (от греч. monos — один, meros — часть) назы¬
ваются низкомолекулярные соединения преимущественно орга¬
нической природы, молекулы которых способны вступать в ре¬
акцию друг с другом или с молекулами других соединений с
образованием высокомолекулярных соединений или полиме¬
ров.Мономерами могут быть соединения, содержащие кратные
связи (алкены и алкадиены, ацетиленовые углеводороды, про¬
изводные ненасыщенных кислот и др.), легко раскрывающие¬
ся циклы (оксиды алкенов, лактамы, лактоны и др.), соедине¬
ния с разнообразными функциональными группами и подвиж¬
ными атомами (дикарбоновые кислоты, аминокислоты, альде¬
гиды, гликоли, фенолы, диамины и др.). При этом необходи¬
мым условием использования низкомолекулярных соединений
в качестве мономеров является их полифункциональность.Функциональность мономеров определяется числом одинар¬
ных связей, которые мономер затрачивает в данной реакции на
образование молекулы полимера. Функциональность зависит
от природы и числа реакционноспособных фрагментов, входя¬
щих в молекулу мономера. Эти фрагменты могут быть одноакт¬
ные (например, -ОН, -СООН, -NH2), двухактные и полиактные,
реагирующие с образованием, соответственно, двух и более свя-318
о_ / \зей (например, С=С, С=С, 0=С, С-С). В соответствии с числом
образуемых связей, то есть функциональностью, мономеры под¬
разделяются на бифункциональные (/=2), трифункциональные
(/=3) и т. д. Очевидно, что низкомолекулярные соединения с
/= 1 не могут быть мономерами.От функциональности мономера существенно зависит стро¬
ение полученного полимера. Взаимодействие бифункциональ¬
ных мономеров дает полимер линейного строения. Реакции, в
которых участвует хотя бы один мономер с функциональнос¬
тью более двух, приводит к образованию полимера разветвлен¬
ного или пространственного строения. Если в реакции хотя бы
одно низкомолекулярное соединение монофункционально, то
с мономером любой функциональности оно не образует поли¬
мера вследствие блокировки его реакционноспособных точек.На рис 15.1 представлены эти случаи взаимодействия моно¬
меров различной функциональности, где О молекула мономе¬
ра, —► функциональность.О* + -«-О —► О—О 0-+ + -*-0> -*• 0-0-0^ - - - - (1)О—► +«-у-* —► 0-0-0+ <-0-+ —► «-0-0 О—0+ (2)•«-0+ +(3)Рис. 15.1. Схема взаимодействия мономеров различной
функциональности:1 — одно из соединений монофункционально, 2 — оба мономера бифунк¬
циональны, 3 — один из мономеров трифункционаленФункциональность мономеров зависит также от особеннос¬
тей строения молекулы (например, пространственной конфи¬
гурации) и условий протекания реакции. Поэтому, трех- и че¬
тырехфункциональные мономеры могут в определенных слу¬
чаях проявлять низшую функциональность и выступать как
бифункциональные соединения или даже вообще не вступать в
реакцию. Например, трехфункциональный фенол (оксибензол)
при взаимодействии с формальдегидом в случае образования
новолака проявляет функциональность, равную двум, а в слу¬
чае образования резола — равную трем. Из двух изомеров буте-319
на 2-метил-пропен-1 способен к полимеризации, а бутен-2 не
полимеризуется вследствие экранирующего эффекта метиль-
ных групп.В соответствии с этим различают молекулярную или струк¬
турную функциональность (/), определяемую числом реакци¬
онноспособных фрагментов в молекуле мономера, и практичес¬
кую или реализуемую^функциональность (/п).Очевидно, что/П<АВ зависимости от назначения мономеры подразделяются на
полимеризационные и поликонденсационные. К полимеризаци-
онным мономерам относятся соединения, содержащие кратные
связи >С=С<, -С=С—, >С=0, -C=N, >С=С=0 (алкены, алкины,
альдегиды, нитрилы, кетены и др.) или раскрывающиеся цик¬
лические группировки >С-С<, Сл-С=0, >Q-p=0, >Q^-£<4d V NH NH(оксиды алкенов, лактамы, лактоны, имины)К поликонденсационным мономерам относятся соединения,
содержащие стабильные валентнонасыщенные функциональ¬
ные группы или отдельные атомы: -ОН, -СООН, -СОС1, -NH2,
-C=N, >С=0, -Н, -С1.Для производства полимеров большое значение имеет чис¬
тота мономеров. Примеси в них могут ингибировать реакцию
синтеза, оборвать рост макромолекул при полимеризации, на¬
рушить соотношение исходных веществ при поликонденсации
и привести к получению полимеров с малой молярной массой и
низкими эксплуатационными свойствами. Поэтому к продук¬
там органического синтеза, используемым в качестве мономе¬
ров, предъявляются высокие требования по чистоте и содержа¬
нию примесей.15.2. Производство бутадиена-1,3 и изопрена15.2.1. Технологические свойства и применение продуктовБутадиен-1,3 (дивинил) С4Н6 представляет бесцветный газ
с температурой кипения -4,4°С, температурой плавления -
108,9°С и плотностью в жидком состоянии 0, 645 т/м3 (при (ГС).
Не растворим в воде, плохо растворим в спиртах, хорошо — в
бензоле, диэтиловом эфире, хлороформе; с некоторыми раство¬
рителями образует азеотропные смеси. Критическая темпера¬
тура бутадиена 152°С. С воздухом бутадиен образует взрывча¬
тые смеси с пределами воспламеняемости 2,0 и 11,5% об. Тем¬320
пература вспышки бутадиена составляет -40°С, температура са¬
мовоспламенения 420°С.Бутадиен легко полимеризуется, причем полимеризация
инициируется пероксидами, образующимися при контакте бу¬
тадиена с воздухом. Тепловой эффект полимеризации зависит
от температуры и составляет от 72, 8 до 125,6 кДж/моль. Вслед¬
ствие этого бутадиен хранится в присутствии ингибиторов, на¬
пример, п-оксидифениламина или я-трет-бутилпирокатехина,
которые удаляются промывкой гидроксидом натрия перед по¬
лимеризацией.Бутадиен в высоких концентрациях обладает наркотическим
действием; в малых концентрациях раздражает дыхательные
пути и слизистую оболочку глаз. ПДК составляет 100 мг/м3.Изопрен (2-метил-бутадиен1,3) С5Н8 представляет бесцвет¬
ную легколетучую жидкость с характерным запахом, с темпе¬
ратурой кипения 34,1°С, температурой плавления -145,9°С и
плотностью 0,681 т/м3. Изопрен не растворим в воде, хорошо
растворим в углеводородах, этаноле, диэтиловом эфире. Обра¬
зует азеотропные смеси с метанолом, этанолом, ацетоном и мно¬
гими другими органическими растворителями. В парах изоп¬
рен образует с воздухом взрывчатые смеси с пределами воспла¬
меняемости 1,67 и 11,5% об. Температура вспышки изопрена
составляет -48°С, температура самовоспламенения 400°С.Изопрен легко полимеризуется и сополимеризуется с други¬
ми непредельными соединениями. В присутствии воздуха об¬
разует пероксиды, инициирующие самопроизвольную полиме¬
ризацию. Вследствие этого изопрен хранят в присутствии ин¬
гибиторов, например, о-нитрофенола или /i-трет-бутилпирока-
техина.Пары изопрена в высоких концентрациях обладает нарко¬
тическим действием, в малых концентрациях раздражают ды¬
хательные пути и слизистую оболочку глаз. ПДК составляет
40 мг/м3.Бутадиен-1,3 и изопрен используются преимущественно как
мономеры в производстве синтетических каучуков различной
природы. Бутадиен-1,3 — для получения бутадиеновых каучу¬
ков и многочисленных сополимеров с другими мономерами (бу-
тадиен-стирольный, бутадиен-нитрильный, бутадиен-винилпи-
ридиновый, бутадиен-метилстирольный и другие каучуки). В
незначительных количествах бутадиен-1,3 применяют в орга¬
ническом синтезе для производства хлоропрена через 1,2-дих-
лор-бутен-3 и цикло-октадиена-1,5. Изопрен — для получения
стереорегулярных изопреновых каучуков различных сортов.11 Химическая технология. Том 2321
В органическом синтезе изопрен применяют для производства
некоторых фармацевтических препаратов и душистых веществ.15.2.2. Промышленные методы производства бутадиена-1,3
и изопренаВ промышленности бутадиен-1,3 и изопрен производятся
многими методами, которые различаются видом используемо¬
го сырья, числом технологических стадий и экономичностью.Бутадиен-1,3 может быть получен:1. Выделением из С±-фракции продуктов пиролиза жидких
нефтепродуктов, содержащей 20—30% бутадиена-1,3;2. Каталитическим дегидрированием н-бу тиле новой фрак¬
ции, выделенной из продуктов пиролиза нефтепродуктовС4Н8 - С4Н6 + Н23. Каталитическим дегидрированием нбутана, выделен¬
ного из попутного нефтяного газаС4Ню > С4Н8 * С4Н6-Н2 -Н24. Каталитическим расщеплением этанола (С.В. Лебедев)2С2Н5ОН — 2Н20 + Н2 + С4Н65. Взаимодействием ацетилена с формальдегидом через бу-
тандиол-1,3 и бутандиол-1,3 (В. Реппе)Н2С2Н2 + 2СН20 - С4Н4(ОН)2 С4Н8(ОН)2 С4Н6-Н206. Из ацетальдегида через альдоль и бутандиол-1,3
(Н.Н. Остромысленский)Н22СН3СНО —► СН3-СН(ОН)-СН2-СНО -> С4Н8(ОН)2 С4Н6-Н20В настоящее время промышленные методы производства бу¬
тадиена-1,3, основанные на использовании этанола и ацетиле¬
на, устарели и представляют только исторический интерес.Изопрен может быть получен:1. Выделением из С5-фракции продуктов пиролиза нефтепро¬
дуктов.2. Каталитическим дегидрированием амиленовой фракции,
выделенной из продуктов пиролиза нефтепродуктовС5Н10 * CgHg-На322
3. Каталитическим дегидрированием изопентанаС5Н12 С5Н10 ^ с5н8-Г12 -Г124. Взаимодействием изобутилена и формальдегида через
4,4-диметил-1,3-диоксан (М.И. Фарберов, М.С. Немцов)С4Н8 + 2СН2О ~► диоксан— 2-метил-бутандиол-2,4 > CsHg—Н2О5. Взаимодействием ацетилена и ацетона через диметил-
ацетиленилкарбинол (А.Е. Фаворский)н2 ,СНзС2Н2 + СН3-С-СН3—сн3-с-с=сн -*•сн3-с-сн=сн2 — с5н8
о он он _Н2°6. Взаимодействием гидропероксида изопентана с изоами¬
леном через эпоксисоединениеСНзСН3-СН-СН2-СН3 + 02 -* СН3-С-СН2-СН3
СНз (JOH(СНз)2С-СН2-СНз+(СНз)2СН=СН-СНз —-^(СН3)2С-СН-СН3 ^(СНз)2С-СН=СН2 ~* с5н8\ / I -НоОо он 27. Димеризацией пропилена через 2-метил-пентен-2 с его пос¬
ледующим пиролизом2СзН6 — СН2=С(СНз)-СН2-СН2-СН3 — СН3-С=СН-СН2-СН3 -»•СН3 ~са*-с5н8Методы производства бутадиена-1,3 и изопрена значитель¬
но различаются по экономической эффективности. В табл. 15.1
приведены сравнительные технико-экономические показатели
основных методов производства бутадиена-1,3 и изопрена. При
этом показатели производства бутадиена из этанола и изопрена
из изобутилена приняты, соответственно, за 100%.Из таблицы следует, что наиболее экономичным является
производство диеновых мономеров выделением их из фракций
С4 и С5 пиролиза жидких нефтепродуктов и дегидрированием
соответствующих алканов и алкенов. Именно эти методы ис¬
пользуются преимущественно в промышленности и удовлетво¬
ряют свыше 50% потребности в этих мономерах.11*323
Таблица 15.1. ТЭП производства бутадиена-1,3
и изопрена (%)Методы производстваКапитал.затратыЭнерго¬емкостьСебесто¬имостьБутадиен-1,3Дегидрирование н-бутана в две стадии727753Дегидрирование н-бутана в одну стадию473439Выделение из фракции С4 пиролиза20414Получение из этанола100100100ИзопренПолучение из изобутилена100100100Дегидрирование изопентана в две стадии1019098Дегидрирование изопентана в одну стадию9711298Выделение из фракции С5 пиролиза766162Димеризация пропилена11214189Получение из ацетилена и ацетона1027296В то же время, из-за сложности состава продуктов реакции и
трудности их разделения, в производстве изопрена находят при¬
менение и синтетические методы его получения из изобутиле¬
на и формальдегида и окислением изопентана.В основе современных промышленных методов производства бута¬
диена-1,3 и изопрена лежат теоретические исследования в области
химии диеновых углеводородов, выполненные в начале XX века. В
1908—12 гг. С.В. Лебедев установил общие закономерности полиме¬
ризации дивинила и изопрена. В 1911 г. И.И. Остромысленский пред¬
ложил способ получения дивинила через альдоль, впоследствии реали¬
зованный в промышленном масштабе только в 1936 г. В 1910—13 гг.
Б.В. Бызов и Ю.С. Залькинд изучили возможность производства ди¬
винила пиролизом нефтяного сырья и в 1922 г. запатентовали этот
метод.В 1926—28 гг. С.В. Лебедев разработал одностадийный метод полу¬
чения дивинила из этанола, положенный впоследствии в основу его
промышленного производства и заложивший базу отечественного про¬
изводства синтетического каучука. В 1949 г. В. Реппе открыл метод
получения бутадиена-1,3 взаимодействием ацетилена с формальдеги¬
дом. Наконец, с 1940 г. начались исследования метода получения ди¬
винила каталитическим дегидрированием «-бутана и н-бутиленаН.Д. Зелинским и А. А. Баландиным, впоследствии успешно внедрен¬
ного в практику.Работы по производству изопрена начаты А.Е. Фаворским, открыв¬
шим в 1905 г. реакцию получения третичных ацетиленовых спиртов
конденсацией ацетилена с карбонильными соединениями. В 1917 г.
Принс доказал принципиальную возможность получения диеновых324
углеводородов из олефинов и формальдегида. В 1928 г. А.Е.Фаворс¬
кий изучил и в 1939 г. предложил метод синтеза изопрена на основе
ацетилена и ацетона, реализованный позже в промышленном масш¬
табе в Италии.В нашей стране до 1948 г. бутадиен-1,3 производился исключитель¬
но из этанола по методу А.В.Лебедева. В 1948 г. было освоено произ¬
водство бутадиена-1,3 из углеводородного сырья на опытном заводе в
Ярославле. В 1960—63 гг. введены в строй крупные установки непре¬
рывного действия по получению бутадиена-1,3 двухстадийным дегид¬
рированием w-бутана на заводах синтетического каучука в Сумгаите,
Самаре, Стерлитамаке и Омске, а в 1967 г. на Ново-Куйбышевском неф¬
теперерабатывающем заводе.В 1974 г. освоено производство бутадиена-1,3 более экономичным
одностадийным дегидрированием н-бутана. К 1980 г. сырьевая база
бутадиена была расширена за счет создания в Перми установки по
выделению индивидуальных бутанов и пентанов из попутных нефтя¬
ных газов. В результате уже к 1970 г. более 50 % бутадиена-1,3 в стра¬
не производится из нефтяного сырья.До 1959 г. бутадиен-1,3 был в нашей стране единственным мономе¬
ром для производства синтетического каучука. В 50-х годах был раз¬
работан метод синтеза изопрена из изобутилена и формальдегида (ди-
оксановый метод), осуществленный в промышленном масштабе
впервые в мире в 1964—65 гг. на Самарском и Волжском заводах син¬
тетического каучука. В1968—70 гг. вводятся в действие крупные мощ¬
ности по производству изопрена двухстадийныМ дегидрированием
изопентана, извлекаемого из попутного газа Среднего Приобья. Поз¬
же на этом сырье организовали производство изопрена на Тобольском
нефтехимическом комбинате.Ниже рассмотрены технологические процессы производства
бутадиена-1,3 одностадийным дегидрированием н-бутана и про¬
изводства изопрена из изобутилена и формальдегида (диокса-
новым методом), дающие представление об особенностях каж¬
дого из этих процессов.15.2.3. Производство бутадиена-1,3 дегидрированием
н-бутанаДегидрирование н-бутана до бутадиена-1,3 представляет об¬
ратимый гетерогенно-каталитический процесс, протекающий
с поглощением тепла через стадию образования изомеров бути¬
лена по схеме:СНз-СН2-СН2-СНзСН2=СН-СН2-СН3СН2=СН-СН=СН2325
Технологический процесс производства бутадиена-1,3 из н-
бутана может выполняться в двух вариантах: в виде двухста¬
дийного процесса с отделением на первой стадии смеси бутиле-
нов и в виде одностадийного процесса без выделения ее. Эти ва¬
рианты различаются природой катализатора, наличием или
отсутствием операции поглощения смеси бутиленов, методом
понижения парциального давления углеводородного сырья.Двухстадийное дегидрирование н-бутана. Первая стадия
процесса дегидрирования описывается уравнением:С4Н10 *=* С4Н8 + Н2 + АЯ АН = 125,7 кДж (а)В результате реакции образуется смесь трех изомеров н-бу-
тилена: бутен-1, транс-бутен-2 и цис-бутен-2 в соотношении,
соответственно, 34, 38 и 28% массовых, поэтому тепловой эф¬
фект дегидрирования представляет среднее из тепловых эффек¬
тов реакций дегидрирования до соответствующего изомера (132,
115 и 117 кДж/моль, соответственно).Реакция дегидрирования катализируется хромоалюминие¬
вым катализатором, промотированным оксидом калия или ок¬
сидами магния, бериллия и циркония. К таким катализаторам
относится, например, катализатор К-16 состава
Cr203Al2(VK20, активный при температуре 570-600°С и обла¬
дающий высокой селективностью (70-75%) и способностью к
регенерации.Вторая стадия процесса дегидрирования описывается урав¬
нением:С4Н8 «=* С4Н6 + Н2 + АН АН = 116 кДж (б)В результате реакции все три изомера н-бутилена превраща¬
ются в бутадиен-1,3, причем тепловой эффект реакции, как и
реакции (а) представляет среднее из тепловых эффектов реак¬
ций дегидрирования соответствующих изомеров (110,122 и 117
кДж/моль, соответственно). На этой стадии дегидрирования
используется кальций-никель-фосфатный катализатор ИМ-
2204 состава CasNi(P04)6 с добавкой 2% оксида хрома (III). Этот
катализатор активен при температуре 580—620°С и не теряет
активности при разбавлении бутиленовой фракции водяным
паром (см. ниже).Термодинамика первой и второй стадий дегидрирования
идентична: обе реакции протекают с выделением тепла и умень¬
шением объема. Поэтому, для сдвига равновесия в сторону обра¬
зования целевых продуктов следует поддерживать высокую тем¬
пературу, снижать парциальное давление углеводородов и уда¬
лять из системы один из продуктов реакции, например, водород.326
абРис. 15.2. Влияние температуры и давления на степень конверсии
углеводородного сырья: а) н-бутаны, б) н-бутиленыР\ < PiНа рис. 15.2 представлена зависимость конверсии н-бутана
в н-бутилены (а) и н-бутиленов в бутадиен-1,3 (б) от температу¬
ры при различном давлении.Температура процесса дегидрирования определяется актив¬
ностью катализатора, но не должна быть выше 70(ГС во избе¬
жание пиролиза углеводородного сырья. Отлагающийся при
пиролизе на поверхности зерен катализатора кокс дезактиви¬
рует его. Поэтому катализатор периодически регенерируют,
выжигая кокс горячим воздухом.Первая стадия дегидрирования проводится при атмосферном
давлении, вторая при парциальном давлении н-бутиленов 9—
13 кПа. Для понижения парциального давления в систему вво¬
дится перегретый водяной пар.На выход целевых продуктов дегидрирования существенно
влияет время контактирования
(или обратная ему объемная ско¬
рость газа), которое уменьшается с
ростом температуры. На рис. 15.3
приведена зависимость выхода н-
бутиленов от времени контактиро¬
вания (т) при различных температу¬
рах.На первой стадии «-бутилены
извлекаются из бутан-бутиленовой
смеси экстрактивной дистилляцией
с акрилонитрилом (АН), а на второйРис. 15.3. Влияние времени
контактирования на выход н-
бутиленов. Ту> Т}327
стадии бутадиен-1,3 из бутилен-бутадиеновой смеси — экстрак¬
цией аммиачным раствором нитрата или ацетата одновалент¬
ной меди. При этом протекает реакция:Си2(КНз)4(СНзСОО)2+С4Нб^С4Нб (NH3)3Cu2 (CH3COO)2+NH3Ввиду близости температур кипения бутилена и бутадиена-
1,3 (-6,2°С и -4,5°С, соответственно) эффективное разделение
их ректификацией невозможно.В табл. 15.2 приведен технологический режим первой и вто¬
рой стадий дегидрирования н-бутана, а на рис. 15.4 принципи¬
альная схема двухстадийного процесса производства бутадие¬
на- 1,3 из н-бутана.Таблица 15.2. Технологический режим дегидрированияСтадияt, °СР, мПаКатализаторОбъемнаяскорость,и"1X,долейед.Л'долейед.Особыеусловия15800,1СГ2О3 ■ AI2O3+ к2о150—2000,40,7Бутилен из¬
влекают ак-
рилонитри-
ломII6200,9—1,3Ca8Ni(P04)6+ СГ2О3150—2500,40,6Введение
пара. Экст¬
ракция ам¬
миачным
раствором
меди{с4н10Н/[С4Н8 '
[С4Н10ДА "{С4Ню}-^ возвращение в цикл{С4Н8}^-в циклС4Н8АНАНАммиачный растворfC4H6]{С4н8} А|c4h8J\комплекс! ^Н20с4нб у►С4Н6J LРис. 15.4. Принципиальная схема двухстадийного процесса
дегидрирования н-бутанаДвухстадийный процесс производства бутадиена-1,3 из бу¬
тана состоит из многих операций, достаточно сложен в аппара-328
турном оформлении, требует большого расхода перегретого
пара, связан с необходимостью использовать громоздкие про¬
цессы разделения контактных газов. При этом выход бутадие-
на-1,3 и «-бутиленов не превышает 50—60% на использован¬
ный «-бутан. Эти недостатки двухстадийного метода привели к
разработке и быстрому внедрению в промышленность более эко¬
номичного процесса производства бутадиена-1,3 одностадий¬
ным дегидрированием «-бутана, который описывается ниже.Одностадийное дегидрирование к-бутана. Одностадийный
процесс дегидрирования «-бутана до бутадиена-1,3 может быть
описан суммарным уравнением:С4Н10 ^ С4Н6 + 2Н2 + АН АН = 247 кДж (в)и складывается из двух последовательных реакций дегидри¬
рования «-бутана до «-бутиленов (реакция а) и «-бутиленов до
бутадиена-1,3 (реакция б), описанных выше. В этом процессе «-
бутилены не выводятся из сферы, реакции и в реакторе создает¬
ся система ««-бутан — «-бутилены — бутадиен-1,3 — водород»,
равновесный состав которой зависит от температуры и давления.
Из рис. 15.5 следует, что при повышении температуры равновес¬
ная концентрация «-бутана (кривая 1) резко падает, равновес¬
ная концентрация «-бутиленов (кривая 2) проходит через мак¬
симум, а содержание бутадиена-1,3 в смеси (кривая 3) возраста¬
ет. При понижении давления (пунктирная кривая) характер
этой зависимости сохраняется, но равновесная концентрация «-
бутана уменьшается, а содержание
бутадиена-1,3 в смеси увеличивает¬
ся. Таким образом, при одностадий¬
ном дегидрировании для увеличе¬
ния выхода бутадиена-1,3 процесс
следует проводить при малом пар¬
циальном давлении компонентов
или при разрежении и при темпера¬
турах более высоких, чем на первой
стадии дегидрирования «-бутана в
двухстадийном процессе.Одностадийный процесс дегид¬
рирования «-бутана осуществляет¬
ся по регенеративному принципу,
при. котором затраты тепла на про¬
ведение эндотермической реакции
дегидрирования в адиабатическом
режиме возмещаются за счет тепла,% мол.► Г РпРис. 15.5. Зависимость
состава равновесной смеси
от температуры и давленияPi>Pi329
выделяющегося на стадии регенерации катализатора при вы¬
жигании отложившегося на нем кокса. В этом процессе разог¬
ретый регенерированный катализатор используется как тепло¬
носитель, а для повышения его способности аккумулировать
тепло, к нему добавляется в отношении 1 : 3 инертный теплоно¬
ситель в виде гранул оксида алюминия, предварительно обо¬
жженных.Жесткие условия чередующихся окислительно-восстанови¬
тельных циклов дегидрирования и регенерации предъявляют
к катализаторам повышенные требования. В одностадийном
процессе используется алюмо-хромовый окисный катализатор
ДВ-ЗМ состава СГ2О3 • AI2O3, активный при температуре около
600°С, ускоряющий обе реакции дегидрирования, прочный и
устойчивый в эксплуатации и хорошо регенерирующийся. Так
как он отравляется парами воды, то понижение парциального
давления углеводородного сырья в процессе достигается не вве¬
дением в систему водяного пара, а проведением дегидрирова¬
ния в вакууме.В табл. 15.3 представлен технологический режим процесса
одностадийного дегидрирования w-бутана до бутадиена-1,3.Таблица 15.3. Технологический режим одностадийногопроцессаПараметр процессаДегидрированиеРегенерацияТемпература, °С565—650620Давление, МПа1,6—Объемная скорость паров, ч“1250—3505000Время процесса, мин.5—95—9Отношение катализатор : теплоноситель1 :31 : 15Технологическая схема производства бутадиена-1,3 односта¬
дийным дегидрированием w-бутана в вакууме (рис.15.6) вклю¬
чает операции:— очистка сырья (бутановой фракции, попутного газа);— каталитическое дегидрирование w-бутана;— сжатие контактного газа и выделение из него фракции С4;— выделение бутадиена-1,3 из фракции С4;— отдувка углеводородов и регенерация катализатора.Реакторный блок установки включает два (или больше) ап¬
паратов, работающих попеременно на дегидрирование сырья и
регенерацию катализатора.330
ГазВоздухА>ГТ ^оI ТопливныйУ(uiuuiiiiiiiiiiiiiii) (nuillllllllillllllll1□13 ;□у-}8ПарГазы на ^
регене- -З*-1рацию f 1311л10\JЛVС4 навыделениебутадиенаРис. 15.6. Технологическая схема одностадийного дегидрирования
н-бутана до бутадиена-1,31 — подогреватель сырья, 2 — лечь, 3 — реакторы, 4 — «закалочный»
аппарат, 5 — скруббер, 6,11 — холодильники, 7 — турбокомпрессор,8 — абсорбер, 9 — десорбер, 10 — стабилизирующая колонна (депропа-
низатор), 12 — топка, 13 — котел-утилизаторЧерез подогреватель 1 н-бутан поступает в печь 2, где нагре¬
вается до 600—620°С и направляется в один из реакторов 3, ко¬
торый работает на дегидрирование. Из реактора контактный
газ, пройдя для «закалки» аппарат 4, подается в скруббер 5, в
котором охлаждается холодным маслом, циркулирующим че¬
рез холодильник 6. Охлажденный в скруббере газ сжимается в
турбокомпрессоре 7 до давления 1,3 МПа и направляется в аб¬
сорбер 8. Из верхней части абсорбера выходит водородсодержа¬
щий топливный газ, а раствор углеводородов в абсорбенте по¬
дается в десорбер 9. Из верхней части десорбера отгоняется
фракция Сз — С4, а абсорбент через холодильник 11 возвраща¬
ется на орошение абсорбера 8. В качестве абсорбента использу¬
ется высококипящая углеводородная фракция С5. Фракция
Сз — С4 из верхней части десорбера 9 поступает в колонну 10331
(депропанизатор), где из нее отгоняется пропан. Оставшаяся
фракция С4 с содержанием бутадиена-1,3 от 11 до 13% массо¬
вых направляется на выделение бутадиена (см. двухстадийный
процесс), а бутан-бутиленовая фракция возвращается в виде
рецикла на дегидрирование, присоединяясь к свежему к-бута-
ну. По окончании цикла дегидрирования поток углеводородно¬
го сырья переключается на другой реактор, а первый продува¬
ется сначала водяным паром для удаления сорбированных ка¬
тализатором углеводородов, а затем для регенерации катализа¬
тора топочными газами с небольшим содержанием кислорода
из топки 12. Теплота газообразных продуктов регенерации ка¬
тализатора используется для выработки технологического пара
в котле-утилизаторе 13.Основной аппарат технологической схемы — реактор дегид¬
рирования (контактный аппарат). Это стальной цилиндр диа¬
метром 6 м и длиной 12—14 м расположенный горизонтально и
футерованный внутри огнеупорным материалом. Внутри реак¬
тора расположены решетки из керамических плит, на которых
размещены слои катализатора.Достоинствами одностадийного процесса дегидрирования н-
бутана до бутадиена-1,3 являются:— значительное сокращение расхода технологического пара;— использование теплоты регенерации катализатора и про¬
ведение реакции дегидрирования в адиабатическом режиме и,
как следствие, простота конструкции реактора и отсутствие
сложного теплообменного оборудования;— исключение второй стадии дегидрирования и операций
разделения бутан-бутиленой фракции.За счет этого относительно невысокие выход бутадиена-1,3
(12—14%) и степень конверсии н-бутана (не превышающая 0,2)
компенсируются меньшими капитальными затратами и энер¬
гоемкостью производства и, как следствие, более низкой, чем в
двухстадийном методе, себестоимостью бутадиена-1,3.15.2.4. Производство изопрена конденсацией изобутилена с
формальдегидомПроцесс производства изопрена из изобутилена и формаль¬
дегида состоит из двух последовательных стадий: конденсации
изобутилена с формальдегидом и разложения полученного ди-
метилдиоксана.1. Конденсация изобутилена с формальдегидом до 4,4-диме-
тил-диоксана-1,3 (ДМД) протекает по уравнению:332
СН2-Оч 7СН3СН3-С=СН2 + 2СН20 ^ О -АЯ (г)сн3 4 сн2-сн2 чсн3Реакция (г) катализируется серной кислотой, которая вво¬
дится в количестве 1,0-1,5% от массы изобутилена. Для пре¬
дотвращения полимеризации изобутилена в системе всегда под¬
держивается избыток формальдегида, который подают в виде
37%-ного водного раствора (формалина). Конденсация прово¬
дится при температуре 85—95°С и давлении около 2 МПа. Это
обеспечивает жидкое состояние реагентов, образующих гетеро¬
генную двухфазную систему. Поэтому интенсивность процесса
конденсации существенно зависит от поверхности контакта уг¬
леводородной и жидкой фаз.В этих условиях степень конверсии изобутилена в ДМД со¬
ставляет 0,85—0,90 при селективности процесса по изобутиле¬
ну 65% и по формальдегиду 80% . В качестве побочных продук¬
тов при конденсации образуются трет-бутанол и 3-метил-бу-
тандиол-1,3.Разложение ДМД протекает через стадию образования 3-ме-
тил-бутандиола-1,3 по уравнению:^СНз-0 уСН3 СН3О С + Н20 ^ СН3-С-СН2-СН2ОН + сн20 (д)сн2-сн2 сн3 онс последующей дегидратацией его до изопрена (2-метил-бута-
диена-1,3):СН3 СНзСН3-С-СН2-СН2ОН ^ СН2=С-СН=СН2 + 2Н20 (е)-ОНОдновременно протекает побочная реакция разложения ДМД
на исходные изобутилен и формальдегид, обратная реакции син¬
теза его (г).Реакция разложения ДМД протекает с поглощением тепла
на гетерогенном фосфатном катализаторе КДВ-15 состава
СаНР04*Саз(Р04)2 и описывается суммарным уравнением:C6Hi202 = С5н8 + СН20 + Н20 + АЯ АН = 146 кДж (ж)Процесс разложения ДМД проводится при температуре 380—
400С в газовой фазе в присутствии перегретого водяного пара,
который повышает селективность реакции (е) и, одновремен¬
но, является источником тепла. В этих условиях степень кон¬
версии ДМД достигает 0,9 при селективности по изопрену 80—
90%.333
остатокРис. 15.7. Технологическая схема производства изопрена
конденсацией изобутилена с формальдегидом:1, 2 —трубчатые реакторы конденсации, 3 — нейтрализатор, 4 —
сепаратор, 5 — колонна отгонки изобутилена, 6 — метанольная колонна,7 — реактор разложения ДМД, 8 — холодильник, 9 — сепаратор, 10,11 —
ректификационные колонныСырьем в производстве изопрена методом конденсации изо¬
бутилена с формальдегидом служит чистый изобутилен или
изобутилен-изобутановая фракция, полученная каталитичес¬
ким дегидрированием изобутана. На рис. 15.7 представлена тех¬
нологическая схема производства изопрена из изобутилена и
формальдегида.Первая стадия процесса (конденсация изобутилена с фор¬
мальдегидом) проводится в двух трубчатых реакторах 1 и 2,
охлаждаемых водой. Изобутиленовая фракция и соединенный
с ней возвратный (циркуляционный) изобутилен подаются в
реактор 2 и из него в реактор 1, в который противотоком посту¬
пает водный раствор формальдегида, подкисленный серной
кислотой. Верхние и нижние части реакторов работают как се¬
параторы, разделяя реакционную массу на углеводородный и
водный слои. В углеводородный слой переходит ДМД, в водном334
слое остается не вступивший в реакцию формальдегид. Углево¬
дородный слой из реактора 1 подается в нейтрализатор 3, где
нейтрализуется и промывается водным раствором гидроксида
натрия, и поступает в сепаратор 4 для отделения от него остат¬
ков воды. Водные слои из нижней части реактора 2, нейтрали¬
затора 3 и сепаратора 4 объединяются и подаются на переработ¬
ку для отделения от них летучих веществ, добавляемых к угле¬
водородному слою. Из сепаратора 4 промытый углеводородный
слой проходит колонну 5, в которой из него отгоняется непро¬
реагировавший изобутилен, возвращаемый в цикл, и поступа¬
ет в ректификационую колонну 6 для отгонки от ДМД более
летучих метанола и трет-бутанола. Освобожденный от метано¬
ла ДМД направляется на вторую стадию (разложение ДМД) в
реактор 7 адиабатического типа со стационарным слоем ката¬
лизатора. В реактор подается также перегретый водяной пар.
Реакционная смесь из реактора конденсируется в холодильни¬
ке S и поступает в сепаратор 0, где разделяется на водный и орга¬
нический слои. Водный слой соединяется с водным слоем пер¬
вой стадии процесса, а органический слой подается последова¬
тельно в ректификационные колонны 10 и 11. В колонне 10 от
него отгоняется изобутилен, образовавшийся при разложении
ДМД, который возвращается на первую стадию процесса, а в
колонне 11 — изопрен от высокомолекулярных побочных про¬
дуктов, образующих кубовый остаток. Для окончательной очи¬
стки изопрен промывается водой, сушится азеотропной пере¬
гонкой и ректифицируется.Полученный продукт содержит не менее 99,2% изопрена и
пригоден для стереорегулярной полимеризации в каучук без
дорогостоящей специальной очистки.15.3. Производство стирола15.3.1. Технологические свойства и применение стиролаСтирол (винилбензол, фенилэтилен) СбНб-СН=СН2 — бес¬
цветная жидкость с характерным сладковатым запахом, с тем¬
пературой кипения 145,2°С, с температурой плавления -30,6°С
и с плотностью 0,906 т/м3. Плохо растворим в воде (0,05% мае.),
образуя с ней азеотропную смесь с температурой кипения 34,8°С,
смешивается во всех отношениях с метанолом, этанолом, диэ-
тиловым эфиром, ацетоном, четыреххлористым углеродом.
Хорошо растворяет различные органические вещества. Крити¬
ческая температура стирола составляет 373°С.335
Стирол легко окисляется кислородом воздуха с образовани¬
ем бензальдегида и формальдегида и перекисей, инициирую¬
щих его полимеризацию. Пары стирола образуют с воздухом с
узком интервале концентраций взрывчатые смеси с пределами
воспламенения 1,1 и 6,1% об. Температура вспышки стирола
равна 34°С, температура воспламенения 490°С.Стирол легко полимеризуется с выделением тепла, особенно
при нагревании, образуя метастирол — стекловидную твердую
массу, которая представляет твердый раствор полистирола в
стироле. Тепловой эффект полимеризации стирола составляет
74,5 кДж/моль. Во избежание самопроизвольной полимериза¬
ции стирол хранится и транспортируется в присутствии инги¬
биторов — гидрохинона, п-трет-бутил-пирокатехина, диоксим-
п-хинона и др., которые перед использованием удаляются пе¬
регонкой продукта в вакууме или промывкой раствором гид¬
роксида натрия.Пары стирола в высоких концентрациях вызывают слезотече¬
ние и раздражение дыхательных путей. ПДК составляет 0,5 мг/
м3, для производственных помещений допускается 5,0 мг/м3.Стирол используют преимущественно как мономер для про¬
изводства полистирола, бутадиен-стирольных каучуков, сопо¬
лимеров с акрилонитрилом, винилхлоридом и другими моно¬
мерами. В меньших количествах применятся в качестве раство¬
рителя полиэфирных пластмасс и для модификации алкидныхполимеров, а также в качестве добавки к моторному топливу./Мировое производство стирола составляет около 12 млн. т в год.15.3.2. Сырье и промышленные методы производства
стиролаСырьем для промышленного производства стирола служит
зтилбензол. Он может быть получен:— алкилированием бензола этиленом,— извлечением из фракций «сырого бензола», полученной
разделением прямого коксового газа, или из ксилольной фрак¬
ции риформинга нефтепродуктов (см. часть II), Таким образом,
сырьевой базой для производства стирола являются, в конеч¬
ном итоге, продукты коксования каменного угля и нефтепере¬
работки.Основным методом промышленного производства стирола
является каталитическое дегидрирование этилбензола. Этим
методом получают более 90% мирового производства стирола.
Менее распространены методы производства из этилбензола336
через гидропероксид этилбензола с одновременным получени¬
ем оксида пропилена (халкон-процесс):С6Н5-СН2-СН3 + 02 — с6н5-сн-сн3ООНСбН5-СН-СН3 + СН2=СН-СН3 — СбН5-СН-СНз+СНз-СН-СН2
ООН он оС6Н5-СН-СН3 - С6Н5-СН=СН2 + Н20;ОНили взаимодействием этилена со стильбеном, полученным окис¬
лением толуола:С6Н5-СН3 + 02 — С6Н5-СН=СН-С6Н5 + 2Н20С6Н5-СН=СН-С6Н5 + С2Н4 — 2С6Н5-СН=СН2и экстрактивной ректификацией из фракции пиролиза бензи¬
на в этилен, содержащей до 35% стирола.На рис. 15.8 представлена взаимосвязь промышленных ме¬
тодов производства стирола из различного сырья.сн2=сн-сн3"► СеН5-(рН-СНз
онСбН5-9Н-СН3
пиролиз °9Н ректификация-► Бензин
С2Н4Нефть
I Ггул—^нбJ2'5-с2н5I-Н20КУФ2 ОКГ риформинг
►ПКГ—СБ-н,*сбн5-сн=сн2о,■С2Н4Рис. 15.8. Промышленные методы производства стиролаКУ — каменный уголь, ПКГ — прямой коксовый газ, ОКГ — обратный
коксовый газ, СБ — сырой бензол, Ф1 — фракция пиролиза бензина,Ф2 — ксилольная фракция нефтепродуктов.Основная масса этилбензола, используемого для производ¬
ства стирола, получается алкилированием бензола этиленом
(реакция Фриделя—Крафтса).Алкилирование бензола представляет гетерогенно-каталити¬
ческую экзотермическую реакцию, описываемую суммарным
уравнением:337
C6H6 + С2Н4 = С6Н5-С2Н5 - АН АН= 108,6 кДж (а)Реакция катализируется хлоридом алюминия и представ¬
ляет собой электрофильное замещение в ароматическом ядре,
протекающее через стадию образования промежуточного ком¬
плекса:А1С13 + НС1 ^ Н+ + А1С14"СН2=СН2 + Н+ ^ СН3-СН2+§О + СНз-СН;++ Н+Так как хлорид алюминия в твердом виде нерастворим в уг¬
леводородах и слабо катализирует реакцию алкилирования, на
практике применяется предварительно приготовленный жид¬
кий катализаторный комплекс хлорида алюминия с диэтилбен-
золом (ДЭБ) и хлоридом водорода (комплекс Густавсона).Комплекс Густавсона — это соль карбкатиона ДЭБ (ст-комп¬
лекс), окруженная сольватной оболочкой из нескольких (1—6)
молекул ДЭБ, и получается пропусканием хлорида водорода при
нагревании через суспензию хлорида алюминия в диэтилбен-
золе.(C2H5)2<D<S •■(л-1)ДЭВВесьма эффективны также катализаторы на основе ионооб¬
менных сорбентов, например, кислотный катионит КУ-2 с ак¬
тивными сульфогруппами, получаемый сополимеризацией сти¬
рола с дивинилом с последующим сульфированием сополиме¬
ра и омылением сульфопродукта щелочью или водой.Алкилирование в присутствии хлорида алюминия по реак¬
ции (а) сопровождается необратимыми побочными реакциями
последовательного введения этильных групп в ядро с образова¬
нием смеси продуктов различной степени алкилирования, важ¬
нейшей из которых является образование диэтилбензола:С6Н6 + 2С2Н4 = С6Н4(С2Н5)2, (б)а также реакции деструкции, окисления и смолообразования.Одновременно с реакциями алкилирования протекают обра¬
тимые реакции переалкилирования полиэтилбензолов, напри¬
мер:С6Н4(С2Н5)2 + С6Н6 ^ 2СбН5-С2Н5 (в)Для подавления реакции (б) и смещения вправо равновесия
реакции (в) алкилирование проводят в избытке бензола, а часть338
NaOH—у^бИбГ^СбИбНСгН4Реактор ►ODДЭБ- ЭБ Г!дав!=Гна ректи¬
фикациюКатализа-- торный - ^
. комплекс.Кислые- продукты *
окисленияРис. 15.9. Принципиальная схема производства этилбензола:ЭБ-этилбензол, ДЭБ-диэтилбензолобразующегося ДЭБ возвращают в ци^л. На рис. 15.9 представ¬
лена принципиальная схема производства этилбензола алкили-
рованием бензола этиленом. Технологическая схема и парамет¬
ры процесса алкилирования бензола рассматриваются ниже на
примере производства изопропилбензола в главе XVI.Процесс алкилирования может проводиться в жидкой или в
паровой фазе, при температуре от 95°С до 450°С и мольном отно¬
шении бензол/этилен от 2:1 до 6:1. Полученный алкилат содер¬
жит 12—35% массовых этилбензола, 55—85% массовых бензо¬
ла и 2,5—8%массовых диэтилбензола. Современные установ¬
ки по производству этилбензола достигают мощности 740 тыс.
т продукта в год. Выход этилбензола в расчете на бензол состав¬
ляет 95%, при расходных коэффициентах на 1 т продукта: бен¬
зол 0,77 т, этилен 0,3 т, хлорид алюминия 0,03 т.История. Стирол впервые был выделен и идентифицирован в 1839 г.
Е. Симоном из стиракса — смолы амбрового дерева. Им же было дано
современное название углеводорода. Ш. Жерар и А. Каур в 1841 г. по¬
лучили стирол разложением коричной кислоты, определили его состав
и дали название «циннамон». В 1845 г. Э. Копп установил тождествен¬
ность обоих веществ. В 1867 г. А. Бертло синтезировал стирол, про¬
пуская через раскаленную трубку смесь паров бензола и ацетилена.
Он же установил присутствие стирола в ксилольной фракции камен¬
ноугольной смолы. Стирол содержится также во многих продуктах тер¬
мической деструкции органических веществ, в продуктах пиролиза
природного газа, крекинга и пиролиза нефтепродуктов и сланцевом
масле.Промышленное производство стирола в нашей стране было орга¬
низовано в 1949 г. на Воронежском заводе синтетического каучука из
этилбензола, получаемого ал копированием бензола. В 60-х годах ус¬339
тановки алкилирования с использованием этилена обратного коксо¬
вого газа мощностью не более 10 тыс. т в год создаются на заводах по
производству синтетического аммиака. Мощность их лимитируется
выпуском аммиака. В 1966—70 гг. строятся уже специальные цехи
по производству этилбензола и стирола на его основе мощностью 40
тыс. т в год на нефтехимических предприятиях. В период 1976—80
гг. вводятся в строй мощные специализированные системы по произ¬
водству стирола из этилбензола в ПО «Нижнекамскнефтехим», на за¬
воде пластмасс в г. Шевченко, в Узловском ПО и др. Мощность этих
систем достигает 300 тыс. т в год, установки автоматизируются и ос¬
нащаются современными АСУ ТП.15.3.3. Производство стирола дегидрированием этилбензолаФизико-химические основы процесса. Дегидрирование этил¬
бензола до стирола представляет собой обратимую эндотерми¬
ческую гетерогенную каталитическую реакцию, описываемую
уравнением:С6Н5-С2Н5^ С6Н5-СН=СН2 + Н2 + АН АН = 124 кДж (г)Реакция катализируется оксидами и сульфидами металлов
восьмой группы периодической системы. В промышленности
применяют железооксидные катализаторы К-22 состава: F2O3 —
55—80%, СГ2О3— 2—28%, К2СО3 — 15—35%. Эти катализа¬
торы обладают достаточной активностью в реакции дегидриро¬
вания и имеют селективность по стиролу до 90%.Энергия активации реакции дегидрирования составляет
152 кДж/моль, поэтому скорость ее сильно зависит от темпера¬
туры. Это также указывает на то, что реакция протекает в ки¬
нетической области. Реакция (г) протекает с поглощением теп-
ла и увеличением объема газообразных продуктов. Следователь¬
но, сдвигу равновесия вправо способствует повышение темпе¬
ратуры и понижение давления — общего и парциального этил¬
бензола. Так, например, при температуре 595С равновесная
степень превращения этилбензола при давлении 0,1 МПа рав¬
на 0,4, а при давлении 10 кПа — 0,8. Вследствие этого процесс
дегидрирования этилбензола проводится при температуре 600°С
и общем давлении ОД МПа, что соответствует парциальному
давлению этилбензола около 10 кПа. Понижение парциально¬
го давления этцлбензола при заданной степени конверсии по¬
зволяет также вести процесс дегидрирования при более низкой
температуре.Для снижения парциального давления этилбензола в реак¬
ционную смесь вводят перегретый водяной пар в массовом от¬
ношении к этилбензолу 2,5:1. Пар, одновременно, играет роль340
теплоносителя, обеспечивая приток тепла для осуществления
эндотермической реакции (а).Реакция дегидрирования этилбензола на железооксидных
катализаторах сопровождается побочными реакциями деструк¬
ции (крекинга) этилбензола и взаимодействия их продуктов,
приводящими к образованию бензола, толуола, а также мета¬
на, этана и оксидов углерода, переходящих в газ: ► С6Нб jC6H5-C2H5 ^ сбн5-сн=сн2 + н2 ►С6Н5-СНз-« Технологическая схема производства стирола дегидрирова¬
нием этилбензола. Технологический процесс производства сти¬
рола из этилбензола состоит из двух основных стадий: дегидри¬
рование этилбензола и выделение стирола-ректификата. Про¬
цесс построен как циркуляционный и предусматривает возвра¬
щение в цикл избытка этилбензола и использование конденса¬
та водяного пара для выработки свежего перегретого пара.Технологическая схема этого процесса представлена на рис.
15.10.Рис. 15.10. Технологическая схема производства стирола
дегидрированием этилбензола:1 — котел-утилизатор, 2 — испаритель, 3 — теплообменник, 4 — трубча¬
тая печь, 5 — реактор, 6 — водяной холодильник, 7 — рассольный
холодильник, 8 — сепаратор отделения газа, 9 — сепаратор отделения
конденсата, 10, 11, 12, 13 — ректификационные колонны341
Свежий и возвратный (циркуляционный) этилбензол смеши¬
ваются с небольшим количеством пара, вырабатываемого в кот-
ле-утилизаторе 1, и проходят последовательно испаритель 2 и
теплообменник 3, обогреваемые горячей реакционной смесью.
Нагретые до 520—530°С пары бензола смешиваются затем с пе¬
регретым до 700°С водяным паром, вырабатываемом в трубча¬
той печи 4, и подаются в реактор 5. Продукты реакции отдают
тепло в теплообменнике 3, испарителе 2 и котле-утилизаторе1 , охлаждаются окончательно в водяном холодильнике 6 и рас¬
сольном холодильнике 7 и поступают в сепаратор 8 для отделе¬
ния конденсата от газа. Углеводородный газ используется на
обогрев печи 4. Конденсат из газового сепаратора 8 поступает в
сепаратор 9, где разделяется на водную и углеводородную фазы.
Водный слой подается в печь 4 и используется для выработки
водяного пара, а углеводородный слой («печное масло») направ¬
ляется на ректификацию. В состав печного масла входят сти¬
рол (до 55% мае.), остаточный этилбензол (40% мае.), побоч¬
ные продукты (бензол 2% и толуол 2% мае.) и смолы. Чтобы
избежать полимеризации стирола в печное масло перед ректи¬
фикацией вводится ингибитор, а ректификацию проводят в ва¬
кууме, что позволяет снизить температуру процесса. Система
ректификации состоит из четырех колонн. Печное масло из се¬
паратора 9 подается в вакуум-ректификационную колонну 10,
где из него отгоняется бензол, толуол и большая часть не всту¬
пившего в реакцию этилбензола. Этот дистиллят поступает в
^колонну 11 и разделяется на бензольно-толуольную фракцию
(бентол) и этилбензол, который возвращается на дегидрирова¬
ние. Кубовая жидкость из колонны 10, содержащая стирол,
подается в вакуум-ректификационную колонну 12, в которой
из нее отгоняются остатки этилбензола с частью стирола. Этот
отгон возвращается на ректификацию в колонну 10. Кубовая
жидкость из колонны 12у представляющая сырой стирол, по¬
ступает на окончательную ректификацию в колонну 13, из ко¬
торой отбирается дистиллят в виде 99,8%-ного стирола.Основным аппаратом технологической схемы является ре¬
актор дегидрирования. Наиболее распространены реакторы
адиабатического типа, в которых тепло, необходимое для про¬
ведения реакции, подводится с перегретым водяным паром.
Адиабатический реактор — это стальной аппарат цилиндричес¬
кой формы с коническими крышкой и дном, футерованный из¬
нутри огнеупорным материалом, диаметром 4 м и общей высо¬
той 7,5 м. Внутри реактора на решетке размещаются слои на¬
садки, обеспечивающей равномерное распределение газового342
потока по сечению реактора, между которыми помещен слой
катализатора. Применяемый в процессе катализатор К-22 спо¬
собен к саморегенерированию и может работать непрерывно в
течение 1—2 месяцев, после чего его регенерируют, подавая в
реактор воздух для выжигания отложившегося на зернах ката¬
лизатора кокса.Технико-экономические показатели процесса:Температура, °С 590—620Объемная скорость подачи сырья, ч'1 0,35—0,50Массовое отношение этилбензол : пар 1,0 : 2,6Конверсия этил бензола, долей ед. 0,45Селективность процесса, долей ед. 0,90Выход стирола на этилбензол, доле ед. 0,4015.4. Производство капролактама15.4.1. Технологические свойства и методы производстваКапролактам (лактам е-аминокапроновой кислоты, 2-оксо-
гексаметиленимин) представляет бесцветное кристаллическое
вещество с температурой плавления 68,8°С, темпе-
/NH ратурой кипения 262,5°С и плотностью 1,02 т/м3
(СН2)^| (при 70°С). Хорошо растворим в воде (525 г в 100 г
С_0 воды), бензоле, ацетоне, этаноле, диэтиловым эфи¬
ре, плохо растворим в алифатических углеводородах. Раство¬
ряется в разбавленной серной кислоте, гидролизуясь до е-ами¬
нокапроновой кислоты. Гигроскопичен. При нагревании с кон¬
центрированными минеральными кислотами капролактам об¬
разует соли. В присутствии каталитических количеств воды,
спиртов, аминов и органических кислот при нагревании поли¬
меризуется с образованием полиамида.Температура вспышки капролактама составляет 135 С, тем¬
пература самовоспламенения 400°С. Капролактам раздражает
кожу, ПДК равна 10 мг/м3. Транспортируется и хранится в
твердом состоянии в мешках, или в виде расплава в цистернах
с обогревом в атмосфере азота.Капролактам получают различными методами из аромати¬
ческого (бензол, толуол, фенол) и неароматического (цикло-
гексан, фурфурол, ацетилен, окись этилена) сырья. Во всех ме¬
тодах промежуточным продуктом является циклогексанонок-
сим, изомеризацией которого получается капролактам. Про¬
мышленное значение имеют следующие методы производства
циклогексаноноксима.343
1. Из фенола через циклогексанол с последующим его дегид¬
рированием до циклогексанона (фенольный метод):Н2 NH2OH<о>-он - 0-°н г о-0 Onoh~Н22. Из толуола через бензойную кислоту с последующим гид¬
рированием ее до циклогексанкарбоновой кислоты и обработ¬
кой последней нитрозилсерной кислотой (толуольный метод):02 Н2 НСК<(0>-СНз <0>-СООН -*■ О-СООН -»■ <^]>=NOH-С023. Из анилина через циклогексиламин с последующим от¬
щеплением аммиака при действии воды (анилиновый метод):Н2 Н20 NH2OH<o>-nh2 - о-о -* O-N0H4. Из циклогексана, выделенного из нефтяного сырья, или
полученного гидрированием бензола, следующими методами:— окислением воздухом через циклогексанол и циклогекса-
нон:„ 02 _ nh2ohО - 0-°н 7 0-° O-N0H-Н2— фотонитрозированием хлористым нитрозилом: k NOC1 , О O-N0H— нитрованием через нитроциклогексан с последующим вос¬
становлением до нитрозоциклогексана (нитроциклогексановый
метод):_ HN03 Н2 Н2<(3 o-n°2 ON0 O=N0H5. Из бензола восстановлением до циклогексана и далее по
схеме 4 (бензольный метод).344
<0>он► <(~)>=NOHНЕФТЬI0-nh2 —► 0=°<0>CH3 ► <(o)-COOH ► ^>COOHРис. 15.11. Взаимосвязь промышленных методов производства
циклогексаноноксима из различного сырьяНа рис. 15.11 представлена взаимосвязь промышленных
методов производства циклогексаноноксима.Из этих методов наиболее распространенным является бен¬
зольный (в схеме на рис, 15.10 показан пунктиром). По данным
1985 года из всего мирового производства капролактама в 2,7
млн. т из бензола было получено 83,5%, из фенола 12,0% и из
толуола 4,5%. По сравнению с фенольным методом бензольный
метод отличается более низкой стоимостью сырья, меньшим
числом стадий и меньшим расходом неорганических реагентов.
Поэтому себестоимость капролактама, полученного бензольным
методом, составляет 85% от себестоимости продукта, произво¬
димого из фенола.Капролактам используют главным образом в качестве моно¬
мера для производства поликапролактама, перерабатываемого
в капроновое волокно и полиамидные пластические массы.
Незначительное количество капролактама применяется в ка¬
честве полупродукта для получения незаменимой аминокис¬
лоты L-лизина (а,е-диаминокапроновая кислота).История. Капроновая кислота СвНцСООН была выделена в 1817—
23 гг. М. Шеврелем в процессе систематического изучения жиров. Син¬
тез и полимеризация капролактама были впервые осуществлены в
конце XIX столетия. Промышленное производство капролактама как
мономера для получения капронового волокна было организовано на
основе исследований В. Карозерса (1930 г.) и Шлака (1940 г.) в конце
40-х годов по фенольному методу. В последующие годы были внедре¬
ны в промышленность методы производства капролактама фотонит-345
розированием (1951 г.), из циклогексанона через капролактон (1966
г.) и другие.В нашей стране на основе работ З.А. Роговина, И.Л. Кнунянца,
А.А. Стрепихеева, Э,В. Хайта и др. в 1949 г. на Дзержинском хими¬
ческом комбинате создается первое в стране производство капролак-
тама из фенола. В 1961—65 гг. подобные производства строятся на хи¬
мических заводах в Чернигове, Рустави, Курске, Кемерово и Барнау¬
ле. В 1963 г. на Лисичанском химическом комбинате организуется про¬
изводство капролактама из бензола через циклогексан. В период
1967—77 гг. создаются новые производства капролактама более эко¬
номичным методом прямого окисления циклогексана до циклогекса¬
нона, который к 1977 г. становится преобладающим.15.4.2. Структурная схема производства капролактама
из бензола через циклогексанНаиболее распространенный в промышленности технологи¬
ческий процесс производства капролактама из бензола вклю¬
чает стадии:— гидрирование бензола,— окисление циклогексана,— дегидрирование циклогексанола,— оксимирование цклогексанона,— изомеризация циклогексаноноксима в капролактам.Структурная схема подобного процесса представлена на рис.15.12.1. Гидрирование бензола до циклогексана протекает над ни¬
келевым катализатором на носителе оксиде хрома (III) при тем¬
пературе 130—220°С и давлении 1 МПа:С6Н6 + ЗН2 ^ C6Hi2 - АНчооРис. 15.12. Структурная схема производства капролактама
из бензола346
2. Окисление циклогексана протекает в жидкой фазе кисло¬
родом воздуха при температуре 140~160°С и давлении 2 МПа в
присутствии катализаторов стеарата магния (Ci7H35COO)2Mg
или нафтената кобальта (СбНпСОО)2Со. При окислении обра¬
зуется смесь циклогексанола и циклогексанона в соотношении
4:1 с суммарным выходом смеси до 75%:C6Hi2 + 0,502 = С6НцОН -АН /С6н12 + 02 = СвНюО + Н20 — АНЧтобы избежать развития реакций глубокого окисления с
распадом цикла степень конверсии циклогексана не должна
превышать ОД дол. единиц.3. Дегидрирование циклогексанола до циклогексанона
протекает при нормальном давлении и температуре 360—
400°С в присутствии цинк-хромового катализатора или при
температуре 260—300°С в присутствии медь-магниевого ка¬
тализатора:С6НцОН ^ С6НюО + Н2 + АНВыход циклогексанона в процессе достигает 95% при степе¬
ни конверсии циклогексанола 0,7—0,8 дол. единиц.415.4.3. Физико-химические основы и технологическая
схема стадий оксимирования и изомеризации
циклогексаноноксима4. Оксимирование циклогексанона до циклогексаноноксима
протекает в избытке водного раствора сульфата гидроксилами-
на и в присутствии аммиака или щелочи при температуре отО С до ЮО С:2 0=° + 2мнгОН • H2S04 + 2NH3** 2 <(^>=NOH + (NH4)2S04 + 2Н20Реакция оксимирования обратима. Равновесие ее сдвигает¬
ся в сторону образования оксима при понижении кислотности
среды, то есть при связывании выделяющейся кислоты щелоч¬
ным реагентом. Реакция протекает в гетерогенной системе, в
диффузионной области и, поэтому, ускоряется путем интенсив¬
ного перемешивания реагентов. Чтобы избежать побочных ре¬
акций конденсации циклогексанона оксимирование проводит¬
ся в две стадии. Сначала процесс ведется в избытке циклогек¬
санона, а затем, в избытке гидроксил амина. Выход циклогек¬
саноноксима в этих условиях приближается к 100%.347
5. Изомеризация циклогексаноноксима в капролактам (Бек-
мановская перегруппировка) протекает в присутствии концен¬
трированной (98% -ной) серной кислоты или 20% -ного олеума
при температуре 125С: С = 00=№Н ^ (СН2)51—-NHРеакция перегруппировки протекает по ионному механиз¬
му через образование катиона с зарядом на атоме азота и может
быть представлена так:Н+ + + +>C=N-OH ^ >C=N-OH ^ >C=N +*- >C=N- ^ -ON-.. -Н20 -Н+ |Н 0S03HПо окончании изомеризации серная кислота нейтрализует-
ся при охлаждении водным раствором аммиака или гидрокси¬
да натрия. При этом образовавшийся сульфат енольной формы
капролактама превращается в капролактам:Технологическая схема стадий оксиминирования и изомери¬
зации циклогексаноноксима в производстве капролактама из
бензола представлена на рис. 15.1.3.В реактор первой ступени оксимирования 1 подаются цик-
логексанон и раствор сульфата гидроксиламина в водном суль¬
фате аммония после второй стадии оксиминирования из сепа¬
ратора 2. Продукты реакции разделяются в сепараторе 3 на вод¬
ный слой — раствор сульфата аммония и органический слой —
раствор оксима в циклогексаноне. Органический слой поступает
в реактор оксиминирования второй ступени 4, в который так¬
же подается свежий раствор сульфата гидроксиламина и амми¬
ачная вода. Реакционная смесь, почти не содержащая цикло-
гексанона, направляется в сепаратор 2, в*котором разделяется
на водно-сульфатный слой, содержащий не вступивший в ре¬
акцию гидроксил амин, и сырой циклогексаноноксим. Водный
слой подается в реактор 1, а циклогексаноноксим поступает на
стадию перегруппировки в реактор 5. Для съема реакционного
тепла смесь в нем циркулирует через выносной холодильник 6,
а олеум вводится в циркулирующую холодную смесь. Продук-348
Рис 15.13. Технологическая схема стадий оксимированияи изомеризации (перегруппировки) в производстве
капролактама из бензола:I — реактор оксимирования I ступени, 2, 3, 8 — сепараторы, 4 — реактор
оксимирования II ступени, 5 — реактор перегруппировки, 6 — выносной
холодильник, 7 — нейтрализатор, 9— экстрактор, 10 — реэкстрактор,II — блок очистки капролактаматы перегруппировки из реактора 5 поступают в нейтрализатор7, куда подается аммиачная вода. Нейтрализованная масса из
нейтрализатора поступает в сепаратор S, в котором водный ра¬
створ сульфата аммония отделяется от «лактамного масла», со¬
держащего 60—65% мае. капролактама, 30—35% мае. воды и
примеси сульфата аммония и побочных продуктов. Для полу¬
чения капролактама высокой степени чистоты лактамное мас¬
ло подвергается очистке в две стадии. На первой стадии капро-
лактам экстрагируется из масла в экстракторе 9 трихлорэтиле-
ном. Полученный экстракт поступает в реэкстрактор 10 у в ко¬
тором капролактам растворяется в воде и отделяется от трих-
лорэтиленового слоя, содержащего примеси. Водный раствор
капролактама из верхней части реэкстрактора направляется на
вторую стадию очистки в блок очистки 11, где он очищается с
помощью ионообменных смол и гидрированием на катализато¬
ре. Очищенный раствор капролактама упаривается и ректифи¬
цируется в вакууме.349
Выход капролактама по циклогексанону составляет 90—
95%. Мощность современных установок по производству кап¬
ролактама колеблется от 90 до 130 тыс. тонн в год. Дальнейшее
использование капролактама зависит от организации производ¬
ства поликапролактама. Если цех полимеризации расположен
на территории предприятия, капролактам транспортируется в
него в виде расплава по обогреваемому трубопроводу. В ином
случае капролактам подвергается кристаллизации и выпуска¬
ется в виде твердого продукта.Контрольные вопросы1. Какие органические соединения называются мономерами?2. Перечислите основные промышленные методы производства бу¬
тадиена-1,3 и изопрена из различного вида сырья.3. В каких условиях и на каких катализаторах протекают первая и
вторая стадии дегидрирования бутана в двухстадийном процессе
производства бутадиена-1,3?4. В чем преимущества одностадийного метода производства бутади¬
ена-1,3 дегидрированием бутана?5. Перечислите особенности производства изопрена методом конден¬
сации изобутилена с формальдегидом и объясните причину высо¬
кой чистоты продукта.6. Из каких последовательных стадий состоит производство стирола
из бензола?7. Каким образом при алкилировании бензола этиленом подавляет¬
ся реакция образования диэтилбензола?8. На каких катализаторах происходит процесс дегидрирования этил-
бензола до стирола?9. Укажите возможные способы производства капролактама из раз¬
личного сырья.10. Приведите структурную схему производства капролактама, исхо¬
дя из бензола.11. В каких условиях протекает реакция оксимирования циклогекса-
нона и изомеризации образовавшегося циклогексаноноксима?
Глава XVIПРОИЗВОДСТВО МОНОМЕРОВ.
ПОЛИКОНДЕНСАЦИОННЫЕ МОНОМЕРЫ16.1. Производство фенола16.1.1. Технологические свойства и применение фенолаФенол (оксибензол, карболовая кислота) СбЩОН — это бес¬
цветное кристаллическое вещество со специфическим «дегтяр¬
ным» запахом с температурой плавления 40,9 *С, температу¬
рой кипения 181,8°С и плотностью 1,032 т/м3. Растворим в воде,
образуя с ней азеотропную смесь с температурой кипения 99,6°С.
Хорошо растворим в этаноле, диэтиловом эфире, бензоле, аце¬
тоне, хлороформе. Обладает слабо кислыми свойствами
(К=1,3’10-10) и растворяется в водных растворах щелочей с об¬
разованием соответствующих фенолятов. Легко окисляется
кислородом воздуха, образуя продукты окисления, окрашива¬
ющие его в розовый, а затем в бурый цвет. В виде паров, пыли и
растворов токсичен. При попадании на кожу фенол вызывает
ожоги, в парах раздражает слизистые оболочки глаз и дыхатель¬
ных путей.Фенол содержится в сточных водах многих производств
(пластических масс, коксохимического, анилинокрасочного и
ДР-)- ПДК фенола в сточных промышленных водах составляет0,001 мг/л.Фенол относится к числу многотоннажных продуктов основ¬
ного органического синтеза. Мировое производство его состав¬
ляет около 5 млн. т. Около половины производимого фенола
используется при получении фенолоформальдегидных полиме¬
ров. Далее, в убывающем порядке, фенол потребляется в про¬
изводствах дифенилолпропана, капролактама, алкилфенолов,
адипиновой кислоты и различных пластификаторов. Фенол
используется также для получения хлор- и нитрозамещенных
фенолов и салициловой кислоты. На основе этих полупродук¬
тов производятся разнообразные красители, пестициды, фар¬
мацевтические препараты (салол, аспирин и др.), присадки к
моторным топливам, маслам и пластмассам (алкилфенолы),
поверхностноактивные вещества. В водных растворах фенол
используется в качестве антисептического средства. На рис.16.1 представлены некоторые направления использования фе¬
нола.351
ЭпоксидныеАнтиоксиданты1 ГербицидыполимерыДефи н илол пропанПластифи¬каторыАлкил-фенолыХимическиеДдипи-волокнаVноваяJкислотаПрисадки]Неионогенные ПАВФЕНОЛИнсектицидыЦиклогек--►Капро¬Химическиесаноллактамволокнафенолоформаль-СалициловаяАнтисептикдегидныекислотаполимеры-КрасителиФунгицидыРис. 16.1. Использование фенола в народном хозяйстве16.1.2. Промышленные методы производства фенолаФенол содержится в фенольной фракции каменноугольной
смолы, а также может быть получен различными синтетичес¬
кими методами, которые различаются природой используемо¬
го сырья, химизмом и экономичностью процесса. Они могут
быть подразделены на:— сульфонатный, состоящий в щелочном плавлении бензол-
сульфокислоты (1);— хлорные, заключающиеся в щелочном или воднопаровом
гидролизе хлорбензола (2);— окислительные, основанные на окислении до фенола бен¬
зола, толуола и циклогексана (3, 4, 5, 6, 7).Из синтетических методов производства фенола промышлен¬
ное значение имеют следующие:— каталитическое окисление толуола (4):2С6Н5-СН3 + 302 — 2С6Н5-СООН + 2Н20
2С6Н5-СООН + 0,502 — С6Н5-СООС6Н4-СООН —— с6н5-он + с6н5-соон + со2— прямое окисление бензола в среде уксусной кислоты (7):С6Н6 + 0,502 - С6Н5ОН— окислительное хлорирование бензола (метод Ф. Рашига) (3):352
С6Нб + 0,502 + НС1 — С6Н5С1 + н2оС6Н5С1 + Н20 -> С6Н5ОН + НС1— кумолъный метод совместного производства фенола и
ацетона через изопропилбензол (6),— из бензола через циклогексан (5).На рис. 16.2 показана взаимосвязь различных методов про¬
изводства фенола, а в табл. 16.1 под теми же номерами приве¬
дены их технико-экономические показатели (в % относитель¬
но сульфонатного метода).КУСректификацияФенольная фракцияС6Н6Cl2 NaOH НС1
► CgHgCl ► CeHsONa ——H2S04 Na2S03 NaOH
► C6H5S03H—►C6H5S03Na 02; HC1 H20
► C6H5C1 C3H6 02
► C6H5=CH(CH3)2 02C6H5-CH3цикло-СбНх202C6H5COOH0202цикло-СбНцОНC6H5OH67Рис. 16.2. Взаимосвязь методов производства фенолаТаблица 16.1. ТЭП производства фенолаПоказательМетод123456Капитальные затраты10083240202208202Стоимость сырья10010558697245Себестоимость100967073765612 Химическая технология. Том 2353
Из таблицы следует, что наиболее экономичным методом
производства фенола является кумольный, позволяющий по¬
лучать одновременно с фенолом другой продукт — ацетон. Про¬
изводство последнего составляет около 3 млн. т в год. Поэтому
на долю кумольного метода приходится в РФ 85%, в США 98%,
в ФРГ 95% и в Японии 100% всего производства фенола.История. Фенол впервые был найден в КУС в 1834 г. Ф. Рунге, а
затем обнаружен в конденсате светильного газа. Элементный состав
фенола был установлен в 1842 г. О.Лораном. В 1843 г. Ш.Жерар полу¬
чил фенол перегонкой салициловой кислоты и ввел в употребление
название «фенол». В 1849 году С. Хант открыл реакцию превращения
анилина в фенол, а в 1889 г. X. Фрид ель установил возможность пря¬
мого окисления бензола до фенола.Первым промышленным методом получения фенола явилось вы¬
деление его из КУС. Однако вследствие низкого выхода продукта (0,05
кг на 1 т угля), он скоро потерял значение и на смену ему пришли син¬
тетические методы производства из ароматического сырья.В нашей стране синтетический фенол до 40-х годов производился
исключительно из бензола щелочным плавлением бензолсульфокис-
лоты, сначала в котлах периодического действия. В 1928—41 гг. осва¬
ивается и внедряется в производство непрерывный сульфонатный про¬
цесс на Рубежанском, Бобриковском, Дорогомиловском и Березняков-
ском химических заводах, а в 1938—42 гг. на Сталиногорском заводе
организуется производство фенола из бензола через хлорбензол.Решающее значение для развития фенольного производства имело
открытие кумольного метода совместного производства фенола и аце¬
тона через гидропероксид изопропилбензола (кумола), теоретические
основы и технология которого были разработаны в нашей стране в
1942—49 гг. П.Г. Сергеевым, Б.Д. Кружаловым, Р.Ю. Удрисом и
М.С. Немцовым. В 1949 г. по этому методу впервые в мире начинается
промышленное производство фенола в г. Дзержинске, а в 1978 г. вво¬
дится в строй завод в г. Уфе. Аналогичное производство было внедре¬
но в Канаде и Франции в 1953 г., в США и ФРГ в 1954 г.16.1.3. Производство фенола кумольным методомТехнологически процесс производства фенола и ацетона по
кумольному методу состоит из двух основных стадий:— алкилирование бензола до изопропилбензола и— окисление изопропилбензола до гидропероксида и его раз¬
ложение.Принципиальная схема этого процесса представлена на рис.16.3.Алкилирование бензола. Изопропилбензол (кумол) получа¬
ется алкилированием бензола пропиленом в присутствии ката¬
лизаторов на основе хлорида алюминия:354
Пропиленовая
фракцияБензолВоздухОтработаннаяфракцияtАл копированиеI[ РазделениеIСмолаОкислениеОтходящиегазыВыделениегидропероксидаРазложениегидропероксида Щ Соли
Фенольная .
водаНейтрализацияГ~Разделение“7 ^■ h2so4- NaOH
СмолаАцетонФенолРис. 16.3. Принципиальная схема производства фенола
по кумольному методуС6Н6+СН2=СН-СН3 - С6Н5-СН(СН3)2 - аЯ (а)АН = 97,8 кДжВ качестве катализатора используется растворимый в угле-
водородах жидкий комплекс Густавсона (15.3.2), получаемый
нагреванием хлорида алюминия с бензолом и изопропилбензо-
лом с добавлением хлорида водорода, или из отходов металли¬
ческого алюминия (ИПБ — молекула изопропилбензола):2 А1 + 6 ИПБ 4- 7НС1 = (ИПБ)б‘А12С1б'НС1 (б)Физико-химические основы и параметры процесса алкили¬
рования бензола пропиленом те же, что и для процесса алкили¬
рования этиленом (15.3.2). Строение образующейся по реакции
(а) алкильной группы подчиняется правилу стабильности кар-
бкатионов: третичный > вторичный > первичный. Поэтому при
алкилировании пропиленом образуется изопропилбензол:н+ + с6н6СНз-СН=СН2 СНз-СН-СНз С6Н5-СН(СН3)2 (в)Технологическая схема производства изопропилбензола ал-
килированием пропиленом представлена на рис. 16.4.В алкилатор 1 подается бензол, осушенный азеотропной рек¬
тификацией, пропилен и каталитический комплекс, приготов¬
ленный в аппарате 2. Продукты алкилирования из алкилатора
поступают в сепаратор 3, где от них отделяется каталитичес¬
кий комплекс, возвращаемый в алкилатор. Углеводородный
слой, состоящий из изопропилбензола, ди- и полиизопропил-12"355
Рис. 16.4. Технологическая схема производства изопропилбензола:1 — алкилатор, 2 — аппарат для приготовления катализатора, 3 — сепара¬
тор, 4 — холодильник, 5,6 — промывные колонны, 7 — подогреватель, 8,9 — ректификационные колонны, 10,13 —холодильники-конденсаторы, 11— абсорбер, 12 — водяной скруббербензолов и непрореагировавшего бензола, охлаждается в водя¬
ном холодильнике 4 и подается на промывку от хлорида алю¬
миния в колонну 5, орошаемую водой, и затем, в колонну б, оро¬
шаемую водным раствором гидроксида натрия. Выйдя из ко¬
лонны 6 нейтрализованная смесь углеводородов (алкилат) по¬
догревается в подогревателе 7 и направляется в систему ректи¬
фикации, состоящую из двух колонн. В колонне 8 отгоняется
избыток бензола, который возвращается на алкилирование, в
колонне 9 — изопропилбензол. Часть кубовой жидкости, со¬
держащей полиизопропил бензолы, направляется из колонны
9 на приготовление каталитического комплекса в аппарат 2.
Газы, выходящие из алкилатора 1, проходят конденсатор 10, в
котором конденсируется бензол, возвращаемый в алкилатор 1,
и поступают в абсорбер 11, орошаемый полиизопропилбензо-
лом, который абсорбирует из них остатки бензола. Затем газы
промываются в скруббере 12 от хлорида водорода и выбрасыва¬
ются в атмосферу.Алкилатор — основной аппарат технологической схемы
представляет собой колонну барботажного типа, заполненную
жидкой реакционной смесью, состоящей из бензола, изопропил¬
бензола и каталитического комплекса. Через смесь барботиру-
ет пропилен.356
Мощность подобных установок алкилирования в производ¬
стве изопропилбензола достигает 360 тыс. т продукта в год.В табл. 16.2 приведены ТЭП производства изопропилбензо¬
ла из бензола.Таблица 16.2. ТЭП производства изопропиблензола
из бензолаПоказательВеличинаПоказательВеличинаМольное отношение Б:П
Температура, °С
Давление, Па
Концентрация
Конверсия СзНз, д. ед.Расход катализатора, кг/т ИПБ2,5:1
70—90
(1—5)*105
35—80
0,99
20Съем ИПБ с 1 м3, кг
Состав алкилата, %
бензол
этилбензол
изопропилбензол
полиизопропилбензолы150—250601—228—337—9Окисление изопропилбензола и разложение гидроперокси¬
да. Получение фенола из изопропилбензола включает операции
окисления изопропилбензола до его гидропероксида, выделе¬
ния гидропероксида из продуктов окисления, разложения гид¬
ропероксида, нейтрализации продуктов разложения и выделе¬
ния из них фенола и ацетона.1. Окисление изопропилбензола (ИПБ) молекулярным кис¬
лородом представляет сложную радикально-цепную каталити¬
ческую реакцию, протекающую в жидкой фазе по уравнению:С6Н5-СН(СНз)2 + 02 = С6Н5-С (СН3)2 - АН (а)ООНКонечным продуктом окисления в этом случае является
гидропероксид изопропилбензола (ГП). Реакция окисления
протекает через образование радикалов со свободной валент¬
ностью на атоме углерода Х> или кислорода > (ХХ> и иници¬
ируется за счет автоокисления ИПБ при введении инициато¬
ров:С6Н5-СН(СН3)2 + 02 - СбН5-С(СНз)2 + НОО-, (б)или при взаимодействии кислорода с катализатором:Со+2 + 02 — Со+3-00- (в)с последующим развитием цепи до образования достаточно ус¬
тойчивого в условиях реакции молекулярного продукта — ГП:С6Н5-С(СН3)2 + 02 - С6н5-С (СН3)2 (г)ОО-357
С6Н5-С(СН3)2 + С6Н5-СН(СНз)2-
00-—* СбНб-С(СНз)2 + СбН5-С(СН3)2 и т. д.IООНКатализаторами реакции окисления ИПБ являются резина¬
ты и нефтенаты кобальта (II) и марганца (II). При этом, одно¬
временно с основной реакцией (а) протекают побочные реакции
образования диметилфенилкарбинола, ацетофенона и муравь¬
иной кислоты, как это показано на схеме:Вследствие этого селективность окисления ИПБ до ГП не пре¬
вышает 95%. С увеличением температуры и степени конверсии
в реакционной массе накапливается ГП и усиливаются побочные
реакции его разложения. Во избежание этого степень конверсии
ИПБ не должна превышать 0,3 дол. единиц. Для нейтрализации
муравьиной кислоты, образующейся в качестве побочного про¬
дукта, окисление проводят в водно-щелочной эмульсии (раствор
карбоната натрия), что позволяет интенсифицировать основную
реакцию образования ГП (а). Поэтому оптимальными условия¬
ми окисления ИПБ до ГП являются: температура 120—130°С,
давление 0,5—1 МПа, pH среды 8,5—10,5. В этих условиях со¬
держание ГП в реакционной смеси составляет 25% масс. Про¬
цесс окисления ИПБ ингибируется такими веществами как фе¬
нолы, алкены и сернистые соединения. Поэтому исходный ИПБ
подвергается тщательной очистке от примесей.2. Разложение гидропероксида изопропилбензола представ¬
ляет экзотермическую реакцию кислотного расщепления, опи¬
сываемую общим уравнением:(СН3)2 = С6Н5ОН + СН3-С-СН3 - АН АН = 316 кДж (д)С6Н5-СН(СН3)2СбН5-С(СНз)2ООН► С6Н5-С(СН3)2
онС6Н5-С(СНз)00- ► с6н5-с-сн3 + сн3о
о
СН30“- сн3он — нсно — нсоон — со2ООН358
Реакция протекает по ионному механизму через следующие
стадии:1) Образование и дегидратация оксониевого иона:С6Н5-С (СН3)2+Н+ С6Н5-С (СН3)2 = С6Н5-С (СНз)2+Н20 (е)ООН ООН2 о+2) Перегруппировка образовавшегося иона с миграцией фе-
нильной группы к атому кислорода:СбН5-С(СНз)2 - С6Н50-С(СН3)2
0+3) Превращение карбкатиона с образованием фенола и аце¬
тона:С6Н50-С(СНз)2 + Н20-С6Н50-С(СНз)2-"С6Н50-С(СНз)2 -+он2 н ОН— СбН5ОН + СНз-С-СНз + н+ (ж)ОРеакция разложения ГП катализируется кислотами и про¬
текает с очень высокой скоростью. На практике для разложе¬
ния используют 0,1%-ный раствор серной кислоты в ацетоне.
Выход целевых продуктов зависит от температуры и достигает
максимума при 50—60°С.Одновременно с основной реакцией (д) протекают побочные
реакции с участием диметилфенилкарбинола и ацетофенона,
присутствующих в техническом ГП. Так например, диметил-
фенилкарбинол дегидратируется до а-метилстирола:С6Н5-С(СНз)2 > С6Н5-С=СН2 + Н20 (з)ОН СН3который выделяется в виде товарного продукта или присоеди¬
няется к циркуляционному возвратному ИПБ.Вследствие экзотермичности и высокой скорости реакции,
при разложении ГП выделяется большое количество тепла. Для
его отвода процесс разложения ведут в среде разбавителя (аце¬
тон или продукты реакции), используя проточно-циркуляци¬
онную схему, и применяют интенсивно работающие выносные
холодильники.На рис. 16.5 представлена технологическая схема про¬
цесса получения и разложения гидропероксида изопропил¬
бензола.359
Рис. 16.5. Технологическая схема окисления ИПБ и разложения ГП:1 — сборник, 2 — теплообменник, 3 — реактор окисления, 4 — холодиль-
ник-конденсатор, 5 — сепаратор-промыватель, 6 — дроссель, 7 — ваку-
ум-ректификационная колонна, 8 — разлагатель, 9 — выносной холодиль¬
ник, 10, 11, 12 — ректификационные колонныСмесь свежего и циркуляционного (оборотного) ИПБ с добав¬
кой ГП для инициирования процесса окисления из сборника 1
поступает в теплообменник 2, где нагревается продуктами окис¬
ления (оксидатом), выходящим из реактора окисления и пода¬
ется в верхнюю часть реактора окисления 3. В нижнюю часть
реактора под давлением 0,4 МПа поступает противотоком очи¬
щенный воздух, барботирующий через жидкость. Избыток воз¬
духа с увлеченными им парами ИПБ и побочных продуктов
(формальдегид и муравьиная кислота) проходит холодильник-
конденсатор 4, где из него выделяются жидкие продукты, и
выбрасывается в атмосферу. Конденсат поступает в сепаратор-
промыватель 5, орошаемый водным раствором щелочи, для уда¬
ления муравьиной кислоты, и разделяется в нем на углеводо¬
родный и водный слои. Углеводородный слой, содержащий
ИПБ, подается в сборник 1 и возвращается в цикл. Выходящий
из нижней части реактора 3 оксидат, содержащий до 30% ГП,
проходит теплообменник 2, дросселируется до давления 40 кПа
в дросселе 6 и подается на ректификацию в вакуум-ректифика-
ционную колонну 7 для отгонки ИПБ и концентрирования ГП.360
Отогнанный ИПБ из колонны поступает в сепаратор 5 и из него
в сборник 1. Сконцентрированный до 85—90% ГП подается на
операцию разложения в разлагатель 8, куда вводится раствор
серной кислоты в ацетоне. Из разлагателя продукты разложе¬
ния после нейтрализации раствором гидроксида натрия направ¬
ляются в систему ректификации, состоящую из нескольких
колонн. При давлении 105 Па в колонне 10 отгоняется ацетон, а
при пониженном давлении в колонне 11 — а-метилстирол и ос¬
татки ИПБ, а в колонне 12 — фенол.Основными аппаратами технологической схемы являются
реактор окисления ИПБ и разлагатель ГП. Реактор окисления
представляет колонну из нержавеющей стали, снабженную
встроенными холодильниками для отвода реакционного тепла
и в нижней части оборудованную системой подачи и барботиро-
вания воздуха. Разлагатель выполнен в виде пустотелой колон¬
ны, снабженной выносным холодильником-конденсатором.
Реакционное тепло отводится за счет испарения ацетона, кото¬
рый водитс,я в колонну с серной кислотой. После конденсации
в холодильнике ацетон возвращается в разлагатель.Состав продуктов разложения ГП (% масс.)Фенол 14 Ацетофенон 1Ацетон 8 а-метилстирол 1Изопропил бензол 7616.1.4. Совместное производство фенола, ацетона
и пропиленоксидаОдним из многотоннажных продуктов промышленного орга¬
нического синтеза является пропиленоксид (окись пропилена)
СН3-СН-СН2, мировое производство которого составляет около"О"3 млн. т в год. Пропиленоксид используется в производстве
пропиленгликолей, полипропиленоксида, пенополиуретанов,
ПАВ. Наиболее эффективным методом производства пропиле¬
ноксида является эпоксидирование пропилена гидропероксид-
ными соединениями.В связи с этим разработка кумольного метода получения фе¬
нола позволила создать кооперированное производство фенола,
ацетона и пропиленоксида, используя в качестве гидроперок-
сидного соединения гидропероксид изопропилбензола. В этом
производстве часть гидропероксида направляется на разложе¬
ние до фенола и ацетона, а остальное количество его поступает
на эпоксидирование пропилена:361
I ► на производство фенола02 С3Н6С6Н5-СН(СН3)2 - С6Н5-С(СН3)2 -* С6Н5-С(СНз)2 + СН3-СН-СН2ООН ОН ОН2В этом производстве побочные продукты эпоксидирования-
(диметилфенилкарбинол и ацетофенон) полностью перераба¬
тываются в исходный изопропил бензол, что существенно сни¬
жает стоимость производимой продукции.16.2. Производство диэтиленгликольтерефталата16.2.1. Свойства и промышленные методы производстваДиэтиленгликольтерефталат (дигликолевый эфир терефта-
левой кислоты, ди(р-оксиэтил)терефталат) ДЭГТ представля¬
ет бесцветное кристаллическое вещество с температуройплавления 110°С. Хорошо растворим в горя-
СОО(СН2)2ОН чей воде, что используется для его очисткиметодом кристаллизации, растворим в раз¬
личных органических растворителях. В про-
СОО(СН ) ОН мышленности ДЭГТ получается тремя мето-
22 дами.1. Переэтерификацией диметилтерефталата этиленглико-
лем:СООСН3 СОО(СН2)2ОН+ 2НО-(СН2)2-ОН 52 lOj + 2CHgOHCOOCHg СОО(СН2)2ОН2. Прямой этерификацией терефталевой кислоты этиленг
ликолем:СОО(СН2)2ОН+ 2НО -(СН2)2-ОН ^ lOj 4- 2Н20СООН СОО(СН2)2ОН362
3. Взаимодействием терефталевой кислоты с окисью этиле¬
на:СООН СОО(СН2)2ОН(О) + 2СН2-СН2 ^ (о)СООН СОО(СН2)2ОНК ДЭГТ, используемому в качестве мономера для производ¬
ства полиэтилентерефталата, предъявляются высокие требова¬
ния по чистоте, которая зависит от чистоты исходного сырья.
Очистка терефталевой кислоты многократной перекристалли¬
зацией ее аммонийных солей или экстракцией под давлением
является трудоемкой и дорогостоящей операцией. Однако ее
диметиловый эфир (диметилтерефталат, ДМТ) может быть лег¬
ко очищен от примесей перегонкой.Вследствие этого метод производства ДЭГТ переэтерифика-
цией ДМТ является в настоящее время преобладающим, а себе¬
стоимость получаемого продукта ниже, чем в других методах.
Основными недостатками метода переэтерификации являются
взрывоопасность и токсичность производства из-за исполь¬
зования в технологическом цикле метанола, а также необходи¬
мость предварительной стадии получения ДМТ.Метод прямой этерификации терефталевой кислоты лишен
этих недостатков. Процесс образования ДЭГТ в нем протекает
с большей скоростью и с меньшим удельным расходом эти-
ленгликоля. Однако получаемый продукт содержит примеси
эфиров с ди- и триэтиленгликолевыми группами состава
-СОО[(СН2)20]пН, которые ухудшают качество получаемых из
ДЭГТ полимеров.В методе производства ДЭГТ из терефталевой кислоты и оки¬
си этилена используется дешевое сырье и не образуется побоч¬
ных продуктов. Поэтому, он является наиболее экономичным
и перспективным, хотя используется в промышленности еще в
ограниченных масштабах.Ниже рассматривается наиболее распространенный метод
производства ДЭГТ переэтерификацией ДМТ этиленгликолем.
Он включает две основных стадии, которые, по существу, яв¬
ляются самостоятельными производствами:— синтез ДМТ и его— переэтерификацию этиленгликолем.363
16.2.2. Производство диметилтерефталатаДиметилтерефталат представляет бесцветное кристалличес¬
кое вещество с температурой плавления 141С и плотностью
1,63 т/м3. Возгоняется при нагревании выше 300°С. Ограни¬
ченно растворим в воде (0,33%), этиленгликоле, ме-
CooCHg таноле, бензоле. Температура воспламенения ДМТ
составляет 578°С. Пылевоздушная смесь ДМТ
[OJ взрывоопасна. Температура вспышки ее равна 146°С.гппгтт Раздражает слизистые оболочки глаз, дыхательных
^ путей и кожу. ПДК равна 0,1 мг/м3.В промышленности ДМТ получается окислением воздухом
смеси n-ксилола и метилового эфира n-толуиловой кислоты до
гс-толуиловой кислоты с последующей этерификацией ее до
ДМТ. Процесс этот описывается следующими уравнениями:СНз-<0>-СНз + 1,5С>2 - СНз-<о)-СООН + Н20 (а)СН3-<0>-СООН + СН3ОН «=* СНз-<0>-СООСНз + Н20 (б)
СНз-<0>-СООСНз + 02 ^ НООС-<0>-СООСНз + Н20 (в)НООС-<о)-СООСНз+СН3ОН^СНзООС-<о)>-СООСНз + Н20 (г)В технологической схеме процессы окисления п-ксилола (ре¬
акция а) и промежуточного продукта окисления метил-п-толу-
илата (реакция в) совмещаются, как и реакции этерификадии
(б и г) в одних аппаратах. Это позволяет сократить число ста¬
дий технологического процесса и исключить промежуточные
операции разделения продуктов.Окисление смеси п-ксилола и метил-л-толуилата, взятых в
отношении 1:2, проводится при температуре 140—180 С и дав¬
лении 0,6—1 МПа воздухом, барботирующим через реакци¬
онную массу. Катализаторами окисления служат бензоат или
нафтенат кобальта. Полученный оксидат содержит 25% /г-то-
луиловой кислоты, 30% метил-п-толуилата, 15% терефтале-
вой кислоты и до 25% монометилового эфира фталевой кис¬
лоты.Этерификация оксидата проводится парами метанола при
температуре 250°С и давлении 2,5 МПа. Пары метанола, прохо¬
дя через реакционную массу, уносят образующуюся воду, спо¬
собствуя смещению равновесия реакций (б) и (г) в сторону обра¬
зования соответствующих эфиров терефталевой кислоты.На рис. 16.6 представлена технологическая схема производ¬
ства диметилтерефталата окислением n-ксилола, включая опе¬364
рации ректификации полученного продукта и его очистки кри¬
сталлизацией из метанола.Выход ДМТ этим методом составляет 85—90%.В колонну-окислитель 1, через которую.снизу барботирует
воздух, подается смесь свежего и возвратного (циркуляционного)
гс-ксилола. Увлеченные током воздуха пары метанола конден¬
сируются в холодильнике-конденсаторе 2 и возвращаются в
колонну. Реакционная масса из колонны собирается в сборни¬
ке 3, из которого перекачивается в эфиризатор 4. В нижнюю
часть эфиризатора поступает парообразный метанол из перегре¬
вателя 5. Выделяющиеся пары метанола и воды конденсируют¬
ся в холодильнике-конденсаторе 6. Из эфиризатора 4 продук¬
ты этерификации дросселируются в дросселе 7 до давления
1,3е 104 Па и поступают в испаритель S, где от смеси эфиров от¬
деляются смолы. Освобожденная от смол смесь подается на рек¬
тификацию. В колонне 9 от нее отделяется метил-л-толуилат,
который конденсируется в холодильнике-конденсаторе 10, и
через сборник 11 возвращается на окисление в окислительную
колонну 1. Эфиры дикарбоновых кислот из куба колонны 9 по-Рис. 16.6. Технологическая схема производства ДМТ из л-ксилола:1 — колонна-окислитель, 2, 6, 10, 13 — холодильники-конденсаторы, 3, 11—
сборники продукта, 4 — эфиризатор, 5 — перегреватель метанола, 7 —
дроссель, в — испаритель, 9, 12 — вакуум-ректификационные колонны,14 — автоклав, 15 — центрифуга365
даются в колонну 12, в которой ДМТ отделяется от менее лету¬
чих примесей. Отогнанный сырой ДМТ конденсируется в хо¬
лодильнике-конденсаторе 13 и поступает на кристаллизацию в
автоклав 14. Выпавший твердый продукт отжимается в цент¬
рифуге 15. Фильтрат метанола направляется через перегрева¬
тель 5 на этерификацию в эфиризатор 4.16.2.3. Производство ДЭГТ переэтерификацией ДМТПереэтерификация ДМТ этиленгликолем представляет об¬
ратимую реакцию алкоголиза, описываемую уравнением:СООСН* СОО(СН2)2ОНРеакция катализируется ацетатами цинка, марганца, ко¬
бальта и свинца; энергия активации ее составляет 40—45 кДж/
/моль.В продуктах реакции переэтерификации не должны содер¬
жаться метил-гликолевый и диметиловый эфиры терефгалевой
кислоты, так как они отрицательно влияют на качество полу¬
чаемого из ДЭГТ полимера. Поэтому, необходимо обеспечить
полноту протекания реакции (д). Для этого процесс проводится
в избытке этиленгликоля с отгонкой образующегося метанола.Эффективность действия катализатора реакции переэтери¬
фикации характеризуется объемом метанола V, отгоняемого в
единицу времени (рис. 16.7). Из рисунка следует, что из двух
катализаторов (ацетата цинка и ацетата кобальта) более эффек¬
тивным является первый. При этом, на обоих катализаторахСООСНдСОО(СН2)2ОН2равновесие реакции переэтери¬
фикации устанавливается за 2—3
часа. На практике, процесс пере¬
этерификации проводится в тече¬
ние 4—5 часов.Температурный режим процес¬
са определяется в значительной2степени температурой кипения
этиленгликоля. Начальная темпе¬
ратура переэтерификации должна
быть ниже температуры кипения
этиленгликоля (197,б°С), обычно
она составляет 180°С. К концу про-Рис. 16.7. Влияние природы
катализатора на скорость
реакции переэтерификации:1 — ацетат цинка, 2 — кобаль¬
та366
цесса для отгонки не вступившего в реакцию избытка этиленг-
ликоля температура повышается до 200°С.Побочными продуктами реакции переэтерификации являют¬
ся низкомолекулярные полиэфиры со степенью полимеризации2—4.Получение и поликонденсация ДЭГТ обычно объединяются
в единое производство. Это позволяет снизить расход тепла на
вторичное расплавление мономера и избежать потерь продукта
при траспортировке. Подобное производство может быть пери¬
одическим или непрерывным.На рис. 16.8 представлена технологическая схема стадии
получения диэтиленгликольтерефталата. ДМТ из бункера 1
через загрузочное устройство 2 поступает в плавильный котел3, снабженный плавильной решеткой и рубашкой, обогревае¬
мыми паром до 160°С. Расплав ДМТ из плавильного котла пере¬
качивается в автоклав переэтерификации 4, обогреваемый ди-
нилом (эвтектическая смесь дифенила и дифенилового эфира),
или перегретым водяным паром. Этиленгликоль и катализатор
смешиваются и подогреваются в аппарате 5 и также подаются в
реактор 4. Расплавленный ДЭГТ из реактора проходит фильтр
6 для отделения механических примесей и, или сразу подается
на стадию поликонденсации, или охлаждается и кристаллизу¬
ется. Отгоняемый в процессе переэтерификации метанол кон-Рис. 16.8. Технологическая схема производства ДЭГТ из ДМТ:1 — бункер, 2 — загрузочное устройство, 3 — плавильный котел, 4 — ре¬
актор, 5 — подогреватель, 6 — фильтр, 7 — конденсатор, 8 — приемник367
денсируется в холодильнике-конденсаторе 7 и собирается в при¬
емнике 8. После ректификации он вновь направляется на про¬
изводство ДМТ.Контрольные вопросы1. Укажите основные направления использования фенола в народном
хозяйстве.2. Какие методы производства фенола имеют промышленное значе¬
ние?3. Почему кумольный метод производства фенола является наиболее
экономичным?4. Какой катализатор используется для алкилирования бензола про¬
пиленом в кумольном методе производства фенола?5. По какому механизму протекает окисление изопропилбензола и
разложение его гидроперекиси?6. Почему в процессе производства диэтиленгликольтерефталата
(ДЭГТ) используется метод переэтерификадии диметилтерефтала-
та этиленгликолем, а не прямая этерификация?7. Из каких стадий складывается производство ДЭГТ ?8. В каких условиях проводится переэтерификация ДМТ этиленгли¬
колем и как обеспечивается полнота протекания этой реакции?
Часть IVПРОИЗВОДСТВО ПОЛИМЕРНЫХ МАТЕРИАЛОВ
Глава XVIIПОЛИМЕРНЫЕ МАТЕРИАЛЫ17.1. Свойства и применение полимерных материаловПолимерными материалами (ПМ) называются одно- или
многокомпонентные системы, основу которых (матрицу) состав¬
ляют высокомолекулярные соединения или полимеры. Состав
ПМ весьма разнообразен и колеблется от почти индивидуаль¬
ных полимеров до весьма сложных систем, включающих
разнообразные компоненты, регулирующие технологические и
эксплуатационные свойства материала. К подобным компонен¬
там относятся различные химически инертные или активные
вещества: растворители, пластификаторы, загустители, краси¬
тели, антипирены, антиоксиданты, термо- и светостабилизато-
ры, антирады, структуро- и порообразователи. Они получили
название наполнителей. Поэтому большинство ПМ можно рас¬
сматривать как наполненные полимеры.Для ПМ характерны широкие возможности регулирования
состава, структуры и свойств, что не всегда может быть достиг¬
нуто в традиционных материалах (металлы, керамика, древе¬
сина). Поэтому, многовариантность свойств обусловливает весь¬
ма широкое и многообразное использования ПМ — от хими¬
ческих волокон и поропластов до твердого ракетного топлива.Полимерные материалы классифицируют по области их ис¬
пользования и назначения, природе полимерной фазы, физи¬
ческим и химическим превращениям, протекающим в ней при
производстве и обработке.По использованию и назначению ПМ делятся на:— пластические массы (пластики) и композиты;— эластомеры (каучуки и резины);— химические волокна и пленки;— полимерные покрытия, клей и герметики.В соответствии с назначением среди них выделяют ПМ об¬
щего назначения и функциональные ПМ (фрикционные, теп¬
ло- и электроизоляционные, электропроводящие, антикоррози¬
онные и т.п.).По природе полимерной фазы (матрицы) ПМ делятся на:— природные (натуральные);369
— химические (искусственные и синтетические).По характеру превращений, протекающих в полимерной
фазе на стадиях производства и переработки в изделия, ПМ де¬
лятся на:— термопластичные;— термореактивные.ПМ отличаются от традиционных материалов, используемых
человечеством с глубокой древности, комплексом особых
свойств, их сочетанием, эффективностью и высокой экономич¬
ностью методов переработки в изделия, практически неогра¬
ниченной и стратегически неуязвимой сырьевой базой.Особые свойства ПМ1. Малая плотность и, как следствие, высокий условный по¬
казатель прочности («весовая прочность») то есть отношение
временного сопротивления на разрыв к плотности стр/р превы¬
шающий аналогичный показатель лучших сортов стали. В табл.17.1 приведены значения этого показателя для различных кон¬
струкционных материалов.Таблица 17.1. Условный показатель прочностиМатериалр. т/м3ар, МПаСТр/рСталь7,81254161Стеклопластик1,8294—686163—381Древесно-слоистый пластик1,43432452. Стойкость к агрессивным средам, атмосферному и радиа¬
ционному воздействиям.3. Высокие радио- и электротехнические свойства, в том чис¬
ле диэлектрические показатели, мало зависящие от темпера¬
туры и частоты электрического поля.4. Широкий диапазон фрикционных и антифрикционных
свойств.5. Специфические оптические свойства, способность пропус¬
кать лучи света в широком диапазоне волн, в том числе, ульт¬
рафиолетовые (70% для полиметилметакрилата против 1—3%
для силикатного стекла).6. Многообразие физико-механических свойств (от жестких
до упругих резиноподобных материалов) и сочетание в одном
материале противоположных качеств, например, твердости и
гибкости («бронированные» полимеры).К недостаткам ПМ относятся:370
— низкая теплостойкость (исключая фторопласты и крем-
нийорганические полимеры, она не превышает 120 С);— недостаточная твердость (6—60 кг/мм2 по Бринелю);— ползучесть и релаксация напряжения;— большое тепловое расширение;— низкая теплопроводность, затрудняющая отвод тепла (в
500—600 раз ниже' теплопроводности металлов).Методы переработки ПМ в изделияВ отличие от переработки металлов и древесины, все методы
переработки ПМ отличаются простотой, низкой энерго- и тру¬
доемкостью, возможностью получить нужное состояние повер¬
хности изделия без дополнительной обработки и практической
безотходностью производства. Так например, коэффициент ис¬
пользования материала при переработке ПМ достигает 0,95—
0,98 в то время как при литье он составляет 0,6—0,8, а при ме¬
ханической обработке металлов всего 0,2—0,6. Поэтому, про¬
цессы изготовления изделий из ПМ являются наиболее эконо¬
мичными.Сырьевая база ПМПМ производят из простых органических соединений — мо¬
номеров, источником которых являются широко распростра¬
ненные и доступные виды сырья: ископаемые угли, нефть, газ,
воздух и известь. Таким образом производство ПМ и изделий
из них практически ограничено только уровнем развития хи¬
мической и нефтехимической промышленности.Перечисленные выше преимущества ПМ по сравнению с тра¬
диционными материалами обусловили все возрастающие мас¬
штабы и опережающий рост их производства по сравнению с
производством металлов.Так уже в период 70—90-х годов XX столетия производство
ПМ увеличилось на 180%, тогда как производство стали вырос¬
ло всего на 23%. Эта разница в темпах роста производства ПМ
и металлов продолжает увеличиваться, что свидетельствует об
определенной исчерпаемости потребления мировым народным
хозяйством черных металлов и все возрастающей потребности
его в ПМ. В результате, уже к 1975 г. в структуре общего по¬
требления конструкционных материалов черные металлы со¬
ставляли 63,7%, цветные металлы 6,3% и полимерные мате*
риалы около 30%.Вследствие низкой себестоимости, экономии трудовых затрат
при производстве готовой продукции и эффективности исполь¬
зования ее, потребителями ПМ стали практически все отрасли
промышленности и народного хозяйства. К ним относятся:371
1) общее машиностроение (крупногабаритные элементы кон¬
струкций, трубы и детали арматуры, зубчатые колеса, ролики,
подшипники, тормозные накладки, электроизоляционные де¬
тали и др,);2) авиастроение (силовые элементы летательных аппаратов,
теплозащитные покрытия, подвесные топливные баки, эле¬
менты остекления, звуко- и теплопоглощающие панели, амор¬
тизаторы, шины, герметики швов, клеи и лакокрасочные ма¬
териалы);3) автомобилестроение (кузова и кабины, тепло- и звукоизо¬
ляционные и декоративные детали, элементы двигателя и шас¬
си, шланги и трубопроводы, наполнители сидений, детали ос¬
текления и осветительных приборов, шины, маслостойкие де¬
тали системы подачи топлива и тормозной системы, лакокра¬
сочные материалы);4) электротехника и радиоэлектроника (изоляция электри¬
ческих машин, аппаратов и кабельных изделий, в том числе,
электродвигателей и генераторов, трансформаторов, комму¬
никационной аппаратуры, конденсаторы, магнитодиэлектри-
ки, полупроводники, компаунды для заливки микросхем,
клеи);5) железнодорожный транспорт (конструкционные элемен¬
ты вагонов, фрикционные, уплотняющие и амортизационные
детали, наполнители и облицовка сидений, прокладки для сцеп¬
ления рельсов со шпалами, светофоры, изоляторы контактных
сетей);6) судостроение (корпуса судов в целом и отдельные судо¬
вые конструкции, уплотнительные материалы для тепло-, зву¬
ко- и виброизоляции, прокладки, герметики, клеи, мастич¬
ные противоскользящие покрытия, лакокрасочные материа¬
лы);7) строительство (конструктивные элементы зданий, сани¬
тарно-техническое оборудование, стеновые панели и перегород¬
ки, кровельные материалы, покрытия полов, дверные и окон¬
ные переплеты, трубопроводы, тепло- и звукоизоляционные ма¬
териалы);8. сельское и водное хозяйство (пленки и элементы конст¬
рукций для культивационных сооружений, материалы для
мульчирования почвы, водорастворимые пленки для посева и
дражирования семян, структурообразователи почв для защи¬
ты от коррозии и аккумуляции влаги, упаковочный материал,
противофильтрационные экраны в мелиорации, водоснабже¬
нии, трубопроводы, облицовка каналов);372
9) пищевая промышленность (конструкционные материалы
и покрытия пищевых машин, тароупаковочные материалы,
консервные лаки и эмали, иониты для обработки молока);10) медицина (изделия медицинской техники, детали
восстановительной хирургии из биоинертных и биоассимили-
руемых полимеров, функциональные узлы аппаратов искусст¬
венного кровообращения, вживляемые стимуляторы сердечной
деятельности, крове- и плазмозаменители, пролонгаторы дей¬
ствия лекарственных препаратов и полимерные лекарственные
вещества).В табл. 17.2 представлено потребление полимерных матери¬
алов в РФ в процентах от их общего производства в различных
отраслях народного хозяйства (по данным 1980 г.).Таблица 17.2. Потребление полимерных материалов
в Российской Федерации, %Отрасль производстваУдельный вес, %Машиностроение27—29Лесная и бумажная промышленность21—22Легкая и пищевая промышленность14—15Строительство13—14Сельское и водное хозяйство4—5Транспорт2,5—3Потребление ПМ в различных отраслях народного хозяйства
неравноценно. Из таблицы следует, что наиболее эффективно
использование ПМ в машиностроении, где 1 т ПМ заменяет 5—
6 т черных и цветных металлов и 3—3,5 т древесины, а эконо¬
мия трудовых затрат достигает 800 человеко-часов на 1 т ПМ.
Наиболее высок удельный вес ПМ в электротехнике и электро¬
нике, потребляющих около 50% всех ПМ, используемых в ма¬
шиностроении. В электротехнике с использованием ПМ про¬
изводится 80% , а в приборостроении до 95% всей выпускаемой
продукции.Внедрение ПМ не только положительно сказывается на со¬
стоянии уже существующих традиционных отраслей народ¬
ного хозяйства. Оно вызвало также качественный скачок в раз¬
витии техники и технологии, определило технический про¬
гресс в современном машиностроении, ракетной и атомной
промышленности, в самолетостроении, телевидении, радио¬
электронике, восстановительной хирургии и медицине в це¬
лом и др.373
f 7.2. Высокомолекулярные соединения как основа ПМПолимерная фаза, составляющая основу ПМ, представляет
собой разнообразные полимерные композиции в виде индиви¬
дуальных полимеров, смесей и сплавов их, блокполимеров и
привитых полимеров, пористых полимеров. Эксплуатационные
и технологические свойства ПМ существенно зависят от состо¬
яния и специфических свойств составляющих полимерную фазу
высокомолекулярных соединений.17.2.1. Фазовые и физические состояния полимеровФазовые состояния. Полимеры могут существовать в крис¬
таллическом, жидком (аморфном) и жидкокристаллическом
(iаморфнокристаллическом) фазовых состояниях, различаю-
щихся степенью упорядоченности частей макромолекул в
структуре полимера. При этом кристаллическая и аморфная
фазы в полимере находятся в состоянии термодинамического
равновесия:Аморфная фаза ^ Кристаллическая фаза - АНСодержание кристаллической фазы в полимере характери¬
зуется степенью кристалличности полимера акр, то есть ве¬
личиной, показывающей, какая часть полимера (по массе или
объему) входит в состав кристаллической фазы. Для отдельных
полимеров степень кристалличности может достигать 0,75—0,90%. При повышении температуры равновесие фазового пе¬
рехода смещается влево, поэтому степень кристалличности по¬
лимера при нагревании уменьшается.Физические состояния. Для аморфных полимеров различа¬
ют три физических состояния: стеклообразное, высокоэласти¬
ческое и вязкотекучее. Физические состояния полимеров свя¬
заны взаимными обратимыми переходами, которые происходят
в определенных интервалах температур:Г, ТТСтеклообразное^Высокоэластическое^Вязкотекучеесостояние (С) состояние (ВЭ) состояние (ВТ)Подобные температурные интервалы перехода от стеклообраз¬
ного состояния к высокоэластическому и от высокоэластическо¬
го к вязкотекучему получили названия, соответственно, темпе¬
ратуры стеклования (Тс) и температуры текучести (Тг).От физического состояния полимеров существенно зависит
такое их механическое свойство как характер деформации при374
приложении нагрузки. Изменение вида деформации полимера
в зависимости от температуры (то есть от изменения физичес¬
кого состояния) выражается так называемой термоме¬
ханической кривой (рис. 17.1). При температуре Т<ТС полимер
находится в стеклообразном состоянии и ему свойственна уп¬
ругая деформация, отвечающая закону Гука:h!=E-o (17.1)где: а — напряжение,Е — модуль упругости.Упругая деформация полимера
(hi) зависит от температуры. При
температуре Т > Тт полимер нахо¬
дится в вязкотекучем состоянии и
ему свойственна пластическая нео¬
братимая деформация, характерная
для вязкого течения вещества (Лз).В интервале температур Тт> Т > Тс
имеет место обратимая высокоэла¬
стическая деформация, не завися¬
щая от температуры (Й2). Этот вид
деформации свойственен исключи¬
тельно полимерам и связан с явле¬
нием релаксации.17.2.2. Особые свойства полимеровК специфическим свойствам полимеров относят их полидис¬
персность, особые физические свойства и свойства растворов,
способность к волокнообразованию, явления релаксации и гис¬
терезиса.Полидисперсность. В отличие от низкомолекулярных соеди¬
нений и природных полимеров синтетические полимеры не яв¬
ляются индивидуальными веществами, а представляют набор
полимергомологов, то есть макромолекул одинакового строения,
но разной степени полимеризации и, следовательно различной
молекулярной массы. Это свойство получило название полидис¬
персности или полимолекулярности. Поэтому молекулярная
масса полимера есть некая средняя молекулярная масса, опре¬
деляемая средней степенью полимеризации:Л/с р tiq-^п (17.2)где: т — молекулярная масса элементарного звена полиме¬
ра, -hРис. 17.1. Термомеханичес¬
кая кривая полимеров375
Л-Срсредняя степень полимеризации, определяемая как
средняя из степеней полимеризации фракций поли¬
мера:7l\ + 712 П,Л-ср(17.3)При одинаковой средней молекулярной массе соотношение
между макромолекулами различных размеров в полимере (то
есть соотношение фракций) различно. Так как короткие моле¬
кулы ведут себя иначе, чем длинные, полимеры с одинаковойсредней молекулярной массой могут
х существенно различаться свойства¬ми. Поэтому, для оценки свойств
полимера большое значение имеет
распределение макромолекул в нем
по степени полимеризации, то есть
мера полидисперсности полимера.
Для ее оценки строят кривые рас¬
пределения, отражающие зависи¬
мость содержания фракции полиме¬
ра со степенью полимеризации п от
степени X полимеризации. Так на¬
пример, из рис. 17.2 следует сделать
вывод, что полимер 2 более полидис-
персен, чем полимер 1, для которо¬
го основное количество мак¬
ромолекул лежит в узком интерва¬
ле степеней полимеризации от п\ до пФизические свойства. Полимеры нелетучи, имеют достаточ¬
но высокие температуры и размытость интервала фазового пе¬
рехода. Для большинства полимеров характеристикой такого
перехода является так называемая температура размягчения,
при которой в процессе нагревания резко возрастает деформи¬
руемость полимера. Для кристаллических полимеров с высо¬
кой степенью кристалличности температура размягчения со¬
впадает с температурой плавления.Свойства растворов. Полимеры ограниченно растворимы в
некоторых растворителях. При этом, растворение начинается
с процесса набухания, то есть проникновения молекул раство¬
рителя между макромолекулами полимера, что сопровождает¬
ся значительным (в 10—15 раз) увеличением объема поли¬
мерной массы. Характеристикой набухания принято считать
степень набухания ан, которая рассчитывается по формуле:Рис. 17.2. Кривые распреде¬
ленияп — степень полимеризации,X — доля фракции со степе¬
нью полимеризации щ, п2 ...376
т - таан=— (17.4)тгде: то и т — массы образца полимера до и после набухания.Набухшие полимеры растворяются в растворителе, образуя
концентрированные растворы с высокой вязкостью. При высы¬
хании растворы полимеров образуют лаки.Волокнообразование. Полимеры линейного строения спо¬
собны образовывать прочные анизотропные высокоориенти¬
рованные в одном (волокна) или в двух (пленки) направлени¬
ях материалы. Свойства этих материалов зависят от размеров,
формы, гибкости и взаимного расположения макромолекул
полимера.Релаксация и гистерезис. Релаксацией называется процесс
запаздывания перехода полимера в новое состояние равновесия
после внешнего воздействия на него, то есть отставание во вре¬
мени реакции полимера (деформации) на действие внешних
напряжений (нагрузки).Релаксации полимера сопутству¬
ет гистерезис — явление, при кото¬
ром кривая деформации при прило¬
жении нагрузки к полимеру не со¬
впадает с кривой деформации при ее
снятии. В результате на графике
« нагрузка-деформация » образуется
так называемая «петля гистерези¬
са» (рис. 17.3). Явление гистерези¬
са сопровождается нагреванием де¬
формируемого полимера.17.3. Основы технологии переработки ПМ в изделияПереработка ПМ в изделия представляет комплекс операций,
с помощью которых из ПМ формуют изделия с заданными фор¬
мой, размерами и свойствами. Наиболее распространенные ме¬
тоды изготовления изделий из ПМ основаны на переводе их по¬
лимерной фазы в вязкотекучее (реже высокоэластическое)
состояние и придании им в этом состоянии определенной кон¬
фигурации путем деформирования различными методами. По¬
этому, для процессов переработки ПМ существенное значение
имеют реологические свойства полимеров, составляющих их
полимерную фазу.Рис. 17.3. Петля гистерезиса:1 — деформация под нагруз¬
кой, 2 — деформация после
снятия нагрузки377
Реологией (от греч. rheos — течение) называется наука, изу¬
чающая деформационные свойства реальных тел, в узком смыс¬
ле — течение вязких и пластичных тел. Основной задачей рео¬
логии является установление функциональной зависимости
между механическим напряжением (а), деформацией (А) и их
изменением во времени (т), то есть F(a, ft, х)Закономерности реологии, устанавливающей соответствие
между деформационным поведением конкретного материала и
его структурой, используются для количественного описания
поведения различных материалов, в том числе полимеров, при
реальных условиях приложения нагрузки.Деформация полимеров в вязкотекучем состоянии представ¬
ляет деформацию сдвига, для которой характерно постоянство
объема деформируемого тела при изменении его формы. Поэто¬
му поведение и характер течения полимеров и низкомолекуляр¬
ных жидкостей существенно различаются.Для низкомолекулярных соединений существует пропорци¬
ональность между напряжением сдвига (а) и скоростью дефор¬
мирования тела (U):а =г\ • U (17.5)где (г|) — коэффициент вязкости.Вязкость низкомолекулярных соединений не зависит от ре¬
жима деформации и остается постоянной с ростом внешнего
напряжения.Для полимеров зависимость (17.5) не
имеет места и при их течении наблюда¬
ется опережающее нарастание скорости
деформации (сдвига) по отношению к
напряжению. Функциональная зависи¬
мость скорости деформации от на¬
пряжения U = /(о) для низкомолекуляр¬
ных соединений и полимеров (кривые
течения) представлена на рис. 17.4.Из кривой течения полимеров сле¬
дует, что при малых и очень больших
скоростях деформации (кривые в зо¬
нах А и Б) полимеры ведут себя анало¬
гично низкомолекулярным соедине-
Рис. 17.4. Зависимость ниям, вязкость которых постоянна. В
скорости деформации от промежуточной области скоростей де¬
напряжения: формации (между зонами А и Б) вяз-1 — полимер, 2 — низко- кость полимеров перестает быть по-
молекулярное соединение стоянной и резко падает. Поэтому ви378
этой области нарушение пропорциональности между скоростью
деформации U и напряжением а зависит уже не только от тем¬
пературы, но и от режима времени деформирования.Существенное значение для процессов переработки ПМ име¬
ет также пластификация полимеров. Под пластификацией по¬
нимают повышение пластичности полимеров при их переработ¬
ке и эластичности при эксплуатации. Сущность пластифика¬
ции состоит в снижении температуры стеклования полимера Тс
и расширении интервала АТ =Тт - Тс. Пластификация полиме¬
ров может быть достигнута различными методами, в связи с чем
различают внутреннюю и внешнюю пластификацию.Внутренняя пластификация заключается в ослаблении
межмолекулярных связей между цепями макромолекул поли¬
мера за счет изменения химической природы элементарных зве¬
ньев в них, что приводит к снижению концентрации полярных
групп, обусловливающих образование этих связей.Так, например, алкилирование иминогруппы в звеньях по¬
лиамида-C-NH ► -ONR-Ц NО Оуменьшает вероятность возникновения водородных связей меж¬
ду макромолекулами и приводит к повышению пластичности
полимера.Внешняя пластификация может быть физической и меха¬
нической. При физической пластификации в полимер вводят¬
ся пластификаторы — низкомолекулярные твердые или жид¬
кие органические соединения с высокой температурой кипения
и низким давлением пара. Пластификаторы экранируют и
сольватируют функциональные группы в звеньях полимера и
снижают потенциальный барьер внутреннего вращения макро¬
молекул, что приводит к увеличению гибкости цепей и сниже¬
нию температуры стеклования. Понижение температуры стек¬
лования пропорционально количеству молей пластификатора,
удерживаемых полимером:ДГС=— (17.6)Мгде: С — массовая концентрация пластификатора;М — молекулярная масса пластификатора.В качестве пластификаторов используются сложные эфиры
фосфорной, фталевой и адипиновой кислот, хорошо совмеща¬
ющиеся с полимерами.379
Механическая пластификация осуществляется путем нагре¬
вания полимера в деформированном состоянии до температу¬
ры выше температуры стеклования и охлаждении под нагруз¬
кой. При этом происходит распределение и ориентация макро¬
молекул в одном (волокна) или двух (пленки) направлениях,
сближение и уплотнение макромолекулярных цепей.Пластификация полимерной фазы в ПМ позволяет суще¬
ственно улучшить их свойства (морозостойкость, огнестойкость
и др.) и расширить температурный интервал использования ПМ.Основными параметрами процессов переработки ПМ в изде-
• лия являются температура, давление и время формования.Нагрев ПМ до определенной температуры обеспечивает пере¬
вод полимерной фазы в вязкотекучее или высокоэластическое
состояние, ускоряет диффузионные и релаксационные процес¬
сы, а для термореактивных полимеров процесс их отверждения.Проведение процессов под давлением приводит к уплотнению
формуемого материала, облегччает создание изделия заданной
конфигурации и способствует выделению из ПМ летучих веществ.Время формования изделия выбирается так, чтобы обеспе¬
чить полноту протекания в полимерной фазе материала необ¬
ходимых физических и химических процессов.Технические методы переработки ПМ весьма разнообразны
и могут быть разделены на две группы. К первой группе отно¬
сятся процессы формования под давлением: прямое прессова¬
ние, литье под давлением, интрузия и экструзия (выдавлива¬
ние), вальцевание, штамповка. Ко второй группе относятся про¬
цессы формования без давления: литье, заливка в формы, на¬
пыление, спекание порошка, ротационное формование.17.4. Краткий исторический очерк развития
производства ПМ«А. М. Бутлеров открытием формаль¬
дегида и изучением его главнейших
свойств как бы впервые открыл дверь в
область высокомолекулярных соедине¬
ний, той категории химических ве¬
ществ, которым суждено в современной
химии и технике играть совершенно ис¬
ключительную по значению роль».А.Е. АрбузовВсе ПМ — это или сложные системы, основой которых являются
полимеры, или различным образом переработанные индивидуальные
полимеры. Поэтому история ПМ есть, по существу, история полиме-380
ров. Она начинается со второй половины XIX столетия и может быть
разбита на два периода.Первый период (1839—1900 гг.) характеризуется использованием
полимеров природного происхождения, натуральных или модифицй-
рованных: природного каучука, целлюлозы, белковых веществ. К это¬
му времени относятся такие важнейшие технические достижения, как
горячая (Ч. Гудьир, 1839 г.) и холодная (А. Паркер, 1846 г.) вулкани¬
зация каучука, получение эбонита (Т. Хэнкок , 1852 г.) и целлулоида
(Д. Хьят, 1872 г.), разработка технологии пироксилинового (1884 г.)
и баллиститного (1888 г.) порохов, изобретение модифицированного
казеина — галалита (1897 г.).С 1884 г. начинается производство искусственных химических во¬
локон на основе целлюлозы: нитрошелка (Г. Шардоне, 1884 г.), вис¬
козного (Ч. Кросс и К. Бидл, 1892 г.), медноаммиачного (Э. Швейцер,
1899 г.). Эти достижения химической науки и технологии были по
достоинству оценены Д.И. Менделеевым, отметившим, что 4необхо¬
димо, чтобы у нас не только поскорее привилось это дело, но и распро¬
странилось широко, потому что страна наша изобилует всякими рас¬
тительными продуктами, не находящими себе применения* (Газета
«Россия» 1890 г.).К этому периоду относятся и первые исследования в области получе¬
ния синтетического каучука: А.М. Бутлерова по синтезу и полимери¬
зации изобутиленов (1867 г.), А.Е. Фаворского по изучению механиз¬
ма полимеризации непредельных углеводородов (1891 г.), А.А. Соло¬
нины (1897 г.), И.Л. Кондакова по получению полимера 2,3-диметил -
бутадиена (1899 г.), работы Ф. Матьюса, Е. Стренжа, В. Перкина (Ан¬
глия), Ф. Гофмана, К. Гарриеса (Германия) по синтезу метил каучука
(1909 г.).Второй период в развитии химии и технологии полимеров начина¬
ется с 1902 г. В этот период, наряду с использованием природных поли¬
меров, интенсивно развивается химия синтетических полимеров,
осуществляется переход от реакций химического превращения при¬
родных полимеров к реакциям их синтеза из мономеров. Делается ре¬
шающий шаг к получению полимеров с заданными свойствами, то есть
к проектированию новых видов ПМ. Второй период в истории поли¬
меров опирается на крупнейшие достижения теоретической и приклад¬
ной органической химии по синтезу мономеров и изучению процессов
их полимеризации и поликонденсации. К ним, в первую очередь, от¬
носятся работы А.В. Лебедева по полимеризации бутадиена (1908—
1912 гг.), И.И. Остромысленского по синтезу каучука (1911—1917 гг.),
Б.В. Бызова по химии и технологии каучука и резины (1913—1915
гг.), Л. Бэкеленда и Г.С. Петрова по синтезу фенолоформальдегидных
полимеров (1906 г.) и другие.Первыми синтетическими полимерами были бакелит (США, 1907
г.) и карболит (Россия, 1913г.), производство которых было организо¬
вано на основе фенола и формальдегида. В 1909 г. Ф. Гофман, исходя
из исследований И.Л. Кондакова, осуществляет синтез каучука поли¬
меризацией 2,3-диметил-бутадиена, на основе которого в 1916 г. в Гер¬
мании организуется его промышленное производство. В 1921 г. осва-381
ивается производство трехмерных полиэфиров (алкидных полимеров)
и в период 20—30-х годов в практику внедряются такие новые виды
поликонденсационных полимеров как мочевиноальдегидные (карба-
мидные) полимеры.Начиная с 30-х годов, ускоренными темпами развивается произ¬
водство различных видов полимеризационных полимеров: полисти¬
рола (1938 г.), поливинилхлорида (1937 г.), поливинилацетата, поли¬
этилена высокого давления (1942 г.) и низкого давления (1953 г.), по-
лиметилметакрилата, политетрафторэтилена, полипропилена.40-е и 50-е годы характеризуются обширными исследованиями в
области химии и технологии полимеров. Это приводит к разработке и
внедрению в промышленность новых видов полимеров и ПМ на их ос¬
нове: как полимеризационных (полипропилен, политрифторхлорэти-
лен), так и поликонденсационных (полиамиды, полиуретаны, подиэ-
поксиды, силиконовые полимеры). Широкое применение получают
различные сополимеры и модифицированные полимеры различного
назначения, которые расширяют области использования ПМ. При этом
возрастает доля полимеров, получаемых методом полимеризации.В результате к концу XX столетия промышленность полимеров и
ПМ на их основе превращается не только в самостоятельную, но и в
одну из ведущих отраслей народного хозяйства.В нашей стране история производства и внедрения полимеров и ПМ
начинаетоя с 1914 г., когда на основе исследований Г.С. Петрова, В.И.
Лисева и К.И. Тарасова было организовано производство фенолоаль¬
дегидных полимеров в г. Орехово-Зуево на заводе, получившим в 1919
г. название «Карболит». В дальнейшем производство ПМ развивалось
в трех основных направлениях: производство пластических масс, про¬
изводство химических волокон и производство синтетических каучу-
ков.Существенную рель в становлении отечественной промышленнос¬
ти полимеров сыграла организация ряда отраслевых институтов и со¬
здание соответствующих кафедр в ряде химических ВУЗов страны:
Ленинградского и Московского НИИ пластмасс, Владимирского НИИ
синтетических смол, Ярославского НИИ синтеза мономеров для СК,
Всесоюзного НИИ СК, НПО «Пластик», кафедр пластмасс в ЛХТИ им.
Ленсовета и МХТИ им. Д.И. Менделеева, кафедр искусственного во¬
локна в ЛХТИ, МВТУ, Ивановском и Красноярском ХТИ, кафедр син¬
тетического каучука в ЛХТИ, МИТХТ им. М.В. Ломоносова, Иванов¬
ском ХТИ и других.Пластические массы. До 40-х годов отечественное производство пла¬
стмасс ограничивалось получением и переработкой модифицированных
полимеров (галалит, целлулоид). Единственным синтетическим поли¬
мером был карболит, производство которого непрерывно расширялось
за счет ввода в строй новых предприятий. В 1938—42 гг. организуется
производство метилметакрилата, поливинилхлорида, карбамидрых по¬
лимеров и аминопластов на заводах в Любочане Московской области,
Владимире, Кусковском, Карачаевском и Ленинградском химических
заводах. В годы войны на базе эвакуированных предприятий строятся
Кемеровский завод «Карболит», Новосибирский химический завод,382
Свердловский и Нижне-Тагильский заводы пластмасс, Челябинский за¬
вод органического стекла и другие. Продукция большинства из них
обеспечивает производство военной техники.В послевоенные годы, совпавшие с интенсивным внедрением ПМ
во все отрасли народного хозяйства, расширяется и меняется ассорти¬
мент продукции, создаются новые производства, реконструируются
действующие предприятия. На Кусковском химическом заводе впер¬
вые в стране организуется производство кремнийорганических поли¬
меров, блочного и эмульсионного полистирола, полимеров на основе
винилацетата, пластификаторов для поливинилхлоридных полиме¬
ров. Осваиваются новые процессы формования изделий из ПМ и но¬
вые виды продукции (стекла «триплекс», полимерные трубы, древес-
но-волокнистые плиты и т. п.).В период 1950—65 гг. вводятся в строй заводы по получению ионо¬
обменных смол (г. Н. Тагил), полиэтилена низкого давления (г. Охта),
полиацеталей (г. Ереван), создаются производства ударопрочного по¬
листирола и его сополимеров, пенополиуретанов (г. Рошаль) и др. В
результате производство пластических масс в стране возрастает с 160
тыс. т в 1955 г. до 800 тыс. т в 1965 г. В последующие годы расширяет¬
ся производство новых термопластичных полимеров и вводятся в строй
крупные специализированные заводы по получению винилацетата, по-
ливинилбутираля, полиэфиров, сополимеров стирола, акрилонитри-
ла и бутадиена в г. Дзержинске, Н. Полоцке и других городах. Объем
производства пластмасс достигает к 1970 году 1670 тыс. т. Одновре¬
менно возрастают единичные мощности установок и внедряются не¬
прерывные процессы. Так, например, мощность установок по произ¬
водству полиэтилена высокой плотности возрастает с 2—3 до 60 тыс. т
в год, полиэтилена высокой плотности с 3 до 70 тыс. т, полистирола с 3
до 30 тыс. т в год.В 1975—1980 гг. вводятся в эксплуатацию завод полиэтилена в г.
Шевченко мощностью 200 тыс. т/год, Прикумский завод полиэтиле¬
на высокой плотности мощностью 200 тыс. т, организуются произод-
ства полиэтилена низкой плотности мощностью 240 тыс. т в г. Северо-
донецке, полипропилена мощностью 30 и 100 тыс. т на Гурьевском и
Томском заводах, ударопрочного полистирола мощностью 150 тыс. т
в г. Днепродзержинске, поливинилхлорида мощностью 250 тыс. т в г.
Зиминске и др.В результате выпуск пластических масс возрастает к 1980 г. до 3,6
млн. т, а в 1987 г. составляет уже 4,5 млн. т.Химические волокна. Первое предприятие по производству хими¬
ческого волокна (вискозы), производительностью 400 кг в сутки, было
организовано в нашей стране в 1909 г. в г. Мытищи и возобновлено на
реконструированной фабрике «Вискоза» в 1927 г. В последующие годы
развивалось производство только искусственных волокон на основе
целлюлозы: вискозного на Могилевском, Клинском, Ленинградском
и Киевском заводах искусственного волокна, медноаммиачного на
Калининском, Ростокинском, Шуйском и Вышне-Волочаевском заво¬
дах, нитрошелка на Урале. В 1940 г. производство искусственных во¬
локон всех видов составило всего 11,1 тыс. т.383
В послевоенные годы реконструируются старые и вводятся в строй
5 новых предприятий, что позволяет довести в 1958 г. выработку ис¬
кусственных волокон до 170 тыс. т.В 1960 г. начинается производство вискозного штапельного волок¬
на на Рязанском заводе, а в 1962—1964 гг. осваивается выпуск корд¬
ной нити на заводах в г. Балашове и г. Светлогорске. С 1954 г. начина¬
ется производство ацетатных волокон на заводах в гг. Серпухове, Кау¬
насе, Фергане и к 1975 г. выпуск их достигает 45 тыс. т в год.С конца 40-х годов в стране быстрыми темпами развивается произ¬
водство синтетических волокон. В 1948 г. введен в строй первый завод
капронового волокна — Клинский комбинат химических волокон на
основе капролактама, получаемого из фенола. В 1961—1965 гг. орга¬
низовано производство полиамидных волокон на новых заводах в
гг.Чернигове, Рустави, Даугавпилсе, Курске, Кемерово и Барнауле.
Объем производства полиамидных волокон достигает в 1975 г. 220 тыс.
т. В 1960 г. начинается промышленное производство полиэфирного во¬
локна из этилентерефталата «лавсан» на Могилевском ПО «Химволок-
но», достигающее в 1980 г. объема 115 тыс. т. В эти же годы был орга¬
низован выпуск полиакрилнитрильного волокна «нитрон» на Сара¬
товском ПО «Нитрон» и затем на заводах химических волокон в гг.
Новополоцке и Навои. В 1980 г. производство его составляет 68 тыс. т.Помимо многотоннажных производств вискозного, ацетатного и
медноаммиачного волокон, начиная с 50-х годов в стране организует¬
ся производство синтетических волокон специального назначения: из
хлорированного поливинилхлорида («хлорин»), поливинилхлоридно¬
го, из фторсодержащих полимеров («фторлон»), полиуретанового.В результате удельный вес синтетических волокон в общем произ¬
водстве химических волокон возрастает с 7% в 1960 г: до 49% в 1980
г. и 60% в 1987 г. При общем объеме производства химических воло¬
кон в 1,517 млн. т в 1987 г. было произведено искусственных волокон0,613 и синтетических 0, 904 млн. т.Синтетические каучуки. Возможность производства синтетичес¬
ких каучукоподобных материалов начала изучаться в нашей стране
еще в 1912—1917 гг. В основу разработок были положены исследова¬
ния С.В. Лебедева, И.И. Остромысленского, Б.В. Бызова. Однако толь¬
ко в 1931 году на опытном производстве была получена первая партия
синтетического каучука в 265 кг на основе бутадиена, синтезирован¬
ного из пищевого этанола по методу С.В. Лебедева. В том же году опыт¬
ная партия бутадиенового каучука была произведена из бутадиена, по1
лученного по методу Б.В. Бызова. Несмотря на более высокую эконо¬
мичность этого метода, вследствие больших расходных коэффициен¬
тов по нефти за основу промышленного производства синтетического
бутадиенового каучука был принят способ С.В. Лебедева.В 1931—1934 гг. были построены и пущены первые заводы синтети¬
ческого натрий-бутадиенового каучука в г. Ярославле (СК-1), г. Воро¬
неже (СК-2) и г. Ефремове (СК-3), а в 1934 г. завод по производству
хлоропренового синтетического каучука по методу А.Л. Клебанского.
В 1940 г. производство хлоропренового каучука было организовано в
г. Ереване. Одновременно отрабатывалась технология производства бу¬384
тадиенового каучука и повышался выход сырья — бутадиена с 40%
до 80% в расчете на используемое сырье.В послевоенные годы промышленность синтетического каучука
интенсивно развивается и переориентируется на нефтяное и газовое
сырье. В 1959—1965 гг. строятся заводы по производству каучука в
г.г. Красноярске, Стерлитамаке, Караганде, Самаре, Омске и Волжс¬
ке, организуется производство бутадиена из нефтяного сырья. Начи¬
нается производство бутадиеннитрильного (СКН) и бутадиенстироль-
ного (а-метилстирольного СКМС) каучуков. Быстрое развитие полу¬
чает производство стереорегулярных синтетических каучуков — изоп-
ренового СКИ-3 (с 1964 г.) и дивинильного СКД (с 1966 г.). Создается
производство совершенно новых видов синтетических каучуков спе¬
циального назначения, таких как этиленпропиленовый, уретановый,
маслонаполненный дивинилстирольный, кремнийорганический.В результате ассортимент синтетических каучуков, выпускаемых
отечественной промышленностью СК, существенно расширяется и
достигает к 1980 г. 160 наименований.Контрольные вопросы1. Какие системы относятся к группе полимерных материалов (ПМ)
и на какие виды они подразделяются?2. Перечислите особые качества ПМ, обусловившие их широкое ис¬
пользование.3. Какими свойствами обладают полимеры, составляющие основу
ПМ?4. В каких физических состояниях могут находиться аморфные по¬
лимеры?5. Что выражает термомеханическая кривая аморфных полимеров?6. В чем заключаются особенности методов переработки ПМ в изде¬
лия и как они связаны с реологическими свойствами полимеров?7. Какое явление называется пластификацией и какое значение оно
имеет для переработки и эксплуатации ПМ?13 Химическая технология. Том 2
Глава XVIIIПРОИЗВОДСТВО ПЛАСТИЧЕСКИХ МАСС8.1- Состав и классификация пластических массПластическими массами (ПлМ) называют полимерные ма¬
териалы, полимерная фаза которых находится в период фор¬
мования изделия в вязкотекучем или высокоэластическом со¬
стоянии, а при эксплуатации в аморфном стеклообразном или
кристаллическом состоянии. Таким образом, рабочим интерва¬
лом температур для ПлМ, в котором они используются как твер¬
дые конструкционные материалы, является Тхр< Тэксп< Тс, где
Тхр температура хрупкости, при которой полимер разрушается
под нагрузкой, не переходя в высокоэластическое состояние.В основу классификации ПлМ положены их состав, отноше¬
ние к нагреванию и природа полимерной фазы.По составу ПлМ делятся на однофазные (гомогенные) и мно¬
гофазные (гетерогенные, наполненные, композиционные). В го¬
могенных ПлМ полимер является основным компонентом, оп¬
ределяющим их свойства, остальные компоненты растворены в
полимерной фазе. В гетерогенных ПлМ полимер представляет
дисперсионную среду (связующее), а введенные в него наполни¬
тели составляют самостоятельные фазы. По природе наполните¬
ля ПлМ делятся на ПлМ с твердым наполнителем и газонапол¬
ненные. Первые могут быть дисперснонаполненными, в которых
наполнитель равномерно распределен в ПлМ, и армированны¬
ми, в которых наполнителем служат волокна, ткань, бумага.
Газонаполненные ПлМ делятся на пенопласты, в которых пу¬
зырьки газа изолированы друг от друга пленкой полимера, и по-
ропласты, которые пронизаны каналами, соединяющими газо¬
вую фазу.По отношению к нагреванию ПлМ подразделяются на тер¬
мопластичные или термопласты, полимерная фаза которых
при горячем формовании изделия не отверждается и ПлМ со¬
храняет способность переходить вновь в вязкотекучее состоя¬
ние при повторном нагреве, и термореактивные или реактоп-
ласты, переработка которых в изделия сопровождается реакци¬
ями образования трехмерной структуры в полимерной фазе (от¬
верждение полимера) и изделие необратимо теряет способность
переходить в вязкотекучее состояние.По природе полимера, составляющего полимерную фазу,
ПлМ делятся на полимеризационные, к которым относятся386
полиэтилен, полипропилен, полистирол, полиакрилонит-
рил, полиметилметакрилат, политетрафторэтилен и поли-
конденсационные, из которых наиболее распространены
феноло-альдегидные или фенопласты и анилино-альдегид-
ные или аминопласты, и модифицированные на основе цел¬
люлозы (целлулоид, этролы).Единой терминологии ПлМ не существует. Одни и те же ма¬
териалы выпускаются в разных странах под разными названи¬
ями, например, полиэтиленДРФ), алкатен (Англия), хифлекс
(США), ротен (Япония).Состав конкретных ПлМ зависит от их назначения и требуе¬
мых свойств и может меняться в широких пределах: от почти
чистого полимера (полиэтилен, полипропилен) до систем, со¬
держащих 50 и более процентов различных добавок. В общем
случае в состав ПлМ входят следующие компоненты, каждый
из которых выполняет определенную функцию.1. Полимеры (связующие, полимерная фаза), создающие ос¬
нову материала.2. Наполнители, обеспечивающие нужные механические
свойства, прочность и повышающие экономичность производ¬
ства.3. Пластификаторы.4. Стабилизаторы, повышающие стойкость к действию вы¬
соких температур, кислорода воздуха, фото- и радиационному
воздействиям (антиоксиданты, антирады, светостабилизаторы)
и уменьшающие способность ПлМ к старению.5. Антипирены, понижающие горючесть ПлМ (инертные и
химически активные, вступающие в реакцию с полимером).6. Красители.7. Отвердители, создающие трехмерную сшитую структуру
полимера.8. Порофоры (порообразователи, вспенивающие вещества),
разлагающиеся при нагревании и вводимые в композиции для
получения газонаполненных пластических масс.9. Смазывающие вещества, предотвращающие прилипание
материала к оборудованию в процессе переработки и изготов¬
ления изделия.Основную массу производимых ПлМ составляют полиолефи-
ны, полистирол, поливинилхлорид, фенолальдегидные и кар¬
бамид ные полимеры. Они составляют около 85% от всего про¬
изводства пластических масс. В табл. 18.1 приведены данные о
мировом производстве ПлМ.13387
Таблица 18.1. Мировое производство ПлМ, млн. тВид ПлМГод196019701987Всего6,930,183,5Полеолефины0,058,138,0Полистирол0,823,65,8Поливинилхлорид1,55,7 Фенолальдегидные0,71.6 Карбамидные0,62,05 В РФ пластические массы производятся на специализирован¬
ных предприятиях в Волгограде, Владимире, Екатеринбурге,
Казани, Кемерове, Москве, Новосибирске, Орехово-Зуеве, Ниж¬
нем Тагиле, С.-Петербурге, Тюмени, Уфе.Ниже рассматриваются производства таких распространен¬
ных пластических масс как полиэтилен высокого и низкого
давления, блочный и блочно-суспензионный полистирол, фе¬
нол формальдегидные полимеры.18.2. Производство полиэтиленаПолиэтилен (~СН2-СН2-)П — карбоцепной термопластич¬
ный кристаллический полимер белого цвета со степенью крис¬
талличности при 20°С 0,5—0,9. При нагревании до температу¬
ры, близкой к температуре плавления он переходит в аморфное
состояние. Макромолекулы полиэтилена (ПЭ) имеют линейное
строение с небольшим количеством боковых ответвлений. ПЭ
водостоек, не растворяется в органических растворителях, но
при температуре выше 70°С набухает и растворяется в аромати¬
ческих углеводородах и галогенпроизводных углеводородов.
Стоек к действию концентрированных кислот и щелочей, од¬
нако разрушается при воздействии сильных окислителей. Об¬
ладает низкой газо- и паропроницаемостью. Звенья ПЭ неполяр¬
ны, поэтому он обладает высокими диэлектрическими свойства¬
ми и является высокочастотным диэлектриком. Практически
безвреден. Может эксплуатироваться при температурах от -70
до +60°С.Свойства ПЭ существенно зависят от способа полимериза¬
ции, в соответствии с чем различают ПЭ высокого давления или
низкой плотности (ПЭВД) и ПЭ низкого давления или высокой
плотности (ПЭНД). Сравнительные свойства этих марок ПЭ при¬
ведены в табл.18.2.388
Таблица 18.2. Свойства полиэтиленаПоказательПЭВДПЭНДМолекулярная масса, тыс. единиц
Плотность, т/м3
Степень кристалличности
Температура плавления, °С
Температура хрупкости, °С
Уд. электрическое сопротивление, Ом-м30—400
0,91—0,93
0,50—0,65
105—108
-80—-120
10’550—8000,95—0,960,75—0,85120—125-100 15010'5Таким образом, ПЭНД отличается от ПЭВД большей плотно¬
стью, более высокой степенью кристалличности, лучшими тем¬
пературными характеристиками и физико-механическими
свойствами.Технология производства ПЭ высокого и низкого давления
различна. ПЭВД получается радикальной полимеризацией эти¬
лена в газовой фазе при температуре 180—300 С и давлении
150—300 МПа в присутствии молекулярного кислорода
(0,003% об.) или пероксида ди трет-бутила. Реакция протекает
через стадию образования пероксида или гидропероксида эти¬
лена с последующим гомолитическим распадом их до би- или
монорадикалов, инициирующих далее процесс полимеризации:СН2=СН2 + 02 — СН2-СН2 — *0СН2-СН20- илио — о
сн2=сн2 + о2 — сн2=сн — сн2=сно- + онООНПолимеризация этилена протекает с выделением большого
количества тепла и описывается общим уравнениемп СН2=СН2 = (-СН2-СН2-)„ -АН АН = 96,4 кДж/мольКонверсия этилена в полиэтилен и свойства полученного по¬
лимера зависят от температуры, давления, концентрации ини¬
циатора и времени полимеризации. При повышении давления
увеличиваются степень конверсии, молекулярная масса, плот¬
ность и механическая прочность полиэтилена. При повышении
температуры степень конверсии падает, а остальные показате¬
ли увеличиваются. Повышение концентрации кислорода при¬
водит к увеличению степени конверсии и снижению молекуляр¬
ной массы полимера. Оптимальное время процесса составляет
1—3 минуты, дальнейшее увеличение его не влияет на степень
конверсии этилена в полиэтилен.389
Технологический процесс полимеризации этилена в присут¬
ствии молекулярного кислорода включает следующие основные
стадии: смешение этилена с возвратным газом и кислородом,
двухстадийное сжатие газовой смеси, полимеризация этилена,
разделение полимера и непрореагировавшего этилена и грану¬
ляция полимера. Технологическая схема подобного процесса
приведена на рис. 18.1.Свежий этилен из хранилища 1 и возвратный этилен из от¬
делителя низкого давления 9 подаются в смеситель 2, куда по¬
ступает кислород. Газовая смесь сжимается в компрессоре пер¬
вого каскада 3, смешивается в смесителе 4 с возвратным этиле¬
ном из отделителя высокого давления 8 и сжимается в комп¬
рессоре второго каскада 5 до давления 150—300 МПа. Пройдя
маслоотделитель 6, газ подается в трубчатый реактор полиме¬
ризации 7. Из него продукты реакции поступают в отделитель
высокого давления 8, где из них выделяется часть не вступив¬
шего в реакцию этилена. Он охлаждается в холодильнике 12 и
направляется в смеситель 4. Полиэтилен в виде расплава из от¬
делителя 8 подается в отделитель низкого давления 9, где от
него при давлении 1,5’1015 Па отделяется остаток этилена, ко¬
торый после охлаждения в холодильнике 11 поступает на сме¬
шение со свежим этиленом. Расплавленный ПЭ поступает на
грануляцию в гранулятор 10, в котором продавливается черезЭтиленВодаКислородгВозвратный этилен12^«ВодаВозвратный этилен гм23 ♦ цьВодаШ18П10пэвдРис. 18.1. Технологическая схема производства ПЭВД:1 —хранилище этилена, 2— смеситель этилена низкого давления, 3 —
компрессор первого каскада, 4 — смеситель этилена высокого давления,
5 — компрессор второго каскада, 6 — маслоотделитель, 7 — трубчатый
реактор, 8— отделитель высокого давления, 9 — отделитель низкого
давления, 10 — гранулятор, 11,12 — холодильники390
фильеры, режется и в виде гранул поступает на дальнейшую
переработку.Современные установки по производству ПЭВД имеют мощ¬
ность до 150 тыс. т в год. Конверсия этилена за один оборот со¬
ставляет около 0,2 при выходе полимера 95—98%. Вследствие
низкой стоимости и достаточно высоких качеств ПЭВД, этим
методом в настоящее время производится около 75% всего по¬
лиэтилена.ПЭНД получается координационно-ионной полимеризацией
этилена в растворе бензина при температуре 70—80°С и давле¬
нии 0,15—0,3 МПа в присутствии комплексных металлоорга¬
нических катализаторов (катализаторов Циглера—Натта). Сре¬
ди них наибольшее распространение получили катализаторы
состава АЦСгНб^СЬТЮЦ.Технологический процесс производства ПЭНД включает сле¬
дующие основные стадии: приготовление катализаторного ком¬
плекса, полимеризация этилена, выделение порошкообразно¬
го полимера и разложение остатков катализатора, промывка и
сушка полиэтилена.Скорость полимеризации и свойства получаемого ПЭНД за¬
висит от температуры, давления и активности катализатора,
которая определяется мольным соотношением диалкилалюми-
ния и тетрахлорида титана. При повышении содержания после¬
днего в контактной массе возрастает скорость процесса и выход
ПЭ, но снижается его молекулярная масса. Для регулирования
молекулярной массы полимера в этилен вводится водород, ко¬
торый играет роль передатчика цепи. Катализаторный комплекс
легко разрушается под воздействием кислорода воздуха и вла¬
ги. Поэтому процесс полимеризации проводится в атмосфере азо¬
та и в среде обезвоженного бензина. Метод приготовления катали¬
заторного комплекса и механизм его действия рассматривается в
главе XX. К недостаткам метода ионной полимеризации относят¬
ся огнеопасность, невозможность регенерации катализатора и
сложность процессов его отмывки и очистки бензина.ПЭ перерабатывается всеми методами, используемыми для
переработки термопластичных полимеров; литьем под давлени¬
ем, экструзией и прессованием. Он легко сваривается, способен
образовывать различные сополимеры. Благодаря широкому
комплексу свойств ПЭ применяется во многих отраслях про¬
мышленности и народного хозяйства: кабельной, радиотехни¬
ческой, химической, легкой промышленности, в медицине и др.
Из ПЭ изготавливаются различные изделия технического назна¬
чения, трубы, кабельная изоляция, упаковочный материал,391
предметы домашнего обихода, парниковые покрытия. Около 70%
ПЭ выпускается для этих целей в виде пленки и листового матери¬
ала. Мировое производство ПЭ составляет свыше 30 млн. т.18.3. Производство полистиролаПолистирол (-CH2-CH-)„ это карбоцепной термопластич-С6Н5ный аморфный полимер с молекулярной массой от 50 до
300 тыс. ед. Макромолекулы полистирола (ПС) имеют преиму¬
щественно линейное строение «голова к хвосту» с небольшим
количеством боковых ответвлений. ПС — бесцветное твердое ве¬
щество с температурой стеклования 93°С, плотностью 1,05 т/м3,
каучукоподобное при температуре 80—150°С. Водостоек, ра¬
створяется в стироле, ароматических углеводородах, хлорпро-
изводных углеводородов, сложных эфирах, нерастворим в али¬
фатических углеводородах, низших спиртах, диэтиловом эфи¬
ре. ПС устойчив к действию разбавленных кислот, щелочей и
растворов солей. Обладает хорошими диэлектрическими свой¬
ствами, не зависящими от температуры.Вследствие низкой теплостойкости (75 С по Мартенсу) ПС мо¬
жет эксплуатироваться при температуре не выше 60°С. В отли¬
чие от полиолефинов он имеет высокую твердость, но весьма хру¬
пок. При этом, хрупкость увеличивается в процессе эксплуата¬
ции вследствие старения материала. Этого недостатка лишен
ударопрочный полистирол (УПС) и сополимеры стирола с акри-
лонитрилом и бутадиеном. При нагревании до температуры
300—400 С ПС деполимеризуется с образованием мономера.ПС получается радикальной полимеризацией стирола в при¬
сутствии инициаторов (пероксиды, динитрил азо-бис-изомасля-
ной кислоты) или без них (термическая полимеризация):пСН2=СН —► (-СН2-СН-)гсПри инициаторе — пероксиде бензоила реакция протекает
через стадию образования бензоатного радикала, присоединя¬
ющегося к группе СН2 молекулы стирола с образованием более
стабильного первичного радикала, начинающего цепь:(С6Н5СОО)2 - 2С6Н5СОО-392
С6Н5СОО* + сн2=сн -► С6Н5-СОО-СН2-СН-По технологическому оформлению процесса различают по¬
лимеризацию в блоке (в массе), в эмульсии, в суспензии или в
растворе. Промышленное значение имеют следующие методы
производства ПС:— блочная полимеризация с неполной конверсией стирола
(непрерывный способ);— суспензионная полимеризация (периодический способ);— блочно-суспензионная полимеризация (периодический
способ).18.3.1. Производство ПС блочным методом (в массе)При блочной полимеризации достижение полной (до 0,99)
конверсии стирола в одном реакторе экономически нерентабель¬
но вследствие длительности процесса и необходимости поддер¬
живать высокую температуру в конце полимеризации. Это при¬
водит к снижению молекулярной массы полимера. Поэтому, в
промышленности в настоящее время применяется метод блоч¬
ной полимеризации с неполной конверсией стирола в каскаде
аппаратов или в аппаратах смешения и вытеснения (интенсифи¬
цированный способ). Этот способ производства является наиболее
распространенным в отечественной промышленности, им получа¬
ется до 80% ПС от всего объема его, производимого в стране.В интенсифицированном способе производства блочного ПС
полимеризация мономера осуществляется сначала до степени
конверсии 0,7—0,8 в двух полимеризаторах смешения, а затем
завершается до степени конверсии 0,95 в реакторе вытеснения
колонного типа.Технологический процесс производства блочного ПС интен¬
сифицированным способом включает следующие стадии: пред¬
варительная полимеризация, окончательная полимеризация,
вакуум-экструзия полимера, гранулирование и складирование
готового продукта.Технологическая схема интенсифицированного способа про¬
изводства блочного ПС представлена на рис. 18.2.Смесь свежего и возвратного регенерированного стирола из
емкости 1 подается в напорный бак 2, откуда, пройдя фильтр 3,
поступает последовательно в форполимеризаторы (реакторы
предварительной полимеризации) 4 и 5. В первом форполиме-
ризаторе при температуре 120°С достигается степень конверсии0,5, во втором при температуре 125 С 0,7—0,8.393
ВодаСтиролВодаа45 1VJT]* ВодаВодаРис. 18.2. Технологическаясхема производства блочного ПС:1 — емкость для стирола, 2 — напорный бак, 3 — фильтр, 4, 5 — форпо-
лимеризаторы, 6, 7, 9, 12 — холодильники-конденсаторы, 8 — полимери-
зационная колонна, 10 — шнек, 11 — вакуум-экструдер, 13 — система
регенерации стирола, 14 — охлаждающая ванна, 15 — гранулятор, 16 —
виброситоРеакционное тепло из форполимеризаторов отводится за счет
испарения части стирола, пары которого конденсируются в хо¬
лодильниках-конденсаторах 6 и 7 и возвращаются в полимери¬
заторы. Из форполимеризатора 5 частично заполимеризовав-
шийся стирол подается в полимеризационную колонну 8, где
при температуре 125—200 С завершается полимеризация до
степени конверсии 0,95. Отвод тепла из колонны осуществля¬
ется с помощью холодильника-конденсатора 9. Во избежание
окисления стирола кислородом воздуха в колонну 8 подается
азот. Расплав ПС шнеком 10 перемещается в вакуум-экструдер11, где из него удаляются пары стирола, которые конденсиру¬
ются в холодильнике-конденсаторе 12. Конденсат стирола из
него направляется в систему регенерации 13 и оттуда в емкость1 на смешение со свежим мономером, Отвакуумированный рас¬
плав ПС проходит охлаждающую ванну 14 и поступает в грану¬
ляционное устройство 15, после чего подвергается классифи¬
кации на вибросите 16.394
18.3.2. Производство ПС блочно-суспензионным методомОсновным недостатком полимеризации в массе являются
затруднения с отводом реакционного тепла. Этот недостаток
устранен в методе суспензионной полимеризации, в котором
отвод тепла облегчен через водную дисперсионную фазу. Это
позволяет в широких пределах изменять условия полимериза¬
ции и получать ПС различного качества.Суспензионная полимеризация стирола протекает в водной
среде в присутствии инициаторов полимеризации, растворимых
в мономере и нерастворимых в воде. Поэтому, реакция осуще¬
ствляется как бы в объеме маленького блока (капли). Инициа¬
торами реакции являются органические перекиси: пероксид
бензоила, трет-бутилпербензоат и др. Для повышения устойчи¬
вости суспензии стирола в воду добавляют стабилизаторы, на¬
пример, гидроксид магния, поливиниловый спирт и др. Полу¬
ченный ПС легко отделяется от водной фазы и осаждается на
дне реактора.Разновидностью суспензионного метода полимеризации яв¬
ляется блочно-суспензионная полимеризация, в которой совме¬
щены преимущества блочной и суспензионной полимеризации.
Он широко применяется для производства ударопрочного ПС и
полимера, предназначенного для получения пенополистирола.
Технологический процесс блочно-суспензионной полимериза¬
ции включает следующие стадии: предварительная полимери¬
зация стирола в массе (получение форполимера), окончатель¬
ная полимеризация форполимера в суспензии, отделение, про¬
мывка и сушка гранул ПС.Технологическая схема производства ПС блочно-суспензи¬
онным способом представлена на рис 18.3.В реактор предварительной полимеризации 1 загружается
стирол и раствор инициатора в стироле и при температуре 80°С
проводится полимеризация до степени конверсии стирола 0,3—0,4. Затем образовавшийся форполимер и водный раствор ста¬
билизатора из аппарата 2 подаются в автоклав-полимеризатор
3, диспергируются в водной фазе и нагреваются при температу¬
ре 90° С до образования гранул. В автоклав вводятся также до¬
полнительно часть инициатора и изопентан, служащий газооб-
разователем при переработке гранул плимера. Полученная сус¬
пензия сливается через сито 4 в сборник 5 и после разбавления
водой подается в центрифугу 6. Отжатые и промытые водой гра¬
нулы ПС сушатся в бврабанной сушилке 7 воздухом, после чего
поступают в бункер 8 и на сито 9 для классификации.395
Рис. 18.3. Технологическая схема производства
блочно-суспензионного ПС:1 — реактор предварительной полимеризации, 2 — аппарат для приготов¬
ления водной фазы, 3 — автоклав-полимеризатор, 4 — сито, 5 — сборник,6 — цетрифуга, 7 — сушилка, 8 — бункер, 9 — ситоПС перерабатывается в изделия всеми способами, использу¬
емыми для переработки термопластичных полимеров и окра¬
шивается органическими красителями. Основным методом фор¬
мования изделий из ПС является литье под давлением, реже
используется экструзия, позволяющая получать пленки и нити
Для повышения теплостойкости и механической прочности в
ПС вводятся минеральные наполнители и стекловолокно.Основными областями применения ПС являются следующие
отрасли промышленности: приборостроительная (комплектую¬
щие детали, конденсаторная пленка), кабельная (изоляция,
нити), строительная (облицовочная плитка, фурнитура), про¬
изводство упаковочных материалов, тары и изделий бытового
назначения.Повышенной ударной прочностью обладают так называемый
ударопрочный полистирол, представляющий сополимеры сти¬
рола и бутадиен-стирольного каучука, получаемые методом
привитой сополимеризации, и сополимеры стирола, акрилонит-
рила и акрилонитрил-бутадиенового каучука, получаемые ме-
ханохимическим методом (АБС-сополимеры, пластик СНП).Широкое применение в качестве тепло- и звукоизолирующе¬
го и упаковочного материала получил газонаполненный поли¬
стирол — пенополистирол. Он получается прессованием смеси
тонкодисперсного ПС с твердым порофором — карбонатом алю¬396
миния, или беспрессовым методом путем введения в стирол на
завершающей стадии полимеризации вспенивающего агента —
изопентана.Мировое производство ПС составило в 1987 году 5,8 млн. т,
из них 3,7 млн. т ударопрочного ПС.18.4. Производство феноло-формальдегидных
полимеровФеноло-альдегидными полимерами называются отвержден¬
ные олигомерные продукты поликонденсации фенолов с альде¬
гидами. Для производства подобных олигомеров в качестве фе¬
нольного сырья используются фенол, крезолы, ксиленолы, п-
mpem-бутилфенол, гидрохинон, в качестве альдегидов — фор¬
мальдегид и фурфурол. Наибольшее промышленное значение
имеют полимеры, полученные из олигомеров на основе фено¬
лов и формальдегида — феноло-формальдегидные полимеры
(ФФАП), производство которых составляет около 95% от общего
объема феноло-альдегидных полимеров. Ниже рассматривает¬
ся производство ФФАП на основе олигомеров, полученных из
формальдегида и простейшего фенола — оксибензола.18.4.1. Физико-химические основы получения олигомеров
ФФАПОлигомеры получаются поликонденсацией фенола и фор¬
мальдегида в водной среде в присутствии катализаторов кис¬
лотного или щелочного характера. Реакция протекает через
стадию оксиметилирования в орто- и параположения феноль¬
ного ядра с образованием соответствующих орто- и парамети-
лолфенолов (оксибензиловых спиртов):ОН ОН ОНСН2ОНДальнейшее течение реакции, следовательно, строение и
свойства получаемых олигомеров, зависят от мольного соотно¬
шения фенола и формальдегида и природы катализатора, то есть
от pH среды.В щелочной среде и при избытке формальдегида протекает и
далее преимущественно реакция оксиметилирования и образу¬
ется смесь фенолоспиртов в соответствии со схемой:397
онHOH2C-^j-CH2OH?н (б)СН2ОНСН2ОН СН2ОНПри этом, на практике условия реакции выбираются так,
чтобы избежать образования триметилолфенола и получить
олигомеры линейного строения.Образовавшиеся по схеме (б) фенолоспирты вступают в ре¬
акцию поликонденсации между собой или с фенолом, образуя
димеры, которые затем взаимодействуют с фенолоспиртами с
образованием олигомеров более высокой степени поликонден¬
сации. Пример поликонденсации на данной стадии показан на
схеме (в).ОН ОН ОН ОН£qJCH2OH + (pjCH2OH ► (pJCH20H (^jch2oh +HaoСИ 2 (в)ОН ОН ОН ОН он ОН(OJCH2oh ((3jCH2OH+ (о] -► (OjCH2OH [оГсн2^о] + н2о
сн2 сн2 ПГ и т.д.СН2ОН СН2ОНОбщее уравнение поликонденсации фенола и формальдеги¬
да в щелочной среде может быть представлено так:(/Z+ZTl+lJCgHsOH + (2/1+771 )СН20 *
->Н-[-С6Н2(ОН)(СН2ОН)-СН2-]я-[С6Нз(ОН)-СН2-]т-С6Н4ОН++(п+т) Н20 (г)где: п — среднее число звеньев, содержащих метилольную
группу,т — среднее число звеньев, не содержащих метилольную
группу.Таким образом, образующиеся в щелочной среде олигомеры,
представляют смесь линейных и разветвленных полимергомо-
логов, содержащих в макромолекуле реакционноспособные
метилольные группы. Среднее число фенольных ядер в макро¬
молекуле олигомера (п + т + 1) составляет 4—10, а число мети-
лольных групп 2—5. Подобные олигомеры при нагревании спо¬398
собны к реакциям сшивания за счет содержащихся в них мети-
лольных групп, то есть представляют термореактивные олиго¬
меры. Они получили название резолов.В кислой среде и при избытке фенола образовавшиеся по схе¬
ме (а) фенолоспирты реагируют с фенолом с образованием ди-
оксидифенилметанов по схеме:Так как в этих условиях скорость поликонденсации значи¬
тельно выше скорости оксиметилирования, то диоксидифенил-
метаны реагируют преимущественно с фенолоспиртами, напри¬
мер ПЛ HVOMO'в результате чего образуются олигомеры преимущественно ли¬
нейного строения с незначительным содержанием метилольных
групп.Общее уравнение поликонденсации фенола и формальдеги¬
да в кислой среде может быть представлено таким образом:где: п = 4—8.Подобные олигомеры практически не содержат метилольных
групп и не способны к реакциям сшивания при нагревании без
добавления специальных отвердителей, то есть являются тер¬
мопластичными олигомерами. Они получили название новола-
ков.Реакции оксиметилирования (а и б) и реакции конденсации
(в, д и е) практически необратимы. Константа равновесия про¬
цесса поликонденсации, условно выражаемого уравнением:ОНОН0Н+Н20ОН+ Н20(д)ОН ОН ОН ОН ОН ОН (е)(0ГСН2“(0) + Н°2НС1^)-*- (§Гсн2“(0ГСН2_(0) +яр,(п +1)С6Н5ОН + яСН20 —— Н-[-СбНз(ОН)-СН2-]п-СбН4ОН + пН20 (ж)399
-Ar-H + О + H-Ar- *2 -Ar-CH2-Ar- + H20 - AHIICH2 AH = 88 кДж/м[CH2] [H20][H]2 [O]имеет порядок 103—104, то есть поликонденсация неравновес¬
ная. Поэтому процесс получения олигомеров осуществляется в
водном растворе формальдегида. Скорость поликонденсации
зависит от температуры и концентрации катализатора (pH сре¬
ды), а молекулярная масса получаемого олигомера от времени.18.4.2. Производство новолачных олигомеровФеноло-формальдегидные новолачные олигомеры в промыш¬
ленности производятся периодическим и непрерывным спосо¬
бами. Технологический процесс включает следующие стадии:
дозировка сырья, поликонденсация, сушка олигомера охлаж¬
дение и измельчение готового продукта.Мольное соотношение фенола и формальдегида составляет
от 1:0,78 до 1:0,86. В качестве катализатора используется со¬
ляная кислота в количестве 0,2—1,5 мае. долей на 100 мае. до¬
лей фенола, что обеспечивает pH среды в пределах 1,5—1,8. На
рис. 18.4 представлена технологическая схема производства
новолачных олигомеров непрерывным способом с использова¬
нием реактора поликонденсации колонного типа.В смеситель 1 подаются расплавленный фенол, формалин
и часть катализатора — соляной кислоты. Остальная часть ка¬
тализатора вводится непосредственно в колонну-реактор 2. Из
смесителя смесь реагентов поступает в первую секцию четы¬
рехсекционной колонны-реактора 2. Процесс образования оли¬
гомера проходит при температуре 100С и давлении 105 Па.
Выходящая из последней секции колонны водно-олигомерная
эмульсия разделяется в сепараторе 3. Надсмольная вода вы¬
водится на очистку, а жидкий олигомер поступает в трубча¬
тый сушильный аппарат 4, обогреваемый паром до темпера¬
туры 140—160 С. Из сушильного аппарата олигомер, пары
воды и летучих веществ подаются в приемник 5. Пары прохо¬
дят холодильник-конденсатор бив виде конденсата, содер¬
жащего до 20% фенола, добавляются к свежему фенолу. Рас¬
плавленный новолак из приемника 5 подается на барабан
крошкообразователя 7, охлаждаемый водой, и после из¬
мельчения поступает в виде чешуек по транспортеру 8 на400 .
Соляная кислота ФенолРис. 18.4. Технологическая схема производства новолачных
олигомеров:1 — смеситель, 2 — реактор колонного типа, 3 — сепаратор, 4 — сушиль¬
ный аппарат, 5 — приемник олигомера, 6, 9 — холодильники-конденсато-
ры, 7 — крошкообразователь, 8— транспортерсклад. Выделяющиеся из колонны 2 летучие продукты кон¬
денсируются в холодильнике-конденсаторе 9 и возвраща¬
ются в колонну.Новолачные феноло-формальдегидные олигомеры предста-
ляют твердые вещества от светло-коричневого до темно-корич¬
невого цвета, плотностью 1,2—1,3 т/м3. Хорошо растворимы
в метаноле, этаноле и ацетоне, растворяются в фенолах и ра¬
створах щелочей. Нерастворимы в ароматических и парафи¬
новых углеводородах и галогенпроизводных углеводородов. Не
отверждаются при длительном хранении и нагревании до
180°С.18Л.З. Производство резольных олигомеровФеноло-формальдегидные резольные олигомеры производят¬
ся в промышленности только периодическим способом. Техно¬
логический процесс включает те же стадии, что и при произ¬
водстве новолачных олигомеров. Однако, в виду возможности401
Рис. 18.5. Технологическая схема производства резопьных
олигомеров:1 — емкость фенола, 2 — емкость формалина, 3 — емкость аммиачной
воды, 4 — варочно-сушильный аппарат, 5 — холодильник-конденсатор,6 — сборник надсмольной воды, 7 — сборник резоласамоотверждения резолов в процессе производства, к режиму
поликонденсации предъявляются жесткие температурные тре¬
бования. Мольное отношение фенола и формальдегида состав¬
ляет от 1:1,1 до 1:2,1. В качестве катализатора используются
водный раствор аммиака, гидроксиды натрия и бария в коли¬
честве 1,2—2,0 мае. долей на 100 мае. долей фенола. На рис.
18.5 представлена технологическая схема производства резоль-
ных олигомеров.Расплавленный фенол, формалин и аммиачная вода из ем¬
костей 1, 2 и 3 соответственно, загружаются в варочно-сушиль¬
ный аппарат 4, обогреваемый паром, к которому присоединен
холодильник-конденсатор 5. На стадии поликонденсации хо¬
лодильник работает как обратный, а на стадии вакуум-сушки
как прямой. После загрузки сырья проводится процесс поли¬
конденсации при температуре 65—75°С, по/6кончании кото¬
рого включается вакуум и производится сушка олигомера. От¬
гоняемая в процессе сушки надсмольная вода собирается в
сборнике 6. По окончании сушки жидкие резолы охлаждают¬
ся в аппарате 4 и сливаются в сборник 7. Твердые резолы в
расплавленном состоянии выгружаются в специальный вагон-
холодильник, где после охлаждения и затвердевания измель¬
чаются.Резольные феноло-формальдегидные олигомеры представля¬
ют твердые или жидкие вещества, цвет которых зависит от при-402
роды катализатора. Олигомеры, полученные в присутствии
аммиачной воды, окрашены в желтый цвет. Плотность твердых
резолов составляет 1,25—1,27 т/м3. Они хорошо растворимы в
метаноле, этаноле и ацетоне. Не растворимы в ароматических
и парафиновых углеводородах и галогенпроизводных углево¬
дородов. Резолы содержат значительное количество свободно¬
го фенола, что снижает их температуру плавления. При хране¬
нии даже при обычной температуре способны переходить в не¬
плавкое нерастворимое состояние.18.4.4. Переработка феноло-формальдегидных олигомеровФеноло-формальдегидные олигомеры являются полупродук¬
тами для производства феноло-формальдегидных пластических
масс (фенопластов). В состав фенопластов, помимо олигомера
(резола или новолака), входят наполнитель, отвердитель (для
ново лаков), катализатор отверждения (для резолов) пластифи¬
катор и красители. В зависимости от природы наполнителя и
его дисперсности фенопласты делятся на прессовочные матери¬
алы и слоистые пластики.К прессовочным материалам относятся:— пресспорошки (наполнитель — древесная мука, каолин,
слюда);— волокниты (наполнитель — хлопковая целлюлоза);— асбоволокниты (наполнитель — асбест);— стекловолокниты (наполнитель — стеклянное волокно).К слоистым пластикам относятся:— текстолит (наполнитель — ткань);— гетинакс (наполнитель — бумага);— древесно-слоистые пластики (наполнитель — слои древе¬
сины).Особую группу фенопластов составляют газонаполненные
пенофенопласты и сотофенопласты.Для превращения фенопластов в феноло-формальдегидные
пластические массы их подвергают термической обработке. Во
многих случаях эта операция совмещается с операцией формо¬
вания изделия. Фенопласты перерабатываются методами горя¬
чего прессования, литья под давлением, экструзией. При этом
происходит отверждение полимерной фазы и образование про¬
странственной сетчатой («сшитой») структуры, в которой
макромолекулярные цепи олигомеров соединены между собой
метиленовыми « мостиками »:403
онОН ОН-ф-СН2 J^CH2-|^p сн2- •
сн2 сн2
• • •~ф-СН2 —TqI—сн2-Col- сн2-•
он ^ онТакие сшитые структуры получили название резитов.Механизм отверждения различен для резольных и новолач-
ных олигомеров. Термореактивные резолы отверждаются при
нагревании в отсутствии отвердителей. При этом продолжает¬
ся реакция поликонденсации и метиленовые мостики между
фенольными ядрами образуются за счет содержащихся в них
метилольных групп. Процесс отверждения ускоряется введени¬
ем ускорителей (катализаторов) в виде оксида кальция или маг¬
ния.Отверждение термопластичных новолаков возможно только
в присутствии отвердителей, то есть веществ, способных созда¬
вать метиленовые мостики. Наиболее распространенным отвер-
дителем новолачных олигомеров является уротропин (гексаме-
тилентетрамин), который при температуре отверждения дис¬
социирует с образованием формальдегида:C6H12N4 = 6СН2О + 4NH3Феноло-формальдегидные пластические массы относятся к
числу весьма распространенных многотоннажных продуктов.
Они используются во многих отраслях промышленности и на¬
родного хозяйства. Из пресспорошков изготавливаются арми¬
рованные и неармированные детали электро- и радиотехни¬
ческих устройств, ненагруженные детали машин, изделия об¬
щетехнического назначения. Из волокнитов производятся эле¬
менты корпусов, шестерни, штурвалы, тормозные колодки.
Фаолит применяется как антикоррозионный материал для из¬
готовления химической аппаратуры. Текстолит и древесно¬
слоистые пластики используются в производстве деталей уз¬
лов трения, крупных конструкционных деталей (шкивы, зуб¬
чатые колеса). Стеклотекстолит применяется в машинострое¬
нии, судостроении и самолетостроении, пено- и сотофенопла-
сты — для изготовления строительных и декоративных эле¬
ментов. Растворы ФФП используются в качестве кислотоупор¬
ных клеев и лаков.404
Контрольные вопросы1. Какие ПМ называются пластическими массами (ПлМ)?2. По каким признакам классифицируются ПлМ?3. Какие наполнители могут входить в состав ПлМ и какова их роль?4. К какому типу ПлМ относятся полиэтилен? полистирол? фенол-
формальдегид ные полимерные материалы?5. Чем различаются полиэтилен низкого давления и полиэтилен вы¬
сокого давления по свойствам и методу получения?6. Какие методы используются в производстве полистирола?7. Чем различаются блочный и блочно-суспензионный способы про¬
изводства стирола?8. Как влияют условия синтеза (pH среды, соотношение компонен¬
тов) на строение и свойства получаемых фенолформальдёгй^ых
олигомеров?9. Какие из фенолформальдегидны?с полимеров являются термопла¬
стичными и какие термореактивными?10. Чем отличается механизм отверждения резолов от механизма от¬
верждения новолаков?11. Какие вещества называются сшивающими агентами (отвердите-
лями)?
Глава XIXПРОИЗВОДСТВО ХИМИЧЕСКИХ волокон19.1. Классификация и использование химических
волоконВолокнами называют материалы, частицы которых пред¬
ставляют гибкие и прочные тела с длиной многократно пре¬
вышающей размеры поперечного сечения, пригодные для из¬
готовления пряжи и текстильных изделий. По происхожде¬
нию волокна делятся на природные (натуральные) и химичес¬
кие.К природным волокнам относят:— волокна растительного происхождения, основу которых
составляет целлюлоза, формирующаяся в процессе развития
растений на поверхности семян (хлопок), в стеблях (лен, пень¬
ка, джут, кенаф, рами), в листьях (сизаль, манильская пенька)
и в оболочках плодов (койр из орехов кокоса);— волокна животного происхождения, состоящие из белка:
кератина (шерсть различных животных) и фиброина и серици-
на (выделения гусениц тутового и других шелкопрядов);— волокна минерального происхождения (асбест).Химическими волокнами (ХВ ) называются волокна, форму¬
емые из органических природных или синтетических полиме¬
ров. По природе исходного полимера они делятся на:— искусственные ХВ, получаемые в результате полимера-
налогичных превращений полимера, при которых он сохраня¬
ет свою полимерную природу:Полимер-1 —* Полимер-2 и
(сырье) (волокно)— синтетические ХВ, получаемые формованием из синте¬
тических полимеров:Мономер —* Полимер —► ВолокноХВ, в зависимости от вида сырья и используемой для их про¬
изводства реакции, подразделяются на несколько групп. На рис.
19.1 приведена классификация искусственных ХВ.Синтетические ХВ по природе полимеров, из которых они
формуются, делятся на гетероцепные и карбоцепные. Каждая
из этих групп, в свою очередь, состоит из большого числа от-406
Искусственные ХВI На основе целлюлозы IИз гидрат-
целлюлозыИз производных
целлюлозы©СЮ® 0Рис. 19.1. Классификация искусственных ХВ:1 — вискозное , 2 — медноаммиачное, 3 — омыленное ацетатное, 4 —
ацетатное (из ди- и триацетата целлюлозы), 5 — казеиновое, 6 — зеино-
вое, 7 — прочие белковыеСинтетические ХВНа основеНа основегетероцепных полимеровкарбоцепных полимеровПоли¬
амидныеПоли¬
эфирныеПоли¬урета¬новыеПоли-винил-галоге-нидныеПоли-винил-спирто-выеПоли-
акр ил-
нитрил ь-
ныеРис. 19.2. Классификация синтетических ХВ:1 — нейлон, 2 — капрон, 3 — сополимеры, 4 — лавсан, 5 — перлон, 6 —
спандекс (лайкра), 7 — политен, 8— моплен, 9— полифайбр, 10 —
хлорин, 11 — ацетохлорин, 12 — тефлон, 13 — винилон, 14 — нитрон,15 — сополимерыдельных представителей. На рис. 19.2 приведена классифика¬
ция синтетических ХВ.Особую группу ХВ составляют искусственные волокна, фор¬
муемые из различных неорганических веществ: соединений
кремния, металлов и их соединений, углерода, бора. На рис.
19.3 приведена классификация неорганических химических
волокон.407
Рис. 19.3. Классификация неорганических ХВ:1 — стекловолокно, 2 — базальтовое, 3 — вольфрамовое, 4 — бериллие-
вое, 5 — молибденовое, 6 — стальное, 7 — оксидные (оксиды алюминия,
бериллия, циркония), 8 — из борида титана, 9 — карбидные (карбиды
бора, кремния), 10 — нитридное (нитрид кремния), 11 — боридное, 12 —
углеродноеХимическое волокно, независимо от природы и способа по¬
лучения, представляет одиночное (элементарное) волокно, ко¬
торое образуется из струйки растворенного или расплавленно¬
го полимера, продавливаемой через отверстие фильеры — ме¬
таллического или стеклянного цилиндра с отверстиями в дне.
Число отверстий в фильере определяет количество элементар¬
ных волокон, а их диаметр — толщину образующегося волок¬
на.Толщина волокна характеризуется показателями: текс, но¬
мер волокна и денье. Текс — масса 1000 м волокна, выражен¬
ная в граммах. Номер волокна — число метров волокна в навес¬
ке массой 1 г. Денье — масса 9000 м волокна, выраженная в
граммах. Чем тоньше волокно, тем выше его номер и меньше
текс и денье.В промышленности химических волокон продукция выпус¬
кается в виде:— мононитей, представляющих одиночное волокно (моно¬
волокно) большой длины и сравнительно низкого номера;— комплексных (филаментных) нитей, состоящих из двух
и более одиночных волокон, соединенных между собой скручи¬
ванием или склеиванием;штапельных волокон из скрученных (методом прядения пос¬
ле формования) коротких отрезков тонких одиночных волокон.Мононити применяются в производстве текстильных изделий
(трикотаж) и изделий технического назначения (автомобильный
корд, рыболовные снасти, транспортерные ленты и др.).408
Комплексные нити, в зависимости от назначения, делятся
на:— текстильные, выпускаемые в виде тонких крученых ни¬
тей для изготовления изделий народного потребления;— технические, представляющие толстые нити повышенной
прочности, применяемые в производстве резинотехнических
изделий и корда.Штапельное волокно перерабатывается в пряжу и использу¬
ется исключительно для текстильных целей.В целом, около 70% всех производимых ХВ потребляется
текстильной промышленностью, производящей предметы на¬
родного потребления.Производство ХВ развивается быстрымй темпами. Это объяс¬
няется как высокими и разнообразными качествами их, так и
экономическими причинами. Для производства и применения
ХВ характерны:— простота и независимость от климатических условий про¬
цессов переработки волокна;— упрощенная технология отделки;— высокая производительность оборудования вследствие
малого числа обрывов нити;— относительно меньшая масса готовых изделий; и более
длительный срок их использования;— практически неограниченная сырьевая база.Все это определило внедрение ХВ в различные отрасли про¬
мышленности и народного хозяйства в целом, и замену ими
натуральных волокон. В табл. 19.1 представлена динамика раз¬
вития производства ХВ.Таблица 19.1. Производство волокон, млн. тВолокноГод19501960197019801988Природные волокна7,711,513,020,416—18Химические волокна1.73,38,314,824—26из них:искусственные1,62,63,43,2—синтетические0,10,74,911,6 Из табл. 19.1 следует, что опережающими темпами развива¬
ется производство синтетических волокон. Так уже в 1975 году
доля их в общем объеме мирового производства ХВ составляла
71,3%, доля вискозного волокна — 25,5% и ацетатного — 3,2%409
При этом, из всех произведенных синтетических волокон 30,6%
составляли полиамидные, 49,0% — полиэфирные и 19,5% —
полиакрилонитрильные волокна. В нашей стране в 1987 г. было
произведено 1,517 млн. т химических волокон, из них синте¬
тических 0,904 млн. т и искусственных 0,613 млн. т
*19.2. Общие принципы получения химических волоконХВ текстильного назначения характеризуются:— очень большим отношением длины к диаметру (более
10000),— высокой прочностью до 1 кПа/мм2,— большим относительным удлинением (более 5%)— эластичностью и быстрым исчезновением деформаций,
возникающих под воздействием нагрузки,— минимальными остаточными деформациями после снятия
нагрузки,— высокой устойчивостью к многократным и знакоперемен¬
ным нагрузкам.Для производства ХВ могут использоваться только полиме¬
ры, состоящие из гибких макромолекул линейной или слабо
разветвленной формы, с высокой молекулярной когезией, обес¬
печивающей прочное сцепление макромолекул под воздействи¬
ем сил межмолекулярного притяжения, молекулярной массой
в пределах 10*103—105 и достаточно узким молекулярно-мас-
совым распределением.Такие волокнообразующие полимеры должны также удов¬
летворять следующим требованиям:— плавиться и переходить в вязкотекучее состояние без раз¬
ложения, или растворяться в растворителях с образованием
концентрированных растворов;— образовывать тонкие жидкие и без нарушения сплошнос¬
ти нити при продавливании расплавов или растворов через фи¬
льеры;— приобретать высокоориентированную структуру при рас¬
тяжении;— кристаллизоваться или образовывать упорядоченные об¬
ласти макромолекул;— не содержать примесей мономеров и инородных включе¬
ний.Производство химических волокон состоит из следующих
стадий:410
1. Приготовление прядильной массы.2. Формование волокна.3. Отделка сформованного волокна.Выбор конкретных способов производства ХВ зависит от
свойств сырья (полимера) и требований к получаемому волок¬
ну.Прядильная масса готовится в виде расплава или высококон¬
центрированного (7—25%) раствора полимера в метаноле, эта¬
ноле, ацетоне, диметилформамиде. Полученный расплав или
раствор фильтруется для удаления твердых примесей, которые
могут забить отверстия фильер, и вакуумируется для удаления
газовых пузырьков, нарушающих/сплошность формуемой
струи. В прядильную массу вводятся добавки: термо- и светос-
табилизаторы, красители, матирующие волокно вещества и др.Формование волокна (прядение) осуществляется на прядиль¬
ных машинах и заключается в продавливании прядильной мас¬
сы через мелкие отверстия в фильере в среду, в которой струй¬
ка полимера затвердевает и образует тонкое волокно. В зависи¬
мости от природы прядильной массы прядение возможно вести
из расплава или из раствора.При прядении из расплава такой средой является холодный
воздух, при обдувании которым струйки полимера он затверде¬
вает. Этим методом формуются волокна из полиамидов, поли¬
эфиров и полиолефинов.При прядении полимера из раствора в летучем растворителе
волокно образуется в результате испарения растворителя в го¬
рячем воздухе («сухое» прядение). При прядении полимера из
раствора в нелетучем растворителе формование волокна проис¬
ходит в осадительной ванне, содержащей реагенты, регенери¬
рующие полимер в виде волокна из раствора («мокрое» пряде¬
ние).В процессе прядения волокна могут подвергаться скручива¬
нию с образованием нитей. Скорость прядения зависит от мето¬
да прядения, толщины и назначения волокна. При прядении
из расплавов она составляет от 10 до 20 м/с; при прядении из
растворов скорость прядения значительно ниже и равна 5—10 м/с при «сухом» методе и всего 0,5—2,0 м/с при «мокром»
методе прядения.Процесс формования волокна может быть двухванным и од¬
нованным. При двухванном процессе физико-химические и
химические процессы протекает раздельно.В первой ванне по¬
лимер высаживается из раствора (коагулирует), во второй — из¬
меняется химический состав полимера. При однованном мето-411
| РастворительПОЛИМЕР —Растворение до
концентрации 7—25%Расплавление приТвердыепримесиТ> 7\тФильтрование
вакуумирование
±| ГазыКрасители
¥ СтабилизаторыПродавливание прядильной
массы через фильеру пря¬
дильной машины в стабили¬
зирующую средуОсадитель¬
ная ваннаПрядение из
раствора{Мокрое —Холодный
воздух |?Прядение
из расплаваСухое
♦РастворительВытягивание"►Отделка^И ВОЛОКНОРис. 19.4. Общая схема производства химических волоконде физико-химические и химические процессы протекают од¬
новременно в одной ванне.Сформованное волокно подвергается операции вытягивания
в пластичном состоянии (ориентированию) для увеличения ме¬
ханической прочности и снижения относительного удлинения,
после чего наматывается на бобины или катушки.Отделка волокна включает операции промывки для удале¬
ния остатков мономера и растворителя, кислот и солей, увлека¬
емых волокном из ванны в процессе формования, сушки, замас¬
ливания для устранения электризации, окраски, а в некоторых
случаях — тепловой обработки в растянутом состоянии с целью
снижения последующей усадки и стабилизации формы пряжи.
На рис. 19.4 представлена общая схема производства ХВ.79.3. Производство вискозного волокнаВискозное волокно представляет искусственное химическое
волокно из гидратцеллюлозы, то есть одной из структурных мо¬
дификаций целлюлозы (СбНюОб)^, которая регенерируется в
процессе формования волокна из раствора. Гидратцеллюлоза412
отличается от природной целлюлозы повышенной гигроскопич¬
ностью, сорбционными свойствами и большей способностью к
гидролизу, этерификации и окислению. Средняя степень поли¬
меризации гидратцеллюлозы в вискозном волокне колеблется
от 300 до 600, что соответствует молекулярной массе 49000—
98000. При формовании вискозного волокна в нем образуются
надмолекулярные структуры, тип которых зависит от условий
формования (характеристик вискозной прядильной массы, со-
става осадительной ванны и др.). Физико-механические свой¬
ства вискозных волокон (ВВ) в значительной степени опреде¬
ляются структурой их наружной оболочки, в которой гидрат-
целлюлоза содержит значительноегашичество поперечных свя¬
зей, что придает волокнам повышенную прочность. Плотность
ВВ составляет около 1,5 т/м3.ВВ не термопластичны и могут кратковременно использо¬
ваться без снижения механических свойств при температуре
100—120С. Устойчивы к действию воды и неполярных орга¬
нических растворителей (бензин, бензол), в которых не набу¬
хают. При действии концентрированных минеральных кислот
при нормальной температуре и разбавленных кислот при нагре¬
вании, а также щелочей в присутствии кислорода воздуха под¬
вергаются деструкции. Сильно набухают в разбавленных ра¬
створах щелочей и растворяются в медноаммиачном растворе.
ВВ неустойчивы к действию микроорганизмов, которые вызы¬
вают их деструкцию.В зависимости от назначения ВВ производятся в виде непре¬
рывных нитей (текстильных и особо прочных кордных) или
штапельного волокна различного типа: обычной прочности, вы¬
сокопрочного, извитого и полинозного (хлопкоподобного). Осо¬
бую группу составляют модифицированные ВВ специального
назначения: повышенной хемостойкости, ионообменные, бак¬
терицидные, кровеостанавливающие и др., а также вискозная
пленка.ВВ имеют хороший внешний вид, легко окрашиваются, об-
ладают лучшими по сравнению с синтетическими волокнами
гигиеническими качествами, отличаются достаточно высоки¬
ми прочностными и усталостными характеристиками, относи¬
тельно дешевы. Вследствие этого ВВ широко используются для
производства текстильных тканей народного потребления и
широкого ассортимента технических изделий. Вискозная плен¬
ка (целлофан) обладает высокой паро- и влагопроницаемостью,
устойчива к действию жиров и масел, вследствие чего исполь¬
зуется в качестве упаковочного материала.413
Производство ВВ состоит из двух последовательных стадий:
получение прядильной массы — вискозы и формование волок¬
на. В качестве сырья используется древесная целлюлоза, содер¬
жащая 95—99% высокомолекулярной волокнообразующей
фракции со степенью полимеризации 800—1100.1. Получение вискозыВискозой (от латинского uiscosus — клейкий) называется
концентрированный раствор Na-соли ксантогената целлюлозы
в разбавленном растворе гидроксида натрия. Получение виско¬
зы включает следующие операции.а) Обработка целлюлозы 20%-ным раствором гидроксида
натрия Смерсеризация) в течение 5—115 минут при температу¬
ре 45—60° С. При этом образуется аддитивное соединение цел¬
люлозы со щелочью (щелочная целлюлоза)[С6Н702(0Н)3]п + пхШОЯ ^[СбН702(0Н)э^ (OH NaOH)*]* (а)и алкоголяты целлюлозы[С6Н702(0Н)з]л + ^Na0H^tC6H702(0H)3-je(0Na)J + пхН20 (б)Одновременно с реакциями (а) и (б) при мерсеризации про¬
исходит набухание целлюлозы и растворение гемицеллюлоз,
что способствует диффузии этерифицирующего агента внутрь
волокна при последующем ксантогенировании щелочной цел¬
люлозы.б) Отжим суспензии для удаления избытка раствора гидро¬
ксида натрия на отжимном прессе до степени отжима (отноше¬
ние масс отжатой щелочной целлюлозы и суспензии) 0,33—0,36.в) Измельчение отжатой щелочной целлюлозы.г) Окислительная деструкция (предсозревание) щелочной
целлюлозы за счет окисления ее кислородом воздуха на транс¬
портере или в специальных аппаратах в течение 1,5—2 часов
при температуре 50—60 С. В процессе предсозревания степень
полимеризации целлюлозы снижается до 400—600.д) Ксантогенирование щелочной целлюлозы обработкой ее
сероуглеродом в количестве 30—50% от массы целлюлозы:[СбН702(0Н)з-^(0Н * NаОН)*]„ + nxCS2 ^^ [С6Н702(0Н)з-*(0С ^ )х]п + п*н20 (в)xSNaОбразовавшийся ксантогенат целлюлозы — Na-соль целлю-
лозоксантогеновой кислоты представляет твердое вещество
оранжевого цвета. Операция ксантогенирования проводится в414
аппаратах — ксантогенаторах в течение 2,5—3 часов при тем¬
пературе 20—30°С. Вследствие токсичности и взрывоопаснос¬
ти сероуглерода ксантогенаторы герметизируются, а процесс
ведется при разряжении и в атмосфере азота.е) Растворение ксантогената в разбавленном растворе гид¬
роксида натрия в течение 1,5—5 часов при температуре 10°С с
образованием вискозы — вязкой прозрачной жидкости оранже¬
вого цвета состава: целлюлоза 6—10%, гидроксид натрия 5—
7%, вода 80—85%.Перед подачей на стадию формования волокна вискоза под¬
вергается фильтрованию и дегазации. Производительность ус¬
тановки по производству вискозысоставляет 25—50 т/сут. Опи¬
санный многостадийный способ производства является длитель¬
ным, связанным с необходимостью использования большого
числа аппаратов различной конструкции и, следовательно, с
высокими капитальными затратами. В настоящее время в про¬
мышленность ВВ внедрен способ производства прядильной мас¬
сы в одном аппарате (BA-аппарат), в котором осуществляются
все стадии получения вискозы. Здесь отсутствует операция от¬
жима щелочной целлюлозы после мерсеризации, что позволя¬
ет использовать щелочь в строго расчетных количествах. Про¬
должительность получения вискозы в BA-аппарате составляет
6—8 часов. Одновременно, на 25% снижаются капитальные
затраты и на 40% затраты труда.2. Формование волокнаВискозное волокно формуется способом мокрого прядения в
осадительную ванну кислого состава. При формовании волок¬
на в прядильной вискозной массе протекают следующие про¬
цессы.а) Высаживание (коагуляция) ксантогената целлюлозы из
раствора в виде пучка тонких параллельных волокон при нейт¬
рализации растворителя ксантогената — гидроксида натрия:2NaOH + H2S04 - Na2S04 + 2Н20 (г)б) Разложение (гидролиз) ксантогената целлюлозы под воз¬
действием компонентов осадительной ванны и регенерация из
него волокон гидрат-целлюлозы:[С6Н702(0Н)з-л(0С ' ) J„ + n*H2S04 - (д)^SNa— [С6Н702(0Н)з]л + пх CS2 + n*NaHS04в) Частичная дегидратация свежесформованных волокон.415
В состав осадительной ванны входят, помимо серной кисло¬
ты, сульфаты натрия и цинка, которые снижают набухание фор¬
муемого волокна и замедляют скорость разложения ксантоге-
ната целлюлозы, способствуя улучшению структуры волокна.Сформованное ВВ подвергается вытягиванию и поступает на
обычные операции отделки. Скорость формования ВВ зависит
от их толщины и назначения и колеблется от 20 до 100 м/мин.
На рис. 19.5 представлена технологическая схема стадии фор¬
мования ВВ из прядильной массы по однованному способу.Отфильтрованный и дегазированный прядильный раствор
(вискоза) из общего вискозопровода 1 забирается зубчатым пря¬
дильным насосом 2, продавливается через свечевой фильтр 3
для окончательной очистки от примесей и по трубке-червяку 4Рис. 19.5. Технологическая схема формования вискозного волокна:1 — вискозопровод, 2 — прядильный насос, 3 — свечевой фильтр, 4 —
червяк, 5 — осадительная ванна, 6 — фильера, 7 — прядильный диск, 8 —
воронка, 9 — сформованное волокно (нить), 10 — центрифугальная
кружка, /1 — электродвигатель (электроверетено) центрифуги416
подается в фильеру 6, погруженную в осадительную ванну 5.
Выходящие из отверстий фильеры струйки вискозы разлагают¬
ся в ванне, образовавшиеся волокна подхватываются прядиль¬
ным диском 7 и через воронку 8, служащую нитеводителем по¬
ступают в центрифугальную кружку 10, вращающуюся со ско¬
ростью 6000—9000 об./мин. Центробежной силой нить отбра¬
сывается к внутренней стенке кружки и образует так называе¬
мый «кулич». На участке от прядильного диска до нижнего
края воронки происходит скручивание элементарных волокон
и образование комплексной нити. По окончании намотки «ку¬
лич» снимается и поступает бработку.19.4. Производство капронового волокнаКапроновое волокно — это синтетическое гетероцепное во¬
локно, сформованное из поли-е-капроамид а (капрона). Оно от¬
носится к группе полиамидных ХВ. Известно под торговыми
названиями капрон, П-6 (РФ), перлон (ФРГ), силон (Чехия),
нейлон-6 (США), амилан (Япония).Поли-е-капроамид [-NH(CH2)5_CO-]n представляет твердое
рогоподобное вещество белого цвета с температурой размягчения
210°С, температурой хрупкости -25°С и плотностью 1,13 т/м3.
Молекулярная масса капрона зависит от условий получения
полимера и лежит в пределах 104—3,5е 104. Степень кристал¬
личности составляет около 0,6.Для капрона характерны высокие химическая и износостой¬
кость, он не деформируется при повышенных температурах,
устойчив к действию большинства растворителей, хорошо ок¬
рашивается. Безвреден и индифферентен по отношению к жи¬
вотным тканям, ферментам и бактериям, вследствие чего не
рассасывается в организме.Поли-е-капроамид используется преимущественно для про¬
изводства капронового волокна, применяемого в текстильной
промышленности, и для изготовления технических тканей.
Помимо этого, из капрона изготавливаются детали машин (зуб¬
чатые колеса, подшипники, крепежные детали) и электроизо¬
ляция. Он перерабатывается прессованием, экструзией, лить¬
ем под давлением. Для производства волокна используется пря¬
дение из расплава.Как правило, поли-£-капроамид производится непосред¬
ственно на заводах синтетического волокна. Поэтому, подобное
производство представляет единый технологический цикл,включающии следующие операции:14 Химическая технология. Том 2417
— подготовка сырья (плавление мономера, приготовление
водного раствора катализатора);— полимеризация капролактама;— обработка полимера (охлаждение и получение полимер¬
ной ленты, ее дробление и сушка);— получение расплава поли-е-капроамида и формование во¬
локна.Полимеризация г-капролактама протекает в расплаве по сту¬
пенчатому механизму через стадию гидролитического раскры¬
тия лактамного цикла под воздействием воды, кислот и раство¬
ров солей:сн2-сн2-сочNH + Н20 — H2N-(CH2)5-COOH (а)СН2-СН2-СЙ2и далее:^СОH2N-(CH2)3-COOH + (n-1) (СН2)5 | - H-[-HN-(CH2)5-CO-]n-OH (б)^NHНаиболее медленной стадией, лимитирующей скорость об¬
разования полимера, является реакция гидролиза лактамного
цикла (а). Для ускорения полимеризации в мономер вводится
катализатор — соль АГ, представляющая продукт взаимодей¬
ствия эквимолекулярных количеств адипиновой кислоты и гек-
саметилендиамина H2N-(CH2)6-NH2*HOOC-(CH2)4-COOH.Чтобы избежать окисления расплавленного поли-г-капроа-
мида процесс ведется в атмосфере азота, подаваемого в аппара¬
туру на стадиях расплавления и полимеризации мономера.Реакция полимеризации (б) обратима, поэтому конверсия
мономера не достигает 100% и в полимере содержится до 10%
мономера и водорастворимых олигомеров, которые удаляются
промывкой продукта горячей водой или вакуумированием.Получение капронового волокна может производиться по
периодической (в автоклавах под давлением) или по непрерыв¬
ной (в реакторах колонного типа при атмосферном давлении)
схеме. На рис.19.6 представлена технологическая схема про¬
изводства капронового волокна непрерывным способом.Твердый капролактам из бункера 1 поступает в плавитель 2,
обогреваемый паром. Расплавленный мономер проходит фильтр
3 и подается в верхнюю часть реактора полимеризации колон¬
ного типа 4 у в который одновременно из аппарата 5 дозируется
50% -ный водный раствор соли АГ. Смесь паров воды и не всту¬
пившего в реакцию капролактама из реактора поступает в хо-418
Рис. 19.6. Технологическая схема производства капронового
волокна непрерывным способом:1 — бункер капролактама, 2 — плавитель капролактама, 3 — фильтр, 4 —
реактор полимеризации, 5 — аппарат для приготовления раствора катали¬
затора, 6 — холодильник-конденсатор, 7 — сборник воды, 8 — охлажда¬
ющий барабан, 9 — крошкообразователь, 10 — экстрактор, 11 — ба¬
рабанная сушилка, 12 — прядильная машина: а — плавильная камера, б —
прядильный насосик, в — фильтр из кварцевого песка, г — фильера, д —
охладительная шахта; 13 — намоточная машиналодильник-конденсатор б, в котором капролактам конденсиру¬
ется и стекает обратно в реактор, а вода собирается в сборнике7. Для предотвращения окисления продуктов в плавитель 2 и
реактор полимеризации 4 вводится азот. Расплавленный поли-
капроамид выдавливается из реактора через щелевидную фи¬
льеру и поступает на охлаждаемый водой барабан 8. Образовав¬
шаяся лента полимера подается в резательный станок 9, где
измельчается в крошку. Из станка крошка поступает в экстрак¬
тор 10, в котором из полимера вымываются водорастворимые
мономер и олигомеры. Промытый поликапроамид высушива¬
ется в сушилке 11 теплым воздухом и подается непосредствен¬
но на формование волокна в прядильную машину 12, или по¬
ступает на склад. Поступившая в прядильную машину крошка
плавится в плавильной камере а, обогреваемой через змеевик,14419
а образовавшийся расплав прядильным насосом-дозатором б
при температуре 250—290 С под давлением 2—6 МПа подается
через фильтр в фильеру г. Выходящие из фильеры струйки рас¬
плавленного полимера охлаждаются в шахте машины д холод¬
ным воздухом и образуют волокна, которые через направляю¬
щие ролики подаются на намоточную машину и затем на даль¬
нейшую обработку. Скорость формования капронового волок¬
на зависит от его толщины и составляет 500—1200 м/мин.19.5- Производство лавсанового волокнаЛавсановое волокно — это синтетическое гетероцепное во¬
локно, сформованное из полиэтилентерефталата. Оно относит¬
ся к полиэфирным химическим волокнам. Известно под торго¬
выми названиями лавсан (РФ), дакрон (США), терилен (Анг¬
лия), эстер (Франция), монтивел (Италия).Полиэтилентерефталат представляет твердое вещество бело¬
го цвета с температурой размягчения 245 С. Плотность поли¬
этилентерефталата (ПЭТФ) зависит
СОО-1 от степени кристаллизации полиме-I ра и равна 1,33 т/м3 для аморфного,
1,40 т/м3 аморфно-кристаллического
(степень кристалличности 0,45) и 1,5 т/м3 кристаллического
(степень кристалличности более 0,65) продукта. Молекулярная
масса полимера, используемого для производства волокна ко¬
леблется от 20000 до 30000.ПЭТФ не растворим в воде и в большинстве органических
растворителей, на растворяется в фенолах, хлороформе, дифе-
нилоксиде и концентрированной серной кислоте. Устойчив к
действию фосфорной, фтористоводородной и органических кис¬
лот. Имеет высокую механическую прочность (в 10 раз превы¬
шающую прочность полиэтилена), хорошие диэлектрические
свойства в интервале температур от -20 С до +80С, влагоустой¬
чив. Термически стоек до температуры 280С. Температура
хрупкости ПЭТФ лежит ниже -50 С.ПЭТФ применяется преимущественно для производства во¬
локна, на что расходуется до 90% всего производимого поли¬
мера. Применяется также для изготовления пленок, исполь¬
зуемых для остекления и электроизоляции, светокопироваль¬
ных материалов, клейких лент и лент для машин. Пленки из
ПЭТФ вырабатываются методом экструзии из расплава с пос¬
ледующей плоскостной двухосной ориентацией и кристалли¬
зацией.Е-СН2-СН2-00С420
Получение лавсанового волокна, как и в случае получения
полиамидных волокон (19.4), представляет единый производ¬
ственный цикл, включающий стадии производства мономера —
ди(Р~оксиэтил)терефталата, его поликонденсации и формова¬
ния волокна. Технологический процесс первой стадии этого
процесса описан в главе XVI.ПЭТФ получается гомополиконденсацией диф-оксиэтил)
терефталата (диэтиленгликольтерефталата, ДЭГТ) по реакции:п Н0-СН2-СН2-ШС-<(0>-С00-СН2-СН20Н 3*
H(H:H2-cH2-[-<xx;-<^o)-coc>-CH2-CH2-]n ;-оос-<(о)>-соо-сн2-сн2-он ++ (п-1) НОСН2-СН2-ОН (в)Реакция поликонденсации ДЭГТ катализируется оксидами
сурьмы (III), кобальта (III) и германия (IV), вводимых в коли¬
честве 0,02—0,04 % от массы мономера. Так как она обратима
(равновесная конденсация), то для получения полиэфира с дос¬
таточно высокой молекулярной массой, выделяющийся низко¬
молекулярный продукт (этиленгликоль) должен непрерывно
отгоняться. Для этого процесс поликонденсации проводится
при высокой температуре (280°С) и вакууме не менее 1,33 кПа.
В этих условиях процесс поликонденсации завершается через
6—8 часов.Последующие операции переработки полученного ПЭТФ в
лавсановое волокно не отличаются от описанных выше опера¬
ций переработки поликапроамида в капроновое волокно пря¬
дением из расплава (19.4).Лавсановое волокно может производиться как периодичес¬
ким, так и непрерывным способом. К достоинствам непрерыв¬
ного метода следует отнести отсутствие отдельных операций
формования и сушки полимерной крошки. Это упрощает кон¬
струкцию прядильной машины, облегчает автоматизацию тех¬
нологического процесса и позволяет получать более однородный
по качеству продукт. На рис. 19.7 представлена технологичес¬
кая схема узла полимеризации ДЭГТ и формования лавсаново¬
го волокна из ПЭТФ.Расплавленный ДЭГТ из узла переэтерификации (16.2.3)
поступает в реактор предварительной поликонденсации 1, в
котором при температуре 270°С и остаточном давлении 4 кПа
образуется низкоплавкая смесь олигомеров со степенью поли¬
конденсации 4—6 и отгоняется выделяющийся этиленгликоль.
Полученный продукт проходит последовательно батарею реак-421
К в/насосуДифенил
1дэгтЭгиленгликольКтДифенил17Т-нннь-IIIуНа переработку
в крошку\ ж ии/ иV1JВолокноI\ШкРис. 19.7. Технологическая схема поликонденсации ДЭГТ
и формования лавсанового волокна:1 — реактор предварительной поликонденсации, 2 — реакторы поликон¬
денсации, 3 — обогреваемый трубопровод, 4— прядильная машина, 5 —
намоточная машинаторов поликонденсации 2, в последнем из которых при темпе¬
ратуре 280С и остаточном давлении 1 мм рт.ст. удаляется оста¬
ток этиленгликоля и завершается процесс поликонденсации.
Образовавшийся расплавленный ПЭТФ по обогреваемому тру¬
бопроводу 3 поступает в прядильную машину 4, из которой
сформованное волокно подается на намоточную машину 5.Производительность установки по производству лавсаново¬
го волокна непрерывным способом составляет 20000 т/год, в то
время, как при периодическом способе производства она не пре¬
вышает 3000 т/год.Мировое производство ПЭТФ составляет свыше 9,5 млн.т.Контрольные вопросы1. На какие группы делятся химические волокна? Приведите при¬
меры.2. Какие требования предъявляются к полимерам, используемым в
производстве химических волокон?3. Из каких основных операций состоит процесс получения химичес¬
ких волокон?4. К какой группе волокон относится вискозное волокно и почему?5. На чем основан процесс превращения целлюлозы в прядильный
раствор (вискозу)?6. Какой метод формования волокна используется в производстве вис¬
козного волокна?422
7. К какой группе волокон относятся капроновое и лавсановое волок¬
на?8. Перечислите основные стадии в процессе превращения капролак¬
тама в капроновое волокно?9. Почему процесс формования капронового волокна ведется в атмос¬
фере инертного газа?10. Из каких стадий состоит процесс производства лавсанового волок¬
на, исходя из ДЭГТ?11. Какой метод формования волокна используется в производстве
капронового и ла ового волокон?
Глава XXПРОИЗВОДСТВО ЭЛАСТОМЕРОВ20.1. Свойства и класификация эластомеровЭластомерами (эластиками) называют полимеры и матери¬
алы на их основе, обладающие высокоэластическими свойства¬
ми во всем диапазоне температур их эксплуатации. Они способ¬
ны к весьма значительным (до тысячи и более процентов) обра¬
тимым деформациям при малых (98 кПа — 9,8 МПа) значени¬
ях напряжений, вызывающих эти деформации. В эластомерах
сочетаются механическая прочность и высокая эластичность,
столь необходимые для изделий, подвергающихся многократ¬
но повторяющимся знакопеременным нагрузкам (например,
автомобильные шины).К эластомерам относятся каучуки и резины. Термином «ка¬
учук» принято обозначать эластомер, состоящий из длинных
гибких макромолекул, которые могут перемещаться друг отно¬
сительно друга при повышении температуры или при действии
механических напряжений. Для каучуков характерно аморф¬
ное состояние, однако при охлаждении или при растяжении они
способны кристаллизоваться. Рабочим физическим состояни¬
ем каучуков является высокоэластическое состояние (17.2.1).
При этом, чем шире интервал эластичности АТ = ТТ - Тс, тем
обширнее температурная область, в которой каучуки могут ис¬
пользоваться в качестве эластомера.Вследствие относительной независимости и подвижности
линейных макромолекул каучуки, в отличие от химических
волокон и пластических масс, обладают способностью к тече¬
нию и необратимым деформациям, которую они сохраняют под
действием механических нагрузок и после охлаждения.Для устранения этого свойства, препятствующего их эксп¬
луатации, каучуки подвергают вулканизации, превращая их в
резины. Так как, при этом макромолекулы каучука не утрачи¬
вают полностью способности к высоким обратимым деформа¬
циям, то полученные вулканизацией резины также являются
эластомерами. Основная масса каучуков используется для из¬
готовления изделий именно в виде резин, полученных вулка¬
низацией твердых каучуков или латексов (водные дисперсии
каучуков).Каучуки по происхождению делятся на натуральный и син¬
тетические. Натуральный каучук представляет продукт рас-424
тительного происхождения, содержащийся в млечном соке (ла¬
тексе) каучуконосных растений. Промышленное значение име¬
ет латекс бразильской гевеи. Макромолекулы натурального
каучука построены из звеньев изопрена, из которых 98—99%
соединены в положении 1,4-цис, а остальные в положении 3,4:Средняя „ рная масса натурального каучука состав¬ляет от 7*104 до 2,5* 106. Он хорошо растворяется в ароматичес¬
ких углеводородах, хлороформе, четыреххлористом углероде,
не растворим в спиртах и ацетоне, стоек к действию воды, раз¬
бавленных кислот и щелочей. Плотность натурального каучу¬
ка равна 0,913 т/м3. Звенья натурального каучука содержат
двойные связи, поэтому он реагирует с кислородом и озоном,
галогенами, хлористым водородом и другими реагентами. При
нагревании выше 220 С и действии кислорода подвергается де¬
струкции.Технологический процесс производства натурального каучу¬
ка состоит из следующих стадий:— добыча латекса и введение в него антикоагулирующих
добавок (аммиак, формальдегид);— фильтрование латекса для отделения самопроизвольно
образовавшихся сгустков каучука;— разбавление латекса водой до концентрации 15—20% ;— коагуляция латекса уксусной или муравьиной кислотой
и выделение каучука;— вальцевание, промывка и сушка каучука.Натуральный каучук вулканизируется серой в присутствииускорителей — сернистых соединений. Полученные вулкани-
заты (резины) имеют хорошую эластичность, высокую износо-
и морозостойкость, обладают хорошими динамическими свой¬
ствами, но нестойки к действию масел и растворителей.Ассортимент синтетических каучуков весьма многообразен.
По применению они подразделяются на :— каучуки общего назначения, применяемые в массовом
производстве изделий, в которых реализуется основное свойство
эластомеров — их эластичность, и— каучуки специального назначения, применяемые для про¬
изводства изделий, которые наряду с эластичностью должны
обладать специальными свойствами — стойкостью к действию
определенных агентов, теплостойкостью, морозоустойчивостьюН3С Н Н3С Н4 / \ /,с=с 'с=с v„ L \ _ - / VН2С\ сн2-сн2 сн2-и др.425
Большинство синтетических каучуков (СК) относится к кар-
боцепным эластомерам, полученным из непредельных углево¬
дородов и их производных. Только некоторые из СК являются
гетероцепными эластомерами и содержат в макромолекуляр-
ной цепи атомы азота, кислорода, серы и кремния.В табл. 20.1 представлены строение свойства и область ис¬
пользования важнейших видов производимых промышленно¬
стью СК.Таблица 20.1. Промышленные синтетические каучукиНаименованиеХимический составСвойстваОбластии маркаи формулапримененияКаучуки общего назначенияБутадиеновыеПолибутадиен нерегу¬СКВ, СКДСРлярного строения(-СН2-СН=СН-СН2-)ЛБутадиеновыеСтереорегулярныйВысокая эластич¬Шины, транспор¬СКД, СКДЛ,полибутадиенность в сочета¬терные ленты,СКДЛПР(-СН2-СН=СН-СН2-)пнии с достаточно
высокой проч¬резиновая
обувь, наружнаяИзопреновыеСтереорегулярныйностью при растя¬оболочка кабе¬СКИполиизопренжении, высокоелей, резинотех¬(-сн2-9=сн-сн2-)псопротивлениенические изде¬СНзк истиранию,
достаточная ус¬лия (РТИ) быто¬
вого и медицинс¬Бутадиен-сти-Сополимеры бутади¬талостная вы¬кого назначения,рольные илиена со стиролом илиносливостьасботехническиеа-метилсти-а-метилстироломизделиярольные(-СН2-СН=СН-СН2-)п--(-СН2-СН-)тс6н5■Каучуки специального назначенияБутил-каучук [Сополимеры изобути- 1 Высокая газоне- |Автокамеры,БКлена с изопреном (3%)проницаемость,теплостойкие[-С(СН3)2-СН2-]П-стойкость к окис-РТИ, антикорро¬-[-СН2-9=СН-СН3-]тлению, тепло изионные покры¬СНзозоностойкостьтияЭтилен-пропи-Сополимеры этиленаСтойкость кАвтокамеры,леновые СКЭПс пропиленомокислению, хи¬теплостойкие[-СН2-СН2-]П -мическая стой¬РТИ, прорези¬-[-СН2-СН(СН3)-]ткость, хорошиедиэлектрическиесвойстваненные ткани,
губчатые рези¬
ны, электроизо-
ляция426
Наименование
и маркаХимический состав
и формулаСвойстваОбластипримененияХлоропреновые
(найрит)Бутадиен-нит-
рильные СКНПолисульфид-ныеКремнийорга-
нические СКТФторкаучукиСКФУретановыеСКУПолихлоропрен
(-СН2-С=СН-СН2-)П
CIСополимеры бутадие¬
на сркрилонитрилом
(-СН2-СН=СН-СН2-)П-
-('СН2-СН-)т
CNПродукты поликон¬
денсации дихлордиэти-
лового эфира с поли¬
сульфидом натрия
(-СН2-СН2-0-СН2-
-CH2-S-S-)nПолиорганосилоксаны(-SiR2-0-SiR2-0-)nСополимеры полнос¬
тью или частично фто¬
рированных олефинов
(-CF2-CFX-)n-
-(-CH2-CF2-)mПолиуретаны(-O-R-O-CO-NH--R-NH-CO-)nМасло-, бензо- и
озоностойкость f
атмосферостой-
костьМасло-, бензо-
и теплостойкостьМасло- и бензо-
стойкость, низ¬
кая влаго- и газо¬
проницаемость,
стойкость к УФТепло- и моро¬
зостойкость, хо¬
рошие диэлект¬
рические свойст¬
ва, озоно и све¬
тостойкость,
физиологичес¬
кая инертностьТеплостойкость,
химическая
инертность,
стойкость к
действию силь¬
ных окислителейИзносостой¬
кость, стойкость
к маслам и озо¬
нуМасло, бензо-
стойкие РТИ, об¬
кладка химичес¬
кой аппаратуры,
нефтеемкостейМаслостойкие
РТИ, резиновая
обувь, электро¬
проводящие
резиныРТИ специально¬
го назначения,
герметизирую¬
щие составыСпециальные
РТИ, изоляция
для электричес¬
ких машин, изде¬
лия медицинско¬
го назначенияРТИ, работаю¬
щие при высоких
температурах и
в агрессивных
средах, обклад¬
ка химической
аппаратурыШины, подошвы
для обуви, тру¬
щиеся детали
машин%Мировое производство СК, резин и латексов на их основе раз¬
вивается значительно более высокими темпами, чем производ¬
ство натурального каучука. Причинами этого являются:— широкий диапазон свойств, присущих каучукам специ¬
ального назначения и возможность их целенаправленного ис-427
пользования во многих областях техники и народного хозяй¬
ства,— успехи в разработке методов синтеза стереорегулярных
СК, успешно заменяющих натуральный каучук в производстве
изделий, которые должны обладать как прочностными так и
эластическими свойствами.В результате с 1940 по 1970 гг. доля СК в общем балансе эла¬
стомеров выросла с 3% до 62% и в настоящее время равняется
75% . Мировое производство СК составило в 1985 году 12 млн. т.
При этом в валовой продукции нефтехимической промышлен¬
ности доля СК и изделий из них равна 40% .Ассортимент СК насчитывает более 80 наименований и ма¬
рок. Наиболее крупнотоннажными СК, используемыми в про¬
изводстве подавляющего большинства резиновых изделий, яв¬
ляются каучуки общего назначения бутадиенстирольные и сте-
реорегулярные изопреновые и бутадиеновые. В табл. 20.2
представлена динамика производства СК в нашей стране.Таблица 20.2. Структура производства СК,%Тип СКГод19651975СКС и СКМС65,038,2СКБ18,85,1Найрит6,14,6СКИ3,228,1скд1,014,6СКН1,53,0Прочие0,92,5Всего100,0100,0Наиболее экономично производство синтетических каучуков
типа СКС и изделий из них по циклу: «нефтепереработка — эта¬
нол — синтетический каучук — шинное производство». В свя¬
зи с этим их получение целесообразно организовывать вблизи
спиртового производства. В РФ основные предприятия по про¬
изводству синтетических каучуков различного назначения рас¬
положены в Архангельске, Волгограде, Воронеже, Ефремове,
Орске, Омске, Перми, Самаре, Саратове, Стерлитамаке, Уфе,
Ярославле.Номенклатура резиновых изделий, изготавливаемых на ос¬
нове СК, насчитывает свыше 50000 наименований. Наиболее428
крупным потребителем СК является шинная промышленность,
в которой используется около половины общего потребления
СК.Основная масса производится методами полимеризации:
эмульсионной радикальной полимеризацией в присутствии
инициаторов и координационно-ионной полимеризацией в ра¬
створе в присутствии стереоспецифических катализаторов. При
этом к мономера^ предъявляют жесткие требования по содер¬
жанию примести, которые могут реагировать с катализатором
или макромолекулами каучука в процессе их роста: кислоро¬
да, воды, карбонил и серусодержащие соединения и др. Поэто¬
му, содержание мономера в исходном сырье не должно быть
менее 99,5%.С целью повышения пластичности каучуков и улучшения
их технологических свойств, а также для придания в после¬
дующем вулканизатам нужных технических свойств, в ка-
учуки общего назначения вводятся масла и сажа. Эти ком¬
поненты смешиваются с каучуками непосредственно в про¬
цессе получения последних. Масло- и саженаполненные СК
позволяют получить резиновые смеси с лучшей клейкостью
и вулканизаты с более высокой прочностью при растяже¬
нии.Ниже рассматривается производство двух основных видов СК
общего назначения бутадиен-стирольного СКС и изопренового
СКИ-3.20.2. Производство бутадиен-стирольного
каучука СКСБутадиен-стпирольные синтетические каучуки (СКС) пред¬
ставляют сополимеры бута¬
диена со стиролом и выпус- (-CH2-CH=CH-CH2-)n-(CH2-CH-)m
каются нескольких марок,различающихся соотноше- (OJнием звеньев мономеров вних. Так, например, СКС-30 содержит 30% стирола и 70% бу¬
тадиена, СКС-45, соответственно, 45% стирола и 55% бутадие¬
на. СКС производятся под торговыми названиями синпол, фи¬
лярен (США), нитолл (Англия), эуропрен (Италия), нипол (Япо¬
ния), бунатекс (ФРГ).СКС имеют нерегулярное строение. Около 80% звеньев бу¬
тадиена соединены в положении 1,4 и около 20% — в положе¬
нии 1,2:429
-СН2-СН=СН-СН2-СН2-СН=СН-СН2- ~сн2-сн~сн2-сн~I Iсн снII IIсн2 сн2Звенья стирола распределены в макромолекулах нерегуляр¬
но. СКС не кристаллизуются при хранении и при деформации.
Температура стеклования их зависит от доли звеньев стирола и
колеблется от -74°С до -13°С. Молекулярная масса СКС лежит
в пределах 150000—400000 и зависит от метода полимериза¬
ции. СКС имеют плотность от 0,9 до 0,98 т/м3, растворяются в
ароматических и алифатических углеводородах, достаточно
стойки к действию разбавленных кислот. Вулканизируются
серой и органическими сульфидами.СКС получается эмульсионной сополимеризацией бутадие¬
на и стирола/iCH2=CH-CH=CH2 + т СН2=СН — (~С4Нб-)п~(СН2-СН~)тI Iс6н5 с6н5по низкотемпературному или высокотемпературному методу.
Наиболее распространен метод низкотемпературной сополиме-
ризации, которым производится основная масса так называе¬
мого низкотемпературного каучука СКС-30. Получаемые сопо¬
лимеризацией в водной эмульсии при температуре 5—8°С эти
каучуки имеют более высокую молекулярную массу, хорошо
совмещаются с другими каучуками и обладают лучшими тех¬
нологическими свойствами по сравнению с высокотемператур¬
ными СКС.Процесс сополимеризации протекает по радикальному меха¬низму в присутствии инициирующей окислитель- снно-восстановительной системы. Она состоит из >—, i 3
инициатора — гидропероксидов изопропилбензо- |ла или гидропероксида «-мента- снз/—v | 3 на, обладающих сильными окис-
СН3~\Ц/~С-ООН лительными свойствами, и активатора-восста-
СН3 новителя, облегчающего распад гидроперокси¬
да. В качестве активатора используется комп¬
лексная соль железа (П) с трилоном Б (этилендиаминтетрааце-
тат натрия). При инициировании ион Fe+2 комплекса перехо¬
дит в ион Fe+3, способствуя радикализации инициатора:RO-OH — RO- + ОНFe+2 + ЮН — Fe+3 + ОН'430
CHgгде R = aIy-c-0*CH3В качестве регуляторов сополимеризации, ограничивающих
рост макромолекул и препятствующих образованию разветвлен¬
ных цепей, используются диксантогены состава RO-C-S-S-C—OR,\ S Sгде R=C2H5, СН(СНз)2 или меркаптаны, например, додецил-
меркаптан C12H25SH.Для остановки процесса сополимеризации в латекс на зак¬
лючительной стадии вводится стоппер — диметилдитиокарбо-
нат натрия (CHa^N-C-SNa или диэтилгидроксиламинS(C2H5)2NOH, а для повышения устойчивости латекса — ве¬
щества, препятствующие агломерации частиц (диспергато-
ры): даксад (Na-соль продукта конденсации формальдегида
с алкилнафталинсульфокислотой) и леканол (Na-соль про¬
дукта конденсации формальдегида с нафталинсульфокисло-
той).Производство СКС строится по непрерывной схеме и состоит
из двух последовательных стадий: получение латекса и выде¬
ление из него каучука.Получение латекса включает следующие операции.1. Приготовление углеводородной фазы смешением свежих
и возвратных бутадиена и стирола в отношении, определяемом
составом (маркой) каучука.2. Приготовление водной фазы растворением в умягченной
воде эмульгаторов — смеси Na-солей смоляных кислот кани¬
фоли и К-солей синтетических жирных кислот (Сю—Cie) с до¬
бавлением электролитов — фосфата натрия и хлорида калия
до рН = 10—11.3. Приготовление отдельных растворов инициатора, актива¬
тора, диспергатора, регулятора полимеризации, стоппера и ан¬
тиоксидантов.4. Сополимеризация мономеров.5. Дегазация латекса (отгонка непрореагировавших мономе¬
ров).На рис. 20.1 представлена технологическая схема пер¬
вой стадии производства СКС — получение латекса низко¬
температурной эмульсионной полимеризацией бутадиена и
стирола.431
СтиролРис. 20.1. Технологическая схема получения латекса СКС:1 — аппарат для приготовления водной фазы, 2 — аппарат для приготовле¬
ния углеводородной фазы, 3 — смеситель фаз, 4 — батарея полимериза¬
торов, 5 — фильтр, 6 — колонна предварительной дегазации (отгонки
бутадиена), 7, 9 — газоотделители (сепараторы), 8 — колонна дегазации
(отгонки стирола), 10 — емкость для латекса, 11 — система очистки
возвратного бутадиена, 12 — система ректификации возвратного стирола,13 — холодильникВодная фаза из аппарата 1 и углеводородная фаза из аппара¬
та 2 поступают в смеситель 3, где эмульгируются. Полученная
эмульсия охлаждается в холодильнике 13 до температуры 15°С
и подается в первый полимеризатор 4, батареи из 12 аппаратов.
Перед первым полимеризатором в эмульсию вводятся заранее
приготовленные растворы инициатора, активатора и регулято¬
ра полимеризации из емкостей, в которых они хранились в ат¬
мосфере азота. На выходе из последнего полимеризатора 4^ где
степень конверсии достигает 0,6 долей единиц, в латекс вводит¬
ся стоппер после чего он, пройдя фильтр 5 для отделения твер¬
дых частиц, направляется на дегазацию. В колонне 6 из латек¬
са удаляется бутадиен, который через сепаратор 7 и систему
очистки 11 возвращается на сополимеризацию. В колонне 8 от¬
гоняется стирол, также возвращаемый через сепаратор 9 и сис¬
тему ректификации 12 в цикл. Освобожденный от изомеров
латекс собирается в емкости 10 и после введения в него антиок¬
сидантов подается на вторую стадию производства — выделение
СКС из латекса.432
Основной аппарат на стадии получения латекса — полиме¬
ризатор представляет автоклав с мешалкой, объемом до 20 м3,
снабженный системой охлаждения в виде рубашки и змееви¬
ка, по которым циркулирует охлаждающий рассол.Выделение каучука из латекса включает следующие опера*
ции:1. Усреднение состава латекса путем смешения отдельных
партий его в общей емкости,2. Коагуляция латекса и формование крошки каучука.3. Сушка и упаковка каучукаКоагуляция латексов и выделение из него каучука СКС про¬
исходит под воздействием смеси 25% -ного раствора хлорида на¬
трия и 2%-ной серной кислоты. Этот коагулянт разрушает
эмульгатор на поверхности капель каучука и нарушает стабиль¬
ность коллоидной системы (эмульсии).На рис. 20.2 представлена технологическая схема второй ста¬
дии производства СКС — выделение каучука из латекса.Усредненный латекс из емкости 1 подается через фильтр 2 в
смеситель 3, куда вводится раствор хлорида натрия и подкис¬
ленный серной кислотой фильтрат из сборника 6 (серум) для
достижения заданной концентрации латекса. Из смесителя ла¬
текс последовательно проходит два аппарата коагуляции 4> в
нижнюю часть которых также вводится серум. Образовавшая¬
ся водная суспензия крошки каучука поступает на виброситоРис. 20.2. Технологическая схема выделения СКС из латекса:1 — усреднительная емкость, 2 — фильтр, 3 — смеситель латекса и коа¬
гулянта, 4 — аппараты коагуляции, 5 — вибросито, 6 — сборник серума,7 — промывной аппарат, 8 — барабанный вакуум-фильтр, 9 — дробилка,10— конвеерная сушилка433
5, на котором крошка отделяется от фильтрата, собираемого
в сборнике 6. Крошка промывается в аппарате 7 водой и в
виде пульпы подается на вакуум-фильтр 8. С барабана филь¬
тра каучук в виде пласта поступает в молотковую дробилку
9 и образовавшаяся крошка после сушки в сушилке 10 и ох¬
лаждения до 40°С подается транспортером на брикетирова¬
ние и упаковку.В последнее время расширяется производство бутадиен-сти-
рольных каучуков в растворе на литийорганических катализа¬
торах. Полученные каучуки, получившие название статисти¬
ческих (ДССК), имеют более регулярное строение и не содер¬
жат в макромолекулах ответвлений и микроблоков из звеньев
стирола. По эластичности и морозостойкости они превосходят
СКС.20.3. Производство изопренового каучука СКИ-3Изопреновый синтетический каучук СКИ-3 представляет поли¬
мер изопрена (2-.метш1-бутадиена-1,3) [-СН2“С(СНз)=СН-СН2-]л.
СКИ-3 имеет стереорегулярное строение. Макромолекулы его состо¬
ят преимущественно из звеньев структуры 1,4-цис с небольшим вклю¬
чением звеньев 1,4-транс и 3,4 и 1,2.Н СНЯI Iн3с н н3с сн2- -н2с-с- -сн2-с-
Y=r/ с=сг с-си,, снII 3 II-н2с сн2- -н2с н СН2 сн21,4-цис 4-транс 3,4 1,2СКИ-3 аморфен при комнатной температуре, но, подобно
натуральному каучуку, кристаллизуется при растяжении и при
температурах ниже 0°С. Молекулярная масса СКИ-3 зависит от
катализатора, применяемого для синтеза, и составляет от0,55’106 до 1-106. СКИ-3 имеет плотность 0,91—0,92 т/м3. Он
растворяется в ароматических углеводородах, хлороформе, че¬
тыреххлористом углероде, циклогексане, сероуглероде. Не ра¬
створим в спиртах и кетонах, стоек к действию воды, но легко
разрушается концентрированными кислотами и щелочами,
окисляется кислородом воздуха. Температура стеклования
СКИ-3 равна -70 С.В табл. 20.3 приведены некоторые характеристики СКИ-3 и
натурального каучука.434
Таблица 20.3. Характеристики каучуков СКИ-3 и НКПоказательСКИ-3НКСодержание звеньев,%1,4-цис96—9998—1001,4-транс0—401,20—203,41—30—2Непредел ьность, %94—9695—98Каучук СКИ-3 по своей структуре и основным свойствам бли¬
зок к натуральному каучуку. Поэтому он может использовать¬
ся вместо него для изготовления практически всех резиновых
изделий общего назначения как в чистом виде, так и в сочета¬
нии с другими СК. СКИ-3 вулканизируется серой в присутствии
ускорителей вулканизации, а также органическими перекися¬
ми.Изопреновый каучук СКИ-3 получается стереоспецифичес-
кой полимеризацией изопрена в растворе в присутствии комп¬
лексных координационных катализаторов Циглера-Натта
А1 (изо С4Нд)з -f ТЮЦ (в мольном отношении 1:1) или литийорга-
нических соединений состава RLi + ROLi + LF. В производстве
более предпочтительны комплексные катализаторы, позволяю¬
щие получить каучук с высоким содержанием звеньев 1,4-цис.Полимеризация протекает с выделением тепла и описывает¬
ся общим уравнениемлС5Н8 = (-С5Н8-)Л - АН АН = 75 кДж/мольПроцесс проводится в среде инертного растворителя — изо¬
пентана, который обеспечивает отвод реакционного тепла и
выделение полученного каучука и состоит из следующих ста¬
дий.1. Приготовление каталитического комплекса в виде суспен¬
зии тетрахлорида титана и триизобутилалюминия в толуоле в
атмосфере азота.2. Полимеризация изопрена в растворе изопентана при тем¬
пературе 20—40"С в атмосфере азота в течение 2—б часов.3. Дезактивация катализатора путем разрушения каталити¬
ческого комплекса действием метанола (стоппер) и стабилиза¬
ция полимеризата. В качестве стабилизатора используется ра¬
створ в метаноле фенил-р-нафтиламина (неозон) или N,N1-flH-
фенил-я-фенилендиамина (ДФФД).4. Промывка полимеризата водой для удаления метанола и
разрушенного катализаторного комплекса.435
5. Выделение каучука из полимеризата и удаление остатка
мономера горячей водой (водная дегазация).6. Обезвоживание и сушка пульпы каучука.На рис. 20.3 представлена технологическая схема производ¬
ства изопренового каучука СКИ-3. Раствор изопрена в изопен -
тане охлаждается в холодильнике 1, орошаемом жидким про¬
паном, и подается в смеситель 2, куда дозируется каталитичес¬
кий комплекс. Полученная смесь последовательно проходит
через батарею из шести полимеризаторов 3i—Зв, в последнем
из которых обеспечивается конверсия изопрена 0,95 долей еди¬
ниц при содержании полимера в растворе до 15%. Из полиме¬
ризатора 3 полимеризат поступает в смеситель 5, куда из аппа-НеозонРис. 20.3. Технологическая схема производства СКИ-3:1 — холодильник, 2 — смеситель, 3 — батарея полимеризаторов, 4 —
аппарат для приготовления раствора стабилизатора, 5 — смеситель, 6 —
сепаратор, 7 — отмывная колонна, 8 — смеситель, 9 — паровой инжек¬
тор, 10 — дегазатор, 11—концентратор, 12 — червячный пресс, 13 —
резательный станок, 14 — вибротранспортер436
рата 4 подается раствор стабилизатора в метаноле. Из смесите¬
ля 5 полимеризат проходит сепаратор 6, где от него отделяется
водный слой, и поступает в колонну 7, в которой окончательно
отмывается от продуктов разложения каталитического комп¬
лекса, после чего поступает на водную дегазацию. В смесителе
8 образуется водная эмульсия, которая через паровой инжек¬
тор 9 подается в дегазатор 10, Пары воды и мономера отводятся
из верхней части дегазатора, а пульпа, содержащая 5% каучу¬
ка, выходит из нижней части его и поступает в концентратор11, где из нее удаляется основная масса воды, после чего пода¬
ется в червячньщ сушильный пресс 12, Образовавшиеся ленты
каучука измельчаются на резательном станке 13 в крошку, ко¬
торая высушивается горячим воздухом на вибротранспортере
14 и поступает на брикетирование.20.4. Переработка каучука в резиновые изделияНатуральный и синтетические каучуки являются основным
компонентом при изготовлении резиновых, резино-тканных и
резино-металлических изделий, объединяемых общим назва¬
нием резино-технические изделия (РТИ) и используемых прак¬
тически во всех отраслях народного хозяйства.Ассортимент РТИ насчитывает десятки тысяч наименований
и типоразмеров. По назначению РТИ принято делить на следу¬
ющие группы.1. Передаточные элементы устройств для перемещения ма¬
териалов (транспортерные ленты, рукава, трубки).2. Гибкие связи передач (приводные ремни, ленты).3. Эластичные соединения узлов машин и аппаратов (муф¬
ты).4. Уплотнители соединений (сальники, кольца, мембраны,
прокладки).5. Амортизирующие детали (буфера, подвески, амортизато¬
ры, втулки).6. Пыле- и грязезащитные детали (чехлы, ковры, колпаки).7. Электроизоляционные изделия и детали (аккумуляторные
баки, пластины, трубки).8. Воздухо- и водоплавательные средства (оболочки аэроста¬
тов, надувные лодки, плоты, понтоны).9. Пневматические строительные конструкции (сборно-раз¬
борные сооружения и здания).10. Средства защиты (противогазы, респираторы, защитные
костюмы).437
11. Изделия бытового назначения, средства санитарии и ги¬
гиены, изделия медицинского назначения, обувь и элементы
для нее.РТИ изготавливаются по единой технологической схеме,
включающей следующие стадии:— приготовление сырой резиновой смеси,— изготовление полуфабрикатов (листов, профильного про¬
ката),— формование или сборка заготовки изделия из полуфабри¬
ката или резиновой смеси,— вулканизация изделий.1. Приготовление резиновой смесиВ состав резиновых смесей входят разнообразные компонен¬
ты, природа и соотношение которых зависят от назначения и
свойств получаемых РТИ. Эти компоненты могут быть разби¬
ты на три группы.Ингредиенты, облегчающие процесс переработки смеси в
изделия:— регенерат (продукт переработки старых РТИ),— пластификаторы (битум, канифоль, парафин, фталаты).Ингредиенты, придающие изделию необходимые свойства:а) наполнители:— активные наполнители или усилители (сажа, кремневая
кислота),— оксиды металлов,— каолин,— инертные наполнители (мел, тальк, сульфат бария).б) противоокислители или противостарители (фенил-р-на-
фтиламин);в) красители:— органические,— неорганические пигменты (оксиды титана, железа, хро¬
ма, ультрамарин).Вулканизирующие инградиенты:— вулканизаторы (сера и другие вещества, в зависимости от
природы вулканизируемого каучука),— ускорители вулканизации (дифенилгуанидин, альтакс,
каптакс),— активаторы вулканизации (оксид цинка, стеарин).Резиновые смеси обладают высокой вязкостью. Поэтому дляполучения однородной массы их готовят не обычным переме¬
шиванием компонентов, а растиранием и продавливанием че¬
рез узкие щели между валками, или через отверстия на специ¬438
альных вальцах или в закрытых резиносмесителях роторного
устройства.2. Изготовление полуфабрикатов и изделий.Для изготовления полуфабрикатов и формования изделий из
резиновых смесей используются разнообразные методы:— каландрование пропусканием смеси через нагретые вал¬
ки аппарата, называемого каландром;— шприцевание выдавливанием смеси через отверстия чер¬
вячной машинь^(й|)есса);— прессование;— штамповка;— прорезинивание ткани на клеепропиточной машине.Изделия сложного профиля собираются из предварительноизготовленных одним из перечисленных методов деталей с пос¬
ледующим склеиванием или подпрессовкой. Отдельные РТИ
получаются из латексов вспениваением (губчатые резины) или
окунанием в латекс форм с последующей сушкой (тонкостен¬
ные изделия).3. Вулканизация.Вулканизация является заключительной и обязательной
операцией в производстве РТИ. Она представляет технологичес¬
кий процесс превращения пластичных каучука или полимер¬
ной фазы сырой резиновой смеси и изделий из них в эластич¬
ный вулканизат — резину. В результате вулканизации проис¬
ходит фиксация формы изделия и оно приобретает необходи¬
мые свойства.Вулканизация каучуков — это частный случай сшивания
линейных полимеров, в процессе которого макромолекулы со¬
единяются поперечными химическими связями с образовани¬
ем пространственной трехмерной вулканизационной сетки. В
подобной структуре макромолекулы не способны к необрати¬
мому перемещению друг относительно друга (деформация
сдвига), вследствие чего резины, в отличие от каучука, теря¬
ют свойства текучести, сохраняя, однако, в широком диапа¬
зоне температур способность к высокоэластической деформа¬
ции.При вулканизации существенно изменяются механические
и физические свойства каучука: увеличиваются плотность,
твердость и механическая прочность, снижается остаточная
деформация, улучшаются динамические свойства (сопротивля¬
емость ударным нагрузкам), уменьшается набухаемость и кау-
чуки теряют способность к самопроизвольному растворению.
Одновременно изменяются влаго- и газопроницаемость, диэлек¬439
трические свойства, теплопровод¬
ность, температура стеклования и
другие свойства эластомера.При этом, изменение свойств вул-
канизата зависит от концентрации
образующихся при вулканизации
поперечных связей, так называемой
степени сшивания каучука или сте¬
пени вулканизации. Она зависит, в
свою очередь, от количества вводи¬
мого в смесь вулканизирующего
агента. Так, если при вулканизации
резиновых смесей, содержащих1,5—3,0% вулканизатора, образу¬
ются мягкие резины, то при содер¬
жании 25—35% — твердый эбонит.
На рис. 20.4 представлена зависи¬
мость эластичности вулканизата от степени вулканизации, Хв.Природа сшивающего агента (вулканизатора) и, следователь¬
но, способ вулканизации зависит от природы каучука. Каучу-
ки, содержащие в молекуле двойные связи (НК, СКС, СКИ,
СКД) вулканизируются серой при 140—160°С (серная или го¬
рячая вулканизация) или, реже, хлористой серой S2CI2 без на¬
гревания (холодная вулканизация). Серные вулканизаты не
обладают достаточно высокой термической и химической стой¬
костью, поэтому, эти каучуки вулканизируют также перокси¬
дами, хинонами, азо- и диазосоединениями, феноло-формаль-
дегидными олигомерами. СК, содержащие функциональные
группы (карбоксилатные, уретановые, хлоропреновый и т.п.)
вулканизируются бифункциональными агентами, реагирую¬
щими с этими группами по реакциям замещения или присое¬
динения (оксиды двухвалентных металлов, соли непредельных
кислот и др.).Вулканизация каучука может также осуществляться воздей¬
ствием ионизирующей радиации (радиационная вулканизация)
и ультрафиолетового облучения (фотовулканизация).Процесс наиболее распространенной серной вулканизации
заключается во взаимодействии серы и каучука с образовани¬
ем полисульфидных связей между макромолекулами последне¬
го >C-Sn-C< и может быть представлен в следующем виде.1. Радикализация молекулы стабильной ромбической серы
под воздействием теплоты или свободного радикала ускорите¬
ля вулканизацииРис. 20.4. Влияние степени
вулканизации на эластич¬
ность вулканизата (резины):I — область мягких резин, I! —
область кожеподобного со¬
стояния, III — область твердых
резин. А —оптимальная
степень вулканизации440
QSg > ‘S'Sg Sg + R* * R-S7-S*где: R’ — радикал ускорителя, например, каптак- ^са (2-меркаптобензтиазола) ^^\q/C”Se2. Образование полисульфидных бирадикаловR-S7-S* —► R-Sx’ + *Sy’, где: х + у ~ 73. Дегидрирование! звеньев каучука с образованием свобод¬
ных макрорадикалов-СН2-СН=СН-СН2- + R-S*-' — -СН2-СН=СН-СН- + R-S*-H4. Присоединение полисульфидного бирадикала к макрора¬
дикалу каучука и образование сшитой структуры1СН9
1 21СН,
1 21сн2СН-
1 +'S$+n -CH-Sy-
- 1 у-<j}H R-SjcHrНСНIIу <рнfiH<рн-fiH?Н2СН1<РН2<fH2?нсн2СН2
1 2сн2?H2+ R-S*.Серная вулканизация проводится при температуре 125—
180 С. Продолжительность процесса зависит от температуры и
давления, состава вулканизируемой смеси и размеров изделия.
Для большинства РТИ она составляет от 5 до 90 минут. Для вул¬
канизации используются аппараты различного типа: вулкани¬
зационные котлы, обогреваемые острым паром, гидравличес¬
кие вулканизационные прессы, с электрообогревом, работаю¬
щие под давлением до 30 МПа, литьевые машины, одно- и мно¬
гоместные форматоры-вулканизаторы и вулканизационные
камеры.Контрольные вопросы1. Какие полимерные материалы относятся к группе эластомеров (ка¬
учуков)?2. Какое физическое состояние является для эластомеров рабочим?3. На какие группы по назначению делятся синтетические каучуки
(СК)?4. К какой группе СК относится бутадиен-стирольный каучук СКС?5. Какой полимеризацией получают основную массу СКС?6. Что такое латекс и какие вещества являются для него коагулято¬
рами?441
7. Чем отличается изопреновый каучук СКИ-3 по своему строению
от СКС и в чем его сходство с натуральным каучуком?8. В присутствии каких катализаторов происходит полимеризация
изопрена в синтетический каучук СКИ-3?9. Из каких операций состоит процесс переработки каучуков в рези¬
новые изделия?10. Что такое вулканизация каучуков и какие реагенты применяются
в качестве сшивающих агентов для каучуков различной природы?
ЛИТЕРАТУРА (ко 2 тому)Антонов ВМЛапидус АС. Производство ацетилена. — М.:
Химия, 1970.Арсентьев ПЛ. и др. Общая металлургия. — М.: Металлур¬
гия, 1986.Башкатов Т.В., Жигалин Я.Л. Технология синтетических
каучуков — М.: Химия, 1980.Белое П.С. Основы технологии нефтехимического синтеза. —
М.: Химия, 1982.Богомолов А.И. и др. Химия нефти и газа. — JI.: Химия,
1986.Брацихин ЕА., Шульгина Э.Б. — Технология пластических
масс. —JL: Наука, 1982.Ворфоломеев С.Л., Калюжный С.В. Биотехнология. — М.:
Высшая школа, 1990.Догадкин Б А. Химия эластомеров. — М.: Химия, 1981.Блинов НЛ. Основы биотехнологии. — М.: Наука, 1995.Жуков М.Ф. и др. Плазмохимическая переработка углей. —
М.: 1990.Караваев М.М. и др. Технология синтетического метано¬
ла. — М.: Химия, 1984.Кирпичников ПА. и др. Химия и технология синтетического
каучука. — JL: Химия, 1987.Kymenoe А.М. и др. Общая химическая технология. — М.:
Высшая школа, 1985.Лебедев НЛ. Химия и технология основного органического
и нефтехимического синтеза. — М.: Химия, 1981.Лейбович Р.Е. и др. Технология коксо-химического производ¬
ства. — М.: Металлургия, 1982.Либенсон Г.А. Основы порошковой металлургии. — М.: Ме¬
таллургия, 1987.Металлургия стали. Под ред. В.И. Явойского и ГЛ. Ойкса. —
М.: Металлургия, 1973.Металлургия черных и цветных металлов. — М.: Металлур¬
гия, 1993.Николаев А.Ф. Технология пластических масс. — М.—Л.:
Химия,1977.Новые процессы органического синтеза. Под ред. СЛ. Чер¬
ных.— М.: Химия, 1989.Общая химическая технология. Под ред. А.Г. Амелина. — М.:
Химия, 1977.443
Основы химической технологии. Под ред. И.П. Мухленова. —
М.: Высшая школа, 1991.Роговин ЗЛ. Основы химии и технологии химических воло¬
кон. — М., т. т. 1,2 Химия, 1974.СассонА. Биотехнология. —М. Мир. 1987.Соколов ГЛ. Внепечное рафинирование стали. — М.: Метал¬
лургия, 1977.Технология пластических масс. Под ред. В.В. Коршака. —
М.: Химия, 1976.Тимофеев B.C., Серафимов JIA. Принципы технологии основ¬
ного органического синтеза. — М. :Химия, 1992.Таиров Ю.М., Цветков В.Ф. Технология полупроводниковых
и диэлектрических материалов. — М.: Высшая школа, 1990.Тулин А.Н. и др. Развитие безкоксовой металлургии. — М.:
Металлургия, 1987.Уайтхерст и др. Ожижение угля. — М.: Химия, 1986.Уткин Н.И. Металлургия цветных металлов. — М.: Метал¬
лургия, 1985.Химия и переработка угля. Под ред: В.Г. Липовича. — М.:
Наука, 1988.ШурАшМ. Высокомолекулярные соединения. — М.: Высшая
школа, 1971.Энциклопедия полимеров, т. т. 1—3. М.: Сов. энциклопедия,
1972—1977.Эрих В.Н. и др. Химия и технология нефти и газа. — JI.: Хи¬
мия, 1985.Яковлев В.И. Технология микробиологического синтеза. —
JL: Химия, 1987.
СОДЕРЖАНИЕЧасть IМЕТАЛЛУРГИЧЕСКИЕ ПРОЦЕССЫ 3Глава IМЕТАЛЛЫ 31.1. Свойства и клао£ификация металлов 31.2. Металлические р9ды 51.3. Металлургические процессы 81.4. Физико-химические основы восстановленияметаллов из руд 10Глава IIПРОИЗВОДСТВО АЛЮМИНИЯ 152.1. Свойства и применение алюминия 152.2. Краткая история развития производства алюминия 172.3. Сырье для производства алюминия 182.4. Общая схема производства алюминия 192.5. Производство глинозема 212.6. Электролитическое производство алюминия 302.7. Очистка и рафинирование алюминия 352.8. Производство криолита и угольных изделий 36Глава IIIЧЕРНЫЕ МЕТАЛЛЫ 393.1. Свойства железа и его сплавов 393.2. Классификация черных металлов 433.3. Масштабы производства и области применениячерных металлов 453.4. Краткая история развития производствачерных металлов 473.5. Железные руды 503.6. Общая схема производства черных металлов 51Глава IVПРОИЗВОДСТВО ЧУГУНА 544.1. Сырье доменной плавки 544.2. Окускование рудной мелочи 564.3. Теоретические основы доменного процесса 604.4. Технологическая схема доменного производства 674.5. Технико-экономические показатели работы печи 694.6. Продукты доменного производства 714.7. Интенсификация доменного процесса 72Глава VПРОИЗВОДСТВО И ОБРАБОТКА СТАЛИ 745.1. Сталелитейное производство 745.2. Выплавка стали в кислородном конвертере 765.3. Выплавка стали в электрических печах 865.4. Выплавка стали в мартеновских печах 925.5. Вторичная обработка стали 945.6. Разливка стали 985.7. Экономика сталелитейного производства 1005.8. Непрерывные сталеплавильные процессы 1015.9. Прямое получение железа 103445
Часть IIПЕРЕРАБОТКА ХИМИЧЕСКОГО ТОПЛИВА 107Глава VIХИМИЧЕСКОЕ ТОПЛИВО 1076.1. Определение, классификация и состав топлив 1076.2. Энергетические характеристики топлив 110Глава VIIПЕРЕРАБОТКА ЖИДКОГО ТОПЛИВА 1147.1. Нефть, ее происхождение и состав 1147.2. Нефтепродукты 1177.3. Краткая история развития переработки нефти 1197.4. Общая схема переработки нефти 1207.5. Подготовка нефти к переработке 1237.6. Первичная перегонка нефти 1267.7. Крекинг нефтепродуктов 1297.8. Каталитический риформинг нефтепродуктов 1437.9. Очистка нефтепродуктов 1497.10. Коксование нефтяных остатков 151Глава VIIIПЕРЕРАБОТКА ТВЕРДОГО ТОПЛИВА 1548.1. Виды и происхождение твердых топлив 1548.2. Каменные угли 1568.3. Коксование каменного угля 1608.4. Улавливание и разделение летучих продуктовкоксования 1748.5. Переработка продуктов коксования 1798.6. Гидрирование твердого топлива 1848.7. Совершенствование процессов переработкитвердого топлива 187Глава IXПРОИЗВОДСТВО И ПЕРЕРАБОТКА ГАЗООБРАЗНОГОТОПЛИВА 1919.1. Классификация и состав газообразных топлив 1919.2. Сырьевые источники природного газообразноготоплива 1939.3. Использование газообразного топлива 1969.4. Переработка нефтяных газов 1999.5. Переработка обратного коксового газа 2049.6. Газификация твердого топлива 2099.7. Конверсия углеводородных газов 2159.8. Производство кислорода и азота разделениемвоздуха 229Часть IIIПРОИЗВОДСТВО ОРГАНИЧЕСКИХ ВЕЩЕСТВ 236Глава XОСНОВНОЙ ОРГАНИЧЕСКИЙ СИНТЕЗ 23610.1. Продукты основного органического синтеза 23610.2. Сырье и процессы основного органического синтеза 23710.3. Краткий исторический очерк развития ООС 24010.4. Значение и перспективы развития ООС 242446
Глава XIПРОИЗВОДСТВО АЦЕТИЛЕНА 24411.1. Технологические свойства и применение ацетилена 24411.2. Промышленные методы производства ацетилена 24611.3. Производство ацетилена из карбида кальция 24711.4. Производство ацетилена из углеводородного сырья 252Глава XIIПРОИЗВОДСТВО СПИРТОВ 25912.1. Производство кетанола 25912.2. Производство этанола 27012.3. Производство высших жирных (синтетических)спиртов и кислот 282Глава XIIIПРОИЗВОДСТВО АЛЬДЕГИДОВ 29413.1. Производство формальдегида и формалина 29413.2. Производство ацетальдегида 299Глава XIVПРОИЗВОДСТВО УКСУСНОЙ КИСЛОТЫ И АНГИДРИДА 30914.1. Технологические свойства и применение 30914.2. История и промышленные методы производствауксусной кислоты и уксусного ангидрида 31014.3. Производство уксусной кислоты окислениемацетальдегида 31214.4. Технологическая схема совместного производствауксусной кислоты и уксусного ангидрида 315Глава XVПРОИЗВОДСТВО МОНОМЕРОВ.ПОЛИМЕРИЗАЦИОННЫЕ МОНОМЕРЫ 31815.1. Мономеры 31815.2. Производство бутадиена-1,3 и изопрена 32015.3. Производство стирола 33515.4. Производство капролактама 343Глава XVIПРОИЗВОДСТВО МОНОМЕРОВ.ПОЛИКОНДЕНСАЦИОННЫЕ МОНОМЕРЫ 35116.1. Производство фенола 35116.2. Производство диэтиленгликольтерефталата 362Часть IVПРОИЗВОДСТВО ПОЛИМЕРНЫХ МАТЕРИАЛОВ 369Глава XVIIПОЛИМЕРНЫЕ МАТЕРИАЛЫ 36917.1. Свойства и применение полимерных материалов 36917.2. Высокомолекулярные соединения как основа ПМ 37417.3. Основы технологии переработки ПМ в изделия 37717.4. Краткий исторический очерк развития производства ПМ 380Глава XVIIIПРОИЗВОДСТВО ПЛАСТИЧЕСКИХ МАСС 38618.1. Состав и классификация пластических масс 38618.2. Производство полиэтилена 38818.3. Производство полистирола 39218.4. Производство феноло-формальдегидных полимеров 397447
Глава XIXПРОИЗВОДСТВО ХИМИЧЕСКИХ ВОЛОКОН 40619.1. Классификация и использование химических волокон 40619.2. Общие принципы получения химических волокон 41019.3. Производство вискозного волокна 41219.4. Производство капронового волокна 41719.5. Производство лавсанового волокна 420Глава XXПРОИЗВОДСТВО ЭЛАСТОМЕРОВ 42420.1. Свойства и классификация эластомеров 42420.2. Производство бутадиен-стирольного каучука СКС 42920.3. Производство изопренового каучука СКИ-3 43420.4. Переработка каучука в резиновые изделия 437ЛИТЕРАТУРА (ко 2 тому) 443Учебное издание
Соколов Ростислав Сергеевич
ХИМИЧЕСКАЯ ТЕХНОЛОГИЯВ двух томахУчебное пособие для студентов высших учебных заведенийТом 2Металлургические процессы
Переработка химического топлива
Производство органических веществ
и полимерных материаловЗав. редакцией А.Н. Соколов
Редактор З.В. Ларкина
Зав. художественной редакцией ИЛ. Пшеничников
Художник Ю.В. Токарев
Компьютерная верстка Г.К. СоколоваКорректор ТЛ. КокореваЛицензия ЛР № 064380 от 04.01.96
Гигиенический сертификат № 77.ЦС.01.952.П.01652.С.98 от 28.08.98
Сдано в набор 08.06.99. Подписано в печать 27.09.99
Формат 60x90/16. Печать офсетная. Уел. печ. л. 28
Тираж 5 000 экз. Заказ № 2305Гуманитарный издательский центр «ВЛАДОС*117571, Москва, просп. Вернадского, 88,Московский педагогический государственный университет
Тел.: 437-99-98, 430-04-92, 437-25-52; тел./факс 932-56-19E-mail: vlados@dol.ru
http://www.vlados.ruГосударственное унитарное предприятие
Издательско-полиграфический комплекс «Ульяновский Дом печати*
432601, г. Ульяновск, ул. Гончарова, 14