


















































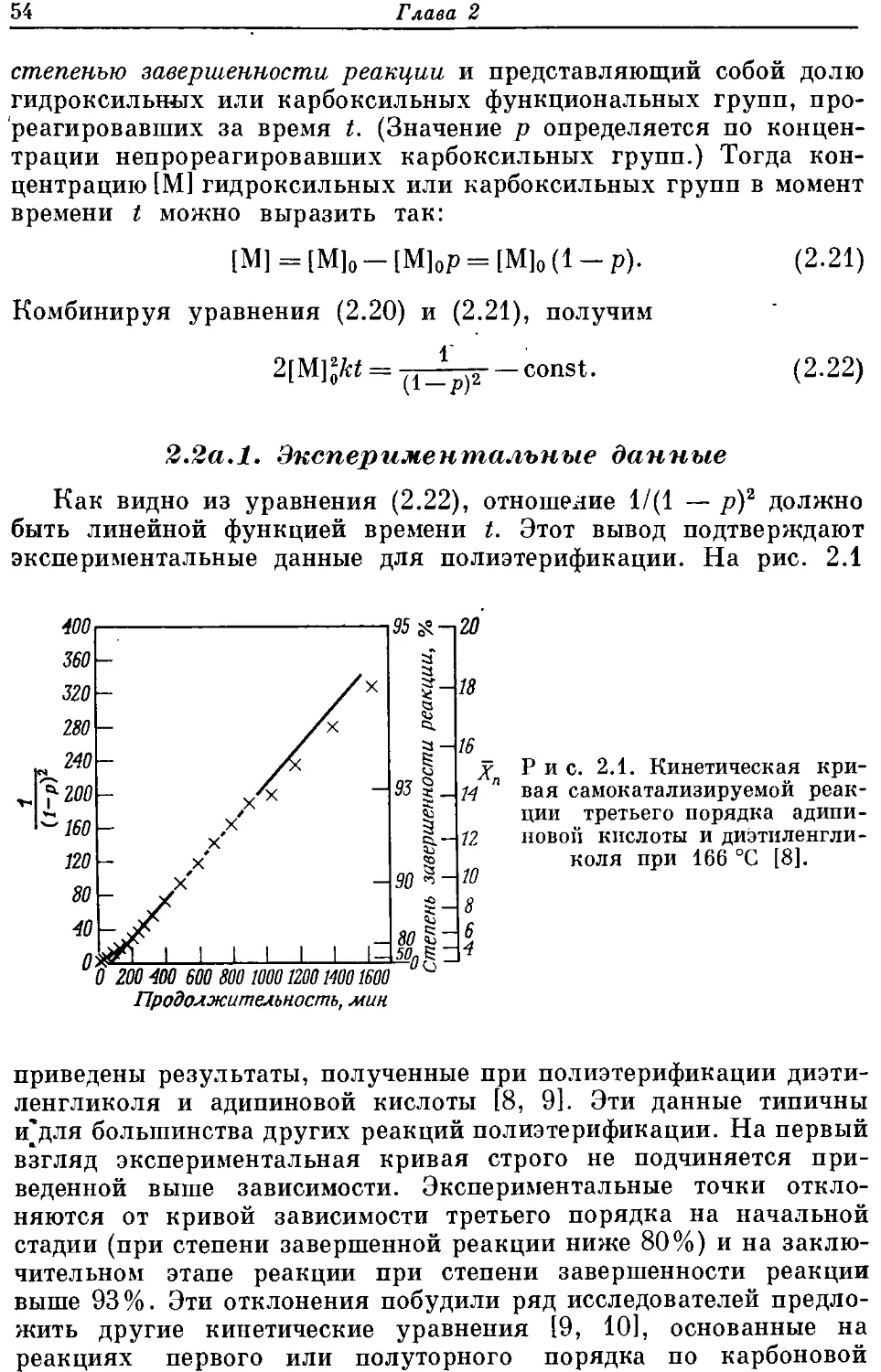















































































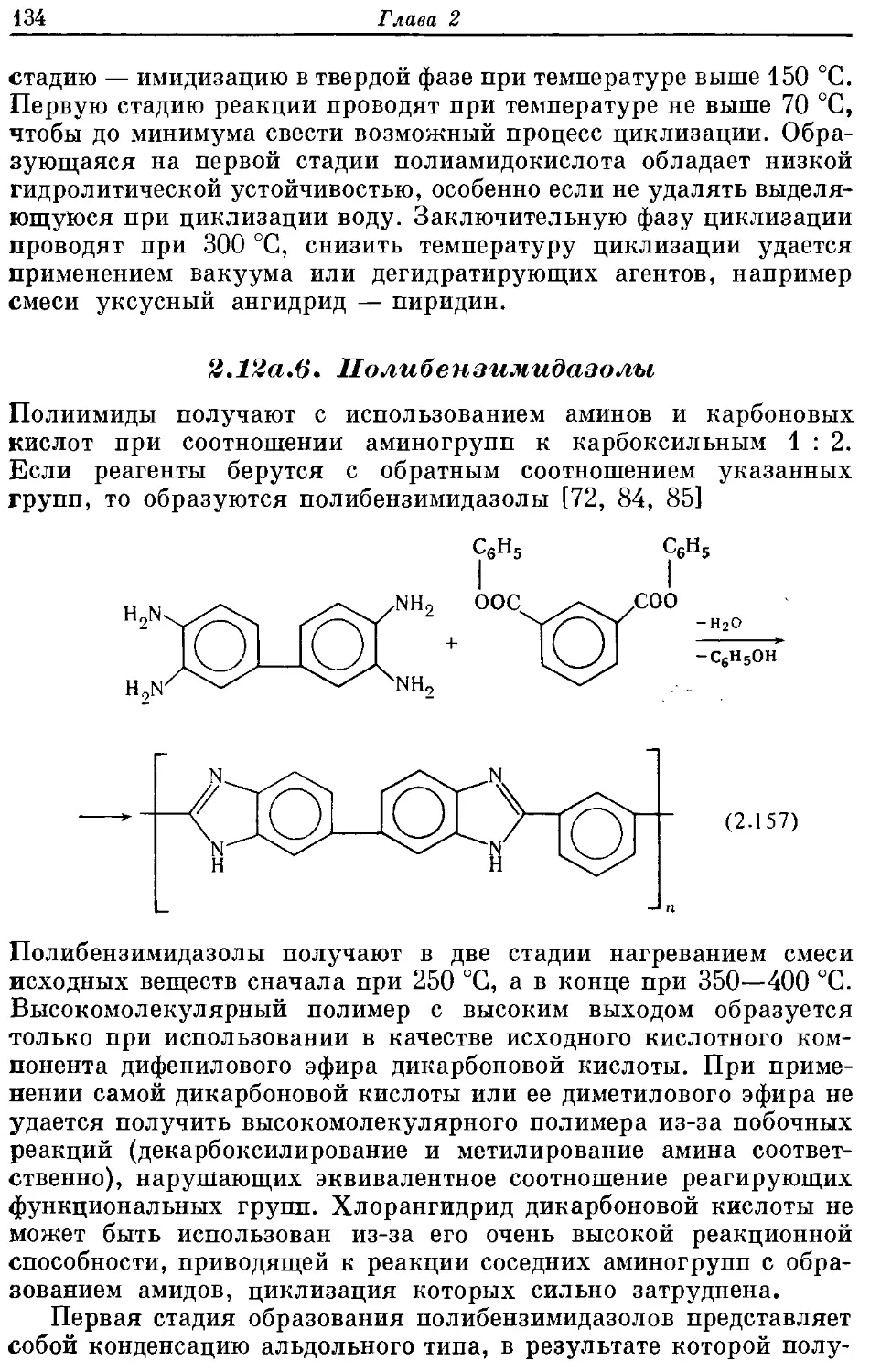



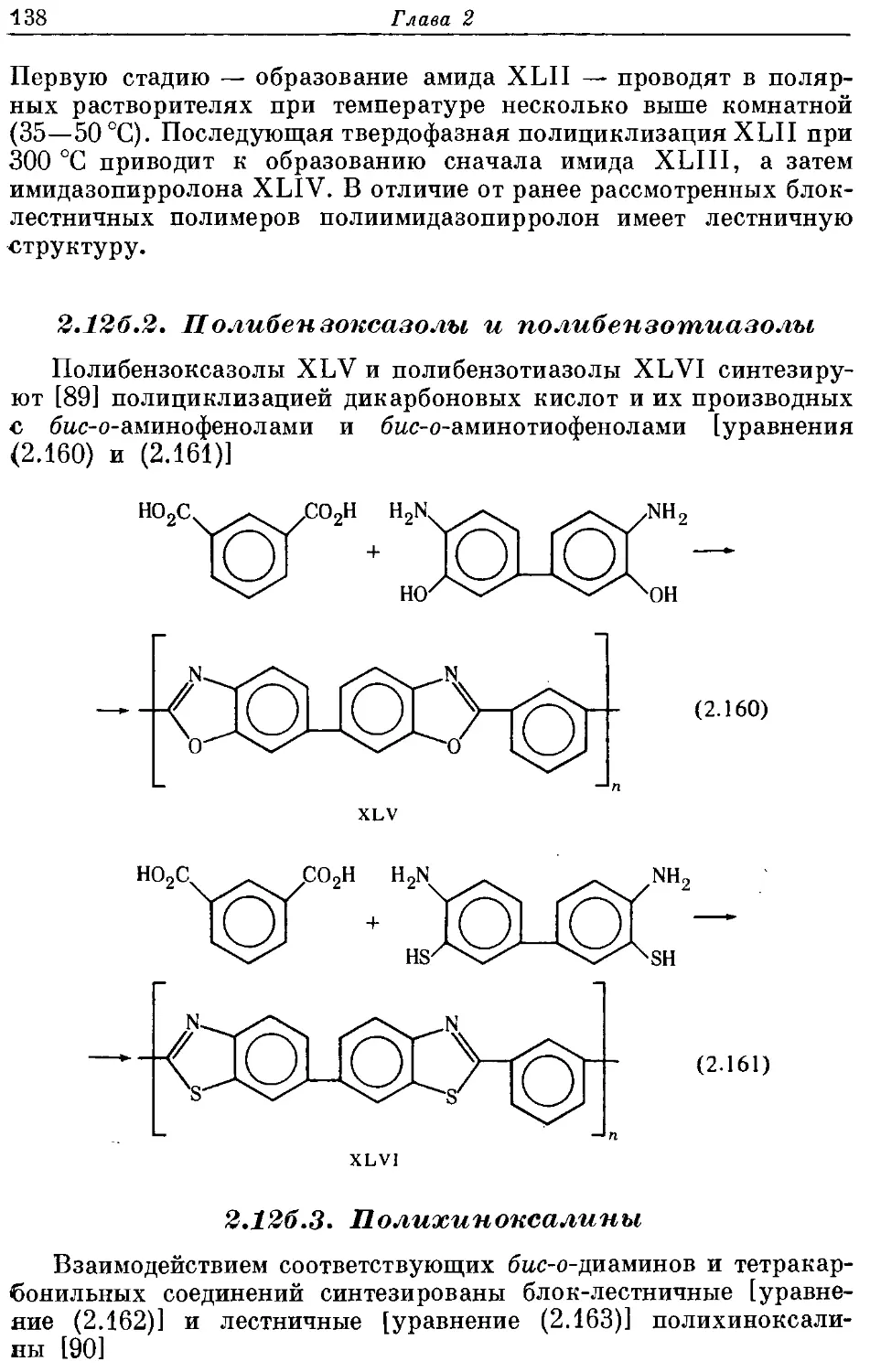

























































































































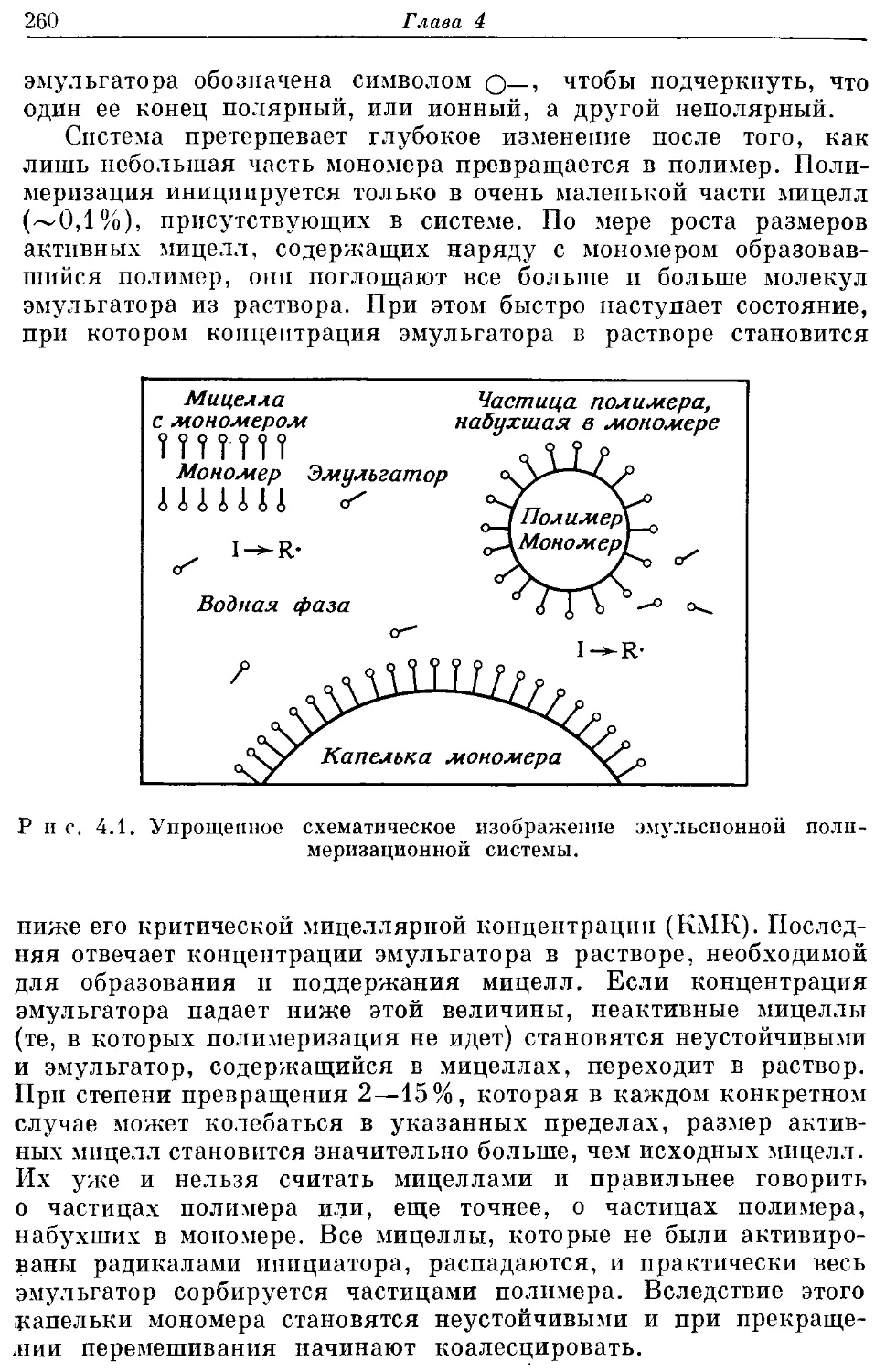




















































































































































































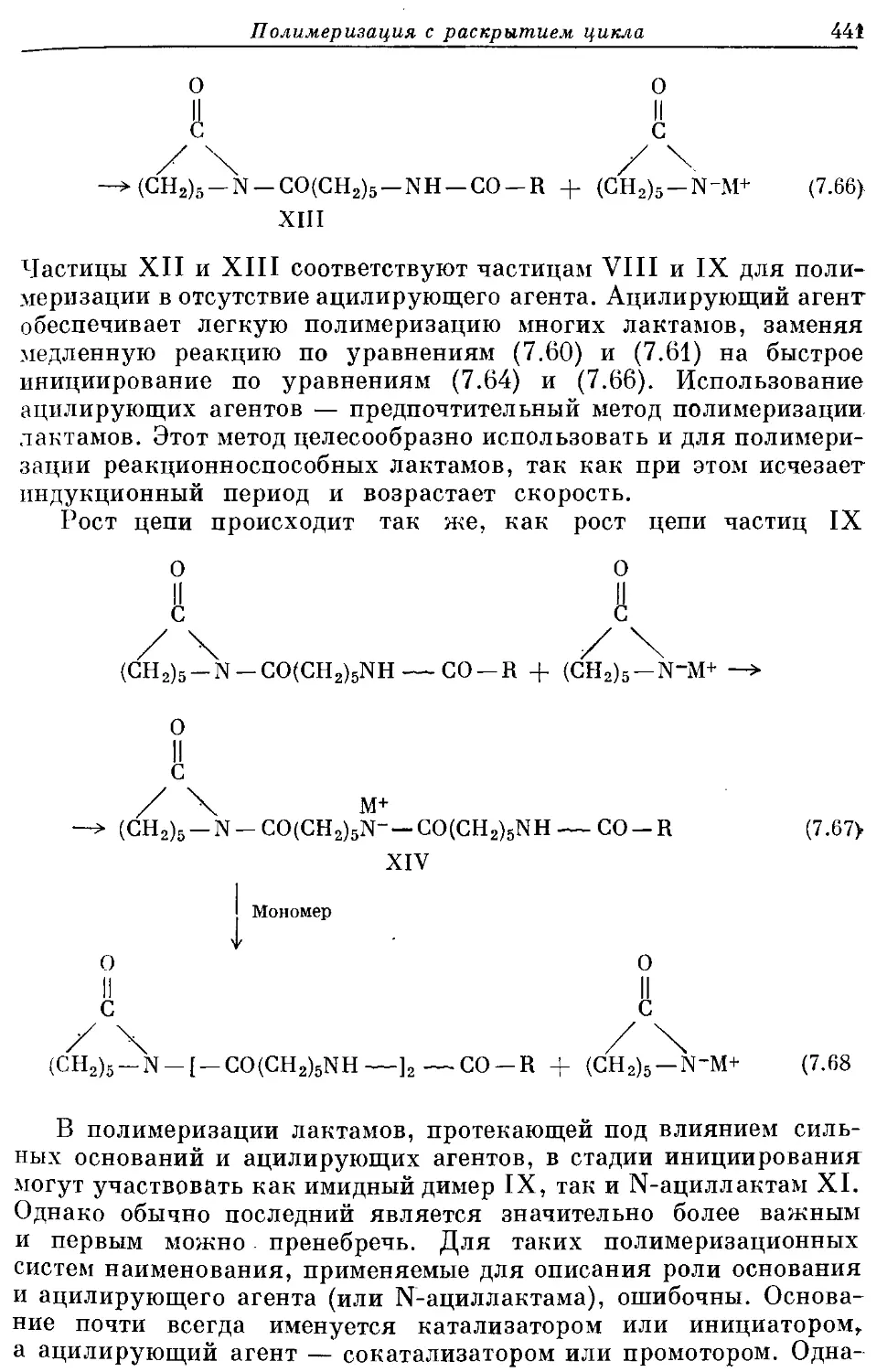










































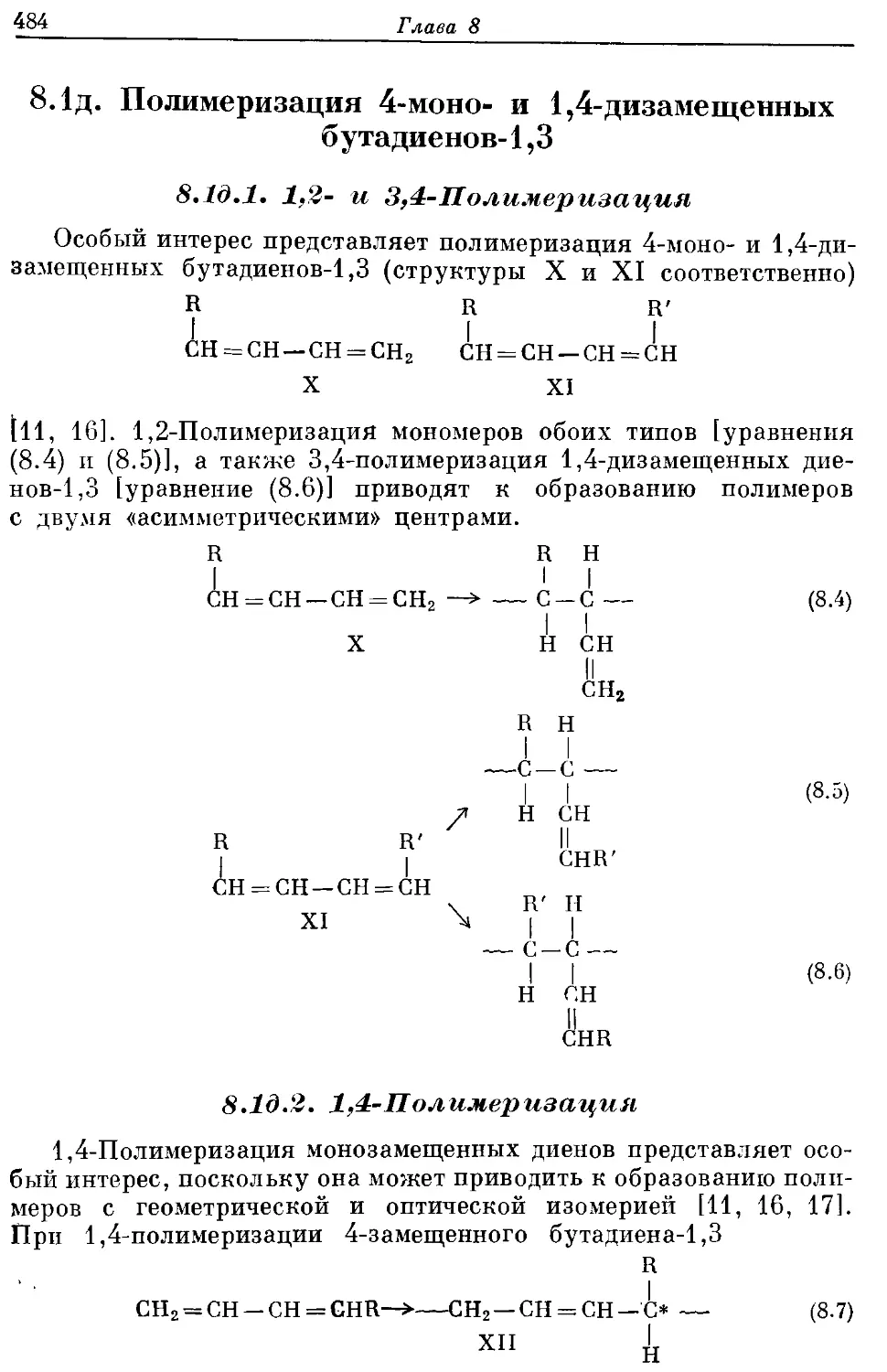































































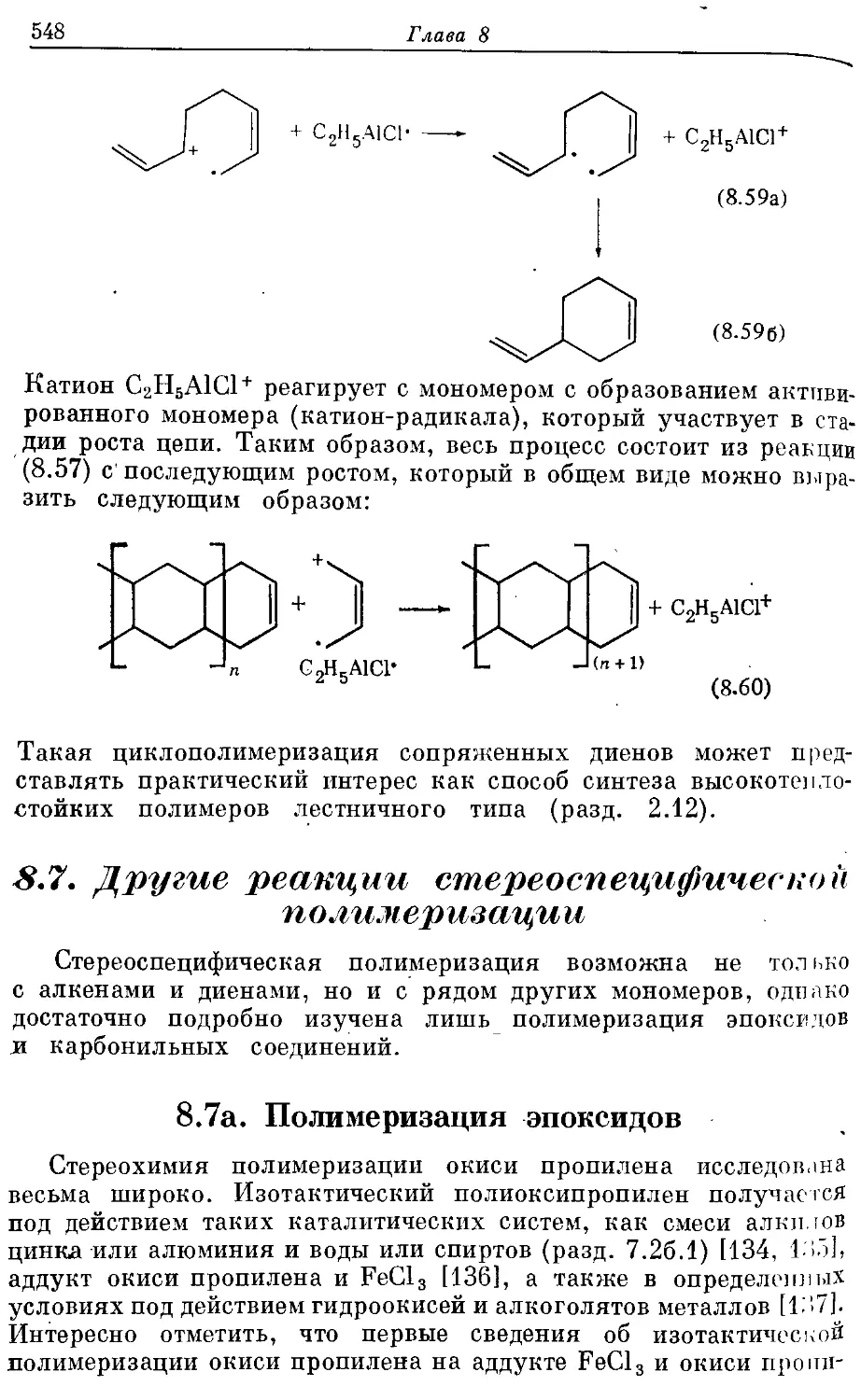




























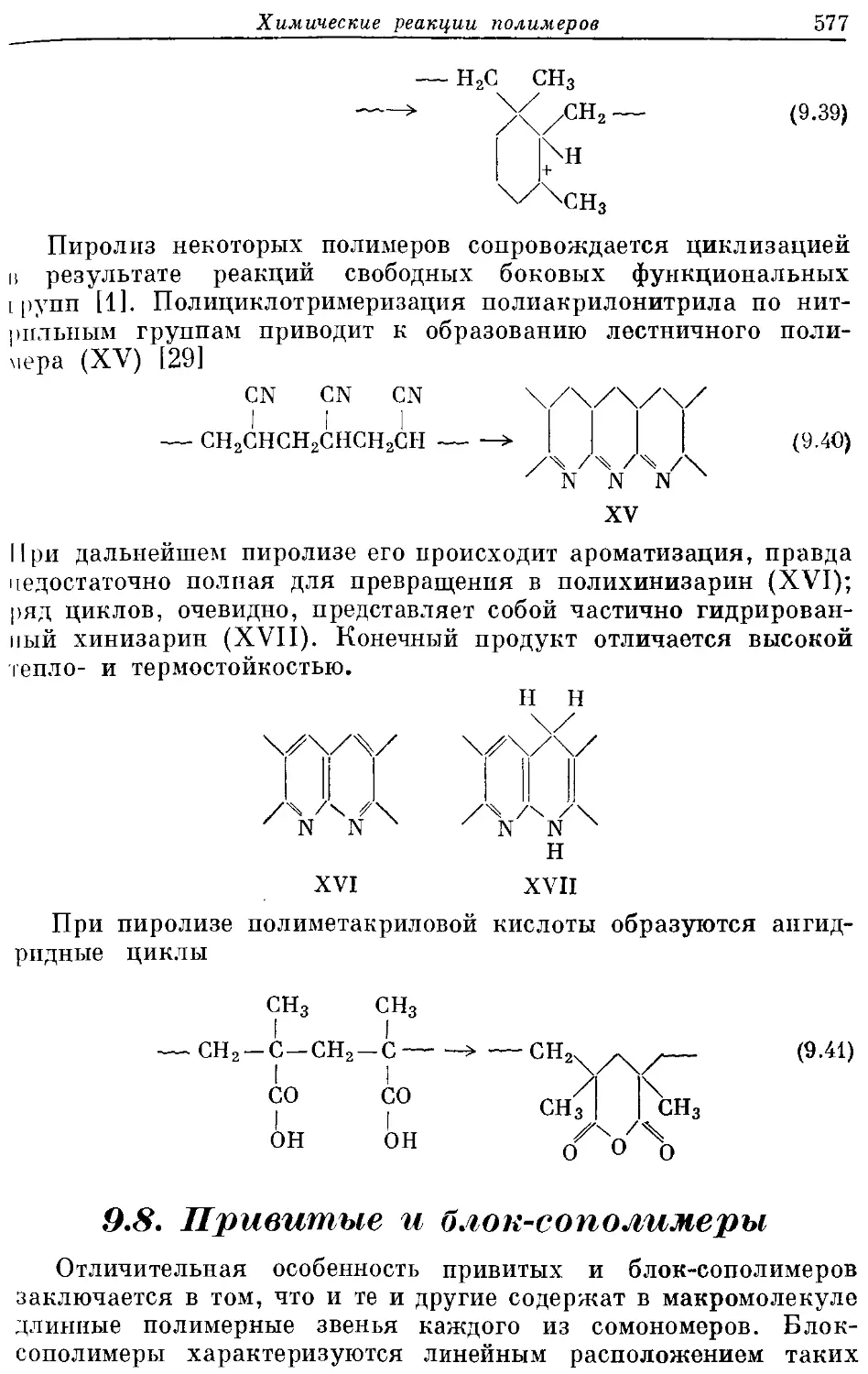









































Текст
ДЖ. ОУДИАН
Основы химии полимеров
Перевод с английского
капд. хим. паук Я. С. Выгодского
и доктора хим. паук Т. М. Фрунзе
Под редакцией
чл.-корр. АН СССР В. В. Коршака
ИЗДАТЕЛЬСТВО «МИР»
МОСКВА 1974
УДК 541.64
Учебник по химии и физике полимеров типа учебника
Бильмейера «Введение в химию и технологию полимеров»,
опубликованного в русском переводе (ИЛ, 1958). Книга напи-
сана с самых современных позиций и с использованием послед-
них достижений в этой области. В ней рассмотрены практи-
чески все области химии высокомолекулярных соединений.
Автор всюду проводит мысль, что в настоящее время уровень
химии высокомолекулярных соединений таков, что есть все
возможности для синтеза полимеров с заданным комплексом
физико-химических свойств. Вопросы и упражнения, приве-
денные в конце каждой главы, способствуют лучшему усвоению
студентами теоретического материала.
Книга предназначена для преподавателей, аспирантов
и студентов химических и химико-технологических вузов; она
будет, безусловно, полезна и научным сотрудникам, занимаю-
щимся синтезом и исследованием полимеров.
Редакция литературы по химии
0253-356
° 041(01)—74
97—73
© Перевод на русский язык, «Мир», 1974
ПРЕДИСЛОВИЕ
Предлагаемая вниманию читателей книга Оудиана «Основы
химии полимеров» охватывает все основные вопросы химии поли-
меров и позволяет получить достаточно ясное представление
о современном уровне наших знаний всех важнейших разделов
полимерной науки. Распределение материала в книге довольно
своеобразно: сначала рассматриваются вопросы номенклатуры
полимеров, а также их физико-химические свойства и применение,
а затем излагаются процессы поликонденсации и полимеризации
с привлечением всего достигнутого полимерной наукой арсенала
средств изучения этих процессов и результатов исследований,
проведенных в последние годы.
К достоинствам книги следует отнести подробное рассмотре-
ние кинетики и механизма реакций синтеза полимеров. Однако
нам пришлось внести некоторые исправления в названия глав
и отдельных разделов книги в соответствии с принятой у нас
номенклатурой. Так, гл. 2 названа «Поликонденсация», хотя
автор обозначил ее как «Ступенчатая полимеризация». Кстати,
автор выделяет обзор эмульсионной полимеризации в отдельную
главу, что несколько нарушает стройность изложения.
Вопросы стереохимии процесса полимеризации рассматри-
ваются автором особо; в отдельной главе обсуждаются механизмы
и кинетика образования различных стереорегулярных полимеров
и их свойства. Химическим превращениям полимеров и, в част-
ности, сшиванию, полимер ан алогичным замещениям в гидроксиль-
ной группе целлюлозы и поливинилового спирта, а также гало-
генированию каучука и полиолефинов посвящена последняя
глава. Там же обсуждаются вопросы получения различными
способами привитых и блок-сополимеров.
6
П редисловие
В основу книги Оудиана положены лекции автора, прочитан-
ные им для студентов Колумбийского университета.
Учебный характер книги подчеркивается наличием контроль-
ных вопросов и упражнений в конце каждой главы, что облегчает
преподавателям и студентам контроль за усвоением материала.
Вместе с тем эта книга будет полезна и широким кругам исследо-
вателей, работающих в научно-исследовательских институтах,
и работникам промышленности, связанным с получением поли-
меров.
При переводе мы дополнили библиографию списком литера-
туры, включающим основные труды советских ученых (поскольку
автор цитирует главным образом работы иностранных специа-
листов) и некоторые последние публикации иностранных авторов.
В. Коршак
ПРЕДИСЛОВИЕ АВТОРА
Книга посвящена физической и органической химии высоко-
молекулярных соединений. Вначале рассмотрены специфические
особенности полимеров, отличающие их от низкомолекулярных
гомологов (гл. 1), а затем дан подробный анализ трех основных
методов образования высокомолекулярных соединений: поликон-
денсации, цепной полимеризации и полимеризации с раскрытием
циклов (гл. 2—5 и 7). При рассмотрении полимеризационных
и поликонденсационных методов образования полимеров большое
внимание обращалось на кинетические и термодинамические
особенности таких процессов, условия их проведения и возмож-
ность использования для синтеза полимеров различных классов.
Не будет большим преувеличением сказать, что возможности
полимерной химии таковы, что уже сейчас можно получать раз-
личные полимеры с заданными структурой и молекулярным весом.
На протяжении всей книги мы стремились подчеркнуть влияние
различных условий проведения реакции на скорость образования,
молекулярный вес и структуру полимеров (разветвленная или
сшитая). Нам хотелось бы также, чтобы после прочтения книги
читатель ощутил сложность и многообразие процессов образова-
ния высокомолекулярных соединений, которыми располагает
в настоящее время химик, работающий в области полимеров.
Когда говорят о многообразии процессов получения полимеров,
то имеют в виду не только возможность использования в синтезе
полимеров исходных веществ различных типов, но и те широкие
перспективы, которые открываются при получении сополимеров
и стереорегулярных полимеров. К числу наиболее важных реак-
ций образования сополимеров относится сополимеризация, кото-
рой и посвящается отдельная глава (гл. 6). Другие процессы
образования сополимеров рассматриваются в соответствующих
8
Предисловие автора
разделах других глав. Специальная глава посвящена стерео-
химии, при этом основное внимание обращено на синтез стерео-
регулярных полимеров путем выбора соответствующих условий
полимеризации. В последней главе кратко рассмотрены некоторые
химические реакции полимеров как интересный способ модифи-
кации существующих и синтеза новых типов полимеров. При
написании книги была использована литература, вышедшая
в свет до июня 1969 г.
В колледжах и университетах США преподавание химии высо-
комолекулярных соединений ведется на довольно низком уровне,
и это несмотря на то, что химия полимеров в настоящее время
является крупнейшим разделом химии. Действительно, прибли-
зительно 30% всех химиков и 50% химиков с высшим образова-
нием в США непосредственно работают в области полимеров *).
Многие начинающие специалисты имеют, по сути дела, только
отрывочные представления в этой области и вынуждены затем
наверстывать упущенное в порядке самообразования. Мы надеемся,
что данная книга поможет изучению курса полимерной химии
студентам химических институтов и университетов, тем более
что в основу ее положены лекции в Колумбийском университете.
Дополненная другими материалами, эта книга также может быть
полезна аспирантам по специальности «химия высокомолекуляр-
ных соединений». К каждой главе приложен список вопросов,
которые должны способствовать лучшему усвоению предмета
студентами.
Дж. Оудиан
*) J. Chem. Educ., 45 (8), 498 (1968).
ОСНОВНЫЕ ОБОЗНАЧЕНИЯ
Р — полимер
М — мономер
S — агент передачи цепи
С — катализатор
Е — эмульгатор
Z — ингибитор
[ ] — концентрация соответствующего компонента
[ ]к — критическая (равновесная) концентрация
R — скорость полимеризации или поликонденсации
Rи — скорость реакции инициирования
Rp — скорость реакции роста
Ro — скорость реакции обрыва
7?Пер — скорость реакции передачи цепи
к и — константа скорости реакции инициирования
кр — константа скорости реакции роста
к0 — константа скорости реакции обрыва
к0. р — константа скорости реакции обрыва рекомбинацией
к0. д — константа скорости реакции обрыва диспропорцио-
нированием
&пер. м — константа скорости реакции передачи цепи на мономер
&пер. р — константа скорости реакции передачи цепи на полимер
/с пер. s — константа скорости реакции передачи цепи на агент
передачи цепи
ki — константа скорости реакции передачи цепи на ини-
циатор
/сДРп — константа скорости реакции деполимеризации
кг — константа скорости реакции гомолитического разло-
жения
kz — константа скорости реакции ингибирования
&конт — константа скорости реакции роста контактных ионных
пар
^сольв — константа скорости реакции роста сольватно разде-
ленных ионных пар
кИзо — константа скорости реакции изотактического при-
соединения
10 Основные обозначения
^СИПДИО — константа скорости реакции синдиотактического при- соединения
с Т’пр — константа передачи цепи — предельная температура, при которой скорости роста
xw Хп р Рравн и деполимеризации равны — средневесовая степень полимеризации — среднечисловая степень полимеризации — степень завершенности реакции — степень завершенности реакции при достижении рав- новесия
Рк — критическая степень завершенности реакции (при гелеобразовании)
ГЛАВА 1
ВВЕДЕНИЕ
Полимерами называются макромолекулы, построенные при-
соединением большого числа маленьких молекул друг к другу.
Небольшие молекулы, которые в комбинации друг с другом обра-
зуют молекулы полимера, называются мономерами, а реакции их
образования — полимеризацией и поликонденсацией. В молекуле
полимера могут содержаться сотни, тысячи, десятки тысяч и более
молекул мономера. Понятие полимер объединяет соединения
с молекулярным весом до нескольких миллионов. Однако боль-
шинство полимеров, которые синтезированы в лаборатории или
нашли практическое применение, имеет молекулярный вес
5000-200 000.
1.1. Типы полимеров и реакций
их образования
До настоящего времени нет единой точки зрения по вопросу
классификации полимеров, что затрудняет изучение предмета
студентами. В процессе развития полимерной науки использова-
лись две классификации полимеров. Одна из них делит все поли-
меры на конденсационные и полимеризационные (аддиционные),
другая — на ступенчатые и цепные полимеры. Ошибки и недо-
разумения обычно возникают из-за того, что понятия одной клас-
сификации механически используют в другой. Термины «кон-
денсационные» и «ступенчатые» иногда считают синонимами,
так же как термины «полимеризационные» и «цепные». Хотя эти
термины действительно во многих случаях равнозначны, их
не следует путать друг с другом, так как в основе их лежат два
различных принципа классификации. Конденсационно-аддицион-
ная классификация имеет в виду главным образом состав или
структуру полимеров, тогда как ступенчато-цепная классифика-
ция основана на механизме реакций полимеризации.
1.1а. Состав и структура полимера
Карозерс [1] в 1929 г. разделил полимеры на конденсационные
и полимеризационные (аддиционные), основываясь на различии
12
Глава 1
в составе полимера и мономера (или мономеров), из которого
синтезирован данный полимер. Конденсационными были названы
полимеры, которые образуются из полифункциональных моно-
меров различными реакциями конденсации, известными в орга-
нической химии и протекающими с выделением низкомолекуляр-
ных продуктов, например воды. Типичным примером такого кон-
денсационного полимера являются полиамиды, получаемые
из диаминов и дикарбоновых кислот с выделением воды по схеме
nH2N-R—NH2 + nHOOC-R' —СООН -+
—> Н —( —NH —R—NHCO —R' —СО —)n —OH-f-(2ra —1)Н2О, (1.1)
где R и R' — алифатические или ароматические группы. Группи-
ровка в скобках в формуле полиамида многократно повторяется
в полимерной цепи и называется поэтому повторяющейся единицей
или элементарным звеном [2]. Состав элементарного звена отли-
чается от состава суммы двух мономеров на молекулу воды.
Полиамид на основе гексаметилендиамина R = (СН2)6 и адипино-
вой кислоты R' = (CH2)4 в настоящее время широко используется
в производстве волокна и пластмасс и хорошо известен под назва-
нием «найлоп-6,6» или «полигексаметиленадипамид». В качестве
других примеров конденсационных полимеров можно привести
сложные полиэфиры, получаемые из дикарбоновых кислот
и диолов с выделением воды:
гаНО,—R —ОН + пНООС —R' —СООН —»
—> НО —( —R —ОСО —R' —СОО —)п —Н + (2га —1)Н2О, (1.2)
и поликарбонаты, образующиеся при взаимодействии аромати-
ческих бисфенолов и фосгена с выделением хлористого водорода
лНО'
ОН + п С1—СО-С1
О-СО--С1 + (2в-1) НС1 .(1.3)
—Лп
В табл. 1.1 приведены наиболее широко известные конденсацион-
ные полимеры и реакции их образования. Как видно из данных
этой таблицы, многие конденсационные полимеры можно полу-
чать из различных исходных веществ. Так, полиамиды можно
синтезировать либо взаимодействием диаминов с дикарбоновыми
кислотами или их хлорангидридами, либо самоконденсацией
аминокислот. Сложные полиэфиры получают из диолов этерифи-
кацией кислотами или реакцией эфирного обмена с диэфирами.
Ряд природных полимеров, и в их числе целлюлоза, шерсть,
крахмал и натуральный шелк, относится к разряду конденса-
Таблица 1.1
Типичные конденсационные полимеры
Полимер Характерная связь Реакция образования
Полиамид -СО —NH — H2N — R-NH2 + HOOC — R' — СООН-> -*Н —( —NH—R-NHCO —R' —СО—)Л-ОН + Н2О H2N-R — NH2 + C1CO-R' — COCI -> -> H—( —NH —R —NHCO —R' —CO —)n —Cl-j-HCl H2N-R — COOH—>-H —( — NH-R-CO-)„-OH + H2O
Белок,шерсть, натураль- ный шелк -СО —NH — Природные полипептидные полимеры, при деструкции образуются смеси различ- ных аминокислот Н —( —NH—R —NHCO —R' —СО —)л —OH + H2O->H2N-R —COOII + + H2N-R'-COOH
Сложный по- лиэфир —СО —О — НО —R —ОН + НООС—R'—СООН -*НО —(-R-OCO—R'—COO-)n-H+H2O НО — R —OH-j-R"OOC — R' — COOR"НО —( — R-OCO— R' — COO — )n—R" — + R"OH HO—R —COOH -> HO —( —R —COO—)л —H+H2O
Полиуретан — О —СО —NH- IIO-R-OII + OCN-R' — NCO( — 0 — R — OCO-NH — R' — NH-CO-)n-
Полисилоксан — Si —О — Cl —SiR2 —Cl -Д HO —SiR2 —0H-*H0 —(-SiR2 —0 —)n —H + H20 — HG1 он Г он
Фенолформаль- дегидный — Аг—СН2 — + CH2° —" + H2° L.
Продолжение табл. 1.1
Полимер Характерная связь Реакция образования
Мочевииофор- мальдегид- ный -NH-CH2- H2N - СО - NH2 + СН2О—HN - СО — NH - СН2 - )п-+Н20 N Г N /Ч /Ч
Меламинофор- мальдегид- ный — NH —СН2— H2N-C C-NH2 + CH2O-> - -HN-C c-nh-ch2- II 1 II 1 NN NN c c I 1 nh2 l nh2
Целлюлоза — О-С — Природный полимер, при деструкции образуется глюкоза — (— CeH 10О5 — )п 1- Н20 CeHi20e
Полисульфид — — Cl - В — Cl Na2Sm -+ — (— R - Sm - )n — NaCl
Полиацеталь — О-СН —0- 1 в R-CH04-H0-R' — ОН—0 —R' — OCHR — H20
Введение
15
ционных полимеров на основании того, что гипотетическая схема
их синтеза предполагает выделение воды: так, можно полагать,
что целлюлоза получается при дегидратации глюкозы. Карозерс
называл конденсационными такие полимеры, в формуле повто-
ряющегося звена которых отсутствует ряд атомов мономера или
мономеров, образующих данный полимер или являющихся про-
дуктами его разложения. В этом смысле целлюлоза действительно
относится к конденсационным полимерам, так как при гидролизе
ее образуется глюкоза, которая содержит повторяющееся звено
целлюлозы плюс элементы воды
СН2ОН
I
СН-0
—о-сн \н—
ОН + (п- 1)Н2О —>
\н-сн
I I
он он
целлюлоза
СН2ОН
I
сн—о
пНО —сн 'сн — он
'сн-сн
I I
он он
(1.4)
глюкоза
По классификации Карозерса полимеризационными полиме-
рами называют полимеры, которые образуются из мономеров без
выделения низкомолекулярных побочных продуктов. В отличие
от конденсационных полимеров элементарный состав такого
полимера и его мономера одинаков. Главные представители адди-
ционных полимеров — полимеры винильных мономеров. При
взаимодействии таких мономеров друг с другом образуются
полимеры, а двойная связь их переходит в насыщенную по схеме
nCH2 = CHY —> —( —СН2 —CHY —)п —, (1.5)
где Y — водород, алкил, арил, нитрил, сложный эфир, карбоксиль-
ная группа, кетон, простая эфирная группа, галоген и др. Другим
типом мономеров, образующих аддиционные полимеры, являются
альдегиды. В табл. 1.2 приведены наиболее распространенные
типы полимеризационных полимеров и мономеры для их синтеза.
По мере развития полимерной науки, разработки новых мето-
дов синтеза полимеров и получения новых полимеров стало оче-
видным, что классификация, предложенная Карозерсом, несовеп-
Таблица 1,2
Типичные полимеризационные полимеры
Полимер Мономер Элементарное звено
Полиэтилен сн2=сн2 СН3 -сн2—сн2- сн3
Полипзобутплен сн2=с 1 СН3 -СН2-С — 1 СНз
Полиакрилонитрил ch2=ch-cn -сн2—сн- 1 CN
Поливинилхлорид СН2=СН — G1 —сн2—сн— 1 С1
Полистирол сн2=сн — сн2-сн-
Свн5 СН3 Свн5 СНз
Полиметил метакрил ат сн2=с 1 СООСНз -СН2-С — 1 СООСНз
П оливини лацетат СН2 = СН —ОСОСНз G1 -сн2—сн— 1 ОСОСНз С1
Поливинилидеихлорид сн2=с 1 С1 F F -СН2-С — 1 С1 F F 1 ।
Политетрафторэтилен с=с 1 1 F F -С-С- 1 1 F F
Полиформальдегид СН2 = О — СН2 —О —
Полиацетальдегид СН = О 1 СН3 — СН —О — 1 СН3 —сн2 ZCH2-
Полпизопрен (натураль- сн2=с—сн=сн2 ^с^сн /
ный каучук) СНз СН3
Введение
17
шенна и нуждается в существенной доработке. Рассмотрим, напри-
мер, полиуретаны, получающиеся при взаимодействии диолов
и диизоцианатов без выделения низкомолекулярного продукта
и НО — R — ОН + nOCN —R' —NCO —>
НО — ( — R — OCONH — R' — NHCO — О — )n_t — R — OCONH — R' — NCO.
(1-6)
По Карозерсу, такие полимеры надо называть аддиционными,
так как состав полимера не отличается от состава мономера.
Однако по структуре полиуретаны ближе к конденсационным
полимерам.
Во избежание неверной классификации полиуретанов (а также
некоторых других полимеров) как полимеров полимеризационного
типа было предложено называть полимеры исходя из химического
строения составляющих их групп [3]. По этому определению
конденсационными называются полимеры, повторяющиеся звенья
которых соединены различными функциональными группами,
например сложноэфирными, амидными, уретановыми, сульфид-
ными или простыми эфирными связями. Тогда структура кон-
денсационных полимеров может быть представлена следующей
формулой:
—R—Z—R—Z—R—Z—R—Z—R—Z—,
I
где В — алифатическая или ароматическая группа, a Z — функ-
циональная группа, например —ОСО—, —NHCO —, —S—,
—OCONH—, —О—, —ОСОО—, —SO2—• Полимеризационные
полимеры не содержат таких функциональных групп в полимер-
ной цепи, а только в боковом ее обрамлении. Согласно такой
классификации, полиуретаны правильнее относить к конденса-
ционным полимерам.
Следует иметь в виду, что не все полимеры, которые по класси-
фикации Карозерса относятся к конденсационным, попадают
в ту же группу при рассмотрении химического строения поли-
мера. Ряд конденсационных полимеров не содержит функцио-
нальных групп в полимерной цепи. Примером являются фенол-
формальдегидные полимеры, образующиеся при взаимодействии
фенола (или замещенных фенолов) с формальдегидом
п СН2О
J 8
Глаеа 1
Эти полимеры не содержат функциональных групп внутри основ-
ной полимерной цепи, но относятся к разряду конденсационных
полимеров, так как при их образовании выделяется вода. Другим
примером служит поли-и-ксилилен, получающийся окислительной
дегидрополиконденсацией и-ксилола
>. С (1.8)
Таким образом, полимер называется конденсационным, если:
1) при синтезе его выделяется низкомолекулярный продукт;
2) в основной полимерной цепи имеются функциональные группы
или 3) в элементарном звене полимера отсутствуют атомы, содер-
жащиеся в мономере (гипотетическом), который может быть
продуктом его деструкции. Остальные полимеры называются
полимеризационными.
1.16. Механизм образования полимеров
Кроме различий в составе и структуре полимеров, Флори 13]
обращает большое внимание на значительную разницу в меха-
низмах их образования. На основании механизма процесса поли-
меризация делится на ступенчатую и цепную. Таким образом,
ступенчатые полимеры — это полимеры, получающиеся ступен-
чатой полимеризацией (поликонденсацией), а цепные полимеры —
это полимеры, образующиеся цепной полимеризацией. По своим
характеристикам эти два процесса сильно различаются. При
этом различие заключается главным образом в разной зависи-
мости параметров реакции от времени. Если говорить более кон-
кретно, то при поликонденсации и полимеризации требуется
различный промежуток времени для получения высокомолеку-
лярных полимеров, т. е. для завершения роста цепи макромоле-
кулы.
Поликонденсация представляет собой ступенчатую реакцию
между функциональными группами исходных веществ [см., напри-
мер, уравнения (1.1)—(1.3) и (1.6) —(1.8)]. При поликонденсации
размер молекулы полимера увеличивается с относительно низкой
скоростью. Сначала из мономера медленно образуется димер,
тример, тетрамер, пентамер и т. д. до тех пор, пока не будет
достигнут сравнительно высокий молекулярный вес, т. е. полимер
будет содержать много молекул мономера. Совершенно иная
картина имеет место при цепной полимеризации, когда высоко-
Введение
19
молекулярный полимер получается почти сразу после начала
реакции.
Цепная полимеризация протекает в присутствии катализа-
тора, который дает частицы инициатора R* с реакционноспособ-
ным центром. Последним может быть свободный радикал, катион
пли анион. Рост цепи при полимеризации идет в результате
присоединения к инициатору большого числа молекул мономера
с образованием на конце молекулы новых реакционноспособных
центров в течение секунды или даже более короткого промежутка
времени. Наиболее типичным примером цепной полимеризации,
несомненно, является полимеризация винильных мономеров,
которую можно описать следующей схемой:
Н НН
ch2=chy I ch2=chy I I
R*------» R —CH2—C*---» R — CH2 — С — CH2 — C* —>
I I I
Y Y Y
Рост цепи полимера прекращается при уничтожении реакционных
центров в результате различных реакций обрыва.
Из сказанного выше не следует, что полимеризация протекает
быстрее поликонденсации. Скорость исчезновения мономера
(т. е. скорость полимеризации) при цепной полимеризации может
быть достаточно высокой и превышать таковую при поликонден-
сации. Различие между двумя процессами заключается только
во времени, необходимом для роста каждой макромолекулы.
Если, например, начать одновременно цепную полимеризацию
и поликонденсацию, то можно заметить ряд характерных особен-
ностей, касающихся их относительных скоростей. Тем не менее
в любой момент времени после начала реакции молекулярный вес
полимера существенно зависит от типа процесса. Если обе реакции
прервать при степени превращения 0,1, 1, 10, 40, 90% и т. д.,
то картина будет одной и той же. При цепной полимеризации
на различных ее стадиях в реакционной смеси всегда имеется
только мономер и высокомолекулярный полимер и отсутствуют
молекулы промежуточных размеров. С увеличением продолжи-
тельности реакции растет лишь число молекул полимера. Моле-
кулярный вес полимера не зависит от степени завершенности
реакции, которая только влияет на выход полимера. В то же время
при поликонденсации высокомолекулярный полимер образуется
только при очень высокой степени завершенности реакции
(более 98%). Таким образом, при поликонденсации и выход,
и молекулярный вес полимера зависят от продолжительности
реакции.
Классификация полимеров на основании механизма реакции,
так же как п на основании структуры и состава, не свободна
от недостатков. Полимеризация с раскрытием цикла таких цикли-
ческих мономеров, как окись пропилена
О
:н—сн2 —> —г—сн2—сн—О —
(1-10)
сн3
п
или е-капролактам
СН2
сн2\о
п | I
СН2 NH
I I
сн2—сн2
—> — ( —NHCH2CH2CH2CH2CH2CO —)„ —,
(1.11)
может протекать по ступенчатому и цепному механизмам в зависи-
мости от условий реакции и типа катализаторов. Структура же
полимера не зависит от механизма реакции. Приведенные примеры
наглядно показывают, что нужно делать различие между класси-
фикациями по механизму образования и структуре полимера.
Термины этих классификаций не взаимозаменяемы. Такие поли-
меры, как простые полиэфиры и полиамиды [уравнения (1.10)
и (1.11)], а также другие полимеры циклических мономеров попа-
дают в разные разряды по двум классификациям. Основываясь
на структуре, их следует относить к конденсационным полиме-
рам, так как они содержат функциональные группы (например,
простую эфирную и амидную группы) в полимерной цепи. Подобно
полиуретанам, их нельзя считать аддиционными полимерами,
хотя это было бы возможно по классификации Карозерса. Поло-
жение еще более усложняется для полимера на основе е-капро-
лактама. Точно такой же полимер можно получить поликонден-
сацией линейного мономера — е-аминокапроновой кислоты. Под-
водя итог вышесказанному, следует еще раз подчеркнуть, что
термины «конденсационный» и «ступенчатый» полимер («конден-
сационная» и «ступенчатая» полимеризация) не синонимы, так же
как не идентичны полностью термины «аддитивный» и «цепной»
полимер или «аддитивная» и «цепная» полимеризация.
1.2. Номенклатура полимеров
Номенклатура полимеров на настоящем ее уровне оставляет
желать много лучшего. Стандартная номенклатурная система,
основанная на химическом строении полимера и аналогичная
ведение
2
принятой для органических соединений, была бы, вероятно,
наиболее приемлемой. К сожалению, названия полимерам часто
давали в соответствии с симпатиями и привычками без какой-либо
единой принятой системы. Нередки примеры, когда один полимер
имеет несколько различных общепринятых названий. Существую-
щие номенклатуры основаны на структуре полимера, исходных
соединениях для его синтеза или торговых наименованиях [2—5].
Ни одна из существующих номенклатур строго не соблюдается.
В настоящее время принимается ряд мер для совершенствования
и стандартизации номенклатуры полимеров.
1.2а. Номенклатура, основанная на названии
исходных соединений
Наиболее простая и общепринятая номенклатура основана
па исходных соединениях для синтеза полимеров. Эта система
применима прежде всего для полимеров, образующихся из одного
мономера. Название таких полимеров складывается из названия
мономера и приставки «поли», которая пишется слитно. Например,
полимеры этилена и ацетальдегида называются соответственно
полиэтилен и полиацетальдегид. Если в молекуле мономера
содержится заместитель или название его состоит из нескольких
слов, то такой мономер ставится в скобки после приставки «поли».
Полимеры З-метилпентена-1, винилхлорида, оксипропилена, хлор-
трифторэтилена и е-капролактама называются соответственно
поли-(3-метилпентен-1), поли(винилхлорид), поли(оксипропилен),
поли(хлортрифторэтилен) и поли-(е-капролактам). В табл. 1.2
приведены другие примеры. Скобки, как правило, опускаются.
Хотя обычно при этом не возникает каких-либо проблем, в неко-
торых случаях наименование полимера без скобок затрудняет
идентификацию его структуры. Так, например, использование
названия «полиоксиэтилен» вместо «поли(оксиэтилен)» может при-
вести к предположению, что полимер имеет одну из следующих
двух структур:
-ТСН2СН2-^-О-(-СН2СН2-)^ ।—(-СН2СН2-)д—j
и HI
тогда как па самом деле речь идет о полимере окиси этилена
— ( —СН2СН2О —)п —.
Ряд полимеров называют по их гипотетическим мономерам.
Например, поливиниловый спирт получают гидролизом поли-
22
Глава 1
винилацетата
СН3СОО
НО
_-СН2-СН-Jn
+ пН2О
-CH2-CH-Jn
-4-пСН3СООН
(1-12)
Назван же он по гипотетическому мономеру — виниловому спирту,
который в действительности существует только в виде енольной
формы ацетальдегида.
Конденсационные полимеры, получаемые из одного мономера,
называют аналогичным способом. Примерами являются поли-
амиды и сложные полиэфиры аминокислот и оксикислот соот-
ветственно. Так, полимер на основе 6-аминокапроновой кислоты
называется поли-(6-аминокапроновой кислотой)
nH2N —СН2СН2СН2СН2СН2—СООН —>
6-амппокапроновая кислота
-( — HN-CH2CH2CH2CH2CH2-CO-)n-
полп-(6-ампнокапроповая кислота)
(1-13)
Следует отметить, что тот же самый полимер, полученный из
е-капролактама, называется поли-(е-капролактамом), причем
такая ситуация, когда один и тот же полимер получен из двух
различных мономеров,— довольно распространенное явление.
1.26. Номенклатура, основанная на химическом
строении
Конденсационные полимеры, синтезированные из двух или
более мономеров, обычно называют в соответствии с химическим
строением их элементарного звена. Название образуется при-
соединением приставки к заключенному в скобки названию струк-
турной группы. Такая структурная группа представляет собой
специфическую группу, определяющую класс полимера: сложный
эфир, амид, уретан и т. д. На Дтом основании полимер из гексаме-
тилендиамина и себациновой кислоты рассматривается как заме-
щенный амид себациновой кислоты НООС(СН2)8СООН и назы-
вается поли(гексаметиленсебацамид). Поли(этилентерефталат) —
полимер на основе этиленгликоля и терефталевой кислоты
и-НООСС6Н4СООН. Полимер на основе триметиленгликоля
и этилендиизоциапата называется поли(триметиленэтиленурета-
ном). Во всех приведенных ниже названиях скобки, как правило,
Введение
23
опускаются
4-HN~(CH2)6— NHCO— (CH2)g—co^ »
полигексаметиленсебаи,амид
полизтиленпгерефталат
-E0—CH2CH2CH2—OCONH—CH2CH2—NHCO^
политриметиленэтиленуретан
Было также предложено [4, 5] называть такие конденсационные
полимеры по названию мономера с приставкой «поли». В этом
случае название мономеров разделяется частицей -со-. По этой
номенклатуре указанные выше три полимера имели бы, соответ-
ственно, следующие названия: поли(гексаметилендиамин-со-себа-
циновая кислота), поли(этиленгликоль-со-терефталевая кислота),
полн(триметиленгликоль-со-этилендиизоцианат). Это предложение
не получило широкого применения и приводится здесь лишь для
того, чтобы показать, насколько запутана номенклатура поли-
меров.
Номенклатура, основанная на химическом строении полимера,
не применяется для полимеров, получаемых полимеризацией,
так как большинство из них названо по их мономеру. Исключе-
ниями являются, пожалуй, полп-1,4-фенилен, полиметилен
и поли-и-ксплилен
поли-1,4-фенилен
полиметилен поли-п-ксилилен
Отметим также, что ряд полимеров полимеризационного типа,
например полиэтилен и полипропилен, называют и по мономеру,
и по химическому строению звена. В этих случаях названия
мономеров и элементарного звена совпадают.
1.2в. Торговые и другие названия полимеров
Для некоторых полимеров принята специальная терминология,
в основу которой положены торговые марки полимеров. Хотя
торговых названий следует избегать, необходимо знать наиболее
общепринятые и часто применяющиеся. Ярким примером номен-
клатуры, основанной па торговых марках, является использова-
24
Глава 1
ние названия «найлон» для полиамидов из незамещенных, нераз-
ветвленных алифатических мономеров. К слову «найлон» добав-
ляют две цифры, первая из которых означает число метиленовых
групп остатка диамина в полиамиде, а вторая — число атомов
углерода остатка дикарбоновой кислоты. Таким образом, поли-
гексаметиленадипамид и полигексаметиленсебацамид — най-
лон-6,6 и найлон-6,10 соответственно. К сожалению, часто при-
меняются различные варианты этих названий, например для
найлона-6,6 в литературе встречаются следующие названия:
найлон-66, 66-найлон, найлон-6/6, 6,6-найлон и 6-6-найлон.
Полиамиды на основе одного мономера называются словом
«найлон» с одной цифрой, означающей число атомов углерода
в элементарном звене полимера. Поли-е-капролактам, или поли-
(6-аминокапроновая кислота),— найлон-6.
Во многих случаях такая номенклатура не дает практически
никакой информации о химическом строении полимера. Например,
полимер, получаемый поликонденсацией формальдегида и фенола,
обозначается как фенолформальдегидный полимер, фенолформ-
альдегидная смола, фенольная смола и фенопласт. Полимеры
формальдегида или других альдегидов с мочевиной или меламином
известны под названием «аминосмолы» или «аминопласты» без
каких-либо конкретных наименований. Поэтому часто бывает
очень трудно определить, какой альдегид или амин использо-
вался для синтеза данного конкретного полимера, который назван
аминопластом. Предпочтительна номенклатура, согласно которой
в названии полимера используются названия обоих мономеров,
например карбамидная (мочевиноформальдегидная) смола или
меламиноформальдегидная смола.
Аналогично полимер
известный как поликарбонат бисфенола A [R = —С(СН3)2 —
уравнение (1.3)], на основании его химического строения более
правильно называть поли-4,4/-изопропилидендифениленкарбонат.
1.3. Структурная форма полимерных
молекул
Есть три основные структурные формы полимерных молекул.
По структурной форме полимеры делятся на линейные, разветвлен-
ные и сшитые. Выше рассматривались только такие полимеры,
Введение
25
в которых молекулы мономера соединены в единую цепь. Такие
полимеры называются линейными полимерами. В определенных
условиях из тех же мономеров могут быть получены полимеры
и в других формах.
Разветвленные полимеры часто образуются при полимеризации
и поликонденсации, однако причины в обоих случаях могут быть
совершенно различными. Разветвленными называются полимеры,
в которых есть боковые ответвления от основной цепи макро-
молекулы, исходящие из различных центральных узловых точек
Линейный
Разветвленный (А)
Разветвленный (Б)
Сшитый
Разветвленный (В)
Р п с. 1.1. Структура линейного, разветвленного и сшитого полимеров.
(центров ветвления). Различие в форме линейного и разветвлен-
ного полимеров наглядно видно из рис. 1.1. Центры ветвления
показаны точками. Как видно из рисунка, существует несколько
типов разветвленных полимеров. Разветвленный полимер может
быть гребнеподобным с длинными (Л) или короткими (5) боко-
выми цепями [6]. Сильно разветвленный полимер может иметь
древовидную структуру (В), в которой боковые цепи также имеют
ветвления. Разветвленность полимера, как правило, оказывает
большое влияние на его физические свойства. Так, разветвление
снижает кристаллизуемость полимера. Разветвленные полимеры
в отличие от линейных не упаковываются легко в кристалличе-
скую решетку.
26
Глава 1
Важно отметить, что термин «разветвленный» не относится
к линейным полимерам, содержащим боковые группы, являю-
щиеся частью мономерной структуры. Разветвленными полиме-
рами называются только такие полимеры, боковые цепи которых
состоят из полных мономерных звеньев. Так, полистирол
— это линейный полимер, так как фенильные группы предста-
вляют собой часть мономера и поэтому не рассматриваются как
боковые цепи. Разветвленный полистирол должен иметь струк-
туру VI, в которой одно или более разветвлений от основной
полимерной цепи имеет также полпстпрольпую структуру
VI
Полимеры, в которых макромолекулы связаны друг с другом
не по концевым группам, называются сшитыми (рис. 1.1). Сшива-
ние может происходить при образовании полимера из соответ-
ствующих мономеров. Оно может также протекать в результате
химической обработки готового полимера. В зависимости от метода
и условий образующиеся между полимерными цепями связи могут
быть различной длины. Можно также регулировать частоту попе-
речных связей, получая слабо и сильно сшитые полимерные
Введение
27
системы. Когда число сшивок становится достаточно большим,
получается трехмерный, или сетчатый, полимер, в котором
полимерные цени соединены друг с другом, образуя одну гигант-
скую молекулу. Наличие небольшого числа поперечных связей
придает полимеру хорошие эластические свойства, что исполь-
зуется при получении каучуков. Полимеры с высокой степенью
сшивания отличаются большой жесткостью и стабильностью
размеров при нагревании и напряжении, что наглядно прояв-
ляется в фенолформальдегидных и мочевиноформальдегидных
полимерах.
1.4. Молекулярный вес
Молекулярный вес имеет первостепенное значение при синтезе
и применении полимера. Именно высоким молекулярным весом
обусловлены многие интересные и часто уникальные свойства
полимеров. Наиболее важные механические свойства полимеров
сильно зависят от их молекулярного веса. Так, например, механи-
ческая прочность начинает проявляться только при молекулярном
весе соединения выше 5000—10 000. Выше этой минимальной
величины механические характеристики полимера резко улуч-
шаются при увеличении молекулярного веса и перестают зависеть
от него только при весьма больших значениях. Во многих случаях
существует некоторый интервал молекулярных весов полимера,
соответствующий его оптимальным свойствам. Это нужно все
время иметь в виду при ознакомлении с реакциями образования
полимеров, которые рассматриваются в этой книге. Ценность
той или иной реакции, того или иного метода во многом зависит
от того, приводят ли они к образованию высокомолекулярного
полимера пли пет. Возможность регулирования молекулярного
веса имеет большое значение и при практическом использовании
различных методов синтеза полимеров.
Понятия молекулярного веса для полимеров и низкомолеку-
лярных соединений не адекватны. От последних полимеры отли-
чаются полидпсперспостыо, или неоднородностью по молекуляр-
ному весу. Если даже синтезированный полимер не содержит
примесей или загрязнений, он не является чистым веществом
в общепринятом смысле этого слова. Самые чистые полимеры
представляют собой смесь молекул различного молекулярного
веса. Полидисперсность полимеров обусловлена статистическими
закономерностями реакций их образования. Когда говорят о моле-
кулярном весе полимера, имеют в виду его средний молекулярный
вес. Для того чтобы полностью охарактеризовать полимер, необ-
ходимо знать его средний молекулярный вес п распределение
по молекулярным весам отдельных молекул данного полимера.
Контроль за молекулярным весом и молекулярповесовым рас-
28
Глава 1
пределением (МБР) часто используется при получении полимера
с заданными физическими свойствами.
Существуют различные методы определения среднего молеку-
лярного веса полимера. К ним относятся светорассеяние, виско-
зиметрия, ультрацентрифугирование и седиментация [6—8]. Моле-
кулярные веса полимеров, определенные различными методами,
как правило, не совпадают. Разница в средних молекулярных
весах обусловлена тем, что зависимость определяемого свойства
от размера молекулы непостоянна. Некоторые методы мало чув-
ствительны к молекулам большого размера, тогда как другими
методами плохо определяются небольшие молекулы. В результате
полученный средний молекулярный вес не совсем верно отражает
количество больших или малых молекул. Ниже рассмотрены наи-
более важные средние молекулярные веса, определяемые суще-
ствующими методами:
1) Среднечисловой молекулярный вес Мп определяется при
оценке таких свойств, как уменьшение температуры замерзания
(криоскопия), повышение температуры кипения (эбуллиометрия),
осмотическое давление и понижение давления паров. Величина Мп
характеризует суммарный вес w всех молекул в образце полимера,
деленный на общее число молекул в полимере, т. е.
Мп
IV
2^
nxmx
(1.14)
2
где сумма охватывает все различные полимерные молекулы
от ж = 1 до ж = оо,а Nx — число молекул с молекулярным весом
Мх> Уравнение (1.14) можно также записать в виде
Ж = 2^м- (1.15>
где Nx — мольная доля (или числовая доля) молекул размером Мх.
2) Средневесовой молекулярный вес М w определяется свето-
рассеянием и выражается как
Mw = 2 tvxMx,
(1.16)
где wx — весовая доля молекул с молекулярным весом Мх.
Величину Mw можно также определить как
2c^/x 2с-^ 2л»мх
1 w~ 2сх с ~ 2*л
(1.17)
где сх — весовая концентрация М х молекул, с — суммарная
весовая концентрация всех макромолекул и выполняются следую-
Введение
29
щие соотношения:
С «г
сх — NXMX,
(1.18)
(1.19)
с = Усх = У ДД/ИХ. (1.20)
3) Средневязкостной молекулярный вес Мv измеряется методом
вискозиметрии и определяется как
М„ = [3 н;.Ж]1/а =
£ ЛгхЛ/“+1 ~11/а
_ y\NxMx _
(1-21)
Где а — константа. При а = 1 средневязкостной и средневесовой
молекулярные веса равны между собой. Однако почти всегда Mv
меньше М w, так как а, как правило, имеет значение 0,5—0,9.
Для более полной характеристики полимера нужно знать
несколько средних молекулярных весов. В этом нет никакой
Рис. 1.2. Распределение
по молекулярным весам в
обычном полимере.
необходимости лишь в том случае, если полимер моподисперсный,
т. е. состоит из молекул одинакового молекулярного веса, так
как все средние молекулярные веса у такого полимера одинаковы.
Совершенно иначе обстоит дело для полидисперсного полимера,
у которого указанные выше три средних молекулярных веса
отличаются друг от друга, если постоянная а в уравнении (1.21)
меньше единицы, что имеет место в действительности. Тщательный
анализ уравнений (1.14)—(1.21) показывает, что с увеличением
доли высокомолекулярной фракции в полимере средние молеку-
лярные веса возрастают в следующем порядке: Мw > Mv > Мп.
Для полидисперсного полимера при расширении молекулярно-
весового распределения различие в средних молекулярных весах
увеличивается. На рис. 1.2 приведена типичная картина моле-
30
Глава 1
кулярновесового распределения полимера. На кривой распре-
деления указаны примерные положения различных средних моле-
кулярных весов.
Для большинства практических целей бывает достаточно оце-
нивать молекулярный вес, измеряя Мп и либо Мw, либо .17
Чаще определяют М„, так как он близок Мw (различие не пре-
вышает 10—20%). В расчетах в большинстве случаев пользуются
Мп и Мw полимера. Первый из них более чувствителен к низко-
молекулярным фракциям, второй — к высокомолекулярным.
Отношение двух средних молекулярных весов М 1С/Мп зависит
от ширины кривой распределения (рис. 1.2) и часто применяется
в качестве критерия полидисперсностп полимера. Для полностью
однородного полимера коэффициент полидпсперсности должен
быть равен единице. Для всех реальных полимеров он всегда
больше единицы и возрастает с увеличением полидисперсностп.
Оценка полимера только по Мп без знания его полидисперс-
ности может привести к большим недоразумениям, так как многие
свойства полимера, и в частности его прочность и вязкость рас-
плава, определяются прежде всего размером той части молекул,
которые составляют основную массу его по весу. Свойства поли-
мера в большей степени зависят от наиболее длинных молекул,
чем от коротких. Рассмотрим, например, гипотетическую смесь,
состоящую на 95% из молекул молекулярного веса 10 000 и на 5%
из молекул молекулярного веса 100. (Низкомолекулярной фрак-
цией в полимере может быть мономер, низкомолекулярный полимер
или просто примесь.) Вычисленные с помощью уравнений (1.14)
и (1.16) Мп и М1С равны соответственно 1680 и 9505. По значе-
нию Мп (1680) можно получить неправильное представление
о свойствах такого полимера. Свойства его определяются глав-
ным образом макромолекулами молекулярного веса 10 000, кото-
рые составляют 95% веса полимера. Средневесовой молекулярный
вес полимера — более объективный показатель свойств, которых
можно ожидать от полимера. Средпечпсловой молекулярный вес
нужен в основном для характеристики полидпсперсности образца
полимера, которая выражается отношением 717 „./'.Т7„.
Кроме различных средних молекулярных весов полимера,
часто желательно и даже необходимо точно знать распределение
по молекулярным весам. Как уже было указано, существует
определенный интервал молекулярных весов, при котором то пли
иное свойство полимера будет оптимальным. Образец полимера
с наибольшей долей макромолекул такого размера представляется
наиболее качественным с точки зреппя этого свойства. Так как
образцы с одинаковым средним молекулярным весом могут иметь
разное молекулярновесовое распределение, то данные о поли-
Введение
31
дисперсности полимера помогут сделать правильный выбор между
различными образцами. Для оценки молекулярновесового рас-
пределения полимера используют различные методы фракциони-
рования [9]. Все они основаны па том, что существует определен-
ная зависимость между каким-либо свойством полимера, напри-
мер растворимостью или проницаемостью, и его молекулярным
весом.
1.5. Физическое состояние
1.5а. Кристалличность и аморфность
От обычных, низкомолекулярных соединений твердые поли-
меры отличаются физическим состоянием или морфологией.
Большинство полимеров проявляют свойства твердых кристалли-
ческих веществ и высоковязких жидкостей [10, И]. На рентгено-
п электронограммах полимеров обнаруживаются четкие рефлексы,
характерные для пространственно упорядоченных, кристалли-
ческих веществ, а также диффузные картины, типичные для
жидкостей. Для обозначения упорядоченных и неупорядоченных
областей в полимере применяются соответственно термины кри-
сталлический и аморфный. Степень кристалличности разных поли-
меров весьма различна. Хотя отдельные полимеры могут быть
полностью аморфными или целиком кристаллическими, боль-
шинство из них характеризуется частичной кристалличностью,
т. е. они являются полукристаллическими.
Истинная природа кристалличности полимеров была пред-
метом многолетней дискуссии. Согласно мицеллярной теории.
распространенной в 30-х годах, полимеры состоят из небольших
упорядоченных, кристаллических областей, называемых кристал-
литами, вкрапленных в неупорядоченную аморфную массу поли-
мера. При этом макромолекулы могут одновременно проходить
через несколько кристаллических областей, так что кристаллиты
образуются в тех случаях, когда сегменты при кристаллизации
располагаются параллельно друг другу. Несколько упорядочен-
ных сегментов одной макромолекулы могут принимать участие
в образовании ряда кристаллитов. Сегменты цепи, расположенные
между кристаллитами, образуют неупорядоченную, аморфную
основу полимера. Схематически это представлено на рис. 1.3.
В конце 50-х годов, когда из растворов полимеров были полу-
чены монокристаллы в форме пластин, названных ламеллами,
возникла ламеллярная теория (или теория складывания цепей).
Согласно этой теории, при кристаллизации макромолекулы накла-
дываются сами на себя, образуя подобие гармошки (рис. 1.4).
Дифракционные картины таких монокристаллов подтверждают
это. Теория складывания цепей применима к большинству поли-
32
Глава 1
меров, кристаллизующихся из раствора и из расплава. По этой
теории полукристаллиты представляют собой кристаллы, полу-
ченные складыванием цепей, с тем или иным количеством дефек-
тов. Структура кристаллов полимеров близка структуре кристал-
лов обычных низкомолекулярных соединений. Причиной дефектов
в таких кристаллах могут быть несовершенность складок, нерегу-
лярность упаковки, запутанность цепей, их перемещения, наличие
свободных концов цепей и сорбированных примесей и др.
К сожалению, полагают, что мицеллярная и ламеллярная тео-
рии взаимно исключают друг друга. С практических позиций
было бы разумно принять такую рабочую модель кристаллич-
ности полимера, которая использовала бы особенности обеих
концепций. Теория складывания цепей наиболее применима для
высококристаллических полимеров, которые можно рассматри-
вать как однофазную кристаллическую систему с дефектами. Для
полимеров со средней или низкой степенью кристалличности
можно с успехом применять мицеллярную теорию, рассматривая
полимер как двухфазную систему, состоящую из кристаллитов,
вкрапленных в некристаллическую, аморфную массу полимера.
Структура же кристаллитов таких полимеров может иметь харак-
Введение
тер складчато-цепной, ламеллы. Степень и тип кристалличности
полимера экспериментально оценивают по изменению плотности
такими методами, как дифракция электронов и рентгеновских
лучей, инфракрасная спектроскопия и ядерный магнитный резо-
нанс. При этом, сравнивая полученные данные с результатами
Рис. 1.4» Складчато-цепная картина кристалличности полимера.
измерений полностью кристаллического и полностью аморфного
образцов полимера, рассчитывают степень кристалличности в весо-
вых или объемных процентах.
1.56. Факторы, влияющие на степень
кристалличности полимеров
Независимо от того, упорядоченным или разупорядоченным
является данный образец полимера, прежде всего необходимо
отметить тенденцию полимеров к кристаллизации. От проявления
этой тенденции во многом зависит практическое использование
полимера, так как кристалличность оказывает существенное
влияние на термические, механические и другие важные свойства
полимеров. Вследствие разной степени кристалличности полимеры
имеют разные свойства, их синтезируют разными способами
и применяют в различных областях. Полезно отметить факторы,
которые обусловливают степень кристалличности различных поли-
меров. Степень кристалличности полимера зависит от склонности
его структуры к упаковке в кристаллическую форму и от вели-
чины вторичных сил (межмолекулярное взаимодействие) поли-
мерных цепей. Упаковка облегчается для полимерных цепей,
имеющих регулярную структуру, некоторую гибкость, компакт-
34
Глава 1
ность и линейную форму. Чем сильнее вторичные силы, тем больше
тенденция полимерных цепей к упорядочиванию и кристаллизации.
Некоторые полимеры имеют высокую кристалличность глав-
ным образом из-за склонности их цепей к упаковке, кристаллич-
ность же других объясняется сильными вторичными взаимодей-
ствиями. Существуют полимеры, кристаллизации которых благо-
приятствуют оба фактора. Например, полиэтилен с точки зрения
склонности к упаковке в кристаллическую форму, по-видимому,
имеет наиболее благоприятную структуру. Очень простая и чрез-
вычайно регулярная структура его позволяет цепям плотно упа-
ковываться без каких-либо ограничений. Кристаллизации поли-
этилена способствует также гибкость его цепей, так как .легко
реализуются конформации, наиболее выгодные для упаковки.
Полиэтилен легко кристаллизуется до высокой степени вследствие
его простой и регулярной структуры, несмотря па то что вторич-
ные взаимодействия в нем малы.
В отличие от полиэтилена другие полимеры имеют менее
простую и менее регулярную форму цепей. Полп-е-капролактам
можно рассматривать как модифицированный полиэтилен, в кото-
ром между каждыми пятью метиленовыми группами содержится
амидная группа. Поли-е-капролактам и другие полиамиды
являются высококристаллическими полимерами. Амидная груп-
па — полярная и обусловливает значительно более сильные меж-
молекулярные взаимодействия (водородные связи) в полиамидах
по сравнению с полиэтиленом; именно этим объясняется хорошая
крпсталлизуемость таких полиамидов. Однако цепи полиамидов
сложнее, чем у полиэтилена, поэтому упаковка цепей таких поли-
меров, обеспечивающая проявление водородных связей, должна
быть затруднена. Ввиду этого степень кристалличности полиамидов
несколько меньше, чем можно было ожидать, основываясь на рас-
смотрении только сильного межмолекулярпого взаимодействия
в них. Кристалличность таких полимеров, как полиамиды, можно
значительно повысить механическим вытягиванием, которое облег-
чает упорядочивание и выравнивание полимерных цепей.
Очень слабую тенденцию к кристаллизации, как правило,
имеют такие полимеры, как полистирол, поливинилхлорид, поли-
метилметакрилат. Это обусловлено структурной нерегулярностью
полимеров этого типа. Боковые заместители повышают жесткость
цепи полимера и затрудняют их упаковку, хотя из-за диполь-
дипольного взаимодействия между такими группами вторичные
силы более ярко выражены, чем в полиэтилене. Плохо кристалли-
зуются также полимеры с жесткими циклическими фрагментами
в основной полимерной цепи, например целлюлоза и полиэтилен-
терефталат. Вследствие очень большого числа поперечных связей,
приводящего к высокой жесткости полимера, не кристаллизуются
фенолформальдегидные и мочевиноформальдегидные полимеры.
Введение
.35
На кристаллпзуемость полимера влияет также гибкость его
цепей. Полимеры с очень гибкими цепями, например полисилок-
сапы и натуральный каучук, неспособны к упаковке. Это обусло-
влено тем, что из-за высокой гибкости цепей не фиксируются
конформации, требуемые для упаковки. Гибкость таких полиме-
ров, как полисилоксаны и натуральный каучук, объясняется гро-
моздкими Si — О и ^нс-олефиновыми группами соответственно.
Такие полимеры практически полностью аморфны и обладают
таким очень важным свойством, как эластичность.
1.5в. Тепловые переходы
Для полимеров существует два основных типа температур
перехода: температура плавления Тап и температура стеклова-
ния ТСТ. Температура плавления — это температура плавления
кристаллической фазы полимера. Температурой стеклования назы-
вается температура, при которой аморфные области полимера
приобретают свойства, характерные для стеклообразного состоя-
ния: хрупкость, жесткость и прочность. Различия между двумя
указанными тепловыми переходами можно легко пояять, рас-
сматривая изменения, происходящие в размягченном, жидком
полимере при его охлаждении. С уменьшением температуры
уменьшаются поступательная, вращательная и колебательная
энергии в молекуле полимера. Когда суммарная энергия моле-
кулы уменьшается до величины, при которой поступательная
и вращательная энергии уже практически отсутствуют, стано-
вится возможной кристаллизация полимера. При этом*, если
удовлетворяются определенные требования симметрии, то моле-
кулы могут принимать упорядоченное расположение и таким
образом реализуется кристаллизация. Температура, при которой
происходит этот процесс, и есть Тил. Однако не у всех полимеров
создаются необходимые условия для кристаллизации. Если тре-
бования симметрии не удовлетворяются, то кристаллизация
не протекает, но по мере дальнейшего снижения температуры
Энергия молекул продолжает уменьшаться. При достижении 7^?
сегментальное движение полимерных цепей прекращается из-за
сильного ослабления вращения связей.
Возможность реализации у полимера одного или обоих тепло-
вых переходов зависит от его морфологии. Полностью аморф-
ные полимеры характеризуются только Тст, тогда как полностью
кристаллические полимеры имеют только ТПЛ. Большинство же
полимеров при Тпп подвергается кристаллизации лишь частично,
подобные полукристаллические полимеры характеризуются темпе-
ратурой плавления и температурой стеклования. Тепловые пере-
ходы легко измерить по изменению таких свойств, как удельный
объем, теплоемкость. На рис. 1.5 приведена зависимость удель-
36
Глава 1
ного объема от температуры для полностью аморфного и пол-
ностью кристаллического полимера (сплошные линии). Темпера-
тура плавления — это переход первого рода с очень резким изме-
нением удельного объема; температура стеклования — это переход
второго рода, характеризующийся лишь изменением температур-
ного коэффициента удельного объема. Соответствующая кривая
для полукристаллического полимера состоит из кривой для кри-
сталлического полимера и пунктирной линии, соответствующей
переходной области в стеклообразное состояние.
Температуры плавления и стеклования полимера влияют
на механические свойства полимера при той или иной температуре
Рис. 1.5. Определение темпера-
туры стеклования и температуры
плавления полимера по измене-
нию его удельного объема.
и определяют температурный интервал его использования.
В табл. 1.3 приведены Тс.г и Тпл для некоторых наиболее распро-
страненных полимеров [12]. Рассмотрим вкратце, как Тст и Тлл
меняются от одного полимера к другому. Можно рассматривать
оба перехода одновременно, так как па них примерно одинаково
влияет химическое строение полимера. Полимеры с низкими Тст,
как правило, имеют низкие Т’пл! высокие Тст и Гпл также обычно
присущи одним и тем же полимерам. Полимерные цепи, которые
нелегко поддаются вращению вокруг связей, необходимому для
стеклообразного перехода, должны, по-видимому, и плавиться
с трудом. Это положение справедливо, так как оба перехода
зависят примерно от одних и тех же факторов. На тот и другой
термический переход, как правило, одинаково влияют молекуляр-
ная симметрия, структурная жесткость и межмолекулярное
взаимодействие [10, 11]. Сильное межмолекулярное взаимодей-
ствие (из-за высокой полярности или водородных связей) приво-
дит к сильным кристаллизационным силам, что обусловливает
высокие температуры Тлл. Сильное межмолекулярное взаимо-
действие понижает также подвижность цепей аморфного поли-
мера, в результате и ТСт имеют высокие значения. Небольшая
Термические переходы в полимерах [12J
Таблица 1.3
Полимер Элементарное звено т СТ’ °C ГПЛ' °с‘
Полидиметилси- -OSi(CH3)2- -123 от —85 до —65
локсан
Полиэтилен — СН2СН2 — -115 137
Полиоксиметилен — СН2О — -85 181
Натуральный кау- — С Н2С(СН3) = СНСН, - -73 14
чук
П олиизобутилен — СН2С(СН3)2- -73 44
Полиоксиэтилен -СН2СН2О- -67 66
Полипропилен — СН2СН(СН3) — -20 176
Поливинилфторид — ch2chf— -20 200
Поливинилиден- -СН2СС12 — -19 190
хлорид
По ливинилацета т — СН2СН(ОСОСН3)- 28
Полихлортрифтор- -CF2CFC1 — 45 ’ 220
этилен
Поли-ё-капролак- -(CH2)5CONH- 50 223
там
П олигексамети- -NH(CH2)6NHCO(CH2)4CO- 53 265
ленадипамид
Полиэтилентере- —ОСН 2СН2ОСО—со— 69 265
фталат Поливинилхлорид -СН2СНС1 — 81 212
Полистирол -СН2СНСвН5- 100 240
Полиметилметак- -СН2С(СН3)(СО2СН3)- 105 200
рилат
Триацетат целлю- СН3СОО -< 105 306
лозы Политетрафтор- СН3СОО ООССНз -cf2cf2- 127 327
этилен
подвижность полимерных цепей, повышенная их жесткость
и высокие Уст характерны для полимеров с несколькими замести-
телями в боковой цепи, как, например, в полиизобутилене и поли-
тетрафторэтилене, пли объемистыми заместителями, как в поли-
стироле. Температура плавления кристаллических полимеров,
полученных из таких жестких цепных макромолекул, как, напри-
мер. у полимеров с циклическими группами в основной цепи,
должна быть также высокой. Высокие Тст и имеют такие полиме-
ры, как ацетат целлюлозы. В то же время полимеры с очень гибки-
ми цепями, например полисилоксаны, имеют низкие Тст и Тпл.
На величину 7'(т и Тил также сильно влияет молекулярная
симметрия полимерных цепей. Полимеры, структурно асимметрич-
ные (например, поливинилхлорид и полипропилен), имеют более
рысокпе 7'ст и Ти:(, чем их симметричные аналоги (поливинили-
денх.торид и полипзобутилен). Асимметричные цепи являются
более полярными и поэтому могут упаковываться плотнее, обеспе-
чивая более сильное межмолекулярное взаимодействие. Хотя
молекулярная структура полимерных цепей влияет одинаково
на 7CT и Тпл, количественные изменения обеих температур пере-
хода не всегда близки друг другу. В табл. 1.3 различные поли-
меры приведены в порядке увеличения их Тст. Как видно из дан-
ных этой таблицы, Тил, как правило, увеличиваются в том же
порядке, но есть много полимеров, Тлл которых не укладываются
в этот ряд. Так, например, полиэтилен и полиоксиметилен имеют
низкие Уст, что обусловлено очень высокой гибкостью их цепей,
в то же время, имея простую и регулярную структуру, они дают
плотноупакованпые кристаллы с высокими Тлл.
Следует отметить, что существуют факторы, которые приводят
к уменьшению тенденции полимера к кристаллизации, но обеспе-
чивают тем не менее повышенные значения Тлл (и также Тст).
Это объясняется тем, что степень кристалличности полимера
определяется кинетическими и термодинамическими факторами,
тогда как температура плавления зависит только от термодина-
мического фактора. Жесткоцепные полимеры медленно или трудно
кристаллизуются, но кристаллическая фаза их имеет высокую
температуру плавления. (Степень кристалличности таких поли-
меров можно существенно повысить механической вытяжкой,
обеспечивающей ориентацию и кристаллизацию полимерных
цепей.) Рассмотрим с этих позиций различия между полиэтиленом
и полпгексаметиленадипамидом. Обладая простой и очень регу-
лярной структурой, полиэтилен кристаллизуется легче и быстрее
полиамида, поэтому его обычно получают с более высокой степенью
кристалличности, чем полиамид (80 и 50% соответственно).
В то же время 7’пл полиамида значительно выше (примерно
на 130 “С), чем у полиэтилена, что обусловлено значительно более
сильным межмолекулярным взаимодействием.
Введение
3
1.6. II рнленен не полилеров
1.6а. Механические свойства
Ряд свойств полимеров, например газопроницаемость, устой-
чивость к действию растворителей и химических реагентов,
электрическое сопротивление, имеют большое значение при опре-
делении возможности использования данного полимера в тех
или иных областях. Однако основным свойством, определяющим
ценность полимера, является его механическое поведение, а именно
деформация и текучесть
под нагрузкой. Механиче-
ские свойства полимера
можно характеризовать его
деформ а ц и о н но-прочност-
ной характеристикой [13].
При этом исследуют поведе-
ние полимера при прило-
жении к нему напряжения
и определяют деформацию
до момента разрушения об-
разца. Полученные ре-
зультаты обычно представ-
Р и с. 1.6. Деформацпопно-
прочпостные характеристики
для типичных эластомера (7),
гибкого пластика (2), жесткого
пластика (3) п волокна (4).
Деформация ЛЬ/L, °/а
ляют в виде кривой зависимости напряжения от удлинения
(деформации). Напряжения выражают в килограммах на квадрат-
ный сантиметр, а удлинение — как относительное увеличение дли-
ны образца полимера (т. е. \ЫЬ, где L — длина исходного об-
разца). [Удлинение можно также выразить в процентах (ДЛ/Л) х
X 100%.] На рис. 1.6 приведены некоторые кривые зависимости
напряжения от деформации. Деформационно-прочностное поведе-
ние полимера характеризуют следующими четырьмя наиболее важ-
ными величинами:
1) Модуль — сопротивление деформации, которое определяется
отношением напряжения, рассчитанного на исходное сечение
образца к относительному удлинению (ДЛ/Л).
2) Разрывная прочность — напряжение, необходимое для
разрушения образца.
3) Разрывное удлинение — деформация в момент разрушения
обпазпа.
40
Глава 1
4) Эластическая деформация — эластичность, оцениваемая
степенью обратимой деформации.
Механические свойства полимера изменяются в широких пре-
делах в зависимости от степени кристалличности, числа попереч-
ных связей, Гст и Гпл. Высокой прочностью и низкими удлине-
ниями характеризуются полимеры с высокой степенью кристаллич-
ности, большим числом поперечных связей пли высокой темпера-
турой стеклования. Наоборот, полимеры с низкой степенью кри-
сталличности или сшивания и низкими ТС1. имеют высокие удли-
нения и низкую прочность. Температурный предел работоспособ-
ности полимера определяется его температурами плавления
и (или) стеклования; аморфный полимер теряет прочность выше 7’ст,
а кристаллический полимер — выше Гпл. ’
Число полимеров, которое можно синтезировать, практически
безгранично. До того как будет синтезирован тот или другой
полимер, необходимо представлять себе отчетливо, каких свойств
можно ожидать от него. Различные сочетания рассмотренных
выше характеристик (кристалличности, степени сшивания, 7'ст
и Гпл) позволяют получить полимер, который можно использовать
как волокно, гибкий пластик, жесткий пластик или эластомер
(каучук) [14—16]. Наиболее распространенными типами изделий,
для которых полимеры применяют в виде указанных выше мате-
риалов, являются одежда (волокно), упаковочные пленки (гибкий
пластик), контактные линзы (жесткий пластик) и резинки (эласто-
мер). Табл. 1.4 дает представление о применении многих обычных
Таблица 1.4
Виды материалов на основе различных полимеров
Эластомеры
Пластики
Волокна
Полиизопрен Полиэтилен
Полиизобутилен Политетрафторэтилен
Полиметилметакрилат
Фенолформальдегидный
Мочевиноформальдегидный
Меламиноформальдегидный •
Полистирол------>
Поливинилхлорид
Полиуретан------>-
-<- Полисилоксан---►
Полиамид------------->-
-<- Сложный полиэфир--->-
ч- Производные целлюлозы
-<- Полипропилен------->-
Полиакрилонитрил
Введение
41
полимеров. Некоторые полимеры могут быть использованы в виде
различных материалов, так как их механические свойства можно
регулировать соответствующими химическими или физическими
методами, как правило, изменяющими степень кристалличности
полимеров. Например, некоторые полимеры находят применение
п в качестве пластиков, и в качестве волокон, тогда как другие —
как в виде эластомеров, так и в виде пластиков.
1.66. Эластомеры, волокна, пластмассы
Различие между волокнами, пластиками и эластомерами легко
понять из рассмотрения типичных деформационно-прочностных
зависимостей, представленных на рис. 1.6. Модуль полимера
определяется по начальному наклону такой кривой, разрывная
прочность и разрывное удлинение — это наиболее высокие значе-
ния напряжения и деформации соответственно. Группу эластоме-
ров составляют полимеры, которые могут подвергаться очень
большим обратимым деформациям (вплоть до 500—1000%) при
сравнительно небольших нагрузках. Этому требованию удовлетво-
ряют часто аморфные полимеры с низкой температурой стеклова-
ния и незначительным межмолекулярным взаимодействием, что
обеспечивает высокую подвижность полимерных цепей. Для того
чтобы деформация быстро снималась (эластичность), необходима
низкая степень сшивания цепей полимера. Эластомер должен
иметь низкий начальный модуль (<10,5 кгс/см2), быстро возра-
стающий с увеличением деформации, обеспечивая таким образом
прогрессивно увеличивающееся сопротивление деформированию,
в противном случае полимер не будет иметь никакой прочности.
Многие эластомеры приобретают такую прочность в результате
частичной кристаллизации при вытягивании. Однако Тпя кристал-
лической фазы должна быть ниже температуры применения
эластомера, для того чтобы кристаллы плавились и деформация
была обратимой после снятия с эластомера нагрузки. Если в про-
цессе вытяжки полимер не кристаллизуется, ему можно придать
прочность путем сшивания или введения неорганического арми-
рующего наполнителя. Типичным эластомером является полиизо-
прен (натуральный каучук). Это аморфный, легко сшивающийся
полимер с низкой Тст (—73 °C), кристаллизующийся при вытяжке
и имеющий низкую Тпя (14 °C). Начальный модуль сшитого
каучука ниже 7 кгс/см2, однако его прочность возрастает до при-
мерно 140 и 210 кгс/см2 при деформации 400 и 500% соответст-
венно. Во всем интервале деформаций его удлинение имеет обра-
тимый характер.
К волокнам относятся полимеры с очень высоким сопротивле-
нием деформированию, такие полимеры характеризуются низкими
удлинениями (10—50%) и очень высокими модулем (более
42
Глава 1
3500 кгс/см2) и разрывной прочностью (более 3500 кгс/см2).
Полимер должен быть высококристаллическим и содержать поляр-
ные группы, обеспечивая сильное межмолекулярное взаимодей-
ствие. Для достижения высокой кристалличности волокна его
подвергают механической вытяжке. Для того чтобы волокно
сохраняло физическую целостность при температурах химической
чистки, стирки и глажения темнература плавления его должна
быть выше 200 СС, но и не превышать 300 °C: в противном случае
волокно нельзя будет формовать из расплава. Полимер должен
растворяться в растворителях, применяющихся для формования
из раствора, но быть устойчивым к действию растворителей, исполь-
зующихся в сухой химчистке. Волокнообразующие полимеры
характеризуются не слишком высокой и не слишком низкой Тст'.
при очень высоких Тс^ затрудняется вытяжка и глажение, тогда
как ткани из полимеров с низкой Тс^ сильно мнутся. Типичным
волокнообразующим полимером является полигексаметиленадип-
амид. При вытяжке происходит значительная кристаллизация,
а водородные связи между амидными группами обеспечивают силь-
ное межмолекулярное взаимодействие и, как следствие этого,
волокно имеет очень высокие разрывную прочность (7000 кгс/см2)
и модуль (49 000 кгс/см2) и низкое удлинение'(менее 20%). Такой
полимер имеет оптимальные значения Тпп и Тст (265 и 53 °C соот-
ветственно).
По механическим свойствам пластики, составляющие третью
большую группу полимеров, занимают промежуточное положение
между эластомерами и волокнами. Различают два типа пластиков:
гибкие и жесткие. Гибкие пластики характеризуются стененью
кристалличности от средней до высокой, их Тил и Тст колеблются
в широких пределах. Они имеют модуль 1400—35 000 кгс/см2,
разрывную прочность от 140 до 700 кгс/см2 и разрывные удлине-
ния от 20 до 800 %. Наиболее часто полимеры, относящиеся к этой
подгруппе, характеризуются нижними пределами модуля и раз-
рывной прочности из только что указанных и наиболее высокими
значениями удлинения. Так, например, типичный гибкий пластик
полиэтилен имеет разрывную прочность 245 кгс/см2, модуль
2100 кгс/см2 и разрывное удлинение 500%. К гибким пластикам
относятся также полипропилен и полигексаметиленадипамид.
Эднако последний может быть использован и как волокно. При
вредней степени кристалличности это пластик, тогда как вытяжка
гревращает его в волокно. Для многих гибких пластиков харак-
терны такие же высокие разрывные удлинения, как и у эластоме-
)ов. Но от последних они отличаются тем, что лишь небольшая
гасть деформации (менее 20%) является упругой. После прохо-
кдения зоны упругой деформации пластик подвергается перма-
гентному удлинению, сохраняя увеличенные размеры после
нятия наппяжения.
Введение
43
Жесткие пластики весьма резко отличаются от гибких. Они
характеризуются большой жесткостью и высоким сопротивлением
деформированию. Модуль жестких пластиков 7000—35 000 кгс/см2,
разрывная прочность 350—850 кгс/см2. Наиболее важным призна-
ком их являются очень низкие удлинения (менее 0,5—3%) перед
разрушением. К этой категории относятся жесткоцепные, аморф-
ные полимеры. Высокая жесткость в ряде случаев обусловлена
большим числом поперечных связей (например, в фенолформаль-
дегидных, мочевиноформальдегидных и меламиноформальдегидных
полимерах), а в других случаях объемистыми боковыми группами,
приводящими к повышению Тст полимера (например, Тст поли-
стирола 100 СС и Тст полиметилметакрилата 105 °C).
Вопросы и упражнения
1.1. Напишите схему реакции образования полимеров из сле-
дующих мономеров:
а) СН2=СН—СО,Н ;
б) СН2СН2СН2О ;
в) H2N-(CH2)5-NH2 + С1СО-(СН2)5-СОС1;
г) НО—(СН ) -со„н,
£ О Z 7
NCO
е) СН2=СН—F .
1.2. Какова структура элементарного звена полимеров, о кото-
рых шла речь в вопросе 1.1? Можно ли для получения тех же
полимеров использовать другие мономеры?
1.3. Назовите полимеры, которые образуются из мономеров,
указанных в вопросе 1.1. Где возможно, дайте для них другие
названия.
1.4. Укажите, какие из полимеров, полученных в вопросе 1.1,
конденсационного типа, а какие полпмерпзационного. Классифи-
цируйте реакции образования этих полимеров на ступенчатые,
цепные пли реакции с раскрытием цикла.
1.5. Как бы вы экспериментально определили, по какому
механизму (цепному или ступенчатому) идет образование поли-
мера из неизвестного мономера X?
1.6. Образец полистирола состоит из ряда фракций:
44
Глава 1
Фракция Весовая доля Молекуляр- ный вес
A 0,10 12 000
Б 0,19 21000
В 0,24 35 000
Г 0,18 49 000
д 0,11 73 000
E 0,08 102 000
Ж 0,06 122 000
3 0,04 146 000
Вычислите среднечисловой и средневесовой молекулярный вес
полимера. Постройте кривую молекулярновесового распределения
(по типу, приведенному на рис. 1.2).
Список литературы
1. Carothers W. Н., i. Am. Chem. Soc., 51, 2548 (1929).
2. Huggins M. L., Corradini P.,' Desreux V., Kratsky 0., Mark H., J. Polymer
Sei., B6, 257 (1968).
3. Flory P. J., Principles of Polymer Chemistry, chap. II, Cornell University
Press, Ithaca, New York, 1953.
4. Fox R. B., Beaman R., Bikales N. M., Block В. P., Cohn W. E., Liring-
ston H. K., Loening K. L., Mercurio A., Schiller A. M., Macromolecules,
1, 193 (1968); Polymer Preprints, 8(1), e (1967).
5. Brandup J., Immergut E. H. (eds.), Polymer Handbook, chap. I, Inter-
science Publishers, John Wiley and Sons, Inc., New York, 1966.
6. Miller M. L., The Structure of Polymers, chaps. 1—3, 7, Reinhold Book
Corp., New York, 1966.
7. Billmeyer F. W., Jr., J. Polymer Sci., C8, 161 (1965).
8. Моравец Г., Макромолекулы в растворе, пзд-во «Мир», М., 1967.
9. Фракционирование полимеров, под ред. М. Кантова, изд-во «Мир», М.,
1971.
10. Gordon М., High Polymers, chaps. 3—7, Addison-Wesley Publishing Co.,
Inc., Reading, Mass., 1963.
11. Шарплез А., Кристаллизация полимеров, изд-во «Мир», М., 1968.
12. Brandup J., Immergut E. H. (eds.), Polymer Handbook, pp. III-32—92,
Interscience Publishers, John Wiley and Sons, Inc., New York, 1966.
13. Nielsen L. E., Mechanical Properties of Polymers, Reinhold Book Corp.,
New York, 1962.
14. Cooper W., Grace N. S., J. Polymer Sci., C12, 133 (1966).
15. Tippetts E. A., Zimmerman J., J. Appl. Polymer Sci., 8, 2465 (1964).
16. Billmeyer F. W., Jr., Textbook of Polymer Science, chaps. 20—22, Inter-
science Publishers, John Wiley and Sons, Inc., New York, 1962.
ГЛАВА 2
ПОЛИКОНДЕНСАЦИЯ
Наиболее распространенные поликонденсационные полимеры
приведены в табл. 1.1, из которой также видно, что все реакции
их образования идут по законам поликонденсации. Эта глава
посвящена подробному рассмотрению закономерностей поликон-
денсации. Синтез полимеров конденсационного типа полимери-
зацией с раскрытием цикла будет рассмотрен в гл. 7. Для син-
теза полимеров поликонденсацией могут быть использованы самые
различные химические реакции. К ним относятся: этерификация,
амидирование, образование уретанов, ароматическое замещение
и другие. Поликонденсация протекает в результате взаимодей-
ствия двух различных функциональных групп, например гид-
роксильной и карбоксильной или изоцианатной и гидроксильной
групп *).
По характеру мономеров поликонденсацию можно разделить
на два типа. К первому типу относятся реакции с использованием
двух различных полифункциональных мономеров, каждый из кото-
рых содержит только один тип функциональных групп. (П олифу нк-
циональным мономером называется мономер с двумя или более
функциональными группами на молекулу.) Ко второму типу
относятся реакции с участием одного мономера, содержащего оба
типа функциональных групп. На примере синтеза полиамидов
можно проиллюстрировать оба типа реакций. Так, полиамиды
можно получить взаимодействием диаминов с дикарбоновыми
кислотами
wH2N —R —NH2 + nHOOC-R'-COOH —>
—> Н —( —NH —В —NHCO —В' —СО —)n —OH-[-(2n —1)Н2О (1.1)
или самоконденсацией аминокислот
»H2N-R —СООН —» H-(-NH-R-CO-)n-OH-L(n-l)H20. (2.1)
*) Исследование закономерностей процессов поликонденсации привело
к установлению двух тппов поликонденсационных реакций — равновес-
ной и неравновесной. В данной главе автор рассматривает в основном
равновесную поликонденсацию. По неравновесным процессам см. книгу
В. В. Коршака, С. В. Виноградовой «Неравновесная поликонденсация»
(изд-во «Наука», М., 1972).— Прим. ред.
Глава 2
Схематически оба типа реакций могут быть представлены следую-
щими уравнениями:
nA — А-рнВ — В —> —(— А — АВ — В — )„—, (2.2)
nA —В —> —( —А —В —)п —, (2.3)
где А и В — функциональные группы различного вида. Законо-
мерности указанных двух типов поликонденсации очень схожи
между собой. Для синтеза высокомолекулярных полимеров
(т. е. полимеров достаточно высокого молекулярного веса, чтобы
быть практически полезными) необходимы те же самые условия
и предосторожности.
2.1. Реакционная способность
фу нкц иональн ыэг г рун н
2.1а. Исходные предпосылки для анализа кинетики
поликонденсации
Изучение кинетики поликонденсации важно с двух точек
зрения. Для практического синтеза высокомолекулярных поли-
меров нужно знать кинетику процесса его образования. С теоре-
тических позиций граница между полимеризацией и поликон-
денсацией во многом определяется особенностями их кинетики.
При поликонденсации происходит медленное, постепенное
нарастание молекулярного веса полимера. В качестве примера
рассмотрим синтез сложного полиэфира из диола и дикарбоно-
вой кислоты. Первой стадией этого процесса является реакция
диола и дикарбоновой кислоты с образованием димера
НО—R —OH-f-HOOC —R' — СООН
—> НО —R —ОСО —R' —СООН-ЬНгО. (2.4)
Затем при взаимодействии двух молекул димера получается
тетрамер
2НО —R —ОСО —IV —СООН —>
НО —R —ОСО —R' —СО2 —R —ОСО —R' —СООН-}-Н2О, (2.5)
а при взаимодействии димера с непрореагировавшим мономером
образуется тример
НО —R —ОСО —R' —СООН + НО —R —ОН —>
—» HO-R —OCO-R' —COOR —ОН + Н2О. (2.6)'
Тетрамер и тример реагируют аналогичным образом сами с собой,
друг с другом, с мономером и димером. Поликонденсация идет,
таким образом, ступенчато, причем молекулярный вес полимера
непрерывно увеличивается. Особенность поликонденсации состоит
П оликонденсация
47
в раннем исчезновении мономера задолго до образования полимера
достаточно высокого молекулярного веса (более 5000—10 000).
В большинстве реакций поликонденсации остается менее 1 %
исходного мономера к моменту образования полимера, состоящего-
в среднем из десяти мономерных единиц. Как будет показано-
в гл. 3, совершенно иное положение имеет место при полимери-
зации.
Скорость поликонденсации является поэтому суммой скоростей
реакций между молекулами различных размеров, т. е. суммой
скоростей следующих реакций:
Мономер -(-Мономер -> Димер,
Димер Д-Мономер -> Тример,
Димер -4-Димер -> Тетрамер.
Тример -(- Димер -> Пентамер,
Тетрамер 4- Мономер -> Пентамер,
Тетрамер -|- Димер Гексамер,
Тетрамер 4-Тример -> Гептамер,
Тетрамер -|- Тетрамер -> Октамер,
Пентамер + Тример -> Октамер
Пентамер Тетрамер -> Нонамер
и т. д.,
(27.а)
которую можно выразить в общем виде как
и-мер 4- m-мер (л + т)-мер. (2.76)
Особенностью поликонденсации является то, что любые две
молекулы в реакционной смеси могут реагировать друг с другом.
Кинетика такой поликонденсации, включающей бесконечное
число отдельных реакций, как правило, с трудом поддается анали-
зу. Анализ становится значительно проще, если принять допуще-
ние, что реакционная способность обеих функциональных групп
бифункционального мономера (например, обеих гидроксильных
групп диола) одинакова, что реакционная способность одной
функциональной группы бифункционального реагента не зависит
от того, прореагировала или нет его другая функциональная
группа, и что реакционная способность функциональной группы
не зависит от размера молекулы, к которой она присоединена
(т. е. не зависит от величин пит). После таких упрощений кине-
тика поликонденсации становится идентичной кинетике анало-
гичных низкомолекулярных реакций. Как будет показано ниже,
кинетика полиэтерификации, например, становится практически
такой же, как и кинетика этерификации уксусной кислоты
этанолом.
4
Глава 2
2.16. Экспериментальные доказательства
Правомерность сделанных выше допущений подтверждается
тем, что константы скорости многих реакций поликонденсации
не зависят от продолжительности реакции и молекулярного веса
полимера. Тем не менее целесообразно оценить более подробно
экспериментальные и теоретические обоснования для таких
предположений. Особенно полезным в этом смысле является
исследование кинетики реакции модельных соединений (неполи-
мерных молекул) [1]. Отсутствие зависимости реакционной спо-
собности функциональной группы от размера молекулы можно
проследить, сопоставляя скорости реакции в гомологическом
ряду соединений, отличающихся друг от друга только молеку-
лярным весом. Рассмотрим, например, данные [2] по константам
скорости, приведенные в табл. 2.1 для этерификации ряда гомо-
логичных монокарбоновых кислот
на
Н(СН2)ПСООН + С2Н5ОН Н(СН2)ПСООС2Н5 + Н2О. (2.8)
Из этих данных хорошо видно, что хотя и наблюдается некото-
рое уменьшение реакционной способности с увеличением размера
Таблица 2.1
Константы скорости этерификации (25 °C) ряда
гомологичных карбоновых кислот [2]
Размер моле- кулы (П) k- 104, Л/(МОЛЬ-С) а
ДЛЯ Н(СН2)ПСООН ДЛЯ (СН2)П(СООН)2
1 22,1
2 15,3 ’ 6,0
3 7,5 8,7
4 7,5 8,4
5 7,4 7,8
6 7,3
8 7,5
9 7,4
И 7,6
13 7,5
15 7,7
17 7,7
Расчет сделан на моль групп СООН.
Поликонденсация
4
молекулы, эффект этот существен только для очень небольших
молекул. Константа скорости реакции очень быстро (при п = 3)
достигает предельной величины, которая остается постоянной
и не зависит от размера молекулы. Для других реакций харак-
терна аналогичная зависимость. Так, константы скорости реакции
полиоксиэтилена (молекулярный вес 393) с концевыми гидро-
ксильными группами и бутанола-1 с фенилизоцианатом
при 30 °C в растворе толуола составляют соответственно
1,5-10"3 и 1,7-10-3 л.'(моль-с) [3]
HO(CH2CH2O)17H + CeH5-NCO —> CeH6-NH-COO-(CH2CH2O)17H, (2.9)
CH3CH2CH2CH2OH + CeH5-NCO —>
—> CeH6-NH-COO-CH2CH2CH2CH3. (2.10)
Положение об отсутствии независимости реакционной способ-
ности функциональных групп от размера молекулы пришло на
смену представлению об уменьшении реакционной способности
функциональных групп с увеличением длины макромолекулярной
цепи, господствовавшему на ранних этапах развития полимерной
науки. Последнее было обусловлено тем, что при поликонденсации
обычно не соблюдалось правило эквивалентности функциональ-
ных групп. В ряде случаев кажущаяся низкая реакционная
способность объясняется плохой или затрудненной растворимостью
исходных веществ более высокого молекулярного веса в одном
гомологическом ряду. Перечисленные эффекты относятся к обыч-
ным ошибкам, которых следует избегать начинающим студентам.
2.1в. Теоретические положения
Существует ряд теорий для доказательства отсутствия влияния
размера молекулы на реакционную способность функциональных
групп [4]. Низкие скорости диффузии молекул большого размера
явились причиной наиболее распространенного неправильного
представления об уменьшении реакционной способности функцио-
нальных групп при переходе от малых молекул к большим. Однако
эффективная реакционная способность функциональной группы
зависит от частоты столкновения групп, а не от скорости диф-
фузии молекулы в целом. Частотой столкновения называется
число столкновений одной функциональной группы с другой
в единицу времени. Подвижность концевой функциональной
группы растущей полимерной цепи значительно больше, чем
подвижность полимерной молекулы в целом. Значительная под-
вижность функциональной группы обусловлена перегруппиров-
ками, которые происходят в близлежащих сегментах полимерной
цепи. Скорость столкновения такой функциональной группы
с соседними группами имеет такую же величину, как и для малых
молекул.
Глава 2
Пониженная подвижность макромолекулы в целом влияет
на распределение столкновений во времени. Низкая скорость
диффузии приводит к тому, что время взаимодействия любых двух
функциональных групп до тех пор, пока из-за диффузии они
не оторвутся друг от друга, будет больше. Так, в любой, произ-
вольно взятый промежуток времени (который больше времени,
необходимого для диффузии группы одного из веществ к другому)
число партнеров, с которыми сталкивается данная функциональ-
ная группа, меньше для концевых групп полимерной цепи, чем
для функциональной группы низкомолекулярного вещества.
Однако наиболее существенным здесь является то, что суммарная
частота столкновений в обоих случаях одинакова. Если бы диффу-
зия функциональной группы одного из реагирующих соединений
к другому была недостаточной для поддержания необходимой
равновесной концентрации пар реагирующих функциональных
групп, то наблюдалась бы пониженная реакционная способность
функциональной группы. В действительности наблюдается обрат-
ная картина, так как при поликонденсации к реакции приводит
одно из каждых 1013 столкновений двух функциональных групп
[1]. В промежуток времени, необходимый для такого числа столк-
новений, скорость диффузии оказывается достаточной для поддер-
жания равновесной концентрации сталкивающихся пар функцио-
нальных групп. Основной вывод из этих рассмотрений заклю-
чается в том, что реакционная способность функциональной
группы не зависит от размера молекулы, к которой она присоеди-
нена. Исключениями являются случаи, когда реакционная способ-
ность групп очень высока и (или) когда молекулярный вес поли-
мера очень велик. В этих случаях поликонденсация лимитируется
диффузией, так как подвижность слишком низка для поддер-
жания равновесной концентрации реагирующих пар и частоты
их столкновений.
2.1г. Эквивалентность функциональных групп
в бифункциональных реагентах
На многих системах была также продемонстрирована эквива-
лентность реакционной способности обеих функциональных групп
в бифункциональных реагентах. В третьей колонке табл. 2.1
представлены данные о скорости этерификации гомологического
ряда дикарбоновых кислот
НООС-(СН2)п-СООН + 2С2Н5ОН —>
С2Н5ООС-(СН2)п-СООС2Н5 + 2Н2О. (2.11)
Приведенные константы скорости являются средними значениями
скорости реакций двух карбоксильных групп каждой дикарбоно-
вой кислоты. Для двух карбоксилов константы скорости одина-
Поликонденсация
5
ковы в пределах экспериментальной ошибки. Другое очень важ-
ное наблюдение состоит в том, что реакционная способность одной
функциональной группы не зависит от того, прореагировала ли
другая группа или нет. Как и в случае монокарбоновых кислот,
реакционная способность карбоксильной группы быстро достигает
некоторой предельной величины.
Из этого нельзя сделать категорического вывода о том, что
реакционная способность функциональной группы не зависит
от присутствия второй такой же группы. Данные показывают,
что при небольших значениях п (4—5) влияние второй группы
проявляется. В большинстве случаев эффект быстро исчезает
по мере возрастания молекулярного веса, когда две функциональ-
ные группы изолируются друг от друга. Таким образом, кон-
станта скорости этерификации дикарбоновой кислоты с п = 6
та же самая, что и для монокарбоновых кислот. На первый взгляд
может показаться, что при поликонденсации бифункциональных
мономеров с низкими значениями п возникают специфические
трудности. Однако на кинетику поликонденсации различия
в реакционной способности функциональных групп би- и моно-
функциональных реагентов не оказывают существенного влияния.
Более важным является то, что реакционная способность функцио-
нальной группы бифункционального реагента не меняется после
того, как прореагировала другая группа, и что реакционная
способность двух функциональных групп одинакова и не меняется
в течение всего процесса.
2.2. Кинетика поликонденсации
В качестве примера кинетики типичной поликонденсации рас-
смотрим полиэтерификацию дикарбоновой кислоты и диола.
Этерификация — хорошо изученный процесс, катализируемый
кислотами, и, без сомнения, аналогичные закономерности харак-
терны для полиэтерификации [5]. Реакция протекает через про-
тонирование карбоновой кислоты
О ОН
II I
— С —0Н4-НА —С —0Н4-А" (2.12)
+
с последующим взаимодействием протонированной формы со спир-
том с образованием сложного эфира
ОН ОН
I ч I
— С — ОН-1----ОН —С-ОН (2.13)
II
5
Глава
ОН О
I А 1!
— С —ОН 7^—С —О--------|-Н2О + Н+ (2.14)
I
— он
В вышеприведенных уравнениях значок — применяется для
обозначения всех кислотных и спиртовых молекул в реакционной
смеси (мономера, димера, тримера, ..., м-мера). Подобно многим
другим реакциям поликонденсации, полиэтерификация — про-
цесс равновесный. Однако на практике для получения высоко-
молекулярных полимеров с высоким выходом реакцию проводят
таким образом, чтобы непрерывно и максимально сдвинуть равно-
весие в сторону образования полимера. В случае полиэтерифика-
ции это легко достигается удалением воды, являющейся низко-
молекулярным продуктом реакции [уравнение (2.14)], что дает
право воспользоваться для расчета кинетики полиэтерификации
уравнениями для необратимых реакций. (Кратко кинетика обра-
тимой поликонденсации рассматривается в разд. 2.66.16.)
Скорость поликонденсации наиболее удобно выражать исходя
из концентрации реагирующих функциональных групп. Так,
кинетику полиэтерификации экспериментально изучают путем
титрования непрореагировавших карбоксильных групп основа-
нием. Скорость поликонденсации Rp можно тогда выразить
в виде скорости исчезновения карбоксильных групп —cZ[COOH]/cZ/.
Для обычной полиэтерификации скорость реакции определяется
скоростью образования соединения II, так как отсутствует
(реакция проводится в неравновесных условиях), а Ад, к2 и к-0
велики по сравнению с к3. Выражение для скорости реакции
можно получить, используя обычные правила расчета кинетиче-
ских уравнений [6]. Скорость полиэтерификации определяется как
ДР - [СООН] = кз ^(0Н)2][0Н], (2.15)
где [СООН], [ОН] и [С(ОН)2] — концентрации карбоксильных,
гидроксильных и протонированных карбоксильных групп соот-
ветственно. Концентрация выражается в молях данных функцио-
нальных групп на литр раствора.
Уравнением (2.15) пользоваться неудобно, так как концентра-
цию протонированных карбоксильных групп экспериментально
определить очень трудно. Более удобное выражение для скорости
реакции можно получить, определив [С(ОН)2] из формулы для
константы равновесия реакции протонирования [уравнение (2.12)]
jz_ _[С(ОН)2][А ] ,р ..
к2 “ [СООНЦНА] ' '
Поликонденсация
53
Ч"
Подставляя затем выражение для [С(ОН)2] в уравнение (2.15),
получим
— d[COOH] Мз[СООН][ОН][НА] /9 17^
dt ~ Л2[А-] ' ( • )
Это уравнение можно переписать в виде
— d[COOH] fcifc3[COOH][OH][H+] / 2.176)
dt k2KHA ’
где &нА — константа диссоциации кислоты НА. В зависимости
от того, добавляется или нет в качестве катализатора сильная
кислота НА, например серная, кинетика реакции сильно меняется.
2.2а. Самокатализируемая поликонденсация
В отсутствие добавок сильной кислоты исходная дикарбоновая
кислота действует как собственный катализатор полиэтерифика-
ции. В этом случае [НА] заменяется [СООН] и уравнение (2.17а)
можно записать в обычной форме [1, 7]
~ 4 = /с [ СООН ]2 [ ОН ], (2.18)
где три константы скорости klt k2, k3 и [А-] объединены в одну,
экспериментально определяемую константу скорости к. Из урав-
нения (2.18) видна роль самокаталитического процесса: это реак-
ция третьего порядка со вторым порядком по карбоновой кис-
лоте, который получается из двух зависимостей первого порядка —
одной для карбоксила как реагента и другой — для карбоксила
катализатора.
Если концентрации двух функциональных групп равны между
собой, что, как правило, характерно для поликонденсации,
то уравнение (2.18) может быть представлено в виде
= /с[М]3 (2.19а)
ИЛИ
= ' (2.196)
где [М] — концентрация гидроксильных или карбоксильных
групп. Интегрирование уравнения (2.196) дает
2/cZ = 2 — const. (2.20)
Константа в уравнении (2.20) равна 1/[М]2, где [М]о — начальная
концентрация гидроксильных или карбоксильных групп (при
t = 0). В это уравнение удобно ввести параметр р, называемый
54
Глава 2
степенью завершенности реакции и представляющий собой долю
гидроксильных или карбоксильных функциональных групп, про-
реагировавших за время t. (Значение р определяется по концен-
трации непрореагировавших карбоксильных групп.) Тогда кон-
центрацию [М] гидроксильных или карбоксильных групп в момент
времени t можно выразить так:
[М] = [М]о —[М]ор = [М]о (1 — р). (2.21)
Комбинируя уравнения (2.20) и (2.21), получим
const. (2.22)
2.2а. 1. Экспериментальные данные
Как видно из уравнения (2.22), отношение 1/(1 — р)2 должно
быть линейной функцией времени t. Этот вывод подтверждают
экспериментальные данные для полиэтерификации. На рис. 2.1
Рис. 2.1. Кинетическая кри-
вая самокатализируемой реак-
ции третьего порядка адипи-
новой кислоты и диэтиленгли-
коля при 166 °C [8].
приведены результаты, полученные при полиэтерификации диэти-
ленгликоля и адипиновой кислоты [8, 9]. Эти данные типичны
и'для большинства других реакций полиэтерификации. На первый
взгляд экспериментальная кривая строго не подчиняется при-
веденной выше зависимости. Экспериментальные точки откло-
няются от кривой зависимости третьего порядка на начальной
стадии (при степени завершенной реакции ниже 80%) и на заклю-
чительном этапе реакции при степени завершенности реакции
выше 93%. Эти отклонения побудили ряд исследователей предло-
жить другие кинетические уравнения [9, 101, основанные на
реакциях первого или полуторного порядка по карбоновой
П оликонденсация
55
кислоте (т. е. второго и двухсполовинного суммарных порядков)
-dfCOQH] [СООН][ОН], (2.23а)
~d[Cooni = к [СООН]3/2 [ОН]. (2.236)
Однако указанные уравнения не выдерживают серьезной кри-
тики 19]. Обработка экспериментальных данных с помощью урав-
нения (2.23а) дает сходимость с расчетной кривой только в области
Р п с. 2.3. Кинетическая кривая
самокатализируемоп полиэтери-
фикации адипиновой кислоты и
диэтиленгликоля при 166 °C в
координатах реакции двухсполо-
винного порядка [8].
Рис. 2.2. Кинетическая кривая са.мо-
катализируемой полиэтерпфпкации ади-
пиновой кислоты и дпэтпленглпколя
при 166 °C в координатах реакции вто-
рого порядка [8].
превращения 50—86%, тогда как выше степени превращения 86%
экспериментальные результаты совершенно не укладываются
на такую зависимость (рис. 2.2). С другой стороны, кривая,
построенная в координатах уравнения полуторного порядка
по карбоновой кислоте (рис. 2.3), совпадает с экспериментальными
данными только до степени превращения примерно 69% и сильно
отклоняется от них выше этой точки. Ни одно из этих двух кине-
тических уравнений не описывает экспериментальные результаты
лучше, чем уравнение третьего порядка. Кривая, построенная
по уравнению третьего порядка, при высоких конверсиях описы-
вает экспериментальные данные значительно лучше, чем другие
кривые. Соответствие данных кривой третьего порядка весьма
удовлетворительно в довольно большом интервале высоких степе-
6
Глава 2
ней превращения. Область высоких степеней превращения имеет
первостепенное значение, так как высокомолекулярный полимер,
как будет показано в разд. 2.2а.2, получается только при высоких
степенях завершенности реакции. Для практики область кинети-
ческой кривой, соответствующая низким конверсиям, малоинте-
ресна.
2.2а.2. Причины отклонений от кривой третьего
порядка
2.2а.2а. Область низких степеней превращения. Два нелиней-
ных участка на кривой, построенной по уравнению реакции
третьего порядка (рис. 2.1), можно объяснить достаточно хорошо.
Рассмотрим начальные стадии реакции. Наблюдаемое в этой
области отклонение от линейной зависимости характерно не только
для реакций поликонденсации. Отклонение от кривой третьего
порядка присуще реакциям этерификации вообще. Так, Флори [8]
обнаружил аналогичное поведение при этерификации в следую-
щих системах:
1) СН3(СН2)4СООН и НОСН2СН2ОСН2СН2ОН,
капроновая кислота диэтиленглпколь
2) СН3(СН2)юСООН и СН3(СН2)юСН2ОН,
лауриновая кислота лауриловый спирт
3) НООС(СН2)4СООН и СН3(СН2)юСН2ОН
адипиновая кпслэта лауриловый спирт
и объяснил отклонения от линейности большими изменениями,
которые происходят в реакционной среде. При степени превраще-
ния 50% концентрация карбоксильных групп уменьшается в два
раза, а средний молекулярный вес, как будет показано ниже,
в этот момент чрезвычайно низкий. При этом резко меняется
полярность реакционной смеси, так как полярные карбоксильные
группы превращаются в значительно менее полярные сложно-
эфирные группы. Однако до сих пор нет ясности в вопросе, как
изменение полярности влияет на скорость этерификации.
Возможно, что способность системы поставлять протон к кар-
боксильной группе [см. уравнение (2.12)1 учитывается неточно
в кинетическом уравнении (2.18). В ряде работ показано, что для
неводных систем или концентрированных кислот кислотность
нельзя оценивать [НА] или [Н+]. Для оценки кислотности среды
в таких системах правильнее пользоваться функцией кислотности
/г0, которая определяется [И] как
, aH+VB ТН+ [НП Тв
Нп = ----- — -----------
Твн+ Твн+
(2.24)
Поликонденсация
7
(2.25)
где ан+ и ун+ — активность и коэффициент активности соответ-
ственно Н+ в системе, а ув/увн+ — отношение коэффициентов
активности основания В и его сопряженной кислоты ВН+. В реак-
ции этерификации карбоксильная группа и ее протонированная
форма должны рассматриваться как В и ВН+ соответственно.
Функцией кислотности h0 следует пользоваться для оценки
кислотности высококонцентрированных систем или неводных
растворов. Она применима и к полиэтерификации, как и к другим
кпслотнокаталитическим поликонденсационным процессам, про-
водящимся в высококонцентрированных неводных растворах.
Определив [Н+] из уравнения (2.24) и подставив выражение для
нее в уравнение (2.176), получаем
..чгогш, Мз[СООН][ОН]йоТ+
— d [СООН]С(ОН)2
dt *2^ндТн+Тсоон
К сожалению, h0 и коэффициенты активности удалось определить
лишь в нескольких случаях. Представляет интерес, что функция
кислотности h0 была с успехом использована для кинетического
анализа поликонденсации формальдегида с фенолом (и другими
ароматическими соединениями) в присутствии кислых катализа-
торов [12].
2.2а.26. Область высоких степеней превращения. По-видимому,,
нелинейность кривых, построенных по уравнению третьего поряд-
ка, на заключительных этапах полиэтерификации не связана
с использованием или неиспользованием функции h0. На этой
стадии процесса концентрация кислоты значительно меньше, чем
на более ранних этапах. Далее, вряд ли оправдано применять
функцию кислотности для описания заключительной стадии
процесса, когда в ней нет необходимости при расчете кинетики
в промежуточной области, соответствующей степени превраще-
ния 80—93%. Более вероятно, что нелинейный характер зависи-
мости при степени превращения выше 93% обусловлен другими
факторами.
При поликонденсации, проводящейся часто при весьма высо-
ких температурах и в вакууме, небольшая часть мономеров может
удаляться из сферы реакции или разлагаться. В случае полиэте-
рификации к таким деструктивным процессам относятся дегидра-
тация диола, декарбоксилирование дикарбоновой кислоты и (или)
другие побочные реакции (разд. 2.66.1). Хотя на начальных ста-
диях процесса такие потери не отражаются, они приобретают
большое значение на заключительных этапах реакции. Так, поте-
ря одного из реагентов в количестве всего 0,3 % может привести
к изменению концентрации до 5% при степени завершенности
реакции 93%. В результате тщательных исследований подобных
58
Глава 2
реакций было показано, что потери такого порядка величины
вполне могут иметь место.
Другой возможной причиной наблюдаемого отклонения от
линейности является вклад обратной реакции. Известно, что
полиэтерификация, как и многие другие реакции поликонденсации,
является равновесной реакцией. С повышением степени завершен-
ности реакции часто становится все более и более затруднительным
сдвиг равновесия в сторону образования полимера. Это обусло-
влено тем, что при повышении степени превращения сильно воз-
растает вязкость реакционной среды. Так, в ходе полиэтерифика-
ции адипиновой кислоты и диэтиленгликоля вязкость возрастает
от 0,015 до 0,30 пуаз (0,0015—0,030 Па-с) [8]. При увеличении
вязкости системы затрудняется отвод воды из сферы реакции, что
может приводить к наблюдаемому понижению скорости реакции
с приближением степени завершенности ее к единице.
2.2а.З. Молекулярный вес полимера
С практических позиций молекулярный вес полимера имеет
первостепенное значение, так как только полимеры определенного
молекулярного веса (>5000—10000) приобретают необходимые
прочностные характеристики. Поэтому весьма важно рассмотреть
изменение молекулярного веса полимера в процессе реакции. Так,
при стехиометрическом соотношении исходных веществ (диола
и дикарбоновой кислоты в рассматриваемом случае) число непро-
реагировавших карбоксильных групп N равно общему числу
молекул, находящихся в системе в момент времени t. Это объяс-
няется тем, что во всех молекулах, больших чем мономер,
на одном конце находится гидроксильная группа, а на другом —
карбоксильная, тогда как в каждой мономерной молекуле дикар-
боповой кислоты имеются две карбоксильные группы, а каждый
мономерный диол не содержит карбоксильных групп вообще.
Остаток диола и дикарбоновой кислоты (по отдельности)
в полимерной цепи называется структурной единицей или моно-
мерной единицей. Повторяющаяся единица, или элементарное
звено цепи, состоит из двух структурных единиц; одной — диола
и одной — дикарбоновой кислоты. В сумме число структурных
единиц в данной системе равно числу бифункциональных моно-
меров, содержащихся в исходной системе. Среднечисловой сте-
пенью полимеризации называется среднее число структурных
единиц на полимерную цепь. Для обозначения среднечисловой
степени полимеризации применяются следующие символы: Хп, Р
и СП. Величина Хп легко определяется как отношение общего
числа молекул, находящихся в исходной системе, к числу молекул
Поликонденсация
59
в системе в момент времени t
Хп = 4 = ^. (2.26)
П TV [М] к /
Из уравнения (2.21) и (2.26) находим
^п=гЬ- (2.27)
Это уравнение связывает степень полимеризации и степень завер-
шенности, первым его предложил Карозерс [13], поэтому иногда
его называют уравнением Карозереа.
Среднечисловой молекулярный вес Мп — это вес образца
полимера, отнесенный к суммарному числу молекул в нем,
МП = МОХП = ^, (2.28)
тде Мо — средняя величина из молекулярных весов двух струк-
турных единиц. При полиэтерификации адипиновой кислоты
и диэтиленгликоля элементарное звено
— ОСН2СН2ОСО(СН2)4СО -,
III
половина его веса равна 86 — это и есть Мо.
Комбинируя уравнения (2.22) и (2.27), получим
2[M]2/c£ = .X’n—const. (2.29)
Поскольку в полученное уравнение время реакции и степень
полимеризации входят соответственно в первой и второй степени,
можно сделать вывод, что с ростом продолжительности реакции
молекулярный вес полимера будет увеличиваться медленно
(исключая ранние стадии процесса). Это значит, что для полу-
чения высокомолекулярных полимеров реакцию нужно вести
достаточно долго. Как отчетливо видно из рис. 2.1 (см. правую
ординату), молекулярный вес полимера повышается очень мед-
ленно со временем. При этом по мере протекания реакции скорость
нарастания Хп во времени уменьшается. С практической точки
зрения время, необходимое для образования высокомолекуляр-
ных полимеров, слишком велико.
2.26. Внешний катализ поликонденсации
Ранее такое медленное нарастание молекулярного веса оши-
бочно объяснялось низкой реакционной способностью функцио-
нальных групп, присоединенных к большим молекулам.
На самом же деле это просто результат особенности кинетики
реакции третьего порядка для прямой реакции полиэтерификации.
GO
Глава 2
Опираясь па такую кинетику, можно получить высокомолекуляр-
ные полимеры в разумное время, используя в реакции небольшие
добавки сильных кислот (например, серной) в качестве катализа-
тора. Тогда [НА] в уравнении (2.17а) или [Н+] в уравнении (2.176) —
это концентрация катализатора. Так как количество его на про-
тяжении реакции не меняется, то уравнение (2.176) можно пере-
писать в виде
~^[М| = А/[М]\ (2.30)
где различные константы уравнения (2.17) объединены в одну,
экспериментально определяемую константу скорости к'. Уравне-
ние (2.30) применимо к реакциям между стехиометрическими
количествами диола и дикарбоновой кислоты.
Интегрируя уравнение (2.30) и подставляя полученный резуль-
тат в уравнение (2.21), получим
[М]о k't = | — const (2.31а)
или
[М]0Л/£ = Хп— const. (2.316)
Данные для поликонденсации диэтиленгликоля и адипиновой
кислоты в присутствии как катализатора тг-толуолсульфокислоты
Р и с. 2.4. Полпэтерифпкация
адипипово ii кислоты и диэтплен-
гликоля при 109 °C в присутствии
как катализатора 0,4 мол. % п-
толуолсульфокпслоты [8].
приведены на рис. 2.4. Полученная кривая описывается уравне-
нием (2.31), причем степень полимеризации линейно возрастает
со временем реакции. Наиболее общим и существенным отличием
каталитической полиэтерификации (рис. 2.4) от пекаталитической
П оликонденсация
61
(рис. 2.1) является значительно более высокая скорость нараста-
ния молекулярного веса полимера (Хп) во времени в первом
процессе. Применение катализатора (кислоты) делает полиэтери-
фикацию значительно более экономичным, легко осуществимым
процессом.
Нелинейность на начальной стадии кривой, приведенной
на рис. 2.4, характерна для этерификации вообще, а не только
для полиэтерификации. Как было показано выше (разд. 2.2а.26),
нелинейность может быть связана с невозможностью применения
функции кислотности h0 вместо концентрации кислоты в урав-
нении скорости реакции. Линейность же кривой в области более
высоких степеней превращения является убедительным доказа-
тельством концепции независимости реакционной способности
функциональных групп от размера молекулы. Из рис. 2.4 видно,
что полиэтерификация подчиняется уравнению второго порядка
по крайней мере до степени полимеризации 90, что соответствует
молекулярному весу приблизительно 10 000. Изменения реак-
ционной способности гидроксильных и карбоксильных групп
не наблюдается, несмотря на значительное увеличение размера
молекулы (и сильное возрастание вследствйе этого вязкости
среды). Аналогичные результаты получены при исследовании
многих других систем. Те же закономерности характерны и для
деструкции полимеров. Так, при кислотно-каталитическом гид-
ролизе целлюлозы [1] размер молекулы не влияет на гидролизуе-
мость полимера вплоть до степени полимеризации 1500 (молеку-
лярный вес 250 000). Концепция отсутствия зависимости реак-
ционной способности функциональных групп от размера молекулы
была много раз подтверждена на многочисленных реакциях
образования и превращения полимеров. Не очень хорошо она
может выполняться только при очень низких или очень высоких
степенях превращения. Имеются данные о таких реакциях поли-
конденсации, в которых мономеры сильно отличаются по своей
реакционной способности от всех получаемых соединений — оли-
гомеров и полимеров, не отличающихся по этому признаку друг
от друга [14, 15].
2.2в. Другие реакции поликонденсации
Кинетика различных реакций поликонденсации в общем под-
чиняется тем же принципам, которые были рассмотрены выше для
полиэтерификации. Число различных кинетических схем, встре-
чающихся в реальных поликонденсационных системах, не отли-
чается большим разнообразием. Для поликонденсации функцио-
нальных групп А и В соответствующих мономеров характерны
следующие основные случаи:
62
Глава 2
1. Некоторые реакции поликонденсации идут с заметной ско-
ростью даже в отсутствие катализатора, например полиамидиро-
вание.
2. Ряд поликонденсационных процессов для достижения необ-
ходимых скоростей требует введения кислого или основного ката-
лизатора, это относится, например, к взаимодействию мочевины,
фенола или меламина с формальдегидом (см. табл. 1.1).
3. Некоторые реакции поликонденсации успешно завершаются
как в присутствии катализатора, так и без него. Приведем в каче-
стве примера образование полиуретанов. Реакцию между диолом
и диизоцианатом проводят часто в присутствии основного катали-
затора. Однако иногда во избежание нежелательных побочных
реакций процесс осуществляют без добавления катализатора.
Независимо от того, к какому конкретному случаю относится
та или иная поликонденсация, кинетические особенности ее
одни и те же. Это реакция первого порядка по группам А и по-
группам В. При стехиометрическом соотношении функциональных
групп кинетика процесса описывается уравнениями (2.30) и (2.31).
Кинетика поликонденсации не зависит от того, проводится ли
реакция с участием мономеров типа А — А и В — В или одного
мономера А — В.
2. 2г. Неэквивалентность функциональных групп
полифункциональных реагентов
Прежде всего заметим, что есть случаи, где первые два допу-
щения, сделанные в разд. 2.1а, оказываются несправедливыми.
Часто неверным является предположение об одинаковой реак-
ционной способности всех функциональных групп в полифунк-
ционалыюм мономере. То же самое можно сказать о допущении,
что реакционная способность функциональной группы не зависит
от того, прореагировала другая группа или нет. Так, оба указан-
ных допущения несправедливы в случае 2,4-толуилендиизоци-
аната [16]
снз
0rNCO
NCO
IV
который широко используется как исходное вещество в синтезе
полиупетанов.
Поликонденсация
63
Образование уретана можно описать следующей схемой:
О 0-
II I
R-N = C + B: В-N = c-В+
| Н'ОП
о-
— В: |
R-NH-COOR' - R-NH-C-B+ (2.32)
I
OR'
где В: — катализатор основного типа (например, третичный амин).
Лимитирующей стадией реакции является нуклеофильная атака
спиртом электрофильной связи углерод — азот изоцианата. Это
подтверждается наблюдением, что реакционная способность изо-
цианатной группы возрастает при введении в молекулу изоцианата
электронооттягивающей группы [17].
Различие в реакционной способности обеих функциональных
групп в 2,4-толуилендиизоцианате может быть связано с несколь-
кими факторами. Для того чтобы они стали более понятными,
следует рассмотреть данные по реакционной способности различ-
ных изоцианатных групп в реакции с н-бутанолом при 40 °C
в присутствии триэтиламина в качестве катализатора (табл. 2.2).
Таблица 2.2
Реакционная способность изоцианатной группы
в реакции с м-С4Н9ОН [18]
Изоцианат Aj-102, л/(моль-с) А1/Ага
Моноизоцианаты Фенилизоцианат 0,53
м-Толилизоцианат 0,26
о-То ли лиз оцианат 0,076
Диизоцианаты •и-Фенилендиизоциа- 7,2 8,4
нат тг-Фенилендиизоциа- 5,3 9,2
нат 2,6-Толуилендиизо- 1,5 6,1
цианат 2,4-Толуилендиизо- 3,3 11,9
цианат
а <4 и Аг в л/(моль-с).
64
Глава 2
Из этих данных видно, что метильный заместитель дезакти-
вирует изоцианатную группу, повышая ее электроноотрицатель-
ность, причем этот эффект сильнее, когда заместитель находится
в орто-положении к ней. Одна изоцианатная группа повышает
активность другой за счет электронооттягивающего эффекта, что
видно из большей реакционной способности диизоциапатов
по сравнению с .моноизоцианатом. Из таблицы также видно, что
после того, как первая изоцианатная группа прореагировала,
реакционная способность второй функциональной группы сни-
жается примерно на целый порядок, что обусловлено значительно
меньшей электронооттягивающей способностью уретановой группы
по сравнению с изоцианатной. Так, в 2,4-толуилендиизоцианате
и-изоцианатная группа более чем в 10 раз реакционноспособнее
о-изоцианатной группы. Это чрезвычайно затрудняет анализ
кинетики такой реакции, если не делает его вовсе невозможным;
действительно, в литературе нет четких кинетических данных
по образованию полиуретанов исходя из 2,4-толуилепдиизоциана-
та. (Следует полагать, что в будущем для решения этой задачи
окажется весьма плодотворным использование электронно-вычис-
лительной техники.)
Важным классом исходных веществ, применяющихся в поли-
конденсации, являются трифункциональные мономеры. Среди
них часто встречаются вещества, функциональные группы в кото-
рых отличаются по своей реакционной способности. Так, при
поликонденсации глицерина (V)
сн2—сн—сн2
он он он
V
с фталевым анигпдридом реагируют не все гидроксильные груп-
пы [19]. Это обусловлено пониженной реакционной способностью
вторичной гидроксильной группы по сравнению с двумя первич-
ными гидроксилами. Однако справедливость этого объяснения
нуждается в дополнительных доказательствах. Действительно,
существуют системы, содержащие трифункциональные реагенты,
которые по прекращении реакции остаются гомогенными раство-
рами. Но есть и системы, содержащие диспергированные частицы
полимера, размером в диаметре от 0,05 до 1 мкм [1, 20]. Такие
частицы называются микрогелями, и они образуются в результате
реакций сшивания (разд. 2.8). Реакционная способность функцио-
нальных групп в трифункциональных мономерах обсуждается
ниже, в разд. 2.10а.2.
Поликонденсация
65
2.3. Реакции циклизации в линейной
поликонденсации
2.3а. Возможные реакции циклизации
Образование линейных полимеров поликонденсацией полифунк-
циональных мономеров сопровождается в ряде случаев побоч-
ными реакциями циклизации. Образование колец возможно при
реакции и по типу А — В, и по типу А — А плюс В — В. Так,
исходные вещества типа А — В, например амино- или оксикис-
лоты, могут образовывать циклический амид (лактам) или сложный
эфир (лактон) при реакции циклической димеризации
R — NH
2H2N- R — СООН —> СО ^СО-ргИгО (2.33)
^NH-R^
2НО —R —СООН —> СО СО-р2Н2О (2.34)
\о —r/
или путем прямой внутримолекулярной циклизации мономеров
Н2ы—R—СО2Н —►HN—R—СО + Н2О,
(2.35)
НО—R—СО2Н —>0—R—СО + Н20
(2.36)
В аналогичные реакции могут вступать реагенты типа А — А
Н02С—R—СО2Н — ОСО—R-CO + Н20,
(2.37)
2 НО„С—R—С09Н —► ОСО—R—COO—R—СО (2.38)
2 2 I________________I
Однако многие реагенты типа А — А содержат такие функцио-
нальные группы, которые не могут реагировать друг с другом
при поликонденсации. Так, например, практически исключена
возможность циклизации за счет реакции между двумя гидро-
ксильными группами диолов или аминогруппами диаминов.
Образование циклических продуктов может иметь место также
в результате циклической димеризации реагентов типа А — А
и В — В. При реакции между диолами и ацетальдегидом, напри-
мер, может наряду с линейным полимером образоваться цикли-
5—01120
66
Глава 2
ческий продукт
НО—R—ОН + СН3СНО
-ео—r—осн-г
/ снз
(2.39)
о—R—о—СН—снз
(2.40)
2.36. Термодинамический и кинетический подход
| От возможного размера кольца, а также от кинетики и термоди-
намики его образования зависит, может ли конкурировать цикло-
образование с реакцией линейной поликонденсации. Из суммы
различных данных складывается представление об относительной
легкости протекания циклизации либо линейной поликонденса-
ции. Данные получают или прямыми исследованиями [1, 21] с уча-
стием бифункциональных мономеров в реакциях циклизации
[см. уравнения (2.33) — (2.40)], или при изучении полимеризации
с раскрытием цикла (гл. 7), или с помощью данных по теплотам
сгорания циклических соединений [22]. Вначале рассмотрим
термодинамическую устойчивость различных циклических соеди-
нений, отличающихся размерами. Хорошее представление о влия-
нии размера цикла на его термодинамическую устойчивость
можно получить из определения теплот сгорания различных
циклоалканов (табл. 2.3). Сравнение теплот сгорания на одну
метиленовую группу в таких циклических соединениях и линей-
ных алканах дает общую оценку термодинамической устойчи-
вости циклов различных размеров [22]. Разности между тепло-
тами сгорания на метиленовую группу циклоалканов и н-алкана
показывают, что термодинамическая устойчивость уменьшается
с увеличением напряжения кольца. Напряжение для 3- и 4-член-
ных колец очень велико и резко уменьшается при переходе
к 5-, 6- и 7-членным циклам, снова увеличивается для 8 — 11-член-
ных циклов, а затем снова уменьшается при переходе к циклам
больших размеров.
Различают два типа напряжений в циклах: угловое напряжение
и отталкивание из-за наличия заместителей. Циклы, состоящие
из менее пяти атомов, сильно напряжены вследствие высокого
углового напряжения, а именно: больших искажений их валентных
углов от нормального тетраэдрического угла. В пятичленных
циклах и кольцах большего размера искажения валентных углов
отсутствуют, так как такие кольца могут существовать в непло-
Поликонденсация
67
Таблица 2.3
Теплоты сгорания и напряженность циклоалканов [22]
п в Теплота сгорания на одну метиленовую группу, ккал/моль Напряженность а на одну метиленовую группу, ккал/моль
L-(ch2)„—।
3 166,6 9,2
4 164,0 6,6
5 158,7 1,3
6 157,4 0,0
7 158,3 0,9
8 158,6 1,2
9 158,8 1,4
10 158,6 1,2
11 158,4 1,0
12 157,7 0,3
13 157,8 0,4
14 157,4 0,0
15 157,5 0,1
16 157,5 0,1
17 157,2 -0,2
и-Алкан 157,4 0,0
а За напряженность принимается разность теплоты сгорания на одну метиленовую
группу данного алкана и н-алкана (157,4 ккал/моль).
скостной форме. В пятичленном кольце напряжения практически
отсутствуют вследствие того, что нет искажений валентных углов.
Для колец большего размера напряжение, обусловленное искаже-
ниями валентных углов, может быть только для плоскостных
структур. Поэтому кольца с числом членов больше пяти суще-
ствуют в более устойчивой некопланарной форме. Шести- и семи-
членные циклы устойчивы, причем из них заметно более устой-
чивыми являются шестичленные кольца. Хотя у колец с числом
членов от восьми до двенадцати напряжения, вызванные искаже-
ниями валентных углов, практически отсутствуют, они все-таки
не являются термодинамически стабильными. Это связано с тем,
что атомы водорода или другие заместители в них находятся
внутри кольца на таких расстояниях, что они отталкивают друг
друга. Рассмотрение систем с числом членов в цикле более две-,1
надцати показывает, что они должны быть весьма стабильными,
так как в них подобное отталкивание уже не имеет места.
Глава /
На основании этого можно составить следующий ряд циклов
различных размеров в порядке увеличения термодинамической
устойчивости:
3 , 4, 8 —11 <7, 12 и выше < 5 < 6.
В том же порядке располагаются циклы, содержащие вместо
некоторых метиленовых групп другие атомы или группы атомов.
Хотя данных для таких циклов, как простые эфиры, лактоны
и лактамы, явно недостаточно, общие положения подтверждаются.
Замена метиленовой группы на кислород, карбонил, азот или
какую-либо группу не влияет практически на валентные углы
в циклических структурах. Напряжение цикла, вызванное оттал-
киванием заместителей, уменьшается при замене метиленовой
группы на менее объемистую карбонильную группу или кислород,
что приводит к повышению термодинамической устойчивости
цикла. Было также показано, что в отличие от водорода другие
заместители в циклических структурах, как правило, повышают
их стабильность, хотя причина этого не совсем понятна.
Кроме термодинамической устойчивости, для определения
соотношения между циклизацией и линейной поликонденсацией
важна также кинетическая разрешимость реакции циклизации.
Кинетическая разрешимость циклизации зависит от возможности
сближения двух функциональных групп, находящихся на разных
концах макромолекулы. По мере увеличения размера потен-
циального цикла мономеры, на основании которых могут быть
получены циклы, обладают множеством конфигураций, лишь
немногие из которых характеризуются близким расположением
двух концов [23]. Возможность образования цикла уменьшается
по мере снижения вероятности встречи двух функциональных
групп. Этот эффект отражается в нежелательном увеличении
энтропии активации.
Возможность протекания циклизации таким образом опре-
деляется соотношением следующих двух факторов: 1) неуклонного
снижения кинетической разрешимости процесса по мере увели-
чения размера кольца; 2) термодинамической устойчивости,
которая возрастает при увеличении размера цикла вплоть до
шестичленного цикла, затем уменьшается при изменении числа
членов в цикле от 6 до 9—11, а затем вновь возрастает при пере-
ходе к еще большим циклам. Итогом совместного действия обоих
факторов является то, что трехчленные циклы получают с большим
трудом, но еще труднее образуются четырехчленные циклы. Это
обусловлено тем, что в первом случае кинетический фактор более
выгодный. Значительно легче замыкаются пятичленные кольца
в основном из-за очень благоприятного термодинамического фак-
тора. Несколько труднее идет образование шестичленных циклов,
что связано главным образом с тем, что более выгодный термо-
Поликонденсация
69
динамический фактор перекрывается ухудшением кинетического
фактора. Сложнее образуются семичленные кольца и еще намного
хуже восьмичленные циклы, что обусловлено ухудшением как
термодинамического, так и кинетического фактора. В этой точке
кинетический фактор становится уже постоянным, и циклизация
определяется исключительно термодинамической ее разреши-
мостью. Циклизация затруднена для образования девяти- и десяти-
членных колец, несколько легче идет для одиннадцати- и двенад-
Р и с. 2.5. Зависимость вероятности циклообразования от размера кольца
цатичленных циклов; и, наконец, для колец большего размера
вероятность циклизации практически остается на одном уровне.
Качественно все эти рассуждения можно выразить одной кри-
вой, которая приведена на рис. 2.5. Вероятность циклизации
высока для пяти- и шестичленных циклов, заметна для семичлен-
ных колец, низка для трехчленных колец и колец, содержащих
более 12 членов, и равна практически нулю для колец всех других
размеров. В практике линейной поликонденсации с возможностью
циклообразования следует считаться, когда применяемые в реак-
ции мономеры могут давать пяти-, шести- или семичленные кольца.
При циклизации согласно уравнениям (2.33), (2.34) и (2.38) это
соответствует мономерам, R в которых может поставлять только
один атом в цикл. При циклизации согласно уравнениям (2.35)
и (2.36) циклизация возможна, когда R дает в цикл 3—5 атомов.
При циклизации по уравнениям (2.37) и (2.40) необходимо, чтобы R
поставляло в цикл только от двух до четырех атомов. Образова-
ние трехчленных циклов практически исключается, так как
обычно использующ иеся мономеры не могут давать такие цикли-
ческие структуры. Кольца очень большого размера (более 12 чле-
нов) очень редко встречаются при поликонденсации. [Совершен-
но иное положение имеет место при цепной полимеризации
(см. разд. 6.66).]
70
Глава 2
2.3в. Другие факторы
Обычно поликонденсацию проводят при высоких концентра-
циях исходных веществ, что благоприятствует образованию линей-
ного продукта. Циклизация — мономолекулярная (внутримоле-
кулярная) реакция, в то время как линейная поликонденсация —
это бимолекулярная (межмолекулярная) реакция. С увеличением
концентрации исходных веществ скорость последней возрастает
значительно быстрее, чем скорость первой реакции. Таким образом,
фактор концентрации реагентов накладывается на рассмотренные
выше кинетический и термодинамический факторы, благоприятно
влияя па протекание линейной поликонденсации. Именно этим
объясняется отсутствие в реальных поликонденсационных систе-
мах колец с числом членов более 12: циклизация практически
не идет, когда при поликонденсации возможно образование
шести- или семичленных колец. Другим обстоятельством, которым
можно воспользоваться для смещения равновесия в сторону линей-
ной поликонденсации, является невозможность в определенных
условиях превращения линейных структур в циклические и наобо-
рот. Это позволяет сдвинуть равновесие между двумя структурами
в сторону образования линейного полимера.
Интересно заметить, что циклизация практически полностью
исключает линейную реакцию, если возможно образование пяти-
членных циклов. Как правило, такая циклизация идет с большей
скоростью, чем линейная поликонденсация других исходных
веществ, где отсутствует циклизация. Однако причины такого
поведения непонятны [1]. (Пятичленные циклы, которые ранее
считались совершенно инертными, в некоторых случаях поли-
меризуются с раскрытием цикла.)
Выше речь шла преимущественно о циклах, содержащих угле-
род, углерод и кислород, углерод и азот. Соотношение между
циклизацией и линейной поликонденсацией может быть и иным
при переходе к циклам, состоящим из других элементов. Так,
в случае силиконов, которые могут образовывать кольца, состоя-
щие из чередующихся атомов кремния и кислорода, например
R R
R R
Поликонденсация
71
циклы с числом членов менее восьми весьма напряженны
из-за большого размера атома кремния, большей длины связи
Si — О и большого валентного угла Si — О — Si. Оптимальным
размером кольца в этом случае является восьмичленный цикл,
хотя судя по кинетическому фактору это предпочтение не вполне
однозначно [24, 25]. Образованию больших колец препятствуют
те же факторы, о которых говорилось выше.
2.4. Регулирование молекулярного веса
полимера при линейной поликонденсации
2.4а. Необходимость стехиометрического контроля
Проблема регулирования молекулярного веса при поликон-
денсации имеет два наиболее важных аспекта. При синтезе поли-
меров, как правило, требуется получить полимер весьма опре-
деленного молекулярного веса, так как свойства полимера сильно
зависят от этого параметра. Одинаково нежелательны полимеры
с большим и меньшим молекулярным весом, чем заданный. Так как
степень полимеризации является функцией продолжительности
реакции, то полимер с заданным молекулярным весом можно полу-
чить, обрывая реакцию на определенном ее этапе (например, путем
охлаждения). Однако полученный таким способом полимер
неустойчив в том смысле, что повторное нагревание может приво-
дить к изменению его молекулярного веса в результате взаимо-
действия функциональных концевых групп.
Во избежание этого реакцию часто проводят при небольшом
избытке одного из компонентов (например, диола или дикарбоно-
вой кислоты). Тогда реакция идет до полного исчерпания одного
из реагентов и на концах цепей будут функциональные группы
одного «сорта»— группы мономера, взятого в избытке. Дальней-
шая поликонденсация становится невозможной, и полимер стаби-
лен к изменениям молекулярного веса. Так, при избытке диамина
в реакции поликонденсации диамина и дикарбоновой кислоты
[уравнение (1.1)1 образуется полиамид с концевыми аминогруп-
пами
Н —( —NH —R —NHOC —R' —СО —)n —NH —R —NH2.
VII
Проведение реакции с избытком кислоты приводит к аналогичному
результату; полиамид (VIII) в этом случае содержит на концах
цепей карбоксильные группы
НО —(СО—R'-CONH —R-NH —)n —СО—R'-COOH,
VIII
72
Глава 2
которые не могут вступать в дальнейшую поликонденсацию из-за
исчерпания исходного диамина.
Молекулярный вес полимера можно регулировать путем про-
ведения реакции в присутствии небольших добавок монофункцио-
нального мономера. Так, для получения полиамидов заданного
и стабильного молекулярного веса в реакции применяются уксус-
ная или лауриновая кислоты. Контроль и ограничение молекуляр-
ного веса полимера монофункциональными добавками основан
на том, что при их взаимодействии с растущей полимерной цепью
на концах цепей отсутствуют функциональные группы, т. е. исклю-
чается возможность дальнейшей реакции. Так, при использовании
в реакции (1.1) бензойной кислоты получается полиамид с кон-
цевыми фенильными группами
CeH5 —СО —( —NH —R —NHCO —R' —СО —)п —ОСОСвН5.
IX
2.46. Количественный подход
Для эффективного регулирования молекулярного веса поли-
мера необходимо проводить реакции при строго соблюдаемом соот-
ношении исходных веществ или в присутствии точно рассчитан-
ного количества монофункционального компонента. Если избыток
одного из компонентов будет достаточно большим, то в результате
получится очень низкомолекулярный полимер. Поэтому важно
знать количественную зависимость между величиной избытка
одного из компонентов и молекулярным весом полимера. Это
необходимо также для того, чтобы оценить количественно влияние
различных реакционноспособных примесей, которые могут при-
сутствовать в реакционной смеси или образоваться в результате
нежелательных побочных реакций. Примеси с функциональными
группами А или В могут резко понижать молекулярный вес
полимера, если нет возможности их количественно оценить.
Рассмотрим теперь, различные реакционные системы, которые
встречаются в поликонденсации.
Тип 1. Для поликонденсации бифункциональных мономеров
А — А и В — В (например, диола и дикарбоновой кислоты или
диамина и дикарбоновой кислоты), проводимой в присутствии
избытка В — В, число функциональных групп А и В равно соот-
ветственно Ад и Ав, которое вдвое превышает число моле-
кул А — А и В — В, находящихся в системе. Стехиометрический
разбаланс выражается как г = N&/N-b- (Отношение г всегда
равно единице или меньше единицы, так как группы В находятся
в избытке.) Суммарное число молекул мономеров составляет
(Ад + Ав)/2 или Ад (1 + 1/г)/2.
Представим теперь понятие степени завершенности реакции р
как долю групп А, прореагировавших за данный промежуток
Поликонденсация
73
времени. Тогда доля прореагировавших групп В составляет гр.
Доля не вступивших в реакцию функциональных групп А и В
равна соответственно (1 — р) и (1 — гр). Суммарное число концов
цепей равно сумме непрореагировавших групп А и В. Так как
каждая полимерная цепь имеет два конца, то число макромолекул
составляет половину от общего количества концов цепей или
[Na (1 - Р) + Nb (1 - гр)]/2.
Среднечисловая степень полимеризации Хп — это общее число
молекул А — АиВ — Вв исходной системе, деленное на число-
молекул полимера
у_________Ад (14- 1/г)/2__ 1 ~4~у (9
п~ [Ад (!-/>)+ Ав(1-гр)]/2 1 + г-2гр ’ V
Уравнение (2.41) показывает, как меняется Хп в зависимости
от стехиометрического разбаланса г и степени завершенности
реакции р. Остановимся на двух крайних формах этой зависи-
мости. При стехиометрическом соотношении двух бифункцио-
нальных компонентов (т. е. когда г = 1,000) уравнение (2.41}
принимает уже знакомый нам вид уравнения (2.27). С другой
стороны, когда реакция проходит на 100% (т. е. р = 1,000),.
уравнение (2.41) можно переписать в виде
*п=|±7. (2.42}
В реальном процессе р может сколько угодно близко приближаться
к единице, но никогда не станет равной единице.
На рис. 2.6 приведена зависимость Хп от избытка одного
из исходных веществ при нескольких степенях завершенности
реакции р, полученная исходя из уравнения (2.41). Для обозна-
чения соотношения исходных веществ в этом рисунке исполь-
зуется г и избыток В — В по отношению кА — А, выраженный
в мольных процентах. Приведенные зависимости дают четкое
представление о том, какие р и г требуются для получения поли-
мера заданного молекулярного веса. Однако при проведении
поликонденсации существуют определенные ограничения в выборе
г и р, обусловленные экономическими соображениями, а также
трудностью очистки ряда исходных веществ, что в известной
степени затрудняет контроль необходимого соотношения реаген-
тов в реакции. Кроме того, следует иметь в виду, что многие
поликонденсационные процессы не доводят до 100%-ной степени
завершенности (т. е. р < 1,000), а обрывают раньше опять-таки
по соображениям экономии. Действительно, время, необходимое
для повышения степени завершенности реакции на заключитель-
ном ее этапе всего на 1%, эквивалентно времени, затраченному
на проведение реакции от начала ее и до степени завершенно-
сти 97—98%. Так, внимательное рассмотрение данных рис. 2.4
74
Глава 2
показывает, что примерно одинаковое время нужно затратить
на повышение степени завершенности реакции от р = 0,97
(•Хп = 33,3) до р = 0,98 (X,, = 50) и на проведение реакции
с начала ее и до р = 0,97.
Рассмотрим ряд примеров, демонстрирующих справедливость
уравнения (2.41) и рис. 2.6. При избытке одного из исходных
веществ 0,1 и 1,0 мол. % (г соответствует 1000/1001 и 100/101)
и степени завершенности реакции 100% Хп равна 2001 и 201
соответственно. При 99 %-ной
степени завершенности реакции
степень полимеризации состав-
ляет уже 96 и 66 соответствен-
но, а при 98 %-ной степени
завершенности реакции умень-
шается соответственно до 49 и
40. Отсюда ясно, .почему поли-
конденсацию почти всегда про-
водят до степени завершенности
не менее чем 98%. Это тем бо-
лее важно, что для проявления
полимером ценных свойств сте-
пень полимеризации его должна
быть как минимум 50—100.
Стехиометрическое отношение г
Рис. 2.6. Зависимость среднечисло-
вой степени полимеризации Хп от
стехиометрического соотношения г
при разных степенях завершенности
реакции р в поликонденсации
А — А и В — В.
Для получения более высокомолекулярных полимеров реакцию
доводят до еще более глубоких степеней превращения, соблю-
дая при этом необходимое соотношение исходных веществ.
С помощью уравнения (2.41) и рис. 2.6 можно найти такие значе-
ния г и р, которые необходимы для получения полимера задан-
ного молекулярного веса. С их помощью можно также рассчитать
аффект от нарушения стехиометрического соотношения исходных
веществ вследствие потерь их за счет испарения или участия
в побочных реакциях. Пользуясь уравнением (2.41), можно также
легко подсчитать точность, требуемую для соблюдения стехио-
метрического соотношения реагентов. Это важно, так как ошибка
в определении количества избытка одного из реагентов влечет
за собой соответствующую ошибку в Хп. Форма кривых рис. 2.6
свидетельствует о том, что эффект от ошибки в оценке г с ростом
Поликонденсация
75
степени полимеризации прогрессивно возрастает. Для получения
еще более высокомолекулярных полимеров необходим все более
строгий контроль за условиями реакции.
Тип 2. Выше уже рассматривалась возможность регулирования
степени полимеризации с помощью добавок монофункциональных
реагентов В при поликонденсации А — А и В — В. В данном
случае справедливы те же уравнения, которые были выведены
для типа 1, разница состоит лишь в том, что здесь г определяется
как
N к
Г~ Ab + 27Vb,
(2.43)
где Ав- — число молекул В в системе, а Ад и Ав имеют тот же
смысл, что и выше. Коэффициент 2 перед Ав- необходим потому,
что по эффекту, оказываемому на рост полимерной цепи, одна
молекула В эквивалентна двум молекулам В — В.
Тип 3. При поликонденсации мономеров типа А — В, например
окси- и аминокислот, стехиометрическое соотношение функцио-
нальных групп поддерживается само собой. В этом случае урав-
нения (2.41) и (2.42) применимы при г = 1. Образующийся полимер
неустойчив с точки зрения изменения молекулярного веса, так как
концевые группы его могут реагировать друг с другом. Для
стабилизации молекулярного веса добавляют монофункциональ-
ный реагент В, при этом становятся применимыми указанные
уравнения при г, равном
__Адв__
ЛАв + 2Ав, ’
(2-44)
где 2АВ- имеет тот же смысл, что и в поликонденсации по типу 2,
а Аав (= Ад = Ав) равно числу молекул А — В. (Для регу-
лирования молекулярного веса полимера здесь нельзя использо-
вать бифункциональные соединения А — А и В — В.)
Кривые, приведенные на рис. 2.6, одинаково хорошо описы-
вают поликонденсацию по типам 1, 2 и 3, все зависит от того, что
отложить на оси х. Если на ней отложено соотношение г, то шкала
применима для всех трех типов реакций. Разница появляется
лишь в том случае, когда на оси абсцисс откладывается количество
добавки в мольных процентах. Так, если отложен избыток В — В,
зависимость справедлива для типа 1. Для реакций, относящихся
к типу 2, где Ад = Ав, и типу 3, на оси х следует откладывать
избыток В в мольных процентах. Масштаб указанных осей отли-
чается в два раза, так как в данном случае две молекулы В — В
эквивалентны одной молекуле В. Соотношение между степенью
полимеризации и соотношением исходных веществ [уравне-
ния (2.41) и рис. 2.6] проверено на большом числе примеров.
Их сппавепливость и ппигопность для пегулипования молекуляр-
76
Глава 2
ного веса полимеров рассмотрена в обзоре Коршака [26] на при-
мере полиамидов, сложных полиэфиров и полибензимидазолов.
Проведение реакций в присутствии избытка А — А или В — В,
равно как и с добавками молекул А или В, подчиняется выше-
рассмотренным закономерностям.
2.5. Молекулярновесовое распределение
в линейной поликонденсации
При полимеризации или поликонденсации образуется смесь
макромолекул различного молекулярного веса. В теоретическом
и практическом отношении представляется интересным обсудить
распределение по молекулярным весам при поликонденсации.
Флори получил молекулярновесовое распределение (МБР), исполь-
зуя статистический подход и основываясь на положении об отсут-
ствии зависимости реакционной способности функциональной
группы от размера молекулы [27, 28]. Ниже рассмотрена по суще-
ству теория Флори, применимая к поликонденсации по
типу А — В, А — А и В — В.
2.5а. Распределение по размерам молекул
Рассмотрим вероятность существования полимерной молекулы,
содержащей х структурных единиц. Это эквивалентно вероят-
ности существования молекулы с (ж — 1) прореагировавшими
группами А и одной непрореагировавшей группой А. Вероятность
того, что группа А прореагировала во время t, определяется как
степень завершенности реакции р. Вероятность того, что (ж — 1)
групп А прореагировали, есть произведение (ж — 1) отдельных
вероятностей, или рх~г. Так как вероятность того, что группа А
не прореагирует, составляет (1 — р), то вероятность Nx суще-
ствования данной молекулы с ж структурными звеньями равна
Nx = p*-i(l-p). (2.45)
Так как Nx соответствует мольной или численной доле ж-меров
(т. е. молекул с ж структурными единицами) в полимерной смеси,
то можно записать
Nx = Npx~l (1 — р), (2.46)
где N — это общее число полимерных молекул, a Nx — число
ж-меров. Если общее число структурных единиц в исходной систе-
ме равно No, то N = No (1 — р) и
^х = А0(1-^)2^-1. (2-47)
Поликонденсация
77
Пренебрегая весом концевых групп, весовая доля wx z-меров
(т. е. весовая доля молекул, содержащих х структурных единиц)
равна wx = xNxIN0 и уравнение (2.47) можно переписать в сле-
дующем виде:
Н7ж = ж(1— р)2рх~1. (2.48)
Выражения (2.46) и (2.48) дают числовое и весовое распределение
по молекулярным весам в линейной поликонденсации при степени
Р и с. 2.7. Распределение ма-
кромолекул по числу при ли-
нейной поликонденсации [28J.
1) р = 0,9600; 2) р —0,9875; 5) р =
=0,9950.
Степень полимеризации х
завершенности реакции р. Эти распределения обычно называют
наиболее вероятными или распределениями по Флори. Кривые
обеих функций распределения для нескольких величин р при-
ведены на рис. 2.7 и 2.8. Из этих данных видно, что число моно-
Степень полимеризации х
Р и с. 2.8. Распределение ма-
кромолекул по весу при ли-
нейной поликонденсации [28].
I) р = 0,9600; 2) р = 0,9875; 3) р =
= 0,9950.
мерных молекул на любой стадии реакции значительно превышает
число любых других полимерных молекул, находящихся в системе.
Хотя число мономерных молекул уменьшается по мере протекания
реакции, они все равно составляют наибольшую фракцию. Иная
картина характерна для весового распределения по молекулярным
весам. Здесь весовая доля молекул, имеющих низкий молекуляр-
ный вес, очень мала и непрерывно уменьшается по мере увеличе-
ния степени завершенности реакции. Максимумы на рис. 2.8
78
Глава 2
наблюдаются при х = —(1/1п р), отвечающем среднечислово!
степени полимеризации, определяемой по уравнению (2.27).
Экспериментально часто молекулярновесовое распределенш
выражается в интегральной форме, представляющей собой зави-
симость от х кумулятивной весовой доли 1Х всех полимерных
Степень полимеризации х
Рис. 2.9. Интегральная кривая
распределения для линейных по-
ликонденсационных полимеров
[28].
7) р = 0,9600; 2) р = 0,9875; 3) р =
= 0,9950.
молекул со степенью полимеризации до х включительно. Поэтому
целесообразно функцию распределения по Флори выразить в вели-
чинах 1Х. Это достигается интегрированием уравнения (2.48),
которое приводит к выражению
/х = 1- [1У(1- (2.49)
Интегральные кривые весового распределения приведены на
рис. 2.9.
2.56. Ширина молекулярно весового распределения
Среднечисловые и средневесовые степени полимеризации Хп
и Xw можно найти из функций распределения по весу и числу
соответственно. Среднечисловой и средневесовой молекулярный
вес определяется по формулам (1.14) и (1.16). Разделив выражение
для Мп [уравнение (1.14)1 на вес Мо структурной единицы, полу-
чают среднечисловую степень полимеризации как
_ У, xNx
Хп = %-^=2^я, (2.50)
где суммирование проводится по всем значениям х. Решая сов-
местно уравнения (2.45) и (2.50), получим
ХП = У ^-‘(1-р). (2 51)
После суммирования имеем
= (2-27>
Поликонденсация
79
т. е. тот же самый результат, который был получен выше. Разде-
лив уравнение (1.16) на Л/о, получим
Xw = S (2.52)
Подставляя уравнение (2.48) в уравнение (2.52), приходим к
X[U=S^-1(1-P)2, (2.53)
а после соответствующих расчетов
= (2.54)
Ширина кривой молекулярновесового распределения тогда опре-
деляется как
-^ = 1 ; Р- (2-55)
Лп
Соотношение XwIXn аналогично отношению МW/Mn, рассмотрен-
ному в разд. 1.4. Оно является мерой полидисперсности полимера.
С увеличением степени завершенности реакции величина XwlXn
возрастает и в пределе при большой степени завершенности
реакции стремится к 2.
С незначительными изменениями вышеприведенные уравне-
ния (2.45)—(2.55) применимы к реакциям поликонденсации,
проводящимся в присутствии монофункционального мономера
или избытка бифункционального мономера [27]. В первом случае
вместо степени завершенности реакции р нужно использовать
вероятность того, что данная функциональная группа прореаги-
рует с бифункциональным мономером. В обоих случаях уравнения
применимы с большой степенью точности при замене р на рг1^.
Функции распределения по числу и весу в таких реакциях, про-
водящихся при нестехиометрическом соотношении исходных
веществ, мало отличаются от тех, которые получены при реак-
циях с эквивалентными соотношениями реагентов.
2.5в. Обменные реакции
Ряд полимеров, например сложные полиэфиры, полиамиды,
полисульфиды, в соответствующих условиях могут вступать
в обменные реакции. Реакция обмена заключается во взаимодей-
ствии концевых функциональных групп одной макромолекулы
с находящейся внутри другой такой же молекулы повторяющейся
связью, например между концевой аминогруппой и амидной
связью полиамида. При таком взаимодействии двух полимерных
so
Глава 2
цепей одна из них может стать длиннее, а другая — короче
Н —(—NH—R—СО—)п —ОН + Н—(—NH—R—СО —)m —ОН ->
—» Н —( —NH-R—СО — )w—ОН 4- Н— (-NH-R-CO-)z—ОН. (2.56)
Если имеет место свободный обмен, то конечное молекулярно-
весовое распределение будет наиболее вероятным распределением,
описываемым уравнениями (2.47) и (2.48) [27, 28]. Свободный
обмен предполагает, что все связи внутри полимерной цепи всех
макромолекул имеют одинаковую возможность для обмена. Это
аналогично концепции независимости реакционной способности
функциональных групп от размера молекулы, примененной
к реакциям обмена. Очевидно, что протекание реакции обмена
в процессе обычной поликонденсации не может влиять на молеку-
лярновесовое распределение. Наиболее вероятное распределение
также будут иметь полимеры, получающиеся при статистическом
расщеплении связей в полимере, например при гидролизе цел-
люлозы.
2.5г. Экспериментальные исследования
Наиболее вероятное расщепление по Флори является доста-
точно твердоустановленным положением, хотя число экспери-
ментальных исследований, подтверждающих его, весьма ограни-
ченно. Для прямого доказательства распределения по Флори
необходимо тщательно расфракционировать полимер, чтобы можно
было построить экспериментальные кривые зависимости Nx, wx
или 1Х от х и сравнить их с теоретическими кривыми. Экспери-
ментальные трудности, возникающие при фракционировании ряда
полимеров различных типов, ограничивают число типов полимеров,
которые можно исследовать. Разработка в последнее время новых
автоматических приборов и методов фракционирования, например
гель-хроматографии [29], должна стимулировать развитие работ
по исследованию молекулярновесового распределения полимеров.
(Это обусловило появление большого количества данных по МБР
полимеризационных полимеров, хотя данных по поликонденса-
ционным полимерам по-прежнему мало.)
Основная часть работ по исследованию молекулярновесового
распределения проведена на полиамидах и сложных полиэфирах.
Из них наиболее подробно исследован, пожалуй, полигексаме-
тиленадипамид. На рис. 2.10 приведена интегральная кривая
молекулярновесового распределения для этого полимера и теоре-
тическая кривая, построенная из распределения Флори [30].
Экспериментальные данные хорошо укладываются на теоретиче-
скую кривую в пределах отклонений, вызванных обычными ошиб-
ками эксперимента. Аналогичные данные для полигексаметилен-
адипамида, подтверждающие распределение по Флори, получены
П оликонденсация
81
также некоторыми другими исследователями [28]. Косвенное
подтверждение наиболее вероятному распределению по Флори
было получено во многих случаях при оценке отношения Xw!Xn.
Для многих реакций поликонденсации это отношение оказалось
равным или близким 2, как это и должно быть согласно уравне-
нию (2.55). Предложены также другие распределения, отличаю-
щиеся от распределения по Флори. Доказательства их правиль-
ности почти всегда основаны на экспериментальных данных,
Рис. 2.10. Интегральная кривая распределения для полигексаметилен-
адината [30].
Сплошная линия — теоретическая кривая для р = 0,9909; точки — экспериментальные
данные.
полученных при неэффективном фракционировании. На основании
обобщения различных данных [28] можно сделать вывод, что
распределение по Флори экспериментально подтверждается лучше
других.
Хотя статистический подход к распределению при поликонден-
сации и оказывается очень эффективным, существует ряд ограниче-
ний применения этого метода. Расчет распределения по размерам
для процессов, протекающих не в реакторах периодического дей-
ствия, а в непрерывной проточной перемешиваемой системе или
в реакторе периодического действия, но с рециркуляцией, весьма
тяжеловесен. Недавно был проведен кинетический анализ таких
систем путем решения уравнений, описывающих образование
образцов одинакового молекулярного веса [31, 32]. Полученные
результаты показывают, что в определенных условиях при обра-
зовании полимеров не в реакторах периодического действия
молекулярновесовое распределение может отличаться от распре-
деления по Флори.
82
Глава 2
2.6. Закономерности поликонденсации
2.6а. Физические характеристики
поликонденсационных систем
Поликонденсация характеризуется несколькими общими при-
знаками. Первостепенное значение в поликонденсации имеет
высокая чистота исходных веществ и их стехиометрическое соот-
ношение, являющиеся основными предпосылками получения
высокомолекулярных полимеров. Для регулирования молекуляр-
ного веса полимера процесс ведут в присутствии определенных
количеств монофункциональных веществ или избытка одного из
бифункциональных реагентов. Другим важным фактором является
температура реакции. Большинство реакций поликонденсации
идет сравнительно медленно при обычных температурах. Поэтому
для ускорения реакции ее часто ведут при высоких температурах
порядка 150—200 °C и выше. Важным обстоятельством является
также равновесность поликонденсационных процессов. Так как
многие поликонденсационные реакции относятся к равновесным
процессам, то для того чтобы максимально сместить равновесие
в сторону образования высокомолекулярного полимера, необходи-
мо использовать соответствующие методы. К ним, в частности,
относится принудительное удаление воды и других низкомолеку-
лярных продуктов реакции из реакционной смеси повышением
температуры реакции или понижением давления.
Поликонденсация в блоке (или в массе) является простейшим
способом проведения поликонденсации, так как для ее осуществле-
ния необходимы только исходные компоненты и, если это тре-
буется, катализатор. Достоинство метода состоит в отсутствии
примесей и простоте выделения полимера. Проведение реакции
в массе особенно выгодно для поликонденсационных процессов,
так как в них высокомолекулярный полимер получается только на
заключительных этапах реакции. Это означает, что вязкость
реакционной системы почти на всем протяжении реакции сравни-
тельно мала и реакционную массу легко перемешивать. Более
того, хотя энергия активации поликонденсации достаточно велика
(например, для процесса получения полиэтилентерефталата или
полигексаметиленадипамида она составляет примерно 20 ккал/моль)
[14, 33], термический контроль за процессом весьма прост, так
как большинство поликонденсаций — мало экзотермические реак-
ции. Обратная картина наблюдается в полимеризации, являющей-
ся, как правило, сильно экзотермическим процессом с высокими
энергиями активации, вязкость реакционной массы в котором
возрастает значительно быстрее. В полимеризации большие про-
блемы представляют перемешивание реакционной среды и терми-
ческий контроль процесса.
Поликонденсация
83
Поликонденсация в массе широко применяется для получения
многих поликонденсационных полимеров. Вместе с тем ряд поли-
кондепсацпонных реакций проводится в растворе, причем раство-
ритель растворяет исходные вещества, позволяет проводить
реакции при очень высоких температурах и ускоряет реакции.
Поликонденсацию иногда проводят в дисперсиях: так, например,
в водной дисперсии из дигалогеналкилов и неорганического суль-
фида получают полисульфиды.
2.66. Различные системы исходных веществ
При поликонденсации для получения одного и того же полиме-
ра могут быть использованы разные исходные вещества (см.
табл. 1.1). Химия реакций, лежащих в основе синтеза различных
поликонденсационных полимеров, рассмотрена в ряде моногра-
фий [33, 34]. Выбор того или иного реагента для получения данно-
го полимера зависит от ряда факторов. К ним относятся доступ-
ность, легкость очистки и свойства (физические и химические)
различных исходных веществ.
Возможность получения высокомолекулярного полимера при
поликонденсации зависит от константы ее равновесия. Если она
не очень велика, то успех во многом определяется тем, насколько
легко можно сместить равновесие в сторону образования полиме-
ра вплоть до завершения реакции. Наряду с легкостью получения
и поддержания стехиометрического соотношения исходных ве-
ществ это является важным обстоятельством, которое надо иметь
в виду при получении поликонденсационных полимеров. В каждом
конкретном случае пути для решения указанных проблем могут
быть различны. Нужно только правильно проанализировать все
особенности данной поликонденсационной системы. Рассмотрим
некоторые реакции поликонденсации, чтобы стало более понятным,
как для получения высокомолекулярных полимеров надо регу-
лировать различные параметры процесса.
2.66.1. Сложные полиэфиры
Сложные полиэфиры можно получать прямой этерификацией
диола и дикарбоновой кислоты
пНО — R — ОН + пНООС — R'—СООН —>
—> НО —( —R —ОСО —R' —COO —)n —H+(2n—1) Н20. (1.2)
Так как полиэтерификация, подобно многим реакциям поликон-
денсации, является равновесной реакцией, для достижения высо-
ких степеней завершенности реакции и получения полимеров
высокого молекулярного веса необходимо непрерывно удалять
воду из сферы реакции. Во избежание нежелательных побочных
84
Глава 2
реакций нужно тщательно соблюдать температурный режим реак-
ции. К таким вредным побочным реакциям относятся дегидратация
диола с образованием ненасыщенных двойных связей или простых
эфиров и пиролиз сложных эфиров [33], например
НОСН2СН2ОН —> Н2О + СН3СНО, (2.57)
2НО —( —R —ОСО —R' —COO —)n —Н —> Н2О +
+ Н — ( — ОСО— R' — COO — R — )п — О — ( — R — ОСО—R' — COO — )n — Н,
(2.58)
--R'— СООСН2СН2 — ОСО — R'--->
—> — R' - СООН + СН2 = СН—ОСО — R' —, (2.59)
НО0С(СН2)4СООН —> СО2 + Н2О + 0 (2-60)
Поликонденсация катализируется протоновыми кислотами или
кислотами Льюиса, наряду с ними применяются также катализато-
ры основного типа, например ацетат кальция, трехокись сурьмы
и тетралкоксиды титана [35]. Катализаторы основного характера
применяются при проведении реакции при более высоких темпе-
ратурах, чтобы избежать нежелательных побочных реакций.
Сложные полиэфиры можно получать также модифицированной
реакцией Шоттена — Баумана, заключающейся во взаимодействии
хлорангидридов дикарбоновых кислот и диолов [см., например,
уравнение (1.3)]. Применение реакции Шоттена — Баумана в меж-
фазной поликонденсации и для получения поликарбонатов рас-
сматривается в разд. 2.6в и 2.116.1 соответственно. Некоторые
сложные полиэфиры получают этерификацией циклических ангид-
ридов, например фталевого ангидрида или малеинового ангидрида,
с многоатомными спиртами или фенолами (см. разд. 2.10).
2.6б.1а. Полиэтилентерефталат. Наиболее часто сложные
Полиэфиры в промышленности получают реакцией эфирного
обмена. Эта реакция, как правило, идет быстрее, чем прямая
полиэтерификация с участием дикарбоновой кислоты, и может
быть использована тогда, когда дикарбоновая кислота плохо
растворима или трудно очищается. Наиболее часто применяются
метиловые эфиры кислот, и реакцию доводят до высоких степеней
завершенности, непрерывно отгоняя из реакционной среды выде-
ляющийся метиловый спирт. В качестве катализаторов поли-
конденсации используются различные слабые основания [35].
Полиэтилентерефталат получают из диметилтерефталата и этилен-
гликоля двухстадийной реакцией эфирного обмена. (Этот полимер
может быть известен читателям под торговым названием лавсан,
майлар или дакрон.) Первая стадия заключается в реакции эфир-
ного обмена с образованием в качестве основного продукта
Поликонденсация
85
бис-(2-оксиэтил)терефталата
НОСН2СН2ОСО
соосн3 + 2 носн2сн2он —►
СООСН2СН2ОН + 2 CH30li ,
(2.61)
а также небольших количеств димера, тримера и тетрамера. Реак-
цию проводят при 105 °C с непрерывной отгонкой выделяющегося
метанола.
На второй стадии температуру поднимают до 260 °C, при этом
идет поликонденсация с выделением этиленгликоля, который
удаляют в вакууме или пропусканием через реакционную смесь
инертного газа
(2.62)
Для получения полимера с высоким молекулярным весом необхо-
димо возможно более полное удаление этиленгликоля, что вызвано
равновесным характером процесса. Если бы не удалялся этилен-
гликоль, то равновесие наступало при низкой степени завершен-
ности реакции (р < 0,7) и полученный продукт имел бы низкий
молекулярный вес (см. рис. 2.6). Характерная особенность реак-
ции эфирного обмена — отсутствие необходимости соблюдения
стехиометрического соотношения исходных веществ в начале
поликонденсации. Стехиометрическое равновесие наступает на
последних этапах второй стадии процесса.
2.66.16. Кинетика равновесной поликонденсации. Хотя, как
правило, равновесная поликонденсация, например вышерассмот-
ренная реакция эфирного обмена, проводится в неравновесных
условиях, интересно рассмотреть кинетику поликонденсации,
проводящейся в условиях, способствующих установлению равно-
весия. (Кинетика обратимой, или равновесной, полимеризации
с раскрытием цикла рассмотрена в гл. 7.) Подробно исследована
кинетика реакции эфирного обмена бпс-(2-оксиэтил)терефталата
86
Глава 2
[14, 36]. Реакцию схематически можно представить следующим
образом:
2 — С6Н5СООСН2С1ЬОН
X h2 -
— С6Н5СООСН2СН2ООССвН5------h НОСН2СН2ОН. (2.63)
XI
Скорость поликонденсации, выражаемая через скорость образова-
ния этиленгликоля d[G]/dt, представляет собой разность скоростей
прямой и обратной реакций
flp = ^ = MM]2-4A:2[P][G], (2.64)
где [М], [Р] и [G] обозначают соответственно концентрации 2-окси-
этилэфирной концевой группы, этилендиэфирных групп (XI) и
этиленгликоля. Коэффициентом 4 в уравнении (2.64) учитывается
статистическая вероятность протекания обратной реакции. Ре-
акция между бифункциональной этилендиэфирной группой и эти-
ленгликолем может идти по четырем эквивалентным направлениям,
тогда как первая реакция между любой гидроксильной группой
и сложноэфирной связью, находящейся у другой 2-оксиэтилэфир-
ной концевой группы, может идти только по одному-единственному
пути.
Концентрацию различных соединений удобно выражать через
степень завершенности реакции р в данный момент времени
[М] = 2(1—р) [R]o, (2.65)
[P]=p[R]o, (2.66)
[G] = (5 + ?-?0)[R]0, (2.67)
где [R]o — начальная концентрация повторяющегося звена XII
— СН2ООС - СвН4—СООСН2 —
XII
и
4 [Н]о
(2.68)
(2.69)
1 2[R]0
где [G]o — начальная концентрация этиленгликоля. Необходимо
заметить, что различные параметры выбраны с таким учетом, чтобы
кинетическая обработка была равно применима к полиэтерифика-
ционным системам, содержащим различные комбинации трех
соединений: X, XI и гликоля. В случае, если в начальной системе
П оликонденсация
87
есть только один бис-(2-оксиэтил)терефталат, то [Р]о — [Gio = О,
[М]о = 2 [R]o; Ро = 0 и q = 0. Подставляя выражения для [М],
[Р] и [G] [уравнения (2.65)—(2.67)] в уравнение (2.64), после ряда
преобразований получим
^- = 4 [R]0{/q(l — р)2— k2p(q + p — р0)}; (2.70)
последнее выражение можно интегрировать в пределах t = 0
(/? = 0) и t = t (р = р), что дает
, К 1 Г (Ро Рравн) [(1 (Р Рравн) + -®1 "> ("2 71^
1 4B[R]o X (р—Рравн) [(1 К)(Ро Рравн)4~#1 J
где Рравн — степень завершенности реакции при равновесии,
К = kjkz п
В = [(2^ + 5-р0)2-4^(^-1)]1/2. (2.72)
Уравнением (2.71) целесообразно пользоваться для определения
скорости поликонденсации, исходя из зависимости между степенью
завершенности реакции и ее продолжительностью, когда известны
К и Рравн [14, 36].
2.66.2. Полиамиды
Закономерности образования полиамидов отличаются от тако-
вых для сложных полиэфиров. Хотя к образованию полиамидов
ведут различные реакции, получают их обычно прямым амидиро-
ванием дикарбоновых кислот и диаминов или самоконденсацией
аминокислот. Для синтеза полиамидов применяется также поли-
меризация с раскрытием цикла лактамов (см. разд. 7.3). Наиболее
важным представителем класса полиамидов является полигекса-
метилен а дип амид, который получают на основе гексаметилендиами-
на и адипиновой кислоты. Для стехиометрического соотношения
исходных веществ предварительно получают 1 : 1-соль адипино-
вой кислоты и диамина, которую часто называют найлоновой солью.
Поликонденсацию проводят путем нагревания водной суспензии,
содержащей 60—80% соли, при температуре примерно 200 °C
в автоклаве под давлением около 15 атм (14,7 НО5 Па)
«адсвд.ад+.ноо^снлсоон - . (2.73)
—>Н —[-NH —(СН2)в —NHCO —(СН2)4-СО —]п —ОН + 2(п —1)Н2О.
Полиамидирование в отличие от полиэтерификации идет хорошо
и без катализатора. Кроме того, при амидировании равновесие
сильно сдвинуто в сторону образования полимера, что также отли-
чает этот процесс от полиэтерификации. Поэтому при амидирова-
нии вплоть до заключительной стадии не заботятся о сдвиге равно-
весия в сторону полимера. Реакцию ведут под давлением в авто-
88
Глава 2
клаве до достижения примерно 80—90%-ной степени завершен-
ности реакции без удаления выделяющейся при реакции воды.
И только затем температуру поднимают до 270—300 °C и путем
непрерывного отвода паров доводят реакцию до конца. На ранних
стадиях процесса реакция протекает в растворе, а заключительный
этап представляет собой поликонденсацию в расплаве, так как
поликонденсацию ведут при температуре, превышающей темпера-
туру плавления полиамида.
2.66.3. Полиуретаны
Синтез полиуретанов имеет свои специфические особенности.
При реакции
пНО — R-OH + nOCN-R' — NCO—»
—» НО —( —R —OCONH — R' — NHCO — O-)n_t — R — OCONH — R' — NCO
(1.6)
возможны различные побочные процессы [33, 34], обусловленные
высокой реакционной способностью изоцианатной группы по
отношению к различным реагентам, в том числе к воде, аминам,
фенолам и карбоновым кислотам. Так, при взаимодействии воды
с изоцианатами образуется двуокись углерода и амин
R — NCO+H2O—>RNH2^CO2. (2.74)
Среди других возможных побочных реакций следует назвать
димеризацию изоцианатов до карбодиимидов
2R —NCO —» R —N = С = N —R-J-CO2 (2.75)
и тримеризацию
О
II
R С ,R
3R-XCO—> | | (2.76)
Z\ /c\
0^ V О
I
R
Из-за таких побочных реакций трудно поддерживать эквимо-
улярное соотношение исходных веществ — диизоцианата и диола.
Поэтому реакцию часто ведут в присутствии небольшого избытка
диизоцианата, покрывающего его участие в побочных реакциях [37].
Указанные побочные реакции играют существенную роль и в опре-
делении ряда других параметров процесса. Как правило, реакцию
осуществляют при сравнительно невысоких температурах (100—
120 °C). Кроме того, во избежание различных побочных процессов
Поликонденсация
89
реакцию ведут в отсутствие добавок третичных аминов, солей
металлов или других оснований, которые являются катализатора-
ми процесса образования уретанов, но могут катализировать
н побочные реакции. Синтез полиуретанов проводят в растворе.
Поликонденсация в расплаве не применяется, так как температура
плавления большинства полиуретанов лежит выше температуры
реакции. Высокие температуры реакции нежелательны также из-
за опасности различных деструктивных процессов, например
R —NH —COO —СН2СН2 —R'—»R —NH24-CO2 + CH2 = CH —R', (2.77)
R-NH-COO — СН2СН2 — R'-»CO2 + R — NH-CH2CH2 — R', (2.78)
n разложения полиуретана на исходные изоцианат и спирт.
2.66.4. Полисилоксаны
Полисилоксаны (или силиконы, как их часто называют) син-
тезируют, исходя из хлорсиланов, например из диметилдихлорси-
лана. Исходный хлорсилан гидролизуется до соответствующего
силанола, который затем подвергают поликонденсации с дегидра-
тацией
СН3 СН3 г СН3-]
I н2о | -н2о I
Cl—Si-Cl----> НО —Si —ОН----->— — О —Si — - (2.79)
I ~HC1 I I
CH3 СН3 L CH3->n
Выбранные условия реакции позволяют не выделять силанол,
а сразу проводят поликонденсацию, которая протекает с большой
скоростью.
В результате такой реакции получается весьма низкомолеку-
лярный полимер, представляющий собой жидкость, которая
используется в качестве смазки. Продукт реакции представляет
собой смесь циклических и линейных полимеров различного разме-
ра. В зависимости от условий реакции состав продуктов может
сильно меняться [38]. При гидролизе силанов в присутствии осно-
ваний образуются высокомолекулярные линейные полимеры.
Проведение процесса в присутствии сильных кислот благоприят-
ствует образованию циклических силоксанов и низкомолекуляр-
ных линейных полимеров. С повышением температуры реакции
возрастает возможность получения полимеров высокого молеку-
лярного веса. Однако твердые высокомолекулярные полисилокса-
ны удается получить только полимеризацией в присутствии ини-
циаторов ионного типа циклических силоксанов с раскрытием
цикла (разд. 7.4). Твердые полисилоксаны применяются в качестве
эластомеров.
so
Глава 2
2.6в. Межфазная поликонденсация
Многие полимеры, которые в промышленности получают высо-
котемпературной поликонденсацией, можно синтезировать также,
используя для этой цели реакцию Шоттен — Баумана, протекаю-
щую с высокой скоростью с использованием хлорангидридов
карбоновых кислот. Так, сложные полиэфиры и полиамиды можно
получить, заменяя дикарбоновую кислоту или ее диэфир на более
реакционноспособное производное ее — дихлорангидрид
лС1СО — R — COCl + nHO — R' — ОН —>
—> _( — СО —R —COO —R' —О —)„—+2пНС1, (2.80)
лС1СО — R — СОС1 + nH2N — R' — NH2 —»
—> — (-СО —R —CONH —R' —NH-)„—+2гаИС1. (2.81)
2.6в.1. Описание процесса
Константы скорости указанных реакций на несколько порядков
больше, чем константы скорости реакций соответствующих кислот
или их дпэфиров. Исследованию этого нового процесса с участием
хлорангидридов дикарбоновых кислот, названного межфазной
поликонденсацией, в последние годы посвящено большое число ис-
следований [39, 401. Поликонденсация двух исходных веществ про-
водится на границе раздела двух жидких фаз, каждая из которых
содержит один из реагентов (рис. 2.11). Полиамидирование осу-
ществляют при комнатной температуре, сливая водный раствор
диамина с органической фазой, содержащей хлорангидрид
дикарбоновой кислоты. Реагенты диффундируют и, встречаясь на
поверхности раздела, реагируют друг с другом. Полимер выпадает
из сферы реакции и непрерывно удаляется в виде пленки или
нити. Скорость поликонденсации определяется диффузией реаген-
тов к границе раздела фаз. От обычной поликонденсации
межфазный процесс отличается и тем, что мономеры, диффун-
дирующие к поверхности раздела фаз, реагируют только с кон-
цевыми группами полимерных цепей. Скорость реакции слишком
высока, чтобы хлорангидрид и диамин могли проникнуть сквозь
полимерную пленку и начать новую полимерную цепь раньше,
чем они прореагируют с концевыми функциональными группами
растущей полимерной цепи. Следовательно, при межфазной поли-
конденсации вероятность образования более высокомолекулярных
полимеров выше, чем при других способах поликонденсации.
Для успешного протекания межфазной поликонденсации необ-
ходимо контролировать ряд ее параметров. Для нейтрализации
выделяющегося кислого продукта в водную фазу добавляют неор-
ганическое основание. Если же не нейтрализовать выделяющийся
хлористый водород, он будет поглощаться исходным диамином,
П оликонденсация
91
превращая его в нереакционноспособную соль, что снижает ско-
рость реакции. При высоких концентрациях неорганического
основания или низких скоростях поликонденсации может идти
гидролиз хлорангидрида. Однако в обычных условиях хлорангид-
рпд очень плохо растворим в воде, а скорость поликонденсации
так высока, что диффузия хлорангидрида сквозь границу раздела
в водную фазу практически исключена.
Очень большое влияние на молекулярный вес полимера оказы-
вает тип органического растворителя, так как показано, что
Сплющенная пленка
Раствор диамина
в воде
Полимерная пленка,
Л. образующаяся на границе
J раздела фаз
Раствор хлорангидрида
дикарбоновой кислоты
в органическом раство-
рителе
Рис. 2.11. Межфазная поликонденсация. Вытягивание пленки полимера
с границы раздела фаз [39].
поликонденсация идет вблизи границы раздела двух фаз со сторо-
ны органической фазы. Наилучшие результаты дает применение
растворителя, в котором растворяется низкомолекулярный поли-
мер и нерастворим полимер с высоким молекулярным весом. При
преждевременном выпадении полимера из реакционной среды не
удается получить полимер желаемого молекулярного веса. Так,
например, ксилол и четыреххлористый углерод являются осади-
телями полигексаметиленсебацината различного молекулярного
веса, тогда как хлороформ не растворяет только высокомолеку-
лярный полимер. При применении в межфазной поликонденсации
первых двух растворителей получаются только низкомолекуляр-
92
Глава 2
ные полимеры. Молекулярновесовое распределение полимеров,
полученных при межфазной поликонденсации, как правило,
отличается от распределения по Флори [26, 40]. Часто встречаются
полимеры с более широким и более узким распределением, чем
наиболее вероятное распределение. Возможно, это обусловлено
частичным фракционированием полимера при его осаждении в ходе
реакции. Указанный эффект, по-видимому, должен сильно зави-
сеть от типа органического растворителя и растворимости образую-
щегося полимера.
2.6в .2. Применение
Межфазная поликонденсация имеет ряд преимуществ перед
другими способами поликонденсации. Одно из них состоит в том,
что чистота реагентов, которая имеет существенное значение
в высокотемпературных процессах, здесь не столь важна, так как
при низких температурах межфазной поликонденсации побочные
реакции за счет примесей не идут столь энергично. При межфазной
поликонденсации не так важно соблюдать эквимолярное соотноше-
ние исходных веществ в каждой из фаз. Стехиометрия автомати-
чески поддерживается на границе раздела двух фаз, где идет
поликонденсация. Благодаря диффузии постоянно обеспечивается
подвод обоих реагентов к границе раздела фаз. Более того, высоко-
молекулярный полимер образуется на границе раздела фаз неза-
висимо от общей степени завершенности реакции, рассчитанной
на суммарное количество обоих реагентов. Общую степень за-
вершенности реакции можно повысить проведением реакции в
перемешиваемой системе, увеличивающей поверхность раздела
фаз.
Межфазная поликонденсация применима для получения разно-
образных полимеров, в том числе полиамидов, сложных полиэфи-
ров, полиуретанов, полисульфонамидов, поликарбонатов, поли-
мочевин, полифосфонамидов и многих других. Кроме тех пре-
имуществ, о которых шла речь выше, к достоинствам межфазной
поликонденсации следует отнести и то, что при низких температу-
рах возможен синтез полимеров, неустойчивых при высоких тем-
пературах, требующихся для проведения других способов поли-
конденсации. При межфазной поликонденсации можно осущест-
влять реакцию между исходными веществами с7 образованием
полимера в форме материала (пленки, покрытия, волокна и др.),
пригодного для использования без дополнительной переработки.
Есть у межфазной поликонденсации и определенные недостатки,
ограничивающие ее практическое применение. К ним относятся
высокая цена хлорангидридов карбоновых кислот, большие
количества растворителя, который надо применять в реакции,
а затем регенерировать, и ряд других.
Поликонденсация
93
2.7. Трехмерная поликонденсация
2.7а. Разветвление
Выше была рассмотрена поликонденсация, бифункциональных
соединений, приводящая к образованию линейных полимеров. При
проведении поликонденсации в присутствии одного или несколь-
ких соединений с числом функциональных групп больше двух
вместо линейного получается разветвленный полимер. При приме-
нении ряда мономеров идет также сшивание с образованием поли-
меров сетчатой структуры, в которых боковые цепи одной макро-
молекулы присоединяются другим концом к другой макромолеку-
ле. На рис. 1.1 сопоставлены структуры линейных, разветвлен-
ных и сетчатых полимеров.
Рассмотрим поликонденсацию вещества А — В в присутствии
небольшого количества мономера Ау, содержащего / функциональ-
ных групп в молекуле. Индекс / означает функциональность моно-
мера. В результате такой поликонденсации получается разветвлен-
ный полимер, у которого f цепей связаны с одной, центральной
точкой ветвления (а именно Ау ветвлений)
-а-ва-ва-ва-ва-ва—ав-ав-ав-ав-ав-ав-ав-ав-ав-ав-ав-а—
I
А
В
А
В
В
I
А
В
I
А
I
ХШ
В конкретном случае для / = 3 поликонденсация А — В в при-
сутствии А — А приводит к образованию структуры XIII. При
внимательном рассмотрении этой структуры видно, что в одной
макромолекуле может быть только один остаток молекулы Ау.
При этом сшитый полимер не образуется, так как невозможна
реакция между полимерными молекулами вышеприведенного типа,
содержащими на концах ветвлений одинаковые функциональные
группы А. (Это не будет справедливо, если группы А могут реаги-
ровать друг с другом. Однако такие случаи крайне редки.)
94
Глава. 2
2.76. Молекулярновесовое распределение
Молекулярновесовое распределение в нелинейной поликонден-
сации рассмотренного выше типа будет намного уже, чем, при
линейной поликонденсации. В этом случае молекул с размерами,
значительно отличающимися от средних, вероятно, должно быть
меньше, чем при линейной поликонденсации, так как в противном
случае статистические возможные ветвления / в молекуле должны
были бы быть или очень длинными, или очень короткими. Из
Распределение макромолекул по весу
конденсации [28].
11 f = 1; 2) / = 2; 3) f = 3; 41 f = h
при трехмерной поли-
[28].
Рис. 2.12.
статистического анализа получены функции распределения [41}
для такой поликонденсации, и его результаты приведены ниже:
Хп^+±-А, (2.82) - (/-1)2р2 + (3/_2)р+1 W~ № + ( Й ’
Ширина распределения характеризуется выражением = 1 + 77-т-т—ЙГ > (2.84) хп (/Р+1 —Р)2 7
которое в пределе при р = 1 становится равным Ф" = 1 +т- (2.85) Ап /
На рис. 2.12 приведено распределение по весу [уравнение (2.83)}
для нескольких величин /. При этом степень завершенности
Поликонденсация
95
реакции выбиралась из расчета постоянной среднечисловой степе-
ни полимеризации, равной во всех четырех случаях 80. С ростом
функциональности мономера Ау распределение по размерам ста-
новится уже. Действительно, как видно из уравнения (2.85), при
р = 1 отношение Xw/Xn уменьшается с 2 при / = 1 до 1,25 при
/ = 4. При значении /, равном 1 или 2, получаются линейные
полимеры, разветвленные же полимеры образуются при / > 2.
Случай, когда / = 1, соответствует поликонденсации типа 3,
рассмотренной в разд. 2.4. При этом полимер имеет распределение
точно такое же, как и наиболее вероятное распределение полиме-
ра, полученного линейной конденсацией (разд. 2.5). Соединение
двух полимерных цепей типа А — В в одну макромолекулу через
молекулу А — А приводит к полимеру со значительно более узким
распределением, чем при обычной поликонденсации мономеров
типа А — В или А — А и В — В.
2.8. Сшивание
Поликонденсация вещества А — В и Ау (при / больше 2) в при-
сутствии вещества В — В приводит не только к разветвлению цепей,
но и к образованию сшитого полимера. В присутствии реагента
В — В ветвления от одной макромолекулы способны реагировать
с ветвлениями другой макромолекулы. Схематическое изображе-
ние сшитого полимера XIV дает представление о том, как две
полимерные цепи соединяются вместе. Мостики, соединяющие
различные цепи, называются поперечными связями или сшивками.
К образованию сшивок ведет взаимодействие двух ветвлений
с различными концевыми функциональными группами, обозначен-
ными стрелками в формуле XIV, например группами А и В
(см. схему на стр. 96).
Сшивание будет иметь место во всех реакциях поликонденса-
ции, в которых участвуют реагенты с функциональностью больше
двух. К ним относятся следующие реакции:
А—А + By—>
А—А + В — В4-Ву~^
АУ + Ву~>
Характерным признаком трехмерной поликонденсации являет-
ся гелеобразование, наступающее в определенный момент реакции.
В этот момент, называемый точкой гелеобразования, вначале наблю-
дается образование геля или нерастворимого полимера. (За точку
гелеобразования принимается момент, в который система теряет
текучесть, т. е. пузырьки воздуха уже не могут передвигаться
в ней.) Гель нерастворим в различных растворителях при повышен-
ных температурах, при которых еще не идет деструкция полимера.
Гель соответствует образованию бесконечной сетки, в которой
96
Глава 2
А
I
В
А
I
В
А
A—BA—BA
-рАВ—АВ—АВ—АВ—АВ—АВ—ВА“ВА-|~А
А
В
I
А
В
.1
В
-*-АВ—АВ—АВ—BA—ВА—ВА-ЧА
В
i
А
В
I
А
В
"j—АВ—А
А
В
полимерные молекулы соединены друг с другом в одну гигантскую
макромолекулу. В сущности гель представляет собой одну молеку-
лу. Золь-фракция полимера растворима в органических раствори-
телях. При продолжении поликонденсации после точки гелеобра-
зования количество геля увеличивается за счет золя и все больше
и больше молекул золя превращаются в гель. В процессе геле-
образования физическое состояние системы резко меняется. Из
вязкой жидкости она превращается в полимер с бесконечно боль-
шой вязкостью.
Реакции сшивания имеют громадное практическое значение.
Сшитые пластики все больше и больше применяются в качестве
конструкционных материалов благодаря их высокой термо- и теп-
лостойкости. Жесткая сетчатая структура их обеспечивает высокую
стабильность размеров таких полимеров в самых различных усло-
виях эксплуатации. Подобные полимеры не текут при нагревании
и называются термореактивными полимерами или реактопласта-
ми. (Пластики, размягчающиеся и текущие при нагревании,
П оликонденсация
97
т. е. несшитые пластики, называются термопластами. Почти все
полимеры, получаемые полимеризацией, являются термопластами,
и лишь небольшое число их подвергается сшиванию.) О практиче-
ской ценности термореактивных полимеров свидетельствует то, что
из примерно 6,8 млн. тонн пластмасс, произведенных в США
в 1968 г., почти 1,6 млн. тонн составляли сшитые пластики [42].
2.8а. Уравнение Карозерса
2.8а. 1. Вывод для системы с эквивалентным
соотношением исходных веществ
Чтобы плодотворно использовать реакцию сшивания, надо
уметь ее регулировать, а для этого необходимо знать соотношение
между гелеобразованием и степенью завершенности реакции.
Карозерс вывел важное соотношение между степенью завершенно-
сти реакции в точке гелеобразования и средней функционально-
стью /ср поликонденсационной системы для случая эквивалентного
соотношения функциональных групп А и В [13]. Вывод уравнения
аналогичен выводу уравнения (2.41). Средней функциональностью
смеси мономеров называется среднее число функциональных групп
на одну молекулу мономера с учетом мономеров всех типов, нахо-
дящихся в системе. Таким образом,
У, Nifi
(2-86)
где — число молекул мономера i с функциональностью ft,
и суммированы все мономеры, находящиеся в реакционной систе-
ме. Следовательно, для системы, состоящей из двух молей глицери-
на (триола) и трех молей фталевой кислоты (дикарбоновой кисло-
ты), на пять молекул исходных „веществ приходится двенадцать
функциональных групп, /ср равно 12/5, т. е. 2,4. Для системы,
содержащей эквимолярные количества глицерина, фталевого
ангидрида и монокарбоновой кислоты, на три молекулы приходит-
ся шесть функциональных групп, /ср равно 6/3, т. е. 2.
В системе, содержащей эквивалентные количества групп А
и В, число молекул мономеров, находящихся в исходной системе,
равно No, а соответствующее суммарное число функциональных
групп составляет А0/ср. Если N обозначить число молекул после
прохождения реакции, то 2(/V0 — N) — это число функциональ-
ных групп, вступивших в поликонденсацию. Тогда степень завер-
шенности реакции р определится как доля израсходованных функ-
циональных групп
98
Глава 2
а степень полимеризации
Y
~1Г •
(2.88)
Комбинируя эти уравнения, получим
2
Хп
2 — Р/ср ’
(2.89)
которое можно переписать в виде
2 2
Р f = .
/ср %nfcp
(2.90)
Уравнение (2.90), называемое часто уравнением Карозерса, связы-
вает степень завершенности реакции, степень полимеризации
и среднюю функциональность реакционой системы.
Важным частным случаем уравнения (2.90) является выраже-
ние для точки гелеобразования, где среднечисловая степень поли-
меризации стремится к бесконечности. Тогда критическая степень
завершенности реакции рк в точке гелеобразования равна
9
РК = ^, (2.91)
так как вторым членом в правой части уравнения (2.90) можно
пренебречь. Уравнением (2.91) можно пользоваться для оценки
степени завершенности реакции, при которой начинается гелеоб-
разование, если известна средняя функциональность смеси данных
реагентов. Так, критическая степень завершенности реакции по-
ликонденсации глицерина и фталевой кислоты (молярное соотно-
шение 2 : 3) равна 0,833.
2.8а.2. Гывод для системы с неэквивалентным
соотнотением, исходных веществ
Уравнения (2.90) и (2.91) применимы только при стехиометри-
ческом соотношении обеих функциональных групп. Для неэквива-
лентной смеси функциональных групп средняя функциональность,
рассчитанная с помощью уравнения (2.86), оказывается сильно
завышенной. Так, рассмотрим крайний случай: смесь 1 моля гли-
церина и 5 молей фталевой кислоты. Используя для расчета урав-
нение (2.86), получим значение /ср, равное 13/15, или 2,17. При
таком значении /, р должен получиться высокомолекулярный поли-
мер. Затем из уравнения (2.91) можно найти, что сшивание должно
начаться при рк =0,922. Оба эти вывода ошибочны. Как было
показано выше (см. разд. 2.4), при резком нарушении стехиометри-
ческого соотношения исходных веществ А и В (г = 0,3) могут
образоваться только очень низкомолекулярные полимеры. Реак-
П оликонденсация
99
ция закончится раньше, чем прореагирует основная масса карбо-
ксильных групп.
Средняя функциональность смесей, содержащих нестехиометри-
ческие соотношения исходных веществ, была оценена [43] и оказа-
лась равной отношению удвоенного количества функциональных
групп вещества, содержащегося в меньшем количестве, чем стехио-
метрическое, к общему числу всех присутствующих в системе моле-
кул. Следовательно, степень завершенности реакции (и сшивание,
если оно может иметь место) зависит от количества вещества, нахо-
дящегося в меньшем количестве. Избыток другого вещества не
оказывает никакого влияния и просто уменьшает функциональ-
ность системы. Для рассмотренного выше случая нестехиометри-
ческого соотношения 1 моля глицерина и 5 молей фталевой кисло-
ты величина /ср составляет 6/6, или 1,00. Такая низкая средняя
функциональность системы соответствует низкой степени поли-
меризации в данном процессе.
Аналогичным способом была рассчитана средняя функциональ-
ность нестехиометрической смеси, содержащей более двух исход-
ных веществ. Рассмотрим, например, тройную смесь N& молей
А/ Nc молей А/с и Ав молей В/в с функциональностями /д, /с
и /в соответственно. В этой системе А/д и А/ содержат одинаковые
функциональные группы (а именно группы А), причем суммарное
число функциональных групп А меньше числа функциональных
групп В (т. е. группы В содержатся в избытке). Средняя функцио-
нальность в такой системе определяется как [43]
/ср 2 (А а/а + ^с/с) Аа + Ас + АВ (2.92а)
или 2г/д/в/с (2.926)
где JC₽ /А/с- г гР/а/в + г (1 — Р)/в/с
Г аа/а +Ас/с Ав/в (2.93)
Ас/с (2.94)
р АГа/а +Ас/с
Величиной г характеризуется отношение групп А и групп В,
причем она всегда равна или меньше единицы. По своему смыслу
г эквивалентен ранее рассмотренному стехиометрическому раз-
балансу. Величиной р характеризуется доля всех функциональ-
ных групп А, принадлежащих реагенту А/ .
С практических позиций интересен случай, когда /с > 2 и /д —
= /в = 2, что соответствует
А/с+А-А+В-В-> (2.95)
100
Глава 2
Для такой системы уравнение (2.92) принимает вид
/ср = /с+Згр+^с (1—р) • (2’96)
Подставив выражение для /ср в уравнение (2.91), получим
+ (2.97)
Необходимо иметь в виду, что степень завершенности реакции рк
в точке гелеобразования относится к степени завершенности реак-
ции по функциональным группам А. Степень завершенности реак-
ции по группам В равна в точке гелеобразования грк.
2.86. Статистическое рассмотрение гелеобразования
Флори [27] и Штокмайер [44] применили статистический под-
ход для того, чтобы вывести уравнение, предсказывающее степень
завершенности реакции в точке гелеобразования. Этот подход
предполагает, что все функциональные группы одинакового типа
одинаково реакционноспособны независимо от молекулярного веса
полимера. Принято также, что реакции конденсации протекают
только между функциональными группами различных молекул.
[Эти допущения необходимо иметь в виду и при использовании
уравнений (2.89)—(2.97).] Для расчета глубины реакции вводится
коэффициент разветвления а, определяемый как вероятность того,
что данная функциональная группа, находящаяся на конце раз-
ветвляющей структурной единицы, окажется связанной с другой
такой же единицей. В случае поликонденсации А — А и В — В
в присутствии Ау это соответствует образованию сегмента типа
А-А + В—В + Ау—>А(/_П —А(В-ВА—А)П-В-ВА—А(/_п, (2.98)
где п может иметь любые значения от нуля до бесконечности.
Полифункциональный мономер Ау рассматривается как разветвля-
ющая структурная единица, тогда как сегменты, заключенные
между ветвлениями, называются цепными сегментами. Развет-
вляющие структурные единицы присоединяются к полимерной
цепи в точках ветвления (рис. 1.1). Бесконечная сетка образуется
тогда, когда из п цепей или цепных сегментов образуется более п
цепей в результате ветвления некоторых из них. Критерием геле-
образования является то, что в данной системе, содержащей
реагент с функциональностью /, по крайней мере один из (/ — 1)
сегментов, отходящих от разветвляющей структурной единицы,
присоединится в свою очередь к другой разветвляющей единице.
Веррятность этого равна 1/(/—1), и критический коэффициент
разветвления ак для образования геля составляет
«к = 7^- (2.99)
Поликонденсация
101
В уравнении (2.99) / — функциональность разветвляющей струк-
турной единицы, т. е. мономера с числом функциональных групп
больше двух. Ее не следует смешивать со средней функционально-
стью /ср в уравнении Карозерса. Если в реакционной системе
присутствует несколько полифункциональных разветвляющих еди-
ниц, то в уравнение (2.99) следует подставить среднее значение /.
При а(/ — 1), равном единице, за цепным сегментом будет
последовательно присоединено в среднем а(/— 1) цепей. Из них
часть а будет заканчиваться центрами ветвления, т. е. цепей станет
больше в а2 (/ — I)2 раз. Процесс ветвления будет и дальше раз-
виваться таким образом, причем число присоединенных цепей
будет прогрессивно расти по мере протекания каждой последую-
щей реакции ветвления. Рост полимера будет ограничен только
границами реакционного аппарата. Если, с другой стороны,
а (/ — 1) меньше единицы, то, вероятно, цепные сегменты не будут
оканчиваться разветвляющими единицами. Для трифункциональ-
ного реагента (/ = 3) критическая величина а равна 0,5.
Определяя вероятность образования цепного сегмента, приве-
денного в уравнении (2.98), найдем зависимость между вероятно-
стью а и степенью завершенности реакции. Степень завершенности
реакции по функциональным группам А и В равна соответствен-
но рд и рв. Обозначим р отношение всех функциональных групп А
в смеси, вступивших и не вступивших в реакцию, которые являют-
ся частью разветвляющих структурных единиц, к общему числу
всех групп А. (Это эквивалентно определению в уравнении (2.94).]
Вероятность того, что группа В прореагирует с разветвляющей
единицей, равна рвр- Вероятность того, что группа В прореаги-
рует с бифункциональным мономером А — А, равна рв(1 — р).
Следовательно, вероятность образования сегмента, приведенного
в уравнении (2.98), равна [/*в(1 —P)Pa1”PbP- Просуммировав
это выражение для всех значений п и найдя сумму, получим
РАРвР
1 —РАРв(1 —Р)
(2.100)
Ра и ръ можно вычислить, применив отношение всех групп А ко
всем группам В и подставив ръ = гр& в уравнение (2.100)
fBP (2.101)
1 ——Р) г — р|(1 — р) v 1
Из уравнений (2.101) и (2.99) легко получить важное выражение
для степени завершенности реакции (по функциональным группам
А) в точке гелеобразования
1
П --- .
ГгД-rof/ —
(2.102)
102
Глава 2
Заслуживают внимания отдельные случаи, когда, например, две
функциональные группы находятся в эквивалентных количествах.
Тогда г\= 1 и рА — рв = р, и уравнение (2.101) принимает вид
---------------------------------- (2.103)
1-р2 (1-Р) •
а уравнение (2.102) превращается в
1 рк = 1Г . (2.104) П + р(/-2)]1/2 V
При поликонденсации без молекул А — А (р = 1) при г < 1 они
принимают вид а = грА-=-^ . (2.105) рк = 1 IT • (2.106) [г + г(/-2)]^ 1
(2.107)
(2.108)
При соблюдении того и другого условий г = р = 1 и уравнения
(2.101) и (2.102) превращаются в
а = р2.
_ 1
[1 + (/-2)]1/2
Для случая, когда в системе присутствуют только мономеры с
/ > 2, вероятность того, что функциональная группа разветвляю-
щей единицы прореагирует с другой разветвляющей единицей,
точно равна вероятности того, что она вообще прореагирует, т. е.
а = р, и поэтому
1
Рк~ f-1
(2.109)
2.8в . Экспериментальное определение точки
гелеобразования
Определение степени завершенности реакции в момент геле-
образования дает различные результаты в зависимости от того,
каким методом пользоваться для ее оценки: статистическим или
уравнением Карозерса. Расчет по уравнению Карозерса и соответ-
ствующему ему уравнению для тройных смесей [уравнения (2.91)
и (2.97) соответственно] дает степень завершенности реакции, при
которой среднечисловая степень полимеризации стремится к бес-
конечности. При таком подходе, очевидно, должны получаться
завышенные результаты, так как в системе имеются полимерные
молекулы со степенью полимеризации, большей Хп, и они должны
достигать точки гелеобразования раньше, чем другие молекулы со
Поликонденсация
103
степенью полимеризации Хп. Статистический подход [уравнение
(2.92)1 позволяет избежать этой ошибки, так как он дает степень
завершенности реакции, при которой кривая распределения поли-
мера по размерам начинает захватывать область бесконечно боль-
ших молекулярных весов [43].
Точку гелеобразования обычно экспериментально определяют
как момент, в который реакционная смесь теряет текучесть, на-
пример когда в ней перестают подниматься пузырьки газа. Экс-
периментальное определение точки гелеобразования для ряда сис-
тем подтвердило справедливость подхода Карозерса и статисти-
ческого анализа. Так, при взаимодействии эквивалентных коли-
честв глицерина (трехатомного спирта) и различных дикарбоновых
кислот точка гелеобразования наблюдалась [45] при степени за-
вершенности реакции 0,765, тогда как расчетное значение, полу-
ченное с использованием уравнений (2.102) (статистический под-
ход) и (2.91) (уравнение Карозерса), равно соответственно 0,709
и 0,833. Флори исследовал ряд систем, состоящих из диэтиленгли-
коля (/ = 2), пропан-1,2,3-трикарбоновой кислоты (/ = 3) и ян-
тарной или адипиновой кислот (/ = 2), при стехиометрическом
и нестехиометрическом соотношениях исходных гидроксильных
и карбоксильных групп. Ряд результатов приведен в табл. 2.4
вместе с данными, вычисленными по уравнениям Карозерса и ста-
тистическим уравнениям.
Таблица 2.4
Определение точки гелеобразования для смеси
пропан-1,2,3-трикарбоновой кислоты, диэтиленгликоля
и адипиновой или янтарной кислоты [46]
[СООН] Г “ [ОН] р Степень .завершенности реакции При точке гелеобразования рк
рассчитана по уравнению (2.91) или (2.97) рассчитана по уравнению (2.Ю2) эксперименталь- но найденная
1,000 0,293 0,951 0,879 0,911
1,000 0,194 0,968 0,916 0,939
1,002 0,404 0,933 0,843 0,894
0,800 0,375 1,063 0,955 0,991
Наблюдаемые значения рк (табл. 2.4) лежат где-то между обеими
расчетными величинами. При *использовании уравнения Каро-
зерса величины рк завышены (причины этого были уже рас-
смотрены). Экспериментальные значения рк очень близки, но всег-
да несколько выше величин, полученных с использованием уравне-
ния (2.102). Это расхождение, как правило, связано с внутримоле-
104
Глава 2
кулярной циклизацией, т. е. с тем, что некоторые функциональные
группы реагируют с образованием внутримолекулярных связей,
что не учитывалось в теоретических расчетах. Вследствие проте-
кания внутримолекулярной реакции часть функциональных групп
не участвует в образовании сетчатой структуры и, следовательно,
для достижения точки гелеобразования реакция должна быть про-
ведена до несколько большей степени превращения. На справедли-
вость этого указывают результаты исследования гелеобразования
при взаимодействии пентаэритрита (/ = 4) и адипиновой кислоты
(/ = 2) при различных их концентрациях [47]. Результаты были
экстраполированы на бесконечно большую концентрацию, при
которой внутримолекулярным процессом можно пренебречь.
Экспериментальная величина 0,578 ± 0,005 очень близка к теоре-
тической величине 0,577. По уравнению Карозерса получается
рк, равная 0,75.
Расхождение между экспериментально найденными величинами
рк и вычисленными с помощью уравнения (2.102) в отдельных
случаях может быть обусловлено тем, что неправильно допущение
об одинаковой реакционной способности всех функциональных
групп одного типа. В качестве примера рассмотрим уже упоминав-
шуюся систему глицерин — фталевая кислота. Расхождение между
наблюдаемыми и расчетными величинами рк (0,709 и 0,765) можно
уменьшить, если ввести поправку на хорошо известную меньшую
реакционную способность вторичной гидроксильной группы гли-
церина.
Хотя оба метода используются для оценки точки гелеобразова-
ния, статистический метод применяется более широко. Последний
метод предпочтительнее, так как с его помощью можно теорети-
чески определить точку гелеобразования для наибольших молекул
полимера. Кроме того, этот метод применим к более широкому
кругу исходных веществ, он позволяет учесть неодинаковую реак-
ционную способность некоторых функциональных групп и наличие
внутримолекулярных реакций. Уравнение (2.102) и следствия из
него широко используются в технологии трехмерных полимеров,
например фенолформальдегидных смол [48], алкидных полимеров
[49—51] и др.
2.9. Молекулярновесовое распределение
в нелинейной поликонденсации
Вывод уравнения, характеризующего молекулярновесовое рас-
пределение для трехмерных полимеров, несколько сложнее, чем
соответствующий вывод для линейных полимеров. Эти выводы рас-
смотрены в ряде работ [27, 44, 52], здесь же будут приведены
только окончательные результаты. Для числа Nx, числовой или
мольной доли Nx и весовой доли wx ж-меров в системе, содержа-
Поликонденсация
105
щей исходные вещества с / > 2, можно записать
"" = [,!(У,-2,. + 2)|] (1 -»)"-2'+2. (2.И0)
jV-“ »’-* << -“)"’2‘+2. (2.И1)
«Ъ = Г-7---X\u7X^ 4.Щ1' I a*"1 (! -a)AT~2x+2- (2.112>
л L (x—1)! (fir — 2г 4-2)! J v '
Среднечисловая сте! тень полимеризации равна Х*~ l-df/2) ’ (2Л13>
а средневесовая Xw~ 1 —(f-l)a ‘ (2.114)
Отсюда коэффициен т полидисперсности (1 Ч-°0 (1—«//2) / 2 1J5) Хп 1 —(/—l)a ’ 1 • /
Эти общие уравнения одинаково применимы к различным поли-
функциональным реакциям, например взаимодействию Ay с By
или АусА — АиВ — В. В зависимости от типа реагентов в соот-
ветствии с уравнениями (2.101)—(2.109) величина а будет опреде-
ляться соответственно параметрами г, f, р и р. Для удобства будет
рассмотрено распределение по размерам продукта реакции экви-
валентных количеств только трифункциональных реагентов, т. е.
где а = р. Сравнение уравнений (2.48) и (2.112) показывает, что
при эквивалентной степени завершенности реакции весовое рас-
пределение у разветвленных полимеров шире, чем у линейных
полимеров. Более того, по мере увеличения функциональности
полифункциональных реагентов распределение у разветвленных
полимеров становится еще более широким. При возрастании вели-
чины а распределение также становится шире. Это хорошо видно
из данных рис. 2.13, на котором приведена весовая доля ж-меров
как функция а в поликонденсации трифункциональных исходных
веществ.
На рис. 2.14 приведены кривые зависимости весовой доли раз-
личных х-меров от а или р при поликонденсации трифункцио-
нальных соединений, а также зависимость весовой доли шг геля,
или оо-мера. При сопоставлении данных рис. 2.13 и 2.14 с анало-
гичными данными для продукта линейной поликонденсации (см.
рис. 2.8) хорошо видно, что весовая доля мономеров всегда выше
доли полимерных молекул любого молекулярного веса (вплоть до
точки пересечения кривых для и\ и шг). С увеличением степени
завершенности реакции доля больших молекул увеличивается за
loti
r.iami 2
счет малых молекул, причем максимум наступает при р, мень-
шем !/2. При этом максимум для высокомолекулярных фракций
сдвинут в сторону больших значений р. В точке гелеобразования
(а = 1/2), где доля сильно разветвленных молекул еще очень
Степень полимеризации х
Р и с. 2.13. Молекулярновесовое
распределение как функция степе-
ни завершенности реакции при
поликонденсации трифункцио-
нальных исходных веществ, где
а = р [52].
а 0,25; 2) а — 0,40; .?) а = 0,50.
мала, распределение становится наиболее широким. Бесконечная
полимерная сетка возникает в точке гелеобразования, и ее весовая
„доля быстро возрастает. Доля золя (состоящего из молекул всех
V и с. 2.14. Весовая доля полимерных молекул различного веса и геля
при поликонденсации трифункциональных исходных веществ, где а = р [52].
сортов, исключая гель) уменьшается, так как наиболее разветвлен-
ные молекулы большей частью вовлекаются в гель. После пере-
сечения кривых для и весовая доля геля становится пре-
обладающей.
П оликонденсация
107
Уширение распределения при увеличении а можно характери-
зовать также отношением XwIXn. Как видно из уравнений (2.113) —
(2.115) с ростом степени завершенности реакции расхождение
между среднечисловой и средневесовой степенью полимеризации
очень быстро возрастает. В точке гелеобразования ширина рас-
пределения, рассчитанная по формуле Xw/Xn, становится очень
большой, так как в этот момент Xw стремится к бесконечности,
а Х„ равно всего четырем (рис. 2.15). После точки гелеобразования
Р и с. 2.15. Средневесовая и сред-
нечисловая степень полимериза-
ции как функция для полимеров,
полученных поликонденсацией
трифупкциональных мономеров.
Ветви кривых после точки геле-
образования. (а = 0,5) относятся
только к золь-фракции [52].
величина Xw!Xn для золь-фракции уменьшается. И наконец, при
а. = р — 1 в системе остается один гель (одна гигантская моле-
кула) и Xw/Xn становится равным единице.
Как уже указывалось выше, поведение систем, содержащих би-
функциональные или трифункциональные исходные вещества, опи-
сывается уравнениями (2.110) — (2.115). Изменение шх в поликонден-
сации бифункциональных реагентов, где коэффициент разветвле-
ния а изменяется при использовании соответствующих количеств
трифункционального мономера, подчиняется примерно тем же зако-
номерностям, что и при поликонденсации одних трифункциональ-
ных мономеров. С увеличением степени завершенности реакции
распределение полимера по молекулярным весам становится шире.
2.10. Технология трехмерной
поликонденсации
Теория процесса трехмерной поликонденсации и параметры,
которые надо в ней регулировать, рассмотрены выше. Контроль за
процессом очень важен при промышленном получении термореак-
тивных пластиков. Это в равной мере относится к проведению
реакции до точки гелеобразования и после нее. Период после
точки гелеобразования обычно называется отверждением. На
свойствах конечного полимера может отрицательно отразиться
как слишком быстрое, так и очень медленное отверждение. Так,
108
Глава 2
если при получении термореактивного пенопласта желатинизация
идет очень медленно, то пена может опасть. С другой стороны, при
формировании армированных и слоистых пластиков слишком
быстрое отверждение может привести к образованию материалов
с низкой прочностью.
Переработка термопластов и термореактивных полимеров в изде-
лия имеет ряд характерных особенностей. В случае термопластов
химическая структура полимера закладывается при его синтезе,
а переработка сводится просто к тому, что переводят полимер при
нагревании в вязко-текучее состояние и под давлением придают
ему желаемую форму изделия. Иная картина характерна для тер-
мореактивных полимеров. Отвержденный термореактивный поли-
мер теряет текучесть, и его нельзя переработать в изделие. Поэто-
му полимер перерабатывают на промежуточной стадии его образо-
вания (такой полимер называется преполимером), завершение же
его синтеза и отверждение идет уже в изделии в процессе перера-
ботки. Так, например, преполимер заливают в форму, а отверждение
его происходит уже в изделии в процессе трехмеризации. Молеку-
лярный вес преполимеров обычно не превышает 500—5000, они
могут быть жидкими или твердыми веществами.
Термореактивные полимеры классифицируют на Л, В и С,
причем в основу такой классификации положено отношение степе-
ни завершенности реакции р для данного полимера и степени
завершенности реакции в точке гелеобразования рк. Полимер А —
это полимер, для которого р < рк, полимер В характеризуется
тем, что система близка к точке гелеобразования (рк), после рк мы
имеем дело с полимером С. Полимер А растворяется и плавится.
Полимер В еще размягчается, но растворяется уже очень плохо.
Полимер С имеет сильно сшитую структуру, и поэтому он не
размягчается и не растворяется. Обычно переработке подвергается
полимер В, хотя в ряде случаев перерабатывают и полимер А.
В результате последующего отверждения полимер переходит
в форму С.
2.10а. Статистические преполимеры
В зависимости от структуры преполимера термореактивные по-
лимеры делятся на две большие категории [53]. Более старыми яв-
ляются термореактивные материалы, получающиеся в результате
статистической реакции бифункциональных мономеров с мономера-
ми с функциональностью, превышающей два. Преполимер получа-
ют, обрывая первую стадию реакции путем охлаждения при задан-
ной степени полимеризации (р < рк), причем такие преполимеры
называются статистическими преполимерами. Поликонденсация
таких преполимеров завершается в процессе второй стадии (пере-
работки), обычно при нагревании.
П оликонденсация
109
2.10а.1. Сложные полиэфиры
Примером статистического типа сшитых полимеров являются
сложные полиэфиры на основе фталевого ангидрида и глицерина.
(2.116)
+ СНОН —-
I
сн2он
Для того чтобы получить более эластичный полимер, в синтезе
полиэфиров применяются диолы и даже монокарбоновые кислоты,
что приводит к уменьшению частоты поперечных связей в полимере
сетчатой структуры. Особого рассмотрения заслуживает случай
использования монокарбоновой кислоты в реакции трифункцио-
нального глицерина и дифункционального фталевого ангидрида.
Для многих систем такого типа пригодны общие схемы расчета,
•когда средня*я функциональность системы равна двум или немного
больше двух. (Для эквимолярной смеси указанных трех реагентов
/ср = 2.) При использовании для расчета таких систем уравнения
Карозерса и аналогичных ему уравнений (см. разд. 2.8а.1 и 2.8а.2)
получаются неверные результаты, согласно которым в таких сис-
темах структурирование не должно идти, тогда как на самом деле
;В этих случаях гелеобразование имеет место.
Такое расхождение обусловлено тем, что при выводе уравне-
ния Карозерса допускают, что гелеобразование начинается, когда
среднечисловой молекулярный вес становится бесконечно боль-
шим. Однако даже для систем с функциональностью, близкой двум,
отдельные полимерные молекулы могут иметь размер, больший, чем
средний, поэтому они будут подвергаться реакции трехмеризации,
если в системе есть трифункциональный реагент. Для определения
точки гелеобразования в таких поликонденсационных системах
выведены уравнения, основанные на статистическом подходе
Ж гелеобразованию. Одним из них является выражение
л=ЬА)Г- <2Л17>
110
Глава 2
где
_ Общее эквивалентное количество ОН-групп .
Общее эквивалентное количество СООН-групп ’ ' '
_ Эквивалентное количество СООН-групп монокарбоновой кислоты
Общее эквивалентное количество СООН-групп ’
(2.1186)
которое особенно полезно [49—51] при расчете систем с алкидны-
ми полимерами (разд. 2.106.3).
2.10а.2. Фенолформальдегидные полимеры
Одноядерные метилолфенолы (XV) образуются на первой ста-
дии реакции фенола (/ = 3) и формальдегида (/ = 2), проводимой
в присутствии оснований как катализаторов
XV6
Наряду с ними получаются двухъядерные (XVI) и полиядерные
(XVII) соединения типа приведенных на стр. 111. Такие смеси,
состав которых определяется соотношением фенола и формаль
дегида и условиями реакции^ называются резольными преполи
мерами. Перед началом второй стадии, проводящейся при
нагревании, резолы обычно нейтрализуют или даже слегка под
кисляют. На второй стадии поликонденсации и при отверждении
идет образование метиленовых связей простых дибензилэфирных
групп между бензольными ядрами, приводящее к возникновению
сетчатой структуры типа XVIII (стр. 111).
Поликонденсация
111
ОН он
HOCH
Соотношение между метиленовыми и простыми эфирными связями
точно не установлено, хотя факт образования обеих не вызывает
сомнений. Повышение температуры благоприятствует образованию
метиленовых мостиков [33, 54].
Получение фенолформальдегидных полимеров — важный про-
мышленный процесс. Однако по ряду причин реакцию очень трудно
количественно обсчитать. В этом случае предположение об одина-
ковой реакционной способности всех функциональных групп
112
Глава 2
в исходных веществах независимо от наличия других функциональ-
ных групп и от того, прореагировали они или нет, представляется
весьма сомнительным. Рассмотрим, например, возможные пути
образования в такой системе триметилолфенола (XVr)
(2.119)
В табл. 2.5 приведены константы скорости различных реакций [55],
из которых видно, что есть существенная разница в реакционной
Таблица 2.5
Поликонденсация фенола
и формальдегида в присутствии
основания [55]
Константа скорости реакции Численное значение константы скорости- 106, л/(моль • С)
fcl 5,3
к 6,2
/с2 8,7
7,3
/с2' .3,8
к3 42
/с3 9,1
способности различных функциональных групп фенола (т. е. раз-
личных положений в ядре). Реакция между фенолом и формаль-
Поликонденсация
113
дегидом включает нуклеофильную атаку формальдегида фенолят-
ным анионом [уравнение (2.120)]. Электронодонорные метилольные
группы, как правило, увеличивают реакционную способность
фенола к дальнейшему замещению. Этот эффект велик для димети-
лолфеполов, особенно для орто-замещенного фенола. Остается не-
объяснимой лишь низкая величина по сравнению с фенолом
Неодинаковой реакционной способностью обладают не только
атомы водорода, находящиеся в различных положениях в феноле,
но и, по-видимому, две функциональные группы в формальдегиде.
Вторая функциональная группа формальдегида является по суще-
ству метилольной группой, так как взаимодействие молекулы
фенола с метилолфенолом или двух молекул метилолфенола, веро-
ятно, идет через следующие соединения:
- он-
(2.121)
К сожалению, сейчас нет прямых кинетических измерений, позво-
ливших бы сопоставить реакционную способность метилольной
группы в такой реакции с реакционной способностью формальде-
гида в исходной реакции. Однако из общих наблюдений, касаю-
щихся того, что количества ди-и полиядерных соединений в резо-
114
Глава 2
лах сильно зависят от условий реакции (температуры, pH и др.),
видно, что две функциональные группы формальдегида неодина-
ковы по своей реакционной способности.
Анализ поликонденсации фенола и формальдегида осложняется
также тем, что реакционная способность функциональной группы
может уменьшаться с увеличением размера моекулы, как это имеет
место в системах, которые подвергаются интенсивной сшивке.
Такие системы перестают быть гомогенными растворами раньше
экспериментально определенной точки гелеобразования. Точке
гелеобразования может предшествовать образование частиц микро-
геля (разд. 2.2г), которые слишком малы, чтобы их можно было
увидеть невооруженным глазом. Функциональные группы в части-
цах микрогеля должны быть сравнительно малореакционноспособ-
ными из-за малой их доступности. Аналогичные выводы справедли-
вы и для реакции после точки гелеобразования.
2.10а.З. МочевиноформаЛьдегидные полимеры
При поликонденсации мочевины и формальдегида в умеренно
основных средах образуются метилольные производные мочевины,
в частности моно- и диметилолмочевина (XIX)
НОСН2 — NH — СО — NH2 НОСН2— NH — СО — NH — СН2ОН,
XIXa XIX6
которые затем в нейтральной или слабо кислой среде превращают-
ся в полимеры сетчатой структуры. Механизм отверждения таких
соединений неясен. Полагают, что реакция идет через образование
линейных (XX) и циклических структур (XXI) 134, 35, 56]
— СН2 —NH —СО —N —СН2 —NH —СО —NH —-
СН2
I
— NH —СО —NH —СН2 —N —СО —NH —СН2 —
XX
сн2
— NH —СО —N N — СО — NH — СН2 —
I I
н2с сн2
I
со
NH
I
сн2
I
— N----
XXI
П оликонденсация
115
Как и для фенолформальдегидных систем, здесь существуют весьма
убедительные данные, не позволяющие считать правомочным пред-
положение об одинаковой и неизменной реакционной способности
всех функциональных групп как мочевины, так и формальдегида
[57]. Аналогичный характер имеют образование и отверждение ста-
тистических преполимеров на основе меламина (XXII)
XXII
и формальдегида.
2.106. Преполимеры известной структуры
Новейшие термореактивные полимеры основаны на использова-
нии специально изготовленных преполимеров более четко установ-
ленной структуры. Такие преполимеры подвергают отверждению
на второй стадии в присутствии, как правило, катализатора или
какого-либо иного реагента. При этом по химизму вторая реакция
обычно отличается от первой. Большим преимуществом подобных
полимеров является то, что процесс их образования на обеих ста-
диях и, что очень важно, структура полимера достаточно просто
регулируются. Области применения и рынок новейших реакто-
пластов увеличиваются более быстрыми темпами, чем расширение
производства традиционных термореактивных полимеров.
Преполимеры известной структуры представляют собой полиме-
ры низкого молекулярного веса с различно расположенными
свободными функциональными группами. Если функциональные
группы находятся на концах цепи преполимера, то такие преполи-
меры называются структурно-концевыми преполимерами (structo-
terminal prepolymer). Преполимер называется структурно-пен-
дантным преполимером (structopendant prepolymer), если функ-
циональные группы расположены вдоль полимерной цепи. Для
обсчета реакций преполимеров указанного строения применим
статистический подход к гелеобразованию, при этом преполимеры
рассматриваются как реагенты с соответствующей функциональ-
ностью [58]. Ниже рассмотрены вкратце некоторые типы таких
преполимеров.
116
Глава 2
2.106il. Диоловые преполимеры
Простые Н(—OR—)ПОН и сложные Н(—OCOR'COOR—)ПОН
полиэфиры широко используются в качестве преполимеров
при получении различных полиуретанов. Простые полиэфиры
^обычно синтезируют, исходя из оксиэтилена и оксипропилена
(см. разд. 7.2); для получения сложных полиэфиров используют
дикарбоновые кислоты и двухатомные спирты, которые берутся
в избытке. При взаимодействии таких преполимеров с концевыми
гидроксильными группами с избытком диизоцианатов получаются
полимеры с концевыми изоцианатными группами, например
пНО — OH + (n + l)OCN — R — NCO —>
—> OCN —( —R —NHCOO — OOCNH — R—)n—NCO, (2.122)
которые можно отверждать различными способами [59]. Так,
в присутствии диамина образуются карбаматные связи, последую-
щая реакция их с концевыми изоцианатными группами при-
водит к получению сшитого полимера, в поперечных мостиках
которого содержатся биуретовые связи
20CN — NCO + H2N — R — NH2 —»
OCN--NCO
-» OCN — NHCONHRNHCONH — NCO -------->
—> OCN — NHCONHRNCONH — NCO
I
CO
I -
NH
) (2.123)
NH
I
CO
I
OCN —NHCONHRNCONH----NCO
2.106.2. Эпоксидные преполимеры
Для получения эпоксидных преполимеров обычно применяют
2,2'-бпс-(4-оксифенил)пропан (бисфенол А) и эпихлоргидрин [601
+ <п + 2> НС1.
(2.124)
В зависимости от того, идет сшивание по концевым эпоксидным
группам или по гидроксильным группам, такие преполимеры
П оликонденсация
117
можно рассматривать как структурно-концевые или структурно-
пендантные преполимеры [61]. Так, отверждение идет преимуще-
ственно по гидроксильным группам при использовании в качестве
отвердителя полиангидрида
I
со
I
/—\ /—\ 0
^oV(^Vc(CH3)2-/(^)\- осн2—сн—сн2-—
Такие преполимеры относятся к структурно-пендантным. При от-
верждении же преполимеров аминами их следует рассматривать
как структурно-концевые преполимеры. Отверждение в последнем
случае идет через катализируемое основаниями раскрытие эпок-
сидного цикла
О
СН2 - СН -СН2 — + H2N - R- NH2 —»
он
он сн2-Ан-сн2—
-> — СН2-СН-СН2-г!г
_ • R ОН (2.126)
I I
N-CH2-CH-CH2 —
I
— СН2-СН-СН2
Ан
Отверждение по эпоксидному циклу можно проводить также путем
полимеризации с раскрытием цикла (см. разд. 7.2).
118
Глава 2
2.106.3. Ненасыщенные сложные полиэфиры
Ненасыщенные сложные полиэфиры также могут быть препо-
лимерами двух типов в зависимости от расположения ненасыщен-
ных двойных связей. Так, типичный структурно-концевой поли-
мер образуется при реакции фталевого ангидрида и глицерина
в присутствии ненасыщенной монокарбоновой кислоты, в качестве
которой часто применяется жирная кислота. В результате полу-
чается преполимер, имеющий на конце молекулы двойную углерод-
углеродную связь,
СН2ОН
+ СНОН + RCH=CH—R — СО2Н —-
СН2ОН
(2.127а)
который отверждается под действием кислорода (разд. 9.2). Слож-
ные полиэфиры такого типа называются алкидными смолами, если
в качестве монокарбоновой кислоты применяется ненасыщенная
кислота жирного ряда [62]; при использовании других ненасыщен-
ных карбоновых кислот полиэфир называется ненасыщенным слож-
ным полиэфиром [63].
Ненасыщенные сложные полиэфиры, содержащие двойные связи
внутри полимерной цепи, можно получить поликонденсацией диола
и фумаровой или малеиновой кислоты (или малеинового ангид-
рида)
НОСН2СН2ОН + НООС-СН = СН-СООН—»
—>—( — ОСН2СН2ООС — СН = СН — СО — )п — (2.1276)
Такие преполимеры отверждают сополимеризацией с ненасыщен-
ными мономерами (см. разд. 9.2).
Поликонденсация
119'
2.106.4. Фенолформальдегидные полимеры
Фенолформальдегидные полимеры, называемые новолака-
ми (XXIII),
получают поликонденсацией фенола и формальдегида в кислых
средах [33, 34]. Во избежание преждевременного отверждения
реакцию ведут с небольшим избытком фенола по отношению
к формальдегиду. В качестве отвердителя на второй стадии реак-
ции применяется гексаметилентетрамин (CH2)0N4; при разложе-
нии последнего образуется формальдегид, который собственно
и образует поперечные метиленовые связи. В качестве низкомоле-
кулярного побочного продукта в этом процессе выделяется аммиак.
Наряду с метиленовыми мостиками в отвержденном полимере
обнаружено некоторое количество поперечных бензиламиновых
поперечных связей. Ниже приведена структура такого сетчатого
полимера (XXIV)
NH
ОН
XXIV
120
Глава 2
2.11. Со-поликон денсация
Поликонденсация обеспечивает богатые возможности для син-
теза самых разнообразных полимеров, свойства которых можно
регулировать в желаемом направлении путем изменения химиче-
ского строения исходных веществ. Применяя вещества с различ-
ными функциональными группами, получают сложные полиэфиры,
полиамиды или полимеры других классов. Кроме того, внутри
полимеров одного класса имеется богатый выбор химических
структур, которые можно использовать в синтезе полимеров.
Рассмотрим, например, полиамиды. Полиамиды с той или иной
химической структурой
— СО —R —СО —NH —R' —NH— —NH —R —СО —
XXV XXVI
можно получить, используя для поликонденсации диамины и ди-
карбоновые кислоты или аминокислоты. Ряд разных полиамидов
дает варьирование природы R и R' в структуре XXV или
R в структуре XXVI. Так, при поликонденсации гексаметилен-
диамина и адипиновой или себациновой кислот получают найлон-
6,6 или найлон-6,10 соответственно. Волокно из найлона-6,10
обладает несколько большей гибкостью, чем волокна из найло-
на-6,6, что обусловлено наличием в первом полиамиде более длин-
ной углеводородной цепи остатка себациновой кислоты.
2.11а. Типы сополимеров
Дальнейшая модификация свойств полимеров возможна при
использовании в их синтезе смесей различных исходных веществ,
обеспечивающих наличие в макромолекулах различных групп
R и R'. Так, возможными модификациями полиамидов XXV
и XXVI являются соответственно полимеры
— СО —R —CONH —R' — NHCO — R" —CONH —R"' — NH — ,
XXVII
— NH —R —СО —NH —R'' —СО—.
XXVIII
Последние полимеры называются сополимерами, а процесс их
образования — сополиконденсацией, или совместной поликонден-
сацией. В отличие от сополимеров полимеры XXV и XXVI назы-
ваются гомополимерами.
В зависимости от способа синтеза возможно образование сопо-
лимеров различных типов. Так, сополиамид, имеющий состав,
изображенный формулой XXVII, может быть регулярно чередую-
щимся сополимером, если группы R, R', R" и R"' регулярно еле-
Поликонденсация
121
дуют друг за другом по всей полимерной цепи, или блок-сополиме-
ром (XXIX), если блоки гомополимера одного состава присоеди-
няются к блоку другого состава, или статистическим сополимером,
если группы R и R" распределены хаотично по полимерной цепи.
Аналогичное строение могут иметь сополиамиды состава XXVIII
— (—СО — R — CONH — R' — NH — )т— (—СО — R" — CONH — Rw—NH—)р—.
XXIX
Регулярно чередующиеся, статистические и блок-сополимеры
схематически можно представить следующим образом:
— АВАВАВАВАВАВАВАВАВАВАВАВ----,
регулярно чередующийся сополимер
— ААВАВВВАВААВАААВВАВВВААВ —,
статистический сополимер
— ААААААВВВВВВААААААВВВВВВ —,
блок-сополимер
где А и В — различные повторяющиеся звенья. Кроме указанных
типов сополимеров, существуют неограниченные возможности
синтеза производных полимеров каждого типа. Широко можно,
варьировать относительную длину и количество повторяющихся
звеньев А и В в сополимере.
2.116. Методы синтеза сополимеров
Существуют различные пути синтеза сополимеров. Многие из
них нашли применение в синтезе полиамидов [64, 65], сложных
полиэфиров [66], поликарбонатов [67, 68], полиуретанов [37, 69]
и других полимеров. Рассмотрим, например, образование сополп-
амидов XXVII. Непосредственно синтезировать регулярно чере-
дующийся сополимер такого состава не представляется возмож-
ным, но возможен синтез сополиамида регулярно чередующейся
структуры путем двухстадийной поликонденсации, если R" = R.
На первой стадии при взаимодействии дикарбоновой кислоты
с диамином в соотношении 2 : 1 получают тример
2п НООС — R — СООН + п H2N — R' — NH2—>
—* мНООС — R —CONH —R' —NHCO —R—СООН, (2.128)
который на второй стадии подвергают взаимодействию с эквимо-
лярным количеством второго диамина
пНООС —R —CONH—R' —NHCO—R—СООН-LnH2N — R"' — NH2—>
—>НО — ( —CO-R — CONH — R' — NHCO — R — CONH — R"' — NH-)n — H+
+ (2n —1)H2O. (2.129)
Блок-сополиамиды легко можно получить путем поликонден-
сации предварительно синтезированных низкомолекулярных гомо-
122
Глава 2
полимеров друг с другом или с другим реагентом. Так, можно
подвергнуть взаимодействию два преполимера, из которых один
имеет на концах макромолекулы аминогруппы, а другой — карбо-
ксильные группы
пНООС —R-CO —(-NH-R'-NHCO —R—СО —)m—ОН +
-|-nH2N—R"' — NH — ( — CO — R"CONH — R'" — NH — )p — H—>
—>HO — [ — CO — R — CO — ( — NH — R' — NHCO — R—CO — )m — NH — R"' —
— NH —( —CO —R" —CONH —R'" —NH —)p —]n —H + (2n —1)H2O (2.130)
или
nHOOC — Am — COOH + nH2N — By NH2 —»
-( — CO — Am — CONH — Bp— NH —)n— .
(2.131)
Обычно молекулярный вес преполимеров не превышает 500—
2000, что соответствует величинам т и р от 1 до 10.
Блок-сополиамиды можно синтезировать также с использова-
нием двух преполимеров с концевыми аминогруппами, подвергая
их взаимодействию с дихлорангидридами дикарбоновых кислот
ziH2N — Am — NH2 + 2mC1CO —R —COC1 + mH2N — B;, — NH2 —>
—> —(—Am— NHCO-R—CONH — Bp — NHCO — R—CONH — )n —.
(2.132)
В реакции с диамином можно также использовать два преполиме-
ра с концевыми карбоксильными группами. Реакции такого типа
уже рассматривались выше при обсуждении реакций образования
преполимеров известной структуры [уравнения (2.122), (2,125),
(2.126)].
Другим путем образования блок-сополиамидов является обмен-
ная реакция между двумя гомополимерами (разд. 2.5в). Так, блок-
сополиамид можно получить нагреванием смеси двух гомополи-
амидов
Am Т Вр А1£>Вж AyBz
(2.133)
где т = w + у, а р = х Д- z.
Статистические сополимеры можно получить поликонденсаци-
ей эквимолярной смеси четырех сомономеров
HOOC-R —СООН
HOOC-R" — СООН
H2N — R' — NH2
H2N- R"' — nh2
> —* Статистический сополимер
XXVII
(2.134)
При использовании в реакции только одной кислоты или одного
только диамина получается статистический сополимер XXVII,
в котором R = R ", или R' = R"'.
Выше речь шла о сополимерах, содержащих одинаковые гете-
росвязи в различных повторяющихся звеньях. Хотя в большинстве
П оликонденсация
123
работ по сополиконденсации рассматриваются сополимеры именно
такого типа, возможны сополимеры с различными связями внутри
повторяющихся звеньев и между ними. Так, при взаимодействии
преполимеров с концевыми аминогруппами и диизоцианатов обра-
зуется блок-сополимер
( —Am —NHCONH —R —NHCONH —Вр —NHCONH —R —NHCONH—)п,
XXX
в котором полиамидные блоки соединены карбаматными связями.
Реакцией эфирного обмена предварительно полученных бис-
эфироамидов с гликолями получают полиэфироамиды
C2H5OOC-R —CONH —R' — NHCO — R —СООС2Н3 ;-НО —R" —ОН
—( — OOC-R-CONH — R' — NHCO — R—COO — R" — )n — . (2.135)
2. Ив. Применение сополиконденсации
Практический интерес представляют статистические и блок-
сополимеры, так как регулярно чередующиеся сополимеры мало-
доступны вследствие трудности их получения. Кроме того, регу-
лярно чередующиеся сополимеры, по-видимому, не обладают каки-
ми-либо новыми свойствами по сравнению со статистическими
и блок-сополимерами. В ряде работ [65, 70] показано, что стати-
стические и блок-сополимеры отличаются по свойствам как друг
от друга, так и от соответствующих гомополимеров. По свойствам
блок-сополимер занимает промежуточное положение между обо-
ими гомополимерами, или, точнее, свойства его находятся в пря-
мой зависимости от весовой доли различных повторяющихся
звеньев в сополимере. Однако свойства статистического сополимера
не находятся в прямой зависимости от весовой доли каждого
сополимера.
Процесс сополиконденсации часто используют для регули-
рования свойств полимеров, при этом в первую очередь затраги-
ваются такие основные характеристики полимеров, как кристаллич-
ность, гибкость, Т’ст и Тил. Как правило, сополимеры менее крис-
талличны, более гибки, чем соответствующие гомополимеры, и име-
ют более низкие температуры плавления и стеклования. Однако
у статистических и блок-сополимеров эти отклонения различны.
Так, температура плавления блок-сополимера лишь ненамного ни-
же, чем у средневесовой смеси двух гомополимеров. В то же время
статистический сополимер плавится намного ниже, чем блок-сопо-
лимер, что обусловлено его большей неупорядоченностью.
На практике сополиконденсация используется значительно
менее широко по сравнению с сополимеризацией (см. гл. 6). Вместе
с тем при сополиконденсации предпочтительным, как правило,
124
Глава 2
является образование блок-сополимеров, а не статистических
сополимеров. Это связано и с тем, что свойства блок-сополимеров
легче прогнозировать. Прекрасным примером практического при-
менения сополиконденсации является разработка эластомерных
волокон спандекс [70]. Частично синтез этих материалов был рас-
смотрен выше (разд. 2.106.1). Преполимеры с концевыми гидро-
ксильными группами
НО —( —RCH2 —О —)n —RCH2OH
XXXI
или НО —( —R' —ООС —R—СО2—)n —R' —ОН
XXXII
получают полимеризацией с раскрытием цикла циклического эфира
(разд. 7.2) или поликонденсацией дикарбоновой кислоты и избыт-
ка диола соответственно.
При взаимодействии такого преполимера с избытком диизоциа-
ната образуется преполимер XXXIII с концевыми изоцианатными
группами
НО —Ат — 0H-J-20CN — R" — NCO—»
OCN -R'"-NHCO2 Ат O2CNH -R'"-NCO, (2.136)
XXXIII
который затем реагирует с низкомолекулярным диолом
OCN — R"'—NHCO2 — Am — O2CNH — Rm— NCO-|-HO-R""-OH —>
XXXIII
—» — ( —O2CNH —R'" —NHCO2 — Am— O2CNH —R'"—NHCO2 —R'"' —)n —
XXXIV
или диамином
(2.137)
OCN — R"'—NHCO2 — Am — O2CNH — R'" — NCO + H2N -R"" - NH2
XXXIII
(— NHCONH - R"' — NHCO2 — Am — O2CNH — R'" — NHCONH - R" " - )n.
XXXV
(2.138)
Конечный полимер содержит полимерные простые эфирные или
сложноэфирные сегменты (сегменты Ат), разделенные уретановыми
(XXXIV) или мочевиноуретановыми (XXXV) сегментами в зави-
симости от природы реагентов, применяющихся на , отдельных
этапах процесса. Полимер сочетает в себе свойства эластомера
и волокна. Волокнообразующим свойствам он обязан уретановым
или мочевиноуретановым сегментам, тогда как простые эфирные
или сложноэфирные сегменты придают ему эластомерные свойства.
П оликонденсация
125
2.12. Новые полипоид енса/ционные реакции
Последние десять-пятнадцать лет ознаменованы стремитель-
ным ростом работ в области синтеза новых, высокотеплостойких
полимеров. В ряде областей могут найти применение высокопроч-
ные, жесткие полимеры, устойчивые к действию растворителей
и агрессивных сред, и полимеры, которые могли бы работать при
высоких температурах (вплоть до 300—500 °C). В подобных поли-
мерах нуждается космическая техника, требуются они и для
создания конструкционных изделий различного назначения. Для
решения поставленных задач проведены большие работы по синте-
зу неорганических, частично неорганических и чисто органических
полимеров с применением различных реакций неорганической
и органической химии. Однако достигнутые успехи сязаны почти
исключительно с получением органических полимеров.
Основная проблема, с которой сталкиваются при синтезе таких
полимеров, заключается в трудности получения высокомолеку-
лярных полимеров (7И не менее 10 000), что совершенно необходи-
мо для создания на их основе высокопрочных изделий. В боль-
шинстве случаев эта трудность обусловлена нерастворимостью по-
лимеров такого типа. Полимеры преждевременно выпадают из
реакционной среды, не успев достигнуть высокого молекулярного
веса, при этом реакция обрывается. Кроме этого, механизм реак-
ций, лежащих в основе образования таких полимеров, известен
далеко не всегда, что значительно затрудняет регулирование про-
цесса с целью повышения выхода и молекулярного веса полимера.
Следует выделить три основных подхода, использующихся при
синтезе теплостойких полимеров. К ним относится получение поли-
меров с высокой кристалличностью, большим числом поперечных
связей и низкой гибкостью цепей [71]. В зависимости от особен-
ностей данного полимера его можно использовать как в отдельно-
сти, так и в сочетании с другими полимерами. Ранее для повышения
верхнего температурного предела работоспособности полимеров
в основном обращали внимание на его кристалличность и трех-
мерное строение. В последние годы основное внимание было сос-
редоточено на синтезе жесткоцепных полимеров, устойчивых
к деформированию и не размягчающихся при повышенных тем-
пературах.
2.12а'. Новые промышленные полимеры
Для повышения жесткости полимерных цепей широко исполь-
зуется введение в них циклических групп, особенно ароматиче-
ских колец, высокая термическая устойчивость которых хорошо
известна. Этот общий принцип положен в основу недавно разрабо-
танных, новых промышленных полимеров с повышенной тепло-
и термостойкостью [72].
126
Глава 2
2.12а.1. П оликарбонаты
Поликарбонатами называются сложные полиэфиры угольной
кислоты. Наиболее широко известный поликарбонат бисфенола А
можно синтезировать [73] поликонденсацией бисфенола и^фосгена
или дифенилкарбоната
В этом случае повышенная жесткость полимера достигается соче-
танием в нем бензольных ядер и объемистых тетразамещенных
атомов углерода.
По своим закономерностям получение поликарбонатов реакци-
ей эфирного обмена в две стадии в расплаве аналогично синтезу
полиэтилентерефталата, рассмотренному в разд. 2.66.1. Первую
стадию проводят при температуре 180—200 °C и остаточном дав-
лении 20—30 мм рт. ст. до степени завершенности 80—90%. Затем
температуру постепенно поднимают до 290—300 °C и заканчивают
реакцию в вакууме (1 мм рт. ст.). Поликонденсацию с участием
фосгена проводят в присутствии оснований. В качестве раствори-
телей наиболее часто используют пиридин или триэтиламин или
смеси их с бензолом, хлорбензолом или хлороформом. Бисфенол А
растворяют в водной щелочи, затем после добавления растворите-
ля в систему вводят фосген. Благодаря органическому раствори-
телю удается предотвратить потери фосгена вследствие гидролиза,
а также преждевременное выпадение низкомолекулярного поли-
мера из раствора.
2.12а.2. Полиамиды
Полиамиды с алициклическими или ароматическими группами
в основной цепи превосходят по теплостойкости алифатические
полиамиды. Ациклические полиамиды (которые часто называют
полициклоамидами), например поли-1,4-циклогексилендиметилен-
П оликоиденсация
127
суберамид
— NHCH2 — / CH2NHCO(CH2)6CO —
XXXVI
получают поликонденсацией соответствующего диамина и дикарбо-
новой кислоты в расплаве, как и полигексаметиленадипамид
(разд. 2.66.2). Вследствие низкой реакционной способности аро-
матических диаминов этот метод неприменим для получения аро-
матических полиамидов [74]. Электронная плотность азота ами-
ногруппы понижена из-за соседства ароматического ядра, что
уменьшает его нуклеофильность по отношению к карбоксильным
группам.
Ароматические полиамиды синтезируют поликонденсацией диа-
мина с такими реакционноспособными производными карбоновых
кислот, как их хлорангидриды; так, например, промышленный
полимер поли-.и-фениленизофталамид
получают гзаимодействием дихлорангидрида изофталевой кисло-
ты с л-фенилендиамином. Исследованы синтез и свойства ряда
других ароматических полиамидов. В их числе — полиамиды,
содержащие нафталиновые ядра и различные гетероциклы, напри-
мер оксадиазольные и тиазольные [74, 75].
2.12а.З. Простые ароматические полиэфиры
Среди большого числа синтетических реакций, применявшихся
для получения простых ароматических полиэфиров, наилучшие
результаты дает использование реакции ароматического нуклео-
фильного замещения и окислительной дегидрополиконденсации
[76]. Реакцией нуклеофильного ароматического замещения в про-
мышленности получают полимер на основе бисфенола А и 4,4'-
128
Глава 2
дихлордифепилсульфона [77]. Реакция проводится с использова-
нием динатриевой или дикалиевой соли бисфенола А
(2.141)
Торговое название полимера — полисулъфон. В соответствии с хи-
мическим строением более логично его называть полиокси-1,4-фе-
ниленсульфонил-1,4 -фениленокси -1,4-фениленизопропилиден-1,4-
фенилен [78].
Фенолят получают in situ при одновременном введении бис-
фенола А и гидроокиси щелочного металла. Реакцию проводят
в апротонных полярных растворителях, например 1М,1\-диметил-
формамиде, диметилсульфоксиде, диметилсульфоне, при повышен-
ных температурах (примерно 150 °C). Обычно ароматические гало-
гены не вступают в реакции нуклеофильного замещения такого
типа. Однако в данном случае атомы хлора в ароматическом
ядре активированы электроноакцепторными сульфоновыми груп-
пами.
Ароматические простые полиэфиры получают также окисли-
тельной дегидрополиконденсацией 2,6-дизамещенных фенолов
Реакцию проводят путем пропускания кислорода через раствор
фенола в органическом растворителе, содержащем также каталити-
ческий комплекс соли одновалентной меди и третичного амина [78].
Производящийся в промышленности полимер на основе 2,6-диме-
Поликонденс ация
129
тилфенола неправильно называется полифениленоксидом. Пра-
вильное его название — поли-2,6-диметилфениленоксид или поли-
окси-2,6-диметил-1,4-фенилен.
В мягких условиях реакция протекает при использовании фе-
нолов с небольшими заместителями. Так, полимер на основе 2,6-
диметилфенола образуется уже при комнатной температуре, тогда
как 2-хлор-6-метилфенол реагирует только при 60 °C. Фенолы
с объемистыми заместителями, например изопропильной или третп-
бутильной группой, вместо линейных полимеров образуют дифено-
хиноны
Данные о механизме окислительной дегидрополиконденсации
весьма немногочисленны [76, 79]. Реакцию можно описать так, как
это сделано в уравнениях (2.144)—(2.148), но экспериментальных
доказательств для такой схемы явно недостаточно. Медно-амин-
ный комплекс действует как катализатор окисления фенола кисло-
родом, в результате чего образуется фенокси-радикал XXXVII.
При конденсации феноксильных радикалов образуется димер
[уравнение (2.144)]. Затем димер окисляется до соответствующего
фенокси-радикала [уравнение (2.145)], который реагирует с дру-
гим таким же радикалом или мономерным фенокси-радикалом
[уравнения (2.146) и (2.147)]. Рост цепи через тример идет таким
же образом через стадии окисления и конденсации, тетрамер же
разлагается [уравнение (2.148)] на два фенокси-радикала (мономер
и тример), которые реагируют по приведенной схеме
130
Глава 2
(2.148)
2.12а.4. Поли-п-ксилилен
Поли-тг-ксилилен получают окислительным пиролизом тг-ксило-
ла [72, 80, 81]
(2.149)
Аналогично получают соответствующие полимеры на основе моно-
и дихлорксилолов. При пиролизе тг-ксилола при 950 °C образуется
ди-тг-ксилилен (XXXVIII). После отделения и очистки ди-тг-кси-
лилен подвергают вакуум-пиролизу при 550—600 °C, в результате
чего образуется тг-ксилилен (XXXIX), который самопроизвольно
полимеризуется в вакууме (менее 1 мм рт. ст.) на поверхности,
температуру которой поддерживают не выше 30 °C. При темпера-
туре выше 30 °C образование полимера не идет. В промышленности
Поликонденсация
13f
XXXVIII
XXXIX
(2.150)
пленки и покрытия на основе поли-тг-ксилилена получают, непо-
средственно осаждая его в вакууме на изделии. Реакция образо-
вания поли-тг-ксилилена протекает с большой скоростью, причем
количественное превращение ди-тг-ксилилена в поли-тг-ксилилен
достигается за доли секунды.
Инициатором реакции является бирадикал (XL), образующий-
ся путем конденсации двух молекул тг-ксилилена, рост цепи идет
через присоединение к нему по обоим концам новых молекул п-
ксилилена
(2.151)
Обрыв растущей цепи полимера происходит в результате рекомби-
нации макрорадикалов или вследствие того, что они настолько
«теряются» в полимерной массе, что становятся недоступными
дальнейшему присоединению молекул тг-ксилилена. На наличие
таких «захороненных» радикалов указывает высокая концентра-
ция их в конечном полимере (приблизительно 5-10~4—10 -10'4 мо-
ля на 1 моль тг-ксилилена). Для отнесения процесса образования
поли-тг-ксилилена из тг-ксилилена к поликонденсационным реак-
циям в настоящее время нет достаточных оснований, так как отсут-
ствуют подробные исследования по механизму и закономерностям
132
Глава 2
этого процесса. Вполне вероятно, что присоединение мономера
к бирадикалу может идти как цепной процесс, причем стадия роста
цепи представляет 1,6-присоединение радикала к тг-ксил илену
R-
Кроме того, процесс инициирования может заключаться в об-
разовании бирадикала XL непосредственно из ди-га-ксилилена
путем разрыва одинарной связи в последнем
(2.153
В результате экспериментальных исследований, выполненных на
монозамещенных ди-тг-ксилиленах, было показано, что процесс
инициирования надо описывать так, как это сделано в уравнениях
(2.150) и (2.151). При пиролизе ацетилди-тг-ксилилена с после-
дующей конденсацией пиролитических паров на охлаждаемой по-
верхности при 90 и 25 °C образуются два различных полимера, ни
один из которых не является полиацетилди-п-ксилиленом. В ре-
зультате пиролиза образуются ацетил-тг-ксилилен и тг-ксилилен
(2.154)
последующая полимеризация которых идет при различных темпе-
ратурах. Полиацетил-и-ксилилен получается при температуре
охлаждаемой поверхности 90 °C, а поли-п-ксилилен — при 25 °C-
Поликонденсация
133
2.12а.5. Ароматические полиимиды
Ароматические полиимиды получают взаимодействием дианги-
дридов тетракарбоновых кислот с диаминами [72, 82, 83], напри-
мер, при поликонденсации пиромеллитового диангидрида и тг-фе-
нилендиамина образуется поли-1,4-фениленпиромеллитимид
Из-за нерастворимости и неплавкости ароматических полиими-
дов высокого молекулярного веса их нельзя получить одностадий-
ным методом. Полимер выпадает в осадок из реакционной среды
(раствора или расплава), не успев достигнуть высокого молекуляр-
ного веса. Поэтому полиимиды получают в две стадии с образовани-
ем на первой в растворе полярных растворителей (N, N-диметил-
ацетамида, NjN-диметилформамида, диметилсульфоксида или N-
метилпирролидона) высокомолекулярных полиамидокислот XLI
(2.156)
Затем из плавкой и растворимой полиамидокислоты формуют из-
делия (пленки, волокна, покрытия), а затем уже проводят вторую
134
Глава 2
стадию — имидизацию в твердой фазе при температуре выше 150 °C.
Первую стадию реакции проводят при температуре не выше 70 °C,
чтобы до минимума свести возможный процесс циклизации. Обра-
зующаяся на первой стадии полиамидокислота обладает низкой
гидролитической устойчивостью, особенно если не удалять выделя-
ющуюся при циклизации воду. Заключительную фазу циклизации
проводят при 300 °C, снизить температуру циклизации удается
применением вакуума или дегидратирующих агентов, например
смеси уксусный ангидрид — пиридин.
2.12а.6. Полибензимидазолы
Полиимиды получают с использованием аминов и карбоновых
кислот при соотношении аминогрупп к карбоксильным 1 : 2.
Если реагенты берутся с обратным соотношением указанных
групп, то образуются полибензимидазолы [72, 84, 85]
(2.157)
Полибензимидазолы получают в две стадии нагреванием смеси
исходных веществ сначала при 250 °C, а в конце при 350—400 °C.
Высокомолекулярный полимер с высоким выходом образуется
только при использовании в качестве исходного кислотного ком-
понента дифенилового эфира дикарбоновой кислоты. При приме-
нении самой дикарбоновой кислоты или ее диметилового эфира не
удается получить высокомолекулярного полимера из-за побочных
реакций (декарбоксилирование и метилирование амина соответ-
ственно), нарушающих эквивалентное соотношение реагирующих
функциональных групп. Хлорангидрид дикарбоновой кислоты не
может быть использован из-за его очень высокой реакционной
способности, приводящей к реакции соседних аминогрупп с обра-
зованием амидов, циклизация которых сильно затруднена.
Первая стадия образования полибензимидазолов представляет
собой конденсацию альдольного типа, в результате которой полу-
Поликонденсация
135
чается полимер, имеющий структуру шиффова основания. Послед-
ний на второй стадии циклизуется с выделением фенола
-CSH«OH
о □
(2.158)
Переработку таких полимеров осуществляют на стадии нецик-
лизованного полимера аналогично тому, как было выше описано
для полиимидов. Вместе с тем переработка полибензимидазолов не-
136
Глава 2
сколько отличается от переработки полиимидов, что обусловлено
растворимостью первых полимеров в некоторых растворителях, на-
пример диметилацетамиде или диметилсульфоксиде, что открывает
возможность формования пленок и волокон из растворов уже
циклизованных полимеров.
2.126. Полимеры в стадии разработки
Из ряда полимеров, рассмотренных в предыдущем разделе,
только полиимиды и полибензимидазолы можно отнести к разряду
высокотеплостойких полимеров, удовлетворяющих современным
требованиям (>300—500 °C). Многие полимеры нельзя считать
полностью жесткоцепными из-за наличия в их основной цепи
одинарных связей. Кроме того, некоторые из таких полимеров име-
ют в своем составе алифатические группы, являющиеся «слабыми
звеньями в цепи». Помимо полимеров, уже производящихся в про-
мышленном масштабе, проведены широкие исследования в области
синтеза ряда других полимеров, которые были бы работоспособны
в интервале температур 300—500 °C и выше [34, 71, 86, 87]. Общий
принцип, которым руководствовались при создании таких поли-
меров, состоит в следующем: максимально возможное насыщение
их структуры циклическими группами, обычно ароматическими
кольцами, т. е. создание так называемых лестничных или блок-
лестничных полимеров. Лестничным (или двоесвязанным) назы-
вается полимер, который имеет непрерывающуюся последователь-
ность колец, соединенных друг с другом двумя связывающими их
атомами (т. е. конденсированную систему циклов). Блок-лестнич-
ный полимер имеет в своем составе одинарные связи между неко-
торыми циклами.
С точки зрения получения жесткоцепного полимера с наиболь-
шей тепло- и термостойкостью блок-лестничная структура менее
предпочтительна по сравнению с чисто лестничным полимером.
Поэтому большие усилия были сосредоточены на получении и ис-
пользовании мономеров, необходимых для создания лестничных
полимеров. При выборе мономеров необходимо учитывать и неко-
торые другие факторы, в частности различия в стабильности разных
циклических систем, от которых сильно зависит термостойкость
полимера. Кроме того, при создании высокотеплостойких полиме-
ров, способных к переработке, иногда необходимо идти на некото-
рое снижение жесткости полимерной цепи. Таким образом, имею-
щиеся в настоящее время высокотермостойкие полимеры синтези-
руют с учетом следующих обстоятельств: доступности исходных
мономеров, термостойкости, физических свойств и перерабатывае-
мости полимера. Для переработки некоторых из таких полимеров
могут быть использованы те же принципы, что и в технологии
полиимидов и полибензимидазолов, так как их синтезируют также
П оликонденсация
137
двухстадийным методом. Поскольку полимеры, образующиеся на
первой стадии синтеза, растворимы и плавки, их можно перера-
батывать в изделия до стадии циклизации. Ниже будут рассмотре-
ны некоторые из известных сейчас полимеров лестничной или
блок-лестничной структуры. Не слишком большим преувеличени-
ем будет сказать, что почти все известные типы карбо- и гетероцик-
лов были апробированы в синтезе таких полимеров.
2. 126.1. Полиимидазопирролоны
Полиимидазопирролоны, или пирроны, получают поликонден-
сацией тетраминов и производных тетракарбоновых кислот [88]
138
Глава 2
Первую стадию — образование амида XLII — проводят в поляр-
ных растворителях при температуре несколько выше комнатной
(35—50 °C). Последующая твердофазная полициклизация XLII при
300 °C приводит к образованию сначала имида XLIII, а затем
имидазопирролона XLIV. В отличие от ранее рассмотренных блок-
лестничных полимеров полиимидазопирролон имеет лестничную
структуру.
2.126.2. Поливен зоксазолы и полибензотиазолы
Полибензоксазолы XLV и полибензотиазолы XLVI синтезиру-
ют [89] полициклизацией дикарбоновых кислот и их производных
с бпс-о-аминофенолами и бпс-о-аминотиофенолами [уравнения
(2.160) и (2.161)]
XLV
(2.160)
2.126.3. Полихиноксалины
Взаимодействием соответствующих бпс-о-диаминов и тетракар-
бонильных соединений синтезированы блок-лестничные [уравне-
ние (2.162)] и лестничные [уравнение (2.163)] полихиноксали-
ны [90]
Поликонденсация
139
(2.163)
2.126.4. Полиоксадиазолы и политриазолы
На основе полигидразидов получены полиоксадиазолы XLVI1
и политриазолы XLVIII [91, 921
(2.164)
2.126.5. Присоединение по реакции Дилъса — Алъдера
Лестничные и блок-лестничные полимеры получают реакцией
Дильса — Альдера [93, 94] в результате 1,4-присоединения нена-
сыщенной двойной связи (диенофила) к диену-1,3
(2.167)
П оликонденсация
141
2.126.6. 1,3-Диполяр ное присоединение
Реакцией 1,3-диполярного присоединения синтезированы раз-
личные блок-лестничные полимеры [95—98]. Примером таких реак-
ций является 1,3-присоединение нитрилоксидной группы: а) к ни-
трильной группе в реакции двух форм такого бпс-нитрилоксида
[уравнение (2.168)]; б) к нитрилу [уравнение (2.169)] или в) к ди-
ацетиленовым соединениям [уравнение (2.170)]. Кроме того, сюда
относится присоединение диполя сиднона к алкенам [уравне-
ние (2.171)] и ацетиленовым соединениям [уравнение (2.172)],
протекающее с одновременным декарбоксилированием, и соответ-
ствующие 1,3-присоединения азидов, нитрилиминов и тетразолов
(2.170;
142
Глава 2
2.126.7. Спирополимеры
Другой путь к получению жесткоцепных полимеров заключает-
ся в синтезе спирополимеров, т. е. полимеров, содержащих цик-
лические группы, соединенные только одним атомом, который,
следовательно, принадлежит обоим циклам. Примерами таких
полимеров являются полиспирокеталь [99], получающийся из
1,4-циклогександиона [уравнение (2.173)], и спирополимер на осно-
ве тетрамина и диангидрида тетракарбоновой кислоты, приведен-
ный в уравнении (2.174) [100]
П оликонденсация
143
2.126.8. Сополимеры
В последнее время синтезированы жесткоцепные сополимеры,
содержащие различные типы гетероциклов, например имид-имида-
зопирролон [уравнение (2.175)] [1011 и оксадиазол-бензиамидазол
[уравнение (2.176)] [102]
(2.175)
144
Глава 2
C6H5NH
2.126.9. Неорганические и элементоорганические
полимеры
Анализ данных по энергиям различных связей приводит к вы-
воду, что по термостойкости полимеры с неорганическими цепями
должны превосходить чисто органические полимеры. Это послужи-
ло предпосылкой проведения многочисленных исследований в об-
ласти синтеза элементоорганических полимеров с неорганическими
основными цепями и органическими цепями в боковом обрамлении,
а также чисто неорганических полимеров [71, 72, 103, 104]. За
незначительными исключениями результаты этих работ не оправ-
дали надежд исследователей. Хотя термостойкость таких полиме-
ров сама по себе и является довольно высокой, необходимо прео-
долеть много барьеров на пути создания полимеров, представляю-
щих практический интерес. Ряд таких полимеров получается
только низкого молекулярного веса. При нагревании многие
линейные системы перегруппировываются в циклы, состоящие из
8, 10, 12 и 14 членов, и утрачивают поэтому механическую проч-
ность. Большинство таких полимеров имеют низкую гидролитиче-
скую стабильность, что приводит к еще большему снижению
молекулярного веса или структурированию полимера (образова-
нию густой сетки). Многие полимеры нельзя переработать с помо-
щью существующих сейчас методов. Все это приводит к тому, что
в настоящее время неорганические и элементоорганические поли-
меры, как правило, существенно уступают органическим полиме-
рам. Однако в будущем с развитием представлений о механизме
образования таких полимеров станет возможным получение этих
полимеров с ценным комплексом свойств.
Наиболее важным положительным исключением из вышеска-
занного являются полисилоксаны. Синтез их был рассмотрен
Поликонденсация
145
в разд. 2.66.4 и будет обсуждаться в разд. 7.4. С целью повышения
термостойкости полисилоксановых цепей разработан лестничный
фенилированный полифенилсилсесквиоксан IL, который получают
гидролизом фенилтрихлорсилана в присутствии оснований [105]
IL
Из других неорганических и элементоорганических полимеров
следует выделить различные полиорганометаллосилоксаны [103,
104], например
R R
— О — Si — О — М — О-Si-О — М —
А А А А
'l 1
R R' R R'
1111
— О —Si —О —М' —О —Si —О —М' —
А А- А А'
LI
I I
R О R О
1111
— О —Si —О —М" —О —Si —О —М" —
I I I I
R О R О
I I
LII
R О R О
— О —Si—O—IJ1"'—О—Si—О—М'"—
А А А А
। ।
LIII
где М = А1, В; М' = Sn или Ge; М" = Ti и М"' = Р или As.
Синтезирован ряд полимеров, содержащих связи В — N, В — О,
В - В, В - Р, В - S, В - С, N - Р, N - S, Р - О, а также их
различные комбинации друг с другом [103, 104, 106]. Другие не-
органические полимеры синтезированы полимеризацией с раскры-
тием цикла (разд. 7.6).
Вопросы и упражнения
2.1. Выведите уравнение для скорости поликонденсации стехио-
метрических количеств адипиновой кислоты и гексаметилендиами-
на. Укажите, какие предположения были сделаны при выводе его.
146
Глава 2
Выведите уравнение для скорости поликонденсации нестехио-
метрических количеств указанных веществ.
2.2. Методом инфракрасной спектроскопии и титрованием ос-
нованием найдено, что в 21,3 г полигексаметиленадипамида имеется
2,5‘Ю-3 моля карбоксильных групп. Вычисленный на основании
этих данных среднечисловой молекулярный вес полимера равен
8520. Какое допущение было сделано при таком расчете? Как
экспериментально можно показать его справедливость? Если
это допущение неверно, как можно экспериментально найти истин-
ный Мп?
2.3. Опишите и изобразите структуры сложных полиэфиров,
образующихся в результате следующих реакций поликонденсации:
a) HOOC-R—СООН + НО — R'-OH,
б) HOOC-R-COOH + HO — R" —ОН,
Jh
в) НООС —R—СООН + НО —R" —ОН+ HO — R' —ОН.
ОН
Зависит ли структура каждого из полимеров от относитель-
ного содержания реагентов в реакционной смеси? Если да, то
объясните, в чем заключается различие.
2.4. Опишите и изобразите структуру сложных полиэфиров,
образующихся в результате следующих реакций поликонденсации:
a) HO-R-COOH,
б) НО —R —СООН + НО — R'-OH,
в) НО — R — СООН + НО — R"—ОН,
I
ОН
г) НО —R —СООН + НО —R' — OH + HO — R" —ОН.
I
ОН
Зависит ли структура полимеров, полученных в реакциях (б),
(в) и (г), от соотношения реагентов? Если да, то объясните, в чем
заключается различие.
2.5. Сравните возможное молекулярновесовое распределение
полимеров, которые образуются при реакциях (2.3а), (2.4а)—
(2.4в).
2.6. Рассмотрите возможность циклизации в процессе поликон-
денсации следующих мономеров:
a) H2N —(СН2)т—СООН,
б) НО —(СН2)2—ОН + НООС —(СН2)т —СООН
П оликонденсация
147
при т от 2 до 10. На какой стадии (стадиях) реакции циклизация
становится возможной? Какие доминирующие факторы в определе-
нии направления в сторону циклизации или в сторону линейной
поликонденсации?
2.7. Вычислите среднечисловую степень полимеризации при
поликонденсации эквимолярных количеств адипиновой кислоты
и гексаметилендиамина при следующих степенях завершенности
реакции: 0,500, 0,800, 0,900, 0,950, 0,970, 0,980, 0,990, 0,995.
2.8. Рассчитайте, какое должно быть начальное соотношение
адипиновой кислоты и гексаметилендиамина для получения поли-
амида с молекулярным весом 15 000 при степени превращения
99,5%. Какие группы находятся на концах цепей такого полимера?
Проведите подобный расчет для полиамида молекулярного ве-
са 19 000.
2.9. Сколько бензойной кислоты нужно добавить в реакцион-
ную смесь эквимолярных количеств адипиновой кислоты и гекса-
метилендиамина для получения полимера молекулярного ве-
са 10 000 при степени превращения 99,5%? Проделайте такой же
расчет для полимеров молекулярного веса 19 000 и 28 000.
2.10. Вычислите степень завершенности реакции при точке
гелеобразования для следующих смесей:
а) фталевый ангидрид и глицерин; стехиометрические коли-
чества;
б) фталевый ангидрид и глицерин в молярном отношении
1,500 : 0,980;
в) фталевый ангидрид, глицерин и этиленгликоль в молярном
отношении 1,500 : 0,990 : 0,002.
г) фталевый ангидрид, глицерин и этиленгликоль в молярном
отношении 1,500 : 0,500 : 0,700.
Сравните точки гелеобразования, оцененные по уравнению Ка-
розерса и уравнению, выведенному путем статистического анализа.
2.11. Напишите уравнение реакции поликонденсации мела-
мина и формальдегида с образованием сшитого полимера.
2.12. Напишите уравнения реакций образования статистиче-
ских сополимеров и блок-сополимеров следующего состава:
148
Глава 2
2.13. Как бы вы синтезировали блок-сополимер, имеющий сег-
менты следующего строения:
+сн2сн2сн2-о-^ и
2.14. Укажите, в чем различие между спирополимерами, лест-
ничными и блок-лестничными полимерами. Приведите примеры
полимеров таких структур.
Список литературы
1. Flory Р. J., Principles of Polymer Chemistry, chap. II, Cornell University
Press, Ithaca, New York, 1953.
2. Bhide В. V., Sudborough J. J., J. Indian Inst. Sci., 8A, 89 (1925).
3. Rand L., Thir B., Reegen S. L., Frisch К. C., J. Appl. Polymer Sci.,
9, 1787 (1965).
4. Rabinowitch E., Trans. Faraday Soc., 33, 1225 (1937).
5. Vancso-Szmercsanyi I., Makay-Bodi E., European Polymer J., 5, 145,
155 (1969).
6. Frost A. A., Pearson, R. G., Kinetics and Mechanism, 2d ed., chap. 9,
John Wiley and Sons, Inc., New York, 1961.
7. Billmeyer F. W., Jr., Textobook of Polymer Science, chap. 8, Interscience
Publishers, John Wiley and Sons, Inc. New York, 1962.
8. Flory P. J., J. Am. Chem. Soc., 61, 3334 (1939).
9. Solomon D. H., J. Macromol. Sci. — Revs. Macromol. Chem, Cl(l), 179
(1967).
10. Tang A., Yao K., J. Polymer Sci., 35, 219 (1959).
11. Hine J., Physical Organic Chemistry, chap. 2, McGraw-Hill Book Com-
pany, Inc., New York, 1962.
12. Imoto M., Tanigaki T., J. Chem. Soc., Ind. (Kogyo Kagaku Zasshi), 66,
517 (1963).
13. Carothers W. H., Trans. Faraday Soc., 32, 39 (1936).
14. Challa G., Makromol. Chem., 38, 105, 123 (1960).
15. LenzR. W., HandlovitsC. E., Smith H. A., J. Polymer Sci., 58, 351 (1962).
16. Lovering E. G., Laidler K. J., Can. J. Chem., 40, 31 (1962).
17. Kaplan M., J. Chem. Eng. Data, 6, 272 (1961).
18. Burkus J. W., Eckert C. F., J. Am. Chem. Soc., 80, 5948 (1958).
19. Kienle R. H., Van der Meulen P. A., Petke F. E., J. Am. Chem. Soc.,
61, 2258 (1939).
20. Solomon D. H., Hopwood J. J., J. Appl. Polymer Sci., 10, 1431 (1966).
21. Carothers W. H., Hill J. W., J. Am. Chem. Soc., 55, 5043 (1933).
22. Eliel E. L., Stereochemistry of Carbon Compounds, chap. 7, McGraw-
, Hill Book Company, Inc., New York, 1962.
23. Jacobson H., Stockmayer W. H., J. Chem. Phys., 18, 1600 (1950).
24. Scott D. W., J. Chem. Phys., 68, 2294 (1946).
25. Brown J. F., Slusarczuk G. M., J. Am. Chem. Soc., 87, 931 (1965).
26. Korshak V. V., Pure and Appl. Chem., 12, 101 (1966).
27. Flory P. J., in [1], chaps. 8 and 9.
Поликонденсация
149
28. Howard G. J., The Molecular Weight Distribution of Condensation Poly-
mers, in «Progress in High Polymers», Robb J. C., Peaker F. W. (eds.),
vol. I, p. 185—231, Iliffe Books, Ltd., London, 1961.
29. Фракционирование полимеров под ред. М. Кантова, изд-во «Мир», М.,
1971.
30. Howard G. J., J. Polymer Sci., 37, 310 (1959).
31. Kilkson H., Ind. Eng. Chem. Fundamentals, 3, 281 (1964).
32. Abraham W. H., Chem. Eng. Sci., 21, 327 (1966).
33. Lenz R. W., Organic Chemistry of Synthetic High Polymers, chaps. 4—8,
Interscience Publishers, John Wiley and Sons, Inc., New York, 1967.
34. Raave A., Organic Chemistry of Macromolecules, chaps. 14—19, Marcel
Dekker, Inc., New York, 1967.
35. Уилфонг P. И., Химия и технология полимеров, № 6, 28 (1962).
36. Stevenson R. W., Nettleton H. R., J. Polymer Sci., A-l(6), 889, 1968.
37. LymanD. J., J. Macromol. Sci.—Revs. Macromol. Chem., 1(1), 191 (1966).
38. Rarry A. J., Reck H. N., Silicone Polymers, in «Inorganic Polymers»,
Stone F.G.A. and Graham W.A.G. (eds.), chap. 5, Academic Press, Inc.,
New York, 1962.
39. Morgan P. W., Kwolek S. L., J. Chem. Ed., 36, 182 (1959); J. Polymer
Sci., 40, 299 (1959).
40. Морган П. У., Поликонденсационные процессы синтеза полимеров,
изд-во «Химия», Л., 1970.
41. Schaefgen J. R., Flory Р. J., J. Am. Chem. Soc., 70, 2709 (1948).
42. Mod. Plastics, 46(1), 25 (1969); Chem. and Eng. News, 47(36), 64A (1969).
43. Pinner S. H., J. Polymer Sci., 21, 153 (1956).
44. Stockmayer W. H., J. Polymer Sci., 9, 69 (1952); 11, 424 (1953); J. Chem.
Phys., 11, 45 (1943).
45. Kienle R. H., Petke F. E., J. Am. Chem. Soc., 62, 1053 (1940), 63, 481
(1941).
46. Flory P. J., J. Am. Chem. Soc., 63, 3083 (1941).
47. Stockmayer W. H., in «Advancing Fronts in Chemistry», Twiss S. B. (ed.),
chap. 6, Reinhold Book Corp., New York, 1945.
48. Strella S., Ribeau A. A., J. Macromol. Chem., 1(3), 417 (1966).
49. Jonason M., J. Appl. Polymer Sci., 4, 129 (1960).
50. Robalek E. G., Moore E. R., Levy S. S, Lee С. C., J. Appl. Polymer Sci.,
8, 625 (1964).
51. Соломон Д. Г., Химия органических пленкообразователей, изд-во
«Химия», М., 1971.
52. Flory Р. J., Chem. Rev., 39, 137 (1946).
53. Rider S. H., Hardy E. E., Prepolymer Technology for Crosslinked Plastics,
in «Polymerization and Polycondensation Processes», Platzer N.A.J. (ed.),
chap. 13, American Chemical Society, Reinhold Book Corp., New York,
1962.
54. Yeddanapalli L. M., Gopalakrishna V. V., Makromol. Chem., 32, 90,
124, 130 (1959).
55. Freeman J. H., Lewis C. W., J. Am. Chem. Soc., 76, 2080 (1954).
56. Vale С. P., Taylor W. G. K., Aminoplastics, Iliffe Books, Ltd., London,
1964.
57. DeJong J. I., deJong J., Rec. Trav. Chim., 72, 139 (1953).
58. Strecker R. A. H., French D. M., Polymer Preprints, 7(2), 952 (1966).
59. Саундерс Дж. X., Фриш К. К., Химия полиуретанов, изд-во «Химия»,
М„ 1968.
60. Edwards G. D., J. Appl. Polymer Sci., 9, 3845 (1965).
61. Skeist I., Somerville G. R., Epoxy Resins, Reinhold Book Corp., New
York, 1958.
62. Martens C. R., Alkyd Resins, Reinhold Book Corp., New York, 1961.
63. Boenig H. V., Unsaturated Polyesters: Structure and Properties, American
Elsevier Publishing Co.. Tnr. Now York 1964.
150
Глава 2
64. Dobinson F., Preston J., J. Macromol. Sci. (Chem.), Al(l), 179 (1967).
65. Saotome K., Sata K., Makromol. Chem., 102, 105 (1967).
66. Coleman D., J. Polymer Sci., 14, 15 (1954).
67. Goldberg E. P., J. Polymer Sci., C4, 707 (1964).
68. Merrill S. H., Petrie S. E., J. Polymer Sci., A3, 2189 (1965).
69. Hicks E. M., Jr., Ultee A. J., Drougas J., Science, 147, 373 (1965).
70. Tippetts E. A., Zimmerman J., J. Appl. Polymer Sci., 8, 2465 (1964).
71. Mark H., Pure and Appl. Chem., 12, 9 (1966).
72. Ли Г., Стоффи Д., Невилл К., Новые линейные полимеры, изд-во
«Химия», М., 1972.
73. Шнелл Г., Химия и физика поликарбонатов, изд-во «Химия», М., 1967.
74. Starr L., J. Polymer Sci., A-l(4), 3041 (1966).
75. Preston J., Black W. B., J. Polymer Sci., C19, 17 (1967).
76. Hay A. S., Fortschr. Hochpolymer-Forsch. (Advan. Polymer Sci.), 4, 496
(1967); Macromolecules, 2(1), 107 (1969).
77. Джонсон P. H., Фарнхам А. Г., Нлендиннинг P. А., Хейл У. Ф., Мер-
риам С. Н., сб. «Новые поликонденсационные полимеры», под ред.
Роговина 3. А., Валецкого П. М., изд-во «Мир», М., 1969, стр. 171.
78. Fox R. В. et al., Polymer Preprints, 8(2), e (1967).
79. McNeils E. J., J. Org. Chem., 31, 1255 (1966).
80. Errede L. A., Szwarc M., Quart. Rev. (London), 12, 301 (1958).
81. Gorham W. F„ J. Polymer Sci., A-l(4), 3027 (1966).
82. Bower G. M., Frost L. W., J. Polymer Sci., Al, 3135 (1963).
83. Sroog С. E., Endrey A. L., Abramo S. V., Berr С. E., Edwards W. M.,
Olivier K. L., J. Polymer Sci., A3, 1373 (1965).
84. Vogel H., Marvel C. S., J. Polymer Sci., 50, 511 (1961); J. Polymer Sci.,
Al, 1531 (1963).
85. Wrasidlo W., Levine H. H., J. Polymer Sci., A2, 4795 (1964).
86. Marvel C. S., J. Macromol. Sci. (Chem.), Al(l), 7 (1967).
87. DeWinter W., J. Macromol. Sci.—Revs. Macromol. Chem., 1(2), 329
(1966).
88. Bell V. L., Jewell R. A., J. Polymer Sci., A-l(5), 3043 (1967).
89. Hergenrother P. M., Wrasidlo W., Levine H. H., J. Polymer Sci., ЗА, 1665
(1965).
90. Stille J. K., Mainen E. L., Macromolecules, 1, 36 (1968).
91. Frazer A. H., Wallenberger F. T., J. Polymer Sci., A2, 1181 (1964).
92. Holsten J. R., Ltlyquist H. R., J. Polymer Sci., A3, 3915 (1965).
93. Bailey W. J., Economy J., Hermes M. E., J. Org. Chem., 27, 3295 (1962).
94. Stille J. K. et al., J. Polymer Sci., A-l(5), 2721 (1967); Macromolecules,
2(1), 85 (1969).
95. Overberger C. G., Fujimoto S., J. Polymer Sci., B3, 735 (1965).
96. Stille J. K., Harris F. W., J. Polymer Sci., B4, 333 (1966), J. Polymer
Sci., A-l(6) 2317 (1968).
97. Stille J. K., Gotten L. D., J. Polymer Sci., B6, 11 (1968).
98. Stille J. K., Bedford M. A., J. Polymer Sci., A-l (6), 2331 (1968); Poly-
mer Preprints, 9(1), 636 (1968).
99. Bailey W. J., Volpe A. A., Polymer Preprints, 8(1), 292 (1967).
100. Heller J., Hodgkin J. H., Martinelli F. J., J. Polymer Sci., B6, 153 (1968).
101. Dunnavant W. R., J. Polymer Sci., B6, 49 (1968).
102. Culbertson В. M., Dietz S., J. Polymer Sci., B6, 247 (1968).
103. Lappert M. F., Leigh G. J., (eds.), Developments in Inorganic Polymer
Chemistry, Elsevier Publishing Co., Amsterdam, 1962.
104. Andrianov K., Metal-Organic Polymers, Interscience Publishers, John
Wiley and Sons, Inc., New York, 1965.
105. Brown J. F., Vogt L. F., Katchman A., Eustace J. W., Kiser К. M.,
Krantz K. W., J. Am. Chem. Soc., 82, 6194 (1960).
106. Stone F. G. A., Graham W. A. G. (eds.), Inorganic Polymers, Academic
Press. Inc.. New York. 1962.
ГЛАВА 3
РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ
В предыдущей главе был рассмотрен поликонденсационный ме-
тод синтеза полимеров. Эта и ряд последующих глав будут посвя-
щены обсуждению полимеризации ненасыщенных мономеров. По-
лимеризация инициируется активными частицами R*, образую-
щимися из некоторого соединения I, называемого инициатором
или катализатором:
I —> R* . (3.1)
При присоединении активной частицы, которой может быть сво-
бодный радикал, катион или анион, к молекуле мономера с рас-
крытием л-связи образуется новый радикал, катион или анион.
Этот процесс неоднократно повторяется по мере присоединения
все новых и новых молекул мономера к непрерывно растущему
активному центру
Н НН
ch2=chy I ch2=chy I I
В* ------> R — CH2 — С*---> R —СН2 —С —СН2 —С* —»
I I !
Y Y Y
В какой-то момент наступает обрыв растущей цепи вследствие
гибели активного центра в результате тех или иных реакций, за-
висящих от природы активного центра и особенностей данного
процесса.
3.1. Природа радикальной полимеризации
3.1а. Сравнение полимеризации и поликонденсации
По своему механизму полимеризация и поликонденсация пред-
ставляют два совершенно различных процесса. Основное отличие
полимеризации заключается в том, что в ней полимер высокого
молекулярного веса образуется в первые моменты реакции. Актив-
152
Глава 3
ный центр (радикал, анион или катион) в момент своего образо-
вания присоединяет по цепному механизму много мономерных
звеньев и поэтому быстро достигает больших размеров. Концен-
трация мономера непрерывно уменьшается на всем протяжении
реакции по мере роста числа молекул высокого молекулярного
веса. На любой стадии процесса в реакционной смеси присут-
ствуют мономер, растущая полимерная цепь и конечный «мертвый»
полимер. В ходе полимеризации молекулярный вес полимера
остается практически неизменным, с увеличением продолжитель-
ности реакции растет лишь степень превращения мономера
в полимер.
Совершенно иное положение имеет место в поликонденсации.
В то время как при полимеризации могут реагировать только
мономер с растущей полимерной цепью, при поликонденсации могут
вступать во взаимодействие любые две частицы, находящиеся
в системе. При поликонденсации мономер исчезает значительно
быстрее, чем при полимеризации, так как первый процесс идет
медленно с постепенным переходом от мономера к димеру, триме-
ру, тетрамеру и т. д. Молекулярный вес возрастает на всем протя-
жении реакции поликонденсации, причем высокомолекулярный
полимер не удается получить, не достигнув высокой степени
завершенности реакции. Поликонденсацию необходимо проводить
в течение большого промежутка времени для получения полимера
с высоким выходом и высоким молекулярным весом.
3.16. Сравнение радикальной и ионной полимеризации
3.16.1. Способность к полимеризации
Способность мономера к полимеризации зависит от термодина-
мического и кинетического факторов. Если полимеризация термо-
динамически не разрешена, то ее невозможно осуществить ни
в каких условиях. Полимеризация возможна, если только раз-
ность свободных энергий AG мономера и полимера величина
отрицательная (разд. 3.96). Отрицательная AG не означает, одна-
ко, что полимеризация пойдет в любых условиях. Возможность
осуществления темодинамически разрешимой полимеризации зави-
сит уже от ее' кинетической разрешимости, т. е. от того, проте-
кает ли данный процесс с заметной скоростью в выбранных усло-
виях реакции. Поэтому, хотя термодинамически возможна поли-
меризация широкого ряда ненасыщенных мономеров, для ее
успешного осуществления часто требуются очень специфические
условия, обеспечивающие кинетическую разрешимость реакции.
Хотя в полимеризации применяются радикальные, катионные
и анионные инициаторы, их нельзя использовать подряд, без
разбора, так как различные мономеры полимеризуются только
Радикальная полимеризация
153
в присутствии инициаторов определенного типа. Мономеры обла-
дают значительной селективностью по отношению к типу актив-
ных центров, которые вызывают их полимеризацию. Большинство
мономеров полимеризуется под действием радикальных инициато-
ров, хотя скорость полимеризации сильно зависит от природы
инициаторов и мономера. Вместе с тем мономеры очень чувстви-
тельны к инициаторам ионного типа. Одни мономеры могут не
полимеризоваться в присутствии катионных инициаторов, тогда
как другие не полимеризуются с анионными инициаторами. Зави-
симость полимеризуемости мономера от типа инициатора иллю-
стрируется данными табл. 3.1, из которой видно, в присутствии
Таблица 3.1
Примеры полимеризации различных ненасыщенных мономеров
Мономер Тип инициирования
радикальный катионный анионный
Этилен + + +
1-Алкилолефины (а-олефины) — — —
1,1-Диалкилолефины — + —
Диены-1,3 + + +
Стирол, а-метилстирол + + +
Галогенированные олефины + — —
Сложные виниловые эфиры
(СН2 = СНОСОВ) + — —
Акрилаты, метакрилаты + — +
Акрилонитрил, метакрилонитрил Д- — +
Акриламид, метакриламид + — +
Простые виниловые эфиры — + —
N-Винилкарбазол + + —
N -Ви нилпирролидон + + —
Альдегиды, кетоны — + +
каких инициаторов образуются полимеры высокого молекулярного
веса. При этом, хотя термодинамически разрешима полимериза-
ция всех мономеров, указанных в этой таблице, кинетически поли-
меризация возможна только в присутствии инициаторов опре-
деленного типа.
Основными типами связей, по которым идет полимеризация,
являются двойная углерод-углеродная связь в алкенах и кисло-
род-углеродная двойная связь в альдегидах и кетонах. При этом
наиболее важное значение имеет полимеризация по углерод-
углеродной двойной связи. Карбонильные соединения не поли-
154
Глава 3
меризуются под действием радикальных инициаторов из-за того,
что карбонильная связь в них поляризована
О 0.-
II I
— с— ** —С— (3.3)
Полимеризация альдегидов и кетонов осуществляется в присутст-
вии анионных и катионных инициаторов (см. гл. 5).
3.16.2. Влияние заместителей
В отличие от карбонильной связи двойная связь в алкенах
принимает участие в полимеризации под действием радикальных
и ионных инициаторов. Такое различие обусловлено тем, что при
присоединении активной частицы инициатора к алкену может
иметь место как гомолитическое, так и гетеролитическое расщеп-
ление л-связи, которое определяется типом активной частицы
+ С—СГ ——С=С *«С—С- (3-4)
II II II
Полимеризации подвержены самые различные соединения с угле-
род-углеродными двойными связями. В табл. 3.1 приведены
мономеры с алкильными, алкенильными, арильными, галогенными,
алкоксильными, сложноэфирными, амидными, нитрильными и ге-
тероциклическими заместителями у алкеновой двойной связи.
От индуктивного и резонансного влияния заместителя зави-
сит, по какому типу протекает полимеризация данного алкена:
радикальному, катионному или анионному. Влияние заместителя
проявляется в изменении электронной плотности двойной связи
и способности его стабилизировать возможный свободный радикал,
анион или катион, образующийся в процессе полимеризации.
Электронодонорные заместители, например алкокси-, алкил-,
алкенил-, фенил-группа, увеличивают электронную плотность двой-
ной углерод-углеродной связи
fi+
СН^ =СН <— Y
и облегчают присоединение ее к частицам катионного типа. Кроме
того, такие заместители стабилизируют растущие катионы за счет
резонанса. Примером процесса такого типа является полимериза-
ция простых виниловых эфиров
Н Н
— СН2 —С+^ — СН2 —с (3.5а)
I II
;о: :о+
r R
Радикальная полимеризация
155
Под влиянием алкоксильного заместителя может происходить
делокализация положительного заряда. Если бы заместитель
отсутствовал (как, например, в этилене), то положительный заряд
локализовался бы у а-углеродного атома. Наличие заместителя
(алкоксильной группы) приводит к стабилизации иона карбония
вследствие делокализации положительного заряда между двумя
атомами: углеродом и кислородом. Аналогичная делокализация
имеет место при наличии фенильного, винильного и алкильного
заместителей, например при полимеризации стирола
Этим обусловлен тот факт, что такие мономеры, как изобутилен,
стирол, метилвиниловый эфир и изопрен, полимеризуются в при-
сутствии катионных инициаторов. Значительно менее выражено
влияние алкильных заместителей в катионной полимеризации,
поэтому только 1,1-дизамещенные алкены полимеризуются по
катионному механизму.
Электроноакцепторные заместители, к которым относятся
нитрильная и карбонильная группы (альдегиды, кетоны, кислоты
или их сложные эфиры), облегчают атаку двойной связи анион-
ными частицами вследствие уменьшения злектронной плотности
такой связи
. , е~
СН®+ = СН-^-Y
Они стабилизируют растущие анионы за счет резонанса, напри-
мер при полимеризации акрилонитрила
Н Н
I I
— сн2——сн2-с (3.6)
I II
с с
III II
N N;-
Растущий карбанион стабилизируется в результате делокали-
зации отрицательного заряда между а-углеродом и азотом нит-
рильной группы. Хотя алкенильные и фенильные группы
обладают электронодонорным индуктивным эффектом, они,
подобно нитрильной группе, могут стабилизировать растущий
анион. Поэтому такие мономеры, как стирол и бутадиен-1,3,
полимеризуются по катионному и анионному механизмам. Гало-
гены обладают электронодонорным резонансным и электроноак-
156
Глава 3
цепторным индуктивным эффектами, однако оба эти эффекта
выражены настолько слабо, что практически никак не отража-
ются на способности к полимеризации галогенсодержащих моно-
меров, например винилхлорида, по анионному и катионному
типам реакций.
В противоположность высокой селективности катионной
и анионной полимеризаций под действием радикальных инициа-
торов полимеризуются соединения с самыми различными угле-
род-углеродными двойными связями. Радикальные частицы нейт-
ральны, требования для атаки л-связи или стабилизации
растущего радикала в таком процессе не являются такими стро-
гими. Растущие радикалы резонансно стабилизированы практи-
чески со всеми заместителями, например
н н
I I
— СН2-С.*->—СН2—С (3.7а)
(3.7в)
Таким образом, почти все заместители могут стабилизировать
растущие радикалы в результате делокализации электрона между
двумя или несколькими атомами. Остальная часть этой главы
посвящена детальному анализу особенностей радикальной поли-
меризации. Ионная полимеризация рассматривается в гл. 5.
3.2. Структурное расположение
мономерных звеньев
3.2а. Возможные способы роста
Растущий радикал может присоединяться к двум различным
атомам С монозамещенных (X = Н) или 1,1-дизамещенных моно-
меров, а именно: по углероду 1
Радикальная полимеризация
157
X X
I I
R.+C = CH2 R-С —СН2.
1| 2 |
Y Y
I
или по углероду 2
X X
I I
R.-f-CH2=C R—сн2-с.
1 2| |
Y Y
II
(3.8)
(3.9)
Если каждое последовательное присоединение мономера к рас-
тущему радикалу идет так, как это указано в уравнении (3.9),
то образуется полимер, содержащий заместители при каждом
втором атоме углерода основной цепи:
X X X X X
1 I I I I
— СН2 —С—СН2 —С —СН2 —С —СН2 —С —СН2 —С —
I I I I I
Y Y Y Y Y
III
Такой тип присоединения обычно называют присоединением «голова
к хвосту» или 1,3-присоединением мономерных звеньев. Наруше-
ние такого порядка присоединения, т. е. рост цепи одновременно
согласно уравнениям (3.8) и (3.9), должен, очевидно, привести
к образованию полимера с 1,2-расположением заместителей в од-
ном или более месте полимерной цепи
Y y v v провост v v
•| । л Л хвосту11 л Л
—сн2—с-сн,—с—сн2—с-с-сн^сн1—с—сн,—с—
2 I 2 I 2 I I 2 2 I 2 I
Y Y Y Y Y Y
„голова
к голове“
IV
1,2-Присоединение часто называется присоединением «голова к го-
лове» или «хвост к хвосту», а также присоединением «голова к голове,
хвост к хвосту». По-видимому, преобладающим должно быть при-
соединение голова к хвосту, так как при последовательных реак-
циях присоединения согласно уравнению (3.9) стерический и ре-
зонансный факторы предпочтительнее. Растущий радикал II,
образующийся при присоединении радикала по углероду 2,
является более стабильным, что обусловлено резонансным эффек-
том заместителей X и Y. Подобная стабилизация радикала I не-
158
Глава 3
возможна, поскольку заместители не соединены непосредственно
с углеродом, имеющим неспаренный электрон. Кроме того, при-
ближение (и последующее присоединение) растущего радикала
к незамещенному атому углерода 2 молекулы мономера стериче-
ски предпочтительнее, чем приближение к замещенному угле-
роду 1.
3.26. Экспериментальные доказательства
Рассмотренные теоретические предпосылки получили экспери-
ментальное подтверждение при исследовании большого числа
полимеров. Марвел с сотр. [1] исследовал внутримолекулярную
циклизацию ряда полимеров. В частности, было изучено образо-
вание циклопропановых циклов при дехлорировании поливинил-
хлорида цинком
— СН2 СН2 СН2 СН2 СН2
\/\/\/\/\ Zn
сн сн сн сн сн— —->
I I I ! I
С1 С1 С1 С1 С1
—> — сн2 сн2 сн2 сн2 сн2
\н — СП \н \н-^сн —
I
С1
(3.10)
и внутримолекулярная циклоконденсация полиметилвинилкетона
— СН2 СН2 —СН2 СН2
\ / \ Основание \ / \
сн2 сн—------------> • сн сн-----
I 1 I 1
СОСНз СОСНз со с
\ z\
СН СНз
(3.11)
Степень превращения при таких реакциях характеризует степень
1,3-присоединения. При 1,2-присоединении максимально возмож-
ная степень циклизации уменьшается. Однако необходимо отда-
вать себе отчет в том, что 100%-ной степени циклизации не уда-
лось бы достигнуть, даже если бы полимер был получен только
1,3-присоединением мономерных звеньев. По законам статистики
некоторое количество функциональных групп останется изолиро-
ванным [например, см. уравнение (3.10)1, что обусловлено реак-
циями функциональных групп, расположенных по обеим сторо-
нам от таких изолированных групп. Последние нереакционноспо-
собны в условиях данной реакции, что и приводит к уменьшению
максимально возможной степени циклизации. Полученные экспе-
риментально значения степени циклизации для двух указанных
реакций совпадают со статистически рассчитанными величинами,
причем отклонения не превышают нескольких процентов.
Радикальная полимеризация
159
Гидролизом поливинилацетата до поливинилового спирта с по-
следующим окислением его до 1,2-гликольных звеньев перйодатом
было доказано [2] наличие в исходном полимере 1—2% звеньев,
присоединенных голова к голове
IOJ
— СН2 —СН-СН —СН2—------> — СН2 —СН +:сн-сн2— . (3.12)
I I II II
ОН ОН 0 0
Методом ЯМР высокого разрешения было показано [3] наличие
в ощутимых количествах присоединения голова к голове в неко-
торых полимерах. Так, в поливинилиденфториде и поливинил-
фториде присоединения такого типа составляют примерно 10
и 30% соответственно.
В общем же присоединение голова к хвосту преобладает при
росте цепи во всех реакциях полимеризации. Однако, когда замес-
тители имеют небольшой размер (и не оказывают заметных стери-
ческих препятствий приближающемуся радикалу) или не харак-
теризуются .сильно выраженным резонансным стабилизирующим
эффектом, как это имеет место в случае атомов фтора, рост цепи
путем присоединения голова к голове может протекать в замет-
ной степени. С ростом температуры полимеризации доля присое-
динения голова к голове увеличивается [2,3]. Повышение темпе-
ратуры приводит к менее избирательному (более статистическому)
росту цепи, но эффект этот не очень значительный. Так, повышение
температуры от 30 до 90 °C при получении поливинилацетата уве-
личивает долю присоединения голова к голове от 1,30 до 1,98%.
3.3. Кинетика полимеризации
3.3а. Последовательность стадий
Радикальная полимеризация является цепной реакцией, состо-
ящей из трех последовательных стадий: инициирования, роста цепи
и обрыва цепи. В свою очередь стадия инициирования состоит из
двух реакций. Первая заключается в образовании свободных
радикалов в результате тех или иных реакций, основная из кото-
рых — гомолитический распад молекулы инициатора I (или ката-
лизатора) на два радикала R •
%
I—>2R., (3.13)
где кг — константа скорости гомолитического распада инициато-
ра. Вторая стадия инициирования заключается в присоединении
такого радикала к первой молекуле мономера с образованием
160
Глава 3
инициирующей частицы растущей цепи Mi •
,?и
R- + M —> М).,
(3.14а)
где М — молекула мономера, а ка — константа скорости стадии
собственно инициирования. Для полимеризации СН2 = CHY
уравнение (2.14а) принимает вид
Н
R.+CH2 = CHY —» R —СН2 —С-
(3.146)
(Радикал R • часто называется инициирующим или первичным
радикалом.)
Процесс роста цепи заключается в последовательном присое-
динении к Mi- большого числа (сотен, а возможно, и тысяч) моле-
кул мономера, т. е. в росте в соответствии с уравнением (3.2).
При каждом присоединении образуется новый радикал той же
природы, что и предыдущий, но длиннее его на одно мономерное
звено. Схематически последовательные реакции присоединения
можно изобразить следующим образом:
hp
мг 4-м —> м2.,
м2-4-м
Мз-4-М
hp
м3.,
или в общем виде
hp
мп-4-м -> мп+1.,
(3.15)
где /ср — константа скорости реакции роста цепи. В результате
реакции роста цепи очень быстро образуется полимер высокого
молекулярного веса. Величина кр для большинства мономеров
порядка 102—104 л/(моль-с), что значительно выше констант ско-
рости других химических реакций.
В некоторый момент реакции наблюдается прекращение роста
полимерной цепи и ее обрыв. Обрыв с уничтожением радикальных
центров происходит в результате бимолекулярной реакции между
радикалами. Два радикала реагируют между собой путем реком-
бинации
НН а0. р НН
— СН, — С-4-.С — СН2—------> — СН2—С —С —СН2— (3.16а)
Y Y Y Y
или, что более редко, путем диспропорционирования, заключаю-
щегося в том, что водород, находящийся в p-положении к радикаль-
ному центру, переносится на другой радикальный центр. В ре-
зультате образуются две полимерные молекулы — одна насыщен-
Радикальная полимеризация
161
ная, а другая ненасыщенная
Н Н Н fe0. д Н НН
— СН2-С-4-.С-С—--------> —СН2—СН + С = С— (3.166)
Y Y Н Y Y
Обрыв может иметь место также вследствие совместного действия
реакций рекомбинации и диспропорционирования. В общем виде
обе реакции обрыва можно представить следующим образом:
Mn'+Mnj* > Mn+m-, (3.17а)
"о. д
Мп- + Мто----+ (3.176)
где /с0.р и /со.д — константы скорости обрыва путем рекомбинации
и диспропорционирования соответственно. Стадию обрыва можно
также выразить как
Mn- + Mm. —> Мертвый полимер, (3.17в)
где не указывается конкретно характер обрыва, причем
*о = *о. р *о. д (3.18)
Название «мертвый полимер» указывает, что рост растущего ради-
кала прекратился. Если бы не энергичное вмешательство реакций
обрыва, рост цепи продолжался бы неопределенно долго вплоть
до полного исчерпания мономера. Как правило, константы скоро-
сти реакций обрыва составляют 106—108 л/(моль>с), т. е. на не-
сколько порядков выше величин констант скорости реакций
роста. Однако, несмотря на это, рост цепи идет, и это обусловлено
как низкой концентрацией радикальных частиц, так и тем, что ско-
рость полимеризации зависит от кп только в степени 1/2.
3.36. Уравнение скорости реакции
Уравнения (3.13)—(3.16) описывают детальный механизм
полимеризации, инициируемой свободными радикалами. Цепной
характер процесса присущ для стадии роста [уравнение (3.15)], на
которой в полимер превращается большое число молекул мономера
в расчете на одну начальную радикальную частицу, образующую-
ся на первой стадии [уравнение (3.13)]. Для вывода кинетического
уравнения полимеризации необходимо предположить, что /ср и к0
не зависят от размера радикала. Аналогичное предположение было
сделано при анализе кинетики поликонденсации. Существуют
достаточно веские экспериментальные факты, подтверждающие,
что хотя скорость полимеризации зависит от размера молекулы,
влияние его перестает ощущаться уже при переходе к пентамеру
или гексамеру.
162
Глава 3
Мономер расходуется на стадии инициирования [уравнени
(3.14)] и на стадии роста [уравнение (3.15)]. Скорость исчезнове
ния мономера, т. е. скорость полимеризации, определяется ка:
=^ = 7?и + 7?р, (3.19
где R и и 7?р — скорости инициирования и роста соответственно
Однако при образовании высокомолекулярного полимера числ<
молекул мономера, участвующих в стадии инициирования, значи
тельно уступает количеству мономера, принимающему участи»
в стадии роста. Поэтому с большой степенью точности первы&
членом в этом уравнении можно пренебречь, и тогда скорост]
полимеризации определяется просто скоростью реакции роста
•=^ = Яр (3.2О;
Скорость роста и, следовательно, скорость полимеризации
представляет собой сумму многих отдельных последовательных
реакций роста. Так как константа скорости реакции для всех
них одинакова, то можно записать
7?р =/ср [М • ] [М], (3.21)
где [М] — концентрация мономера, а [М •] — суммарная концен-
трация всех цепных радикалов, т. е. всех радикалов с размером
Mi и больше.
Поскольку в уравнение (3.21) для скорости полимеризации
входит концентрация радикалов, в таком виде пользоваться им
нельзя. Концентрацию радикалов измерить трудно, так как она
очень низка (приблизительно 10-8 М). Для того чтобы исключить
этот член из уравнения (3.21), вводится допущение о стационар-
ном состоянии, согласно которому концентрация радикалов снача-
ла. возрастает, а потом очень быстро достигает постоянной вели-
чины. В ходе полимеризации скорость изменения концентрации
радикалов быстро становится равной нулю, а это равносильно
положению', что скорость инициирования 7?и и скорость обрыва
По радикалов равны между собой, т. е.
йи = 2ММ.]а (3.22)
Теоретически справедливость допущения стационарного состоя-
ния доказана в работе [4]. Экспериментально оно было подтверж-
дено на многих реакциях полимеризации. В обычных полимери-
зационных процессах оно наступает очень быстро, спустя
несколько секунд после индукционного периода.
Правая часть уравнения (3.22) отражает скорость реакции
обрыва. При этом не имеет значения, идет ли обрыв путем диспро-
порционирования или рекомбинации, так как оба они подчиняют-
Радикальная полимеризация
163
ся одним и тем же кинетическим зависимостям. Коэффициент 2
в уравнении скорости реакции обрыва, который можно часто
встретить в уравнениях реакций уничтожения пары радикалов
путем их рекомбинации, применяется просто из соображений
удобства. Его часто ставят также в кинетических уравнениях
реакций образования пары радикалов [см., например, уравне-
ние (3.13)]. (При изучении литературы по полимерам необходимо
иметь в виду, что коэффициент 2 ставится не всегда.) Представляя
уравнение (3.22) в виде
<3-23>
и подставляя его в уравнение (3.21), получим
Яр = /ср[М](-^)1/а. (3.24)
Из уравнения (3.24) видно, что скорость полимеризации зависит
от скорости инициирования только в степени 1/2. Следовательно,
удвоение скорости инициирования не приводит к увеличению ско-
рости полимеризации в два раза; она возрастает только в 2 раза.
Такое поведение объясняется бимолекулярным механизмом обры-
ва радикалов.
Экспериментально скорость полимеризации можно оценить
путем определения изменения какого-либо параметра системы —
плотности, показателя преломления, вязкости, светопоглощения.
К числу наиболее точных и чувствительных методов относится
измерение плотности. В ходе полимеризации многих мономеров
плотность возрастает на 20—25%. На практике измеряют объем
полимеризующейся системы, проводя реакцию в дилатометре,
который представляет собой сосуд специальной конструкции
с капиллярной трубкой, дающий возможность измерить неболь-
шие изменения объема с очень большой точностью. Таким спосо-
бом устанавливают степень превращения мономера, составляющую
всего несколько сотых процента.
3.4. Инициирование
Вывод уравнения (3.24) является весьма общим, так как реак-
ция образования радикалов [уравнение (3.13)] не конкретизирует-
ся. Скорость инициирования обозначают Rn. В полимеризации
используются различные инициаторы (катализаторы). (Термин
катализатор здесь используется не в классическом смысле слова,
а лишь для того, чтобы подчеркнуть, что под влиянием одной
молекулы катализатора очень большое число молекул мономера
превращается в полимер.) Для образования радикалов можно
использовать различные методы — термические, фотохимические
и окислительно-восстановительные [5]. Вещества, применяющиеся
164
Глава 3
в качестве инициаторов, должны удовлетворять ряду требований:
быть легко доступными соединениями, устойчивыми при комнат-
ной и пониженных температурах, но разлагаться с образованием
радикалов с заметной скоростью при не очень высоких темпера-
турах (< 150 °C).
3.4а. Термический распад инициаторов
3.4а.1. Типы инициаторов
Термический гомолитический распад инициаторов — наиболее
широко применяющийся в промышленности и исследовательской
практике способ образования радикалов для инициирования
полимеризации. Реакции полимеризации, инициируемые таким спо-
собом, часто называют реакциями термически катализируемой
полимеризации. В качестве инициаторов можно использовать
довольно ограниченный круг соединений. К ним относятся веще-
ства с энергией диссоциации связей порядка 25—40 ккал/моль
(105-103—168 -103 Дж/моль.) (Соединения с более высокими или
более низкими значениями энергии диссоциации будут разлагать-
ся слишком медленно или слишком быстро.) Этому требованию
удовлетворяют лишь несколько классов соединений, содержащих
связи O — O,S — 8, N — О. Вместе с тем из них только перекиси
находят широкое практическое применение в качестве источника
радикалов. Другие классы соединений или недостаточно доступны
или мало стабильны. Широко используется несколько типов пере-
кисей. К ним относятся ацилперекиси, например перекись ацети-
ла и перекись бензоила
0 0 0
II II II
СНз-С-О — 0 — С — СНз - -> 2СН3 — С — о. (3.25а)
0 II СвН5-С-О- 0 II — 0 —С —С6Н5 0 II —» 2СвН5—С —0- (3.256)
алкилперекиси, например перекись кумола и ш/?еш-бутила
СН3 СНз СН3
1 С6Н5-С-О- 0 —А —СвН5 1 —» 2С6Н5—С —0. (3.25в)
Анз СН3 । СН3
СН3 1 СН3 СН3
СИ3 —С — 0 1 -o-i-снз । —» 2СН3-С-О. (3.25г)
СН3 Ан3 СН3
Радикальная полимеризация
165
гидроперекиси, например гидроперекись mpem-бутила и кумола
СНз СН3
СН3—С —О —ОН -» СНз-J-О- 4- -ОН (3.25д)
I I
СН3 СНз
СНз СН3
С-Н5—С —О —ОН —» С6Н5-С —О- + -ОН (3.25е)
I I
СНз СН3
и перэфиры, например трет-бутилпербензоат
О СН3 0 СН3
CeH5—С —О —О —С—СН3 -* CeH5 —С —О-+-О —С —СНз (3.25ж)
СНз ^Н3
Кроме перекисей, пожалуй, только азосоединения нашли широкое
практическое применение, и среди них наиболее распространен
2,2'-азо-бнс-изобутиронитрил (АИБН)
СН3 СНз СН3
СН3 —С —N = N —С —СН3 —> 2СН3 —С-4-N2 (3.25з)
CN CN (^N
В отличие от перекисей легко протекающая диссоциация азосоеди-
нений не вызвана наличием в них слабых связей: энергия диссо-
циации связи С — N достаточно высока [приблизительно
70 ккал/моль (293 *103 Дж/моль)]. Легкое разложение азосоедине-
ний обусловлено образованием в процессе их распада очень
устойчивых молекул азота.
Различные катализаторы применяются при разных темпера-
турах, определяющихся скоростью их разложения. Так, азо-бпс-
изобутиронитрил обычно применяют при 50—70 °C, перекись
ацетила — при 70—90 °C, перекись бензоила — при 80—95 °C
и, наконец, перекись дикумола и перекись mpem-бутила — при
120—140 °C. [В области температур, в которой используется тот
или иной катализатор, константа скорости разложения его состав-
ляет, как правило, 10-4—10-6 с-1 (табл. 3.10).] Широкое исполь-
зование перекисей в качестве инициаторов связано с устойчиво-
стью многих из них в широком интервале температур. Различия
в скоростях разложения разных инициаторов обясняются разным
химическим строением их и образующихся из них радикалов.
Прайер [5] рассмотрел влияние структуры перекисей на их реак-
ционную способность.
166
Глава 3
3.4a.'i. Кинетика инициирования и полимеризации
Скорость термического гомолитического распада инициатора Rr
(уравнения (3.13) и (3.25)] определяется как
ВГ = 2ДГ[1], (3.26)
где [I] — концентрация инициатора, а / — эффективность иници-
атора. Эффективностью катализатора называется доля образую-
щихся при гомолизе радикалов, которые принимают участие
в инициировании полимеризации. Величина / обычно меньше
единицы, что обусловлено участием радикалов в побочных реак-
циях. Как уже указывалось, коэффициент 2 и в этом уравнении
применяется для удобства.
Выше уже говорилось, что реакция инициирования полимери-
зации состоит из двух стадий [уравнения (3.13) и (3.14)]. В боль-
шинстве случаев вторая стадия (присоединение первичного радика-
ла к мономеру) намного быстрее первой. Лимитирующей стадией
является гомолитический распад инициатора со скоростью
Ки = 2/^(1] (3.27)
Подставляя уравнение (3.27) в уравнение (3.24), получим
ЯР = ЫМ](^)1/2 (3.28)
3.4а.3. Влияние концентрации инициатора на
скорость полимеризации
Уравнение (3.28) описывает наиболее общий случай радикаль-
ной полимеризации. Оно показывает, что скорость полимеризации
зависит от корня квадратного из концентрации инициатора. Эта
зависимость подтверждена на большом числе систем мономер —
инициатор в широком интервале [М] и [I]. На рис. 3.1 приведена
типичная кривая, иллюстрирующая такую зависимость [6—8].
В определенных условиях наблюдаются отклонения от этой за-
висимости. При очень высоких концентрациях инициатора 7?р мо-
жет зависеть от [I] в степени меньше х/2. Это, однако, нельзя счи-
тать отклонением от уравнения (3.28), так как данный эффект
может быть обусловлен уменьшением / с увеличением концентрации
инициатора. Кроме того, реакция обрыва может свестись к реак-
ции между радикалом растущей полимерной цепи и первичным
радикалом [9]
Mn-+R. ->Mn-R (3.29)
Такой путь обрыва цепи и связанное с ним изменение зависимости
7?р от [I] может наблюдаться в так называемой тромсдорфовской
Радикальная полимеризация
Ю/
области полимеризации (разд. 3.10). Могут встретиться случаи,
когда 7?р зависит от [I] в степени больше 1/2. Это может иметь
место опять-таки в области Тромсдорфа, если полимерные ради-
Р и с. 3.1. Квадратичная зависимость ско-
рости полимеризации от концентрации ини-
циатора [1].
1 — полимеризация метилметакрилата в присут
ствии перекиси бензоила при 50 °C [61; 2 — по-
лимеризация винилбензоата в присутствии АИБН
при 60 °C [7, 8].
калы не подвержены реакциям обрыва или в определенных усло-
виях реакции передачи цепи или ингибирования (разд. 3.7).
3.4а.4. Влияние концентрации мономера
на скорость полимеризации
Согласно уравнению (3.28), зависимость скорости полимериза-
ции от концентрации мономера должна быть линейной. В боль-
шинстве случаев эксперимент подтверждает это. На рис. 3.2 пока-
Р и с. 3.2. Зависимость скорости поли-
меризации Нр метилметакрилата от
концентрации мономера [М]. Инициа-
тор — смесь mpem-бутилпербензоата и
дифенилтиомочевины [10].
вана линейная зависимость скорости полимеризации метилметакри-
лата от его концентрации (10]. Известно, однако, много реакций
полимеризации, характеризующихся большей зависимостью ско-
168
Глава 3
рости полимеризации 7?р от [М]. Так, скорость полимеризации
стирола в растворе бензола при 120 °C в присутствии трет-
бутиловых перэфиров как инициатора зависит от концентрации
стирола в степени 3/2 [И]. При полимеризации стирола в при-
сутствии перекиси бензола в толуоле при 80 °C показатель степени
при концентрации мономера в уравнении скорости полимеризации
по мере уменьшения [М] возрастает [12]. При [М], равном 1,8, он
равен 1,18 и увеличивается до 1,36 при [М] = 0,4. Указанные эффек-
ты могут быть обусловлены наличием зависимости скорости
инициирования от концентрации мономера. При выводе уравне-
ния (3.28) было сделано допущение, что 7?н не зависит от [М].
Однако в ряде случаев величина [М] оказывает влияние на
скорость инициирования. Так, эффективность мономера / может
пропорционально изменяться с концентрацией мономера
/ = /'[М], (3.30)
что должно приводить к линейной зависимости Ни от [М] и к полу-
торной зависимости 7?р от [М] [при соответствующей подстановке
уравнения (3.30) в уравнения (3.27) и (3.28)]. (В разд. 3.4е при-
ведены примеры, подтверждающие такое поведение.) Аналогичные
результаты получаются, если при инициировании лимитирующей
стадией становится второй его этап [уравнение (3.14)], а не пер-
вый [уравнение (3.13)].
Для ряда систем наблюдалось отклонение процесса иницииро-
вания от обычной двухстадийной схемы, выраженной уравнениями
(3.13) и (3.14). Так, например, в системе гидроперекись трет-
бутила — стирол только незначительная часть всего процесса
инициирования относится к гомолитической реакции первого
порядка [уравнение (3.25е)], ответственной за полный распад
такой гидроперекиси в отсутствие стирола. В присутствии стирола
гомолитический распад гидроперекиси идет с большей скоростью.
Повышение скорости разложения гидроперекиси mpem-бутила
в присутствии стирола обусловлено протеканием молекулярно
индуцированной реакции гомолиза, которая является реакцией
первого порядка по стиролу и по гидроперекиси [13]. Тогда реак-
цию инициирования можно представить в следующем виде:
М + 1-*М. +R-, (3.31)
что и приводит к полуторной зависимости 7?р от [М]. Отклонения
от линейной зависимости 7?р от [М] возможны также вследствие
изменения механизма реакций обрыва цепи, который может вклю-
чать реакцию с первичными радикалами [см. уравнение (3.29) и
разд. 3.10г)].
Радикальная полимеризация
169
3.46. Фотохимическое инициирование
Полимеризацию можно инициировать фотохимически [14, 15].
В этом случае радикалы образуются в результате ультрафиолето-
вого облучения чистого мономера, а также мономера, содержащего
катализатор или фотосенсибилизатор.
3.46.1. Чистый мономер
При фотолизе некоторые мономеры переходят в возбужденное
состояние путем поглощения кванта света
M + (3.32)
Возбужденные частицы М* в результате последующего гомолиза
распадаются на радикалы
М* —> R«+R'-, (3.33)
способные инициировать полимеризацию остального мономера.
Природа радикалов R- и R'• выяснена недостаточно точно. Так,,
для фотохимической полимеризации стирола (основная полоса
поглощения которого находится при 0,25 мкм) для разрыва связи
предлагаются следующие пути [16]:
С6Н5-СН = СН2 —> С6Н5- + -СН = СН2 (3.34)
или
СвН5 —СН = СН2 —» СвН5 —СН = СН + Н. (3.35)
Инициирование бирадикалами
С6Н5-СН = СН2 —» СвН5 — СН — СН2 (3.36)
маловероятно (разд. 3.4г).
Скорость фотохимического инициирования равна
7?и = 2/Л = 2Ф1а, (3.37)
где 1а — число квантов света (эйнштейны), поглощенного в еди-
ницу времени (секунда) в единице объема (литр), а / — число пар
радикалов, образовавшихся при поглощении одного кванта света.
Вместо / в фотополимеризации употребляется индекс Ф, который
называется квантовым выходом инициирования. Интенсивность
поглощенного света составляет
7а = е/0[М], (3.38)
где 10 — интенсивность кванта света, падающего на мономер,
а е — коэффициент экстинкции для определенной длины погло-
щенного света. Подставив уравнение (3.38) в уравнение (3.37),
170
Глава 3
получим
Ди = 2Фе/0[М], (3.39)
последнее можно подставить в уравнение (3.24) и получить тогда
следующее выражение для скорости фотополимеризации:
Др = кр [М]3/2 (^М1/2- (3.40)
Интенсивность света, испускаемого различными источниками,
обычно выражается в килокалориях в секунду или эргах в секун-
ду, поэтому, прежде чем подставлять ее в уравнение (3.40), нужно
сначала перевести ее в эйнштейны. Для этого необходимо знать
длину волны % или частоту v испускаемого света. Энергия в эйн-
штейнах равна Nhv или Nhdk, где h — постоянная Планка, с —
скорость света, N — число Авогадро.
При использовании уравнения (3.40) принимают, что интен-
сивность падающего света практически постоянна по всей толщине
реакционного сосуда. Однако это справедливо только при слабом
поглощении света или проведении реакции в тонкостенных сосу-
дах. В большинстве же реакций полимеризации поглощение света
нельзя не учитывать, так как /0 и 1а изменяются с толщиной слоя.
В этих условиях 1а можно определить, пользуясь законом Бера
/ = /ое-^м]Ь) (3.4!)
где I — интенсивность падающего света в точке реакционного
юсуда, отстоящей от стенки его на расстояние Ъ. Тогда интен-
сивность поглощенного света определяется как
Za = /0[l-e-s[M]i>], (3.42)
де Ъ — уже толщина поглощаемого слоя. Скорость полимериза-
ции можно определить из уравнений (3.24), (3.37) и (3.42)
7?Р = ММ] (^-f2 , (3.43)
Др = /ср [М] (Ф/°[1-^~е[М]Ь] )1/2 • (3-44)
Часто предпочитают прямое измерение 1а для данной полиме-
шзационной системы использованию уравнения (3.44). Определе-
гие интенсивности света называется актинометрией [5]. Опреде-
[ения проводятся с помощью актинометра, основной частью
юторого является термоэлемент, состоящий из большого коли-
[ества термопар, соединенных в блоки, при этом горячие спаи
акрепляют в зачерненной поверхности, которая поглощает весь
[адающий свет, превращая его в тепло. Для измерения 1а термо-
лемент помещают непосредственно за стенкой реакционного
осуда и определяют разность между интенсивностями света,
Радикальная полимеризация
171
прошедшего через пустой сосуд и сосуд с реакционной смесью.
Для определения 1а можно использовать также химический акти-
нометр. В основу работы такого актинометра положена химиче-
ская реакция, квантовый выход которой предварительно опреде-
ляют путем калибрования с помощью термоэлемента. Такой реак-
цией, в частности, является разложение щавелевой кислоты
(в присутствии солей уранила)
(СООН)2 СО2+СО + Н2О, (3.45)
квантовый выход которой составляет 0,5. Контроль за протекани-
ем реакции осуществляют, оттитровывая непрореагировавшую
щавелевую кислоту перманганатом. Интенсивность поглощенного
света легко можно вычислить, зная количество продуктов разло-
жения и продолжительность облучения. Применяется также другой
химический актинометр, в основу которого положен фотохими-"
ческий гидролиз хлоруксусной кислоты
С1СН2СООН + Н2О НОСН2СООН + НС1 (3.46)
с квантовым выходом 1,0.
3.46.2. Фотолитическая диссоциация инициаторов
В фотоинициировании можно также применять радикалы, обра-
зующиеся при термическом гомолизе. Как правило, в результате
фотолитического и термического гомолиза получаются одинаковые
радикалы. При фотохимическом методе в качестве инициаторов
можно использовать значительно большее число соединений, чем
при полимеризации в присутствии инициаторов, образующихся
при нагревании. Термический гомолиз соединений идет при доста-
точно высоких температурах и сопровождается образованием
самых различных радикалов, так как различные связи разрыва-
ются чисто статистически. Фотохимическое инициирование можно
использовать для полимеризации в присутствии карбонильных
соединений, например кетонов
бензоина
О О
II hv ||
R—С — R' R — C- + -R',
О ОН О ОН
И । hv И I
С6Н5-С-0НСвН5 С6Н5-С. + -СНСеН5,
(3.47)
(3.48)
а также галогеналкилов, металлоорганических соединений и сое-
динений других типов.
В этом случае скорость инициирования определяется как
7?и = 2Фе/0 [I], (3.49)
172
Глава 3
а скорость полимеризации тогда равна
7?р = /ср[М](^^Щ-)1/2, (3.50)
или
Др = [М] , (3.51)
или в зависимости от типа данной системы к ней применимо урав-
нение (3.43).
3.46.3. Фотосенсибилизаторы
К методу фотохимического инициирования относится также
применение фотосенсибилизаторов, вызывающих гомолиз мономе-
ра или инициатора, которые не переходят в возбужденное состоя-
ние при облучении светом обычно применяющейся частоты. Сенси-
билизатор Z возбуждается
Z + /iv->Z*, (3.52)
а затем передает свою энергию соединению С
Z* + C Z-J-C*, (3.53)
в результате последующего разложения которого в возбужденном
состоянии образуются радикалы
C*-^R.-|-R'- (3.54)
Следовательно, хотя С* нельзя получить непосредственным облу-
чением С светом с частотой v, некоторое поглощение энергии все-
таки происходит, так как при соответствующей частоте Z* способен
передавать энергию С, которая и поглощается С. Наиболее часто
в радикальных процессах в качестве сенсибилизаторов применяют
бензофенон. Для этой же цели используют различные красители,
например флуоресцеин, эозин и некоторые другие фотосенсибили-
заторы. Л
При фотосенсибилизированной] полимеризации скорость ини-
циирования и полимеризации можно характеризовать уравнения-
ми (3.49)—(3.51), подразумевая под [I] концентрацию сенсибили-
затора. Однако при низких концентрациях мономера или низких
квантовых выходах в таком процессе Ди может зависеть от [М],
и в этом случае для зависимости Др от [М] уравнения (3.50) и (3.51)
неприменимы.
3.46.4. Общие наблюдения
Как видно из уравнений, которые были рассмотрены выше,
в фотохимической полимеризации скорость процесса зависит от
корня квадратного из интенсивности света. Это было продемон-
Радикальная полимеризация
173
стрировано на многих мономерах с использованием различных
фотоинициирующих систем. Вместе с тем зависимость 7?р от [М]
определяется и типом фотоинициирования. На зависимость _ffp от
концентрации инициатора или фотосенсибилизатора также влия-
ют условия эксперимента.
Хотя практического значения фотохимическая полимеризация
почти не имеет, исследована она довольно подробно. Практическо-
го применения фотоинициирование не находит из-за доступности
инициаторов, разлагающихся при самых различных температурах.
Вместе с тем с теоретической точки зрения фотохимическая поли-
меризация представляет большой интерес. Для определения инди-
видуальных констант скорости кр и кй метод фотохимической
полимеризации незаменим (разд. 3.8).
3.4в. Инициирование с помощью ионизирующего
облучения
Благодаря тому что в последние годы радиоактивные источники
становятся все более и более доступными, появилась возможность
использования их для инициирования полимеризации. К радио-
активным частицам относятся электроны ([1-лучи), нейтроны, а-
частицы (Не2+), тогда как рентгеновские и у-лучи относятся
к электромагнитному излучению. Под действием ионизирующего
излучения в веществе идут более сложные процессы, чем под
действием света [17]. Качественно химические эффекты от раз-
личных типов облучения одинаковы, но в количественном отноше-
нии они отличаются друг от друга. Молекулярное возбуждение
с последующим образованием радикалов протекает так же, как
при фотолизе, но из-за более высоких энергий ионизирующего
излучения процесс этот сопровождается ионизацией соединения С
с выбросом электрона по схеме
С-[-Излучение -> С+ + е
(3.55)
Именно поэтому такое излучение и называется ионизирующим
излучением. При диссоциации катиона может затем образоваться
радикал
С+ —» А-+В+
(3.56)
Выброшенный электрон может присоединяться к катиону В+ с об-
разованием другого радикала
В+ + е В-
(3.57)
К образованию радикалов могут также приводить следующие
последовательные реакции, инициируемые захватом С выброшен-
174
Глава 3
ного электрона:
С4е->С-, (3.58]
С-—>В. + А-, (3.59]
А’ А- +е (3.60)
При радиолизе олефинов образуются катионы, анионы и свобод-
ные радикалы по вышеприведенным схемам. Такие частицы могут
инициировать полимеризацию. От условий реакции зависит,
инициируется полимеризация радикалами, катионами или анио-
нами. В большинстве случаев полимеризация под действием
ионизирующего излучения является радикальным процессом.
Ионы обладают устойчивостью, необходимой для инициирования
полимеризации, только при низких температурах. При комнатной
или более высоких температурах они, как правило, нестабильны
и диссоциируют с образованием радикалов. Радиолитическая
радикальная полимеризация имеет те же самые кинетические
характеристики, что и фотолитический процесс. Ее можно про-
водить также с использованием инициаторов или других соедине-
ний, способных разлагаться при облучении.
3.4г. Чисто термическое инициирование
Многие мономеры самопроизвольно полимеризуются при на-
гревании без добавок специального катализатора. При исследо-
вании ряда таких реакций было показано, что в результате нагре-
вания мономеров образуются радикалы. В большинстве случаев
такие реакции полимеризации инициируются при термическом
разложении примесей, присутствующих в мономере. Тщательно
очищенные мономеры не полимеризуются в результате само-
инициируемой чисто термической реакции. Твердо установлено
лишь, что стирол и метилметакрилат самополимеризуются при
нагревании. По-видимому, аналогично должны вести себя и заме-
щенные стирола и метилметакрилата. Скорость термической
самоинициируемой полимеризации намного ниже, чем соответ-
ствующих реакций полимеризации, проводящихся в присутствии
таких инициаторов, как АИБН, но в определенных условиях
может быть все-таки заметной. Так, скорость термической поли-
меризации стирола при 60 °C не превышает 0,1 % в час, а при
127 °C — 14% в час, тогда как скорость полимеризации метил-
метакрилата в этих же случаях составляет лишь 1 % от скорости
для стирола [4]. Скорость чисто термической полимеризации необ-
ходимо учитывать при исследовании полимеризации, проводя-
щейся при температурах, при которых вклад самоинициирования
достаточно велик.
Исследование термической полимеризации стирола не дало
четкого ответа на вопрос о механизме инициирования. Скорость
Р адикальная полимеризация
17'
полимеризации пропорциональна [Стирол]2 [18]. Это значит, чтс
инициирование имеет второй порядок по стиролу, так как
в соответствии с уравнением (3.24) изменяется пропорциональнс
[М]7?и2- Предл о;кено бимолекулярное инициирование с образо-
ванием бирадикалов
2С6Н5 —СН = СН2 —> С6Н5 —СНСН2СН2СН —СвН5 (3.61)
Однако с этим предположением трудно согласиться, так как в реак-
ции с бирадикалами, по-видимому, должны получаться только
низкомолекулярные соединения вследствие возможности цикли-
зации по радикальным концам. Это подтверждается рядом безу-
спешных попыток инициирования полимеризации бирадикалами,
образующимися при фотолитическом гомолизе циклических пере-
кисей, дисульфидов, а также азосоединений [16]. Более вероятно,
что бимолекулярное инициирование идет по схеме Уоллинга [4]
2СвН5 — СН = СН2 СвН5 —СН —СН3 + СН2 = ССвН5 (3.62).
Картина инициирования при термической полимеризации ста-
новится еще более запутанной, если принять во внимание работу
[19], согласно которой при проведении полимеризации в растворе
бромбензола инициирование является реакцией третьего порядка
по мономеру. Выдвинуто предположение о тримолекулярном
инициировании
СН3
ЗСвН5—СН = СН2 —> СвН5—СН —СН3 + СвН5—С = СН — СНСвН5, (3.63}
но оно кажется маловероятным вследствие того, что тримолекуляр-
ные реакции в растворе вообще встречаются весьма редко. Более
обоснованной и реальной выглядит следующая схема реакций
при инициировании:
2СвН5—СН = СН2 —» СвН5—СНСН2СН2СН —С6Н5, (3.64}
С6Н5 —СНСН2СН2СН —СвН5 + свн5 —сн = сн2 —>
—> СвН5—СН —СНз + СвН5 —СНСН2СН = СН —СвН5 (3.65}
3.4д. Инициирование окислительно-
восстановительными системами
В результате многих окислительно-восстановительных реакций
генерируются радикалы, которые можно использовать для ини-
циирования полимеризации. Такой тип инициирования называется
окислительно-восстановительным инициированием, окислительно-
восстановительным катализом или окислителъно-восстановителъ-
176
Глава 3
ной активацией. Главным преимуществом инициирования такого
типа является то, что радикалы образуются с достаточной скоро-
стью при весьма низких температурах (0—50 °C). Это позволяет
варьировать температуру полимеризации в более широких преде-
лах, чем при полимеризации с термическим распадом инициаторов.
Для окислительно-восстановительного инициирования могут быть
использованы различные окислительно-восстановительные реак-
ции с участием как неорганических, так и органических соеди-
нений.
Одной из наиболее известных и хорошо изученных окислитель-
но-восстановительных систем, производящих радикалы, является
реактив Фентона, представляющий собой смесь перекиси водорода
и иона двухвалентного железа
H2O2 + Fe2+ —» НО- + НО. -j-Fe3+
(3.66а)
Ион двухвалентного железа ускоряет разложение и многих других
соединений, в том^числе и органических перекисей различного
типа
Fe2+
ROOR----> RO- + RO-
(3.666)
Fe2+
ROOH----> HO- + RO.
(3.66b)
О [О
II Fe2+ ||
ROOCR' ----> R'CO- + RO.
(3.66г)
Во многих случаях вместо иона двухвалентного железа могут быть
использованы другие восстановители, например Cr2+, V2+, Ti3+,
Со2+ и Сп+. Большинство окислительно-восстановительных систем
представляют собой водные или эмульсионные системы.
Инициирование окислительно-восстановительными системами
с участием перекисей ацилов можно проводить в органических
средах, используя амины в качестве восстановителей. Представля-
ет интерес система, состоящая из перекиси бензоила и NjN-ди-
этиланилина, преимуществом которой по сравнению с обычным
термическим распадом перекиси является значительно большая
скорость образования радикалов. Так, константа скорости гомо-
литического распада кг чистой перекиси бензоила в реакции поли-
меризации стирола составляет 1,33 ПО-4 с-1 при 90 °C, тогда как
в случае окислительно-восстановительной системы, состоящей и;
перекиси бензоила и NjN-диэтиланилина она составляет
1,25>10-2 л/(моль-с) при 60 °C и 2,29ПО"3 л/(моль-с) при 30 °C [21].
Генерирование радикалов в этой окислительно-восстановитель-
ной системе протекает, по-видимому, через первоначальное ион-
Радикальная полимеризация
177
пое замещение анилина в перекиси
О
II
С13Н5—N — R -|- — С — О
I
R
О
о—с—свн5
R О О
I II II
С0115 —N+ —О —С —С6Н5 С6Н5-С-О- -»
I
R -1
С0Н5 — N — R +
О О
II II
С0Н5-С-О. + свн5-с-о-
(З.ббд)
R
К числу других исследованных окислительно-восстановитель-
ных реакций относятся:
1. Восстановление персульфатного иона ионами Fe, Ag, суль-
фита или тиосульфата, например
-O3S —О-О —SO;+Fe2+ —> Fe3++ SO^+SOv, (3.67a)
— 03S —О —0 —SO7+S2O|--> SO^+SO^-- + -S2OJ (3.676)
2. Использование таких восстановителей, как HSO", S0|“,
320з- и S2O2_ в сочетании с окислителями, например Cu2+, Fe3+,
СЮ; и Н2О2.
3. Окисление тиомочевины ионами BrO;, MnO4, S2O2- и Fe3+,
а также Н2О2 [22].
4. Окисление различных органических соединений ионами
Се4+ или V5+ [23—25], например
R —СН2—ОН + Се4+ —» Се3+ + Н+ + R — СН — ОН . (3.68)
Кинетика полимеризации, инициируемой окислительно-восста-
новительными системами, как правило, бывает двух типов. Боль-
шинство реакций такого типа по своим закономерностям не отли-
чается от других реакций полимеризации, и вся разница состоит
в источнике радикалов на стадии инициирования. Скорости иници-
ирования и полимеризации в таких процессах описываются урав-
нениями, близкими тем, которые были представлены выше. Так,
при инициировании окислительно-восстановительной системой
перекись бензоила — диалкиланилин справедливы следующие
кинетические уравнения:
7?и = [Перекись] [Амин], (3.69)
7?р = [М] (^[Перекись] [Амин] р . (3,70)
Все отличие последних уравнений от рассмотренных выше для
других способов инициирования заключается в отсутствии в них
178
Глава 3
коэффициента 2 в уравнении скорости инициирования. Это объяс-
няется тем, что одна молекула перекиси бензоила дает только
один радикал (СоЩСОг-), который может инициировать полимери-
зацию. Катион-радикал амина, также образующийся при такой
реакции, не инициирует полимеризацию, что было доказано элемен-
тарным анализом полимера, показавшим отсутствие азота. Катион-
радикал амина разрушается в результате какой-то побочной реак-
ции. Уравнения (3.69) и (3.70) описывают также многие другие
системы, характеризующиеся зависимостью первого порядка по
окислителю и восстановителю. При инициировании согласно
уравнениям (3.66а) — (3.66г) образуется только один радикал на
каждую молекулу катализатора.
Некоторые реакции полимеризации, инициируемые окислитель-
но-восстановительными системами, отличаются по механизму
обрыва от других полимеризаций, причем отличие заключается
в том, что обрыв идет не в результате бимолекулярной реакции,
а вследствие взаимодействия растущих радикалов с компонентами
окислительно-восстановительной системы. Это существенным
образом меняет кинетику процесса. Так, при ппциировании систе-
мой спирт — Се4+ [уравнение (3.68)1 обрыв наступает вследствие
следующей реакции:
Мп«+Се41-—> Се3+ + Н+-|-Мертвый полимер (3-71)
Растущий радикал теряет водород с образованием полимера, имею-
щего на конце ненасыщенную двойную связь. Скорости иницииро-
вания и обрыва определяются как
7?и = /сг[Се4+] [Спирт], (3.72а)
Во = кЛ [Се4+] [М • ] (3.726)
Вводя допущение о стационарном состоянии (т. е. 7?п = 7?0), мож-
но получить следующее выражение для концентрации радикалов:
[М-] = fer[Спирт] . (3 73)
/с0
Подставляя это выражение в уравнение (3.21), получим следующее
уравнение скорости полимеризации:
fcrfcp [М][Спирт]
ЯР =
(3-74)
к0
3.4е. Эффективность инициатора
3.4е.1. Определение f
Если провести материальный баланс по количеству инициато-
ра, который распадается в процессе полимеризации, и сравнить
его с количеством, участвующим в инициировании, то окажется,
Радикальная полимеризация
179
что инициатор расходуется весьма неэффективно. Такой непроиз-
водительный расход инициатора вызван следующими двумя
реакциями.
Первая из них заключается в индуцируемом разложении ини-
циатора под влиянием растущих радикалов, например
0 0 О О
II II II II
мп • + С6Н5 — СО — ОС — С6Н5 —» М п — ОС — С6Н5 + С6Н5 — СО • (3.75)
Эта реакция называется передачей цепи на инициатор и рассмат-
ривается ниже (разд. 3.66). В результате такого распада инициа-
тора концентрация радикалов не изменяется, так как вновь обра-
зующийся радикал (С6Н5СОО •) инициирует новую полимерную
цепь. Однако в результате такой реакции расходуется лишний
инициатор. Молекула инициатора разлагается, а увеличения
количества превращенного в полимер мономера не происходит.
Второй реакцией является побочная реакция радикалов, обра-
зовавшихся на первой стадии процесса инициирования. Некоторые
первичные радикалы, образовавшиеся на первой стадии распада,
например при реакциях
О О
С6Н5СО - ОС — С6Н5 —» 2С6Н5СОО •, (3.76а)
Ее2+ + С6Н5С(СНз)2ООН-» Fe3++HO- + C6H5C(CH3)2O. (.3.766)
вступают в реакции с образованием нейтральных молекул вместо
того, чтобы инициировать полимеризацию. Именно эту реакцию
имеют в виду, когда обсуждают эффективность инициатора /.
Эффективность инициатора определяется как доля радикалов из
образовавшихся на первой стадии разложения инициатора, кото-
рые участвуют в инициировании полимеризации. Эффективность
инициатора не включает расхода его в результате индуцирован-
ного разложения. Это следует учитывать при рассмотрении вели-
чин /, приведенных в литературе. Очень часто при расчетах /
индуцированным разложением пренебрегают и не вводят на него
соответствующую поправку. Такие значения / следует рассматри-
вать как кажущиеся или практические, так как они отвечают
суммарной эффективности инициирования от начальной концен-
трации инициатора. Однако лучше всего для количественной
обработки индуцированного разложения инициатора восполь-
зоваться методом, рассмотренным в разделе 3.66.3.
3.4е.2. Механизм инициирования при f <Z1
Большинство применяемых инициаторов имеет эффективность
порядка 0,3—0,8. Для того чтобы понять, почему эффективность
инициатора не достигает единицы, рассмотрим следующие реак-
18
Глава 3
ции, которые протекают при полимеризации в присутствии пере-
киси бензоила
С6Н5СОО - ООССвН5 (2СвН5СОО •),
(3.77)
(2С6Н5СОО ) _» (С6Н5СООС6Н5 + СО2), (3.78)
(2С6Н5СОО.)+М—»СвН5СОО. + СвН5СООМ-, (3.79)
(2С6Н5СОО-) —> 2СвН5СОО-, (3.80)
С6Н5СОО.-|-М—»СвН5СООМ-, (3.81)
С6Н5СОО- —>С6Н5- +СО2, (3.82)
CgHj- -J-М CjHsM-,
(3.83)
С6Н5- +СвН5СОО- -» С6Н5СООС6Н5, (3.84)
2С6Н5- —> СвН5 —С6Н5
(3.85)
Скобки ставятся для того, чтобы показать, что растворитель захва-
тывает радикалы и удерживает их в течение некоторого проме-
жутка времени, как в «клетке», прежде чем они диффундируют из
него. Уравнение (3.77) отражает первую стадию разложения ини-
циатора на два радикала, которые удерживаются в клетке раство-
рителя. В клетке растворителя радикалы могут рекомбинировать
[процесс, обратный уравнению (3.77)1, реагировать друг с другом
[уравнение (3.78)1, с мономером [уравнение (3.79)1 или диффунди-
ровать из нее [уравнение (3.80)]. Кроме того вне клетки раствори-
теля радикалы могут реагировать с мономером [уравнение (3.81)]
или в результате разложения согласно уравнению (3.82) давать
радикал, который может вступать в различные реакции [уравне-
ния (3.83)—(3.85)1.
На эффективность инициатора рекомбинация первичных ради-
калов [уравнение (3.77)1 не оказывает никакого влияния. Иници-
ирование полимеризации описывается уравнениями (3.79), (3.81)
и (3.83). Эффективность инициатора уменьшается в результате
протекания реакций (3.78), (3.84) и (3.85), так как продукты
таких реакций — устойчивые соединения, которые не могут обра-
зовывать радикалы. При этом наибольшее влияние на уменьшение
/ оказывает разложение радикала внутри клетки растворителя
[уравнение (3.78)1. Очень наглядным в этом смысле является рас-
смотрение временных зависимостей различных реакций [4]. Сред-
нее время жизни соседних радикалов составляет 10"10—10-9 с.
Поскольку константа скорости радикал-радикального взаимодей-
ствия равна 107 л/(моль-с) и выше, а концентрация радикалов
в клетке растворителя достигает 10 М, то вероятность протекания
реакции (3.78) достаточно велика. Реакция (3.79) не может кон-
курировать с реакцией (3.78), так как реакции присоединения
радикалов идут со значительно более низкой скоростью [констан-
та скорости таких реакций 10—105 л/(моль-с)1.
Радикальная полимеризация
1 1
Понижение эффективности инициаторов в результате протека-
ния реакций, аналогичных уравнению (3.78),— явление достаточно
общее почти для всех инициаторов. Так, например, ацетокси-ра-
дикалы, образующиеся при разложении перекиси ацетила, декар-
боксилируются быстрее, чем бензоатные радикалы, и при взаимо-
действии радикалов внутри клетки растворителя получаются
устойчивые продукты, неспособные к образованию радикалов
СН3СОО - ООССНз —» (2СН3СОО •) —» (СН3СООСН3 + СО2) (3.86а)
(СН3СН3 + 2СО2) (3.866)
щреш-Бутоксильные радикалы перекиси ди-шреш-бутила в ре-
зультате внутримолекулярного отщепления выделяют метильные
радикалы, а затем последние димеризуются; аналогично ведут
себя кумилокси-радикалы перекиси дикумила или гидроперекиси
кумола [5]
СН3 О
СН3 — С-О. —»СН3 — й-СНз + СН3. (3.87)
I *
сн3 сн3сн3
Гомолиз АИБН идет как согласованный процесс с одновременным
разрывом двух связей С — N, выделением молекулы азота и обра-
зованием 2-циано-2-пропильных радикалов. При взаимодействии
таких радикалов друг с другом получаются тетраметилсукциноди-
нитрил и диметил-Й-(2-циано-2-изопропил)кетенимин
CN CN
I I
(СН3)2С — N = N — С(СН3)2 —»
CN CN CN
। L ।
-> [2(СН3)2С. -|-N2] -» [(CH3)2C-C(CH3)2 + N2] (3.88а)
| CN
I
[(CH3)2C = C = N — C(CH3)2 + N2] (3.886)
Эффективность инициатора обычно не зависит от концентрации
мономера и от протекания реакций, выраженных уравнениями
(3.82)—(3.85). К величине /, меньшей 1, приводят в основном
реакции, идущие внутри клетки растворителя. Интересно заметить,
что / АИБН при полимеризации стирола уменьшается по мере
увеличения вязкости реакционной среды [26]. По-видимому, повы-
шение вязкости способствует увеличению времени жизни радика-
лов в клетке, что и обусловливает повышение роли реакций (3.88а)
и (3.886).
Как только радикал вырывается из клетки растворителя, реак-
ция его с мономером начинает превалировать над всеми другими
1 2
Глава 3
реакциями. Если даже реакция (3.82) имеет место, то непосред-
ственно за ней будет протекать реакция (3.83), а не взаимодей-
ствие радикалов между собой [уравнения (3.84) и (3.85)]. Это
обусловлено тем, что концентрация мономера, составляющая
0,1—10 М, намного превышает концентрацию радикалов (10-7—
10-9 М). Однако при низких концентрациях мономера иногда
можно наблюдать зависимость / от концентрации мономера
Р и с. 3.3. Влияние концентрации стирола
па эффективность инициатора АИБН [27].
(рис. 3.3). Эффективность инициатора очень быстро повышается
с ростом [М], но также довольно быстро достигает предела.
Эффективность инициатора зависит также от природы раство-
рителя, в котором ведут полимеризацию. Так, f АИБН [28] и пере-
киси бензоила [29] как инициаторов ниже в четыреххлористом
углероде, чем в ароматических углеводородах. Это связано с тем,
что, кроме взаимодействия с мономером, радикалы могут реагиро-
вать с растворителем. Более подробно этот вопрос будет рассмот-
рен в разд. З.бв. Эффективность инициатора зависит также от
химического строения мономера. Так, например, / АИБН при
полимеризации метилметакрилата, винилацетата, стирола, винил-
хлорида и акрилонитрила возрастает с 0,6 до 1,0 [30]. Этот поря-
док отвечает относительным скоростям присоединения радикалов
к различным мономерам.
3.4е.3. Экспехзименталъные методы оценки f
Для экспериментальной оценки / существуют различные мето-
ды. Один из них основан на определении и сравнении параметров
процесса распада инициатора и образования полимера. Распад
инициатора лучше всего исследовать в процессе полимеризации
мономера. Независимое исследование распада инициатора в отсут-
ствие мономера может привести к ошибочным результатам,
поскольку скорость разложения чистого инициатора может отли-
чаться от скорости его распада в присутствии мономера. (Ср. моле-
кулярно-индуцированный гомолиз гидроперекиси mpezn-бутила,
рассмотренный в разд. 3.4а.4.) За распадом АИБН сравнительно
Радикальная полимеризация
просто проследить по выделению азота. Измерив среднечисловой
молекулярный вес полимера, можно определить / путем сопостав-
ления количества образовавшихся радикалов и числа-полимер-
ных молекул. Необходимо только знать, как идет обрыв цепи,
поскольку при рекомбинации растущих радикалов два фрагмента
инициатора расходуются на одну макромолекулу, тогда как при
диспропорционировании — только один такой фрагмент. При
расчете числа образовавшихся радикалов следует учитывать так-
же возможность индуцированного распада, который идет
в заметной степени при применении в полимеризации многих
перекисей, но практически не играет никакой роли при исполь-
зовании в качестве инициаторов азонитрилов.
Другим методом определения эффективности инициатора явля-
ется анализ полимера на присутствие в нем осколков инициатора.
При проведении такого анализа возникает ряд трудностей, связан-
ных с низкой концентрацией определяемых концевых групп,
содержащих остатки инициатора. Так, в полимере молекулярного
веса 50 000 концевые группы составляют только 0,1 % веса поли-
мера. Очень хорошие результаты дает применение меченых инициа-
торов, например АИБН, содеращего 14С [26], и персульфата калия
с 35S, которые легко обнаружить в полимере даже в очень неболь-
ших количествах.
Третий метод основан на использовании некоторых ингибито-
ров, которые реагируют стехиометрически с радикалами, прекра-
щая их рост. Часто применяют такие ингибиторы, как стабильный
радикал дифенилппкрилгидразил (ДФПГ), получающийся окис-
лением дифенилпикрилгидразипа
Обрыв под действием ДФПГ протекает, по-видимому, следующим
образом:
(3.90)
1 4
Глава 3
За реакцией можно весьма просто следить спектрофотометрически,
так как радикал ДФПГ темно-фиолетового цвета, а продукт реак-
ции светло-желтый или даже бесцветный. Для определения числа
радикалов можно также применять треххлористое железо.
R-+FeCl3B-»JRCl + FeCl2 (3.91)
В качестве ингибиторов применяют также дурохинон и бензохи-
нон. Большой недостаток последнего метода в том, что реакция
ингибитора и радикала часто идет неколичественно. Радикал
ДФПГ, например, является очень эффективным ингибитором во
многих системах. В его присутствии прекращается полимериза-
ция винилацетата [31] и стирола даже при концентрации ингиби-
тора ниже 10“4 М. Однако так ДФПГ ведет себя не во всех
случаях.
Особый интерес представляет метод Тобольского [32—34] (так
называемый метод мертвых концов), позволяющий одновременно
определять кг и /. Метод относится к реакциям полимеризации,
при которых концентрация инициатора снижается до такого
низкого уровня, при котором время полужизни растущих поли-
мерных цепей становится соизмеримым с временем полужизни
инициатора. При этом полимеризация останавливается, в резуль-
тате чего наблюдается некоторая предельная величина степени
превращения мономера в полимер при бесконечно большой
продолжительности реакции. Рассмотрим такую реакцию поли-
меризации, которая инициируется в результате термического раз-
ложения инициатора. Скорость исчезновения инициатора равна
-2=£Ш. = М1], (3.92)
В результате интегрирования этого уравнения имеем
[1] = [1]ое-Ч (3.93)
где [1]0 — концентрация инициатора в момент начала полиме-
ризации. Подставляя уравнение (3.93) в уравнение (3.28), после
соответствующей перестановки получаем
(3-94)
[1V1] \ К0Кр /
а после его интегрирования
- 1п ЖГ = - In (1 - р) = 2fcp ( 4^ )1/2 (1 (3.95)
|М]0 ' кокг '
где р — степень превращения мономера в полимер, равная ([М]0 —
— [М])/[М]0. При больших временах реакции (<-> оо) величины
[М] и р достигают предельных значений [М] □= ирх соответственно,
а уравнение (3.95) принимает вид
-ln-g^= -1п(1-Роо) = 2/ср (^тТ2. (3-96)
L-LVlJo \ KqK^ /
Радикальная полимеризация
1 5
После деления уравнения (3.95) на уравнение (3.96), перестановки
и логарифмирования обеих частей получим
1пГ1--| = 2=м- (3 97у
L 1П(1 — Poo) J 2 ' >
Величину kr затем легко найти по наклону кривой зависимости
In ^1 —от продолжительности реакции. Ларис. 3.4 при-
ведена такая зависимость для полимеризации изопрена при раз-
личных температурах в присутствии АИБН как инициатора [34].
Рис. 3.4. Определение эффективности инициатора по методу Тобольского
для полимеризации изопрена в присутствии АИБН [34].
Величину / можно рассчитать по уравнению (3.28) или (3.96), если
известно отношение kvlk4z для мономера.
В результате математической обработки стало возможно исполь-
зование этого метода для различных систем [35, 36], и в частности
для реакций полимеризации, которые доходят до конца, для
реакций полимеризации, в которых имеет место в заметной степени
индуцированный распад инициатора, а также для реакций с бимо-
лекулярным распадом инициатора (например, в системе перекись
бензоила — диалкиланилин).
3.5. Молекулярный вес
(3.5а. Длина кинетической цепи
Длину кинетической цепи v в радикальной полимеризации
определяют как среднее число молекул мономера, приходящихся
на один образовавшийся активный центр. Эту величину, очевидно,
можно найти из отношения скорости полимеризации и скорости
186
Глава 3
инициирования или скорости обрыва, так как последние две
скорости равны между собой
Яр Яр
Из уравнений (3.21), (3.22) и (3.98) получим
fcp [М]
v ~ 2*0 [М.]-
Затем из уравнений (3.99) и (3.21), находим
[Мр
2/с0Яр
Для термической полимеризации уравнение (3.100) после подста-
новки в него уравнения (3.28) принимает следующий вид:
кР [М]
V~ 2(/Mo[IJ)1/2 '
(3.98)
(3.99)
(3.100)
(3.101)
Уравнения (3.100) и (3.101) — очень важные характеристики
радикальной полимеризации. Длина кинетической цепи обратно
пропорциональна концентрации радикалов или скорости полиме-
ризации. Увеличение концентрации радикалов и скорости ради-
кальной полимеризации приводит к образованию макромолекул
меньшего размера. Длина кинетической цепи при постоянной
температуре полимеризации определяется природой мономера
и не зависит от способа инициирования. Таким образом, для
любого мономера при одинаковых [М •] или Лр длина кинетиче-
ской цепи не будет зависеть от того, инициируется полимеризация
термическим, окислительно-восстановительным или фотохими-
ческим методом, а также от природы инициатора.
3.56. Способ обрыва
Среднечисловая ^степень полимеризации Хп определяется как
среднее число молекул мономера, содержащихся в молекуле поли-
мера, и зависит от длины кинетической цепи. Если обрыв расту-
щих цепей наступает в результате рекомбинации, то конечный
мертвый полимер состоит из двух длин кинетических цепей
Xn = 2v. (3.102а)
При обрыве в результате диспропорционирования длина кинетиче-
ской цепи эквивалентна среднечисловой степени полимеризации
Xn=-v.
(3.1026)
Радикальная полимеризация
187
Среднечисловой молекулярный вес полимера равен
Мп = М0Хп, (3.103)
где Мо — молекулярный вес мономера.
Механизм обрыва экспериментально определяют путем оценки
числа осколков инициатора па молекулу полимера. Для этого
необходимо измерить молекулярный вес полимера и общее число
осколков инициатора в нем. Хотя не для всех полимеров можно
провести такие исследования, можно сказать, что обрыв боль-
шинства полимерных радикалов наступает в результате реакции
рекомбинации. Исследования, выполненные на небольших али-
фатических радикалах, например этильном, изопропильном
и щрет-бутильном, показали, что реакция диспропорционирова-
ния начинает играть большую роль в случае разветвленных
радикалов при высоких температурах полимеризации. Реакция
рекомбинации разветвленных радикалов стерически затруднена.
Протеканию реакции диспропорционирования благоприятствует
также увеличение количества Р-водородных атомов, участвующих
в реакции передачи цепи. Так, если при полимеризации стирола
обрыв идет только в результате рекомбинации, то для полимериза-
ции метилметакрилата характерен обрыв как путем рекомбинации,
так и вследствие диспропорционирования. Степень диспропорцио-
нирования в последнем случае возрастает с температурой, потому
что энергия активации диспропорционирования примерно на
5 ккал/моль (21-103 Дж/моль) больше энергии активации реком-
бинации. При проведении реакции при 60 °C 60% реакций обрыва
протекает за счет диспропорционирования [37]. Важную роль
диспропорционирование играет при полимеризации винилацета-
та [38].
3.6*. Реакция передачи цепи
3.6а. Влияние реакций передачи цепи
Для многих полимеризационных систем характерно, что реаль-
ный молекулярный вес полимера оказывается более низким, чем
тот, который предсказан на основании реакций обрыва вследствие
рекомбинации или диспропорционирования. Этот эффект обуслов-
лен прежде всего протеканием обрыва вследствие реакции переноса
водорода или другого атома или группы атомов на растущий
полимер от различных соединений, находящихся в реакционной
системе: мономера, инициатора или растворителя. Такие реакции
замещения радикалов называются реакциями передачи цепи; они
могут быть представлены следующим образом:
Mn. -LXA--^>Mn'-X + A., (3.104)
188
Глава 3
где ХА может быть мономер, инициатор, растворитель или другое
вещество, а X — атом, который переносится на полимер. Реакция
передачи цепи упоминалась ранее (разд. 3.4е.1) в связи с рассмот-
рением индуцированного разложения инициатора.
Скорость реакции передачи цепи равна
•Япер = ^пер [М • ] [ХА], (3.105)
где /спер — константа скорости реакции передачи цепи. В резуль-
тате реакции передачи цепи образуется новый радикал А-, кото-
рый повторно инициирует полимеризацию по схеме
Д.+М-^М.. (3.106)
Реакция передачи цепи приводит к уменьшению размера цепи
полимера. Влияние передачи цепи на скорость полимеризации
зависит от того, насколько скорость реинициирования близка
скорости роста начального растущего радикала. В табл. 3.2 пред-
ставлены основные возможные примеры такого влияния. Когда
Таблица 3.2
Влияние передачи цепи на величины Rp и Хп
Пример Относительные константы ско- рости передачи цепи, роста и реинициирования Изменение Rp Изменение Хп
1 ^р » knep, ^р Не изменяется Уменьшается
2 kp ^пер, ка ^р Не изменяется Сильно уменьши-
ется
3 ^р » ^пер> < ^р Уменьшается Уменьшается
4 ^р ^Пер, ка < кр Сильно уменына- Сильно уменьши-
ется ется
реинициирование идет быстро (примеры 1 и 2), скорость полиме-
ризации не изменяется. В единицу времени расходуется одно и то
же число молекул мономера, приводя к образованию большего
числа молекул полимера меньшего размера. Относительное умень-
шение Хп зависит от величины константы скорости реакции
передачи цепи. Когда константа скорости реакции передачи цепи
намного превышает константу скорости роста (пример 2),
образуется очень низкомолекулярный полимер (Xn х 1—5), назы-
ваемый теломером. Когда реинициирование идет медленнее, чем
рост цепи (пример 3 и 4), наблюдается уменьшение Rp и Хп. Сте-
пень уменьшения Rp определяется отношением величин кр и кТ[рр.
Рассмотрим подробнее пример 1, а примеры 3 и 4 обсуждаются
в разд. 3.7 (пример 2 — теломеризация — выходит за рамки
предмета рассмотрения этой книги).
Радикальная полимеризация
189
В реакции полимеризации с передачей цепи, описываемой при-
мером 1, длина кинетической цепи остается неизменной, так как
число полимерных молекул, образующихся в расчете на одну
длину кинетической цепи, изменяется. Среднечисловая степень
полимеризации в этом случае уже не равна v или 2v, как при
диспропорционировании или рекомбинации соответственно. Реак-
ция передачи цепи играет большую роль, так как от нее сильно
зависит молекулярный вес полимера, который может изменяться
в нежелаемом направлении. Вместе с тем с помощью регулируемой
реакции передачи цепи можно получить полимер заданного моле-
кулярного веса.
Степень полимеризации тогда правильно определять как отно-
шение скорости полимеризации к сумме всех реакций обрыва
(т. е. обычных реакций обрыва и всех реакций передачи цепи).
В общем случае для полимеризации с термическим разложением
инициатора и обрывом вследствие рекомбинации и передачи цепи
на мономер, инициатор и некоторое соединение S (называемое
агентом передачи цепи) среднечисловая степень полимеризации
определяется из уравнения (3.105) и равна
Лр
Хп ~ (ад + fcnep. м[М-] [МЦ-*пер. s[M-l [S] +fcnep. I [М-] [I] * (3’107)
Первый член в знаменателе относится к реакции рекомбинации,
остальные три отражают обрыв вследствие передачи цепи на моно-
мер, агент передачи цепи и инициатор соответственно. Константа
передачи цепи С для данного вещества определяется как отноше-
ние константы скорости передачи цепи /спер растущего радикала
на это вещество к константе скорости роста радикала кр. Тогда
константы скорости передачи на мономер, агент передачи цепи
и инициатор равны соответственно
fcnep'M’. Г fcnep'S . Г
СМ — —Г- ь, bs — —т---, Ci = —т--• (3.108)
Л-р /Ср
Из уравнения (3.21), (3.27), (3.28), (3.107), (3.108) получим
уравнение
fl k0Rn rqi k0Ri,'
K=w-+C“;+Csw+C-S^- <ЗЛ09>
которое показывает количественную зависимость между различ-
ными реакциями передачи цепи и среднечисловой степенью поли-
меризации. Для определения констант передачи цепи могут быть
использованы различные методы.
190
Глава 3
3.66. Передача цепи на мономер и инициатор
3.66.1. Определение См и С\
Представляют интерес два частных случая уравнения (3.109)
Когда агент передачи цепи отсутствует, член [S] исчезает и урав-
нение (3.109) принимает вид
хп ^2р[м]2 м ' 1 к^кг[М]з •
Используя уравнение (3.28), уравнение (3.110а) можно предста-
вить в следующем виде:
1 'k°Rp , r . r II]
хп ^[.М]2 ' м-' Чмг
Уравнение (3.110а) представляет собой квадратичную зависи-
мость 1 /Хп от 7?р; соответствующие кривые для полимеризации
(3.110а)
(3.1106)
Рис. 3.5. Зависимость степени поли-
меризации стирола от скорости поли-
меризации. Показано влияние передачи
цепи на инициатор при 60 °C [39].
Инициатор: 1 — гидроперекись трет-бутила.
2 — гидроперекись кумола; з — перекись бен-
зоила; 4 — АИБН.
стирола в присутствии различных инициаторов приведены па
рис. 3.5 [39]. Начальный участок каждой кривой прямолинеен^
но при более высоких концентрациях инициатора, и поэтому при
более высоких значениях _Яр, кривая отклоняется от линейной
зависимости, так как возрастает доля реакций передачи цепи.
Отрезок, отсекаемый линейной частью кривых, дает величину См-
Наклон линейной части определяется отношением krJkp [М]2, из
которого можно определить важное отношение koik?, так как
концентрация мономера известна. Для систем, в которых переда-
чей цепи па инициатор можно пренебречь (например, при поли-
меризации в присутствии АИБН), зависимость 1/Хп от Нр является
линейной во всем интервале Др.
Существует несколько методов определения Ср После некото-
рых преобразований и деления на T?D уравнение (3.110а) принимает
Радикальная полимеризация
191
вид
I____„ I 1 _ ftp . CIfcoflP
CMj - fc2[Mj2 + к*/ММ]3 •
(3.111)
Зависимость левой части уравнения (3.111) от Rp представляет
прямую с наклоном, равным (Сгк0/кр/кг [М]3). Тогда константу
передачи цепи на инициатор можно определить из наклона этой
Р п с. 3.6. Определение констант пе-
редачи цепи на инициатор при поли-
меризации стирола в растворе бен-
зола при 70 °C в присутствии гидро-
перекиси mpem-бутила [13] в каче-
стве инициатора.
линии, так как другие величины известны или могут быть рас-
считаны через другие известные величины с помощью уравнения
(3.28). Если передачей цепи на мономер можно пренебречь, то
уравнение (3.1106) преобразуется к виду
[ХГ- ^[м]2]=СгТмТ’ (ЗЛ12)
Зависимость левой части этого уравнения от [1]/[М] представляет
прямую линию, наклон которой равен Сг. На рис. 3.6 представ-
лена соответствующая кривая для полимеризации стирола в при-
сутствии гидроперекиси трет-бутила [13].
З.вб.2. Константы передачи цепи на мономер
При полимеризации стирола в присутствии перекиси бензоила
величины Сы и С1; определенные с помощью рассмотренных выше
методов, составляют 0,00006 и 0,055 соответственно [40]. Вклад
передачи цепи на мономер в таком процессе незначителен. Вместе
с тем константа передачи цепи на перекись бензоила достигает
значительной величины и возрастает с увеличением концентрации
192
Глава 3
инициатора. Все эти эффекты отражены на рис. 3.7, из которого
хорошо виден также вклад различных реакций передачи цепи
в процессе полимеризации. На кривой 1 представлено общее число
полимерных молекул в расчете на 105 мономерных (стирольных)
звеньев. По разнице между соседними кривыми можно судить
Рис. 3.7. Роль различных реак-
ций обрыва в полимеризации сти-
рола в присутствии перекиси бен-
зоила при 60 °C [40].
1 — инициирование перекисью бензои-
ла; 2 — передача цепи на перекись
бензоила; з — передача цепи на мо-
номер.
о том, какое число полимерных молекул обрывается вследствие
протекания реакции рекомбинации, передачи цепи на перекись
бензоила и передачи цепи на стирол.
Для большинства мономеров константы передачи цепи на
мономер невелики (табл. 3.3) [41]. Величина См для многих моно-
Таблица 3.3
Константы передачи цепи на мономер [41]
Мономер
См-101 при 60 °C
Акриламид
Акрилонитрил
Метилакрилат
Метилметакрилат
Стирол
Винилацетат
Винилхлорид
0,6
0,26-0,3
0,036-0,325
0,07—0,18
0,6-1,1
1,75—2,8
6,25а
а
Пии 30 °C.
Радикальная полимеризация
193
меров составляет 10~5—10-4, поэтому самопроизвольная реакция
передачи цепи на мономер практически не влияет на молекуляр-
ный вес полимера, образующегося при такой полимеризации.
В реакции
Н
Mn'.'+CHZ=C—>МП — Н + СН2=С. (3.113)
Y Y
передача цепи на мономер идет в очень незначительной степени,
так как такая реакция должна идти с разрывом прочной виниль-
ной С — Н-связи. Наиболее высокими величинами См характери-
зуются реакции полимеризации винилацетата и винилхлорида.
В случае винилацетата большое значение См объясняют участием
в реакции передачи цепи ацетоксильной группы. Соответствующе-
го объяснения для процесса полимеризации винилхлорида пока
не найдено. Возможно, что большие значения См для винилацетата
и винилхлорида являются следствием высокой реакционной
способности растущих радикалов.
Хотя большие значения См для винилацетата и винилхлорида
и оказывают влияние на повышение молекулярного веса полимера
в процессе полимеризации, тем не менее молекулярный вес таких
полимеров не так низок, как об этом говорится в ряде публикаций.
Согласно уравнению (3.110) при См порядка 2-10~4—5-10~4 пре-
дельная среднечисловая степень полимеризации должна состав-
лять 2000—5000, что соответствует Мп для поливинилацетата
и поливинилхлорида приблизительно 140 000 и 450 000. Особенно
противоречивы данные для винилацетата. Приводящаяся в ряде
работ, например в [4], величина См = 0,002 явно завышена (выше
других значений для этого параметра примерно на порядок).
3.66.3. Константы передачи цепи на инициатор
Различные инициаторы имеют различные константы передачи
цепи (табл. 3.4). Принято считать, что азонитрилы не участвуют
в реакциях передачи цепи, поэтому полимеризация в их присут-
ствии не осложняется протеканием реакций передачи цепи даже
при высоких концентрациях инициатора. (Однако недавно появи-
лось сообщение, из которого следует, что и АИБН может иметь
весьма заметную константу передачи [9].) Многие перекиси харак-
теризуются весьма значительными константами передачи цепи.
При использовании в качестве инициаторов перекисей диалкилов
и диацплов передача цепи (индуцированное разложение переки-
си) идет как реакция замещения
Mn-+RO—OR—>Mn — OR + RO-, (3.114)
где R — алкил пли ацил. Перекиси ацилов имеют более высокие
константы передачи цепи по сравнению с перекисями алкилов,
194
Глава 3
Таблица 3.4
Константы передачи цепи на инициатор [41J
Инициатор Cl для полимеризации при 60 °C
стирола метилметакрилата
2,2' -Азо-б ис-изобутиронитрил 0 0
Перекись тпрет-бутила 0,0003-0,0013 —
Перекись кумола 0,01 а —
Перекись лауроила 0,024 6 —
Перекись бензоила 0,048-0,055 0,02
Гидроперекись пгрепг-бутила 0,035 1,27
Гидроперекись кумола 0,063 0,33
d При 50 °C.
6 При 70 °C.
что обусловлено более слабыми связями О — О в перекисях аци-
лов. Наиболее сильными агентами передачи цепи среди инициато-
ров являются обычно гидроперекиси. Передача цепи идет через
отрыв атома водорода]
Mn-+ROO — — H + ROO- (3.115)
Наглядное представление о влиянии передачи цепи на инициатор
можно получить из рис. 3.5 [39]. Уменьшение молекулярного веса
вследствие передачи цепи на инициатор значительно меньше рас-
считанного на основании Сг, так как на Хп влияет величина
Cj[I[/[M] [уравнение (3.1106)], а при’низких концентрациях иници-
атора (10“2—10-4 М) отношение [1]/[М] обычно колеблется в интер-
вале 10-3—10-5.
З.бв. Передача цепи на специальный агент
3.6в.1. Определение Cs
Второй частный случай уравнения (3.109) относится к процес-
сам, в которых наиболее важной является передача цепи на агент
передачи цепи. В отдельных случаях им может быть растворитель,
в других — специально добавленное вещество. В таком случае
третий член в правой части уравнения (3.109) вносит наибольший
вклад в величину степени полимеризации. В определенных усло-
виях полимеризации можно найти значение Се для различных
Радикальная полимеризация 195
агентов передачи цепи [42]. При низких концентрациях инициато-
ров или при использовании инициаторов с низкими значениями С\
(например, АИБН) последним членом в уравнении (3.109) можно
пренебречь. Регулируя соответствующим образом концентрацию
инициатора в ходе полимеризации, можно поддерживать постоян-
ным отношение 7?Р/[М]2, тогда первый член в правой части урав-
нения (3.109) будет величиной постоянной. В этих условиях
уравнение (3.109) принимает вид
где (1/Хп)о — это значение ИХп в отсутствие агента передачи
цепи. Величина (1/Хп)о представляет собой сумму первого, второго
Рис. 3.8. Влияние различных аген-
тов передачи цепи на молекулярный
вес полистирола. Температура по-
лимеризации 100 “С [42].
[Агент передачи цепи]
[Стирол]
и четвертого членов правой части уравнения (3.109). Тогда Cs
можно определить по наклону кривой зависимости 1/Хп от [S]/[M].
Такие кривые приведены на рис. 3.8 для нескольких агентов пере-
дачи цепи, использующихся при полимеризации стирола [18].
Как было показано, этим способом можно определять в раз-
личных процессах для разных соединений.
По другому способу определения Cs, являющемуся собственно
видоизмененным первым способом, строят зависимость не 1/Хп,
а И/Хп- (1/Хп)0] от [S]/[M]. В условиях, когда отношение
/?р/[М]2 непостоянно, можно откладывать (l/Xn—k0Rp/kl М2) от
[S1/IM]. При этом получают прямую линию с наклоном Cs. Для
определения Cs можно воспользоваться отношением скоростей
передачи цепи [уравнение (3.105)1 и роста цепи [уравнение (3.21)]
^[S]/^ fcnep. s [S] [S] . „
d[M]/dt fcp[M] “Gs [M] ’
Cs находят как наклон кривой зависимости отношения скоростей
исчезновения агента передачи цепи и мономера d [S]/d [М] от
IS]/[M1.
3.6в.2. Структура соединении и реакционная
способность
Константы передачи цепи для различных соединений приведены
в табл. 3.5. На основании данных этой таблицы можно сделать
некоторые выводы о зависимости между структурой и реакцион-
ной способностью различных соединений в радикальных реакциях
замещения. Алифатические углеводороды, например циклогексан,
с прочными связями С — Н характеризуются низкими констан-
тами передачи цепи. По этой же причине низкую Са имеет бензол.
По-видимому, передача цепи на бензол идет путем присоединения
к нему радикала
Мп-+^____Ч —> Мп— /____Ч (3.118)
Толуол, этилбензол и изопропилбензол имеют более высокие Cs,
чем бензол, что обусловлено наличием в них слабых бензильных
атомов водорода. Бензильная связь С —Н разрывается легко,
так как при этом образуется резонансно стабилизированный радикал
При переходе к mpem-бутилбензолу константа передачи цепи
уменьшается, так как в последнем уже нет бензильных связей
С — Н. н-Бутилхлорид и н-бутилбромид, подобно другим алифа-
тическим соединениям, имеют низкие константы передачи цепи.
Вместе с тем из-за передачи цепи в н-бутилиодиде на атом иода это
соединение характеризуется высокой константой передачи цепи,
что обусловлено низкой прочностью связи С — I.
Радикальная полимеризация
97
Таблица 3.5
Константы передачи цепи для различных соединений [41]
Агент передачи цепи Cs.104 для полимеризации при 60 °C
стирола винилацетата
Бензол 0,023 1,2
Циклогексан 0,031 7,0
Гептан 0,42 17,0 а
Толуол 0,125 21,6
Этилбензол 0,67 55,2
Изопропилбензол 0,82 89,9
трет-Бутил бензол 0,06 3,6
и-Бутилхлорид 0,04 10
и-Бутилбромид 0,06 50
Ацетон 0,40 6 11,7
Уксусная кислота 0,20 6 1,1; 10
н-Бутиловый спирт 0,40 6 20
Хлороформ 0,5 150
и-Бутилиодид 1,85 800
Бутиламин 0,5 —
Триэтиламин 7,1 370
и-Бутилдисульфид 24 10 000
Четыреххлористый углерод 90 9 600
Четырехбромистый углерод 22 000 28 700 в
трет-Бутилмеркаптан 37 000 —
и-Бутилмеркаптан 210 000 480 000
а При 50 °C.
6 При 80 °C.
в При 70 °C.
Так как при передаче цепи на карбоновые кислоты, карбониль-
ные соединения, простые эфиры, амины и спирты, происходит раз-
рыв связи G — Н, константы передачи цепи у указанных соедине-
ний близки константам Cs для алифатических углеводородов.
Некоторые вторичные и третичные соединения характеризуются
более высокими Cs, чем углеводороды.
Дисульфиды, содержащие слабые связи S — S, имеют высокие
константы передачи цепи
Мп- + RS—SR -> Mn —SR]+ RS- (3.120)
Высокие значения для четыреххлористого и четырехбромистого
углерода обусловлены наличием в них слабых галоген-углерод-
ных связей. Разрыв таких связей облегчается еще и тем, что воз-
никающие тригалогенуглеродные радикалы очень стабильны
| Cl —С —С11 | С1 = С—С11 *-► | С1—С=С11| Cl—С—С11 (3.121)
|ii| |ii| |С1| |С1.
При этом, поскольку связь С — Вг слабее связи С — С1, четырех-
бромистый углерод имеет более высокую константу передачи,
чем четыреххлористый углерод. В то же время, поскольку в хлоро-
форме при передаче цепи рвется связь С — Н, он имеет низкую Cg.
Наиболее высокие константы передачи среди изученных соеди-
нений имеют тиолы, что обусловлено низкой прочностью свя-
зи S - Н.
Интересно проследить, как меняется С$ для различных соеди-
нений при полимеризации разных мономеров. Абсолютная вели-
чина Cs для всех изученных соединений сильно зависит от природы
полимеризующегося мономера. Как это очень хорошо видно из
данных табл. 3.5, многие агенты передачи цепи на 1—2 порядка
активнее в полимеризации винилацетата, чем в полимеризации
стирола. Это обусловлено большей реакционной способностью
растущего винилацетатного радикала. Как правило, константа
передачи для данного соединения возрастает с увеличением реак-
ционной способности радикала. Различные мономеры располагают-
ся в порядке уменьшения реакционной способности радикалов
в следующий ряд: винилхлорид > винилацетат > акрилонит-
рил > метилакрилат > метилметакрилат > стирол > бутадп-
ен-1,3. Более подробно реакционная способность радикалов рас-
сматривается в гл. 6.
Если агенты передачи цепи — неполярные соединения, то их
реакционная способность обычно не зависит от природы мономера.
Однако при переходе к полярным соединениям это положение теря-
ет свою справедливость. В табл. 3.6 представлены данные для
четыреххлористого углерода и триэтиламина с несколькими моно-
мерами. Мономеры расположены в порядке уменьшения реак-
ционной способности в реакциях передачи цепи на нейтральные
передатчики цепи, например углеводороды. Как видно из этой
таблицы, электронодонорный триэтиламин характеризуется по-
вышенной реакционной способностью в реакциях полимеризации
электроноакцепторных мономеров, например акрилонитрила,
метилакрилата и метилметакрилата. Электроноакцепторный агент
передачи цепи — четыреххлористый углерод, — напротив, обла-
дает повышенной реакционной способностью по отношению к элек-
тронодонорным мономерам (винилацетату и стиролу). Полагают,
что повышение реакционной способности при реакции передачи
цепи происходит в результате стабилизации соответствующих
Радикальная полимеризация
199
Таблица 3.6
Полярные эффекты в реакции передачи цепи при полимеризации
при 60 °C [41]
Мономер CS- 104 ftnepa
для ССЦ для (CaHshN для ССЦ для (C2H5)3N
Винилацетат 9600 370 2500 85
Акрилонитрил 0,85 3800 0,17 760
Метилакрилат 1,256 400 0,26 б 84
Метилметакрилат 2,4 1900 0,12 98
Стирол 90 7,1 1,6 0,13
а ftnep рассчитаны по уравнению (3.64) с использованием значений hp из табл. 3.9.
6 Cg при 8 0 СС. В рассвете ftnep использовано значение hp при 60 °C.
переходных состояний за счет таких полярных структур, в кото-
рых имеет место частичный перенос заряда между электронодо-
нором и электроноакцептором [4]
— СН2— СН-|-Н—СН—N(CH3)2 «-►
I I
CN СН3
сн2—сн
:n
Н СН —N(CH3)2 (3.123)
СН3
Полярные эффекты такого типа весьма распространены в реакциях
свободных радикалов. При этом, как правило, реакционная способ-
ность электронодонорного радикала выше в реакциях с электроно-
акцепторными соединениями, чем в реакциях с электронодонор-
ными соединениями. Для электроноакцепторных радикалов спра-
ведлива обратная зависимость. В разд. 6.36.3 обсуждается влия-
ние полярных факторов в реакциях присоединения радикалов.
200
Глава 3
З.вв.З. Применение агентов передачи цепи
С помощью уравнений (3.109), (3.110) и (3.116) можно количе-
ственно оценить влияние передачи цепи на мономер, инициатор
и растворитель на молекулярный вес образующегося полимера.
Еще большее значение агенты передачи цепи имеют как регулято-
ры молекулярного веса полимера в процессе полимеризации.
С помощью уравнения (3.116) можно найти концентрацию агента
передачи цепи, которая требуется для получения полимера задан-
ного молекулярного веса. Когда агенты передачи цепи применяют
для этой цели, их называют регуляторами или модификаторами.
Большую практическую ценность представляют передатчики цепи,
имеющие константы передачи больше единицы, поскольку такие
вещества можно вводить в реакцию в небольших количествах. Так,
в промышленности получение бутадиенстирольных каучуков про-
водят путем эмульсионной полимеризации в присутствии мер-
каптанов, например н-додецилмеркаптана. Практический интерес
представляет также образование очень низкомолекулярных поли-
меров реакцией передачи цепи (теломеризация). Заполимеризо-
ванный в хлороформе низкомолекулярный полиэтилен после
фторирования концевых групп используется в качестве исходного
соединения для получения фторсодержащих смазок. Низко-
молекулярные полимеры сложных акриловых эфиров применяются
как пластификаторы. В то же время бензол, имеющий низкую
константу передачи цепи, применяется в качестве растворителя
для получения высокомолекулярного полиэтилена.
Необходимо также подчеркнуть, что исследования реакций
передачи цепи дали дальнейшее подтверждение концепции неза-
висимости реакционной способности функциональных групп от
размера молекулы. Следовательно, степень полимеризации того
пли иного полимера можно изменять применением различных
агентов передачи цепи или введением одного и того же передатчика
цепи в разных концентрациях. При этом, как было показано,
константа скорости роста кр не зависит от Хп. Кроме того, кон-
станта передачи цепи /сПРр для данного агента передачи цепи так-
же не зависит от размера растущего радикала.
3.6г, Передача цепи на полимер
Выше не рассматривалась возможность передачи цепи на моле-
кулы полимера. При реакции передачи цепи на полимер в послед-
нем образуются радикальные центры. В результате последующей
полимеризации мономера по этому центру получается разветвлен-
Радикальная полимеризация
01
ный полимер, например
Y
Мп.+—сн2-с—-»мп-н +
н
Y м Y
— СН —С — —» — СН2 —С—
I
Мт —
(3.124)
При определении констант Cj, См и Cs влияние реакции передачи
цепи на полимер не учитывают, но это не отражается на точности
результатов, так как константы оценивают при низких степенях
превращения мономера в полимер. В этих условиях концентрация
полимера низка, и поэтому реакцией передачи цепи на полимер
можно пренебречь.
На практике же, когда полимеризацию доводят до высоких
степеней завершенности реакции, реакцию передачи цепи на поли-
мер нельзя не учитывать. Эта реакция оказывает существенное
влияние на физические свойства и, следовательно, области при-
менения полимера. Как было показано в гл. 1, разветвление резко-
снижает кристаллизуемость полимера.
Определение константы передачи Ср для передачи цепи на
полимер сопряжено со значительными трудностями. Ср нельзя
просто определить путем введения нового члена Ср [Р]/[М] в урав-
нение (3.109), как это делается во многих учебниках. При передаче
цепи на полимер степень полимеризации не обязательно должна
уменьшаться. При каждом акте передачи цепи образуется развет-
вленная макромолекула большего размера, чем исходная моле-
кула полимера.
Оценка Ср включает весьма трудоемкое определение числа
ветвлений, образующихся в процессе полимеризации, с отнесением
их к числу заполимеризованных молекул мономера. Это можно
осуществить путем проведения полимеризации в присутствии опре-
деленного количества полимера известного молекулярного веса.
Продукт такой реакции содержит молекулы трех различных
типов:
Тип 1. Неразветвленные молекулы исходного полимера.
Тип 2. Неразветвленные молекулы полимера, образующегося
при полимеризации мономера.
Тип 3. Разветвленные молекулы полимера, получающегося
в результате реакции передачи цепи радикалов молекул типа 2 на
молекулы типа 1.
Число новых полимерных молекул, образующихся в системе,
определяет число молекул типа 2. Общее число ветвлений получа-
ют исходя из материального баланса системы при предположении,
что размер ветвлений эквивалентен размеру молекул типа 2.
Экспериментальный анализ такой системы осложняется одно-
временным протеканием других реакций передачи цепи (на моно-
мер, инициатор или другие соединения). Для оценки констант
Глава 3
А
передачи цепи на полимер были апробированы другие методы, но
все они не являются адекватными. Так, например, Ср для поли-
винилацетата определяли путем деструктивного гидролиза [43].
При этом предполагали, что передача цепи идет по ацетоксиметиль-
ной группе с образованием полимерных ветвлений следующего
чипа:
— СН2-СН —
I
О
I
с=о
I
сн2—мп—
V
которые можно отделить от исходного полимера путем гидролиза
сложноэфирной связи. Однако известно также, что передача цепи
идет не только по ацетоксиметильным группам.
Из-за таких трудностей определения Ср в литературе можно
найти очень немного достоверных данных для этой константы.
При этом для одного и того же полимера часто приводятся вели-
чины, значительно отличающиеся друг от друга [40]. Поэтому
для получения правильного представления о важности реакций
передачи цепи на полимер часто лучше провести исследование
модельных низкомолекулярных соединений (например, этил-
-бензола или изопропилбензола). Рассмотрение более или менее
надежных данных Ср, полученных при исследовании модельных
соединений, показывает, что во многих случаях даже при высоких
степенях завершенности реакции доля реакций передачи цепи на
полимер не является достаточно большой. Для многих полимеров,
например полистирола и полиметилметакрилата, Ср равна при-
мерно 10-4.
Флори [44] вывел уравнение
Р = -Ср [1 + (|) 1п(1-р)], (3.125)
выражающее зависимость между плотностью ветвления р, кон-
стантой передачи Ср и степенью завершенности реакции р. Плот-
ность ветвления р представляет собой число ветвлений на одну
молекулу вступившего в полимеризацию мономера. При Ср =
= 1-10-4 и степени завершенности реакции 80% по уравнению
(3.125) можно подсчитать, что на каждые 104 молекул полимери-
зующегося мономера приходится одно ветвление, что хорошо под-
тверждается экспериментальными данными [16]. В обычной реак-
ции полимеризации стирола при степени превращения 80% одно
ветвление приходится на каждые 4-103—10-103 мономерных звень-
ев, причем это показано для полимера с молекулярным весом
Радикальная полимеризация
2 3
105—106. Следовательно, примерно одна из десяти полимерных
молекул имеет ветвление [45].
Полимеры, характеризующиеся очень реакционноспособными
радикалами, например поливинилацетат, поливинилхлорид и по-
лиэтилен, имеют более высокую степень ветвления. При получе-
нии поливинилацетата Ср составляет примерно 2-10—4—5-Ю"4.
Кроме того, как было выше отмечено, винилацетат характеризует-
ся высокой величиной См (табл. 3.3). При передаче цепи на моно-
мер образуется радикал
Н
сн2 = с—о—со—сн2,
VI
который может инициировать полимеризацию и участвовать
в реакциях роста ряда других растущих радикалов. Это приводит
Рис. 3.9. Степень ветвления при
полимеризации винилацетата при
60 °C [46].
к образованию разветвленного полимера. Интенсивное ветвление
в поливинилацетате объясняется тем, что ветвления возникают при
каждом акте передачи цепи на мономер или полимер. На рис. 3.9
показано, как меняется степень ветвления поливинилацетата
в зависимости от степени завершенности полимеризации [46].
Особенно быстро она возрастает на заключительных этапах про-
цесса.
Степень ветвления полиэтилена сильно зависит от температуры
полимеризации и других условий реакции и может достигать
значений, составляющих 30 ветвлений на 500 мономерных звеньев.
/204
Глава 3
Полиэтилен содержит в основном короткие ветвления — этиль-
ные и бутильные, — количество которых примерно одинаково,
количество же более длинных ветвлений примерно в пятьдесят
раз меньше. Бутильные и этильные ветвления, по-видимому,
образуются в результате следующих реакций:
Н
СН СН2 Передача цепи
I I-------------------:
сн2 сн2
сн2
— сн
I
сн2 сн2—сн3
сн2
(3.126а)
I сн2=сн2
сн2
сн2 н
I' I
сн сн-сн2-сн3
СН3
I
сн2
——СН СН-СН2-СН3^^~
\н2
(3.126в)
(3.1266)
Бутильные ветви образуются в результате внутримолекулярной
передачи цепи, в которой радикальный конец отрывает водород
от пятой метиленовой группы (считая от конца растущего радика-
ла) [уравнение (3.126а)]. Этильные ветви появляются при после-
дующем присоединении одной молекулы мономера [уравнение
(3.1266)] и реакции передачи цепи по уравнению (3.126в).
3.7 . Ингибирование и замедлен не
3.7 а. Типы ингибиторов и замедлителей
В присутствии ряда веществ полимеризация мономеров подав-
ляется. Действие таких веществ основано на том, что они реагиру-
ют с инициатором и растущими радикалами, превращая их в не-
радикальные соединения или радикалы с очень низкой реакцион-
ной способностью, недостаточной для поддержания реакции роста
цепи. По эффективности такие вещества делятся на ингибиторы
и замедлители. Ингибиторы уничтожают все имеющиеся в системе
радикалы, в результате чего полимеризация не идет до полного
исчерпания ингибитора. Замедлители менее эффективны и ней-
трализуют только часть радикалов. В их присутствии полимери-
зация не прекращается, но протекает с более низкой скоростью.
Следовательно, различие между ингибитором и замедлителем носит
Радикальная полимеризация
205
скорее количественный, чем качественный характер. Рис. 3.10
отражает действие таких соединений в процессе термической
полимеризации стирола [47]. Полимеризация не идет в присут-
ствии бензохинона, типичного ингибитора, в течение индукцион-
ного, или ингибирующего, периода (кривая 2). В конце этого
периода, когда весь бензохинон израсходован, полимеризация про-
текает с обычной скоростью, как и без ингибитора (ср. с кривой!).
В присутствии нитробензола, являющегося замедлителем, ско-
рость полимеризации снижается, но никакого индукционного
Рис. 3.10. Ингибирование и за-
медление в процессе чисто тер-
мической полимеризации стирола
при 100 °C [47].
Ингибитор: 1 — отсутствует; г — бен-
зохинон (0,1%); з — нитробензол
(0,5%); 4— нитрозобензол (0,2%).
Продолжительность, мин
периода не наблюдается (кривая 5). Более сложный характер имеет
полимеризация в присутствии нитрозобензола GeH6NO (кривая 4).
Вначале он действует как ингибитор, но после окончания индук-
ционного периода, по-видимому, работает как замедлитель. Такое
поведение наблюдается и в ряде других систем. Тот факт, что
полимеризация не очень хорошо очищенных мономеров не идет
с постоянной скоростью, объясняется, очевидно, ингибированием
и замедлением. Примеси, содержащиеся в мономерах, могут дейст-
вовать как ингибиторы или замедлители. Кроме того, в промыш-
ленные мономеры часто добавляют ингибиторы, чтобы предотвра-
тить преждевременную полимеризацию в процессе хранения и пере-
возки мономера. Перед началом полимеризации такие ингибиторы
обычно удаляют или ведут полимеризацию в присутствии соот-
ветствующего избытка инициатора.
Соединения различных типов работают как ингибиторы и за-
медлители. К их числу относятся стабильные радикалы, которые
не могут инициировать полимеризацию, но в состоянии реагиро-
вать с радикалами. Таким радикалом является дифенилпикрил-
гидразил, действие которого рассматривалось в разд. 3.4е.3.
Соотношение числа оборванных кинетических цепей под действием
такого радикала и числа радикалов ДФПГ равно 1:1. Этим ради-
калом, собственно, и ограничивается число стабильных радика-
лов, имеющих практическую ценность в качестве ингибиторов.
Й06
Глава 3
Широкое распространение в качестве ингибиторов получили
соединения, реагирующие с растущими радикалами с образовани-
ем радикалов с низкой реакционной способностью. Среди них,
пожалуй, наибольшее значение имеют хиноны. В присутствии
бензохинона полностью прекращается полимеризация ряда моно-
меров, и в том числе винилацетата [31] и стирола [16]. Поведение
хинонов в таком процессе достаточно сложно. Сильно колеблется
отношение числа оборванных кинетических цепей и числа израс-
ходованных молекул хинона. Для бензохинона оно в случае
многих мономеров равно 1:1. Хлоранил (2,3,5, 6-тетрахлор-
бензохинон) является эффективным ингибитором при полимериза-
ции винилацетата, но он сополимеризуется со стиролом (гл. 6)
г
(3.127)
снижая общую скорость полимеризации [48]. Механизм ингибиро-
вания хинонами изучен не очень хорошо [5, 16]. Обрыв растущих
радикалов под действием хинонов, возможно, идет через присое-
динение радикалов к атому кислорода или циклу хинона
н' хм,
Радикальная полимеризация
207
хотя не исключаются также реакции диспропорционирования!
(3.128г)
Соотношение радикалов и ингибитора зависит от дальнейшего поведе-
ния радикалов, образовавшихся в результате реакций (3.128а)—
(3.128г). Эти радикалы могут вступать в реакции диспропорциони-
рования и димеризации как друг с другом, так и с растущими
радикалами. При диспропорционировании регенерируется одна
молекула ингибитора в расчете на каждую пару радикалов ингиби-
тора. В случае протекания димеризации соотношение радикалов
и ингибитора должно быть равно 1:1, тогда как при протекании
двух других реакций оно должно быть 2 : 1. В экспериментальной
практике наблюдались оба соотношения. Сложный характер'
поведения хинонов подтверждается тем, что при полимеризации
метилметакрилата и метилакрилата они выступают в роли
замедлителей.
Гидрохинон и-ОНС6Н4ОН и другие диокспбензолы, например
тпретп-бутилпирокатехин, действуют как ингибиторы только в при-
сутствии кислорода. Их ингибирующий эффект обусловлен окис-
лением таких соединений в хиноны [48]. В некоторых случаях
ингибиторами являются полиалкилзамещенные в ядро фенолы
и анилины. Действие таких ингибиторов, вероятно, основано на
реакции отрыва водорода
(3.129)
и последующей конденсации феноксильного радикала (по кисло-
роду или ядру) с другим полимерным радикалом. Незамещенные-
фенол и анилин — довольно слабые ингибиторы.
Растущие радикалы обрываются под действием ди- и трини-
троароматических соединений, хотя ингибирование в этом случае-
08
Глава 3
не такое сильное, как в присутствии хинонов. Нитросоединения
действуют как ингибиторы только при полимеризации винилаце-
тата [4] и в нескольких других случаях. Они замедляют полимери-
зацию стирола [47] и не оказывают никакого влияния на полиме-
ризацию акрилатов и метакрилатов [49]. От природы мономера
зависит отношение числа оборванных радикалов и числа израс-
ходованных молекул замедлителя. Обрыв растущих радикалов,
возможно, включает радикальную атаку ароматического ядра
и нитрогруппы
(3.130а)
(3.1306)
При использовании 1,3,5-тринитробензола указанное отношение
достигает 5—6.
Способность ингибировать полимеризацию проявляют также
различные соединения других классов, в том числе кислород, сера,
углерод и хлорное железо (разд. 3.4е.3) [16]. Интересным ингиби-
тором является молекулярный кислород, который реагирует
с растущими радикалами с образованием сравнительно неактив-
ного перокси-радикала
Мп-+О2 -+ МП-ОО. (3.131)
Вместе с тем известно, что кислород инициирует полимеризацию
многих мономеров. Так, некоторые промышленные способы поли-
меризации этилена основаны на использовании кислорода в качест-
ве инициатора. В этом случае инициирование может идти вслед-
ствие термического разложения перокси-радикала [уравнение
(3.131)] или других окисленных соединений, присутствующих
в реакционной системе.
3.76. Кинетика ингибирования или замедления
Для расчета кинетики замедленных реакций полимеризации
рассмотрим схему, состоящую из обычного инициирования [урав-
нение (3.14)], роста цепи [уравнение (3.15)] и обрыва цепи [уравне-
ние (3.16)], дополнив ее реакцией ингибирования
ь,
Mn-+z 4m„ + Z., (3.132)
Радикальная полимеризация
9
где Z — ингибитор. Кинетику процесса можно несколько упрос-
тить, если допустить, что радикалы ингибитора не реинициируют
полимеризацию и что в результате обрыва не регенерируются
молекулы исходного ингибитора.
В стационарном состоянии
Ra- k0 [М • ]2- kz [Z] [М• ] = 0. (3.133)
Из уравнения (3.133) и уравнения (3.8) находим
, T?p[Z]fc2
^[Ml2 + *Р[М]
(3.134)
Уравнение (3.134) используется для того, чтобы скоррелировать
данные по скоростям полимеризации, проводящейся в присутствии
ингибиторов [51]. Рассмотрение этого уравнения показывает, что
7?р обратно пропорциональна отношению kzlkv констант скоро-
сти ингибирования и роста. Это отношение часто называют констан-
той ингибирования, т. е.
/с?
Z===T~
(3.135)
Легко заметить также, что /?р зависит от 7?и в степени от 1/2 до
1,0, которая определяется относительной величиной первых двух
членов в уравнении (3.134). Представляют интерес два крайних
случая уравнения (3.134). Если второй член значительно меньше
первого, то полимеризация не тормозится и уравнение (3.134)
сводится просто к уравнению (3.24).
Если замедление выражено очень сильно (kzlkv > 1), обычной
реакцией бимолекулярного обрыва можно пренебречь. Тогда пер-
вый член в уравнении (3.134) опускают и оно принимает вид
или
др [£] fez
feP [М]
или
fep [М] R„_ — d[M]
Лр— kz[Z] ~ dt
(3.136a)
(3.1366)
Как видно из уравнения (3.1366), скорость замедленной полимери-
зации зависит линейно от скорости инициирования. Кроме того,
7?р обратно пропорциональна концентрации ингибитора. Про-
должительность индукционного периода прямо пропорциональна
концентрации ингибитора.
210
Глава 3
С увеличением продолжительности реакции концентрация ин-
гибитора уменьшается и [Z] в любой момент времени равна
[Z] = [Z]0—(3.137)|
где [ZJO —начальная концентрация Z, t — время и у — число
образованных радикалов на одну молекулу ингибитора. Из урав-
нений (3.135), (3.1366) и (3.137) получим
— rf[M] ДИ[М]
di z ([ZJq —
или после перестановки
— 1 __ z [Z]o zt
d In [M]/dt Ra у
(3.138a) ।
(3.1386)1
Кривая зависимости левой части уравнения (3.1386) от продолжи-'
тельности реакции представляет прямую линию, причем z и у>
Рис. 3.11. Ингибирование поли-
меризации винилацетата дурохино-
ном. Инициатор — перекись бензо-
ила; температура 45 °C [31].
Три кривые — для различных концентра-
ций дурохинона.
можно найти по отрезку, отсекаемому на оси ординат, и наклону
прямой, если [Z]o и Ни известны. Экспериментальное определение
этих величин сопряжено с большими трудностями, вызванными
малыми значениями измеряемых скоростей полимеризации, осо-
бенно при больших z. На рис. 3.11 представлена такая зависи-
мость для ингибированной полимеризации винилацетата [31].
В табл. 3.7 приведены значения z для различных систем. Нетрудно
заметить, что константы ингибирования, как и константы переда-
чи цепи, сильно зависят от реакционной способности растущего
радикала. Так, 1,3,5-тринитробензол является ингибитором при
полимецизации винилацетата, характеризующегося высокореак-
Радикальная полимеризация
211
Таблица 3.7
Константы ингибирования в реакциях полимеризации при 50 °C [41]
Ингибитор Мономер z = kz/kv
Нитробензол Метилакрилат 0,00464
Стирол 0,326
Винилацетат 11,2
1,3,5-Тринитробензол Метилакрилат 0,204
Стирол 64,2
Винилацетат 404
п-Бензохинон Акрилонитрил 0,91
Метилметакрилат 4,5а
Стирол 518
ДФПГ Метилметакрилат 2 000 6
FeCl3 в диметилформамиде Акрилонитрил 3,3а
Стирол 536 а
Винилацетат ~ 800 в
Кислород Метилметакрилат 33 000
Стирол 14 600
Сера Метилметакрилат 0,075 6
Винилацетат 470 6
Анилин Метилакрилат 0,0001
Винилацетат 0,015
Фенол Метилакрилат 0,0002
Винилацетат 0,012
а При 60 °C.
6 При 44 °C.
в Определена при 60 °C из отдельных величин Л „ и А, .
р
ционноспособным растущим радикалом, и лишь замедляет поли-
меризацию с менее реакционноспособным метилакрилатным рас-
тущим радикалом.
При внимательном анализе уравнения (3.1386) видно, что
в случае сильного замедлителя (z > 1) скоростью полимеризации
можно пренебречь, пока концентрация ингибитора не станет
достаточно низкой. При низкой концентрации ингибитора рост
цепи начинает конкурировать с реакцией ингибирования. Это ста-
новится более ясно при рассмотрении выражения
lZl __ Z tZ] /О л ОП\
d [М] - [М] ’ (O.ldV)
получающегося делением уравнения для скорости исчезновения
ингибитора (3.132) на уравнение для скорости исчезновения моно-
212
Глава 3
мера (3.15). В результате интегрирования уравнения (3.139) имеем
где [Z]o и [М] о — начальные концентрации. Как видно из этого
уравнения, прежде чем мономер может начать полимеризоваться,
должен исчерпаться весь ингибитор. Уравнением (3.140) можно
также воспользоваться для определения константы ингибирования
как наклона кривой зависимости 1g [Z] от 1g [М].
3.7в. Аутоингибирование аллильных мономеров
Особым, интересным случаем ингибирования является внутрен-
нее ингибирование, или аутоингибирование, аллильных мономе-
ров СН2 = СН — СН2У. Аллильные мономеры, например аллил-
ацетат, полимеризуются с необычно низкой скоростью, при этом
неожиданно обнаруживается линейная зависимость скорости от
концентрации инициатора. Кроме того, степень полимеризации,
которая не зависит от скорости полимеризации, оказывается
очень низкой и не превышает в случае аллилацетата 14. Указанные
эффекты обусловлены деструктивными реакциями передачи цепи
(пример 4 в табл. 3.2). Растущий радикал в таких реакциях поли-
меризации очень реакционноспособен, тогда как аллильная связь
С — Н (связь С — Н в a-положении к двойной связи) в мономе-
ре — сравнительно слабая, что и обусловливает легкость проте-
кания реакции передачи цепи на мономер
Н Н
— сн2-с. +CH2 = CH-CY —> — СН2-СН2 +СН2 = СН-СУ
I Н I t н
ch2y ch2y I
CH2-CH = CY
н
(3.141)
Слабость аллильной связи С — Н объясняется высокой резонанс-
ной устойчивостью образующегося аллильного радикала. Дест-
руктивная реакция передачи цепи успешно конкурирует с нор-
мальным ростом цепи, причем обрыв цепей имеет место в резуль-
тате передачи цепи после присоединения очень небольшого числа
Мономерных звеньев. Экспериментальные доказательства разрыва
аллильной С — Н-связи при реакции передачи цепи были полу-
чены [52] при исследовании полимеризации дейтерированного
аллилацетата СН2 = СН — CD2OCOCH3. Этот мономер поли-
меризуется в 1,9—2,9 раза быстрее нормального аллилацетата,
причем степень полимеризации полимера в этом случае в 2,4 раза
выше, чем у второго полимера аллилацетата. Этого и следовало
Радикальная полимеризация
21
ожидать, если учесть, что связь С — D прочнее связи С — Н из-за
более низкой пороговой энергии [4], поэтому деструктивная реак-
ция передачи цепи для дейтерированного мономера идет в мень-
шей степени.
Аллильные радикалы слишком устойчивы, чтобы инициировать
полимеризацию, поэтому, когда совершается акт передачи цепи,
кинетическая цепь обрывается. Дальнейшая судьба аллильных
радикалов не ясна. Реакция (3.141) эквивалентна обрыву ингиби-
тором, которым в данном случае является сам мономер. В такой
реакции полимеризации рост и обрыв цепи будут характеризовать-
ся одними и теми же кинетическими уравнениями линейной зави-
симости от концентрации инициатора и мономера, так как в реак-
циях участвуют одни и те же реагенты в одинаковых соотношениях.
Степень полимеризации определяется просто как отношение кон-
стант скоростей роста и обрыва цепи и не зависит от концентрации
инициатора. При полимеризации некоторых аллильных мономе-
ров обрыв может идти не только вследствие деструктивной реак-
ции переноса аллильного атома водорода, но и в результате реак-
ции радикального замещения по схеме
Mn.+CH2 = CH —CH2OCOR Mn-CH2 —CH = CH2 + RCOO- . (3.142)
Тем не менее при полимеризации аллильных мономеров основная
реакция обрыва цепи — отрыв водорода.
Низкая реакционная способность а-олефинов, например про-
пилена, или 1,1-диалкилолефинов, например изобутилена, в ради-
кальной полимеризации, возможно, обусловлена реакцией пере-
дачи цепи с участием аллильных атомов водорода. Следует отме-
тить, что для метилметакрилата и метакрилонитрила, также
имеющих аллильные связи С — Н, реакция передачи цепи рассмо-
тренного выше типа не характерна, что обусловлено низкой реак-
ционной способностью растущих радикалов у таких мономеров.
Сложноэфирные и нитрильные группы стабилизируют радикалы,
что уменьшает реакционную способность последних в реакциях
передачи цепи. При этом склонность таких мономеров к полимери-
зации повышается. В результате радикальной полимеризации
таких мономеров образуются высокомолекулярные полимеры.
3.8. Определение констант абсолютных
скоростей
3.8а. Кинетика нестационарного состояния
В радикальной полимеризации встречаются пять различных
типов констант скоростей реакций: инициирования, роста, обрыва,
передачи цепи и ингибирования. С помощью данных, характери-
зующих процесс полимеризации в стационарном состоянии, можно
определить только константу скорости инициирования кг (или ка
в случае термического инициирования). Из уравнения (3.24) можно
найти отношения kpllto* или /ср//с0, так как 7?р, 7?и и [М] можно
измерить. Подобным образом вышерассмотренными способами
определяют константу передачи цепи кпер/кр и константу ингиби-
рования kz/kp. Однако на основании данных, полученных при
исследовании кинетики стационарной стадии полимеризации, не
удается рассчитать индивидуальные константы кр, к0, каер и kz.
Определить их можно, используя данные по кинетике полимери-
зации в нестационарном состоянии.
Рассмотрим, например, фотохимическую полимеризацию. Со-
гласно представлениям Флори [53] и Уоллинга [4], скорость изме-
нения концентрации радикалов равна разности скоростей их
образования и обрыва
= 7?и-2к0 [М-р (3.143)
Различные параметры в этом уравнении относятся к нестационар-
ному состоянию; соответствующие характеристики для стацио-
нарного состояния имеют индекс $. Для стационарного состояния
уравнение (3.143) принимает вид
(7?и)в = 2fc0 [М.]1 (3.144)
Комбинация уравнений (3.143) и (3.144) дает
^-=.-2*0([M.]s2 -[М-]2) (3.145)
Введем теперь параметр т8, который означает среднее время жизни
растущего радикала в стационарном состоянии. Время жизни ради-
кала равно отношению концентрации радикалов в стационарном
состоянии к скорости их исчезновения в стационарном состоянии
Ts = 2ММ-Д = 2к0 (М-]8 (3’146)
Из уравнений (3.146) и (3.21) в стационарном состоянии находим
fep [М]
2/с0 (Лр)«
(3.147)
Индивидуальные константы кр и к0 можно было бы определить,
если бы было известно xs. Исследования в условиях нестационар-
ного состояния и ставили своей целью определение rs. В резуль-
тате интегрирования уравнения (3.145) имеем
<зл48>
Радикальная полимеризация
2 5
где i0 — постоянная интегрирования, которая выбирается из
расчета [М •] = 0. Из уравнений (3.146) и (3.148) находим, что
™л(тят) = ‘~^ <зл49>
ИЛИ
J& = _^_ = th (—°). (3.150)
[M-]s (Яр), \ т, /
Для вывода уравнений (3.149) и (3.150) читатель должен знать,
что такое гиперболический тангенс и обратный гиперболический
тангенс. В этих уравнениях t — время облучения. Рассмотрим
теперь процесс полимеризации с чередующимися периодами осве-
щения и затемнения. Только в самый первоначальный момент реак-
Р и с. 3.12. Зависимость [М-]/[М-], от времени при прерывистом осве-
щении [53].
ции концентрация радикалов равна нулю, но в любой последую-
щий момент времени концентрация радикалов больше никогда не
снижается до нуля: в начале периода освещения (который соот-
ветствует концу периода затемнения) она равна некоторой вели-
чине [М >]г > 0. Поэтому уравнение (3.149) принимает вид
т,
— arcth (гтт-г)
и
arcth (УгтФ) — arcth (тМФ) = ~
(3.151)
(3.152)
где [M -h — концентрация радикалов после периода освещения за
время i. Величину [М di также можно определить как концентра-
цию радикалов в начале периода затемнения. Кривая ОАЕ на
рис. 3.12 показывает рост концентрации радикалов в течение пери-
ода освещения, расчет проводился с помощью уравнения (3.152)
для нулевой начальной концентрации радикалов при t0 = 0.
216
Глава 3
После выключения света во время t концентрация радикалов
в период затемнения уменьшается по следующей зависимости:
2&О[М.]2; (3.153)
после интегрирования получим
= ’ (3.154)
где [М •] — концентрация радикалов после окончания следующего
отрезка времени, соответствующего продолжительности затемне-
ния. Умножив уравнение (3.154) на [М-]s и подставив его в уравне-
ние (3.146), получим
[М-]8 [М-] [М-]8 _ г Ts (3.155a)
[M-h
или (Я₽)8 (Яр), t' (3.1556)
(Яр)1 Ts
Снижение числа радикалов согласно уравнению (3.155) отражает-
ся кривой ABF на рис. 3.12.
Один из способов определения ts с помощью уравнения (3.1556)
основан на постэффекте и состоит в том, что измеряют скорость,
полимеризации (7?p)s при стационарных условиях при постоянном
освещении. Затем освещение заканчивают и определяют 7?р как
функцию продолжительности t' периода затемнения. Строят кри-
вую зависимости (7?p)s/7?p от t' согласно уравнению (3.1556),
наклон которой равен 1/ts. Хотя экспериментальное определение
ts таким способом вызывает некоторые трудности, связанные
с измерением очень малых скоростей реакции, этот способ доволь-
но распространен [54]. Однако возможно использование и быст-
рых методов детектирования, позволяющих следить за полимери-
зацией в период освещения до наступления стационарного состоя-
ния. Использовался, в частности, импульсный фотолиз, в котором
высокая концентрация радикалов достигается за счет интенсив-
ного импульса света от разрядной трубки [55].
3.86. Метод вращающегося сектора
Наиболее распространенным методом определения ts является
иетод вращающегося сектора, основанный на чередовании перио-
job освещения и затемнения. Согласно этому методу исследуют
сависимость между скоростью полимеризации и продолжительно-
стью цикла (складывающегося из одного периода освещения
I одного периода затемнения). Рассмотрим частный случай, когда
>тношение г продолжительности периода затемнения t' к периоду
Радикальная полимеризация
217
освещения t равно 3. Если продолжительность цикла очень велика
но сравнению с т3 (редкие вспышки), то Rp в период полного осве-
щения будет равна (7?p)s, а в период полного затемнения будет
превращаться в нуль. Это связано с тем, что отрезок времени,
в течение которого концентрация радикалов достигает стационар-
ного состояния или снижается до нуля, мал по сравнению с t и t'
соответственно. Средняя скорость полимеризации Rv в течение
одного полного цикла будет составлять лишь одну четвертую часть
от скорости в стационарном состоянии, так как система освещает-
ся только одну четвертую часть времени, т. е.
= (3.156)
Если продолжительность цикла уменьшается (частые вспышки),
то количество радикалов в процессе затемнения не успевает
снизиться до нуля и в течение периода освещения не достигается
их стационарная концентрация (см. рис. 3.12). Средняя величина
концентрации радикалов за один цикл становится больше (7?p)s/4t
так как убыль ее за время затемнения меньше, чем прирост в про-
цессе периода освещения. При очень быстрых циклах устанавли-
вается некоторая стационарная концентрация радикалов (кривая
OGH на рис. 3.12), такое состояние эквивалентно полимеризации
при непрерывном освещении с интенсивностью света, уменьшен-
ной в 1/(1 + г) раза. Отношение средней скорости полимериза-
ции (7?р) о» при очень высокой или бесконечной скорости вращения
сектора к скорости полимеризации для стационарного состояния
равно
(^п)оо 1
-,ДР. =----(3.157)
("p)s (1 + г)1^2 ’
а при г = 3 оно равно 1/2. Таким образом, средняя скорость состав-
ляет от х/4 до х/2 скорости для стационарного состояния (Rp)s,
когда частота циклов или частота вспышек 1/(2 + rt) возрастает от
очень маленькой до очень большой величины относительно 1/ts.
Математическая обработка метода прерывистого освещения
впервые была осуществлена Брирсом, Чепменом и Уолтерсом [561
и теперь применена для рассматриваемого случая. После ряда
циклов концентрация радикалов начинает колебаться около
постоянного значения концентрации радикалов [М в конце
каждого периода освещения с продолжительностью t и около
постоянной концентрации [М -]2 в конце каждого периода затем-
нения с продолжительностью t' = rt. Используя уравнения (3.152)
и (3.155а), находим
"Mh(fS)-,rclh(S)=v <ЗЛ58>
218
Глава 3
И
1M-L [M-h
[М-]2 [M-]i
(3.159)
Для данных значений г и t/xs максимальные и минимальные вели-
чины отношений [M’]t/[M*]S и [M-]2/[M-]s можно рассчитать
с помощью _у равнений (3.158) и (3.159). Средняя концентрация
радикалов [М •] в течение одного или нескольких циклов опреде-
ляется как
t г
[М«] = f [М-]сЙ+ ( \M-]dt',
о о
(3.160)
где первый интеграл (для периода освещения) находится из урав-
нения (3.157), а второй интеграл (для периода затемнения) дается
уравнением (3.159). Определение интегралов [57] в уравнении г
(3.160) приводит к
(М-] n-х Г-1 щ т’1п /IM-h/lM-h+lM-MM-H-l
Из уравнений (3.161), (3.158) и (3.159) для данного значения г
можно найти отношение [М-]/[М-]s как функцию t/xs. Полу-
логарифмическая анаморфоза такой зависимости при г = 3 при-
Р и с. 3.13. Полулогарифмическая
зависимость 2[M-]/[M-]sot t!xs [57].
ведена на рис. 3.13. В соответствии с вышерассмотренным
концентрация радикалов уменьшается от половины ее значения
в стационарном состоянии при частых вспышках до одной четвер-
ти при редких вспышках.
Для определения величины ts для данной системы враща-
ющийся сектор помещают между источником света и реакционным
сосудом. Величина сектора и определяет значение г. Вначале
в отсутствие сектора определяют скорость полимеризации для
стационарного состояния. Затем измеряют Rp в присутствии сек-
тора, вращая его со все возрастающей скоростью. Продолжитель-
ность цикла, а также t и ? определяются скоростью вращения
Ра икальная полимеризация
219
сектора. Затем строят графическую зависимость 7?p/(7?p)s или
/?р'(/?р)оо [связанных между собой уравнением (3.157)] от лога-
рифма t. После этого теоретическую кривую (например, рис. 3.13)
для того же значения г сравнивают с экспериментальной кривой,
для чего первую накладывают на верхнюю часть эксперименталь-
ной кривой и передвигают ее вдоль абсциссы до наилучшего их
совпадения. Указанные кривые отличаются между собой по абсцис-
се па величину lgxs, так как на абсциссе теоретической кривой от-
ложена разность lg£ — lgxs. На рис. 3.14 приведены теоретическая
Р п с. 3.14. Зависимость отноше-
ния скорости полимеризации ме-
такриламида при прерывистом
освещении со световыми периода-
ми t (секунды) к скорости поли-
меризации при очень высокой
(бесконечно большой скорости)
вращения сектора от продолжи-
тельности t [58].
Сплошная линия — теоретическая кри-
вая; точки — экспериментальные дан-
ные.
и экспериментальная кривые для полимеризации метакриламида
в водном растворе, инициируемой фотохимическим распадом
перекиси водорода [58].
Определив экспериментально т8, рассчитывают кр, к0, кпер и kz.
Из уравнений (3.24) и (3.147) находят отношения кр/$2 и кр/к0. Из
них можно рассчитать индивидуальные константы кр и к0. С по-
мощью уравнений (3.21) и (3.22) рассчитывают [М-]s и Ro. По
константам передачи цепи и ингибирования находят А'П(!р и kz.
3.8в. Значения параметров реакции полимеризации
В табл. 3.8 приведены значения концентраций, скоростей
и констант скоростей фотополимеризации метакриламида и наряду
с ними указаны пределы изменения различных параметров ради-
кальной полимеризации вообще. При этом экспериментально опре-
делены Ra, (Rp)s, [М], [I], кр/кгр2, ts и кр/к0, тогда как остальные
параметры находили расчетным путем из соответствующих урав-
нений. Как видно из данных этой таблицы, найденные параметры
укладываются в область, характерную для реакций радикальной
полимеризации. По величине кр (~103) на много порядков превы-
шает константы скорости обычных химических реакций (напри-
мер, как видно из табл. 2.1, 2.2 и 2.5, константы скорости этери
220
лава
Таблица 3.8
Параметры реакции радикальной полимеризации
Параметр Единица измерения Общий порядок величин Значения параметров при фотополимери- зации метакрил- амида [58]
моль/(л-с) -10-8—10-ю 8,75-10-9
кг с-1 10-4— 10-е —
[I] моль/л 10-2—10-4 3,97-10-2
[M-]s моль/л Ю-7—10-9 2,30-10-8
(^p)s моль/(л-с) Ю-4— ю-в 3,65-10-в
[MJ моль/л 10 -10-1 0,20
Zfp л/(моль-с) 102 —104 7,96-102
Ro моль/(л-с) Ю-8—10-ю 8,73-10-9
л/(моль-с) 106 —108 8,25-10е
Ts с 10-1—10 2,62
*p/*o — 10-4—10-е 9,64-10-®
Л1/2/(МОЛЬ.С)1''2 1 — 10-2 2,77-10-1
фикации, образования уретанов и фенолформальдегидных полиме-
ров составляют соответственно приблизительно 10~3, 10~2 и 10-6).
Поэтому рост цепи идет очень быстро и высокомолекулярный по-
лимер образуется практически мгновенно. Однако вследствие еще
больших значений к0 (~107) полимеризационная система характе-
ризуется быстрым обрывом растущей цепи, низкой концентрацией
радикалов (~10-8 М) и низкой продолжительностью жизни ради-
калов. В исследованных условиях продолжительность жизни
радикалов метакриламида составляла 2,62 с, но в других случаях
она может быть и намного меньше. Интересно, что эксперименталь-
но найденная Ra (8,75-Ю"9) очень близка к расчетной Ro
(8,73-10-9), что подтверждает справедливость предположения
о стационарном состоянии.
Константы скорости роста и обрыва цепи были определены для
процессов полимеризации многих мономеров. Для некоторых
наиболее распространенных мономеров они представлены в
табл. 3.9, причем мономеры указаны в порядке, соответствующем
уменьшению реакционной способности их радикалов (см. разд.З.бв).
Значения кр хорошо укладываются в такой порядок, тогда как
в ряду к0 есть некоторые отклонения. Это связано с тем, что, как
уже было указано, реакционная способность радикала зависит не
только от его собственной активности, но также и от природы
вещества, с которым он реагирует.
Радикальная полимеризация
22
Таблица 3.9
Кинетические параметры радикальной полимеризации [4, 41]
Мономер /<р- 10-з при 60 °C, л/(моль-с) Е а, ккал/моль (4,186- 103 Дж/моль) o' А R О £ о а feo-10-i при 60 °C, л/(моль-с) Ео б, ккал/моль (4,186-103 Дж/моль) o' А R О S R* Т О
Винилхлорид 12,3 3,7 0,33 2300 4,2 600
Винилацетат 2,30 6,3 3,2 2,9 3,2 3,7
Акрилонитрил 1,96 3,9 — 78,2 3,7 —
Метилакрилат 2,09 7,1 10 0,95 5,3 15
Метилметакрилат 0,705 4,7 0,087 2,55 1,2 0,11
Стирол 0,145 7,3 0,45 2,9 1,9 0,058
Бутадиен-1,3 0,100 9,3 12 — — —
а Расчет сделан на моль полимеризующегося мономера,
б Расчет сделан на моль растущих радикалов.
3.9. Энергетические характеристики
3.9а. Энергия активации и частотный фактор
Зависимость между температурой, скоростью и степенью поли-
меризации имеет большое значение при выборе способа проведе-
ния полимеризации. С увеличением температуры реакции возра-
стает скорость полимеризации и уменьшается молекулярный вес
образующегося полимера. Эта зависимость представлена на
рис. 3.15 для чисто термической полимеризации стирола [59].
Однако количественную зависимость между указанными парамет-
рами установить непросто, так как 7?р и Хп зависят от констант
кГ, кр, и к0 сложным образом. Каждую из указанных констант
можно связать с температурой через уравнение Аррениуса
k = Ae~E/RT (3.162а)
или
In & = In А-^г , (3.1626)
где А — частотный фактор, Е — энергия активации Аррениуса,
Т — температура в градусах Кельвина. Построив кривую зависи-
222
Глава 3
мости In к от ИТ, можно определить Е и А из наклона такой кри-
вой и отрезка, отсекаемого на оси ординат. Энергии активации
роста Ер и обрыва цепи Ео при полимеризации ряда мономеров
приведены в табл. 3.9. Интересно заметить, что изменения в зна-
чениях Ир, частотного фактора для стадии роста, значительно
Р и с. 3.15. Зависимость ско-
рости полимеризации (7) и мо-
лекулярного веса полимера (2)
от температуры при чисто тер-
мической полимеризации сти-
рола [59].
больше, чем изменения Ер, что свидетельствует о том, что стери-
ческие эффекты, по-видимому, играют основную роль в определе-
нии абсолютной величины кр. Поэтому стерически более затруд-
ненные мономеры (например, метилметакрилат) имеют более низ-
кие кр и Лр по сравнению с менее стерически затрудненными моно-
мерами. По своей величине Лр, как правило, значительно ниже,
чем частотный фактор в обычных бимолекулярных реакциях
(1011—1013), что может быть обусловлено значительным пониже-
нием энтропии при полимеризации. Изменения величины Ло,
частотного фактора для стадии обрыва цепи, в общем подчиняются
тем же закономерностям, что и для Лр, но по абсолютным значе-
ниям Ло, как правило, выше Лр.
3.9а.1. Скорость полимеризации
Рассмотрим энергию активации различных реакций радикаль-
ной полимеризации. При термическом способе инициирования ско-
рость полимеризации зависит от отношения трех констант скорости
кр{кг1к0)112 [см. уравнение (3.28)]. Путем решения трех уравне-
ний Аррениуса для отдельных этапов реакции находят темпепа-
Радикальная полимеризация
223
турную зависимость для такого соотношения
1 Гь / кг \1/2П I Гл ( \1/2“| 1£р + (£г/2) — (£о/2)] R
lnLMir) J= 1п 1_Лр ( а? ) J--------------RT-------• <зл63>
Суммарная энергия активации для скорости полимеризации ER
равна [Е^ + (Ег/2)—(Ео/2)]. Из уравнений (3.28) и (3.163) нахо-
дим
1п7?р = 1п[лр (4;)1/2] + In [(/ [11)1/2[М]]~§-• (3.164)
Er и Лр (Иг/И0)1/2 можно найти по наклону кривой зависимости
In Rp от 1/Т и отрезку, отсекаемому ею на оси ординат соответ-
ственно (аналогично рис. 3.15).
Для большинства применяющихся инициаторов энергия акти-
вации распада инициатора Ег составляет примерно 30 —
35 ккал/моль (125,5-103—146,5 ИО3 Дж/моль) (табл. 3.10) [41].
Таблица 3.10
Термический распад инициаторов
Инициатор Ег hr-105 Г, °C
2,2'-Азо-бис-изобутиронитрил 29,5 0,85 60
Перекись ацетила 32,5 8,7 80
Перекись бензоила 29,7 4,7 85
Перекись кумола 40,7 1,6 115
Перекись mpem-бутпла 35,1 3,0 130
Гидроперекись mpem-бутила 40,8 0,43 155
Для большинства известных мономеров Ер и Ео находятся в интер-
вале 5—10 ккал/моль (20,9-103—41,8-103 Дж/моль) и 2 —
5 ккал/моль (8,4-103—20,9-103 Дж/моль) соответственно (табл. 3.9
и [41]), поэтому суммарная энергия активации ER для большин-
ства реакций полимеризации, инициированных в результате
термического распада инициатора, составляет примерно
20 ккал/моль (83,6 -103 Дж/моль).Это отвечает двух- или трехкратно-
му увеличению скорости при повышении температуры на 10 °C. При
использовании других способов инициирования положение резко
меняется. Так, например, выше было показано, что инициирование
в присутствии окислительно-восстановительных систем (Fe2+ в со-
четании с тиосульфатом или гидроперекисью кумола) идет при
более низких температурах, чем термическая полимеризация. Вли-
яние способа инициирования проявляется и на энергии активации
процесса инициирования. Так, при окислительно-восстановитель-
2 4
Глава 3
ном инициировании Ег составляет только 10—15 ккал/моль
(41,8-103—62,5 ПО3 Дж/моль), что примерно на 20 ккал/моль
(83,6-103 Дж/моль) меньше энергии активации термического ини-
циирования 1601. В результате этого ER для окислительно-вос-
становительной полимеризации не превышает 10 ккал/моль
(41,8-103 Дж/моль), что приблизительно в два раза меньше, чем
Ен для полимеризации в присутствии других инициаторов.
На процесс инициирования в чисто фотохимической полимери-
зации температура не оказывает никакого влияния (Ег = 0), так
как распад инициатора вызывается квантами света. Суммарная
энергия активации фотохимической полимеризации составляет
только 5 ккал/моль (20,9 -103 Дж/моль). Это показывает, что фото-
химическая полимеризация в отличие от других реакций ради-
кальной полимеризации мало чувствительна к изменению темпе-
ратуры. Однако влияние температуры на фотохимическую полиме-
ризацию не является таким простым, как это кажется на первый
взгляд, так как большинство применяющихся в таком про-
цессе инициаторов также может подвергаться чисто термическому
распаду. Поэтому при повышенных температурах, кроме фото-
химического распада инициаторов, может в заметной степени идти
их термическое разложение. В таких случаях необходимо учиты-
вать термическое и фотохимическое инициирование. Энергия
активации инициирования и кажущаяся энергия активации чисто
термической полимеризации являются величинами того же поряд-
ка, что и при полимеризации в присутствии инициаторов, однако
эта реакция протекает со значительно меньшей скоростью, что
объясняется низкой вероятностью инициирования такой поли-
меризации вследствие крайне малых величин (104—106) частотного
фактора.
3.9а.2. Степень полимеризации
Для оценки влияния температуры на молекулярный вес поли-
мера, образующегося при термической полимеризации в присут-
ствии инициаторов, где передача цепи не играет существенной роли,
следует рассмотреть соотношение kvl(krlk0)112, так как от него за-
висит степень полимеризации [уравнение (3.101)]. Температурный
коэффициент, характеризующий изменение молекулярного веса
с температурой, определяется из соотношения
г *р -| г 1 [2?Р“ (£г/2)-(£о/2)1
1п 1( W/.J =1,1 Lsvs]------------ГТ-------• <зл65>
где [£р —(£г/2)—(£0/2)] = Е^п представляет собой энергию акти-
вации образования полимера со степенью полимеризации Хп.
При бимолекулярном обрыве для Хп справедливы уравнения
Радикальная полимеризация
2
(3.101) и (3.102а), тогда можно записать
1пХп = 1пГ Лр -1+1П Г-^L-I-^. (3.166)
Ь(Лгл0) /aJ rt
Ехп Равна примерно —15 ккал/моль (—62,5 -103 Дж/моль), следо-
вательно, при повышении температуры молекулярный вес умень-
шается. Уравнение (3.166) справедливо также для чисто терми-
ческой полимеризации (см. рис. 3.15). При чистой фотополимери-
зации Е-^ положительна и равна приблизительно 5 ккал/моль
(20,9-103 Дж/моль), так как Ет равна нулю; следовательно,
Хп несколько увеличивается с повышением температуры. Во всех
других случаях Хп понижается с температурой.
Если в процессе полимеризации передача цепи играет сущест-
венную роль, то для расчета Хп следует воспользоваться уравне-
нием (3.109). Температурная зависимость Хп может быть весьма
сложной, что определяется относительным вкладом различных
членов уравнения (3.109). Если лимитирующей реакцией является
передача цепи на агент передачи цепи S [уравнение (3.116)], то
Таблица 3.11
Активационные параметры передачи цепи
в полимеризации стирола при 60 °C [18, 42, 61]
Агент передачи цепи - (Е - Е ч),ккал/моль 1/ 11 (4,186 • 103 дж/моль) (dy/S -<1эпу) gT
Циклогексан 13,4 3,1
Бензол 14,8 3,9
Толуол 10,1 1,7
Этилбензол 5,5 -0,55
Изопропилбензол 5,5 -0,47
трет-Бутплбензол 13,7 3,8
и-Бутилхлорид 14 4
и-Бутилбромид 11 2
н-Бутилиодид 7 1
Четыреххлористый углерод 5 1
226
Глава 3
получаем
-In Г Ж ( J----=lnJ^ =1п^^— £p-~/°ep-S.
L [S] \ xn (^n)o'J ^nep. S -^nep-S TIT
(3.167)
Здесь E^n равна (Ev — Enev, s); определяется она с помощью кри-
вой зависимости любой из двух форм левой части уравнения (3.167)
от ИТ. Обычно j&nep. s превышает Ev на 5—10 ккал/моль (20,9 X
X 103—41,8 -103 Дж/моль), причем более активные агенты переда-
чи цепи имеют более низкие энергии активации [18, 42, 61]. Из
данных табл. 3.11 видно, что разность (Ev — Епер s) в большинстве
случаев равна —5 4----20 ккал/моль (—20,9 -103 4--83,6 X
X 103 Дж/моль), поэтому молекулярный вес уменьшается с повы-
шением температуры. Частотный фактор для реакции передачи
цепи обычно выше, чем для реакции роста, поэтому низкая кон-
станта передачи цепи для данного агента передачи цепи объяс-
няется только высокой энергией активации.
3.96. Термодинамика полимеризации
3.96.1. Значение AG, АН и Д5
Термодинамические характеристики полимеризации (Дб, ДЯ,
Д5) имеют большое значение для понимания влияния структуры
мономера на процесс полимеризации. Далее, зная ДЯ, можно
путем подбора соответствующего теплового режима процесса под-
держивать на заданном уровне 7?р и Хп и получать полимер жела-
емого молекулярного веса. Величины Дб, АН и Дб в полимериза-
ции представляют собой разность свободных энергий, энтальпий
и энтропий соответственно одного моля мономера и одного моля
повторяющихся звеньев в молекуле полимера. Термодинамика
полимеризации определяется только стадией роста, так как стадии
инициирования и обрыва цепи являются единичными актами,
тогда как на стадии роста цепи идет большое число реакций при-
соединения.
Полимеризация алкенов — экзотермический (отрицательная
АН) и экзоэнтропийный (отрицательная Дб) процесс. Экзотерми-
ческий характер полимеризации можно объяснить тем, что про-
цесс заключается в экзотермическом превращении л-связей
мономеров в о-связи полимеров. Отрицательная AS полимериза-
ции обусловлена тем, что при переходе от мономера к полимеру
уменьшается число степеней свободы. Следовательно, полимери-
зация благоприятна с точки зрения энтальпийного фактора, но
невыгодна по энтропийным соображениям. Как видно из табл. 3.12,
АН различных мономеров изменяется в довольно широких преде-
лах [62]. AS изменяется в более узком интервале: от —25 до
Радикальная полимеризация
227
—30 кал/(моль«К) [—104,6 4-------124,5 Дж/(моль* К)]. Для
определения ДЯ и Д5 применяют различные методы [63].
К ним относятся прямые калориметрические измерения ДЯ поли-
меризации, определение разницы между теплотами сгорания
мономера и полимера, измерение константы равновесия полиме-
ризации. В общем с точки зрения термодинамики полимеризация
алкенов является весьма выгодным процессом. Изменение свобод-
ной энергии Дб, определяемое по формуле
Дб = ДЯ—ГДб, (3.168)
величина отрицательная, так как отрицательный член TAS пере-
крывается, т. е. меньше также отрицательного члена АН.
Здесь, вероятно, имеет смысл вернуться к вопросу о термодина-
мической и кинетической разрешимости полимеризации (разд. 3.16).
Согласно данным табл. 3.12, термодинамически разрешима поли-
Таблица 3.12
Теплота и энтропия полимеризации при 25 °C [62]
Мономер -ДНа, ккал/моль (4,186-103 Дж/моль) -AS®, кал/(моль-К) [4,186 Дж/(моль-К)]
Этилен 22,7 24
Пропилен 20,5 в 27,8
Бутен-1 20,0 в 26,8
Изобутилен 12,3 28,8
Бутадиен-1,3 17,4 20,5
Изопрен 17,8 24,2
Стирол 16,7 25,0
а-Метилстирол 8,4 24,8
Винилхлорид 22,9
Винилиденхлорид 18,0
Винилиденфторид 30
Тетрафторэтилен 37,2 26,8
Акриловая кислота 16,0
Акрилонитрил 18,4
Малеиновый ангидрид 14
Винилацетат 21,0 26,2
Метилакрилат 18,8
Метилметакрилат 13,5 28,0
чеокий палимерЧаеТ превраП1ению жидкого мономера в аморфный илн слабокристалли-
кристал^ескГй ТпоПлимеРрЩ''НИЮ мономера (концентрация 1 М) в аморфный нли слабо-
в Из работы [41].
28
Глава 3
меризация любых мономеров с двойными углерод-углеродными
связями. При этом, хотя относительная термодинамическая раз-
решимость для каждого мономера зависит от его химического стро-
ения (наличия заместителей), Дб? отрицательна во всех случаях,
что делает полимеризацию выгодной. Однако нельзя только на
основании термодинамической разрешимости полимеризации по-
добрать экспериментальные условия ее осуществления. Как видно
из табл. 3.1, кинетическая вероятность полимеризации в значи-
тельной степени определяется химическим строениэм мономера,
так как от последнего зависит, какой тип инициатора — радикал,
катион или анион — является наиболее подходящим в том или
ином конкретном случае. Иногда для осуществления термодина-
мически возможной полимеризации могут потребоваться очень
специфические катализаторы. Это относится, например, к а-оле-
финам, на основе которых не удается получить высокомолекуляр-
ные полимеры в присутствии обычных радикальных или ионных
инициаторов. Полимеризация таких мономеров стала возможной
только после разработки Циглером и Натта новых катализаторов
координационного типа (гл. 8).
3.96.2. Влияние химического строения мономера
Рассмотрим влияние природы мономера на энтальпию полиме-
ризации. Величина \Н этилена —22,7 ккал/моль (—95,0 X
X 103 Дж/моль) очень близка разности ~20—22 ккал/моль
(-~83,7 -103—92,1 -103 Дж/моль) энергий л-связи в алкенах и
о-связив алканах. Для других мономеров ДЯ сильно зависит от их
химического строения, что обусловлено:
1) разной резонансной стабилизацией мономера и полимера
из-за неодинакового сопряжения или гиперконъюгации;
2) разными пространственными напряжениями в мономере
и полимере, вызванными изменениями валентных углов, формы
связей или взаимодействий между не связанными между собой
атомами;
3) разными водородными связями или диполь-дипольными
взаимодействиями в мономере и полимере.
Многие заместители стабилизируют мономер, но не оказывают
заметного влияния на устойчивость полимера, так как резонанс
возможен только в мономере. Следствием этого является умень-
шение экзотермического эффекта полимеризации. Так, из-за гипер-
конъюгации алкильных групп с С = С -связью ДЯ полимериза-
ции пропилена и бутена-1 понижается. Сопряжение двойной
связи С=С с такими группами, как бензольное кольцо (стирол
и а-метилстирол), двойная связь диенов (бутадиен, изопрен),
карбонильная связь (акриловая кислота, метилакрилат, метил-
метакрилат), нитрильная (акрилонитрил), приводит к стабилиза-
Радикальная полимеризация
9
ции мономеров и понижению энтальпии их полимеризации. В то
же время при полимеризации мономеров с ослабленным сопряже-
нием заместителей с С = С-связью, например винилацетата, ДЯ
становится близкой энтальпии полимеризации этилена.
Эффект 1,1-дизамещения проявляется в пониженных значениях
ДЯ. Это связано со стерическими напряжениями в полимере,
обусловленными взаимодействием заместителей при регулярно
чередующихся (одинаково расположенных) атомах углерода в по-
лимерной цепи
1,3 ~ взаимодействие
VII
На этой схеме основная полимерная цепь находится в плоскости
страницы, тогда как заместители Н и Y расположены над пло-
скостью листа и под ней, причем пунктирные линии ведут к заме-
стителям, находящимся под плоскостью листа, а треугольники —
к заместителям поверх листа. Подобные взаимодействия, называ-
емые 1,3-взаимодействиями, ответственны за понижение ДЯ
в таких мономерах, как изобутилен, а-метилстирол, метилмет-
акрилат и винилиденхлорид. Особенно сильный эффект имеет
место в случае а-метилстирола, теплота полимеризации которого
—8,4 ккал/моль (—35,4-Ю3 Дж/моль) — наименьшая среди всех
изученных мономеров.
В некоторых случаях понижение ДЯ обусловлено уменьшени-
ем водородных связей или дипольного взаимодействия в результате
полимеризации. Существенная стабилизация таких мономеров,
как акриловая кислота и акриламид, обусловлена сильным меж-
молекулярным взаимодействием. В полимере оно не играет замет-
ной роли, так как из-за стерических факторов взаимодействие
заместителей не может проявиться в достаточной степени.
Не так просто объяснить наблюдаемые значения ДЯ при
полимеризации винилхлорида, винилиденхлорида и тетрафтор-
этилена. Учитывая повышенную резонансную стабилизацию таких
мономеров и повышенные стерические напряжения в полимерах
на их основе, следовало, по-видимому, ожидать, что ДЯ полиме-
ризации указанных мономеров должна быть ниже, чем ДЯ поли-
меризации этилена. Однако полимеризация винилхлорида со-
провождается примерно таким же тепловым эффектом, что
и полимеризация этилена, а винилиденфторид и тетрафторэтйлен
30
Глава 3
характеризуются очень высокими значениями —АН полимери-
зации [30 и 37,2 ккал/моль (125,6 *103 и 155,7 «103 Дж/моль) соот-
ветственно]. Кроме того, высокое значение—Деполимеризации
винилиденхлорида становится еще более удивительным, если
учесть, что этот мономер относится к 1,1-дизамещенным производ-
ным этилена. Возможно, что такие высокие—АН обусловлены
резонансной стабилизацией полимера вследствие межмолекуляр-
ной ассоциации (диполь-дипольное взаимодействие).
В то время как ДЯ колеблется в довольно широких пределах
в зависимости от природы мономера, Д5 не чувствительна к хими-
ческому строению мономера и для всех них составляет примерно
—25 -=--30 кал/(моль-К) [—104,6 4--125,5 Дж/(моль-К)] [41,
62]. Как уже было выше указано, доля TAS в полимеризации
невелика и мало меняется при переходе от одного мономера к дру-
гому. Действительно, величина ГДS при 50 °C для всех мономеров
изменяется в интервале 8,1—9,7 ккал/моль (33,9-10®—58,6 X
X Ю3 Дж/моль). Исследования изменения энтропии в процессе
полимеризации проводились на примере нескольких мономеров [63,
64], и было показано, что AS обусловлена главным образом поте-
рей поступательной энтропии мономера. Потери вращательной
и колебательной энтропии мономера практически компенсируются
выигрышем вращательной и колебательной энтропии полимера.
Следовательно, AS полимеризации — это практически поступа-
тельная энтропия мономера, которая мало зависит от природы
мономера.
3.96.3. Полимеризация 1,2-дизамегценных
производных этилена
1,2-Дизамещенные производные этилена, например малеиновый
ангидрид, стильбен, и 1,2-дихлорэтилен
О
О = С^ \=О
С = С С6Н5СН = СНС6Н5 С1СН = СНС1
Н Н
характеризуются очень низкой способностью к полимеризации,
что обусловлено особенностями стерической структуры этих
мономеров, но в основе такого поведения лежат другие причины,
нежели те, которые затрудняют полимеризацию 1,1-дизамещен-
ных производных этилена.
Полимеры на основе 1,2-дизамещенных производных этилена
(схема VIII) содержат один заместитель на каждый атом С основной
полимерной цепи. Как показано на моделях, в таких соединениях
напряжения не очень сильные. Стерическое напряжение, вызван-
Радикальная полимеризация
231
VIII
ное взаимодействием заместителей Y, в таких структурах значи-
тельно меньше, чем в полимерах на основе 1,1-дизамещенных про-
изводных этилена (ср. структуры VII и VIII). Так, АН для малеи-
нового ангидрида составляет —14 ккал/моль (—58,6 *103 Дж/моль),
что должно приводить к более легкой полимеризуемости такого
мономера по сравнению с 1,1-дизамещенными производными
этилена.
Очень низкая склонность 1,2-дизамещенных этиленов к поли-
меризации связана прежде всего не с термодинамическими факто-
рами, а с кинетической разрешимостью процесса. Подход расту-
щего радикала к молекуле мономера стерически затруднен.
Поэтому рост цепи идет очень медленно из-за взаимодействий между
0-заместителем растущей частицы и обоими заместителями входя-
щей молекулы мономера
При этом, естественно, возрастает роль реакций обрыва или
передачи цепи.
3.9в. Равновесие полимеризация—}
деполимеризация. Предельная температура
Большинство реакций полимеризации характеризуется темпе-
ратурой, при которой они становятся обратимыми, т. е. рост
цепи [уравнение (3.15)1 правильнее писать в следующем виде:
ftp
Mn-+M —Мп+1., (3.169)
&пеп
3
Глава 3
где клеп — константа скорости обратной реакции, называемой
деполимеризацией. Таким образом, из-за наличия равновесия
полимеризация — деполимеризация роль температуры в полиме-
ризации достаточно сложна. В процессе полимеризации мономера
с повышением температуры скорость полимеризации увеличивает-
ся, так кай*возрастает (разд. 3.9а.1). Однако при более высоких
температурах константа скорости деполимеризации, которая вна-
чале равнялась нулю, становится все более и более заметной при
Рис. 3.16. Зависимость ftp [М]
и Агдеп от температуры при по-
лимеризации стирола [63].
дальнейшем повышении температуры. Наконец, при какой-то
температуре, называемой предельной температурой Тпр, скорости
полимеризации и деполимеризации становятся равными. На
рис. 3.16 это иллюстрируется на примере полимеризации стирола.
При предельной температуре суммарная скорость образования
полимера превращается в нуль.
Равновесие полимер —мономер, определяемое с помощью урав-
нения (3.169), зависит от температуры процесса, причем с повы-
шением температуры равновесие сдвигается влево, так как прямая
реакция экзотермична. Изотерма реакции
AG = AG° + /77’lnK (3.170)
может быть использована для анализа равновесия полимериза-
ция — деполимеризация. Величина AG° представляет собой AG
мономера и полимера в соответствующих стандартных состояниях.
Для мономера это жидкость или одномолярный раствор. За поли-
мер в стандартном состоянии принимается аморфный или слабо-
кристаллический полимер или его одномолярный раствор (в рас-
чете на повторяющееся звено полимера). В состоянии равновесия
AG = 0 по определению и из уравнений (3.170) и (3.168) можно
найти, что
AG° = A#°—TbS° = — RT\nK. (ЗЛ71\
Радикальная полимеризация
33
Константа равновесия определяется как кр/кяеп или
К [Мп+г] =_1_ (3.172)
Л [МП.][М] [М] <• f
Комбинация уравнений (3.171) и (3.172) дает
= Д5° + Я1п[М]к (3'173а)
ИЛИ
•Ш[М]к = ^-^. (3.1736)
Уравнение (3.173) выражает зависимость равновесной (критиче-
ской) концентрации мономера [М]к от предельной температуры
полимеризации Тпр. Поскольку Д№ — отрицательная величина,
с повышением температуры концентрация мономера в равновесии
с полимером возрастает, т. е. зависимость In [М]к от 1/Т представ-
ляет прямую линию с отрицательным наклоном \H°IR и отсекае-
мым отрезком —&S°/R. Это говорит о том, что существует ряд
предельных температур, соответствующих различным равновесным
концентрациям мономера. Для любого раствора концентрации
[М]к есть своя Тир, при которой полимеризация не идет. (Для
каждой [М]к существует кривая, подобная приведенной на
рис. 3.16, для которой кяея = кр [Ml при соответствующей Тпр.)
Иными словами, полимеризация данного раствора мономера при
заданной температуре идет до установления равновесия, т. е. до
тех пор, пока концентрация мономера не станет равной [М]к,
отвечающей его Гцр. Следовательно, для получения высокомолеку-
лярного полимера при повышенных температурах процесс необхо-
димо вести при высоких начальных концентрациях мономера.
Существует верхняя предельная температура, выше которой
полимер нельзя получить, даже если исходить из чистого мономе-
ра, а не из его раствора. Здесь следует обратить внимание чита-
теля на то, что в литературе часто встречается только одно значе-
ние Уцр. Из вышесказанного должно быть ясно, что для каждой
концентрации мономера есть своя собственная Тпр. Встречающая-
ся в литературе величина Гцр обычно относится к Тпр для чистого
мономера или в некоторых случаях для одномолярного раствора
мономера.
Для большинства алкенов равновесие рост — деструкция
сдвинуто сильно вправо в обычных условиях полимеризации, т. е.
реакцию полимеризации удается довести до высоких степеней
завершенности. В табл. 3.13 указаны критические концентрации
мономеров при 25 °C, а также предельные температуры для
чистых мономеров. Как видно из этих данных, в полимерах имеет-
ся небольшое количество остаточного мономера, определяемое
с помощью уравнения (3.173). Кроме того, есть ряд мономеров,
Таблица 3.13
Равновесие полимеризация— деполимеризация
[65—68]
Мономер [М]к при 25 °C, моль/л тпр ДЛЯ чистого моно- мера, °C
Винилацетат МО"»
Метилакрилат 1•IO"» —
Стирол 1-10-’ 310
Метилметакрилат 1-Ю-з 220
а-Метилстирол 2,2 61
у которых указанное равновесие сдвинуто влево. Так, 2,2 М
раствор а-метилстирола не полимеризуется при 25 °C. Чистый
<х-метилстирол не полимеризуется при 61 °C. Метилметакрилат
занимает в этом смысле промежуточное положение. Чистый мономер
полимеризуется при температурах вплоть до 220 °C, но с количе-
ственным выходом полимер получить не удается. Так, при поли-
меризации метилметакрилата при НО °C [М]к составляет 0,139 М
[41]. Как будет показано ниже, уравнения (3.173а) и (3.1736)
справедливы также для процессов ионной полимеризации
и полимеризации с раскрытием цикла. При более низких темпе-
ратурах ^ионной полимеризации удается заполимеризовать моно-
меры, которые из-за низких предельных температур не полимери-
зуются по радикальному механизму. Поэтому, когда говорят, что
удалось заполимеризовать мономер, который считался ранее
неполимеризующимся, это значит, что разработан способ про-
ведения полимеризации такого мономера при температуре, лежа-
щей ниже его предельной температуры.
Не следует думать, что проведение полимеризации и исполь-
зование полимера при температурах, превышающих предельную,
лишено всякого смысла. Если ^образовавшийся полимер вывести
из сферы реакции, то при условии отсутствия в нем активных
центров он будет устойчивым и не будет деполимеризоваться, до
тех пор пока такие активные центры не образуются в нем в резуль-
тате термического, химического, фотохимического или другого
воздействия. При наличии активных центров начнется процесс
деполимеризации, который будет продолжаться до того момента,
пока концентрация мономера не станет равной [М]к для данной
температуры. Однако на практике термическое поведение полиме-
ров является более сложным. При температурах, лежащих ниже
предельной температуры, в полимере, кроме деполимеризации,
часто развиваются другие деструктивные процессы.
Радикальная полимеризация
235
3.10. Аутоускорение
3.10а. Ход процесса
Особенностью радикальной полимеризации является ее ауто-
ускорение на глубоких стадиях превращения мономера в полимер.
Основываясь на общих положениях, следовало ожидать умень-
шения скорости полимеризации с ростом продолжительности
реакции, так как при этом снижается концентрация мономера
Рис. 3.17. Аутоускорение в процессе полимеризации метилметакрилата
при 50 °C в растворе бензола в присутствии перекиси бензоила как инициа-
тора при различных концентрациях мономера в растворе [69].
(а также концентрация инициатора). Однако многие реакции
полимеризации характеризуются прямо противоположным поведе-
нием, а именно увеличением скорости полимеризации по мере
образования полимера. На рис. 3.17 приведены типичные кривые,
отражающие это явление на примере полимеризации метилмет-
акрилата в растворе бензола [69]. Полимеризация 10%-ного рас-
твора метилметакрилата подчиняется обычным закономерностям.
При полимеризации чистого мономера наблюдается очень сильное
увеличение скорости полимеризации. Такое поведение получило
название гель-эффект. В более ранних работах для него можно
встретить также такие обозначения, как эффект Тромсдорфа
и эффект Норриша — Смита.
Появление гель-эффекта обусловлено уменьшением константы
скорости реакций обрыва при углублении полимеризации. По
мере протекания полимеризации вязкость системы увеличивается,
и обрыв становится все менее и менее вероятным. Хотя рост цепи
также замедляется, этот эффект выражен в значительно меньшей
степени, так как kv ниже к0 примерно на 4—5 порядков. При
обрыве цепи реагируют два макрорадикала, тогда как в росте цепи
участвует один макрорадикал и одна маленькая молекула моно-
мера. Поэтому прежде всего высокая вязкость системы оказывает
влияние на первый процесс, а не на второй. Это приводит к росту
отношения /ср/Тсо2, что, согласно уравнению (3.24), сопровождается
увеличением 77 р с увеличением степени завершенности реакции.
Вторым следствием такого поведения является повышение моле-
кулярного веса полимера с увеличением степени превращения
в соответствии с уравнением (3.98). Эти выводы получили экспе-
риментальное подтверждение в результате измерения зависимости
кр и к0 от степени завершенности реакции. В качестве примера
в табл. 3.14 приведены данные для полимеризации метилметакри-
Таблица 3.14
Влияние степени превращения на полимеризацию
метилметакрилата при 22,5 °C (70]
Степень превраще- ния, % Скорость, %/ч т, с fep ho.10-5 (hp/fc^/z-ioa
0 3,5 0,89 384 442 5,78
10 2,7 1,14 234 273 4,58
20 6,0 2,21 267 72,6 8,81
30 15,4 5,0 303 14,2 25,5
40 23,4 6,3 368 8,93 38,9
50 24,5 9,4 258 4,03 40,6
60 20,0 26,7 74 0,498 33,2
70 13,1 79,3 16 0,0564 21,3
80 2,8 216 1 0,0076 3,59
лата [70]. Как видно из данных этой таблицы, до степени превра-
щения примерно 50% кр мало меняется, тогда как к0 при этом
уменьшается почти на два порядка. Отношение кр/к}!,2 и скорость
полимеризации сначала одновременно быстро вырастают, а затем
уменьшаются, так как на заключительных этапах реакции кр тоже
начинает зависеть от вязкости системы. Необходимо обратить
внимание на то, что продолжительность жизни радикалов также
возрастает по мере увеличения степени превращения.
3.106. Диффузный фактор
Согласно ранним работам, эффект Тромсдорфа определялся
как момент в процессе, при котором реакция обрыва начинает
лимитироваться диффузией. Однако это определение явно неудач-
но, так как обрыв лимитируется диффузией почти на всех стадиях
Радикальная полимеризация 237
реакции полимеризации. Норт с сотр. [71] считает, что обрыв
идет в следующие три стадии:
1) поступательная диффузия двух растущих радикалов на-
встречу друг другу
(Мп.—мт.) (3.174)
Й2
X
2) перегруппировка двух цепей таким образом, чтобы их ради-
кальные концы находились друг от друга на расстоянии, обеспе-
чивающем возможность химической реакции между ними, это
достигается сегментальной диффузией цепей (т. е. движением
отдельных участков или сегментов полимерной цепи относительно
ДРУГ друга)
(Мл. — -мт.) (Мп./Мт.) (3.175)
X XI
3) химическая реакция двух концевых радикалов
h
(Мп./Мт.) —> Мертвый полимер (3.176)
XI
Обычно реакция (3.176) идет очень быстро, т. е. к /с4, и легко
показать, что
(3.177)
при допущении, что концентрации X и XI равны стационарным.
Отсюда вытекают два крайних случая обрыва. При медленной
поступательной диффузии к3 к2 и
n0 = fct[M.]ar (3.178)
При медленном сегментальном движении к2 к3 и
й Мз [М-]2
л0=* — (3.179)
Следовательно, для двух крайних случаев экспериментально
определяемая к0 соответствует к^ и кгк31к2 соответственно.
Теоретические положения Норта с сотрудниками получили
экспериментальное подтверждение. Было изучено влияние вязко-
сти на полимеризацию ряда метакрилатов при 30 °C и невысоких
степенях превращения, причем вязкость регулировали добавками
диизооктилфталата и ацетата сахарозы [71]. С увеличением вязко-
сти наблюдали непрерывное уменьшение отношения krJk„. В более
238
Глава 3
поздней работе [72] было показано, что в процессе обрыва самой
медленной стадией является сегментальная диффузия, т. е. про-
цесс обрыва лимитируется реакцией (3.175), и скорость реакции
выражается уравнением (3.179).
З.Юв. Влияние условий реакции
Степень превращения, при которой аутоускорение начинает
играть заметную роль, определяется условиями реакции и тща-
тельностью ее осуществления. В результате проведения таких
исследований было показано, что многие мономеры, в том числе
стирол [35] и различные метакрилаты [71, 73], характеризуются
повышением отношения кг./к,;2 уже на ранних стадиях полимери-
зации, подтверждая, что реакция обрыва лимитируется диф-
фузией.
Степень превращения, при которой наблюдается аутоускорение,
и величина его зависят от молекулярного веса образующегося
полимера. Так, поскольку на основе винилацетата получаются
только низкомолекулярные полимеры (из-за реакций передачи
цепи), то гель-эффект при такой полимеризации выражен не так
сильно, как при полимеризации других мономеров. На гель-
эффект влияет также температура полимеризации. При увеличении
температуры вязкость реакционной среды уменьшается. В резуль-
тате этого гель-зффект наступает позже и аутоускорение не такое
сильное. Аналогичные эффекты наблюдаются при проведении
полимеризации в растворе и в присутствии агентов передачи цепи.
Растворители понижают вязкость реакционной среды, под дей-
ствием агентов передачи цепи снижается молекулярный вес поли-
мера, что также сопровождается уменьшением вязкости среды.
На рис. 3.17 показано, как влияет растворитель на полимериза-
цию метилметакрилата.
При использовании растворителей, не растворяющих полимер
(например, метанол для полистирола, гексан для полиметил-
метакрилата), наблюдается заметное увеличение скорости поли-
меризации. Аналогичный эффект имеет место в случае полимери-
зации мономеров, полимеры которых не растворяются или лишь
слабо растворяются в собственных мономерах. Это относится,
в частности, к полимеризации акрилонитрила, винилхлорида,
трифторхлорэтилена и винилиденхлорида [74]. Эффект ускорения
полимеризации в таких гетерогенных системах имеет много общего
с гель-эффектом и обусловлен тем, что к0 уменьшается в большей
степени, чем kv. Растущие полимерные радикалы скручиваются
в клубки вследствие нерастворимости их в растворителях, при-
меняющихся в синтезе полимеров, и в исходных мономерах. Это
приводит к уменьшению вероятности обрыва растущей полимер-
ной цепи, тогда как рост цепи может продолжаться.
Радикальная полимеризация
239
3.10г. Зависимость скорости полимеризации
от инициатора
Вследствие гель-эффекта в реакциях гомогенной полимериза-
ции и гетерогенности ряда реакций радикальной полимеризации
имеют место отклонения от обычной зависимости /?р от корня
квадратного из концентрации инициатора. Показатель степени
при концентрации инициатора в таких зависимостях колеблется
между Vз и 1 [43, 74]. Это свидетельствует о том, что обрыв, веро-
ятно, обусловлен протеканием следующих двух реакций: обычной
реакции второго порядка и реакции первого порядка относительно
полимерных радикалов. Истинный механизм второй реакции
остается неясным, можно лишь предположить, что при этом идут
реакции передачи цепи на мономер, полимер или другие вещества,
содержащиеся в реакционной системе. По мере протекания поли-
меризации роль реакций обрыва первого порядка возрастает,
а показатель степени в зависимости 7?р от R„ достигает 1/2. При
этом показатель степени при R„ в зависимости степени полимери-
зации от 7?и колеблется между —V2 и —1- В условиях гетеро-
генной полимеризации полимерные радикалы могут быть настолько
скрученными, что обрыв их в обычных условиях становится прак-
тически невозмояйным. На присутствие в полимере, образующем-
ся при фотополимеризации, захваченных свободных радикалов
указывает то, что полимеризация не заканчивается в течение
нескольких дней после окончания облучения.
В тех случаях, когда обычный обрыв не играет существенной
роли, возможна реакция обрыва в результате соединения поли-
мерных радикалов и первичных радикалов, образующихся в ре-
зультате реакции (3.13),
Мп- +R. ->МП — R (3.180)
Нетрудно показать, что при обрыве согласно уравнению (3.180)
7?р не зависит от концентрации инициатора, а зависит от [М]2.
Косвенные доказательства указанной зависимости были полу-
чены при проведении полимеризации стирола в присутствии
добавок полистирола, увеличивающего вязкость реакционной
системы [26]. Было показано, что скорость полимеризации при высо-
кой вязкости зависит от [М]2 и [I]0’3. При описании таких систем
лучше всего пользоваться кинетическими схемами, применяющи-
мися в эмульсионной полимеризации (гл. 4).
Не исключено также, что при обрыве цепи в области Тромс-
дорфа и реакциях гетерогенной полимеризации идут оба про-
цесса. На это указывает то, что Rv часто зависит от концентрации
инициатора в степени 0,4—0,8, что может быть связано с одно-
временным протеканием двух реакций обрыва — одной первого
и одной нулевого порядков.
240
Глава 3
3.11. Молекулярновесовое распределение
3.11а. Начальные стадии полимеризации
Молекулярновесовое распределение при радикальной полиме-
ризации имеет более сложный характер, чем в поликонденсации.
В радикальной полимеризации обрыв может иметь место в резуль-
тате нескольких реакций: диспропорционирования, рекомбинации
и передачи цепи. Положение еще больше усложняется в результа-
те того, что молекулярный вес образующегося полимера в каждом
случае зависит от степени завершенности реакции, что связано
с изменением в процессе полимеризации как концентраций моно-
мера и инициатора, так и констант скорости реакций обрыва
и роста. Молекулярновесовое распределение можно сравнительно
просто оценить для начальных стадий полимеризации, где все
кинетические параметры ([М], [I], kv и к0) практически постоянны
175—77]. В этих условиях молекулярный вес полимера не зависит
от степени превращения мономера.
Рассмотрим пример, когда на одну кинетическую цепь при-
ходится одна молекула полимера. Это соответствует полимери-
зации, в которой обрыв идет или путем диспропорционирования,
или путем передачи цепи, или в результате обеих этих реакций.
Приведенный пример аналогичен линейной поликонденсации
(разд. 2.5), поэтому с помощью уравнений (2.45), (2.47), (2.48),
(2.27), (2.54) и (2.55) можно найти числовую долю, весовое и число-
вое распределение, среднечисловую и средневесовую степень
полимеризации и ширину распределения.
Особенность радикальной полимеризации в этом случае заклю-
чается в следующем: р определяется здесь как вероятность того,
что растущий радикал продолжает расти, а не обрывается. Тогда р
равно отношению скорости роста к сумме скоростей вс ех реакций,
в которые может вступать растущий радикал
z’==Rp+7?0 + 7?nep (ЗЛ81)
где Яр, Ro и Япер— скорости роста, обрыва диспропорциониро-
ванием и передачи цепи соответственно. Есть и другая особен-
ность, которую необходимо учитывать при использовании таких
уравнений в радикальной полимеризации. Если в случае поли-
конденсации эти уравнения применимы ко всей реакционной сме-
си, то при описании полимеризации они относятся только к поли-
мерной части реакционной смеси.
Анализ вышеуказанных уравнений показывает, что высоко-
молекулярный полимер (т. е. высокие Хп и Xw) образуется, если р
очень близка единице. Этого и следовало ожидать, принимая во
Радикальная полимеризация
241
внимание положения, о которых шла речь в разд. 3.5. Распреде-
ления, описываемые уравнениями (2.45), (2.47) и (2.48), графи-
чески были представлены на рис. 2.7—2.9. Когда степень завер-
шенности реакции приближается к единице, ширина распределе-
ния в пределе равняется двум.
При обрыве рекомбинацией, в процессе которой одна поли-
мерная молекула образуется из двух кинетических цепей, рас-
пределение по размерам сужается. Можно показать, что для
такой полимеризации справедливы следующие уравнения:
7VX = (1 —р)2 (ж—1) рж-2, (3.182)
wx=-^x(l— р)3 (ж — 1) рх~2, (3.183)
Хп=^-р, (3.184)
(3.185)
(3.186)
Хп
В данном случае Ro в уравнении (3.181) относится к скорости
обрыва рекомбинацией, a Rnep равна нулю. Распределение в этом
случае похоже на распределение при трехмерной поликонден-
сации (разд. 2.7) при / = 2. В пределе при р, стремящемся к еди-
нице, ширина распределения будет равна 1,5.
Если полимерные молекулы образуются в результате различ-
ных реакций обрыва, то распределение по размерам находится
соответствующим суммированием обеих функций распределения.
В этом случае весовое распределение можно рассчитать по следу-
ющей формуле:
= (1 —p)2px-i-|-| (1-Л)ж(1 — р)3(ж-1) рх~2, (3.187)
где А — доля молекул полимера, образующихся в результате
реакций диспропорционирования и передачи цепи [78].
3.116. Высокие степени завершенности
полимеризации
На практике большинство реакций радикальной полимериза-
ции проводится до глубоких степеней превращения. Распределе-
ние по молекулярным весам при углублении процесса становится
более широким, чем на начальных стадиях полимеризации или
поликонденсации. По мере протекания полимеризации концен-
трации мономера и инициатора уменьшаются. В соответствии
242
Глава 3
с уравнением (3.101) молекулярный вес полимера зависит от
отношения [М]/[1]1/2. Обычно [I] уменьшается быстрее, чем [М],
поэтому с ростом степени превращения молекулярный вес поли-
мера возрастает. При очень высоких степенях завершенности
полимеризации молекулярновесовое распределение достаточно
широкое, причем отношение Xw/Xn находится в интервале 2—5.
Для практических целей такое широкое распределение явно
невыгодно, поскольку большинство полимеров проявляют опти-
мальные свойства в весьма определенном интервале молекуляр-
ных весов. Поэтому промышленная технология полимеризации
часто предусматривает дополнительное многократное введение
инициатора и/или мономера в ходе реакции, чтобы не допустить
образования полимера с широким молекулярновесовым распреде-
лением.
Еще более широкое молекулярновесовое распределение наблю-
дается в реакциях полимеризации, характеризующихся аутоуско-
рением. Большое увеличение отношения kJk„" (см. табл. 3.12),
сопровождающее гель-эффект, приводит к сильному увеличению
размеров макромолекул, образующихся в процессе такой поли-
меризации. При наличии гель-эффекта значение Xw!Xn может
достигать 5—10. Если передача цепи на полимер играет суще-
ственную роль, то получаются разветвленные полимеры с крайне
широким распределением. Так, при передаче цепи на полимер
отношение Xw/Xn может возрасти до 20—50. Такое широкое
распределение обусловлено тем, что вероятность образования
ветвлений приблизительно пропорциональна молекулярному весу
полимера. Следовательно, по мере углубления полимеризации
разветвленные молекулы имеют тенденцию к еще большему раз-
ветвлению. Обычно расширение распределения, вызванное гель-
эффектом и передачей цепи на полимер, пытаются свести к мини-
муму, но сделать это эффективно не так просто. Так, например,
при проведении полимеризации при низких температурах сни-
жается вероятность протекания реакции передачи цепи на полимер,
но сильно возрастает роль гель-эффекта.
Для описания широких распределений были предложены раз-
личные функции распределения, но все они оказались мало удач-
ными. Следует, надеяться, что применение электронно-вычисли-
тельной техники будет очень полезным для учета влияния степени
завершенности процесса на [М], [I], /ср и к0 [79, 80].
3.12. Влияние давления
Влияние давления на полимеризацию, которое исследовано
явно недостаточно [4,81—83], имеет большое практическое значе-
ние. Выпускаемый в очень большом объеме (более 1 580 000 т)
Радикальная полимеризация
243
полимер — полиэтилен низкой плотности — получают полимери-
зацией при весьма высоких давлениях [1000—3000 атм (9,8 х
х 1Q7__ 29,4-Ю7 Па)[. Высокие давления могут оказывать большое
влияние на скорость полимеризации и молекулярный вес образу-
ющегося полимера. Обычно с повышением давления скорость
полимеризации и молекулярный вес возрастают. Это хорошо видно
Р и с. 3.18. Влияние давления на скорость полимеризации (Q) и молеку-
лярный вес полимера (#) при полимеризации стирола в присутствии перекиси
бензоила при 60 °C [83]. 7?p/(7?D)i — отношение скорости полимеризации
при данном давлении р к скорости полимеризации при 1 атм (9,8 104 Па).
из данных рис. 3.18, на котором демонстрируется влияние давле-
ния на молекулярный вес полистирола и скорость полимеризации
стирола в присутствии перекиси бензоила в качестве инициато-
ра [85].
(3.188)
3.12а. Объем активированного комплекса
Зависимость Rv и Хп от давления (и от температуры) опреде-
ляется изменением под влиянием давления следующих трех
констант скоростей: кг, kv и к0. В количественной форме зависи-
мость между константой скорости и давлением Р выглядит как
d In к —ДГ’^
~dP~= RT~ '
где AV* _ объем активированного комплекса, т. е. изменение
объема (см3/моль) при образовании из исходных мономеров акти-
вированного комплекса. Отрицательное значение AV* объясняется
тем, что объем активированного комплекса меньше объема исход-
ных реагентов. Из этого следует, что константа скорости поли-
меризации растет с увеличением давления. Так, при AV* =
244
Глава 3
= — 10 см3/моль скорость реакции повышается в 60 раз при
проведении полимеризации при комнатной температуре и давле-
нии 10 000 атм (9,8-107 Па). Положительное значение АУ* соот-
ветствует уменьшению константы скорости реакции с повышением
давления в случае, когда объем активированного комплекса
больше объема мономеров.
При полимеризации в присутствии инициаторов АУ* — вели-
чина положительная, так как распад их протекает как моно-
молекулярная реакция, сопровождающаяся повышением объема
при переходе инициатора в переходное состояние. С увеличением
давления скорость инициирования незначительно уменьшается.
Так, при повышении давления от 1 до 1500 атм (9,8-104—147 X
X 10е Па) константа скорости распада АИБН уменьшается при-
близительно на 30%. Аналогичные результаты были получены
при исследовании других инициаторов [82, 83], так как А У*
составляет, как правило, 5—10 см3/моль. Соответствующая вели-
чина для стадии роста АУр" отрицательна, так как относится
к бимолекулярным реакциям, сопровождающимся уменьшением
объема при переходе к активированному комплексу; например,
АУ* при полимеризации стирола составляет приблизительно
—13,4 см3/моль [85]. При бимолекулярном обрыве АУ* является
положительной величиной, несколько превышающей соответ-
ствующую величину АУ*. На первый взгляд АУ* должна быть
отрицательной величиной, так как обрыв цепи идет с уменьшением
объема при взаимодействии двух радикалов в переходном состоя-
нии. Однако, поскольку стадия обрыва цепи лимитируется диф-
фузией, уменьшение к0 с ростом давления обусловлено повышени-
ем вязкости реакционной среды [85].
3.126. Скорость полимеризации
В соответствии с уравнениями (3.28) и (3.188) влияние давле-
ния на скорость полимеризации определяется зависимостью между
отношением /ср (/cr/Zc0)1/2 и давлением, которая выражается сле-
дующим образом:
d In [ftp (А?г/^о) /2] —АУ^
dP = ВТ ’
где А Уд — изменение объема на стадии роста, равное
АУ* , АУ*
АУ)Г=^ + АУ*------
Для большинства мономеров А У* — величина отрицательная,
равная —15 ------------20 см3/моль, причем скорость полимеризации
(3.189)
(3.190)
Радикальная полимеризация
245
возрастает с увеличением давления (рис. 3.18). Действительно,
полимеризация винилацетата, характеризующаяся АУ^ =
_ __ 17 4 см3/моль, при температуре 50 °C и давлении 1000 атм
(9,8-107 Па) протекает примерно в 4—5 раз быстрее, чем в обыч-
ных условиях [86]. Было показано [82, 83, 87, 88] также, что
с повышением давления возрастает скорость полимеризации эти-
лена, пропилена, стирола и других мономеров.
3.12в. Степень полимеризации
Зависимость степени полимеризации от давления определяется
влиянием давления на соотношение /ср/(/сг/с0)1/2
d In [fcB/(M0)1/2] — А1Г
------------------------ = (3-191)
где
±_ AV* AV*
АУ± = ДР*л__£_---------(3.192)
В большинстве случаев АУ^п — отрицательная величина,
равная —20 -j 25 см3/моль; следовательно, молекулярный вес
полимера возрастает при повышении давления (рис. 3.18). Однако
увеличение молекулярного веса с ростом давления далеко не
беспредельно. Сначала он возрастает, но довольно быстро дости-
гает предельной величины для данного давления, причем даль-
нейшее повышение давления практически не отражается на
молекулярном весе полимера. В самом деле, начальное повыше-
ние молекулярного веса полимера с увеличением давления
намного меньше, чем можно было ожидать исходя из высоких
значений АУ± . По абсолютной величине АУ^ больше, чем
АУд, поэтому с повышением давления Хп должна расти значи-
тельно быстрее, чем 7?р, однако на практике наблюдается обрат-
ная картина (рис. 3.18). Причина такого поведения становится
понятной, если проанализировать различные параметры, входя-
щие в уравнение для определения молекулярного веса полимера
[уравнение (3.109)]. Подставляя в него различные данные для
полимеризации стирола при атмосферном давлении, видим, что
при этом важен только первый член правой части этого уравне-
ния, учитывающий бимолекулярный обрыв [83, 84]. По мере
увеличения давления См уменьшается вследствие увеличения kv,
но при этом еще более резко убывает член, отражающий бимоле-
кулярный обрыв. Следовательно, вклад См с повышением давле-
ния все время возрастает, и при очень высоких давлениях, когда
246
Глава 3
роль передачи цепи на полимер сводится к минимуму, основное
значение приобретает передача цепи на мономер.
С повышением давления скорость реакций передачи цепи дол-
жна, по-видимому, возрастать, так как передача цепи — бимо-
лекулярный процесс с отрицательной величиной ДУ*. Вместе
с тем изменение констант передачи цепи от давления определяется
относительным влиянием давления на константы скорости роста
и передачи цепи. Если важна только передача цепи на агент
передачи цепи, зависимость С< от давления выражается следую-
щим образом:
dlnCs d In (/спер, s/^p) (АЕПер. s /g
dP ~ dP RT ’ (Р-1УО)
где ЛУгЙр. s — изменение объема при реакции передачи цепи.
Для некоторых агентов передачи цепи ДУ^р. s меньше, чем
ДУ^, тогда с повышением давления Cs увеличивается; для других
соединений характерна обратная зависимость. Так, например,
константа скорости передачи цепи на триэтиламин при полимери-
зации стирола уменьшается в пять раз при повышении давления
от 1 до 4400 атм (от 9,8-104 до 43,1 -10е Па) [88]. Вместе с тем
в аналогичных условиях Cs для четыреххлористого углерода
уменьшается только примерно на 15%. Интересно заметить, что
в процессе получения полиэтилена роль реакции внутримолеку-
лярной передачи цепи [уравнение (3.126)1 с повышением давле-
ния уменьшается [89].
Это значит, что шри повышении давления число ветвлений
в полиэтилене уменьшается. Роль реакции передачи цепи и число
ветвлений зависят также от температуры, причем с ростом тем-
пературы вклад таких реакций и число ветвлений в полимере
увеличиваются.
3.13. Способы проведения полимеризации
Радикальная полимеризация проводится в гомогенных и гете-
рогенных условиях, причем в основе такой классификации лежит
фазовое состояние исходной реакционной смеси, т. е. является
она гомогенной или гетерогенной. Интересно заметить, что неко-
торые гомогенные системы превращаются по мере протекания
полимеризации в гетерогенные системы, что обусловлено нерас-
творимостью образующегося полимера в реакционной среде.
Полимеризация в массе и растворе относится к гомогенным про-
цессам, тогда как суспензионная и эмульсионная полимеризации
представляют собой примеры гетерогенной полимеризации. Рас-
смотрению эмульсионной полимеризации посвящена специальная
глава (гл. 4). Остальные способы проведения полимеризации будут
Радикальная полимеризация
247
рассмотрены ниже. Все мономеры могут быть заполимеризованы
различными способами. Вместе с тем было обнаружено, что для
полимеризации того или другого мономера в промышленных
масштабах эффективен только один или в крайнем случае два
способа. В ряде монографий подробно обсуждается технология
полимеризационных процессов [90, 91].
3.13а. Полимеризация в блоке (или в массе)
Полимеризация в блоке или массе — простейший способ поли-
меризации чистого мономера, при котором не происходит загряз-
нения готового продукта (полимера). Однако полимеризация
в блоке трудно поддается регулированию, что связано с особен-
ностями радикальной полимеризации вообще. Вследствие высокой
экзотермичности процесса, высоких энергий активации его,
а также тенденции к проявлению гель-эффекта сильно осложняет-
ся отвод тепла от сферы реакции. При полимеризации в блоке
нужно очень внимательно следить за температурным режимом
реакции. Кроме того, так как вязкость реакционной системы ста-
новится высокой даже при низких степенях завершенности реак-
ции, процесс необходимо вести при сильном и эффективном
перемешивании. Вследствие высокой вязкости реакционной массы
и экзотермического эффекта трудно поддерживать температуру
полимеризации на заданном уровне. Местные перегревы могут
приводить к деструкции полимера и потемнению его; кроме того,
из-за увеличения роли реакции передачи цепи на полимер стано-
вится более широким молекулярновесовое распределение. В от-
дельных случаях при очень сильном увеличении скорости поли-
меризации на заключительном этапе ее проведения реакция может
выйти из-под контроля со всеми вытекающими отсюда послед-
ствиями.
Из-за только что перечисленных трудностей радикальная
полимеризация в массе не находит очень широкого практического
применения. Вместе с тем она используется при полимеризации
этилена, стирола, метилметакрилата [90, 91]. Вопросы отвода
тепла решаются путем обрыва процесса на ранних стадиях или
проведением полимеризации в несколько стадий. Полимеризация
этилена идет при высоких давлениях [1000—3000 атм (9,8 X
X 107—29,4-107 Па)] и температурах, превышающих температуру
плавления полиэтилена. Система считается гомогенной, так как
полимер обычно растворим в этилене или сильно набухает в нем.
В одном из непрерывных процессов образования полиэтилена
применяются очень длинные и узкие трубчатые реакторы (напри-
мер, трубы диаметром менее 25 мм и длиной более 30 м). Таким
методом удается получить полимер с относительно узким
молекулярновесовым распределением, что достигается сравни-
248
Глава 3
тельно простым контролем температурного режима процесса,
многократным введением инициатора и обрывом реакции на низ-
ких степенях ее завершенности (10—20%). Полиэтилен отделяют
от реакционной смеси, а непрореагировавший мономер снова
пускают в производственный цикл.
При полимеризации стирола и метилметакрилата проблема
теплоотвода решается проведением реакции в две стадии. В фор-
полимеризаторах, представляющих собой реакторы с перемеши-
ванием, при 80 °C проводят полимеризацию стирола до содержа-
ния в нем полимера 30 %. Затем вязкий раствор или сироп полимера
в мономере сливают в полимеризационную колонну. Полимериза-
ционная колонна представляет собой цилиндрический аппарат,
состоящий из различных секций с индивидуальным обогревом,
который выбирается с таким расчетом, чтобы максимальная
температура была у днища колонны, а наиболее низкая — у ее
верха. По мере передвижения реакционной массы сверху вниз
глубина полимеризации возрастает и достигает на выходе из
колонны 98—100%.
Подобным же образом изготавливают листы, стержни, трубы
и изделия других профилей из полиметилметакрилата. Частично
полимеризованный мономер (форполимер) заливают в соответ-
ствующую форму и затем нагревают на воздушной или водяной
бане с постепенным повышением температуры. Этим достигается
не только более простой отвод тепла, но и получение изделия
заданных размеров. В процессе полимеризации метилметакрилата
происходит сильная усадка (~21%), которую удается снизить
заливкой в формы форполимера метилметакрилата.
3 136. Полимеризация в растворе
Полимеризация мономера в растворе лишена многих недо-
статков блочной полимеризации. При полимеризации в растворе
устраняется возможность местных перегревов, поскольку выде-
ляющееся тепло идет на нагревание растворителя, выполняющего
роль разбавителя. Растворитель обеспечивает также более легкое
перемешивание реакционной массы, так как вязкость ее стано-
вится меньше. Однако наличие в полимеризационной системе
растворителя имеет и определенные недостатки. Так, при проведе-
нии полимеризации в ряде растворителей большую роль может
приобрести реакция передачи цепи на растворитель. Кроме того,
полимер может быть загрязнен остатками растворителя, который
не всегда легко удаляется из полимера. Полимеризация в растворе
применяется для получения поливинилацетата, полиакрило-
нитрила и различных полиакрилатов. Иногда полимеризацию
этилена проводят в растворе с добавками воды или бензола,
облегчающими отвод тепла реакции.
Радикальная полимеризация
249
Полимеризация акрилонитрила в водном растворе проводится
в присутствии инициаторов окислительно-восстановительного типа.
В этом случае полимеризация быстро становится гетероген-
ной. Полиакрилонитрил выпадает из раствора в виде мелкодис-
персного порошка.
3.13в. Полимеризация в суспензии
Гетерогенная полимеризация в суспензии широко используется
для синтеза различных полимеров. Таким способом получают
поливинилхлорид, полистирол, полиметилметакрилат, поливинил-
ацетат. Этот процесс часто называют бисерной, жемчужной или
гранульной полимеризацией. При полимеризации по этому способу
мономер диспергируют в воде в виде мелких капелек, диаметр
которых 0,01—0,5 см. Устойчивость дисперсии достигается меха-
ническим перемешиванием или введением в реакционную систему
специальных добавок (стабилизаторов). К добавкам, предотвра-
щающим агломерацию капелек мономера, относятся водораство-
римые полимеры, например желатина, метилцеллюлоза, поливи-
ниловый спирт, электролиты и водонерастворимые неорганиче-
ские соединения (каолин, силикаты магния и гидроокись алю-
миния).
Для полимеризации в суспензии применяются инициаторы,
растворимые в мономере. Такие инициаторы часто называют
маслорастворимыми катализаторами. При полимеризации в сус-
пензии каждую отдельную капельку мономера можно рассматри-
вать как изолированную блочную полимеризационную систему.
Кинетика полимеризации внутри капельки мономера не отли-
чается от кинетики полимеризации в массе. Преимуществом поли-
меризации в суспензии является хороший отвод тепла. Недостаток
этого способа состоит в том, что полимер может быть загрязнен
остатками стабилизатора, а также необходима промывка и сушка
полученных гранул полимера.
3.13г. Твердофазная полимеризация
Полимеризацию ряда мономеров можно осуществлять в твердой
фазе. Эти реакции обычно проводятся при облучении мономера
в кристаллическом состоянии ниже его температуры плавле-
ния [92]. Некоторые из таких процессов протекают по радикаль-
ному механизму, другие — по ионному. Заслуживает внимания
тот факт, что полимеризация отдельных мономеров идет легче
в твердом состоянии, чем жидком. В твердофазной полимеризации
большую роль может играть кристаллическая структура мономера,
но механизм этого явления пока не ясен. Для выяснения механиз-
250
Глава 3
ма твердофазной полимеризации необходимо проведение широких
исследований. Промышленного значении этот способ полимериза-
ции в настоящее время не имеет.
3.14. Полимеризация диенов
В зависимости от химического строения диены (мономеры,
содержащие две двойные связи в молекуле) могут полимеризо-
ваться различными путями. Диены делятся на сопряженные
и несопряженные в зависимости от того, находятся в сопряжении
друг с другом две двойные связи или нет. Сопряженные диены
называются также диенами-1,3. При полимеризации несопряжен-
ных диенов нередко образуются разветвленные и сшитые поли-
меры. Несопряженные диены часто применяют в реакциях сополи-
меризации с винильными мономерами для сшивания последних
(разд. 6.6а). Ряд несопряженных диенов полимеризуется по осо-
бому механизму (циклополимеризация) путем внутримолекуляр-
ной реакции (разд. 6.66).
Особого внимания заслуживает полимеризация сопряженных
диенов. При полимеризации диенов-1,3 (бутадиена-1,3, изопрена
и хлоропрена)
СН2 = СН —СН = СН2 СН2=С —СН = СН2 СН2 = С —СН = СН?
I I
СН3 С1
бутадиен-1,3 изопрен хлоропрен
идут реакции двух типов. В первом случае протекает обычная
полимеризация по одной из двух двойных связей. Во втором слу-
чае в полимеризации своеобразно и согласованно участвуют обе
двойные связи. Так, при присоединении первичного радикала
к такому диену-1,3, как, например, бутадиен-1,3, образуется
аллильный радикал, имеющий две эквивалентные резонансные
формы XII и XIII
R —СН2 —СН —СН = СН2
XII
R.-|-CH2 = CH —сн = сн2 —» $ (3.194)
1 2 3 4 R—СН2 —СН = СН —СН2
XIII
Рост цепи может идти путем присоединения мономера по угле-
роду 2 (рост цепи через XII) или по углероду 4 (рост цепи
через XIII). Указанные реакции полимеризации называются
1,2-полимеризацией и 1,4-полимеризацией, причем образующиеся
Радикальная полимеризация
251
в результате полимеры имеют следующее строение:
г_сн2—СН
I
сн = сн2
- -(-СН2-СН = СН-СН2-)П-
п
1,2-полпмеризация’ 1,4-полимеризация
и называются 1,2-полибутадиен-1,3 и 1,4-полибутадиен-1,3 соот-
ветственно. Оба полимера являются ненасыщенными, причем
в 1,2-полимере ненасыщенные двойные связи находятся в боковой
цепи, а в 1,4-полимере они расположены в основной цепи. Поли-
меризация диенов-1,3 подробно рассматривается в разд. 8.6.
Вопросы и упражнения
3.1. При рассмотрении различных полимеров на основе алке-
нов напрашиваются следующие выводы:
а. Известные полимеры получены главным образом на основе
этилена, 1-монозамещенных производных этилена и 1,1-диза-
мещенных производных этилена. Полимеры на основе 1,2-диза-
мещенных производных этилена практически неизвестны.
б. Большинство полимеризационных процессов идет по ради-
кальному механизму и лишь сравнительно немного реакций
протекает по ионному механизму.
Почему? На каком основании сделаны эти выводы или они
являются в достаточной степени случайными? Обсудите этот
вопрос.
3.2. Напишите химическое уравнение полимеризации акрило-
нитрила в присутствии гидроперекиси кумола в качестве ини-
циатора.
3.3. Полистирол, полученный в присутствии как инициатора
АИБН, меченного 14С, имеет среднюю степень полимеризации
1,52-104. Активность АИБН составляет 9,81 -107 частиц в 1 мин
в сцинтилляторе. Если активность 3,22 г полистирола составляет
203 частицы в 1 мин, то в результате какой реакции обрыва обра-
зовался данный полимер? Проведите необходимый расчет.
3.4. Рассмотрим полимеризацию стирола при 60 °C в присут-
ствии перекиси ди-шрет-бутила в качестве инициатора. Для
раствора стирола (1,0 М) и перекиси (0,01 М) начальные скорости
инициирования и полимеризации составляют 4,0-10'11
и 1,5-10~7 моль/(л-с) соответственно. Рассчитайте значения (fkr),
начальную длину кинетической цепи и начальную степень поли-
меризации. Покажите, как часто в среднем проходит реакция
передачи цепи на один инициирующий радикал перекиси. Какое
должно быть молекулярновесовое распределение, т. е. величи-
на Xw/Xn. При расчетах пользуйтесь следующими константами
252
Глава 3
передачи цепи:
См = 8,0-1(Г5,
cI = 3,2.io-4,
СР = 1,9- 1(Г4,
Cs = 2,3.10~e.
3.5. Дли каких-то конкретных целей полистирол, получаю-
щийся в условиях, указанных в вопросе 3.4, имеет слишком
высокий молекулярный вес. Сколько нужно ввести н-бутилмер-
каптана в реакционную систему для получения полистирола
молекулярного веса 85 000? Определите скорость полимеризации
в присутствии меркаптана.
3.6. Рассмотрим реакцию полимеризации, описанную
в вопросе 3.4. Каким путем, кроме увеличения концентрации
мономера, можно повысить скорость полимеризации? Сравните
различные способы по их влиянию на молекулярный вес полимера.
3.7. Напишите химические уравнения реакций передачи цепи
на гексан, бензол, изопропилбензол, пропанол, иодистый бутил,
четырехбромистый углерод, н-бутилмеркаптан и ди-н-бутилсуль-
фид. Объясните, чем вызваны различия в константах передачи
указанных соединений при полимеризации винилацетата
(табл. 3.5).
3.8. Полимеризация метилакрилата в растворе (1М в бензоле)
проводится в присутствии фотосенсибилизатора под действием
света (X 0,313 мкм) от дуговой ртутной лампы. Свет погло-
щается системой со скоростью 1,0НО8 эрг/(л-с). Рассчитайте
скорость инициирования и полимеризации, если квантовый выход
при образовании радикалов составляет 0,50.
3.9. Рассмотрим следующие данные для полимеризации 0,2М
раствора мономера Z,
Номер опыта [АИБН] Интенсив- ность УФ-све- та [Сенсибили- затор] Т, °C яр.юз Хп
1 2А 0 0 50 0,30 200
2 2А 0 0 80 ?
3 2А в С 50 0,45 ?
4 0 2В С 50 0,30 200
5 А В С 50 ? ?
6 2А 2В С 50 ? ?
7 0 2В С 80 ? ?
8 4А 8В С 50 ? ?
9 4А 8В С 80 ? ?
Радикальная полимеризация
253
Подставьте в таблицу вместо знаков вопроса соответствующие
величины, если Ev и Ео для мономера Z равны 7,3 и 1,9 ккал/моль
соответственно. Укажите, какие допущения были вами сделаны
при расчетах.
3.10. Большинство реакций радикальной полимеризации харак-
теризуется зависимостью половинного порядка скорости поли-
меризации от скорости инициирования или концентрации
инициатора [I]. Укажите, в каких условиях радикальная поли-
меризация будет описываться уравнениями следующего порядка:
а) первый порядок;
б) нулевой порядок;
в) между половинным и нулевым порядком;
г) между половинным и первым порядком.
Дайте четкое объяснение тому, как разный механизм реакции
приводит к различным кинетическим порядкам реакции. Выведите
соответствующие кинетические уравнения.
3.11. Насколько широким будет молекулярновесовое распре-
деление при обрыве полимеризации на начальной стадии вслед-
ствие реакции рекомбинации? Обсудите, как отразятся на моле-
кулярновесовом распределении следующие факторы:
а) передача цепи через н-бутилмеркаптан;
б) высокая степень завершенности реакции;
в) передача цепи через полимер;
г) аутоускорение.
В тех случаях, когда наблюдается тенденция к более широкому
молекулярновесовому распределению, укажите, как путем изме-
нения условий полимеризации можно уменьшить роль такой
тенденции.
3.12. Рассчитайте, как изменится скорость и степень полиме-
ризации метилметакрилата при увеличении давления от 1 до
2500 атм. Инициатор АИБН, температура 50 °C, А Уд =
= —19,0 см3/моль, АУ* = 3,8 см3/моль.
Список литературы
1. Marvel С. S. et al., J. Am. Chem. Soo., 60, 280 (1938); ibid., 61 ,(3241 (1939).
2. Friedlander H. N., Harris H. E., Pritchard J. G., Polymer Sci., A-l(4),
649 (1966).
3. Wilson C. W., Santee E. R., Jr., J. Polymer Sci., C(8), 97, (1965).
4. Уоллинг Ч., Свободные радикалы в растворе, изд-во «Мир», М., 1960.
5. Прайер У., Свободные радикалы, Атомиздат, М., 1970.
6. Schulz С. V., Blaschke F., Z. Physik Chem. (Leipzig), B51, 75 (1942).
7. Santee G. F., Marchessault R. H., Clark H. G., Kearny J. J., Stannett V.,
Makromol. Chem., 73, 177 (1964).
8. Vrancken A., Smets G., Makromol. Chem., 30, 197 (1959).
9. Pryor W. A., Fiske T. R., Macromolecules, 2, 62 (1969).
10. Sugimura T., Minoura Y., J. Polymer Sci., A-l(4), 2735 (1966).
11. Misra G. S., Mathiu V. R. B., Makromol. Chem., 100, 5 (1967).
254
Глава 3
12. Horikx М. W., Hermans J. J., Polymer Sci., 11, 325 (1953).
13. Walling C., Heaton L., J. Am. Chem. Soc., 87, 38 (1965).
14. Burnett G. M., Melville H. W., Proc. Roy. Sos. (London), A189, 456, 494
(1947).
15. Dainton F. S., Sisley W. D., Trans. Faraday Soc., 59, 1369 (1963).
16. George M. H., Styrene, in «Vinyl Polymerization», G.E. Ham (ed.), vol. 1,
part I, chap. 3, Marcel Dekker, Inc., New York, 1967.
17. Chapiro A., Radiation Chemistry of Polymer Systems, chap. IV, Inter-
science Publishers, John Wiley and Sons, Inc., New York, 1962.
18. Gregg R. A., Mayo F. R., Disc. Faraday Soc., 2, 328 (1947).
19. Mayo F. R., J. Am. Chem. Soc., 75, 6133 (1953).
20. Burnett G. M., Loan L. D., Trans. Faraday Soc., 51, 214 (1955).
21. O’Driscoll K. F. et al., J. Polymer Sci., A3, 283, 1567 (1965).
22. Mukherjee A., Pal(Mitra) R., A marendra, Biswas M., Maiti S., J. Polymer
Sci., A-l(5), 135 (1967).
23. Saccubai S., Santappa M., J. Polymer Sci., A-l(7), 643 (1969).
24. Kaeriyama K., Polymer, 10, 11 (1969).
25. Subramanian S. V., Santappa M., Makromol. Chem., 112, 1 (1968); J. Poly-
mer Sci., A-l(6), 493 (1968).
26. DeSchrijver F., Srnets G., J. Polymer Sci., A-l(4), 2201 (1966).
27. Bevington J. C., Trans. Faraday Soc., 51, 1392 (1955).
28. Walling C., J. Polymer Sci., 14, 214 (1954).
29. Nozaki K., Bartlett P. D., J. Am. Chem. Soc., 68, 1686 (1946).
30. Arnett L. M., Peterson J. H., J. Am. Chem. Soc., 74, 2031 (1952).
31. Bartlett P. D., Kwart H., J. Am. Chem. Soc., 72, 1051 (1950).
32. Tobolsky A. V., Rodgers С. E., Brickman R. D., J. Am. Chem. Soc., 82,
1277 (1960).
33. Bohme R. D., Tobolsky A. V., Dead-End Polymerization, in «Encyclopedia
of Polymer Science and Technology», vol. 4, p. 599, John Wiley and Sons,
Inc., New York, N.Y., 1966.
34. Gobran R. H., Berenbaum M. B., Tobolsky A. V., J. Polymer Sci., 46, 431
(1960).
35. O’Driscoll K. F., White P. J., J. Polymer Sci., Bl, 597 (1963); A3, 283 (1965).
36. O’Driscoll K. F., McArdle SA., J. Polymer Sci., 40, 557 (1959).
37. Bevington J. C., Melville H. W., Taylor R. P., J. Polymer Sci., 12, 449
(1954): 14, 463 (1954).
38. Funt B. L., Pasika W., Can. J. Chem., 38, 1865 (1960).
39. Baysal B.. Tobolsky A. V., J. Polymer Sci., 8, 529 (1952).
40. Mayo F. R., GreggR. A., Matheson M.:S., J. Am. Chem. Soc., 73, 1691 (1951).
41. Brandup J., Immergut E. H. (eds.), Polymer Handbook, Interscience
Publishers, John Wiley and Sons, Inc., New York, 1966.
42. Gregg R. A., Mayo F. R., J. Am. Chem. Soc., 70, 2373 (1948L
43. Lindeman M. K., The Mechanism of Vinyl Acetate Polymerization, in
«Vinyl Polymerization», Ham G. E. (ed.), vol. 1, part I, chap. 4, Marcel
Dekker, Inc., New York, 1967.
44. Flory P. J., J. Am. Chem. Soc., 69, 2893 (1947).
45. Bevington J. C., Guzman G. M., Melville H. W., Proc. Roy. Soc. (London),
A221, 453, 547 (1947).
46. Stein D. J., Makromol. Chem., 76, 170 (1964).
47. Schulz G. V., Chem. Ber., 80, 232 (1947).
48. Georgieff К. K., J. Appl. Polymer Sci., 9, 2009 (1965).
49. Tabata Y., Ishigure K., Oshima K., Sobue H., J. Polymer Sci., A2, 2445
(1964).
50. Ashmore P. G., Catalysis and Inhibition of Chemical Reactions, p. 304—
310, Butterworth and Co. (Publishers) Ltd., London, 1963.
51. Melville H. W., Watson W. F., Trans. Faraday Soc., 44, 886 (1948).
52. Bartlett P. D., Tate F. A., J. Am. Chem. Soc., 75, 91 (1953).
Радикальная полимеризация 255
53 Flory Р. J--, Principles of Polymer Chemistry, chap. IV, Cornell University
Ithaca, 1953.
r™ 5 II’., North A. M„ J. Am. Chem. Soc., 81, 1339 (1959).
L’ Knrrish R. G. W., Thrush B. A., Quart. Rev. (London), 10, 149 (1956).
L’ Briers F. ' Chapman D. L., Walters E., J. Chem. Soc., 1926, 562.
47' Matheson M. S., Auer E. E., Bevilacqua E. B., Hart E. J., J. Am. Chem.
Soc 71, 497 (1949).
58 Dainton F. S., Sisley W. D., Trans. Faraday Soc., 59, 1369 (1963).
59' Roche A., Price С. C., in «Styrene: Its Polymers, Co-Polymers, and Deriva-
tives» Boundy R. H„ Boyer R. F. and Stoesser S. M. (eds.), unpublished
data in Table 7—1, p. 216, Reinhold Book Corp., New York, 1952.
60 Barb W. G., Baxendale J. H., George P., Hargrave K. R., Trans. Faraday
’ Soc., 47, 462, 591 (1951).
61. Gregg R. A., Mayo F. R., J. Am. Chem. Soc., 75, 3530 (1953).
62i Joshi R. M., Zwolinski B. J., Heat of Polymerization and Their Structural
and Mechanistic Implications, in «Vinyl Polymerization», Ham G. E. (ed.),
vol. 1, part I, chap. 8, Marcel Dekker, Inc., New York, 1967.
63. Daintoon F. S., Ivin K. J., Quart. Rev. (London), 12, 61 (1958).
64. Dainton F. S., Ivin K. J., Trans. Faraday Soc., 46, 331 (1950).
65. Wall L. A., Sos. Plastic Eng. /., 16, 1 (I960).
66. McCormick H., J. Polymer Sci., 25, 488 (1957).
67. Worsfold D. J., Bywater S., J. Polymer Sci., 26, 299 (1957).
68. Cook R. E., Dainton F. S., Ivin K. J., J. Polymer Sci., 29, 549 (1958).
69. Schulz G. V., Ilaborth G., Makromol. Chem., 1, 106 (1948).
70. Hayden P., Melville H. W., J. Polymer Sci., 43, 201 (1960).
71. North A. M. et al., I. Polymer Sci., Al, 1311 (1963); Trans. Faraday Soc.,
57, 859 (1961); 60, 960 (1964).
72. Ito K., J. Polymer Sci., A-l(7), 827 (1969).
73. Mangaraj D., Patra S. K., Makromol. Chem, 104, 125 (1967).
74. Jenkins A. D., Occlusion Phenomena in the Polymerization of Acrylo-
nitrile and Other Monomers, in «Vinyl Polymerization», Ham G. E. (ed.),
vol. 1, part I, chap. 6, Marcel Dekker,' Inc., New York, 1967.
75. Тенфорд Ч., Физическая химия полимеров, изд-во «Химия», М., 1965.
76. Flory Р. J., Principles of Polymer Chemistry, chap. VIII, Cornell Univer-
sity Press, Ithaca, 1953.
77. Peebles L. H„ Jr., p. 11—421 in [41].
78. Smith W. B., May J. A., Kim C. W., J. Polymer Sci., A-2(4), 365 (1966).
79. Duerksen J. H..Hami elec A. E., Polymer Preprints, 8(1), 344 (1967).
80. Zeman R. J., Amundson N. R., Chem. Eng. Sci., 20, 331, 637 (1965).
81. Zutty N. L., Burkhart R. D., Polymer Synthesis at High Pressures, in
«Polymerization and Polycondensation Processes», Platzker N. A. J.»(ed.),
chap. 3, American Chewical Society, Reinhold Book Corp., New York, 1962.
82. Weale К. E., Quart. Rev. (London), 16, 267 (1962).
83. Weale К. E., Chemical Reactions at High Pressures, chaps. 8 and 9, E. and
F.N. Spon, London, 1967.
84. Merrett F. M., Norrish'.R. G. W., Proc. Roy. Soc. (London), A206, 309 (1951).
85. Nicolson A. E., Norrish R. G. W., Disc. Faraday Soc., 22, 104 (1956).
86. Nakamoto H., Ogo Y., Imoto T., Makromol. Chem., Ill, 93 (1968).
87. Mortimer G. A., Arnold L. C., J. Polymer Sci.. A2, 4247 (1964).
88. Toohey A. C., Weale К. E., Trans. Faraday Soc., 58, 2446 (1962).
89. Woodbrey J. C., Ehrisch P., J. Am. Chem. Soc., 85, 1580 (1963).
90. Smith W. M., Manufacture of Plastics, vol. 1, p. 16—32, 81—87, 100—118,
225—246, 308—323, 364—381, 414—432, Reinhold Book Corp., New York,
1964.
91. Brydson J. A., Plastics Materials, p. 100—102, 159—161, 231—234, 251 —
254, D. Van Nostrand Co., Princeton, 1966.
92. Morametz H.. J. Polymer Sci., A-l(4), 2487 (1966).
ГЛАВА 4
ЭМУЛЬСИОННАЯ ПОЛИМЕРИЗАЦИЯ
Эмульсионная полимеризация является одним из способов
проведения радикальной полимеризации в эмульсии мономера.
По механизму и закономерностям она отличается от рассмотренной
выше суспензионной полимеризации (см. разд. 3.13в), причем
различными являются тип и размер частиц, в которых идет поли-
меризация, а также природа инициатора.
4.1. Описание процесса
4.1а. Применение
Впервые эмульсионная полимеризация начала применяться
в период второй мировой войны в производстве синтетического
бутадиенстирольного каучука, потребности в котором стали очень
велики, так как источники натурального каучука оказались
отрезанными. В настоящее время эмульсионная полимеризация
является основным методом получения полимеров на основе сопря-
женных диенов: бутадиена, изопрена и др. Широко применяется
эмульсионная полимеризация винилацетата, винилхлорида, акри-
латов, метакрилатов и их смесей.
Эмульсионная полимеризация имеет ряд несомненных преиму-
ществ. Так, при проведении полимеризации в эмульсии легко
регулировать процесс. В отличие от полимеризации в блоке
в эмульсионном процессе термические и вязкостные параметры
не доставляют особых хлопот. В ряде случаев после окончания
эмульсионной полимеризации полимер можно непосредственно
использовать для формования различных изделий (после введе-
ния других ингредиентов), минуя стадию его выделения. Это
относится к изготовлению покрытий, лаков, красителей и т. п.
Между эмульсионным и другими способами полимеризации
существует большое различие и в кинетике процессов. Для всех
методов полимеризации, кроме эмульсионного, наблюдается
обратная зависимость скорости полимеризации от молекулярного
веса полимера [уравнение (3.100)], что существенно ограничивает
практическую возможность регулирования молекулярного веса,
Эмульсионная полимеризация
257
например получения полимера молекулярного веса 2 000 000
вместо полимера молекулярного веса 200 000 или, наоборот,
полимера молекулярного веса 200 000 вместо 20 000. Синтезиро-
вать полимер существенно более низкого молекулярного веса,
не изменяя скорости полимеризации, можно путем применения
агентов, вызывающих реакции передачи цепи. Значительно же
повысить молекулярный вес полимера можно, только уменьшив
скорость полимеризации за счет снижения концентрации инициа-
тора или температуры реакции. Эмульсионная полимеризация
представляет в этом отношении уникальный метод синтеза поли-
меров, так как позволяет повысить молекулярный вес полимера
без снижения скорости полимеризации. Преимущество эмуль-
сионной полимеризации, вытекающее из ее механизма, заклю-
чается в возможности достижения как высоких скоростей реакций,
так и высоких молекулярных весов полимеров. Молекулярный
вес полимера и скорость полимеризации можно изменять в широ-
ких пределах независимо друг от друга.
4.16. Качественная характеристика
4.16.1. Компоненты и их ориентация в мицелле
Физическое описание эмульсионной полимеризации основано
главным образом на качественных представлениях Харкинса [1]
и количественной обработке данных Смитом и Эвартом [2, 3].
В табл. 4.1 приведен типичный состав смеси при эмульсионной
Таблица 4.1
Эмульсионная полимеризация смеси
стирола и бутадиена [4]
Компонент Количество, вес. ч.
Стирол 25
Бутадиен 75
Вода 180
Эмульгатор (Dresinate 5
731)
Меркаптан 0,5
NaOH 0,061
Гидроперекись кумола 0,17
FeSO4 0,017
Na4P2O7-10H2O 1,5
Фруктоза 0,5
258
Глава 4
полимеризации [4], применявшейся сначала в технологии произ-
водства бутадиенстирольного каучука (GR-S) и сохранившей
свое значение для большинства эмульсионных систем. Основными
компонентами в эмульсионной полимеризации являются мономер
(или мономеры), диспергирующая среда, эмульгатор и водораство-
римый инициатор. Диспергирующей средой называется жидкость,
в которой различные компоненты образуют под влиянием эмуль-
гатора эмульсию. Обычно ею является вода. Эмульгатор — это
поверхностноактивное вещество, действие которого основано
на том, что в его составе есть гидрофильные и гидрофобные сег-
менты. В эмульсионной системе могут быть и другие компоненты.
Так, для регулирования молекулярного веса полимера в выше-
приведенном составе смеси применяют меркаптан, участвующий
в реакциях передачи цепи. В качестве инициатора эмульсионной
полимеризации используют окислительно-восстановительную
систему: гидроперекись — ион Fe2+, а роль фруктозы, по-види-
мому, сводится к регенерации иона двухвалентного железа вос-
становлением трехвалентного железа, образующегося в процессе
инициирования [уравнение (З.ббв)]. Пирофосфат натрия приме-
няют для улучшения растворимости солей железа в сильно щелоч-
ной реакционной среде. Во время реакции эмульсию интенсивно
перемешивают.
Остановимся теперь на состоянии различных компонентов
в эмульсионной системе. Небольшая часть эмульгатора раство-
ряется в воде. Основное же его количество агрегирует в неболь-
шие коллоидальные кластеры или мицеллы. Между частицами
эмульгатора в растворе и в мицеллах существует динамическое
равновесие. По данным светорассеяния коллоидные мицеллы
имеют стержнеподобную форму [5]. Каждая мицелла состоит
из 50—100 молекул эмульгатора и в длину имеет 0,1—0,3 мкм,
диаметр же мицеллы равен удвоенной длине молекулы эмульга-
тора. Молекулы эмульгатора в мицелле располагаются таким
образом, что углеводородные концы их направлены к центру
мицеллы, а их ионные группы — к воде. Число и размер мицелл
зависит от соотношения между количествами эмульгатора и моно-
мера. При повышении количества эмульгатора увеличивается
число частиц меньшего размера, т. е. при этом возрастает поверх-
ность мицелл.
При введении мономера, нерастворимого или плохо раствори-
мого в воде, лишь очень небольшая часть его растворяется и пере-
ходит в раствор. Несколько большее количество мономера прони-
кает во внутреннюю, углеводородную область мицелл. Это дока-
зано рентгеновскими исследованиями и с помощью метода свето-
рассеяния, которые указывают на увеличение размера мицелл
после добавления мономера [6]. Основная часть мономера диспер-
гирует в виде капелек, размер которых определяется интенсив-
Элулъсионная полимеризация 259
постью перемешивания. Диаметр таких капелек обычно не менее
1 мкм Таким образом, в стандартной эмульсионной полимериза-
цпонной системе капельки мономера по размеру несколько больше
мицелл, в которых есть мономер. Так, если концентрация мицелл
обычно составляет 1018,'мл. то в 1 мл содержится не менее 1010—1011
капелек мономера. Различие между мицеллами и отдельными
капельками мономера заключается и в том, что первые имеют
значительно большую поверхность. Оценку размера и количе-
ства частиц в эмульсионной системе проводят с помощью элек-
тронной микроскопии, светорассеяния, ультрацептрифугирования
п других методов [(>—8].
4.16.2. Место протекания полимеризации
Инициатор находится в водной фазе, которая и является
местом образования радикалов инициатора. Обычно скорость
образования инициатора составляет примерно 1013 радикалов
па 1 мл в секунду. (15 эмульсионной полимеризации вместо Rn
часто применяется символ р.) Теперь остановимся на том, где про-
текает полимеризация. Несомненно, что в растворе идет поли-
меризация мономера, однако вклад ее очень невелик из-за низкой
концентрации мономера в растворе. В капельках мономера
не может происходить полимеризации, так как использующиеся
инициаторы нерастворимы в органическом мономере. Такие
инициаторы известны под названием маслоперастворимых инициа-
торов. Все это отличает эмульсионную полимеризацию от суспен-
зионной. В последней применяются маслорастворимые инициа-
торы и реакция идет в капельках мономера. Экспериментальная
проверка подтвердила то, что полимеризация в капельках моно-
мера в эмульсионном процессе не наблюдается. Если прервать
эмульсионную полимеризацию на какой-то промежуточной стадии,
то можно выделить капельки мономера и подвергнуть их анализу.
Таким способом было показано, что в капельках мономера содер-
жится лишь незначительное количество полимера (<0,1%).
Полимеризация идет почти исключительно внутри мицелл.
В них «встречаются» органический (маслорастворимый) мономер
и водорастворимый инициатор. Протеканию реакции в мицеллах
благоприятствует также высокая концентрация в них мономера
и значительно большее, чем в капельках мономера, отношение
поверхности к объему. По мере протекания полимеризации мицел-
лы растут за счет поступления мономера сначала из водной фазы,
а затем из капелек мономера. Схематическое изображение эмуль-
сионной полимеризации показано на рис. 4.1. Система состоит
из частиц трех типов: капелек мономера, неактивных мицелл,
в которых не обнаружено полимеризации, и активных мицелл,
являющихся местом протекания полимеризации. Молекула
260
Глава 4
эмульгатора обозначена символом q_, чтобы подчеркнуть, что
один ее конец полярный, или ионный, а другой неполярный.
Система претерпевает глубокое изменение после того, как
лишь небольшая часть мономера превращается в полимер. Поли-
меризация инициируется только в очень маленькой части мицелл
(~0,1 %), присутствующих в системе. По мере роста размеров
активных мицелл, содержащих наряду с мономером образовав-
шийся полимер, они поглощают все больше и больше молекул
эмульгатора из раствора. При этом быстро наступает состояние,
при котором концентрация эмульгатора в растворе становится
Р и с. 4.1. Упрощенное схематическое изображение эмульсионной полп-
меризационной системы.
ниже его критической мицеллярной концентрации (КМК). Послед-
няя отвечает концентрации эмульгатора в растворе, необходимой
для образования и поддержания мицелл. Если концентрация
эмульгатора падает ниже этой величины, неактивные мицеллы
(те, в которых полимеризация не идет) становятся неустойчивыми
и эмульгатор, содержащийся в мицеллах, переходит в раствор.
При степени превращения 2—15 %, которая в каждом конкретном
случае может колебаться в указанных пределах, размер актив-
ных мицелл становится значительно больше, чем исходных мицелл.
Их уже и нельзя считать мицеллами и правильнее говорить
о частицах полимера или, еще точнее, о частицах полимера,
набухших в мономере. Все мицеллы, которые не были активиро-
ваны радикалами инициатора, распадаются, и практически весь
эмульгатор сорбируется частицами полимера. Вследствие этого
капельки мономера становятся неустойчивыми и при прекраще-
нии перемешивания начинают коалесцировать.
Эмульсионная полимеризация
261
В гомогенных условиях полимеризация идет в частицах поли-
мера, когда концентрация мономера в частицах постоянная
за счет диффузии мономера из капелек. В процессе полимеризации
число полимерных частиц не меняется. С увеличением размера
полимерных частиц величина капелек мономера уменьшается.
Наконец, при степени превращения 50—80% капельки мономера
совершенно исчезают, а частицы полимера содержат весь непро-
реагировавший мономер. По мере уменьшения концентрации
мономера в частицах полимера скорость полимеризации непре-
рывно понижается. Каждый полимер представляет собой частицы
диаметром 0,05—0,2 мкм, промежуточные по размеру между
исходными мицеллами и капельками мономера.
4.2. Кинетика процесса
4.2а. Скорость полимеризации
Теоретические расчеты Смита и Эварта [2, 3] позволяют
вывести кинетические уравнения эмульсионной полимеризации.
Эти расчеты основаны на сравнении скоростей рекомбинации
и диффузии радикалов в воде. Было показано, что радикалы,
инициирующие эмульсионную полимеризацию, не погибают
в водной фазе, а являются достаточно долго живущими для того,
чтобы проникнуть в частички полимера. Поэтому практи-
чески все радикалы, образовавшиеся в водной фазе, достигают
частичек полимера.
Для анализа эмульсионной полимеризации удобно рассматри-
вать стационарное состояние, когда исчезают последние неактив-
ные мицеллы мономера, а число частиц полимера становится
постоянным. В эмульсионной полимеризации число частиц поли-
мера N составляет примерно 1013—1015 на 1 мл водной фазы,
а скорость инициирования — 1012—1014 радикалов на 1 мл водной
фазы в секунду. Обычно при концентрации частиц полимера
1014 частиц/мл и = 1013 радикалов/(мл-с) радикал диффунди-
рует в каждую полимерную частицу в среднем раз в 10 с. Внутри
частицы полимера радикал участвует в росте цепи со скоростью гр,
зависящей от константы скорости реакции роста кр и концентрации
мономера [М] в частице:
гр = кр [М]. (4.1)
Как правило, концентрация мономера достаточно высока, так
как во многих случаях равновесное набухание частицы в мономере
достигает 50 вес. %. Концентрация мономера порядка 5 М встре-
чается достаточно часто.
Рассмотрим теперь, что происходит при попадании радикала
в частицу, в которой уже имеется радикал. Можно подсчитать,
262
Глава 4
что молярная концентрация радикалов в частице полимера соста-
вляет 10-6 М, что выше соответствующей величины при гомоген-
ной полимеризации. Время жизни радикала не превышает
нескольких тысячных секунды. При попадании второго радикала
в частицу полимера немедленно наступает бимолекулярный
обрыв. Поэтому в полимерной частице может быть или один
радикал, или ни одного. Наличие двух радикалов в одной частице
равнозначно их отсутствию, настолько быстро идет реакция
обрыва. В течение последующих 10 с до появления следующего
(третьего) радикала частица является неактивной, затем она снова
активизируется, идет рост цепи, пока в частицу не попадет еще
Рис. 4.2. Зависимость сте-
пени превращения от про-
должительности эмульсион-
ной полимеризации стиро-
ла при различных концен-
трациях лаурата калия [9].
Загрузка: воды 180 г, сти-
рола 100 г, K2S2O8 0,5 г.
Содержание эмульгатора, моли:
1) 0,0035; 2) 0,007;.з) 0,014.
один радикал. Такие циклы — рост цепи и неактивность полимер-
ной частицы — повторяются до исчерпания всего количества
мономера.
Каждая полимерная частица половину времени активна,
а в остальное время она «спит». Другими словами, в любой произ-
вольно взятый момент половина частиц полимера содержит один
радикал и участвует в реакции роста цепи, тогда как другая
половина частиц неактивна. Число радикалов на частицу п,
усредненное для всех частиц, составляет половину. Суммарная
скорость полимеризации (в расчете на 1 см3 эмульсии) равняется
сумме NI2 частиц, полимеризующихся согласно уравнению (4.1).
Это выражается как
Из уравнения (4.2) видно, что скорость полимеризации непосред-
ственно зависит от числа полимерных частиц в системе, но не зави-
сит от скорости образования радикалов R„. Это справедливо,
пока в системе есть радикалы. Естественно, что полимеризация
прервется, если образование радикалов прекратится. Повыше-
ние R и приводит просто к увеличению скорости чередования
в каждой частице полимера активных и неактивных моментов.
На рис. 4.2 приведены данные для эмульсионной полимериза-
ции стирола [9] при трех различных концентрациях эмульгатора.
Эмульсионная полимеризация
263
С увеличением концентрации эмульгатора скорость полимериза-
ции возрастает, что связано с увеличением числа N. Все кривые,
приведенные на рис. 4.2, весьма типичны для эмульсионной
полимеризации. Есть начальная стадия, протекающая с само-
ускорением, зависящим от свойств данной системы. Это отвечает
промежутку времени, необходимому для образования мицелл.
Когда число активных мицелл становится постоянным, зависи-
мость между скоростью полимеризации и временем становится
линейной, до тех пор пока сохраняется избыток капелек моно-
мера и [М] остается постоянной. При высоких степенях превра-
щения с уменьшением [М] скорость снижается.
4.26. Степень полимеризации
Среднечисловую степень полимеризации в эмульсионном про-
цессе можно вычислить путем анализа реакции в изолированной
Р и с. 4.3. Зависимость сред-
невязкостного молекулярного
веса полистирола от степени
превращения стирола в эмуль-
сионной полимеризации при
различных концентрациях лау-
рата калия [9]. Загрузка: во-
ды 180 г, стирола 100 г,
0,3 г.
Содержание эмульгатора, моли:
1) 0,0035; 2) 0,007; 3) 0,014.
полимерной частице. Скорость ги, при которой первичный радикал
проникает в полимерную частицу, определяется как
тг- 0-3)
Такой же вид имеет выражение для скорости обрыва, так как
цепь обрывается непосредственно после попадания радикала
в полимерную частицу, в которой идет рост полимерной цепи.
Тогда степень полимеризации представляет собой отношение
скорости роста полимерной цепи к скорости проникновения пер-
вичных радикалов в полимерную частицу, т. е.
Д = = (4.4)
ги и
На рис. 4.3 приведена зависимость средневязкостного молеку-
лярного веса от степени превращения мономера при эмульсион-
Глава 4
бой полимеризации стирола. Повышение молекулярного веса
полимера с ростом концентрации эмульгатора отвечает зависи-
мости, выраженной уравнением (4.4).
Следует обратить внимание на то, что степень полимеризации
в эмульсионной полимеризации адекватна длине кинетической
цепи. Хотя обрыв наступает в результате бимолекулярного
взаимодействия, один из реагирующих радикалов — первич-
ный — существенно не влияет на размер мертвой полимерной
молекулы. При выводе уравнения (4.4) исходят из отсутствия
обрыва вследствие передачи цепи. Если передача цепи имеет
место, то степень полимеризации становится равной
где 2 гпер — сумма скоростей всех реакций передачи цепи. Ско-
рость передачи цепи в полимерной частице выражается урав-
нением
гпер = ^пер [ХА], (4.6)
аналогичным уравнению для скорости передачи цепи в гомогенной
полимеризации [уравнение (3.105)].
Как и скорость полимеризации, степень полимеризации про-
порциональна N, но в отличие от нее степень полимеризации
зависит от 7?и. При сопоставлении уравнений (4.2) и (4.4) и соот-
ветствующих уравнений для гомогенной радикальной полимериза-
ции [уравнения (3.24) и (3.100)] выявляются специфические
особенности эмульсионного процесса. В гомогенной полимериза-
ции скорость полимеризации можно резко увеличить путем
повышения скорости инициирования, что, однако, вызывает
понижение молекулярного веса полимера. Иное положение имеет
место в эмульсионной полимеризации, где скорость и степень
полимеризации можно повысить увеличением числа частиц поли-
мера, не изменяя скорости инициирования. Эти выводы получили
экспериментальное подтверждение в ряде работ [3, 10—12].
4.3. Число полимерных частиц
Степень и скорость полимеризации во многом определяются
числом полимерных частиц, которое входит в первой степени
в уравнения (4.2) и (4.4). Выше уже обсуждался механизм образо-
вания частиц полимера в мицеллах. Радикалы инициатора входят
в небольшую часть мицелл и растут в полимерных частицах,
поглощая эмульгатор из других (неактивных) мицелл. Число
образовавшихся частиц полимера зависит от числа исходных
мицелл (которое в свою очередь определяется концентрацией
Эмульсионная полимеризация 265
эмульгатора) и скорости образования радикалов. Смит и Эварт
[2] вывели соотношение
;V=A:(^)2/5(an[E])3/\ (4.7)
в котором к — константа (0,37—0,53), р — скорость увеличения
объема полимерной частицы (рассчитывается из гр и геометрии
молекулы), [Е1 — концентрация эмульгатора в мицеллах и ап —
пограничная поверхность, занятая молекулами эмульгатора
в мицеллах (все величины в единицах системы СГС).
Путем подстановки выражения для N [уравнение (4.7)] в урав-
нения (4.2) и (4.4) находим зависимость 7?р и Хп от скорости обра-
зования радикалов в степени 2/6 и концентрации эмульгатора
в степени 3'5. Такая зависимость 7?р от Ra не противоречит
выводу об отсутствии зависимости скорости полимеризации
от скорости образования радикалов. От скорости образования
радикалов зависит число получающихся частиц полимера, которое
в свою очередь определяет скорость полимеризации. Однако, как
только в эмульсионной полимеризации достигается стационарное
состояние по отношению к N (т. е. когда N постоянно и все неак-
тивные мицеллы разрушились), скорость образования радикалов
перестает оказывать какой-либо эффект на скорость полимериза-
ции при условии, что инициирование не прекращается. Далее,
необходимо иметь в виду, что число частиц полимера можно повы-
сить увеличением концентрации эмульгатора, не затрагивая при
этом скорости образования радикалов. Таким образом, практи-
чески высоких скоростей полимеризации можно достичь при
высоких начальных скоростях образования радикалов и высокой
концентрации эмульгатора. Уравнение (4.7) было эксперимен-
тально проверено при исследовании ряда систем [13], причем
наиболее подробно изучена эмульсионная полимеризация стирола.
Концентрация эмульгатора, которая фигурирует в уравне-
нии (4.7), является мицеллярной концентрацией. Если для дан-
ного эмульгатора достижима сравнительно высокая критическая
мицеллярная концентрация, на величину [Е] нужно вводить
соответствующую поправку, учитывающую количество раство-
ренного эмульгатора. Хотя при концентрации эмульгатора ниже
КМК мицелл в системе нет, частицы полимера образуются
в заметном количестве, которое может составлять примерно 10%
от их числа при КМК. Механизм возникновения таких частиц
включает, по-видимому, осаждение образующихся в растворе
олигомеров или низкомолекулярных полимеров [14].
Как уже было указано, после исчезновения мицелл число
частиц полимера остается постоянным. Это было показано на мно-
гих системах, несмотря на то что стабильность полимерных частиц
становится ниже вследствие увеличения их суммарной поверх-
266
Глава 4
ности и одновременного уменьшения ее доли, покрытой эмуль-
гатором. Вероятно, во многих случаях это не имеет решающего
значения. Однако при полимеризации других мономеров, например
винилхлорида, стабильность снижается настолько, что начи-
нается их слипание, т. е. с увеличением степени завершенности
реакции N уменьшается.
4.4. Закономерности эмульсионной
п олимериз ации
4.4а. Инициирование
В эмульсионной полимеризации используются водораствори-
мые инициаторы, такие, как персульфат калия или аммония,
перекись водорода, некоторые азосоединения, например 2,2'-азо-
бис-(2-метил-4-карбоксибутиронитрил). Применяются также пере-
киси, частично растворимые в воде: перекись янтарной кислоты
и гидроперекись третичного бутила. Наиболее часто эмульсион-
ную полимеризацию ведут в присутствии окислительно-восстано-
вительных систем (разд. 3.4д), состоящих из персульфата и соли
двухвалентного железа
S20iT + Fe2+ —> Fe3+4-S0£- + S0j. (3.67а)
или гидроперекиси кумола и соли двухвалентного железа
СН3 СН3
I I
СвН5 —С —00H + Fe2+ —> Fe3+ + HO~ + CeH5 —с-0. (4.8)
СН3 СН3
Кинетика эмульсионной полимеризации предполагает последова-
тельное проникновение радикалов в частицы полимера. Это тре-
бование легко удовлетворяется при применении водорастворимых
инициаторов, типа упомянутых выше. Вместе с тем подобные
кинетические закономерности наблюдаются при полимеризации
в присутствии инициаторов, растворимых в углеводородах,
например гидроперекисей, при термическом разложении каждой
молекулы которых образуются два радикала. Хотя при разложе-
нии таких инициаторов внутри частицы образуется два радикала,
полимерные радикалы, по-видимому, возникают только последо-
вательно один за другим, но механизм этого явления не ясен.
Одно возможное объяснение может быть связано с диффузией
радикалов из полимерных частиц [15]. Так, при термическом
разложении гидроперекиси кумола можно предположить, что
неорганический радикал НО • диффундирует из полимерной
частицы, тогда как другой, органический радикал С6Н5С(СН3)2О •
остается в ней, инициируя полимеризацию. Возможно также,
Эмульсионная полимеризация
267
что разложение инициатора идет на границе раздела фаз поли-
мерная частица — вода; образующийся при этом радикал НО •
диффундирует в водную фазу, а органический радикал — в поли-
мерную частицу. В большей степени этот эффект может наблю-
даться в системах инициаторов, состоящих из инициатора, раство-
римого в органических жидкостях, и водорастворимого активатора,
например в смеси гидроперекиси кумола и соли двухвалентного
железа.
4.46. Эмульгаторы
В эмульсионной полимеризации применяют как анионные, так
и неионные эмульгаторы (или поверхностноактивные вещества,
как их часто называют). Эмульгаторы катионного типа «отрав-
ляют» многие инициаторы, поэтому их применяют только в очень
редких случаях [16]. В основном используют поверхностноактив-
ные вещества анионного типа, а в числе их — алкилсульфаты,
алкпларилсульфонаты и фосфаты. Многие из них, например
лаурилсульфат натрия, являются мылами, и под таким названием
эмульгаторы часто встречаются в литературе. К неионным эмуль-
гаторам относятся некоторые гидрофильные соединения: окси-
этплцеллюлоза, поливиниловый спирт и ряд производных поли-
оксиэтилена. Количество поверхностноактивных веществ в типич-
ной смеси для эмульсионной полимеризации составляет 1—5%
веса мономеров. При [Е];>8—10% N перестает зависеть от коли-
чества эмульгатора.
Большое значение в эмульсионной полимеризации имеет
качество воды. Как правило, реакцию проводят с использованием
деионизованной воды. Посторонние ионы могут губительно влиять
на процесс инициирования и действие эмульгатора.
4.4в. Рост и обрыв
Эмульсионная полимеризация идет в полимерной частице,
концентрация полимера в которой весьма велика в течение всего
времени реакции. На заключительных этапах реакции такая
система имеет много общего с полимеризацией в блоке, что позво-
ляет предположить проявление эффекта Тромсдорфа. Определив
IM] и Д’, можно вычислить константу скорости роста цепи
из уравнения (4.2).
Большие трудности вызывает определение константы скорости
обрыва [12, 17]. Для некоторых мономеров кр в эмульсионной
и гомогенной полимеризации близки между собой. Так, кр стирола
^’етДл^кРилата в эмУльсионной полимеризации при 50 °C равны
206 и 472 [18], а в гомогенной полимеризации при 60 °C они состав-
ляют соответственно 145 и 2090 (см. табл. 3.9). Не очень большое
расхождение в кр для двух процессов неудивительно и связано
268
Глава 4
с тем, что при высоких степенях превращения кр в гомогенной
полимеризации изменяется незначительно (см. табл. 3.12).
Иным зависимостям подчиняются реакции обрыва. Константы
скорости обрыва к0 в эмульсионной полимеризации примерно
в 1000 раз ниже, чем в гомогенной полимеризации [18, 19]. В обоих
процессах реакция обрыва значительно более чувствительна
к вязкости среды, чем реакция роста цепи.
4.4г. Энергетика
По-видимому, суммарная теплота эмульсионной полимериза-
ции эквивалентна теплоте полимеризации в расплаве и растворе.
Так, теплоты эмульсионной полимеризации акриловой кисло-
ты, метилакрилата и метилметакрилата составляют 18,4, 18,6
и 13,6 ккал/моль соответственно [20]. При гомогенной полимери-
зации тех же мономеров выделяется соответственно 18,5, 18,8
и 13,5 ккал/моль (см. табл. 3.10). Поскольку суммарная теплота
полимеризации определяется в основном теплотой стадии роста
цепи, на нее заметно не влияет различие в механизмах гомогенной
и эмульсионной полимеризаций.
Хотя влияние температуры на процесс эмульсионной поли-
меризации изучено недостаточно, можно считать, что с неболь-
шими отклонениями оно имеет тот же характер, что и в гомогенной
полимеризации. С ростом температуры суммарная скорость
полимеризации повышается вследствие увеличения кр и N.
N увеличивается в результате повышения [Е], вызванного умень-
шением критической мицеллярной концентрации эмульгатора.
(Число частиц увеличивается также вследствие повышения ско-
рости образования радикалов при более высоких температурах.)
Однако при увеличении температуры несколько понижается кон-
центрация мономера в частицах. Так, при повышении темпера-
туры от 30 до 90 °C [М] для стирола уменьшается примерно на 15%
[2]. Суммарная энергия активации в эмульсионной полимериза-
ции включает энергии активации роста цепи и образования ради-
калов, а также КМК и [М]. Для некоторых исследованных систем
суммарная энергия активации в эмульсионной полимеризации
мало отличается от таковой в гомогенной полимеризации. Так,
суммарная энергия активации полимеризации метилметакрилата
в блоке и эмульсии составляет соответственно 13,2 и 12,2 —
13,0 ккал/моль [21].
4.4д. Инверсионные эмульсионные системы
В общепринятом варианте эмульсионной полимеризации гид-
рофобный мономер эмульгируется в воде в присутствии эмуль-
гатора, причем полимеризация инициируется водорастворимыми
Эмульсионная полимеризация
269
инициаторами. Возможен и обратный вариант, согласно которому
водный раствор гидрофильного мономера, например акриловой
кислоты или акриламида, эмульгируется в гидрофобной органи-
ческой жидкости в присутствии эмульгатора [22]. В качестве
инициаторов полимеризации здесь применяются вещества, раство-
римые в воде или органических растворителях. По механизму
полимеризации такие инверсионные эмульсионные системы почти
не отличаются от обычных систем. Единственное важное разли-
чие между ними заключается в значительно меньшей стабиль-
ности частиц в инверсионных эмульсиях по сравнению с обыч-
ными эмульсиями, что обусловлено, по-видимому, разной приро-
дой электростатических сил, действующих в обеих системах.
4.5. Отклонения от теории Смита —
Эварта
Как правило, в большинстве случаев на практике поведение
систем, которые называют эмульсионными, плохо подчиняется
теории Смита — Эварта (см. выше). Согласно этой теории [уравне-
ния (4.2), (4.4) и (4.7)], должны выполняться следующие соотно-
шения:
BP<XN, [1]2/б, [Е]3/5, (4.9)
N<X [1]2/б, [Е]3/5, (4.10)
XnOCN, [Е]’/з, [1]_3/5, (4.11)
где [I] — концентрация инициатора.
Даже при беглом просмотре литературы [22—26] обнаружи-
вается, что во многих работах встречаются различные отклонения
от поведения по Смиту — Эварту. К ним относятся такие кажу-
щиеся аномальными результаты, как отсутствие зависимости N
от [I], 7?р от [I] или Хп от [I]. Другие авторы приводят данные,
согласно которым показатель степени зависимости 7?р от N значи-
тельно ниже, например 0,2. В ряде систем число полимерных
частиц зависит от [Е] в степени 3; известны случаи, когда показа-
тель степени в этой зависимости меняется от х/2 до 3, а также
системы, где N зависит от [Е] в степени меньше х/2. Еще более
удивительными являются обратные зависимости между Rv и N,
Вр и [Е], а также Хп и [Е].
Отдельные аномальные результаты появляются из-за непра-
вильной обработки экспериментальных данных и при неправиль-
ном толковании теории Смита — Эварта. Другие можно объяс-
нить невозможностью применения этой теории к ряду систем [27].
Некоторые из аномальных результатов лучше всего описываются
более новой теорией эмульсионной полимеризации, разработанной
Медведевым и Шейпкер [28].
270
Глава 4
4.5а. Величина п
Рассмотрим некоторые отклонения от теории Смита — Эварта.
Одним из допущений в этой теории является то, что среднее число
радикалов (п) на одну полимерную частицу равно 0,5. Однако
если размер частиц превышает 0,1—0,15 мкм, то такая частица
может захватывать сразу больше одного радикала. При этом,
поскольку среднее число радикалов больше 0,5, скорость поли-
меризации увеличивается и становится выше величины, которая
получается из расчета по уравнению (4.2). С этим эффектом осо-
бенно следует считаться при больших размерах частиц и высоких
степенях превращения. При высокой степени превращения размер
частиц увеличивается, к0 уменьшается, что и приводит к повыше-
нию п. Для частиц большого размера возможно увеличение п
и при невысоких степенях превращения. Так, при полимеризации
частиц стирола размером 0,7 мкм при степени превращения 90°,о п
возрастает всего лишь с 0,5 до 0,6. В то же время для частиц раз-
мером 1,4 мкм при степени превращения 80% п равно уже 1,
а при 90%-ной конверсии — более 2 [13]. (Величину п можно
получить путем независимых измерений _Rp, N, кр и [М].) Многие
аномалии, о которых шла речь выше, обусловлены именно этим
эффектом. При расчетах с помощью уравнений (4.2) и (4.4) полу-
чаются неверные соотношения между различными параметрами
реакции, если п фактически не равно 0,5.
4.56. Теория Медведева — Шейнкер
При внимательном рассмотрении литературных данных оказы-
вается, что во многих реакциях эмульсионной полимеризации,
особенно при более высоком содержании полимерных частиц
(> 2-1014 — 3-1014), число полимерных частиц и скорость поли-
меризации связаны с большой степенью точности следующими
соотношениями:
МОС[Е]3, [1]°, (4.12)
Bpa№/5, [Е]1/2, [I]12. (4.13)
В таких системах механизм эмульсионной полимеризации
должен быть иной. Реакция в этом случае, очевидно, протекает
на поверхности частицы, а не внутри ее. Медведев и Шейнкер
[27, 28] предположили, что радикалы не могут проникнуть в объем
частицы из-за ее высокой вязкости, поэтому полимеризация идет
на поверхности частицы. Эту теорию подтверждают результаты
Эмульсионная полимеризация
271
экспериментальных исследований, согласно которым скорость
полимеризации линейно зависит от суммарной поверхности всех
частиц [29].
Осуществление полимеризации на поверхности частиц в таких
системах подтверждается тем, что степень разветвления поли-
стирола. полученного эмульсионной полимеризацией, примерно
такая же, как и полистирола, синтезированного блочной поли-
меризацией [30]. Если бы полимеризация протекала внутри
полимерной частицы, то на протяжении всей реакции концентра-
ция полимера вблизи реакционного центра была бы высокой.
В таких условиях следовало ожидать большего разветвления
в результате реакций передачи цепи на полистирол, чем при
блочной полимеризации. На поверхности же частицы концентра-
ция полимера намного ниже, поэтому при полимеризации на
поверхности разветвление не должно протекать в заметной
степени.
Теория Медведева и Шейнкер достаточно хорошо описывается
уравнениями (4.12) и (4.13), хотя некоторые детали процесса
пока не ясны. Скорость полимеризации линейно зависит от сум-
марной поверхности частиц, которая в свою очередь сравнительно
мало чувствительна к числу полимерных частиц. Так, исходя
из геометрии сферических частиц при десятикратном увеличении
N суммарная поверхность частиц лишь удваивается. В итоге
в уравнение, связывающее Яр и N, N входит в степени, значи-
тельно меньшей единицы, которая требуется теорией Смита —
Эварта. Кроме разной зависимости Вр от N, теории Смита —
Эварта и Медведева — Шейнкер совершенно по-разному описы-
вают влияние концентрации эмульгатора и инициатора на N
[ср. уравнение (4.10) и (4.12)]. Кубическая зависимость N от [Е]
вытекает из линейной зависимости суммарной поверхности от кон-
центрации эмульгатора.
Неправильно было бы считать, что указанные две теории
эмульсионной полимеризации должны обязательно исключать
друг друга. Обе теории применимы, по-видимому, в разных
условиях в зависимости от того, где протекает реакция: на поверх-
ности частицы или внутри ее. К сожалению, далеко не ясно,
на каком этапе реакция переходит из глубины частицы на поверх-
ность или наоборот. По всей вероятности, теория Смита — Эварта
лучше описывает процессы, в которых полимер растворим в соб-
ственном мономере (например, стироле). В этом случае реакция
идет во всем объеме частицы, включая, несомненно, и ее поверх-
ность. Реакции, в которых полимер нерастворим в его собственном
мономере (например, винилхлорид, винилиденхлорид, акрило-
нитрил), обычно лучше описываются теорией Медведева — Шейн-
кер. И наконец, как уже было показано выше, теория Медведева—
Шейнкер более применима при высоком числе частиц.
Глава 4
4.5в. Влияние растворимости в воде
Зависимости, вытекающие из теории Смита — Эварта, выпол-
няются лучше всего при полимеризации таких мономеров, как
стирол, бутадиен, изопрен, которые очень плохо растворяются
в воде ( < 0,1 вес. %). Положение меняется при переходе к моно-
мерам, заметно растворяющимся в воде. К ним относятся винил-
хлорид, метилметакрилат, винилацетат, метилакрилат и акрило-
нитрил, растворимость в воде которых составляет 1,1, 1,5, 2,5
5,6 и 8,5% соответственно [23]. При полимеризации таких моно-
меров часто наблюдаются отклонения от теории Смита —
Эварта. Более удачно эмульсионная полимеризация таких моно-
меров описывается в рамках теории Медведева — Шейнкер.
В зависимости от условий реакции система может подчиняться
закономерностям той или другой теории.
При эмульсионной полимеризации мономера, растворимого
в воде, реакция осуществляется и в растворе, и в мицеллах.
При реакции в растворе образующийся полимер (или макроради-
кал) выпадает в осадок и может при этом захватывать часть эмуль-
гатора, находящегося в системе. Вследствие понижения при
этом [Е] уменьшается N. С другой стороны, в выпадающих в осадок
полимерных частицах может идти эмульсионная полимеризация
[26]. Источником образования полимерных частиц могут быть
частички выпавшего полимера и мицеллы мономера. Таким обра-
зом, весь полимеризационный процесс складывается из полимери-
зации в растворе, эмульсионной полимеризации с участием исход-
ных мицелл мономера и эмульсионной полимеризации в части-
цах осадка полимера. (Много общего с таким эмульсионным
процессом имеет гомогенная полимеризация, протекающая с выпа-
дением полимера из реакционной среды. Разница заключается
лишь в том, что в последней отсутствуют исходные мицеллы
мономера и эмульгатор.)
На поведение таких эмульсионных систем влияет соотношение
между тремя указанными полимеризационными процессами, кото-
рое в свою очередь может сильно зависеть от конкретных условий
реакции. Поэтому не удивительно, что эмульсионная полимери-
зация мономеров с заметной растворимостью в воде в одних слу-
чаях удовлетворительно описывается теорией Смита — Эварта,
в других — теорией Медведева — Шейнкер, а в третьих — обе
теории оказываются неподходящими.
4.5г. Ингибирование эмульгатором
Ряд аномалий в эмульсионной полимеризации, а именно
обратные зависимости между 2?р и N, Нр и [Е], Хп и [Е], нельзя
объяснить ни с позиций теорий Смита — Эварта, ни Медведева —
Эмульсионная полимеризация
273
Щейнкер. Все три указанные аномальные зависимости были
установлены при полимеризации этилена [241. При эмульсионной
полимеризации винилацетата в присутствии ряда эмульгаторов
наблюдалась обратная зависимость между 7?р и [Е] [31]. В этих
системах эмульгатор замедляет полимеризацию, участвуя в реак-
ции передачи цепи. В заметной степени эта реакция имеет место
только с высокоактивными радикалами этилена и винилацетата.
Наличие реакции передачи цепи является еще одним доказатель-
ством правильности теории Медведева — Шейнкер, так как
передача цепи на эмульгатор может идти только на поверхности
частицы, а не внутри ее.
4.5д. Молекулярновесовое распределение
В соответствии с теорией Смита — Эварта молекулярный вес
полимера, образующегося при эмульсионной полимеризации,
изменяется в процессе реакции по закону, описываемому уравне-
нием (4.4). На рис. 4.3 приведены кривые зависимости молекуляр-
ного веса полистирола от степени завершенности реакции. [Моле-
кулярный вес сначала быстро возрастает (при повышении степени
превращения от 0 до 20%), так как число частиц повышается.
При дальнейшем росте степени завершенности реакции молеку-
лярный вес растет уже не так быстро, причем повышение молеку-
лярного веса может быть связано с изменением 7?и. На заключи-
тельных этапах реакции возможно некоторое снижение молеку-
лярного веса, вызванное уменьшением [М] и /ср.
Теоретически молекулярновесовое распределение полимера,
полученного эмульсионной полимеризацией, должно быть более
узкое, чем в соответствующем полимере, полученном гомогенной
полимеризацией.
Можно найти такие условия полимеризации, в которых N,
[М], кр и R и сохраняются неизменными в течение почти всей
реакции, и тогда достигается относительно узкое молекулярно-
весовое распределение. Только что сказанное хорошо иллюстри-
рует кривая 1 на рис. 4.3. Однако во многих реакциях эмульсион-
ной полимеризации изменения различных параметров или откло-
нение от поведения по Смиту — Эварту приводят к получению
полимера с более широким молекулярновесовым распределением,
чем при гомогенной полимеризации.
Вопросы и упражнения
4.1. Перечислите компоненты смеси для эмульсионной поли-
меризации (на макроскопическом уровне). Укажите положитель-
ные и отрицательные стороны эмульсионной полимеризации
по сравнению с полимеризациями в блоке и растворе.
274
Глава 4
4.2. Нарисуйте микроскопическую картину эмульсионной:
полимеризации по Харкинсу, Смиту и Эварту. Где находятся
мономер, инициатор и эмульгатор? Опишите изменения, которые-
происходят в системе при достижении 100%-ной степени превра-
щения.
4.3. Каковы характерные особенности эмульсионной полиме-
ризации, отличающие ее от гомогенной полимеризации? Сравните
оба процесса по теплотам полимеризации и по влиянию темпера-
туры реакции на скорость полимеризации.
4.4. Вычислите скорость и степень полимеризации стирола
в эмульсии при 60 °C для N = 3,2-1013 частиц на 1 мл, [М] =
= 5,071/ и Ви = 1,1 -1012 радикалов/(мл-с). Примите, что система
подчиняется теории Смита — Эварта и что кр такая же, как
и в гомогенной полимеризации.
Список литературы
1. Harkins W. D., J. Am. Chem. Soc., 69, 1428 (1947).
2. Smith W. V., Ewart R. W., J. Chem. Phys., 16, 592 (1948).
3. Smith W. V., J. Am. Chem. Soc., 70, 3695 (1948).
4. Vandenberg E. J., Hulse G. E., Ind. Eng. Chem., 40, 932 (1948).
5. Bradford E. B., Vanderhoff J. W., J. Polymer Sci., Cl, 41 (1963).
6. Debye P., Anacker E. W., J. Phys. Coll. Chem., 55, 644 (1951).
7. Kratohvil J. P., Anal. Chem., 36, 485R (1964).
8. Brodnyan J. G., J. Colloid Sci., 15, 76, 563 (1960).
9. Williams D. J., Bobalek E. G., J. Polymer Sci., A-l(4), 3065 (1966).
10. Smith W. V., J. Am. Chem. Soc., 71, 4077 (1949).
11. Bovey F. A., Kolthoff I. M., Medalia A. I., Meehan E. J., Emulsion Poly-
merization, Interscience Publishers, John Wiley and Sons, Inc., New York,
1955.
12. Van Der Hoff В. M. E., Kinetics of Emulsion Polymerization, in «Poly-
merization and Polycondensation Processes», Platzer N. A. J. (ed.), chap. 1,
American Chemical Society, Reinhold Book Corp., New York, 1962.
13. Gerrens H., Fortschr. Hochpolymer.-Forsch. (Advan. Polymer Sci.), 1, 234
(1959).
14. French D. M., J. Polymer Sci., 32, 395 (1958).
15. Patsiga R., Litt M., Stannett V., J. Phys. Chem., 64, 801 (1960).
16. Mast W. C., Fisher С. H., Ind. Eng. Chem., 41, 790 (1949).
17. Vanderhoff J. W., Bradford E. B., Tarkowski H. L., Wilkinson B. W.,
J. Polymer Sci., 50, 265 (1961).
18. Gerrens H., Polymer Preprints, 7(2), 699 (1966).
19. Ley G. J. M., Schneider C., Hummel D. O., Polymer Preprints, 7(2), 725
(1966).
20. McCurdy K. G., Laidler K. J., Can. J. Chem., 42, 818 (1964).
21. Ставрова С. Д., Маргаритова M. Ф., Медведев С. С., Высокомолек.
соед., 7, 792 (1965).
22. Vanderhoff J. W., Bradford E. B., Tarkowski H. L., Shaffer J. B.,
Wiley R. M., Inverse Emulsion Polymerization, in «Polymerization and
Polycondensation Processes», Platzer N.A.J. (ed.), chap. 2, American
Chemical Society, Reinhold Book Corp., New York, 1962.
Эмульсионная полимеризация
275
23. Lindemann М. К., The Mechanism of Vinyl Acetate Polymerization, in
«Vinyl Polymerization», Ham G. E. (ed.), vol. 1, part I, chap. 4, Marcel
Dekker, Inc., New York, 1967.
24. Stryker H. K., Mantell G. J., Helin A. F., J. Polymer Sci., 11, 1 (1957).
25. Mantell G. J., Stryker H. K., Helin A. F., Jamieson D. R., Wright C. R..
J. Polymer Sci., 10, 1845 (1956).
26. Izumi Z., J. Polymer Sci., A-l(5), 469 (1967).
27. Garden J. L., J. Polymer Sci., A-l(6), 623, 643, 665, 687 (1968)
28. Шейнкер А., Медведев С. С., ДАН СССР, 97, 111 (1954).
29. Brodnyan J. G., Cala J. A., Konen T., Kelley E. L., J. Colloid Sci 18
73 (1963). ’’ ’
30. Helin A. F., Stryker H. K., Mantell G. J., J. Appl. Polymer Sci. 9 1797
(1965). ’ ’
31. Okamura S., Motoyama T., J. Polymer Sci., 58, 221 (1962).
ГЛАВА 5
ИОННАЯ ПОЛИМЕРИЗАЦИЯ
Ионная полимеризация по двойным связям углерод — углерод
и углерод — кислород была кратко описана в гл. 3. В настоящей
главе этот тип полимеризации будет рассмотрен более подробно.
Основное внимание уделено полимеризации по двойной углерод-
углеродной связи под влиянием катионных и анионных катали-
заторов (разд. 5.2 и 5.3). В последнем разделе главы рассмотрена
полимеризация по другим ненасыщенным связям. В целях удоб-
ства в данную главу включено несколько примеров реакций
полимеризации, не относящихся к ионной полимеризации.
Полимеризация, инициируемая координационными соедине-
ниями и твердыми металлами или окислами металлов, обычно
тоже бывает ионной по своей природе. Полимеризация такого
рода называется стереоспецифической, или координационной,
полимеризацией и будет рассмотрена в гл. 8. Данные о полимери-
зации диенов также не включены в настоящую главу. Циклополи-
меризация несопряженных диенов, так же как и 1,2- и 1,4-по-
лимеризация сопряженных диенов, обсуждается в гл. 6 и 8.
Л.1. Сравнение радикальной и ионной
полимеризаций
Почти все мономеры способны к радикальной полимеризации,
тогда как ионная полимеризация протекает в высшей степени
избирательно (см. табл. 3.1).
Катионная полимеризация по существу ограничивается кру-
гом мономеров с электронодонорными заместителями, такими,
как алкоксигруппы, фенил, винил и 1,1-диалкил. Анионная поли-
меризация протекает в случае мономеров, содержащих электроно-
акцепторные группы, например нитрильную, карбоксильную,
фенильную и винильную. Избирательность ионной полимериза-
ции обусловлена весьма строгими требованиями стабилизации
анионных и катионных растущих частиц (разд. 3.16.2). Использо-
вание катионной и анионной полимеризации в промышленности
довольно ограниченно вследствие отмеченной выше высокой селек-
тивности ионной полимеризации по сравнению с радикальной,
а также вследствие развития координационной полимеризации.
Ионная полимеризация
277
Вследствие экспериментальных трудностей исследования ион-
ной полимеризации она изучена в меньшей степени, чем радикаль-
ная полимеризация. Часто остается неясной природа реакционной
среды в ионной полимеризации, так как в процессе участвуют
гетерогенные неорганические катализаторы. Далее, в большинстве
случаев чрезвычайно трудно получать воспроизводимые данные
по кинетике ионной полимеризации, поскольку она протекает
с очень высокими скоростями и весьма чувствительна к присут-
ствию малых концентраций сокатализаторов, примесей и других
веществ. Скорость ионной полимеризации обычно больше скорости
радикальной полимеризации.
Катионная и анионная полимеризации имеют много общего.
Обе зависят от образования и роста ионных частиц, положительных
в одном случае и отрицательных в другом. Хотя можно ожидать,
что ионы с малой стабильностью (с высокой энергией) будут реа-
гировать с олефиновой двойной связью с большими скоростями,
они либо не образуются, либо легко разрушаются. Образуются
и имеют достаточную продолжительность жизни для роста цепи
только ионы с довольно высокой стабильностью. Энергия, необ-
ходимая для образования пары ионов из нейтральной молекулы,
очень велика. Так, для появления в газовой фазе ионов трет-
бутила и этилкарбония из соответствующих алкилхлоридов необ-
ходимо 155 и 195 ккал/моль (648,83 ПО3—816,27-К)3 Дж/моль)
соответственно [1]. Такие ионы слишком неустойчивы, и раньше,
чем произойдет полимеризация, их необходимо стабилизировать
путем сольватации и использования низких температур.
Хотя для сольватации ионов весьма желательны растворители
с высокой полярностью, они часто по ряду причин не могут быть
использованы. Сильно полярные гидроксильные растворители
(вода, спирты) реагируют с большинством ионных катализаторов,
разрушая их. Другие полярные растворители, такие, как кетоны,
препятствуют инициированию полимеризации, образуя устой-
чивые комплексы с катализаторами. Кроме того, такие раствори-
тели не могут быть жидкими при температурах реакции, лежащих
ниже —100 °C. Поэтому ионную полимеризацию обычно проводят
в мало полярных растворителях, таких, как хлористый метил,
дихлорэтилен, пентан и нитробензол. В этих растворителях нет
таких изолированных ионов, как в водных растворах. Ионы в та-
ких системах присутствуют в виде прочно связанных ионных пар.
Так, при катионной полимеризации около растущего катионного
конца цепи в течение всего периода его жизни имеется отрица-
тельный противоион. Аналогично около растущего анионного
конца цепи при анионной полимеризации имеется положи-
тельный противоион. Расстояние между ионом и его противо-
ионом возрастает с увеличением сольватирующей способности
среды.
278
Глава 5
Для ионной полимеризации характерно большое разнообразие
способов инициирования и обрыва цепи. В отличие от радикальной
полимеризации обрыв цепи при ионной полимеризации никогда
не происходит в результате бимолекулярной реакции двух цепей,
несущих одинаковый заряд. Обрыв цепи обычно происходит
вследствие мономолекулярной реакции растущей цепи или при
передаче цепи на мономер или растворитель.
5.2. Катионная полимеризация алкенов
5.2а. Инициирование
Ниже описаны различные катализаторы, которые могут быть
использованы для инициирования катионной полимеризации
мономеров с электронодонорными заместителями. Более детальные
данные по этим и многим другим применяемым катионным ини-
циаторам приведены в разд. 7.26.1.
5.%а.1. Протонные кислоты
Протонные кислоты могут быть использованы для инициирова-
ния катионной полимеризации путем протонирования олефина.
Метод основан на использовании кислот, которые обладают доста-
точной силой, чтобы образовать протонированные частицы в необ-
ходимой концентрации
HA + RR'C = CH2 -» RR'C — СН3(А)-, (5.1)
однако анион кислоты не должен отличаться сильной нуклеофиль-
ностью, иначе он прореагирует с протонированным олефином
с образованием ковалентной связи, что приведет к обрыву цепи
А
+ I
RR'C —СН3(А)- —> RR'C —СН3 (5.2)
[Метод изображения ионных частиц как в уравнениях (5.1) и (5.2),
так и во всей этой главе имеет целью подчеркнуть, что ионные
частицы обычно существуют не в виде отдельных ионов, а в виде
ионных пар. Скобки у анионного фрагмента применяются, чтобы
показать, что отрицательный противоион остается вплотную около
положительной частицы.]
Сильная нуклеофильность аниона обычно ограничивает исполь-
зование большинства сильных кислот в качестве катионных ката-
лизаторов. Галогенные кислоты часто не применяют для этой
цели из-за высокой нуклеофильности иона галогена. Однако
другие сильные кислоты, такие, как хлорная, серная и фосфорная,
находят применение в процессах полимеризации некоторых моно-
Ионная полимеризация
279
мепов. Молекулярные веса полимеров, полученных при этом,
редко превышают несколько тысяч. Низкомолекулярные поли-
меры полученные из углеводородов, используют в качестве
дизельного топлива, газолина, смазочных материалов и ряда
других продуктов. В качестве катализаторов обычно применяют
фосфорную или серную кислоту, нанесенную на инертный мате-
риал, или кислоты Льюиса; процесс проводят, как правило,
при 200-300 °C.
При полимеризации кумарона (бензофурана) и индена
кумарон инден
в присутствии серной кислоты образуются полимеры (молеку-
лярный вес до 1000), которые применяют для получения поверх-
ностных покрытий, клеев, плиток для полов, парафинированной
бумаги и др.
5.2а.2. Кислоты Льюиса
Катализаторы Фриделя — Крафтса — А1С13, BF3, SnCl4, ZnCl2,
TiBr4 и другие кислоты Льюиса — используют при низких темпе-
ратурах для получения продуктов высокого молекулярного веса.
Самым важным промышленным полимером большого молекуляр-
ного веса, получаемым катионной полимеризацией, является
бутилкаучук, сополимер изобутилена и диена-1,3 (обычно изопре-
на). Полимеризацию проводят приблизительно при —100 °C
в хлорированных растворителях с использованием в качестве
катализатора А1С13. Все катализаторы Фриделя — Крафтса, при-
меняемые в катионной полимеризации, требуют присутствия
какого-либо сокатализатора, который действует как донор про-
тона. Катализатором может быть вода, органическая кислота
и даже органический углеводород. Так, изобутилен не.чувстви-
телен к сухому трехфтористому бору, но мгновенно полимери-
зуется при добавлении следов воды [2]. Сокатализатор (вода),
очевидно, реагирует с катализатором (трехфтористый бор), обра-
зуя комплекс катализатор — сокатализатор, который затем про-
должает протонировать олефин и инициировать рост цепи
BF3 + H2O H+(BF3OH)-, (5.3а)
Н+ (BF3OH)"-|- (СН3)2С = СН2 —> (CH3)SC+(BF3OH)- (5.4а)
280
Глава 5
Процесс инициирования можно выразить в общем виде следующим
образом:
к
C + RH — H+(CR)-, (5.36)
H+(CR)~ + M ---> HM+(CR)“, (5.46)
где G, RH и M — катализатор, сокатализатор и мономер соот-
ветственно.
Общепринято мнение, что катализаторы Фриделя — Крафтса
нуждаются в сокатализаторах, хотя точно это было показано лишь
в нескольких случаях. Детальному изучению необходимости при-
сутствия сокатализаторов сильно мешают экспериментальные
Рис. 5.1. Влияние концентрации
воды на скорость катализируемой
SnCl4 полимеризации стирола в СС14
при 25 °C.
Концентрация катализатора, М: 1) 0,08;
2) 0,12.
трудности, связанные с приготовлением катализаторов и моно-
меров, полностью свободных от веществ, которые могут действо-
вать как сокатализаторы.
Активность комплекса катализатор — сокатализатор зависит
от его способности отдавать протон. Так, было установлено, что
скорость полимеризации изобутилена с SnCl4 в качестве катали-
затора возрастает с увеличением кислотности сокатализатора.
Скорость уменьшается в ряду: уксусная кислота > нитроэтан >
> фенол > вода. Этиловый и mpem-бутиловый спирты не оказы-
вают сокаталитического влияния [3]. В большинстве случаев
максимальной скорости полимеризации соответствует некоторое
определенное отношение сокатализатора к катализатору. Выше
и ниже этого оптимального значения отношения сокатализа-
тор/катализатор скорость полимеризации уменьшается. Это видно
из данных рис. 5.1 для катализируемой четыреххлористым оловом
полимеризации стирола в четыреххлористом углероде водой
в качестве сокатализатора [4]. Оптимальное значение отношения
Ионная полимеризация
281
сокатализатора к катализатору существенно изменяется для
различных комплексов сокатализатор — катализатор и даже для
одного и того же комплекса в различных растворителях. Так,
полимеризация стирола протекает с максимальной скоростью
при значениях [H2O]/[SnCl4], равных 0,002, в четыреххлористом
углероде и 1,0 в смеси 30% нитробензола и 70% четыреххлори-
стого углерода [4, 5]. Эти результаты считают свидетельством
того, что полимеризация в среде полярного нитробензола катали-
зируется дигидратом хлорида олова или его эквивалентами
(например, гидратом трихлороловянной кислоты SnGl3OH-Н2О),
а в среде неполярного четыреххлористого углерода — моногид-
ратом. Другое объяснение этого явления приводится в разд. 5.2в.2.
5.2а.З. Другие катализаторы
При взаимодействии алкилгалогенидов с катализаторами Фри-
деля — Крафтса может образовываться ион карбония, способный
инициировать катионную полимеризацию, например
RCl + SnCl4 -» R+ (SnCl5)- (5.5)
Возможностью протекания реакции этого типа можно объяс-
нить наблюдаемое в некоторых случаях катионной полимеризации
явление сокатализирующего действия хлорированных углево-
дородов, используемых в качестве растворителей [4]. Известно
также множество других катионных катализаторов, таких, как
I2, Gu2+, ионы оксония, и излучение высокой энергии [4,6—8].
Молекулярный иод инициирует полимеризацию путем реакции
I2 + I2 1+(13)- (5.6)
(Отрицательный противоион, возможно, представляет собой ком-
бинацию ионов I-, 1“ и Ij.) Ион Gu2+, вероятно, инициирует поли-
меризацию либо путем окисления л-связи (переход электрона
от мономера к Си2+), либо присоединением по л-связи. Излучение
высокой энергии — гамма-лучи, электроны и нейтроны — может
инициировать как радикальную, так и ионную полимеризацию
в зависимости от условий реакции, т. е. в зависимости от темпера-
туры и применяемых в данном случае мономера и растворителей.
«
5.26. Рост цепи
Инициирующая ионная пара (состоящая из иона карбония
и его отрицательного противоиона), образовавшаяся на стадии
инициирования [уравнение (5.4)], продолжает расти путем после-
довательного присоединения молекул мономера
Н — [ — СН2С(СН3)2-]+ (BF3OH)-+(CH3)2C = CH2 -»
-> н—[-СН2С(СН3)2— ]n-CH2C+(CH3)2(BF3OH)- (5.7а;
282
Глава 5
ИЛИ
hp
HM+(CR)- + M----> HMnM+(CR)~. (5.76)
Предполагают, что это присоединение происходит внедрением
мономера между ионом карбонил и его отрицательным проти-
воионом.
Реакция роста в некоторых случаях может осложняться реак-
циями внутримолекулярной перегруппировки. Перегруппировки
или изомеризации ионов карбонил через гидридные (Н:_) или
карбанионные (R:~) сдвиги хорошо известны в органической
химии. Доля реакций перегруппировки при катионном росте
цепи зависит от относительной стабильности растущего и под-
вергающегося перестройке иона карбонил и от относительных
скоростей реакций роста и перестройки. Высокие стабильность
растущего иона и скорость реакции роста благоприятствуют росту
цепи без перегруппировки для большинства мономеров, полиме-
ризуемых по катионному механизму. Однако при полимеризации
некоторых олефинов перегруппировка все же наблюдалась. Так,
продукт полимеризации З-метилбутена-1 содержит как нормаль-
ные (I), так и подвергшиеся перегруппировке (II) повторяющиеся
звенья
— СН2 —СН — _сн2—СН2—С(СН3)2—
I
СН(СН3)2
I II
в количествах, меняющихся в зависимости от температуры реак-
ции [9]. Полимеризацию такого рода часто называют изомериза-
ционной полимеризацией. Изомеризация нормального растущего
иона карбонил (III) происходит путем гидридного сдвига, проис-
ходящего раньше, чем присоединение следующей мономерной
единицы
Н
I
— СН2-С+ — СН2 —СН2 —С(СН3)2
(СН3)2С:Н
III IV
Изомерный ион IV представляет собой третичный ион карбонил
и является более стабильным, чем нормальный ион III, предста-
вляющий собой вторичный ион карбонил. Продукт содержит боль-
шей частью изомеризованные повторяющиеся единицы, но при
повышенных температурах происходит в некоторой степени
и нормальный рост, обусловленный кинетическими причинами.
В полимере, полученном при —130 и —100° С, содержится соот-
ветственно 0 и 30% нормальных повторяющихся единиц.
Ионная полимеризация
283
Среди других мономеров, подвергающихся изомеризационной
полимеризации, можно назвать 4-метилпентен-1, 5-метилгексен-1
и 6-метилгептен-1 [10, И]. Изомеризационная полимеризация
4 4-диметплпентена-1 происходит в результате перемещения
метильной группы, тогда как в а- и Р-пинене протекает пере-
стройка кольца [11].
5.2в. Обрыв цепи
5.2в.1. Отсутствие обрыва кинетической цепи
Передача цепи на мономер — одна из наиболее частых реакций
обрыва цепи при катионной полимеризации. Эта реакция заклю-
чается в переходе комплекса сокатализатор — катализатор к моле-
куле мономера, сопровождающемся образованием концевой нена-
сыщенной группы в макромолекуле полимера
Н—[ —СН2С(СН3)2- ]n -CH2C+(CH3)2(BF3OH)- + СН2 = С(СН3)2 -»
-» (CH3)3C+(BF3OH)-+H-[-CH2C(CH3)2-]n-CH2C(CH3) = CH2 (5.8а)
или
ftnep.М
HMnM+(CR)--|-M-------> Mn+i + HM+(CR)-. (5.86)
Следует отметить, что кинетическая цепь при этом не обры-
вается, так как происходит регенерирование инициирующей
ионной пары. Каждый единичный комплекс катализатор — сока-
тализатор приводит к образованию многих макромолекул.
Передача цепи на мономер при катионной полимеризации
оказывает значительно большее влияние на рост полимерной цепи,
чем при радикальной полимеризации.
Поскольку передача цепи на мономер кинетически неотличима
от роста, относительные скорости передачи и роста даются отно-
шением *пер. м//ср, которое представляет собой константу передачи
цепи для мономера См. Значение См определяет молекулярный
вес полимера, если обрыв цепи в результате других процессов
незначителен. Чем больше значение См, тем меньше молекуляр-
ный вес образующегося полимера.
Другим достаточно важным типом реакции передачи цепи на
мономер является реакция отрыва гидрид-иона от мономера
растущей частицей
H-[-CH2C(CH3)2-]n_CH2C(CH3)2(BF3OH)-+CH2 = C(CH3)2 -»
-» СН2 = С(СН3) - CH2(BF3OH)- 4- н — [ -СН2С(СН3)2-]„ - СН2СН(СН3)2
X (5.8в)
Обе эти реакции передачи цепи на мономер кинетически неотли-
чимы, но первая из них [уравнение (5.8а)] приводит к образованию
284
Глава 5
ненасыщенной концевой группы, тогда как вторая [уравнение
(5.8в)] дает насыщенную концевую группу. Обрыв цепи может
происходить также путем перестройки растущей ионной пары.
Этот тип обрыва часто называется самопроизвольным обрывом или
передачей цепи на противоион. Первоначальный комплекс катали-
затор — сокатализатор регенерируется выделением из ионной
пары, и полимерная макромолекула остается с ненасыщенной
концевой группой
Н —[ —СН2С(СН3)2 —]n —CH2C(CH3)2(BF3OH)_ ->
-> H+(BF3OH)- + H-[-CH2C(CH3)2-]n-CH2C(CH3) = CH2 (5.9а)
или
*о
НМПМ+(СВ)"----> Mn+i + H+(CR)-. (5 96)
Обрыв цепи такого рода сходен с обрывом цепи путем передачи
цепи на мономер тем, что кинетическая цепь не обрывается и каж-
дая частица комплекса катализатор — сокатализатор способствует
образованию большого числа полимерных молекул.
5.2в.2. Обрыв кинетической цепи
Обрыв цепи в результате комбинации растущего иона карбония
с противоионом происходит в том случае, когда последний доста-
точно нуклеофилен, так что происходит образование ковалентной
связи
%
HMnM+(CR)-----> HMnMCR, (5.10а)
как, например, при полимеризации стирола, катализируемой
трифторуксусной кислотой
Н - (СН2СНСвН5- )n -CH2CHCeH5(OCOCF3)- ->
—» Н —( —CH2CHCeH5 —)n —CH2CHCeH5 —OCOCF3. (5.106)
Иногда растущий ион может соединяться с анионным фрагмен-
том противоиона, например
Н — [ — СН2С(СН3)2 - ]„- CH2C(CH3)2(BF3OH)-
-» н — [-СН2С(СН3)2—]n-CH2C(CH3)2OH + BF3. (5.10в)
Обрыв цепи путем комбинирования отличается от других видов
обрыва тем, что он приводит к уменьшению концентрации ком-
плекса катализатор — сокатализатор.
Помимо передачи цепи на мономер в различных частных случа-
ях полимеризационных систем может иметь существенное значение
и ряд других реакций передачи цепи. Различные агенты переноса
(обозначенные символами S или ХА, как в гл. 3), присутствующие
Ионная полимеризация
285
качестве растворителя, примесей или намеренно добавленные
в систему, могут обрывать растущую полимерную цепь путем
переноса и образования ковалентной связи с отрицательным
фрагментом А"
fenep.S
IIM71M+(CR)- + XA----> HMnMA4-XCR. (5.11)
Вода, спирты, кислоты, ангидриды, простые и сложные эфиры,
а также амины обладают различной способностью передачи цепи
[12]. В присутствии большинства из этих агентов передачи проте-
кает реакция (5.11), которая представляет собой важнейший
способ обрыва. Возможно, что обрыв цепи аминами происходит
не путем передачи цепи, а путем образования стабильных четвер-
тичных попов, которые переакционноспособпы, например
IIMnM+(CR)- + :NR3 —> HMnMN+R3CR~. (5.12)
Ароматические соединения и алкилгалогепиды также в некоторых
случаях действуют как агенты передачи цепи. Передача цепи
на ароматические соединения происходит как путем передачи
отрицательного фрагмента, так и алкилированием ароматических
ядер, в зависимости от природы заместителей в ароматическом
соединении.
Наличие передачи цепи на полимер, происходящей в различ-
ной степени, тоже было отмечено. Передачей цепи на полимер,
вероятно, объясняется образование только низкомолекулярных
полимеров из таких а-олефинов, как пропилен. Растущие ионы
карбония представляют собой реакционноспособные вторичные
ионы карбония, которые могут отрывать третичные гидридные
ионы от полимера.
+ Н II +
— СН2 — CR-|----СН2 —С — —* — СП, —CR-!--------СН, —С— (5.13)
II R II R
Бензохинон действует как ингибитор вследствие передачи прото-
нов от растущих частиц или комплекса катализатор — сокатали-
затор к молекуле хинона
2HMnM+(CR)- + O=^2^\= О Mn+1 + [HO-/ Oh]2+(CR2)
~ (5.14)
Следует отметить, что многие из веществ, которые действуют
как сокатализаторы (например, вода), являются также хорошими
агентами передачи цепи. В этом одна из причин того, что на кривых
зависимости скорости полимеризации (а также степени поли-
меризации) от концентрации сокатализатора обычно наблюдаются
максимумы.^ Максимум на таких кривых (рис. 5.1) соответствует
оптимальной концентрации комплекса катализатор — сокатали-
затор. Выше оптимальной концентрации сокатализатора при-
обретает большое значение передача цепи на сокатализатор.
286
Глава 5
5.2г. Кинетика ионной полимеризации
5.2г.1. Различные кинетические положения
Кинетика процесса в целом существенно зависит от способа
обрыва цепи в каждой данной системе. Рассмотрим случай моно-
молекулярного обрыва цепи в результате перегруппировки
растущей ионной пары [уравнение (5.9)]. Кинетическая схема
инициирования, роста и обрыва цепи включает реакции (5.3),
(5.4), (5.7) и (5.9). Уравнение скорости полимеризации для этого
процесса выводят аналогично уравнению скорости радикальной
полимеризации (гл. 2).
Скорости инициирования, роста и обрыва выражаются урав-
нениями
КИ = ОИ[С][КН][М],
7?р = *P[HM+(CR)-][M],
R0 = *0[HM+(CR)-],
(5.15)
(5.16)
(5-17)
где [HM+(CR)~] представляет собой общую концентрацию всех
растущих ионных пар, а К — константа равновесия для реакции
(5.36).
Допуская постоянство концентрации растущих частиц в стацио-
нарном состоянии, т. е. считая равными скорости инициирования
и обрыва, имеем
[HM+(CR)“] ^h[C][RH][M]
(5.18)
ко
Комбинируя уравнения (5.16) и (5.18), получим для скорости
полимеризации выражение
XMn[C][RH][M]2
Лр
Среднечисловая степень
(5.19)
(15.20)
п
7с0
полимеризации выражается как
Др _ 7ср[М]
До к0
Уравнения (5.19) и (5.20) применимы также в тех случаях, когда
обрыв цепи происходит комбинированием растущего иона карбо-
ния со своим противоионом, так как реакция (5.10) кинетически
неотличима от реакции (5.9). Однако следует иметь в виду, что
в то время, как концентрация комплекса катализатор — соката-
лизатор в первом случае остается неизменной, во втором случае
она уменьшается с увеличением степени превращения. Если же
обрыв происходит путем передачи цепи на мономер [уравне-
Ионная полимеризация
287
ние (5.8)], то концентрация [HM+(CR) ] выражается как
[HM+(CR)-]=*yi[RH1 , (5.21)
«пер М
а скорость и степень полимеризации как
XMP[C][RH][M]
Р ^пер.М
X _ 1
П ^пер.М Cjj ’
(5.22)
(5.23)
где См — константа передачи цепи на мономер.
Для случая, когда преобладает передача цепи на агент пере-
дачи S [уравнение (5.11)],
[HM+(CR)-]=-^-f[-[R^— > (5.24)
Я’пер. SL^J
а скорость и степень полимеризации дается уравнениями
WP[C][RH][M]2 Лр *nep.s[S] X ым1 [М1 п fcnep. S [S] Cs[S] ’ (5.25) (5.26)
где Cs — константа передачи цепи на данный агент.
Для скорости полимеризации были выведены различные
выражения, исходя из допущения, что определяющей стадией
в процессе инициирования является реакция (5.4). Если это
не так и определяющей для скорости полимеризации является
реакция (5.3), то скорость инициирования становится независи-
мой от концентрации мономера. Каждое из выражений [уравне-
ния (5.19), (5.20), (5.22), (5.23), (5.25), (5.26)] для скорости и сте-
пени полимеризации будет давать зависимость от [М] на один
порядок ниже, а кИ будет константой скорости для реакции (5.3)
вместо реакции (5.4). Во многих случаях реакция (5.4) определяет
скорость, и равновесие для реакции (5.3) полностью сдвинуто
вправо. В таких случаях скорость инициирования выражается как
7?и = ка [RH] [М] при избытке С (5.27а)
или
ЯИ = МС][М] при избытке RH. (5.276)
Тогда соответственно должны быть видоизменены различные
кинетические выражения.
/ч «3>\)аВ/Еения скорости катионной полимеризации — (5.19),
(5.22), (5.25) — указывают на весьма существенное различие
между процессами катионной и радикальной полимеризации.
Так, при радикальной полимеризации зависимость Rp от Ra
288
Глава 5
имеет порядок 1/2, тогда как при катионной — первый. Различие
между этими типами полимеризации является следствием разных
способов обрыва. При радикальной полимеризации обрыв расту-
щих цепей — реакция второго порядка, тогда как при ионной
полимеризации — реакция первого порядка.
5.2г.2. Справедливость допущения стационарного
состояния
Возможно, что для многих случаев катионной полимеризации
допущение постоянства [HM+(CR)_] не совсем правильно. Иногда
ионная полимеризация протекает так быстро, что стационарное
состояние не достигается. Некоторые из этих реакций (например,
полимеризация изобутилена в присутствии А1С13 при —100 °C)
могут завершаться в течение секунд или минут. Даже при более
медленной реакции стационарное состояние может быть достиг-
нуто только на поздних стадиях полимеризации. Так, стацио-
нарное состояние при катализируемой ReCls полимеризации
стирола, протекающей при 0 °C, достигается только после
103—104 с, когда степень превращения мономера составляет
приблизительно 20—30% [14].
Еще одним ограничением при использовании различных кине-
тических уравнений является неопределенность представления
о том, гомогенны или гетерогенны многие системы катализатор —
мономер — растворитель. Однако было установлено, что,
несмотря на отмеченные затруднения, приведенные выше кине-
тические уравнения обычно справедливы [3, 12].
5.2д. Абсолютные константы скорости
5.2д.1. Экспериментальные методы
Индивидуальные константы скорости (Ли, Zcp, к0, каер) при
ионной полимеризации не исследованы так широко, как при
радикальной полимеризации. По величине степени полимери-
зации можно определить отношения kolkv и кпе^/к^. Рассмотрим
общий случай, когда возможен мономолекулярный обрыв цепи
и обрыв при передаче цепи как на мономер, так и на агент пере-
дачи. Величина, обратная Хп, полученная из уравнений (5.20),
(5.23) и (5.26), дается уравнением
1_____ко__I ^пер. М । ^пер. S [S]
“ fcp[M] кР fep[M]
или
1 ^8 [8]
(5.28а)
(5.286)
Ионная полимеризация
289
Уравпение (5.28) для катионной полимеризации аналогично
уравнению (3.109), описанному выше для радикальной полимери-
3аЦТр'и отношения констант скорости (Аг0/*р, *пер. м^р и *пер. s^p)
могут быть получены соответствующими методами. В отсутствие
агентов передачи цепи зависимость 1/Хп от 1/[М] дает прямую
линию, наклон которой соответствует kjkp, а на оси ординат,
отсекается отрезок, равный &Пер.м^р или ^м- В присутствии аген-
та передачи цепи knep. s/kp или Cs соответствует наклону прямой,
выражающей зависимость 1/Хп от [S]/[M1.
Для определения индивидуальных констант скорости необ-
ходимо определение константы скорости роста. Величина кр была
рассчитана при использовании данных по скорости и степени
полимеризации, определенных в условиях нестационарного состоя-
ния [15, 16]. В большинстве примеров, однако, кр получают прямо
пз уравнения (5.16), используя эксперименты, выполненные при
условиях, где предполагается, что
[HM+(CR)-] = [C], (5.29)
или где [HM+(CR)~] может быть определена экспериментально.
Условие уравнения (5.29) удовлетворяется в таких системах,
в которых скорость процесса инициирования значительно превы-
шает скорость роста.
Прямое определение концентрации промежуточных реакцион-
ных соединений [HM+(CR)_] возможно в катионной полимериза-
ции, так как в этом случае концентрации достаточно велики. (Это
резко отличается от случая радикальной полимеризации, где
активные промежуточные соединения присутствуют в очень
малых концентрациях.) Для аналитических целей часто приме-
няются спектроскопические методы. В некоторых случаях ионы
карбония бывают окрашены, например полистирильный ион.
Однако прямое определение [HM+(CR)_] часто неоднозначно,
поскольку не удается точно установить полную идентичность
частиц (разд. 5.2е).
5.20.2. Сравнение скоростей катионной
и радикальной полимеризаций
В табл. 5.1 приведены различные кинетические параметры
катализируемой серной кислотой полимеризации стирола в дихлор-
этане при 25 °C. Интересно сравнение этих данных с соответст-
вующими данными для радикальной полимеризации (табл. 3.8
и 3.9). Вполне очевидно, что реакции роста и обрыва цепи
при катионной полимеризации протекают на порядок медленнее.
Однако обычно скорости катионной полимеризации больше скоро-
290
Глава 5
стей радикальной полимеризации. Величину 7?р определяю'
не индивидуальные константы скорости, а отношения кр1$г длт
радикальной полимеризации и кр/к0 (или, возможно, кр/кпер\
для катионной.
Для стирола отношение кр1к1ог равно -~10“2 (табл. 3.9), тогде
как кр1к0 равно -~102 (табл. 5.1). Таким образом, скорость катион-
ной полимеризации на 4 порядка выше.
То, что отношение константы скорости роста к сумме констан!
скоростей всех видов обрыва цепи обычно составляет величину
порядка 102, характерно для катионной полимеризации.
Более высокую скорость катионной полимеризации обусловли-
вает также и то, что концентрация растущих частиц в этом случае
значительно больше, чем при радикальной полимеризации. Кон-
центрация HM+(GR)_ близка к концентрации катализатора, кото-
рая обычно составляет величину порядка 10~3 М. При радикаль-
ной полимеризации [М •] находится в области 10-7—10-9 М.
Таблица 5.1
Кинетические параметры полимеризации
стирола, катализируемой H2SO4 {16]
Параметр Значение
[H2SO4], м ~ IO-3
fcp, л/(моль-с) 7,6
^'пер. м» л/(моль’С) 1,2-10-1
fc0 (самопроизвольный обрыв), с-1 4,9-10-2
к0 (комбинация), с-1 6,7-Ю-з
5.2д.З. Величины См и Cs
Константы передачи на мономер приведены в табл. 5.2 и 5.3
для различных случаев полимеризации стирола и изобутилена
[17—21]. Из этих данных видно, что, как правило, значения См
для стирола на один или два порядка больше, чем для изобутилена.
Это находится в соответствии с большей активностью изобутилена
в отношении роста цепи. Хотя и нет достаточно обширных данных
по этому вопросу, но обычно мономеры с большими значениями кр
имеют меньшие значения См или кпер. м/&р. Вопрос об относи-
тельной реакционной способности различных мономеров при
катионной полимеризации будет рассмотрен в гл. 6. Обычно
наблюдается увеличение реакционной способности в ряду вини-
ловые эфиры < изобутилен < стирол, изопрен.
Ионная полимеризация
291
Таблица 5.2
Константы передачи цепи на мономер при катионной
полимеризации стирола
Катализатор Растворитель Темпера- тура, °C см-102 Литера- тура
SnCl4 c6He 30 1,88 17
SnCl4 30% CC14 — 70% CeH5NO2 0 0,'51 18
SnCl4 C2H5Br -63 0,02 19
TiCl4 CeHe 30 2,0 20
TiCl4 30% (CH2C1)2 —70% C6He 30 1,5 20
TiCl4 CH2C12 -60 0,04 12
TiCl4 CH2C12 -90 <0,005 12
FeCl3 C6H6 30 1,2 19
BF3-(C2H5)2O c6H6 30 0,82 19
Таблица 5.3
Константы передачи цепи на мономер при катионной полимеризации
изобутилена в различных растворителях [21]
Каталитическая система Температура, °C CM;104
H-CeHi4 CHCI3 CH2C12
TiCl4-H2O -20 10,3 12,0 21,2
-50 2,6 3,00 6,60
—78 0,80 1,00 1,52
TiCl4-Cl3CCO2H -20 6,6 25,2 26,9
-50 2,00 4,48 5,68
-78 0,72 1,56 2,44
SnCl4-Cl3CCO2H -20 60,0 60,0
-50 17,0 22,8 36,0
-78 6,7 8,1 5,7
Значения Cs для некоторых соединений при полимеризации
стирола [17—19, 22—25] приведены в табл. 5.4.
Большие константы передачи характерны для агентов передачи
цепи, которые имеют слабо связанный отрицательный фрагмент
(например, СН30~ в СН3ОН) или легко алкилируются (напри-
мер, СН3ОС6Н5). Однако значения Cs для агента передачи цепи
сильно изменяются при замене катализатора или растворителя.
292
Глава 5
Таблица 5.4
Константы передачи цепи при катионной полимеризации стирола
Катализатор Растворитель Темпера- тура, °C Агент передачи цепи Cs. 102 Литера- тура
SnCl4 ceHe 30 с6н6 0,22 19
h2so4 . (CH2C1)2 25 изо-с3н7сбн5 4,5 22
SnCl4 CeHj2 0 изо-С3Н7С6Н5 0,60 23
SnCl4 30% CC14- 0 СН3ОС6Н5 162 18
- 70%C6H5NO2
SnCl4 CC14 30 СС14 0,52 24
SnCl4 ceH6 30 СН3ОН 312 17
SnCl4 (CH2C1)2 30 СН3ОН 90 17
SnCl4 ceHe 30 СН3СО2Н 350 17
SnCl4 (CH2C1)2 30 СН3СО2Н 40 17
BF3 ceHe 30 СН3СО2Н 144 17
SnCl4 ceHe 30 (СН3СО)2О 960 17
SnCl4 c6He 30 СН3СО2С2Н5 120 17
H2SO4 (CH2C1)2 25 (С2Н5)2О 19 22
CC13CO2H Нет 25 Бензохинон 24 25
5.2е. Влияние реакционной среды
Уже отмечалось, что активными промежуточными соедине-
ниями при катионной полимеризации обычно являются не сво-
бодные ионы карбония, а сравнительно прочно связанные ионные
пары. Для любой растущей частицы —ВА можно представить
ряд различных состояний от одного крайнего состояния полной
ковалентности (V) до другого — совершенно свободных ионов
(VIII).
--ВА —В+(А)~ —В+ЦА- —В++А- (5.30)
V VI VII VIII
Промежуточные состояния включают прочно связанную, или
контактную, ионную пару VI и разделенную растворителем, или
солъватно разделенную, ионную пару VII. Последняя представляет
собой ионы, которые частично разделены молекулами раствори-
теля. Часто целесообразнее рассматривать ионные полимеризую-
щиеся системы как состоящие из двух типов растущих частиц —
ионной пары и свободного иона VIII, находящихся в равновесии
друг с другом. Ковалентной частицей V обычно можно пренебречь,
так как она нереакционноспособна. Точное строение ионной пары
Ионная полимеризация
293
(т. е. соответствует оно строению VI или VII) зависит от специ-
фических условий реакции.
Характер реакционной среды играет наиболее важную роль
в катионной полимеризации. Так, природа отрицательного проти-
воиона и растворителя может в большой степени изменять ход
полимеризации, меняя относительные концентрации ионной пары
и свободного иона. Поскольку свободный ион растет быстрее, чем
ионная пара, наблюдаются значительные различия в скорости
полимеризации при изменении среды. Замена противоиона и раст-
ворителя влияет также на энергию связи между растущим ионом
и его противоионом (так же как расстояние между ними), а эти
изменения оказывают влияние на скорость роста ионной пары,
так как внедрение мономера между растущим ионом карбония
и противоионом облегчается при уменьшении энергии связи
между этими двумя ионами. Таким образом, изменение раствори-
теля и противоиона влияет на ход полимеризации благодаря
изменениям концентрации свободных ионов и природы ионной
пары.
В идеальном случае следует выражать наблюдаемую скорость
ионной полимеризации в виде суммы скоростей полимеризации
ионной пары и свободного иона — каждой со своей собственной
константой скорости роста. Тогда влияние изменений реакционной
среды может быть выражено как изменения концентрации свобод-
ного иона и константы скоростей роста. Однако такой анализ
реакции полимеризации экспериментально слишком сложно осуще-
ствить. Поэтому в большинстве статей, посвященных изучению
влияния реакционной среды, не приводят количественного отраже-
ния основных причин изменений, наблюдаемых в системе. Влия-
ние реакционной среды обычно описывают как изменение значе-
ний Zcp, определенных экспериментально по уравнениям (5.19),
(5.22) и (5.25). Определенные таким образом значения /ср пред-
ставляют собой сумму значений кр для ионной пары и свободного
иона, взятых пропорционально к относительным количествам
обеих растущих частиц. Такие значения кр лучше называть
эффективными константами или псевдоконстантами скоростей
роста.
5.2е.1. Влияние растворителя
При увеличении сольватирующей способности реакционной
среды обычно наблюдается сильное повышение скорости и степени
полимеризации. Влияние растворителя выражается двумя путями,
о-первых, концентрация свободных ионов растет с увеличением
сольватирующей способности, и поэтому наблюдается увеличение
р, поскольку свободный ион растет быстрее, чем ионная пара,
о-вторых, повышение сольватирующей способности реакционной
среды приводит к возрастанию /сп для ионной пары благодаря
294
Глава И
облегчению условий разделения иона карбония с его противоионом
и тем самым созданию возможности для внедрения мономера
и роста цепи. Как было отмечено выше, влияние растворителя
обыкновенно характеризуют изменением эффективной константы
скорости роста. В табл. 5.5 приведены данные по катализируемой
Таблица 5.5
Влияние растворителя на катионную
полимеризацию стирола, инициированную
НС1О4 [26]
Растворитель Диэлектри- ческая про- ницаемость Е ftp при 25 °C, л/(моль-с)
СС14 2,3 0,0012
40% СС14 —60% (СН2С1)2 5,16 0,40
20% СС14—80% (СН2С1)2 7,0 3,2
(СН2С1)2 9,72 17,0
хлорной кислотой полимеризации стирола, проведенной в раство-
рителях с различной сольватирующей способностью [26]. Кон-
станта скорости роста увеличивается на 5 порядков при переходе
от четыреххлористого углерода, имеющего низкую диэлектриче-
скую проницаемость (е = 2,3), к 1,2-дихлорэтану с высокой
диэлектрической проницаемостью (е = 9,7). (Хотя диэлектриче-
скую проницаемость обычно используют для характеристики
сольватирующей силы растворителя, однако она не обязательно
является количественной мерой. Важное значение имеют также
величины удельной сольватации и поляризуемость.)
Изменение кр происходит параллельно с соответствующими
изменениями энергии активации и частотного фактора реакции.
Необходимость сольватации становится совершенно очевидной
при проведении полимеризации в среде с низкой диэлектрической
проницаемостью. Кроме пониженных скоростей полимеризации,
в плохо сольватирующих средах часто наблюдается более высокий
кинетический порядок по одному из реагентов. Скорость полиме-
ризации может иметь увеличенный порядок зависимости по моно-
меру, катализатору или сокатализатору, соответствующий соль-
ватации ионной пары этими компонентами. Так, скорость полиме-
ризации стирола, катализируемой SnCl4, зависит от [М]2 в растворе
в бензоле и от [М]3 в СС14 [17]. Четыреххлористый углерод —
плохой сольватирующий агент по сравнению с бензолом, и более
высокий порядок по концентрации стирола объясняется тем, что
стирол участвует в сольватации растущего иона и его противоиона.
При больших концентрациях мономерного стирола (а также
Ионная полимеризация
295
в чистом стироле) порядок по стиролу понижается до 2, так как
реакционная среда становится эквивалентной бензольной системе.
При катализе ди- и трихлоруксусной кислотой полимеризации
чистого стирола или стирола в растворах в 1,2-дихлорэтане
п нптроэтане наблюдается сольватация ионной пары катализато-
ром [25]. Кинетический порядок по концентрации хлоруксусной
кислоты возрастает от единицы в сильно полярном нитрометане
до двух в менее полярном дихлорэтане и до трех в чистом стироле.
Следует отметить, что растущие ионные пары, очевидно, одина-
ково сольватируются бензолом или чистым стиролом, когда
катализатором является четыреххлористое олово, но не в случае
катализа хлоруксусной кислотой. Причиной этого являются
различия в полярности и требованиях к сольватации для этих
двух каталитических систем (разд. 5.2е.2).
5.2е.2. Влияние противоиона
Характер отрицательного противоиона также может оказы-
вать влияние на катионную полимеризацию. Чем больше и чем
слабее связан противоион, тем легче происходит рост цепи. Влия-
ние противоиона, так же как и влияние растворителя, может
быть очень широким. Так, эффективная константа роста цепи
для полимеризации стирола при 25 °C в растворе в 1,2-дихлорэтане
возрастает от 0,003 при катализе иодом до 0,42 и 17,0 при катализе
SnCl4-H2O и НС1О4 соответственно [26, 27].
Подробных данных по этому вопросу нет, но можно предполо-
жить, что во многих случаях влияние противоиона обусловлено
изменениями частотного фактора при реакции роста. Значе-
ние Ар для полимеризации, катализируемой хлорной кислотой,
составляет около 107—109, т. е. находится в той же области,
что п при радикальной полимеризации (табл. 3.9). Однако Лр
уменьшается на много порядков (до ~102) для полимеризации,
катализируемой иодом [28]. Это свидетельствует о том, что проти-
воион 1з расположен очень близко от растущего иона карбония
п прочно связан с ним. Способность мономера внедряться в расту-
щую цепь сильно затруднена. О прочности связи 1~ с растущим
центром с очевидностью свидетельствует также отсутствие влия-
ния внешнего электрического поля (постоянного тока) на поли-
меризацию [7]. Приложенное электрическое поле увеличивает
скорость и степень катионной полимеризации в тех случаях, когда
имеется некоторое разделение ионной пары. Под действием элек-
трического поля происходит дальнейшее увеличение расстояния
между ионом карбония и его противоионом. Результатом этого
является увеличение кр, Rp и Хп. (Такого рода влияние электри-
ческого поля не следует путать с использованием электрического
поля для инициирования полимеризации путем электролитиче-
296
Глава 5
ского образования радикалов, анионов или катионов [29],
т. е. с так называемой электролитической, или электроинициируе-
мой, полимеризацией.) Аналогичным образом влияние раствори-
теля на Лр максимально, когда разделение ионной пары начинает
становиться ощутимым.
5.2е.З. Псевдокатионная полимеризация
В последние годы Плеш с сотр. [30, 31] сделал вывод, иго
многие случаи так называемой катионной полимеризации не про-
текают через промежуточные соединения ионной природы —
ионные пары, сольватно разделенные ионы или свободные
ионы. Оказывается, что при определенных условиях реакции
растущие промежуточные частицы являются ковалентными соеди-
нениями. Такие реакции называют псевдокатионной полимери-
зацией.
Механизм полимеризации стирола, катализируемой хлорной
кислотой, был обсужден с позиций как катионной, так и псевдо-
катионной полимеризации [30]. При больших концентрациях
хлорной кислоты имеет место быстрая полимеризация, проте-
кающая через'образование ионов или ионных пар. Псевдокатиои-
ная полимеризация происходит при низких значениях отноше-
ния [НС1О4]/[Стирол]. Скорость псевдокатионной реакции
на несколько порядков меньше скорости катионной. Очевидно,
активным промежуточным соединением при псевдокатионной
полимеризации стирола является ковалентно связанный сложный
эфир хлорной кислоты. Полимеризация протекает путем внедре-
ния мономера по связи С — О
Н
I
--СН2 —С —ОСЮз + СН2 = СНС6Н5 —>
с6н5
н н
I I
-- СН2 —С-СН2 —С —ОС1О3 (5.31)
I I
С6н5 С6Н5
Доводы в защиту псевдокатионного механизма полимеризации
кажутся для многих систем достаточно обоснованными. Например,
отсутствие влияния электрического поля на полимеризацию,
катализируемую иодом, согласуется с предположением о кова-
лентной природе растущего промежуточного соединения. Призна-
ние псевдокатионного механизма не является столь драматиче-
ским отказом от катионного механизма, как кажется с первого
взгляда. Оно представляет собой наиболее подходящее описание
Ионная полимеризация
297
крайнего случая катионной полимеризации при условиях (реак-
ционная среда и катализатор), когда ионную пару правильнее
рассматривать как связанную ковалентной связью.
5.2ж. Энергетика
Катионная полимеризация, так же как и радикальная, являет-
ся экзотермической реакцией, так как она связана с превраще-
нием л-связей в о-связи. Теплота полимеризации для любого
данного мономера по существу постоянна независимо от типа
инициирования (если мономер может полимеризоваться и по
катионному, и по радикальному механизму).
Из уравнений (5.19) и (5.20) можно получить энергии актива-
ции для процесса образования полимера с данной скоростью ER
и степенью полимеризации Ej^ в виде
Er — Ея — Ер — Ео,
Е* =Ер — Е0,
п
(5.32)
(5.33)
где ЕИ, Ер и £0 — энергии активации стадий инициирования,
роста и обрыва цепи соответственно. Ео будет заменяться на Епер,
если обрыв происходит путем передачи цепи. Рост цепи заклю-
чается в присоединении иона к мономеру в мало полярной среде
и не требует большой энергии активации. Значения Ея и Ео
в большинстве случаев бывают больше, чем Ер. Суммарное значе-
ние энергии активации ER находится в пределах — 5 4-
— 10 ккал/моль ( —20,93-103 4- + 41,86-103 Дж/моль). Для
многих полимеризующихся стадий величина ER отрицательна
и наблюдается довольно необычное явление — увеличение скорости
полимеризации с понижением температуры. Отрицательная энер-
гия активации для скорости полимеризации не является, однако,
всеобщим законом. Знак и величина ER изменяются для разных
мономеров. Даже для одного и того же мономера величина ER
может значительно различаться в зависимости от применяемых
катализатора, сокатализатора и растворителя. Данные табл. 5.6
показывают, что для полимеризации стирола ER может меняться
от 8,5 до +14 ккал/моль (—35,58-103 4—1-58,60-103 Дж/моль).
Изменения Еп обусловлены различиями в Ея, Ер и Ео, вызван-
ными различиями в катализаторах и сольватирующей активности
реакционной среды. Следует отметить, что независимо от знака
абсолютные величины ER обыкновенно меньше, чем при радикаль-
ной полимеризации. Скорости катионной полимеризации не
меняются так сильно в зависимости от температуры, как скорости
радикальной полимеризации.
298
Глава 5
Таблица 5.6
Энергии активации катионной полимеризации стирола [25]
Каталитическая система Растворитель Er. ккал/моль (4,186-103 Дж/моль)
TiCl4-H2O а (СН2С1)2 -8,5
TiCl4-CCl3CO2H а С6н5сн3 -1,5
СС13СО2Н С2Н5Вг 3,0
SnCl4-H2O С6н6 5,5
СС13СО2Н (СН2С1)2 8,0
СС13СО2Н ch3no2 14,0
а Данные работы [13].
Энергия активации Е%п образования полимера со степенью
полимеризации Хп всегда отрицательна, так как Ео больше Ev
Температура, °C
-30 -78 -120 -146
во всех случаях независимо от спо-
соба обрыва. Это означает, что сте-
пень полимеризации уменьшается
с повышением температуры поли-
меризации. Величина Е-^п имеет
большие отрицательные значения,
когда обрыв осуществляется в ре-
зультате реакций передачи цепи,
чем когда он происходит путем са-
мопроизвольного обрыва или путем
комбинации, поскольку реакция
передачи имеет более высокие энер-
гии активации. С повышением тем-
пературы полимеризации прекра-
щение роста цепи определяется
Рис. 5.2. Зависимость Хп от темпера-
туры для катализируемой А1С13 поли-
меризации изобутилена [32].
в основном реакциями передачи цепи. Кроме того, с изменением
температуры может происходить изменение характера передачи
цепи. На рис. 5.2 показана логарифмическая зависимость Хп
от ИТ для полимеризации изобутилена в дихлорэтане, катализи-
Ионная полимеризация
299
руемой А1С13 [32]. Приблизительно при —100 °C наблюдается
изменение наклона кривой, при этом значения Е*п меняются
от —5.6 до —0,73 ккал/моль (—23,44 *103 -4 30,76 -103 Дж/моль).
Это объясняют тем, что механизм обрыва цепи изменяется: ниже
__Ю0° С происходит передача цепи на мономер, а выше этой тем-
пературы — передача цепи на растворитель.
J.3. Анионная полимеризация алкенов
Для инициирования анионной полимеризации используют
основные катализаторы [33]. В числе их — ковалентные или
ионные амиды металлов, алкоголяты, алкилы, арилы, гидро-
окиси и цианиды. Реакция инициирования включает присоедине-
ние отрицательного фрагмента В:-, такого, как NH” или R-,
с образованием карбаниона
Н Н
I I
В;- + СН2=С —> В—СН2—С;- (5.34)
Y Y
Анионная полимеризация может быть инициирована за счет
процесса прямого перехода электрона от некоторого донора
к мономеру с образованием анион-радикала
н н
е + СН2 = С .СН2 — С;- (5.35)
4-
Рост цепи происходит присоединением мономера к растущему
аниону.
Анионная полимеризация имеет очень много характерных
черт, общих с катионной полимеризацией, хотя обладает и рядом
существенных отличий. Растущий анион, как и в случае катион-
ной полимеризации, представляет собой не свободный ион, а ион-
ную пару. Анионная полимеризация, как правило, быстро проте-
кает при низких температурах. Однако она не столь чувстви-
тельна к температуре. В большинстве случаев анионная полиме-
ризация имеет положительные значения ER, поэтому легко идет
и при комнатной температуре, а также выше ее.
Обрыв цепи обычно происходит переносом положительной
частицы, например протона, от растворителя или какого-либо
агента переноса, хотя известны и другие способы обрыва. Во мно-
гих случаях анионной полимеризации вообще отсутствуют реакции
обрыва. Полимеризация, протекающая без обрыва цепи, суще-
ственно отличается от катионной и радикальной полимеризаций.
300
Глава 5
Реакционная способность различных катализаторов при ини-
циировании различается в зависимости от их основности. Такие
мономеры, как акрилонитрил и метилметакрилат, которые имеют
сильные электроноакцепторные заместители, могут полимеризо-
ваться со слабо основными катализаторами типа гидроксильных
ионов и цианидов. Однако для полимеризации таких мономеров,
как стирол или бутадиен-1,3, имеющих относительно слабые
электроноакцепторные заместители, требуются сильные основа-
ния, вроде амидных ионов или алкильных анионов. Рассмотрим
некоторые наиболее хорошо изученные инициирующие системы
для анионной полимеризации.
5.3а. Инициирование амидами металлов
Первым детальным исследованием анионной полимеризации
было изучение полимеризации стирола амидом калия в жидком
аммиаке [34]. Эта полимеризация — один из небольшого числа
примеров ионной полимеризации, протекающей со свободными
ионами, а не с ионными парами. Причиной этого является высокая
диэлектрическая проницаемость и большая сольватирующая
способность жидкого аммиака. Положение будет иным в раствори-
телях с меньшими диэлектрической проницаемостью и сольвати-
рующей способностью, например в эфирах и углеводородах.
Полимеризацию проводили при температуре кипения аммиака
(—33 °C). Было установлено, что скорость полимеризации растет
с увеличением концентрации амида и квадрата концентрации
мономера и уменьшается при добавлении иона калия. Анализ
показал, что полимер содержал один атом азота на молекулу
и не имел ненасыщенных связей. Кроме того, молекулярный вес
полимера не зависел от концентрации ионов калия и амида. Меха-
низм полимеризации в этой системе прямо следует из этих и других
экспериментальных данных. Инициирование происходит путем
диссоциации амида калия и присоединения образующегося амид-
ного иона к мономеру
к
KNH2 K+ + H2N:~
Н
I
H2N;--i-C6H5CH=CH2---> H,N —СН2—С;-
Скорость инициирования выражается уравнениями
7?n = MH2N;-] [М]
или
kaK [М] [KNH2]
[К+]
(5.36)
(5.37)
(5.38а)
(5.386)
Ионная полимеризация
301
Рост цепи
П
H2N-M;+M —H2N-M„M-
(5.39)
происходит со скоростью
Вр = kv [М-] [М], (5.40)
где [М_] — общая концентрация растущих анионных центров.
Обрыв цепи в этой системе не происходит путем перехода
гпдридного иона (Н“) к мономеру, о чем свидетельствует отсут-
ствие ненасыщенности в полимере. Не реализуется также мало-
вероятная возможность комбинирования растущего иона с ионом
калия, т. е. амид калия не расходуется в ходе полимеризации.
Обрыв цепи происходит в результате передачи цепи на раствори-
тель
Н
I kjiep. S
’"H2N-(-CH2GHG6H5-)n-GH2-G:-+NH3------------>
С6Н5
H2N-(-CH2CHC6H5-)n-CH2CH2C6H5 + H2N:- (5.41а)
или
H2N —MnM--|-NH3 —>5H2N-MnMH+H2N:- (5.416)
с регенерацией инициирующего амидного иона. Скорость обрыва
дается уравнением
tfnep = *nep.S[M-][NH3]. (5.42)
Уравнение скорости полимеризации можно вывести обычным
путем из уравнений (5.38), (5.40) и (5.42) при допущении постоян-
ства [М_]
OHfcp[M]2[KNH2]
Яр~ fcnep. S [К+] [NH3]
или
_ Mp[M]2[H2N:-n
Р fcnep. S [NH3]
(5.436)
Этот механизм полимеризации был подтвержден также для дру-
гих мономеров, таких, как метакрилонитрил и этилакрилат [35],
а также для стирола. Для обычного эксперимента, когда кон-
центрации амидных ионов и ионов калия равны (т. е. когда не
производится специального добавления К+), скорости иницииро-
вания и полимеризации даются несколько иными выражениями
Ra = каК^ [М] [KNH2]1/2,
R кИК^кр [М]2 [KNH2]1''2
Р fcnen. S [NH3]
(5.44)
(5.45)
302
Глава 5
Среднечисловая степень полимеризации в этой системе, полу-
ченная делением уравнения (5.40) на (5.42), выражается как
X = М [MJ (5.46Ъ
П ^пер. S [NH3] Cs[NH3] ’ Р
где Cs = Апер, s^p — константа передачи цепи на аммиак.
Рассмотрим теперь влияние температуры на скорость и степень
полимеризации. Как видно из уравнения (5.46), энергия актива-
ции Е-^п образования полимера с некоторой степенью полимери-
зации Хп равна (Ер — Епер). Было найдено [33], что Е^. для
полимеризации стирола, катализированной амидом калия, имеет
величину —4 ккал/моль (16,74-103 Дж/моль). Как и в катионной
полимеризации, энергия активации реакции обрыва больше
энергии активации реакции роста, и при повышении темпе-
ратуры наблюдается понижение степени полимеризации. Энер-
гия активации для общей скорости полимеризации ER дается
выражением (Е и + Ер — £пер) и равна приблизительно
9 ккал/моль (37,67-103 Дж/моль), так как £'„ = 13 ккал/моль
(54,42-103 Дж/моль). Скорость полимеризации в этой системе
уменьшается с понижением температуры. Теплота диссоциации
KNH2 Д77и также определяет значение £я, поскольку константа
ионизации амида калия входит в уравнение скорости полимериза-
ции [уравнения (5.43а) или (5.45)]. Однако в этой системе значе-
ние Д/7„ очень близко к нулю, и им можно пренебречь.
5.36. Полимеризация, протекающая без обрыва цепи
Часто анионная полимеризация протекает в условиях, когда
отсутствуют реакции обрыва и рост происходит до полного исчер-
пания мономера. Анионные центры остаются неизменными, потому
что перенос протона (или другой положительной частицы)
от растворителя не имеет места. Такие полимерные анионы назы-
ваются живущими полимерами. Живущие полимеры образуются
при использовании растворителей (например, тетрагидрофуран,
1,2-диметоксиэтан, диоксан), неспособных обрывать растущий
анион в результате передачи цепи.
Отсутствие обрыва цепи в таких реакциях легко наблюдать.
Многие растущие карбанионы окрашены. Если система хорошо
очищена, т. е. отсутствуют любые примеси, то цвет карбаниона
сохраняется в течение всей полимеризации и не исчезает и не изме-
няется при 100%-ной степени превращения. Последующее доба-
вление к живущей полимерной системе новой порции мономера
вызывает дополнительную полимеризацию. Добавленный мономер
полимеризуется также количественно. Молекулярный вес живу-
Ионная полимеризация
303
щего полимера увеличивается соответственно, так как число
полимерных молекул не изменяется. На рис. 5.3 такой процесс
показан на примере полимеризации метилметакрилата, иницииро-
Р и с. 5.3. Зависимость молекулярного
веса полимера от степени превращения
при анионной полимеризации метилмет-
акрилата [36].
I — полимеризация, инициированная
Zn(C2H6)2; 2 — полимеризация живущих анио-
нов при добавлении второй порции мономера.
ванной комплексом бутиллития и диэтилцинка [36]. Инициирую-
щей частицей в этом случае является C4H~[LiZn(C2H5)2l+.
5.36.1. Инициирование путем переноса электрона
Шварц с сотр. [37] исследовал интересную реакцию полимери-
зации, инициируемую ароматическими анион-радикалами, такими,
как Na-нафталин и Na-бифенил. Эти инициаторы являются
и радикалами, и анионами. Полимеризация стирола Na-нафтали-
ном в тетрагидрофуране при температурах от —78 до 0 °C была
первой детально изученной живущей полимерной системой [38, 39].
Инициирование в этой реакции происходит путем первоначаль-
ного образования активного катализатора — нафталинового ион-
радикала
еакция заключается в переходе электрона от натрия к нафта-
лину. атем нафталиновый анион-радикал (окрашенный в зеленый
цвет) переносит электрон к мономерному стиролу с образованием
304
Глава 5
стирильного анион-радикала
Na + СН=СН9
I 2
с6н5
СН—сн2 СН-СН2] Na
С6Н5 С6Н5
(5.48)
Стирильный анион-радикал показан как резонансный гибрид форм,
в которых анионные и радикальные центры попеременно находятся
у а- или ^-углеродных атомов.
Для нескольких полимеризационных систем мономерные
анион-радикалы могут расти и по радикальному, и по анионному
механизму, т. е. с обоих активных концов. Однако в обычных
случаях такие анион-радикалы подвергаются димеризации с обра-
зованием дианионов
2[С6Н5СН —СН2
IX
С6Н5СН —CH2]-Na+
X
- С6н5
Na+ -:С—СН2 — СН2 — с;-
- н н
С6Н5-|
Na+
(5.49)
Эта реакция доказана изучением спектров электронного пара-
магнитного резонанса, которое показало полное отсутствие
в системе радикалов. Димеризация происходит через резонансную
форму X, а не IX, с образованием более стабильного стирильного
карбаниона. Образовавшиеся таким образом стирильные дианионы
окрашены в красный цвет и участвуют в реакции роста по анион-
ному механизму с обоих концов
Na+
CeHs СбЩ-
:i!-ch2-ch2-c:-
А Ь -
Na++(n+7n)C6H6CH=CH2 —>
—> Na+
C6H6
. I
:с-сн2-(-снс6н5-сн2-)
СбНб-.
(-СНг-СНСбНб-О^-СНг-с:-
н -
(5.50)
Na+
Кроме инициирования анион-радикалами, инициирование
путем переноса электрона наблюдалось при применении опре-
деленных щелочных металлов в жидком аммиаке. В этих условиях
процесс инициирования может протекать по двум различным
механизмам. В некоторых системах, например при полимеризации
Ионная полимеризация
305
иоола и метакрилонитрила калием, инициирование осущест-
вляется образующимся в системе амидным ионом [34, 35]. Эти
оеакции аналогичны реакциям, инициируемым амидами щелочных
металлов. В других системах полимеризация не инициируется
амидными ионами. Так, полимеризация метакрилонитрила
в жидком аммиаке, инициированная литием, протекает со значи-
тельно большей скоростью, чем полимеризация, инициирован-
ная амидом лития [40]. Считают, что механизм инициирования
включает образование сольватированного электрона
Li + NH3 Li+(NH3) + ₽(NHj)
(5.51)
Такие аммиачные растворы имеют характерный глубокий
синий цвет. Затем сольватированный электрон переносится
к мономеру с образованием анион-радикала
?(NH3) + CH2=CHY (СН —CHY CH-CHY)-(NH3) (5.52)
Апион-радикал продолжает расти таким же образом, как
описано выше для процесса инициирования Na-нафталином.
Возможно, что при полимеризации щелочными металлами в виде
дисперсий в углеводородах инициирование происходит также
в результате переноса электрона.
.5.36.2. Инициирование алкилами металлов
Разнообразные металлоорганические соединения (алкилы
металлов) могут инициировать анионную полимеризацию. Наи-
более широко применяют такие алкилы металлов, как бутилли-
тий [41] и трифенилметилнатрий. Бутиллитий в некоторой степени
используют и в промышленном производстве полибутадиена
и полиизопрена (гл. 8). Полимеризация, инициируемая алкилами
металлов (например, бутиллптием), протекает через присоедине-
ние катализатора к мономеру
Y
C4H9LiJ-CH2 = CHY С4Н9-СН2 —C;-(Li+) (5.53)
I
Н
с последующим ростом цепи
Y
^H9-CH2-C:-(Li+) + reCH2 = CHY —>
н
Y
С4Н9- (— СН2СН Y — )п - СН2 — C;-(Li+) (5.54)
н
306
Глава 5
5.36.3. Скорость полимеризации
Скорость полимеризации в системах, где нет обрыва цепи,
обычно выражают просто как скорость роста
7?р = *р [М-] [М], (5.55>
где [М“] — общая концентрация растущих живущих анионных
концов. Концентрацию живущих концов можно определить,
спектрофотометрически или при взаимодействии с соответствую-
щим агентом передачи цепи. Если в системе полностью отсут-
ствуют агенты передачи цепи, [М~] задается концентрацией ката-
лизатора, т. е. концентрацией или Na-нафталина, или бутиллития,
или, наконец, в некоторых случаях — лития. Живущие концы
являются моноанионами при полимеризации в присутствии бутил-
лития и дианионами при полимеризации, инициируемой перено-
сом электрона. Анионы этих двух типов называют анионами
с одним или соответственно с двумя живущими концами.
Во многих случаях скорость анионной полимеризации слишком
велика и не поддается экспериментальному определению такими
методами, как дилатометрия. Например, полимеризация стирола
Na-нафталином в тетрагидрофуране полностью завершается
в течение нескольких секунд. Для изучения таких реакций
быстрой полимеризации были разработаны струевые методы —
метод капиллярной струи и метод остановленной струи [42].
По первому методу растворы мономера и частиц с живущими
концами находятся в двух отдельных сосудах. Оба раствора
передавливают в смесительную камеру и затем в капиллярную
трубку, где и протекает полимеризация. Реакцию обрывают
на выходе из капилляра, выливая реакционную смесь в раствори-
тель, содержащий агент обрыва с высокой константой передачи
цепи (например, воду). Продолжительность реакции определяется
отношением объема капилляра к скорости течения. Таким образом
удается детально изучать очень короткие реакции (от ~0,05 до 2 с).
Степень превращения и, следовательно, 7?р и А'р, получают, опре-
деляя содержание полимера или остаточного мономера в резко
охлажденной реакционной смеси.
Интересно понять причины более быстрого протекания реак-
ции во многих случаях анионной полимеризации по сравнению
с их радикальными аналогами. Это можно сделать путем сравне-
ния кинетических параметров в соответствующих уравне-
ниях (3.21) и (5.55). Значение кр в анионной полимеризации стирола
очень близко к значению А'р в радикальной полимеризации,
хотя оно может быть на порядок выше при проведении полимери-
зации в сильно полярном растворителе [37]. Большое различие
в скоростях анионной и радикальной полимеризаций большей
частью обусловлено различиями концентраций растущих анионов
Ионная полимеризация
307
и радикалов. Концентрация растущих радикалов при типичной
радикальной полимеризации составляет 10-8—10-7 М, тогда как
концентрация растущих анионов равна 10-3—10-2 М. Таким
образом, скорость анионной полимеризации в 104—10’ раз больше
скорости радикальной полимеризации.
5.36.4. Влияние реакционной среды
Константа скорости роста и скорость анионной полимеризации
сильно зависят от природы как растворителя, так и противоиона.
Так, данные табл. 5.7 демонстрируют ярко выраженное влияние
Таблица 5.7
Влияние растворителя на анионную полимеризацию
стирола [37]
Растворитель Диэлектри- ческая про- ницаемость е ftp, л/(моль-с)
Бензол 2,2 2
Дпоксап 2,2 5
Тетрагидрофуран 7,6 550
1,2-Диметоксиэтан 5,5 3800
растворителей на полимеризацию стирола, катализируемую
Na-нафталином (3-10-3 М) при 25 °C. Константа скорости роста
возрастает на два порядка в тетрагидрофуране и на три порядка
в 1,2-диметоксиэтане по сравнению с константами скорости в бен-
золе или диоксане. Полимеризация протекает значительно быстрее
в более полярных растворителях. То, что диэлектрическая прони-
цаемость не является количественной мерой сольватирующей
способности растворителя, доказывается большей скоростью
реакции в 1,2-диметоксиэтане по сравнению с реакцией в тетра-
гидрофуране (ТГФ). Большая скорость в первом случае должна
быть связана со специфическим сольватирующим эффектом эфир-
ной группы.
В нескольких случаях наличие сольватации при анионной
полимеризации проявляется также в отклонениях от нормальных
выра,кений скорости. Так, полимеризация метилметакрилата
утиллитием в толуоле при —60 °C имеет второй порядок 7?р
от концентрации мономера [36]. В неполярном толуоле мономер
участвует в сольватации растущих ионов и ионных пар. При
проведении полимеризации в смеси растворителей диоксан—толуол
308
Глава 5
(более полярной, чем толуол) наблюдается обычный для завися
мостп 7?р от [М] первый порядок.
Значения /ср в табл. 5.7 были вычислены из уравнения (5.55)
Как было указано ранее (разд. 5.2е), полученную таким образоь
для ионной полимеризации кр необходимо считать смешанно!
или псевдоконстантой скорости роста. Шварц с сотр. [43, 44
выражает скорость полимеризации для живущих полимерных
систем как сумму скоростей полимеризации для ионных naj
Р“(С+) и свободных растущих ионов Р_. Так, можно написать
Яр = &р-(с+) IP’ (С+)] [М] + 7ср_ [Р-] [М], (5.56)
где fcp-(c+) и /ср- — константы скорости роста ионной пары и сво-
бодного иона соответственно; [Р"(С+)] и [Р-] — концентрации
двух растущих частиц; [М] — концентрация мономера, С+ — поло-
жительный противоион.
Обе растущие частицы находятся в равновесии
Р-(С+) Р-+СЛ (5.57)
управляемом константой диссоциации К, определяемой как
1Z [Р ][С+] /Г rm
Л ““ [Р~(С+)[ (5.5о)
или
'С = -|Ё^)Г <5-59>
если [Р“] = [С+].
Сочетание уравнений (5.56) и (5.59) дает
» К1/2*Р.
[M][P-(C+)J =/ср-(С+>-Ь [Р-(с+)]1/2 • (о'60)
Правая часть уравнения (5.60) будет равна /ср в уравнении (5.55)
при допущении, что концентрация свободных анионов мала.
Так и бывает обычно, и [Р_(С+)] может очень близко выражаться
общей концентрацией живущих ионов (т. е. концентрацией ката-
лизатора).
Сравнение уравнений (5.55) и (5.60) показывает, что зависи-
мость экспериментально определенных значений кр от [Р“(С+)]-1/2
должна быть линейной. На рис. 5.4 приведена эта зависимость для
полимеризации стирола в тетрагидрофуране при 25 °C с различ-
ными противоионами. Величины К1>2кР- и &р-(с+) получают
из графиков этих зависимостей как наклон кривой и как отре-
зок, отсекаемый ею на оси ординат, соответственно. Константу дис-
социации К можно рассчитать из данных по проводимости и ско-
рости полимеризации. Из полученных величин и значений К1,г кР-
легко найти константу скорости роста для свободного аниона
Ионная полимеризация
309
, ,о 44] В табл. 5.8 приведены результаты такого анализа данных
дЛЯ’ полимеризации стирола в тетрагидрофуране. Указаны также
значения *р-(с+) Для полимеризации стирола в диоксане.
Таблица 5.8
Влияние противоиона на анионную полимеризацию стирола [43, 44|
Противо- ион Полимеризация в тетрагидрофуране Полимеризация в диоксане
АР- (С+) Ар- К- 107 ftp- hp- (С+)
Li' 160 30,3 2,2 0,94
Na- 80 25,2 1,5 3,4
К- 60-80 18,0 0,8 6,5-104 19,8
Rb+ 50-80 6,7 0,1 21,5
Са+ 22 3,0 0,02 24,5
Результаты полимеризации в тетрагидрофуране свидетель-
ствуют о значительно более высокой реакционной способности
свободного аниона по сравнению с любой ионной парой. Вели-
чина кР- составляет 6,5 л/(моль-с) и в 102— 103 раз больше,
Р п с. 5.4. Зависимость эксперименталь-
но наблюдаемой константы скорости ро-
ста цепи полистирола от концентрации
живущих концов для различных про-
тивоионов [43].
чем /ср-(С+). Константы диссоциации ионных пар увеличиваются
при переходе от цезия к литию (в качестве противоионов). Этот
порядок соответствует порядку возрастания сольватации проти-
воиона. Малый по размерам Li+ сольватируется в наибольшей
степени. Как показывают значения /ср-(С+), реакционная способ-
ность различных ионных пар возрастает в том же порядке. Дан-
ные табл. 5.8 также показывают, что реакционная способность
310
Глава 5
ионных пар в диоксане обычно меньше, чем в тетрагидрофуране,
что объясняется меньшей сольватирующей способностью диоксана.
Однако интересно отметить, что порядок реакционной способности
для различных ионных пар в диоксане является обратным этому
порядку в тетрагидрофуране. В плохом растворителе — дпокса-
не— сольватация противоиона ничтожна. Наиболее реакцион-
носпособна ионная пара с наименьшей кулоновской энергией свя-
зи между анионом и противоионом. Энергия связи уменьшается,
а реакционная способность возрастает с увеличением размера
противоиона. Внедрение мономера между анионом и его проти-
воионом облегчается при уменьшении энергии связи между
этими ионами.
Влияние растворителя на /сР-(с+) в количественной форме
выразили Байуотер и Уорсфолд [45]. Была установлена корреля-
Р и с. 5.5. Зависимость константы ско-
рости роста для ионной пары полпсти-
рил,1л+ от диэлектрической проницае-
мости реакционной среды [45].
ция значений /ср-(с+) для полистириллития в различных смесях
тетрагидрофуран— бензол от диэлектрической проницаемости
среды (на рис. 5.5). Такой вид зависимости, т. е. зависи-
мость логарифма константы скорости от (s — 1),'(2е -j- 1),
является стандартным методом выражения влияния раствори-
теля на реакцию, в которой участвуют полярные частицы [46].
Следует отметить, что отношение (в — 1)/(2е -ф- 1) увеличивается
с увеличением 8. Положительный наклон графика зависимости
согласуется с результатами, ожидаемыми для реакций, которые
протекают с переходными состояниями, имеющими более высокую
полярность, чем реагенты [46]. Переходное состояние при росте
цепи заключается в разделении растущего аниона и противоиона,
когда молекула мономера внедрится между ними. Это разделение
облегчается в возрастающей степени при увеличении диэлектри-
ческой проницаемости реакционной среды.
Ионная полимеризация
311
,5.36.5. Степень полимеризации
Средиечпсловая степень полимеризации для живущих поли-
меров выражается просто отношением концентраций мономера
и живущих концов
- [М]
Лп— [М-]
в обычном случае, когда весь катализатор превращается в расту-
щие живущие анионные концы, уравнение (5.61) превращается в
(5-62а)
(5.61)
или
(5.626)
[С]
в зависимости от способа инициирования. Уравнение (5.62а)
применимо для полимеризации, инициируемой переходом электро-
на, так как каждая полимерная молекула получается из двух
молекул катализатора (через дианионные растущие частицы). При
других процессах инициирования (например, бутиллитием), когда
на образование одной полимерной молекулы расходуется одна
молекула катализатора, применимо уравнение (5.626).
Следствием отсутствия реакции обрыва цепи при полимериза-
ции является значительная монодисперсность (т. е. Мп та Mw)
образующегося при определенных условиях полимера. Если
инициирование происходит достаточно быстро, так что все актив-
ные центры начинают расти почти одновременно, то полимерные
молекулы будут расти в течение одного и того же времени. Рас-
величине будет определяться распределением
пределение по
Пуассона [47]
которое может
(5.63а)
(5.636)
X W 1 j__Хп
Хп ~ (Хп + 1)2 ’
быть упрощено
Хп хп
Уравнение (5.63) показывает, что для всех полимеров, кроме
очень низкомолекулярных, распределение по величине цепи очень
узкое с Xw/Xr, близким 1. Это было установлено для многих
систем. Так, например, при полимеризации стирола Na-нафтали-
ном отношение М W/Mn полученного полимера оказалось в узкой
о ласти 1,06—1,12. Теоретические расчеты показывают, что
распределение по молекулярным весам должно быть более узким.
312
Глава 5
чем у полимеров, полученных радикальной полимеризацией, даже
если скорость инициирования на несколько порядков ниже ско-
рости роста [48]. Метод полимеризации, протекающей с образова-
нием живущих полимеров, представляет собой уникальный способ
синтеза стандартных полимерных образцов известного и точно
определенного молекулярного веса. Интересно отметить, что
в работах последнего времени [49, 50] описаны некоторые реакции
инициирования катион-радикалами, происходящие аналогично
инициированию анион-радикалами. Это обещает расширение воз-
можностей получения стандартных образцов полимеров с узким
молекулярновесовым распределением.
Хотя при полимеризации, протекающей с образованием живу-
щих полимеров, не должен происходить обрыв цепи, присутствие
активных загрязнений, примесей может изменить это положение.
Обычно принимают специальные меры для очистки системы
от кислорода, двуокиси углерода и воды. Кислород и двуокись
углерода присоединяются к растущим анионам, образуя перокси-
и карбокси-анионы. Эти анионы обычно недостаточно активны
для продолжения роста, и кинетическая цепь обрывается. Вода
и другие вещества с кислыми атомами водорода (например, карбо-
новая кислота) обрывают цепь переносом протона. Константа
переноса для воды при полимеризации стирола Na-нафталином
при 25 °C равна приблизительно 10 [51], поэтому присутствие
воды в заметных количествах может ограничивать молекулярный
вес полимера. Однако константа переноса для этанола равна
только 10"3. Присутствие этанола не мешает образованию высоко-
молекулярного полимера, так как реакция обрыва протекает
очень медленно. На практике живущие концы полимеров при
желании обрывают (обычно при 100%-иой степени превращения)
добавлением таких агентов переноса, как вода.
5.36.6. Ассоциация
При реакциях полимеризации, инициируемых литийорганиче-
скими соединениями в неполярных растворителях (бензол, гексан
и толуол), возможна ассоциация различных литпйорганических
частиц, присутствующих в системе. Этот процесс осложняет
течение реакции, так как только неассоциированные частицы
активны в полимеризации. Влияние ассоциации очень важно;
оно было широко изучено [41, 52, 53] на примере полимеризации
стирола, инициируемой бутиллитием. Ассоциация оказывает
влияние как на скорость инициирования, так и на скорость роста
цепи. При низких общих концентрациях бутиллития (ниже
~10“4 М) ассоциация мала или вообще отсутствует, и скорости
инициирования и роста имеют первый порядок по бутиллитию.
Однако при больших концентрациях бутиллития происходит
Ионная полимеризация
313
ассоциация. Было найдено, что при этих условиях скорости
инициирования и роста пропорциональны концентрации бутилли-
тпя в степени х/6 и х/2 соответственно. Если концентрация бутилли-
тпя выше 0,02 М, то Ra и /?рне зависят от общей концентрации
бутиллития.
Эти результаты показывают, что для бутиллития и растущей
ионной пары, находящихся в равновесии с соответствующими
гексамером и димером, имеют место следующие равновесия:
К1
(С4Н9Ы), --» 6С4Н9Ы, (5.64)
Кг
(С4НЯ—M~Li+)->. ~-> 2C4H9-M"Li+ (5.65)
Концентрации недиссоциированного бутиллития и недиссо-
цпрованной ионной пары определяются уравнениями
[C4H9Li] = »[(C4H9Li)e]Ve, (5.66)
[С4Н9- M-L1+] = ^/2[С4Н9- M-Li+]4 (5.67)
которые объясняют зависимость Ra и /?р от концентрации С4Н9Ы
в степени 1/6 и 1/2 соответственно.
Ассоциация катализатора и растущих центров в неполярных
средах обусловливает очень низкие скорости полимеризации.
Типичная полимеризация стирола бутиллитием в бензоле проте-
кает на несколько порядков медленнее, чем соответствующая
полимеризация Na-нафталином. При проведении полимеризации
с C4H9Li в таких полярных растворителях, как тетрагидрофуран,
ассоциация полностью исчезает и полимеризация протекает значи-
тельно быстрее. Ассоциацию можно также предотвратить доба-
влением основания Льюиса, которое координирует с катализато-
ром [52—54]. Возможно, что диэтилцинк в описанной выше ката-
литической системе бутиллитий — диэтилцинк (разд. 5.36) при-
меняется с этой целью. Повышение температуры также уменьшает
степень ассоциации.
5.36.7. Энергетика
Судя по данным для температурной зависимости скорости
полимеризации, протекающей без обрыва цепи, энергия актива-
ции ER этого процесса сравнительно невысока и положительна.
Для живущих полимеров ER равна просто энергии активации
роста. Скорость полимеризации относительно мало чувствительна
к температуре, по с повышением температуры увеличивается,
роме того, энергия активации заметно изменяется в зависимости
от применяемого при полимеризации растворителя, как это было
в случае катионной полимеризации. Так, например, энергия
314
Глава 5
активации роста в системе стирол — Na-нафталин равна
9 + 3 ккал/моль (37,67-103 + 12,56-103 Дж/моль) при проведении
реакции в диоксане и только около 1 ккал/моль (4,18 -103Дж/моль)
при реакции в тетрагидрофуране [42, 55]. Если в системе отсут-
ствуют агенты передачи цепи, то молекулярный вес полимера,
полученного полимеризацией без обрыва, не зависит от темпера-
туры. Однако в присутствии агентов передачи цепи положение
может измениться.
Большинство имеющихся в литературе данных по энергии
активации представляют собой значения эффективной константы
скорости роста. Пользуясь описанным в разд. 5.36.4 способом
кинетического анализа, Шварц с сотрудниками сумел определить
отдельно значения энергии активации для стадии роста через
свободные анионы и для стадии роста через ионные пары. Особенно
интересны недавние исследования [56] живущей полимерной
системы: полистирол в тетрагидрофуране и 1,2-диметоксиэтане
с натриевыми и цезиевыми противоионами. Энергия активации
роста через свободные ионы равна 5—6 ккал/моль (20,93 ПО3—
—25,11 -103 Дж/моль), причем значение ее не зависит ни от проти-
воиона, ни от растворителя. Частотный фактор роста свободного
иона также одинаков в обоих растворителях. Истинное значение Ар
равно 108—109 л/(моль-с), т. е. немного больше, чем обычное
значение его для процесса радикальной полимеризации. Энергия
активации стадии роста Ер через ионную пару полистирилцезий
примерно такая же, как и в случае роста через свободный ион,
и тоже не зависит от растворителя. Частотный фактор Ар для
этой ионной пары равен приблизительно 105 л/(моль -с), т. е. на
несколько порядков меньше, чем для свободного иона, что опре-
деляет значительно более высокие значения константы скорости
роста свободного иона.
Неожиданными оказались результаты этого исследования в от-
ношении энергии активации стадии роста Ер через ионную пару
полистирилнатрий. Энергия активации полимеризации в тетра-
гидрофуране мала и отрицательна [—1,5 ккал/моль
(—6,28-IO3 Дж/моль)] в интервале температур от —80 до +25 °C.
Однако при полимеризации в 1,2-диметоксиэтане энергия актива-
ции роста ионной пары проходит через максимум при ~0 °C,
причем имеет положительные значения ниже и отрицательные
значения выше этой температуры. Такое поведение в 1,2-диметок-
сиэтане объясняют присутствием заметных количеств как прочно
связанных (контактных) ионных пар, так и разделенных раство-
рителем (сольватно разделенных) ионных пар. [Эти два типа
ионных пар соответствуют структурам VI и VII в (5.30).] Иное
поведение в тетрагидрофуране свидетельствует об отсутствии
заметного количества разделенных растворителем ионных пар
в этом менее полярном растворителе.
Ионная полимеризация
315
Константа скорости роста ионной пары /сР-(С+), определенная
в 1,2-диметоксиэтане, является суммарной величиной констант
скорости роста контактной (/сКОНт) и сольватно разделенной (&Сольв)
ионных пар согласно уравнениям
&Р-(С+) = Х^СОЛЬВ -К (1 — X) ^-конт (5.68)
или
7. ^контТ ^СОЛЬВ-^И сп\
*Р-(С+) =----, (о.ЬУ)
где х и (1 — X) — фракции сольватно разделенных и контактных
ионных пар соответственно, а Ка — константа, характеризующая
равновесие между обоими типами ионных пар:
Кп
— Р’(С+) ~ — Р~ II с+. (5.70)
Наблюдаемая энергия активации ЕИ выражается уравнением
Е» = ЕС0ЛЪВ + ~^, (5.71)
где £С0Льв — энергия активации роста разделенной растворите-
лем ионной пары, а ДЯИ — теплота реакции (5.70). Применяя
этот способ анализа экспериментальных данных, Шварц с сотруд-
никами смог установить, что величина А'СОЛь» равна примерно
половине величины константы скорости роста свободного аниона.
Было также установлено, что число разделенных растворителем
ионных пар равно примерно 2-1СГ3—3-10~3.
Таким образом, показано, что эффективная константа скорости
роста в принципе может быть (хотя и с экспериментальными труд-
ностями) представлена как сумма констант скорости роста сво-
бодных ионов, сольватно разделенных ионных пар и контактных
ионных пар.
о.4. У становле ние механизма
полимеризации
Иногда бывает не ясно, по какому механизму (радикальному,
катионному или анионному) инициирует полимеризацию данная
каталитическая система. Такой вопрос может, например, воз-
никнуть при полимеризации, инициируемой радиоактивным
излучением. Механизм инициирования определенным катализато-
ром можно установить, сравнивая характерные особенности
данной полимеризации с особенностями полимеризации, проте-
кающей под влиянием известных радикальных, катионных или
анионных инициаторов:
316
Глава 5
1. Ионная полимеризация, как правило, протекает при более
низкой температуре, чем радикальная полимеризация. Несмотря
на то что температура ионной полимеризации обычно ниже О °C,
есть много примеров ионной полимеризации, протекающей при
температуре, несколько превышающей 0° С. Но радикальная
полимеризация почти всегда протекает при температурах, значи-
тельно превышающих 50 °C. Кроме того, энергия активации
ионной полимеризации всегда меньше энергии активации соот-
ветствующей радикальной полимеризации и во многих случаях,
действительно, может иметь отрицательную величину.
2. Ионная полимеризация отличается заметной чувствитель-
ностью к изменениям полярности и сольватирующей способности
реакционной среды. При радикальной полимеризации такого
влияния нет.
3. Добавление в полимеризующуюся систему таких извест-
ных акцепторов радикалов, как радикалы дифенилфосфата,
обрывает полимеризацию, если она протекает по радикальному
механизму. На ионную полимеризацию такое добавление не влияет.
Однако надо к выбору акцептора радикалов подходить осторожно,
чтобы не использовать соединение, которое влияет и на ионную
полимеризацию. Например, выбор гидрохинона в качестве акцеп-
тора радикалов был бы неудачным, так как он может ингибиро-
вать также и катионную полимеризацию.
4. Можно определить константу передачи цепи на раствори-
тель или специально добавленное соединение. Значение этой
константы можно затем сравнить с константой передачи на то же
вещество при полимеризации того же мономера под влиянием
известных инициаторов радикальной, катионной или анионной
полимеризации.
5. Для установления различий между радикальной и ионной
полимеризацией можно использовать также закономерности
сополимеризации (гл. 6).
5.5. Полимеризация по карбонильной
группе
Полимеризация по карбонильной группе приводит к образо-
ванию полимеров, содержащих ацетальные повторяющиеся струк-
турные единицы:
R R R R
I III
С = О—>—С —О —С —О —С—О— (5.72)
I III
R' R' R' R'
Кроме углерод-углеродной двойной связи, связь С = О является
единственной ненасыщенной связью, полимеризацию по которой
Ионная полимеризация
317
удалось успешно осуществить [57]. Из соединений такого типа
значительно более широко изучена полимеризация формальдегида.
Несмотря на то что формальдегид был успешно заполимеризован
более 100 лет назад, только сравнительно недавно удалось получить
высокомолекулярные полимеры из других карбонильных соеди-
нений [58].
Этот прогресс в области полимеризации по карбонильной связи
является результатом обнаружения и использования влияния
предельной температуры (Т’пр) на процесс полимеризации
(разд. 3.9в). Карбонильные мономеры (за исключением формальде-
гида) имеют низкие предельные температуры. В табл. 5.9 указаны
Таблица 5.9
Предельные температуры и концентрации мономеров при
полимеризации альдегидов [58]
Мономер ТПР. °C Концентрация мономера
Формальдегид (газообразный) Формальдегид (в растворе СН2С12) Ацетальдегид (жидкий) Пропионовый альдегид (жид- кий) Трихлоруксусный альдегид (в растворе пиридина) 126 30 -31 -31 12,5 1 атм 0,06 М Чистый мономер Чистый мономер 0,1 М
предельные температуры и равновесные концентрации мономера
для полимеризации некоторых альдегидов в газообразном и жид-
ком состоянии, а также в растворе [58]. Предельные температуры
почти всех карбонильных мономеров соответствуют комнатной
темпертуре (или бывают заметно ниже). Все альдегиды, кроме
формальдегида, при предельной температуре (в области от —20 до
—30 °C находятся в жидком состоянии. Низкие значения ТПр
для полимеризации карбонильных соединений в первую очередь
связаны с Д/7. Энтропия полимеризации по двойной связи С = О
приблизительно такая же, как и для полимеризации по алкеновой
двойной связи. Однако энтальпия полимеризации по карбониль-
ной связи значительно меньше. Например, А/7 полимеризации
ацетальдегида [59] равна только ~7 ккал/моль (29,30 -103 Дж/моль)
но сравнению с обычной величиной 15—20 ккал/моль (62,79-103 —
о. >,72-103 Дж/моль) для полимеризации по углерод-углеродной
двойной связи (табл. 3.12).
Как это стало теперь ясно, большинство ранних попыток поли-
меризации карбонильных соединений проведено при слишком
318
Глава 5
высоких температурах. Проведение реакции при температурах
выше Tnv не приводит к образованию полимера вследствие неблаго-
приятного сдвига равновесия цикл — полимер. Успешная поли-
меризация карбонильных мономеров с образованием высокомоле-
кулярных продуктов протекает при температурах ниже предель-
ных равновесных температур мономеров. Как катионные, так
и анионные катализаторы всех типов, применяемых при полиме-
ризации алкенов, можно использовать для инициирования поли-
меризации соединений с карбонильной двойной связью. Основ-
ные закономерности этой полимеризации аналогичны законо-
мерностям ионной полимеризации алкеновых мономеров. Однако'
число работ по детальному изучению механизма полимеризации
карбонильных соединений невелико.
5.5а. Анионная полимеризация
Полимеризацию соединений с карбонильной группой прово-
дили в присутствии различных основных катализаторов. Основ-
ность катализатора, требующаяся для инициирования полимери-
зации, зависит от характера заместителей у карбонильной группы.
5.5а.1. Полимеризация формальдегида
Карбонильная группа формальдегида весьма чувствительна
к нуклеофильной атаке, и этот мономер можно полимеризовать
почти с любыми основными катализаторами. К эффективным
катализаторам полимеризации формальдегида относятся алкилы
металлов, алкоголяты, феноляты, фосфины, арсины и азотсодер-
жащие гетероциклы [60].
Одна из наиболее полно изученных реакций — полимеризация
формальдегида с образованием полиоксиметилена под влиянием
третичных аминов. Считают, что истинной инициирующей частицей
при этом является четвертичная аммонийная соль, образующаяся
при реакции амина с водой, спиртом или каким-либо другим сока-
тализатором [61]. Полимеризация с водой в качестве сокатализа-
тора протекает по следующей схеме:
инициирование
к
R3N + H2O (R3NH+)OH", (5.73)
(R3NH+)OH--]-CH2 = O —» HO — CH2-O-(R3NH+), (5.74)
рост цепи
kp
HO-(-CH2-O-)n-CH2-O-(R3NH+) + CH2 = O —>
—> HO —( —CH2—0 —)n+i—CH2—O_(RSNH+), (5.75)
Ионная полимеризация
319
обрыв цепи путем передачи на R'OH
fenep
НО - (- СН2 - О - )n - СН2 - O-(R3NH+) + R'OH->
—>HO-(-CH2-O-)n-CH2-OH + (R3NH+)OR' . (5.76)
Приведенный механизм полимеризации аналогичен общим схе-
мам, предложенным для ионной полимеризации алкенов. Такие
кинетические выражения, как уравнения (5.25) и (5.26), при-
годны, по-видимому, и для полимеризации формальдегида. Однако
детальных экспериментальных исследований, подтверждающих
кинетику этого процесса, недостаточно.
Агентом передачи цепи R'OH может быть любое соединение,
которое способно передавать протон растущему аниону. Являю-
щаяся катализатором вода тоже может служить агентом передачи
цепи. Некоторые агенты передачи цепи оказывают влияние на ско-
рость полимеризации. Так, муравьиная и уксусная кислоты
в одинаковой степени снижают молекулярный вес полиоксимети-
лена, но уксусная кислота уменьшает и скорость полимеризации
[60]. Муравьиная кислота на скорость полимеризации не влияет.
Влияние уксусной кислоты на Rv вызвано, очевидно, пониженной
активностью инициатора, регенерируемого при реакции переноса.
Ацетат, соответствующий (R3NH+)OR“, должен подвергаться
обратимому гидролизу [аналогично реакции (5.73)] в большей
степени, чем формиат, так как основность ацетатного иона выше.
5.5а.2. Полимеризация других карбонильных
мономеров
Полимеризация других карбонильных мономеров происходит
подобным же образом, однако основность требующегося катали-
затора может быть существенно различной. Для инициирования
полимеризации алифатических альдегидов, таких, как и-масляный
альдегид, требуются сильные основания [62]. Индукционный эффект
алкильного заместителя дестабилизирует растущий ион
R
— С — о-,
н
увеличивая электронную плотность кислорода. Таким образом,
слабые основания, такие, как амины, нельзя использовать для
полимеризации альдегидов. Хорошими катализаторами для этих
мономеров являются алкилы и алкоголяты щелочных металлов.
Кетоны вследствие индукционного влияния двух алкильных групп
полимеризуются с большим трудом. Без сомнения, низкая реак-
320
Глава 5
ционная способность кетонов по отношению к полимеризации
обусловлена и стерическими факторами. Единственным исключе-
нием является полимеризация ацетона в присутствии координа-
ционных катализаторов (гл. 8).
Замещение водорода алкильной группы алифатического альде-
гида на галогены увеличивает способность мономера к полимери-
зации. Трихлорацетальдегид (хлораль) легко полимеризуется
такими слабыми основаниями, как пиридин и щелочные тиоциа-
наты [63]. Полимеризация трифторацетальдегида бутиллитием
при —78 °C заканчивается менее чем за одну секунду [64]. Благода-
ря положительному индукционному эффекту галогенов растущий
анион
С13С
— с-о-
Н
стабилизируется вследствие уменьшения электронной плотности
кислорода. Это влияние еще более ясно видно в случае полимери-
зации фтортиокарбонильных соединений, таких, как тиокарбо-
нилфторид
F
C = S — ( —CF2—S—)п — (5.77)
F
и гексафтортиоацетон
CF3
(L-s-*
।
CF3
г CF3
I
-C-S —
I-
L CF3
(5.78)
Тиокарбонилфторид полимеризуется в присутствии следов слабых
оснований, например диметилформамида [65]. Полимеризация
гексафтортиоацетона — яркий пример влияния предельной тем-
пературы на полимеризацию: очень низка, полимеризация
гладко протекает при —110° С.
5.56. Катионная полимеризация
Соединения с карбонильной двойной связью полимеризуются
также в присутствии кислотных катализаторов. Кислотность при-
меняемого для катионной полимеризации катализатора обычно
больше, чем основность катализатора, необходимая для анионной
полимеризации. Протонные кислоты — соляная кислота, уксусная
Ионная полимеризация
321
кислота и кислоты Льюиса — эффективные катализаторы катион-
ной полимеризации карбонильных мономеров. Стадии иницииро-
вания и роста при полимеризации, инициируемой протонными
кислотами, можно представить следующим образом:
R R
I I
О = СГНА-> НО-С+(А") (5.79)
I I
н н
R R
I I
н _(-О —CHR-),,-O-C+(A-) + O = C
А А
R
I
-->H-(-O-CHR-)n+i-O—С+(А~) (5.80)
Н
Реакция обрыва, вероятно, заключается в передаче цепи на воду
II.I и какую-либо другую присутствующую частицу
R
I
Н-(--O-CHR-)n+1-O-C+(A-) + H2O—»
i
R
I
—> H —( —О —CHR —)n+i —О —C —OH-j-HA (5.81)
Н
Механизм полимеризации, катализируемой кислотами Льюиса,
не выяснен окончательно. В частности, не ясно, требуются ли
в этом случае сокатализаторы. Кажется, что в ряде случаев поли-
меризация происходит таким образом, как и катионная полиме-
ризация алкенов, и требует применения сокатализаторов обычного
типа (например, воды). Однако для полимеризации хлораля,
катализируемой А1Вг3, предполагают [66] протекание стадии
инициирования следующим образом:
СС13
С13С —СНО-|-А1Вгз—»Вг3АЮ—С+ (5.82)
II
Аналогичный механизм процессов инициирования, происходящих
оез сокатализаторов. был предложен для полимеризации в при-
сутствии и других кислот Льюиса. Однако это утверждение
322
Глава 5
имеет большой недостаток, поскольку рост цепи при инициирова-
нии такими частицами, как указано в уравнении (5.82), должен
происходить со все возрастающим разделением и локализацией
положительных и отрицательных зарядов по мере увеличения
размеров растущей частицы XI
СС13 СС13
_ I I
Вг3А1О —С —( —О —СНСС13 —)п —О —С+
i н
XI
Такого затруднения не возникает, если предположить [66], что
реакцией инициирования является
СС13
СС13 — СНО + 2А1Вг3 —> Вг2А1О — d+(А1Вг4)" (5.83)
Н
[Возможно, реакция, описанная уравнением (5.83), протекает
в две стадии: первоначальное образование комплекса альдегида
с катализатором 1:1 с последующим взаимодействием этого
комплекса со второй молекулой катализатора.] Это соответствует
обычному типу механизма катионной полимеризации (разд. 5.2),
в которой катализатор ведет себя одновременно и как сокатали-
затор.
5.5в. Радикальная полимеризация
Как правило, карбонильная двойная связь не чувствительна
к радикальным инициаторам. Такое поведение объясняется двумя
причинами: сильной поляризованпостью карбонильной группы
и тем, что большинство радикалов образуется при температурах,
превышающих предельные температуры карбонильных мономе-
ров. Однако есть несколько примеров полимеризации карбониль-
ных мономеров под влиянием радикальных инициаторов. Была
осуществлена полимеризация трифторацетальдегида под действием
перекиси бензоила при 22 °C. Полимеризация протекает медленно,
так что 90%-ная степень превращения достигается через 18 ч [64].
Однако полимеризация фтортиокарбонильных мономеров, таких,
как тиокарбонилфторид, под влиянием окислительно-восстанови-
тельной системы триалкилбор — кислород происходит быстро
ПрИ —78 °C [65]. Радикалы образуются [67] согласно реакциям
R3B + O2—»R2BOOR, (5.84)
R2BOOR + 2R3B —» R2BOBR2 + R2BOR +2R• (5.85)
Полимеризация по радикальному механизму для этих моно-
меров возможна потому, что электроноакцепторные заместители
Ионная полимеризация
323
у карбонильной и тиокарбонильной групп уменьшают их поляр-
ность (и поляризуемость). Большая склонность тиокарбонильной
двойной связи к радикальной полимеризации связана с меньшей
электроотрицательностью серы по сравнению с кислородом.
Тиокарбонильная группа менее полярна, чем карбонильная,
п более чувствительна к атаке радикалом. Успех радикальной
полимеризации тиокарбонильных мономеров в большой степени
определяется также низкими температурами применяемого про-
цесса инициирования.
5.5г. Ступенчатая полимеризация
Полимеризацию некоторых карбонильных мономеров осуще-
ствили по ступенчатому механизму, используя водные или спирто-
вые растворы мономеров. Наиболее широко изучена полимеризация
формальдегида [60]. Реакция заключается в ступенчатой полиме-
ризации гем-диола или полуформаля (в зависимости от того,
проводят реакцию в воде или спирте). Процесс можно изобразить
следующим образом:
инициирование
ЙОН4-Н2СО—»ЙОСН2ОН, (5.86)
рост
н2со н2со
ЙОСН2ОН ---* ЙО(СН2О)2Н Т-» ЙО(СН2О)3Н и т. д., (5.87а)
или в общем виде
н2со
ЙО —( —СН2О—)n —Н --» ВО-(-СН2О-)п+1-Н, (5.876)
где R — атом водорода для полимеризации в воде и алкил для
полимеризации в спиртовом растворе. Этот тип механизма поли-
меризации по кинетическим закономерностям аналогичен поли-
меризации с раскрытием цикла (гл. 7).
5.5д. Блокирование концевых групп
Полиацетали, полученные полимеризацией карбонильных моно-
меров, как правило, нестабильны при умеренных температурах
вследствие низких предельных температур. Они легко деполиме-
ризуются с образованием мономера. Таким образом, может
казаться, что этот класс полимеров не имеет практического значе-
ния. Однако влияние низких предельных температур полиацеталей
удалось ликвидировать превращением их активных концевых
гидроксильных групп в нереакционноспособные эфирные связи
путем взаимодействия с галогенангидридами или ангидридами
324
Глава 5
. кислот, например
RCOC1 или
НО-(-СН2О-)„-СН2ОН
—> RCOO — ( — СН2О — )п — CH2OCOR. (5.88)
Эта реакция называется реакцией закрытия или блокирования
концевых групп. В результате этого реакционноспособная частица
(т. е. анион или катион) на концах цепи не образуется, и при
предельной равновесной температуре полимера не происходит
деполимеризации. Другими словами, полимерные цепи защищены
на концах от деполимеризации. Для этой цели можно применять
целый ряд блокирующих реагентов, таких, как ацетил- и бензоил-
хлорид или уксусный ангидрид. Во многих случаях путем блоки-
рования концевых групп удается значительно повысить (на сотни
градусов) температуру разложения полиацеталей. [Для предотвра-
щения деполимеризации можно использовать также сополиме-
ризацию (разд. 7.6).]
3.6. Различные мономеры
5.6а. Мономеры с двумя различными
полимеризующимися группами
Большинство мономеров содержит только одну способную
к полимеризации связь. Однако есть мономеры с двумя такими
связями в молекуле. Полимеризация подобных мономеров может
приводить к образованию более чем одной полимерной структуры.
Примером такого поведения является полимеризация диенов.
Существует также несколько мономеров, которые в отличие
от диенов содержат полимеризующиеся связи двух различных
типов. К мономерам такого типа относятся
(СН3)2С = С = О и СН2 = СН-СН = О.
диметилкетен акролеин
Полимеризация диметилкетена и акролеина интересна потому,
что различие в реакционной способности обоих способных к поли-
меризации групп может приводить в зависимости от условий
реакции к образованию различных полимерных структур.
5.6а,1. Диметилкетен
Диметилкетен полимеризуется под влиянием анионных ини-
циаторов. Реакция может происходить путем полимеризации
по двойной алкеновой связи с образованием поликетопа
п(СН3)2С = С = О ----
о
II
— С(СН3)2 —С —
(5.89)
Ионная полимеризация
325
или полимеризацией по карбонильной связи с образованием
ненасыщенного полиацеталя
СН3 СН3 -
п(СНз)2С = С = О —>
(5.90)
пли полимеризацией обеих групп с образованием ненасыщенного
полиэфира
2п(СН3)2С = С = О —>—
О
II
— С(СН3)2-С-О-С---
(5.91)
Хотя в каждом случае полимеризации образуются обе, а то и все
три структуры, можно получить полимер с преобладанием повто-
ряющейся единицы одного типа [68, 69]. Полимеризации по более
полярной карбонильной двойной связи способствуют полярные
растворители. Полимеризации по менее полярной алкеновой
связи либо с образованием одного поликетона, либо с частичным
образованием полиэфира способствуют неполярные растворители.
Па образование поликетона благоприятно влияют противоионы
Li, Mg и Al, а на образование полиэфира — Na и К.
5.6а.2. Акролеин
Полимеризация акролеина была изучена в присутствии ини-
циаторов радикальной, катионной и анионной полимеризаций
в широком диапазоне температур [70]. Радикальная полимериза-
ция протекает исключительно по двойной связи с образованием
полиальдегида
пСН2=СН —ГСН2 —СН —"1 — (5.92)
I I
СНО [ СНО _|п
При катионной полимеризации под влиянием обычных типов
катализаторов (например, BF3, H2SO4) образуются полимеры,
содержащие повторяющиеся единицы двух типов
+ т)СН2 = СН — СН = О —>
-СН2 — CH--I г—О —СН—"I
н 1 т сн
II
п L сн2
(5.93
что соответствует полимеризации и по алкеновой, и по карбо-
нильной связям. Отношение п/т может несколько изменяться
326
Глава 5
но случаев образования полимеров с п или т, равным пулю,
не наблюдали. Аналогичное поведение установлено для анионной
полимеризации в присутствии таких соединений, как бутпллптип
или Na-нафталин. Единственным исключением является полимери-
зация с натрием или цианистым натрием в тетрагпдрофурапе пли
толуоле при температуре от —50 до —40 СС, которая протекает
полностью по карбонильной группе
пНС = О
I
сн-сн2
-сн-о —-|-
I
CH = CH2Jn
(5.94
Сходство акролеина с бутадиеном-1,3 (сопряжение алкеновых
и карбонильных двойных связей) позволило некоторым исследова-
телям предположить возможность 1,4-полимеризацпи акролеина
гаСН2 = СН —СН = О —> — ( —СП2 — СИ = СП —О —)п — , (5.95)
по .тгого до сих пор не наблюдали.
5.66. Акриламид
Полимеризация акриламида (или метакриламида) в присут-
ствии сильных оснований — натрия, литийоргапических соеди-
нений и алкоголятов — приводит к образованию полимеров со
структурой
аСН2=СН —СО —NH2—> —( —СН2 —СН2 —СО —NH —)п — , (5.96)
что совершенно неожиданно. Полиамидная структура полимера
была подтверждена данными количественного гидролиза полимера
до 3-аминопропионовой кислоты.
Механизм полимеризации заключается в присоединении анион-
ного катализатора по двойной связи мономера
B- + CH2 = CHCONH2—>ВСН2 —CHCONH2, (5.97а)
сопровождаемом отщеплением протона от другой молекулы моно-
мера с образованием амидного аниона ХП
всн2—chconh2+ch2=chconh2—»
—»BCH2CH2CONH2 + CH2 = CHCONH . (5.976)
XII
Амидный анион инициирует полимеризацию через ряд аналогич-
ных реакций путем присоединения к мономеру
CH2 = CIICONH + CII2=CHCONH2 —> CH2 = CHCONHCH2CHCONH2i (5.98а)
CH2 = CHCONHCH2CIICONH2 + CH2 = CHCONU2—>
—> CH2=CHCONHCH2CH2CONH2 + CII2 = CHCONH. (5.986)
Ионная полимеризация
327
Рост цепи происходит аналогичным образом
€H2 = CHCONH-(-CH2CH2CONH-)n-H + CH2=CHCONH-^
XIII
—> CH2 = CHCONH —СН2—CHCONH —( —CH2CH2CONH—)n —Н 2222^
-> CH2 = CHCONH-(-CH2CH2CONH-)n+1-H + CH2 = CHCONH. (5.99)
Эта полимеризация протекает совершенно иначе, чем любой
ранее описанный тип полимеризации. Центр роста здесь
не является ионом или радикалом, а представляет собой алкено-
вую двойную связь на конце растущей частицы XIII. Помимо
этого, к центру роста присоединяется не мономер, а анион моно-
мера XII. О таком механизме реакции свидетельствуют данные
анализа, доказывающие наличие алкеновых концевых групп,
и наблюдение, что реакция представляет собой ступенчатый
процесс (повышение молекулярного веса полимера в процессе
полимеризации). Этот механизм очень близок к механизму анион-
ной полимеризации циклических амидов, протекающей с раскры-
тием цикла (разд. 7.3а). По аналогии с последним механизмом
анион мономера можно назвать активированным мономером. Всю
реакцию полимеризации в целом часто называют полимеризацией
с переносом водорода.
5.6в. Изоцианаты
Изоцианаты под влиянием анионных инициаторов, таких, как
металлоорганические соединения, натрий и цианистый натрий,
могут полимеризоваться по двойной связи С = N [72]
ziR — N = С = О —>
(5.100)
Вследствие низкой предельной температуры полимеризации основ-
ным продуктом этой реакции при температурах выше —20 °C
является циклический тример
3RNCO—»
(5.101)
328
Глава 5
5.6г. Диазоалканы
Полимеризация диазоалканов, таких, как диазометан
MCH2N2 — (— СН2 — )п J- aN2,
(5.102*
может протекать под влиянием BF3 и других кислот Льюиса [731.
Предложенный для этой реакции механизм включает следующую
последовательность реакций:
ВГз - + -Nz +
ch2n2-----> f3bch2n = N------> f3bch2
ch2
I
F2B-CH2F
-n2
ch2n2
I CH2N2
f2b—ch2f *-----f2bch2f
V CH2N2
F2B-CH2CH2F------> F2B —( —CH2 —)n —F
(5.103)
Этот тип полимеризации интересен потому, что позволяет получить
чисто линейный полиэтилен без каких-либо разветвлений. Такой
линейный полимер можно использовать в качестве стандартного
образца для проведения исследований по влиянию разветвления
на свойства полимера.
5.6д. Мономеры с тройной связью
Описано [74, 75] несколько примеров полимеризации нитрилов
(5.104)
и ацетиленов
«RC ~ СН —> —
R
— (! = СН-п-
(5.105)
hRC * N —
с ионными катализаторами. Однако структура и молекулярный
вес полученных продуктов не определены достаточно точно. Поли-
мерные структуры, изображенные уравнениями (5.104) и (5.105),
могут представлять интерес с точки зрения их потенциальных
полупроводниковых свойств.
Ионная полимеризация
32»
Вопросы и упражнения
5.1. Рассмотрите следующие системы мономеров и катализа-
торов:
Катализаторы Мономеры
(С6Н3СО2)2 С6Н5СН = СН2
(СН3)3ССООН + Fe2+ CH2 = C(CN)2
Na-Нафталпп СН2 = С(СН3)2
H2SO4 СН2 = СН-О-н-С4Н9
BF3 СН2 = СН-С1
H-C4HgLi СН2 = С(СН3)-СО2СН3
СН2 = О
cf2=s
Что является инициирующей частицей или активным центром
каждого из этих катализаторов? Напишите уравнения. Какие
катализаторы могут быть использованы для полимеризации
каждого из перечисленных мономеров? Поясните ваши ответы.
Какие общие условия реакции (т. е. температура, растворитель)
требуются для осуществления реакции в каждом случае?
5.2. Рассмотрите катионную полимеризацию изобутилена
в бензоле в присутствии TiCl4 в качестве катализатора и Н2О
в качестве сокатализатора. При определенных условиях реакции
было установлено, что экспериментальная скорость полимериза-
ции дается уравнением
Яр=[Т1С14][Изобутплен][Н2О]0
Обрыв цепи происходит путем перегруппировки растущего центра
с образованием полимера, имеющего ненасыщенную концевую
группу, и комплекса катализатор — сокатализатор. Напишите
механизм этой полимеризации и выведите уравнения скорости
и степени полимеризации.
При каких условиях реакции скорость полимеризации может
иметь:
а) первый порядок по [Н2О];
б) нулевой порядок по [TiCl4J;
в) второй порядок по [Изобутилен];
г) второй порядок по [Н2О1?
5.3. Скорость полимеризации возрастает с повышением темпе-
ратуры. По радикальному или ионному механизму протекает эта
реакция? Какие экспериментальные методы, кроме влияния тем-
пературы, можно использовать для определения типа иницииро-
вания?
5.4. Полимеризация метилметакрилата, инициируемая Na-наф-
талином, проводится в растворе в воде, в тетрагидрофуране
и в нитробензоле. В каком растворителе скорость полимеризации
330 Глава 5
будет максимальной? Обсудите влияние растворителя на кон-
центрацию и идентичность растущих частиц.
5.5. 1,5 М раствор стирола в тетрагидрофуране полимеризуется
при 25 °C под влиянием Na-нафталина концентрации 3,2-IO-5 М.
Вычислите начальную скорость полимеризации, используя дан-
ные табл. 5.7. Вычислите степень полимеризации при 100%-ной
степени превращения. Сравните значение Rp со значением этой
величины, вычисленным по уравнению (5.60) с использованием
данных табл. 5.8.
5.6. 2,0 М раствор стирола в дихлорэтилене полимеризуется
при 25 °C в присутствии 4,0-10-4 М серной кислоты. Вычислите
начальную степень полимеризации. Какой будет степень поли-
меризации, если раствор мономера содержит изопропилбензол
концентрации 8,0-IO-5 М? Для вычислений используйте данные
табл. 5.1 и 5.4.
Список литературы
1. Берстол М., Трелоар Ф., Карбониевые ионы, в кн. «Катионная поли-
меризация», под ред. П. Плеша, гл. 1, изд-во «Мир», М., 1966.
2. Evans A. G., Meadows G. W., Trans. Faraday Soc., 46, 327 (1950).
3. Плеш Л., Изобутилен, в кн. «Катионная полимеризация», под ред.
П. Плеша, гл. 4, изд-во «Мир», М., 1966.
4. Colclough R. О., Dainton F. S., Trans. Faraday Soc., 54, 886, 894 (1958).
5. Overberger C. G., Ehrig R. J., Marcus R. A., J. Am. Chem. Soc., 80, 2456
(1958).
6. Tazuke S., Nakagawa K., Okamura S., J. Polymer Sci., B3, 923 (1965).
7. Sakurada I., Ise N., Tanaka Y., Hayashi Y., J. Polymer Sci., A-l(4), 2801
(1966).
8. Potter R. C., Johnson C. L., Metz D. J., Bretton R. H., J. Polymer Sci.,
A-l(4), 419 (1966).
9. Kennedy J. P., Minckier L. S., Wanless G., Thomas R. M., J. Polymer Sci.,
A2, 2093 (1964).
10. Ketley A. D., J. Polymer Sci., B2, 827 (1964).
11. Kennedy J. P., Langer A. W., Jr., Fortschr. Hochpolymer.-Forsch. (Advan.
Polymer Sci.), 3, 508 (1964).
12. Матизон А., Стирол, в кн. «Катионная полимеризация», под ред.
П. Плеша, гл. 6, изд-во «Мир», М., 1966.
13. Plesch Р. Н„ J. Chem. Soc., 1953, 1653.
14. Kamachi М., Miyama Н., J. Polymer Sci., A3, 1337 (1965).
15. Burton R. E., Pepper D. C., Proc. Roy. Soc. (London), A263, 58 (1961).
16. Hayes M. J., Pepper D. C., Proc. Roy. Soc., A263, 63 (1961); Proc. Chem.
Soc 1958 228
17. Okamura S., Higashimura T., J. Polymer Sci., 21, 289 (1956); Chem. High
Polymers (Tokyo), 13, 342, 397 (1956) and 17, 57 (1960).
18. Endres G. F., Overberger C. G., J. Am. Chem. Soc., 77, 2201 (1955).
19. Biddulph R. H., Plesch P. H., J. Chem. Soc., 1960, 3913.
20. Sakurda Y., Higashimura T., Okamura S., J. Polymer Sci., 33, 496 (1958).
21. Imanishi Y., Higashimura T., Okamura S., Chem. High Polymers (Tokyo),
18, 333 (1961).
22. Jenkinson D. H., Pepper D. C., Proc. Roy. Soc. (London), A263, 82 (1961).
23. Overberger C. G., Endres G. F., Monad A., J. Am. Chem. Soc., 78, 1969
(1956).
24. Okamura S., Higashimura T., J. Polymer Sci., 21, 289 (1956).
Ионная полимеризация
331
25 Brown С. Р., Mathieson A. R., J. Chem. Soc., 1957, 3608, 3612, 3620, 3631
and 1958, 3445.
26 Pepper D. C., Reilly P. J., J. Polymer Sci., 58, 639 (1962).
2?" Kanoh N., Gotoh A., Higashimura T., Okamura S., Makromol. Chem.,
63, 115 (1963).
28 Kanoh N., Higashimura T., Okamura S., Makromol. Chem., 56, 65 (1962).
29^ Funt B. L., Electrolytically Controlled Polymerizations, in Peterlin A.,
Goodman M., Okamura S., Zimm В. H. and Mark H. F. (eds.), Macromole-
cular Reviews, vol. 1, Interscience Publishers, John Wiley and Sons, Inc.,
New York, 1967.
30. Gandini A., Plesch P. H., J. Polymer Sci., B(3), 127 (1965); The Chemistry
of Polymerization Processes, p. 107—114, 122—129, Society of Chemical
Industry, London, 1966.
31. Plesch P. II., Polymer Preprints, 7(2), 492 (1966).
32. Kennedy J. P., Squires R. G., Polymer, 6, 579 (1965).
33. Szwarc M., Ann. N.Y. Acad. Sci., 155(2), 400 (1969).
34. Higginson W. C., Wooding N. S., J. Chem. Soc., 1952, 760, 1178.
35. Overberger C. G., Yuki H., Urakawa N., J. Polymer Sci., 45, 127 (1960).
36. L'Abbe G., Smets G., J. Polymer Sci., A-l(5), 1359 (1967).
37. Szwarc M., Smid J., The Kinetics of Propagation of Anionic Polymeriza-
tion and Copolymerization, in «Progress in Reaction Kinetics», Porter G.
(ed.), vol. 2, chap. 5, Pergamon Press Ltd., Oxford, 1964.
38. Szwarc M., Waak R., Rembaum A., Coombes J. D., J. Am. Chem. Soc.,
79, 2026 (1957).
39. Szwarc M., Levy M., Milkovich R., J. Am. Chem., 78, 2656 (1956).
40. Overberger C. G., Pearce E. M., Mayes IV., J. Am. Chem. Soc., 34, 109 (1959).
41. Bywater S., Fortschr. Hochpolymer.-Forsch. (Advan. Polymer Sci.), 4, 66
(1965).
42. Geacintov C., Smid J., Szwarc M., J. Am. Chem. Soc., 84, 2508 (1962).
43. Bhattacharyya D. N. Lee C. L., Smid J., Szwarc M., J. Phys. Chem., 69,
612 (1965).
44. Bhattcharyya I). N., Smid J., Szwarc M., J. Phys. Chem., 69, 624 (1965).
45. Bywater S„ Worsfold D. J., J. Phys. Chem., 70, 162 (1966).
46. Frost A. A., Pearson R. G., Kinetics and Mechanism, 2d ed., chap. 7, John
Wiley and Sons, Inc., New York, 1961.
47. Flory P. J., J. Am. Chem. Soc., 62, 1561 (1940).
48. Gold L., J. Chem. Phys., 28, 91 (1958).
49. Ledwith A., Ann N.Y. Acad. Sci., 155(2), 385 1(969).
50. Tokwia N., Hagai T., Sonoyama Y., Tetrahedron Letters, 17, 1145 (1965).
51. l'orlschr. Hochpolymer.-Forsch. (Advan. Polymer Sci.), 2,
52. Worsfold D. J., Bywater S., Can. J. Chem., 38, 1891 (1960).
53. О Driscoll K. F., Tobolsky A. V., J. Polymer Sci., 35, 259 (1959).
54. Erusalimskii B. L. Krasinosel'skaya I. S., Makromol. Chem., 123, 80,
(1969). v ’
^llen G- et al., Polymer, 2, 151 (1961); J. Polymer Sci., 48, 189 (1960).
3b. hAiimomura T., Smid J., Szwarc M., Polymer Preprints, 8(2), 897 (1967).
Z" Л1пр»<гбм'^Ж1’965гегг/еа Полимеризация альдегидов и окисей, изд-во
58. Brandrup J Immergut E. H. (eds.), Polymer Handbook, p. 11-389, Inter-
na rzle?Czi Publishers, John Wiley and Sons, Inc., New York, 1966.
5У. Vogl O. Bryant W. M. D., J. Polymer Sci., A2, 4633 (1964).
bU. Brown N., J Macromol. Sci. (Chem.), Al(2), 209 (1967).
' 1 ’’ ‘^acBonatd R. N., Punderson J. O., J. Appl. Polymer Sci.,
1, 160 (1959).
62. Vogl О J Macromol. Sci. (Chem.), Al(2), 243 (1967).
b3. Rosen I., J. Macromol. Sci. (Chem.), Al(2), 267 (1967).
332
Глава 5
64. Busjield W. К., Whalley E„ Can. J. Chem., 43, 2289 (1965).
65. Sharkey W. H., J. Macromol. Sci. (Chem.), Al(2), 291 (1967).
66. Rosen I., Hudgin D. E., Sturm C. L., McCain G. H., Wilmjelm R. M.,
J. Polymer Sci., A3, 1535, 1545 (1965).
67. Barney A . L., J. M. Bruce, Jr., Coker J. TV., Jacobson N. W., Sharkey W. H.,
J. Polymer Sci., A-l(4), 2617 (1966).
68. Yamashita Y., Nunomoto S., Makromol. Chem., 58, 244 (1962).
69. Pregaglia G. F., Minaghi M., Cambini M., Makromol. Chem., 67, 10 (1963).
70. Schulz R. C., Polymerization of Acrolein, in «Vinyl Polymerization»,
Ham G. E. (ed.), vol. 1, part I, chap. 7, Marcel Dekker, Inc., New York,
1967.
71. Bush L. W., Breslow D. S., Macromolecules, 1, 189 (1968).
72. Shashoua V. E., Sweeny W., Tietz R. F., J. Am. Chem. Soc., 82, 866 (1960).
73. Bawn С. E. H., Ledwith A., Matthies P., J. Polymer Sci., 34, 93 (1959).
74. Luttinger L. B., Colthup E. C., J. Org. Chem., 27, 3752 (1962).
75. Kabanov V. A., Zubov V. P., Kovaleva V. P., Kargin V. A., J. Polymer
Sci., C4, 1009 (1963).
ГЛАВА 6
СОПОЛИМЕРИЗАЦИЯ
Для осуществления поликонденсации в большинстве случаев,
например при синтезе полигексаметиленадипинамида или поли-
этилентерефталата, необходимы два реагирующих вещества или
мономера, и полученный продукт содержит в цепи структуры
двух различных типов. При полимеризации для получения поли-
мера требуется наличие только одного мономера. Однако можно
проводить полимеризацию смеси двух мономеров для получения
полимерных продуктов, содержащих две различные структуры
в полимерной цепи. Такой тип полимеризации, когда одновре-
менно полимеризуются два мономера, называется сополимериза-
цией, а образующийся продукт называется сополимером. Важно
подчеркнуть, что сополимер не является сплавом или смесью
двух гомополимеров, а содержит в каждой макромолекуле звенья
обоих мономеров. Процесс можно изобразить следующим образом:
Mj + Ma—»
•— MjMaMaMjMaMaMzMiMjMaMaMjMjMaMiMjMiMaMaMiMi —. (6.1)
Оба мономера входят в сополимер более или менее произвольно,
причем содержание их в полимере определяется их относительной
концентрацией и реакционной способностью. Можно также про-
водить одновременную сополимеризацию смеси трех и более
различных мономеров. Такие процессы полимеризации обычно
называют многокомпонентной сополимеризацией. Применительно
к системам из трех мономеров употребляется термин терполиме-
ризация.
(>.1. Общие положения
6.1а. Значение сополимеризации
Процессы сополимеризации важны по многим причинам. Значи-
тельная часть сведений о реакционной способности мономеров,
радикалов, попов карбонпя и карбанионов в полимеризации
получена в результате изучения сополимеризации. Особенно
полезно изучение поведения мономеров в реакциях сополимериза-
ции для выяснения влияния химической структуры на реакцион-
334
Глава 6
ную способность. Сополимеризация важна также с технологиче-
ской точки зрения. Она в значительной степени увеличивает воз-
можности получения полимерных продуктов со специфическими
заданными свойствами. При полимеризации одного мономера
(гомополимеризации) количество возможных продуктов сравни-
тельно ограниченно. Сополимеризация позволяет осуществить
синтез практически неограниченного числа различных продуктов
путем изменения природы и относительных количеств звеньев
двух мономеров, содержащихся в сополимере.
Огромные возможности процесса сополимеризации видны
на примере модификации свойств полистирола. Полистирол —
хрупкий пластик с низкой ударной вязкостью и малой устойчи-
востью к действию растворителей. По этим причинам он имеет
сравнительно ограниченную практическую ценность. При сополи-
меризации и терполимеризации стирола свойства полимера улуч-
шаются, что значительно увеличивает его практическую полез-
ность. В результате годовое производство полимерных продуктов,
содержащих стирол, все время увеличивается. Сополимеры
и терполимеры стирола находят применение не только в качестве
пластиков, но и в качестве эластомеров. Так, например, сополи-
меризация стирола с акрилонитрилом приводит к увеличению
ударной прочности сополимера и повышению стойкости его
к растворителям, тогда как при сополимеризации с бутадиеном
образуются сополимеры, обладающие эластическими свойствами.
Терполимеризация стирола с акрилонитрилом и бутадиеном улуч-
шает все эти свойства одновременно.
Другим примером большой важности сополимеризации
является изменение свойств наиболее распространенного пла-
стика — полиэтилена. При сополимеризации этилена с неболь-
шими количествами другого мономера, такого, как бутен-1, винил-
ацетат или акрилат, получают сополимеры этилена, которые отли-
чаются большей гибкостью, чем сам полиэтилен. Сополимериза-
ция с большими количествами мономеров может превратить
полиэтилен из пластика в эластомер, как, например, недавно
разработанные сополимеры этилена с пропиленом. Существуют
и многие другие примеры технологической целесообразности
процессов сополимеризации.
6.16. Типы сополимеров
В сополимере, описанном уравнением (6.1), структурные
звенья двух мономеров распределены вдоль полимерной цепи
беспорядочно. Продукты с таким типом структуры называют
статистическими сополимерами. Известны еще три другие основ-
ные структуры сополимера. Это сополимеры, структура которых
отличается чередованием звеньев, а также блочные и привитые.
Сополимеризация
335
регулярно чередующийся сополимер содержит звенья обоих моно-
меров в эквимолярных количествах и с правильным чередованием
их вдоль цепи
— MtMaMjMaMjMaMiMaMiMaMiMaMiMaMi— .
I
Блочные и привитые сополимеры отличаются от статистиче-
ских и чередующихся сополимеров тем, что цепи их образованы
длинными последовательностями звеньев каждого мономера.
Блок-сополимер представляет собой линейный полимер с длин-
ными последовательностями (блоками) одинаковых звеньев, раз-
деленными блоками других звеньев:
— М1М1М1М1М1М1М1М1М1М1М1М1М1М2М2М2М2М2М2М2— .
II
Привитой сополимер, называемый также иногда графт-сополи-
мером, является разветвленным сополимером, у которого основ-
ная цепь состоит из звеньев одного мономера, а к ней присоеди-
нены одна или больше боковых цепей, построенных из звеньев
другого мономера
-М jM [М jM jM jM [M jM jM ]M jM jM jM jM jM jM jM jM jM ]M jM jM jM f
sp
2*7.
2*7.
147.
247.
И7 *
гЛ/,
$4r
in
В настоящей главе рассмотрена главным образом сополимери-
зация двух мономеров, протекающая одновременно и сопровождаю-
щаяся образованием статистических и регулярно чередующихся со-
полимеров. Привитые сополимеры, а также большей частью и блок-
сополимеры при этом не образуются. Для получения их исполь-
зуются обычно^другие методы. Блок-сополимеры частично затра-
гиваются в этой главе, но более полно будут рассмотрены в гл. 9.
Общепринятой номенклатуры для сополимеров в настоящее
время нет L1J. Так, сополимер, полученный из стирола и метил-
метакрилата, можно назвать поли(метилметакрил ат-со-стирол) или
просто сополимер стирол-метилметакрилат. Структурные разли-
чия между статистическими, регулярно чередующимися, блочными
336
Глава 6
и привитыми сополимерами можно обозначать соответственно
приставками: -со-, -чер-, -б- и -пр-, например поли(метилметакри-
лат-со-стирол), поли(метилметакрилат-чер-стирол), поли(метилме-
чакрилат-б-стирол), поли(метилметакрилат-гар-стирол) *).
6.2. Состав сополимера
6.2а. Уравнение состава сополимера
Константы сополимеризации
Установлено, что состав сополимера в большинстве случаев
отличается от состава исходной смеси мономеров, из которой этот
сополимер получен. Другими словами, разные мономеры имеют
различную способность к сополимеризации. Уже давно заметили,
что относительная склонность мономеров к сополимеризации
не совпадает с отношением скоростей их гомополимеризации [2].
Некоторые мономеры более реакционноспособны при сополиме-
ризации, чем можно было ожидать, судя по скорости их гомо-
полимеризации; другие мономеры, наоборот, менее реакционно-
способны. Наконец, что особенно интересно, ряд мономеров,
например малеиновый ангидрид, стильбен и эфиры фумаровой
кислоты, легко вступает в реакции радикальной сополимеризации,
хотя эти мономеры очень мало или даже совсем не способны к гомо-
полимеризации.
Таким образом, состав сополимера нельзя определить по скоро-
стям гомополимеризации мономеров. Факторы, определяющие
состав сополимеров, установлены работами ряда исследователей
[3—5], причем в основу было положено предположение, что
реакционная способность растущей цепи (которая может быть
свободным радикалом, ионом карбония или карбанионом) при
сополимеризации зависит от строения всей цепи. Рассмотрим
сополимеризацию двух мономеров Mj и М2. Несмотря на то что
радикальная сополимеризация изучена значительно более широко
[6, 7] и является более важной, чем ионная сополимеризация,
рассмотрим общий случай сополимеризации, отвлекаясь от
ее инициирования. Сополимеризация двух мономеров приводит
к образованию растущих частиц двух типов: один тип — с Mt
на растущем конце и другой тип — с М2 на конце цепи. Эти типы
растущих частиц можно представить как М! и М*, где звездочка
означает радикал, ион карбония или карбанион (в зависимости
от механизма инициирования). Если предположить, что реак-
ционная способность растущей частицы зависит только от конце-
вого звена, то возможны четыре варианта реакций роста. Каждый
*) Вопросы номенклатуры и классификации блок- и привитых сопо-
лимеров освещены в обзорах Г. С. Колесникова (см. список дополнитель-
ной литературы).— Прим. ред.
Сополимеризация
337
из мономеров - М( и М2 — может присоединиться к растущей
цепи с концевым звеном М* или М2, т. е.
+ (6.2)
МТ + М2^>М!, (6.3)
+ (6.4)
М? + М2^>М!, (6.5)
где кц — константа скорости роста цепи, оканчивающейся моно-
мерным звеном Мь при реакции с мономером Mt; к12 — константа
скорости роста цепи, оканчивающейся Mt, при реакции с моно-
мером М2 и т. д. Рост активного центра путем присоединения
того же мономера [например, реакции (6.2) и (6.5)] часто назы-
вают гоморостом; рост активного центра присоединением другого
мономера [реакции (6.3) и (6.4)] называют перекрестным ростом.
Мономер М\ расходуется по реакциям (6.2) и (6.4), тогда как
мономер М2 исчезает по реакциям (6.3) и (6.5). Скорости исчезно-
вения обоих мономеров, которые совпадают с их скоростями вклю-
чения в сополимер, определяются выражениями
+ (6.6)
- ™ = ^[М*ПМ2) + *22[М2*][М2]. (6.7)
Разделив уравнение (6.6) на уравнение (6.7), получим отношение
скоростей вступления обоих мономеров в сополимер, т. е. состав
сополимера будет выражаться уравнением
d[Mt] __ g
d[M2] л12[мшм2Ш~[м*нм2] • *•
Для того чтобы исключить неизвестные абсолютные значения кон-
центраций растущих частиц М* и М* из уравнения (6.8), для
каждой из растущих частиц предполагается стационарное состоя-
ние концентраций М* и М*. Концентрации частиц М* и М* оста-
ются постоянными, если скорости их взаимного превращения
одинаковы. Другими словами, скорости реакций (6.3) и (6.4)
равны
/с21 [М2] [MJ =/с12 [М*] [М2]. (6.9)
Уравнение (6.9) можно преобразовать и, объединяя его с уравне-
нием (6.8), получить
<ИМ,1 —а",Ы‘|г+*-1МЯ1М,|
11мн — *12 1М2]__________ /п 4 рч
d [М2] ЛгИМШМгЦ-ЛгДМШМ!] ’
(R Разделив числитель и знаменатель правой части уравнения
(Ь.10) на k2l [МД [М2] и подставив в полученное выражение
О О Л л л л л
338
Глава 6
значения rt и г2, которые равны
к и к 22
И Г2"’
В конце концов получают
d[Mt] [Mt] (г, [М11 + [М2])
d[M2] [М2]([М1] + г2[М2]) •
(6.11)
(6.12)
Уравнение (6.12) известно как уравнение сополимеризации или
уравнение состава сополимера. Несмотря на то что для решения
этого уравнения необходимо сделать допущение стационарного
состояния системы, некоторые исследователи [8, 9] показали, что
то же самое выражение можно получить статистическим методом
без каких-либо допущений стационарности состояния. Состав
сополимера — это молярное отношение звеньев двух мономеров
в сополимере d[MiI/d[M2]• Это отношение выражено в уравне-
нии (6.12) через концентрации обоих мономеров в исходной смеси
[MJ и [М2] и параметры п и г2, которые называют константами
сополимеризации *). Параметр г [см. выражение (6.11)1 есть отно-
шение константы скорости присоединения растущей частицей
мономера молекулы такого же типа к константе скорости присоеди-
нения ею другого мономера.
Склонность двух мономеров к сополимеризации выражается
величинами г. Значение т\ > 1 соответствует тому, что М* пред-
почтительно присоединяет Mi, тогда как значение Г1<1 указывает
на то, что М* предпочтительно присоединяет М2. Значение т\ = О
означает, что М4 не способен к гомополимеризации.
Уравнение сополимеризации можно выразить не через кон-
центрации, а через мольные доли. Если и /2 — мольные доли
мономеров Mi и М2 в исходной смеси, a Ft и F2 — мольные доли М4
и М2 в сополимере, тогда
/ =1-/ [Ml]
71 72 [М11+[М2[
и
Р ___ Л р _____ d [Ml]
1- 2 — d [MiJ + d [М2] •
(6.13)
(6.14)
Комбинация уравнений (6.13) и (6.14) с уравнением (6.12) дает
р Г1^ + /1/2
1 Г1/1 + 2/1/2 + г2/2
(6.15)
Уравнение (6.15) выражает состав сополимера как мольную долю
мономера М\ в сополимере, и часто им бывает более удобно поль-
зоваться, чем уравнением (6.12).
*) Коэффициенты сополимеризации rt и г2 часто называют также отно-
сительными активностями мономеров.— Прим. ред.
Сополимеризация
339
6.26. Пределы применимости уравнений
сопо л име ризации
Уравнение сополимеризации экспериментально проверено для
огромного числа мономерных систем. Оно в равной степени при-
менимо к радикальной, катионной и анионной полимеризациям,
хотя значения констант сополимеризации и г2 Для каждой пары
мономеров могут очень сильно различаться в зависимости от типа
инициирования. Так, например, они соответственно равны 0,52
Р и с. 6.1. Зависимость «мгновен-
ного» состава сополимера 1-\ от
состава смеси мономеров для
системы стирол — метилметакри-
лат при катионной (1), радикаль-
ной (2) и анионной (3) сополиме-
ризации, инициируемой SnCI4,
перекисью бензоила и натрием в
жидком аммиаке соответственно
[И].
Содержание стирола в смеси
мономеров fi, мол. доля
и 0,46 при радикальной сополимеризации, 10Ju 0,1 при катион-
ной и 0,1 и 6 при анионной [10, 11]. Данные, приведенные
на рис. 6.1, показывают, что различия значений Г! и г2 приводят
к большим различиям в составе сополимера в зависимости от типа
инициирования [11]. Ионная сополимеризация всегда значительно
более селективна, чем радикальная сополимеризация. Метил-
метакрилат, как и можно было ожидать, активнее в анионной
сополимеризации и весьма мало активен в катионной, тогда как
стирол — наоборот. Так, например, состав сополимера, полу-
ченного из эквимолярной смеси стирола и метилметакрилата,
примерно 1 : 1 при радикальной сополимеризации, при катионной
сополимеризации сополимер содержит в основном стирол, а при
анионной — метилметакрилат. Высокая селективность ионной
сополимеризации ограничивает ее практическое применение,
так как лишь незначительное количество мономеров способно
к ионнои сополимеризации, а следовательно, число сополимерных
продуктов, которые могут быть получены, весьма ограниченно.
С другой стороны, практически все мономеры могут участвовать
в радикальной сополимеризации, что дает возможность синтези-
ровать множество разнообразных сополимеров.
340 Глава 6 1
1
Для всех типов инициирования (радикального, катионного;
или анионного) величины относительной активности мономеров
и, следовательно, состав сополимера не зависят от многих пара-
метров реакции. Поскольку константы скоростей инициирования:
и обрыва не входят в уравнение сополимеризации, состав сополи-
мера не зависит от различий в скоростях этих стадий, а также
от отсутствия или наличия ингибиторов и веществ, способных
к передаче цепи. Для широкого диапазона условий состав сополи-
мера не зависит от степени полимеризации. Единственным усло-
вием этого общего положения является то, что сополимер должен
обладать высоким молекулярным весом. Кроме того, состав
сополимера не зависит от конкретного выбора инициатора при
условии, что механизм инициирования остается тем же. Так,
сополимер имеет одинаковый состав, если он получен радикаль-
ной сополимеризацией, независимо от того, как осуществлялось
инициирование: термическим разложением таких катализаторов,
как азо-бмс-изобутиронитрил или перекиси, фотолизом, радио-
лизом или с помощью окислительно-восстан >вительных систем.
6.2в. Типы сополимеризации
Наблюдаются различные типы сополимеризации в зависимости
от значений констант сополимеризации мономеров. Процессы
сополимеризации можно подразделить на три типа в зависимости
от величины произведения констант сополимеризации. Для пер-
вого типа произведение rtr2 равно 1, для второго оно больше 1
и для третьего — меньше 1.
6.2в.1, Идеальная сополимеризация: = 1
Идеальной называется сополимеризация, для которой г(г2 = 1.
Идеальная сополимеризация наблюдается, когда обе растущие
частицы, М* и М*, обладают одинаковой способностью присоеди-
нять оба мономера. Для этих условий
^22 ___ ^12
Zc2i кц
1
ИЛИ Л> =------
Г!
(6.16)
и относительные скорости включения мономера в сополимер
не зависят от строения звена, находящегося на конце растущей
молекулы. Для идеальной сополимеризации при комбинировании
уравнений (6.16) с (6.12) или (6.15) получается уравнение сополи-
меризации в виде
или
d[M±] _ MMd
d[M2] [М2]
р____rif 1
‘“пШ ’
(6-17)
(6.18)
Сополимеризация
341
Ит еальный тип сополимеризации наиболее часто встречается при
ионной (как анионной, так и катионной) сополимеризации.
В том случае, когда = r2 = 1, оба мономера имеют одина-
ковую активность по отношению к обеим растущим частицам. Сос-
тав сополимера остается тем же самым, что и состав исходной сме-
си мономеров, причем распределение мономерных звеньев вдоль
цепи сополимера носит случайный, статистический» характер.
Если константы сополимеризации различны, т. е. о > 1, arj< 1
или ft < 1, а гz > 1, один из мономеров обладает более высокой
Содержание МА в смеси моно-
меров fj, мол. доля
активностью по отношению к обеим растущим молекулам. Сополи-
мер в этом случае содержит больше звеньев более активного
мономера, расположенных в цепи опять-таки статистически.
На рис. 6.2 приведена зависимость состава сополимера от
исходной смеси мономеров при различных значениях [12].
Термин «идеальная сополимеризация» был введен Уоллом [5] для
того, чтобы показать аналогию между кривыми, изображенными
на рис. 6.2, и кривыми равновесия между жидкостью и паром
в идеальных смесях жидкостей. В случае, когда rt >• 1, полимер
богаче мономером М15 а при г4 < 1 мономера Mj в полимере мень-
ше. Важное практическое следствие идеальной сополимеризации
состоит в том, что с увеличением различия в величинах относи-
тельной активности мономеров все труднее получить полимер,
содержащий заметное количество обоих мономеров. Это является
одной из причин того, что ионная сополимеризация имеет сравни-
тельно небольшое практическое применение. Например, при
ri — U и г2 = 0,1 полимер, содержащий заметное количество М2,
получить нельзя. Гак, из исходной смеси мономеров, содержащей
мол. /о М2 (/2 = 0,8), образуется сополимер, содержащий только
, мол. /о М2 (F2 = 0,185). Только при незначительной разнице
342
Глава 6
между rt и г2 (например, при п = 0,5—2) из исходной смеси
мономеров различного состава получаются полимеры, содержащие
значительные количества обоих мономеров.
6.2в.2. Сополимеризация, приводящая к образованию
регулярно чередующихся сополимеров: г1 = г2 = О
При условии щ = г2 = 0 (и одновременно г{г2 = 0) оба моно-
мера входят в состав сополимера в эквимолярных количествах,
и при этом звенья располагаются упорядоченно, чередуясь вдоль
полимерной цепи. Сополимеры этого типа называются регулярно
чередующимися. Растущая частица каждого из двух типов преиму-
щественно присоединяет молекулы другого мономера, т. е. М*
присоединяет только М2, a М* присоединяет только Мр Уравнение
сополимеризации в этом случае упрощается
или
Л =0,5. (6.20)
Сополимер имеет чередующуюся структуру I независимо от соста-
ва исходной смеси мономеров. Реакция радикальной сополимери-
зации во многих случаях имеет тенденцию к протеканию по это-
му типу.
На практике большинство сополимерных систем занимает про-
межуточное положение между этими предельными случаями:
идеальной и чередующейся сополимеризацией. При уменьшении
величины произведения т\г2 от единицы до нуля возрастает
тенденция к чередованию звеньев мономеров. Наиболее совершен-
ное чередование происходит, когда и щ и г2 равны нулю. Стрем-
ление к чередованию увеличивается тем больше, чем меньше
становятся значения щ и г2. Различные типы сополимеризации
можно наблюдать, рассматривая процесс, в котором г2 остается
постоянной и равной 0,5, a п уменьшается от 2 до 0. На рис. 6.3
представлена зависимость состава сополимера от состава исход-
ной смеси в этих случаях. Кривая для г1 = 2 соответствует идеаль-
ному типу сополимеризации, описанному ранее. При уменьшении
П возрастает тенденция к преимущественному присоединению
растущими частицами любого типа другого мономера. Увеличе-
ние тенденции к чередованию определяется степенью приближен-
ности произведения щг2 к нулю. Большое практическое значение
имеет тот факт, что сополимер, содержащий значительные коли-
чества обоих мономеров, может быть получен из мономерных сме-
сей разнообразного состава. Однако, когда произведение rtr2 очень
мало или равно нулю, стремление к чередованию становится слиш-
ком большим и число возможных составов сополимера опять ста-
Сополимеризация
343
новится ограниченным. В предельном случае, когда и п и г2
равны нулю, можно получить лишь регулярно чередующийся
сополимер состава 1.1.
Кривые, представленные на рис. 6.3, иллюстрируют интерес-
ную особенность реакций сополимеризации, имеющих тенденцию
к образованию чередующихся сополимеров. Если обе величины —
г и Г2__меньше единицы, то кривые зависимости от /1 пере-
секают прямую, соответствующую случаю 1'\ = В точках пере-
сечения составы сополимера и смеси мономеров одинаковы, и сопо-
лимеризация протекает без изменения состава исходной смеси
Рис. 6.3. Зависимость «мгновен-
ного» состава сополимера от
состава смеси мономеров для
указанных значений г, при
г2= 0,5 [6].
мономеров. Такой вид процесса называют азеотропной сополиме-
ризацией. Математически условия, при которых протекает азео-
тропная полимеризация, можно выразить путем комбинирования
уравнения (6.12) с уравнением
d[Mt] [Mt]
d[M2] - [М2] ’
что соответствует равенству
(MJ _ (r2—1)
[М21 (п-1) ’
(6.21)
или в мольных долях
. (1-г2)
71 (2 — г1 — г2) •
(6.22)
Особое положение наблюдается, когда одна из констант сополи-
меризации значительно больше другой. Так, при т\ г2 (т. е.
Л^>1 и r2 1) растущие частицы обоих типов присоединяют пре-
имущественно мономер Mj и имеется тенденция к последователь-
ной гомополимеризации] обоих мономеров. Мономер М\ будет
344 Глава 6
полимеризоваться, до тех пор пока не израсходуется, после
чего начнет гомополимеризоваться мономер Мг. Пример такого
типа сополимеризации — радикальная сополимеризация стирола
с винилацетатом, константы сополимеризации которых 55 и 0,01
(разд. 6.36.1 и 6.3в.1).
а.2в.З. Блочная сополимеризация: г1, r2Z> 1
Если и т\ и г2 больше единицы (и, следовательно, пг2 > 1), то
наблюдается тенденция к образованию блок-сополимера (струк-
тура II), в цепи которого имеются блоки обоих мономеров. Этот
тип сополимеризации встречается лишь в немногих случаях сопо-
лимеризации, инициируемой координационными катализаторами
(разд. 8.4г.2). Предельный случай, когда и и г2 много больше
единицы, т. е. когда протекает одновременная и независимая гомо-
полимеризация двух мономеров, наблюдается только в одной или
двух системах [13].
6.2г. Изменение состава сополимера в зависимости
от степени превращения
Различные формы уравнения сополимеризации [(6.12) и (6.15)}
соответствуют «мгновенному» составу сополимера, т. е. составу
сополимера, образовавшегося из данной смеси исходных мономе-
ров при очень малых степенях превращения (<5%), при которых
состав мономерной смеси мало отличается от состава исходной сме-
си мономеров. Во всех случаях сополимеризации, кроме азеотроп-
ной сополимеризации, состав сополимера отличается от состава
смеси мономеров. Состав смеси мономеров изменяется, так как
один из мономеров реагирует быстрее и входит в состав макро-
молекулы в большем количестве. Так, с увеличением степени
превращения происходит накопление в мономерной смеси менее
реакционноспособного мономера. Это приводит к аналогичным
превращениям и в составе сополимера с увеличением степени
превращения. Для того чтобы определить «мгновенный» состав
сополимера, т. е. состав сополимера в данный момент времени, как
функцию степени превращения для каждой данной смеси мономе-
ров, нужно пользоваться интегральной формой уравнения сопо-
лимеризации. Различные попытки прямого интегрирования урав-
нения (6.12) не дали положительных результатов [4, 5]. Интег-
ральная форма уравнения (6.12), выведенная Майо и Льюисом [4],
была недавно преобразована [14].
Наиболее часто пользуются методом расчета, разработанным
Скейстом [15], а также некоторыми вариациями этого метода [16,
17]. Рассмотрим систему, содержавшую первоначально общее
Сополимеризация
345
количество М молей обоих мономеров, в которой образующийся
сополимер содержит больше звеньев мономера М1? чем исходная
смесь мономеров (т. е. Ft > /t). Когда dM молей мономеров
вступит в реакцию сополимеризации, сополимер будет содержать
FidM молей мономера 1, а смесь мономеров будет содержать
(j/__dM) (/i — dh) молей этого мономера. Материальный баланс
по мономеру 1 требует, чтобы число молей этого мономера, всту-
пившее в реакцию сополимеризации, было равно разнице числа
молей его в смеси мономеров до и после реакции, или
Mfi -(М- dM) (fi - dfi) = FidM. (6.23)
Уравнение (6.23) можно преобразовать (пренебрегая членом
dfidM, величина которого мала) и привести к интегральной формя
м ft
f dM , М С dft
J тг=1"-к= ) (6'24)
Мо (/1)0
где Мо и (Л)о — исходные значения М и
Уравнение (6.15) позволяет вычислить значение Ft как функ-
цию ft для данной пары и г2- Полученные данные затем можно
использовать в виде (Ft — ft) для графического или числового
интегрирования уравнения (6.24) в пределах от (/i)0 до Д. Таким
образом, можно получить изменения состава смеси мономеров
и состава сополимера в зависимости от степени превращения
(которая определяется как 1 — М/Мо).
Недавно была предложена [16, 17] более простая форма инте-
грирования уравнения (6.24)
<б-25>
которая связывает глубину превращения с изменениями состава
мономерной смеси. Индекс «нуль» относится к исходным количе-
ствам, а другие символы имеют следующие значения:
«=т^.
_ (1 —Г1Г2) с (1 -г2)
(1 — rj)(l—г2) ’ (2-4—г2)'
Уравнение (6.25) было использовано для вычисления изменений
в составе смеси мономеров и составе сополимера в ходе реакции.
ычисления легко провести с помощью вычислительной машины,
причем программу можно составить так что результаты будут
получены прямо в виде графиков [17].
Гб 24^СК?к 9^0 ПРИМСРОВ иллюстрируют применимость уравнений
м₽п ч И Ь’^)’ На рис. 6.4 приведен пример радикальной сополи-
ри ации стирола с метилметакрилатом. Образующийся сополи-
346
Глава 6
мер несколько богаче метилметакрилатом, чем исходная смесь
мономеров, так как относительная активность метилметакрилата
Рис. 6.4. Зависимость
«мгновенного» состава сме-
си мономеров и сополиме-
ра от степени превращения
для системы стирол (MJ —
метилметакрилат (М2) при
(А)о = 0,80, (/2)0 = 0,20 и
г, = 0,53, г2 = 0,56 [6].
немного больше, чем стирола. По мере увеличения степени пре-
вращения смесь мономеров обогащается стиролом, что ведет к воз-
Р и с. 6.5. Зависимость «мгно-
венного» состава сополимера
Ft от состава исходной смеси
мономеров fi и степени превра-
щения для системы стирол
(М,) — 2-винилтиофен (М2) при
г, = 0,35 и г2 = 3,10 [6].
растанию содержания стирола в сополимере при высоких степенях
превращения. На рис. 6.4 показан также средний состав всего
сополимера, соответствующий определенной конверсии, как функ-
Сополимеризация
347
пия степени превращения. Сополимер среднего состава содержит
больше стирола, чем метилметакрилата, но все же меньше, чем
образец «мгновенного» состава.
Содержание стирола в сополи-
мере , мол. доля
Рис. 6.6. Распределение
сополимера по составу при
100%-ной степени превра-
щения для системы сти-
рол — 2-винилтиофен при
указанных значениях содер-
жания стирола в исходной
смеси мономеров [7].
Изменение состава сополимера в зависимости от степени пре-
вращения для различных смесей сомономеров можно представить
на трехмерном графике, пример которого приведен на рис. 6.5 для
Р и с. 6.7. Зависимость
«мгновенного» состава сопо-
лимера от состава исход-
ной смеси мономеров fa и
степени превращения для
системы стирол (Mj) — ди-
этилфумарат (М2) при г.=
= 0,30 и г2 = 0,07 [6].
si
£ е
| § 5
5 2
чо *
а
1,0
0,8
0,6
0,4
00
0,2
0
1,0
/ 0,8
X
о/о
Xх
а
е
и
процесса радикальной сополимеризации стирола (МО с 2-винил-
™°Феном (М2). Это идеальная сополимеризация с = 0,35 и г2 =
— , U. Вследствие большей реакционной способности 2-винил-
348
Глава 6
тиофена сополимер, образующийся на первых стадиях реакции,,
оказывается обогащенным этим мономером. По мере протекания
реакции смесь мономеров, а вследствие этого и сополимер, все
в большей степени обогащаются стиролом. Это показано на рис. 6.6,
который иллюстрирует распределение сополимеров по составу при
100%-ной степени превращения для нескольких различных исход-
ных смесей.
Соответствующие данные для процесса образования чередую-
щихся сополимеров при радикальной сополимеризации стирола
&100
<8
I
8
5г
5г
сз
§
S
е
8
5J
51
£
8-
"5
О
S’
о
(^0=0,80
(^0=0,60
80 -
60 -
40 -
20
О
80
60 -
40
20
О
80
60
40
20
О 0,2 0,4 0,6 0,8 0 0,2 0,4 0,6 0,8 1,0
(^0^0,57
(fl)o=O,5O
(^0=0,40
(^0=0,20
(М^ с диэтилфумаратом
(М2) при = 0,30 и гг =
= 0,07 приведены на рис.
6.7 и 6.8. Эта система под-
вергается азеотропной со-
полимеризации при содер-
жании стирола 57 мол.%.
Если состав исходной смеси
мономеров близок к азеот-
ропному составу, то рас-
пределение сополимеров по
составу получается очень
узким; только при высоких
степенях превращения на-
блюдается сдвиг в сторону
чистого полистирола или
Содержание стирола в сополи-
мере Ft, мол. доля
Рис. 6.8. Распределение по
составу сополимера при 100%-
ной степени превращения для
системы стирол — диэтилфума-
рат при указанных значениях
содержания стирола в исход-
ной смеси мономеров [7].
чистого полифумарата в зависимости от того, меньше или больше
57 мол. % стирола содержит исходная смесь мономеров. Распре-
деление сополимера по составу становится все шире по мере от-
клонения состава исходной смеси от «азеотропного» состава.
Для практического применения продуктов сополимеризации
обычно желательно иметь сополимеры с возможно более узким
распределением по составу. Для достижения этой цели применяют
два метода. Первый заключается в прекращении процесса сополи-
меризации до достижения 100%-ной конверсии, а второй —
в поддержании постоянного состава смеси мономеров путем не-
прерывного добавления более реакционноспособного мономера.
Сополимеризация
349
Согласно этим методам, для получения продукта с нужными свой-
ствами необходимо определение требуемого распределения сополи-
меров по составу (например, рис. 6.6 или 6.8).
6.2д. Экспериментальное определение констант
сополимеризации
Все способы определения и г2 заключаются в эксперимен-
тальном определении состава сополимера, полученного из не-
скольких различных смесей сомономеров. Для анализа сополиме-
ров используют введение меченых атомов, а также такие методы,
как элементный анализ, ультрафиолетовая, инфракрасная и ЯМР-
спектроскопия.
При более старом, но широко используемом методе определе-
ния констант сополимеризации процесс проводят при малых сте-
пенях превращения (менее 5%). Полученные экспериментальные
данные можно анализировать несколькими путями. Один путь
заключается в нанесении на график экспериментально получен-
ных значений состава сополимера в зависимости от соотношения
мономеров в исходной смеси и подборе методом проб и ошибок
«лучших» значений г( и г2, причем для сравнения пользуются тео-
ретической кривой. Это очень трудоемкий путь, и обычно бывает
проще решить одно из уравнений — (6.12) или (6.15). При этом
точность полученных значений и г2 зависит от более или менее
удачного выбора соотношения исходных мономеров, которое дол-
жно быть наиболее чувствительно к изменению значений г. При
втором методе [4] определения констант г1 и г2 используют урав-
нение (6.12) в следующем преобразованном виде:
_ _[Mj] Гй [М2] [. , ri Я Гб 27^
Данные о составе сополимера и смеси мономеров для каждого опы-
та с заданными значениями [MJ и [М2] подставляют в уравнение
(6.27) и г2 выражают в виде функции (при произвольно выбран-
ных значениях последней). По данным каждого опыта строят
прямую линию. Пересечение этих прямых, полученных при разных
соотношениях мономеров, дает величину «наилучших» значений
и г2. Отклонения, наблюдаемые для точек пересечения этих пря-
мых, являются мерой ошибки эксперимента при получении данных
по составу сополимера и смеси мономеров.
Олень полезный метод [18] основан на использовании уравне-
ния (6.15), преобразованного в виде
ависимость левой части этого уравнения от коэффициента при г}
дает графически прямую линию, тангенс угла наклона которой
350
Глава 6
соответствует rlt а пересечение с осью ординат дает г2. Использо-
вание вычислительной техники позволило обсудить относительные
достоинства различных способов анализа данных по составу сопо-
лимера для определения наилучших значений и r2 [49].
Значения и г2 были недавно определены из данных по составу
сополимера в зависимости от глубины реакции. В работе [20] было
использовано приведенное выше уравнение (6.25). Эксперимен-
тальные данные по изменению состава сополимера в зависимости
от степени превращения выражают в виде зависимости или /2 от
1—(М/Мо), причем получаются кривые, подобные кривым, приве-
денным на рис. 6.4. При помощи вычислительной техники получа-
ют наилучшие значения г1 и г2, которые при подстановке в урав-
нение (6.25) должны давать ту же кривую.
6.2е. Микроструктура сополимеров
Уравнение сополимеризации описывает состав сополимера на
макроскопическом уровне, т. е. суммарный состав образца сопо-
лимера, полученного из данной смеси мономеров. При этом оста-
ются невыясненными два вопроса относительно микроструктуры
сополимера.
Первый вопрос касается точного распределения звеньев обоих
мономеров вдоль полимерной цепи. В каждом частном случае
средний состав сополимера слагается из многих различных микро-
структур. Так, например, сополимер состава Д = 0,6, /2 = 0,4
может иметь регулярную структуру
— М1М1М1М2М2М1М1М1М2М2М1М1М1М2М2М1М1М1М2М2 — ,
статистическую структуру
— М1М2М1М2М2М1М1М1М1М2М2М1М1М2М1М1М2М1М1М2 —
или структуру блок-сополимера
— MiMiMiMiMxMiMiMiMiMiMiMiMaMaMaM.jM.iM.iMgM.i — .
Статистический анализ уравнения сополимеризации предска-
зывает, что процесс сополимеризации, как правило, протекает
с образованием статистического сополимера. Тенденция к образо-
ванию продуктов с регулярно чередующимися звеньями наблюдает-
ся только при использовании сомономеров, у которых произве-
дение rtr2 близко к нулю. Блок-сополимеры образуются лишь при
определенных условиях (разд. 6.2в.З). (Возможность получения
длинных блоков из звеньев одного мономера существует также
в том случае, если смесь исходных мономеров содержит в большом
количестве более реакционноспособный мономер.)
Сополимеризация
351
Хотя эти теоретические выводы общеприняты, эксперименталь-
ное подтверждение статистической природы сополимеризации
достаточно сложно. Оно требует экспериментального определения
относительных количеств трех возможных типов связей между
звеньями мономеров в сополимере (а именно Mt — М2 — М2,
д! — М2) и сравнения их с относительными количествами этих
типов связей, вычисленными на основе статистического распре-
деления. Для некоторых сополимеров оказались эффективными
химические методы. Используют, например, образование лакто-
нов при гидролизе сополимеров винилацетата с акриловой кисло-
той или малеиновым ангидридом, дехлорирование цинком сопо-
лимеров винилхлорида с винилацетатом, а также окисление
гидролизованных сополимеров винилацетата с виниленкарбона-
том. Эта последняя реакция приводит к образованию альдегид-
ных концевых групп и муравьиной кислоты
Н1О4
— СН2—СН —( —СН —СН —)т —СН2 —СН----------»
ОН Ан Ан Ан
—» — СН2—СН + (2т-1)НСООН4-НС —СН2-СН — (6.29)
п 11 1
о о он
Число звеньев виниленкарбоната, связанных с другими звень-
ями той же структуры (т. е. связи Mi— Mi), определяют по коли-
честву муравьиной кислоты [21]. В последнее время для подтвер-
ждения статистического распределения мономерных звеньев в со-
полимере используется ядерно-магнитный резонанс высокого раз-
решения [22, 23]. Так, методом ЯМР были определены относитель-
ные количества связей Mi — Mi, М2 — М2 и Mt — М2 в сополимере
этилена с винилхлоридом (анализ различного типа протонов ме-
тиленовой группы, характерных для каждой связи). Во всех слу-
чаях экспериментальные данные подтверждают статистической
распределение звеньев мономеров в сополимере.
Вторым неясным вопросом, касающимся композиционной одно-
родности сополимера, является возможность присутствия в об-
разце сополимера, полученного при любой данной степени прев-
ращения, макромолекул различного состава. Стокмайер [24] ука-
зал, что распределение сополимера по составу обычно соответствует
очень узкой гауссовой кривой практически для всех сополи-
меров, кроме низкомолекулярных сополимеров и идеальных
случаев сополимеризации, когда распределение довольно широ-
кое по сравнению с распределением сополимеров с чередующи-
мися звеньями. Например, вычислено, что в идеальном сополи-
мере^ содержащем в среднем 50 мол. % каждого компонента,
при Хп = ЮО 12% макромолекул содержат менее 43% любого
г
Глава 6
гомеров, тогда как при Хп = 10 000 только 12% молекул
х»держат менее 49% любого мономера. Эти теоретические выводы
Стокмайера были экспериментально подтверждены в работе [25].
6.2ж. Многокомпонентные системы
Терполимеризация (совместная полимеризация трех мономе-
ров) очень важна с практической точки зрения. Выше уже были
отмечены те преимущества, которые дает сополимеризация стиро-
ла с акрилонитрилом или бутадиеном. Радикальная терполпмери-
зация стирола с акрилонитрилом и бутадиеном создает еще больше
возможностей изменения свойств конечных продуктов. Существует
и много других практических путей использования терполимери-
зации. В большинстве случаев два сомономера присутствуют в со-
полимере в больших количествах, придавая ему основные требуе-
мые свойства, а третий сомономер вводят в небольших количествах
для модификации в каком-либо специфическом направлении. На-
пример, разработанные недавно этиленпропиленовые эластомеры
терполимеризуют с некоторым количеством диена для получения
продукта с небольшим числом поперечных связей.
Количественная обработка данных терполимеризации очень
сложна, так как для этого процесса необходимо учесть скорости
девяти реакций роста
Реакция Скорость реакции
Mf- -J-Mf -> Mt-
Mj- +м2->м2-
Mi.'-M3->M3-
M2- -j-Ml -> Ml-
Мг • -J- M2 —> M2 •
M2- -j-Mg—fr-Mg-
M3-+M1^M1-
Mg - -)-M2—>M2-
Mg- 4-Мз^Мз-
/?и = кц [Mi-] [Mt]
T?i2 = ki2 [Mj-] [Ma]
^13 = ^13 [Ml • ] [Mg]
= ^21 [M2- ] [Ml]
Й22 = k22 [M2 • ] [M2]
R23 = &23 [M2 • ] [Mg]
Kg» — ^31 [Mg• ] [MJ
К32 = ks2 [M3 • ] [M2]
K33 = &зз[Мд-] [Mg]
(6.30)
и шесть констант сополимеризации мономеров
&22
Г2,“^Г’
Г — кз3
г32 — р~ •
#32
^11
rl2=w
_ к22
г2з — р~ ’
й23
'is —~r~ 1
K13
r ksa
r3l~k3i ’
(6.31)
Выражение для состава терполимера можно, получить аналогич-
но тому, как это сделано в случае двойных сополимеров [27]. Ско-
Сополимеризация
353
оостп исчерпания трех мономеров выражаются уравнениями
d[MJ_ dt = 2?п-]- J?21 + Rsi, (6.32a)
d [М2] _ dt = R12R?2 + ^32, (6.326)
d [M8] dt = R13 + ^23 + Rs3- (6.32b)
Допущение постоянства концентраций радикалов Mf-> М2-и М3-
можно выразить как
Л12 + 2?1з = +^3i, (6.33а)
^?21 + ^23 —' ^12 + -^32> (6.336)
7?з1Ч Йз2-^з + В23. (6.33b)
Совместное решение уравнений (6.32) и (6.33) при использовании
соответствующих выражений для скоростей индивидуальных
реакций R из уравнений (6.30) приводит к следующему сложному
уравнению состава сополимера:
d [MJ : d [М2] : d [М3] =
I /-31^21 Г21Г32 Г31Г23 J Г12 Г13 J
+ ([Мг]+ж+т :
UT2T31 r12r32 r32r13j ll * * a r21 r23 J
: ЫМ + М + ЖШнЖ + Ш . (6.34)
l Г13'21 r23ri2 r13r23 J I 3J T31 r32 J V '
Более простое уравнение для состава терполимера было получено
[28] при выражении условия постоянства концентрации ниже-
следующими зависимостями
Т?12 = К21, (6.35а)
•^23 = -^321 (6.356)
7?3i = J?13 (6.35в)
вместо зависимостей, приведенных в уравнении (6.33). Комбини-
рование уравнений (6.35) и (6.32) приводит к выражению для
состава сополимера в следующем виде:
d [М J : d [М2]: d [М3] = [MJ {[MJ } :
1 4 21 r23 J r13 r31 r32 J
(6.36)
Хэм [29, 30] предположил, что вероятность образования цепочки
из определенной последовательности мономепов опинякопя n ют-
354
Глава 6
чае как прямого, так и прямо противоположного чередования
мономеров. Это можно выразить как равенство двух произведе-
ний вероятностей Р, например
Р цР 13Р = Р 23Р si.P а (6.37)
для случая образования последовательностей мономеров M2MiM3M2
и M2M3MiM2. Член Р характеризует вероятность того, что опреде-
ленная растущая цепь будет присоединять определенный мономер.
Например, — вероятность того, что растущая цепь, имеющая
на конце Mi, будет присоединять мономер М2, выражается урав-
нением
р _ Д12_______
12 Pi 1 + Рц + Р13'
(6.38)
Традиционное уравнение терполимеризации (6.34) можно выра-
зить теперь в терминах вероятности
(6.39a)
(6.396)
d [Mi] _ Р21Р31 + Р32Р21 + Р23Р31
d [М2] Р12Р32 + Р31Р12 + Р13Р32 ’
d [MJ____P21P31 + P32P21 + P23P31
d [М3] P13P23 + P21P13 + P12P23
и, преобразовав затем соответствующим образом, получить в еле-
дующем виде:
[Mt] J ГЛ1 1 1. [М2] ] [М3] I
d[Mi] r21 s iiviij -f- l r12 ПЗ J
d [М2] [M2] |1MiL + [m2] + [М3] 1
Г12 l Г21 r23 J
[Mil [М3] -I
d [Mi] Г31 ( 1 y. 1 I r12 Г13 J
d[M3] [M3] r 13 {^-^U[M3]} l Г31 r32 J
Рассмотрение уравнений (6.36) и (6.40) показывает их полную
равноценность.
Традиционное уравнение (6.34) и упрощенное уравнение [((>.36)
или (6.40)] терполимеризации можно использовать для предсказа-
ния состава терполимера, исходя из значений констант сополиме-
ризации мономеров в двухкомпонентных системах Mi/M2, М1/М3
и М2/М3. В табл. 6.1 приведены рассчитанные таким образом
и определенные экспериментально составы терполимеров для
некоторых трехкомпонентных систем. Составы, вычисленные по
любому из уравнений терполимеризации, хорошо согласуются
с составами, определенными экспериментально [31]. Ни одно из
этих уравнений не имеет в этом отношении преимуществ. Оба
уравнения были успешно распространены на многокомпонентный
системы из четырех и более мономеров [28, 31—33]. Совпадение
Сополимеризация
355
вычисленных и экспериментально определенных составов сополи-
меров для систем из четырех мономеров также показано в табл. 6.1.
Таблица 6.1
;азанные и определенные экспериментально составы сополимеров,
полученных радикальной терполимеризацией [28, 31]
Преды
Состав исходной смеси мономеров Состав терполимера, мол.%
система мономер содержание, мол. % определено эксперимен- тально вычислено по уравнению
(6.34) (6.36) или (6.40)
1 Стирол 31,24 43,4 44,3 44,3
Метилметакри- лат 31,12 39,4 41,2 42,7
Винилидеи- хлорид 37,64 17,2 14,5 13,0
2 Метилметакри- лат 35,10 50,8 54,3 56,6
Акрилонитрил 28,24 28,3 29,7 23,5
Винилиден- хлорид 36,66 20,9 16,0 19,9
3 Стирол 34,03 52,8 52,4 53,8
Акрилонитрил 34,49 36,7 40,5 36,6
Влнилпден- хлорид 31,48 10,5 7,1 9,6
4 Стирол 35,92 44,7 43,6 45,2
Метилметакри- лат 36,03 26,1 29,2 33,8
Акрилонитрил 28,05 29,2 26,2 21,0
5 Стирол 20,00 55,2 55,8 55,8
Акрилонитрил 20,00 40,3 41,3 41,4
6 Винилиден- хлорид 60,00 4,5 2,9 2,8
Стирол 25,21 40,7 41,0 41,0
Метилметакри- лат 25,48 25,5 27,3 29,3
Акрилонитрил 25,40 25,8 24,8 22,8
Винилиден- хлорид 23,91 8,0 6,9 6,9
(С позиций Хэма оказывается возможным предсказать ход сопо-
лимеризации системы Mj/Мз, исходя из данных для систем Mj/Мг
и М2/М3; см. разд. 6.36.5.)
356
Глава 6
Уравнения состава трехкомпонентных и многокомпонентных
систем поддаются решению, как правило, только в тех случаях,
когда все константы сополимеризации имеют конечные значения.
Если же хотя бы один из мономеров не способен к гомополимери-
зации, уравнения обычно становятся неопределенными. Для неко-
торых частных случаев сополимеризации были выведены различ-
ные видоизмененные выражения, базирующиеся как на традицион-
ном, так и упрощенном видах уравнений.
6.3. Радикальная сополимеризация
В предыдущих разделах речь шла об общих положениях про-
цесса сополимеризации, без учета механизма роста цепи. Рас-
смотрим теперь некоторые специфические особенности радикаль-
ной сополимеризации.
6.3а. Влияние условий реакции
В дополнение к известному факту независимости констант
сополимеризации мономеров от стадий инициирования и обрыва
цепи было показано, что при радикальной сополимеризации эти
константы почти совсем не зависят и от реакционной среды. Так,
например, значения г и состав сополимера для системы стирол —
метилметакрилат не меняются при проведении реакции в блоке,
эмульсии или в таких разных по диэлектрической проницае-
мости растворителях, как бензол и ацетонитрил, или даже
в метаноле, в котором сополимер нерастворим [6]. При эмульсион-
ной [26] или суспензионной полимеризации иногда образуются
сополимеры, состав которых отличается от состава сополимера,
полученного в блоке или растворе; такое явление наблюдается,
когда состав сополимера в месте реакции (капельки мономера при
суспензионной полимеризации и мицеллы при эмульсионной)
отличается от состава в общем объеме реакционной системы. Это
может происходить в следующих случаях: один из мономеров
имеет ощутимую растворимость в суспендирующей среде; один из
мономеров предпочтительно адсорбируется образующимся в ходе
реакции полимером; диффузия одного из мономеров в мицеллы
при эмульсионной полимеризации происходит слишком медленно.
Значения констант сополимеризации мономеров в этих системах
остаются неизменными, а расхождения в составе сополимера
обусловлены просто изменением значений [MJ и [М2] в месте
реакции. Эти явления необходимо учитывать на практике, когда
нужно получить сополимер определенного состава.
Влияние температуры на константы сополимеризации мономе-
ров относительно мало [34]. Например, значения и г2 для сопо-
лимера стирола с метилметакрилатом равны 0,52 и 0,46 при 60 °C
Сополимеризация
357
О 59 и 0,54 при 131 °C. Константа сополимеризации — это
отношение двух констант скорости роста, а поэтому изменение г
с температурой зависит от разницы энергий активации этих двух
процессов. Этот эффект невелик, поскольку энергии активации
радикальных реакций роста относительно малы, и различия между ,
значениями их для разных реакций обычно незначительны.
Повышение температуры приводит к уменьшению селективности
сополимеризации, так как константы сополимеризации обоих
сомономеров приближаются к единице (т. е. = r2 = 1) и пред-
почтительность присоединения определенных мономеров к опре-
деленным радикалам уменьшается. Наибольшее влияние темпе-
ратура оказывает на те системы, у которых значения г заметно
отличаются от единицы.
Хотя процессы полимеризации и сополимеризации при высоком
давлении имеют промышленное значение, данных о влиянии дав-
ления на константы сополимеризации в литературе очень мало.
Повышение давления уменьшает селективность сополимеризации,
изменяя значения г в направлении идеальной сополимеризации
[35], т. е. давление действует так же, как и температура. Напри-
мер, для сополимера метилметакрилата с акрилонитрилом про-
изведение равно 0,16 при 1 атм (9,8-104 Па), 0,54 при 100 атм
(9,8-106 Па) и 0,91 при 1000 атм (9,8-Ю7 Па). Этот эффект был
приписан увеличению реакционной способности радикала без
изменения реакционной способности мономера. В ряде других
систем, в том числе и при сополимеризации этилена, применение
высокого давления сдвигает процесс в направлении условий
идеальной сополимеризации [36]. Однако для некоторых систем
при увеличении давления получены значения rtr2 2—3 [37].
6.36. Реакционная способность
Константы сополимеризации для многих мономеров при ради-
кальной сополимеризации приведены в табл. 6.2. Эти данные поле-
зны для изучения зависимости между строением и реакционной
способностью в реакциях радикальной полимеризации [38]. Ак-
тивность мономера по отношению к радикалу зависит как от
активности мономера, так и от активности радикала. Относитель-
ные активности мономеров и соответствующих им радикалов
можно вычислить при анализе данных по константам сополимери-
зации мономеров, рассматривая величину, обратную константе
сополимеризации (1/г). Эта величина равна отношению скорости
реакции радикала с другим мономером к скорости реакции его
со своим собственным мономером
1 __ ^12
~ кп
(6.41)
Таблица 6.2
Константы сополимеризации мономеров при радикальной
сополимеризации [38]
Ml Г1 м2 Г2 Г, °C
Акриловая кисло- 1,15 Акрилонитрил 0,35 50
та 0,25+0,02 Стирол 0,15+0,01 60
2 Винилацетат 0,1 70
Акрилонитрил 0,35 Акриловая кисло- 1,15 50
та
0,02 Бутадиен-1,3 0,3 40
0,14+0,04 т pem-Бути лвини- 0,0032+0,0002 60
ловый эфир
0,7+0,2 Этилвиниловый 0,03+0,02 80
эфир
0,02+0,02 Изобутилен 1,8+0,2 50
1,5+0,1 Метилакрилат 0,84+0,05 50
0,150+0,080 Метилметакрилат 1,224+0,100 80
0,61+0,04 Метилвинилкетон 1,78+0,22 60
0,04+0,04 Стирол 0,40+0,05 60
4,2 Винилацетат 0,05 50
2,7+0,7 Винилхлорид 0,04+0,03 60
0,91+0,10 Вини лиденхл орид 0,37+0,10 60
0,113+0,002 2-Винил пиридин 0,47+0,03 60
Аллилацетат 0 Метилметакрилат 23 60
0,00 Стирол 90+10 60
0,7 Винилацетат 1,0 60
Бутадиен-1,3 0,3 Акрилонитрил 0,2 40
0,75+0,05 Метилметакрилат 0,25+0,03 90
1,35+0,12 Стирол 0,58+0,15 50
8,8 Винилхлорид 0,035 50
Диэтилфумарат 0 Акрилонитрил 8 60
0,070+0,007 Стирол 0,30+0,02 60
0,444+0,003 Винилацетат 0,011+0,001 60
0,12+0,01 Винилхлорид 0,47+0,05 60
Диэтилмалеат 0 Акрилонитрпд 12 60
0 Метилметакрилат 20 60
0,0+0,1 Стирол 5+1,5 70
0,043+0,005 Винилацетат 0,17+0,01 60
0,009+0,003| Винилхлорид 0,77+0,03 60
Продолжение табл. 6..
Ml Г1 м2 Т, °C
Фхмаронитри л 0,01+0,01 Метилметакрилат 3,5±0,5 79
0,01+0,01 Стирол 0,23+0,01 60 '
0,00 Винилацетат 0,14 —
Малеиновый ан- 0,046+ 0,052 Додецилвпнило- — 0,046±0,054 50
гидрид вый эфир
0,02 Метилакрилат 2,8+0,05 75
0,02 Метилметакрилат 6,7±0,2 75
0,015 Стирол 0,040 50 .
0,003 Винилацетат 0,055±0,015 75
0,008 Винилхлорид 0,296+0,07 75
Метакриловая 0,526 Бутадиен 0,201 50
кислота
0,7+0,05 Стирол 0,15+0,1 60
20 Винилацетат 0,01 70
0,58+0,05 2-Винилпиридин 1,55±0,10 70
Метилакрилат 0,05+0,05 Акриламид 1,30±0,05 60
0,67+0,1 Акрилонитрил 1,26±0,1 60
0,05+0,02 Бутадиен-1,3 а 0,76±0,04 5
3,3 Этилвиниловый 0 60
эфир
0,504 Метилметакрилат 1,91 130
0,15+0,05 Стирол 0,7±0,1 60
9 Винилацетат 0,1 60
4 Винилхлорид 0,06 45
0,20+0,09 2-Винилпиридин 2,03+0,49 60
Метилметакрилат 0,46±0,026 Стирол 0,52+0,026 60
20+3 Винилацетат 0,015±0,015 60
10 Винилхлорид 0,1 ' 68
2,53+0,01 Винилиденхлорид 0,24+0,03 60
а-Метилстирол 0,1+0,02 Акрилонитрил 0,06±0,02 75
0,010+0,01 Бутадиен-1,3 1,6+0,5 12,8
0,038+0,003 Малеиновый ан- 0,08+0,03 60
гидрид
0,14+0,01 Метилметакрилат 0,50±0,03 60
0,38 Стирол 2,3 —
360 Глава 6
• — Продолжение тпабл. 6.2
Г1 м2 Г2 т, °C
Метилвинилкетон 0,35+0,02 Стирол 0,29+0,04 60
7,00 Винилацетат 0,05 70
8,3 Винилхлорид 0,10 70
1,8 Винилиденхлорид 0,55 70
Стильбен 0,03+0,03 Малеиновый ан- гидрид 0,03+0,03 60
0 Стирол 11,2+1,2 60
Стирол 80+40 Этилвиниловый эфир 0 80
1,38+0,54 Изопрен 2,05±0,45 50
55±10 Винилацетат 0,01±0,01 60
17±3 Винилхлорид 0,02 60
1,85+0,05 Винилиденхлорид 0,085+0,010 60
0,55 2-Винилпиридин 1,14 60
Тетрафторэтилен 1,0 Хлортрифторэти- лен а 1,0 60
0,85 Этилен а 0,15 80
<0,3 Изобутилен а 0,0 —
Винилацетат 3,0+0,1 Этилвиниловый эфир 0 60
0,23+0,02 Винилхлорид 1,68+0,08 60
0,1 Винилиденхлорид 6 68
а Эмульсионная полимеризация.
В табл. 6.3 приведены значения 1/г, вычисленные из данных
табл. 6.2. Цифры в вертикальных колонках показывают актив-
ность мономеров по отношению к одному и тому же определенному
радикалу. Так, в первой колонке приведены данные активности
ряда мономеров по отношению к радикалу бутадиена, во второй —
к радикалу стирола и т. д. Следует отметить, что данные в гори-
зонтальных рядах табл. 6.3 нельзя сравнивать между собой.
Сравнивать можно только данные в каждой вертикальной
графе.
Сополимеризация
361
Таблица 6.3
Относительная активность мономеров (1/г) при реакции
с различными полимерными радикаламиа
Мономер Относительная активность мономера по отношению к полимерному радикалу
бута- диена стира- ла винил- аце- тата винил- хлори- да метилмет- акрилата метил- акрилата акрило- нитрила
Бутадиен 1,7 29 4 20 50
Стирол 0,4 100 50 2,2 6,7 25
Метилметакрилат 1,3 1,9 67 10 2 6,7
Метплвпнилкетон 3,4 20 10 1,7
Акрилонитрил 3,3 2,5 20 25 0,82 1,2
Метилакрилат 1,3 1,4 10 17 0,52 0,67
Вппилиденхлорпд 5,4 10 0,39 1,1
Винилхлорид 0,11 0,059 4,4 0,10 0,25 0,37 .
Винилацетат 0,019 0,59 0,050 0,11 0,24
а Значения 1/т вычислены из данных табл. 6.2.
6.36.1. Влияние сопряжения (резонансный эффект)
В табл. 6.3 мономеры расположены в порядке изменения их
активности. Порядок изменения активности мономеров остается
приблизительно постоянным во всех вертикальных графах неза-
висимо от того, к взаимодействию с каким радикалом относятся
эти данные. Встречающиеся исключения связаны с резко выра-
женной тенденцией некоторых пар мономеров к образованию регу-
лярно чередующихся сополимеров. Результаты, приведенные
в табл. 6.3, так же как и другие аналогичные данные [6], показы-
вают, что заместители увеличивают активность мономера по отно-
шению к атаке радикала, причем по силе воздействия замести-
тели можно расположить в следующую последовательность:
-С6Н5, — СН = СН2 > -CN, —COR > —СООН, —COOR >
— Cl > —OCOR, —R > —OR, —Н .
Эта последовательность соответствует порядку повышения ре-
зонансной стабилизации радикала, образуйщегося из данного
мономера, ПРИ сопРя'кении с данным заместителем. Наиболее
эффективны в отношении повышения стабилизации радикалов
заместители, содержащие ненасыщенные связи, л-злектроны кото-
рых легко вступают в сопряжение с радикалом. Эффективность
таких заместителей, как галоген, ацетоксигруппа и эфирная
362
Глава 6
группа, при стабилизации радикалов последовательно уменьшает-
ся в этом ряду, так как только несвязанные электроны галогена
или кислорода способны к взаимодействию с радикалом. Эффек-
тивность различных заместителей в отношении повышения актив-
ности мономера изменяется приблизительно в 50—200 раз в зави-
симости от активности радикала. Чем менее активен атакующий
радикал, тем больше проявляется разница в активности различных
мономеров. Влияние второго заместителя, находящегося в поло-
жении 1 (например, у винилиденхлорида), оказывается почти
аддитивным.
Ряд активности радикалов можно получить умножением вели-
чины 1/г на соответствующую константу скорости роста при
гомополимеризации (&ц). При этом получаются величины /с12 для
различных комбинаций радикал — мономер (табл. 6.4). Значе-
Таблица 6.4
Константы скорости (fci2) реакций полимерных радикалов
с различными мономерами а
Мономер (Mi) Константы скорости реакции полимерного радикала <21 61
бута- диена стиро- ла метил- мет- акри- лата акри- лонит- рила метил- акри- лата винил- аце- тата винил- хлори- да
Бутадиен 100 246 2820 98 000 41 800 357 000 2,39 -1,05
Стирол 40 145 1550 49 000 14 000 230 000 615 000 1,00 -0,80
Метилметакри- 130 276 705 13 100 4 180 154 000 123 000 0,74 0,40
лат Акрилонитрил 330 435 578 1960 2 510 46 000 178 000 0,60 1,20
Метилакрилат 130 203 367 1 310 2 090 23 000 209 000 0,42 0,60
Винилхлорид 11 8,7 71 720 520 10 100 12 300 0,044 0,20
Винилацетат 2,9 35 230 230 2 300 7 760 0,026 -0,22
а Значения feis вычислены из данных табл. 3.9 и 6.3.
ния ki2 в вертикальных колонках табл. 6.4так же, как и в табл. 6.3,
дают порядок активности мономера. Данные в горизонтальных
рядах дают порядок активности радикалов по отношению к дан-
ному мономеру. (Приведенные в табл. 6.4 величины Qi й будут
рассмотрены в разд. 6.36.4.)
Как и в случае активности мономеров, порядок активности
радикалов остается приблизительно одинаковым независимо от
того, к какому мономеру он относится. Порядок расположения
заместителей по увеличению активности радикалов оказывается
Сополимеризация
363
обратным порядку расположения их в случае увеличения актив-
ности мономеров. Заместитель, повышающий активность мономе-
ра, одновременно стабилизирует и понижает активность соответ-
ствующего радикала. Данные табл. 6.4 показывают, что влияние
заместителя на активность радикала значительно больше, чем
его влияние на активность мономера. Например, радикал винил-
ацетата приблизительно в 100—1000 раз более активен, чем радикал
стирола, по отношению к данному мономеру, тогда как стирол
Р и с. 6.9. Изменение потен-
циальной энергии в направ-
лении координаты реакции
при реакции полимерного
радикала и мономера в за-
висимости от расстояния
между радикалом и ненасы-
щенным атомом углерода
мономера. Индекс s указы-
вает на присутствие заме-
стителя, способного к резо-
нансной стабилизации. Энер-
гии активации представле-
ны сплошными стрелками,
теплоты реакции — штрих-
пунктирными [6].
Расстояние между радикалом и нена-
сыщенным атомом углерода
как мономер только в 50—100 раз активнее, чем винилацетат
(мономер) по отношению к данному радикалу. Сравнение констант
скорости роста гомополимеризации (/ср) винилацетата и стирола
показывает, что эти два эффекта почти полностью компенсируют
ДРУГ друга. Как видно из табл. 3.9, кр винилацетата только
в 16 раз больше кр стирола.
Взаимное влияние радикала и мономера при определении
скорости реакции между ними еще более ясно можно видеть из
диаграммы [39], приведенной на рис. 6.9. На этом рисунке пока-
зано изменение потенциальной энергии, сопровождающее реак-
цию радикала с мономером, как функции расстояния между ато-
зань1' °°РазУюш'ими новую связь. Эти изменения энергии пока-
для четырех возможных реакций между резонансно стабили-
рованными и нестабилизированными мономерами и радикалами
R«+M->R., (6.42)
R • + Ms —> Rs-, (6.43)
364
Глава 6
Rs • J- Ms —> Rs-,
Rs. +M->R-,
(6.44)?
(6.45)1
где наличие или отсутствие индекса s указывает на присутствие
или отсутствие соответственно заместителя, способного к резо-
нансной стабилизации. Винилацетат и стирол представляют собой
примеры мономеров М и Ms соответственно; радикалы винил-
ацетата и стирола являются примерами R-и Rs- соответственно.
На рис. 6.9 представлены две серии кривых потенциальной
энергии. Одна серия из четырех кривых показывает изменение
энергии радикала и мономера при их сближении; серия из двух
кривых характеризует стабильность образующейся связи (или по-
лимерного радикала). Точки пересечения этих кривых представ-
ляют переходные Состояния при реакциях мономера с радикалом
[уравнения (6.42)—(6.45)1; мономер, радикал и продукт реакции
имеют одинаковую энергию. Энергии активации и теплоты реак-
ции представлены сплошными и штрихпунктирными стрелками
соответственно. Из данных рисунка видно, что замещение оказы-
вает значительно большее влияние на понижение активности
радикала, чем на повышение активности мономера.
Рис. 6.9 показывает также, что константы скорости различных
реакций мономер — радикал возрастают в следующем ряду
систем:
Rs-+M < Rg-+Ms < R-+M < R-+Ms,
(6.45) (6.44) (6.42) (6.43)
так как энергии активации этих реакций изменяются в противопо-
ложном направлении. (При этом предполагается, что в значениях
энтропии активации нет существенных различий — вполне вероят-
ное допущение для стерически незатрудненных мономеров.) Этот
порядок активности представляет в общем виде данные табл. 6.3
и 6.4 и многие данные по гомополимеризации. Ясно, что скорость
роста при гомополимеризации мономеров, не имеющих стабилизи-
рующих заместителей (например, винилхлорида или винилацетата),
будет больше скорости роста для мономеров, имеющих стабилизи-
рующие заместители (например, стирола) [реакции (6.42) и (6.44)1.
С другой стороны, сополимеризация будет лучше протекать между
двумя мономерами, имеющими стабилизирующие заместители,
или между мономерами, не имеющими их. Сополимеризация моно-
мера, имеющего стабилизирующий заместитель, с мономером, не
имеющим такового (например, стирола с винилацетатом), приводит
к одновременному протеканию реакций, выраженных уравнения-
ми (6.45) и (6.43). Но реакция (6.45) очень медленная, поэтому
сополимеризация стирола с винилацетатом идет очень плохо:
радикал стирола слишком мало активен, для того чтобы при-
соединять малоактивный мономер — винилацетат.
Сополимеризация
365
6.36.2. Влияние стерических затруднений
(стерический эффект)
Скорости реакций радикал — мономер зависят также и от сте-
рических затруднений. Это легко проследить по данным активно-
сти ди-, три- и тетразамещенных этиленов при сополимеризации.
В табл.' 6.5 показаны значения kl2 для реакций различных хлор-
Таблица 6.5
Константы скорости (fci2) реакций полимерных
радикалов с различными мономерами а
Мономер Константа скорости реакции полимерного радикала
винил- ацетата стирола акрило- нитрила
Винилхлорид 10 100 8,7 720
Винилпденхлорид 23 000 78 2200
1/ ис-1,2-Дпхлорэтплен 370 0,60
жранс-1,2-Дпхлорэтплен 2 300 3,9
Трихлорэтилен 3 450 8,6 29
Тетрахлорэтилен 460 0,70 4,1
а Значения вычислены из данных табл. 3.9 и 6.2 [38].
этиленов с радикалами винилацетата, стирола и акрилонитрила.
Влияние второго заместителя на активность мономера оказывает-
ся почти аддитивным, если оба заместителя находятся в положе-
нии 1,1, т. е. в «-положении. Однако если второй заместитель
находится в p-положении к первому, то активность мономера
снижается вследствие стерических препятствий между мономером
и радикалом, к которому он присоединяется. Так, активность
винилиденхлорида выше в 2—10 раз, а активность 1,2-дихлорэти-
лена ниже в 2—20 раз, чем активность винилхлорида.
Хотя активность 1,2-замещенных этиленов при сополимериза-
ции мала, она все же больше, чем их активность при гомополи-
меризации. В разд. 3.96.3 было отмечено, что неспособность
1,2-замещенных этиленов к гомополимеризации обусловлена сте-
рическими препятствиями, связанными с наличием р-заместителей
у атакующего радикала и заместителя у мономера. Активность
1,2-замещенных этиленов при сополимеризации объясняется от-
сутствием P-заместителей у атакующих радикалов (например,
у радикалов стирола, акрилонитрила и винилацетата).
36G
Глава 6
Сравнение цис- и тпранс-1,2-дихлорэтиленов показывает, что<
активность транс-изомера в 6 раз больше активности цис-пзоме-:
ра [40]. Такое соотношение активностей сохраняется для всех!
цис- и транс-1,2-замещенных этиленов. ^нс-Изомер, который-
бывает обычно и менее стабильным соединением, менее активен
при взаимодействии с радикалом. Различие в активности объяс-
няют неспособностью ^нс-изомера в переходном состоянии суще-
ствовать в виде плоской конформации, что необходимо для резо-
нансной стабилизации вновь образующихся радикалов замести-
телями [6].
Дальнейшей иллюстрацией различного влияния замещения в 1-
и 2-положения у этилена являются данные по активности три-
и тетрахлорэтиленов. Трихлорэтилен более активен, чем оба
изомера 1,2-дихлорэтилена, но менее активен, чем винилиденхло-
рид. Тетрахлорэтилен менее активен, чем трихлорэтилен, что
аналогично различию в активностях винилхлорида и 1,2-дихлор-
этилена. Исключением из общего правила — уменьшения актив-
ности при полизамещении — являются полифторэтилены. Тетра-
фторэтилен и хлортрифторэтилен имеют повышенную активность,
что объясняется, вероятно, малыми размерами атомов фтора.
6.36.3. Влияние полярности (полярный эффект)
Ранее было отмечено, что расположение мономеров в ряд по
активности не совсем одинаково в случае различных радикалов
(табл. 6.3 и 6.4). Аналогично этому и расположение радикалов по
активности несколько меняется в зависимости от мономера.
Активность мономера нельзя рассматривать независимо от актив-
ности радикала, и наоборот. В определенных парах мономеров
наблюдают повышенную активность, обусловленную, вероятно,
взаимодействиями радикала с мономером. Этот эффект весьма
обычен для радикальной сополимеризации и связан с тенденцией
пар сомономеров к чередованию. Мерой этой тенденции является
отклонение значения произведения г{г2 от единицы и приближение
его к нулю. Можно расположить мономеры по значению про-
изведения rtr2 констант сополимеризации их с другими мономера-
ми в таком порядке, что чем дальше друг от друга находятся два
мономера, тем больше их склонность к чередованию (табл. 6.6).
Например, акрилонитрил вступает в идеальную сополимеризацию
с метилвинилкетоном (г^г2 = 1,1) и образует регулярно чередую-
щиеся сополимеры с бутадиеном (г^з = 0,006).
Порядок расположения мономеров в табл. 6.6 основан на
полярности двойной связи. Мономеры с электронодонорными за-
местителями расположены в левой верхней части таблицы, а мо-
номеры с электроноакцепторными заместителями — в правой ниж-
ней. Значения произведения rtr2 постепенно уменьшаются по мере
Сополимеризация
367
«каления мономеров в таблице друг от друга. На основании
опведенных данных можно сделать вывод о повышении тенден-
* мономеров к чередованию по мере увеличения разницы поляр-
ности Наличие полярных влияний между электронодонорными
и электроноакцепторпымп частицами при радикальных реакциях
было отмечено при обсуждении реакций передачи цепи
(оазд. 3.96. 3). Наиболее ярким примером влияния полярности
является легкая сополимеризация мономеров, которые очень мало
или даже совсем не способны к гомополимеризации. Малеиновый
ангидрид и диэтилфумарат не гомополимеризуются, но легко об-
разуют сополимеры со строго чередующимися звеньями с такими
электронодонорными мономерами, как стирол и виниловые эфиры
[41]. Сополимеризация малеинового ангидрида (электроноакцептор)
и стильбена (электронодонор) протекает, несмотря на то что оба
мономера практически не способны к гомополимеризации
и СН = СН 4- п сн = сн
I I
CgHg CgHs
СО СО
—СН —СН — СН-СН —
С6Н5 СвН5 СО СО
- (6.46>
О
о
□ п
6.6 можно видеть, что величину
данных табл.
Однако из
тенденции мономера к образованию регулярно чередующихся
сополимеров нельзя объяснить только влиянием полярности.
Например, винилацетат менее чем стирол склонен к чередовании}
с акрилонитрилом, но к чередованию с диэтилфумаратом — более
чем стирол. Причиной таких отклонений от прямой корреляции
тенденции к чередованию и полярности мономера являются, веро-
ятно, стерпческие эффекты.
Увеличение активности при радикальных реакциях в результа-
те полярных влияний объясняют тем, что взаимодействие электро-
ноакцепторного радикала с электронодонорным мономером илц
электронодонорного радикала с электроноакцепторным мономе-
ром приводит к уменьшению энергии активации реакции ради-
кал — мономер. Наблюдаемое обычно отсутствие влияния раство-
рителя на константы сополимеризации при радикальном процессе-
указывает на то, что полярное взаимодействие радикала с моно-
мером происходит не в основном состоянии, как предполагал
раис 142], а в переходном состоянии, когда расстояния между
реагирующими атомами очень близки к действительным длинам
связей, так что растворитель отсутствует в переходном состоянии.
V днако полученные недавно данные [43] показывают, что значе-
ri и г2 несколько изменяются в зависимости от полярности
растворителя.)
ги ^ЫЛ0 ВЫСказапо предположение [6, 44], что уменьшение энер-
и переходного состояния происходит в результате переноса
Значений rtr2 йри радикальной Сополимеризации а
Виниловые
эфиры 6
(е=-1,3)
Бутадиен
Стирол
(е =
= -0,80)
0,55 Винилацетат (е - -0,22)
0,34 0,39 Винил- хлорид (е=0,20)
0,24 0,30 1,0 Метил-
мстакрп-
. I а т
(е -0,40)
Таблица 6.6
<0,1 0,16 0,6 0,96 0,61
0,10 0,35 0,83
0,0004 0,006 0,016 0,21 0,11 0,18
0 0,021 0,0049 0,056
0,002 0,006 0,00017 0,0024 0,11
а Значения rjr2 вычислены из данных табл. 6.2, [38] и [41].
б Этил-, изобутил- или додецилвиниловые эфиры.
Винили-
денхло-
рид
Метил-
винил-
кетон
(е=0,68)
...
Акрило-
нитрил
(<?=!, 20)
0,56
Диэтил-
фумаратI
(е = 1,25)!
Малеиновый
ангидрид
(е =2,25)
370
Глава 6
электрона от электронодонорной частицы к электроноакцептор-
ной, например, при реакции мономера стирола с радикалом мале-
инового ангидрида
или аналогичным образом при реакции радикала стирола с моно-
мером — малеиновым ангидридом
Позднее возникла другая гипотеза [45]: сополимеризация
с образованием чередующихся сополимеров происходит скорее
в результате гомополимеризации эквимолярного комплекса (1 : 1)
электронодонорного и электроноакцепторного мономеров, чем
путем присоединения донорного и акцепторного мономеров к ра-
стущей цепи. Если это так, то можно ожидать, что образование
таких комплексов удается катализировать увеличением электро-
ноакцепторной способности мономера-акцептора путем комплексо-
образования со специально добавленным подходящим соединением.
Это было подтверждено на самом деле на примере сополимериза-
ции стирола с метилметакрилатом, когда в результате добавле-
ния сесквихлорида этилалюминия получен сополимер со значи-
тельно более высокой степенью чередования звеньев. Кроме того,
такие добавки увеличивают и скорость сополимеризации.
6.36.4. Схема Q — e
Были предприняты различные попытки установить количест-
венные зависимости между структурой и реакционной способно-
стью соединений при реакциях радикал — мономер. Успех в этом
направлении очень желателен, так как это позволило бы пред-
сказать константы сополимеризации для таких пар мономеров,
которые еще не подвергались сополимеризации. Найденная Гам-
метом зависимость, связывающая активность мономеров с кон-
стантами о и р, характеризующими полярность заместителей
Сополимеризация
371
реакции, очень хорошо оправдала себя для мета- и пара-
П Т1Шенпь1Х стиролов [6]. Более общей является так называемая
замещ _ g Алфрея и Прайса [46, 47]. Эти авторы предполо-
СХе^' чт0 константу скорости для реакции радикала с мономе-
ром например для реакции радикала Mj- с мономером М2, можно
выразить как
fci2 = J’i<?2exp( —е^г),
(6.49)
г е i>A _ мера общей активности радикала, Q2 — мера общей ак-
тивности мономера, а е( и е2 - меры полярности радикала и моно-
мера соответственно. Величины Q и Р определяют резонансные
эффекты в мономере и радикале. Если допустить, что одно и то
же значение е относится и к мономеру, и к его радикалу (т. е. е4
определяет полярность Mj и Мр, а е2 определяет полярность М2
и М2«), то можно написать выражения для кп,к 22 и ки аналогично
уравнению (6.49). Объединяя соответствующим образом эти урав-
нения, получим выражения констант сополимеризации в следую-
щем виде:
О = -^-ехр[ —ejet —е2)], (6.50)
V2
r2 = -^-exp [ —е2(е2 —Cj)], (6.51)
которые связывают активность мономера и радикала с параметрами
(Л. и е2.
В основу схемы Q — е [уравнения (6.50) и (6.51)] положено
теоретически неудовлетворительное предположение Прайса [42]
о том, что тенденция к чередованию обусловлена электростати-
ческими взаимодействиями зарядов мономера и радикала. Были
предприняты попытки [48] подвести под эту схему серьезную
теоретическую основу. Так, например, расчеты молекулярных
орбиталей показали связь величин Р и Q с энергией локализации
мономера, а величина е мономера оказалась связанной с его
сродством к электрону. Однако ни один из этих теоретических
подходов до сих пор не был успешно реализован количественно.
В настоящее время лучше всего рассматривать схему Q — е как
эмпирический путь установления количественных соотношений
для активности мопомерв. Активность мономера входит в пара-
метр Q, который описывает резонансный эффект (и в некоторой
степени — стерический эффект), и в параметр е, который описывает
полярный эффект.
Рассмотрим теперь использование схемы Q — е для предсказа-
ния констант сополимеризации мономеров. Значения Q и е для
различных мономеров были вычислены, исходя из их значений г
и величин Q и е для произвольно выбранного в качестве стан-
дартного вещества стирола. Стиролу приписаны значения Q = 1
372
Глава 6
и е = — 0,80 (первоначально для стирола выбрали значение е =
= — 1,0, но позднее эта величина была изменена на —0,80, чтобы
улучшить корреляцию значений Q и е со значениями г). Значения
Q и е вычислены для большого числа мономеров [38]; для неко-
торых из них в табл. 6.7 приведены средние значения Q и е.
Таблица 6.7
Средние значения Q и е для различных
мономеров [38]
Мономер Q е
трепг-Б ути л виниловый 0,15 -1,58
эфир
Этилвиниловый эфир 0,032 -1,17
Бутадиен 2,39 -1,05
Стирол 1,00 -0,80
Винилацетат 0,026 -0,22
Винилхлорид 0,044 0,20
Винилиденхлорид 0,22 0,36
Метилметакрилат 0,74 0,40
Метилакрилат 0,42 0,60
Мети лвинил кетон 0,69 0,68
Акрилонитрил 0,60 1,20
Диэтилфумарат 0,61 1,25
Малеиновый ангидрид 0,23 2,25
Практическое использование схемы Q — е для предсказания
значений г1 и г2 для новых пар сомономеров ограниченно. Причи-
на этого состоит в том, что значения Q и е для каждого мономера
неоднозначны как с экспериментальной, так и с теоретической
точки зрения. Точность вычисленных значений Q и е часто очень
мала вследствие неточности экспериментально определенных кон-
стант сополимеризации мономеров. Это можно видеть по данным
табл. 6.8, где приведены значения е, вычисленные для акрилони-
трила по различным значениям и г2, полученным для одной
и той же пары сомономеров (акрилонитрил — винилацетат или
акрилонитрил — винилхлорид). Далее, значения Q и е мономера
значительно изменяются в зависимости от того, с каким мономе-
ром он сополимеризуется (табл. 6.8). Это связано с недостатками
самой схемы Q — е, которая плохо учитывает стерические факторы,
влияющие на активность мономеров при определенных комбина-
циях радикал — мономер. В этом отношении показательно, что
значения Q — е часто бывают бессмысленными при сополимериза-
ции 1,1-дизамещенных мономеров или вообще всех замещенных
Сопол имеризация
373
Таблица 6.8
Изменения величин Q и е (42]
Мономер (Mi) Сомономер
Акрилонитрил Стирол 0,44 1,20
Винилацетат 0,37 0,90
Винилацетат 0,67 1,0
Винилхлорид 0,37 1,3
Винилхлорид 0,75 1,6
Винилхлорид Стирол 0,024 0,2
Метилакрилат 0,035 0,0
мономеров, кроме монозамещенных этиленов. Допущение равен-
ства величин е для мономера и его радикала также неверно. По-
пытки уточнения схемы Q — е применением различных значений е
для мономера и радикала не привели к успеху.
Несмотря на эти недостатки, схема Q — в представляет собой
вполне приемлемый качественный и даже полуколичественный
подход к вопросу о влиянии структуры мономера на его актив-
ность. Ею пользуются, чтобы получить общее представление
о поведении, которое можно ожидать от не изученной ранее пары
сомономеров, и особенно она полезна для предсказания поведе-
ния сомономеров при терполимеризации. Значения Q — е исполь-
зуют также, чтобы с более количественных позиций обсуждать
данные по активности мономеров, такие, например, как в табл. 6.4
и 6.6. Ясно, что мономеры в табл. 6.6 расположены в порядке
значений е. Этот порядок определяет полярность мономеров.
Относительную важность резонансного и полярного эффектов при
определении активности мономеров легче понять, рассматривая
данные табл. 6.4 с точки зрения значений Q — е мономеров. Раз-
личные радикалы можно разделить на две группы: относительно
неактивные радикалы (стирол и бутадиен) и активные радикалы
(все остальные). В случае активных радикалов слабые влияния
полярности практически не имеют значения, и активность мономе-
ров зависит в первую очередь от резонансных факторов. Значе-
ния к12 возрастают с увеличением значений Q для мономера.
и Дикады бутадиена и стирола относительно мало активны
влт^°ЭТ0МУ „оказываются чувствительными к слабым полярным
обла™™' ДВа РаДикала, имеющие отрицательные значения е,
Д Ют ПОВЬ1Ц1енной активностью по отношению к таким мономе-
сительЯК МетилметакРилат и акрилонитрил, которые имеют отно-
пмппаи° высокие положительные значения е. Однако влияние
сного эффекта оказывается более важным, чем влияние
374
Глава 6
полярности; именно он определяет величину активности мономе-
ра. Так, мономеры по их активности относительно радикалов сти-
рола и бутадиена можно разделить на две группы: мономеры
с большими значениями Q и высокой активностью и мономеры
с малыми значениями Q и низкой активностью.
6.36.5. Определение активности из произведена я
вероятностей
Хэм [29, 30, 33, 49] использовал уравнение (6.37), т. е. про-
изведение вероятностей, для предсказания поведения пары сомо-
номеров при сополимеризации исходя из данных по сополимери-
зации каждого из сомономеров в отдельности с каким-либо общим
мономером или, другими словами, для предсказания поведения
мономеров Mj/Мг/Мз при терполимеризации по данным системам
Mj/M2 и М2/М3. При терполимеризации эквимолярных количеств
трех мономеров уравнение (6.37) после подстановки вместо чле-
нов Р соответствующих выражений [уравнения (6.38)] принима-
ет вид
/ г13 \ / г21 \ / г32 \ =
' г1з + г12г1з + г12 / \ ^21 + г23г21 + ^23 / \ Г32 + Г32г31 + Г31 /
= (__________Н2_______) /________С31_______ \ . (6.52'
' г12+г12г1з + г13 / ' Г31 + Г31Г32Ч-Г32 ' \ г2з + г23г21 + Г21 /
Это выражение можно упростить
г12г 23г 31 — Г13Г 32Г 21
(6.53а)
и преобразовать в уравнение
Лз= (6.536)
г,31 Г21Г32
при помощи которого вычисляют отношение констант сополимери-
зации для системы Mi/M3 из данных для систем Mt/M2 и М2/М3.
В дальнейшем этот метод был расширен с целью получения
возможности вычисления значений индивидуальных констант
сополимеризации (а не только их отношения), а также для лучше-
го понимания активности мономеров. На основании теоретических
соображений и данных экспериментального исследования многих
терполимерных систем Хэм пришел к выводу, что произведения
вероятностей P21Pi3P32 и Т’23/>31/>12 должны иметь одно из двух
единых значений — или 0,037 или 0,006. Если все три мономера
системы содержат сопряженные связи (как, например, мономеры
с фенильными, винильными, нитрильными, карбонильными или
карбоксильными заместителями), то произведение вероятностей
равно приблизительно 0,037. Это число получено при допущении,
что активность мономеров при реакции с различными радикалами
Сополимеризация
375
инакова, так что произведение вероятностей равно (Ч3)3, или
О 037 Если для одного пли двух мономеров вероятность боль-
ше Vg, то вероятность для других (или другого) мономеров (или
мономера) должна быть достаточно мала, чтобы дать в итоге то же
самое значение произведения вероятностей. Если два из трех
мономеров содержат сопряженные связи, а один — слабо сопря-
женные (мономеры с такими заместителями, как галоген, ацеток-
спгруппа, эфирная или алкильная группа), то произведение веро-
ятностей будет 0,006. Это значение получено при допущении, что
р = Р23 (обе величины малы), Р31 = Р32 (обе величины велики),
н г12 = ?’21 = 1, r32^23 = 1, г31г13 = 1, причем М3 — слабо сопря-
женный мономер.
Тогда уравнение (6.37) можно написать в следующем виде:
РцР1зРз2 = Р 23Р цР 12 — 6.54)
где еР — константа, характеризующая тройную систему и имею-
щая величину либо 0,037, либо 0,006. Комбинируя уравнения
(6.52)—(6.54). можно определить значения г13 и г31 по константам
сополимеризации мономеров для систем Mi/M2 и М2/М3. Левая
и правая части уравнения (6.52) равны оР. В сочетании с уравне-
нием (6.53) это приводит к двум уравнениям с двумя неизвестными
г13 и г31. Совместное решение этих уравнений дает индивидуальные
константы сополимеризации мономеров.
Этот метод Хэма был предметом больших споров [50, 51]. Метод,
конечно, имеет недостатки. До сих пор нет полной количествен-
ной проверки правильности уравнений (6.37) и (6.53), а также
значения & (табл. 6.9) экспериментальными методами. Точность
имеющихся данных недостаточна для безусловного подтверждения
равенства произведений вероятности [уравнение (6.37)] или равен-
ства произведений констант сополимеризации [уравнение (6.53)].
Это можно видеть просто при сравнении величин оР в табл. 6.9
для систем СТ/МА/АН, СТ/АН/АК и ММА/МА/ВХ. Оба значе-
ния еР — я 0,006, и 0,037 — полезны для качественных или
полуколичественных предсказаний, но оставляют желать много
лучшего в смысле количественного предсказания активности
мономера. Допущения, сделанные при теоретическом выводе этих
величин, вероятно, мешают их количественному применению,
днако, несмотря па все эти соображения, метод произведения
вероятностей Хэма весьма полезен, как и схема Q — е, для при-
изительной оценки активности таких мономеров, для которых
нет экспериментальных данных. При этом следует отметить, что
соб “ ч ~ 6 И л1етоД произведения вероятностей связаны между
°и’ та связь неоднократно обсуждалась [49, 50]. Оба подхода
мпжЯСуЮТСЯ друг с Другом. На самом деле, уравнение (6.53)
° П°Лучить пепосРеДственно как из схемы Q — е, так и по
методу произведения вероятностей.
376
Глава 6
Таблица 6.9
Вероятность образования фрагментов с различным
чередованием мономеров [49]
Мономеры а Р23Р31Р12 Рг1Р1зРз2
СТ/ММА/АН 0,053 0,0454
СТ/МА/АН 0,00504 0,00435
0,0157 0,00284
СТ/ММА/В'Х 0,042 0,023
ММ/АН/В'Х 0,0262 0,0467
СТ/ММА/ВП 0,051 0,0562
СТ/ММА/МА 0,0348 0,046
СТ/АН/АК 0,0491 0,0199
0,0164 0,085
ММА/МА/ВХ 0,00782 0,0055
0,0131 0,00315
АН/МА/ВХ 0,0168 0,0167
а Принятые обозначения: АК — акриловая кислова, АН — акрило-
нитрил, МА — метилакрилат, ММА — метилметакрилат, СТ — стирол,
ВХ — винилхлорид, В'Х — винилиденхлорид, ВП — 2-винилпиридин.
6.Зв. Скорость сополимеризации
Скорость сополимеризации в отличие’ от состава сополимера
зависит как от стадий инициирования и обрыва, так и от стадии
роста цепи. Обычно оба мономера энергично реагируют с иниции-
рующими радикалами, и скорость инициирования не зависит от
состава смеси реагентов. Для вывода уравнений скорости сопо-
лимеризации были использованы два пути.
6.3в.1. Обрыв цепи, контролируемый химическими
реакциями
Обычно предполагают [6, 11], что реакции обрыва определяют-
ся химическими факторами. Сополимеризация состоит из четырех
реакций роста [уравнения (6.2)—(6.5)] и трех реакций обрыва
М,
fto(ll)
ГМ1----------?
м2-+м2-
fto(22)
Мг +м2-
feo(12)
(6.55)
(6.56)
(6.57)
Сополимеризация
377
тветствующих взаимодействию двух одинаковых радикалов
киавнения (6.55) и (6.56)] и перекрестному взаимодействию двух
различных радикалов [уравнение (6.57)].
Р Общая скорость сополимеризации определяется суммой четы-
рех скоростей роста
d[Mi]-H[M2]_
Яр — dt
= kti [Mi • ] [MJ + fc12 [Mj • ] [M2] + k22 [M2 • ] [M2] + k2i [M2 • ] [M J.
(6.58)
Для того чтобы исключить из уравнения (6.58) концентрации ради-
калов, сделаны два допущения стационарности состояния. Пола-
гают, что концентрации радикалов каждого типа постоянны
fc2i[M2-][Mj = fc)2[M1.][M2], (6.59)
как это было сделано [уравнение (6.9)] при выводе уравнения
состава сополимера. Постоянство концентраций предполагается
также для общей концентрации радикалов
Яп — 2/с0()1) [М]-]2-) 2/с0(12) [Mi*] [М2-] -р 2/сО(22) [М2-]2. (6.60)
Исключив из уравнения (6.68) концентрации радикалов путем
объединения этого уравнения с уравнениями (6.59) и (6.60) и ис-
пользуя затем определения т\ и г2, получают для скорости сопо-
лимеризации выражение
R (ri [MJ2 + 2 [MJ [М2] + г2 [М2р)
₽ {^1 [М1]2 + 2Фг1г2б2б1 [Mi] [M2] + rg6| [M2]2}V8 ’ < •
где
б>=(^г<)1/2’ (6-62а)
(6.626)
кт )^- (6-62в)
б это величины, обратные знакомому отношению к-е/(2к0)1^ для
гомополимеризации индивидуальных мономеров; ср — отношении
половины константы скорости перекрестного обрыва к среднему
ометрическому значению констант скоростей обрыва одинако-
х радикалов. Множитель 1/2 в формуле для ср присутствует
нее°мУ’ что перекрестный обрыв статистически в два раза выгод-
’ Ч6М °ОРЬ1В за счет одинаковых радикалов. Значение <р <1
поттоЗЫВает’ что пеРекрестный обрыв неблагоприятен, а <р > 1
означает, что он предпочтителен.
378
Глава 6
Значения 6] и 62 получают из данных по гомополимеризации,
а и г2 — из данных по сополимеризации. Экспериментальное
определение скорости сополимеризации позволяет затем вычис-
лить значение ср по уравнению (6.61). Значения ср для некоторых
пар мономеров приведены в табл. 6.10 наряду со значениями их
Таблица 6.10
Значения ф и rir2 при радикальной
сополимеризации [6]
Система сомономеров ф Гр*2
Стирол — бутилакрилат 150 0,07
Стирол — метилакрилат 50 0,14
Метилметакрилат — га-метокси-
стирол 24 0,09
Стирол— метилметакрилат 13 0,24
Стирол — га-метоксистирол 1 0,95
Г1Г2- Из этих данных видно, что значение ср больше единицы, т. е.
перекрестный обрыв предпочтителен. Одновременно с тенденцией
к перекрестному обрыву наблюдается и тенденция к перекрестному
росту (т. е. к чередованию), поскольку с возрастанием ср значение
rtr2 приближается к нулю. Это позволило прийти к выводу, что
тенденция к перекрестному обрыву определяется влиянием поляр-
ности. Взаимодействие радикалов различной полярности усили-
вается благодаря эффектам переноса электрона подобно тому, как
зто было описано для тенденции к чередованию несходных моно-
меров.
Следствием предпочтительности перекрестного обрыва является
уменьшение скорости сополимеризации сравнительно со средней
скоростью гомополимеризации обоих мономеров. Большие скоро-
сти перекрестного роста при сополимеризации, приводящей к обра-
зованию чередующихся сополимеров, компенсируются большими
скоростями перекрестного обрыва, что, согласно уравнению (6.61),
обусловливает уменьшение средних скоростей сополимеризации.
Это можно видеть из данных рис. 6.10, где показаны вычисленные
скорости сополимеризации системы стирол — метилметакрилат
для значений ср 1 и 13.
Предсказать характер зависимости общей скорости от состава
смеси мономеров для сополимеризации, протекающей с сильно
выраженным чередованием звеньев, трудно вследствие противо-
положных влияний больших значений ср и малых значений про-
изведения Г1Г2. Интересный случай сополимеризации идеального
типа представляет система стирол — винилацетат (разд. 6.2в.2)
Сополимеризация
379
. Предполагается, что значение г2 очень малой выражение
(6 61) приближенно можно записать как
ЯР= ([MJ + ^-) (6.63)
откуда видно, что добавление очень малых количеств стирола
к винилацетату приводит к значительному уменьшению скорости
поста. Стирол существенно ингибирует полимеризацию винилаце-
тата. Радикалы винилацетата быстро присоединяют стирол, т. е.
Р п с. 6.10. Зависимость скорости
радикальной сополимеризации
стирола с метилметакрилатом (при
60 °C в присутствии азо-бис-
изобутиро нитрила) от состава
исходной смеси мономеров [52].
Обе кривые—теоретические, вычислен-
ные для различных значений <р; точ-
ки — экспериментальные данные.
1,00 0,80 0,60 0,40 0,20 0,00
Содержание стирола в смеси
мономеров, мол. доля
превращаются в радикалы стирола даже при низких концентраци-
ях последнего благодаря высокой активности и радикала винил-
ацетата, и мономерного стирола. Радикалы же стирола с винил-
ацетатом реагируют очень медленно, а со стиролом не реагируют
совсем, так как он присутствует в очень малых концентрациях.
Результатом этого является почти полное прекращение поли-
меризации.
б.Зв.2. Об21ыв, контролируемый диффузией
Норт с сотр. [53, 54] указал, что обработка данных по со-
полимеризации при помощи фактора ср вряд ли правильна, так
как твердо установлено, что обрыв при радикальной полимери-
зации обычно контролируется диффузией, поэтому нельзя толко-
вать ср только с точки зрения химических взаимодействий ради-
ных концов. Зависимость скорости обрыва от поступательной
3 106Мд1,11110*1 Диффузии полимерных цепей обсуждалась в разд,
тот 10ГДа И значения ф? отличные от единицы, нужно также
(Ьузи ВЙТЬ °- учетом изменений поступательной и сегментной диф-
в нек И'епеи’ °бусловливаемых их строением. Наблюдение, что ср
орых системах изменяется при изменении состава сополи-
380
Глава 6
мера, подтверждает эти соображения и делает сомнительным;:
использование уравнения (6.61) с единственным значением <р.
Более подходящее кинетическое выражение для скорости
сополимеризации, учитывающее влияние диффузии, получено [54}<
при рассмотрении обрыва как реакции
Мр+М,- 1 h
1 1 I О<12>
Мр+М2- >-------> Мертвый полимер, (о.оЧ^
М2- М2- J
где константа скорости обрыва &0(i2) зависит от состава сополиме-
ра. Тогда условие стационарности состояния для общей концен-
Содержание винилацетата,
мол. доля
Рис. 6.11. Зависимость кон-
станты скорости обрыва fc0<12>
от содержания винилацетата
при радикальной сополимери-
зации винилацетата с метил-
метакрилатом [54].
1 — вычислено по уравнению (6.66);
2 — вычислено по уравнению (6.67).
трации радикалов принимает вместо уравнения (6.60) форму
Ли = 2/сО()2> ([Мр] + [М2.])2. (6.65)
Объединяя уравнения (6.58), (6.59) и (6.65) с определениями для
т-! и г2, получим скорость сополимеризации в следующем виде:
_ (г! [Mip + 2 [Mt] [М2] + г2 [М2]2)
'
Можно ожидать, что константа скорости обрыва Л:0(12) будет
являться функцией константы скрости обрыва двух соответ-
ствующих реакций гомополимеризации. В идеальном случае эта
зависимость может принять вид
^о(12> — 4-/’2Л:О(22), (6.67)
гДе ^о(12) — средняя величина значений А:0(11) и /сО(22), выра-
женных через состав сополимера (мольные доли F, и F2).
Хотя применимость уравнений (6.66) для корреляции данных
по скорости сополимеризации не была широко изучена, но она
была установлена несколькими исследованиями [54, 55]. Так, на
рис. 6.11 показаны экспериментально определенные значения
Сополимеризация
381
ъ [т. е. значения, вычисленные по уравнению (6.66)] для
сополимеризации винилацетата с метилметакрилатом (кривая 1).
Ожидаемые отклонения значений Zc0(i2) в зависимости от состава
смеси мономеров (и сополимера) можно вычислить по уравне-
нию (6.66), поскольку уравнение (6.61) с единственным значением <р
неприменимо. На рис. 6.11 показаны также значения /с0(12),
вычисленные по уравнению (6.67) из состава сополимера и кон-
стант скорости обрыва для двух процессов гомополимеризации
(кривая 2). Уравнение (6.67) оказывается правильным качествен-
но, но не количественно. Очевидно, выбранный метод соотнесе-
ния значений констант скоростей реакций обрыва /с0()1) и /с0<22>
непосредственно с составом сополимера не является правильным.
Этот метод обработки данных был усовершенствован на системе
стирол — метилметакрилат [55]. Предположим, что обрыв
в результате реакции между цепями, содержащими на радикаль-
ных концах два стирольных звена, будет медленнее, чем обрыв
при взаимодействии цепей, последним звеном которого является
радикал стирола, предпоследним — метилметакрилат. Было по-
стулировано, что стерические взаимодействия между бензольными
кольцами соседних мономерных звеньев приводят к замедлению
сегментной диффузии вследствие ограничения вращения вокруг
оси макромолекулы. Модификация уравнения (6.67), произведен-
ная с целью включения в него трех типов концевых частиц (метил-
метакрилата, стирола и стирола с метилметакрилатом в качестве
предпоследнего звена), позволила успешно предсказывать зна-
чения /^0(12)*
6.4. Ионная сополимеризация
Ионная сополимеризация в некоторых отношениях существен-
но отличается от радикальной.
Ионная сополимеризация значительно селективнее, чем ради-
кальная. Число пар сомономеров, способных к сополимеризации
по катионному или анионному механизму, сравнительно ограни-
ченно вследствие более широкого диапазона значений реакционной
способности мономеров при ионной сополимеризации [56, 57].По
катионному механизму сополимеризуются мономеры, имеющие
электронодонорные заместители, а по анионному — электроно-
акцепторные. У способных к ионной сополимеризации пар сомоно-
меров наблюдается тенденция к сополимеризации по идеальному
типу (разд. 6.2в.1), если произведение г(г2 близко к единице
и активность обоих мономеров по отношению к обоим растущим
ионным центрам приблизительно одинакова. Тенденция к образова-
нию регулярно чередующихся сополимеров обычно полностью отсут-
ствует. Всего в нескольких случаях сополимеризации величина
произведения гхг2 превышает единицу. Так, имеется сравнительно
382
Глава 6
мало пар сомономеров, образующих сополимеры, содержащие
большие блоки, построенные из звеньев одного и другого моно-
меров.
Другой характерной особенностью ионной сополимеризации
является чувствительность констант сополимеризации мономеров
к изменению инициатора, реакционной среды или температуры,
тогда как при радикальной сополимеризации константы сополи-
меризации практически не зависят от условий реакции.
6.4а. Катионная сополимеризация
6.4а.1. Активность мономеров
Влияние заместителя на активность мономеров при катионной
сополимеризации зависит от того, в какой степени этот замести-
тель увеличивает электронную плотность двойной связи, и от его
способности к резонансной стабилизации образующегося иона
карбония. Однако порядок активности мономеров при катионной
сополимеризации (так же, как и при анионной) далеко не так
легко установить, как при радикальной сополимеризации. На
активность мономера часто более сильно влияют условия реакции,
чем строение мономера. В литературе описано сравнительно мало
работ, где изучали бы активность широкого круга сомономеров
в одинаковых условиях (растворитель, противоион, температура
реакции).
Наиболее широким исследованием активности мономеров были
работы, посвященные сополимеризации различных мета- и пара-
замещенных стиролов с другими стирольными мономерами (сти-
рол, tx-метилстирол и и-хлорстирол) [56, 58, 59]. Была проведена
корреляция относительной активности различных замещенных
стиролов с константами ст Гаммета. Графическая зависимость
lg l/fj от константы ст, характеризующей заместитель, выражается
прямой линией, как это и должно быть по методу Гаммета. Вели-
чина ст представляет собой количественную меру суммы резонанс-
ного и полярного влияний данного заместителя. Электроноакцеп-
торные и электронодопорные заместители имеют соответственно
положительные и отрицательные константы ст. Заместители увели-
чивают активность стирола приблизительно в том же порядке,
в котором возрастают их электронодонорные свойства, как это
видно из следующего ряда (в скобках приведены значения ст):
п-ОСНз > n-СНз > п-Н > га-С1 > Ж-С1 > ji-NO2
( — 0,27) ( — 0,17) (0) ( + 0,23) ( + 0,37) ( + 0,71)
Хотя метод Гаммета наиболее применим для установления ко-
личественной связи между активностью мономера и его структу-
рой, применим он только для замещенных стиролов. Однако боль-
Сополимеризация
383
ший интерес обычно представляют относительные активности та-
ких общеупотребительных мономеров, как изопрен, акрилонитрил
пропилен, изобутилен и т. д. Соответствующие количественные
исследования этих мономеров сравнительно редки и несистематич-
ны. Обычно наблюдается снижение активности мономеров в сле-
дующем ряду:
виниловые эфиры > изобутилен > стирол, изопрен.
В этом ряду мономеры расположены в порядке, которого можно
ожидать на основании электронодонорной способности различных
заместителей. Мономеры с электроноакцепторными заместителями,
такие, как акрилонитрил, метилметакрилат и винилхлорид, обла-
дают незначительной активностью при катионной сополимери-
зации.
Изучение сополимеризации показало, что влияние стерических
факторов, аналогичное тому, которое наблюдали при радикаль-
ной полимеризации, имеет место и при катионной полимериза-
ции. В табл. 6.11 показано влияние метильного заместителя стиро-
Таблица 6.11
Стерические эффекты при
сополимеризации а- и р-метилстиролов
[MJ с n-хлорстиролом |М2] под
влиянием SnCl4 в четыреххлористом
углероде при О °C [60—62]
Ml н Г2
Стирол 2,50 0,30
а-Метилстирол 15,0 0,35
тдаис-р-Метил-
стирол 0,32 0,74
гщс-р-Метилстирол 0,32 1,0
ла в а- и fJ-положении 160—62]. Метильная группа в «-положении
увеличивает активность мономера вследствие своего электроно-
донорного влияния. Пониженная активность Р-метилстирола по
сравнению со стиролом показывает, что стерические затруднения,
обусловленные ^-заместителем, превышают его полярное влияние,
увеличивающее электронную плотность двойной связи. Кроме
того, оказывается, что тракс-р-метилстпрол более активен, чем
^ас-изомер, хотя эта разница значительно меньше, чем в случае
радикальной сополимеризации (разд. 6.36.2). Следует отметить,
что 1,2-замещенные алкены имеют при катионной сополимериза-
ции конечные значения г в отличие от радикальной сополимериза-
ции, когда г этих мономеров равны нулю (табл. 6.2). При катион-
ной сополимеризации 1,2-дизамещенные алкены имеют тенденцию
к гомополимеризации, хотя в радикальных реакциях эта тенден-
ция отсутствует.
6.4а.2. Влияние растворителя и противоиона
Ранее было показано, что скорость катионной сополимериза-
ции может сильно изменяться при использовании различных раст-
ворителей и различных противоионов (разд. 5.2е). На константы
сополимеризации мономеров изменение растворителя или про-
тивоиона также может оказывать большое влияние, хотя это влия-
ние довольно сложно. Зависимость активности мономера от харак-
тера растворителя и противоиона проявляется примерно в 50%
исследованных систем. Огромное влияние, которое могут иметь
растворители, иллюстрируют представленные в табл. 6.12 данные
Таблица 6.12
Влияние растворителя и противоиона на константы
сополимеризации [63]
Г1 изобутилена Г2 п-хлорсти- рола Растворитель Катализатор
1,0 1,0 Гексан (8 = 1,8) А1Вг3
14,7 0,15 Нитробензол (е = 36) А1Вг3
8,6 1,2 Нитробензол (8 = 36) SnCl4
по сополимеризации изобутилена с п-хлорстиролом [63]. Кон-
станты сополимеризации, катализируемой бромистым алюминием,
имеют значения г = 1,0, г2 = 1,0 при проведении реакции в гек-
сане и г( = 14,7, г2 = 0,15 — в нитробензоле. Изменение значе-
ний констант сополимеризации объяснили преимущественной
сольватацией или комплексообразованием растущего свободного
иона и/или ионной пары в неполярной среде (гексан) более поляр-
ным мономером (n-хлорстирол). В результате повышенной кон-
центрации" n-хлорстирола в месте реакции вычисленные констан-
ты сополимеризации его имеют более высокие значения. В поляр-
ном нитробензоле растущие частицы полностью сольватированы
растворителем без участия n-хлорстирола, и изобутилен может
проявить свою повышенную активность.
Влияние растворителей на константы сополимеризации нельзя
рассматривать независимо от применяемого противоиона. Здесь,
как это было и в случае различных растворителей, в некоторых
Сополимеризация
385
11С1емах сомономеров наблюдается изменение значений г, и г2
в зависимости от катализатора, а в других — нет. Так, для систе-
Ь1 изобутилен — и-хлорстирол (табл. 6.12) значения г, и г2 при
латал язе бромистым алюминием и четыреххлористым оловом
„аз;п|хшы. Взаимовлияние растворителя и противоиона видно из
данных табл. 6.13 для сополимеризации стирола с п-метилстиро-
Таблица 6.13
Влияние растворителя и противоиона на состав сополимера
при сополимеризации стирола с w-метилстиролом а [64]
Содержание стирола в сополимере, %
Катализатор толуол (е - 2,4) 1,2-дихлор- этан (е = 9,7) нитробензол (е = 36)
SbCl5 46 25 28
А 1Хз 34 34 28
TiCl4, SnCI4, BF3-O(C2H5)2, 28 27 27
SbCl3
C13CCOOH 27 30
I2 17
а Соотношение мономеров в смеси 1:1.
лом [64]. Катализаторы расположены в порядке их активности
при гомополимеризации. Пятихлористая сурьма — самый силь-
ный катализатор, а иод — самый слабый. Этот порядок связан
с прочностью ионной пары.
Данные, приведенные в табл. 6.13, показывают, что состав
сополимера нечувствителен к силе катализатора при использова-
нии сильно полярных растворителей (1,2-дихлорэтан и нитробен-
зол). а также нечувствителен к полярности растворителей для
всех катализаторов, кроме самого сильного (SbCl5). Содержание
стирола в сополимере уменьшается с повышением полярности
растворителя при катализе SbCl5. Содержание стирола снижается
также по мере уменьшения силы катализатора при проведении
реакции в малополярном растворителе (толуоле). Эти результаты
можно объяснить с точки зрения влияния растворителя и противо-
иона па расстояние между ионами ионной пары и на селективное
комплексообразование ионной пары с одним из мономеров. В систе-
w стирол — и-метилстирол последний сомономер одновременно
и более полярный, и более активный. В слабом растворителе
(толуоле) мономеры конкурируют друг с другом и с растворителем
3S6 Глава 6
за комплексообразование с ионной парой. Более полярный н-ме.
тилстирол участвует в образовании комплекса в большей степени
п потому преимущественно включается в сополимер. Селектив-
ность возрастает с увеличением прочности ионной пары. В более
сильных растворителях противоион не оказывает существенного
влияния на реакцию, так как мономеры не могут конкурировать
с растворителем. При сополимеризации, катализируемой Sb(J.
увеличение сольватирующей способности реакционной среды так-
же уменьшает способность мономеров конкурировать с раствори-
телем за образование комплекса с ионной парой. Состав сополиме-
ра в таком случае в первую очередь определяется химической
активностью мономеров.
6.4а.З. Влияние температуры
Температура оказывает большее влияние на константы сополи-
меризации при катионном процессе, чем при радикальном, что
обусловлено изменением энергий активации реакций роста в боль-
шем интервале. Однако в этом случае отсутствует общая для всех
мономеров зависимость г от температуры, как это было при ради-
кальной сополимеризации. Некоторые значения г возрастают
с температурой, другие уменьшаются. Для различных пар сомоно-
меров наблюдались различные комбинации эффектов. Так, напри-
мер, для системы изобутилен — стирол увеличилась в 1,5 раза,
а г2 — в 3 раза при повышении температуры реакции от —90 до
—30 СС [65]. С другой стороны, для системы а-метилстирол —
/г-хлорстирол при изменении температуры от —78 до 0°С величи-
на ту уменьшилась от 28 до 15, а г2 увеличилась от 0,12 до 0,33 [61].
6.46. Анионная сополимеризация
6.46.1. Активность мономеров
Активность мономеров при анионной сополимеризации противо-
положна активности их при катионной сополимеризации. Актив-
ность увеличивается под влиянием электроноакцепторных замести-
телей, понижающих электронную плотность двойной связи или
стабилизирующих образующийся карбанион. Хотя имеющихся
данных недостаточно, установлено [57, 66], что по степени увели-
чения активности под влиянием заместителей последние можно
расположить в следующем порядке:
— CN > —COOR > —С6Н5, — СН = СН2 > —Н.
Мономеры с электронодон орными заместителями очень мало актив-
ны при анионной сополимеризации.
Сополимеризация
387
Основные особенности анионной сополимеризации очень сход-
ны с особенностями катионного процесса. В большинстве случаев
анионной сополимеризации наблюдается определенная тенденция
к «идеальному» поведению [67]. Стерические эффекты в некоторых
парах сомономеров приводят к возникновению тенденции к чере-
дованию. Так, система стирол — н-метилстирол [67] подчиняется
условиям идеальной сополимеризации с = 5,3, г2 = 0,18
и Гг __ 0,95, тогда как система стирол — а-метилстирол обнару-
живает определенную тенденцию к чередованию [68] с = 35,
г _ 0,003 и 7-р'г = 0,1. Стерическое влияние добавочного замести-
теля в а-положенпи затрудняет присоединение а-метилстирола
к аниону а-метилстпрола. Тенденция к образованию регулярно
чередующихся сополимеров наиболее сильно выражена в случае
сополимеризации стерически затрудненных 1,1-дифенилэтилена
и тра«с-1,2-дпфенилэтилена с бутадиеном, изопреном и 2,3-диме-
тилбутадиепом [69].
6.46.2. Влияние растворителя и противоиона
Константы сополимеризации и состав сополимера во многих
случаях анионной сополимеризации меняются при изменении про-
тивоиона или растворителя. В табл. 6.14 приведены данные для
Таблица 6.14
Влияние растворителя и противоиона
на состав сополимера при
сополимеризации стирола с изопреном
[70]
Растворитель Содержание стирола в сополимере, %
Na+ Li+
Без растворителя 66 15
Бензол 66 15
Триэтиламин 77 59
Этиловый эфир 75 68
Тетрагидрофуран 80 80
в попя СТИРОЛ — изопрен [70]. Растворители расположены
ной сопКе ИХ сольватиРУющей способности. Как и в случае катион-
теля не ЛимеРизаЦи0> ясно, что влияние противоиона и раствори-
ьзя рассматривать независимо друг от друга. Для прочно
388
Глава 6
связанного противоиона лития наблюдается сильно выраженное
влияние растворителя. В плохих растворителях сополимер обога-
щен менее активным изопреном, поскольку изопрен преимуще-
ственно образует комплексы с ионом лития. (Образование комплек-
сов диенов-1,3 с литием обсуждается в разд. 8.66.) Влияние раство-
рителя на менее прочно связанный ион натрия меньше.
Особенный интерес представляет сополимеризация метилмета-
крилата и стирола под влиянием лития в эмульсии и бутилли-
тия [71]. Блочная сополимеризация эквимолярной смеси обоих
мономеров с литием в эмульсии приводит к образованию сополиме-
ров с высоким содержанием стирола, тогда как при проведении
реакции в растворе в тетрагидрофуране содержание стирола
в сополимере ниже. Если при блочной сополимеризации в качестве
катализатора применяют бутиллитий, в сополимере практически
не содержится стирола. Для объяснения этих результатов было
постулировано, что инициирование при катализе литием проис-
ходит путем переноса электрона с образованием ион-радикалов,
радикальные концы которых растут с образованием сополимера
стирола с метилметакрилатом состава 50 : 50, тогда как анионные
концы растут с образованием полиметилметакрилата, практически
не содержащего стирола. Влияние тетрагидрофурана при литиевой
полимеризации обусловлено его ускоряющим влиянием на анион-
ную полимеризацию без существенного влияния на скорость
радикальной сополимеризации. Катализ бутиллитием представ-
ляет собой чисто анионное инициирование и приводит к образо-
ванию сополимера, являющегося почти чистым полиметилметакри-
латом.
При анионной сополимеризации стирол вступает в сополимер
лишь постольку, поскольку инициирование происходит на стиро-
ле и затем рост идет за счет стирола [72]. Растущие анионы поли-
метилметакрилата не имеют тенденции к присоединению стирола.
Однако анионы стирола легко присоединяют метилметакрилат.
Это различие связано с малой основностью аниона полиметил-
метакрилата. После присоединения к анионам стирола молекул
метилметакрилата стирол больше не присоединяется к этим цепям.
В результате этого вскоре после инициирования все растущие цепи
представляют собой полиметилметакрилат, и заметного присоеди-
нения стирола не происходит. Это поведение соответствует случаю
(разд. 6.2в.2), когда 1 и г2 <^1, и наблюдается при анион-
ной сополимеризации пар мономеров, анионы которых сильно
различаются по основности. Основность анионов изменяется парал-
лельно изменению значений е мономеров. Так, мономеры с близки-
ми значениями е, например стирол (е = —0,80) и бутадиен (е =
.= —1,05), вступают в сополимеризацию, тогда как мономеры
с различными значениями е, например стирол и метилметакри-
лат (е = 0,40), в первую очередь склонны к образованию в ка-
Сополимеризация
389
честве начального продукта гомополимера более активного
мономера.
Более новая работа 173] показала, что при реакции, иниции-
руемой литием, не происходит радикального роста, поскольку
в полученном сополимере не обнаружены статистические соче-
тания звеньев стирола и метилметакрилата. Сополимер состоит
из блок-сополимера двух мономеров и, возможно, некоторого
количества гомополимера метилметакрилата. Тот факт, что при
проведении сополимеризации в блоке сополимер обогащен стиро-
лом, объясняют нерастворимостью противоиона лития. Ион лития
представляет собой часть нерастворимой литиевой частицы, и рост
происходит на поверхности этой частицы. Стирол сильнее абсорби-
руется этими поверхностями, чем метилметакрилат, благодаря
своей высокой л-электронной плотности. В результате этого сопо-
лимеризация происходит при очень высоких концентрациях сти-
рола, и полученный продукт оказывается обогащенным этим моно-
мером. Общее содержание стирола в сополимере уменьшается
с конверсией, так как растущий конец цепи становится раствори-
мым и реакция протекает уже в растворе, где концентрация сти-
рола меньше. Растущие цепи стирола присоединяют метилметакри-
лат, и в результате образуется блок-сополимер. Поскольку анионы
полиметилметакрилата не способны к перекрестной реакции роста
со стиролом, содержание метилметакрилата в блок-сополимере
возрастает со степенью превращения. При проведении реакции
в более полярном растворителе, таком, как тетрагидрофуран, где
противоион литий растворяется лучше, содержание стирола
в сополимере также немного понижается, но этот эффект невелик.
Однако очень большое уменьшение содержания стирола наблю-
дается, когда в качестве катализатора используется бутиллитий.
В этом случае нет твердых частиц лития и рост происходит
в растворе.
в.о. Отклонения от уравнения состава
сополимера
Вывод уравнения состава сополимера [уравнение (6.12) или
(6.15)] основан на двух важных предположениях — кинетическом
и термодинамическом. Согласно первому предположению, реак-
ционная способность растущей частицы не зависит от природы
мономерного звена, предшествующего концевой группе; согласно
второму — различные реакции роста необратимы (отсутствует
деполимеризация). Отклонения от количественного поведения
системы, предсказанного уравнением состава сополимера, при
определенных условиях реакции объясняют несостоятельностью
того или другого предположения.
390
Глава 6
6.5а. Кинетический эффект предпоследнего звена
Поведение некоторых систем сомономеров показывает, что
природа предпоследнего звена растущей цепи влияет на реакцион-
ную способность растущей частицы. Этот эффект, который часто
называют эффектом предпоследнего звена, выражается в специфи-
ческой сополимеризации, в результате которой получают противо-
речивые значения констант сополимеризации для различных соот-
ношений смеси мономеров. Такое явление наблюдали во многих
случаях радикальной сополимеризации, когда мономеры содержа-
ли очень объемистые пли сильно полярные заместители. Так, Хэм
и Фордайс [74] сообщили, что при сополимеризации стирола (Mj)
с фумаронитрилом (М2) макромолекулы, богатые фумаронитрилом
и имеющие на конце стирол, отличаются значительно пониженной
активностью по отношению к фумаронптрилу. Это объясняется
стерпческимп и полярными отталкивающими эффектами между
предпоследним фумаронптрильным звеном растущей цепи и моле-
кулой присоединяющегося фумаронитрила.
Математическая обработка эффекта предпоследнего звена [75,
30] при сополимеризации такого рода включает восемь возможных
реакций роста
— MiMr+Mj
— MjMr + Mz
— M2M2.+Mj
— М,М2. + М2
—-M2Mr + M2
— MXM2. + Mj
— M4M2. + M2
kin
>
*412
&221 — M2M2Mr
*1222 — М2М2М2
*1211 —
*1212 — М2м,м2
*421 —
*1122 — MiM2M2.
(6.68)
с четырьмя константами сополимеризации
Ми
/Сц2 ’
^222
^221 ’
O =
- _ Mil
1 ^212 ’
' _ *<122
2~ М21
(6.69)
Таким образом, каждый мономер характеризуется двумя констан-
тами сополимеризации. Одна из этих констант относится к расту-
щим частицам, в которых предпоследнее и последнее звенья одина-
ковы. Другая представляет растущие частицы, в которых эти
Сополимеризация
391
звенья разные. В последнем случае константы сополимеризации
имеют верхний индекс (г\ и гг).
Способом, подобным тому, который был использован при выво-
де уравнения (6.12), было получено [75] уравнение состава сопо-
лимера с учетом эффекта предпоследнего звена
r;x(rtx+i)
' ^[^11 (г(Х + 1)
d [М21 4 , г2 (Г2~Ь^)
+ Х(т’2-\-Х)
г-1е Д' = [MJ/[М2]. Для системы стирол — фумаронитрил, в кото-
рой фумаронптрил не способен к гомополимеризации (г2 = г2 =
- 0), уравнение (6.70) упрощается [76] в выражение
__________ Л _______ (rjX -|~ 1)______/С г?Л \
d[Al2]________________________________(г(Х + 1) • 1 '
(6.70)
Значения, полученные при расчете по этому уравнению, удовлет-
ворительно совпадают с экспериментальными данными по соста-
ву сополимера (рис. 6.12) при г4 = 0,072 и г{ = 1,0 [77].
Эффект предпоследнего звена наблюдали и в других системах,
в тон числе при радикальной сополимеризации стирола с метил-
Рис. 6.12. Кинетический эффект
предпоследнего звена при ради-
кальной сополимеризации стиро-
ла (У,) с нитрилом фумаровой
кислоты (М2). Зависимость состава
сополимера как {(d [MJ/d [М2]) —
-11- от состава мономерной сме-
си А' [17].
т — вычислено по уравнению (6.71) при
’< = 0.072 и Г1 = 1,0; вычислено по
уравнению (6.73) при г, = 0,08, г[ =
= 0,3 п 1-1 = 4,0.
метакрилатом, а-метилстирола с акрилонитрилом и других пар
мономеров [30, 74, 78, 79]. Несмотря на то что ионная сополиме-
ризация изучена не столь широко, но и здесь в некоторых случаях
ооиаружепо влияние предпоследнего звена. Так, при сополимери-
зации стирола с винплпиридином, происходящей с переносом
электрона, вшшлпиридин быстрее присоединяется к цепям, имею-
Щпм па конце винилпиридин, если предпоследнее звено стироль-
ное [80]. Если предпоследним звеном является винилпиридин,
392
Глава 6
то он, очевидно, действует как ловушка электронов п тем самым
препятствует образованию связи с приближающимся мономером.
Проявляется ли эффект предпоследнего звена в той или иной систе-
ме, не всегда можно легко установить. Часто точность эксперимен-
тальных данных недостаточно высока, для того чтобы сделать
возможным использование уравнения (6.70).
Данные, полученные для системы стирол — фумаронитрил,
показали, что более удаленные звенья (т. е. звенья, предшествую-
щие предпоследнему звену) также влияют па активность мономера.
Хэм [77] учел этот эффект путем дальнейшего расширения уравне-
ния состава сополимера введением большего числа констант сопо-
лимеризации для каждого мономера. Для системы стирол (Mi)—
фумаронитрил (М ) константы сополимеризации
кщ , _____ ^11211 '•_А’21211 ii-
'1 7 , '4 » , • I 7 I / — )
«412 *11212 *21212
представляются важными, так как последовательность звеньев
МгМ^! имеет тенденцию к чередованию с M2Mj, о чем свидетель-
ствует невозможность получения сополимера, содержащего более
40 мол. % фумаронитрила. В этом случае уравнение состава сопо-
лимера имеет вид [77]
d [Mil
d [М2]
(r[X -|-1) |
r'lX+i
(6.73)
1 =
Из рис. 6.12 видно, что экспериментальные данные для системы
стирол — фумаронитрил точнее соответствуют уравнению (6.73)
при значениях = 0,08, = 0,3, г)' = 4,0, чем уравнению (6.71).
Это особенно очевидно при высоком содержании фумаронитрила
в смеси мономеров, где экспериментальные данные плохо согла-
суются с уравнением (6.71).
6.56. Деполимеризация в процессе сополимеризации
В противоположность кинетическому методу Хэма Лоури [81]
рассматривал отклонения от уравнения состава сополимера с тер-
модинамической точки зрения. Отклонение состава сополимера
от рассчитанного в некоторых случаях сополимеризации при таком
подходе объясняют склонностью одного из мономеров (М2)
к деполимеризации. Существенным различием кинетического
и термодинамического подходов является то, что последний пред-
полагает возможность изменения состава сополимера в зависи-
мости от концентрации мономеров. Если концентрация мономе-
ра М2 будет меньше его равновесной (критической) концентра-
ции [М]к при определенной температуре реакции, то концевые
Сополимеризация
393
звенья М2 будут склонны к деполимеризации. В результате этого
содержание мономера 2 в сополимере будет уменьшаться. Кинети-
ческий подход Хэма не предусматривает какой-либо зависимости
состава сополимера от концентрации мономеров. Далее, термоди-
намический подход отличается от кинетического тем, что он под-
черкивает зависимость состава сополимера от температуры, так
как равновесие процессов полимеризация — деполимеризация за-
висит от температуры.
Здесь будут рассмотрены такие процессы сополимеризации,
в которых только один из мономеров имеет склонность к деполи-
меризации. Системы, в которых оба мономера могут деполимеризо-
ваться, очень сложны с точки зрения математической обработки
и содержат много неизвестных параметров. Лоури рассматривает
систему, в которой мономер Mj абсолютно не способен к деполиме-
ризации независимо от типа предыдущего звена цепи, тогда как
мономер М2 не склонен к деполимеризации только в том случае,
если он присоединен к звену М4. При этом возможны два случая
в зависимости от способности мономера М2 к деполимеризации:
1) М2 склонен к деполимеризации, если он присоединен к дру-
гому звену М2
--MjM2M2* + (6.74)
2) мономер М2 склонен к деполимеризации, только если он
присоединен к последовательности из двух или более звеньев М2
— MjM2M2M? -------M!M2M.r + M2 (6.75)
Таким образом, при первом условии не деполимеризуется
только — М1М2, тогда как при втором условии не деполимери-
зуются ни — MiM*, ни— MiM2M*. Уравнение состава сополимера
при сополимеризации, включающей деполимеризацию согласно
Двум вышеназванным случаям, было выведено Лоури [81]. Уравне-
ние состава для первого случая описывается выражением
d[Mi] (п [M1] + [M2])(l-g) ,f 7fiv
d[M2] [М2] ’ k }
где а определяется как
_ ( 1 + К [М2] + ffill) - [( 1 + К [М2])2-4Х [М2]]1/2
• ’
(6-77)
К константа равновесия для уравнения (6.74).
394
Глава 6
Состав сополимера
rf [Ml]
d ]М2]
для второго случая дается уравнением
1 \2
(6.78)
с а, определяемой по
по формуле
уравнению (6.77), и у, рассчитываемой
а-ГМ 1Д-^ „
Л [МгН---z----а
г2
(6.79)
У
где К — константа равновесия уравнения (6.75).
, Уравнения (6.76) и (6.78) были недавно проверены экспери-
ментально на нескольких примерах [82, 83]. Была исследована
радикальная сополимеризация систем стирол — а-метилстирол,
стирол — метилметакрилат, акрилонитрил — а-метилстирол и
анионная сополимеризация винилмезитилена (2,4,6-триметилсти-
рола) с а-метилстиролом. Сополимеризацию этих систем изучали
при различных соотношениях исходной смеси мономеров, различ-
ных температурах реакций и различных концентрациях мономе-
ров. При изменении условий реакции в направлении, способ-
ствующем деполимеризации (повышение температуры, понижение
концентрации мономера М2), наблюдается переход от «нормаль-
ного» протекания сополимеризации [т. е. в соответствии с уравне-
нием (6.12)] к «ненормальному» протеканию ее (т. е. соответственно
уравнениям (6.76) и (6.78)]. Так, при радикальной сополимериза-
ции стирола с а-метилстиролом [82] при повышении температуры
реакции от 0 до 100 °C наблюдается значительное уменьшение
содержания а-метилстирола в сополимере. При повышенной тем-
пературе увеличивается деполимеризация а-метилстирола, обус-
ловленная влиянием предельной температуры (разд. 3.9в). Этот
эффект проявляется в наибольшей степени для смесей мономеров,
богатых а-метилстиролом. Данные, полученные для этой системы,
соответствуют ожидаемым в предположении механизма, соответ-
ствующего второму случаю деполимеризации.
Интересна анионная сополимеризация системы винилмезити-
лен — а-метилстирол [83]. Сначала эту сополимеризацию изучали
при —78 °C и при высоких концентрациях мономеров для опреде-
ления констант сополимеризации в условиях, когда деполимериза-
ция ничтожна. При повышении температуры до 0 °C деполимери-
зация оставалась незначительной, пока концентрация винил-
мезитилена [М2] была существенно выше равновесной концентра-
ции его [М2]к при этой температуре. При 0 °C [М2]к равна
0,75 моль/л. При уменьшении концентрации винилмезитилена
ниже равновесной и постоянном отношении [MJ/[M2] деполимери-
зация становится заметной и содержание винилмезитилена
Сополимеризация
395
сополимере уменьшается (рис. 6.13). Теоретические кривые
я этой сополимеризации для первого и второго случаев были
вычислены по уравнениям (6.76) и (6.78) с использованием значе-
ний [MJ, [М2], г2 и К. Значение К в этих вычислениях было
получено из данных по равновесию полимеризация — деполиме-
ризация в процессе гомополимеризации мономера М2. Можно
Рис. 6.13. Влияние деполимеризации на состав сополимера при анионной
сополимеризации впнплмезитилена (Mj) с а-метилстиролом (М2) при О °C,
когда /2 = 0,91 [83].
Пунктирные кривые — теоретические кривые, вычисленные для первого и второго слу-
чаев три г, = 0,20 и г, = 0,72); сплошная кривая — экспериментальные данные, и
видеть, что механизм, соответствующий второму случаю, значи-
тельно больше согласуется с экспериментальными данными, чем
механизм, соответствующий первому случаю. Это наблюдается
для большинства изученных систем.
Было также отмечено, что теоретическая кривая еще лучше
совпадает с экспериментальными данными, если допустить, что
мономер Mj тоже имеет склонность к деполимеризации. Лоури [81]
математически обработал эту систему, но полученный им результат
в настоящее время трудно применять для количественных иссле-
дований.
6.6. Сополимеризация диенов
6.6а. Сшивание цепей
Диеновые мономеры часто используют при сополимеризации
Для получения сшитых структур. Реакция в общем аналогична
поликонденсации, протекающей с участием трифункциональных
реагентов (разд. 2.8). В этих случаях сополимеризации сшивание
может происходить пли в ходе сополимеризации, или после нее
в зависимости от относительной активности обеих двойных связей
396
Глава 6
диенового мономера. Степень сшивания зависит от активности
двойных связей диена и от количества его в реакционной смеси
по отношению к другому мономеру. Точка, при которой происхо-
дит сшивание, была рассчитана математически для некоторых
представителей различных типов диенов [84, 85]. Во всех случаях
предполагается, что диен присутствует в небольших количествах,
так как именно так бывает обычно на практике.
Первый пример, который следует рассмотреть,— это сополиме-
ризация мономера А с диеновым мономером ВВ, когда все двойные
связи (т. е. двойная связь А и обе двойные связи В) имеют одина-
ковую активность. К системам такого типа относятся системы
метилметакрилат — диметакрилат этиленгликоля (ДМАЭГ), ви-
нилацетат — дивиниладипинат (ДВА) и стирол — дивинилбен-
зол (ДВБ). Точка гелеобразования, или критическая степень
завершенности реакции рк (т. е. количество прореагировавших
групп А), необходимая для начала гелеобразования, определяется
в этом случае уравнением
_ [А] + [В]
Рн — —
[В] xw
(6.80)
где [А] и [В] — концентрации двойных связей А и В (но не кон-
центрации мономеров) и X w — средневесовая степень полимериза-
ции линейных цепей. За Xw обычно принимают средневесовую
степень полимеризации, которая наблюдалась бы при полимериза-
ции мономера А в отсутствие диена ВВ. Уравнение (6.80) показы-
вает, что при полимеризации этого типа происходит интенсивное
сшивание. Так, в системе метилметакрилат — диметакрилат эти-
ленгликоля, содержащей 0,05 мол. % ДМАЭГ, гелеобразование
наблюдалось [86] при 12,5%-ной завершенности реакции. Было
Таблица 6.15
Образование поперечных связей при
сополимеризации стирола с дивинилбензолом
Содержание диви нил бензол а, мол. доли Точка гелеобразования рк
вычислено по уравнению (6.80) определено эксперименталь- но [8 7]
0,004 0,21 0,16
0,008 0,10 0,14
0,02 0,042 0,076
0,032 0,026 0,074
0,082 0,010 0,052
0,30 0,0042 0,045
Сополимеризация
397
(6.82)
установлено, что уравнение (6.80) применимо для предсказания
точки гелеобразования для сополимеризации различного типа
[86, 87]. В табл. 6.15 приведены данные для сополимеризации
системы стирол — дивинилбензол. Уравнение лучше всего удов-
летворяет системам, содержащим диеновый мономер в низких
концентрациях; при повышении концентрации диена полезность
уравнения уменьшается. При увеличении концентрации диена
предсказываемые уравнением (6.80) точки гелеобразования соот-
ветствуют более низким степеням превращения, чем наблюдаемые
экспериментально, причем эти расхождения увеличиваются. Такое
расхождение приписывалось потере диенового мономера вслед-
ствие внутримолекулярной циклизации (разд. 6.66).
Второй пример — это сополимеризация мономеров А и ВВ,
у которых активности двух групп не одинаковы и равны соответ-
ственно т\ и г2. В этом случае критическая степень завершенности
реакции определяется уравнением
= _(г1[А]2 + 2[А][В] + г2[Вр)2
Рк Xw [В] ([А] + [В]) (г2 [В] + [А]Р ’
которое упрощается до выражения
v = -Щ_
Р" [В] xw
при LA] [В]. Если двойные связи диена более активны, чем
двойная связь другого мономера (т. е. г2 > г(), то сшивание про-
исходит на ранних стадиях полимеризации. Сшивание задержи-
вается до более поздних стадий, если > г2. Для систем, где
Г1 > г2 п /д > 1, сшивание при данной степени завершенности
реакции не столь интенсивно, как при сополимеризации, проте-
кающей по уравнению (6.80), где гх — г2.
Третий пример — это сополимеризация мономера А с диеном
ВС, когда группы А и В имеют одинаковую активность, а актив-
ность группы С значительно меньше. К такому типу сополиме-
ризации относится система метилметакрилат — аллилметакрилат,
гДе А п В — это две метакрилатные группы, а С — аллильная
группа. Если г — отношение активностей групп С и В, то для ра-
дикалов, получаемых из групп А и В, можно написать следующее
соотношение:
= кАС = kAC ^ВС ^ВС /р оо\
кАА кАВ кВА ^ВВ
На первых стадиях реакции образующийся сополимер состоит
из прореагировавших друг с другом групп А и В и содержит непро-
реагировавшие группы С в виде боковых привесков. Вследствие
низкой активности групп С сшивание происходит только на срав-
нительно поздних стадиях. Критическая степень завершенности
398
Глава 6
реакции в этом случае определяется как
Л = 1-ехр (-^-) , (6.84)
где q — мольная доля диена в исходной смеси мономеров.
При выборе сшивающей системы следует учитывать ряд пере-
менных, чтобы контролировать процесс. Так, гелеобразование
можно задержать, получая несшитый продукт с высокой степенью
полимеризации, который способен к последующему сшиванию.
Гелеобразование можно задержать также, уменьшая количество
диена, снижая степень полимеризации применением агентов пере-
носа цепи или выбирая диен, у которого активность одной из групп
значительно меньше активности другой группы. Степень сшивания,
конечного продукта также контролируется этими переменными.!
Так, интенсивное сшивание с образованием плотно сшитых струк-i
тур происходит в отсутствие агентов передачи цепи и при исполь-
зовании повышенных количеств диена, имеющего двойные связи
одинаковой активности.
Часто встречается особое положение, когда реакция одной из
двойных связей диена уменьшает активность остающейся двойной
связи. Если уменьшение активности велико, то оно приводит,
к заметному замедлению сшивания. В этом случае система стано-
вится аналогичной примеру, когда активность группы С значитель-
но меньше активности групп А и В. Наиболее важным процессом,
в котором происходит сильное уменьшение активности второй
группы после реакции первой, является сополимеризация дие-
нов -1,3, когда полимеризация в положения 1,4 приводит к обра-
зованию остаточных двойных связей 2,3, имеющих меньшую
активность. Эти связи используют для осуществления последующе-
го сшивания по реакциям, описанным в разд. 9.2.
Сшивание может происходить также в ходе гомополимеризации
диенов. Иногда это — нежелательное явление, причиняющее боль-
шие затруднения при полимеризации диенов-1,3. По мере проте-
кания реакции имеется возможность, сшивания вследствие сополи-
меризации непрореагировавшего мономера с образовавшимися
двойными связями 2,3 прореагировавшего мономера. Число сши-
вок р, образовавшихся в первоначально линейном продукте в рас-
чете на мономерное звено, зависит от степени завершенности,
как было показано в работе [88], следующим образом:
р= _(1) ^1 + (±)1п(1-^)] , (6.85)
где г — константа сополимеризации диена-1,3, представляющая
собой отношение констант скоростей присоединения растущего
радикала к мономерному диену и двойной связи 2,3 в полимере.
Поскольку точка гелеобразования будет достигнута при р, рав-
Сополимеризация
399
м 1 Хи-, уравнение (6.85) для критической степени завершен-
ности реакции приобретает вид
<6-8в>
-1/(1 ' / I— ' i Г\ ' —1
Установлено, что бутадиен-1,3 при 50 °C имеет значение г,
равное 7,4 -103, т. е. сшивание в этом случае в заметной степени
происходит только на поздних стадиях реакции.
6.66. Сополимеризация, происходящая с чередованием
внутри- и межмолекулярных реакций
Сополимеризация несопряженных диенов (например, аллилфта-
лата, бпс-аллилкарбопата этиленгликоля, диаллилмалеината)
п триенов (например, триаллилцианурата), происходящая с обра-
зованием сильно сшитых термореактивных продуктов, является
практически важным процессом. Это относится также к исполь-
зованию таких мономеров для сшивания ряда сополимерных
спетом (разд. 6.6). (Математическая обработка [90] процесса сши-
вания в несопряженных диенах аналогична обработке, описанной
выше для сопряженных диенов.) Эффективность реакции сшивания
почти всегда оказывается меньше, т. е. определяемая экспери-
ментально точка гелеобразования всегда соответствует более высо-
ким степеням завершенности реакции, чем это предсказывается
теорией. Происходит потеря диенового и триенового мономера,
которую приписывают внутримолекулярной циклизации диена
или трпена [91, 92]. Это явление встречается не только при гомо-
п сополимеризации диенов и триенов, но и при трехмерной поли-
конденсации (разд. 2.8в).
Конкуренцию между внутри- и межмолекулярными реакциями
при полимеризации диенов и триенов можно изобразить следую-
щим образом:
СН2
II
СН
СН2 = СН — Z ______
сн2
II
сн
сн2 -I
II
сн
I
Z
I
сн2—сн
— 1-сн2сн-1п-
IV
—> Сшивание пу-
тем полимери-
зации по двой-
ным связям
боковых групп
I
Z
Внутримолекулярная
циклизация
(6.87)
400
Глава 6
Z
— сн2-сн 'сн.
V \н2
I СН2
Межмоле- ,
кулярный Д
рост С.п
Z Z
/ \ | Межмолекулярный рост
— СН2-СН СН —СН2 —СН-------------------------> (6.88)
Хсщ
VI
| Внутримолекулярная
I циклизация
-сн2-сн\н—
\н2
(6.89)
где Z — такие структурные группы, как бензольное кольцо
в дивинилбензоле или триметиленовая группа в гептадиене-1,6.
Полимеризация за счет межмолекулярных реакций [уравнение
(6.87)] происходит с образованием линейных полимеров, содержа-
щих двойные связи в боковых группах. Полимеризация этих
двойных связей либо с мономером, либо с двойными связями дру-
гих цепей приводит к образованию конечного сшитого продукта.
Степень сшивания зависит от факторов, описанных в разд. 6.6а.
Циклизация происходит, когда растущая частица IV реагирует
со своей двойной связью, находящейся в виде подвеска. (Анало-
гичные схемы реакций описывают внутримолекулярную циклиза-
цию при полимеризации триенов или при сополимеризации
систем, в которых одип из сомономеров является диеном или три-
епом.)
Важность реакции внутримолекулярной циклизации стала
особенно ясна после работ Батлера с сотр. [93, 94], установивше-
го, что радикальная полимеризация четвертичных аммониевых
солей дпаллпла приводит к образованию растворимых несшитых
полимеров с очень малой остаточной ненасыщенностью или даже
вообще без нее. В этом случае нет заметной тенденции к росту
радикала VI за счет межмолекулярного взаимодействия; основной
реакцией является внутримолекулярная циклизация. Реакция
идет по уравнению (6.89); тенденции протекания реакции (6.88)
Сополимеризация
401
не наблюдается. Полимер представляет собой полностью линейный
продукт с циклами в основной цепи
СН — СН
(6.90)
Полимеризация этого типа называется полимеризацией с чередо-
ванием внутри- и межмолекулярных реакций или циклополиме-
ризацией. Ее можно рассматривать как последовательное чере-
дование стадий внутримолекулярного и межмолекулярного
роста.
Широкие исследования, проведенные Батлером и Марвелом
с сотрудниками, а также рядом других исследователей [91—98],
показали, что именно размер кольца, которое может образоваться
из данного мономера, определяет предпочтительный для этого
мономера механизм реакции: межмолекулярный рост или внутри-
молекулярную циклизацию. Степень циклизации обычно увели-
чивается в зависимости от размера цикла в следующем порядке:
степень циклизации для пяти- и шестичленных циклов значитель-
но больше, чем для семичленных, а степень циклизации последних
несколько больше, чем для циклов большего размера. Если могут
образовываться пяти- или шестичленные циклы, протекает почти
исключительно циклополимеризация. Такое явление наблюдали
для большого числа мономеров, в том числе для 2,6-диметилен-
пимелиновой кислоты и ее производных, ангидрида акриловой
кислоты, замещенных гексадиенов-1,5 и гептадиенов-1,6, а также
для гептадиина-1,6. При этом следует отметить, что аналогичные
результаты получены при радикальной, анионной и катионной
полимеризации.
Степень циклизации довольно сильно уменьшается при перехо-
де к циклам из семи и более членов. Однако вопреки ожиданиям
степень циклизации для мономеров, способных образовывать
при полимеризации такие циклы, достаточно велика. Так, степень
циклизации (выраженная как процентное содержание зациклизо-
ванных звеньев) для диаллиловых эфиров, при полимеризации
которых образуются циклы, содержащие до 17 членов [91], состав-
ляет 15—20%. Еще более неожиданно, что при полимеризации
о-диаллилфталата получается полимер со степенью циклизации
риолизительно 40%. Циклическая структура в этом случае
402
Глава 6
представляет собой одиннадцатичленное кольцо
соосн2сн=сн2
соосн2сн=сн2
Аналогичные наблюдения были сделаны при исследовании других
мономеров.
Далее было установлено, что при полимеризации диенов-1,4,
например дивинилового эфира и некоторых метилзамещенных
мономеров типа диен-1,4-онов-З, также образуются продукты
с малой остаточной ненасыщенностью и незначительной степенью
сшивания [98]. В этих случаях происходит внутримолекулярная
циклизация, но механизм ее иной, так как здесь образовывались бы
неустойчивые четырехчленные циклы. Реакция протекает по схеме
где Z представляет собой группы СН2, О, S или СО. Циклизация
происходит через последовательность реакций уравнения (6.92)
с димерным радикалом (или ионом при ионной полимеризации) VII.
Может происходить также вторичная циклизация согласно урав-
нению (6.93), в результате которой образуются бициклические
структуры VIII.
Сополимеризация
403
Здесь полезно вспомнить описанную в разд. 2.3 конкуренцию
тппейиой поликонденсации и циклизации. При образовании более
чем семичленных циклов циклизация не способна конкурировать
с синтезом линейных полимеров. Даже для большинства реаген-
тов, которые способны давать при циклизации шести- и семичлен-
ные циклы, можно сдвинуть процесс в сторону линейной поли-
конденсации, благодаря способности к взаимопревращению цикли-
ческих и линейных структур. Различие между поведением при
полимеризации и при поликонденсации частично связано с тем,
что циклические структуры при полимеризации не способны
к деполимеризации при условиях реакции, т. е. циклические
структуры в этом случае не способны к взаимопревращениям
с линейной структурой. Но основным различием в этих двух слу-
чаях является то, что радикалы IV и VII (или соответствующие
ионные частицы при ионной циклополимеризации) значительно
более активны в отношении циклизации при полимеризации, чем
соответствующие реагенты при поликонденсации. Причина этого
не установлена, но это может быть просто связано со стерическими
затруднениями, возникающими между группой —СН = СН2 расту-
щей частицы IV и приближающейся молекулой мономера.
6.7. Другие случаи сополимеризации
6.7а. Сополимеризация алкенов
Установлено, что многие соединения, в том числе двуокись
серы, окись углерода, фосфины, нитрозосоединения, кислород
и хиноны, которые в силу затруднений, обусловленных полярны-
ми или стерическими эффектами, не способны к гомополимериза-
цин, легко вступают в радикальную сополимеризацию с алкенами,
образуя полимерные сульфоны [99, 100], кетоны [101], фосфины
[102], аминоокиси [103, 104], перекиси [105] и эфиры [106]
СН, = CHR + SO2 —> - (- CH2CHR _SO2 —)„ —, (6.94)
CH2 = CHR + CO — (—CH2CHR —СО —)n —, (6.95)
ch2=chr + r;p — (—ch2chr —PR3—)n—, (6.96)
R' -I
CH2 = CHR + R'~no —— CH2CHR —N — O —_ n—, (6.97)
CH2-_CHR^O2 —^>—( —CH2CHR —O —O —)n —, (6.98)
(6.99)
404
Глава 6
Вероятно, наиболее изученной реакцией является реакция
с двуокисью серы. Относительно электроотрицательные алкены
например этилен, а-олефины, 1,2-дизамещенные олефины, аллило-
вые эфиры, винилхлорид и стирол, легко сополимеризуются
с двуокисью серы. Такие мономеры, как метилакрилат, имеющие
электронооттягивающие заместители, не дают таких полимеров
вследствие полярного отталкивания с электроположительной дву-
окисью серы. Многие из простых олефинов образуют чередующие-
ся полисульфоны эквимолярного состава [99, 107] вследствие
полярного взаимодействия электроотрицательного алкена
с электроположительной двуокисью серы. Другим объяснением
тенденции к образованию чередующихся структур в этом случае
является то, что SO2 образует с олефином комплекс 1:1, который
затем реагирует как мономерная единица.
Мономеры, содержащие объемистые заместители, такие, как
стирол и винилхлорид, дают полисульфоны, содержащие меньше
SO2, чем алкенового мономера. Это обусловлено, очевидно, стери-
ческими препятствиями, создаваемыми заместителем алкена и гро-
моздкой молекулой двуокиси серы. Расчет кинетики и состава
сополимера для этих систем осложнен наличием деполимеризации
(разд. 6.66) в ходе реакции [107]. Влияние деполимеризации
повышается по мере увеличения степени замещения мономера
и увеличения размеров заместителя; соответственно увеличивается
и содержание серы в сополимере. Если мономер имеет два объе-
мистых заместителя у одного атома углерода, как, например,
2-метилбутен-2, то сополимеризации не происходит.
Очевидно, в других системах также имеется определенная
тенденция к чередованию, хотя подробных данных для этих систем
нет. Так, реакция м&кду CF3NO и G2F4 приводит к образованию
чередующегося сополимера состава 1 : 1 [104], и константы сополи-
меризации этилена и окиси углерода равны соответственно 0,04
и 0 [100]. Реакции алкенов с хинонами и перекисями недостаточно
хорошо изучены с точки зрения стехиометрии продуктов. Эти две
реакции лучше отнести к реакциям замедления, или ингибирова-
ния, вследствие очень малых скоростей сополимеризации
(разд. 3.7а).
6.76. Сополимеризация карбонильных мономеров
Хотя гомополимеризация карбонильных мономеров исследова-
на очень широко (разд. 5.5), по сополимеризации этих мономеров
имеется всего несколько работ
RCHO + R'CHO CHR-O-CHR'-O-),, —. (6.100)
Проведена катионная сополимеризация ацетальдегида с формаль-
дегидом и рядом других алифатических альдегидов [108, 109].
Сополимеризация
405
Описана [110, 111] анионная сополимеризация для формальдеги-
да, ацетальдегида и различных хлорзамещенных ацетальдегидов.
Многие из этих исследований неполны или недостаточно точны,
для того чтобы дать ясную картину реакции. В некоторых случаях
не ясно, образуются ли статистические или блок-сополимеры.
В других случаях оказалось важным влияние специфического
катализатора. В зависимости от применяемого катализатора пара
сомономеров может давать гомополимер, блок-сополимер или ста-
тистический сополимер. Общей тенденцией при сополимеризации
карбонильных мономеров является стремление к идеальному про-
цессу. Однако у пар мономеров, один из которых имеет объемистые
заместители, наблюдается ясно выраженная тенденция к образова-
нию сополимеров с регулярно чередующимися звеньями. Так, при
анионной сополимеризации ацетальдегида и хлораля при —78 °C
= 0,18 п г2 = 0,00) обнаруживается четкая тенденция к чере-
дованию [110].
Сополимеризация карбонильных мономеров с алкенами изуче-
на еще меньше, чем сополимеризация различных карбонильных
мономеров. Более или менее подробно исследована радиационная
сополимеризация стирола с формальдегидом [112]. Судя по тому,
что скорость сополимеризации формальдегида с различными алке-
нами уменьшается в ряду
а-метилстирол > стирол > метилметакрилат > акрилонитрил,
можно предположить, что реакция протекает по катионному
механизму. Система формальдегид (MJ — стирол (М2) при —78 °C
ведет себя как идеальная система с = 52 и г2 = 0. Продукт
реакции, состоящий в основном из формальдегида, очень интере-
сен с практической точки зрения, так как обладает пониженной
склонностью к термической деструкции по сравнению с чистым
полиформальдегидом. Очевидно, небольшое количество присут-
ствующих в цепи сополимера звеньев стирола препятствует про-
цессу деполимеризации.
Вопросы и упражнения
6.1. Обсудите различия в структуре статистических, регулярно
чеР*;ДУЮщихся, привитых и блок-сополимеров.
Какое различие между «идеальным» поведением системы
Ри сополимеризации и стремлением к образованию сополимеров
ч|РеДующимися звеньями?
па,, ' ‘ Рассмотрите следующие константы сополимеризации для
Различных пар мономеиов:
406
Глава 6
Пример п г2
1 0,1 0,2
2 0,1 10
3 0,1 3
4 0 0,3
5 0 0
6 0,8 2
7 1 15
Каков в каждом примере состав сополимера, полученного при
малых степенях превращения из эквимолярных смесей обоих
мономеров?
6.4. Пользуясь значениями и г2 из табл. 6.2, постройте кри-
вые, выражающие зависимость начального состава сополимера
от состава смеси мономеров при радикальной сополимеризации
метилакрилата с метилметакрилатом и стирола с малеиновым
ангидридом. Примерами идеальной или регулярно чередующейся
сополимеризации являются эти процессы?
6.5. Вычислите состав терполимера, который может быть полу-
чен при радикальной сополимеризации раствора, содержащего
акрилонитрил, стирол и бутадиен-1,3 в концентрациях 0,47, 0,47
и 0,06/И соответственно.
6.6. Обсудите влияние активности мономера и радикала на
наблюдаемую активность реакций радикала с мономером. В каком
порядке можно расположить следующие мономеры на основании
их активности: стирол, винилацетат, метилметакрилат, акрило-
нитрил? В каком порядке изменяется активность радикалов, полу-
ченных из этих мономеров?
6.7. Расположите следующие мономеры в порядке возрастания
склонности к образованию чередующихся сополимеров при ради-
кальной сополимеризации с бутадиеном-1,3:
а) «-бутилвиниловый эфир,
б) метилметакрилат,
в) метилакрилат,
г) стирол,
д) малеиновый ангидрид,
е) винилацетат,
ж) акрилонитрил.
Объясните относительную склонность мономеров к чередова-
нию в этих системах.
6.8. Вычислите константы сополимеризации мономеров для
систем стирол — бутадиен и стирол — метилметакрилат, ноль-
Сополимеризация
407
зуясь значениями Q и е из табл. 6.7. Сравните полученные резуль-
таты со значениями и г2, приведенными в табл. 6.2.
6.9. Пользуясь методом произведения вероятностей [уравне-
ния (6.52) — (6.54)], вычислите константы сополимеризации моно-
меров системы стирол — бутадиен. Сравните полученные резуль-
таты с результатами, полученными при помощи схемы Q — е,
и со значениями, приведенными в табл. 6.2.
6.10. Какой качественный состав сополимеров можно ожидать
при катионной сополимеризации систем, перечисленных в вопро-
се 6.7? Расположите сополимеры в порядке увеличения содержа-
ния в них бутадиена. Из всех ли перечисленных пар сомономеров
будут образовываться сополимеры? Объясните. Что будет проис-
ходить при анионной сополимеризации?
6.11. Обсудите влияние температуры, растворителя и катали-
затора на константы сополимеризации мономеров при ионном
процессе. Сравните влияние этих факторов при ионной и радикаль-
ной сополимеризации.
6.12. Каковы различия между двумя подходами (кинетическое
влияние предпоследнего звена и деполимеризация), используемы-
ми для объяснения наблюдаемых отклонений от значений, вычис-
ленных по уравнению состава сополимера.
6.13. Обсудите качественно ход радикальной сополимеризации
для каждой из перечисленных ниже систем сомономеров с точки
зрения степени превращения, при которой ожидается гелеобразо-
вание:
а) стирол — дивинилбензол,
б) метилметакрилат — аллилметакрилат,
в) винилацетат — диметакрилат этиленгликоля,
г) метилметакрилат — дивиниладипат,
д) стирол — бутадиен.
6.14. Продукт, полученный при полимеризации 4-метилгепта-
диена-1,6, не содержит ненасыщенных связей. Какова его хими-
ческая структура?
Список литературы
1. lox R. В. et al., Polymer Preprints, 8(1), e(1967); 8(2), e(1967).
"• Staudinger H., Schneiders J., Ann., 541, 151 (1939).
3. Alfrey T., Jr., Goldfinger G., J. Chem. Phys., 12, 205 (1944).
4. Mayo F. R., Lewis F. M., J. Am. Chem. Soc., 66, 1594 (1944).
5. Wall F. T., J. Am. Chem. Soc., 66, 2050 (1944).
6. Уоллинг Ч., Свободные радикалы в растворе, гл. 4, изд-во «Мир», М./1960.
'тГ1}1!п,еуег р- > Textbook of Polymer Science, chap. 11, Interscience
r л?,.®’ John Wiley and Sons, Inc., New York, 1962.
. Melville H. W., Noble B., Watson W. F., J. Polymer Sci., 2, 229 (1947).
10 joldlinger G., Kane T., J. Polymer Sci., 3, 462 (1948).
Ji ^andler Y., Compt. rend., 230, 539 (1950).
12 MPperrP- C" Quart- Rev- (London), 8, 88 (1954).
. Mayo F. R., Walling C., Chem. Rev., 46, 191 (1950).
408
Глава 6
13. Gumbs R., Penczek S., Jagur-Grodzinski J., Szwarc M., Macromolecules
2, 77 (1969).
14. Kruse R. L., J. Polymer Sci., B5, 437 (1967).
15. Skeist I., J. Am. Chem. Soc., 68, 1781 (1946).
16. Meyer V. E., Lowry G. G., J. Polymer Sci., A3, 2843 (1965).
17. Meyer V. E., Chan R. K. S., Polymer Preprints, 8(1), 209 (1967).
18. Fineman M., Ross S. D., J. Polymer Sci., 5, 259 (1950).
19. Tidwell P. W., Mortimer G. A., J. Polymer Sci., A3, 369 (1965).
20. Meyer V. E., J. Polymer Sci., A-l(4), 2819 (1966).
21. Marder H. L., Schuerch С., J. Polymer Sci., 44, 129 (1960).
22. Yamashita У., Ito K., Ikuma S., Kida H., J. Polymer Sci., B6, 219 (1968).
23. Wilkes С. E., Westjahl J. C., Backderf R. H., Polymer Preprints, 8(1),
386 (1967).
24. Stockmayer W. H., J. Chem. Phys., 13, 199 (1945).
25. Phillips G., Carrick W., J. Am. Chem. Soc., 84, 920 (1962), J. Polvmer
Sci., 59, 401 (1962).
26. Пиблс Л. Г., Сополимеризация акрилонитрила, в кн. «Сополимериза-
ция», под ред. Дж. Хэма, гл. 11, изд-во «Химия», М., 1971.
27. Alfrey Т., Jr., Goldfinger G., J. Chem. Phys., 12, 332 (1944).
28. Valvassori A., Sartori G., Fortschr. Hochpolymer-Forsch. (Advan. Polymer
Sci.), 5, 28 (1967).
29. Ham G.' E., J. Polymer Sci., A2, 2735 (1964); J. Macromol. Chem., 1,
403 (1966).
30. Хэм Дж. E., Теория сополимеризации, в кн. «Сополимеризация», под
ред. Дж. Хэма, гл. 1, изд-во «Химия», М., 1971.
31. Walling С., Briggs Е. R., J. Am. Chem. Soc., 67, 1774 (1945).
32. Alfrey Т., Jr., Goldfinger G., J. Chem. Phys., 14, 115 (1946).
33. Ham G. E., J. Polymer Sci., A2, 4191 (1964).
34. Lewis F. M., Walling C., Cummings W., Briggs E., Mayo F. R., J. Am.
Chem. Soc., 70, 1519 (1948).
35. Burkhart R. D., Zutty N. L., J. Polymer Sci., 57, 593 (1962); Al, 1137
(1963).
36. Thompson B. R., Raines R. H., J. Polymer Sci., 41, 265 (1959).
37. Brown F. E., Ham G. E., J. Polymer Sci., A2, 3623 (1964).
38. Brandup J., Immergut E. H., (eds.), Polymer Handbook, Interscience
Publishers, John Wiley and Sons, Inc., New York, 1966.
39. Evans M. G., Discussions Faraday Soc., 2, 271 (1947).
40. Dawson T. L., Lundberg R. D., Welch F. J., J. Polymer Sci., A-l(7), 173
(1969).
41. Baldwin M. G., J. Polymer Sci., A3, 703 (1965).
42. Price С. C., J. Polymer Sci., 3, 772 (1948).
43. Ito T., Otsu T., J. Macromol. Sci. (Chem.), A3, 197 (1969).
44. Walling C. et al., J. Am. Chem. Soc., 70, 1537, 1544 (1948).
45. Gaylord N. et al., J. Polymer Sci., B6, 743, 749 (1968); B7, 145 (1969);
Polymer Preprints, 10(2), 546, 554 (1969).
46. Alfrey T., Jr., Price С. C., J. Polymer Sci., 2, 101 (1947).
47. Алфрей T., мл., Юнг Л. Дж., Схема Q — е, в кн. «Сополимеризация»,
под ред. Дж. Хэма, гл. 2, изд-во «Химия», М., 1971.
48. O'Driscoll К. F., Yonezawa Т., J. Macromol. Sci.— Revs. Macromol.
Chem., 1(1), 1 (1966).
49. Ham G. E., General Aspects of Free Radical Polymerization, in «Vinyl
Polymerization», Ham G. E. (ed.), vol. 1, part I, chap. 1, Marcel Dekker,
Inc., New York, 1967.
50. Mayo F. R., J. Polymer Sci., A-2, 4207 (1964).
51. O'Driscoll K. F., J. Polymer Sci., B-3, 305 (1965).
52. Walling С., J. Am. Chem. Soc., 71, 1930 (1949).
53. North A. M., Polymer, 4, 134 (1963).
Сополимеризация
409
54. Atherton J. N., North A. M., Trans. Faraday Soc., 58, 2049 (1962).
55^ O'Driscoll K. F., Wertz W., Husar A., Polymer Preprints, 8(1), 380 (1967).
56^ Кеннеди Дж. П., Катионная сополимеризация, в кн. «Сополимеризация»,
под ред. Дж. Хэма, гл. 8, изд-во «Химия», М., 1971.
57. Мортон М., Анионная сополимеризация, в кн. «Сополимеризация»,
под ред. Дж. Хэма, гл. 9, изд-во «Химия», М., 1971.
58. Dunphy J. F., Marvel С. S., J. Polymer Sci., 47, 1 (1960).
59. Кандалл Р., Сополимеризация, в кн. «Катионная полимеризация», под
ред. П. Плеша, гл. 15, изд-во «Мир», М., 1966.
60. Overberger С. G., Ehrig R. J., Tanner D., J. Am. Chem. Soc., 76, 772 (1954).
61. Overberger C. G., Tanner D. H., Pearce E. M., J. Am. Chem. Soc., 80,
4566 (1958).
62 Overberger C. G., Arnold L. H., Taylor J. J., J. Am. Chem. Soc., 73, 5541
(1951).
63. Overberger C. G., Kamath V. G., J. Am. Chem. Soc., 81, 2910 (1959).
64. O’Driscoll K. F., Yonezawa T., Higashimura T., J. Macromol. Chem.,
1, 17 (1966).
65. Rehner J., Zapp R. L., Sparks W. J., J. Polymer Sci., 9, 21 (1953).
66. Szwarc M., Smid J., Kinetics of Propagation of Anionic Polymerization
and Copolymerization, in «Progress in Reaction Kinetics» Porter G. (ed.),
vol. 2, chap. 5, Macmillan Company, New York, 1964.
67. Shima M., Bhattacharyya D. N., Smid J., Szwarc M., J. Am. Chem. Soc.,
85, 1306 (1962).
68. Bhattacharyya D. N., Lee C. L., Smid J., Szwarc M., J. Am. Chem. Soc.,
85, 533 (1963).
69. Yuki H., Kosai K., Murahashi S., Hotta J., J. Polymer Sci., B2, 1121
(1964).
70. Kelley D. J., Tobolsky A. V., J. Am. Chem. Soc., 81, 1597 (1959).
71. O’Driscoll K. F., Tobolsky A. V., J. Polymer Sci., 31, 115, 123 (1958).
72. O’Driscoll K. F., J. Polymer Sci., 57, 721 (1962); B3, 43 (1965).
73. Overberger C. G., Yamamoto N., Polymer Preprints, 7(1), 115 (1966).
74. Fordyce R. G., Ham G. E„ J. Am. Chem. Soc., 73, 1186 (1951).
75. Merz E., Alfrey T., Jr., Goldfinger G., J. Polymer Sci., 1, 75 (1946).
76. Barb W. G., J. Polymer Sci., 11, 117 (1953).
77. Ham G. E., J. Polymer Sci., 45, 177 (1960).
78. Guyot A., Guillot J., J. Macromol Sci. (Chem.), Al(5), 793 (1967).
79. Hecht J. K., Ojha N. D., Macromolecules, 2, 94 (1969).
80. Lee C., Smid J., Szwarc M., Trans. Faraday Soc., 59, 1192 (1963).
81. Lowry G. G., J. Polymer Sci., 42, 463 (1960).
82. OHdriscoll K. F., Gasparro F. P., J. Macromol. Sci. (Chem.), Al(4), 643
83. Ivin K. J., Spensley R. H., J. Macromol. Sci. (Chem.), Al(4), 653 (1967).
84. Алфрей T., Боер Дж., Марк Г., Сополимеризация, гл. 9, 10, ИЛ, М.,.
1953.
85. Billmeyer F. W., Jr., Textbook of Polymer Science, Interscience Publishers,
John Wiley and Sons, Inc., 1962, p. 348—353.
8b. Walling С., J. Am. Chem. Soc., 67, 441 (1945).
8/. Storey В. T., J. Polymer Sci., A3, 265 (1965).
«8. Flory P. J.t J. Am. Chem. Soc., 69, 2893 (1947); Principles of Polymer
oQ Chemistry, chap. IX, Cornell University Press, Ithaca, 1953.
»У. Morton M., Salatiello P. P„ J. Polymer Sci., 6, 225 (1951).
’ Bamford С. H., Barb W. G., Jenkins A. D., Onyon P. F., The Kinetics of
vinyl Polymerization by Radical Mechanisms, chap. 7, Butterworth and
o, <Publishers) Ltd., London, 1958.
09’ n , Simpson W., Proc. Roy. Soc. (London), A238, 154 (1956).
Gordon M., Roe R. J., J. Polymer Sci., 21, 57, 75 (1956).
butler G. B., Ingley F. L., J. Am. Chem. Soc., 73, 894 (1951).
410
Глава 6
94. Butler G. В., Angelo В. J., J. Am. Chem. Soc., 79, 3128 (1957).
95. Butler G. B., Pure and Appl. Chem., 4, 299 (1962); Polymer Preprints
8(1), 35 (1967).
96. Marvel C. S., Vest B. D., J. Am. Chem. Soc., 79, 5771 (1957), 81, 984
(1959).
97. Marvel C. S., Stille J. К., J. Am. Chem. Soc., 80, 1740 (1958).
98. Gibbs W. E., Barton J. M., The Mechanism of Cyclopolymerization of
Nonconjugated Diolefins, in «Vinyl Polymerization», Ham G. E. (ed.),
vol. 1, part I, chap. 2, Marcel Dekker, Inc., New York, 1967.
99. Dainton F. S., Ivin K. J., Quart. Rev. (London), 12, 61 (1958).
100. Matsuda M., lino M., Macromolecules, 2, 216 (1969).
101. Colombo P., Kukacka L. E., Fontana J., Chapman B. N., Steinberg M
J. Polymer Sci., A-l(4), 29 (1966).
102. Laszkiewicz B., J. Appl. Polymer Sci., 8, 2117 (1964).
103. Crawford G. H., Bice D. E., Landrum В. F., J. Polymer Sci., Al, 565 (1963).
104. Green J., Mayes N., Cottrill E., J. Macromol. Sci. (Chem.), Al(7), 1387
(1967).
105. Griesbaum K., Oswald A. A., Naegele W., J. Org. Chem., 29, 1887 (1964).
106. Hauser C. F., Zutty N. L., Polymer Preprints, 8(1), 369 (1967).
107. Hazell J. E., Ivin K. J., Trans. Faraday Soc., 58, 176, 342 (1962); 61,
2330 (1965).
108. Vogl O., J. Macromol. Sci. (Chem.), Al(2), 243 (1967).
109. Mark H. F., Ogata N., J. Polymer Sci., Al, 3439 (1963).
110. Iwata T., Wasai G., Saegusa T., Furukawa J., Makromol. Chem., 77,
229 (1964).
111. Bosen I., J. Macromol. Sci. (Chem.), Al(2), 243 (1967).
112. Castille Y. P., Stannett V., J. Polymer Sci., A-l(4), 2063 (1966L
ГЛАВА 7
ПОЛИМЕРИЗАЦИЯ С РАСКРЫТИЕМ ЦИКЛА
Кроме рассмотренных выше методов полимеризации и поликон-
денсации существует еще способ получения полимеров, который
приобрел большое значение в последние годы. Это — полимери-
зация с раскрытием цикла, т. е. полимеризация циклических моно-
меров, таких, как циклические эфиры, ацетали, сложные эфиры,
амиды и силоксаны. Процесс с раскрытием цикла представляет
промышленный интерес во многих случаях, включая полимериза-
цию окисей этилена и пропилена (R = —Н и —СН3 соответствен-
но)
О
nH2C--CHR —> — ( —СН2—СН —О —)п — (7.1)
I
R
t-капролактама
^сн2х
сн2 NH
I | — -f-NHCH?CH2CH?CH?CH.,CO-h (7.2)
СН., СО
\ /
хсн2—сн2
и 3,3-бис-(хлорметил) оксетана
п
СН2С1
0-сн2 г I
| I — о — сн2 — с — сн2 —
И2С— С(СН2С1)2 |
СН2С1
(7.3)
(Для четырехчленного циклического эфира вместо названия оксе-
тан может быть применен термин оксациклобутан.) Названия та-
ких полимеров обычно базируются на названии исходного моно-
мера, например полиоксиэтилен, полиоксипропилен, поли-е-капро-
лактам и поли-3,3-бпс-(хлорметил)оксетан. Полимеры, полу-
ченные полимеризацией с раскрытием цикла, обычно классифици-
412
Глава 7
руют *) как конденсационные полимеры на основании того, что-
в полимерной цепи их имеются функциональные группы, такие,
как эфирные и амидные (разд. 1.1а).
7.1. Общие характеристики
Разнообразные циклические мономеры были успешно заполи-
меризованы с раскрытием цикла [1]. Кроме тех классов мономе-
ров, которые были перечислены выше, сюда относятся цикличе-
ские амины, сульфиды, полисульфиды и даже алканы. Легкость
полимеризации данного циклического мономера зависит от актив-
ности функциональной группы в цикле, от использованного ката-
лизатора и от размера цикла. Хотя все три фактора обсуждаются
на протяжении всей этой главы, целесообразно рассмотреть здесь
влияние размера кольца. Влияние размера цикла на способность
мономера к полимеризации, как правило, зависит от факторов,
рассмотренных ранее в разд. 2.3. Изменение активности цикли-
ческих мономеров как функция размера цикла обычно противо-
положна зависимости, указанной на рис. 2.3 для способности
к циклизации. Способность к полимеризации высока для циклов,
содержащих 3, 4, а также 7—11 членов, и мала для пяти- и шести-
членных циклов. В современной практике полимеризация с рас-
крытием цикла обычно ограничивается мономерами, содержащими
менее 9 членов вследствие недостаточной активности циклических
мономеров большого размера.
7.1а. Механизм и кинетика полимеризации
Полимеризация с раскрытием цикла была инициирована как
ионными катализаторами, так и катализаторами, представляющи-
ми собой молекулярные частицы. Инициирование приводит
к раскрытию цикла с образованием инициирующей частицы М*,
которая может быть как ионом, так и нейтральной молекулой
в зависимости от катализатора.
Это можно в общем виде представить как
R— Z + С — М* ,
где Z — функциональная группа мономера, а С — ионный или
молекулярный инициатор. К ионной полимеризации с раскрытием
*) Классификацию полимеров правильнее производить на основании
их строения, а не способа получения. В настоящее время известно немало
случаев, когда один и тот же полимер может быть получен как методом по-
ликонденсации, так и методом полимеризации. Поэтому для классификации
используют в качестве основного принципа строение главной цепи поли-
мера. Полимеры, главная цепь которых состоит только из атомов углерода,
называются кар боцеиными, а полимеры, главная цепь которых состоит
из атомов различных элементов, называются гетероцепными.— Прим. ред.
Полимеризация с раскрытием цикла
413
цикла относится полимеризация, инициируемая такими катализа-
торами, как Na, RO-, НО-, Н+ и BF3. Основным катализатором
молекулярного типа является вода. Ионные инициаторы обычно
активнее, чем молекулярные. Так, лишь более активные цикли-
ческие мономеры способны к полимеризации под влиянием инициа-
торов молекулярного типа. Большинство мономеров требует при-
менения более сильных ионных инициаторов. Ионная полимериза-
ция с раскрытием цикла имеет много общего (например, влияние
растворителя и противоиона) с обычной ионной полимеризацией.
Инициирующая частица М* растет за счет последовательного
присоединения с раскрытием цикла многих мономерных молекул
М* + nR~Z - M-f-R—Z),* (7.5)
Природа процесса роста цепи в полимеризации с раскрытием цикла
имеет поверхностное сходство с этим процессом при цепной поли-
меризации. Только мономер присоединяется к растущей цепи
на стадии роста. Частицы, большие чем мономер, не реагируют
с растущими цепями. Однако полимеризации с раскрытием цикла
могут быть присущи черты как обычной полимеризации, так
и поликонденсации или обеих вместе. Отнесение полимеризации
с раскрытием цикла к цепному или ступенчатому процессу может
быть сделано двумя путями. Один путь — это экспериментальное
наблюдение кинетических закономерностей, описывающих поли-
меризацию; второй путь — исследование распределения обра-
зующегося высокомолекулярного полимера во времени. Послед-
няя характеристика является основной, отличающей полимериза-
цию от поликонденсации. При полимеризации полимер с высоким
молекулярным весом образуется на протяжении всей реакции
в противоположность медленному увеличению молекулярного веса
при поликонденсации. Большинство (но не все) процессов полиме-
ризации с раскрытием цикла протекает как ступенчатая полимери-
зация, в которой молекулярный вес полимера увеличивается посте-
пенно в течение всего процесса. Высокомолекулярный полимер
образуется лишь на поздних стадиях реакции.
Независимо от того, протекает полимеризация с раскрытием
Цикла по цепному или ступенчатому механизму, ее кинетика может
подчиняться уравнениям, сходным с уравнениями как для обыч-
ной полимеризации, так и для поликонденсации. Многие процессы
полимеризации с раскрытием цикла осложнены наличием равнове-
сия полимеризация — деполимеризация. Различные случаи будут
проиллюстрированы в данной главе при детальном рассмотрении
кинетики некоторых типичных процессов полимеризации с раскры-
414
Глава 7
7.2. Циклические эфиры
Эфирные связи отличаются большой прочностью и, согласно
классификации Льюиса, имеют основной характер. В результате
полимеризация с раскрытием цикла может быть инициирована
лишь катионными частицами. Эпокиси (трехчленные окиси) явля-
ются исключением из этого правила. Эпокиси полимеризуются под
воздействием как анионных, так и катионных инициаторов, благо-
даря высокой степени напряженности в маленьком трехчленном
цикле. Инициирование полимеризации циклических эфиров моле-
кулярными частицами в сущности неизвестно вследствие малой
активности эфирной связи.
Полимеризация простых циклических эфиров (т. е. эфиров,,
содержащих одну эфирную связь) практически ограничена поли-
меризацией эфиров с 3-, 4- и 5-членными циклами. Изучение цикли-
ческих эфиров большего размера проводили на примере цикли-
ческих ацеталей (разд. 7.26.4). Активность циклических эфиров-
разного размера следует обычно предполагаемому порядку. Цикли-
ческие эфиры, содержащие менее 5 или более 6 членов, полимери-
зуются относительно легко. Пятичленные циклические эфиры
полимеризуются труднее. Замещенные пятичленные циклические
эфиры и ацетали обычно неактивны; например, 2-метилтетрагидро-
фуран не полимеризуется. Влияние заместителей на увеличение
устойчивости и уменьшение активности циклических соединений
хорошо известно. Шестичленные циклические эфиры, такие, как
тетрагидропиран (I) и 1,4-диоксан (II)
О О
/ х / х
совершенно неактивны. Полимеризация не протекает в широком:
интервале исследованных условий реакции.
7.2а. Анионная полимеризация эпоксидов
7.2а.1. Характеристики реакции
Анионная полимеризация эпоксидов, таких, как окиси этиле-
на и пропилена, может быть инициирована гидроксидами, алкокси-
дами, окислами металлов, металлоорганическими соединениями
и другими основаниями [2, 3]. Так, полимеризация окиси этилена
Полимеризация с раскрытием цикла
41&
с катализатором М+А_ включает стадию инициирования
О
Н2С^СН2 + М+А- —> А—СН2СН2О-М+, (7.6)
за которой следует стадия роста цепи
О
А-СН2СН2О-М+ + Н2С/—''сН2 —> А —СН2СН2ОСН2СН2О"М+. (7.7а)
Последнюю реакцию в общем виде можно представить следующим
образом:
О
А - (- СН2СН2О - )п - СН2СН2О-М+ + ->
—> А — ( — СН2СН2О — )п+1 — СН2СН2О-М+ (7.76)
Часто при полимеризации эпоксидов процесс имеет характеристи-
ки полимеризации с образованием живущих полимеров, при кото-
рой не происходит обрыва цепи в отсутствие специально добавлен-
ных агентов.
Полимеризация несимметричного эпоксида, такого, как окись
пропилена, предполагает возможность двух различных мест
в эпоксидном цикле (атомы углерода 1 и 2) для нуклеофильной
реакции с раскрытием цикла. В таком случае возможно образова-
ние двух растущих частиц в зависимости от места протекания
реакции
СН3
I
О —СН —СН2—О~К+ (7.8)
/ \ Я 2 1
СН3 —СН —СН2 СН3
2 1 \ I
— СН2 — СН — О-К+ (7.9)
1 2
Хотя с первого взгляда может показаться, что структура обра-
зующегося при этом полимера будет различной, это не так. Струк-
тура полимера одинакова в обоих случаях, за исключением незна-
чительной разницы в концевых группах. Однако исследования
каталитического присоединения небольших молекул, таких, как
вода и спирты, к эпоксидному кольцу (с образованием гликолей
или эфироспиртов соответственно) указывают на то, что вследствие
меньших стерических затруднений атака идет почти исключительно
и а углерод 2 [уравнение (7.9)].
Анионная полимеризация эпоксидов обычно является ступен-
чатой полимеризацией, при которой молекулярный вес полимера
нарастает медленно по мере его образования. Но выражения для
скорости и степени полимеризации удивительно' похожи на эти же
выражения для процесса образования живущих полимеров. Так,
416
Глава 7
скорость полимеризации окиси этилена, катализируемой метила-
том натрия [1], может быть представлена как
АД = [СЛ1.О] = Л.р[СГ1з0-Ха ] [С.Н.О]. (7.10)
Однако из рассмотрения относительных скоростей реакций ини-
циирования и роста цепи следует, что не все процессы анионной
полимеризации эпоксидов протекают ступенчато. Полимеризация
окиси пропилена, катализируемая едким калием, является, как
установлено [5]. цепной реакцией. Молекулярный вес образующего-
ся полимера остается практически постоянным на протяжении
всей реакции. В данном случае реакция осложняется тем, что
является, ио-видимому, поверхностной, т. е. протекает на поверх-
ности гидроокиси калия.
Для обычного случая, когда молекулярный вес полимера
медленно повышается по мере протекания процесса, степень поли-
меризации в момент времени t может быть представлена отноше-
нием концентрации прореагировавшего мономера к начальной
концентрации катализатора, т. е.
Т7 _ [С2Ь14О]() — [СгНдО];
[CHjO-Na+Jo 1 '
пли, в общем виде,
RI]o^1Mb , (7.12)
I’-d
где пуль в индексе обозначает начальные концентрации, [С] —
концентрация катализатора.
7.Ча.2. Реакции обмен а
Многие процессы полимеризации эпоксидов, такие, как реак-
ции, катализируемые алкоголятамп и гидроокисями, проводятся
в присутствии спирта (обычно того, алкоголят которого исполь-
зуется). Спирт применяют для получения гомогенной системы
путем растворения катализатора. При этих условиях возможна
обменная реакция между растущей цепью и спиртом
R — (—ОСН2СН2-)„O-Na + + ROII
R —( —OCII2CH2-)n —OTI-;-RO-Xa+. (7.13)
Подобные реакции обмена возможны также между вновь образо-
вавшимся по уравнению (7.13) полимерным спиртом и другими
растущими цепями
R — (— ОСН2СН2 — )„ — OII-LR — ( —ОСПоСПп — — О-.\а+
( — ОСП2СТТ2 — )л—O-Na+-|-R —( —ОСТ12СН2—)„ —ОН, (7.14) '
П олимеризация с раскрытием цикла
417
где (п + т) = (х + у)- Эти реакции обмена, эквивалентные реак-
циям передачи цепи, будут уменьшать молекулярный вес поли-
мера. Степень полимеризации в этом случае будет определяться
уравнением
i[M]0-[M]t
[C]o + [ROH]0 ’
(7-15)
так как каждую молекулу спирта можно считать соответствующей
частице катализатора, которая обеспечивает образование растущих
цепей.
Ряд несколько отличных кинетических ситуаций может возник-
нуть в связи с относительными скоростями реакций инициирова-
ния, обмена и роста цепи. Если скорость инициирования значи-
тельно больше скорости роста, то инициирование оканчивается
в основном прежде, чем начнется рост цепи, и полимеризация про-
текает согласно уравнению (7.10) вплоть до ее завершения. Все
полимерные цепи начинают расти в одно и то же время и молеку-
лярновесовое распределение является очень узким, как и в случае
реакции образования живущих полимеров. Однако, если скорость
инициирования мала, то некоторые цепи начинают расти, в то вре-
мя как другие еще не инициированы. Полимеризация протекает
с начальным периодом, во время которого скорость реакции увели-
чивается по мере превращения катализатора в растущие частицы,
а затем становится постоянной. Молекулярновесовое распределе-
ние расширяется, так как цепи растут в течение различных про-
межутков времени. Влияние реакции обмена зависит от относи-
тельной кислотности добавленного и полимерного спиртов. Реак-
ция обмена протекает на протяжении всей полимеризации, если
кислотность этих двух спиртов приблизительно одинакова. Ско-
рость полимеризации в этом случае не изменяется, в то время как
степень полимеризации уменьшается, а молекулярновесовое рас-
пределение становится шире.
Если же добавленный спирт ВОН значительно более кислый,
чем полимерный, то он весь прореагирует с первыми же образо-
вавшимися растущими частицами
ROCH2CH2O-Na+ + ROH —> ROCH2CH2OH + RO"Na+, (7.16)
прежде чем начнется реакция полимеризации. Молекулярновесо-
вое распределение будет в этом случае узким, так же как в отсутст-
вие спирта; разница заключается в том, что небольшое количество
мономера превращается в спирт ROCH2CH2OH. Начальная ско-
рость полимеризации будет, вероятно, уменьшаться, так как реи-
ниципрование с помощью RO_Na+ обычно происходит медленно
вследствие относительно высокой кислотности ROH. Скорость
лимеризации будет возрастать и затем станет постоянной, когда
сь спирт вступит в реакцию обмена [уравнение (7.16)].
418
Глава 7
Когда спирт ROH менее кислый, чем полимерный спирт, на
скорости полимеризации это не сказывается. Обмен идет на
последних стадиях полимеризации, сопровождаясь расширением
молекулярновесового распределения.
7.2а.З. Передача цепи на мономер
При полимеризации эпоксидов образуются полимеры низкого
молекулярного веса: обычно менее 5000 и редко — выше 10 000.
Причиной этого ограничения является относительно низкая актив-
ность эпоксидного кольца по отношению к росту цепи при анион-
ной полимеризации и наличие нежелательной реакции передачи
цепи на мономер. Последнее особенно существенно в замещенных
окисях этилена, таких, как окись пропилена [6, 7]. Реакция
передачи цепи включает отщепление водорода от алкильного
заместителя в эпоксидном цикле и последующее очень быстрое
расщепление кольца с образованием аниона аллилового эфира
СН3 О
I / \ ftnep. М
— CH2-CH-O-Na+ + CH3 —СН —СН2-----—>
СН3 О
—> —СН2-СН — ОН + ЩС^СН-CH2]Na+ (7.17)
О
/ \ Очень быстро _
Н2С — СН-CH2Na+----------> CH2 = CH-CH2O~Na+. (/.18)
Математическая обработка данных о влиянии реакции передачи
цепи на мономер на степень полимеризации несколько отличается
от ранее описанной для случая передачи цепи в радикальной
и ионной полимеризации (разд. 3.6 и 5.2д.1). В случае, когда
реакция обмена протекает очень быстро [уравнение (7.13)]. ско-
рость исчезновения мономера может быть представлена суммой
скоростей реакций роста и передачи цепи
=4Л = ^р + ^пер.м)[М][С1о- (7.19)
Увеличение концентрации полимерных цепей [N] выражается
скоростью реакции передачи цепи
^=Аер.м[М] [CJo- (7-20)
Разделив уравнение (7.20) на уравнение (7.19), учитывая, что
константа передачи цепи на мономер См = /спер. м/^р и проинте-
грировав, получим
[N] = [N]o + T^-([M]o-[M]), (7.21)
1 ~^м
где [N]o — концентрация полимерных цепей в отсутствие реакции
передачи цепи на мономер.
Полимеризация с раскрытием цикла
419
Степени полимеризации при отсутствии и наличии передачи
цепи (Хп)о и Хп соответственно выражаются
= (7.22)
(7.23)
Уравнение (7.22) точно соответствует уравнению (7.12), так как
[Мо эквивалентна [С]о. Из уравнений (7.21) — (7.23) получим
1 __ 1 । см
Хп (Хп)0^1 + См'
(7-24)
Уравнение (7.24) указывает, что зависимость i/Xn от 1/(Хл)0 будет
линейной с отсекаемым на оси ординат отрезком См/(1 + См )•
Рис. 7.1. Влияние на молекуляр-
ный вес полимера реакции переда-
чи цепи на мономер при полимери-
зации окпсп пропилена в диоксане
в присутствии метилата натрия при
70 и 93 °C [6].
На рис. 7.1 показана такая зависимость для полимеризации окиси
пропилена в присутствии метилата натрия. Были получены кон-
станты скорости реакции передачи цепи на мономер, равные 0,013
и 0.027 при 70 и 93 °C соответственно. Эти значения в 104 — 10е раз
больше, чем обычные константы передачи цепи на мономер
(табл. 3.3 и 3.3), и обусловливают наблюдаемое сильное ограниче-
ние достигаемых молекулярных весов полимеров.
7.26. Катионная полимеризация
Рост цепи при катионной полимеризации циклических эфиров
ычно рассматривается как протекающий через образование
ретичного иона оксония (III). напнимен пни полимепизапии 3.3-
420
Глава 7
бш?-(хлорметил)оксетана (R = СН2С1)
(7.25)
где А- — противоион. Рост цепи представляет собой нуклеофиль-
ную атаку атома кислорода мономера на а-углеродный атом иона
оксония. Многие полимеризационные процессы являются равно-
весными, в которых полимерные цепи достигают ограниченной
величины, соответствующей равновесию рост цепи — распад цепи.
7.26.1. Инициирование
Каталитические системы, применяемые при катионной полиме-
ризации алкенов (разд. 5.2а), могут быть использованы для полу-
чения растущей частицы — третичного иона оксония [1, 2, 8, 9].
7.26.1а. Протонные кислоты. Очень сильные протонные кисло-
ты, такие, как концентрированная серная, трифторуксусная или
фторсерная кислоты, инициируют полимеризацию, образуя вначале
вторичный ион оксония
(7.26)
который реагирует с другой мономерной молекулой с образовани-
ем третичного иона оксония
Этот тип инициирования ограничен нуклеофильностью аниона
А-, образующегося из протонной кислоты. У не очень сильных
кислот анион достаточно нуклеофилен, чтобы конкурировать
с мономером за протон или за вторичный ион оксония. Так, даже
серная кислота при концентрации 10 мол. % недостаточно сильна,
чтобы полимеризовать тетрагидрофуран [8]. Ион бисульфата более
Полимеризация с раскрытием цикла
421
нуклеофилен, чем тетрагидрофуран; протонированный тетрагидро-
фуран реагирует предпочтительно с бисульфат-противоионом
(7.28)
вместо того, чтобы образовать третичный ион оксония. Присут-
ствие воды при реакции полимеризации, инициируемой сильными
кислотами, может также привести к обрыву реакции, так как
нуклеофильность ее достаточна для конкуренции с мономером
за вторичный ион оксония.
7.26.16. Кислоты Льюиса. Для инициирования полимеризации
могут быть использованы кислоты Льюиса, такие, как BF3
и SnCl4. Эти катализаторы почти всегда требуют присутствия воды
(или какого-либо другого соединения с кислотным водородом)
в качестве сокатализатора. Взаимодействие между катализатором
и сокатализатором приводит к образованию комплекса, например
BF3 + H2O —> H+(BF3OH)-, (7.29)
который инициирует полимеризацию, действуя, как протонная
кислота в уравнениях (7.26) и (7.27). Для небольшого числа ката-
лизаторов присутствие сокатализатора не является обязательным.
Так, сообщалось [8, 9], что PF5 и SbCl5 инициируют полимериза-
цию тетрагидрофурана, хотя всегда трудно экспериментально
установить отсутствие следов воды или других примесей, которые
могут действовать как сокатализаторы. Если инициирование дей-
ствительно протекает в отсутствие сокатализатора, то это может
происходить благодаря диспропорционированию катализатора
с образованием катионных частиц, например
2PF5 -» PFt(PF6)“, (7.30а)
которые инициируют полимеризацию по уравнению
PF+(PF6r + о'
(7.306)
Хотя при инициировании полимеризации таким катализатором,
как PF5, присутствие сокатализатора и не обязательно, известно,
что добавление воды, действующей как сокатализатор, значитель-
но увеличивает скорость реакции [10]. График зависимости ско-
рости полимеризации тетрагидрофурана от концентрации воды
422 Глава 7 1
------------------------------------------------------------------j
при катализе PF5 аналогичен графику, приведенному на рис. 5.1;
для полимеризации стирола, катализируемой системой SnCl4 — '
Н2О. Скорость полимеризации возрастает с увеличением [Н2О]
до максимума, а затем, при больших концентрациях воды, умень-
шается. Это уменьшение приписывают разложению катализатора
под действием воды.
7.2б.1в. Ионы карбония. Для полимеризации циклических эфи-
ров применяют также ионы карбония. Реакция инициирования
протекает в той же последовательности, которая показана в урав-
нениях (7.26) и (7.27), за исключением того, что катализатором
вместо Н+А~ является R+A~. Для получения ионов карбония
используются среди прочих [9, 11, 12] следующие реакции:
О О
НС1О4 + (СН3С)2О —» СН3С+(С1О4)- + СН3СООН, (7.31)
О О
CH3(!ci + SnCl4 —» СН3(У+(8пС15)-, (7.32)
C6H5CH2Cl + FeCl3 —> C6H5CHJ(FeCl4)-, (7.33)
ROSO3R + BF3 —> R4ROSO3BF3)“. (7.34)
Стадия инициирования с некоторыми ионами карбония вклю-
чает, очевидно, отщепление гидрид-иона от мономера вместо
присоединения к мономеру [13]. Так, предположение, что
инициирование полимеризации тетрагидрофурана частицей
(C6H5)3C+(SbCle)~ протекает следующим образом:
(7.35а)
(7.356)
подтверждается быстрым образованием трифенилметана в количе-
стве, эквивалентном количеству катализатора. Рост цепи протека-
ет через третичный ион оксония, образовавшийся по уравнению
(7.356). Предполагают [14, 15], что инициирование п-хлорфенил-
диазонийгексафторфосфатом протекает по аналогичному механиз-
му.
7.2б.1г. Ионы оксония. Поскольку именно третичный ион оксо-
ния является активной растущей частицей, Меервейн с сотр. [91
использовал предварительно приготовленные третичные ионы оксо-
ния, такие, как тетрафторборат триэтилоксония (C2H5)3O+(BF4)“,
для инициирования полимеризации циклических эфиров. Ини-
П олимеризация с раскрытием цикла
423
ципровапие ионом оксония включает, возможно, реакцию обмена
алкила с мономером
(C2H5)3O+(BF4r + О
С2Н5-\
№4
+ (С2Н5)2О (7.36)
7.2б.1д. Промоторы. Многие комбинации кислот Льюиса
и активных циклических эфиров (таких, как эпоксиды или окса-
циклобутан) обычно в сочетании с сокатализатором с успехом были
применены для инициирования полимеризации таких относительно
неактивных циклических эфиров, как тетрагидрофуран [8, 10, 13].
Инициирование, очевидно, протекает через образование вторично-
го п третичного ионов оксония из активного эфира, которые затем
реагируют как действительные инициирующие частицы. Напри-
мер. в случае использования эпоксида при полимеризации тетра-
гидрофурана (ТГФ) реакция протекает по следующей схеме:
НОСН2СН2 —О
СН2
hoZ
ТГФ
СН2
ТГФ
| ТГФ
Рост цепи
Н(—ОСН2СН2)2
ТГФ
Рост цепи
Активные циклические эфиры, применяющиеся как компоненты
каталитической системы, называются промоторами (или соката-
лизаторами). Промотор вводится, конечно, в малых количествах
относительно полимеризующегося циклического эфира.
Одни из наиболее часто употребляемых промоторов — эпихлор-
гидрин. Механизм образования инициирующей частицы при ис-
пользовании эпихлоргидрина и некоторых катализаторов может
отличаться от описанного выше. Так, тетрафторборат триэтилоксо-
ния был получен Меервейном при взаимодействии эпихлоргидрина
с эфиратом трехфтористого бора. Очевидно, суммарная реакция
424
Глава 7
имеет вид [9.15]
О
6(C2H5)2O:BF3 + ЗН2С----СН — СН2С1
—> 3(С2Н5)30н (BF4)- + 2BF3 + Г С2Н5ОСН2 - СН - О “] В
(.7.38)
CIHC1 _з
7.2б.1е. Металлоорганические катализаторы. В качестве ката-
лизаторов применяют также металлоорганические соединения,
например диэтилцинк и триэтилалюмипий [16—18]. Их исполь-
зуют обычно в присутствии воды или спирта. Такие промоторы,
как эпихлоргидрин, также могут быть введены в каталитическую
систему вместе с водой или спиртом. Природа инициирующих
и растущих частиц сильно различается в зависимости от каталити-
ческой системы и условий реакции. В некоторых случаях реакция
протекает по обычному механизму с оксониевым ионом. < )дпако
в большинстве случаев, видимо, имеет место координационный
механизм, включающий такие каталитические частицы, как
СаН5—ZnOZn—С2Н5 и (С2Н5)2А1ОА1(С2Н5)2, образовавшиеся при
взаимодействии воды или спирта с металлоорганическими соеди-
нениями. Механизм координационной полимеризации обсуждает-
ся в гл. 8.
7.26.2. Обрыв цепи
Во многих случаях катионной полимеризации циклических
эфиров образуются долгоживущие растущие частицы, в результа-
те конечный продукт имеет узкое молекулярновесовое распреде-
ление. Так, полимеризация З,3-бнс-(хлорметил)оксетана, катали-
зируемая триизобутилалюминием, дает продукт с величиной
MwIMn, равной 1,09 [19]. Однако обрыв цепи может происходить
по множеству различных реакций; некоторые из них аналогичны
реакциям обрыва при катионной полимеризации алкенов (см.
разд. 5.2в). Передача цепи на полимер посредством алкильного
обмена— это обычный способ, при котором растущая цепь обры-
вается, а кинетическая цепь не изменяется. Реакция идет через
обмен алкила между растущим центром (ионом оксония) и эфир-
ным кислородом полимерной цепи
+ /Х1 /(СН2)4 —
— О(СН2)4-О + О' ->
а\/ (СН2)4
о
+ /(СН2)4 — /_>
-»—О(СН2)4-О(СН2)4-О' -----> 5
д\сн2)4— j
Полимеризация с раскрытием цикла
425
— О(СН2)4-О(СН2)4-О(СН2)4— + | б-(СН2)4 — (7.39)
х/А
Передача цепи может быть как межмолекулярной, так и внутри-
молекулярной. В любом случае результатом является расширение
молекулярновесового распределения по сравнению с узким рас-
пределением, наблюдаемым для живущих полимеров [14].
Катионная полимеризация окиси этилена интересна тем, что
может включать два вида равновесия. Наличие равновесия поли-
мер — циклический димер в сочетании (или вместо) с равновесием
полимер — мономер установлено [1, 2] при образовании 1,4-диок-
сана при полимеризации окиси этилена и деполимеризации поли-
оксиэтилена. Циклический димер диоксана образуется путем
внутримолекулярной реакции расширения кольца, за которой
следует стадия вытеснения
Оксациклобутан также подвергается аналогичной реакции. Пред-
полагают, что эта реакция приводит к образованию устойчивого
16-членного иона оксония, который неспособен к дальнейшему
росту
(ОСН2СН2СН2)4—0„
А
+ _(СН9)ЧО(СН2)3.
0СН9СН9СН9~О /О
А (СН2)3О(СН2)3
(7.41)
Из реакционной смеси выделен циклический тетрамер. Он обра-
зуется по реакции вытеснения [аналогично реакции (7.40)] при
воздействии нуклеофила, такого, как мономер, противоион, сока-
тализатор или специально добавленный реагент. Такой способ
рыва цепи не характерен для замещенных оксациклобутанов,
Как 3>3-бпс-(хлорметил)производные, в связи с возникающи-
при циклизации большими стерическими затруднениями,
426
Глава 7
обусловленными большим размером заместителей. Полимеризация
3,3-бнс-(хлорметил)оксетана осложнена нерастворимостью поли-
мера в реакционной смеси. Считается, что обрыв цепи в этом слу-
чае является результатом того, что в твердом полимере активный
оксониевый ионный центр становится недоступным для участия
в дальнейшем росте цепи.
Обрыв цепи может происходить в различной степени посред-
ством соединения растущего оксониевого иона либо с противоио-
ном, либо с анионной частью противоиона, что более вероятно.
Так, выше было отмечено, что использование протонной кислоты
в качестве катализатора лимитируется нуклеофильностью аниона
кислоты. Переход аниона от противоиона, например
ОСН2СН2—О
BF3OH
ОСН2СН2ОСН2СН2ОН + BF3 (7.42)
происходит до различной степени в зависимости от устойчивости
противоиона. Такие противоионы, как (PF6)~ и (SbCl6)“, не склонны
вызывать обрыв цепи посредством перехода галогена, тогда как
галогениды алюминия и титана обладают значительной способ-
ностью к такому виду обрыва; другие противоионы, например
(BF4)~ и (FeCl4)~, занимают промежуточное положение [1, 2, 14,
20].
Обрыв цепи может также происходить в результате передачи
цепи на сокатализатор (вода, спирт) или специально добавленный
агент. Например, было обнаружено [21], что действие ацикличе-
ских эфиров как агентов передачи цепи аналогично действию поли-
меров [уравнение (7.39)].
О(
А
7.26.3. Кинетика
7.26.За. Сходство с поликонденсацией. Катионная полимериза-
ция циклических эфиров аналогична поликонденсации с точки
зрения зависимости выхода полимера от времени. С увеличением
степени превращения возрастает размер полимерных молекул.
Полимеризация с раскрытием цикла
427
Между тем молекулярный вес полимера не обязательно продолжа-
ет повышаться во время всего процесса полимеризации, как это
происходит при поликонденсации. Процесс поликонденсации не
сопровождается реакцией обрыва, если в нем участвуют стехиомет-
рические количества функциональных групп. Процессы полимери-
зации с раскрытием цикла отличаются от поликонденсации тем,
что .могут включать реакции обрыва. Таким образом, хотя молеку-
лярный вес увеличивается при повышении степени превраще-
ния, этот рост может прекратиться задолго до полной кон-
версии.
Сходство между поликонденсацией и полимеризацией с раскры-
тием цикла становится еще более явным, если рассмотреть величи-
ны констант скорости роста цепи при полимеризации различных
циклических эфиров.
Таблица 7.1
Некоторые параметры полимеризации
тетра гидрофура на [17]
Параметр Величина
[М], моль/л ](С2Н5)3А1], моль/л [Н2О]/[(С2Н5)3А1] [Эпихлоргидрин], моль/л [М*], моль/л fcp, л/(моль-с) 12,1 0,179 0,500 0,159 2,4-Ю-3—2,6-Ю-з 1,28-10-2
В табл. 7.1 приведены значения некоторых основных пара-
метров блочной полимеризации тетрагидрофурана при О °C
с использованием в качестве каталитической системы триэтил-
алюминия, эпихлоргидрина и воды [17]. Эти величины можно
сравнить со значениями соответствующих величин для обычной
полимеризации и поликонденсации (табл. 2.1, 3.8, 5.1 и 5.7), чтобы
установить природу полимеризации циклических эфиров. Так
величина кр для тетрагидрофурана почти совпадает с соответ-
ствующей величиной для полиэтерификации и намного меньше
Для различных случаев обычной полимеризации. Близкие значе-
ния кр были получены для различных процессов полимериза-
ции эпоксидов, оксетанов и тетрагидрофурана [2, 14, 15, 20].
7.26.36. Скорость полимеризации. Уравнения, описывающие
катионную полимеризацию циклических эфиров с раскрытием
цикла, можно представить в нескольких видах. Многие процессы
полимеризации эфиров характеризуются кинетическими выраже-
428
Глава 7
ниями, очень близкими к уравнениям, описывающим полимериза-
цию алкенов (разд. 5.2г.1), например уравнения (5.19), (5.20),
(5.22), (5.23), (5.25), (5.26). В некоторых процессах, где реакция
обрыва цепи не играет существенной роли, можно использовать
кинетические зависимости, аналогичные зависимостям для про-
цессов с участием живущих полимеров (разд. 5.36.2 и 5.36.5),
например
Яр = А:р[М*][М], (7.44)
где [М*] — концентрация растущего иона оксония.
Процессы полимеризации с раскрытием цикла, которые проте-
кают без обрыва цепи и для которых характерно равновёсие рост—
деполимеризация, могут быть описаны различными способами [17,
18, 22]. (Нижеследующее описание равновесной полимеризации
с раскрытием цикла применимо также для других типов равно-
весной полимеризации, например для полимеризации алкенов
или карбонильных мономеров.) Равновесие рост — деполимериза-
ция может быть выражено следующим уравнением:
%
М* + М ----> МЙ+ь (7.45)
йЯеп
которое аналогично уравнению (3.169). Скорость полимеризации
определяется разностью между скоростями роста и деполимериза-
ции
= = (7.46)
В условиях равновесия скорость полимеризации равна 0 и урав-
нение (7.46) обращается в выражение
fcp[M]K = ^en, (7.47)
где [М]„ — равновесная (критическая) концентрация мономера
[так же, как в уравнении (3.173)].
Из уравнений (7.46) и (7.47) можно получить следующее выра-
жение для скорости полимеризации:
Г^М = А;р[М*](1М]-[М]к), (7.48)
интегрирование которого дает
ta(->5^)-‘pIM*l'. Р-49»
Полимеризация с раскрытием цикла
429
где [iMJo — начальная концентрация мономера. (Соответствующее
рассмотрение степени полимеризации в равновесном процессе
дано в разд. 7.3в.2.)
Уравнения (7.48) и (7.49) могут быть использованы для опре-
деления константы скорости роста цепи. Равновесная концентра-
ция мономера [М]к определяется аналитически или из графика
зависимости константы скорости роста цепи от исходной кон-
центрации мономера (как пересечение прямой с осью [М]о).
Рис. 7.2. Определение рав-
новесных концентраций мо-
номера [М]к для катализи-
руемой (C2H5)3O+BFj поли-
меризации тетрагидрофура-
на в дихлорэтане при О °C
[23].
Рис. 7.3. Исчезновение мо-
номера при полимеризации
тетрагидрофурана в присутст-
вии (С2Н5)3А1 — Н2О при
О °C [17].
На рис. 7.2 приведен график полимеризации тетрагидрофурана
в растворе дихлорэтана при О °C с использованием тетрафторбора-
та триэтилоксония в качестве инициатора [23].
Кроме того, по данным полимеризации можно построить зави-
симость левой части уравнения (7.49) от времени, причем полу-
чается прямая линия, наклон которой соответствует &Р[М*]
(рис. 7.3). График на рис. 7.3 представляет собой прямую линию,
за исключением начального периода (степень превращения ме-
нее 5%), соответствующего индукционному периоду образования
некоторого количества растущих частиц. Поскольку величину
1М*] для живущего полимера можно получить из измерений сред-
нечислового молекулярного веса, константу скорости роста легко
Глава 7
оценить. Этот метод является общим способом определения вели-
чины /ср, приведенной в табл. 7.1. Концентрация растущих частиц
и скорость полимеризации при реакции, инициированной много-
компонентной системой, обычно зависят от концентрации каждого
компонента инициирующей системы. Рис. 7.4 и 7.5 иллюстрируют
Рис. 7.5. Зависимость кон-
центрации растущего нона ок-
сония от концентрации эпи-
хлоргидрина при полимериза-
ции тетрагидрофурана в ци-
клогексане, катализируемой
системой А1 (С2Н5)3 — эпихлор-
гидрин — Н2О, при 0 °C.
Концентрация А1(С2Н5)3 0,924
моль/л, молярное соотношение
Н2О/А1(С2Н5)3 равно V2 [17].
Р и с. 7.4. Зависимость кон-
центрации растущего иона ок-
сония от концентрации
А1(С2Н5)3 при полимеризации
тетрагидрофурана в циклоге-
ксане, катализируемой систе-
мой А1(С2Н5)з — эпихлоргид-
рин — Н2О, при О °C. Концен-
трация эпихлоргидрина 0,185
моль/л, молярное соотноше-
ние Н2О/А1(С2Н5)3 равно V2
[17].
этот эффект для зависимости [М*1 от концентрации катализатора
и промотора при полимеризации тетрагидрофурана в присутствии
системы триэтилалюминий — вода — эпихлоргидрин.
Полимеризация с раскрытием цикла
431
7.26.4. Циклические ацетали
7.2б.4а. Триоксан. Различные циклические ацетали также спо-
собны к каталитической полимеризации с раскрытием цикла.
Триоксан — циклический тример формальдегида — легко подвер-
гается полимеризации с образованием полиоксиметилена, который
получается и при полимеризации формальдегида
О —СН2
Н2с/ ^0 —СН2О —)п — (7.50)
'О-СН2
Подробно механизм этой полимеризации не был выяснен *). Необ-
ходимость в сокатализаторах точно не установлена, хотя, очевид-
но, сильные кислоты Льюиса, такие, как BF3 и TiCl4, не нуждают-
ся в сокатализаторах. Полимеризацию триоксана при иницииро-
вании BF3 можно представить следующим образом [24]:
О-СН2
/ \ ВГз
Н2С\ ^0------>
О —СН2
сн2-о
F3B —0^ ^СНг
СН2-0
F3B — ОСН2ОСН2 — О — СН2
4 +
f3b—осн2осн2—о=сн2
V
(7.51)
Здесь ион оксония IV подвергается реакции открытия цикла
с образованием иона карбония V, который, как считают, и являет-
ся растущей частицей. Ион карбония образуется при полимериза-
ции триоксана (вместо одного только иона оксония, как это имеет
место при полимеризации циклических эфиров) вследствие резо-
нансной стабилизации соседним атомом кислорода.
Процесс роста цепи состоит из повторения реакций, заклю-
чающихся во взаимодействии иона карбония с мономером и после-
дующем открытии цикла
F3B - (- ОСН2ОСН2ОСН2-)- п - ОСН2ОСН2ОСН2 +
СН2-0
0\ \н2
сн2—о
сн2—о
F3B-(-OCH2OCH2OCH2-)n+1-6^ ^сн2
сн2—о
—> F3B — ( — ОСН2ОСН2ОСН2 — )п+1—ОСН2ОСН2ОСН2 (7.52)
*) Кинетика и механизм полимеризации триоксана были изучены
Н. С. Ениколоповым (см. список дополнительной литературы к гл. 7
[3]). — Прим. ред.
432
Глава 7
Этот механизм страдает тем же недостатком, что и описанный
ранее (разд. 5.56) для полимеризации хлораля, катализированной
А1Вг3. С ростом цепи постоянно увеличивается расстояние между
зарядами растущих частиц. (Частицы, которые содержат противо-
положные заряды на своих концах, обычно называют цвиттер-
ионами,.') Это требует, чтобы оба конца растущего карбониевого
иона оставались в относительной близости друг к другу. Кроме
этого, возможен механизм, включающий вторую молекулу BF3
[подобный механизму, описанному уравнением (5.83)]. Это рас-
суждение неприменимо для полимеризации с участием системы
катализатор — сокатализатор. В этих случаях отсутствуют цвит-
тер-ионные частицы. Например, полимеризация с П+А~ в качестве
инициатора протекает следующим образом:
О —СН2 О —СН2 Н
/ \ Н+А~ / \ + /
Н2С\ ^0--------> Н2С\ о£ НОСН2ОСН2ОСН2 —>
о-сн2 о-сн2а
Мономер
------->
сн2-о
НОСН2ОСН2ОСН2—С)/ ^сн,
А СН2—ОХ
—> НОСН2ОСН2ОСН2ОСН2ОСН2ОСН2 (7.53)
А
В дополнение к равновесию полимер — мономер, которое
может иметь место, полимеризация триоксана осложняется нали-
чием равновесия полимер — формальдегид
— ОСН2ОСН2ОСН2 Tt — ОСН2ОСН2 + СН2О. (7.54)
+ +
Полимеризация триоксана обычно протекает с большими индук-
ционными периодами, которые обусловлены установлением равно-
весной концентрации формальдегида. Индукционные периоды
могут быть уменьшены или даже ликвидированы путем добавления
формальдегида к исходной реакционной смеси.
Обрыв цепи происходит описанными ранее способами
(разд. 7.26.2). Дополнительное осложнение при полимеризации
триоксана — обрыв цепи путем внутримолекулярного перехода
гидрид-иона
— ОСН2 —О О —>—ОСН2 —О О —>
\н2 ^сн7 \нз
+ +
—>—0СН2-0 +СНз, (7.55)
\н
Полимеризация с раскрытием цикла
433
результатом чего является образование метоксильных и формиль-
ных концевых групп в полимере. В некоторых случаях при поли-
мерлзации триоксана *) наблюдается образование тетраоксана [25]
СН2—О
+ o' ^СНг
(ОСН2)зОСН2ОСН2ОСН2 -> | I + — ОСН2ОСН2 (7.56)
сн2 о
\-сн^
Z.26.46. Циклические формали. Циклические формали имеют
структуру
О
сн2\н2)т
VI
Различные представители этой группы, такие, как 1,3-диоксолан
(т - 2), 1,3-диоксепан (т = 4) и 1,3-диоксокан (иг = 5), легко
подвергаются полимеризации. Реакция протекает аналогично по-
лимеризации триоксана [26]. Например, для 1,3-диоксолана
/СН2
ОСН2СН2ОСН24-О о —»
СН2 —СН2
сн2
—» —ОСН2СН2ОСН2 — б о —»
^СЩ-СНг
—»—ОСН2СН2ОСН2ОСН2СН2ОСН2 (7.57)
Энергетические характеристики
7,26.5а. Влияние температуры. Влияние температуры на ско-
рость и степень полимеризации циклических эфиров и ацеталей
значительно меняется в зависимости от специфики реакционной
системы. Изменения, обусловленные характером мономеров,
растворителей, катализаторов, сокатализаторов и промоторов,
оказываются обычно аналогичными изменениям, наблюдаемым
при ионной полимеризации алкенов (разд. 5.2ж и 5.36.7). Повы-
*) Важным этапом процесса полимеризации триоксана является «пере-
1ача цепи с разветвлением», установленная Н. С. Ениколоповым (см. список
Дополнительной литературы к гл. 7 131).— Прим. ред.
434
Глава 7
шение температуры почти всегда приводит к увеличению скорости
полимеризации (т. е. 2?Hp положительна). Суммарная энергия
активации полимеризации представляет собой величину поряд-
ка 5—15 ккал/моль (20,9-103— 62,8-103 Дж/моль), хотя в зависи-
мости от условий реакции могут иметь место значительные откло-
нения. Так, значение ERp для полимеризации 3,3-бнс-(хлорметил)-
оксациклобутана в растворе хлористого метила, инициированной
BF3 — Н2О, составляет 18,0 ккал/моль (75,4-103 Дж/моль), тогда
как для радиационной полимеризации в кристаллическом состоя-
нии ERp равна 3—4 ккал/моль (12,6-103—16,7-10% Дж/моль)
[27, 28]. Эта разница может быть объяснена предпочтительной
ориентацией молекул мономера при твердофазной полимеризации.
Аналогичные'эффекты наблюдались при полимеризации триокса-
на, хотя различия в значениях ERp для разных систем катали-
затор — сокатализатор часто бывают большими, чем различия,
обусловленные полимеризацией в растворе и в твердом состоя-
нии [29].
Влияние температуры на степень полимеризации более сложно.
В большинстве случаев повышение температуры реакции ведет
к уменьшению молекулярного веса полимера вследствие возра-
стания скоростей реакций обрыва. В табл. 7.2 показано это влия-
ние на примере полимеризации оксациклобутана в присутствии
BF3 - Н2О [30].
Таблица 7.2
Влияние температуры на полимеризацию
окса циклобутана а [30]
Темпера- тура, “С Характери- стическая вязкость полимера, ДЛ/г Суммарная степень превраще- ния моно- мера, % Содержание тетрамера, %
-80 2,9 95 4
0 2,1 94 10
50 1,3 64 66
100 1,1 62 62
а Мерой молекулярного веса полимера является харак-
теристическая вязкость.
Уменьшение молекулярного веса полимера в этом случае
объясняется увеличением реакций обрыва цепи путем внутримоле-
кулярной циклизации [«кусание своего хвоста», см. уравнение
(7.41)], о чем свидетельствует увеличение выхода тетрамера.
В других случаях полимеризации повышение температуры может
не оказывать влияния на реакцию обрыва цепи, но скорость роста
Полимеризация с раскрытием цикла
435
может увеличиваться. В результате этого наблюдается повышение
молекулярного веса полимера с температурой. На рис. 7.6 показан
интересный случай полимеризации тетрагидрофураиа, прй которой
вначале, при низких температурах, молекулярный вес возрастает
с повышением температуры, а затем при более высоких температу-
Р н с. 7.6. Влияние темпера-
туры на молекулярный вес по-
лимера при полимеризации те-
трагидрофурана в присутствии
BF3. Концентрация BF3 10%
[31].
Ч 3>5
| 3'°
| о 2,5
е Е
2,0
р СО
к ьо
> 0,5
4 °.
Температура, °C
рах уменьшается [31]. Низкие температуры относительно мало
влияют на реакции обрыва, но это влияние резко возрастает при
более высоких температурах.
Данные, приведенные в табл. 7.2, показывают, что суммарная
степень превращения оксациклобутана в полимер уменьшается
Р и с. 7.7. Зависимость равновесной
концентрации мономера от темпера-
туры при полимеризации тетрагид-
рофурана в присутствии CeHjN.tBFT
[14].
с повышением температуры реакции. Очевидно, это обусловлено
термической деструкцией катализатора [30]. Такой же общий ход
реакции наблюдается при полимеризации большинства цикли-
ческих эфиров и ацеталей, но причина этого иная. Повышенная
температура обычно понижает степень превращения мономера
в полимер. Равновесие рост цепи — деполимеризация [уравнение
(7.4о)] при повышении температуры сдвигается налево. Равновес-
ная концентрация мономера при этом, согласно уравнению
(3.1756), возрастает (АЯ отрицательна). Пример такого влияния
показан на рис. 7.7 для блочной полимеризации тетрагидрофура-
на, катализируемой бензолдиазонийгексафторфосфатом.
436
Глава 7
-----------------—-----------.---------------------------------
7.26.56. Термодинамика полимеризации. Значения энтальпий
и энтропии полимеризации для различных циклических мономер
ров приведены в табл. 7.3 [32—34]. Одни из этих данных пред-*
ставляют собой экспериментально определенные величины; другие
Таблица 7.3
Теплоты и энтропии полимеризации циклических эфиров
и ацеталей [32—34]
Мономер Число членов в цикле —ДИ, ккал/моль (4,187-Ю3 Дж/моль) —AS, кал/(моль • К) [4,187 Дж/(моль«К)]
Окись этилена 3 22,6
Оксациклобутан 4 16,1
3,3-бис-(Хлорме- 4 20,2 19,9
тил)оксацикло-
бутан
1,3-Диоксолап 5 6,2
Тетрагидрофурап 5 5,3 18
Тетрагидроппран 6 0,4
.н-Диоксаи 6 0,0
1,3-Диоксепан 7 3,6 11,5
1,3-Диоксокан 8 12,8
Формальдегид 7,4 19
Хлораль 9 23
были вычислены полуэмпирическими методами. Данные для поли-
меризации формальдегида и хлораля также включены в табл. 7.3.
Некоторое, хотя и ограниченное сравнение значений А// и AS
для алкенов, карбонильных мономеров и циклических мономеров
можно сделать, пользуясь данными табл. 7.3 и 3.12.
Значения АН для трех- и четырехчленных циклических моно-
меров сравнимы с этими величинами для алкенов, причем они зна-
чительно превышают АН для обоих карбонильных соединений.
Превращение л-связи карбонила в ст-связь менее экзотермично,
чем соответствующее превращение л-связи алкена. Однако изме-
нение энтропии для обоих типов мономеров приблизительно одина-
ково. Значения AS более упорядоченных циклических мономеров
показывают, что упорядочивание при полимеризации их оказы-
вается меньшим, чем в случае нециклических мономеров.
Значения АН для различных циклических, эфиров и ацеталей
хорошо соответствуют порядку этих величин, который можно
ожидать, исходя из соображений относительной стабильности как
функции размера кольца (разд. 2.36 и рис. 2.3). Трех- и четырех-
Полимеризация с раскрытием цикла
437
членные циклические мономеры полимеризуются с наибольшим
экзотермическим эффектом. Затем наблюдается быстрое уменьше-
ние ЬН для пяти- и шестичленных колец, и далее — повышение
цля восьмичленного цикла. Нулевые значения А// для шестичлен-
пых циклических мономеров согласуются с тем фактом, что эти
циклы еще не были заполимеризованы. Как было указано
в разд. 2.36, заместители в цикле обычно увеличивают стабиль-
ность его по сравнению с незамещенным соединением. Например,
в то время как тетрагидрофуран полимеризуется в присутствии
множества катализаторов, удалось успешно заполимеризовать
лишь небольшое число замещенных тетрагидрофуранов.
7.3. Циклические амиды
Полимеризацию циклических амидов
nCO-NH-(CH2)^ -*-HNH-(CH2)mCO^
(7.58)
можно инициировать основаниями, катионными частицами и во-
дой. Из циклических амидов хорошо изучена полимеризация
лактамов — соединений, образующихся при внутримолекулярном
амидировании аминокислот. (Другим типом циклических амидов
являются соединения, образованные при димеризации аминокис-
лоты.) Лактамы называют обычно тривиальными названиями, на-
пример: Р-пропиолактам или 2-азетидинон для четырехчленного
цикла, у-бутиролактам или 2-пирролидон для пятичленного цикла,
6-валеролактам или 2-пиперидон для шестичленного цикла, 6-гек-
санолактам или е-капролактам для семичленного цикла, 7-гепта-
нолактам или энантолактам для восьмичленного цикла и 8-окта-
лолактам или каприллактам для девятичленного цикла.
7.3а. Анионная полимеризация
?.3а.1. Инициирование сильными основаниями
Сильные основания, такие, как щелочные металлы, гидриды
металлов, амиды металлов и металлоорганические соединения,
инициируют полимеризацию лактамов, образуя анион лактама [1],
например для капролактама с металлом
(СН2)5-NH-f-M (CH2)5-N-M+ + i/2H2
(7.59а)
438
Глава 7
или с производными металлов
С=О
С = 0
(СН2)6 —NH + B-M+ (CH2)5-N-M+4-BH
VII
(7.596)
Использование более слабых оснований типа гидроокисей и алко-
голятов дает худшие результаты, так как достаточно высокую
концентрацию аниона при этом удается обычно получить лишь
при условии удаления продукта ВН, чтобы сдвинуть равновесие
вправо.
На второй стадии инициирования анион лактама VII реагирует
с мономером путем трансамидирования с раскрытием цикла
О
II
О
с
/ \ /. т 7 х Медленно
(СН2)5—N~M+ + HN—(СН2)5---------<
(СН2)5—N—СО(СН2)-.\М+
VIII
(7.60)
Анион первичного амина VIII в противоположность аниону лакта-
ма VII не стабилизирован за счет сопряжения с карбонильной
группой. Он отличается высокой реакционной способностью
и быстро реагирует с мономером, отрывая от него протон
(CH2)5-N-CO(CH2)5N-M+ + (CH2)5-NH
(СН2)5 — N — CO(CH2)5NH2 + (CH2)5-N-M+
IX
VII
(7.61)
и образуя имидный димер IX, N-капроилкапролактам, с одновре-
менным регенерированием аниона лактама.
Имидный димер IX был выделен [35], и было показано% что он
действительно является инициирующей частицей, необходимой
для начала роста цепи. Большие индукционные периоды, наблю-
дающиеся при полимеризации лактамов, обусловлены медленной
реакцией, предшествующей образованию этого димера [уравне-
ние (7.60)]. Имидный димер необходим для полимеризации вслед-
ствие того, что амидная связь недостаточно активна (т. е. недоста-
точно электроноакцепторна) по отношению трансамидирования
анионом лактама. Присутствие в имидном димере карбонильной
П олилеризация с раскрытием цикла
439
группы, соединенной с атомом азота амидной группы кольца, уве-
личивает электроноакцепторные свойства амидной связи. Это при-
водпт к увеличению активности амидного цикла по отношению
к нуклеофильному аниону лактама. Рост цепи представляет собой
реакцию между растущей имидной частицей X и анионом лактама
О
С
(CII2)5-N-CO(CH2)5NH-----и
X
(СН2)5-N-M+
о
м+
—> (CH2)S—N —СО(СН2)6 —N- —CO(CH2)6NH— (7.62)
с последующим быстрым обменом протона с мономером
/ \ М+
(CH2h-N — СО(СН2)5 — N-—CO(CH2)5NH--(-
О
(CH2)5-N-[-CO(CH2)6NH —]
+ (СН2)5 —N-M+
(7.63)
причем происходит регенерация аниона лактама и растущего
имида X.
Можно видеть, что катализируемая основаниями полимериза-
ция лактамов существенно отличается от других видов полимери-
зации в двух отношениях. Во-первых, растущий центр не является
радикалом, анионом или катионом, а представляет собой цикли-
ческую амидную связь. Во-вторых, не мономер присоединяется
к растущей цепи, а его анион — активированный мономер [36].
Этот механизм полимеризации чрезвычайно похож на описанный
выше механизм анионной полимеризации акриламида (разд. 5.66).
Для такой полимеризации концентрации обоих растущих частиц
и активированного мономера определяются концентрацией осно-
вания. Новым следствием такой реакции является то, что скорость
роста каждой растущей цепи зависит от концентрации основания.
тог°, если равновесйе обмена протона [уравнения (7.61)
11 ('.63)] далеко сдвинуто направо, то скорость роста каждой цепи
440
Глава 7
будет полностью независимой от концентрации мономера, а будет
зависеть только от концентрации активированного мономера,
определяемой концентрацией основания.
7.За.2. Применение ацилирующих агентов
7.3а.2а. Механизм реакции. Применение одних только силь-
ных оснований для полимеризации лактамов весьма ограниченно.
Это объясняется наличием упомянутых выше больших индукцион-
ных периодов, а также тем, что только самые реакционноспособные
лактамы, такие, как капролактам или энантолактам, полимери-
зуются в этих условиях. Менее активные лактамы, например
шестичленный пиперидон, не могут быть заполимеризованы
в присутствии одних сильных оснований. Эти мономеры слишком
мало активны для образования требуемого имидного димера.
Оба препятствия были преодолены путем образования имида при
взаимодействии мономера с ацилирующими агентами — хлоран-
гидридами и ангидридами кислот, изоцианатами, неорганическими
ангидридами и др. [37—39]. Так, капролактам может быть быстро
превращен в N-ацилкапролактам XI при реакции с хлорангидри-
дом
(СН2)5 —NH
О
RCOC1
(СН2)5—N—СО —R
(7-64)
XI
N-Ациллактам может быть синтезирован in situ или же получен
заранее и затем добавлен к реакционной системе.
Инициирование заключается во взаимодействии N-ацилкапро-
лактама с активированным мономером, сопровождаемом быстрым
обменом протона с мономером
(CH2)5-N-CO-R + (CH2)6-N-M+ -*
О
С
(CH2)6-N-CO(CH2)5-N-—СО-R
(7.65)
XII
Полимеризация с раскрытием цикла
444
О
—* (СНг)5 —N —СО(СН2)5 —NH —СО —R +
О
С
(CH2)5-N-M+
(7.66}
XIII
Частицы XII и XIII соответствуют частицам VIII и IX для поли-
меризации в отсутствие ацилирующего агента. Ацилирующий агент
обеспечивает легкую полимеризацию многих лактамов, заменяя
медленную реакцию по уравнениям (7.60) и (7.61) на быстрое
инициирование по уравнениям (7.64) и (7.66). Использование
ацилирующих агентов — предпочтительный метод полимеризации
лактамов. Этот метод целесообразно использовать и для полимери-
зации реакционноспособных лактамов, так как при этом исчезает
индукционный период и возрастает скорость.
Рост цепи происходит так же, как рост цепи частиц IX
О
О
(СН2)5 —N —CO(CH2)6NH— СО —R +
(СН2)5— N-M+
О
м+
-> (СН2)5 — N — CO(CH2)5N- — CO(CH2)6NH — СО - R
(7.67>
XIV
Мономер
(СН2)6 - N - CO(CH2)6NH —]2 — СО — R
(7.68
В полимеризации лактамов, протекающей под влиянием силь-
ных оснований и ацилирующих агентов, в стадии инициированиям
могут участвовать как имидный димер IX, так и N-ациллактам XI.
Однако обычно последний является значительно более важным
и первым можно пренебречь. Для таких полимеризационных
систем наименования, применяемые для описания роли основания
и ацилирующего агента (или N-ациллактама), ошибочны. Основа-
ние почти всегда именуется катализатором или инициатором,
а ацилирующий агент — сокатализатором или промотором. Одна-
442
Глава 7
ко ясно, что действительным инициатором является ацилирующий
агент или N-ациллактам. Основание более точно было бы называть
активатором [36], так как его назначением является образование
активированного мономера (аниона лактама).
7.3а.2б. Скорость полимеризации. Подробные кинетические
исследования полимеризации лактамов этого типа не проводи-
лись, однако можно отметить
основные кинетические харак-
теристики этой реакции. Ско-
рость полимеризации можно
выразить уравнением типа
Rv = k [I] [АГ], (7-69)
где [I] и [М~]—концентрации
растущих имидных частиц н
активированного мономера со-
ответственно. Поэтому ско-
Р и с. 7.8. Зависимость выхода и
молекулярного веса полимера, об-
разующегося при полимеризации
а-пирролидона при 40J С, от кон-
центраций N-бензоил-а-пирроли-
дона и калия [38].
Концентрация калия, мол.%: 1) 0,61;
2) 4,32.
рость полимеризации должна возрастать с увеличением кон-
центрации основания и N-ациллактама, так как при этом будут
увеличиваться [М-] и [I] соответственно. На рис. 7.8 и 7.9 пока-
заны эти зависимости для полимеризации пиперидона и пирроли-
дона с калием и N-ациллактамами. В качестве индикатора скоро-
сти реакции здесь использована степень превращения мономера в
полимер в данный момент времени. Степень превращения возрастае г
с увеличением концентрации калия, достигает максимума, и затем
уменьшается при более высоких концентрациях калия. Снижение
степени превращения может быть связано с гидролизом Х-ацпл-
лактама основанием.
7.3а.2в. Степень полимеризации. По мере повышения процента
превращения степень полимеризации увеличивается, как при
поликонденсации. Зависимость Хп от концентрации активирован-
ного мономера и растущих частиц аналогична этой зависимости
для процесса образования живущего полимера. Размер макромо-
лекул растет с увеличением концентрации основания и уменьше-
нием концентрации N-ациллактама. Рис. 7.8 и 7.9 иллюстрируют
эти эффекты для полимеризации пиперидона и пирролидона.
Полимеризация с раскрытием цикла
443
В связи с вопросом о степени полимеризации возникает вопрос
о способах обрыва цепи. Во многих случаях полимеризации не
имеется агентов обрыва цепи и рост
продолжается до исчерпания моно-
лера. Соединение с активным (кис-
лым) водородом, присутствующее
в виде примеси или добавленное
преднамеренно, приводит к обрыву
цепи вследствие реакции с активи-
рованным мономером или растущим
анионом XIV.
Распределение по молекулярным
весам в этих процессах полимериза
цпи бывает обычно широким вследст-
вие реакций разветвления, которые
происходят на поздних стадиях про
цесса [40]. При уменьшении кон-
центрации мономера и увеличении
концентрации амидных групп, вклю-
ченных в полимерную цепь, воз-
растает тенденция растущего анио-
Р и с. 7.9. Зависимость выхода и молеку-
лярного веса полимера, о бразующегося при
полимеризации а-пиперидона при 40° С,
от молярного соотношения калий/N-ane-
тил-а-пиперидон [38].
Концентрация калия, мол.%: 1) 2,58; 2) 5,16;
3) 7,74.
на XIV к реакциям разветвления за счет взаимодействия с конце-
выми циклическими амидными группами полимерной цепи
О
/ \ М+ / \
(СН,) 5 - N -CO(CH2)sN- - CO(CH2)5NH — СО - R + (СН2)6 - N
О
(CH2)5-N-CO(CH2)5-N-CO(CH2)5NH — CO-R
со
(7.70)
(<1н2)5
I
N-M+
)
444
Глава 7
Реакции протонного обмена с мономером имеют меньшее значение
на этом этапе [уравнение (7.68)]. Было найдено [40], что амины
сужают молекулярновесовое распределение, что может быть
обусловлено взаимодействием аминов с концевым амидным циклом,
в результате которого образуются мертвые полимерные цепи
(т. е. цепи без концевых циклических структур), не способные
к реакции по уравнению (7.70).
7.36. Катионная полимеризация
7.36.1. Полимеризация в присутствии протонных
кислот
Для инициирования полимеризации лактамов были использо-
ваны различные протонные кислоты, такие, как фосфорная, соля-
ная, бромистоводородная, и карбоновые кислоты [41—43]. Поли-
меризация протекает с инициированием по уравнениям
(СН2)5—NH + HClTt (СН2)5-NH2
+
С1-
(7.71)
/с<^ \ /с\
(СН,)-— NH., +\hN—(СН„),
ZD Z \ * * ZD
cr v
(CH2)6 —NH —CO(CH2)5NH2
+
Cl-
(7.72)
с последующим ростом цепи
О О
— NH(CH2)6CO — NH-(CH2)5+HN-(CH2)5
+
Cl-
— [— NH(CH2)5CO-]2-HN-(CH2)5
+
Cl-
(7.73)
Эффективность кислоты при инициировании полимеризации не-
посредственно зависит от ее кислотности [44]. Как реакция роста,
Полимеризация с раскрытием цикла
445
так и реакция инициирования [уравнения (7.72) и (7.73)] включают
нуклеофильную атаку атома азота мономера на карбонил лактам-
ного цикла. Реакция несколько похожа на реакцию при катализе
основаниями. В обоих типах полимеризации активность лактамно-
го карбонила к нуклеофильной атаке (мономером или активиро-
ванным мономером) увеличивают, делая его частью имидной
структуры. Нуклеофильный агент (мономер) в катализируемой
кислотами полимеризации менее нуклеофилен, чем нуклеофильный
агент (активированный мономер) при полимеризации, катализи-
руемой основаниями. Эта разница в нуклеофильности компенси-
руется в первой реакции увеличением активности лактамного
карбонила протонированием.
Кислоты Льюиса, такие, как SnCl4, также были использованы
для инициирования полимеризации лактамов [45]. Реакция заклю-
чается в образовании комплекса SnCl4 с мономером (1 : 2)
2Мономер-|-8пС14 —* [(Мономер)28пС14] —> НС1+ Продукт. (7.74)
Этот комплекс, идентифицированный методом ИК-спектроскопии,
разлагается с образованием НС1 и неидентифицированного про-
дукта. Полимеризация катализируется НС1 по реакциям, приве-
денным в уравнениях (7.71) и (7.73).
7.36.2. Полимеризация в присутствии аминов
Интересна полимеризация капролактама при действии таких
аминов, как анилин или бензиламин [46]. Эта полимеризация вклю-
чает катионное инициирование аммонийной солью, образованной
при реакции между амином и лактамом. Общая схема реакции
представлена ниже
RNH2 + HA RNHJA"
(7.75)
RNH3+A- + (CH2)5- NH
—> RNHCO(CH2)5NHJA-,
(7.76)
0
О
II
c
RNII—-CO(CH2)5NHJA- + (cK)5-^NH -»
—> RNH — CO(CH2)5NHCO(CH2)5NHJA-, (7.77)
гДе лактам. Правильность этого механизма подтверждается
тем фактом, что непосредственное применение солянокислого ами-
на йриводит к полимеризации без индукционного периода, причем
зависимость Rri от концентрации аммонийной соли пнепставляет
446
Глава 7
собой зависимость первого порядка. Однако полимеризация с ами-
нами имеет половинный порядок по концентрации амина, так как
реакция (7.75) равновесна, и концентрация аммонийной соли
зависит от концентрации амина в степени х/г. Кроме того, при
катализируемой аминами реакции имеется индукционный период,
необходимый для создания определенной концентрации аммоний-
ной соли.
Реакции инициирования и роста требуют переноса двух прото-
нов от аммонийной соли к азоту лактама одновременно с открыти-
ем лактамного кольца. Это может происходить согласно уравне-
ниям
О ОН
II I
С С —NHR
RNHt-f-(CH2)5-NH-^(CH^)5^NH2 —>.RNH —СО(СН2)5 —NHJ (7.78)
Необходимость переноса двух протонов объясняет, почему третич-
ные амины оказались полностью неэффективными в процессах
полимеризации лактамов. И первичные, и вторичные амины могут
быть использованы для этой цели, хотя эффективность вторичных
меньше, чем первичных, вследствие стерических затруднений.
Для каждого класса аминов инициирующая способность прямо
зависит от основности амина [44, 46].
7.Зв. Катализ водой
7.3в.1. Механизм реакции
Установлено, что вода — полезная добавка при промышлен-
ной полимеризации капролактама. (Другим методом, имеющим
промышленное значение, является катализ основаниями.) Гидро-
лиз лактама в аминокислоту — начальная реакция этого процесса
(СН2)5 —NH-J- Н2О —> HOOC(CH2)5NH2 (7.79а)
XV
Следующую реакцию ранее рассматривали [47, 48] как реакцию
раскрытия цикла при взаимодействии аминокислоты с лактамом
О
HOOC(CH2)5NH2 + (CH2}5-NH—>HOOC(CH2)5NHCO(CH2)5NH2 (7.796)
XVI
Полимеризация с раскрытием цикла
447
с ростом цепи, продолжающимся таким же образом, наряду
с ростом путем поликонденсационных реакций между концевыми
аминами и карбоксильными группами различных частиц, таких,
как XV и XVI. Более удобно инициировать эту реакцию добавле-
нием аминокислоты непосредственно перед началом реакции.
Позднее для катализируемой водой полимеризации лактама
был предложен механизм, подобный механизму полимеризации,
катализируемой аминами [44]. Обычно считают, что аминокислота,
такая, как XV, существует в виде цвиттер-иона
Тогда можно утверждать, что полимеризация инициируется элек-
трофильной атакой четвертичного азота цвиттер-иона на мономер
точно таким же образом, как при реакции, катализируемой ами-
нами
(CH2)5-NH + H2O —± -O2C(CH2)5NH3+
(7.80)
с ростом цепи, протекающим аналогично
— CO(CH2)5NH+4-HN-(CH2)5 —Zt — CO(CH2)5NHCO(CH2)5NHJ (7.81)
7.Зв.2. Степень полимеризации
Эта полимеризация — процесс равновесный, и ее кинетика
должна рассматриваться соответствующим образом, т. е. согласна
уравнению (7.48) для скорости полимеризации. Количественную
зависимость степени (равновесной) полимеризации от различных
параметров реакции можно рассматривать способом, описанным
Тобольским [49, 50]. Этот путь обычно применим для многих
равновесных реакций полимеризации. Для катализируемой водой
полимеризации лактама КИ и Кр имеют различные значения, при-
чем предполагается, что Кр не зависит от размера растущей части-
цы. (Последнее утверждение — еще один пример положения об
отсутствии зависимости реакционной способности функциональ-
ных групп от длины цепи полимера.) Концентрация [М„] растущих
цепей размера, определяемого п, дается уравнением
[М*]=ки [С]к [М]к (Яр [М]к)”-1
(7.82)
где С катализатор (вода). Индекс «к» относится к равновесным
(критическим) концентрациям. Общая концентрация полимерных
молекул [N] получается суммированием уравнения (7.82) по цепям
448
Глава 7
всех размеров
*и[С]к[М]к
1-Яр[М]к
(7.83)
тогда как общая концентрация [W] мономерных сегментов, содер-
жащихся в полимере, дается выражением
г wi = 51 п [M+i —
LVVJ ?J nUVi„J-(1_Kp[M]K)2 •
(7-84)
Степень полимеризации полимера получим, разделив уравнение
(7.84) на (7.83):
у [W] 1
п [N] 1-Кр[М]к
Материальный баланс по мономеру и катализатору дается урав-
нениями
(7.85)
(7.86)
(7-87) .
где индекс «0» относится к начальным концентрациям. Комбинация
уравнения (7.84) с (7.86) и уравнения (7.83) с (7.87) дает
[М]0 = [М]к(14-КиХ*[С]к), (7.88)
[С]О = [С]К(1 +КиХп [М]к). (7.89)
Комбинация же уравнений (7.85) — (7.87) также приводит к оче-
видной зависимости
[M]0 = [M]K + [W],
[Clo = [C]K-HN],
у _ [М]р-[М]К
n [Clo-[C]K
Все эти уравнения показывают зависимость степени полимериза-
ции от начальной и равновесной концентраций мономера и ката-
лизатора и от констант равновесия инициирования К„ и роста Кр.
Молекулярный вес полимера увеличивается с уменьшением Ки
и [С]о и увеличением Кр и [М]о. Так, на рис. 7.10 показано изме-
нение Хп в зависимости от начальной концентрации воды для поли-
меризации 8-капролактама [50, 51]. Пунктирная кривая пред-
ставляет собой теоретическую кривую, полученную из уравнений
(7.84), (7.88), (7.89) и (7.90) следующим образом. Ка и Кр могут
быть получены из одной или более серий экспериментальных
данных для Хп, [С]о, [М]о и [М] с использованием различных
комбинаций уравнений. Значения [М]к затем вычисляют по урав-
нению (7.85) для различных принятых величин Хп. Соответствую-
(7.90)
Полимеризация с раскрытием цикла
449
щие значения [С]о рассчитывают из выражения
[М]0-[М] (791)
КИ [М]в
1+ки[М]вхп _
полученного комбинацией уравнений (7.89) и (7.90). Величины Хп
п [С]о используют для построения теоретической кривой. Как
Р и с. 7.10. Зависимость степени поли-
меризации полимера, образующегося при
полимеризации е-капролактама при
220 °C, от начальной концентрации во-
ды [50].
Сплошная линия — экспериментальные дан-
ные; пунктирная линия — теоретическая кри-
вая.
видно из данных рис. 7.10, экспериментальная кривая очень
хорошо совпадает с теоретической.
7.3г. Реакционная способность
Реакционная способность лактамов по отношению к полимери-
зации зависит в первую очередь от термодинамической устойчи-
вости цикла и от типа инициирования. Термодинамическая ста-
бильность лактамных циклов различного размера следует тем же
общим закономерностям, что и другие циклические соединения
(разд. 2.36). В табл. 7.4 приведены теплоты и энтропии полимери-
зации некоторых лактамов [32, 52]. Общий порядок величин Д/7
близок к их значениям для полимеризации циклических эфиров
(табл. 7.3) и порядку стабильности циклоалканов (табл. 2.2).
Относительный порядок реакционной способности лактамов
несколько изменяется в зависимости от типа инициирования.
Сведения о зависимости реакционной способности от размера
цикла весьма ограниченны. Имеющиеся данные приведены
в табл. 7.5 [47, 53, 54]. Порядок активности лактамов в полимери-
зации очень близок к ожидаемому (разд. 2.36), хотя и имеются
определенные колебания. Реакционная способность замещенных
циклических амидов оказалась, как и можно было ожидать, более
низкой, чем реакционная способность незамещенных мономеров
450
Глава 7
Таблица 7.4
Теплоты и энтропии полимеризации лактамов [32, 52J
Мономер Число членов в цикле —дн, ккал/моль (4,187-103 Дж/моль) —AS, кал/(моль-К) (4,187 Дж/моль-К)
2-Пирролпдон 5 1,3 7,3
2-Ппперидон 6 1,1 6,0
Капролактам 7 3,0 1,1
Энантолактам 8 5,7 4,0
Каприллактам 9 9,6
12-Додекалактам 13 1,5
Таблица 7.5
Реакционная способность лактамов к полимеризации
в зависимости от размера кольца
Тип инициирования Ряд реакционной способ- ности циклов различного размера Литература
Ацилирующий агент 7>5>6 53
НС1 8>7>11»5,6 54
Вода 7>8«9»5,6 47
[47]. Однако влияние заместителей на понижение реакционной
способности тем больше, чем меньше размер цикла. Замещение
у атома азота также оказывает более сильное влияние на сниже-
ние активности лактама, чем замещение у углеродных атомов.
Последний эффект является, возможно, результатом стерических
затруднений. Неожиданно низкие реакционные способности 9-
и 11-членных лактамов при полимеризации в присутствии соответ-
ственно воды и НС1 могут быть связаны с возможностью их суще-
ствования в транс(или (штпн)-конформации XVII или г^нс(или
снн)-конформации XVIII
XVII
(штпн-Конформация (в которой карбонильный кислород и амидный
водород находятся по разные стороны от амидной связи С—N)
является более стабильной формой для амида. Однако большин-
ство циклов, содержащих 7 и менее членов, способны существо-
Полимеризация с раскрытием цикла
451
пять только в менее стабильной смн-конформации. По мере увели-
чения цикла амидная связь приобретает способность в различной
степени переходить в анти-конформацию. Циклы с 10 и более
членами полностью существуют в виде антпн-конформации, тогда
как 8- и 9-членные циклы могут находиться частично как в син-,
•гак и в анти-конформациях. Таким образом, несколько изменен-
ный порядок реакционной способности циклических амидов может
быть обусловлен различиями в реакционной способности син-
11 антпн-форм этих амидов.
Интересно отметить, что 5- и 6-членные лактамы значительно
более реакционноспособны в условиях анионной полимеризации,
чем в условиях полимеризации, инициируемой НС1 или водой.
Это можно объяснить с точки зрения различия механизма реакции
в этих случаях. При анионной полимеризации лактамное кольцо
атакуется более нуклеофильным агентом, чем при катионной
полимеризации. В случае полимеризации под влиянием воды имеет
место электрофильная атака на лактамный цикл. Благодаря низ-
кой электронной плотности амидной группы, эта реакция проте-
кает не столь легко, как реакция с сильным нуклеофилом.
В результате всего этого анионная полимеризация обычно являет-
ся более быстрой и менее селективной реакцией, чем обе другие
реакции. Лактамы, не способные к полимеризации в присутствии
воды или НС1, легко полимеризуются по анионному механизму.
Эти различия в реакционной способности проявляются также
при сравнении необходимых для полимеризации температур. Для
полимеризации, инициируемой водой или кислотой, так же, как
и для других случаев катионной полимеризации, обычно необхо-
димы температуры порядка 250 °C. Анионная полимеризация про-
текает при значительно более низких температурах.
7.4. Циклосилоксаны
Линейные полисилоксаны, или, как их часто называют, сили-
коны, могут быть синтезированы путем анионной или катионной
полимеризации циклических силоксанов. Наиболее часто приме-
няется полимеризация циклического тетрамера — октаметилцикло-
тетрасилоксана
СН3
I
СН3 о —Si-СНз г СН3
^sf" \> СН3-н>— — Si-O —
/II/ I
СН3 О Si L СН3
\-О/ ^СНз
СНз ХСН3
(7.92)
452
Глава 7
7.4а. Анионная полимеризация
Анионная полимеризация циклических силоксанов может быть
инициирована окисями и гидроокисями щелочных металлов, сила-
нолятами, такими, как триметилсиланолят калия (CH3)2SiOK,
и другими основаниями [1, 55]. Как инициирование
А + SiR2^OSiR2->J
A-t-SiR2O^SiR2O ;
(7.93)
так и рост цепи
.0——
/ Л *з
---SiR./r + SiR„-f-OSiR,V =₽=*= —•(•SiRoOArSiRoO (7.94)
Z Л Z >J Ь, X т i-
включает нуклеофильную атаку аниона на циклический мономер
аналогично тому, как это происходит при анионной полимеризации
эпоксидов. Рост цепи прекращается путем добавления агентов
обрыва.
Интересным аспектом этой полимеризации является наблюде-
ние [56], что ДЯ реакции близка к нулю и, что особенно удивитель-
но, Д£— величина положительная, равная ~1,6 кал/(моль>К)
16,7 Дж/(моль»К)]. Основное влияние при этой полимеризации
оказывает возрастание энтропии (неупорядоченности) в ходе поли-
меризации. Положительное значение Д5 — очень редкий случай
при полимеризации. Единственными описанными примерами поло-
жительного значения Д5 являются случаи полимеризации цикли-
ческих октамеров серы и селена [32]. Все другие процессы полиме-
ризации сопровождаются уменьшением энтропии вследствие умень-
шения неупорядоченности полимера по сравнению с его мономером.
Положительную Д5 для реакций полимеризации циклических
силоксанов, серы и селена можно объяснить большой гиб-
костью линейных полимерных цепей, обусловленной большими
размерами атомов, образующих эти цепи. Такая гибкость ведет
к большим степеням свободы в линейных полимерах по сравнению
с циклическими мономерами.
7.46. Катионная полимеризация
Катионную полимеризацию циклических силоксанов проводят
с протонными кислотами и кислотами Льюиса [1, 57]. Как ини-
циирование [уравнение (7.95)] так и рост цепи [уравнение (7.96)]
происходит через третичный ион оксония, который перестраивается
в переходный ион силикония
Полимеризация с раскрытием цикла
453
А"
О------НО R
I х _ 1 I + _
SiR2-<-OSiR24^ + Н+А SiR2“<-OSiR243 H-f-OSiR^OSrA ,
R
R
^OSiR24jOSi+A“
R
(7.96)
Доказательств действительного существования иона силикония
в приведенном выше механизме нет. Имеющиеся по этой полиме-
ризации данные могут равным образом быть истолкованы с пози-
ций механизма, включающего электронную атаку третичного иона
оксопия на мономер (без перегруппировки в ион силикония),
аналогично полимеризации циклических эфиров
А~ А-
OS iR9—О — 2 1 Мономер + ► -'~COSiR2)4~0
SiR2’f-OSiR2' )g SiR2-f~OSiR2^3 (7.97)
Интересно, что было высказано предположение о существовании
иона силикония в качестве промежуточного продукта и в другом
случае полимеризации. Так, полагают [58], что катализируемая
кислотами полимеризация 1,1,3,3-тетраметил-1,3-дисилациклобу-
тапа
СН2
п (CH3)2Si Si(CH3)2—>
\н2
г СН3
— Ji —СН2 —
- Jh3
(7.98)
протекает через промежуточное соединение XIX
СН3
+Si — CH2Si(CH3)2 — Clly
сн3
УТУ
454
Глава 7
7.5. Другие циклические мономеры
7.5а. Сложные циклические эфиры
Сложные циклические эфиры полимеризуются с раскрытием
цикла и образованием гетероцепных полиэфиров под влиянием
множества катионных и анионных катализаторов. Лактиды (слож-
ные циклические эфиры, образующиеся путем межмолекулярной
димеризации оксикислоты), например
О
/° —\
тгО = С С = О—»
—СН2 —С —О —_|2п —
(7.99)
так же как лактоны
О
О
«СО —R—>— L— R — С — 0-
(7.100)
были изучены в ряде работ [1, 59, 60]. Реакционная способность
различных циклических эфиров изменяется в том же порядке,
что и реакционная способность лактамов за одним исключением.
Реакционная способность пяти- и шестичленных лактонов прямо
противоположна реакционной способности соответствующих лакта-
мов. Пятичленный лактон (у-бутиролактон) не полимеризуется
в противоположность шестичленному 6-валеролактону.
Катионная полимеризация происходит путем атаки на мономер
растущего центра, который представляет собой либо ион оксо-
ния XX, либо ацилий-ион XXI, образованный мономолекулярным
раскрытием иона оксония
О
Я О
/ \ II
—— О —(СН2)т — О(СН2)т —С+
XX XXI
Возможно, что анионная полимеризация протекает следующим
образом:
О
О
С
СО(СН2)тО -|-0 — (CH2)m > CO(CH2)mOC(CH2)mO (7.101)
Этот механизм согласуется с тем фактом, что омыление сложных
эфиров основаниями обычно протекает путем расщепления связи
ацил — кислород, т. е. связи между карбонильным углеродом
П олимеризация с раскрытием цикла
455
и некарбонильным кислородом. Однако для реакций некоторых
лактонов было предположено [1] расщепление связи алкил —
кислород, и тогда полимеризация их может протекать через обра-
зование аниона карбоксилата как растущего центра
О О
!1 А
—• (CH2)mCO + О_____(СН2)5 > —(СН2)тСО2(СН2)5СОО (7.102)
7.56. Циклические амины
Циклические амины, называемые иминами, полимеризуются
в полпамины посредством кислотного, а не основного катализа
|1, 61, 62]. Низкая реакционная способность по отношению
к основному катализу является следствием нестабильности амин-
ного аниона (например, — CH2CH2NH для этиленимина) как
растущей частицы. Трехчленные имины (или азиридины) — един-
ственные мономеры этого класса, которые имеют значение. Высо-
кая степень напряженности трехчленного цикла проявляется
и чрезвычайно быстрой полимеризации. Так, довольно бурной
может быть полимеризация этиленимина при комнатной темпера-
туре. Разнообразные катионные частицы, в том числе обладающий
мягким действием бензил-катион (из бензилхлорида), катализиру-
ют эту полимеризацию.
Катионная полимеризация иминов протекает аналогично поли-
меризации циклических эфиров. Растущей частицей является ион
иммония и рост цепи протекает через нуклеофильную атаку моно-
мером
f СН2 ,CH2 +/СН2
— n( | + nA —> —nhch2ch2-n(; I (7.ЮЗ)
н хсн2 н хсн2 н хсн2
Разветвление при полимеризации иминов происходит либо
путем атаки иминной концевой группы одной полимерной цепи
на иммониевый центр растущей частицы
СН2 —СН2 СЙ2 —СН2
+/Т 2 \ / \ / сн2~сн2
— N. | + N---->—NHCH2CH2N-------------->
н\сн2 +
— nhch2ch2n —
сн2
I
СН2 (7.104)
+NH
СЩ —СН2
456
Глава 7
или атакой вторичной аминной группы полимерной цепи на иммо-
ниевый центр
+ /СН2
— | 4- — CH2CH2NH----> — CH2CH2NH— (7.105)
НХСН2 |
СН2
сн2
I
NH
Обрыв цепи может происходить при отрыве протона от иммониево-
го иона противоионом или любым атомом азота полимерной цепи
или, наконец, атомом азота мономера. Было также высказано
предположение о протекании реакции обрыва, происходящей
с расширением цикла путем «кусания своего хвоста» [аналогично
уравнению (7.40) для окиси этилена] и образованием в результате
этого концевого относительно нереакционноспособного пиперази-
нового кольца [61]
+"/ сн2 /~\
— NHCH2CH2Nz I -»—N NH-j-HA (7.106)
н\6н2 \_/
7.5в. Циклические сульфиды
Циклические сульфиды полимеризуются под действием кислот-
ных катализаторов [1, 6, 3]
п R—S —- -f-R—S-^ (7‘107)
По аналогии с полимеризацией простых циклических эфиров пред-
полагается, что растущая частица является циклическим сульфо-
пиевым ионом XXII
ххп
—R—S~
XXIII
Трехчленные циклические сульфиды также были заполимери-
юваны [64] основными катализаторами, очевидно, через растущий
сульфидный ион XXIII. Как и для эфиров, неизвестны случаи
шионной полимеризации циклических сульфидов с числом членов
5олее 4.
Полимеризация с раскрытием цикла
457
Полимеризация циклических сульфидов включает также ка-
тионную полимеризацию тритиана, циклического тримера тиоформ-
альдегида
S
( -»-(-CH2-S-)n- (7.108)
S S
и радикальную полимеризацию циклических дисульфидов и ром-
бической серы
(7.109)
s—s\
1 П.110)
s s
\—sz
7.5г. N-Карбоксиангидриды а-аминокислот
N-Карбоксиангидриды а-аминокислот, называемые также окса-
золидиндионами-2,5 или ангидридами Лейхса, полимеризуются
в присутствии основных катализаторов с образованием поли-
амидов
Н. /СО.
\
r/| 0-^ —(—COCHRNH —)п— (7.111)
HN. /
\со
Стадия роста, которая включает одновременно декарбоксили-
рование, протекает через анион мономера, подобно анионной
полимеризации лактамов, когда в качестве катализаторов приме-
няются сильные основания [36, 65]
СО СО
/ \о / \о
— NHCHRCO—N I + -N | —>
\ /С0 \ /С0
С С
l/ \ Н7 \
со
/ \о Мономер
~>— NHCHRCON —CHRCO —N I -----
I \ .СО ~с°2
COj
458
Глава 7
СО
NHCHRCONHCHRCO —N
(7.112)
С
Н R
Полимеризация в присутствии слабых оснований типа первичных
аминов протекает иным путем — включающим нуклеофильную
атаку аминной группы на мономер [64, 65]
/СО.
/ XNH
RNH2 + O I
-со2
—---> RNHCOCHRNH2 (7.113а)
RNH— COCHRNH
-co2
-» RNH — COCHRNHCOCHRNH2
(7.1136)
7.5д. Различные случаи полимеризации
Различные циклические экзо-иминосоединения были заполиме-
ризованы в присутствии катионных катализаторов [66]. Реакция
заключается в присоединении к иминной связи с одновременным
раскрытием цикла
0-
п R—N=c
Y-
—- -ONR—СО—V—СН2СН2-^
(7.114)
Эту реакцию наблюдали в случае иминокарбонатов (Y = О), 2-ими-
но-1,3-оксазолидинов (Y = NR) и 2-иминотетрагидрофуранов
(Y = СН2). Подобной же является и полимеризация циклических
эмдо-иминоэфиров [67]
N
Z \
nR-C (CH2)m
Хчо/
Г COR
_-N-(CH2)m-Jn-
(7.115)
П олимеризация с раскрытием цикла
459
Были также заполимеризованы некоторые циклоалканы, такие,
как производные циклопропана и норборнена [1, 8]
7.6. Полимеризация неорганических или
.элементоорганических мономеров
Как отмечалось в разд. 2.12в.9, значительный интерес пред-
ставляет синтез неорганических или элементоорганических полиме-
ров. Некоторые из изученных полимеров были получены полиме-
ризацией с раскрытием цикла [69].
7.6а. Полифосфазены
Наиболее тщательно изученной полимеризацией с раскрытием
кольца неорганического мономера является полимеризация цикли-
ческого тримера дихлорфосфазена (или фосфонитрилхлорида,
как его обычно называют). Некатализируемая полимеризация
протекает при температурах выше 250 °C, тогда как применение
катализаторов, таких, как карбоновые кислоты или их соли,
металлы п спирты, позволяет осуществлять полимеризацию при
более низких температурах [70, 71]. В зависимости от специфи-
ческих условий реакции имеет место в различной степени развет-
вление и сшивание макромолекул. Очевидно, при некатализируе-
мой полимеризации [70] инициирование происходит путем терми-
ческого разрыва в мономере связи N—Р
С1 С1
/ -
-----> N N
ci2p p'ci2
Ч /
N
(7.118)
460
Глава 7
за которым следует нуклеофильная атака аниона азота на мономер
С1 С1
ci2p = npci2np=n-+
ii
C12P
V
Cl
-» Cl2P = N-(—PC12N—)3-PCl2NP = N- (7.119)
Cl
Рост идет аналогичным образом
Cl Cl
Cl
Cl2P = N-(-PCl2N-)n J = N-+
| C12P PC12
Cl 4NZ
Cl
+ I
C12P = N-(-PC12N—)n+3-P = N- (7.120)
Cl
Обрыв происходит путем межмолекулярного нуклеофильного
замещения иона хлора с последующим связыванием этого иона
с положительной фосфорной концевой группой
N Xn
С12Р\^РС12
(7.121)
C12P\N^PC12
Полимеризация с раскрытием цикла
461
Разветвление происходит путем соответствующей межмолекуляр-
ной реакции между анионом азота одной полимерной цепи
п связью Р—С1 другой цепи, тогда как сшивание, вероятно, воз-
никает вследствие реакций типа
С1 С1
— p = N-----1---N = PC12— -»— P = N— (7.122)
Cl —N — PC13 —
Каталитическая полимеризация включает инициирование путем
нуклеофильной атаки катализатора на мономер с образованием
аниона азота, подобного инициирующей частице в некатализи-
руемой реакции. Для инициирования анионом бензоата это пред-
ставляется следующим образом:
С6Н5СОСГ +
ci24n/pci2
N
Cl io—COCcHg
-CGH5COCI
+ cr
С12*Чк-/РС12
О Cl
II I
C1P=NPC12NP=N“
(7.123)
7.66. Другие случаи полимеризации
Полимеры фосфонитрилхлорида обладают некоторыми полез-
ными с практической точки зрения свойствами. В частности, они
являются неплохими эластомерами. Были предприняты попытки
полимеризовать другие циклические неорганические мономеры,
но без серьезных практических успехов. Такие результаты можно
объяснить главным образом плохим пониманием механизма реак-
ции. В настоящее время многие системы дают только полимеры
низкого молекулярного веса и с плохо определенной структурой.
Ясное понимание реакции позволит в будущем синтезировать более
полезные материалы.
Циклосилазаны, как, например, гексаметилциклотрисилазан
l(CH3)2SiNH]3, полимеризуются [72] с образованием продуктов
низкого молекулярного веса в присутствии каталитических коли-
честв бромистого аммония при 140 °C. Структура полимера нели-
нейна [ Si(CH3)2NH—имеется большое количество атомов
азота, к которым присоединены по три атома кремния. Полимер
462
Глава 7
содержит циклическую (XXIV) и разветвленную (XXV) структуры
(СН3)2
/Si\
— Si(CH3)2—N N —
(CH3)2ii si(CH3)2
\N/
H
XXIV
--N —Si(CH3)2—NH —Si(CH3)2 —N —Si(CH3)2 —
I 1
(CH3)2Si I
| XXV
7.7. Сополимеризация
По сополимеризации различных циклических мономеров опу-
бликовано значительное количество работ. В них описана сополи-
меризация пар циклических мономеров, содержащих как одинако-
вые функциональные группы (например, группы двух циклических
эфиров или двух циклических силоксанов), так и различные
(например, группы эфира и лактона), а также сополимеризация
циклических мономеров с алкенами. Очень немногие из этих
реакций представляют в настоящее время практический интерес.
Исключением является сополимеризация триоксана с небольшими
количествами циклического эфира для повышения термостойкости
полиоксиметилена. Такой сополимер более стабилен в отношении
термической деполимеризации, чем гомополимер триоксана [24].
Поскольку в сополимере сегменты циклического эфира беспоря-
дочно распределены по цепи полиоксиметилена, случайные тер-
мические расщепления не приводят к полной деполимеризации
макроцепи.
Многие реакции сополимеризации циклических мономеров
довольно сложны по ряду причин. В большинстве случаев имеет
место равновесие рост — распад цепи. Это требует соответствую-
щей обработки экспериментальных данных (разд. 6.56), что,
однако, не всегда делается. Кроме того, сополимерные композиции
обычно чрезвычайно чувствительны к условиям реакции. Изме-
нения типа противоиона, растворителя, температуры могут оказы-
вать большое влияние на сополимеризацию. Применяемая ини-
циирующая система особенно важна для сополимеризации моно-
меров, содержащих различные функциональные группы. Принци-
пиальные различия в природе растущих центров (ион карбония,
оксониевый ион, имид, карбанион, ацилий-ион и т. д.) для различ-
ных типов мономеров во многих случаях препятствуют протеканию
сополимеризации.
Полимеризация с раскрытием цикла
463
7.7а. Мономеры, содержащие одинаковые
функциональные группы
Была изучена сополимеризация пар циклических эфиров [73],
лактамов [74], лактонов [75] и циклических силоксанов [76].
Интерпретация данных по реакционной способности мономеров
чрезвычайно сложна вследствие большого влияния как особен-
ностей данной пары сомономеров, так и условий реакции. Напри-
мер, вследствие низкой реакционной способности циклические
эфиры, содержащие более четырех членов, не способны к анионной
сополимеризации, однако легко полимеризуются по катионному
механизму. При успешно протекающей сополимеризации поведе-
ние мономеров в этом процессе часто можно рассматривать с точки
зрения влияния размеров цикла на реакционную способность
мономера по отношению к атаке растущими частицами.
В табл. 7.6 приведены некоторые данные по катионной сополи-
меризации различных циклических эфиров и ацеталей. Основы-
Таблица 7.6
Относительная активность циклических мономеров
при сополимеризации3 [73]
Ml м2 П Г2
3,3-бис-(Хлорметил)ок- Эпихлоргидрин 3-2 0,3-0,5
сетан Тетрагидрофуран 0,82+0,05 1,00+0,05
Тетрагидрофуран 6 0,01+0,01 1,80+1,01
2-Метилтетрагидрофуран 2,7+0,25 0,05+0,01
Тетрагидропиран 1,66 0
1,4-Диоксан 2,12 0
1,3-Диоксан 13 0
1,3-Диоксолап 1,5+0,1 0,65+0,05
1,3-Диоксолан Эпихлоргидрин 0,08 1,2
Оксетан 0,0125 165
Тетрагидрофуран 0,25+0,05 28+4
Тетрагидрофуран в 0,24+0,03 195+10
Триоксан г 1,75 0,57
Т етрагидрофу ран Эпихлоргидрин 3,85+0,05 0,00+0,05
1,3-Диоксолан 28+4 0,25+0,05
“Катализатор BF3- (С2Н5)2О при 0° С.
о В присутствии А1(С2Н5)з с эпихлоргидрином в качестве промотора.
в В присутствии SnC, Ц.
г Ион силикония при 70° С.
464
Глава 7
ваясь на соображениях, рассмотренных в разд. 2.36, следует ожи-
дать, что склонность этих циклических мономеров к раскрытию
цикла будет уменьшаться с изменением размера цикла в ряду 4 >
> 3 > 5 > 6. Однако реакционная способность мономера зависит
также от способности к образованию иона оксония, поскольку
именно его образование происходит на стадии роста. Данные
работы [73] указывают, что основность мономеров снижается
с изменением размера кольца в ряду 4 > 5 > 6 > 3 для цикли-
ческих формалей, основность которых в общем случае ниже, чем
основность соответствующих циклических эфиров. Рассмотрение
данных табл. 7.6, так же как и соответствующих значений 1/г1?
указывает, что оба эти эффекта имеют примерно одинаковое
значение.
Для циклических формалей, менее реакционноспособных чем
эфиры, реакционная способность мономеров снижается с измене-
нием размера цикла в ряду 4 > 5 > 3 > 6. Вышеупомянутое
влияние каталитической системы на сополимеризацию ясно видно
из данных по относительной реакционной способности мономеров
при сополимеризации тетрагидрофурана с 3,3-бмс-(хлорметил)ок-
сетаном и 1,3-диоксоланом.
Способность 2-метилтетрагидрофурана, тетрагидропирана, 1,3-
диоксана и 1,4-диоксана вступать в сополимеризацию интересна
в связи с тем, что эти мономеры не способны к гомополимерпзации
при большинстве условий. Необычное поведение этих мономеров
при сополимеризации с формальной точки зрения аналогично
поведению при радикальной сополимеризации таких моно-
меров, как малеиновый ангидрид, стильбен и диэтилфумарат
(разд. 6.36.3), однако причина этого иная. Способность к сополи-
меризации этих циклических мономеров может быть обусловлена
высокой реакционной способностью (нестабильностью) их расту-
щих частиц. Рассмотрим последовательность стадий при сополиме-
ризации
/""Ч ^2 /""Ч МI /~Ч
~'~0-М1 ----MjO—м2 ---------- ~'~М1М2О-М1 , (7.124)
XXVI
где М2 — циклический мономер, не обладающий способностью
к гомополимерпзации. При гомополимеризации равновесие рост —
распад для М2 полностью сдвинуто влево. Однако в присутствии
второго мономера Mj частицы XXVI, отличающиеся высокой
реакционной способностью, могут присоединять Mi прежде, чем
произойдет распад. Результатом этого является сополимериза-
ция Mi и М2. Такое положение наблюдается при сополимеризации
0-пропиолактона = 18 ± 2) с у-бутиролактоном (г2 = 0,36 ±
Полимеризация с раскрытием цикла
465
_|_ 0,10) в присутствии в качестве катализатора смеси триэтилалю-
лшшй — вода, в то время как к гомополимеризации у-бутиролак-
тон не способен [75]. Однако способность у-бутиролактона к сопо-
лимеризации существенно зависит от характера каталитической
системы. Использование гидроокиси калия, системы диэтилцинк—
вода п изопропилат аммония не приводит к сополимеризации, при
этом образуется только гомополимер р-пропиолактона.
Анионная сополимеризация (в присутствии ацетильных ини-
циаторов) е-капролактама (М^ и а-пирролидона (М2) демонстри-
рует некорректность выводов на основании значений т\ и г2, полу-
ченных при использовании стандартного уравнения сополимериза-
ции [уравнение (6.12)]. Значения относительной реакционной
способности мономеров, вычисленные по этому уравнению, оказа-
лись равными = 0,75 и г2 = 5,0. Отсюда был сделан вывод,
что а-пирролидон почти в восемь раз более активен, чем е-капро-
лактам [74]. Однако детальное изучение [77] этой системы пока-
зывает, что строение образующегося сополимера не определяется
константами скоростей ktl, kiz, kZ2 и k2l для различных реакций
роста гомополимера и сополимера. Трансацилирование, заклю-
чающееся в обмене мономерных сегментов в инициирующих
п растущих имидах
с—N(CH2)3CO + N(CH2)5CO
с—N(CH2)5CO + N(CH2)3CO
(7.125)
происходит со значительно большими скоростями, чем другие
реакции роста. Состав сополимера, таким образом, зависит от
равновесия реакции трансацилирования в соответствии с уравне-
нием
<*[МД] Х0[МГ]
d [М2] [М2 ]
(7.126)
где [М7] и [М7] — концентрации анионов лактамов. Отношение
[MJ/tM^] определяется относительной кислотностью обоих моно-
меров по равновесию
Mj-f-M, 3---» М2 + Му.
(7.127)
Из уравнений (7.126) и (7.127) получим
d[Mf] KgKoIMd
d[M2] [М2]
(7.128)
466
Глава 7
Значения Ка и Ко были определены независимо и равны 0,4
и 0,3 соответственно. Большую реакционную способность «-пирро-
лидона в сополимеризации с е-капролактамом можно объяснить
большей кислотностью «-пирролидона (приблизительно в 2,5 раза)
и большей нуклеофильностью аниона а-пирролидона в равновесии
трансацилирования (приблизительно в 3,3 раза).
7.76. Мономеры, содержащие различные
функциональные группы
Сополимеризация мономеров, содержащих различные функцио-
нальные группы, в высшей степени избирательна. В рассматри-
ваемом типе сополимеризации некоторые взаимодействия блоков
сомономеров легко могут оказаться невыгодными вследствие боль-
ших различий в типах, участвующих в реакции растущих центров,
а часто также вследствие различий в энергиях связей, подвер-
гающихся разрыву в ходе роста цепи. Сополимеризация между
некоторыми типами мономеров оказывается невозможной, между
другими — трудно осуществимой, тогда как в некоторых случаях
можно ожидать, что сополимеризация пойдет сравнительно легко.
Так, комбинации лактама с мономером другого типа, таким, как
лактон, эпоксид или алкен, несовместимы из-за различий в меха-
низме их роста *). Анион, образующийся из второго мономера,
будет быстро обрывать цепь, отщепляя протон от мономерного
лактама. Сополимеризации, таким образом, не произойдет; в луч-
шем случае получится гомополимер лактама и/или гомополимер
другого мономера (низкого молекулярного веса).
Реакции анионной сополимеризации между лактонами и эпокси-
дами протекают легко [78]. Сюда относятся реакции сополимериза-
ции 0-иропиолактона = 15,0) с окисью стирола (г2 = 0,1),
Р-пропиолактона (г] = 9,0) с эпихлоргидрином (г2 =• 0,2), а также
б-валеролактона (г] = 5,3) с окисью пропилена (г2 = 0,25).
Эти реакции сополимеризации демонстрируют почти идеальное
поведение (г ~ 1) с тенденцией образования блоков полилактонов.
Способность этих двух типов мономеров к анионной сополимери-
зации обусловлена тем, что растущие центры обоих мономеров
одинаковы — алкоксидные анионы. Большая активность лактонов
в сополимеризации является следствием большей реакционной
способности сложноэфирной связи по сравнению с реакционной
способностью простой эфирной связи по отношению к нуклеофиль-
ной атаке.
Сополимеризация циклических оксидов с лактонами проводит-
ся при катионном инициировании. Рост цепи при взаимодействии
*) Известны случаи сополимеризации лактамов с лактонами по анион-
ному механизму (см. список дополнительной литературы к гл. 7 [1, 7, 11]).—
Прим. ред.
Полимеризация с раскрытием цикла
467
иона оксония простого циклического эфира с оксоииевым XX
птп ацилиевым XXI ионом лактона происходит сравнительно
легко. Таким образом, эфират трехфтористого бора инициирует
[73] сополимеризацию 0-пропиолактона (rj = 0,06) с 3,3-бнс-(хлор-
метпл)оксетаном (г2 = 38) и 0-пропиолактона (гх = 0,4) с тетра-
гпдрофураном (г2 =" 2,9). Большая реакционная способность
циклического эфира является следствием большей легкости обра-
зования в этом случае иона оксония (вследствие высокой основ-
ности) по сравнению с лактоном. Четырехчленный циклический
простой эфир проявляет большую склонность вступать в сополи-
мер в виде длинных блоков. Сополимеризация между алкенами
п циклическими мономерами требует тщательного подбора мономе-
ров и условий реакции, так как типы растущих центров обоих
мономеров часто сильно различаются [1]. Так, катионная сополи-
меризация простых циклических эфиров с алкенами невозможна
вследствие того, что ион оксония недостаточно электрофилен для
атаки двойной связи алкена. Однако оказывается возможной
катионная сополимеризация алкена с триоксаном, тетраоксаном
и циклическими формалями, такими, как 1,3-диоксолан [1, 73, 79].
Сополимеризация происходит потому, что растущие центры обоих
мономеров одинаковы — ионы карбония. С другой стороны, такие
лактоны, как |3-пропиолактон, не сополимеризуются катионным
методом с алкенами. Это показывает, что растущим центром в поли-
меризации лактонов является не ацилий-ион XXI, а ион оксония
XX. Стирол не удалось сополимеризовать ни с эпоксидами, ни
с лактонами при использовании основных катализаторов, хотя
были получены блок-сополимеры с окисью этилена. Алкоксидный
ион (растущий центр как для эпоксидов, так и для лактонов) яв-
ляется, очевидно, недостаточно основным для того, чтобы вступить
в реакцию с двойной связью стирола. Был изучен ряд других
случаев сополимеризации. Осуществлена сополимеризация таких
пар мономеров, как этиленимин — 0-пропиолактон [80], триок-
сан — тритиан [81], этиленимин — окись углерода [82], малеиновый
ангидрид — окись пропилена [83] и окись пропилена — двуокись
серы [84[. Два последних примера сополимеризации
О
со ''со -l О
'сН=СП СН-СНСНз-*
СНз
— СО —СН = СН —СОО —СН —СН2О —
о о
11 / \
& = О4-СН-СНСНз —
О СНз
У ।
— S — О — СН — СН2О —
(7.129)
(7.130)
468
Глава 7
интересны тем, что они относятся к ионным реакциям, в которых
рост цепей малеинового ангидрида и двуокиси серы протекает
иначе, чем при радикальном процессе полимеризации этих
мономеров (разд. 3.6.7а).
Хотя все описанные в этом разделе реакции сополимеризации
являются ионными, известны и некоторые примеры радикальной
сополимеризации [1], однако число мономерных пар, способных
к росту по радикальному механизму, незначительно. В качестве
примера можно привести описанную выше радикальную сополиме-
ризацию этиленимина, окиси углерода и этилена под влиянием
динитрила азо-бпс-изомасляной кислоты и у-излучения [85].
Вопросы и упражнения
7.1. Рассмотрите следующие мономеры и катализаторы:
Катализаторы н-С4Н9Ы BF3 H2SO4 NaOC2H5 Н2О Мономеры Окись пропилена Пирролидон б-Валеролактон Этиленимин Октаметиленциклотетра - силоксан Пропиленсульфид Триоксан Оксациклобутан
Какие катализаторы могут быть использованы для полимери-
зации каждого из перечисленных мономеров? Обсудите необходи-
мость, если она имеется, применения сокатализаторов или других
реагентов в сочетании с катализатором. Напишите механизм
полимеризации для каждого случая.
7.2. Обсудите влияние размера цикла на склонность цикли-
ческого мономера к полимеризации с раскрытием цикла.
7.3. Анионная полимеризация окиси пропилена обычно приво-
дит к образованию полимеров со сравнительно низким молекуляр-
ным весом. Объясните причины этого явления.
7.4. Рассмотрите полимеризацию окиси пропилена под влияни-
ем метилата натрия при 70 °C. Концентрации окиси пропилена
и метилата натрия равны 0,80 и 2,0 ПО-4 М соответственно. Вычис-
лите среднечисловой молекулярный вес полимера при 80 %-ной
степени превращения, учитывая влияние реакции переноса цепи
на мономер.
7.5. Опишите при помощи уравнений роль промоторов при
катионной полимеризации циклического эфира.
7.6. Полимеризация тетрагидрофурана, характеристики кото-
рой приведены в табл. 7.1, представляет собой равновесную поли-
меризацию с [М]к = 1,7 М. Вычислите начальную скорость поли-
Полимеризация с раскрытием цикла 469
меризации и скорость полимеризации при 20%-ной степени пре-
вращения.
7.7. Какова роль ацилирующего агента и активированного
мономера при анионной полимеризации лактамов?
7.8. Рассмотрите равновесную полимеризацию е-капролактама,
инициированную водой при 220 °C. Вычислите равновесные значе-
ния Л'и и если [С]о = 0,352, [М]о = 8,79 и [М]к = 0,484,
а степень полимеризации при равновесии равна 152.
7.9. Опишите при помощи уравнений реакции «кусания своего
хвоста», протекающие с расширением цикла при полимеризации
простых циклических эфиров, ацеталей и аминов.
Список литературы
Patsiga R. A., J. Macromol. Sci.-— Revs. Macromol. Chem., Cl(2), 223
(1967).
Фурукава Дж., Саегуса T., Полимеризация альдегидов и окисей, гл. III —
VII, изд-во «Мир», М., 1965.
Gurgtolo А. Е., J. Macromol. Sci.— Revs. Macromol. Chem., 1(1), 39 (1966).
Gee G., Higginson W. С. E., Merrall G. T., J. Chem. Soc., 1959, 1345.
Steiner E. C., Pelletier R. R., Trucks R. О., J. Am. Chem. Soc., 86, 4678
(1964).
GeeG., Higginson W. С. E., Taylor K. J., Trenholme M. W., J. Chem. Soc.,
1961, 4298.
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
И.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
24.
25.
26.
27.
28.
29.
30.
31.
Snyder W. H., Meisinger A. E., Jr., Polymer Preprints, 9(1), 382 (1968).
Burrows R. C., Polymer Preprints, 6(2), 600 (1965).
Meerwein H., Delfs D., Morshel H., Angew. Chem., 72, 927 (1960).
Sims D., Makromol. Chem., 98, 135, 245 (1966).
Okada M., Yamashita Y., Ishii Y., Makromol. Chem., 80, 196 (1964).
Bawn С. E. H., Bell R. M., Ledwith A., Polymer, 6, 95 (1965).
K^ntz I., J. Polymer Sci., A-l(5), 193 (1967); Polymer Preprints, 9(1),
Dreyfuss M. P., Dreyfuss P., J. Polymer Sci., A-l(4), 2179 (1966).
Истхем А., Эпоксидные соединения, в кн. «Катионная полимеризация»
под ред. П. Плеша, гл. 10, изд-во «Мир», М., 1966.
Tsuruta Т., Inoue S., Tsubaki К., Makromol. Chem., Ill, 236 (1968).
Imai H., Saegusa T., Matsumoto S., Tadasa T., Furukawa J., Makromol.
Chem., 102, 222 (1967).
Saegusa T., Imai H., Matsumoto S., J. Polymer Sci., A-l(6), 459 (1968).
Kubisa P., Brzezinski J., Penczek S., Makromol. Chem., 100, 286 (1967).
Fortschr. Hochpolymer.-Forsch. (Advan. Polymer Sci.),
Penczek I., Sazanov Y. N., Penczek S., Makromol. Chem., 100, 156 (1967).
Beste L F., Hall H. K„ Jr., J. Phys. Chem., 68, 269 (1964).
n • ' £'’ Tobolsky A. V., J. Polymer Sci., A3, 3261 (1965).
m B'’ McAndrew F. В., J. Macromol. Sci. (Chem.), Al(2), 231 (1967).
”lkl T Higashimura T., Okamura S, J. Polymer Sci., B5, 583 (1967).
lamashita Y., Okada M., Suyama K., Makromol. Chem., Ill, 277 (1968).
penczek I., Penczek S., Makromol. Chem., 67, 203 (1963).
ль Ptr° o' Penczek S., J. chim. phys., 59, 696 (1962).
amura S., Kobayashi E., Higashimura T., Makromol. Chem., 88, 1 (1965).
“ose J. B., J. Chem. Soc., 542, 547 (1956).
Burrows R. C„ Crowe B. F., J. Appl. Polymer Sci., 6, 467 (1962).
470
Глава 7
32. Brandrup J., Immergut E. H. (eds.), Polymer Handbook, p. 11—363,
Interscience Publishers, John Wiley and Sons, Inc., New York, 1966.
33. Joshi R. M., Zwolinski B. J., Heats of Polymerization and Their Structural
and Mechanistic Implications, in «Vinyl Polymerization», Ham G. E.
(ed.), vol. 1, part I, chap. 8, Marcel Dekker, Inc., New York, 1967.
34. Plesch P. H., Westermann P. II., Polymer, 10, 105 (1969).
35. Rothe V. M., Reinisch G., Jaeger W., Schopov I., Makromol. Chem., 54,
183 (1962).
36. Szwarc M., Pure and Appl. Chem., 12(1), 127 (1966); Fortschr. Hochpoly-
mer.-Forsch. (Advan. Polymer Sci.), 4, 1 (1965).
37. Yasumoto T., J. Polymer Sci., A3, 3301 (1965).
38. Tani H., Konomi T., J. Polymer Sci., A-l(4), 301 (1966).
39. Scelia R. P., Schonfeld S. E., Donaruma L. G., J. Appl. Polymer Sci.,
8, 1363 (1964).
40. Mottus E. H., Hedrick R. M., Butler J. M., Polymer Preprints, 9(1), 390
(1968).
41. Rothe V. M., Reinisch G., Jaeger W., Schopov I., Makromol. Chem., 54,
183 (1962). .
42. Doubravsky V. S., Geleji F., Makromol. Chem., 113, 270 (1968).
43. Kagiya T., Kishimoto H., Narisawa S., Fukui K., J. Polymer Sci., A3,
145 (1965).
44. Hay J. N., J. Polymer Sci., B5, 577 (1967).
45. Amass A. J., Hay J. N., Makromol. Chem., 103, 244 (1967).
46. Burnett G. M., Hay J. N., MacArthur A. J., Polymerization of Caprolac-
tam, in «The Chemistry of Polymerization Processes», p. 139—156, Mono-
graph 20, Society of Chemical Industry, London, 1966.
47. Cubbon R. С. P., Makromol. Chem., 80, 44 (1964).
48. Hermans P. H., Heikens D., J. Polymer Sci., 44, 429, 437 (1960).
49. Tobolsky A. V., J. Polymer Sci., 25, 220 (1957).
50. Tobolsky A. V., Eisenberg A., J. Am. Chem. Soc., 81, 2302 (1959); 82, 289
(1960).
51. Wiloth F., Z. Physik. Chem., N. F. (Frankfurt), 4, 66 (1955).
52. Dachs K., Schwartz E., Angew. Chem. (Intern. Ed.), 1, 430 (1962).
53. Yoda N., Miyake A., J. Polymer Sci., 43, 117 (1960).
54. Okata N., J. Polymer Sci., Al, 3151 (1963).
55. Барри А., Бек X., Силиконовые полимеры, в кн. «Неорганические поли-
меры» под ред. Ф. Стоуна и В. Грэхема, гл. 5, изд-во «Мир», М., 1965.
56. Lee С. L., Johannson О. К., J. Polymer Sci., A-l(4), 3013 (1966).
57. Kendrick Т. С., J. Chem. Soc., 1965, 2027.
58. Levin G., Carmichael J. B., J. Polymer Sci., A-l(6), 1 (1968).
59. Chujo K., Kobayashi H., Suzuki J., Tokuhara S., Tanabe M., Makromol.
Chem., 100, 262 (1967).
60. Ohse V. H., Cherdron H., Makromol. Chem., 95, 283 (1966), 97, 139 (1966).
61. Jones G. D., MacWilliams D. C., Braxtor N. A., J. Org. Chem., 30, 1994
(1965).
62. Джонс Дж., Соединения азота, в кн. «Катионная полимеризация под
ред. П. Плета, гл. 14, изд-во «Мир», М., 1966.
63. Stille J. К., Empen J. A., J. Polymer Sci., A-l(5), 273 (1967).
64. Cooper W., Morgan D. R., Wragg R. T., Eur. Polym. J., 5(1), 71 (1969).
65. Terbojevich M., Pizziolo G., Peggion E., Cosani A., Scofjone E., J. Am.
Chem. Soc., 89, 2733 (1967).
66. Mukaiyama T., Fujisawa T., Nohira H., Hijugaji T., J. Org. Chem., 27,
3337 (1962).
67. Levy A., Litt M., J. Polymer Sci., A-l(6), 57 (1968).
68. Micheletti F. W., Keaveney W. P., J. Polymer Sci., A3, 895 (1965).
69. Олкок Г., Гетероциклические соединения и полимеры на их основе, гл. С
и 8, изд-во «Мир», М., 1970.
Полимеризация с раскрытием цикла
471
7(1, У1 ас Callum. J. R., Werninck A., J. Polymer Sci., A-l(5), 3061 (1967).
7L Paddock N. L., Quart. Rev. (London), 18, 168 (1964).
72. Kruger C. R., Rochow E. G., J. Polymer Sci., A2, 3179 (1964).
73 Yamashita Y., Tsuda T., Okada M., Iwatsuki J. Polymer Sci., A-l(4),
2121 (1966).
74. Kobayashi Matsuya K., J. Polymer Sci., Al, 111 (1963).
75. Tada K., Numata Y., Saegusa T., Furukawa J., Makromol. Chem., 77,
220 (1964).
76. Merker R. L., Scott M. J., J. Polymer Sci., 43, 297 (1960).
77. Bar-Zakay S., Levy M., Vofsi D., J. Polymer Sci., A-l(5), 965 (1967).
78. Cherdron V. II., Ohse H., Makromol. Chem., 92, 213 (1966).
79. Higashimura T., Tanaka A., Miki T., Okamura S., J. Polymer Sci., A-l(5),
1927, 1937 (1967).
80. Kagiya T., Narisawa S., Manabe K., Fukui К., J. Polymer Sci., B3, 617
(1965).
81. Lando J. B., Prayer P., J. Polymer Sci., B6, 285 (1968).
82. Kagiya T., Narisawa S., Ichida T., Fukui K., Yokota H., Kondo M., J. Po-
lymer Sci., A-l(4), 293 (1966).
83. Kern R. J., Schaefer J., J. Am. Chem. Soc., 88, 3627 (1966).
84. Schaefer J., Kern R. J., Katnik R. J., Macromolecules, 1, 107 (1968).
85. Kagiya T., Maruta I., Ichida T., Narisawa S., Fukui K., J. Polymer Sci.,
A-1(5), 1645 (1967).
ГЛАВА 8
СТЕРЕОХИМИЯ ПОЛИМЕРИЗАЦИИ
И ПОЛИКОНДЕНСАЦИИ
В полимерах встречается несколько типов структурной изоме-
рии. Существуют полимеры с одинаковым суммарным химическим
составом, но с различным взаимным расположением атомов или
групп атомов. Такие изомерные полимеры можно получать при
использовании в их синтезе изомерных мономеров; к таким полиме-
рам относятся, например, поливиниловый спирт, полиацетальде-
гид и полиоксиэтилен
ОН I Г СН3
J I
—сн2-сн —Jn— —L—сн-о—
_[-сн2сн2-о-]п-
поливиниловый спирт полиацетальдегид полиоксиэтилен
Изомерны также полиметилметакрилат и полиэтилакрилат
--сн2-сн
СООС2Н5 Jn
г СНз -
I
— — сн2-с--------
СООСНз-'п
полиметилметакрилат полиэтилакрилат
Поли-е-капролактам и полигексаметиленадипамид
- [ - CO(CH2)5NH - ]п- - [ -NH(CH2)6NHCO(CH2)4CO - ]п -
являются изомерами, хотя исходные вещества для их синтеза,
строго говоря, нельзя называть изомерами: е-капролактам имеет
такой же суммарный состав, что и смесь (1 : 1) гексаметилендиа-
мина и адипиновой кислоты. Аналогично этому сополимер (1 : 1)
этилена и бутена-1 изомерен полипропилену.
Изомерные полимеры можно получать также из одного моно-
мера, если есть несколько путей его полимеризации. Образующий-
ся в небольших количествах при полимеризации полимер с распо-
ложением мономеров голова к голове изомерен полимеру с обыч-
ным присоединением голова к хвосту (см. структуры III и IV
в разд. 3.2а). Изомерные полимеры можно получить при полимери-
зации мономеров с двумя полимеризующимися группами, если есть
возможность направлять полимеризацию по одному или другому
Стереохимия полимеризации и поликонденсации
473
пути. Примерами такого типа изомерии являются 1,2- и 1,4-поли-
меры диенов-1,3 (разд. 3.14 и 8.6), отдельные реакции полимериза-
ции кетенов и акролеина по двойным углерод-углеродным и карбо-
нильным связям (разд. 5.6а), а также синтез полимеров линейной
п циклической структуры па основе несопряженных диенов
(разд. 6.66). Все указанные примеры структурной изомерии пред-
ставляют интерес, так как изомерные полимеры значительно
отличаются по своим физико-химическим свойствам.
В этой главе основное внимание будет сосредоточено на другом
типе изомерии в полимерах; стереоизомерии, обусловленной осо-
быми условиями синтеза полимеров. Важность этого вопроса
вытекает из того большого влияния, которое стереоизомерия ока-
зывает на ряд важнейших свойств полимеров. На стереоизомерию
при полимеризации алкенов обратил внимание еще Штаудингер [1]
в 1932 г. Однако только в последние пятнадцать лет было показа-
но, что полимеризацию можно провести в таких условиях, что
при каждом акте роста цепи будет проявляться возможность сте-
реоизомерии. Кроме того, ранее практически не рассматривали
вопроса синтеза полимеров с упорядоченной пространственной
конфигурацией, в процессе которого стереоизомерия повторяю-
щихся элементарных звеньев обеспечивала создание упорядо-
ченных, или регулярных, а не хаотических структур. К числу важ-
нейших достижений полимерной химии за последнее десятилетие
следует отнести выяснение существования стереоизомерии в поли-
мерах. Но наиболее существенно то, что Циглер [2] и Натта [3, 4J
сумели найти удобный метод синтеза полимеров с высоким содер-
жанием стереорегулярной структуры [5—11]. Здесь уместно заме-
тить, что мы теперь только начинаем делать то, чем природа зани-
мается в течение столетий и тысячелетий. В природе встречаются
стереорегуляфные полимеры различных типов, к ним относятся
натуральный каучук, целлюлоза, крахмал и различные биополи-
меры.
8.1. Типы стереоизомерии в полимерах
Стереоизомерия в полимерах, как и во всех органических соеди-
нениях, бывает двух типов: геометрическая и оптическая, причем
своим появлением опа обязана различным расположениям (конфи-
гурациям) атомов или групп атомов в молекуле [12]. Геометри-
ческая изомерия возникает вследствие различного расположения
заместителей при двойной углерод-углеродной связи или у цикли-
ческих структур. Оптическая изомерия обусловлена различной
конфигурацией заместителей при насыщенном атоме углерода,
термин «конфигурация» не следует путать с термином «конформа-
ция». Под конформацией понимается различное расположение
атомов или групп атомов в молекуле, которое обусловлено простым
вращением вокруг одинарных связей. Примерами различных кон-
формаций полимеров являются полностью вытянутый плоский
зигзаг, хаотически свернутый клубок, спираль и пластинчато
сложенные цепи. При этом молекулы с различным расположением
называют конформационными изомерами. Последние могут пре-
вращаться друг в друга при простом вращении связей. Вместе
с тем конфигурационная изомерия определяется изомером с различ-
ным расположением атомов или групп атомов, которые нельзя
превратить друг в друга без разрыва и повторного образования
химических связей.
8.1а. Полимеризация монозамещенных этиленов
Центр стерическои изомерии
Изомерия наблюдается при полимеризации любых алкенов,
содержащих заместитель у углерода при двойной связи. При
полимеризации монозамещенного этилена СН2=СНВ (где R —
заместитель) образуются полимеры, в которых каждый второй
атом углерода полимерной цепи теоретически можно считать асим-
метрическим. Каждый из таких «асимметрических» атомов углеро-
да, обозначаемых С*, схематически выглядит следующим образом:
Н
I
— С*------
R
I
Здесь асимметрия, по-видимому, может быть обусловлена тем, что
атом углерода С* имеет четыре различных заместителя: Н, R и два
полимерных сегмента (— и -----) различной длины. Хотя на
какое-то время эта точка зрения и получила распространение, сейчас
такие атомы углерода, как С*, уже не считают истинно асимметри-
ческими [13, 14]. Настоящая асимметрия в атоме (или молекуле),
т. е. асимметрия, которая проявляется в оптической активности,
определяется лишь несколькими первыми атомами любого замести-
теля, присоединенными к кажущемуся асимметрическому атому.
Первые же несколько атомов двух полимерных сегментов, при-
соединенные к С*, одинаковы, и поэтому С* не может быть асим-
метрическим атомом. R результате такие полимерные структуры,
как I, не проявляют оптической активности. К такому же выводу
приводит и следующее рассуждение: при С* есть плоскость
внутренней симметрии, так как два полимерных сегмента эквива-
лентны, следовательно, внутренняя компенсация обусловливает
отсутствие оптического вращения.
Стереохимия полимеризации и поликонденсации
475
Хотя С* не является истинно асимметрическим центром, он
представляет собой центр стерической изомерии при полимериза-
ции CH2 = GHR. Каждый такой центр, который для удобства мож-
но называть псевдоасимметрическим или «асимметрическим» цент-
ром, может существовать в одной из двух различных конфигура-
ций. Предполагая, что основная цепь полимера —(—СН2СНК—)„
представляет собой вытянутую плоскую зигзагообразную конфор-
мацию, для каждого «асимметрического» атома углерода можно
представить две различные конфигурации, отличающиеся распо-
ложением группы R по отношению к плоскости углеродной цепи
полимера. Если считать, что плоскость углеродной цепи полимера
находится в плоскости страницы, то группы R будут располагаться
сверху или снизу такой плоскости. Две конфигурации обычно
называются R и ^-конфигурациями или D- и L-конфигурациями,
хотя такими символами, строго говоря, пользоваться нельзя,
так как они относятся к обозначению истинной асимметрии, кото-
рая в данном случае отсутствует. Рекомендуют [13, 14] конфигура-
ции при псевдоасимметрических центрах обозначать буквами г-
и я-, но это пока не получило распространения.
8.1а.2, Т античность
Регулярность, с которой последовательные «асимметрические»
центры показывают одинаковую конфигурацию, определяет сум-
марный порядок или тактичность полимерной цепи. (Термин
«тактичность» часто применяется на практике для обозначения
кристалличности образца полимера.) Если группы R у следующих
друг за другом «асимметрических» атомов углерода хаотически
расположены сверху и снизу плоскости зигзагообразной основной
полимерной цепи, то полимер не имеет никакого порядка и назы-
вается атактическим. Возможны два типа упорядоченных, или
тактических, структур полимера: изотактический и синдиотакти-
ческий. Для изотактического полимера характерно то, что центр
стерической изомерии в каждом повторяющемся звене имеет одну
и ту же конфигурацию. Все группы будут расположены по одну
сторону от плоскости углеродной цепи полимера: все сверху или
снизу от нее. Синдиотактическим называется полимер, у которого
центры стерической изомерии в каждом повторяющемся звене цепи
имеют конфигурации, противоположные конфигурациям соседних
с ним звеньев. В молекуле такого полимера R- и 5-конфигурации
регулярно чередуются вдоль полимерной цепи, или, иными слова-
ми, группы R располагаются над и под плоскостью полимерной
цепи в регулярно чередующемся порядке. На рис. 8.1 приведены
рассмотренные типы полимерных структур. При этом применяются
два типа изображений. Слева структуры изображаются так же,
476
Глава 8
как это сделано в разд. 3.96.2; с их помощью хотят показать, что
углеродная цепь находится в плоскости страницы, а заместители R
и Н расположены над этой плоскостью и под ней. Справа приведе-
ны соответствующие фишеровские проекционные формулы [12].
Вертикальными линиями в них изображены связи, направленные
за плоскость страницы, горизонтальные черточки отвечают связям,
направленным от плоскости страницы к глазу. Конфигурация
у каждого атома углерода основной полимерной цепи в фишеров-
ских формулах изображена для полимера в эклиптической кон-
z формации, тогда как в реальном
Н RH RH RH R
Н НН НН НН Н
изотакт ический
Н RR НН RR Н
Н НН НН НН Н
синдиотактический.
Н RH R R НН R
Н НН НН НН Н
атактический
полимере мы имеем дело с зигза-
гообразной конформацией.
Реакции полимеризации,
протекающие с образованием
тактических структур (изотак-
тических или синдиотактиче-
ских), называются реакциями
стереоспецифической полимери-
зации, а такие полимеры —
стереорегулярными полимерами.
(Здесь термины «стереорегуляр-
Р и с. 8.1. Различные структуры по-
лимеров моцозамещенных производ-
ных этилена — (— СН2— CHR—)п—.
Пунктирными линиями и треуголь-
никами обозначены связи при заме-
стителях, находящихся, соответствен-
но, ниже и выше плоскости углерод-
углеродной связи.
ный» и «стереоспецифический» имеют практически один и тот
же смысл.) Для наименования или обозначения различных про-
странственных изомерных полимеров пользуются инструкцией,
рекомендованной Международным союзом по чистой и при-
кладной химии [14]. Перед собственно названием химической
структуры полимера для обозначения его тактической структуры
ставится слово «изотактический» или «синдиотактический», напри-
мер, изотактический полипропилен. С этой же целью перед
формулой полимера ставятся приставки it- и st-, например
И-[СН2СН(СН3)]П, si-[CH2CH(CH3)]n. Если же перед названием
полимера нет этих терминов, это означает, что мы имеем дело
с атактическим полимером, например, если написано полипропи-
лен, то это атактический полипропилен.
Стереохимия полимеризации и поликонденсации
477
8.16. Полимеризация дизамещенных этиленов
8.16.1. Полимеры 1,1-дизамещенных этиленов
Наличие и тип тактичности для полимеров дизамещенных эти-
ленов зависит от положения и природы заместителей. При полиме-
ризации 1,1-дизамещенных этиленов (CH2=CRR') стереоизомерия
отсутствует, если группы R и R' одинаковые, как, например,
в изобутилене или винилиденхлориде. Если группы R и R' имеют
разную природу (например, —СН3 и —СООСН3 в метилметакри-
лате), то имеет место такая же стереоизомерия, как и в полимерах
монозамещенного этилена. Метильные группы могут быть над
плоскостью основной полимерной цепи или под ней (изотактиче-
ский полимер), могут находиться в регулярно чередующемся
порядке то сверху, то снизу плоскости цепи (синдиотактический
полимер) или располагаться хаотически (атактический полимер).
Наличие второго заместителя не оказывает никакого влияния на
стереоизомерию, так как пространственное положение первого
заместителя автоматически фиксирует положение второго замести-
теля. Второй заместитель располагается изотактически, если пер-
вый заместитель — изотактический; синдиотактически, если пер-
вый заместитель синдиотактический, и атактически, если пер-
вый заместитель — атактический.
8.16.2. Полимеры 1,2-дизамещенных этиленов
Иными особенностями характеризуется полимеризация 1,2-ди-
замещенных этиленов RCH=CHR', например, пентена-2 (R=CH3,
R' = С2Н5). В результате полимеризации таких соединений обра-
зуется полимер
имеющий два центра стерической изомерии (два «асимметрических»
атома углерода). При этом возникает возможность проявления
оитактичности, определяющейся различными сочетаниями тактич-
ности для обоих центров [15]. На фиг. 8.2 представлены четыре
различные стереорегулярные структуры такого типа. Диизотак-
тические структуры образуются в тех случаях, когда каждый из
Двух различных центров стерической изомерии является изотак-
тическим; дисиндиотактические структуры возникают тогда,
когда каждый из двух различных центров — синдиотактический.
трео-Диизатактическая структура отвечает диизотактическому
имеру, в котором два центра стерической изомерии находятся
противоположных конфигурациях. При зигзагообразном гра-
478
Глава 8
Формулы Ньюмена
Н R'H К'11 R’H R'
HRHRHRH R
трео-диизотакпшческий
R-----Н
Н----R'
R-----Н
Н-----R'
Н
H R’H R'H R'H R'
R HR HR HR H
aputripiiduajrjmanmu-
ческий
H R' R' H H R' R' H
эритро-дисиндиотакти-
ческий
H R'R' НН R'R' Н
трео - дисиндиотакти-
ческий
Р п с. 8.2. Стереорегулярныс структуры полимеров 1,2-дизамещенных про-
изводных этилена — (—CHR — CHR'—)п —.
фическом изображении оба заместителя — R и R' — расположены
по одну сторону от плоскости основной полимерной цепи. В фи-
шеровской проекции R и R' располагаются по разные стороны от
линии, представляющей полимерную цепь. эритро-Дииэотакти-
ческой структурой характеризуется диизотактический полимер,
у которого конфигурации при обоих центрах стерической изомерии
одинаковые. Различие между эритро- и трео-диизотактическими
структурами наглядно видно на формулах Ньюмена (рис. 8.2),
изображающих последовательные атомы углерода в полимерной
цепи в эклиптических конформациях [12].
трео-Дисиндиотактическими и эритро-дисиндиотактическими
полимерами называются дисиндиотактические полимеры с противо-
положными и одинаковыми конфигурациями по обеим сторонам
соответственно. Внимательное рассмотрение двух таких ди сип-
Стереохимия полимеризации и поликонденсации
479
дпотактических структур показывает, что они полностью идентич-
ны и отличаются только концевыми группами. Таким образом,
практически есть лишь один дисиндиотактический полимер. Но-
менклатура для дитактических полимеров [13, 14] строится на тех
;ке принципах, что и для монотактических полимеров. Так, раз-
личными стереорегулярными изомерами полипентена-2 были бы
иг/л?о-диизотактический полипентен-2, эрптро-дпизотактический
полипентеп-2 и дисиндиотактический полипентеп-2, в формулах
их перед [СН(СН3)СП(С2Н5)]П ставятся соответственно приставки
tit- eit- и st-.
Можно также представить себе стереорегулярные структуры
полимеров 1,2-дизамещенных этилена с атактическим расположе-
нием одного заместителя и изотактическим или синдиотактическим
другого, а также с изотактически расположенным одним замести-
телем и синдиотактически расположенным другим. Однако в на-
стоящее время такие предположения достаточно абстрактны, так
как при существующих методах синтеза факторы, которые могут
приводить к упорядочению или разупорядочению одного замести-
теля, оказывают аналогичный эффект и на другой заместитель.
8.1 в. Полимеризация карбонильных соединений
и полимеризация с раскрытием цикла
Стереоизомерия может проявляться не только у полимеров
алкенов, но и в ряду других полимеров. Образование упорядочен-
ных структур возможно при полимеризации карбонильных моно-
меров (RCHO и RCOR') и ряда циклических мономеров с раскры-
тием цикла. Так, например, полимеры на основе ацетальдегида
и окиси пропилена могут иметь изотактическую и синдиотакти-
ческую структуру (рис. 8.3 и 8.4).
Полиацетальдегпд, подобно всем вышеописанным полимерам
алкенов, не содержит истинно асимметрических центров (так как
у него есть внутренняя плоскость симметрии), поэтому он не может
обладать оптической активностью. Однако у полиоксипропилена
внутренняя плоскость симметрии отсутствует, так как он имеет
несимметричное повторяющееся звено —СН2СН(СН3)О—, поэтому
в его структуре III есть истинно асимметрический центр — атом
углерода С*
Н
I
— СН2 —С* —О-----
В отличие от структур I и II соседство углерода и кислорода
в структуре III обусловливает асимметричность этого атома С,
поэтому полиоксипропилен в изотактической форме может обла-
н сн3 н сн3 н сн3 н сн3
ХЛЛХ/
изотактический
H-f~CH3
О
нЧ—сн3
о 3
н—I—сн,
о
н—|—сн,
о 3
I
н сн, сн, Н Н СН3 СН3 н
» о о 9.0
Х.ХКХ.
х о о о о
синдиотактический
Н—I—СН,
О 3
СН,—I—Н
О
Н—I— СН3
О
СН3-Н Н
О
I
Рис. 8.3. Стереорегулярные структуры полиацетальдегида
—[—СН(СН3)О—]л—.
н сн н н н сн н н
изотактический
Н—I— Н
Н—I—СН,
О 3
Н—I—Н
н—I—СН3
о
Н—I—Н
Н—|—СН3
о
Н—|— н
н—|—сн3
о
СН3 НН Н СН3НН Н
синдиотактический
Рис. 8.4. Стереорегулярные структуры
- [- СН2СН(СН3)О-]Л -.
н—I—н
сн3—|—н
о
н—I— н
н—I— сн3
о
н—|—н
сн3—I—н
о
н—I—н
н—|—сн5
о
I
полиоксипропилена
Стереохимия полимеризации и поликонденсации
481
чать оптической активностью. Синдиотактический полиоксипропп-
лен. очевидно, не обладает оптической активностью; вследствие
чередования конфигураций у следующих друг за другом асим-
метрических атомов вдоль полимерной цепи оптическая активность
пропадает. Необходимо отметить, что стереорегулярность полиме-
ров. полученных полимеризацией с раскрытием цикла, не обуслов-
лена образованием асимметрических (или «асимметрических»)
центров в результате возникновения новых связей в процессе
полимеризации, как это имеет место при полимеризации алкенов
или карбонилсодержащих мономеров. Наоборот, в случае поли-
меризации с раскрытием цикла асимметрический центр содержится
уже в исходном мономере (например, окиси пропилена).
С этих позиций следует рассматривать и стереохимию поликон-
денсации. Образование в процессе поликонденсации новых связей
почти никогда не вызывает появления асимметрических или
«асимметрических» центров. Так, ни группы —О— и С = О в слож-
ных полиэфирах, ни группы —NH— и С=О в полиамидах не
являются центрами стерической изомерии. В таких полимерах
стереорегулярность может быть обусловлена действием тех же
факторов, что и в полимерах, получаемых полимеризацией с рас-
крытием цикла, т. е. она возможна, если хотя бы в одном из исход-
ных соединений есть центр стерической изомерии. Такие мономеры
весьма редко используют в поликонденсации. Еще более важно то,
что даже при применении таких мономеров образованию стереоре-
гулярных полимеров не способствуют типичные для поликонденса-
ции весьма жесткие условия.
8.1г. Полимеризация бутадиена-1,3 и 2-замещенных
бутадиенов-1,3
8.1г.1. 1,2- и 3,4-Полимеризация
При полимеризации диенов-1,3, например бутадиена, изопрена
и хлоропрена, возможно образование полимеров с оптической
('•Н2 — СН —СН = СН
бутадпен-1,3
СН3
сн2 = с—сн=сн2
изопрен
CI
СН2 = С — СН = СН2
хлоропрен
и геометрической изомерией [6—8,10,11,16]. Полимеризация таких
мономеров по одной из двойных связей приводит к возможности
проявления оптической изомерии. Так, при 1,2-полимеризации
бутадиена может получаться изотактический, синдиотактический
и атактический 1,2-полибутадиен
482
Глава 8
СН2 = СН —СН = СН2 —>
г—СН2 —СН——
I
сн
II
L СН2 -1п
IV
(8.1)
Следовательно, образуются полимеры таких же типов, что и при
полимеризации монозамещенных олефинов, поэтому строение их
можно выразить формулами, приведенными на рис. 8.1, где R =
= —СН = СН2. При полимеризации хлоропрена и изопрена число-
возможных структур еще больше, так как при замещении по раз-
ным двойным связям образуются различные изомеры. В отли-
чие от полимеризации бутадиена при 1,2- и 3,4-полимерпзации
хлоропрена и изопрена образуются разные продукты. Кроме того,
в этом случае возможно образование шести структур, отвечающих
соответственно двум изотактическим, двум синдиотактическим
и двум атактическим полимерам. Так, например, полиизопрен
имеет структуры V и VI с тремя различными формами для каждой
структуры (рис. 8.1, R = —СН = СН2 и —С(СН3) = СН2 соответ-
ственно).
СН3
СН2 = СН — с = сн2
г- СНз-
I
— — СН2 —С —
сн
II
L- сн2-
V
1,2-полимеризация
г — СН2 —СН —
С—СН3
СН2
VI
3,4-полимеризация
8.1г.2. 1,4-Полимеризация
(8.2)
Большой практический интерес представляют продукты 1,4-
полимеризации (разд. 3.14) сопряженных диенов-1,3, например
изопрена
СН3 Г снз 1
I I
СН2 = ССН = СН2 —> - L —СН2—С = СН-СН2—Jn- (8.3)
VII
В результате 1,4-полимеризации получается полимер VII,
содержащий повторяющуюся двойную связь ^)С=С^. Такая
двойная связь является центром стерической изомерии, так как
Стереохимия полимеризации и поликонденсации
483
становится возможным существование цис- и траяс-конфигураций.
В ifwc-конфигурации полимерные сегменты, связанные с атомами
углерода двойной связи, располагаются по одну сторону от двой-
ной связи (VIII), тогда как в траке-конфигурации они находятся
по разные стороны ее (IX)
— СН2 СН2 — — СН2 Н
Хс - Хс = с,/
СЩ \ СН^ ^СНг —
цис транс
VIII IX
Если все двойные связи в полимере имеют одинаковую конфигу-
рацию, то можно говорить о существовании двух различно упоря-
доченных (тактических) полимерных структур — геометрических
транс-1,4-полимер
цис-1,4-полимер
Рис. 8.5. цис- и транс-1,4-Полимеры 2-замещенного бутадиена-1,3
— (— СН2— СН = CR — СН2 —.
изомеров. На рис. 8.5 приведены структуры цис- и транс-ио.таме-
ров 2-замещенного бутадиена. Для обозначения их стереохимии
употребляются приставки цис-1,^- и транс-1,4-, например цис-1,4-
полиизопрен и тра«с-1,4-полиизопрен. При статистическом распо-
ложении mpawc-конфигураций получается атактический полимер,
чес Д°ВаНИе 11,ис~ и тжяс-конфигураций, сходное с синдиотакти-
встречаетаСП°Л0ЖеНИеМ В П0ЛимеРах алкенов, почти никогда не
484
Глава 8
8.1д. Полимеризация 4-моно- и 1,4-дизамещенных
бутадиенов-1,3
8.1д.1. 1,2- и 3,1-Полимеризация
Особый интерес представляет полимеризация 4-моно- и 1,4-ди-
замещенных бутадиенов-1,3 (структуры X и XI соответственно)
R R R'
I I I
СН = СН —СН = СН2 сн = сн—сн=сн
X XI
[11, 16]. 1,2-Полимеризация мономеров обоих типов [уравнения
(8.4) и (8.5)], а также 3,4-полимеризация 1,4-дизамещенных дие-
нов-1,3 [уравнение (8.6)] приводят к образованию полимеров
с двумя «асимметрическими» центрами.
R R Н
I । I
сн = сн—сн = сн2 — с-с —
I I
X н сн
II
сн2
(8.4)
R R'
I I
сн = сн—сн = сн
XI
R Н
I I
—с—с —
I I
н сн
II
CHR'
(8-5)
R' Н
I I
— С-С —
I I
н сн
II
CHR
(8.6)
8.16.2. 1,4-Полимеризация
1,4-Полимеризация монозамещенных диенов представляет осо-
бый интерес, поскольку она может приводить к образованию поли-
меров с геометрической и оптической изомерией [И, 16, 17].
При 1,4-полимеризации 4-замещенного бутадиена-1,3
R
СН2 = СН — CH = CHR—»—СН2 —СН = СН —С* — (8.7)
XII I
Стереохимия полимеризации и поликонденсации
485
возможно образование девяти дитактическихполимеров,составлен-
ных из разных геометрических и оптических изомеров. Двойные
связи могут иметь цис-, транс- или статистические конфигурации,
каждая из них в свою очередь может сочетаться с изотактической,
синдиотактической или атактической конфигурацией при асим-
метрическом атоме углерода (С*). Так, в качестве примера на
рис. 8.6 приведены все изотактические транс-структуры и все
сппднотактпческ е транс-структуры, образующиеся в результате
н н н
изо-транс-тактический
Н Н СН3 Н СН3Н Н Н СН3
синдио-транс-тактический
Рис. 8.6. Две стереорегулярные структуры 1,4-полипентадпена-1,3
— [— СН2 — СН = СН — СН(СН3) —.
1,4-полпмерпзацпи пептадиена-1,3. В названии полимеров с опти-
ческой и геометрической изомерией сначала отражается оптическая
изомерия; например, название изо-транс-тактический 1,4-поли-
пентадпен-1,3 или Д-[транс-СН2СН = СНСН(СН3)]п относится ко
всем изотактическим транс-структурам, а для всех синдиотакти-
ческих rn/тнс-структур 1,4-полипептадиепа-1,3 применяется
ооозначепие С1П1дио-7нранс-тактический 1,4-полипентадиен-1,3 или
s(-[mpaHc-CH2CH = CllCH(CH3)]n. 1,4-Полимеризация 1,4-дпзаме-
щенных бутадиенов-1,3 приводит к полимеру XIII
R R
I
СН=СН-СН=СН —
трео- цис-
эритро транс
К ~~ R'
I II I
-сн-сн=сн—с—с—сн=сн—СИ— >
Н Н (8.8)
X111
ся^чпр111 мо,1'ет быть тритактическим, так как в его повторяющем-
не имеется три центра стерической изомерии: один центр
486
Глава 8
геометрической и два центра оптической изомерии [17]. В этом
случае для обозначения двух асимметрических атомов углерода,
находящихся в положении 1,2 в полимерной цепи, по аналогии
с полимерами 1,2-дизамещенного этилена, полимер XIV, напри-
мер, называют эрг/тро-диизо-игранс-тактпческим 1,4-полиметил-
сорбатом пли eiZ-[z?zpawc-CH(CH3)CH = CHCH(CO2CH3)]n.
XIV
1,4-Полпмеризация 4-моно- и 1,4-дизамещенных бутадиенов также
представляет особый интерес, так как оптическая изомерия в струк-
турах XII и XIII обусловлена действительной асимметрией. При-
соединение двойной связи непосредственно к центру стерической
изомерии делает его чисто асимметрическим атомом углерода.
Поэтому в таких полимерах возможна оптическая активность.
8.1е. Циклополимеры
К проявлению стерической изомерии способны полимеры,
содержащие кольца, связанные с основной цепью [10, И, 14, 18].
У таких полимеров есть два центра истинной асимметрии — два
углеродных атома, по которым цикл присоединен к основной
цепи. На этом основании к образованию полициклогексепа из
(8.9)
циклогексена применимы те же самые положения, что и к полиме-
ризации 1,2-дизамещенного алкена. При полимеризации каждый
атом углерода, находящийся при двойной связи, становится асим-
метричным. На рис. 8.7 приведены четыре возможные упорядочен-
ные структуры. эритро-Структурами являются структуры для
которых характерна i^izc-конфигурация остатков полимерных це-
пей, присоединенных к циклу; для игрео-структур характерна
щракс-конфигурация таких остатков.
Стереохимия полимеризации и поликонденсации
487
эригпро-диизотактический
трео-диизотактический
эригпро-дисиндио тактический
трео-дисиндиотпактический
Р и с. 8.7. Стереорегулярные структуры полициклогексена. Жирными и тон-
кими черточками при положениях 1 и 2 показаны связи, расположенные
выше и ниже плоскости кольца соответственно.
8.2, Свойства стереорегулярных
полимеров
8.2а. Значение стереорегулярности полимеров
При практическом использовании многих полимеров их изоме-
рия играет большую роль. Свойства полимеров во многом изме-
няются при переходе от неупорядоченных к упорядоченным струк-
турам и зависят также от типа упорядоченности структуры (син-
диотактическая или изотактическая, цис или транс). Упорядочен-
ные полимеры наиболее сильно отличаются от неупорядоченных
по физическим свойствам. (Это представляет особый интерес в слу-
чае Потимеров. упорядоченность которых обусловлена оптической
изомерией. Па свойства полимеров оптическая изомерия оказыва-
ет значительно больший эффект, чем на свойства низкомолекуляр-
488
Глава 8
ных соединений. Низкомолекулярные оптические изомеры отли-
чаются друг от друга только направлением вращения плоскости
поляризованного света и биологической активностью.)
8.2а. 1. Изотактический, синдиотактический
и атактический полипропилен
Влияние регулярности в полимерах на их свойства обусловлено
разной кристаллизуемостью полимеров различных структур.
Атактические полимеры — аморфные (некристаллические) мягкие
материалы с очень низкой механической прочностью. Соответ-
ствующие же изо- и синдиотактические полимеры являются, как
правило, высококристаллическими веществами. Упорядоченные
структуры могут упаковываться в кристаллическую структуру,
а неупорядоченные — нет. Кристалличность обусловливает высо-
кую механическую прочность полимера, повышенную химическую
устойчивость, стойкость к действию растворителей и влияет па
другие свойства полимера. Первым примером практического ис-
пользования стереорегулярных полимеров является полипропи-
лен. Атактический полипропилен не имеет практического приме-
нения, тогда как изотактический полипропилен, характеризующий-
ся высокой температурой плавления, прочностью, кристаллич-
ностью, находит все более и более широкое применение в пластмас-
сах и волокнах [19].
В настоящее время практическое значение приобретают изотак-
тические поли-4-метилпентен-1 и полибутен-1. В отличие от изо-
тактических полимеров, изученных довольно подробно, синдиотак-
тические полимеры исследованы значительно в меньшей степени.
Это обусловлено сравнительной простотой получения изотакти-
ческих полимеров, тогда как известно значительно меньше мето-
дов и реакций образования синдиотактических полимеров. Опре-
деленным исключением из этого правила является синдиотакти-
ческий полипропилен, ряд свойств которого изучен [20]. Синдио-
тактический полипропилен, как и изотактический, легко кристал-
лизуется, но в отличие от последнего имеет несколько более низкую
плотность, более низкую (на 20 °C) температуру плавления, лучше
растворим в серном эфире и углеводородах.
8.2а.2. цис- и транс-1,4-По лид иены-1,3
Геометрическая изомерия 1,4-полидиенов-1,3 обусловливает
резкое различие в свойствах цис- и транс-изомеров. В табл. 8.1
приведены температуры плавления и стеклования стереорегуляр-
ных цис- и транс-1,4-полибутадиена п 1,4-полиизопрена [21].
Подобно тому как это имеет место в низкомолекулярных соедине-
ниях, п в частности бутепе-2, транс-полимеры имеют более высо-
Стереохимия полимеризации и поликонденсации
489
Таблица 8.1
Температуры плавления
и стеклования некоторых
1,4-полидиенов [21]
Полимер Изомер Т СТ’ °C т 1 пл’ СС
1,4-Полибута- цис -108 1
диен транс -18 141
1,4-Полиизо- цис — 73 14
прен транс -53 65
кие 7'ст и Тпл, чем соответствующие цис-полимеры. Это объясняет-
ся большей симметричностью и более плотной упаковкой тпранс-
пзомеров. Различия в физических характеристиках цис- и транс-
полпмеров приводят к существенным отличиям в их свойствах.
При температурах, лежащих выше температуры плавления, цис-
п транс-1,4-полиизопрены являются каучуками, при более низких
температурах — пластиками, поэтому цис-1,4-полиизопрен в обыч-
ных условиях применяется как эластомер в отличие от транс-1,i-
по.тинзопрена, который имеет значительно более высокую темпе-
ратуру плавления (табл. 8.1). Заслуживает внимания тот факт,
что в природе 1,4-полиизопрен встречается в цис- и /иранс-форме.
В каучуке дерева гевеи более 98% двойных связей находится
в цис-форме. Каучук, добываемый с других деревьев (главным
образом в Центральной Америке и Малайе), дает гуттаперчу, или
балату, которая представляет собой преимущественно транс-
изомер.
S.2a.3, Целлюлоза и амилоза
Стереоизомерия оказывает существенное влияние на свойства
изомерных полисахаридов — целлюлозы и крахмала (рис. 8.8).
Оба они являются полимерами глюкозы, звенья которой связаны
В них глюкозидными связями по углеродам 1 и 4. Их структура
аналогична структурам, изображенным на рис. 8.7, разница состо-
ит лишь в том, что шестичленные циклы глюкозы показаны в фор-
ме кресла (форма кресла отвечает действительной конформации
шее гпчленных колец, тогда как плоскостная структура, приведен-
ная ,,а рПС 3.7, на самом деле не СущестВует). Эти полисахариды
личаются только конфигурацией при углероде 1. Вследствие
этого в повторяющемся звене целлюлозы содержатся два остатка
‘ юзы. тогда как в звене крахмала — только один. Согласно
НОМОЙ К Л л тт-по «
Цетл! ’^пользующейся в стереорегулярных полимерах,
юлоза имеет пгрео-дисиндиотактическую структуру, а крах-
49(1
Глава 8
мал является э/шигро-диизотактическим полимером. По номенкла-
туре, применяющейся в химии углеводов, целлюлоза состоит
из D-глюкопираиозных цепей, связанных ^-глюкозидными связя-
ми в положениях 1,4, а крахмал — из D-глюкопиранозных цепей,
связанных а-глюкозидпыми связями в положениях 1,4. В целлю-
лозе связь 1,4-игранс-диэкваториальная, а в крахмале — цис-
целлюлоза
экваториально-аксиальная. Благодаря такой структуре целлюло-
за представляет собой более высококристаллический полимер,
чем крахмал. По сравнению с последним целлюлоза имеет более
высокую механическую прочность, низкую растворимость и повы-
шенную устойчивость к гидролизу. Это обусловливает использова-
ние целлюлозы в качестве конструкционного материала как при-
родой (в растениях), так и человеком. Хотя крахмал и не приме-
няется как конструкционный материал, он выполняет важную
функцию как составной элемент питания растений и животных.
Применение крахмала для этих целей основано па его легкой гпд-
ролпзуемости, обусловленной особенностями его структуры.
Стереохимия полимеризации и поликонденсации
491
8.26. Анализ стереорегулярности
8.26.1. Экспериментальные методы
Для оценки стереорегулярности или тактичности полимеров
применяются различные методы [22]. Часто критерием стереорегу-
ляриости полимера является его степень кристалличности. Тож-
дественность кристалличности и тактичности достаточно обоснова-
на, хотя количественно эти понятия не всегда равноценны.
В кристаллическую часть полимера могут входить не все стерео-
регулярные макромолекулы. Способность к кристаллизации неко-
торых коротких стереорегулярных цепей может быть весьма пони-
жена. В ряде случаев не поддаются определению таким методом
короткие стереорегулярные сегменты в массе атактического поли-
мера. Вместе с тем короткие атактические сегменты в тактической
цепи могут быть приняты за тактические участки. (Полимеры,
содержащие участки с разным типом тактичности, называются
стереоблок-полимерами, если такие сегменты имеют достаточную
длину.) Наиболее удачными аналитическими способами оценки
тактичности являются методы, позволяющие определить конфигу-
рацию. Из них наилучшие результаты, особенно при исследовании
продуктов полимеризации диенов-1,3, дает метод инфракрасной
спектроскопии. В различных продуктах полимеризации (1,2-,
3,4-, цис-1,4; транс-1,4) двойные связи по-разному замещены, что
приводит к различным полосам поглощения в ИК-спектрах. При
этом становится возможной прямая количественная оценка
различных стереоизомеров. С помощью ИК-спектроскопии мож-
но анализировать также тактичность полимеров, обусловлен-
ную оптической изомерией, однако часто таким методом вместо
тактичности определяется кристалличность. Для количественного
анализа оптической изомерии в полимерах наилучшие результаты
flQ19р])именение мет°Да ядерпо-магнитного резонанса (ЯМР)
8.26.2. Оценка тактичности
Для характеристики степени синдиотактичности или изотак-
тичности применяются, как правило, два способа [20]. Один из
них, предложенный Натта,— это диадная тактичность, опреде-
ляемая как доля соседних мономерных звеньев (диад) с изотакти-
ческим или сипдпотактпческпм взаимным расположением. Схема-
тическое изображение изотактических и синдиотактических диад
приведено ниже:
изотактические синдиотактические
xva XV6
492
Глава 8
причем горизонтальными линиями обозначены сегменты полимер-
ной цепи, а каждая вертикальная черточка отображает конфигура-
цию у центра стерической изомерии в мономерном звене. Доли
изотактических и синдиотактических диад обозначаются символа-
ми I и s соответственно (необходимо иметь в виду, что в литературе
для обозначения диадной тактичности применяются также симво-
лы I и S).
Другое определение тактичности, предложенное Бовеем
и Тирсом [24], основано на триадной тактичности. В соответствии
с ним изотактическую, синдиотактическую и гетеротактическую
триады изображают следующим образом:
изотактические
XVIa
синдиотактические
XVI6
гетеротактические
XVIe
которые обозначаются I, S и Н соответственно. При этом диадные
доли i и s не всегда тождественны триадным долям I и S, но между
ними может быть взаимная связь. По определению диадные
и триадные фракции в сумме должны давать единицу
г 4-s=l, (8.10)
/ + 5 + Я=1. (8.11)
Комбинируя уравнения (8.10) и (8.11), получим
г = / + 1/2Я, (8.12)
s = S + i/2H. (8.13)
Возможность определения диадной или триадной тактичности
методом ЯМР зависит от природы полимера и чувствительности
ЯМР-спектрографа. Приборы более высокого разрешения позво-
ляют анализировать тактичность не только диад и триад, но и более
длинных последовательностей [25]. Так, с помощью ЯМР высокого
разрешения была недавно оценена тетрадная тактичность поли-
метилметакрилата [26].
Ясно, что с помощью определения любых двух триадных фрак-
ций можно, воспользовавшись уравнениями (8.10) — (8.13), пол-
ностью охарактеризовать и триадные, и диадные структуры в поли-
мере. Определив же только одни диадные фракции, нельзя оценить
триадпую структуру.
Стереохимия полимеризации и поликонденсации 493
Атактическую структуру имеет полимер, в котором i = s =
_ 0 5 ПрИчем распределение диад в полимере имеет статисти-
ческий характер. При статистическом распределении выполняются
соотношения
Z=i2,
S = s2, (8.14)
Я = 2is,
причем у атактического полимера I = 0,25, S = 0,25, Я = 0,50.
Чисто изотактический полимер имеет i = I = 1 и совершенно
пест атлетическое распределение, т. е. все диады и триады распола-
гаются изотактически. Соответственно полностью синдиотакти-
ческий полимер характеризуется 8 = 5 = 1. При нестатистпче-
ком распределении с i s =£ 0,5 (/ 7^= 8 0,25) имеет место раз-
личная степень синдиотактичности или изотактичности. Изотак-
тичность преобладает, когда i > 0,5, а синдиотактичность — при
s > 0.5. Такие полимеры можно отнести к статистическим такти-
ческим сополимерам в том смысле, что их можно рассматривать как
статистпческие сополимеры изотактических и синдиотактических
диад или триад. Если распределение диад или триад уже нельзя
считать чисто статистическим, а оно ближе к упорядоченному,
полимер становится тактическим стереоблок-полимером, в кото-
ром различные области полимерных цепей содержат в достаточном
для кристаллизации количестве изотактические или синдиотакти-
ческие диады, связанные друг с другом. Такие полимерные цепи
содержат синдиотактические или изотактические блоки, связанные
с сегментами, атактичность которых может быть достаточна для
кристаллизации или нет.
8.3. Факторы, влияющие на образование
етереорегулярных полимеров
при полимеризации алкенов
Проанализировав типы возможных стереоизомеров в полиме-
рах, остановимся теперь на тех движущих силах, которые опре-
деляют возможность протекания стереоспецифической полимери-
зации. Вначале рассмотрим стереохимию полимеризации алкенов.
Степень стереоспецифичности в процессе полимеризации опреде-
ляется соотношением скоростей присоединения к полимерной
цепи мономеров с той же конфигурацией, что и у предыдущего
мономерного звена, и мономеров с обратной конфигурацией.
Движущие силы, определяющие относительные скорости ука-
занных двух способов присоединения, различны и зависят от
™го, является ли активный конец растущей полимерной цепи
о одным или координационно связанным (ассоциированным)
494
Глава 8
с катализатором. Для свободно растущих концов цепи, которые
не координированы, возможны оба способа присоединения. Сте-
реорегулярность конечного полимера определяется главным обра-
зом температурой реакции, от которой собственно и зависит соот-
ношение скоростей двух способов присоединения. Положение
в корне меняется, когда конец растущей полимерной цепи коорди-
нируется с катализатором. В этом случае возможен только один
(любой из двух) вариант присоединения, определяющийся конфи-
гурацией координационного комплекса, включающего, как прави-
ло, конец растущей полимерной цепи, инициатор и мономер.
В таких условиях вследствие координации присоединение мономе-
ра идет стереоспецифически.
8.3а. Радикальная полимеризация
При радикальной полимеризации, как правило, концы расту-
щей полимерной цепи свободны. Плоская или близкая к ней три-
гональная конфигурация атома углерода не является специфи-
ческой, так как есть свободное вращение вокруг концевой угле-
род-углеродной связи [в уравнении (8.15) это показано круглыми
стрелками]. Это означает, ч*го конфигурация мономерного звена
в полимерной цепи определяется не в момент присоединения его
к радикальному центру, но только после присоединения к расту-
щей цепи следующей молекулы мономера. Схематически этот
процесс выглядит следующим образом:
причем константы скорости образования синдиотактического [урав-
нение (8.15а)] и изотактического [уравнение (8.156)] полимеров
соответственно обозначены /сСиндио и /сИзо. Стереорегулярность
конечного полимера зависит от того, реализуется или нет этот
Стереохимия полимеризации и поликонденсации
495
порядок при всех последующих актах присоединения мономера.
Количество и тип стереорегулярности определяются отношением
/Ссиндио /кИ30. Когда это отношение равно нулю, получается изо-
тактический полимер; синдиотактический полимер образуется,
если это отношение равно бесконечности, и атактический, если
оно равно единице. Промежуточное значение /сСиндио /кИ30 (между 1
и бесконечностью) соответствует распределению полимерных моле-
кул, содержащих в разных пропорциях атактические и синдиотак-
тические сегменты.
8.3а.1. Энергетика образования синдиотактических
и изотактических структур
Величина /сСиндио /&изо зависит от разности F свободных энергий
активации синдиотактического /'синдиоИ изотактического РИ30 при-
соединений:
^’спндио ( —tiF^ 1 ) /о л а\
= ехр { ) = ехр { ------) , (8.16).
г«е AF* = А/^индио - \F *>,
AS* = AS*H„H0-AS*0, (8.17)
АЯ* = АЯсиндио АЯ1130,
причем Д/^ндио, АЯ^ндио, А Асин ди О представляют свободную энер-
гию активации, энтальпию и энтропию синдиотактического присое-
динения, а АТ^о, АЯ^0, AS^o — соответствующие характеристики
изотактического присоединения. Хаггинс [27] показал это еще
в 1944 г., предположив, что стереорегулярность должна быть
функцией температуры, причем образованию упорядоченных
структур способствует понижение температуры.
Фордхам [28] математически показал справедливость этих
зависимостей; для этого он, используя данные по низкомолекуляр-
ным гомологам полимеров, вычислил ожидаемые различия в акти-
вационных энтальпии и энтропии AS* синдиотактических
и изотактических полимеров. При этом различия в энергиях весьма
невелики и составляют от —1 до —2 ккал/моль (—4,18-103ч~
----8,36-103 Дж/моль) для АЯ* и от 0 до —1 энтр. ед. для AS*.
Следовательно, то, что синдиотактическая конформация более
выгодна по сравнению с изотактической, обусловлено главным
образом энтальпийным фактором. Небольшие различия в энергиях
синдиотактических и изотактических полимеров наблюдались так-
же при изучении температурной зависимости тактичности для ряда
мономеров. Так, АЯ* и AS* составляют —1,07 ккал/моль
( 4,48-103 Дж/моль) и —0,99 энтр. ед. для метилметакрилата [29]
496
Глава 8
и —0,31 ккал/моль (—1,30-103 Дж/моль) и —0,6 энтр. ед. для ви-
нилхлорида [30].
Несколько большая вероятность образования синдиотактиче-
ской конформации по сравнению с изотактической вызвана стери-
ческим и/или электростатическим отталкиванием заместителей
полимерной цепи. Отталкивание между заместителями R или,
более точно, между группами R концевых и предпоследних звеньев
растущей цепи минимально в переходном состоянии при стадии
роста (и в конечном полимере), когда они располагаются в чере-
дующейся последовательности синдиотактической конформации.
Температура полимеризации, °C
Р и с. 8.9. Зависимость синдиотактичности от температуры при радикаль-
ной полимеризации винилхлорида [32].
В изотактическом полимере такие стерические и электростати-
ческие отталкивания между группами R максимальны. Однако
имеются факты, подтверждающие, что не всегда предпочтитель-
ность синдиотактической конформации связана с меньшим оттал-
киванием между группами R концевых и предпоследних групп.
! * Расчетным путем было показано [31], что в случае полиметил-
метакрилата растущий радикальный конец не может, как выше
указывалось, свободно вращаться; энергетический барьер для
вращения вокруг углерод-углеродной связи превышает
20 ккал'моль (83,72-103 Дж/моль); энергия же активации роста
цепи составляет всего лишь 6,3 ккал/моль (26,37 -103 Дж/моль).
В этом случае предпочтительное образование синдиотактического
полимера, по-видимому, обусловлено отталкиванием между груп-
пой R па конце растущей полимерной цепи и такой же группой
в составе повой, внедряющейся молекулы мономера.
Вследствие различия в активационных энтальпиях синдиотак-
тической и изотактической конформаций с понижением температу-
ры полимеризации вероятность образования синдиотактических
структур возрастает. Как видно из рис. 8.9, содержание синдиотак-
тической фракции s в поливинилхлориде возрастает от 0,51 до 0,67
при понижении температуры от 120 до —78 °C [32]. (В цитируемой
Стереохимия полимеризации и поликонденсации
497
работе низкотемпературная полимеризация проводилась в присут-
ствии триалкилбора как инициатора, причем условия были анало-
гичны условиям, указанным в разд. 5.5в.) Соответствующие дан-
ные для полиметилметакрилата приведены в табл. 8.2. С уменьше-
нием температуры различие в энергиях синдиотактических и изо-
тактических конформаций оказывает прогрессивно возрастающее
влияние на стереоспецифичность полимеризации; при высоких
температурах эти эффекты отсутствуют.
Поскольку радикальную полимеризацию обычно проводят при
повышенных температурах, то большинство полимеров, получаю-
щи*хся этим методом, являются атактическими. Это, конечно,
не означает, что полимеры синдиотактической структуры совер-
шенно не образуются. Степень синдиотактического расположения
сильно зависит от природы полимера. Так, сопоставление данных
рис. 8.9 и табл. 8.2 показывает, что тенденция к образованию
синдиотактических полимеров у метилметакрилата выражена
в большей степени, чем у винилхлорида, что, несомненно, является
Таблица 8.2
Влияние температуры на тактичность
в радикальной полимеризации
метилметакрилата [29]
Температура, °C Доля синдиотакти- ческой фракции s
-40 0,86
60 0,76
100 0,73
150 0,67
250 0,64
следствием большей степени замещения в молекуле первого поли-
мера, приводящей к более сильному отталкиванию между замести-
телями соседних мономерных звеньев. При этом, если поливинил-
хлорид, который в промышленности получают при 60 °C, — прак-
тически атактический полимер (i = s = 0,5), при полимеризации
метилметакрилата при температурах до 100 °C образуются поли-
меры с заметной синдиотактичностью (73% при 100 °C, табл. 8.2).
Вместе с тем полученный в аналогичных условиях полиметилакри-
лат — практически атактический полимер, тогда как полиакрило-
нитрил обладает заметной синдиотактичностью (примерно 70%
при 100 °C) [33, 34].
498
Глава 8
8.3а.2. Распределение по размеру стереорегулярных
блоков
Полимер неоднороден не только по молекулярным весам, но
и по размеру стереорегулярных блоков в нем. Другими словами,
полимер с синдиотактичностью s характеризуется средней длиной
блоков, состоящих из мономерных звеньев с синдиотактической
конфигурацией, причем синдиотактические блоки имеют разные
размеры. От отношения констант скоростей роста кс,Лг1Я1ТО/
зависит как средний размер блока, так и распределение их по раз-
мерам [28]. Вероятность образования синдиотактического блока
из мономерных звеньев отвечает вероятности существования бло-
ка, в котором начальное синдиотактическое расположение повто-
ряется (п — 1) раз, прежде чем за ним последует участок с изо-
тактической структурой. Вероятность _Р?Индио равна
-Рсиндио = Р’‘~Ч (8.18)
где
*синдио , (8
«-синдиоФ^-изо
« = д +к --=1 -Р- (18-196)
«СИНДИОП^ «-изо
Параметры аир эквивалентны i и з соответственно. Последние
обозначения взяты из экспериментальных работ, тогда как первые
впервые появились в теоретических исследованиях. Средний раз-
мер синдиотактического блока пСиндио определяется как
оо
^синдио — 2 прп ха. (8.20а)
П=1
После суммирования получим
— сс 1
ПСИНДИО — (1_р)2 = "о" (8.206)
Доля Р?индио блоков с размером п в образце полимера равна
Рсиндио — ~ = лга р . (8.21)
^синдио
На рис. 8.10 приведено распределение’синдиотактических бло-
ков по размерам, рассчитанное с помощью уравнения (8.21) для
/Ссиндио/^изо = 1, 10 и 100. С увеличением отношения &СИНдИ0/&изо'
(т. е. доли синдиотактичности) синдиотактические блоки становят-
ся длиннее, причем длина некоторых из них равна длине всей
полимерной молекулы. Распределение по размерам*последователь-
ностей широкое, даже у атактического полимера. В атактическом
Стереохимия полимеризации и поликонденсации 499
полимере примерно половина макромолекулы состоит из синдио-
тактически расположенных трех и более мономерных звеньев
П лишь около четверти цепи состоит из синдиотактических после-
довательностей меньшей длины. С ростом синдиотактичности поли-
мера распределение по размерам блоков становится все более
п более широким. В полимерах с высокой степенью синдиотактич-
ности в значительном количестве имеются короткие синдиотакти-
ческие последовательности. При не очень высокой синдиотактич-
ности в полимере есть некоторая доля атактической фракции.
Длина синдиотактического блока п
Р и с. 8.10. Распределение по длинам синдиотактических блоков при^раз-
личных значениях отношения /сСиндисАизо [28].
Это хорошо объясняет, почему даже при достаточно высокой- сте-
пени синдиотактичности полимер, например полиметилметакрилат,,
не способен к интенсивной кристаллизации.
8.36. Ионная и координационная полимеризация
8.36.1. Эффект координации
В ионной полимеризации с участием свободных растущих
частиц факторы, определяющие стереохимию процесса, аналогич-
ны тем, которые наблюдаются в радикальной полимеризации.
Синдиотактическая конфигурация становится предпочтительной
при понижении температуры реакции. Если же между инициато-
ром, концом растущей полимерной цепи и мономером имеет место
сильная координация, то положение в корне меняется. Благодаря
-особенностям строения (конфигурации) координационного комплек-
са (включающего конец растущей цепи, инициатор и мономер)
500
Глава 8
внедрение новой мономерной молекулы в полимерную цепь воз-
можно только стереоспецифическим образом. Обычно при росте
цепи одно или другое расположение запрещается, так как коорди-
нация становится ведущей движущей силой стереоспецифичности
полимеризации. Отношение (^Сиядио/А:изо) стремится к нулю и
стереоспецифическая полимеризация приводит к образованию
изотактического полимера. В ряде случаев координация может
обусловливать возникновение синдиотактической структуры, но,
как правило, она направляет процесс в сторону образования изо-
тактического полимера. В табл. 8.3 приведен ряд примеров различ-
Таблица 8.3
Примеры стереоспецифической полимеризации
Мономер Условия полимеризации Структура полимера Лите- ратура
Бутен-1 TiCl3, (C2H5)2Zn в гептане при 50 °C Изотактическая 35
Изобутилвинило- вый эфир BF3-(C2H5)2O в пропане при -60 ч—80 °C » 36, 37
Метилакрилат CeH5MgBr или w-C4H9Li в СвН5СН3 при —20 °C » 33
Метилметакрилат zt-C4H9Li в СвН5СН3 при -78° С » 26
Метилметакрилат (C2H5)2AlN(CeH5)2 в СвН5СН3 при —78 °C Синдиотактиче- ская 26
Пропилен TiCl4, (С2Н5)2А1 в гептане при 50 °C Изотактическая 35
Пропилен VC14, А1(изо-С4Нд)2С1, анизол в СвН5СН3 при —78 °C Синдиотактиче- ская 38
ных реакций высокостереоспецифической полимеризации [26, 33,
35—38]. В большинстве случаев стереоспецифичность, оцененная
с помощью изотактических или синдиотактических диад, превыша-
ет 90%, а в некоторых случаях опа достигает 99%.
Вероятно, первым известным примером координационно-на-
правленной стереоспецифической полимеризации является катион-
ная полимеризация изобутилвинилового эфира, осуществленная
Шильдкнехтом в 1947 г. [36]. Он наблюдал образование высоко-
кристаллического полимера при проведении полимеризации в
интервале температур —60 4------80 °C в присутствии эфирата
трехфтористого бора в качестве катализатора. Всю важность
этого открытия в то время оценить не могли, так как кристаллич-
ность тогда связывали только с синдиотактической структурой.
Стереохимия полимеризации и поликонденсации 501
]] лишь в 1956 г. методом рентгеноструктурного анализа было
доказано, что полимер имеет изотактическую структуру [37].
В полной мере стереоспецифическая полимеризация была оце-
нена в 1954 г., когда Циглер в ФРГ и Натта в Италии открыли
новые катализаторы полимеризации, показавшие уникальную
стереорегулирующую способность [2—4]. Научное и практическое
значение этих работ было подчеркнуто присуждением Натта
п Циглеру Нобелевской премии по химии за 1963 г. Их речи при
вручении им премии являются превосходным введением в область
стереоспецифической полимеризации.
Работы Натта и Циглера стимулировали разработку большого
числа каталитических систем, состоящих из металлоорганических
соединений или гидридов металлов групп 1—3 и галогенидов или
других производных переходных металлов групп 4—7 периоди-
ческой системы.
Полимеризацию проводят в углевородных растворителях,
например гептане. В числе наиболее часто применяющихся метал-
лоорганических соединений элементов групп 1—3 следует назвать
триэтилалюмйний, диэтилалюминийхлорид и диэтилцинк; приме-
рами соединений переходных металлов являются треххлористый
титан, треххлористый ванадий, циклопентадиепилтитандихлорид,
триацетилацетонат хрома. Такие каталитические системы в лите-
ратуре часто встречаются под названиями: катализаторы Циглера,
Циглера — Натта, Натта или координационные катализаторы.
Последнее название наиболее удачно, так как оно включает, кроме
перечисленных выше, также катализаторы других типов, в присут-
ствии которых стереоспецифическая полимеризация идет по иден-
тичному механизму. К числу последних относятся н-бутиллитий,
фепилмагнийбромид, эфират трехфтористого бора.
8.36.2, Механизм стереоспецифического
присоединения
Катализаторы координационного типа выполняют двойную
функцию. Во-первых, они являются источником частиц, ини-
циирующих полимеризацию. Во-вторых, кроме частицы инициато-
ра, при разложении катализатора образуется фрагмент с исключи-
тельно сильной координационной способностью. При координации
указанного фрагмента катализатора (который можно рассматри-
вать как противоион по отношению к растущим частицам) с концом
растущей цепи и вновь присоединяющимся мономером мономер
ориентируется таким образом, чтобы имело место стереоспецифи-
ческое присоединение. Для объяснения образования изотакти-
ческих полимеров под действием катализаторов координационного
типа предложено много различных механизмов [5—11]. Наиболее
удовлетворительным из них можно считать рассмотрение стерео-
502
Глава 8
специфической полимеризации как согласованной многоцентровой
реакции. На рис. 8.11 приведена общая схема анионно-координа-
ционной полимеризации, в которой конец полимерной цепи можно
рассматривать как отрицательно заряженную частицу, а осколок
катализатора G имеет частичный положительный заряд (аналогич-
ный механизм можно представить и в катионно-координационной
Рис. 8.11. Механизм стереоспецифической полимеризации с изотактиче-
ским присоединением.
полимеризации, причем все отличие заключается в том, что указан-
ные частицы несут противоположные заряды). Осколок катализа-
тора G координируется с концом растущей полимерной цепи
и вновь присоединяющейся молекулой мономера.
Полагают, что на конце растущей полимерной цепи имеется
тетраэдрический атом углерода, причем роль четвертого замести-
теля выполняет осколок катализатора.
В ходе присоединения к полимерной цепи мономер ориенти-
руется и «удерживается в соответствующем положении» благодаря
координации. Одновременно с разрывом координационной связи
между катализатором и растущей полимерной цепью образуется
связь между концом цепи и новым мономерным звеном, а также
между этим звеном и осколком катализатора. Рост цепи протекает
через переходное циклическое четырехчленное состояние путем
внедрения мономера между катализатором и вновь входящими
мономерными звеньями. Таким образом, катализатор можно фор-
мально рассматривать как матрицу или шаблон, на котором
Стереохимия полимеризации и поликонденсации
503
существляется последовательно ориентация и изотактическое
присоединение новых мономерных звеньев. Движущей силой
образования изотактических структур служит взаимное отталки-
вание осколка катализатора G и заместителя R в мономере. Это
заставляет мономер приближаться к реакционному центру только
в одной из двух возможных ориентаций, что, таким образом, и
Приводит к образованию изотактической структуры.
Синдиотактическая структура при стереоспецифической поли-
меризации возникает лишь в очень ограниченном числе случаев,
когда возможна координация катализатора с мономером в обеих
его ориентациях (т. е. R и S). Тогда природа движущей силы для
образования синдиотактической структуры является такой же,
что и в описанной выше низкотемпературной полимеризации,
а именно взаимное отталкивание заместителей соседних мономер-
ных звеньев. Координация усиливает подобные взаимодействия
так же, как, вероятно, и понижение температуры.
Выбор того или иного катализатора для осуществления стерео-
специфической полимеризации, направленной на образование изо-
тактического полимера, во многом определяется природой моно-
мера. С увеличением координационной способности мономера
по отношению к катализатору возрастает вероятность соответ-
ствующего стерпческого расположения мономера при присоедине-
нии его к полимерной цепи. Способность мономера вступать
в координацию зависит прежде всего от его полярности. Она
наиболее велика для мономеров с полярными функциональными
группами, которые могут активно участвовать в координаций
(например, акрилатов, метакрилатов и простых виниловых эфи-
ров). Этилен, а-олефипы (например, пропилен и бутен-1) и другие
алкены, не содержащие полярных заместителей, обладают низкой
координационной способностью. Поэтому для получения изотак-
тических полимеров на основе неполярных мономеров необходимы
катализаторы с очень сильной стереорегулирующей способностью.
Для этого требуются гетерогенные катализаторы Циглера —
патта, которые представляют наиболее серьезное препятствие
на пути~ синдиотактического присоединения мономера к концу
mod^^R1 П0ЛимеРн°й цепи при полимеризации неполярных моно-
как п присутствии гомогенных (растворимых) катализаторов,
торыхРслИЛ°' °бРазУются атактические полимеры и лишь в неко-
пии потгЛУЧаЯХ Си|,лио1якгические полимеры. При полимериза-
ппелпосктРНЫХ м°НОмеРОв гетерогенность не является необходимой
мне каталК°И °°Разовапия изотактических полимеров. Раствори-
ческих полИЗаТ°РЬ1 можно использовать для получения изотакти-
зуются в ппИиХРте™„Т0ГДа КЙК С1гадиотактические полимеры обра-
жуточное поло' ВИИ только Растворимых катализаторов. Проме-
ми занимав ®eHHe межДУ полярными и неполярными мономера-
ирол и диены-1,3, так как фенильные и алкениль-
504
Глава 8
ные группы не обладают большой полярностью. Изотактические
полимеры на основе таких мономеров образуются при полимериза-
ции в присутствии как гомогенных, так и гетерогенных катали-
заторов.
8.4. Полимеризация неполярных алкенов
по Циглеру—Натта
Катализаторы Циглера — Натта образуют важную и значи-
тельную группу катализаторов. Они являются единственными^
катализаторами полимеризации ос-олефинов, в частности пропиле-
на и бутена-1. Следует лишний раз подчеркнуть, что, как уже гово-
рилось в гл. 3 и 5, а-олефины не полимеризуются под действием
радикальных и ионных катализаторов. Широкое признание ката-
лизаторы Циглера — Натта приобрели в связи с тем, что с их
использованием связано получение изотактического полипропиле-
на и линейного полиэтилена высокой плотности. Хотя, очевидно,
в применении к симметричному этилену понятие оптической изо-
мерии неприменимо, в присутствии стереоспецифических катали-
заторов образуется полиэтилен, по свойствам отличающийся от
полиэтилена, полученного радикальной полимеризацией. Поли-
этилен, синтезированный в присутствии таких новых каталити-
ческих систем, значительно менее разветвлен, что обусловлено как
более низкими температурами полимеризации, так и уменьшением
роли реакций передачи цепи. Более линейное строение такого
полимера делает его лучше, обусловливает более высокую кристал-
лизуемость и большую плотность полимера. По прочности и ряду
других показателей высокоплотный полиэтилен превосходит поли-
этилен низкой плотности, получающийся радикальной полимери-
зацией.
Со времени открытия Натта и Циглером стереоспецифических
катализаторов разработано и изучено большое число катализато-
ров такого типа. Описаны сотни и даже тысячи различных катали-
тических систем, состоящих из металлоорганических соединений
металлов групп 1—3 и соединений переходных металлов групп 4—
8. Примеры их приведены в табл. 8.4. В зависимости от активно-
сти и стереоспецифичности катализатор проявляет себя неодина-
ково с разными мономерами. В этой главе термин активность
применяется исключительно для характеристики скорости полиме-
ризации; в этом смысле этот термин используется далеко не всегда.
Порядок расположения различных катализаторов Циглера —
Натта в зависимости от влияния их на скорость полимеризации
и на стереоспецифичность часто не одинаковый. Многие катализа-
торы с высокой активностью (высокой скоростью полимеризации)
показывают низкую стереоспецифичность. Свойства катализатора
Стереохимия полимеризации и поликонденсации
505
Таблица 8.4
Компоненты катализаторов Циглера — Натта
Соединения металлов первой — третьей групп Соединения переходных металлов
(С2Н5)3А1 TiCl4
(С2Н5)2А1С1 TiCl3
(С2Н5)2А1Вг TiBr3
(С2Н6)А1С12 VC14
(изо-С4Нд)3А1 VC13
(С2Н5)2Ве (C5H5)2TiCl2
(C2H5)2Mg (CH3COCHCOCH3)3V
G4H9Li Т1(ОС4Нд)4
(C2H5)2Zn Ti(OH)4
(C2H5)4Pb VOC13
[(C6H5)2N]3A1 MoCl5 '
C6H5MgBr MoCl4
(C2H5)4AlLi CrCl3
ZrCl4
CuCl
WCle
MnCl2
NiO
Циглера — Натта настолько чувствительны к изменению природы
составляющих его (одного или другого) компонентов, что этим
можно часто воспользоваться для того, чтобы «сделать на заказ»
катализатор, который был бы наиболее подходящим для данного,
конкретного мономера. Соответствующим подбором компонентов
катализатора можно добиться очень высокой стереоспецифичности
и одновременно достаточно высокой скорости полимеризации.
К сожалению, подбор компонентов для каталитических систем
часто носит чисто эмпирический характер из-за недостатка подроб-
ных данных о механизме стереорегулирования с помощью таких
катализаторов.
8.4а. Механизм полимеризации Циглера — Натта
Для объяснения стереоспецифичности катализаторов Цигле-
Ра — Натта предложено много различных механизмов, число
которых уступает лишь количеству исследованных каталитических
систем. Различие между ними часто сводится к отдельным, хо/я
и важным деталям, экспериментальная проверка которых невоз-
можна. Кроме того, в большинстве своем тот или иной механизм,
-506
Глава 8
разработанный для данной каталитической системы, нельзя пере-
носить на другой тип катализатора. При рассмотрении особенно-
стей механизма полимеризации Циглера — Натта основное внима-
ние будет обращено на анализ таких характеристик, которые
одинаково справедливы для большинства каталитических систем.
Наибольший интерес вызывают катализаторы титан-алюминиевого
типа и среди них система (C2H5)3Al-TiGl3, которая наиболее деталь-
но изучена.
Подробные сведения о ряде других каталитических систем
можно почерпнуть из прекрасных обзоров [9, И, 20, 40—42].
При рассмотрении механизма полимеризации Циглера — Нат-
та возникает ряд общих вопросов. Какова природа растущих
частиц? Идет рост по связи углерод — переходный металл или
по связи углерод — металл групп 1—3? Какова химическая
и физическая природа истинного или активного катализатора,
инициирующего полимеризацию, т. е. что скрывается за G на
рис. 8.11?
8.4а.1. Химическая природа растущих частиц
Относительно природы растущих частиц в литературе существу-
ет изрядная путаница. Становится все более очевидным, что тип
растущего центра при полимеризации с использованием катализа-
торов Циглера — Натта зависит от состава катализатора и моно-
мера. Многие полимеризационные процессы, которые относят
к полимеризации Циглера — Натта, в действительности обычные
свободнорадикальные, катионные или анионные реакции полиме-
лизаци I, идущие, как правило, не стереоспецифично. Катализато-
ры Циглера — Натта в ряде случаев составлены из компонентов,
которые способны инициировать соответствующую ионную поли-
меризацию и часто применяются (см. гл. 5) для этой цели. Так,
например, под действием бутиллития, являющегося компонентом
металла первой — третьей групп катализатора Циглера — Натта,
идет анионная полимеризация мономеров с электронодефицитнымй
двойными связями, например акрилатов и метакрилатов. Напро-
тив, мономеры с электронообогащенными двойными связями,
например простые виниловые эфиры, могут полимеризоваться по
катионному механизму в присутствии катализаторов, являющихся
сильными электроноакцепторными соединениями, например Т1С14,
VCl4 и RA1C12. Некоторые компоненты катализаторов Циглера —
Натта могут реагировать с образованием свободных радикалов;
например, при взаимодействии TiCl4 и R3A1 идут следующие реак-
ции:
TiCl4 + R3Al —> RTiCl3 + R2AlCl, (8.22)
TiCl4 + R2A1C1 —> RTiCl3 + RA1C12, (8.23)
Стереохимия полимеризации и поликонденсации 507
TiCl4-j-RAlCl2 RTiCl3 + А1С13, (8.24)
RTiCl3 —> TiCl3+R’, (8.25)
R-+TiCl4 —> TiCl3 + RCl, (8.26)
2R- —> Рекомбинация и диспропорционирование (8.27)
В определенных условиях радикалы, образующиеся в результате
реакции (8.25) или других реакций, могут выступать в роли ини-
циаторов радикальной полимеризации некоторых мономеров. Тен-
денция к протеканию радикальной полимеризации возрастает при
применении каталитических систем с участием кислорода [9],
например R3B — О2, которая была уже рассмотрена в разд. 5.5в.
Говоря о полимеризации по Циглеру — Натта, почти всегда
подразумевают стереоспецифическую полимеризацию. Стереоспе-
цифичность имеет место лишь тогда, когда реакция протекает по
координационному механизму. Для объяснения стереоспецифи-
ческой полимеризации в присутствии катализаторов Циглера —
Натта был предложен ряд механизмов, и в том числе анионно-
координационный, катионно-координационный и радикально-коор-
динационный. Экспериментальные данные четко опровергают
радикально-координационный механизм. Можно привести ряд
аргументов, подтверждающих ионный механизм [9]:
1. Радикальные агенты передачи цепи не влияют на молекуляр-
ный вес полимера.
2. Время жизни растущих частиц значительно болыпэ, чем
при радикальной полимеризации, и близко тому, какое характерно
для полимеризации с образованием живущих полимеров.
3. Можно получать блок-сополимеры путем регулярно чере-
дующегося введения в реакцию двух различных мономеров.
4. При варьировании природы различных компонентов ката-
литических систем меняется отношение реакционной способ-
ности мономеров в сополимеризации.
Наиболее общепринятым механизмом стереоспецифической
полимеризации Циглера — Натта в случае а-олефинов (и других
неполярных алкенов) является анионно-координационный про-
цесс, близкий изображенному на рис. 8.11. В более простой форме
его можно представить следующим образом:
5+ 5-
R—CH.77777.CH,
\ \ « •
2\ V
~^снк-сн-;щц-с
3- 5+
XX
проц Мери3аг/о!1 ПРИСУЩИ черты как анионного, так и катионного
цессов 143]. Реакция заключается в нуклеофильной атаке
508
Глава 8
зарождающимся карбанионом на конце полимерной цепи двойной
связи мономера (реакция 2 в XX) и электрофильной атаке катио-
ном катализатора л-электронов алкена (реакция 1 в XX). Считают,
что обе- стадии идут согласованно, т. е. практически одновре-
менно.
На анионный характер полимеризации указывает то, что
скорость полимеризации сс-олефинов уменьшается в следующем
ряду: этилен > пропилен > бутен-1 [44]. При полимеризации,
идущей через превращение мономера в соответствующий ион
карбония, следовало бы ожидать обратного порядка. При при-
соединении карбаниона к мономеру атака идет по сс-углероду
с образованием менее замещенного (и более устойчивого) карб-
аниона. Влияние «-заместителей заключается в том, что из-за
стерического фактора затрудняется приближение карбаниона
и, как результат, с увеличением размера заместителя реакционная
способность мономера в полимеризации уменьшается. Дополни-
тельные доказательства анионной природы растущих полимерных
цепей получены в результате исследований с применением для
обрыва цепи меченого метанола. Получающийся в результате
реакции обрыва полимер радиоактивен при применении СН3О—3Н,
тогда как при обрыве метанолом, содержащим 14С, образуется
нерадиоактивный полимер. На примере полимеризации мономеров
с одинаковым стерическим фактором можно проследить вклад
катионов в анионно-координационный механизм. В ряду мета-
и пара-замещенных стиролов реакционная способность их в поли-
меризации возрастает с увеличением электронодонорной способ-
ности заместителя [45]. Таким образом, анионно-координацион-
ный механизм предполагает внедрение молекулы мономера по
карбанионному центру, причем лимитирующей стадией реакции
является электрофильная атака осколком катализатора (реак-
ция 1 в XX).
8.-La.2. Рост через связь углерод—переходный
металл
В качестве центров, на которых идет рост цепи, указывают
связи переходный металл — углерод и углерод — металл
групп 1—3. Вместе с тем имеющиеся в литературе данные, кото-
рые получены при исследовании зависимости изотактичности или
скорости полимеризации от состава катализатора Циглера —
Натта, не вполне однозначны. Анализ данных свидетельствует
скорее о росте через связь углерод — переходный металл. Допол-
нительные доказательства получены при исследовании полимери-
зации в присутствии каталитических систем, не содержащих
компонента с металлом групп 1—3. Было показано, что ряд ката-
лизаторов, содержащих только переходный металл, является
Стереохимия полимеризации и поликонденсации
509
эффективными стереоспецифическими катализаторами [40]. Это
относится, в частности, к полимеризации этилена и пропилена
[46—48] под действием:
1) TiCl2, измельченного в шаровой мельнице;
2) переходных металлов, например Ti, V, Nb, измельченных
в шаровой мельнице;
3) измельченных в шаровой мельнице смесей переходных
металлов с галогенидом (Ti + C2HsBr), с галогеном (Ti + 12)
пли водородом (Ti + Н2);
4) смесей самих переходных металлов и их соединений, напри-
мер Ti -j- TiGl3 и TiGlз 4- GH3TiGl3.
Полимеризация с катализаторами, не содержащими металлов
групп 1—3, протекает так же, как и в присутствии каталитических
систем, включающих соединения металлов этих групп, разница
заключается лишь в меньшей активности первых. Оба процесса
не отличаются и по кинетике. Изотактические полимеры обра-
зуются как в присутствии катализаторов, являющихся соедине-
ниями переходных металлов, так и под действием каталитических
систем, содержащих соединения металлов обоих типов.
8.4а.З. Биметаллический и монометаллический
механизм
Для активных частиц катализатора Циглера — Натта пред-
ложен ряд структур. Разнообразие выдвинутых структур связано
с многочисленностью экспериментально наблюдаемых [например,
при реакциях (8.22)—(8.26)] соединений, а также тех соединений,
которые, возможно, образуются при взаимодействии между собой
компонентов катализатора Циглера — Натта. По числу металли-
ческих центров предложенные структуры можно разделить на две
большие группы: биметаллические и монометаллические соеди-
нения. При взаимодействии хлорида титана и алкилалюминия,
например TiGl4 и (C2HS)3A1, это могут быть соединения XXI и XXII
Соединение XXII образуется при алкилировании хлорида
титана металлоорганическим соединением. Символом □ в XXII
ооозначена незанятая, или вакантная, орбиталь октаэдрического
510
Глава 8
титана. Соединение XXI представляет собой координационный
комплекс соединений алюминия и титана, образующийся при
взаимодействии исходных компонентов катализатора с обменом
групп R и G1. Заключая группы R и С1 в скобки в XXI, показы-
вают, что точное размещение лигандов А1 и Ti не известно. Число
СНз
'сн2АЛ?
Рис. 8.12. Биметаллический механизм стереоспецифической полимериза-
ции [49].
и тип лигандов, связанных с каждым металлическим центром,
зависит от природы компонентов катализатора и их взаимного
соотношения.
Для иллюстрации двух возможных механизмов роста можно
воспользоваться обоими типами активных частиц. Биметалли-
ческий механизм, предполагающий рост цепи по обоим металли-
ческим центрам, описан рядом исследователей [11, 40—42, 49—53].
На рис. 8.12 представлена схема такого механизма. При этом
отличие его от механизма, изображенного на рис. 8.11, заклю-
чается в том, что на рис. 8.12 расшифрована структура осколка
катализатора G. Мономер координируется с катализатором,
а затем внедряется по поляризованной титап-углероднсй связи.
Монометаллический механизм, предполагающий рост по одному
активному металлическому центру, показан на рис. 8. 13 [54].
Б этом случае мономер координируется по вакантной орбитали,
октаэдрического комплекса переходного металла, а затем внед-
ряется в полимерную цепь по связи переходный металл — углерод.
Стереохимия полимеризации и поликонденсации
511
Ппп этом регенерируется вакантная орбиталь с ориентацией,
отличающейся от исходной. Если бы рост цепи шел через такие
соединения, то это привело бы к образованию синдиотактиче-
ского полимера [20]. Для^протекания изотактической полимери-
сн2
| /С1 сн2=снсн3
С1—Ti—□ ---------*-
С1
сн2
|/С1 снсн3
С1С1
/ I сн2
С1 С1
□
: С1
Cl—Ti—СН'
/|
С1 С1
СНг
.снсн3
СНСН3
I /С1 1
I сн
С1—Ti" г
С1С1
сн,
I
снсн3
сн2
Cl—Ti—-□
/|
С1С1
Рис. 8.13, Монометаллический механизм
зации [54].
стереоспецифической полимери-
зации необходимо, чтобы полимерная цепь мигрировала в свое
исходное положение с регенерацией исходной вакантной орбитали.
В настоящее время невозможно экспериментально доказать тот
или иной механизм. За£исключением стадии миграции, мономе-
таллический механизм выглядит проще, чем биметаллический.
Предложен монометаллический механизм, не требующий стадии
512 ' Глава 8
миграции [40]. Он заключается в непосредственном внедрении,*
мономера по связи переходный металл — углерод без координа-^
ции, но с сохранением исходной геометрии катализатора. Отсут-|
ствие координации мономера с катализатором в таком механизме ’
однако, не выглядит очень убедительным, так как общепринято;
считать, что стереоспецифичность немыслима без координации.^
Был предложен ряд других механизмов, являющихся в той пли]
иной степени вариантами биметаллического и монометалличе-=
ского механизмов [И, 40—42]. В некоторых случаях с трудом]
можно установить различие между обоими механизмами; напри-
мер, для некоторых биметаллических катализаторов предложен
монометаллический механизм, который предполагает рост цепи-
исключительно по связи переходный металл — углерод [55].
Кроме того, невозможно, чтобы оба механизма, биметаллический
и монометаллический, работали при полимеризации Циглера —
Натта. Может оказаться, что при применении одних каталитиче-
ских систем рост цепи идет по биметаллическому механизму, тогда
как с другими имеет место монометаллический механизм.
Прп ознакомлении с механизмами, описанными в литературе,
следует иметь в виду, что большинство из них сильно детализиро-
вано, причем в основу такой детализации, часто положены лишь
предположения, а также различия, которые являются семан-
тическими или которые по прошествии ряда лет стали вызы-
вать большие сомнения. В качестве примера сравним биметалли-
ческий механизм Натта [49] с механизмом Патата и Зинна [50]
в том виде, в котором они интерпретированы в работах [11, рис. 34
и 35] и [40, рис. 15 и 16]. По данным работ [11] и [40] механизм
Натта поразительно похож на механизм Патата — Зинна, хотя,
если Проанализировать оригинальные работы, то между нпми
есть существенные различия.
8.4а.4. Физическая природа катализатора
Известны катализаторы Циглера — Натта гетерогенного
и гомогенного типов. Как указывалось выше, только в присут-
ствии гетерогенных катализаторов Циглера — Натта получаются
изотактические полимеры на основе сс-олефинов, хотя известно
несколько примеров образования и синдиотактических структур.
8.4а.4а. Взаимодействие лиганда с мономером при изотактиче-
ской полимеризации. Считают, что рост цепи на гетерогенных ката-
лизаторах протекает на активных участках кристаллической
поверхности нерастворимого соединения переходного металла
(например, TiCl3).
Такие активные центры возникают при реакции растворимого
соединения металла групп 1—3 [например, А1(С2Н6)3 или
А1(С2Н5)2С1] с поверхностью TiCl3. По химическому строению
Стереохимия полимеризации и поликонденсации
513
иные участки идентичны биметаллическим или монометалли-
аескцм соединениям, рассмотренным в разд. 8.4а.З. Ориентация
молекулы мономера при присоединении ее к растущей цепи зави-
сит не только от химического строения катализатора, но и от типа
кристаллической структуры последнего. Движущая сила для
изотактического роста определяется суммой стерических и элек-
тростатических взаимодействий между заместителем вновь входя-
щего мономера и лигандами переходного металла на активных
участках кристаллической поверхности. Мономер может прибли-
зиться к связи переходный металл — углерод только в одной-
едпнственной конфигурации. Вследствие вращения связи пере-
ходный металл — углерод взаимодействия между входящим моно-
мером и мономерным звеном на конце полимерной цепи сводятся
к минимуму, в результате чего заместитель при концевом моно-
мерном звене отодвигается от вакантной орбитали металла.
Изотактическое присоединение такого типа было предложено [40]
называть моделью взаимодействия лиганд — мономер.
Известей ряд других моделей изотактического присоединения,
но, очевидно, пи одна из них не имеет общего применения. Так
полагают [56], что при спиральном росте цепи на кристалличе-
ской поверхности образуется изотактический полимер, что
обусловлено взаимодействием мономера с такой спиралью. Однако
при полимеризации пропилена при таких высоких температурах,
при которых полипропилен не существует в спиральной форме,
может тем не менее получаться высокоизотактический полимер
[57]. Таким образом, спираль не является обязательным усло-
вием для изотактического присоединения. Многие исследователи
подчеркивали роль соединения металла групп 1—3 в каталитиче-
ской системе. Натта [44] предположил, что биметаллические
катализаторы асимметричны и координируются с мономерами
только в одной конфигурации. Родригес и Ван Лой [55] связывают
изотактическое присоединение с взаимодействиями между моно-
мером и частью биметаллического катализатора, относящейся
к .металлу групп 1—3. Основываясь на том, что присутствие соеди-
нения металла групп 1—3 не обязательно в изотактической поли-
меризации (разд. 8.4а.2), Бур [40] отрицает такие модели. Однако
эта модель не противоречит модели взаимодействия лиганд —
мономер, если в полимеризации Циглера — Натта справедлив
как моно-, так и биметаллический механизм.
8.4а.4б. Кристаллическая структура катализатора. Изотак-
тическая полимеризация очень чувствительна к типу кристалли-
ческой структуры поверхности катализатора. Для того чтобы
кристалл был электронейтрален, в любой координационной
решетке в отличие от кристаллической решетки молекулы должен
ряд свободных лигандных мест. Кристаллы a-TiCl3 построены
элементарных кристаллических пластин (XXIII). каждая
514
Глава 8
из которых содержит слой титана, заключенный между двумя
XXIII
слоями хлора [58]. Атомы титана и хлора в XXIII изображаются
в виде маленьких черных и больших белых шаров соответственно.
Два слоя атомов хлора чередуются со слоем титана вдоль главной
кристаллической оси. Атомы титана расположены в промел.уктах
между атомами хлора, образуя октаэдрическую решетку, тогда
Рис. 8.14. Кристаллическая структура TiRCl43 для изотактического при-
соединения [60].
как сами атомы хлора плотно упакованы в гексагональной решетке.
Каждый третий атом титана в решетке отсутствует. Такие тита-
новые участки, каждый из которых содержит вакантное место,
и являются активными центрами (TiRCl4O) при полимеризации
Циглера — Натта. Они образуются на титановых участках
в результате алкилирования, идущего с заменой атома хлора
на группу R. Обычно считают [42, 55, 59], что активные центры
(вакантные места титана) расположены на краях элементарных
пластин, а не в главной плоскости кристалла. Это подтверждается
микроскопическими исследованиями роста полимера на ребрах
кристалла [55].
На рис. 8.14 показано, как меняется кристаллическая струк-
тура в процессе изотактической полимеризации (в рамках моде-
ли взаимодействия лиганд — мономер) [60]. Это соответствует
Стереохимия полимеризации и поликонденсации
515
еденной на рис. 8.13 схеме процесса с алкилированными
пРр’ВИнениями титана TiRGl4D (ХХП) в качестве активных участ-
С°в на поверхности a-TiCl3. Одна растущая полимерная цепь,
Кзображенная в виде большого черного шара, приведена
на рис. 8.14 (маленькими черными шарами обозначены атомы
титана, а большими белыми — хлорные лиганды). Структуры,
представленные на рис. 8.14,а и 8.14,6, отличаются геометрией
активного титанового участка. При этом все различие состоит
в том, что растущая полимерная цепь и орбитальное вакантное
место'в них поменялись местами. Это соответствует различным
геометрическим структурам, показанным на рис. 8.13 для актив-
ного центра до и после присоединения молекулы мономера
к растущей цепи. Оба положения неэквивалентны, причем для
полимерной цепи предпочтительна ориентация, приведенная
на рис. 8.14,а, в результате которой она дальше отстоит от поляр-
ной поверхности TiGl3, чем при другой ориентации. Суммарная
степень изотактической полимеризации зависит от отношения
скорости миграции полимерной цепи к скорости внедрения моно-
мера в цепь. Изотактический процесс идет, если миграция проте-
кает быстрее, чем внедрение. Для каждого конкретного случая это
отношение определяется природой мономера и каталитической
системы, а также температурой полимеризации. Так, полимериза-
ция пропилена при 50—100 °C в присутствии (C2H6)3Al-cc-TiCl3
идет как изотактический процесс. С понижением температуры
можно ожидать увеличения роли синдиотактического присоеди-
нения, так как при этом создаются более благоприятные условия
для внедрения мономера, что обусловлено более сильной коорди-
нацией мономера и катализатора. Тенденция к образованию син-
диотактического полимера наблюдалась при полимеризации про-
пилена только при очень низких температурах 70 °C) [42].
При повышенных температурах (выше 50—100 °C), очевидно,
процесс должен быть сдвинут в сторону нестереоспецифической
(атактической) полимеризации из-за ослабления координации
между мономером и катализатором.
Сильная зависимость изотактического присоединения от кри-
сталлической структуры катализатора становится очевидной при
рассмотрении различных кристаллических форм одного и того же
катализатора. Известно, что TiCl3 существует в четырех различ-
ных формах: обычно использующейся a-форме, которая была
описана выше, 0-, у- и 6-формах. (0-TiCl3 коричневого цвета,
огда как остальные формы окрашены в фиолетовый цвет.) Три-
п Риды титана в а-, у- и 6-формах практически не отличаются
соепП°ВеДе™Ю’ В ПРИСУТСТВИИ катализаторов на основе таких
обуслНеПИИ П0ЛУчаютСя изотактические полимеры [61]. Это
форм g?-QHOrR ^лизостью кристаллических структур указанных
w р Ibl, 62]. T-TiCl 3 имеет слоистую структуру, аналогия-
516
Глава 8
ную структуре a-TiCl3, но в первой атомы хлора плотно упакованы
в кубической решетке, а во второй — в гексагональной. Для
6-TiCl3 характерна смешанная гексагональная и кубическая
плотноупакованная слоистая структура.
Отличия в поведении P-TiCl3 обусловлены особенностями
кристаллической структуры этой формы, представляющей собой
узлы линейных цепей TiCl3. Схематически его структура пред-
ставлена в виде XXIV (вид сверху) и XXV (вид спереди) [58].
При такой структуре создается поверхность, в которой одна поло-
вина атомов титана имеет два вакантных места, а другая — только
одно вакантное место. Титановые участки с двумя вакантными
местами ответственны за нестереоспецифические свойства p-TiCl3.
Они образуют центр со свободной конфигурацией, так как напра-
вление одного из атомов хлора и/или растущей полимерной цепи
не будет строго фиксировано. Эта ситуация прямо противоположна
той, которая наблюдается для других трихлоридов титана (одно
вакантное место на активное положение), в которых все атомы
хлора и полимерная цепь строго фиксированы в жесткой конфи-
гурации у титанового центра. Образованию в небольшом количе-
стве изотактического полимера в присутствии [3-TiCl3, очевидно,
способствуют титановые центры только с одним вакантным местом.
8.4а.4в. Взаимодействие мономер — мономер в синдиотакти-
ческой полимеризации. Высокосиндиотактический полимер в при-
сутствии катализаторов Циглера — Натта удалось получить
только при полимеризации пропилена [20, 40]. Синдиотактиче-
ские фракции обнаружены также при полимеризации бутадиена
в определенных условиях. Для получения синдиотактического
полипропилена были использованы гетерогенные и гомогенные
катализаторы, но только в присутствии растворимых катализато-
ров удалось синтезировать высокосиндиотактический полипропи-
лен, не содержащий изотактической структуры. Степень синдиотак-
Стереохимия полимеризации и поликонденсации
517
тпчностп в полимеризации на гетерогенных катализаторах оказа-
чась низкой. В растворимых катализаторах Циглера — Натта,
необходимых для образования синдиотактического полипропи-
лена. в качестве компонента переходного металла могут исполь-
зоваться практически только соединения ванадия. (Ряд раствори-
мых катализаторов, основанных на соединениях титана,
например дициклопент адиенилтитандихлорид, применялся
в полимеризации этилена и других мономеров, не дающих син-
диотактических полимеров.)
Катализатор, образующийся на основе VC14 и (С2Н5)2А1С1
в присутствии третьего компонента (анизола),— один из наиболее
эффективных катализаторов для получения синдиотактического
полипропилена [63]. Вместо VC14 могут применяться также ацетил-
ацетопат ванадия и различные ванадаты [VO(OR)XC13_X, где
х — 1—3], но онп характеризуются меньшей стереоспецифич-
ностью. Присоединение анизола или других электронодонорных
веществ, например фурана и диэтилового эфира, к каталитиче-
ским системам, повышает их синдиотактическую стереоспецифич-
ность, по механизм этого явления остается неясным. Синдиотак-
тичность увеличивается с понижением температуры, поэтому для
получения полимеров с высокой степенью синдиотактичности
полимеризацию проводят обычно при температурах ниже —40 °C
и наиболее часто при —78 °C. Как правило, такие катализаторы
также нужно готовить при низких температурах, поскольку при
получении пх или при последующем нагревании выше темпера-
туры ~—40 °C многие из них становятся гетерогенными и не дают
больше синдиотактического полимера.
Движущая сила для синдиотактического присоединения
на катализаторах Циглера — Натта имеет ту же природу (см.
разд. 8.36.2), что и при низкотемпературной радикальной и ионной
некоордпнационной полимеризации: отталкивание заместителей
у мономерного звена па конце цепи и у вновь входящего мономера.
Обычно это называют моделью взаимодействия мономер — мономер
для синдиотактического присоединения [40]. На рис. 8.15 приве-
дено схематическое изображение этой модели для синдиотактиче
ского присоединения [20, 64]. За исключением двух важных
особенностей, опа очень близка соответствующей модели для
изотактического присоединения (рис. 8.13).
В случае гомогенного катализатора возможна координация
мономера (и внедрение его в полимерную цепь) в любой из двух
возможных конфигураций. Стереоспецифичность катализатора
синдиотактической полимеризации Циглера — Натта вызывается
теми же факторами, которые проявляются при понижении тем-
пературы реакции в иекоординационной полимеризации. Окру-
жение ванадиевого центра исключает вращение [уравнение (8.28)]
311 металл — активный конец растущей полимерной цепи
518
Глава 8
СН
-□
XXVII
(8.28)
что обеспечивает реализацию взаимодействия между входящиэй
и концевым мономерным звеном (XXVI) и приводит к синдиотак-3
тическому присоединению. |
На гетерогенных катализаторах взаимодействие мономер —J
мономер сводится к минимуму из-за вращения около связи|
1
i
Ьх .сн, 5
/нс
--II
сн2
СН3
Н2С=С „
/X СН3 ! ,Х CHj
х—V-ch2ch-ch2-AA *Нг СН~5х-^-сн2сн-сн2-ЛЛ
/I /I
Х X х х
Р и с. 8.15. Модель взаимодействия мономер — мономер для синдиотакти-
ческого присоединения [64].
металл — растущая полимерная цепь. Химическая и кристалли-
ческая структура гетерогенных катализаторов обеспечивает изо-
тактическое расположение путем разрешения только одной конфи-
гурации для координации и присоединения мономера к XXVII.
Стереохимия полимеризации и поликонденсации
519
Ог чичис между изотактической и синдиотактической моделями
заключается также в том, что в первой необходима миграция
получерной цепи в ее исходное лигандное положение, прежде
чем произойдет следующий акт роста. Синдиотактический рост
имеет место при любом положении лиганда.
S.-ta.5. Нисправление раскрытия двойной связи
Хотя в большинстве реакций полимеризации при раскрытии
двойной связи образуется тпранс-форма, механизм реакций Циг-
лера __ Натта связан, как правило, с возникновением цнс-формы.
Стереохимическая структура полимеров на основе 1-монозаме-
1 и с. 8.16. ци<-Раскрытпе трансЛ,2-дпзамещенного этилена с образова-
нием трео-дипзотактического полимера [И].
Щепных этиленов не зависит от направления, в котором раскры-
в.ичся двойная связь. Кроме того, дитактичпость полимеров
па основе 1,2-диза.мещенных этиленов определяется не только
с хг, как раскрывается двойная связь, но также и структурой
мономера (цис или транс). При диизотактической полимеризации
ш/шнс-мономера с цис-раскрытием двойной связи образуется
рео-диизотактическая структура. Это видно из рис. 8.16, где
520
Глава 8
показано, что углерод-углеродная связь после присоединения
мономера к полимерной цепи вращается, чтобы было исключено
1,2-взаимодействие в полностью заслоненной конформации [111.
Аналогично этому при ^пс-раскрытии tyuc-мономера образуется
эршпро-диизотайтический полимер. тпранс-Раскрытие обоих моно-
меров должно, очевидно, привести к противоположным результа-
там. Натта [5] наблюдал ^пс-раскрытие двойной связи при изо-
тактической полимеризации цис- и щранс-1-дейтеропропиленов.
цис- и тпрянс-Мономеры дают эритро- и трео-диизогактпческне
структуры соответственно. В последнее время было показано [63],
что синдиотактическая полимеризация также идет с грш-раскры-
тием двойной связи. При сополимеризации цис- и 7пр«нс-1-дейтеро-
пропиленов с гексадейтеропропиленом образуются транс- и гот-
сиидиотактические структуры XXVIII и XXIX соответственно.
8.46. Влияние природы компонентов катализатора
Циглера — Натта
Стереоспецифичность и активность катализатора Циглера —
Натта сильно зависят от природы и соотношения различных ком-
понентов катализатора Циглера — Натта.
Ряд имеющихся в литературе данных трудно интерпретировать,
так как неизвестна природа активных центров и, кроме того,
изменения в составе каталитической системы часто влекут за собой
неадекватные изменения каталитической активности и стерео-
специфичности .
8.46.1. Компонент, в котором участвует
переходный металл
Для активных центров катализаторов на основе титана пред-
полагают существование следующих валентных состояний: 4+,
3+ и 2+. Наибольшее число данных свидетельствует, что трех-
валентпый титан соответствует активному валентному состоянию.
Катализаторы на основе тригалогенидов титана (за исключением
Стереохимия полимеризации и поликонденсации 521
_—--------
rpjQi ) являются более стереоспецифическими (изотактиче-
Р' ^1И)3п0 сравнению с тетра- и дигалогенидами. Наиболее неактив-
С л из них TiCl2 активируется при измельчении в шаровой мель-
^пце благодаря реакции диспропорционирования до TiGl3 и Ti [47].
Валентное состояние переходного металла зависит не только
от "природы соединения, в котором он находится, но и от соот-
ношения последнего и компонента металла групп 1—3. Анализ
системы TiCl4-(C2H5)3Al показывает, что активность катализатора
изменяется пропорционально концентрации трехвалентного соеди-
нения, которая в свою очередь зависит от соотношения алюминия
и титана [66]. Под действием алюминийалкила в зависимости
от соотношения алюминия и титана четырехвалентный титан
восстанавливается до трехвалентного, а затем и до двухвалент-
ного состояния. При отношении Al/Ti, меньшем ~3, в температур-
ном интервале —70 ч- +70 °C восстановление до двухвалентного
титана идет в незначительной степени, при более высоких отно-
шениях Al/Ti оно протекает значительно интенсивнее, вследствие
чего каталитическая активность системы уменьшается [66].
Показано, что при полимеризации на каталитической системе
VC14-R2A1C1 трехвалентное состояние ванадия также является
наиболее активным [67]. При рассмотрении вместо Ti и V других
переходных металлов могут наблюдаться другие зависимости.
Изменение природы лиганда или переходного металла в соеди-
нении переходного металла, являющемся одним из компонентов
каталитической системы, может сильно отразиться на стерео-
специфичности и активности катализатора. При полимеризации
пропилена при температуре 75 °C в присутствии различных соеди-
нений титана в паре с (С2Н5)3А1 степень изотактичности снижается
в соответствии с уменьшением стереорегулирующей способности
в следующих рядах [58, 68]:
a-TiCl3> TiBr3> Til3,
TiCl4 « TiBr4 « Til4, (8.29}
TiCl4 > TiCl2(OC4H9)2 > Ti(OC4H9)4 « Ti(OH)4.
При изменении природы самого переходного металла происходят
следующие изменения в стереорегулирующей способности:
TiCl4 » VC14 « ZrCl4 MoCl4,
a-TiCl3> VC13> ZrCl3> СгС13. ' ?
*PW Различпых хлоридов титана в той же самой реакции поли-
Р ацпи имеет место следующий порядок активности:
а- у- или 6-TiCl3> TiCl2> TiCl4 ж 0-TiCl3. (8.31}
ностиЗЛПЧИЯ В катал11Тической активности и стереоспецифич-
проявляются только при использовании наиболее стерео-
522
Глава 8
специфических или активных соединений переходных металлов^
Так, катализаторы, содержащие четырехвалентные переходные
металлы, мало чувствительны к изменениям структуры. Кристал^
лическая структура, стерические размеры, электроотрицательЗ
ность лигандов и переходного металла влияют на стереоспеци-1
фичность и активность катализатора. При уменьшении электро^
отрицательности переходного металла (способствующей лучшей
поляризуемости связи металл — растущая цепь) за счет измене-
ния природы переходного металла, его валентного состояния
или его лиганда стереоспецифичность катализатора Циглера —
Натта увеличивается. С ростом размера лиганда стереоспецифич-
ность уменьшается, что, вероятно, обусловлено понижением коор-
динационной способности. Количественная взаимосвязь между
влиянием этих и некоторых других факторов, с одной стороны,
и кристаллической структурой переходного металла и активных
центров, с другой, на стереоспецифичность и активность катали-
затора в настоящее время четко не установлена.
8.46.2. Компонент, содержащий металл групп 1 — 3
Хотя присутствие соединения металла групп 1—3 и необяза-
тельно для создания катализатора Циглера —Натта, оно оказы-
вает очень существенное влияние на активность и стереоспецифич-
ность катализатора [40, 69]. Для проведения реакции полимери-
зации с высокой скоростью и высокой степенью изотактичности
почти всегда необходимо наличие соединения металла этих групп
в паре с соединением переходного металла. Эффект природы метал-
Таблица 8.5
Влияние природы металла первой — третьей групп
на стереорегулярность полипропилена а
Соединения металлов первой — третьей групп Содержание изотактиче- ской фракции % Литература
(С2Н6)2Ве 93-95 61
(С2Н5)3А1 80-85 61
(C2H5)2Mg 78-85 70
(C2H5)2Zn 62 69
C,H9Li 58 69
CsHiiNa 56 69
а Соединение переходного металла TiCls, температура полимериза-
ции 70 °C.
6 Доля полимера, нерастворимого в кипящем растворителе: дпбути-
ловом эфире для полимера, полученного в присутствии (C-2Hj)2Zn, и ге-
птане во всех других случаях.
Стереохимия полимеризации и поликонденсации 523
а групп 1—3 зависит также от типа соединения переходного
металла, но тем не менее некоторые выводы можно все-таки сделать.
’ табл. 8.5 показывают, как влияет природа металла
_________3 на стереоспецифичность при использовании в качестве
соединения переходного металла TiCl3 [61, 69, 70]. Стереоспеци-
фичность уменьшается в ряду
Be>A]>Mg>Lis=Na«Zn. (8.32)
Катализаторы на основе Pb, Sn, Ge, Sb, Hg и Cd не показывают
никакой активности.
Для соединений металлов групп 1—3, в которых сам металл
остается постоянным, а варьируется только природа органиче-
ского радикала, стереоспецифичность катализатора уменьшается
по мере увеличения размера органических групп, например
(С2Нз)3А1> (СвНз)зА1 > (изо-СдН^зАЕ (8.33)
Стереоспецифичность увеличивается при замене одного из органи-
ческих радикалов на любой галоген (кроме фтора)
(С2Н5)2АП > (С2Н5)2А1Вг> (С2Н5)2А1С1 > (С2Н5)3А1 > (C2H5)2A1F. (8.34)
Замена второй алкильной группы на галоген сопровождается
уменьшением стереоспецифичности.
Найти подходящее объяснение этим фактам весьма трудно
по причинам, указанным в предыдущем разделе. Особенно затруд-
нительна, по-видимому, попытка объяснить влияние природы
соединения металла групп 1—3 на стереоспецифичность в рамках
монометаллического механизма полимеризации. Порядок стерео-
специфичности, наблюдающийся для различных компонентов,
не совпадает, как предполагалось вначале, с рядом зтих же соеди-
нений для реакций восстановления или алкилирования соедине-
нии переходных металлов. Несколько легче объяснить многие
экспериментальные данные с позиций биметаллического меха-
низма полимеризации Циглера — Натта [69]. Можно полагать,
что сила биметаллического катализатора XXI должна быть
максимальной, когда металл групп 1—3 близок по размеру
и электроотрицательности переходному металлу, что имеет место
в случае Be и Al. С увеличением размера мостиковой алкильной
группы сила катализатора уменьшается. Один атом галогена
молекуле алкилалюминия приближает алюминий по электро-
гепаЦаТеЛЬП°СТИ К ™Тану; Под влиянием ?ке второго атома гало-
Эти ^‘Г1ОА11П-Г11Я уже становится более электроположительным,
сове ° ъяспеиия оставляют желать много лучшего, так как в них
затг>РШеНи° пе Учитывается кристаллическая структура катали-
524-
Глава 8
8.46.3. Третий компонент
В двухкомпонентную каталитическую систему Циглера _
Натта в качестве третьего компонента добавляют [40, 58, 69]
такие соединения, как О2, Н2, спирты, Н2О, амины, галогенида
металлов (КС1, NaF), органические галогенсодержащие вещества.
CS2, COS, фенолы, простые эфиры, фосфины, ароматические соеди-
нения (анизол, азулен) и гексаметилфосфамид. (Некоторые из них
в виде примесей могут содержаться в обычных каталитических
системах.) Влияние этих добавок на активность и стереоспеци
фичность катализатора во многом определяется природой добавки
и остальных компонентов катализатора. В присутствии одних
добавок увеличивается активность и/плп стереоспецифичности
катализатора, тогда как другие оказывают прямо противоположи
ный эффект. Некоторые добавки способствуют увеличению стерео
специфичности, но снижают активность катализатора. Ряд других
добавок влияет на молекулярный вес образующегося полимер;,
затрагивая или не затрагивая активность и/или стереоспецифии
ность катализатора.
Роль этих добавок недостаточно выяснена. В ряде случае!,
третий компонент реагирует с одним или обоими соединениями
металлов, например
TiCl3 + H2O —> Ti(OH)Cl2-j-HCl, (8.35)
АШз + ROH —> R2A1OR + RH, (8.36)
превращая их в соединения с другой каталитической активностью
и/или стереоспецифичностью. Повышенную стереоспецифичность
а-Т1С13-А1(С2Н8)С12 в полимеризации пропилена при добавлении
гексаметилфосфамида (ГМФА) объясняют [71] перегруппировкой
неактивного А1(С2Н8)С12 в активный А1(С2Н5)2С1 по схеме
2А1(С2Н5)С12 + ГМФА —> А1(С2Н5)2С1 + А1С13-ГМФА. (8.37)
Однако это объяснение явно неудовлетворительно, так как система
а-Т1С13-А1(С2Н8)С12-ГМФА более стереоспецифична по сравнению
с системой a-TiCl3-Al(C2H8)2Cl [72].
Многие соединения, применяющиеся в качестве третьего ком-
понента, являются донорами злектронов. Часто влияние третьего
компонента на активность и стереоспецифичность катализатора
объясняют координацией их с одним или двумя соединениями
металлов. Однако остается непонятным, как это приводит в одних
случаях к повышению каталитической активности и/или стерео-
специфичности, а в других случаях — к противоположным
эффектам.
Стерео.ги’мия полимеризации и поликонденсации
525
8.4в. Кинетика
Кинетика полимеризации Циглера — Натта достаточно сложна,
как и другие аспекты этого процесса. При этом кинетика гомо-
генной полимеризации обсчитывается, как правило, легче, чем
кинетика гетерогенной реакции. Для гомогенных процессов
применимы те же закономерности, что и для некоординационной
ионной полимеризации (гл. 5). Необходимо только учитывать
реакции обрыва. Хотя законы кинетики гомогенных реакций
в ряде случаев были весьма успешно перенесены на гетерогенную
полимеризацию Циглера — Натта, это следует признать скорее
исключением, чем правилом. Для корректной обработки гетеро-
генных процессов необходимо пользоваться уравнениями для
гетерогенной кинетики [73]. Общие закономерности такого под-
хода будут кратко рассмотрены ниже [9, 58, 74, 75].
8.4в.1. Схема реакции
Возможно рассмотрение полимеризации как адсорбции моно-
мера и металлоорганического соединения MeR металла групп 1—3
из раствора поверхностью соединения переходного металла
(обозначенной жирной вертикальной линией)
(8.38)
(8.39)
с последующим инициированием в результате реакции между
адсорбированными частицами
(8.40)
и ростом цепи в результате аналогичной реакции
(8.41)
526
Глава 8
Обрыв может иметь место в результате передачи цепи
на мономер
(8.42)
самопроизвольного внутреннего переноса протона
Me—М —R
te н I МеН*
''пер.п
(8.43)
пли передачи цепи Me — R,
(8.44
а также реакции с мономером с образованием неактивной частицы.
(8.45)
Возможны также другие реакции обрыва, например передача
цепи на переходный металл катализатора или соединения, содер-
жащие активный атом водорода (Н2, Н2О, ROH и т. д.).
8.4в.2. Уравнения скорости реакции
Для того чтобы вывести уравнение скорости реакции, учиты-
вающее вышеприведенные последовательные стадии полимериза-
ции [уравнения (8.38)—(8.44)], пользуются механизмом Ленг-
мюра — Хишпельвуда [73], предложенным для реакции между
двумя адсорбированными частицами (Me — R и М), которые кон-
курируют между собой за одни и те же активные центры на поверх-
ности твердого катализатора. Принято [73], что адсорбция [урав-
нения (8.38) и (8.39)] быстро достигает равновесного состояния,
которое поддерживается на всем протяжении реакции. Доли
поверхности катализатора бмеп. и 6м, покрытые MeR и М соот-
Стереохимия полимеризации и поликонденсации 527
—
ветственно, определяются с помощью изотерм Ленгмюра
л_____________________________[MeR]______ /о лсч
°MeR-1 + Ki[MeR]+K2[M]’ к ,
а_________^2 [М]_____/й А7\
м- 1 + Kt [MeR] + K2 [М]-' 1 '
Пренебрегая стадией инициирования, скорость исчезновения
мономера па единицу активных центров определяется как
= (*р + ^пер. м) 9м [Р*], (8-48)
гдР fl’*] — концентрация растущих полимерных цепей. В стацио-
нарном состоянии можно считать равными скорость инициирова-
ния и сумму скоростей всех реакций обрыва. Тогда
А'ц9мрН0М ~ ^пер. Н [Р* 1 "Г ^пер. Р [Р*] ^MeR 4“ &о. М [Р*]9м- (8.49)
Из уравнений (8.46)—(8.49) скорость полимеризации опреде-
ляется как
_<7[М| (^р + ^пер. м) 4i [MeR] KiK% [М]2
dt ~ I г Г1И1_1_Г гМоРПЗ/ь , ^пер. P^l [MeR] + A:0. мК2[М] 1
Ц —А2[М]-|-2ЩМей])3 ^пер.н+ P1 + K2[M]+KliMeR] )
(8.50)
Соответствующее выражение для степени полимеризации полу-
чается обычным приемом путем деления скорости роста на сумму
скоростей всех реакций обрыва. Уравнение (8.50) легко можно
упростить, пренебрегая одной или несколькими стадиями в выше-
приведенной схеме реакции. Так как MeR намного полярнее
мономера, то возможен процесс, в котором адсорбция мономера
очень низка (Ai^> Х2), тогда К2 в уравнении (8.50) можно пре-
небречь. В ряде систем преобладают реакции обрыва одного
типа, поэтому можно опустить соответствующие параметры для
Других реакций обрыва.
8.4в.З. Экспериментальная кинетика
Часто кинетика реальных реакций полимеризации Циглера —
Натта описывается весьма сложными кривыми (рис. 8.17). Кривая 1
ооычпо получается при участии в реакции сравнительно больших
частиц соединений переходного металла. Полагают, что частицы
катализатора состоят из агрегатов более мелких кристаллов,
ным Мехаиическом давлении, оказываемом растущими полимер-
v цепямп’ большие частицы разрушаются, вследствие чего
шаетИЧИВаеТСЯ повеРхность и число активных частиц и повы-
Ся скорость полимеризации во времени [76]. Величина Rn,
528
Глава 8
становится постоянной, когда разрушаются самые маленькие
кристаллы. Если в начальный момент реакции частицы катализа-
тора имеют меньший размер (в результате предварительного
измельчения), то промежуток времени, необходимый для дости-
жения стационарного состояния, сокращается (кривая 2).
Некоторые реакции полимеризации характеризуются резким
повышением 7?р с последующим быстрым ее уменьшением до ско-
рости, при которой полимеризация входит в стационарный режим
(кривая 5). Такое поведение свидетельствует о том, что в катали-
заторе Циглера — Натта существуют различные типы полимери-
зационных центров. Некоторые, менее активные центры на поверх-
ности твердого катализатора в начальный период- реакции могут
Р п с. 8.17. Типичные кинетиче-
ские кривые при полимеризации
Циглера — Натта.
дезактивироваться [76]. Высокая начальная скорость реакции
может быть обусловлена также действием растворимых, нестерео-
специфических каталитических частиц, образующихся вначале
при реакции между различными компонентами катализатора.
Количество их во времени уменьшается, что приводит к тому, что
скорость начинает соответствовать скорости гетерогенной поли-
меризации в стационарном режиме [77]. Для некоторых полимери-
зационных систем наблюдается непрерывное уменьшение скорости
на всем протяжении реакции, кроме начального ее периода
(кривая 4), что можно объяснить разложением катализатора
при проведении полимеризации при достаточно высокой темпера-
туре. Такое поведение может быть обусловлено также тем, что
реакция роста лимитируется диффузией, т. е. тем, что стадией,
определяющей скорость полимеризации, становится диффузия
мономера к центру полимеризации через уже образовавшийся
полимер. На справедливость этого предположения указывает
повышение скорости полимеризации с ростом скорости перемеши-
вания реакционной смеси [78].
Стереохимия полимеризации и поликонденсации
529
8.4вЛ. Кинетические параметры
В табл. 8.6 приведены различные кинетические параметры
олпмерпзацип Циглера — Натта [79]. Число растущих центров
составляет лишь небольшую долю (~0,1 — 1%) от общего количе-
Таблица 8.6
Кинетические параметры
полимеризации этилена в присутствии
(C2H5)2AlCl-y-TiCl3 при 40 °C [79]
Параметр
Значение
[Р*], моль/моль TiCl3
Ар, л/(молЬ‘С)
А'о. ль с"1
^'пер. М, л/(моль-с)
^пер. Н2- л/(моль-с)
10-2
80
3-10-4
1-10-2
3
ства атомов переходного металла, находящихся в системе.
По. порядку величины константы скоростей различных реакций
близки к соответствующим константам для некоординационной
ионной полимеризации (табл. 5.1 и 5.7). В реакции полимериза-
ции, кинетические параметры которой приведены в табл. 8.6,
обрыв происходит главным образом при передаче цепи на водород
(^пер Н2). П олимеризация Циглера — Натта очень близка поли-
меризации с образованием живущих полимеров по времени жизни
растущих цепей, которое колеблется здесь от минут до часов.
(Время жизни растущих цепей в полимеризации в присутствии
растворимых катализаторов значительно меньше, чем при поли-
меризации на гетерогенных катализаторах. В первом случае оно
редко превышает 5—10 мин.) Время жизни активных центров еще
ольше и во многих случаях составляет несколько дней.
Полимер, образующийся при полимеризации на гетерогенных
катализаторах Циглера — Натта, имеет достаточно широкое
молекуляриовесовое распределение. Отношение MwIMn дости-
ает обычно 5—20 у полиэтилена [79] и 5—15 у полипропилена [80].
акое широкое распределение может быть обусловлено рядом
длит°Р01,: Разлпчн°й активностью отдельных активных центров,
РУю Л„ПОСтыо достижения стационарного состояния, лимити-
п°лиз1И Р°‘11,ю Диффузионного фактора на заключительных этапах
еризации. Гомогенная полимеризация Циглера — Натта
530
Глава 8
1
характеризуется значительно более узким молекулярновесовьщ
распределением.
Энергия активации большинства реакций полимеризации Циг_
лера — Натта находится в интервале 3—15 ккал/моль (12,56 -103-^
62,79-Ю3 Дж/моль) и складывается из энергий активацщ
стадий инициирования, роста и обрыва цепи, а также теплот ста-
дий адсорбции.
Хотя скорость полимеризации возрастает с повышением тем-
пературы, обычно процесс ведут при температурах не выше
70—100 °C. При более высоких температурах полимеризация
не идет стереоспецифично, причем скорость ее вследствие разло-
жения катализатора даже понижается.
8.4г. Области применения катализаторов
Циглера — Натта
В присутствии катализаторов Циглера — Натта полимери-
зуются многие мономеры [40], в том числе этилен, а-олефины
(пропилен, бутен-1, 4-метилпентен-1, винилциклогексан) и стирол.
1,1-Дизамещенные этилены, например изобутилен, полимери-
зуются в присутствии таких катализаторов, но только по некоор-
динационному ионному механизму. Из-за стерических препят-
ствий все 1,2-дизамещенные этилены, за исключением 1-дейтеро-
пропиленов (разд. 8.4а.5), не полимеризуются. Полимеры на основе
некоторых 1,2-дизамещенных этиленов удалось получить путем
изомеризации мономеров в 1-замещенный этилен с последующей
полимеризацией, например, таким путем из бутена-2 получен
полибутен-1. Полимеризация диенов на катализаторах Циглера —
Натта рассматривается в разд. 8.6. Ряд мономеров ацетиленового
типа также полимеризуется под действием катализаторов Циг-
лера — Натта. 5
Многие полярные мономеры, например винилацетат, винил-,
хлорид, акрилаты и метакрилаты, полимеризуются под действием
катализаторов Циглера — Натта, но не по анионно-координа-
ционному механизму. Реакция идет по некоординационному,
(радикальному или ионному) механизму.
Ряд полярных мономеров, особенно содержащих такие электро-
нодонорные атомы, как азот и кислород, не полимеризуются в при-'
сутствии катализаторов Циглера — Натта. Эти мономеры дезак-^
тивируют катализатор, образуя с ним прочные комплексы или
реагируя необратимо с одним из его компонентов [81]. Известна
лишь несколько примеров успешной стереорегулярной полимери-
зации полярных мономеров на катализаторах Циглера — Натта?
Стереоспецифическую полимеризацию мономеров, координирую-
щихся с катализатором, иногда удается осуществить путем прове-
Стереохимия полимеризации и поликонденсации
531
„кипи в растворителе (например, диметилформамиде или
дения Р офуране), который сам образует комплекс с катализато-
тетраг ^ожет gbITb вытеснен из него мономером. Иногда исполь-
р°м, ®°енее аКТивнуто форму катализатора. Полярные мономеры
ЗУЮТ заполимеризовать на катализаторах Циглера — Натта,
моЯ<н молярные атомы или группы экранированы объемистыми
еСЛИстителями. Так, мономеры с гидроксильными или аминными
Зпуппами полимеризуются, если эти группы превратить в группы
' siR2 и — NR2 соответственно [81].
8.4г.1. Циклоалкены
Хотя циклоалкены являются по существу 1,2-дизамещенными
алкенами, они полимеризуются в присутствии катализаторов
Циглера_ Натта, что, видимо, связано с определенной напря-
женностью их циклов [40, 82]. При этом полимеризация может
идти с раскрытием двойной связи или с раскрытием цикла, напри-
мер в случае циклобутана
(8.51)
СН = сн
I I
сн2-сн2
(8.52)
-----> -(-CH2-CH = CH-CH2-)n-
XXXI
В результате каждой из этих реакций возможно образование
Двух различных стереоизомеров. Структура XXX может суще-
ствовать в двух изомерных формах: эриигро-диизотактической
и эДитро-дисиндиотактической (разд. 8.16.2). Полимер со струк-
час°И XXX называют [14] полициклобутиленомером-2 или, более
тиел°' ПР°СТ0 полицпклобутеном. При полимеризации с раскры-
Риз П'ИКЛа получается такой же полимер, что и при 1,4-полиме-
1^Цс^П'ИИ бутадиепа-1,3, который, следовательно, может быть
ваблю*1™ m^awc-1I30MeP0M- При полимеризации циклобутена
При пи™” обРазование всех четырех изомерных полимеров,
ванадия МеНеНИИ В качестве катализатора соединений хрома,
связи [1g1 89>ДИо1 полимеРизаЦия идет исключительно по двойной
мер с эпи На хромовых катализаторах получается поли-
тРо-диизотактической структурой, эритпро-дисиндиотак-
532
Глава 8
тический полициклобутен образуется в результате эмульсионц0^
полимеризации с родиевым катализатором [85]. При использова
нии катализаторов на основе вольфрама, титана и молибде11а
идет преимущественно полимеризация с раскрытием цикла с обра.
зованием в различных соотношениях цис- и транс-изомеров
На рутениевых катализаторах протекает исключительно подпив
ризация с раскрытием цикла.
Аналогично протекает полимеризация других циклоалкенов
однако она может осложняться вмешательством других процессов’
Так, при полимеризации З-метилциклобутена-1 раскрывается
связь между атомами углерода 1 и 4
•СН—СН=СН-СН,-
I 2
СН,
о
сн=сн
(8.53)
•сн2—с=сн—сн2-
снз
а полиизопрен не образуется. Полимеризация мономеров с двумя
двойными связями часто сопровождается одновременной транс-
аннулярной миграцией, зависящей от природы катализатора.
Например, для циклооктадиена-1,5
(8-54)
8.4г.2. Сополимеризация
Сополимеризация с применением катализаторов Циглера —
Натта имеет ряд интересных особенностей [40, 86, 87]. При сополи-
меризации мономеров, способных к гомополимеризации на катали-
заторах Циглера — Натта, образуются статистические сополи-
меры. Реакционная способность мономеров в сополимеризаци11
Стереохимия полимеризации и поликонденсации
533
яет с их поведением в гомополимеризации, а именно
совпад пр0Ппдек ;> бутен-1. Как и можно было ожидать,
ЭТЙЛепенпя реакционных способностей мономеров часто зависят
°ТНО„„т.,т каталитической системы; правильнее сказать, что
пт
_ п ?•., влияет природа соединения переходного металла
(табл* 8 7). Отношение реакционных способностей обычно не зави-
Таблица 8.7
Реакционная способность различных мономеров
в сополимеризации Циглера — Натта
Mi м2 Каталитическая систе- ма а Г1 T2 Литера- тура
Этилен Пропилен TiCl3-Al(C6H13)3 15,7 0,110 88
TiCl4-Al(C6H13)3 33,4 0,032 88
VC13-A1(C6H13)3 5,61 0,145 89
Этилен Бутен-1 VC14-A1(C6H13)3 29,6 0,019 90
VC13-A1(C6H13)3 67,0 0,043 90
Пропилен Бутеп-1 VCl4-Al(CeH13)3 4,39 0,227 91
VC13-A1(C6H13)3 4,04 0,252 91
а Системы с УШз и TiCl3 гетерогенны; другие системы — коллоидные дисперсии.
сит от природы соединения металлов групп 1—3. Это является
лишним подтверждением (см. выше, разд. 8.4а) большей роли
соединения переходного металла. Относительные количества
обоих компонентов катализатора могут иногда влиять на т\ и г2>
все определяется природой растворителя и другими условиями
реакции. На строение и состав сополимеров, получающихся
сополимеризацией па катализаторах Циглера — Натта, большее
влияние оказывает физическое состояние катализатора. При
использовании растворимых катализаторов получаются атакти-
ие, аморфные полимеры. Практическое значение предста-
ют аморфные сополимеры этилена и пропилена, характери-
пУолЩИеСЯ пптеРСС111,пш эластомерными свойствами. Такие каучуки
меп аЮТСЯ B^T'IKaiIII3a4Ini, если смесь этилена и пропилена сополи-
УЮт в присутствии небольших добавок диенов, например
Дициклопентаднеиа (XXXII)
XXXII
534
Глава 8
При использовании гетерогенных катализаторов наблюдает^
тенденция к образованию нестатистических полимеров, содерИу
щих длинные последовательности одного или другого сомономера
что связано с большей селективностью гетерогенных активны^
центров полимеризации.
Хотя 1,2-дизамещенные этилены не гомополимерпзуются
цис- и транс-бутен-2 вступает в реакцию сополимеризации с эти'
леном на катализаторах Циглера — Натта [18]. Стерические цре.
пятствия, которые исключают возможность гомополимерпзации
бутена-2, перестают играть такую решающую роль, когда бутеп-2
присоединяется к растущей цепи с концевой этиленовой группой.
Таким образом, благодаря тенденции к образованию регулярно
чередующихся сополимеров этилен и бутен-2 вступают в реакцию
сополимеризации. (Эта тенденция в данном случае обусловлена
уменьшением стерических препятствий; при радикальной поли-
меризации она связана с полярными эффектами.) Однако вслед-
ствие значительно более низкой реакционной способности бутена-2
по сравнению с этиленом необходимы специальные условия для
получения сополимеров с заметным содержанием бутена-2. Регу-
лярно чередующийся сополимер с соотношением сомономеров,
близким к 1, удается получить, проводя реакцию с высоким
исходным соотношением бутена-2 и этилена в присутствии раство-
римых или коллоидно-диспергированных (низкоселективных) ката-
лизаторов. В результате подобной сополимеризации удалось
выделить кристаллическую фракцию полимера (путем экстракции
растворителем). Натта с сотр. [93] полагает, что фракция имеет
структуру аршпро-диизотактического типа.
Последовательным введением двух сомономеров в реакцион-
ную систему удалось синтезировать блок-сополимеры, содержа-
щие длинные блоки обоих мономеров [94, 95]. При этом необхо-
димо использовать гетерогенные катализаторы, так как время
жизни растущих цепей в гомогенной полимеризации слишком
мало, чтобы было возможно последовательное присоединение
(разд. 8.4в.4). Полученные блок-сополимеры отличаются различ-
ной степенью кристалличности и тактичности. Кристаллические
сополимеры представляют практический интерес.
8.4д. Окисные катализаторы на носителях
Этилен и некоторые другие алкены полимеризуются пр11
использовании окисных катализаторов на носителях [58, 96 —981-
Такие катализаторы представляют собой дисперсию окиси пере'
ходного металла (например, СгО3, МоО3, V2O5 и NiO3). начесен-
ную на другое вещество, называемое носителем; в качестве послеД"
него наиболее часто используют А12О3, активированный угол1”
SiOo, глину. Носитель обеспечивает возможность практического
Стереохимия полимеризации и поликонденсации
535
чьзоваппя катализаторов с небольшим размером частиц
?СП°оэтому большой поверхностью). Окись металла на носителе
Й BHPvercH (т. е. превращается в катализатор полимеризации)
аутем восстановления водородом при высоких температурах или
бпаботкп такими восстановителями, как ЫВН4 или Nall, которые
называются промоторами или активаторами. В процессе акти-
вации катализатор может взаимодействовать с металлом носителя.
Полимеризация этилена на окислах металлов на носителе
представляет промышленный интерес, так как образующийся
полиэтилен, полученный полимеризацией на катализаторах
Циглера __ Натта, имеет линейное строение. То, что указанные
типы катализаторов одинаково влияют на строение образующихся
полимеров, является, очевидно, следствием идентичности меха-
низмов пх полимеризации и природы активных центров. Актива-
ция путем восстановления окиси металла на носителе очень близка
к алкилированию (восстановлению) соединения переходного
металла, одного из компонентов катализатора Циглера — Натта.
Собственно, если не обращать внимания на носитель, то боль-
шинство таких систем, состоящих из катализатора (окисла металла)
и восстановителя, попадает под определение катализатора Цигле-
ра — Натта. От последних они отличаются лишь меньшей актив-
ностью. Полимеризацию на окисных катализаторах проводят при
сравнительно высоких температурах (100—200 °C). Многие моно-
меры (например, стирол), полимеризующиеся на катализаторах
Циглера — Натта, неактивны в присутствии окислов металлов
на носителе. Такие катализаторы отличаются также очень низкой
стереорегулирующей способностью. Если частично кристалличе-
ский полипропилен еще можно получить на таких катализаторах,
то при полимеризации большинства других а-олефинов образуются
только аморфные или очень слабо кристаллические полимеры.
Стереоспецифическая полимеризация
полярных алкенов
нап ТеРеоспеЦиФическая полимеризация полярных мономеров,
в РИМеР метакрилатов и простых виниловых эфиров, возможна
шениределснном интервале температур при определенных соотно-
когдаЯХ ,Л1ОиомеРа> катализатора и растворителя. В условиях,
Р°вань\КТПППЫе цептРы растущих частиц свободны (не координи-
Приятн ° поип'кением температуры создаются более благо-
(разд g1® Условия для образования синдиотактических структур
МеРизап ЭТ° спРавеДЛИВ0 для всех радикальных реакций поли-
Дящихся1 П Д 1Я ионных полимеризационных процессов, прово-
ТаЧеских В СПЛЬН0 сольватирующих средах. Образование изотак-
полимеров может иметь место в слабо сольватирующих
536
Глава 8
средах, где активные частицы растущих цепей коордиш1руюТСя
с противоионом. Поскольку полярные мономеры обладают бо.тее
сильной координирующей способностью по сравнению с неполдр.
ными алкенами, степени координации, необходимой для изотак-
тического присоединения, можно достичь в присутствии гомоген-
ных катализаторов. Используют также гетерогенные катализа-
торы, но в отличие от полимеризации неполярных алкенов в этом
процессе их присутствие не обязательно. Многие условия поли-
меризации, о которых шла речь выше (гл. 5), могут приводить
к образованию изотактических полимеров. Ниже рассмотрена
стереоспецифическая полимеризация некоторых, наиболее широко
известных полярных мономеров, причем особое внимание будет
обращено на синтез изотактических полимеров.
8.5а. Полимеризация метилметакрилата
Изучена [99, 100] стереоспецифическая полимеризация раз-
личных акрилатов и метакрилатов (табл. 8.8 и 8.9). Для сравне-
Таблица 8.8
Влияние природы растворителя и противоиона
при температуре полимеризации 0 °C на тактичность
полиметилметакрилата [99]
Растворитель Содержание изотактической фракции в полимере, полученном в присутствии различных противоионов, %
M-C4H9L1 M-CjHiiNa н-СдНпК
Толуол 81 72 56
Диметилформамид 30 21 37
Пиридин 24 35 40
ния в табл. 8.9 приведены данные для радикальной полимериза-
ции соответствующих мономеров. Тенденции, которые прослежи-
ваются при анализе этих данных, справедливы и для реакции
полимеризации других мономеров (в том числе и для полимери-
зации Циглера — Натта неполярных алкенов). В полярных раство-
рителях противоион не находится в непосредственной близости
к растущему центру, поэтому он не может оказывать влияния
(стереохимического) на присоединение следующей молекулы моно-
мера. С увеличением температуры возрастает атактичность полу-
чаемого полимера, тогда как понижение температуры благоприят-
ствует образованию синдиотактических структур.
Стереохимия полимеризации и поликонденсации
537
Таблица 8.9
Влияние природы растворителя, противоиона и температуры
полимеризации на тактичность полиметилметакрилата [100]
— Диадная тактичность Триадная тактичность
Полпмеризэцпонная система г 1 8
Радикальная полимериза- ция в блоке при 60 °C K-C4H9Li при —78 °C, со- держание тетрагпдрофу- рапа в толуоле, % 0,24 0,76 0,08 0,033 0,59
0 0,85 0,15 0,78 0,16 0,06
2,5 0,46 0,54 0,30 0,31 0,39
5 0,41 0,59 0,24 0,34 0,42
10 (C2H5)2A1N(C6H5)2 в С6Н5СН3, температура, °C 0,30 0,70 0,13 0,35 0,52
-78 0,09 0,91 0,01 0,14 0,85
-40 0,13 0,87 0,03 0,20 0,77
0 0,15 0,85 0,03 0,23 0,74
В неполярных растворителях реакция может протекать по меха-
низму анионно-координационной полимеризации. Под влиянием
противоиона каждая новая молекула мономера присоединяется
к полимерной цепи изотактическим способом. При этом степень
изотактического расположения зависит от координационной спо-
собности противоиона. Различные щелочные металлы по коорди-
национной способности располагаются в следующем порядке:
2> Na > К, который отвечает их последовательности, выте-
кающей из вышеприведенных рассуждений (разд. 5.36.4). Наиболь-
шей координационной и стереоспецифической способностью обла-
дает ион лития, самый маленький из них. Другим крайним случаем
противоион большого размера, образующийся из
НИЮ 5 2ALX(CgH5)2’ под влиянием которого тенденция к образова-
изотактического полимера проявляется очень слабо даже
с темПР°ВеДе1111П рСаКЦПИ В неполяРных растворителях. Вместе
хим Разл15*Р противоиона нельзя рассматривать в отрыве от его
в ди скон природы. Так, полимеризация метилметакрилата
1лА1НПЛ0В0М :’ФиРе ПРИ комнатной температуре в присутствии
ной к4 ДаСТ 91 % изотактической структуры [101] благодаря силь-
оордипациоппой способности лития.
538
Глава 8
В ряде реакций полимеризации на степень изотактичности
влияет также фрагмент катализатора, играющий роль инициатора.
Так, например, при полимеризации метилметакрилата фенолят
лития менее стереоспецифичен по сравнению с соответствующим
тиофенолятом [102]. Этот результат достаточно неожиданный, так
как активный центр в обоих инициаторах одинаковый. Возможно,
что причина такого поведения состоит в разной координации
непрореагировавшего инициатора с концом растущей цепи и его
противоионом. Координационная способность тиофенолята выше,
чем у фенолята, что приводит к образованию более жестких
и стереорегулярных концевых групп цепи. При переходе от одной
полимеризационной системы к другой зависимость изотактич-
ности от температуры реакции может сильно меняться. Повыше-
ние температуры обычно приводит к уменьшению синдиотактич-
ности. Вместе с тем при использовании высокостереоспецифиче-
ских катализаторов колебания температуры в широком интервале
(до 50 °C) практически не влияют на тактичность.
Предложены различные механизмы для объяснения образо-
вания изотактического полиметилметакрилата [6, 103]. К ним
относится ужесточение концевых групп растущей цепи за счет
координации противоиона с концевой и соседней с ней группами
с образованием следующей структуры:
СН3 осн.
СН
I
—сн9—с-сн2—с—с
2 I ~ II
CH..O--Ck : О
3 Ж).....G
. +
t ХХХШа
сн3 сн3 осн3
'-СН,—С—сн9—с=с
I I
сн3о С о
Ж...G"" “
хххшб
Механизм стереоспецифического действия под влиянием такого
жесткого конца полимерной цепи (XXXIII) сходен с механизмом,
принятым в настоящее время для роста цепей в стереоспецифиче-
ской полимеризации Циглера — Натта. Молекула мономера
координируется с жесткой структурой XXXIII через противоион,
а затем стереоспецифически присоединяется путем внедрения
по связи металл — углерод.
Стереохимия полимеризации и поликонденсации
539
8.56. Полимеризация простых виниловых эфиров
Изотактическая полимеризация простого винилового эфира
идет по катионно-координационному механизму, который анало-
гичен анионно-координационному механизму с той лишь разницей,
что в первом растущий центр — ион карбония, а не карбанион,
и противоион •— анион вместо катиона. Полимеры с различной
степенью изотактичности образуются при использовании гомоген-
ных и гетерогенных катализаторов разного типа: SnCl4-CCl3COOH,
Д1(С.,П5)С12, A12(SO4)3-H2SO4, эфират трехфтористого бора [104—
106]. Растворитель, температура и другие условия реакции влияют
на тип и степень стереоспецифичности так же, как и в вышерас-
смотренных случаях. Для получения синдиотактических и изо-
тактических полимеров простых виниловых эфиров нужно строго
контролировать условия реакции.
Для получения изотактических полимеров простых виниловых
эфиров неприменимы катализаторы типа Циглера — Натта, кото-
рые используют для синтеза полимеров на основе большинства
других полярных мономеров. Поэтому необходимо подобрать
такую каталитическую систему, которая обеспечила бы образова-
ние катализатора катионно-координационного типа с необходимой
координационной способностью. Подробно исследованы катали-
заторыпа основе соединений ванадия, например УС14-(изо-С4Н9)зА1.
Стереоспецифичность полимеризации простых виниловых эфи-
ров очень чувствительна к составу указанной каталитической
системы [107], что типично для реакций полимеризации Циглера —
Натта вообще.
8.5в. Полимеризация стирола
По полярности стирол занимает промежуточное положение
между неполярными и полярными алкенами. Благодаря отсутст-
вию в его молекуле сильно полярных или реакционноспособных
Функциональных групп стирол стереоспецифически полимери-
зуется на катализаторах Циглера — Натта. Вместе с тем стирол
подвергается стереоспецифической полимеризации в присутствии
не только катализаторов Циглера — Натта, но и других катали-
заторов, что обусловлено некоторой полярностью его бензольного
яДра. Стереоспецифическая полимеризация стирола в присутствии
лития и его алкилов идет по тому же механизму, что и полимери-
зация метилметакрилата [108].
540
Глава 8
8.6. Стереоспецифическая полимеризация
диенов-1,3
8.6а. Радикальная полимеризация
Из-за большего набора возможных стереоизомерных структур
полимеризация диенов-1,3 — более сложный процесс, чем полиме-
ризация алкенов (разд. 8.1г и 8.1д). В табл. 8.10 представлены
Таблица 8.10
Структура полимеров, полученных нз диенов радикальной
полимеризацией [109—111]
Мономер Температура полимеризации, °C Содержание структуры, %
цис-1 ,4- транс-1, к- 1,2- 3,4-
Бутадиен-1,3 -20 6 77 17
-10 9 74 17
5 15 68 17
20 22 58 20
100 28 51 21
175 37 43 20
233 43 39 18
Изопрен -20 1 90 5 4
-5 7 82 5 5
10 11 79 5 5
50 18 72 , 5 5
100 23 66 5 6
203 19 69 3 9
структуры полимеров, полученных радикальной полимеризацией
бутадиена-1,3 и изопрена [109—111]. Как видно из данных этой
таблицы, предпочтительно идет 1,4-полимеризацпя, а не 1,2-
или 3,4-полимеризация, причем доля транс-!,4-полимеризации
выше доли цис-i,4-полимеризации. Исследование зависимости
структуры полибутадиена от температуры полимеризации пока-
зало, что по энтальпии активации транс-!, 4-присоединение
выглядит предпочтительнее цис-присоединения (несмотря на то,
что последнее характеризуется более выгодной энтропией актива-
ции, достигающей 7 энтр. ед.), транс-!,4-Присоединение выгод-
нее 1,2-присоединения по энтальпийному фактору при одинаковой
энтропии активации.
Полимеризация диенов-1,3 предполагает делокализацию ради-
кала между углеродами 2 и 4 концевого мономерного зве-
Стереохимия полимеризации и поликонденсации
541
на (XXXIV)
-^~сн2—сн—сн—сн2
12 3 4
XXXIV
Преимущественный рост в положение 1,4, а не в положение 1,2
связан главным образом с меныпими стерическими затруднениями
при углероде в положении 4 по сравнению с углеродом, находя-
щимся в положении 2. Это различие становится еще более ощути-
мым при введении заместителя к углероду 2, поэтому в случае
изопрена (табл. 8.10) и хлоропрена [16] 1,4-полимеризация идет
в еще большей степени. Преимущественное протекание трансЛ,^-
полимеризацпи, а не цисЛ,4-полимеризации связано с конформа-
цией мономера. Диены-1,3 существуют главным образом в s-транс-
конформацпп (XXXVa), а не в s-^wc-конформации (XXXV6) [112].
СН2
У
СН —СН
н2с
s-транс
XXXVa
Н2С СН2
%
СН —СН
s-цис
XXXV6
Присоединение мономерного диена к растущему центру проте-
кает с сохранением конформации мономера, поэтому конечный
полимер имеет преимущественно транс-конфигурацию.
8.66. Анионная и координационная полимеризация
В зависимости от того, является ли растущий центр при анион-
ной полимеризации диенов-1,3 свободным ионом или он координа-
ционно связан с противоионом, образуются полимеры различной
структуры. В табл. 8.11 приведены данные, полученные при
полимеризации бутадиена [ИЗ, 114]. В случае реакции через
свободный анион предпочтительна 1,2-полимеризация, а не 1,4-по-
ли.мерпзацпя. Анионный центр при углероде 2 делокализован
не так сильно, как соответствующий радикал, поскольку двойная
связь алкена скорее электронодонорная, чем электроноакцеп-
торпая.
Аналогичные результаты получены при полимеризации изо-
прена (табл. 8.12) [115], разница состоит лишь в том, что в послед-
нем случае в основном идет 3,4-полимеризация, а не 1,2-полиме-
ризация вследствие более низкой электронной плотности двойной
542
Глава 8
Таблица 8.11
Влияние природы растворителя и противоиона на структуру
полибутадиена
Катализатор Содержание структуры, %
цис-1,4- транс-1,4- 1,2-
Полимеризация в пентане [113]
Li 35 52 13
Na 10 25 65
К 15 40 45
Rb 7 31 62
Cs 6 35 59
Полимеризация в тетрагидрофура-
не [114]
Li-нафталин 0 4 96
Na-нафталин 0 9 91
К-нафталин 0 18 82
Rb-нафталин 0 25 75
Cs-нафталин 0 25 75
Таблица 8.12
Влияние природы растворителя и противоиона на структуру
полиизопрена [115]
Катализатор Растворитель Содержание структуры, %
цис-1,4- транс-1 1,2- 3,4-
Пентан 93 7
90% пентана — 10% тетра- гидрофурана — 26 9 65
Тетрагидрофуран — — 26 74
Li Пентан 94 — — 6
Li (С2Н5)2О — 49 5 46
Na Пентан — 43 6 51
Na Тетрагидрофуран — — 18 82
Cs Пентан 4 51 8 37
связи в положении 3,4 по сравнению с электронной плотностью
двойной связи в положении 1,2.
При полимеризации в неполярных растворителях возрастает
роль 1,4-полимеризации, причем эффект наиболее сильно прояв-
ляется в присутствии иона лития, обладающего наибольшей
Стереохимия полимеризации и поликонденсации 543
коОрдпнационной способностью. Более того, при применении
Штин становится возможной уже цис-1,4-полимеризация, несмотря
ца то что диены существуют преимущественно в х-транс-конфи-
гурации. При получении полиизопрена в присутствии литиевого
соединения идет практически только цис-1,4-полимеризация. Пред-
почтительное протекание цис-1,4-полимеризации связано, вероят-
но, с образованием л-комплекса XXXVI между ионом лития
и диеном с фиксацией диена в г^с-форме до и после присоединения
его к растущей цепи [115]
СН—CR
Z ч
н2с сн2
\ у
/
Li+
XXXVI
О’Дрисколл с сотр. [116], который детально исследовал меха-
низм полимеризации диенов-1,3, полагает, что противоион коор-
динируется одновременно с аллильным карбанионом на конце
растущей цепи и вновь входящей молекулой мономера. Образую-
щееся при этом промежуточное соединение (XXXVII)
3 2
СН—CR w
4 / \...\
~сн2 ; /сн2
3 2
XXXVII
подвергается 1,4-полимеризации путем образования связи между
углеродом 1 аллильного карбаниона и углеродом 4 мономера.
В случае сильной координации в XXXVII (например, в неполяр-
ных растворителях или в присутствии лития) вращение вокруг
связи между углеродами 2 и 3 на конце цепи невозможно, что
и приводит к цис-1,4-полимеризации. В слабо координированном
комплексе, который допускает вращение вокруг связи, идет транс-
и цис-1,4-полимеризация. Если координация очень слабая, то
имеет место 3,4-полимеризация вследствие образования связи
544 Глава 8
между углеродом 3 аллильного карбаниона и углеродом 4 моно-
мера.
Стереоспецифическая полимеризация диенов-1,3 проводилась
в присутствии ряда других каталитических систем [16, 82]. Еще
в 1946 г. Мортон [117] использовал гетерогенный алфиновый
катализатор, состоящий из аллилнатрия, изопропилата натрия
и хлористого натрия. Под влиянием алфинового катализатора
возрастает относительная доля 1,4-полимеризации по сравнению
с 1,2-полимеризацией, но эти катализаторы не являются очень
стереоспецифическими, поскольку они не оказывают особого
предпочтения ни цис-1,4-, ни транс-1,4-полимеризации. В то же
время при использовании катализаторов Циглера — Натта полу-
чаются очень интересные результаты, так как по стереоспецифич-
ности они в этом случае превосходят литиевые катализаторы.
В табл. 8.13 указаны структуры полимеров, полученных при
полимеризации в присутствии различных катализаторов [49, 118—
127]. Исключительно высокая стереоспецифичность катализаторов
Циглера — Натта при полимеризации бутадиена проявляется
даже более сильно, чем при полимеризации мономеров винильного
типа. При полимеризации бутадиена возможно образование
четырех различных стереоспецифических полимерных структур
(цис-1,4, транс-1,4, st-1,2 и it-1,2).
Как видно из данных табл. 8.13, путем подбора катализатора
каждый стереоизомер можно получить практически без примесей
полимеров другой структуры. Стереоспецифичность катализаторов
определяется типом их координации с концом растущей цепи
и вновь входящим мономером, а также свойствами образующегося
промежуточного соединения (аналогичного XXXVII).
Для стереоспецифической полимеризации бутадиена, кроме
катализаторов Циглера — Натта, можно также использовать
другие катализаторы на основе соединений переходных металлов,
в том числе различные хелатные соединения и л-комплексы,
например бис-(л-кротилникельиодпд)
XXXVIII
Среди различных катализаторов, приведенных в табл. 8.13,
есть растворимые и гетерогенные системы. Известны катализаторы,
которые можно использовать в водных системах (обычно эмуль-
Стереохимия полимеризации и поликонденсации 545
Таблица 8.13
Стереоспецифичность при полимеризации бутадиена
Каталитическая система Структура полимера Лите- ратура
-—
TiCli- АШз
Al/Ti=0,5 91% транс-1,4- 118
Al/Ti = 1 50% цис-1,4-; 45%транс-1,4- 118
ТЩ. А11<з 95% цис-1,4- 119
Ti(OHh- A1R3 — 100% 1,2- 118
VC13- A1(C2H5)3 99% транс-1,4- 118
VOCI3 или VCI4, А1(С2Н5)3 или AI(G2H5)2G1 95—98% транс-1,4- 118]
У(Ацетилацетонат)3, А1(С2Н5)3 Cr(CAC6Hs)6, А1(С2Н3)3 — 90% st-1,2- 49
А1/Сг = 2 — 100% st-1,2- 118
А1/Сг> 10 — 100% И-1,2- 118
Со2(СО)8, МоС15 99% at-1,2- 120
Co(SCN)2[P(C6H5)3]2, [(C2H5)2A1]2SO4 — 100% st-1,2- 121
CoCL 90% цис-1,4- 122
Хелаты Со, А1(С2Н5)3, п-С1С6Н4СН2С1 99,5% цис-1,4- 123
NiCl2 95% цис-1,4- 122
бис-(л-Кротилникельхлорид) 92% цис-1,4- 124
бис-(.т-Кротилникельиодид) 94% транс-1,4- 124
(л-Циклооктадиен)2№, Ш (л-Цш;лооктадиен)2№, CF3COOH? ~ 100% транс-1,4- 125
CF3COOH/Ni « 1 — 100% цис-1,4- 126
RbCI3, додецилбензолсульфонат натрия 96—99% транс-1,4- 127
спях); это катализаторы на основе родия и других металлов вось-
мой группы [18]. Стереоспецифичность различных катализаторов
сильно зависит от химического строения компонентов катализа-
тора и их взаимного соотношения, от условий реакции и природы
растворителя. Так, при изменении состава каталитической системы
Ci(Ci;H5CN)6-A1(C2H5)3 вместо чисто синдиотактической 1,2-поли-
меризации идет исключительно изотактическая 1,2-полимериза-
Ция. В присутствии смеси бис-циклооктадиенникеля и HI проте-
кает транс-1,4-полимеризация. При замене HI на CF3COOH
направление полимеризации зависит от соотношения обоих ком-
понентов катализатора. Когда они берутся в эквимолярных коли-
чествах, имеет место цис-1,4-полимеризация. При отношении
кислоты и никеля больше единицы в полимере содержатся в оди-
наковом количестве цис-1,4- и транс-1,4-структуры, которые, как
полагают, следуют регулярно друг за другом [126].
546
Глава 8
Высокая стереоспецифичность катализаторов проявляется
также при полимеризации других диенов-1,3, например изопреца
и пентадиена-1,3 (табл. 8.14) [118, 128, 129]. Последний мономер
Таблица 8.14
Стереоспецифичность в полимеризации изопрена и пентадиена-1,3
Каталитическая система Структура полимера Литера- тура
Полимеризация изопрена
Т1С14, А1(С2Н5)3
Al/Ti > 1 96% цас-1,4- 118
Al/Ti < 1 95% траис-1,4- 118
Ti(OR)4, А1(С2Н5)3 95% 3,4- 118
VC13, А1(С2Н5)3 99% траис-1,4- 118
УО(Ацетилацетонат)3, А1(С2Н5)3 90% 3,4- 128
Полимеризация пентадиена-1,3
Т1С1з' или VC13, АШ3 it-транс-! ,4-а 118
Ti(OR)4, А1(С2Н5)з 85% it-цис-! ,4-а 129
118
У(Ацетилацетонат)3, А1(С2Н5)2С1 50% транс-!,4-; 129
50% 1,2-а
Со(Ацетилацетонат)3, А1(С2Н5)2С1 129
бензол 85—90% st-цис-! ,4-6 129
гексан sM, 2-6 129
а Из цис- и транс-пентадиена-1,3.
6 Из тракс-пентадиена-1,3; чкс-мономер не полимеризуется на кобальтовых ката-
лизаторах.
интересен тем, что сам он существует в цис- и транс-форме,
а кроме того, при его 1,4-полимеризации образуются оптические
и геометрические изомеры. Сильная стереоспецифичность различ-
ных катализаторов становится очевидной уже потому, что одни
и те же структуры получаются из цис- и транс-пентадиена-1,3.
Структура полимера определяется конфигурацией аллильной
концевой группы и сильным координационным взаимодействием,
при этом начальная конфигурация мономера не имеет решающего
значения. Полимеризация на кобальтовых катализаторах является
в этом смысле исключением, так как транс-пентадиен-1,3 поли-
меризуется на них, а г^нс-изомер — нет.
Осуществлена также стереоспецифическая полимеризация
1,4-дизамещенных бутадиенов. Тритактические полимеры с эритро-
диизо-транс-тактической структурой получены полимеризацией
сложных эфиров транс-транс-изомеров сорбиновой кислоты
Стереохимия полимеризации и поликонденсации
547
,-стирплакриловой кислоты [130]
где В = СН3, С6Н6, a R = СН3, С2Н6, С4Нд.
8.6в. Катионная полимеризация
При катионной полимеризации диенов-1,3 обычно образуются
сравнительно низкомолекулярные полимеры циклической струк-
туры 1131]. Степень ненасыщенности полимеров, полученных
1,4- или 1,2(или 3,4)-полимеризацией колеблется от 80% при
полимеризации с соответствующими катионными катализаторами
практически до нуля (полимеризация на таких комплексных
катализаторах, как TiCl4-(C2H6)2AlCl или TiCl4-C2H6AlCl2 при
высоких отношениях Ti/Al [131, 132]). Полимеризация на послед-
них катализаторах не относится к полимеризации Циглера —
Натта. Многие обычные катализаторы Циглера — Натта
являются катионными катализаторами, если соединения переход-
ного металла берутся в значительном избытке. Циклические
структуры при полимеризации диенов-1,3 образуются в резуль-
тате различных внутри- и межмолекулярных реакций переноса
и присоединения.
Для объяснения практически однозначного образования
циклополимеров при использовании C2H6AlCl2-TiCl4 Гейлорд
с сотр. [132,133] предложил механизм с переносом заряда, в основу
которого положена реакция типа Дильса — Альдера. Механизм
включает стадии инициирования и роста
Катализатор — С2Н5А1С1+ (8.56)
548
Глава 8
|| + c2h5aici------►
(8.59б)
Катион С2Н6А1С1+ реагирует с мономером с образованием активи-
рованного мономера (катион-радикала), который участвует в ста-
дии роста цепи. Таким образом, весь процесс состоит из реакции
(8.57) с’ последующим ростом, который в общем виде можно выра-
зить следующим образом:
Такая циклополимеризация сопряженных диенов может пред-
ставлять практический интерес как способ синтеза высокотенло-
стойких полимеров лестничного типа (разд. 2.12).
8.7. Другие реакции стереоспецифической
полимеризации
Стереоспецифическая полимеризация возможна не тол ько
с алкенами и диенами, но и с рядом других мономеров, однако
достаточно подробно изучена лишь полимеризация эпоксидов
и карбонильных соединений.
8.7а. Полимеризация эпоксидов
Стереохимия полимеризации окиси пропилена исследована
весьма широко. Изотактический полиоксипропилен получается
под действием таких каталитических систем, как смеси алкилов
цинка или алюминия и воды или спиртов (разд. 7.26.1) [134, 135],
аддукт окиси пропилена и FeCl3 [136], а также в определенных
условиях под действием гидроокисей и алкоголятов металлов [137].
Интересно отметить, что первые сведения об изотактической
полимеризации окиси пропилена на аддукте FeCl3 и окиси пропп-
Стереохимия полимеризации и поликонденсации 549
а относятся к 1955 г. [136], когда появилась и первая работа
ттатта, посвященная синтезу изотактического полипропилена.
Много различных предположений было сделано относительно
природы инициирующих частиц в различных каталитических
системах. Так, инициатору каталитической системы FeCl3
и окиси пропилена приписывается строение [138]
ДОСН2СНСН3)тС1
Cl —Fe
\(ОСН2СНСН3)ПС1
XXXIX
где т+ п = 5. В системах алкил металла — спирт предпола-
гается образование следующих стерео регулирующих частиц:
R
О
RZn —О —ZnR RO —Zn" 'zn—OR
V
i
XL XLI
Однако нет экспериментальных данных, подтверждающих нали-
чие таких структур. Механизм реакции роста также не выяснен,
рассмотрена возможность протекания процесса как по катионно-
координационному, так и по анионно-координационному меха-
низмам.
8.76. Полимеризация карбонильных мономеров
В результате стереоспецифической полимеризации ацеталь-
дегида в присутствии анионно-координационных каталитических
систем образуется изотактический полимер [139]. Такие катали-
тические системы состоят из алкилов цинка и алюминия обычно
в сочетании с водой, спиртом или другими веществами; в качестве
катализаторов применяются также алкилы, гидриды и алкоголяты
Щелочных металлов [140—142]. Аналогичные исследования выпол-
нены для других членов ряда альдегидов, например масляного
альдегида; стереорегулярные полимеры несимметричных кетонов
До сих пор неизвестны.
&& Оптическая активность в полимерах
В изотактических полимерах оптическая активность прин-
ципиально возможна благодаря конфигурации асимметрического
атома как в основной полимерной цепи, так и в боковых группах.
550
Глава 8
Примерами первых являются изотактический полипропилен
полиацетальдегид, полиоксипропилен. В качестве примера поли
меров второго типа приведем поли-З-метилпентен-1
— СН2 —СН —
I
сн3—с-н
I
С2н3
XLII
с асимметрическим атомом углерода в боковой изобутильной груп-
пе. Обычная изотактическая полимеризация таких мономеров ле
приводит к образованию оптически активных полимеров.- Неак-
тивность изотактических полимеров винильных и карбонильных
мономеров, центры потенциальной активности которых находятся
в основной полимерной цепи, объясняется существованием у каж-
дой полимерной молекулы внутренней плоскости симметрии
(разд. 8.1а.1 и 8.1в). Изотактический полиоксипропилен оптически
неактивен по другой причине. В обычном полиоксппропплепе
имеется одинаковое число макромолекул только с R- и только
с 5-конфигурацией, что обусловлено статистической природой
начальной стадии образования изотактической цепи с R- или
5-конфигурацией. По той же причине неактивен изотактический
полимер З-метилпентена-1: половина молекул полимера имеет|
^-конфигурацию боковой группы, а другая половина — 5-конфи-
гурацию. Оптически активные полимеры можно синтезировать
путем использования оптически активных мономеров или опти-
чески активных катализаторов [143 —145].
8.8а. Полимеризация на оптически активных
катализаторах
При использовании катализатора, содержащего оптически
активный компонент, полимеризация изотактически полимери-
зующихся рацемических мономеров, например рацемических
оксипропилена или З-метилпентена-1, часто протекает селективно
с вовлечением в процесс преимущественно одного антипода. Такой
полимер, состоящий преимущественно из одного изомера, обла-
дает оптической активностью. Так, например, оптически актив-
ный полиоксипропилен можно получить с использованием ката-
литической системы диэтилцинк-й-борнеол [146], а различные
поли-а-олефины — с использованием оптически активного 2-метил-
бутиллития вместе с TiCl4 или TiCl3 [143]. Этот процесс известен
под названием стереоселективной полимеризации. По имеющейся
номенклатуре в названии оптически активных полимеров часто
используется приставка R- и 5-, например поли-/?-оксипропилен
Стереохимия полимеризации и поликонденсации
551
й полп-^-оксипропилен, или —[Т?-СН2СН(СН3)О —] п—
я __ [5-СН2СН(СН3)О —]п —. Направления вращения света
ложно показать с помощью знаков «+» и «—» или букв d и Z,
которые ставятся перед названием полимера. Для обозначения
полирацематов используют слово рацемический, например раце-
мический полиоксипропилен.
Стереоселективная полимеризация заключается в таком взаимо-
действии оптически активного катализатора с рацемической
смесью мономеров, которое сопровождается селективным при-
соединением к растущей полимерной цепи только одного из анти-
подов. На ход и степень стереоселективной полимеризации влияют
состав катализатора, природа его оптически активной составляю-
щей н ее оптическая чистота. Истинный механизм стереоселектив-
ной полимеризации неизвестен, так как существует лишь незна-
чительная корреляция между конфигурациями катализатора
п мономера.
Кроме термина «стереоспецифическая полимеризация», иногда
применяют другое, не совсем правильное, название — асимметри-
ческая индукция. Последний термин относится, вообще говоря,
к образованию оптически активного полимера на основе мономера,
не имеющего асимметрического центра, под влиянием находя-
щегося в реакционной среде оптически активного центра. Это
относится, в частности, к 1,4-полимеризации 1-замещенных
п несимметрично 1,4-дизамещенных диенов-1,3 в присутствии
оптически активных катализаторов. Таким способом получены
оптически активные изо-транс-тактический 1,4-полиметил-
Р-шшилакрилат, изо-г^нс-тактический 1,4-полипентадиен-1,3 иэри-
/нро-диизо-тракс-тактический 1,4-полибутилсорбат, причем в ка-
честве катализатора применяли бутиллитий-й-борнилат натрия,
трпэтпл-алюминий-/-тетраментилат титана и бутиллитийЛ-ментил-
этпловый эфир соответственно [147—149].
Другим примером асимметрической индукции является поли-
меризация несимметрических циклических алкенов (разд. 8.1е).
Оптически активные полимеры бензофурана (XLIII)
XL III
получены на катионных катализаторах, состоящих из кислоты
Льюиса (например, А1С13) и оптически активного основания
Льюиса [например, В-фенилаланина и ментил-О8п(С2Н6)3]
1143, 144].
552
Глава 8
8.86. Полимеризация оптически активных
мономеров
По-видимому, полимеризация оптически активных мономе-
ров — наиболее широко использующийся способ синтеза опти-
чески активных полимеров. Этим способом получены полимеры
на основе оптически активных сс-олефинов, например 3- и 4-метп,i-
пентенов-1, простых виниловых эфиров, например ментил- и бор-
нилвиниловых эфиров [150], акриловых и метакриловых эфиров
оптически активных спиртов [151, 152], окиси пропилена [1531.
альдегидов [154] и других мономеров. В результате изотактической
полимеризации оптически активного мономера на стереоспецифи-
ческом катализаторе образуются полимеры с асимметрическими
центрами в полимерной цепи с той же самой конфигурацией.
Полимеризация оптически активных мономеров представляет
интерес не только потому, что таким способом удается получиib
оптически активные полимеры, но и потому, что оптическая
активность мономера может оказывать влияние на тактичное i ь
полимера. В отдельных случаях при полимеризации оптически
активного мономера может быть получен изотактический поли-
мер даже при использовании нестереоспецифических катализа-
торов. Так, в результате полимеризации рацемической окиси
пропилена в присутствии нестереоспецифического' катализатора
КОН образуется атактический полимер, тогда как при использо-
вании оптически активного мономера на том же катализаторе
получается' оптически активный изотактический полимер [155].
При применении стереоспецифического катализатора FeCl3 изо-
тактические полимеры образуются на основе как рацемических,
так и оптически активных мономеров, но только последние дащт
оптически активные полимеры. Асимметрический центр в моно-
мере, введенный в полимер, ориентирует следующий мономер
таким образом, чтобы он имел ту же самую конфигурацию.
Особый интерес представляет сополимеризация рацемического
3,7-диметилоктена-1 с оптически активным З-метилпентеном-1
в присутствии TiCl4-Zn(u3o-C4H9)2 как катализатора [156]. Сополи-
мер, очевидно, является оптически активным относительно обоих
сомономеров, так как оптически активные 3-метилпентеновые
звенья предпочтительно присоединяют только один из антиподов
рацемического сомономера. Подобный эффект асимметрического
центра мономера характерен и для некоторых радикальных реак-
ций сополимеризации. Так, при сополимеризации малеинового
ангидрида с оптически активным сс-метилбензилметакрилатом
в присутствии азо-бпе-изобутиронитрила образуется сополимер,
обладающий оптической активностью даже после гидролиза
[уравнение (8.61)] оптически активной сс-метилбензильной группы
[157]
Стереохимия полимеризации и поликонденсации
553
О
ОсГ со СНз
1 I
_сн-сн-сн2-с*-
I.
со
I
о
I
СН3 —С* — н
I
c6H5 J
XLIV
ОС со сн3
I I I
— сн—сн—сн2—с*— -
I
со
<{)Н _ п
(8.61)
XLV
Вопросы и упражнения
8.1. В чем заключается различие между оптической и гео-
метрической изомерией? Какая связь между ними и молекуляр-
ной структурой исходных соединений? Какие различия в свой-
ствах полимеров обусловлены оптической и геометрической
изомерией? В чем своеобразие полимеров по сравнению с низко-
молекулярными соединениями с точки зрения влияния стерео-
изомерии на свойства?
8.2. Что понимается под термином «тактичность»? Какие суще-
ствуют способы оценки тактичности?
8.3. Изобразите структурные формулы возможных стереорегу-
лярных полимеров на основе следующих мономеров:
1) СН2=СН—СН3,
2) СН2 = С(СН3)2,
3) сн3-сн=сн-сн3,
4) СН2=С(СН3)(С2Н5),
5) СН3-СН = СН —С2Н5,
6) сн2 = сн —сн = сн2,
7) сн3-сн = сн — сн = сн2,
8) СН2 = С(СН3) —СН = СН2,
9) СН3 —СН = С(С1)-СН = СН2,
10) СН3 —СН = СН —С(СН3) = СН2,
И) сн3 —сн=сн—сн = сн —сн3,
о
15) СН3-СН^СН-СН3,
о
16) СН3-СН^СН-С2Н&
17) СН3 —ОНО,
18) (СН3)2СО.
19) СН3-СО-СС13,
12) СН2 = С(СН3)-С(СН3) = СН2, /\/СНз
13)H2N-(CH2)6-NH2+HOOC-(CH2)4-COOH, 21) | ||
14) СН22>сн-СН3,
Назовите различные структуры полимеров в соответствии с номен-
клатурой, изложенной в этой главе.
554
Глава 8
8.4. Что можно сказать о механизмах синдиотактического
и изотактического присоединения при полимеризации пропилена?
Укажите, как условия полимеризации влияют на каждый вид
стереоспецифического присоединения. Рассмотрите вопрос о стерео-
специфичности радикальной, ионной и координационной полиме-
ризаций.
8.5. От каких условий реакции и других факторов зависит, как
идет полимеризация бутадиена: в 1,2-, г^нс-1,4- или транс-1,4-по-
ложение?
8.6. Какие из стереорегулярных полимеров на основе моно-
меров, указанных в вопросе 8.3, могут проявлять оптическую
активность? Как можно синтезировать оптически активные поли-
меры такой структуры? Объясните, почему ряд стереорегулярных
полимеров не может проявлять оптической активности.
8.7. Объясните, в чем заключается различие между терминами
стереорегулярный, стереоспецифический и стереоселективный.
8.8. Что понимают под термином «асимметрическая индукция»?
8.9. Опишите монометаллический и биметаллический меха-
низмы полимеризации Циглера — Натта. Что подтверждает эти
механизмы и что противоречит им?
8.10. Рассмотрите вопрос о применении гомогенных и гетеро-1
генных координационных катализаторов в реакциях полимериза-1
ции пропилена, изопрена, стирола, метилметакрилата и бутил-?
винилового эфира; в чем своеобразие при использовании катали-
заторов различных типов в полимеризации указанных мономеров?
Список литературы
1. Staudinger Н., Die Hochmolekularen Organischen Verbindungen, Sprin-
ger — Verlag, Berlin, 1932.
2. Ziegler K., Holzkamp E., Breil H., Martin H., Angew. Chem., 67, 426,
541 (1955).
3. Natta G., Pino P., Corradini P., Danusso F., Mantica E., Mazzanti G.,
Moraglio G., J. Am. Chem. Soc., 77, 1708 (1955).
4. Natta G., Makromol. Chem., 16, 213 (1955); J. Polymer Sci., 16, 143 (1955);
35, 94 (1960).
5. Natta G.. Science, 147, 261 (1965).
6. Bawn С. E. H., Ledwith A., Quart. Rev. (London), 16, 361 (1962).
7. Lehr M. H., Stereoregular Polymers, in «Survey of Progress in Chemistry»,
Scott A. F. (ed.), vol. 3, Academic Press, Inc., New York, 1966.
8. Ketley A. D. (ed.), The Stereochemistry of Macromolecules, vols. 1 and 2,
Marcel Dekker, Inc., New York, 1967.
9. Beich L., Shindler A., Polymerization by Organometallic Compounds,
Interscience Publishers, John Wiley and Sons, Inc., New York, 1966.
10. Miller M. L., The Structure of Polymers, chap. 8, Reinhold Book Corp.,
New York, 1966.
11. Goodman M., Concepts of Polymer Stereochemistry, in «Topics in Stereoche-
mistry», Allinger N. L. and Eliel E. L. (eds.), vol. 2, p. 73—156, Inter-
science Publishers, John Wiley and Sons, Inc., New York, 1967.
Стереохимия полимеризации и поликонденсации 555
12. Робертс Дж., Касерио М. К., Основы органической химии, изд-во
«Мир», М., 1968.
13. Cahn R. S., Ingold С. К., Prelog V., Experientia, 12, 81 (1956).
14. Huggins М. L., Natta G.', Desreux V., Mark H., Pure and Appl. Chem.,
12, 645 (1966).
15 Natta G., Peraldo M., Farina M., Bressan G., Makromol. Chem., 55, 139
(1962).
16. Cooper W„ Vaughan G., Recent Developments in the Polymerization of
Conjugated Dienes, in «Progress in Polymer Science», Jenkins A. D. (ed.),
vol. 1, Pergamon Press Ltd., 1967.
17. Pino P., Fortschr. Hoch poly met.-Forsch. (Advan. Polymer Sci.), 4,
399 (1966).
18. Natta G., Dall’Asta G., Mazzanti G., Motroni G., Makromol. Chem., 69,
163 (1963).
19. Compostella M., The Manufacture and Commercial Applications of Stereo-
regular Polymers, in [8] vol. 1, chap. 6.
20. 'Youngman E. A., Boor J., Jr., Syndiotactic Polypropylene, in «Macromo-
lecular Reviews», Peterlin A., Goodman M., Okamura S., Zimm В. H.,
Mark H. F. (eds«), vol. 2, p. 33—69, Interscience Publishers, John Wiley
and Sons, Inc., New York, 1967.
21. Brandrup J., Immergut E. H. (eds.), Polymer Handbook, chap. III-l;
III-61, Interscience Publishers, John Wiley and Sons, Inc., New York,
1966.
22. Ketley A. D. (ed.), The Stereochemistry of Macromolecules, vol. 3, Marcel
Dekker, Inc., New York, 1968.
23. Natta G., J. Polymer Sci., 16, 143 (1955).
24. Bovey F. A., Tiers G. V. D., J. Polymer Sci., 44, 173 (1960).
25. Bovey F. A., Accounts of Chem. Research, 1(6), 175 (1968).
26. Hatada K., Ota K., Yuki H., J. Polymer Sci., B5, 225 (1967).
27. Huggins M. L., J. Am. Chem. Soc., 66, 1991 (1944).
28. Fordham J. W. L., J. Polymer Sci., 39, 321 (1959).
29. Fox T. G., Schnecko H. W., Polymer, 3, 575 (1962).
30. Bovey F. A., Hood E. P., Anderson E. W., Kornegay R. L., I. Phys. Chem.,
71, 312 (1967).
31. Ваш С. E. H. et al., J. Polymer Sci., C4, 427 (1963).
32. Talamini G., Vidotto G., Makromol. Chem., 100, 48 (1967).
33. Matsuzaki K., Uryu T., Ishida A., Ohki T., Takeuchi M., J. Polymer Sci.,
A-l(5), 2167 (1967).
34. Murano M., Yamdera R., J. Polymer Sci., B5, 333 (1967).
35. Boor J., Jr., J. Polymer Sci., Cl, 237 (1963).
36. Schildknecht С. E., Zoss A. O., McKinley C., Ind. Eng. Chem., 39, 180
(1947).
37. Natta G., Bassi I., Corradini P., Makromol. Chem., 18—19, 455 (1956).
38. Zambelli A., Natta G., Pasquon I., J. Polymer Sci., C4, 411 (1963).
39. Ziegler K., Angew. Chem. 76, 545 (1964).
40. Boor J., Jr., The Nature of the Active Site in the Ziegler-Type Catalyst,
in «Macromolecular Reviews», Peterlin A., Goodman M., Okamura S.,
Zimm В. H., Mark H. F. (eds.), John Wiley and Sons, Inc., New York,
1967.
41. Hoeg D. F., The Mechanism of Ziegler-Natta Catalysis. I. Experimental
Foundations, in [8] vol. 1, chap. 2.
42. Cosse P., The Mechanism of Ziegler-Natta Polymerization. II. Quantum-
Chemical and Crystal-Chemical Aspects, in [8], vol. 1, chap. 3.
43. Kennedy J. P., Langer A. W., Jr., Fortschr. Hochpolymer.— Forsch.
(Advan. Polymer Sci.), 3, 508 (1964).
44. Natta G., J. Inorg. and Nuclear Chem., 8, 589 (1958).
45. Danusso F., Chim. Ind. (Milan), 44, 611 (1962).
556
Глава 8
46. Matlack А. S., BreslowD. S., J. Polymer Sci., A3, 2853 (1965).
47. Werber F. X., Benning C. J., Wszolek W. H., Ashby G. E., J. Polymer
Sci., A-l(6), 743 (1968).
48. Karapinka G. L., Smith J. J., Carrick W. L., J. Polymer Sci., 50, 143
(1961).
49. Natta G., J. Polymer Sci., 48, 219 (1960).
50. Patat F., Sinn H., Angew. Chem., 70, 496 (1958).
51. Eirich F., Mark H., J. Colloid. Sci., 11, 748 (1956).
52. Huggins M. L., J. Polymer Sci., 48, 473 (1960).
53. Furukawa J., Tsuruta T., J. Polymer Sci., 36, 275 (1959).
54. Cossee P., J. Catalysis, 3, 80 (1964).
55. Bodriguez L. A. M., van Looy H. M., J. Polymer Sci., A-l(4), 1951, 1971
(1966).
56. Coover H. W., Jr., J. Polymer Sci., C4, 1511 (1963).
57. Longi P., Mazzanti G., Boggero A., Lache A. M., Makromol. Chem., 61,
63 (1963).
58. Lenz H. W., Organic Chemistry of Synthetic High Polymers, chap. 15,
Interscience Publishers, John Wiley and Sons, Inc., New York, 1967.
59. Kollar L., Simon A., Kallo A., J. Polymer Sci., A-l(6), 937 (1968).
60. Arlman E. J., Cossee P., J. Catalysis, 3, 99 (1964).
61. Natta G., Chim. Ind. (Milan), 42, 1207 (1960).
62. Natta G., Corradini P., Allegra G., J. Polymer Sci., 51, 399 (1961).
63. Natta G., Pasquon I., Zambelli A., J. Am. Chem. Soc., 84, 1488 (1962).
64. Boor J., Youngman E. A., J. Polymer Sci., A-l(4), 1861 (1966).
65. Zambelli A., Giongo M. G., Natta G., Makromol. Chem., 112, 183 (1968).
66. Kollar L., Simon A., Osvath J., J. Polymer Sci., A-l(6), 919 (1968).
67. Lehr M. H., Macromolecules, 1, 178 (1968).
68. Natta G., Pino P., Mazzante G., пат. США 3197452 (1965).
69. Coover H. W., Jr., McConnell H. L., Joyner F. B., Relationship of Cata-
lyst Composition to Catalyst Activity for the Polymerization of a-Olefins,
in «Macromolecular Reviews», Peterlin A., Goodman M., Okamura S.,
Zimm В. H., Mark H. F. (eds.), vol. I, p. 91—118, Interscience Publishers,
John Wiley and Sons, Inc., New York, 1967.
70. Cooper W., Stereospecific Polymerization, in «Progress in High Polymers»,
Robb J. C., Parker F. W. (eds.), vol. 1, p. 279—333, Academic Press,
Inc., New York, 1961.
71. Zambelli A., DiPietro J., Gatti G., J. Polymer Sci., Al, 403 (1963).
72. Combs H. L., Slonaker D. F., Joyner F. B., Coover H. W., Jr., J. Polymer
Sci., A-l(5), 215 (1967).
73. Ашмор H., Катализ и ингибирование химических реакций, изд-во
«Мир», М., 1966.
74. Natta G., J. Polymer Sci., 34, 21 (1959).
75. Heich L., Stivala S. S., J. Polymer Sci., Al, 203 (1963).
76. Natta G., Pasquon I., The Kinetics of the Stereospecific Polymerization
of a-Olefins, in «Advances in Catalysis and Related Subjects», Eley D. D.,
Seiwood P. W., Weisz P. B. (eds.), vol. XI, p. 2—67, Academic Press,
Inc., New York, 1959.
77. Overberger C. G., Jarovitsky P. A., Mukamal H., J. Polymer Sci., A-l(5),
2487 (1967).
78. Lehmann G., Gumboldt A., Makromol. Chem., 70, 23 (1964).
79. Grieveson В. M., Makromol. Chem., 84, 93 (1965).
80. Bier G., Hoffman W., Lehmann G., Seydel G., Makromol. Chem., 58, 1
(1962).
81. Giannini U., Brucker G., Pellino E., Cassata A., J. Polymer Sci., B5, 527
(1967).
82. Marconi W., The Polymerization of Dienes by Ziegler —Natta Catalysts
in [8], vol. 1, chap. 5.
Стереохимия полимеризации и поликонденсации
557
83. Boor J., Jr., Youngman Е. A., Dimbat М., Makromol. Chem , 90, 26
(1966).
84. Natta G., Dall’Asta G., Bassi J. W., Carella G., Makromol. Chem, 91, 87
(1966).
85. Natta G., Dall’Asta G., Motroni G., J. Polymer Sci., B2, 349 (1964).
86. Pasquon I., Valvassori A., Sartori G., The Copolymerization of Olefins
by Ziegler — Natta Catalysts, in [8], vol. 1, chap. 4.
87. Хэм Дж. E., Сополимеризация, изд-во «Мир», М., 1971.
88. Natta G., Valvassori A., Mazzanti G., Sartori G., Chim. Ind. (Milan), 40,
896 (1958).
89. Natta G., Mazzanti G., Valvassori A., Sartori G., Chim. Ind. (Milan), 40,
717 (1958).
90. Natta G., Mazzanti G., Valvassori A., Pajaro G., Chim. Ind. (Milan), 41,
764 (1959).
91. Mazzanti G., Valvassori A., Sartori G., Pajaro G., Chim. Ind. (Milan),
42, 468 (1960).
92. Cunningham R. E., J. Polymer Sci., A-l(4), 1203 (1966); A-l(5), 243, 251
(1967).
93. Natta G.,Allegra G., Bassi I. W., Corradini P., Ganes P., Makromol. Chem.,
58, 242 (1962).
94. Coover H. W., Jr., McConnell R. L., Joyner F. B., Slonaker D. F.,
Combs R. L., J. Polymer Sci., A4, 2563 (1966).
95. Uagemayer H. J., Edwards M. B., J. Polymer Sci., C2, 731 (1964).
96. Werber F. X., Fortschr. Hochpolymer.-Forsch. (Advan. Polymer Sci.),
1, 180 (1959).
97. Friedlander H. N., Smith W. E., Ross R.J., J. Polymer Sci., 48, 17
(1960).
98. Carrick W. E., Klinker R. W., Bonner E. F., Wartman L. H., Rugg F. M.,
Smith J. J., J. Am. Chem. Soc., 82, 3883 (1960).
99. Braun D., Hemer M., Johnson U., Kern W., Makromol. Chem., 51, 15
(1962).
100. Hatada K., Ota K., Yuki H., J. Polymer Sci., B5, 225 (1967).
101. Fujimoto T., Kawabata N., Furukawa J., J. Polymer Sci., A-l(6), 1209
(1968).
102. Hirahara T., Nakano T., Minoura Y., J. Polymer Sci., A-l(6), 485 (1968).
103. Cram D. J., Kopecky K. R., J. Am. Chem. Soc., 81, 2748 (1959).
104. Ketley A. D., Stereospecific Polymerization of Vinyl Ethers, in [8], vol. 2,
chap. 2.
105. Ohsumi Y. et al., J. Polymer Sci., A-l(5), 849 (1967').
106. Higashimura T., Ohsumi Y., Kuroda K., Okamura S., J. Polymer Sci.,
A-l(5), 863 (1967).
107. Vandenberg E. J., Heck R. F., В reslow D. S., J. Polymer Sci., 41, 519 (1959).
108. Braun D., Stereospecific Polymerization of Vinyl-Type Monomers and
Dienes by Alkali-Metal-Based Catalysts, in [8], vol. 2, chap. 1.
109. Condon F. E., J. Polymer Sci., 11, 139 (1953).
110. Richardson W. S., J. Polymer Sci., 13, 229 (1954).
111. Pollock D. J., Elyash L. J., DeWitt T. W., J. Polymer Sci., 15, 86 (1955).
112. Craig D., Ship man J. J., Fowler R. В., J. Am. Chem. Soc., 83, 2885 (1961).
113. Tobolsky A. V., Rogers С. E., J. Polymer Sci., 40, 73 (1959).
114. Rembaum A., Ells F. R., Morrow R. C., Tobolsky A. V., J. Polymer Sci.,
61, 155 (1962).
115. Sterns R. S., Forman L. E., J. Polymer Sci., 41, 381 (1959).
116. O'Driscoll K. F., Yonezawa T., Higashimura T., Macromol. Chem., 1, 1
(1966).
117. Morton A. A., Lampher E. G., J. Polymer Sci., 44, 233 (1960).
118. Natta G. et al., Chim. Ind. (Milan), 40, 362 (1958), 41, 116, 398, 526, 1163
(1959); Makromol. Chem., 77, 114, 126 (1964).
558
Глава 8
119. Saltman W. M., Link T. H., Ind. and Eng. Chem. Prod. Res. and Dev
3, 199 (1964).
120. Otsuka S., Kawakami M., Angew. Chem., Intern. Ed. Engl., 2, 618 (1963).
121. Iwamoto M., Yuguchi S., J. Polymer Sci., B5, 1007 (1967).
122. Anderson W. S., J. Polymer Sci., A-l(5), 429 (1967).
123. Takahashi A., Takahashi K., Hirose T., Kambara S., J. Polymer Sci
B5, 415 (1967).
124. Matsumoto T., Furukawa J., J. Polymer Sci., B5, 935 (1967).
125. Durand J. P., Dawans F., Teyssie P., J. Polymer Sci., B5, 785 (1967).
126. Durand J. P., Teyssie P., J. Polymer Sci., B6, 299 (1968).
127. Rinehart R. E., Smith H. P., Witt H. S., Romeyn H., Jr., J. Am. Chem.
Soc., 83, 4864 (1961); 84, 4145 (1962).
128. Wilke G., Angew. Chem., 68, 306 (1956).
129. Natta G. et al., J. Polymer Sci., 51, 463 (1961); Bl, 67 (1963), Makromol.
Chem., 51, 229 (1962); 53, 52 (1962); European Polymer J., 1, 81 (1965);
5, 1 (1969).
130. Natta G., Farina M., Donati M., Peraldo M., Chim. Ind. (Milan), 42,
1363 (1960).
131. Купер У., Полиены, в кн. «Катионная полимеризация», под ред.
П. Плеша, гл. 8, изд-во «Мир», М., 1966.
132. Gaylord N. G. et al., J. Polymer Sci., A-l(4), 2493 (1966), A-l(6); 125
(1968).
133. Gaylord N. G., Svestka M., J. Polymer Sci., B7, 55 (1969).
134. Tsuruta T„ Inoue S., Tsubaki K., Makromol. Chem., Ill, 236 (1968).
135. Saegusa T., Imai H., Matsumoto S., J. Polymer Sci., A-l(6), 459 (1968).
136. Pruitt M. E„ Raggett J. M., пат. США 2706182 (1955).
137. Tsuruta T., Stereospecific Polymerization of Epoxides, in [8], vol. 2,
chap. 4.
138. Colclough R. O., Gee G., Higginson W. С. E., Jackson J. B., Litt M.,
J. Polymer Sci., 34, 171 (1959).
139. Pregaglia G. F., Binaghi M., in [8], vol. 2, chap. 3.
140. Natta G., Mazzanti G., Corradini P., Bassi I. W., Makromol. Chem., 37,
156 (1960).
141. Furukawa J., Saegusa T., Fujii H., Kawasaki A., Imai H., Fujii Y., Makro-
mol. Chem., 37, 149 (1960).
142. Tani H., Yasuda H., J. Polymer Sci., B7, 17 (1969).
143. Pino P., Fortschr. Hochpolymer-Forsch. (Advan. Polymer Sci.), 4, 393
(1965).
144. Farina M., Bressan G., «Optically Active Stereoregular Polymers», in
[8], vol. 3, chap. 4.
145. Goodman M., Abe A., Fan Y-L., Optically Active Polymers, in «Macromole-
cular Reviews», Peterlin A., Goodman M., Okamura S., Zimm В. H.,
Mark H. F. (eds.), vol. 1, p. 1—34, Interscience Publishers, John Wiley
and Sons, Inc., New York, 1967.
146. Kumata Y., Furukawa J., Saegusa T., Makromol. Chem., 105, 138 (1967).
147. Tsunetsugu T., Fueno T., Furukawa J., Makromol. Chem., 112, 220 (1968).
148. Natta G., Valenti S., Makromol. Chem., 67, 225 (1963).
149. Natta G., Donati M„ Makromol. Chem., 43, 251 (1961).
150. Liquori A. M., Pispisa B., J.., Polymer Sci., B5, 375 (1967).
151. Sobue H., Matsuzaki K., Nakano S., J. Polymer Sci., A2, 3339 (1964).
152. Yuki H. et al., J. Polymer Sci., A-l(6), 829 (1968).
153. Tsuruta T., Inoue S., Yoshida N., Yodota Y., Makromol. Chem., 81, 258
(1965).
154. Abe A., Goodman M., J. Polymer Sci., Al, 2193 (1963).
155. Price С. C., Spector R., J. Am. Chem. Soc., 87, 2069 (1965).
156. Ciardelli F., Carlini C., Montagnoli G., Macromolecules, 2, 296(1969).
157. Beredjick N., Schuerch C., J. Am. Chem. Soc., 80, 1933 (1958).
ГЛАВА 9
ХИМИЧЕСКИЕ РЕАКЦИИ ПОЛИМЕРОВ
В предыдущих главах были рассмотрены полимеризационные
и полпконденсационпые методы синтеза полимеров. Кроме них,
для получения новых полимеров используется метод химической
модификации известных полимеров [1], которая включает такие
реакции, как получение сложных и простых эфиров целлюлозы,
гидролиз поливинилацетата, хлорирование полиэтилена, а также
сшивание ненасыщенных сложных полиэфиров, 1,4-полидие-
нов-1,3 и полисилоксанов. Рассмотрению таких реакций посвя-
щена эта глава, причем основное внимание будет уделено реак-
циям, нашедшим практическое применение.
Не будут обсуждаться различные реакции деструкции (хими-
ческие, термические, фотолитические, радиационные), которые
имеют большое, но самостоятельное значение в химии полимеров.
9.1. Реакционная способность полимеров
Полимеры вступают в те же самые реакции, что и их низко-
молекулярные гомологи. Ацетилирование гидроксильных групп
целлюлозы протекает практически так же, как и ацетилирование
этилового спирта, хлорирование полиэтилена подчиняется тем же
закономерностям, что и хлорирование гексана. Обычно принимают,
что реакционная способность функциональных групп в полимере
и низкомолекулярном соединении одинакова. Это уже знакомая
нам концепция независимости реакционной способности функцио-
нальных групп от размера молекулы (разд. 2.1), на которой
основан анализ кинетики поликонденсации. Однако можно при-
вести много примеров реакций, в которых скорость и максималь-
ная степень превращения для функциональных групп полимеров
существенно отличаются от таковых для соответствующих гомоло-
гов низкого молекулярного веса. Как правило, в реакциях поли-
меров скорость и степень превращения ниже, хотя известны
и обратные явления.
Зависимость реакционной способности функциональной группы
от размера молекулы определяют следующие факторы [1,2].
560
Глава 9
1. Кристалличность. При проведении реакции в условиях,
способствующих сохранению кристаллической фазы в полимере,
реакционноспособны функциональные группы только аморфной
части полимера. Функциональные группы в кристаллической
фазе, как правило, недоступны химическим реагентам, и реак-
ция лимитируется аморфными областями. Полимер и его низко-
молекулярный гомолог реагируют одинаково при определенных
температурах в растворе соответствующих растворителей.
2. Изменение растворимости. Аномальное поведение иногда
наблюдается в реакции с полимером, проводящейся сначала
в гомогенных условиях, но сопровождающейся изменением физи-
ческой природы системы. В результате реакции может получаться
полимер, уже нерастворимый в реакционной среде или образую-
щий очень вязкую систему. В таких случаях степень превращения
достигает некоторой максимальной, предельной величины, причем
с ростом степени завершенности реакции скорость ее может даже
уменьшаться. В отдельных случаях наблюдалось и повышение
скорости реакции при уменьшении растворимости полимера в ходе
процесса, что вызвано специфическими особенностями определен-
ных реакций.
3. Изоляция функциональных групп. В тех случаях, когда при!
реакции на полимере необходимо участие двух соседних функцио-J
нальных групп, существует максимально возможная степень:
завершенности реакции, которая определяется количеством изоли-<
рованных единичных функциональных групп между парами
реагирующих групп. Этот эффект был уже рассмотрен в разд. 3.26
на примере таких реакций, как дехлорирование поливинилхлорида
и окисление поливинилового спирта перйодатами. Для неравно-
весных реакций рассчитанная максимальная степень превращения
составляет 86,5% [3], что хорошо согласуется с экспериментально
найденными значениями для многих систем. Для равновесных
реакций эта величина становится выше, но время реакции, необ-
ходимое для ее полного завершения, часто практически недости-
жимо.
4. Эффект соседней группы. На реакционную способность
функциональной группы может влиять соседняя с ней функцио-
нальная группа [1, 4]. В отдельных случаях прореагировавшая
функциональная группа изменяет реакционную способность
соседней свободной функциональной группы. Такие эффекты
обусловлены стерическими и/или электростатическими взаимо-
действиями между соседними группами и могут приводить как
к замедлению, так и к ускорению реакции. Так, при омылении
ряда гомо- и сополиметакрилатов вскоре после начала реакции
наблюдается аутокаталитический эффект. Вслед за первоначаль-
ным образованием карбоксилатных анионов гидролиз сложно-
эфирных групп идет не в результате атаки гидроксильной группы,
Химические реакции полимеров
561
а под действием соседних
идет через промежуточное
карбоксилатных анионов [5]. Реакция
образование циклического ангидрида.
сн3 сн3
•^сн,—с—сн,—с----
со со
I I
о о
(9.1)
Повышение скорости реакции под влиянием соседней группы,
известное под названием анхимерное ускорение, проявляется
главным образом тогда, когда возможно промежуточное замыка-
ние 5- и 6-членных циклов. Такой эффект имеет место и в низко-
молекулярных бифункциональных соединениях, например слож-
ных эфирах янтарной кислоты. Известны также отрицательные
эффекты соседней группы. Пониженные скорости реакции и менее
чем 100%-ная степень превращения наблюдаются в некоторых
случаях вследствие электростатического отталкивания между
химическим агентом и реагирующей функциональной группой,
когда оба они имеют одинаковый заряд. Гидролиз полиметакрил-
а.чпда в сильноосновных растворах протекает не более чем на 70%
вследствие отталкивания гидроксильного иона карбоксилатным
анионом, когда метакриламидные фрагменты окружены гидроли-
зованными группами [6].
Влияние соседней группы определяется не только природой
функциональной группы и типом реакции, но и стереохимией
соседней группы. Так, изотактический полиметилметакрилат
гидролизуется быстрее синдиотактического или атактического
полимеров [7]. В изотактическом полимере соседние функциональ-
ные группы расположены наиболее благоприятно с точки зрения
их взаимодействия друг с другом и образования промежуточного
Циклического ангидрида.
5. Конформация макромолекул. Реакционная способность
Функциональной группы зависит также от конформации полимер-
ных цепей. От того, какая форма полимерной цепи — клубок
или спираль — и насколько плотный или рыхлый клубок, может
562
/
Глава 9
зависеть возможность атаки функциональной группы химическим
реагентом. Реализация тех или иных конформаций зависит от сте-
реохимии полимерных цепей и природы применяющегося в реак-
ции растворителя.
9.2. Сшивание
Некоторые реакции сшивания, например отверждение продук-
тов взаимодействия диолов и эпоксидов, уже были описаны
в разд. 2.10а и 2.106. В этом разделе будет рассмотрено отвержде-
ние ненасыщенных сложных полиэфиров, сшивание 1,4-полидие-
нов-1,3, полиолефинов и полисилоксанов. Изучение кинетики
подобных процессов часто весьма затруднено из-за нераствори-
мости конечного трехмерного продукта. Во многих случаях такие
процессы идут с понижением скорости во времени и не с очень
высоким выходом.
9.2а. Отверждение ненасыщенных сложных
полиэфиров
9.2а.1. Сополимеризация
По своим закономерностям отверждение ненасыщенных слож-
ных полиэфиров имеет много общего с сшиванием других форполи-
меров (разд. 2.10а и 2.106). Низкомолекулярным полимером (как
правило, в виде вязкой жидкости) заполняют нужную форму
и превращают его в трехмерное твердое вещество сшиванием.
Отверждение ненасыщенных сложных полиэфиров достигается
сополимеризацией их по двойным связям с такими ненасыщенными
мономерами, как стирол, метилметакрилат, винилтолуол или
диаллилфталат [8—10].
— ООС —СН = СН —СОО-h СН2 = СНА —>
— ООС-СН — СН — СОО-оос-сн—сн-соо—
-» (Ан2-CHA)m (СН2 — СНА)т
I I
— ООС —СН-СН-СОО----ООС —СН —СН —СОО —
I
(9.2)
Механические свойства сшитого полимера зависят от числа
и длины поперечных связей. Последние в свою очередь опреде-
ляются природой мономера или, точнее, особенностями сополи-
меризации полиэфира и мономера. Так, при сополимеризации
полиэфира, содержащего остатки фумаровой кислоты, со стиролом
Химические реакции полимеров
563
бпазгется более твердый и прочный полимер, чем при сополи-
° ппзацпп с метилметакрилатом. При сополимеризации поли-
хууарата со стиролом образуются более регулярно чередующиеся
полимеры, чем при взаимодействии полифумарата с метил-
метакрилатом (г, 2,2, г2 = 0,062 вместо г, = 0,50, г2 = 1,05)
[91 В результате метилметакрилат дает небольшое число длинных
поперечных связей (т велико в I), тогда как при сополимеризации
со стиролом образуется большое число коротких поперечных
связей (т мало). Высокоплотные полимеры получаются при
использовании в качестве сомономера аллильных соединений,
которые при полимеризации дают короткие поперечные связи
(разд. 3.7в).
Интересно сравнить результаты по отверждению полималеатов
и полифумаратов. Приведенные в табл. 6.2 отношения реакцион-
ных способностей свидетельствуют о том, что двойная связь
малеиновых фрагментов менее склонна к сополимеризации, чем
двойная связь фумаратов. С этих позиций отверждение полималеа-
тов сополимеризацией с непредельными мономерами должно
было бы быть малоэффективным. Однако, поскольку при образо-
вании полиэфира имеет место изомеризация до глубоких степеней
превращения (~90%) остатков малеиновой кислоты в фумаро-
вую [11]. то отверждение идет весьма легко.
9.2а.2. Сшивание кислородом
Ненасыщенные сложные полиэфиры, содержащие непредель-
ные связи в остатках жирной кислоты, например олеиновой (II)
пли линолевой (III), называются «алкидами».
СНз—(СН2)7—СН = СН —(СН2)7—соон
II
СН3—(СН2)4 —СН = СН —СН = СН —(СН2)7 —соон
III
Алкиды отверждаются окислением кислородом воздуха. Процесс
называется высыханием. Пропиточные составы и другие поверх-
ностные покрытия, основанные на жирных кислотах или их гли-
цератах. сшиваются аналогичным образом.
Сшивание идет по-разному в зависимости от того, изолирован-
ные (как в олеиновой кислоте) или сопряженные (как в линолевой
кислоте) двойные связи содержат алкиды [8, 12]. Сшивание по изо-
лированным двойным связям идет через промежуточное образо-
вание аллильной гидроперекиси
о2
— СН2 —СН = СН—-------> —СН — СН = СН— (9.3)
ion
564
Глава 9
с последующим ее разложением по схеме
— ООН —>—О- + НО., (9Д)
2—ООН —>—ОН----------ОО- + Н2О, (9.5)
——O.-J-—— Н — •+-—ОН, (9.6)
НО-Н----Н —>------|~Н2О, ' (9.7)
2---(9.8)
— ОН------—> —о—, (9.9)
2---О-—»-----О — О—, (9.10)
где — ООН и —Н — окисленные и неокисленные формь^
макромолекул. Для повышения скорости отверждения реакцию;
проводят в присутствии катализаторов — ионов металлов^
(кобальта, марганца, железа, олова, цинка), вводимых в составе!
солей карбоновых кислот. Ионы металлов ускоряют разложение’
гидроперекисей аналогично тому, как это происходит в окисли-;
тельно-восстановительных системах (разд. 3.4е). Ионы могут
также катализировать образование гидроперекисей. Относитель-'
ные количества углерод-углеродных, простых эфирных и пере-
кисных сшивок зависят от условий отверждения.
Сшивание сопряженных систем обычно идет через промежу-
точное образование и разложение циклических перекисей
СН
02
— сн = сн—сн = сн—------->—сн сн
сн—
o'
— СН — СН = СН — СН— (9.11)
00-
дающих радикалы, инициирующие сшивание в результате 1,4-по-
лимеризации полимерных молекул.
9.26. Сшивание эластомеров на основе диенов-1,3
1,4-Полимеры изопрена, бутадиена й хлоропрена и ряд их
сополимеров (бутадиенакрилонитрильный, бутадиенстирольпый,
изопренизобутиленовый) составляют большую группу полимеров,
использующихся в качестве эластомеров. Сшивание является
совершенно обязательным требованием для получения эластоме-
ров, обладающих быстрым и полным восстановлением размеров
после снятия напряжения. В технологии эластомеров процесс
сшцвания называется вулканизацией. Вулканизация таких гомо-
Химические реакции полимеров
565
иолимеров диенов-1,3 и их сополимеров осуществляется с помощью
серы, перекисей или ионизирующего облучения [13]. В промыш-
ленности для вулканизации таких полимеров применяется прак-
тически только сера.
9.26.1. Вулканизация серой
Хотя со времени открытия Гудьиром в 1839 г. вулканизации
каучука серой прошло уже более ста лет, механизм этой
реакции до конца не понят [1, 13, 14]. Ранее для нее предлагали
свободнорадикальный механизм, но есть экспериментальные
доказательства ионного пути [15, 16]. Ингибиторы радикального
действия не влияют на вулканизацию серой, при исследовании
сшитых систем методом электронного парамагнитного резонанса
свободны? радикалы в них не обнаружены. Вместе с тем вулкани-
зация серой ускоряется органическими кислотами и основаниями,
а также высокополярными растворителями. Ионный процесс
сшивания можно описать цепной реакцией, включающей перво-
начальное образование сульфониевого иона (IV) при взаимодей-
ствии полимера с поляризованной серой или ее ионной парой:
д е+ б-
S8----> Sm --.Srl или S++S"
| — сн2-сн=сн-сн2--
— СН2-СН — СН — СН2--FS- (9.12)
+s'm
IV
Сульфониевый ион реагирует с полимером с гидридным отще-
плением [уравнение (9.13а)] или переносом протона [уравне-
ние (9.136)].
— СН2-СН2—СН —СН2 —
I
Sm
Полимер
— СН2-СН-СН-СН2—----------->+ (9.13а)
+$т —сн—сн = сн-сн2 —
V
Полимер
+ (9.136)
— СН2-СН-СН = СН------1--сн2-сн2-сн-сн2— v '
I VI
s
Полимерные катионы V и VI подвергаются сшиванию, реаги-
руя сначала с серой, а затем присоединяясь к двойной связи
566
Глава 9
полимера. При последующем протонном [уравнение (9.15а)]
или гидридном перемещении [уравнение (9.156)] регенерируются
соответствующие полимерные катионы. Для полимерного катио-
на V это можно представить следующим образом:
+ Se
— GHGH = СНСН2 —--------> — GHCH = GHGH2 —
j, Полимер
— CHCH=CHCH2------
| —CHCH=CHCH2 —
‘"’m I
I ‘"’m
---CH2C = CHCH2— Полимер I
+ .<---------CH2-CH —CHCH2 (9.14)
— CH2CH2 —CHCH2— VII
+
(9.15a)
j, Полимер
— CHCH = CHCH2 —
I
I
— CH2CHCH2CH2 —
+
— CH —CH = CHCH2— (9.156)
9.26.2. Ускоренная вулканизация серой
Вулканизация диеновых полимеров нагреванием с серой —
очень малоэффективный процесс, причем приблизительно 50 ато-
мов серы вводятся в полимер в составе одной мостиковой группы.
Сера расходуется на образование длинных полисульфидных
мостиков (большие т в VII), вицинальных поперечных связей
(VIII), которые оказывают такое же влияние на свойства полиме-
ров, что и одинарные связи, и внутримолекулярных циклических
сульфидов (IX)
— СН2СН —СНСН2 —
— СН2(^Н —Ансн2 —
VIII
сн
— сн2сн \н
s —(1н-сн2 —
IX
Химические реакции полимеров
567
11а практике вулканизация серой проводится в присутствии
различных добавок, ускоряющих процесс и повышающих его
эффективность. К таким добавкам, называемым ускорителями,
относятся различные серусодержащие соединения, например
гпурамсульфиды (X), дитиокарбаматы (XI) и бензтиазолы (ХП),
а также, соединения типа арилгуанидинов
S S
II II
R2NC —S—S —CNR2
X
s
II
(R2NC — S)2Zn
XI
хп
При применении лишь одних ускорителей не достигается очень
большого увеличения эффективности сшивания. Максимальный
эффект имеет место только при использовании в определенном
сочетании ускорителей, окисей металлов и органических кислот.
Последние обычно называют активаторами, среди них наиболее
распространены окись цинка и стеариновая кислота. Анализ
сшитых полимеров показывает, что при вулканизации в присут-
ствии смеси активаторов и ускорителей расход серы резко умень-
шается. Эффективность сшивания не превышает в отдельных
случаях 2 атомов серы на одну мостиковую связь. Большинство
сшивок представляют собой моно- или дисульфидные мостики
с очень низким содержанием вицинальных и циклических суль-
фидных групп [14—18]. Несмотря на очевидные преимущества
такой ускоренной вулканизации, о механизме ее известно очень
немногое.
9.2в. Сшивание полиолефинов и поли силоксанов
Сшивание полиэтилена, сополимеров этилена и пропилена
п полисилоксанов проводят путем смешивания их с перекисями,
например с перекисью дикумила или ди-тпретп-бутила, с после-
дующим нагреванием такой смеси [13]. Сшитый полиэтилен
остается твердым материалом даже при температурах, при кото-
рых обычный полиэтилен плавится и течет. Особое значение
сшивание приобретает для получения эластомеров из полисило-
ксанов и сополимеров этилена и пропилена. Сшивание пере-
кисями включает образование полимерных свободных радикалов
в результате отщепления водорода радикалами, образующимися
при разложении перекиси. Сшивание происходит путем реком-
568
Глава 9
бинации двух радикалов
ROOR 2RO-,
RO.-I--СН2СН2-----> ROH + —СН2СН —, ('и-,
— СН2СН —
2—СН2СН— | (9.18;
— СН2СН —
Максимальная эффективность сшивания в таком процессе
составляет одну сшивку на молекулу разложившейся перекиси,
что значительно меньше, чем при полимеризации, где один радикал
дает начало превращению большого числа молекул мономера.
В действительности же эффективность сшивания часто бывает
ниже из-за различных побочных реакций инициатора (разд. 3.4е.2)
и полимерного радикала. Так, если полимерный радикал обра-
зуется не вблизи другого полимерного радикала,' то сшивание
будет затруднено, так как процесс идет в очень вязкой, твердой
системе. Возможны также такие побочные реакции, как разрыв
цепи, отщепление водорода, соединение с радикалом инициатора
и т. п. Наряду с высокой стоимостью перекисей все это сущест-
венно снижает практическую ценность сшивания перекисями.
Для повышения эффективности сшивания перекисями могут
быть использованы различные способы. Более эффективного
сшивания полисилоксанов можно достичь введением небольшого
количества винильных групп в обычные полидиметилсилоксановые
или полифенилметилсилоксановые полимеры, что достигается
сополикондепсацией соответствующих соединений с небольшими
количествами винилметилсиланола
СН2 СН2
СН3 СН СН3
НО — Si — ОН + НО — Si —ОН -» —- O-Si-0-Si-O— (9.19)
<4н3 (^Н3 СН3 СН3
Сшивание перекисями такого сополимера значительно более
эффективно, чем сшивание гомополимера диметплсиланола
(табл. 9.1) [19]. Процесс сводится к цепной реакции полимериза-
ции боковых винильных групп полисилоксановых цепей. Анало-
гичную модификацию эластомерных сополимеров этилена п про-
пилена проводили путем синтеза тройных сополимеров с неболь-
шим содержанием несопряженных диенов, например дицпклопен-
тадиена и гексадиена-1,4 [20, 21]. Образующиеся сополимеры
содержат свободные двойные связи, которые также можно уско-
ренно вулканизовать серой, аналогично тому как это делается для
1,4-полидиенов-1,3.
Химические реакции полимеров
569
Таблица 9.1
Эффективность сшивания полидиметилсилоксаиа
перекисью 2,4-дихлорбензоила [19]
Количество винильного сомономера, мол. % Число сшивок на молекулу разложившейся перекиси при концентра- ции перекиси (%)
0,74 1,47
0,0 0,31 0,19
0,1 0,80 0,42
0,2 1,00 0,63
9.2г. Другие процессы сшивания
Для модификации полимеров используется и ряд других
реакций сшивания [1, 13, 14]. Трехмеризацию эластомеров
на основе сополимеров винилиденфторида и гексафторпропилена
проводят нагреванием их с диаминами и окислами. Процесс
включает- дегидрофторирование с последующим присоединением
диамина [уравнение (9.20)] и/или реакцией Дильса — Альдера
[уравнение (9.21)]
CF3
I -HF
—- CH2CF2CH2CF2CF2CF—-----> —CH = CFCH2CF2CF(CF3)'—
XIII
h2n-r-nh2
— CH = CFCH = CFCF(CF3) —
XIII
CH — CF — CH2CF2CF(CF3) —
/ XCF(CF3) —
(9.21)
— CH2CFCH2CF2CF(CF3) —
NH
R
I
NH
— CH2CFCH2CF2CF(CF3) —
(9.20)
Для сшивания полиуретановых эластомеров применяются пере-
киси, диолы и диамины. Для уменьшения сминаемости целлю-
570
Глава 9
лозные волокна сшивают путем взаимодействия гидроксильных
групп целлюлозы с диметилолмочевиной, диметилолэтиленмочевц-
ной, мочевино- и меламиноформальдегидными форполимерами
и другими реагентами.
В последние годы широкое распространение для ряда поли-
меров получило радиационное сшивание [22]. Реакция анало-
гична сшиванию под действием перекисей, за исключением того
что полимерные радикалы образуются в результате воздействия
на полимер ионизирующего облучения (например, электронов,
у-лучей и нейтронов). (В процессе сшивания могут также идти
ионные реакции с образованием соединений ионного типа.)
До настоящего времени радиационная сшивка практически
не используется в промышленности, исключение составляет,
пожалуй, модификация полиэтилена и его сополимеров.
9.3. Реакции целлюлозы
Специфический комплекс свойств, присущих целлюлозе, пе(
позволяет использовать природный полимер без соответствую-^
щей обработки. Очень сильное межцепное взаимодействие, обус-j
ловленное водородными связями, приводит к тому, что целлюлоза '
как высококристаллический нерастворимый полимер разлагается
при высоких температурах не плавясь. Из-за этого нативную '
целлюлозу нельзя перерабатывать в те или иные изделия. Приме-
нение же ее в очень больших количествах при производстве
волокон и пластмасс основано на использовании различных хими-
ческих превращений, приводящих к изменению ее свойств [23, 24].
9.3а. Растворение целлюлозы
Формование целлюлозы в пленки и волокна основано на пре-
вращении ее в растворимые производные. Для этого измельчен-
ную целлюлозу (древесную пульпу) обрабатывают при комнатной
температуре концентрированным водным раствором щелочи
(обычно 18%-ным NaOH). Большая часть NaOH удерживается
в свободном виде в набухшем полимере, часть его расходуется
на образование алкоголятов целлюлозы.
Избыток щелочи отжимают и выдерживают массу в течение
нескольких дней для ускорения окислительной деструкции цепей
до нужной степени полимеризации. Затем щелочную целлюлозу
обрабатывают сероуглеродом, превращая в ксантогенат целлю-
лозы [уравнение (9.22)]. Как правило, степень ксантогенирования
целлюлозы в промышленности весьма низка и составляет при-
мерно 0,5 ксантогенатной группы на три гидроксильные группы,
что обусловлено тем, что реакция происходит в гетерогенных
Xимические реакции полимеров
571
S
(9.23)
условиях. Однако этого оказывается достаточно для придания
целлюлозе растворимости. Пленки и волокна получают формо-
ванием вязкого щелочного раствора ксантогената целлюлозы
в ванне с серной кислотой. Серная кислота гидролизует ксанто-
генат до ксантогеновой кислоты, которая, будучи неустойчивой,
разлагается (без выделения) с регенерацией целлюлозы [урав-
нение (9.23)], нерастворимой в водной среде [25]. В результате
образуется пленка или волокно целлюлозы. Весь этот процесс
называется вискозным методом.
В некоторых случаях применяется другой, медноаммиачный
метод. Он заключается в растворении целлюлозы в аммиачном
растворе окиси меди. Растворимость целлюлоза приобретает,
по-видимому, в результате образования комплекса между ионом
меди и гидроксильными группами целлюлозы. Регенерацию цел-
люлозы проводят обработкой сформованного волокна или пленки
кислотой или щелочью.
9.36. Образование сложных эфиров
В промышленности производятся ацетат, пропионат, ацето-
пропионат, ацетобутират и нитрат целлюлозы. Наиболее важным
из них до настоящего времени остается ацетат целлюлозы, кото-
рый получают ацилированием целлюлозы уксусной кислотой
в присутствии сильного кислотного катализатора (обычно H2SO4
572
Глава 9
или НСЮ4)
Целл —ОН-|-НОСОСН3 Т4* Целл —OCOCH3-j-Н20. (9.24)
Равновесие реакции смещают вправо, связывая воду уксусным
ангидридом (или хлористым ацетилом), который добавляется
в реакционную смесь.
По свойствам диацетат целлюлозы часто бывает лучше, чем
полностью ацилированный продукт (триацетат). Прямой синтез
диацетата не получил широкого распространения, так как обычно
из-за гетерогенности процесса образуется смесь продуктов. Часть
целлюлозных цепей может проацилироваться полностью, тогда
как другие могут быть совершенно незатронуты. Поэтому ди аце-
тат получают регулируемым гидролизом триацетата. Триацетат
растворим в реакционной смеси, что обеспечивает возможность
получения однородного продукта. Регулируемый гидролиз триаце-
тата проводят водой или разбавленной уксусной кислотой. При
этом, хотя затруднения, вызываемые гетерогенностью процесса
этерификации, удается преодолеть путем гидролиза триацетата,
конечный продукт может быть неоднородным по распределению
ацетильных групп между тремя возможными реакционноспособ-
ными положениями в молекуле целлюлозы, из которых два —
вторичные гидроксильные группы, а третья — первичный гидро-
ксил. Более реакционноспособной является сложноэфирная группа
первичного спирта, поэтому при гидролизе триацетата остается
преимущественно диацетат по вторичным гидроксилам.
9.Зв. Образование простых эфиров
Метиловые и этиловые эфиры целлюлозы получают взаимо*
действием щелочной целлюлозы с соответствующим алкилгало-
генидом
Целл —ОН + NaOH+ RC1 —> Целл —OR + NaCl + H2O. (9.25)
9.4. Реакции поливинилацетата
Из поливинилацетата, на основе которого изготавливаю^
пластмассы, получают два полимера, которые нельзя сннтези^
ровать непосредственно из мономеров, так как последние неизй
вестны [24]. Один из них — поливиниловый спирт — образуется!
при алкоголизе поливинилацетата метанолом или этанолом J
СН3 (
СО (
I 1
О ОН 5
I СНзОН I J
— СН2 —СН—------------>— СН2 —СН— (9.26
2 -СНзСООСНз 2 ' '
Химические реакции полимеров
573
Скорость реакции повышают кислоты и основания, при этом
предпочтение отдается основаниям, в присутствии которых реак-
ция идет с большей скоростью и отсутствуют побочные процессы.
При взаимодействии поливинилового спирта с альдегидом
образуется поливинилацеталь
СН2 СН2
/ \ RCHO / \
— СН,СН СН—-------->—СН2СН СН— (9.27)
'I I “Н2° I I
ОН ОН 0 0
\н
I
R
Реакцию обычно проводят в водном растворе в присутствии кислого
катализатора. Образование ацеталя не идет с количественным
выходом вследствие образования изолированных единичных гид-
роксильных групп между ацетальными циклами. Наиболее важ-
ными представителями этого класса полимеров являются формаль
и бутираль (R = — Ни — С3Н7 соответственно).
9.3. Галогенирование
9.5а. Натуральный каучук
Хлорирование и гидрохлорирование натурального каучука —
промышленные процессы, которые проводят в гомогенных усло-
виях с использованием растворов каучука в хлорсодержащих
пли ароматических растворителях [1, 14]. Процесс гидрохлори-
рования идет по ионному механизму, причем присоединение про-
текает по правилу Марковникова с образованием продукта,
содержащего галоген у третичного углеродного атома [уравне-
ние (9.28)]. Входе реакции происходит также частичная циклиза-
ция протонированного промежуточного соединения (XIV) (разд. 9.7)
СН3 СН3
। н+ I
— СН2С = СН —сн2 —-->сн2 —с —сн2—сн2 —
+ XIV
|а-
сн3
I
— СН2 — С — СН2 — СН2— (9.28)
С1
Хлорирование натурального каучука включает несколько
различных реакций, соотношение между которыми зависит от
574
Глава 9
условий процесса. Основная реакция состоит в замещении
аллильного водорода [уравнение (9.29)]. Выделяющийся хло-
ристый водород может вызывать циклизацию (разд. 9.7) галоге-
нированного продукта [уравнение (9.30)]. Циклическое соедине-
ние затем галогенируется присоединением галогена по двойной
связи или замещением аллильного водорода. Имеются также
данные о частичном сшивании в процессе галогенирования,
ио механизм сшивания не ясен
СН3 СН3
I I ' ci2-
СН9—с=сн—сн9—сн,—С=СН— СН —ИгГ*"
сн3 сн3
—* — СНС1— С=СН—СН2—СНС1—С=СН~ сн2— (9.29)
HC1
XV
(9.30)
9.56. Насыщенные полиуглеводороды
Изучено хлорирование полиэтилена, полипропилена, поли-
изобутилена и ряда других насыщенных полиуглеводородов
и их сополимеров [1,26]. Эта свободнорадикальная реакция
— СН2----|-СЬ -»—СН-------[-НС1, (9.31)
— СН-----|-С12 —* — СНС1--|-С1. (9.32)
•
ускоряется при нагревании в присутствии радикальных инициа-
торов и под действием ультрафиолетового облучения. Хлориро-
ванию подвергают твердый полимер или его раствор. Гетеро-
генная реакция идет с низкой степенью превращения, обычно
с поверхности образца полимера. Этот метод целесообразно
использовать в тех случаях, когда требуется изменить поверх-
ностные характеристики полимера и сохранить неизменными
механические свойства полимера в целом.
С этой реакцией имеет много общего и сульфохлорирование
полиэтилена [27]
Химические реакции полимеров
575
SO2C1
CI2SO2 I
— СН2СН2— ——> — СН2СН — (9.33)
— HL1
приводящее к образованию важного в практическом отношении
эластомера. Вулканизация сульфохлорированного продукта
заключается в образовании поперечных сшивок, состоящих
из сульфонатов металлов или сложных эфиров сульфокислот.
SO2C1
I
— СП2СН —
РЬО2
но-н-он
— СН2СН —
I
so2
I
о
I
R
I
О
I
so2
— СН2СН— (9.35)
— СН2СН —
I
so2
I
о
I
РЬ
I
о
I .
SO 2
I
— СН2СН —
(9.34)
9.6. Ароматическое замещение
Полистирол вступает во все реакции замещения, характерные
для бензола [1]. Ароматическое замещение используется для
создания на основе производных полистирола ионообменных
полимеров, для чего в бензольные ядра вводят сульфокислотные
или четвертичпоаммонийпые группировки. В качестве исходного
полимера используют сшитый сополимер стирола и дивипил-
бепзола, обеспечивающий нерастворимость конечного продукта;
из самого полистирола получались растворимые ионообменпики.
Катионообменные полимеры образуются при сульфировании
[уравнение (9.36)1, анионообменные полимеры синтезируют хлор-
метплированием сополимеров стирола и дивинилбензола с после-
дующим взаимодействием с третичным амином [уравнение (9.37)]
576
Глава 9
(9-37)
SO3H
9.7. Циклизация
В разд. 2.12 уже была рассмотрена внутримолекулярная
циклизация как способ образования высокотеплостойких блок-
лестничных или частично лестничных полимеров. К повышению
теплостойкости приводят и некоторые другие реакции циклиза-
ции. Натуральный каучук и другие 1,4-полидиены-1,3 цикли-
зуются при обработке протонными кислотами [1]. Реакция идет
через присоединение протона по двойной связи полимера [урав-
нение (9.38)] с последующей циклизацией в результате атаки
двойной связи соседнего звена ионом карбония [уравнение (9.39)1.
Ряд би- и полициклических конденсированных звеньев полу-
чается, когда при циклизации атакуются не соседние, а более
отдаленные повторяющиеся звенья. При этом среднее значение
числа конденсированных циклов в лестнице не превышает 2—4,
что обусловлено стерическими затруднениями, реакциями пере-
дачи цепи и образованием изолированных непрореагировавших
звеньев между парами зациклизовавшихся повторяющихся моно-
мерных единиц [28]. На практике циклизация используется для
превращения каучука в твердый пластик специального назначе-
ния. Подобная циклизация как побочный процесс наблюдается
при хлорировании и гидрохлорировании натурального каучука,
протекающих в присутствии кислот (добавленных или образую-
щихся в процессе реакции)
СН3 СН3
I I н+
--СН2С-СНСН2СН2С = СНСН2— —>
СН3 СН3
I I
—> — СН2С —СН2СН2СН2С = СНСН2—-» (9.38)
Химические реакции полимеров
577
Пиролиз некоторых полимеров сопровождается циклизацией
в результате реакций свободных боковых функциональных
1 рупп [11. Полициклотримеризация полиакрилонитрила по нит-
рпльпым группам приводит к образованию лестничного поли-
мера (XV) [291
CN CN CN \/\/\/\/
— СН2СНСН2СНСН2СН — —> | | | | (9.40)
/-Ч /-Ч /-ч /\
z N N N
XV
При дальнейшем пиролизе его происходит ароматизация, правда
недостаточно полная для превращения в полихинизарин (XVI);
ряд циклов, очевидно, представляет собой частично гидрирован-
ный хинизарин (XVII). Конечный продукт отличается высокой
тепло- и термостойкостью.
Н Н
\Z\Z4/ \Z\Z\/
II HI II
/ч /\ /\ /-Ч /\
z N N 4 ZNNX
Н
XVI XVII
При пиролизе полиметакриловой кислоты образуются ангид-
ридные циклы
СН3 СН3
I I
— СН2-С—СН2-С-
I I
со со
I I
он он
9.8. Привитые и блок-сополимеры
Отличительная особенность привитых и блок-сополимеров
заключается в том, что и те и другие содержат в макромолекуле
длинные полимерные звенья каждого из сомономеров. Блок-
сополимеры характеризуются линейным расположением таких
578
Глава 9
звеньев, тогда как в привитом сополимере к длинной цепи, образо-
ванной одним мономером, как ответвления присоединяются
цепочки, состоящие из второго мономера (разд. 6.16). Разработано
весьма много методов синтеза таких сополимеров [1, 30, 31];
наиболее интересные из них будут рассмотрены ниже.
9.8а. Привитые сополимеры
Для получения привитого сополимера необходимо в присут-
ствии полимеризующегося мономера создать в макромолекуле
реакционноспособный центр. Большинство методов образования
привитых сополимеров основано на радикальной полимеризации,
ионная сополимеризация распространена значительно меньше.
9.8а.1. Передача цепи
При радикальной полимеризации мономера в присутствии
полимера прививка может идти в результате образования радикала
в основной полимерной цепи за счет реакции передачи цепи
от полимеризующегося мономера. В большинстве случаев, хотя
и не всегда, передача цепи предполагает отрыв атома водорода.
Так, например, идет прививка стирола к полибутадиену
— СН2СН = СНСН2----1--СН2СНС6Н5 —>
— СНСН = СНСН2----1--СН2СН2СвН5, (9.42)
— СНСН = СНСН2-----(-пСН2 = СНС6Н5 —>
— СНСН = СНСН2— (9.43)
(СН2СНС6Н5)П
XVIII
Этот процесс нашел очень широкое практическое применение для
модификации свойств полибутадиена. Привитой сополимер (XVIII)
называют полибутадиен-пр-стиролом, причем первым в названии
указывается мономер, образующий основную полимерную цепь,
а вторым — прививаемый мономер.
9.8а.2. Ультрафиолетовое и ионизируюгцее
облучение
Полимерные радикалы образуются также при облучении
системы полимер — мономер ультрафиолетовым светом (часто
в присутствии фотосенсибилизатора) или ионизирующей радиа-
цией. Например, при воздействии на систему полиэтилен —
стирол ионизирующего излучения в полиэтилене возникают сво-
бодные радикалы, инициирующие привитую полимеризацию
Xимические реакции полимеров
579
стирола
Облучение
— сн2сн2—------------> — СН2СН —
I сн2=снс6н5
— СН2СН — (9.44)
(СН2СНС6Н5)П
с образованием полиэтилен-пр-стирола. Привитую сополимериза-
цию, особенно при инициировании облучением, проводят, как
правило, в гетерогенных условиях, когда твердый полимер нахо-
дится в жидком мономере в набухшем состоянии. От соотношения
скоростей прививки и диффузии зависит, идет реакция с поверх-
ности полимера или во всем его объеме [32].
При получении сополимеров в результате реакций с передачей
цепи и методами, основанными на облучении, образуется смесь
продуктов, состоящая из привитого сополимера, исходного поли-
мера и гомополимера прививаемого мономера. Относительное
содержание указанных трех продуктов определяется природой
мономера, полимера и условиями реакции. При реакции с пере-
дачей цепи эффективность прививки (т. е. доля привитой сополи-
меризации по отношению к гомополимеризации) зависит от того,
насколько предпочтительнее участие данного радикала в пере-
даче цепи, чем в ее росте. Эффективность прививки для данной
системы полимер — мономер можно оценить довольно точно,
исходя из констант скоростей реакций роста цепи мономера
и передачи цепи на полимер. Последнюю, правда, определить
бывает очень трудно, но можно воспользоваться соответствующей
константой скорости передачи цепи для соответствующего соеди-
нения (например, этилбензола вместо полистирола). Эффектив-
ность прививки при облучении зависит от относительных ско-
ростей образования радикала полимера и мономера. Обычно это
соответствует, по крайней мере качественно, константам передачи
цепи на полимер и мономер (или энергиям связи для наиболее
слабых связей в обоих соединениях). Так, прививка к поливинил-
хлориду обычно более эффективна, чем к полиэтилену, поскольку
в первом есть более слабая связь углерод — хлор. При использо-
вании ионизирующего излучения эффективность прививки
может несколько отличаться от ожидаемой из-за сравнительно
мало селективной природы радиолитического расщепления связи
п радиолитической стабильности ароматических структур в целом.
Радиолитический метод можно осуществлять в две стадии.
Сначала полимер облучают в отсутствие мономера, а затем смеши-
вают с мономером. Привитая сополимеризация инициируется
небольшим числом долгоживущих радикалов, захваченных твер-
дым полимером. Этим способом получают привитой сополимер,
580
Глава 9
не содержащий гомополимера. Однако скорость реакции в этомз
случае ниже, чем тогда, когда полимер и мономер облучают вместе.^
Последний процесс называют совместной радиационной прививкой, j
а первый известен как процесс с предварительным облучением.'
К разновидности прививки с предварительным облучением отно-
сится облучение полимера в присутствии кислорода, способст-
вующего образованию перекисных и гидроперекисных групп,
которые затем при нагревании в присутствии мономера разла-
гаются до полимерных радикалов.
9.8а.3. Окислительно-восстановительные системы
Инициирование окислительно-восстановительными системами
может быть наиболее эффективным методом проведения привитой
сополимеризации. Полимеры с гидроксильными группами, напри-
мер поливиниловый спирт и целлюлоза, подвергают окислительно-
восстановительной реакции с ионом церия или другими окисли-
телями, в результате чего образуются полимерные радикалы,
способные инициировать полимеризацию [33].
— СН2СН----|-Се4+ —> —СН2С---|-Н+-|-СеЗ+ . (9.45)
I I
ОН он
При такой прививке гомополимеры не образуются, так как боль-
шинство мономеров не реагирует с ионом церия. Однако приме-
нение окислительно-восстановительного инициирования ограни-
чено только небольшим кругом полимеров, содержащих специфи-
ческие функциональные группы.
9.8а.4. Другие привитые системы
В литературе встречаются и другие примеры привитой сополи-
меризации. Многие из них предполагают синтез специальных
полимеров с такими функциональными группами, по которым
можно осуществлять прививку. Так, взаимодействием целлюлозы
с этиленсульфидом можно ввести в нее меркаптогруппы
S
Целл —ОН + СНг — СНг —> Целл —OCH2CH2SH, (9.46)
по которым можно проводить прививку реакцией с передачей
цепи. Для тех же целей в целлюлозу реакцией с хлорангидридом
акриловой кислоты вводят двойные связи
-HC1
Целл-ОН + СН2 = СНСОС1---> Целл — ОСО — СН = СН2 . (9.47)
Химические реакции полимеров
581
Изопропильные группы в замещенном полистироле можно
превратить в гидроперекисные группы
(9.48)
9.86. Блок-сополимеры
Выше были рассмотрены некоторые способы получения блок-
сополимеров. Их можно синтезировать сополиконденсацией поли-
меров с различными функциональными группами (разд. 2.11).
Ряд блок-сополимеров, представляющих практический интерес,
получают сополимеризацией по методу Натта — Циглера
(разд. 8.4г.2), когда и г2 значительно больше единицы
(разд. 6.2в.З). Универсальным методом является анионная поли-
меризация (разд. 5.36) с образованием живущих полимеров, причем
возникший полимер инициирует полимеризацию другого мономера
М2
— —> -~М1М1М1М1М1М1М2М2М2М2 (9.49)
Многократным повторением такого приема можно получить сопо-
лимер с несколькими регулярно чередующимися блоками каждого
из мономеров. Этот способ применим к блок-сополимеризации
алкенов, циклических мономеров и др., а также смесей мономеров
различных типов в определенных сочетаниях, например стирола
и формальдегида, стирола и окиси этилена, б-валеролактона
и окиси пропилена (разд. 6.76 и 7.7).
9.86.1. Механохимическый способ
Блок-сополимеры можно получать из полимерных радикалов,
которые в отличие от использующихся в привитой сополимериза-
ции имеют радикальные центры на концах макромолекул. Послед-
ние образуются при механохимическом расщеплении связи в про-
цессе измельчения или вальцевания полимера [1, 30]. Блок-сопо-
лимеры образуются при вальцевании смеси двух гомополимеров
или смеси полимера и мономера
— MjMi —
— М2М2 —
-— MfMj —
Измельчение ____Mj. "|
* — М2. J
— MjM2 —
— MtMt —
— М2М2 —
Измельчение М2
----------> — Mi- ---------MiM2 —
(9.50)
(9.51)
582
Глава 9
В первом случае получается смесь блок-сополимера и двух гомо-
полимеров, так как полимерные радикалы рекомбинируются
статистически, во втором — образуется практически один блок-
сополимер.
9.86.2. Другие методы
Блок-сополимеры можно получать из полимеров с подвижными
связями на концах макромолекул. Так, например, сначала поли-
меризацией стирола в присутствии четырехбромистого углерода
получают полистирол, содержащий на концах цепей атомы брома.
Затем под действием ультрафиолетового облучения разрывают
связь углерод — бром, а образовавшиеся свободные радикалы
используются в качестве инициатора полимеризации другого
мономера
свг4 уф м2
---->—Mi — Br -—>—М. —MiM2— (9.52)
Для получения блок-сополимеров можно применять дифунк-
циональные инициаторы, если удается провести разложение
одной группы инициатора, не затронув другую
Mi
НОО —R —ООН —> HOORO- HOORO —Mi----->
м2
ORO —Mi------>—М2 —ORO —Mi— (9.53)
Вопросы и упражнения
9.1. Рассмотрите факторы, обусловливающие различия в реак-
ционной способности функциональных групп в полимерах и в их
низкомолекулярных гомологах.
9.2. Какие реакции можно использовать для сшивания следую-
щих полимеров:
а) сложного полиэфира на основе этиленгликоля и малеино-
вого ангидрида;
б) сложного полиэфира на основе этиленгликоля, фталевой
кислоты и олеиновой кислоты;
в) 1,4-полиизопрена;
г) полиэтилена;
д) полидиметилсилоксана?
Напишите уравнения указанных реакций.
9.3. Напишите уравнения реакций образования
а) ацетата целлюлозы;
б) нитрата целлюлозы;
в) метилцеллюлозы;
г) поливинилформаля.
Химические реакции полимеров
583
9.4. Напишите уравнения реакций
а) хлорирования полиэтилена;
б) хлорирования 1,4-полиизопрена;
в) сульфохлорирования полиэтилена;
г) синтеза анионообменного полимера на основе полистирола;
д) циклизации 1,4-полиизопрена под действием НВт.
9.5. Напишите формулы
а) блок-сополимера пропилена и стирола;
б) привитого сополимера стирола и метилметакрилата;
в) привитого сополимера метилметакрилата и стирола;
г) регулярно чередующегося сополимера метилметакрилата
и стирола.
Приведите уравнения реакций образования этих сополимеров.
Список литературы
1. Химические реакции полимеров, под ред. Е. Феттеса, изд-во «Мир», М.,
1967.
2. Gaylord N. G„ J. Polymer Sci., С24, 1 (1968).
3. Flory P. J., J. Am. Chem. Soc., 61, 1518 (1939).
4. Моравец T., Макромолекулы в растворе, изд-во «Мир», М., 1967.
5. Bender М. L., Neveu М. С., J. Am. Chem. Soc., 80, 5388 (1958).
6. Moens J., Smets G., J. Polymer Sci., 23, 931 (1957).
7. Chapman С. B., J. Polymer Sci., 45, 237 (1960).
8. Соломон Д. Г., Химия органических пленкообразователей, изд-во
«Химия», М., 1971.
9. Burnett G. М., Polymer Preprints, 8(1), 96 (1967).
10. Learmonth G. S., Pritchard G., Beinhardt J., J. Appl. Polymer Sci., 12, 619
(1968).
11. Vancso-Szmercsanyi I., Maros-Greger K., Makay-Bodi E., J. Appl. Polymer
Sci., 53, 241 (1961).
12. Wexler H., Chem. Rev., 64, 591 (1964).
13. Вулканизация эластомеров, под ред. Т. Аллигера и И. Сьетуна, изд-во
«Химия», М., 1967.
14. Bateman L. (ed.), The Chemistry and Physics of Rubber-Like Substances,
John Wiley and Sons, Inc., New York, 1963.
15. Bateman L., Moore C. G., Porter M., J. Chem. Soc., 1958, 2866.
16. Bateman L., Moore C. G., Porter M., Saville B., Chemistry of Vulcanization,
Bateman L. (ed.), op. cit., Chap. 15.
17. Bateman L., Glazebrook B. W., Moore C. G., J. Appl. Polymer Sci., 1,
257 (1959).
18. Gregg E. C., Jr., J. Polymer Sci., C24, 303 (1968).
19. Bobear W. J., Rubber Chem. Technol., 40, 1560 (1967).
20. Amberg L. O., Brown B. F., Rubber World, 147(3), 52 (1963).
21. Wolfe J. B., Jr., J. Appl. Polymer Sci., 12, 1167 (1968).
22. Chapiro A., Radiation Chemistry of Polymeric Systems, chaps. IX, X,
Interscience Publishers, John Wiley and Sons, Inc., New York, 1962.
23. Ott E., Spurlin H. M., Grafflin M. W. (eds.), Cellulose and Cellulose
Derivatives, Interscience Publishers, John Wiley and Sons, Inc., New York,
1954.
584 Список литературы.
24. Brydson J. A., Plastics Materials, Iliffee Books Ltd., London. I960.
25. Dyer J., Phifer L. H., Macromolecules, 2(2), 111, 118 (1969).
26. McGuchan R., McNeil I. C., J. Polymer Sci., A-l(6), 205 (1968).
27. Nersasian A., Anderson D. E., J. Appl. Polymer Sci., 4, 74 (I960).
28. Golub M. A., Heller, Can. J. Chem., 41, 937 (1963).
29. Monahan A. R., Polymer Preprints, 7(2), 204 (1966).
30. Цереза P., Блок- и привитые сополимеры, изд-во «Мир», М.. 1964.
31. Баттерд Г., Трегер Д., Свойства привитых и блок-сополимеров, изд-во
«Химия», Л., 1970.
32. Odian G„ Kruse R. L., J. Polymer Sci., C22, 691 (1969).
33. Ogiivara Y., Kubota H., J. Polymer Sci., A-l(6), 1489 (1968).
СПИСОК ДОПОЛНИТЕЛЬНОЙ ЛИТЕРАТУРЫ
К главе 1
1. Энциклопедия полимеров, т. 1, изд-во «Сов. энциклопедия», М., 1972.
2. Коршак В. В., Общие методы синтеза высокомолекулярных соединений,
Изд-во АН СССР, М„ 1953.
3. Стрепихеев А. А., Деревицкая В. А., Слонимский Г. Л., Основы химии
высокомолекулярных соединений, 2-е изд., изд-во «Химия», М., 1966.
4. Лосев И. Л., Тростянская Е. Б., Химия синтетических полимеров, 3-е изд.
изд-во «Химия», М., 1971.
5. Каргин В. А., Слонимский Г. Л., Краткие очерки по физпкохимии поли-
меров, 2-е изд., изд-во «Химия», М., 1967.
6. Рафиков С. Р., Павлова С. А., Твердохлебова И. И., Методы определения
молекулярных весов и полидисперсности высокомолекулярных соеди-
нений, изд-во «Наука», М., 1963.
7. Тагер А. А., Физико-химия полимеров, изд-во «Химия», М., 1968.
8. Шур А. М., Высокомолекулярные соединения, 2-е изд., изд-во «Высшая
школа», М., 1971.
9. Каргин В. А., Роль структурных явлений в формировании свойств поли-
меров, в сб. «Прогресс полимерной химии», под ред. В. В. Коршака,
изд-во «Наука», М., 1969.
10. Колесников Г. С., Полимеризация и поликонденсация, МХТИ им. Менде-
леева, М., 1970.
К главе 2
1. Коршак В. В., Виноградова С. В., Равновесная поликонденсация, изд-во
«Наука», М., 1968.
2. Коршак В. В., Виноградова С. В., Неравновесная поликонденсация,
изд-во «Наука», М., 1972.
3. Соколов Л. Б., Поликонденсационный метод синтеза полимеров, изд-во
«Химия», М., 1966.
4. Энциклопедия полимеров, т. 1, изд-во «Сов. энциклопедия», М., 1972.
5. Серенсон У., Кемпбел Т., Препаративные методы химии полимеров, ИЛ,
М„ 1963.
6. Технология пластических масс, под ред. В. В. Коршака, изд-во «Химия»,
М., 1972.
7. Коршак В. В., Термостойкие полимеры, изд-во «Наука», М., 1968.
8. Коршак В. В., Химическое строение и температурные характеристики
полимеров, изд-во «Наука», М., 1970.
9. Коршак В. В., Виноградова С. В., Гетероцеппые полиэфиры, Изд-во
АН СССР, М„ 1958.
10. Коршак В. В., Виноградова С. В., Полиарилаты, изд-во «Наука», М.,
1964.
11. Коршак В. В., Фрунзе Т. М., Синтетические гетероцепные полиамиды,
Изд-во АН СССР, М., 1962.
12. Паттон Т. К., Технология алкидных смол, изд-во «Химия», М., 1970.
586 Список дополнительной литературы
13. Пакен А. М., Эпоксидные соединения и эпоксидные смолы, изд-во
«Химия», Л., 1962.
14. Васнев В. А., Выгодский Я. С., Успехи в области синтеза полиамидов
и полиэфиров, в сб. «Новые поликонденсационные полимеры», под ред.
3. А. Роговина и П. М. Валецкого, изд-во «Мир», М., 1969.
15. Адрова Н. А., Бессонов М. И., Лайус Л. А Рудаков А. П., Полиимиды —
новый класс термостойких полимеров, изд-во «Наука», Л., 1968.
16. Кардаш И. Е., Телешов Э. Н., Синтез, свойства и применение высоко-
термостойких гетероциклических полимеров, Итоги пауки, сер. хим.,
ВИНИТИ, 1971.
17. Николаев А. Ф., Синтетические полимеры и пластические массы на их
основе, изд-во «Химия», Л., 1966.
18. АскадскийА. А., Физико-химия полиарилатов, изд-во «Химия», М., 1968.
19. Петров Г. С., Левин А. Н., Термореактивные смолы и пластические
массы, Госхимиздат, М., 1959.
20. Виноградова С. В., Коршак В. В., Успехи химии, 39, 679 (1970).
21. Кучанов С. И., Письмен Л. М., Высокомолек. соед., А13, 791 (1971);
А14, 131 (1972).
22. Кудрявцев Ю. П., Окислительная дегидрополиконденсация — новый
метод синтеза полимеров с тройными связями, в сб. «Прогресс полимер-
ной химии», под ред. В. В. Коршака, изд-во «Наука», М., 1969.
23. Тарасова Г. И., Механизм элементарных актов равновесной поликонден-
сации, в сб. «Прогресс полимерной химии», под ред. В. В. Коршака,
изд-во «Наука», М., 1969.
24. Коршак В. В., Тепляков М. М., Успехи в создании полимеров с азоль-
ными циклами, в сб. «Прогресс полимерной химии», под ред. В. В. Кор-
шака, изд-во «Наука», М., 1969.
25. Коттер Р., Матцнер М., Полициклизация, изд-во «Мир», М., 1972.
26. Кронгауз Е. С., Синтез полимеров реакцией 1,3-диполярного циклопри-
соединения, в сб. «Прогресс полимерной химии», под ред. В. В. Коршака,
изд-во «Наука», М., 1969.
27. Андрианов К. А., Кононов А. М., Полимеры со связью кремний — азот,
в сб. «Прогресс полимерной химии», под ред. В. В. Коршака, изд-во
«Наука», М., 1969.
28. Коршак В. В., Дорошенко Ю. Е., Ренард Т. Л., Полимеры лестничного
типа, в сб. «Химия и технология высокомолекулярных соединений»
под ред. В. В. Коршака, т. 3, ВИНИТИ, М., 1971.
29. Korshak V. 7., Teplyakov М. М., J. Macromol. Sci., Revs. Macromol. Chem.,
C5, 409 (1971).
К главе
1. Багдасаръян X. С., Теория радикальной полимеризации, 2-е изд., изд-во
«Наука», М., 1966.
2. Энциклопедия полимеров, т. 1, изд-во «Сов. энциклопедия», М., 1972.
3. Френкель С. Я., Введение в статистическую теорию полимеризации,
изд-во «Наука», Л., 1965.
4. Бреслер С. Е., Ерусалимский Б. Л., Физика и химия макромолекул,
Изд-во АН СССР, М,—Л., 1965.
5. Вацулик П., Химия мономеров, ИЛ, М., 1960.
6. Технология пластических масс, под ред. В. В. Коршака, изд-во «Химия»,
М., 1972.
7. Кинетика радикальной полимеризации виниловых соединений, ИЛ,
М„ 1961.
8. Серенсон У., Кемпбел Т., Препаративные методы химии полимеров,
ИЛ. М.. 1963.
Список дополнительной литературы
587
9. Гладышев Г. П., Полимеризация винильных мономеров, Изд-во АН
КазССР, Алма-Ата, 1964.
10. Колесников Г. С., Синтез винильных производных ароматических и гете-
роциклических соединений, Изд-во АН СССР, М., 1960.
11. Абкин А. Б., Герасимов Г. Н., Высокомолек. соед., А13, 240 (1971).
12. Цурута Тэйдзи, Реакции получения синтетических полимеров, Гос-
химиздат, М., 1963.
13. Tudos F., Acta Chim. Hung., 43, 397 (1965); 44, 403 (1965).
К главе 4
1. Багдасаръян X. С., Теория радикальной полимеризации, 2-е изд., изд-во
«Наука», М., 1966.
2. Энциклопедия полимеров, т. 1, изд-во «Сов. энциклопедия», М., 1972.
3. Технология пластических масс, под ред. В. В. Коршака, изд-во «Химия»,
М., 1972.
4. Николаев А. Ф., Синтетические полимеры и пластмассы на их основе,
изд-во «Химия», Л., 1966.
5. Гладышев Г. П., Полимеризация винильных мономеров, Изд-во АН
КазССР, Алма-Ата, 1960.
6. Колесников Г. С., Полимеризация и поликонденсация, МХТИ им. Мен-
делеева, М., 1970.
7. Письмен Л. М., Кучанов С. И., Высокомолек. соед., А13, 1055 (1971).
К главе 5
1. Бреслер С. Е., Ерусалимский Б. Л., Физика и химия макромолекул,
гл. 5, изд-во «Наука», М.— Л., 1965.
2. Гладышев Г. П., Полимеризация винильных мономеров, стр. 225—316,
Изд-во АН Каз.ССР, Алма-Ата, 1964.
3. Ерусалимский Б. Л., Ионная полимеризация полярных мономеров,
изд-во «Наука», Л., 1970.
4. Ерусалимский В. Л., Некоторые вопросы катионной полимеризации,
в сб. «Прогресс полимерной химии», под ред. В. В. Коршака, стр. 48—73,
изд-во «Наука», М., 1969.
5. Колесников Г. С., Полимеризация и поликонденсация, МХТИ им. Мен-
делеева, М., 1970.
6. Медведев С. С., Успехи химии, 37, 1923 (1968).
7. Сидельковская Ф. И., Химия а-винилпирролидона и его полимеров,
изд-во «Наука», М., 1970.
8. Френкель С. Я., Введение в статистическую теорию полимеризации,
гл. 5, изд-во «Наука», М.— Л., l^eS.
9. Шварц М., Анионная полимеризация, изд-во «Мир», М., 1971.
10. Reich L., Schindler A., Polymerization by organometallic compounds,
Ch. 7, 8, Interscience Publishers, J. Wiley and Sons, N.Y.— London —
Sydney, 1970.
, К главе 6
1. Геворкян А. В., Конформационные и термодинамические свойства макро-
молекул в тройных полимерных системах, Усп. химии, 41, 401 (1972).
2. Колесников Г. С., Полимеризация и поликонденсация, МХТИ им. Менде-
леева, М., 1970.
3. Колесников Г. С., Цзен Хань-мин, Привитые сополимеры, Усп. химии,
31. 1025 (1962).
588
Список дополнительной литературы
4.. Колесников Г. С., Яралов Л. К., Блок-сополимеры, Успехи химии, 34
454 (1965).
5. Тростянская Е. Б., Бабаевский П. Г., Формирование сетчатых полимеров
Успехи химии, 40, 117, 1971.
6. Баттерд Г., Трегер К., Свойства привитых и блок-сополимеров, изд-во
«Химия», Л., 1970.
7. Берлент У., Хофман А., Привитые и блок-сополимеры, ИЛ, М.. 1963.
8. А'э.и Дж., Сополимеризация, изд-во «Химия», М., 1971.
9. Цереза Р., Блок- и привитые сополимеры, изд-во «Мир», М., 1964.
К главе 7
1. Бреслер С. Е., Ерусалимский Б. Л., Физика и химия макромолекул,
стр. 378, изд-во «Наука», М,— Л., 1965.
2. Гембицкий П. А., Жук Д. С., Каргин В. А., Полиэтиленимин. изд-во
«Наука», М., 1971.
3. Ениколопов Н. С., Вольфсон С. Я., Химия и технология полиформальде-
гида, изд-во «Химия», М., 1968.
4. Ениколопов Н. С., Иржак В. И., Розенберг Б. А., Успехи химии. 35,
714 (1966).
5. Коршак В. В., Фрунзе Т. М., Данилевская Л. Б., Курашев В. В.. Пзв.
АН СССР, 3, 637 (1968).
6. Сазонов Я., Полимеризация лактонов, Успехи химии, 37, 1084, (1968).
7. Соколова Г. С., Волохина А. В., Кудрявцев Г. И., Высокомолек. соедин.,
9. Б, 395 (1967).
8. Фрунзе Т. М., Курашов В. В., Сополимеризация циклических соединений,
Успехи химии, 37, 1600 (1968).
9. Kinetics and mechanisms of polymerization, vol. 2, Ringopening polymeri-
zation, К. C. Frisch, S. L., Reegen (eds.), M. Dekker, N.J., London, 1969.
10. Reich L., Schindler A., Polymerization by organometallic compounds.
Ch. 9, Interscience Publishers, J. Willey and Sons, N.Y.—London —
Sydney, 1970.
11. Komoto H., Makromolek. Chem., 115, 33 (1968).
К главе 8
1. Бирштейн T. М., Птицын О. Б., Конформация макромолекул, изд-во
«Наука», М., 1964.
2. Пространственные эффекты в органической химии, под ред. М. Нью-
мена, ИЛ, М., 1960.
3. Натта Д. и др., Химия и технология полимеров, № 9, 9 (1961); № 2, 5
(1963); № 7, 10 (1964).
4. Медведев С. С., Успехи химии, 37, 1923 (1968).
5. Гейлорд Н., Марк Г., Линейные стереорегулярные полимеры, ИЛ, М.,
1962.
6. Долгоплоск Б. А., Полимеризация диенов под влиянием л-аллильных
комплексов, изд-во «Наука», М., 1968.
7. Кристаллические полиолефины, под ред. Р. А. Раффа и К. В. Дока,
т. 1, изд-во «Наука», М., 1970.
8. Кренцель Б. А., Новое в синтезе стереорегулярных полимеров, в сб.
«Успехи химии полимеров», изд-во «Наука», М. — Л., 1966.
9. Энциклопедия полимеров, т. 1, изд-во «Сов. энциклопедия», М., 1972.
10. Коршак В. В., Слонимский Г. Л., Виноградова С. В., Аскадский А. А.,
Васильев А . В., Васнев В. А., Бычко К. А., ДАН СССР, 199, 607 (1971).
Список дополнительной литературы 589
ц. Долгоплоск Б. А., Высокомолек. соед., А13, 325 (1971).
12" Шварц М., Анионная полимеризация, изд-во «Мир», М., 1971.
13. Дануссо Ф., Успехи химии, 39, 304 (1970).
К главе 9
1. Роговин 3. А., Шорыгина Н. Н,, Химия целлюлозы и ее спутников, Гос-
химиздат, М.— Л., 1953.
2. Роговин 3. А., Основы химии и технологии производства химических
волокон, т. 1, изд-во «Химия», М., 1964.
3. Энциклопедия полимеров, т. 1, изд-во «Сов. энциклопедия», М., 1972.
4 Каргин В. А., Берестнева 3. Я., Калашников В. Г., Успехи химии,
36, 203 (1967).
5. Плата Н. А. Шибаев В. П., Журн. ВХО им. Д. И. Менделеева, 9, № 6,
638 (1964).
6. Колесников Г. С., Яралов Л. К., Успехи химии, 34, 454 (1965).
7. Плата Н. А., Шибаев В. П., Высокомолек. соед., А13, 410 (1971).
8. Плата П. А., Литманович А. Д., Высокомолек. соед., А14, 2503 (1972).
9. Лившиц Р. М., Роговин 3. А., Привитые сополимеры целлюлозы и ее
производных, в сб. «Прогрессполимерной химии», под ред. В. В. Кор-
шака, изд-во «Наука», М., 1969.
10. Methoden der Organischen Chemie (Houben-Weil), Makromolekulare Stoffe,
Teil 1, 2, Georg Thieme Verlag, Stuttgart, 1961.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсолютная константа скорости
поликонденсации 49, 63, 112
полимеризации анионной 307,
309-310, 315
— катионной 288—289, 294
— радикальной 213—221, 289—
230
— стереоспецифической 529
— с раскрытием цикла 213—221
Агент передачи цепи 189
константы скорости 197
структура и реакционная способ-
ность 196—199
Азеотропная сополимеризация 343
Азо-бас-изобутиронитрил (АИБН)
как инициатор 165, 223
Азосоединения как инициаторы 165
Акриламид, полимеризация 192, 326
327
Акрилонитрил
полимеризация 192, 199, 211,
221, 227, 497
сополимеризация 352, 355, 359,
361, 362, 372, 373, 376, 405
Акролеин, полимеризация 325—326
Активатор 535, 567
Активация окислительно-восстанови-
тельная 175—176
Активность
катализатора 504
мономеров 382—384, 386—387
— определение из произведения
вероятностей 374—376
Активированный комплекс 243
Активированный мономер 327, 439
Актинометрия 170
Алкены, сополимеризация 403—404
Алкидные смолы 118, 563
Алкиды, высыхание 563—564
Аллильная полимеризация 212—213
Альдегиды, полимеризация 317
Амиды циклические, полимеризация
437, 444—449
Амилоза 489—490
Амины циклические, полимеризация
455—456
Аморфность 31—33
Анионная полимеризация 299—315
акриламида 326—327
акролеина 325—326
ассоциация 312—313
бутадиена-1,3 300, 305
влияние температуры 302, 313
диметилкетена 324—325
изоцианатов 327
инициирование 299—301, 303—
305
карбонильных мономеров 316—
320
кинетика 300—302, 306—310
константа скорости 306—310. 315
метакрилонитрила 301, 305
метилметакрилата 303
молекулярновесовое распределе-
ние 311—312
мол. вес полимеров 302, 311—312
обрыв 301
параметры активации 302, 313—
315
протекающая без обрыва цепи
302—315
с раскрытием цикла N-карбок-
сиангидридов а-аминокислот
457-458
•--с образованием сополиме-
ров 462—468
---циклических амидов, ини-
циирование сильными основа-
ниями 437—440
---------использование ацили-
рующих агентов 440—442
— — — — кинетика 442
— — — — молекулярный вес
442-443
— — циклических эфиров 454 —
455
Предметный указатель
591
— — циклосилоксана 452
— — эпоксидов, кинетика 412—
413, 416—419
— — — перенос цепи на моно-
мер 418—419
— — •— реакции обмена 416—
417
— — — характеристики 414—
415
сравнение с радикальной поли-
меризацией 152—156, 276—
278, 315—316
стереоспецифическая 502—503,
541___547 i
стирола 300, 303—305, 306, 307,
309, 310, 314
формальдегида 317
этилакрилата 301
Анион-радикал 299
Анхимерное ускорение 561
Ароматическое нуклеофильное заме-
щение 127
Аррениуса энергия активации 221
Асимметрическая индукция 551
Асимметрический центр 474—475
Атактический полимер 475—476, 493
Аутоингибирование 212—213
Аутоускорение 235—239
Ацетали циклические, полимериза-
ция 431—433
Ацетилен, полимеризация 328
Ацилперекиси как инициаторы 164
Бисерная (жемчужная) полимериза-
ция см. Полимеризация в суспен-
зии
Блокирование (закрытие) концевых
групп 324
Блок-лестничные полимеры 136
Блок-сополимеры 121, 335, 344, см.
также Сополимеризация, Сополи-
меры
Бутадиен
полимеризация 221, 227, 257,
305, 481-486, 540, 542, 545
сополимеризация 352, 358, 359,
361, 362, 372
Бутен, полимеризация 227
Бутиловый спирт, поликонденсация
с изоцианатами 63
1,3-Взаимодействие 229
Винилацетат
полимеризация 192, 199, 203,
206, 210, 211, 221, 227
сополимеризация 358, 359, 360,
361, 362, 364, 372, 373
Винилиденфторид, полимеризация
227
Винилиденхлорид
полимеризация 227
сополимеризация 355, 358, 359,
361, 365, 372, 376
Виниловые эфиры
полимеризация 539
сополимеризация 368—369
Винилтиофен, сополимеризация 346,
347
Винилхлорид
полимеризация 192, 221, 227, 496
сополимеризация 358, 359, 360,
361, 362, 364, 372, 373, 376
Вискозный метод растворения целлю-
лозы 571
Внутримолекулярная реакция рас-
ширения кольца 425, 433, 456
Волокно 40—42, 92, 124
Время жизни растущего радикала
214
определение методом вращающе-
гося сектора 216—219
Вулканизация эластомеров 564—567
Галогенирование 573—575
Гелеобразование 95
Гель 95—96
Гель-эффект 235—236
Гетеротактические триады 492
Гибкий пластик 39, 40, 42
Гидроперекиси как инициаторы 165,
223
Гомополимеры 120, 334, 343
Гоморост 337
Гранульная полимеризация см. По-
лимеризация в суспензии
Дакрон 84, см. также Полиэтиленте-
рефталат
Двоесвязанные полимеры 136
Деполимеризация
при радикальной полимериза-
ции 231—234
при сополимеризации 392—395
Диадная тактичность 491
Диазоалканы, полимеризация 328
Диены
полимеризация 250—251, 400—
403, 540—548
стереоизомеры 474—479, 540—
547
592 Предметный
указатель
mpeo-Диизотактический полимер 478,
519
зритро- Диизотактический полимер,
478, 520, 531, 534
Дилатометр 163
Дильса — Альдера полимеризация
140—141, 547—548
Диметилкетен, полимеризация 324—
325
Диолы, полимеризация 116
1,3-Диполярное присоединение 141 —
142
mpeo-Дисиндиотактический полимер
477, 478—479
зрипгро-Дисиндиотактический поли-
мер 477, 478, 531
Диспропорционирование 160, 187
Дитактичность 477—£9, 519
Диффузный фактор 236—238
Дихлорэтилен, сополимеризация 365,
366
Диэтилфумарат, сополимеризация
347, 348, 358
Жесткий пластик 40, 43
Живущий полимер 302 см. также
Анионная полимеризация
Закономерности поликонденсации
82—92
Замедлители 204
Изобутилен
полимеризация 227, 290, 291, 298
сополимеризация 360, 384
Изомеризационная полимеризация
282
Изомеризация 282—283
Изомерия конфигурационная 474
Изомеры конформационные 474, 561
Изопрен
полимеризация 185, 227, 290,
305, 482, 540, 542, 546
сополимеризация 378
Изотактические триады 492
Изотактический полимер 475—476,
480, 491—493
Изоцианаты, полимеризация 327
Инверсионные эмульсионные систе-
мы 268—269
Ингибирование радикальной полиме-
ризации 204—213
аутоингибирование 211—212
кинетика 208—212
механизм 204—208
эмульгатором 272—273
Ингибиторы
определение 204
радикалов 183—184
типы 205—208
Индукционный (ингибирующий) пе-'-
риод 162, 205
Инициаторы '
влияние на скорость полимерй-.
зации 166—167 j
индуцированное разложение 179,'
. 190—194 i
определение 151
фотолитическая диссоциация 171 =
172
эффективность, механизм 179—
182
— определение 166, 178—179
---- экспериментальное 182—
—185
Инициирование полимеризации
анионной, алкилами металлов 305
— амидами металлов 300—301
— влияние температуры 313
— при переносе электрона 303—
305
катионной, влияние температу-
ры 297—299
— ионом карбония 281
— кислотами Льюиса 279—281
— протонными кислотами 278—
279
— с раскрытием цикла 420—424,
444—447, 452—453
радикальной 159—160, 163—185
— влияние аутоускорения 235—
236
— — давления 242—246
— ионизирующим излучением
173—174
— кинетика 160, 166—168, 170,
172, 177—178, 182, 184—185,
208—212
— окислительно-восстановитель-
. ными системами 175—178, 580
— параметры активации 221 —
222, 243—246
— термическое 164—169, 174—
175
---влияние на скорость кон-
центрации инициатора 166—
167
-----------мономера 167—168
— фотохимическое 169—173
— эффективность 166, 178—185
эмульсионной 266—267
Предметный указатель
593
Ионизирующее излучение 173—174,
578-580
Лонная полимеризация 276—328
стереоспецифическая 499—504
Лонная сополимеризация 381—389
Лонные пары
инициирующие 281
контактные 314
сольватно разделенные 314
Карбония ионы 422—423
Карозерса уравнение 59, 97 —100 см.
также Сшивание
Катализ окислительно-восстанови-
тельный 175
Катализаторы см. также Инициато-
ры; Инициирование
алфиновые 544
координационные, влияние при-
роды компонентов 520—524
— гетерогенные и гомогенные
503-504
— компоненты: металлы групп
1—3 501, 504-505, 522-523
переходные металлы 501,
504-505, 520-522
— кристаллическая структура
513-516
— механизм стереоспецифиче-
ского присоединения 501—504
— определение 501, 504—505
— третий компонент 524
— физическая природа 512—520
маслорастворимые 249
металлоорганические 424
окисные на носителях 534—535
оптически активные 550—551
Катионная полимеризация 278—299
акролеина 325—326
влияние противоиона 295—296
— растворителя 293—295
— температуры 297—299
— электрического поля 295
дпазоалканов 328
изобутилена 291, 298
ингибирование 285
инициирование 278—281
— механизм 321—322
карбонильных мономеров 320—
322
кинетика 286—288
константа скорости 288—289, 294
З-метилбутена-1 282
мол. вес полимеров 288—289
обрыв цепи 283—285
передача цепи 283—284, 291—292
псевдокатиопные реакции 296—
297
с раскрытием цикла с образова-
нием сополимеров 462—468
— — циклических амидов, ини-
циирование 444—446
— — — — молекулярный вес
447—449
---------равновесная 447—449
---------термодинамика 449—
451
— — — аминов 455—456
— — — ацеталей и формалей
431-433
— — — иминосоединений 458
— — — простых эфиров, ини-
циирование 420—424
------------кинетика 426—430
------------константа скорости
427-428
— — — обрыв цепи 424—
426
— — _ — _ перенос цепи 424
— ———— равновесная 428
— — - - — — энергетика 433—
437
— — — сложных эфиров 454—
455
— — — сульфидов 456—457
— — цпклоалканов 459
— — циклосилазанов 461
— — циклосилоксанов 452—453
рост цепи 281—283
сравнение с радикальной поли-
меризацией 152—156, 276—
278, 289—290, 315-316
стереоспецифическая 502, 547—
548
стирола 280, 284, 290, 291, 292
294, 296, 298
Кинетика
поликонденсации 46—47, 51—64
полимеризации анионной 300___
302, 306—310
— катионной 286—292
— равновесной 85—87, 232 —
234, 426—430, 447-449
— радикальной 159—168, 170
172, 177—178, 182, 208—212 '
— с раскрытием цикла 412—413,
416—419, 426-430, 442
— стереоспецифической 525—
530
— эмульсионной 261—264
сополимеризации 376—381
Кинетическая цепь, длина 185—
186
594
Предметный указатель
Классификация полимеров
по механизму полимеризации
18—20
по структуре 11—18
Конденсационные полимеры 11 —14,17
Константа скорости см. также Абсо-
лютная константа скорости
гомолитического разложения 166
176, 177, 184, 185, 220, 223, 243
деполимеризации 231, 232, 428
ингибирования 209, 211, 213—
216
инициирования 160, 213—216,
264
обрыва 160, 161, 186, 187, 213—
216, 220, 243
передачи цепи 187, 189, 192, 194,
213-216, 264, 291, 418-419
поликонденсации 49, 63, 112
присоединения изотактического
494, 495, 498
— синдиотактического 494, 495,
498
роста 160, 213—216, 220, 231,
243, 263—264, 306, 428
— ионных пар контактных 315
— — — сольватно разделенных
315
сополимеризации, эксперимен-
тальное определение 349—350
D- и L-Конфигурация 475
R- и 5-Конфигурация 475
Конформация
влияние на реакционную способ-
ность 561
определение 473
— транс 541
— цис 541
Концевые группы 71
Коэффициент разветвления 100
критический 100
Крахмал 12, 489—490
Кристаллиты 31
Кристалличность
влияние на механические свой-
ства 39—41, 488
— на реакционную способность
полимера 560
природа 31—33
Критическая мицеллярная концен-
трация эмульгатора 260, 265
Критическая степень завершенности
реакции 98
Ксантогенирование 570—571
Лавсан 84, см. также Полнэтпленте-
рефталат
Лактамы, реакционная способность
при полимеризации 449—450
Ламеллы 31—32
Лестничные полимеры 136
Линейные полимеры 25, 93
Льюиса кислоты 279 — 281, 421— 422
Майлар 84, см. также Полиэти.тенте-
рефталат
Медведева — Шейнкер теория 270—
271
Медноаммиачный метод растворения
целлюлозы 571
Межфазная поликонденсация 90—92
Me л аминоформа л ьдегпдны е полиме-
ры 14, 40, 43, 62, 115
Мертвый полимер 161, 178, 184---185
Метакриламид, полимеризация 219,
227
Метакрилонитрил, полимеризация
301, 305
Метилакрилат
полимеризация 192, 199, 211,
221, 227
сополимеризация 358, 359. 361,
362, 372, 373, 376
Метплбутен, полимеризация 282
Метилметакрилат
полимеризация 192, 194, 199,
211, 221, 227, 235, 236, 303,
497, 500, 536—538
сополимеризация 339, 346, 355,
358, 359, 361, 362, 372, 376,
378, 405
Метилстирол
полимеризация 227
сополимеризация 359, 382, 383,
384, 395, 405
Метод мертвых концов 184—185
Механизм полимеризации, методы
установления 315—316
Механические свойства полимеров
влияние на практическое исполь-
зование 40—43
зависимость от кристалличности
40—41
характеристика 39
Мпкрогель 64, 114
Мицеллы 31 — 32, 257
Модификатор 200
Модуль, определение 39—40
Молекулярновесовое распределение
76-81
влияние реакций обмена 79—80
наиболее вероятное 77
определение 29—31
II редметный указатель
595
в полимеризации анионной 311 —
312
— радикальной при высокой сте-
пени завершенности 241 — 242
— — на начальной стадии 240—
241
— эмульсионной 273
по Флори 77 — 78
ширина 78—79
Молекулярный вес
влияние температуры 224—226
значение 27
при поликонденсации линейной
72—81
— — количественный подход
72-75
— — необходимость стехиомет-
рического контроля 71—72
— нелинейной 104—107
— разветвленной 93—95
при полимеризации анионной
302, 311—312
— катионной 288—289
— равновесной 447—449
— радикальной, бимолекуляр-
ный обрыв 185—187
--влияние давления 242—246
------- передачи цепи 187—204
— с раскрытием цикла 416, 417,
419, 442—444, 447—449
— эмульсионной 263—264
распределение весовое 77
— числовое 77
средневесовой 28
средневязкостный 29—30
среднечпсловой 28
Мономерная единица 58
Мономеры
монофункциональные 72
ненасыщенные, полимеризация
по различным механизмам 153
определение 11
полифункциональные 44
Мочевиноформальдегидные полимеры
14, 40, 43, 62
синтез 114—115
Мыла как эмульгаторы 267
Найлон 24, 87, см. также Полиами-
ды
Напряжение в циклах
отталкивание из-за наличия за-
местителей 66
угловое 6(5—68
Неорганические полимеры 144—145,
459-462
Нитрилы, полимеризация 328
Новолакп 119 см. также Фенолфор-
мальдегидные полимеры
Номенклатура полимеров 20—24
основанная на названии исход-
ных соединений 21—22
— стереорегулярности 475—479,
482—483, 485—487
— торговом названии 23—24
— химическом строении 22—23
Норриша — Смита эффект 235
Обмена реакции 79—80, 416—418
Образование полимеров 11 — 20
механизм 18—20
Обрыв цепи
при полимеризации анионной,
влияние температуры 313
— катионной, влияние темпера-
туры 297—299
----без обрыва кинетической
цепи 283—284
----с обрывом кинетической
цепи 284—285, 290—292
---- с раскрытием цикла 424—
426
— радикальной 159—161, 236—
239
— эмульсионной 267—268
при сополимеризации, контроли-
руемый диффузией 379—381
— — химическими реакциями
376—379
Объем активированного комплекса
243
Окислительная дегпдрополиконден-
сация 128
Окислительно-восстановительные си-
стемы как инициаторы
полимеризации 175—178
сополимеризации 580
Оксония ион 419
Оптически активные полимеры 549—
553
Пентадиен, полимеризация 546
Передача цепи
при полимеризации анионной
с раскрытием цикла 418—419
— катионной 283—284, 291—292
— — на полимер посредством
алкильного обмена 424
— радикальной, агентами пере-
дачи цени 189, 194—199
---- влияние на молекулярный
вес и скорость 187—190
596
Предметный указатель
— — внутримолекулярная 204,
246
— — деструктивная 212—213
----на инициатор 179, 190, 193,
194
----константа 189, 191 —194
---- на мономер 190—193
— — с образованием разветвлен-
ных полимеров 200—204
— — параметры активации 223,
225, 243 — 246
---- применение 200
----на полимер 200—204
— — на растворитель 187 —189,
194—199
— эмульсионной 273
при получении привитых сопо-
лимеров 578
Перекрестное взаимодействие ради-
калов 377
Перекрестный рост 337
Перенос электрона 303—305
Перэфпры как инициаторы 165
Пирроны 137
Пластмассы 40—43, 334
Плотность ветвления 202
Поверхностноактпвные вещества 267
Повторяющаяся единица 12
Полиамидокислоты 133—134
Полиамиды 12, 13, 34, 38, 40, 42, 62
ароматические 126—127,133—134
синтез 87—88, 126—127
сополимеры 120—123
Поли-6-аминокапроновая кислота 22
Полиакрилонитрил 16, 40, 497, 577
Полиацетальдегид 16, 21, 472, 479—
481, 550
Полиацеталь 14
Полпбензимидазолы 134—136
Полибензоксазолы 138
Полибензотиазолы 138
Поли-3,3-бнс-(хлорметил)оксетан 411
1,2-Полибутадпен 481, 540
1,4-Полибутадпен 488, 540
Полибутен-1 488, 530
Поливинилацетат 16, 22, 37, 159
разветвленный 202 — 203
реакции 572—573
Поливинилиденфторид 159
Поливинилпденхлорид 16, 37, 38
Поливиниловый спирт 21, 158, 159,
472, 572
Поливинилфторид 37, 159
Поливинилхлорид 16, 21, 34, 37, 38,
40, 158
разветвленный 203
стереоизомерия 496
Полигексаметпленадипамид 22, 37,1
38, 40, 42 |
молекулярновесовое распределе- i
нпе 80—81 1
Полигексаметилендиамин - со-себаци- I
новая кислота 23 !
Полигексаметиленсебацамид 23
Полидиены 251, 481—483
сшивание 564—567, 652
Полидпметилсилоксап 37, 568—569
Пол п-2,6-диметилфени леноксид 128
Полиимидазоппрролоны 137—138
Полпимиды 133—134
Полиизобутилен 16, 37, 40
хлорирование 574
Полиизопрен (натуральный каучук)
16, 37, 40, 41
стереоизомерия 481, 482, 489
хлорирование 573—574
циклизация 573, 576—577
Поли-4,4' - пзопропилидендифенилен-
карбонат 24, 126
Поли-е-канролактам 20, 21, 34, 37,
411, 472
Поликарбонаты 24, 126
Поликонденсация 45—145
адипиновой кислоты и диэтилеи-
гликоля 54, 55, 60
Поликонденсация
в блоке, (в массе) 82
бутилового спирта 63
в дисперсии 83
изоцианатов 63
кинетика катализированного
процесса 59—62
— некатализпрованного процес-
са 51—53
— равновесного процесса 85—
87
— самокатализируемого процес-
са 53—59
константа скорости 49, 63, 112
межфазная 90—92
механизм образования полиме-
ров 18—20
молекулярновесовое распределе-
ние 76—82, 94—95, 104—107
неравновесная 45
с образованием разветвленных
полимеров 93
— сшитых полимеров 95 —104
равновесная 45, 85—87
в расплаве 88
в растворе 83
реакционноспособные функцио-
нальные группы 45—51, 62—
64, 111-114
Предметный указатель
597
регулирование молекулярного
веса 71 — 75
термодинамические параметры
66 — 69
трехмерная 93—95
условия проведения процесса
82 — 85, 87 — 92
циклизация 65—69
Поли-и-ксилилен 18, 23, 13U—132
Полимеризация
в блоке (в массе) 247 — 248
ио карбонильной группе ионная
316-322
— — радикальная 322—323
— — ступенчатая 323
с переносом водорода 327, 439
последовательная 343
с раскрытием цикла 20, 411—468
----тетрагидрофурана 427, 429,
430, 436
в растворе 248—249
в суспензии 249
в твердом состоянии 249—250
с чередованием внутри- и меж-
молекулярных реакций 400—
403
в эмульсии 256—273
Полимеры
типы 11 — 18
транс 482—483
химические реакции 559—582
цис 482—483
Полиметакрпловая кислота 577
Полпметилвинплкетон 158
Полиметилен 23
Полиметилметакрилат 16, 34, 37, 40,
43, 472
стереоизомерия 497, 499
Полп-З-метилпентен-1 21
Поли-4-метилпентен-1 488
1,4-Полнметилсорбат 486
Полиоксадиазолы 139—140
Полиоксиметилен 37, 38
Полиоксипропилен 20, 21, 411, 480—
481, 548, 550
Полиоксиэтилен 21, 37, 411, 472
Полпорганометаллосилоксаны 145
1,4-Полипентадиен-1,3 485
Полипентен-2 479
Полипропилен 37, 38, 40, 42
стереорегулярный 476, 488—489,
504, 513, 516, 550
хлорирование 574
Полисплоксаны 13, 35, 37, 40, 144—
145
синтез 89—90, 451 — 454
сшивание 567—568
Полистирол 16. 54, 37, 38, 41), 43, 14)5
реакции ароматическою замеще-
ния 575
сополимеры 334, 339
Полисульфид 4об—45/
Полисульфон
Политетрафторэтилен 16, 3/, 40
Полнтрпазолы 139—140
11оли(трпметиЛ1’нгликоль-со'эт11лен.
диизоцпанат) 23
Поли(триметиЛЬ'нэтиленуретцц) 22
Полиуретаны 13.1 /, 40, 6-
синтез 88—°!, Но
сополимеры 124
сшивание °6J
11олп-1,4-фенПЛеи -3
Полп-.ч-фенплени.чофталамид 127
Поли-1 4-фен11Ленпиромеллптцмид
133 . , 1/г
11 олифенплспЛ^есквиоксап 14э
П о л пформа л ьдегпд 16
Полпфосфазены 459—461
Полпфосфонптрилхлорид 4о!)—460
Полихпноксалины 138 13.)
Полихлортрифторэтилен 21, 37
Полицпклобутен 530 531
Полициклогексен 487
Полиэтерификация 46, 51, 83—85
см. также Поликонденсация
адипиновой кислоты и диэтилед-
гликоля 54, 55, 60
Полиэтилакрилат 4/2
Полиэтилен 16’ “1’ ’ 42
разветрлеянып 203 —04
синтез 243, 504
сополимеры 334 оЗЗ 334
сшивание м7—568
хлорирование о74
П о лпэтнлекгликол ь-со-терефта левая
кислота 23
Полпэтилеятерефталат _3, 34, 37,
84-85
кинетика образования 85—87
Полиэфироам/Щы 123
Полиэфиры 12, 40
линейные »о—S3
ненасыщенные 118
__ реакции с кислородом 563—.
564
__ сшивание при сополдмериза-
цип 562 563
статистически сшитые 109—110
Полукристаллические полимеры 31
Полярный эффект 199, 366-370
Последователь! ki я гомополпмериза.
цпя 343
Ностэффект 210
598
Лредметный указатель
Преполимеры 115 —119
диоловые 116
определение 108
статистические 108—119
структурно-концевые 115
структурно-иендантные 115
эпоксидные 116—117
Промоторы 423, 424, 535
Пропилен, полимеризация 227, 500,
530
Противоионы 277, 295—296
Протонные кислоты 420—421
Процесс с предварительным облуче-
нием 580
Псевдокатионная полимеризация
296—297
Псевдоконстанта скорости 293
Поперечная связь 95
Равновесие полимеризация — депо-
лимеризация 231 — 234, 428—430,
447—449
Равновесная концентрация мономе-
ров 234, 428, 447—449
Радикалы
время жизни 214
первичные 160, 166, 239
Радикальная полимеризация 151 —
253
акриламида 192
акрилонитрила 192, 199, 211,
221, 227
акролеина 325—326
аутоускорение 235—239
бутадиена-1,3 221, 227
бутена-1 227
винилацетата 192, 199, 203, 206,
210, 211, 221
винилиденфторида 227
винилиденхлорида 227
винилхлорида 192, 221, 227
влияние давления 242—246
— заместителя 154—156, 228—
231
— полярности 199
— температуры 222, 232—234
1,3-дпенов 250—251 см. также
Диены
ингибирование 204—213
ингибиторы 183—184, 204—213
инициирование 159—160, 163—
185
— ионизирующим излучением
173—174
— механизм 172 — 182
— окислительно-восстановитель-
ное 175—178
— определение 178—179,182—185
— термическое 164—169, 174—
175, 222, 223
— фотохимическое 169—173
изобутилена 227
изопрена 185, 227
карбонильных мономеров 322—
323
кинетика 159—168, 170, 172,
177—178, 208—212
— нестационарного состояния
213—219
кинетическая длина цени 185 —
186
метакриламида 219
метилакрилата 192, 199, 211,
221, 227
метплметакрила 192, 194, 199,
211, 221, 227, 235, 236
метилстирола 227
молекулярновесовое распределе-
ние 240—242
мол. вес 185—204
обрыв цени 159, 160—161
— — бимолекулярный 186 —189
параметры активации 221—222,
243—246
передача цепи 187—204
природа 151 —156
присоединение «голова к голове»
(1,2-присоединение) 157
— «голова к хвосту» (1,3-ири-
соединение) 157
пропилена 227
рост цепи 154—156, 159, 160
скорость 239
влияние температуры 222—224
сравнение с ионной полимериза-
цией 152-156, 276-278
— с поликонденсацией 18—20,
151 — 152
стереоспецифическая 494—499,
540—541
стирола 190, 191, 192, 194, 199,
205, 211, 212, 227, 232, 243,
257
теплота 227
термодинамика 226—234
— влияние строения мономера
228—231
тетрафторэтилена 227
факторы, влияющие на образо-
вание стереорегулярных поли-
меров 194—497; 535-537
эмульсионная 253—273
энтропия 227
этилена 208, 227
Предметный, указатель
599
1’азветвлепня
при поликонденсации 93
— молекулярновесовое распре-
деление 94—95
при полимеризации радикальной
200—204
— с раскрытием цикла 424—425,
443—444
Разветвленные полимеры 25—26,
93
Разветвляющая структурная единица
100
Разрывная прочность, определение
39—40
Разрывное удлинение, определение
39-40
Распределение по размеру стереоре-
гулярных блоков 498—499
Растворимость полимеров 560
Реактопласты 96
Реакционная способность
мономеров 357—376, 449—450,
см. также Соотношение реак-
ционных способностей моно-
меров при сополимеризации
полимеров 559—562
функциональных групп 46—51,
62, 64, 111 — 114, 199
Реакционноспособный центр, методы
образования
ионизирующее облучение 579—
580
механохпмический 581—582
окислительно - восстановитель -
иые системы 580
ультрафиолетовое облучение 578
579, 582
Регуляторы 200
Резола полимеры ПО—111
Резонансный эффект 361—364
Рекомбинация 160, 187
Рост цепи при полимеризации
анионной, влияние противоиона
309
— — растворителя 307, 310
— константа скорости 306—310,
315
катионной, влияние противо-
иона 292, 295-296
— — растворителя 293—295
— изомеризация 282—283
— реакционноспособность моно-
мера 290—292
радикальной, влияние замести-
телей 154—156
— возможные способы 156—158
— константа скорости 219—221
— параметры активации 221,
244—245
эмульсионной 267—268
Сетчатая структура 93
Сетчатый (трехмерный) полимер 27
Синдиотактические триады 492
Синдиотактический полимер 475 —
476, 480, 492
Слита — Эварта теория 265
отклонения 269—273
Совместная радиационная прививка
580
Сокатализатор 279
Соотношение реакционных способно-
стей мономеров при сополимериза-
ции
анионной 386—389
— влияние растворителя и про-
тивоиона 387—389
катионной 382—386
— влияние растворителя и про-
тивоиона 384—386
---- температуры 386
радикальной, влияние полярно-
сти 366—370
----сопряжения 361—364
— — стерических затруднений
365—366
---- условий реакции 356—357
— количественная зависимость
от структуры (схема Q — е)
371 — 374
— из произведения вероятностей
374—376
— из ряда активностей радикала
361 — 364
— тенденция к чередованию 370
расчет 349—350
циклических мономеров 463
Сополпконденсация 120—124
Сополимеризация 333—405
азеотропная 343
акрилонитрила 352, 355, 358,
361, 362, 372, 373, 376, 405
алкенов 403—404
анионная 381 — 382, 386—389
бутадиена 352, 358, 359, 361,
‘362, 372
винилацетата 358, 359, 360, 361,
362, 364, 372, 373
винплиденхлорпда 355, 358, 359,
361, 365, 372, 376
винилтпофена 346, 347
винилхлорида 358, 359, 360, 361,
362, 364, 372, 373, 376
600
Предметный указатель
дненов
сшивание 395 — 399
циклополпмеризацпя 400—
403
днхлорэтилена 365, 366
диэтилфумарата 347, 348, 358
значение 333-334, 389-390
идеальная 340—342
изобутилена 360, 384
изопрена 378
карбонильных мономеров 404—
405
катализируемая катализаторами
Циглера — Натта 532—534
катионная 381—386
метилакрилата 358, 359, 361,362,
372, 373, 376
метилметакрилата 339, 346, 355,
358, 359, 361, 362, 372, 376,
378, 405
метилстирола 359, 382, 383, 384,
395, 405
многокомпонентная 333, 352 —
356
с образованием блок-сополиме-
ров 344
— регулярно чередующихся со-
полимеров 342—344
радикальная алкенов 403—405
— реакционная способность мо-
номеров п радикалов 357—376
— скорость 376 — 381
с раскрытием цикла 462—468
стереоблок 491, 493
стирола 334, 346, 347, 352, 355,
358, 359, 360, 361, 362, 364,
372, 373, 376, 378, 383, 387,
391, 405
при сшивании 562 — 563
трихлорэтилена 365, 366
уравнение 336 — 338, 343, 345,
349, 357, 389—395
формальдегида 404—405
хлорстпрола 383, 384
циклических мономеров 462—
468
эффект предпоследнего звена
390-392
Сополимеры
блок 121 — 123, 335, 344, 534,
581 — 582
«мгновенный» состав 344
микроструктура 350—352
номенклатура 120—121, 334
335
определение 120, 333
привитые 335, 578—581
регулярно чередующиеся 120—
123, 335, 342—343, 534
синтез 121 —123
статистические 121 —123, 334,
533
стереоблок 491
— статистические 493
уравнение состава 336—338
Спандекс 124
Спирополимеры 142—143
Способы проведения полимеризации
82, 246-250
Средняя функциональность 97—100
Статистические преполимеры 108 —
119
Стационарное состояние 162
Степень кристалличности 31
влияние различных факторов
33—35
Степень полимеризации 58, 186—187,
224—226, 245—246, 311, 416, 417,
419, 442 — 444, 447—449, см. также
Молекулярный вес; Молекулярно-
весовое распределение
Стереоблок-нолимеры 491, 493
Стереоизомерия 473—486 см. также
Стереоспецифическая полимериза-
ция
анализ 491—493
отсутствие при поликонденсации
481
в полимерах дпенов-1,3 481—483
— дизамещеиных этиленов 477 —
479
— карбонилов 479—481
— монозамещенных этиленов
474-475
— циклических 486—487
при полимеризации с раскры-
тием цикла 479—481
Стереорегулярные полимеры 476
значение 487—490
Стереоселективная полимеризация
550
Стереоспецифическая полимеризация
472-553
акрилонитрила 497
но анионному механизму 541 —
547
бутадиена 481 — 486, 540, 542,
'545
винилхлорида 496
влияние различных факторов при
радикальном механизме 483—
497, 535-537
1,3-диенов 540—548
изопрена 482, 540, 542, 546
Предметный указатель
601
по ионному механизму 499—504,
535-538
карбонильных мономеров 549
кинетические кривые 528
по координационном}’ механиз-
му 499—504
— — биметаллическому и моно-
металлическому 509—512
— — взаимодействие лиганда
с мономером 512—513
— — мономера с мономером
516—520
— — кинетика 525—530
— — компоненты каталитиче-
ской системы 520—524
---- константа скорости 529
— — молекулярновесовое рас-
пределение 529—530
— — направление раскрытия
двойной связи 519—520
----рост через связь углерод —
переходный металл 508—509
---- схема реакции 525—526
----химическая природа расту-
щих частиц 506—508
— — энергия активации 530
метилметакрилата 497, 500, 536
538
модель изотактического присое-
динения 512—513
— синдиотактического присоеди-
нения 516—520
неполярных и полярных мономе-
ров 503—504, 530—531, 535—
539
окисные катализаторы на носи-
телях 534—535
определение 476
оптически активных мономеров
552—553
пентадиена 546
пропилена 500, 530
простых виниловых эфиров 539
по радикальному механизму 540
541
распределение по размеру стерео-
регулярных блоков 498—499
стирола 530, 539
циклоалкенов 486, 531—532
эпоксидов 548—549
этилена 529, 533
Стерический эффект 365—366
Стехиометрический разбаланс 72
Стирол
полимеризация 190, 191, 192,
194, 199, 205, 211, 221, 227,
232, 243, 257, 262, 263, 280,
284, 290, 291, 292, 294, 296,
298, 300, 303—305, 306, 307,
309, 310, 314, 530, 539
сополимеризация 334, 339, 346,
347, 352, 355, 359, 360, 361,
362, 364, 372, 373, 376, 378,
383, 387, 391, 405
Структура полимеров
«голова к голове» 157
«голова к хвосту» 157
методы исследования 158—159
Структурная единица 58
Структурная форма полимерных мо-
лекул 24—27
Ступенчатая полимеризация 323
Ступенчатые полимеры 1Г
Сульфиды циклические, полимериза-
ция 456—457
Схема Q — е 370—374
Сшивание
кислородом 563—564
ненасыщенных полиэфиров 562—
' 563
перекисями 567—568
при поликонденсации, молеку-
лярновесовое распределение
104—107
— статистическое рассмотрение
100—102, 108—115
— технология 107—119
— уравнение Карозерса 97—100
экспериментальные результаты
102—104, 109—110
1,4-полидиенов 564—567
полиолефинов 567
полисилоксанов 567—568
радиационное 570
серой 565—566
при сополимеризации 562—563
— диенов 395—399
фторполимеров 568—569
Сшитый полимер 25—27, 95—96, см.
также Сшивание
Тактичность
анализ 491—493
определение 475, 491—493
Твердофазная полимеризация 249—
250
Теломер 188
Температура
плавления 35—38, 123, 489
предельная 233—234, 317
стеклования 35—38, 123, 489
Тепловые переходы 35—38
602
Предметный указатель
Теплота
полимеризации лактамов 450
— радикальной 227
Термическая самоинициируемая по-
лимеризация 174—175
Термический гомолитический распад
инициаторов 164
Термически катализируемые реакции
164
Термодинамика полимеризации
катионной 297 — 299
радикальной 226—234
с раскрытием цикла 436—437,
450
Термопласты 97
Термореактивные полимеры 96
классификация 108
Термостойкие полимеры
неорганические 144—145, 459—
462
полиамиды 126—127
полибензимидазолы 134—136
полибензоксазолы 138
полибензотиазолы 138
полиимидазопирролоны 137—138
полиимиды 133—134
поликарбонаты 126
поли-п-ксилилен 130—132
полпоксадиазолы 139—140
полисилоксаны 89, 144-—145
политиазолы 139—140
полпхпноксалины 138—139
получение по реакции Дильса —
Альдера 140—141
— в результате 1,3-дпполярного
присоединения 141 —142
спиро 142—143
Терполпмеризация 333, 533, 568
Тетрагидрофуран, полимеризация
427, 429, 430, 436
Тетрафторэтилеп, полимеризация 227
Тобольского метод определения эффек-
тивности инициатора 184—185
Толилизоцианат, реакция с «-бута-
нолом 63
Толуилендиизоцианат, поликонден-
сация 63
Точка гелеобразования 95
экспериментальное определение
102—104
Трансанулярная миграция 532
Трехмерный полимер см. Сетчатый
полимер
Триады тактические 492
Триоксан 431
Трихлорэтилен, сополимеризация
365, 366
Троммсдорфа эффект 235
Уравнение сополимеризации
влияние степени превращения
344—349
для многокомпонентных систем
352-356
применимость 339 — 340
для разных типов 340—344
соотношения реакционноспособ-
ностей мономеров 338, 349—
350, 357-361
Уравнение состава сополимера 336—
338
отклонение, влияние деполиме-
ризации 392—395
— кинетический эффект предпо-
следнего звена 390—392
Ускоритель 567
Фенплендипзоцианат, поликонденса-
ция 63
Фенилпзоциапат, реакция с «-бута-
нолом 63
Фенолформальдегидные полимеры 13
17, 24—40, 43, 62
синтез 109—ПО, 119
Флори распределение по молекуляр-
ному весу
теория 76—80
экспериментальные данные 80—
81
Формали циклические, полимериза-
ция 433
Формальдегид
полимеризация 318—319
сополимеризация 404—405
Фотосенсибилизаторы 172
Фотохимическая полимеризация 169
—173, 214—219, 578—579
Фриделя — Крафтса катализаторы
279
Функциональность мономера 93
Функциональные группы 45—50, 62
—64, 111 — 114, 198—199
Функция кислотности 56
Химические реакции полимеров 559
— 582
ароматического замещения 575
блок-сополимеров 581 — 582
галогенирование 573—575
привитых сополимеров 578—581
поливинилацетата 572—573
целлюлозы 570—572
циклизация 574, 576 — 577
Предметный указатель
603
Хлорстирол, сополимеризация 383,
384
Цвиттер-ионы 432
Центр стерической изомерии 474
Целлюлоза 14, 15
реакции 570—572
структура 489—490
триацетат 37
Цепные полимеры 11
Цепные сегменты 100
Циглера — Натта катализаторы, см.
Катализаторы, координационные
Циглера — Натта полимеризация
см. также Стереоспецифическая по-
лимеризация
механизм 505—520
неполярных алкенов 504—535
Циклизация 574, 576—577
внутримолекулярная 103—104,
158
в линейной поликонденсации
65-69
при сшивании 104
Циклические мономеры, реакцион-
ная способность при сополимери-
зации 463
Циклоалканы, полимеризация 459
Циклополимеризация 401
Циклополимеры 486—487
Частотный фактор 221, 295, 314
Чередование при сополимеризации
342-344
Шоттена — Баумана реакция 84
Эластическая деформация, определе-
ние 40
Эластомер 40—43, 124, 334
Электролитическая полимеризация
296
Электрон сольватированный 305
Элементарное звено 12, 58
Эмульгатор 258, 267
как ингибитор 272—273
Эмульсионная полимеризация 256—
273
бутадиена 257
инверсионные системы 268—269
ингибирование 272—273
инициирование 266—267
кинетика 261 — 264
компоненты 257 — 259, 266—267
место протекания процесса 259—
261
молекулярновесовое распределе-
ние 273
молекулярный вес 263—264
обрыв цепи 267—268
передача цепи 273
применение 25—257
рост цепи 267—268
стирола 257, 262, 263
теория Медведева — Шейнкер
269, 270—271
Смита — Эварта 265
--- отклонения 269—273
число частиц полимера 261, 264
266
эмульгатор 258, 267, 272—273
энергетика 268
Энергия активации
поликонденсации 82
полимеризации анионной 302,
313—315
— катионной 297—298
— радикальной 221 — 226
— с раскрытием цикла 433—435
— стереоспецифической 530
Энтропия
полимеризации лактамов 450
— радикальной 227
Эффективная константа скорости 293
Эффект координации 499—501
Эффект предпоследнего звена 390
Эффект соседней группы 560—561
Эфиры
простые 424, 430, 572
сложные 454—455, 571—572
Эпоксидные преполимеры 116—117
Эпоксиды
полимеризация анионная 414—
419
— стереоспецифическая 548—549
Этилакрилат, полимеризация 301
Этилен, полимеризация 208, 227, 529,
533
СОДЕРЖАНИЕ
Предисловие...................................................... 5
Предисловие автора .............................................. 7
Основные обозначения............................................. 9
Глава 1. Введение ............................................. И
1.1. Типы полимеров п реакций их образования................. И
1.1а. Состав п структура полимера......................... 11
1.16. Механизм образования полимеров...................... 19
1.2. Номенклатура полимеров ................................ 20
1.2а . Номенклатура, основанная на названии исходных сое-
динений ................................................. 21
1.26. Номенклатура, основанная на химическом строении 22
1.2в. Торговые и другие названия полимеров................ 23
1.3. Структурная форма полимерных молекул .................. 24
1.4. Молекулярный вес....................................... 27
1.5. Физическое состояние................................... 31
1.5а. Кристалличность и аморфность .................. . 31
1.56. Факторы, влияющие па степень кристалличности поли-
меров ............................................... 33
1.5в. Тепловые переходы .................................. 35
1.6. Применение полимеров................................... 39
1.6а. Механические свойства............................... 39
1.66. Эластомеры, волокна, пластмассы..................... 41
Вопросы и упражнения ........................................... 43
Список литературы............................................... 44
Глава 2. Поликонденсация........................................ 45
2.1. Реакционная способность функциональных групп .... 46
2.1а. Исходные предпосылки для анализа кинетики поли-
конденсации ........................................ 46
2.16. Экспериментальные доказательства................... 48
2.1 в. Теоретические положения .......................... 49
2.1г. Эквивалентность функциональных групп в бифункцио-
нальных реагентах ................................... 50
2.2. Кинетика поликонденсации............................... 51
2.2а. Самокаталпзпруемая поликонденсация.................. 53
2.2а.1. Экспериментальные данные . ................. 54
Содержание 605
2.2а.2. Причины отклонений от кривой третьего порядка 56
2.2а.З. Молекулярный вес полимера .................. 58
2.26. Внешний катализ поликонденсации .................... 59
2.2в. Другие реакции поликонденсации...................... 61
2.2г. Неэквивалентность функциональных групп нолпфупк-
циоиальпых реагентов . ..................... 62
2.3. Реакции циклизации в линейной полпкоидеисацпн .... 65
2.3а. Возможные реакции циклизации ....................... 65
2.36. Термодинамический и кинетический подход ..... 66
2.Зв. Другие факторы...................................... 70
2.4. Регулирование молекулярного веса полимера при линейной
поликонденсации .......................................... 71
2.4 а. Необходимость стехиометрического контроля .... 71
2.46 . Количественный подход............................. 72
2.5. Молекулярповесовое распределение в линейной поликон-
денсации . 76
2.5а. Распределение по размерам молекул .................. 76
2.56. Ширина молекулярновесового распределения .... 78
2.5в. Обменные реакции . 79
2.5г. Экспериментальные исследования...................... 80
2.6. Закономерности поликонденсации . ....................... 82
2.6а. Физические характеристики поликонденсационных
систем .................................................. 82
2.66. Различные системы исходных веществ.................. 83
2.66.1. Сложные полиэфиры ........................... 83
2.66.2. Полиамиды . 87
2.66.3. Полиуретаны ................................. 88
2.66.4. Полисилоксаны................................ 89
2.6в. Межфазная поликонденсация . ........................ 90
2.6в.1. Описание процесса . ....................... 90
2.6в.2. Применение............................ 92
2.7. Трехмерная поликонденсация . ........................... 93
2.7а. Разветвление ....................................... 93
2.76. Молекулярновесовое распределение............. 94
2.8. Сшивание................................................ 95
2.8а. Уравнение Карозерса.......................... 97
2.8а.1. Вывод для системы с эквивалентным соотноше-
нием исходных веществ ............................... 97
2.8а.2. Вывод для системы с неэквивалентным соотно-
шением исходных веществ ...................... 98
2.86. Статистическое рассмотрение гелеобразования .... 100
2.8в. Экспериментальное определение точки гелеобразования 102
2.9. Молекулярновесовое распределение в нелинейной поликон-
денсации .................................................... 104
2.10. Технология трехмерной поликонденсации ................. 107
2.10а. Статистические преполимеры ........................ 108
2.10а.1. Сложные полиэфиры . 109
2.10а.2. Фенолформальдегидные полимеры........ 110
2.10а.З. Мочевиноформальдегидпые полимеры . ... 114
606
Содержание
2.106. Преполимеры известной структуры .................. 115
2.106.1. Дполовые преполимеры....................... 116
2.106.2. Эпоксидные преполимеры..................... 116
2.106.3. Ненасыщенные сложные полиэфиры .... 118
2.106.4. Фенолформальдегидные полимеры . .... 119
2.11. Сополпкондепсацпя..................................... 120
2.11а. Тины сополимеров.................................. 120
2.116. Методы синтеза сополимеров ....................... 121
2.11в. Применение соноликонденсацпи...................... 123
2.12. Новые поликоиденсационные реакции .................... 125
2.12а. Новые промышленные полимеры....................... 125
2.12а.1. Поликарбонаты.............................. 126
2.12а.2. Полиамиды ................................. 126
2.12а.З. Простые ароматические полиэфиры............ 127
2.12а.4. Поли-71-ксилплен .......................... 130
2.12а.5. Ароматические полиимиды.................... 133
2.12а.6. Полибеизпмидазолы.......................... 134
2.126. Полимеры в стадии разработки ..................... 136
2.126.1. Полипмидазоппрролоиы . .................... 137
2.126.2. Полпбензоксазолы и полибензотиазолы . . . 138
2.126.3. Полпхпноксалины ........................... 138
2.126.4. Полиоксадиазолы и политриазолы ........... 139
2.126.5. Присоединение по реакции Дильса — Альдера 140
2.126.6. 1,3-Диполярное присоединение .............. 141
2.126.7. Спирополимеры......................... 142
2.126.8. Сополимеры ................................ 143
2.126.9. Неорганические и элементоорганические поли-
меры . ........................................... 144
Вопросы и упражнения ........................................... 145
Список литературы............................................... 148
Глава 3. Радикальная полимеризация ............................. 151
3.1. Природа радикальной полимеризации................ . . . 151
3.1 а. Сравнение полимеризации и поликонденсации .... 151
3.16 . Сравнение радикальной и ионной полимеризации . . . 152
3.16.1. Способность к полимеризации ................ 152
3.16.2. Влияние заместителей................... 154
3.2. Структурное расположение мономерных звеньев............ 156
3.2 а. Возможные способы роста .......................... 156
3.26 . Экспериментальные доказательства............. 158
3.3. Кинетика полимеризации .............................. 159
3.3 а. Последовательность стадий......................... 159
3.36 . Уравнение скорости реакции ....................... 161
3.4. Инициирование.......................................... 163
3.4а. Термический' распад инициаторов.................... 164
3.4а.1. Типы инициаторов............................ 164
3.4а.2. Кинетика инициирования и полимеризации 166
3.4а.3. Влияние концентрации инициатора иа скорость
полимеризации ................................. 166
3.4а.4. Влияние концентрации мономера па скорость
полимеризации ................................. 167
Содержание 607
3.46. Фотохимическое инициирование ...................... 169
3.46.1. Чистый мономер . ..................... 169
3.46.2. Фотолитическая диссоциация инициаторов . . . 171
3.46.3. Фотосенсибилизаторы . 172
3.46.4. Общие наблюдения .......................... 172
3.4в. Инициирование с помощью ионизирующего облучения 173
3.4г. Чисто термическое инициирование .................. 174
3.4д. Инициирование окислительно-восстановительными систе-
мами ............................................. 175
3.4е. Эффективность инициатора ......................... 178
3.4е.1. Определение f.............................. 178
3.4е.2. Механизм инициирования при f < 1 ..... 179
3.4е.3. Экспериментальные методы оценки f.......... 182
3.5. Молекулярный вес.................................... 185
3.5а. Длина кинетической цепи .......................... 185
3.56. Способ обрыва .................................... 186
3.6. Реакция передачи цепи ................................ 187
3.6 а. Влияние реакций передачи цепи ................... 187
3.66 . Передача цепи на мономер и инициатор........ 190
3.66.1. Определение См и Cj ....................... 190
3.66.2. Константы передачи цепи на мономер .... 191
3.66.3. Константы передачи цепи на инициатор . . . 193
З.бв. Передача цепи на специальный агент.......... 194
3.6в.1. Определение С$............................. 194
З.бв.2. Структура соединений и реакционная способ-
ность ........................................ 196
З.бв.З. Применение агентов передачи цепи .......... 200
3.6г. Передача цепи на полимер ......................... 200
3.7. Ингибирование и замедление............................ 204
3.7а. Типы ингибиторов и замедлителей .................. 204
3.76. Кинетика ингибирования или замедления............. 208
3.7в. Аутоингибирование аллильных мономеров ..;... 212
3.8. Определение констант абсолютных скоростей............. 213
3.8а. Кинетика нестационарного состояния ............... 213
3.86. Метод вращающегося сектора ....................... 216
3.8в. Значения параметров реакции полимеризации .... 219
3.9. Энергетические характеристики . ...................... 221
3.9а. Энергия активации и частотный фактор.............. 221
3.9а.1. Скорость полимеризации . 222
3.9а.2. Степень полимеризации...................... 224
3.96. Термодинамика полимеризации....................... 226
3.96.1. Значение AG, ДН и AS....................... 226
3.96.2. Влияние химического строения мономера . . . 228
3.96.3. Полимеризация 1,2-дпзамещенных производных
этилена . ......................................... 230
3.9в. Равновесие полимеризация — деполимеризация. Пре-
дельная температура ................................ 231
3.10. Аутоускорение........................................ 235
3.10а. Ход процесса..................................... 235
3.106. Диффузный фактор ................................ 236
З.Юв. Влияние условий реакции........................... 238
608
Содержание
3.10г. Зависимость скорости полимеризации от инициатора 239
3.11. Молекулярновесовое распределение.................... 240
3.11а. Начальные стадии полимеризации ................. 240
3.116. Высокие степени завершенности полимеризации . . , 241
3.13. Влияние давления.................................... 242
3.12а. Объем активированного комплекса................. 243
3.126. Скорость полимеризации.......................... 244
3.12в. Степень полимеризации........................... 245
3.13. Способы проведения полимеризации ................... 246
3.13а. Полимеризация в блоке (или в массе)....... . 247
3.136. Полимеризация в растворе ....................... 248
3.13в. Полимеризация в суспензии....................... 249
3.13г. Твердофазная полимеризация . 249
3.14. Полимеризация диепов . 250
Вопросы и упражнения.......................................... 251
Список литературы............................................. 253
Глава 4. Эмульсионная полимеризация........................... 256
4.1. Описание процесса.................................. 256
4.1 а. Применение..................................... 256
4.16 . Качественная характеристика .................... 257
4.16.1. Компоненты и их ориентация в мицелле . . . 257
4.16.2. Место протекания полимеризации............ 259
4.2. Кинетика процесса ................................... 260
4.2 а. Скорость полимеризации.......................... 260
4.26 . Степень полимеризации . ........................ 263
4.3. Число полимерных частиц.............................. 264
4.4. Закономерности эмульсионной полимеризации ........... 266
4.4а. Инициирование .................................. 266
4.46. Эмульгаторы ..................................... 267
4.4в. Рост и обрыв .................................... 267
4.4г. Энергетика................:...................... 268
4.4д. Инверсионные эмульсионные системы ............... 268
4.5. Отклонения от теории Смита — Эварта ................. 269
4.5а. Величина и....................................... 270
4.56. Теория Медведева — Шейнкер ...................... 270
4.5в. Влияние растворимости в воде..................... 272
4.5г. Ингибирование эмульгатором....................... 272
4.5д. Молекулярновесовое распределение . .............. 273
Вопросы и упражнения.......................................... 273
Список литературы............................................. 274
Глава 5. Ионная полимеризация................................. 276
5.1. Сравнение радикальной и ионной полимеризаций......... 276
5.2. Катионная полимеризация алкенов ..................... 278
5.2а. Инициирование ................................... 278
5.2а.1. Протонные кислоты .... 278
5.2а.2. Кислоты Льюиса............................ 279
Содержание 609
5.2а.З. Другие катализаторы......................... 281
5.26. Рост цепи ......................................... 281
5.2в. Обрыв цепи........................................ 283
5.2в.1. Отсутствие обрыва кинетической цепи .... 283
5.2в.2. Обрыв кинетической цепи..................... 284
5.2г. Кинетика ионной полимеризации ..................... 286
5.2г.1. Различные кинетические положения............ 286
5.2г.2. Справедливость допущения стационарного состоя-
ния ......................................... 288
5.2д. Абсолютные копстаиты скорости ..................... 288
5.2д.1. Экспериментальные методы.................... 288
5.2д.2. Сравнение скоростей катионной и радикальной
полимеризаций ............................... 289
5.2д.З. Величины См и С§ ........................... 290
5.2е. Влияние реакционной среды ......................... 292
5.20.1. Влияние растворителя'....................... 293
5.2е.2. Влияние противоиона ........................ 295
5.20.3. Псевдокатионпая полимеризация............... 296
5.2ж. Энергетика......................................... 297
5.3. Анионная полимеризация алкенов ....................... 299
5.3 а. Инициирование амидами металлов ................... 300
5.36 . Полимеризация, протекающая без обрыва цепи . . . 302
5.36.1. Инициирование путем переноса электрона . . . 303
5.36.2. Инициирование алкилами металлов ............ 305
5.36.3. Скорость полимеризации...................... 306
5.36.4. Влияние реакционной среды ........ 307
5.36.5. Степень полимеризации....................... 311
5.36.6. Ассоциация ................................. 312
5.36.7. Энергетика ................................. 313
5.4. Установление механизма полимеризации....................315
5.5. Полимеризация по карбопильпоп группе .................. 316
5.5а. Анионная полимеризация............................. 318
5.5а.1. Полимеризация формальдегида................. 318
5.5а.2. Полимеризация других карбонильных мономе-
ров ......................................... 319
5.56. Катионная полимеризация............................ 320
5.5в. Радикальная полимеризация.......................... 322
5.5г. Ступенчатая полимеризация......................... 323
5.5д. Блокирование концевых групп........................ 323
. .6. Различные^мономеры.................................... 324
5.6а. Мономеры с двумя различными полимеризующимися
группами ................................................. 324
5.6а.1. Диметилкетен................................ 324
5.6а.2. Акролеин................................... 325
5.66. Акриламид.......................................... 326
, 5.6в. Изоцианаты .............................. 327
5.6г. Диазоалкаиы........................................ 328
5.6д. Мономеры с тройной связью ......................... 328
Вопросы и упражнения............................................ 329
Список литературы . .............................:.............. 330
610
Содержание
Глава 6. Сополимеризация............................. 333
6.1. Общие положения ....................................... 333
6.1а. Значение сополимеризации........................... 333
6.16. Типы сополимеров................................... 334
6.2. Состав сополимера ..................................... 336
6.2а. Уравнение состава сополимера ...................... 336
6.26. Пределы применимости уравнений сополимеризации 339
6.2в. Типы сополимеризации............................... 340
6.2в.1. Идеальная сополимеризация: Г]Г2= 1 .... 340
6.2в.2. Сополимеризация, приводящая к образованию
регулярно чередующихся сополимеров: ?’!= z'2= 0 342
6.2в.З. Блочная сополимеризация: 1, г2> 1 . . . 344
6.2г. Изменение состава сополимера в зависимости от степени
превращения............................................... 344
6.2д. Экспериментальное определение констант сополимери-
зации .................................................... 349
6.2е. Микроструктура сополимеров......................... 350
6.2ж. Многокомпонентные системы.......................... 523
6.3. Радикальная сополимеризация............................ 356
6.3 а. Влияние условий реакции .......................... 356
6.36 . Реакционная способность .......................... 357
6.36.1. Влияние сопряжения (резонансный эффект) 361
6.36.2. Влияние стерических затруднений (стерическип
эффект)............................................. 365
6.36.3. Влияние полярности (полярный эффект) . . . 366
6.36.4. Схема Q — е................................ 370
6.36.5. Определение активности из произведения вероят-
ностей ............................................. 374
6.Зв. Скорость сополимеризации........................... 376
6.3в.1. Обрыв цепи, контролируемый химическими реак-
циями .............................................. 376
6.Зв.2. Обрыв, контролируемый диффузией ..... 379
6.4. Ионная сополимеризация ................................ 381
6.4а. Катионная сополимеризация.......................... 382
6.4а.1. Активность мономеров....................... 382
6.4а.2. Влияние растворителя и противоиона......... 384
6.4а.З. Влияние температуры........................ 386
6.46. Анионная сополимеризация........................... 386
6.46.1. Активность мономеров....................... 386
6.46.2. Влияние растворителя и противоиона......... 387
6.5. Отклонения от уравнения состава сополимера............. 389
6.5 а. Кинетический эффект предпоследнего звена.......... 390
6.56 . Деполимеризация в процессе сополимеризации .... 392
6.6. Сополимеризация диенов..................'.............. 395
6.6 а. Сшивание цепей.................................... 395
6.66 . Сополимеризация, происходящая с чередованием внутри-
и межмолекулярных реакций............................... 399
6.7. Другие случаи сополимеризации ..............,.......... 403
6.7а. Сополимеризация алкенов ........................... 403
6.76. Сополимеризация карбонильных мономеров ............ 404
Содержание 611
Вопросы и упражнения.......................................... 405
Список литературы............................................. 407
Глава 7. Полимеризация с раскрытием цикла.................... 411
7.1. Общие характеристики................................. 112
7.1а. Механизм и кинетика полимеризации ............... 412
7.2. Циклические эфиры.................................... 414
7.2а. Анионная полимеризация эпоксидов ................ 414
7.2а.1. Характеристики реакции .................. 414
7.2а.2. Реакции обмена........................... 416
7.2а.З. Перенос цепи на мономер ................. 418
7.26. Катионная полимеризация.......................... 419
7.26.1. Инициирование . ........................ 420
7.26.2. Обрыв цепи............................... 424
7.26.3. Кинетика................................. 426
7.26.4. Циклические ацетали . .. ................ 431
7.26.5. Энергетические характеристики............ 433
7.3. Циклические амиды.................................... 437
7.3а. Анионная полимеризация..............•........... 437
7.3а.1. Инициирование сильными основаниями .... 437
7.За.2. Применение ацилирующих агентов........... 440
7.36. Катионная полимеризация.......................... 444
7.36.1. Полимеризация в присутствии протонных кислот 444
7.36.2. Полимеризация в присутствии аминов .... 445
7.Зв. Катализ водой.................................... 446
7.3в.1. Механизм реакции ........................ 446
7.Зв.2. Степень полимеризации.................... 447
7.3г. Реакционная способность . ..................... 449
7.4. Циклосилоксаны ...................................... 451
7.4 а. Анионная полимеризация . ...................... 452
7.46 . Катионная полимеризация . ..................... 452
7.5. Другие циклические мономеры ......................... 454
7.5а. Сложные циклические эфиры ....................... 454
7.56. Циклические амины . 455
7.5в. Циклические сульфиды ............................ 456
7.5г. N-Карбоксиангидриды а-аминокислот ............... 457
7.5д. Различные случаи полимеризации................... 458
7.6. Полимеризация неорганических или элементоорганических
мономеров.................................................. 459
7.6 а. Полифосфазены .................................. 459
7.66 . Другие случаи полимеризации .................... 461
7.7. Сополимеризация . 462
7.7а. Мономеры, содержащие одинаковые функциональные
группы . 463
7.76. Мономеры, содержащие различные функциональные
группы............................................ 466
Вопросы и упражнения.......................................... 468
Список литературы............................................. 469
612
Содержание
Глава 8. Стереохимия полимеризации и поликонденсации........... 472
8.1. Типы стереоизомерии в полимерах........................ 473
8.1а. Полимеризация мопозамещенных этиленов.............. 474
8.1а.1. Центр стерической изомерии ......... 474
8.1а.2. Тактичность.......................... 475
8.16. Полимеризация дизамещенных этиленов................ 477
8.16.1. Полимеры 1,1-дпзамещениых этиленов .... 477
8.16.2. Полимеры 1,2-дизамещенных этиленов .... 477
8.1в. Полимеризация карбонильных соединений и полимериза-
ция с раскрытием цикла . ............... 479
8.1г. Полимеризация бутадиена-1,3 и 2-замещенпых бутадпе-
иов-1,3............................................ 481
8.1г.1. 1,2- и 3,4-Полимеризация .................. 481
8.1г.2. 1,4-Полимеризация.......................... 482
8.1д. Полимеризация 4-мопо- и 1,4-дизамещенных бутадие-
пов-1,3 484
8.1Д.1. 1,2- и 3,4-Полимеризация .................. 484
8.1д.2. 1,4-Полпмеризация.......................... 484
8.1е. Циклополимеры...................................... 486
8.2. Свойства стереорегулярных полимеров ................... 487
8.2а. Значение стереорегулярпости полимеров.............. 487
8.2а.1. Изотактический, синдиотактический и атакти-
ческий полипропилен ............................... 488
8.2а.2. цис- и транс-i,4-Полидиены-1,2............. 488
8.2а.З. Целлюлоза и амилоза ....................... 489
8.26. Анализ стереорегулярпости.......................... 491
8.26.1. Экспериментальные методы .................. 491
8.26.2. Оценка тактичности......................... 491
8.3. Факторы, влияющие па образование стереорегулярных поли-
меров и полимеризация алкенов . ........................... 493
8.3а. Радикальная полимеризация.......................... 494
8.3а.1. Энергетика образования синдиотактических и изо-
тактических структур .............................. 495
8.За.2. Распределение по размеру стереорегулярных
блоков ............................................ 498
8.36. Ионная и координационная полимеризация ............ 499
8.36.1. Эффект координации . ...................... 499
8.36.2. Механизм стереоспецифического присоединения 501
8.4. Полимеризация непоЛярпых алкенов по Циглеру — Патта 504
8.4а. Механизм полимеризации Циглера — Натта............. 505
8.4а. 1. Химическая природа растущих частиц....... 506
8.4а.2. Рост через связь углерод — переходный металл 508
8.4а.З. Биметаллический и монометаллический меха-
низм .............................................. 509
8.4а.4. Физическая природа катализатора ........... 512
8.4а.5. Направление раскрытия двойной связи .... 519
8.46. Влияние природы компонентов катализатора Циглера—
Н атта .................................................. 520
8.46.1. Компонент, в котором участвует переходный
металл ...................................... 520
Содержание 613
8.46.2. Компонент, содержащий металл групп 1—3 522
> 8.46.3. Третий компонент......................... 524
8.4в. Кинетика.......................................... 525
8.4в.1. Схема реакции............................. 525
8.4в.2. Уравнения скорости реакции ............... 526
8.4в.З. Экспериментальная кинетика................ 527
8.4в.4. Кинетические параметры.................... 529
8.4г. Области применения катализаторов Циглера — Натта 530
8.4г.1. Циклоалкены . 531
8.4г.2. Сополимеризация........................... 532
8.4д. Окисные катализаторы на носителях ................ 534
8.5. Стереоспецифическая полимеризация полярных алкенов 535
8.5а. Полимеризация метилметакрилата.................... 536
8.56. Полимеризация простых виниловых эфиров ........... 539
8.5в. Полимеризация стирола . .......................... 539
8.6. Стереоспецифическая полимеризация диенов-1,3 ..... 540
8.6а. Радикальная полимеризация......................... 540
8.66. Анионная и координационная полимеризация .... 541
8.6в. Катионная полимеризация........................... 547
3.7. Другие реакции стереоспецифической полимеризации . . . 548
8.7а. Полимеризация эпоксидов........................... 548
8.76. Полимеризация карбонильных мономеров ............. 549
8.8. Оптическая активность в полимерах..................... 549
8.8а. Полимеризация на оптически активных катализаторах 550
8.86. Полимеризация оптически активных мономеров .... 552
Вопросы и упражнения . .................................. 553
Список литературы . ........................................... 554
Глава 9. Химические реакции полимеров.......................... 559
9.1. Реакционная способность полимеров.................... 559
9.2. Сшивание.............................................. 562
9.2а. Отверждение ненасыщенных сложных полиэфиров . . . 562
9.2а.1. Сополимеризация .......................... 562
9.2а.2. Сшивание кислородом....................... 563
9.26. Сшивание эластомеров па основе диенов-1,3........ 564
9.26.1. Вулканизация серой........................ 565
9.26.2. Ускоренная вулканизация серой ............ 566
9.2 в. Сшивание полиолефинов и полисилоксапов .......... 567
9.2 г. Другие процессы сшивания......................... 569
9.3. Реакции целлюлозы..................................... 570
9.3а. Растворение целлюлозы . 570
9.36. Образование сложных эфиров . ................. 571
9.Зв. Образование простых эфиров ....................... 572
9.4. Реакция поливинилацетата . ........................... 572
9.5. Галогенирование ..................................... 573
9.5 а. Натуральный каучук ............................. 573
9.56 . Насыщенные полиуглеводороды . ................... 574
9.6. Ароматическое замещение . 575
9.7. Циклизация ........................................... 576
9.8. Привитые и блок-сополимеры............................ 577
614 Содержание
9.8а. Привитые сополимеры................................. 578
9.8а. 1. Передача цепи.............................. 578
9.8а.2. Ультрафиолетовое и ионизирующее облучение . 578
9.8а.З. Окислительно-восстановительные системы . . . 580
9.8а.4. Другие привитые системы .................... 580
9.86. Блок-сополимеры .................................. 581
9.86.1. Механохимический способ .................... 581
9.86.2. Другие методы ............................. 582'
Вопросы и упражнения ........................................... 582.
Список литературы . 583
Список дополнительной литературы ............................... 584