



















































































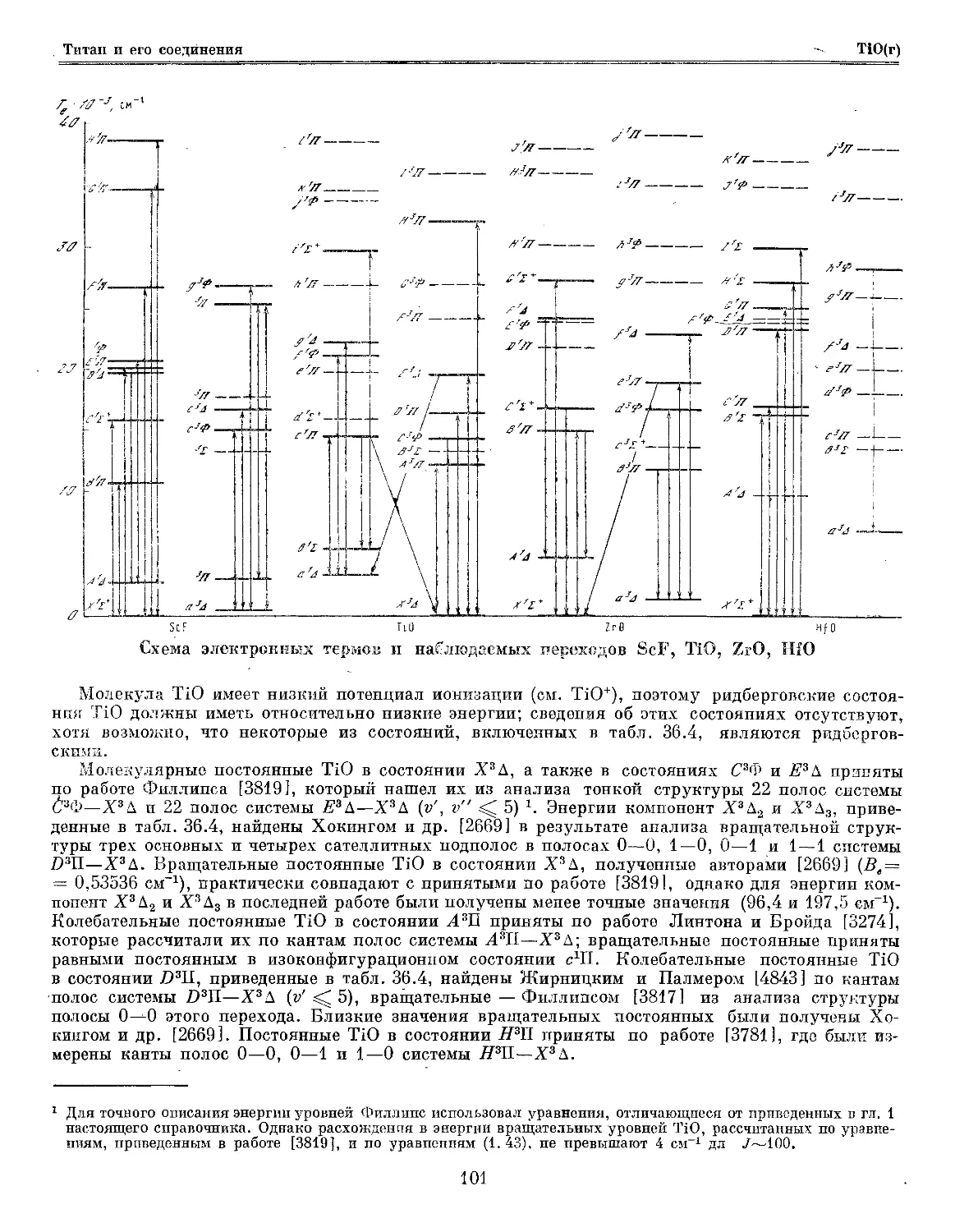

















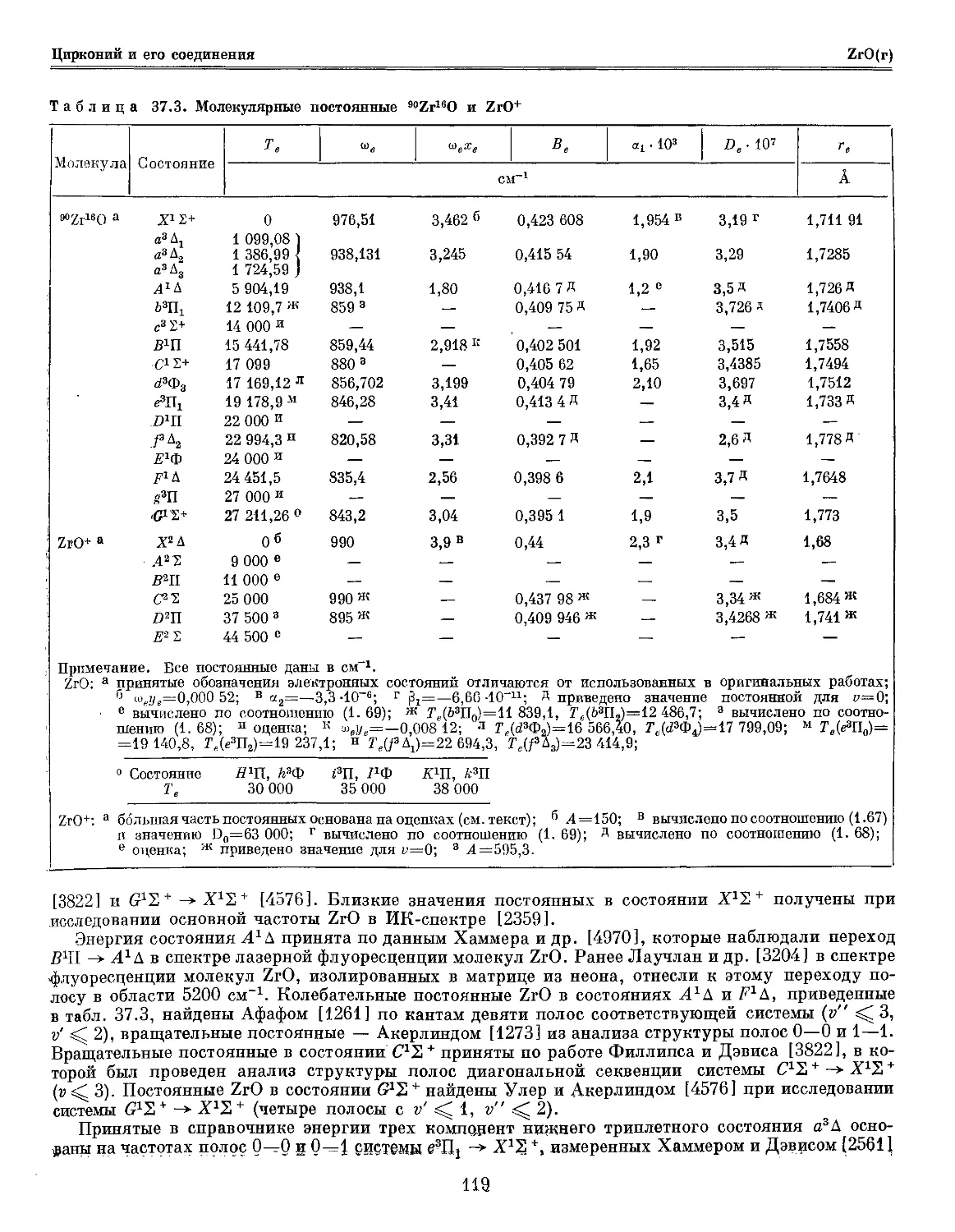
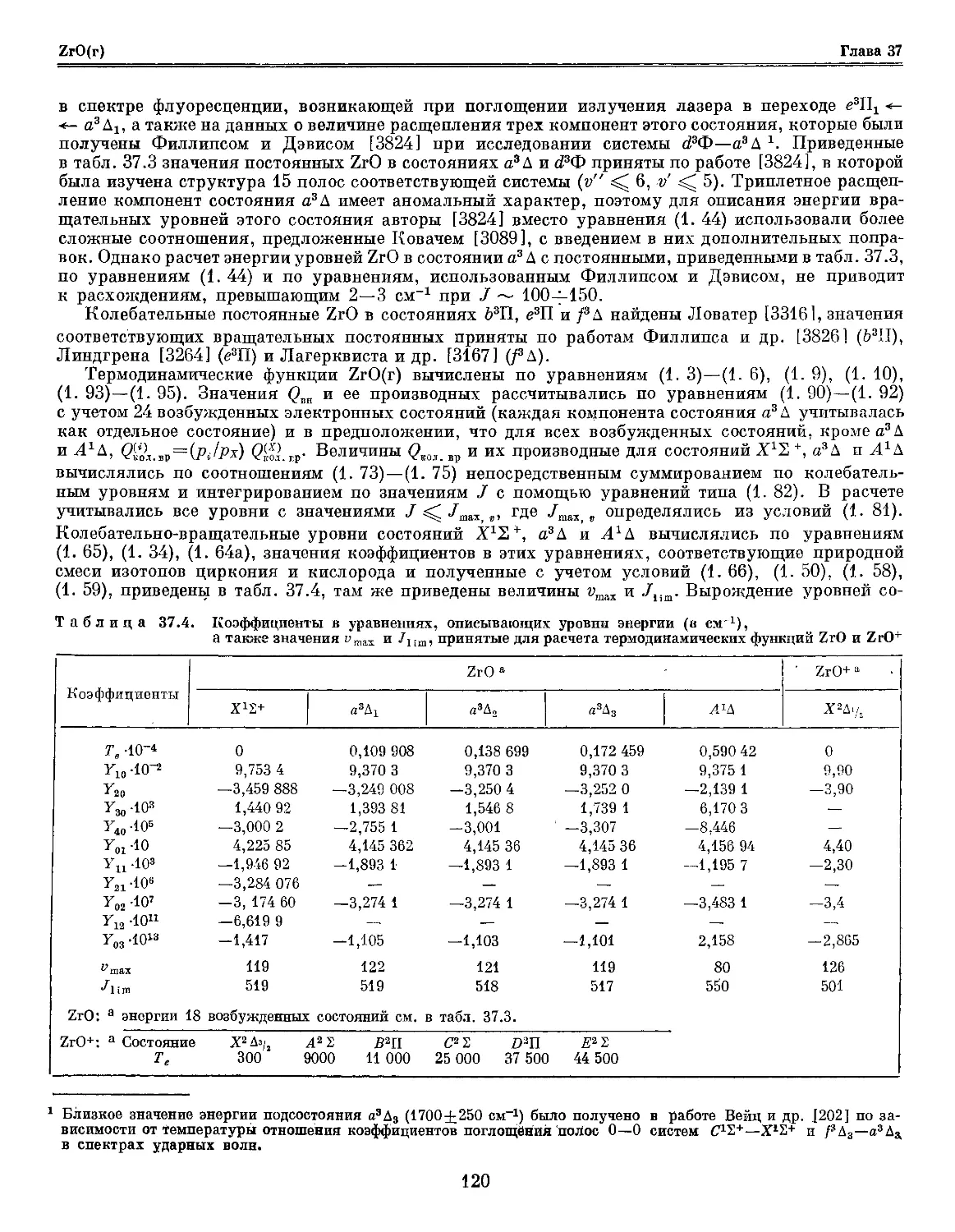
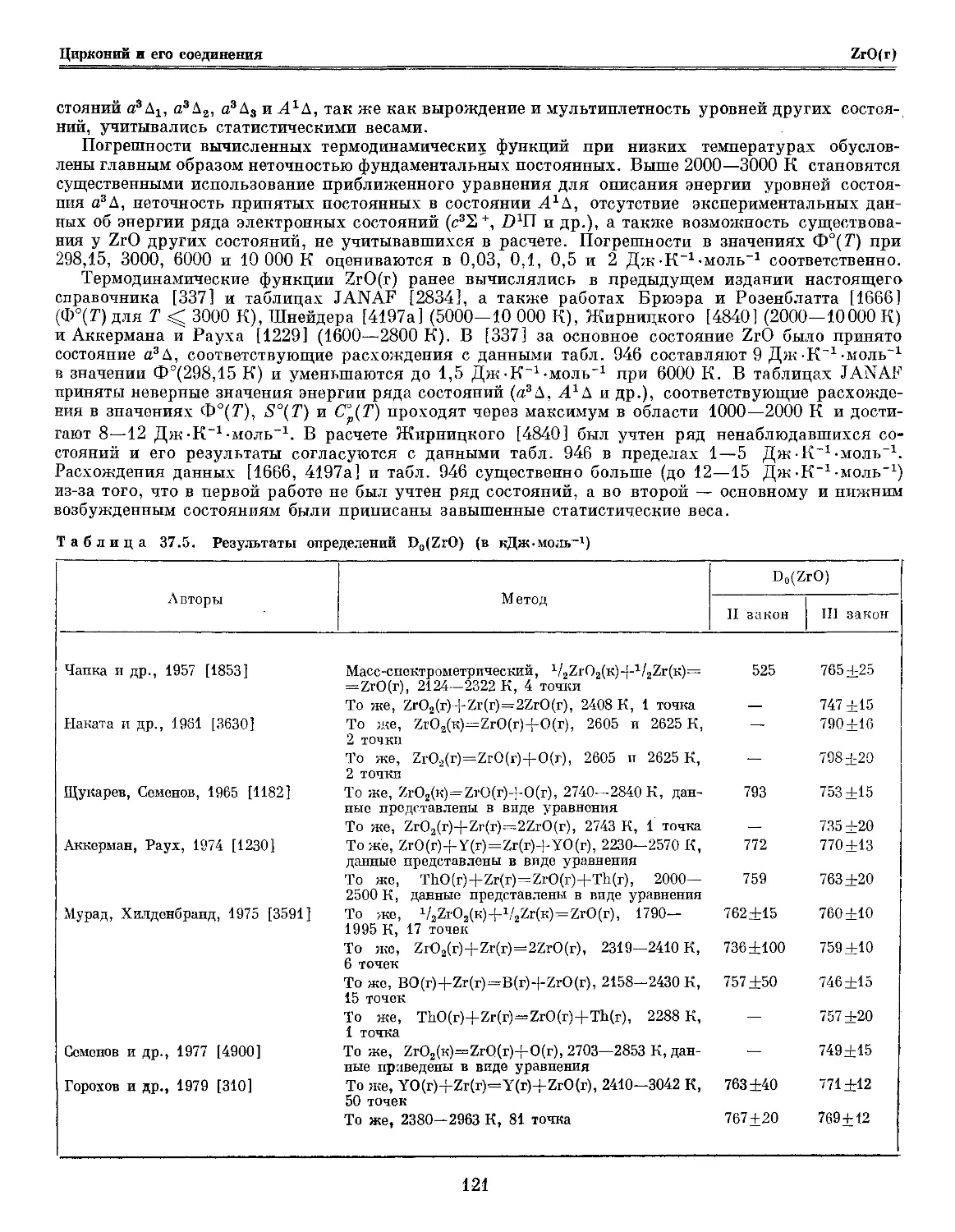




















































































































































































































































































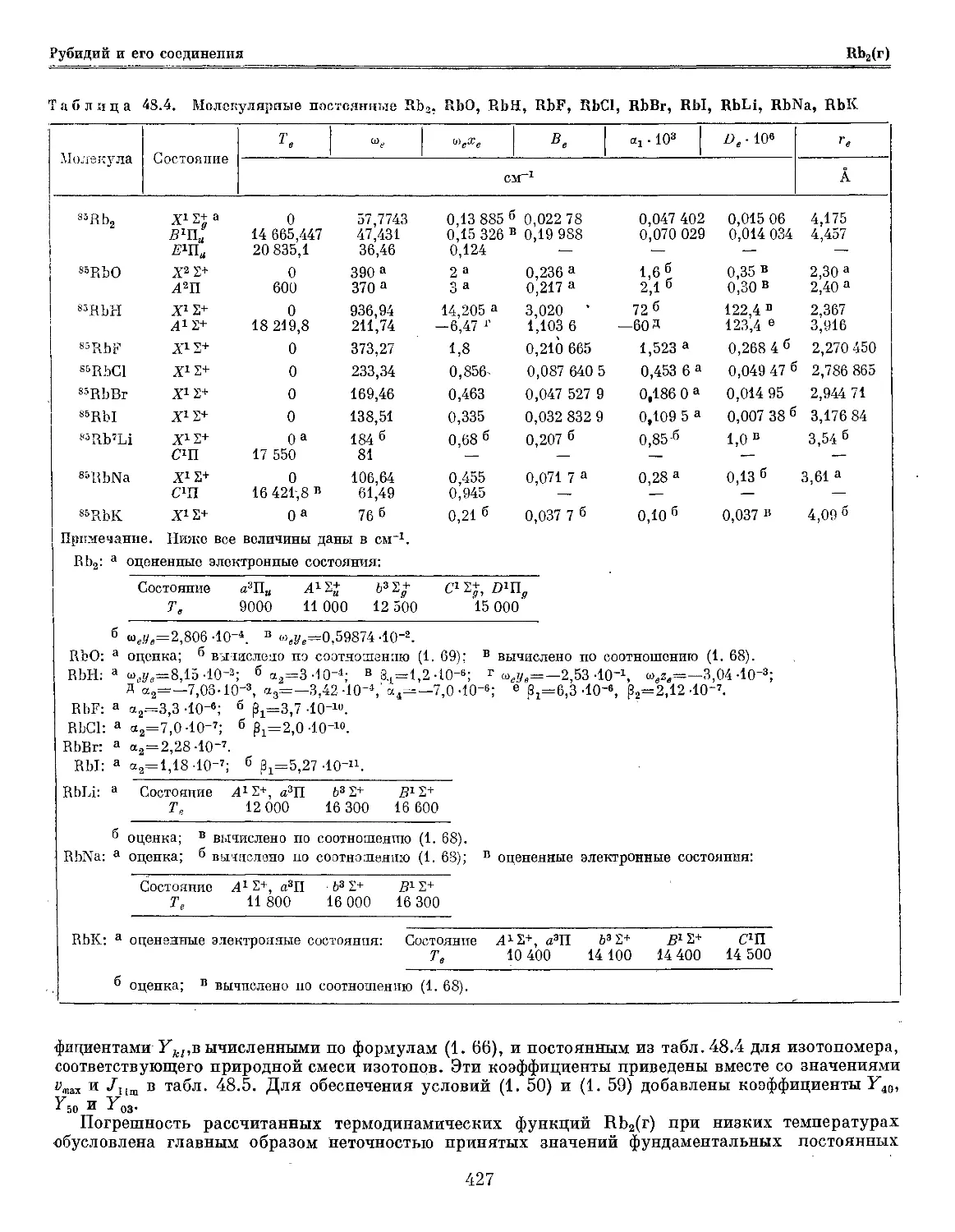
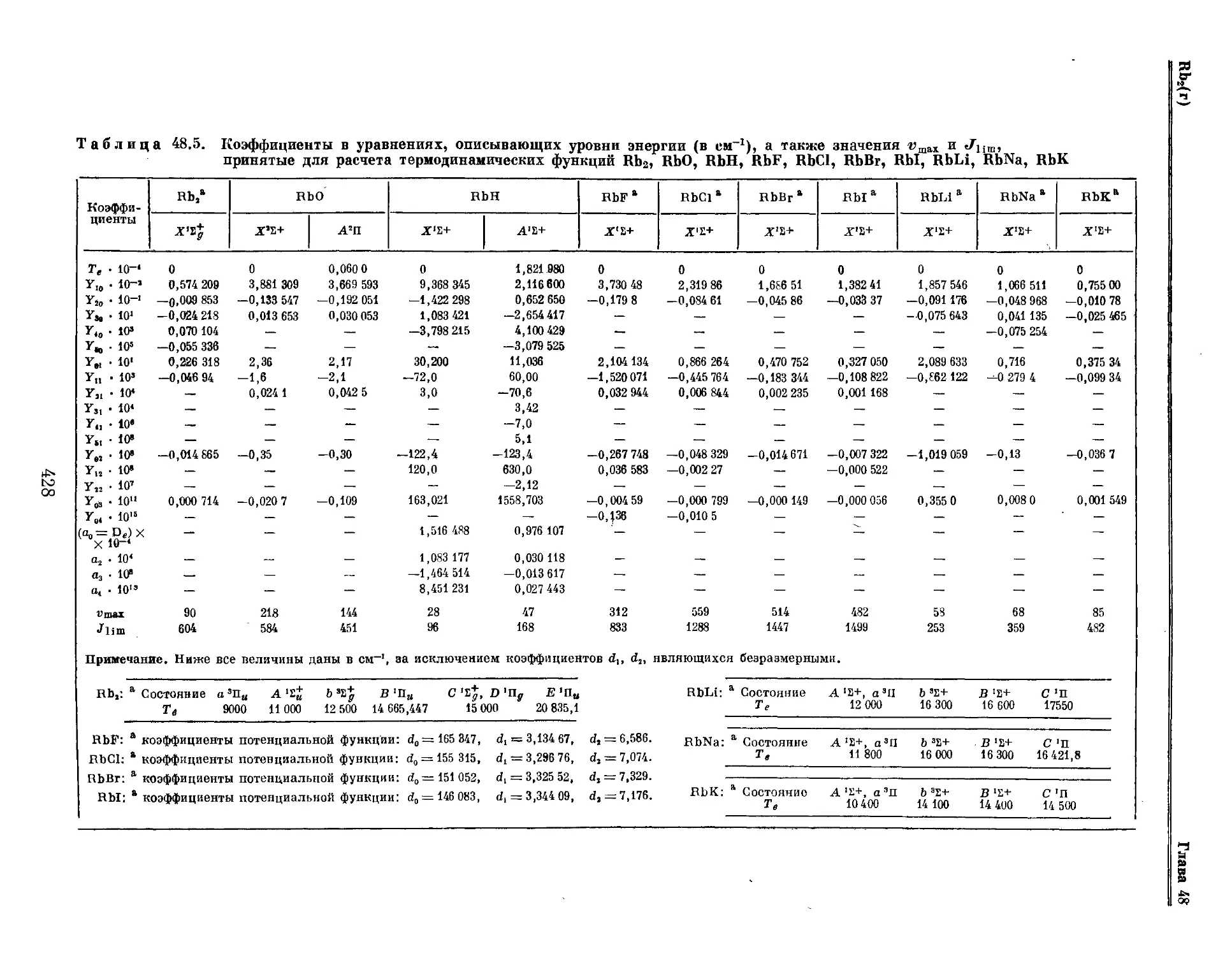



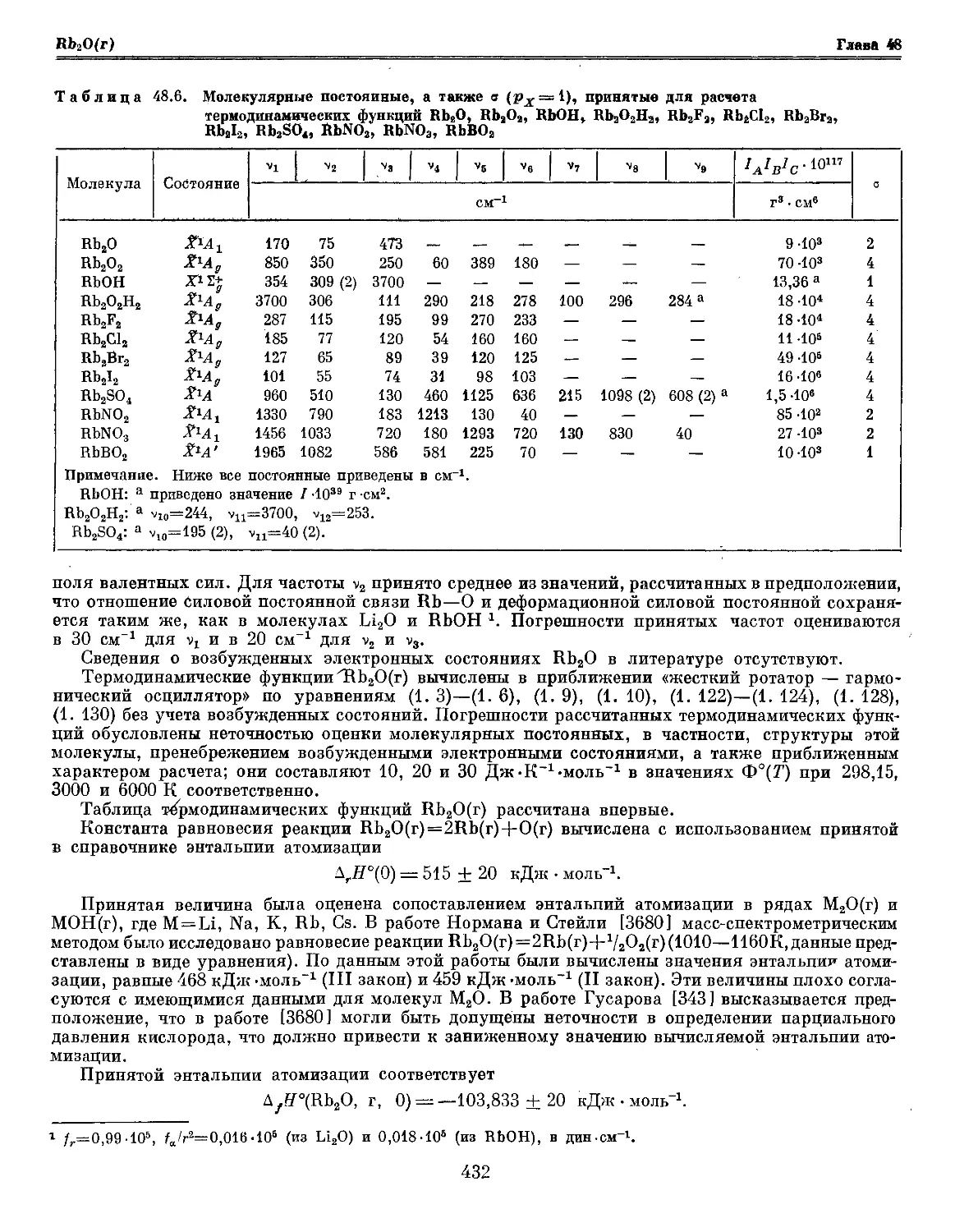




















































































































































































Текст
АКАДЕМИЯ НАУК СССР
ИНСТИТУТ ВЫСОКИХ ТЕМПЕРАТУР
ГОСУДАРСТВЕННЫЙ ИНСТИТУТ ПРИКЛАДНОЙ ХИМИИ
ТЕРМОДИНАМИЧЕСКИЕ
СВОЙСТВА
ИНДИВИДУАЛЬНЫХ
ВЕЩЕСТВ
СПРАВОЧНОЕ ИЗДАНИЕ
В ЧЕТЫРЕХ ТОМАХ
Издание третье, переработанное и расширенное
Согласовано с Государственной службой
стандартных справочных данных
РЕДАКЦИОННАЯ КОЛЛЕГИЯ:
В. П. ГЛУШКО (ответственный редактор),
Л.-З^ГУРВИЧ (зам. ответственного редактора),
Г. А. 6ЕРГМАН, И. В. ВЕЙЦ, В.А.МЕДВЕДЕВ,
Г. А. ХАЧКУРУЗОВ, В. С. ЮНГМАН v
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 198
ТЕРМОДИНАМИЧЕСКИЕ
СВОЙСТВА
ИНДИВИДУАЛЬНЫХ
ВЕЩЕСТВ
Том IV
ЭЛЕМЕНТЫ
Сг, Mo, W, V, Nb, Та, Ti, Zr, Hf, Sc,
Y, La, Th, U, Pn, Li, Na, K, Rb, Cs
И ИХ СОЕДИНЕНИЯ
Книга 1
ВЫЧИСЛЕНИЕ
ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
ИЗДАТЕЛЬСТВО «НАУКА» МОСКВА 1982
УДК 536-|54J.l
Термодинамические свойства индивидуальных веществ. Справочное издание: В 4-х т.
/ Л. В. Гурвич, И. В. Вейц, В. А. Медведев и др. —3-е изд., перераб. и расширен. —
Т. IV. Кн. 1. —М.: Наука, 1982. — 623 с.
Издание состоит из четырех томов, каждый том — из двух книг: в первой описаны методы
расчета, во второй книге помещены таблицы термодинамических свойств.
В первой книге IV тома изложены результаты критического выбора постоянных, необходимых
для расчета термодинамических свойств (молекулярных постоянных, энтальпий образования
и фазовых переходов, теплоемкости, энергии диссоциации и т. д.) соединений Сг, Mo, W, V, Nb,
Та, Ti, Zr, Hf, So, Y, La, Th, U, Pu, Li, Na, K, Rb и Cs, описаны расчеты таблиц
термодинамических свойств и дана оценка их достоверности для каждого вещества. Для ряда газов
рассмотрены свойства при повышенных давлениях.
Л. В. ГУРВИЧ
И. В. ВЕЙЦ
В. А. МЕДВЕДЕВ
Г. А. БЕРГМАН
В. С. ЮНГМАН
Г. А. ХАЧКУРУЗОВ
В. С. ИОРИШ
О. В. ДОРОФЕЕВА
Е. Л. ОСИНА
АВТОРЫ
П. И. ТОЛМАЧ
И. Н. ПРЖЕВАЛЬСКИЙ
И. И. НАЗАРЕНКО
Н. М. АРИСТОВА
Е. А. ШЕНЯВСКАЯ
Л. Н. ГОРОХОВ
A. Л. РОГАЦКИЙ
М. Е. ЕФИМОВ
B. Я. ЛЕОНИДОВ
Ю. Г. ХАИТ
А. Г. ЕФИМОВА
С. Э. ТОМБЕРГ
A. В. ГУСАРОВ
Н. Э. ХАНДАМИРОВА
Г. Н. ЮРКОВ
Л. Р. ФОКИН
Л. Ф. КУРАТОВА
B. Г. РЯБОВА
2602040600-052
055 @2)-82
Подписное к»даяие
¦Q Издательство «Наука», 1982 г.
Эта книга не может быть полностью или частично
воспроизведена или размножена, введена в
информационно-поисковую систему или передана
по линиям связи в любой форме или любыми
средствами (в том числе электронными
устройствами и на магнитных носителях информации)
без письменного разрешения издательства
«Наука»,
ОГЛАВЛЕНИЕ
От редколлегии 6
Предисловие к IV тому 7
Глава 30. Хром и его соединения 9
Глава 31. Молибден и его соединения . 2И
Глава 32. Вольфрам и его соединения .......... 39
Глава 33. Ванадий и его соединения 57
Глава 34. Ниобий и его соединения 71
Глава 35. Тантал и его соединения 83
Глава 36. Титан и его соединения 93
Глава 37. Цирконий и его соединения 114
Глава 38. Гафний и его соединения . , 127
Глава 39. Скандий и его соединения 137
Глава 40. Иттрий и его соединения . 147
Глава 41. Лантан и его соединения 158
Глава 42. Торий и его соединения '.-'. 169
Глава 43. Уран и его соединения 181
Глава 44. Плутоний и его соединения , 231
Глава 45. Литий и его соединения 244
Глава 46. Натрий и его соединения 309
Глава 47. Калий и его соединения 365
Глава 48. Рубидий и его соединения 422
Глава 49. Цезий и его соединения 463-
ПРИЛОЖЕНИЯ
Приложение 3. Термодинамические свойства газов при повышенном
давлении 510
Приложение 5. Термодинамические свойства водных ионов в
стандартном состоянии 517
Приложение 6. Приближенная оценка энергии электронных
состояний атомов и ионов . . 518
Приложение 7. Вычисление (>К01.вр через потенциал межатомного
взаимодействия 519
Приложение 8. Вычисление энтальпии сублимации при сложном
составе пара 521
Литература 523
Уточнение выбора постоянных, принятых в I и III томах (Приложение 9) 602
Формульный указатель веществ . 606
Список исправлений к томам I — III 621
ОТ РЕДКОЛЛЕГИИ
Публикацией четвертого тома завершается третье издание справочника «Термодинамические
свойства индивидуальных веществ». При подготовке этого издания было проанализировано около
17 000 работ; на основании критического анализа этих работ, многочисленных расчетов и оценок
были выбраны молекулярные, термодинамические и термохимические постоянные, необходимые
для вычисления таблиц термодинамических свойств свыше 1100 веществ — соединений 50
элементов (О, Н, F, Cl, Br, I, He, Ne, Ar, Кг, Хе, Rn, S, N, Р, С, Si, Ge, Sn, РЬ, В, Al, Ga, In,
Tl, Cr, Mo, W, V, Nb, Та, Ti, Zr, Hf, Sc, Y, La, Th, U, Pu, Be, Mg, Ca, Sr, Ba, Li, Na, K,
Rb и Gs), а также изотопов водорода и лития (D, T, 6Li и 7Li). Рассчитаны для широкого
интервала температур 278 таблиц термодинамических свойств конденсированных веществ и 1057
таблиц термодинамических свойств газов в стандартном состоянии (для 230 газов вплоть до 10 000
или 20 000 К). Для 30 веществ в конденсированном состоянии и 240 газов таблицы
термодинамических свойств публикуются впервые, для большинства остальных веществ выполненные расчеты
позволили существенно повысить точность табулированных величин и расширить интервал
температур по сравнению с данными, известными в литературе.
Подготовка I тома этого издания была завершена в 1976 г. Анализ литературы,
опубликованной после 1976 г., показал, что только для нескольких веществ (Щ, НО2, HOg, DO2, DOj, HOC1,
SF5, SFJ, BC1) результаты новых исследований позволяют существенно уточнить рассчитанные
термодинамические свойства. Для этих веществ в Приложении 9 первой книги этого тома
приведены обоснования выбора новых значений постоянных, а в Дополнении 2 второй книги —
новые таблицы термодинамических свойств. В Дополнение 2 включены также новые таблицы
термодинамических свойств H2F2, SiO, DO", Хе2, SH, SH~, SF~, G2F2Gla, SiO2, (к, >K)SiC (к, ж), A1F~. Для
двух первых расширен температурный интервал, для остальных — устранены ошибки,
допущенные в расчетах. Этим таблицам приписаны прежние номера с буквой «а».
Для удобства читателей в четвертый том включен формульный указатель, охватывающий
весь материал справочника. В обеих книгах четвертого тома даны сводные таблицы исправлений
и опечаток, обнаруженных в соответствующих книгах томов I—IV.
На базе методических разработок, алгоритмов и программ, созданных при подготовке этого
издания, в 1981 г. в Институте высоких температур АН СССР был введен в эксплуатацию Банк
данных по термодинамическим свойствам индивидуальных веществ — ИВТАНТЕРМО [4868а].
Принципиальная особенность ИВТАНТЕРМО, отличающая его от известных зарубежных и
отечественных банков данных, заключается в том, что все накапливаемые в нем таблицы
термодинамических свойств не являются компиляцией материалов, опубликованных в различных
справочниках, а вычисляются с помощью комплекса специальных программ на основании
постоянных, отбираемых, в результате критического анализа и обработки всей первичной
литературы. Создание такого Банка позволило не только существенно повысить точность и обеспечить
полную внутреннюю согласованность данных, накапливаемых и выдаваемых ИВТАНТЕРМО,
но и сделало возможной публикацию таблиц термодинамических свойств III и IV томов этого
издания непосредственное магнитных лент, выдаваемых ИВТАНТЕРМО. Намеченное развитие
ИВТАНТЕРМО в 1983—1990 гг. позволит наряду с уточнением материалов этого издания
рассчитать свойства соединений Cr, Mo, W, V, Nb, Та, Ti, Zr, Hf, Sc, Y, La, Th, U, Pu с
галогенами, серой, азотом и углеродом, а также соединений Mn, Fe, Co, Ni, Cu, Zn, Gd, Se, Те, As,
Sb, Bi и широкого круга органических веществ. На основании этих материалов редколлегия
предполагает опубликовать в 1985—1990 гг. три новых тома книги «Термодинамические свойства
индивидуальных веществ». Редколлегия будет признательна читателям за критические
замечания и предложения, а также информадию,о проводимых; иди,планируемых исследованиях
термодинамических свойств индивидуальных веществ. Все замечания, предложения и
соответствующую информацию следует направлять по адресу: 127412 Москва, Коровинское шоссе, Институт
высоких температур АН СССР, Отдел химической термодинамики.
Академик В. П. Глушко
ПРЕДИСЛОВИЕ К IV ТОМУ
В IV томе настоящего издания рассматриваются термодинамические свойства 12 переходные
металлов (Cr, Mo, W, V, Nb, Та, Ti, Zr, Hf, Sc, Y, La) и их оксидов, трех актинидов (Th, U,
Pu), их оксидов и соединений урана со фтором, а также пяти щелочных металлов (Li, Na, К, Rb,
Cs), их соединений с кислородом, водородом, галогенами и солей ряда кислородных кислот. Всего
в IV томе рассмотрено 296 веществ. Во второй книге этого тома приведено 138 таблиц
термодинамических свойств конденсированных веществ и 266 таблиц термодинамических свойств газов
в стандартном состоянии (для 94 газов вплоть до 10 000 К).
Материалы первой книги IV тома изложены в 20 главах, каждая из которых содержит
сведения о соединениях одного элемента. Расположение материалов в главах первой книги и
соответствующих таблиц во второй книге четвертого тома следуют стандартному термохимическому
порядку (см. рисунок в формульном указателе настоящего тома). Помимо двадцати глав
первая книга этого тома содержит 5 приложений и указатель веществ, рассмотренных во всех
четырех томах этого издания. Приложение 3 является окончанием соответствующего
приложения трех предыдущих томов, и в нем представлены данные, позволяющие учесть отклонения свойств
пяти газов при высоких давлениях от их свойств в стандартном состоянии, а также значения
критических постоянных для 23 веществ. В Приложении 5 в дополнение к величинам, приведенным
в Приложении 5 третьего тома, рассматривается выбор энтальпии образования иона N0^ в
водном растворе. В Приложении 6 описан использованный в справочнике приближенный способ
оценки ненаблюдавшихся уровней энергии атомов и ионов. В Приложении 7 изложена
использованная в IV томе методика расчета колебательно-вращательной суммы по состояниям
двухатомных молекул через потенциал межатомного взаимодействия. Приложение 8 посвящено
изложению подхода к расчету энтальпий сублимации щелочных металлов и их галогенидов, для
которых характерен сложный состав пара в широком интервале температур.
В текстах глав и приложений первой книги нумерация таблиц и уравнений поглавная, так же
как в предыдущих трех томах; нумерация таблиц термодинамических свойств, включенных во
вторую книгу, сплошная и является продолжением нумерации таблиц предыдущих томов.
Во второй книге IV тома, помимо ее основной части, включающей 401 таблицу
термодинамических свойств, имеются два Дополнения. В одном из них включены таблицы термодинамических
свойств UF6(k) и Na2SO4(K Ш), а также 2 таблицы с вириальными коэффициентами и
критическими постоянными для 23 веществ, рассмотренных в IV томе. В другом Дополнении приведены
новые таблицы термодинамических свойств 20 веществ, рассмотренных в томах I—III.
Подготовка IV тома была завершена в середине 1981 г., при этом была использована
литература, опубликованная до конца 1980 г. Более поздние публикации учитывались, как правило,
только в тех случаях, когда полученные в них данные позволяли существенно уточнить расчеты
соответствующих таблиц термодинамических свойств.
Четвертый том подготовлен сотрудниками Отдела химической термодинамики ИВТАН (главы
39—49, приложения 3, 5, 7, 8, а также разделы по термодинамическим свойствам 12
двухатомных газов в главах 30—38, всего 240 веществ) при участии сотрудников группы по расчетам
термодинамических свойств ГИПХ (главы 30—38, кроме двухатомных газов, Приложение 6 — всего
56 веществ). Основная авторская работа по IV тому выполнена Л. В. Гурвичем, И. В. Вейц,
В. А. Медведевым, Г. А. Бергманом, В. С. Юнгманом, Г. А. Хачкурузовым. Кроме того, выбор
молекулярных постоянных и подготовку соответствующих текстов проводили Е. А. Шенявская,
О. В. Дорофеева, Е. Л. Осина, И. И. Назаренко, В. С. Иориш, И. Н. Пржевальский,
Ю. Г. Хаит, Л. Ф. Куратова при участии Н. Б. Щербака, расчеты термодинамических функций
газов — В. С. Иориш, О. В. Дорофеева, Е. Л. Осина, И. И. Назаренко, И. Н. Пржевальский,
Ю. Г. Хаит, Л. Ф. Куратова при участии Н. Б. Щербака, выбор постоянных и расчеты
термодинамических функций веществ в конденсированном состоянии — П. И. Толмач, Н. М.
Аристова, Г. Н. Юрков, Л.Ф. Куратова, А. Л. Рогацкий, при участии М.В. Волковой Л. Р.
Фокина, В. Я. Малецкого и Т. Г. Кибисовой, выбор термохимических величин — Л. Н. Горохов,
А. Л. Рогацкий, М. Е. Ефимов, В. Я. Леонидов, А. Г. Ефимова, С. Э. Томберг, В. С. Иориш,
А. В. Гусаров, Н. Э. Хандамирова, Е. А. Шенявская, М. В. Волкова, Л. Р. Фокин при уча-
Предисловие к IV тому
стии Ю. С. Ходеева, В. Г. Рябовой, И. В. Сидоровой, А. М. Емельянова, М. Ф. Московской,
B. Я. Малецкого, А. Т. Пятенко, Приложение 3 подготовили В. Ф. Байбуз, В. Ю. Зицерман в
Л. Р. Фокин, Приложение 5 — В. А.Медведев, Приложение 6 — И. Н. Пржевальский,
Приложение 7 — В. С. Иориш и Н. Б. Щербак и Приложение 8 — Н. Э. Хандамирова.
Основная техническая работа, связанная с проведением расчетов выполнена И. Г. Байбуз,
подготовкой рукописи — Н. Р. Симагиной, B.C. Павловой и В. В. Чепик, составлением списка
литературы — Е. С. Маевской, А. И. Варшавской и В. С. Шмелевой, оформлением таблиц —
C. А. Равинской и В. С. Шмелевой.
Общее редактирование IV тома выполнено В. П. Глушко и Л. В. Гурвичем, редактирование
разделов по молекулярным постоянным и глав 45—49 — И. В. Вейц, разделов по
термодинамическим свойствам веществ в конденсированном состоянии — Г. А. Бергманом, разделов по
термохимическим постоянным — В. А. Медведевым, приложений первой книги и материалов
второй книги тома — В. С. Юнгманом, предварительных материалов глав 30—38,
подготовленных в ГИПХ, — Г. А. Хачкурузовым.
При подготовке материалов четвертого тома некоторые разделы были обсуждены с советскими
и зарубежными специалистами. Редколлегия признательна профессору Э. Э. Шпильрайну и
кандидату технических наук Д. Н. Кагану за помощь в подготовке разделов по
термодинамическим свойствам пяти щелочных металлов в конденсированном состоянии, профессорам
А. А. Мальцеву и В. П. Спиридонову и кандидату химических наук Е. 3. Засорину (химфак
МГУ), доктору Д. Хилденбранду (Стэнфордский исследовательский институт США), профессору
Э. Ф. Веструму (Мичиганский университет США), докторам Д. Гарвину и Д. Д. Уагману
(НБС США) за предоставление результатов их исследований до публикации соответствующих
данных.
Основной табличный материал второй книги воспроизводит данные, рассчитанные и
накопленные в автоматизированном банке ИВТАНТЕРМО, созданном Отделом химической
термодинамики совместно с Отделом вычислительной техники ИВТАН. Редколлегия отмечает большую
и эффективную помощь, оказанную авторскому коллективу в создании ИВТАНТЕРМО и
подготовке второй книги этого тома коллективом Отдела вычислительной техники ИВТАН
(руководитель Г. П. Малюжонок).
Глава 30
ХРОМ И ЕГО СОЕДИНЕНИЯ
Сг(к, ж), Сг, Сг+, СгО,
СгО3, CrOj, Сг2О3(к, ж)
В настоящей главе рассматриваются хром и его кислородные соединения — всего 7 веществ.
Для двух веществ приведены данные об их термодинамических свойствах в конденсированном
состоянии, для 6 — в газообразном состоянии. Термодинамические свойства трех газов (Сг, Сг+
и СгО) рассчитаны до 10 000 К, остальных — до 6000 К. Термодинамические свойства всех
веществ рассчитаны для природной смеси изотопов хрома (атомный вес 51,996). Однако различие
постоянных разных изотопомеров учитывалось только для СгО(г) ввиду меньшей точности
расчетов свойств других соединений.
Для элементарного хрома в Приложении 3.10 приведены сведения о его критических
постоянных.
В справочнике представлены данные только для одного оксида хрома в конденсированном
состоянии. В литературе отсутствуют сведения об отклонениях состава этого соединения от ста
хиометрического, однако можно предполагать, что они незначительны [1030]. При температурах,
близких к температуре плавления, и в восстановительных условиях Сг2О3(к) превращается в Сг3О4,
имеющий структуру шпинели. Известны также малостабильные соединения, существующие
в конденсированном состоянии: Сг3О, СгО, СгО2, Сг5О15, Сг2О5, CrgO21, CrO3 и, возможно, Сг3О8
(см. [549а, 1023]), которые не рассматриваются в справочнике. В нем не рассмотрены также
малоустойчивые газообразные полимерные оксиды (Сг3О9, Сг4О12, Сг5О16), а также из-за высоких
потенциалов ионизации соответствующих окислов молекулярные ионы типа СгО+ и СгО,.
Сг(к, ж). Термодинамические свойства кристаллического и жидкого хрома в стандартном
состоянии при температурах 100—4700 К приведены в табл. 873.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 30.1. За стандартное состояние Сг(к) в справочнике принят поликристаллический хром.
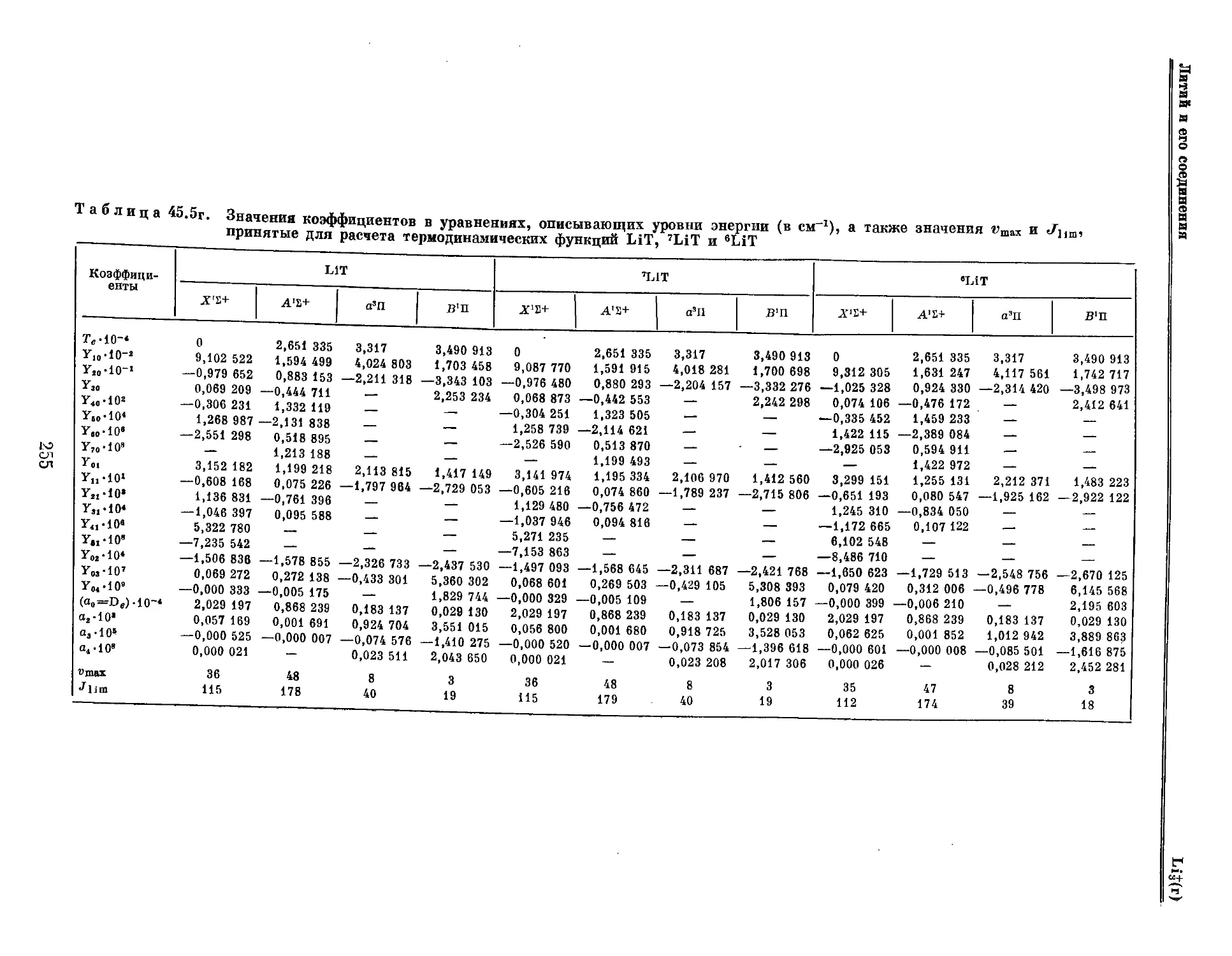
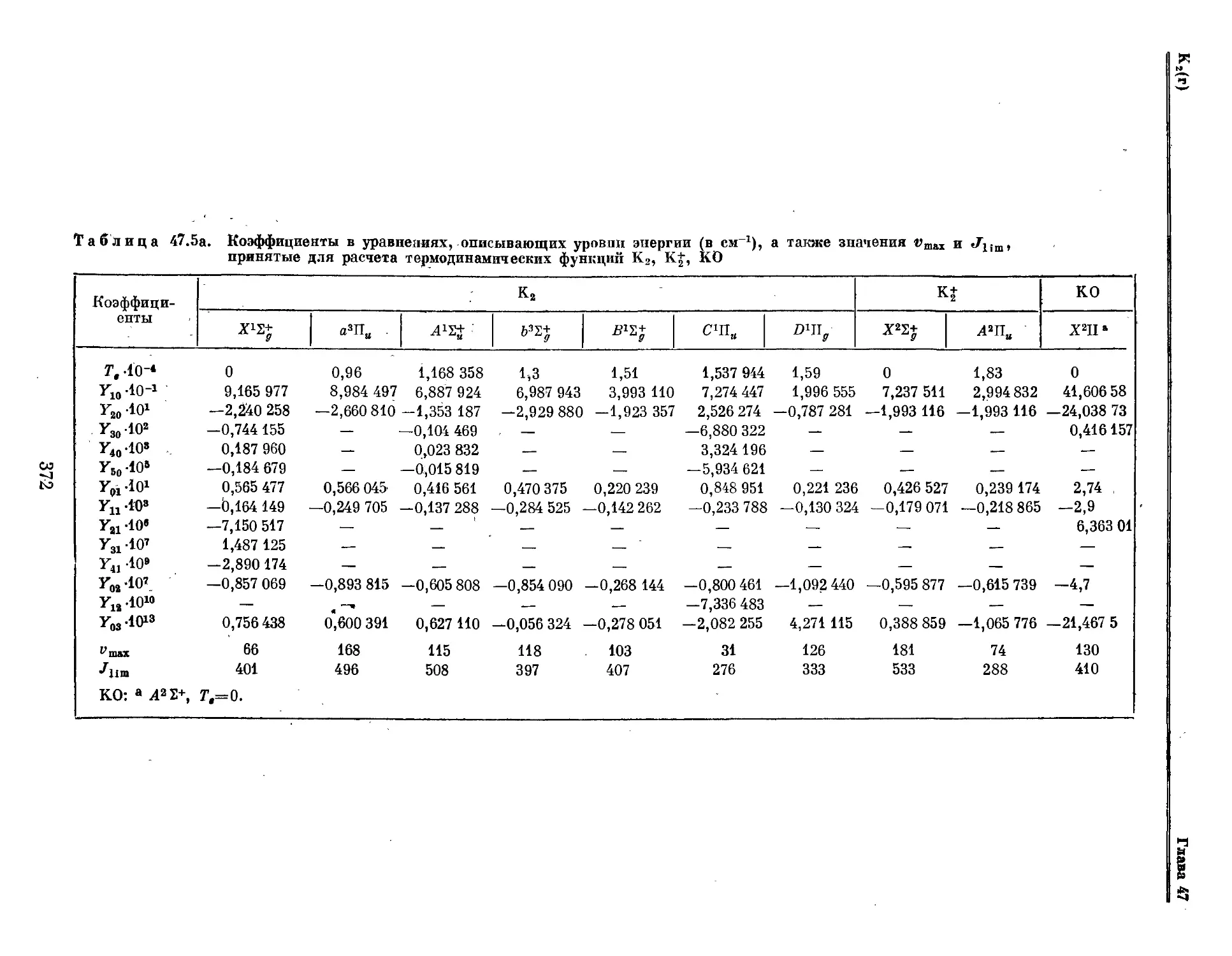
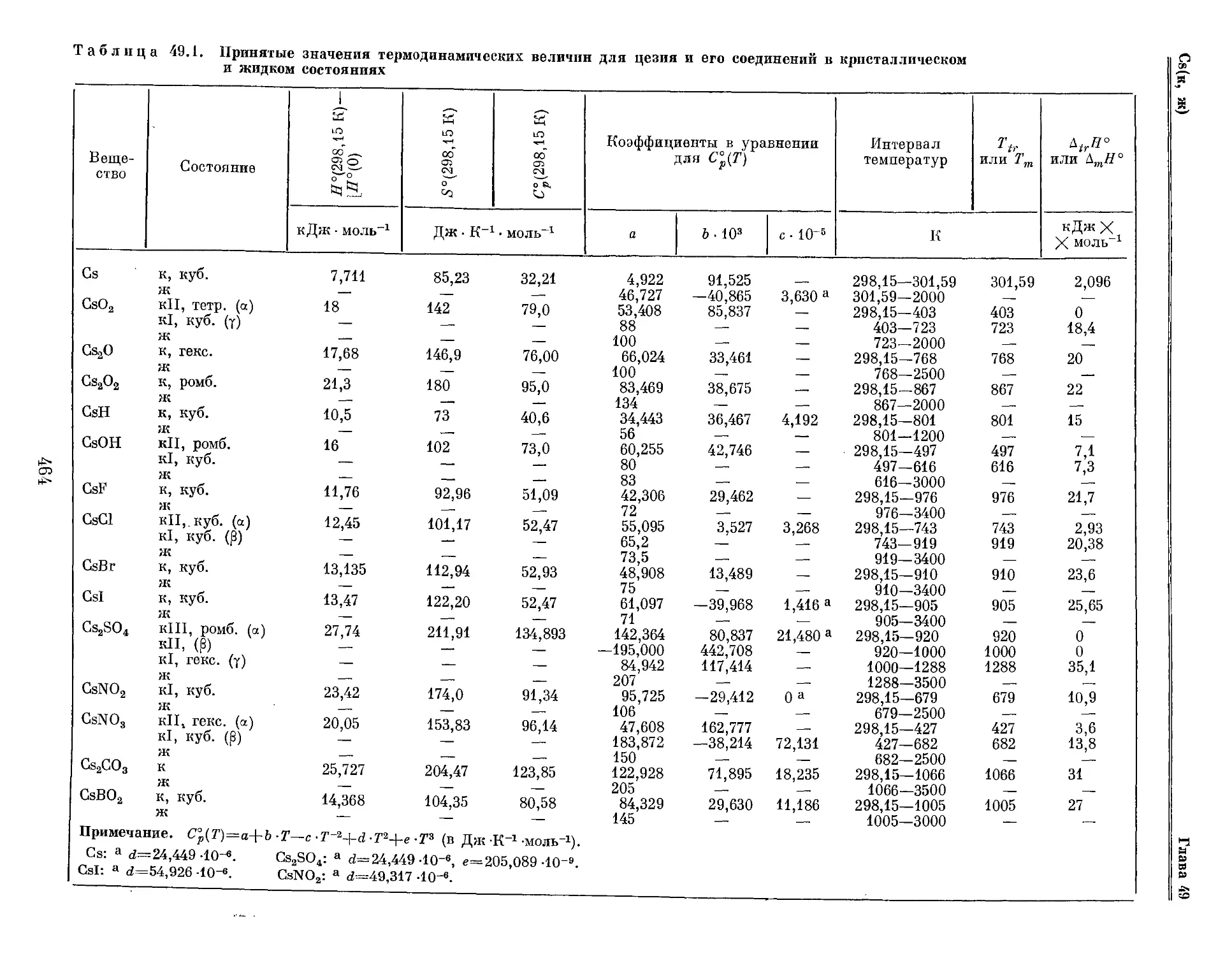
Таблица 30.1. Принятые значения термодинамических велнчин|для|хрома и его соединений
в кристаллической и жидком состояниях
Вещество
Состояние
in
По
оо --'
О5 О
С*<! (щ
?|
КДЖ'МОЛЬ
5° B98,15 К)
С° B98,15 К)
Дж • К •
• МОЛЬ
Коэффициенты в уравнении
для С'р (Г)
а
Ь-10»
СЮ-
Интервал
температуры
Tir или
К
AjrH или
кДж-
• моль
Сг кП, ромб. 4,05 23,56 23,55 14,550 30,187 — 298,15—311,5 311,5 0,001
к1, куб. — — — 19,032 15,125 — 311,5—400 — —
к1, куб. — — — 31,916 —19,912 4,174» 400—2180 2180 21,3
ж — — — 50 — — 2180-4700 — —
Сг2О3 кГ, гекс. 15,3 81,1 120,08 1926,054 -12 841,984 0? 298,15—306 306 0
к1, гекс. — — — 55756,402 —357 836,600 0б 306—310 — —
к1, гекс. — — — 2031,709 —11635,704 0в 310—335 — —
к1, гекс. — — — 134,440 —12,620 28,398 г 335—2705 2705 125
ж — — — 170 — — 2705-4500 — —
Примечание С°р(Т)=а-\-Ь -Т— с-T^-^-d-Т^-^-е-Т3 (в Дж К-моль).
Сг: а й=25,408 -КГ», «=—5,075 -10~e.
Cra03: а <2=22 756,059 ЛОГ»; б d=575 300 •«-«•, В <2=17 572,8 -МГ*; Г i=8,438 ЛОГ*.
€г(к, ж) Глава 30
При температурах от 0 до 124 К он является антиферромагнетиком с тетрагональной структурой,
от 124 до 311,5 К — антиферромагнетиком с ромбической структурой [801] и в интервале 311,5—
2180 К — парамагнетиком с кубической объемно-центрированной структурой 1. При 0,082 К
хром переходит в сверхпроводящее состояние [1286].
Термодинамические функции Сг (к) при Т ^ 298,15 К вычислены на основании измерений
теплоемкости образцов хрома чистотой 99,9—99,998%, выполненных Уолкотом [4750] A,2—
20 К), Клузиусом и Францозини [1880, 1881] A4—274 К), Бомоном и др. [1445] B68—324 К),
Вильямсом и др. [4731] B05—380 К). Магнитный переход Сг(к) из первого
антиферромагнитного состояния во второе при 124 К [801] не связан с какими-либо особенностями температурной
зависимости теплоемкости. Погрешности приведенных в табл. 30.1 значений S° B98,15 К) и
Н° B98,15 К)—Н° @) оцениваются в 0,2 Дж-К^-моль и 0,04 кДж-моль. Теплоемкость
Сг (к) при Т < 298,15 К измерялась также в работах [3160] A73—873 К), [4294] G1—80 К),
[1317] E6-291 К), [2193] A,8-4,2 К), [3951] A,5-4,2 К), [981] @,2-1,2 К), [3803]
{1,4-4,2 К), [479] E4-300 К), [2659] @,06-1,2 К), [4677] B80-330 К), [354, 1346]
(^ 273 К), где были получены менее точные данные. Прецизионные измерения теплоемкости
монокристаллического хрома (чистота 99,996%) в интервале 80—345 К были выполнены Полововым
[801]. Им было найдено, что температурные зависимости теплоемкости монокристаллического
и поликристаллического хрома существенно отличаются, что, по-видимому, обусловлено наличием
дополнительной электронной составляющей теплоемкости монокристаллического хрома.
Приведенные в табл. 30.1 уравнения для теплоемкости в интервалах 298,15—311,5 и 311,5—
400 К найдены на основании измерений теплоемкости в работах Бомона и др. [1445] и Вильямса
и др. [4731]. Температура фазового перехода из антиферромагнитного состояния в
парамагнитное (точка Нееля) принята равной 311,5 ± 0,1 К по температуре максимума теплоемкости,
найденной в работах [801, 1445, 4731], а также температуре скачка энтальпии по данным [1469].
Исследованиями Мацумото, Мицуи [5014] и Бенедиктсона и др. [1469] установлено, что фазовый
переход Сг(к) при 311,5 К является переходом первого рода. Энтальпия фазового перехода
принята равной 0,001 +0,0001 кДж-моль~х на основании калориметрических измерений [1469]
на образцах чистотой 99,996 и 99,999%. Эта величина подтверждается значениями, полученными
в работах [801, 5014].
Приведенное в табл. 30.1 уравнение теплоемкости Сг(к) в интервале 400—2180 К получено,
совместной обработкой измерений энтальпии, выполненных Бюстом и др. [4786] D73—1773 К),
Халтгреном и Ландом [2753] E00—1500 К), Конуэем и Хейном [1912] A173—2173 К),
Чеховским и Жуковой [1122] E61—2096 К), и измерений теплоемкости в работах Армстронга и
Грейсон-Смита [1346] E00 — 1100 К) и Кольхааса и др. [3062] E00—1800 К). Результаты этих
измерений недостаточно хорошо согласуются между собой, ввиду чего точность описания
энтальпии хрома выведенным уравнением составляет 1 и 2% при 1500 и 2000 К. Энтальпия Сг(к)
измерялась также в работах [3992] (85-293 К), [4182] F3-373 К), [42151 C73-867 К) [4579]
B73 - 1873 К), [2815] F13-1339 К), [930] B95-1500 К), [577] B97—1873 К), [507] F00—
2030 К), а теплоемкость — в работах [3102] (964—1598 К), [3145] B98—800 К). Эти данные
в справочнике не использовались как существенно менее точные.
Результаты измерений температуры плавления хрома значительно различаются вследствие
его высокой реакционной способности вблизи Тт и влияния примесей кислорода. Принятое
в справочнике значение B180 + 20 К) получено усреднением наиболее надежных измерений
образцов хрома высокой чистоты в работах Грубе и Кнабе [2510] B163 ± 10 К), Патмена и др.
[3900] B168 К), Блума и др. [1583] B179 ± 10 К), Пана и др. [752] B166 + 10 К), Недумова
и Григоровича [710] B206 К, образец содержал 99,999% Сг). Менее точные значения были
найдены в работах [914, 1582, 1747, 2475, 2594]. Результаты ранних определений Тт рассмотрены
в работе [2510]. Энтальпия плавления Сг B1,3 ± 0,4 кДж-моль) принята по данным Недумова
и Григоровича [710], полученных методом безконтактного ДТА. Менее точное значение АтН
A5,3 кДж-моль) определено Умино [4579] с использованием образца, содержавшего 5%
примесей.
В литературе имеются сообщения о фазовых переходах хрома в области 1300—2100 К, основывающиеся на
исследованиях сплавов хрома с другими металлами (см., например, [321, 4384]. Эти данные не получили
подтверждения в рентгенографических [192, 950] и дилатометрических исследованиях [913] образцов хрома
высокой чистоты. Аномалия теплоемкости хрома при 1640 К, отмеченная при калориметрических измерениях
Мартином (см. [3102]), в последующих работах [354, 3062, 3102] не была обнаружена.
10
Хром и его соединения
Сг(к, ж)
Энтальпия Сг(ж) не измерялась. В справочнике теплоемкость Сг(ж) оценена равной 50 +
+ 8 кДж-моль с учетом принятых значений теплоемкости Сг(к, Тт), ТЛ(ж) и У(ж).
Погрешности вычисленных значений Ф° (Т) при 298,15, 1000, 3000 и 4500 К оцениваются равными 0,15,
0,7, 3 и 6 Дж-К^-моль.
Ранее термодинамические функции Сг(к, ж) вычислялись в справочниках JANAF [2835],
Шика [4181] (до 6000 К), Халтгрена и др. [2752], Сталла и Зинке [4421] и Келли [2960]
(до 3000 К). Различия в значениях Ф° (Т), приведенных в табл. 873 и в [2835], составляют 0,03—
4,1 Дж-К^-моль.
В настоящем справочнике в качестве стандартного состояния хрома принято кристаллическое
состояние и в соответствии с этим
Д/ПСг, к) = 0.
Давление пара хрома в реакции Сг(к, ж) = Сг(г) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = Д8Я°(Сг, к, 0) = 395 + 2 кДж • моль.
В табл. 30.2 приведены значения А,Н°(Сс, к, 0), вычисленные по результатам измерений
давления насыщенного пара хрома. В соответствии с результатами масс-спектрометрических
исследований [2921, 2922] принималось, что пары хрома одноатомны. Приведенные в таблице
погрешности учитывают только воспроизводимость измерений. При обработке данных,
полученных методом испарения с открытой поверхности в вакуум, принималось, что коэффициент
Таблица 30.2. Результаты определений А,ЕГ°(Сг, к, 0) (в кДж-моль)
Авторы
Гринвуд, 1909 [2478, 2479]
Баур, Бруннер, 1934 [1436]
Шпейзер и др., 1950 [4361]
Гульбрансен, Эндрю, 1952 [2519]
Дроварт, 1957 [2112] (см. [462])
Бурлаков, 1957 [153]
Мак-Кейб и др., 1958 [3440]
Винтайкин, 1958 [218]
Несмеянов, Де Дык Ман, 1960 [714,
716]
Кубашевский, Хеймер, 1960 [3126]
Пилоян и др., 1960 [789]
Тульбрансен, Эндрю, 1961 [2520]
Игнатьев, Лебедев, 1961 [456]
Винтайкин, 1962 [219, 220]
Федоров и др., 1963 [1048]
Альфред, Майлз, 1964 [1290]
Диксон и др., 1965 [2073, 2074]
Амоненко и др., 1966 [30]; Ковтув
и др., 1962 [526]
Метод
Точек кипения, 2473 К, 1 точка
То же, 1614—2166 К, 7 точек
Испарение с открытой поверхности, 1285—
1505 К, 14 точек
То же, 1164—1284 К, 9 точек
Масс-спектрометрический, 1360—1630 К,
в работе указано только найденное значение
Испарение с открытой поверхности, 1294—
1511 К, 17 точек
Эффузионный, 1383—1507, 9 точек
То же, с использованием радиоактивного
изотопа, 1370—1670 К а
То дае, 1319—1529 К, 10 точек
Эффузионный, 1450—1675 К а
То же, 1503,5 и 1578,5 К, 2 точки
Испарение с открытой поверхности, 1275—
1375 К, 8 точек
Эффузионный, 1425—1575 К, 4 точки
То же, с использованием радиоактивного
изотопа, 1373—1573 К а
То же, 1459—1650 К а
Эффузионный, 1378—1600 К а
То же, 1561—1808 К, 14 точек
[ Испарение из цилиндрического тигля с
отверстием, 1455—1625 К, 8 точек
а Данные представлены в виде уравнения.
Д8Я°(Сг,
II закон
_
199 ±20
402 ±9
411 ±17
420+15
393 ±15
419 ±25
373
400 ±9
389
361
404 ±30
418±6
389
407
382
423 ±20
433 ±13
к, 0)
III закон
341
336 ±14
395,3+0,5
397,5 ±0,7
—
384,5 ±0,9
395,8±1,0
395,8±1,0
390,0±0,5
394,8±1,0
375,7 ±4,5
400,4 ±0,7
388 ±2
394
393,6 ±1,1
391,2±1,0
397,0+1,6
388,0±1,6
И
Сг(г), Сг*(г) =__=====m=__=m==_= Глава 3ft
испарения хрома равен единице. Точное значение этой величины не известно. Определения <хсг
на основании эффузионных измерений [30, 526, 714, 716] (асг «* 0,5) были признаны в [53, 210,
661] недостаточно корректными. Сравнение данных по давлению пара, полученных методом
испарения с открытой поверхности [2519, 2520, 4361] и эффузионным методом [218, 220, 716, 1048,
2074, 3126, 3440], приводит к значению асг = 0,7, тогда как в работе [3440] было найдено асг я»
т 0,8-^1,0.
Принятое в справочнике значение Д,Я°(Сг, к, 0) является средневзвешенным из данных
[218, 219, 220, 456, 714, 716,1048,1290,2073, 2074, 3126,3440 ], полученных эффузионным методом,
и в работах [2519, 2520, 4361] — методом испарения с открытой поверхности. При этом
учитывалась погрешность, обусловленная неточностью использованных в расчетах термодинамических
функций, приводящая к погрешностям в энтальпии сублимации от 1,1 до 2,1 кДж-моль (для
измерений, выполненных при 1300 и 2500 К соответственно).
Сг(г). Термодинамические свойства газообразного хрома в стандартном состоянии при
температурах 100—10 000 К приведены в табл.874.
Для расчета термодинамических функций использованы 127 уровней энергии х атома Сг с
суммарным статистическим весом 2298 в интервале 0—54 414,7 см, т. е. ниже первого потенциала
ионизации. Эти уровни соответствуют валентным электронным конфигурациям ...3d5As (к ней
относится основной уровень 7S3), ...3d44s2, ...3d6, ...3d4l (I = 1, 2, 3), ...3d4sAl A = 1,2) и
...3d34s24p. Принятые величины основаны на данных Шугара и Корлисса [4437], а также на
оценках для ряда ненаблюдавпшхся электронных состояний. Для электронных конфигураций ...3db4l
(I = 0, 1, 3) оценка выполнена методом, изложенным в Приложении 6, а для конфигураций ...3d44s2
и ...3d6 — сравнением энергии соответствующих состояний Cr, Mo, W, Мп+ и Fe+2.
Термодинамические функции Сг(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Т ^ 3000 К обусловлены главным образом неточностью фундаментальных
постоянных и не превышают 0,02—0,03 Дж-К^-моль. При более высоких температурах
становятся заметными ошибки из-за неточности оценки энергии ряда уровней и пренебрежения рид-
берговскими состояниями. Соответствующие погрешности могут достигать 0,1 и 0,3 Дж -К -моль
в значениях Ф°(Т) при 6000 и 10 000 К соответственно.
Ранее таблицы термодинамических функций Сг(г) вычислялись в справочниках JANAF [2835],
Шика [4181] (Т < 6000 К), Сталла и Зинке [4421] (Г < 3000 К) и в работах Хилзенрата и др.
[2652] (Т < 10 000 К), Кольского и др. [3067] (Г < 8000 К), Каца и Маргрейва [2934] (Т <
^ 2000 К) по уровням энергии, рекомендованным Мур [3550]. При Т ^ 3000 К расхождения
результатов этих расчетов с данными табл. 874 не превышают 0,02 Дж-К^-моль, за
исключением расчета [3067], для которого из-за использования завышенного значения R расхождения
втрое больше. При более высоких температурах расхождения возрастают и достигают 0,03 [2835
и 0,15 [2652] Дж-К-моль в значениях Ф°(Т) при 6000 и 10 000 К соответственно из-за учета
в настоящем справочнике ряда ненаблюдавшихся уровней энергии Сг.
Энтальпия образования
bfH°(Cr, г, 0) = 395±2 кДж^моль
соответствует принятой в справочнике энтальпии сублимации хрома.
Сг+(г). Термодинамические свойства газообразного положительного иона хрома в стандартном
состоянии при температурах 298,15—10 000 К приведены в табл. 875.
Для расчета термодинамических функций использован 81 уровень энергии иона Сг+ в
интервале 0—78 005 см с суммарным статистическим весом 1670. Эти уровни соответствуют
электронным конфигурациям ...3d5 (к ней относится основной уровень eS^lj), ...3d44Z (.' = 0, 1, 2)'
и ...ЗсР4в2. Принятые величины основаны на данных Шугара и Корлисса [4437], а также на
оценках для ряда ненаблюдавшихся электронных состояний. Для электронной конфигурации ...3d*4s
1 В отличие от принятой в I, II и III томах настоящего справочника формы изложения в этом томе в целях:
экономии места не публикуются таблицы уровней энергии атомов и ионов переходных металлов и актинидов.
Соответствующая информация хранится в Банке данных ИВТАНТЕРМО и при необходимости может быть
получена по запросу.
12
Хром и его соединения
СгО(г)
оценка выполнена методом, изложенным в Приложении 6, а для конфигураций ...3d5 и ...
сравнением энергии соответствующих состояний Cr+, Mo+, W+, V, Nb, Та.
Термодинамические функции Сг+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Т ^ 6000 К обусловлены главным образом неточностью фундаментальных
постоянных и не превышают 0,02—0,03 Дж-К^-моль. При более высоких температурах
становятся заметными ошибки из-за неточности оценки ряда уровней энергии, и соответствующие
погрешности могут достигать 0,05 Дж-К^-моль в Ф°A0 000 К).
Ранее термодинамические функции Сг+(г) вычислялись Хилзенратом и др. [2652] (Т ^ 10 000 К)
и Грином и др. [2473] (Т ^ 50 000 К). При Т ^ 8000 К расхождения результатов этих расчетов
с данными табл. 875 не превышают 0,02 Дж-К~1-моль~1. При более высоких температурах из-за
учета в настоящем справочнике ряда ненаблюдавшихся уровней энергии расхождения несколько
возрастают и достигают 0,08 Дж-К^-моль в значениях Ф°A0 000 К).
Константа равновесия реакции Сг+(г) + е(т) = Сг(г) вычислена с использованием значения
ДгЯ°@) = —652,869 ± 0,004 кДж -моль, соответствующего принятому в справочнике
потенциалу ионизации
|/0(Сг) = 54 575,6 ± 0,3 см = 652,869 ± 0,004 кДж . моль.
Это значение найдено Хьюбером и др. [4977] как предел ридберговской серии ...3db(9S)np 7P
(га = 4-^-38) в спектре атома Сг. Близкие значения 10(Ст), равные 54 565 см [3550] и 54 570 см
[2988, 3549, 4437], получены из более коротких серий и менее точны.
Принятому значению /0(Сг) соответствует
Д,Я°(Сг+, г, 0) = 1047,869 + 2,0 кДж . моль.
СгО(г). Термодинамические свойства газообразного оксида хрома в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 876.
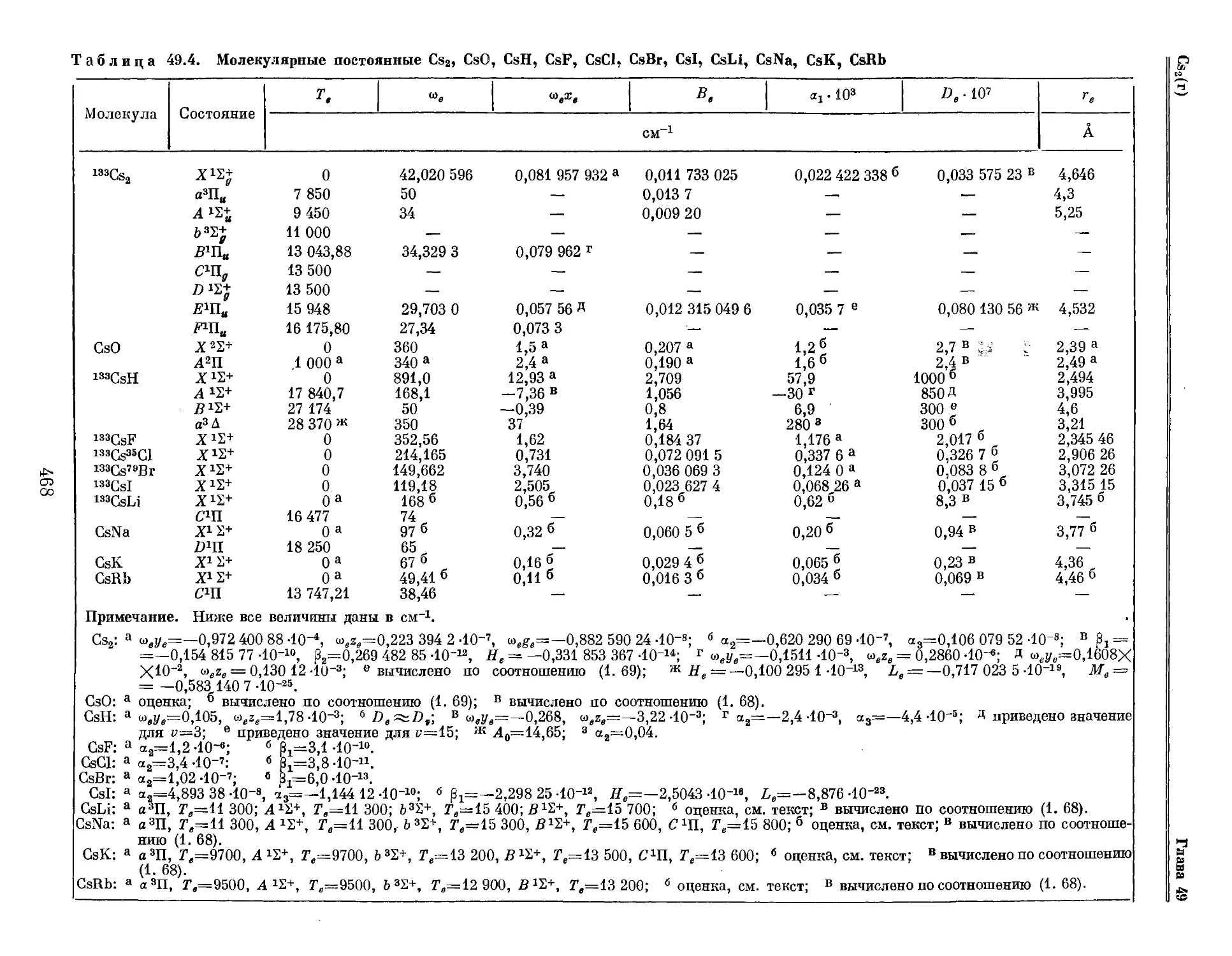
В табл. 30.3 приведены молекулярные постоянные СгО, принятые в. справочнике на
основании результатов исследований электронного спектра этой молекулы и приближенных оценок.
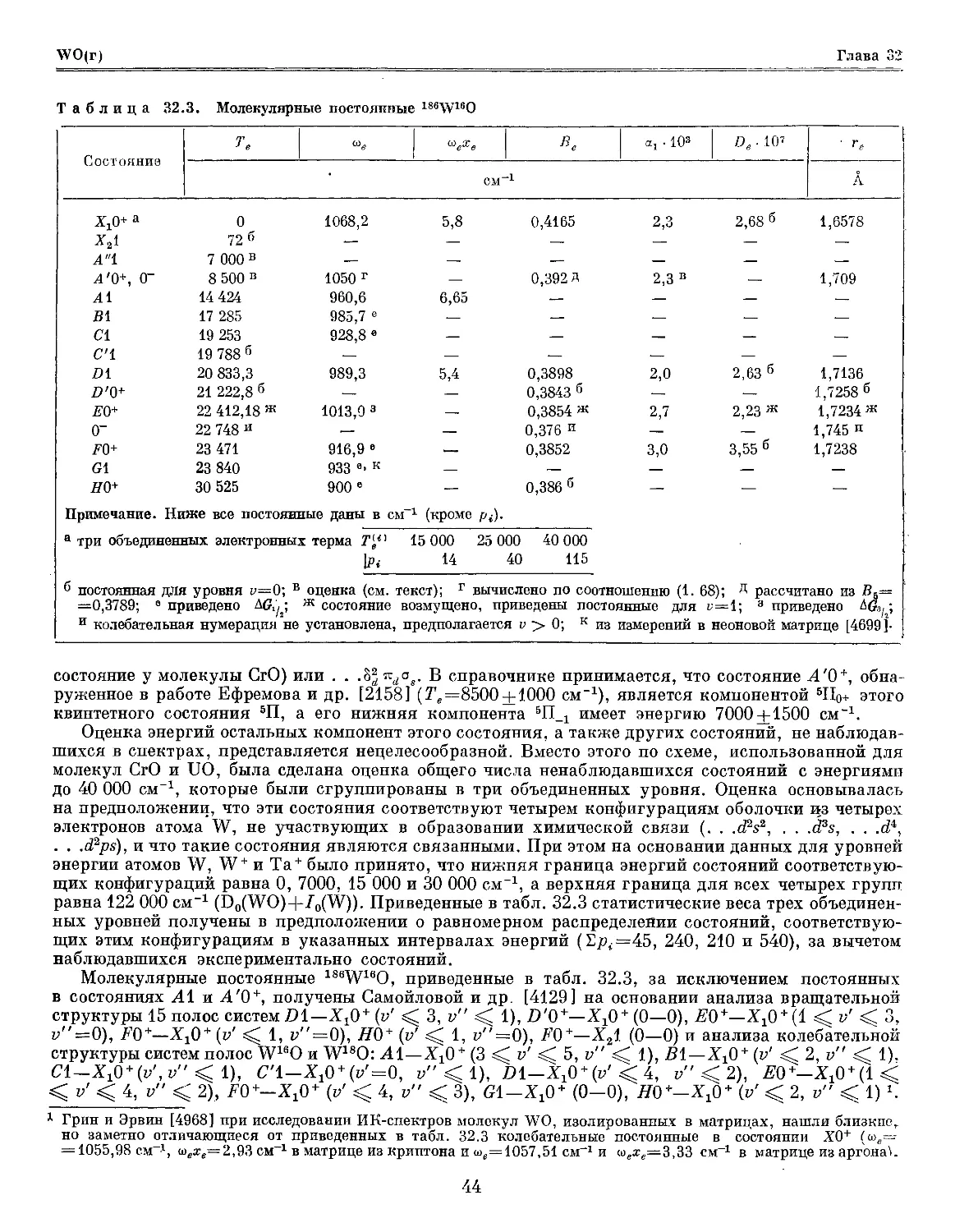
Таблица 30.3. Принятые значения молекулярных постоянных 82Сг160
VjOu 1ОННИ0
XilL.1
Х5Шт
хтг .
¦Х*Па
х*п3
Л*2+, а3 2+
Ь3Ф, с3Д
В5Пб
Примечание.
Те
0
56,1
117,0
182,6
252,2
11 000 а
14 000 а
16 697,0 в
899,07
898,76
898,50
897,93
897,39
—
—
752,81
шеХе
6,64
6,65
6,72
6,70
6,70
—
—
10,12
Ниже все постоянные приведены в см,
в.
ЕМ
0,526 643
—
—
0,473 712
кроме pt.
ах ¦ 108
4,434
—
—
5,483
Do. 101
г.
А
7,317 1,6179
— —
—
7,881 1,7059
а оценка; б приведены постоянные для подсостояния 5П!, значение Т, относительно подсостояния ХъИ-1, Л~54,4;
в объединенные
Т„ 20 000
Pi 20
электронные уровни
30 000 40 000
50 70
В спектре СгО проанализирована только одна система полос ВЪП—Х5П [903, 1569, 2002, 2234,
2374, 2395, 2670, 2988, 3663]. Наблюдавшиеся в работе [2374] в спектре испускания полосы в
области 8300—8500 А были предположительно отнесены к переходу в основное состояние этой
молекулы. Имеются косвенные данные (многочисленные возмущения в состоянии В5П [2670]) о
существовании у СгО триплетных состояний с энергиями, меньшими, чем энергия этого состояния.
13
СгО(г)
Глава 30
К молекуле СгО отнесены также полосы с максимумами интенсивности при 2920, 2970 и 3075 А,
наблюдавшиеся в работе [903] в спектре поглощения продуктов импульсного фотолиза смеси
Сг(СОN + О2 + Аг.
Оценка энергий неидентифицированных электронных состояний СгО была выполнена в
предположении, что основное состояние ХЬЛ соответствует конфигурации •••^l^dad, а состояние
Z?8II — конфигурации •••^'!Zaa,- Это предположение основано на том, что согласно
неэмпирическому расчету Пинчмела и Сампа [3843] основным состоянием молекулы МпО является
состояние 62+ конфигурации ¦••§1^|а<г, а у атомов Сг, Сг+, Сг+2 и V+ конфигурации ...cP^s2 или ...d"^
лежат выше соответствующих конфигураций ...d?s или ...dn.
Были рассмотрены электронные состояния молекулы СгО, которые могут соответствовать
трем конфигурациям электронной оболочки из четырех электронов атома Сг, не участвующих
в образовании химической связи (d4, d3s, d2s2), и сделано допущение, что ниже состояния В5П
расположены .только 4 терма: 52+(...S|tc|), 32+(...§2с|), 3Ф(...§31гй) и 3Д(...8|ай). Это позволила
отнести полосы, наблюдавшиеся Гаттерером и др. [2374] в области 8300—8500 А, к переходу
Л52+ -> Х5П, а состояние -4в2+(...8|тг|), имеющее общий диссоциационный предел с Х5П,
рассматривать как нижнее возбужденное состояние СгО. Энергии состояний а32+, Ь3Ф и с3Л были
оценены в предположении, что они обусловливают возмущения, наблюдавшиеся в системе полос
ВЬЛ—Х5П [2670]; их погрешности могут достигать 2000—3000 см.
В связи с тем что обоснованная оценка энергий остальных связанных валентных состояний
СгО невозможна, при подготовке справочника была оценена плотность электронных состояний
этой молекулы с энергиями до 40 000 см. Предполагалось, что в этой области энергий
существуют только состояния, соответствующие приведенным выше конфигурациям оболочки из
четырех электронов атома Сг. Приведенные в табл. 30.3 суммарные статистические веса трех
объединенных электронных термов получены из условия равномерного распределения электронных
состояний этих конфигураций в интервалах-энергий от 0 до 90 000 см B,pt = 180), от 17 000
до 90 000 см {2р( = 240) и от 30 000 до 90 000 см Bр; = 45) соответственно, их погрешности
могут достигать коэффициента 2.
Молекулярные постоянные СгО в состояниях Х5П и В5П, приведенные в табл. 30.3, были
определены Хоккингом и др. [2670] на основании анализа вращательной структуры 6 полос
системы ВЪП—ХЪИ (г/, v" ^ 2). Принятые колебательные постоянные несущественно отличаются
от вычисленных по кантам полос cv',v" <^ 8 в работах [2234, 2395], а также от рекомендованных
Хьюбером и Герцбергом [2736]. В то же время принятые вращательные постоянные существенно
отличаются от постоянных, найденных Ниномийя [3663] в результате приближенного анализа
структуры полос 0—0, 1—0 и 0—1 той же системы и рекомендованных в справочнике [2736].
Термодинамические функции СгО(г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBS и ее производных вычислялись по уравнениям A. 90)—A. 92)
с учетом 12 возбужденных электронных состояний (в том числе четырех компонент состояния
ХЪП ) Q^l05$[*С
р (
ХЪП и трех объединенных состояний) и в предположении, что -
= 0,5р$[*>_вр.
Статистическая сумма по колебательно-вращательным уровням энергии подсостояния Х6!!^ и ее
производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням энергии и интегрированием по значениям / с помощью уравнений типа A. 82).
В расчете учитывались все колебательно-вращательные уровни энергии с значениями / ^ Лпах.ш
последние находились из условий A.81). Энергия уровней вычислялась по уравнениям A. 65),
A. 34) и A. 64а), их вырождение учитывалось статистическим весом 2. Значения г;шах и /цт,.
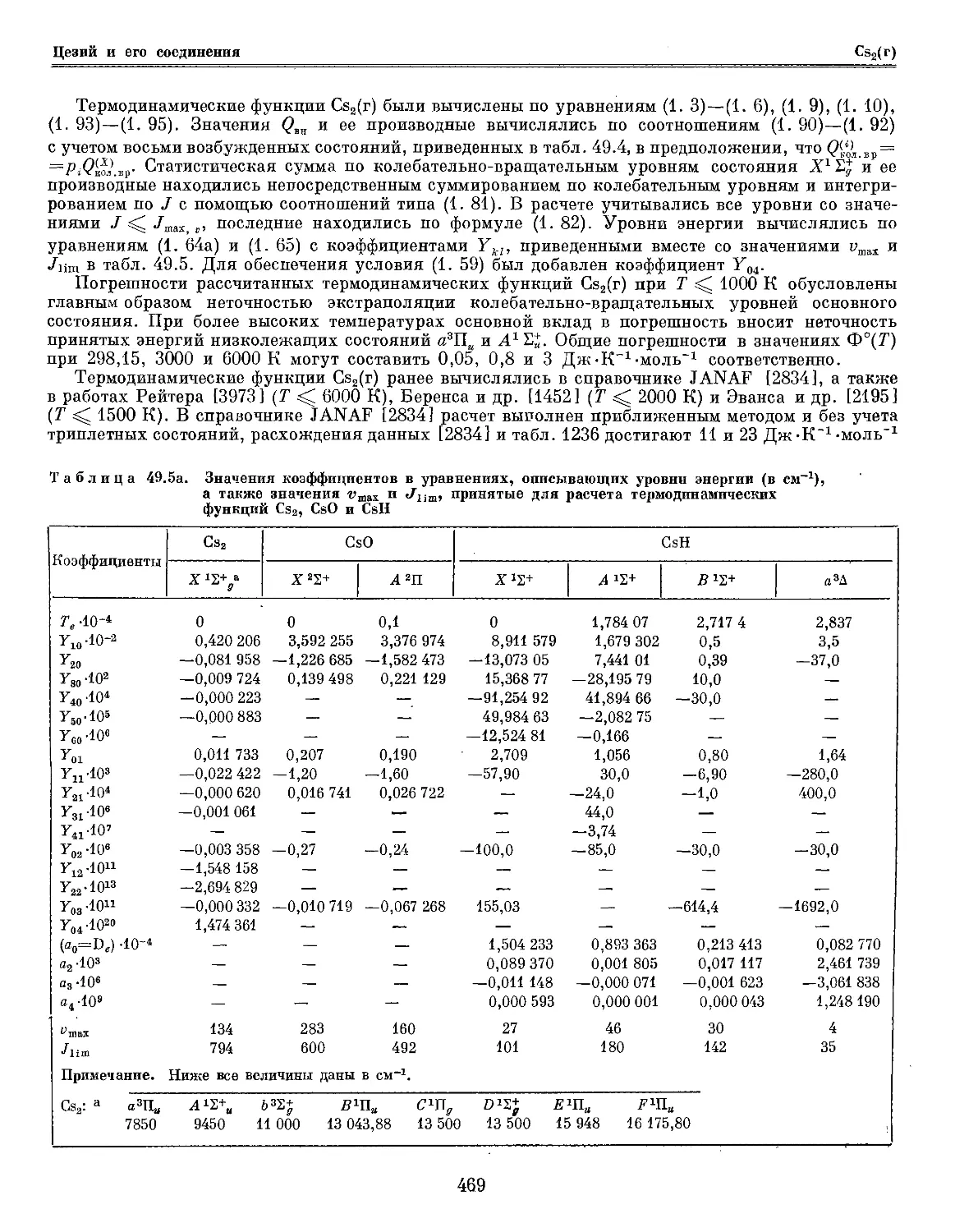
Таблица 30.4. Значения коэффициентов, описывающих уровни энергии (в см),
а также значения г»шах и </1ш, принятые для расчета термодинамических функций СгО
Коэффициент
Те 0
У10 -10 8,980 53
У20 —6,954 51
Коэффициент
Y АО2
1 30 1и
У01-10
Уц 103
1,783 96
5,242 55
—4,403 9
Коэффициент
Yn -10'
Y02 -Ю'
Уоз Ю13
х5п_,
5,03
-7,2508
-2,122
а Энергии возбужденных состояний, включая четыре компоненты состояния Х5П (см.
Коэффициент
"шах
Jim
табл. 30.3).
119
349
14
Хром и его соединения
СгО(г>
а также величины коэффициентов в этих уравнениях, полученные с учетом условий A. 50) и A. 59),
но без учета различия постоянных изотопомеров молекулы СгО, приведены в табл. 30.4.
Погрешности рассчитанных термодинамических функций СгО(г) при низких температурах
обусловлены пренебрежением особенностями энергии вращательных уровней квинтетного
основного состояния этой молекулы. Выше 3000 К становятся существенными, в особенности в
значениях С°(Т), погрешности из-за отсутствия экспериментальных данных об энергии
возбужденных электронных состояний. Погрешности в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К
оцениваются в 0,15, 2, 5 и 10 Дж-К^-моль"".1.
Ранее термодинамические функции СгО(г) вычислялись в таблицах JANAF [2835], Шнейде-
ром [4197а] (Г = 1000 Ч- 9000 К) и Брюэром и Розенблаттом [1666] (значения Ф°(Г) для Т <
<^ 3000 К). Расхождения данных [2835] и табл. 876 при низких температурах обусловлены тем, что
авторы [2835] не учли мультиплетное расщепление состояния Х5П; соответствующие расхождения
в значенияхФ°B98,15 К)иЯ°B98,15 К)—Я°@) составляют4,06 Дж-К^-моль. и 1,02кДж-моль"
В б 10003000 К 1 ДК11 6000 К
( )( Д
В области 1000—3000 К расхождения не превышают 1 Дж-К-моль~1, но к 6000 К достигают 1,
5 и 12 Дж-К^-моль в значениях Ф°(Т), S°(T) и Ср(Т) из-за того, что в таблицах [2835] учтено
только одно возбужденное состояние СгО. Расхождения с данными [4197а] имеют аналогичные
причины и примерно такую же величину, при 9000 К они достигают 3 и 11 Дж-К^-моль в
значениях Ф°(Г) и S°(T) соответственно. В работе [1666] для молекулы СгО принята неверная
систематика нижних электронных состояний, что привело к существенно завышенным значениям
Ф°(Г).
Константа равновесия реакции СгО(г) = Сг(г) + О(г) вычислена с использованием принятой
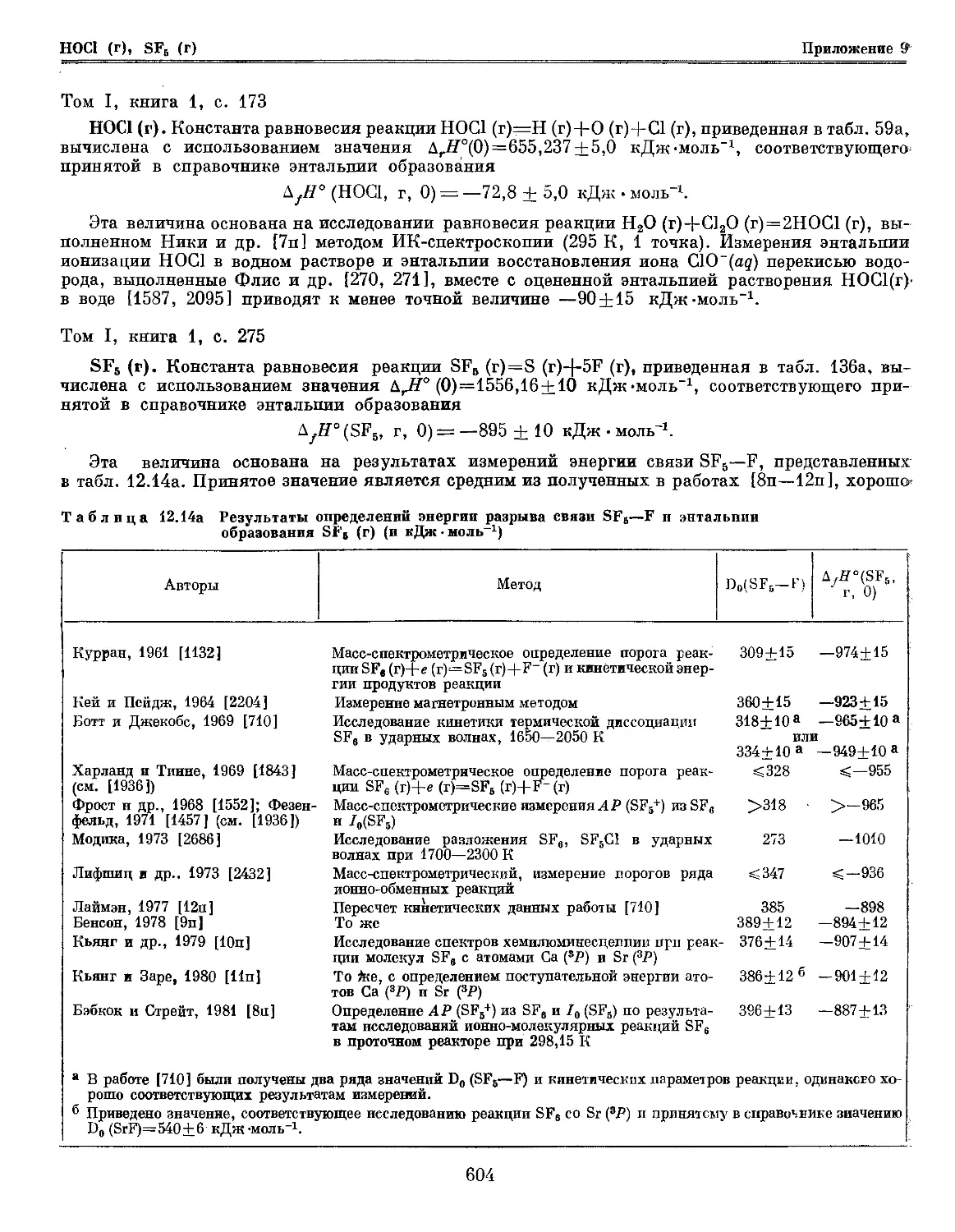
в справочнике энергии диссоциации
ДгЯ°@) = D0(CrO) = 457 + 7 кДж • моль« 38 200 + 600 см.
Это значение основано на результатах масс-спектрометрического исследования равновесия
реакции АЮ(г) + Сг(г) = А1(г)г + СгО(г) B242—2690 К, 12 точек), выполненного Гороховым
и др. [310, 463] и находится в согласии с приведенными в табл. 30.5 термохимическими
величинами, рассчитанными по результатам исследований равновесия различных реакций с участием
газообразных СгО, СгО2 и СгО3 [1135, 2492, 4991]. Менее точные значения, согласующиеся с при-
Таблица 30.5. Термохимические величины, использованные в расчетах энтальпий образования О02(г)
и СгО3(г) (в кДж-моль)
Авторы
Метод
Термохимическая
величина (III закон)
Гримли др., 1961 [2492]
Чижиков и др., 1972 [1135]
Ким, Белтон, 1974 [4991]
Горохов и др., 1979 [310, 463]
а 2\fH°(Cr0, г, 0)—A
г t>fH°(Cr0, г, 0)-
Масс-спектрометрический, исходное вещество Сг2О3,
СгО2(г)+Сг(г)=2СгО(г), 1839—2059 К, 13 точек
То же, СгО2(г)+О(г)=СгО(г)+О2(г)
Масс-спектрометрический, исходные вещества Сг2О3
и О2, СгО2(г)+Сг(г)=2СгО(г), 1890—1942 К, 3 точки
То же, СгО2(г)+О(г)=СгО(г)+О2(г)
То же, СгО3(г)=СгО2(г)+О(г)
То же, СгО3(г)=СЮ(г)+О2(г)
То же, Сг2О3(к)+8/2О2(г)=2СгО3(г)
Масс-спектрометрический, исходное вещество Сг2О3,
СгО2(г)+Сг(г)=2СгО(г), 1840—2000 К, данные
представлены в виде уравнения
Метод протока, Сг2О3(к)+3/2О2(г)=2СгО3(г), 1597—
1706 К, 3 точки
Масс-спектрометрический, исходное вещество Сг2О3,
СгО2(г)+Сг(г)=2СгО(г), 1786-1959 К, 9 точек
То же, исходные вещества Сг2О3 и А12О3, АЮ(г)+
+Сг(г)=СгО(г)+А1(г), 2242—2690 К, 12 точек
г, 0); 6AfH°(CiO, г, 0)—AfH°(CTO2, г, 0); в bfH°(Cr02, г, 0)--
477,2+7,0а
284+12 б
492+16 а
294+11 б
211+10 в
505±15 г
— 320+37 Д
489+34 а
—324 +28 Д
480,6+7,2 а
184,8+7,2 е
, г, 0);
г, 0);
г, 0); е AfH°(CrO, г, 0).
15
СгО2(г)
Глава 30
нятым в пределах погрешностей их определения, получены методом фотометрии пламен Лагер-
квистом и Хульдтом [5003] E10 ± 42 кДж -моль) и Зегерсом и др. [5075] D73 ± 10 кДж -моль),
а также на основании данных Ванга и др. [5068] и Гримли и др. [2492] для равновесия реакции
СгаО8(к) = 2СгО(г) + О(г) D76 ± 17 кДж-моль, 1504—1821 К, 15 точек) и Бальдуччи и др.
[4921] для реакции WO3(r) + Сг(г) = W02(r) + СгО(г) D42 + 25 кДж-моль, 1865-2340 К,
8 точек).
Принятому значению D0(CrO) соответствует
Д/йго(Сг0, г, 0) = 184,783 + 7,2 кДж • моль.
СгО2(г). Термодинамические свойства газообразного диоксида хрома в стандартном состоянии
при температурах 100—6000 К приведены в табл. 877.
Молекулярные постоянные, использованные для расчета термодинамических функций и
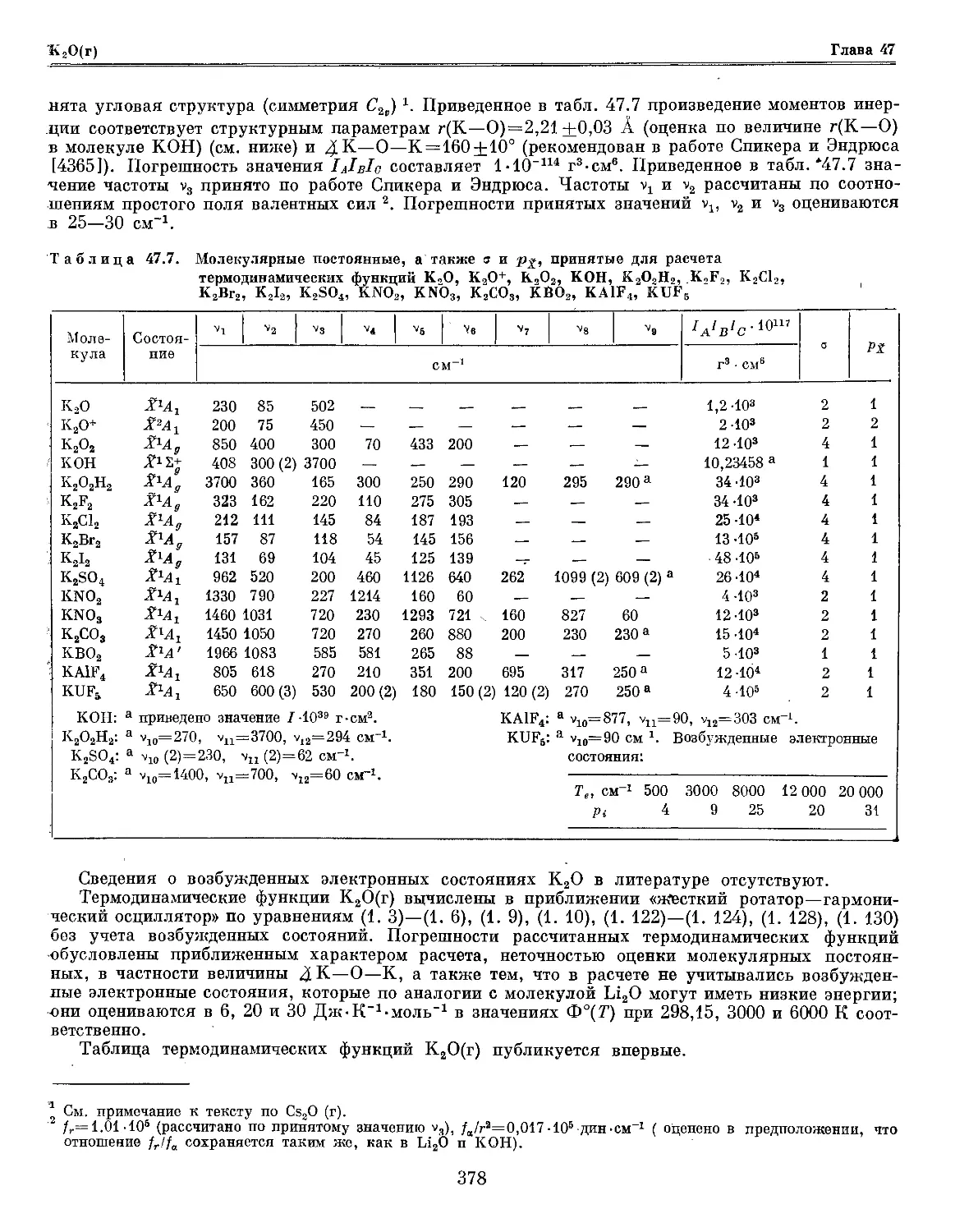
приеденные в табл. 30.6, приняты на основании результатов исследования ИК-спектра СЮ2 [926,
928], полуэмпирического расчета [1065] и оценок. В основном электронном состоянии %аА2 [1065]
молекула СгО2 имеет угловую структуру (симметрия С2„) с углом 109 ± 10°, найденным
Серебренниковым и Мальцевым [928] по величине изотопического сдвига частоты vs в ИК-спектре этой
молекулы, изолированной в матрице из Аг. Произведение моментов инерции, приведенное
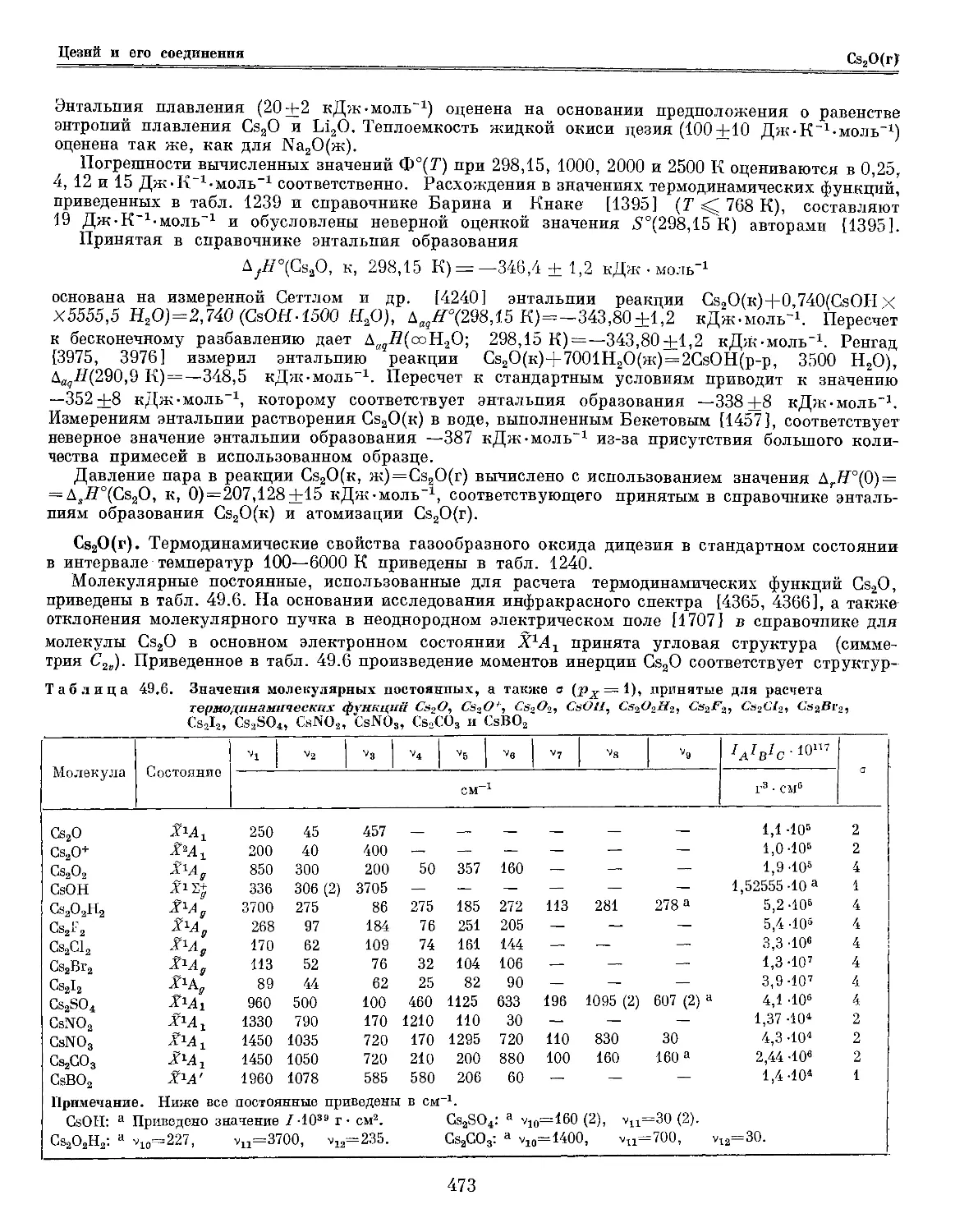
Таблица 30.6. Значения молекулярных постоянных, а также о и рх , принятые для расчета
термодинамических функций СгО2, CrO3, CrOj
Молекула
Состояние
•
*3
СМ
/aVc.io»t
г3 • см"
а
Рх
СгОа %*А3* 911 530 968 — 3-Ю2 2 3
СЮ3 X*Ai* 800 400 980B) 350B) 2-Ю8 6 1
CrOj &А[а 800 400 900B) 300B) 2-Ю3 6 2
Примечание. Ниже все постоянные приведены в см*1.
СгО„: а T0(A4J=m0, Т0(Ё*А2, 6ГМ1)=1О 000, Г0E3В1)=15 000, Г0(?»А1( F3Ba)=20 000, Г0(<?3В2)=25 000.
СгО3: а Г0(ЛМ2)=25 000, Т0(В»Аа)=Э& 000.
CrOj: а Гв(Л*Л1)=10 000, Т0(В^А1)=2Ъ 000.
в табл. 30.6, рассчитано для этой структуры и значения г(Сг—О) = 1,60 ± 0,05 А (оценено
по величине гДСгО) и г(Сг—0) в других кислородных соединениях хрома); погрешность
принятого значения составляет 1-10 ш г3-см8. Основные частоты приняты по рекомендации
Серебренникова и Мальцева [926, 928], которые наблюдали в ИК-спектре молекул СгО2, изолированных
в матрице из аргона, частоты vx и vs, а также рассчитали значение v3 в приближении поля
валентных сил. Погрешности принятых значений могут достигать 10—20 см для vx и v3 и 50 см для
v2. Приведенные в таблице энергии семи нижних электронных состояний СгО2 получены Хаитом
и Хачкурузовым [1065] в результате полуэмпирического расчета в рамках теории
кристаллического поля; их погрешности оцениваются в 3000—5000 см.
Термодинамические функции СгО2(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122) — A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом семи возбужденных электронных состояний по уравнениям A. 168)—A. 170).
Погрешности рассчитанных термодинамических функций обусловлены приближенным характером
расчета и неточностью принятых постоянных, главным образом энергий электронных состояний.
Погрешности значений Ф°(Г) при 298,15, 3000 и 6000 К составляют 1, 4 и 8 Дж-К^-моль.
Ранее термодинамические функции СгО2(г) вычислялись в справочниках JANAF [2835] и
Шика [4181], работах Брюэра и Розенблатта [1665] (до 3000 К), Нагараяна [3618] (до 2000 К)
и Гордона и Робинсона [2446] (до 6000 К). В работах [1665, 2446, 3618] предполагалось, что
молекула СгОа имеет линейную структуру, а в справочнике [4181], принималось, что молекула
СгОа имеет синглетное основное состояние. Расхождения данных табл. 877 и [2835] составляют
2—4 Дж-К^-моль в значениях ФО(Г) и S°(T) и обусловлены различием принятых значений
основных частот я тем, что авторы таблиц JANAF не учитывали возбужденных состояний; рас-
16
Хром и его соединения СгО3(г)
хождения с данными [1665J и [4181] достигают 14 и 8 Дж-К*1-моль~1 соответственно в
значениях Ф°(Т).
Константа равновесия реакции СгО2(г) = Сг(г) + 2О(г) вычислена с использованием
значения ДГЯ°(О) = 994,566 + 15 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH° (СгО2, г, 0) = —106 ±15 кДж • моль.
Эта величина найдена одновременно с энтальпией образования СгО3(г) согласованием
представленных в табл. 30.5 термохимических величин методом наименьших квадратов. Приведенные
в таблице термохимические величины вычислены по уравнению III закона. Указанные
погрешности учитывают воспроизводимость измерений, погрешности термодинамических функций и
сечений ионизации. При оценке погрешности принятой величины были учтены также погрешности
использованных в расчетах термохимических величин. В работе Гримли и др. [2492] были
измерены парциальные давления Сг, СЮ, СгО2, СгО3, О и О2 в парах над Сг2О3(к). Однако, как
показали Брюэр и Розенблатт [1666], для Сг(г) были получены завышенные значения. Поэтому
в табл. 30.5 из данных [2492] представлено только пять реакций, не включающих Сг(г), и изомо-
лекулярная реакция СгО2(г) + Сг(г) = 2СгО(г).
Обработка результатов исследования испарения Сг2О3(к) с открытой поверхности [5068] A504—
1821 К, 15 точек) с использованием данных [2492] по составу пара над Сг2О3(к) приводит к
значительно менее точной величине (—109 + 35 нДж-моль), совпадающей с принятой в пределах
погрешностей.
СгО3(г). Термодинамические свойства газообразного триоксида хрома в стандартном состоянии
при температурах 100—6000 К приведены в табл. 878.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 30.6. Спектр и структура молекулы СгО3 экспериментально не изучались. На
основании сравнения с молекулой МоО3 и теоретического рассмотрения электронных состояний СгО3
в справочнике принимается, что в основном электронном состоянии X1-А'\ молекула СгО3 имеет
структуру правильного треугольника с атомом хрома в центре (симметрия Dsh). Приведенное
в табл. 30.6 произведение моментов инерции соответствует значению г(Сг—О) = 1,58 + 0,03 А,
оцененному по величине г(Сг—О) в молекулах Сг3О9 и Сг4О12, найденной Ивановым [452] при элек-
тронографическом исследовании пара над СгО3 г. Погрешность значения IaIbIo составляет
3-10~11В г3-см8. Приведенные в табл. 30.6 основные частоты СгО3 вычислены по уравнениям поля
валентных сил по силовым постоянным, оцененным по соотношениям /г = АДСгО), /а/г2=0,05/г
и /. /г2 = 0,15/г, предложенным в работах [568, 2633]. Корректность расчета подтверждается
близостью вычисленных значений v2, v3 и v4 к максимумам полос при 485, 995 и 350 см,
наблюдавшихся в ИК-спектре пара над СгО3(ж) и отнесенных Ямпольским и Мальцевым [1194] к
частотам колебаний молекул (СгО3)и (га = 3, 4, 5). Погрешности принятых значений основных
частот СгО3 оцениваются в 50—80 см. Приведенные в таблице энергии двух возбужденных
электронных состояний СгО3 оценены Хаитом и Хачкурузовым [1065] на основании
полуэмпирического расчета в рамках теории кристаллического поля. Их погрешности могут достигать 5000 см.
Термодинамические функции СгО3(г) вычислены по уравнениям A.3)—A.6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом двух возбужденных электронных состояний по уравнениям A. 168)—A. 170).
Погрешности вычисленных величин обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета. Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К
составляют 2, 4 и 8 Дж-К^-моль.
Ранее термодинамические функции СгО3(г) вычислялись в справочниках JANAF [2835], Шика
[4181], а также Гордоном и Робинсоном [2446] (до 6000 К) по оцененным молекулярным
постоянным в предположении, что молекула СгО3 имеет плоскую симметричную структуру Z)SA.
Расхождения данных табл.878 и [2835] составляют 1—4 Дж-К^-моль в значениях Ф°(Т).
Константа равновесия реакции СгО3(г) = Сг(г) + 30(г) вычислена с использованием
значения Дг#°@) = 1453,349 + 15 кДж -моль", соответствующего принятой в справочнике
энтальпии образования
Д/Я°(СгО3, г, 0) = —318 ± 15 кДж • моль.
1 В работах [452, 2797] показано, что в молекулах МоО3 и Мо2О6 величина г(Мо—О) одинакова.
2 Заказ М 1590 17
СгО3~(г), Сг2О3(к, ж) Глава 30
Эта величина определена одновременно с значением АуЯ°(СгО2, г, 0) согласованием
приведенных в табл. 30.5 термохимических величин методом наименьших квадратов (см. текст по СгО2(г)).
Погрешность принятой величины включает воспроизводимость измерений, погрешности
термодинамических функций, термохимических величин и сечений ионизации.
СгО~(г). Термодинамические свойства газообразного отрицательного иона триоксида хрома
в стандартном состоянии при температурах 298,15 — 6000 К приведены в табл.879.
Молекулярные постоянные, использованные при расчете термодинамических функций, при-
дены в табл. 30.6. Спектр и структура СгО" экспериментально не исследовались. На основании
теоретического анализа электронных состояний СгО~ (см. [1065]) в справочнике принимается,
что в основном электронном состоянии Ж2А[ ион CrOg имеет плоскую симметричную структуру
(симметрия Dah). При сохранении симметрии структуры многоатомные отрицательные ионы и
соответствующие им нейтральные молекулы имеют близкие по величине структурные параметры
и основные частоты колебаний. Поэтому приведенные в таблице значения постоянных СгО^ в
основном состоянии приняты приближенно такими же, как для молекулы СгО3, с учетом, что их
погрешности должны быть существенно больше E-Ю15 г3-см6 в значении 1А1в1с и до 100 см
в значениях основных частот). Энергии двух нижних возбужденных состояний СгО" оценены Хай-
том и Хачкурузовым [1065] на основании полуэмпирического расчета в рамках теории
кристаллического поля.
Термодинамические функции СгО~(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128), A. 130) с учетом двух возбужденных электронных состояний по
уравнениям A. 168) — A. 170). Погрешности рассчитанных величин обусловлены в основном
неточностью оценки молекулярных постоянных и приближенным характером расчета, они составляют 3,
8 и 12 Дж-К^-моль-.1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций СгОд(г) публикуется впервые.
Константа равновесия реакции СгО~(г) = Сг(г) + 30(г) + е(г) вычислена с использованием
значения Дгй"°@) = 1758,349 + 34 кДж-моль, соответствующего принятому в справочнике
сродству молекулы СгО3 к электрону
А0(СтО3) = —305 ± 30 кДж-моль.
Эта величина основана на данных работ Миллера [3506], где измерялась константа
равновесия реакции НСгО3(г) + е(т) = СгО~(г) + Н(г) (фотометрия пламен, 1815 — 2475 К), и Кима
и Белтона [4991], где измерялись константы равновесия реакций: Сг2О3(к) + 3/2О2(г) = 2СгО3(г)
и Сг2О3(к) + О2(г) + Н2О(г) = 2НСгО3(г) (метод протока, 1572—1856 К). Погрешность
принятого значения учитывает неточность измерений констант равновесия и использованных в расчете
термодинамических функций и термохимических величин. В работе [3506] было рекомендовано
менее точное значение —390 + 100 кДж-моль, основанное на данных [3506] для реакции
НСгО3(г) + е(т) = СгОд(г) + Щг), на данных Гримли и др. [2492] для реакции СгО3(г) = Сг(г)+
+ 30(г) и на данных Булевич и Педли [1718] для реакции Сг(г) + ЗН2О(г) = НСгО3(г) + 2Н2(г) +
+ Щг).
Принятому значению сродства к электрону соответствует
l±fB°{Ci:O-, г, 0) = —623 ± 34 кДж моль.
Сг2О3(к, ж). Термодинамические свойства кристаллического и жидкого триоксида дихрома
в стандартном состоянии при температурах 100—4500 К приведены в табл. 880.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 30.1. В стандартном состоянии Сг2О3(к) имеет гексагональную структуру типа корунда.
При 306 К наблюдается фазовый переход второго рода (точка Нееля), не сопровождающийся
изменением кристаллической структуры.
Термодинамические функции Сг2О3(к) при Т <^ 298,15 К вычислены по измерениям
теплоемкости, проведенным Андерсоном [1317] E6—335 К) на образце Сг2О3 фирмы Кальбаум
(химический состав не указан). Экстраполяция теплоемкости ниже 56 К проводилась по уравнению Ср(Т)=~-
= DC61,5/Т) + 2ЕE04,1/Т) + 2ЕG20/Т), которое аппроксиммирует данные [1317] в интервале
56—103 К с погрешностью ~1% и приводит к значению S°E6 К) = 2,3 Дж-К~1-моль~1. С
данными Андерсона [1317] удовлетворительно согласуются измерения энтальпии, проведенные Рус-
селом [4104] (82—322 К). Измерения теплоемкости Сг2О3(к) в работах Волгера [4619] (90—350 К)
18
Хром и его соединения СгаО3(к, ж)
и Брюса и Каннелла [1694] B91—323 К) систематически выше данных Андерсона [1317] в
среднем на 10%. Решающим доводом в пользу выбора данных Андерсона [1317] является их
согласованность с измерениями энтальпии, проведенными Келли и др. [2962] D78—1742 К) (см. ниже);
однако поскольку причины расхождений между данными [1317] и [1694, 4619] остались не
установленными, погрешности значений ?°B98,15 К) и #°B98,15 К) — Н°@), приведенных в табл. 30.1,
оцениваются в 5 Дж-К^-моль и 0,8 кДж-моль соответственно.
Теплоемкость Сг2О3 в области ^-аномалии B98,15—335 К) аппроксимирована тремя
параболами (см. табл. 30.1), одна из которых в интервале 298,15—306 К описывает восходящую кривую,
а две другие при 306—310 К и 310—335 К — нисходящую кривую с минимумом теплоемкости
при 335 К. Все три уравнения основаны на данных Андерсона [1317]. Температура точки Нееля
Сг2О3(к), при которой теплоемкость имеет максимум C06 + 1 К), принята на основании ее
определения различными методами в работах [1317, 1694, 4848, 4976, 4982, 5015]. Близкое значение
было получено в работе [4619].
В интервале 335—2740 К для теплоемкости Сг2О3(к) принято приведенное в табл. 30.1
уравнение, полученное совместной обработкой результатов измерений энтальпии, выполненных Келли
и др. [2962] D78—1742 К), и значений энтальпии в интервале 1740—2740 К, полученных
экстраполяцией данных [2962]. Весьма неточные измерения энтальпии Сг2О3(к) [616] D41—1428 К)
в справочнике не использовались.
Измерения температуры плавления Сг2О3 приводят к значениям, лежащим в интервале 2125—
2705 К г. Это связано с тем, что в большей части исследований не соблюдались условия,
обеспечивающие неизменность стехиометрического состава Сг2О3 при плавлении. Вартенберг и Профет
[4660] показали, что для Сг2О3 существует сильная зависимость Тт от атмосферы, в которой
ведутся измерения, и от продолжительности процесса плавления. В работах Вартенберга и Эк-
харда [4658] и Тромбе и др. [4549, 4550] были приняты специальные меры, обеспечивающие
быстрое плавление образца и уменьшающие изменение его состава при плавлении. Этими авторами
получены практически совпадающие значения 2703 + 10 К и 2705 К. Учитывая, однако, что и
в указанных работах могли иметь место небольшие изменения состава при плавлении 2,
погрешность принятого значения B705 К) увеличена до 30 К. Энтальпия плавления Сг2О3A25 +
+ 10 кДж -моль) принята на основании энтропии плавления Ti2O3 и А12О3, имеющих, как и
Сг2О3, структуру типа корунда, и на основании величины Дот/7°(Сг203) = 125,5 кДж-моль,
вычисленной Есиным и Захаровым [414] по диаграмме плавкости системы СаО—Сг2О3.
Энтальпия и теплоемкость Сг2О3(ж) не измерялись. Принятое значение теплоемкости A70 +
+ 25 Дж-К^-моль) было оценено с учетом принятых в этом справочнике теплоемкостей
А12О3(ж) и Т1203(ж). Погрешности приведенных в табл. 880 значений Ф°(Т) при 298,15, 1500,
3000 и 4500 К оцениваются в 4, 10, 18 и 25 Дж-К~1-моль соответственно. Ранее
термодинамические функции Сг2О3(к, ж) вычислялись в справочниках JANAF [2835] ^цо 4500 К) и Келли
[2960] (до 2000 К). Расхождения между значениями Ф°(Г), приведенными в [2835, 2960] и
в табл.880, не превышают 1 Дж-К^-моль.
Константа равновесия реакции Сг2О3(к, ж) = 2Сг(г) -f- 30(г) вычислена с использованием
значения Дг#°@) = 2665,126 + 4,3 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
b.fH°{Qx%03, к,?298,15?К)= —1140,6 + 1,7 кДж • моль.
Результаты определения энтальпии образования Сг2О3(к) различными методами представлены
в табл. 30.7. Принятое значение основано на калориметрических измерениях энтальпии сгорания
хрома чистотой 99,95%, выполненных Ма [3343]. В расчетах учтены поправки на примеси,
неполноту сгорания хрома и на изменение внутренней энергии кислорода с давлением. Однофаз-
ность образующегося при сгорании оксида была доказана рентгенографически. Принятое
значение согласуется с менее точными калориметрическими измерениями Голутвина и Цзинь-Куя
[303]. Калориметрические измерения в работах [3519, 4061, 4071] ненадежны. В пределах
погрешностей с принятым значением согласуются также результаты наиболее надежных исследо-
1 Результаты определений 7тш(Сг,О3'), Гопублпкованные до 1963 г., приведены в [41Р8]. Позднее значение Тт
определялось Портным и др. [815] B567 + 50 К).
2 Рентгенографический анализ застывшего расплава, проведенный в [4658], показал наличие в нем малого
количества Сг3О4. Такие же изменения могли иметь место в работах [4549, 4550], в которых плавление Сг,О3
проводилось на установке с солнечным нагревом на воздухе, а -*г ¦? : тмосфере кислорода.
19
Сг2О3(к, ж)
Глава 30
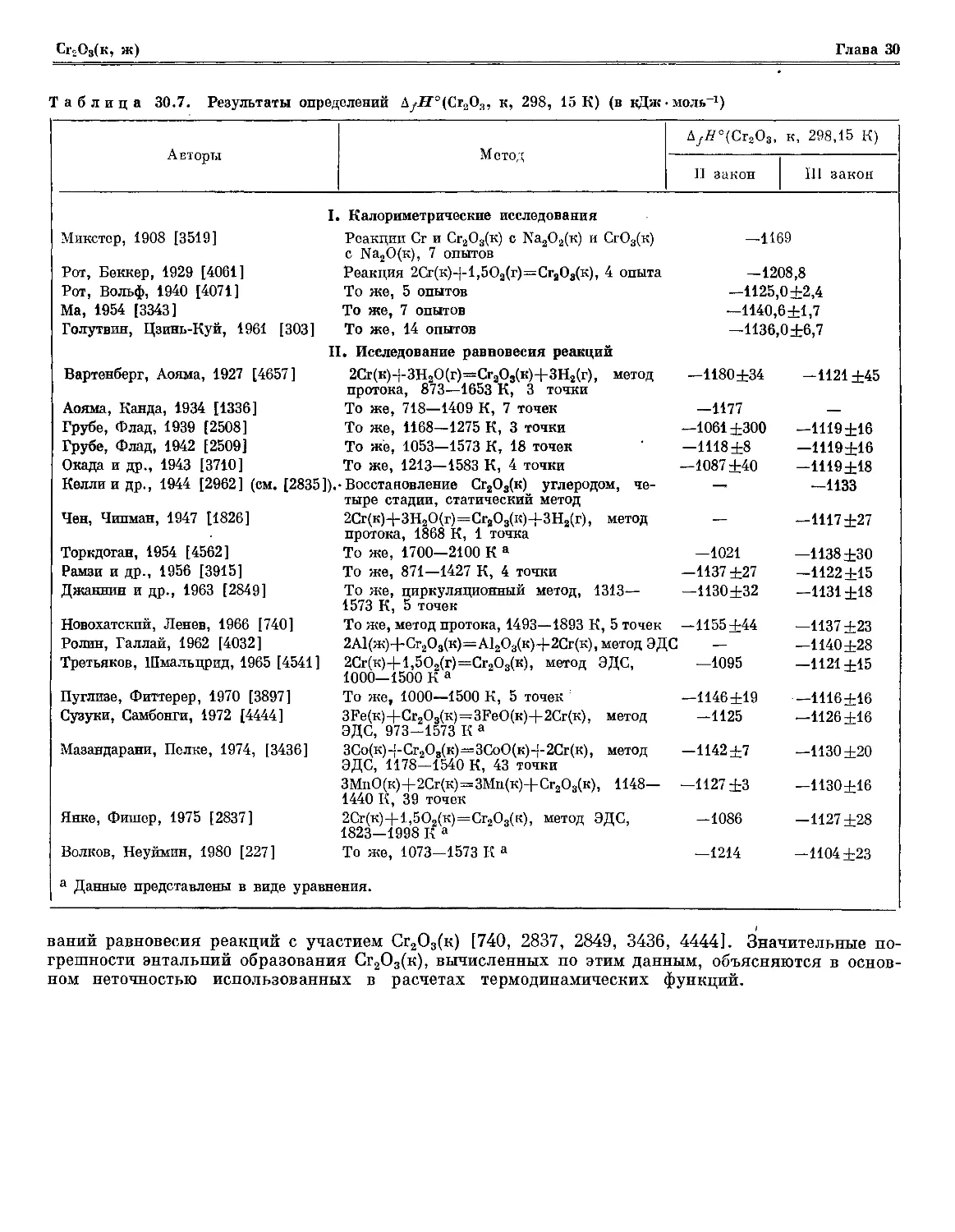
Таблица 30.7. Результаты определений Д^-Н"°(Сг3О3, к, 298, 15 К) (в кДж
Авторы
I.
Микстер, 1908 [3519]
Рот, Беккер, 1929 [4061]
Рот, Вольф, 1940 [4071]
Ма, 1954 [3343]
Голутвин, Цзинь-Куй, 1961 [303]
11
Вартенберг, Аояма, 1927 [4657]
Аояма, Канда, 1934 [1336]
Грубе, Флад, 1939 [2508]
Грубе, Флад, 1942 [2509]
Окада и др., 1943 [3710]
Келлии др., 1944 [2962] (см. [2835]).
Чен, Чипман, 1947 [1826]
Торкдоган, 1954 [4562]
Рамзи и др., 1956 [3915]
Джаннин и др., 1963 [2849]
Новохатский, Ленев, 1966 [740]
Ролин, Галлай, 1962 [4032]
Третьяков, Шмальцрид, 1965 [4541]
Пуглизе, Фиттерер, 1970 [3897]
Сузуки, Самбонги, 1972 [4444]
Мазандарани, Пелке, 1974, [3436]
Янке, Фишер, 1975 [2837]
Волков, Неуймин, 1980 [227]
Метод
Калориметрические исследования
Реакции Сг и Сг2О3(к) с Na2O2(K) и СгО3(к)
с Na2O(K), 7 опытов
Реакция 2Сг(к)+1,5Оа(г)=СгаО3(к), 4 опыта
То же, 5 опытов
То же, 7 опытов
То же, 14 опытов
. Исследование равновесия реакций
2Сг(к)+ЗН2О(г)=Сг2О3(к)+ЗН2(г), метод
протока, 873—1653 К, 3 точки
То же, 718—1409 К, 7 точек
То же, 1168—1275 К, 3 точки
То же, 1053—1573 К, 18 точек
То же, 1213—1583 К, 4 точки
Восстановление Сг2О3(к) углеродом,
четыре стадии, статический метод
2Сг(к)+ЗН2О(г)=Сг2О3(к)+ЗН2(г), метод
протока, 1868 К, 1 точка
То же, 1700—2100 К а
То же, 871—1427 К, 4 точки
То же, циркуляционный метод, 1313—
1573 К, 5 точек
То же, метод протока, 1493—1893 К, 5 точек
моль)
AfH°(Cr2Oa,
II закон
к, 298,15 К)
III закон
-1169
—1208,8
—1125,0 ±2,4
—1140,
-1136,
—1180±34
—1177
—1061 ±300
—1118±8
—1087 ±40
—
—
-1021
—1137 ±27
-1130±32
—1155 ±44
2А1(ж)+Сг2О3(к)=А12О3(к)+2Сг(к), метод ЭДС —
2Сг(к)+1,502(г)=Сг203(к), метод ЭДС,
1000 1500 К а
Л \^ \J \У Л U \S \S J, I
To же, 1000—1500 К, 5 точек :
3Fe(K)+Сг2О3(к)=3FeO (к)+2Сг(к), метод
ЭПС 973—1574 К а
ЗСо(к)+Сг2О3(к)=ЗСоО(к)+2Сг(к), метод
ЭДС, 1178—1540 К, 43 точки
ЗМпО(к)+2Сг(к)=ЗМп(к)+СгаО8(к), 1148—
1440 К, 39 точек
2Сг(к)+1,502(к)=Сг203(к), метод ЭДС,
1823 1998 К а
То же, 1073—1573 К а
а Данные представлены в виде уравнения.
—1095
—1146 ±19
-1125
-1142±7
—1127 ±3
—1086
-1214
В ±1,7
0±6,7
—1121 ±45
-1119±16
—1119±16
—1119 ±18
-1133
—1117 ±27
—1138±30
—1122 ±15
—1131 ±18
—1137 ±23
—1140 ±28
—1121 ±15
—1116±16
—1126 ±16
—ИЗО ±20
—1130±16
—1127 ±28
—1104 ±23
ваний равновесия реакций с участием Сг2О3(к) [740, 2837, 2849, 3436, 4444]. Значительные
погрешности энтальпий образования Сг2О3(к), вычисленных по этим данным, объясняются в
основном неточностью использованных в расчетах термодинамических функций.
Г л а в а 31
МОЛИБДЕН И ЕГО СОЕДИНЕНИЯ
Мо(к, ж), Мо, Мо+, МоО, МоО2(к), МоО2, МоО3(к, ж), МоО3, МоО;, Мо2О6» Мо309, Мо4О12,
Мо6О15
В настоящей главе рассматриваются молибден и его кислородные соединения — всего 10 веществ.
Для трех веществ представлены данные о термодинамических свойствах в конденсированном
состоянии, для 10 веществ — в газообразном состоянии. Термодинамические свойства трех
газов (Мо, Мо+, МоО) рассчитаны для температур до 10 000 К, остальных газов — до 6000 К.
Термодинамические свойства всех веществ приведены для природной изотопической смеси
молибдена (атомный вес 95,94) и кислорода, однако различие постоянных разных изотопомеров этих
веществ в расчетах не учитывалось из-за отсутствия необходимых данных и пренебрежимо
малого влияния этого эффекта на свойства соединений молибдена.
Для элементарного молибдена в Приложении 3.10 приведены сведения о критических
постоянных.
Рассматриваемые в справочнике кристаллические оксиды молибдена имеют достаточно
большие области гомогенности, МоОх 93 — MoO2il0 и МоО2 95—МоО3 (см. [549а]).'; Известны и
другие оксиды молибдена в кристаллическом состоянии (Мо3О, Мо2О6, Mo4O1]L, Mo5O14 и т. п.),
которые остаются малоизученными (см. [1030]). Эти соединения в справочнике не рассматриваются.
Не рассматриваются также ионизированные кислородные соединения молибдена, посколъкур'по-
тенциалы ионизации оксидов превышают 65 000 см
Мо(к, ж). Термодинамические свойства кристаллического и ж ,-дкого молибдена в стандартном
состоянии при температурах 100—6000 К приведены в табл. 881.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 31.1. За стандартное состояние Мо(к) в интервале 0—2896 К принята кубическая
модификация (структурный тип a-Fe). При 0,916 К х молибден переходит в сверхпроводящее состояние.
При Т ^ 298,15 К термодинамические функции Мо(к) вычислены на основании измерений
теплоемкости образцов Мо чистоты ]>99,9%, выполненных Рорером и др. [4047] @,4—4 К), Уол-
Табпица 31.1.
Принятые значения термодинамических величин для молибдена и его соединений
в кристаллическом и жидком состояниях
Вещество
Состояние
х
Ю о*
оо-?Г
го tq
о
кДж-моль
и
ю
оо"
оз
о
со
ю
оо"
го
CNI
о а,
О
Дж ¦ К • моль
Коэффициенты в
уравнении для С°р{Т)
а
Ь ¦ 103'
с • Ю-5
Интервал
температур
или
Т
К
или
кДж-
•МОЛЬ
Мо к, куб. 4,58 28,57 23,932 27,462 —2,406 2,770а 298,15-2896 2896 40
ж — . — — 40 — — 2896—6000 — —
МоО2 к, монокл. 8,328 46,51 55,91 74,522 —0,937 17,050а 298,15-3000 — —
МоО, к, ромб. 12,59 77,66 75,07 73,515 34,903 7,868 298,15—1075 1075 48,7
ж — — — 127 - — 1075—3000 - —
Примечание. C°p{T)=a+b -T— c-T~2+d-r2 (в Дж -К -моль).
Мо: а &=3,416 -М-". МоО2: а й=9,534-10-»
1 Это значение установлено прецизионными измерениями на образцах молибдена чистотой ~99,999% [392,
4045, 4046, 4047].
21
Мо(к, ж) Глава 31
котом [4750] A—20 К), Горовицем и Даунтом [2713] A—20 К), Клузиусом и Францозини [1879]
A0—273 К) с учетом табулированных данных Дитмарса и др. [2083] для Т > 273,15 К.
Измерения Хриплович и Паукова [1090] F—303 К) при Т > 30 К согласуются с принятыми данными.
Исследования теплоемкости Мо(к) при Т <^ 298,15 К выполнялись также в работах [4296] A6—
283 К), [2714] A,3—10 К), [3950] A К), [1699] A,0-4,5 К), [3563] A,5—25 К), [982] @,3-
1,2 К). Данные, полученные в этих работах, согласуются с принятыми, но менее точны. В
работах [1920, 4491] допущены систематические ошибки. Погрешности приведенных в табл. 31.1
значений ?°B98,15 К) и Я°B98,15 К) — #°@) оцениваются в 0,2 Дж -К -моль"-1 и 0,03 кДж -моль-1.
В интервале 298,15—2896 К термодинамические функции Мо(к) вычислены с
использованием приведенного в табл. 31.1 уравнения для теплоемкости, полученного совместной обработкой
измерений энтальпии, проведенных Бронсоном и др. [1679] C23—774 К), Егером и Веенстрой
[2818, 2820] C73-1773 К), Котеном [3085] B98-2650 К), Лазаревой и др. [613] A155-2466 К),
Кириллиным и др. [501, 504, 1118, 3015, 3016] (972—2614 К), Конуэйем и др. [1913] (см. [2663])
A266—2632 К), Чеховским и Петровым [511, 1127, 1128, 1824] (972—2834 К), Чеховским и др.
[1120, 1825] B097-2874 К), Исихарой и Дугласом [2083, 2788] A170-2102 К), Эттингом и Нав-
ратилом [3694] F96—1380 К), Бондаренко -и др. [1608] A200—2500 К), Березиным и др. [118,
1121, 1482], Дугласом и Дитмарсом [2083, 2109] C23—1173 К), Шейнером и др. [4248] A928—
2883 К), Литвиненко и др. [631] A269—2304 К), Бетцом и Фробергом [1536] B202—2857 К),
и значений энтальпии, вычисленных по измерениям теплоемкости Чеховским и Калинкиной
[1125] C00—900 К). Чистота образцов Мо(к) во всех этих работах была ~99,9%. Принятое при
Т > 298,15 К уравнение для теплоемкости Мо(к) позволяет аппроксимировать результаты
измерений [118, 501, 613, 631, 1127, 1536, 1608, 1679, 1825, 1913, 2109, 2788, 2818, 3085, 3694, 4248]
с точностью ^1% х и находится в согласии с результатами измерений теплоемкости, точность
которых существенно ниже, чем точность избранных измерений энтальпии. В частности, данные
[3318] систематически занижены, а данные [788, 1043, 3939, 4081] и [382] систематически
завышены (при Т > 2600 К). Измерения Цезерляна [2083] при Т > 2400 К также являются
систематически завышенными. Менее точные измерения энтальпии для менее чистых образцов Мо(к)
были получены в работах [3958] C72 К), [2020] C66—717 К), [4419] C73—923 К), [3992] B93 К),
[4786] C73—1773 К) и [4394] C73 и 718 К). Помимо упомянутых выше калориметрических
измерений [1125, 1920], теплоемкость Мо(к) при 71 > 298,15 К измерялась также импульсными
методами в работах [3939] A273-2893 К), [4081] A550-2200 К), [4490, 4491] A00-2880 К),
[1779] A300—1600 К), [1797] A900—2800 К), [382] B100—2900 К), [667] A200—2700 К), [1043]
A500-2600 К), [787] A500—2200 К), [2083] A500—2800 К) и модуляционными методами в
работах [3318] A288-2015 К), [574, 575, 3100] A300-2500 К), [788] (800-1500 К), [667] E00-
2700 К), [1093] D50—1200 К), [640, 1053, 1055, 4906] A000-2500 К) и [5016] A300-2500 К).
Температура плавления Мо B896 + 9 К) принята в соответствии с рекомендацией МПТШ-68
[2779], где она принята в качестве вторичной реперной точки (см. также [4879]). Это значение
основано на результатах определений Тт(Мо), полученных Уортингом [4773] B889 + 20 К),
Райли [4002, 4003] B896 + 10 К), Рудым и Прогульским [4084] B897 + 9 К), Цезерляном
и др. [1796] B894 + 10К), Латта и Фрикселлом [3202] B896 + 15 К), Кенисариным и др. [496,
2968] B899 + 9 К) для образцов Мо(к) чистоты 99,9—99,97%. Принятое значение Тт(Мо)
согласуется со значениями, определенными Паном [752] B898 + 10 К для образца чистоты 99,9%
и 2908 + 10 К для образца чистоты 99,99%). Энтальпия плавления Мо и теплоемкость Мо(ж)
в справочнике приняты соответственно равными 40 + 2 кДж-моль и 40 + 2 Дж-К^-моль
на основании измерений энтальпии Мо(ж) Бетцом и Фробергом [1536, 1538] B838—3383 К)
методом левитационной калориметрии (с учетом принятых в справочнике данных для Мо(к)). Менее
точные измерения энтальпии Мо(ж) этим методом были выполнены Тревертоном и Маргрейвом
[4542] B692—3112 К) и Березиным и др. [118, 1121, 1482] B892—2929 К) и методом импульсного
нагрева в работах [382] B900—3700 К), [4248] B883-4450 К), [4245] B890—7000 К). С
принятым значением Дт#°(Мо) согласуются менее надежные значения, полученные в работах [4542]
C5 кДж-моль^1), [118, 1121, 1482] C7 кДж-моль), [4248] C6 кДж-моль), [665, 667]
(ЗвнДж-моль), [382, 617] D0 кДж-моль), [619, 894] D1 кДж-моль), [4244] D8 кДж-моль).
Ошибочное значение Дт/Г°(Мо) было получено в работе [559] B5,5 кДж-моль). В работах [382,
4248] получены аномально высокие значения С"(Мо, ж) E8 и 70 Дж-К^-моль), соответствую-
1 Данные работы [1125] для теплоемкости Мо(к) систематически занижены на 0,5 Дж-К^-моль.
22
Молибден и его соединения
Мо(к, ж)
щие, по-видимому, неравновесному состоянию Мо(ж). Завышенное значение С^(Мо, ж) также
следует из данных [4245] D8 Дж-К^-моль), тогда как из данных [4542] следует заниженное
значение C4 + 4 Дж-К^-моль).
Погрешности приведенных в табл. 881 значений Ф°(Т) при 298,15, 2000, 3000 и 6000 К равны
0,1, 0,5, 0,9 и 3,7 Дж-К^-моль-1.
Ранее термодинамические функции Мо(к, ж) вычислялись в справочниках Халтгрена и др.
[2752] (до 5000 К), JANAF [2834] (до 6000 К), Шика [4181] (до 4965 К), Сталла и Зинке [4421]
и Келли [2960, 2963] (до 3000 К), а также Дитмарсом и др. [2083] (до 2800 К), Жагриной и др.
[420] (до 2892 К) и Брюэром и Ламоро [1662] (до 4000 К). Различия в значениях
термодинамических функций Мо(к), приведенных в табл. 881 и в [420, 1662, 2083, 2752, 2834], малы и не
превосходят 0,3 Дж-К^-моль в значениях Ф°(Г). Расхождения в значениях термодинамических
функций Мо(ж) существенно больше и достигают --—1,5 Дж -К -моль для данных [2752] и [2834].
В настоящем справочнике в качестве стандартного состояния молибдена принято
кристаллическое состояние и в соответствии с этим
bfH°(Mo, к) = 0.
Давление пара молибдена в реакции Мо(к, ж) = Мо(г) вычислено с использованием
принятой в справочнике энтальпии сублимации
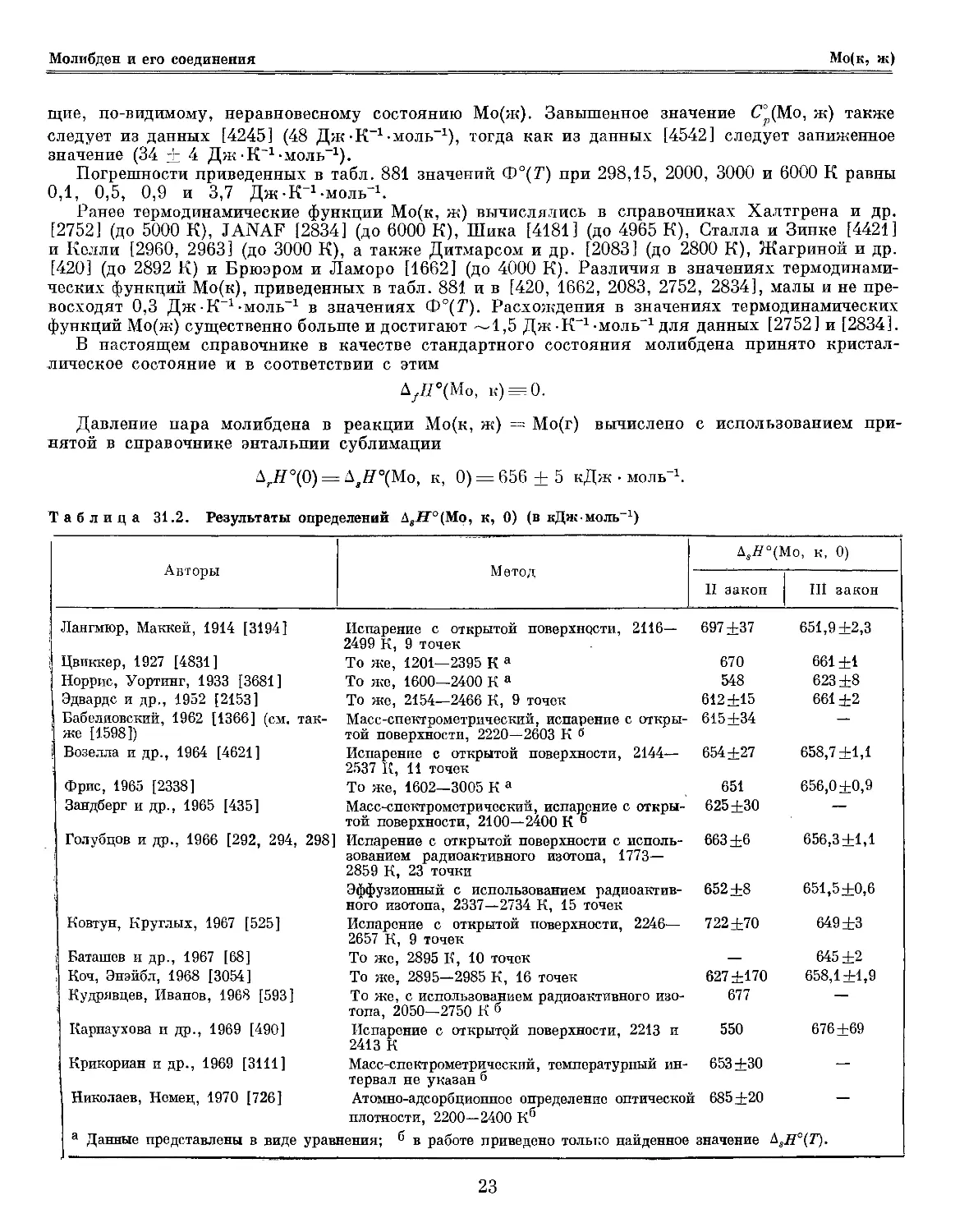
Дг#°@) = ДвЯ°(Мо, к, 0) = 656 + 5 кДж • моль
Таблица 31.2. Результаты определений AsH°(Mo, к, 0) (в кДж-моль)
-1
Авторы
Метод
Д5Я°(Мо, к, 0)
II закон
III закон
Лангмюр, Маккей, 1914 [3194] Испарение с открытой поверхности, 2116— 697+37 651,9+2,3
2499 К, 9 точек
Цвиккер, 1927 [4831] То же, 1201—2395 Ка 670 661+1
Норрис, Уортинг, 1933 [3681] То же, 1600—2400 Ка 548 623+8
Эдварде и др., 1952 [2153] То же, 2154—2466 К, 9 точек 612+15 661+2
Бабелиовский, 1962 [1366] (см. так- Масс-спектрометрический, испарение с откры- 615+34 —
же [1598]) той поверхности, 2220—2603 К б
Возелла и др., 1964 [4621] Испарение с открытой поверхности, 2144— 654+27 658,7+1,1
2537 К, 11 точек
Фрис, 1965 [2338] То же, 1602—3005 Ка 651 656,0+0,9
Зандберг и др., 1965 [435] Масс-спектрометрический, испарение с откры- 625+30 —
той поверхности, 2100—2400 К "
Голубцов и др., 1966 [292, 294, 298] Испарение с открытой поверхности с исполь- 663±6 656,3+1,1
зованием радиоактивного изотопа, 1773—
2859 К, 23 точки
Эффузионный с использованием радиоактив- 652±8 651,5+0,6
ного изотопа, 2337—2734 К, 15 точек
Ковтун, Круглых, 1967 [525] Испарение с открытой поверхности, 2246— 722+70 649+3
2657 К, 9 точек
Баташев и др., 1967 [68] То же, 2895 К, 10 точек — 645±2
Коч, Энэйбл, 1968 [3054] То же, 2895—2985 К, 16 точек 627±170 658,1+1,9
Кудрявцев, Иванов, 1968 [593] То же, с использованием радиоактивного изо- 677 —
топа, 2050-2750 К б
Карнаухова и др., 1969 [490] Испарение с открытой поверхности, 2213 и 550 676+69
2413 К
Крикориан и др., 1969 [3111] Масс-спектрометрическяй, температурный ин- 653+30 —
тервал не указан б
Николаев, Немец, 1970 [726] Атомно-адсорбционное определение оптической 685+20 —
плотности, 2200—2400 К6
а Данные представлены в виде уравнения; б в работе приведено только найденное значение hsH°(T).
23
Мо(г), Мо+(г) Глава 3t
В табл. 31.2 приведены значения энтальпии сублимации молибдена, вычисленные на
основании измерений давления его насыщенного пара. В расчетах принималось, что пары молибдена
одноатомны [435]. При расчетах по данным [3194] использовались исправленные [2894]
значения температуры. Приведенные в табл. 31.2 погрешности соответствуют только
воспроизводимости измерений. Принятая величина является средневзвешенной из результатов работ [292,
294, 298, 2153, 2338, 3054, 3194, 4621]. Ее погрешность оценена с учетом неточности
термодинамических функций и возможных систематических ошибок.
Мо(г). Термодинамические свойства газообразного молибдена в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 882.
Для расчета термодинамических функций использованы уровни энергии атома Мо в
интервале 0—57 000 см, соответствующие валентным электронным конфигурациям ..Adb5s (к этой
конфигурации относится основной уровень '?), ...4d*5s2, ..Ade, ..Ad55l (I = 1-^-4), ...4d*5s5Z (l~
— 1, 2) x и имеющие суммарный статистический вес 4235. Принятые величины основаны на
данных Мур [3550], а также на оценках для ряда ненаблюдавшихся электронных состояний. Для
электронных конфигураций ...ЫЪЫ (I = 1-^4) и ...4d45s5Z (I = 1, 2) оценка выполнена по
методике, изложенной в Приложении 6, а для конфигураций ...4d45s2 и ..Ad6 — сравнением энергии
соответствующих состояний Gr, Mo, W и V+, Nb+ и Та+. Оцененные уровни имеют энергию от
29 000 см" и выше с суммарным статистическим весом 2082. Погрешность оценки энергии лежит
в пределах 100—4000 см.
Термодинамические функции Мо(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Т ^ 3000 К обусловлены неточностью фундаментальных постоянных, а при
более высоких температурах — неточностью оценки ряда уровней энергии и пренебрежением
ридберговскими состояниями. Погрешности значений ФС(Г) при 3000, 6000 и 10 000 К
составляют 0,02, 0,3 и 1 Дж-К^-моль".
Ранее термодинамические функции Мо(г) вычислялись в справочниках JANAF [2834], Шика
[4181 ] и Сталла и Зинке [4421 ] (до 3000 К), а также в работе Хилзенрата и др. [2652] (до 10 000 К).
При Т ^ 3000 К термодинамические функции Мо(г), приведенные в табл. 882 и в перечисленных
работах, согласуются в пределах 0,02—0,03 Дж-К^-моль. При более высоких температурах
расхождения возрастают вследствие того, что в настоящем справочнике учтены ненаблюдавшиеся
уровни энергии Мо; расхождения составляют до 0,04 и 0,01—0,7 Дж-К^-моль в значениях
Ф°(Л с данными [28341 и [26521 соответственно.
Энтальпия образования
^H0(Mo, г, 0) = 656 ± 5 кДж • моль,
соответствует принятому в справочнике значению энтальпии сублимации молибдена.
Мо+(г). Термодинамические свойства газообразного положительного иона молибдена в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 883.
Для расчета термодинамических функций использованы уровни энергии Мо+ в интервале
0—76 468 см, соответствующие электронным конфигурациям ..Ad5 (к ней относится основной
уровень eS), ..Ads5s2, ..Ad?5l3 (I = 0, 1), ..Ads5s5p x и имеющие суммарный статистический вес
3158. Принятые величины основаны на данных Мур [3550], а также на оценках для ряда
ненаблюдавшихся электронных состояний. Для электронных конфигураций ...4d*5Z (Z = 0, 1) и ..Ad35s5p
оценки выполнены по методике, изложенной в Приложении 6, а для конфигураций ...4d35s2 и
..Ad6 — сравнением энергии соответствующих состояний Сг+, Мо+, W+ и изоэлектронных
атомов V, Nb и Та. Оцененные уровни имеют энергию от 21 000 см" и выше с суммарным
статистическим весом 1546. Погрешность оценок лежит в пределах 500—4000 см.
Термодинамические функции Мо+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Т ^ 3000 К обусловлены неточностью принятых значений фундаментальных
постоянных и не превосходят 0,02—0,03 Дж-К^-моль, при более высоких температурах —
неточностью оценки ряда уровней энергии. В значениях Ф°(Т) при 6000 и 10 000 К они
составляют 0,1 и 0,4 Дж-К^-моль.
1 См. примеч. 1 к тексту по Сг(г).
24
Молибден и его соединения
МоО(г)
Ранее термодинамические функции Мо+(г) вычислялись в справочнике JANAF [2834], а также
Хилзенратом и др. [2652] (до 10 000 К) и Грином и др. [2473] (до 50 000 К). При Т < 3000 К
различия в значениях термодинамических функций, приведенных в этих работах и в табл. 883,
не превосходят 0,02 Дж-К^-моль. При более высоких температурах возникают расхождения
вследствие того, что в настоящем справочнике учтен ряд ненаблюдавшихся уровней энергии Мо+.
Эти расхождения составляют 0,04 и 0,3 Дж-К^-моль в значениях Ф°(Г) при 6000 и 10 000 К.
Константа равновесия реакции Мо+(г) + е(г) = Мо(г) вычислена с использованием
значения ЬгН°@) = —685,0 ± 0,1 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации атома Мо
/0(Мо) = 57 260 + 10 см = 685,0 + 0,1 кДж-моль.
Принятая величина основана на рекомендации Мур [3550] и расчетах пределов ридбергов-
ских серий ..Adb{fiS)ns'7S, ..Adb(eS)ns5S и ...4d5F.S)red7.D атома Мо, выполненных по данным [3550]
при подготовке справочника. Раух и Аккерман [3944] методом электронного удара получили
менее точное значение 697 ± 10 кДж-моль.
Принятому значению соответствует
bfH°(Mo+, г, 0) = 1341,000 ±5,0 кДж моль.
МоО(г). Термодинамические свойства газообразного оксида молибдена в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 884.
В табл. 31.3 приведены молекулярные постоянные МоО, принятые в справочнике на
основании приближенных оценок и крайне ограниченных экспериментальных данных.
Многочисленные попытки получения электронного спектра свободных молекул МоО [903, 2374, 2720, 3834,
4448] остались безуспешными. В спектре молекул МоО, изолированных в матрице из аргона,
в видимой области наблюдались два электронных перехода A4 325 и 14 450 см), а в ИК-области—
основные частоты молекул Мо1вО и Мо18О [1426, 2633]. Квантовомеханические расчеты этой
молекулы не проводились.
Таблица 31.3. Молекулярные постоянные МоО
Состояние
• 103
De ¦ 107
А
а3 Е+
Ь3ф,
В6П
0
130 е
260 е
390 е
520 е
11 000 е
14 000 е
14 500
902 б
4,4 б
0,42 в
Л Л
1,72 е
Примечание. Ниже все постоянные (кроме р{) даны всм1.
а Объединенные электронные термы Г»' 20 000 30 000 40 000
Pi 20 40 40;
б вычислено по соотношению A. 67) и значениям AG,, =893,5, D0=45 700; в вычислено по соотношению A. 38);
г вычислено по соотношению A. 69); Д вычислено по соотношению A. 68); е оценка (см. текст.)
По аналогии с молекулой СгО в справочнике принимается, что молекула МоО имеет ХЪП
основное состояние с конфигурацией ¦¦¦^dad, а наблюдавшиеся в работе [2633] электронные
переходы интерпретированы как переходы 55П0 <- ХШ^ и 55n_j <- Х5П_Х. Оценка энергий
ненаблюдавшихся электронных состояний МоО была выполнена так же, как для СгО. При этом
предполагалось, что ниже состояния ВЧ1 с конфигурацией .••§|^(го« расположены только 4
электронных состояния (A5~E+, a32+, Ь3Ф и с3Д), энергии которых были приняты такими же, как у мо-
25
МоО(г)
Глава 31
лекулы СгО. Погрешности этих величин не превышают 3000 см х. Суммарные статистические
веса трех объединенных электронных термов, которые приведены в табл. 31.3, учитывают
электронные состояния МоО в области энергий до 40 000 смГ1, принадлежащие электронным
конфигурациям, соответствующим четырем электронам атома Mo (...<24, ...d3s и ...d2s2) и имеющим не
более даух разрыхляющих электронов. Погрешности приведенных статистических весов могут
иметь коэффициент 2—3.
Колебательные постоянные МоО в состоянии Х6П основаны на величине Д&/2 = 893,5 см,
найденной Бейтсом и Грином [1426] при исследовании ИК-спектра молекул Мо16О,
изолированных в матрице из аргона. Погрешность принятого значения ш, оценивается в 15 см.
Значение^ (МоО) = 1,72 + 0,05 А было оценено на основании сопоставления величины г(М—О)
в молекулах СгО3 и MoOs и значения гДСЮ) в состоянии ХЪЛ 1. Постоянная спин-орбитальной
связи A30 + 30 см) оценена в результате сравнения ее величины в молекулах оксидов металлов
с факторами расщепления нижних мультиплетных состояний атомов соответствующих металлов.
Термодинамические функции МоО(г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBn и ее производных вычислялись по уравнениям A. 90)—A. 92)
с учетом 12 возбужденных электронных состояний (в том числе четырех компонент состояния
Х5И и трех объединенных термов) в предположении, что Q(J0\ вР = 0,5/?;(?№>вр.
Статистическая сумма по колебательно-вращательным уровням энергии подсостояния Х«П_Х и ее
производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням энергии и интегрированием по значениям / с помощью уравнений типа A. 82).
В расчете учитывались все колебательно-вращательные уровни с значениями /г^/тах,„, п0~
следние находились из условий A. 81). Энергия уровней вычислялась по соотношениям A. 65),
A. 34), A. 64а), вырождение уровней учитывалось статистическим весом 2. Значения
коэффициентов YM в этих уравнениях, а также величины г;шах и /1(ш приведены в табл. 31.4;
коэффициент Y03 получен из условия A. 59), остальные идентичны соответствующим постоянным из
табл. 31.3.
Т а б л и ц а 31.4. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см)
в состоянии Jrfn_!, а также значения г>шах и JUm, принятые для расчета
термодинамических функций МоО (гI
У 01 • Ю
Yu.103
' lim
9,02 —4,4 4,2 —3
1 Энергии возбужденных состояний см. в табл. 31.3.
—2,67
101
447
Погрешности рассчитанных термодинамических функций при низких температурах
обусловлены главным образом отсутствием экспериментальных данных о величине вращательной
постоянной, межъядерного расстояния и постоянной спин-орбитальной связи молекулы МоО
в основном состоянии. При высоких температурах добавляются погрешности из-за отсутствия
данных о возбужденных состояниях этой молекулы. Погрешности в значениях Ф°(Т) при 298,15,
3000, 6000 и 10 000 К оцениваются в 1, 3, 6 и 10 Лж-К^-моль.
Термодинамические функции МоО(г) раньше вычислялись в таблицах JANAF [2834], а также
работах Краснова и др. [5683 (Т < 3000 К), Брюэра и Розенблатта [1666] (Ф°(Т) до 3000 К) и
Де Мария и др. [2032] (Т = 2000-^-2500 К). Все расчеты были выполнены по оцененным
постоянным. Расхождения данных JANAF и табл. 884 составляют от 1 до 4 Дж-К^-моль в значениях
Ф°(Г) и до 9 и 13 Дж-К^-моль в значениях S°(T) и С°(Т), они обусловлены пренебрежением
возбужденными состояниями МоО в [2834]. Расхождения с данными [568]
и 18—20 Дж-К^-моль-1 в значениях Ф°(Т) и S°(T), с данными [1666]
5 Дж-К^-моль в значениях ФС(Г).
составляют 12—16
и [2032] - 2-5 и
1 Оценка величины ге(МоО) по re(WO) представляется некорректной, поскольку молекула WO имеет
основное состояние другой симметрии с резко отличающейся величиной ше.
26
Молибден и его соединения
МоО2(к)
Константа равновесия реакции МоО(г) = Мо(г) + О(г) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#°@) = D0(MoO) = 547 + 15 кДж . моль« 45 700 ± 1300 см.
Результаты определений энергии диссоциации МоО представлены в табл. 31.5. Приведенные
в таблице погрешности учитывают воспроизводимость измерений, неточность использованных
в расчетах термодинамических функций, термохимических величин и сечений ионизации.
Принятая величина основана на исследованиях изомолекулярной реакции в работе Горохова и др.
[310, 409, 463]. Менее точные величины были получены при исследовании равновесий
гетерогенных реакций [2032, 2115], что, возможно, связано с пониженной активностью веществ в
конденсированном состоянии (см. [1666, 1844]).
Таблица 31.5. Результаты определений D0(MoO) (в кДж-моль")
Авторы
Де Мария и др., 1960 [2032, 2115]
Горохов и др., 1977 [310, 409, 463]
Метод
Масс-спектрометрический, МоО(г)=Мо(к)+ О(г),
2265-2470 К, 7 точек
То же, ЗМо(к)+А12О3(к)=ЗМоО(г)+2А1(г), 2265—
2470 К, 7 точек
То же, Mo(r)+YO(r)=MoO(r)+Y(r), 2708—2842 К
6 точек
Do(MoO)
II закон
266 ±350
170+430
, 468+280
III закон
518+16
501 +20
547+15
В работах Чоудери и др. [1844] и Брюэра и Ламоро [1662] были рассмотрены данные по
энтальпиям образования газообразных оксидов молибдена. Эти авторы выбрали практически
совпадающие значения D0(MoO) ;=» 600 ± 30 кДж-моль, основанные на исследованиях
равновесия реакции 2МоО2(г) = МоО3(г) + МоО(г) [1844, 2032, 2115]. Пересчет данных работ [1844,
2032, 2115] с использованием принятых в настоящем справочнике термодинамических функций,
сечений ионизации и термохимических величин приводит к значениям D0(MoO), равным 557 ±
± 40 кДж-моль [2032, 2115] и 583 + 35 кДж-моль [1844], что находится в значительно
лучшем согласии с принятой в справочнике величиной. В настоящем справочнике результаты
исследований этого равновесия используются при выборе энтальпий образования МоО2(г) и МоО3(г).
Включение принятого в справочнике значения D0(MoO) в совместный расчет с другими данными
по исследованиям реакций с участием МоО, МоО2 и МоО3 (см. текст по МоО2), показывает его
корректность, в то время как значение D0(MoO) = 600 кДж-моль плохо согласуется с этими
данными.
Принятой энергии диссоциации соответствует
г, 0) = 355,783 + 16 кДж • моль.
МоО2(к). Термодинамические свойства кристаллического диоксида молибдена стехиометриче-
ского состава в стандартном состоянии при температурах 100—3000 К приведены в табл. 885.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 31.1. За стандартное состояние МоО2(к) в справочнике принимается моноклинная
модификация х. Данные о существовании МоО2 в жидком состоянии не известны.
Термодинамические функции МоО2(к) при Т <[ 298,15 К вычислены на основании измерений
теплоемкости, проведенных Кингом [ЗОЮ] E3—296 К) на образце, состав которого был
охарактеризован только на содержание молибдена (точно совпадает с расчетным значением).
Экстраполяция теплоемкости ниже 53 К проводилась с использованием дробно-рациональной функции шестой
степени [1066], аппроксимирующей данные [ЗОЮ] с погрешностью не выше 1%, и привела к
значению ?°E3 К)=1,65 Дж-К-моль. Погрешности принятых значений 5го B98,15 К) и
1 Сведения о полиморфизме МоО2 отсутствуют. Принятое в справочнике [2834] предположение об
антиферромагнитном превращении МоО2 при Т < 50 К не подтверждено нейтронодиффракционным исследованием [2394],
я также измерениями электрического сопротивления монокристалла МоО2 в интервале 4,2—300 К [4028].
27
МоО2(г) Глава 31
#°B98,15 К)—#°@) (см. табл. 31.1) оцениваются в 0,2 Дж-К^-моль и 0,03 кДж-моль
соответственно.
При Т >¦ 298,15 К для теплоемкости МоО2(к) принято уравнение (см. табл. 31.1), полученное
при совместной обработке измерений энтальпии, проведенных Кингом и др. [3013] C99—1801 К,
образец тот же, что и в работе [ЗОЮ]) и Мезаки и др. [3495] D54—1084 К, химический анализ
образца МоО2 не приводится), с учетом значений, найденных графической экстраполяцией данных
[3013] при 2400 и 3000 К. В этом расчете данные [3495] учтены с существенно меньшим весом,
поскольку они имеют большой разброс. Погрешности приведенных в табл. 885 значений Ф°(Г)
при 298,15, 1000, 2000 и 3000 К оцениваются в 0,15, 1,2, 3 и 6 Дж -К -моль.
Ранее термодинамические функции МоО2(к) вычислялись в справочниках JANAF [2834]
(до 3000 К) и Шика [4181] (до 2000 К). Расхождения термодинамических функций МоО2(к),
приведенных в [2834] и табл. 885, достигают 3,5 Дж-К~1-моль~1 в значениях S°(T) и ФС(Г); они
объясняются тем, что авторы [2834] увеличили энтропию МоО2, предполагая существование
антиферромагнитного превращения ниже 50 К (см. примечание на с. 27).
Принятая в справочнике энтальпия образования
к, 298,15 К) = —589,3 ±0,6 кДж • моль
основана на калориметрических измерениях Стаскиевича и др. [4379] энтальпий сгорания
Мо(к) и МоО2(к). В этой работе были использованы образцы высокой чистоты и проведен
тщательный анализ продуктов сгорания. В аналогичном исследовании Ма [3344] получено близкое
значение —588,4±0,5 кДж-моль. Однако этот результат является менее надежным, поскольку
в работе [3344] не были проведены достаточно точные анализы исходных веществ и продуктов
сгорания. Принятое значение Ду#°(МоО2, к) находится в согласии с менее точными данными,
полученными методом ЭДС для реакций: 2Ре095О(к)+Мо(к)=МоО2(к)+1,9Ре(к) [388, 428,
1388, 1495, 2930, 3038, 3938], 2ШО(к)+Мо(к)=МоО2(к)+2Ш(к) [3938], Мо(к)+О2(г) =
=МоО2(к) [428, 2803, 2837] и 2СоО(к);+Мо(к)=МоО2(к)+2Со(к) [2803] при 1000—2000 К
и при исследованиях равновесия реакций МоО2(к)+2Н2(г)=Мо(к)+2Н2О(г) [168, 1819, 2431,
3543, 4522, 4594] и МоО2(к)+2СО(г)==Мо(к)+2СО2(г) [2418] при 1000-1400 К.
Калориметрические измерения [3521] энтальпий реакций с Na2O2(K) и Na2O(K) приводят к ошибочным
результатам.
Давление пара в реакции МоО2(к)=МоО2(г) вычислено с использованием значения
Дг#°@) = As-ff°(MoO2, к, 0)=571,366 + 15 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования МоО2 в кристаллическом и газообразном состояниях.
МоО2(г). Термодинамические свойства газообразного диоксида молибдена в стандартном
состоянии при температурах 100—6000 К приведены в табл. 886»
Молекулярные постоянные, принятые для расчета термодинамических функций, приведены
в табл. 31.6. На основании исследования ИК-спектра молекул МоО2, изолированных в матрице
из Аг, выполненного Серебренниковым и Мальцевым [926], и результатов полуэмпирического
расчета в рамках модели кристаллического поля, выполненного Хаитом и Хачкурузовым [1065],
в справочнике принимается, что в основном электронном состоянии Х3А2 молекула МоО2 имеет
угловую структуру (симметрия С2в). Приведенное в табл. 31.6 произведение моментов инерции
соответствует структурным параметрам г (Мо—О)=1,71+0,05 А (оценка по величине г(Мо—О)
в молекулах Мо63~, Мо2О6, Мо3О9) и 2}О—Мо—О = 108 ±10° (найден в работе [926 ] по изотопическому
смещению частоты v3). Погрешность значения IaIbIc оценивается в 2-Ю15 г3-см6. Значение
частоты v3, приведенное в таблице, найдено авторами [926]. Частота v2 оценена по величине частоты
деформационного колебания в молекулах диоксидов других переходных металлов; частота vx
рассчитана по уравнениям поля валентных сил и силовым постоянным, соответствующим принятым
значениям v2 и v3 х. Погрешности приведенных в табл. 31.6 значений частот оцениваются в 50 см
для vx и v2 и 15 см для v3.
1 С принятым значением v3 согласуется частота 950 см, наблюдавшаяся Никитиным и Мальцевым [723] в ИК-
спектре паров над МоО2(к) и МоО3(к), хотя авторы [723] не исключают возможность ее отнесения к
молекуле МоО3. Хьюэтт и др. [2633] при исследовании ИК-спектров оксидов молибдена, изолированных в матрицах
из Аг и Ne, отнесли интенсивную полосу в области 900 см к антисимметричному колебанию v3 и слабую
полосу при 948 см — к симметричному колебанию v2 молекулы Мо02. Однако это отнесение представляется
ошибочным, так как оно противоречит данным [926].
28
Молибден и его соединения
Мо02(г)
Приведенные в табл. 31.6 энергии семи возбужденных электронных состояний получены в
работе [1065] на основании полуэмпирического расчета. Для состояния С3А2 принятая энергия
хорошо согласуется с частотой перехода, наблюдавшегося в работах [1426, 2720] A1 550—11 631 см)
и отнесенного авторами [1065] к переходу С3А2—Х3Аг. Погрешность принятого значения
То (С3А) оценивается в 300 см остальных состояний в 4000—5000 см
о (С3А2) оценивается в 300 см, остальных состояний в 4000—5000 см
Таблица 31.6.
Значения молекулярных постоянных, а также а и рх, принятые для расчета
термодинамических функций MoO,, MoO,?, MoOg", Mo2O0, Mo3O9, Мо4О]2,
Молекула
МоО2
МоО3
MoOg
Мо2О6
Мо3О9
Мо4О12
Мо5О15
МоО2: а То{
МоО3: а То(
MoOj: a То
Состояние
хч2*
%lA'i a
%2А[а
&А,
^A±
%гАг
Х*Аг
Л1^1)=6000,
<?3В2)=25 000
13Л2)=20 000,
(АЫ])=10 000
V
925
824
800
970
970
970
970
ТоA
см
т0
, т
1
B)
C)
D)
E)
.
{ВЧ
ч2
520
380
350
900 B)
900 D)
900 D)
900 E)
=10 000,
12)=30 000
42)=20 000
V3
963
915 B)
900 B)
700 B)
815 B)
800 D)
800 E)
Т0(С*А2
см.
см.
см
340
300
685
600
600
600
)=11
B)
B)
B)
C)
D)
E)
500
350
200
200
200
B)
A4)
A9)
B4)
03В
200
100
100
100
з)=15
8
G)
D)
G)
A0)
ООС
v,
Г3 • CM9
— 6,2 -102
— 3 -103
— 3 -103
100 5 -106
— 1,5 -10'
— 9 -10'
— 5 -10s
, Т0(Ё*В2, F*At)=
а
2
6
6
4
3
4
5
20 000,
Рх
3
1
2
1
1
1
1
Термодинамические функции МоО2(г) вычислены по уравнениям A. 3)—A. 6), A. 10), A. 122)—
A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический осциллятор» с
учетом семи возбужденных электронных состояний по уравнениям A. 168)—A. 170). Погрешности
вычисленных значений термодинамических функций обусловлены приближенным характером
расчета и неточностью принятых значений молекулярных постоянных, главным образом энергий
возбужденных состояний. Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К равны 1, 4
и 6 Дж-К^-моль.
Ранее термодинамические функции МоО2 (г) вычислялись в справочниках JANAF [2835]
и Шика [4181] и в работах Брюэра и Розенблатта [1665], Краснова и др. [568] и Брюэра и Ламоро
[1662] (до 3000 К). В расчете [1665] принималось, что молекула МоО2 имеет линейную структуру,
а в расчетах [568, 4181] — что эта молекула имеет синглетное основное состояние.
Соответствующие расхождения этих расчетов с данными табл. 886 достигают 9—16 Дж-К-моль.
Расхождения результатов расчетов [2835] и [1662] с данными табл. 886 в основном обусловлены неполным
учетом возбужденных состояний Мо02 в этих расчетах, особенно существенным выше 2000 К;
они составляют 1—3 и 1—4 Дж-К-моль соответственно в значениях Ф°(Г).
Константа равновесия реакции МоО2(г)=Мо(г)+2О(г) вычислена с использованием значения
кгН°@) —1162,566 + 16 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(МоОа, г, 0) = —13 ± 15 кДж • моль.
Эта величина найдена одновременно с энтальпией образования МоО3(г) согласованием
приведенных в табл. 31.7 термохимических величин методом наименьших квадратов. В этом расчете
использовались только наиболее надежные измерения, выделенные курсивом. Указанные погрешности
учитывают воспроизводимость измерений, неточность термодинамических функций,
термохимических величин и сечений ионизации, использованных в расчетах. Приведенные в таблице
термохимические величины были вычислены по уравнению III закона, однако расчеты по II закону
в большинстве случаев также привели к близким значениям. В таблицу не включены результаты
исследований некоторых гетерогенных равновесий в работах [2032, 2115], так как они приводят
к менее точным величинам, плохо согласующимся с другими измерениями. Ненадежность этих
данных отмечалась в работах [1666, 1844]. Принятая величина подтверждается данными работы
[1466]. В работах [1732, 2705, 2834] дан критический анализ измерений [1573, 3854].
29
МоО3(к, ж)
Глава 31
Таблица 31.7. Результаты определений Ду-НГ°(МоО2, г, 0) и ДуН°(МоО3, г, 0) (в кДж-моль)
Авторы
Метод
Термохимическая
величина
(III закон)
Блэкберн и др., 1958 [1573] (см.
также [1570])
Эффузионный, МоО2(к)=МоО2(г), 1820—2031 К, 7
точек •
Берне и др., 1960 [1732]
Планте, 1960 [3853, 3854] (см.
[1769, 2834])
То же, 8/2МоО2(к)=МоО3(г)+1/2Мо(к), 1820—2031 К,
7 точек а
Масс-спектрометрический, МоО2(к)=МоО2(г), 1568—
1779 К, 12 точек
То же, 3/2Мо02(к)=Мо03(г)+1/2Мо(к), 1483^1779 К,
33 точки
Эффузионный, МоО3(к)=МоО2(г), 1611—1802 К, 6 точек
То же, 3/2МоО2(к)=МоО3(г)-|-1/2Мо(к), 1592-1918 К,
119 точек
Де Мария и др., 1960 [2032, 2115] Масс-спектрометрический, 2МоО2(г)=МоО(г)+МоО3(г),
2284—2470 К, 5 точек
То же, МоО(г)+О(г)=МоО2(г), 2265—2420 К, 6 точек
То же, МоО2(г)+А1(г)=:А1О(г)+МоО(г), 2265—2470 К,
6 точек
То же, СаО(г)+МоО2(г)=МоО3(г)+Са(г), 2397—2414 К,
3 точки
То же, SrO(r)+MoO2(r)=MoO3(r)+Sr(r), 1990—2236 К,
7 точек
То же, Sr2O(r)+2O(r)+Mo(K)=2Sr(r)+MoO8(r), 2149 К
Протока, взаимодействие Мо(к) с газовыми смесями
Н2О—Н2—Аг, МоО2(к)=МоО2(г), 1475—1775 К, 6 точек а
То же, Мо(к)+ЗН2О(г)=МоО3(г)+ЗН2(г), 1475—1775 К,
5 точек а
Франклин, Стикни, 1971 [2316] (см. Масс-спектрометрический, Мо(к)-Р/2О2(г)—МоО3(г),
"" """" 1900—2200 К, 4 точки
То же, Мо(к)+О2(г)=МоО2(г), 1900—2200 К, 4 точки
Масс-спектрометрический, 2МоО2(г)=МоО(г)+МоО3(г),
1462—1908 К, 8 точек, в работе приведено только
значение ДГ#°B98К)=—35+13
Масс-спектрометрпческий, МоО2(к)=МоО2(г), 1659—
1737 К, 15 точек
То же, 3/2МоО2(к)==МоО3(г)-1-1/2Мо(к), 1659—1737 К,
16 точек
Дроварт и др., 1964 [2116] (см.
также [2113])
Белтон, Иордан, 1965 [1466]
также [4382])
Чоудери и др., 1975 [1844]
Чижиков и др., 1976 [1132] (см.
также [476, 1131])
Балдуччи и др., 1977 [1376]
То же, МоО2(г)+РО2(г)=Р0(г)+МоО3(г), 1760-2025 К,
5 точек
а Вычислено с использованием данных [1732] о составе пара над МоО2(к); б Д^#°(МоО2, г, 0);
D A^0(Mo03, г, 0); Г Д^ЧМоОз, г, 0)-2Д/Я°(МоО2, г, 0); Д Д^МоО,,, г, 0)-AfH°(№O2,
-41±12б
—384+15 в
—15+12 б
—357+12 в
-0,4 б
-375 в
343+25 г
—22+18 б
—34+20 б
—366 +25 Д
—360 +25 Д
—326+25 в
—11+15 б
—371+15 в
—358+10 Б
—10+10 б
—369+25 г
—11+12 f>-
—316+12 в
—343 +15 Д
г, 0).
МоО3(к, ж). Термодинамические свойства кристаллического и жидкого триоксида молибдена
стехиометрического состава в стандартном состоянии при температурах 100—3000 К приведены
в табл. 887.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 31.1. За стандартное состояние МоО3(к) принимается ромбическая модификация слоистого
типа. Фазовых переходов у МоО3(к) нет (см., например, [30133).
TepiH ^^амические функции МоО3(к) при Т <С 298,15 К вычислены по измерениям
теплоемкости, проведенным Смитом и др. [4320] A8—300 К, по оценке авторов погрешности составляют
0,2% при Т ^> 50 К и ><~0,5% при более низких температурах). Экстраполяция теплоемкости
ниже 18 К приводит к значению 5°A8 К)=0,60 Дж-К~1-моль~1. Погрешности принятых значений
5°B98,15 К) и #°B98,15)—#°@) (см. табл. 31.1) оцениваются в 0,5 Дж-К-^моль и 0,05 кДж-
30
Молибден и его соединения МоО3(г).>
•моль соответственно. Менее точные измерения теплоемкости МоО3(к) в работе Зельтца и др.
[4232] G0—299 К) в справочнике не использовались.
В интервале 298,15—1075 К для теплоемкости МоО3(к) принято уравнение (см. табл. 31.1),
полученное по измерениям энтальпии в работе Кинга и др. [3013] C99—1064 К, образец содержал
~99,9% МоО3). Менее точные измерения выполнены Косгровом и Снайдером [1940] B73—1068 К,
результаты в среднем на 2% выше данных [3013]) и Буске и др. [1632] C79—971 К, метод ДСК,
результаты представлены только уравнением).
Температура плавления МоО3 A075 + 5 К) принята на основании калориметрических измерений
Кинга и др. [3013] энтальпии МоО3(к, ж) в интервале 1065—1085 К. В работах [327, 328, 878,
973, 1940, 2676, 2810, 3479, 3998] она определена равной 1069 К методом термического анализа,
который, по-видимому, фиксировал начальную стадию плавления образцов МоО3, состав которых
не был охарактеризован. Близкие значения были получены в работах [445] A068 К) и [55] A071 К).
Энтальпия МоО3(ж) измерялась Кингом и др. [3013] A081—1394 К) и существенно менее точно
Косгровом и Снайдером [1940] A069—1300 К). На основании данных [3013] энтальпия
плавления МоО3 принята равной 48,7±4,0 кДж-моль и теплоемкость МоО3(ж) равной 127 +
+5 Дж-К~1-моль~1. Принятое значение Аш^° согласуется в пределах точности определения
с найденным в работе [1940] E2,5 кДж-моль).
Погрешности приведенных в табл. 887 значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К
оцениваются равными Q,4, 1, 6 и 13 Дж-К~1-моль.
Ранее термодинамические функции МоО3(к, ж) вычислялись в справочниках JANAF [2834]
(до 2000 К) и Шика [4181 ] (до 1500 К) по данным [3013, 4320]. Различия в значениях
термодинамических функций, приведенных в [2834, 4181 ] и в табл. 887, незначительны и обусловлены различием
методов аппроксимации исходных данных.
Принятая в справочнике энтальпия образования
^fH°(MoO3, к, 298,15 К) = —744,6 + 0,5 кДж • моль
основана на калориметрических измерениях Стаскиевича и др. [4379] энтальпий сгорания Мо(к)
и МоО2(к). В этой работе были использованы образцы высокой чистоты и проведен тщательный
анализ продуктов сгорания. В аналогичном исследовании Ма [3344] получено близкое значение
—745,4+0,5 кДж-моль. Однако этот результат является менее надежным, поскольку в работе
[3344] не было проведено достаточно точных анализов исходных веществ и продуктов сгорания.
Существенно менее точные значения были получены в ранних калориметрических измерениях
энтальпии сгорания молибдена [2028, 3554, 3645], так как в них не было учтено образования в
продуктах сгорания МоО2(к). К весьма неточному значению приводят калориметрические измерения
[3521] энтальпий реакций Мо(к) и МоО2(к) с Na2O2(K). Равновесие восстановления МоО3(к)
водородом, исследованное Васильевым и др. [168, 4594] G73—883 К), интерпретировано ими как
происходящее в две стадии: МоО3(к)'+Н2(г)=МоО2(кI+Н2О(г) и МоО2(к)+2Н2(г)==Мо(к)+2Н2О(г).
Однако проведенные при подготовке настоящего справочника расчеты показали, что эта
интерпретация для первой стадии, видимо, ошибочна.
Давление пара в реакции МоО3(к, ж)=МоО3(г) вычислено с использованием значения ДГ^Г°(О) =
= Д8Я°(МоО3, к, 0)=379,587 ±15 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования МоО3 в кристаллическом и газообразном^; состояниях.
МоО3(г). Термодинамические свойства газообразного триоксида молибдена в|'стандартном
состоянии при температурах 100—6000 К приведены в табл. 888.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 31.6. На основании результатов электронографического [452] и спектрального [924, 927J
исследований, а также теоретического анализа электронных состояний МоО3 [1065] в справочнике
принимается, что в основном электронном состоянии Х1А[ молекула МоО3 имеет плоскую
симметричную структуру (симметрия D3h) 1. Приведенное в табл. 31.6 произведение главных моментов
инерции соответствует г(Мо—О) = 1,711 ±0,008 А, найденному Ивановым при электронографиче-
ском исследовании пара МоО3 [452]. Погрешность значения IjJbIc не превышает 1 •1015г3-см°.
Из приведенных в табл. 31.6 частот рсновных колебаний только частота v3 наблюдалась
1 Заключение авторов работы [2633] о пирамидальной структуре молекулы МоО3 основывается на неправильной
интерпретации ЙК-спектров паров триоксида молибдена, изолированных в матрицах инертных газов.
31
Мо03~(г) __ ' Глава 31
Серебренниковым и Мальцевым [924, 927] в ИК-спектре молекул МоО3, изолированных в матрице
из Аг. Другие частоты рассчитаны в приближении поля валентных сил по оцененным в работах
[568, 924, 927, 2633J значениям силовых постоянных х. Погрешность значения v3 оценивается
в 15 см, погрешность частоты vx в 75 см, частот v2 и v4 — в 50 см. В пределах погрешности
с принятым значением v3 согласуются частоты 922 и 916 см, наблюдавшиеся Хьюэттом и др.
[2633] в ИК-спектре продуктов высокотемпературного окисления молибдена. Приведенные
в табл. 31.6 энергии двух возбужденных электронных состояний МоО3 оценены Хаитом и Хачку-
рузовым [1065] на основании полуэмпирического расчета в рамках теории кристаллического
поля. Погрешность этих величин оценивается в 10 000 см.
Термодинамические функции МоО3(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом двух возбужденных электронных состояний по соотношениям A. 168)—A.170).
Погрешности вычисленных величин обусловлены приближенным характером расчета и неточностью
принятых значений молекулярных постоянных (главным образом частот колебаний и энергий
возбужденных состояний). Они составляют 2, 6 и 10 Дж -К -моль в значениях Ф°(Т) при 298,15,
3000 и 6000 К.
Ранее термодинамические функции MoOs(r) вычислялись в справочниках JANAF [2835 ] и Шика
[4181], а также Брюэром и Ламоро [1662] и Красновым и др. [568] (до 3000 К) в предположении,
что молекула МоО3 имеет плоскую структуру [568, 2834, 41811 или структуру правильной пирамиды
[1662]. Расхождения данных [2835] и табл. 888 не превышают 2—4 Дж-К"-моль~1 в значениях
Ф°(Т); расхождения с данными [1662] составляют ~6 Дж-К~1-моль.
Константа равновесия реакции МоО3(г)=Мо(г)-)-ЗО(г) вычислена с использованием значения
Дг#°@)=1756,349 + 16 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —360 ± 15 кДж • моль.
Эта величина определена одновременно с значением Дуйг°(МоО2, г, 0) согласованием
приведенных в табл. 31.7 термохимических величин методом наименьших квадратов (см. текст по МоО2(г)).
Погрешность принятой величины учитывает воспроизводимость измерений, погрешности
термодинамических функций, термохимических величин и сечений ионизации.
МоОд(г). Термодинамические свойства газообразного!отрицателъного иона триоксида
молибдена в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 889.
Молекулярные постоянные, принятые для расчета термодинамических функций, приведены
в табл. 31.6. Спектр и структура MoOj экспериментально не исследовались. На основании
теоретического рассмотрения электронных состояний МоО", выполненного Хаитом и Хачкурузовым
[1065], в справочнике принимается, что в основном электронном состоянии Х2А{ эта молекула
имеет плоскую структуру (симметрия D3h). Приведенные в табл. 31.6 значения постоянных МоО~
оценены по постоянным молекулы МоО3 на основании предположения, что у соединений
переходных металлов присоединение электрона ведет к уменьшению частот колебаний и увеличению
межатомных расстояний. Приведенное в табл. 31.6 значение IaIbIo имеет погрешность 1 -10~114 г3-см6,
а частоты колебаний погрешности до 100 см. Энергии двух низших электронных состояний
МоО~, приведенные в табл. 31.6, оценены Хаитом и Хачкурузовым [1065] на основании
полуэмпирического расчета для модели кристаллического поля; погрешности этих величин равны ~5000
и 10 000 см соответственно.
Термодинамические функции MoOj(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом двух возбужденных электронных состояний по уравнениям A. 168)—A. 170).
Погрешности вычисленных величин обусловлены главным образом неточностью оцененных
значений молекулярных постоянных, а также приближенным характером расчета; они составляют
4, 10 и 15 Дж-К-^моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций МоО~(г) публикуется впервые.
Константа равновесия реакции МоОз(г)=Мо(г)+ЗО(г)-|-е(г) вычислена с использованием
значения Дг#°@)=2041,349 + 30 кДж-моль, соответствующего принятому в справочнике сродству
молекулы МоО3 к электрону
1 /г=6,4+0,5, />2=0,3+0,1, ^//^=0,9+0,3 (XW-5 дин-см).
32
Молибден и его соединения Ма2О6(г)
40(МоО3) = — 285 + 25 кДж-моль.
,Эта величина основана на данных Енсена и Миллера [2856] (см. также [3507]), которые
исследовали константы равновесия реакций НаМоО4(г)+е(г)=НМоО^(г)+Н(г) и НМоО~(г)+Н(г) =
=МоО~(г)+Н2О(г) (фотометрия пламен, 1815—2475 К), и Белтона и Иордана [1466], которые
исследовали константу равновесия реакции Мо(к)+4Н2О(г)=Н2МоО4(г)+ЗН2(г) (метод протока,
1473—1773 К). Погрешность принятого значения учитывает неточность измерений констант
равновесия, погрешности использованных в расчетах термодинамических функций и термохимических
величин.
Принятая величина подтверждается исследованиями равновесия реакций в пламенах,
выполненными Поллом и Пиккарди [5032] (см. также [4856]), где было получено .40(МоО3) =
=—263 кДж-моль. Исследования реакции МоО3(к)+Н2О(г)=Н2МоО4(г), выполненные Глем-
зером и Хеслером [2419] (метод протока, 873—963 К), при их комбинировании с данными работы
[2856] приводят к завышенному по абсолютной величине значению —374 кДж-моль, что, по-
видимому, связано с неточностью в определении характера реакции [2419].
Принятому значению сродства к электрону соответствует
AfH°(MoO-, г, 0) == —645,000 + 29 кДж • моль.
Мо2О6(г). Термодинамические свойства газообразного гексоксида димолибдена в стандартном
состоянии при температурах 100—6000 К приведены в табл. 890.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 31.6. На основании результатов электронографического исследования, выполненного
Ивановым [452], в справочнике принимается, что в основном электронном состоянии ХгА молекула
Мо2О6 имеет циклическую структуру с двумя группами МоО2, расположенными в плоскости,
перпендикулярной плоскости кольца Мо2О2 (симметрия D^I. Приведенное в табл. 31.6 произведение
моментов инерции вычислено для структурных параметров: г(Мо—Овнеш) = 1,711+0,08 А, г(Мо—
-Омост) = 1,892+0,025 А, ^Овнеш-Мо-Овнет=120 + 10°и ^ОИОСТ-Мо-Омоог-90 + 10°, полученных
Ивановым [452] при обработке электронограмм с учетом данных для А12С16, А12Вг6 и Fe2Cl6 [439,
2159, 2568]. Погрешность значения IaIbIc оценивается в 1 • 10~11а г3-см6. В ИК-спектре молекул
Мо2О6, изолированных в матрицах из Ne и Аг, Хьюэтт и др. [2633] наблюдали три полосы в
областях 976 (970), 694 F85) и 350 C49) см 2, а Серебренников и Мальцев [927] в матрице из Аг
зарегистрировали две полосы с максимумами 697 и 685 см, отнесенные по аналогии с полосами
молекул (МоО3)„ (п 1> 3) к валентному колебанию Мо—О групп МоО2 и к колебанию кольца Мо2О2.
Молекула Мо2О6 имеет 18 основных частот колебаний типов симметрииМгD), ВоE), АиA), Ви(8).
Приведенные в табл. 31.6 усредненные значения близких по типу и величине основных частот
с соответствующими степенями вырождения рассчитаны по уравнениям поля валентных сил.
Силовые постоянные были оценены на основании результатов исследований ИК-спектра Мо2О6
[452, 2633] и силовых постоянных других кислородсодержащих соединений молибдена [1072,
3103, 3324, 3325]. Последние корректировались по результатам обработки по методам II и III
законов экспериментальных данных по равновесию реакций с участием газообразных оксидов Мо.
Погрешности приведенных в табл. 31.6 частот оцениваются в 100 см для vx—v4, 70 см для v5
и 50 см для v6, v7.
Термодинамические функции Мо2О6(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности вычисленных величин
обусловлены неточностью принятых значений основных частот и приближенным характером
расчета. Погрешности в значениях Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 15, 30 и
40 Дж-К-^моль.
Таблица термодинамических функций Мо2О6(г) публикуется впервые.
Константа равновесия реакции Мо2О6(г)=2Мо(г)+6О(г) вычислена с использованием значения
ДгЯ°@)=3932,698+41 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^^МозОе, -г, 0) == —1140 ± -40 кДж • моль.
1 Структура Мо,Ов аналогична структуре димерных молекул галогенидов алюминия А12Х6 (см. т. III, кн. i).
2 В работе [2633] не было выполнено отнесение наблюдаемых полос к конкретным колебаниям.
3 Заказ Kt 1590 33
Мо3О9(г)
Глава 31
Результаты измерений энтальпии образования Мо2О6(г) представлены в табл. 31.8. Приведенные
в этой таблице погрешности учитывают только воспроизводимость измерений. Как видно из
таблицы, результаты измерений, выполненных в разных работах, различаются больше, чем это можно
объяснить их воспроизводимостью, что объясняется сложностью исследованных реакций. Принятая
величина основывается прежде всего на наиболее точных значениях, полученных Бернсом и др.
[1732]. Найденная по данным этой работы величина совпадает со средневзвешенной из всех
приведенных в таблице величин, вычисленных по уравнению III закона. Основным источником
погрешности принятой величины является неточность термодинамических функций Мо2О6(г).
Таблица 31.8. Результаты определений ДуН°(Мо2Ов, г, 0) (в кДж-моль)
Авторы
Метод
AfH°(Мо2Ов, г, 0)
II закон
III закон
Блэкберн и др., 1958 [1573] (см. так- Эффузионный, ЗМоО2(к)=Мо2Ов(г)+Мо(к), 1820 1249+80 —1160+4
же [1570]) 2031 К, 6 точек а
Берне и др., 1960 [1732] Масс-спектрометрический, ЗМоО2(к)=Мо2О6(г)+ —1143+40 —1137+1
+Мо(к), 1568—1779 К, 12 точек
Белтон, Иордан, 1965 [1466] Протока, 2Мо(к)+6Н2О(г)=Мо2О6(г)+6Н2(г), —1066+100 —1171+11
1475—1775 К, 6 точек а
Норман, Стели, 1965 [3679] Масс-спектрометрический, сублимация МоО2(к), —1193+40 —
2МоО3(г)=Мо2Ов(г), 1664—1850 К
То же, сублимация МоО3(к)+В2О3(к), 2МоО3(г)= —1222+40 —
=Мо2Ов(г), 1667-1890 К
Чижиков и др., 1976 [476, 1131,1132] То же, ЗМоО2(к)=Мо2О6(г)+Мо(к), 1659—1737 К, —786+100 —1119+3
15 точек
а Вычислено с использованием данных [1732] о составе пара над МоО2(к)
Мо3О9(г). Термодинамические свойства газообразного ноноксида тримолибдена в стандартном
состоянии при температурах 100—6000 К приведены в табл. 891.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 31.6. В справочнике на основании результатов исследования структуры молекул Сг3О9
и W3O9 в работе Иванова [452] принимается, что в основном электронном состоянии ХгАх молекула
Мо3О9 имеет циклическую неплоскую структуру (симметрия C3v), образованную тремя группами
МоО2, связанными между собой тремя атомами кислорода. Ранее в электронографических
исследованиях, выполненных Засориным и др. [5074], Егоровой и Рамбиди [2159], принималось, что
молекула Мо3О9 имеет плоское кольцо Мо3О3 (симметрия D3k). Приведенное в табл. 31.6
произведение моментов инерции вычислено по структурным параметрам г(Мо—ОЕнеган) = 1,70+0,03 А,
г(Мо-ОМОС1)-1,90±0,02 А, ^Огаеша-Мо-Овнешн=112 + 6°, ^Омои-Мо-Омост=110+3°, ^Мо-
—Омоот—Мо=128 + 2°, которые основаны на исследованиях структуры молекул Мо3О9, МоО3,
Мо2О6, W3O9 в работах [452, 2159]; погрешность значения IaIbIg оценивается в 3-Ю11 г3-смв.
Молекула Мо3О9 имеет 20 основных частот колебаний типов симметрии АгG), А2C) и ЕA0).
Приведенные в табл. 31.6 усредненные значения близких по типу и величине основных частот
Мо3О9 с соответствующими кратностями вырождения основаны на результатах приближенного
расчета по уравнениям поля валентных сил с использованием силовых постоянных
кислородсодержащих соединений Мо [1071, 3103, 3324, 3325]. Правильность выбора силовых
постоянных валентных колебаний связей Мо—Овиеш и Мо—Омоет подтверждается согласием вычисленных
значений частот с частотами, наблюдаемыми в ИК-спектре газообразного Мо3О9 [723, 2781 ] и
молекул Мо3Ой, изолированных в матрицах инертных газов [782, 2633]. Силовые постоянные
деформационных колебаний корректировались с учетом результатов расчетов по II и III законам рАвнове-
сий реакций с участием газообразных оксидов Мо. Погрешности приведенных в табл. 31.6
значений частот Мо3Оэ оцениваются в 20 см для vx и v3, 100 см для v2, v4 и 50 см для v- и vs.
Термодинамические функции Мо3Оэ(г) вычислены по уравнениям A. 3) — A-6), A. 9), A. 10),
A. 122)—A. 124), A. 128), A. 130) в приближении «жесткий ротатор — гармонический осциллятор»
без учета возбужденных электронных состояний. Погрешности вычисленных величин определяются
34
Молибден и его соединения
Мо3О9(г)
Таблица 31.9. Результаты определений Ау?Г°(Мо3О9, г, 0) (в кДж • моль)
Авторы
Файзер, 1931 [2232]
Уено, 1941 [4572] (см. [1507],
а также [476])
Ария и др., 1953 [43]
Зеликман и др., 1956 [446]
Берковиц и др., 1957 [1507] (см.
также [2777])
Бабаджан, 1957 [55]
Блэкберн и др., 1958 [1573] (см.
также [1570])
Джонс и др., 1958 [2890]
Аккерман и др., 1960 [1245]
Берне и др., 1960 [1732]
Хёрбе и др., 1961 [2705]
Глемзер, Хеслер, 1962 [2419]
Гулбрансен и др., 1963 [2521]
Белтон, Иордан, 1965 [1466]
Казенас, 1968 [473] (см. также [475,
478])
Чижиков и др., 1976 [1132] (см.
также [476, 1131])
Норман, Стели, 1965 [3679]
Алешко-Ожевская и др., 1978 [25]
Метод
Измерение скорости испарения МоО8(к),
ЗМоО3(к)=Мо3О9(г), 883—1023 К, 6 точек
Точек кипения, ЗМоО3(ж)=Мо3О9(г), 1074—
1427 К, 8 точек
Эффузионный, ЗМоО3(к)=Мо3О9(г), 883—973 К,
3 точки
Протока, ЗМо03(к)=Мо309(г), 941—987 К, 5 точек
То же, ЗМоО3(ж)=Мо3О9(г), 1094—1274 К,
5 точек
Точек кипения, ЗМоО3(ж)=Мо3О9(г), 1179—
1430 К, 6 точек
Масс-спектрометрический, ЗМоО3(к)=Мо8О9(г),
861—973 К, 4 точки
Протока, ЗМоО3(к)=Мо3О9(г), 953—1054 К,
6 точек
То же, ЗМоО3(ж)=Мо3О9(г), 1074—1134 К, 3 точки
Эффузионный, ЗМо03(к)=Мо309(г), 808—958 К,
15 точек
То же, 9/2МоО2(к)—Мо309(г)+3/2Мо(к), 1820—
2031 К, 6 точек
Испарение с открытой поверхности, ЗМоО3(к)=
=Мо3О9(г), 811—1034 К, 3 точки
Протока, ЗМоО3(к)=Мо3О9(г), 981—1061 К, 5 точек
Масс-спектрометрический, 9/2МоО2(к)=Мо3О9(г)+
+3/2Мо(к), 1634—1779 К, 7 точек
Эффузионный, ЗМоО3(к)=Мо3О9(г), 873—983 К,
12 точек
Протока, ЗМоО3(к)=Мо3О9(г), 900—1030 К,
данные представлены в виде уравнения
Эффузионный, ЗМоО3(к)=Мо3О9(г), 873—973 К,
6 точек
Протока, ЗМо(к)+9Н2О(г)=Мо3О9(г)+9Н2(г),
1475—1775 К, 5 точек
Масс-спектрометрический, ЗМоО3(к)=Мо3О9(г),
800—1000 К, данные представлены в виде
уравнения
То же, 9/2Мо02(к)=Мо3Ое(г)-Р/2Мо(к), 1659—
1737 К, 15 точек
То же, сублимация МоО2(к), ЗМоО3(г)=Мо3О9(г),
1664 1850 К
То же, сублимация смеси МоО3(к) и В2О3(к1,
ЗМоО3(г)=-Мо3О9(г), 1667—1890 К
То же, ЗМоО3(к)=-Мо3О9(г), 848—935 К
Ду#°(Мо3О9, г, 0)
II закон
-1945 ±6
-1882 ±11
—1965
—1944 ±17
—1911 ±16
—1888±11
—1871 ±6
-1855 ±27
-1808±900
-1888 ±13
—2076 ±70
—2014 ±180
—1863 ±6
-1908±80
—1850 ±13
-1894
—1904 ±56
-1811 ±20
—1922
—2083 ±35
—1979 ±50
-2008 ±80
—1864 ±8
III закон
-1899 ±3
-1897 ±2
-1883 ±10
—1897+1
-1895 ±2
—1897+1
—1893+2
—1891 ±2
-1893 ±8
-1903 ±1
—1891 ±7
—1833 ±9
—1893 ±1
—1877 ±2
-1896 ±1
—1891
—1899 ±2
—1927+17
—1897
-1879±1
—
—
в основном неточностью принятых значений молекулярных постоянных и приближенным
характером расчета, они составляют 20, 40 и 60 Дж-К-моль в значениях Ф°(Т) при 298,15, 3000
и 6000 К.
Таблица термодинамических функций Мо3О9(г) публикуется впервые.
Константа равновесия реакции Мо3О9(г)=ЗМо(гL-9О(г) вычислена с использованием значения
ДгЯ°@)=6079,047+43 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(Mofi9, г, 0) = —1890 + 40 кДж • mojhTV
Результаты определений энтальпии образования Мо3О9(г) представлены в табл. 31.9. При про-
35 3*
Mo4Oi2(r) _____ Глава 31
ведении расчетов состав пара над МоО2(к) и Мо03(к, ж) принимался по данным работ [1057, 1732].
Приведенные в таблице погрешности учитывают только воспроизводимость измерений. Как видно
из таблицы, за исключением выпадающих величин, вычисленных по данным [1466] и [2890],
остальные величины находятся в удовлетворительном согласии. Принятая величина является
средней, основной вклад в ее погрешность вносит неточность термодинамических функций Мо8О9(г).
Брюэр и Ламоро [1662] на основании исследований состава и давления пара над МоО3(к, ж)
[1245, 1507, 2521, 2705] рекомендовали значение Ь;Н°(Мо3Оа, г, 0) =—1859 + 25 кДж-моль,
которое заметно отличается от величины, принятой в настоящем справочнике. Еще больше
расходятся энтальпии образования Мо4О12(г) и Мо5О15(г), рекомендованные в [1662] и в настоящем
справочнике (см. ниже). Рекомендации [1662], видимо, основываются в основном на расчетах
по уравнению II закона и поэтому представляются менее надежными. Чарлю и Клеппа [1815]
калориметрически измерили энтальпию испарения МоО3(к) в высокотемпературном проточном
калориметре и нашли значения энтальпий испарения, равные 53,6±3,1 кДж-моль и 51,3±
+ 1,5 кДж-моль при 1086 и 1119 К соответственно. В состав насыщенного пара МоО3(ж) входят
Мо3О9, Мо4О12 и Мо5015. Данные этой работы приводятся Брюэром и Ламоро [1662] в
подтверждение выбранных ими значений энтальпии образования Мо3О9, Мо4О12 и Мо5О16. Действительно,
расчет энтальпии испарения МоО3(ж) с использованием рекомендованных в [1662] энтальпий
образования приводит к величинам, близким к полученным Чарлю и Клеппой [1815 ]. В то же время
принятые в настоящем справочнике термодинамические функции и энтальпии образования
указанных ассоциированных молекул приводят к худшему согласию с приведенными в [1815]
энтальпиями испарения. Объяснить это расхождение неточностью термодинамических функций,
принятых в настоящем справочнике, трудно из-за удовлетворительного согласия результатов расчетов
по уравнениям II и III законов (см. табл. 31.9 и 31.10). С другой стороны, измеренные в работе [1815]
энтальпии испарения МоО3(ж) нуждаются в корректировке. Детальный анализ работы [1815]
был проведен Рогацким и Томберг [4896], которые показали, что в условиях измерений [1815]
пары МоО3(ж) в момент выхода из калориметрической системы были сильно разбавлены газом-
носителем и парциальные давления компонентов пара были значительно меньше равновесных
величин. Такое отклонение от равновесных значений сопровождается изменением состава пара
и работой изотермического расширения. Авторы [4896] провели приближенные расчеты
соответствующих поправок и получили значение энтальпии испарения, которое находится в хорошем
соответствии с энтальпиями образования, принятыми в настоящем справочнике.
Мо4О12(г). Термодинамические свойства газообразного додекоксида тетрамолибдена
стандартном состоянии при температурах 100—6000 К приведены в табл. 892.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 31.6. Структура и спектр Мо4О12 экспериментально не исследовались. По аналогии с
молекулами Сг4О12 и W4O12 [452] в справочнике принимается, что в основном электронном состоянии
Х1^ молекула Мо4О12 имеет циклическую структуру симметрии С4„, образованную четырьмя
группами МоО2, соединенными друг с другом атомами кислорода. Приведенное в табл. 31.6
произведение главных моментов инерции вычислено по структурным параметрам Мо4О12, принятым равными
соответствующим параметрам Мо3О9 *. Погрешность IaIbIc оценивается в 2 -1010 г3 -см8. Молекула
Мо4О12 имеет 32 основные частоты симметрии ЛхG), А2C), 5хG), В2E) и #A0). На основании
результатов расчетов основных частот молекул Мо2О6 и Мо3О9 были оценены приближенные значения
частот однотипных колебаний молекулы Мо4О12, которые были объединены в 6 групп и усреднены.
В табл. 31.6 приведены усредненные таким образом значения основных частот с соответствующими
степенями вырождения 2. Их погрешности оцениваются в 100 см для vx—у4 и 50 см для v5 и v6.
1 В электронографйчёском исследовании паров СгО3(к) и W03(k), выполненном Ивановым [452], найдено, что
молекулы Сг3О9 и Сг4О12 и молекулы W3O9 и W4Oi2 имеют практически совпадающие по величине структурные
параметры одинаковых типов, аа исключением диэдрического угла 6, характеризующего не компланарность
;1атомов металла и кислорода в кольце МПОЯ. После/гаий определяется соотношением
sin (f/2) — cos A80/ra) - sin (ft/2)
cos sinA80/re) • cos(p/2; »
где n — числоггрупп':МО2 в молекула МяО3я, р = 2[Ом00Т—М—Омоот, ? = 2[М—Омоот—М.
2 В ИК-спектре паров МоО3 (к), изолированных в матрице из Ne, Хьюэтт и др. [2633] предположительно
приписали полосы 973, 896, 884, 866 и 514 см либо молекуле Мо4О12, либо Мо6О1б без их отнесения к опреде-
36
Молибден и его соединения
Мо„О12(г)
Термодинамические функции Мо4О12(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128), A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных величин обусловлены преимущественно неточностью принятых
значений молекулярных постоянных и приближенным характером расчета; они составляют 40,
60 и 80 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций Мо4О12(г) публикуется впервые.
Константа равновесия реакции Мо4О12(г)=4Мо(г)-{-12О(г) вычислена с использованием
значения АгН°@)=8195,396+45 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(Мо4О12, г, 0) = —2610 + 40 кДж • моль.
Результаты определений энтальпии образования Мо4О12(г) представлены в табл. 31.10.
Поскольку данные для Мо5О15(г) были получены в тех же работах, что и для Мо4О12(г), в таблицу
включены результаты определений энтальпии образования Мо6О16(г). Состав пара над МоО3(к)
принимался по данным работы [1507]. Приведенные в таблице погрешности учитывают только
воспроизводимость измерений. Принятая величина является средней из результатов согласующихся
Таблица 31.10. Результаты определений AyJ2"°(Mo,iOj;2, г, 0) и A^ff^MosO^, г, 0), основанные
на исследованиях равновесия реакций 4МоО3(к)=Мо4О12(г) и 5МоО3(к)—МОбО1в(г)
Авторы
Метод
Файзер, 1931 [2232] Измерение скорости испарения
МоО3(к), 883—1023 К, 6 точек
То же и метод точек кипения,
Ю74—1427 К, 8 точек
Уено, 1941 [4572] (см. Эффузионный, 883—973 К, 3 точки
[1507], а также [476])
Ария и др., 1953 [43] Протока, 941—987 К, 6 точек
Зеликман и др., 1956 То же, 1094—1274 К, 5 точек
Д/#°(Мо4О1а, г, 0)
II закон
—2633 ±6
—2564 + 9
-^2652+35
—2632+20
—2591+15
[446] Точек кипения, 1179—1430 К, 6 точек —2565+12
Берковиц и др., 1957 Масс-спектрометрический, 905—
[1507] (см. также [2777]) 973 К, 3 точки
Бабаджан, 1957 [55] Протока, испарение М0О3(к), 953—
1054 К, 6 точек
То же, испарение МоО3(ж), 1074—
1134 К, 2 точки
Блэкберн и др., 1958 Эффузионный, 808—958 К, 15 точек
[1573] (см. также [1570])
Джонс и др., 1958 [2890] Испарение с открытой поверхности,
811—1034 К, 4 точки
Аккерман и др., 1960 Протока, 981—1061 К, 5 точек
Г12451
Хорбе и др., 1961 [2705 ] Эффузионный, 873—983 К, 12 точек
Глемзер, Хеслер, 1962 Протока, 873—973 К, 14 точек
[24191
Гулбрансен и др., 1963 Эффузионный, 873—973 К а
[2521]
Казенас, 1968 [473] (см. Масс-спектрометрический, 800—
также [475, 478]) 1000 К а
Алешко-Ожевская и др., То же, 848—935 К а
1978 [25]
а Данные представлены в виде уравнения.
—2641 ±28С
—2540+22
—2499+900
—2576+17
—2699+18С
—2546+8
—2537+13
—2581 ±2
—2588+50
—2618
—2551 + 12
III закон
—2615 ±1
—2624±5
—2598+7
—2612+1
—2618+3
—2625+4
—2609+4
—2607+3
—2614+10
—2616+2
-2550+6
-2610+3 П!
-2610+2
—2606+1
—2613+2
—2615
Д/Я°(Мо6О16, г, 0)
II закон
—3321 ±7
—3246+8
—3339+20
-3323+18
—3276+14
—3245+10
—3206+600
—3227+30
—3184+900
—3263+14
—3390+180
—3235+5
—3225+13
—3268+1
—3280+50
-3300+3
—3216+15
III закон
—3315+1
—3328+7
-3298+5
-3313+1
—3322+4
—3331+6
—3309+6
—3309+3
—3316 + 11
—3316+2
—3250+5
—3311+3
-3311+2
—3307+1
—3314+2
-3326+3
ленным типам колебаний. В дальнейшем эти данные не уточнялись. Данные [2633] находятся в согласии со
значениями частот Мо4О12 и Мо6О15, принятыми в табл. 31.6.
37
Мо6О15(г) Глава 31
между собой измерений [43, 55, 446, 473, 1245, 1507, 1573, 2232, 2419, 2521, 2705].Ее погрешность
определяется в первую очередь неточностью термодинамических функций.
Брюэр и Ламоро [1662] на основании данных работ [1507, 1815, 2521, 2705, 4572]
рекомендовали существенно отличающееся значение —2533 + 25 кДж-моль. Обсуждение этого выбора
см. в тексте по Мо3О9(г).
Мо5О15(г). Термодинамические свойства газообразного пентадекоксида пентамолибдена в
стандартном состоянии при температурах 100—6000 К приведены в табл. 893.
Молекулярные постоянные, использованные в расчете термодинамических функций, приведены
в табл. 31.6. Структура и спектр Мо5О15 экспериментально не исследовались. По аналогии с
другими полимерными молекулами (МО3)В в справочнике принимается, что в основном электронном
состоянии Х1А1 молекула Мо5О15 имеет циклическую структуру, образованную пятью группами
МоО2, соединенными друг с другом пятью атомами кислорода (симметрия Cbv). Приведенное
в табл. 31.6 значение произведения главных моментов инерции вычислено по тем же структурным
параметрам, что для молекул Мо3О9 и Мо4О12 *; его погрешность оценивается в 1-Ю09 г3-см6.
Молекула Мо5О15 имеет 32 основные частоты типов симметрии Л1(8), А2B), ЕхA0) и i?2A2). На
основании результатов расчетов основных частот молекул Мо2О6 и Мо3О9 были оценены приближенные
значения частот однотипных колебаний молекулы Мо5О1б, которые были объединены в 6 групп и
усреднены. В табл. 31.6 приведены полученные таким образом значения с соответствующими
степенями вырождения. Их погрешности оцениваются в 100 см для vx—v4 и 50 см для v5 и v6.
Термодинамические функции Мо6О1Б(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128), A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных функций
обусловлены преимущественно неточностью принятых значений молекулярных постоянных и
приближенным характером расчета и составляют 50, 70 и 90 Дж -К -моль в значениях Ф°(Т) при 298,15,
3000 и 6000 К.
Таблипа термодинамических функций Мо6О15(г) публикуется впервые.
Константа равновесия реакции Мо5О15(г)=5Мо(г)+15О(г) вычислена с использованием
значения ДгЯ°@) = 10291,745+ 56 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/ИМоАб, г, 0) = — 3310 + 50 кДж.моль.
Результаты определений энтальпии образования Мо5О15(г) представлены в табл. 31.10.
Принятая величина является средней из результатов согласующихся между собой измерений [43, 55,
446, 473, 1245, 1507, 1573, 2232, 2419, 2521, 2705]. Ее погрешность определяется в первую очередь
неточностью термодинамических функций Мо5О15(г).
Брюэр и Ламоро [1662] на основании данных [1245, 1507, 1815, 2521, 2705] рекомендовали
существенно отличающееся значение —3213+42 кДж-моль. Обсуждение этого выбора
см. в тексте по Мо3О9(г).
См. примеч. 1 к тексту по Мо4О12.
Глава 32
ВОЛЬФРАМ И ЕГО СОЕДИНЕНИЯ
W(k, ж), W, W+, WO, WO2(k), WO2, WO3(k, ж), WO3, WO",
wso6, w3o9, w4o12, w5o15
В настоящей главе рассматриваются вольфрам и его кислородные соединения — всего 10
веществ. Для трех веществ представлены данные о термодинамических свойствах в
конденсированном состоянии, для 10 веществ — в газообразном состоянии. Термодинамические свойства трех
газов (W, W+, WO) рассчитаны до 10 000 К, остальных газов — до 6000 К. Термодинамические
свойства всех веществ приводятся для природной изотопической смеси вольфрама (атомный вес
183,85) и кислорода, однако различие постоянных разных изотопомеров всех этих веществ в
расчетах не учитывалось как из-за отсутствия соответствующих данных, так и из-за пренебрежимо
малого влияния этого фактора на свойства соединений вольфрама.
Для элементарного вольфрама в Приложении 3.10 приведены сведения о критических
постоянных.
Рассматриваемые в справочнике кристаллические оксиды имеют достаточно широкие (при
1100-1600 К) области гомогенности WO1>95—WO2j04 [178, 1030] и WO2>9—WO3,0 [186, 1030].
Известны другие оксиды вольфрама в кристаллическом состоянии; в литературе описаны
соединения W18O49, W24Oe8, W20O58, W24O70 и т. д. до W50O148 (см. [549а, 1030]). Существование субоксида
W3O до настоящего времени надежно не установлено. Высказывались также предположения
о существовании других газообразных оксидов, в частности W3O8 (см. [1220]), и WeO18, см. [1472],
однако их содержание в равновесных условиях должно быть незначительным и они не
рассматриваются в справочнике. Не включены в справочник также ионизированные оксиды вольфрама,
так как потенциалы ионизации соответствующих оксидов превышают 70 000 см.
W(k, ж). Термодинамические свойства кристаллического и жидкого вольфрама в стандартном
состоянии при температурах 100—6000 К приведены в табл. 894.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 32.1. За стандартное состояние W (к) в интервале 0—3695 К принимается стабильная куби-
Таблица 32.1.
Принятые значения термодинамических величин для вольфрама и его соединений
в кристаллическом и жидком состояниях
о
G
ffl
W
wo2
wo3
Состояние
к, куб.
ж
к, монокл.
кН1, монокл
кШ, монокл.
кП, ромб.
к1, тетр.
ж
ю
ооо"
5н
кДш-моль
15 К)
ОС
СОТ
о
Со
Дж ¦ К-*
4,97 32,64
— —
8,711 50,64
13,280 81,64
* — —
— —
Примечание. C°n(T)=a+b -T—c -T~2+d -Г2+е
W: a
d=4,728 Л0гв,
15 К)
00
от
Cs|
• моль
24,27
—
55,78
79,705
—
•г3+/-г4
е=—1,509 -КГ9, /=0,234-Ю2.
Коэффициенты п
уравнении для С° {Т)
а
6-Ю3
с-10~5
26,101 —2,266 1,367 а
45 — —
66,377 13,263 12,935
91,320 —38,957 —
71,075 35,895 4,312
71,075 35,895 4,312
80,963 16,285 —
129 — —
(в Дж -К -моль)
Интервал
температур
Е-Г
3
К
298,15—3695
3695-6000
298,15-3000
298,15—325
325—612
612—1013
1013-1747
1747-4000
3695
—
—
—
612
1013
1747
—
ч
о о
кДжХх
46
—
—
—
0
1
73
—
39
W(k, ж) Глава 32
ческая объемно-центрированная модификация (a-W) г. При 0,0160 К W(k) переходит в
сверхпроводящее состояние. Температура перехода найдена Триплеттом и др. [4544] для образца W(k)
чистоты 99,99%.
Термодинамические функции W(k) при Т <^ 298,15 К вычислены на основании измерений
теплоемкости образцов чистотой 99,999 и 99,99%, выполненных Триплеттом и др. [4544] @,35—
25 К) и Клузиусом и Францозини [1879] A0—273 К). Менее точные измерения теплоемкости
проведены в работах [3185] B6-91 К), [4834] (92—290 К), [4289] A,5—3,5 К), [2714] B-4 и 15—
20 К), [3950] @,2-1 К), [4750] A,2—20 К), [4639] D-15 К), [2054] A3-93 К). Погрешности
значений 5°B98,15 К) и #°B98,15 К)—#°@), приведенных в табл. 32.1, составляют
0,3 Дж-К-моль и 0,04 кДж-моль.
В интервале 298,15—3695 К для теплоемкости W(k) принято приведенное в табл. 32.1
уравнение, полученное совместной обработкой измерений энтальпии, проведенных Вюстом и др. [4786]
C73-1779 К), Егером и Розенбомом [2811, 2812] E80-1570 К), [2813, 2814] F01—1883 К),
Магнусом и Хольцманом [3342] C73—1176 К), Бронсоном и др. [1679] C28—774 КJ, Хохом и
Джонстоном [2667] A376—2905 К), Чеховским и др. [1129] F30—2244 К), Кириллиным и др.
[502, 503] B286—2622 К), [509, 510] B690—3104 К), Турчанином и др. [1034] A452-2401 К),
[1033] E01—1400 К), Хейном и Флагеллой [2603] (см. [2663]) A208—3248 К), Фредриксоном
и др. [2325] A333—1861 К), Лейбовицем и др. [3227] B834—3526 К), Шпильрайном и др. [66,
4274] B183-3190 К), Моризуром и др. [3564] A573—2723 К) и Дитмарсом [2082] C73—1173 К)
для образцов W(k) чистоты ^ 99,8%. В этом расчете все температуры были приведены к
температурной шкале 1968 г. и с наибольшим весом учитывались данные работ [66, 502, 503, 509, 510,
1129, 1679, 2082, 2603, 2813, 2814, 3227, 3342, 4274]. Разности между значениями энтальпии,
вычисленными по уравнению табл. 32.1, и данными [502, 503, 509, 510, 1033, 1129, 1679, 2082,
2603, 2813, 2814, 3227, 3342] не превосходят 1%. Известны также весьма неточные измерения
энтальпии в работах [3958] C72 К), [2020] C66—696 К) и [3341 ] F75—1175 К). Измерения
энтальпии W(k) Уэстом и Исихарой [4709] до 2600 К остались недоступны при подготовке справочника.
Теплоемкость W (к) при Т > 298,15 К измерялась калориметрическим методом [4193] C00—
1900 К), модуляционным методом [1926] A073—2173 К), [3845] F13—1623 К), [4771] A200—
2400 К), [2357] A416—2465 К), [4326] B368—2485 К), [1593] B371-2486 К), [4834] F40-2220,
1150-2521 К), [571, 575, 576, 3100] A500-3600 К), [3318] A200-2400 К), [565] A300-3000 К),
[52, 1055] A100—2400 К), [517] A100—2000 К), методами импульсного нагрева [4081] A400—
3000 К), [1265] A200—3600 К), [381, 2078] B600—3500 К), [1787] A900—3600 К), [1786] B000-
3600 К), [1043] B700—3200 К) и по остыванию вольфрамовой проволоки в вакууме [448] B300—
3600 К). Точность этих измерений значительно ниже, чем точность измерений энтальпии. Принятое
в справочнике уравнение для С°„ (W. к) при Т > 298,15 К находится в согласии с измерениями
теплоемкости и существенно превосходит большую их часть по точности. В связи с этим можно
утверждать, что калориметрические измерения Шмидта и др. [4193] систематически занижены
в пределах 0,4—1,0 Дж-К-моль. Данные Аффорти и Лаллемена [1265] сильно завышены при
Т <2000 К и Т > 2800 К. Данные Цезерляна и Мак-Клара [1786, 1787] систематически
завышены при Т >¦ 2800 К и расходятся с принятыми в справочнике при 3600 К на 6 Дж-К-моль".
Данные Крафтмахера [571, 575, 576, 3100] занижены в интервале 2000—2800 К и сильно завышены
при Т > 3000 К.
Температура плавления W(k) принята равной 3695+15 К по измерениям Цезерляна [1783]
для образцов W(k) чистоты 99,9%. Это значение рекомендовано в качестве вторичной реперной
точки температурной шкалы 1968 г. [2779]. Оно подтверждается результатами, полученными в
работах [4084] C703+26 К), [1293] C689±20 К), [4810] C687+25 К), [4772] C695 К), [1271]
C697 К), [4406] C683 К). Менее точные, преимущественно заниженные значения Tm(W, к),
получены в работах [381, 2310, 2610, 2611, 2894, 3047, 3190, 3846, 4770]. Энтальпия плавления W(k)
принята равной 46+4 кДж>моль согласно определению ее Вучем и др. [4774] по кривой
затвердевания и охлаждения свободно парящего в вакууме жидкого вольфрама (метод электромагнитной
левитации). Совпадающее значение D6 кДж-моль) было получено Шейнером и др. [4247] методом
1 Известна еще нестабильная модификация W, также имеющая объемно-центрированную кубическую структуру,
но значительно больший параметр кристаллической решетки, называемая J3-W. Эта форма образуется в
специальных условиях (см. [780]) и при нагревании до 900 К необратимо превращается в a-W.
* Измерения [1679] при 253 и 278 К не использовались.
40
Вольфрам и его соединения
W(k, ж)
импульсного нагрева. Этим же методом были получены значительно большие значения ДШЛ"О(\У, к),
равные 61,4 [381, 2078], 55 [619, 667, 894] и 51 кДж-моль [4243], что связано, по-видимому,
с неравновесным состоянием вольфрама в соответствующих экспериментах. Энтальпия W(jk)
измерялась методом импульсного нагрева Дихтером и Лебедевым [381] C655—4500 К) и Зейделем
и Китцелем [4245] C680—7500 К). В работе [381] получены аномально высокие значения
теплоемкости \У(ж) A01—75 кДж-моль), что свидетельствует о неравновесном состоянии вольфрама.
Из данных работы [4245] для C°p(W, ж) следует значение, равное 57 кДж-моль (см. также [4243]).
Оно также представляется завышенным, поскольку данным [4245] для энтальпии Мо(ж)
соответствует значение С°(Мо, ж), на 20% превосходящее установленное по калориметрическим
измерениям (см. текст по Мо(к, ж)). С учетом этого результата, а также принятых в справочнике значе-
ний С^,(Мо, ж) и C°p(W, к, Тт), теплоемкость ^У(ж) принимается равной 45 + 5 кДж-моль.
Погрешности приведенных в табл. 894 значений Ф°(Г) при 298,15, 1500, 3000 и 6000 К равны
0,1, 0,5, 1 и 3,2 Дж-К-^моль.
Ранее таблицы термодинамических функций W(k, ж) вычислялись в справочниках JANAF
[2834], Халтгрена и др. [2752], Шика [4181] (до 6000 К), Стаяла и Зинке [4421], Келли [2960]
(до 3000 К), а также в работах [510] и [3227] (до 3600 К). Различие в значениях Ф°(Г) для W(k),
полученных в этих расчетах и в табл. 894, не превосходят 0,2 Дж-К-моль~1; расхождения в
значениях Ф°E000 К), приведенных в табл. 894 и в справочниках [2752, 2834, 4181], составляют 2,3,
0,5 и 1,3 Дж-К^-моль.
В настоящем справочнике в качестве стандартного состояния вольфрама принято
кристаллическое состояние и в соответствии с этим
AfH°(W, к) = 0.
Таблица 32.2. Результаты определений A,Jff°(W, к, 0) (в кДж-моль)
Авторы
Лангмюр, 1913 [3189]
Фонда, 1923 [2285]
Цвшдаер, 1925 [4829]
Уолин, Уитни, 1936 [4634]
Форсайт, Адаме, 1937 [2309],;;
Рейман, 1938 [3963]:
Горбатый, Шуппе, 1957 [305]
Котляр, Андреева, 1960 [562]|
ш
Зандберг и др., 1965 [435]
Дидмор, 1965 [2009] (см. [3855])
Мармер и др., 1965 [660]
Шварц и др., 1965 [4452] ;¦
Голубцов, 1966 [292, 294, 298]
Сасаки и др., 1970 [4143, 4144]
Планте, Сисэмс, 1973 [3855]
Метод
Испарение с открытой поверхности, 2515—
3058 К, 12 точек
То же, 2816 К, 11 точек
То же, 2387—3128 К, 14 точек :
То же, 2838—3168 К, 7 точек
То же, 1901—3659 К, 18 точек
То же, 2821—3495 К, уравнение
То же, с использованием радиоактивного
изотопа, 2404—2805 К, 5 точек
Испарение с открытой поверхности, 2604—
13005 К, данные представлены в виде уравнения
Масс-спектрометрический, испарение с
открытой поверхности, 2600—2900 К а>!
Испарение с открытой поверхности, 2500—
?3000 К а
То же, 3239 К, 1 точка
То же, 2578—3189 К, 10 точек
То же, с использованием радиоактивного
изотопа, 2066—3209 К, 24 точки
Эффузионный, с использованием
радиоактивного изотопа, 2654—3125 К, 7 точек
Масс-спектрометрический, испарение с
открытой поверхности, 2860—3010 К а
Испарение с открытой поверхности, 2567—
3067 К, 41 точка
а В работе приведено только найденное значение Д8#°(Г).
А8Н°(
II закон
856 ±35
—
896+10
851+24-
874+3
S 818 •
767+13
778
819+40
905+40 ~:
896+30
863+7
856+12
% 886 ±26
851 ±10
W, к, 0)
III закон
844,0+2,1
819,6+4,5
846,1+3,0
869,3+1,1
849,4 ±2,2
847
> 838+5
?: 839
—
854,4 ±2,0;
843 !¦
851,5±1,8
852,7 ±1,0
850,7+0,4
—
859,2±0,5
41
W(r), W+(r) Глава 32
Давление пара вольфрама в реакции W(k, H?)=W(r) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = \H°(W, к, 0) = 850 + 5 кДж • моль.
В табл. 32.2 представлены значения энтальпии сублимации вольфрама, вычисленные на
основании измерений давления его насыщенного пара. В соответствии с результатами масс-спектро-
метрических исследований [435, 1855] в расчетах предполагалось, что пары вольфрама
одноатомны. При обработке данных [3189, 3963, 4829] были приняты во внимание скорректированные
значения температур (см. [2894, 4452]); значения температур, приведенные в работах [2309, 4634],
были исправлены на основании измерений излучательной способности вольфрама [615].
Приведенные в.таблице погрешности учитывают только воспроизводимость измерений. Причины,
приведшие к более высокому значению энтальпии сублимации в работе [3855], обсуждаются ее
авторами. Принятая величина является средневзвешенной из результатов работ [292, 294, 298, 2309,
3189, 3963, 4452, 4829]. Ее погрешность оценена с учетом неточности термодинамических функций
и возможных систематических ошибок. При этом с наибольшим весом учитывались значения,
вычисленные по измерениям, выполненным Голубцовым [292, 294, 298] и Шварцем и др. [4452].
W(r). Термодинамические свойства газообразного вольфрама в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 895.
Для расчета термодинамических функций использованы уровни энергии * атома W в интервале
О—64 000 см, соответствующие валентным электронным конфигурациям . . .5d46s2 (к ней
относится основной уровень 5D0), . . .5d6, . . .5d56l (i=0-^-2), . . .5d46s6l (J=l+-3) и имеющие
суммарный статистический вес 5358. Принятые величины основаны на экспериментальных данных Лоуна,
Корлисса [3208] и Корлисса [1934], а также на оценках для ряда ненаблюдавшихся электронных
состояний. Для электронных конфигураций . . .5d&Ql A=0—2) и . . .5d46s6Z (Z=l-+-3) оценка была
выполнена по методике, изложенной в Приложении 6, а для конфигураций . . .5cZ46s2 и . . .5de —
сравнением энергии однотипных электронных состояний Сг, Мо и W. Оцененные уровни имеют
энергию от 16 000 см и выше с суммарным статистическим весом 4986. Погрешность этих оценок
лежит в пределах 100—4000 см.
Термодинамические функции W(r) вычисленыпо уравнениям A. 3)—A. 6), A. 9), A.10), A. 23)—
A.25) непосредственным суммированием по уровням энергии. Погрешности вычисленных величин
при Т ^ 2000 К обусловлены неточностью принятых значений фундаментальных постоянных,
а при более высоких температурах — отсутствием экспериментальных данных для большей части
учитывавшихся уровней и пренебрежением ридберговскими состояниями. В значениях Ф°(Т)
при 3000, 6000 и 10 000 К они составляют 0,4, 2 и 3 Дж-К^-моль.
Ранее термодинамические функции W(r) вычислялись в справочниках JANAF [2834], Шика
[4181 ] и Сталла и Зинке [4421] (до 3000 К), а также в работах Хилзенрата и др. [2652] и Гурвича
и др. [339] (до 10 000 К). Расхождения результатов всех расчетов при Т ^ 2000 К не превосходят
0,04 Дж-К^-моль в значениях Ф°(Г) и S°(T). При более высоких температурах Возникают
расхождения в связи с тем, что в настоящем справочнике учтены ненаблюдавшиеся электронные
состояния атома W и более полные экспериментальные данные, полученные в работах [1934, 3208].
Расхождения данных табл. 895 и расчетов [339, 2652 и 2834] составляют 0,4, 0,5 и 0,5 Дж-К^-моль
в значении Ф°F000 К) и 2 и 3 Дж-К^-моль в значении Ф°A0 000 К).
Энтальпия образования
AfH°(W, г, 0) = 850 + 5 кДж ¦ моль
соответствует принятому значению энтальпии сублимации вольфрама.
W+(r). Термодинамические свойства газообразного положительного иона вольфрама в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 896.
Для расчета термодинамических функций использованы уровни энергии х W+ в интервале
"О—70 000 см, соответствующие валентным электронным конфигурациям . . .5<i46s (к ней
относится основной уровень 6Z>V2), . . Ыъ, . . .5d36s2, . . .bd^p, . . .5d36sQl A=1, 2) и имеющие
суммарной статистический вес 3842. Принятые величины основаны на экспериментальных данных Лоуна
[3207], а также на оценках для ряда ненаблюдавшихся электронных состояний. Для электронных
1 См. примеч. 1 к тексту по Сг (г).
42
Вольфрам и его соединения WO(r)
конфигураций . . .Ъй^Ш A=0, 1) и . . .5d36s6l A=1, 2) оценка выполнена по методике, изложенной
в Приложении 6, а для конфигураций . . .Ыь и . . .5d36s2 — сравнением энергии однотипных
состояний ионов Cr+, Мо+, W+ и изоэлектронных атомов V, Nb, Та. Оцененные уровни имеют
энергию 15 000 см и выше с суммарным статистическим весом 3564. Погрешность этих оценок лежит
в пределах 50—4000 см.
Термодинамические функции W+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 23)—
A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных величин
обусловлены преимущественно неточностью оценки ненаблюдавшихся уровней энергии; в
значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К они составляют 0,02, 0,4, 2 и 3 Дж-К^-моль.
Ранее термодинамические функции W+(r) вычислялись в справочнике JANAF [2834], а также
в работах Хилзенрата и др. [2652], Гурвича и др. [339] (до 10 000 К) и Грина и др. [2473]
(до 50 000 К). При Т ^ 2000 К результаты всех расчетов Ф°(Т) согласуются в пределах
0,03 Дж-К-моль. При более высоких температурах благодаря учету более полных сведений
об электронных уровнях W+ данные табл. 896 выше результатов остальных расчетов;
соответствующие расхождения составляют 0,5, 0,6, 0,6 и 0,2 Дж-К^-моль в значении Ф°F000 К)
с данными [339, 2473, 2652, 2834] и 1,6 и 2,1 Дж-К-^моль в значении Ф°A0 000 К) с данными
[339] и 12473, 2652].
Константа равновесия реакции W+(r)+e(r)=W(r) вычислена с использованием значения
ДгЯ°@)=—770+10 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации атома W
/0(W)=770+10 кДж-моль-х=64 400+800 см.
Принятая величина была найдена Лапортом и Маком [3196] по первым двум членам ридбергов-
ской серии . . .bd%s(9D)ns bD атома W, соответствующим п=6 и 7, и подтверждена более точными
расчетами, проведенными при подготовке справочника с использованием данных [3208, 3550].
Раух и Аккерман [3944] методом электронного удара получили существенно более низкое
значение, равное 730 кДж-моль.
Принятому значению соответствует
+, г, 0) = 1620,000 + 11 кДж • моль".
WO(r). Термодинамические свойства газообразного оксида вольфрама в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 897.
В табл. 32.3 приведены молекулярные постоянные WO, принятые в справочнике на основании
анализа результатов исследований спектров этой молекулы и приближенных оценок, выполненных
при подготовке справочника. Экспериментальные исследования [4129, 4699] показали, что в
молекуле WO характер связи моментов количества движения близок к случаю с Гунда. В электронном
спектре поглощения в газовой фазе [903, 2158, 4129] и в низкотемпературной матрице [4699]
проанализировано 11 систем полос, связанных с основным состоянием: А'0+—Хг0 + [2158],
А1-Х,0\ В1~Хг0+, С1-Хх0+ [4129, 4699], C'l—XjO* [4129], Dl—Xfi* [4129, 4699],
D'0+-X10+ [4129], ?0+-Х10+, F0+—Хг0\ Gl—X10+ [4129, 4699] и Н0+—Х10+ [4129]. В
результате анализа возмущений в системе D1—Хх0+ в работе [4129] было обнаружено также состояние 0".
Кроме того, две полосы в спектре поглощения в газовой фазе авторы [4129] предположительно
отнесли к переходам из компоненты Х21 основного состояния: F0+ <— Х21 и D1 <— Х21.
Большое число неотнесенных полос, наблюдавшихся в спектрах поглощения [4129] и
испускания [790, 2374, 2375, 4617], свидетельствует о существовании других стабильных возбужденных
состояний молекулы WO.
Нижней конфигурации электронной оболочки . . .§|а?, помимо основного состояния, должны
соответствовать еще два состояния (ХГ и 1Е+). У молекулы VO (см. гл. 33) разность энергий изо-
конфигурационных состояний 2Г и 4?+ (. . .S*0,) составляет около 9500 см, а у молекулы МпО
разность энергий состояний 4Г и вЕ+ (конфигурация . . .8|я|ай) превышает 30 000 см [3843] г.
Поэтому в справочнике принимается, что состояния ХГ и 12+, изоконфигурационные с основным
состоянием WO, имеют энергии больше 10 000 см. Первым возбужденным состоянием WO (в
символике термов для случая Гунда а) должно быть состояние 5П с конфигурацией . . .§| ndod (основное
1 У иона Та+ разность энергии нижних синглетного и триплетного термов конфигурации . . ,d2s2 составляет
10 000 см.
43
WO(r)
Глава 32
Таблица 32.3.
Состояние
Молекулярные постоянные
гр
¦
СМ
в.
ал ¦ 103
De ¦ 10'
' ге
А
А'0+, 0"
А1
В1
а
а
DI
D'0+
Е0+
(Г
F0+
Gl
о
72 б
7 000в
8 500в
14 424
17 285
19 253
19 788 б
20 833,3
21 222,8 б
22 412,18 ж
22 748 и
23 471
23 840
30 525
1068,2
1050 г
960,6
985.7 е
928.8 о
989,3
1013,0 3
916.9 е
933 е- к
900 е
5,8
6,65
5,4
0,4165
0,392 Д
0,3898
0,3843 б
0,3854 ж
0,376 и
0,3852
0,386 б
2 3
2,3 в
2,0
2,7
3,0
2,68 (
2,63 б
2,23 ж
3,55 б
Примечание. Ниже все постоянные даны в см 1 (кроме р().
а три объединенных электронных терма T{j) 15 000 25 000
\Pi 14 40
40 000
115
1,6578
1,709
1,7136
1,7258 б
1,7234 ж
1,745 и
1,7238
постоянная для уровня у=0; в оценка (см. текст); г вычислено по соотношению A. 68); Д рассчитано из В5=
=0,3789; е приведено bGi)j ж состояние возмущено, приведены постоянные для и=1; 3 приведено Дба^;
и колебательная нумерация не установлена, предполагается v > 0; к из измерений в неоновой матрице [4699]-
состояние у молекулы СгО) или . . .b\ndas. В справочнике принимается, что состояние А'0+,
обнаруженное в работе Ефремова и др. [2158] (Г,=8500+1000 см), является компонентой 5П0+ этого
квинтетного состояния бП, а его нижняя компонента 8П_Х имеет энергию 7000+1500 см.
Оценка энергий остальных компонент этого состояния, а также других состояний, не
наблюдавшихся в спектрах, представляется нецелесообразной. Вместо этого по схеме, использованной для
молекул СгО и UO, была сделана оценка общего числа ненаблюдавшихся состояний с энергиями
до 40 000 см, которые были сгруппированы в три объединенных уровня. Оценка основывалась
на предположении, что эти состояния соответствуют четырем конфигурациям оболочки из четырех
электронов атома W, не участвующих в образовании химической связи (. . .cPs2, . . .d3s, . . .d4,
. . ,d2ps), и что такие состояния являются связанными. При этом на основании данных для уровней
энергии атомов W, W+ и Та+ было принято, что нижняя граница энергий состояний
соответствующих конфигураций равна 0, 7000, 15 000 и 30 000 см, а верхняя граница для всех четырех групп
равна 122 000 см (D0(WO)+/0(W)). Приведенные в табл. 32.3 статистические веса трех
объединенных уровней получены в предположении о равномерном распределении состояний,
соответствующих этим конфигурациям в указанных интервалах энергий (Ер<=45, 240, 210 и 540), за вычетом
наблюдавшихся экспериментально состояний.
Молекулярные постоянные 18eW16O, приведенные в табл. 32.3, за исключением постоянных
в состояниях А\ и А'0+, получены Самойловой и др. [4129] на основании анализа вращательной
структуры 15 полос систем Z)l—Z10+ (v' < 3, v" < 1), D'0+—X10+ @—0), E0+—Xj0*(i < v' < 3,
v"=0), Л)+-Хх0+ (v' < 1, v"=0), H0+ (v' < 1, v"=0), F0+—X2i @-0) и анализа колебательной
структуры систем полос W16O и W18O: А1—Хх0+C< и' < 5, v" < 1), Bl—X10+ (v' < 2, v" < 1).
Cl—ar1O + (p',i;"<l), С'1-ХхО + (г;'=О, v" < 1), Dl-X^^v' < 4, v" < 2), Я0+-Х10 + A <
< v' < 4, v" < 2), F0+—Xx0+ (v' < 4, v" < 3), Gi—Xx0+ @-0), H0+-Xx0+ (vr < 2, v" < 1) К
1 Грин и Эрвин [4968] при исследовании ИК-спектров молекул WO, изолированных в матрицах, нашли близкиег
но заметно отличающиеся от приведенных в табл. 32.3 колебательные постоянные в состоянии Х0+ (<»>,,=
= 1055,98 см~1, шеже=2,93 см в матрице из криптона и «„=1057,51 см и шеа;с=3,33 см в матрице из аргона\.
44
Вольфрам и его соединения
WO(r)
Молекулярные постоянные WO в состоянии Х21 приняты такими же, как в состоянии Хх0+.
Для энергии состояния Х21 принято значение, полученное в работе [4129] на основании отнесения
двух полос в спектре поглощения 186W18O D284,2 и 4823,1 А) к переходам из этого состояния.
Однако нельзя исключить, что такая интерпретация спектра является неверной, а энергия этого
состояния существенно выше. В связи с тем что возмущения, наблюдавшиеся в большинстве
возбужденных состояний, затрудняют определение колебательных постоянных WO в этих состояниях,
энергии состояний Bl, Cl, F0+, G1 и Н0+ были вычислены из приведенных в [4129] значений То
и Д&д (для состояния G1 использовалось значение Д&/„, полученное Велтнером и Мак-Леодом
[4699]).
Молекулярные постоянные в состоянии А'0 + основаны на результатах анализа вращательной
структуры полосы 5—4 системы А'0+—Хг0+ в работе Ефремова и др. [2158]. При расчете
значения Ве величина а1 принималась такой же, как в основном состоянии. Погрешности
соответствующих значений Те, шв и Ве оцениваются в 1000, 150 и 5-Ю" см соответственно.
Термодинамические функции WO(r) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBK и ее производных вычислялись по уравнениям A. 90)—A. 92) с
учетом 17 возбужденных состояний (включая состояние Х2\ и три объединенных уровня) и в
предположении, что QiH.BV=PiQ{^. „p- Статистическая сумма по колебательно-вращательным уровням
энергии состояния Х±0+ и ее производные находились по уравнениям A. 73)—A. 75)
непосредственным суммированием по колебательным уровням и интегрированием по значениям / с
помощью уравнений типа A. 81). В расчете учитывались все уровни с значениями / ^ /шах о,
последние находились из условий A. 81). Энергия уровней вычислялась по уравнениям A. 65),
A. 34) и A. 64а). Коэффициенты в этих уравнениях, полученные с учетом условий A. 50) и A. 59),
но без учета различия постоянных изотопомеров молекулы WO, а также значения t>max и /Р]
ведены в табл. 32.4.
Таблица 32.4.
Коэффициенты в уравнениях, описывающих уровни энергии (в см) в состоянии
а также значения vmscx и Jiim, принятые для расчета
термодинамических функций WO(r) a
У,п-Ю3
'lim
1,06898 —6,18026 1,1627 4,165 —2,30
1 Энергии возбужденных состояний см. табл. 32.3.
—2,68
—2.74
149
487
Погрешности рассчитанных термодинамических функций W0(r) при низких температурах
обусловлены только неточностью энергии состояния Х21; при температурах выше 2000 К
становятся существенными, в особенности в значениях С°р(Т), погрешности из-за неточности оценки
энергии и числа ненаблюдавшихся возбужденных электронных состояний. Погрешности в
значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,05, 2, 5 и 12 Дж-К^-моль.
Термодинамические функции W0(r) ранее вычислялись в таблицах JANAF [2834], Брюэром
и Розенблаттом [1666] (Ф°(Г) для Т < 3000 К) и Де Мария и др. [2032] (Ф°(Т) для Г=2000^-
2500 К). Расхождения данных [2834] и табл. 897 при низких температурах обусловлены тем, что
авторы [2834] пренебрегли расщеплением компонент Хх0+ и Х21 основного состояния и
использовали оцененное (и существенно завышенное) значение re(WO), а при высоких температурах —
тем, что они учитывали только три возбужденных состояния. Эти расхождения при 298,15 К
составляют 3,2 и 1,5 Дж-К^-моль и 0,12 нДж-моль в значениях Ф°(Г), S°(T) и Л°(Т)—На@)
соответственно, а при 6000 К — 1,3, 8 и 17 Дж-К^-моль в значениях Ф°(Г), S°(T) и С°р(Т).
Данные [1666] отличаются от приведенных в табл. 897 на 3—13 Дж-К^-моль из-за того, что
в этой работе была принята некорректная систематика электронных состояний WO и неверное
значение re(WO).
Константа равновесия реакции WO(r)=W(r)+O(r) вычислена с использованием принятой
в справочнике энергии диссоциации
ДгЯ°@) = D0(WO) = 695 + 12 кДж • моль «* 58 100 ± 1000 см.
45
WO2(k)
Глава 32
Результаты определений энергии диссоциации WO представлены в табл. 32.5. Приведенные
в таблице погрешности учитывают воспроизводимость измерений, неточность термодинамических
функций, термохимических величин и сечений ионизации. Результаты приведенных в таблице
измерений находятся в хорошем согласии. Принятое в справочнике значение является
средневзвешенным, причем наибольший вес придавался измерениям [310, 463] и [2032, 2115] (реакция
WO(r)=W(K)-fO(r)). Менее надежны исследования [2032, 2115] реакции с А12О3, хотя их
надежность больше, чем в случае молибдена (см. [1665, 1666]).
Таблица 32.5. Результаты определений D0(WO) (в кДж-моль)
Авторы
Де Мария, Дроварт и др., 1960
[2032, 2115] (см. также [2033])
Горохов и др., 1978 [310, 463]
Метод
Масс-спектрометрический, WO (г)=W(k) + О (г),
2191—2478 К, 8 точек
То же, 3W(K)+Al2O8(K)=3WO(r)+2Al(r), 2191—
2478 К, 7 точек
То же, YO(r)+.W(r)=Y(r)+WO(r), 2538—2963 К,
38 точек
Do
II закон
857 ±200
819 +700
803+100
WO)
III закон
694 ±14
681 ±15
696+12
Принятому значению соответствует
&fH°(WO, г, 0) = 401,783 + 13 кДж • моль.
WO2(k). Термодинамические свойства кристаллического диоксида вольфрама стехиометриче-
ского состава в стандартном состоянии при температурах 100—3000 К приведены в табл. 898.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 32.1. За стандартное состояние W02(k) принята моноклинная модификация (структурный
тип МоО2) г. Плавление WO2 не наблюдалось, так как при 1800—2000 К W02(k) диспропорциони-
рует с образованием W(k) и высших оксидов вольфрама (WO2 72(к), W03(k)) [2834, 3812].
Термодинамические функции WO2(k) при Т ^ 298,15 К вычислены на основании измерений
теплоемкости, проведенных Кингом и др. [3013] E3—297 К, чистота образца 99,8%).
Экстраполяция теплоемкости ниже 53 К с использованием дробно-рациональной функции шестой степени:
[1066], аппроксимирующей данные [3013] с погрешностью ~0,2%, приводит к значению ?°E3 К)=
=2,5 Дж-К^-моль. Принятые в справочнике значения ?°B98,15 К) и Я°B98,15 К) —Я°@>
(см. табл. 32.1) имеют погрешности 0,3 Дж-К~1-моль~1 и 0,04 кДж-моль соответственно.
При Т > 298,15 К термодинамические функции W02(k) вычислены с помощью уравнения для
теплоемкости (см. табл. 32.1), полученного на основании результатов измерений энтальпии в
работе Кинга и др. [3013] C96—1802 К). Это уравнение аппроксимирует данные [3013] в
пределах 1%. Теплоемкость W02(k) при Т > 298,15 К измерялась Сейлом [4121] C40—1000 К)
методом ДСК. При Т > 500 К данные [4121] находятся в согласии с принятым уравнением, но при
Т ^ 500 К они систематически больше (до 5% при 350 К) рассчитанных по этому уравнению.
Погрешности приведенных в табл. 898 значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К
оцениваются в 0,2, 0,7, 2 и 5 Дж-К~1-моль~1.
Ранее термодинамические функции W02(k) вычислялись в справочниках JANAF [28341
(до 3000 К) и Шика [4181] (до 2000 К); расхождения между значениями Ф°(Т), приведенными
в [2834] и табл. 898, не превышают 0,6 Дж-К^-моль.
Принятая в справочнике энтальпия образования
i/°(W02, к, 298,15 К) = —588,1 + 1,5 кДж • моль
основана на калориметрических измерениях Ма [3346] энтальпии сгорания W02(k). Работа [3346]
отличается высокой точностью измерений, использованием чистых образцов, близких по составу
к стехиометрическому, и высокой полнотой сгорания. Принятая величина подтверждается
результатами калориметрических измерений Гриффиса [2488] (—584+5 кДж-моль) и Чарлю и Клеппы
1 WC (к) не имеет фазовых переходов, как это следует из измерении электрического сопротивления [4028] D,2 —
300 К),' теплоемкости и энтальпии [3031, 4121] E3 — 1802 К).
46
Вольфрам и его соединения
WO2(r>
[1817] (—585+5 кДж-моль х). Измерения [697, 2029, 3257] привели к заниженным значениям
энтальпии образования W02(k) (•—550-^—570 кДж-моль) вследствие недостаточного контроля
за полнотой сгорания. Энтальпия образования W02(k) может быть вычислена на основании
результатов исследований равновесия реакций восстановления W02(k) водородом и оксидом углерода
[179, 180, 1634, 1819, 1820, 1841, 2347, 2488, 2489, 3390, 3841, 3965, 4263, 4746, 4747, 4748, 4749]
и электрохимического окисления W(k) методом ЭДС [185, 271, 272, 623, 859, 860, 1388, 1495,
2930, 3244, 4011]. Результаты ранних исследований равновесия реакции восстановления W02(k)
содержат систематические ошибки, связанные с термодиффузией. Последняя была устранена
в работах Гриффиса [2489] и Пиэрре и др. [3841], измерения которых приводят к значениям,
совпадающим с принятым (—588,5+2,0 и —588,0+2,5 кДж-моль). К близким, но менее точным
значениям, приводят измерения ЭДС, проведенные в работах [178, 185, 271, 272, 623, 859, 2930,
3244, 4011].
Давление пара в реакции WO2(K)=WO2(r) вычислено с использованием значения Ь.ГН° @) =
= AS#°(WO2, к, 0)=615,159+15 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования WO2 в кристаллическом и газообразном состояниях.
WO2(r). Термодинамические свойства газообразного диоксида вольфрама в стандартном
состоянии при температурах 100—6000 К приведены в табл. 899.
Молекулярные постоянные, принятые для расчета термодинамических функций, приведены
в табл. 32.6. На основании результатов исследования ИК-спектра изотопомеров WO2 в матрицах
инертных газов [4699, 4968] в справочнике принимается, что в основном электронном состоянии
Х1А1 молекула WO2 имеет угловую структуру (симметрия C2v) 1. Приведенное в табл. 32.6
произведение главных моментов инерции соответствует структурным параметрам r(W—О) = 1,71+0,05 А
(оценка по величине r(W—О) в молекулах (WO3)n, см. ниже) и ^О—W—0 = 114+10° (рассчитан
Грином и Эрвином [4968] по изотопическим сдвигам частот); его погрешность составляет
З-Ю15 г3-см6.
Таблица 32.6. Значения молекулярных постоянных, а также о и рх, принятые для расчета
""""" термодинамических функций WO2, WO3, WO3, W2O6, W3O9, W4Oi2, W6OlB
Молекула
wo2
WO3
WO3
W2O6
w3o9
w4o12
W5O15
WO2: a
WO3: a
WOj: a
Состояние
X^AX a 975
X1A1 a 950
X4[ aj 800
ХЫд 980 B)
1141 960 G)
X1Ax 960 (8)
1хАг 960 A0)
Г0(Л3Л2)=5000, Т0(ВЬ
TO{HSBX, /3B1)=20 000
-
510
370
300
900 B)
810 B)
900 D)
900 E)
л п\ А
см.
v3
937
918 B)
900 B)
800 B)
600 C)
600 D)
600 E)
=10 000,
Г0(Л3,42)=15 000, Г0(г3Л2)=20 000 см.
Г0(Л:М1)=5000, ГО(Й2Л2)=15 000 см.
CM-1
_
320 B)
300 B)
700 B)
200 A4)
200 A9)
200 B4)
T0(D3B2
_
— —
— —.
350 B) 200 G)
100 D) —
100 G) —
100 A0) —
)=12 672, T0(EUj
V7
We-»"
Г3 • CM6
— 6,4 -102
— 3-103
— 3 -10s
100 1 -10»
— 4 -10'
— 3 -108
— 1,5 -10B
)=12 810, TO(F*B17 &
a
2
6
6
4
3
4
5
Aj)=i
Px
1
1
2
1
1
1
1 '
5 000,
Найденные в работе [4968] основные частоты v3 и v3 приведены в табл. 32.6. Частота v2
вычислена по уравнениям поля валентных сил по силовым постоянным /г и f , соответствующим
частотам V, и v3, и силовой постоянной / /г2==1,1 • 105 дин-см'1, оцененной по аналогии с молекулами
ScO, и VO,
С принятым значением v3 согласуется частота 928 см", наблюдавшаяся Велтнером
и Мак-Леодом [4699] в ИК-спектре молекул W02, изолированных в матрице из Ne. Слабый пик
1 Велтнер и Мак-Леод [4699] предполагали, что основным электронным состоянием WO2 является состояние Ы^
Позднее Хьюэтт и др. [2633] пришли к заключению, что основным должно быть синглетное М^состояние. Этот
вывод подтверждается результатами полуэмпирического расчета, выполненного Хаитом и Хачкурузовым
[1065]. Однако нельзя исключить, что основным электронным состоянием является сильно расщепленное 3А1-
состояние.
47
WO2(r)
Глава 32
992 см 1, отнесенный в работе [4699] к частоте vlt очевидно, принадлежит другому компоненту
пара. Погрешности значений vx и v3 оцениваются в 20 см, погрешность значения v2 — в 50 см.
Приведенные в табл. 32.6 энергии электронных состояний молекулы WOS получены Хаитом
и Хачкурузовым [1065] на основании полуэмпирической оценки для модели кристаллического
поля. Состояния с энергиями 12 672 и 12 810 см, наблюдавшиеся Велтнером и Мак-Леодом [4699]
в спектре поглощения молекул WO2 в матрице из неона, отождествлены в работе [1065] с
состояниями &В% и Ё3А1. Погрешности значений ?у) состояний D3B2 и Ё3А1 оцениваются в 200 см,
остальных состояний в 3000—5000 см.
Термодинамические функции W02(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом девяти возбужденных электронных состояний по уравнениям A. 168)—A. 170).
Погрешности рассчитанных термодинамических функций обусловлены приближенным характером
расчета и неточностью принятых значений молекулярных постоянных, в том числе типа основного
состояния. Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 2, 4 и
10 Дж-К^-моль.
Ранее термодинамические функции WO2(r) вычислялись в справочниках JANAF [2834] и
Шика [4181], а также Брюэром и Розенблаттом [1665] (до 3000 К). Расхождения данных [2834]
и табл. 899 составляют 12—7 Дж-К-моль в значениях Ф°(Т) и обусловлены тем, что авторы
JANAF считали основное состояние WO2 триплетным и использовали другие молекулярные
постоянные. Расхождения с данными [4181 ] не превышают 5 Дж-К~1-моль~1 в значениях Ф°(Т).
Константа равновесия реакции WO2(r)=W(r)-f-2O(r) вычислена с использованием значения
Дг-й°@)=1311,566+16 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/nWOa, г, 0) = 32 + 15 кДж • моль.
Эта величина найдена одновременно с энтальпией образования WO3(r) согласованием
приведенных в табл. 32.7 термохимических величин методом наименьших квадратов. Указанные в таблице
Таблица 32.7. Результаты определений A/H°(W02, г, 0) и A/Jff°(W03, г, 0) (в кДжмоль1)
Авторы
Метод
Термохимическая
величина
(III закон)
Аккерман, Торн, 1958 [1241] (см.
также [1242, 1243, 1245])
Чапка и др., 1959 [1852]
Де Мария и др., 1960 [2032, 2115]
(см. также [2033])
Дроварт и др., 1964 [2116] (см.
также [2113])
Франклин, Стикни, 1971 [2316] (см.
также [4185])
Балдуччи и др., 1977 [1377]
г, 0);
Эффузионный, MgO(K)+V8W(K)=VsWO,(r)+Mg(r), —324±20 а
1922—2019 К, 8 точек
Масс-спектрометрический, BeO(r)+WO2(r)=Be(r)-j- —343 ±18 б
WO3(r), 2245 К, 1 точка
Тоже, WO2(r)+O(r)=WO3(r), 2245 К, 1 точка —342 ±15 б
Тоже, W(K)+3O(r)=WO3(r), 2245 К, 1 точка —315 ±15 а
То же, WO(r)+O(r)=WO2(r), 2191—2478 К, 7 точек 20+19 в
То же, WOs(r)=W(K)+3O(r), 2191-2478 К, 11 точек -324+15 а
То же, WO2(r)+O(r)=WO3(r), 2191—2478 К, 11 точек —346±17 б
То же, W03(r)+Al(r)=A10(r)+W02(r), 2191—2478 К, —362+15 б
10 точек
То же, MgO(r)+WO2(r)=WO3(r)+Mg(r), 2109—2228 К, -359±25 б
5 точек
Тоже, GaO(r)+WO2(r)=WO3(r)+Ca(r), 2331—2337 К, —342 ±90 б
2 точки
То же, SrO(r)+WO2(r)=WO3(r)+Sr(r), 2329 К, 2 точки -366+50 б
То же, взаимодействие кислорода с поверхностью воль- —303 ±13 а
фрама, W(K)+3/2O2(r)=WO3(r), 2000—2300 К, 3 точки
То же, W(K)+O2(r)=WOa(r), 2000—2900 К, 10 точек 38±16 в
То же, WOs(r)+Eu(r)=EuO(r)+WO2(r), 1920-2220 К, -344±15 б
15 точек *
г, 0)-A/H°(WO2, г, 0); в bfH°(\4O2, г, 0).
48
Вольфрам и его соединения WO3(k, ж)
погрешности учитывают воспроизводимость измерений, неточность использованных в расчете
термодинамических функций, термохимических величин и сечений ионизации. Приведенные в
таблице величины были вычислены по уравнению III закона. Расчеты по уравнению II закона
приводят к менее точным значениям, которые в пределах их погрешностей в большинстве случаев
совпадают с приведенными.
WO3(k, ж). Термодинамические функции кристаллического и жидкого триоксида вольфрама
стехиометрического состава в стандартном состоянии при 100—4000 К приведены в табл. 900.
Значения постоянных, принятые в расчете термодинамических функций, приведены
в табл. 32.1. За стандартное состояние W03(k) в справочнике приняты в интервале. 0—246 К —
моноклинная модификация, в интервале 246—293 К — триклинная модификация, в интервале
293—612 К — другая моноклинная модификация, в интервале 612—1013 К — ромбическая
модификация и в интервале 1013—1747 К — тетрагональная модификация (см. [1009])Л
Термодинамические функции W03(k) при Т <^ 298,15 К вычислены на основании измерений
теплоемкости, проведенных Беволо и др. [1541] A—55 К, чистота образца ~99,8%), Зельтцем
и др. [4232] F3—299 К, чистота образца не охарактеризована) и Кеи и др. [1889] 2 A20—790 К„
монокристаллический образец), с учетом энтальпии превращения при 246 К @,159+0,010 кДж-
• моль [1889]) 3. Температура этого перехода B46+5 К) определена в [1889]. Близкие
значения получены в работах [1481, 1966, 4474, 4475]. Превращение WO3 при 293+10 К [1481*
1889, 1966, 4474, 4475] является фазовым переходом второго рода ввиду близости параметров
кристаллических решеток низкотемпературной и высокотемпературной модификаций. Погрешности
вычисленных значений ?°B98,15 К) и #"B98,15 К) —Я°@) (см. табл. 32.1) оцениваются
в 1 Дж-К-моль и 0,15 кДж-моль. Менее точные данные по теплоемкости W03(k) при Т <^
< 298,15 К получены также Савадой [4154] A06—296 К) и Кингом и др. [3013] E3—297 К).
При Т > 298,15 К теплоемкость W03(k) аппроксимирована несколькими уравнениями
(см. табл. 32.1). В интервале 298, 15—325 К по данным Кеи и др. [1889] принято уравнение,
описывающее нисходящую ветвь Х-аномалии теплоемкости с максимумом при 293 К. Для
теплоемкости моноклинной модификации C25—612 К) и ромбической модификации WO3 F12—1013 К)
принято уравнение, выведенное совместной обработкой измерений энтальпии, проведенных
Кингом и др. [3013] D00—1003 К), и измерений теплоемкости, выполненных Сейлом [4121] методом
ДСК D00—1000 К); при этом данные [4121] учитывались с меньшим весом. Полиморфный переход
при 612+10 К [1889] сопровождается очень малой энтальпией перехода (&frH <^ 0,03 кДж-моль
[1889]),"находящейся в пределах погрешности измерений энтальпии W03(k) в работе [3013J.
Поэтому энтальпия этого перехода была принята равной нулю. Данные по теплоемкости W03(k),
полученные в работах [1889, 4154],-неточны и не учитывались.
Температура полиморфного перехода A013 ±10 К) принята на основании измерений
электрического сопротивления монокристаллов WO3 в работе Савада и Даниэлсона [4157 ]; она согласуется
с результатами оптических [2799, 4158], термографических [792, 1278, 1810, 2248, 2601, 2954,
3861], дилатометрических [2277, 3861, 4154] и калориметрических [4153, 4154] исследований.
В работе [3013] температура полиморфного перехода W03 принята равной 1050 К; однако по
измерениям энтальпий нельзя достаточно точно определить эту величину. Энтальпия тетрагональной
модификации WO3 измерялась Кингом и др. [3013] A044—1704 К); выведенные по этим данным
уравнения для теплоемкости (см. табл. 32.1) и энтальпии приводят к принятому в справочнике
значению Д,г#=1,0 ±0,4 кДж «моль. Отличие от значений, полученных в [3013] A,7 кДж -моль),
и [2834] A,5 кДж-моль), обусловлено тем, что в настоящем справочнике для ромбической
модификации учтены измерения Сейла [4121].
Температура плавления A747+10 К) принята по данным Кинга и др. 13013] для энтальпии
WOs(k, ж) в интервале 1704—1755 К. Близкие значения получены в работах [2676, 2810, 3998]
и заниженные значения A705 и 1709 К) в работах 11810, 3240]. Энтальпия "\^О3(ж) измерялась
в работе [3013] A755—1838 К, 4 точки). На основании этих данных в справочнике приняты
значения энтальпии плавления G3±2 кДж-моль) и теплоемкости \У03(ж) A30±15 Дж-К-моль~1).
* По измерениям диэлектрической постоянной и электропроводности фазы WO3^ в интервале 4,2 — 300 К Леф-
ковиц и др. [3224] обнаружили фазовые переходы при 40, 70, 130 и 170 К, которые не отражаются, однако,
на теплоемкости WO3 (к).
8 Данные работ [1541,1889] приведены только на графике, при атом данные [1889] представлены в Дж • К *-моль~*г
а не в кал-К моль, как это ошибочно указано в работе [1889].
* Близкое значение @,163 кДж-моль~*) получено в работе [1481].
49
WO3(r) Глава 32
Погрешности приведенных в табл. 900 значений Ф°(Т) при 298,15, 1000, 3000 и 4000 К
оцениваются в 1,5, 2,5, 10 и 15 Дне «К.-моль.
Ранее термодинамические функции W03(k, ж) вычислялись в справочниках JANAF [28341
(до 3000 К) и Шика [4181] (до 2000 К). Расхождения между значениями Ф°(Г), приведенными.
в [2834] ив табл. 900, достигают 5 Дж-К~1-моль~1 вследствие различия принятых исходных
данных, в том числе теплоемкости при Т < 298, 15 К.
Принятая в справочнике энтальпия образования
к, 298,15 К) = —841,3 + 2,0 кДж • моль
является средневзвешенной из значений, основанных на измерениях энтальпии сгорания
вольфрама в калориметрической бомбе в работах Ма [3346] (—841,8+2,0 кДж-моль) и Хаффа и др.
[2746] (—839,6 +_3,0 кДж-моль). При вычислении поправок на неполноту сгорания в [3346]
предполагалось, что в продуктах сгорания содержатся только W03(k) и W(k). Возможность
наличия в продуктах сгорания W02(k) и* других оксидов вольфрама была учтена при оценке
погрешности этих измерений. В ранних калориметрических исследованиях [2027, 2029, 3256, 3554, 46861
для энтальпии образования W03(k) были получены заниженные по абсолютной величине значения
(—810-f-—825 кДж-моль), что объясняется неполным сгоранием и недостаточной чистотой
использованных образцов. Величины, полученные в калориметрических измерениях Гриффиса;
12488] (—833 кДж-моль) и Мюллера и др. [3584, 3585] (—843 кДж-моль), в пределах их
погрешности (~10 кДж-моль) совпадают с принятой. Энтальпия образования W03(k) может быть-
вычислена на основании результатов исследования равновесий процессов восстановления W03(k)'
до W02(k) водородом [179, 180, 1633, 1635, 1841, 2489, 3390], а также исследования равновесия
процесса электрохимического окисления W02(k) до WO3(k) методом ЭДС [4011]. Эти расчеты
приводят к значениям энтальпии образования W03(k) от —836 до —848 кДж-моль~1с погрешностью
~10 кДж-моль.
В ранних исследованиях реакций с участием W03(k) [1819, 1820, 2347, 3965, 4263, 4746, 4747v
4749] неточно определялись составы промежуточных оксидов вольфрама. Васильева и др.
исследовали равновесие реакций W03(k) методом ЭДС [178, 185—187, 271, 272, 590]. Целью этих работ
являлись измерения парциальных давлений кислорода над оксидами вольфрама различного
состава.
Давление пара в реакции W03(k, ж)=\\гО3(г) вычислено с использованием значения; ДгЯ°@) =
— ASH°(WOS, к, 0)== 521,587 +15 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования WO3 в кристаллическом и газообразном состояниях.
WO3(r). Термодинамические свойства газообразного триоксида вольфрама в стандартном-
состоянии при температурах 100—6000 К приведены в табл. 901.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 32.6. На основании результатов исследования изотопического смещения частоты v3
в ИК-спектре молекул WO3, изолированных в матрице из Аг [4968], в справочнике принимается,
что в основном состоянии XхАх молекула WO3 имеет плоскую структуру правильного треугольт-
ника (симметрия D3ft) -1. Приведенное в табл. 32.6 произведение моментов инерции рассчитано
для значения r(W—О)=1,70 +0,03 А, основанного на данных о структуре молекул W3O9, MoO3,
Мо3О9; полученных Ивановым и др. [452, 2797] при электронографическом изучении паров над
оксидами вольфрама и молибдена. Погрешность значения IaIbIc оценивается в 5-Ю15 г3-см8.
Приведенные в таблице основные частоты WO3 основаны на значениях частоты v3, полученных
Грином и Эрвином [4968] для четырех изотопомеров молекул WO3, изолированных в матрице-
из аргона. Принятое значение v3 найдено в работе [4968] для изотопомера 184W16O3, его
погрешность с учетом матричного сдвига не превышает 15 см; значения остальных частот рассчитаны
по уравнениям поля валентных сил на основании силовых постоянных, найденных по данным
Грина и Эрвина, если принять /гг=0,1 /г, /ei(/r?=0,l /а/г2 и /у/г2—0,15 /г 2. Погрешности принятых
значений оцениваются в 100 см для vx и 50 см для v2 и v4.
Приведенные в табл. 32.6 энергии двух возбужденных электронных состояний WO3 получены-
Предположение Дикстера и др. [2085] о пирамидальной структуре молекулы WO3 основывалось на
неправильной интерпретации ИК-спектров паров над WO3 (к), полученных этими авторами и в работе [4699] (см. [4968)].
(fr-7rr)=7,0.10» дин-см-ь C/«/i*-/ei/j*)=0,3.ib» дин-ear*.
50
Вольфрам и его соединения WOj(r)
Хаитом и Хачкурузовым [1065] на основании полуэмпирической оценки для модели
кристаллического поля, их погрешности оцениваются в 10 000 см.
Термодинамические функции WO3(r) вычислены по уравнениям A.3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом двух возбужденных электронных состояний по соотношениям A. 168)—A. 170).
Погрешности вычисленных величин обусловлены неточностью оцененных значений молекулярных
постоянных и приближенным характером расчета. В значениях Ф°(Г) при 298,15, 3000 и 6000 К
они составляют 2, 5 и 12 Дж-К~1-моль~1.
Ранее термодинамические функции W03(r) вычислялись в справочниках JANAF [2834] и
Шика [4181], а также Моханом [3533] (до 1700 К). Все расчеты выполнены по оцененным
значениям молекулярных постоянных WO3 без учета возбужденных электронных состояний.
Расхождения в функциях, приведенных в табл, 901 и в справочниках [2834, 4181], составляют
3—9 Дж-К~1-моль-1 в значениях Ф°(Т) и S°(T).
Константа равновесия реакции WO3(r)=W(r)+3O(r) вычислена с использованием значения
Дг#°@) = 1905,349+16 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —315 + 15 кДж • моль.
Эта величина определена одновременно с значением AfH°(W02, г, 0) согласованием
приведенных в табл. 32.7 термохимических величин методом наименьших квадратов (см. текст по W02(r)).
Погрешность принятой величины учитывает воспроизводимость измерений, погрешности
термодинамических функций, термохимических величин и сечений ионизации.
\У0з(г). Термодинамические свойства газообразного отрицательного иона триоксида
вольфрама в стандартном состоянии при температурах 298,15—6000 К приведены в табл. 902.
Молекулярные постоянные, принятые для расчета термодинамических функций, представлены
в табл. 32.6. Спектр и структура WOJ экспериментально не исследовались. На основании
полуэмпирического анализа электронных состояний WOg (см. [1065]), в справочнике принимается,
что в основном электронном состоянии Х2А[ ион WOg имеет плоскую симметричную структуру
(симметрия D3h). Многоатомные отрицательные ионы и соответствующие им нейтральные
молекулы, имеющие одинаковую конфигурацию, должны иметь близкие по величине структурные
параметры и частоты колебаний. Поэтому молекулярные постоянные WO3 были оценены по
постоянным, принятым для молекулы WO3. Приведенное в табл. 32.6 значение IaIbIc (его
погрешность 1-Ю"14 г3-см6) соответствует r(W—О) = 1,70+0,05 А, приведенные в той же таблице
значения основных частот имеют погрешности ~ 100 см. Энергии двух электронных состояний WOg
получены Хаитом и Хачкурузовым [1065] на основании приближенной оценки для модели теории
кристаллического поля; погрешности этих величин оцениваются в 3000 и 10 000 см.
Термодинамические функции WOg (г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом двух возбужденных электронных состояний по уравнениям A. 168)—A. 170).
Погрешности вычисленных величин обусловлены в основном неточностью оцененных значений
молекулярных постоянных и приближенным характером расчета. В значениях Ф°(Т) при 298,15, 3000
и 6000 К ойи составляют 4, 10 и 15 Дж-К-моль.
Таблица термодинамических функций WOg(r) публикуется впервые.
Константа равновесия реакции WOg(r)=W(r)+3 О(г)+е(г) вычислена с использованием
значения Дг#°@)=2230,349+ 34 кДж-моль, соответствующего принятому в справочнике сродству
молекулы WO3 к электрону
) = —325+30 кДж-моль-1.
Эта величина основана на работах Дженсена и Миллера [4984] (см. также [3507]), где
измерялись константы равновесия реакции H2WO4(r)+e(r)=HWO4(r)+H(r) и HWO~(r)-j-H(r) =
=WO~(r)+H2O(r) (фотометрия пламен, 2475 К) и Белтона и Мак-Керрона [4924], где измерялись
константы равновесия реакции W(K)+4H2O(r)=HaWO4(r)+3H2(r) (метод протока, 1473—1773 К).
Погрешность принятого значения учитывает неточность измерений констант равновесия реакций,
погрешности использованных в расчетах термодинамических функций и термохимических
величин. Принятая величина подтверждается исследованиями захвата электрона молекулами WO3,
51
W2O6(r) ' Глава 32
выполненными Шуи и др. [4269] методом электростатического зондирования (—347 кДж-моль).
Исследования реакции WO8(K);+H2O(r)=H2WO4(r), выполненные Глемзером и Хеслером [2419]
(метод протока, 873—963 К), при их комбинировании с данными работы [2856] приводят к менее
надежному значению —353 кДж-моль (см. текст по MoOg).
Принятому значению сродства к электрону соответствует
г> °) = —640 ± 34 кДж>моль.
W2Oe(r). Термодинамические свойства газообразного гексоксида дивольфрама в стандартном
состоянии при температурах 100—6000 К приведены в табл. 903.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 32.6. Структура и спектр молекулы W2O6 экспериментально не исследовались х.
По аналогии с молекулой Мо2Ов в справочнике принимается, что в основном электронном
состоянии ХхАд молекула W2O6 имеет циклическую структуру (симметрия D.2h). Приведенное в табл. 32.6
произведение главных моментов инерции соответствует структурным параметрам r(W—OBHeiirH) =
=1,70+0,021, r(W-OM0oI)=1,90+0,02 A, ^OBH6TOI-W-OBHemH=120±10°, ^OM0CI-W-OM0eI =
=90 + 10°, которые были оценены на основании данных, полученных в электронографическом
исследовании Иванова [452] для молекул W3O9, W4O12, и структурных параметров Мо2О6,
принятых в настоящем справочнике. Погрешность значения 1А1в1с составляет 3-1012 г3 см6.
Молекула W2O6, так же как Мо2О6 (см. е. 33), имеет 18 основных частот колебаний. Приведенные
в табл. 32.6 усредненные и объединенные значения близких по типу и величине основных частот
W2O6 с соответствующими степенями вырождения вычислены по уравнениям поля валентных сил
с использованием силовых постоянных Мо2О6 и предложенного в работе [1071] соотношения между
силовой постоянной и длиной связи W—О. Силовые постоянные деформационных колебаний
корректировались с учетом результатов обработки по уравнениям II и III законов данных о
равновесии реакций с участием газообразных оксидов вольфрама. Погрешности приведенных в табл. 32.6
частот W2O6 оцениваются в 100 см для ч1—v4, 70 см для v5 и 50 см для vg, v7.
Термодинамические функции W2O6(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных значений обусловлены преимущественно неточностью
оцененных значений молекулярных постоянных, а также приближенным характером расчета. В
значениях Ф°(Т) при 298,15, 3000 и 6000 К они составляют 15, 25 и 40 Дж-К^-моль.
Ранее термодинамические функции WaO6(r) вычислялись авторами справочника JANAF
[2834] по оцененным значениям молекулярных постоянных, существенно отличающимся от
принятых в настоящем справочнике. Расхождения в значениях Ф (Т), приведенных в табл. 903 и
в [2834], составляют от 6 до 38 Дж-К-моль.
константа равновесия реакции W2O6(r)=2W(r)+6O(r) вычислена с использованием значения
ДгЯ°@) =4380,698+36 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(W2O6, г, 0) = —1200 + 35 кДж • моль.
Результаты определений энтальпии образования W2O6(r) представлены в табл. 32.8.
Указанные погрешности учитывают воспроизводимость измерений, неточность термохимических
величин, сечений ионизации и термодинамических функций, кроме W2O6(r). Неточность
термодинамических функций W2O6(r) учтена при оценке погрешности принятой величины. Принятая
величина является средневзвешенной из наиболее точных значений, вычисленных по данным
[1133, 1220, 1430]. Больший вес придавался работе [1430], где было доказано, что пары находятся
в равновесии с конденсированной фазой и детально исследована диссоциативная ионизация.
Результаты измерений в работах [1573, 3679] менее точны и согласуются с принятой величиной
в пределах их погрешностей (см. также [4572]). Основной вклад в погрешность принятой величины
вносит неточность термодинамических функций W2O6(r).
1 Р ИК-спектре пара WO3 (к), изолированного в матрице из Ne, Велтнер и Мак-Леод [4699] предположительно
отнесли три интенсивные полосы при 1007, 989 и 694 см, а также слабые полосы при 666, 646 и 618 см
к молекулам W2Oe или WgOB. Поскольку при матричной изоляции пара WO3 (к) возможно образование
комплексов, не существующих в паре, приведенные данные следует рассматривать как предварительные и
нуждающиеся в проверке. Отметим, что полосы при 989 и 694 см~1, отнесенные в работе [4699] к молекуле W2Oe?
находятся в близком согласии со значениями vs и v4, приведенными в табл. 32.6.
52
Вольфрам и его соединения
W3O»(r)
Таблица 32.8. Результаты определений A^2Z"°(W2Oe, г, 0) (в кДж-моль)
Авторы
Блэкберн и др., 1958 [1573]
(см. также [1570, 1571])
Аккерман, Раух, 1963 [1220]
Бэттлс, 1964 [1430]
Норман, Стейли, 1965 [3679]
Чижиков и др., 1976 [1133]
Метод
Д^Я°(\У2Ов, г, 0)
II закон
Зффузионный, 3WO2(K)=W2O6(r)+W(K), 1326— —1365 ±60
1660 К, 7 точек, состав пара принимался по [1430]
Масс-спектрометрический, 3W02(K)=W20e(r)-l- —1157
+W(k), 1452-1652 К а
То же, 3WO2(K)=W2O6(r)+W(K), 1420—1529 К, —1147+10
8 точек
То же, испарение WO3 в смеси с другими оксидами, ¦—1186
2WO8(r)=W2Oe(r), 1852 и 2063 К, 2 точки
То же, 3WO2(K)=W2Oe(r)+W(K), 1400-1600 К а —1181
а Данные представлены в виде уравнения.
III закон
—1220+15
—1194+10
—1199+8
—1121+00
-1209+10
W3O9(г). Термодинамические свойства газообразного ноноксида тривольфрама в стандартном
состоянии при температурах 100—6000 К приведены в табл. 904.
Молекулярные постоянные, использованные в расчете термодинамических функций,
приведены в табл. 32.6. На основании результатов электронографических исследований [216, 452,
2567, 2797] в справочнике принимается, что в основном электронном состоянии ХгАг
молекула W3O9 имеет циклическую структуру симметрии С3г, образованную тремя группами WO2,
связанными друг с другом тремя атомами кислорода. Приведенное в табл. 32.6 произведе
ние моментов инерции соответствует структурным параметрам W3O9: r(W—Овнешн) =1,703 +
±0,006 A,r(W-OMOCr)=l,899+0,005, А ^ Овнешн-\У-Овнешн=112±5°, ^ OMOCI-W-OMOCT=110+3°,
J}W—OMOCT—W=128,0+0,5°, найденным Ивановым и др. [452, 2797] в результате электроногра-
фического исследования паров над \УО2(к). Погрешность IaIbIc составляет 1-1010 г3-см6.
Молекула W3O9 имеет 20 основных частот колебаний типов симметрии AX(J), А%C) и ?'A0).
Приведенные в табл. 32.6 объединенные и усредненные значения близких по типу и величине основных
частот W3O9 с соответствующими степенями вырождения оценены в результате расчета по
уравнениям поля валентных сил и силовым постоянным, близким к силовым постоянных Мо3О9. Выбор
силовых постоянных корректировался учетом двух основных частот молекулы W3O9, найденных
Ямпольским и Мальцевым [1193] при исследовании ИК-спектра газообразного W3O9 (полосы
с максимумами при 960 и 810 см отнесены в работе [1193] к валентным колебаниям связи W—О
в группах WO2 и колебаниям кольца W3O3). Силовые постоянные деформационных колебаний
корректировались с учетом результатов обработки равновесия реакций с участием газообразных
оксидов вольфрама по уравнениям II и III законов. Погрешности рассчитанных таким образом
частот оцениваются в 100 см для vx—v3 и в 50 см" для v4 и v5 1.
Термодинамические функции W3O9(r) вычислены по уравнениям A.3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных функций обусловлены преимущественно неточностью
принятых значений основных частот, а также приближенным характером расчета. В значениях Ф°(Т)
при 298,15, 3000 и 6000 К они составляют 20, 40 и 60 Дж-К^-моль.
Ранее термодинамические функции W3O9(r) вычислялись авторами справочника JANAF [2834J
в предположении, что молекула W3O9 имеет симметрию D3k (плоское кольцо) с использованием
оцененных молекулярных постоянных. Расхождения в значениях Ф°(Г), приведенных в табл. 904
и в [2834], составляют 17—2 Дж-К^-моль.
Константа равновесия реакции W3O9(r)=3W(r)-J-9O(r) вычислена с использованием
значения Дг#°@)=6771,047±52 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д, #°(WSO9> г, 0) = —2000 + 50 кДж ¦ моль.
* Отнесение полос, наблюдаемых Велтнером и Мак-Леодом [4699] в ИК-спектре паров над WO3 (к),
изолированных в матрице из Ne, к молекуле WsOe является ошибочным (см. [2633]).
53
W4O12(r)
Глава 32
Результаты определений энтальпии образования W3O9(r) представлены в табл. 32.9.
Исследования равновесия реакций с участием \УО3(к) [26, 474, 1220, 1504, 1571, 1573, 3493, 4572]
в таблицу не включены, так как испарение W03(k) происходит инконгруэнтно [1220, 1430].
Указанные в таблице погрешности учитывают воспроизводимость измерений, неточность
термохимических величин, сечений ионизации и термодинамических функций, кроме термодинамических
функций W3O9(r); неточность последних учтена при оценке погрешности принятой величины.
Как видно из таблицы, за исключением работы [3679], результаты измерений оказались в хорошем
согласии. Наиболее точные измерения были выполнены Бэттлсом [1430] и Аккерманом и Рау-
хом [1220] и при вычислении средневзвешенного им придавался наибольший вес. Погрешность
рекомендованной величины определяется неточностью термодинамических функций W3Og(r).
Таблица 32.9. Результаты определений A^ff°(W3O9, г, 0) (в кДж-моль)
Авторы
Метод
bfH°(Wa09, г, 0)
II закон
III закон
Берковиц и др., 1957 [1504] Масс-спектрометрический, сублимация W03(k), — —2005+50
3W2O6(r)=2W3O9(r), 1395 К, 1 точка а
Блэкберн и др., 1958 [1573] (см. так- Эффузионный, сублимация W02(k), 3W2O6(r)= —2040±60 —1998+50
же [1570, 1571]) =2W3Oe(r), 1326-1660 К, 7 точек6
То же, 9WO2(K)=2W3Oe(r)+3W(K), 1326—1660 К, —2208+60 —2028+20
7 точек б
То же, сублимация WOs(k), 3W2Oe(r)==2W3O9(r), -2073+50 -1999+50
1316—1583 К, 9 точек а
Мейер и др., 1959 [3493] Протока, сублимация W03(k), 3W2Oe(r)=2W3O9(r), —2107+40 —1995+50
1391—1521 К, 24 точки а
Аккерман, Раух, 1963 [1220] Масс-спектрометрический, 9WO2(K)=2W3O9(r)+ —2005 —1991+15
+3W(k), 1452-1652 К в- г
То же, сублимация W03(k), 3W2O6(r)=2W3O9(r), —2080 —2004+50
1302-1502 К в- г
Бэттлс, 1964 [1430] То же, 9WO2(K)=2W3O9(r)+3W(K), 1420-1529 К, —1988±10 —2007+10
13 точек
Норман, Стейли, 1965 [3679] То же, испарение WO3 в смеси с другими окси- —2006 —1907+100
дами, 3WO3(r)=W3O9(r), 1862 и 2063 К, 2 точки
Казенас, Цветков, 1967 [474] (см. То же, сублимация W03(k), 3W2O,(r)=2W3O9(r), —2085 —2011+50
также [473, 478]) 1301-1502 №• г
Чижиков' и др., 1976 [1133] То же, 9WO2(K)=2W3O9(r)+3W(K), 1400—1600 К в —2079 —2000+15
а Состав пара принимался по данным [1220]; *> состав пара принимался по данным [1430]; в данные
представлены в виде уравнения; г принималось, что обнаруженный в этих измерениях ион W3Og" является продуктом
диссоциативной ионизации, а не ионизации соответствующей нейтральной молекулы (см. [26, 1133, 1245,
1430, 1472]). . -
W4O12(r). Термодинамические свойства газообразного додекоксида тетравольфрама в
стандартном состоянии при температурах 100—6000 К приведены в табл. 905.
Молекулярные постоянные, использованные при расчете термодинамических функций,
представлены в табл. 32.6. На основании результатов электронографического исследования пара
W02(k), выполненного Ивановым [452], в справочнике принимается, что в основном электронном
состоянии ХгАх молекула W4O12 имеет циклическую структуру симметрии Civ, образованную
четырьмя группами WO2, связанными друг с другом четырьмя атомами кислорода. В работе [452]
найдены следующие значения структурных параметров W4O12: r(W—Овнешн) = 1,703+0,005 А,
KW-OM0cI) = 1,899+0,003 A, J$ Овнешн-\У-Овнешн=112,4+4,2°, ^ OMOOI-W-OMOCT-110,2 + l,2°,
-^ W—Омост—W=128,8+3,2°. Им соответствует приведенное в табл. 32.6 значение произведения
главных моментов инерции, погрешность которого составляет 6-10~110 г3-см6. Предварительные
данные о возможных значениях некоторых основных частот молекулы W4O12 были получены Велт-
нером и Мак-Леодом [4699] при исследовании ИК-спектра паров над W03(k), изолированных
в матрице из Ne. Эти данные представляются ненадежными и в справочнике не использовались.
54
'Вольфрам и его соединения
W40ls(r)
Молекула W4O12 имеет 32 основные частоты типов симметрии A-JJ)% Аг(Ъ), 2?iG), -В2E) и #A0).
Значения основных частот были оценены на основании принятых частот однотипных колебаний
молекул W2O6 и W3O9, а затем объединены в пять групп и усреднены. В табл. 32.6 приведены
усредненные таким образом значения основных частот W4O12 с соответствующими степенями
вырождения. Их погрешности оцениваются в 100 см для vx—v3 и 50 см для v4 и v5.
Термодинамические функции W4O12(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
{1. 122)—A.124), A.128), A.130) в приближении «жесткий ротатор — гармонический осциллятор».
Погрешности рассчитанных функций обусловлены главным образом неточностью оцененных
значений молекулярных постоянных и приближенным характером расчета, они составляют 40,
<№ и 80 Дж -К -моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции W4012(r) вычислялись в справочнике JANAF [2834] в
предположении, что молекула W4O12 имеет симметрию Dik (плоское кольцо). Расхождения в функциях,
приведенных в табл. 905 и в [2834], составляют 23—11 Дж-К-моль в значениях Ф°(Т).
Константа равновесия реакции W4O12(r)=4W(rL-12 О(г) вычислена с использованием
значения Дг#°@)=9161,396+63 кДж-моль, соответствующего принятой в справочнике энтальпии
•образования • . ¦
AH^WOiz, г, 0) = — 2800 + 60 кДж • моль'1.
Результаты определений энтальпии образования W4O12(r) представлены в табл. 32Л0.
Исследования равновесия реакций с участием WQs(k) [26, 4572] не включены в эту таблицу, так как
испарение WQs(k) происходит инконгруэнтно [1220, 1430.] Приведенные в таблице погрешности
учитывают воспроизводимость измерений, неточности термохимических величин, сечений
ионизации и термодинамических функций, кроме термодинамических функций W4Ow(r); погрешность
шоследних учтена при оценке погрешности принятого значения. Результаты измерений разными
методами и разных реакций находятся в хорошем согласии между собой. Принятая величина
является средневзвешенной из приведенных в таблице данных, причем оолыпий вес п$й|цавалея
наиболее точным измерениям Бэттлса [1430] и Аккермана и Рауха [1220]. Погрешность
рекомендованной величины определяется неточностью термодинамических функций W4O12(r). ;
"Таблица 32.10. Результаты определений Xj>S°(WiO12, г, 0) (в кДж-моль)
Авторы
Метод
AfH°(WtO12, г, 0)
II вакон
III 8ЭКОН
Берковиц и др., 1957 [1504] Масс-спектрометрический, сублимация W03(k), — —2793±55
4W3O9(r)=3W4O12(r), 1395 К, 1 точка а
Блэкберн и др., 1958 [1573] (см. Эффузионный, сублимация WO2(i<), 2W2Oe(r)= —2644+80 —2777+70
также [1570, 1571]) =W4O12(r), 1326—1660 К, 7 точек6
То же, 6WO2(K)=W4O12(r)+2W(K), 1326-1660 К, 2975±60 2818±20
7 точек б
То же, сублимация W03(k), 2W2O6(r)=W4O12(r), -2850+70 —2785±70
1316-1583 К, 9 точек а
Мейер и др., 1959 [3493] Протока, сублимация W03(k), 2W2O6(r)=W4O12fг), —2911 ±60 —2777 ±70
1389-1519 К, 24 точки а
Аккерман, Pays, 1963 [1220] Масс-спектрометрический, сублимация W02(k), —2863 —2792+70
2W2Oe(r)=W4O12(r), 1302—1502 К в
То же, 6WO2(k)=W4O12(r)+2W(K), 1452—1652 К в, —2775 —2807 +13
см. [1430]
Бэттлс, 1964 [1430] То же, сублимация W03(k), 4W3O9(r)=3W4O12(r), —2771+55 —2791 ±55
1335—1420 К, 9 точек
То же, 6WO2(K)=W4O12(r)+2W(K), 1451—1529 К, -2735+30 —2796±12
10 точек
Казенас, Цветков, 1967 [474] (см. То же, сублимация W03(k), 2W20e(r)=W4012(r), —2872 —2807+70
также [473, 478]) 1301-1502 К в
Чижиков и др., 1976 [1133] То же, 6WO2(K)=W4O12(r)+2W(K), 1400-1600 К в -3000 —2786±20
а Состав пара принимался по данным [1220]; б состав пара,принимался по данным [1430]; в данные
представлены в виде уравнения.
55
W5pl6(r) Глава 3?
W5O15(r). Термодинамические свойства газообразного пентадекоксида пентавольфрама в
стандартном состоянии при температурах 100—6000 К приведены в табл. 906.
,Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 32.6. Структура и спектр WBO15 экспериментально не исследовались. По аналогии
с W3O9 и W4Ola в справочнике принимается, что в основном электронном состоянии Х1А1
молекула W5O15 имеет циклическую структуру симметрии С5с, образованную пятью группами WO2,
соединенными друг с другом пятью атомами кислорода. Для- структурных параметров W5O15
приняты те же значения, что для W4O12. Им соответствует приведенное в табл. 32.6 произведение
главных моментов инерции, погрешность которого оценивается в 3-1009 г3-см6. Молекула W5O15
имеет 32 основные частоты типов симметрии Аг (8), А2B), -?\A0) и 2?2A2). Значения основных
частот были оценены на основании принятых частот однотипных колебаний молекул W2O6 и W3O9
и затем усреднены и объединены в пять групп. В табл. 32.6 приведены усредненные таким образом
значения с соответствующими степенями вырождения. Их погрешности оцениваются в 100 см
для уг—v3 и 50. см для v4 и Vg.
Термодинамические функции W5O15(r) вычислены по уравнениям A. 3)—A.6), A. 9), A. 10),.
A. 122)—A. 124), A. 128), A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности вычисленных величин обусловлены в основном неточностью оцененных
значений молекулярных постоянных и приближенным характером расчета. В значениях Ф°(Г)
при 298,15, 3000 и 6000 К они составляют 50, 70 и 90 Дж-К^-моль-1.
Таблица термодинамических функций W5O15(r) публикуется впервые.
Константа равновесия реакции W5O15(r)=5W(r)-|-15O(r) вычислена с использованием
значения Дг/У°@) = 11 481,745±103 кДж-моль, соответствующего принятой в справочнике энталь-
нии образования
ЬуН°№ъО15, г, 0) = —3530 + 100 кДж • моль.
Принятая величина является средней из представленных в табл. 32.11 результатов
исследований. В приведенных в таблице работах были измерены отношения парциальных давлений
компонентов реакций, при вычислении констант равновесия были использованы значения
парциальных давлений W3O9 и W4O12 по данным работы [1220]. Оценки энтальпии сублимации W03(k)
с образованием W5O15(r), выполненные в работах [26, 1504], находятся в согласии с принятой
величиной. Ее погрешность определяется в первую очередь неточностью термодинамических
функций W5015(r). ;
Таблица 32.11. Результаты определений A^ir°(W6Ol5, г, 0) (в кДж-моль)
Авторы
Метод
bfH°(WbOn, г, 0)
III закон
Берковиц и др., 1957 [1504] Масс-спектрометрический, 5W3O9(r)=3W5O15(r), 1494 К, —3551
1 точка
То же, 5W4O12(r)=4W6O15(r), 1494 К, 1 точка —3550
Алешко-Ожевская и др., 1980 [26] ' То же, 5\У3О9(г)=3\У6О15(г), 1387 К, 1 точка —3517
То же, 5W4O12(r)=4W5O16(r), 1387 К, 1 точка -3519
Глава ЪЪ
ВАНАДИЙ И ЕГО СОЕДИНЕНИЯ
V(k, ж), V, V+, VO(k, ж), VO, VO2, V2O3(k, ж),
V2O4(k, ж), V2O5(k, ж), VAo
В настоящей главе рассматриваются ванадий и его кислородные соединения — всего 8 веществ.
Для пяти веществ приведены данные о термодинамических свойствах в конденсированном
состоянии и для пяти — в газообразном состоянии. Термодинамические свойства V(r), V+(r), VO(r)
рассчитаны до 10 000 К, VO2(r) и V4O10(r) — до 6000 К. Термодинамические свойства всех веществ
рассчитаны для природной смеси изотопов ванадия (атомный вес 50,9414), однако ввиду
преобладающего содержания в этой смеси изотопа 51V различие постоянных разных изотопомеров
в расчетах не учитывалось.
Для элементарного ванадия в Приложении 3.10 приведены сведения о его критических
постоянных.
В справочнике рассмотрены четыре основных, наиболее стабильных и изученных
кристаллических оксида ванадия стехиометрического состава. Области гомогенности этих оксидов
характеризуются пределами: VO0)90—VO1>24 (при 1073 К), V203|ooo—V203,oi5 (при 1400 К), V2O399—
V204,oo) V2O4>94—V2O5 00. Известны также кристаллические мало устойчивые субоксиды ванадия:
V9O, V4O, V2O (см. [183, 1288]), а также высшие оксиды ванадия — члены гомологических рядов
У„О2и 1 (фазы Магнели), соответствующие значениям п от 3 до 10 [1322, 2712, 3154] и VBO2n+1,
где п=2, 3 и 6 [3077].
Масс-спектрометрические исследования показали (см. [1062, 1134, 1472, 1505, 2216]), что
в газообразном состоянии существуют также оксиды V2O4, V40g, V6O12, V6O14, V6O15, которые не
рассматриваются в справочнике ввиду их низкой стабильности. Не рассматриваются также
ионизированные оксиды, поскольку потенциалы ионизации оксидов ванадия превышают 70 000 см.
V(k, ж). Термодинамические свойства кристаллического и жидкого ванадия в стандартном
состоянии при температурах 100—5400 К приведены в табл. 907.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 33.1. В справочнике за стандартное состояние V(k) принята кубическая
объемно-центрированная модификация (структурный тип a-Fe) x. При 5,435 К ванадий переходит в
сверхпроводящее состояние [711, 1802, 3237, 3796, 4229, 4766]. Термодинамические функции V(k) при
Т<298,15 К вычислены на основании измерений, проведенных Кораком и др. [1925] A,1—5,4 К),
Радебау и Кеезомом [3908] @,5—5,4 К), Леуполдом и др. [3237] B—20 К), Паном и др.
[753] B3—35 и 185—205 К), Клузиусом и др. [1882] B2—274 К). Для Т<20 К принятые
значения теплоемкости основаны на данных [1925, 3237, 3908], в интервале 20—50 К они получены
усреднением данных [753, 1882], в интервале 50—270 К использованы данные [1882],
подтвержденные измерениями [753] и [1956] A75—265 К). В интервале 270—298,15 К значения
теплоемкости получены плавной интерполяцией между данными [1882] и результатами измерений
теплоемкости ванадия в работах Чеховского и Калинкиной [1119, 1124] C07—617 К) и Кольхааса
и др. [3062] C20—1800 К). Учитывая чистоту исследованных образцов ванадия, составляющую
в перечисленных работах 99,5—99,9%, погрешности принятых значений теплоемкости при
Т<^20 К, в интервале 20—50 К и при Г>50 К оцениваются в 3, 4 и 1% соответственно.
Погрешности значений <S°B98,15 К) и #°B98,15 К)—#°@), приведенных в табл. 33.1, составляют
0,3 Дж-К-моль~1 и 0,05 кДж-моль. Измерения теплоемкости V(k) при низких температурах
в работах [1316, 1558, 1828, 4257, 4258, 4750, 4766] менее точны и не учитывались в расчетах.
1 В ряде работ были отмечены аномалии в температурной зависимости физических свойств образцов ванадия
разной степени чистоты в интервале 180—325 К, в том числе фазовый переход при 230 К в тетрагональную
модификацию [1056], и аномалия теплоемкости при 195 К [753]. Согласно [753] причиной этих аномалий
являются малые количества примесей (в частности, водорода). Измерения теплоемкости более чистых образцов,
ванадия [1882, 1956] не обнаружили каких-либо аномалий.
57
V(k, ж)
Глава 33
Таблица 33.1.
Принятые значения термодинамических величин для ванадия и его соединений
в кристаллическом и жидком состояниях
Вещество
V
VO
v,o,
v2o4
v,o.
Состояние
к, куб.
ж
к, куб.
ж
к, гекс.
ж
kII, mohokj
к1, тетр.
ж
к, ромб.
ж
1
И
СП J-'
кДж • моль
4,58
5,98
17,13
I. 16,72
21,21
ю
оо"
СП
о
со
К
оо"
СП
о~а,
Дж • К • моль-1
28,67 24,48
33,51 38,42
94,49 100,96
93,18 112,13
130,5 127,77
Коэффициенты в
нении для С
а
25,806
45,6
52,920
70
144,310
160
364,962
144,694
180
190,878
190
Примечание. С°р{Т)=а+ЪТ— cT^+dT2 (в Дж-К-моль).
V: а й=3,245 • 10~6. V2O4: a d=3548,895 -Ю..
V2O3: а d=10,892 • 10~e. V2O5: а d=95,232 ¦ 10.
6-Ю3
0,909
13,113
—18,663
—1906,105
19,061
—92,779
урав-
c-10-s
1,676 ?
16,365
34,450«
0а
28,737
39,034 а
Интервал
температур
Ё
К
1 298,15—2220
2220—5400
298,15-2063
2063—5000
1 298,15-2230
2230—5000
298,15—338,7
338,7—1818
1818-5000
298,15-954
954-5000
-
2220
2063
2230
338,7
1818
954
а=°а,
? 8
кДж-
•моль 1
23,0
50
140
9,15
112
64
В интервале 298,15—2220 К для теплоемкости V(k) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии, выполненных Егером и Веенстрой [2820] E65—
1828 К), Березиным ш др. [123, 124] A345—2173 К), Гатерсом и др. [2373] A700—2190 К) и
Зейделем и др. [4243] B200 К), и результатов измерений теплоемкости, проведенных Чеховским
и Калинкиной [1119, 1124] C07—892 К) и Кольхаасом и др. [3062] C20—1800 К) на образцах
ванадия чистоты 99,7—99,94%. Это уравнение аппроксимирует данные [123, 124, 2373, 2820,
4243] для энтальпии V(k) с погрешностью 3% и данные [1124, 3062] (при 2Х1600 К) и [1802]
^при 1600—2000 К) для теплоемкости в пределах 1 %. Энтальпия V(k) измерялась также в работах
J2233] D80—1890 К), [301] D89—1486 К), а теплоемкость — в работах [711] B93—1173 К),
[51, 1055] A000—1900 К) и [1802] A500—2100 К). Однако в работах [51, 301, 1055, 2233] имеют
место систематические ошибки, а данные [711] представлены только на графике.
Температура плавления V(k) принята равной 2220 + 10 К на основании измерений Кенисарина
[496] B206+4 К) и Пана и др. [752] B226 + 10 К), выполненных на образцах чистотой 99,94 и
99,99%. В работах [496, 752, 3721, 4084, 4408] при измерениях на образцах чистотой 99,6—99,8%
были получены значения от 2163 до 2202 К и показана существенная зависимость этой величины
«от чистоты образцов (см. также [896, 1036, 1256, 1751, 2215, 3465, 4732]).
Энтальпия V(m) измерялась методом левитационной калориметрии Тревертоном и Маргрей-
вом [4543] B205—2638 К) и Березиным и др. [118, 120, 124, 1483] B084-2325 К) и методом
импульсного нагрева Зейделем и др. [4243, 4245] (Тт — 6000 К) и Гатерсом и др. [2373] (Тт —
4500 К). Чеховской [1119] пересчитал данные Березина и других; на основании значений
энтальпии V(jk), рекомендованных в [1119], и приведенного в табл. 33.1 уравнения для теплоемкости V(k)
получены принятые в справочнике величины &mH°(V, к) =23,0+1,9 кДж-моль и C°(V, ж) =
=45,6 + 2,8 Дж-К~1-моль. С принятым значением hmH°(V, к) согласуются энтальпии
плавления, найденные в работах [124] B3,1+0,5 кДж-моль), [118, 1483, 1484] B3,0+0,7 кДж-моль),
[2373] B1,9 кДж-моль). Менее точные значения AmH°(V, к) были получены в работах
[4543] A7,3 кДж-моль),, [1617] х A8,2 кДж-моль), [2215] A8,0 кДж-моль) и [4243]
1 Боннел и др. [1617] внесли изменения в данные [4543] после учета тепловых потерь на излучение и уточнения
коэффициента нзлучательной способности У(ж).
58
Ванадий и его соединения
V(r)
B7,5 кДж-моль). Для теплоемкости У(ж) значения, близкие к принятому, были получены в
работах [118, 1483J D6,2 + 2,9 Дж-К -моль) и [1617] D6,8 + 1,2 Дж-К-моль). Менее точные
значения получены в работах [4543] D8,7±1,5 Дж-К-моль~1) и [4245] C9,7 Дж-К-моль~1).
Завышенное значение C°(V, ж) получено в [2373] F5 Дж-К~1-моль~1), по-видимому, вследствие
неравновесного состояния образца при импульсном нагреве.
Погрешности приведенных в табл. 907 значений Ф°(Г) при 298,15, 1000, 3000 и 5000 К равны
0,2, 0,5, 1,5 и 5 Дж-К^-моль.
Ранее термодинамические функции V(k, ж) вычислялись в справочниках JANAF [2835] (до
4500 К), Шика |4181] (до 6000 К) и Халтгрена [2752] (до 3800 К) с использованием менее точных
данных для V(k) и оцененных данных [4543] для У(ж). Различия в значениях Ф°(Г), приведенных
в табл. 907 и в [2835], составляют от 0,05 до 6 Дж-К-моль~1.
В настоящем справочнике в качестве стандартного состояния ванадия принято кристаллическое
состояние и в соответствии с этим
bfH°{V, к) = 0.
Давление пара ванадия в реакции V(k, ж)=У(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = \H°(V, к, 0) = 514 + 2 кДж • моль.
В табл. 33.2 представлены значения AeH°(V, к, 0), вычисленные на основании измерений
давления насыщенного пара. В соответствии с данными [2920) принималось, что пары ванадия
одноатомные. Приведенные в таблице погрешности учитывают воспроизводимость измерений и
неточность сечений ионизации. Принятое значение является средневзвешенным из совпадающих
Таблица 33.2. Результаты измерений Д{?Г°(У, к, 0) (в
Авторы
Метод
A,H°(V, к, 0)
II закон
III закон
Эдварде и др., 1951 [2152] Испарение с открытой поверхности, 1668—1885 К, 506+9 513,1 ±0,4
12 точек
Сексер, 1962 [4161] (см. [2215, 2752]) Эффузионный, 1771—1880 К, измеренные давле- — 513,3
ния не приведены, указано только полученное
значение ЬеН°(Т)
Фарбер, Сривастава, 1973 [2215] Масс-спектрометрический, 1900—2155 К, 9 точек 504+14 514,7±6,8
То же, 2198—2412 К, 5 точек 487 ±62 515,9 ±7,0
Стормс и др., 1973 [4407] То же, 1445—1945 К, измеренные давления не 537 —
приведены, указано только полученное
значение А4Я°(Г)
в пределах погрешностей результатов работ [2152, 221§, 4161]. При этом данным [2215]
придавался меньший вес из-за систематической ошибки в измерениях температуры. Погрешность
принятого значения оценена с учетом воспроизводимости измерений, неточности термодинамических
функций и сечений ионизации.
V(r). Термодинамические свойства газообразного ванадия в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 908.
Для расчета термодинамических функций использованы уровни энергии х атома V в интервале
от 0 до 54 000 см с суммарным статистическим весом 4708. Эти уровни соответствуют валентным
электронным конфигурациям . . .3d34s2 (к ней относится основной уровень *#>/,), . . .3d34p2,
. . .3#, . . .3cZ44Z (Z=0+-3), . . .3d34s4Z (Z=l-j-3) и лежат ниже первого потенциала ионизации.
Принятые величины основаны на данных, рекомендованных Шугаром и Корлиссом [4438] и
дополненных Дэвисом и Эндрю [2004] для состояний 4(?, *Р и *Р(а), соответствующих конфигурации
. . .3d5, а также на результатах оценок энергий не наблюдавшихся экспериментально состояний
методами, изложенными в Приложении 6. Оцененные уровни имеют энергию от 17 000 см и выше
См. примеч. 1 к тексту по Сг (г).
59
У" (г), УО(к, ж) Глава 3$
с суммарным статистическим весом 2078. Погрешность оценки энергии лежит в пределах 100—
4000 см.
Термодинамические функции V(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),.
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Г^ЗООО К обусловлены в основном неточностью фундаментальных постоянных
и не превышают 0,02 Дж-К-моль в значениях Ф°(Г) и S°(T). При более высоких
температурах погрешности возрастают из-за неточности принятых значений уровней энергии и
пренебрежения ридберговскими состояниями и при 6000 и 10 000 К достигают соответственно 0,2 и
1 Дж-К^-моль" в значениях Ф°(Т).
Ранее термодинамические функции V(r) вычислялись в таблицах JANAF [2835], справочнике
Шика [4181], справочнике Сталла и Зинке [4421] G^3000 К) и в работах Хилзенрата [2652]
(Г<10 000К), Кольского и др. [3067] (Г<2000 К) и Каца и Маргрейва [2934] (Г<6000 К).
Расхождения данных табл. 908 и всех остальных расчетов при Г<3000 К не превышают
0,06 Дж-К~1-моль~1 в значениях Ф°(Т) и S°(T). При более высоких температурах в связи с тем,
что в настоящем справочнике были учтены уровни энергии, не наблюдавшиеся экспериментально,,
расхождения возрастают, но не превышают 0,16 Дж-К~1-моль в значении Ф°F000 К) для
данных [2652, 2835] и 0,5 Дж-К-^моль в значении Ф°A0 000 К) с данными [2652].
Энтальпия образования V(r)
bfH°(V, г, 0) = 514 + 2 кДж • моль
соответствует принятой энтальпии сублимации ванадия.
V+(r). Термодинамические свойства газообразного положительного иона ванадия в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 909.
Для расчета термодинамических свойств использованы уровни энергии г иона V+ в интервале
0—80 000 см с суммарным статистическим весом 1672. Эти уровни соответствуют электронным
конфигурациям . . .3d4 (к ней относится основной уровень 5ZH), . . .3d24s2, . . .3<P4l (Z=l,2) и
. . .3d4sip. Энергии этих состояний основаны на рекомендации Шугара и Корлисса [4438].
Энергия состояния . . .3d4 1-S'(fe) получена Мэни [3365] и Мешковым [3486а] в результате
полуэмпирических расчетов. Энергия состояния r . .3d24s2 1S оценена на основании сравнения уровней энергии,
принадлежащих электронной конфигурации . . .3J24s2 атома Ti и иона V+. Погрешности
оцененных значений составляют соответственно 1000 и 5000 см.
Термодинамические функции V+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин обусловлены главным образом неточностью фундаментальных постоянных и не
превышают 0,02—0,03 Дж-К^-моль в значениях Ф°(Т) и S°(T).
Ранее термодинамические функции V+(r) вычисляли Хилзенрат и др. [2652] (до 10 000 К>
и Грин и др. [2473] (до 50 000 К) по данным Мур [2053]. Расхождения в функциях, приведенных
в табл. 909 и в [2473, 2652], при Т<5000 К не превосходят 0,01 Дж-К^-моль. ПриГ>5000К
это различие увеличивается и достигает при 10 000 К 0,1 и 0,3 Дж-К-моль~1 в значениях S°(T\
С°(Т)
Р
Константа равновесия реакции V+(r)+e(r)=V(r) вычислена с использованием значения
ДгЯ°@)=—650,3+1,2 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(V)=54 360+100 см-^650,3+1,2 кДж-моль.
Это значение основано на расчетах Меггере и Рассела [3473] и авторов настоящего справочника.
В этих расчетах использовались спектральные данные для первых двух членов ридбёрговских
серий атома V и оцененные значения квантовых дефектов.
Принятому значению /0(V) соответствует
Д/Я°(У+, г, 0) = 1164,3 + 2,3 кДж • моль.
VO(k, ж). Термодинамические свойства кристаллического и жидкого оксида ванадия стехио-
метрического состава в стандартном состоянии при температурах 100—5000 К приведешь
в табл. 910.
См. примеч. 1 к тексту по Сг(г).
60
Ванадий и его соединения _ УО(к; ж)
Значения постоянных, использованные при вычислении термодинамических функций,
приведены в табл. 33.1. В справочнике за стандартное состояние VO(k) принимается кубическая
модификация (структурный тип NaQ) с беспорядочным расположением вакансий в обеих подрешетках^
¦общее содержание которых в VO(k) определяется в 16—17% (см., например, [1023, вып. 2, с. 213]).
Термодинамические функции VO(k) при Г<^298,15 К вычислены по данным Черняева и др.
11116] по теплоемкости образца VO099 в интервале 59—299 К. Экстраполяция теплоемкости
ниже 59 К проведена с использованием дробно-рационального уравнения шестой степени,
выведенного по данным [1116] и значению 6d=600 К, которое было оценено на основании значений
(Ь(ТЮ, к)=650+50 К и 0B(NbO, к)=550 К, полученных Оказом и Кеезомом [3711];
экстраполяция привела к значению S°(VO, к, 59 К) = 1,5 Дж-К^-моль. Погрешности принятых значений
5°B98,15К) и #"B98,15 К)—Л°@) (см. табл. 33.1) оцениваются в 0,4 Дж-К^-моль и
0,05 кДж-моль соответственно 1. В интервале 298,15—2063 К для теплоемкости VO(k) принято
уравнение (см. табл. 33.1), полученное совместной обработкой данных по энтальпии, полученных
Черняевым и др. [1113] D00—1300 К) для УОж(к), а;=0,86, 1,105, 1,18 и 1,24 и приведенных к
составу VO100, и значений Н°(Т)—#°B98,15 К), определенных графической экстраполяцией
данных [1113] с учетом данных Матизена и Ефремовой [670] по энтальпии ТЮ(к) в интервале 1268—
2027 К. Менее надежные измерения энтальпии VO(k) в работах [3725] C94—1698 К) и [1191]
D55—1048 К) не учитывались.
Температура плавления VO принята равной 2063+10 К по данным Александера и Карлсона
[1288] для образца с содержанием примесей ~0,1%. Менее точные значения получены в работах
[1648] B103 К) и [4414] B033 К). Энтальпия плавления E0+10 кДж-моль) оценена на
основании предположения о равенстве энтропии плавления VO и TiO. Для теплоемкости У0(ж) принято
оцененное значение 70 Дж-К-моль, равное экспериментально определенному для ТЮ(ж)
<см. с. 99).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 3000 и 5000 К оцениваются
в 0,3, 1, 8 и 15 Дж-К~1-моль соответственно. Расхождения между термодинамическими
функциями VO(k, ж), приведенными в табл. 910 и справочниках [2835] (до 3500 К) и [2960] (до 2000 К)
составляют при 298,15, 2000 и 3000 К соответственно 2,4, 4 и 23 Дж'К~1-моль~1, что объясняется
учетом в настоящем справочнике более надежных данных [1113] и различием в оценке
теплоемкости У0(ж).
Принятая в справочнике энтальпия образования
bfH°(VO, к, 298,15 К) = —430,8 + 5,5 кДж • моль
получена в результате согласования методом наименьших квадратов приведенных в табл. 33.3
данных. При их вычислении были использованы измеренные Ма и Келли [3348] энтальпии
реакций сгорания V(k) с образованием смеси V2O4 и У2ОБ E опытов), V2O3(k) — с образованием смеси
Таблица 33
.3. Энтальпии сгорания V(k),
калориметрических измерен
Реакция
2V(K)+2O2(r)=V2O4(Kl,
2V(K)+V2O2(r)=V2O5(K)
V2O3(K)+V2O2(r)=VaO4(Kl,
V2O3(K)+O2(r)=V2O6(K)
2VO(K)+V2O2(r)=V2O3(K)
гетр.)
тетр.)
V2O3(k) и VO(k)
шй Ма и Келли
, основанные на результатах
[3348]
ДГЯ°B98,15К), кДж-моль-1
—1423,5 ±5,0
—1551,0 ±8,8
—206,6 ±1,0
—334,3 ±1,0
—335,2 ±9,2
V2O4 и V2O5 E опытов) и VO(k) — с образованием смеси V2O3 и V2O4. Принималось, что продукты
сгорания являются смесью соответствующих стехиометрических оксидов. Погрешности энтальпий
реакций сгорания вычислялись по методике, предложенной Корниловым [542]. Погрешность при-
1 Результаты измерений теплоемкости VO (к) в работе Тодда и Бонниксона [4521] E5—296 К) не учитывались,
так как они завышены на —20% по сравнению с данными [1116] вследствие наличия значительного количества
примесей У2ОЯ в исследованном образце.
61
VO(r)
Глава 33-
нятого значения энтальпии образования VO(k) учитывает неточность измерений в работе [33481?
и возможное отклонение от стехиометрического состава образующихся при сгорании оксидов-
ванадия. Принятое значение согласуется в пределах погрешностей с существенно менее
точными величинами, основанными на измерениях энтальпий сгорания УОа.(к) в работах [2351
(—417 кДж-моль) и [699] (—426+13 кДж-моль) и на результатах тензиметрического
исследования реакции восстановления V2O3 углеродом до VO(k) в работе [904] (—427 кДж-моль-1).
Ранние попытки определить AjH°(VO, к) в работах Микстера [3522, 3525] привели к ошибочным
результатам (—265 и —303 кДж-моль). Давление пара в реакции VO(k, ж)=УО(г) вычислено»
с использованием значения АгН°@) = &SH°(VO, к, 0)=576,642+11 кДж-моль",
соответствующего принятым в справочнике энтальпии образования VO(k) и энергии диссоциации VO(r). .
VO(r). Термодинамические свойства газообразного оксида ванадия в стандартном состоянии'
в интервале температур 100—10 000 К приведены в табл. 911.
В табл. 33.4 приведены молекулярные постоянные VO, принятые на основании анализа
результатов исследований спектров этой молекулы, неэмпирического квантовомеханического расчет*
[1749] и приближенных оценок. В электронном спектре VO проанализированы две системы полос:
54?-**Z4E- [2234, 2374, 2898, 3166, 3205, 3350, 3987, 3988] и Л4П =** Х4Е" [4696]. Кроме того,
к молекуле VO были отнесены многочисленные неинтерпретированные полосы, наблюдавшиеся,
в ИК-области [3026, 3162, 3163, 3165, 4696] и в ультрафиолете [979, 1672].
Таблица 33.4. Молекулярные постоянные 51V16O
Состояние
Те
X*S~a О6 1011,3
АЩ 12 655,8в 910,9
B*S- 17 494,3 863,35
?4П 23 980 833 Д
Примечание. Ниже все постоянные даны
а Оцененные электронные состояния:
Состояние
Состояние
4500
С4Ф, 22~
22 000
Ь2Г
9500
2П, 2Ф
26 000
6 Х=1,371, п=0,0112, Т2=0,0111; в А„
в см г.
С2Д
11 000
27 000
-63; г
Ь>ехе
СМ
4,86
5,0
5,35
ве
0,54825
0,5144
0,4953
2Ф, 2Н 2П, 2П
14 500 16 500
2П, 2Ф
28 000
приведено
32 000
значение Do;
ах • 103
3,52
4
3,5
18 000
2 Б+ 2 Д 2
36 000
Д приведено
De ¦ 107
6
6Г
6
Д
значение Д
ге
А
1,5893
1,6408
1,6721
Результаты исследований вращательной структуры полос в работах [3987, 3988., 3989] и
спектров молекул VO, изолированных в матрицах [2927, 4696], а также теоретический расчет
[1749] однозначно свидетельствуют, что основным состоянием этой молекулы является
состояние 4Е~ (. . .а§2) с большим сверхтонким расщеплением. Квантовомеханический расчет Карлсона
и Мозера [1749] показал, что нижними у этой молекулы являются состояния конфигураций
. . .зв8|, . . -ag§,z, • • -ЗЦГСр) • • -81°^) • • •as5d7tp и °Аар- Несмотря на то, что рассчитанные энергии
состояний Л4П и S4S~ существенно превышают (на 28 000 и 31 000 см) найденные
экспериментально, результаты этого расчета позволяют оценить энергии ненаблюдавшихся состояний,
принадлежащих перечисленным электронным конфигурациям. При оценке было принято, что
состояния Х4Е~, 44П и 2?42~ соответствуют конфигурациям . . .°s§^, . . .S|tc и . . .8|о , а
найденное в спектре низкотемпературной матрицы состояние D интерпретировано как состояние-
4П (. . .os§rf'rp)- Результаты расчета [1749] были использованы для оценки: 1) относительного
расположения состояний, принадлежащих конфигурациям . . .°вЪ\ и частично . . .as8drc ; 2) разности,
энергий состояний 4Д (. . .а8§йор)и4П (. . .csbdKp); 3) величины дублет-квартетного расщепления;
термов одной конфигурации; 4) энергии состояния с2Д (. . . ag§da ) после внесения поправки на
62
Ванадий и его соединения
УО(г>
неучтенное взаимодействие конфигураций. Следует отметить, что принятая энергия состояния
с2Д (. . .°2гЬл) близка к разности энергий состояний ?4П (. . .0,8^) и Л4П (. . .8|тер) и
совпадает с разностью энергий. центров тяжести термов, принадлежащих конфигурациям . . .s2d и
. . .(Ps у Ti+. На основании этих оценок в табл. 33.4 наряду с экспериментальными данными
представлены оцененные энергии еще 19 возбужденных электронных состояний; погрешности
приведенных энергий этих состояний оцениваются в 4000 см.
Колебательные постоянные в состояниях Х4Е~ и Б4Е~ приняты по работе Харрингтона и др.
[2572]; они были получены по началам полос (z/, v"^.8), рассчитанным по кантам, измеренным
Маханти [3350] и Лаудом и Калсулкаром [3205] с использованием вращательных постоянных,
найденных Лагерквистом и Селином [3166]. Для состояния Х4?~ они близки к полученным в
работе [3166] по началам полос (г/, г/'<^2) системы 54?~—Х4?~. Вращательные постоянные в
состоянии Х4?~ были получены Ричардсом [3987] из анализа структуры полос подсистемы
441Ъ/2—Х4?~. Они хорошо согласуются с найденными в работе [3166] в результате анализа полос
(г/, и"г?^2) системы 54?~—Х4?~ и результатами анализа полосы 0—0 системы Л4П—Х4?~.
Колебательные постоянные в состоянии А*П приняты по данным [3987], где они были
получены для компоненты 4П5/;. Они близки к рассчитанным в работах [2948, 3205] по кантам полос
(с v'^.5) системы АЧ1—Х*?~. Вращательные постоянные в состояниях А*П и BiH~ приняты по
данным [3987, 4609] и [3988] соответственно1.
Термодинамические функции VO(r) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производных вычислялись по уравнениям A. 90)—A. 92) с
учетом 22 возбужденных электронных состояний в предположении, что QHl.Bi=z(piIPx)Qi^ вр-
См'.вр и ее производные находились по уравнениям "A.73)—A.75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью уравнений типа
A. 82). В расчете учитывались все уровни состояния Х4?~ с значениями /^/шах „; последние
определялись из условий A. 81). Колебательно-вращательные уровни энергии состояния Х4?~
вычислялись по уравнениям A. 65), A. 34), A. 64а), их мультиплетность учитывалась
статистическим весом 4. Значения vm3j7L и /1Ш, а также величины коэффициентов в этих уравнениях,
полученные с учетом условий A. 50), A. 59), приведены в табл. 33.5»
Таблица 33.5.
Значения коэффициентов в уравнениях, описывающих уровни энергии (в см1),
а также значения «шах и <7ш в состоянии X* ?~, принятые для расчета
термодинамических функций VO(r) a
Узо ¦ W4
Уо1 • 10
¦ и
• 103
02 '
У03 • 10»
1.01131 —4.8638 4.1679 —1.206 5.4825 —3.52 —6.0 —1.045
Значения Те для возбужденных состояний см. табл. 33.4.
99
408
Погрешности рассчитанных термодинамических функций VO(r) при низких температурах
обусловлены приближенным учетом мультиплетности вращательных уровней состояния Х*1,~
и использованием уравнения A. 34) для аппроксимации их энергий. При температурах выше
1000 К становятся существенными ошибки, особенно в значениях С°р(Т), из-за использования
оцененных энергий состояний а2?~, Ь2Г и других. Погрешности в значениях Ф°(Г) при 298,15, 3000,
6000 и 10 000 К оцениваются в 0,05, 2, 4 и 8 Дж-К^-моль.
Термодинамические функции VO(r) ранее вычислялись в таблицах JANAF [2835], работах
Брюэра и Розенблатта [1666] (Ф°(Т) для Г<3000 К), Шнейдера [4197а] (Т=2000~Ю 000 К).
Расхождения данных [2835, 4197а] и табл. 911 достигают 3 и 8 Дж-К^-моль в значениях
Ф°F000 К) и ?°F000 К), а также 10 Дж-К^-моль в значениях С°р(Т) для [2835]; они
обусловлены главным образом тем, что в обоих расчетах учитывались только два возбужденных состоя-
д Приведенное в справочниках [2736, 4453] значение Ве соответствует эффективному значению для подсостоянпя
4П5,, оно существенно отличается от истинного. Значение Л0=0,5124 см вычислено Везетом [4609] в
результате анализа всех четырех компонент перехода *П—*2.
63
V0#)
Глава 33
ния. В расчете [1666] принята неверная схема электронных термов VO; соответствующие данные
превышают приведенные в табл. 911 на 5—14 Дж-К^-моль.
Константа равновесия реакции VO(r)=V(r)-j-O(r) вычислена с использованием принятого
в справочнике значения
Дг#°@) = D0(VO) = 612 + 10 кДж • моль я« 51 200 + 850 см.
Принятая величина основана на масс-спектрометрических исследованиях обменных реакций,
выполненных Полпенсом и др. [1923] и Фарбером и др. [2216]. В работе [1923] была исследована
реакция VO(r)-)-;Ge(r)=V(r)+GeO(r) V1927—2145 К, 8 точек), этим измерениям соответствуют
значения D0(VO)=t=611,6 кДж-моль (III закон) и 601 кДж-моль (II закон). В работе [2216]
исследовалась реакция AlO(r)+V(r)=Al(r)+VO(r) B270 К, 1 точка), из этих данных получено
D0(VO)=616,2 кДж-моль (III закон). При оценке погрешности рекомендованной величины были
учтены воспроизводимость измерений, погрешности использованных в расчетах
термодинамических функций, термохимических величин и сечений ионизации.
В работе Берковица [1505] масс-спектрометрическим методом была изучена реакция VO(k) =
=VO(r). Полученным для этой реакции величинам соответствуют значения D0(VO)=650 +
+30 кДж-моль (III закон, 1872 и 1873 К) и 627 кДж-моль (II закон, измерение интенсивно-
стей ионов VO+ в интервале 1680—1950 К). Эти данные уступают по точности результатам
исследований обменных реакций.
Принятой энергии диссоциации соответствует
г, 0) = 148,783 + 10 кДж . моль.
VO2(r). Термодинамические свойства газообразного диоксида ванадия в стандартном состоянии
при температурах 100—6000 К приведены в табл. 912.
Молекулярные постоянные, принятые для расчета термодинамических функций, приведены
в табл. 33.6. В соответствии с результатами исследования Серебренниковым и Мальцевым [925]
Таблица 33.6.
Значения молекулярных постоянных, а также а и р2, принятые для расчета
термодинамических функций \О2 и V4O10
Молекула
Состояние
v2
v3
VO2 Я2А2а 983 538 1034
V4O10 XU-l 200(8) 300(8) 500D)
Примечание. Ниже все постоянные даны в см.
VO2: а 7'0(^М1)=10 000, Г0(б2В1)=15 000, Т0(С2А
=30 000.
V4O10: a v,=600 C), vg=800 C), v9=1050 A).
V4
CM
626 C) 828 C)
l!)=18 000, ТофЧ
v.
1030 C) a
32)=25 000,
Vb^c-io117
rs • см6
3,2 -103
8-10»
Г0(ё2Б2)=28 000
a
2
12
rp /I
0\
Px
2
1
*2#i)==
изотопического сдвига частоты v3 в ИК-спектре молекул VO2, изолированных в матрице из Аг,
в справочнике принимается, что в основном состоянии Х2А2 эта молекула имеет угловую структуру
(симметрия С2„) и -^О—V—0=95+10°. Приведенное в таблице значение IaIbIc рассчитано для
этой структуры и величины r(V—О) = 1,60+0,03 А, оцененной на основании значений re(VO)s
re(Nb0) и r(Nb—О) в молекуле NbO2. Погрешность принятого значения произведения моментов
инерции VO2 оценивается в 5-10~11в гэ-смв. Приведенные в табл. 33.6 основные частоты VO2
найдены Серебренниковым и Мальцевым [925] (см. выше); их погрешности могут достигать 10—
20 см. Энергии нижних возбужденных электронных состояний VO2 приняты по оценке Хаита
и Хачкурузова [1065], выполненной с помощью полуэмпирического варианта теории
кристаллического поля с использованием параметров, полученных для VO, TiO2 и ТаО2. Погрешности
соответствующих величин составляют 4000—5000 см.
Термодинамические функции VO2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом шести возбужденных электронных состояний по соотношениям A. 168)—A. 170).
64
Ванадии и его соединения
V2O3(k,
Погрешности вычисленных значений обусловлены приближенным характером расчета и
неточностью принятых молекулярных постоянных. Погрешности в значениях Ф°(Г) при 298,15, 3000
и 6000 К могут достигать 1, 2 и 4 Дж-К^-моль.
Ранее термодинамические функции VO2(r) вычислялись в справочниках JANAF [2835] и
Шика [4181J, а также Брюэром и Розенблаттом [1665] (до 3000 К). Расхождения данных табл. 912
и [2835] составляют 1—4 Дж-К^-моль в значениях Ф°(Т) и обусловлены тем, что в [2835]
были приняты другие значения v2 и v3 и не учитывались возбужденные электронные состояния.
Расчеты [1665, 4181 ] основывались на предположении о линейной структуре молекулы VO2 и
соответствующие расхождения достигают 8 — 14 Дж-К^-моль в значениях Ф°(Г).
Константа равновесия реакции VO2(r)=V(r)+2O(r) вычислена с использованием значения
ДгЯ°@)=1237,566 +15 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(VO2, г, 0) = — 230 ± 15 кДж • моль.
Принятая величина основана на приведенных в табл. 33.7 данных, указанные погрешности
учитывают воспроизводимость измерений и неточность использованных в расчетах
термодинамических функций, термохимических величин и сечений ионизации. Принятая величина является
средневзвешенной из совпадающих в пределах погрешностей измерений в работах [1062, 1505,
2998]. Остаются неясными причины, приведшие к заниженному по абсолютной величине
значению AfH°, вычисленному по данным работы [2216].
Таблица 33.7. Результаты определений Д/Н"°(УО2, г, 0) (в кДж-мояь-1)
Авторы
Берковиц, 1957 [1505]
Киллингбек, 1965 [2998]
Францева и др., 1969 [1062]
Фарбер и др., 1972 [2216]
Метод
Масс-епектрометрический, 2VO(k)=V(r)+VO2(r),
комбинирование измерений VO(K)=VO(r) A872
и 1873 К, 2 точки) и VO2(r)+V(r)=2VO(r)
A945 К, 2 точки)
Эффузионный, V2O3(K)=VO2(r)+VO(r), 1922—
2270 К, 58 точек
Масс-спектрометрический, 0,5 V2O4(k)=VO2 (г),
1799—1863 К, 3 точки
То же, VaO8(K)=VOa(r)+VO(r), 2031—2177 К,
5 точек
Масс-спектрометрический, 2AlO(r)-[-V(r)=
= 2Al(r)+VO2(r), 2270 К, 1 точка
AfH°(VO2, г, 0)
II закон
-163+27
—173
—204
—
III закон
—236+30
—210+18
—235+12
—232+18
—184
V2O3(k, ж). Термодинамические свойства кристаллического и жидкого триоксида диванадия
в стандартном состоянии при температурах 100—5000 К приведены в табл. 913.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 33.1. В интервале 0—174 К за стандартное состояние в справочнике принята моноклинная
модификация V2O3(k), а в интервале 174—2230 К — гексагональная модификация (структурный
тип а-А12О3) *.
При 7^298,15 К термодинамические функции V2O3(k) вычислены на основании измерений
теплоемкости, проведенных Березовским и Пауковым [125, 132] A2—301 К, образец V2O3 001+0 004
с общим содержанием примесей <Х),15%). При Т^.17 К теплоемкость V2O3(k) определялась по
уравнению С°(Г)=38,5-10~6-Г3 (Дж-К-моль), основанному на данных [125, 132], полученных
в интервале 12—30 К. Температура фазового перехода V2O3(k) принята равной 174 К по
максимуму теплоемкости в измерениях Березовского и Паукова [125, 130, 132]. Для менее чистых
образцов V2O3 и для образцов, состав которых значительно отличается от стехиометрического, в ряде
Х Бри температурах 400—600 К происходит постепенный переход от корундовой фазы V2O3 (к) а-типа к
корундовой фазе 8-типа, которые незначительно различаются в структурном отношении. Об этом переходе см. [1889
2281].
65
Глава 33
работ были получены значения Tir в интервале 150—170 К и даже 113—128 К (см., например.,
13143, 4571, 4649]). Энтальпия фазового перехода V2O3(k) при 174 К принята равной 1,87 +
+0,04 кДж-моль на основании данных Березовского и Паукова [125, 132]. Близкая величина
получена в работе [125] по данным [1316] A,84 кДж-моль), а также в работах [680, 681]
A,90 кДж-моль) и [1889] A,91 кДж-моль). Менее точные значения были получены в работах
[1078, 2910, 2977, 2978].
Погрешности значений ?°B98,15 К) и #°B98,15 К)—#°@), приведенных в табл. 33.1,
оцениваются в 0,4 Дж-К^-моль и 0,06 кДж-моль. При 2"^298,15 К теплоемкость V203vk)
измерялась также в работах [1316] E7-287 К), [1325] @,1-0,4 К), [3470] D-20 К), [3469] @,3-10 К),
[1078] A23-184 К), [2977, 2978] A00-700 К), [1888, 1889] A40-590 К), [680, 681] F0-300 К),
[355] (80—300 К). Измерения [1325, 3469, 3470] для образцов V2O3 высокой чистоты (99,99%)
находятся в согласии с данными [125, 132], в остальных работах исследовались более грязные
образцы V2O3.
В интервале 298,15—2230 К для теплоемкости V2O3(k) принято уравнение (см. табл. 33.1),
полученное совместной обработкой результатов измерений энтальпии в работах Кука [1915]
C69—1760 К, образец состава V2O3012), Яковлевой и Красиловой [1191] D45—1048 К) и Слю-
саря и др. [942, 943] E74—2218 К,'образец состава V2O302, содержание примесей 0,073%). Это
уравнение описывает экспериментальные данные с точностью в 3—4%. При Т >• 298,15 К
теплоемкость V2O3(k) измерялась также в работах [2823] C43-573 К), [2978] B98,15—700 К) и [18891
B98,15—600 К). Данные [2823] весьма неточны, а данные [1889, 2978] находятся в согласии с
принятым уравнением.
Температура плавления V2O3(k) принята равной 2230+20 К по данным Слюсаря и др. [942,
943] на основании результатов измерений энтальпии V2O3(k, ж) до 2276 К. Практически
совпадающее значение было получено Фридрихом и Ситтигом [2336] B240 К). Энтальпия плавления V2O3(k)
принята равной 140+10 кДж-моль в предположении, что величина AmS(V2O3, к) близка к энтро-
пиям плавления V2O4(k) и V2O5(k), равным 62 и 67 Дж-К-моль~1.
Теплоемкость У2О3(ж) оценена равной 160+10 Дж-К~1-моль~1 на основании принятой в
справочнике величины C°(V2O3, к) при температуре плавления A56 Дж-К~1-моль) и результатов
оценки C°(V2O3, ж) по методике, описанной в I томе настоящего издания A70 Дж-К-моль).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 3000 и 5000 К оцениваются в 0,3,
2, 10 и 20 Дж-К^-моль.
Ранее термодинамические функции V2O3(k, ж) вычислялись в справочниках JANAF [2835]
(до 3500 К) и Келли [2960, 2963] (до 2000 К) на основании данных [1316, 1915] для V2O3(k) и
оценок для У2О3(ж). Расхождения в значениях Ф°(Т), приведенных в табл. 913 и [2835], составляют
2,4—6 Дж-К^-моль.
Константа равновесия реакции V2O3(k, ж)=2У(г)+30(г) вычислена с использованием
значения АгН°@)=2980,096 +6,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(V2Os, к, 298,15 К) =—1216,8 + 5,0 кДж • моль.
Таблица 33.8. Результаты определений ДуЯ"°(У2О3, к, 298,15 К) (в кДж-моль-1)
Авторы
Микстер, 1912 [3522]
Руфф, Фридрих, 1914 [4088] ,
Симонсен, Улих, 1940 [4284]
Вольф, Ария, 1959 [234]
Ма, Келли, 1961 [3348]
Чарлю, Клеппа, 1973 [1816]
Сухушина, 1973 [1004]
Метод
Калориметрический, V~03(K)+2Na20,(K)+Na20(K)=
=3Na2O -V2O5(k) и V2O6(K)+3Na2O(K)=3Na2O -V2O6(k)
To же, V2O3(K)+O2(r)+Na2O2(K)=V2O5-Na2O2(K) и
V2O5(K)+Na2O2(K)=V2OB -Na2O2(K)
To же, измерение энтальпии сгорания V2O3(k), 3 опыта
То же для V2O3,0i8> 2 опыта
То же для V(k), V2O3(k), VO(k) при 303,15 К, 15 опытов
То же, У2О3(к)+О2(г)=У2О5(к), 910 К, 5 опытов
ЭДС, 1300 К, b,H°(V2O3+x, к, 298,15 К)=—1216,8+
+410,4 -х
AfH°(V2O3, к,
298,15 К)
—1152
—987
—1229+10
—1232+17
—1216,8 ±5,0
—1220,8+6,5
—1216,8±6,0
66
Ванадий и его соединения V2O4(k, ж)
Результаты определений энтальпии образования V2O3(k) приведены в табл. 33.8.
Приведенные в таблице погрешности учитывают воспроизводимость измерений и неточность
использованных в расчетах термодинамических функций и термохимических величин. Принятая величина
основана на результатах согласования методом наименьших квадратов результатов исследований
Ма и Келли [3348] (см. табл. 33.3 и текст по VO(k)). Эта величина подтверждается измерениями
Сухушиной [1004] и Чарлю и Клеппы [1816]. Менее точные, но совпадающие в пределах их
погрешностей величины были получены в работах [235, 4284]. В работах [3522, 4088] исследовались
сложные реакции, видимо, сопровождавшиеся неучтенными побочными процессами. Исследования
равновесия реакций V2O3(k) со смесями Н2+Н2О и СО+СО2 [39, 486, 700, 2933, 3052, 3480, 4362]
не позволяют получить достаточно надежные значения энтальпии образования V2Os(k), так как
в них не был установлен фазовый состав конденсированной фазы.
V2O4(k, ж) . Термодинамические свойства кристаллического и жидкого тетраоксида диванадия
в стандартном состоянии при температурах 100—5000 К приведены в табл. 914.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 33.1. За стандартное состояние в справочнике в интервале 0—338,7 К принята
моноклинная модификация УаО4(к), а в интервале 338,7—1818 К — тетрагональная модификация.
Термодинамические функции V2O4(k) при 1^298,15 К вычислены на основании измерений
теплоемкости монокристаллического образца V2O4?004±0 004 с содержанием примесей <10,13%,;
выполненных Березовским и Лукащуком [127] F—355 К). При 74^15 К использовалось
уравнение C°(J)=4,18-10-6-J3 ^Дж-К^-моль), основанное на данных [127] в интервале В—45 К.
Неточность принятых значений теплоемкости оценивается в 5% в интервале 0—20 К и от 1 до
0,1% _ в интервале 20—298,15 К. Погрешности принятых значений ?"B98,15 К) и Я°B98,15 К)—
—#°@) (см. табл. 33.1) оцениваются в 0,2 Дж-К^-моль и 0,03 нДж-моль. Теплоемкость
V2O4(k) при низких температурах измерялась также Андерсоном [1316] F1—279 К, образец, по-
видимому, содержал примесь V2O3), Ридером и др. B0—370 К, опубликовано в [1494, 3785],.
данные представлены на графике и согласуются с принятыми), Чандрашекхаром и др. [1807]
A50—730 К), Кеи и др. [1889] B00-400 К), Березовским и др. [125, 760] A2—358 К) и
Суриковым и др. [680, 681, 999] B90—380 К). Данные [125, 760] для порошкообразного образца
V204,oo4±o,oo4 c содержанием примесей ^0,15% согласуются с данными Березовского и Лукащука
[127] для СДУ2О4, к). Результаты измерений теплоемкости V2O4(k), проведенных в работах [999,
1807, 1889] методом дифференциальной сканирующей калориметрии, менее точны, чем данные
[125, 127], и приведены только на графиках.
В интервале 298,15—338,7 К термодинамические функции V2O4(k) вычислены с использованием
приведенного в табл. 33.1 уравнения для С°р{Т), основанного на данных работы [127].
Температура полиморфного превращения C38,7+0,2 К) принята на основании измерений
теплоемкости [127] и электропроводности [3022, 4142] монокристаллических образцов высокой
чистоты. Принятое значение подтверждается несколько менее точными значениями C37—341 К),
полученными в ряде работ [125, 760, 999, 1379, 2910, 2939, 3077, 3130, 3512, 3562, 3844, 4463, 4537].
Энтальпия полиморфного превращения принята равной 9,15+0,15 кДж-моль как разность
энтальпий моноклинной и тетрагональной модификации V2O4(k) при 338,7 К, вычисленных на
основании измерений теплоемкости [127] и энтальпии [1915] в интервале 310—380 К. Близкие
значения (8,53—10,13 кДж-моль) получены в работах [1807, 1889, 1915, 2910, 3844, 3874],
В работах [125, 680, 760, 2939, 3032, 3890, 3891, 3922, 3960] получены заниженные значения
C,85—7,45 кДж-моль).
В интервале 338,7—1818 К термодинамические функции V2O4(k) вычислены с использованием
приведенного в табл. 33.1 уравнения для С°р(Т), полученного совместной обработкой результатов
измерений энтальпии тетрагональной модификации V2O4(k), выполненных Куком [1915] C57—
1578 К), и ее теплоемкости — Березовским и Лукащуком [127] C46—355 К). Это уравнение
аппроксимирует данные [127, 1915] с погрешностями меньше 1%.
Температура плавления A818+20 К) и энтальпия плавления (Й2+2 кДж-моль) приняты
на основании измерений энтальпии V2O4(k, ж) в работе [1915] A830—1857 К) и значения
энтальпии V2O4(k) при 1818 К. В работе [2336] было получено значение Гт=1910 К для образца со
значительным содержанием V2O3.
67
У2О5(к, ж) Глава 33
Теплоемкость У2О4(ж) оценена равной 180+10 Дж-К-моль~1, на основании значений тепло-
емкостей V2O4(k, Тт), У2О3(ж) и У2О5(ж). Измерения энтальпии У2О4(ж) [1915] выполнены в
ограниченном интервале температур и приводят к значению C°(V2O4, ж)=213 Дж-К~1-моль.
Погрешности рассчитанных значений Ф°(Т) при 298,15, 1000, 3000 и 5000 К оцениваются в 0,2,
1,5, 12 и 27 Дж-К^-моль-1.
Ранее термодинамические функции V2O4(k, ж) вычислялись в справочниках JANAF [2835]
(до 3000 К) и Келли [2960, 2963] (до 2000 К) на основании данных [1316, 1915]. Расхождения
менаду значениями Ф°(Г), приведенными в табл. 914 и в [2835], составляют 6—13 Дж-К^-моль.
Константа равновесия реакции V2O4(k, ж)=2У(г)+4 О(г) вычислена с использованием
значения Дгйго@)=3437,928+6,4 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A^°(V2O4, к, монокл., 298,15 К) = —1432,6 ± 5,0 кДж • моль.
Результаты определений энтальпии образования V2O4(k, монокл.) представлены в табл. 33.9.
Приведенные в таблице погрешности учитывают воспроизводимость измерений и неточность
использованных в расчетах термохимических величин. При сгорании ванадия и его окислов
образуется тетрагональная модификация V2O4, при 298,15 К термодинамически менее стабильная по
сравнению с моноклинной. Поэтому приведенные в табл. 33.9 измерения, в которых исследовались
реакции с V2O4(k, тетр.), были пересчитаны с учетом принятой в справочнике энтальпии перехода
между этими модификациями. Последняя основана на результатах согласования методом
наименьших квадратов результатов исследований Ма и Келли [3348] (см. табл. 33.3 и текст по VO (к)).
Измерения Чарлю и Клеппы [1816] и Симонсена и Улиха [4284] подтверждают принятое значение.
Таблица 33.9. Результаты определений Af-ff°(V2O4, к, монокл., 298, 15 К)
Авторы
Метод
Д/Я°(У3О4, к,
ионокл., 298,15 К)
Микстер, 1912 [3522] Калориметрический, V2O4(K)+3Na2O2(K)=3Na2OX —1384
X У2О5(к)+О2(г) и V2O5(K)+3Na2O(K)=3Na2O -V2O5(k)
Симонсен, У лих, 1940 [4284] То же, V2O4(k, тетр)+1/2О2(г)=У2О6(к), 4 опыта —1432,4+7,0
Ма, Келли, 1961 [3348] То же, измерение энтальпий сгорания V(k), V2O3(k), —1432,6+5,0
VO(k), 303,15 К, 15 опытов
Чарлю, Клеппа, 1973 [1816] То же, V2O4(k, тетр.)+1/2О2(г)=У2О5(к), 910 К, —1436,8+6,0
5 опытов
В работе [3522] были исследованы сложные реакции, которые, по-видимому, сопровождаются
побочными процессами. Исследования равновесия реакций с участием V2O4(k) [2268, 3480, 4362]
не позволяют вычислить достаточно надежные значения энтальпии образования V2O4(k), так как
в них не был установлен фазовый состав конденсированной фазы.
V2O5(k, ж). Термодинамические свойства кристаллического и жидкого пентаоксида диванадия
стехиометрического состава в стандартном состоянии при температурах 100—5000 К приведены
в табл. 915.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 33.1. За стандартное состояние V2O5(k) в справочнике принята ромбическая модификация.
Термодинамические функции V2O5(k) при Г<^298,15 К вычислены с использованием уравнения
С^(Г)=3,20-10~4-Г3 (Дж-К^-моль) в интервале 0—15 К и уравнения С* (Г) в виде дробно-
рациональной функции шестой степени в интервале 15—298,15 К г. Эти уравнения получены на
основании измерений Суховея и др. [1005, 1006] A1—311 К, образец состава V20B>0o с содержанием
примесей <Ч),1%). Погрешность принятых значений теплоемкости оценивается в 5% в интервале
0—20 К и от 1 до 0,1% в интервале 20—298,15 К. Погрешности приведенных в табл. 33.1 значений
?"B98,15 К) и #°B98,15 К)—#°@) составляют 0,3 Дж-К^-моль и 0,04 кДж-моль.
Теплоемкость V2O5(k) измерялась также Андерсоном [1316] E7—290 К) с использованием образца,
содержащего ~0,5% примесей, но не охарактеризованного точным отношением кислорода к ме-
1 Это уравнение аппроксимирует данные [1005, 1006] в интервале 15—50 К с точностью 1,0—0,2% и в интервале
50 — 298,15 К с точностью 0,2 — 0,02%.
68
Ванадий и его соединения V4Oi0 (г)
таллу. Полученные в [1316] значения C°(V2O5, к) систематически превосходят на 2% данные
[1005, 1006].
В интервале 298,15—954 К термодинамические функции V2O5(k) вычислены по уравнению для
С°р(Т), полученному обработкой результатов измерений энтальпии, выполненных Слюсарем и др,
[942, 943] E77—938 К) на образце, характеризующемся строго стехиометрическим составом
(содержание примесей <Ч),1%). Энтальпия V2O5(k) при Г>298,15 К измерялась также Куком
[1915] C72—921 К) для образца с содержанием примесей ~0,1% и субстехиометрическим
соотношением кислорода к металлу.
Температура плавления (954+5 К) принята на основании измерений,, проведенных на
образцах V2O5(k) стехиометрического состава [942, 943, 1778, 3077, 4417]. В более ранних определениях
Тт(У2®ы к) (см. [1722, 2072, 2424, 4198]) были получены преимущественно меньшие значения
(931—943 К), соответствующие субстехиометрическим составам образцов. Энтальпия V2O5
измерялась Куком [1915] (946—1513 К) и Слюсарем и др. [942, 943] (960—1095 К). На основании
этих данных в справочнике принято: ДИД°(У2О5, к) = 64+5 кДж-моль и C°p(V2O5, ж)=190 +
+4 Дж-К-моль. Погрешности приведенных в табл. 915 значений Ф°(Т) при 298,15, 1000*
3000 и 5000 К оцениваются в 0,2, 2, 15 и 30 Дж-К^-моль-1.
Ранее термодинамические функции V2O6(k, ж) вычислялись в справочнике JANAF [2835]
(до 3000 К) и Келли [2960,2963] (до 2000 К) по данным [1316, 1915]. Расхождения в
значениях Ф°(Г), приведенных в табл. 915 и [2835], составляют 1—8 Дж-К^-моль.
Константа равновесия реакции V2O6(k, ж)=2У(г)+5 О(г) вычислена с использованием
значения АгН°@)=3803,260+6,8 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(V2O6, к, 298,15 К) = —1551,0 + 5,5 кДж • моль.
Результаты определений энтальпии образования V2O6(k) представлены в табл. 33.10.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, а [в случае работы
[3348] — погрешности, выявленные при согласовании полученных в ней величин для других
оксидов ванадия. Принятая величина основана на результатах согласования методом наименьших
квадратов измерений Ма и Келли [3348] (см. табл. 33.3 и текст по VO(k)). Результатам работы
[3348] отдается предпочтение, несмотря на то, что в некоторых других работах (см., например,
[230]) была получена лучшая воспроизводимость измерений. Этот выбор обеспечивает взаимную
согласованность значений, принятых для оксидов ванадия. Принятая величина подтверждается
совпадающими с ней в пределах погрешностей измерениями Волковой, Гельда [230], Голутвина,
Козловской [300] и Симонсена, Улиха [4284]. В более ранних работах [3522, 3609, 4088, 4093]
были получены существенно менее точные значения. ¦
Таблица 33.10. Результаты определений АуН°(\2Оъ, к, 298, 15 К) (в кДж-моль)
Авторы
Симонсен, Улих, 1940 [4284]
Вольф, Ария, 1959 [234]
Морозова, Егер, 1960 [699]
Голутвин, Козловская, 1960 [300]
Ма, Келли, 1961 [3348]
Волкова, Гельд, 1963 [230]
Метод
Калориметрический, 2У(к)+2,5О2(г)=У2О6(к), 3 опыта
То же, 2V(K)+2,5O2(r)=V2O5(K), 2 опыта
То же, 2У(к)+2,5О2(г)=У2О6(к), 1 опыт
То же, 2У(к)+2,5О2(г)=У2О5(к), 6 опытов
То же, измерение энтальпий сгорания V(k), V2O3(k),
VO(k), 303, 15 К, 15 опытов
То же, 2У(к)+2,5О2(г)=У2О6(к), 8 опытов
AfH°(V2O5, к,
298,15 К)
—1560,6+8,5
—1573,2±8,5
—1560,6
—1556+11
—1551,0+5,5
—1556+4,0
V4O10(r). Термодинамические свойства газообразного декаоксида тетраванадия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 916.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 33.6. На основании выполненного Битти и др. [1437] исследования ИК-спектра
молекул V|6O10 и Vi8O10, изолированных в азотной матрице, в настоящем справочнике принимается,
что в основном состоянии "К1 А молекула V4O10 имеет тетраэдрическую структуру (симметрия Td),
69
V4O10(r)
Глава 33
аналогичную структуре молекулы Р4О10. Приведенное в табл. 33.6 произведение моментов инерции
V4O10 вычислено с использованием структурных параметров: r(V=O)=l,55+0,05 A, r(V—О) =
= 1,72+0,05 А, -$0—V—0 = 117 + 5°, ^V—О—V=124 + 10°, оцененных на основании
соответствующих данных для молекул Р4О10, РО и VO (см. гл. 14 и табл. 33.4). Погрешность принятого
значения 1л1в1с составляет 2-Ю11 г3-см6. Приведенные в табл. 33.6 значения основных частот
основаны на результатах их определения в работе [1437] и оценках, базирующихся на
соответствующих частотах молекулы Р4О10. Битти и др. [1437] наблюдали в ИК-спектре молекул V4O10,
изолированных в матрице из азота, две интенсивные полосы с максимумами при 1029,9 и 828,4 см
и слабую полосу при 626 см, которые они отнесли к колебаниям концевой связи V=O (v16),
колебанию, связанному с группировкой V—О—V(v14) и деформационному колебанию v13.
Сравнение с соответствующими частотами Р4О10 (см. т. I, табл. 14.6) показывает, что значения частот
V4O10 составляют ~75+-80% от значений соответствующих частот Р4О10. С этим коэффициентом
были оценены значения остальных частот, приведенных в таблице1. Погрешности
экспериментально наблюдаемых частот с учетом матричного сдвига составляют 10—20 см, погрешности
оцененных частот могут достигать 20 %.
Термодинамические функции V4O10(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 10), A. 122)—A. 124), A. 128) и A. 130).
Погрешности рассчитанных функций обусловлены неточностью принятых молекулярных постоянных
и приближенным характером расчета. При 298,15, 3000 и 6000 К погрешности в значениях Ф°(Г)
оцениваются в 20, 40 и 60 Дж-К~1-моль~1.
Ранее термодинамические функции V4O10(r) вычислялись Фарбером и др. [2216] A000—1300 К)
по молекулярным постоянным, близким к принятым в справочнике. Расхождения в функциях,
приведенных в работе [2216] и в табл. 916, не превышают 2 Дж-К-моль~1 в значениях Ф°(Т).
Константа равновесия реакции V4O10(r)=4V(r)+10O(r) вычислена с использованием значения
ДГЯ°@)=7323,830±40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(У4О10, г, 0) = —2800 ± 40 кДж • моль".
Принятое значение основано на результатах исследований равновесия реакции V2O5(k, ж) =
=1/2V4O10(r), представленных в табл. 33.11. Приведенные погрешности учитывают
воспроизводимость измерений, неточность сечений ионизации и оцененные систематические погрешности
измерений (в работе [802] не были приняты меры, обеспечивающие достижение насыщенного давления
паров). Принятая величина основана на совпадающих результатах работ [802 (после учета
термодиффузии), 920, 2216], причем основной вес придавался наиболее точным измерениям Семенова
и др. [920]. Основным источником погрешности этой величины является неточность
термодинамических функций.
Таблица 33.11. Результаты определений к^Н°(\т.\О1В, г, 0) (в кДж • моль-1)
Поляков,
Авторы
1946 [802]
Семенов и др., 1970 [920]
Чижиков
Фарбер и
и др., 1970 [1134]
др., 1972 [2216]
Протока, 973—1473 К, 5 точек
То же, с учетом термодиффузии
Протока, 1215—1530 К, 7 точек
Масс-спектрометрический, 835—
представлены в виде уравнения
То же, 1003—1205 К, 11 точек
(см.
940 К
[749])
, данные
Д/Яо^Ою, г, 0)
II закон
—3083+20
—2815+55
—2789 ±4
—2869
-2820+4
III закон
—2843+20
—2805+7
—2795,7+1,5
—2779 ±10
-2799+7
Частоты, близкие по величине, в табл. 33.8 объединены и приведены с суммарным статистическим весом.
Глава 34
НИОБИЙ И ЕГО СОЕДИНЕНИЯ
Nb(K, ж), Nb, Nb+, NbO(K, ж), NbO, NbO2(K, ж), NbO2, Nb2O5(K, ж)
ЗВ настоящей главе рассматриваются пять веществ — ниобий и его основные кислородные
соединения в конденсированном и газообразном состояниях. Для четырех веществ представлены
данные о свойствах в конденсированном состоянии — это ниобий и три наиболее стабильных и
изученных оксида стехиометрического состава (NbO, NbO2 и Nb2O5). Они имеют области гомогенности
в пределах NbO094—NbOx 04, NbOl94—NbO209 и Nb2O4 485—Nb2O6>0. Для четырех веществ в
справочнике приводятся данные о свойствах в газообразном состоянии, причем для трех (Nb, Nb+,
NbO) — при температурах вплоть до 10 000 К.
Для элементарного ниобия в Приложении 3.10 приведены сведения о его критических постоян-
аых.
В справочнике не включены данные для субоксидов ниобия (Nb2O, Nb4O, Nb6O), см. [549а],
и высших оксидов (Nb12O29, Nbl9046, Nb22O62, Nb47Oll6, Nb53O1S2l), см. [3623, 3656, 4170]. В
литературе упоминается также соединение Nb2O3, см., например, [549а], однако специальное
исследование [3656] показало, что индивидуальная фаза такого состава не существует.
Nb(K, ж). Термодинамические свойства кристаллического и жидкого ниобия в стандартном
•состоянии при температурах 100—6000 К приведены в табл. 917.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 34.1. За стандартное состояние Nb(K) в интервале 0—2750 К принята кубическая
модификация (структурный тип a-Fe). При 9,28 К Nb(K) переходит в сверхпроводящее состояние г.
Таблица 34.1. Принятые значения термодинамических величии для ниобия и его соединений
в кристаллическом и жидком состояниях
Вещество
*
Nb
NbO
NbO2
" Nb2O6
Состояние
к, куб.
ж
к, куб.
ж
кП, тетр.
к1, тетр.
ж
к, монокл.
ж
-К)-
ю
&5 1
кДжХ
X моль 1
5,22
—
7,2
9,267
—
22,24
—
Примечание. СЦТ)=а+Ь -Т—с
Nb: a
NbO2: *
Nb2O6: a
d=2,101 -10"
1 d=66,400-l
<2=-798,74S
(Гв.
К
ю
оо"
стэ
SS
о
К
,15
00
ОЗ
о а.
Дж • К • моль"
36,27 24,435
— —
46 41
— —
54,29 57,660
—
_ _
137,1 131,95
— —
j. -{-а ± -\-е ± j-y
Коэффициенты s
НИИ ДЛЯ С°
а
Ь-103
26,472 —0,408
41,8 —
42,184 9,93С
65 —
89,363 —61,170
85,468 1,339
100 —
—25,754 648,992
200 - —
ура вне-
(Г)
с • 10~5
Интервал
температур
или
Т
1 m
К
1,869 а 298,15-2750
— 2750—6000
3,684 298,15—2217
— 2217—5000
17,217 а 298,15—1082
20,541 1082—2360
— 2360—5000
—21,868 а 298,15—1783
— 1783—5000
•Г* (в Дж-К-моль).
! -КГ», е=423,495 -1(Г8, /=—77,695 -КГ1*.
2750
—
2217
—
1082
2360
—
1783
—
или
кДжХ
X моль
31
—
50
—
2,5
75
—
101
—
1 Температура перевода Nb в сверхпроводящее состояние определена Леуполдом и др. [3237] для монокристал-
личестгого образца, очищенного зонной плавкой. В ряде других работ (ссылки см. в [1009]) получены более
низкие значения Tfr для менее чистых образцов ниобия.
71
Nb(K, ж) Глава 34
При Т <1 298,15 К термодинамические функции Nb(K) вычислены на основании измерений
теплоемкости, выполненных Клузиусом и др. [1882] A1—273 К) и Рахменкуловым и Пауковым
[854] A1-346 К), а также Шеном и др. [4257, 4258] @,7—1,4 и 0,3-25 К), Сенозаном [4233]
@,25—25 К), Леуполдом и др. [2658, 3236, 3237] @,5-10, 8,95-11,5, 1,1-11,5 К), Да Сильва
и др. [1991, 1992] A—10 К) и Ватанабэ [4669] D—17 К) для образцов №э(к) чистоты 99,7—99,99%.
Погрешность соответствующих значений теплоемкости 1ЧЬ(к) в интервале 10—298,15 К оценивается
в —0,5%. Погрешности приведенных в табл. 34.1 значений ?°B98,15 К) и Я°B98,15 К)—#°@)
равны 0,2 Дж-К~1-моль и 0,02 кДж-моль. Теплоемкость менее чистых образцов N1d(k)
измерялась в работах [1689] F4-76 К), [1688] B,5-20 К), [1690] B,5-11 К), [1843] A,5-30 К)г
[1618] «2 К), [1619] «1,7 К), [3563] (9-19 К), [1993] @,2-2,8 К), [2678] @,4-4,2 К),
[1702] A,2-20 К), [3445] C,2-9,2 К), [4233, 4257] @,3-25 К), [1739] F0-340 К).
В интервале 298,15—2750 К термодинамические функции ЫЬ(к) вычислены на основании урав-,
нения для С°р(Т) (см. табл. 34.1), полученного совместной обработкой результатов измерений
энтальпии, выполненных Егером и Веенстрой [2818, 2820] C73—1775 К), Гельдом и Кусенко [2681
D33-1840 К), Хокинсом и Орром [2593] C58-1416 К), Кириллиным и др. [505] E92-1939 К),.
Конуэем и Хейном [1912] A300—2000 К), Березиным и др. [118, 123, 4254] A650—2707 К) и Бет-
цом и Фробергом [1537] B031—2699 К) для образцов Nb(K) чистотой лучше 99,5%. При этом
результаты измерений энтальпии Nb(K), полученные в работах [505, 1912] для температур 2039—
2591 К и 2100—2600 К соответственно, не использовались как явно завышенные. Не учитывались
также недостаточно точные измерения энтальпии ]ЧЬ(к) в работах [3175] D54—1885 К), [527]
D04—2677 К) и [4247] B300, 2650, 2740 К). Принятое уравнение для С°р{Т) при Т > 298,15 К
согласуется с результатами измерений теплоемкости калориметрическим методом [854] (до 346 К)
и [916, 4890] C70—1291 К), а также модуляционным методом [641, 1054, 3318]. В ряде работ,
см., например, [573, 575, 1055, 1781, 3100], получены неверные значения теплоемкости Nb(K) x.
Температура плавления ниобия 2750 + 10 К принята по согласующимся между собой измерениям
Цезерляна [1782] B750 К, 99,9% Nb), Пемслера [3798] B744 К, 99,87%Nb), Хаузнера [25891
B752 К), Руди и Прогульского [4084] B752 К, 99,7% Nb), Кенисарина и др. [121, 496] B742 К,,
99,8% Nb), а также результатам измерений Скофилда [4203] для образца ниобия, содержащего 2%
примеси тантала B745 К). Принятая величина совпадает с рекомендованной в качестве вторичной
реперной точки МПТШ-68, редакция 1975 г. [4888а] B750 К). В нескольких работах были получены
более высокие значения ^(Nb) — на образцах, чистота которых авторами работ оценивалась
выше 99,9% Nb: [3964] B783 К), [563, 752] B797 К) и [4481] B862 К). Однако эти данные
недостаточно надежны в методическом отношении. Менее надежные измерения ^(Nb) проведены также
в работах [804, 898, 1069, 2892, 2986, 4027, 4406, 4726, 4730].
Энтальпия №э(ж) измерялась методом левитационной калориметрии Боннелом и др. [1617]
B738—3292 К), Березиным и др. [118, 4254] B742—2753 К) и Бетцом и Фробергом [1537, 1538]
B708—3159 К) и методом импульсного нагрева Шейнером и др. [4247] B740—4110 К). Наиболее
точные и полные данные получены Бетцом и Фробергом [1537, 1538]. С ними согласуются
результаты несколько менее точных измерений, выполненных Боннелом и др. [1617]. Результаты
измерений [118, 4254] занижены по сравнению с данными [1537, 1617] на 6,5 кДж-моль,
по-видимому, из-за недостаточно полного учета тепловых потерь. Данные Шейнера и др. [4247]
систематически завышены при Т > 2900 К, по-видимому, вследствие неравновесности Nb(w)
в условиях эксперимента. Принятые значения Am#°(Nb, к)=31,0±1,2 кДж-моль и C°(Nb, ж) =
=41,8 + 1,7 Дж-К'Моль основаны на данных [1537, 1538] для энтальпии №)(ж) и уравнении
для C°(Nb, к). Значение Am#°(Nb, к) находится в близком согласии с величинами, полученными
в работах [1537] C0,5 кДж-моль) и [1795] C1,5 + 1,6 кДж-моль). Менее точные значения
найдены в работах [665, 667, 1617, 3387] C3-34,6 кДж-моль) и [118, 893, 894, 1784, 4247, 4254,
4255] B7—29 кДж-моль). Принятое значение C°(Nb, ж) согласуется с основанным на данных
[1617] D0,6 + 5,3 Дж-К-моль~1). Измерения [4247] приводят к более высокому значению
E6,5 Дж-К-^моль). Данные для C°(Nb, ж), полученные в работе [4889] B800—5000 К),
соответствуют, по-видимому, неравновесному состоянию Ш>(ж).|
Погрешности приведенных в табл. 917 значений Ф°(Т) при 298,15, 1000, 3000 и 6000 К
составляют 0,2, 0,4, 1 и 4 Дж-К-моль~1.
1 В интервале 2600—2700 К в работе [4889] получены аномально большие значения теплоемкости Nb (к),
соответствующие, по-видимому, неравновесному состоянию Nb (к).
72
Ниобий и его соединения
Nb(K, ж)
Ранее термодинамические функции Nb(K, ж) вычислялись в справочниках JANAF [2835JT
Халтгрена и др. [27521, Шика [4181], а также Сталла и Зинке [4421], Келли [2960, 2963]
(до 3000 К) и в работе Кириллина и др. [506] (до 2740 К). Во всех справочниках принимались
оцененные значения энтальпии плавления и теплоемкости ]МЬ(ж), которые меньше принятых
в настоящем справочнике соответственно на 4 кДж-моль и на 8 Дж-К~1-моль~1. Расхождения
между значениями термодинамических функций Nb(K, ж), приведенными в [2835] и в табл. 917,
составляют 0,1—2 Дж-К~1-моль~1 в значениях Ф°(Г). Соответствующие расхождения между
табл. 917 и данными справочников [2752, 2960, 4181, 4421] также не превосходят 2 Дж-К~1-моль~1.
В настоящем справочнике в качестве стандартного состояния ниобия принято кристаллическое
состояние и в соответствии с этим
к) = 0.
Давление пара ниобия Nb(K, ж)=Ш)(г) вычислено с использованием принятой в справочнике
энтальпии сублимации
ДгЯ°@) = As#°(Nb, к, 0) = 720 ± 10 кДж • моль.
Результаты расчетов энтальпии сублимации ниобия на основании измерений давления его паров
представлены в табл. 34.2. В этой таблице в качестве погрешностей приведены воспроизводимости
Таблица 34.2. Результаты определений Asfi"°(Nb, к, 0) (в кДж-моль)
Авторы
Метод
ЫЪ, к, 0)
II закон III закон
Рейман, Грант, 1936 [3964]
Шпейзер и др., 1959 [4360]
Власов, 1960 (см. [714])
Вернер, Вейкфнлд, 1962 [4744]
Щукарев и др., 1962 [1186]
Кауфман, Клогерти, 1963 [2937] (см.
[2834, 4403])
Шир, Файн, 1965 [4179]
Францева, 1968 [1061]
Коч и др., 1968 [3055]
Стормс и др., 1969 [4403]
Крикориан и др., 1969 [3111]
Гупта, Гингерич, 1979 [2533]
Испарение с открытой поверхности, 2363—2674 К,
данные представлены в виде уравнения
То же, 2367—2600 К, 16 точек
Эффузионный с использованием радиоактивного
изотопа Nb, 2554—2685 К, 4 точки
Испарение с открытой поверхности, 2203—
2604 К, данные представлены в виде уравнения
Масс-спектрометрический, испарение с открытой
поверхности, 2246—2389 К, 10 точек
Испарение с открытой поверхности, 2456—2691 К,
8 точек, измеренные давления не указаны
Измерения термоэмиссионного тока Nb+ с
поверхности Nb и работы выхода электронов,
1990—2175 К
Масс-спектрометрический, 2758—2964 К
Испарение с открытой поверхности, 2745—3146 К,
15 точек
Масс-спектрометрический, интервал температуры
не указан
Испарение с открытой поверхности NbC, Nb2C
с учетом активности Nb, 2738 и 2752 К
Масс-спектрометрический, испарение с
поверхности, измеренные давления и интервал
температуры не указаны
Масс-спектрометрический, 2290—2677 К, 9 точек
780
737,8
769+30
595+217
765
729 ±7
696 ±44
719+19
733+27
635+45
735+6
—
712+22
707+27
718,7+1,2
798,0+7,3
728,3
—
731
—
730,3+3,4
—
733,7+4,1
752+10
710.3+8.
измерений, в случае масс-спектрометрических измерений в них также включены погрешности
сечений ионизации. Наиболее точные и детальные измерения были выполнены методом испарения
с открытой поверхности [3055, 4360, 4403, 4744] и масс-спектрометрическим методом [2533]. Их
результаты расходятся больше, чем это можно объяснить приведенными в таблице погрешностями,
что свидетельствует о наличии систематических ошибок. Можно предполагать, что коэффициент
испарения ниобия меньше единицы и соответственно энтальпии сублимации, вычисленные по
данным, полученным методом испарения с открытой поверхности, — завышены. Однако никаких дока-
73
(г), Nb+(r) Глава 34
зательств справедливости такого предположения нет. Принятая величина является средней из
полученных двумя указанными методами. Ее погрешность оценена с учетом отмеченной выше
противоречивости имеющихся данных и неточности использованных в расчетах термодинамических
функций.
Nb(r). Термодинамические свойства газообразного ниобия в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 918.
Для расчета термодинамических функций использованы уровни энергии х атома Nb с
суммарным статистическим весом 7460 в интервале 0—55 000 см, т. е. ниже первого потенциала
ионизации. Эти уровни соответствуют валентным электронным конфигурациям . . .4d45s (к ней относится
основной уровень ЮЧг), . . .4d35s2, . . Ad35p2, . . Ad\ . . Ad*5l ((Z=l-f-4), . . Ad?5s5l (?=l-f-4).
Принятые величины основаны на рекомендации Мур [3550] и на оценках для ряда ненаблюдав-
шихся электронных состояний по методам, изложенным в Приложении 6. Оцененные уровни имеют
энергию от 13 000 см и выше с суммарным статистическим весом 5118. Погрешности оценки
энергии лежат в пределах 500—4000 см.
Термодинамические функции Nb(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Т <^ 2000 К обусловлены преимущественно неточностью фундаментальных
постоянных, а при более высоких температурах — неточностью оценки энергии ряда уровней
и пренебрежением ридберговскими состояниями. Погрешности значений Ф°(Г) и S°(T) при Т <
< 2000 К составляют 0,02 Дж-К^-моль. При 3000, 6000 и 10 000 К погрешности значений
Ф°(Г) оцениваются в 0,1, 0,5 и 1 Дж-К-моль.
Ранее термодинамические функции Nb(r) вычислялись в таблицах JANAF [2835], справочниках
Сталла и Зинке [4421] до 3000 К и Шика [4181] до 6000 К, а также в работах Каца и Маргрейва
,12934] до 2000 К, Кольского и др. [3067] до 8000 К и Хилзенрата и др. [2652] до 10 000 К. Все
расчеты основаны на данных, приведенных в справочнике Мур [3550]; при этом в [2835] уровни
с энергией выше 14 899,26 см были усреднены по интервалам 4000 см с соответствующими
суммарными статистическими весами. При Т <[ 3000 К результаты всех расчетов, за исключением
[3067], согласуются с данными табл. 918 в пределах 0,02—0,03 Дж-К-моль. В случае [3067]
из-за использования завышенного значения R расхождения в 2—3 раза больше. При более высоких
температурах расхождения возрастают вследствие того, что в настоящем справочнике учтены нена-
блюдавшиеся уровни энергии, и составляют для [2652] 0,2 и 0,8 Дж-К-моль~1 в Ф°(Т) при 6000
и 10 000 К.
Энтальпия образования Nb(r)
AfH°{Nb, г, 0) = 720 ±10 кДж.моль
соответствует принятой энтальпии сублимации Ш)(к).
гй>+(г). Термодинамические свойства газообразного положительного иона ниобия в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 919.
Для расчета термодинамических функций использованы уровни энергии х иона Nb+ с
суммарным статистическим весом 2893 в интервале 0—78 000 см. Эти уровни относятся к валентным
электронным конфигурациям . . Adi (к ней относится основной уровень 5ZH), . . Ad25s2, . . АйяЫ
(/=0-^-2) и . . Ad25s5p. Принятые величины основаны на рекомендации Мур [3550] и на оценках
для ряда ненаблюдавшихся электронных состояний. Для электронных конфигураций . . Ad'35l
(I=0-^-2) и . . Ad2bsbp оценка выполнена методами, изложенными в Приложении 6, а для
конфигураций . . Ad* и . . Ad25s2 — сравнением энергии соответствующих состояний Ti, Zr, Hf и V+,
Nb , Ta+. Оцененные уровни имеют энергию от 20 000 см и выше с суммарным статистическим
весом 1720. Погрешности этих оценок лежат в пределах 500—5000 см". Состояния Nb+,
относящиеся к другим валентным конфигурациям, имеют высокую энергию и не принимались во
внимание при расчете термодинамических функций.
Термодинамические функции Nb+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т <^ 3000 К обусловлены главным образом неточностью значений фундаментальных
1 См. примеч. 1 к тексту по Сг (г).
74
Ниобий и его соединения 1Ш)(к, ж)
постоянных и не превосходят 0,02—0,03 Дж -К -моль. При более высоких температурах
погрешности возрастают из-за неточности оценок ненаблюдавшихся уровней энергии. При 6000 и 10 000 К
-они составляют 0,03 и 0,1 Дж-К^-моль в значениях Ф°(Т) и 0,1 и 0,3 Дж-К-моль~1 в
значениях S°(T).
Ранее термодинамические функции Nb+(r) вычисляли Хилзенрат и др. [2652] (до 10 000 К)
и Грин и др. [2473 ] (до 50 000 К) на основании данных Мур [3550 ]. Расхождения между значениями
термодинамических функций, приведенных в табл. 919 и в работах [2473, 2652], обусловлены
более полным учетом уровней энергии Nb+ в настоящем справочнике. Они составляют 0,05—
0,2 Дж-К^-моль в значениях Ф°(Т).
Константа равновесия реакции Nb+(r)+e(r)=Nb(r) вычислена с использованием значения
Дг//°@) =—664 + 8 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации атома Nb
70(Nb)=664±8 кДж-моль-1я«55 500±700 см.
Эта величина была рекомендована Каталано и Рико [1767] на основании сравнительного
анализа спектров элементов с достраивающейся 4й-оболочкой. Погрешность принятой величины
оценена по значению /0(Nb)=55 200 + 700 см, подученному при подготовке настоящего справочника
ъ результате расчета по двум членам (п=Ъ и 6) ридберговской серии . . .4d4EZ>)«s eD в спектре Nb
* использованием разностей смежных значений квантовых дефектов, найденных по данным [3550]
для Rb, Sc, Mo, Pd и Ag. Раух и Аккерман [3944] методом электронного удара получили
существенно более низкое значение, равное 637 кДж-моль.
Принятому значению 70(Nb) соответствует
bfH°(Nb+, г, 0) = 1384 ±13 кДж.моль.
NbO(K, ж). Термодинамические свойства кристаллического и жидкого оксида ниобия стехио-
метрического состава в стандартном состоянии при температурах 298,15—5000 К приведены
¦в табл. 920.
Значения постоянных, использованные для вычисления термодинамических функций,
приведены в табл. 34.1. В справочнике за стандартное состояние NbO(K) принята кубическая
модификация (структурный тип NaCl) с упорядоченным расположением вакансий в подрешетках, общее
содержание которых определяется в 25% [3656]. При 1,38 К NbO(K) переходит в сверхпроводящее
состояние [2751]. Теплоемкость NbO(K) при низких температурах измерялась только в работе
Оказа и Кеезома [3711] в узком интервале температур от 0,4 до 1,8 К. Поэтому в настоящем
справочнике термодинамические функции NbO(K) при Т <3 298,15 К не рассчитывались. Приведенные
в табл. 34.1 значения С;B98,15 К)=41 + 1 Дж-К^-моль, S°B98,15 К)=46+5 Дж-К^-моль,
,Я°B98,15 К)—//°@)=7,2+0,5 кДж-моль оценены на основании графической интерполяции
по принятым значениям этих величин для №Ь(к), NbO2(K) и Nb2O5(K). Соответствующее значение
¦С°B98,15 К) согласуется с зеличиной, вычисленной Гельдом и Кусенко [268] D1,25 Дж-КХ
Хмоль) на основании измерений энтальпии NbO(K) в интервале 420—1700 К. Значение
5°B98,15 К) совпадает с величиной, оцененной авторами таблиц JANAF [2835], и находится
в соответствии с данными по равновесиям реакций с участием NbO(K).
В интервале 298,15—2217 К для теплоемкости NbO(K) принято уравнение, полученное
совместной обработкой результатов измерений энтальпии, выполненных Гельдом и Кусенко [268]
.D20—1700 К) для образца NbO1>01(K), однофазность которого была подтверждена
рентгенографическим исследованием, и значений энтальпии NbO(K), найденных графической экстраполяцией
данных 1268) в интервале 1700—2217 К.
^Температура плавления B217 + 15 К) принята как среднее из измерений, проведенных
Эллиоттом [2170] B222* К) и Колчиным и Сумароковой [537] B212 + 15 К) (с учетом поправки на
перерасчет к шкале МПТШ-68). Энтальпия плавления NbO(K) оценена равной 50 + 10 кДж-моль
в предположении, что AmS(NbO, к^Д^ТЮ, к) =23,2 Дж-К^-моль.
Теплоемкость ЫЬО(ж) принята равной теплоемкости NbO(K) при Тт F5 + 7 Дж-К^-моль).
Погрешности приведенных в табл. 920 значений Ф°(Г) при 298,15, 1500 и 3000 и 5000 К
оцениваются в 3, 5, 10 и 15 Дж-К~1-моль.
Ранее термодинамические функции NbO(K, ж) вычислялись в справочниках JANAF [2835]
(до 3000 К) и Шика [4181] (до 6000 К). Расхождения функций, приведенных в табл. 920 и в [41813,
75
NbO(r)
Глава Ж
составляют примерно 4 Дж-К^-моль в значениях Ф°(Т) и S°(T). Значения S°(T), приведенные
в табл. 920 и в [2835], согласуются в пределах 0,4 Дж-К^-моль при Т <; 2000 К. При 3000 К
расхождения достигают 14 Дж-К~1-моль~1 из-за различия принятых значений &тН°¦
Принятая в справочнике энтальпия образования
&fH°(NbO, к, 298,15 К) = —406 + 6 кДж • моль
основана на результатах исследований, приведенных в табл. 34.3. Указанные в таблице
погрешности учитывают только воспроизводимость измерений. Наиболее надежные калориметрические
измерения выполнены Морозовой, Гецкиной [698] и Шефером, Лидмайером [4174], и принятая
Таблица 34.3. Результаты определений A^ifo(Nb0, к, 298, 15 К) (в кДж-моль)
Авторы
Морозова, Гецкина, 1959 [698]
Кусенко, Гельд, 1960 [610]
Шефер, Лидмайер, 1964 [4174]
Лаврентьев и др., 1961 [612]
Дробышев, Резухина, 1965 [387]
Уоррелл, 1965 [4769]
Стил, Алкок, 1965 [4383]
Игнатович, Девис, 1968 [2767]
Барри, 1968 [1403]
Хираока, Сано, 1971 (см. [2657])
Васильева и др., 1975 [188]
Соммер, Катер, 1975 [4344]
Метод
Калориметрический, 21\тЬО(к)+3/2О2(г)=
=Nb2O5(K), 3 опыта
То же, число опытов не указано
То же, NbO(K)+3/2Cla(r)=NbOCl3, 3 опыта
ЭДС, Nb(K)+FeO(K)=NbO(K)+Fe(K), 1114—
1346 К, 12 точек
То же, 1244-1378 К а
То же, Nb(K)+1/BTa305(K)=2/5Ta(K)+Nb0(K
1050—1300 К а
То же, NbO2(K)+Nb(K)=2NbO(K), 1050—
1300 К а
То же, Nb(K)+FeO(K)=Fe(K)+NbO(K),
1273 К, 1 точка
То же, Nb(K)+NiO(K)=Ni(K)+NbO(K),
1273 К, 1 точка
То же, NbO(K)+Nb2O5(K)=3NbOa(K),
1273 К, 1 точка
То же, Nb(K)+FeO(K)=Fe(K)+NbO(K),
1074 "Н7Ч К а
j-\J 1 О iulO XX
То же, 1000—1400 К а
AfH°(NbO,
II закон
—406
-397
к, 298,15 К)
III закон
7+7,9
7+2,1
-405,8+5,8
-415,7+10,4
-416,7
), —425,9
—429
—
—
-420,6
—425,4
То же, Nb(K)+i/3Cr203(K)=Nb0(K)+2/sCr(K), —418,8
1177 1388 К а
То же, Nb(K)+FeO(K)=Fe(K)+NbO(K),
({ой 1241 К а
-L х ij\J Х^ Ил. х\
То же, 1089-1426 К а
а Данные представлены в виде уравнения.
-414,2
—424,3
—423,8+0,7
—418,7
-420,8
—422+19
-419,0
-418,4
-421,3
-418,9
—416,4
-418,9
-418,4
-419,8
величина является средней из полученных в этих работах. Большая группа исследований методом
ЭДС приводит к хорошо согласующимся между собой результатам, погрешность которых,
связанная с неточностью термодинамических функций, составляет 4—6 кДж -моль. Возможным
объяснением различия величин, вычисленных из калориметрических измерений и из измерений методом
ЭДС, является то, что в последних использовались электроды, полученные спеканием Nb и NbO,
и измерения могут относиться к оксиду нестехиометрического состава.
Давление пара в реакции NbO(K, ж)=№Ю(г) вычислено с использованием значения АгН°@) =
= AsH°(NbO, к, 0)=615.422 + 17 кДж-моль, соответствующего принятым в справочнике
энтальпии образования NbO(K) и энергии диссоциации NbO(r).
NbO(r). Термодинамические свойства газообразного оксида ниобия в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 921.
В табл. 34.4 приведены молекулярные постоянные, принятые в справочнике на основании
анализа результатов исследований электронного и ИК-спектров NbO, спектра ЭПР и приближенных
Г-7 ft
/О
Ниобий в его соединения
АШ
В4Ф
от
0
15 000 в> г
17 000
18 500в
21385,3
21400в
989,0
850,5
3,83
3,37
0,4321
0,4001
2,1
1,9
а оцененные электронные состояния:
2,6
NbO(r)
Таблица 34
Состояние
.4.
Молекулярные
Те
постоянные
93Nb160
СМ
?е а± ¦ 103
De ¦ 107
ге
А
2,2б 1,690
1,757
Состояние а2 2 Ь2 А
Т„ см 4500 7000
С2Г 2ф5 2Н 2Д B) 2Д5 2ф 2 S-? 2^ 2ф
9500 17 000 19 000 22 000 26 000
*Д 2Г, 3А B) 22+
28 000 31 000 39 000
приведено значение Do; в см. текст; г А=700 см 1.
•оценок. В электронном спектре NbO проанализирована только одна система полос С42 ~ ^* Z42 ~
[1678, 3929, 3930, 3931, 3932, 3933, 3987, 4574]. Кроме того, в спектре испускания были
зарегистрированы многочисленные неинтерпретированные полосы, отнесенные к молекуле NbO [2374,
3933, 3934], а в спектре поглощения молекул NbO, изолированных в низкотемпературной матрице,
помимо системы С42~—Х42~, были выделены 6 систем или подсистем полос, обозначенных как
переходы А, А', В, С, D и X [1678]. Квантовомеханические расчеты молекулы NbO не
проводились. Исследования спектров показали, что основным состоянием молекулы NbO является
состояние 42~ с большим сверхтонким расщеплением. Энергии ненаблюдавшихся состояний,
аналогичных состояниям молекулы VO и принадлежащих конфигурациям .. .cjsS|, . . .0^, .. .§|тс , . . .о|а , ...
•3А*ар (см- табл. 33.4), были оценены в предположении, что: 1) состояния
о соответственно; 2) со-
А% и
- иС42-
р
принадлежат электронным конфигурациям .
и
Ъ\ор
р
стояние, вызывающее возмущение в состоянии С42~, — это состояние D41 (. . . °,Sdn ) — верхнее
состояние системы X, наблюдавшейся в спектре низкотемпературной матрицы; 3) верхние
состояния систем А, А' ж В — компоненты квартетного состояния Л4П_1/2, ^44IIi/2, ^441Ь/2 конфигурации
. . .Ъ\т. , а верхние состояния систем С и D — состояния 54Фз/г (. . .°„о"й71р) ий22+ (. . .°s§2).
Принятая энергия возбуждения состояния 62Д (. . .°1ЪЛ) получена усреднением разности энергий
состояний D4I1 (. . •°s5rfirc,o) и Л4П(. . .^Тср) и разности центров тяжести конфигураций . . .s2d и . . .sd2
иона Zr+, изоэлектронного двухзарядному иону ниобия. Предполагается, что погрешности
оцененных энергий возбуждения составляют 2000—4000 см.
Молекулярные постоянные NbO в состояниях Х42~ и С*2" приняты по работе Улер [4574],
которая нашла их в результате анализа вращательной структуры полос системы С*И~т— Х42~
<г/'< 2, г/ < 3). Позднее в работах Брома и др. [1678], Ричардса [3987] и Грина и др. [2467]
было показано, что предложенная Улер интерпретация этого перехода как перехода 2Д—2Д
ошибочна, однако молекулярные постоянные были определены верно. Те же постоянные
рекомендованы в справочнике Хьюбера и Герцберга [2736].
Термодинамические функции NbO(r) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBn и ее производных вычислялись по уравнениям A. 90)—A. 92) с
учетом 22 возбужденных электронных состояний в предположении, что <?^.вр=0,25 p(QW вр.
Величина QW вр и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью уравнений
типа A. 82). В расчете учитывались все уровни состояния Х42~ с значениями / <^ / s;
последние определялись из условий A. 81). Колебательно-вращательные уровни энергии состояния
Х42~ вычислялись по уравнениям A. 65), A. 34),-A. 64а), их мультиплетность учитывалась
статистическим весом 4. Значения г>тах и. /ит, а также величины коэффициентов в этих уравнениях,
полученные с учетом условий A. 50), A. 59), приведены в табл. 34.5.
77
NbO2(K, ж)
Глава 34
Таблица 34.5. Коэффициенты в уравнениях, описывающих уровни энергии (в см-1),
а также значения vm!a и J\im в состоянии X* 2~,
принятые для расчета термодинамических функций NbO(r)a
Ую-Ю-2
9.890
Примечание
^20
-3,83
а Энергии
Уо1 • 10
4,321
возбужденных
Уп-
—2
состояний
103
1
см.
у02
2
табл. 34
¦107
,2
.4.
Уоз-
7
L013
,7
128 508
Погрешности рассчитанных термодинамических функций NbO(r) при низких температурах
обусловлены приближенным учетом мультиплетности вращательных уровней состояния Х42~
и использованием уравнения A. 34) для аппроксимации их энергий. При температурах выше-
1000 К становятся существенными ошибки, в особенности в значениях Ср(Т) из-за использования
оцененных значений энергии состояний а2Е~, Ь2Д и других. Погрешности в значениях Ф°{Т\
при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,05, 2, 4 и 8 Дж-К^-моль.
Термодинамические функции NbO(r) ранее вычислялись в таблицах JANAF [2835], работах
Брюэра и Розенблатта [1666] (Ф°(Т) для Т < 3000 К) и Шнейдера [4197а] (Г=1000Ч-10 000 К).
Расхождения данных [2835, 4197а] и табл. 921 обусловлены тем, что авторы этих работ учитывали-
только два возбужденных состояния NbO, наблюдавшиеся в спектрах. Соответствующие
расхождения пренебрежимо малы до 1000 К, но достигают 4 и 10 Дж -К -моль в значениях Ф0(Г) и S°(T}
при 6000 К (до 9 и 10 Дж -К -моль в значениях этих функций при 10 000 К с данными [4197а]).
В работе [1666] принята неверная схема электронных состояний NbO и рассчитанные значения
Ф°(Т) больше приведенных в табл. 921 на 1—11 Дж-К~1-моль~1.
Константа равновесия реакции NbO(r)=Nb(r)+O(r) вычислена с использованием принятого
в справочнике значения
ДгЯ°@) = D#(NbO) = 755 + 12 кДж^'моль ^ 62 700 + 1000 см.
Принятое значение основано на совпадающих в пределах их погрешностей эффузионных и масс-
спектрометрических измерениях Щукарева и др. [1187] и Францевой [1061] (NbO(K, ж)=ШЮ(г),.
1873—2473 К, данные представлены в виде уравнения) и масс-спектрометрических измерениях
Горохова и др. [310] (YO(r)+Nb(r)=Y(r)+NbO(r), 2605—2988 К, 20 точек). По данным этих
измерений были вычислены значения D0(NbO), равные 753 + 18 кДж-моль (III закон) и 776±
+25 кДж-моль (II закон) [1061, 1187] и 756±12 кДж-моль (III закон) и 716 + 50 кДж-моль
(II закон) [310].
Принятой энергии ди°социации соответствует
г, 0) = 211,783 ± 16 кДж . моль.
1ЧЬ02(к, ж). Термодинамические свойства кристаллического и жидкого диоксида ниобия
стехиометрического состава в стандартном состоянии при температурах 100—5000 К приведены
в табл. 922.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 34.1. За стандартное состояние 1ЧЬО2(к) в справочнике приняты в интервале 0—1082 К
тетрагональная объемноцентрированная модификация [3389, 3903] и в интервале 1082—2360 К —
тетрагональная модификация типа рутила [4115, 4116].
При Т ^ 298,15 К термодинамические функции NbO2(K) вычислены на основании измерений
теплоемкости, выполненных Венгером и Кеезомом [4703] B,5—30 К, для монокристаллического
образца чистоты >99,9%) и Кингом [ЗОЮ] E3—296 К, для образца чистоты 99,9%).
Интерполяция и согласование данных [ЗОЮ] и [4703] проводились с использованием дробно-рациональной
функции шестой степени [1066], аппроксимирующей эти данные в пределах 0,5 и 5%
соответственно. Погрешности вычисленных значений 5°B98,15 К) и -?Г°B98,15 К)—.ffc@) (см. табл. 34.1)
оцениваются в 0,3 Дж-К-моль~1 и 0,04 кДж-моль. Теплоемкость монокристаллического»
образца NbO2(K) измерялась также в работе [2765] B—20 К) и ее результаты находятся в согласии
с данными [4703].
Приведенные в табл. 34.1 уравнения для теплоемкости NbO2(K) в интервалах 298,15—1082 К
и 1082—2360 К получены совместной обработкой результатов измерений энтальпии, проведенных
78
Ниобий и его соединения
NbO2(K,
Гельдом и Кусенко [268] D57—1005 К и 1084—1717 К) и Кингом и Кристенсеном [3012] D00—
1000 К и 1090—1800 К)г. Температура полиморфного превращения NbO2(K) принята равной.
1082 +1 К на основании результатов рассеяния нейтронов на монокристаллах NbO2 очень высокой
чистоты [3901, 3902, 4250] 2. В работах [268, 2838, 3012, 4113, 4116, 4170] существенно менее
точными методами и для менее чистых образцов были получены значения r<r(Nb02, к) в пределах
1040—1090 К. Принятое значение энтальпии превращения B,5+0,9 кДж-моль) является
разностью энтальпий двух модификаций при 1082 К, вычисленных в соответствии с принятыми
уравнениями для энтальпии двух модификаций NbO2.
Температура плавления B360 + 50 К) принята на основании измерений Колчина и Сумароковой
[537] B357 + 15 К) в условиях вакуума для образцов субстехиометрических составов с учетом
данных Брауэра и Литтке [1647] для Гт(ТЮ2) в вакууме и в атмосфере кислорода. Менее точные'
значения Тт(Ш02) были получены Эллиоттом [2171 ] B188 К) и Ортнером и др. B275 К, см. [2838]).
Принятое значение энтальпии плавления G5+8 кДж-моль) получено Щукаревым и др. [1187]
как разность энтальпий разложения NbO2(K) и ШЮ2(ж) с образованием NbO(r). Последние
вычислены по II закону на основании измерений ионного тока NbO+ при масс-спектроскопическом
исследовании пара над NbO2(K) и ШH2(ж) в интервале 1700—3100 К. С принятым значением
согласуется величина &mH°(NbO2, к), оцененная в предположении, что Д <S°(NbO2, к)?«Д S°(VO2, к) —
=31,3 Дж-К^-моль-1 [1915].
Теплоемкость ИЬО2(ж) оценена равной 100 + 10 Дж^К^-моль по принятой в справочнике
методике (см. гл. 3). Погрешности вычисленных значений Ф°(Т) при 298,15, 1500, 3000 и 5000 К
оцениваются в 0,2, 1,5, 6 и 15 Дж-К~1-моль соответственно.
Ранее термодинамические функции NbO2(K, ж) вычислялись в справочниках JANAF [2835]
(до 3000 К) и Шика [4181] (до 6000 К) на основании данных [ЗОЮ, 3012] и оцененных значений
Д„,Я° и C°(NbO2, ж). Расхождения в функциях, приведенных в табл. 922 и в [2835], составляют
0,2—4 Дж-К^-моль в значениях Ф°(Т).
Принятая в справочнике энтальпия образования
A//°(NbO2, к, 298,15 К) = —795,0 ±1,3 кДж • моль
Таблица 34.6. Результаты определений AfH°(NbO2, к, 298,15 К) (в кДж• моль)
Авторы
Сю, 1939 [4434, 4435]
Грубе и др., 1939 [2511]
Шефер, Брейль, 1952 [4171]
Ма, 1958 [3345]
Морозова, Гецкина, 1959 [698]
Кусенко, Гельд, 1959 [608, 610, 611 ]
Уоррелл, 1964 [4768]
Хираока и др., 1969 [2656, 2657]
Метод
Статический, Nb2O6(i!)+H2(r)=2NbO2(K)+
+Н2О(г), 1258-1443 К, 8 точек
Протока, та же реакция, 1128—1345 К,
4 точки
Протока, та же реакция, 1173 К, 1 точка
Калориметрический, NbO2(K)+1/4Oa(r)=
=l/2Nb205(K), 303,15 К, 6 опытов
То же, 291,15 К, 3 опыта
То же, 293,15 К
Статический, Nb2O6(K)+C(K)=2NbO2(K)+
+СО(г), 1074—1175 К, 6 точек
ЭДС, 2NbO2(K)+V3Cr2O3(K)=Nb2O5(K)+
+2/3Сг(к), 1050—1300 К, 6 точек, данные
представлены в виде графика
A/^°(NbO2, к, 298,15 К)
II закон
III закон
—801,3±6,7 —798,6+2,1
—798,3 ±2,4 —793,5 ±1,9
— —796,5±1,8
—795,0 ±1,3
—793,7 ±4,0
—791,6±2,5
-816+13 —796,8±1,8
—793,5 —784,4 ±3,0
1 Данные [268, 3012] для области температур вблизи Ttr A040—1080 К) в этом расчете не использовались.
2 Кинг и Кристенсен [3012] на основании выполненного ими исследования энтальпии NbO2 (к) предположили
существование фазового перехода при 1200 К. Однако измерения энтальпии NbO2(K) в работе Гельда и
Кусенко [268], а также измерения электропроводности и термоэлектрической ЭДС при 196—1273 К [2838]
свидетельствуют об отсутствии фазового перехода при 1200 К. Тщательные рентгенографические исследования^
структуры NbO2 в интервале 298—1300 К, проведенные Сакатой и др. [4113, 4114, 4116], позволили
заключить, что переход от низкотемпературной модификации к высокотемпературной при 1082 К в структурном
отношении происходит не полностью и завершается при 1173 К.
79
NbO2(r)
Глава 34
основана на измерениях, представленных в табл. 34.6. Приведенные в таблице погрешности
учитывают воспроизводимость измерений, а также неточность использованных в расчетах
термохимических величин и термодинамических функций. Как видно из таблицы, результаты измерений
находятся в удовлетворительном согласии. Наиболее точные измерения были выполнены Ма
[3345], и принятая величина основывается в первую очередь на этой работе.
Давление пара в реакции NbO2(K, ж)=Г\ГЬО2(г) вычислено с использованием значения Дг-йГ°(О) =
= As-fiTo(NbO2, к, 0)=592,365+ 10 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования NbO2 в кристаллическом и газообразном состояниях.
NbO2(r). Термодинамические свойства газообразного диоксида ниобия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 923.
Молекулярные постоянные NbO2, принятые для расчета термодинамических функций,
представлены в табл. 34.7. Они основаны на результатах электронографического исследования,
выполненного Ерохиным и др. [2191], и интерпретации этих экспериментальных данных в работе Гер-
пшкова и др. [2392]. Авторы [2392] показали, что молекула NbO2 в основном состоянии ХгАх
Таблица 34.7.
Значения молекулярных постоянных, а также о
термодинамических функций NbO2
и рх, принятые для расчета
Состояние
= 26 000 см
8000,
-1
960
Т.(&А1) =
СМ"
530
14 000,
^3
1010
Вх) = 15 000,
III- 10117
г3 ¦ см6
6,Ы02
ге(Же2) = 2зооо,
0
2
Рх
2
имеет угловую структуру (симметрия С2с) с равновесными структурными параметрами
-2»0—Nb—О = 101,6±3,3° и r(Nb—О)=1,713+0,005 А, а также определили постоянные силового
поля и основные частоты этой молекулы. Приведенное в табл. 34.7 значение IJbIc рассчитано по
этим структурным параметрам, его погрешность оценивается в 2-1016 г3-см6. Для основных
частот v2 и v3 в справочнике приняты значения, найденные авторами [2392]; для частоты v2 оно
хорошо согласуется с данными Серебренникова и Мальцева [928, 929] о величине частоты
деформационного колебания в молекулах ScOa E16 см), VO2 (см. табл. 33.6) и МпО2 E26 см).
Погрешности в этих значениях оцениваются в 30 и 50 см соответственно. Для частоты va в работе [2392]
было получено значение 854 + 50 см, которое представляется заниженным при сопоставлении
значений v3 и vx у молекул VO2, GrO2 и МпО2. Приведенное в табл. 34.7 значение vx оценено на
основании величины v3 и соответствующих данных для диоксидов других переходных металлов,
его погрешность оценивается в 50 см. К близкой величине (985 см) приводит расчет по
уравнениям поля валентных сил и принятым значениям v2, v3 и -=^0—Nb—О. Приведенные в табл. 34.7
энергии возбужденных электронных состояний оценены Хаитом и Хачкурузовым [1065] с помощью
полуэмпирической методики, основанной на представлениях теории кристаллического поля.
Использованные в расчете параметры основывались на значениях соответствующих параметров
молекул NbO, TiO2 и ТаО2. Погрешности приведенных энергий оцениваются в 4000—5000 см.
Термодинамические функции NbO2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124) и A. 130) в приближении «жесткий ротатор — гармонический осциллятор»
•с учетом шести возбужденных электронных состояний по соотношениям A. 168)—A. 170).
Погрешности рассчитанных функций, обусловленные приближенным характером расчета и неточностью
принятых молекулярных постоянных, составляют 0,2, 2 и 4 Дж-К-моль в значениях Ф°(Т)
при 298,15, 3000 и 6000 К.
Ранее термодинамические функции NbO2(r) вычислялись в справочнике JANAF [2835] и Шика
[4181], а также Брюэром и Розенблаттом [1665] (до 3000 К). Расчеты [1665, 4181] были выполнены
для линейной структуры молекулы NbO2. Расхождения данных табл. 923 и [2835] составляют
0,2—3 Дж-К-моль в значениях Ф°(Г) и обусловлены тем, что в расчете [2835] использованы
отличающиеся значения постоянных и не учтены возбужденные электронные состояния NbOa.
80
Ниобий и его соединения
NbA(K, ж)
Константа равновесия реакции NbO2(r)=Nb(r)*{-2O(r) вычислена с использованием значения
ДГЯ°(О)=1411,566 + 14 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
j, г, 0) = —198 ± 10 кДж • моль.
В табл. 34.8 представлены результаты исследований этой величины. Приведенные в таблице
погрешности учитывают воспроизводимость измерений и неточность термодинамических функций,
термохимических величин и сечений ионизации. За исключением выпадающей работы [295]
остальные измерения согласуются в пределах их погрешностей. Принятое значение является
средневзвешенным из результатов этих работ. ,
Таблица 34.8. Результаты определений Д^2зГ°(]ЧЬО2, г, 0) (в кДж• моль)
Авторы
Метод
4/5°(Nb0a> г, 0)
II закон
III з«кон
Щукарев и др., 1959 [1184] Масс-спектрометрический, испарение с открытой —162 — '
поверхности, NbO2(K)=NbO2(r), 1500—1880 К, ¦¦'¦-.
приведено значение Д8Я°(П закон)
Голубцов п др., 1960 [295] Эффузионный, NbO2(K)=Nb0a(r), 1489—1905К, — 697±3 —267±25
9 точек
Щукарев и др., 1962 Ц185] Эффузионный, NbO2(K)=NbO2(r), 1938-2122 К, —191 ±40 —198±10
10 точек
Щукарев и др., 1962 [1186] Масс-спектрометрический, испарение с открытой —189 —
поверхности, 2NbO(K)=Nb(K)+NbO2(r),
1773—1973 К, приведено значение Дг#°@)
Щукарев и др., 1966 [1061, 1187] Масс-спектрометрический, NbO»(K)=NbO2(r), —196 —199+10
1773—2173 К, 25 точека . ~
То же, NbO2(Ht)=NbO2(r), 2173—2373 К, 14 точек а —282 —196+14
То же, 2NbO(K)=Nb(K)+NbO2(r), 1873—2203 К, —187 —189+28
20 точек а
То же, 21ЧЬО(ж)=Щк)+Ш)О2(г), 2203—2473 К, —218 —183±35
10 точек а 1
Горохов и др., 1979 [310] Масс-спектрометрический, 2YO(r)+Nb(r)= —163+80 —203+18
=NbO2(r)+2Y(r), 2570—2988 К, 10 точек
То же, ZrO2(r)+Nb(r)=Zr(r)+NbO3(r), 2876 и — —208±25
2988 К, 5 измерений
а Данные представлены в виде уравнения.
Nb О (к, ж). Термодинамические свойства кристаллического и жидкого пентоксида диниобия
стехиометрического состава в стандартном состоянии при температурах 100—5000 К приведены
в табл. 924.
Значения постоянных, принятые для вычисления термодинамических функций, приведены
в табл. 34.1. За стандартное состояние Nb2O5(K) в интервале 0—1783 К в справочнике принимается
моноклинная модификация (Я-форма) Ч
При Т s^ 298,15 К расчет термодинамических функций ]\Ъ2О5(к) основывается на измерениях
теплоемкости, проведенных Кингом [3008] E3—297 К) 2. Экстраполяция теплоемкости ниже 53 К
проводилась с использованием дробно-рациональной функции шестой степени [1066],
аппроксимирующей данные [3008] в пределах ~1% и приводящей к значению ?°E3 К) = 11,1 Дж-К^х
Хмоль. Погрешности приведенных в табл. 34.1 значений ?°B98,15 К) и #"B98,15 К)—Н°@)
1 ДК^ 01 До
Хмоль. р
оцениваются в 1 Дж-К^-моль и 0,1 кДж-моль соответственно.
В интервале 298,15—1783 К для теплоемкости Nb2O6(K) принято уравнение (см. табл. 34.1),
полученное совместной обработкой результатов измерений энтальпии, проведенных Орром [3724]
* Известно еще по крайней мере десять модификаций Nb2O5(K) (см. [2425, 4173]), которые, как показано в
работах [3858, 3971, 4173, 4651], в термодинамическом отношении являются менее стабильными.
^PW B 1300S1 представлял собой моноклинную модификацию и содержал менее 0,1% примесей.
!¦..:• 81
Ш»2О5(к, ж)
Глава 34
C81—1751 К) и Гельдом и Кусенко [268, 611] D61—1742 К) на образцах моноклинной
модификации с содержанием примесей ~0,13%. Данные об энтальпии Nb2O6(K), полученные Крюссом и
Нильсоном [3122] C73—718 К) для образца, не охарактеризованного по химическому составу
и содержанию фаз, не использовались. Они систематически занижены по сравнению с данными
[268, 3724]. Принятое в справочнике уравнение для C°(Nb2O5, к) согласуется с измерениями
теплоемкости ШJО5(к), выполненными Басили и др. [1420] методом ДТА D10—940 К).
Температура плавления Nb2O5(K) принята равной 1783 + 10 К на основании данных Орра
[3724] A787 К), Гельда и Кусенко [268, 611] A782 + 10 К), Джонгеджана и Уилкинса [2901]
A782 + 5 К) и Андреевой и Келлер [32] A795±20К). В работах [2696, 2766, 3534, 3863, 3887,
3969, 4055, 4056] были получены значения в пределах 1753—1768 К, что объясняется авторами
[2901] переходом Nb2O6(K) в другую кристаллическую модификацию. Для энтальпии плавления
Nb2O5(K) и теплоемкости ЙЬ2Ов(ж) в справочнике приняты значения, равные 101+3 кДж-моль
и 200 + 10 Дж-К-моль на основании измерений энтальпии ШJО5(ж) при 1795, 1797, 1811,5
и 1830 К в работах Орра [3724] и Гельда и Кусенко [268]. Погрешности вычисленных значений
•моль
-1
соответст-
Ф°(Г) при 298,15, 1000, 3000 и 5000 К оцениваются в 0,5, 2, 12 и 30 Дж-К-
венно.
Ранее таблицы термодинамических функций Nb2O5(K, ж) вычислялись в справочниках JANAF
[2835] (до 5000 К), Шика [4181] (до 6000 К), Келли [2960,2963]. Расхождения данных табл. 924
и [2835] составляют 0,04—16 Дж-К-моль в значениях Ф°(Т).
Константа равновесия реакции Nb2O5(K, ж)=2]\Ъ(г)+5О(г) вычислена с использованием
значения ДгЯ°@) =4561,010+ 20 кДж-моль, соответствующего энтальпии образования Nb2O5(K)
Ь,Н°(т2О5, к, 298,15 К) = —1897 ±3 кДж • моль.
Эта величина была принята на основании данных, представленных в табл. 34.9. Указанные
погрешности учитывают только воспроизводимость измерений. Принятое значение является
средним из двух величин, найденных Корниловым и др. [543, 545]. В этих работах было тщательно
изучено влияние примесей и их содержание (при вычислении соответствующих поправок
использовались данные справочника [1009]). Близкие, но менее точные значения были получены в работах
[609, 2735, 2756]. Величины, вычисленные по данным [610, 698, 701, 1449, 3610], ненадежны из-за
низкой чистоты образцов.
Таблица 34.9. Результаты определений &^Н°С^Ь2Оь, к, 298,15 К) (в кДж-моль-1)
Авторы
Метод
AfH°(m20s, к,
298,15 К)
Мутман и др., 1907 [3610]
Беккер и Рот, 1933 [1449]
Хамфри, 1954 [2756]
Морозова и Гецкина, 1959 [698]
Морозова и Столярова, 1960 [701]
Кусенко и Гельд, 1960 [610]
Кусенко и Гельд, 1960 [609]
Хыобер и др., 1961 [2735]
Корнилов и др., 1962 [543]
Корнилов и др., 1964 [545]
Калориметрический, измерение энтальпии сгорания —1835 ±80
ниобия, 3 опыта
То же (чистота образца 98,8%), 3 опыта —1938+9
То же (чистота образца 99,69%), 9 опытов —1904+2
То же (чистота образца 98,59%), 3 опыта —1977+.7
То же, порошкообразный образец —1980+3
То же, компактный образец —1903+3
То же (чистота образца 98,52%) —1918+20
То же (чистота образца 99,01%) —1904+3
То же (чистота образца 99,45%), 8 опытов —1900+7
То же (чистота образца 99,35%), 8 опытов —1897,9+3,0
То же (чистота образца 98,47%), 5 опытов —1895,8±2,5
Г л а в а 35
ТАНТАЛ И ЕГО СОЕДИНЕНИЯ
Та(к, ж), Та, Та+, ТаО, ТаО2, Та2Об(к, ж)
Ъ настоящей главе рассматривается тантал и его кислородные соединения — всего 5 веществ.
Для двух веществ приведены данные об их термодинамических свойствах в конденсированном
состоянии, для четырех — в газообразном состоянии. Термодинамические свойства трех газов
(Та, Та+, ТаО) рассчитаны для температур до 10 000 К, одного (ТаО2) — до 6000 К.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов тантала (атомный
вес 180,9479). Однако различие постоянных разных изотопомеров в расчетах не учитывалось
ввиду того, что природная изотопическая смесь содержит практически один стабильный изотоп
этого элемента.
В Приложении 3.10 приведены критические постоянные элементарного тантала.
Основным оксидом тантала в кристаллическом и жидком состояниях является Та2О5, который
в зависимости от условий получения может быть в разных полиморфных формах (см. [3971, 4650]).
Область гомогенности Та2О5(к) достаточно надежно не определена: в ряде работ (см. [4416]) было
установлено, что она очень узка, но в работах [3135, 4650] для нее указан интервал от Та2О4>7
до Та2О50. Известны также субоксиды тантала ТавО, Та4О, Та2О (см. [549а]) как промежуточные
продукты его окисления. Потенциалы ионизации ТаО и ТаО2 превышают 65 000 см, и поэтому
соответствующие ионы не рассматриваются в справочнике.
Та(к, ж). Термодинамические свойства кристаллического и жидкого тантала в стандартном
состоянии при температурах 100—6000 К приведены в табл. 925.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 35.1. За стандартное состояние Та(к) в интервале 0—3295 К принята кубическая
модификация (структурный тип a-Fe). При 4,475 К Та(к) переходит в сверхпроводящее состояние *.
Таблица 35.1. Принятые значения термодинамических величин для тантала него соединений
в кристаллическом и жидком состояниях
Вещество
Та
Ta2Os
Состояние
к, куб.
ж
кП, ромб.
к1, монокл
ж
К)-
ю
оо-
аэ о
аз 1
кДжХ
X моль
5,68
—
22,95
—
Примечание. С°(Г)=а+Ь -Т—с
Та: а
d=—9,551 -Ю"', е=2,283
LO
оо"
со
о
?
Ю
оо"
СО
CNJ
о"».
Дж • К • моль
41,52 .25,29
— —
141,9 134,935
— —
•Г+е2 -Г2+е -Ts (в Дж •
¦ю-».
Коэффициенты
в уравнении
для С°р(Т)
а
Ь-103
с ¦ Ю--*
21,531 14,123 —0,299а
44 — —
151,254 31,062 22,739
210 — —
220 — —
К-моль).
Интервал
температур
или
т
х m
К
298,15—3295
3295—6000
298,15-1633
1633-2150
2150—5500
3295
—
1633
2150
—
или
кДжХ
X моль
36
—
2
120
* Это значение Ttr получено Леуполдом и др. [3237] для образца чистоты > 99,99%. Близкие значения были
получены в работах [1712] D,483 К) и [4257] D,45 К) для образцов Та(к) высокой чистоты. Об отсутствии у Та(к)
фазовых переходов при более высоких температурах свидетельствуют результаты рентгенографических иссле-
* [951] A10—400 К), [2155] B93—2473 К), [191] B93—2973 К).
.8 3
ta(K, ж) Глава 35
' Термодинамические функции при Т ^ 298,15 К вычислены на основании измерений
теплоемкости образцов тантала высокой чистоты Шеном [4257] (см. [2752]) @,3—25 К), Леуполдом и
др. [3237] D—20 К) и Стерретом и Уоллесом [4395] A1—543 К). Погрешности вычисленных
значений ?°B98,15 к) и #°B98,15 К) — #°@) (см. табл. 35.1) оцениваются в 0,2 Дж• К • моль
и 0,01 кДж'Моль. Менее точные измерения теплоемкости при Т < 298,15 К были выполнены
в работах [2957] E3-298 К), [2953] A-4 К), [3482] C,0-5,5 К), [4766] A,7-5 К), [4750] B—
20 К), [1884] A0-273 К), [4720] A,3-25 К).
В интервале 298,15—3295 К термодинамические функции Та(к) вычислены с использованием
уравнения для С°р(Т) (см. табл. 35.1), полученного совместной обработкой результатов измерений
энтальпии, выполненных Магнусом и Хольцманом [3342] E47—1175 К), Егером и Веенстрой
[2818, 2820] E73—1831 К), Хохом и Джонстоном [2667] A366—2950 К), Конуэем и Хейном
(см. [2663], 1316—2626 К), Гусевой и др. [350] A367—2154 К), Эттингом и Навратилом [3694]
E33—1384 К), Березиным и Чеховским [122] B440—3238 К), Шейнером и др. [4248] B200—
3270 К), а также на основании значений энтальпии при 300—543 К, вычисленных по данным Стер-
рета и Уоллеса [4395] для теплоемкости. В расчете наименьший вес придавался данным работ
{350, 2667, 2820] как наименее точным. Теплоемкость Та(к) при Т > 298,15 К измерялась в
работах [3188] A650-2800 К), [3939] F00-3200 К), [3318] A200-2400 К), [572, 3100] A200—
2900 К),-[4491] C00-3000 К), [1263] F00-3100 К), [47, 50, 1055] A100-2300 К), [50, 638,
1055] A100-2400 К), [50, 1031, 1055] A500-2900 К), [1791] A900-3200 К), [618] B300-3260 К),
[2400] C23—973 К). Точность этих измерений существенно ниже точности измерений
энтальпии; систематически заниженными являются данные [2400] (при Т ^ 473 К), [4491] (при Т <С
< 1200 К) и [1263] (при Т < 1800 К), а данные [3188, 3939] (при Т > 1600 К) и [618] (при Т >
^= 3100 К) завышены.
Температура плавления C295 + 15 К) принята на основании измерений Руди и Прогуль-
ского [4084] C293 + 14 К) и Пемслера [3798] C296 + 15 К) для образцов чистоты —99,9%.
С принятым значением согласуются данные [4001, 4002] C285 + 15 К), [1037] C279 + 20 К),
13042, 3047] C299 К), [2985] C309 К). Более низкие значения получены в работах [2589] C274 +
± 10 К) [3202] C261 +15 К), [3188] C258 +20 К), а также в работах [1835,2308,3847,
4730]. Энтальпия плавления C6 + 2 кДж-моль) принята на основании измерений методом
импульсного нагрева в работах Шейнера и др. [4248] C6 кДж-моль) и Савватинского и др.
[619, 894] C6,6 + 2,2 кДж-моль). С принятым значением согласуется значение, найденное
тем же методом Мартынюком и др. [667] C3 кДж-моль), а также оценки, основанные на
величине kmS ряда переходных металлов (Hf, Nb, Mo, W).
Теплоемкость Та(ж) определялась методом импульсного нагрева Лебедевым и Можаровым
[618] и Шейнером и др. [4248]. В работе [618] применялись импульсы тока большой мощности
и получены данные для Та(ж) в неравновесном состоянии, о чем свидетельствуют аномальные
значения теплоемкости Та(ж), изменяющиеся в пределах от 85 до 23 Дж • К • моль в интервале
3258—4200 К. Аномально высокое значение С°р(Та., ж) было получено и в работе [4248]
F9 кДж-моль). В справочнике теплоемкость Та(ж) принята равной 44+4 кДж-моль на
основании принятых значений С°(Та, к, Тш), С°р(Ш, ж) и Cp(W, ж). Погрешности приведенных
в табл. 925 значений Ф°(Т) при 298,15, 1000,' 3000 и 6000 К составляют 0,1, 0,5, 1,2 и
5 Дж-К^-моль'1.
Ранее термодинамические функции Та(к, ж) вычислялись в справочниках JANAF [2835]
Халтгрена и др. [2752] (до 6000 К), Шика [4181] (до 5700 К), Сталла и Зинке [4421] и Келли
[2960, 2963] (до 3000 К). Расхождения между значениями Ф°(Т) для Та(к), приведенными
в табл. 925 и в [2835], не превышают 0,3 Дж-К^-моль; для Та(ж) эти расхождения возрастают
до 4,5 Дж-К^-моль при 6000 К ввиду различной оценки теплоемкости Та(ж).
В настоящем справочнике в качестве стандартного состояния тантала принято
кристаллическое состояние и в соответствии с этим
Д,#°(Та, к)==0.
Давление пара тантала в реакции Та(к, ж) = Та(г) вычислено с использованием принятой
в справочнике энтальпии сублимации
АгН°@) = Д,Я°(Та, к, 0) = 782 + 3 кДж • моль.
84
Тантал и его соединения
Та(г)
Таблица 35.2. Результаты определений Д51Г°(Та, к, 0) (в кДж • моль)
Авторы
Лангмюр, Молтер, 1939 [3187]
Фиске 1942 [2264]
Эдварде и др., 1951 [2151]
Горбатый, Шуппе, 1957 [304]
Котляр, Андреева, 1960 [562]
Гебхарт и др., 1962 [2384]
Бабельёвский, 1962 [1366] (см. также
[1367])
Зандберг и др., 1965 [435]
Голубцов, Несмеянов, 1965 [292, 294,
298]
Сасаки и др., 1970 [4143, 4144]
Метод
Испарение с открытой поверхности, 1999—3178 К
6 точек
То же, 2606—2815 К, 6 точек
То же, 2628—2953 К, 6 точек
Д5Я°(Та, к, 0)
II закон
770+1
643+100
791 +30
То же, с использованием радиоактивного изотопа, 765+25
2073—2404 К, 6 точек
, Испарение с открытой поверхности, 2404—2805 К
данные представлены в виде уравнения
То же, 2905—3156 К, 8 точек
Масс-спектрометрический, испарение с
открытой поверхности, 2578—3042 К
То же, 2400-2600 К
Испарение с открытой поверхности с
использованием радиоактивного изотопа, 1877—2975 К,
12 точек
Эффузионный, 2393—2739 К, 14 точек
Масс-спектрометрический, испарение с открытой
поверхности, 2550—2770 К
668+50
973+20
714+15
683+30
791+5
787+16
774+12
III закон
775,9+1,0
754,3+3,0
781,6+1,0
780,6+1,0
774+5
796+4
—
—
788,9+0,8
784,0+0,7
—
В табл. 35.2 представлены значения энтальпии сублимации тантала, вычисленные по резуль^
татам измерений давления его насыщенного пара. В соответствии с данными [435, 1367] прини^
малось, что пары тантала одноатомные. Приведенные в таблице погрешности учитывают только
воспроизводимость измерений. Принятое значение Д8#°(Та, к, 0) является средневзвешенным
из наиболее надежных данных, полученных в работах [292, 294, 298] (для рост ^ 5-1(Г7 Торр)
и [304, 2151, 3187]. Средневзвешенное вычислялось с учетом воспроизводимости измерений, нег
точности термодинамических функций Та(к) и отклонения полученного в данной работе значе^
ния от среднего. При этом больший вес придавался эффузионным измерениям Голубцова и Her
смеянова [292, 294, 298].
Та(г). Термодинамические свойства газообразного тантала в стандартном состоянии при тем*
пературах 100—10 000 К приведены в табл. 926.
Для расчета термодинамических функций использовано 84 уровня энергии г атома Та с сум-
марным статистическим весом 4904 в интервале 0—63 000 см, т. е. ниже первого потенциала
ионизации. Эти уровни соответствуют валентным электронным конфигурациям ...5d?6s2 (к ней
относится основной уровень 4^з/г), ...5d5, ...5d3Qp2, ...5d*Ql (I = 0,1), ...5d36s6l (I = 1-^-3) и
...5d26s2Ql (I = 1,2). Принятые величины основаны на данных Мур [3550] и на оценках для ряда
ненаблюдавшихся электронных состояний. Для электронных конфигураций ...ЫЩ1 (I = 0,1),
...5d36.s6Z (I = 14-3) и ...5d2Qs26l (I = 1,2) оценка выполнена методом, изложенным в Приложен
нии 6, а для электронных конфигураций ...5й6, ...5d36s2 и ...5d36p2 — сравнением энергии
соответствующих состояний V, Nb, Та, Сг+, Мо+ и W+. Оцененные уровни имеют энергию от 28 000 см
и выше с суммарным статистическим весом 4056. Погрешность оценки энергии лежит в пределах
500—4000 см.
Термодинамические функции Та(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Т <^ 3000 К обусловлены главным образом неточностью принятых значений
фундаментальных постоянных, а при более высоких температурах — неточностью оценки
энергий ненаблюдавшихся уровней и пренебрежением ридберговскими состояниями. В значениях
Ф°(Г) при 3000, 6000 и 10 000 К погрешности составляют 0,03, 0,6 и 1 Дж-К^-моль.
Ранее термодинамические функции Та(г) вычислялись в справочниках JANAF [2835], Шика
[4181] и Сталла и Зинке [4421] (до 3000 К), а также в работе Хилзенрата и др. [2652] (до 10 000 К)
* См. примеч. 1 к тексту по Сг(г).
85
Та+(г), ТаО(г) Глава 35
на основании всей совокупности данных Мур [3550] для уровней энергии атома Та. Расхождения
между значениями термодинамических функций Та(г), приведенными в табл. 926 и в других
расчетах, при Т < 2000 К не превосходят 0,05 Дж• К • моль в значениях Ф°(Г) и S°{T). При более
высоких температурах расхождения обусловлены тем, что в разных расчетах учитывалось
разное число уровней энергии. Расхождения данных табл. 926 и [2835] достигают 0,1 и
0,2 Дж-К^-моль в значениях Ф°F000 К) и 6"оF000 К), расхождения с данными [2652]
составляют 0,3 и 2,5 Дж-К^-моль при 10 000 К.
Энтальпия образования
A/i°(Ta, г, 0) = 782 "+ 3 кДж • моль
соответствует принятой энтальпии сублимации Та(к).
Та+(г). Термодинамические свойства газообразного положительного иона тантала в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 927.
Для расчета термодинамических функций использовано 34 уровня энергии г иона Та+ с
суммарным статистическим весом 1215 в интервале 0—55 000 см. Эти уровни соответствуют
валентным электронным конфигурациям ...5d3Qs (к ней относится основной уровень bFx), ...5d4, ...5d26s2
и ...Ь<Р(зр. Принятые величины основаны на данных Мур [3550] и на оценках для ряда ненаблю-
давшихся электронных состояний. Для электронных конфигураций ...5d36l (I = 0,1) оценка
выполнена методом, описанным в Приложений 6, а для электронных конфигураций ...5<24 и ...5d26s2—
сравнением энергии соответствующих состояний Ti, Zr, Hf, V+, Nb+, Ta+. Оцененные уровни
имеют энергию от 15 900 см и выше с суммарным статистическим весом 997. Погрешность оценки
энергии лежит в пределах 50—4000 см.
Термодинамические функции Та+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин обусловлены преимущественно неточностью оценок энергий ненаблюдавшихся
уровней. В значениях Ф°(Г) при 3000, 6000 и 10 000 К они составляют 0,1, 0,5 и 1 Дж-К^-моль.
Ранее таблицы термодинамических функций Та+(г) вычисляли Хилзенрат и др. [2652] (до
10 000 К) и Грин и др. [2473] (до 50 000 К) на основании всей совокупности уровней энергии
иона Та+, приведенных в справочнике [3550]. Расхождения между термодинамическими
функциями Та+(г), приведенными в табл. 927 и в работах [2473, 2652], обусловлены тем, что в
настоящем справочнике учтены ненаблюдавшиеся уровни энергии, соответствующие валентным
электронным конфигурациям, и не учтены уровни, не отнесенные в [3550] к определенным
электронным конфигурациям; расхождения составляют 0,01—0,7 Дж• К • моль в значениях Ф°(Т).
Константа равновесия реакции Та+(г) + е(т) = Та(г) вычислена с использованием значения
&гН°@) = —761 + 3 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Та) = 761+3 кДж-моль «* 63 600 + 200 хм.
Это значение рекомендовано Мур [3550] на основании расчета предела серии ...5ds6sns (n =
= 6-|-8L^ по формуле Ридберга. Оно представляется более надежным по сравнению с
результатом аналогичного расчета Ван ден Берга и др. [1486] для я = 6и7 F2 700 + 700 см). Раух
и Аккерман [3944] методом электронного удара получили существенно более низкое значение,
равное 715 кДж-моль.
Принятому значению 10(Т&) соответствует
Д,Яс(Та+, г, 0) = 1543,0 ± 4,2 кДж • моль.
ТаО(г). Термодинамические свойства газообразного оксида тантала в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 928.
В табл. 35.3 приведены молекулярные постоянные ТаО, принятые в справочнике на
основании анализа результатов исследований электронного спектра этой молекулы [1675, 1823, 3884,
3886, 4697] (см. также [2736, 4453]) и приближенных оценок. В спектре испускания в газовой
фазе изучены 18 переходов в состояния X\bi2 жХ\Ь1г [1823, 3886] (из них 10 в состояние Х\Ь^.
В спектре поглощения молекул ТаО, изолированных в матрицах из Аг [1675, 4697],
зарегистрированы все переходы из состояния Х\Ь>^, кроме трех, и еще 10 переходов, не проанализирован-
1 См. примеч. 1 к тексту по Сг(г).
86
Тантал и его соединения
Таблица 35.3. Молекулярные постоянные 181Та16О
1 /~\ /"* fl f"b ГТ ТТ ТТ Q
О OL 1ОЯНИО
те %
Xj2 Д3/2 0
Х|Д5/22 3 504,39
Л/22 10 908
Ап12 11062 а
.4 12134
BVa 12 900
С3/2 13 612
D4t 14 437
Е1/2 15 928
f% ¦
i<? 77G
1028,69
1030,81
933 б
—
939 в
931 б
942 б
943 в
935
922
3,51
3,59
—
—
—
—
—
5
—
ве
см"
0,402 839
0,403 584
0,387 291 а
0,389а .
—
0,386 851 а
0,387 547 а
—
0,386 18
—
aj. • 103
1,82
1,87
—
—
—
—
—
—
—
—
De ¦ 107
2,450 а
2,503
2,668 а
—
—
2,674 а
2,624 а
3,26
—
А
1,687 46
1,685 91
1,721 0 а
1,717 а
_
1,721 98 а
1,720 44 а
1,723 48
—
0%
Q'hU
ТЧ2
18 000 а
20 868
20 925
22 196
22 396
22 981,58
23 411
24 127
25 657
26 186
26 342
26 736,50
27 356
29 306
32 445
35 864
35 955
36 615 а
36 785 а
910
944
892
902
903,06
887,70 в
890,31 в
900 б
898 б
913
902,06
896,09 в
895
885 б
871 б
890 б
3
3,3
3,56
4,5
4,08
0,380 81
0,377 42
0,377 06
0,377 207 а
0,381 304 а
0,377 49
0,381 834
0,383 93 а
0,375 36 а
0,376 88 а
0,371 5 а
0,375а
1,92
1,95
1,84
1,78
2,19
2,756 а
2,706 а
2,635 а
2,649 а
2,745 а
2,573 а
2,744 а
2,89 а
2,79 а
2,70 а
3,3 а
3,3 а
1,735 59
1.743 36
1.744 20
1,743 86 а
1,734 46 а
1.743 20
1,733 26
1,728 52 а
1,74814 й
1.744 61 а
1;757 2 а
1,749 а
а Приведено значение для уровня у=0; б вычислено по соотношению A. 68); в приведено значение
объединенных электронных терма:
три
7<<\ см"
Pi
15 000
7
25 000
7
35 000
7
ных в газовой фазе из-за их низкой интенсивности. Большое число переходов, наблюдавшихся
в спектрах ТаО, обусловлено тем, что характер связи моментов количества движения в этой
молекуле приближается к случаю Гунда с. Сравнение с молекулами VO и NbO и результаты
расчета VO в работе [1749] позволяют предполагать, что в области энергий до 40 000 см молекула
ТаО должна иметь существенно больше электронных состояний, чем те 29 состояний, которые
отождествлены в экспериментальных исследованиях. Для конфигураций ...°^8Й, ...°,8| А
8|,
.ashda ,
...8|«
и
s | ^ которым принадлежат состояния VO и NbO, включенные в табл. 33.4
и 34.4, это 50 состояний в символике, принятой для случая Гунда с B3 состояния в обозначениях
для случая Гунда а). Конфигурации ...°*hd у молекулы ТаО принадлежат состояния Х?Д»/,
ф б
д у у ) ф d
и Х|Д«/г. Нижними состояниями конфигурации
!i
р ?,
и |/г. фурц ,|, по-видимому, должны быть состояния
А!а шАп/2 (компоненты состояния iI,~, которое является основным у молекул VO и NbO) с энер_
\жй. около 11 000 см'1. Эта величина согласуется с разностью энергий конфигураций ...ds2 и ...<?^
у иона Ш+ (~11 000 см), изоэлектронного Та+2, что позволяет сделать вывод об отсутствии у мо
87
ТаО(г)
Глава 35
лекулы ТаО ниже состояния Ап{2 других состояний, кроме Х12Дз/2, Х2аД=/3 и А"г1г. Поэтому
в справочнике принимается, что все электронные состояния ТаО, ненаблюдавшиеся
экспериментально, расположены в области от 11 000 до 40 000 см. Поскольку молекула ТаО имеет
нечетное число электронов, во всех этих состояниях квантовое число 2 =? 0. Основания для
корректной оценки энергии каждого из этих состояний отсутствуют, поэтому они представлены в табл. 35.3
в виде трех групп состояний со средними значениями энергии.
Приведенные в табл. 35.3 вращательные постоянные ТаО во всех электронных состояниях,
а также колебательные постоянные в состояниях Х2Дз/2, Х2Д5/г, Кп1<г, L1/^, Ж/2, Р3/2, (?'5/2
получены Читамом и Барроу [1823] на основании анализа вращательной структуры 18 систем
полос х. Энергии состояний F3/>2, Gs/2, H1/^, I, J, КЪ12, Os/2 и Q4%, колебательные постоянные ТаО
в этих состояниях, а также в состоянии i?*/a найдены Велтнером и Мак-Леодом [4697] при
исследовании электронного спектра молекул ТаО, изолированных в матрице из Ne. Погрешности
соответствующих значений Те оцениваются в 100 см. Энергии состояний А и ZI/2 рассчитаны по
кантам полос 0—0 переходов А -> Х3/2 и Dxl2 ->- Х3/^ наблюдавшихся в газовой фазе в работе
[1823]. Их погрешности могут составлять до 30 см. Согласно данным [1823], многие
возбужденные состояния ТаО возмущены.
Термодинамические функции ТаО(г) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения (?вн и ее производных вычислялись по уравнениям A. 90)—A. 92) с
учетом 31 возбужденного состояния, в том числе трех объединенных уровней энергии, и в
предположении, что (?кй.вр = 0,5 QW вр. Статистическая сумма по колебательно-вращательным уровням
энергии состояния -Х^Дз^ и ее производные находились по уравнениям A. 73)—A. 75)
непосредственным суммированием по колебательным уровням и интегрированием по значениям / с
помощью уравнений типа A. 82). В расчете учитывались все колебательно-вращательные уровни
энергии этого состояния с значениями / ^/шаХ) „, последние определялись из условий A. 81).
Уровни энергии состояния Х^»^ вычислялись по соотношениям A. 65), A. 34) и A. 64а), их
вырождение учитывалось статистическим весом 2. Значения vmix и /Ит, а также коэффициентов
в этих уравнениях, полученные с учетом условий A. 50) и A. 59), приведены в табл. 35.4.
Таблица 35.4. Значения коэффициентов в уравнениях, описывающих уровни энергии (в смг1),
а также значения vma% и «7цт, принятые для расчета
термодинамических функций ТаО (г)
Коэффициент
Коэффициент
3k
Коэффициент
Т.
о
1,027 43
-2,864 51
-8,730 96
У01-10
4,028 39
—1,820
—2,450
'lim
—1,3151
116
550
а Значения Т, для возбужденных состояний см. табл. 35.3.
Погрешности рассчитанных термодинамических функций ТаО(г) при температурах до 1000 К
обусловлены главным образом использованием уравнения A. 34) для описания вращательных
уровней состояния ХхДз/г, при более высоких температурах становятся существенными ошибки
из-за применения приближенной методики для учета состояния Хг^1г, а выше 3000 К — из-за
неполноты данных о возбужденных электронных состояниях этой молекулы. Погрешности в
значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,05, 0,1, 2 и 5 Дж-К^-моль.
Термодинамические функции ТаО(г) ранее вычислялись в таблицах JANAF [2835], работах
Брюэра и Розенблатта [1666] (Ф°(Г) для Т < 3000 К), Шнейдера [4197а] (Т = 2000^-10 000 К).
Данные [2835] и табл. 928 практически совпадают (в пределах 0,1—0,5 Дж-К^-моль) в зна-
1 Исследование спектра магнитного кругового дихроизма молекул ТаО в матрицах из Аг, выполненное Бриттеном
и др. [1675], подтвердило интерпретацию всех состояний, предложенную Читамом и Барроу, за исключением
состояния Е, уровни которого сильно возмущены. В справочнике в соответствии с работой [1675] принимается,
что в этом состоянии Q=1/2. По работе [1675] приняты также значения Q для тех состояний ТаО, переходы
в которые изучены только в матрицах.
88
Тантал и его соединения
ТаО2(г)
чениях Ф°(Т) и S°(T). Учет ряда ненаблюдавшихся состояний молекулы ТаО в настоящем
справочнике оказался существенным в значениях S°(T) выше 5000 К и С(Т) выше 4000 К @,8 и
5 Дж• К • моль при 6000 К соответственно). В работе [4197а], по-видимому, допущены ошибки
в расчетах и соответствующие расхождения достигают 7—8 Дж • К • моль при Т = 2000—
-6000 К.
Константа равновесия реакции ТаО(г) = Та(г) 4т О(г) вычислена с использованием
принятой в справочнике энергии диссоциации
Дг#°@) = Do(TaO) = 785 + 20 кДж • моль я» 65 600 + 1700 см.
Результаты исследований энергии диссоциации ТаО(г) представлены в табл. 35.5. Указанные
в таблице погрешности учитывают воспроизводимость измерений, неточность использованных
в расчетах термодинамических функций, сечений ионизации и термохимических величин. За
Таблица 35.5. Результаты определений D0(TaO) (в кДж • моль)
Авторы
Метод
D0(TaO)
II закон III закон
Инграм и др., 1957 [2776]
Уолш и др., 1960 [4644]
Крикориан, Карпентер, 1965
[3110]
Сму и др., 1976 [4330]
Масс-спектрометрический, 0,6Та(к)+0,2Та2Ов(ж)=
=ТаО(г), 2148—2275 К, 5 точек
То же, ZrO2(K)+Ta(K)=ZrO(r)+TaO(r), 2288 К, 1 точка
Эффузионный, Ьа2О3(к)+Та(к)=2ЬаО(г)+ТаО(г),
2050—2250 К
Масс-спектрометрический, Та(к)+ТаО2(г)=2ТаО(г),
2268 К, 1 точка
То же, 0,6Та(к)+0,2Та2О5(к, ж)=ТаО (г), 1924—
2268 К, 5 точек
То же, 2139—2512 К, 17 точек
То же, 2137—2527 К, 20 точек
То же, 1943—2082 К, 12 точек
То же, 1902—2079 К, 13 точек
То же, Та(к)+РО(г)=ТаО(г)+Р(г), 1960—2411 К,
22 точки а
То же, Та(к)+2А1О(г)=ТаО(г)+А12О(г), 2092—
2507 К а
То же, Ta(K)+TiO(r)=TaO(r)+Ti(r), 2043—2466 К а
То же, 2ТаО(г)=Та(к)+ТаО2(г), 1902—2527 К,
16 точек
То же, YO(r)+Ta'(r)=Y(r)+TaO(r), 2380—2777 К,
35 точек
То же, 2407—2988 К, 18 точек
а Данные представлены в виде уравнения.
617 ±400 820 ±15
Горохов и др., 1979 [310]
886 ±180
—
771 ±32
829 ±13
798 ±14
797 ±40
782 ±30
788 ±16
790 ±35
813 ±15
802 ±15
803 ±25
784+27
891 ±20
895 ±25
778 ±15
704 ±13
783 ±15
783+15
785 ±15
784 ±15
801 ±14
783+50
806 ±15
793 ±15
768 ±17
779+15
исключением величин, полученных в наиболее ранних работах [2776, 3110, 4644], остальные
данные удовлетворительно согласуются между собой. При вычислении средневзвешенного больший
вес придавался результатам исследований обменных реакций с YO, TiO и РО. Исследованиям
равновесия реакции 0,6Та(к) + 0,2Та2ОБ(к, ж) = ТаО(г) придавался меньший вес, так как
компоненты конденсированной фазы могли иметь активности, меньшие единицы.
Принятой энергии диссоциации соответствует
Д//°(ТаО, г, 0) = 243,783 + 20 кДж • моль.
ТаО2(г). Термодинамические свойства газообразного диоксида тантала в стандартном состоянии
при температурах 100—6000 К приведены в табл. 929.
Молекулярные постоянные, принятые для расчета термодинамических функций, приведены
в табл. 35.6. На основании отклонения молекулярного пучка ТаО2 в неоднородном электрическом
89
TaO2(r)
Глава 35
Таблица 35.6. Значения молекулярных постоянных, а также о и рх,
принятые для расчета термодинамических функций ТаО2
Состояние
^3
см
U1 в1 с ¦10
г3 • см6
а
Рх
12А2 950 510 1000 7-102 2 2
Примечание. Т,(А*А{) =4000, Г.^Мд) = 8000, ГДС2^) = И 615, Те{В*В2) = 16 232, Т,(Ё*В{) = 18 000,
Te(F*B2) = 19 000 см.
поле, установленного в работе [2935], в справочнике принимается, что в основном состоянии
Х2А2 молекула ТаО2 имеет угловую структуру (симметрия С2е). Приведенное в табл. 35.6
значение IaIbIc рассчитано по оцененным структурным параметрам (г(Та—О) = 1,72 + 0,03 А,
^.0—Та—О = 100 + 15°), его погрешность составляет 2-1015 г3-сме. Структурные параметры
и основные частоты ТаО2 в состоянии Х2АХ были оценены на основании соответствующих
постоянных молекул VO2, NbO2, VO, NbO и ТаО; погрешности принятых значений основных частот
могут достигать 50 см. Ранее Велтнер и Мак-Леод [4697] в ИК-спектре окислов тантала,
изолированных в матрицах из Ne и Аг, наблюдали переходы в области 912 и 971 см, однако
предложенное авторами этой работы их отнесение к молекуле ТаО2 вызывает сомнение (см. [925]). В
электронном спектре окислов тантала Велтнер и Мак-Леод [4697] наблюдали переходы в области
11 615 и 16 232 см. Хаит и Хачкурузов [1065] выполнили оценку энергии нижних электронных
состояний ТаО2, так же как это было сделано для молекул VO2 и NbO2, и отнесли переходы,
наблюдавшиеся в работе [4697], к переходам в состояния C2B± и DiB% молекулы ТаО2. В табл. 35.6
приведены энергии нижних электронных состояний этой молекулы, принятые по работам [1065,
4697]. Погрешность энергий состояний С2ВХ и D*B2 оценивается равной 50 см, для остальных
состояний 3000—5000 см.
Термодинамические функции ТаО2(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом шести возбужденных электронных состояний по соотношениям A. 168)—A. 170).
Таблица 35.7. Результаты определений Ду?Гс(Та02, г, 0) (в кДж ¦ моль)
Авторы
Инграм и др., 1957 [2776]
Сму и др., 1976 [4330]
Емельянов и др., 1978 [463]
а Данные представлены в виде
Метод
Масс-спектрометрический, 0,2Та(к)-(-0,4Та2О6(к, ж)=
=ТаО2(г), 2019—2206 К, 6 точек
Масс-спектрометрический, 0,2Та(к)-{-0,4Та2О5(к, ж)=
=ТаО2(г), 1880-2155 К, 17 точек
То же, 1911—2108 К, 20 точек
То же, 2139—2512 К, 17 точек
То же, 2137—2527 К, 20 точек
Масс-спектрометрический, Та(к)+ЗАЮ(г)=ТаО2(г)+
+А1(г)+А12О(г), 2092—2507 К, 20 точек а
Масс-спектрометрический, Та(к)-{- 2ТЮ (г)=ТаО2(г)+
+2Ti(r), 2043—2466 К, 37 точек а
Масс-спектрометрический, Та(к)+2РО(г)=ТаО2(г)-{-
+2Р(г), 1960—2411 К, 22 точки а
Масс-спектрометрический, 2УО(г)+Та(к)=ТаО2(г)-|-
+2.Y(r), 2497-2988 К, 15 точек
То же, 2360—2856 К, 24 точки
уравнения.
Д/Я°(ТаО2
II закон
-384 ±250
—167+24
—159+15
-200+15
—170±15
-176±30
—203 +20
—144 ±15
—125 ±55
—188 ±30
, г, 0)
III закон
—166 ±16
—157 ±11
—157 ±11
-154±15
—154 ±14
—174 ±40
—200 ±25
—187 ±15
—154 ±28
-157 ±25
90
Тантал и его соединения Та2ОБ(к, ж)
Погрешности вычисленных значений термодинамических функций ТаО2(г) обусловлены
приближенным характером расчета и неточностью принятых значений молекулярных постоянных.
Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 2, 4 и 8 Дж• К • моль.
Ранее термодинамические функции ТаО2(г) вычислялись в справочниках JANAF [2835] и
Шика [4181], а также в работе Брюэра и Розенблатта [1665] (до 3000 К). Расчеты [1665, 4181]
выполнены для линейной структуры молекулы ТаО2 и соответствующие расхождения с данными
табл. 929 достигают 15 и 17,5 Дж• К • моль в значениях Ф°(Г). Расхождения с данными [2835]
обусловлены разной оценкой основных частот и структурных параметров ТаО2, а также темл что
в настоящем справочнике полнее учитывались возбужденные электронные состояния ТаО2; они
составляют 1—3 Дж-К^-моль в значениях Ф°(Г).
Константа равновесия реакции ТаО2(г) = Та(г) + 20 (г) вычислена с использованием
значения АгН°@) = 1445,566 + 25 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
г, 0) = —170 + 25 кДж • моль.
Это значение основывается на результатах исследования энтальпии образования ТаО2(г),
представленных в табл. 35.7. Погрешности, приведенные в таблице, учитывают воспроизводимость
измерений и неточность использованных в расчетах термодинамических функций,
термохимических величин и сечений ионизации. Как видно из таблицы, разброс величин, полученных в
разных работах, значителен. Принятая величина является средневзвешенной, причем больший вес
придавался исследованиям обменных реакций с РО, TiO, YO и АЮ. Исследованиям равновесия
реакции 0,2Та(к) + 0,4Та2О5(к) = ТаО2(к) придавался меньший вес, так как компоненты
конденсированной фазы могли иметь активности, меньшие единицы.
Та2О6(к, ж). Термодинамические свойства кристаллического и жидкого пентоксида дитантала
стехиометрического состава в стандартном состоянии при температурах 100—5500 К приведены
в табл. 930.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 35.1. За стандартное состояние Та2Об(к) в интервале 0—1633 К принята ромбическая (Р)
модификация [4388], а в интервале 1633—2150 К — моноклинная (а) модификация [4389] г.
При Т < 298,15 К термодинамические функции Та2О5(к) вычислены на основании измерений
теплоемкости, проведенных Келли [2956] E3—294 К) на образце, о котором в [2956] сообщается
только, что он был тщательно очищен. Рентгенографический анализ этого образца, выполненный
Орром [3724], показал, что он представлял ромбическую модификацию Та2О5. Экстраполяция
теплоемкости ниже 53 К проводилась по уравнению CAT) = 3?A83,8/Г) + 6?'B56,1/Г) +
+ 6?F13,8/Т) + 6?A400,5/Г) + 3?E45,6/Г), которое аппроксимирует экспериментальные
данные [2956] с точностью 0,6% 2. Этому уравнению соответствует значение 5°E3 К) =
= 24,3 Дж-К^-моль. Погрешности принятых значений 5°B98,15 К) и #°B98,15 К) - #°@)
(см. табл. 35.1) оцениваются в 2 Дж• К • моль и 0,3 кДж-моль г соответственно.
В интервале 298,15—1633 К для теплоемкости ромбической модификации Та2О5 принято
уравнение (см. табл. 35.1), полученное на основании измерений в работе Орра [3724] C96—1803 К)
энтальпии того же образца, что и в [2956]. Температура полиморфного превращения A633 +
+ 10 К) принята по результатам рентгенографического исследования Рейсмана и др. [2695,
3972], подтвержденным в работе [4650]. Близкие, но менее точные значения получены в
работах [3161, 3534] A593 + 20 К), [3000] A573 К) и более высокое значение A723 + 25 К) — в
работе [4430].
Энтальпия превращения ?S-Ta2O5 в а-Таа05 экспериментально не определялась; по ряду
косвенных соображений 8 можно предположить, что она мала и лежит в пределах от 0 до 4 кДж-моль,
поэтому в справочнике она принята равной 2+2 кДж-моль. Теплоемкость моноклинной
модификации B10 + 10 Дж • К • моль) оценена с учетом измерений энтальпии в работе Орра
13724], экстраполированных до температуры плавления. Температура плавления а-Та2О5 B150 +
+ 20 К) принята по данным Рейсмана и др. [2695, 3972] B145 + 10 К) и Уэринга и Рота [4650]
1 Для Та2О6 (к) известно также несколько метастабильных модификаций (см. [3971, 4650, 4981]).
2 Исследование свойств Та2О3 (к) при Т < 50 К [3135] не обнаружило каких-либо фазовых переходов.
» Медленность протекания (8—а)-превращения (см. [3972, 4430]); измерения энтальпии Та2О5 (к) в работе Орра
[3724] (У=1678 и Г=1803' К) не обнаружили перехода в пределах погрешности измерений (~2 кДж-моль).
91
Та2О5(к, ж)
Глава 35-
B160 + 20 К), полученным на образцах с содержанием примесей менее 1 %. Близкие значения:
были получены в работах [4097 ] B148 К) и [5041 ] B153 К) для менее чистых образцов Та2О5.
Существенно более низкие значения, полученные в работе [2071 ] A963, 2013 и 2083 К),
соответствуют, как это было показано в работах [2695, 3972], плавлению низкотемпературной
ромбической модификации Та2О5. Энтальпия плавления Та2О5 не измерялась. Принятое в справочнике^
значение A20 + 20 кДж-моль) оценено в предположении, что ДЯ1?°(Та206) ?« Am-Sro(Nb20B)=
= 56,4 Дж-К^-моль. Теплоемкость Та2О6(ж) B20 + 20 Дж • К • моль) оценена на основании
расчета по неэмпирическому уравнению, принятому в справочнике, а также сравнения со
значениями C;(Nb2O8, ж) и С;(Та2О5, к, Тп).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1500, 3000 и 5500 К оцениваются в 2,
6, 16 и 35 Дж-К^-моль.
Ранее термодинамические функции Та2О5(к, ж) вычислялись в справочниках JANAF [2835]
(до 5000 К), Шика [4181] (до 6000 К) и Келли [2960] (до 2000 К) на основании данных [2956] ж
[3724] для ромбической модификации. Расхождения между значениями Ф°(Т), приведенными
в [2835] и табл. 930, составляют 1—4 Дж-К^-моль.
Константа равновесия реакции Та2О5(к, ж) = 2Та(г) + 5О(г) вычислена с использованием
значения ДГ?Г°(О) = 4836,800 + 7,2 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
kfHo(Ta20b, к, ромб., 298,15 К) = —2049+.4 кДж • моль..
Эта величина основана на измерениях энтальпии образования Та2О6 (к, монокл.),
представленных в табл. 35.8. Приведенные в таблице погрешности учитывают только воспроизводимость-
измерений. При сгорании тантала образуется моноклинная модификация Та2О5 [543, 544, 2734Г
4495], которая при 298,15 К термодинамически менее стабильна, чем ромбическая. Наиболее
точные и детальные измерения, выполненные Корниловым и др. [543, 544], позволяют выбрать
для энтальпии образования моноклинной модификации значение —2047,2 + 1,7 кДж-моль.
Эта величина подтверждается большинством приведенных в табл. 35.8 данных, в первую очередь
работами [754, 2734, 2756]. При вычислении энтальпии образования ромбической модификации
принималось значение Д/г#°B98,15 К) = 2 + 2 кДж-моль (см. выше).
Т а б л и
Мутман
ц а 35.8. Результаты
Авторы
и др., 1907 [3610]
Муз, Парр, 1924 [3554]
Зивертс
Беккер,
Нейман
Хамфри
и др., 1930 [4287]
Рот, 1933 [1449]
и др., 1934 [3646]
1954 [2756]
Смирнова, Ормонт, 1955—1956
9541
Хьюбер
и др., 1963 [2734]
Корнилов я др., 1982—64 [543,
Панова
и др., 1978 [754]
определении
Д7Л-°(Т
а2С>5, к,
монокл., 298, 15 К) (в кДж
Мето;
I,
• МОЛЬ)
AJ7°(Ta2O5, к,
монокл., 298,15 К)
Калориметрический, 2Та(к)+6/202(г)=Та206(к,
монокл.)
То же
То же
То же
То же
То же
[952, То же
То же
(чистота
(чистота
(чистота
(чистота
(чистота
(чистота
(чистота
(чистота
образца
образца
образца
образца
образца
образца
образца
образца
Та
Та
Та
Та
Та
Та
Та
Та
99,76%)
99,96%)
99,9%),
99,9%),
99,6%),
99,89%)
99,8%)
99,8%),
544] То же (чистота образцов Та 99,64 и 98
То же
(чистота
образца
Та
99,96%)
7 опытов
3 опыта
3 опыта
6 опытов
, 6 опытов
12 опытов
,15%), 15 опытов
-1294
-2086 ±15
—2010 ±7
—2033 ±2
—2092 ±7
-2045,4 ±4,5
—2008,3 ±3,3
—2037 ±6
—2047,2±1,7
—2043,9 ±6,3
Исследования реакции
(к, ромб.) методом ЭДС [1389, 1390,
2767, 3981, 4869] приводят к значениям энтальпии образования, близким к принятому. Однако»
их погрешности, вычисленные с учетом неточности термодинамических функций и отклонения
от стехиометрического состава, превышают 10 кДж-мольГ1.
Глава 36
ТИТАН И ЕГО СОЕДИНЕНИЯ
Т1(к, ж), Ti, Ti+, ТЮ(к, ж), ТЮ, ТЮ+, ТЮ2(к, ж), ТЮ2(к, анатаз), ТЮ2,
Т12О3(к, ж), Т13О5(к, ж), Ti4O7(K, ж)
В настоящей главе рассматриваются титан и его оксиды в конденсированном и газообразном
состояниях. Термодинамические свойства четырех газов (Ti, Ti+, TiO, TiO+) рассчитаны до 10 000 К,
одного (ТЮ2) — до 6000 К. Потенциал ионизации TiO2 достигает 10 эВ, поэтому
соответствующий ион в справочнике не рассматривается. В конденсированном состоянии существует большое
число оксидов титана, состав которых в большей или меньшей степени может отклоняться от
стехиометрического. В настоящей главе рассматриваются основные, наиболее стабильные и
относительно хорошо изученные оксиды стехиометрического состава: TiO, TiO2 (рутил и анатаз),
Ti2Og, Ti3O5 и Ti4O7. Низкотемпературная модификация ТЮ(к), стабильная при Т ^ 1265 К,
имеет узкую область гомогенности (TiO09—TiOlx), в отличие от высокотемпературных
модификаций, для которых характерна широкая область гомогенности (ТЮ08—ТЮ1>3),
увеличивающаяся с возрастанием температуры. Области гомогенности ТЮ2(к, рутил) и Ti2O3(k)
характеризуются соответственно следующими интервалами: ТЮ1973—ТЮ2000 И Ti2O2 9s—Ti2O3 02 [1324].
Ti3O5 и Ti4O7 имеют чрезвычайно узкие области гомогенности. Известны также субоксиды титана
TieO, Ti4O, Ti2O — вещества малоустойчивые, и высшие оксиды — члены гомологического ряда
Ti,,O2»-n гДе п ^ 4 (фазы Магнели), которые имеют структуру, близкую к структуре рутила.
Свойства оксидов ряда TiBO2K_-[ сильно сближаются по мере увеличения п. Некоторые
термодинамические свойства соединений этого ряда удалось определить вплоть до п = 30 (см. [4756]).
Термодинамические свойства всех веществ приведены для природной изотопической смеси
титана (атомный вес 47,90) и кислорода. Однако различие молекулярных постоянных
индивидуальных изотопических модификаций не учитывалось ввиду отсутствия соответствующих
данных для других соединений. Для элементарного титана в Приложении 3.10 приведены сведения
о его критических постоянных.
ТЦк, ж). Термодинамические свойства кристаллического и жидкого титана в стандартном
состоянии при температурах 100—5600 К приведены в табл. 931.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 36.1. В справочнике за стандартное состояние Ti(K) в интервале 0—1156 К принята
гексагональная (а) модификация (структурный тип Mg), а в интервале 1156—1944 К — кубическая
(Р) модификация (структурный тип a-Fe). При 0,39 К [629] Ti(K) переходит в сверхпроводящее
состояние х.
При Т < 298,15 К термодинамические функции Ti(K) вычислены на основании согласующихся
между собой результатов измерений теплоемкости, выполненных Нейпом и др. [3049], Хейком
.и Кейпом [2550], Агарвалом и Беттертоном [1268] A,2—4,5 К), Авеном и др. [1365] D—16 К),
Уолкоттом [4751] A,2—21 К), Думмером [2125] @,9—12 К), Котеном и Джонстоном [3085, 30861
A5—306 К), Клузиусом и Францозини [1878] A4—272 К) и Сталинским и Биганским [4376]
(B7—360 К) на образцах Ti чистотой 99,5%—99,9%. Погрешности принятых значений 5°B98,15)
и Я°B98,15 К)—#°@) (см. табл. 36.1) оцениваются в 0,10 Дне-К-моль и 0,013 кДж-моль.
С принятыми значениями теплоемкости Ti удовлетворительно согласуются данные [2607] A,5—
16 К), [1902] B,3—5,3 К), а также данные [1267] C—18 К) и [4846] B,2-330 К), приведенные
-только на графике. Менее точные данные были получены в работах [2925] E3,5—295 К), [2193]
^1,8-4,2 К) и [1727] B2,5-200 К) 2.
11 При высоких давлениях известны тетрагональная модификация титана кШ (тройная точка I—II—III при
80 кбар) и кубическая модификация kIV (при давлении > 350 кбар) [1021].
3 Попытка обнаружить скачок теплоемкости, обусловленный переходом Ti в сверхпроводящее состояние,
предпринятая Ваттом [1429] @,24—4,2 К), не удалась, возможно, из-за недостаточной чистоты исследованного
образца.
93
ТЦк, ж)
Глава 36
Таблица 36.1. Принятые значения термодинамических величин для титана и его соединений
в кристаллическом и жидком состояниях
Вещество
Ti
ТЮ
тю2
TiO2
Ti2O3
Ti3O6
Ti4O7
Состояние
1
ю
00 S-
С73 о
о
fe I
кДжХ
X моль
kII, гекс. (а) 4,824
kI, куб. (Р) —
ж —
кШ, монокл. (a
6,175
kII, куб. (p) -
kI, куб. Or) —
ж —
к, тетр. (рутил) 8,68
ж —
к, тетр. (анатаз) 8,63
к1, гекс. 14,09
к1, гекс. —
к1', гекс. —
кГ, гекс. —
ж —
КН, МОНОКЛ. (а) 22,63
к1, ромб. (Р) —
ж —
к, трикл. 35,63
ж —
И
Ю ¦
СО
Оз
о
Дж-
К
ю
оо"
аз
С\]
1 о в.
К X
X моль
30,72
—
—
34,79
—
—
—
50,62
—
50,08
77,29
—
—
—
126,2
—
—
201,48
—
Примечание. C%(T)=a+b -T—c -T^+d Л
Ti
TiC
Ti2O3
Ti3O5
25,06
—
—
39,905
—
—
—
55,08
—
55,27
95,81
—
—
—
150,83
—
—
216,81
—
Коэффициенты в
а'
160,773
16,908
46,8
68,496
51,328
48,187
70
63,196
100
65,204
27,221
473,311
772,269
106,589
170
8416,051
167,964
260
246,224
362
Г2+е-Г3+/-Г* (в
a d= 1444,158-Ю-», е=—1343,516 -Ю"»,
: а о!=31,626-10~6.
а d=3105,313 -Ю1; б d=2020,758 -10"*.
а d=30982,509 -КГ».
ДЛЯ СрA
Ъ • 103
-693,569
5,884
—
—40,652
12,100
11,678
—
11,820
—
10,972
230,048
—2127,303
—2268,375
33,367
—30133,084
42,610
—
48,007
—
уравнении
)
с • 10~**
22,566 а
об
—
17,140 а
11,641
-17,298
—
10,347
—
11,739
0 а
Об
—35,160
—
1809,144 а
__
—
38,871
—
Дж-К^-моль).
/=467,200 -Ю2; б ?
2=3,192 -10~6
Интервал
температур
К
298,15-1156
1156-1944
1944-5600
298,15—1265
1265—1810
1810—2030
2030-5000
298,15-2185
2185-5000
298,15-2000
298,15-400
400—464
464-580
580-2110
2110-5000
298,15-448
448-2050
2050—5000
298,15—1960
1960—5000
или
Т
1156
1944
—
1265
1810
2030
—
2185,
—
—
464
.—
2110
—
448
2050
—
1960
—
ИЛИ
кДжХ
X моль
3,8
14,6
—
3,6
2,7
47,1
—
68
—
—
0
—
92
—
9,6
167
—
180
—
Термодинамические функции Ti(K) в интервале 298,15—1156 К вычислены с использованием
приведенного в табл. 36.1 уравнения для теплоемкости, которое получено совместной обработкой
результатов измерений энтальпии a-модификации Ti в работах Егера и др. [2816, 2817] D93—
1143 К), Котена [3085] A050-1154 К), Серебренникова и Гельда [931] B94—1153 К), Гельда и
Путинцева [269] D00—1500 К) и измерений теплоемкости в работах Бэкхерста [1370] (873—1153 К),
Сталинского и Биганского [4376] B98,15—360 К), Новикова и др. [728] C75—807 К), Кольхааса
и др. [3062] C20—1150 К), Беляковой [94] B93—1123 К) и Зарецкого и Пелецкого [437]
G06—1154 К); наибольший вес придавался данным работ [94, 269, 437, 931, 3062, 3085], а при
температурах 1060—1156 К, где измерения разных авторов весьма противоречивы,
использовались только данные Зарецкого и Пелецкого, которые приняли меры для сохранения чистоты
титана и наиболее надежно установили резкий рост его теплоемкости в этой области.
Существенно менее точные измерения [47, 48, 147, 734, 1055, 3685] не принимались во внимание.
Температура полиморфного превращения A156 + 4 К) найдена в работах Бургерса и др.
[1725, 2017] A156 ± 20 К), Мак-Квиллона [3466, 3467, 3468] A156 + 1 К), Голутвина [299]
A156 К), Дувеца [2133] A156 ± 5 К), Руди и Прогульского [4084] A156 К) для образцов
чистотой лучше 99,9%. С принятым значением согласуются данные [2154, 4222] A157 К), [549, 1265]
A154 К), [437, 888] A155 К), [1860, 2220, 2483, 2564, 4202] A159 К); несколько более высокие
значения были получены авторами [1649, 1794, 3062, 3696, 4767] (от 1161 до 1168 К). Энтальпия
полиморфного превращения C,8 + 0,4 кДж-моль) принята как разность значений H°(H5Q К)—
94
Титан и его соединения ТЦк, ж)
#°B98,15 К) для а- и ^-модификаций Ti, соответствующих приведенным в табл. 36.1
уравнениям для CAT). В пределах погрешности она согласуется с результатами определений в
работах [269, 299, 931, 1370, 1649, 1794, 2569, 2816, 2817, 3062, 3085, 4222].
При температурах 1156—1944 К термодинамические функции вычислены с помощью
уравнения для теплоемкости (см. табл. 36.1), полученного на основании измерений энтальпии в
работах Егера и др. [2816, 2817] A210-1476 К), Котена [3085] A156-1800 К), Гельда и Путинцева
[269] A200—1500 К) и Каца и др. [119, 493, 1485] A736—1908 К) и измерений теплоемкости
в работах Кольхааса и др. [3062] A156—1350 К), Шестопала [1150] A400—1450 К), Цезерляна
и Миллера [1792] A500—1900 К) и Зарецкого и Пелецкого [437] A156—1641 К). Энтальпия и
теплоемкость ^-модификации Ti измерялись также в работах [4222] A156—1233 К),
[1370] A156-1353 К), [299] A156-1402 К), [931] A156—1923 К), [2682] A156—1345 К),
[432] A156-1300 К), [47, 48, 1055] A156-1700 К), [665, 666] A156 К), [94] A173-1223 К),
[889] A156—1944 К). В них были получены неточные и противоречивые данные, обусловленные
высокой химической активностью этой модификации титана, в частности, ее повышенной
способностью адсорбировать газы. Принятое значение температуры плавления A944 ± 5 К)
является средним из результатов определений в работах Скофилда и Бэкона [4204] A938 + 10 К),
Скофилда [4201] A948 ± 10 К), Фэррэра и Марголина [2218] A943 ± 10 К), Руди и Прогуль-
ского [4084] A944 + 8 К), Ориани и Джонса [3721] A948 + 6 К), Цезерляна и Миллера [1793]
[1945 + 5 К), Дирдорфа и Хайеса [2011] A943 ± 10 К), Бикердике и Хьюза [1556] A945 ±
+ 8 К), Кенисарина и др. [119, 496, 1485] A941 + 4 К), полученных для образцов чистотой
выше 99,9%. Для менее чистых образцов Ti и менее точными методами Тт определялась также
в работах [1726] B067 К), [2219] A998 К), [2564] A996 К), [1256] A976 К), [1441, 3434] A956 К),
[549] A951 К), [1265] A856 К). Энтальпия плавления A4,6 + 1,0 кДж-моль) принята по
разности энтальпии ТЦж) и Ti(K) при 1944 К. Близкие значения получены в работах [119, 14851
A4,2 + 0,5 кДж-моль) и [493] A3,8 + 0,5 кДж-моль), а также в работе [1617] A5,0 +
+ 0,2 кДж-моль, пересчет данных работы [4543], авторы которой прлучили несколько
меньшее значение A3,2 + 0,5 кДж-моль)). Более высокие значения были получены методом
смешения [405, 674, 559] A7,2 ±1,2 кДж-моль) и методом импульсного нагрева [665, 666, 667]
A8 кДж-моль). Энтальпия Т1(ж) измерялась методом левитационной калориметрии Тревер-
тоном и Маргрейвом [4543] A969-2315 К) и Кацем [493] A947-2049 К) (см. также [119, 1485])
и методом импульсного нагрева Зейделем и Китцелем [4245] A943—5100 К). Наиболее надежные
данные были получены в работе [493] и им соответствует принятое в справочнике значение
теплоемкости D6,8 + 5 Дж-К^-моль). В пределах погрешности оно согласуется со значениями,
полученными в работах [4543] D5,5 Дж^К^-моль), [1617] D6,5 Дж-IT1 -моль), [119, 493,
1485] D7,1 Дж-К-моль). Данным [4245] соответствует завышенное значение E1,3 Дж-К X
Хмоль).
Погрешности приведенных в табл. 931 значений Ф°(Т) при 298,15, 1000, 3000 и 5500 К
уцениваются в 0,07, 0,5, 3 и 7 Дж-К^-моль соответственно.
Ранее термодинамические функции Ti(K, ж) вычислялись в справочниках JANAF [2834] (до
4500 К), Халтгрена и др. [2752], Шика [4181] (до 3600 К), Келли [2960] и Сталла и Зинке [4421]
(до 3000 К). Все расчеты основаны на существенно менее точных экспериментальных данных для
Ti(K) и на оцененных свойствах Т1(ж). Расхождения в значениях Ф°(Г), приведенных в [2834]
и в табл. 931, составляют от 0,05 до 2,6 Дж-К^-моль.
В настоящем справочнике в качестве стандартного состояния титана принято
кристаллическое состояние и в соответствии с этим
Давление пара титана в реакции Ti(K, ж) = Ti(r), вычислено с использованием принятой
в справочнике энтальпии сублимации
Дг#°@) = A,#°(Ti, к, 0) = 471+2 кДж • моль.
В табл. 36.2 представлены результаты расчета энтальпии сублимации титана на основании
измерений разных авторов. В соответствии с данными [2154, 2920, 4779] в расчетах принималось,
что пары титана одноатомны и что коэффициент испарения с открытой поверхности в вакуум
95
Ti(r)
Глава 36
\
Таблица 36.2. Результаты определений Ag-H"°(Ti, к, 0) (в
Авторы
Блочер, Кемпбелл, 1949 [1580]
Карпентер, Мейер, 1951 [1758]
Эдварде и др., 1953 [2154]
Моррисон, 1960 [3567]
Кузьмин, Палатник, 1962 [601]
Кузьмин, Моисеева, 1964 [600]
Штрассмайер, Штарк, 1967 [4409]
Коч и др., 1967 [3056]
Кудрявцев, Иванов, 1969 [594] (см.
также [593])
Николаев, Немец, 1970 [726]
By, Уолбек, 1971 [4779]
Баталов, 1971 [67]
Метод
Испарение с открытой поверхности, 1525—1844 К,
10 точек
То же, 1641—1823 К,а 6 точек
То же, 1626—1766 К,а 7 точек
То же, 1450—1700 К,а- б
То же, 1657—1840 К,а- 6
То же, для системы Ti—Nb, 1590—1790 К,а б
Испарение с открытой поверхности,1156—1774 К,
8 точек
То же, 1956—2196 К, 10 точек
То же, с использованием радиоактивного
изотопа, 1473—1873 К, 3 точки
Атомно-адсорбционные измерения оптической
плотности пара, 1630—1900 К б
Эффузионный, 1830—1923 К, 16 точек
Измерение электросопротивления нагреваемой
проволоки, 1000—1200 К
As#°(Ti, к, 0)
II закон
III закон
474+18 469,3 ±1,2
475+67 469,3 ±1,6
475 ±11 471,2+0,2
484 472,2
478 469,2
529 —
472+37 464,2+1,4
424+43 465,4±1,6
448 —
467 ±21 —
473±83 470,9+1,2
466+10 —
а Температура измерений пересчитана в соответствии с принятым значением излучательной способности ТЦк)
(¦^о.оБа =0,43) [2154]; " Данные представлены в виде уравнения. ,
равен единице. Приведенные в таблице погрешности отражают только воспроизводимость
измерений.
Принятое значение основано на наиболее точных измерениях Эдвардса и др. [2154]
(испарение с открытой поверхности) и By и Уолбека [4779] (эффузионный метод). Его погрешность
включает как воспроизводимость экспериментальных данных, так и неточность использованных в
расчетах термодинамических функций. С принятым значением в пределах погрешности согласуются
измерения скорости испарения с открытой поверхности в работах [601, 1580, 1758, 3567]; данные
[67, 726, 3056, 4409] менее точны, но также не противоречат этой величине.
Ti(r). Термодинамические свойства газообразного титана в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 932.
Для расчета термодинамических функций использованы уровни энергии х атома Ti в
интервале 0—54 800 см^1, соответствующие валентным электронным конфигурациям ...3d24s2 (к ней
относится основной уровень3^), ...3d4p2, ...3d4, ...3d4l(l = 0^-3), ...3d4s4l(l = 1-^-3) и ...3dAs4p
и имеющие суммарный статистический вес 4512. Принятые величины основаны на данных,
рекомендованных Шугаром и Корлиссом [4439] в результате анализа спектральных исследований,
а также на оценках для ненаблюдавшихся уровней энергии, выполненных по методике,
изложенной в Приложении 6 (для состояния ...3d24s2 lS принята величина, полученная Мэни [3365] в
результате полуэмпирического расчета).
Термодинамические функции Ti(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при Т ^ 3000 К обусловлены преимущественно неточностью фундаментальных
постоянных, а при более высоких температурах — неточностью оценки энергии ряда уровней и
пренебрежением ридберговскими состояниями. Погрешности значений Ф°(Т) при 3000, 6000 и
10 000 К составляют 0,02, 0,05 и 0,5 Дж-К^-моль.
Ранее термодинамические функции Ti(r) вычислялись в таблицах JANAF [2834],
справочниках Шика [4181] и Сталла и Зинке [4421] (до 3000 К), в работах Жирницкого [4836] (до
15 000 К), Хилзенрата и др. [2652] (до 10 000 К), Кольского и др. [3065, 3067] (до 8000 К), Гил-
леса и Уитли [2406] (до 5000 К) и Каца и Маргрейва [2934] (до 2000 К). Кроме того, в работе
1 См. примеч. 1 к тексту по Сг (г).
96
Титан и его соединения Ti+(r)
Черниховского [1112] были рассчитаны значения Qai до 6000 К. При Т <^ 3000 К результаты
всех расчетов (за исключением расчета [3067], в котором принято завышенное значение R, и
[4836], где, по-видимому, допущена ошибка при вычислении поступательной составляющей)
согласуются между собой и с данными табл. 932 в пределах 0,02—0,03 Дж-К^-моль. При
более высоких температурах расхождения возрастают до 0,07 Дж-К^-моль в значении ?оF000 К)
с данными [2834] и достигают 0,2 и 1,5 Дж-К^-моль при 10 000 К с данными [2652] в
значениях Ф°(Т) и S°(T); это обусловлено тем, что в справочнике учтено большое число ненаблюдав-
шихся уровней энергии. В работе [4836] учтены ридберговские состояния (га ^ 9);
соответствующие расхождения имеют противоположный знак и составляют при 10 000 К (после коррекции
QUOJ 1, 1 и 17 Дж-К^-моль для Ф°(Т), S°(T) и С'/Т).
Энтальпия образования
&fH°(Ti, г, 0) = 471 ±2 кДж моль-1
соответствует принятой в справочнике величине энтальпии сублимации.
Ti+(r). Термодинамические свойства газообразного положительного иона титана в стандартном
состоянии при температурах 298,15—10 000 К приведены в табл. 933.
Для расчета термодинамических функций использованы уровни энергии 1 иона Ti+ в
интервале о—78 000 см, соответствующие электронным конфигурациям ...3d24s (к ней относится
основной уровень 4i\), ...3d4l (I = 1,2), ...3d3, ...3d4s2 и ...3d4s4p и имеющие суммарный
статистический вес 970. Принятые величины основаны на данных, рекомендованных Шугаром и Кор-
лиссом [4439], а также на оценках ненаблюдавшихся уровней энергии для конфигураций ...3dHl
(I = 1,2) и ...3d4s4p, выполненных по методике, изложенной в Приложении 6 (для состояния
...3d32D принята величина, полученная Мэни [3365] согласно полуэмпирическому расчету).
Термодинамические функции Ti+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25). Погрешности вычисленных величин при Т ^ 5000 К обусловлены главным
образом неточностью фундаментальных постоянных и не превосходят 0,02 Дж-К •моль в
значениях Ф°(Т) и S°(T), при более высоких температурах становятся существенными ошибки из-за
использования оцененных значений энергии ряда уровней, которые могут достигать 0,3 Дж-К-1Х
Хмель в Ф°(Т).
Ранее термодинамические функции Ti+(r) вычислялись в таблицах JANAF [2834], а также
в работах Грина и др. [2473] (до 50 000 К), Жирницкого [4836] (до 15 000 К) и Хилзенрата
и др. [2652] (до 10 000 К). Кроме того, в работе Черниховского [1112] были рассчитаны значения
Q3I до 6000 К. Результаты всех расчетов, за исключением [4836], согласуются с данными табл. 933
в'пределах 0,02—0,03 Дж-К^-моль в значениях Ф°(Т) при Г ^ 5000 К; при более высоких
температурах возникают небольшие расхождения (до 0,1 и 0,2 Дж-К^-моль в S°(T) и С"р(Т)
при 10 000 К для [2652]) вследствие того, что в настоящем справочнике учтены многие ненаблю-
давшиеся электронные состояния. С работой [4836] имеют место нерегулярные расхождения (до
2 Дж-К^-моль); практически полное совпадение значений Q3I, вычисленных в настоящем
справочнике и в [4836], свидетельствует о численных ошибках в расчете этого автора.
Константа равновесия реакции Ti+(r) + е (г) = Ti(r) вычислена с использованием значения
Дг#°@) = —659,1 + 1,2 кДж-моль^1, соответствующего принятому в справочнике потеншталу
ионизации i
/0(Ti) = 659,1 ± 1,2 кДж моль я* 55 100]± 100 см
Это значение основано на данных Шугара и Корлисса [4439] для первых членов ридбергов-
ских серий ...3d2isnd ЪН (п = 4, 5) и ...3d2AsndbG(n — 4, 5) атома Ti и величине 0,02 + 0,02 для
разности смежных значений квантовых дефектов, оцененной по соответствующим данным для
атомов Са, Сг и Мп. Принятое значение согласуется с найденными ранее по спектральным данным
в работах [4105] E5 138 см) и [1768] E5 010 см); близкие величины были получены также
методом электронного удара F56 + 10 кДж -моль" [3943] и 654 + 20 кДж-моль [3944]').
Принятому значению соответствует
Д^ТГ", г, 0)= 1130,1 +2,3 кДж-моль-1.
1 См. примеч. 1 к тексту по Сг (г).
97
ТЮ(к, ж) Глава 36
ТЮ(к, ж). Термодинамические свойства кристаллического и жидкого оксида титана стехио-
метрического состава в стандартном состоянии при температурах 100—5000 К приведены
в табл. 934.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 36.1. В справочнике за стандартное состояние ТЮ(к) в интервале 0—1265 К принята
моноклинная (а) модификация, в интервале 1265—1810 К — кубическая (|3) модификация, а в
интервале 1810—2030 К — кубическая (у) модификация (структурный тип NaCl) *. При Т <^ 0,5 К
согласно данным [3711] ТЮ(к) переходит в сверхпроводящее состояние.
Термодинамические функции при Т <; 298,15 К вычислены па основании измерений
теплоемкости, выполненных Оказом и Кеезомом [3711] @,2—2,0 К, суммарное содержание примесей
в образце —0,02%), Березовским и Пауковым [4854] (8,5—305 К, образец состава TiO0 99±0 02
с суммарным содержанием примесей ~0,03%) и Шомейтом [4270] E3—296 К, примеси 0,1%
TiC и 0,7% SiO2, на примеси введена поправка). Данные [4854] и [4270] согласуются между
собой в пределах 0,5% и с учетом измерений [3711] приводят к значениям 51ОB98,15 К) и
JGT°B98,15 К)—Н°@), представленным в табл. 36.1, погрешности которых оцениваются в 0,15 ДжХ
XК • моль и 0,02 кДж-моль соответственно2. Теплоемкость ТЮ(к) исследовалась также
Черняевым и др. [1114, 1115] E0-300 К) и Бейером [1542] E-300 К).
В интервале 298,15—1265 К термодинамические функции вычислены по уравнению для
теплоемкости (см. табл. 36.1), полученному совместной обработкой результатов наиболее надежных
измерений энтальпии а-модификации TiO, выполненных Нейлором [3637] C57—1254 К,
образец такой же, как в [4270]), Ефремовой и Матизеном [419] F84—1265 К, образец такой же, как
в [4854]) и Ерофеевой и др. [413] D93—1082 К). Данные работы [413] менее точны и
учитывались с меньшим весом. Погрешность аппроксим ции значений C°p(TiO, к) принятым уравнением
составляет 1% при 298,15 < Т < 1000 К и 1,5% при 1000 < Т < 1265 К.
Температура фазового перехода между а- и [3-модификациями A265 + 15 К) принята по
результатам измерений энтальпии ТЮ(к) авторами [419, 3637], она согласуется с данными [2045,
2653, 2904/4205, 4496, 4497, 4666, 4667] 3. Энтальпия а<^р превращения C,6 ± 1,2 кДж-моль)
определена как разность между значениями энтальпии [3- и а-модификаций TiO при 1265 К.
Близкое значение b.trH было получено Нейлором [3637] C,43 кДж-моль).
В интервалах 1265—1810 и 1810—2030 К термодинамические функции ТЮ(к) вычислены по
уравнениям для СЦТ) (с-г табл. 36.1), полученным т-! основании измерений энтальпии [3-TiO
в работах Нейлора 13637] A267—1772 К) и |3- и т-ТЮ в работе Ефремовой и Матизена [419]
A268—1809 К, 1814—2027 К). Эти уравнения воспроизводят экспериментальные данные с
погрешностью, не превосходящей 0,6%. Температура перехода между [3- и у-модификациями A810 +
+ 10 К) основана на данных [419], а энтальпия этого перехода B,7 + 0,9 кДж-моль) принята
по разности энтальпии у- и |3-модификаций при 1810 К.
Плавление TiO согласно измерениям [1720, 2060, 3664, 4205], которые обобщены в диаграмме
состояния системы Ti—О [4632], происходит в интервале 2020—2046 К через образование
гетерогенной системы. С этим представлением согласуются измерения энтальпии ТЮ(к, ж) в этой
области, выполненные Ефремовой и Матизеном [419J4. В справочнике температура плавления
TiO принята 2030 ± 15 К как середина интервала, в котором по данным [419] происходит
возрастание энтальпии, обусловленное инконгруэнтным плавлением TiO. Это значение
согласуется в пределах погрешности с полученными в работах [2006] B022 К) [1659, 1720] B036 ±
± 25 К), [4205] A996-2036 К), [3664] B026 и 2046 К). Энтальпия плавления D7,1 + 4,0 кДжХ
Хмоль) принята как разность значений энтальпии ТЮ(ж) и у -модификации при Тт. Теплоем-
1 При стехиометрическом составе TiO его «титановая* и «кислородная» подрешетки содержат —15% вакансий
(см. [1030]). Согласно данным многочисленных исследований (см., например, [419, 2189, 2653, 4496, 4497, 4.647,
4666—4668]) эти вакансии расположены упорядочено в a-TiO и полностью разупорядочены в 7-TiO; в
отношении степени упорядоченности вакансий в (З-ТЮ литературные данные противоречивы. При высоких давлениях
концентрация вакансий в ТЮ(к) уменьшается.
2 В связи со способностью ^-модификации ТЮ(к) легко закаливаться и несовместимостью термохимических
данных в системе Ti—О [2401, 3068, 4633] в работах [2664, 2834] принималось, что ТЮ(к) имеет остаточную
энтропию при Т=0. Однако позднее работы [2903, 3712] показали ошибочность этого предположения.
3 Противоречивые данные о характере (а— Ш-перехода обсуждаются в ряде работ (см. [419, 1720, 2904, 4205,
4497]).
4 Согласно диаграмме состояния системы Ti—О, предложенной в работе [2904], ТЮ(к) плавится конгруэнтно.
98
Титан и его соединения
TiO(r)
кость ТЮ(ж) G0 + 5 Дж-К х-моль х) основана на измерениях энтальпии в интервале 2050—
2224 К, выполненных Ефремовой и Матизеном [419].
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 3000 и 5000 К оцениваются в 0,1,
0,5, 3 и 10 Дж-К^-моль соответственно.
Ранее термодинамические функции ТЮ(к, ж) вычислялись в справочниках JANAF [28351
(до 4500 К) и Шика [4181 ] (до 6000 К) на основании данных [3637, 4270] для ТЮ(к) и оценок
термодинамических свойств ТЮ(ж). Данные [2835] отличаются от приведенных в табл. 934 на 0,03—8
Дж • К-моль" в значении Ф°(Т). В [4181] получены неверные значения функций вследствие
введения поправки на остаточную энтропию ТЮ(к) и учета несуществующего фазового перехода
при Т < 50 К.
В справочнике принимается энтальпия образования
AfH°(TiO, к, монокл., 298,15 К) = —542 + 4 кДж • моль.
Результаты определений этой величины представлены в табл. 36.3. Принятая величина
является средней взвешенной из результатов калориметрических измерений Чарлю и др. [1818]
Таблица 36.3. Результаты определений AfH°(TiO, к, 298,15 К) (в кДж • моль-1)
Авторы
Метод
AfH°(TlO, к, 298,15 К)
II закон III закон
Хамфри, 1951 [2754]
Ария и др., 1957 [41] (см. также
[40, 233, 261])
Сузуки, Самбонги, 1972 [4444]
Калориметрический, TiO(K)+1/2O2(r)=TiO2(K,
рутил), 303 К, 7 опытов
Калориметрический, реакции сгорания
TiO0,29-TdO2,00, 293 К
—518,7
—526
—559+5 —540,6+5,0
-628+40 —539+10
ЭДС, измерения AG(O2) для TiO! „—ТЮ2 „, —560+7 —543,2+7,0
1173—1873 К, 8 изотерм, расчет ArG°(T) для
реакции ТЮ(к)+1/2О2(г)=ТЮ2(к, рутил)
То же для реакции ТЮ(к)+1/4О2(г)=
=1/2Ti2O3(K)
Сузуки, Самбонги, 1972 [4444]; Ко- ЭДС, расчет ДГ€°(Г) реакции Ti(K)+V202(r)=
марек, Силвер, 1962 [3068] =ТЮ(к) по ДС(О2) для ТЮЖ при 0,6 < х < 1,0,
1173—1873 К [4444] и по Д#(О2) и Д?(О2)
при х < 0,4, 964—1281 К [3068]
То же, при 0,334 < х < 1,0 [4444, 3068] и —639+40 —537+10
Д/го(ТЮ0,т) [3349]
Чарлю и др., 1974 [1818] Калориметрический, реакция ТЮ(к)+1/2О2(г)= —543,2+4,6
=ТЮ2(к, рутил), 1100 К, 6 опытов I
и измерений ЭДС, выполненных Сузуки и Самбонги [4444] для системы Ti—О в интервале
составов TiO—TiO2. Она подтверждается менее точными данными [3068, 3349, 4444] для системы
Ti—О в интервале составов Ti—TiO. Электрохимические измерения в работе [2664] для TiO^^i»
A275—1445 К, 5 опытов) недостаточны для точного определения AfH°(TiO, к) г.
Калориметрическим данным [41, 2754] соответствуют заниженные по абсолютной величине значения
энтальпии образования, по-видимому, вследствие фазовой неоднородности и отклонения от стехиоме-
трического состава образцов ТЮ(к).
Давление пара в реакции ТЮ(к, ж) = TiO(r) вычислено с использованием значения АгН°@) =
= Д #°(ТЮ, к, 0) = 588,793 + 11 кДж-моль, соответствующего принятым в справочнике
величинам Д,#°(ТЮ, к) и D0(TiO).
ТЮ(г). Термодинамические свойства газообразного оксида титана в стандартном состоянии
при температурах 100—10 000 К приведены в табл. 935.
Молекулярные постоянные TiO, принятые в справочнике на основании результатов
исследований электронного спектра и приближенных оценок, представлены в табл. 36.4. В электрон-
1 Еслгт пренебречь зависимостью AG (О2) от г в системе ТЮТ<1 5, эти данные могут быть интерпретированы как
соответствующие реакции ЗТЮ (K)=Ti (n)+Ti2O, (к), что ведет к Д/#° (TiO, к, 298,15 К)=— 527 + 7кДж-моль~1.
99
ТЮ(г)
Глава
Т а б jhii
Молекула
¦Ti"O а
ТЮ+а
S а 36.4. Молекулярные постоянные TiO и ТЮ+
•
Состояние
Xs Дх
Xs ^
х3д3
ахД
Ы-Х+
А3И±
В3 2+
С3Ф2
с1!!
D3U
Я3Д2
/!ф
i12+
#3П
Х2Д
шехе
см^1
0 1
97,8 1009,028 4,4978б
202,6 j
3 448,32 1016,3 3,93
5 663,5 1023,8 4,64
12 026,0 е 924,2 5,1
13 000ж — —
14 264,2 з 867,779 3,9422
14 769,9 918,7 3,75
16 247,9л 870,2 3,36
19 526,9м 838,2567 4,7592н
21 338,8 916,7 4,33
22 575,0 890 п _
30 050 853,9 4,7
32 000 Д 1040 Р —
0б
1060
4,8 в
Ве
0,535 412
0,537 60
0,547 53 Д
0,513
0,507 39
0,513 37
0,506 13 Д
0,489 888
0,523 01
0,502 21Д
0,489 2
—
0,56
а, ¦ 103
3,011 в
2,98
—
2,9
3,145 ll
2,91
—
3;062 °
3,13
—
2,5
3,4 г
De ¦ 10'
6,029 г
5,98 Д
6,04 Д
6
.
6,918 к
6Д
6,79 Д
6,627
3,87
6,4 Д
6Д
—
6Д
ге
о
А
1,6202
1,6169
1,6022 Д
1,654
1,6644
1,6546
1,6639 Д
1,6938
1,6393
1,6729 Д
1,695
—
1,58
Примечание. Все постоянные даны в см1.
TiO: a принятые обозначения электронных состояний отличаются от использовавшихся в оригинальных работах;
6 ш«г/е=—0,0107; в а2=1,1 -Ю; г В=— 3,4-1 ~9; Д приведено значение постоянной при v=0;
е Те(А3п0)=11 941,9, Уе(Л3П2)=12 313,3; ж оценка, другие оцененные состояния:
Состояния d12+ еШ
Те 16 000 20 000
24 000
27 000
34 000
РП, кЩ, 141
36 000
)=14 089,9, ГС(С3Ф4)=14 436,1; иа2=1
м Те{Е3\)= 19 427,1, ГС(?3Д3)=19 622,1; н шеуе
A. 68); Р приведено значение Дб^о.
П-5. К 0_
= —2,3-10 9; л приведено значение
о а ,=—3-10""
вычислено по соотношению
TiO+: a все постоянные оценены; оцененные состояния А2П, Вг
Т. 11 000
cm
42 000
49 000
б 4=50; в вычислено по соотношению A. 67) и значению Do=58 300; г вычислено но соотношению A. 69);
Д вычислено по соотношению A. 68).
ном спектре TiO наблюдалось двенадцать систем полос, связанных с переходами между шестью
синглетными и шестью триплетными состояниями этой молекулы1: А3П.^Х3& [3274, 3458], С3Ф^*
[1849, 3315, 3816, 3819], D3U^>X3A [1899, 1904, 2669, 2947, 3817, 3818, 3819, 4843],
3Д [1848, 3819, 3821, 4698], Н*п*ьХяЬ [3458, 3747, 3781], сЧЬ^.Д [2987, 3277, 3291,
3815, 3819, 4450, 4785, 4838, 4840], /1Ф^а1Д[3272, 3276, 3315, 3815, 4450, 4838, 4841], ^Д-^Д
[3271, 3277], i1S+->61E+ [3263, 3277, 3820], йгП-*Ьх2:+ [3808, 3809], сгП-»Х3Д [1677] и ^Дз-*агД
[3275, 4880]. Анализ конфигураций электронной оболочки TiO (см. [1661]), а также сравнение
с изоэлектроиной молекулой ScF свидетельствуют, что молекула TiO в области до 40 000 см
должна иметь значительное число валентных электронных состояний, еще не наблюдавшихся
в ее спектре. В табл. 36.4 представлены 22 электронных состояния, главным образом состояния,
соответствующие конфигурациям электронной оболочки ...оД., ...оз, ...ая, ...о,- ¦¦¦°s°
...о.,а , ...oatzd. Они включают 10 состояний, не наблюдавшихся в спектрах. Энергии этих
состояний были оценены на основании имеющихся данных для молекул ScF, TiO, ZrO (см. рисунок),
а также вероятной величины разности энергии изоконфигурационных триплетных и синглетных
состояний. Погрешности оцененных величин могут достигать 3000—4000 см.
1 В справочнике принята символика состояний ТЮ, отличающаяся от использованной в оригшпльных работах.
100
Титаи и его соединения
ТЮ(г)
ScF TiO ZrD
Схема электронных термов и наблюдаемых переходов ScF, TiO, ZrO
HfO
HfO
Молекула TiO имеет низкий потенциал ионизации (см. ТЮ+), поэтому ридберговские
состояния TiO должны иметь относительно низкие энергии; сведения об этих состояниях отсутствуют,
хотя возможно, что некоторые из состояний, включенных в табл. 36.4, являются ридбергов-
скими.
Молекулярные постоянные TiO в состоянии Х3А, а также в состояниях С3Ф и Е3 А приняты
по работе Филлипса [3819], который нашел их из анализа тонкой структуры 22 полос системы
<^3Ф—Х3А и 22 полос системы Е3А—Х3А (г/, v" ^ 5) г. Энергии компонент Х3А2 и Xs А3,
приведенные в табл. 36.4, найдены Хокингом и др. [2669] в результате анализа вращательной
структуры трех основных и четырех сателлитных подполос в полосах 0—0, 1—0, 0—-1 и 1 — 1 системы
/KП—Х3А. Вращательные постоянные TiO в состоянии Х3А, полученные авторами [2669] (Ве =
— 0,53536 см), практически совпадают с принятыми по работе [3819], однако для энергии
компонент Xs А2 и Х3А3 в последней работе были получены менее точные значения (96,4 и 197,5 ем).
Колебательные постоянные TiO в состоянии А3И приняты по работе Линтона и Бройда [3274],
которые рассчитали их по кантам полос системы АЧ1—Х3А; вращательные постоянные приняты
равными постоянным в изоконфигурационном состоянии сЧ1. Колебательные постоянные TiO
в состоянии /KП, приведенные в табл. 36.4, найдены Жирницким и Палмером [4843] по кантам
¦полос системы D3H—Х3А (г/ <^ 5), вращательные — Филлипсом [3817] из анализа структуры
полосы 0—0 этого перехода. Близкие значения вращательных постоянных были получены Хо-
кингом и др. [2669]. Постоянные TiO в состоянии Н311 приняты по работе [3781], где были
измерены канты полос 0—0, 0—1 и 1—0 системы НШ—Х3А.
1 Для точного описания энергии уровней Филлипс использовал уравнения, отличающиеся от приведенных в гл. 1
настоящего справочника. Однако расхождения в энергии вращательных уровней TiO, рассчитанных по
уравнениям, приведенным в работе [3819], и по уравнениям A. 43), не превышают 4 см дл /—100.
101
TiO(r)
Глава 36
Энергия нижнего синглетного состояния ахД длительное время была предметом дискуссий
и для нее предлагались значения от 581 см [3816] до 4000 см [1750]. Приведенное в табл. 36.4
значение найдено Кобылянским и др. [4880] по частоте интеркомбинационного перехода Е3А3-*а}А
в спектре лазерной флуоресценции, его погрешность не превышает 0,5 см. Близкие значения
были получены в работах Линтона и Бройда [3274, 3275] и Брома, Бройда [1677]. Вращательные
постоянные TiO в синглетных состояниях приняты по рекомендации Линтона и Сингхала [3277],
которые с учетом полученных Линтоном [3272] постоянных TiO в нижнем синглетном
состоянии ааД заново обработали результаты анализа систем c1IL—a1A [3815], сЧТ—&Х2+ [3808, 3809],
/1ф_а1д [3277], g1^—агД [3271], г12+—5ХЕ+ [3263, 3820]. Колебательные постоянные в этих
состояниях, приведенные в табл. 36.4, получены Филлипсом [3819] совместной обработкой данных по
началам полос систем сЧ1—а1 А (г/, v" < 3) [3815], сШ—&Х2+ (г/ < 2, v" < 1) [3809], /ХФ—а1 А
(г/, г/'<4) [3271, 3276], ^Д-^Д [3271], i1S+-61S+ (г/ < 2, v" < 0) [3263].
Термодинамические функции ТЮ(г) вычислены по уравнениям A. 3)—A.6), A.9), A.10),
A. 93)—A. 95). Значения -Qm и ее производных рассчитывались по уравнениям A.90)—A. 92)
с учетом 21 возбужденного электронного состояния и в предположении, что Q^ вр = 0,5р,()№ вр.
Величина <?<2.вр и ее производные находились по уравнениям A. 73)—-A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням с
помощью уравнений типа A. 82). В расчете учитывались все уровни энергии с значениями 1 <С/ ^
^ Лпах vi гДе Jnna „ определялись из условий A. 81). Колебательно-вращательные уровни
состояния Xs Д вычислялись по уравнениям A. 65), A. 43), A. 64а) 1ш, значения коэффициентов
в этих уравнениях, полученные с учетом условий A. 50), A. 58), A. 59), а также величины г>Ш1И
и /,.ш приведены в табл. 36.5. Различие молекулярных постоянных разных изотопических
модификаций TiO во внимание не принималось. Вырождение уровней состояния Х3&, так же как
вырождение и мультиплетиость уровней других электронных состояний, учитывалось
статистическим весом.
Таблица 36.5.
Коэффициенты в уравнениях, описывающих уровни энергии (в см !),
а также значения »шах и е7цт, принятые для расчета термодинамических функций TiO и TiO+
| Коэффициенты
т
У10 -Ю
уа0
У.0-104
Yi0 -10б
Уо1 -10
Уц-10»
TiO: a Состояние
Те
Состояние
Те
TiO+: a Состояние
Те
тю,
0
1,009
—4,582
-9,869
1,342
5,354
—3,011
а1 Д
3448,32
ein
20 000
Х2Д5/
100
218
58
396
871
12
б12+
5663,5
21 338,
TiO+, Х2Д
0
1,060
-4,8
—
—
5,60
-3,4
12 026,0
3 22 575
42П, В2 2+ СЩ
11 000 42 000
k
Коэффициенты
V \ 0^
У12 -10»
У03-1013
А
В3 2+ С3Ф сЧ1
13 000 14 264,2 14 765
F3n МП, С3Ф г12+
TiO, Х3Д
1,1
-6,029
-3,4
-4,443 182
114
428
50,65
tP2+
),9 16 000
Я3П
24 000 27 000 30 050 32 000
49 000
В3П
16 247,
/1ф
34 000
TiO+, Х2Д3, а
-6,0
—
—8,88
110
416
—
?3Д
9 19 526,9
km, рп, гхп
36 000
Погрешности вычисленных значений термодинамических функций TiO (г) при низких
температурах обусловлены недостаточной точностью имеющихся данных (см. выше) о величине
расщепления компонент основного состояния Х3А, а также использованием уравнения A. 43) для
энергии вращательных уровней состояния Х3Д. При температурах выше 2000 К становятся
существенными неточность оценки энергии ряда электронных состояний TiO, прежде всего состояний в32+
1 За постоянную спин-орбитального взаимодействия основного состояния принималась величина, равная
0,25 [F3 C)-F1 A)] (см. табл. 36.5).
102
Титан и его соединения
ТЮ(г)
и d1E+, использование приближенной методики учета возбужденных состояний, в частности,
триплетных состояний с энергиями меньше 20 000 см, а также неполнота данных о
возбужденных состояниях этой молекулы. Погрешности в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К
достигают 0,05, 0,1, 0,5 и 2 Дж-К^-моль, в значении #"B98,15 К)—#°@) — 0,01 кДж-моль.
Термодинамические функции TiO(r) вычислялись ранее в таблицах JANAF [2834], а также
в работах Климова и др. [513] (уравнение для Ф°(Т) при 298,15—6000 К), Шнейдера [4197а],
Жирницкого и Черниховского [4835, 4842] (соответственно от 2000 и 3000 К до 10 000 К), Броун-
штейна и Юркова [151] (Т < 1000 К), Папоушека [3767] (до 2000 К), Брюэра и Розенблатта
[1666] (Ф°(Т) до 3000 К). Несмотря на отличающиеся энергии синглетных состояний ТЮ,
принятые в [2834] и в настоящем издании, и меньшее число возбужденных электронных состояний,
учитывавшихся авторами таблиц JANAF, расхождения результатов этих двух расчетов
благодаря частичной компенсации ошибок не превышают 1,5 Дж-К^-моль в значениях Ф°(Т) и S°(T).
В работе [518] не учтены расщепление компонент состояния Х3Д и ряд возбужденных состояний,
соответствующие расхождения меняют знак с ростом Т и составляют —3,5 и +2,5 Дж-К^-моль
при 298,15 и 6000 К. Расхождения данных [4835, 4842] и табл. 935 составляют 2 Дж-К^-моль
при 3000 К и уменьшаются с ростом температуры; частично они обусловлены неверным
значением ГДа^Д), принятым в этом расчете. Расхождения с данными [4197а] достигают 3 и 7 Дж-К~*Х
Хмоль" в значениях Ф°A0 000 К) и 5°A0 000 К), поскольку Шнейдер не учитывал ряд элек-
Таблица 36.6. Результаты определений D0(TiO) (в кДж-моль)
Авторы
Метод
Do (ТЮ)
И закон III закон
ровз и др., 1955 [2506]
;ерковиц и др., 1957 [1506)
Дроварт и др., 1969 [2114]
Хемпсон, Гиллес, 1971 [2562]
By, Уолбек, 1972 [4780]
Лиу, Уолбек, 1975|[3286]
Хилденбранд, 1976 [2642]
Уитли и др., 1977'[4718]
Шелдон, Гиллес, 1977 [4256]
Дюбуа, Гоул, 1977 [2122]
Хайдеман и др., 1980 [4971]
Эффузиошшй, ТЮ(к)=ТЮ(г), 1847-1968 К,
8 точек а
<654
677
664
Масс-спектрометрический, ТЮ(к)=ТЮ(г), 2027—
2072 К, 2 точки
Масс-спектрометрический, TiO(r)+Y(r)=Ti(r)-f-
YO(r) 2313 К, 1 точка
То же, TiO(r)+Sc(r)=Ti(r)+ScO(r), 1951—246? К,
29 точек а
То же, TiO(r)+Sc(r)=Ti(r)+ScO(r), 2145- 629+150
2318 К, 9 точек
То же, TiO(r)+Ge(r)=Ti(r)+GeO(r), 2145— 605+80
2418 К, 9 точек
Эффузионный, 2Tia08(K)=Ti305(K)+Ti0(r), 1937— 690±70
2044 К, 15 точек
Масс-спектрометрический, 2Т1аО3(к)=Т13ОБ(к)+ 747 ±200
+ТЮ(г)
То же, TiO(r)+Al(r)=Ti(r)+AlO(r), 1874—2023 К, 671 ±25
7 точек
То же, TiO(r)+Ba(r)=Ti(r)+BaO(r), 1975— 664±30
2135 К, 7 точек
Эффузионный, ТЮ(к)=ТЮ(г), 1921—1998 К, <684
5 точек а
Эффузиошшй, ТЮ(к)=ТЮ(г), 1806 К, 1 точка —
Масс-спектрометрический, ТЮ(к)=ТЮ(г), 1694— 678±25
'К6
1919:
<693
689 ±15
667 ±12
667 ±12
667+11
670 ±11
678 ±25
J74±25
660 ±9
670 ±11
667|
667+25
Исследование хемилюминесценции в
пересекающихся молекулярных пучках Ti с О2, NO2 и N2O
Масс-спектрометрический, ТЮ(к)=ТЮ(г), 1693— 687 ±20
1895 К, 9 точек
669 ±13
673 ±12
а Расчет проводился в предположении о существовании в паре только ТЮ. Поскольку в действительности в состав
пара входит также и Ti, эти величины являются вэрхшшн пределами возможных значений,
б Данные приведены в виде уравнений.
103
ТЮ+(г) Глава 36
тронных состояний TiO, с данными [1666] 4—10 Дж-К -моль, так как в этой работе трем
нижним состояниям были приписаны неоправданно большие статистические веса.
Константа равновесия реакции TiO(r) = Ti(r) -f О(г) вычислена с использованием принятого
в справочнике значения энергии диссоциации
ДД°@) = D0(TiO) = 668 ± 10 кДж • моль «* 55 800 ± 800 см.
Результаты определений этой величины представлены в табл. 36.6. Приведенные в этой
таблице погрешности вычислены с учетом воспроизводимости измерений, неточности использованных
в расчетах термодинамических функций, термохимических величин и сечений ионизации. Как
видно из таблицы, результаты определений D0(TiO) в разных работах и разными методами
согласуются между собою в пределах их погрешностей. Принятое значение является средним
взвешенным из полученных при исследовании обменных реакций с YO, ScO, BaO, GeO и АЮ, его
погрешность в существенной степени определяется неточностью энергий диссоциации этих
молекул. Данные, полученные при изучении процессов сублимации ТЮ(к) и Ti3O3(K),
представляются менее надежными (см. [4256]).
Принятой энергии диссоциации соответствует
г, 0) = 49,783±10 кДж-моль.
ТЮ+(г). Термодинамические свойства газообразного ионизированного оксида титана в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 936.
Молекулярные постоянные ТЮ+, принятые в справочнике, приведены в табл. 36.4. Спектр
молекулы TiO+ экспериментально не исследовался, а квантовомеханические расчеты не
проводились. Приведенные в табл. 36.4 постоянные получены при подготовке справочника на
основании приближенных оценок и известных постоянных изоэлектронных молекул и ионов. В
спектрах изоэлектронных молекул ScO и TiN, а также в спектрах YO, ZrN, YF+, LaO и LaF+
наблюдались переходы, связанные с состояниями 22+ (...°я,), 2Д(...§(Я_1Ы), 2Щ...к^) и 22(...а^).
Спектры молекул ScO, YO, LaO и TiN аналогичны: имеют основное состояние X22+(...<\,s),
первое возбужденное — Л2Д(...8(),_1) d), а энергии состояний В2Щ...кгр) и Са2+(...о ) у всех этих
молекул близки по величине. Спектры молекулярных ионов ScF+, YF+ [4260] и LaF+ [4259]
существенно отличаются от спектров соответствующих изоэлектронных нейтральных молекул.
Следует отметить, что энергии переходов, наблюдавшихся в спектрах этих ионов, 2П—2Д (YF+
и LaF+), 2П—22(ScF+) и 2Ф—2A(LaF+), удовлетворительно согласуются с рассчитанными по
приближенной методике, предложенной Грином [2462] для оценки энергии электронных состояний
молекул ScO, YO и LaO. В этой методике принимается, что в низколежащих состояниях
молекулы ScO неспаренный электрон находится на молекулярных орбиталях, практически
идентичных орбиталям атома Sc, что позволило оценить энергии молекулярных орбиталей из энергий
атомных орбиталей, а последние — как центры тяжести термов атома, принадлежащих
соответствующим конфигурациям. Рассчитанные таким образом энергии электронных состояний
молекул ScO, YO, LaO, ScF+, YF+ и LaF+ отличаются от найденных экспериментально не более чем
на 20%.
Энергии состояний :'2+(o4s), 8A(83d) и 2П(тг4 ) молекулы TiO+ были|оценены по этой методике.
Разность энергий состояний !Д и 42)т рассчитывалась из э.-r гий центров тяжести конфигураций
...3d24s и ...3d4s2 иона Ti+, разность энергий состояний 2П и 22+ определялась по энергиям
центров тяжести состояний конфигураций ...3d2AD)Ap и ...3cZ2AZ)Ls. Согласно выполненным
оценкам молекула ТЮ+ должна иметь основное состояние Х2Д. Эта же симметрия основного
состояния TiO+ была предсказана ранее Карлсоном и Мозером [1750] при обсуждении результатов кван-
товомеханического расчета ScF. Энергия состояния v!2II(itm) принята по разности энергий
состояний 3П, 41 конфигурации °И8гс(я_1)й и 3Д, гА конфигурации от§i!t_1} d молекул TiO и ZrO.
Энергия состояния Д22+(а4в) оценена в предположении, что у TiO+ разность энергий состояний
°22+ (%) и С2П(тсир) такая же как у молекул TiN и ZrN [1424, 2102, 12128]. Погрешности в
энергиях возбужденных состояний TiO+, приведенных в табл. 36.4, могут 'достигать 25% от их
величины.
Значения постоянных ше и Bt молекулы TiO+ в состоянии Х2Д были оценены в предположении,
что их отношение к аналогичным постоянным молекулы TiO в состоянии Х3Д такое же, как у ионов
YF+, LaF+ и соответствующих нейтральных молекул в аналогичных состояниях. Погрешности
104
Титан и его соединения ТЮ2(к, ж)
ч.
принятых значений постоянных могут достигать 50—75 и 0,02—0,03 см. Постоянная
спин-орбитальной связи TiO+ в состоянии Х2Д принята такой же, как в состоянии .Х3Д молекулы TiO,
ее погрешность оценивается в 25 см.
Термодинамические функции ТЮ+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBS и ее производных рассчитывались по' уравнениям A. 90)—A. 92)
с учетом пяти возбужденных состояний (подсостояние Х2Дб/2 рассматривалось как возбужден-
ное состояние с Те—2А) и в предположении, что Q$,.vV ~ 0>5p»(?S.Ep- Величина (J^. ВР и
ее производные (для подсостояния Х2Дз;г) вычислялись по уравнениям A. 73)—A. 75)
непосредственным суммированием по колебательным уровням и интегрированием по вращательным
уровням с помощью уравнений типа A. 82). В расчетах учитывались все уровни энергии со
значениями / ^ /msX) с, где /шах> „ находились из условий A. 81). Колебательно-вращательные уровни
состояния Х2Дзг вычислялись по уравнениям A. 65), A. 34), A. 64а). Значения коэффициентов
в этих уравнениях, тождественные соответствующим постоянным из табл. 36.4, а также величины
ушах и /lim, приведены в табл. 36.5. Вырождение и мультиплетность уровней энергии всех
состояний учитывались статистическими весами.
Погрешности вычисленных термодинамических функций ТЮ+(г) при низких температурах
обусловлены главным образом отсутствием экспериментальных данных о вращательной
постоянной и постоянной спин-орбитальной связи в состоянии Х2Д, при высоких температурах —
отсутствием достоверных данных об энергии возбужденных электронных состояний. Погрешности
в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К оцениваются в 1,' 3, 5 и 8 Дж^К^-моль.
Погрешность величины Я°B98,15 К)—'Н°ф) составляет 0,15 кДж-моль.
Таблица термодинамических функций ТЮ*(г) публикуется впервые.
Константа равновесия реакции ТЮ+(г) + е(т) = Ti(r) + О(г) вычислена с использованием
значения Дг7/°@) = 38,0 + 22 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0(ТЮ) = 630 + 20 кДж-моль-1.
Принятая величина основана на измерениях потенциала ионизации TiO, выполненных Рау-
хом, Аккерманом [3943] F18 + 10 кДж-моль) и Хилденбрандом [2642] F46 + 10 кДж-моль).
Менее точные значения были получены в работах [1375, 2150, 3487, 4256].
Принятому значению соответствуют
Д ,Я°(ТЮ+) = 679,783 + 22 кДж • моль,
Do(TiO+) == 697,1 ± 22 кДж моль яи 58 300 + 2000 см.
ТЮ2(к, ж). Термодинамические свойства кристаллического и жидкого диоксида титана стехио-
метрического состава в стандартном состоянии при температурах 100—5000 К приведены
в табл. 937.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 36.1. Известны три кристаллические модификации TiO2: тетрагональные рутил и анатаз
и ромбический брукит. В справочнике за стандартное состояние ТЮ2(к) принят рутил,
являющийся термодинамически наиболее стабильной формой х. При высоких температурах анатаз и
брукит самопроизвольно и необратимо превращаются в рутил. Однако скорости этих
превращений при Т < 900 К ничтожно малы [1954, 3924].
Термодинамические функции ТЮ2(к) при Т < 298,15 К вычислены на основании результатов
измерений теплоемкости рутила, выполненных Сандином и Кеезомом [4135] @,3—20 К), Даг-
дейлом и др. [2124] A2—270 К), Шомейтом [4271 ] E2,5—297,7 К) и Мицухаши и Такахаши [3517]
(80—1100 К).2 В работе [2124] численные данные приведены только для Т <^ 50 К. При Г^>80 К
наиболее точные измерения выполнены в работе [3517], данные Шомейта в интервале 80—200 К
[4271] систематически ниже данных [3517] (до 2,5%), а при Т > 200 К согласуются с последними
в пределах 0,6%. Поэтому данные [2124, 4271] использовались только для Т < 80 К, их
погрешность оценивается в 1—3%, погрешность данных [3517] при Т — 80-^-298,15 К составляет 0,5%.
1 При высоких давлениях (> 200 кбар) термодинамически стабильной становится ромбическая модификация
( ТЮ2(к) со структурой а-РЬО2 [2832, 3262].
'* В работах [351*7, 4135] исследовались монокристаллические образцы чистотой 99,99%, а в работах [2124.
4271] — порошкообразные образцы чистотой 99,7%.
105
TiO2(K, ж)
Глава 36
Погрешности рассчитанных значений 5°B98,15 К) и Я°B98,15 К)—Я°@) (см. табл. 36.1),
составляют 0,3 Дж-К^-моль и 0,05 кДж-моль. Теплоемкость рутила измерялась также в работах
[2951] A—20 К) и [34491 F9—295 К). В сообщении [2951] результаты измерений не приведены;
в работе [3449] получены систематически завышенные значения по сравнению с данными [3517,
4271].
В интервале 298,15—2185 К для теплоемкости ТЮ2(к) принято уравнение (см. табл. 36.1),
полученное совместной обработкой результатов измерений энтальпии рутила Ыейлором [3637]
C93,5—1746 К), Артуром [1351] C97-1072 К), Литцом [3261] E78-1283 К), Слюсарем и др.
[944] F45—1972 К) и значений энтальпии, основанных на измерениях теплоемкости рутила Ми-
цухаши и Такахаши [3517] C00—1100 К). Цоследние данные учитывались с наибольшим весом,
а данные [1351, 3637] — с наименьшим. Это уравнение описывает экспериментальные данные
с погрешностью, не превосходящей ~2%.
Температура плавления B185 + 10 К) принята по измерениям Кутюра [1945], выполненным
на образце чистотой 99,9%; менее точное значение Тт было получено Брауэром и Литтке [1647]
B146 + 15 К). Результаты более ранних определений, рассмотренные в [673, 4198], а также
данные [944] относятся к рутилу нестехиометрического состава. Энтальпия ТГО2(ж) измерялась
Слюсарем и др. [944] B014—2035 К) в условиях, когда равновесный состав существенно отличался
от стехиометрического. В справочнике энтальпия плавления F8 + 8 кДж-моль) принята на
основании приближенных расчетов этой величины по данным о плавлении бинарных оксидных
систем с участием ТЮа(к) [2835], а также ее оценки на основании предположения о равенстве
энтропии плавления изоструктурных диоксидов титана и ванадия F8,4 кДж-моль).
Полученное в работе [944] значение F2,2 кДж-моль) соответствует нестехиометрическому составу
диоксида титана. Теплоемкость ТЮ2(ж) A00 + 10 Дж-К~1-моль~1) оценена по принятому в
справочнике эмпирическому правилу (см. т. I, с. 79) с учетом теплоемкостей У02(ж) и ТЮ2(к) при Тт.
Погрешности приведенных в табл. 937 значений Ф°(Г) при 298,15, 1500, 3000 и 5000 К
оцениваются в 0,2, 1, 6 и 13 Дж-К^-моль.
Ранее термодинамические функции ТЮ2(к, ж) вычислялись в справочниках JANAF [2834,
2835] (до 5000 и 4000 К), Шика [4181 ] (до 6000 К) и Келли и Ма [2960, 2964] (до 2000 К). В
большинстве расчетов использовались менее точные и полные данные о свойствах ТЮ2(к).
Расхождения в значениях Ф°(Т), приведенных в [2835] и табл. 937, для ТЮ2(к) не превышают
1 Дж-К • моль, а для ТЮ2(ж) составляют 6 Дж-К^-моль.
Принятая в справочнике энтальпия образования
A/ff°(TiO2, к, рутия, 298,15 К) = —944,0 + 0,8 кДж • моль
основана на результатах наиболее надежных определений, представленных в табл. 36.7. Эта
величина является средневзвешенной из значений, полученных в работах [2754] и [3349], в
которых использовались образцы титана высокой чистоты и было показано, что в продуктах
сгорания содержался только рутил. Близкие, но менее точные значения были получены в работах
[3156,41]. Энтальпия сгорания титана определялась также в работах [3520,3523, 3645,4062,
4285, 4685], в которых не контролировалась полнота сгорания образцов и не исследовался
фазовый состав продуктов сгорания. В них были получены значения, лежащие в интервале от —912
до —946 кДж-моль.
Т а б л и
Хамфри
Ария и
и, а 36.7. Основные
Авторы
, 1951 [2754]
др., 1957 [41]
Ма и др., 1957 [3349]
Кибетт,
[2835])
Маргрейв, 1965 [3156]
результаты измерений Д^Н
Калориметрический,
7 опытов
°(ТЮ2, к, рутил, 298,15 К) (в кДжмоль-1)
Метод
сжигание титана (99,72%),
То же, чистота образца не указана, 2 опыта
То же, титан 99,89%
То же, титан 99,86%
То же, титан 99,85%
(см. Калориметрический,
-TiF4(K)+O2(r)
, 5 опытов
, 6 опытов
, 7 опытов
TiO2(K, pyTiMi)+2F2(r)=
AfH° (TiO2, к, рутил,
298,15 К)
—943,58+0,96
—941+2
-944,16 ±0,20
—944,12+0,33
—942,74+0,88
-941 +4
106
Титан и его соединения
ТЮ2(к, анатаз)
Энтальпия образования ТЮж(к, рутил) в области гомогенности A,973 < х <^ 2,000) может
быть представлена уравнением:
Х, к, рутил, 298,15 К) = 1,374 — 472,687ж кДж • моль,
основанным на измерениях энтальпии сгорания Ti(K) в кислороде до оксидов ТЮх(к, рутил) при
1,9852 <^ х ^ 1,9944 [3349] и принятом в справочнике значении Д *-йГ°(ТЮ2, к, рутил, 298,15 К).
Измерения Пикара и Жерданьяна [3833] и Алкока и др. [1285] подтверждают это уравнение.
Измерения парциальных энтальпий растворения О2(г) в ТЮх(к, рутил) [177] приводят к
несколько отличающимся значениям. '
Давление пара в реакции ТЮ2(к, ж, рутил) = ТЮа(г) вычислено с использованием
значения ДгД"°@) = AsH°(Ti02, рутил, 0) = 619,174 + 20 кДж-моль, соответствующего принятым
в справочнике энтальпиям образования ТЮ2(к, рутил) и ТЮ2(г).
ТЮ2(к, анатаз). Термодинамические функции анатаза при температурах 100—2000 К
приведены в табл. 938.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 36.1. При Т <[ 298,15 К термодинамические функции вычислены на оснований измерений
теплоемкости анатаза, проведенных Шомейтом [4270] E2,5—296 К, чистота образца 99,3%),
погрешности которых оцениваются в 2—0,5%. Экстраполяция этих данных ниже 52 К с помощью
дробно-рациональной функции третьей степени [1066] приводит к значению iS°E2 К) =
= 2,25 Дж-К^-моль. Погрешности приведенных в табл. 36.1 значений 5°B98,15 К) и
#°B98,15 К)—Я°@) оцениваются в 0,6 Дж-К"-моль-1 и 0,07 кДж-моль^1. В интервале 298,15—
2000 К термодинамические функции анатаза вычислены по уравнению для теплоемкости (см.
табл. 36.1), выведенному совместной обработкой данных по энтальпии, полученных Нейлором
[3637] D16—1305 К) и Литцем [3261] E80—1001 К). Это уравнение описывает значения С°р(Т)
в интервале 298,15—1000 К с погрешностями, не превышающими 1—-2%, а в интервале 1000—
2000 К 2—5%. Погрешности приведенных в табл. 938 значений Ф°(Т) при 298,15, 1000 и 2000 К
составляют 0,3, 2 и 3 Дж-К^-моль.
Ранее термодинамические функции анатаза вычислялись в справочниках JANAF [2834, 2835]
(до 2500 и до 2000 К) и Келли [2960] (до 1300 К). Расхождения между значениями Ф°(Г),
приведенными в табл. 938 и в [2835], составляют 0,2—0,7 Дж-К-моль.
Таблица 36.8. Результаты определений энтальпии превращения анатаза в рутил при 298,15 К (в кДж-моль)
Авторы
Рот, Беккер, 1931 [4062]
Рао, 1961 [3921]
Кибетт, Маргрейв, 1965 [3156] (см.
[2835])
Вальдик, 1966 [4583]
Навротский, Клеппа, 1967 [3636] (см.
[3516])
Роби, Вальдбаум, 1968 [4021, 3516]
Бобыренко и др., 1972 [140]
Мицухаши, Клеппа, 1979 [3516]
Метод
Калориметрический, Т1(к)-р/2О2(г)=ТЮ2(к,
анатаз), 5 опытов
ДТА, 903 К
Калориметрический, сжигание анатаза и рутила
во фторе
Измерение Ttr при р=3,8-^24 кбар. Расчет по урав- ,
нендю Клаузиуса—Клайперона и экстраполяция
к р=1 бар
Калориметрический, измерение энтальпий
растворения в расплаве 3Na2O • 4МоО3 анатаза (9 опытов) и
рутила F опытов), 968 К
Измерение Tfr в зависимости от давления. Расчет
по уравнению Клаузиуса—Клайперона
Расчет на основе приближенных модельных
представлений по смещению полос в УФ-спектрах
поглощения анатаза и рутила
Калориметрический, измерение энтальпий
растворения в расплаве 3Na.2O • 4Мо03 анатаза A2 опытов)
и рутила A2 опытов), 971 К
ДТА и ДСК, 1360 К
Д,ГЯ° B98,15 К)
—29,0+4,2
—0,75+0,53
-8,4+6,0
—11,3
-7,0+1,0
-0,4
—6,9
—3,62+0,97
-3,4+1,4
107
ТЮ2(г)
Глава 38
Для энтальпии образования анатаза в справочнике принято значение
bfH°(Ti02, к, анатаз, 298,15 К) = —940,4 ± 1,3 кДж • моль.
Это значение основано на измерениях энтальпии превращения анатаза в рутил, выполненных
Мицухапга и Клеппой [3516] (см. табл. 36.8). Попытки определения этой величины в других
работах, приведенных в табл. 36.8, привели к существенно менее точным значениям.
Давление пара в реакции ТЮа(к, анатаз) = TiOs(r) вычислено с использованием значения
&гН°@) = Д4/7°(ТЮг, анатаз, 0) = 615,524 + 20 кДж -моль, соответствующего принятым в
справочнике энтальпиям образования ТЮ2(к, анатаз) и ТЮ2(г).
ТЮ2(г). Термодинамические свойства газообразного диоксида титана в стандартном состоянии
при температурах 100—6000 К приведены в табл. 939.
Молекулярные постоянные, приведенные в табл. 36.9, приняты на основании результатов
исследования поведения молекулярного пучка TiO2 в неоднородном электрическом поле [2935],
Таблица 36.9. Значения молекулярных постоянных, а также з и рх, принятые для расчета
термодинамических функций ТЮ2
Состояние
^3
CM
962
см, p = 'S; T
см, р = 3; Т.
550
, (В'Я2) = Те (С1
ФВХ) == 30 000
935
fi,) =22 000 см-
см, р = 1.
1А1В1 с-10^
Г3 • CMG
а
3,2 ¦ 102 2 [
-1, р = 4; Гв фгВ2) = 24 000 иг1, р = J;
ИК-спектра молекул TiO2, изолированных в матрице из Ne [3458] и приближенных оценок, вы
полненных при подготовке справочника. Приведенное в таблице произведение главных моментов
инерции в состоянии ХМ, рассчитано по структурным параметрам r(Ti—О) = 1,63+0,03 А (оценка
по величинам r.(TiO), ге(Ш0) и r(Nb—О) в молекуле NbO2) и -^O—Ti—0 = 110+15° (найден
Мак-Интайром и др. [3458] по изотопическому сдвигу частоты v3). Погрешность принятого
значения I/JpJc оценивается в 1 • 1035 г3-см6. Приведенные в таблице частоты vx и v3 найдены в работе
[3458] в ИК-спектре молекул TiO2, изолированных в матрице из Ne; принятое значение v,
подтверждается результатами исследования электронного спектра TiO2, выполненного авторами
[3458] *. Ранее Велтнер и др. [4698] при исследовании ИК-спектра TiO2 получили для v3 близкое
значение (940 см). Погрешности принятых значений vt и v3 могут достигать 10—20 см.
Приведенное в таблице значение v2 оценено на основании силовых постоянных TiO2, рассчитанных
ОО ( 0)
по принятым значениям vx, v3 и
2
Ti—О (в предположении, что/п.=0,1/г), а также величины
0
x 3 ( р /п/г) 2
в молекулах диоксидов других переходных металлов, его погрешность оценивается в 50 см.
В электронном спектре молекул TiO2, изолированных в матрице из Ne [3458], обнаружен
переход в области 5295 А. В справочнике для схемы нижних электронных состояний ТЮ2 приняты
результаты полуэмпирических оценок, выполненных Хаитом и Хачкурузовым [1065] для модели
кристаллического поля. Переход, наблюдавшийся в электронном спектре TiO2, был отнесен
авторами [1065] к переходу в первое возбужденное состояние АЪВЪ энергии пяти следующих состояний
рассчитаны с использованием параметров, найденных по экспериментальным данным для молекулы
TiO. Погрешность приведенного в таблице значения Te(AsB1) оценивается в 500 см, значений
энергии других состояний — в 4000 см.
Термодинамические функции ТЮ2(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом шести возбужденных электронных состояний по соотношениям A. 168)—A. 170).
Погрешности вычисленных значений обусловлены приближенным характером расчета и неточ-
1 В работе [928] отнесение полосы 962 см J к частоте vt молекулы TiO2 поставлено под сомнение, поскольку
у.молекул диоксидов переходных металлов V, VI, VII п VIII групп, как правило. vx < v3. Однако данные
работ [2355, 3268, 3458], по-видимому, свидетельствуют, что у диоксидов металлов IV и III групп vx > v3.
108
Титан и его соединения
Ti2O3(K, ж)
ностыо принятых молекулярных постоянных. Погрешности значений Ф°(Г) при 298,15, 3000 и
6000 К оцениваются в 1, 3 и 5 Дж-К~1-моль~1; в том числе из-за неточности постоянных в состоянии
ХЫХ в 1—2 Дж-К^-моль-1.
Термодинамические функции ТЮ2(г) вычислялись в справочниках JANAF [2835], Шика
[4181] и в работе Брюэра и Розенблатта [1666] (до 3000 К). Расчеты [1666, 4181] основывались
на предположении о линейной структуре молекулы ТЮ2 и отличаются от данных табл. 939 на
21 Дж-К-моль~1 в значениях Ф°(Г). Расхождения данных [2835] и табл. 939 составляют
2—6 Дж-К-моль в значениях Ф°G') и обусловлены различием принятых значений v2 и тем,
что в настоящем справочнике учтены возбужденные состояния TiO2.
Константа равновесия реакции TiO2(r)=Ti(r)-f-2O(r) вычислена с использованием значения
Д,.Я°@) = 1284,566-f20 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
&fB°(Ti0.z, г, 0) = —320 + 20 кДж • моль".
Результаты определений энтальпии образования ТЮ2(г) представлены в табл. 36.10.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, неточность использованных
в расчетах термодинамических функций, термохимических величин и сечений ионизации. За
исключением двух величин, найденных из констант равновесия реакции TiO2(r)=TiO(r)-f-O(r),
результаты остальных работ согласуются между собой в пределах их погрешностей. Выпадение
результатов измерений по указанной выше реакции, возможно, объясняется трудностью точного
определения парциального давления атомарного кислорода. Принятая величина является средневзвешенной
из приведенных в табл. 36.10 значений (за исключением отмеченных выше двух выпадающих
величин). При вычислении средневзвешенного значения результаты исследований испарения
ТЮ2(к, ж) брались с меньшими весами, так как в условиях этих измерений состав
конденсированной фазы мог отличаться от стехиометрического.
Таблица 36.10. Результаты
Авторы
эаределений LfH° (TiO2, г, 0) (в кДж-моль)
. Метод
A/ff°(TiO2, г, 0)
II закон
III закон
Берковиц и др., 1957 [1506] Масс-спектрометрический, ТЮ2(к)=ТЮ2(г), 1881 К, — —313+15
3 точки
Семенов, 1969 [919] Масс-спектрометрический, ТЮ2(к, ж)=ТЮа(г), —347 —314 + 13
1850—2540 К, данные представлены в виде уравнения
Эффузионный,ТЮ2(ж)=ТЮ2(г), 2263—2538К, 6точек —373±80 —308±15
Масс-спектрометрический, ТЮ2(г)=ТЮ(г)+О(г), —154+60 —262+20
2263—2538 К, 6 точек
Хемпсон, Гиллес, 1971 [2562] Масс-спектрометрический, TiO2(r)+Ti(r)=2TiO(r), —288+100 —324+20
2145—2318 К, 5 точек
Балдуччи и др., 1972 [1375] То же, 2165—2500 К, 6 точек —352+25 —315±25
Масс-спектрометрический, ТЮ2(г)=ТЮ(г)+О(г), —287+50 —257+20
2015—2240 К, 12 точек
Хнлденбранд, 1976 [2642] Масс-спектрометрический, ¦TiO2(r)+Ti(r)=2TiO(r), —329+45 —324+20
1975—2161 К, 10 точек
.То же, 2039—2222 К, 8 точек —330±35 —323+20
Ti2O3(K, ж). Термодинамические свойства кристаллического и жидкого триоксйда дититана
в стандартном состоянии при температурах 100—5000 К приведены в табл. 940.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 36.1. В справочнике за стандартное состояние Т12О3(к) принята гексагональная
модификация (структурный тип корунда), которая имеет фазовый переход второго рода типа
полупроводник—полуметалл. При этом переходе меняются параметры решетки Ti2O3 без изменения ее
симметрии. Фазовый переход сопровождается аномалией теплоемкости в интервале 400—480 К с
максимумом при 464 К [1405].
Термодинамические функции Ti2O3(k) при Т <^ 298,15 К .вычислены по измерениям
теплоемкости образцов Ti2Og высокой чистоты, проведенным Сьестрендом и Кеезомом [4305, 306] @,4—
109
Ti2O.(K, ж) Глава 36
20 К) и Березовским и Пауковым [125, 758, 759] A1,5—305,5 К). Погрешности вычисленных
значений ?°B98,15 К) и Я°B98,15 К)—Я°@) (см. табл. 36.1) оцениваются в 0,4 Дж-К^-моль
и 0,07 кДж-моль. Менее точные данные по теплоемкости Т12О3(к) при Т <^ 298,15 К получены
в работе Шомейта [4270] E3—296 К, примеси 0,3% TiG и 0,3% SiO2), в которой был обнаружен
максимум теплоемкости при 242 К, ке найденный позднее авторами [759] на существенно более
чистых образцах Ti2O8, а также в работе Барроса и др. [1405] A48—713 К, примеси 0,02%), в
которой методом ДСК получены значения теплоемкости, завышенные относительно данных [759,
1393] на 10 Дж-К^-моль.
Теплоемкость Т12О3(к) в интервале 298,15—580 К, где наблюдается широкая ^-аномалия,
описана тремя уравнениями (см. табл. 36.1), из которых два первых описывают восходящую ветвь
Х-кривой, а третье уравнение — нисходящую ветвь. Эти уравнения основаны на результатах
измерений теплоемкости Ть,О3(к) в работе Барроса и др. [1405], исправленных (уменьшенных)
на отмеченную выше систематическую ошибку в 10+1 Дж-К^-моль. Уравнение для
теплоемкости при 580—2110 К получено совместной обработкой результатов измерений энтальпии в
работах Нейлора [3637] F61—1750 К) и Слюсаря и др. [944] F72—2084 К) и значений энтальпии
при 1800—2110 К, определенных графической экстраполяцией данных [3637] г. Погрешность
аппроксимации значений C°(Ti2O3, к) принятыми уравнениями оценивается в 2% при Г=298,15-^-
—580 К и в 3% при Г=580—2110 К. Менее точные данные по теплоемкости Ti2O3(K) при Т >
> 298,15 К получены в работах [3674] C20-520 К), [1889] C60-590 К) и [1393] D93-1082 К).
Измерения энтальпии Ti2O3(k) [413] D93—1082 К), представленные только на графике,
удовлетворительно согласуются с данными [944, 3637].
Температура плавления Ti2O3 B110+20 К) принята по измерениям, проведенным Нишимура
и Кимура [3664] B106+20 К), Брауэром и Литтке [1647] B096+15 К), Карлсоном B115+10 К,
см. [4632]) и Слюсарем и др. [944] (~2100 К) 2. Существенно менее точные значения получены
в работах [2060, 2908, 4205]. Энтальпия плавления (92+16 кДж-моль) принята как разность
энтальпии Т12О3(,ж) при 2110 К по данным [944] и энтальпии Т12О3(к) при 2110 К, рассчитанной
по принятым уравнениям для теплоемкости. Теплоемкость Т12О3(ж) A70+20 Дж-К~1-моль~1)
была оценена по эмпирической формуле, принятой в справочнике (см. т. I, с. 79) и с учетом
величины C;(Ti2O3, к) при 2110 К.
Погрешности значений Ф°B"), приведенных в табл. 940, при 298,15, 1500, 3000 и 5000 К
оцениваются в 0,4, 4, 13 и 25 Дж'К~1-моль~1 соответственно.
Ранее термодинамические функции Ti,O3(K, ж) вычислялись в справочниках JANAF [2835]
и Шика [4181 ] (до 3500 и до 6000 К), а также в справочниках [1038, 2960] (до 2000 К). В таблицах
Таблица 36.11. Результаты определений h.jll° (Ti2O3, к, 298,15 К) (в кДж/моль)
Авторы
Метод
A/ffo(Ti20s, к, 298,15 К)
II закон
III закон
Хамфри, 1951 [2754] , Калориметрический, Ti2O3(K)+i/2O2(r) = —1519,5 + 2,0
= 2ТЮ2(к, рутил), 303 К, 7 точек
Ария и др., 1957 [41] То же, 298,15 К, 2 точки —1525 + 10
Сузуки, Самбонги, 1972 [4444] ЭДС, намерения AG(O2) Для TiC^ 5—TiO2 0. —1522 —1524,7+6,0
1173—1873 К, 8 изотерм
Чарлю и др., 1974 [1818] Калориметрический, Т12О3(к) + i/2O2(r) = —1520,4+5,0
= 2ТЮ2(к, рутил), 1100 К, 6 точек
Соммер, Катер, 1975 [4344] ЭДС, 3Ti2O3(K)+1/2O2(r)=2Ti3O8(K), 1089— —1520 —1517,3+6,5
1426 К, данные представлены в виде уравнения
Васильева и др., 1981 [184] То же, 1290—1350 К, данные представлены —1519 —1518+6
в виде уравнения
1 В работе [3637] исследовался такой же образец Ti2O3 (к), как и в [4270], а в работе [944] — такой же, как
и в [759].
2 В статье [944] приведено значение Гт=2083 К, что соответствует средней температуре, наблюдавшейся в
двухфазной области перед полным плавлением Ti2O3 (точнее, она равна 2088 К). Согласно данным этой же работы
[944] Ti2O3 полностью расплавляется при Т ^ 2100 К.
110
Титан и его соединения Ti3O5(k, ж)
JANAF [2835] аномалия теплоемкости при 464 К рассматривалась как фазовый переход I рода;
расхождения с данными табл. 940 составляют 0,5 и 1 Дж-К~1-моль~1 в значениях Ф°(Т) при 2000
и 3000 К соответственно. Остальные расчеты выполнены по менее надежным данным и без учета
работ [759, 944].
Константа равновесия реакции Т12О3(к, ж)=2Т1(г)+ЗО(г) вычислена с использованием
значения Дг#°@)=3193,768+4,5 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
t\fH°(ThOs, к, 298,15 К) = —1520,0 + 2,0 кДж • моль.
Результаты определений энтальпии образования Ti2O3(k) представлены в табл. 36.11. В
пределах указанных погрешностей все данные согласуются между собою. Принятая величина является
средневзвешенной из значений, приведенных в этой таблице, при этом основной вес придавался
результатам калориметрических измерений [1818, 2754].
Т13ОБ(к, ж). Термодинамические свойства кристаллического и жидкого пентаоксида трититана
стехиометрического состава в стандартном состоянии при температурах 100—5000 К приведены
в табл. 941.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 36.1. В справочнике за стандартное состояние Т13ОБ(к) в интервале 0—448 К принята
моноклинная (а) модификация, а в интервале 448 — 2050 К—ромбическая (J3) модификация
(см. [1353, 2802]) К
Термодинамические функции Ti3O5(k) при Т <^ 298,15 К вычислены по результатам измерений
теплоемкости в работе Березовского и др. [126] A1,5—303 К) для образца Ti3O5, содержавшего
4% Ti4O7 и 0,02% других примесей. В расчеты внесена поправка на примесь Ti4O7. Ввиду разброса
данных [126] при Т <^ 30 К, экстраполяция теплоемкости в интервале 0—30 К проводилась
по уравнению С°(Г)=7,66-10~6Г3 (Дж-К-моль), выведенному на основании I и IV серий
измерений, как более надежных в интервале 30—55 К. Значение S°C0 К) найдено равным
0,8 Дж-К^-моль-1. Погрешности вычисленных значений ?°B98,15 К) и #"B98,15 К)—#°@)
оцениваются в 1,3 Дж-К^-моль и 0,2 кДж-моль соответственно. Менее точные измерения
теплоемкости Ti3O5(k) проведены Шомейтом [4270] E3—296 К) для образца, содержащего 0,2% TiC
и 0,7% SiOa. Эти данные систематически выше измерений [126] (на 1—4%) и не принимались во
внимание.
В интервале 298,15—448 К термодинамические функции Ti3O5(k) вычислены на основании
уравнения для С°р(Т) (см. табл. 36.1), полученного по данным Нейлора [3637] C26—1340 К).
Температура превращения D48+5 К) принята по результатам прецизионных измерений
электропроводности и рентгенографических и ДТА-исследований образца Ti3O6 высокой степени
чистоты О 99,99%) в работе Рао и др. [3923]. Практически совпадающее значение D50 К)
получено Нейлором [3637]. Близкие, но менее точные значения Tir были определены в работах [1889,
2980, 2981, 3578]. Энтальпия превращения (9,6+0,8 кДж-моль) принята как разность значений
#сD48 К)—Я°B98,15) для а- и р-модификаций Ti3O5, полученных по данным [944, 3637]. С этим
значением согласуются результаты калориметрических измерений (9,4 и 10,4 кДж-моль) в
работах [3637] и [2350]. Результаты измерений методом ДТА [3923] и [1889] F,7 и 6,1 кДж-моль)
не учитывались.
В интервале 448—2050 К для теплоемкости принято уравнение (см. табл. 36.1), полученное
совместной обработкой результатов измерений энтальпии Ti3O5, проведенных Нейлором [3637]
C26—1340 К) и Слюсарем и др. [944] E80—1994 К) после введения поправок на примеси 2. Данные
по теплоемкости Ti3O5, измеренные методом ДСК в работе [1889] D00—600 К), не использовались,
так как они приводят к сильно заниженным величинам.
Температура плавления Ti3O6 B050 +20 К) принята на основании данных Уолбека и Гиллеса
[4632] B050+10 К) и Слюсаря и др. [944] B043 К). Существенно менее точные значения были
получены в работах [2060] B173 К), [3664] A953 К), [1647] A983 К) и [2423] B130 К). Энтальпия
Т13О5(ж) измерялась Слюсарем и др. [944] при 2116 К. Энтальпия плавления A67+6 кДж-моль)
была определена исходя из этих данных, принятого в справочнике значения C°(Ti3O5, ж) и вели-
1 Известна еще метастабильная моноклинная (?) модификация Ti3O5, изоструктурная V8O5 [1352].
2 В работе [3637] исследовался такой же образец Ti3O5, как и в [4270], а в работе [944] — такой же, как и в [126].
111
Т140,(к, ж)
Глава 36
чины #°B050 К)—#°B98,15 К) для Ti3O5(K). Теплоемкость Т130Б(ж) оценена равной 260 +
+10 Дж-К~1-моль на основании значения C°(Ti3O5, к, Тт), соответствующего принятому
уравнению для C°(Ti3O5, к, Р), и значения, полученного по принятой в справочнике методике оценок
(т. I, с. 79).
Погрешности приведенных в табл. 941 значений Ф°(Т) при 298,15, 1500, 3000 и 5000 К
оцениваются равными 0,6, 6, 18 и 40 Дж-К-моль~1.
Ранее термодинамические функции Ti3O5(k, ж) вычислялись в справочниках JANAF [2835]
(до '4000 К) и Шика [4181] (до 6000 К). Расхождения значений Ф°(Т), приведенных в табл. 941
и в [2835, 4181], составляют до 10 и 4 Дж-К-моль~1 соответственно.
Константа равновесия реакции Ti3O5(K, ж)=ЗТ1(г)+5О(г) вычислена с использованием
значения АгН°@)=5090,368 +6,7 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(Ti3Os, к, 298,15 К) = —2457,0 ±3,0 кДж • моль.
Результаты определений энтальпии образования Т13О5(к) представлены в табл. 36.12. Как
видно из таблицы, наиболее точные измерения были проведены Хамфри [2754], и принятое
значение основано на этой работе. Измерения в работах [41, 858, 1818, 3486, 4444] привели к
существенно менее надежным значениям, совпадающим в пределах их погрешностей с принятым. При
исследовании равновесий в работе [2906] использовалась недостаточно точная методика и не были
учтены возможные побочные реакции (см. [3737]).
Таблица 36.12. Результаты определений А./Н0 (Ti3O5, к, 298,15 К) (в кДж-мбль)
Авторы
Хамфри, 1951 [2754]
Ария и др., 1957 [41]
Резниченко и др., 1963 [858]
Сузуки и Самбонги, 1972 [4444]
Меррит и др., 1973 [3486]
Чарлю и др., 1974 [1818]
Джунея и др., 1980 [2906]
Метод
Калориметрический, Ti3O5(K)+1/2O2(r)—ЗТЮ2(к,
рутил), 7 опытов
То же, 2 опыта
Протока, 3TiO2(K)+H2(r)=Ti3O5(K)+H3O(r),
1328—1553 К, 6 точек
ЭДС, измерения ДС(О2) для ТЮ1>67—TiO2 00 при
1173—1873 К, 8 изотерм
Гравиметрический, измерения Д?(О2) для TiOx e7—
ТЮ2>0 при 1304 К
Калориметрический, Ti3O5(K)+1/2O2(r)=3TiO2(K)
при 1100 К, 3 опыта
Статический, 21ГлаО8(к)+С(графит)=ЗгП2О3(к)+
СО(г), 1180—1330 К, данные представлены в виде
уравнения
Д/Я°(Т13Об1 к, 298,15 К)
II закон
III закон
-2457,0+3,0
—2466+9
-2467 ±5 -2456+10
—2458 —2468+11
_ —2465+9
—2458 ±14
-2435 -2501 +20
Ti4O7(K, ж). Термодинамические свойства кристаллического и жидкого гептоксида тетратитана
стехиометрического состава при температурах 100—5000 К приведены в табл. 942.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 36.1. В справочнике за стандартное состояние Ti4O7(k) принята триклинная модификация,
стабильная от 0 до Тт. Фазовые переходы при 142 К (типа полупроводник—полупроводник)
и при 154 К (типа полупроводник—металл) х не изменяют симметрию его кристаллической решетки.
Термодинамические функции Т14О7(к) при Т <^ 298,15 К вычислены по измерениям
теплоемкости в работе Березовского и Паукова [128, 129] A1,6—304 К), скорректированным на
содержание 7% Ti3O5 в исследованном ими образце 2, и с учетом энтальпий фазовых переходов при 142
Фазовые переходы Ti4O- (к) исследовались в ряде работ, принятые в справочнике температуры этих переходов
установлены в работах [128, 1889, 3169, 4189].
При Т < 14 К в работе [128] были получены систематически заниженные значения Cj,(Ti4O,, к). При
вычислении термодинамических функций Ti4O7 (к) в интервале 0—16 К использовалось уравнение С^,(Г) = 8,8 -10~5Х
XT3 (Дж-К^-моль), основанное на данных работы [128] при 14 < Г < 24 К.
112
Титан и его соединения
Ti4O7(K, ж)
и 154 К, которые по измерениям Шленкера и др. [1889, 3169, 4189] равны 0,40+0,2 и 1,96 +
+0,02 кДж-моль. В работе [128] фазовый переход при 142 К не наблюдался, а^ля энтальпий
перехода при 154 К были получены более низкие значения A,52 и 1,56 кДж-моль). Это различие
обусловлено, по-видимому, темт что в [1889, 4189] исследовался монокристаллический образец
высокой чистоты, а авторами [128] — порошкообразный образец, содержавший 7% Ti3O5.
Однако измерения теплоемкости были выполнены Березовским и Пауковым более надежным методом,
чем в работах [1889, 4189], и это, вероятно, является причиной значительных систематических
расхождений между полученными данными х. Погрешности приведенных в табл. 36.1 значений
5°B98,15 К) и Я°B98,15 К)—#°@) оцениваются в 2 Дж-К^-моль и 0,3 кДж-моль.
В интервале 298,15—1960 К для теплоемкости Ti4O7(k) принято уравнение (см. табл. 36.1),
найденное совместной обработкой результатов измерений энтальпии Ti4O7, проведенных Слюсарем
и др. [944] F79—1937 К) и скорректированных на содержание 7% Ti3O6 в исследованном образце,
и значений энтальпии Ti4O7 при 358 и 442 К, вычисленных по данным Шленкера и др. [1889, 4189]
для теплоемкости и уменьшенных на 8,5% для их согласования с данными [128].
Температура плавления Ti4O7 A960+20 К) принята на основании измерений Брауэра и Литтке
[1647] A955 К), Хемпсона и Гиллеса [2562] A943 К) и Слюсаря и др. [944] A993 К). В работах
[1647, 2562] значение Тт определялось по началу плавления и могло быть несколько заниженным,
в работе [944] оно находилось на основании калориметрических измерений энтальпии образца,
содержавшего 7 % Ti3O5, в связи с чем полученное значение Тт является ненадежным. Энтальпия
Т14О7(ж) измерялась Слюсарем и др. [944]. Принятые в справочнике значения энтальпии
плавления Ti4O7 A80+40 кДж-моль) и теплоемкости жидкого Ti4O7 C62+20 Дж-К~1-моль) основаны
на трех измерениях энтальпии Т14О7(ж) в работе Слюсаря и др. [944] B020—2235 К).
Погрешности приведенных в табл. 942 значений Ф°(Г) при 298,15, 1500, 3000 и 5000 К
оцениваются в 1, 7, 30 и 60 Дж-К^-моль.
Ранее термодинамические функции Ti4O7(K, ж) вычислялись в справочнике JANAF [2835]
(до 4000 К) на основании существенно менее полных и точных данных. Расхождения между
значениями Ф°(Т), приведенными в [2835] и табл. 942, составляют от 1 до 12 Дж-К~1-моль.
Константа равновесия реакции Ti4O7(k, ж)=4Т1(г)+7О(г) вычислена с использованием
значения ДгЯ°@)=7000,428+17 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^Т^О,, к, 298,15 К) = —3403 + 15 кДж • моль.
Результаты определений энтальпии образования Ti4O7(k) представлены в табл. 36.13.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, неточность
использованных в расчетах термодинамических функций и термохимических величин. Принятая величина
является средневзвешенной из приведенных в табл. 36.13 данных, за исключением работы [2906],
где могли протекать неучтенные побочные реакции (см. [3737]).
Таблица 36.13. Результаты определений XfH°(TUO7, к, 298,15 К) (в кДж-моль)
Авторы
Сузуки, Самбонги, 1972 [4444]
Меррит и др., 1973 [3486]
Васильева и др., 1977 [175, 176]
(см. также [189])
Васильева и др., 1977 [176]
Джунея и др., 1980 [2906]
Метод
ЭДС, измерения Д<?(О2) для TiOa 75—TiO2l00,
1173—1873 К, 8 изотерм
Гравиметрический, измерения Д(?(О2) для ТЮХ 75—
ТЮ2 „о, 1304 К
ЭДС, 8Ti3O5(K)+O2(r)=6Ti4O7(K), 1220—1350 К а
ЭДС, Ti4O7(K)+V2O2(r)=4TiO2(K, рутил), 1150—
1273 К, 5 изотерм
Статический, 3Ti4O,(K)+C(K, графит)=4Тл3Ов(к)+
+СО(г), 1180-1330 К а
а Данные представлены в виде уравнения.
ДуЯ° (Ti4O7
II закон
—3396
—
—3394
-3413
—3364
к, 298,15 К)
III закон
—3407+20
—3406+18
—3395+25
—3402+16
—3422+50
1 В работах [1889, 4189] данные о теплоемкости Ti4OT(K) в интервале 10—430 К приведены только на графике.
8 Заказ W» 1590
Глава 37
ЦИРКОНИЙ И ЕГО СОЕДИНЕНИЯ
Zr(K, ж), Zr, Zr+, ZrO, ZrO+, ZrOa(K, ж), ZrO2
В настоящей главе рассматриваются цирконий и его кислородные соединения — всего 5 веществ.
Для двух веществ приведены сведения об их термодинамических свойствах в конденсированном
состоянии и для пяти — в газообразном состоянии. Термодинамические свойства четырех газов
(Zr, Zr+, ZrO, ZrO+) рассчитаны для температур до 10 000 К и одного (ZrO2) — до 6000 К.
В Приложении 3.10 для элементарного циркония приведены сведения о критических
постоянных.
Термодинамические свойства всех веществ приведены для природной смеси изотопов циркония
(атомный вес 91,22) и кислорода, однако различие молекулярных постоянных разных изотопоме-
ров учитывалось только для ZrO (г).
В конденсированном состоянии основным оксидом циркония является ZrO2, который имеет
узкую область гомогенности (ZrO2985—ZrO2000), известны также субоксиды Zr6O, Zr3O, Zr2O,
которые мало устойчивы и плохо изучены. В литературе обсуждался вопрос о существовании
кристаллических оксидов ZrO и Zr2O3, однако при давлениях до 100 кбар эти оксиды не являются
устойчивыми соединениями (см. [29]).
Zr(K, ж). Термодинамические свойства кристаллического и жидкого циркония в стандартном
состоянии при температурах 100—6000 К приведены в табл. 943.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 37.1. В справочнике за стандартное состояние Zr(K) в интервале 0—1140 К принята гек-
Т аб л
ство
ff
со
в
Zr
ZrO2
ица 37.1. Принятые значения термодинамических величин для циркония и его соединений
в кристаллическом и жидком состояниях
Состояние
1
ю
oo'S
го
1н
кДж-моль
к11, гекс. (а) 5,523
к1, куб. (?) -
ж —
кШ, монокл. 8,751
кП, тетр. —
к1, куб. —
298,15 К)
о
298,15 К)
¦Те,
Дж • К • моль
38,98 25,40
50,39 55,92
Примечание. С°р(Т)=а+Ъ -Т—с-Г* (в Дж-К-моль).
Коэффициенты
нении для С
а
6-103
23,584 10,008
9,929 11,527
42,6 —
68,329 9,081
78,1 —
80 —
100 —
в урав-
пР(Т)
С • 10-в
1,038;
—69,922
13,438
Интервал
температур
или
Т
х m
К
298,15-1140
1140—2133
2133—6000
298,15—1445
1445—2620
2620—2983
2983—6000
1140
2133
1445
2620
2983
или
кДж-
• МОЛЬ
3,87
13,7
8,4
13
90
сагональная (а) модификация (структурный тип Mg), а в интервале 1140—2133 К — кубическая
(Р) модификация (структурный тип a-Fe) г. При 0,546 К [4329] a-Zr переходит в сверхпроводящее
состояние.
1 При давлениях, превосходящих 20 кбар, стабильна гексагональная модификация Zr высокой плотности (см.
[1021]).
114
Цирконий и его соединения Zr(K, ж$
При Т <^ 298,15 К термодинамические функции a-Zr вычислены на основании согласующихся
между собой результатов измерений теплоемкости, выполненных Нейпом и др. [3049] A,2—4,5 Щ%
Думмером [2125] @,9-12 К), Коллинзом и Хо [1903] A,5-6 К), Уолкотом [4751] A,15-20 К),
Скиннером и Джонстоном [4311] A4—298 К), Тоддом [4518] E3—297 К) и Бурком и др. [17271
B0—200 К). Менее надежные данные, полученные в работе [2193] A,8—4,2 К), не учитывались.
Погрешности принятых значений теплоемкости a-Zr в интервале 20—298,15 К оцениваются в ~1 %.
Погрешности вычисленных значений 5°B98,15 К) и Н°B98,15 К)—Н°@) (см. табл. 37Л) составляют
0,3 Дж-К^-моль и 0,04 кДж-моль.
В интервале 298,15—1140 К термодинамические функции модификации a-Zr вычислены с
использованием приведенного в табл. 37.1 уравнения для С"р(Т), полученного совместной обработкой
измерений энтальпии, проведенных Егером и Веенстрой [2821] E05—1074 К), Кофлином и Кингом
[1944] C90—1125 К), Скиннером [4308] C00—1140 К), Аденштедтом [1255] C73 К), Дугласом
и Виктором [2111] C73—1123 К), а также измерений теплоемкости, выполненных Фольмером
и др. [4620] C00—1140 К), Федоровым и Смирновым [1047] B93—1073 К) и Новиковым и др.
[728] C56—992 К). При этом наибольший вес придавался данным [2111, 2821] и наименьший —
[1047, 4620]. Менее точные измерения энтальпии выполнены в работах [3527] B73—373 К), [3957?
D04—1309 К), [2249] D72—1140 К), а измерения теплоемкости — в работах [4222] C33—1140 К),
[3025] B98-1140 К), [47] A100 К), [889] A100, 1135 К), [3590] C00-850 К), [148] G00-1135К)
и [438] (900-1139 К) К
Температура а *± [3 превращения Zr принята равной 1140+5 К — среднему из значений,
полученных в работах [4618] A135+5 К), [2019] A138+10 К), [2133] A139+10 К), [4308, 4312]
A144+5 К), [2091] A136+10 К), [3048] A143+4 К), [4222] A144+2 К), [3157] A144+10 К),
[3025] A139+10 К), [263] A135 +10 К) и [1799] A147+10 К), в которых исследовались наиболее
чистые образцы циркония (>99%) и применялись наиболее надежные методы. В ряде работ
(см., например, [277, 4140, 4620]) были получены менее точные данные. Энтальпия полиморфного
превращения C,87+0,3 кДж-моль) определена как разность значений энтальпий JJ-и а-модифи-
каций Zr при 1140 К. С этим значением AtrH° согласуются результаты наиболее надежных
калориметрических измерений [1944] C,85 кДж-моль), [2111] C,74 кДж-моль), [1649, 46201
C,98+0,04 кДж-моль), измерений импульсным методом [1799] C,98+0,2 кДж-моль") и
методом ДТА [2569] D,0 кДж-моль). Менее точные значения (в пределах 2,95—4,5 кДж-моль)
определены в работах [277, 666, 3168, 4222, 4308, 4618].
В интервале 1140—2133 К для теплоемкости ^-модификации Zr принято уравнение, выведенное
совместной обработкой данных по энтальпии, полученных Кофлином и Кингом [1944] A148—
1371 К), Скиннером [4308] A140—1798 К), Дугласом и Виктором [2111] A143—1173 К), и данных
по теплоемкости Фольмера и др. [4620] A140—1700 К), Арутюнова [47, 1055] A300—2100 К)
и Цезерляна и Ригини [1798] A500—2100 КJ. Наибольший вес придавался последней работе;,
в которой теплоемкость измерялась методом импульсного (субсекундного) нагрева наиболее чистого
образца циркония (99,98%). Погрешность этих измерений авторы [1798] оценивают в 0,7%, считаяж
что примененный ими метод исключает влияние абсорбции газов на полученные результаты.
Существенно менее надежные данные об энтальпии Р-модификации Zr были получены в работах
[2821] A173—1273 К), [3957] A140—1309 К), [2249] A140—1307 К) и о теплоемкости — в
работах [4222] A140—1233 К), [3025] A140—2118 К), [480] A300—2000 К), [438] (предварительные
данные), [889] A100-2100 К).
Температура плавления B133+5 К) принята как среднее значение из измеренных Дирдор-
фом и Хайесом [2011] B131+10 К), Сара [4140] B136+10 К), Аккерманом и Раухом [1224]
B134+4 К), Чарлесвортом [1224, 1814] B132+8 К), Цезерляном и Ригини [1800, 1801] B128+8 К)
и Аккерманом и др. [1217] B133+3 К). В этих работах применялись наиболее точные методы
и исследовались наиболее чистые образцы (99,9—99,99%). Tm(Zv) определялась также в работах
[1255, 4222, 4830], где были получены значения 2121—2126 К, и в работах [1811, 3721, 4084]х
где были получены значения 2144—2152 К.
Энтальпия Zt(tk) измерялась Боннелом и др. [1616,1617] методом левитационной калориметрии
при 2233—3048 К. Результатам этих измерений в интервале 2233—2997 К A2 точек) соответствует
1 О данных работы [438] авторам справочника известно только по предварительному сообщению.
2 Теплоемкость ^-модификации Zr при 1140 К принималась равной 28,45 Дж-К^-моль и определялась
экстраполяцией к 1140 К усредненных значений C°(Zr, к, C), основанных на данных [47, 1798, 1944, 4308, 4620]
в области температур, близких к Ttr,
115 .
Zr(K, ж)
Глава 37
C°(Zr, ж)=42,6+1,8 Дж-К^-моль. G учетом значения #°B133 К)—#°B98,15 К), рассчитанного
для Zr(K) по постоянным табл. 37.1, данные [1616, 1617] приводят к принятой в справочнике
энтальпии плавления A3,7+4,0 кДж-моль'1). С этим значением согласуются величины, полученные
в работе [1617] (М.гЗГкДж-моль-1) по энтальпии H°(TJ—Я°B98,15 К) Zr(«) и Zr(ic) и в работе
[274] A4,6 кДж-моль) по энтальпиям сублимации и испарения циркония. Попытки прямого
определения величины АтН°Bт, к) методом смешения в калориметре с жидким магнием [406,
559] B0,9 кДж-моль) и методом импульсного нагрева [663—667] B1,5 кДж-моль), по-видимому,
привели к неверным величинам.
Погрешности вычисленных значений Ф°(Г) при 298,15, 2000, 4000 и 6000 К оцениваются в 0,2,
1,3, 4 и 6 Дж-К^-моль.
Ранее термодинамические функции Zr(K, ж) вычислялись во втором издании настоящего
справочника [337] и в справочниках [2752, 2834, 2960, 4181, 4421] (до 3000—6000 К). Расхождения
между значениями Ф°E000 К), приведенными в табл. 943 и в [337] и [2834], составляют 2 и
11
1 Дж-К~1-моль~1.
В настоящем справочнике в качестве стандартного состояния циркония принято
кристаллическое состояние и в соответствии с этим
Давление пара циркония в реакции Zr(K, ж)=Zr(г) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = A,#°(Zr,. к, 0) = 598 + 5 кДж • моль.
Эта величина основана на приведенных в табл. 37.2 величинах, которые были вычислены по
результатам измерений давления пара циркония; в расчетах принималось, что пары циркония
одноатомны [1226]. Приведенные погрешности учитывают только1 воспроизводимость измерений.
Таблица 37.2. Результаты определений AsH"°(Zr, к, 0) (в кДж-моль)
Авторы
Метод
!\sli° (Zr, к, 0)
II закон
III закон
Цвиккер, 1928 [4833] (см. [2018]) Испарениес открытой поверхности, 1600—2130 К, 256 579+30
данные представлены в виде уравнения
Скиннер и др., 1951 [4310] То же, 1949—2054 К, 12 точек 588+16 609,0+0,5
Чапка и др., 1957 [1854] (см. [2435]) Масс-спектрометрический, 2135—2332 К, 4 точки, — 557 +22
данные представлены в виде графика
Федоров, Смирнов, 1962 [1046, 3811] Эффузионный с использованием радиоактивного 200+89 522+9
(см. [1044]) изотопа, 1340—1738 К, 10 точек
Трулсон, i дстейн, 1964 [4555, 4556] Масс-спектрометрический, 1971— 2115 К, 13 точек 580+35 593,8-*-0 6
То же, 2151—2277 К, 8 точек 559+50 594,0+0,8
Старостина и др., 1967 [976] То же, 2000 К, 1 точка — 594
То жеа 600 —
Коч, Энейбл, 1968 [3054] Испарение с открытой поверхности, 2233—2900 К, 621 ±40 620,7+2,7
16 точек
Федоров и др., 1968 [1049] Эффузмонный с использованием радиоактивного 244+54 493+37
изотопа, 1531—1892 К, 4 точки
Аккерман, Раух, 1972 [1226] Масс-спектрометрический, 2140—2550 К, учтена 599+5 599,6+6,0
растворимость W в Zr^) [1224], данные
представлены в виде уравнения
Микульская п др., 1972 [296, 297, Иопарение с открытой поверхности, с использова- 532+12 594,3+2,0
678] нием радиоактивного изотопа, 1703—2063 К,
17 точек б
Стормс, Гоффин, 1973 [4404] Масс-спектрометрический, 1673—2041 К а 630 —
Сумин, Пе&зулаев, 1973 [995] Испарение с открытой поверхности, 2133— 608 —
2500 К а
а В работе приведено только полученное по уравнению II закона значение As#°(r); б исправлены имеющиеся
в работе [297] опечатки. Измерения при 1373—1643 К, 6 точек, в расчетах не использовались.
' 116
Цирконий и его соединения Zr(r), Zr+(r)
Расхождения между результатами наиболее точных измерений в работах [297, 1226, 3054, 4310,
4555] превышают их погрешности; принятая величина является средневзвешенной из них. В
работах [976, 995, 1854, 4404, 4833] были получены менее надежные величины, совпадающие с
принятой в пределах их погрешностей. При оценке погрешности рекомендуемого значения были
учтены неточность термодинамических функций и сечений ионизации.
&
Zr(r). Термодинамические свойства газообразного циркония в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 944.
Для расчета термодинамических функций использованы уровни энергии г атома Zr в интервале
0—55 000 см, соответствующие валентным электронным конфигурациям . . Ad25s2 (к ней
относится основной уровень SF2), . . Ad%5f, . . Ad\ . . Ad35l (Z=0^-4), . . Ad25s5l (l=l~A) и
. . Adbs25l (Z=l—4) и имеющие суммарный статистический вес 4245. Принятые величины основаны
на данных, рекомендованных Мур [3550] в результате анализа спектральных исследований,
а также на оценках ненаблюдавшихся уровней энергии перечисленных конфигураций,
выполненных методом, изложенным в Приложении 6. Оцененные уровни имеют энергию 21 100 см и выше
с суммарным статистическим весом 2610. Погрешности оцененных величин лежат в пределах 100—
4000 см.
Термодинамические функции Zr(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т ^ 3000 К обусловлены главным образом неточностью фундаментальных постоянных
и составляют 0,02—0,03 Дж-К^-моль. При более высоких температурах возникают
погрешности из-за приближенной оценки ненаблюдавшихся уровней энергии и пренебрежения
ридберговскими состояниями; погрешности в Ф°(Г) при 6000 и 10 000 К могут достигать
0,05 и 0,5 Дж-К^-моль.
Ранее термодинамические функции Zr(r) вычислялись в предыдущем издании настоящего
справочника [337], в справочниках JANAF [2834], Шика [4181] и Стаяла и Зинке [4421] (до 3000 К),
а также в работах Жирницкого [4837 ] до 15 000 К, Хилзенрата и др. [2652 ] до 10 000 К, Кольского
и др. [3066, 3067] до 8000 К и Каца и Маргрейва [2934] до 2000 К. Во всех расчетах, как и в
настоящем справочнике, были использованы уровни энергии атома Zr, рекомендованные в [3550].
В расчетах [337, 4837] были выполнены приближенные оценки ненаблюдавшихся ридберговских
состояний для к^11 [337] и п ^ 10 [4837]. При Т <^4000 К результаты всех расчетов (кроме
[3067]) практически совпадают между собой и с данными табл. 944. При Т >4000 К расхождения
возрастают. Данные [337] и табл. 944 согласуются при 6000 К в пределах 0,02 Дж-К^-моль
для Ф°(Т), расхождения с данными [2834] достигают0,05 Дж-К -моль в значении Ф°F000 К),
расхождения данных [2652, 4837] и табл. 944 не превышают 0,1 и 0,8 Дж-К~1-моль в значениях
Ф°(Г) при 6000 и 10 000 К.
Энтальпия образования газообразного циркония
&fH°(Zi-, г, 0) = 598 + 5 кДж • моль
соответствует принятой в настоящем справочнике энтальпии сублимации циркония.
Zr+(r) .Термодинамические свойства газообразного положительного иона циркония в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 945.
Для расчета термодинамических функций использованы уровни энергии г иона Zr+ в интервале
0—79 032 см, соответствующие валентным электронным конфигурациям . . Ad25s (к ней
относится основной уровень 4Л/г), . . Ad3, . . Ad5s2, . . Ad25l A=1, 2) и . . Ad5s5p и имеющие
суммарный статистический вес 908. Принятые величины основаны на данных, рекомендованных Мур
[3550],и на оценке для ненаблюдавшихся уровней конфигурации . . Ad3 (энергия 76 000+5000 см,
статистический вес 84).
Термодинамические функции Zr+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т ^ 5000 К обусловлены неточностью фундаментальных постоянных и не
превосходят 0,02—0,03 Дж-К~1-моль. Выше 5000 К становятся существенными погрешности из-за при-
> См. примеч. 1 к тексту по О(г).
117
ZrO(r) Глава 37
ближенной оценки энергии ряда уровней и пренебрежения ридберговскими состояниями; они
могут достигать 0,1 Дж-К-моль~1 в значении Ф°A0 000 К).
Ранее таблицы термодинамических функций Zr+(r) вычислялись в справочнике JANAF [2835],
а также в работах Грина и др. [2473] до 50 000 К, Жирницкого [4837] до 15 000 К и Хилзенрата
и др. [2652] до 10 000 К. Все расчеты основаны на уровнях энергии, рекомендованных Мур 13550];
в расчете [4837] дополнительно были учтены оцененные ридберговские уровни с п ^ 10. При
Т <d 3000 К результаты всех расчетов и данные табл. 945 согласуются в пределах 0,02 Дж-К-1Х
Хмоль. Расхождения с данными [2835] достигают 0,6 Дж-К~1-моль~1 в значении Ф°F000 К)
вследствие ошибки в расчете этих авторов, с данными [2652] и [4837] — 0,02 и 0,10 Дж-К~1-мпль
в значении Ф°A0 000 К).
Константа равновесия реакции Zr+(r) +e(r)=Zr(r) вычислена с использованием значения
Дг/?°@) = —656 + 8 кДж-моль, соответствующего принятому в справочнике потенциалу иониза-
щии
/0(Zr)=656+8 кДж-моль?«54 800+700 см.
Последний основан на данных Мур [3550] для первых двух членов ридберговских серий
... Ad?bsns3F и Ad3nsbF (п=Ъ, 6) атома Zr и величине 0,08±0,04 для разности смежных значений
квантовых дефектов, оцененной по соответствующим данным для Rb, Sr, Mo, Pd и Kg. Принятое
значение согласуется с принятыми в справочнике величинами /0(Ti) и /0(Hf), а также со
значением, найденным в работе [1767] E5 145 см). Ранее в работе [2990] по спектральным данным
было получено более высокое значение E6 077 см), а в работах [3943] и [3944] — заниженные
значения 6,4 и 6,6 эВ F17 и 634 кДж-моль).
Принятому значению соответствует
i\fH°(ZT+, г, 0) = 1254 ± 10 кДж ¦ моль.
ZrO(r). Термодинамические свойства газообразного оксида циркония в стандартном состоянии
три температурах 100—10 000 К приведены в табл. 946.
Молекулярные постоянные 90Zr160, принятые в справочнике на основании анализа результатов
исследований электронного спектра этой молекулы и приближенных оценок, приведены в табл. 37.3.
В электронном спектре ZrO наблюдалось 10 систем полос, связанных с переходами между шестью
синглетными и пятью триплетными электронными состояниями1: S41 ?± Щ+ [1374, 2003
2374, 3600, 3601, 3823, 4698, 4790], СгЪ+ -*Z12+ [2374, 3600, 3601, 3822, 4453] GXI.+ -*Z12 +
1202, 1262, 3600, 4576, 4698], ВШ ->АХ\ [3204, 3600, 4970], FXL -+А1*. [1261, 1273], Ь3П -> а3Д
[3826], #Ф**а3Д [3167, 3316, 3317, 3795, 3824, 4168, 4468, 4480], е3П**а3Д [1262,3264,4575],
/3Д«*а3Д [1262, 2003, 3167] и е3П -+ X1!,+ [2561, 3204]. Анализ конфигурации электронной
оболочки ZrO (см., например, [1661]) и данные для молекул YF, ScF и TiO (см. рис. на с. 101)
позволяют предполагать, что молекула ZrO должна иметь ряд валентных состояний с энергиями
12 000—40 000 см, которые еще не наблюдались в спектрах. Кроме того, благодаря низкому
потенциалу ионизации эта молекула должна иметь ридберговские состояния с относительно
низкими энергиями. Количественные квантовомеханические расчеты ZrO отсутствуют. В табл. 37.3
помимо 11 валентных состояний, переходы между которыми изучены экспериментально, включено
еще 10 состояний, аналогичных наблюдавшимся в спектрах ScF и TiO. Энергии этих состояний
@ыли оценены при подготовке справочника на основании энергий известных изоконфигурационных
еостояний, а также имеющихся данных для молекулы TiO и величин синглет-триплетного
расщепления изоконфигурационных состояний TiO и ZrO. Погрешности оцененных таким образом
величин могут достигать 3000—5000 см2.
Молекулярные постоянные ZrO в состояниях ХХ2 + и В1!!, приведенные в табл. 37.3, получены
Фяллипсом и Дэвисом [3823] из анализа вращательной структуры 18 полос системы В1!!—Х12 +
f»'' ^ 5, г/ ^ 7). Они хорошо согласуются с результатами исследования этой системы в работе
Ц374], а для состояния ХХ2+ — с постоянными, найденными из анализа систем
*¦ В справочнике принята символика электронных состояний, отличающаяся от использованной в оригинальных
работах.
* Состояния с энергиями до 30 000 см (кроме состояния G1 2+) могут быть приписаны конфигурациям. . . of,
... . Cgbd, . . . а8тср, . . . ag op, . . .bdTip, . . .bd <3p, . . . a87trf. Если состояние G1 2+ соответствует конфигурации . . .SJ,
at области 15 000—25 000 см должны существовать еще два состояния: ' 2~ и гГ,
118
Цирконий и его соединения
ZrO(r)
Таблиц
Молекула
а 37.3. Молекулярные постоянные 9
Состояние
Те
90Zri6O a Xi 2+ о
¦а^ 1 099,08 )
а3Д2 1 386,99 {
а*Аа 1 724,59 )
А1 А 5 904,19
Ь3П! 12 109,7 ж
с3 2+ 14 000 и
.ВЧ1 15 441,78
С12+ 17 099
й3Ф3 17 169,12 л
е3Пг 19 178,9 м
X»1!! 22 000 и
/3Д2 22 994,3 н
Е^Ф 24 000 и
ЯД 24 451,5
g3n 27 000 и
•G1^+ 27 211,26°
ZrO+a Х2Д 0б
•Л2!! 9 000 е
вт 11 ооо е
С2 25 000
?>2П 37 500 3
?2 2 44 500 е
Примечание. Все постоянные даны
ше
'Zr^O и ZrO+
<»ехе
ве
СМ
976,51 3,462б 0,423 608
938,131 3,245 0,415 54
938,1 1,80 0,416 7 Д
8593 — 0,409 75 Д
— — , —
859,44 2,918к 0,402 501
8803 — 0,405 62
856,702 3,199 0,404 79
846,28 3,41 0,413 4 Д
— — —
820,58 3,31 0,392 7 Д
— — —
835,4 2,56 0,398 6
— — —
843,2 3,04 0,3951
990 3,9в 0,44
— — —
— — —
990ж — 0,437 98ж
895ж — 0,409 946ж
— — —
в см.
aL ¦ 103
1,954 в
1,90
1,2 е
—
,
1,92
1,65
2,10
—
—
—
2,1
1,9
2,3 г
—
—.
—
—
—
ZrO: a принятые обозначения электронных состояний отличаются от использованных
б («^=0,000 52; в сB=—3
Ве ¦ 107
3,19 г
3,29
3,5 Д
3,726 л
—
3,515
3,4385
3,697
3,4 Д
—
2,6 Д
—
3,7 Д
—
3,5
3,4 Д
—
—
3,34 ж
3,4268 ж
—
Ге
А
1,711 91
1,7285
1,726 Д
1,7406 Д
—
1,7558
1,7494
1,7512
1,733 Д
—
1,778 Д
—
1,7648
—
1,773
1,68
—
—
1,684 ж
1,741 ж
—
в оригинальных работах;
,3 -10 6; г 3,=—6,60 -10 и; Д приведено значение постоянной
е вычислено по соотношению A. 69); ж~ ГеF3П„)=11 839,1, Те(ЬЧ
шению A. 68); и оценка;
=19 140,8, ГДе3П2)=19 237
0 Состояние НЩ, к3ф
Те 30 000
к меуе=— 0,008 12; л Г,,(й3Ф2)=16 566
1; н Te(fiA1)=22 694,3, Г„(/3Д3)=23 4
?3п, /1ф кт, km
35 000 38 000
12)=12 486,7;
> 40 Т (й3Ф4)
14,9;
t для у=0;
3 вычислено по соотно-
=17 799,09;
м Те(е3П0)=
ZrO+: а большая часть постоянных основана на оценках (см. текст); *> -4 = 150; в вычислено по соотношению A.67)
я значению Do=63 000; г
вычислено по соотношению A. 69); "
е оценка; ж приведено значение для v=0; 3 .4=595,3.
вычислено
ю соотношению A. 68);
[3822] и (?Х2 + -^Х1^" [4576]. Близкие значения постоянных в состоянии ХХ2 + получены при
исследовании основной частоты ZrO в ИК-спектре [2359].
Энергия состояния А1 А принята по данным Хаммера и др. [4970], которые наблюдали переход
ВЧ! -> А1 Д в спектре лазерной флуоресценции молекул ZrO. Ранее Лаучлан и др. [3204] в спектре
^флуоресценции молекул ZrO, изолированных в матрице из неона, отнесли к этому переходу
полосу в области 5200 см. Колебательные постоянные ZrO в состояниях ЛХД и F1^., приведенные
в табл. 37.3, найдены Афафом [1261] по кантам девяти полос соответствующей системы (v" ^ 3,
v' ^ 2), вращательные постоянные — Акерлиндом [1273] из анализа структуры полос 0—0 и 1—1.
Вращательные постоянные в состоянии СХ2 + приняты по работе Филлипса и Дэвиса [3822], в
которой был проведен анализ структуры полос диагональной секвенции системы G*2+ -> ZX2+
(v <^ 3). Постоянные ZrO в состоянии G^S + найдены Улер и Акерлиндом [4576] при исследовании
системы G12+ -> X1!, + (четыре полосы с »' ^ 1, v" <J 2).
Принятые в справочнике энергии трех компонент нижнего триплетного состояния а3А
основаны на частотах полос 0—0 и 0-^1 еидтемы ^Ilj -> ХХ2 +, измеренных Хаммером и Дэвисом [2561 \
119
ZrO(r)
Глава 37
в спектре флуоресценции, возникающей при поглощении излучения лазера в переходе
¦*— а3 Аг, а также на данных о величине расщепления трех компонент этого состояния, которые были
получены Филлипсом и Дэвисом [3824] при исследовании системы й3Ф—а3Д г. Приведенные
в табл. 37.3 значения постоянных ZrO в состояниях а3А и Й3Ф приняты по работе [3824], в которой
была изучена структура 15 полос соответствующей системы (v" ^ 6, v' ^ 5). Триплетное
расщепление компонент состояния а3 А имеет аномальный характер, поэтому для описания энергии
вращательных уровней этого состояния авторы [3824] вместо уравнения A. 44) использовали более
сложные соотношения, предложенные Ковачем [3089], с введением в них дополнительных
поправок. Однако расчет энергии уровней ZrO в состоянии а3 Д с постоянными, приведенными в табл. 37.3,
по уравнениям A. 44) и по уравнениям, использованным Филлипсом и Дэвисом, не приводит
к расхождениям, превышающим 2—3 см при / ~ 100-^150.
Колебательные постоянные ZrO в состояниях Ь3И, е3П и /3Д найдены Ловатер [3316], значения
соответствующих вращательных постоянных приняты по работам Филлипса и др. [3826] (ЬЧ1),
Линдгрена [3264] (е3П) и Лагерквиста и др. [3167] (/3Д).
Термодинамические функции ZrO(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QSB и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом 24 возбужденных электронных состояний (каждая компонента состояния а3Д учитывалась
как отдельное состояние) и в предположении, что для всех возбужденных состояний, кроме а3Д
и A1 A, Q<ih.vV=(Pi/px) Qiol.if Величины QK0]l вр и их производные для состояний X1!, +, а3Д и А1 А
вычислялись по соотношениям A.73)—A.75) непосредственным суммированием по
колебательным уровням и интегрированием по значениям / с помощью уравнений типа A. 82). В расчете
A 81)
учитывались все уровни с значениями
щ ур ( ) р
/тах „, где /шах „ определялись из условий A. 81).
Колебательно-вращательные уровни состояний ХЧЗ*, а3Д и А1 А вычислялись по уравнениям
A. 65), A. 34), A. 64а), значения коэффициентов в этих уравнениях, соответствующие природной
смеси изотопов циркония и кислорода и полученные с учетом условий A. 66), A. 50), A. 58),
A. 59), приведены в табл. 37.4, там же приведены величины г?шах и Jlsm. Вырождение уровней со-
Таблица 37.4. Коэффициенты в уравнениях, описывающих уровни энергии (в см-1),
а также значения ушах и /iim, принятые для расчета термодинамических функций ZrO и ZrO+
Коэффициенты
ZJ2+
т„ -ю-* о
У10 -Ю 9,753 4
У20 -3,459 888
У30 -103 1,440 92
У40 -105 -3,000 2
У01 -10 4,225 85
Уи -103 —1,946 92
У21-106 —3,284 076
У02-107 —3,174 60
У12-10и -6,619 9
У03-Ю« -1,417
Ушах Н9
/i,m 519
ZrO: a энергии 18 возбужденных
ZrO+: a Состояние Х2Дз/2
аЧ-t
0,109 908
9,370 3
—3,249 008
1,393 81
—2,755 1
4,145 362
—1,893 1
—
—3,274 1
—.
-1,105
122
519
состояний см.
42 s вт
Те 300 9000 11 000
ZrOa
а3Д2
0,138 699
9,370 3
—3,250 4
1,546 8
—3,001
4,145 36
—1,893 1
—
—3,274 1
-1,103
121
518
в табл. 37.3.
С2 2 В3П
25 000 37 50(
а3Д3
0,172 459
9,370 3
—3,252 0
1,739 1
' -3,307
4,145 36
—1,893 1
—
—3,274 1
—1,101
119
517
Я2 2
) 44 500
А1 А
0,590 42
9,375 1
—2,139 1
6,170 3
-8,446
4,156 94
—1,195 7
—
—3,483 1
2,158
80
550
' ZrO+a
0
9,90
—3,90
—
—
4,40
—2,30
—
-3,4
—
-2,865
126
501
1 Близкое значение энергии подсостояния а3Д3 A700+250 см~1) было получено в работе Вейц и др. [202] пс
висимости от температуры отношения коэффициентов поглощения nojioc 0—0 систем Сг^+—Хг^+ и fA3—
по за-
в спектрах ударных волн.
120
Цирконий и его соединения
ZrO(r)
стояний а?Ах, а3Д2, а3Д3 и А1 А, так же как вырождение и мультиплетность уровней других
состояний, учитывались статистическими весами.
Погрешности вычисленных термодинамических функций при низких температурах
обусловлены главным образом неточностью фундаментальных постоянных. Выше 2000—3000 К становятся
существенными использование приближенного уравнения для описания энергии уровней
состояния а3Д, неточность принятых постоянных в состоянии А1 А., отсутствие экспериментальных
данных об энергии ряда электронных состояний (с32 +, D1!! и др.), а также возможность
существования у ZrO других состояний, не учитывавшихся в расчете. Погрешности в значениях Ф°(Г) при
298,15, 3000, 6000 и 10 000 К оцениваются в 0,03, 0,1, 0,5 и 2 Дж-К^-моль соответственно.
Термодинамические функции ZrO(r) ранее вычислялись в предыдущем издании настоящего
справочника [337] и таблицах JANAF [2834], а также работах Брюэра и Розенблатта [1666]
(Ф°(Г) для Т < 3000 К), Шнейдера [4197а] E000-10 000 К), Жирницкого [4840] B000-10000 К)
и Аккермана и Рауха [1229] A600—2800 К). В [337] за основное состояние ZrO было принято
состояние а3Д, соответствующие расхождения с данными табл. 946 составляют 9 Дж-К-моль
в значении Ф°B98,15 К) и уменьшаются до 1,5 Дж-К~1-моль~1 при 6000 К. В таблицах JANAF
приняты неверные значения энергии ряда состояний (а3Д, А1 А и др.), соответствующие
расхождения в значениях Ф°(Т), S°(T) и CAT) проходят через максимум в области 1000—2000 К и
достигают 8—12 Дж-К~1-моль~1. В расчете Жирницкого [4840] был учтен ряд ненаблюдавшихся
состояний и его результаты согласуются с данными табл. 946 в пределах 1—5 Дж-К~1-моль~1.
Расхождения данных [1666, 4197а] и табл. 946 существенно больше (до 12—15 Дж-К~1-моль)
из-за того, что в первой работе не был учтен ряд состояний, а во второй — основному и нижним
возбужденным состояниям были приписаны завышенные статистические веса.
Таблица 37.5. Результаты определений D0(ZrO) (в кДж-моль)
Авторы
Метод
Do(ZrO)
II закон III закон
Чапка и др., 1957 [1853]
Наката и др., 1981 [3630]
Щукарев, Семенов, 1965 [1182]
Аккерман, Раух, 1974 [1230]
Масс-спектрометрический, 1/2Zr02(K)+1/2Zr(K)=
=ZrO(r), 2124—2322 К, 4 точки
То же, ZrO2(r)+Zr(r)=2ZrO(r), 2408 К, 1 точка
То же, ZrO2(K)=ZrO(r)+O(r), 2605 и 2625 К,
2 точки
То же, ZrO2(r)=ZrO(r)+O(r), 2605 и 2625 К,
2 точки
То же, ZrO2(K)=ZrO(r)+O(r), 2740—2840 К,
данные представлены в виде уравнения
То же, ZrO2(r)+Zr(r)=2ZrO(r), 2743 К, 1 точка
То же, ZrO(r)+Y(r)=Zr(r)+YO(r), 2230—2570 К,
данные представлены в виде уравнения
То же, ThO(r)+Zr(r)=ZrO(r)+Th(r), 2000—
2500 К, данные представлены в виде уравнения
Мурад, Хилденбранд, 1975 [3591] То же, V2Zr02(K)+1/2Zr(K)=Zr0(r), 1790-
1995 К, 17 точек
То же, ZrO2(r)+Zr(r)=2ZrO(r), 2319—2410 К,
6 точек
Тоже, BO(r)+Zr(r)=B(r)+ZrO(r), 2158-2430 К,
15 точек
То же, ThO(r)+Zr(r)=ZrO(r)+Th(r), 2288 К,
1 точка
Семенов и др., 1977 [4900] Тоже, ZrO2(K)=ZrQ(r)+O(r), 2703—2853 К,
данные приведены в виде уравнения
Горохов и др., 1979 [310] То же, YO(r)+Zr(r)=Y(r)+ZrO(r), 2410—3042 К,
50 точек
То же, 2380—2963 К, 81 точка
525
765 +25
—
—
793
772
759
762+15
736+100
757+50
—
—
763+40
767+20
747 +15
790+16
798+20
753+15
735 ±20
770+13
763+20
760+10
759+10
746+15
757+20
749 ±15
771+12
769 + 12
121
ZrO+(r) Глава 37
Константа равновесия реакции ZrO(r)=Zr(r)+O(r) вычислена с использованием принятой
в справочнике энергии диссоциации
Дг#°@) = D0(ZrO) = 760 ± ЮкДж-- моль «63 500 + 800 см.
Результаты определений энергии диссоциации ZrO представлены в табл. 37.5. Приведенные
в таблице погрешности учитывают воспроизводимость измерений, неточность термодинамических
функций, термохимических величин и сечений ионизации, использованных в расчетах. За
исключением работы [3630] результаты измерений согласуются в пределах их погрешностей. Принятое
значение является средневзвешенным из величин, вычисленных по данным работ [310, 1182,
1230, 3591].
Принятому значению энергии диссоциации соответствует
^H°(ZiO, г, 0) = 84,783 ± И кДж • моль.
ZrO+(r). Термодинамические свойства газообразного ионизированного оксида циркония
ъ стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 947.
Молекулярные постоянные ZrO+, принятые в справочнике на основании анализа результатов
'исследований спектров и приближенных оценок, приведены в табл. 37.3. В спектре ZrO+
наблюдалась и проанализирована только одна система полос 2П—22 + [1373, 3825]. Квантовомеханиче-
ские расчеты этой молекулы не проводились, поэтому симметрия ее основного состояния и
систематика возбужденных состояний неизвестны. В спектрах изоэлектронных молекул YO, YF+
[4260] и ZrN [468, 1424], а также в спектрах молекул ScO, TiN [2102, 2128], LaO и LaF+ [4260]
наблюдались переходы, связанные с состояниями 22 *(<*„,), 2ДC(Я_1, а)' 2ЩппР) и 2-^ +(°пР)-
Приведенные в табл. 37.3 постоянные ZrO+ основаны на результатах исследований спектров этих молекул
и приближенных оценках, выполненных так же, как для TiO+. Оценка для иона Zr+ разности
энергий атомных орбиталей . . .Ър—. . .5s, . . .5s—. . Ad, . . .Ър—. . Ad' из центров тяжестей
термов, принадлежащих конфигурациям . . Ad25s, . . Ad25p, . . Adbs% и . . Ad5s5p, приводит
к заключению, что основным состоянием ZrO+, по-видимому, должно быть состояние 2Д(. . . §4^)»
а наблюдавшийся электронный переход 2П -> 22 не аналогичен переходу аП(л6 )—22(a6s) в
молекуле YO. В рамках использованной схемы этот переход должен быть интерпретирован как переход
р
Энергия состояния В2Щкы) принята равной разности энергий состояний 3П, 1П конфигурации
ansn(n-D d и 3Д> 1д конфигурации °„§(я_1} d в молекулах TiO и ZrO, а разность энергий состояний
Е22,(а5р) и ?>alT(ir5^) принята такой же, как у молекул TiN и ZrN. Погрешности оцененных энергий
возбуждения могут достигать 25%.
Значения постоянных ше и Ве в состоянии Х2Д, приведенные в табл. 37.3, оценены так же, как
для TiO+ на основании постоянных молекулы ZrO в состоянии а3Д и отношения значений этих
постоянных в аналогичных состояниях у молекул YF, LaF и ионов YF+, LaF+. Погрешности в
оцененных значениях могут достигать 50 и 0,03 см соответственно. Постоянная спин-орбитальной
связи ZrO+ в состоянии Х2к принята такой же, как в состоянии а3 Д молекулы ZrO, ее погрешность
оценивается в 50 см. Постоянные в состояниях D2H и С22 + приняты по работе Балфура и Линд-
грена [1373], которые нашли их в результате анализа вращательной структуры полосы 0—0
системы D2H—С22. Существенное отличие этих постоянных от значений, найденных ранее в работе
[3825], обусловлено тем, что авторы [1373] смогли уточнить анализ.
Термодинамические функции ZrO+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом шести возбужденных электронных состояний (подсостояние Х2Дз,2 рассматривалось как
возбужденное состояние с Те=2А) и в предположении, что Q^l SB=0,5ptQ^}mBV. Статистическая
сумма по колебательно-вращательным уровням энергии подсостояния Х2А^/2 и ее производные
находились по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным
уровням и интегрированием по вращательным уровням с помощью уравнений типа A. 82). В
расчете учитывались все уровни энергии с значениями / ^ 7шах „, последние определялись из
условий A. 81). Колебательно-вращательные уровни состояния Х2А.у3 вычислялись по уравнениям
A. 65), A. 34) и A. 64а); коэффициенты в этих уравнениях, тождественные соответствующим
постоянным из табл. 37.3, приведены в табл. 37.4 вместе со значениями vmSi% и/1т; значение Yos по-
122
Цирконий и его соединения ZrO2(K, ж)
лучено из условия A. 59). Вырождение уровней состояния -X2Ai/z, а также вырождение и мульти-
плетность уровней возбужденных состояний учитывались статистическими весами.
Погрешности рассчитанных термодинамических функций ZrO+(r) при низких температурах
обусловлены отсутствием экспериментальных данных о величине вращательной постоянной и
постоянной спин-орбитальной связи в состоянии Х2Д, они составляют до 2 Дж-К~1-моль~1 в
значении Ф°B98,15 К) и до 0,03 кДж-моль в значении #°B98,15 К) — #°@). Выше 2000 К
основные погрешности обусловлены отсутствием экспериментальных данных о систематике
электронных состояний ZrO+. Погрешности в значениях Ф°(Г) при 3000, 6000 и 10 000 К могут достигать 3,
5 и 8 Дж-К-моль~1.
Таблица термодинамических функций ZrO+(r) публикуется впервые.
Константа равновесия реакции ZrO+(r)+e(r)=Zr(r)-|-O(r) вычислена с использованием
значения Дг#°@) = 130,0+ 32 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(ZrO)=630 + 30 кДж-молъ.
Принятая величина основана на измерениях потенциала появления иона ZrO+ в масс-спектраль-
ных исследованиях Рауха и Аккермана [3943] F00±10 кДж-моль), Мурада и Хилденбранда
[3591] E80±20 кДж-моль) и Горохова и др. [310] E90 + 20 кДж-моль). В работах [3943]
я [310] для величины /0(Zr) по потенциалу появления иона Zr+ были получены значения,
которые почти на 40 кДж -моль меньше рассчитанного по спектральным данным (см. с. 118). В связи
ю этим принятое в справочнике значение /0(ZrO) и его погрешность увеличены по сравнению с
данными [310, 3591, 3943].
Принятой величине соответствуют
A^°(ZrO+, г, 0) = 714,783 + 32 нДж-моль,
D0(ZrO+) = 786 + 34 кДж ¦ моль ж 65 700 + 3000 см.
2гО2(к, ж). Термодинамические свойства кристаллического и жидкого диоксида циркония
в стандартном состоянии при температурах 100—6000 К приведены в табл. 948.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 37.1. В справочнике за стандартное состояние ZrO2(K) в интервале 0—1445 К принята
моноклинная модификация, в интервале 1445—2620 К тетрагональная модификация и в
интервале 2620—2983 К — кубическая модификация.
При Т<С2Ш,15 К теплоемкость ZrO2(K) измерял Келли [2958] E4—295 К) с
использованием образца чистоты ~99%; точность измерений оценена в [2958] в 0,3%. Экстраполяция
теплоемкости ниже 54 К, проведенная по уравнению C°p(T)=D{343AIT)+E(M6,2lT)+E(8№/T),
аппроксимирующему данные [2958] с погрешностью в 0,5%, привела к значению 6'°E4 К) =
=2,3 Дж-К~1-моль~1. Вычисленные по данным [2958] (с учетом поправок на содержание
основных примесей) значения S°B98,15 К) и Я°B98,15 К)—Я°@) (см. табл. 37.1) имеют погрешности
0,4 Дж-К~1-моль~1 и 0,04 кДж-моль соответственно.
В интервале 298,15—1445 К для теплоемкости Zi*O2(k) принято уравнение (см. табл. 37.1),
полученное совместной обработкой результатов измерений энтальпии ZrO2(K, монокл.),
проведенных Кофлином и Кингом [1944] C97-1433 К), Чеховским и др. [1123] A159-1350 К),
Егером и Веенстрой [2821] F73—1074 К), Артуром [1351] D68—1098 К) и Цагарейшвили и Гве-
лесиани [1095] C98—1372 К); наибольший вес придавался данным [1123, 1944]. Принятое
уравнение аппроксимирует данные [1123, 1944] в пределах 1% и данные [1095, 1351, 2821] — в
пределах 1,5—4%.
Температура полиморфного перехода моноклинной модификации ZrO2 в тетрагональную
принята равной 1445 + 10 К на основании результатов исследования, выполненного Мамптоном
я Ройем [3589] методами рентгенографии и ДТА на образце, содержавшем менее 3 -10~4 мольных %
НЮ2 г. Значения Тт, близкие к принятому, были получены в работах [508, 595, 1123, 2075, 2787,
3980, 4755]. Энтальпия перехода (8,4 + 1,1 кДж-моль) вычислена как разность энтальпий
тетрагональной и моноклинной модификаций ZrO2(K) при 1445 К. Энтальпия тетрагональной модифи-
Авторы [3589] показали, что в образцах ZrO2 с повышенным содержанием НГО2 температура полиморфного
превращения увеличивается, с чем согласуются значения этой величины, полученные в работах [141, 255, 989,
1002, 1095, 1435, 1944, 1974, 2230, 2420, 2455, 2655, 3327, 3599, 3693, 3782].
123
ZrO2(K, ж) Глава 37
нации ZrO2(K) описывалась уравнением #°(Г)—#°B98,15 К)=G8,1+0,2)Г—B0 625+330) ДжХ
Хмоль, основанным на калориметрических измерениях Чеховского и др. [508, 1123] A574—
2414 К). Соответствующее этому уравнению значение теплоемкости ZrO2(K, тетр.), приведенное
в табл. 37.1, использовано для расчета термодинамических функций ZrO2(K) в интервале 1445—
2620 К '. Энтальпия тетрагональной модификации ZrO2(K) измерялась также в работах [1944]
A481-1841 К), [3787] (см. [1234]), [1095] A504-1615 К), [481] A520-2410 К). Измерения
Пирса и Осмента [3787] согласно [1234] являются неверными. Результаты измерений [481, 1095,
1944] согласуются между собой, но систематически занижены по сравнению с полученными в
работе [1123] примерно на 3 кДж-моль. (
Температура превращения тетрагональной модификации ZrO2 в кубическую B620 + 20 К)
определена термографическим и рентгенографическим методами Коллонге и др. [1905] B623 К)
и рентгенографическим методом Уолтеном [4755] B619 К). Менее точные значения (от 2558 до
2650 К) были найдены в работах [141, 977, 1001, 3945, 4322, 4418, 4614]. Энтальпия
превращения тетрагональной модификации ZrO2 в кубическую A3 + 2 кДж-моль) получена Сухаревским
[1001] по уравнению для границы между тетрагональной и кубической фазами в системах
ZrO2—CaO, ZrO2—MgO, основанному на данных [1003, 2076]. Теплоемкость кубической
модификации ZrO2 оценена равной 80 + 2 Дж-К~1-моль~1 на основании сравнения теплоемкостей
моноклинной и тетрагональной модификаций ZrO2 при 1445 К и данных [141, 4086] для плотности и
параметров кристаллической решетки трех модификаций ZrO2. Принятое значение температуры
плавления ZrO2 B983 + 15 К) получено Аккерманом и др. [1217] и подтверждается результатами
определений на образцах ZrO2 чистотой не ниже 99,8% [977, 1905, 2278, 3200, 3667, 3668, 3670,
4098]. Результаты измерений в более ранних работах [673, 4198] менее точны, так как получены
для образцов, содержащих значительное количество примесей. Энтальпия плавления ZrO2
(90+5 кДж-моль) принята как среднее между значением 93 кДж-моль, вычисленным в
предположении, что &mS°(Zr02, K) = AmiS°(VO2, к)=31,3 Дж-К~1-моль~1 [1915] и значением
87 кДж-моль, вычисленным Келли [4990] по данным о плавлении двухкомпонентных систем
с участием ZrO2.
Теплоемкость ZrO2^K) A00 + 10 Дж-К~1-моль~1) оценена по эмпирическому соотношению,
принятому в справочнике (см. т. I, кн. 1, с. 79).
Погрешности приведенных в табл. 948 значений Ф°(Г) при 298,15, 3000 и 6000 К составляют
0,3, 3 и 15 Дж-К~1-моль".
Ранее термодинамические функции ZrO2(K, ж) вычислялись во втором издании настоящего
справочника [337], в справочниках JANAF [2834] и Шика [4181] (до 6000 К), а также в работе
[1234] (до 2800 К). В справочниках [337, 2834, 4181] не учитывалось существование кубической
модификации ZrO2(K) и предполагалось, что тетрагональная модификация стабильна до
температуры плавления. Расхождения данных [337] и табл. 948 составляют от 0,05 до 5,5 Дж-К-моль~1
в значениях Ф°(Т); соответствующие расхождения с данными [2834] достигают 8,5 Дж-К-моль
при 6000 К.
Принятая в справочнике энтальпия образования
A/9"°(Zr02, к, монокл., 298,15. К) = —1100,3 ±0,7 кДж • моль
является средневзвешенной из значений, вычисленных по согласующимся результатам измерений
энтальпии сгорания циркония в кислороде, выполненных Корниловым и др. [548] (—1100,31 +
±0,67 кДж-моль) и Хьюбёром и др. [2733] (—1100,14 + 2,1 кДж-моль). Приведенные в
работах [548, 2733] величины были пересчитаны с учетом поправок на неполноту сгорания и
примеси в исходных образцах по методике, изложенной в работе Корнилова [545]. В работах [2756,
3646, 4061, 4063, 4065, 4286, 4687] были получены менее точные значения, что объясняется
примесями в исходных образцах и неполнотой сгорания.
Энтальпия образования ZtO2x(k) в пределах области гомогенности исследовалась
методом ЭДС Васильевой и Грановской [181, 182, 270], а для ZrOj 9932 — калориметрическим методом
Корниловым и Ушаковой [547]. По этим измерениям было получено уравнение
,, к, монокл., 298,15 К) = —1100,83 + 369,95а;,
1 В работе [1123] образец ZrO2 длительное время выдерживался при Т > 1445 Kt что обеспечивало его полный
переход в тетрагональную модификацию.
124
Цирконий и его соединения
ZrOa(r)
которое в пределах 0^3^0,15 имеет погрешность +1,3 кДж-моль. Калориметрические
измерения парциальных энтальпий растворения кислорода в ZrO^ [1631] подтверждают это
уравнение.
Давление пара в реакции ZrO2(K, >K)=ZrO2(r) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДД°@) = ЬД°B1О.л, к, 0) = 780 ± 15 кДж • моль.
Результаты определений этой величины представлены в табл. 37.6. Приведенные в таблице
погрешности учитывают воспроизводимость измерений, неточности термодинамических функций,
Таблица 37.6. Результаты определений As-ff°(ZrO2 к, 0)
Авторы
Метод
As#°(ZrO2, к, 0)
II закон
III закон
Чапка и др., 1957 [1853] Масс-спектрометрический, ZrO2(K)=ZrO2(r), 2331— — 786+25
2480 К, 5 точек
То же, 2200—2504 К, 4 серии 764 —
Наката и др., 1961 [3630] Эффузионный, ZrO2(K)=ZrO2(r), 2410—2830 К, 727 749
10 точек, предполагая единственным продуктом
испарения ZrO2(r)
То же, 2605 и 2625 К, 2 точки, с учетом образо- — 788+20
вания ZrO
Щукарев, Семенов, 1965 [1182] Масс-спектрометрический, ZrO2(K)=ZrO2(r), 781 794+15
2740—2841 К,. данные представлены в виде
уравнения
Аккерман и др., 1975 [1234] Протока, в присутствии О2, ZrO2(K)=ZrO2(r), 736+18 778+10
1925—2820 К, 15 точек
Масс-спектрометрический, ZrO2(K)=ZrO2(r), 10 се- 745 —
рий измерений, Гср=2500 К
Семенов и др., 1977 [4900] Масс-спектрометрический, ZrO2(K)=ZrO2(r), 693+200 800+15
2703—2803 К, 4 точки
Горохов и др., 1979 [310] Масс-спектрометрический, 2YO(r)+Zr(r)=2Y(r)+ 777+70 770+20
+ZrO2(r), 2380—3042 К, 74 точки
термохимических величин и сечений ионизации, использованных в расчетах. За исключением
величины, вычисленной по данным работы [3630] без учета действительного состава пара,
остальные приведенные в таблице величины согласуются в пределах их погрешностей. При
вычислении средневзвешенного больший вес придавался наиболее надежным измерениям,
выполненным в работах [310, 1234]. При оценке погрешности принятой величины учитывался разброс
результатов измерений в разных работах.
ZrO2(r). Термодинамические свойства газообразного диоксида циркония в стандартном
состоянии при температурах 100—6000 К приведены в табл. 949.
Молекулярные постоянные, принятые для расчета термодинамических функций, приведены
в табл. 37.7. На основании результатов изучения отклонения молекулярного пучка ZrO2 в не-
Таблица 37.7. Значения молекулярных постоянных, а также в и рх, принятые для расчета
термодинамических функций ZrO2
Состояние
1 Те(А3Вг) = 15 000
re(E3i?!)=22 000
см",
СМ,
1
900
3;
см
530
(Ё*В2) =
T.(C1Bi) =
25 000 см-1,
884
18 000
г3
6
см, р =
С
¦ смв
¦ 102
= 4; Т^П1
а
2
В2)=20000
см,
Рх
1
1;
125
ZrO3(r) Глава 37
однородном электрическом поле [2935] в справочнике принимается, что в основном состоянии Х1А1
молекула ZrO2 имеет угловую структуру (симметрия С2с). Приведенное в таблице значение IaIbIc
рассчитано по структурным параметрам r(Zr—О)=1,73+0,05 А (оценка по величинам re(Zr0)r
r,(NbO) и r(Nb—О) в NbO2, см. выше) и -^ О—Zr—О=109±15° (оценено на основании величины
Jj О—М—О в молекулах МО2, где M=Sc, Ti, V, Nb, Сг, Mo, W, . . .). Погрешность принятого
значения IaIbIc составляет 2-10~115 г3-см6. В ИК-спектре молекул ZrO2, изолированных в
матрице из Аг, Линевский [3268] наблюдал две частоты (884 и 818 см), интерпретированные им как
частоты Vi и v3 соответственно. Сопоставление значений этих частот с известными частотами
молекул ТЮ2, VO2, NbO2, CrO2 и МоО2, а также значений силовых постоянных в этих молекулах и
соответствующих двухатомных окислах позволяет предполагать, что частота 818 см,
наблюдавшаяся Линевским, не может рассматриваться как основная частота ZrO2. Приведенные
в табл. 37.7 значения основных частот основаны на данных для диоксидов других переходных
металлов и данных [3268]; погрешности принятых значений оцениваются в 10% от их величины.
Приведенные в табл. 37.7 энергии возбужденных электронных состояний ZrO2 получены Хай-
том и Хачкурузовым [1065] на основании полуэмпирической оценки для модели кристаллического
поля. В расчете учитывались результаты аналогичных оценок электронных состояний TiO2 и
ТаО2, для которых известны отрывочные экспериментальные данные, и электронных состояний
молекулы ZrO. Погрешности принятых энергий возбужденных электронных состояний ZrO2
оцениваются в 5000 см.
Термодинамические функции ZrO2(r) вычислены до уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом по соотношениям A. 168)—A. 170) шести возбужденных электронных состояний.
Погрешности вычисленных значений термодинамических функций обусловлены приближенным
характером расчета и неточностью принятых молекулярных постоянных. Погрешности
значений Ф°(Т) при 298,15, 3000 и 6000 К оцениваются в 2, 4 и 6 Дж-К-^моль.
Ранее термодинамические функции ZrO2(r) вычислялись в предыдущем издании
справочника [337], в справочниках JANAF [2834] и Шика [4181], а также в работах 11665, 3619] (до 3000
и до 5000 К). Расчеты [337, 1665, 4181] выполнены для линейной модели этой молекулы,
соответствующие расхождения с данными табл. 949 составляют 23—15 Дж -К -моль в значениях Ф°(Т).
Расхождения с данными [2834] E—10 Дж-К~1-моль~1 в значениях Ф°(Г)) обусловлены тем, что
авторы таблиц JANAF приняли заниженное значение v2 A37 см) и не учитывали возбужденные
электронные состояния ZrO2. |--,
Константа равновесия реакции ZrO2(r) = Zr(r)+2O(r) вычислена с использованием
значения АгН°@) = 1406,412 + 16 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации &О2(к). Этим величинам также соответствует
&fH°(ZrO2, г, 0) = —314,846 + 15 кДж • моль
-1
Глава 38
ГАФНИЙ И ЕГО СОЕДИНЕНИЯ
Hf(K, ж), Hf, Hf+, НЮ, НЮ+, НЮ2(к, ж), НЮ2
В настоящей главе рассматриваются гафний и его кислородные соединения — всего 5 веществ.
Для двух веществ приведены сведения об их термодинамических свойствах в конденсированном
состоянии и для пяти — в газообразном состоянии. Термодинамические свойства четырех газов
(Hf, Hf+, НЮ, НЮ+) рассчитаны до 10 000 К, одного (НЮЯ) -до 6000 К.
Для элементарного гафния в Приложении ЗЛО приведены сведения о критических постоянных.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов гафния
(атомный вес 178,49) и кислорода, однако различие молекулярных постоянных разных изотопо-
меров учитывалось только для HfO(r).
В конденсированном состоянии основным оксидом гафния является НЮ2, который имеет очень
узкую область гомогенности. Известны также три кристаллических субоксида гафния: Hf6O,
Hf4O, Hf3O — вещества малоустойчивые и недостаточно изученные.
Ш(к, ж). Термодинамические свойства кристаллического и жидкого гафния в стандартном
состоянии при температурах 100—6000 К приведены в табл. 950.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 38.1. За стандартное состояние Ш(к) в интервале 0—2016 К в справочнике принята
гексагональная (а) модификация (структурный тип Mg) и в интервале 2016—2506 К — кубическая (р)
Таблица 38.1. Принятые значения термодинамических величин для гафния и его соединений
в кристаллическом и жидком состояниях
о
Щ
н
о
f
CD
н
Hf
HfO2
Состояние
кП, гекс. (а)
к1, куб. (р)
ж
кШ, монокл.
кП, тетр.
к1, куб.
ж
1
ю .—.
-ГЧ О
оо ' '
0, 1
кДж-моль
5,845
—
9,81
—
—
—
ю
оо"
оэ
о
со
ю
со"
СП
&¦
Дж • К • моль
Коэффициенты з
нении для Сс
а
6-Ю3
43,56 25,69 22,701 8,301
- — 91,061 -58,936
_ _ 44 —
59,4 60,255 69,271 11,565
_ _ 98,4 —
_ _ ЮО —
_ _ Ю5 —
Примечание. C°p(T)=a+b -T—c -T^+d -Г2 (в Дж-К-моль)
Hf: a
d=15,011 ЛОГ»
з урав-
с ¦ 10~5
—0,457
0а
—
11,080
,—
—
—
Интервал
температур
ИЛИ
т
1 m
К
298,15—2016
2016—2506
2506—6000
298,15-2100
2100-2793
2793—3073
3073—6000
2016
2506
—
2100
2793
3073
—
ИЛИ
кДжХ
Хмоль х
5,4
26
—
12
18
96
—
модификация (структурный тип a—FeI. При 0,374 К [4329] Hf переходит в сверхпроводящее
состояние.
При Т ^298,15 К термодинамические функции Ш(к) вычислены на основании измерений
теплоемкости, выполненных Веструмом [5070в] E—350 К) на образце гафния чистотой 99,95%
1 При давлениях, превосходящих 180 кбар, стабильна гексагональная модификация Hf высокой плотности (см.
[1021]).
127
Ш(к, ж) Глава 38
с погрешностью 0,1% при Г>25 К и ~4% при Т^ЮК (в предварительном сообщении Оллеса
и Веструма [1296] для величины >SOB98,15 К) приведено отличающееся значение —42,72 ДжХ
хК-моль). Погрешности значений 5°B98,15 К) и #°B98,15 К)—Н°@), рекомендованных
Веструмом (см. табл. 38.1), оцениваются в 0,2 Дж-К~1-моль~1 и 0,03 кДж-моль соответственно.
Данные по теплоемкости гафния, полученные в работах [3049] A,2—4,5 К), [1903] A,5—6 К)
[4750, 4751] A,5—20 К) и [1727] A0—200 К), менее точны и не учитывались (в пределах их
погрешности они не противоречат данным [5070в]). Измерения [1965] A3—210 К) ошибочны (резко
завышены).
В интервале 298,15—2016К функции вычислены с использованием уравнения для С°(Т)
(табл. 38.1), полученного совместной обработкой измерений энтальпии в работах Аденштедта [1255]
C73 К), Хокинса и др. [2592] C89—1346 К) и Голутвина и Масленниковой [302] B98—1400 К,
исключая выпадающие данные при 893 и 957 К), а также измерений теплоемкости в работе Це-
зерляна и Мак-Клара [1788] A500—1800 К). При обработке были введены поправки на содержание
основных примесей в образцах гафния. В расчете с наибольшим весом учитывались данные
[1255, 2592]1. Полученное уравнение находится в хорошем соответствии сданными [775] A400—
1900 К), а также с предварительными результатами измерений энтальпии гафния в работе
Чеховского и др. [1126] A230—2026 К). Данные для теплоемкости Hf, полученные в работах [47,
49, 889, 1055], систематически завышены на ~2 Дж-К^-моль"-1, а данные [2677] A850—2000 К)
сильно занижены.
Температура полиморфного превращения B016 + 20 К) принята по работе Крикориана и
Уоллеса [3108], которая основывается на данных [2012, 3108], полученных методом термического
анализа. Работа [3108] выделяется среди многих других определений Ttr(ili, к) наиболее точным
учетом влияния примесей и наиболее точными температурными измерениями. Принятое
значение подтверждается измерениями [1398» 1423, 1451, 1753, 1789, 1790, 2012, 2013, 2396, 2399,
3804, 4283] A997—2033 К). К несколько более высоким значениям B040—2053 К) приводят
данные [891, 2092, 2677, 4043]. В ранних работах [2132, 4830] были получены более низкие значения
A600—1800 К), а в ряде работ [2221, 2398, 4051, 4481, 4484, 4485] более высокие значения
B150—2270 К) вследствие диффузии примесей в образец в течение эксперимента [4043, 4283].
Принятая в справочнике энтальпия полиморфного превращения E,4 + 0,5 кДж-моль) является
средней из найденных методом термического анализа Пелецким и Дружининым [775] E,176 кДжХ
Хмоль) и методом импульсного нагрева Мартынюком и др. [666, 667] E,2 кДж-моль) и Це-
зерляном и Мак-Кларом [1789, 1790] E,9+0,6 кДж-моль). В работе [2677] методом
термического анализа получено значение 2,22 кДж -моль.
Уравнение для С°(Т) в интервале 2016—2506 К основано на измерениях Цезерляна и Мак-
Клара [1788] B150—2400 К) теплоемкости [З-модификации Hf, скорректированных на содержание
основных примесей в исследовавшемся образце. Надежность этих данных подтверждается
согласием измерений теплоемкости а-модификации Hf в работе [1788] и измерений энтальпии в работах
[302, 1255, 2592]. Результаты менее точных определений теплоемкости |3-модификации авторами
[775, 2677] также согласуются с данными [1788]. В работе [47] для C^(Hf, к, C) были получены
завышенные величины.
к Температура плавления B506 + 10 К) принята как среднее из значений, полученных для
наиболее чистых образцов (98,9—99,8%) Дирдорфом и Хайесом [2011] B499+30 К), Карлсоном и
др. [1754] B512 К) и Крикорианом и Уоллесом [3108] B508 + 20 К). При оценке погрешности
учтены измерения Спеддинга [4352] B508 К, см. [2752]), Скиннера и др. [4309] B502 К,
см. [2752]), авторов [3905] B507 К, см. [3108]), Чарлесворта [1814] B500 К, см. [1224]) иАккер-
мана и Рауха [1224] B494 + 5 К). Существенно менее точные значения были получены в работах
[317, 1255, 1650, 2018, 2092, 2398, 3282, 3804, 4085, 4141, 4283, 4481, 4484, 4485, 4504] B250-
2563 К), в которых исследовались образцы, содержащие значительное количество Zr и других
примесей. Многие из этих измерений были несовершенны в методическом отношении. Энтальпия
плавления Hf B6+5 кДж-моль) принята по работе Аккермана и Рауха [1226] и основана на
измерениях давления пара над жидким и кристаллическим гафнием. Близкая величина была
оценена Кацем и Чеховским [494, 2931]. Теплоемкость Ш(ж) экспериментально не определялась.
Принятое в справочнике значение D4+4 Дж-К-моль~1) оценено на основании предположения,
1 Данные по энтальпии Hf, полученные Фипдхаузом и Лонгом [2249] E34—1884 К) и использованные в [2752],
остались недоступными авторам справочника.
128
Гафний и его соединения
Hf(r)
что АтСар(Ш, K)^AmC°(Zr, к); оно подтверждается оценкой по эмпирическому уравнению Брайса
[1668J. Погрешности приведенных в табл. 950 значений Ф°(Г) при 298,15, 2000, 4000 и 6000 К
оцениваются в 0,2, 1, 4 и 7 Дж-К~1-моль.
Ранее таблицы термодинамических функций Ш(к, ж) вычислялись в справочниках Сталла и
Зинке [4421] (до 3000 К), Шика [4181] и Халтгрена и-др. [2752] (до 5000 К). В справочниках
[4181, 4421] использованы неточные данные и грубые оценки. Расчет [2752] основан на данных,
близких к принятым в настоящем справочнике, и расхождения в значениях Ф°(Т) составляют
0,01-1,7 Дж-К^-моль-1.
В настоящем справочнике в качестве стандартного состояния гафния принято
кристаллическое состояние и в соответствии с этим
AfH°(Ef, к) = 0.
Давление пара гафния в реакции Ш(к, ж)=Ыг(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = As#°(Hf, к, 0) = 622 + 5 кДж • моль.
В табл. 38.2 приведены результаты вычислений энтальпии сублимации гафния, основанные
на измерениях давления его насыщенного пара. Согласно масс-спектральным данным [1226,
3750] пары гафния являются одноатомными. Приведенные в таблице погрешности отражают только
воспроизводимость измерений. Принятая величина является средневзвешенной из значений,
Таблица 38.2. Результаты определений AsBT°(Hf, к, 0) (в кДж-моль)
Авторы
Цвиккер, 1928 [4833] (см. [2018])
Крупка, 1962 [3121]
Лайон, 1962 [3329]
Пэниш, Рейф, 1963 [3750]
Киблер и др., 1964 [2984] (см. [1226])
Блекберн, 1964 [1572] (см. [1226])
Мак Клейн, 1964 [3443] (см. [2834])
Коч и др., 1968 [3055]
Микульская, 1972 [296, 678]
Аккерман, Раух, 1972 [1226]
а В работе не приведены измеренные
представлены в виде уравнения.
Метод
Испарение с открытой поверхности
Испарение с открытой поверхности и эффузион-
ный, HfB2(K)=Hf(r)+2B(r), 2175—2500 К а
Испарение с открытой поверхности а
То же, 2069—2277 К, 6 точек
То же, 2035—2325 К а
То же, 2200—2360 К а
То же, 2200—2363 К а
То же, 2505—2815 К, 17 точек
То же, с применением радиоактивных, изотопов,
1473—2213 К б
Масс-спектрометрический, 1950—2200 К б
То же, 2510—2830 К, учтена растворимость W
в 1К(ж) [1224] б
Д,Я (Hf
II закон
594
—
616
666 ±95
610 ±10
537 ±24
—
595±30
588
624
628
к, 0)
III закон
601
592 ±30
612,7
608 ±4
620,8±1,7
624±15
624 ±3
622,1 ±1,3
606
621,2+4,0
620,5 ±6,4
давления, даны только вычисленные авторами значения ^tH°(T); *> данные
основанных на измерениях Аккермана и Рауха [1226] и Коча и др. [3055]. При оценке ее
погрешности учтена неточность термодинамических функций. В пределах погрешностей принятая
величина согласуется со значениями, основанными на данных [1572, 2984, 3443]. Более низкие
значения, полученные по данным [296, 678, 3121, 3329, 3750, 4833], [обусловлены недостаточной
чистотой образцов и неточностью измерений температуры.
Hf(r). Термодинамические свойства газообразного гафния в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 951.
Для расчета термодинамических функций были использованы уровни энергии 1 атома Hf
в интервале 0—54 000 см, соответствующие валентным электронным конфигурациям . . .5da6s2
(к ней относится основной уровень 3F2), . . .5d26p2, . . .5d6s6p2, . . .5d4, . . .5d36l A=0-^-5),
. . .5d26s6Z (l—i—5), . . .5d6s26Z (Z=0-^5) и имеющие суммарный статистический вес 2981. При-
1 См. примеч. 1 к тексту по Сг(г).
129
Hf+(r) Глава 38
нятые величины основаны на рекомендациях Меггерса и Мур [3472] и на оценках для ненаблю-
давшихся уровней этих конфигураций по методике, изложенной в Приложении 6. Оцененные
уровни имеют энергию от 27 000 см и выше с суммарным статистическим весом 1386.
Погрешность оценки энергии лежит в пределах 500—4000 см. Сделанные в [4788] уточнения данных
об уровнях энергии несущественны для расчета термодинамических функций Hf(r).
Термодинамические функции Hf(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин при 7^3000 К обусловлены неточностью принятых значений фундаментальных
постоянных, а при более высоких температурах — неточностью оценки ненаблюдавшихся уровней
энергии и пренебрежением ридберговскими состояниями. Погрешности значений Ф°(Т) при 3000,
6000 и 10 000 К составляют 0,03, 0,1 и 1 Дж-К-^моль.
Ранее термодинамические функции Hf(r) вычислялись в справочниках Сталла и Зинке [4421]
до 3000 К и Шика [4181] до 6000 К, а также в работах Поланда и др. [3862] до 7000 К, Хилзен-
рата и др. [2652] до 10 000 К и Жиршщкого [4839] до 15 000 К. Данные [2652, 3862, 4181, 4421]
и табл. 951 согласуются в значениях Ф°(Т) в пределах 0,04 Дж -К -моль при Г^ 3000 К ив
пределах 0,15 и 0,6 Дж -К-моль при 6000 и 10 000 К (имеющиеся расхождения обусловлены тем,
что в справочнике использованы более полные спектральные данные). В расчете [4839] учтены
ридберговские уровни с «г^12 и расхождения в значениях Ф°(Т) с табл. 951 достигают от 0.1
до 1,8 Дж-К^-моль в области 3000—10 000 К.
Энтальпия образования
г, 0) = 622 + 5 кДж • моль
соответствует принятому в справочнике значению энтальпии сублимации гафния.
Hf+(r). Термодинамические свойства газообразного положительного иона гафния в стандартном
состоянии при температурах 298,15—10 000 К приведены в табл. 952.
Для расчета термодинамических функций использованы уровни энергии 1 Hf+ в интервале
0—63 727,6 см, соответствующие валентным электронным конфигурациям . . .5d6s2 (к ней
относится основной уровень 2?>з/2), . . .5d3, . . .5d?Ql (Z=0,l) и . . .5d6s6p и имеющие суммарный
статистический вес 600. Принятые величины основаны на рекомендациях Мур [3550] и на оценке
энергии ненаблюдавшихся уровней . . .5d^ASNs 2S B2 000+4000 см) и . . .5cZ2A5Nj?
гР E0 000+4000 см), сделанной но методике, изложенной в Приложении 6.
Термодинамические функции Hf+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т7^ 4000 К обусловлены в основном неточностью принятых значений
фундаментальных постоянных и не превышают 0,02—0,03 Дж-К~1-моль. При более высоких температурах
становятся существенными неточность оценки уровня 2S (см. выше) и пренебрежение
ридберговскими состояниями; погрешность значения Ф°A0 000 К) может достигать 0,1 Дж-К-моль~1.
Ранее термодинамические функции Hf+(r) вычислялись Хилзенратом и др. [2652] до 10 000 К,
Жирницким [4839] до 15 000 К и Грином и др. [2473] до 50 000 К. Расчеты основаны на уровнях
энергии, приведенных в [3550], в том числе и не отнесенных к определенным электронным
состояниям. Расхождения в значениях Ф°(Т), S°(T) и С°р(Т), приведенных в табл. 952 и в работах [2473,
2652], не превышают 0,04, 0,01 и 0,01 Дж-К-моль соответственно при 10 000 К. В работе [4839]
были учтены оцененные ридберговские уровни энергии, соответствующие значениям п ^С 10;
расхождения [4839] и табл. 952 не превышают 0,3 Дж-К-моль в значениях Ф°(Т) и S°(T).
Константа равновесия реакции Hf+(r)+e(r)=Hf(r) вычислена с использованием значения
Дг/7°@) =—654+7 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Ш)=654 + 7 кДж-моль-1«54 700 + 600 см.
Эта величина рекомендована Меггере и Мур [3472] на основании расчета Шугара по методике,
описанной в работе [4436], и по данным авторов [3472] для уровней энергии состояний 3F атома Hf,
относящихся к электронным конфигурациям . . .5d26s2, . . .5d26sDi^Os и . . .5d?6sBFOs. Аккер-
ман и Раух [3944] на основании измерений методом электронного удара [3943] с учетом поправок
* См. примеч. 1 к тексту по Сг(г).
130
Гафний и его соединения
НГО(г)
на термическое возбуждение атомов Hf [1237 ] и образование ионов Hf+ в возбужденных состояниях
получили более низкое значение (—629 кДж-моль).
Принятому значению потенциала ионизации соответствует
AfH°(m+, г, 0) = 1276,000 + 8,6 кДж • моль.
НЮ(г). Термодинамические свойства газообразного оксида гафния в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 953.
Молекулярные постоянные 180Hf16O, принятые в справочнике на основании анализа
результатов исследований электронного спектра этой молекулы, ИК-спектров молекул, изолированных
в матрицах, и приближенных оценок, приведены в табл. 38.3. В электронном спектре НЮ изученцг
Таблица 38.3. Молекулярные постоянные 180Ш16О и НЮ+
Молекула
Состояние
Те
шщ1бо а х12+ б 0
а3А1 5 000
а3Д2 6 000
а3Д3 7 000
А1 А 9 000
Д!2+ 16 616,
СШ 17 562
Dm 23 554
Gin 25 230
к3ф 27 240
Я12+ 27 413
/!2+ 30 032
НГО+ а X2 2+ 6 0
ш
шехе
974,09 3,228
г 910 Д —
г 910 Д —
г 910 Д —
г
92 914,24 3,38
22 907,01 3,38
42 872,60 3,31
94 866,93 3,68
к . 820г —
849,4 3,67
7 е 852,3Л —
1025 4,4 в
Примечание. Ниже все постоянные в см.
НЮ: а принятые обозначения
б Состояние Ь3П
Те 13 000
электронных состояний
с3 2+ <13Ф е3П
14 000 18 000 20 000
В
см х
0,386
0,377
0,378
е
«1 • Ю3
537 1,724
6 е —
1 е
0,378 8 е —
0,377
0,377
0,368
0,368
0,369
0,365
0,370
0,41
—
985 1,827
799 3 1,850 3
75 3 1,810
98 3 1,975 3
9 е —
3 1,88
106 2,071
2Г
De ¦ 10'
2,438 в
2,8 е
2,6 е
2,6 е
—
2,587 ж
2,624 3
2,628 3
2,68 3
3 е
2,70 й
2,764 е
зд
отличаются от использовавшихся в
f A
22 000
в р,=—6 -Ю0; г оценка; Д вычислено по соотношению A
v=0\ !K Sx=—1,6 -10~9;
=—2,3 -1(ГЭ; к оценено
ЕЩ, Р-А, g3n
24 000
68); е приведено
г3п, лф
35 000
А
1,723 07
1,741 е
1,742 е
1,743 е
1,742 4
1,742 9
1,764 2
1,763 6
1,761 е
1,771 6
1,760 9
1,67 е
литературе; |
/Зп, хт
38 000 ;
значение постоянной при
3 приведено среднее значение постоянной для компонент Х-удвоения; и fjx= i
на основании принятой энергии состояния а3 Д2 и частоты средней подполосы три- '
плетного перехода; л приведено значение AGi/,.
HfO+: a все постоянные оценены
(см. текст); б Состояние
Те
А2 А
12 000
в вычислено по соотношению A. 67) п значению D0=60 000;
числено по соотношению
в2и с2п
23 000 36 000
?>22+
43 000
г вычислено по соотношению
A. 68); е вычислено по соотношению A. 38).
A.69); Д вы-
только 7 систем полос, связанных с переходами между семью синглетными и двумя триплетнымк
состояниями ь 512+^Х1Е+, С1П^Х^+ [2147, 2374, 4698], ?>1П**Х1Е+, G1n<*Z1S+, H1E+^*X1L*
[467, 2147, 2374, 4698], РЪ+^Х1!!* [2147, 2374, 4698] и /г3Ф-*а3Д [2374, 4698, 4704]. Анализ
этих систем полос не может рассматриваться как окончательный; в молекуле НГО характер связи
должен приближаться к случаю Гунда с и поэтому переходы, имеющие вид переходов между
синглетными состояниями, могут быть связаны с компонентами 0+ и 1 триплетных состояний. Так,
Эдвинссон [2145] высказал предположение, что состояния, интерпретированные как В1!,* и С1!!,,
являются компонентами 0+ и 1 триплетного состояния 3П. Квантовомеханические расчеты НЮ
отсутствуют, что также затрудняет определение схемы электронных термов этой молекулы. Однак©
сравнение с молекулами TiO, ZrO и ThO, а также анализ возможных конфигураций электронной
оболочки молекулы HfO позволяют предполагать существование у этой молекулы большого числ*
электронных состояний с низкими энергиями возбуждения. Поскольку в спектре поглощения HfO
1 В справочнике принята символика состояний НГО, отличающаяся от использованной в оригинальных работах,
131
НГО(г)
Глава 38
(см. [467]) наблюдались переходы только из состояния 1S+, в справочнике принимается, что
состояние 1S+ является основным (конфигурация . . .а;I, а состояния а3Д и А1А(. . .°sod) имеют
энергии выше 3000—4000 см. Принятые энергии этих состояний оценены на основании данных
для молекул ZrO и ТЮ, их погрешности оцениваются в 2000 см. Если предположение Эдвинс-
сона справедливо, молекула HfO не должна иметь электронных состояний в области от 9000 до
16 600 см, однако такой случай представляется маловероятным, и в справочнике для НЮ
принята схема термов аналогичная реализующейся у молекулы ZrO (а также у ThO, за исключением
состояний Ь3Ф, ВгФ, Й3Д, С1 А, которые, по-видимому, принадлежат конфигурациям . . .osof
и . . .os§f). Энергии соответствующих ненаблюдавшихся электронных состояний Ь3П (. . .
3 3 ^ 3
+(. . .а,ар), «РФ, ЕЩ), е3Щ. . .яр8„), /3Д,
,apbd), ?3П(. . .о,Л(?) и других, приведенных
в табл. 38.3, были оценены на основании этого предположения. Погрешности оцененных величин
могут достигать 4000—5000 см.
Молекулярные постоянные 180HfieO в состоянии X1 S+ и шести возбужденных синглетных
электронных состояниях приняты по работе Эдвинссона и Пилена [2147]. Авторы [2147]
исследовали вращательную структуру 28 полос шести систем. Наиболее полные данные были получены
при анализе системы ДШ-^Р 2 + A1 полос с значениями!/, v" ^4) и СЧГ-^Х1 ? + E полос с v' ^2,
у"<^1). Значения молекулярных постоянных, приведенные в таблице, были рассчитаны по
методу Аслунда на основании волновых чисел 5083 линий.
В работе [2147] проанализированы также полосы 0—0 трех переходов, которые
предположительно были интерпретированы как три подсистемы перехода /г3Ф—>а3А. Найденные авторами
[2147] значения вращательных постоянных приведены в табл. 38.3. В работе отмечается, что
имеется возмущение уровней состояний /IП, СХП (взаимное), а также /12+ и /г3Ф3.
Термодинамические функции НЮ(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qss и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом 23 возбужденных электронных состояний (компоненты состояния а3Д учитывались как
QHl Qil В ?*
у у
отдельные состояния) и в предположении, что QHl_B9=Pi Q
iol. *P-
Величина
и ее
производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по значениям J с помощью уравнений типа A. 82). В расчете
учитывались все уровни энергии с значениями / ^ /шах „, где /шах „ определялись из условий
A. 81). Колебательно-вращательные уровни состояния Х1!,* вычислялись по уравнениям A. 65),
A. 64а). Значения коэффициентов в этих уравнениях, приведенные в табл. 38.4, соответствуют
природной смеси изотопов гафния и получены с помощью соотношений A. 66) по постоянным,
принятым в табл. 38.3 с учетом условий A. 50) и A. 59). В той же таблице приведены значения
' lim-
Таблица 38.4.
Коэффициенты в уравнениях:, описывающих уровни энергии (в см-1), и значения vB
и JUm, принятые для расчета термодинамических функций HfO(r) и ШО+(г)
Коэффициенты
HfO,
НЮ+,
Х22+а
Т, 0 0
Ую-10 0,974 332 1,0250
У20 —3,252180 -4,4
Узо -Ю3 3,478 907 —
Коэффициенты
HfO,
у4о -105 —6,202 859
Уо1 -10 3,866 950
Уп -103 —1,725 057
У02 -107 -2,439 993
HfO: a Энергии возбужденных состояний см. в табл. 38.3.
НЮ+: а Энергии возбужденных состояний см. в табл. 38.3.
ШО+,
Х2И+ »
_
4,10
-2,0
—3,0
Коэффициенты
Ух, -Ю10
У03 -1013
^тах
HfO,
-6,006 134
—1,060 530
ИЗ
549
ШО+,
X2S+ а
—3,234
124
523
Колебательно-вращательные уровни состояния ХХЕ+ с энергиями до 4000—5000 см хорошо
описываются уравнениями с постоянными, приведенными в табл. 38.4. Поэтому до 1000 К
погрешности рассчитанных функций в основном обусловлены неточностью фундаментальных
постоянных. С ростом температуры становятся существенными погрешности из-за отсутствия экспери-
1 Это подтверждается исследованием спектра HfO в матрицах из Ne и Аг, выполненным Велтнером и Мак-Леодом
[4698].
132
Гафний и его соединения
НЮ+(г)
ментальных данных о возбужденных электронных состояниях, прежде всего а3Д и Л1 Д.
Погрешности в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,03, 0,5, 3 и
б Дж-К^-моль.
Термодинамические функции HfO(r) ранее вычислялись в работах Жирницкого [4840] (от 2000
до 10 000 К), Брюэра и Розенблатта [1666] (Ф°(Г) для Т <3000 К), Шнейдера [4197а] (Г < 8000 К)
и Аккермана и Рауха [1230] (Т=2200+-2800 К). В работе [1666] принята неверная систематика
электронных состояний НЮ, и рассчитанные значения Ф°(Г) на 11—21 Дж-К~1-моль~1
превышают данные табл. 953. Данные [4840] для Ф°(Г) и S°{T) согласуются с табл. 953до Г<3000 К.
При более высоких температурах расхождения быстро возрастают и достигают 3 и 7 Дж-К~хХ
Хмоль соответственно; в значениях С°р(Т) расхождения составляют 5—8 Дж-К~1-моль во всем
интервале температур, меняя знак в области 1700 К.
Константа равновесия реакции HfO(r)=Hf(r)-)-O(r) вычислена с использованием принятой
в справочнике энергии диссоциации
ДгЯ°@) = D0(HfO) = 795 + 15 кДж • моль" ш 66 500 + 1300 см.
Эта величина основана на представленных в табл. 38.5 данных. Приведенные в таблице
погрешности учитывают воспроизводимость измерений, неточности термодинамических функций,
Таблица 38.5. Результаты определений D0(HfO) (в кДж-моль)
Авторы
Метод
Do(HfO)
II закон III закон
Пэниш, Рейф, 1963 [3750]
Щукарев, Семенов, 1965 [1182]
Аккерман, Раух, 1974 [1230]
Семенов и др., 1977 [4900]
а Данные представлены в виде уравнения.
Эффузионный, с использованием радиоактивного
изотопа, НЮ2(к)=НЮ(г)+О(г), 2246—2536 К,
12 точек
Масс-спектрометрический, Hf О2(к)=Hf О (г)-(- О (г),
2600 К, 1 точка
То же, НЮ2(к)=НЮ(г)+0(г), 2703—2950 К а
То же, HfO(r)+Y(r)=Hf(r)+YO(r), 2180—
2590 К а
То же, HfO(r)+Zr(r)=ffi(r)+ZrO(r), 2140-
2610 К а
То же, ThO(r)+Hf(r)=Th(r)+HfO(r), 2200-
2550 К а
Эффузионный, HfO2(K)=HfO(r)+O(r), 2773—
2950 К, 9 точек
737+200 779+20
745
726
820
807
807
915+100
808+25
794+15
788+20
795+20
813+20
термохимических величин и сечений ионизации, использованных в расчетах. За исключением
выпадающих масс-спектрометрических измерений в работах [3750, 4900], остальные величины
согласуются между собой в пределах их погрешностей. Принятая величина является
средневзвешенной. Принятой энергии диссоциации соответствует
^fHo(m0, г, 0) = 73,783 + 16 кДж-моль.
НЮ+(г). Термодинамические свойства газообразного ионизированного оксида гафния в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 954.
Молекулярные постоянные, использованные при расчете термодинамических функций,
приведены в табл. 38.3. Спектр молекулы НЮ+ не исследовался, и принятые значения постоянных
основаны на приближенных оценках, выполненных так же, как для TiO+ (см. выше). В спектрах
молекул, имеющих 9 электронов на внешней оболочке (см. ScO, YO, LaO, а также LaF+ [4260],
TiN [2102, 2128], YF+ [4260] и ZrN [468, 1424]), наблюдались переходы, связанные с
состояниями Z2S+(. . .os), 2Д(. . .8Й), 2П(. . . пр) и 2Е+(. . .ар). Относительные энергии состояний
Х2?+(. . .aGs), Л2Д(. . .Ььа) и С2П(. . . квр) молекулы ИЮ+ были оценены по разностям энергии
атомных орбиталей 5d—65, 6р—6s, 6р—5d иона Hf+ в результате расчета центров тяжестей тер-
133
НЮ2(к, ж) Глава 38
мов, принадлежащих конфигурацияк . . .5d6s2, . . ,5d26s, . . .5d26p и . . .5d6s6p. При этом в
отличие от TiO+ и ZrO+, оказалось, что основным состоянием НЮ+, по-видимому, является
состояние Х2?+. Энергия состояния В2Щ. . . тс5 J была оценена по разности энергий возбуждения
состояний 3П, 1П конфигурации . . . o^t,^, а и 3Д, ХД конфигурации . . -а„Ь(п_Г) d в молекулах TiO
и ZrO и принятой энергии состояния ^42Д. Разность энергий состояний ZJS+(. . .<з6р) и С2П(. . . пе )
принята такой же, как у молекул TiN и ZrN. Погрешности в оцененных энергиях возбуждения
могут достигать 25—30% от их величины.
Постоянные ше и Ве молекулы НЮ+ в состоянии Х2?+, приведенные в табл. 38.3, оценены
по постоянным молекулы HfО в состоянии X1 ? + и отношениям этих постоянных для молекул LaF +
и LaF в состояниях 2Д и а3Д соответственно. Погрешности принятых значений могут достигать
70 и 0,03 см.
Термодинамические функции НЮ+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A.93)—A.95). Значения (?вн и ее производных рассчитывались по уравнениям A.90)—A.92)
с учетом четырех возбужденных электронных состояний и в предположении, что (?$л.«р~
=0,5pi(?W вA. Статистическая сумма по колебательно-вращательным уровням энергии
состояния Х2? + и ее производные находились по уравнениям A. 73) — A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по вращательным уровням с помощью
уравнений типа A.82). В расчете учитывались все уровни с значениями 7 ^ /шах, „> последние
определялись из условий A. 81). Уровни энергии состояния X2L + вычислялись по уравнениям A. 65),
A. 34) и A. 64а); коэффициенты в этих уравнениях, тождественные соответствующим постоянным
из табл. 38.3, а также значения итлх и /lim приведены в табл. 38.4. Коэффициент Y03 был получен
из условия A. 59). Мультиплетность уровней основного состояния учитывалась статистическими
весами. Различие молекулярных постоянных разных изотопических модификаций НЮ+ в
расчетах не учитывалось.
Погрешности рассчитанных термодинамических функций НЮ+(г) при низких температурах
обусловлены главным образом отсутствием экспериментальных данных о вращательной
постоянной в состоянии X2 ?+. При температурах выше 1000—2000 К становятся существенными
погрешности из-за отсутствия экспериментальных данных о числе и энергии возбужденных
электронных состояний НЮ+. Погрешности в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К
оцениваются в 0,5, 3, 5 и 8 Дж-К~1-моль~1 соответственно.
Таблица термодинамических функций НЮ+(г) публикуется впервые.
Константа равновесия реакции НЮ+(г)+е(г) = Ш(г) + О(г) вычислена с использованием
значения Ari/°@) = 65,0+35 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(НЮ)=730+30 кДж-моль,
найденному Раухом и Аккерманом [3943] методом электронного удара. Погрешность принятого
значения увеличена по сравнению с рекомендацией [3943] с тем, чтобы учесть неопределенность
в заселенности возбужденных состояний НЮ, а также тот факт, что величина /0(Hf), найденная
Раухом и Аккерманом (см. выше), на 25 кДж-моль отличается от принятой в справочнике.
Принятому потенциалу ионизации соответствуют
i\fH°{Ei0+, г, 0) = 803,783 ± 35 кДж • моль,
D0(HfO+) = 719,000 + 35 кДж • моль« 60 000 ± 3000 см.
ШО2(к, ж). Термодинамические свойства кристаллического и жидкого диоксида гафния
в стандартном состоянии при температурах 100—6000 К приведены в табл. 955.
Значения постоянных, принятые для расчета термодинамических функций НГО2(к, ж),
приведены в табл. 38.1. В справочнике за стандартное состояние НЮ2(к) в интервале 0—2100 К
принята моноклинная модификация, в интервале 2100—2793 К тетрагональная модификация и
в интервале 2793—3073 К кубическая модификация х. При Г<^298,15 К теплоемкость НЮ2(к)
измерялась Тоддом [4520] E2—296 К) на образце с содержанием 1,66% ZrO2 и 0.37% других
примесей. Экстраполяция теплоемкости НЮ2 ниже 52 К проводилась с использованием
уравнения С°(Г)=?B63,1/Г)+?D90,3/Г)-Ь?G76,4/Г)+?A911,3/Т), которое аппроксимирует данные
1 При высоких давлениях (> 50 кбар) стабильна ромбическая модификация НГО2.
134
Гафний и его соединения НЮ2(к, ж)
[4520] с точностью ~0,5% и приводит к значению S°E2 К)=4,0 Дж-К-моль~1. Вычисленные
по данным [4520] (с учетом поправки на содержание примеси ZrO2) значения 5ОB98,15 К) и
#°B98,15 К)—#°@) (см. табл. 38.1), имеют погрешности 1 Дж-К^-моль и 0,1 кДж-моль
соответственно. Для теплоемкости НЮ2(к) в интервале 298,15—2100 К принято уравнение,
полученное на основании результатов измерений энтальпии диоксида гафния в работе Орра [3723]
C83—1807 К), выполненных на том же образце, который исследовался Тоддом г. 'Погрешности
аппроксимации теплоемкости этим уравнением оцениваются в 1% в интервале 298,15—1800 К
и в 2% в интервале 1800—2100 К. Существенно менее точные данные об энтальпии НЮ2(к, монокл.)
были получены Пирсом и Осментом [3787] F50—1658 К) 2.
Температура полиморфного перехода моноклинной модификации в тетрагональную
B100+20 К) принята по измерениям Шевченко и др. [977, 1148, 1149] методом ДТА. Ранее этот
метод применялся в работах [141, 1435, 1972, 3703, 3980, 4098, 4375, 4378, 4755] в условиях, не
обеспечивающих сохранение стехиометрического состава диоксида гафния. Теплоемкость
тетрагональной модификации (98,4+2,7 Дж-К~1-моль~1) и энтальпия перехода в эту модификацию
A2,0+8,0 кДж-моль) приняты на основании данных об энтальпии тетрагональной модификации,
полученных Пирсом и Осментом [3787] A911—2506 К) 3, и энтальпии моноклинной модификации
при Ttr.
Температура превращения тетрагональной модификации в кубическую B793+20 К) также
принята по измерениям Шевченко и др. [977, 1148, 1149], выполненным методом ДТА. Она близка
к средней между значениями, найденными Ногути [3666] B697 и 2875 К) тем же методом для
менее чистого образца НЮ2. Измерения этой величины в работе Богданова и др. [141 ] являются
неверными. Принятые значения энтальпии перехода тетрагональной модификации в кубическую
A8+7 кДж-моль) и теплоемкости последней A00+10 Дж-К~1-моль~1) основываются на
измерениях энтальпии кубической модификации при 2757 и 2894 К в работе [3787] (см. [4181]) 4.
Температура плавления НЮ2 C073+25 К) определена методом ДТА Шевченко и др. [977,
1148, 1149]. Близкие значения были получены в работах [1868] C035+25 К), [3391] C048 К),
136671 C030±20 К), а менее надежные величины — в работах [977, 1149, 1972, 2609, 4098].
Энтальпия плавления (96+8 кДж-моль) оценена в предположении, что AmS°(HiO2, к) =
= AmS°(VO2, к)=31,3 Дж-К~1-моль (см. ZrO2(K, ж)), а теплоемкость жидкого диоксида гафния
A05+10 Дж-К^-моль") оценена по соотношению, принятому в справочнике (см. т. I, с. 79).
Погрешности приведенных в табл. 955 значений Ф°(Т) при 298,15, 2000, 4000 и 6000 К
оцениваются соответственно равными 0,8, 2, 10 и 18 Дж-К~1-моль~1. Ранее термодинамические
функции НЮ2(к, ж) вычислялись в справочнике Шика [4181 ] (до 6000 К), где принималось, что НЮ2
существует только в форме моноклинной и тетрагональной модификации 5. Расхождения между
значениями Ф°(Г), приведенными в табл. 955 и в [4181], не превосходят 0,8 Дж-К-моль~1.
Принятая в справочнике энтальпия образования
A/r(HfO2! к, монокл., 298,15 К) = —1115,6 + 2,5 кДж • моль
является средневзвешенной из четырех значений, основанных на измерениях Хьюбера и Холли
[2741, 3075] (—1112,1+5,0 и —1114,9+3,2 кДж-моль) и Корнилова и Ушаковой [1041, 3075]
(—1116,6+2,5 и —1116,4+4,0 кДж-моль). В этих работах измерялась энтальпия сгорания
гафния в кислороде в калориметрической бомбе. Поправки на неполноту сгорания и на примеси
в исходных образцах были вычислены по уточненной методике, предложенной в работе [545].
Авторы этих работ провели рентгенофазовый анализ и показали, что HfO2 в продуктах сгорания
существует только в виде моноклинной модификации. Энтальпия образования НЮ2(к)
определялась также в работах [546, 2738, 2755, 4063]. Измерения Хамфри [2755] привели к значению
—1111,1+3,0 кДж-моль, близкому к принятому. Данные, полученные в остальных работах,
менее точны (см. [546, 1041, 2741, 3075]).
1 Данные [3723] скорректированы на содержание ZrO2 в образце.
2 Результаты измерений, выполненных Пирсом и Осментом [3787], приведены в справочнике [4181]. *
3 Использованный образец ШО2 содержал значительное количество примесей и температура его превращения
была, по-видимому, меньше 1900 К.
* В справочнике [4181] эти данные отнесены к тетрагональной модификации НЮо(к).
5 В справочниках [1038, 2960] приведена таблица значений #°G)— Я°B98,15 К) и 5°(Г)—5°B98,15 К) для
моноклинной модификации НЮ2(к) до 1800 К, составленная Орром [3723].
135
НЮ2(г)
Глава 38
Давление пара в реакции НЮ2(к', ж)=НЮ2(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
к, 0) = 900 ±30 нДж-моль.
Эта величина основана на масс-спектрометрическом измерении давления пара НЮ2(к),
выполненном Щукаревым и Семеновым [1182] B703—2953 К, данные представлены в виде
уравнения). По этим измерениям были вычислены значения As#°(ffiO2, к, 0), равные 902+30 кДж-моль
(расчет по III закону) и 920 кДж-моль (расчет по II закону). Погрешность принятого значения
учитывает неточность термодинамических функций НЮ2(к, ж) и НЮ2(г), использованных в расчете.
НЮ2(г). Термодинамические свойства газообразного диоксида гафния в стандартном состоянии
при температурах 100—6000 К приведены в табл. 956.
Спектры и структура молекулы НЮ2 не исследовались; молекулярные постоянные,
приведенные в табл. 38.6, были оценены при подготовке справочника на основании принятых постоянных
Таблица 38.6. Значения молекулярных постоянных, а также б и рх, принятые для расчета
термодинамических функций ШО2
Состояние
а fe(^35i) = 15 000
Те(Ё3В1) = 23 000
СМ,
см,
850
Р =
= 3;
3;
V2
СМ
500 850
Те{В3В2)=Те(С1В1) = 18 000 см-1,
T^Bi) = 26 000 см, р=1.
гз
8
/с•10"'
• см*
• 102
4; Te(D1B
а
2
г) =21
000 см,
1
P=U
молекул МО2, где M=Ti, Zr, V, Nb, Та, Сг, Mo, W. Приведенное в таблице значение IaIbIc в
состоянии Ж1А1 вычислено для структурных параметров r(Hf—О)=1,75+0,03 А (оценка по
величинам г,(НЮ), re(Nb0) и r(Nb—О) в NbO2, см. выше) и -4 О—Hf—0 = 110+15° (оценка по величине
-$ О—М—О в молекулах диоксидов переходных металлов); его погрешность составляет
3-Ю15 г3-см6. Погрешности принятых значений основных частот оцениваются в 10% от их
величины.
Приведенные в таблице 38.6 энергии возбужденных электронных состояний получены Хаитом
и Хачкурузовым [1065] на основании полуэмпирической оценки для модели кристаллического
поля. В расчете использовались параметры, найденные для молекулы НЮ, а также известные
экспериментальные данные о первых возбужденных состояниях TiO2 и ТаО2. Погрешности
принятых энергий возбужденных электронных состояний оцениваются в 5000 см.
Термодинамические функции Ш02(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом шести возбужденных электронных состояний по соотношениям A.168)—A.170).
Погрешности вычисленных значений термодинамических функций обусловлены неточностью
оценки молекулярных постоянных и приближенным характером расчета. Погрешности
значений Ф°(Т) при 298,15, 3000 и 6000 К составляют 2, 5 и 8 Дж-К^-моль.
Ранее термодинамические функции НЮ2(г) вычислялись в справочнике Шика [4181 ] и в работе
[1666] (до 3000 К); оба расчета были выполнены для линейной структуры молекулы НЮ2 по
оцененным постоянным основного состояния и без учета возбужденных электронных состояний.
Расхождения в значениях Ф°(Т), приведенных в табл. 956 и в работах [1666, 4181], составляют
от 23 до 13 Дж-К^-моль.
Константа равновесия реакции HiO2(r)=Hf(r)+2O(r) вычислена с использованием значения
Дг#°@)=1326,449+30 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации НЮ2(к). Этим величинам также соответствует
AfH°(HfO2, г, 0) = —210,883 + 30 нДясмоль.
Глава 39
СКАНДИЙ И ЕГО СОЕДИНЕНИЯ
Sc(k, ж), Sc, Sc+, ScO, ScO% ScO2, Sc2O, Sc2O2, Sc2O3(k, ж)
В настоящей главе рассматриваются скандий и его кислородные соединения — всего 8 веществ.
Для двух веществ приведены данные об их термодинамических свойствах в конденсированном
состоянии, для семи — в газообразном состоянии. Термодинамические свойства четырех газов
(Sc, Sc+, ScO, ScO+) рассчитаны до 10 000 К, остальных трех — до 6000 К.
Для элементарного скандия в Приложении 3.10 приведены сведения о критических постоянных.
Sc2O3 является единственным оксидом скандия, стабильным в широком интервале температур
в конденсированном состоянии; практически он не имеет области гомогенности. В справочнике
не рассматриваются ионизированные многоатомные кислородные соединения скандия, так как
ввиду относительно невысокой стабильности Sc2O(r) и Sc2O2(r) можно предполагать, что в
равновесных условиях соответствующие ионы не должны присутствовать в заметных количествах.
Более вероятно, что ион ScO2 может играть определенную роль в некоторых системах, однако
отсутствие данных о величине ^40(Sc02) делает затруднительным расчет термодинамических свойств
этого соединения.
Sc (к, ж). Термодинамические свойства кристаллического и жидкого скандия в стандартном
состоянии при температурах 100—5100 К приведены в табл. 957.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 39.1. За стандартное состояние скандия в интервале 0—1609 К принята гексагональная
плотноупакованная а-модификация (структурный тип Mg), а в интервале 1609—1814 К —
объемно-центрированная кубическая [3-модификация (структурный тип a-Fe).
Таблица 39.1. Принятые значения термодинамических величин для скандия и его соединений
в кристаллическом и жидком состояниях
о
EH
О
1
at
CQ
Sc
Sc2O
Ш
К
я
о
о
о
U
1
ю
5-2-
й °
a, I
к Дж • моль
к11, гекс.(а) 5,207
к1, куб. (р) -
ж —
з к, куб. 13,93
JK —
К
ю
оо"
аз
?i
о
Дж ¦ К-'
34,77
—
—
76,8
—
LO
98,1
Сх!
о а.
• МОЛЬ
25,51
—
—
94,22
—
Примечание. C°(T)=a+b -T— с -T~2+d-T2+e -Г3 (в Дж
Sc: a d= 17,720 -10~6, e=— 3,867 -lO"9.
Sc2O3: a a
Коэффициенты в
НИИ ДЛЯ С°
а
6-Ю3
29,847 -12,297
44,2 —
44 —
115,235 12,770
200 —
К -моль).
=1,603 -10.
уравне-
(Т)
с ¦ 10
1,905 а
—
—
22,192 а
—
Интервал
температур
К
298,15-1609
1609-1814
1814-5100
298,15—2762
2762—6000
Ttr
Т
К
1609
1814
—
2762
—
кДж-
•моль 1
4,022
14,1
—
127
—
При Г<^298,15 К термодинамические функции вычислены на основании данных о теплоемкости
скандия, полученных Флотоу и Осборном [2272] @,9—23 К, образец чистотой 99,67%) и Герштей-
номидр. [2393] F—350 К, образец чистотой 99,9%, погрешность измерений 2% при 10 К, 1% при
20 К и 0,2% при Г>40 К). Погрешности вычисленных значений ?°B98,15 К) и Я°B98,15 К)—Н°ф)
137
Sc(k, ж)
Глава 39
(см. табл. 39.1) оцениваются в 0,15 Дж-К~х-моль х и 0,01 кДж-моль х соответственно.
Результаты измерений в работах [3326] @,15—3 К), [4559] A—20 К) и [2514] A—20 К)
удовлетворительно согласуются с принятыми значениями; данные [4692] E2—296 К), [937] C—300 К) и
[3545] A,7—4,2 К) менее точны и не учитывались.
Уравнение для теплоемкости a-Sc в интервале 298,15—1609 К получено обработкой
результатов измерений энтальпии, выполненных Деннисоном и др. [2048] B73—1812 К, образец
чистотой 99,62%), их погрешность составляет ~0,5% х. С принятым уравнением в пределах
погрешности согласуются данные о теплоемкости, полученные Герштейном и др. [2393] B98—350 К).
Измерения теплоемкости скандия [652] A100—1600 К), погрешности которых составляют ~6%,
в расчетах не учитывались. Температура полиморфного превращения A609+3 К) принята по
согласующимся между собой данным [1442, 2048, 2513]. Результаты менее точных измерений
[708] A623 К), [3378] A646 К) и [652] A723 К) не учитывались. Энтальпия превращения D,02 +
0,4 кДж-молъ), теплоемкость ^-модификации Sc D4,2+1,0 Дж-К~1-моль~1) и энтальпия
плавления A4,1+1,0 кДж-моль) основаны на измерениях энтальпии, проведенных Деннисоном и
др. [2048]. Температура плавления A814+3 К) принята по практически совпадающим между
собой данным [353, 1441, 1442, 2048, 2513, 4356] с учетом поправки на переход к температурной
шкале МПТШ-68 (+2°). Более низкие значения были получены в работах [708] A808 К), [652]
A803 К), [3378] A795 К). Ввиду отсутствия экспериментальных данных по теплоемкости и
энтальпии жидкого скандия значение C°(Sc,jk) принято приближенно равным теплоемкости Р-модифика-
ции D4+5 Дж-К-моль~1).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 3000, и 5000 К оцениваются
в 0,1, 0,2, 3, и 6 Дж-К~1-моль соответственно.
Термодинамические функции Sc(k, ж) вычислялись ранее в справочнике Халтгрена [2752]
(до 3200 К), они согласуются с данными табл. 957 в пределах 0,2 Дж-К-моль.
В настоящем справочнике в качестве стандартного состояния скандия принято
кристаллическое состояние и в соответствии с этим
Давление пара скандия в реакции Sc(k, ж)=Эс(г) вычислено с использованием значения
h.rH°@)= Asi7°(Sc, к, 0)=375,905+3 кДж-моль, соответствующего принятой в справочнике
энтальпии сублимации
V/°(Sc, к, 298,15 К) = 377,7+ 3,0 кДж • моль.
В табл. 39.2 представлены результаты исследований энтальпии сублимации скандия.
Принятая величина является средневзвешенной из всех данных, за исключением работы [487], в
которой, по мнению ее авторов, были получены недостаточно надежные результаты. Наиболее
достоверными являются измерения [1219, 2540, 4356], которым придавались наибольшие веса.
Таблица
Спеддинг и
Карелин и
Аккерман,
Крикориан,
Де Мария i
Хаберманн,
Верхаген и
39.2. Результаты
Авторы
др., 1960 [4356]
др., 1962 [487]
Раух, 1962 [1219]
1963 [3109]
i др., 1963 [2034]
Даан, 1964 [2540]
др., 1965 [4606]
определений Agff° (Sc, к, 298,15 К) (в кДж-моль")
Метод
Эффузионный, 1340—1748 К, 16 точек
То же, 1301—1644 К, 13 точек
Эффузионный, 1555—1780 К, 6 точек
Масс-спектрометрический, 1420—1845 К, 2 серии
Эффузионный, 1478—1979 К, 16 точек
Масс-спектрометрический, 1255—1572 К, данные
представлены в виде уравнения
Эффузионный, 1521—1813 К, данные
представлены в виде уравнения
Масс-спектрометрический, 1200—1530 К, (см.
[2752])
As#"(Sc, *
II закон
360+10
350+8
350+15
341+3
391+20
348
386
—
?, 298,15 К)
III закон
376,9+0,7
371,7+1,2
375,2+1,6
381,5+1,3
381,4+2,5
379,4+0,3
379,5
В работе [2048] приведены только сглаженные значения Н°(Т)— Я°B98,15 К) с шагом в 100 К.
138
Скандий и его соединения Sc(r), Sc+(r)
Приведенные в таблице погрешности отражают только воспроизводимость измерений.
Неточность термодинамических функций конденсированной фазы скандия приводит к дополнительной
погрешности в значении энтальпии сублимации, достигающей 0,7 кДж-моль для измерений
при 1820 К.
Sc(r). Термодинамические свойства газообразного скандия в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 958.
Для расчета термодинамических функций использованы уровни энергии х атома Sc с
суммарным статистическим весом 1816 в интервале 0—52 000 см, т. е. ниже первого потенциала
ионизации. Эти уровни соответствуют валентным конфигурациям . . .3dAs2 (к ней относится основной
уровень 2D%), . . .3d3, . . .3d4l A=0—3), . . .3dAsAl A=1, 2, 3), . . As4l A=1, 2). Принятые
величины основаны на данных Шугара и Корлисса [4440], а также на оценках для ряда ненаблюдав-
шихся электронных состояний, принадлежащих электронным конфигурациям . . .3d2(x5L/),
. . .3d24l A=2, 3) и . . .3d4s4/; оценки были выполнены по методике, изложенной в
Приложении 6. Оцененные уровни имеют энергию 45 700 см и выше с суммарным статистическим весом 816.
Погрешность оцененных величин лежит в пределах 400—4000 см.
Термодинамические функции Sc(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т ^ 5000 К обусловлены главным образом неточностью фундаментальных постоянных
и не превышают 0,02—0,03 Дж«К~1-моль~1. При более высоких температурах становятся
заметными ошибки из-за приближенной оценки энергии ряда уровней и пренебрежения ридберговскими
состояниями. Соответствующие погрешности могут достигать 0,05 и 0,2 Дж«К~1-моль в
значениях Ф°(Т) при 6000 и 10 000 К соответственно.
Ранее термодинамические функции Sc(r) вычислялись в справочнике Сталла и Зинке [4421 ]
(до 3000 К), а также в работах Каца и Маргрейва [2934] (до 2000 К), Кольского и др. [3067]
(до 8000 К) и Хилзенрата и др. [2652] (до 10 000 К). Результаты расчета [2652] при 77<6000 К
совпадают с данными табл. 958 в пределах 0,03 Дж-К-моль~1; при увеличении температуры
расхождения возрастают до 0,3, 1,6 и 6 Дж-К-моль~1 при 10 000 К в значениях Ф°(Г), S°(T)
и С°р(Т) соответственно. Эти расхождения обусловлены использованием в расчете [2652] неточных
данных по уровням энергии из справочника Мур [3550]. Расхождения с данными [2934] настолько
велики (до 6 Дж-К~1-моль в значениях Ф°B000К)), что это можно объяснить только ошибкой
в этом расчете.
Энтальпия образования
bfH°(Sc, г, 0) = 375,905 + 3 кДж • моль
соответствует принятой в справочнике энтальпии сублимации скандия.
Sc+(r). Термодинамические функции газообразного положительного иона скандия в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 959.
Для расчета термодинамических функций использованы уровни энергии г иона Sc+ с
суммарным статистическим весом 415 в интервале 0—89 950 см. Эти уровни соответствуют валентным
электронным конфигурациям . . .3dAs (к ней относится основной уровень 3D±), . . As2, . . .3d2,
. . Лр'2, . . AsAl A = 1, 2) и . . .3dAl A = 1, 2, 3). Принятые величины основаны на данных Шугара
и Корлисса [4440]; энергии двух ненаблюдавшихся уровней . . Ар2 *S и . . .4s B5LеР.О были
оценены сравнением соответствующих состояний Y+, La+ и Sc+ и приняты равными 80 000 +
+ 1000 см и 85 000+1000 см соответственно. Другие валентные состояния иона Sc+ имеют
высокие энергии и не учитывались в расчете термодинамических функций.
Термодинамические функции Sc+(r) вычислены по уравнениям A.3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Г^8000 К обусловлены главным образом неточностью фундаментальных постоянных
и не превышают 0,02—0,03 Дж-К~1-моль. Погрешности из-за приближенной оценки энергии
уровней и пренебрежения рядом состояний с большой энергией, в том числе ридберговскими
состояниями, становятся заметными только при Г>8000 К; соответствующая погрешность в
значении Ф°A0 000К) оценивается в 0,05 Дж-К^-моль.
См. пргтмеч. 1 к тексту по Сг (г).
139
ScO(r)
Глава 39
Ранее термодинамические функции Sc+(r) вычислялись в работах Грина и др. [2473] до
50 000 К и Хилзенрата и др. [2652] до 10 000 К. Результаты этих расчетов практически совпадают
с данными табл. 959 (расхождения не превышают 0,01—0,02 Дж-К-моль~1 в значениях Ф°(Г)).
Константа равновесия реакции Sc+(r)+e(r)=Sc(r) вычислена с использованием значения
ДгЯ°@) =—633,088+0,006 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Sc)=52 922,0±0,5 см-^633,088 + 0,006 кДж-моль.
Эта а еличина найдена Гартоном и др. [2370] из ридберговской серии . . .3d4sAZ?)/2/> 2Рзv
При этом авторы [2370] показали, что известное ранее в литературе более низкое значение
52 750 см [3549] было ошибочным. Принятому значению /0(Sc) соответствует
A//°(Sc+, г, 0) = 1008,993 + 3,0 кДж • моль.
ScO(r). Термодинамические свойства газообразного оксида скандия в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 960.
Молекулярные постоянные, принятые в справочнике на основании анализа результатов
исследований электронного спектра ScO, приведены в табл. 39.3. В спектре ScO изучены три системы
Таблица 39.3. Молекулярные постоянные ScO и ScO+
Молекула
Состояние
см
А
5Sc160
В2 2+ 3
ScO4
0
15 028,16
15 134,81
16 485,8
16 604,8
20 646,29
0
975,74
844,1
848,0
879,1
881.56
825,47
1000 б
4,2 б
4,25
5,75
5,13 е
5,5
4,21
4,2 в
0,515 08
0,48 в
0,504 71
0,484 68
0,53 г
3,3
3,6
3,9
3,2
ЗД
6
6Д
6,54 ж
6,74
6 е
1,6656
1,73
1,6826
1,7170
1,65 б
Примечание. Все постоянные даны в см 1 (кроме pf).
ScO: a y=0; ^ <uej/e=6-10; в оценка; г рассчитано по соотношению A.69); Д рассчитано по
соотношению A.68); е шеуе=3 -10~3; ж приведено значение постоянной для г--=0; 3 объединенные электронные
термы:
Те
Pi
30 000
22
40 000
30
ScO+: a объединенные электронные термы:
Т
1 в
Pi
20 000
12
30 000
24
40 000
40
б оценка; в вычислено по соотношению A. 67) и значению D0=59 000; г вычислено по соотношению A. 38);
Д вычислено по соотношению A. 69); е вычислено по соотношению A. 68).
полос: Л2Ш*Х22 + [1274, 1804, 2880, 3474, 4413, 4700], В22,+^±Х*2+ [1249, 1274, 1804, 2880,
3474] и Л'2Д->Х22+ [1803, 1804] х. Кроме того, в спектре хемилюминесценции ScO [1804]
наблюдались неинтерпретированные переходы в области 2700—3800 А, Карлсон и др. [1748] в
результате неэмпирического расчета ScO без учета взаимодействия конфигураций нашли, что в области
энергий до 48 000 см эта молекула должна иметь 14 связанных дублетных и квартетных
возбужденных состояний. Предсказанная авторами [1748] энергия нижнего возбужденного
состояния А'2А удовлетворительно согласуется с результатами последующих исследований спектра ScO
[1804], однако для состояний Л2П и, особенно, /?22+ были получены существенно завышенные
величины. Таким образом, расчет Карлсона и др. [1748] не дает значений энергии ненаблюдав-
1 Ранее в результате неправильной интерпретации тонкой структуры полос переходов А—X и В—X в работе
[1274] принималось, что нижнее состояние этих переходов имеет симметрию 4 S+. В работах [1249, 2929, 4413.
4700] было показано, что сложная структура полос обусловлена большим сверхтонким расщеплением уровней
состояния 2 ?+ из-за взаимодействия моментов спина ядра атома Sc (i— 7/2) п сппна ^.-электрона.
140
Скандий и его соединения
ScO(r)
шихся состояний, но позволяет предполагать в согласии с результатами исследования спектра
хемилюминесценции существование у ScO ряда состояний в области выше 26 000 см. Для учета
этих состояний в табл. 39.3 включены два объединенных электронных терма с суммарным
статистическим весом, равным сумме статистических весов 11 электронных состояний.
Приведенные в табл. 39.3 колебательные постоянные в состояниях Х22 + и А2П найдены Ча-
леком и Гоулом [1804] по началам полос системы А2П—Х22 + (г/, v" ^ 8), вычисленным из кантов,
измеренных Меггерсом и Вилером [3474!. В расчете использовались вращательные постоянные ScO
в состояниях Х22 + и /12П, полученные в работах [1249, 1274] и [4413] соответственно. Эти
колебательные постоянные хорошо описывают полосы с г/ ^ 12 и v" ^ 11, наблюдавшиеся в спектре
хемилюминесценции [1804]. Однако их точность, по-видимому, невелика, так как значение AG"/,,
вычисленное по ним (967,36 см), существенно отличается от полученного Акерлиндом [1274]
непосредственно по началам полос 0—0 и 0—1 системы i?22+—Х22 + (964,95 см). Принятые
вращательные постоянные в состоянии Х22 + рассчитаны из значений Во и Do, найденных Адамсом и
др. [1249] и Стрингатом и др. [4413], и значения al7 полученного Акерлиндом [1274] в результате
анализа полос 0—0 и 0—1 системы i?22 +—Ха2+. Вращательные постоянные в состоянии А2П
получены Стрингатом и др. [4413] на основании анализа структуры полосы 0—0 системы
Л2П—Х22+.
Колебательные постоянные в состоянии 522 + найдены Акерлиндом [1274] по началам полос
системы 522 +—Х22 + (г/ ^ 2), вращательные постоянные приняты по результатам анализа
структуры полосы 0—0 этой системы в работе [1249] и значения al5 полученного Акерлиндом.
Колебательные постоянные и энергия состояния А'2 А рассчитаны по кантам полос системы
Л'2Д—Х22 + (z/, v" ^2), измеренных Чалеком и Гоулом [1803] в спектре хемилюминесценции.
Постоянная Ве оценена в предположении, что относительное изменение межъядерного
расстояния при переходе электрона с орбитали о ;s на орбиталь 5(я_1) а примерно одинаково для оксидов и
галогенидов одного элемента.
Термодинамические функции ScO(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производных рассчитывались по уравнениям A.90)—A. 92)
с учетом пяти возбужденных состояний и в предположении, что QiH^^—O^PiQ^^. Величина
Qml. вр и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям 7 с помощью уравнений
типа A. 82). В расчете учитывались все уровни энергии со значениями / ^ /ша!С) „, где /ШШС) v
определялись из условий A.81). Колебательно-вращательные уровни энергии состояния Х22 +
вычислялись по уравнениям A. 65), A. 34), A. 64а), мультиплетность уровней учитывалась
статистическим весом 2. Значения коэффициентов в этих уравнениях приведены в табл. 39.4, они
соответствуют принятым молекулярным постоянным ScO с учетом условий A. 50) и A. 59).
Таблица 39.4. Коэффициенты в уравнениях, описывающих уровни энергии (в см),
а также значения ушах и е/цш, принятые для расчета термодинамических функций
ScO(r) и SeO+(r)
Коэффициент
ScO,
Х+ а
ScO+,
_\rlv+ а.
Те 0 0
У10 -10 9,755 608 10,00
У20 -4,143 9 -4,20
У30-103 1,770 7 —
Примечание. Ниже все постоянные в
Коэффициент
ScO,
X 2S+ а
ScO+,
X1S'1" а
У40 -105 -2,087 —
Уо1-10 5,150 8 • 5,3
Уи 103 —3,3 —3
У02 -10' —6,0 —6
см (кроме р{).
Коэффициент
У03 -Ю13
^юах
ScO: а Состояние А'* А АШ В2 2+
Г<" 15 081 16 545 20 646 30 000 (р=22) 40 000 (р=30)
ScO+: а возбужденные состояния см. в табл. 39.3.
ScO,
Z2S+a
-2,99
111
443
ScO,+
X1H+i
-3,53
118
444
Погрешности рассчитанных термодинамических функций при температурах до 3000—4000 К
обусловлены главным образом неточностью известных колебательных постоянных в состоянии
Х22 + (см. выше); при более высоких температурах становится существенной приближенная оценка
141
ScO+(r)
Глава 39
числа и энергии ненаблюдавшихся состояний с энергиями выше 25 000 см х. Погрешности в
значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,05, 0,1, 0,3 и 1 Днс-К^-моль.
Ранее термодинамические функции ScO вычисляли Брюэр и Розенблатт [1666] (Ф°(Г),
Г<3000К), Шнейдер [4197а] (Г=2000—10 000 К), Юдин и др. [1188] (Г<3000К), Вардиа
[4591] (Ф°(Г), Г=1000^-8000К) и Сму и др. [4331] (Ф°(Т), Т=1800^-2200 К). Данные [1666]
превышают величины, приведенные в табл. 960, на 9 —13 Дж-К~1-моль~1, так как в этой работе
принят неверный статистический вес основного состояния и сделано допущение о существовании
возбужденного состояния с энергией ~200 см. Результаты расчета [4197а] согласуются с
данными табл. 960 в пределах 1 Дж-К-моль~1 вплоть до 5000 К, при более высоких Т
расхождения возрастают до 4 Дж-К-моль~1. В расчете [1188] допущены ошибки и использован
приближенный метод, соответствующие расхождения составляют 6 —13 Дж-К~1-моль".
Константа равновесия реакции ScO(r)=Sc(r)+O(r) вычислена с использованием принятого
в справочнике значения
Дг#°@) = D0(ScO) = 677 + 7 кДж • моль & 56 600 + 600 см.
Эта величина основана на представленных в табл. 39.5 данных. Указанные в таблице
погрешности учитывают воспроизводимость измерений, неточность термодинамических функций, энталь-
Та блица 39.5. Результаты определений D0(SeO) (в кДж-моль-1)
Л вторы
Метод
Do(ScO)
II закон
III закон
Семенов, 1965 [918] Масс-спектрометричесюш, Sc,O3(K)=2ScO(r)+ — 676+25
+О (г), 2551—2567 К, 3 точки"
Сму и др., 1965 [4331] Масс-спектрометрический, ScO(r)+Y(r)=Sc(r)+ 669 674,9+8,6
+ YO(r), 1761—2584 К, данные представлены в виде
графтка
Масс-спектрометричесюш, ScO(r)+La(r)=Sc(r)+ 677 677 + 8,6
~LaO(r), 1805—2200 К, данные представлены
в виде графика
Коппенс и др., 1967 [1923] Масс-спектрометрический, GeO(r)+Sc(r)=Ge(r)+ 675+16 669,7+6,5
+ScO(r), 1961—2145 К, 9 точек
Эймс и др., 1967 [1310] Масс-спектрометрический, Sc(r)+LaO(r)=La(r)+ 681 684,3+8,6
+ScO(r), 1870—2080 К, данные представлены
в виде уравнения
Эффузионный, Sc2O3(K)=2ScO(r)+O(r), 2555— 666+90 678,5+6,0
2693 К, 5 точек
Чалек, Гоул, 1977 [1804] Исследование хемилюминесценции в пересекаю- > 688+10
щихся пучках Sc и О2 (NO2, N2O, O3)
пий образования и сечений ионизации. Исследования равновесия реакции Sc2O3(K)=2ScO(r) +
+О(г) [918, 1310] и четырех обменных реакций с участием ScO [1310, 1923, 4331] приводят к
величинам, удовлетворительно согласующимся между собой. Принятое значение является
средневзвешенным из приведенных в табл. 39.5. Ему соответствует
Д/nScO, г, 0) = —54,312+ 7,6 кДж • моль.
ScO+(r). Термодинамические свойства газообразного ионизированного оксида скандия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 961.
Спектр молекулы ScO+ не исследовался, а ее квантовомеханические расчеты не проводились.
Молекулярные постоянные, принятые в справочнике и приведенные в табл. 39.3, были оценены
при подготовке справочника на основании имеющихся данных для молекул изоэлектронного
ряда (СаО—SrO—ВаО, см. т. III) с учетом особенностей, свойственных молекулам, содержащим
переходной металл, а также постоянных молекулы ScO.
В справочнике принято, что молекула ScO+, так же как молекула СаО, имеет основное
состояние Х1И+ с конфигурацией . . .8о237т:4, возникающей при удалении электрона с орбитали 9а
молекулы ScO. Нижние возбужденные состояния ScO+ должны соответствовать конфигурациям,
142
Скандий и его соединения ScO2(r)
связанным с переходами электронов с орбиталей 8 с и Зтс на орбитали 9<з и 1§, которые, по
существу, являются атомными 4s- и 3d- орбиталями Sc+. Единственный пример перехода,
связанного с возбуждением переноса заряда, наблюдавшийся в оксидах переходных металлов, — это
переход 62—62 в молекуле МпО с энергией около 18 000 см [3843]. Энергии электронных
конфигураций . . .8°23^39о, . . .8с2Зи31§, . . .8аЗтт:49о и . . .8аЗтт:41§ B0 000—40 000 см) были
оценены на основании результатов квантовомеханического расчета ScO [1748] с учетом того, что
в этом расчете не принималась во внимание корреляционная энергия, и в предположении, что
энергии молекулярных орбиталей мало изменяются при ионизации молекулы. Это приводит к
близости энергии орбиталей 1§ и 9 а у молекулы ScO+ в отличие от изоэлектронной молекулы СаО.
В расчете [1748] в области до 48 000 см не получено ни одного состояния с конфигурациями,
содержащими два электрона на Згс-орбитали. В молекулах изоэлектронного ряда СаО—SrO—ВаО
разность энергий конфигураций . . . <з2^2 и . . .оя3 составляет около 20 000 см. Эта величина
была использована для оценки относительного расположения состояний, соответствующих
конфигурациям . . .тт:2а§ и . . .я3о, . . ,т:38 в молекуле ScO + . Аналогия с атомными состояниями Sc+,
а также результаты квантовомеханических расчетов ScF и TiO [1748, 1750] позволили принять,
что состояния с конфигурацией . . .и2о2 и тем более . . .и2§2 имеют энергии, существенно выше
энергий состояний конфигурации . . .тс2а§, и могут не учитываться в справочнике. Ввиду
невозможности корректной оценки энергии индивидуальных состояний ScO + были оценены и приведены
в табл. 39.3 их суммарные статистические веса в области 20 000—40 000 см. Погрешности этих
оценок могут достигать 50 %.
Значения постоянных и>е и ге молекулы ScO+ в состоянии Х1И+ были оценены из
соответствующих постоянных ScO с учетом того, что орбиталь 9 а, с которой удаляется электрон при
ионизации, является несвязывающей или слегка разрыхляющей. Погрешности принятых значений
оцениваются в 50 см и 0,05 А.
Термодинамические функции ScO+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Q%B и ее производных находились по уравнениям A. 90)—A. 92) с
учетом трех объединенных электронных термов в предположении, что Q^o\ щ—PiQ^l. вр.
Статистическая сумма по колебательно-вращательным уровням энергии состояния Z1^ + и ее
производные рассчитывались по уравнениям A. 73)—A. 75) суммированием по колебательным уровням и
интегрированием по значениям / с помощью уравнений типа A. 82). В расчете учитывались все
уровни с значениями / ^ /maXi „, последние определялись из условий A. 81). Уровни энергии
состояния Хг2 + вычислялись по уравнениям A. 65) и A. 64а); коэффициенты Ykl в этих уравнениях
приведены в табл. 39.4 вместе со значениями г7шах и /lim, они тождественны постоянным ScO+ из
табл. 39.3, за исключением величины Yo3, полученной из условия A. 59).
Погрешности рассчитанных термодинамических функций до 3000 К обусловлены главным
образом отсутствием экспериментальных данных о постоянных ScO+ в состоянии ХХ2 +, при более
высоких температурах — приближенной оценкой вклада возбужденных электронных состояний.
Погрешности в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,5, 2, 5 и
8 Дж-К^-моль.
Термодинамические функции ScO+(r) публикуются впервые.
Константа равновесия реакции ScO+(r)+e(r)=Sc(r)-f О(г) вычислена с использованием
значения ДгЯ°@)=66,912+35 кДж-моль, соответствующего принятой в справочнике энергии
диссоциации
D0(ScO+) == 700 + 35 кДж • моль & 59 000 + 3000 см.
В литературе отсутствуют экспериментальные данные о потенциале ионизации молекулы ScO
и энергии диссоциации ScO+. Принятое значение энергии диссоциации основано на
предположении, что потенциал ионизации молекул ScO, YO, ЬаОна20—40 кДж -моль меньше потенциала
ионизации атома соответствующего металла. Принятому значению соответствуют
bfH°(Sc0+, г, 0) = 555,776 + 35 кДж • моль,
70(ScO) = 610,088 + 36 кДж • моль.
ScO2(r). Термодинамические свойства газообразного диоксида скандия в стандартном состоянии
при температурах 100—6000 К приведены в табл. 962.
143
Sc2O(r)
Глава 39
Таблица 39.6. Значения молекулярных постоянных, а также с и
термодинамических функций ScO3, Sc2O и Sc2O2
принятые для расчета
Молекула
ScO2
Sc2O
V2
v.
v4
CM
905
600
1000
516
250
500
905
1000
1000
130
500
1 Т Г IDllv
1a'b'c ¦1U
Г3 • CM6
— 4 -102
— 1 -10s
1100 6 -103
Q
2
2
4
Px
2
1
1
Молекулярные постоянные, использованные для расчета термодинамических функций ScO2,
приведены в табл. 39.6. В соответствии с результатами исследования ИК-спектра молекул ScO2,
изолированных в матрице из Аг [925], в справочнике принимается, что в основном состоянии Х2А
молекула ScO2 имеет нелинейную структуру (симметрия С2в). Приведенное в табл. 39.6
произведение моментов инерции вычислено по структурным параметрам r(Sc—О) = 1,70 + 0,05 А (оценено
по величине /%(ScO)) и -? О—Sc—0 = 110 + 20° (оценено по величине ^ О—М—О в молекулах
диоксидов других переходных металлов). Погрешность значения IaIbIc составляет 2-1015 г3-см6.
Основные частоты ScO2, приведенные в табл. 39.6, определены Серебренниковым и Мальцевым
[925] из ИК-спектра молекул ScO2, изолированных в матрице из Аг (авторы [925] предположили
совпадение частот vx и v3). Погрешности в значениях частот с учетом возможного матричного
сдвига оцениваются в 20—30 см. Возбужденные электронные состояния ScO2 в справочнике
не рассматривались.
Термодинамические функции ScO2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и
A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных, приближенным характером расчета и пренебрежением возбужденными электронными
состояниями; они составляют 2, 4 и 8 Дж -К -моль" в значениях Ф°(Т) при 298,15, 3000 и 6000 К
соответственно.
Таблица термодинамических функций ScO2(r) публикуется впервые.
Константа равновесия реакции ScO2(r)=Sc(r)+2 О(г) вычислена с использованием принятой
в справочнике энтальпии атомизации
Дг/7°@) = 1280 ± 90 кДж • моль.
Принятая величина была оценена на основании отношения D0(MO2) : D0(MO) = 1,89,
найденного Кордесом и Гингеричем [5000] в результате масс-спектрометрического исследования
равновесия реакций в системах Се—О, Nd—О, Gd—О и Но—О. Принятой величине соответствует
энтальпия образования
A/ff°(Sc0a, г, 0) = —410,529+ 90 кДж • моль.
Sc2O(r). Термодинамические свойства газообразного оксида дискандия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 963.
Молекулярные постоянные, использованные для расчета термодинамических функций Sc2O,
приведены в табл. 39.6. Экспериментальные данные о строении и спектрах Sca0 в литературе
отсутствуют. По аналогии с молекулами Ga2O, In2O и Т12О в справочнике принимается, что в
основном электронном состоянии ХХА молекула Sc2O имеет нелинейную структуру (симметрия C2v).
Приведенное в табл. 39.6 произведение моментов инерции вычислено по структурным
параметрам r(Sc—О) = 1,75+0,1 А (оценено в предположении, что r(Sc2O)/r(ScO);«r(M2O)/r(MO), где М =
=А1, Ga, In) и -3 Sc—О—Sc=145+20° (принят таким же, как в молекулах Ga2O, In2O, Т12О).
Погрешность значения IaIbIc составляет 2-Ю14 г3-см6. Принятые значения основных частот
были вычислены в приближении валентного силового поля, если принять, что для молекулы Sc2O,
так же как и для молекул Ga2O и 1п2О, справедливы соотношения /r//ce«i0,8, /rr//r«i0,15,
fjr2 : /,.«0,05. Погрешности в значениях vx, v2 и v3 оцениваются в 100, 50 и 150 см соответственно.
Возбужденные электронные состояния Sc2O в справочнике не рассматривались.
144
Скандий и его соединения Sc2O2(r), Sc2O,(r, ж)
Термодинамические функции Sc2O(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124). A. 128) и
A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета; они составляют 6, 8 и 10 Дж-К-моль"х в
значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно (погрешности из-за неточности
молекулярных постоянных имеют величину ~5 Дж-К-моль).
Термодинамические функции Sc2O(r) (значения Ф°(Т) для 2^=1700-^2600 К) вычислялись
в работах [4331, 5000]. Расхождения данных [4331] и табл. 963 составляют 3 Дж-К -моль
в значениях Ф°(Т), они обусловлены разной оценкой молекулярных постоянных. В работе [5000]
принято существенно более низкое значение v2 A04 см) и pj=6, соответствующие расхождения
достигают 22 Дж-К-моль в значениях Ф°(Г).
Константа равновесия реакции Sc2O(r)=2Sc(r)+O(r) вычислена с использованием значения
Дг#°@)=1018,593 ±40 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д,Я°(Sc2O, г, 0) = —20 ± 40 кДж • моль.
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции
Sc2O(r)=ScO(r)+Sc(K), выполненном Сму и др. [4331]. В работе сообщена i только энтальпия
реакции АгН°@) =—29 ±40 кДж'Моль. С учетом разности в термодинамических функциях,
использованных в [4331 ] и принятых в настоящем справочнике, из этой величины было найдено
значение АгН°ф) = —34±40 кДж-моль, которому соответствует принятая энтальпия
образования.
Sc2O2(r). Термодинамические свойства газообразного диоксида дискандия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 964.
Молекулярные постоянные, использованные для расчета термодинамических функций Sc2O2,
приведены в табл. 39.6. В литературе отсутствуют какие-либо сведения о молекулярных
постоянных Sc2O2. По аналогии с молекулами Li2O2 и А12О2 в справочнике принимается, что в основном
электронном состоянии Х1А молекула Sc2O2 имеет ромбическую структуру (симметрия D2h).
Приведенное в табл. 39.6 произведение моментов инерции вычислено по структурным параметрам:
r(Sc—О) = 1,85 ±0,1 А, (оценка по принятым в справочнике величинам г(М—О) в молекулах AlOt
А120, А12О2 и ScO, Sc2O), r(O—О)=1,5±0,1 А (принято таким же, как в молекулах А12О2 и Li2O2).
Погрешность в значении IaIbIg составляет 2-101* г3-смв. Основные частоты рассчитаны в
приближении валентного силового поля; постоянная /sc-o принята такой же, как в молекулах Sc2O
и ScO2, остальные постоянные оценены по постоянным Li2O2 (см. [4796]). Погрешности принятых
значений оцениваются в 40 см для v4, 70 см для v2, v5 и 150 см для vx, v3 и v6. Возбужденные
электронные состояния Sc2O2 в справочнике не рассматривались.
Термодинамические функции Sc2O2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A. 10), A. 122)—A. 124), A. 128)
и A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета, они составляют 6, 10 и 15 Дж-К~1«моль
в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций Sc2O2(r) публикуется впервые. "
Константа равновесия реакции Sc2O2(r)==2Sc(r)+2O(r) вычислена с использованием
принятой в справочнике энтальпии атомизации
Дг#°@).= 1730 ± 60 кДж • моль.
Принятая величина была оценена сопоставлением энергий связей в молекулах МО, М2О и
М2О2, где M=Sc, Y, La. Близкое значение, 1736 кДж»моль, было оценено Кордесом и Гинге-
ричем [5000]. Принятой величине соответствует
bfH°(Sc2O2, г, 0) = —484,624 + 60 кДж.моль.
Sc2O3(k, ж). Термодинамические свойства кристаллического и жидкого триоксида
дискандия в стандартном состоянии при температурах 100—6000 К приведены в табл. 965.
Значения постоянных, принятые для расчета термодинамических функций, приведены
Ю Заказ К. 1590 145
Sc2Os(k, ж) Глава 39
в табл. 39.1. В справочнике за стандартное состояние Sc2O3(k) в интервале 0—2762 К принята
кубическая модификация (структурный тип Т12О3) [2441] 1. . .
При Г-<298,15 К термодинамические функции Sc2O3 вычислены по результатам измерений
теплоемкости, выполненных Уиллером и Кингом [4693] E3—296 К) на образце, содержавшем
менее 0,1% примесей. Экстраполяция теплоемкости ниже 53 К проводилась в этой работе по
уравнению Cp=D(M3lT)+2E{470lT)+2EG77tT) и привела к значению ?°E3 К)=2,2 Дж-К^Х
Хмоль. Погрешности принятых по [4693] значений ?°B98,15 К) и #°B98,15 К)—#°@) (см.
табл. 39.1) оцениваются в 0,4 Дж-К~1-моль и 0,04 кДж-моль соответственно.
Уравнение для теплоемкости в интервале 298,15—2762 К (см. табл. 39.1) было получено
совместной обработкой результатов измерений энтальпии в работах Цагарейшвили и др. [1096)
C85—1607 К, 13 точек, содержание примесей в образце 0,5%), Панкраца и Келли [3754] C94—
1799 К, 17 точек, чистота образца 99,9% Sc2O3) и Шпильрайна и др. [1168, 4276] A570—2759 К,
17 точек, чистота образца 99,9% Sc2O3). При обработке погрешности данных [1096] были оценены
в 0,5%, данных [3754] — в 1%, а погрешности данных [1168, 4276], имеющих большой разброс,
ввиду трудностей измерений при столь высоких температурах оценены в 2 %. Это уравнение
приводит к значениям для энтальпии Sc2O3(k), которые несколько меньше данных Цагарейшвили и
др. [1096] (в среднем на 0,8% в интервале 780—1607 К) и больше данных Панкраца и Келли
13754] (в среднем на 1,2%). Данные Шпильрайна и др. [1168, 4276] при 1570—1774 К больше
рассчитанных по уравнению (на 3—5%), в интервале 1972—2484 К меньше (в среднем на 3%),
а в интервале 2585—2759 К расхождения резко увеличиваются, достигая 6,2 и 7,5% при 2748 К
и 2759 К 2.
Температура плавления Sc2O3 B762+20 К) принята по рекомендации [1168, 4276], основанной
на собственных данных этих авторов B762 К) и результатах измерений Трзебятовского и Хорина
[4557] B763 К). Данные других авторов [1024, 1029, 1408, 1947, 3669, 4199, 4792] менее точны и
приводят, как правило, к более низким значениям Тт из-за наличия примесей в исследованных
образцах и склонности Эс2О3(ж) к переохлаждению. Энтальпия плавления A27+8 кДж-моль)
принята как разность энтальпий кристаллического и жидкого Sc2O3 в точке плавления,
вычисленных по принятым в справочнике уравнениям. Резкое различие с величиной 100+8 кДж-моль,
рекомендуемой Шпильрайном и др. [1168, 4276], объясняется тем, что в настоящем справочнике
для энтальпии Sc2O3(k) принимается уравнение, выведенное по данным [1096], [3754] и [1168,.
4276], в то время как в работах [1168, 4276] применено графическое усреднение измерений
энтальпии Sc2O8(k), которое приводит к резко завышенным значениям энтальпии и теплоемкости в точке
плавления. Теплоемкость 8с2Р3(ж) B00+20 Дж-К-моль) вычислена по результатам девяти
измерений энтальпии 8с2О3(ж) в работах [1168, 4267] B773—2930 К).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 3000 и 6000 К оцениваются в 0,3,.
1, 4 и 20 Дж-К^-моль.
Термодинамические функции Sc2O3(k), вычисленные в справочнике Барина и Кнакке [13951
(до 2500 К), отличаются от приведенных в табл. 965 на 4 Дж-К^-моль в значении Ф° B500 К)
из-за того, что авторы [1395] приняли для теплоемкости Sc2O3 при Г>298,15 К оцененное
значение. Таблица термодинамических функций 8с2О3(ж) публикуется впервые.
Константа равновесия реакции Sc2O3(k, ж)=28с(г)+ЗО(г) вычислена с использованием
значения Д^0^)=3391,252+6,3 кДж-моль, соответствующего принятой в справочнике:
bfH°(Sc20a, к, 298,15 К) = —1908,6 ± 2,0 кДж-моль.
Эта величина основана на измерениях энтальпии сгорания скандия в кислороде, выполненных
Хьюбером и др. [2730] и подтвержденных Ма [3347]. В работе [2730] были также проведены
измерения энтальпии растворения скандия и Sc2O3(k) в соляной кислоте, что привело к заметно
отличающемуся значению —1917,5+3,0 кДж-моль. Авторы работы предположили, что это различие
может быть связано с наличием неучтенных примесей в использованном образце оксида скандия.
* При высоких давлениях (р > 1,3-106 атм) и температурах A273 К) получена моноклинная модификация Sc203
(структурный тип В — Sm2O3) [3961].
1 Возможно, что эта особенность двух измерений, проведенных вблизи температуры плавления, обусловлена
частичным плавлением образцов.
Глава 40
ИТТРИЙ И ЕГО СОЕДИНЕНИЯ
Y(k, ж), Y, Y+, YO, YO+, YO2, Y2O, Y2O2, YA,(K, ж>
В настоящей главе рассматриваются иттрий и его кислородные соединения — всего 8 веществ.
Для двух веществ приведены данные об их термодинамических свойствах в конденсированном
состоянии, для семи веществ — в газообразном состоянии. Термодинамические свойства четырех
газов (Y, Y+, YO, YO+) рассчитаны до 10 000 К, остальных трех — до 6000 К.
Для элементарного иттрия в Приложении 3.10 приведены сведения о критических постоянных.
Y2O3 является единственным оксидом иттрия, стабильным в конденсированном состоянии в
широком интервале температур; он практически не имеет области отклонений от стехиометрического
состава. В справочнике не рассматриваются многоатомные ионизированные соединения иттрия.
Энергии диссоциации соответствующих нейтральных молекул невелики, а их потенциалы
ионизации достаточно большие, поэтому можно предполагать, что в равновесных условиях такие
вещества, как Y2O+(r) или Y2O2(r), не должны присутствовать в заметных количествах. Более
вероятно образование в этих условиях YO^(r), однако отсутствие данных о величине A 0(YO2)
затрудняет расчет термодинамических свойств этого газа.
Y(k, ж). Термодинамические свойства кристаллического и жидкого иттрия в стандартном
состоянии в интервале 100—5700 К приведены в табл. 966.
Значения постоянных, принятые для расчета термодинамических функций иттрия,
приведены в табл. 40.1. За стандартное состояние иттрия в интервале 0—1755 К принята
гексагональная плотноунакованная а-модификация, а в интервале 1755—1801 К кубическая
объемно-центрированная ^-модификация.
Таблица 40.1.
Принятые значения термодинамических величин для иттрия и его соединений
в кристаллическом и жидком состояниях
cq
я
о
а
И
Y
Y20,
Состояние
kII, гекс. (а)
к1, куб. (8)
ж
к11, куб. (С)
к1, гекс. (Я)
ж
B98,15 К) —
Я°@)
Sj 1
кДж-моль
5,965
16,69
Примечание. С°(Г)=а+Ь -Г— с Л
Y2O3: a d=—33,498 -10~e, e=9,20S
298,15 К)
О
ДжК-
44,43
99,16
Г*-Н ¦'Р+е
) -Ю-».
298,15 К)
IT1
• МОЛЬ
26,52
102,50
•Г3 (Дж-IC1
Коэффициенты в
уравнении для Ср(Т)
а
Ъ- 10s
с • Ю-5
23,896 7,619 —0,313
35 — —
39,7 — —
107,301 47,250 14,360а
160 — —
200 — —
моль).
Интервал
температур
или
Т
К
298,15—1755
1755—1801
1801—5700
298,15—2550
2550—2712
2712-6000
1755
1801
2550
2712 f
или
кДж-
• моль
4,97
11,32
54
81
При Т <С 298,15 К термодинамические функции вычислены по данным о теплоемкости иттрия,
полученным Дженнингсом и др. [2851] A5—300 К, образец с примесями 0,5%, точность
измерений ~0,3% при Т >30К) и Уэлсом и др. [4695] A,5—16 К, образец с примесями 0,2%).
Погрешности вычисленных значений ?°B98,15 К) и Я°B98,15 К)—Н°@) (см. табл. 40.1) оцениваются
147
Y(k, ж)
Глава 40
в 0,2 Дж«К~1-моль~1 и 0,02 кДж-моль соответственно. Результаты измерений теплоемкости Y
в работах [3545] A,7—4,2 К) и [3563] A,5—20 К) менее точны и не учитывались.
. Уравнение для теплоемкости а-модификации в интервале 298,15—1755 К (см. табл. 40.1)
получено совместной обработкой результатов измерений теплоемкости, выполненных Дженнинг-
сом и др. [2851] B98—350 К), и данных по энтальпии, полученных Бергом и др. [1488] C74—
1948 К) и Велти и др. [4701] D01—1300 К). При обработке погрешность данных [2851]
оценивалась в 0,3%, данных [1488] — в 1% и данных [4701] — в 2%. Результаты менее надежных
измерений теплоемкости в работах Савицкого и др. [897] B73—1825 К), Суганеева и др. [991] C73—
673 К) и Новикова и др. [730, 731] не учитывались.
Температура полиморфного превращения A755+5 К) определена в работах [1440, 1442, 2513].
Менее надежные значения, найденные авторами [897, 2140, 2539, 3672] A732—1763 К), не
учитывались. Энтальпия полиморфного превращения D,97+0,2 кДж-моль) и теплоемкость ^-иттрия
C5+1 Дж'К^-моль), а также энтальпия плавления A1,32+0,60 кДж-моль) приняты на
основании результатов исследования энтальпии иттрия в работах Берга и др. [1487, 1488] C74—
1948 К); при выборе последней величины учитывались измерения энтальпии У(ж), выполненные
Стретзом и Баутиста [4411]. Температура плавления A801+5 К) определена в работах [353,
1440, 1442, 2513, 3672]; данные [897, 1981, 2140, 2539, 3613] A723—1788 К) не учитывались.
Теплоемкость жидкого иттрия C9,7'+0,5 Дж-К~1-моль~1) рассчитана из данных по
энтальпии, полученных Стретзом и Баутиста [4411] A799—2360 К).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1500, 3000 и 5500 К оцениваются
в 0,15, 1, 2 и 6 Дж-К~1-моль~1 соответственно. Термодинамические функции, приведенные
в табл. 966, отличаются от данных справочника Халтгрена [2752] (Т<^3700 К) не более чем на
0,1—0,7 Дж-К~1-моль в значениях Ф9(Т); расхождения с данными справочника Келли [2960]
(Г < 3000 К) составляют 0,4—2 Дж-К-моль в значениях Ф°(Т).
В настоящем справочнике в качестве стандартного состояния иттрия принято кристаллическое
состояние и в соответствии с этим
Давление пара иттрия в реакции Y(k, ж)=У(г) вычислено с использованием значения
Дг#°@)= Д##°(У> к, 0)=422,908+3,0 кДж-моль, соответствующего принятой в справочнике
энтальпии сублимации
Д,#°(У, к, 298,15 К) = 423,8
нДнсмоль
Результаты расчетов энтальпии сублимации по данным о давлении его насыщенного пара
представлены в табл. 40.2. Принятая величина является средневзвешенной из наиболее надежных
измерений, выполненных Аккерманом и др. [1219, 1239], Хаберманном и Дааном [2540] и
Круглых и др. [588, 589]. Более низкие значения энтальпий сублимации получены в работах [488,
489, 717], по-видимому, из-за использования недостаточно чистых образцов иттрия.
Таблица 40.2. Результаты
Авторы
Карелин и др., 1962 [489, 717]
Карелин и др., 1962 [488]
Аккерман, Раух, 1962 [1219]
Де Мария и др., 1963 [2035]
Хаберманн, Даан, 1964 [2540]
Круглых и др., 1964 [588, 589]
Аккерман и др., 1970 [1239]
определений A.gH°(Y, к, 298,15 К) (в кДж-моль)
Метод
Эффузионный, 1361—1761 К, 10 точек
То же, 1405—1733 К, 11 точек
То же, 1774—2103 К, 7 точек
Масс-спектрометрический, 1690—2050 К, две серии
То же, 1331-^-1760 К, данные представлены в виде
уравнения
Эффузионный, 1780—2185 К, данные
представлены в виде уравнения
То же, 1433—1753 К, 10 точек
То же, 1789—2055 К, 11 точек
Масс-спектрометрический, 1336—1994 К, восемь
серий
Asff°(Y,
II закон
314±15
355 ±4
415 ±3
407 ±10
425
430
352+30
419+18
411 ±4
к, 298,15 К)
III закон
358,0+3,0
404,2+2,5
424,0+0,6
—
—
424,2+0,3
421,4 ±2,9
421,2 ±0,7
—
148
Иттрий в его соединения Y(r), Y+(r)
Приведенные в таблице погрешности учитывают только воспроизводимость измерений.
Неточность термодинамических функций Y(k, ж) приводит к дополнительной погрешности в энтальпии
сублимации, достигающей для измерений, выполненных при 2000 К, —2 кДж-моль.
Y(r). Термодинамические свойства газообразного иттрия в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 967.
Для расчета термодинамических функций использованы уровни энергии г атома Y в
интервале 0—46 000 см, т. е. ниже первого потенциала ионизации, с суммарным статистическим
весом 1036. Эти уровни соответствуют валентным электронным конфигурациям . . .4d5s2 (к ней
относится основной уровень 2D%), . .Ad3, . . .5s25Z A=1, 2), . . Adbsbl A=1, 2), . . Ad25l A=0, 1, 2),
Принятые величины основаны на рекомендации Мур [3550] и на оценках для ряда ненаблюдав-
шихся уровней энергии. При этом энергии уровней, соответствующих конфигурации . . Ad3,
а также состояния 2Р конфигурации . . Ad2CPMp были оценены на основании сравнения с
имеющимися данными для Sc и La, а энергии уровней, соответствующих конфигурациям . . Ad5s5d,
. . Ad2CFMd, 4d2A5'Ms и . . Ad2ASMp, — по методике, изложенной в Приложении 6. Оцененные
уровни имеют энергию 25 000 см и выше с суммарным статистическим весом 379. Погрешности
в энергиях оцененных таким образом уровней могут составлять от 1000 до 5000 см'1. Уровни
энергии, относящиеся к другим валентным конфигурациям атома Y, лежат выше первого потен- <
циала ионизации и не учитывались при расчете термодинамических функций.
Термодинамические функции Y(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
величин при Т <^ 5000 обусловлены главным образом неточностью фундаментальных постоянных
и не превышают 0,02—0,03 Дж»К-моль. При более высоких температурах становятся
заметными погрешности из-за приближенной оценки энергии ряда уровней и пренебрежения ридбер-
говскими состояниями. Соответствующие погрешности могут достигать 0,05 и 0,2*Дж-К~1Х
Хмоль в значениях Ф°(Т) при 6000 и 10 000 К.
Ранее термодинамические функции Y(r) вычислялись в справочнике Сталла и Зинке [4421 ]
(до 3000 К) и в работах Каца и Маргрейва [2934] (до 2000 К), Кольского и др. [3067] (до 8000 К)
и Хилзенрата и др. [2652] (до 10 000 К). При Г^.6000 К результаты всех расчетов, за исключением
[3067], согласуются с данными табл. 967 в пределах 0,02—0,04 Дж-К^-моль. Для расчета
[3067] из-за использования завышенного значения R расхождения приблизительно вдвое больше.
При более высоких температурах расхождения возрастают из-за различия числа учитывавшихся
в расчетах уровней. Для [2652] расхождения достигают 0,3, 1,2 и 3 Дж-К'^моль
соответственно в Ф°(Т), S°(T) и С°р(Т) при 10 000 К.
Энтальпия образования
bfH°(Y, г, 0) = 422,908 ±3 кДж • моль
соответствует принятой в настоящем справочнике энтальпии сублимации иттрия.
Y+(r). Термодинамические свойства газообразного положительного иона иттрия в стандартном
состоянии при температурах 298,15^-10 000 К приведены в табл. 968.
Для расчета термодинамических функций использованы уровни энергии1 иона Y+ в
интервале 0—75 000 см с суммарным статистическим весом 733. Эти уровни соответствуют валентным
электронным конфигурациям . . .5s2 (к ней относится основной уровень XS), . . Ad2, . . .5р2,
. . .5s5Z A=1, 2), . . Adbl (?=0-^-4) и . . AdAf. Принятые величины основаны на рекомендации
Мур [3550] и на оценках для ряда ненаблюдавшихся уровней. При этом энергии уровней XS
конфигурации . . Ad2 и . . .5р2 были оценены сравнением с данными для Sc+ и La+, а уровней,
соответствующих конфигурациям . . AdBDLf, . . AdBD)bf и . . AdBD)bg, •—по методике,
изложенной в Приложении 6. Оцененные уровни имеют энергию от 28 000 см и выше с суммарным
статистическим весом 462. Погрешность оцененных таким образом энергий может составлять
приблизительно 4000—5000 см. Уровни энергий, соответствующие другим валентным электронным
конфигурациям, лежат выше первого потенциала ионизации иона Y+ и не учитывались в
справочнике.
Термодинамические функции Y+(r) вычислялись по уравнениям A.3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
1 См. примеч. 1 к тексту по Сг(г).
149
YO(r)
Глава 40
величин почти во всем интервале температур обусловлены главным образом неточностью
фундаментальных постоянных и не превышают 0,02—0,03 Дж-К-моль". Только при Г>8000 К
погрешности из-за приближенной оценки ненаблюдавшихся уровней и пренебрежения ридбер-
говскими состояниями становятся заметными; соответствующая погрешность в значении
Ф°A0 000 К) составляет приблизительно 0,05 Дж-К^-моль.
Ранее термодинамические функции Y+(r) вычислялись в работе Грина и др. [2473] до
50 000 К и Хилзенрата и др. [2652] до 10 000 К. Результаты этих расчетов практически совпадают
с данными табл. 968, и расхождения не превышают 0,01—0,02 Дж-К^-моль в значениях Ф°(Т).
Константа равновесия реакции Y+(r)+e(r)=Y(r) вычислена с использованием значения
ДгЯ°@)=—599,855+0,012 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0(Y)=50 144+1 см'^599,855+0,012 кДж-моль.
Эта величина найдена Гартоном и Ривсом [2369], которые исследовали в спектре поглощения
несколько протяженных ридберговских серий и показали, что принятое ранее в литературе более
высокое значение 51 447 см [3549] было ошибочно.
Принятому значению /0(Y) соответствует
A;H°{Y+, г, 0) = 1022,763 + 3,0 кДж • моль.
YO(r). Термодинамические свойства газообразного оксида иттрия в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 969.
Молекулярные постоянные, принятые в справочнике на основании анализа результатов
исследований электронного спектра этой молекулы, приведены в табл. 40.3. В спектре YO изучены
Таблица 40.3. Молекулярные постоянные YO и YO+
Молекула
Состояние
D, •
СМ"
А
89YWQ
YO+
.Х-2 2+а
0
14 531,2
14 870,4
16 529,75
20 793,35
0
862,0
794,0
794,9
820,337
758,73
900 б
2,86 б
3,23 1
3,3 /
3,377 е
3,97
3,3 в
0,388 907
0,376 1
0,386 78
0,373 14
0,41 г
1,745
1,74
2,03
2,49
2Д
3,04 в
3,4 г
3.3 ж
3.4 г
1,7882
1,818
1,7931
1,8255
1,75 б
Примечание. Ниже все постоянные даны в см х (кроме pf).
YO: а то=— 4,9 10~6; б <o,y,=6 -10~3; в 81=—1,2-10~8; #„=— 2,6 10~13; г вычислено по соотношению A.
Д А„=428,31+0,60 (w+V,); е (^„=1,4-«Г2; ж рх=—1,4-Ю"8, #„=-2,7 Ю8;
3 объединенные электронные термы:
Г"» 30000
Pi 22
40 000
30
YO+: a объединенные электронные термы:
Pi
20 000
12
30 000
24
40 000
40
б оценка; в вычислено по
соотношению A.67) и D0(YO+)=
=61 300; г вычислено по соотношению A. 38); Д вычислено по соотношению A. 69); е вычислено по
соотношению A. 68).
три системы полос: Л2Ш*Х2Е + [1513, 1514, 1804, 2192, 2880, 3273, 3474, 4266, 4577, 4578],
?22+<±:X2S+ [1513, 1514, 1804, 2192, 2880, 3474, 4266, 4577, 4578] и 4'2Д->Х2?+ [1803]. Кроме
того, в спектре хемилюминесценции YO [1804] в области 2800—3900 А наблюдались неиденти-
фицированные переходы, связанные по крайней мере с одной системой полос, которая по аналогии
с LaO была предположительно отнесена авторами этой работы к переходу С2П—Х2Е+.
Теоретические расчеты YO не проводились. Однако, так же как ScO, эта молекула должна иметь
связанные- дублетные и квартетные валентные состояния с энергиями 20 000—40 000 см. О
существовании состояния 22+ свидетельствуют гомогенные возмущения, обнаруженные в системе
150
Иттрий и его соединения
YO(r>
+—X2S+. Для учета этих состояний в табл. 40.3 включены два объединенных электронных
терма, статистические веса которых приняты такими же, как для мрлекулы ScO.
Колебательные постоянные YO в состояниях X2S+ и Л2П, приведенные в табл. 40.3, получены
Чалеком и Гоулом [1804] по началам полос системы А2П—Х22,+ (и', и" ^10), которые были
рассчитаны по кантам, измеренным Меггерсом и Вилером [3474]. В расчете использовались
вращательные постоянные YO, найденные Улер и Акерлиндом [4578]. Принятые колебательные постоянные
удовлетворительно описывают частоты полос с значениями г/ ^ 13 и v" ^ 12, измеренные
в спектре хемилюминесценции [1804]; для состояния Х22 + они хорошо согласуются с значениями,
лолученными Бернаром и Гравином [1514] при исследовании системы полос Z?2S+—X22+.
Вращательные постоянные в состояниях Х2Е+, 48Пи522+ приняты по работам Бернера и др.
[1513] и Бернара и Гравина [1514]. В работе [1513] они были получены совместной
обработкой результатов анализа структуры полос 0—0 и 1—1 систем А2П—Х22 + и В2Е +—Х2Е+ и
независимой обработкой полос секвенции Ai>=0 системы А2П—Х2? + с 2<J v<^5. В работе [15141
был выполнен анализ тонкой структуры семи полос системы 5аЕ+— Х2Е+. При расчете энергии
уровней состояния А2Л авторы [1513] использовали выражение в матричном виде, что позволило
учесть большое количество малых параметров, связанных со спин-орбитальным взаимодействием
и взаимодействием с состоянием B2Z + (параметры Х-удвоения). Тот же метод расчета был
применен Линтоном [3273] при обработке результатов измерений полосы 0—0 системы Л2П—Х2Е*
авторами [4578]. Вращательные постоянные YO в состояниях Х2Е+ и А2П, полученные в
работах [1513, 1514, 3273, 4578], прекрасно согласуются.
Молекулярные постоянные в состоянии А'2А приняты по работе Чалека и Гоула [1803]. Авторы
ЦЭОЗ] выполнили анализ колебательной структуры системы полос А'2А—Х2?+ и рассчитали
вращательные постоянные на основании измеренных интервалов между В.- и ф-кантами с
использованием вращательных постоянных состояния Х2?+ из работы [4578].
Большое количество полос системы JS2E+—Х2? + с р'^15 наблюдалось в спектре
хемилюминесценции [1804]. Волновые числа полос с i/>4 приближенно описываются постоянными ш#=
=722,5 и (o,z<1=7,39 см, рассчитанными из данных, полученных в работе [2880]. Уровни
3 2 + й б Т(В2 +)
<1
возмущены, по-видимому, другим состоянием типа 2 ? + с энергией больше Т0(В2 S +) и меньше
, и=4)=24 050 см.
Термодинамические функции YO(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QM и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом пяти возбужденных состояний и в предположении, что Q^,.t9=0,5piQ^ „р. Величина
р
v и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью уравнений типа
{1. 82). В расчете учитывались все уровни с значениями /^/Ш1„ последние определялись из
условий A. 81). Колебательно-вращательные уровни энергии состояния Х2Е + вычислялись по
уравнениям A. 65), A. 34) и A. 63), мультиплетность уровней учитывалась статистическим
весом 2. Значения коэффициентов в этих уравнениях, полученные с учетом условий A. 50) и A. 59)
из принятых молекулярных постоянных YO, приведены в табл. 40.4 вместе со значениями ь>шахи /l
Таблица 40.4. Коэффициенты в уравнениях, описывающих уровни энергии (в сыт1),
а также значения v^^ и J\im, принятые для расчета
термодинамических функций YO(r) и YO+(r) ,
lim.
Коэффициент
YO,
YO+,
Хг2+»
Те 0 0
У10 -КГ8 8,619 50 9,00
У20 -2,853 98 -3,3
У30-103 6,600 7 —
У40 -Ю6 —6,661 2 —
Примечание. Ниже все постоянные
Коэффициент
YO,
Ха?+»
YO+,
У01-10 3,88907 4,1
Уи -10s —1,745 —2
Уоа -10' —3,04 —3
У12-108 1,2 —
даны в см (кроме р).
Коэффициент
YO,
Х22+»
Уоз-Ю1» -2,6
У04-10» 1,04
г>шах И9
/пш 525
YO: a Состояние А'2А АЩ В22+
Т, 14 700 16 530 20 793 30 000 (р=22) 40 000 (р=30)
YO+: a возбужденные состояния см. в табл.40.3.
YO+,
—3,88
135
512
151
YO(r)
Глава 40
Погрешности рассчитанных термодинамических функций YO(r) при температурах до 2000—
3000 К обусловлены фактически только неточностью фундаментальных постоянных. При более
высоких температурах становятся существенными погрешности из-за отсутствия
экспериментальных данных об электронных состояниях с энергиями больше 21 000 см. Погрешности в
значениях Ф°(Г) при 298.15, 3000, 6000 и 10 000 К оцениваются в 0,02, 0,05, 0,3 и 1 Дж-К'^моль.
Термодинамические функции YO(r) ранее вычислялись в работах Шнейдера [4197а] (Г==
=2000—10 000,К), Брюэра и Розенблатта [1666] (Ф°(Г) для Г<3000К), Юдина и др. [11881
(Т<3000 К), Вардиа [4591] (T=1000-f-8000 К) и Аккермана и Рауха [1228] (Ф°(Г) для Т=
=1600-^-2800 К). При Т <[ 5000 К данные Шнейдера согласуются с приведенными в табл. 969 в
пределах 1 Дж-К^-моль. При более высоких температурах расхождения возрастают до 2 и 6 Дж X
хК-моль~1 в значениях Ф°(Т) и S°(T) главным образом из-за того, что в работе [4197а] не
учитывалось состояние А'2А. Расхождения данных [1188, 1666] и табл. 969 имеют разные знаки
и составляют 6—12 и 5—13 Дж-К-моль соответственно. Авторы [1666] приняли неверную
систематику электронных состояний YO и получили завышенные значения, а авторы [1188]
допустили ошибки в расчете и получили заниженные значения термодинамических функций.
Таблица 40.5. Результаты определений D0(YO) (в кДж-моль)
Авторы
Метод
Do(YO)
II закон
III закон
Уолш и др., 1960 [4644]
Аккерман и др., 1964 [1236]
Сму и др., 1965 [4331]; Куппенс и
др., 1967 [1923]
Сму и др., 1965 [4331]
Эймс и др., 1967 [1310]
Аккерман, Раух, 1971 [1222]
Уй, Дроварт, 1970 [4582] .
Алкок, Пелег, 1967 [1283]
Эймс и др., 1967 [1310]
Аккерман, Раух, 1973 [1228]
Лиу, Уолбек, 1974 [3285]
Чалек, Гоул, 1977 [1804]
Эффузионный, Y2O3(K)=2YO(r)+O(r), 2500— 752
2700 К, 3 точки
Эффузионный, Y2O3(k)=2Y0(r)+O(r), 2509— 748±70
2720 К, 11 точек
Масс-спектрометрический, комбинация данных 715+.30
реакций GeO(r)+Sc(r)—Ge(r)+ScO(r), см. табл. 39.5,
и ScO(r)+Y(r)=YO(r)+Sc(r), 1761—2584 К,
5 точек
То же, SiO(r)+La(r)=LaO(r)-fSi(r), см. 716+100
табл. 41.5, и LaO(r)+Y(r)=YO(r)+La(r), 1880— '
2294 К, 5 точек
То же, BO(r)+La(r)=B(r)+LaO(r), см. табл. 41.5, 830±300
и BO(r)+Y(r)=YO(r)+p(r), 2226—2482 К„5 точек
То же, YO(r)-t-La(r)=Y(r)+LaO(r), с использо-, 725
ванием значения D0(LaO), полученного
фотоионизационным методом (см. [3285])
Тоже * • 696
Тоже \ 703
Масс-спектрометрический, YO(r)+B(r)=Y(r)+ 722
+ВО(г), 2226—2482 К, данные представлены
в виде уравнения1
Торзионный, Y2O3(KH2YO(r)+O(r), 2250— —720
2475 К, измеренные давления даны только в виде
графика
Эффузионный, Y2O3(K)=2YO(r)+O(r), 2492—
2697 К, 14 точек
Масс-спектрометрический, Y2Os(k)=2Y0(r)-f-0(r),
2217—2660 К, 12 точек, данные представлены
в виде уравнения
Эффузионный, Y2O3(K)=2YO(r)+O(r), 2502—
2634 К, 7 точек
Масс-спектрометрический, Y2O3(K)=2YO(r)-fO(r),
2342-2614 К, 28 точек
Исследования хемилюминесценции в пересекаю- > 729+10
шихся пучках Y с О2 (NO2, N2O, O3)
712,7+7,2
694,9+6,0
708,7+7,1
714+10
705+12
713,1+7,8
717,1+7,3
711,6+7,3
703+11
-709
750+150
712
707 + 150
716
715,3+6,9
706,3+7,5
719,5+6,3
715,7 ±7,0
В работе [4582] имеются неточности в коэффициентах уравнения lg. Кр=А 1Т-\-В. Приведенные в этой работе
график и другие данные позволяют предположить, что в действительности эти коэффициенты соответствуют
уравнению 4,576 \gKp=AlT+B.
152
Иттрия а его соединения YO+(r)
Константа равновесия реакции YO(r)=Y(r)+O(r) вычислена с использованием принятого
в справочнике значения
Дг#°@) = D0(YO) = 714 ± 7 кДж • моль« 59 700 + 600 см.
Энергия диссоциации YO является общепринятым стандартом в исследованиях обменных
реакций, и ее выбор проводился с привлечением данных, полученных из всех известных авторам
справочника термохимических циклов. Принятая величина основана на результатах измерений,
приведенных в табл. 40.5. Указанные в таблице погрешности учитывают воспроизводимость
измерений, неточность использованных в расчетах термодинамических функций, энтальпий
образования и сечений ионизации. Основную погрешность в значения, полученные при изучении
равновесия обменных реакций, вносит неточность использованных энергий диссоциации других
молекул F—8 кДж-моль), а в значения, полученные при изучении диссоциации Y2O3(k),* —
неточность термодинамических функций этого вещества и его энтальпии образования (по 4 «"Дж-моль).
Наиболее точные измерения константы равновесия реакции Y2O3(K)=2YO(r)-f-O(r) были
выполнены в работах [1310, 3285, 4644]; среднее из четырех величин, полученных в этих работах,
составляет 716+7 кДж-моль. Величина, вычисленная по уравнению III закона из данных
[1228], основана на калибровке, в которой было использовано значение парциального
давления YO, среднее между полученными в работах [1236] и [1310], поэтому она не является
независимой. Работы [1236, 1283] по неизвестным причинам приводят к более низким значениям.
Исследования обменных реакций [1923, 4331, 4582] являются основным путем определения
величины D0(YO). Комбинирование результатов этих измерений с данными по энергиям
диссоциации других молекул дает четыре величины (см. табл. 40.5), средневзвешенное из которых
составляет 708+7 кДж-моль.
Значение D0(LaO)=793,7+3,0 кДж-моль, полученное Парром методом фотоионизации,
известно только по краткому упоминанию в работе [3285]. При вычислении D0(YO) по
результатам исследования равновесия реакции YO(r)+La(r.)=Y(r)+LaO(r) [1222, 1310, 4331] принималось,
что погрешность D0(LaO) составляет 6,0 кДж-моль. Среднее из трех величин, полученных этим
путем, равно 714+7 кДж-моль. . - ,
Таким образом, результаты определений энергии диссоциации YO приводят к
удовлетворительно согласующимся значениям. Принятое в справочнике значение является
средневзвешенным из приведенных в табл. 40.5 величин за исключением данных [1228, 1236,1283,1804]. Ему
соответствует
bfH°(YO, г, 0) = —44,309 + 7,6 'кДж • моль.
YO+(r). Термодинамические свойства газообразного ионизированного оксида иттрия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 970.
Спектр молекулы YO+ не исследовался, а ее квантовомеханические расчеты не проводились.
Молекулярные постоянные YO +, приведенные в табл. 40.3, были оценены при подготовке
справочника так же, как для ScO+. При этом энергии и статистические веса электронных состояний YO +
приняты такими же, как у ScO+ в предположении, что аналогия в электронных состояниях,
наблюдающаяся в ряду нейтральных молекул ScO—YO—LaO, должна проявляться и у их ионов.
Различие в энергиях §in_1)d- и см-брбиталей, которое можно ожидать у ионов оксидов, судя по
спектрам ионизованных атомов, а также нейтральных молекул, должно сказываться только на
относительном расположении состояний с энергией возбуждения свыше 20 000 см и учитывается
погрешностью приписанных им статистических весов, равной 50—60%.
Значения постоянных ше и ге молекулы YO+ в основном состоянии Х12+ были оценены из
соответствующих постоянных YO с учетом tofo, что орбиталь obs, с которой удаляется электрон
при ионизации нейтральной молекулы, является несвязывающей или слегка разрыхляющей.
Погрешности этих постоянных оцениваются в 50 см и 0,05 А соответственно.
Термодинамические функции YO+(r) вычислены по уравнениям A.3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения (?вн и ее производных находились по уравнениям A. 90)—A. 92) с
учетом трех объединенных электронных термов в предположении, что Q^\% *v~PiQim. Sf
Статистическая сумма по колебательно-вращательным уровням энергии состояния Хг2 + и ее производные
рассчитывались по уравнейиям A. 73)—A. 75) суммированием по колебательным уровням и
интегрированием по значениям / с помощью уравнений типа A: 82). В расчете учитывались все уровни,
с значениями /^/шаХI>, последние определялись из условий A.81). Уровни энергии состояния
153
YO2(r)
Глава 40
+ вычислялись по уравнениям A. 65) и (Д. 64а); коэффициенты Ykl в этих уравнениях
приведены в табл. 40.4 вместе с значениями г;шах и /1)ш, они тождественны постоянным YO+ из табл. 40.3,
за исключением величины Y03, полученной из условия A. 59).
Погрешности рассчитанных термодинамических функций до 3000 К обусловлены главным
образом отсутствием экспериментальных данных о постоянных YO+ в состоянии ХХ2+, при более
высоких температурах — приближенной оценкой вклада возбужденных электронных состояний.
Погрешности в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,5, 2, 5 и
8 Дж-К^-моль-1.
Термодинамические функции YO+(r) публикуются впервые.
Константа равновесия реакции YO+(r)+e(r)=Y(r)+O(r) вычислена с использованием
значения Дгй"°@)=134+17 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации молекулы YO
/0(YO)=580±15 кДж.моль.
Это значение было принято на основании измерений потенциала появления YO+ в масс-
спектрометрическом исследовании Лиу и Уолбека [3285]. В более ранней работе Аккермана и
Рауха [1222] была получена существенно меньшая величина E30+50 кДж-моль), которая
представляется заниженной при сравнении ее с потенциалом ионизации атома Y (см. выше).
Принятому значению соответствуют
?LfH°(YO+, г, 0) = 535,691 + 17 кДж.моль~\
D0(YO+) = 733,855 ± 17 кДж • моль я* 61300 + 1400 см.
YO2(r). Термодинамические свойства газообразного диоксида иттрия в стандартном состоянии
при температурах 100—6000 К приведены в табл. 971.
Спектры и структура молекулы YO2 экспериментально не исследовались. Молекулярные
постоянные, использованные для расчета" термодинамических функций YO2 и приведенные
в табл. 40.6, оценены при подготовке справочника. По аналогии со ScO2 было принято, что в основ-
Таблица 40.6. Значения молекулярных постоянных, а также s и рх, принятые для расчета
термодинамических функций YO2, Y2O и Y2O2
Молекула
YO2
Y2O
Y2O2
v2
v.
CM
800
500
1000
450
200
350
780 —
900 —
950 130
400
We-117
Г3 • CM*
— 7,5 -102
— 6,5 -103
1000 2,8 -10*
с
2
2
4
Px
2
1
1
ном электронном состоянии %2А молекула YO2 имеет нелинейную структуру (симметрия C2t).
Приведенное в табл. 40.6 произведение моментов инерции вычислено по структурным параметрам
r(Y—О)=1,80+0,05 А (оценка по величине re(YO)) и -? О—Y—0 = 110+20° (оценка по величине
-$ О—М—О в молекулах диоксидов переходных металлов). Погрешность значения IaIbIc
составляет 5-Ю18 г3-см6. Основные частоты YO2 вычислены в приближении валентного силового
поля в предположении, что для молекулы YO2, так же как для ScO2 и других диоксидов
переходных металлов, приближенно справедливы соотношения /r/&,«0,85, /rr//r=0,l, fjr% : /r«0,15.
Погрешности в значениях \t и v3 оцениваются в 100 см, v2 — в 75 см. Возбужденные
электронные состояния YO2 в справочнике не рассматриваются.
Термодинамические функции YO2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A.10), A. 122)—A. 124), A.128) и
A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета и составляют 3, 5 и 8 Дж-К-моль
в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций YO2(r) публикуется впервые.
Константа равновесия реакции YO2(r)=Y(r)-f-2O(r) вычислена с использованием принятой
в справочнике энтальпии атомизации
154
Яттрий и его соединения Y2O(r), Y2O2(r)
ДГЯ°(О) = 1350 + 90 кДж • моль"*.
Принятое значение было оценено на основании отношения D0(MOa) : D0(MO) = 1,89,
найденного Кордесом и Гингеричем [5000] в результате масс-спектрометрического исследования
равновесия реакций в системах Се—О, Nd—О, Gd—О и Но—О. Принятой величине соответствует
bfH°{YO2, г, 0) = —433,526 + 90 кДж • моль.
Y2O(r). Термодинамические свойства газообразного оксида дииттрия в стандартном состоянии
при температурах 100—6000 К приведены в табл. 972.
Экспериментальные данные о строении и спектрах молекулы Y2O в литературе отсутствуют.
Молекулярные постоянные, использованные для расчета термодинамических функций и
приведенные в табл. 40.6, оценены при подготовке справочника. По аналогии со Sc2O в справочнике
принимается, что в основном электронном состоянии Х1А молекула Y2O имеет нелинейную структуру
{симметрия С2в). Приведенное в таблице произведение моментов инерции вычислено по
структурным параметрам r(Y—О)=1,85+0,1 А, -^ Y—О—Y=145 + 20°, оцененным таким же образом,
как и для Sc2O. Погрешность в значении IaIbIc составляет 5 -Ю~1и г3-см6. Основные частоты Y2O
были получены в приближении валентного силового поля с помощью силовых постоянных,
оцененных так же, как для Sc2O. Погрешности в значениях vx, v2 и v3 оцениваются в 100, 50 и 150 см
соответственно. Возбужденные электронные состояния Y2O в справочнике не рассматриваются.
Термодинамические функции Y2O(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по Сравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и
A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета; они составляют 6, 9 и 12 Дж-К~1-моль в
значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции Y20(r) (значения Ф°(Т) при температурах 1700—2600 К)
вычислялись в работах Сму и др. [4331 ] и Кордеса и Гингерича [5000]. Расхождения данных табл. 972
и расчета [4331] не превышают 12 Дж-К-моль в значениях Ф°(Г). В работе [5000] принято
существенно более низкое значение v2 (81,5 см) и Рх=6, что явилось причиной расхождений,
не превышающих 9 Дж-К-моль~1.
Константа равновесия реакции Y2O(r)=2Y(r)+O(r) вычислена с использованием значения
АгН°@)=1090,599+31 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
&fH°(Y2O, г, 0) = 2 + 30 кДж • моль.
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции
Y2O(r)=YO(r)+Y(K), выполненном Сму и др. [4331]. В работе приведена только энтальпия
реакции (—46+30 кДж-моль). С учетом разности термодинамических функций, использованных
в расчетах [4331] и принятых в настоящем справочнике, было получено близкое значение
(—47+30 кДж-моль), которому и соответствует принятая энтальпия образования.
Y2O2(r). Термодинамические свойства газообразного диоксида дииттрия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 973.
В литературе отсутствуют сведения об исследованиях спектров и структуры молекулы Y2O2.
Молекулярные постоянные, принятые для расчета термодинамических функций и приведенные
в табл. 40.6, оценены при подготовке справочника так же, как для молекулы Sc2O2. По аналогии
с 1Л2О2 и А12О2 принято, что в основном электронном состоянии ХХА молекула Y2O2 имеет
ромбическую структуру (симметрия D.ih). Приведенное в табл. 40.6 произведение моментов инерции
вычислено по структурным параметрам r(Y—О) = 1,95+0,1 А, г(О—О) = 1,5±0,1 А.
Погрешность значения 1А1в1с составляет 1-1013 г3-см6. Основные частоты рассчитаны в приближении
поля валентных сил; силовая постоянная /у_о принята такой же, как в молекулах Y2O и YO2,
остальные постоянные были оценены по постоянным 1Л2О2 (см. [4796]). Погрешности могут
достигать 40 см для v4, 76 см для v2 и v5 и 150 см для vl7 v3 и ve. Возбужденные электронные
состояния Y2O2 не рассматриваются.
Термодинамические функции Y2O2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)-A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Погрешности рассчитанных значений обусловлены неточностью оценки молекулярных
155
Y,O3(k, ж) Глава 40
постоянных и приближенным характером расчета; «они составляют 6, 12 и 18 Дж-К-моль"'1
в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Термодинамические функции Y2O2(r) (значения Ф°(Т) при температурах 1700—2600 К)
вычислялись ранее в работах Сму и др. [4331] и Кордеса и Гингерича [5000]. Расхождения
данных табл. 973 и [4331] не превышают 4 Дж-К-моль~1. В работе [5000] принято существенно
более низкое значение va E4 см) и рх=4, что явилось причиной расхождений, достигающих
24 ДжК^-моль.
Константа равновесия реакции Y2O2(r)=2Y(r)+2O(r) вычислена с использованием значения
АгН°@) = 1889,382+50 кДж-моль", соответствующего принятой в справочнике энтальпии
образования
AfH°(Y2O2, г, 0) = —550 + 50 кДж • моль.
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции
Y2O2(r)+Y(r)=Y2O(r)+YO(r), выполненном Сму и др. [4331]. В работе приведена только
энтальпия этой реакции G5+40 кДж-моль). С учетом разности термодинамических функций,
использованных в расчетах [4331] и принятых в настоящем справочнике, было получено значение
Дг#°@)=84+40 кДж-моль, которому соответствует принятая энтальпия образования.
Y2O3(k, ж). Термодинамические свойства кристаллического и жидкого триоксида дииттрия
в стандартном состоянии при температурах 100—6000 К приведены в табл. 974.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 40.1. В справочнике за стандартное состояние Y2O3(k) принята в интервале 0—2550 К
кубическая модификация kII (С—Y2O3, структурный тип Т12О3 [2245]), а при Т > 2550 К —
гексагональная модификация к1 (Н—Y2O3, [2282]) х.
При Т <[ 298,15 К термодинамические функции Y2O3(k) вычислены для теплоемкости,
полученной Голдстейном и др. [2436] A6—298 К) на образце чистотой 99,9%. Погрешность этих
измерений оценивается в 0,3% при 1">30К. Экстраполяция теплоемкости ниже 16 К
приводит к значению ?°A6 К) = 0,47 Дж-К^-моль. Погрешности принятых по данным [2436] зна-
чений 5°B98,15К) и #°B98,15 К)-Г(О) (см. табл. 40.1) составляют 0,3 Дж-К-моль и
0,04 кДж-моль *.
При Т ^> 298,15 К измерения энтальпии Y2O3(k) проводились Панкрацем и др. [3755] C98—
1799 К', 29 точек, чистота образца 99,99%), Яшвили и др. [1197] C89—1615 К, 13 точек, чистота
образца 99,94%), Фомичевым и др. [4906а] A194—2468 К, 17 точек, чистота образца —99,99%)
и Шпильрайном и др. [4275] A993—2691 К, 17 точек, чистота образца ~99,99%). Данные трех
первых работ согласуются между собой в пределах 1 % (за исключением двух точек, найденных
в работе [4906а] при 1194 и ,1286 К, которые лежат ниже данных [1197, 3755] на 2—4%).
Результаты измерений энтальпии С—Y2O3 в работе Шпильрайна и др. [4275] в интервале 1993—
2379 К систематически завышены в среднем на 12% относительно данных [4906а], причина этих
расхождений не установлена. Принятое в справочнике уравнение для теплоёмкости С—Y2OS
(см. табл. 40.1) получено совместной обработкой данных [1197, 3755, 4906а] 2 с оценкой
погрешности всех этих измерений в 1%. Принятое уравнение описывает экспериментальные измерения
в пределах указанной погрешности, при этом данные [1197] в среднем на 0,7% больше, данные
[3755] — на 0,5% меньше вычисленных по уравнению, а данные [4906aJ равномерно рассеяны
в разные стороны со средним отклонением в 0,8%.
Температура превращения B550 + 70 К) принята в результате усреднения результатов
измерений, проведенных Фоэксом и Траверсом [2283] B520—2570 К), Лопато и др. [633] B620 К),
Жмакиным [425] (перелом на кривой электропроводности Y2O3 при 2520 + 20 К) и Шпильрайном
и др. [4275] B550 К, калориметрические измерения). Температура плавления в последние годы
определялась неоднократно ввиду предполагаемого использования этой величины в качестве
вторичной реперной точки МПТШ. Фоэкс [2280] на основании измерений, выполненных на
образцах чистотой лучше 99,99% в 10 лабораториях разных стран и приведших к значениям в ин-
1 Панкрац и др. [3755] на основании измерений энтальпии Y,OS указали на «незначительную термическую
аномалию» Y2O3 вблизи 1330 К с энтальпией перехода 1,3 кДжмоль. Поскольку природа этой аномалии не
изучена, а существование фазового перехода при 1330 К не подтверждено в последующих исследованиях, этот
переход в справочнике не учитывался. При высоких давлениях C5 кбар) и температурах (823 К) отмечено
образование моноклинной модификации кШ (В—Y2O3) [4975].
При совместной обработке не были учтены точки при 1194 п 1286 К [4906а].
156
Иттрий я его соединения Y2O3(k, ж)
тервале 2682—2741 К, рекомендовал средневзвешенную величину B712 ± 12 К), принятую
в справочнике. Позднее в работе [4307] было получено несколько более высокое значение 2735 +
± 19 К. .
Единственные измерения энтальпии H-Y203 и У203(ж) в интервалах 2609—2691 и 2707—
2919 К проведены Шпильрайном и др. [4275]; по этим данным рассчитаны приведенные в табл. 40.1.
значения теплоемкости A58 + 10 Дж-К^-моль и 190 ±15 Дж-К^-моль соответственно).
Энтальпия превращения E4 + 8 кДж-моль) и энтальпия плавления (81 + 10 кДж-моль)
основаны на измерениях [4275], которые были скорректированы с учетом разницы в энтальпии
С—Y2O3, найденной в работах [4275] и [4906а] 1. Погрешности вычисленных значений Ф°(Т)
при 298,15, 1000, 3000 и 6000 К оцениваются в 0,2, 1,2, 4 и 20 Дж-К^-моль.
Термодинамические функции Y3b3(K), вычисленные в справочнике Барина и Кнакке [1395] (Т ^ 2693 К),
основаны на данных [2436] и [3755] и отличаются от приведенных в табл. 974 на 2,5 Дж-К^-моль
в значении Ф°B700 К). Термодинамические функции У2О3(ж) публикуются впервые.
Константа равновесия реакции Y2O3(k, ж) = 2Y(r) + 30(г) вычислена с использованием
значения &гН°@) = 3482,902 + 7,7 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
&.fH°(Y2Os, к, 298,15 К) = —1905 + 4 кДж . моль.
Принятая величина основана на измеренной Хьюбером и др. [2731] энтальпии сгорания
иттрия в кислороде. Ее погрешность несколько увеличена по сравнению с рекомендацией авторов
измерений, если принять во внимание неточность расчета поправки на примеси.
Имеющиеся в литературе данные по энтальпиям растворения Y(k) в соляной кислоте [1607,
4357] и Y2O3(k) в соляной кислоте [289, 3546J противоречивы и относятся к разным
концентрациям и температурам, что затрудняет их использование для расчетов энтальпии образования
Y2O3(k). Получаемые из этих измерений приближенные величины лежат в интервале от —1823
до —1892 кДж-моль.
Бо и Моррис [1596] методом ЭДС исследовали реакции Y2O3(k) + 3/2С(к) = 2Y(k) + 3/2СО2(г)
A012-1121 К, 8 точек) и Y2O3(k) = 2Y(k) + 3/2O2(r) A018-1118 К, 7 точек). Расчеты для
первой из этих реакций привели к значениям энтальпии образования, равным —1948 + 2 кДж-моль
(II закон) и —1977 ± 2 кДж-моль (III закон). Для второй реакции было получено —2022 +
+ 2 кДж-моль (II закон) и —1995 + 2 кДж-моль (III закон). Эти величины представляются
менее надежными по сравнению с калориметрическими измерениями [2731].
1 Авторы [4275] провели только графическую обработку своих данных и рекомендовали значения AtrH=25 +
+8 кДж-моль и ДтЯ=84+8 кДжмоль, отличающиеся от принятых в справочнике.
Глава 41
ЛАНТАН И ЕГО СОЕДИНЕНИЯ
Ьа(к, ж), La, La+, LaO, LaO+, LaO2, La2O, La2O2, La2O3(K, ж)
В настоящей главе рассматривается лантан и его кислородные соединения — всего 8 веществ..
Для двух веществ приведены данные об их термодинамических свойствах в конденсированном
состоянии, для семи веществ — в газообразном состоянии. Термодинамические свойства четырех
газов (La, La+, LaO, LaO+) рассчитаны до 10 000 К, остальных трех — до 6000 Й.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов лантана (атомный
вес 138,905). Однако различие в молекулярных постоянных индивидуальных изотопомеров этих
веществ в расчетах не учитывалось ввиду пренебрежимо малого влияния этого эффекта на
точность расчета термодинамических свойств соединений лантана.
Для элементарного лантана в Приложении 3.10 приведены сведения о его критических
постоянных.
La2O3 является единственным оксидом лантана, стабильным в конденсированном состоянии
в широком интервале температур; он практически не имеет области отклонения от стехиометри-
ческого состава. В справочнике не рассматриваются многоатомные ионизированные соединения
лантана. Энергии диссоциации соответствующих нейтральных молекул невелики, а их
потенциалы ионизации достаточно большие, поэтому можно предполагать, что в равновесных условиях
такие вещества, как La2O+(r) или LajO^r) не должны присутствовать в значительных
количествах. Более вероятно образование в этих условиях LaO^r), однако отсутствие данных о
величине А0(Ьэ.О2) затрудняет расчет свойств этого газа.
La(K, ж). Термодинамические свойства кристаллическою и жидкого лантана в стандартном
состоянии при температурах 100—5600 К приведены в табл. 975.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 41.1. За стандартное состояние лантана в интервале 0—550 К принята гексагональная
Таб
н
О
а-
ф
РЭ
La
La2G
лица 41.1. ]
1
Состояние
Принятые значения термодинамических величин для
» кристаллическом и жидком состояниях
1
in
(Л о
&з 1
кДж-моль"
кШ, гекс. (а) 6,657
к11, куб. (В) -
к1, куб. (?) —
ж —
з кШ, гекс. (А) 19,84
к11, гекс. (Я) —
к1, куб. (X) -
ж —
К
оо"
щ.
О
Дж • К-1
56,7
127,32
Примечание. C°(T)=a+b-T—c-T~2+d-Ti (в
La: а d=6,438 -Ю"».
оо"
оэ
см
• моль
! 27,11
108,80
Коэффициенть
нения для С
а
26,411
25,521
39,5
32,8
119,604
160
160
200
Дж-К^-моль").
6-10»
2,343
1,384
14,514
лантана
i урав-
°р(Т)
с ¦ 10-е
3,135 а
13,452
и его соединениЁ
Интервал
температур
или
тт
К
298,15-550
550—1134
1134—1193
1193—5600
298,15-2313
2313—2383
2383—2586
2586—6000
550
1134
1193
2313
2383
2586
А1гН
или
кДж-
• моль
0,36
3,12
6,2
0
0
125
158
Лантан и его соединения La (к, ж}
а-модификация, в интервале 550—1134 К кубическая р-модификация (структурный тип Си) и
в интервале 1134—1193 К кубическая f-модификация (структурный тип a-Fe).
Расчет термодинамических функций Ьа(к) при Т < 298,15 К1 основан на измерениях
теплоемкости, выполненных Лоунасмаа и Сандстремом [3312] C—25 К) и Паркинсоном и др. [37741
D,5—180 К), а также на измерениях энтальпии в работе Берга и др. [1488] B98—1373 КJ.
Значения теплоемкости в интервале 180—298,15 К были получены интерполяцией данных [37741
и [1488]. Измерения теплоемкости при Т < 10 К в работах [1512, 2256, 2865, 2866, 2867, 37481
менее надежны и не учитывались.
Уравнение для теплоемкости в интервале 298,15—550 К (см. табл. 41.1) выведено по
результатам измерений энтальпии в работе Берга и др. [1488].
Температура полиморфного (а->-|3) превращения E50 + 40 К) получена усреднением данных
[31, 404, 1488, 2513, 4355, 4358], его энтальпия @,36 + 0,13 кДжчмоль) принята по работе
[1488].
Уравнение для теплоемкости |3-La (см. табл. 41.1) в интервале 550—1134 К основано на
результатах измерений энтальпии, выполненных Бергом и др. [1488]; при обработке погрешность этих
данных оценивалась в 0,3%. Данные по энтальпии, полученные Егером и др. [2809] F23—998 К),
а также по теплоемкости, найденные Мардыкиным и др. [653] и Новиковым и Мардыкиным [7321
A200—1600 К), имеют большие погрешности и при выводе этого уравнения не учитывались.
Температура полиморфного .(Р"->Т) превращения A134 ± 5 К) принята по работам [1488,
4355, 4358]; результаты менее надежных измерений [31, 404, 4354] A123—1153 К) не учитывались.
Энтальпия этого перехода C,12 + 0,13 кДж-моль) получена в работе Берга и др. [1488].
Теплоемкость f-La C9,5 + 1,5 Дж-К^-моль) в интервале 1134—1193 К также рассчитана из
измерений энтальпии, выполненных Бергом и др. [1488].
Температура плавления A193 + 1 К) принята по хорошо согласующимся между собой
данным [1488, 2513, 4358]; менее точные измерения [31, 404, 4551] A203—1158 К) не учитывались.
Энтальпия плавления F,2 ± 0,2 кДж-моль) найдена Бергом и др. [1488].
Теплоемкость жидкого лантана C2,8 + 2 Дж-К^-моль) рассчитана по результатам
измерений энтальпии Ьа(ж), выполненных Стретзом и Баутиста [4412] A250—2420 К); в пределах
погрешности она согласуется с данными Берга и др. [1488] C4,3 Дж-К^-моль, 1193—1375 К).
Измерения Банчилла и Филиппова [63] A200—1800 К), представленные только в виде
графической зависимости Ср(Т), не, учитывались.
Погрешности вычисленных значений Ф°(Г) при 298,15, 2000, 4000 и 5500 К оцениваются в 0,6,
3, 6 и 8 Дж-К^-моль соответственно. Термодинамические функции Ьа(к, ж), приведенные'
в табл. 975 и справочнике Халтгрена и др. [2752] (Т ^4000 К), согласуются в пределах 0,2—
1 Дж-К^-моль в значениях Ф°(Г); расхождения с данными Келли [2960] (Т ^3000 К)
достигают 1,7 Дж-К-моль при 3000 К.
В настоящем справочнике в качестве стандартного состояния лантана принято его
кристаллическое состояние и в соответствии с этим
AfH°(La, к) = 0.
Давление пара лантана в реакции Ьа(к, ж) == La(r) вычислено с использованием значения
Дг#°@) = Д,Я°(Ьа, к, 0) = 430,343 + 6,0 кДж-моль, соответствующего принятой в
справочнике энтальпии сублимации
Д,Я°(Ьа, к, 298,15 К) = 430 + б^кДж . моль.
Результаты расчетов энтальпий сублимации лантана по данным о давлении его насыщенного
пара представлены в табл. 41.2. Принятая величина является средневзвешенной из наиболее
надежных измерений, выполненных Хаберманном и Дааном [2540] и Аккерманом и Раухом [1219].
В работах [1856, 1980, 4353] были использованы менее чистые образцы лантана, что, возможно,
и привело к заниженным значениям энтальпий сублимации. В работе [1147], посвященной
исследованию давления пара лантана, экспериментальные данные не приводятся, авторы отмечают
лишь их хорошее согласие с результатами работы [2540].
1 При Т < 298,15 термодинамические функции Ьа(к) рассчитаны для смеси а-и ^-модификаций, так как
большинство литературных данных получено для такой смеси.
• Ввиду недоступности этой работы полученные в ней данные взяты из справочника Халтгрена [2752].
159
La(r), La+(r)
Глава 41
Таблица 41.2. Результаты определений L,H° (La, к, 298,15 К) (в кДж-моль^1)
Авторы
Метод
А8Н° (La, к, 298,15 К)
II закон
III закон
Спеддинг, Даан, 1954—1955 [1980, Эффузионный, 1640—2000 К, данные представ- 352 422,0+5,0
4353] лены в виде уравнения
Чапка и др., 1956 [1856] Масс-спектрометрический, 1723—1873 К, 4 точки 393 421,1+3,2
Аккерман, Раух, 1962 11219] Эффузионный, 1866—2167 К, 8 точек 430^19 425,9±1,0
Масс-спёктрометрический, 1655—2050 К, две серии 436 ±5 —
Хаберманн, Даан, 1964 [2540] Эффузионный, 1874—2182 К, 40 точек, данные 434 431,0±0,2
представлены в виде уравнения
Приведенные в таблице погрешности учитывают только воспроизводимость измерений.
Неточность термодинамических функций Ьа(к, ж) приводит к дополнительной погрешности в
энтальпии сублимации, достигающей 6 кДж-моль для измерений при 2000 К.
La(r). Термодинамические свойства газообразного лантана в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 976.
Для расчета термодинамических функций использованы уровни г атома La с суммарным
статистическим весом 2952 в интервале 0—42 000 см, т. е. имеющие энергии, меньше энергии
первого потенциала ионизации. Эти уровни соответствуют валентным электронным конфигурациям
...5d6s2 (к ней относится основной уровень 2/)з/г), ...5сР, ...5d26l (I = 0, 1, 2), ...5d6sQp, ...Qs26p,
...4/6s2, ..AfQsQp, ..Afbd&s и 4/5d2. Принятые величины основаны на данных Мартина и др. [3411],
а также на оценках для ряда ненаблюдавшихся электронных состояний. Оцененные уровни имеют
энергию от 26 000 см и выше с суммарным статистическим весом 1988. Погрешность оценки
энергии лежит в пределах 500—4000 см.
Термодинамические функции La(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных значений при Т ^ 5000 К обусловлены главным образом неточностью фундаментальных
постоянных и не превышают 0,02—0,03 Дж-К^-моль. При более высоких температурах
становятся заметными погрешности из-за приближенной оценки ненаблюдавшихся состояний и
пренебрежения ридберговскими состояниями. Соответствующие погрешности могут достигать
0,1 и 0,3 Дж-К^-моль в Ф°(Т) при 6000 и 10 000 К соответственно.
,г^4Ранее термодинамические функции La(r) вычисляли Сталл и Зинке [4421] (до 3000 К),
Кольский и др. [3067] (до 8000 К) и Хилзенрат и др. [2652] (до 10 000 К). В расчете [3067] было
использовано неверное значение R, и этим объясняются расхождения с данными табл. 976 @,06—
0,07 Дж-К^-моль) даже при низких температурах. Расхождения с данными расчета [2652]
несуществены при низких температурах, но возрастают с увеличением температуры, достигая
2,5 Дж-К^-моль в значениях Ф°(Т) вследствие различия в числе учитываемых уровней.
Энтальпия образования
t\fH°(La, г, 0) = 430,343 ± 6,0 кДж. моль
соответствует принятому в справочнике значению энтальпии сублимации лантана.
La+(r). Термодинамические свойства газообразного положительного иона лантана в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 977.
Для расчета термодинамических функций использованы уровни энергии La+ в интервале
0—85 000 см с суммарным статистическим весом 1076 х. Эти уровни соответствуют валентным
электронным конфигурациям ...5d2 (к ней относится основной уровень 3F2), ...6s2, ...6р2, ...4/2,
...6s6Z (I = 1, 2), ...5<Ш (l = 0-^3), ...4/6Z (Z = 0, 1), ...5d4/, ...5d5l (I = 3, 4). Принятые
величины основаны на данных Мартина и др. [3411 ], а также на оценках ряда ненаблюдавшихся
уровней. Энергии состояний 1S(...5di) и 1D(...QsBS)Qd) оценены по аналогии с соответствующими
величинами для Sc+, а уровни, соответствующие конфигурациям ...5d5f, ...5d5g, ...5d5f, — по ме-
1 См. примеч. 1 к тексту по Сг(г).
160
Лантан и его соединения
ЬаО(г)
тодике, изложенной в Приложении 6. Оцененные уровни имеют энергию от 20 000 см х и выше
с суммарным статистическим весом 466. Погрешности оцененных величин лежат в пределах 100—
2000 см.
Термодинамические функции La+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных величин обусловлены главным образом неточностью фундаментальных постоянных и
составляют 0,02—0,03 Дж-К^-моль, за исключением самых высоких температур, для которых
становятся заметными ошибки из-за приближенной оценки ряда уровней и пренебрежения ридбер-
говскими состояниями. Соответствующая погрешность в значении Ф°A0 000 К) может достигать
0,1 Дж-IT1-моль".
Ранее термодинамические функции La+(r) вычислялись Грином и др. [2473] (до 50 000 К) и
Хилзенратом и др. [2652] (до 10 000 К). Результаты этих расчетов практически совпадают с
данными табл. 977 во всем интервале температур.
Константа равновесия реакции La+(r) + е(г) = La(r) вычислена с использованием значения
АгН°@) = —538,092 + 0,060 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации атома лантана
/0(La) = 44 981 ± 5 см = 538,092 + 0,060 кДж-моль.
Это значение рекомендовано Мартином и др. [3411] на основании спектральных данных,
полученных в работе Гартона и Вильсона [4965].
Принятому значению /0(La) соответствует
A/ff°(La+, г, 0) = 968,435 ± 6,0 кДж • моль.
LaO(r). Термодинамические свойства газообразного оксида лантана в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 978.
Молекулярные постоянные, принятые в справочнике на основании анализа результатов
исследований электронного спектра LaO, приведены в табл. 41.3. В электронном спектре LaO
изучены 6 систем полос: АЧ1 -> Х22+ [1275, 2591, 2859, 3475, 4211], 5а2+ ^ Х22+ [201, 1275, 1369,
Таблица 41.3. Молекулярные постоянные LaO и LaO+
Молекула
Состояние
i39Lai6o x2 2+ а
А '2 Д3/
А'2 &*,
А2Щг
-42П5/,
В2 2+
С2П
DB2+)
FB 2+) м
LaO+ X12+а
Примечание. Ниже все
LaO: a ?=2,57 ЛОГ»; б
по соотношению
0 817,26
7 493,4 768,20
8190,1 773,87
12 662,8 762,57
13 525,2 761,60
17 878,88 734,59
22 631,3 3 /792,37
\ 801,14
26 962,2 790,8
28 015,8 857,6
0 850б
"А
3,097 б
2,990в
3,229 Д
2,224
2,235
2,064
2,919 и
3,291 к
1,9
3,6
2,6 в
постоянные даны в см (кроме pt)
@^=4,06 -10; в и>„у
„=2,96 -10
A.69); ж приведено значение ?>0;
* ^=3,5 -Ю"8; м объединенные электронные термы
LaO+: a объединенные электронные термы Г<"
Pi
б оценка; в вычислено по соотношению
Д вычислено по соотношению A. 69); е
20 000
12
в.
СМ
0,3526
1 0,3439
j
\ 0,3470
/
0,3414
1 0,3511
/
0,3643
0,3618
0,36 г
; г ^=2,7.10"
3 Л0=221,44;
Г<<> 30 000
Pi 22
aj-103
1,4
1,6
1,4 е
1,7
1,6
1,9
2,4
1,6 Д
2,6
2,94 г
2,6 ж
2,5»
2,97 *
зд
2,59
3е
8; Д «,„,=1,31 .«Г»
40 000
30
30 000 40 000
24 40
A. 67) и D0(LaO+)=69 000;
вычислено по соотношению
87 -10~2; K
ге
А
1,8257
1,8487
1,840
1,8554
1,8296
1,796
1,802
1,80 б
е вычислено
о)ег/е=2,31 -10;
г вычислено по соотношению A. 38);
A. 68).
11 Заказ MS 1590
1G1
LaO(r) Глава 4f
2591, 2859, 3475, 4428, 4429], СШ ** X22+ [201, 2460, 2461, 2591, 2859, 3475, 4211], ?>22 + -» X22+
[1745, 1746, 2439, 2591, 2859, 3475], F*2+ -> X22 + [1745,^2859, 3475, 4428] и С2П -* A'2 A [2459,
2460, 2591, 4211 ]. Молекула LaO, так же как ScO, должна обладать другими дублетными, а также
квартетными состояниями в области 25 000—40 000 см, которые до настоящего времени не
наблюдались в спектрах. Сильные возмущения уровней состояния Б22 + с и>4 позволяют
предполагать существование в области 21 000 см еще одного состояния 22+. Для учета ненаблюдав-
шихся состояний в табл. 41.3 включены два объединенных электронных терма, статистические
веса которых приняты на основании данных для ScO.
В основном состоянии LaO имеет место большое сверхтонкое расщепление, обусловленное
взаимодействием спина ядра атома La (i = 7/2) со спином электрона. Это расщепление явилось
причиной неправильной интерпретации основного состояния как состояния 42+ в работах [1275,
1744]. Впоследствии вращательная структура переходов В—X и С—X была проанализирована
вновь и было показано [1369, 2461], в согласии с результатами исследования спектра
электронного парамагнитного резонанса молекул LaO, изолированных в матрице при 4 К [2928, 4700],
что основным состоянием молекулы является состояние 22+.
Колебательные постоянные LaO в состояниях Х2Ъ+, А'2 А и С2П приняты по работе Грина
[2460]. Они были определены по началам полос систем С2П—Х22+ (г/, v" <I 10) и С2И—А'2А
(г/, v" <J 5), которые были рассчитаны по кантам полос с использованием значений
вращательных постоянных, найденных авторами работ [1275,2459]. Приведенные в табл. 41.3
вращательные постоянные в состояниях Х22+, АЧ1 и 522+ найдены Акерлиндом [1275] на основании
анализа вращательной структуры полос систем Л2ГЬ,—Х22+ @—0 и 1—0), A2Hij—Х22+ @—0
и 0—1) и ?22+—Х22+ @—0, 1—0 и 0—1). Найденные позднее Грином [2461] значения Во и Da
в состоянии Х22+ хорошо согласуются с принятыми. Вращательные постоянные в состояниях
А'2А ш С2П получены Грином [2459] из анализа структуры полос 0—0 и 1—1 системы
С2П—А'2А. Принятые колебательные постоянные LaO в состояниях А2Н и #22+
рассчитаны Шунвельдом и Сандарамом [4211] по началам полос систем А2П—Х22+ (г/ ^ 13)
и 522+—Х22+ (г/ ^ 10), которые были вычислены ими с помощью вращательных постоянных
по кантам полос, измеренным Маггерсом и Вилером [3475]. Близкие значения колебательных
постоянных для состояния 522+ были получены Суарэ [4428] из постоянных молекулы La18O,
найденных в результате анализа системы полос Z?22+—Х22+ этой молекулы х.
Приведенные в таблице постоянные LaO в состояниях ZJ2+ и F2H+ получены в работах Ка-
ретта и Хударта, которые выполнили анализ колебательной структуры систем ZJ2+ -> Х22+
(г/ ^ 4) и JP2+ -> Х22+ (г/ ^ 5) [1745] и частичный анализ тонкой структуры полос 0—0, 1—0,
1—1 и 2—1 обеих систем [1746].
Термодинамические функции LaO(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QBB и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом 10 электронных состояний (компоненты состояний А'2А и .42П рассматривались как
отдельные состояния) в предположении, что (?<^ гр = 0,5р^^_вр. Величина <?S. вр и ее ПР°"
изводные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по значениям / с помощью уравнений типа A. 82). В
расчете учитывались все уровни энергии с значениями / ^ /ma3t „, последние определялись из
условий A. 81). Колебательно-вращательные уровни состояния Х22+ вычислялись по
уравнениям A. 65), A. 34) и A. 64а), их мультиплетность учитывалась статистическим весом 2.
Коэффициенты в этих уравнениях, приведенные в табл. 41.4, получены с учетом условий A. 50) и
A. 59) по принятым молекулярным постоянным LaO, если не принимать во внимание различие
в постоянных изотопомеров этой молекулы. В той же таблице приведены значения wmax и /ljm.
Погрешности рассчитанных термодинамических функций при температурах до 2000—3000 К
обусловлены главным образом неточностью фундаментальных' постоянных. При более высоких
температурах становятся существенными погрешности из-за приближенного учета вкладов
состояний А'2А и А2П, а также отсутствия экспериментальных данных о состояниях с энергией
выше 28 000 см. Погрешности в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются
в 0,03, 0,05, 0,5 и 2 Дж-К^-моль.
1 Принятые значения вращательных постоянных в пяти электронных состояниях LaO хорошо согласуются с
найденными в 1981 г. Бернаром и Сибаи [1515] в результате совместного анализа тонкой структуры девяти полос,
принадлежащих четырем электронным переходам.
162
Лантан и его соединения
LaO(r)
Таблица 41.4., Коэффициенты в уравнениях, описывающих уровни энергии (в см-1),
а также значения *тах и <J\im , принятые для расчета термодинамических функций
LaO(r) и LaO+(r)
Коэффициенты
LaO,
LaO+,
Коэффициенты
LaO,
LaO+,
Коэффициенты
LaO,
LaO+,
Те
1 20
У
-102
О
8,165
-2,768 25
1, 451 44
О
9,50
-2,6
5V105
У01-10
Уи -Ю3
У Ю'
-8,521 0
3,526 0
—1,40
-2,6
3,6
1,6
3
У*. 1011
'iim
а Возбужденные электронные состояния см. в табл. 41.3.
2,62
136
561
1,12
162
585
Термодинамические функции LaO(r) вычисляли ранее Брюэр и Розенблатт [1666] (Ф°(Т) для
Т < 3000 К), Шнейдер [4197а] (Т = 4000—10 000 К), Вардиа [4591 ] (Qm для Т = 1000—8000 К),
Бернар и Сибаи [1515] (<?„ для Т = 1000-^8000 К) и Сму и др. [4331] (Ф°(Т) для Т'= 1800-f-
-f-2300 К). В работе [1666] приняты неправильные статистический вес основного состояния и
система электронных термов LaO, результаты этого расчета завышены по сравнению с данными
табл. 978 на 6—9 Дж-К^-моль. В расчетах [4197а] и [4591] не был учтен ряд возбужденных
состояний этой молекулы, в том числе состояние А'2А, соответствующие расхождения с данными
табл.978 составляют 2—6 и 6—10 Дж-К^-моль в значениях Ф°(Т) и S°(T).
Таблица 41.5. Результаты определений D0(LaO) (в кДж-моль)
Авторы
Метод
Do(LaO)
II закон
III закон
Чапка и др., 1956 [1856] Масс-спектрометрический, 1/3Ьа(г)+1/3Ьа203(к)= 675 780+13
=LaO(r), 1775—1865 К, 4 точки
Уолш и др., 1960 [4644] Эффузиошшй, Ьа2О3(к)=2ЬаО(г)+О(г), 2230— 875 804+15
2440 К, 3 точки \
Гольдстейн и др., 1961 [2437] Эффузионный, Ьаа03(к)=2Ьа0(г)+0(г), 2234— 851+25 805,6+7,5 '
2441 К, 26 точек <
Масс-спектрометрический, La2O3(K)=2LaO(r)+O(r), 783+25 —
Тср=2140К ;
Сму и др., 1965 [4331] Масс-спектрометрический, YO(r)+La(r)=LaO(r)-f 783+12 794,6+9
-f-Y(r), 1880—2294 К, данные представлены в виде
графика
Эймс и др., 1967 [1310] Масс-спектрометрический, LaO(r)+Y(r)=La(r)-b 811 790,6±8
+УО(г), 1890—2220 К, данные представлены в виде
уравнения
Коппенс и др., 1967 [1923] Масс-спектрометрический, SiO(r)+La(r)=Si(r)+ 785+100 795+11
-f LaO(r), 1877—2088 К, 5 точек
Коппенс и др., 1968 [1924] Масс-спектрометрический, LaO(r)+B(r)=La(r)+ 900+300 786+12
+ВО(г), 2146-2270 К, 5 точек
Бенецех, Фоэкс, 1969 [1470] Протока, La2O3(K)=2LaO(r)+O(r), эксперимен- 811 832
тальные данные не приведены
Аккерман, Раух, 1971 [1222] Масс-спектрометрический, YO(r)+La(r)=Y(r)+ 805 796±8
-f LaO(r), 1783—2184 К, данные представлены
в виде уравнения
Масс-спектрометрический, La2O3(K)=2LaO(r)-|- 713 802,5+9
+О(г), 2258—2427 К, 5 точек, данные
представлены в виде уравнения
Парр, 1974 (см. [3285]) Фотоионизационный 793,7+3,0
Гоул, Чалек,' 1976 [2440] Исследование хемилюминесценции в пересекаю- 790,1 ±4,2 !
щихся пучках La и О2 (NO2, N2O)
163
LaO+(r) Глава 41
Константа равновесия реакции LaO(r) = La(r) + О(г) вычислена с использованием принятого
в справочнике значения
Дг#°@) = D0(LaO) = 796 + 7 кДж • моль« 66 500 + 600 см.
Принятая величина основана на результатах исследований, приведенных в табл. 41.5.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, неточность
термодинамических функций, энтальпий образования и сечений ионизации, использованных в расчетах.
Наиболее точные измерения констант равновесия реакции Ьа2О3(к) = 2LaO(r) + О (г)
выполнены в работах [1222, 2437, 4644], средневзвешенное из полученных по этим данным величин
составляет 804,3 ± 7,0 кДж-моль. Исследования обменных реакций [1222, 1310, 1923, 1924,
4331] привели к хорошо согласующимся между собою значениям, средневзвешенное из них
составляет 793,0 + 7,0 кДж-моль. В работе [3285] со ссылкой на частное сообщение Парра
сообщается, что в результате фотоионизационных измерений для энергии диссоциации LaO им было
найдено значение 794 + 3 кДж -моль, однако отсутствие каких-либо деталей об этих
измерениях снижает степень доверия к этой величине и ее погрешности.
Принятая величина является средневзвешенной из приведенных в табл. 41.5 значений за
исключением результатов работ [1470, 1856, 2440]. Ей соответствует
Д/?°(ЬаО, г, 0)==—118,874 + 9,2 кДж • моль.
LaO+(r). Термодинамические свойства газообразного ионизированного оксида лантана в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 979.
Спектр LaO+ не исследовался, а квантовомеханические расчеты этой молекулы не проводились.
Молекулярные постоянные LaO+, приведенные в табл. 41.3, оценены при подготовке
справочника так же, как для ScO+. Энергии и статистические веса электронных состояний LaO+ приняты
такими же, как у молекулы ScO+ в предположении, что аналогия электронной структуры,
наблюдающаяся в ряду нейтральных молекул ScO—YO—LaO, должна проявляться и у их ионов.
Различие в энергиях Ььа- и о6,-орбиталей, которое можно ожидать у ионов оксидов на основании
спектров ионизированных атомов этих металлов, а также спектров нейтральных оксидов, должно
сказаться только на относительном расположении состояний с энергией возбуждения выше
20 000 см и учитывается оценкой погрешности статистических весов этих состояний E0—60%).
Значения постоянных ш, и г, в состоянии ХгЛ+ были оценены из соответствующих постоянных
молекулы LaO с учетом того, что орбиталь °es, с которой удаляется электрон при ионизации
нейтральной молекулы, является несвязывающей или слегка разрыхляющей. Погрешности принятых
значений оцениваются в 50 см~х и 0,05 А соответственно.
Термодинамические функции LaO+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения <?вя и ее производных находились по уравнениям A. 90)—A. 92) с
учетом трех объединенных электронных термов в предположении, что (?<?>. вр = ptQ?} вр.
Статистическая сумма по колебательно-вращательным уровням энергии состояния Х1^* и ее
производные рассчитывались по уравнениям A. 73)—A. 75) суммированием по колебательным
уровням и интегрированием по значениям / с помощью уравнений типа A. 82). В расчете учитывались
все уровни с значениями / <J /raaif „ последние определялись из условий A. 81). Уровни
энергии состояния Х1Е+ вычислялись по уравнениям A. 65) и A. 64а); коэффициенты Ykl в этих
уравнениях приведены в табл. 41.4 вместе с значениями vm№ и /1]ш, они тождественны постоянным
LaO+ из табл. 41.3, за исключением величины Y03, полученной из условия A. 59).
Пот^еткяиета. доеякятаяяю. та\мкэдкв.шжяйских. ^унющй. до ЗОШ^у. обусловлены главным
образом отсутствием экспериментальных данных о постоянных LaO+ в состоянии X1S+, при
более высоких температурах — приближенной оценкой вклада возбужденных электронных
состояний. Погрешности в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,5, 2, 5,
в 8 Дж-К^-моль'1.
Таблица термодинамических функций LaO+(r) публикуется впервые.
Константа равновесия реакции LaO+(r) + е(г) = La(r) + О(г) вычислена с использованием
значения ДгЯ°@) = 286,908 ± 31 кДж-моль \ соответствующего принятой в справочнике энер-
тии диссоциации
D0(LaO+) = 825 ± 30 кДж -моль « 69 000 ± 2500 см.
164
Лантан и его соединения
LaO2(r), LaaO(r)
Эта величина была оценена на основании предположения, что потенциал ионизации LaO меньше
потенциала ионизации атома La на 30 + 30 кДж-моль, как это следует из данных для оксидов
ряда других металлов, в том числе Се, Рг, Nd. Принятому значению соответствуют
Д/Я°(ЬаО+, г, 0) = 390,218 + 31 кДж • моль,
/0(LaO) = 509,092 + 32 кДж • моль.
Чапка и др. [1856] по потенциалу появления LaO+ в масс-спектре паров над Ьа2О3(к) получили
значение 463 ± 50 кДж -моль, которое представляется слишком низким.
LaO2(r). Термодинамические свойства газообразного диоксида лантана в стандартном состоянии
при температурах 100—6000 К приведены в табл. 980.
Спектры и структура молекулы LaO2 экспериментально не исследовались. Приведенные
в табл. 41.6 и использованные для расчета термодинамических функций молекулярные
постоянные LaO2 были оценены при подготовке справочника. По аналогии со ScO2 принято, что в
основном электронном состоянии Х2А молекула LaO2 имеет нелинейную структуру (симметрия Си).
Таблица 41.6.
Значения молекулярных постоянных, а также о и рх, принятые для расчета
термодинамических функций LaO2, La2O и La2O2
Молекула
LaOg
LaaO
LaaOa
780
450
1000
420
150
300
4
CM"
750
850
900
130
•'б
400
950
[Ah1 С ¦lom
Г3 • CM*
l-10s
1,9 -10*
7,7 -10*
a
2
2
4
Px
2
1
1
Приведенное в табл. 41.6 произведение моментов инерции вычислено по структурным
параметрам r(La—О) = 1,85 + 0,05 А, -$0—La—О = 110 + 20°, оцененным таким же образом, как
и в случае ScO2. Погрешность значения IaIbIc составляет 7-Ю18 г3-смв. Основные частоты LaO2
вычислены по уравнениям валентного силового поля в предположении, что для этой молекулы,
так же как для ScO2 и молекул других диоксидов переходных металлов, приближенно
выполняются соотношения fr/k, ?« 0,85, frrlfr «0,1, fjr2 : /r « 0,15. Погрешности принятых значений
vx и v3 оцениваются в 100 см, v2 — 75 см. Возбужденные электронные состояния LaO2 в
справочнике не рассматриваются.
Термодинамические функции LaO2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и
A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета и составляют 4, 6 и 10 Дж-К^-моль
в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций LaO2(r) публикуется впервые.
Константа равновесия реакции LaO2(r) = La(r) + 2О(г) вычислена с; использованием
принятой в справочнике энтальпии атомизации*
:, Лг#°@) = 1500 ± 90|кДж /моль.
Это значение было оценено из условия D0(MO2) : D0(MO) = 1,89, найденного Кордесом и
Гингеричем [5000] в результате масс-спектрометрического исследования равновесий в системах
Се—О, Nd—О, Gd—О и Но—О. В пределах погрешностей оно совпадает с оценкой, сделанной
авторами этой работы. Принятой величине соответствует
"* ^ г, 0) = —576,091 ±90.кДж.моль.
La2O(r). Термодинамические свойства газообразного оксида дилантана в стандартном состоянии
при температурах 100—6000 К приведены в табл. 981.
Экспериментальные данные о строении и спектрах молекулы La2O в литературе отсутствуют.
Молекулярные постоянные, использованные для расчета термодинамических функций La2O и
165
La2O2(r) Глава 41
приведенные в табл. 41.6, были оценены при подготовке справочника так же, как для Sc2O, и
в предположении, что в основном электронном состоянии ХгА молекула La2O имеет нелинейную
структуру (симметрия С2г). Приведенное в табл. 41.6 произведение моментов инерции вычислено
по структурным параметрам r(La—О) = 1,90 + 0,1 А и -^La—О—La = 145 + 20°.
Погрешность значения IaIbIg составляет 1-1013 г3-см6. Основные частоты La2O вычислены по
уравнениям валентного силового поля при использовании тех же соотношений между силовыми
постоянными, что и для Sc2O. Погрешности в значениях v2, v2 и v3 оцениваются в 100, 50 и 150 см
соответственно. Возбужденные электронные состояния La2O в справочнике не рассматриваются.
Термодинамические функции La2O(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), (i. 122)—A. 124), A. 128)
и A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета и составляют 6, 10 и 15 Дж • К • моль
в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции La2O(r) (значения Ф°(Т) при температурах 1700—2600 К)
вычислялись в работах Сму и др. [4331 ] и Кордеса и Гингерича [5000]. Расхождения данных табл. 981
и расчета [4331] составляют 20 Дж-К^-моль и, по-видимому, объясняются ошибкой в
вычислениях авторов этой работы. Расхождения с данными [5000] составляют 22 Дж • К • моль и
объясняются тем, что в этой работе принято более низкое значение v2F7 см) и рх=6.
Константа равновесия реакции La2O(r) = 2La(r) + О(г) вычислена с использованием
значения /irHo@) = 1154,469 + 32 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
-1
AfH°(La20, г, 0) = —47 + 30 кДж • моль
Принятая величина основана на масс-спектрометрическом исследовании равновесия реакции
La2O(r) = LaO(r) + La(K), выполненном Сму и др. [4331]. В работе сообщена только энтальпия
этой реакции (—113 + 30 кДж-моль).- С учетом разницы в термодинамических функциях,
использованных авторами [4331 ] и принятых в настоящем справочнике, было получено значение
А,Л°@) =—72 +30 кДж-моль, которому соответствует принятая энтальпия образования.
La2O2(r). Термодинамические свойства газообразного диоксида дилантана в стандартном
состоянии при температурах 100—6000 К приведены в табл. 982.
Спектры и структура молекулы La2O2 экспериментально не исследовались. Молекулярные
постоянные La2O2, приведенные в табл. 41.6 и использованные для расчета термодинамических
функций, были оценены при подготовке справочника так же, как для Sc2O2, и в предположении,
что в основном электронном состоянии ХгА эта молекула имеет ромбическую структуру
(симметрия D2h). Приведенное в табл. 41.6 произведение моментов инерции вычислено по структурным
параметрам г (La—О) = 2,0 +0,1 А, г (О—О) = 1,5 + 0,1 А. Погрешность значения IaIbIc
составляет 4-1013 г3-см6. Основные частоты рассчитаны в приближении поля валентных сил;
силовая постоянная /ьа-о принята такой же, как в молекулах La2O и LaO2, остальные постоянные
были оценены по постоянным Li2O2 (см. [4796]). Погрешности принятых значений оцениваются
в 50 см для v4, 100 см для v2, v3, ve и 150 см для vx и vs. Возбужденные электронные
состояния La2O2 в справочнике не рассматриваются.
Термодинамические функции La2O2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A.-3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128)
и A. 130). Погрешности рассчитанных значений обусловлены неточностью принятых
молекулярных постоянных и приближенным характером расчета и составляют 8, 15 и 20 Дж-К^-моль
в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Термодинамические функции La2O2(r) (значения Ф°(Т) при температурах 1700—2600 К) вы
числялись в работах Сму и др. [4331 ] и Кордеса и Гингерича [5000]. Расхождения данных табл. 982
и [4331] достигают 31 Дж-К^-моль; расхождения с данными [5000] составляют 29 Дж-К-1Х
Хмоль и обусловлены тем, что авторы этой работы приняли более низкое значение v4 и рх = 4.
Константа равновесия реакции La2O2(r) = 2La(r) + 2О{г) вычислена с использованием
значения Д^^О) = 1969,252 + 51 кДж-моль, соответствующего принятой в справочнике энталь--
пии образования ч •
АуВ^Ъа^, г, 0) = — 615 + 50 кДж • моль, • : ¦•
166
Лантан и его соединения
Ьа2О3(к, ж)
Это значение основано на масс-спектрометрическом исследовании равновесия реакции La2O2(rL-
+ La(r) = La2O(r) + LaO(r), выполненном Сму и др. [4331]. В работе сообщена только
энтальпия этой реакции D6 + 40 кДж-моль). С учетом разницы в термодинамических функциях,
использованных авторами [4331] и принятых в справочнике, было получено значение Д,.#о@)=
= 19+40 кДж-моль, которому соответствует принятая энтальпия образования.
Ьа2О3(к, ж). Термодинамические свойства кристаллического и жидкого триоксида дилантана
в стандартном состоянии при температурах 100—6000 К приведены в табл. 983.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 41.1. За стандартное состояние Ьа2О3(к) в интервале 0—2313 К в справочнике принимается
гексагональная модификация D-La2O3), при 2313—2383 К — другая гексагональная
модификация G/-La2O3), а при 2383—2586 К — кубическая модификация (Z-La2O3) (см. [1009]).
При Т <^ 298,15 К термодинамические функции Ьа2О3(к) рассчитаны на основании измерений
теплоемкости, проведенных Джастисом и Веструмом [2909] E—346 К) на образце чистотой
99,997%, погрешности измерений оцениваются авторами работы в 0,1% при Т > 25 К, 1% при
10 К и ~5% при 5 К. Экстраполяция теплоемкости ниже 5 К приводит к значению S°E К) =
= 0,01 Дж-К-^моль. Погрешности принятых по [2909] значений 5°B98,15 К) и Я°B98,15 К)—
#°@) (см. табл. 41.1) составляют 0,2 Дж• К • моль и 0,03 кДж-моль соответственно.
Существенно менее точные данные по теплоемкости Ьа2О3(к) получены в работах [2436] A7—300 К)
и [3014] E4—296 К); при Т < 80 К они выше принятых на 1—5%, а при Т > 100 К — ниже
в пределах 0,5—1%.
При Т > 298,15 К для теплоемкости Л-Ьа2О3 принято уравнение (см. табл. 41.1),
полученное совместной обработкой результатов измерений энтальпии, выполненных Кингом и др. [3014]
C99—1797 К), Яшвили и др. [1198] C90—1626 К), Бломеке и Циглером [1581] C84—1172 К) и
измерений теплоемкости в работе Джастиса и Веструма [2909] B98—346 К) г; погрешности
данных [1198, 3014] оценены в 0,5%, данных [1581] — в 1%, а данных [2909] — в 0,2%.
Принятое уравнение описывает исходные данные в пределах указанных погрешностей.
Температуры полиморфных превращений La2O3 B313 + 20 К и 2383 + 20 К) приняты по
данным Ногути и Мизуно [3669] и Фоэкса и др. [2283]. Энтальпии этих переходов
экспериментально не определялись и были приняты равными 0. Теплоемкости двух высокотемпературных
модификаций (H-L&203 и X-La2O3), существующих в сравнительно узких интервалах
температур B313—2383 К и 2383—2586 К), оценены равными 160+15 Дж-К^-моль"". Температура
плавления B586 + 10 К) принята по хорошо согласующимся между собой данным Фоэкса [2279]
B588 К), Сата и Кшора [4147] B581 К), Тресвятского и др. [4540] B583 К) и Кутюра и др. [1947]
B593 К), эти измерения проведены на образцах чистотой 99,9%—99,99%. Менее точные
определения Тт выполнены в работах [693, 3669, 4661]. Энтальпия плавления A25 + 25 кДж-моль^1)
Таблица 41.7. Результаты определений A^jET°(La2O3, к, 298,15 К) (в кДж-моль)
Авторы
Метод
AfH°(La203, к, 298,15 К)
Мутманн, Уэйсс, 1904 [3607]
Кремерс, Стивене, 1923 [3106]
Муз, Парр, 1924 [3554]
Рот и др., 1940 [4072]
Хьюбер, Холли, 1953 [2737]
Матиньон, 1906 [3418]
Вартенберг, 1959 [4655]
Фицгиббон и др., 1965 [2265]
Гвелисиани, Яшвили, 1967 [265]
Измерение энтальпии сгорания лантана в кисло- —1857,7
роде, 2 измерения
То же, 298,15 К, 1 измерение —1912,1
То же, 298,15 К, 4 измерения —1907,1
То же, 294 К, 6 измерений —2255+17
То же, 298,15 К, 5 измерений —1793,1+2,2
Измерения энтальпии растворения Ьа2О3(к) в со- —1789,0
ляной кислоте, 289 К, 2 измерения
То же, 298,15 К, 6 измерений —1799,0+4,2
Измерение энтальпии растворения Ьа2О3(к) и Ьа(к) —1794,2+2,7
в соляной кислоте, 298,15 К, 2 серии опытов, 6 и —1794,8+2,7
5 измерений
То же, 298,15 К, 7 измерений —1797,9+5,4
1 Эти данные вводились в совместную обработку в виде пяти значений Н°(Т)— #°B98,15 К) при Г=310—350 К
(см. [2909]).
167
La2O3(K, ж) Глава 41
оценена, если принять энтропию плавления La2O3 как среднее значение между значениями
энтропии плавления Sc2O3 и суммы htrS и Дте5 для Y2O3 E1 Дж'К^-моль)г. Теплоемкость Ьа2О3(ж)
B00 + 30 Дж-К^-моль) принята равной экспериментальному значению C°(Sc2O3, ж),
измеренному в работе [4275].
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 3000 и 6000 К оцениваются в 0,15,
0,7, 8 и 30 Дж'К^-моль соответственно. Термодинамические функции Ьа2О3(к), вычисленные
в справочнике Барина и Кнакке [1395] (до 1200 К), отличаются от приведенных в табл. 983 на
1 Дж-К^-моль в значении Ф°A200 К). Таблица термодинамических функций Ьа2О3(ж)
публикуется впервые.
Константа равновесия реакции Ьа2О3(к, ж) = 2La(r) + 30(г) вычислена с использованием
значения Дг#°@) = 3388,738 + 12 кДж'Моль, соответствующего принятой в справочнике
энтальпии образования
^fHo{Laa03, к, 298,15 К) = —1794,2 ± 2,0 кДж • моль.
В табл. 41.7 приведены результаты измерений этой величины. При вычислении энтальпии
образования по данным [3418, 4655] было использовано значение энтальпии растворения La
в соляной кислоте, полученное в работе [2265]. Принятое значение является средневзвешенным
из результатов измерений [265, 2265, 2737], совпадающих в пределах их погрешностей.
1 Оцененную таким образом энтальпию плавления La2O3 следует рассматривать'как сумму авталыгаи плавления
и энтальпий полиморфных превращений.
ТЬ(к
Глава 42
ТОРИЙ И ЕГО СОЕДИНЕНИЯ
, ж), Th, Th+, ThO, ThO+, ТЬО2(к, ж), ThO2, ThO+, ThO"
В настоящей главе рассматриваются торий и его кислородные соединения — всего 7 веществ.
Для двух веществ представлены сведения об их термодинамических свойствах в
конденсированном состоянии и для всех веществ — в газообразном состоянии. Для всех газов их
термодинамические свойства рассчитаны до 10 000 К. Для элементарного тория в Приложении 3.10
приведены его критические постоянные.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов тория
(атомный вес 232,0381) и кислорода. Однако различие постоянных разных изотопомеров
соединений тория в расчетах не учитывалось из-за их пренебрежимо малого влияния на вычисляемые
величины. Термодинамические свойства диоксида тория представлены для соединения стехио-
метрического состава. Согласно данным [4913а] область гомогенности этого соединения очень
узка (ТЬ.О1>98—ТЮ2) вплоть до Т ~ 2000 К.
ТЬ(к, ж). Термодинамические функции кристаллического и жидкого тория в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 984.
Значения постоянных, принятые для расчета термодинамических функций ТЬ(к, ж),
приведены в табл. 42.1. За стандартное состояние Th в интервале 0—1650 К принята гранецентриро-
ванная кубическая а-модификация (структурный тип_меди), а в интервале 1650—2023 К —
объемно-центрированная кубическая ^-модификация (структурный тип a-Fe).
Таблица 42.1.
Принятые значения термодинамических величин для тория
в кристаллическом и жидком состояниях
и его соединений
iCTBO
Я
га
Th
ThO,
Состояние
1
?Г
?q °
о 1
к Дж • моль
KlI, куб. (а) 6,35
к1, куб. (Р) -
к, куб. 10,56
ж —
Примечание. Ср(Т)=а+Ъ -Т—е ¦
ThO2
a d=0,743 -Ю-*.
к
ю
.о?
о
со
Дж-К-1
51,83
—
—
65,23
—
T^+d-T1 (в
S.15K)
ОЗ
ь-
• МОЛЬ
26,23
—
—
61,76
—
Коэффициенты в
уравнении для С° (Т)
а
6-103
с • Ю-5
23,435 8,945 —0,114
15,702 11,950 —
46,0 — —
71,567 6,345 10,458 а
110 — —
Дж-К^-моль).
Интервал
температуры
или
Тт
К
298,15—1650
1650—2023
2023-6000
298,15—3623
3623—6000
1650
2023
—
3623
—
или
Дт#
кДж-
• моль 1
3,5
13,8
—
90
—
Наиболее полные измерения теплоемкости ТЬ(к) при Т <[ 298,16 К выполнены Гриффелом
и Скочдополом [2487] A8—300 К, образец, содержавший Ni @,04%), О @,04%), S @,025%),
а также А1, Са, Cd, Fe, Mg, Zn (не более 0,01% каждого) и Накамурой и др. [3629] (80—1000 Кг
метод лазерной калориметрии, образец 99,95% чистоты). Результаты измерений [3629] при 298,15 К
на 4,2% ниже данных [2487], при 80 К они практически совпадают.
Термодинамические функции ТЬ(к) вычислены в интервале 0—1,374 К по измерениям Уол-
кота и Хейна [4752] @,1—1,37 К), в интервале 1,374—4 К по согласующимся между собой дан-
169
ТЪ(к, ж)
Глава 42
ным работ Смита, Уолкота [4328] A,4—20 К), Гордона и др. [2444] (Г < 4 К), Шмидта и Вольфа
[4192] B—11 К); в интервале 4—10 К проведено усреднение данных [4328] и [4192]. Результаты
измерений [4328], [4192] и [2487] при 20 К отличаются приблизительно на 10%, поэтому
значения теплоемкости в интервале 20—35 К получены графической интерполяцией данных этих
трех работ. При 35—80 К использованы данные [2487] и в интервале 80—298,15 К — измерения
авторов [3629]. Погрешности принятых значений 5°B98,15 К) и #°B98,15 К)—Н°ф)
оцениваются в 0,4 Дж• К • моль и 0,020 кДж-моль соответственно.
Уравнение для теплоемкости а-ТЪ в интервале 298,15—1650 К (см. табл. 42.1) выведено
совместной обработкой данных по теплоемкости, полученных Накамурой и др. [3629] (80—1000 К),
и хорошо согласующихся с ними данных по энтальпии, полученных Левинсоном [3243] A269—¦
1650 К). При обработке погрешности измерений в обеих работах оценивались в 0,5%.
Результаты менее точных измерений теплоемкости в работах Егера и Веенстры [2819] C00—1200 К),
Уоллеса [4641] B98—1293 К) и Митькиной [682] C23—973 К) не учитывались.
Температура полиморфного превращения 1650 + 20 К принята в результате усреднения
данных [429, 1834, 1837, 2140, 3464, 4464]; измерения [1008, 1380, 1836, 3441, 3962, 4741]A473-
1683 К) менее надежны и не учитывались. Энтальпия полиморфного превращения C,5 + 0,1 кДж X
Хмоль) рассчитана из уравнений для изменения энтальпии а- и ^-модификаций Th, принятых
в настоящем справочнике.
Уравнение для теплоемкости [3-Th в интервале 1650—2023 К (см. табл. 42.1) выведено из
данных по энтальпии, полученных в работе Левинсона [3243] A667—1991 К).
Температура плавления B023 ± 10 К) принята по измерениям [1224, 1479, 1837, 2140, 3072]
в согласии с рекомендацией справочника [3695]. Менее надежные данные [1008, 3377, 3464] A973—
2115 К) не учитывались. Энтальпия плавления A3,8 +1,3 кДж-моль) рассчитана по
уравнениям для изменения энтальпии жидкого и кристаллического тория.
Теплоемкость Тд(ж) D6,0 + 0,5 Дж-К^-моль) основана на данных по изменению
энтальпии жидкого тория, полученных в работе Левинсона [3243] B069—2100 К).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 3000 и 6000 К оцениваются в 0,3,
0,5, 2,5 и 7 Дж • К • моль соответственно. Te-рмодинамические функции, приведенные в табл. 984
и справочниках МАГАТЭ [3695] (Т ^5100 К) и Халтгрена и др. [2752] (Т< 5500 К), отличаются
при 298,15 К на 1 Дж-К^-моль; при высоких температурах расхождения достигают 3 Дж-К 1Х
Хмоль-1 с [3695] и 7 Дж-IT1-моль с [2752].
В настоящем справочнике в качестве стандартного состояния тория принято кристаллическое
состояние и в соответствии с этим
к) = 0.
Давление пара тория в реакции ТЬ(к, ж) = Th(r) вычислено с использованием значения
krH°@) = &sH°(Th, к, 0) = 602,153 ±6,0 кДж-моль, соответствующего принятой в
справочнике энтальпии сублимации
Д8#°(Тп, к, 298,15 К) = 602+ 6 кДж • моль.
Результаты исследований этой величины представлены в табл. 42.2. Приведенные в таблице
погрешности учитывают только воспроизводимость измерений. Принятое значение основано на
измерениях, выполненных Аккерманом и Раухом [1226]. В более ранних работах были получены
Таблица 42.2. Результаты
Авторы
Цвиккер, 1928 [4832]
определений Ae.ff°(Th, к, 298,15 К) (в кДжмоль)
Метод
Испарение с открытой поверхности, 1667—2500 К а
Голдуотер, Дэнфорс, 1960 [2438] То же, 1700—2100 а
Дарнелл и др., 1960 [1990]
Аккерман, Раух, 1972 [1226]
а Данные представлены в виде
То же, 1757—1956 К, 20 точек
Эффузионный, 2380—2494 К, 7 точек
Масс-спектрометричёский, 2023—2460 К а
уравнения.
Д,#°(Th,
II закон
522
584
573+35
697+40
596+5
к, 298,15 К)
III закон
546 ±42
518+90
583,7+1,0
602,4+2,5
603+10
170
Торий и его соединения Th(r). Th+(r)
заниженные значения энтальпии сублимации, по-видимому, из-за влияния примеси кислорода
или остаточного давления кислорода, которые приводили к образованию ThO(r) и увеличению
летучести тория. Влияние кислорода было изучено в работе Дарнелла и др. [1990] и особенно
тщательно в работе Аккермана и Рауха [1226].
При оценке погрешности принятого значения учтена неточность термодинамических функций,
приводящая к погрешности в энтальпии сублимации около 5 кДж-моль.
Th(r). Термодинамические свойства газообразного тория в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 985.
Использованные в расчете термодинамических функций уровни энергии х атома Th приняты
по данным Залубаса [5073]. В этой работе был выполнен анализ спектра тория в области 2345—
29 662 А и классифицировано около 9500 линий, связанных с переходами между 576 уровнями
энергии в интервале 0—44 438 см с суммарным статистическим весом более 4000. Из этих
уровней только 94 ^с суммарным статистическим весом 670) отнесены к определенным электронным
конфигурациям; для остальных уровней приведены лишь значения /. Всего идентифицировано
10валентных электронных конфигураций: ...6d?7s2 (к ней относится основной уровень8/^), ¦ ¦¦6ds7s,
...5f7s*7p, ...6d7s7p*, ...bjbdlslp, ...5/6d7s2, ...6d7s?7p, ...Qd?7s7p, ...5f6<P7s, ...№7р. Полный
статистический вес всех уровней энергии, соответствующих этим конфигурациям, составляет
немногим более 5000. Согласно оценке Брюэра [4935] имеется еще несколько валентных
электронных конфигураций атома Th, которым должны соответствовать уровни с энергией выше 22 000—
23000 см; число этих уровней велико, но можно предположить, что большей частью они лежат
выше первого потенциала ионизации атома Th. По-видимому, основная часть валентных уровней
энергии, лежащих ниже потенциала ионизации, находится вместе с 94 идентифицированными
уровнями среди 576 термов, приведенных Залубасом [5073]. Возможно, что некоторая часть
этих уровней относится к ридберговским состояниям. На основании оценок Брюэра [4935] и
отнесения, выполненного в [5073], можно полагать, что в области ниже 10 000 см валентные
состояния атома Th определены в [5073] практически полностью.
Термодинамические функции Th(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии атома Th. Погрешности
вычисленных значений определяются неполнотой данных о валентных уровнях атома Th с
энергией выше 10 000 см, при высоких температурах добавляются ошибки из-за пренебрежения
ридберговскими состояниями. Общая погрешность в значениях Ф°(Т) может достигать 0,05, 0,2,
0,5 и 2 Дж-К^-моль при 298,15, 3000, 6000 и 10 000 К соответственно. В значениях S°{T)
погрешности будут в 2—3 раза больше.
Ранее таблицы термодинамических функций Th(r) вычислялись в справочнике МАГАТЭ [3695 ]
(до 6000 К), а также в работах Фебера и Херрика [2227] (до 6000 К) и Поланда и др. [3862] (до
7000 К). Эти расчеты были выполнены на основании сведений об уровнях энергии атома Th,
известных в литературе на время данного расчета. Наибольшее число уровней E23) использовано
в расчете [3695]; расхождения в значениях Ф°F000 К), приведенных в табл. 985 и этой работе,
составляют 0,2 Дж-К^-моль. В наиболее старом расчете [3862] величина Ф°G000 К) отличается
от данных табл. 985 на 4,5 Дж-К^-моль.
Энтальпия образования
bfH°(Th., г, 0)^602,153 +6 кДж-моль-1
¦соответствует принятой в справочнике энтальпии сублимации тория.
Th+(p). Термодинамические свойства газообразного положительного иона тория в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 986.
Использованные для расчета термодинамических функций Th+ уровни энергии х приняты
ло рекомендации Залубаса и Корлисса [4811]. В этой работе был выполнен анализ спектра в
области 2000—25 400 А и классифицировано около 6500 линий, отнесенных к 470 уровням Дона
Th+ в интервале 0—60 000 см с суммарным статистическим весом почти 2500. Эти уровни
отнесены к состояниям (большей частью смешанным), принадлежащим валентным электронным кон-
1 См. примеч. 1 к тексту по Сг (г),
171
ThO(r) Глава 42
фигурациям ...6d7s2, ...&P7*. ...6d3, ...5/27s, ...bflslp, ...5f6d, ...5/6d7s, ...5/7s2, ...5/6d7s, ./\
...6d7s7p, ...7p7s2, и они включают практически все состояния, соответствующие этим
конфигурациям. Основной уровень / = 3/2 соответствует смешанной конфигурации ...6d27s + ...6tf7s2.
Согласно оценке Брюэра [4936] другим валентным электронным конфигурациям соответствуют
состояния Th+ с более высокой энергией. Так, несколько уровней с энергией выше 52 000 см
были отнесены в [4811] предположительно к конфигурациям ...bflp и ...5f7p2.
Термодинамические функции Th+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных значений при Т <[ 5000 К обусловлены главным образом неточностью фундаментальных
постоянных и не превышают 0,02—0,03 Дж-К^-моль в значениях Ф°(Г) и S°(T). При более
высоких температурах возникают ошибки из-за неполноты данных о высоких валентных
электронных состояниях и пренебрежения ридберговскими состояниями. В значениях Ф°(Г) при 6000
и 10 000 К они оцениваются в 0,05 и 1 Дж-К^-моль.
Таблицы термодинамических функций Th+(r) ранее в литературе не публиковались.
Константа равновесия реакции Th+(r) -f- е(г) = Th(r) вычислена на основании значения
Arff°@) = —585 + 12 кДж-моль, соответствующего принятому в настоящем справочнике
потенциалу ионизации атома Th
/0(Th) = 585 + 12 кДж-моль.
Эта величина D9 000 + 1000 см) была оценена Шугаром [4436] интерполяцией
соответствующих спектральных данных для атомов других актинидов. Она хорошо согласуется со значением
580 + 10 кДж-моль, полученным Раухом и Аккерманом [3943] методом электронного удара,
а также с оценкой Меггерса и др. [5017] E0 000 см). Величины, полученные методом
поверхностной ионизации в работах Ионова и Митцева [4874] F70 + 6 кДж-моль) и Смита и Хер-
тела [5048] G20 + 30 кДж-моль), а также из измерений в дуговой плазме в работе Авни и Клейна
[4920] G10 + 30 кДж'Моль), представляются слишком высокими при сравнении с соответствую*-
щими данными для других актинидов.
Принятому значению /0(Th) соответствует
г, 0) = 1187,153 + 13 кДж • моль.
ТЬО(г). Термодинамические свойства газообразного оксида тория в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 987.
Молекулярные постоянные ThO, принятые в справочнике на основании результатов
исследования электронного спектра этой молекулы, ИК-спектра молекул ThO, изолированных в
матрицах инертных газов, а также приближенных оценок, представлены в табл. 42.3. В электронном
спектре ThO проанализировано 12 систем полос1: с3П0+(Л1Е+) -> X1!,*, сЧТ^В1!!) ХХ+
3 +(СШ) -* X1 Е+, Dm -* X1 Е+, Е1Е+ -* X1?+, gT^F1Е+) -> X1Е+ [2149 ],
[2146], WA^Mm) -> Z1E+, &ЩКЧ1) -* ZX2+ [1623], РЩЬШ) -+ Z1E+, ()
+ [1624], /3Ф2(?3Ф2) -> a'A^JSPAi) [2146, 2148, 2149]. В настоящем справочнике, как и
в других работах, для электронных состояний ThO использованы обозначения, принятые для
случая Гунда а, хотя тип связи в этой молекуле должен быть ближе к случаю Гунда с. Согласна
Эдвинссону [2145] последним объясняется то, что в спектре ThO идентифицировано мало
переходов между триплетными состояниями, так как многие из состояний, отнесенных к типу 1П,
вероятно, являются компонентами 3Пг и 3ДХ триплетных состояний. Эти представления
использованы в настоящем справочнике при оценке ненаблюдавшихся электронных состояний ThO.
Основное и нижнее состояния атома Th соответствуют электронным конфигурациям с
заполненной /-оболочкой, и поэтому они аналогичны соответствующим состояниям атомов Ti, Zr и
Ш (см. выше). Состояния атома Th с конфигурацией ...5/6d7s, т. е. изоконфигурационные
основному состоянию атома Се, имеют энергию 7795 см и выше. Это позволяет предполагать, что
молекула ThO наряду с электронными состояниями, аналогичными имеющимся у молекул TiO,
ZrO и НЮ (в том числе основным состоянием ХХЕ+) 2, должна иметь ряд возбужденных состояний,
1 В скобках дана номенклатура электронных состояний ThO, использованная в оригинальных работах.
2 Симметрия основного состояния ThO подтверждается тем, что частота, наблюдавшаяся в ИК-спектре молекул
ThO, изолированных в матрицах инертных газов, хорошо согласуется с постоянными этой молекулы в состоянии
172
Торий и его соединения
ThO(r)
аналогичных известным состояниям молекулы СеО (см. [4916а, 4921а]). На основании этих
соображений были оценены энергии 23 ненаблюдавшихся связанных валентных электронных
состояний (или их компонент) молекулы ThO с энергиями меньше 40 000 см (см. табл. 42.3).
Погрешности оцененных величин составляют 4000—6000 см, однако нельзя исключить также
возможность существования и других состояний ThO в этой области. Результаты изучения спектра ThO
в области 3800—8000 А, представленные в атласе Гаттерера и др. [2374], по-видимому,
подтверждают выполненные в настоящем справочнике оценки 1.
Таблица 42.3. Молекулярные постоянные ThO и ThO+
Молекула
Состояние
232"Т1и1бГ) Y1 У+ а б
дЗ Д (ff& Д }
с3П0+ (А12+)
С3П! (ВЧ1)
?41 (?>Ч1)
Я1 Е+ (?12+)
/Зф2 (б8ф2)
Я3П0+ (.F1 2+)
g8]^ (/41)
/^ (ЛТП)
G4I (ЯЧ1)
/41 (ЛЧ1)
г3П (ЛГЧ1)
232ThleO+ X2 2+ а
Примечание. Все постоянные
те
со
Ш X
<7j • 103
De ¦ 107
см
0
5 321
10 625,5
11 155,57
14 520,36
15 974,53
16 353,60
18 037,9
18 337,56 в
19 586,29
21 752,22
22 683,50
24 881,5
27 754,7
0
п;аны в см.
895,77
864 г
846,4
842,8
835,1
839,2
829,26
816
—
800,85
861,41
800
847,7
823
940 б
2,39
2,4
2,44
2,18
2,39
2,50
2,30
2,4
—
1,47
5,27
2,27
5,77
1,74
Зв
ThO: а в скобках приведены обозначения состояний, принятые в j
состояний:
Состояние а3 Д2
Те 6800
Состояние ЯФ, /3Ф,
Г, 21 500
а8 Д3 Ь3Ф
3300 10 000
зд, 1Д, зг
24 000
А1^
10 500
12+
27 000
сЗд
11700
Зф 1ф
30 000
0,332 644 1,302
0,326 427 1,26
0,323 044 1,294
0,324 306 1,294
0,322 036 1,285
0,323 620 1,33
0,323 090 1,303
0,318191 1,28
0,321 397 в —
0,329 678 в 1,87
0,325 806 1,394
0,318 639 1,28
0,325 531 2,44
0,320 5 1,0
0,35 1,5 г
1,833 в
1,864 в
1,866 в
1,912 в
1,902
1,924 в
1,990 в
1,936 в
2,042 в
2,289 в
2,055 в
2,016 в
2,766
1,96 г
1,9 Л
г
А
1,8403
1,8578
1,8675
1,8638
1,8704
1,8658
1,8673
1,8816
1,872 а
1,8486
1,8595
1,8803
1,8603
1,8749
1,79 б
гатературе; " оцененные энергии возбужденных
ВгФ й3Д <
12 000 13 000 15
1П 12+ зд з
32 000 36
в приведено значение постоянной для v=0; г вычислено по соотношению A. 68).
ThO+: a Состояние А2Ф
Те 9000
В2 Д С2П
12 000 23 000
Dm
31000
?2 2+
38 000
б оценка; в вычислено по соотношению A. 67) и значению Е
Д вычислено по соотношению A. 68).
;1Д /3Ф3
000 20 00(
п, in
D00
s П
) 2100
„=72 000; г вычислено по соотношению A. 69);
Молекулярные постоянные ThO в состояниях Хх?+, с3П0+, с3!!^ es2+, D4\, ЕХ1,+, ^3П0+,
приведенные в табл. 42.3, найдены Эдвинссоном и др. [2149] на основании анализа вращательной
структуры полос соответствующих переходов (для состояний X12+ и eT^ v <^ 2, для состояния
Е1!,* v ^ 3, для остальных состояний v ^ 1). Поскольку в состоянии #3П0+ при v = 1
наблюдаются возмущения, для этого состояния в таблице приведены постоянные для v = 0. Принятые
3 /3Ф %U Э [2146]
д ущ, р р
постоянные в состояниях а3дх, /3Ф2 и g%U1 получены Эдвинссоном и др. [2146], которые
выпол3^XX+ A " ^ 2)
р р
нили анализ вращательной структуры полос системы g3^—XXE+ (v1, v" <^ 2) и полосы 2—2
системы /3Ф2—а3Дц а также использовали данные работы [2149], где были проанализированы
полосы 0—0 и 1—1 системы /—а. Эдвинссон и др. [2146] показали, что уровни v — 0,. 1 и 3 состоя-
1 Предложенное Вентинком и Спиндлером [4704] отнесение к ThO некоторых групп полос, приведенных в этом
атласе, представляется сомнительным.
173
ThO(r)
Глава 42
ния g3Hi возмущены колебательными уровнями v = 2, 3 и 4 верхнего состояния системы /—а.
Хотя авторы [2146] отнесли систему/—а к переходу ХА—ХФ, такое отнесение противоречит
наблюдавшемуся соотношению интенсивностей в Л- и Р-ветвях. Позднее Эдвинссон [2145] отнес
эту систему к переходу 3Ф2—3ДХ, что находится в хорошем согласии со всеми экспериментальными
данными и принимается в настоящем справочнике. Молекулярные постоянные в состояниях (ЭТ!
и h3Ax приняты по работе Борнстедта и Эдвинссона [1623], в которой был выполнен анализ
вращательной структуры четырех полос системы С?1!!—ХХЕ+ (v', v" ^ 1) и четырех полос системы
/13Д2—Х1!4" (г/ ^ 1, v" ^2), в состояниях /41 и ?3П — по работе Борнстедта и др. [1624],
которые исследовали вращательную структуру полос 0—0 и 1—1 систем /41—Х1Б+ и i3!^—Х1^.
Во всех четырех состояниях имеют место заметные возмущения.
Термодинамические функции ТЪО(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом 35 возбужденных электронных состояний (компоненты ряда триплетных состояний
учитывались как отдельные состояния, при этом для состояний с3П0+ и ?3П0+ статистический вес
привр
и ее
произнимался равным 2) и в предположении, что QHI, вр = PiQml.sr Величина р
водные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по значениям /с помощью уравнений типа A. 82). В
расчете учитывались все уровни энергии с значениями / ^ /тах е, где /тах е определялись из
условий A. 81). Колебательно-вращательные уровни состояния Х*2 + вычислялись по
уравнениям A. 65), A. 34), A. 64а); значения коэффициентов в этих уравнениях, полученные с учетом
условий A. 50) и A. 59), а также величин vm&% и JUm приведены в табл. 42.4. Вырождение и муль-
типлетность возбужденных состояний учитывались статистическими весами.
Таблица 42.4.
Коэффициенты в уравнениях,
а также значения г?тах и J^i
ThO(r) и ThO+(r)
описывающих уровни энергии (в см~1),
, принятые для расчета термодинамических функций
Коэффициенты
Те
У10-10г»
Узо-Ю3
Уи-10
ThO, ХхЕ+а
ThO+, Х22+а
0 0
8,946 9,40
—1,845 36 —3,00
—5,898 —
3,326 44 3,50
а Энергии возбужденных состояний см. в табл. 42.3.
Коэффициенты
Уц-10»
^оз-Ю14
¦^Ит
ThO, Ал12+а
—1,302
—1,833
-4,12
143
633
ThO+, Х22+а
-1,5
-1,9
—10,18
156
608
Колебательно-вращательные уровни молекулы ThO в состоянии ХХ2+ при небольших
значениях квантового числа v хорошо описываются постоянными, приведенными в табл. 42.3.
Поэтому погрешности рассчитанных термодинамических функций при температурах до 500—1000 К
обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах становятся существенными ошибки из-за отсутствия экспериментальных данных
о большом числе возбужденных электронных состояний этой молекулы. Погрешности в
значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются в 0,03, 0,2, 2 и 4 Дж-К^-моль.
Термодинамические функции ThO(r) ранее вычислялись Абрамовицем и др. [1204а], а также
в работах Брюэра и Розенблатта [1666] (Ф°(Г) для Т < 3000 К), Шнейдера [4197а] D000<
< Т < 10 000 К) и Рэнда [3916] (Т < 3000 К). В расчетах [1204а, 4197а] учитывалось
существенно меньшее число возбужденных состояний, чем в настоящем справочнике.
Соответствующие расхождения с данными табл. 987 в значениях Ф°(Т) и S°{T) растут с температурой и
достигают 6 и 13 Дж-К^-моль соответственно при 6000 К. В работе [1666] была принята
ошибочная систематика электронных состояний молекулы ThO и получены значения, которые на
14—23 Дж-К^-моль превышают приведенные в табл. 987.
Константа равновесия реакции ThO(r) = Th(r) + О(г) вычислена с использованием
принятой в справочнике энергии диссоциации
ДгЯ°@) = D0(ThO) == 868 + 10 кДж • моль-1« 72 600 + 800 см.
174
Торий и его соединения
ТЬО+(г)
Результаты измерений энергии диссоциации ThO представлены в табл. 42.5. Приведенные
в таблице погрешности учитывают воспроизводимость измерений, неточность
термодинамических функций, термохимических величин и сечений ионизации. Результаты измерений, за
исключением работы [1182], где могли появиться ошибки вследствие неточного определения
парциального давления кислорода, находятся в удовлетворительном согласии. Принятая величина
является средневзвешенной. При ее вычислении несколько больший вес придавался
исследованиям обменных реакций с YO, SiO и LaO.
Таблица 42.5. Результаты определений D0(ThO) (в кДж • моль)
Авторы
Метод
Do (ThO)
II закон
III закон
Аккермйн и др., 1963 [1238] Эффузионный, 1/2ТЬО2(к)+1/2ТЬ(ж)=ТЬО(г), 2337— — 873+20
2381 К, 5 точек
Щукарев, Семенов, 1965 [1182] Масс-спектрометрический, ТЬО2(к)=ТЬО(г)+О(г), 1008 931
2573-2973 К а
Аккерман, Раух, 1973 [1227] Эффузионный, 1/2ТЬО2(к)+1/2ТЬО(ж)=ТЬО(г), 2080— — 860+22
2214 К, 7 точек
Масс-спектрометрический, 1/2ТЬО2(к)+1/2ТЬС(ж)= 872+10 873+10
=ТЪО(г), 2020-2420 К а
То же, Тп02(к)=ТЬО(г)+О(г), 2400—2800 К а 875 874+15
То же, YO(r)+Th(r)=ThO(r)+Y(r), 1930—2280 К а 881+.12 876+12
Хилденбранд, Мурад, 1974 [2649] То же, 1/2ТЬО2(к)+1/2ТЬ(к)=ТЬО(г), 1782-1940 К, 848±20 867+10,
i4i „ 10 точек
То же, ThO(r)+Si(r)=Th(r)+SiO(r), 2064—2212 К, 835±90 871 ±12
12 точек
Нойберт, Змбов, 1974 [3643] То же, ThO(r)+ La(r)=Th(r)+LaO(r), 1759—1961 К, 864±40 860±10
18 точек
а Данные представлены в виде уравнения.
Принятой энергии диссоциации соответствует
, г, 0) = —19,064 + 12 кДж • моль.
ТЬО+(г). Термодинамические свойства газообразного положительного иона оксида тория
в стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 988.
Спектр молекулы ТЪО+ не исследовался, а ее квантовомеханические расчеты не проводились.
Постоянные, приведенные в табл. 42.3, получены при подготовке справочника на основании
приближенных оценок и известных данных для сходных молекул. В спектрах молекул, имеющих,
так же как ThO+, девять электронов на внешней оболочке (ScO, YO, LaO, см. выше, YF+ [4260],
LaF+ [4259], TiN [2102, 2128], ZrN [468, 1424]), наблюдались переходы, связанные с
состояниями а2+(ом), 2Д(&(Я_1)Й), an(v)' 2S+(%)- кРоме тог°. в спектре LaF+ изучен переход,
связанный с состоянием 2Ф(у4/)' Аналогичные пять состояний, а также состояние 2П(тсм)
включены в табл. 42.3.
Энергии состояний X22+(o7s), Л2Ф(срБ/), B2A(\d), D2U(n,p) (см. табл. 42.3) были оценены по
разностям энергий атомных орбиталей 5/ и 7s, Ы и 7s, 1р и 7s иона Th+, найденным по термам,
принадлежащим конфигурациям ...6d27s, ...5/6d2, ...6d27p, ...6d7s2, ...5/7s2, ...7s27/>, ...5f7s, ...bfU,
...5/7s7p, ...5/6d7s, ...5f6d7p, ...Qdlslp. Средние энергии этих конфигураций определялись по
гистограмме, приведенной в работе [4811]. На основании этих оценок за основное состояние
ThO+ было принято состояние 22+(о,8). Энергия состояния С2П(тс6(г), приведенная в таблице,
рассчитана по разносги энергий состояний 8П, 41 конфигурации ...оЯ1Г1г(я_1)й и состояний Зд,
ХД конфигурации •••ав88(я_1, л в молекулах TiO и ZrO, а также оцененной энергии состояния В2А.
Разность энергий состояний i?22+(a7p) и 2JП(л7р) принята такой же, как у молекул TiN и ZrN.
Предполагается, что погрешности оцененных энергий составляют 4000—6000 см.
175
ТЪО2(к, ж) Глава 42
Молекулярные постоянные ThO+ в состоянии Х22 + были оценены в предположении, что их
отношение к аналогичным постоянным ThO в состоянии ХХ2+ такое же, как отношение
постоянных LaF+ и LaF, а также YF+ и YF в состояниях 2Д и а3Д соответственно. Погрешности
принятых значений шв и Ве составляют 50 и 0,02 см.
Термодинамические функции ThO+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения <?вн и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом пяти возбужденных состояний в предположении, что (?{С2. вр = 0,5р^^]вр.
Статистическая сумма по колебательно-вращательным уровням энергии состояния Х22+ и ее
производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по / с помощью уравнений типа A. 82). В расчете
учитывались все уровни с/^/шах_ „, последние определялись из условий A.81). Колебательно-
вращательные уровни состояния Ха2+ вычислялись по уравнениям A. 65), A. 34), A. 64а).
Значения коэффициентов в этих уравнениях, тождественные постоянным из табл. 42.3, а также
величины ушх и /Iim приведены в табл. 42.4. Мультиплетность уровней состояния Х22+
учитывалась статистическим весом 2.
Погрешности рассчитанных термодинамических функций ThO+(r) при низких температурах
обусловлены главным образом неточностью оценки вращательной постоянной (до 0,5 Дж-К^Х
Хмоль в Ф°(Г) и S°(T)). Выше 1000 К становятся заметными погрешности из-за отсутствия
достоверных данных о числе и энергии возбужденных состояний ThO+. Погрешности в
значениях Ф°(Г) при 3000, 6000 и 10 000 К составляют 3, 5 и 8 Дж-К^-моль.
Таблица термодинамических функций ThO+(r) публикуется впервые.
Константа равновесия реакции ThO+(r) + е(г) = Th(r) + О(г) вычислена с использованием
значения Дг/Р@) = 278,0 + 21 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0(ThO) = 590 ± 20 кДж-моль-1.
Это значение принято' на основании масс-спектрометрического исследования методом
электронного удара, выполненного Раухом и Аккерманом [3943] E89 + 10 кДж-моль); оно
хорошо согласуется с найденным Хилденбрандом и Мурадом [2649] E79 + 10 кДж-моль) в
результате масс-спектрометрического изучения системы Th—Si—О. Его погрешность оценивалась
с учетом возможного влияния теплового возбуждения и немоноэнергетичности электронов.
Принятому значению /0(ThO) соответствуют
AfH°(ThO+, г, 0) = 570,936 ± 23 кДж • моль,
D0(ThQ+) = 863 ± 25 кДж • моль ж 72 000 ± 2000 см.
ТЪО2(к, ж). Термодинамические свойства кристаллического и жидкого диоксида тория в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 989.
Значения постоянных, использованные для расчета термодинамических функций ТЬОг(к, ж),
приведены в табл. 42.1. В справочнике за стандартное состояние ТЬО2(к) в интервале 0—3623 К
принимается кубическая модификация (структурный тип флюорита) [4315]. При Т <[ 298,15 К
термодинамические функции вычислены по результатам измерений теплоемкости, выполненных
Осборном и Веструмом [3728] A0—305 К, содержание тория 87,93—87,54%, погрешности
измерений 3% при 10 К, 1% при 14 К и 0,1% выше 25 К). Экстраполяция ниже 10 К была
выполнена по уравнению Дебая и дала S°A0 К) = 0,046 Дж -К-моль. Принятые значения 5°B98,15 К)
и Я°B98,15 К)—#°@) (см. табл. 42.1) имеют погрешности 0,2 Дж-К^-моль и 0,02 кДж-моль
соответственно. В работе Хюттига и др. [2762] приведены результаты измерений средней
теплоемкости ThO2 в интервалах 85—196, 196—271 и 276—373 К.
При Т > 298,15 К для теплоемкости ТпО2(к) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии в работах Саусарда [4349] E23—1787 К, чистота
образца 99,4%), Хоха и Джонстона [2666] A456—2753 К) и Виктора и Дугласа [4610] C23-
1173 К, чистота образца 99,95%). При обработке погрешность данных [4610] оценивалась в 0,3%,
данных [4349] —в 0,5% и данных [2666] —в 1%. Измерения энтальпии, выполненные ранее
Егером и Веенстрой [2819] F71—1664 К, чистота образца не указана), не принимались во
внимание ввиду существенных отклонений от результатов более точных работ [2666, 4349, 4610].
176
Торий и его соединения
ТЬО2(к, ж)
Температура плавления C623 ± 100 К) принята по работам Ламбертсона и др. [3172] C573 +
± 100 К), Бенца [1476] C663 + 100 К) и Чикаллы и др. [1831] C573 К); в работах [2380, 4087,
4662] были получены значения 3323 + 25, 3323 и 3803 К соответственно. Энтальпия плавления
(90 ± 10 кДж-молгГ1), приведенная в табл. 42.1, найдена Ламбертсоном и др. [3172] на
основании изучения равновесия в системе UO2—ThO2 (чистота ThO2 — 99,99%). Значение AmH°(UO2),
рассчитанное авторами [3172], хорошо согласуется с принятым в настоящем справочнике. В
работе Оты и Саты [3705], изучавших систему BeO—ThO2 методами ДТА и рентгенографии,
приведена оцененная величина Дт#°(ТпО2) = 61,5 + 9,2 кДж-моль.
Сведения о теплоемкости жидкого ТЮ2 в литературе отсутствуют. В справочнике принято
оцененное значение 110 + 15 Дж-К^-моль.
Погрешности вычисленных значений Ф°G7) при 298,15, 2000, 3000 и 6000 К оцениваются в 0,15,
1, 2 и 11 Дж-К^-моль соответственно. Расхождения между термодинамическими функциями
T1iO3(k, ж), приведенными в табл. 989 и в справочниках [1395, 2960], не превышают 0,4 Дж-К-1Х
Хмоль в значениях Ф°(Т) во всем интервале температур.
Таблица 42.6. Результаты определений ХуН° (ThO2, к, 298,15 К) (в кДж • моль)
Авторы
Вартенберг, 1909 [4653]
Шавеньо, 1911 [1821]
Микстер, 1914 [3526]
Рот, Беккер, 1931 [4058, 4063]
Хьюбер и др., 1952 [2742]
Смирнов, Ивановский, 1957 [4901]
Метод
Калориметрический, сожжение тория в
кислороде, 293 К
То же
То же
То же, 5 опытов
То же, 298,15 К, 16 опытов
ЭДС, ТпО2(к)+С(к)+2С12(г)=ТпС14(ж)+СО2(г),
928—1233 К, данные представлены в виде
уравнения
AjH° (ThO2, к,298,15 К)
II закон
III закон
— 1364
—1380,7
—1347,2
—1224,2+5,5
—1226,4±3,5
—1265 —1128±20
Принятая в справочнике энтальпия образования
^/H0{^:Ю!i, к, 298,15 К) = —1226,4 + 3,5 кДж-моль-1
основана на измерениях, представленных в табл. 42.6. Наиболее точные измерения были
проведены Хьюбером и др. [2742]; совпадающее в пределах погрешности, но менее точное значение, было
получено Ротом и Беккером [4058, 4063]. Исследования равновесий с участием ТЬС14(ж) [4901]
при расчете по уравнению III закона приводят к величинам, имеющим сильный температурный
ход, что, видимо, свидетельствует о систематических погрешностях в этих измерениях. Принятая
величина совпадает с рекомендацией КО ДАТА—MCHG [1887].
Давление пара в реакции ТпО2(к,ж)=ТпО2(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Д,Я°(ТпО2, к, 0) = 770 + 15 кДж • моль.
Результаты определений энтальпии сублимации ТЮ2(к) представлены в табл. 42.7.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, неточность
термодинамических функций, термохимических величин и сечений ионизации. Как видно из таблицы, за
исключением выпадающей величины, вычисленной по данным [2665], результаты всех остальных измерений
находятся в хорошем соответствии. Обработка эффузионных измерений [1238, 1988, 4939]
проводилась в предположении, что в состав пара входит только ТЮ2. Однако, как показали Аккерман
и Раух [1227], отношение общего давления над ThO2 (включая ТЮ и О) к давлению ThO2
составляет при 2500 К около 1,3. Учет образования ТЮ в указанных выше эффузионных измерениях
увеличивает вычисленные энтальпии сублимации на 5—6 кДж-моль, что приводит к еще
лучшему согласию приведенных в таблице данных. Принятое значение является средневзвешенным,
причем для эффузионных измерений учитывался сложный состав пара.
177
ThO2(r)
Глава tt
Таблица 42.7. Результаты определений AeJ5T0 (ThOa, к, 0) (в кДж • моль)
Авторы
Метод
ASH° (ThO2, к, 0)
II закон
III закон
Шапиро, 1952 [4249] Испарение с открытой поверхности, ТЬО2(к)= 750 772+15
=ТЪО2(г), 2062—2257 К а
Хох, Джонстон, 1954 [2665] Эффузионный, ТпО2(к)=ТпО2(г), 2389—2676 К, 684±50 715+15
15 точек
Дарнелл, Мак-Коллам, 1961 [1988] То же, ТЬО2(к)=ТпО2(г), 2268—2593 К, 13 точек 729+50 766+12
Аккерман и др., 1963 [1238] То же, ТЬО2(к)=ТЬО2(г), 2180—2871 К, 13 точек 748±25 |764+15
Воронов и др., 1963 [248] Испарение с открытой поверхности, ТпО,(к)= — 746+40
=ТЬО2(г), 1803-2233 К а
Щукарев, Семенов, 1965 [1182] Масс-спектрометрический, ТпО„(к)=ТЬО2(г), " 736 780+16
2573-2773 К а
Аккерман, Раух, 1973 [1227] То же, ТЬО2(к)=ТЬО2(г), 2400—2800 К а, исполь- 726 770+16
зованы данные работы [1238]
Хилденбранд, Мурад, 1974 [2649] То же, ThO2(r)+Th(r)=2ThO(r), 2160—2176 К, — 780+25
3 точки
Аккерман и др., 1975 [1234] То же, ThO(r)+ZrO2(r)=ZrO(r)-fThO2(r), 2300— 771 780+25
2700 К а
Белов, Семенов, 1980 [76] То же, ТЬ02(к)=Тп02(г), 2480—2860 К а 683 —
Кайерс и др., 1980 [4939] Эффузионный, ТЬО2(к)=ТЬО2(г), 2057—2421 Ка 815 758+20
а Данные представлены в виде уравнения.
ThO2(r). Термодинамические свойства газообразного диоксида тория в стандартном состоянии
при температурах от 100 до 10 000 К приведены в табл. 990.
Молекулярные постоянные ThOa использованные для расчета термодинамических функций,
приведены в табл. 42.8. На основании результатов исследования ИК-спектров молекул ТЮ2,
изолированных в аргоновой матрице [2355, 3269], а также отклонения молекулярного пучка
в электрическом поле [2935 ], в справочнике принимается для ThO2 нелинейная структура
(симметрия C2v). Приведенное в табл. 42.8 произведение моментов инерции вычислено для значений
r(Th—О)=1,82+0,05 А (оценка по величине гДТЬО)) и -$О—Th—0=122,5+10° (найден Габель-
ником и др. [2355] из изотопного сдвига частоты v3 в ИК-спектрах молекул Th16O2 и Th18O2,
изолированных в матрице из Аг). Погрешность значения IaIsIc составляет 3-10~11Б г3-см6. Частоты v1
и v3, приведенные в табл. 42.8, найдены в ИК-спектрах молекул Th16O2, изолированных в матрице
из Аг [2355]. Их погрешности с учетом матричного сдвига оцениваются в 15 см. Значение
частоты v2 принято на основании расчета в приближении валентного силового поля в предположении,
что /а/г2 : /г=0,04, как это имеет место в случае UO2. Погрешность этого значения оценивается
в 50 см.
Таблица 42.8. Значения молекулярных постоянных, а также о и
термодинамических функций ThO2, ThOJ и
принятые для расчета
Молекула
CM
Г3 • CM6
G
Px
ThO2a 787 220 735 8,3 -103 2 1
ThOJa 750 200 700 1-103 2 2
ThO2a 750 200 700 1-103 2 2
a Ниже приведены энергии Т% (в см) и статистические веса р{ возбужденных электронных состояний.
ThO2: 20 000 B), 21 000 B), 22 000 B), 23 000 B), 26 000 B), 28 000 B)
ThOt: 20 000 C), 25 000 C)
ThO2:: 2000 B), 5000 B), 7000 B)
178
Торий и его соединения
Экспериментальные данные об электронном спектре ThO2 отсутствуют. Молекула ThO2 имеет
заполненную электронную оболочку и вряд ли может иметь низколежащие возбужденные
состояния. Поскольку ионы ТЪ4+ и U6+ имеют одинаковую электронную конфигурацию, в табл. 42.8
для ThO2 приведены такие же электронные состояния, как для молекулы U03.
Термодинамические функции ThO2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124),A. 128),
A. 130) с учетом шести возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью оценки молекулярных постоянных и
приближенным характером расчета; они составляют 2, 5, 8 и 12 Дж-К~1-моль в значениях Ф°(Т) при
298,15, 3000, 6000 и 10 000 К.
Таблицы термодинамических функций ТЮ2(г) вычислялись ранее в работах Абрамовица
[1200] и Грина [2463] (Г < 6000 К). Отдельные значения Ф'(Т) для Т < 3000 К вычислены в
работах [1665, 2649]. Расхождения данных [2463] и табл. 990 растут с температурой и достигают
6 Дж-К~1-моль~1 в значении ФсF000 К), они обусловлены различием в оценке возбужденных
состояний ThO2. Расхождения с данными [1200], [2649] и [1665] достигают 8, 11 и 16 Дж-К"хХ
Хмоль в значениях Ф°(Т) соответственно, они объясняются пренебрежением возбужденными
состояниями и отличием в принятых структурных параметрах и частоте v2. Рэнд [3916] рассчитал
термодинамические функции ThO2(r) для различных моделей этой молекулы и указал на
значительное расхождение результатов расчета с данными по испарению, а также плохое согласие
расчетов по второму и третьему законам. Удовлетворительное согласие достигалось только для
линейной модели, которая противоречит результатам спектральных исследований [2355, 3269].
В настоящем справочнике это противоречие практически устранено. ^3
Константа равновесия реакции ThO2(r)=Th(r)+2O(r) вычислена с использованием значения
ДГЯ°(О)=1547,647+ 16 кДж-моль, соответствующего принятой в справочнике^энтальпии
образования и сублимации ТЬО2(к). Этим величинам также соответствует
ДуЯо(Тп02, г. 0) = —451,928 ±И5|кДж. моль-V,
ТЪО^г). Термодинамические свойства газообразного положительногоУ[иона|?диоксида тория
в стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 991. Т|
Молекулярные постоянные ТЮ?, использованные для расчета термодинамических функций»
приведены в табл. 42.8. Экспериментальных данных о строении и частотах колебаний ThOJ в
литературе нет. По аналогии с UOJ и РиО?, которые имеют такую же структуру, как и
соответствующие нейтральные молекулы UO2 и РиО2, в справочнике для ThOJ принимается нелинейная
структура (симметрия С2с). Ионизация ThO2 происходит при удалении электрона с заполненной
электронной оболочки, поэтому так же, как в СО2, она должна сопровождаться некоторым увеличением
межатомного расстояния и уменьшением основных частот по сравнению с ThOa. Приведенное
в табл. 42.8 произведение моментов инерции вычислено для значеьий r(Th—O)=i,85+0,05 A
и -§О—Th—0=120+20°. Погрешность значения IaIbIc оценивается в 5-Ю16 г3-смв, значений
основных частот — в 100 см для vx и v3 и 50 см~х для v2. Отсутствие электронов на 5/-орбиталях
в ThOJ позволяет ожидать, что эта молекула не имеет низколежащих электронных состояний.
Приведенные в табл. 42.8 энергии возбужденных состояний ThOJ оценены из сравнения принятых
в справочнике значений энергий и суммарного статистического веса электронных состояний
молекул СО2, СО+ и ТЮ2.
Термодинамические функции ThOJ(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) ги
A. 130) с учетом двух возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью оценки принятых молекулярных постоянных,;
включая число и энергии возбужденных состояний, и приближенным характером расчета; они
составляют 4, 7, 10 и 15 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К.
Таблица термодинамических функций ТЬО^(г) публикуется впервые. 1
Константа равновесия реакции ThOJ(r) +e(r)=Th(r)+2O(r) вычислена с использованием
значения ДгЯ°@)=707,647+35 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(ThO2)=840+30 кДж-моль-1.
179
Глава 42
Это значение принято на основании масс-спектрометрического исследования методом
электронного удара, выполненного Раухом и Аккерманом [3943] (839+15 кДж-моль), и хорошо
согласуется с найденным Хилденбрандом и Мурадом [2649] G70+100 кДж-моль) в результате масс-
спектрометрического изучения равновесия в системе Th—Si—О. Погрешность принятого значения
оценена с учетом возможного влияния теплового возбуждения и немоноэнергетичности электронов.
Принятому значению /0(ThO2) соответствует
Д,Я °(ТЪО+а, г, 0) = 388,072 ± 34 кДж . моль.
ТЮ~(г). Термодинамические свойства газообразного отрицательного иона диоксида тория
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 992.
Молекулярные постоянные ThOj", использованные для расчета термодинамических функций,
приведены в табл. 42.8. Экспериментальные данные о строении и основных частотах ThO~ в
литературе отсутствуют. Так же как для ThO2, в справочнике для ThOjT принята нелинейная структура
(симметрия С2с). Можно ожидать, что появление электрона на 5/-орбитали атома Th в ThO^ должно
привести к увеличению межатомного расстояния и уменьшению частот колебаний (как, например,
это имеет место при переходе от UO|+ к UOJ и от UFe к UF~). Приведенное в табл. 42.8
произведение моментов инерции вычислено для структурных параметров r(Th—О) = 1,85+0,05 А и
^О—Th—0 = 120 + 20°. Погрешность значения IaIbIc составляет 5• 10~115 г3-см6, погрешности
приведенных в табл. 42.8 основных частот оцениваются в 100 см для vx и v3 и 50 см для v2.
Поскольку ионы Th3+ и U5+ имеют одинаковую электронную конфигурацию, возбужденные
состояния ThO~ с энергиями, меньше величины /„(ThOj), приняты идентичными состояниям UF5
(см. примеч. к тексту по UO2(r)).
Термодинамические функции ThO^r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и
A. 130) с учетом трех возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью принятых молекулярных постоянных и
приближенным характером расчета; они составляют 4, 8, 12 и 20 Дж-К-моль в значениях Ф°(Г)
при 298,15, 3000, 6000 и 10 000 К.
Таблица термодинамических функций ThO^"(r) публикуется впервые.
Константа равновесия реакции ThO2(r)=Th(r)-j-2O(r)-]-e(r) рассчитана с использованием
значения Дг.ЕГо@)=1637,647+52 кДж-моль, соответствующего принятой в справочнике величине
сродства к электрону
Л0(ТпО2) = — 90+50 кДж-моль-1.
Эта величина принята такой же, как для молекулы UOa, которая, в связи с отсутствием
экспериментальных данных, была оценена по величине J.0(UF4), и поэтому является грубо
ориентировочной. Ей соответствует
г, 0) = —541,928 + 52 кДж. моль.
Глава 43
УРАН И ЕГО СОЕДИНЕНИЯ
U(k, ж), U, U+, UO, UO+, UO2(k, ж), UO2, UOJ, UO~, UO3(k), UO3, UO", U3O8(k),
U4O9(k), UF, UF+, UF-, UF2, UFJ, UF~, UF3(k, ж), UF3, UF3+, UF", UF4(k, ж) UF4,
UF4+, UF4~, UF5(k, ж), UF5, UF*, UFJ, UF6(k, ж), UF6, UF", UOF, UO2F, UOF2,
UO2F2(k), UO2F2, UOF3, U0F4
В настоящей главе рассматриваются уран и его соединения с кислородом и фтором — всего 34
вещества. Для 10 веществ приведены данные о термодинамических свойствах в конденсированных
состояниях и для 32 веществ — в газообразном состоянии. Термодинамические свойства восьми
газов (UFe, UFe, UOF, UO2F, UOF2, UO2F2, UOF3 и U0F4) рассчитаны для температур до 6000 К,
остальных газов — до 10 000 К 1. Для элементарного урана и гексафторида урана в
Приложении 3.10 приведены данные об их критических постоянных.
Термодинамические свойства урана и всех его соединений вычислены для природной
изотопической смеси урана (атомный вес 238,029), кислорода и фтора. Однако различие постоянных разных
изотопомеров этих веществ в расчетах не учитываются из-за пренебрежимо малой величины
соответствующих поправок п недостаточной точности всех расчетов из-за неполноты
экспериментальных данных о молекулярных и термохимических постоянных.
В справочнике приводятся данные о свойствах в конденсированных состояниях только для
четырех наиболее устойчивых окислов. UO2(k) ниже 700 К имеет узкую область гомогенности,
однако с ростом температуры она расширяется до UO20—UO224 при 1400 К. Три остальных оксида
имеют заметные области гомогенности уже при температурах, незначительно превышающих 400—
500 К. Для U4O9 область гомогенности достигает при 1300 К интервала UO2>23—UO2 26, у p-U3Og
и <x-UO3 границы этой области смещены в сторону дефицита кислорода (при Т ~ 1200 К
соответственно иО2вз—UO2e6 и UO2e5—UO3) (см. [1023]). В литературе имеются указания о
существовании в конденсированном состоянии других оксидов: UO (при Т > 1200 К), U3O7 и метастабиль-
ных фаз ряда U4O10, U5O12 и т. п. Эти соединения в справочнике не рассматриваются ввиду их
стабильности только в узкой области температур. Не рассматриваются также положительно
заряженные газообразные ионы UOg и UFJ ввиду высоких потенциалов ионизации (больше 90000 см)
соответствующих нейтральных молекул.
Ч(к, ж). Термодинамические свойства кристаллического и жидкого урана в стандартном
состоянии при температурах 100—6000 К приведены в табл. 993.
Значения постоянных, принятые для расчета термодинамических функций U(k, ж),
представлены в табл. 43.1. В справочнике за стандартное состояние урана в интервале 0—942 К принята
ромбическая а-модификация. в интервале 942—1049 К тетрагональная Р-модификация и в
интервале 1049—1408 К объемно-центрированная кубическая у-модификация.
При Т <^ 298,15 К термодинамические функции U(k) вычислены по измерениям теплоемкости,
проведенным Флотовым и Осборном [2271] A,7—23 К, образец с содержанием примесей не более
0,006%, погрешность измерений —1% при 1,7 К и ~0,4% при Г > 2 К) и Флотовым и Лором
[2269] E,7—348 К, образец чистотой 99,99%, погрешность измерений 1% при 14 К и —0,2%
при Т > 30 К). Значения 6ГОB98,15 К) и #°B98,15 К)—#°@), принятые в справочнике
(см. табл. 43.1), совпадают с рекомендованными КОДАТА—МСНС [1887]. Погрешности этих
значений оцениваются в 0,2 Дж-К~1-моль~1 и 0,02 кДж-моль соответственно. С принятыми
данными по теплоемкости удовлетворительно согласуются несколько менее точные измерения
Джонса и др. [2899] A5—300 К) и Накамура и др. [3629] (80—1000 К). Данные ряда других
авторов [1885] A0—300 К), [2552] D,2—80 К), [3221] и [1957] не учитывались ввиду того, что
они либо менее точны, либо представлены только на графиках 2.
Уравнение для теплоемкости oc-U в интервале 298,15—942 К (табл. 43.1) выведено совместной
обработкой данных по энтальпии, полученных Джиннингсом и Корручини [2407] B73—1173 К)
1 Данные о равновесном составе системы U+3F2 ПРИ Т «S 10 000 К см. в [338, 2536].
2 В работах [1957, 2552, 3221] были найдены небольшие аномалии теплоемкости урана при 23, 37 и 40 К,
существование которых не подтвердилось в исследованиях [2269, 2899].
181
Таблица 43.1. Принятые значения термодинамических величин для урана в его соединений
в кристаллическом и жидком состояниях
«3
Моле
и
ио2
ио3
и3о8
и4о9
UF3
UF4
UF5
UFe
U02F2
Состояние
Kill, ромб, (а
к11, тетр. (Ь)
к1, куб. (у)
ж
к, куб.
ж
К, МОНОКЛ. (~()
к11, ромб.
к1, гекс.
кШ, куб.
к11, куб.
кН, куб.
Kl
к, гекс.
ж
К, МОНОКЛ.
ж
кИ.тетр. ф)
к1, тетр. (а)
ж
к, ромб,
ж
к, гекс.
1
кДж-молгГ1
) 6,364
11,28
14,585
42,74
50,75
18,38
22,55
26,15
31,565
19,7
ю
со'
го
0
<о
Дж-K-i
50,2
77,03
96,11
282,55
334,13
123,4
151,67
179,5
227,8
135,5
298,15 К)
• МОЛГтГ1
27,665
63,60
81,67
238,00
293,34
95,10
116,02
132,00
166,75
103,20
Коэффициенты в уравнении
а
6-Ю3
28,207 -5,792
42,4 —
38,3 —
42,144 3,232
96,525 —28,515
131. —
105,737 -20,855
1 020,690 —2 008,423
248,481 54,852
56 515,012 -183 412,408
-11243,263 20 449,286
380,311 —90,112
298,738 83,914
106,539 0,705
134 -
138,865 —32,068
167 —
125,159 30,208
125,159 30,208
167 -
-53,920 608,187
145,550 134,000
106,238 28,326
Примечание. Cp(T)=a+b -T—c -T-2+dT2-\-eT3 (в Дж -К моль-1).
U: a d=28,481-10-«. U3O8: a d=0, «=3011,753 Л0~9; б d=-3,847 ЛОГ».
. UOa: a d=16,996-Ю-*. U4O9: a d=0, e=391 818,144 -КГ»; б d=80,580-Ю.
UO3: a d= 19,086 -10-*. UF4: a d=27,988-КГ*.
для Ср(Т)
с ¦ Ю-5
1,197 а
-20,699
23,054 а
17,375 а
234,413 а
4,863 б
10 597,735 »
-5 464,611
66,257 б
25,730
10,355
14,020 а
1,925
1,925
—34,970
10,208
Интервал
температур
Tir
или
Т
К
298,15—942
942—1049
1049—1408
1408—6000
298,15—3123
3123-6000
298,15-3000
298,15—483
483-3000
298,15—348
348-380
380—1398
1398-3500
298,15—1768
1768—3200
298,15—1309
1309—2500
298,15—398
398-621
621-1800
298,15—337,2
337,21-500
298,15-2100
942
1049
1408
3123
—
483
348
1398
1768
1309
398
621
1 337,21
—
&»в°
или
Ьп.В°
кДж •
• моль
2,78
4,73
8,72
78
—
0
0
2,34
36,8
47
8
35
19,193
—
Уран и его соединения
Муром и Келли [3552] D74,9—1276,4 К), Марчиданом и Чопеком [3370] B98—1500 К), а также
измерений теплоемкости в работах Накамура и др. [3629] B98—1000 К). При обработке
погрешность измерений [2407] оценивалась в 0,3%, [3552] и [3629] — в 0,7% и [3370] — в 1%. Менее
надежные измерения теплоемкости в работах Митькиной [682] C23—873 К) и Фёдорова и Смирнова
[1047] B98—1273 К) не учитывались.
Температура полиморфного (а -» Р) превращения (942+2 К) принята по наиболее тщательным
измерениям Блюменталя [1587] (чистота образца 99,992%; определение Ttr по кривым
нагревание — охлаждение); принятое значение согласуется с полученным Марчиданом и Чопеком [3370].
Данные [1588, 1736, 1985, 2134, 3144, 3552] (875—955 К) не учитывались. Энтальпия превращения
B,78 + 0,17 кДж-моль) рассчитана из уравнений, принятых для теплоемкости а- и Р-модифика-
ций; в пределах погрешности она согласуется с данными [2407] B,82 кДж-моль), [3552]
B,85 кДж-моль). В работе [3370] было получено более низкое значение B,56 кДж-моль).
Теплоемкость [3-U D2,4+0,4 Дж-К~1-моль~1) в интервале 942—1049 К рассчитана из данных
по энтальпии, полученных Джиннингсом и Корручини [2407] (949,7—997,0 К); принятое значение
практически совпадает с измерениями теплоемкости в работе [3629] (953—1000 К); измерения
Мура и Келли [3552] D6 Дж-К~1-моль~1) представляются менее надежными в этом интервале
температур и не учитывались.
Температура полиморфного превращения A049+2 К) принята по работе Блюменталя [1587]
и согласуется в пределах погрешности с результатами, полученными в работах [3552] и [3370].
Данные [1588, 1736, 1985, 2134, 3144] A027—1045 К) не учитывались. Энтальпия полиморфного
превращения D,73±0,20 кДж-моль) рассчитана из уравнений, выведенных по данным Джин-
нингса и Корручини [2407] и описывающих энтальпию [3- и у-модификаций урана. Она согласуется
в пределах погрешности с величиной, найденной Муром и Келли [3552] D,87 кДж-моль);
измерения Марчидана и Чопека [3370] D,18 кДж-моль) не учитывались.
Теплоемкость f-U C8,3 + 0,80 Дж-К~1-моль) в интервале 1049—1408 К рассчитана из данных
по энтальпии, полученных Джиннингсом и Корручинии [2407] и Муром и Келли [3552]; принятое
значение согласуется с измерениями энтальпии в работе Левинсона [3242] A205—1394 К).
Температура плавления A408+2 К) принята по работе [1587], в работах [1588, 1736, 1984,
3144, 4385] получены практически совпадающие значения; данные работ [1295, 3370] A363—
1405 К) не учитывались. Энтальпия плавления (8.72+0,4 кДж-моль^ рассчитана по результатам
измерений энтальпии жидкого урана в работе Левинсона [3242] с учетом уравнений, принятых
для энтальпии U(k). Измерения [3370] F,98 кДж-моль) и [4385] (9,22 кДж-моль)
представляются менее точными.
Уравнение для теплоемкости 11(ж) выведено совместной обработкой данных по энтальпии
жидкого урана, полученных Левинсоном [3242] A415—1579 К) и Стефенсом [4385] A428—1348 К);
при обработке погрешности измерений [3242] и [4385] оценивались в 1,5 и 3% соответственно.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 3000 и 6000 К оцениваются в 0,15,
0,4, 3 и 8 Дж-К-моль~1 соответственно.
Термодинамические функции U(k, ж) вычислялись ранее в справочниках МАГАТЭ [3695]
(до 5000 К), Халтгрена и др. [2752] (до 5000 К) и Келли [2960] (до 3000 К). Данные [3695]
и табл. 993 согласуются в пределах 0,2—0,5 Дж-К^-моль в значениях Ф°(Г) вплоть до 5000 К.
Расхождения данных [2752, 2960] и табл. 993 не превышают 0,2 и 1 Дж-К-моль~1.
В настоящем справочнике в качестве стандартното состояния урана принято кристаллическое
состояние и в соответствии с этим
A/ff°(U, к) = 0.
Давление пара урана в реакции U(k. ж)=U(r) вычислено с использованием значения АгН°@) —
= Д8#°(и, к, 0)=534,865+8,0 кДж-моль, соответствующего принятой в справочнике энтальпии
сублимации
Д8Я°(и, к, 298,15 К) = 535 + 8 кДж • моль.
В табл. 43.2 приведены результаты расчетов энтальпии сублимации по данным о давлении пара
урана. Приведенные в таблице погрешности учитывают только воспроизводимость измерений.
Помимо приведенных в этой таблице работ, измерения давления пара урана проводились также,
авторами [1282, 1962, 1963, 2051, 4293], однако полученные результаты оказались неточными из-за
влияния примеси кислорода в исходных образцах, остаточного давления газов в измерительных
183
U(r)
Глава 43
Таблица 43.2. Результаты определений \SH°(U, к, 298,15 К) (в кДж • моль)
Авторы
Метод
Д,Я°(и, к, 298,15 К)
II закон
III закон
Раух, Торн, 1954 [3946] Эффузионный, 1630—1970 К, 5 серий, при раз- 484 472,2+1,1
личающихся количествах кислорода
Де Мария и др., 1960 [2032] Эффузионный, 1876—1909 К, 3 точки — 537+10
Патторе и др., 1969 [3784] Масс-спектрометрический, 1794—2339 К, 47 точек 545+7 а 541,7+0,5 а
Аккерман, Раух, 1969 [1221] Эффузионный, 1980—2416 К, 19 точек 531+12 а 531,7+0,7 а
а Аккерман и Раух [1225] исследовали растворимость вольфрама и тантала в жидком уране. Указанные величины
включают связанные с этим поправки, вычисленные авторами работы [1225].
приборах, способности урана проникать через стенки контейнера и распространяться по
поверхности контейнера. В работе [3946] было изучено и учтено влияние кислорода на измеряемое
давление пара, однако в ней были получены завышенные давления вследствие проникновения урана
через стенки тигля (см. [1221, 2032]).
Результаты наиболее надежных измерений [1221, 2032, 3784] различаются больше, чем это
можно объяснить воспроизводимостью измерений, что затрудняет выбор рекомендуемого значения.
Принятая величина является средневзвешенной из данных этих работ, причем наибольший вес
придавался измерениям [1221] (см. также [1225] и [3784]). При оценке погрешности
рекомендованного значения были учтены неточности термодинамических функций, сечений ионизации и
расхождение результатов измерений в работах [1221] и [3784].
Исследования испарения систем U—О, U—N, U—С и U—Ри—С также позволяют определить
энтальпию сублимации урана. Получаемые при таких расчетах величины лежат в интервале
525—535 кДж-моль (см. [3695]). Поскольку исследуемые в этих случаях процессы являются
более сложными, чем испарение урана, вычисляемые энтальпии сублимации, по-видимому,
уступают по точности лучшим работам, в которых исследовалось испарение чистого металла.
U(r). Термодинамические свойства газообразного урана в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 994.
Для расчета термодинамических функций использовано 1596 уровней энергии г атома U в
интервале 0—42 062,897 см, приведенных в работе Блэза и Радзиемского [4930], их суммарный
статистический вес равен 17 932. Авторы [4930] обобщили все известные к 1975 г. результаты
исследований энергетических уровней атома U, выполнили тщательный анализ спектра урана в
области 3500—9000 А, полученного с высоким разрешением, и классифицировали около 35 000 линий,
относящихся к U и в меньшей степени к U+. Из 1596 уровней энергии только 192 уровня (суммарный
статистический вес 2473) были отнесены авторами [4930] к определенным электронным
конфигурациям, в том числе к 10 валентным (. . .5fs6d7s2, которой принадлежит основной уровень bL6,
. . .5/36d27s, . . .5f7s7p, . . .5fQd7s, . . .5/47s2, . . .5/26d27s2, . . .5f7s27p, . . .5fs6d7s7p, . . .5f6d7p
и . . .5/36d27p) и к двум ридберговским (. . .5/36d7s8s и . . .5fs6d7s8p). Для остальных 1404 уровней
энергии в работе [4930] даны лишь значения /.
Согласно оценке Брюэра [4935], выполненной на основании анализа имеющихся спектральных
и термодинамических данных, а также некоторых теоретических представлений, атом U должен
иметь еще несколько валентных конфигураций, для которых возможны относительно
низкорасположенные уровни энергии. Общее число уровней, соответствующих всем валентным
конфигурациям, очень велико, а их полный статистический вес превышает 5-Ю5. Однако подавляющая часть
этих уровней лежит выше первого потенциала ионизации атома U. Так, полный статистический
вес уровней, принадлежащих семи относительно изученным и низколежащим электронным
конфигурациям (энергия нижнего уровня меньше 20 000 см), составляет всего 68 068, причем
значительное число даже этих уровней имеет энергию выше потенциала ионизации. В литературе
предпринимались попытки [2535,; 3917., 3909] оценить ненаблюдавшиеся уровни атома U для расчета
См, примеч, 1 к тексту по Сг (г).
184
Уран и его соединения U+(r)
суммы по состояниям. Наиболее интересной из них является работа Радзиемского и Мака [3909],
в которой выполнен расчет ab initio уровней энергии атома U и его ионов. Однако численные
данные об уровнях энергии в этой работе не приведены. Хотя большинство принятых в справочнике
уровней атома U не имеет отнесения к определенным электронным конфигурациям, согласно
выводам работы [3909] они вполне адекватны валентным состояниям с энергиями ниже 20 000 см.
Область более высоких энергий изучена значительно хуже.
Термодинамические функции U(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по экспериментально наблюдавшимся уровням
энергии. Погрешности вычисленных значений обусловлены неопределенностью данных об уровнях
энергии, главным образом выше 20 000 см, они могут быть оценены для Ф°(Г) на основании
сравнения с данными [3909] и составляют 0,03, 0,5, 1 и 4 Дж-К^-моль при 298,15, 3000, 6000 и
10 000 К соответственно. Для S°(T) погрешности должны быть в 2—3 раза больше. Следует
подчеркнуть, что значения термодинамических функций могут быть только выше приведенных
в табл. 994.
Ранее термодинамические функции U(r) вычислялись в справочниках МАГАТЭ [3697]
(до 6000 К), Сталла и Зинке [4421] (до 3000 К), а также в работах Кендалла [2967] (до 5000 К),
Гурвича с сотрудниками [338, 2535] (до 20 000 К), Фебера и Херрика [2227] (до 6000 К), Рэнда
[3917] (до 10 000 К) и Рассера и Реми [3940] (до 15 000 К). За исключением [3917] в расчетах были
использованы только экспериментальные данные по уровням энергии U, известные к моменту
проведения расчета. В настоящем справочнике использовано большее число уровней по сравнению
с другими расчетами, поэтому табулированные величины больше известных в литературе, причем
с ростом температуры расхождения увеличиваются. Эти расхождения составляют в значениях
Ф°A0 000 К) от 2 до 9 Дж-К-моль. В работе Рэнда [3917] учтен вклад в термодинамические
функции ненаблюдавшихся уровней атома U с использованием модели плотности распределения
этих уровней по Гауссу (ранее Гурвич и Юнгман [2535 ] использовали равновесное распределение
уровней). Вычисленные Рэндом [3917] термодинамические функции U(r) при температурах выше
2000—3000 К больше, чем в табл. 994, и расхождения достигают 8 Дж-К^-моль в значениях
Ф°A0 000 К). В работе Радзиемского и Мака [3909] на основании результатов квантовомеханиче-
ского расчета уровней для 21 электронной конфигурации атома U был выполнен расчет Qn
(с ограничением по энергии на величине первого потенциала ионизации). Значение Ф°A0 000 К),
рассчитанное по данным [3909], отличается от данных табл. 994 на 4 Дж-К-моль~1. По-видимому,,
методика, использованная Рэндом, дает завышенные величины.
Энтальпия образования
Д/Я^и, г, 0) = 534,865 + 8,0» кДж • моль
соответствует принятой в настоящем справочнике величине энтальпии сублимации урана
(см. выше).
U+(r). Термодинамические свойства газообразного положительного иона урана в стандартном
состоянии при температурах 298,15—10 000 К приведены в табл. 995.
Для расчета термодинамических функций использованы 273 уровня энергии г U+ в интервале
0—45 533,5 см, рекомендованные Стейнхаузом и др. [5052], с суммарным статистическим весом
3152. Из этих уровней только 71 с суммарным статистическим весом 749 отнесены к определенным
электронным конфигурациям: . . .5/37s2 (к этой конфигурации относится основной уровень 4/»/),
. . .5f6d7s, . . .5/*7s, . . .5f6d\ . . .bflp, . . .5/46d, . . .5/26d27s, . . .5f 6d7s2, . . .5/W, . . .5f7s7p,
...5f6d7p. Для остальных уровней определены только значения /. Согласно оценке Брюэра
[4936] к перечисленным электронным конфигурациям принадлежат наиболее низкие валентные
состояния U+; нижний уровень каждой из них имеет энергию меньше 30 000 см. Число уровней
энергии, соответствующих этим конфигурациям, весьма велико, их полный статистический вес
составляет почти 80 000. Однако подавляющая их часть имеет настолько высокие энергии, что их
вклад в термодинамические функции при температурах до 10 000 К достаточно мал. Радзиемский
и Мак [3909] на основании результатов квантовомеханических расчетов считают, что валентные
состояния U+ исследованы достаточно полно вплоть до 15 000 см.
1 См. примеч. 1 к тексту по Сг(г).
185
UO(r) Глава 43
Термодинамические функции U+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по экспериментально определенным уровням
энергии. Погрешности вычисленных значений обусловлены неполнотой данных об уровнях
энергии, главным образом выше 15 000 см, и были оценены путем сравнения с расчетом [3909!.
Они составляют 0,1, 1, 2 и 5 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К
соответственно. Для S°(T) погрешности должны быть в 2—3 раза больше. Следует подчеркнуть,
что истинные значения термодинамических функций не могут быть меньше приведенных в табл. 995.
Ранее таблицы термодинамических функций U+(r) вычислялись в работах Гурвича и др. [338,
2535] (до 20 000 К) и Рассера и Реми [3940] (до 15 000 К). Кроме того, Радзиемский и Мак [3909]
рассчитали статистическую сумму по электронным состояниям U+ в интервале 100—15 000 К.
Расчеты [338, 2535, 3940] основаны на экспериментальных данных об уровнях энергии U+ и
расхождения их результатов с данными табл. 995 обусловлены только разницей в числе
учитывавшихся уровней. Расхождения растут с температурой и достигают 2—3 Дж-К^-моль" в значении
Ф°(Ю 000 К). Расчет Радзиемского и Мака [3909] выполнен на основании уровней энергии,
полученных в квантовомеханическом расчете для 16 электронных конфигураций и ограниченных
энергией 100 000 см. Соответствующие значения расходятся с данными табл. 995 даже при низких
температурах, что свидетельствует о невысокой точности квантовомеханического расчета низко-
лежащих уровней энергии. При 3000 К расхождение составляет 1 Дж-К~1-моль~1. При более
высоких температурах расхождения проходят через нуль и достигают 5 Дж-К~1-моль при
10 000 К.
Константа равновесия реакции U+(r)+e(r) = U(r) вычислена с использованием значения
Дг#°@) =—597,632±0,050 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(U)=49 958,1+4 см-^597,632+0,050 кДж-моль.
Принятое значение найдено Соларцем и др. [5050] экстраполяцией ридберговских серий
. . .5f7s2np {п <; 52) и . . .5f7s2nf (п ^ 44), впервые зарегистрированных в спектре U с помощью
лазерной техники. Это значение значительно точнее полученных ранее методами поверхностной
ионизации D9 000+650 см [4852] и49 900±500 см [5048]), электронного удара D9 300±400 см
[5011], 49 900±500см-х [4871] и 49 200 + 300см [3943]), короткой экстраполяции спектральных
данных D8 800 + 600 см [5057]) и фотоионизации D9 900±16 см [4983]).
Принятому значению io(U) соответствует
\\jH°(\}+, г. 0) = 1132,497 +'
UO(r). Термодинамические свойства газообразного оксида урана в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 996.
В табл. 43.3 приведены молекулярные постоянные, принятые в справочнике на основании
результатов исследований ИК-спектров молекул UO, изолированных в матрицах из Аг [1202,
2352, 2353, 4964], и приближенных оценок, выполненных при подготовке справочника.
Электронный спектр UO не изучен, несмотря на неоднократные попытки его получения, а квантовомехани-
ческие расчеты этой молекулы не проводились. Благодаря наличию у атома U частично
заполненных /- и d-оболочек молекула UO должна иметь сложную систему электронных термов, так же
как это имеет место у ThO, CeO и РгО (см., в частности, [4951, 4952, 5009, 5046]). Оценка энергии
многих сотен связанных валентных состояний UO, принадлежащих конфигурациям молекулярной
оболочки, соответствующим нижним конфигурациям атома U, практически невозможна. В связи
с этим была выполнена оценка суммарных статистических весов электронных состояний,
расположенных в вобранных интервалах энергий в области до 40 000 см, с использованием схемы,
предложенной Йоргенсеном [4987 ] при рассмотрении низколежащих состояний оксидов и галогенидов
переходных металлов, которая не противоречит отрывочным данным по спектрам оксидов лантани-
дов. В молекуле UO характер связи моментов количества движения должен быть близок к случаю
Гунда с, поэтому статистические веса электронных состояний равны 2 (Q > 0) или 1 (&=0). В
соответствии с принятой схемой основное состояние UO, вероятно, принадлежит конфигурации
. . -ф^0,, или . . .ср^З^, таким образом, для значения Q в основном состояниивозможны оба случая.
В справочнике принимается, что основное состояние является вырожденным, так как даже при
2=0 расщепление компонент 0+ и 0" должно быть небольшим. Суммарные статистические веса
связанных возбужденных состояний UO были оценены на основании следующих предположений:
186
Уран и его соединения
UO(r)
Таблица 43.3. Молекулярные постоянные UO, UO+, UF, UF+ и UF~
Молекула
Состояние
аг ¦ 103 De ¦ 107
СМ"
А
238U16O
238^160+
238|J19p
2381J19p+
238TJ19p-
X
X
X
X
X
840 б
900 б
600 б
650 б
570 б
2.6 б
3,1 в
1.7 в
9 В
2,4»
0,340 в
0,35 г
0,23 г
0,24 г
0,23 г .
1,5 г
1,6Д
1,0 Д
1,0 Д
1,0Д
2.3 Д
2,1 е
1.4 е
1,3 е
2,0 е
1,82 е
1,80 б
2,03 б
2,00 б
2,05 б
Примечание. Ниже все постоянные (кроме р,:) в см г.
UO: a
Г 1000
Pi 15
3000
45
5000
70
10 000
150
20 000
300
30 000
700
40 000
700
б вычислено по соотношению A. 67) и значениям ^Gi/2 =835 и DO=65 000; в вычислено по соотношению
A. 38); г вычислено по соотношению A. 69); Д вычислено по соотношению A. 68); е оценка.
UO+: а
Pi
2000
2
4000
4
10 000
12
20 000
20
30 000
20
40 000
60
б оценка; в вычислено по соотношению A. 67) и Do=65 000; г вычислено по соотношению A. 38);
Д вычислено по соотношению A. 69); е вычислено по соотношению A. 68). v
UF: a T'*1 1000 3000 5000 10 000 20 000 30 000 40 000
Pi 6- 12 20 120 300 500 500
б оценка; в вычислено по соотношению A.67) и значению Do=54 000; г вычислено по соотношению
A. 38); Д вычислено по соотношению A. 69); е вычислено по соотношению A. 68).
UF+
UF'
1000
Pt
3000
20
5000
30
10 000
60
20 000
120
30 000
200
40 000
250
б оценка; в вычислено по соотношению A. 67) и значению D0(UF+)=54 000; г вычислено по
соотношению A. 38); Д вычислено по соотношению A. 69); е вычислено по соотношению A. 68).
а Ге=5000, pi=& и 7^=8000, л=12; б оценка; в вычислено по соотношению A. 67) и D0(UF~)=34 500;
г вычислено по соотношению A.38); д вычислено по соотношению A.69); е вычислено по
соотношению A. 68).
а) связь в UO осуществляется за счет перехода двух электронов атома U (рассматривались три
нижних конфигурации . . .5/36d7s2, . . .5/36d27s и . . .5/47s2) на связывающие а- и тс-орбитали
молекулы, которые при этом оказываются замкнутыми (. . .и4о2); б) остальные внешние молекулярные
орбитали заполняются за счет четырех остающихся электронов атома U; формально они
рассматривались как принадлежащие иону U+2, находящемуся в одной из шести нижних конфигураций
(. . ./3d, . . ./3s, . . ./*, . . .f2d2, . . .f2ds, . . ./2s2); в) на орбиталях ср^, 8^, bd эти электроны не участвуют
в образовании связей с ар- и тср-электронами атома О и, следовательно, являются несвязывающими
или слабосвязывающими, а на орбиталях п„ a nd, orf, as — разрыхляющими; г) состояния,
имеющие больше двух разрыхляющих электронов, рассматривались как отталкиватёльные или
слабосвязанные и не учитывались; д) принималось, что в области энергий до 20 000 см плотность
связанных состояний растет примерно линейно, а от 30 000 до 115 000 см (величина D0(UO)-{-
+/0(U)), где вносят вклад одновременно все 6 конфигураций U+2, она постоянна. Погрешности
приведенных в табл. 43.3 статистических весов могут достигать коэффициента 2—3 1.
Межъядерное расстояние U—(i в основном состоянии, приведенное в табл. 43.3, было оценено
на основании сопоставления величины ге(МО) в молекулах LnO (см. гл. 41 и [4878]), ThO, WO,
а также величины г(М—F) в молекулах UF6 и WF6; его погрешность составляет 0,05 А. В ИК-
спектре молекул оксидов урана, изолированных в матрице из Аг, который исследовался в ряде
работ, Гарстенс и др. [4964], Габельник и др. [2352, 2353], Абрамович и Аквиста [1202] отнесли
1 Оценка по этой схеме суммарных статистических весов объединенных электронных термов молекул TiO и ZrO
в тех же интервалах энергии показала ее удовлетворительную точность, отклонения оцененных величин от
соответствующих данных табл. 36.3 и 37.3 не превышали коэффициента 1,5—2 и в среднем составляли около 20%.
187
UO(r)
Глава 43
к молекуле 238UI6O частоту1 820 см Ч Для оксидов лантанидов исследования их ИК-спектров в
матрицах приводят к значениям Д&/2, которые на 10—15 см меньше полученных при изучении
электронных спектров свободных молекул. Значение 820 см представляется несколько
заниженным для UO и при его сравнении с колебательными постоянными молекулы ThO. Приведенные
в табл. 43.3 колебательные постоянные UO соответствуют значению Д&/„=835 + 15 см,
принятому на основании этих соображений.
Термодинамические функции UO(r) рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения ()вн и ее производных вычислялись по уравнениям
A. 90)—A. 92) с учетом семи объединенных электронных термов с приписанными им
суммарными статистическими весами в предположении, что Qf?}x BP=0,5Pj(?{^ вр- Величина QW вр и
ее производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием
по колебательным уровням и интегрированием по значениям / с помощью уравнений типа
A. 82). В расчете учитывались все колебательно-вращательные уровни основного
состояния с значениями / ^ /шах „, последние определялись из условий A.81). Энергия уровней
вычислялась по уравнениям A. 65), A. 34) и A. 64а), их вырождение учитывалось статистическим
весом 2. Значения коэффициентов в этих уравнениях, полученные по принятым молекулярным
постоянным с учетом условий A. 59), приведены в табл. 43.4 вместе с значениями vmax и /Иш-
Таблица 43.4.
Значения коэффициентов в уравнениях, описывающих уровни энергии (в см х),
а также значения vmax и </]im, принятые для расчета термодинамических функций
UO, UO+, UF, UF+ и UF-
Коэффициенты
Те
У1в-1(П
Уо1-10
Yu-iO3
а Энергии и
UO
X (пфО)
0а
8,40
-2,6
3,4
-1,5
UO+
X (пфО)
оа
9,00
—3,1
3,5
-1,6
UF
X (пфО)
0а
6,00
-1,7
2,3
-1,0
UF+
X(Q^0)
0а
6,50
-2,0
2,4
-1,0
статистические веса объединенных
UF-
Х(йф0)
0
5,70
—2,35
2,28
-1,0
Коэффициенты
Уоз-Ю«
^тах
электронных термов
UO
X (ОфО)
ио+
X (пфО)
-2,3 -2,1
-0,487 -1,2
161 144
594 572
см. в табл. 43.3.
UF
Х(пф0)
-1,4
—0,229
175
642
UF+
X (пфО)
-1,3
—0,55
161
622
UF-
X (йфО)
—2
L —1,593
120
517
Погрешности рассчитанных термодинамических функций UO(r) при низких температурах
обусловлены главным образом неопределенностью типа основного электронного состояния этой
молекулы. Выше 2000 К становятся существенными ошибки из-за использования в расчете
результатов приближенной оценки числа и энергии возбужденных электронных состояний. Последние
особенно существенны в значениях Ср(Т) и Н°(Т)—Н°@). Погрешности в значениях Ф°(Т) при
298,15, 3000, 6000 и 10 000 К оцениваются в 4, 8, 12 и 16 Дж-К^-моль"-1.
Термодинамические функции UO(r) ранее вычислялись в работах Грина [2463] (Т ^ 6000 К),
Брюэра и Розенблатта [1666] (Ф°(Г) для Т < 3000 К), Абрамовица и др. [1200] (Т < 6000 К),
атакжеПаттореидр. [3783] и Де Мария и др. [2032] (Ф°(Т) и Н°(Т)—Я°@) для :Г=1500-^-2500 К).
Расхождения данных табл. 996 и [2463] составляют от 6 до 25 Дж-К~1-моль в значениях Ф°(У)
и S°(T); при Т — 298,15 К они обусловлены тем, что Грин принял />х=1, выше 1000 К — тем, что
он учел меньше возбужденных состояний. В работе [1200] возбужденные состояния вообще не
учитывались, и при 6000 К расхождения достигают 35 и 48 Дж-К-моль~1 в значениях Ф°(Т) и
S°(T). Данные [3783] согласуются с табл. 996 в пределах 3 Дж-К~х
Константа равновесия реакции UO(r) = U(r)+O(r) вычислена
в справочнике энергии диссоциации
ДгЯ°@) = D0(UO) = 750 ± 15 кДж • моль яй 62 700 ± 1300 см.
Результаты определений энергии диссоциации UO представлены в табл. 43.5; приведенные
в таблице погрешности учитывают воспроизводимость измерений, неточность термодинамических
функций, термохимических величин и сечений ионизации. Данные, полученные при исследованиях
¦моль .
с использованием принятой
1 В более ранних работах [1204, 3212] к молекуле UO были отнесены частоты в области 770 см~*.
188
Уран й его соединения
Ш+(г)
Таблица 43.5. Результаты определений D0(UO) (в кДж • моль)
Дроварт
Дроварт
Паторе и
Куппенс
Аккерман
Стайгер,
Юне и др
а Данные
Авторы
и др., 1966 [2118]
и др., 1967 [2117]
др., 1968 [3783]
и др., 1968 [1924]
и др., 1969 [1235]
Кейтер, 1975 [5051]
., 1981 [5071]
представлены в виде
Метод
Масс-спектрометрическвй, 1/2UO2(k
1705—2098 К, 20 точек
То же,
То же,
То же
5 точек
То же,
То же,
UO(r)+Si(r)=U(r)+SiO(r),
1/2иО2(к)+1/2и(ж)=иО(г),
UO(r)+B(r)=U(r)+BO(r),
1/2иО2(к)+1/2и(ж)=UO(r),
YO(r)+U(r)=Y(r)+UO(r),
13 точек а
То же,
данные
уравнения.
UO(r)+La(r)=U(r)+LaO(r)
-р/2и(ж)=Ш(г
2530—2310 К а
1700—2150 К а
2238—2418 К,
1540—2315 К а
2148-2369 К,
, 1530—1900 К,
представлены в виде графика
II закон
), 763+40
756
753
658+150
781
777
796
(UO)
III закон
746+20
752 + 25
746+20
756+20
735 +25
762+20
762+20
равновесия реакций с участием конденсированной фазы, приводят к более низким значениям
D0(UO), чем обменные газофазные реакции. Принятая величина является средневзвешенной; при
ее вычислении больший вес придавался исследованиям обменных реакций. Погрешность
принятой величины оценена с учетом неточности термодинамических функций, которая соответствует
погрешности в энергии диссоциации около 10 кДж-моль, и расхождения результатов измерений
в разных работах.
Принятой величине соответствует
AfH°(UO, r, 0) = 31,648 ±17 кДж-моль-1.
UO+(r). Термодинамические свойства газообразного ионизированного оксида урана в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 997.
Спектр UO+ не исследовался, а квантовомеханические расчеты этой молекулы не проводились.
Молекулярные постоянные UO+, приведенные в табл. 43.3, основаны на оценках, выполненных
при подготовке справочника так же, как для молекулы UO. Молекула UO+ имеет нечетное число
электронов, поэтому во всех электронных состояниях, включая основное, которое, вероятно,
принадлежит конфигурации . . .ср^ S^, Q >0. Суммарные статистические веса связанных
валентных электронных состояний UO+ в области энергий до 40 000 см были получены с учетом
состояний, которые соответствуют двум конфигурациям электронной оболочки U+3 (. . ./3 и . . .fd).
Приведенные в табл. 43.3 постоянные <яе и ге в основном состоянии UO+ были оценены на основании
принятых значений соответствующих постоянных UO. Экспериментальные данные для молекул
YF, LaF, ZrO и соответствующих ионов, возникающих при удалении одного несвязывающего
или слаборазрыхляющего электрона нейтральной молекулы, свидетельствуют, что ионизация
сопровождается увеличением величины ше на 5—13% и уменьшением примерно на 1—3%
величины ге. Это согласуется с наблюдавшимся экспериментально существенным возрастанием (на 15%)
частоты v3 при ионизации молекулы UO2 (см. ниже). Погрешности принятых в справочнике
значений (ое и ге оцениваются в 50 см и 0,05 А соответственно.
Термодинамические функции UO+(r) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения @вн и ее производных вычислялись по уравнениям A. 90)—A. 92) с учетом
шести объединенных электронных термов с приписанными им суммарными статистическими
весами и в предположении, что ()<?>_ BP=0,5pf()W_вр. Величина <?S-BP и ее производные находились
по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням
и интегрированием по значениям / с помощью уравнений типа A. 82). В расчете учитывались
все уровни основного состояния с значениями / <^ /maXj 5, последние определялись из условий
A. 81). Колебательно-вращательные уровни энергии основного состояния вычислялись по
уравнениям A. 65), A. 34) и A. 64а), их вырождение учитывалось статистическим весом 2. Значения
189
UO2(k, ж) Глава 43
коэффициентов в этих уравнениях, полученные с учетом условий A. 59), приведены в табл. 43.4
вместе со значениями г?шги и /11ш.
Погрешности рассчитанных термодинамических функций UO+(r) обусловлены полным
отсутствием экспериментальных данных о молекулярных постоянных этого соединения. Корректная
оценка их величины при Т > 3000 К затруднительна, особенно в значениях С°(Т). Погрешности
в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К оцениваются в 2, 4, 8 и 12 Дж-К^-моль.
Таблица термодинамических функций UO+(r) публикуется впервые.
Константа равновесия реакции UO+(r)+e(r) = U(r)+O(r) вычислена с использованием
значения Д^^О)=205,00+ 25 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(UO)=545 + 20 кДж-моль.
Принятая величина основана на результатах измерений методом электронного удара,
выполненных Раух ом и Аккерманом [3943] E40 +20 кДж-моль) и Манном [5011] E52 + 20 кДж -моль).
Близкое, но менее точное значение 550+40 кДж-моль было получено в работе [3783]. В работах
[2032, 5071 ] были получены заметно отличающиеся значения. Однако если их скорректировать
с учетом разности принятого в настоящем справочнике и найденных в этих же работах значений
/0(U), то исправленные значения (~530 кДж-моль) оказываются близкими к принятому.
Погрешности оценивались с учетом воспроизводимости измерений и возможного влияния теплового
возбуждения.
Принятой величине соответствуют
,+, г, 0)= 576,648 ± 26 кДж . моль*1,
D0(UO+) = 802,632 ± 25 кДж • моль «* 57 100 + 2000 см.
UO2(k, ж). Термодинамические свойства кристаллического и жидкого диоксида урана в
стандартном состоянии при температурах 100—6000 К приведены в табл. 998.
Значения постоянных, использованные для расчета термодинамических функций U02(k, ж),
представлены в табл. 43.1. В справочнике за стандартное состояние UO2(k) в интервале 0—3123 К
принимается кубическая модификация (структурный тип флюорита) [2498]. При Г=30,44 +
+0,01 К [2758] имеет место переход I рода парамагнетик—антиферромагнетик, при котором
симметрия решетки остается кубической, но вблизи точки Нееля найден небольшой скачок
постоянной решетки (Да ~ 0,00002 А) [3088]. В работе [1586] дана теоретическая модель,
обосновывающая характер перехода и рассчитана его скрытая энтальпия @,06 кДж-моль). Расчет энтальпии
превращения по данным [2758] приводит к значению 0,12 кДж-моль. Температура перехода
определялась также в работах [1645, 1948, 2212, 2320, 4711, 4714, 4722, 4735], соответствующие
значения лежат в пределах 30,4—30,8 К.
При Т < 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работах Веструма и Хунтцикера [2758, 4711 ] E—350 К). Содержание урана и кислорода
в образце составляло 88,20+0,17 и 11,87+0,01 вес.% соответственно, при теоретическом
содержании 88,15 и 11,85%. Мольное отношение содержания кислорода к урану (O/U) было равно 2,002 +
+0,001. Погрешности измерений теплоемкости оценены авторами [2758] в 5% при 5 К, 1% при
10 К и меньше 0,06% при Т >25 К. Погрешности значений S°B98,15K) и Н° B98,15 К)—#°@),
приведенных в табл. 43.1, составляют 0,2 Дж-К~1-моль~1 и 0,02 кДж-моль. Измерения [2899]
A5—300 К) выполнены на более грязном образце, в работе [3959] B73—373 К) получено
завышенное значение С;B98,15 К)=69,9 Дж-К-^моль.
• При Т >¦ 298,15 К для теплоемкости 1ГО2(к) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии в работах Мура и Келли [3552] D83—1463 К), Попова
и др. [806] D33-876 К), Конуэя и Хейна [1911] A173-2623 К), Ограда и Лири [3697] A338-
2303 К), Хейна и др. [2604, 2606] A174-3107 К), Лейбовицаи др. [3229] B557—3083 К), Энгела
[2176] C00—1000 К), Грёнволда и др. [2499] C00—1000 К), Фредриксона и Часанова [2322]
F75—1434 К). При обработке погрешность данных [2499] оценивалась в 1%, данных [2322,
190
Уран и его соединения
Ш2(к, ж)
1,5% и данных [806, 2176, 3697] — в 2%. Измерения [1264,
1266]
2604, 2606, 3229, 3552] — в
не учитывались, поскольку они существенно отличаются от более точных данных
Температура плавления UO2 C123 + 20 К) принята по результатам наиболее тщательных
определений [1381, 2161, 2604, 3202, 3331, 3332, 3333]; менее точные значения получены в работах
[1320, 1428, 1585, 1831, 1847, 2589, 3170, 3201, 3287, 3398, 3654, 4180].
Энтальпия плавления G8 + 3 кДж-моль) рассчитана из уравнений для энтальпии
кристаллического и жидкого UO2. Принятое значение в пределах погрешности согласуется с величинами,
найденными экспериментально [2604, 3172, 3228] G6,2, 74,1 и 79,1 кДж-моль). В работах
[2504, 2946] на основании измерений скорости охлаждения расплавленного UO2 получено значение
105,9 кДж-моль.
Для теплоемкости жидкого UO2 A31+4 Дж-К~1-моль~1) принято значение, полученное Рэндом
и др. [3918] в результате совместной обработки экспериментальных данных Лейбовица и др.
[32283 C173—3523 К, 6 измерений) и недоступных авторам настоящего справочника данных Хейна
и Флагеллы C123—3264 К). В опубликованной работе Хейна и др. [2604] результаты семи
измерений для жидкой фазы представлены только в графическом виде и поэтому не учитывались.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 3000 и 6000 К оцениваются в 0,15,
1, 2 и 14 Дж•К~1-моль~1 соответственно. Расхождения между термодинамическими функциями,
приведенными в табл. 998 и в справочниках Барина и Кнакке [1395] (Т ^ 3000 К) и Келли [2960]
(Т < 2000 К), не превышают 1,4 Дж-К^-моль в значениях Ф°(Т).
Таблица 43.6. Результаты определений A.fH° (UO2, к, 298,15 К) (в кДж-моль-1)
Авторы
Метод
AfH°(UO2, к, 298,15 К)
II закон
III закон
I. Калориметрические измерения
Микстер, 1912 [3524] Реакции UO2 и U3O8 с Na2O2, 3 измерения —1071,1
Сжигание в кислороде, ЗиО2(к)+О2(г)=и3О8(к), —1086,6
5 измерений
Хьюбер и др., 1952 [2742] То же, UO2t00e, 12 измерений —1085,6+1,0
Фарр и др., 1959 [2217] То же, UO2i014 —1087,3+1,0 ¦
Хьюбер и др., 1963 [2732] То же, 1ГО2,025 —1088,0±1,0
Стормс, Хьюбер, 1967 [4405] То же, UO2,039 —1089,4±1,0
Хьюбер, Холли, 1969 [2739] То же, UO2,055, 8 измерений —1085,6+0,8
Расчет зависимости Д,Я° от состава 1ГО2+Ж по —1085,0+3,0
данным [2217, 2732, 2739, 2742, 4405]
Бурдезе, Аббатнста, 1958 [1723] Растворение в азотной кислоте, 7 измерений —1077,0+1,0
Джонсон, Стил, 1981 [2875] Сжигание во фторе UO2,000 и UO2l023, 5 измерений —1085,1 ±2,5
Жердань и др., 1963 [2387] Высокотемпературная калориметрия, и(к)+О2(г)= —1087 ±9
=1Ю2(к), 40 измерений
II. Другие методы
Кьюккола, 1962 [3023] ЭДС, 3UO2(K)+O2(r)=U3O8(K), 1073—1473 К, — —1087,2
Дг<?B98,15 К)=—267,5
Усизима и др., 1962 [3575, 4567] ДТА, ЗШ2(к)+02(г)=и308(к), 503 и 653 К —1087,3
Маркин и др., 1963 [3393] ЭДС, ЗиО2(к)+О2(г)=и3О8(к), 1373 и 1473 К - -1082,8
Гашич и др., 1968 [2371] ДТА, ЗШ2(к)+02(г)=и308(к), 673-803 К, 7 из- —1071
мерений
1 Измерения энтальпии UO2(k) обнаруживают аномальный рост теплоемкости, начиная с ~2000 К. что, по мнению
ряда авторов, связано с образованием точечных дефектов. В работе Шварца [4451] обсуждается модель,
основанная на предположении, что доминирующим типом разупорядочения в UO., при высоких температурах являются
дефекты Френкеля в кислородной подрешетке. В работах [1770, 2565, 3335, 4215] аномальный рост теплоемкости
вблиз(Гтемпературы плавления объясняется переходом валентных электронов в свободное состояние.
191
UO2(k, ж)
Глава 43
В справочнике принята энтальпия образования
Д^ПШа, к, 298,15 К) = —1085,0 ±1,0 кДнс • моль,
основанная на приведенных в табл. 43.6 измерениях. Наиболее точные калориметрические
измерения были выполнены Хьюбером и Холли [2739] (сожжение в кислороде) и Джонсоном и Стилом
[2875] (сожжение во фторе) и принятое значение основывается на этих работах, оно совпадает
с рекомендацией КОДАТА—МСНС [1887]. Исследования равновесий реакций с участием UO2(k)
приводят к величинам, совпадающим в пределах их погрешностей с принятой.
Давление пара в реакции UO2(k, ж) = 1Ю2(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
\Ho(P) = A,H°(XJ02, к, 0) = 603+ 20 кДж-моль.
Результаты определений этой величины представлены в табл. 43.7. Приведенные в таблице
погрешности учитывают воспроизводимость измерений, неточность термодинамических функций,
термохимических величин и сечений ионизации, использованных в расчетах. Как показали Ак-
керман и др. [1233], измерения давления паров UO2(k) эффузионным методом при давлениях,
превышающих 10~4 атм, могут быть искажены вследствие отклонений от молекулярного характера
истечения. Поэтому наиболее высокотемпературные измерения в работах [1218, 3704, 4865] не
учитывались при проведении расчетов. Результаты расчетов энтальпии сублимации UO2(k) из данных
по давлению пара и равновесию обменных реакций приводят к величинам, совпадающим в
пределах погрешностей, средневзвешенные для этих двух серий исследований совпадают F08 ±20
и 598+30 кДж-моль). Принятая в справочнике величина является средневзвешенной из всех
приведенных в табл. 43.7 значений.
Таблица 43.7. Результаты определений Д8?ГОAГО2, к, 0) (в кДж • моль)
Авторы
Метод
Д8#°AЮ2, к, 0)
II закон III закон
Рен, Цефола, 1944 [5035]
Аккерман и др., 1956 [1218]
Иванов и др., 1963 [453]
Воронов и др., 1963 [248]
Озе, 1966 [3704]
Горбань и др., 1967 [4865]
Патторе и др., 1968 [3783]
Тетенбаум, Хант, 1970 [4500]
Риди, Хасанов, 1972 [5034]
Аккерман и др., 1980 [1233]
Де Мария и др., 1960 [2032]
Дроварт и др., 1967 [2117]
Купене и др., 1968 [1924]
Аккерман и др., 1969 [1235]
Горохов и др., 1974 [313]
Эффузионный, UO2(K)=UO2(r), 1873-2273 К,
5 точек
То же, UO2(K)=UO2(r), 1600—280Э а
Испарение из открытого цилиндра, 1Ю2(к)=
=Ш2(г), 1930—2160 К, 5 точек
Испарение с открытой поверхности, 1)О2(к)=
=U02(r), 1723—2573 К а
Эффузионный, и02(к)=и02(г), 2278—2768 К,
14 точек
То же, UO2(K)=UO2(r), 1850—2600 К а
Масс-спектрометрический, UO2(K)=UO2(r),
1890—2420 К а
Протока, UO2(K)=UO2(r), 2080—2705 К а
То же, UO2(K)=UO2(r), 2615—3391 К, 5 точек
Масс-спектрометрический, UO2(K)=UO2(r),
1800-2460 К а
То же, U02(r)+U(r)=2UO(r), 1876-1909 К,
3 точки
То же, UO2(r)+U(r)=2UO(r), 1890—2500 К б
То же, UO2(r)+Si(r)=UO(r)+SiO(r), 1890-
2500 К б
То же, UO2(r)+B(r)=UO(r)+BO(r), 2155—
2418 К, 19 точек
То же, UO2(r)+U(r)=2UO(r), 1540-2315 К а
То же, UO2(r)+U(r)=2UO(r), 1984—2358 К,
13 точек
645+50
565
601
660 ±50
623
653+50
656
627
646
657 +100
627
—
656
639
578+40
661
646+40
615+20
606 +25
624+25
604+20
603+30
605+20
609+25
585+30
610+20
602+40
602+40
589+35
589+30
614+40
606+40
Юнэ и др., 1981 [5071]
а Данные представлены в виде уравнения; б данные представлены в виде графика.
То же, UO2(r)+La(r)=UO(r)+LaO(r), 1630—
1901 К б
627
596+30
192
Уран и его соединения
Ш2(г)
UO2(r). Термодинамические свойства газообразного диоксида урана в стандартном состоянии
при температурах от 100 до 10 000 К приведены в табл. 999.
Молекулярные постоянные UO2, использованные для расчета термодинамических функций,
приведены в табл. 43.8. Исследования ИК-спектров молекул UO2, изолированных в
низкотемпературных матрицах (отсутствие частоты симметричного валентного колебания vx и величина
изотопного сдвига частоты v3), в работах Габельника, Грина и др. [2352, 2353, 24711 свидетельствуют, что
молекула UO3 в основном состоянии имеет линейную структуру симметрии Z)^1. Приведенный
в"табл. 43.8 момент инерции вычислен для значения r(U—О) = 1,80+0,05 А, оцененного по
величине межъядерных расстояний в молекулах SO, SO2, SO3 и UO, UOS. Погрешность значения /
составляет 1 -Ю9 г-см2. Основные частоты молекулы UO2 приняты по результатам исследования
ИК-спектра молекул UO2, изолированных в матрице из Аг [2353, 2471]; частоты v2 и v3
наблюдались экспериментально, для vx принято значение, вычисленное авторами [2353] по рассчитанным
ими силовым постоянным 2. Погрешности принятых значений оцениваются в 50, 10 и 15 см
соответственно для vl5 v2 и v3. Приведенные в табл. 43.8 энергии и статистические веса
возбужденных состояний UO2 приняты идентичными состояниям молекулы UF43.
Таблица 43.8. Значения молекулярных постоянных, а также в и рл, принятые для раечета
термодинамических функций UO2, UOJ, UOj, UO3 и UO3
Молекула
v2
v3
^5
ve
СМ
/ • 10S9
Г • CM2
a
UO2a 765 225B) 776 — — — 17 2 1
Ш?а 820 250B) 880 — — — 16 2 2
UOi"а 700 200 B) 700 — — — 18 2 2
U<V 844 746 186 853 212 152 3,2-1036 2 1
UOja 750 700 170 800 200 150 4-Ю36 2 2
а Нише приведены энергии и статистические веса Т{ (р{) для объединенных электронных состояний, Т\ — в см:
UO,: 500D), 2000C), 3000A), 4000E), 6000(8), 7000C), 9000A4), 11000A1), 12 000D), 14 000E),
17 000 A0), 19 000 A5), 22 000 F);
UOJ, UOj: 2000 B), 5000 B), 7000 B), 9000 B), И 000 B), 14 000 B);
UOi-: 200 D), 400 D), 4500 A2), 7000 B0);
UO3: 20 000 B), 21 000 B), 22 000 B), 23 000 B), 26 000 B), 28 000 B).
6 Приведено значение IAIBIC "Ю117 г3 -см6.
Термодинамические функции UO2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126) и A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом 13 возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчи-
* В первых исследованиях ИК-спектров молекул UO2, изолированных в матрицах [1201, 1204, 3212], принималась
нелинейная структура UO2. Однако в работах [1202, 1763, 2352, 2353] было показано, что авторы [1201, 1204,
3212] допустили ошибки в отнесении частот. В работе [2935] было обнаружено небольшое отклонение
молекулярного пучка UO2 в неоднородном электрическом поле, однако из-за значительной заселенности возбужденных
состояний при температуре эксперимента этот результат не позволяет сделать вывод о строении молекулы UO2.
3 Абрамович и Аквиста [1201] для частоты v2 молекулы UieO2 нашли значение 81 см, однако в более поздних
исследованиях [2470, 2471] это отнесение не подтвердилось.
3 Число и энергии валентных электронных состояний молекул, образованных атомами с внешними d - или
/-электронами, определяются главным образом этими электронами, за исключением того, что понижение симметрии
молекулы приводит к расщеплению и некоторому сдвигу уровпей. В справочнике при рассмотрении
возбужденных состояний молекул с внешними 5/-электронами учитывалась лишь конфигурация 5/-орбиталей; влияние
симметрии молекулы принималось во внимапие в том случае, если оно приводит к изменению статистического веса
основного состояния (см. UF4 (г)). Изменение природы лиганда и симметрии поля ведут к различиям в спектрах
молекул с одинаковой конфигурацией 5/-орбиталей. Однако это учитывалось в справочнике только при наличии
соответствующих экспериментальных данных. Поэтому, например, системы электронных состояний молекул
UFS, UFJ, UOJ и UOj (конфигурация 5/1) приняты одинаковыми. По той же причине они приняты
одинаковыми для молекул UO3 и UF4 (конфигурация 5/2).
193
TJOt(r) =^ Глава 43
тайных значений обусловлены неточностью принятых молекулярных постоянных, главны^:
образом числа и энергии возбужденных состояний, а также приближенным характером расчета; они!
составляют 4, 8, 15 и 20 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000, 6000и 10 000 К
(погрешность из-за неточности структурных параметров и основных частот не превышает 2 Дж-К-1Х
Хмель).
Таблицы термодинамических функций UO2(r) вычислялись ранее в работах Абрамовича и др.
[1200] и Грина [2463] (Т < 6000 К). Отдельные значения Ф°(Т) для Т < 3000 К вычислены
в работах [1665, 2032, 3783]. Расхождения данных [2463] и табл. 999 не превышают 3 Дж-К"хХ
Хмоль в значениях Ф°(Т) и обусловлены разной оценкой числа и энергии возбужденных
электронных состояний. Расхождения с результатами других расчетов достигают в значениях Ф°(Г):
9 Дж-К^-моль для [1200, 1665] и 21—26 Дж-К^-моль для [2032, 3783]. В работе [1200]
возбужденные электронные состояния не учитывались, однако при высоких температурах это
компенсируется тем, что для частоты v2 было принято низкое Значение (81 см); в работах [2032,
3783] для молекулы UO2 была принята нелинейная структура.
Константа равновесия реакции UO2(r)=U(r)-|-2O(r) вычислена с использованием значения
АгН°@)=1504,665 + 22 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации UO2(k). Этим величинам также соответствует
г, 0) = —476,234 + 20 кДж • моль.
1ГО2(г). Термодинамические свойства газообразного положительного иона диоксида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1000.
Молекулярные постоянные UO2, использованные для расчета термодинамических функций,
приведены в табл. 43.8. Грин и др. [2466] исследовали методом матричной изоляции ИК-спектры
продуктов реакции UO и UO2 с NO и NO2, в результате которых в матрице образуется UO2 вместе
с NO2 или NO". Отсутствие частоты симметричного валентного колебания vx в ИК-спектрах,
а также проведенный в работе [2466] анализ нормальных координат показали, что в матрице UOJ
имеет линейную структуру. На основании этих результатов в справочнике принята линейная
структура UOj (симметрия Dmh). Предполагая, что ионизация UO2 наряду с обнаруженным
возрастанием частоты v3 сопровождается уменьшением межатомного расстояния, приведенный
в табл. 43.8 момент инерции UOJ вычислен для r(U—О)=1,75+0,05 А, которое несколько меньше^
чем в молекуле UO2. Погрешность значения / составляет 2-109 г-см2. Основные частоты UOJ,
приведенные в табл. 43.8, основаны на результатах исследования ИК-спектров молекул UOJ,
изолированных в матрице из Аг [2466]. Для частоты v3 принято среднее значение из
наблюдавшихся в системах UieO2+NO2 и U^OJ+NO", для vx — среднее значение, вычисленное авторами
[2466] по найденным ими силовым постоянным, для v2 — значение, рассчитанное при подготовке
справочника в предположении, что /а/г2 : /г=0$4, как это имеет место для молекулы UO2.
Погрешности частот оцениваются в 80, 50 и 30 см соответственно.
Экспериментальные данные об электронном спектре UO2 в газообразном состоянии отсутствуют;
имеются только отрывочные сведения о спектрах поглощения UOJ в растворах и расплавах [1465,
4231]. Мак-Глинн и Смит [3455] провели расчет электронных состояний иона UO2 с учетом спин-
орбитального взаимодействия и эффекта поля лигандов. Авторы [3455] приняли, что 12 из 13
валентных электронов UO2 полностью заполняют молекулярные орбитали а+, а*, пи и пд, которые
получаются из 2р0 — орбиталей, а остающийся электрон занимает одну из орбиталей сра, §и?
этв, ав, возникающих из 5/и-орбитали, и получили для UO2 следующие электронные состояния
(в скобках приведены значения энергий в см): 2Ф*/2@), 2Дз/гB000), 2фг/гF752), 2Д=,д8168),
2Пу2A8 180) 2Пз,2B1 100), 2?+B4 000). Результаты этого расчета не согласуются с
экспериментальными данными работы [1465], в которой к UOJ было отнесено поглощение при 10 638 и 13 550 см,
и, поскольку количественные результаты расчетов по модели поля дигандов являются очень
ненадежными, в справочнике (см. примечание к UO2(r)), электронные состояния UO2 оценены по
экспериментальным и более надежным расчетным данным для соединений, содержащих ион U6 + в полях
различной симметрии и в окружении лигандов различного типа (см. текст по UF5(r)). Поэтому
в табл. 43.8 для UO2 приняты энергии электронных состояний, идентичные энергиям состояний
молекулы UF5. Погрешности этих значений оцениваются в 1000—5000 см.
Термодинамические функции UO2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126) и A. 129)
194
Уран и его соединения ТГО?(г), иО3(кУ
с учетом шести возбужденных состояний до уравнениям A. 168)—D* 170). Погрешности
рассчитанных значений обусловлены неточностью принятых молекулярных посшоявиых, прежде всего
энергий электронных состояний, и приближенным характером расчета; они составляют 4, 8, 12 в
16 Дж-Кт1.модь~х в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Таблица термодинамических функций UO^r) публикуется впервые.
Константа равновесия реакции иО?(г)+е(г)=и(г)-1-2О(г) вычислена с исшшьзованием
значения A,_ff°@)=979,665 +34 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(UO2)=525+25 кДж-моль-1.
Принятая величина основана на результатах измерений методом электронного удара, выпо'л-
ненных Раухом и Аккерманом [3943] E21 +20 кДж • моль) и Манном [5011 ] E31 +20 кДж •
Близкое, но существенно менее точное значение E31+^40 кДж-моль) было найдено в
Патторе и др. [3783]. Юнэ и др. [5071] получили значение 540 кДж-моль, что также близко
к принятому. Погрешности оценивались с учетом воспроизводимости измерений и возможного
влияния теплового возбуждения. .
Принятой величине соответствует
Д^^Ш!, г, 0) = 48,766+ 33 кДж-моль. :
UO^r). Термодинамические свойства газообразного отрицательного иска диоксида урхща
в стандартном состоянии нри температурах от 298,15 до 10 000 К приведены в табл. 1001.
Молекулярные постоянные UOg, использованные для расчета термодинамических -функций,,
приведены в табл. 43.8. Экспериментальные исследования и теоретические расчеты структуры в
спектра UOa отсутствуют. По аналогии с молекулами UO2 и UO? в справочнике принимается, что»
UO^ имеет линейную структуру (симметрия /?со*). Приведенный в таблице момент инерции вычислен
для значения r(U—О)=1,85+0,05 А, принятого несколько большим, чем в молекуле UO2.
Погрешность значения / составляет 2-109 г-см2. Основные частоты UOj, приведенные в табл. 43.8,
оценены для модели валентного силового поля в предположении, что силовые постоянные1
изменяются при переходе от UO2 к UOj так же, как при переходе от SO2 к SO2 и от UO? к UO2.
Погрешности принятых значений частот оцениваются в 100, 50 и 100 см. Приведенные в табл. 43.8
энергии возбужденных состояний UOj приняты такими же, как для UF3 (см. примеч. к тексту по UO2(r))»
за исключением того, что отброшены уровни, имеющие энергии выше иотенидала
ионизации UOj.
Термодинамические функции UOj(r) вычислены в приближении «жесткий ротатор t~-
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126) и,
A. 129) с учетом четырех объединенных электронных термов по уравнениям A. 168)—A. 170).
Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных, а также приближенным характером расчета и составляют 7, 10, 16 и 20 Дж-К~1-моль~х
в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Таблшщ термодинамических функций UO2(r) публикуется впервые. ,.-
Константа равновесия реакции иОг(г)=11(г)-|-2О(г)-|-е(г) рассчитана с использованием
значения ДгЯ°@)==1594,665 + 55 кДж-моль, соответствующего принятому в справочнике сродству
к электрону
Л0(Ш2)=— 90+50 кДж-моль.
Эта величина была оценена на основании принятых в справочнике значений ^0(UF4), Л0(МоО3)
и -40(WO3), а также величин 40(MoFe) и ^0(WF6) (см. UOg(r)).
Принятому значению соответствует
г, 0) =—566,234 + 55 кДж • моль.
UO3(k). Термодинамические свойства кристаллического триоксида урана в стандартном
состоянии при температурах 100—3000 К приведены в табл. 1002.
195
ЦО3(к) Глава 43
Значения постоянных, использованные для расчета термодинамических функций U03(k),
представлены в табл. 43.1. В справочнике во всем интервале температур за стандартное состояние
иО3(к) принимается моноклинная у-модификация [2179] как наиболее устойчивая х.
При Т •<¦ 298,15 К термодинамические функции y-UO3 вычислены по данным Веструма [4711],
который измерил теплоемкость а-, [3- и y-UO3 модификаций в температурном интервале 5—350 К.
Погрешности измерений оценены Веструмом в 1% при 10 К, 0,2% при 20 К и в несколько сотых
процента при температурах выше 20 К. Принятые значения ?°B98.15 К) и ?Т°B98,15 К)—Н°@),
приведены в табл. 43.1 и имеют погрешности 0,4 Дж-К^-моль и 0,05 кДж-моль. Результаты
измерений теплоемкости UO3 в работе Джонса и др. [2899] A4—295 К) выше данных [4711 ] на 3—
4% во всем'интервале температур.
При Т?> 298,15 К для теплоемкости UO3(k) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии и теплоемкости в работах Мура и Келли [3552] D16—
886И) й Попова и др. [806] C92—673 К). Содержание урана в образцах составило 83,02 и 83,0%
сбответственно; при обработке погрешности данных [3552] оценивались в 1%, данных
1806]-в 2%.
Экспериментальные сведения о температуре плавления и существовании U03 в жидком
состоянии в литературе отсутствуют. Термическая устойчивость кристаллических модификаций UO3
на воздухе изучена в работе Хоэкстры и Зигеля [2672]. Для всех пяти модификаций конечным
продуктом разложения является U3O8. Температура начала диссоциации на воздухе по данным
[2672] составляет 870 К для a-U03, 820 К для j3-UO3 и 950 К для y-UO3. 8- и s-UO3 при —740 К
переходят в 7-UO3 и при дальнейшем нагревании разлагаются до U3O8 вблизи 920 К. Учитывая, что
при высоких парциальных давлениях кислорода UO3(k), по-видимому, существует при более
высоких iтемпературах, в настоящем справочнике расчет термодинамических функций UO3(k)
проводился до 3000 Kf когда равновесное давление кислорода достигает 100 атм.
Погрешности табулированных значений Ф°(Г) при 298,15, 1000 и 3000 К могут достигать 0,3,
1 и 16 Дне-К-моль соответственно. Расхождения между термодинамическими функциями,
приведенными в табл. 1002 и справочниках [1395] (Г < 900 К), [2960] (Т < 1000 К), не
превышают 2,6 Дж-К-моль~1 в значениях Ф°(Т) и обусловлены уточнением теплоемкости UO, в
работе [4711].
Принятая в справочнике энтальпия образования
к, 298,15 К) = —1223.8 + 2,0 кДж • моль
основана на измерениях энтальпии растворения оксидов урана в сернокислом растворе сульфата
церия, выполненных Фицгиббоном и др. [2266] (см. пересчет в [1009]). Она совпадает с
рекомендацией КОДАТА-МСНС [1887].
Джойсон и О'Хара [2874] измерили энтальпии растворения у-1Ю3(к) и UF6(k) в
плавиковой кислоте, откуда для реакции у-иО3(к)+61Щадг)=иРв(к)+ЗН2О(ж) было получено
A,iPB98,15 К)=180,79±0,48 кДж-моль. Этой величине соответствует значение bfH°(t-UO3f
к, 298,15'К) =—1223,9+4,3 кДж-моль, совпадающее с принятым, но. имеющее значительно
большую погрешность из-за неточности использованной в расчетах энтальпии образования F~(aq)
[1887]. Эта работа подтверждает надежность и взаимную согласованность принятых в справочнике
энтальпий образования у-1Ю3(к) и UF6(k). Эти величины связаны еще одним термохимическим
циклом, включающим энтальпии растворения обоих соединений и UO2F2(k) в плавиковой кислоте
и в воде (см. ХГОа^Ук))-
Обработка данных Кордфунке и Алинга [1929] по равновесию реакции 6UO3(k)=2U3O8(k)+
+О2(г) (статический метод, 785—873 К, 11 точек) приводит к значениям —1230+7 кДж-моль
(II закон) и —1223,7+3,0 кДж-моль (III закон).
UO3 существует в пяти кристаллических модификациях: а [2458], р [2015], г [2179], 8 [4637], е [524] и
аморфном состоянии, различные условия получения, существования и термическая устойчивость которых изучены в
целом ряде работ (см.: Торопов и др. [1023]). На образование той или иной модификации UO3 оказывают влияние
структура исходного вещества, температура прокаливания, а в случае термического разложения — скорость
нагревания; тем не менее данных об условиях равновесия различных модификаций друг с другом пока нет.
Авторами [71, 4719] показано, что термическая устойчивость модификаций UO3 возрастает в ряду аморфный UO3,
S-UO3, a-UO3, Y-UO3, однако, согласно Кордфунке [1927], самой устойчивой является a-UO3. В работе [4282]'
приведены параметры решетки полиморфной модификации UO3, полученной при давлении 30 кбар и температуре
1373 К.
196
Уран и его соединения ~ UO3(r)
Помимо наиболее устойчивой при комнатной температуре у-модификации UO3 известны также
а-, Р-, 8- и е-модификации. Энтальпии перехода этих модификаций в T~UO3 были измерены
калориметрически в работах Хоэкстры и Зигеля [2672] иКордфунке [1928].
Давление пара триоксида урана в реакции у-1Ю3(к)=и03(г) вычислено с использованием
значения Дг^°@)= ^sH°(y-\JO3, к, 0)=423,998+15 кДж-моль, соответствующего принятым в
справочнике энтальпиям образования UO3 в кристаллическом и газообразном состояниях.
1ГО3(г). Термодинамические свойства газообразного триоксида урана в стандартном состоянии
при температурах от 100 до 10 000 К приведены в табл. 1003.
Молекулярные постоянные UO3, использованные для расчета термодинамических функций,
приведены в табл. 43.8. В справочнике на основании результатов исследования ИК-спектров
молекул UO3 и их изотопомеров в матрицах из Аг в работах Габельника и др. [2354] и Грина и др.
[2471 ] принимается, что молекула UO3 имеет Т-образную структуру (симметрия С2в) с
неэквивалентными связями U—О. Приведенное в табл. 43.8 произведение моментов инерции вычислено
по структурным параметрам r(U—О)=1,79 +0,05 A, r(U—О') = 1,76 + 0,05 А, ->Ю'—U—О'=180°
(где U—О — неэквивалентная связь, U—О' — две эквивалентные связи), оцененным в работе
[2354] с помощью эмпирических соотношений, связывающих величины г и /г. Погрешность
значения IaIbIc составляет 1-Ю14 г3>смв. Основные частоты UO3 приняты по результатам исследования
ИК-спектров молекул UO3, изолированных в матрице из Аг [2354, 2471]. Для частоты vlt не
активной в ИК-спектре, принято значение, полученное авторами [2354] по рассчитанным ими силовым
постоянным этой молекулы. Погрешности приведенных в табл. 43.8 основных частот оцениваются
в 50 см для vx и в 10—15 см для остальных частот.
Экспериментальные данные об электронных состояниях молекулы UO3 отсутствуют. Молекула
UO3 имеет замкнутую электронную оболочку и поэтому не должна иметь низколежащие
возбужденные состояния. Большое число экспериментальных и теоретических работ посвящено спектру
поглощения уранил-иона UOf+, который имеет так же, как UO3, замкнутую электронную оболочку
и незаполненные 5/-орбитали. Электронный спектр этого иона обусловлен переносом электрона
с 2р0- орбиталей на 5/и-орбитали. Согласно результатам анализа спектра поглощения UO|+b работе
Деннинга и др. [2047] и теоретического расчета Вуда и др. [4757] ион UO|+ в области 20 000—
28 000 см имеет шесть возбужденных состояний с р{=2, которые являются компонентами
состояний li3Afa2p0) (S5/u) и 1>3O(a2p0Vcf5/u), расщепляющихся вследствие спин-орбитального
взаимодействия. В молекуле UO3 электронные состояния должны быть обусловлены переносом электрона
с молекулярных орбиталей, образуемых 2]эо-орбиталями, на орбитали 2a1-fa2-t-2fc1-j-2b2t которые
дает 5/и-орбиталь в поле симметрии C2v. Пренебрегая различием в характере расщепления /-орбита-
лей в полях симметрии Dmh и C2t, энергии электронных состояний UO3 были приняты (см. табл. 43.8)
такими же, как для иона UOf+ [2047, 4757]. Погрешность этих значений оценивается в 5000 см;.
Термодинамические функции UO3(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128), A. 130) в приближении «жесткий ротатор—гармонический [осциллятор»
с учетом шести возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью принятых молекулярных постоянных и приближенным
характером расчета и составляют 2, 6, 10 и 15 Дж-К^-моль в значениях ФС(Г) при 298,15,
3000, 6000 и 10 000 К.
Термодинамические функции UO3(r) вычислялись ранее в работе Грина [2463] (до 6000 К),
а также в работах [2032, 3783] (значения Ф°(Т) при Т=1500-^2500 К). Расхождения данных
табл. 1003 и [2463] достигают 7 Дж-К~1-моль~1 в значениях Ф°(Т) и обусловлены главным образом
тем, что Грин учел в расчете неоправданно большое число электронных состояний. Расхождения
с результатами расчетов [2032], [3783] составляют 16 и 6 Дж-К-мрль~1 в значениях Ф°B500 К)
и обусловлены тем, что авторы этих работ приняли другую структуру молекулы U08 и оцененные
значения частот.
Константа равновесия реакции UO3(r)=U(r)+3O(r) вычислена с использованием значения
Дг#с@)=2070,214+17 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д ^°(UO3> г, 0) = —795 ± 15 кДж • моль.
Эта величина основана на данных, приведенных в табл. 43.9. Указанные в таблице погрешности
учитывают воспроизводимость измерений, неточность термодинамических функций^ термохими-
197
tJOj(p)
Глава 43
Таблица
}43ЛК 'Результаты
Авторы
определений Ду
Н° (Ш).„
р, 0) ^в
Метод
кДж
• моль 1)
II
закон
Зз, г
III
- 0)
закон
Аккерман и др., 1960 [1245]
Де Мария и др., 1960 [2032]
Дроварт и др., 1967 [2117]
Юнэ и др., 1981 {5071]
—815 —795 ±15
Протока, 1/3и3О8(к)+1/бО2(г)=иО3(г), 1200—
1370 К, данные представлены в виде уравнения
Масе-спектрометрическнй,и03(г)+и0(г)=2и02(г), —768+200 —772+50
2200—2322 К, 6 точек
То же, UO3(r)+UO(r)=2UO2(r), 2080—2500 К а
То же, UO3(r)+UO(r)=2UOs(r), 1920-2340 К а
-926
—922
—807 ±50
—789 ±50
а Данные представлены в виде графика.
ческнх величин и сечений ионизации. Наиболее точные измерения были выполнены Аккерманом
и др. [1245] и принятая в справочнике величина основана на этой работе. В работах [2032, 2117,
5071] были нолучены величины, совпадающие с принятой в пределах их погрешностей.
1Юз(г). Термодинамические свойства газообразного отрицательного иона триоксида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1004.
Молекулярные постоянные UO3, использованные для расчета термодинамических функций,
приведены в табл. 43.8. Экспериментальных данных о строении и спектре иона UOg в литературе
нет. По аналогии с UO3 в справочнике принимается, что в основном электронном состоянии UO
имеет Г-образную структуру симметрии C2ll, причем так же, как при переходе от UO2 к j
имеет место некоторое увеличение межатомных расстояний и уменьшение основных частот в UOg
по сравнению с UO3 x. Приведенное в табл. 43.8 произведение моментов инерции вычислено для
структурных параметров r(U—O)= 1,85+0,05 A, r(U—О')=1,82+0,05 А, ^0'—U—О' = 180°,
где (U—О)— неэквивалентная связь, (U—О') — две эквивалентные связи. Погрешность значения
IaIbIg составляет 2-10~114 г3-см6. Погрешности в оцененных значениях основных частот могут
достигать 10—20%. Электронные состояния UOg приняты идентичными состояниям UF5 (см. примеч.
к тексту по UO2(r)).
Термодинамические функции 1Юз(г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом шести возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчи-
тандых значений обусловлены грубой оценкой всех молекулярных постоянных и приближенным
характером расчета, они могут достигать 5, 10, 15 и 20 Дж-К^-моль в значениях Ф°(Г) при
298,15, ЗОЮО, 6000 и 10 000 К.
Таблица термодинамических функций UOg(r) публикуется впервые.
Константа равновесия реакции UOj(r)=U(r)+3O(r)+e(r) вычислена с использованием
значения ДгЯ°@)=2570,214+53 кДж-моль, соответствующего принятому в справочнике сродству
к электрону
A 0(UO3) = — 500 + 50 кДж-моль.
Эта величина была оценена по значению ^0(UF6) в предположении, что разность ^4O(UF6)—
—A0(\JOg) приближенно равна разности соответствующих величин для гексафторидов и триокси-
дов молибдена и вольфрама. Для тртоксидов использовались значения, принятые в гл. 31 и 32,
для гексафторидов — значения A0(MoF6) =—347 + 20 кДж-моль [4855а] и ^40(WFe) =—338 +
+20 кДж-моль4-1 [2385aj:
Принятому значению соответствует
г, 0) = —1295 + 52 кДж-моль.
В ряду UO|+—UOJ—UO2—UOg основные частоты уменьшаются; у UO|f электронная оболочка имеет замкнутую
кЪнфирУра^ию 5/*и евя9ь в этом шве, по-видимому, наиболее прочная, а допалнительные 5/-электроны в U0JE/1),
UO2E/a) и UQ$ EЯ> занимают несаязывающие или слаборазрыхляющие орбита ли.
193
Уран и его соединения и3О8{и)/:
U3O8(k). Термодинамические свойства кристаллического октокеида триурана в стандартном
состоянии при температурах 100—3000 К приведены в табл. 1005.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 43.1. В справочнике за стандартное состояние U3O8(k) в интервале 0—483 К принимается
ромбическая модификация a-U3O8 [3302], а в интервале 483—3000 К гексагональная
модификация a'-U3O8 [3305] 1.
При Т < 298,15 К термодинамические функции U3Og вычислены по результатам измерений
теплоемкости в работе Веструма и Грёнволда [4713] E—350 К); погрешность измерений оценена
авторами [4713] в 5% при 5 К, 1% при 10 К и 0,1% выше 25 К. На кривой теплоемкости Веструм
и Грёнволд обнаружили А-образную аномалию с максимумом при 25,3 + 0,1 К и энтальпией
перехода 0,05 кДж-моль. Температура перехода согласуется с результатами измерений
магнитной восприимчивости [1350, 3213]. Леаск и др. [3213], измеряя магнитную восприимчивость
U3O8 в интервале 1,5—44 К, нашли острый максимум при 4,2 К и незначительные эффекты при 8
и 25,3 К. Последнее значение совпадает с температурой максимума аномалии теплоемкости, а
эффект при 8 К невелик и не получил подтверждения в [4713]. Острый максимум магнитной
восприимчивости при 4,2 К очень четкий, что позволяет принять Ttr= 4,2 + 0,1 К. Принятые
значения 5°B98,15 К) и Я°B98,15 К)—Я°@) приведены в табл. 43.1, их погрешности оцениваются
в 0,5 Дж-К^-моль и 0,1 кДж-моль соответственно. Измерения средней теплоемкости U3O8(k),
выполненные Донатом [2094] B73—373 К) и Русселом [4104] (82—314 К), имеют значительный
разброс и не учитывались.
При Т >¦ 298,15 К для теплоемкости U3O8(k) принято уравнение, полученное на основании
результатов измерений теплоемкости в работах Попова и др. [806] C80—875 К), Гирдхара и
Веструма [2412] C03—555 К), Инабы и др. [2775] C10—970 К) и измерений энтальпии в работах
Марчидана и Чопека [3369] C32—1227 К), Маглича и Герака [3339] B73—927 К). На кривых
теплоемкости авторы [2412] и [2775] обнаружили А-аномалию с максимумом при 482,7 и 483 К
соответственно. Маглич и Герак [3339] наблюдали небольшую аномалию в температурной
зависимости энтальпии при 481 К. По данным рентгенографических исследований [2190, 2775, 3305,
3537, 3538, 3539] переход при 483 К обусловлен превращением низкотемпературной ромбической
a-модификации в высокотемпературную гексагональную модификацию. До точки превращения
данные [806, 2412, 2775, 3339] хорошо согласуются между собой и с данными Веструма и
Грёнволда [4713]. Выше 483 К результаты измерений крайне противоречивы. Попов и др. [806]
отметили аномальный ход теплоемкости в интервале 593—673 К, и объяснили его изменением
структуры U3O8. Инаба и др. [2775] зафиксировали еще две Х-образные аномалии теплоемкости при
568 и 850 К с энтальпиями превращения 0,15 и 0,31 кДж-моль; эти переходы авторы связывают
с процессами разупорядочения. Хомяков и др. [1081 ], измерившие теплоемкость в интервале 641 —
1233 К, обнаружили два превращения при 1043 и 1213 К с энтальпиями 1,1 и 4,6 кДжмоль.
Следует отметить, что невоспроизводимость кривых нагревания и охлаждения в этой работе
достигает 10%, имеют место также значительный разброс и незакономерность в температурной
зависимости теплоемкости, затрудняющие обработку данных [1081]. Пауэре и др. [3875]
определили среднюю теплоемкость U3O8 в интервале 523—1373 К и нашли ее постоянной до 1250 К,
небольшой рост теплоемкости наблюдался от 1250 до 1373 К. В силу противоречивости данных
и того обстоятельства, что ни одно из превращений обнаруженных в [806, 1081, 2775], не нашло
подтверждения при измерениях энтальпии в работах [3339, 3369, 3875], переходы U3O8 при
температурах выше 483 К не учитывались.
1 К настоящему времени в литературе нет единого мнения относительно числа и природы полиморфных
модификаций U3O8. При нормальном атмосферном давлении и температуре известны три различных формы ромбической
модификации (a-, -8-h8-U3O8). из которых наиболее устойчивой и изученной является a-модификация.
Существование 8-и„О8 [1313, 2925, 3537, 3538] подвергается сомнению авторами [1098, 2615, 3304], полагающими, что В-
U3O8 не является самостоятельной модификацией, а представляет смесь фаз a-U3O8 (в незначительном
количестве) и р-ия08 (основная фаза).
Б работе Цмгуаова и др. \\Ш] получена более плотная и устойчивая при высоком давлений кубическая
фаза U3O8. Вильсон [4740] обнаружил полиморфную модификацию y-U3Os при давлениях > 16 000 атм и
температурах около 673 К, кристаллизующуюся в гексагональной сингонни. В [4740] показано, что при комнатной
температуре а- и 3-U3O8 переходят с повышением давления до —.100 000 атм в почти «аморфное» состояние.
Аморфная фаза U3O8 появляется и при 25 000 атм в интервале температур 473—573 К. Стпб и Брукляхер [4381]
описали орторомбическую модификацию, полученную из a-U3O8 под давлением более 50 атм и названную ими
р-и3о8.
199
UgOe(K). Л Глава 4?
t
Уравнение теплоемкости для a-U3O8 в интервале 298,15—483 К выведено с
использованием значений С;B98,15 К) = 238, С;C60 К) = 257,3, Q420 К) =4267,4 и С;D83 К) =
= 289,5 Дж*К-моль~1. Вторая и третья величины получены усреднением хорошо согласующихся
между собою данных [806, 2412, 2775]; значение теплоемкости в точке перехода приведено в
работе [2412]. Уравнение теплоемкости для a'-U3O8 в интервале 483—1309 К получено на
основании измерений энтальпии в работах [3369] и [3339] с учетом данных по теплоемкости [806, 2775].
При этом значения С°E00 К) = 273 и С°G00 К) = 284 Дж-К^-моль, использованные при
выводе уравнения, получены совместной обработкой данных [3369] и [3339] (их погрешности
оценены в 2%). Значения С°A000 К) = 299 и С°A300 К) = 313 Дж^К^-моль получены
корректировкой результатов совместной обработки по данным о теплоемкости, полученным в работах
[806, 2775], которые имеют более высокие значения.
Температура превращения D83 + 1 К) принята по хорошо согласующимся измерениям [529Г
2412, 2775, 3305, 3339, 3537, 3538, 3539, 4216]; результаты, полученные в работах [857, 2190,
2673, 2789, 3684], представляются ненадежными. Энтальпия превращения, которое по своему
характеру близко к фазовому переходу второго рода, принята равной нулю. Имеющиеся в
литературе определения энтальпии перехода, выполненные интегрированием аномальной части
теплоемкости, приводят к различающимся значениям @,06 [3537,35381,0,14 [2775] и 0,17 кДж-моль-
[2412]).
Экспериментальные данные о температуре плавления и существовании U3O8 в жидком
состоянии в литературе отсутствуют. Термическая устойчивость U8O8 на воздухе изучена в работах
[152, 2069, 2070, 3328, 4108]. Потеря кислорода отмечается уже,при —770 К [2070], однако до
1723 К количество кислорода в оксиде все еще соответствует формуле UO2 e3, при 1773 К
начинается интенсивное разложение U3O8 с образованием летучих оксидов урана, а при 2173—2223 К
образец испаряется почти целиком за короткий промежуток времени [152]. По данным
рентгенографического анализа [152] начиная с 1773 К вплоть до температуры плавления в равновесии
с кислородом при атмосферном давлении находится U4O9. Учитывая, что при высоких парциальных
давлениях кислорода U3O8(k), по-видимому, существует и при более высоких температурах, в
настоящем справочнике расчет термодинамических функций U3O8(k) проводился до 3000 К —
температуры, при которой равновесное давление кислорода достигает 100 атм.
Погрешности вычисленных значений Ф°(Г) при 298,15, 2000 и 3000 К оцениваются в 0,1, 11
и 20 Дж-К^-моль соответственно. Расхождения между термодинамическими функциями,
приведенными в табл. 1005 и в справочнике Барина и Кнакке [1395], не превышают 1,5 Дж -К -моль"*
в значениях Ф°(Г) при Т < 900 К.
Константа равновесия реакции U3O8(k) = 3U(r) + 8О(г) вычислена с использованием
значения Дг#°@) = 7142,579 + 24 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
bfH0(UsOu, к, 298,15 К) = — 3574,8 ±2,5 кДж • моль.
Принятая величина основана на прецизионных измерениях энтальпии сгорания урана в
кислороде, выполненных Хьюбером и Холли [2739] (см. табл. 43.10). Работы [808, 2742] привела
к значениям, совпадающим с принятым в пределах их погрешности. Принятое в справочнике?
значение совпадает с рекомендацией КОДАТА—MGHC [1887].
Таблица 43.10. Результаты определений b.fH° (U3O8, к, 298,15 К) (в кДж-моль)
Авторы
Метод
, к, 298,15 К)
Микстер, 1912 [3522] Калориметрический, сжигание урана в кислороде —3536
То же, измерение энтальпий реакций U и USO8 —3589
с Na2O2
Хьюбер и др., 1952 [2742] То же, сжигание урана в кислороде, 20 измерений — 3570,9±7,0
Попов, Иванов, 1957 [808] То же, 17 измерений —3583,6±13
Хьюбер, Холли, 1969 [2739] То же, 8 измерений —3574,8±2,5
200
Уран и его соединения U4O9(k)
U4O9(k). Термодинамические свойства кристаллического ноноксида тетраурана в стандартном-
состоянии при температурах 100—3500 К приведены в табл. 1006.
Значения постоянных, использованные для расчета термодинамических функций, приведены-
в табл. 43.1. Б справочнике за стандартное состояние U4O9(k) во всем интервале температур
принимается кубическая модификация (структурный тип флюорита) [1463, 1464, 4734].
При Т <; 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работах Флотова и др. [2275] A,6—23 К) и Осборна и др. [3730] E—310 К). Принятые-
значения ?"B98,15 К) и #°B98,15 К)—Н°@) приведены в табл. 43.1 и имеют погрешности 0,7 Дж X
XК-моль и 0,1 кДж-моль соответственно. Сообщение авторов работ [1350, 3213] о наличии;
магнитного превращения U4O9(k) при 6,4 К опровергнуто Флотовым и др. [2275], которые
установили отсутствие аномалии теплоемкости в этой области.
При Т >¦ 298,15 К для теплоемкости U4O9(k) приняты четыре уравнения, выведенные на
основании результатов измерений теплоемкости в работах Веструма и др. [4716] A90—399 К), Гото-
и Найто 124481 B97—515 К), Грёнволда и др. [2499] C00—1000 К), Инаба и Найто [2774] A80—
470 К) и измерений энтальпии в работе Маклеода [3336] (845—1568 К). U4O9 претерпевает два
фазовых перехода при 348 и 1398 К, относящихся к переходам типа порядок—беспорядок.
Превращение при 348 К обусловлено перераспределением атомов кислорода в кристаллической решетке
без изменения пространственной группы. Высокотемпературный фазовый переход при 1398 К
определен Маклеодом [3336] как разупорядочение—диспропорционирование и сопровождается
превращением U4O9 в неупорядоченную фазу состава UO2+;t. (основная фаза) и фазу U02elf (при-
сутствует в качестве небольших примесей). Уравнение теплоемкость в интервале 298,15—348 К,
описывающее восходящую ветвь Х-кривой, выведено с использованием значений теплоемкости,
полученных усреднением согласующихся между собой данных [2499, 2774, 3336, 4716]:
С;B98,15 К) = 293,34, С;C10 К) - 302, С;C25 К) = 323 Дж-К^-моль и Я°C48 К)—
—Я0B98,15 К) = 16,77 кДж-моль. Результаты измерений [2448] в указанном интервале
температур не учитывались, поскольку А-кривая в этой работе смещена в область более низких
температур и максимальное значение теплоемкости приходится на 330 К. Уравнение теплоемкости
для ниспадающей ветви ^-кривой в интервале 348—380 К выведено по данным Веструма и др.
[4716], которые измерили теплоемкость в области Х-аномалии с погрешностью 0,1% на хорошо
аттестованном образце. Данные [2499] и [2774] в этом интервале температур не учитывались,
так как обнаруживают резкое падение теплоемкости выше 348 К, а погрешности измерений [2774]
выше 2%. При выводе уравнения были использованы значения теплоемкости, полученные в
работе [4716]: С;C50К) = *374,9, С;C60 К) = 335 и С°C80 К) = 311,8 Дж-К^-моль.
Уравнение теплоемкости в интервале 380—1398 К получено в результате совместной обработки данных
[2448, 2499, 2774, 3336], при этом погрешности данных [2499] оценены в 0,5%, данных [2774,
3336] — в 1% и данных [2448] — в 2%. Для температур выше 1398 К для смеси фаз UO2+a. и ЬО2ж<п
принято уравнение, выведенное Маклеодом [3336] на основании измерений энтальпии.
Поправкой на негомогенность образца можно пренебречь, так как она составляет ~0,01%.
Температура перехода порядок—беспорядок C48 ± 2 К) принята по результатам
калориметрических измерений Веструма и др. [4716] и Грёнволда и др. [2499]; менее точные значения
получены в работах [1459, 1460, 2090, 2448, 2498, 2774, 2790, 2791, 3413, 3621, 3622, 3624].
Температура перехода разупорядочение—диспропорционирование A398 + 5 К) принята по
согласующимся между собой результатам [855,856,1459, 1460, 3260, 3336, 4019]. Энтальпия ^-превращения
при 348 К составляет 2,64 + 0,20 к Дж-моль по результатам калориметрических определений
[2448, 2499, 3336, 4716]; изотермический вклад в энтальпию перехода принят равным нулю.
Энтальпия перехода при 1398 К B,34 ± 0,10 кДж-моль) принята по данным Маклеода [3336].
Поскольку, высокотемпературное превращение развивается в достаточно узком интервале (—10°),.
принятое значение отнесено за счет изотермической энтальпии превращения. Сведения о
температуре плавления и существовании U4O9 в жидком состоянии в литературе отсутствуют.
Термическое разложение начиная с ~1273 К сопровождается обеднением конденсированной фазы
кислородом от U4O9 до UO2. Учитывая, что при высоких парциальных давлениях кислорода U4O9(k),
по-видимому, существует при более высоких температурах, в настоящем справочнике расчет
термодинамических функций U4O9(k) проводился до 3500 К, когда равновесное давление кислорода»
достигнет 100 атм.
201
UF(r) Глава 43
t
Погрешности вычисленных значений Ф°(Т): при 293,15, 1QQQ и 3000 К оцениваются в 0,4, 3
и 22 Дж -К -моль соответственно. Расхождения между термодинамическими функциями U4O9(k),
приведенными в табл. 100в\и в справочнике Барина и Кнакке [1395] (Т ^ 900 К), достигают
3,4 Дж-К^-моль в значениях Ф°(Г) и обусловлены уточнением теплоемкости U409 в работах
[2499, 2774, 3336].
Константа равновесия реакции U4O9(k) = 4U(r). -+- 9О(т) вычислена с использованием
значения Ar.ffo@) = 8858,732. ± 32 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
bfH°{\}fi9, к, 298,15 К) = —4512 + 7 кДж • моль.
Эта величина основана на калориметрических измерениях энтальпии реакций взаимодействия
U4O9(k), UO8(k) a UO2(k) с сернокислым раствором сульфата церия, выполненных Фицгиббоном
и др. [32661. Из этих измерений с учетом отличия состава использованных образцов от стехиоме-
чрического (см. [1009]), для реакции 0,920UQa 9ва(к) + 3,080UO2028(k) = U4O9(k) было
найдено значение ДГЯСB98,15 К) = —33,7 + 6,3 кДж-моль. Поправки, учитывающие изменение
энтальпии образования UO2{k) в UO3(k) при переходе к указанным нестехиометричеекш*
составам, приняты по данным справочника [1009].
Бурдезе и Аббатиста [1723] измерили энтальпии растворения U4O9(K), UO2(k) и U3O8(k) в
азотной кислоте. Этим данным соответствуют два значения энтальпии образования U4O9(k):
—4514,0 кДж-моль (цикл с UOZ) и —4527,7 КДж-моль (цикл с U8©8). He исключено, что в
условиях опытов [1723] наряду с NO образовывались другие окислы азота, которые не учитывались
при обработке экспериментальных данных, что могло сказаться на результатах.
В литературе известны работы [1334, 2130, 2131, 3023, 3393, 4019, 4431, 4432, 4433], в
которых исследовались равновесия реакций, включающих U4O9(k). Расчеты по этим работам приводят
к значениям энтальпии образования U4O9(k) значительно менее точным по сравнению с принятым
в справочнике. Наиболее точные из этих измерений [2130, 2131, 3023, 3393] проведены методом
ЭДС (реакция 4UO2(k) -\- 1/2О2(г) = U4O9(k)) и приводят к средней! величине Д ,ff°(U4O9* K>
298,15 К) = -4513 ± 10 кДж-моль.
UF(r). Термодинамические свойства газообразного фторида урана в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 1007.
Спектры UF не исследовались, а квантовомеханические расчеты этой молекулы не
проводились. Приведенные в табл. 43.3 молекулярные постоянные UF приняты по результатам
приближенных оценок. Оценки были выполнены так же, как для молекулы UO. Молекула UF имеет
нечетное число электронов, поэтому во всех электронных состояниях, включая основное,
которое, по-видимому, принадлежит конфигурации ...ф*8/о2 или cp^S^S^, квантовое число 2^0.
При оценке суммарных статистических весов групп электронных состояний принималось, что:
а) связв в UF осуществляется за счет перехода одного электрона атома U на связывающую о-ор-
биталь; б) все остальные орбитали заполняются за счет остающихся пяти электронов, которые
рассматриваются как принадлежащие иону U+, находящемуся в одной из шести электронных
конфигураций (...fs2, ...fsd$, ...f*s, ...f(P, ...fd2s, .../W); в) состояния, имеющие во внешней оболочке
больше одного разрыхляющего электрона, рассматривались как несвязанные или слабосвязанные;
г) принималось, что состояния, соответствующие двум первым конфигурациями4", имеют энергии
от 0 до 105 000 см (величина, равная D0(UF) +/0(U)), соответствующие следующим четырем
конфигурациям — от 3000, 7000, 12 000 и 12 000 см до того же предела. Погрешности
оцененных статистических весов могут достигать коэффициента 2—3.
Приведенное в табл. 43.3 значение r,(UF) = 2,03 ± 0,03 А в основном состоянии оценено на
основании значений r(U—F) в молекулах UFe и UF4 (см. ниже), величины r(M—F) в молекулах
ThF4 B,14 А [398]) и CeF4 B,035 А [784]), PrFa и HoF3 B,09 и 2,03 А [4872]), ге (LaF) = 2,027 А
[4453] и ^(HoF) = 1,9399 А [2376]. Данные [11} для LaF3 и NdF3 не использовались, так как
они содержат явные ошибки. Принятое значение и>„ = 600 + 30 см основано на значении
силовой постоянной молекулы UFe (см. ниже). Оно находится в разумном согласии с известными
данными для молекул LnF [2376, 4453].
Термодинамические функции UF(r) рассчитаны по уравнениям A.3)—A.6), A.9), A. 10),
A. 93)—A. 95). Значения @вя и ее производных вычислялись по уравнениям A. 90)—A. 92) с уче-
202
Уран и его соединения UF+(r)
том семи объединенных электронных термов в предположении, что , Q^.вр.= 0,5p-QW вр .
Величина (?S.bP. и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью
уравнений типа A. 82). Б расчете учитывались все колебательно-вращательные уровни основного
состояния с значениями / ^ Jma,x,Pt последние определялись из условий A. 81). Энергия
уровней вычислялась по уравнениям A. 65), A.34) и A. 64а), их вырождение учитывалось
статистическим весом 2. В табл. 43.4 приведены значения ушвх и /Ит, а также коэффициентов в этих
уравнениях, полученные по принятым молекулярным постоянным с учетом условия A. 59).
Погрешности рассчитанных термодинамических функций UF(r) обусловлены приближенной
оценкой всех постоянных этой молекулы, при Т > 1000 К основной источник ошибок —
отсутствие данных о возбужденных электронных состояниях UF, они особенно значительны в С(Т)
и Нь(Т)—Н°@). Погрешности в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К оцениваются
в 2, 4, 8 и 12 Дж-К^-моль.
Термодинамические функции UF(r) ранее вычислялись в работах Гурвича и др. Е338, 2536]
(до 10 000 К), Галкина и др. [742а] (Т < 5000 К). Расхождения данных [338, 2536] И табл. 1007
составляют 0,3—10 и 1—17 Дж-К~1-моль в значениях Ф°(Г) и S°(T); они обусловлены
уточнением оценки постоянных UF в основном состоянии и увеличением числа учитываемых
электронных состояний. Расхождения с данными [742а] изменяются от —6 до +20 Дж-К>моль
в значениях Ф°B1); при низких температурах основной источник ошибок в этой работе —
пренебрежение вырождением уровней основного состояния, выше 2000 К — пренебрежение
возбужденными состояниями UF.
Хилденбранд [4974] на основании масс-спектрометричеекого изучения равновесия реакций
UF(r) нашел <S°(UF, г, 2300 К) = 348 + 8 Дж-К^-моль, это значение хорошо согласуется
с приведенным в табл. 1007 C47 + 4 Дж-К~1-моль~1).
Константа равновесия реакции UF(r) = U(r) + F(r) вычислена с использованием принятой
в справочнике энергии диссоциации
ДД^О) = D0(tJF)-=s 660 + 15 кДж • моль?» 55 200 ± 1200 схГ1.
Принятая величина является средневзвешенной из результатов масе-сняктрометрических
исследований, проведенных Хилденбрандом [4974] (U(r) + BaF(r) = UF(r) + Ва(г), 2222—2505 К,
ДД°B250 К) = -74,4 + 3,0 (II закон); UF(r) + BaF(r) = UF2(r) + Ва(г), 2034—2466 К,
ДгЯ°B250 К) = 20,1 + 2,5 (Ы закон), сообщены только полученные значения &,№(!)) и
Гороховым и др. [4868] BUF2(r) = UF3(r) + UF(r), 1616 и 1667 К, 2 точки). По этим данным были
вычислены значения D5(UF), равные 665 + 15 кДж-моль (II закон), 657 ;+- 28 кДж-моль
(II закон) [4974] и 650 + 50 кДж-моль (III закон) [4868] х. Погрешность принятой энергии
диссоциации в основном обусловлена неточностью термодинамических функций, использованных
в расчетах.
Принятой энергии диссоциации соответствует
г, 0) = —47,860 + 17 кДж • моль.
UF+(r). Термодинамические функции газообразного ионизированного фторида урана в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 1008.
Спектры UF+ не исследовались, а квантовомеханические расчеты этой молекулы отсутствуют.
Приведенные в табл. 43.3 молекулярные постоянные UF+ основаны на результатах приближенных
оценок, выполненных так же, как для молекул UO и UF. UF+ имеет четное число электронов,
поэтому эта молекула может иметь электронные состояния как с 2 > 0, так и с 2 = 0. Можно
предполагать, что основное состояние UF+, так же как у молекулы UO, принадлежит
конфигурации ...tp^o, или ...y2fbfas. Это не позволяет сделать однозначный вывод о том, равно ли
квантовое число 2 в, основном состоянии нулю или нет. В справочнике принято, что основное состояние
1 После завершения подготовки этого тома авторы получили рукопись статьи Лау и Хнлденбранда [5003а], в
которой изложены окончательные результаты масс-спектрометрического исследования низших фторидов урана,
известного ранее по предварительному сообщению [4974]. Приведенные в этой работе значения Do (UF),
AfH° (UF2, г), AfH° (UF3, г), lSfH° (UF4, r), /0 (UF), Io (UF2), /0 (UF4) близки к принятым в справочнике.
203
UF~(r) Глава 4$
UF+ является вырожденным (Q =^ 0), так как если 2=0, разность энергий" компонент 0+ и О"
вряд ли превышает 100—200 см.
Оценка суммарных статистических весов групп электронных состояний UF+ была выполнена
так же, как для молекулы UF. Рассматривались состояния, возникающие из шести конфигураций
электронной оболочки U+2: ...fd, .../4 (в области от 0 до 150 000 см, последняя величина равна
D0(UF+) + /0(U+)), ...fs (выше 5000 см), ...fd2, ...fds и .../V (выше 30 000 см). При этом
исключались те состояния, которые соответствуют конфигурациям электронной оболочки UF+,
имеющим больше одного электрона на разрыхляющих орбиталях Эту, о/5 nd, od, os. Погрешности
приведенных в табл. 43.3 значений статистических весов оцениваются коэффициентами 2—3.
Значения постоянных ш„ и г, молекулы UF+ в основном состоянии, приведенные в табл. 43.3,
были оценены на основании принятых значений постоянных молекулы UF и предположения, что»
ионизация UF сопровождается уменьшением величины гд и возрастанием величины щ, как это-
имеет место в случае YF и LaF. Погрешности оцененных величин составляют 50 см и 0,06 А.
Термодинамические функции UF+(r) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
A.93)—A. 95). Значения (?вн и ее производных вычислялись по уравнениям A. 90)—A. 92) с
учетом семи объединенных электронных термов в предположении, что Q^JJL_BV= 0,5р,@<*]-вр.
Величина (?ю].вР и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью
уравнений типа A. 82). В расчете учитывались все колебательно-вращательные уровни энергии
основного состояния с значениями / <! /ши „, последние определялись из условий A. 81). Энергия
уровней находилась по уравнениям A. 65), A. 34), A. 64а), их вырождение учитывалось
статистическим весом 2. В табл. 43.4 приведены значения vm&x и /]]т, а также коэффициентов в этих
уравнениях, полученные по принятым молекулярным постоянным UF+ с учетом
условия A. 59).
Погрешности рассчитанных термодинамических функций при температурах до 1000—2000 К
обусловлены главным образом отсутствием данных о величине 2 в основном электронном
состоянии UF+, при более высоких температурах — о возбужденных электронных состояниях этой
молекулы. Погрешности в значениях ФС(Г) при 298,15, 3000, 6000 и 10 000 К оцениваются в 4,
8, 12 и 15 Дж-К^-моль.
Термодинамические функции UF+(r) ранее вычислялись в работе Гурвича и др. [338, 25361
(до 10 000 К). Расхождения данных [338, 2536] и табл. 1008 составляют от —4 до 22 и от —6 до
30 Дж-К-моль в значениях Ф°(Т) и S°(T)f они обусловлены уточнением оценок
молекулярных постоянных UF+ в настоящем издании.
Константа равновесия реакции UF +(г) + е(г) = U(r) + F(r) вычислена с использованием
значения А,Ло@) = 60,0 + 52 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(UF) = 600 + 50 кДж-моль.
Принятая величина основана на результатах измерений методом электронного удара в работе
Горохова и др. [4868] F30 + 20 кДж'Моль) и предположении, что потенциалы ионизации
двухатомных соединений урана должны быть меньше величины /0(U) г. Ей соответствуют
A^UF*, г, 0) = 552,140 ±53 кДж. моль,
D0(UF+) = 657,632 + 52 кДж • моль яа 55 000 ± 4000 см.
UF~(r). Термодинамические свойства газообразного отрицательно заряженного фторида урана
в стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 1009.
Молекулярные постоянные, использованные для вычисления термодинамических функций,
приведены в табл. 43.3. Спектры молекулы UF" не исследовались, а ее квантовомеханические
расчеты не проводились. Поэтому систематика электронных состояний и молекулярные
постоянные UF" были оценены при подготовке справочника так же, как для молекулы UF. Оценка
плотности электронных состояний с энергиями меньше потенциала ионизации TJF" основана на
предположении, что все они связаны с электронными конфигурациями, соответствующими конфигу-
1 См. примеч. 1 к тексту по UF (г).
204
Уран и его соединения ,. UF2(r)
рации атома U ...fds*, где / — «р/> 8/, d — bd, a s — о( — молекулярные орбитали. Состояния,
¦соответствующие конфигурациям хотя бы с одним разрыхляющим электроном (я:,, о, или ой)
во внешней оболочке молекулы, не рассматривались. Принималось, что стабильные состояния
распределены равномерно вплоть до энергии 84 500 см (D0(UF~) + -^o(U)), и учитывались
состояния с энергиями меньше величины потенциала ионизации молекулы UF". При такой оценке
суммарный статистический вес состояний с энергиями до 8000 см равен 20, его погрешность
оценивается в 50%. На основании рассмотрения возможных электронных конфигураций принято, что
в основном состоянии UF" Q =^= 0.
Молекулярные постоянные UF" в основном состоянии оценены по принятым постоянным
молекулы UF с учетом тенденции к увеличению межъядерного расстояния и уменьшению
колебательной частоты при увеличении числа несвязывающих электронов во внешней оболочке
молекулы при одном и том же заряде ядер (см. UF+, TiO+, ZrO+ и НЮ +). Погрешности оцененных
значений ше и В, составляют 50 и 0,01 см.
Термодинамические функции UF"(r) рассчитаны по уравнениям A. 3)—A. 6), A. 9), A. 10),
•\1. 93)—A. 95). Значения Q3B и ее производных вычислялись по уравнениям A. 90)—A. 92).
Статистическая сумма по колебательно-вращательным уровням энергии основного состояния и ее
производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
.колебательным уровням энергии и интегрированием по значениям / с помощью уравнений типа
'{1. 82), вырождение вращательных уровней учитывалось статистическим весом 2. В расчете
учитывались все колебательно-вращательные уровни с значениями / ^ Jm&Xt „, последние
находились из условий A. 81). Энергия уровней вычислялась по соотношениям A. 65) и A.64а).
Значения коэффициентов Ykl в этих уравнениях, а также величин 1>шах и JUm приведены в табл. 43.4;
коэффициент Y03 получен из условия A. 59), остальные идентичны соответствующим постоянным
шз табл. 43.3.
Погрешности рассчитанных термодинамических функций при низких температурах
обусловлены главным образом неточностью оценки постоянных UF" в основном состоянии, при высоких
температурах — отсутствием данных о возбужденных электронных состояниях этой молекулы.
Погрешности в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К оцениваются в 4, 8, 12 и
20 Дж-К^-моль.
Термодинамические функции UF"(r) ранее вычислялись в работе Гурвича и др. [338, 2536]
{до 10 000 К). Расхождения данных этой работы и табл. 1009 (в пределах от 0,1 до 5,7 и от 0,8
до 14 Дж-К^-моль в значениях Ф°(Г) и S°(T) соответственно) обусловлены уточнением оценки
числа учитываемых возбужденных состояний UF" при подготовке настоящего справочника.
Константа равновесия реакции UF"(r) = U(r) + F(r) + е(г) вычислена для значения ДгЯ°@) =
= 760 + 52 кДж-моль, которое соответствует принятому в справочнике сродству к электрону
А0(Ш) — —100 ± 50 кДж-моль.
Эта величина была оценена на основании сопоставления значений ^40(UFB+1) и D0(UFB—F")
в молекулах UF—UFe и UF"—UFJT. Принятой величине соответствуют
г, 0) = —147,86 ±53 кДж . моль,
D0(UF~) = 431,99 ± 52 кДж моль« 36 000 ± 4300 см.
UF (г). Термодинамические функции газообразного дифторида урана в стандагтномсостоянии
при температурах от 100 до 10 000 К приведены в табл. 1010.
Молекулярные постоянные UF2, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальных данных о строении и основных частотах молекулы
TJF в литературе нет. По аналогии с молекулами дигалогенидов лантанидов и UC12, нелинейная
•структура которых установлена из ИК-спектров молекул, изолированных в низкотемпературных
матрицах х [2025, 2581], в справочнике для UFa принята в основном состоянии нелинейная струк-
тура'(симметрия C2t). Приведенное в таблице произведение моментов инерции соответствует зна-
Валентные углы рассчитаны по изотопическим сдвигам (Д С1—М—01=130^-140° для SmCl2) EuGl2, и YbCl2
[2025, 2581]') или относительным интенсивностям полос vu и v3 (EuF2 A10°), UG12 A00°) [2581]).
205
UF2(r)
Глава 43"
Таблица 43.11. Значения мвлекулярных постоянных, а также а и рх, принятые для расчета
термодинамических функций UF2, UF+, UFj-, UF3, UFJ, VF*,. UF4, UFJ, UFj, UF6, UF+
DFj, UFe и UFe
Молекула
^2
•
¦
¦'б
CM
г3 • см6
с
Px
UF2a 530 115 510 ______ 4-Ю3 2 1
UF|a 530 115 510 ______ 4.10s 2 2
UFsa 530 115 510 ______ 4-ЮЗ 2 2
UF3a 570 80 ! 520B) 130B) _____ 1,8-104 3 2
UF?a 570 80 520B) 130 B) — — — ¦ — — 1,8 -104 3 1
UFja 570 80 520B) 130 B) — - — - — 1,8-104 3 1
UF4a 600 540 145 140 150 540 170 550 145 4,7-104 2 1
UF+a 600 540 145 140 150 540 170 550 145 4,7-104 2 2
UF^a 600 540 145 140 150 540 170 550 145 4,7-104 2 2
UF5a 649 572 130 515 60 200 593B) 200B) 180B) 7,7-10* 2 2
VF? 650 570 130 515 60 200 590B) 200B) 180B) 7,7-10* 2 1
UFg-a 650 570 130 515 60 200 590B) 200B) 180B) 7,7-10* 2 1
UFe 667 534B) 626C) 186C) 200C) 143C) — — — 1,28-105 24 1
UF^a 600 500B) 600C) 170C) 180C) 120C) — — — 1,28-105 24 2
a Ниже приведены значения Г* (р() для возбужденных электронных состояний, Tfy — в см:
UF2: 200 F), 500 F), 2000 B5), 4000 B5), 6000 B5), 8000 B5), 10 000 B5), 15 000 E0), 20 000 E0);
UFJ, UF3: 200 D), 400 D), 4500 A2), 7000 B0), 10 000 C5), 15 000 C5), 20 000 C5);
UF2: 1000 F), 3000 A2), 5000 B0), 7000 E0);
UFJ, UF4, UFg": 500 D), 2000 C), 3000 A), 4000 E), 6000 (8), 7000 C), 9000 A4), 11 000 A1), 12 000 D), 14 000 E),
17 000 A0), 19 000 A5), 22 000 F);
UFj: 200 F), 500 F), 2000 B5), 4000 B5), 6000 B5), 8000 B5);
UF?, UF5: 2000 B), 5000 B), 7000 B), 9000 B), 11 000 B), 14 000 B);
UFj: 200D), 400D), 4500A2), 7000B0), 9000B5);
UFj: 4500 D), 7000 B), 13 000 D), 14 000 B).
чениям r(U—F) = 2,10 + 0,1 А (принято таким же, как в UF3) и 4F—U—F = НО + 20° (оценка-
по EuF2 [2581]). Погрешность значения IaIbIc составляет 2-Ю14 г3-см6. Приведенные
в табл. 43.11 основные частоты UF2 вычислены для модели валентного силового поля. Значение
/г=2,8-105 дин-см было оценено по силовым, постоянным молекул UFe [3452], UC16 [3064] и
UC12 [2581]. Постоянные /гг = 0,2 и /а/г2 = 0,07-105 дин-см оценены в предположении, что их
отношение к fr должно быть примерно таким же, как для молекулы EuF2 [2581 ]. Погрешности
в vx и v3 оцениваются в 100 см, v2 — в 30 см.
Экспериментальные данные о возбужденных электронных состояниях молекулы UF2, а также
соединений, содержащих ион U2+, отсутствуют. Эта молекула должна иметь большое число
электронных состояний, отвечающих расщеплению термов свободного иона U2+ в поле лигандов
симметрии С2р. Приведенные в табл. 43.11 электронные состояния молекулы UF2 оценены в
предположении, что два нижних уровня отвечают усредненным компонентам терма основного
состояния ьЬв (конфигурация 5/36d), а остальные распределены примерно равномерно в области до>
20 000 см, причем их суммарный статистический вес в этой области принят таким же, как для
молекулы UF+. Из-за отсутствия каких-либо экспериментальных данных оценка электронных
состояний молекулы UF2 является самой грубой среди всех многоатомных фторидов урана.
Погрешность энергии объединенных электронных термов может достигать 2000—3000 см, а
суммарное значение статистических весов может в 2-^3 раза отличаться от принятого.
Термодинамические функции UF2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A.124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены главным образом неточностью принятых молекулярных постоян-
206
Уран и его соединения
ных, а также приближенным характером расчета, они оцениваются в 40, 15, 20 и 25 Дж-К"хХ
Хмель в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UF2(r) вычислялись ранее в работе Гурвича и др. [338, 2536],
а также в справочнике Галкина и др. [742а] (Т ^ 5000 К). Расхождения с результатами расчета
[338, 2536] наиболее существенны при низких температурах (достигают 13 Дж-К^-моль в
значениях ФС(Т)) и обусловлены главным образом уточнением оценки энергии и числа возбужденных
электронных состояний. Расхождения с расчетом [742а], составляющие 27—48 Дж-К~1-моль
в значениях Ф°(Т), вызваны разницей в принятых структурных параметрах, значении v2, а также
тем, что авторы [742а] не учитывали возбужденные состояния UF2.
Хилденбранд [4974] на основании масс-спектрометрического изучения равновесия реакций
с участием UF2(r) нашел 5°(иГ3., г, 2200 К) = 454 + 12 Дж-К~1-моль; это значение находится
в удовлетворительном согласии с приведенным в табл. 1010 D42,1 + 14 Дж-К~1-Моль"г).
Константа равновесия реакции UF2(r) = U(r) + 2F(r) вычислена с использованием шачения
ДгЯ°@) = 1224,415 + 26 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
^H°(UF2, г, 0) = —535 + 25 кДж • моль.
Результаты определений энтальпии образования UF2(r) представлены в табл. 43.12.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, неточности
использованных в расчетах термодинамических функций, термохимических величин и сечений ионизации.
Таблица 43.12. Результаты определений X^H°(UF2, г, 0) (в кДж • моль)
Авторы
Метод
&fH°(UF2, г, 0)
II закон
III закон
Змбов, 1969 [4825] Масс-спектрометрический, UF4(r)+2Ca(r)=UF2(r)+2CaF(r), —614+60 — 579+.40
1201—1313 К, 13 точек, результаты измерений
опубликованы в виде кратких тезисов
Хилденбранд, 1981 [4974] Масс-спектрометрический, U(r)+2BaF(r)=UF2(r)+2Ba(r), —542+30 —
получено комбинированием реакций U(r)+BaF(r)=UF(r)-j-
+Ва(г), ДгЯ°B250К)=-74,4±3,0, 2222-2505 К и
UF(r)+BaF(r)=UF2(r)+Ba(r), ДгЯ°B250 К)=20,1±2,5,
2034—2466 К, сообщены только измеренные \Н°(Т)
Масс-спектрометрический, UF2(r)-f BaF(r)=UF3(r)+Ba(r), —511+35 —
ДгЯ°B250 К)=—34,7 ±2,0, 2034—2466 К, сообщена только
измеренная ДГЯ°(Г)
Горохов и др., 1982 [4868] Масс-спектрометрический, 2UF3(r)=UF4(r)+UF2(r), 1403— —533+35 —537+40 ,
1616 К, 26 точек
В пределах указанных погрешностей результаты измерений согласуются между собою.
Принятая величина является средневзвешенной из приведенных в таблице величин, включая расчеты
по уравнению II закона по данным работ [4974] и [4868] 1.
UFj"(r). Термодинамические свойства газообразного положительного иона дифторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1011.
Молекулярные постоянные UF|, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UF+ в литературе
отсутствуют. По аналогии с UF2 в справочнике принимается, что UF+ в основном электронном
состоянии имеет нелинейную структуру (симметрия С2в) 2. Приведенное в табл. 43.11 произведение-
1 См. примеч. 1 к тексту по UF (г).
2 Изменение электронного строения может привести к изменению геометрической структуры молекул UF? и UF~
по сравнению с нейтральной молекулой UFK. Однако из-за полного отсутствия экспериментальных данных
структурные параметры и частоты ионных молекул фторидов урана принимаются в справочнике идентичными
соответствующим постоянным нейтральных молекул, хотя по аналогии с оксидами урана можно предполагать, что
удаление электрона у молекул UFB с га=2ч-4 должно сопровождаться некоторым уменьшением величины г (U—F)
и увеличением частот валентных колебаний.
207
iUFi(r) Глава 43
моментов инерции и основные частоты UFJ приняты аналогичными соответствующим постоянным
молекулы UF2. Погрешность значения IaIbIo оценивается в 3-Ю14 г3-см6, значений vx и v3 —
в 100 см, значения v2 — в 50 см. Возбужденные состояния UF? приняты идентичными
состояниями UF3 (см. 3-е примеч. к тексту по UO2(r)).
Термодинамические функции UFj(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)-A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчитанных
значений обусловлены главным образом неточностью оценки молекулярных постоянных, а также
приближенным характером расчета; они составляют 10, 15, 20 и 25 Дж-К~1-моль в значениях
Ф°(Т) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UFj(r) вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождения с результатами этого расчета составляют 9—3 Дж-К-моль в значениях Ф°(Т)
и обусловлены уточнением оценки молекулярных постоянных в настоящей работе.
Константа равновесия реакции UF+(r)+e(r)=U(r)ij-2F(r) вычислена с использованием значения
АгН°@)=624,415 + 57 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(UF2)=600 + 50 кДж-моль.
Принятая величина 10 основана на измерениях методом электронного удара, выполненных
Гороховым и др. [4868] F30+20 кДж-моль), и на предположении, что потенциал ионизации
у UF2, так же как у UO, должен быть меньше величины /0(U). Менее надежное значение G04 +
+ 50 кДж-моль) было получено Змбовым [4825] *. Принятому потенциалу ионизации
соответствует
^fHo(\}Щ, г, 0) = 65,0 + 56 кДж • моль.
UFj(r). Термодинамические свойства газообразного отрицательного иона дифторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1012.
Молекулярные постоянные UFj, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре,UFj в литературе
отсутствуют. По аналогии с UFJ в справочнике принимается, что UFj в основном электронном
состоянии имеет нелинейную структуру (симметрия С2г). Приведенные в табл. 43.11 произведение
моментов инерции и основные частоты аналогичны соответствующим постоянным UF2 (см. примеч.
к тексту по UF+ (г)).Погрешность значения IaIbIo оценивается в 3-Ю14 г3-см8, значений v —
в 100, 50 и 100 см.
Возбужденные состояния UFg приняты идентичными состояниям UF (см. 3-е примеч. к тексту
по UO2(r)), за исключением того, что отброшены уровни, имеющие энергии выше потенциала
ионизации UFj.
Термодинамические функции UF^(г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A.122)—A. 124), A. 128), A. 130)
с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчитанных
значений обусловлены главным образом неточностью оцененных молекулярных постоянных,
а также приближенным характером расчета, они составляют 10, 15, 20 и 30 Дж-К~1-моль в
значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UF^(r) вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождения с результатами этого расчета составляют 3—9 Дж-К-моль~1 в значениях Ф°(Т)
и обусловлены уточнением оценки молекулярных постоянных в настоящем справочнике.
Константа равновесия реакции UF2(r) = U(r)+2F(r)+e(r) вычислена с использованием
значения АгН°@)=1359,415+50 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A^°(UF2, г, 0) = —670 + 50 кДж • моль.
Принятая величина была получена на основании оценки значения D0(UF—F~)=370+50 кДжХ
Хмоль по величине D0(UFB—F") в молекулах UF-, UFj, UF7 и UF~.
Принятой величине соответствует
A0(UF2) =—135 + 56 кДж-моль.
1 См. примеч. 1 к тексту по UF (г).
208
Уран и его соединения
UF3(k, ж)
UF3(k, ж). Термодинамические свойства кристаллического и жидкого трифторида урана
в стандартном состоянии при температурах 298,15—3200 К приведены в табл. 1013.
Значения постоянных, использованные для расчета термодинамических функций UF3(k, ж),
представлены в табл. 43.1. В справочнике за стандартное состояние UF3(k) в интервале 0—1768 К
принимается гексагональная модификация [3210].
Измерения теплоемкости UF3(k) при низких температурах выполнены Веструмом и до сих пор
не опубликованы, но известны по справочнику [2344]. Принятые по этим данным значения
5°B98,15 К) и #°B98,15 К)—#°@) (см. табл. 43.1) имеют погрешности 0,4 Дж-К^-моль и
0,06 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости UF3(k) принято уравнение, предложенное Кордфунке
и Оуэлтисом на основании измерений энтальпии в интервале 398—866 К и известное также по
справочнику [2344].
Температура плавления UF3 A768 + 20 К) принята по измерениям Д'Эя и Мартина [2068],
выполненным на образце, содержащем в качестве примеси менее 0,2% двуокиси урана. Энтальпия
плавления C6,8 + 2,0 кДж-моль) определена в работе Хрипина и др. [1087], изучавших фазовые
отношения в бинарной системе UF4—UF3 методами дифференциально-термического и рентгено-
фазового анализов. Образец трифторида урана был получен восстановлением паров UF4
металлическим цирконием и содержал в качестве примеси около 3 мае. % двуокиси урана и 0,01 мае. %
циркония.
Для теплоемкости UF3(ffl) ввиду отсутствия экспериментальных данных принято значение
134+20 Дж-К-моль~1, равное теплоемкости жидкого трифторида церия.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,3, 1, 6 и 14 Дж-К-моль соответственно. Расхождения между термодинамическими
функциями UF3(k), приведенными в табл. 1013 и в справочнике Барина и Кнакке [1395] (Т ^ 1000 К),
достигают 6,4 Дж -К -моль в значениях Ф°(Т). Они обусловлены тем, что авторы [1395] в своих
расчетах использовали оцененные уравнения теплоемкости, приведенные в работе Кнакке и др.
[3044]. Термодинамические функции UF3(k), вычисленные в справочнике [2344] до 800 К,
практически совпадают с данными табл. 1013.
Принятая в справочнике энтальпия образования
b.fH\\}?z, к, 298,15 К) = —1508,7 ±3,0 кДж.моль
основана на представленных в табл. 43.13 данных. Эта величина является средневзвешенной из
результатов последних и наиболее точных термохимических измерений, выполненных Кордфунке
Оуэлтисом [1933] и Вижбенга и др. [4725]. Близкое значение было найдено по измерениям Хей-
Т а б лица '43.13. Результаты определений A^ar°(UF3, к, 298,15 К) (в кДж-моль)
л
Авторы
Метод
AfH°(UF3, к, 298,15 К)
II закон III закон
Хойс, Эган, 1966 [2630]
Маркин и др., 1967 [3392]
Ханаев, Хришш, 1970- [1068]
Кордфунке, Оуэлтис, 1981
[1933]
Хейман, 1967 [2597]
Вижбенга и др., 1982 [4725]
ЭДС, A1F3(k)+U(k)=A1(k)+UF3(k), 873 К, 1 точка
То же, U(K)+3/aMgF2(K)=3/2Mg(K)+UF3(K), 873 К,
1 точка
То же, A1(k)+UF3(k)=A1F3(k)+U(k), 873 К, 1 точка
То же, U(K)+3/2MgF2(K)=<V2Mg(K)+UF3(K), 873 К,
1 точка
Калориметрия растворения, UO2Cl2(K)+3FeCl2(K)-f-
+3HF(p-p)+HCl(p-p)=UF3(K)+3FeCl3(K)+2H2OM,
\Н°{323 K)=126,7+l,0
То же, UCl4(K)+FeCl2(K)+3HF(p-p)=UF3(K)+FeCl3(K)+
+ЗНС1(р-р), ДГЯ°C23К)=—28,3 ±1,1
То же, 3|2U3O8(k)+3(HF-16H2O)=UF3(k)+V2UO3(k)+
+3/2Н20(ж), ДГЯ°B98 К)=107,1 +2,7
Фторная калориметрия, UF3(K)+s/2F2(r)=UF6(r),
^^B98 К)=—641,8±2,0
То же, UF3(K)+3/2F2(r)=UFe(K), ДГЯ°B98 К)=—688,9+3,1
—1498±8
-1495 ±8
—1500 ±8
—1497 ±8
-1484,9 ±3,3
-1483,9 ±2,9
(-1517,0)
-1508,5 ±5,5
-1506,8±2,9
-1508,8±3,6
209
UF3(r) Глава 43
мана [2597], если использовать в расчете принятое в справочнике значение энтальпии образования
UF6(k), а не найденное в этой же работе Хеймана (см. текст по выбору энтальпии образования
UF6(k)). Результаты исследований равновесий [2630, 3392] в пределах их погрешностей
согласуются с принятым значением.
Работа Ханаева и Хрипина [1068] приводит к заметно меньшей по абсолютному значению
величине, однако она включает реакции с UC14 и UO2G12, что может привести к некоторым
дополнительным погрешностям из-за недостаточной надежности энтальпий образования этих веществ Ч
В работе Паркер [3773] отмечается хорошее согласие разности энтальпий образования UF4(k)
(см. ниже) и UF3(k), вычисленных по результатам этих и других измерений Ханаева и др. и
полученных в других работах, и высказывается предположение о возможной систематической
погрешности при измерении энтальпий некоторых из этих реакций.
Давление пара в реакции UF3(k, ffi) = UF3(r) вычислено с использованием значения Дг#°@) =
= A3H°(XJF3, к, 0)=447,479+20 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования UF3 в кристаллическом и газообразном состояниях. р
UF3(r). Термодинамические функции газообразного трифторида урана в стандартном
состоянии при температурах от 100 до 10 000 К приведены в табл. 1014.
Молекулярные постоянные UF3, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и основных частотах молекулы
UF3 отсутствуют. По аналогии с тригалогенидами Sc, Y, La и лантанидов, пирамидальная
структура которых установлена на основании исследований ИК-спектров [2582, 2588] и электроно-
графических данных [281, 282, 284, 356, 357] 2, в справочнике для UF3 в основном состоянии
принята пирамидальная структура (симметрия С3о). Приведенное в табл. 43.11 произведение моментов
инерции рассчитано по структурным параметрам r(U—F)=2,10+0,05 А и -=$F—U—F=110 + 5°.
Значение r(U—F) оценено по значениям r(M—F) в молекулах PrF3 B,09 А [4872а]), UF4 B,068 А)
и CeF4 B,035 А [784]) 3. Погрешность значения IaIbIc составляет 5-1014 г3-см6. Основные
частоты молекулы UF3 были вычислены в приближении модели валентного силового поля по
значениям силовых постоянных /г=3,0-105 дин-см (промежуточное между значениями для UF4
и UF2), /rr=0,25-105 дин-см и /а/г2=0,07 -105 дин-см" (оценены, если положить, что их
отношение к /г такое же, как в LaF3 [2582, 2588]). Погрешности в значениях частот оцениваются в 15—
20% от их величины.
Приведенные в табл. 43.11 электронные состояния UF3 оценены по экспериментальным данным
о спектрах поглощения кристаллического NdF3 [1757], иона U+3 в расплаве LiCl—KG1 [1755]
и иона U+3 в решетке CaF2 [3485], которые интерпретированы на основе атомных состояний,
соответствующих конфигурации /3, с учетом расщепления благодаря спин-орбитальному
взаимодействию и влиянию полей лигандов различной симметрии. На основании данных [3485] оценены
компоненты расщепления двух нижних термов 4/»/2 и 4/п/г в поле симметрии С2„ в области 0 и
4500 см соответственно; их усредненные значения приняты в качестве трех первых
возбужденных состояний UF3. На основании данных [1755] и [1757] было принято, что остальные
электронные состояния UF3 распределены приблизительно равномерно в области до 20 000 см, а
суммарный статистический вес этих состояний составляет ~150. Погрешности в объединенных значениях
1 При проведении ряда расчетов в справочнике использованы энтальпии образования некоторых соединений
урана, рекомендованные в работе Паркер и др. [2344, 3773], в том числе А/Н° (UC14, к, 298,15 К)=—1018,8 +
+2,5 кДж-моль. Эта величина получена из расчетов для разных реакций и представляется достаточно
обоснованной. Однако в более ранних работах и в недавно опубликованной работе Суглобова и Чиркста [992] (см.
дискуссию [993, 1064]) было получено значительно отличающееся значение (—1051,9+4,2 кДж-моль). Оно
противоречит ряду данных (см. [1064, 2344]), однако если использовать данные [992] в термохимических циклах,
включающих UF4(k), UF3(k) и UC14(k), получается существенно лучшее соответствие с наиболее надежными
измерениями другими методами (рассчитанные таким образом величины даны в скобках в табл. 43.13 и 43.15).
2 Первые электронографические данные [10, 11, 12], а также ИК-спектры [4694, 4706] тригалогенидов лантанидов
были интерпретированы как отвечающие плоской структуре симметрии Dsh или не противоречащие такой
структуре. Решающим аргументом в пользу пирамидального строения этих молекул явились электронографическпе
исследования Гиричева и др. [281, 282, 284, 356, 357], в которых было показано, что ScF3 и ряд трихлоридов
лантанидов имеют пирамидальную равновесную конфигурацию.
8 В электронографпческом исследовании структуры молекул LaF3 и NdF3 [11] было найдено г (La—F)=r (Nd—F)=
=2,22A; по-видимому, в этой работе были допущены систематические ошибки.
210
Уран и его соединения
UFj(r)
энергий возбуждений оцениваются в 200 см г для первых двух состояний, в 2000—3000 см —
для остальных.
Термодинамические функции UF3(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический осциллятор»
с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчитанных
значений обусловлены главным образом неточностью принятых молекулярных постоянных и
приближенным характером расчета; они составляют 10, 15, 20 и 25 Дж-К~1-моль~1 в значениях
Ф°(Т) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UF3(r) вычислялись ранее в работе Гурвича и др. [338, 2536]
(Т <; 10 000 К) и справочнике Галкина и др. [742а] (Т ^ 5000 К). Расхождения результатов
этих расчетов и настоящего справочника достигают соответственно 12 и 33 Дж-К^-моль в
значениях Ф°GТ) и обусловлены отличием оцененных молекулярных постоянных в основном
состоянии, а для [742а], кроме того, пренебрежением возбужденными электронными состояниями.
Хилденбранд [4974] на основании результатов масс-спектрометрического изучения равновесия
реакций с участием UF3(r) нашел 5°(UF3, г, 2200 К) = 521+ 14 Дж-К~1-моль~1, это значение
хорошо согласуется с приведенным в табл. 1014 E12,9 + 13 Дж-К~1-моль~1).
Константа равновесия реакции UF3(r) = U(r)+3F(r) вычислена с использованием значения
Дг#°@) = 1826,690 + 22 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —1060 + 20 кДж • моль.
Результаты определений энтальпии образования UF3(r) представлены в табл. 43.14. При
вычислении погрешностей приведенных в этой таблице величин учтены воспроизводимость измерений,
Та
блица 43.14. Результаты определений A/J3"°(UF3, г, 0) (в кДж-моль)
Авторы
Метод
Д7Я° (UF3, г, 0)
II закон III закон
Ходеев, 1964 [1079]
Змбов, 1969 [4825]
Хилденбранд, 1981 [4974]
Горохов и др., 1981 [4867]
Электронный удар, UF4+t=UFJ+F+2e —1113+30
Масс-спектрометрическпй, UF4(r)+Ca(r)=UF3(r)+CaF(r), —1041+25 —1070+25
1201—1313 К, 13 точек
То же, UF,(r;+Ca(r)=UF3(r)+CaF(r), 1533-1745 К, -1054+25 —
АгЯ°A640 К)=81,6+2,5
То же, UF3(K)=UF3(r), 1252—1469 К, 25 точек —1101+20 —1062+20
неточности использованных в расчетах термодинамических функций, сечений ионизации и
термохимических величин. Как видно из таблицы, результаты измерений энтальпии образования UF3(r)
в четырех работах тремя методами привели к значениям, совпадающим в пределах их
погрешностей. Принятая величина является средневзвешенной из величин, полученных в работах [4825,
4867, 4974] К
g(). Термодинамические свойства газообразного положительного иона трифторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1015.
Молекулярные постоянные UF+, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UFJ в литературе
отсутствуют. По аналогии с UF3 в справочнике принимается, что UF+ в основном электронном
состоянии имеет пирамидальную структуру (симметрия С3„) (см. примеч. к тексту по UF*(r)).
Приведенные в табл. 43.11 произведение моментов инерции и основные частоты аналогичны
соответствующим постоянным молекулы UF3. Погрешность значения 1aIbIc оценивается в1-1013г3-смв,
значений v,- — в 20—25% от их величины. Электронные состояния UFJ, приведенные в табл. 43.11,
приняты идентичными состояниям UF4 (см. 3-е примеч. к тексту по UO2(r)).
1 См. примеч. 1 к тексту по UF(r).
211
UFi(r), UF4(k, ж) Глава 43
Термодинамические функции UFg(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A .3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчитанных
значений обусловлены главным образом неточностью принятых молекулярных постоянных и
приближенным характером расчета; они составляют 15, 20, 25 и 30 Дж-К~1-моль в значениях
Ф°(Т) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UFg (г) вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождения данных табл. 1015 и результатов этого расчета составляют 6—8 Дж-К~1-моль~1
в значениях Ф°(Т) и обусловлены уточнением оценки постоянных в настоящем издании.
Константа равновесия реакции UFg(r)+e(r) = U(r)+3F(r) вычислена с использованием
значения ДгЯ°@) = 1056,69 + 29 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(UF3)=770 + 20 кДж-моль.
Принятая величина основана на измерениях методом электронного удара в работах Ходеева
[1079] G72 + 15 кДж-моль, квазимоноэнергетические электроны) и Горохова и др. [4868] G72 +
+20 кДж-моль). Менее точное значение 820 + 50 кДж-моль было получено в работе Змбова
[4825]. Принятой величине соответствует
\fH°(\JF$, г, 0) = —290,000 + 25 кДж • моль.
UFg (г). Термодинамические свойства газообразного отрицательного иона трифторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1016.
Молекулярные постоянные UFg, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UFg в литературе
отсутствуют. По аналогии с UF3 в справочнике принимается, что UFg в основном электронном
состоянии имеет пирамидальную структуру (симметрия CSv). Приведенные в табл. 43.11
произведение моментов инерции и основные частоты иона UF^ аналогичны соответствующим постоянным
молекулы UF3 (см. примеч. к тексту по UFJ(r)). Погрешность значения IaIbIc оценивается
в 1-1013 г3-см6, значений v,- — в 20—30% от их величины. Электронные состояния UFg,
приведенные в табл. 43.11, приняты идентичными состояниям UF2 (см. 3-е примеч. к тексту по UO2(r)) за
исключением того, что они ограничены на энергии примерно равной потенциалу ионизации UFg.
Термодинамические функции UF~(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчитанных
значений обусловлены главным образом неточностью принятых молекулярных постоянных и
приближенным характером расчета; они составляют 20, 25, 30 и 40 Дж-К-моль~1 в значениях Ф°(Г)
при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UFg (г) вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождение данных табл. 1016 и результатов этого расчета составляют 14—16 Дж-К-моль
в значениях Ф°(Т) и обусловлены уточнением оценки постоянных в настоящем издании.
Константа равновесия реакции UFg(r)=U(r)+3F(r)+е(г) вычислена с использованием значения
Дг#°@)=1946,69 + 54 кДж-мольГ1, соответствующего принятой в справочнике энтальпии
образования
bfH°{UF5, г, 0) = —1180 + 50 кДж • моль.
Это значение было получено на основании принятого значения Aj,#°(UF3, г) и оценки энергии
связи UF2—F~ C95 + 50 кДж-моль) по величине энергии связи UF(J—F~ в молекулах UF~
UF; и UFJ.
Принятому значению соответствует
A0(UF3) =—120+54 кДж-моль.
UF4(k, ж). Термодинамические свойства кристаллического и жидкого тетрафторида урана
в стандартном состоянии при температурах 100—2500 К приведены в табл. 1017.
212
Уран и его соединения UF4(k, ж)
Значения постоянных, использованные для расчета термодинамических функций, представлены
в табл. 43.1. В справочнике за стандартное состояние UF4(k) в интервале 0—1309 К принимается
моноклинная модификация (структурный тип ZrF4) [3197, 4807] г.
При Т <С 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работах Бернса и др. [1730] A,3—20 К) и Осборна и др. [3729] E—304 К). В обеих
работах использован чистый образец с содержанием примесей не более 0,04 вес. %. Погрешность
измерений теплоемкости составляла 2% при 1,3 К, 0,6% при 2 К, 0,2% при 2,4—16 К, 0,4%
при 20 К и 0,1 % при температурах выше 25 К. Авторы [1730] обнаружили аномалию теплоемкости
с максимумом при 6,4 К, имеющую характерную форму аномалии типа Шоттки. Значения
?°B98,15 К) и #°B98,15 К)—#°@), приведенные в табл. 43.1, имеют погрешности 0,4 Дж-К^Х
Хмоль и 0,06 кДж-моль" соответственно. Измерения теплоемкости в работе Брикведде и др.
[1669] B0—350 К), выполненные на образце, содержащем ~2% примеси UO2F2, не учитывались.
При Т > 298,15 К для теплоемкости UF4(k) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии в работах Кинга и Кристенсена [ЗОН] C98—1347 К),
Дворкина [2136] D98—1377 К) и Кордфунке C84—849 К, цитируется по справочнику [2344]).
При обработке погрешность данных [2136] и данных Кордфунке оценивалась в 0,5%, данных
[ЗОН] — в 1%. Гальченко и др. [262] измерили теплоемкость UF4 в интервале температур 373—
673 К на образце, содержащем около 2% UO2F2; результаты представлены в графическом виде,
что не позволило включить их в совместную обработку. Измерения энтальпии в широких
интервалах температур вплоть до точки плавления [2136, 2344, ЗОИ] не обнаружили каких-либо
аномалий UF4 в твердом состоянии. На этом основании Дворкин [2136] подвергает сомнению данные
Хрипина и др. [1084, 1085, 1086, 1087] и Некрасовой и др. [712], которые исследовали бинарные
системы с UF4 методами ДТА и рентгенофа*зового анализа и пришли к выводу о наличии
полиморфных превращений UF4 при 1113 и 1183 К соответственно. Указания на возможность полиморфного
превращения UF4(k) имеются также в работах [1139] A154 К) и [368] A103 К). Дворкин,
анализируя результаты известных ему работ [712, 1084, 1085, 1086, 1087], отмечает, что эвтектической
точке в системе UF4—CaF2, согласно [712], отвечает температура 1113 К, а температура эвтектики
для системы UF4—UO2 по данным [1086] составляет 1183 К. Следует отметить, что попытки
авторов [1085] закалить высокотемпературную модификацию UF4(k) не увенчались успехом.По данным
[1085] энтальпия полиморфного превращения составляет 14,23 кДж-моль, а по данным [1139]
18,14 кДж-моль. Поскольку маловероятно, чтобы такой тепловой эффект не был зафиксирован
при измерениях энтальпии в работах [ЗОИ] и [2136], в справочнике возможность этого
полиморфного превращения не принималась во внимание.
Температура плавления UF4 A309 + 2 К) принята по хорошо согласующимся между собой
данным [369, 1087, 1414, 2897, 3186, 4507, 4508, 4675]. Энтальпия плавления D7,0 + 0,8 кДж-моль)
принята по калориметрическим измерениям Дворкина [2136], выполненным на тщательно
очищенном образце UF4. В работе [ЗОИ] получено более низкое значение D2,8 кДж-моль).
Приближенные значения энтальпии плавления приведены в работах [3186] F7,4 кДж-моль) и
[1087] D3,5 кДж-моль).
Теплоемкость-UF4(ж) A67 + 5 Дж-К~1-моль~1) принята на основании измерений энтальпии
в работах [ЗОН] A334—1347 К, 2 измерения) и [2136] A328—1377 К, 4 измерения).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 0,3, 1
и 4 Дж-К-моль~1 соответственно. Расхождения между термодинамическими функциями
UF4(k, ж), приведенными в табл. 1017 ив справочниках [2344] (Т <; 1400 К) и [1395] (Т < 1700 К),
не превышают 0,1 и 1 Дж-К~1-моль~1 в значениях Ф°(Т) во всем интервале температур.
Принятая в справочнике энтальпия образования
A/?f°(UF4, к, 298,15 К) =—1920,5 + 3,0 кДж • моль
является средневзвешенной из результатов измерений Кордфунке, Оуэлтиса [1933] и Вижбенги
и др. [4725]. В этих работах были проведены наиболее точные измерения. В табл. 43.15
представлены результаты определений энтальпии образования UF4(k), полученные в этих, а также других
работах. Приведенные погрешности вычислены с учетом воспроизводимости измерений, неточности
термодинамических функций и энтальпий образования, использованных в расчетах.
1 Новая более плотная кубическая модификация UF4 получена в результате ударного сжатия в работах [69, 366].
При ее нагревании происходит обратное превращение в моноклинную модификацию при 600 К.
213
UF4(k, ж)
Глава 43
Таблица 43.15. Результаты определений Ayff°(UF4, к, 293,15 К) (в кДж-моль)
Авторы
Джонс, Уолш, 1945 [2863]
Доманж, Вольхютер, 1949
[2093]
Вдовенко и др., 1963 [198];
Севидзк, Браун, 1960 [4152]
Маркин и др., 1967 [3392]
Либби, 1944 [3252]
ш
Зельдес, 1946 [4818] (см. [492])
Ханаев, 1968 [1067] (см.
также [256, 649])
Хью и др., 1980 [2727]
Волков и др., 1980 [228]
Кордфунке, Оуэлтис, 1981
[1933]
Хейман, 1967 [2597]
Вижбенга и др., 1982 [4725]
а См. текст по A^°(UF3, к).
Метод
I. Исследования равновесий
Протока, UO2(K)+4HF(r)=UF4(K)+2H2O(r), 873—
1073 К, 3 точки
То же, 473—773 К, 5 точек
Исследования равновесий в растворах,
UF4-2,5H2O(K)+a9=U+4(a?)+4F-(as)+2,5H2O(H<),
Дг(?°B92 К)=154,4 (см. [3773])
ЭДС, Mg(K)+2UF4(K)=MgF2(K)+2UF3(K), 873 К,
1 точка
ЭДС, A1(k)+3UF4(k)=A1F3(k)+3UF8(k), 873 К,
1 точка
ЭДС, U(k)+3UF4(k)=4UF3(k), 873 К, 1 точка
П. Калориметрические измерения
Калориметрический, растворение U(SO4J, UF4 -H2O
в воде и др. данные
То же, UCl4(K)+4KF(p-p)=UF4(K, гидратир)+
+4КС1(р-р)
То же, UF4(K)+4HCl(p-p)=UCl4(K)+4HF(p-p),
ДГ#ОB98К)=—208,2+0,7 (см. [3773])
То же, растворение UF4 в HF+HC1+A1C]S+H2O и
др. данные
То же, UCl4(K)+4KF(p-p)=UF4(K)+4KCl(p-p),
АГЯ°B98 К)=—205,0+3,3
То же, растворение в сернокислых растворах церия,
U3O8(k)+4(HF-16H2O)=UF4(k)+2UO3(k)+2H2OM,
ДгЯ°B98К)=-77,1+2,1
Фторная калориметрия, проточный калориметр,
UF4(K)+F2(r)=UF6(r), ДГ#°B98,К)=-237,7 +0,7
Фторная калориметрия, сжигание в бомбе, UF4(k)+
+F2(r)=UF6(K), ДГЯ°B98 К)=-276,3+4,2
A/77°(UF4, к, 298,15 К)
II закон
III закон
—1872 —1922+15
—1803 —1900±20
— —1877
— —1918,3+7,0
— -1919+10
— —1922,1+5,0
—1866
—1858
—1892,4+3,8
(-1925,5) а
—1894,5+8,0
—1884,1+5,0
(—1917,2) а
—1920,0±3,7
—1910,9+2,2
—1921,4±4,6
Таблица 43.18. Результаты определений jAsff°(UF4, к, 0) (в кДж ¦ моль)
Авторы
Район, Твичелл, 1947 (см. [3186])
Кац, Рабинович, 1954 [492]
Попов и др., 1959 [809, 810]
Лангер, Бланкенпшп, 1960 [3186]
Акишин, Ходеев, 1961 [4845]
Чудинов, Чопоров, 1970 [1139]
Хилденбранд, 1977 [2644]
Ходеев и др., 1981 [4909]
Нагараян и др., 1980 [5023]
Метод
Эффузионный, 1015—1136 К, данные
представлены в виде уравнения
Метод не указан, 1033 К, 1 точка
Протока, 1148—1273 К, 5 точек
Точек кипения и статический, 1291—1575 К,
22 точки
Масс-спектрометрический, 900—1000 К, данные
представлены в виде уравнения
Эффузионный, 828—1280 К, 93 точки
Торзионный, 980—1137 К, данные представлены
в виде уравнения
Эффузионный, 1060—1137 К, 4 точки
Точек кипения и переноса, 1169—1427 К, 29 точек
Asff°(UF4, к, 0)
II закон
III закон
337 318,1
— 340,1
308±40 318,9+1,4
307+8 316,2+0,4
307 314,3
283±10 310,5+0,5
315 314,0
304+80 309,0+1,6
313+10 316,2+0,4
Уран и его соединения . UF4(ry
Давление пара в реакции UF4(k, JK) = UF4(r) вычислено с использованием принятого в
справочнике значения энтальпии сублимации
ДгЯ°@) = AS#°(UF4) к, 0) = 315 ± 15 кДж • моль.
Принятая величина основана на приведенных в табл. 43.16 результатах измерений давления
пара UF4(k, ж). Указанные в этой таблице погрешности учитывают только воспроизводимость
измерений. За исключением выпадающей величины, полученной в работе [492], остальные данные
достаточно хорошо согласуются между собою. Наиболее точные и детальные измерения были
проведены в работах [1139, 2644, 4845, 5023], и на них основана рекомендуемая величина. Погрешность
принятой величины определяется главным образом неточностью термодинамических функций
ивд. ММ
Щ UF4(r). Термодинамические функции газообразного тетрафторида урана в стандартном
состоянии при температурах от 100 до 10 000 К приведены в табл. 1018.
Молекулярные постоянные UF4, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Строение молекулы UF4 в газовой фазе в настоящее время окончательно
не установлено. В электронографическом исследовании Ежова и др. [398, 399] было определено
только расстояние U—F, однако на основании анализа величины константы асимметрии было
высказано предположение о нететраэдрическом (симметрия С2р) строении UF4. Соотношение между
расстояниями U—F и F. . .F, полученное в электронографическом исследовании Петрова [784],
не противоречит конфигурации правильного тетраэдра. Однако по величине найденных амплитуд
колебаний автор [784] пришел к выводу об искаженной тетраэдрической структуре UF4 с
одинаковыми связями U—F B,068+0,005 А) и неравноценными расстояниями F. . .F (-^Fx—U—F2=
= -^F3—U—F4=99,4+2,5°, -^Fx—U—F4 = 115,l±2,5°). Экспериментальным данным одинаково
хорошо удовлетворяют две такие модели — симметрии Du и C3v. Модель симметрии C2v в работе
[784] не рассматривалась, хотя полученные Петровым данные не противоречат такой модели.
С учетом этого замечания, аналогии с молекулой SF4 и данных Ежова и др. [4870а] для молекулы
WC14 в справочнике принимается, что молекула UF4 в основном состоянии имеет структуру
искаженного тетраэдра (симметрия С2с) 1. Произведение моментов инерции, приведенное в табл. 43.11,
вычислено по структурным параметрам r(U—F1)=2,02+0,05 A, r(U—Fs)=2,12+0,05 A, ^?г—
—U—F2=105 + 10°, -$F3—U—F4=115 + 10° (пары атомов Fx, Fa'и Fs, F4 лежат во взаимно
перпендикулярных плоскостях), которые были оценены при подготовке справочника. Погрешность
значения IaIbIc оценивается в 1 -10~113 г3 -см6. Значения основных частот, приведенные в табл. 43.11,
получены на основе расчета в приближении валентного силового поля по силовым постоянным
(в 105 дин-см~1)/г=3,1, /rr=0,3, fjr2—0,1, оцененным из сравнения силовых постоянных молекул
SF6, SF4 и UF6. Рассчитанные значения основных частот валентных колебаний U—F E40—550 см)
близки к наблюдавшимся в ИК-спектрах молекул UF4, изолированных в матрицах [3141, 3142]
E10—550 см). Погрешности в значениях частот оцениваются в 100 см для vlt v2, v6, v8 и 40 см
для v3—v5, v7 и v9.
Экспериментальные данные о возбужденных состояниях молекулы UF4 в литературе
отсутствуют. Пренебрегая влиянием природы лиганда, можно ожидать, что электронный спектр UF4
должен быть близок к электронному спектру молекулы UC14, который интерпретирован в рамках
теории поля лигандов [2512] 2. Почти все наблюдаемые переходы в спектре UC14 объяснены на
основании атомных состояний, соответствующих конфигурации /2 и претерпевающих расщепление
вследствие спин-орбитального взаимодействия и влияния поля лигандов симметрии Td.
Приведенные в табл. 43.11 энергии электронных состояний UF4 оценены по данным для электронных
состояний UC14 [2512]. Поскольку при переходе от симметрии Td (UC14) к C2v (UF4) терм основного
состояния Г5 (рх=3) должен расщепляться, статистический вес основного электронного состоя-
1 Кунае и др. [3142] в ИК-спектрах молекул UF4, изолированных в различных низкотемпературных матрицах,
в области валентных колебаний U—F наблюдали большое число полос. Анализ спектров привел авторов [3142]
к выводу, что большое число полос обусловлено матричными эффектами и не может рассматриваться как
свидетельство низкой симметрии этой молекулы. Поэтому они приняли, что в газовой фазе UF4 имеет структуру
правильного тетраэдра (симметрия Td) пли плоского квадрата (симметрия Dih). С учетом интерпретации электронного
спектра UC14 в работе [25121 на основе симметрии Тл Кунзе и др. отдают предпочтение первой структуре. Для
модели правильного тетраэдра интерпретирован фотоэлектронный спектр газообразного UF4 [2139], однако
характер наблюдаемых полос не позволил исключить возможность небольшого нарушения симметрии Тd.
2 Отметим, что спектр свободного иона U*+ (см. [4789]) оказался очень близок спектру UC14.
215
UFt(r) Глава 43
ния принят равным 1, а для первого возбужденного состояния статистический вес увеличен
с 2 до 4. Погрешности в усредненных значениях энергий возбужденных состояний
оцениваются в 500—2000 см.
Термодинамические функции UF4(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» с учетом 13 электронных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью принятых молекулярных постоянных и приближенным
характером расчета; они составляют 8, 12, 16 и 20 Дж-К-моль~1 в значениях Ф°(Г) при 298,15,
3000, 6000 и 10 000 К.
Хилденбранд [2644] на основании выполненных им измерений давления насыщенного пара
UF4(k) нашел <S°(UF4, г, 1050 К)=499,2 +4 Дж-К^-моль в хорошем согласии с значением
497,8+12 Дж-К^-моль, соответствующим данным табл. 1018.
Термодинамические функции UF4 вычислялись ранее в работе Гурвича и др. [338, 2536] и
справочнике Галкина и др. [742а] (Т ^ 5000 К). Расхождения результатов этих расчетов и данных
табл. 1018 достигают 30 и 50 Дж-К«моль соответственно в значениях Ф°(Г); они обусловлены
тем, что для молекулы UF4 принималась симметрия Td, а также другие значения молекулярных
постоянных. В расчете [742а], кроме того, не учитывались возбужденные электронные состояния
молекулы UF4.
Константа равновесия реакции UF4(r)=U(r)-f-4F(r) вычислена с использованием значения
Дг#°@)=2448,001+17 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации UF4(k) 1. Этим величинам также соответствует
A^UFi, г, 0) = —.1604,036 ±15 кДж • моль.
^r). Термодинамические свойства газообразного положительного иона тетрафторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1019.
Молекулярные постояннее UFJ, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UF+ в литературе
отсутствуют. По аналогии с UF4 в справочнике принимается, что UFJ в основном электронном
состоянии имеет структуру искаженного тетраэдра (симметрия С2е) (см. примеч. к тексту по UF?(r)).
Приведенные в табл. 43.11 произведение моментов инерции и основные частоты UFJ приняты
такими же, как постоянные молекулы UF4. Погрешность значения IaIbIg оценивается
в 2-Ю13 г3-см6, значений vl5 v2, v6, v8 — в 150 см, v3—v5, v7, v9 — в 50 см. Электронные
состояния UF+, приведенные в табл. 43.11, приняты идентичными состояниям UF5 (см. 3-е примеч.
к тексту по UO2(r)).
Термодинамические функции UFj(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)-A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом шести возбужденных электронных состояний по уравнениям A. 168)—A. 170).
Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных постоянных,
в частности структуры UF*, и приближенным характером расчета; они составляют 10, 15, 20
и 30 Дж-К-^моль в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UF* вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождения с результатами этого расчета составляют 22—28 Дж-К-моль~1 в значениях Ф°(Т)
и обусловлены тем, что в работе [338, 2536] для молекулы UF* принята симметрия правильного
тетраэдра Td, а также некоторым уточнением оценки частот в настоящем издании.
'Константа равновесия реакции UT4^ (г) -^е(г) =\3 (,г) -V4F(г) вычислена с использованием
значения Дг#°@)=1488,001+44 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(UF4)=960+40 кДж-моль.
Результаты измерений этой величины представлены в табл. 43.17. Полученные величины имеют
широкий разброс, значительно превышающий их погрешности. В работе Дайка и др. [2139]
методом фотоэлектронной спектроскопии был установлен сложный характер процесса ионизации
UF4 и найдено два значения потенциала ионизации (в кДж-моль): 918 (адиабатический) и 996
(вертикальный). Низкая величина сечения отрыва 5/и-электрона, не участвующего в образовании
1 См. примеч. 1 к тексту по UF(r).
216
Уран и его соединения UF4(r), UF5(k, ж)
Таблица 43.17. Результаты определений J0(UF4) (в кДж • моль)
Авторы
Метод
Ходеев, 1964 [1079] Электронный удар, квазимонокинетические электроны 1052+20
Змбов, 1969 [4825] Электронный удар 1129+50
Хилденбранд, 1977 [2644] То же 961+10
Дайк и др., 1980 [2139] Фотоэлектронная спектроскопия 957±40
Горохов и др., 1982 [4868] Электронный удар 1052±20
химических связей в молекуле UF4, могла привести к затрудениям при регистрации начального
участка кривой эффективности ионизации и к завышенным значениям потенциала ионизации,
полученным в работах [1079, 4868]. Принятая величина основана в первую очередь на результатах
исследования Дайка и др. [2139], подтверждаемого измерениями Хилденбранда [2644] Ч Ей
соответствует
kfH°(UFt, г, 0) = —644,036 + 43 кДж • моль.
^). Термодинамические свойства газообразного отрицательного иона тетрафторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1020.
Молекулярные постоянные UFJ, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UF^ в литературе
отсутствуют. По аналогии с UF4 в справочнике принимается, что UF^ в основном электронном
состоянии имеет структуру искаженного тетраэдра (симметрия С2„) (см. примеч. 1 на с. 215).
Приведенные в табл. 43.11 произведение моментов инерции и основные частоты UF" приняты
тождественными соответствующим постоянным молекулы UF4. Погрешность значения IaIbIc оценивается
в 2-Ю"13 г3»смв, значений vx, v2, v6, vg — в 150 см, v3—v5, v7, v9 — в 50 см. Возбужденные
состояния UF^, приведенные в таблице, приняты идентичными состояниям UF3 (см. 3-е примеч.
к тексту по UO2(r)), но ограничены энергией равной потенциалу ионизации UFJ.
Термодинамические функции UF^r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом четырех объединенных электронных термов по уравнениям A. 168)—A. 170).
Погрешности рассчитанных значений обусловлены неточностью оценки молекулярных постоянных,
в частности структуры UF^ в основном состоянии, и приближенным характером расчета; они
составляют 15, 20, 25 и 35 Дж-К-^моль в значениях Ф°(Т) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UF^ вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождения результатов этого расчета и данных табл. 1020 составляют 26—29 Дж-К~1-моль~1
в значениях Ф°(Т), они обсуловлены тем, что в работе [338, 2536] для UF~ принята структура
правильного тетраэдра, а также некоторым уточнением оценки постоянных в настоящем издании.
Константа равновесия реакции UF^(r)=U(r)+4F(r)-f е(т) вычислена с использованием значения
Дг#°@)=2563,965+50 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
, г, 0) = —1720 + 50 кДж • моль.
Эта величина основана на результатах масс-спектрометрического исследования реакции
UF7(r)+UFg(r)=2UF~(r) A203—1293 К, И точек), выполненного Пятенко и др. [4891, 4894].
По этим измерениям были найдены значения —1722+50 кДж-моль (III закон) и —1787 +
+150 кДж-моль (II закон). Принятой величине соответствует
=—115,964+52 кДж-моль.
UF5(k, ж). Термодинамические свойства кристаллического и жидкого пентафторида урана
в стандартном состоянии при температурах 100—1800 К приведены в табл. 1021.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 43.1. В справочнике за стандартное состояние UF6(k) в интервале 0—398 К принимается
1 См. примеч. к тексту по UF(r).
217
UF5(k, ж) Глава 43
тетрагональная ^-модификация [2169, 4806], а в интервале 398—621 К — высокотемпературная
а-модификация, также обладающая тетрагональной симметрией [4106, 4806].
При Т <^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в интервале 25—310 К, выполненным Брикведде и приведенным в книге Каца и
Рабиновича [492]. Исследованный образец a-UF5 содержал только 82,82% пентафторида урана, и поэтому
полученные данные являются весьма приближенными. Поскольку ниже 398 К устойчивой
является р-модификация, значение энтропии при 298,15 К принято по справочнику МАГАТЭ [2344],
авторы которого рассчитали 5°B98,15 К) для а- и P~UF5. Эти расчеты основаны на данных Агрона
по равновесному давлению пара гексафторида урана над а- и [3-UF5 при их диспропорционирова-
нии. Экспериментальные данные со ссылкой на недоступную авторам настоящего справочника
публикацию Агрона приведены в книге Каца и Рабиновича [492]. Погрешности значений
?°B98,15 К) и Я°B98,15 К)—Н°@), приведенных в табл. 43.1, оцениваются в 12 Дж-К^-моль
и 0,4 кДж-моль.
Сведения об измерениях теплоемкости или энтальпии UF6(k) при высоких температурах в
литературе отсутствуют. При Т > 298,15 К для теплоемкости обеих модификаций UF5(k) принято
уравнение, предложенное Кнакке и др. [3044]. Температура полиморфного превращения C98 +
+3 К) получена авторами [492] из уравнений, описывающих давление пара гексафторида урана
над а- и P~UFS при диспропорционировании последних. Энтальпия полиморфного превращения
(8+4 кДж-моль"*1) принята по оценке [2344].
Температура плавления UF6 F21+3 К) измерена Вольфом и др. [4753]. Образец, полученный
взаимодействием тетрафторида урана и газообразного гексафторида урана, представлял собой
чистый a-UF6. Энтальпия плавления C5+10 кДж-моль) оценена на основании предположения,
что энтропии плавления UF5 и UF6 равны E7 Дж-К~1-моль~1). Расчет энтальпии плавления
из данных Вольфа и др. [4753] о давлении пара над твердым E55—619 К) и жидким F22—685 К)
UF5 D7 кДж-моль) ненадежен, так как пар над UF5 (к, ж) имеет сложный состав.
Теплоемкость ОТ6(ж) A67+25 Дж-К^-моль) принята по оценке Кнакке и др. [3044].
Погрешности вычисленных значений Ф°(Т) при 298,15 и 1500 К оцениваются в 10 и 40 Дж- К X
Хмоль соответственно. Расхождение между термодинамическими функциями UF5(k, ж),
приведенными в табл. 1021 и в справочнике Барина и Кнакке [1395] (Т ^ 900 К), достигают 18 ДжХ
XК-моль в значениях Ф°(Г) и обусловлены существенным уточнением оценки энтропии
UF5(k) при 298,15 К авторами справочника [2344].
Принятая в справочнике энтальпия образования
i±fH0(p-UF&, к, 298,15 К) = —2083,0 + 6,3 кДж • моль
основана на измеренных О'Хара и др. [5076] энтальпиях реакций UF6(k), U02(k) и UO3(k) с
.сернокислыми растворами Ge(SO4J. Ранее Агрон [1270] исследовал равновесие реакций диспропорциони-
рования а- и C-UF6(k) с образованием U4F17(k), U2F9(k) и UFB(r). Эти измерения были обработаны
авторами справочника МАГАТЭ [2344], которые получили A/ff°(P-UF6, к, 298,15 К) = —2075,5 +
+ 8,4 кДж'• моль, что находится в согласии с результатами работы [5076].
В работе [5076] по разности энтальпий растворения а- и |3-UF5(k) в сернокислых растворах
Ce(SO4J было получено A/E°(a-UF6, к, 298,15 К) = — 2075,5 ± 6,7 кДж • моль.
Давление пара в реакции UF5(k, ж) = иГ5(г) вычислено с использованием значения
t\rH°@) = \H°($-UFb, к, 0) = 135,724 + 26 кДж • моль.
Вольф и др. [4753] исследовали скорость потери веса UF5(k, ж) в токе UFe (метод протока,
556 — 685 К, 18 точек). Значительное (до 3 атм) давление UF6 предотвращало диспропорциони-
рование UF5(k, ж). Кляйншмидт и Хилденбранд [4993] исследовали масс-спектрометрическим
методом реакцию 2UF6(r) = U2Fl0(r) и нашли, что в условиях опытов [4753] в парах содержится
до 95% димерных молекул. Позднее Лейтнакер [5005] учел также возможность образования
U2Fu(r), что уменьшило долю парциального давления UF6 в насыщенном паре примерно до 2%.
Расчет энтальпии сублимации по скорректированным таким образом измерениям [4753] приводит
к значению AS#°(C-UF5, к, 0) = 167 + 20 кДж • моль. Хотя это значение и принятое в
справочнике совпадают в пределах погрешностей, величина P(UF6, к, 600 К), приведенная в табл. 1021,
почти на порядок превышает суммарное давление насыщенного пара, измеренное в работе [4753].
Это свидетельствует о противоречивости результатов измерений давления и состава пара над
218
Уран и его соединения UF5(r)
UF5(k, ж) [4753, 4993, 5005] и энтальпий образования UF5 в кристаллическом и газообразном
состояниях (см. соответствующие тексты).
В справочнике отдано предпочтение измерениям энтальпий образования UF6(k) и UFB(r).
Однако эта точка зрения не бесспорна. Так, в частности, принятая в справочнике энтальпия
образования UF8(r) вычислена с использованием значения энтальпии образования UF4(r). Последняя
зависит от принимаемого значения для энтальпии образования UF4(k). Как отмечают О'Хара
и др. [5076], в неопубликованной работе Джонсона для энтальпии образования UF4(k) получено
значение —1910,6 + 2,0 кДж • моль. В справочнике принято значение, которое на 10 кДж • моль
меньше этой величины. Использование новой величины, измеренной Джонсоном, привело бы
к значениям kfH°(UF5, г, 0) = —1935 кДж • моль-1 и AS#°(C-UF6, к, 0) = 146 кДж • моль, что
лучше согласуется с измерениями давления пара UF8(k, ж).
UF5(r). Термодинамические функции газообразного пентафторида урана в стандартном
состоянии при температурах от 100 до 10 000 К приведены в табл. 1022.
Молекулярные постоянные UF5, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Строение молекулы UF6 в газовой фазе в настоящее время окончательно
не установлено. Несколько полос, наблюдаемых в ИК-спектре газообразного UF5 [3001, 3002,
3003], не удалось интерпретировать однозначно. Исследования ИК-спектров и спектров КР
молекул UF6, изолированных в низкотемпературных матрицах [2895, 2896], а также расчет ИК-
спектра поглощения UF5 [3117] позволяют предполагать, что в матрице молекула UF5 имеет
структуру квадратной пирамиды симметрии С4„. Неэмпирический расчет геометрии UF8 в основном
состоянии, выполненный Вадтом и Хеем [4625], показал, что структуры симметрии Civ и D3h
отличаются по энергии всего на 4 кДж-моль. В соответствии с этим в справочнике принимается,
что в основном электронном состоянии молекула UF5 имеет равновесную структуру квадратной
пирамиды (симметрия Civ) с переходом в конфигурацию тригональной бипирамиды симметрии
D3h при колебании х. Небольшой барьер, ограничивающий внутримолекулярную перегруппировку,
учтен введением низкочастотного деформационного колебания v5.
Приведенное в табл. 43.11 произведение моментов инерции соответствует значениям r(U—F) =
=2,02+0,03 А (оценено как среднее между величиной r(U—F) в молекулах UF4hUF6) и -$FaK0—U—
—FBKB=100+5° (принят по расчету [4625], совпадает с оценками авторов [2895, 3117]).
Погрешность значения IaIbIc составляет 1-Ю"*113 г2-смв.
Основные частоты UF5, за исключением v5, приняты по работе Джонса и Экберга [2895]: для
валентных колебаний v1, v2 и v7—это значения частот, наблюдавшихся в спектре молекул UF6,
изолированных в матрице из Ne, а для остальных частот — значения, рассчитанные по модели
валентного силового поля 2. Для частоты деформационного колебания v6, соответствующего
переходу между конфигурациями симметрии Civ и D3h, принято значение, оцененное по величине этой
частоты в других молекулах пентагалогенидов, имеющих такой же небольшой барьер
внутримолекулярных перегруппировок [1522, 2037, 2786]. Погрешность в значении v5 оценивается в 20 см,
частот v2, v2 и v7 — в 15 см", v3, v6, vg и v9 — в 40 см, v4 — в 70 см.
Экспериментальные данные о возбужденных состояниях молекулы UF5 в литературе
отсутствуют. Приведенные в табл. 43.11 электронные состояния UF5 оценены на основании результатов
неэмпирического расчета [4625] и экспериментальных данных для соединений, содержащих ион
U8+ в полях лигандов симметрии С2о [2345] и Di [4230] 8.
Термодинамические функции UF5(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)— Д. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и A. 130)
с учетом шести возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности рассчи-
1 Хотя для равновесной конфигурации UF5 принята симметрия Civ, в табл. 43.11 приведено число симметрии 2;
это обусловлено тем, что переход от конфигурации Cis к D3h осуществляется через промежуточную структуру
симметрии С2„.
2 Значения частот, полученные в работах [2895, 3117] для структуры симметрии Civ, хорошо согласуются с
расчетом Раиса и др. [3986], основанном на результатах исследования спектра испускания газообразного UF6.
3 В расчете [4625] было показано, что молекула UF5 имеет основное состояние Х2В2 и возбужденные состояния
2Е, 2В1; 2Аг и 2Е, которые соответствуют нахождению одного электрона на молекулярных орбиталях &2,
¦е, Ьг, а] и е, образующихся из 5/-орбитали атома U в поле симметрии C4v. После завершения подготовки главы
43 было опубликовано исследование спектра испускания газообразного UF6 в работе Раиса и др. [3986],
которое позволило определить энергию самого высокого (/—/)-перехода в UF6 A4 647 см), близкую к величине
дриведенпой в таблице.
219
Глава 43
танных значений обусловлены неточностью использованных молекулярных постоянных, в том
числе неоднозначностью принятой структуры, и приближенным характером расчета, в частности
учетом внутримолекулярной перегруппировки как гармонического колебания; они составляют
12, 20, 25 и 35 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UF6(r) вычислялись ранее в работе Гурвича и др. [338, 2536]
(Т < 10 000 К) и в справочнике Галкина и др. [742а] {Т < 5000 К). Расхождения данных [338,
2536] и табл. 1022 достигают 9—11 Дж-К^-моль в значениях Ф°(Г), они обусловлены некоторым
уточнением оценки постоянных UF6 в настоящем издании. Расхождения с результатами расчета
[742а] достигают 26—46 Дж-К^-моль в значениях Ф°(Г) из-за того, что авторы этого расчета
не учитывали возбужденных состояний UFB, а для основного состояния приняли структуру
симметрии D3h.
Хилденбранд [2644] на основании экспериментального изучения равновесия реакций с участием
UF5(r) нашел iS°(UF5, г, 1100 К)=550+4 Дж-К^-моль, это значение хорошо согласуется
с приведенным в табл. 1022 E50+15 Дж-К^-моль).
Константа равновесия реакции UF5(r) = U(r)+5F(r) вычислена с использованием значения
Дгйго@)=2866,24+26 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —1945 + 25 кДж • моль.
Это значение основывается на результатах масс-спектрометрического исследования Хилден-
бранда [2644] (UF5(r)+Ag(r)=UF4(r)+AgF(r), 1012—1158 К, 12 точек). Константам равновесия,
измеренным Хилденбрандом, соответствуют AjH°(\JF5, г, 0) =—1944+25 кДж-моль (III закон)
и —1946+20 кДж-моль (II закон). В этой же работе методом электронного удара была
исследована реакция UF6+e=UFg+F+2e, откуда было получено значение —1935 кДж-моль, близкое
к вычисленным по результатам исследования указанного выше равновесия. Приведенная
погрешность учитывает воспроизводимость экспериментальных данных и неточность использованных
в расчетах величин, прежде всего термодинамических функций UF6(r) и UF4(r). Данные [2641]
подтверждаются измерениями равновесия реакции UF4(r) -j-CuF2(r) = UF5(r) -|-CuF(r) в работе
Горохова и др. [4866].
g. Термодинамические свойства газообразного положительного иона пентафторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1023.
Молекулярные постоянные UF*, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UF* в литературе
отсутствуют. По аналогии с UF5 в справочнике принимается, что UF* в основном электронном
состоянии Х1А1 имеет структуру квадратной пирамиды (симметрия Civ), находящейся в равновесии
с конфигурацией тригональной бипирамиды (симметрия DSh), от которой она отделена низким
барьером (промежуточная структура симметрии C2t). Приведенные в табл. 43.11 произведение
моментов инерции и основные частоты UFg приняты аналогичными соответствующим постоянным
молекулы UF6. Погрешность значения IaIbIc оценивается в 2-10~113 г3-см6, значений vl7 v2, v4,
v7 — 100 см, v3 — 30 см, v5 — 20 см, v6, v8, v9 — 50 см.
Молекула UFg имеет заполненную электронную оболочку, и в справочнике принимается, что
она так же, как молекула UF6, не имеет низколежащих возбужденных электронных состояний.
Термодинамические функции UFj(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130).
Погрешности рассчитанных значений обусловлены главным образом неточностью принятой
структуры молекулы и других молекулярных постоянных, а также приближенным характером расчета;
они могут достигать 15, 25, 30 и 40 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000, 6000 и
10 000 К.
Термодинамические функции UF*(r) вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождения результатов этого расчета и данных табл. 1023 составляют 10 Дж-К-моль в
значениях Ф°(Т) и обусловлены уточнением оценки постоянных этой молекулы в настоящем издании.
Константа равновесия реакции UFj(r)+e(r)=U(r)+5F(r) вычислена с использованием значения
Дг^Гс@)=1776,24+33 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(UF5)=1090+20 кДж-моль-1.
220
Уран и его соединения
UFi(r)
Принятая величина была получена Хилденбрандом [2644] методом электронного удара. UF5
получался непосредственно в эффузионной камере по реакции UF4(K)+UF6(r)=2UF5(r). Принятой
величине соответствует
bfH°(XJFt, г, 0) = —855,0+-32 кДж • моль.
UFg(r). Термодинамические свойства газообразного отрицательного иона пентафторида урана
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1024.
Молекулярные постоянные UF7, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UF~ в литературе
отсутствуют. По аналогии с молекулой UF5 в справочнике принимается, что UF" в основном
электронном состоянии имеет структуру квадратной пирамиды (симметрия С4р), в равновесии с которой
находится конфигурация тригональной бипирамиды (симметрия D3h), а соответствующий переход
осуществляется через промежуточную структуру симметрии C2v. Приведенные в табл. 43.11
произведение моментов инерции и основные частоты UF~ аналогичны соответствующим постоянным
молекулы UF5. Погрешность значения IaIbIc оценивается в 2-10~113 г3-см6, значений
v, и v7 —
100 см", v3 — 30 см, v6 — 20 см \ ve, v, и v, — 50 см. Возбужденные состояния UF5,
приведенные в табл. 43.11, приняты идентичными состояниям молекулы UF4(r) (см. 3-е примеч. к тексту
по UO2(r)).
Термодинамические функции UF6(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом 13 объединенных электронных термов по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью принятой структуры UFg и других
молекулярных постоянных, а также приближенным характером расчета; они могут достигать 15, 25,
35 и 50 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Термодинамические функции UFr(r) вычислялись ранее в работе Гурвича и др. [338, 2536]
(Т ^ 10 000 К). Расхождения с результатами этого расчета составляют 10—12 Дж-К^-моль
в значениях Ф°(Г) и обусловлены уточнением оценки постоянных UFJ в настоящем издании.
Константа равновесия реакции UF~(r)=U(r)+5F(r)+e(r) вычислена с использованием
значения Дгй^°@)=3201,24+22 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(UF5, г, 0) = — 2280 + 20 кДж • моль.
Результаты определений энтальпии образования UFg(r) представлены в табл. 43.18.
Приведенные в таблице погрешности учитывают воспроизводимость измерений, неточность использованных
Таблица 43.18, Результаты определений Д/Я° (UFjr, г, 0) (в кДж-моль)
Комптон
Пятенко
Сидоров
же [725,
Аэторы
, 1977,[1909]
и др., 1980 [4891,
и др., 1980 [4280]
4278])
4892]
(см. так-
Метод
Измерение порогов реакций UFe(r)+M(r)=
= UFj(r)+M+(r)+F(r)
То же, принимая, что вблизи порога протекает
реакция UFe(r)+M(r)=UF5(r)+MF+(r)
Масс-спектрометрический, UFf(r)+AlF3(K)=
=UF4(K)+AlFj(r), 1135 и 1145 К, 2 точки
Масс-спектрометрический, UF4(K)+F~(r)=UFj(r),
1073—1116 К, 7 точек
AfH° (UFg , г, 0)
II закон
III закон
—2331 ±50
—2277+50
— —2267+45
—2125+100 —2283+20
в расчетах термодинамических функций и термохимических величин. Приведенные в таблице
величины (если принять по данным работы [1909] величину, соответствующую реакции с
образованием MF+, как более обоснованную) согласуются в пределах их погрешностей. Принятая
величина является средневзвешенной. Ей соответствует сродство UF8 к электрону
40(UF5) =—335,0+32 кДж-моль.
221
UF6(k, ж) Глава 43
UF6(k, ж). Термодинамические свойства кристаллического и жидкого гексафторида урана
в стандартном состоянии при температурах 100—500 К приведены в табл. Д.66.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 43.1. За стандартное состояние UF6(k) в интервале 0—337,21 К принята ромбическая
модификация; сведения о полиморфизме UF6 отсутствуют. Термодинамические функции UF6(k) при
Т <^ 298,15 К вычислены на основании измерений теплоемкости, проведенных Брикведде и др.
[1669] A4—370 К, образец чистотой 99,98%, погрешности измерений ~0,3%). Экстраполяция
теплоемкости ниже 14 К проводилась по уравнению С° =3DG3/T)JrEA10/T), которое приводит
к значению 5°A8 К)=6,5 Дж-К^-моль-1. Погрешности значений 5°B98,15 К) и #°B98,15 К)—
—Н°@) (см. табл. 43.1) оцениваются в 0,6 Дж-К~1-моль и 0,08 кДж-моль. Менее точные
данные по теплоемкости UF8(k) [3288] B23—298 К) не учитывались.
Уравнение для теплоемкости UF6(k) в интервале 298,15—337,21 К (см. табл. 43.1), температура
плавления C37,21+0,10 К) и энтальпия плавления A9,193+0,050 кДж-моль) приняты по
данным Брикведде и др. [1669]. Измерения Левеллина [3288] приводят к более высоким
значениям теплоемкости (при 333 К на 3,3%). Близкие, но менее точные значения Tm(XJFe) получены
авторами [3713, 4683]. Калориметрическое определение Am//(UFe) в работе [3288] привело к
значению 19,03+0,20 кДж-моль, согласующемуся с принятым в пределах погрешности измерений.
Уравнение для теплоемкости иР6(ж) (см. табл. 43.1) выведено по данным, полученным Брикведде
и др. [1669] C37,2—370 К); погрешности этих данных оцениваются в 0,5%. Менее точные
измерения Левеллина [3288] C43—373 К), выполненные на образце UF6, чистота которого не
охарактеризована, приводят к более высоким значениям С°р(Т) (в среднем на 5%).
Погрешности приведенных в табл. Д.66 значений Ф°(Г) при 298,15 и 500 К оцениваются в 0,4
и 3 Дж-К~1-моль~1 соответственно. Термодинамические функции UF0(k, ж) вычислялись в
справочнике Барина и Кнакке [1395] (до 500 К), расхождения этих данных и данных табл. Д.66 не
превышают 1 Дж-К-моль.
Принятая в справочнике энтальпия образования
bfH°(UFe, к, 298,15 К) =—2197,7 + 1,8 кДж • моль
основана на калориметрических измерениях, представленных в табл. 43.19. В течение ряда лет
Таблица 43.19. Результаты определений AtH ° (UF6, к, 298,15 К) (в кДж • моль)
Авторы
Сеттл и др., 1963 [4239]
Хейман, 1967 [2597]
Паркер, 1976 [3772] (см. также
[2344, 3773])
Джонсон, 1979 [2869]
Метод
\fH° (UFe, к, 298,15 К)
Сжигание урана во фторе в калориметрической —2186,7 40,8
бомбе, 7 опытов
То же, в проточной системе, с образованием —2184,9+2,9
UF6(r), 5 опытов. Учтена As#°(UFe, к)
Расчет из термохимических циклов, включающих —2197,0+2,0
энтальпии растворения UF6, UO3 и UO2F2 в
плавиковой кислоте и в воде
Сжигание урана во фторе в калориметрической —2197,7 ±1,8
бомбе, 7 опытов
полученные в работах [4239] и [2597] совпадающие величины не вызывали сомнений в их
надежности. Однако Паркер [3772] (см. также [2344, 3773]) при анализе литературных данных по
энтальпиям образования соединений урана обнаружила противоречие данных по энтальпиям
образования и растворения в плавиковой кислоте и в воде y-UO3(k), UF6(k) и UO2F2(k) и показала, что
наиболее вероятной причиной этого противоречия является заниженное по абсолютной величине
примерно на 10 кДж-моль значение энтальпии образования UF6(k), найденное в работах [2597,
4239]. Повторное измерение энтальпии сгорания урана во фторе, выполненное Джонсоном [2869],
показало полную обоснованность анализа, проведенного Паркер. Найденная в работе [2869]
величина принимается в справочнике. Причины, обусловившие ошибки измерений [2597, 4239],
остались не установленными.
Принятая энтальпия образования UF6(k) подтверждается также расчетами Паркер [3772,
3773], основанными на термохимическом цикле, включающем энтальпию диссоциации нитрида
222
Уран и его соединения
UF6(r)
урана [2675] и его энтальпию сгорания во фторе [3701], и аналогичном цикле с сульфидом урана
[3634, 3701]. Им соответствуют значения — 2199+8 кДж-моль и —2202+13 кДж-моль.
Измерения энтальпии гидролиза UF6(k), выполненные Лонгом и Дэвидсоном [3292], привели
к значению —2188+25 кДж-моль.
Давление пара в реакции UF6(k, №") = UF6(r) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = AS#°(UF6, к, 0) = 54,0 ± 1,0 кДж • моль.
Результаты исследований энтальпии сублимации представлены в табл. 43.20. Приведенные
Таблица 43.20. Результаты определений А8ЙГ° (UFe, к, 0) (в кДж-моль)
Авторы
Руфф, Хайнцельманн, 1911 [4090]
Амфпетт,
Амфлетт
Вайншто!
Кигоши,
Леве длин
Оливер и
Томас, 1941 [см. [492])
и др., 1948 [1314]
;, Крист, 1948 [4683]
1950 [2992]
, 1953 [3288]
др., 1953 [3713]
Метод
Точек кипения, 310—330 К, 5 точек
Компенсационный, 294—304 К, 6 точек
То же, 298—329 К, 18 точек
То же, 286—323 К, 11 точек
То же, 295—336 К, 8 точек
То же, 338—358 К, 8 точек
Статический, 273—308 К, данные
представлены в виде уравнения
Протока и статический, 278—373 К, 28 точек
Компенсационный и точек кипения, 278—
333 К, 9 точек
То же, 337—365 К, 12 точек
ASH°
II закон
46+15
57+3
54,3+1,4
54,7 ±1,1
53,3+1,2
55,1+1,6
54,4+1,1
?0,4+2,0
54,9+0,5
56,1+1,5
(UFe, к, 0)
III закон
53,6+0,3
54,09+0,04
53,85+0,02
54,08+0,03
53,98+0,05
53,86+0,03
54,08+0,05
53,93+0,04
54,04 ±0,05
53,79+0,04
в таблице погрешности учитывают только воспроизводимость измерений. За исключением данных
работы [4090], где, вероятно, использовался недостаточно чистый образец, результаты измерений
находятся в хорошем соответствии. Принятая величина является средней из всех данных, за
исключением [4090]. Основной вклад в погрешность принятой величины вносит неточность
термодинамических функций UF6(r).
UF6(r). Термодинамические свойства газообразного гексафторида урана в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 1025.
Молекулярные постоянные UF6, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Согласно электронографическим [2416, 3006, 4207, 4226] и спектральным
[1418, 1559, 1625, 1728, 1813, 1861, 1862, 1949, 2339, 2376, 3452, 3502, 3746, 4684] данным
молекула UFe в основном электронном состоянии ХгА1д имеет структуру правильного октаэдра
(симметрия Oh). Приведенное в табл. 43.11 произведение моментов инерции вычислено для значения
r(\j—F) = 1,999+0,003 А, найденного Сейпом [4226] в электронографическом исследовании;
погрешность в его значении составляет 2-1014 г3-см6. Значения основных частот UFe,
приведенные в таблице, приняты на основании результатов исследования ИК-спектра и спектра КР
газообразного 238UFe в работе Мак-Дауэлла и др. [3452]. Данные этих авторов хорошо согласуются
с другими спектральными исследованиями, перечисленными выше. Погрешности принятых
значений частот оцениваются в 2—3 см.
Молекула UFe имеет заполненную электронную оболочку и поэтому у нее не ожидается
существование низколежащих электронных состояний. Согласно экспериментальным и
теоретическим данным [1620, 1621, 2049, 3060, 3249, 3435, 3448, 3502, 4372] нижние возбужденные состояния
UF6 обусловлены (р—/)-переходами и имеют энергии выше 25 000 см, поэтому они не
рассматриваются в справочнике.
Термодинамические функции UF6(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122) A. 124), A. 128), A. 130) в приближении «жесткий ротатор—гармонический осциллятор»
без учета возбужденных электронных состояний. Погрешности рассчитанных величин опреде-
223
UFe(r) Глава 43
ляются преимущественно приближенным характером расчета и составляют 4, 12 и 20 Дж-К~1Х
Хмоль в значениях Ф°(Т) при 298,15, 3000 и 6000 К; погрешности, обусловленные неточностью
принятых значений молекулярных постоянных, не превышают 1 Дж-К~1-моль~1 во всем интервале
температур.
Термодинамические функции UF6 (г) вычислялись ранее в справочниках [742а] (Т ^ 5000 К),
[2344] (Т < 3000 К), работах Гурвича и др. [338, 2536] (Т < 10 000 К), ([2376, 3617,
3620, 4441] (Т ^ 500-f-2000 К). Расхождения данных табл. 1025 с результатами
перечисленных расчетов составляют 0,05—2 Дж-К -моль и обусловлены некоторым различием принятых
молекулярных постоянных.
Константа равновесия реакции UF6(r)=U(r)+6F(r) вычислена с использованием значения
Дг#°@)=3140,941 + 8,5 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации UF6(k). Этим величинам также соответствует
Д/HUFe, г, 0) = — 2142,426 + 2,1 кДж • моль.
UF~(r). Термодинамические свойства газообразного отрицательного иона гексафторида урана
в стандартном состоянии при температурах от 298,15 до 6000 К приведены в табл. 1026.
Молекулярные постоянные UF~, использованные для расчета термодинамических функций,
приведены в табл. 43.11. Экспериментальные данные о строении и спектре UFJ7 в литературе
отсутствуют. По аналогии с газообразным UFe и кристаллическими соединениями, содержащими
ион UF~ [3967, 4231], в справочнике для UF~ принята октаэдрическая структура (симметрия Oh).
Приведенное в табл. 43.11 произведение моментов инерции вычислено для значения r(U—F) =
=2,0+0,05 А (оценка по величине r(U—F) в молекуле UFe). Погрешность значения IaIbIc
оценивается в 2-Ю13 г3-см6. Значения основных частот, приведенные в табл. 43.11, оценены по
основным частотам молекулы UFe и кристаллического иона UF" [3967, 4231, 4798]. Погрешности в этих
значениях составляют 100 см для Vj, v2, v3 и 40 см для v4, v6 и v6.
Неэмпирический расчет UF" [2595], а также исследования спектров поглощения GsUF6 [3967]
и различных соединений, содержащих ион U5+ в октаэдрическом окружении лигандов [4231],
показали, что UFJ7 имеет основное состояние X2Aiu и возбужденные состояния 2F2u и 2Flu,
соответствующие нахождению одного электрона на молекулярных орбиталях а2и, /2м и /1и,
образующихся из 5/-орбиталей атома U в поле симметрии Oh. Наличие спин-орбитального взаимодействия
приводит к расщеплению вырожденных состояний и поэтому в кристаллах обнаружено четыре
перехода — с основного уровня Г7 BА2и) на возбужденные Г8, Т'7 BF2m) и Г^, Г6 BF1u). Приведенные
в табл. 43.11 энергии электронных состояний UFg найдены для CsUFe[3967] и хорошо согласуются
со значениями, рассчитанными в работе [2595]. Погрешности принятых величин оцениваются
в 1000-2000 см.
Термодинамические функции UF~(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом четырех возбужденных электронных состояний по уравнениям A. 168)—A. 170).
Погрешности рассчитанных значений обусловлены неточностью принятых молекулярных
постоянных и приближенным характером расчета; они составляют 10, 25 и 35 Дж-К^-моль в значениях
Ф°(Г) при 298,15, 3000 и 6000 К.
Термодинамические функции UF~(r) вычислялись ранее в работе Гурвича и др. [338, 2536].
Расхождения данных табл. 1026 с результатами этого расчета составляют 7—11 Дж-К~1-моль-1
в значениях Ф°(Т) и обусловлены уточнением оценки постоянных в настоящем издании.
Константа равновесия реакции UFg(r) = U(r)+6F(r)+e(r) вычислена с использованием значения
АгН°@)=3678,515 +36 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
^fH0(UFъ, г, 0) = —2680 + 35 кДж • моль.
Результаты измерений этой величины представлены в табл. 43.21. Указанные в таблице
погрешности учитывают воспроизводимость измерений, неточность использованных в расчетах
термодинамических функций и термохимических величин. Приведенные в таблице величины совпадают
в пределах указанных погрешностей, и принятая величина является средневзвешенной. Ей
соответствует сродство UF6 к электрону
A0(UFe)=—537,574+35 кДж-моль.
224
Уран и его соединения
UOF(r)
Таблица 43.21. Результаты определений Д^2Т° (UFg, г, 0) (в кДж • моль)
Авторы
Метод
AfH° (UFg, г, 0)
II закон
III закон
Бишам, 1976 [1439] Измерение порога реакции UFjr(r)+BF3(r)= —2716+50
=UF5(r)+BFj(r)
Сидоров и др., 1980 [4280] Масс-спектрометрический, UFi-(r)+K(r)=UFs(r)+ — —2662+35
+KF(r), 1087 К, 1 точка (см. пересчет в [4891,
4894])
Пятенко и др., 1981 [4891, 4894] Масс-спектрометрический, UFe(r)+UF4(r)= —2658+75 —2690+35
=UF5(r)+UF5(r), 1045—1100 К, 7 точек
UOF(r). Термодинамические функции газообразного оксид-фторида урана в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 1027.
Молекулярные постоянные UOF, использованные для расчета термодинамических функций,
приведены в табл. 43.22. Экспериментальные данные о строении и спектрах молекулы UOF в
литературе отсутствуют. По аналогии с молекулой SOF [4955] в справочнике принимается, что в
основном электронном состоянии молекула UOF имеет нелинейную структуру (симметрия Cg).
Таблица 43.21
Молекула
!. Значения молекул ирных постоянных, а также з и рх, принятые для расчета
термодинамических функций UOF, UO2F, UOF2, UO2F2, U0F3 и UOF4
v2
V4
•*7
^8
см"
We-1»1"
r3 ¦ cm'
a
Px
UOFа 750 500 150 — — — — — — 2 -103 1 2
UO2Fa 800 500 180 170 750 150 — — — 7-103 1 2
UOFaa 800 550 180 130 500 150 — — — 1,2-10* 1 1
UO2F2 830 550 200 150 150 850 180 600 200 2,2-10* 2 1
UOF3 a 800 550 150 600 B) 180 B) 130 B) — — — 3 -104 3 2
UOF4 830 650 150 500 200 180 600B) 200B) 170B) 6-Ю4 4 1
a Ниже приведены значения энергий Т\ (в см) и статвесов р{ возбужденных электронных состояний:
UOF: 200 D), 400 D), 4500 A2), 7000 B0), 10 000 C5), 15 000 C5), 20 000 C5);
UO2F, UOF3: 2000 B), 5000 B), 7000 B), 9000 B), 11 000 B), 14 000 B);
UOF2: 500D), 2000C), 3000A), 4000E), 6000(8), 7000C), 9000A4), 11000A1), 12 000D), 14 000E),
17 000 A0), 19 000 A5), 22 000 F).
Приведенное в табл. 43.22 произведение моментов инерции вычислено по структурным параметрам
r(U—O)=l,80+0,05 A, r(U—F)=2,10+0,05 A, ^O—U—F=110+10°, которые были оценены
по структурным параметрам молекул SOF [4955], SO2, SF2, SO2F2 и UO2F2, UO2 и UF2.
Погрешность значения IaIbIo составляет 8-Ю16 г3-см6. Приведенные в таблице основные частоты UOF
приняты на основании сравнения основных частот молекул SOF A215, 763 и 396 см) [4955],
SO2, SF2 с частотами молекул UO2 и UF2, а также с принятыми в справочнике основными частотами
UO2F2 и UOF2. Погрешность значений vx и v2 оценивается в 100 см, v3 — в 50 см. Энергии
возбужденных электронных состояний UOF приняты идентичными энергиям состояний UF3 (см. 3-е
примеч. к тексту по UO2(r)), их погрешности могут достигать 25% от их величины.
Термодинамические функции UOF(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом семи возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью принятых молекулярных постоянных и приближенным
характером расчета; они составляют 6, 10 и 15 Дж-К^-моль в значениях Ф°(Г) при 298,15,
3000 и 6000 К соответственно (погрешность из-за неточности оценки структурных параметров и
15 Заказ W 1590
225
UO2F(r), UOF2(r) Глава 43
основных частот в основном состоянии составляют 3—4 Дж-К~1-моль~1 во всем интервале
температур).
Таблица термодинамических функций UOF(r) публикуется впервые.
Константа равновесия реакции UOF(r)=U(r)+O(r)+F(r) вычислена с использованием
принятой в справочнике энтальпии атомизации
Arff °@) = 1400 ± 100 кДж • моль.
Это значение было оценено на основании величины энергии связей в газообразных оксидах,
фторидах и оксид-фторидах урана. Имеющиеся экспериментальные данные по энтальпиям
образования газообразных U0F4 и UO2F2 свидетельствуют, что в молекулах оксид-фторидов энергии
связи больше, чем в оксидах и фторидах той же валентности.
Принятой энтальпии атомизации соответствует
A/1°(UOF, г, 0)=—541,077 ±100 кДж.моль.
UO2F(r). Термодинамические функции газообразного диоксид-фторида урана в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 1028.
Молекулярные постоянные UO2F, использованные для расчета термодинамических функций,
приведены в табл. 43.22. Экспериментальные данные о строении и спектрах молекулы UO2F в
литературе отсутствуют. По аналогии с молекулой SO2F [4932] в справочнике принимается, что в
основном электронном состоянии молекула UO2F имеет пирамидальную структуру (симметрия Cg).
Приведенное в табл. 43.22 произведение моментов инерции вычислено по структурным параметрам
r(U-O) = 1,80+0,05 A, r(U—F)=2,0+0,10 A „^0—17—0 = 100+15°, 4O-U-F=110+10°,
принятым такими же, как в молекуле UO2F2 (структурные параметры молекул SO2F, SOF2 и SO2F2
близки по величине). Погрешность значения IaIbIc составляет 2-1014 г3-см6. Приведенные в
таблице основные частоты молекулы UO2F оценены по принятым в справочнике значениям основных
частот молекул UOF2, UO3 и UF3. Погрешности в значениях vl5 v2 и v6 могут достигать 100 см,
в значениях v3, v4 и v6 — 50 см. Система возбужденных электронных состояний UO2F принята
идентичной с молекулой UF5 (см. 3-е примеч. к тексту по UO2(r)), погрешности приведенных
в табл. 43.22 значений Те могут достигать 1500—4000 см.
Термодинамические функции UO2F(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A.-122)—A. 124), A. 128), A. 130)
с учетом шести возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью принятых молекулярных постоянных и приближенным
характером расчета и составляют 6, 12 и 15 Дж-К-моль~1 в значениях Ф°(Г) при 298,15, 3000
и 6000 К соответственно.
Таблица термодинамических функций UO2F(r) публикуется впервые.
Константа равновесия реакции UO2F(r)=U(r)+2O(r)+F(r) вычислена с использованием
принятой в справочнике энтальпии атомизации
Дг#°@) = 2100 + 100 кДж • моль.
Это значение было оценено на основании сопоставления энергий связи в газообразных оксидахе
фторидах и оксид-фторидах урана (см. UOF(r)).
Принятой энтальпии атомизации соответствует
, г, 0) = — 994,294 ± 100 кДж • моль.
UOF2(r). Термодинамические функции газообразного оксид-дифторида урана в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 1029.
Молекулярные постоянные UOF2, использованные для расчета термодинамических функций,
приведены в табл. 43.22. Экспериментальные данные о строении и спектрах молекулы UOF2 в
литературе отсутствуют. В справочнике по аналогии с молекулой SOF2 принимается, что в
основном электронном состоянии молекула UOF2 имеет пирамидальную структуру (симметрия Cs).
Приведенное в табл. 43.22 произведение моментов инерции вычислено по структурным параме-
трам r(U-O) = 1,80 + 0,05 A, r(U-F) = 2,10 + 0,05 A, ^O-U-F = 110 + 10°, ^F-U-F =
= 115 + 5°. Величины углов между связями основаны на соответствующих данных для молекул
SF2, SO^Fa, SOF2 и UF2, UO2F2. Из сопоставления значений r(S—О) и r(S—F) в молекулах SO2F2
226
Уран и его соединения UO2F2 (к)
и SOF2 величина r(U—О) в молекуле UOF2 принята такой же, как в молекуле UO2F2, а значение
r(U—F) несколько большим, чем в молекуле UO2F2. С этой оценкой согласуется предположение
о близости величины r(U—О) в молекулах UO2 и UOF2 и величины r(U—F) в молекулах UFg ж
UOF2. Погрешность значения IaIbIc составляет 3-1014 г3-см6. Приведенные в таблице
основные частоты молекулы UOF2 оценены по основным частотам UF3, UO3, UO2F2, SO2F2 и SOF2.
Погрешности в значениях vx, v2, v5 оцениваются в 100 см, в v3, v4 и v6 — 50 см. Схема
электронных термов UOF2 принята такой же, как для молекулы UF4 (см. 3-е примеч. к тексту по UO2(r)).
Погрешности приведенных в табл. 43.22 значений Т'е оцениваются в 300—5000 см.
Термодинамические функции UOF2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом 13 возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью принятых молекулярных постоянных и
приближенным характером расчета и составляют 10, 15 и 20 Дж-К^-моль в значениях Ф°(Г) при
298,15, 3000 и 6000 К.
Таблица термодинамических функций UOF2(r) публикуется впервые.
Константа равновесия реакции UOF2(r) = U(r) + О(г) + 2F(r) вычислена с использованием
принятой в справочнике энтальпии атомизации
Дг#°@) = 2050 + 100 кДж • моль-1.
Эта величина оценена по принятым энергиям связей в газообразных оксидах, фторидах и
оксид-фторидах урана (см. текст по UOF(r)).
Принятой энтальпии атомизации соответствует
AfH°(UOF2, г, 0) = —1113,802 ±100 кДж ¦ моль.
UO2F2(k). Термодинамические свойства кристаллического диоксид-дифторида урана в
стандартном состоянии при температурах 100—2100 К приведены в табл. 1030.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 43.1. В справочнике за стандартное состояние UO2F2(k) в интервале 0—2100 К принимается
гексагональная модификация [1357, 4805].
При Т <С 298,15 К термодинамические функции вычислены по данным Уокера и Чени [4624],
которые измерили теплоемкость UO2F2 в интервале 13—418 К на образце с содержанием примесей
менее 0,07%. Погрешности измерений оценены авторами [4624] в 1% ниже 40 К и 0,1% — при
температурах выше 40 К. Экстраполяция теплоемкости к Г = 0 по функции Дебая приводит
к значению 5°A3 К) = 0,98 Дж-К^-моль. Принятые значения ?°B98,15 К) и #"B98,15 К)—
—Я°@) (см. табл. 43.1) имеют погрешности 0,8 Дж-К^-моль и 0,06 кДж-моль соответственно.
Уравнение для теплоемкости UO2F2(k) при Т ]> 298,15 К основано на результатах измерений
энтальпии в работе Кордфунке и др. [1931] C75—811 К); при обработке погрешность данных
[1931] оценивалась в 0,5%.
Сведения о температуре плавления и существовании UO2F2 в жидком состоянии в литературе
отсутствуют. В книге Каца и Рабиновича [492], со ссылкой на недоступные авторам
справочника работы, сообщается, что термическое разложение UO2F2 наблюдается уже при 473 К, однако
термическая диссоциация в основном протекает при 1123—1173 К (в вакууме). В работах Ферриса
и др. [2241, 2242] (973—1223 К) и Кнакке и др. [3045] A033—1073 К), изучавших термическую
устойчивость UO2F2, а также в масс-спектрометрическом исследовании Горохова и Смирнова
[4866], было показано, что наряду с диссоциацией имеет место частичное испарение. В настоящем
справочнике расчет термодинамических функций UO2F2(k) выполнен до 2100 К — температуры,
при которой равновесное давление продуктов диссоциации и испарения достигает 100 атм.
Погрешности табулированных значений Ф°(Т) при 298,15, 1000 и 2000 К оцениваются в 0,6,
2 и 12 Дж-К~1-моль соответственно. Расхождения между термодинамическими функциями
UO2F2(k). приведенными в табл. 1030 и в справочниках МАГАТЭ [2344] (Т < 400 К) и Барина
и Кнакке [1395] (Т < 1500 К), не превышают 0,1 и 1 Дж-К^-моль в значениях ФЦТ).
Принятая в справочнике энтальпия образования
2, к, 298,15 К) = —1653,6 + 2,5 кДж • моль
основана на калориметрических измерениях энтальпий растворения y-UO3(k), UF6(k) и UO2F2(k)
б плавиковой кислоте и в воде, представленных в табл. 43.23. В таблице приведены уравнения
227
UO2Fa(r)
Глава 43
Таблица 43.23. Результаты определений энтальпий растворения T-UO3 (к), UFe (к) и UO2F2 (к)
в плавиковой кислоте и в воде (в кДж • моль'1)
Авторы
Исследованные реакции
ДГЯ,° B98,15 К)
Видавский и др., 1965 [212, 908]
Видавский, Ипполитова, 1971 [214]
Кордфунке, Оуэлтис, 1977 [1932]
Джонсон, О'Хара, 1978 [2874]
Попов и др., 1957 [811]
Джонсон, О'Хара, 1978 [2874]
Попов и др., 1957 [811]
Супоницкий и др., 1971 [997]
Кордфунке, Оуэлтис, 1977, 119321
Y-U03(k)+6(HF-267H20)=(UO2F2+H2O) р-р 4 (HF • -76,6
400Н2О); —102,8+0,5, 7 и 3 измерения^
T-UO3(K)+19(HF-20H2O)=(UO2F2+H2O)"p-p 17 (HF • —80,7
22,4Н2О);—107,2+0,3, 5 измерений
T-UO3(k)+48(HF-264H2O)=(UO2F2+H2O) р-р 46 (HF- —79,7
276Н2О);—105,9+0,3, 4 измерения
T-UO3(k)+27,3(HF-115,25H2O)=(UO2F2+H2O) р-р 25,3 —80,1
(HF-124,4H2O);—106,2 ±0,4, 6 измерений
UF6(k)+1602H2O=(UO2F2+4HF) р-р 4 (HF-400Н2О); -258,9
—208,1+1,3, 505 К, 5 измерений
UF6(k)+21,33(HF-147,80H2O)=(UO2F2+4HF) р-р 25,33 —260,9
(HF-124,4H2O);—208,3 ±0,3, 6 измерений
UO2F2(k)+4(HF -400H2O)=UO2F2 р-р 4 (HF-400HaO); -33,0
—33,9 ±0,8, 305 К, 5 измерений
UO2F2(K)+571HaO=UO2F2 р-р E7ШаО); —29,8, 4 изме- —29,8
рения
UO2F2(k)+47,6(HF-264H2O)=UO2Fa р-р 47,6 (HF- -33,9
264Н2О);—33,9±0,2, 5 измерений
реакций, исследованных в каждой из работ, и найденные в них значения энтальпии реакции.
В последней колонке даны величины, относящиеся к реакциям с участием растворов HF и Н20(ж)
в их стандартных состояниях и к UO2F2 в состоянии раствора исследованной в данной работе
концентрации, которые были рассчитаны Паркер [997, 2344, 3045]. Как можно видеть из таблицы,
результаты измерений энтальпии растворения указанных соединений оказались в
удовлетворительном согласии. Комбинирование попарно результатов измерений с наиболее близкими
конечными концентрациями растворов UO2F2 позволяет составить три термохимических цикла и
найти значения энтальпии образования UO2F2(k): —1652,3 +2,6 кДж-моль [212, 811, 908],
—1654,5 + 2,4 кДж-моль" [1932] и —1653,9" + 3,5 кДж-моль [811]. Принятое значение является
ередним из этих величин. Основным источником его погрешности является неточность
использованных в расчетах энтальпий образования y-UO3(k), UF6(k) и F~(aq).
Исследования равновесия реакции 3UO2F2(k) = 2/3U3O8(k) + UFe(r) + 1/3О2(г), выполненные
Кнакке и др. [3045] A033—1132 К, 9 точек) приводят к значениям энтальпии образования
UO2F2(k), равным —1648 + 5 кДж-моль-1 (III закон) и —1694 + 100 кДж-моль (II закон),
которые совпадают в пределах погрешности с принятым.
Давление пара в реакции UO2F2(k) = UO2F2(r) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = AS#°(UO2F2, к, 0) = 300 ± 12 кДж • моль.
Принятая величина основана на совпадающих в пределах погрешностей измерениях,
выполненных Гороховым и Смирновым [4866] (масс-спектрометрический метод, 929—1094 К, 44 точки)
и Кнакке и др. [3045] (эффузионный метод, 1033—1132 К, 9 точек). По данным этих авторов были
найдены значения 302 + 12 и 296 + 15 кДж-моль" (III закон) и 287 + 15 и 275 + 100 кДжХ
Хмоль (II закон) соответственно. В работе [3045] было принято, что основным процессом
является 3UO2F2(k) = 2/3U3O8(k) + UFe(r) + x/3O2(r), а сублимация UO2F2(k) без разложения
происходит в меньшей степени. Однако принятый механизм разложения оказался неверным, как
это было показано в работе [4866].
UO2F2(r). Термодинамические функции газообразного диоксид-дифторида урана в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 1031.
Молекулярные постоянные UO2F2, использованные для расчета термодинамических функций^
приведены в табл. 43.22. Экспериментальные данные о строении и спектрах молекулы UO2F2
в литературе отсутствуют. По аналогии с молекулами CrO2F2 [2367] и SO2F2 в справочнике при-
228
Уран и его соединения UOF3(r)
нимается, что в основном электронном состоянии Х1А1 молекула UO2F2 имеет структуру
искаженного тетраэдра (симметрия C2v), в которой центральный атом урана связан с парами атомов
кислорода и фтора, расположенными во взаимно перпендикулярных плоскостях. Приведенное
в табл. 43.22 произведение моментов инерции вычислено по структурным параметрам r(U—О) ==
= 1,80 ± 0,05 A, r(U—F) = 2,0 + 0,05 A ^O-U—О = 100 + 10°, ^F—U-F = 120 + 10°,
которые были оценены по структурным параметрам родственных молекул. Значения углов
приняты близкими к их величине в молекуле GrO2F2 [1443, 2367] х. На основании данных для
молекул SO2F2 и S0F4, XeO2F2 и XeOF4 значения r(U—О) и r(U—F) в молекуле UO2F2 были приняты
такими же, как в UOF4. Погрешность значения IaIbIo составляет 5-Ю14 г3-смв. Приведенные
в табл. 43.22 основные частоты молекулы UO2F2 оценены на основании сравнения основных
частот молекул SO2F2, SO2, SF2 и CrO2F2 [1693, 2661, 4377], CrF2 [4587], CrO2 с частотами,
принятыми для молекул UO2, UO3, UF2, UF4, U0F4. Погрешности в значениях частот vb va, v6 и v8
оцениваются в 100 см, остальных пяти частот — в 50 см. Возбужденные электронные
состояния молекулы UO2F2 в справочнике не рассматриваются, поскольку нижнее состояние
молекулы CrO2F2 имеет энергию ~20 000 см.
Термодинамические функции UO2F2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных электронных состояний. Погрешности рассчитанных значений
обусловлены неточностью оценки молекулярных постоянных и приближенным характером
расчета, они составляют 6, 15 и 20 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К^
в том числе из-за неточности молекулярных постоянных 5—7 Дж-К~1-моль~1 во всем интервале
температур. '
Таблица термодинамических функций UO2F2(r) публикуется впервые. ,
Константа равновесия реакции UO2F2(r) = U(r) + 2О(г) + 2F(r) вычислена с
использованием значения АгН°@) = 2532,41 + 14 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации UO2F2(k). Этим величинам также соответствует
a г, 0) = —1349,429 + 12 кДж.моль.
UOF3(r). Термодинамические функции газообразного оксид-трифторида урана в стандартном
состоянии при температурах от 100 до 6000 К приведены в табл. 1032.
Молекулярные постоянные UOF3, использованные для расчета термодинамических функций,
приведены в табл. 43.22. Экспериментальные данные о строении и спектрах молекулы UOF3 в
литературе отсутствуют. По аналогии с молекулами СгОС13 [5008] и VOF3 [4916] в справочнике
принимается, что в основном электронном состоянии молекула UOF3 имеет структуру
искаженного тетраэдра (симметрия C3v). Приведенное в табл. 43.22 произведение моментов
инерции вычислено по структурным параметрам r(U—О) = 1,80 + 0>05 A, r(U—F) = 2,0 + 0,10 А,
^F—U—F = 110 + 10°, оцененным по структурным параметрам молекул VOF3 [4916], VFg
[4897], VO2 и молекул UF6, UO2, а также других оксид-фторидов урана. Погрешность значения
IaIbIo составляет 1-Ю13 г3-см6. Приведенные в таблице основные частоты молекулы UOF3
получены на основании сравнения основных частот молекул VOF3 [4948], VF6 [5042] с
основными частотами UF6, UF3, UF4, UO2 и U0F4. Погрешности значений vlt v2 и v4 оцениваются
в 100 см, значений v3, v5 и v6 — в 40—50 см. Схема возбужденных электронных
состояний UOF3 принята такой же, как у молекулы UF5 (см. 3-е примеч. к тексту по UO2(r)).
Термодинамические функции UOF3(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) с учетом шести возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью оценки молекулярных постоянных и
приближенным характером расчета; они составляют 10, 20 и 30 Дж-К-моль~1 в значениях Ф°(Г) при
298,15, 3000 и 6000 К соответственно.
Молекулы CrO2F2 и SO2F2 имеют одинаковые структуры искаженного тетраэдра симметрии С2г1, однако
соотношение величины углов О—X—О и F—X—F (где Х=Сг и S) в этих молекулах противоположное. Аналогичная
ситуация имеет место и для других диоксяд-дигалогенидов переходных металлов и элементов основных подгрупп
[1443]. Молекулы, содержащие атомы переходных металлов, в некоторых случаях могут иметь структуры, вообще
отличающиеся от структуры аналогичных молекул, содержащих атомы основных подгрупп, как например,
W0F4 и SOF4. Поэтому в справочнике при оценке конфигурации молекул UO,F2 и U0F4 отдавалось предпочтение
оценкам по постоянным оксид-фторидов переходных металлов, а не молекул SO2F2 и SOF4.
229
WOF4(r) Глава 43
Таблица термодинамических функций UOF3(r) публикуется впервые.
Константа равновесия реакции UOF3(r) = U(r) + О(г) + 3F(r) вычислена с использованием
«ринятой в справочнике энтальпии атомизации
Д,Я°@) = 2520 + 100 кДж • моль.
Эта величина получена сопоставлением энергий связей в газообразных оксидах, фторидах и
эксид-фторидах урана (см. текст по UOF(r)).
Принятой энергии атомизации соответствует
bfH°(UOFs, г, 0) = —1506,527 + 100 кДж • моль.
U0F4(r). Термодинамические функции газообразного оксид-тетрафторида урана в стандартном
гостоянии при температурах от 100 до 6000 К приведены в табл. 1033.
Молекулярные постоянные UOF4, использованные для расчета термодинамических функций,
дриведены в табл. 43.22. Экспериментальные данные о строении и основных частотах молекулы
UOF4 отсутствуют. По аналогии с молекулами UF5, MoOF4 [2770] и WOF4 [4022] в
справочнике принимается, что в основном электронном состоянии Х1А1 молекула UOF4 имеет структуру
квадратной пирамиды (симметрия С4с) (см. примеч. к тексту по UF?(r)). Приведенное в табл. 43.22
ироизведение моментов инерции вычислено по структурным параметрам r(U—О) = 1,80 + 0,05 А,;
w(XJ—F) = 2,0 + 0,05 А, -40—U—F = 105 + 10°. Валентный угол принят таким же, как
в молекулах MoOF4 [2770] и W0F4 [4022]. Из сравнения соответствующих расстояний в
молекулах MoF6 [4227] и MoOF4 [2770], WFe [4227] и WOF4 [4022], SFe и SOF4 [2522, 3691],
XeF6 и XeOF4 можно ожидать, что значение r(U—F) в молекуле U0F4 будет таким же, как
в молекуле UFe или на 0,01—0,02 А больше. Величина r(U—О) оценена по отношению
значений г(М—О) и r(M—F) в молекулах M0F4, где М = Mo, W, S и Хе, а также принятым
значениям r(U—О) в молекулах UOB. Погрешность значения IaIbIo составляет 1-Ю13 г3-см6.
Приведенные в табл. 43.22 основные частоты молекулы UOF4 получены на основании
сравнения -соответствующих частот молекул WOF4 [1289], WF6 [3451] и WO3 с частотами UFe
ж UO3, а также кристаллических соединений, содержащих ион UOF7 [2905] (отнесение
основных частот этих соединений проведено в предположении симметрии Civ). Погрешности
значений vlt v2, v4 и v7 оцениваются в 100 см, остальных частот — в 50 см. Возбужденные
электронные состояния U0F4 в справочнике не рассматриваются, так как благодаря
заполненной электронной оболочке этой молекулы они должны иметь энергии не меньше 20 000 см.
Термодинамические функции UOF4(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
fl. 130) без учета возбужденных электронных состояний. Погрешности рассчитанных значений
обусловлены неточностью принятых молекулярных постоянных и приближенным характером
расчета; они составляют 12, 20 и 25 Дж-К^-моль'1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций U0F4(r) публикуется впервые.
Константа равновесия реакции UOF4(r) = U(r) + О(г) + 4F(r) вычислена с использованием
значения .ДгЯ°@) = 2870,748 + 26 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
г, 0) = —1780 + 25 кДж . моль.
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции
?O2F2(r) + 2UFB(r) + 0,5О2(г) = 3UOF4(r) A014—1112 К, 14 точек), выполненном Гороховым
и Смирновым [4866]. Приведенная погрешность учитывает воспроизводимость измерений,
неточность термодинамических функций и других величин, использованных при обработке
экспериментальных данных по уравнению III закона. Расчет по уравнению II закона приводит к
значению —1660 кДж-моль.
Г л а в а 44
ПЛУТОНИЙ И ЕГО СОЕДИНЕНИЯ
Ри(к, ж), Pu, Pu+, PuO, PuO+, PuO2(k, ж), PuO2,
PuO+, PuO; и Ри2О3(к, ж)
В настоящей главе рассматриваются плутоний и его кислородные соединения — всего 8 веществ.
Для трех веществ приведены сведения о термодинамических свойствах в конденсированном
состоянии, для семи — в газообразном состоянии. Термодинамические свойства всех газов
рассчитаны для температур вплоть до 10 000 К.
В Приложении 3.10 для элементарного плутония приведены критические постоянные.
Термодинамические свойства плутония и его соединений рассчитаны для изотопа 239Ри
(атомная масса 239,052): хотя плутоний имеет изотопы с большей продолжительностью жизни, этот
изотоп получил наиболее широкое применение. В то же время различие в термодинамических
свойствах разных изотопов плутония и его соединений при Т ^> 500 К меньше неточности
современных сведений об этих свойствах.
В справочнике рассматриваются два наиболее устойчивых оксида плутония в
кристаллическом состоянии. C-Ри2О3 имеет очень узкую область гомогенности (PuO150—PuO151). Диоксид
плутония при Т < 700 К также имеет узкую область гомогенности, однако при более высоких
температурах эта область расширяется и для Т > 900 К составляет РиО2 в1—РиО2 00 (см. [1030],
с. 231).
Известен также малоустойчивый кристаллический оксид PuO, который может быть
стабилизирован примесями углерода. При Т > 573 К существует также соединение РиО1>в1,- а в поле
твердых растворов PuO1>ei—PuO2 — узкая двухфазная область с границами РиО170 и РиО1>71.
Ри(к, ж). Термодинамические свойства кристаллического и жидкого плутония в стандартном
состоянии при температурах 100—5600 К приведены в табл. 1034.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 44.1. В справочнике за стандартное состояние плутония в интервале 0—395 К принята
Таб л
Вещество
ица 44.1. Принятые значения термодинамических величин для
в кристаллическом и жидком состояниях
Состояние
Н° B98,15 К)-
—Н° @)
кДж-моль 1
S0 B98,15 К)
ю
со"
О5
СМ
о а,
Дж-К^-моль
плутония и его соединении
Коэффициенты
в уравнении для
СР°{Т)
а
&.103
с-10-5
Интервал
температур
или
Т
К
или
кДжХ
Хмолъ *
Pu kVI, монокл.(о) 6,909 54,49 31,19 42,342 -71,621 0а 298,15-395 395 3,45
KV, монокл.(З) — — — 25,062 22,277 - 395-485 485 0,56
kIV, ромб.(т) - — — 21,504 28,451 — 485—588 588 0,60
к1И, куб.(8) - — — 37,15 — — 588-731 731 0,085
кП, тетр.(8') — — — 35,6 — — 731—752 752 1,86
к1, куб.(е) — — — 34,4 — — 752—913 913 2,76
ж — — — 42,5 — — 913—5600 — —
РиО2 к, куб. 10,78 66,13 66,25 83,319 11,684 18,270 298,15—2663 2663 67
ж — — — 131 — — 2663-6000 — —
РиаО3 к, куб. 22,57 163,02 116,98 169,466 -79,980 25,459 298,15-350 - -
к, куб. — — — 122,953 28,548 15,012 350—2358 2358 ИЗ
ж — — — 200 — — 2358—5500 — —
Примечание. C°p(T)=a+b-T— c-T^+d-P+e-T3 (Дж-К-моль).
Pu: a d=0; е=384,934-10-».
231
Ри(к, ж) Глава 44
моноклинная (простая) а-модификация, в интервале 395—485 К — ^-моноклинная (объемно-
центрированная), в интервале 485—588 К — ромбическая гранецентрированная у-модификацият
в интервале 588—731 К — кубическая 8-модификация (структурный тип Си), в интервале 731—
752 К — тетрагональная 8'-модификация (структурный тип у-Mn) и в интервале 752—913 К —
кубическая е-модификация (структурный тип a-Fe).
Естественная радиоактивность Ри приводит к разупорядочиванию кристаллической решетки
твердого тела, в результате чего измерения теплоемкости могут приводить к значениям,
завышенным по сравнению с истинными х, и к появлению ложных максимумов на кривой С (Т) [3220т
4133].
Измерения теплоемкости при Т < 298,15 К проводились для изотопов 239Ри [2443, 3220,
4130, 4134, 4488] и 242Ри [2443, 4132]. Несмотря на разность в периодах полураспада 2з9Ри
B,4-104 лет) и 242Pu D-105 лет), при корректных измерениях мольные теплоемкости двух
изотопов должны быть одинаковыми. Однако в ранних работах Санденау [4130, 4134] A1—300 К),
Ли и др. [3220] A3—300 К), Тейлора и др. [4488] и Санденау и Гибней [4132] A5—373 К) для
этих изотопов были получены значения, различающиеся ,на 10—15%.
Анализ перечисленных работ приводит к выводу, что наиболее надежными являются данные
Гордона и др. [2443], полученные для 242Ри (содержание примесей 0,004%, интервал измерений
4—300 К) и 239Ри (содержание примесей 0,005%, интервал 7,2—70 К); воспроизводимость
соответствующих измерений по оценке авторов [2443] составляла ~0,4%. Результаты для двух
образцов совпали в пределах воспроизводимости измерений (за исключением интервала 40—60 К,
где наблюдались расхождения ~1%, что, вероятно, обусловлено аномалией в ходе Ср(Т), на
которую указывалось в работах [3220, 4133]). Полученные в работе Гордона и др. [2443] данные
по теплоемкости образца 242Ри представлены в виде таблицы сглаженных значений в интервале
6—298 К; погрешность этих данных оценена авторами [2443] в 1,2% при Т < 40 К и в 0,8% при
более высоких температурах.
Термодинамические функции Ри(к) при Т < 298,15 К рассчитаны по значениям теплоемкости,
полученным авторами [2443] для изотопа 242Ри, которые были приняты в справочнике для
изотопа 239Ри. Экстраполяция теплоемкости ниже 6 К приводит к значению S°F К) = 0,106 Дж-К-1Х
Хмель. Погрешности значений ?°B98,15 К) и #°B98,15 К)—#°@), приведенных в табл. 44.1,.
оцениваются в 0,8 Дж-К^-моль и 0,08 кДж-моль соответственно2.
Уравнение для теплоемкости a-Pu в интервале 298,15—395 К (см. табл. 44.1) основано на
данных Санденау и Гибней [4132] B98,15—373 К), скорректированных с учетом измерений
Гордона и др. [2443] 3. Измерения теплоемкости Ри в работе Кей и Лоусби [2942] B60—873 К) при
выводе уравнения для теплоемкости a-Pu не учитывались, так как они не согласуются с
измерениями теплоемкости a-Pu при Т <^ 298,15 К и приводят к завышенным значениям C5,5—
37 Дж-К~1-моль при 300—340 К). Однако для [З-Ри и f-Pu измерения Кей и Лоусби [2942]
являются единственными, ввиду чего уравнения в интервалах 395—485 и 486—588 К,
приведенные в табл. 44.1, основаны на этих данных. Для теплоемкости 5-Ри, 8'-Ри и е-Ри в интервалах
588—731, 731—752 и 752—913 К приняты постоянные значения C7,15 + 0,5; 35,6 + 0,5 и 34,4 +
+ 0,5 Дж-К-моль~1 соответственно); они получены усреднением результатов измерений
теплоемкости в работах Кей и Лоусби [2942] и Энгеля [2174] G03—783 К).
Данные по температурам и энтальпиям полиморфных превращений и плавления плутония
многочисленны и противоречивы (см. табл. 44.2 и 44.3), поскольку на эти константы влияют
кинетические факторы, связанные с чистотой исследованных образцов, изотопным составом
образцов и т. п. Приводимые авторами погрешности измерений характеризуют только неточность
определения этих констант для конкретных исследованных образцов 4. В справочнике выбор при-
Влияние радиоактивного излучения на термодинамические свойства радиоактивных веществ отметили в 1961 г.
Копытин и Гагаринский [540].
2 В работе Тейлора и др. [5059а] была измерена теплоемкость S-239Pu, стабилизированного примесями Ga (от
0,75% до 1,8%) в интервале 20—300 К. Обработка этих экспериментальных данных с экстраполяцией
содержания примесей Ga к нулю и пересчетом их на a-Pu приводит к значению S" B98,15 К)=51,3 + 3 Дж-К^-моль,
которое в пределах погрешности (+6%) согласуется с значением, приведенным в табл. 44.1, но значительно
ниже рассчитанного по данным [4132] E6,2 Дж-К^-моль).
3 Экспериментальные значения теплоемкости, найденные в [4132], были уменьшены на 3,7% (разница в данных
[2443] и [4132] при 298.15 К).
4 Это относится и к данным [5037], полученным для очень чистых, но не достаточно полно охарактеризованных
в отношении их термической предыстории образцов плутония (в работе [5037] отсутствует описание
эксперимента).
232
Плутоний и его соединения
Ри(к, ж>
Таблица 44.2. Результаты определений температуры фазовых превращений плутония (в К)
Авторы
Джитт, 1955 [2858] 395+2
Дин и др., 1958 [2010] 394
Санденау, Гибней, 1958 [5038а] 396,7 ±0,3
Конобеевский и др., 1958 [3070] 395
Паскар, 1959 [3778] 396+6
Ли, Мардон, 1961 [3219] 399
Крамер и др., 1961 [1953] 395+2
Абрамсон, 1961 [1205] 383+4
Кей, 1961 [2941] 394
Энгель и др., 1962 [2177] 400
Хилл, 1962 [2651] 385 ±1
Кей, Лоусби, 1964 [2942] 392
Андерсон и др., 1965 [1321] 390+0,5
Сприт, 1966 [4369] 385
Энгель, 1967 [2174] —
Прюньер и др., 1970 [3893,3894,3895] 392+2
Гольдберг, Массальски, 1970 [2432] 388
Роллон, Галлегос, 1981 [5037] 399,6+0,6
Принятые значения 395+5
478+3
481
475+2
484
488+3
482+3
479+3
476 ±6
484
480
457+2
477
—
457
—
477+6
458
489,7+0,9
485+5
Ttr
Т^5
593+5
589
591 ±2
588
588+2
589+2
592+6
586 ±7
588
593
583
584
—
588
—
588+2
583
592,6+0,2
588+3
Вн>8'
725+4
728
729 ±2
713
—
729
724+4
733+5
732
733
.—
731
—
729
741+2
730 ±2
725
736 ±2
731 ±5
В'->е
749 ±5
752
757+3
733
752 ±7
757+2
749+4
752 ±4
753
754
—
753
—
748
754
752+4
753
755,7+1,0
752+5
Т
2 m
г ->-ж
913+2
908
914 ±2
914+2
913
—
911
913
—
911
—
913
—
—
—
912,1+0,8
913+2
Таблица 44.3. Результаты определений энтальпии фазовых превращений плутония (в кДж-моль г)
Авторы
Конобеевский и др., 1958 [3070]
Санденау, Гибней, 1958 [5038а]
Паскар, 1959 [3778]
Кей, 1961 [2941]
Энгель и др., 1962 [2177]
Кей, Лоусби, 1964 [2942]
Энгель, 1967 [2174]
Прюяьер и др., 1970 [3893, 3894, 3895]
Роллон, Галлегос, 1981 [5037]
Принятые значения
а—> р
3,2±0,5
3,92+0,04
5,0
4,00 ±0,04
3,39 ±0,02
3,36 ±0,04
—
3,53 ±0,10
0,70+0,10
—
0,59
0,59 ±0,06
0,52 ±0,06
0,64+0,06
—
0,52 ±0,03
0,80+0,10
—
0,54
0,65 ±0,06
0,57 ±0,03
0,52±0,02
—
0,65+0,02
3,503 ±0,140 0,486 ±0,038 0,655 ±0,03С
3,45 ±0,08
0,56 ±0,08
0,60 ±0,06
8->8'
~0
—
<0,17
0,07 ±0,04
0,10 ±0,03
0,08 ±0,04
0,03 ±0,01
—.
) 0,103 ±0,041
0,085 ±0,04С
В' -> е
1,6±0,3
—
1,6
1,97+0,04
1,85 ±0,03
1,86 ±0,04
1,92 ±0,03
—
1,617 ±0,187
1,86 ±0,20
е ->Ж
3,30
—
2,2
3,9
2,74 ±0,03
2,90+0,04
—
—
2,644 ±0,175
2,76 ±0,10
нятых значений Ttr и Д<ГЯ, так же как и в справочных изданиях [2752, 3695], проводился
усреднением данных большинства определений, исключая выпадающие значения.
Теплоемкость жидкого плутония D2,5 + 1,0 Дж-К~1-моль~1) принята по согласующимся
между собой экспериментальным данным Тейера и Роббинса [4501] (измерения энтальпии в
интервале температур 913—2200 К), Энгеля [2175] и Лоусби [3290] (в двух последних работах
определялась теплоемкость Ри(ж)).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 3000 и 5500 К оцениваются в 0,5,.
2, 6 и 12 Дж-К^-моль соответственно.
Термодинамические функции Ри(к, ж), приведенные в табл. 1034, согласуются с данными
справочника МАГАТЭ [3695] (Г < 4000 К) в пределах 0,9—2 Дж-К^-моль в значениях Ф°(Т)
и с данными Халтгрена и др. [2752] (Т ^ 4000 К) в пределах 1,6—3,4 Дж-К-моль; расхожде -
233
Pu(r)
Глава 44
ния обусловлены в первую очередь различием принятых значений энтропии 5°B98,15 К),
поскольку авторы в [2752, 3695] не учитывали данные Гордона и др. [2443].
В настоящем справочнике в качестве стандартного состояния плутония принято
кристаллическое состояние и в соответствии с этим
&fH°(Pu, к) = 0.
Давление пара плутония в реакции Ри(к, ж) = Ри(г) вычислено с использованием значения
ДГЯ°(О) = AsJ?°(Pu, к, 0) = 348,710 + 8,0 кДж-моль, соответствующего принятой в
справочнике энтальпии сублимации
\Н°(Ри, к, 298,15 К) = 348+ 8 кДж • моль.
Принятая величина является средневзвешенной из приведенных в табл. 44.4. Наибольший
вес придавался работам [1231, 1641]. данные работы [2008] не учитывались. Приведенные в
таблице погрешности учитывают только воспроизводимость измерений. При оценке погрешности
Таблица 44.4. Результаты определений А3Н° (Pu, к, 298,15 К) (в кДж • моль)
Авторы
Метод
Д„Я° (Pu, к, 298,15 К)
II закон
III закон
Фипс и др., 1956 [3829] Эффузионный, 1392—1793 К а 356 347,0±1,5
Даутон, Уилкинсон, 1956 [2008] То же, 1723 К, 1 точка — 369,1
Мюлфорд, 1965 [3579] То же, 1133—1792 К, 45 точек 352±4 347,3 ±0,4
Кент, 1969 [2976] Масс-спектрометрический, 1426—1658 К, см. кор- 355 357,2
ректировку в [3695]а
Аккерман, Раух, 1975 [1231] Эффузионный, 1209—1619 К, 17 точек 348+5 350,6+0,3
Бредбери, Озе, 1979 [1641] То же, 1724—2219 К, 16 точек 348+8 345,5+0,5
а Данные представлены в виде уравнения.
принятой величины были учтены неточность термодинамических функций, приводящая к
погрешности в энтальпии сублимации около 6 и 12 кДж-моль для измерений, выполненных при 1500
и 2000 К соответственно, и неточность использованных в расчетах сечений ионизации [2976].
Ри(г). Термодинамические свойства газообразного плутония в стандартном состоянии при
температурах 100—10 000 К приведены в табл. 1035.
Для расчета термодинамических функций использованы уровни энергии * атома Pu с
суммарным статистическим весом свыше 9100 в интервале 0—43 823 см, т. е. ниже первого потенциала
ионизации. Эти уровни приняты главным образом по рекомендации Фреда [4962], дополненной
(в незначительной степени) данными Стриганова [4904] и Ричардса и Риджли [5036]. Из них
только немногим более одной трети (суммарный статистический вес около 3600) отнесено к 11
валентным электронным конфигурациям ...5/67s2 (к ней относится основной уровень 7F0), ...bfQxlll
(I = 0, 1), ...bflslp, ...5fQd\ ...5f7s*7p, ...6f6d7s7l A=0, 1), ...5/56d27Z (I = 0, 1), ...5/W7*2
и 2 ридберговским ...5/67s8s и ...5f6d7s8s. Для остальных уровней известны только значения /.
Отнесенные к перечисленным конфигурациям уровни энергии составляют небольшую часть
уровней, которые должны соответствовать этим конфигурациям. Так, полный Статистический вес всех
уровней, соответствующих семи низколежащим валентным электронным конфигурациям (энергия
нижнего уровня меньше 21 000 см), составляет 551 551, в то время как суммарный
статистический вес наблюдавшихся уровней, отнесенных к этим же конфигурациям, равен 3400. Несмотря
на отдельные попытки [2535, 3917], проблема оценки ненаблюдавшихся уровней энергии Pu
еще не решена. Можно лишь полагать, что уровни энергии атома Pu, наблюдавшиеся в интервале
О—15 000 см, достаточно полно представляют все валентные состояния, которые должны лежать
в этой области, а подавляющая часть ненаблюдавшихся уровней перечисленных конфигураций
имеют энергии выше первого потенциала ионизации.
1 См. примеч. 1 к тексту по Сг(г).
234
Плутоний и его соединения Ри+(г)
Термодинамические функции Ри(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A.23)—A.25) непосредственным суммированием по уровням энергии атома Ри. Погрешности
вычисленных значений обусловлены главным образом неполнотой данных об энергетических
уровнях атома Ри. Для Ф°(Т) погрешности достигают величин 0,05, 1, 6 и 10 Дж-К-моль
при 298,15, 3000, 6000 и 10 000 К соответственно, причем вычисленные в настоящем справочнике
значения термодинамических функций могут быть только заниженными.
Ранее таблицы термодинамических функций Ри(г) были вычислены в справочнике МАГАТЭ
[3695] (Т < 6000 К), работах Бентона [1475] (до 5000 К), Фебера и Херрика [2227] (Т < 6000 К),
Гурвича и Юнгмана [2535 ] (расчет выполнен для 244Ри и Т < 20 000 К) и Рэнда [3917 ] (до 10 000 К).
За исключением работы [3917], во всех расчетах были использованы только экспериментально
определенные уровни энергии атома Ри, и расхождения результатов расчетов обусловлены
разным числом уровней, известных к моменту проведения расчета. Так, в работе 1966 г. [2535]
величина Ф°A0 000 К) отличается от данных табл. 1035 на 9,5 Дж-К~1-моль~1, а в расчете 1976 г.
[3695] — на 0,25 Дж-К^-моль. В работе Рэнда сделана попытка учесть вклад ненаблюдав-
шихся уровней энергии, соответствующих низколежащим электронным конфигурациям, на
основании использования модели распределения уровней энергии по Гауссу. Рассчитанные таким
образом значения термодинамических функций значительно превышают приведенные в табл. 1035,
соответствующие расхождения в значениях Ф°(Т) возрастают от 2 Дж-К~1-моль~1 при 3000 К
до 18,4 Дж-К^-моль при 10 000 К. Однако такой подход, по-видимому, приводит к сильно
завышенным значениям термодинамических функций (см. U(r)).
Энтальпия образования
Д^^Ри, г, 0) = 348,710 + 8,0 кДж • моль
соответствует принятой в справочнике энтальпии сублимации плутония.
Ри+(г). Термодинамические свойства газообразного положительного иона плутония в
стандартном состоянии при температурах 298,15—10 000 К приведены в табл. 1036.
Использованные в расчете термодинамических функций уровни энергии х иона Ри+ приняты
по рекомендации Фреда [4962], который составил сводку 843 уровней энергии в интервале 0—
47 644 см; суммарный статистический вес этих уровней равен 6826. Только 146 уровней с
суммарным весом 168 отнесены к девяти валентным электронным конфигурациям ...5/e7s (к ней
относится основной уровень 8^/2), ...5f6d, ...5f7p, ...bflsll (I = 0, 1), ...bf&dll (I = 0, 1), ...5/66d2
и ...5/46da7s, а для остальных уровней известны только значения /. Число всех уровней энергии,
соответствующих этим конфигурациям, чрезвычайно велико (их полный статистический вес
составляет 420 420); кроме того, согласно оценке Брюэра [4936], имеется еще несколько валентных
конфигураций иона Ри+, для которых соответствующие уровни энергии пока не идентифицированы.
Однако значительная часть этих уровней имеют высокие энергии возбуждения. Для шести низко-
лежащих электронных конфигураций, у которых энергия нижнего уровня не превышает 22 000 см,
полный статистический вес соответствующих состояний равен 171 600. Большая часть этих
уровней также должна иметь высокие энергии. Так как в настоящее время отсутствуют надежные
данные о системе уровней энергии иона Ри+, в справочнике для расчета термодинамических
функций принимаются все известные экспериментально уровни энергии независимо от того, отнесены
они к каким-либо электронным конфигурациям или нет. (Можно лишь предполагать, что по
крайней мере до 15 000 см известные уровни энергии представляют валентные состояния Ри+ лучше,
чем в области более высоких энергий.)
Термодинамические функции Ри+(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности
вычисленных значений определяются значительной неполнотой данных по уровням энергии и могут
достигать в значениях Ф°(Г>при Т = 298,15, 3000, бООи и 10 000 К величин 0,05, 1, 5 и 10 Дж-К"хХ
Хмоль, причем вычисленные в справочнике величины могут быть только заниженными.
Ранее таблица термодинамических функций Ри+(г) была рассчитана Гурвичем и Юнгманом
12535] (до 20 000 К). Расчет [2535] был выполнен для 2МРи+, и в нем было учтено всего
100 уровней в интервале 0—38 530 см. Расхождения настоящего расчета с данными [2535J при
комнатной температуре обусловлены разницей в значении атомного веса (около 0,1 Дж-К-моль),
а при более высоких температурах — различным числом уровней энергии, принятых в расчетах
(до 5,4 и 10,8 Дж-К^-моль в значениях Ф°(Г) и S°(T) соответственно при 10 000 К).
1 См. примеч. 1 к тексту по Сг(г).
235
PuO(r)
Глава 44
Константа равновесия реакции Ри+(г) + е(г) = Ри(г) вычислена с использованием значения
А.ГН°(О) = —585,0 + 2,5 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации атома плутония
/0(Ри) = 585,0 + 2,5 кДж-моль-1«48900±200 см.
Эта величина принята по оценке Шугара [4436, 5057], выполненной экстраполяцией
сериальных закономерностей. С ней хорошо согласуется значение 582 + 10 кДж-моль, найденное
методом поверхностной ионизации в работе Четверикова и др. [4910]. Более низкие значения
были получены ранее Бертло [4927] E31 кДж-моль) из измерений температуры в дуге,
содержащей Ри, Бошем и др. [4923] E60 кДж-моль) по аналогии с ТЪ и Смитом и Хертелом [5048]
методом поверхностной ионизации E51 кДж-моль, исправлено на принятое в настоящем
справочнике значение потенциала ионизации урана).
Принятому значению /0(Ри) соответствует
Д//°(Ри+, г, 0) = 933,71 + 8,4 кДж • моль.
РиО(г). Термодинамические свойства газообразного оксида плутония в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 1037.
В табл. 44.5 приведены молекулярные постоянные РиО, принятые в справочнике на
основании результатов исследования ИК-спектра молекул РиО, изолированных в матрице из аргона
и криптона [2469], а также приближенных оценок. Электронный спектр РиО не исследовался,
Таблица 44.5. Молекулярные постоянные
Молекула
239ри16О
239pu16Q+
PuO: a Te, см
Pi
б вычислено
A. 68); Д
PuO+: a Те, см-1
Pi
б оценка; г
шению A.
Состояние
X (Рх=2) «
X (&ф0) а
1000 5000
10 40
830
900 б
10 000
80
по соотношению A. 38); Е
оценка.
1000 5000
6 20
10 000
50
РиО
и РиО+
«А
3
3,5 в
20 000 30 000
1000 1000
Ве
см
0,34 б
0,35 г
40 000
1000
вычислено по соотношению A
20 000 30 000
80 80
40 000
80
ai-103
1,6 в
1,8Д
Z>e-10'
ге
А
2,3 г 1,82 Д
2,1 е 1,80 б
. 69); г вычислено по соотношению
вычислено по соотношению A. 67) и значению D0(PuO+)=57 000 см; г вычислено по соотно-
38); Д вычислено по соотношению A. 69); е вычислено по соотношению A. 68).
а квантовомеханические расчеты этой молекулы не проводились. Оценка статистических весов
и энергии электронных состояний РиО была выполнена так же, как для молекулы UO. При этом
предполагалось, что у РиО заполняются электронные орбиталп, соответствующие следующим
конфигурациям электронной оболочки Pu+2: .../6, ...fbd и ...f^s1. Состояния, соответствующие
электронным конфигурациям РиО, которые содержат больше двух разрыхляющих электронов,,
рассматривались как несвязанные. Принималось, что связанные состояния распределяются
равномерно в области энергий до 104 000 см (последняя величина равна D0(PuO) + /0(Pu)), причем
состояния, возникающие из конфигураций ...f5d и .../Bs, имеют энергию выше 10 000 см.
В принятой схеме основное состояние РиО может принадлежать конфигурациям ...ср^ или
Йчй. Нижние состояния обеих конфигураций должны иметь Q = 0. Поскольку обе
конфигурации могут иметь практически совпадающие энергии, в справочнике принимается, что для
основного состояния р = 2 (или S ^= 0).
Колебательные постоянные РиО в основном состоянии, приведенные в табл. 44.5, основаны
на результатах исследования ИК-спектра молекул 239ри1вО и 239Ри18О, изолированных в
матрицах из Аг и Кг, в работе Грина и Риди [2469]. Авторы [2469] нашли по 4 частоты в области 816—
1 Состояния, соответствующие
другим конфигурациям иона Ри+2, должны иметь энергии выше 40 000 см»
236
Плутоний и его соединения
РиО(г)
825 и 772—781 см для двух изотопомеров в матрице из Аг х и по одной частоте (817,27 и 773,62 си)
в матрице из Кг. На основании данных для двух изотопомеров Грин и Риди рассчитали значения
колебательных постоянных РиО и нашли ше = 820,68 + 1,0 и чз„х, = 1,70 + 0,5 см (для
матрицы из Кг) и 828,2 + 1 и 3,0 f 0,5 см (для матрицы из Аг). Поскольку для оксидов ланта-
нидов (и ThO) измерения основных частот в матрицах благородных газов привели к значениям,
которые на 10—15 см меньше полученных при исследованиях их электронных спектров в
газовой фазе, в табл. 44.5 для со, принято округленное верхнее значение из полученных авторами
[2469], его погрешность оценивается в 10 см. Приведенное в таблице значение гДРиО) принято
таким же, как для молекулы UO на основании имеющихся данных об изменении величины
re(Ln0) в ряду лантанидов (см. [4878] 2). Погрешность этого значения составляет 0,05 А.
Термодинамические функции PuO(r) рассчитаны по уравнениям A. 3)—A. 6),, A. 9), A. 10),
{1. 93)—A. 95). Значения (?вн и ее производных вычислялись по уравнениям A. 90)—A. 92)
с учетом шести объединенных электронных состояний в предположении, что Q^Mt вр = 0,5р$&1 в .
Статистическая сумма по колебательно-вращательным уровням энергии основного состояния и
ее производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием по
колебательным уровням и интегрированием по значениям / с помощью уравнений типа A. 82).
В расчете учитывались все уровни с значениями / ^ /щах, г-> последние находились из условий
{1. 81). Энергия уровней вычислялась по уравнениям A. 65) и A. 64а), если принять их
статистический вес равным 2. В табл. 44.6 приведены соответствующие значения vmiX и /Ilm, а также
коэффициентов в этих уравнениях» которые были получены с учетом условий A. 50) и A. 59).
Таблица 44.6. Значения коэффициентов, описывающих уровни энергии (в см), а также значения vmiX
и Jlim, принятые для расчета термодинамических функций РиО и РиО+
Коэффициенты
5VW
Уи-10
Yu-103
а Энергии возбужденных
РиО
X (Q ф 0)
8,30
—3
3,4
-1,6
СОСТОЯНИЙ СМ. В
РдО+
9,00
-3,5
3,5
-1,8
табл. 44.5.
Коэффициенты
5V107
Уоз-Ю13
PuO
X (Q ф 0) »
-2,3
-1,2
137
544
PuO+
-2,1
-1,7
128
541
Погрешности рассчитанных термодинамических функций РиО(г) при температурах до 500 К
обусловлены главным образом отсутствием данных о типе основного электронного состояния РиО.
При более высоких температурах становятся существенными ошибки из-за грубой оценки числа
и энергии возбужденных состояний этой молекулы. Погрешности в значениях Ф°(Т) при 298,15,
3000, 6000 и 10 000 К оцениваются в 3, 6, 10 и 15 Дж-К^-моль.
Термодинамические функции РиО(г) ранее вычислялись в работах Грина и др. [2463, 4968а],
которые отличаются разным учетом возбужденных состояний этой молекулы. Значения Ф°(Т)
и S°(T), рассчитанные в [4968а], больше приведенных в табл. 1037, соответствующие
расхождения уменьшаются с ростом температуры и составляют от 15 до 9 Дж-К~1-моль~1. Результаты
расчетов [2463] меньше табулированных в настоящем справочнике, расхождения растут с
увеличением температуры от 3 до 7 Дж-К^-моль в значениях Ф°(Т) и от 0,6 до 16 Дж-К^-моль
в значениях S°(T).
Константа равновесия реакции РиО(г) = Ри(г) + О(г) вычислена с использованием
значения ДД°@) = Do(PuO) = 655,493 + 26 кДж-моль
принятой в справочнике энтальпии образования
54 800 + 2000 см х, соответствующего
&fH°{PuO, г, 0) == —60 + 25 кДж • моль
-1
1 Появление четырех частот Грин и Риди объяснили разными местами захвата молекул РиО в твердом Аг: нагрев
матрицы показал, что интенсивность частот 822,3 и 778,19 см растет, а двух других — падает.
2 Эта оценка подтверждается совпадением (в пределах 0,02 А) величины г (М—F) в молекулах UF6 и PuF6.
237
PuO+(r) Глава 44
Принятая величина основана на результатах исследования испарения оксидов плутония в
работе Аккермана и др. [1216], которые согласуются с более ранним исследованием Фиппса и др.
[3830]. В обеих работах измерения были выполнены эффузионным методом. В работе [1216] было
показано, что при испарении из танталовой ячейки, т. е. в восстановительных условиях,
достигается стационарное состояние, соответствующее реакции ^-щ Ри2О3(к) = РиО(г) -)-^— РиО1в1(к).
Из этих данных, используя принятые значения термодинамических свойств Ри2О3(к) и
РиО1в1(к), авторы [1216] нашли Д/?°(РиО, г) = —121 300—50,6- Т Дж-моль A600—2150 К).
Расчет энтальпии образования РиО(г) по этим данным затруднен тем, что в настоящем
справочнике не рассматриваются фазы переменного состава, в том числе и РиО161, а также тем, что
принятые в [1216] термодинамические свойства Ри2О3(к) заметно отличаются от принятых в
настоящем справочнике. С учетом этого различия и в предположении, что термодинамические свойства
РиО1в1 (к) могут быть скорректированы на ту же величину, что и Ри2О3(к), при подготовке
настоящего справочника было получено уравнение Д^(?°(РиО, г) = —113 200—46,6- Т Дж-моль.
Этому уравнению соответствуют значения Д/Я°(РиО, г, 0) = —60 + 25 кДж-моль (III закон)
и —88 кДж-моль (II закон).
РиО+(г). Термодинамические свойства газообразного положительного иона оксида плутония
в стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 103S.
Спектры РиО+ не исследовались, а теоретические расчеты этой молекулы не проводились.
Приведенные в табл. 44.5 молекулярные постоянные РиО+ были оценены при подготовке
справочника так же, как для молекул РиО и UO+. Поскольку РиО+ имеет нечетное число электронов,
во всех электронных состояниях этой молекулы, в том числе в основном, Q ^> 0. При оценке
суммарных статистических весов объединенных электронных состояний учитывались состояния,
соответствующие конфигурациям электронной оболочки РиО, которые могут возникать из
конфигурации /6 иона Ри+3. При этом состояния, имеющие конфигурации с тремя (или более)
разрыхляющими электронами, рассматривались как несвязанные. Предполагалось, что все
связанные состояния этих конфигураций лежат в области до 150 000 см (величина, равная D0(PuO+)+
+ /o(Pu+)) \
Приведенные в табл. 44.5 значения постоянных ше и ге молекулы РиО+ в основном состоянии
были оценены на основании предположений, использованных для оценки постоянных UO+ и
ThO+ (см. выше). Их погрешности оцениваются в 70 см и 0,07 А соответственно.
Термодинамические функции РиО+(г) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QBn и ее производных вычислялись по уравнениям A. 90)—
A. 92) с учетом шести объединенных электронных уровней и в предположении, что 0$х вр =
= 0,5ptQW вр. Статистическая сумма по колебательно-вращательным уровням энергии
основного состояния и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью
уравнений типа A. 82). В расчете учитывались все уровни с значениями / ^ /шах v, последние
определялись из условий A. 81). Энергия уровней основного состояния рассчитывалась по
уравнениям A. 65), A. 34) и A. 64а), их вырождение учитывалось статистическим весом 2. В табл. 44.6
приведены соответствующие значения 1>шах и /1Ш, коэффициентов в этих уравнениях,
тождественных принятым молекулярным постоянным РиО+, а также коэффициента У03., полученного из
условия A. 59).
Погрешности рассчитанных термодинамических функций РиО +(г) при температурах до 500 К
обусловлены главным образом отсутствием данных о постоянных в основном состоянии, при
более высоких температурах — грубой оценкой числа и энергии возбужденных состояний этой
молекулы. Погрешности в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К оцениваются в 2,
7, 12 и 15 Дж-К^-моль.
Таблица термодинамических функций РиО+(г) публикуется впервые.
Константа равновесия реакции PuO+(r) + е(г) = Ри(г) + О(г) вычислена с использованием
значения АгН°@) = 95,493 + 36 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0(РиО) = 560 + 25 кДж-моль.
1 Состояния, соответствующие другим конфигурациям иона Ри+3, должны иметь энергии выше 40 000 см.
238
Плутоний и его соединения РиО2(к, ж}
Принятая величина была измерена методом электронного удара в работе Блекб&рна и др.
(см. [2463, 3943]). Ей также соответствуют
A,ff °(PuO+, г, 0) = 500 + 35 кДж • моль,
D0(PuO+) = 680,493 + 35 кДж • моль «* 56 900 ± 3000 см.
РиО2(к, ж). Термодинамические свойства кристаллического и жидкого диоксида плутония
в стандартном состоянии при температурах 100—6000 К приведены в табл. 1039.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 44.1. В справочнике за стандартное состояние РиО2(к) в интервале 0—2663 К
принимается кубическая модификация (структурный тип флюорита [3395, 4186]).
При Т < 298,15 К термодинамические функции РиО2 вычислены по данным Флотова и др.
[2274], которые исследовали теплоемкость 244РиО2 от 4 до 25 К и теплоемкость 242РиО2 в
интервале 12—350 К. По данным рентгенофазового анализа состав образцов отвечал стехиометриче-
скому. Точность измерений теплоемкости оценена авторами [2274] для 244РиО2 в 18% при 10 К
и 12% при 25 К, а для 242РиО2 в 1% при 12—24 К и 0,4% при Т > 24 К.
«Температурная зависимость теплоемкости РиО2 не имеет каких-либо аномалий. Принятые
значения ?°B98,15 К) и Н° B98,15 К)—#°@) приведены в табл. 44.1 и имеют погрешности
0,3 Дж-К^-моль и 0,04 кДж-моль соответственно1.
При Т > 298,15 К для теплоемкости принято уравнение, полученное совместной обработкой
результатов измерений теплоемкости и энтальпии РиО2(к) в работах Флотова и др. [2274]
B98,15—350 К), Крюгера и Севиджа [3120] A92—1404 К) и Энгеля [2176] C00—1100 К). При
обработке погрешность данных [2176, 2274 и 3120] оценивалось в 3, 0,3 и 1% соответственно.
Температура плавления B663 + 20 К) принята по результатам наиболее тщательных
определений, выполненных Лайоном и Бейли [3331, 3333]. Менее точные значения были получены
в работах [1829, 1830, 1832, 1833, 3842, 4102]. Энтальпия плавления F7 + 10 кДж-моль)
оценена с учетом значения энтропии плавления UO2 (&Sm = 25 Дж-К^-моль), изоструктурного .
с РиО2. Близкое значение G0,3 кДж-моль) было оценено Эпштейном [2183].
В связи с отсутствием экспериментальных данных для теплоемкости РиО2(ж) принято
значение A31 + 15 Дж'К^-моль), равное теплоемкости иОа(ж).
Погрешности вычисленных значений Ф°(Т) при 298,15, 2000, 3000 и 6000 К оцениваются в 0,2,
3, 7 и 20 Дж-К^-моль соответственно.
Таблица термодинамических функций РиО2(к, ж) публикуется впервые.
В справочнике для энтальпии образования принято значение
AfH°(PuO2, к, 298,15 К) = —1055,8 ± 0,8 кДж.моль.
Энтальпия сгорания а-плутония в кислороде измерялась Поповым и Ивановым [808], Холли
и др. [2684] и Джонсоном и др. [2870]. В этих работах были найдены значения —1056,0 +
+ 4,6 кДж-моль (две серии, 6 и 7 опытов), —1058,0 +1,6 кДж-моль (детали эксперимента
не опубликованы) и —1055,79 + 0,71 кДж-моль (два образца плутония, 11 опытов)
соответственно. Найденные величины совпадают в пределах их погрешностей. Наиболее тщательные
измерения были выполнены Джонсоном и др. [2870] и принятое значение основывается на этой
работе.
Давление пара диоксида плутония в реакции РиО2(к, ж) = РиО2(г) вычислено с
использованием принятой в справочнике энтальпии сублимации
ДгЯ°@) = Д,#°(РиО2, к, 0) = 640 + 20 кДж • моль.
Принятая величина основана на результатах эффузионных исследований испарения оксидов
плутония, выполненных Аккерманом и др. [1216]. При испарении из вольфрамовых A646—2066 К)
и рениевых A755—2048 К) ячеек, т. е. в нейтральных условиях, стационарное состояние дости-
Санденау [4131] провел семь серпй измерений теплоемкости в интервале 13—325 К. Ему не удалось добиться
воспроизводимости результатов: в первых сериях на кривой теплоемкости наблюдались два пика при 20—40 К
и ~~70 К; в последующих сериях первый пик, уменьшаясь по величине, сместился в область более низких
температур, а второй исчез. Вычисленное Санденау значение S° B98,15 К)=68,2+0,8 Дж-К^-моль" примерно
на 2 Дж-К^-моль больше данных [2274]. Наиболее вероятным источником ошибки в работе [4131]
является нарушение кристаллической решетки РиО2 излучением изотопа 238Ри с малым периодом полураспада.
239
РиОа(г)
Глава 44
галось при составе РиО1ва (при 2033 К). С учетом образования в парах РиО(г) (~9%), авторы
нашли для РиО2(г) уравнение ДуС°(РиО2, г) =—473 200 + 18,2- Т Дж-моль. Комбинация
этого уравнения с приведенным в работе [1216] уравнением для A^G°(PuO2, к) приводит к
соотношению Д,6?°(РиО2т к) == 570 700 — 58,4- Т Дж-моль. Последнему соответствую! значения
Д8#°(Ри02, к, 0) = 642 + 20 кДж-моль (III закон) и 606 нДж-моль" (II закон).
Менее детальные исследования испарения оксидов плутония были выполнены Мюлфордом
и Ламаром [3580] (эффузионный метод, 1984—2379 К, 25 точек, состав равновесной
конденсированной фазы не был установлен, Д4Я°(РиО2, к, 0) = 628 кДж-моль (III закон) и 608 кДж-моль
(II закон)), Пардью и Келлером [3769] (метод протока, газ носитель — кислород, 1770—2060 К,
8 точек, Д8Я°(Ри02, к, 0) = 674 кДж-моль (III закон) и 462 кДж-моль (II закон)) и Мессье-
ром [3491] (эффузионный метод, состав конденсированной фазы РиО1>83, 2071—2376 К, 17 точек,
Д8#°(РиО2, к, 0) = 648 кДж-моль (III закон) и 643 кДж-моль (II закон)). Расчеты
энтальпии сублимации РиО2(к) по данным работ [3491, 3580, 3769] были проведены без учета
отклонения состава конденсированной фазы от стехиометрического и присутствия в газовой фазе РиО.
РиО2(г). Термодинамические свойства газообразного диоксида плутония в стандартном
состоянии при температурах от 100 до 10 000 К приведены в табл. 1040.
Молекулярные постоянные РиО2, использованные для расчета термодинамических функций,
приведены в табл. 44.7. Результаты исследования ИК-спектров молекул РиО2„ изолированных
Таблица 44.7. Значения молекулярных постоянных, а также а и
термодинамических функций PuOa, PuOJ и PuOj
принятые для расчета
Молекула
v,B)
v3
CM
/•1039
Г-CM2
a
Pi
PuO2a 746 220 794 17 2 1
PuOJa 800 250 900 16 2 2
РиОга 700 200 750 18 2 2
a Приведены энергии Т\ (в см) и статистические веса pt.
PuO2: 200F), 500F), 2000B5), 4000B5), 6000B5), 8000B5), 10 000B5), 15 000E0), 20 000E0).
PuOJ: 200 D), 400 D), 4500 A2), 7000 B0), 10 000 C5), 15 000 C5), 20 000 C5).
PuOj: 1000 F), 3000 A2), 5000 B0), 7 000 E0).
в матрицах, в работе Грина и Риди [2469] (отсутствие в спектрах Ри16О2 и Ри18О2 частоты
симметричного валентного колебания vx, а также анализ нормальных координат РиО2 и его изотопо-
меров) свидетельствуют, что РиО2 имеет линейную структуру (симметрия D^). Приведенный
в табл. 44.7 момент инерции вычислен для значения r(Pu—О) = 1,80 + 0,07 А, принятого
несколько меньшим величины re(PuO) (см. UO2(r)). Погрешностьв значении /составляет 2-109 г -см2.
Значения основных частот валентных колебаний приняты по работе Грина и Риди (значение vx
вычислено авторами [2469] на основе найденных ими силовых постоянных). Частота v2, также
ненаблюдавшаяся в спектре, рассчитана для модели валентного силового поля в предположении,
что /a/r2 : /г = 0,04, как это имеет место для UO2. Погрешности принятых значений vlt v2 и v3
оцениваются в 50, 50 и 15 см.
Экспериментальные данные об электронном спектре РиО2 отсутствуют. В справочнике на
основании предположения, что ионы U2+ и Ри4+ имеют одинаковые нижние электронные
конфигурации, принимается, что молекулы UF2 и РиО2 имеют одинаковые электронные состояния в
области до 20 000 см (см. примеч. 3 к тексту по UO2(r)).
Термодинамические функции РиО2(г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126) и
A. 129) с учетом девяти возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены главным образом неточностью принятых молекулярных
постоянных, а также приближенным характером расчета; они составляют 8, 12, 15 и 20 Дж-К~1Х
240
Плутоний и его соединения PuO2+(r), PuO2~(r>
Хмоль в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К. Основной вклад в эти погрешности
вносит неточность оценки возбужденных электронных состояний.
Таблица термодинамических функций РиО2 (г) вычислялась ранее в работе Грина [2463], где
электронные состояния оценивались по модели, предложенной этим автором. Расхождения
данных [2463] и табл. 1040 составляют 12—17 Дж-К^-моль в значениях Ф°(Г).
Константа равновесия реакции РиО2(г) = Ри(г) + 2О(г) вычислена с использованием
значения АгН°@) = 1253,265 + 22 кДж-моль, соответствующего принятым в справочнике
энтальпии образования и сублимации РиО2(к). Этим величинам также соответствует
г, 0) = —410,989 ± 20 кДж • моль.
з(г). Термодинамические свойства газообразного положительного иона диоксида плутония
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1041.
Молекулярные постоянные PuOJ, использованные для расчета термодинамических функций,
приведены в табл. 44.7. Экспериментальные данные о строении и ИК-спектре РиО? весьма
ненадежны. Грин и Риди [2469 ] при исследовании ИК-спектров окислов плутония, изолированных
в матрице из Аг, предположили возможность отнесения двух групп полос к частоте v3 молекул
Pu16O2, Pu16O18O+ и Pu18OJ, имеющих линейную структуру. На основании этих данных и
аналогии с молекулами PuO2, UOa и UOJ в справочнике для PuOj принята линейная структура
(симметрия jDoo*)- В предположении, что ионизация РиО2 наряду с обнаруженным возрастанием
частоты v3 сопровождается уменьшением величины r(Pu—О), приведенный в табл. 44.7 момент
инерции вычислен для значения r(Pu—О) = 1,75 + 0,10 А, несколько меньшего, чем в
молекуле РиО2. Погрешность в значении / составляет 3-109 г-см2. Основные частоты, приведенные
в таблице, оценены, если принять для силовых постоянных значения /г = 6,43-10* и frr =
= —0,41 • 105 дин-см (средние между полученными Грином и Риди [2469] для двух групп
полос, отнесенных к PuOj) и соотношение /a/r2 : fr = 0,04, справедливое для UO2. Погрешности
в значениях vx и v3 оцениваются в 150 см, частоты v2 — в 70 см. В справочнике принимается,
что РиО^ имеет в области до 20 000 см те же электронные состояния, как молекула UF3 (см.
примеч. 3 к тексту по UO2(r)).
Термодинамические функции PuOJ(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126)
и A. 129) с учетом семи возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений обусловлены неточностью оценки молекулярных постоянных, главным
образом числа и энергии возбужденных состояний, и приближенным характером расчета; они
составляют 6, 9, 12 и 15 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Таблица термодинамических функций PuOJ (г) публикуется впервые.
Константа равновесия реакции PuOJ(r) + е(г) = Ри(г) + 2О(г) вычислена с использованием
значения Дг#°@) = 353,265 + 29 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
70(Ри02) = 900 + 20 кДж-моль.
Эта величина была измерена методом электронного удара в работе Блекбёрна и др. (см. [2463,
3943]). -Ей также соответствует
bfH°(PuO$, г, 0) = 489,011 +28 кДж.моль.
з(г). Термодинамические свойства газообразного отрицательного иона диоксида плутония
в стандартном состоянии при температурах от 298,15 до 10 000 К приведены в табл. 1042.
Молекулярные постоянные PuOj, использованные для расчета термодинамических функций,
приведены в табл. 44.7. Экспериментальных данных о строении и частотах колебаний РиО^ в
литературе нет. Так же как для РиО2 и PuOJ, в справочнике принимается, что молекула РиО7 имеет
линейную структуру (симметрия Dmh). Добавление электрона на 5/-орбиталь в PuOg,
по-видимому, должно привести к ослаблению связи в PuOj по сравнению с РиО2, сопровождающемуся
увеличением межатомного расстояния и уменьшением частот колебаний. Приведенный в табл. 44.7
момент инерции вычислен для значения r(Pu—О) = 1,85 + 0,1 А. Погрешность в значении /
оценивается в 4-Ю9 г-см2. Значения основных частот приняты несколько меньшими, чем в
молекуле РиО2. Погрешности в их величинах оцениваются в 150 см для vx и v3 и 70^см~х для v2.
241
Ри2О8(к, ж) Глава 44
В справочнике принимается, что в области энергий, меньших ее потенциала ионизации,
молекула PuOj имеет такое же число возбужденных состояний, как молекула UF (в предположении^,
что нижние конфигурации электронных оболочек U+ и Ри3+ одинаковы (см. также 3-е примеч.
к тексту по UO2(r)).
Термодинамические функции РиО^г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126)
и A. 129) с учетом четырех возбужденных состояний по уравнениям A. 168)—A. 170).
Погрешности рассчитанных значений обусловлены неточностью оценки молекулярных постоянных,
в особенности электронных состояний, а также приближенным характером расчета; они
оцениваются равными 8j 12,15 и 20 Дж-К^-моль-1 в значениях Ф°(Г) при 298,15, 3000, 6000 и 10 000 К.
Таблица термодинамических функций PuOj (г) публикуется впервые.
Константа равновесия реакции РиО^(г) = Ри(г) + 20 (г) + е(г) вычислена с использованием
значения Д,ЛУ°(О) = 1343,265 + 55 кДж-моль, соответствующего принятому в справочника
сродству к электрону
Л0(РиО2) = —90 + 50 кДж-моль.
Принятое значение было оценено равным аналогичной величине для UO2.
Принятому сродству к электрону соответствует
г, 0) = —500,989 ± 54 кДж • моль.
Ри2О3(к, ж). Термодинамические свойства кристаллического и жидкого триоксида диплутония
в стандартном состоянии при температурах 100—5500 К приведены в табл. 1043.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 44.1. В справочнике за стандартное состояние Ри2О3(к) в интервале 0—2358 К принимается
гексагональная ^-модификация, как наиболее устойчивая [2361, 3442] Ч
При Т < 298,15 К термодинамические функции вычислены на основании результатов
измерений теплоемкости в работе Флотова и Тетенбаума {2276] (8—350 К). При синтезе образца был
использован изотоп 242Ри, период полураспада которого существенно больше, чем у 239Ри (см.
Ри(к, ж)). Погрешность измерений при Т > 25 К авторы [2276] оценили в 0,5%. На кривой
температурной зависимости теплоемкости Флотов и Тетенбаум [2276] обнаружили аномалию А-типа
с пиком при 17,65 К и LtrH — 0,133 + 0,0008 кДж-моль. Подобная аномалия вблизи 19 К,
связанная с антиферромагнитным упорядочением, была обнаружена Мак-Картом и др. [3442]
при изучении структурных и магнитных свойств Ри2О3. Принятые значения ?°B98,15 К) и
Я°B98,15 К)—#°@) приведены в табл. 44.1 и имеют погрешности 0,6 Дж-К^-моль и 0,09 кДжХ
Хмоль соответственно.
При Т > 298,15 К теплоемкость Ри2О3(к) аппроксимирована двумя уравнениями.
Уравнение в интервале 298,15—350 К выведено на основании найденных в работе [2276] значений
С;B98,15 К) = 116,98, С;C20 К) = 119,01 и С;C50 К) = 120,69 Дж-К^-моль. Поскольку
для Т >¦ 350 К экспериментальные данные о термодинамических свойствах Ри2О3(к, ж)
отсутствуют, уравнение для теплоемкости в интервале 350—2358 К было получено на основании
значения С*C50 К) и оцененных значений С;A000 К) = 150, С;B358 К) = 190 Дж-К^-моль.
Оценки были выполнены в соответствии с характером температурной зависимости теплоемкости
РиО2(к) (см. выше).
Температура плавления Ри2О3 B358 + 25 К) принята по измерениям Чикалы и др. [1833].
Энтальпия плавления A13 + 20 кДж-моль) оценена по энтропии плавления А12О3 (Дт? =
= 48 Дж-К-моль~1), для которой известны надежные экспериментальные данные.
Теплоемкость жидкого Ри2О3 B00 ± 30 Дж-К-моль) принята равной этой величине для 8с2О3(ж),
которая известна из экспериментальных измерений.
Погрешности рассчитанных термодинамических функций Ри2О3(к, ж) из-за отсутствия
экспериментальных данных для Т >• 350 К весьма велики и составляют в значениях Ф°(Г) при 298,15,
1000, 3000 и 5500 К 0,4, 10, 35 и 50 Дж-К^-моль соответственно.
Таблица термодинамических функций Ри2О3(к, ж) публикуется впервые.
1 Так называемая кубическая (а) модификация состава PuOj Б1в при Т > 650 К, по-видимому, диспропорциони-
рует на р-Ри2О3 и PuOlj61 (см. [5032а]).
242
Плутоний и его соединения Ри2О3(к, ж)
Константа равновесия реакции Ри2О3(к, ж) = 2Ри(г) + ЗО(г) вычислена с использованием
значения ДГЯ°(О) = 3103,498 + 26 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
^Н°(РщО8, к, 298,15 К) = —1670 + 20 кДж • моль.
Принятая величина является средней из результатов исследования реакции 2Ри2О3(к) +
-j- О2(г) = 4РиО2(к) в работах [4946, 5012]. Маркин и Рэнд [5012] (см. также [5032а]) методом
ЭДС измерили парциальные свободные энергии кислорода над оксидами плутония в области
отношений О : Ри = 1,53 -4- 2,00 и температур 973 + 1413 К. Интегрированием парциальной
мольной энергии Гиббса как функции от состава авторы [5012] нашли значения Дг17°A150 К) =
= —823,8 кДж-моль и Дг?сA150 К) = —180 Дж-К^-моль. Полученному значению
ДгЯ°A150 К) соответствует &.fHo(Pu20ai ка 298,15 К) = —1685 + 25 кДж-моль, а значениям
энтальпии и энтропии реакции — величина Дг(г°A150 К) = —616,9 кДж-моль. Это позволяет
вычислить по уравнению III закона с использованием принятых в справочнике
термодинамических функций значение &fH°(Pu2Os, к, 298,15 К) = —1660 + 20 кДж • моль.
Шеро и др. [4946] с помощью микрокалориметра измерили парциальные мольные энтальпии
растворения кислорода в оксидах плутония в области отношений О : Ри = 1,50 -f- 2,00 при 1373 К.
Интегрирование измеренных парциальных' энтальпий растворения как функции от состава
оксидов позволило рассчитать значение Д,#ОA373 К) = —879 + 20 кДж-моль, которому
соответствует Ь^Н^РщО^ к, 298,15 К) = —1654 + 20 кДж-моль1. В этой же работе [4946] были
калориметрически измерены энтальпии сгорания РиО1E7 и PuO1Ml до РиО2. Экстраполируя эти
данные, можно получить ДуЯо(Ри2О3) кв 298,15 К) = —1681 +20 кДж-моль.
В работе [4946] приведено существенно отличающееся значение —1685 кДж-моль 1; расхождения результатов
обработки, по-видимому, обусловлены различием использованных в расчетах термодинамических функций.
Г л а в а 45
ЛИТИЙ И ЕГО СОЕДИНЕНИЯ
1л(к, ж), Li, Li+, Li2, Li?, LiJ, LiO, 1Л2О(к, ж), Li2O, Li2O+, 1Л2О2(к), Li2O2, ЫН(к, ж), LiH,
LiD(K, ж), LiD, 1ЛТ(к, ж), LiT, 1Л0Н(к, ж), LiOH, Lia02H2, LiF(K, ж), LiF, Li2F2, Li3F3,
LiG1(k, ж), LiCl, Li2Cl2, Li3Cl3, 1ЛВг(к, ж), LiBr, Li2Br2, Li3Br3, 1Л1(к, ж), Lil, Li2I2, Li3I3,
L12SO4(k, ж), LiNO2(K, ж), LiNO2, LiNO3(K, ж), LiNO3, 1Л2СО3(к, ж), 1ЛВОа(к, ж), LiBO2,
LiAlF4, Li3AlF6(K, ж), eLi(K, ж), eLi, 6Li+, 6ЫН(к, ж), eLiH, 6LiD(K, ж), eLiD, 6LiT, 7Li, 7Li%
7LiH, 7LiD, 7LiT
В настоящей главе рассматриваются литий и его соединения с кислородом, водородом
(дейтерием, тритием), галогенами, соли лития и некоторых кислородсодержащих кислот, а также его
фторалюминаты — всего 32 вещества. Для 17 веществ в справочнике представлены сведения об
их свойствах в конденсированном состоянии, для 29 веществ — в газообразном состоянии.
Термодинамические свойства всех соединений рассчитаны для природной изотопической смеси лития
(атомный вес 6,941) и других элементов. Кроме того, в справочник включены термодинамические
свойства чистых изотопов лития 6Li и 7Li, а также их соединений с водородом и его изотопами х.
В Приложении 3.10 представлены сведения о критических постоянных лития.
Ы(к, ж). Термодинамические свойства кристаллического и жидкого лития в стандартном
состоянии при температурах 100—3000 К приведены в тао*л. 1044.
Значения постоянных,; использованные для расчета термодинамических функций,
представлены в табл. 45.1. В справочнике за стандартное состояние 1Л(к) в интервале 0—80 К принята
гексагональная плотноупакованная модификация (структурный тип Mg), а в интервале 80—
453,69 К — кубическая объемно-центрированная модификация (структурный тип Na).
При Т ^ 298,15 Тх термодинамические функции вычислены по результатам удовлетворительно
согласующихся между собой измерений теплоемкости лития в работах Мартина [3403, 3406]
B0—300 К) и Филби и Мартина [2250] C—30 К) с учетом полиморфного («мартенситного»)
превращения лития при 80 К. Измерения теплоемкости в работах [4295] A5—300 К), [4020] A,5—
20 К), [3405] @,4—1,5 К) и [3399] @,35—2 К), по-видимому, менее точны и не учитывались.
Погрешности принятых значений ?°B98,15 К) и .ffoB98,15 К)—Н°@), совпадающих с
рекомендованными |КОДАТА—МСНС [1887], оцениваются в 0,2 Дщ-К^-моль и 0,04 кДж-моль
соответственно.
В интервале 298,15—453,69 К для теплоемкости 1л(к) принято уравнение, полученное на
основании результатов измерений энтальпии лития Дугласом и др. [2110] B73—443 К,
погрешность ~0,3%). С температурой плавления D53,69 + 0,03 К), принятой по данным [2110], хорошо
согласуется значение, найденное Мартином [3403] D53,65 + 0,10 К). Менее точные данные
получены в работах [2955,, 2999, 3311, 4128]. Энтальпия плавления C,00 + 0,02 кДж-моль)
принята по работе [2110]; измерения [1971, 2999, 4128] выполнены с гораздо большей погрешностью,
чем в работе [2110], и не учитывались.
Уравнение для теплоемкости жидкого лития выведено совместной обработкой результатов
измерений энтальпии в семи работах; при этом погрешность данных Дугласа и др. [2110] D58—
1169 К) оценена в 0,3%, Новикова и др. [729] D87—921 К) — в 0,7%, Шпильрайна и Ковер-
дяева [1170] G88—1573 К) и Аченера и Фишера [1212] E23—1373 К) — в 3%, а данных Ред-
монда и Джонса F43—1361 К), Бейтса и Смита G73—1273) 2 и Пчелкина [844] D73—1573 К) —
в 5%?3. Приведенному в табл. 45.1 уравнению соответствует уменьшение теплоемкости от
* Ввиду относительно невысокой точности данных о свойствах лития и его соединений в конденсированных
состояниях среди рассмотренных соединений отсутствуют 7Li (к, ж), 71ЛН (к, ж), 7LiD (к, ж), 71ЛТ (к, ж) ив1ЛТ (к, ж).
В пределах достоверности имеющихся данных их свойства могут быть приравнены к свойствам соответствующих
веществ с природной смесью изотопов лития.
2 Результаты измерений, выполненных Редмондом и Джонсом и Бейтсом и Смитом, цитируются в монографии
Шпильрайна и др. [1179].
^Данные [844] по теплоемкости Ы(ж) были пересчитаны и вводились в обработку в виде 12 значений энтальпии
в|указанном интервале температур.
244
Литий н его соединения
Li(K, ж)
Таблица 45.1. Принятые значения термодинамических величин для лития и его соединений
в кристаллическом и жидком состояниях
Веще-.
ство
Li
Li2O
Li2O2
LiH
LiOH
LiD
LiT
LiF
LiCl
LiBr
Lil
Li2SO4
LiNO2
LiNO3
Li2CO3
LiBO2
Li3AlFe
«Li
«LiH
«LiD
Состояние
kI, куб.
ж
к , куб.
ж
к, гекс.
к, куб.
ж
к, тетр.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
1
К
ю
СМ о
кДжХ
Хмоль
4,632
—
7,251
—
10,5
3,883
—
7,406
—
4,535
—
4,87
—
6,473
—
9,3
—
10,476
—
11,36
—
к11,монокл.(а) 18,631
к1, куб. ф)
ж
к11
к1, гекс.
ж
к, гекс.
ж
к, монокл.
ж
к, монокл.
ж
KIV, ромб.(Р)
кШ, тетр. (?)
к11, куб. (8)
к1(е)
Ж
к1, куб.
ж
к, куб.
ж
к, куб.
ж
_
—
12,1
—
12,8
—
15,18
—
8,88
—
32,15
,—
_
—
4,35
—
3,638
—
4,242
—
Примечание. Ср(Т)=а-\-Ь-Т—с
Li: ¦•
Li2O: !
LiF: l
Li2SO4:
» d= 2,628 -10-*.
» d=27,56O -10-*
» d= 1,738 -10-*.
a d=—5608,736
К
Ю
">Н
ОО
см
о
Дж-К
К
ОО
О!
СЧ
Хмоль
29,12
—
37,61
—
58
20,60
—
42,76
—
23,56
—
26,1
—
35,66
—
59,27
—
74,01
—
86,71
—
113,97
—
—
88
—
104
—
90,12
—
51,27
—
187,9
—
—
26,9
—
18,97
—
21,68
—
•10-в, «=3143,987 •
24,86
—
54,10
—
75
28,950
—
49,58
—
34,60
—
36,9
—
41,80
—
47,99
—
49,83
—
51,04
—
117,57
—
—
63
—
89
—
98,32
—
59,8
—
202,53
—
_
—
24,0
—
28,25
—
33,60
—
+- e T* (в
ю-».
Коэффициенты в уравнении
для С°(Т)
1,309
31,227
76,666
104
57,158
21,000
55
51,381
87,3
36,131
56
38,980
56
44,561
63,8
45,568
63,2
40,706
65,3
43,637
63,2
—644,732
218
205
—8,539
80
90
87,544 •
149
58,423
185
58,984
144,4
187,815
285
295
278
386
0,449
31,227
20,010
55
35,524
56
56,287
—5,265
—13,634
—
59,842
43,729
—.
32,796
—
29,158
—
26,982
—
13,502
—
19,393
—
26,054
—
24,831
—
3531,798
—
—
239,944
- —
-78,557
—
150,438
—
49,813
—
171,935
—
—
—
56,287
—5,265
44,704
—
29,733
—
Дж К моль-1)
LiNO3:
Li3AlF6:
«Li:
с•10~s
—6,017
—2,050 a
18,624 a
—
—
4,523
—
10,293
—
9,089
—
9,000
—
6,170 a
—
2,987
—
—1,205
—
__
—
—110,716 a
—
i—
—
0a
—
4,406
—
12,477
—
27,896 a
—
—
—
-6,017
—2,050 a
4,523
—
9,591
—
a d=279,863 -10-«.
a d=—58,
a d=2,628
113 -10-».
•ю-».
Интервал
температур
К
XX.
2S8,lE-453,69
453,69—3000
298,15—1726
1726—4000
298,15—1000
288,15—865
965—2000
298,15—746
746—3500
298,15—867
967—2000
298,15—968
968—2000
298,15—1122
1122—3500
298,15—883
883—3200
298,15—823
823—3200
298,15—742
742—3000
298,15—848
848—1131
1131—3500
298,15—369
369—495
495—1500
298,15—526
526—2000
298,15—1005
1005—3500
298,15—1122
1122—3000
298,15—788
788—873
873—978
978—1058
1058—4000
298,15—453,5
453,5—3000
298,15—965
965—2000
298,15—967
967—2000
или
T
или
кДжХ
Хмоль *
453,69 3,0
,—
1726
—
965
—
746
—
967
—
968
—
1122
—
883
—
823
—
742
—
848
1131
—
369
495
—
526
—
1005
—
1122
—
788
873
978
1058
.—
453,5
.—
965
—
967
—
—
35,6
—
—
21,8
—
20,9
—
22
—
22 .
—
27,08
—
19,75
.—
17,66
—
14,64
—
23,9
9,3
1,7
9,2
25,0
—
41,4
53,9
—
3,0
3,0
7,7
87,9
—
3,0
—
21,8
—
22
—
245
Li(k, ж)
Глава 45
С;D53,65 К) = 30,4 Дж-К^-моль до С;A000 К) = 29,2 Дж-К^-моль (минимум Q, а за-
. Погрешности
2% при 2000 К
тем ее плавное, но ускоренное увеличение до С°C000 К) = 37,8 Дж-К«моль
-1% при 1000 К,,
описания значении энтальпии этим уравнением оцениваются в
и -5% при 3000 К.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К составляют 0,15,
0,4, 0,8 и 2 Дж-К^-моль соответственно. Значения термодинамических функций Ы(к, ж),
рассчитанные в справочниках JANAF [2834], Шпильрайна и др. [1179] и Халтгрена и др. [2752],
до 1000 К несущественно (в пределах 0,3 Дж>К-моль в значениях Ф°GТ)) отличаются от
приведенных в табл. 1044. При Т > 1000 К эти расхождения увеличиваются с ростом температуры
из-за того, что в настоящем справочнике принято ускоренное возрастание теплоемкости 1л(ж)
(вместо предположения о постоянстве теплоемкости [2752Ж 2834] или о ее медленном линейном
увеличении [1179] при Т > 1200 К).
В справочнике в качестве стандартного состояния лития принято кристаллическое состояние
и в соответствии с этим
Д/Г°Aл, к,) = 0.
Давление пара лития в реакции 1л(к, ж) = Li(r) вычислено с
дг#°@) = 157,735 ± 1,0 кДж-моль
сублимации
Д,Я°Aл, к, 298,15 К) = 159,3 + 1,0 кДж • моль.
Принятая величина основана на приведенных в табл. 45.2 результатах измерений давления
насыщенного пара лития. Приведенные в таблице погрешности учитывают только
воспроизводимость измерений. Расчет энтальпии сублимации проводился для модели идеальной смеси
химически реагирующих атомарной и молекулярной компонент. Поправка на отклонение от идеаль-
Таблица 45.2. Результаты определений Д,2Г° (Li, к, 298,15 К) (в кДж-моль)
использованием значения
соответствующего принятой в справочнике энтальпии
Авторы
Метод
Д,Я°(Ы, к, 298,15 К)а
II закон
III закон
Руфф, Йохансен, 1905 [4091] Точек кипения, 1673,1 К, 1 атм, 1 точка — 164,12
Крёнер, 1913 [3119] Статический, 903-1173 К, 6-10-»—7-Ю"» атм, 160±23 164,4±1,8
6 точек
Хартман, Шиейдер, 1929 [2575] Точек кипения, 1204—1353 К, 2-Ю"*—1-10"» 162,6±4,1 159,00 + 0,19
атм, 8 точек
Богро, 1930 |1601, 1602] Эффузионный, 732-836 К, 1 -10-"—2 -10-* атм, 139±30 160,4+1,4
8 точек
Льюис, 1931 [3248] « То же, 852—927 К, 1 -Ю^4—8 -Ю атм, 5 точек 164,1 ±6,4 150,94±0,34
Мошэра, 1939 [3430, 3431] То же, 735—915 К, 1-Ю-6—2-Ю"* атм, 16 точек 164,1 ±9,7 160,61 ±0,55
Боданекий, Шанс, 19S5 [1603,1604] Точеккипения, 1374—1881 К,0,1— 4,8атм, 16точек 161,4±1,1 159,54+0,13
Рягаи и др., 1965 [4000] Статический, 1307—1806 К, 7-Ю—3,3 атм, 161,06 ±0,83 159,34 ±0,14
22 точки
Аченер, Фишер, 1967 [1211] То же, 1308—1535 К, 6-10~2—0,5 атм, 8 точек 161 ±12 159,88±0,54
Шшшьрайи, Белова, 1968 [1163] То же, 1178—2156 К, 1 -Ю—.15,7 атм, 59 точек 160,7 ±1,2 159,06 ±0,12
Анисимов, Воляк, 1969 [38] То же, 1057—1646 К, 3 -Ю-3—1,23 атм, 98 точек 160,34±0,54 159,60 ±0,03
Поттер и др., 1970 [3869] Торзионный6 — 161,6±2,5
Шанс и др., 1971 [4184] Точек кипения, 1448—1875 К, 0,2—4,8 атм, 161,3±4,3 159,23±0,17
40 точек
By, 1976 [4776] Масс-спектрометрический, 553—683 К, 2-Ю-3-0— 161,1 ±1,6 161,55±0,11
1 -10~7 атм, 11 точек
Бэналла, 1977 [1610] Точек кипения, 1795,2—1965,4 К, 3,04—8,3 атм, 181,9±9,8 158,09±0,8 6
6 точек
а Приведенные величины рассчитаны с учетом образования в насыщенном паре димерньга молекул; б в работе
приведено только значение Д4ЯОB98,15 К), вычисленное с использованием функций из справочника [2834].
Приведенное значение было пересчитано с учетом разности термодинамических функций, принятых в [2834] и в
настоящем справочнике.
246
Литий и его соединения
ТТабдица 45.3. Уровни энергии атомов Li и Li+,
принятые для расчета термодинамических
функций
ного состояния, оцененная по варианту метода смеси, разработанному Фокиным [1060],
оказалась пренебрежимо малой и в расчетах не учитывалась. Метод соответствующего расчета изложен
в Приложении 8.
Рекомендованное значение является средневзвешенным из наиболее надежных измерений
давления пара в работах [1603, 1604, 2575, 4184] (метод точек кипения) и в работах [38, 1163»
4000] (различные варианты статического метода). Всего для расчета средневзвешенного были
использованы 243 точки, лежащие в интервале 1204—2156 К. Погрешность принятой величины
обусловлена в основном неточностью термодинамических функций лития в конденсированном
состоянии. Соответствующие погрешности достигают 0,6 кДж-моль для измерений,
выполненных при 1200 К, и 2,3 кДж-моль для измерений при 2200 К. Принятое значение энтальпии
сублимации совпадает с рекомендованным КОДАТА—МСНС [1887]. Результаты измерений
давления пара в работах [1211, 1601, 1602, 1610, 3119, 3248, 3430, 3431, 3869, 4091, 4776] менее
точны и не учитывались при подготовке справочника.
Li (г). Термодинамические свойства газообразного лития в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 1045.
Уровни энергии атома Li* использованные при расчете термодинамических функций,
приведены в табл. 45.3, в которой даны валентные состояния Li, соответствующие электронным
конфигурациям ...2s и ...2р. Приведенные в этой
таблице значения основаны на рекомендации
Мур [3550], а также на результатах
спектральных измерений Йоханссона [2861]. При этом
для состояния гР принято значение,
усредненное для двух подсостояний по [2861].
Термодинамические функции Li(r) вычислены
по уравнениям A. 3)—A. 6), A. 9), A. 10),
{1.23)—A.25) непосредственным суммированием
по уровням энергии. Погрешности
вычисленных значений термодинамических функций при
Т <С 5000 К определяются только неточностью
фундаментальных постоянных и не превышают
0,02 Дж-К-^моль в значениях Ф°(Т) и S°(T).
При более высоких температурах погрешности возрастают из-за того, что в расчете не
учитывались ридберговские состояния и при Т = 6000 и 10 000 К могут достигать 0,05 и 0,3 Дж-К"хХ
Хмоль в значениях Ф°(Г) и 0,3 и 1 Дж-К-моль в значениях S°(T).
Ранее термодинамические функции Li(r) вычислялись в предыдущих изданиях настоящего
справочника [337, 340], в таблицах JANAF [2834], Мадера [3338] и Мак-Брайд и др. [3439]
{до 6000 К), а также в работах Гурвича и др. [335], Хилзенрата и др. [2652] и Рейтера [3973]
(до 10 000 К), Кольского и др. [3067] (до 8000 К) и Эванса и др. [2195] (до 2500 К). Результаты
расчетов при Т <[ 5000 К удовлетворительно согласуются с данными табл. 1045; расхождения
в значениях Ф°(Т), как правило, не превышают 0,01 Дж-К~1-моль~1. Исключение составляют
работы [3067, 3973], для которых они достигают 0,06 Дж-К«моль. При Т > 5000 К
термодинамические функции Li(r), вычисленные во всех работах, кроме [3338], возрастают с температурой
быстрее, чем данные табл. 1045, вследствие того, что практически во всех опубликованных
расчетах в той или иной степени учитывались ридберговские состояния атома Li. Однако
применение разных методик выбора числа учитываемых состояний привело к значительному разбросу
полученных результатов. Максимальные расхождения имеют место с данными [335, 3973],
которые достигают 6—7 Дж-К^-моль в Ф°A0 000 К).
Энтальпия образования
Д,#°(Li, г, 0) = 157,735 ± 1,0 кДж • моль
соответствует принятой энтальпии сублимации лития.
Li+(r). Термодинамические свойства газообразного положительного иона лития в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 1046.
В табл. 45.3 приведено только основное состояние Li+5j относящееся к типу 1S; все
возбужденные состояния Li+ являются ридберговскими.
Li
Li+
Электронная
конфигурация
lsa2s
№2р
lsa
Состояние
*Р
СМ
0
14 903,88
0
Pi
2
6
1
247
1Л2(г) Глава 45
Термодинамические функции Li+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
при условии, что In (?вн = 0. Погрешности вычисленных значений термодинамических функций
во всем интервале температур обусловлены только неточностью фундаментальных постоянных
и не превышают 0,01—0,02 Дж-К^-моль.
Ранее термодинамические функции Li+(r) вычислялись в предыдущих изданиях настоящего
справочника [337, 340] и в таблицах JANAF [2834] (до 6000 К), а также в работах Хилзенрата
и др. [2652] до 10 000 К и Грина и др. [2473] до 50 000 К. Результаты этих расчетов совпадают
сданными табл. 1045 в пределах 0,01 Дж-К^-моль (за исключением Ф°A0 000 К) и S°A0 000 К)
в расчете [2652], где по-видимому, для этих величин допущена ошибка в 0,2 Дж-К-моль~1).
Константа равновесия реакции Li+(r) + е(т) = 1Л(г) вычислена с использованием значения
Д^^О) = —520,222 + 0,001 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0(Li) = 43 487,15 ± 0,10 см = 520,222 + 0,001 кДж-моль.
Это значение было получено Йоханссоном [2861 ] на основании измерений энергии 2D
состояний атома Li, соответствующих четырем членам (п = 3-^-6) ридберговской серии ...nd. Оно
рекомендовано также Мур [3549] с погрешностью 0,005 см. Однако в настоящем справочнике
погрешность принятого значения увеличена из-за расхождений между вычисленными и
экспериментально определенными значениями энергии других состояний атома Li, которые были
изучены в работе [2861].
Принятому значению соответствует
bfH°(lA+, г, 0) = 677,957 ±1,0 кДж • моль.
Li2(r). Термодинамические функции газообразного дилития в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 1047.
Молекулярные постоянные 7Lia, приведенные в табл. 45.4, приняты на основании анализа
результатов спектральных исследований [2627, 3150] и квантовомеханических расчетов [3715,
3716, 4672]. В спектре молекулы Li2 изучено шесть систем полос, связанных с переходами между
основным состоянием X12+< и возбужденными состояниями А1!,* [1301, 3150, 4784], ВхИи [2578,
2627, 3298, 3459, 4782, 4783], СЧЗ,,, DXU.U [1407, 4300, 4586, 4597, 4599, 4601], гЦ [1520, 15211
и хПр [1520]. Согласно многочисленным теоретическим расчетам [1337, 2314, 2348, 3071, 3153,.
3364, 3587, 3715Я 3716, 4012, 4672] молекула Li2 должна иметь еще ряд устойчивых
возбужденных триплетных и синглетных состояний, из которых пять (а3 2*, &3ПИ, с3 2*, 1Ид и Х2р
коррелируют с атомами в состояниях 2S + 2S и 2S + 2P. Молекулярные состояния, коррелирующие
с более высокими состояниями атомов (в том числе экспериментально изученные С1!!,,, D1!!^
Х2* и 1Пу), не рассматриваются в справочнике.
Молекулярные постоянные в состояниях X1!,* и BlIiu приняты на основании детального
анализа большого числа полос (г/ ^ 14 и v" ^ 18) системы В1!!,, =е? X1!,*, которые были изучены
Хесселом и Видалом [2627] в спектрах поглощения и лазерной флюоресценции1. Постоянные
в состоянии А1 2* получены Кушем и Хесселом [3150] из анализа полос с v' ^ 25 и v" ^ 14
системы А1!,^ *~ Х1!,*. Значения энергий возбуждения, частот колебаний и вращательных
постоянных для состояний а3 2*, bsUu, c3E? и хИд приняты равными округленным значениям
соответствующих величин, полученных в неэмпирических расчетах Олсона и Коновалова [3715, 3716].
Постоянные для возбужденного состояния 12^ приняты по результатам расчета методом
псевдопотенциала в работах Уотсона и др. [4672]. Погрешности рассчитанных значений Те оцениваются
в 1000 см (для состояния а3Е? — в 50 см), значений шея Ве — в 10 и 20% от их величин.
Термодинамические функции Li2(r) были вычислены по уравнениям A. 3)—A. 6), A'. 9), A. 10),
A. 93)—A. 95). Значения (?вн и ее производных вычислялись по соотношениям (l.tH)—A. 92)
с учетом семи возбужденных электронных состояний, в предположении, что для состояний с
энергией выше 20 000 см Q$3, вр = PiQi*l вр. Статистические суммы по
колебательно-вращательным уровням энергии состояний X1!,*, a32*t ЬаНи, А1!,* и с8Е^ и их производные находились
1 Согласно работе [2627] потенциальная кривая Li2 в состоянии ВхПи имеет барьер высотой 410+50 см над,
диссоциационным пределом Li BS)+Li BP).
248
Литий и его соединения Li+2(r)
по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным и
вращательным уровням, ограниченным предельной кривой диссоциации. Уровни энергии вычислялись по
уравнениям A. 63), A. 64а) и A. 65) с коэффициентами Ykl, приведенными в табл. 45.5 вместе
со значениями ушах и /Иш, а также коэффициентами а,- в уравнении, аппроксимирующем
предельную кривую диссоциации. Величины Ykl для изотопической модификации, соответствующей
природной смеси изотопов Li, были получены при помощи соотношений A. 66) по постоянным
молекулы 7Li2 из табл. 45.4. Для обеспечения условий A. 59) и A. 50) для состояний X1 ?* и
А12* было изменено значение YOi и добавлены коэффициенты Y80 и Yeo соответственно, а для
состояний а3?*, Ь3Ии и с3?* введен коэффициент Y03. Мульташлетность и вырождение состояний
а3?*, bsliu и с3 2* учитывались статистическими весами 3, 6 и 3 соответственно.
Погрешности рассчитанных термодинамических функций Li2(r) при низких температурах
(до 1000 К) обусловлены главным образом неточностью аппроксимации
колебательно-вращательных уровней основного состояния уравнениями с принятыми значениями постоянных, а также
неточностью физических постоянных. При более высоких температурах основной вклад в
погрешность вносит отсутствие экспериментальных данных об энергии триплетных состояний 63ПИ и
с3?*. Погрешности в значениях Ф°(Т) могут достигать 0,02j 0,1 и 0,5 Дж-К^-моль при 298,15,
3000 и 6000 К соответственно.
Термодинамические функции Li2(r) ранее вычислялись в предыдущих изданиях настоящего
справочника [337, 340], справочниках JANAF [2834], Мак-Брайд и др. [3439] и Мадера [3338],;
а также в'работах Эванса и др. [2195], Фебера и Херрика [2228], Артыма [44] (Т < 10 000 К),'
Рейтера [3973] (Т < 10 000 К) и Шнейдера [4197а] (Т = 600-^-2400 К). Расхождения данных
[2834, 3439], полученных по приближенным методикам и без учета возбужденных электронных
состояний, и табл. 1047 не превышают 0,8 и 2,5 Дж-К~1-моль в значениях Ф°(Г) и S°(T),
поскольку разные источники ошибок в этих работах частично компенсировали друг друга.
Расхождения с данными [337, 2228] достигают 3 и 12 Дж-К-моль в значениях Ф°(Г) и S°(T) в
обусловлены тем, что в обоих расчетах не учитывались триплетные состояния Li2. Наилучшее
согласие с данными табл. 1047 имеет место для расчета Артыма [44] (в пределах 0,3 и 1 Дж-К"хХ
Хмоль в значениях этих функций).
Константа равновесия реакции Li2(r) = 2Li(r) вычислена на основании принятой в
справочнике энергии диссоциации
ДгЯ°@) = D0(Li2) == 8445 ± 100 см «101 + 1 кДж • моль,
полученной Хесселом и Видалом [2627] экстраполяцией колебательных уровней энергии
состояния X1!,* (примерно на 2400 см) с учетом асимптотического поведения потенциала вида V(r) =
= De — С6/г6. Принятое значение находится в согласии с несколько менее точными величинами,
полученными более далекой экстраполяцией в работах Стволли [4422] (8440 см), Веласко и др.
[4599] (8275 см), а также с найденными Льюисом [3248] (8307 см) и By [4775, 4776] (8900 ±
+ 350 см) при исследовании равновесия реакции диссоциации Li2(r). Завышенное значение,,
полученное в работе Лумиса и Нусбаума [3298] короткой экстраполяцией уровней состояния
В1Ии (9232 см), обусловлено наличием барьера на потенциальной кривой этого состояния,
который не принимался во внимание авторами работы.
Принятой энергии диссоциации соответствует
Д/Я°(Ыа,в[г, 0) = 214 + 2,2 кДж • моль.
Li^r). Термодинамические свойства газообразного положительного иона дилития в
стандартном состоянии при температурах 298,15—6000 К приведены в табл. 1048.
В табл. 45.4 приведены молекулярные постоянные 7Lij, принятые на основании результатов
квантовомеханических расчетов и приближенных оценок. Неэмпирические расчеты молекулы
Lij показали, что двум первым диссоциационным пределам Li+(XS) + LiBS) и Li+(x?) + ЫBР)
соответствуют два стабильных электронных состояния X2L* и Л2П„ [4973, 4997, 5022].
Колебательные и вращательные постоянные Li* в этих состояниях, приведенные в табл. 45.4, вычислены
по результатам расчета Коновалова и Розенкрантц [4997]. Погрешности принятых значений сое,
Ве и Те (А2Ии) оцениваются в 5, 0,01 и 400 см соответственно.
Термодинамические функции LiJ(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)г
A. 93)—A. 95). Значения (?м и ее производных рассчитывались по уравнениям A. 90)—A. 92)
249
Li+2(r)
Глава 45
Таблица 45.4. Молекулярные постоянные 'Li2, 'Li+ 'LiO, «LiH, 7LiH, «LiD, 'LiD, 'LiF, 'LI3*Cl, 'Li81Br
и 'Lil
МолекуСостоя-
в.
¦ 102
De ¦ 105
CM
A
'Li2
'LiO
6LiH
'LiH
6LiD
'LiD
'LiF
'Li38Cl
'Li81Br
'Lil
вт
Xi 2+
Л12+
вт
а3П
вт
Л* 2+
а3П
вт
XI2+
О
8 300
12 000
14 068,307
16 700
20 436,323
20 500
22 000
0
23 400
0а
2 330
0
26 515,21
33170
34 912
0
26 515,14
33170
34 912,18
0
26 513,45
33 170
34 909
0
26 513,35
33 170
34 909,13
О
О
О
О
351,419 285
60
340
255,467 30
240
270,870 406
140
80
262
98
851,5
866,7
1419,843
243,758 7
627
217,7
1405,86
239,999 3
620,89
215,5
1074,192
185,100 9
475
201,1
1055,085
183,758 6
466,52
197,45
910,25
640,22
563,16
498,16
2,587 587 2 а 0,672 606 550
2,8 0,268
2,5 0,695
1,581 360 6 г 0,497 423 76
2,1
3,093 675 ж
1,6
1,0
1,93 а
0,871Д
12,5
6,7
23,488 2 а
—23,600 86 г
53
43,3
23,371 8 а
—23,913 40 г
52,38
42,4
13,453 1 а
—13,822 72 г
30,8
46,58
13,162 04 а
-12,450 02 г
29,71
44,916 3
8,104
4,49
3,53
3,39.
0,500
0,557 537 91
0,345
0,278
0,491 б
0,298 б
1,203
1,352
7,668 8
2,911 3
5,11
3,45
7,516 1
2,849 5
5,01
3,383
4,390 7
1,677 7
2,95
1,94
4,235 09
1,611 2
2,84
1,904
1,345 26
0,706 522
0,555 403
0,443 186
0,707 902 б
1,6
0,77
0,548 064 86Д
0,66
0,836 521 3
0,60
0,55
0,54°
0,39°
1,51
1,99
22.5 б
—3,205 5 Д
66
102
21,894 б
—3,589 8 Д
64,0
98.6 а
9,688 б
—0,898 Д
30
45
9,4711 б
—1,171 5 Д
28
42,5
2,029
0,801
0,564
0,409
0,986 374 в 2,673
2.1 4,23
1.2 2,63
0,746 860 53 е 3,108
0,87
0,935 393 и
0,84
1,3
0,69 г
1,1 г
0,96 б
1,3 б
88.19 в
195,06 е
135
156
87,7 в
111,5 е'
130,0,ж
150,0 и
29,06 в
27,67 е
45
47
27.20 в
28,5 е
42 ж
44 й
1,175
0,340 9
0,215 9
0,144 8
3,10
2,936
3,73
4,16
3,127
4,014
1,695
1,599
1,595 8
2.590 О
1,95
2,38
1,595 3
2.591 О
1,95
2,378 !
1,595 2
2,580 6
1,95
2,38
1,594 9
2,585 7
1,95
2,378 6
1,563 892
2,020 671
2,170 4
2,391 9
Примечание. Ниже все постоянные даны в см~х.
'Li2: а о>Л=-5,301 28-10, <oeze= 1,195 55-10-*, о)е9е=—1,225 15-10~5, <м,=-3,778 1-10-', »Л=— 7,034 4-10-";
6 а2=-1,3081-10-8, а3=4,3904-10-в, а4=2,8655-10-', а5=1,4153-10-8, ав=2,0281-10-10; Bpj=— 2,9333X
ХЮ-8,Р2=7,644.10-9,Рз=1.2669.10-9,^=1,3843-10-10,В5=6,300-10-12, ^1,0404-Ю-13, He=i,55№-10-™,
fti=—1,039-Ю-13, Ь2=3,718-Ю-13, ^=7,124-10*, Л4=6,065-10-15, Л5=2,0716-10-», ?„=3,254-Ю-",
8Х=— 0,635-10-», 82=1,9718-10-17; г ШдУе=з,127 908 9-Ю, о)А=3,463 643 2-Ю, ueqe=—1,188 920 2Х
ХЮ~в; Д аа=2,005 072 7-Ю, а3=1,558 119 9-10-', а.=—1,122 497 0-10"8;' е ^=6,542 245 2-Ю-8, fi2=
=6,728 066 9-Ю-10, C3=—2,673 592 5-10"u, Яе=9,750 894 3-101, ftx=l,643 478 5-Ю2; ж шеуе=
=—1,077 54-Ю-3, й>А=3,65 074-10-3, ш,а.=1,550 68-10-*, а>А=4,248 44-10-8, mete=—1,05149-10"';
3 <xa=—1,731 71-10-*, а3=-1,4135-10-5, а,=—3,2263-10-«, а5=—2,2243-10"', ae=-7,3690-10"9;' и р1=
=—3,461 -10-', Р2=—1,8077-10-', Р3=-6,044-10-8, р,=—9,522-10"9, рв=-8,122-Ю0, p6=-3,5729-10-",
Р,=—6,708-Ю-", Яе=6,269-Ю-11,fe1=— l,469-10-10,ft2=—1,0205-10-10,Л3=—2,780-10-",^=—3,506-10-12,
А6=—1,9906-Ю-13, fee=—4,272-Ю-15, Z,e=—1,175-Ю-14, Sj=-4,065-10-14, S2=—2,556-10"Ч 83=-6,215Х
XlO-i5, 84=—6,074-Ю-16, 8в=—1,923-10-", ДГ,=—1,863-10-18, ^=-5,255-10-18, fi2= -3,233 -Ю'18,
fi3=-7,263-10-19, ^=-5,887-10-2», (л5=—1,0483-101,^=—0,843-Ю2, пх= -2,659-Ю2, га2=— 1,581Х
Х1О-22, ге3=—3,329-Ю-23, ге4=—2,115• 1Q-24.
250
Литий и его соединения Li|(r), 1Л\{т)
Примечание к таблице 45.4 (окончание)
'Lit: а шег/е=3,13 -10~3; б вычислено по соотношению A.38); в вычислено по соотношению A. 69); г
вычислено по соотношению A. 68); Д шеуе=—1,12-10~2.
LiO: а А=112; б вычислено по соотношению A. 68).
"L1H: а адв=0,14119; б а2=34,022-10-4; в ^=1,96-10, Я0=8,64-10-8; г шл/„=—2,114 970, co,z,=—1,244 ОЗХ
X1O~\ шеде=—4,4751-Ю-3, шА=-6,7457-10~5; Д а2=— 4,851-Ю"8, а3=— 9,057.10-*; е приведено
значение Do, Я1=28,2-10"8.
7ЫН: а a>,ye=0,21496; а>А=0,0056; б а =2,719-10-3, а3=9,4.10-5; в B,=2,2-10-5, P2=6,8-1O-', Я0=9,92-10-8;
Г ы„у,=*— 2,251 97, ш„г,=—0,143 271, а>е?е=—5,573.10, a>ese=—9,0978-Ю-5; Д а2=-6,2762-10-», а3=
=—2,7045-Ю; е приведено значение Do, Яо=6,6-10"'; ж вычислено по соотношению A. 68); 3 а2=
=—0,045, ^=—2,2-Ю; и Я,=3-10-«, /^=3,9-10-*.
eLiD: а а>9г/е=6,149-10-3; б а2=6,240-10-4; в ?х=*6,52Л0-*, Я0=2,0-10-8; г <о,у,=—1,051 26, <o,z,=-5,8566X
Х10-2, 0),^=— 2,0925-Ю-3, шА=-3,228-10; Д <х2=— 1,0069-Ю-3, as=— 3,229-Ю; е приведено
значение Dlt Я!=3,93 -Ю'8.
^LiD: а ш,уе=0,Ю7 780 8, »3z,=5,527 79-Ю-3, co9ge=2,655 13-Ю-4, шА=6,1875.10-«; б а2=2,0521-10-3, а3=
=2,1894-10-4, а4=1,2909-10-5, a5=2,034-10-'; в р1=4,83-10-в, Яо=1,68-10-8; г <овуе=-0,818 099,
u)eze=—0,036 63, а)г?,=—10,4532-Ю-4, шА=—1,282 55-10-*; Д а2=—1,3744-10-3, а3=— 2-10; е
приведено значение Dx, ^=6,6-Ю'8; ж вычислено по соотношению A. 68); * швг/е=3,509; и рх=—6,3-Ю,
Яо=1,3-10-«, ^5,6-Ю-6, Л2=—4,2.10-«.
LiF: а a2=l,557 74-10, a3=3,469 07-10; б ^=1,244-10-'.
LiCl: а а2=3,966 08-10-*; б р1==1,901-10-8.
LiBr: а »,г/е=0,02.
ЫГ. а to,j,=0,08.
с учетом одного возбужденного состояния. Величины Qmi. Бр и их производные для состояний
Х2Е+ и ^42ПЯ вычислялись по соотношениям A. 73)—A. 75) непосредственным суммированием
по колебательным уровням и интегрированием по вращательным уровням с помощью уравнений
типа A. 82). В расчете учитывались все уровни со значениями / ^ /шах> „, где /шах „
определялись из условий A. 81). Колебательно-вращательные уровни вычислялись по уравнениям
A. 65), A. 63а) со значениями коэффициентов Уы, приведенными вместе с величинами ушах и
Jlim в табл. 45.5а. Для соблюдения условия A. 59) добавлен коэффициент Y03. Все коэффициенты
соответствуют природной смеси изотопов лития и были получены по уравнениям A. 66) и
постоянным молекулы 'Lij. Мультиплетность и вырождение состояний Х2Е* и АШи учитывались
статистическими весами 2 и 4 соответственно.
Погрешности вычисленных термодинамических функций обусловлены главным образом
неточностью принятых молекулярных постоянных и составляют 0,2, 0,5 и 1 Дж-К'^моль в
значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций Li^r) публикуется впервые.
Константа равновесия реакции 1л2(г)+е(г)— 2Li (г) вычислена с использованием значения
АгН°@) = —398,2 + 5,1 кДж-моль-^ соответствующего принятому в справочнике потенциалу
яонизации
/0(Li2) = 499,2 + 5,0 нДж-моль «« 41 730 + 420 см.
Это значение было получено Мэйзе и др. [5013] методом двухфотонной лазерной фотоионизации
и хорошо согласуется с результатом менее точных фотоионизационных измерений [2312] D97 +
+ 10 кДж.моль), но заметно отличается от найденных методом электронного удара [411] D77 +
+ 10 кДж-мояь) и [4776] D67 +10 кДж-моль).
Принятому значению соответствуют
^Н°(Щ, г, 0) = 713,670+ 5,5 кДж • моль,
ZH(LiJ) = 122,022 + 5,1 кДж • моль «< 10 200 + 420 см.
Ыд(г). Термодинамические свойства газообразного положительного иона трилития в
стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 1049.
Молекулярные постоянные, использованные в расчете термодинамических функций Li3,
приведены в табл. 45.9. Экспериментальные данные о строении и спектрах Lij в литературе отсут--
251
Таблица 45.5а. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см), а также значения vmax
принятые для расчета термодинамических функций Li2, LiJ, LiO, LiF, LiCI, LiBr и Lil
Коэффициенты
T,
YM
У»о
У»
y«
У50
Ум
Уго
у»
Уо,
Уц
V
V
y«i
Уи
У«
Уо*
У«
у»
У«
У52
Ув2
Уо.
У13
Узз
У»з
у
Уо.
а, •
а3 •
а, •
10-'
• ю-3
. 103
• 10*
¦ 10s
• 10'
• 10»
• 10to
. 101
. 101
• 10»
. 10»
. 107
. 10"
. 1O10
. 105
• 10»
. 10»
• 10"
. 1010
• 10"
. 10"
.10»
. 10»
. 1013
• 101*
. 1015
. 10"
¦ 1O1S
= De)
105
108
10u
«шах
n
Li2: a
LiO: a
L
iF: a
LiCI: a
LiBr: a
Lil: a
Х1Ъд
0
3,533 021
-2,607 617
—7,884 989
2,848 907
—4,968 951
2,299 221
-5,909 321
5,758 413
6,798 748
-0,719 407
—1,336 524
-4,509 970
2,959 403
—1,469 555
2,117195
—1,007 807
-3,013 187
—7,894 491
1,315 466
-1,445 112
6,612 198
—1,097 842
0,160 730
1,078 830
3,881332
-7,477 031
6,399 854
—2,197 754
—1,972 241
• 10-* 0,862 023
1,285 876
-0,060 762
0,001 238
41
156
0,83
0,603 233
—2,830 257
—
—
—
_
—
_
2,708 961
—1,626 004
—
_
_
.—
—2,145 631
_
_
—
-2,702 028
.—
—
_
0,032 143
8,366 755
-5,182 055
1,280 228
10
46
Состояние В'Пи ¦sj
Г„ см-1 20 436,3 20 500
A = 112 см-1.
коэффициенты потенциальной
коэффициенты потенциальной
коэффициенты потенциальной
коэффициенты потенциальной
Li2a
Ь3Па
1,2
3,418 321
-2,527 015
.
—
_
—
—
_
7,025 103
-0,782 515
_
_
_
—
-1,226 075
_
_
_
_
—
0,179 754
_
_
_
_
_
_
—
_
_
—
67
179
'%
22 000
функции с
1,406 831
2,568 307
-1,592 674
2,218 166
0,380 216
-0,383 342
0,035 420
-0,001 462
_
5,027 989
-0,556 973
2,048 641
-0,160 055
-0,115 928
0,009 921
_
—0,763 089
6,720 421
-0,694 854
-0,027 761
_
_
—
0,100 704
-1,706 481
_
—
_
-1,397 793
—
—
—
—
72
194
ia = 153 977
функции d0 — 145 035
функции с
функции
о = 142 756
i0 = 139 9S8
1,
2,
-2,
5,
-0,
-0,
0
CM
CM*
ЗМ",
см-
67
412 932
122 693
_
_
—
—
—
054 030
670 727
—
—
_
—
_
888 904
_
—
_
_
—
—
094 244
—
_
—
—
—
_
—
—
—
—
56
160
d, = 2
d1==2
d, =2
d, = 5
L
X'lig
0
2,634 118
-1,950 855
3,180 87
_
4,963 056
—0,548 776
-0,704 992
_
_
0,068 768
_
_
—
_
—
—
—
-
84
203
,700 64, d,=
,71213, d2 =
716 17, d2 =
,728 97, d2 =
A
2
0
-0
-11
3
-0
-1
0
:
34
985 281
880 412
382 02
—
.
_
_
_
012 201
396 338
_
—
.
,123 901
_
—
_
,382 055
—
—
_
—
—
_
—
-
3?
112
= 5,101.
= 4,958.
: 4,985.
= 4,210.
X
0
8,
-7,
22,
12,
—1,
0,
-0,
-0
LiO
412 163
822 171
072 22
_-
_
119 33
526 850
975 721
_
974 310
_
_
_
105 233
_
_
—
—
_
—
—
—
82
245
A
0
8
-6,
13,
13,
—2
7,
0
'S+
233 0
677 559
009 345
602 29
—
_
_
_
62040
012 207
522 050
319 379
—
078 158
—
126
258
LiPa
2
0
9
-8
13
2
15
-1
12
,138 35
,167 95
_
,558 74
,052 74
,824 3
,193 97
,589
_
_
—
115
366
LlGla
xw
0
6,423 67
-4,52017
_
_
7,112 69
—0,089 046
4,019 55
-0,345 465
1,933 4
—
—
—
123 -
428
LiBra
0
5
-3
20
5,
-0,
-0,
656 65
56148
2681
—
_
603 56
571 584
—
_
—
219 716
_
_
—
—
—
_
—
—
—
—
—
112
443
Lil1
ХЧ+
0
5,007 04
-3,424 71
81,2319
—
_
_
_
4,477 24
-0,415 315
_
_
_
_
—
-0,147 781
_
_
_
—
—
—
_
_
_
—
_
_
_
—
—
—
—
117
415
Таблица 45.56. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см"), а также значения vm№ a Jlm,
принятые для расчета термодинамических функций LiH, 7LiH и 6LiH
N5
Коэффициенты
Тв-10-*
У,о-10-»
У.о-10-1
У«о
У40-Ю2
У.о-Ю3
у«-ю5
У,о-Ю'
Уо,
У„
Y.i-10»
Уз,-Ю'
Г„-10«
У»,-10»
У„2-103
У„,-Ю«
Ум-Ю«
(ae=D,)-i0-*
а,-Ю'
о3-104
а4-10'
Jlim
0
1,406
—2,340
0,255
—1,747
1,119
—3,478
—
7,530
—0,224
6,487
—9,229
7,256
— 1,524
—0,859
0,094
—0,001
2,029
891
288
541
606
312
191
238
553
699
757
561
622
926
438
084
197
0,032 626
—0,000
0,000
23
74
715
070
2,651
0,246
2,109
—1,642
7,602
—1,880
0,707
2,556
2,864
0,027
—4,345
0,843
—
—
—0,901
0,371
—0,016
0,868
0,000
—2,000
—
32
U5
LiH
335
447
762
004
175
390
411
344
808
775
156
131
025
007
855
239
965
009
а'П
3,317
0,622
—5,282
—
—
—
5,049
—0,663
—
—
—1,327
—0,590
0,183
0,527
—0,101
0,076
5
26
000
076
512
686
861
827
721
137
713
670
571
В'П
3,490
0,263
—7,986
8,319
—
—
3,385
— 1,007
—.
—
—1,391
7,307
5,959
0,029
2,026
—1,922
6,655
2
12
913
287
328
601
424
646
057
713
090
130
504
631
741
0
1,405
—2,337
0,255
— 1,742
1,115
—3,464
7,520
—0,224
6,470
—9,198
7,227
—1,517
—0,857
0,094
—0,001
2,029
0,032
—0,000
0,000
23
74
937
115
022
872
522
066
031
097
122
509
091
400
596
055
078
197
537
712
070
А'?+
2,651
0,246
2,106
—1,638
7,581
—1,874
0,704
2,544
2,860
0,027
—4,333
0,840
—0,898
0,369
—0,016
0,868
0,000
—0,000
—
31
115
'LIH
335
280
902
666
577
023
538
235
925
719
383
277
584
500
764
239
962
009
a3n
3,317
0,621
—5,275
5,.042
—0,662
—1,324
—0,588
0,183
0,526
—0,101
0,076
5
26
000
654
451
842
511
229
322
137
283
257
157
В'П
3,490
0,263
—7,975
8,302
'
3,380
— 1,005
—1,387
7,278
5,926
0,029
2,021
—1,914
6,619
2
12
913
109
502
689
835
598
288
035
845
130
013
823
724
0
1,420
—2,385
0,263
—1,816
1,174
—3,685
7,677
—0,231
6,743
—9,686
7,689
—1,631
—0,893
0,100
554
963
058
488
727
843
206
159
410
712
784
333
820
076
—0,001171
2,029
0,033
—0,000
0,000
23
74
197
912
758
076
A'S+
2,651
0,248
2,150
—1,690
7,901
—1,973
0,749
2,735
2,920
0,028
—4,516
0,884
—0,936
0,393
—0,018
0,868
0,001
—0,000
—
30
114
«LiH
335
840
938
308
812
486
644
267
721
593
418
874
539
156
210
239
003
010
asn
3,317
0,628
-5,385
—
—
—
—
—
5,148
—0,683
—
—
—1,380
—0,625
0,183
0,548
—0,107
0,082
5
26
000
117
713
241
390
163
987
137
512
739
726
В'П
3,490
0,265
—8,142
8,564
—
—
—
—
3,451
—1,037
—
—
—
—
—1,445
7,743
6,438
0,029
2,106
—2,037
7,190
2
12
913
844
197
344
497
289
885
990
101
130
378
414
750
Таблица 45.5в. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см), а также значения vmb,% и Jiim,
принятые для расчета термодинамических функций LiD, 7LiD и eLiD
Коэффициенты
Т..10-
Уи-Ю-»
Уго-Ю
Узо
У40-Ю2
Убо*!
Y...10*
Yoi
Y..-10'
у"-ю*
Ум-Ю'
у"-ю«
(аГ=Ое)-10-'
аа-10»
а4-108
«шах
Jlim
Z'B+
0
1,056
—1,319
0,108
—0,555
2,671
—6,232
—
4,245
—0,950
2,062
—2,202
1,300
—2,051
—2,733
0,016
—0,001
2,029
0,103
—0,001
0,000
31
99
356
377
171
447
159
350
298
536
005
617
257
211
128
922
095
197
695
281
071
А'ЗИ
2,651
0,185
1,189
—0,695
2,416
—4,487
1,267
3,439
1,615
0,117
— 1,381
0,201
—
—
—2,863
0,066
—0,017
0,868
0,003
—0,000
—
41
154
LID
335
043
414
062
222
417
565
278
084
574
034
207
756
478
027
239
067
016
о'П
3,317
0,467
—2,978
—
—
—
—
—
2,846
—2,810
—
—
—
—
—4,220
—0,105
—
0,183
1,677
—0,182
0,077
7
34
082
161
846
130
271
848
137
245
175
351
В'П
3,490
0,197
—4,502
3,521
—
—
—
—
1,908
—4,265
—
—
—
—
—4,421
1,309
6,019
0,029
6,440
—3,445
6,723
3
16
913
688
427
695
589
376
237
423
743
130
897
042
483
ХЧ+
0
1,055
—1,316
0,107
—0,552
2.655
—6,187
—
4,235
—0,947
2,052
—2,189
1,290
—2,034
—2,720
0,016
—0,001
2,029
0,103
—0,001
0,000
31
99
085
204
781
779
130
499
090
НО
100
400
900
000
0
8
084
197
197
272
070
A'S+
2,651
0,184
1,186
—0,692
2,404
—4,460
1,258
3,410
1,611
0,117
—1,374
0,2
—
—
—2,85
0,066
—0,016
0,868
0,003
—0,000
—
41
154
T^D
335
820
553
556
616
490
443
420
200
150
400
864
239
052
016
а'П
3,317
0,466
—2,971
—
2,84
—2,8
—
-4,2
—0,105
—
0,183
1,669
—0,180
0,076
7
34
520
086
137
189
864
609
В'П
3,490
0,197
—4,491
3,509
1,904
—4,25
—
—4,4
1,3
5,962
0,029
6,409
—3,420
6,659
3
16
913
450
6
051
130
960
249
047
0
1,074
—1,365
0,113
—0,594
2,908
—6,902
4,392
—1,000
2,207
485
053
836
571
381
296
268
322
246
—2,398 228
1,440 028
—2,310
—2,925
0,018
—0,001
2,029
0,110
—0,001
0,000
30
97
694
642
741
255
197
999
419
081
A'S+
2,651
0,188
1,230
—0,731
2,586
—4,885
1,403
3,874
1,670
0,123
—1,478
0,219
—
—3,065
0,073
—0,019
0,868
0,003
—0,000
-
40
151
•LiD
335
219
590
466
413
940
822
355
997
732
310
076
471
624
510
239
283
018
о'П
3,317
0,475
—3,081
—
—
2,945
—2,957
—
—4,517
—0,117
0,183
1,795
—0,201
0,088
7
34
098
264
402
313
535
226
137
386
758
631
В'П
3,490
0,201
—4,658
3,706
—
—
—
1,974
—4,488
—
—4,732
1,450
6,897
0,029
6,894
—3,815
7,704
2
16
913
081
298
146
663
778
656
179
636
130
576
367
006
а лица 45.5г. Значения' ««ФФвднтон в уравнениях, описывающих уровни энергии (в см), а также значения гшл% и J,
принятые для расчета термодинамических функций LiT, 7LiT и 6ЫТ
Коэффициенты
ел
У«о
Уго
Уо,
Уц
Уи
у»
У«
•10е
•10»
•10»
•10«
•10«
•10«
у„2-ю«
a2-10«
9,102 522
—0,979 652
0,069 209
—0,306 231
1,268 987
—2,551 298
3,152 182
—0,608 168
1,136 831
—1,046 397
5,322 780
—7,235 542
—1,506 836
0,069 272
—0,000 333
2,029 197
0,057 169
—0,000 525
0,000 021
2,651 335
1,594 499
0,883 153
-0,444 711
1,332 119
-2,131 838
0,518 895
1,213 188
1,199 218
0,075 226
-0,761 396
0,095 588
-1,578 855
0,272 138
-0,005 175
0,868 239
0,001 691
-0,000 007
3,317
4,024 803
—2,211 318
2,113 815
1,797 964
2,326 733
0,433 301
—
0,183 137
0,924 704
—0,074 576
0,023 511
S
40
ВЧ]
3,490
1,703
—3,343
2,253
—
—
—
—
1,417
—2,729
—
—
—
—2,437
5,360
1,829
0,029
3,551
—1,410
2,043
3
19
913
458
103
234
149
053
530
302
744
130
015
275
650
ЛГ'2И
0
9,087
—0,976
0,068
—0,304
1,258
—2,526
—
3,141
—0,605
1,129
—1,037
5,271
—7,153
—1,497
0,068
—0,000
2,029
0,056
—0,000
0,000
36
115
770
480
873
251
739
590
974
216
480
946
235
863
093
601
329
197
800
520
021
A'SH
2,651
1,591
0,880
—0,442
1,323
—2,114
0,513
1,199
'LiT
-
335
915
293
553
505
621
870
493
1,195 334
0,074
—0,756
0,094
—
—1,568
0,269
—0,005
0,868
0,001
—0,000
—
48
179
860
472
816
645
503
109
239
680
007
а?п
3,317
4,018
—2,204
—
—
—
—
281
157
2,106 970
—1,789
—
—
—2,311
—0,429
—
0,183
0,918
—0,073
237
687
105
137
725
854
0,023 208
8
40
3
1
—3
2
1
2
—2
5,
1
0,
3,
—1
2
ВЧ
,490
,700
,332
,242
,412
,715
,421
308
806
029
528
,396
017
3
19
I
913
698
276
298
560
806
768
393
157
130
053
618
306
Х'Я+
0
9,312
305
—1,025 328
0,074
—0,335
1,422
—2,925
3,299
—0,651
1,245
—1,172
6,102
—8,486
—1,650
0,079
—0,000
2,029
0,062
—0,000
0,000
35
112
106
452
115
053
151
193
310
665
548
710
623
420
399
197
S25
601
026
А'Е+
2,651
1,631
•LiT
335
247
0,924 330
—0,476
1,459
—2,389
0,594
1,422
1,255
0,080
—0,834
0,107
—1,729
0,312
—0,006
0,868
0,001
—0,000
—
47
174
172
233
084
911
972
131
547
050
122
513
506
210
239
352
008
3,317
4,117 561
—2,314 420
_
2,212 371
—1,925 162
—2,548 756
—0,496 778
0,183 137
1,012 942
—0,085 501
0,028 212
8
39
3
1
—3
2
1
—2
—2
6
2
0
3
—1
2
ВЧ1
,490
,742
,498
,412
,483
,922
670
145
195
029
889
616
452
з
18
913
717
973
641
223
122
125
568
603
130
863
875
281
LiO(r) Глава 45
ствуют. Квантовомеханические расчеты [4954, 4967, 4969, 5030] показали, что молекула LiJ
в основном электронном состоянии Xx.4j имеет структуру правильного треугольника (точечная
группа Du). Произведение моментов инерции, приведенное в табл. 45.9, вычислено на основании
значения r(Li—Li) = 3,084 + 0,01 А, полученного в наиболее точном неэмпирическом расчете
Гола и др. [4967]. Его погрешность не превышает 3-Ю16 г3-см6. Основные частоты vx и v2
приняты по результатам полуэмпирического расчета Эллисона и Дель Донне [4954], погрешности
этих значений могут достигать 10—20% от их величины.
Термодинамические функции Lij(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A.122)—A.124), A.128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор».
Погрешности рассчитанных термодинамических функций обусловлены неточностью принятых
значений молекулярных постоянных, а также приближенным характером расчета и составляют 2,
6 и 8 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций LiJ(r) публикуется впервые.
Константа равновесия реакции LiJ(r) + е (г) = 3Li(r) вычислена с использованием значения
АгН°@) = —277,795 + 10 кДж-моль, соответствующего принятой величине энтальпии
образования
A,ffo(LiJ, г, 0) = 751 ± 10 кДж • моль.
Эта величина основана на квантовомеханическом расчете Гола и др. [4967], которые
получили для изменения энтальпии в реакции Lig(r) = Li?(r) + Li(r) значение Дг#°@) = 120 +
+ 10 кДж-моль (с учетом нулевых колебательных энергий Lig и LiJ).
LiO(r). Термодинамические свойства газообразного оксида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1050.
Молекулярные постоянные LiO, принятые на основании результатов экспериментальных
исследований и квантовомеханических расчетов, приведены в табл. 45.4. Теоретические расчеты [3284,
4803], исследования отклонения молекулярного пучка LiO в неоднородном электрическом поле
[1489] и изучение микроволнового спектра LiO [2333] показали, что основным электронным
состоянием этой молекулы является состояние Х2П{ (электронная конфигурация ..ЛоЧп3),
коррелирующее с атомами LiBS) и O(SP). Согласно расчету [3284] молекула LiO имеет по крайней
мере одно стабильное состояние Л2Е+ (электронная конфигурация ...4axlu4) с очень низкой
энергией.
Молекулярные постоянные LiO в состояниях Х2П{ и42Е+ приняты по квантовомеханическому
расчету Иошимина [4803], выполненному на расширенном базисе и с учетом конфигурационного
взаимодействия. Погрешности принятых значений Те, ше и В„ не превышают 100, 10 и 0,02 см
соответственно. Значение Д&/2 = 826,5 см, соответствующее колебательным постоянным из
табл. 45.4, существенно превышает частоты 745 см [4721] и 700 см [4364], наблюдавшиеся
в ИК-спектре молекул LiO, изолированных в матрицах из криптона и азота. Вероятно, это
объясняется большим матричным сдвигом, возможным для сильно полярной молекулы LiO.
Постоянная спин-орбитального взаимодействия в состоянии Х2П.{, приведенная в табл. 45.4, получена
Фреундом и др. [2333] при исследовании микроволнового спектра LiO.
Термодинамические функции LiO(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?„„ и ее производные вычислялись по соотношениям A. 90)—
A. 92) с учетом состояний Х2П( и Л2Е+. Компонента X2Hi/2 основного состояния учитывалась как
возбужденное состояние с энергией Те = А и с условием, что <?к01- вр (Х2Пу3) = (?кол вр (X2H^2).
Статистические суммы по колебательно-вращательным уровням энергии состояний ХЧЬ/, и А2И +
и их производные находились непосредственным суммированием по колебательным уровням и
интегрированием по / с помощью соотношений типа A. 81). В расчете учитывались все уровни
со значениями / ^ /Шах, «> последние находились по формуле A. 82). Уровни энергии
вычислялись по уравнениям A. 64а) и A. 65) с коэффициентами Ykl, полученными для природной смеси
изотопов лития по соотношениям A. 66) и данным табл. 45.4. Для обеспечения условий A.50)
и A. 59) добавлены коэффициенты Y30 и Y03 для обоих состояний. Постоянные Ykl вместе
со значениями ушах и /llm приведены в табл. 45.5 а. Мультиплетность и вырождение состояний
Х2Пз/2, Х2Пч2 и А2И + учитывались статистическим весом 2.
256
Литий и его соединения
1л2О(к, ж)
Погрешности рассчитанных термодинамических функций LiO(r) обусловлены главным
образом неточностью молекулярных постоянных основного электронного состояния Х2И{, энергии
возбуждения состояния A2L+ и отсутствием данных о других состояниях; они составляют 0,1,
1 и 3 Дж-К^-моль-1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции LiO(r) ранее вычислялись в предыдущих изданиях настоящего
справочника [337, 340], а также в справочниках JANAF [2834], Мак-Брайд и др. [3439] и
работах Уилкинса и др. [4729] и Брюэра и Розенблатта [1666]. Расхождения данных [2834] и табл. 1050
не превышают 3 и 5 Дж-К-моль~1 в значениях Ф°(Т) и S°(T), они обусловлены главным образом
тем, что авторы [2834] не учитывали состояние А2!,. Расхождения с результатами других расчетов
достигают 8—9 Дж-К~1-моль~1 в значениях Ф°(Г), в частности из-за выбора другой симметрии
для основного состояния этой молекулы.
Константа равновесия реакции LiO(r) = Li(r) + О(г) вычислена с использованием принятого
в справочнике значения
Дг#°@) = D0(LiO) = 332 ± 8-,кДж • моль^ 27 750 ± 700 см.
Принятая величина является средней из приведенных в табл. 45.6 результатов исследований
равновесия реакций с участием LiO(r), за исключением выпадающей величины, вычисленной
Таблица 45.6. Результаты определений P0(LiO) (B кДж • моль)
Авторы
Метод
D0(LiO)
II закон
III закон
Берковиц и др., 1959 [1503] Масс-спектрометрический, LiO(r)=Li(r)-j-V2O2(r), 354+90 322+9
Щ 1346—1470 К, 6 точек
Уайт и др., 1963 [4721] То же, Li2O(r)=Li(r)+LiO(r), 1500 К, 1 точка — 332+13
|f То же, 1430—1630 К, И точек 312+50 —
Даугерти и др., 1971 [2101] Спектрофотометрия в пламенах CO+N2-j-O2, LiO(r)+ — 339+13
г. +СО(г)=П(г)+СО2(г), 2295 К, 2 измерения
Хилденбранд, 1972 [2641] Масс-спектрометрический, Li(r)+O2(r)=LiO(r)-f О(г), 334+70 332+10
1772—1913 К, 10 точек
То же, Li2O(r)+O(r)=2LiO(r), 1772—1913 К, 9 точек 332±50 338+8
То же, Li2O(K)=LiO(r)+Li(r), 1500 К, 1 точка — 335+8
Кудо и др., 1978 [3134] То же, Li2O(r)=Li(r)+LiO(r), 1352—1663 К, 16 точек 349±25 361+15
Кимура и др., 1980 [3005] То же, LiO(r)=Li(r)+1/2O2(r), 1329—1507 К, 9 точек 392+30 329+8
по данным [3134]. Приведенные в таблице погрешности включают воспроизводимость измерений
и погрешности, связанные с неточностью использованных в расчетах термодинамических функций,
энтальпий образования и сечений ионизации.
Принятой энергии диссоциации соответствует
r, 0) = 72,518 + 8,1|кДж • моль.
1Л2О(к, ж). Термодинамические свойства кристаллического и жидкого оксида дилития
в стандартном состоянии при температурах 100—4000 К приведены в табл. 1051.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние 1Л2О(к) в интервале 0—1726 К принята
кубическая модификация (структурный тип флюорита, CaF2).
При Т ^ 298,15 К термодинамические функции 1Л2О(к) вычислены по результатам измерений
теплоемкости, проведенных Джонстоном и Бауэром [2882] A6—320 К, образец чистотой 99,74%,
погрешности измерений при 20К~2%, при Т > 45 К 0,2%). Экстраполяция теплоемкости
по закону Дебая привела к значению ?°A6 К) = 0,013 Дж-К-моль. Погрешности принятых
значений 5°B98,15 К) и Я°B98,15 К)-Я°@) (см. табл. 45.1) составляют 0,13 Дж-К-^моль
и 0,02 кДж-моль*1 соответственно Ч
1 Принятое значение <S°B98,15 К) на 0,29 Дж-К^-моль ниже величины, полученной в работе [2882], так как
при интегрировании в интервале 100—200 К авторами [2882] была допущена арифметическая ошибка, которая
не была исправлена в справочнике [2834].
257
Ы2О(к, ж) Глава 45
Уравнение для теплоемкости Ы2О(к) в интервале 298,15—1726 К выведено совместной
обработкой результатов измерений энтальпии, проведенных Кандыбой и др. (см. [337, т. 1, с. 876])
(985—1667 К), Шомейтом и Коэном [4272] D25—1045 К) и Родигиной и Гомельским [883] C73—
1073 К). При обработке погрешности перечисленных выше данных оценивались в 2, 0,5 и 1%
соответственно. Значения теплоемкости, рассчитанные по принятому уравнению, хорошо
согласуются с измерениями Танифужи и др. [4473] C06—1073 К, адиабатический сканирующий
калориметр).
Температура плавления A726 + 3 К) принята по согласующимся между собой
термографическим данным [1627] A726 К) и [3762] A729 К). Измерения в работах [1663, 3766] проводились
на менее чистых образцах и привели к заниженным значениям A700—1705 К); данные более
ранних работ [1343, 3118] A843—1900 К) ненадежные и не учитывались. Энтальпия плавления C5,6 +
+ 2 кДж-моль) рассчитана из данных по энтальпии жидкой окиси лития, полученных
Кандыбой и др. (см. [337]) A746—2015 К) с учетом уравнения, принятого для энтальпии 1л20(к).
Значение ЬтН°, полученное методом ДТА в работе [1627] C5,6 кДж-моль"), согласуется с
принятым значением. Теплоемкость 1Л20(ж) A04 + 5 Дж-К~1-моль~1) принята по результатам
четырех измерений энтальпии в работе Кандыбы и др. (см. [337]) A746—2015 К).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000, 3000 и 4000 К оцениваются
в 0,1, 0,5, 2, 6 и 10 Дж-К~1-моль~1 соответственно.
Ранее термодинамические функции 1л20(к, ж) вычислялись в двух первых изданиях
настоящего справочника [340] и [337] (до 5300 К) и в таблицах JANAF [2834] (до 4000 К).
Термодинамические функции, приведенные в [337] и табл. 1051, согласуются в пределах 0,4 Дж-К~1-моль~1
в значениях Ф°(Т). Расхождения с данными [2834] достигают 3 Дж-К~1-моль~1 (в значениях
Ф°C000 К)), что обусловлено в основном различием принятых значений АтН° и C^(Li2O, ж).
В справочнике принята энтальпия образования
^H°{Li2O, к, 298,15 К) = — 597,88 + 0,30 нДж-моль.
Эта величина основана на измерениях энтальпии растворения 1л20(к) в воде, представленных
в табл. 45.7. Получение чистогр образца Li2O вызывает большие затруднения. Наиболее тщательно
Таблица 45.7. Результаты измерений A.aqH°(Li20, к->ооН2О, 298,15 К) (в кДж • моль)
Авторы
Конечная концентрация, температура
измерений
Дй,Я°(ооН2О, 298,15 К)
Форкран, 1907 [2296] 220 Н2О, 3 измерения —134,5
Колесов и др., 1959 [534] 2200 Н2О, 292,9 К, 5 измерений —132,02+0,35; (—133,42)
Джонсон и др., 1975 [2873] 298,15 К, 6 измерений8 —133,21 +0,17
а Измерялась энтальпия реакции 1л2О(к)+1,509AЛОН -1106,84 Н2О)=3,509(ЫОН -475,70 Н2О).
образцы готовились в работах Колесова и др. [534] и Джонсона и др. [2873]. В работе [534]
образец был получен прокаливанием карбоната лития в течение 70 часов в вакууме при 973 К.
Анализ на карбонат-ион проводился только качественно. В работе [2873] степень превращения
карбоната в окись была исследована количественно и было показано, что при прокаливании в
условиях, близких к имевшим место в работе [534], в образце остается около 0,35 мол. % карбоната.
Более длительным нагреванием при 1023 К удалось снизить содержание карбоната до 0,10%.
Как видно из таблицы, результаты измерений в работах [534] и [2873] различаются больше, чем
это можно объяснить их погрешностями. Однако если принять, что в образце, использованном
в [534], было 0,35% карбоната (величина в скобках), то согласие оказывается хорошим.
Принятое в справочнике значение энтальпии образования 1Л2О(к) было вычислено с использованием
величины AaqH°(ooH20; 298,15 К) = —133,25 + 0,17 кДж-моль, средневзвешенной из
работ [2873] и [534] (скорректированная величина).
Менее точные величины были найдены по измерениям Бекетова [1455] (растворение 1Л2О,
содержавшего до 12% карбоната, в соляной кислоте, —620 кДж-моль) и Фазолино (см. [2834])
(сжигание лития в кислороде, —590 + 18 кДж-моль). Исследования равновесия реакции
21ЛОН(к, ж) = Ы20(к) + Н2О(г) были выполнены Джонстоном [2883] (статический метод, 793—
258
Литий и его соединения
Ы2О(г)
—1176 К, 10 точек, —598,7 кДж-моль х), Дитмарсом и Джонстоном [2084] (эффузионный метод,,
648—795 К, 18 точек, —563.2 кДж-моль) и Грегори и Мором [2481] (эффузионный метод, 530—
660 К, —592,5 кДж-моль). Как видно из соответствующих значений энтальпии образования
1Л2О(к) (расчет по уравнению III закона), эти измерения приводят к плохо согласующимся
величинам, что, возможно, связано с не учитываемыми в расчетах коэффициентами активности
компонентов конденсированной фазы в расплаве, а также высокой агрессивностью расплава.
Давление пара в реакции 1Л2О(к, ж) = Li2O(r) вычислено с использованием принятой в
справочнике энтальпии сублимации
[.; ДгЯ°@) = ДД"(LijO, к, 0) = 425 + 10 кДж • моль,
основанной на приведенных в табл. 45.8 результатах исследований давления пара. В расчетах по
данным работ [715, 1663, 2646, 4721 ] было учтено частичное испарение 1Л2О в виде Li и О2.
Приведенные в табл. 45.8 погрешности включают только воспроизводимость измерений. Результаты
Таблица 45.8. Результаты определений Д8Ж°(Ь13О, к, 0) (в кДж-моль)
Авторы
Брюер, Маргрейв, 1955 [1663]
Берковиц и др., 1959 [1503]
Несмеянов, Белых, 1960 [715]
Хилденбранд и др., 1963 [2646]
Уайт и др., 1963 [4721]
Кудо и др., 1978 [3134]
Икеда и др., 1979 [2771]
Кимура и др., 1980 [3005]
Метод
Эффузионный, 1541—1669 К, 3 точки
Масс-спектрометрический, 1248—1534 К, 13 точек
Эффузионный, 1383—1506 К, 9 точек
Торзионный, 1494—1669 К, 21 точка
Эффузионный, 1477—1662 К, 25 точек
Масс-спектрометрический, 1352—1663 К, 16 точек
То же, 1316—1608 К, 32 точки
То же, 1225—1507 К, 15 точек
То же, LiaO(r)=2Li(r)+0,5O2(r), 1329—1507 К,
9 точек
Д,Я°AЛ2О, к, 0)
II закон
283+200
471 ±12
377+38
386+19
402 ±20
424 ±10
402+16
458±16
439+55
III закон
421+14
453 ±1
406,1+1,1
427,8 ±0,8
427,2 ±1,2
420,9 ±0,5
425,0 ±1,0
458,2±1,0
461,1 ±2,0
масс-спектрометрических измерений включают дополнительную погрешность порядка 6 кДжХ
Хмоль, связанную с неточностью используемых в расчетах сечений ионизации. Как видно из
таблицы, результаты измерений разделяются на две группы. Первая группа [715, 1663, 2646а
2771, 3134, 4721] приводит к средневзвешенному значению ~425 кДж-моль. По данным работ
[1503, 3005] получена более высокая величина ~457 кДж-моль. Более убедительными
представляются результаты работ первой группы. Погрешность рекомендованного" значения оценена
с учетом неточности термодинамических функций.
1Л2О(г). Термодинамические свойства газообразного оксида дилития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1052. ?
Молекулярные постоянные, использованные для расчета термодинамических функций Li2O,
приведены в табл. 45.9. На основании исследований инфракрасного спектра [4238, 4364, 4366,
4721] и отклонения молекулярного пучка в электрическом поле [1708], а также квантовомеха-
нических расчетов [1713, 2507, 4628] для молекул Li2O в основном электронном состоянии X1}}*
принята линейная симметричная структура (симметрия Dmh) 1. Приведенный в табл. 45.9 момент
инерции соответствует значению r(Li—О) = 1,60 + 0,03 А, найденному Толмачевым и др. [1020]
в результате электронографического исследования пара Li2O. Погрешность принятого значения /
составляет 4'10~40 г«см2. Приведенные в табл. 45.9 основные частоты v2 и v3 найдены Сешадри
и др. [4238, 4721] при исследовании ИК-спектра молекул 1Л2О, изолированных в матрице из Кг.
С принятой величиной v3 удовлетворительно согласуется частота v3, измеренная Спикером и Энд-
рюсом [4364] в ИК-спектре Li2O в матрице из N2(945,6 см). Частота vx рассчитана по
уравнениям простого поля валентных сил. Погрешности принятых значений v2 и v3 с учетом матричного
р
сдвига оцениваются в 20 см, а погрешность частоты
в 100 см
1 Результаты электронографического исследования [1020] не противоречат этому предположению. !'т;\, ''. |;.
259
Ii2O+(r)
Глава 45
Таблица 45.9.
Молекулярные постоянные, а также о и р%, принятые для расчета термодинамических
функций Li+, Li2O, Li2O+, Li2O2, LiOH, Li^O2H2, LiNO2, LiNO3, LiBO2 и LiAIF4
Молекула
Состояние
^2
CM
Li? fr-Ai 370 260B) —
Li2O Z1 Ц a 720 112 B) 986
LiaO+ X22+ 650 100B) 900
Li2O2 %xAg 850 740 640
LiOH %ГЦ 850 370B) 3700
Li2O2H2 P-Ag | 3700 700 450
LiNO2 %XA{ 1330 790 530
LiNOg %хАг 1524 1011 736
LiBO2 XlA' 1990 1098 580
1 LiAlF4 &At 817 608 560
Li2O: a аЩа, АЩи, Ге=10 000 см", о=2, р=8; 53ПИ
—
—
100
—
320
1205
528
573
220
вти, т
=18 000 см, а=1, р=6; Cm, Г„=18 000 см, о = 1
Li2O+: а приведено значение / -1039 г -см2.
LiOH: а приведено значение / -1039 г -см2.
Li2O2Ha: a v,=300, v8=310, ve=310, vlo=52O, v
LiNO3: a v,=350, v8=817, v9=150 см.
LiAlF4:a v,=644, v8=350, v9=450, vlo=9H, %
n=3700,
u=160,
—
796
—
440
350
1264
472
390
„=14 000,
, p=2, °
v12=670
v12=316
_
—
—
446
—
310 a
150
765 a
108
200 a
см, o=2
fit 10117
г3 • см6
3,3 -102
5,9б
7а
1,4-102
2,363 а
5,1 -102
1,9 -102
1,2-103
2,3 -102
2-104
0
6
2
2
4
1
4
2
2
1
2
, p=8; Li—Li-O, 3П,
приведено значение / -1039 г -си
CM~l.
см.
Pt
1
1
2
1
1
1
1
1
1
1
Te=
i2.
Теоретический расчет Гроу и Питцера [2507] показал, что молекула должна иметь 4
стабильных возбужденных состояния, а также изомер LiLiO и его возбужденное электронное состояние.
Поскольку значение потенциала ионизации /0(Li2O), полученное в работе [2507], на 14 500 см"
ниже найденного экспериментально авторами [2641], энергии электронных состояний,
приведенных в табл. 45.9, увеличены по сравнению с найденными в [2507] пропорционально
отношению /0(эксп.)//0(расч.), их погрешности могут достигать 3000—6000 см.
Термодинамические функции 1Л2О(г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A.10), A. 122)-A. 124), A.126),
A. 129) с учетом шести возбужденных электронных состояний по уравнениям A. 168.)—A. 170).
Погрешности рассчитанных термодинамических функций обусловлены неточностью принятых
молекулярных постоянных, а также приближенным характером расчета и составляют 4, 10 и
15 Дж-К-моль~1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно (в том числе 3—
8 Дж-К-1-моль~1 из-за неточности молекулярных постоянных).
Ранее термодинамические функции 1л2О(г) рассчитывались в предыдущих изданиях
настоящего справочника [337, 340], справочниках JANAF [2834], Мак-Брайд и др. [3439], а также
в работах Уилкинса и др. [4729] (до 5000 К) и Гордона [2445] (до 6000 К). Во всех расчетах, кроме
[2834], для молекулы LiaO была принята угловая структура по работе Акишина и Рамбиди [13],
иГрасхождения с данными табл. 1052 достигают 11—30 Дж-К~1-моль. Расхождения с данными
[2834] (до 10 Дж-К~1-моль в значении Ф°F000 К)) обусловлены тем, что авторы таблиц JANAF
не учитывали возбужденных состояний молекулы Li2O.
Константа равновесия реакции Li2O(r) = 2Li(r) + О(г) вычислена с использованием
значения АгН°@) = 728,779 + 10 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации 1л2О(к). Этим величинам также соответствует
Д,Яe(LiaO, г, 0) = —166,526 + 10 кДж . моль.
1Л2О+(г). Термодинамические свойства газообразного положительного иона дилития в
стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 1053.
Молекулярные постоянные, использованные для расчета термодинамических функций Li2O+,
приведены в табл, 45.9. Экспериментальные данные о структуре и частотах колебаний Li2O +
в литературе отсутствуют. В настоящем справочнике по аналогии с молекулой Li2O принимается,
260
Литий и его соединения 1Л2О2(к)
что Li2O + в основном электронном состоянии Х2Е+ имеет линейную симметричную структуру
(симметрия Dmh). Приведенные в табл. 45.9 постоянные были оценены по принятым постоянным
молекул Li2O, Н2О, Н2О+ в предположении, что соотношение между постоянными Li2O и Li2O*
такое же, как у Н2О и Н2О+. Момент инерции Li2O+ рассчитан со значением r(Li—О) = 1,7 +
+ 0,1 А, его погрешность оценивается в 1-Ю9 г-см2. Погрешности в значениях vx, v2 и v3
составляют 200,25 и 250 см. Молекула Li2O+, так же как молекула LiO, должна иметь электронные
состояния с низкими энергиями возбуждения. Однако обоснованная оценка их энергии
невозможна, и в связи с невозможностью корректной оценки постоянных Li2O+ даже в основном
состоянии возбужденные состояния этой молекулы в справочнике не рассматриваются.
Термодинамические функции 1Л2О+(г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126),
A. 129) без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены грубой оценкой молекулярных постоянных Li2O+,
пренебрежением возбужденными состояниями, а также приближенным характером расчета; они
составляют 10, 15 и 20 Дж-К^-моль-1 в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций Li2O+(r) публикуется впервые.
Константа равновесия реакции Li2O+(r) -j- е(г) = 2Li(r) + О(г) вычислена с использованием
значения АгН°@) = 128,779 + 22 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации Р^Ш к* i ЙУйй
Л /0AЛ2О) — 600*+ 20 кДж-моль.
Принятая величина была получена Хилденбрандом [2641] методом электронного удара.
Менее точные значения 656 и 666 кДж-моль были найдены этим же методом в работах Берковица
и др. [1503] и Уайта и др. [4721].
Принятому потенциалу ионизации соответствует]
AfH°(L\2O\ г,г|0) = 433,474 ± 22 кДж • моль.
1Л2О2(к). Термодинамические свойства кристаллического диоксида дилития" в стандартном
состоянии при температурах 298,15—1000 К приведены в табл. 1054.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние 1л2О2(к) в интервале 0—1000 К принята
гексагональная модификация [2288] *. Сведения о термодинамических свойствах 1Л2О2(к) (за
исключением данных по энтальпии образования) в литературе отсутствуют. Теплоемкость и
термодинамические функции при 298,15 К, приведенные в табл. 45.1, оценены так же, как для К2О2
(см. ниже). Принятое значение 5°B98,15 К) согласуется в пределах погрешности с оценкой в
таблицах JANAF [2834] E6,5 + 6 Дж-К^-моль). Погрешности принятых значений 5°B98,15 К)
и .#ОB98,15 К)—Н°@) оцениваются в 8 Дж-К^-моль ш 0,8 кДж-моль соответственно.
Для теплоемкости 1л2О2(к) в интервале 298,15—1000 К принято уравнение (см. табл. 45.1),
выведенное по двум значениям теплоемкости С°B98,15 К) = 75 + 8 Дж-К-моль и С°A000К) =
= 117 + 12 Дж-К^-моль; последнее оценено так же, как для К2О2(к).
Погрешности вычисленных значений Ф°(Т) при 298,15, 500 и 1000 К оцениваются в 3,
5 и 12 Дж-К~1-моль~1 соответственно. Расхождения между значениями Ф°(Т), приведенными
в справочнике JANAF [2834] и табл. 1054, составляют —1,5 Дж-К~1-моль~1 и обусловлены
разной оценкой ?°B98,15 К).
В справочнике принята энтальпия образования
ЬцОа'Ч 298,15 К) = — 632,5 + 4,0 даДж • моль,
основанная на калориметрических измерениях, выполненных Форкраном [2290], и
исследовании равновесия реакции 21л2О2(к) = 21Л2О(к) + О2(г) в работе Центнершвера и Блюменталя [1591,
1774]. Форкран измерил энтальпию растворения 1Л2О2(к) в воде (—30,1 кДж-моль) и энтальпию
реакции между растворами Н2О2 и LiOH (—27,3 кДж-моль), что дает для реакции 2LiOH(p-p;
200Н2О) + Н2О2(р-р; 200Н2О) = 1л2О2(к) + 2Н2О(ж), АГН° = 2,8 кДж-моль. Этой величине
соответствует A^°(Li2O2, к, 298,15 К) = —632 + 4 кДж-моль. Центнершвер и Блюменталь
1 Данные Роде и др. [882J о полиморфном превращении Li2O2 при 498 + 10 К не были подтверждены в более
позднем исследовании Нотца и Баха [3683].
261
1Л2О2(г) Глава 45
провели измерения динамическим и компенсационным методами в интервале 315—473 К, 60
точек. По этим данным были получены значения —632,9 + 4,0 кДж-моль (III) и —616 + 6 кДжХ
^)
Давление пара диоксида дилития в реакции] 1Л2О2(к) = 1Л2О2(г); вычислено с
использованием энтальпии сублимации
ДгЯ°@) = Д,Я°AЛаОа к, 0) = 350,054 + 20 кДж • моль,
соответствующей принятым в справочнике энтальпиям образования 1Л2О2 в кристаллическом и
газообразном состояниях.
1Л2О2(г). Термодинамические свойства газообразного диоксида дилития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1055.
Молекулярные постоянные, использованные для расчета термодинамических функций 1Л2О2,
приведены в табл. 45.9. Структура Li2O2 экспериментально не исследовалась. На основании кван-
товомеханических расчетов [2924, 3802, 4796, 4915], а также изучения ИК-спектров [1326, 4364,
4366] для молекулы Li2O2 в основном электронном состоянии &хАд принята плоская
ромбическая структура со связью кислород—кислород в цикле (симметрия D2h). Приведенное в табл. 45.9
произведение моментов инерции рассчитано по структурным параметрам r(Li—0) = 1,73 + 0,05 А,
г(О—О) = 1,50 + 0,05 А, найденным Йейтсом и Питцером [4796] в результате квантовомехашг
ческого расчета с использованием расширенного базиса и поляризационных функций. С
принятыми структурными параметрами удовлетворительно согласуются данные расчетов [2924, 3802]
и оценки, выполненные Эндрюсом [1326]. Погрешность величин IaIbIc составляет 3-1018 г3-смв.
Приведенные в табл. 45.9 частоты валентных колебаний v6, ve найдены Эндрюсом [1326] в ИК-
спектре молекул Li2O2, изолированных в матрице из Аг. С ними согласуются частоты, найденные
в работах [4364, 4366], а также результаты квантовомеханического расчета [4796]. Частота вне-
нлоскостного колебания v4, так же как частоты деформационного v2 и валентного v3 колебаний
Li2O2, приняты по результатам расчета [4796] *. Для частоты колебания О—О (v1) принято
значение, являющееся средним из частоты, найденной Хюбертом и Озином [2743] в спектре
комбинационного рассеяния образца, полученного при конденсации паров лития и кислорода при 4 К
(802 см), частоты колебания иона О22 в кристалле [2194, 2209] G90 см) и значения vx = 954 см,
рассчитанного Йейтсом и Питцером [4796] 2.
Погрешности в значениях vlt v4, v5 и v6 оцениваются в 20—25 см, частот v2 и v3 — в 100—
150 см.
Согласно квантовомеханическому расчету [4796] энергии возбужденных электронных
состояний Li2O2 превышают 24 000 см и они не рассматриваются в справочнике, так же как изомер
е линейной структурой (Те — 17 000 см) [2924].
Термодинамические функции Li2O2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных электронных состояний. Их погрешности обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета и
составляют 6, 12 и 18 Дж-К~1-моль~1 в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Li2O2(r) вычислялись в справочнике JANAF [2834], а также
в работе By и др. [4778] (Т = 1400^-1700 К). Расхождения данных [2834] и табл. 1055
достигают 23 Дж-К-моль в значениях Ф°(Г) из-за использования в таблицах JANAF неверных
молекулярных постоянных Li2O2 по работе [4721].
Константа равновесия реакции Li2O2(r) = 21Л(г) + 2О(г) вычислена с использованием
значения ДгЯ°@) = 1084,036 + 20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
kfH°(Li202 г, 0) = —275 ± 20]кДж .моль.
* В предположении, что слабая полоса 297,5 см 1, наблюдавшаяся Эндрюсом [1326], относится не к основному
колебанию v4, а к обертону C v4), результаты расчета [4796] и эксперимента [1326] согласуются между собой.
Область частот ниже 200 см в работе [1326] не исследовалась.
* Уайт и др. [4238, 4721] отнесли к молекуле Li2O2 полосы при 330 и 530 см, однако Эндрюс [1326] показал, что
эти полосы принадлежат димеру (LiO2J.
262
Литий и его соединения
1ЛН(к,ж)
Таблица 45.10. Результаты определений АуД"°AЛ2О2, г, 0) (в кДж • моль г)
Авторы
Метод
A/H°(hlz02, г, 0)
11 закон
III закон
Уайт и др., 1963 [4721] Масс-спектрометрический, Li3O(r)+1/aO2(r)=Li2O2(r), — —280+20
1710 К, 1 точка
Кудо и др., 1978 [3134] То же, Li20(r)+Li0(r)=Li202(r)+Ii(r), 1482— —148+80 —285+20
1663 К, 9 точек
By и др., 1979 [4778] То же, Li2O2(r)+Li2(r)=2LiaO(r), 1528-1663 К, 7 точек -428+60 -370+25
Кимура и др., 1980 [3005] Тоже, Li2O2(r)=2Li(r)+O2(r), 1329—1507 К, 9 точек —341+85 —272+12
Принятая величина основана на данных, приведенных в табл. 45.10. Указанные в таблице
погрешности учитывают воспроизводимость измерений, неточность термодинамических функций,,
термохимических величин и сечений ионизации. За исключением работы [4778], которая
вызывает сомнения (в таблице экспериментальных данных, приведенной в этой работе, имеются
неточности), результаты измерений совпадают в пределах их погрешностей. Принятое значение
является средневзвешенным из данных трех работ, его погрешность в существенной степени
определяется неточностью термодинамических функций 1Л2О2(г).
1ЛН(к, ж). Термодинамические свойства кристаллического и жидкого гидрида лития в
стандартном состоянии при температурах 100—2000 К приведены в табл. 1056.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние 1ЛН(к) в интервале 0—965 К принята
кубическая модификация (структурный тип NaCl) 1.
Термодинамические функции 1ЛН(к) при Т ^ 298,15 К вычислены на основании данных
Йейтса и др. [4795] по теплоемкости 7LiH 2 в интервале 5—320 К. Примеси других элементов по
данным масс-спектрометрического анализа составляют в сумме 0,003%. Погрешность этих
измерений оценивается авторами работы в 2% в интервале 50—320 К и до 10% при более низких
температурах. Погрешности приведенных в табл. 45.1 значений ?°B98,15 К) и Я°B98,15 К)—Н°@)
составляют 0,4 Дж-К~1-моль~1 и 0,06 кДж-моль соответственно.
Менее точные измерения теплоемкости на образце с природным изотопическим составом
(чистота образца 99,8%) проведены Кострюковым [560] C,7—296 К); эти данные в интервале 50—
100 К удовлетворительно согласуются с данными [4795] (в пределах -—1%), однако при Т <^
<^ 50 К они лежат значительно выше измерений [4795] (при 20 К — на 30%), а при Т > 100 К—
систематически ниже (при 200 К — на 2%, а при 296 К — на 3,5%). Данные Гюнтера [2529]
по теплоемкости ЫН(к) в интервале 74—96 К ошибочны. Измерения теплоемкости, выполненные
Бьюси и Бивеном C0—300 К), не опубликованы и известны по отчету [1733], в котором
отсутствуют первичные экспериментальные данные.
Уравнение для теплоемкости ЫН(к) в интервале 298,15—965 К (см. табл. 45.1) получено
совместной обработкой данных по энтальпии, полученных в работе Родигиной и др. (см. [1177, с. 35]),
C68—664 К, 13 точек), Шпильрайна и др. [1169] E21—950 К, 7 точек), Новикова и др. (см. [1177,
с. 36]) D27—937 К, 15 точек) и Вильсона и др. [4739] C63—925 К, 16 точек); при этом
погрешность измерений Родигиной и др. (см. [1177, с. 35]) и [1169] оценена в 1%, а данных Новикова
и др. (см. [1177, с. 36]) и [4739] —в 2%.
Температура плавления (965+2 К) принята по измерениям Мессера и Леви [3489]. Эти авторы
исследовали 10 образцов LiH чистотой 99,6—99,9% LiH и, экстраполировав свои результаты
к стопроцентному составу LiH, нашли Т'от=964,5 К; поправка на пересчет этой величины к шкале
МПТШ-68 составляет+0,4 К. Измерения, проведенные ранее Мессером и др. [3490], а также
1 Согласно теоретическим расчетам (см. обзор [1190]) гидрид лития при высоких давлениях должен претерпевать
фазовые переходы — сначала в кубическую модификацию со структурным типом CsCl, а затем в металлическое
состояние. Экспериментальные данные о таких переходах отсутствуют.
2 Расхождения между теплоемкостью гидрида лития природного изотопического состава (e> M1LiH) и
теплоемкостью 7LiH согласно [1190] значительно меньше погрешности измерений, ввиду чего соответствующие
поправки в справочнике не вводились.
263
ЫН(к, ж)
Глава 45
данные других работ [2864, 3490, 4691] приводят к более низким значениям температуры
плавления (958—962 К) из-за наличия примесей в исследованных образцах LiH x. Энтальпия
плавления B1,8+0,3 кДж-моль) вычислена как разность между принятыми значениями Н°(Тт)—
.ffoB98,15 К) для 1лН(к) и 1лН(ж). С этой величиной в пределах ее погрешности согласуются
результаты калориметрических измерений [1169] B2,2+0,3 кДж-моль") и [1177, с. 36] B1,8 +
+0,8 кДж-моль); менее точные значения получены в работах [4739] B0,8 кДж-моль). [3490]
B1,3 кДж-моль-1) и [4690] B3,1 кДж-моль).
Теплоемкость жидкого гидрида лития E5 + 4 Дж-К~1-моль~1) принята в результате
усреднения данных по энтальпии 1ЛН(ж), полученных в работах [1169] (962—1325 К), [1177, стр. 35]
(989—1385 К) и [4739] (988—1027 К) 2.
Погрешности значений Ф°(Т) для 1ЛН(к, ж) при 298,15, 1000, 1500 и 2000 К оцениваются
в 0,3, 0,6, 1,8 и 4 Дж-К^-моль.
Значения термодинамических функций 1ЛН(к), вычисленные во II издании настоящего
справочника [337], согласуются с приведенными в табл. 1056 в пределах 0,8 Дж-К-моль~1 в
значениях Ф°(Г). Для жидкого гидрида лития соответствующие расхождения с ростом температуры
изменяют знак на обратный и достигают 5 Дж-К~1-моль при 2000 К. Термодинамические
функции ЫН(к, ж), вычисленные в справочнике JANAF [2834] (до 2000 К), отличаются от данных
табл. 1056 не больше, чем на 2 Дж-К^-моль в значениях Ф°(Т).
Принятая в справочнике энтальпия образования
5 К) = —90,65 ± 0,20 ] кДж • моль
оенована на приведенных в табл. 45.11 калориметрических измерениях энтальпии растворения
ЬаН(к) и 1л(к) в воде и на приведенных в табл. 45.12 результатах исследования равновесия
21лН(к, ж)=21Л(ж)+Н2(г). Расчет энтальпии образования по калориметрическим измерениям
проводился двумя путями: по энтальпии растворения LiH в воде и по разности энтальпай
растворения LiH и Li в воде. Результаты всех четырех калориметрических исследований совпадают
в пределах их погрешностей. Однако наиболее точное значение было получено Ганном и Грином
[2525, 2527], и принятая величина основана на этой работе.
Таблица 45.11. Результаты калориметрических определений Л_^Л(ЫН, к, 298,15 К) (в кДж-моль)
Авторы
Гунц, 1896 [2531, 2532]
Моерс, 1920 [3530]
Мессер и др., 1955 [3488]
Ганн, Гран, 1958 [2525, 2527]
Метод
Измерение энтальпии растворения ЫН(к)
и Li (к) в воде
То же, 3 и 3 измерения
То же, 8 и 6 измерений
То же, 8 и 7 измерений
Д/Я°(ЬШ, i
1-й путь
—91,1
—90,7 ±2,8
—90,0±1,1
—90,65+0,21
е, 298,15 К)
2-й путь
—89,8
—89,8+4,0
-89,3+1,5
—90,63+0,26
Большинство из приведенных в табл. 45.12 величин вычислено по давлениям в монотекти-
ческой области (см. [1177]) без учета отклонения термодинамических свойств Li(k) и LiH(K) в
равновесной смеси от свойств чистых компонентов. Только в работе [4604] была сделана попытка
приближенно учесть эти отклонения при расчете константы равновесия. Приведенные в табл. 45.12
погрешности характеризуют только воспроизводимость измерений. Как видно из таблицы,
исследования равновесия 2LiH(K, ж)=2Li(ж)+H2(г) находятся в удовлетворительном согласии с ре-
1 Согласно диаграмме состояния системы Li—LiH (см. [1190]) температура плавления фазы LiHi.j в интервале
100—98% LiH понижается от 965 до 958 К, затем в монотектической области C0—98% LiH) постоянна
и равна Р58 + 1 К.
2 Шпильрайн и Якимович [4910а] по тем же данным вывели трехчленное уравнение для теплоемкости ЫЩж),
по которому она падает от С°р (965 К)=61,8 Дж-К^-моль до С°р A150 К)=50,7 Дж-К-моль, а затем воз-
"растает до С°р A300 К)=54,5 Дж-К^-моль. Учитывая разброс данных по энтальпии
и необходимость
экстраполяции теплоемкости
нято постоянное значение.
в область более высоких температур, в справочнике для C°(LiH, ж)
при264
Литий и его соединения
LiH(r)
Таблица 45.12. Результаты определений A^?r0(LiH, к, 298,15 К) по исследованиям равновесия реакции
2ЫН(к, ж)=21Л(ж) Н) Д1
Авторы
Гунц, 1896 [2532]
Хард, Мур, 1935 [2759]
Гибб и др., 1954 (см. [1177])
Хойман, Салмон, 1956 [2629]
Мессер и др., 1955 [3488]
Новиков и др., 1959 (см. [1177])
Уэлч, 1961 (см. [1177])
Джонсон и др., 1966 [2864]
Бергштейн и др., 1968 [1496]
Шпильрайн, Якимович, 1972 [1177]
Ихл, By, 1974 [2768] I
Велецкис и др., 1974 [4604]
Смит, Ланд, 1975 [(см. [4603])
Шпильрайн и др., 1977 [1180]
Кацуда и др., 1977 [2932]
Смит, Вебб, 1977 [5049]
Велецкис, 1979 [4603]
Метод
Статический, 1ЛН(к), 953 К, 1 точка
То же, ЫН(к), 782—926 К, 5 точек
То же, LiH(K), 851—958 К а
То же, 1ЛН(ж), 973—1273 К а
То же, ЫН(ж), 973—1077 К, 7 точек
То же, ЫН(ж), 973 К, 1 точка
То же, ПН(ж), 963—1364 К, 13 точек
Протока, ЫН(к), 888—945 К, 4 точки
То же, 1ЛН(ж), 965—1061 К, 4 точки
ЭДС, ЫН(к), 675-885 К, 47 точек а
Го же, 1лН(к), 500—900 К а
Аяализ литературных данных, ЫН(к), 773—
358 К а
То же, 1ЛН(ж), 958—1273 К а
Маоо-спектрометрический, ЫН(к), 725—800 К,
8 точек
Тензиметрический, 1ЛН(ж), 983—1176 К, 6точека
Статический, 1лН(ж), 973—1173 К, 3 точки
То же, ЫН(ж), 963—1196 К, 31 точка
Циркуляционный, LiH(k), 813—960 К а
Го же, ЫН(ж), 965-1113 К а
Статический, 1лН(к), 733—961 К а
То же, 1ЛН(ж), 961—1023 К а
То же, 1лН(к), 904—967 К, 7 точек
То же, 1ЛН(ж), 967—1132 К, 15 точек
а Данные представлены в виде уравнения.
ДЯ°(ЫН
II закон
_
—89+4
-96,7+1,2
—88
-95+6
—
—80+5
—95+18
—102+15
—84
-92,3
—94,5
—101
—84+7^
—91,8
-99+25
—95,2+0,8
—70
—94
—96
—96
—95,5+1,5
—94,6+0,8
к, 298,15 К)
III закон
—88,5
-98,8+0,9
-89,7+0,6
—88,5+1,1
-89,2+0,4
—89,9
—91,6+0,8
—89,5+0,8
—88,6+1,0
—90,9+0,8
—89,1+1,0
—90,0+0,5
—88,0+2,0
—83,7±0,3
-91,0+0,2
-89,3+2,2
-88,5+0,2
—90,2+1,8
—91,6+0,7
—90,9+0,6
—89,8+0,2
—89,8+0,2
-89,2+0,2
зультатами более точных калориметрических измерений. Неточность термодинамических функций
увеличивает погрешности величин, приведенных в табл. 45.12 примерно на 0,7 кДж-моль.
Давление пара гидрида лития в реакции 1лН(к, ж)==1ЛН(г) вычислено с использованием
энтальпии сублимации Arif°@)=A8i/°(LiH, к, 0)=225,110+1,0 кДж-моль", соответствующей
принятым в справочнике энтальпии образования 1ЛН(к) и энергии диссоциации LiH(r).
1ЛН(г). Термодинамические свойства газообразного гидрида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1057.
В табл. 45.4 приведены молекулярные постоянные 7LiH, принятые на основании анализа
результатов исследований электронных спектров и квантовомеханических расчетов этой
молекулы. Согласно теоретическим расчетам [2086, 2087] молекула LiH имеет 4 связанных валентных
состояния X1!,* (...2а2), Л1Е+(...2аЗо), а3П(...2а1тс) и 51П(...2а1и), коррелирующих с атомами
LiB6')+HB6r) и LiBP)+HB^). В электронном спектре 7LiH наблюдались две системы полос А1!,+*^
^ХЧ+ [1959, 1960, 3250, 3627, 3628, 3726] и ВгП <- Хг1,+ [4597]. Исследован также ее
колебательно-вращательный спектр [2831, 3033].
Молекулярные постоянные 7LiH в основном состоянии приняты по данным Орта и Стволли
[3726], полученным на основании анализа 65 полос системы ^41Е+**Х1Е+ с v" ^ 12,
выполненного Крофордом и Йоргенсеном [1959] (z/'=0-^3), Ли и Стволли [3250] (ь>"=4,5) и самими
авторами [3726] (y"=2-f-12). Постоянные De, px и [32, представленные в табл. 45.4, рассчитаны
по значениям Dv из работы [3726]. Зависимость постоянной Hv от v нерегулярная и плохо
аппроксимируется полиномом, поэтому в табл. 45.4 приведено значение Но. Постоянные состоя-
265
1ЛН(г) Глава 45
ния А1!,* получены Ли и Стволли [3250] в результате анализа 33 полос системы А1!,* *а X1!,*
с v' -\ 15 г. Энергии состояний АХИ + и В1!! приняты по данным [4611]. Молекулярные
постоянные в состоянии ВгП получены Веласко [4597] на основании анализа вращательной структуры
шести полос системы ВгП <~ X1!,* с г/=0^-2.
Состояние а3П экспериментально не исследовано. Молекулярные постоянные, приведенные
в табл. 45.4, получены в результате квантовомеханического расчета Докена и Хинце [2087].
Энергия возбуждения оценена на основании энергии диссоциации молекулы LiH в этом
состоянии, найденной в работе [2087] с учетом возможной систематической погрешности в значениях
энергий диссоциации других состояний гидрида лития. Ее погрешность может составить 200 см.
Приведенные в табл. 45.4 молекулярные постоянные 7LiH обеспечивают весьма точный
расчет термодинамических функций LiH(r). Однако из всех изотопомеров этой молекулы наиболее
полно изучен 7LiD. При этом колебательные уровни состояния Х1Б+ молекулы 7LiD исследованы
почти до диссоциационного предела. Энергия последнего наблюдавшегося уровня при v"=26
составила 95% от энергии диссоциации. В связи с этим целесообразно для расчета
термодинамических функций всей группы изотопических разновидностей гидрида лития использовать
молекулярные постоянные 7LiD, пересчитав их по изотопным соотношениям A. 66).
Термодинамические функции LiH (г) были рассчитаны по уравнениям A.3)—A.6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн и ее производные вычислялись по уравнениям A. 90)—A. 92)
с учетом трех возбужденных состояний. Значения статистических сумм по
колебательно-вращательным уровням энергии состояний ХХ2+, А1!,*, а3П и В1]! и их производные находились по
уравнениям A. 70)—A. 75) непосредственным суммированием по уровням энергии. В расчетах
учитывались все уровни, ограниченные предельными кривыми диссоциации этих состояний.
Колебательно-вращательные уровни вычислялись по уравнениям A. 65), A. 63) и A. 64а).
Значения коэффициентов Ykl в этих уравнениях, использованные в расчетах, приведены в табл. 45.5;
они были получены для изотопической модификации LiH, соответствующей природной смеси
изотопов по формулам A. 66) из коэффициентов Ykl молекулы 7LiD, которые приведены в той же
таблице. В табл. 45.56 приведены также значения ymaI, /Ит и коэффициенты уравнений,
аппроксимирующих предельные кривые диссоциации. Мультиплетность и вырождение состояний а3И
и В1!! учитывались статистическими весами 6 и 2 соответственно.
Погрешности вычисленных термодинамических функций LiH(r) при Т <^ 3000 К обусловлены
в основном неточностью фундаментальных постоянных и не превышают 0,02 Дж-К~1-моль.
При Т ^> 3000 К становится существенной неточность экстраполяции высоких колебательно-
вращательных уровней учитываемых электронных состояний и погрешность в значении Ф°F000 К)
может достигать 0,1 Дж-К'^моль.
Ранее термодинамические функции LiH(r) вычислялись во II издании настоящего
справочника [337], таблицах JANAF [2834], справочниках Мак-Брайд и др. [3439], Мадера [3338], Хара
и др. [2537], Хаффа и др. [4978], а также в работах Горбова и др. [306], Уилкинса и др. [4729],
Шнейдера [4197а]. Все перечисленные расчеты выполнялись по молекулярным постоянным
основного электронного состояния, близким к принятым в настоящем справочнике. Расхождения
данных справочников [337, 2834, 3338, 3439] с приведенными в табл. 1057 не превышают 0,4 ДжХ
хК^-моль в значениях Ф°(Г), но достигают 2 и 0,8 Дж-К^-моль в S°F000K) для [337, 2834]
и 5,6 Дж-К^-моль в С°F000 К) для [2834]. Они обусловлены тем, что в этих справочниках
использовались менее точные методы расчета и учитывалось меньшее число возбужденных состояний.
Расчет [306] выполнен но методике, совпадающей с принятой в настоящем справочнике и
расхождения @,3 Дж-К~1-моль в значении Ф°F000 К)) обусловлены тем, что в расчете [306] не
учитывалось состояние я3П.
Константа равновесия реакции LiH(r) = Li(r)+H(r) вычислена с использованием значения
D0(LiH) = 19 588,1+1,0;см-1=234,326+0,012™кДж.моль. .
Эта величина принята для изотопомера LiH природного изотопического состава на основании
энергии диссоциации молекулы 7LiHA9 588,6+1,0 см), найденной в работе Вея и Стволли
14673] при обработке данных Веласко [4597] по предиссоциации 7LiH при вращении в
состоянии В1!!. Близкие значения были получены ранее Веласко [4597] A9 591+2 см, пре-
1 Состояние Л1Х+, как у всех гидридов щелочных металлов, аномально. Кривые зависимостей Bv и AG,+i/ от v
растут с ростом v и, пройдя через максимум, падают (см. [3588]).
266
Литий и его соединения IAD(k, ж)
диссоциация при вращении на уровнях v—О, /=8; у=1, /=5 и v=2, /=2 состояния BLI1) и
в квантовомеханическом расчете [3494] A9 400 см).
Принятой энергии диссоциации соответствует
?\fH°(Lm, г, 0) = 139,443 ±1,0 кДж.моль.
LiD(k, ж). Термодинамические свойства кристаллического и жидкого дейтерида лития в
стандартном состоянии при температурах 100—2000 К приведены в табл. 1058.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние ЬШ(к) в интервале 0—967 К принята
кубическая модификация (структурный тип NaCl).
Термодинамические функции ЫВ(к) при Т <^ 298,15 К вычислены по данным Йейтса и др.
[4795] по теплоемкости LiD в интервале 4,8—320 К; погрешности этих измерений оцениваются
авторами в 2% при Т > 50 К и до 10% при Т < 50 К г. Погрешности принятых по работе [4795]
значений ?°B98,15 К) и #°B98,15 К)—#°@) (см. табл. 45.1) оцениваются в 0,6 Дж-К^-моль-1
и 0,08 кДж-моль соответственно.
Экспериментальные данные о термодинамических свойствах LiD(k, ж) при Т > 320 К
отсутствуют. В связи с этим в справочнике использованы результаты расчетов теплоемкости и
энтальпии 1лБ(к), проведенные Якимовичем и др. [1190, 4910в] на основании экспериментальных
данных по теплоемкости 1лН(к) (до 965 К) и 1лБ(к) (до 320 К) с учетом изотопического эффекта.
Уравнение для теплоемкости LiD(k) в интервале 298,15—967 К вычислено по значениям
?;B98,15 К), #°F00 К)—#"B98,15 К)=13,29 кДж-моль и #°(900 К)—#"B98,15 К) =
=30,2 кДж-моль; последние два значения приняты по работе [1190]. Погрешности значений
теплоемкости, вычисленных по этому уравнению, оцениваются в —3% при 900 К.
Температура плавления (967+2 К) принята по работе Велецкиса [4602], который определил
температуру монотектического плавления LiD равной 962 К. Пересчет последнего значения к
температуре плавления LiD выполнен Якимовичем [49106] на основании данных по диаграмме
состояния LiH—Li. Энтальпия плавления B2+1 кДж-моль) и теплоемкость жидкого LiD E6 +
+5 Дж-К~1-моль~1) оценены с учетом экспериментальных данных для LiH, которые были
несколько увеличены и округлены.
Погрешности табулированных значений Ф°(Т) для LiD(K, ж) при 298,15, 1000, 1500 и 2000 К
оцениваются в 0,4, 1,3, 2,5 и 4 Дж-К-моль соответственно.
Таблица термодинамических свойств LiD(K, ж) публикуется впервые.
В справочнике принята энтальпия образования
A^°(LiD, к, 298,15 К) = —90,97 + 0,20 кДж • моль,
основанная на калориметрических измерениях энтальпии растворения LiD(K) и Li(K) в воде,
выполненных Ганном и Грином [2527] и скорректированных впоследствии Ганном [2525].
Разность измеренных величин дает для реакции Li(K)+HD(r)=LiD(K)+1/2H2(r) значение
Дг#°B98,15 К) =—91,29+0,22 кДж-моль, чему соответствует приведенная выше энтальпия
образования.
В табл. 45.13 приведены значения энтальпии образования LiD(K), вычисленные по
результатам исследования равновесия реакции 2LiD(K, ж)=2Li(ж)+D2(г). Приведенные в таблице
погрешности учитывают только воспроизводимость измерений. Неточность термодинамических
функций LiD(K, ж) приводит к дополнительной погрешности в энтальпии образования,
составляющей 1,1 кДж-моль для измерений в области 900 К и 1,8 кДж-моль для измерений при
1100 К. Как видно из таблицы, эти исследования приводят к значениям, совпадающим в
пределах погрешностей с принятым на основании калориметрических измерений. Это совпадение
свидетельствует в частности о достаточной точности принятых в справочнике значений
термодинамических функций LiD(K, ж).
Давление пара дейтерида лития в реакции LiD(K, ж)=^О(г) вычислено с использованием
значения Дг#о@) = As#°(LiD, к, 0)=227,665+1,0кДж-моль, соответствующего принятым в
справочнике энтальпии образования L1D(k) и энергии диссоциации LiD(r).
Причина аномального хода теплоемкости LiD (к) в интервале 7—15 К (Х-кривая с максимумом при 12,8 К)
не выяснена. Вклад этой аномалии в энтропию LiD пренебрежимо мал (менее 0,01 Дж-К^-моль).
267
LiD(r)
Глава 45
Таблица 45.13. Результаты определений A^ff°(LiD, к, 293,15 К) по исследованиям равновееая реакции
2LiD(k, ж) = 2ХЛ(ж)+D2(f) (в кДж • моль)
Авторы
Хойман, Саломон, 1956 [2629]
Велецкис, и др., 1974 [4604]
Смит и др., 1976 (см. [4602, 4603])
Велецкис, 1977 [4602]
Смит, Вебб, 1977 [5049]
Велецкис, 1979 [4603]
Метод
Статический, LiD (ж), 973—1073 К, 3 точки
Тешиметрический, ЬШ(ж), 978—1144 К, 5 точек,
данные представлены в виде уравнения
Статический, LiD(jft), 973—1173 К, 3 точки
То же, LiD(K), 905—955 К, 8 точек
То же, ЬЮ(ж), 965—1152 К, И точек
То же, ЫБ(к), 733—961 К, 7 точек
То же, ьт(ж), 961—1023 К, 5 точек
То же, LiD(K), 901—963 К, 6 точек
То же, LiD (ж), 963—1124 К, 12 точек
AfHo(LiV, г
II закон
-89,5
t, 298,15 К)
III закон
—89,6+1,5
—94,1 —91,8+2,0
—91,7
—89,9+3,0
—95,4+0,2
—98,8
-96,0
—99,7+3,0
—94,7+1,0
-88,8 + 2,0
—89,9+0,2
—89,1+0,2
—91,2+0,8
—90,0+0,2
—89,8+0,3
—89,3+0,2
LiD(r). Термодинамические функции газообразного дейтерида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1059.
В табл. 45.4 приведены молекулярные постоянные 7LiD, выбранные на основании анализа
спектральных данных и квантовомеханических расчетов этой молекулы. Систематика нижних
электронных состояний 7LiD такая же, как молекулы 7LiH. В электронном спектре молекулы
7LiD наблюдались две системы полос A1L+ ** ZXE+ [1958, 1960, 2180, 2181, 3250] и ВЩ <- Х1!,*
[4423, 4597]. Исследован также микроволновой спектр этой молекулы [3791].
Колебательные и вращательные постоянные в состоянии Х1И + (кроме De, px и Но) получены
Энненом и Оттингером [2181] в результате анализа большого числа линий системы АХ11+ ** ХХЕ +
в спектре лазерной флуоресценции, связанных с переходами на уровни су" ^ 26. Расчет
постоянных проводился с учетом условий A. 50) и В„тях+1 = 0. Постоянные De, $г и Но этого
состояЛ1 Л С [50]
e г о
ния и все молекулярные постоянные состояния Л1!!" найдены Ли и Стволли [3250] в результате
анализа вращательной структуры 35 полос системы А1!!* ** Х111 + с v" ^ 4 и 1 ^ v' ^ 19 с
использованием данных Крофорда и Йоргенсена [1958]. Колебательные постоянные в состоянии
ВЩ основаны на измерениях начал полос системы ВЩ <-Х1И + су'<СЗв работах Веласко [4597]
и Стволли и др. [4423]. Энергия состояния В1!! принята по данным [4611]. Постоянные Ве и <х±
рассчитаны авторами [4423] на основании данных [4597] (z/=0-^-2) и собственных измерений
(у' = 3). Постоянные De, pit He, hx и h2 найдены Веласко [4597]. Молекулярные постоянные LiD
в состоянии asH приняты по результатам квантовомеханического расчета Декена и Хинце [2087].
Энергия возбуждения основывается на энергии диссоциации этого состояния, вычисленной в
работе [2087], с учетом систематической ошибки расчета энергий диссоциации в других состояниях
этой молекулы. Погрешность величины Те(а3П) оценивается в 200 см.
Термодинамические функции LiD(r) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения <?вни ее производные вычислялись по уравнениям A. 90)—A. 92)
с учетом трех возбужденных состояний, приведенных в табл. 45.4. Статистические суммы по
колебательно-вращательным уровням состояний X1L+, А1!!*, а?П и ВЩ и их производные находились
по уравнениям A. 70)—A. 75) непосредственным суммированием по уровням энергии. В расчете
учитывались все уровни, ограниченные предельными кривыми диссоциации этих состояний.
Колебательно-вращательные уровни вычислялись по уравнениям A. 65), A. 63) и A. 64а).
В табл. 45.5в приведены значения коэффициентов для изотопомера LiD, соответствующего
природной смеси изотопов лития, вычисленные по соотношениям A. 66) и постоянным молекулы 7LiD.
Там же приведены величины уш, /Иш и коэффициенты а(, аппроксимирующие предельные
кривые диссоциации. Для обеспечения выполнения условий A. 50) и A. 59) добавлены коэффициенты
У X1 F У A1 Oi
Д у ( ) ( ) фф
У04 для состояния X1S+, F70 и У04 — для состояния A1!,*, Y03 — для состояния а3П и YOi
для состояния В1!!. Мультиплетность и вырождение состояний а3П и ВЩ учитывались
статистическими весами 6 и 2 соответственно.
Погрешности вычисленных термодинамических функций LiD(r) при Т ^ 3000 К обусловлены
в основном неточностью фундаментальных постоянных и не превышают 0,02 Дж-К~1-моль~ж
268
Литий н его соединения 1ЛТ(к, ж)
в значениях Ф°(Г).. При более высоких температурах становится существенной неточность
экстраполяции колебательно-вращательных уровней состояний Х1!!*, Л12+и а3П, и погрешность в
значении Ф°F000 К) достигает 0,1 Дж-К~1-моль.
Ранее термодинамические функции LiD(r) вычислялись в справочнике Хара и др. [2537] по
молекулярным постоянным основного состояния, близким к принятым в справочнике.
Расхождения с данными табл. 1059 возрастают с ростом температуры и достигают 0,7, 1 и 1,9 Дж-К^-моль
в значениях Ф°(Г), S°(T) и С°р(Т) соответственно. Они обусловлены тем, что авторы [2537]
использовали приближенную методику расчета и не учитывали возбужденные электронные
состояния.
Константа равновесия реакции LiD(r)=Li(r)+D(r) вычислена с использованием значения
V7°@)=D0(LiD) = 19 767,0+1,0 см-^гЗбДбб+0,012 кДж-моль.
Эта величина соответствует значению D0GLiD) = 19 767,6+1,0 см, найденному Стволли
и др. [4423] по предиссоциации этой молекулы при вращении в состоянии В1!!, v—3, /=2.
Близкое, но существенно менее точное значение B37,9+1,5 кДж-моль) было вычислено по
результатам масс-спектрометрического исследования равновесия реакции Lia(r)+D2(r)=2LiD(r) F23—
—683 К, 8 точек), выполненного Ихлом и By [2769].
Принятой энергии диссоциации соответствует Д^Я0 (LiD, г, 0)=141,076+ 1,0 кДж-моль".
1ЛТ(к, ж). Термодинамические свойства кристаллического и жидкого тритида лития в
стандартном состоянии при температурах 100—2000 К приведены в табл. 1060.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние 1лТ(к) в интервале 0—968 К г/ринята
кубическая модификация (структурный тип NaCl).
Какие-либо экспериментальные данные по термодинамическим свойствам 1ЛТ(к, ж) в
литературе отсутствуют. В справочнике термодинамические функции ЫТ(к) в интервале 0—968 К
вычислены с использованием результатов расчетов теплоемкости и энтальпии 1лТ(к), выполненных
Якимовичем и др. [1190, 4910в] на основании экспериментальных данных по теплоемкости ЬШ(к)
(до 965 К) и ЫБ(к) (до 320 К) и с учетом изотопического эффекта. Погрешности оцененных
значений ?°B98,15 К) и Я°B98,15 К)—Я°@) (см. табл. 45.1) оцениваются в 1 Дж-К^-моль-1 и
0,1 кДж-моль соответственно. Уравнение для теплоемкости 1лТ(к) в интервале 298,15—
968 К было получено по значениям С;B98,15 К) (см. табл. 45.1), Я°F00 К)—Я°B98,15 К) =
= 13,88 кДж-моль-1 и Я°(900 К)—Я°B98,15 К)=31,14 кДж-моль-1. Погрешности значений
теплоемкости, вычисленных по этому уравнению, могут достигать 3—5% при 900 К. Температура
плавления LiT (968+2 К) принята по рекомендации Якимовича [49106]. Значения энтальпии
плавления B2+1 кДж-моль) и теплоемкости жидкого LiTE6+6 Дж-К^-моль-1) оценены с
учетом соответствующих экспериментальных данных для LiH. Погрешности рассчитанных
значений Ф°(Г) для 1ЛТ(к, ж) при 298,15, 1000, 1500 и 2000 К оцениваются в 0,7, 2,5, 5 и 8 Дж-К~хХ
Хмоль соответственно.
Таблица термодинамических функций 1лТ(к, ж) публикуется впервые.
В справочнике принята энтальпия образования
к, 298,15 К) = — 91,2 + 0,5 кДж • моль.
Эта величина была оценена на основании прецизионных калориметрических данных об
энтальпии образования 1лН(к) и LiD(k) в предположении, что тенденция роста абсолютных
значений энтальпии образования при переходе от LiH к LiD сохраняется в ряду LiH—LiD—LiT.
Энтальпия образования LiT(K) может быть найдена также на основании результатов
исследований равновесия реакции 21ЛТ(к, ж)=2Li(ж)+T2(г), представленных в табл. 45.14. В
работах [2629] и [4603] измерения были проведены в монотектической области (см. [1177]), а в
работе Смита и Ланда (см. [4603]) измерялись давления, относящиеся к другой части фазовой
диаграммы и сильно отличающиеся от равновесных. Результаты работ [2629, 4603] совпадают в
пределах их воспроизводимости, среднее значение равно AfH°(LiT, к, 298,15 К)=—89,7+0,4 кДжХ
Хмоль. Его погрешность с учетом неточности термодинамических функций должна быть
увеличена до 4,0 кДж-моль. Сопоставление значений энтальпии образования LiH(K), NaH(K),
КН(к) и LiD(K), полученных из прецизионных калориметрических измерений и вычисленных по
имеющимся в литературе исследованиям равновесия реакций диссоциации в монотектической
269
LiT(r), LjOH(k, ж) Глава 45
Таблица 45.14. Результаты определений Ayff°(LiT, к, 298,15 К) по исследованиям равновесия реакции
21ЛТ(к, ж) = 21Л(ж) + Т2(г) (в кДж ¦ моль)
Авторы
Хойман, Саломон, 1956
Смит, Ланд, 1975 (см.
Велецкис, 1979 [4603]
[2629]
[4603])
Метод
Статический, 1ЛТ(ж), 973—1074 К, 6 точек
То же, 1лТ(ж), 973—1273 К, 4 точки
То же, ЫТ(к), 869—961 К, 10 точек
То же, LiT (ж), 961—1118 К, 10 точек
ДуЯ°AЛТ,
II закон
—91,7
-98,5+2,0
—94,0+1,0
к, 298,15К)
III закон
—89,3+0,3
-96+10
-90,2+0,2
—89,7+0,2
области (см. соответствующие тексты), показывает, что значения, вычисленные по последним
данным, оказываются примерно на 1,2 кДж-моль больше. С этой поправкой результаты
исследований равновесия диссоциации LiT приводят к значению —90,9+4,0 кДж-моль, которое
согласуется с принятой величиной.
Давление пара тритида лития в реакции 1лТ(к, ж)=ЫТ(г) вычислено с использованием
значения ArH°@) = AsH°(LiT, к, 0)=229,019+1,1 кДж-моль-1, соответствующего принятым в
справочнике энтальпии образования 1лТ(к) и энергии диссоциации LiT(r).
LiT(r). Термодинамические функции газообразного тритида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1061.
Экспериментальные данные о молекулярных постоянных 7LiT отсутствуют. Расчет
термодинамических функций LiT(r) проводился так же, как для LiD(r) на основании значений Ykl, ymax,
/lim и а(, вычисленных по изотопическим соотношениям A. 66) и соответствующим величинам
для молекулы 7LiD и приведенных в табл. 45.5 г.
Хотя расчет термодинамических функций LiT(r) проводился по постоянны , полученным с
помощью изотопических соотношений и постоянным молекулы 7LiD, погр' яости рассчитанных
величин, практически такие же, как для LiD(r). При Т ^ 3000 К эти погрешности обусловлены
в основном неточностью фундаментальных постоянных и не превышают 0,03 Дж-К~1-моль~1
в значении Ф°(Т). При более высоких температурах становится существенной неточность расчета
энергии колебательно-вращательных уровней и погрешность в значении Ф°F000 К) может
достигать 0,1 Дж-К^-моль.
Ранее термодинамические функции LiT(r) вычислялись в справочнике Хара и др. [2537].
Расхождения данных [2537] и табл. 1061 возрастают с ростом температуры и достигают 0,7, 1
и 1,8 Дж-К^-моль" в значениях Ф°(Т), S°(T) и С°р(Т) соответственно. Они обусловлены тем,
что авторы [2537] использовали приближенный метод расчета и не учитывали возбужденных
электронных состояний LiT.
Константа равновесия реакции LiT(r)=Li(r)-|-T(r) вычислена с использованием принятого
в справочнике значения
Ar#°@)=D0(LiT)=19 839,2+2,0 см-1=237,329 +0,024 нДж-моль.
Эта величина была рассчитана по принятым в справочнике значениям D0GLiD) и
колебательным постоянным (см. табл. 45.4) в предположении, что DeGLiT)=DeGLiD).
Принятой энергии диссоциации соответствует
Д,Яв(ЫТ, г, 0) = 141.885 + 1,0 кДж ¦ моль.
1лОН(к, ж). Термодинамические свойства кристаллического и жидкого гидроксида лития
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1062.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние ЫОН(к) в интервале 0—746 К принята
тетрагональная модификация (структурный тип SnO) х. При Т <[ 298,15 К термодинамические
В работе Перфильевой и Решетникова [783] было показано, что LiOH существует только в виде одной
кристаллической модификации, а имеющиеся в литературе данные [873, 874, 1074] о полиморфном превращении LiOH
при температурах 670—687 К ошибочны.
270
Литий и его соединения 1Л0Н(к,. ж)
функции ЬЮН(к) вычислены по результатам измерений теплоемкости, выполненных Бауэром
и др. [1434] A5,91—301,63 К) на образце, содержавшем менее 0,01% примесей. Погрешность
измерений оценивается в 0,5% при Т ~ 20 К и в 0,2% при Т > 45 К. Экстраполяция теплоемкости
по уравнению C°p=DB85/T) приводит к значению ?°A6 К)=0,117 Дж-К^-моль. Погрешности
принятых значений 5ГОB98,15 К) и #°B98,15 К)—Н°@) (см. табл. 45.1) оцениваются в 0,4 Дж-Х
ХК~1-моль и 0,04 кДж-моль соответственно.
Для теплоемкости ЫОН(к) в интервале 298,15—746 К принято уравнение (см. табл. 45.1),
найденное обработкой результатов измерений энтальпии в работе Шомейта и Коэна [4272] D18—
691 К, образец содержал следы Li2CO3, погрешность измерений оценивалась в 1%); выпадающие
результаты трех измерений вблизи точки плавления при 725—732 К не принимались во
внимание. Измерения энтальпии ЫОН(к) в работе Тай и др. [4564] B98—713 К), проведенные на менее
чистом образце (99,8% LiOH; количество примеси Н2О не определяли), приводят к значениям
теплоемкости, которые примерно на 5% выше рассчитанных по принятому уравнению (в работе
[4564] численные результаты измерений не приведены).
Температура плавления G46+2 К) принята по согласующимся между собой данным [744,
814, 866, 1260, 1895, 3497, 3498, 3499, 4272]. Результаты менее точных измерений [1074, 4166]
G35 К) и [370, 380, 867, 870, 873, 874, 1039] G50 К) не учитывались. Энтальпия плавления B0,9 +
+1,3 кДж-моль) принята по калориметрическим измерениям Шомейта и Коэна [4272] с учетом
принятых уравнений для энтальпии кристаллического и жидкого LiOH. Это значение согласуется
с термографическими данными [1260, 3499] B0,3 кДж-моль) и калориметрическими измерениями
Пауэрса и Блейлока B1 кДж-мольI. Данные Решетникова и Баранской [866] B2,1 кДж-моль")
и Тай и др. [4564] B4,3 кДж-моль), по-видимому, менее точны и не учитывались.
Теплоемкость жидкой LiOH (87,3+4,0 Дж-К-моль) принята по результатам измерений
энтальпии, проведенных Шомейтом и Коэном [4272] G48—879 К). С принятым значением
удовлетворительно согласуются данные Пауэрса и Блейлока (85,4 Дж-К^-моль) (см. [2421]).
Данные Тай и др. [4564] (см. выше) приводят к резко уменьшающимся с ростом температуры
значениям теплоемкости от С°р(823 К)=94,3 до С°(873 К)=87,7 Дж-К-моль и не принимались во
внимание.
Погрешности вычисленных значений Ф°G') при 298,15, 1000, 2000 и 3000 К оцениваются в 0,3,
0,7, 5 и 8 Дж-К^-моль соответственно.
Термодинамические функции LiOH(K, ж), приведенные в 1-м и 2-м изданиях настоящего
справочника, справочнике JANAF [2834] и в табл. 1062, хорошо согласуются до 1000 К; при более
высоких температурах расхождения в значениях Ф°(Т) возрастают до 1 Дж-К~1-моль и
обусловлены различием принятых значений Cp(LiOH, ж). L
Принятая в справочнике энтальпия образования
,AfH°(L\OE, к, 298,15 К) :=—484,90 ± 0,25 кДж-моль-
основана на измеренной Решетниковым [864] энтальпии растворения LiOH(k) в воде (см.
табл. 45.15). Результаты измерений в более ранних работах менее точны, в основном вследствие
недостаточной чистоты использованных образцов.
Таблица 45.15. Результаты измерений ДагЕГ°AЛОН, к-»соН2О, 298,15 К) (в кДж-моль)
Авторы
Конечная концентрация, температура
измерений
ДягЯ°(ооН2О, 298,15 К)
Трюшо, 1884 [4553] Детали измерений не указаны —24,99
Форкран, 1906 [2294] 1 ! ' ' ' т"ш Н3О, 297,15 К, 1 измерение Г —19,90
Уеда, 1933 [4568, 4569] , 400 Н2О, 298,15 К, 4 измерения —21,104
Решетников, 1961 [864] 400 Н2О, 298,15 К, 7 измерений —23,581+0,20
Давление пара гидроксида лития в реакции LiOH(K, ж)=ЬЮН(г) вычислено с
использованием значения Дг#°@) = A,#°(LiOH, к, 0)=236,099+8 кДж-моль, соответствующего принятым
в справочнике энтальпиям образования LiOH в кристаллическом и газообразном состояниях.
1 Результаты измерений Пауэрса и Блейлока цитируются по справочнику Гмелина [2421].
271
Li ОН (г)
Глава 45
LiOH(r). Термодинамические свойства газообразного гидроксида лития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1063.
Молекулярные постоянные, использованные для расчета термодинамических функций LiOH,
приведены в табл. 45.9. Исследование микроволнового спектра 7Li160H и 7Li160D, выполненное
Фройндом и др. (см. [3313]), а также квантовомеханические расчеты [1, 450, 713, 978, 1022, 1105,
1292, 1713, 1762, 2021, 2026, 2079, 3476, 3641] показывают, что в основном электронном состоянии
ZXE+ молекула LiOH линейна (симметрия Cmh). Вращательным постоянным i?00»0GLi18OH) =
=1,178897 +1-10 и ?0o°0GLiieOD) = l,049973+l-10-6 см (см. [3313]) соответствуют значения
ro(Li—О) = 1,595+0,001 А и г0(О—Н)=0,926+0,001 А. Момент инерции, приведенный в табл. 45.9,
рассчитан по этим значениям структурных параметров для природной изотопической смеси LiOH.
Погрешность значения 10 составляет 3-Ю2 г-см2. Экспериментальные данные об основных
частотах молекулы LiOH отсутствуют. Приведенные в табл. 45.9 значения частот vt и v2 вычислены
по уравнениям простого поля валентных сил с оцененными значениями силовых постоянных
(/ы-о=2,13-105 дин-см, перенесено из Li2O, поскольку силовые постоянные /м-о в оксидах М2О
и гидроксидах МОН калия, рубидия и цезия близки, и /а/г1г2=0,04-105 дин-см
экстраполировано по зависимости jjr-ir% для молекул МОН, где M=Na, К, Rb, Cs). Частота v3 принята
приближенно равной соответствующей частоте GsOH. Погрешности значений v1; va и v3 оцениваются
в 100, 50 и 100 см.
Согласно неэмпирическому расчету [450] энергия изомера HLiO на 31 000 см превышает
энергию LiOH. Сравнение с изоэлектронной молекулой LiF свидетельствует, что энергии других
возбужденных состояний LiOH должны быть больше 20 000 см.
Термодинамические функции LiOH(r) вычислены по уравнениям A.3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126), A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета и составляют 1, 4 и 8 Дж-К^-моль в значениях Ф°(Г) при
298,15, 3000 и 6000 К.
Ранее термодинамические функции LiOH(r) вычислялись в первом [340] и втором [337]
изданиях настоящего справочника, в справочниках JANAF [2835] и Мак-Брайд и др. [3439], а также
Уилкинсом и др. [4729] (Ф°(Г) для Т < 5000 К и Я°B98,15 К) — Я°@)) и Берковицем и др. [1508]
(Ф°(Т) и S°(T) для Г=500-|-1400 К). Термодинамические функции LiOH, вычисленные в
справочнике JANAF [2835] и в работе Уилкинса и др. [4729] для линейной модели молекулы, отличаются
от приведенных в табл. 1063 на 0,4—2,7 Дж-К~1-моль~1 в значениях Ф°G7), что объясняется
различием принятых значений r(Li—О) и частот. Расхождения с результатами расчетов [337, 340, 1508,
Таблица 45.16. Результаты определений Aj.ff°(LiOH, г, 0) (в кДж • моль х)
Авторы
Метод
ДуЯ^ЫОН, г, 0)
11 закон
III закон
Смит, Сагден, 1953 [4323] Сдектрофотометрия в пламенах На+воздух, — —230
LiOH(r)=Li(r)+OH(r), 2200 К, 1 точка
Берковиц и др., 1960 [1508] Масс-спектрометрический, 1л2О(к)+Н2О(г)= —243+15 —247 + 8
=2ЫОН(г), 1119—1448 К, 18 точек
Зегерс, Алкемаде, 1965 [4816, 4817] Спектрофотометрия в пламенах С2Н2+воздух, —263 —
LiOH(r)=Li(r)-f ОН(г), детали экспериментов не
опубликованы
Енсея, Педли, 1966 [2854, 2855] То же, в пламенах H2+O2+N2, Li(r)+H2O(r)= — —238
=LiOH(r)+H(r), 2475 К, 1 точка
Мак Эван, Филлипс, 1967 [3453] То же, детали экспериментов не опубликованы — —238
Коттон, Дженкинс, 1969 [1941] То же, 2370 К, 10 измерений — —243±9
Келли, Педли, 1971 [2965] То же, в пламенах Н2+О2+СО2, 1950—2750 К, —238 —239+8
33 точки, данные представлены в виде уравнения
272
Литий и его соединения Li2O2H2(r), 1лР(к, ж)
3439], в которых принималась угловая модель молекулы LiOH, достигают 13 Дж-К^-моль
в значениях Ф°(Т) х.
Константа равновесия реакции LiOH(r) = Li(r)+O(r)+H(r) вычислена с использованием
значения Ari7o@)=863,552+8 кДж-моль", соответствующего принятой в справочнике энтальпии
образования
г, 0) = —243 + 8 кДж • моль.
В табл. 45.16 приведены результаты исследований равновесия реакций с участием LiOH(r).
Принятое значение является средним из результатов наиболее детальных исследований [1508,
1941, 2965]. При оценке погрешностей учтены воспроизводимость измерений, неточность сечений
ионизации и неточность термодинамических функций, приводящие к погрешностям в энтальпии
образования от 4 кДж-моль (измерения при 1400 К) до 7 кДж-моль (измерения при 2400 К).
1Л2О2Н2(г). Термодинамические свойства газообразного димера гидроксида лития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1064.
Молекулярные постоянные, использованные для расчета термодинамических функций Li2O2H2,,
приведены в табл. 45.9. Спектр и структура молекулы Li2O2H2 экспериментально не исследовались.
Молекулярные постоянные Li2O2H2 оценены сравнением с соответствующими постоянными К2О2На,;
Rb2O2H2, Cs2O2H2 и постоянными димеров фторидов щелочных металлов M2F2, принятыми в
настоящем справочнике. Произведение моментов инерции IaIbIc вычислено в предположении, что в
основном электронном состоянии ХхАд молекула Li2O2H2 имеет плоскую ромбическую структуру со
связями ОН, расположенными на одной прямой (симметрия D2h), и структурными параметрами
^ О—Li—0=105+5°, r(Li—О)=1,78+0,05 А, г(О—Н)=0,97+0,03 А. Погрешность значения
IaIbIc составляет 1 • 10~115 г3-см6. Погрешности частот Vj и vn оценены равными 100 см, всех
остальных — 10% 2 от их величины. Сведения о возбужденных электронных состояниях Li2OaH3 в
литературе отсутствуют.
Термодинамические функции 1Л2О2Н2(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены неточностью принятых значений молекулярных постоянных, а также приближенным
характером расчета и составляют 5, 12 и 20 Дж-К-моль в значениях Ф°(Т) при 298,15, 3000
и 6000 К.
Ранее термодинамические функции 1Л2О2Н2 вычислялись в справочниках JANAF [2835] и
Мак-Врайд и др. [3439] для молекулы симметрии C2h (с атомами водорода, находящимися в
трансположении относительно плоскости OLiOLi) и с другими значениями частот колебаний, особенно
четырех деформационных частот МОН A120 см в [3439] и 1250 см в [2835]). В связи с этим
расхождения в значениях Ф°(Г), приведенных в настоящем справочнике и в справочниках [2835]
и [3439], достигают 31 и 27 Дж«К-моль соответственно.
Константа равновесия реакции 1л2О2Н2(г)=21Л(г)+2О(г)+2Н(г) вычислена с использованием
значения Д,.#о@)=1956,104+20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(Li2O2H2, г, 0) = — 715 + 20 кДж • моль.
Принятая величина основана на результатах масс-спектрометрического исследования
равновесия реакции Li2O2H2(r)=2LiOH(r) A119—1448 К, 19 точек), выполненного Берковицем и др.
[1508]. Из этих измерений были вычислены значения —713+18 кДж-моль (III закон) и —737 +
+30 кДж-моль (И закон). Близкое, но менее точное значение —753+50 кДж-моль было
найдено по результатам исследования реакции 1Л2О(к)+Н2О(г)=1ЛаО2Н2(г) методом переноса A095
и 1145 К, 2 точки) в работе Берковиц—Маттук и Бюхлера [1510].
LiF(u, ж). Термодинамические свойства кристаллического и жидкого фторида лития в
стандартном состоянии при температурах 100—3500 К приведены в табл. 1065.
1 Значения термодинамических функций, вычисленные по аппроксимационным формулам, предложенным
Кулагиным и др. [602] для угловой модели LiOH, также отличаются от данных табл. 1063 на 4—13 Дж-К^-моль
в значениях Ф°(Т).
2 Отнесение частот к различным типам колебаний см. в тексте по КаО2Н2.
18 Заказ Н 1590 273
LiF(K, ж) Глава 45
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние LiF(K) в интервале 0—1122 К принимается
кубическая модификация LiF (структурный тип NaCl)x.
При Т <1 298,15 К термодинамические функции вычислены по данным, полученным
усреднением хорошо согласующихся между собой результатов измерений теплоемкости LiF в работах
Клузиуса и др. [1877, 1883] A9-272 К и 10—111 К), Стрелкова и др. (см. [337]) A8-300 К),
Джонса и Мартина [2893, 3400] B—30 К) и Скейлса [4165] B—7 К). Значения ?"B98,15 К)
и #"B98,15 К) —Я°@), приведенные в табл. 45.1, имеют погрешности 0,12 Дж-К'^моль и
0,012 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости 1ЛР(к) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии в работах Дугласа и Девера [2107] C73—1169 К),
Воскресенской и др. [253] F72—1411 К) и Маклеода [3337] E17—1371 К), с учетом данных
по теплоемкости [253] C18—658 К). При обработке погрешность данных [2107, 3337] оценивалась
в 0,2%, а данных [253] — в 0,4%. Температура плавления A122+1 К) принята по измерениям
Дугласа и Девера [2107] A121,25 К), проведенным на образце, содержавшем менее 0,1 % примесей,
с учетом поправки на переход к шкале МПТШ-68 (+0,80 К). С выбранным значением
удовлетворительно согласуются данные [161, 314, 497, 668, 1710, 2545, 2691, 3083, 4013] A120—1123 К).
Энтальпия плавления B7,08+0,20 кДж-моль) принята по согласующимся между собой
измерениям [2107] B7,06 кДж-моль) и [253] B7,10 кДж-моль); измерения [3337] привели к более
низкому значению B6,7 кДж-моль).
Теплоемкость 1ЛБ\ж) F3,8+1 Дж-К-моль) принята по результатам измерений энтальпии
[253] A128—1411 К, 9 измерений). Немногочисленные данные [2107] A127—1169 К, 3 измерения)
и [3337] A151—1371 К, 5 измерений) приводят к более высоким значениям F4,8 и
66,8 Дж-К^-моль соответственноJ.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
0,2, 1,5 и 4 Дж-К-моль соответственно.
Значения термодинамических функций LiF(k, ж), опубликованные ранее в справочниках [337]
(Т < 3400 К), [2834] (Т < 3000 К) и [2960] (Т < 1500 К), согласуются с данными табл. 1065
в пределах 0,3 Дж-К-моль~1 в значениях Ф°(Т).
Принятая в справочнике энтальпия образования
к, 298,15 К) = —618,3 ± 0,7 кДж.моль
основана на приведенных в табл. 45.17 измерениях энтальпии растворения LiF(K) в воде. Для
дальнейших расчетов принимается средневзвешенное значение Дяг#°(ооН20; 298,15 К)=4,45 +
+ 0,20 кДж-моль, которому и соответствует принятая величина. Измерения энтальпии растворе-
Таблица 45.17. Результаты определений AarH"°(LiF, к-»-соН2О, 298,15 К) (в кДж-моль)
Авторы
Конечная концентрация, температура
измерений
ДагЯ°(ооН2О, 298,15 К)
Капустинский, Стаханова, 1956 [484] 1600 Н2О, 298,15 К, 1 измерение 4,55
Колесов, Скуратов, 1961 [533] 3800 Н2О, 294,65 К, 10 измерений8 4,59±0,20
3800 Н2О, 294,65 К, 5 измерений6 4,70±0,20
Вулф, 1962 (см. [2834]) 298,15 К 4,60+0,42
Стефенсон и др., 1964 [4387] 1400—2780 Н2О, 298,15 К 4,48+0,21
Кокс, Харроп, 1965 [1950] 6310-8800 Н2О, 298.15К, 5 измерений 4,24+0,17
Гермейн и др., 1979 [2390] 10884 Н2О, 298,15 К, 8 измерений 4,30±0,21
а Калориметр с изотермической оболочкой. *> Калориметр с адиабатической оболочкой.
1 Сообщения о фазовых превращениях LiF при 1035 и 1100 К (метод ДТА) [516, 739] не были подтверждены
в последующих работах.
2 Маклеод [3337] на основании своих измерений рекомендовал уравнение, согласно которому теплоемкость
LiFEK) круто растет от С°р A122 К)=63,4 Дж-К^-моль-* до С° A400 К)=70,8 Дж• К • моль" и С°р B000 К) =
= 87,5 Дж-К^-моль (экстраполяция).
274
Литий и его соединения
LiF(r)
ния, выполненные Форкраном [2300, 2301], относятся к 288 К и их приближенный пересчет
к 298 К приводит к более низкому значению стандартной энтальпии растворения.
Давление пара в реакции LiF(K, }K)=LiF(r) вычислено с использованием принятой в
справочнике энт альпии сублимации
Дг#°@) = \H°(LiF, к, 0) = 275,0 ± 3,0 кДж . моль.
Принятая величина основана на измерениях давления насыщенного пара LiF(K, ж). Насыщен^
яый пар фторида лития содержит молекулы LiF, Li2F2 и Li3F3. На основании данных [8, 28, 309,
2166] и принятых в справочнике. термодинамических свойств при rm(LiF)=1122 К содержание
этих соединений составляет 45, 43 и 12 мол.%. Методика выбора термохимических величин для
индивидуальных компонентов насыщенного пара галогенидов палочных металлов изложена в
Приложении 8.
Результаты расчетов энтальпии сублимации LiF(K) в виде мономерных молекул представлены
в табл. 45.18. Приведенные в таблице погрешности учитывают только воспроизводимость измере-
Таблпца 45.18. Результаты определений Ag-ff°(LiF, к, 0)
Авторы
Вартенберг, Шульц,Ч921 [4663]
Руфф и др., 1922 [4096]
Эйзенштадт и др., 1958 [2166]
Сенс, Стоун, 1958 [4235]
Пью, Барроу, 1958 [3896]
Евсеев и др., 1959 [395]
Акишин и др., 1959 [8];ТГорохов,
1972 F3091
Хилденбранд и др., 1964 [2647]
Алиханян и др., 1972 [28]
Топор, 1976 [4527]
Дабрингхауз, Мейер, 1977 [1982]
Гримли и др., 1978 [2493]
Метод
Точек кипения, 1626—1820 К, .5 точек
То же, 1671—1936 К, 14 точек
Эффузионный, 1072—1158 К, 7 точек
Протока, 1134—1433 К а
Торзионно-эффузионный, 1001—1126 К, 48 точек
Эффузионный, 926—1052 К, 12 точек
Масс-спектрометрический, 1037—1120 К а
Торзионно-эффузионный, 997—1159 К, 24 точки а
Масс-спектрометрический, 1102 К, 1 точка
Статический, 1387—1483 К, 17 точек
Масс-спектрометрический, 926—1093 К а
То же, 1058—1091 К, 6 точек а
а Данные представлены в виде уравнения.
As#°(LiF, к, 0)
II закон
288+24
288+6
275 ±12
275
а 282
241+18
283 ±6
280+8
—
290+8
268+6
270+6
III закон
273,1 ±1,2
270,9 ±0,5
282,6 ±1,0
276,7+0,9
275,1 ±1,1
262,7 ±0,9
—
274,9 ±0,5
274,7 ±3,0
275,2 ±0,2
—
ний. Принятое в справочнике значение является средневзвешенным из данных [2647, 3896,
4235, 4527, 4663]. Погрешность принятого значения оценена с учетом воспроизводимости
измерений, расхождения результатов измерений в разных работах, неточности термодинамических
функций и неточности вычисления парциального давления мономера.
LiF(r). Термодинамические свойства газообразного фторида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1066.
Приведенные в табл. 45.4 молекулярные постоянные LiF приняты на основании анализа
результатов исследований микроволнового [3790, 4595, 4717] и ИК-спектров [60, 170, 4613] этой
молекулы, а также квантовомеханических расчетов [1626, 2430, 2912, 3461, 3462]. Электронный спектр
LiF не исследовался. Неэмпирические расчеты [1626, 2912], выполненные с учетом
конфигурационного взаимодействия, показали, что первое возбужденное электронное состояние этой
молекулы имеет энергию около 50 000 см. Полуэмпирические оценки постоянных LiF были
выполнены в работах [558, 567, 2700, 3531, 3532, 3749, 3780, 4009, 4443].
Приведенные в табл. 45.4 постоянные LiF в состоянии X1 ? + получены Пирсоном и Горди [3790]
обработкой результатов измерений микроволновых переходов / -* /+1 (/ ^ 6) на колебательных
уровнях с v=0, 1, 2 и 3. Соответствующие постоянные юе и шехе были получены не непосредственно
по экспериментальным данным, а рассчитаны с помощью соотношений Данхема. Они хорошо
согласуются с результатами исследования ИК-спектра [4613] ((«,,=910,34, о>л=7,929 см).
275
Li2F2(r)
Глава 45
Термодинамические функции LiF(r) были вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производные вычислялись без учета возбужденных состояний
по методике, изложенной в Приложении 7. Использованные в расчете коэффициенты
потенциальной функции d0, йг и d2 приведены в табл. 45.5а и получены по принятым молекулярным постоянным
LiF. Расчет был выполнен для природной изотопической смеси изотопов лития.
Погрешности рассчитанных термодинамических функций LiF(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным учетом
изотопного состава фторида лития. При более высоких температурах дополнительный вклад
в погрешность вносит неточность примененного способа экстраполяции потенциальной функции.
Погрешности в значениях Ф°(Г) составляют 0,03, 0,05 и 0,1 Дж-К^-моль при 298,15, 3000 и
6000 К соответственно.
Термодинамические функции LiF(r) ранее вычислялись в предыдущих изданиях настоящего
справочника [337, 340], в таблицах JANAF [2834], справочниках Хаффа и др. [4978], Мак-Брайд
и др. [3439], Мадера [3338], а также в работах Уилкинса и др. [4729], Альтмана [1303] (Т ^
< 6000 К), Кучерека и Папоушека [3132] (Т < 3500 К), Брюэра и Брэккет [1660] (Ф°(Г), Т <
< 2000 К), Уилкинса [4728] (Т < 6000 К). Расхождения данных [337, 340, 2834, 3439] и табл. 1066
не превышают 0,5 и 1,3 Дж-К-моль в значениях Ф°(Г) и S°(T) соответственно и объясняются
тем, что в этих справочниках использовался приближенный метод расчета.
Константа равновесия реакции LiF(r)=Li(r)+F(r) вычислена с использованием значения
Ar#o@)=D0(LiF)=575,739+3,3 кДж-моль~1л;48 130+280 см, соответствующего принятым
в справочнике энтальпии образования и сублимации LiF(K). Этим величинам также соответствует
A^°(LiF, г, 0) = —340,729 + 3,1 кДж • моль.
Булевич и др. [1719] и Пейдж и Сагден [3745] на основании спектрофотометрического
исследования равновесия реакции образования LiF(r) (пламена H2+O2+N2, 1500—2600 К) получили
близкие, но неточные значения энергии диссоциации E70+35 и 590+30 кДж-моль).
Li2F2(r). Термодинамические свойства газообразного дифторида дилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1067.
Молекулярные постоянные, использованные для расчета термодинамических функций,
приведены в табл. 45.19. Результаты изучения ИК-спектра [1203, 2331, 3034, 3266, 3956, 4190, 4335,,
Таблица 45.19. Значения молекулярных постоянных, а также о и р%, принятые для расчета
термодинамических функций Li2F2, Li3F3, Li2CI2, Li3Cl3, Li2Br2, Li3Br3, Li2I2, Li3I3
Молекула
Li2F2
Li3F3
Li2Cla
Li3Cl3
Li2Br2
Li3Br3
Li2I2
Li3I3
Состояние
¦
XUg 646
I1A1 507
1У, 450
lUi 380
l1Ag 410
XlAx 352
X^Ag 365
yi л о\ а
v2
445
307
290
169
180
104
130
74
v3
411
747
330
527
280
463
258
410
"
287
267
174
162
150
140
160
127
v5
CM
553
736 B)
352
533 B)
303
479 B)
248
427 B)
641
509 B)
480
335 B)
430
270 B)
375
229 B)
v7
258 B)
—
188 B)
—
176 B)
—
161 B)
.
154 B)
—
94B)
—
81B)
73B)
IAIBIC. 101"
Г3 • CM8
460
9-103
64-102
21-10*
42-103
28-105
18-10*
14.10"
о
4
6
4
6
4
6
4
6
PS
1
1
1
1
1
1
1
1
4338] и электронографических исследований структуры [14, 961, 963, 964], а также теоретические
расчеты [144, 961, 1195, 1421, 1605, 1696, 4100, 4689] показали, что в основном состоянии XхА
молекула Li2F2 имеет плоскую ромбическую структуру (симметрия D2h). Линейный изомер LiFLiF
согласно неэмпирическому расчету [965] неустойчив по отношению к деформационному колебанию
и является седловой точкой на потенциальной поверхности. Произведение моментов инерции,
приведенное в табл. 45.19, отвечает структурным параметрам r(Li—F) = l,74+0,01 А14 F—Li—F=
=104,4+2,2°, найденным Соломоником [961, 963, 964] при электронографическом исследовании
276
Литий и его соединения
паров hi2F21. Погрешность значения IaIbIc составляет 2-Ю16 г3-см6. Приведенные в табл. 45.19
основные частоты v4, v6 и ve найдены Снелсоном [4335 ] при исследовании ИК-спектра молекул Li2F2,
изолированных в матрице из Ne, они хорошо согласуются со значениями, полученными в более
тяжелых матрицах [1203, 2331, 3266, 3956, 4190, 4338] 2. Для частот vx, v2 и v3, ненаблюдавшихся
экспериментально, в табл. 45.1 приведены значения, вычисленные Соломоником [961, 963]
с использованием ионной модели 3. Погрешности принятых значений частот v4—v6 оцениваются
в 20 см, а погрешности рассчитанных значений vx, v2, v3 могут достигать 100 см.
Термодинамические функции Li2F2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных состояний и изомеров этого соединения. Погрешности рассчитанных
величин обусловлены неточностью принятых молекулярных постоянных, а также приближенным
характером расчета, они составляют 4, 10 и 12 Дж-К-моль в значениях Ф°(Г) при 298,15,
3000 и 6000 К соответственно.
Ранее термодинамические функции Li2F2(r) рассчитывались в справочниках JANAF [2834]
и Мак-Брайд и др. [3439], а также в работах Сивина и др. [1975] (до 2000 К), Уилкинса и др.
[4729] (до 5000 К) и Краснова и др. [569] (до 5000 К). Расхождения данных [2834, 3439] и
табл. 1067 достигают 2 и 4 Дж-К~1-моль в значениях Ф°(Т) соответственно; они обусловлены
различием принятых постоянных Li2F2. Результаты расчетов [569, 1975] практически совпадают
с приведенными в табл. 1067 (расхождения не превышают 1 Дж-К^-моль).
Константа равновесия реакции Li2F2(r)=2Li(r)-f 2F(r) вычислена с использованием значения
Дг#°@)=1401,020 +8,3 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Ayff°(Li2F2, г, 0) = —931 + 8 кДж • моль.
Принятая величина основана на результатах исследования равновесия реакции LiF(K, ж)+
-f-LiF(r)=Li2F2(r), приведенных в табл. 45.20, где в качестве погрешностей указана воспроизводи-
Таблица 45.20. Результаты определений энтальпии реакции LiF(K, ж) + LiF(r) = Li2F2(r)
(в кДж • моль)
Авторы
Эйзенштадт и др., 1958 [2166]
Акишин и др., 1959 [8]; Горохов,
1972 [309]
Алиханян и др., 1972 [28]
Дабрингхауз, Мейер, 1977 [1982]
Гримли и др., 1978 [2493]
Метод
Эффузионный с селектором скоростей, 1072—
1158 К, 7 точек
Масс-спектрометрический с двойной эффузионной
ячейкой, 1111 и 1118 К, 2 точки
Масс-спектрометрический, 1037—1120 К а
Масс-спектрометрический с двойной эффузионной
ячейкой, 1102 К, 1 точка
Масс-спектрометрический, 926—1093 К а
То же, 1058—1098 К, 6 точек а
а Данные представлены в виде уравнения.
ДГЯ
II закон
7+20
—
22,2+2,0
—
26 + 10
37+8
"(О)
III закон
28,5+0,7
25,9+2,0
—
22,5
—
—
мость измерений. В справочнике для последующих расчетов принимается значение АгН°@)=-
= 25,0+7,0 кДж-моль, являющееся средневзвешенным из этих данных, причем больший вес
придавался измерениям, выполненным с применением двойных эффузионных ячеек [8, 28, 309].
Приведенная погрешность учитывает неточность термодинамических функций Li2F2.
3 В работе [14] было получено неверное значение г (Li—F)=l,68+0,02 А из-за пренебрежения содержанием
в паре мономерных молекул и наличия систематической ошибки (см. [961]).
2 Клемперер и Норрис [3034] в ИК-спектре паров фторида лития при температурах 2300—2800 К наблюдали
широкие размытые полосы в области 460 и 640 см и отнесли их к колебаниям v5 и ve молекулы Li2F2.
Значение частоты v6 совпадает с частотой, найденной в матрице, а полоса в области 460 см, очевидно, не
принадлежит молекуле Li2F2.
3 Частоты колебаний и межъядерные расстояния димерных молекул галогенидов щелочных металлов
рассчитывались также в работах [144, 1605, 1696, 4554, 4689].
277
LisF3(r) Глава 45
В работах [4883, 4898] было изучено равновесие реакции Li2F2(r)=2LiF(r) A017—1139 К,
7 измерений). Обработка этих данных по методу III закона приводит к значениям ДгЯ°@)=260 +
+12 кДж-моль и 255+12 кДж-моль, которые хорошо согласуются с величиной 250 +
+8 кДж-моль, соответствующей принятым в справочнике энтальпиям образования Li2F2(r)
и LiF(r). В работах [28, 309, 1509, 2166, 5042а] приведены данные, позволяющие найти значение
энтальпии этой же реакции по уравнению II закона B57+15 кДж-моль).
^ Li3F3(r). Термодинамические свойства газообразного трифторида трилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1068.
Молекулярные постоянные, использованные для расчета термодинамических функций Li3F3(r),
приведены в табл. 45.19. Структура молекулы Li3F3 экспериментально не изучалась. Исследования
инфракрасного спектра [2331, 4335], а также результаты расчета, выполненного Уэлчем и др.
[4689] с использованием ионной модели, показывают, что в основном электронном состоянии Х1А1
эта молекула имеет плоскую гексагональную конфигурацию (симметрия D3h). Приведенное
в табл. 45.19 произведение моментов инерции соответствует значению r(Li—F)=l,61+0,l А,
найденному Уэлчем и др. [4689] х, его погрешность оценивается в 5-Ю14 г3-см6. Приведенные
в табл. 45.19 значения частот v4, v5, v6 найдены Снелсоном [4335] в ИК-спектре молекул Li3F3,
изолированных в матрице из Ne. С учетом матричного сдвига они согласуются со значениями,
полученными в работах [1203, 2331] при исследовании спектра молекулы Li3F3, изолированных
в матрице из Аг2. Для частоты v7 принята величина, оцененная Соломоником [961, 962] на
основании данных, [1203, 2331 ]3. Частоты колебаний vx—v3, v8, которые неактивны в ИК-спектре,
вычислены Соломоником [961, 962] по уравнениям простого поля валентных сил. Погрешности
принятых значений v4, v5, v6, v7 с учетом возможного матричного сдвига оцениваются в 15—30 см,
а частот vx—v3, v8 в 20—25% от их величин.
Термодинамические функции Li3F3(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных электронных состояний. Их погрешности обусловлены неточностью
принятых молекулярных постоянных, а также приближенным характером расчета; они составляют 8,
15, 25 Дж-К-^моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Li8F3(r) рассчитывались в справочниках JANAF [2834],
Мак-Брайд и др. [3439], а также в работе Соломоника и Даниловой [962] (до 5000 К). Результаты
расчетов [2834] и [962] согласуются с данными табл. 1068 в пределах 1—2 Дж-К~1-моль~1 в
значениях Ф°(Т). Данные [3439] из-за неверной оценки основных частот Li3F3 отличаются от табл. 1068
на 20—30 Дж-К^-моль-1.
Константа равновесия реакции Li3F3(r)=3Li(r)-f 3F(r) вычислена с использованием значения
Дг#°@)=2223,030+10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
&fH°(Li3Fs, г, 0)=:—1518 + 10 кДж • моль.
Принятая величина основана на результатах исследований равновесия реакции 2L1F(k, ж)+
+LiF(r)=Li3F3(r), приведенных в табл. 45.21. В справочнике для последующих расчетов
принимается значение Дг#°@)=55+12 кДж-моль, являющееся средневзвешенным из имеющихся
данных, причем больший вес придавался измерениям с двойными эффузионными ячейками [8, 28,
309 ]. Приведенная погрешность учитывает неточность термодинамических функций Li3F3(r). Масс-
спектрометрическое исследование равновесия реакции Li3F3(r)=3LiF(r) в работе. [4898] A059 К,
i
Неэмпирический расчет Свепстона и др. [4449] подтвердил циклическую структуру Li3F3. Найденному при
оптимизации геометрии Li3F3 искажению шестичленного кольца соответствует изменение термодинамических
функций, не превышающее 2 Дш-К^-моль.
2 Авторы [1203] ошибочно отнесли наблюдаемые ими особенности в ИК-спектре продуктов испарения LiF в
областях 270—255 и 261—246 см к линейному димеру Li2F2. В работах [961, 2331] показано, что эти полосы
связаны с колебаниями молекулы Li3F3.
3 В работах Абрамовпца и др. [1203] и Фрейберга и др. [2331] не обнаружено полосы, соответствующей полосе
230 см~1, полученной в Ne-матрице [4335], и в то же время найдены полосы 245,7 см [1203] и 244,0 см"
[2331], которых нет в Ne-матрице. Это расхождение может быть объяснено трудностями получения спектра
в области, члизкой к пределу чувствительности прибора (в работе [4335] около 200 см), в связи с чем
полоса 230 см не рассматривается в настоящем справочнике.
278
Литий и его соединения
ЫС1(к, ж)
Таблица 45.21. Результаты onpeji
(в кДж • моль)
Авторы
елений энтальпии реакции 2LiF(u, ж) + LiF(r) = Li3F3(r)
Метод
ДгЯ°@)
II закон
III закон
Эйзенштадт и др., 1958 [2166] Эффузионный с селектором скоростей, 1072— 96+60 63,0+2,0
1158 К, 7 точек
Акшпин и др., 1959 [8]; Горохов, Масс-спектрометрический с двойной эффузион- — 52,0
1972 [309] ной ячейкой, 1118 К, 1 точка
Масс-спектрометрический, 1037—1120 Ка 72+8 —
Алиханян и др., 1972 [28] Масс-спектрометрический 1102 К, 1 точка — 57,0
Дабрингхауз, Мейер, 1977 [1982] Масс-спектрометрический, 926—1093 Ка 35+10 —
Гримли и др., 1978 [2493] То же, 1058—1098 К, 6 точек а 65+8 —
а Данные представлены в виде уравнения.
3 измерения) приводит к значению Дг.й"о@)=490+20 кДж-моль (III закон) для этой реакции;
это значение близко к значению, соответствующему принятым в справочнике энтальпиям
образования Li3F3(r) и LiF(r) D96+16 кДж-моль).
1ЛС1(к, ж). Термодинамические свойства кристаллического и жидкого хлорида лития в
стандартном состоянии при температурах 100—3200 К приведены в табл. 1069.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние ЫС1(к) в интервале 0—883 К принимается
кубическая модификация (структурный тип NaCl).
При Т ^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости, выполненных в работах Шерли [4267] A4—320 К) и Мойера [3572] C—5 К). Данные Сталла
и др. [4420] A5—298 К) по точности несколько уступают данным [4267] и при температурах
вблизи 298 К, по-видимому, завышены примерно на 1%. Измерения средней теплоемкости 1ЛС1,
выполненные Слонимом и Хюттигом4 [4314] (85—270 К), неточны и не учитывались.
Принятые значения ?°B98,15 К) и #°B98,15 К) — #°@) (см. табл. 45.1) имеют погрешности
0,15 Дж-К~1-моль и 0,015 кДж-моль соответственно.
При Т ^> 298,15 К для теплоемкости принято уравнение, найденное совместной обработкой
данных по энтальпии 1лС1(к), полученных Дугласом и др. B98—850 К) (см. [2834]) и Родигиной
и сотр. [884] C67—878 К). При обработке погрешность данных Дугласа оценивалась в 0,7%,
данных [884] — в 2%.
Температура плавления (883+2 К) принята по согласующимся между собой результатам
измерений в работах Родигиной и др. [884] (883+1 К), Хендлера и др. [2545] (883+2 К), Богача и
Трзебятовского [1599] (883 К), Шмитц-Дюмонта и Шмитца [4196] (882 К); Сафонова и др. [909]
(885 К). Измерения авторов [20, 154, 163, 367, 436, 441, 848, 1567, 1711, 2267, 2409,
3991, 4663], по-видимому, вследствие трудностей, связанных с получением чистых и сухих
образцов LiCl, привели к менее надежным результатам (879—887 К). Энтальпия плавления
A9,75+0,20 кДж-моль) принята по согласующимся данным Родигиной и др. [884]
A9,54 кДж-моль), Дугласа A9,74 кДж-моль; цитируется по [2834])
[2137]
A9,92 кДж-моль). Заниженные значения были получены в результате криометрических
определений [4819] A5,1 кДж-моль) и [1576] A6,1+0,6 кДж-моль); в работе [4663] была
вычислена приближенная величина (~20,9 кДж-моль).
Теплоемкость 1ЛС1(ж) F3,2+1 Дж-К-моль) получена на основании обработки
экспериментальных данных [884] по энтальпии (883—1076 К, 10 измерений). По неопубликованным данным
Дугласа (см. [2834]) теплоемкость 1ЛС1(ж) значительно уменьшается в интервале 900—1100 К.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
0,5, 2 и 5 Дж-К^-моль.
Расхождения между термодинамическими функциями, приведенными в табл. 1069 и в
справочниках [337] (Т < 3200 К), [1395] (Т < 1600 К) и [2834] (Т < 2000 К), не превышают
1 Дж-К^-моль в значениях Ф°(Г).
279
LiCI(K, ж)
Глава 45
Таблица 45.22. Результаты определений Дв-Я"оAЛС1, к^-соН2О, 298,15 К) (в кДжмоль)
Авторы
Томсен, 1883 [4511]
Пиккеринг, 1888 [3839]
Бонефуа, 1897 [1612, 1614]
Хейг, 1912 [2548]
Ланге, Дюрр, 1926 [3177]
Майер, 1927 [3353]
Вассерман, 1930 [4665]
Аскю и др., 1934 [1354]
Ланге, Мартин, 1937 [3179]
Самойлов, 1951 [899, 900]
Самойлов, Буслаева, 1960 [902]
Дракин, Чжан Ю-Линь, 1964 [385]
Крестов, Абросимов, 1967 [579]
Цветков, Рабинович, 1969 [1097]
Воробьев и др., 1974 [246]
Падунова, 1975 [748]
Джоли, Перашон, 1977 [2884, 2885]
Конечная концентрация, температура
измерений
200—300 Н2О, 291,65 К, 3 измерения
200 Н2О, 291,15 К, 1 измерение
220 Н2О, 288,15 К, 1 измерение
400 Н2О, 294,15 К, 3 измерения
3—1400 Н2О, 298,15 К, 78 измерений
230 Н2О, 292,65 К, 1 измерение
460—700 Н2О, 291,45 К, 4 измерения
1400 Н2О, 293,15 К, 1 измерение
280 Н2О, 298,15 К, 1 измерение
925 Н2О, 298,15 К, 1 измерение
925 Н2О, 298,15 К, 3 измерения
230—700 Н2О, 298,15 К, 4 измерения
273,15—373,15 К
500 Н2О, 298,15 К, 1 измерение
420 и 1800 Н2О, 298,15 К, 7 измерений
760 Н2О, 298,15 К
1040 Н2О, 298,15 К, 1 измерение
ДагЯ°(оэН2О,
298,15 К)
—36,38
—36,97
—36,38
—36,87+0,17
—36,25
—37,14+0,13
—36,33
-37,50
-36,74
—36,78+0,42
—36,96+0,20
-36,92+0,37
—37,58+0,30
—37,15+0,10
—36,80+0,25
—35,96+0,38
Принятая в справочнике энтальпия образования
bfH°(LiC\, к, 298,15 К) =—408,54 ± 0,20 кДж • моль
основана на измерениях энтальпии растворения 1ЛС1(к) в воде, представленных в табл. 45.22.
Как видно из таблицы, результаты измерений расходятся больше, чем это можно объяснить
воспроизводимостью измерений, что свидетельствует о наличии систематических ошибок, вероятно
связанных с гигроскопичностью этой соли. Придавая основной вес измерениям, выполненным
Воробьевым и др. [246], Ланге, Дюрром [3177] и Дракиным, Чжан Ю-Линем [385], в
справочнике для дальнейших расчетов используется значение kaqH° (ooHaO; 298,15 К) = —37,00 +
+0,15 кДж-моль.
Таблица 45.23. Результаты определений AsiT°(LiCl, к, 0) (в кДж • моль)
Авторы
Руфф, Мугдан, 1921 [4094]
Вартенберг, Шульц, 1921 [4663]
Майер, 1925 [3352]
Нива, 1938 [3665]
Кангро, Викинг, 1938 [2919]
Миллер, Куш, 1957 [3504]
Несмеянов, Сазонов, 1959 [720]
Милн, Клейн, 1960 [3510]
Хилденбранд и др., 1964 [2647]
Шнып, Новиков, 1972 [1156]
Горохов, 1972 [309]
Бурылев, Миронов, 1976 [158]
Шэфер, Вагнер, 1979 [4175]
Метод
Точек кипения, 1318—1598 К, 7 точек
То же, 1442—1657 К, 12 точек
Статический, 1154—1525 К, 10 точек
Эффузионный, 783—823 К, 5 точек
Протока, 1133—1263 К, 3 точки
Эффузионный, 870 К, 2 точки
То же, 808—889 К, 19 точек
Масс-спектрометрический, средняя температура
800 К а
Торзионно-эффузионный, 798—921 К а
Точек кипения, 1250—1460 К а
Масс-спектрометрический, 823—868 К а
Точек кипения, 1202—1421 К а
Масс-спектрометрический, 721—872 К а
а Данные представлены в виде уравнения.
As#°(LiCl, к, 0)
II закон
222+10
218+5
212+25
205 ±6
199±18
—
212+7
233
202 ±6
200
204
218
217
III закон
210,9+0,9
214,9+0,3
216,3+2,0
214,6+0,6
216,5+2,5
221+14
219,6+0,2
—
216,2+0,6
211,8+0,9
—
211,7+1,4
—
280
Литий и его соединения LiCl(r)
Давление пара в реакции 1лС1(к, ж)=1лС1(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = ДД°(ЫС1, к, 0) = 215,0 + 4,0 кДж • моль.
Принятая величина основана на измерениях давления и состава насыщенного пара 1ЛС1(к, ж).
Насыщенный пар хлорида лития содержит молекулы LiCl, Li2Cl2, Li3Cl3. На основании данных
[309, 3504] и принятых в справочнике термодинамических свойств содержание этих соединений
при Г)ИAЛС1)=883 К составляет 20, 74 и 6 мол.% соответственно. Методика определения
термохимических величин для индивидуальных компонентов насыщенного пара галогенидов щелочных
металлов изложена в Приложении 8.
Результаты расчетов энтальпии сублимации 1лС1(к) в виде мономерных молекул представлены
в табл. 45.23. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятое в справочнике значение является средневзвешенным из данных табл. 45.23. Его
погрешность оценена с учетом воспроизводимости измерений, расхождения результатов измерений
в разных работах, неточности термодинамических функций и неточности вычисления парциального
давления мономерных молекул.
LiCl(r). Термодинамические свойства газообразного хлорида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1070.
Приведенные в табл. 45.4 молекулярные постоянные LiCl приняты на основании анализа
результатов исследований микроволнового [1876, 3253, 3790] и ИК-спектров [3035, 3036] этой
молекулы. Электронный спектр LiCl не исследовался. По аналогии с молекулой LiF принимается,
что валентные возбужденные электронные состояния LiCl имеют энергии больше 40 000 см.
Полуэмпирические оценки постоянных LiCl были выполнены в работах [558, 566, 567, 2700, 4009].
Приведенные в табл. 45.4 колебательные и вращательные постоянные LiCl в состоянии ХгЕ +
получены Пирсоном и Горди [3790] обработкой результатов измерений микроволновых переходов
/ -»• /+1 (/ <^ 12) на колебательных уровнях с v=0, 1, 2 и 3. Соответствующие постоянные ше
и юехе были получены не непосредственно по экспериментальным данным, а рассчитаны с помощью
соотношений Данхема. Они хорошо согласуются с результатами исследования ИК-спектра [3035]
(све=641±3, (вЛ=4,2+0,3 см).
Термодинамические функции LiCl(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 45.5а и получены по принятым
молекулярным постоянным LiCl. Расчет был выполнен для природной изотопической смеси изотопов
лития и хлора.
Погрешности рассчитанных термодинамических функций LiCl(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным учетом
изотопного состава хлорида лития. При более высоких температурах дополнительный вклад в
погрешность дает неточность принятого способа экстраполяции потенциальной функции.
Погрешности в значениях Ф°(Г) составляют 0,03, 0,05 и 0,2 Дж-К^-моль при 298,15, 3000 и 6000 К
соответственно.
Термодинамические функции LiCl(r) ранее вычислялись в предыдущих изданиях настоящего
справочника [337, 340], в таблицах JANAF [2834], справочниках Мак-Брайд идр. [3439], Мадера
[3338], а также в работах Уилкинса и др. [4729], Альтмана [1303] (Т <; 6000 К), Брюэра и Брэк-
кет [1660] (Ф°(Т), Т < 2000 К), Папоушека и Валентовой [3768] (Т < 3500 К), Уилкинса [4728]
(Т < 6000 К). Расхождения данных [337, 2834, 3439] и табл. 1070 не превышают 0,5 и
2 Дж-К-1-моль~1 в значениях Ф°(Т) и S°(T) соответственно и объясняются тем, что в этих
справочниках использовался приближенный метод расчета.
Константа равновесия реакции LiCl(r) = Li(r)+Cl(r) вычислена с использованием значения
Af.//°@)=D0(LiCl)=470,973 + 4,l кДж-моль яа 39 370±340 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации Ь>1С1(к), Этим величинам также соответствует
kfH°(LiCl, г, 0) = —193,618 ± 4,0 кДж • моль.
С приведенным значением энергии диссоциации согласуются в пределах их погрешности
значения, найденные Берковицем и др. [1500] при фотоионизационных измерениях D60 кДж-моль)
281
Ы2С12(г)
Глава 45
и Булевич и др. [1719] при спектрофотометрическом исследовании равновесия реакции
образования LiCl в пламенах D62+13 кДж-моль).
1Л2С12(г). Термодинамические свойства газообразного дихлорида дилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1071.
Молекулярные постоянные, использованные для расчета термодинамических функций Li2Cl2,
приведены в табл. 45.19. Результаты изучения ИК-спектра [2331, 3034, 4190, 4338] и электроно-
графические исследования [14, 1433] показывают, что молекула Li2Gl2 в основном электронном
состоянии ХхАд имеет плоское ромбическое строение (симметрия Du). Приведенное в табл. 45.19
произведение моментов инерции соответствует структурным параметрам r(Li—С1)=2,23+0,03 А
и ^ С1—Li—С1=108,4 + 1,5°, найденным Бауэром и др. [1433] при электронографическом
исследовании паров Li2Cl2 г. Погрешность значения IaIbIc оценивается в 1-Ю14 г3-см6. Приведенные
в табл. 45.19 значения частот v5, vg найдены Шликом и Шнеппом [4190] и Снелсоном и Питцером
[4338], а величина v4 — Фрейбергом и др. [2331] в ИК-спектрах молекул Li2Gl2, изолированных
в матрице из Аг. Найденные Клемперером и Норрисом [3034] в ИК-спектре газообразного Li2Cl2
при высоких температурах частоты v5=335 и ve=460 см в пределах погрешностей согласуются
с принятыми значениями. Для ненаблюдавшихся экспериментально частот vx—v3 приняты
значения, рассчитанные Соломоником [961, 963] с использованием ионной модели. Погрешности
экспериментально наблюдаемых частот v4, v5, ve с учетом матричного сдвига оцениваются в 20 см,
а частот vx, v2, v3 — в 60—90 см.
Термодинамические функции Li2Cl2(r) вычислены в приближении «жесткий ротатор — гармо-
лический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний и возможных изомеров (см. LiF(r)). Их погрешности
обусловлены неточностью принятых молекулярных постоянных, а также приближенным
характером расчета и составляют 4, 10 и 12 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции Li2Gl2(r) рассчитывались в справочниках JANAF [2834]
и Мак-Брайд и др. [3439], а также в работах Бауэра и др. [1433] (до 5000 К) и Краснова и др. [569]
(до 3000 К). Расхождения данных [2834, 3439] и табл. 1071 достигают 4 Дж-К-моль~1 в
значениях Ф°(Т) и объясняются некоторым отличием в принятых значениях основных частот.
Результаты расчета [569] практически совпадают с данными табл. 1071 (расхождения не превышают
1 Дж-К^-моль.
Константа равновесия реакции Li2Cl2(r)=2Li(r)+2Cl(r) вычислена с использованием значения
ДгЯ°@) = 1149,710+6,3 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
kfH°(Lifil2, г, 0) = —595 + 6 кДж • моль.
Таблица 45.24. Результаты определений энтальпии реакции ЫС1(к, ж) + LiCI(r) = Li2Cl2(r)
(в кДж • моль)
Авторы
Метод
Миллер, Куш, 1957 [3504] Эффузионный, с селектором скоростей, 870 К,
1 точка
Милн, Клейн, 1960 [3510] Масс-спектрометрический, средняя температура
~800 К
Горохов, 1972 [309] Масс-спектрометрический с двойной эффузионной
ячейкой, 890 К, 1 точка
Масс-спектрометрический, 823—868 К а
Шефер, Вагнер, 1979 [4175] То же, 721—872 К а
а Данные представлены в виде уравнения.
дгд
II закон
-3,3
19+14
7
°@)
Ш закон
7,1
—
7,3
—
—
1 В работе [14] было получено значение г (Li—С1)=2,17+0,01 А, по-видимому, из-за систематической ошибки
и пренебрежения сложным составом пара (см, [961]),
282
Литий и его соединения
Li3CI3(r)
Принятая величина основана на результатах исследований равновесия реакции 1ЛС1(к, ж)+
+LiCl(r)=Li2Cl2(r), приведенных в табл. 45.24. В справочнике для дальнейших расчетов
принимается среднее значение ДгЯ°@)=7+7 кДж-моль; его погрешность учитывает неточность
термодинамических функций, использованных в расчетах.
В работах [309, 1509, 3504, 3510] приведены данные, позволяющие рассчитать по уравнению
II закона значение энтальпии реакции Li2Cl2(r)=2LiCl(r), Дг#°@)=205+20 кДж-моль. Эта
величина в пределах погрешности совпадает с величиной 208+10 кДж-моль, вычисленной на
основании принятых в настоящем справочнике энтальпий образования 1Л2С12(г) и LiCl(r).
1Л3С13(г). Термодинамические свойства газообразного трихлорида трилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1072.
Молекулярные постоянные, использованные для расчета термодинамических функций Li3Cl3,
приведены в табл. 45.19. Структура и спектр молекулы Li3Cl3 экспериментально не исследовались.
По аналогии с молекулой Li3F3 для Li3Cl3 в основном электронном состоянии ХХА^ принята
плоская гексагональная конфигурация (симметрия D3h). Приведенное в табл. 45.19 произведение
моментов инерции соответствует значению r(Li—С1)=2,11+0,1 А, рассчитанному Уэлчем и др.
[4689] с использованием ионной модели молекулы. Погрешность значения IaIbIc, оценивается
в 1-1012 г3-см6. Основные частоты Li3Cl3 приняты по расчету Соломоника [961, 962], основанному
на оцененных значениях силовых постоянных х. Погрешности принятых основных частот могут
достигать 20—25% от их величины.
Термодинамические функции Li3Cl3(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных электронных состояний. Их погрешности обусловлены неточностью
принятых молекулярных постоянных, а также приближенным характером расчета; они могут
достигать 10, 20 и 30 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Li3Cl3(r) рассчитывались в справочнике JANAF [2834],
а также в работе Соломоника и Даниловой [962] (до 5000 К). Расхождения данных [2834] и
табл. 1072 составляют 20—40 Дж-К-моль и обусловлены резким различием в выборе основных
частот 1Л3С13. Результаты расчета [962] согласуются с данными табл. 1072 в пределах
Константа равновесия реакции Li3Cl3(r)=3Li(r)-j-3Cl(r) вычислена с использованием значения
Дгй"°@)=1805,065+10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH0(U3G]s, г, 0) = — 973 + 10 кДж • моль.
Принятая величина основана на результатах исследования равновесия реакции 21ЛС1(к, ж)+
+LiCl(r)=Li3Cl3(r), приведенных в табл. 45.25. Для дальнейших расчетов принимается значение
Таблица 45.25. Результаты определений энтальпии реакции 21ЛС1(к, ж) + LiCI(r) = Li3Cl3(r)
(в кДж • моль)
Авторы
Метод
Миллер, Куш, 1957 [3504] Эффузионный с селектором скоростей, 870 К,
1 точка
Милн, Клейн, 1960 [3510] Масс-спектрометрический, средняя температура
800 К а
Горохов, 1972 [309] Масс-спектрометрический с двойной эффузионнок
ячейкой, 890 К, 1 точка
Масс-спектрометрический, 823—868 К а
Шефер, Вагнер, 1979 [4175] То же, 721-872 К а
а Данные представлены в виде уравнения.
II закон
—
31
[ —
55+15
14
III закон
38,3
—
37,0
—
—
Частоты колебаний, рассчитанные в таком же приближении для Na3F3 и K3F3 [961, 962],
результатами исследований ИК-спектров паров фторидов натрия и калия [2793].
283
были подтверждены
1ЛВг(к, ж) Глава 45
Дг#°@)=38+13 кДж-моль, среднее из данных [309] и [3504]; его погрешность учитывает
неточность термодинамических функций Li3Cl3.
1лВг(к, ж). Термодинамические свойства кристаллического и жидкого бромида лития в
стандартном состоянии при температурах 100—3200 К приведены в табл. 1073.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние ЫВг(к) в интервале 0—823 К принимается
кубическая модификация (структурный тип NaCl).
При Т ^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости LiBr, проведенных Пауковым и др. [757] A2—318 К). Исследованный образец содержал
менее 0,04% влаги, 0,04% магния и менее 0,01% примесей других металлов. Экстраполяция
теплоемкости ниже 12 К приводит к значению S°{12 K)=0,15 Дж-К~1-моль~1. Погрешности принятых
значений ?°B98,15 К) и Я°B98,15 К) — Н° @) (см. табл. 45.1) оцениваются в 0,2 Дж-К^-моль-1
и 0,02 кДж-моль соответственно. Данные Слонима и Хюттига [4314] по средней теплоемкости
LiBr (85—370 К) не точны и приводят к заниженным результатам.
Измерения энтальпии LiBr при Т > 298,15 К проводились только в работах Бибаса и Леонарди
[1554] D50—900 К) и Дворкина [2135] G23—883 К). В работе [1554] приведены только
уравнения для теплоемкости, расчеты по которым приводят в интервале 300—761 К к очень низким
значениям (значение С°B98,15 К), вычисленное по этому уравнению, на 22% ниже величины,
полученной в работе [757]). При Т ^> 761 К теплоемкость резко возрастает, что наряду с эффектом
«предплавления», наблюдавшимся в работе [1554], по-видимому, свидетельствует о загрязненности
исследованного образца. Работа Дворкина [2135] осталась неопубликованной и известна по
таблицам JANAF [2834], где приводятся значения #°(823 К)—#°B98,15 К)=29,29 кДж-моль-1 и
C;(LiBr, ж)=65,3 Дж-К^-моль.
При Т > 298,15 К для теплоемкости LiBr(K) принято уравнение, полученное на основании
значений С°B98,15 К) (см. табл. 45.1), С°D00 К)=51,88 Дж-К^-моль (получено экстраполяцией
данных [757]) и Я°(823 К)—if°B98,15 К). Результаты измерения теплоемкости LiBr(K) в старой
работе Хюттига и Велинга [2763] B98—825 К) не точны и приводят к завышенному значению
С;B98, 15 К).
Температура плавления LiBr (823+2 К) принята по согласующимся между собой измерениям
Вартенберга и Шульца [4663] (822 К), Джиннингса и Фипса [2409] (824 К), Шмитц-Дюмонта
и Шмитца [4196] (822 К) и Агаева [5] (823 К); несколько более низкие значения были получены
в работах [1554, 1576, 1577, 2876, 4138] (819—821 К). Энтальпия плавления A7,66 +
+0,40 кДж-моль) принята по данным Дворкина и Бредига [2137]. Результаты измерений
Бибаса и Леонарди [1554] A3,7 кДж-моль) занижены вследствие наблюдавшегося «предплавления»
образца.
Теплоемкость LiBr^) F5,3+2 Дж-К-моль~1) определена Дворкиным (см. [2834]). Значение
85,5 Дж-К~1-моль~1, найденное в работе [1554], явно завышено и не согласуется с данными по
теплоемкости других жидких галогенидов щелочных металлов.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,15,
1, 4 и 8 Дж-К~1-моль соответственно. Термодинамические функции LiBr(K, ж), приведенные
в справочниках [2834] (Т < 3000 К) и [1395] (Г < 1500 К) и табл. 1073, согласуются в пределах
0,5—1 Дж-К^-моль.
В справочнике принята энтальпия образования
r, к, 298,15 К) = —351,16 + 0,27 кДж • моль,
Таблица 45.26. Результаты определений Аа„Ш°(ЫВг, к-^-соН2О, 298,15 К) (в кДж • моль")
Авторы
Конечная концентрация, температура
измерений
Да5Я°(соН20, 298,15 К)
Бодиско, 1889 [143] 160 Н2О, 290,15 К, 1 измерение —48,69
Бонефуа, 1900 [1613] 160 Н2О, 287,15 К, 1 измерение —49,45
Майер, 1927 [3353] 460 Н2О, 290,15 К, 1 измерение —49,55
Ланге, Шварц, 1928 [3183] 3,2—250 Н2О, 298,15 К, 44 измерения —48,82+0,20
Джоли, Перашон, 1977 [2884, 2885] 2130 Н2О, 298,15 К, 1 измерение —48,31 ±0,50
284
Литий и его соединения
LiBr(r)
основанная на приведенных в табл. 45.26 результатах измерений энтальпии растворения ЫВг(к)
в воде. Для дальнейших расчетов используется значение Даг?Г°(соН20, 298,15 К)=—48,80 +
+0,20 кДж-моль, основанное на наиболее детальных исследованиях Ланге и Шварца [3183].
Погрешность измерений в этой работе увеличена по сравнению с оценкой самих авторов с учетом
возможной систематической ошибки (см. [3771]).
Давление пара в реакции 1лВг(к, ж) = 1лВг(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = Д,#°AЛВг, г, 0) = 201,3+ 4,0 кДж • моль.
Принятая величина основана на измерениях давления и состава насыщенного пара 1ЛВг(к, ж).
Насыщенный пар бромида лития содержит молекулы LiBr, Li2Br2 и Li3Br3. На основании данных
[309, 3504] и принятых в справочнике термодинамических свойств при 21шAлВг)=823 К
насыщенный пар состоит из 26, 66 и 8 мол. % этих молекул. Методика вычисления термохимических величин
для индивидуальных компонентов насыщенного пара галогенидов щелочных металлов изложена
в Приложении 8.
Результаты расчетов энтальпии сублимации ЫВг(к) в виде мономерных молекул представлены
в табл. 45.27. Приведенные в таблице погрешности отражают только воспроизводимость
измерений. Принятое в справочнике значение является средневзвешенным из приведенных в табл. 45.27.
Таблица 45.27. Результаты определений As-H"°(LiBr, к, 0) (в кДж • моль)
Авторы
Вартенберг, Шульц, 1921 [4663]
Руфф, Мугдан, 1921 [4094]
Миллер, Куш, 1957 [3504]
Горохов, 1972 [309]
Метод
Точек кипения, 1387—1590 К, 7 точек
То же, 1283—1538 К, 9 точек
Эффузионный, 803 К, 1 точка
Масс-спектрометрический, 744—823 К, данные
представлены в виде уравнения
As#°(LiBr, к, 0)
II закон
III закон
207+7 201,3 ±0,6
225+25 198,4+1,8
— 202,3
200+7 —
Его погрешность оценена с учетом воспроизводимости измерений, расхождения результатов
измерений в разных работах, неточности термодинамических функций и определения парциального
давления мономерных молекул.
LiBr(r). Термодинамические свойства газообразного бромида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1074.
Приведенные в табл. 45.4 молекулярные постоянные LiBr приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3035, 3036] и УФ- [558, 1524, 1525]
спектров этой молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [558, 1524,
1525] в справочнике принимается, что валентные возбужденные электронные состояния имеют
энергии больше 35 000 см.
Приведенные в табл. 45.4 вращательные постоянные LiBr в состоянии ХХ2 + получены в работах
Хонига и др. [2700] и Раска и Горди [4101] обработкой результатов измерений микроволновых
переходов. Поскольку в работе [2700] были исследованы переходы на возбужденных
колебательных уровнях, а в работе [4101] только на уровне с v=0, постоянные Ве и а.х (Y01 и У1г) приняты
по работе [2700]. В то же время число вращательных уровней, переходы с которых
зарегистрированы в [4101], больше, чем в [2700], что позволило авторам [4101] получить постоянную У02.
Приведенные в табл. 45.4 колебательные постоянные LiBr были получены Клемперером с сотр.
[3035, 3036], которые исследовали ИК-спектр поглощения и идентифицировали переходы @—1),
A-2), B-3) и C-4).
Термодинамические функции LiBr(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Qm и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 45.5 и получены по принятым моле-
285
Li2Br2(r) Глава 45
кулярным постоянным LiBr. Расчет был выполнен для природной изотопической смеси изотопов
лития и брома.
Погрешности рассчитанных термодинамических функций LiBr(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным учетом
изотопного состава бромида лития. При более высоких температурах дополнительный вклад
в погрешность вносят неточность использованного способа экстраполяции потенциальной
функции, а также пренебрежение возбужденными электронными состояниями. Погрешности в
значениях Ф°(Т) составляют 0,03, 0,05 и 0,3 Дж-К^-моль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции LiBr(r) ранее вычислялись в табл. JANAF [2834], в работах
Уилкинса и др. [4729] (Т < 6000 К), Альтмана [1303] (Т < 6000 К), Папоушека и Валентовой
[3768] (Т < 3500 К), Раиса и Клемперера [3985] (Т < 2000 К), Брюэра и Брэкет [1660] (Ф°(Т),
Т < 2000 К), Уилкинса [4728] (Т < 6000 К). Расхождения данных [1303, 2834, 4728] и табл. 1074
не превышают 0,5 и 1,5 Дж-К~1-моль~1 в значениях Ф°(Г) и S°(T) соответственно, они объясняются
тем, что в этих работах использовался приближенный метод расчета.
Константа равновесия реакции LiBr(r)=Li(r)+Br(r) вычислена с использованием значения
A,.#o@)=D0(LiBr)=419,102+4N,2 кДж-моль~1«35 000+350 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации ЫВг(к). Этим величинам также
соответствует
Ь;Н°(ЫВг, г, 0) =—143,444+ 4,0 кДж • моль.
Су и Рейли [5056] методом фотофрагментарной спектроскопии нашли значение D0(LiBr) =
=415,1 +4,2 кДж-моль, согласующееся в пределах погрешностей с приведенным выше. Близкое,
но существенно менее точное значение было получено Булевич и д^.. [1719] D21+13 кДж-моль)
при спектрофотометрическом исследовании равновесия реакции образования LiBr(r) в пламенах.
Li2Br2(r). Термодинамические свойства газообразного дибромида дилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1075.
Молекулярные постоянные, использованные для расчета термодинамических функций Li2Br2,
приведены в табл. 45.19. Исследования ИК-спектра [2331, 3034, 4190] и электронографическое
исследование структуры [14] показывают, что молекула Li2Br2 в основном электронном состоянии
Х*-Ад имеет плоское ромбическое строение (симметрия D2k). Приведенное в табл. 45.19 произведение
моментов инерции соответствует структурным параметрам г(Ы—Вг)=2,36+0,05 Аи4 Вг—Li—
—Вг=116+5°, рассчитанным Соломоником для ионной модели молекулы [961, 963] Ч Погрешность
значения IaIbIc оценивается в 1-1013 г3-см6. Приведенные в табл. 45Л9 частоты v5, v6 найдены
Шликом и Шнеппом [4190], а частота v4 — Фрейбергом и др. [2331 ] в ИК-спектрах молекул Li2Br2,
изолированных в матрицах из Аг и Кг соответственно. С учетом принятых погрешностей эти
значения согласуются с частотами v5=295 и v6=413 см, полученными Клемперером и Норрисом [3034]
в ИК-спектре газообразного Li2Br2 при высоких температурах. Для ненаблюдавшихся
экспериментально частот vl5 v2: v3 приняты значения, вычисленные Соломоником [961, 963] с
использованием ионной модели молекулы. Погрешности частот v4, v6, ve составляют 15—20 см, а значений vlt
v2 и v3 могут достигать 40—80 см.
Термодинамические функции Li2Br2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A.10), A. 122)—A. 124), A.128),
A. 130) и без учета возбужденных состояний и возможной изомерии (см. Li2F2). Погрешности
рассчитанных величин обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета, они составляют 5, 10 и 15 Дж-К-моль~1 в значениях Ф°(Г)
при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Li2Br2(r) рассчитывались в справочнике JANAF [2834],
а также в работах Бауэра и др. [1433] (до 5000 К) и Краснова и др. [569] (до 3000 К). Расхождения
данных [569, 2834] и табл. 1075 не превышают 3 и 1 Дж-К^-моль в значениях Ф°(Т).
1 Данные работы [14] (г (Li—Br)=2,35+0,02 A, /L Вг—Li—Вг=110+4°) не принимались во внимание, так как
ее авторы не учитывали сложный состав пара и, кроме того, возможна систематическая ошибка в измерениях
(ем. [961]).
286
Литий и его соединения LisBr3(r), Ш(к, ж)
Константа равновесия реакции Li2Br2(r)=2Li(r)-j-2Br(r) вычислена с использованием значения
ДгЯ°@)=1030,316+6,3 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(Li2Br2, г, 0) = —479 + 6 кДж • моль.
Принятая величина основана на результатах исследования равновесия реакции 1ЛВг(к)-Ь
+1лВг(г)=1Л2Вг2(г), проведенного Гороховым [309] (масс-спектрометрический метод с
использованием двойной эффузионной ячейки, 816 К, 1 точка, Дгйго@) = 11,4 кДж-моль) и Миллером
и Кушем [3504] (эффузионный метод с использованием селектора скоростей, 803 К, 1 точка,
Дг#°@)=7,0 кДж-моль). В справочнике принимается среднее значение ДгЯ°@)=9 +
+6 кДж-моль, его погрешность учитывает неточность термодинамических функций,
использованных в расчетах.
Li3Br3(r). Термодинамические свойства газообразного трибромида трилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1076.
Молекулярные постоянные, использованные для расчета термодинамических функций Li3Br3,
приведены в табл. 45.19. Структура и спектр молекулы Li3Br3 экспериментально не исследовались.
По аналогии с молекулой Li3F3 для Li3Br3 в основном электронном состоянии Х1А1 принимается
плоская гексагональная конфигурация (симметрия DSh). Приведенное в табл. 45.19 произведение
моментов инерции соответствует значению г(Ы—Вг)=2,28+0,1 А, рассчитанному Уэлчем и др.
[4689] с использованием ионной модели. Погрешность значения IaIbIc оценивается
в 1 • Ю~112 г3-сме. Приведенные в табл. 45.19 основные частоты рассчитаны Соломоником [961 962]
по оцененным им значениям силовых постоянных х. Погрешности принятых частот достигают 20—
25% от их величин.
Термодинамические функции Li3Br3(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6). A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Погрешности результатов расчета обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета, они
составляют 10, 20 и 30 Дж-К~1-моль"'1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Li3Br3(r) рассчитывались Соломоником и Даниловой [962]
(до 5000 К); результаты этого расчета согласуются с данными табл. 1076 в пределах 1 —
2 Дж-К^-моль.
Константа равновесия реакции Li3Br3(r)=3Li(r)+3Br(r) вычислена с использованием значения
ДгЯ°@)=1626,974+10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(Li3Br3, г, 0) = —800 ± 10 кДж • моль.
Принятая величина основана на результатах исследований равновесия реакции 2ЫВг(к)+
+LiBr(r)=Li3Br3(r), выполненных Гороховым [309] (масс-спектрометрический метод с
использованием двойной эффузионной ячейки, 816 К, 1 точка, ДгЯг°@)=34,2 кДж-моль) и Миллером и
Кушем [3504] (эффузионный метод с использованием селектора скоростей, 803 К, 1 точка, ДгЯ°@) =
=30,8 кДж-моль). В справочнике принято среднее значение Дг#°@)=33+11 кДж-моль,
его погрешность учитывает неточность термодинамических функций Li3Br3(r).
1Л1(к, ж). Термодинамические свойства кристаллического и жидкого иодида лития в стандарт"
ном состоянии при температурах 100—3000 К приведены в табл. 1077.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние Ы1(к) в интервале 0—742 К принимается
кубическая модификация (структурный тип NaCl).
При Т <^ 298,15 К термодинамические функции вычислены на основании измерений
теплоемкости, проведенных Хриплович и др. [1089] E—316 К) и Бергом [1491] B—20 К). Результаты
измерений в этих работах при 5—10 К существенно расходятся (на 20% при 7 К), но в интервале
Ю—20 К они хорошо согласуются в пределах экспериментальной погрешности. Причины
расхождений не ясны, так как в обеих работах измерения проводились на чистых образцах. Расчет термо-
1 Рассчитанные в [961, 962] в таком же приближении частоты Na3F3 и K3F3 удовлетворительно согласуются
с результатами исследований ИК-спектров паров фторидов натрия и калия [2793].
287
1Л1(к, ж) Глава 45
динамических функций при Т <^ 20 К был выполнен по усредненным данным, а в интервале 20—
298,15 К — по данным [1089]. Принятые значения 5°B98,15 К) и Я°B98,15 К)—#°@)
(см. табл. 45.1), имеют погрешности 0,2 Дж-К^-моль и 0,02 кДж-моль соответственно. Ранее
Слоним и Хюттиг [4314] измерили среднюю теплоемкость 1л1(к) (85—366 К, образец чистотой
99,5%).
При Т > 298,15 К для теплоемкости 1Л1(к) принято уравнение, выведенное на основании
величин С;B98Д5 К)=51,04 Дж-К^-моль-1 [1089] и Я°G42 К)-Я°B98 К)=25,10 кДж-моль
[2135]. Неопубликованная работа Дворкина [2135] известна по справочнику JANAF [2834].
Хюттиг и Велинг [2763] измерили среднюю теплоемкость Lil (к) в интервалах 276—359 и 277—
373 К. На основании этих данных Келли [2960] вывел уравнение для теплоемкости, согласно
которому С°B98,15 К) на 6,5% выше величины, полученной в работе [1089].
Температура плавления Lil G42 +2 К) принята по согласующимся между собой измерениям
Джиннингса и Фипса [2409] G40 К), Дворкина и Бредига [2137] G42 К), Сридхара и др. [4371]
G43 К); менее точные результаты получены в работах [910, 1000, 3230, 3283, 4196, 4791].
Энтальпия плавления A4,64+0,40 кДж-моль) принята по данным Дворкина и Бредига [2137].
Теплоемкость 1л1(ж) F3,2+2 Дж-К^-моль) принята по результатам измерений энтальпии
жидкой фазы G42—802 К) в работе Дворкина [2135].
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,2,
1, 4 и 8 Дж-К~1-моль. Различия между термодинамическими функциями 1л1(к, ж),
табулированными в настоящем справочнике и таблицах JANAF [2834] (Т ^ 2500 К), не превышают
1 Дж-К^-моль в значениях Ф°(Г) и обусловлены тем, что в настоящем издании учтены
экспериментальные данные [1089] по теплоемкости 1л1(к) при Т <^ 316 К.
Принятая в справочнике энтальпия образования
к, 298,15 К) = —273,2+ 0,5 кДж ¦ моль
основана на приведенных в табл. 45.28 результатах измерения энтальпии растворения Ы1(к)
в воде. Для энтальпии растворения принимается значение Да?Я°(соН20, 298,15 К) =—62,2 +
+0,5 кДж-моль, среднее из данных [2884, 2885, 4781].
Таблица 45.28. Результаты определений кщН0 (Lil, к->гаН2О, 298,15 К) (в кДж • моль)
Авторы
Конечная концентрация, температура
измерений
ДагЯ°(ооН2О; 298,15 К)
Бодиско, 1888 [142, 1594, 1595] 308 Н2О, 290,15 К, 1 измерение —63,04
Мознер, 1897 [3570] 800 Н2О, 288,15 К, 1 измерение —63,76
Бекетов, Бекетов, 1904 [1458] 230 Н2О, 291,15 К, 2 измерения —62,77
Майер, 1927 [3353] (см. [4314]) 800 Н2О, 290,15 К, 1 измерение —63,17
By, Фридман, 1966 [4781] 5550 Н2О, 298,15 К, 1 измерение —62,55
Джоли, Перашон, 1977 [2884, 2885] 3280 Н2О, 298,15 К, 1 измерение —61,94
Давление пара в реакции 1л1(к, ж)=1л1(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = Д,Я°AЛ1, к, 0) = 190 ±8 кДж-моль-1.
Принятая величина основана на измерениях давления и состава насыщенного пара над 1Л1(к)
и энергии диссоциации Lil. В состав насыщенного пара иодида лития входят молекулы Lil, Li2I2
и Li3I3. Согласно данным [309] и термодинамическим свойствам этих соединений, принятым
в справочнике, при jT)n(LiI)=742 К их содержание в насыщенном паре составляет 30, 68 и 2 мол.%
соответственно. Методика расчета термохимических величин для индивидуальных компонентов
насыщенных паров галогенидов щелочных металлов изложена в Приложении 8.
Результаты расчетов энтальпии сублимации LH(k) в виде мономерных молекул представлены
в табл. 45.29. Там же приведены значения, соответствующие энергиям диссоциации Lil, найденным
в работе [5055] методом фрагментарной спектроскопии и в работе [1719] на основании спектрофото-
метрических измерений. Принятое значение основано на данных, приведенных в таблице; его по-
288
Литий и его соединения
Таблица 45.29. Результаты определений AsjST°(LiI, к, 0) (в кДж • моль)
Авторы
Вартенберг, Шульц, 1921 [4663]
Фридман, 1955 [2337]
Горохов, 1962 [308]
Горохов, 1972 [309]
Булевич и др., 1961 [1719]
Су, Рейли, 1979 [5055]
•
Метод
Точек кипения, 1213—1413 К, 6 точек
Масс-спектрометрический, 633—742 К, 12 точек
То же, 742—758 К, 7 точек
То же, 724 К, 1 точка
То же, 626—725 К, данные представлены в виде
уравнения
AsH°(LiI, к, 0)
II закон
III закон
208+22 182±4
185+4 —
172+5 —
— 178+8
195 ±4 -
Спектрофотометрическое исследование реакции — 194+13
Li+HI=LiI+H в пламенах, 1500-2600 К
Фотофрагментарная спектроскопия, D0(LiI)=
=341 ±4 кДж -моль-1
197+4
грешность оценена с учетом расхождений величин, полученных в разных работах,
воспроизводимости измерений, а также неточности термодинамических функций.
Lil(r). Термодинамические свойства газообразного иодида лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1078.
Приведенные в табл. 45.4 молекулярные постоянные Lil приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3035, 3036] и УФ- [1539] спектров этой
молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1539]
в справочнике принимается, что валентные возбужденные электронные состояния Lil имеют
энергии больше 29 000 см.
Приведенные в табл. 45.4 вращательные постоянные Lil в состоянии Х1?+ получены в работах
Хонига и др. [2700] и Раска и Горди [4101] обработкой результатов измерений микроволновых
переходов. Поскольку в работе [2700] были исследованы переходы на возбужденных
колебательных уровнях, а в работе [4101] только переходы в состояния с v=0, постоянные Ве и ах (F01 и
УХ1) приняты в работе [2700]. В то же время число вращательных уровней, переходы с которых
зарегистрированы, в работе [4101] больше, что позволило авторам получить постоянную Y02.
Приведенные в табл. 45.4 колебательные постоянные Lil были получены Клемперером с сотр.
[3035, 3036], которые исследовали ИК-спектр поглощения и идентифицировали переходы @—1),
A-2), B-3) и C-4).
Термодинамические функции Lil(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения QSB и ее производные вычислялись без учета возбужденных электронных
состояний по методике, изложенной в Приложении 7. Использованные в расчете коэффициенты
потенциальной функции d0, dx и d2 приведены в табл. 45.5 и получены по принятым молекулярным
постоянным Lil. Расчет был выполнен для природной изотопической смеси изотопов лития.
Погрешности рассчитанных термодинамических функций Lil (г) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава иодида лития. При более высоких температурах дополнительный вклад
в погрешность вносят неточность принятого способа экстраполяции потенциальной функции,
а также пренебрежение возбужденными электронными состояниями. Погрешности в значениях
Ф°G) составляют 0,03, 0,05 и 0,5 Дж-К^-моль-1 при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции Lil(r) ранее вычислялись в таблицах JANAF [2834], в работах
Уилкинса и др. [4729] (Т < 6000 К), Альтмана [1303] (Т < 6000 К), Папоушека и Валентовой
[3768] (Т < 3500 К), Раиса и Клемперера [3985] (Т < 2000 К), Брюэра и Брэкет [1660] (Ф°(Т),
Т < 2000 К), Уилкинса [4728] (Т < 6000 К). Расхождения данных [1303, 2834, 4728] и табл. 1078
не превышают 0,8 и 2,5 Дж-К^-моль в значениях Ф°(Г) и S°(T) соответственно и объясняются
тем, что в этих работах использовался приближенный метод расчета.
Константа равновесия реакции LiI(r)=Li(r)+I(r) вычислена с использованием значения
Ar#°@)=D0(LiI)=348,228±8,l кДж • моль -x?»29 100+700 см', соответствующего принятым
289
Li2I2(r), Li3I3(r) Глава 45
в справочнике энтальпиям образования и сублимации 1Л1(к). Этим величинам также
соответствует
bfH°(Lil, г, 0) = —83,330 + 8 кДж • моль.
Результаты исследований энергии диссоциации Lil в работах [1719, 5055] использованы при
выборе энтальпии сублимации 1Л1(к) (см. выше).
1ЛЛ2(г). Термодинамические свойства газообразного дииодида дилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1079.
Молекулярные постоянные, использованные для расчета термодинамических функций Li2I2r
приведены в табл. 45.19. Исследование инфракрасного спектра [3034], а также электронографи-
ческое исследование структуры [14] показали, что молекула Li2I2 в основном электронном
состоянии ХгАд имеет плоское ромбическое строение (симметрия D2k). Приведенное в табл. 45.19
произведение главных моментов инерции соответствует структурным параметрам r(Li—I)=2,59+0,08 A
и -$ I—Li—1=118+5°, найденным Соломоником в результате теоретического расчета с
использованием ионной модели [961, 963] *. Погрешность значения IaIbIb составляет 5-1013 г3-смв.
Приведенные в табл. 45.19 значения частот v5, ve найдены Клемперером и Норрисом [3034] в ИК-
спектре газообразного Li2I2 при высоких температурах. Для ненаблюдавшихся экспериментально
частот vx—v4 приняты величины, рассчитанные Соломоником [961, 963] для ионной модели
молекулы. Погрешности значений v5 и ve оцениваются в 15—20 см, а частот vx— v4 — в 20—25%
от их величины.
Термодинамические функции Li2Ia(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных состояний и возможных изомеров (см. Li2F2(r)). Погрешности
рассчитанных величин обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета, они составляют 6, 12 и 16 Дж»К-моль~1 в значениях Ф°(Т) при 298,15,
3000 и 6000 К соответственно.
Ранее термодинамические функции Li2I2(r) рассчитывались в таблицах JANAF [2834], а также
в работах Бауэра и др. [1433] (до 5000 К) и Краснова и др. [569] (до 3000 К).
Расхождения данных [2834], [569] и табл. 1079 не превышают 4 и 2 Дж.К~1-моль
соответственно в значениях Ф°(Г). Они обусловлены уточнением оценки постоянных Li2I2 в работе [961,.
963]. Расчет [1433] основан на неверных оценках и соответствующие расхождения достигают
12 Дж-К^-моль.
Константа равновесия реакции Li3I2(r)=2Li(r)-f2I(r) вычислена с использованием значения
AjiTXO)=887,796±7,3 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
e(LiJa, г, 0) == —358 + 7 кДж • моль.
,,
Принятая величина основана на результатах исследования равновесия реакции Ы1(к)+
+LiI(r)=Li2Is(r), выполненного масс-спектрометрическим методом Гороховым [309] G24 К,
i точка). На основании этих измерений было получено значение ДгЯ°@)=7 + 7 кДж-моль
(III закон); его погрешность оценена с учетом неточности термодинамических функций. Расчет
этой же величины по температурной зависимости интенсивностей соответствующих ионных токов
(II закон), измеренных в работах Горохова [308] F26—725 К, данные представлены в виде
уравнения) и Фридмана [2337] F33—742 К, 12 точек), приводит к значениям 6 + 7 кДж-моль и 3 +
+10 кДж-моль соответственно.
Li3Is(r). Термодинамические свойства газообразного трииодида трилития в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1080.
Молекулярные постоянные, использованные для расчета термодинамических функций Li3I3,
приведены в табл. 45.19. Структура и спектры молекулы Li3I3 экспериментально не исследовались.
По аналогии с молекулой LisF3 для Li3I3 в основном электронном состоянии ]t1A1 принята плоская
* Данные [14] (г (Li—1)=2,54+0,03 А и X I—Li—1=116+4°) не принимались во внимание, так как в этой
работе возможна систематическая ошибка (см. [961]), а при расшифровке электронограмм не учитывался
сложный состав пара.
290
Литий и его соединения Li2SO4(K, к;)
гексагональная конфигурация симметрии D3h. Приведенное в табл. 45.19 произведение моментов
инерции соответствует r(Li—F)=2,40+0,l А, рассчитанному Уэлчем и др. [4689]. Погрешность
значения IaIbIc составляет 5- 1011 г3-см6. Приведенные в табл. 45.19 основные частоты
рассчитаны Соломоником [961, 962] по оцененным значениям силовых постоянных. Погрешности
принятых значений частот могут достигать 20—25% от их величин.
Термодинамические функции Li3I3(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и A. 130)
без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены неточностью
принятых молекулярных постоянных, а также приближенным характером расчета, они
составляют 12, 25 и 35 Дж-К^-моль-1 в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Li3I3(r) рассчитывались Соломоником и Даниловой
[962] (до 5000 К). Результаты расчета [962] согласуются с данными табл. 1080 в пределах
1,5 Дж-К^-моль в значениях Ф°(Г).
Константа равновесия реакции Li3I3(r)=3Li(r)+3I(r) вычислена с использованием значения
Дг#°@)=1400,694+10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д//°(Ы313, г, 0) = —606 + 10 кДж • моль.
Принятая величина основана на результатах исследования равновесия реакции 2Ы1(к)+
+LiI(r)=Li3I3(r), выполненного Гороховым [309] масс-спектрометрическим методом G24 К,;
1 точка, ДГ-ЙГО@)=32+10 кДж-моль). Основным источником погрешности, этой величины
является неточность термодинамических функций Li3I3(r), использованных в расчетах.
Li2SO4(K, ж). Термодинамические свойства кристаллического и жидкого сульфата дилития
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1081.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние 1Л2ЗО4(к) в интервале 0—848 К принята
моноклинная а-модификация [1280], а в интервале 848—1131 К кубическая ^-модификация
[2305].
При Т ^ 298,15 К термодинамические функции Li2SO4(K) вычислены по результатам измерений
теплоемкости, выполненных Пауковым [756] A2—311 К, образец чистотой —99,9%). Среднее
отклонение экспериментальных точек от сглаженной кривой не превышает ~0,7% в интервале
12—20 К и 0,12% в интервале 50—300 К. Графическая экстраполяция теплоемкости приводит
к значению <5°A2 К)=0,17 Дж-К^-моль. Погрешности принятых значений 5°B98,15 К) и
#°B98,15 К)—Я°@) (см. табл. 45.1) оцениваются в 0,2 Дж-К^-моль и 0,03 кДж-моль
соответственно.
Уравнение для теплоемкости a-Li2SO4 в интервале 298,15—848 К (см. табл. 45.1) выведено
совместной обработкой данных по теплоемкости, полученных Шмидт [1153] B98,15—770 К) и
результатов измерений энтальпии, проведенных Соколовым и др. [957] G81—844 К, 11 точек),
Дениелоу и др. [2042, 2043, 2044] D00—839 К, 6 точек), и Воскресенской и Банашек [249] C60—
743 К, 30 точек); погрешности измерений [957] оценивались в 0,5%, измерений [1153] и [2042,
2043, 2044] — в 1%, а погрешности наименее точных данных [249] — в 2,5%. При выводе
уравнения для теплоемкости Li2SO4(K) не учитывались измерения теплоемкости в работе Съюблома [43Q4]
C93—1073 К), поскольку они превышают данные других авторов на ~10% во всем интервале
температур, и измерения энтальпии в работе Кларка [1866] D88—906 К), которые лежат на 16—
17% ниже данных других работ.
Температура полиморфного превращения 848+3 К принята по согласующимся между собою
данным, полученным различными методами [19, 21, 84, 226, 522, 1058, 2764, 3581, 3671]
(термический анализ) [20, 62, 96, 103, 105, 285, 363, 624, 4195] (визуально-политермический метод) и [1866,
2042, 2043, 2044] (калориметрия). Результаты менее надежных измерений [200, 249, 622, 851,
2566, 2778] (839—865 К) не учитывались. Энтальпия полиморфного превращения 23,9 +
+ 1,0 кДж-моль рассчитана с помощью уравнений, принятых для теплоемкости а- и р-модифика^
ций Li2SO4. Поскольку использованное в справочнике уравнение для теплоемкости a-Li2SO4
передает наблюдавшийся экспериментально ускоренный рост теплоемкости вблизи точки
превращения, принятое значение b.trH° меньше тех измерений этой величины, в которых указанная
аномалия не учитывалась B5,6 кДж-моль [2042.,2043, 2044], 25,0 нДж-моль [1866], 27,3 нДж-моль
[249], 29,3 кДж-моль-1 [4220]).
291
LiNO,(K, ж)
Глава 45
Теплоемкость f3-Li2SO4 в интервале 848—1131 К B18+2 Дж-К^-моль) рассчитана из хорошо
согласующихся между собой данных по энтальпии этой модификации, полученных в работах [957]
(854—1127 К), [2042, 2043, 2044] (849-1119 К) и [249] (888—1132 К).
Температура плавления A131+3 К) принята по согласующимся между собой работам [18, 19,
21, 62, 84, 96, 103, 105, 249, 285, 363, 530, 622, 851, 852, 1058, 2042, 2043, 2764, 2825, 3671, 3805,
4195, 4895]. Энтальпия плавления (9,3 + 0,6 кДж-моль) принята по работам Дениелоу [2042,
2043, 2044], принятое значение в пределах погрешности согласуется с данными [3805]
(9,9 кДж-моль); менее надежные измерения, проведенные в работах [3671] (8,5 кДж-моль),
[1866] G,4 кДж-моль-1), [249] G,7 кДж-моль) и [4220] A4 нДж-моль), не учитывались.
Значение Cp(Li2SO4, ж)=205+4 Дж-К~1-моль~1 рассчитано из данных по энтальпии жидкого
сульфата лития, полученных в работах [249] A148—1271 К), [957] A132—1270 К) и [2042, 2043,
2044] A138-1540 К).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,15,
1,5, 5 и 13 Дж-К^-моль соответственно.
Термодинамические функции Li2SO4(K, ж), вычисленные в справочнике JANAF [2835] (Т ^
^ 3000 К), согласуются в пределах 0,2 Дж-К^-моль в значениях Ф°(Г) с приведенными
в табл. 1081.
Константа равновесия реакции Li2SO4(K, ж)=2Ы(г)+8(г)+4О(г) вычислена с использованием
значения Дг#°@)=3000,978+2,1 кДж-моль, соответствующего принятой в справочнике
энтальпии образования Li2SO4(K)
Ь,НОA,\?О4, к, 298,15 К) =—1436,0 + 0,5 кДж • моль.
Эта величина основана на приведенных в табл. 45.30 результатах измерений. Наиболее точные
измерения энтальпии растворения Li2SO4(K) [1097, 4570] и измерения энтальпии реакции 1ЛС1(к)
Таблица 45.30. Результаты определений A^ff°(Li2SO4, к, 298,15К) (в кДж-моль)
Авторы
Пиккеринг, 1885 [3836]
Пиккеринг, 1886 [3837]
Томсен, 1886 [4511]
Уеда, 1933 [4570]
Цветков, Рабинович, 1969 [1097]
Томсен, 1886 [4511]
Жуковский, 1911 [4827]
Бэрани, Адами, 1966 [1387]
Фикалора и др., 1968 [2247]
Метод
Калориметрический, измерение энтальпии
растворения Li2SO4(k) в воде, разведение 400 Н2О, 295,8 К
То же, 400 Н2О, 290,7—295,9 К
То же, 200 Н2О, 291 К
То же, 400 Н2О, 298,15 К
ЭДС, 2Li(K)+Hg2SO4(K)=Li2SO4(K)+2HgM,
293—303 К
fcrf %SU tJ ¦_/ tx* J- Ir ^
Калориметрический, измерение энтальпии
растворения Li2SO4(K) в воде, разведение 500 Н2О, 298,15 К
Калориметрический, измерение энтальпии реакции
2ЫОН(р-р, 200 H2O)+H2SO4(p-p, 400 H2O)=Li2SO4
(р-р, 800 Н2О)+2Н2О (р-р)
То же, 2Li(K)+H2SO4(p-p)=Li2SO4 (р-р)+Н2(г)
То же, 2LiCl(K)+H2SO4(p-p, 7,068H2O)=Li2SO4(KL
+18,394 Н2О(ж)+2НС1(р-р, 12,73Ш2О), 303 К
Масс-спектрометрический, Li2SO4(K, ж)=2Ы(г)+
+SO2(r)+O2(r), 950—1250 К, II закон
A/^°(Li2SO4, к, 298,15 К)
—1436,4
-1436,0
—1436,7
—1435,9+0,5
—1425,8
-1436,5+0,5
—1435,0
—1420,6
—1434,7+0,9
-1386+50
с серной кислотой [1387] привели к совпадающим в пределах погрешностей величинам. При выборе
рекомендуемой величины больший вес придавался измерениям энтальпии растворения [1097,
4570], подтверждаемым более ранними аналогичными измерениями [3836, 3837, 4511].
Принятой энтальпии образования соответствует значение AaqH°(Li2SOll, к -»• соН20,
298,15 К) = —30,51+0,50 кДж-моль, вычисленное для реакции Li2S04(K)+ooH20=2Li+(a<?)+
LiNO2(K, ж). Термодинамические свойства кристаллического и жидкого нитрита лития в
стандартном состоянии при температурах 298,15—1500 К приведены в табл. 1082.
292
Литий и его соединения
LiNO2(K, ж)
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние LiNO2(K) в интервале 0—369 К принимается
низкотемпературная а-модификация с неизученной структурой, а в интервале 369—495 К —
высокотемпературная гексагональная ^-модификация, относящаяся к структурному типу Cs[ICL]
[1765].
Ввиду отсутствия экспериментальных данных по теплоемкости LiNO2 при Т <^ 298,15 К
значения 5°B98,15 К) и Я°B98,15 К)—Н°@), приведенные в табл. 45.1, были получены с помощью
приближенных оценок. Значение iS°B98,15 К) получено усреднением оценок, выполненных по
аддитивным схемам Келли [3771], Латимера [614] и Цагарейшвили [1094], а #"B98,15 К)—#°@)
оценено по методу Карапетьянца [485] на основании сопоставления экспериментальных данных
в рядах нитриты—метабораты щелочных металлов. Погрешности принятых величин могут
достигать 10 Дж-К~1-моль~1 и 1 кДж-моль соответственно.
Для теплоемкости a-LiNO2 в интервале 298,15—369 К принято уравнение, выведенное на
основании значений С°B98,15 К)=63 и С°C69 К)=80 Дж-К~1-моль~1, оцененных с учетом характера
температурной зависимости теплоемкостей для нитритов натрия и калия, определенных
экспериментально. В интервале 369—495 К для теплоемкости p~LiNO3 принято постоянное значение
80 Дж-К^-моль.
Температура фазового превращения LiNO2 C69 +2 К) принята по результатам измерений Вер-
хотурова и др. [209] и Проценко и др. [822, 828, 837], выполненных для хорошо аттестованных
образцов. В работах [587, 819, 824, 836] найдены более низкие значения C53—366 К),
по-видимому, из-за недостаточной чистоты образцов. Температура плавления D95+2 К) также принята
по работам Верхотурова и др. и Проценко и др.; менее точные значения были получены в работах
[676, 823, 824, 837, 2089] D91—498 К). Энтальпии фазового превращения A,7±0,4 кДж-моль-1)
и плавления (9,2+1,2 кДж-моль) приняты по единственной экспериментальной работе
Верхотурова и др. [209], выполненной термографическим методом. Теплоемкость 1лГ*Ю2(ж) (90 +
+10 Дж-К~1-моль~1) оценена с учетом экспериментальных данных о теплоемкостях жидких
нитритов натрия и калия.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000 и 1500 К оцениваются в 7, 15 и
20 Дж-К^-моль-1.
Таблица термодинамических функций LiNO2(k, ж) публикуется впервые.
Таблица 45.31. Результаты определений Ay-ffo(LiNO2, к, 298,15 К) (в кДж • моль)
Авторы
Центнершвер, Блюменталь, 1936
[1773, 1775] (см. также [3123])
Доде, 1936, 1937 [2088, 2089]
Доде, 1937 [2089]
Бюро, 1937 [1724]
Андреева, Бондаренко, 1979 [33]
Верхотуров и др., 1979 [208]
Метод
Исследование равновесия реакции 2LiNO3(jK)=
=21лГЮ2(ж)+О2(г) компенсационным методом,
628—708 К, 13 точек
Калориметрический, LiOH(p-p)+HNO2(p-p)=
=LiNO2(p-p)+H2O(p-p), температура и
концентрации не указаны, ДГЯ°=—44,6
Калориметрический, растворение LiNO2(K) в воде
400 Н2О, 285 К, 3 измерения, ДГЯ°=—9,20
То же, 400 Н2О, 293 К, 1 измерение, ДГЯ°=—9,96
То же, 2,6—106 Н2О, 298 К, 19 измерений
Электронный удар, D0(Li—NO2)=468
A/5°(LiNO2,K,298,15K)
—386 (II) -427 (III)
—367,7 ±0,8
—368,1 ±0,8
—367,9 ±0,8
—369,6+1,0
—445
Принятая в справочнике энтальпия образования
^fH0(Li№2, к, 298,15 К) = —368,3 + 1,0 кДж -моль
основана на измерениях, приведенных в табл. 45.31. Погрешность величин, полученных
калориметрическим методом, определяется в основном неточностью использованных в расчетах значений
энтальпии образования NOj(ag) и HNO2(p-p) (см. Приложение 5). Пересчет результатов
измерений энтальпии растворения к стандартной температуре проводился с использованием значения
° ДК^1
=-68 Дж-К
^
моль
293
LiNO3(r), LiNO3(K, ж) Глава 45
Исследования равновесия реакции 21лГТО3(ж)=21л1ЧО2(ж)+О2(г) в работах [1773, 1775, 3123],
как и в случае нитритов других щелочных металлов, приводят к ненадежной величине. Также
неточно значение, полученное методом электронного удара [208].
Принятое значение является средним из результатов калориметрических измерений [33, 1724,
2088, 2089]. Ему соответствует
AeJ#°(LiNOa, к-*соН20, 298,15 К) = —10,5 + 0,5 кДж -моль'1.
Давление пара в реакции LiNO2(K, ж)=1Л]МО2(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = Asff°(LiNO2, к, 0) = 165 + 10 кДж • моль,
вычисленной по данным Верхотурова и др. [205, 207]. В этих работах масс-спектрометрическим
методом (двойная эффузионная ячейка) было установлено, что пар состоит в основном из
мономерных молекул (количество димеров при 600 К составляет 2,5 мол. % 1). Измерения интенсивностей
ионных токов проводились в интервале 554—607 К и при 600 К. Методом полного изотермического
испарения было определено давление мономера в парах. Принятая величина вычислена по
уравнению III закона по результатам измерения при 600 К. Ее погрешность обусловлена в основном
неточностью термодинамических функций 1ЛГТО2(ж) и LiNO2(r). Уравнение II закона приводит
к близкому значению A62 кДж-моль).
LiNO2(r). Термодинамические свойства газообразного нитрита лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1083.
Молекулярные постоянные, использованные для расчета термодинамических функций LiNO2,
приведены в табл. 45.9. Экспериментально структура молекулы LiNOa не исследовалась.
Произведение моментов инерции IaIbIo в основном электронном состоянии Х1А1 вычислено для плоской
циклической структуры (симметрия С2е,) с углом -^ О—N—0 = 115+5° и межатомными
расстояниями r(N—О)=1,25 +0,05 А (оценено по структурным параметрам молекул RbNO2 и
CsNO2) и r(Li—О)=1,8+0,1 А (перенесено из LiNO3). Погрешность значения IaIbIc составляет
6-10"ш г3-смв.
Из всех частот LiNO2, приведенных в табл. 45.9, только частота v4, связанная с
антисимметричным колебанием N—О, наблюдалась экспериментально в ИК-спектре молекул LiNO2,
изолированных в матрице из Аг [3508]. Остальные частоты оценены на основании соответствующих частот
иона NOj (v1=vN_0), молекул CsNO2 (v2=vo_n-o), LiNO3 (v3=vL1_o) и Li2SO4 К=^хл_0_м2 и v6=
= vHeiuoo«)' Погрешность значения v4 оценивается в 15—20 см, частот vx и v2 — в 30 см,
остальных частот в 20—25% от их величин. Сведения о возбужденных электронных состояниях LiNO2
в литературе отсутствуют.
Термодинамические функции LiNO2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124)г A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета, они составляют 4, 10 и 12 Дж-К~1-моль~1 в значениях Ф°(Л
при 298,15, 3000 и 6000 К.
Таблица термодинамических функций LiNO2(r) публикуется впервые.
Константа равновесия реакции LiNO2(r)=Li(r)-|-N(r)+2O(r) вычислена с использованием
значения Дг?Г°@)=1319,871+10 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации LiNO2(k).
Этим величинам также соответствует
г, 0) = —197,752 ± 10 кДж.моль.
LiNO3(K, ж). Термодинамические свойства кристаллического и жидкого нитрата лития в
стандартном состоянии при температурах 298,15—2000 К приведены в табл. 1084.
1 Авторы работы [1706] пришли к ошибочному заключению о преобладании в паре димерных молекул; это было
обусловлено тем, что они не провели детального анализа происхождения основного иона в масс-спектре (Li+).
s Частота v5 у нитритов меньше, чем у сульфатов, и разница увеличивается при переходе от Cs к Li.
294
Литий и его соединения LiNO3(K, ж)
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние L1N03(k) в интервале 0—526 К принимается
гексагональная модификация (структурный тип кальцита СаСО3 [2261] г.
Сведения о низкотемпературных измерениях теплоемкости LiNO3 в литературе отсутствуют.
Келли [2960], обработав результаты измерений энтальпии, выполненных Гудвином и Кальмусом
[2442] в интервале 442—576 К, вывел уравнение для теплоемкости, в соответствии с которым
в справочнике принимается значение С°B98,15 К). Приведенные в таблице 45.1 значения
<S°B98,15 К) и ZPB98,15 К)—Н°@) оценены: первое — по приближенным аддитивным схемам
Латимера [614] и Цагарейшвили [1094], а второе — по методу Карапетьянца [485] при
сопоставлении экспериментальных данных в рядах нитраты — карбонаты щелочных металлов. Погрешности
этих значений оценены в 10 Дж-К~1-моль~1 и 1 кДж-моль соответственно. Значение
S°B98,15 K)=88 Дж-К-моль~1, вычисленное Лесуром и др. [3233] на основании результатов
электрохимических исследований расплава LiNOs—NaNO'3—AgCl, представляется завышенным.
При Т > 298,15 К термодинамические функции вычислены на основании результатов изучения
энтальпии LiNO3 в работах Гудвина и Кальмуса [2442] D42—576 К), Тая и др. [4564] D33—573 К)
ж Камимото [2915] B99—552 К) (в последних двух работах результаты измерений, погрешность
которых оценивается авторами в 1 и 1,5%, представлены в виде мелкомасштабных графиков).
Для теплоемкости кристаллической фазы в работах [2442] и [2915] приняты постоянные значения§
111,7 и 105 Дж'К>моль соответственно. По данным [4564] теплоемкость монотонно растет.
В справочнике для теплоемкости 1лгГО3(к) в температурном интервале 298,15—526 К принимается
уравнение, полученное на основании величин С°B98,15 К) (см. табл. 45.1), С°D33 К)=106 я
С!D73 К)=113 Дж-К^-моль [4564].
Температура плавления E26+0,5 К) принята по измерениям [1858, 2098, 2099, 2319, 2411,
4217, 4303], выполненным на достаточно чистых образцах. Энтальпия плавления B5,0 +
+0,5 кДж-моль) принята по согласующимся между собой результатам калориметрических работ
[2442, 2915, 3037, 4217] B5,52, 24,94, 25,26 и 24,60 кДж-моль соответственно). Определения
энтальпии плавления, выполненные методом криоскопии и ДТА [2098, 2319, 3983], приводят к более
высоким значениям B5,68, 26,92 и 26,78 кДж-моль соответственно). Необъяснимо большая
величина &тН получена в работе [4564] C6,74 кДж-моль).
Теплоемкость 1лК03(ж) A49+5 Дж-К-моль~1) принята по согласующимся между собой
данным [4564] E25—573 К) и [2915] E25—552 К). Заниженное значение A12,5 Дж-К^-моль),
полученное в работе [2442], обусловлено, по мнению Камимото [2915], содержанием примесей
в образце, очистка которого затруднена вследствие его гигроскопичности.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, и 2000 К оцениваются в 8, 13
и 20 Дж-К~1-моль~1. Расхождения между термодинамическими функциями 1л1ЧО3(к, ж),
приведенными в табл. 1084 и в справочнике Келли [2960] (Т <^ 600 К), не превосходят 0,13 Дж-К-моль
в значениях Ф°(Г) для LjNO3(k) и достигают 0,6 Дж-К^-моль для 1лЖK(ж), что обусловлено
уточнением теплоемкости 1л]ЧО3(ж) в работах [2915] и 14564].
Таблица 45.32. Результаты измерений AajIT^LiNOs, к-х»Н20, 298,15 К) (в кДж-моль
Авторы
Томсен, 1886 [4511]
Пиккеринг, 1888 [3839]
Хейг, 1912 [2548]
Недельякович, 1968 [3638]
Ояри, 1971 [746]
Шпакова и др., 1973 [1161]
Конечная концентрация, температура
измерений
100 Н2О, 293,4 К, 2 измерения
206 Н2О, 291,2 К, 1 измерение
460 Н2О, 294,2 К, 3 измерения
400 Н2О, 296,7 К, 4 измерения
400 Н2О
2775 Н2О, 298,15 К
Да2#°(соН2О, 298,15 К)
—2,43+0,21
—2,62+0,10
—2,64+0,21
—2,640+0,075
—2,76
—2,619+0,042
В справочнике принята энтальпия образования
,, к, 298,15 К) = —482,7 + 0,5 кДж • моль,
* Имеющееся в работе [1776] указание на фазовое превращение при 453 К не подтверждено в других работах.
295
LiNO3(r) Глава 45
основанная на приведенных в табл. 45.32 результатах измерений энтальпии растворения LiNO3(K).
Наиболее достоверные измерения были проведены в работах [1161] и [3638]. Для
дальнейших расчетов принимается средневзвешенное значение Дяг#°(соН20, 298,15 К) = —2,62 +
+ 0,05 кДж-моль, которому соответствует принятая энтальпия образования. Совпадающее
значение, —482,7 кДж-моль, было вычислено на основании измеренной Ричардсом и Роу [3994]
энтальпии нейтрализации растворов LiOH и HNO3.
Давление пара в реакции ЬШ03(к, ж)=1ЛГ\ГОа(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = As#°(LiNO3, к, 0) = 170 + 10 кДж • моль.
Принятая величина основана на масс-спектрометрическом исследовании, выполненном Бага-
ратьян и Никитиным [56, 58]. Опыты с двойной эффузионной ячейкой показали, что основным
компонентом пара является мономер (количество димера при 700 К составляет 12%). При 606,8 К
методом изотермического испарения было найдено парциальное давление мономера, что позволило
вычислить по уравнению III закона принятое в справочнике значение. Его погрешность
обусловлена в основном неточностью термодинамических функций 1ЛГТО3(ж) и LiNO3(r). Измерения
интенсивностей ионных токов, выполненные в интервале 605—655 К, приводят к значению
192 кДж-моль. В работе этих же авторов методом электронного удара была найдена энергия
связи D0(Li—NO3)=530+20 кДж-моль, которой соответствует значение Д81го@) = 180 +
+20 кДж-моль, совпадающее в пределах погрешности с принятой величиной.
LiNO3(r). Термодинамические свойства газообразного нитрата лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1085.
Молекулярные постоянные, использованные для расчета термодинамических функций LiNO3(r),
приведены в табл. 45.9. На основании неэмпирических квантовомеханических расчетов [434, 1298,
3553, 4809] и результатов электронографических исследований паров RbNO3 [605] и CsNO3 [605а]
в справочнике принимается, что в основном электронном состоянии X1Аг молекула LiNO3 имеет
плоскую бидентатную циклическую конфигурацию (симметрия C2v) x с геометрическими
параметрами: -3 O4rN-O4=115+10°, ^N-0^=1,29+0,04 A, r(N—Овнешн)=1,21+0,04 А и гAл-Оц) =
=1,8+0,1 А. Погрешность вычисленного по этим параметрам произведения моментов инерции
IaIbIc составляет 5-1015 г3-см6 2.
Приведенные в табл. 45.9 значения частот валентных колебаний vx, v2 и v5, деформационных
(неплоского v8 и плоских v3, ve) колебаний иона NO~ и значение частоты v4=vLi_o получены Смир-
лом и Девлином [4333] в ИК-спектре молекул LiN03, изолированных в Аг-матрице 3. Близкие
значения основных частот иона NO" наблюдались в многочисленных исследованиях спектров
кристаллов, расплавов и растворов LiNO3 [803, 1685, 1686. 1715, 1717, 2059, 2100, 2456, 2476, 2783, 2784,
2828, 2829, 2841, 3231, 3251, 3505., 3625, 4503, 4638, 4733, 4737, 4738, 4801], а также молекул LiNO3,
изолированных в матрицах из Н2О, NH3, CO2 и СС14 [3864, 4319, 4333]. Для частот v7 и v9,
характеризующих колебания иона Li+ относительно иона NO3, приняты значения соответствующих частот
LiNO2. Погрешности принятых значений v7 и v9 оцениваются в 50 см, остальных частот в 15—
20 см. Сведения о возбужденных электронных состояниях LiNO3 в литературе отсутствуют.
Термодинамические функции LiNO3(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний и возможных изомеров. Погрешности рассчитанных
термодинамических функций обусловлены неточностью принятых значений молекулярных постоянных,,
прежде всего структурных параметров, а также приближенным характером расчета, они
составляют 6, 12 и 15 Дж-К^-моль-1 в значениях Ф°(Г) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций LiNO3(r) публикуется впервые.
1 Согласно расчету [4809] бидентатная конфигурация симметрии С2„ выгоднее монодентатной и тридентатной
соответственно на —3500 и 13 000 см.
2 Ходченков и др. [1080] при злектронографическом исследовании паров LiNO3 нашли, что молекула LiNO3 имеет
монодентатную конфигурацию симметрии Ct. Однако Бюхлер и Штауфер [1706] в результате масс-спектрометри-
ческого исследования показали, что мономер и димер существуют в парах примерно в одинаковых количествах
наряду с заметным количеством продуктов разложения. Поэтому данные [1080] представляются ошибочными-
* В соответствии с рекомендацией Битти и др. [1444] в справочнике принято обратное отнесение приведенных
в работе [4333] частот Vj и v5.
296
Литий и его соединения 1л2СО3(к, ;к>
Константа равновесия реакции LiNO3(r)=Li(r)+N(r)+3O(r) вычислена с использованием
значения Дг?Г°@)=1672,413+10 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации LiNO3(k). Этим величинам соответствует также
г, 0) = —303,511 ±10 нДж-моль.
1Л2СО3(к, ж). Термодинамические свойства кристаллического и жидкого карбоната дилития
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1086.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние 1Л2СО3(к) в интервале 0—1005 К принята
моноклинная модификация [2156].
При Т <^ 298.15 К термодинамические функции 1Л2СО3(к) вычислены по результатам
измерений теплоемкости в работе Брауна и Латимера [1691] A6—300 К, образцы чистотой 99,8%).
Экстраполяция теплоемкости проводилась по закону Дебая и привела к значению ?сA6,7 К) =
=0,22 Дж-К^-моль. Погрешности принятых значений ?°B98,15 К) и Я°B98,15 К)—Я°@>
(см. табл. 45.1), составляют 0,3 Дж-К~1-моль~1 и 0,05 кДж-моль соответственно.
При Т > 298,15 К измерения энтальпии 1Л2СО3 проводили Янц и др. [2845] E48—1150 К)
и Ролин и Рекапе [4035] F72—1071 К) х. В справочнике обработка данных по энтальпии 1л2СО3(к)
проводилась без учета полиморфных превращений, существование которых остается недоказанным.
Уравнение для теплоемкости в интервале 298,15—1005 К (см. табл. 45.1). выведено на основании
значений С;B98,15 К), Я°G00 К)-#°B98,15 К)=52,8 кДж-моль и Я°A000 К)-#°B98,15 К) =
= 108,5 кДж-моль; последние два значения получены усреднением данных Ролина и Рекапе
[4035] и Янца и др. [2845], различающихся между собой на ~2% 2. Результаты измерений [4035],
проведенные вблизи термической аномалии Li2CO3 F72—764 К), не учитывались.
Температура плавления A005+3 К) принята по согласующимся между собой измерениям,
проведенным в работах [78, 377, 423, 442, 703, 871, 948, 3763, 3766]. Данные [851, 880, 2843, 2845,
3029, 3733, 3797, 3968, 4035, 50366], в том числе калориметрические измерения [2845, 4035],
приводят к более низким значениям (996—999 К), по-видимому, из-за примесей в исследованных
образцах. Энтальпия плавления D1,4+4,0 кДж-моль) вычислена по уравнениям, принятым
для теплоемкости жидкого и кристаллического карбоната дилития. Принятое значение
согласуется в пределах погрешности с величинами, полученными в работах [2845] D4,8 кДж-моль) и
[4035] C7,7 кДж-моль-1).
Теплоемкость 1Л2СО3(ж) A85+6 Дж-К~1-моль~1) рассчитана из данных по энтальпии,
полученных Янцем и др. [2845] (996—1150 К) и исправленных в справочнике [2834]. Данные по
энтальпии 1л2СО3(ж), полученные Ролиным и Рекапе [4035] (999—1071 К), приводят к нереально
высокому значению С°р(ЬцСО3, ж)=297 Дж-К-моль, по-видимому, из-за того, что они получены
в очень узком интервале температур.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,2,
2, 10 и 20 Дж-К~1-моль соответственно. Расхождения между термодинамическими функциями
1Л2СО3(к, ж), приведенными в справочнике JANAF [2834] (до 2000 К) и в табл. 1086, достигают
3,5 Дж-К^-моль в значениях Ф°(Г); они обусловлены различием принятых значений
теплоемкости и энтальпии плавления.
Константа равновесия реакции 1л2СО3(к, ж)=21л(г)+С(г)+ЗО(г) вычислена с использованием
значения Дг/7°@)=2972,947+2,3 кДж-моль, соответствующего принятой в справочнике
энтальпии образования 1л2СО3(к)
bfH°(Li2COs, к, 298,15 К) = —1214,1 + 1,0 кДж • моль.
Принятая величина основана на измерениях энтальпии растворения 1л2СО3(к) в воде,
выполненных Брауном и Латимером [1691] (разведение Li2CO3-1900 Н2О,; ДагЯB98,15 К)=—14,14 +
+0,13 кДж-моль) и Монтгомери и др. [3548] (разведение Li2CO3-725H2O, ДЯ(г#B98,15 К) =
=—14,81+0,04 кДж-моль). Пересчет этих величин к стандартному состоянию был проведен
1 В таблицах JANAF [2834J на основании данных Рейсмана [3968] принимается, что 1Л2СО3(к) имеет два
полиморфных превращения F23+3 и 683+3 К). Смирнов и др. [948], Папен [3763], а также авторы [851, 3733J
поставили под сомнение это предположение, объяснив наблюдавшиеся эффекты плавлением эвтектик карбоната
дилития с примесями (Li2O, LiOH и т. п.).
2 Данные Янца и др. [2845] использовались с исправлениями, приведенными в [2834J.
297
1лВ02(к, ж) Глава 45
с учетом поправки на гидролиз (см. [4099]) и оцененных значений энтальпии разведения,
полагаемых приближенно равными энтальпии разведения растворов Li2SO4(k). Из этих работ для реакции
Ь12С03(к)+соНаО=21Л(ад)+СОз2И) было найдено AaqH°(Li2COs, к -* ооН20; 298,15 К) =
=—18,05+0,90 кДж-моль, чему соответствует принятое значение энтальпии образования
1Л2СО3(к).
Измерения энтальпии реакции взаимодействия растворов 1л2СО3(к) с соляной кислотой,
проведенные Мюллером [3582], и растворов LiOH и СО2, проведенные Форкраном [2297], приводят
к близким, но менее точным величинам (—1215,5 и —1210,1 кДж-моль соответственно).
Исследования равновесий реакций с участием 1л2СО3(к, ж) [947, 2723, 2844, 2883, 2961, 3113, 3214, 3397]
не позволяют получить надежных значений энтальпии образования Li2CO3 вследствие сложного
состава продуктов реакции и отсутствия данных по коэффициентам активности их компонентов.
1ЛВО2(к, ж). Термодинамические свойства кристаллического и жидкого метабората лития
в стандартном состоянии при температурах 100—3000 К приведены в табл. 1087.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. В справочнике за стандартное состояние 1лВО2(к) в интервале 0—1122 К принимается
моноклинная а-модификация [4808] *.
При Т <¦ 298,15 К термодинамические функции вычислены на основании данных по
теплоемкости, полученных в работах Турдакина и Тарасова [1032] E5—300 К) и Сталла и др. [4420]
A5—300 К, образец содержал не менее 99,6% основного продукта и в виде примесей — карбонаты
лития и натрия» а также остаточную влагу). В расчетах для интервала 15—55 К использованы
данные [4420], а в интервале 55—298,15 К — значения, полученные усреднением данных [1032,
4420]. Экстраполяция теплоемкости ниже 15 К по уравнению Дебая приводит к величине S°A5 К)=
=0,05 Дж-К^-моль. Погрешности принятых значений 5°B98Д5 К) и #°B98,15 К)—#°@)
(см. табл. 45.1) оцениваются в 0,6 Дж-К^-моль и 0,08 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости 1лВОа(к) принято уравнение, полученное на основании
результатов измерений энтальпии, выполненных Мак-Дона льдом B88—1116 К) (работа не
опубликована и известна по таблицам JANAF [2835]). При выводе уравнения были использованы
значения С;B98,15К) (см. табл. 45.1), Я°F00 К)-Я°B98,15 К)=22,45 кДж-молъ-1 и Я°B000 К)-
#°B98Д5 К)=61,15 кДж-моль-1 [2835].
Температура плавления A122+2 К) принята по работам Састри и Хуммеля [4145] A122+2 К)
и Буазиза и др. [1627, 3366] A122 К). Измерения температуры плавления в [1627, 3366., 4145]
проводились на весьма чистых образцах и сопровождались рентгенографическими исследованиями.
В ряде работ были получены менее точные результаты A109—1123 К). Энтальпия плавления
C3,9+0,4 кДж-моль) принята по данным Мак-Дональда (см. [2835]). В работах [3807, 4812,
4813] на основании криоскопических измерений получены менее точные значения C4,7 и
-31,0 кДж-моль).
Теплоемкость 1ЛВО2(ж) A44,4+3 Дж-К-моль~1) принята по результатам измерений энталь-
лии, проведенных Мак-Дональдом (см. [2835]) A118—1707 К).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,4,
1, 3 и 8 Дж-К-моль~1. Термодинамические функции 1лВО2(к, ж), приведенные в таблицах JANAF
[2835] (Т ^ 3000 К), согласуются с данными табл. 1087 в пределах 0t7 Дж-К^-моль в
значениях Ф°(Г).
Принятая в справочнике энтальпия образования
к, 298,15 К) = —1022,9 + 2,5 кДж • моль
основана на измерениях энтальпии растворения в 2N азотной кислоте ЫВО2(к)а (Шартсис, Каппе
[4252], ДГ#°B98,15К)=—45,2+2,1 кДж-моль-1), В2О3 (стекл.) (Шульц и др. [HSfl],
Ari?0B98,15 K) = —38,5+0,8 кДж-моль) и вычисленной по принятым в справочнике данным
энтальпии растворения 1Л2О(к) в 2N азотной кислоте (Дг#°B98,15 К) =—244,9+0,5 кДж-моль).
1 Кроме моноклинной модификации известны еще две другие устойчивые при высоких давлен-иях [1808, 1809,
3366, 3384, 3385]: тетрагональная ^-модификация (структурный тип халькопирита) и ^-модификация с низкой
симметрией. Полиморфизм 1лВО2(к) остается не вполне изученным. Авторам [3366] удалось получить J3- и у-
модификации при обычном давлении, по-видимому, в метастабильном состоянии.
2 Величина найдена линейной экстраполяцией к составу LiBO2 энтальпий растворения соединений nLi2O-B2Os,
полученных в [4252].
298
Литий и его соединения
1лВО2(г)
В справочнике [2835] со ссылкой на неопубликованную работу Зинке приведено значение
энтальпии растворения 1лВ02(к) в азотной кислоте, совпадающее с измеренным в работе [4252].
Давление пара метабората лития в реакции 1лВ02(к, ж)=1ЛВ02(г) вычислено с использованием
принятой в справочнике энтальпии сублимации
ДгЯ°@) == Д8#оAЛВ02, к, 0) = 366 + 6 кДж • моль.
Результаты измерений давления пара и равновесия реакций с участием LiBO2(r) представлены
в табл. 45.33. Погрешности учитывают воспроизводимость измерений и неточность использованных
Таблица 45.33. Результаты определений AsJET°(LiBO3, к, 0) (в кДж • моль)
Авторы
Бюхлер, Берковиц-Маттук, 1963 [1703'
Хилденбранд и др., 1963 [2646]
Горохов и др., 1969 [311, 642]
Енсен, 1969 [2853]
Метод
Масс-спектрометрическин, 1160 К, 1 точка
То же, 1000—1220 К, 16 точек
Торзионный, 1121—1280 К, 22 точки
Эффузионный, 1323 К, 1 точка
Масс-спектрометрический, 1016—1112 К, 4 серии
То же, 1175—1273 К, 3 серии
Спектрофотометрия в пламенах H2+Oa+N2,
Li(r)+HBO2(r)=LiBO2(r)+H(r), 2020—2560 К,
данные представлены в виде уравнения
Д,Я°AЛВО2, к, 0)
II закон
340+13
362 ±11
—
332+10
351±10 .
362
III закон
378
—
365,7 ±5,0
368,4
—
—
371+20
в расчетах термодинамических функций. Наиболее детальные измерения были выполнены Хил-
денбрандом и др. [2646], и на них основана принятая в справочнике величина. Близкое значение
было найдено в эффузионных измерениях Горохова и др. [311, 642]. Основным источником
погрешности принятой величины является неточность термодинамических функций.
ЫВО2(г). Термодинамические свойства газообразного метабората лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1088.
Молекулярные постоянные, использованные для расчета термодинамических функций LiBO2,
приведены в табл. 45.9. Электронографические исследования [15, 16, 402, 403], изучение
ИК-спектра молекул LiBO2, изолированных в матрицах из Аг и Кг [4237], и неэмпирические
расчеты равновесной геометрии [2036] показывают, что молекула LiBO2 в основном состоянии %ХА'
имеет угловую конфигурацию с линейным фрагментом О—В=О (симметрия Cs). Значение угла
Li—О—В, рекомендованное в работах [15, 16, 2036, 4237], изменяется в пределах от 90 до 125°.
Расчет [2036] показал, что разность энергии угловой и линейной конфигураций составляет около
600 см, и, следовательно, ион Li+ может почти свободно перемещаться вокруг иона ВО2.
Учитывая заметные отличия равновесного и эффективного значений ^ М—О—В в метаборатах щелочных
металлов, а также ограниченные возможности элейтронографического метода в определении угла
Li—О—В, в справочнике для -^ Li—О—В принято эффективное значение 130+30°, равное
соответствующему углу в молекуле CsBO2, где он был определен более точно, чем в других
метаборатах.
Произведение моментов инерции, приведенное в табл. 45.9, рассчитано по структурным
параметрам r(Li—О)=1,76+0,05 А (оценка Ежова [397]), г(В—О)=1,29+0,05 А и г(В=0)=1,19 +
+0,05 А (рекомендованы Ежовым и др. [402] на основании измеренного среднего значения
г(В—О)=1,26+0,01 А [403] и рассчитанной величины разности г(В—О)—r(B=O) ^ 0,10 А).
Погрешность значения IaI&Ic оценивается в 2-10~115 г3-см6. Основные частоты, приведенные
в табл. 45.9, приняты по работе Сешадри и др. [4237] после пересчета их для природной смеси
изотопов В и Li. В ИК-спектре высокотемпературного пара над LiBO2 наблюдались' частоты v1?
v3 и v4 [1705] и vx—v5 [648], значения которых хорошо согласуются между собой, но на 20—70 см
отличаются от частот, наблюдаемых в ИК-спектре в матрицах [4237]. С учетом данных для пара
[648, 1705] и возможных матричных сдвигов погрешности частот vx—ve оценены равными 20—
30 см.
299
LiAlF4(r) Глава 45
Термодинамические функции LiBO2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и
A. 130) без учета возбужденных состояний и особенностей движения иона Li+ относительно ВОт.
Погрешности рассчитанных термодинамических функций определяются приближенным характером
расчета и неточностью принятых молекулярных постоянных, они составляют 5, 8 и
10 Дж-К^-моль-1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции LiBO2(r) были рассчитаны в таблицах JANAF [2835];
расхождения данных [2835] и табл. 1088 не превышают 2,5 Дж-К^-моль в значениях Ф°(Г).
Константа равновесия реакции LiBO2(r) = Li(r)+B(r)+2O(r) вычислена с использованием
значения ?±ГН°((У) = 1862,545 +8,3 кДж-моль, соответствующего принятым в справочнике энтальпиям
образования и сублимации 1ЛВО2(к).
Этим величинам также соответствует
bfH°(WBO%, г, 0) = — 651,244 + 6,5 кДж • моль.
LiAlF4(r). Термодинамические свойства газообразного тетрафторалюмината лития в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 1089.
Молекулярные постоянные, использованные для расчета термодинамических функций LiAlF4,
приведены в табл. 45.9. На основании спектроскопических [1978, 2750] исследований, а также
квантовомеханического расчета Куртисса [1973] для молекулы LiAlF4 в основном электронном
состоянии ХгАх в справочнике принята бидентатная структура (симметрия С2г). Геометрическая
структура LiAlF4 экспериментально не изучалась. Приведенное в табл. 45.9 произведение моментов
инерции вычислено по структурным параметрам г(А1—F)BHeniH=r(Al—F)M0CT=1,70+0,05 А
(оценено по молекулам NaAlF4, KA1F4 и CsAlF4), r(Li—F) = l,74+0,1 А (оценено по Li2F2) и ~$ FBHemH—
Al—F»Hemi=-$ fmc,—Al—FMOCT=109,5+10°1. Погрешность величины IaIbIc оценивается
в 5-101* г3-см6. Основные частоты, приведенные в таблице, основаны на исследованиях ИК-спек-
тров молекул LiAlF4, изолированных в матрицах из Ne в работе Сивина и др. [1978] и в матрицах
из Аг и N2 в работе Хаглена и др. [2750]2. Спектры, полученные в работах [1978, 2750], хорошо
согласуются между собой, за исключением трех частот в областях 220, 270 и 611 см. Низкие
частоты B20 и 270 см) наблюдались Сивином и др. [1978], но не были обнаружены в матрицах
из Аг и N2 [2750]. Авторы [2750] объяснили это тем, что их спектральный прибор не позволял
исследовать область частот ниже 250 см. Поглощение в области 610 см наблюдалось в обеих
работах, однако авторы [1978] не отнесли его к молекуле LiAlF4. Хаглен и др. [2750] наблюдали
слабое поглощение в области 616—623 см (Аг) и 628—630 см (N2) в спектрах тетрафторалюмина-
тов всех щелочных металлов, причем частота менялась при изменении щелочного металла. Это
позволило авторам [2750] отнести частоты колебаний в области 616—630 см к молекулам MA1F4.
В справочнике основные частоты приняты по работе [2750], авторы которой оценили частоты
свободных молекул LiAlF4 на основании результатов своего исследования и данных [1978].
Для ненаблюдавшихся в спектрах частот v5, v6 (рассчитаны в работе [2750] для модели поля
валентных сил) и v8 (рассчитана в работе [1978]) приняты значения, отличающиеся от
рекомендованных авторами [2750], так, чтобы сохранилось закономерное изменение частот однотипных
колебаний в ряду молекул LiAlF4, NaAlF4, KA1F4, RbAlF4 и CsAlF4 (для NaAlF4, KA1F4, RbAlF4,
CsAlF4 частоты v6 и v8 наблюдались в работе [2750] экспериментально). Значение частоты v6,
рассчитанное Хагленом с соавторами, представляется завышенным. Ее значение было принято
в справочнике на основании соответствующих частот A12F6 и AlFj (см. гл. 21). Погрешности
основных частот LiAlF4, наблюдавшихся в спектре, оцениваются в 15—20 см, частот vB, ve и v8 —
в 80, 50 и 80 см соответственно. Куртисс [1973] на основании квантовомеханического расчета
показал, что молекула LiAlF4 кроме бидентатной конфигурации имеет тридентатную и моноден-
татную структуры; эти конфигурации в справочнике не рассматриваются.
1 Согласно расчетам Куртисса [1973] и Жилинского и др. [424], г (Al—F)Menra, г (Al—F)M0CT и 2L Р,в,шн-a1~
F,mnra, 2L Fhoot—Al—FH00T неравноценны. В справочнике длины связей и валентные углы приняты эквивалент"-
ными, поскольку в электронографических исследованиях тетрафторалюмияатов натрия, калия и цезия
неравноценность межъядерных расстояний не обнаружена, а неэквивалентность валентных углов найдена лишь в работе
Вайда и др. [4585] для молекулы KA1F4.
2 В работе [3447] в ИК-спектрах паров над 3LiF • A1F3 и Na,AlF6 наблюдались две широкие полосы в области
775—783 см~* и 853—880 см, которые предположительно были отнесены к молекулам LiAlF4 и NaAlF4.
300
Литий и его соединения
Li3AlFe(K, ж)
Термодинамические функции LiAlF4(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний и возможных изомеров этой молекулы. Погрешности
рассчитанных величин обусловлены приближенным характером расчета и неточностью принятых
молекулярных постоянных; они составляют 8,15 и 25 Дж-К~1-моль~1 в значениях Ф°(Т) при 298,15,
3000 и 6000 К соответственно.
Ранее термодинамические функции LiAlF4(r) рассчитывались в таблицах JANAF [2834],
а также в работах Сивина и др. [1976] (до 2000 К), Снелсона и Сивина [4336] (до 2000 К), Морозова
и др. [696] (до 5000 К) и Хаглена и др. [2750] (до 2000 К). Расхождения данных [2834], [2750]
и табл. 1089 не превышают 3 и 0,3 Дж-К^-моль в значениях Ф°(Г) соответственно. Расхождения
с данными [696, 1976, 4336] достигают 5, 7 и 8 Дж-К^-моль-1 в значениях Ф°(Т).
Константа равновесия реакции LiAlF4(r)=Li(r)+Al(r)-f-4F(r) вычислена с использованием
значения Дг/У°@)=26441181+12 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
г, 0) = —1850 ±12 кДж • моль"
Результаты определений этой величины представлены в табл. 45.34. Приведенные в таблице
Таблица 45.34. Результаты определений Ayfi"°(LiAlF4, г, 0) (в кДж-моль)
Авторы
Метод
Ayff°(LiAlF4, г, 0)
II закон
III закон
Портер, Целлер, 1960 [3872] Масс-спектрометрический, Li2F2(r)+AlF3(r)= — —1825+12
=LiF(r)+LiAlF4(r), 1000 К, 1 точка
Хилденбранд, 1963 [2650] (см. Торзионный, Li3AlFe(K)=2LiF(K)+LiAlF4(r), —1727 —1858+10
[2834, 3926]) 987—1058 К а
То же, 1/3Li3AlFe(K)+2/3AlF3(K)=LiAlF4(r), 854— —1833 -1849,9+8,0
980 К, 3 серии измерений а
Рао, 1970 [3926] Эффузионный, V3Li3AlF6(K)+2/3AlF3(K)=LiAlF4(r), —1841+9 -1848,3+7,5
872—979 К, 34 точки
Сидоров и др., 1973 [935] Масс-спектрометрический, Li3AlF6(K)=2LiF(K)+ — —1843+10
+LiAlF4(r), 946 К, 1 точка
То же, 1/3Li3AlF6(K)+2/3AlF3(K)=LiAlF4(r), - -1844+10
946 К, 1 точка
То же, LiAlF4(r)=LiF(r)+AlF3(r), 946 К, 1 точка — —1845+10
Дьюинг, 1980 [2067] Пересчет неопубликованных литературных дан- — —1856+13
ных, полученных методом точек кипения, 1лР(ж)-[-
+AlF3(K)=LiAlF4(r), 1293 К, 5 измерений,
учтены активности компонентов
а Измеренные константы равновесия не указаны. Энтальпии образования получены пересчетом приведенных в [2834 ]
величин АГЯ°B98 К) с учетом различия термодинамических функций, принятых в [2834] и в настоящем
справочнике.
погрешности учитывают воспроизводимость измерений, неточность термодинамических функций
и термохимических величин, использованных в расчетах. Приведенные в таблице величины
(за исключением работы [3872], где были допущены неточности в отнесении ионных токов, см. [933])
совпадают в пределах их погрешностей. Принятая величина является средневзвешенной и близка
к наиболее полным и точным измерениям в работах [2650, 3926]. См. также работы [1159, 1704,
2749, 3659, 4279], где обсуждаются имеющиеся данные по термохимии этой молекулы.
Li3AlF6(K, ж). Термодинамические свойства кристаллического и жидкого гексафторалюмината
трилития в стандартном состоянии при температурах 100—4000 К приведены в табл. 1090.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 45.1. За стандартное состояние Li3AlFe(K) в интервале 0—788 К принята ромбическая
^-модификация, в интервале 788—873 К тетрагональная у-модификация, при 873—978 К
кубическая 8-модификация, а при 978—1058 К е-модификация (см. [1009, вып. 10]).
301
Li8AlFe(K, ж)
Глава 45
При Т <^ 298,15 К термодинамически^ функции Li3AlF6(K) вычислены по измерениям
теплоемкости, выполненным Фурукава и др. [23511 A5—380 К) на образце ^-модификации, полученном
сплавлением стехиометрических количеств LiF и A1F3. Образец содержал заметные примеси натрия
@,1—1%), никеля @,04%) и менее 0,1% примесей других металлов; поправки на содержание
примесей авторы не вводили. Погрешность измерений оценивается в 1% при 15 К и ~0,2%.
при Т > 100 К. Экстраполяция теплоемкости ниже 15 К по закону Дебая дала S°A5 К) =
=0,18 Дж-К^-моль [2351]. Погрешности принятых по работе [2351] значений 5°B98,15 К)
и #°B98,15 К)—#°@) (см. табл. 45.1), оцениваются в 0,4 Дж-К^-моль и 0,06 кДж-моль
соответственно.
При Т >¦ 298,15 К для теплоемкости P-Li3AlFe принято уравнение, полученное совместной
обработкой согласующихся между собой данных Фурукава и др. [2351] B98,15—380 К) и
результатов неопубликованных измерений энтальпии, проведенных Дугласом и Нефером C23—973 К) %
на том же образце Li3AlF6.
Температуры трех полиморфных превращений G88+10 К, 873+10 К и 978+5 К) и энтальпии
этих переходов C,0+0,3, 3,0+0,4 и 7,7+0,7 кДж-моль соответственно) приняты по
результатам калориметрических измерений Володкович и др. [231, 232]. Для температур превращения
эти данные близки к измерениям [2368, 2692, 4975а], но отличаются от рекомендаций справочника
JANAF [2834], где для температур двух первых превращений на основании рентгенографических
данных [2474] приняты более низкие значения G48 и 848 К). Принятое значение энтальпии
(Р—т)-превращения хорошо согласуется с данными Дугласа и Нефера C,2 кДж-моль). Значения
теплоемкостей у- и 8-модификаций B85 и 295 Дж-К-моль~1), стабильных в относительно узких
интервалах температур, оценены экстраполяцией уравнения для теплоемкости ^-модификации.
Холм и Гренволл [2688] провели измерения энтальпии г-модификации (975—1041 К) и жидкого»
Li3AlF6 A073—1120 К), по этим данным в справочнике принимаются значения теплоемкостей
B78 и 386 Дж-К^-моль) и энтальпии плавления (87,9+1,5 нДж-моль). С последней
удовлетворительно согласуются значения, полученные в менее точных работах [1566] (82,4 кДж-моль)
и [4033] (90 кДж-моль). Температура плавления A058+3 К) принята по данным Бьёрджа и
Енсена [1566]; близкие значения получены в работах [2368, 3426] A056 К) и [3359, 4033] A055 К).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000, 3000 и 4000 К оцениваются-
в 0,3, 3, 20, 40 и 60 Дж-К-^моль.
Термодинамические функции, приведенные в справочнике JANAF [2834], для P-Li3AlF6
практически совпадают с данными табл. 1090. С ростом температуры расхождения увеличиваются и
составляют 9 и 37 Дж-К~1-моль~1 в значениях Ф°(Т) при 2000 и 3000 К соответственно, что
обусловлено использованием в настоящем справочнике данных [231, 232, 2688].
Таблица 45.35. Результаты определений A^ff°(LisAIF,, к, 298,15К) (в кДж-
Авторы
Метод
МОЛЬ)
Aytf^LLjAlF,,, к, 298,15 К)
II закон
III закон
Сидоров и др., 1973 [935] Масс-спектрометржческий, Li3AlFe(K)=3LiF(K)+ — —3387+10
+AlF3(r), 946 К, 1 точка
То же, V8LisAlF,(K)=i/sAlFs(K)+LiF(r), 946 К, - -3402 ±10
1 точка
Дышшг, 1976 [2066] ЭДС, 3LiF(K)+AlF3(K)=LigAlFe(K), 745-957 К, —3390+5 -3391,3+3,5
12 точек
Грин и др., 1968 [2477] Калориметрический, калориметр с микропечью, —3387,9+3,5
3LiF(K)+AlF3(K)=Li3AlFe(K), Д.Я°B98,15 К)=
=-22,6±2,1
Хонг, Клеппа, 1978 [2698] Калориметрический, двойной высокотемпературный —3401 ±12
калориметр, 3LiF(m)+AlF3(K)=Li3AlF6(K),
ДгЯ°A320К)=16,7±2,0
1 Данные известны по таблицам JANAF [2834] а введены в обработку в виде четырех значений И0 (Т)—Н° B98,15К)'
в интервале 400—700 К. Данные [2351] использованы в виде восьми значений Н° (Т)—Н° B98,15 К) в
интервале 310—380 К.
302
Литий и его соединения 6Щк, ж>
Константа равновесия реакции Li3AlFe(K, 5K)=3Li(r)+Al(r)+6F(r) вычислена с использованием
значения АГН°(О)=4641,015 +6,7 кДж-моль, соответствующего принятой в справочнике энталь-~
пии образования
A/ff°(Li8AlFe, к, 298,15 К) = —3389,6 ± 4,0 кДж • моль.
Принятая величина основана на приведенных в табл. 45.35 данных. Как видно из этой таблицы^
результаты измерений разными методами совпадают в пределах их погрешностей (указанные,
в таблице погрешности учитывают воспроизводимость измерений и неточность использованных
в расчетах термодинамических функций и термохимических величин). Значительная погрешность,
стандартной энтальпии образования Li3AlFe, вычисленной по данным работы [2698], объясняется
неточностью значений энтальпий компонентов исследованной реакции, использованных при
пересчете от 1320 К к стандартной температуре. Принятая величина является средней из наиболее
точных определений, проведенных в работах [2477] и [2066].
61л(к, ж). Термодинамические свойства кристаллического и жидкого лития-6 в стандартном
состоянии при температурах 100—3000 К приведены в табл. 1091.
Значения постоянных, использованные для расчета термодинамических функций,
представлены в табл. 45.1. Как и в Случае кристаллического лития природного изотопического состава,;
для Т <^ 105 К в справочнике за стандартное состояние принята гексагональная модификация
(структурный тип Mg), а при Т > 105 К — кубическая модификация (структурный тип NaI.
При Т <^ 298,15 К термодинамические функции лития-6 вычислены по согласующимся между
собой результатам измерений теплоемкости, проведенных Мартином [3405] @,4—1,5 К), _ Мартином,
[3401 ] B0—300 К) и Филби и Мартином [2250] C—30 К). Во всех этих работах исследовался обра-~
зец, содержавший 99,3+0,1% изотопа 6Li, 0,7+0,1% изотопа 7Li, а также примеси кальция
@,25%) и других металлов (—0,1%). Авторы оценивают погрешности измерений в 1—2% при
3_30 К и в ~0,2% для Т > 80 К. Расчет термодинамических функций проводился с учетом мар-^
тенситного превращения 6Li. Погрешности принятых значений ?°B98,15 К) и #°B98,15 К)—#°@)
(см. табл. 45.1) оцениваются в 0,2 Дж-К^-моль и 0,04 кДж-моль.
При Т > 298,15 К экспериментальные данные о теплоемкости или энтальпии eLi (к, ж) в
литературе отсутствуют. В справочнике для в1л(к) принято уравнение (см. табл. 45.1), приводящее-
к значениям теплоемкости, которые на 0,857 Дж-К-моль меньше принятых для лития
природного изотопического состава. Эта величина получена при сравнении экспериментальных значений
теплоемкости 6Li и e>941Li в интервале 250—298,15 К, разность которых сохраняется практически
постоянной.
Температура плавления D53,5+0,2 К) принята по данным Крофорда и Монтгомери [1961],
в пределах погрешности она совпадает с температурой плавления природной смеси изотопов
лития D53,69+0,03 К). Данные по энтальпии плавления и теплоемкости жидкого лития-6 в
литературе отсутствуют; в справочнике они приняты такими же, как для лития-6,941. Погрешность,
этих значений для eLi не превышает 3—5%. Погрешности вычисленных значений Ф°(Т) при 298,15V
1000, 2000 и 3000 К оцениваются в 0,15, 1, 2, и 4 Дж-К-моль соответственно.
Таблица термодинамических функций *1л(к, ж) публикуется впервые.
В настоящем справочнике в качестве стандартного состояния лития-6 принято кристалличе-.
ское и жидкое состояния и в соответствии с этим
A/TfLi, к, ж)==0.
Давление пара лития-6 в реакции 61л(к, ж)=е1Л(г) вычислено с использованием энтальпии
сублимации
ДгЯ°@) = Д,Я°FЫ, к, 0) = 158,0 + 1,5 кДж • моль.
Эта величина была вычислена на основании соотношения рFЫ) : pGLi)=1,002+_04004 при,
~1000 К, полученного Траугером и др. (см. [4878ал 4887в]J.
1 Температура «мартенситного» превращения A05 + 10 К) определена в работе Мартина [3401] как температура
максимума избыточной теплоемкости ?Li этого процесса.
а Принятые в справочнике термодинамические свойства 6Ы(к, ж, г) приводят к тому, что при Т > 1000 К дав^
ление насыщенного пара 6Ы(ж) превышает давление 7Li (ж); при Т < 800 К имеет место обратное соотношение
и при Т,п это различие составляет 10%. Экспериментальные данные об отношении р (eLi): р GLi) при
температурах выше и ниже 1000 К в литературе отсутствуют.
303
*Li(r), 8Li+(r), 6LiH(k, ж) Глава 45
6Li(r). Термодинамические свойства газообразного лития-6 в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 1092.
Уровни энергии атома eLi, использованные для расчета термодинамических функций, приняты
такими же, как для изотопа 7Li (см. табл. 45.3). Термодинамические функции eLi(r) вычислены
по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 23)—A. 25) непосредственным суммированием по
уровням энергии. Погрешности вычисленных значений при Т ^ 5000 К определяются в основном
неточностью значения R и не превышают 0,02 Дж-К~1-моль~1 в Ф°(Т) и S°(T); при более высоких
температурах погрешности возрастают из-за того, что в расчете не учитывались ридберговские
состояния и при 6000 и 10 000 К они могут достигать 0,05 и 0,3 Дж-К-моль~1 в значениях
Ф°(Г) и 0,3 и 1,0 Дж-К^-моль-1 в значениях S°(T).
Таблица термодинамических функций 6Li(r) публикуется впервые.
Энтальпия образования eLi(r)
bfH°(*Li, г, 0) = 158,0 ±1,5 кДж.моль
принята равной энтальпии сублимации 61л(к) (см* выше).
6Li+(r). Термодинамические свойства газообразного положительного иона лития-6 в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 1093.
Термодинамические функции 6Li+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
при условии, что QS1[—1-, так как основное электронное состояние 6Li+ относится к типу XS, а все
возбужденные состояния являются ридберговскими и имеют высокие энергии. Погрешность
вычисленных значений термодинамических функций во всем интервале температур обусловлена
только неточностью фундаментальных постоянных и не превышает 0,01—0,02 Дж-К-моль.
Таблица термодинамических функций 6Li+ публикуется впервые.
Константа равновесия реакции 6Li+(r)+e(r)=6Li(r) вычислена с использованием значения
АгН°@)=—520,068+0,002 кДж-моль, соответствующего потенциалу ионизации
/0(eLi)=43 474,3+0,2 см=520,068+0,002 кДж-моль.
Эта величина была принята на основании оценки разности потенциалов ионизации 'Li и 6Li
A2,9 см), выполненной в работе Мака (см. [3550]).
Принятому значению соответствует
Д/Я°(вЫ+, г, 0) = 678,068±1,5 кДасмоль.
61ЛН(к, ж). Термодинамические свойства кристаллического и жидкого гидрида лития-6 в
стандартном состоянии при температурах 100—2000 К приведены в табл. 1094.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. За стандартное состояние вЬШ(к) в интервале 0—965 К, как и в случае гидрида лития
природного изотопического состава F|941LiH), принята кубическая модификация со структурным
типом NaCl. Какие-либо экспериментальные данные по термодинамическим свойствам 61лН(к, ж)
в литературе отсутствуют. Термодинамические функции 61лН(к) в интервале 0—965 К были
вычислены с использованием результатов расчетов теплоемкостей в1лН(к) и 'ЦН(к) с учетом
изотопического эффекта1, выполненных Якимовичем и Мельниковой [1190]. Для нахождения
приведенных в табл. 45.1 постоянных 61лН(к) были использованы принятые в справочнике значения для
гидрида лития природного изотопического состава и вычисленные авторами [1190] разности в
значениях других термодинамических функций 61лН(к) и 71лН(к). Погрешности полученных таким
образом для 6LiH значений 5°B98,15 К) и Я°B98,15 К)—#°@) оцениваются в 0,8 Дж-К^-моль
и 0,12 кДж-моль соответственно. Полученное уравнение для теплоемкости 61ЛН(к) в интервале
298,15—965 К (см. табл. 45.1) приводит к значениям, которые при 298,15 К отличаются от
соответствующих данных для 1лН(к) на 0,7 Дж-К~1-моль~1, а при 965 — на 0,13 Дж-К~1-моль.
Принятые в справочнике температура плавления 6LiH (965+3 К), энтальпия плавления B1,8 +
+ 1 кДж-моль) и теплоемкость 6ЫН(ж) E5+5 Дж-К-моль) практически совпадают с
экспериментальными данными для гидрида лития природного изотопического состава.
Погрешности рассчитанных значений Ф°(Г) 61лН(к, ж) оцениваются в 0,6, 2 и 5 Дж-К~1-моль~1
при 298,15, 1000 и 2000 К. Вычисленные Якимовичем и Мельниковой [1190] значения термоди-
1 Аналогичные расчеты для 7LiH (к) и 7LiD (к), проведенные в этой же работе [1190], основаны на
экспериментальных данных по теплоемкости этих изотопических модификаций.
304
Литий и его соединения 'LiH(r), *LiD(K, ж)
намических функций в1ЛН(к) различаются с приведенными в табл. 1094 на 0,3 Дж-К~1-моль
в значении 5°(900 К), поскольку расчеты [1190] термодинамических функций изотопических
модификаций LiH не были полностью приведены в соответствие с экспериментальными данными по
теплоемкости 1лН(к).
В справочнике принята энтальпия образования
°(«1ЛН, к, 0) = —85,5 ± 0,8 кДж • моль.
Это значение было оценено на основании принятой выше величины &fH°(LiH., к) и изменения
энтальпии образования в ряду 1ЛН(к)—LID(k)—1ЛТ(к).
Давление пара гидрида лития-6 в реакции в1ЛН(к, ж)=в1ЛН(г) вычислено с использованием
энтальпии сублимации Д^^О) —Д,#°(в1лН, к, 0)=225,286+1,7 кДж-моль, соответствующей
принятым в справочнике энтальпии образования в1ЛН(к) и энергии диссоциации 6LiH(r).
6LiH(r). Термодинамические свойства газообразного гидрида лития-6 в стандартном состоянии
в интервале температуры 100—6000 К приведены в табл. 1095.
В табл. 45.4 приведены молекулярные постоянные eLiH, принятые на основании анализа
спектральных данных и постоянных молекулы 71ЛН. В спектре eLiH изучена только одна система
полос А1!,*—ХХЕ+ [3250, 3628, 4600]. Молекулярные постоянные 6LiH в состояниях ХХ2+ и
41Е+, приведенные в таблице, получены Ли и Стволли [3250] на основании анализа вращательной
структуры 57 полос системы AlT,+—X1I,+ (i/ ^ 16 и v" <^ 7). Энергия состояния А1!,* найдена
в работе [4611]. Молекулярные постоянные в состояниях i^n и а3П рассчитаны по соотношениям
A. 66) и постоянным молекулы 7ЫН.
Расчет термодинамических функций 61ЛН(г) проводился так же, как для LiH(r), на основании
значений Ykl, i^, /llm и а(, вычисленных по изотопным соотношениям A. 66) и соответствующим
величинам, приведенным в табл. 45.5 для молекулы 7LiD.
Погрешности рассчитанных термодинамических функций 61ЛН(г.) составляют 0,02, 0,03 и
0,1 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К (см. выше LiH(r)).
Константа равновесия реакции eLiH(r)=eLi(r)+H(r) вычислена на основании значения
D0FLiH) = 19 581 ±1,0 см = 234,248 ± 0,012 кДж i моль;
Эта величина была принята на основании значений D0GLiH) и колебательных постоянных обоих
изотопомеров (см. табл. 45.4) в предположении, что D,(eLiH)=D,GLiH).
Принятой энергии диссоциации соответствует
r, 0) = 139,786 ±1,5 кДж.моль.
6IAD(k, ж). Термодинамические свойства кристаллического и жидкого дейтерида лития-6
в стандартном состоянии при температурах 100—2000 К приведены в табл. 1096.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 45.1. За стандартное состояние eLiD(K) в интервале 0—967 К принята кубическая
модификация со структурным типом NaCl. Экспериментальные данные по теплоемкости и энтальпии
6ЬШ(к, ж) в литературе отсутствуют, поэтому термодинамические функции были вычислены
с использованием результатов расчетов теплоемкости 61ЛБ(к) и 7ЬШ(к) с учетом изотопического
эффекта в работе Якимовича и Мельниковой [1190]. Приведенные в табл. 45.1 постоянные 6LiD(k),
были найдены по соответствующим данным для LiD(K) и вычисленным в [1190] разностям значений
теплоемкости и других термодинамических функций 61лБ(к) и 7LiD(K ). Погрешности
соответствующих значений 5°B98,15 К) и #"B98,15 К)—Я0 @) оцениваются в 1 Дж-К-^моль и 0,15 кДжХ
X моль соответственно. Полученное таким же способом уравнение для теплоемкости 61лБ(к) в
интервале 298,15—967 К (см. табл. 45.1) приводит к значениям, которые отличаются от данных по
теплоемкости ЫБ(к) при 298,15 К на 1,0 Дж-К^-моль, а при 900 К на 0,17 Дж-К^-моль.
Принятые в справочнике значения температуры плавления (967 +5 К), энтальпии плавления B2 +2 кДж X
Хмоль) и теплоемкости 61лБ(ж) E6+6 Дж-К~1«моль) оценены равными соответствующим
постоянным ЬШ(к, ж) с некоторым увеличением погрешностей этих величин.
Погрешности рассчитанных значений Ф°(Т) для 6LiD(K, ж) при 298,15, 1000, 1500 и 2000 К
оцениваются в 0,7, 2, 5 и 7 Дж-К-^моль соответственно.
Таблица термодинамических функций eLiD(K, ж) публикуется впервые.
305
8LiT(r), Ш(г) =================^ Глава 45
В справочнике принята энтальпия образования
bfH°FLiD, к, 0) = —86,5 ± 0,8 кДж > моль.
Это значение оценено на основании принятой выше величины Д J/°(LiD, к) и изменения1
энтальпии образования в ряду 1лН(к)—1ЛБ(к)—ЫТ(к).
Давление пара в реакции 6LiD(k, ж)=в1л?)(г) вычислено с использованием значения ДгЯ°@)=
A4#°FLiD, к, 0)=227,948+1,7 кДж-моль, соответствующего принятым в справочнике
энтальпии образования 6ЬШ(к) и энергии диссоциации eLiD(r).
6LiD(r). Термодинамические свойства газообразного дейтерида лития-6 в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1097.
В табл. 45.4 приведены молекулярные постоянные 6LiD, принятые на основании анализа
спектральных данных и постоянных молекулы 7LiD. В электронном спектре eLiD наблюдалась одна
система полос Аг1,+—Х1!* [32503. Исследован также микроволновый спектр этой молекулы
13791 ]. Приведенные в таблице постоянные eLiD в состояниях X12 + и А1 ? + найдены Ли и Стволли
{3250] из анализа вращательной структуры 45 полос системы А1!,*—Х12 + (i/ ^ 17 и v" ^ 7).
Молекулярные постоянные в состояниях 2?ХП и а3П рассчитаны по изотопным соотношениям*
A. 66) и постоянным молекулы 7LiD.
Расчет термодинамических функций 6LiD(r) проводился так же, как для LiH(r), на основании;
приведенных в табл. 45.5 значений Ykl, vmax, /,1ш и а(, которые были получены по изотопным
соотношениям A. 66) и соответствующим постоянным 'LiD. Погрешности рассчитанных
термодинамических функций 6LiD(r) принимаются такими же, как для LiD(r).
Таблица термодинамических функций eLiD(r) публикуется впервые.
^Константа равновесия реакции 6LiD(r)=eLi(r)+D(r) вычислена на основании значения
D0(eLiD)=19 758+1,0 cm=236,359±0,012 кДж-моль.
Эта величина была рассчитана по принятым в справочнике значениям D0GLID) и колебательных
постоянных этих изотопомеров (см. табл. 45.4) в предположении, что De(eLiD)=DeGLiD).
Принятой энергии диссоциации соответствует
A/H°(eLiD) = 141,448 ±1,5 кДж.моль.
eLiT(r). Термодинамические свойства газообразного тритида лития-6 в стандартном состоянии*
в интервале температур 100—6000 К приведены в табл. 1098.
Экспериментальные данные о молекулярных постоянных eLiT отсутствуют. Расчет
термодинамических функций eLiT(r) проводился так же как, для LiT(r), на основании значений Ykl, уша1,
Jllm и а4, приведенных в табл. 45.5 и вычисленных по изотопическим соотношениям A. 66) и
соответствующим величинам для 7LiD. Погрешности рассчитанных термодинамических функций
eLiT(r) принимаются такими же, как для LiT(r).
Таблица термодинамических функций eLiT(r) публикуется впервые.
Константа равновесия реакции eLiT(r)=eLi(r)+T(r) вычислена с использованием принятого*
в справочнике значения
D0(eLiT)=19 828,8+2,0 см-^237,205 +0,024 нДж-моль.
Эта величина была рассчитана по принятым в справочнике значениям D0GLiD) и колебательных
постоянных (см. табл. 45.4) и в предположении, что De(eLiT)=DeGLiD).|
Принятой энергии диссоциации соответствует]
bjH%<LiT, г, 0) = 142,274 ±1,5 кДж.моль.
71л(г). Термодинамические свойства газообразного лития-7 в стандартном сострянии в
интервале температур 100—10 000 К приведены в табл. 1099.
Уровни энергии атома 7Li, использованные при расчете термодинамических функций,
приведены в табл. 45.3. Термодинамические функции 7Li(r) вычислены по уравнениям A.3) —A. 6),
A. 9), A. 10) A. 23)—A. 25) непосредственным суммированием по уровням энергии.
Погрешности вычисленных значений при Т ^ 5000 К определяются в основном неточностью фундаменталь-
306
Литий и его соединения 7Li+(r), 7LiH(r), 7LiD(r)
ных постоянных и не превышают 0,02 Дж-К~1-моль в значениях Ф°(Г) и S°(T); при более
высоких температурах погрешности возрастают из-за того, что в расчете не учитывались ридбергов-
ские состояния и при 6000 и 10 000 К могут достигать 0,05 и 0,3 Дж-К-моль~1 в значениях
Ф°(Т) и 0,3 и 1,0 Дж-К^-моль в значениях S°(T).
Таблица термодинамических функций 'Li(r) публикуется впервые.
В справочнике принята энтальпия образования
Ы, г, 0) = 157,74 ±1,2 кДж • моль.
Эта величина была оценена на основании значения энтальпии сублимации природной смеси
изотопов лития, если принять 71л(к, ж) за стандартное состояние.
7Li+(r). Термодинамические свойства газообразного положительного иона лития-7 в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 1100.
Термодинамические функции 7Li+(r) были вычислены по' уравнениям A. 3)—A. 6), A. 9),
A. 10) при условии, что (?вн=1,.так как 7Li+ имеет основное электронное состояние'XS, а все
возбужденные состояния являются ридберговскими и имеют высокие энергии. Погрешность
вычисленных значений термодинамических функций во всем интервале температур обусловлена только
неточностью фундаментальных постоянных и не превышает 0,01—0,02 Дж>К~1-моль.
Таблица термодинамических функций 7Li+(r) публикуется впервые.
Константа равновесия реакции 7Li+(r)+e(r)=7Li(r) вычислена с использованием значения
Дг#о@)=—520,222+0,001 кДж-моль, соответствующего потенциалу ионизации
/0GLi)=43 487,15+0,10 см-х=520,222 +0,001 нДж-моль,
принятому равным потенциалу ионизации атома Li (см. выше).
Принятому значению соответствует
•bjHyhi*, г, 0) = 677,962 + 1,2 кДж • моль.
71ЛН(г). Термодинамические свойства газообразного гидрида лития-7 в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1101.
В табл. 45.4 приведены молекулярные постоянные 7LiH, принятые на основании анализа
результатов исследований электронного спектра и квантовомеханических расчетов. Выбор
постоянных описан в тексте по LiH(r). Расчет термодинамических функций 7LiH(r) проводился так же,
как для LiH(r), на основании значений Ykl, vmia, Jlim и ait вычисленных по изотопическим
соотношениям A. 66) и соответствующим величинам для 7LiD, (см. табл. 45.5). Погрешности
рассчитанных величин такие же, как для LiH(r).
Таблица термодинамических функций 7LiH(r) публикуется впервые.
Константа равновесия реакции 7LiH(r)=7Li(r)+H(r) вычислена с использованием значения
D0GLiH)=19 588,6+1,0 см-^234,332 +0,012 кДж-моль,
принятого по работе Вэй и Стволли [4673]. Авторы [4673] обработали экспериментальные данные
Веласко [4597] по предиссоциации при вращении молекулы 7LiH в состоянии ВШ. на уровне
v—2, /=2. Принятое значение хорошо согласуется с полученными ранее менее точными
величинами 19 591+2 см [4597] и 19 400 см [3494] (см. выше LiH(r)).
Принятой энергии диссоциации соответствует
Д/ГG1ЛН, г, 0) = 139,442 ±1,2 кДж-моль.
7LiD(r). Термодинамические свойства газообразного дейтерида лития-7 в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1102.
В табл. 45.4 приведены молекулярные постоянные 7LiD, принятые на основании^анализа
результатов исследований электронного спектра и квантовомеханических расчетов этой молекулы
(см. выше текст по LiD(r)). Расчет термодинамических функций 7LiD(r) был выполнен так же,
как для LiD(r), на основании значений Ykl, vmax Jllm и а,, приведенных в табл. 45.5. Погрешности
рассчитанных термодинамических функций 7LiD(r) принимаются такими же1 как у LiD(r).
Таблица термодинамических функций 7LiD(r) публикуется впервые.
307
'LiT(r) Глава 45
Константа равновесия реакции 7LiD(r)=7Li(r)+D(r) вычислена на основании значения
D0GLiD)=19 767,6+1,0 см-^236,473+0,012 кДж-моль'1,
принятого по работе Стволли и др. [4423], в которой авторы обработали данные по предиссоциации
при вращении молекулы 7LiD в состоянии В1!! х (ем. выше текст по LiD(r)). Принятая величина
согласуется с менее точным значением, полученным ранее Веласко [4597] и результатом масс-спектро-
метрических измерений [2769].
Принятой энергии диссоциации соответствует
Д_,# °GLiD, г, 0) = 141,074 + 1,2 кДж • моль.
'LiT(r). Термодинамические свойства газообразного тритида лития-7 в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1103.
Экспериментальные данные о молекулярных постоянных 'LiT отсутствуют. Расчет
термодинамических функций 7LiT(r) проводился так же, как для LiD(r), на основании значений Ykl,
vm&%, /Ит и а,-, приведенных в табл. 45.5 и вычисленных по изотопическим соотношениям A.66)
и соответствующим величинам для молекулы 7LiD. Погрешности рассчитанных термодинамических
функций 71ЛТ(г) принимаются такими же, как для LiT(r).
Таблица термодинамических функций 71лТ(г) публикуется впервые.
Константа равновесия реакции 7LiT(r)=7Li(r)+T(r) вычислена с использованием принятого
в справочнике значения
D0GLiT)=19 839,9+2 см~1=237,338±0,024 нДж-моль.
Эта величина была рассчитана по принятым в справочнике значениям D0GLiD) и колебательных
постоянных 7LiD и 7LiT,, (см. табл. 45.4) в предположении, что DeGLiT)=D#GLiD).
Принятой энергии диссоциации соответствует
г, 0) = 141,881 + 1,2 кДж • моль.
Погрешность принятого значения Do GLiD), возможно, больше указанной. Его расчет по величине D0GLiH)
приводит к значению 19 761,7 см~*. Расхождение в б см трудно объяснить отклонением от приближения Борна—
Оппенгеймера.
Глава 46
НАТРИЙ И ЕГО СОЕДИНЕНИЯ
Na(u, ж), Na, Na+, Na2, NaO, NaO2(K, ж), Na2O(K, ж), Na2O, Na2O+, Na2O2(K, ж), Na2O2,
NaH(K, ж), NaH, NaOH(K, ж), NaOH, Na2O2H2, NaF(K, ж), NaF, Na2F2, Na3F3, NaCl(K, ж),
NaCl, Na2Cl2, Na3Cl3, NaBr(K, ж), NaBr, Na2Br2, NaI(K, ж), NaI,Na2I2, Na2SO4(K, ж),
Na2SO4(KlII), Na2SO4, NaNO2(ie, ж), NaNO2, NaNO3(K, ж), NaNO3, Na2CO3(K, ж), NaBO2(K, ж),
NaBO2, NaAlF4, Na3AlF6(K, ж), Na6Al3FI4(K, ж), NaLi
В настоящей главе рассматриваются соединения натрия с кислородом, водородом, галогенами,
а также некоторые соли натрия и кислородсодержащих кислот, фторалюминаты натрия и его
соединение с литием — всего 30 веществ. Для 17 веществ представлены сведения об их свойствах
в конденсированном состоянии (для сульфата натрия, помимо свойств в стандартном состоянии,
рассмотрены свойства метас.табильной модификации кШ). Для 26 веществ даны их свойства
в газообразном состоянии, причем для Na и Na+ до 10 000 К, а для остальных — до 6000 К.
В Приложении 3.10 приведены сведения о критических постоянных элементарного натрия
и NaCl, а также постоянных, позволяющих учитывать особенности свойств Na при
высоких давлениях.
В справочнике не рассматриваются многие соединения натрия, образование которых можно
ожидать в системах, содержащих натрий вместе с другими элементами, по крайней мере в
ограниченном интервале температур. К таким соединениям принадлежат сульфид и нитрид натрия, его
хлоралюминаты, ионы типа Na2F+ и NaF2~ и некоторые другие.
Na(K, ж). Термодинамические свойства кристаллического и жидкого натрия в стандартном
состоянии при температурах 100—2500 К приведены в табл. 1104.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние Na(K) в интервале 0—51 К принята
гексагональная плотноупакованная модификация (структурный тип Mg), а в интервале 51—371,01 К —
кубическая объемно-центрированная модификация.
При Т <^ 298,15 К термодинамические функции вычислены по согласующимся между собой
измерениям теплоемкости образцов натрия (чистоты ~99,9%) в работах Мартина [3402] B0—
300 К), [3404] @,4—1,5 К) и Филби и Мартина [2250] C—30 К), с учетом полиморфного «мартен-
ситного» превращения натрия при 51 К. Расчеты были проведены тремя путями: а) по
экспериментальным данным [2250, 3404] по теплоемкости смеси гексагональной E0%) и кубической E0%)
модификаций натрия и значению энтальпии превращения этого образца в кубическую
модификацию (Tjr=51 К); б) по данным для теплоемкости кубической модификации натрия в интервале
0—51 К [2250, 3402] и в) по данным для гексагональной модификации натрия в интервале 0—51 К,
вычисленным в работе [2250, 3402], и значению энтальпии превращения гексагональной
модификации Na в кубическую. Все расчеты приводят к совпадающим значениям #°B98,15 К) — Н°@)
для стандартного состояния натрия; для энтропии согласующееся значение E1,30 Дж-К~1'Моль)
получено двумя первыми способами, третий привел к менее точному значению E1,48|Дж-К~1Х
Хмель) вследствие использования вычисленных значений теплоемкости Na(reKc) и энтальпии
мартенситного превращения. Данные других измерений теплоемкости Na [4296] A7—118 К),
[3835 ] B—25 К), [1998] E0—320 К) менее точны и не учитывались. Принятые значения ?°B98,15 К)
и Я°B98,15 К)—#°@) совпадают с рекомендованными КОДАТА—МСНС [1887]. Их погрешности
оцениваются в 0,2 Дж-К'^моль и 0,02 кДж-моль соответственно.
В интервале 298,15—371,01 К для теплоемкости Na(K) принято уравнение, выведенное
совместной обработкой результатов измерений энтальпии Na(K), выполненных Джиннингсом и др.
[2408] B73,15—367 К, чистота образца 99,9%) и результатов измерений теплоемкости,
проведенных Мартином [3408] в интервале 300—370 К (чистота образца 99,99%), погрешности этих данных
оценены в 0,3 и 0,2% соответственно.
Температура плавления 371,01+0,03 К и энтальпия плавления 2,598+0,005 кДж-моль,
приняты по прецизионным измерениям Мартина [3408]. С принятым значением Тт
удовлетворительно согласуются данные [2103] C70,97 К), [1259] C70,98 К), [3735] C71,05 К), [2408] C70,95К),
[1643] C70,96 К), [2427, 3735, 3736] C71,05+0,02 К); менее точные измерения [2491, 3978, 4191J
309
Na(K, ж)
Глава 46
Таблица 46.1. Принятые значения термодинамических величин для натрия и его соединений
в кристаллическом и жидком состояниях
о
Вещест!
Na
NaO2
Na2O
Na2O2
NaH
NaOH
NaF
NaCl
NaBr
Nal
Na2SO4
Na2SO4
NaNO2
NaNO3
Na2CO3
NaBO2
Состояние
kI, куб.
ж
kI, куб.
ж
кШ, куб. (y)
kII (В)
Kl (a)
Ж
kII, тетр. C)
К1(а)
Ж
к, куб.
ж
kII, ромб, (а)
к1, монокл. (В)
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
kV, ромб.
kIV, ромб.
к1, гекс.
ж
кШ, ромб.
к1, ромб.
к1, ромб.
кГ, ромб,
ж
kII, гекс. (а)
kII, гекс. (а)
к1, гекс. ф)
ж
1
in
й
к s
6,46
18,3
12,4
15,694
6,259
10,49
8,482
10,60
11,590
12,26
23,204
23,535
16,5
17,23
кШ, монокл.(аJ0,75
kII, монокл. (8) —
к1, гекс. (y) —
ж —
к, гекс.
ж
11,632
)8,15К)
О
¦ 4
)8,15К)
(Та,
]Дш • К-1 •
• МОЛЬ *
i
51,30
115,9
75,04
94,77
40,00
64,43
51,16
72,15
86,93
98,56
149,58
154,92
106
116,4
135,0
73,53
28,23
72,13
69,12
89,33
36,40
59,54
46,82
50,50
51,38
52,09
128,04
129,3
69
93,05
112,3
65,94
Коэффициенты в уравнении
для С°р(Т)
а
—23,346
38,121
62,702
100
66,178
100
100
100
87,626
ИЗ
134
31,398
56
—7,934
86
82,283
60,378
70,1
47,121
67,9
43,845
68
41,968
67
108,829
108,829
208,118
204
10,437
—251,422
4506,004
133
100
—493,991
222,890
140
141
64,118
64,118
36,836
195
45,658
145
Ь- 103
120,736
—19,493
31,623
26,102
38,631
35,340
158,243
1,195
—16,377
7,219
20,291
25,188
110,080
110,080
—131,471
311,175
732,660
—16 616,760
4 577,216
—1 457,760
161,604
161,604
136,800
97,410
с • 10~Б
Интервал
температур
или
Т
К
—13,848 298,15—371,01 371,01
0,688 а 371,01—2300 —
— 298,15—800 800
— 800—2000 —
4,303 298,15—1023 1023
— 1023-1243 1243
— 1243-1405 1405
— 1405-3500 —
8,724 298,15—785 785
— 785—948 948
— 948-2500 —
4,920 298,15—911 911
— 911—1000 —
—18,040 298,15-572 572
— 572-596 596
—10,932 596—3000 —
9,155 а 298,15-1269 1269
— 1269—3400 —
—0,209 а 298,15-1074 1074
— 1074-3400 —
—1,320 298,15-1020 1020
- — 1020—3400 —
—2,322 298,15-934 934
— 934-3400 —
12,098 298,15-458 458
12,098 458—514 514
0а 514—1157 1157
— 1157-3500 —
—23,189 298,15—509 509
—90,653 298,15-400 -
0а 400—436,7 436,7
— 436,7—557 557
— 557—2500 —
0а 298,15—500 .—
0б 500-549 549
— 549-579,6 579,6
— 579,6—2000 —
— 298,15—623 623
— 623—752 752
— 752-4131 1131
— 1131—3000 —
4,651 а 298,15-1239 1239
. — 1239—3000 —
ИЛИ
кДж-
• моль 1
2,598
20,5
1,3
9
36
5,36
17
26
5,85
7,8
34,25
28,2
26,23
23,68
0,3 '
10,9
23,5
—
3,2
14,94
0
15,05
0,8
2,1
28
33,5
310
Натрий и его соединения
Na(K, ж)
Таблица 46.1. (окончание)
Вещество
Na3AlFe
NajAlgFj^
Состояние
1
oo'S
О 1
t!
kII, монокл.(а) 38,07
Kl, куб. (8) —
ж —
, к, тетр. 80,83
Примечание. C°v(T)=a+b -T—c ¦
Na: '
NaF: <
NaCl:
Na2SO4:
оо"
to
ю
оо"
Дж • К"' •
• МОЛЬ
238,2
—
—
515,2
—
T~2+d
1 B=10,240 -Ю-*. NaNO2:
1 <2=18,267 -10-*. I
1 B=11,156-lO. ]
» B=132,353 -Ю"».
SfaNO3:
4aBO2:
215,90
—
—
454,00
—
•Я+е-
а с2=С
Коэффициенты в уравнении
для С°р(Т)
а
Ь-103
с • Ю-5
192,516 122,980 11,807
218,196 66,358 —
391 — —
507,907 92,630 72,470
974 — —
Т3 (в Дж -К -моль).
, е=34984,382 -10"».
а d=—11964,863-Ю-*, «=10788,787-Ю-8;
a d=— 39,691 -10-».
Интервал
температур
или
Т
1 m
К
298,15—838
838-1286
1286-4000
298,15—1010
1010-3000
6 d=2631,960
838
1286
—
1010
—
AirH
или
кДж-
• моль *
9,15
114,4
—
229
—
не учитывались. В работе Джиннингса и др. [2408] получено значение Д„Д°=2,602 +0,008кДжХ
Хмоль, хорошо согласующееся с принятым. Результаты определений этой величины в работах
11670, 2491, 3281, 3358, 3978, 4197] менее точны и не принимались во внимание.
Уравнение для теплоемкости жидкого натрия выведено совместной обработкой результатов
измерений энтальпии (или теплоемкости) Ка(ж) в шести работах; погрешности данных Джиннингса
и др. [2408] D23—1170 К), Фредриксона и Часанова [2324] E54—1505 К) и Мартина [3408J
{380—470 К) оценены в 0,3%, данных Шпильрайна и др. [1174] E73—1273 К) и Новикова и др.
[729] C84—1359) — в 2%, а данных Пчелкина [844] D73—1573 К) — в 5%К Согласно
приведенному в табл. 46.1 уравнению теплоемкость Na^) уменьшается от С C71 К)=31,8 Дж-К~1-моль~1
до С° (950 К)==28,8 Дж-К^^моль (минимум теплоемкости), а затем ускоренно возрастает,
достигая при 2000 К 40 Дне «К-моль. Точность описания измерения энтальпии для жидкого
натрия можно оценить в ~0,5% при 1000 К, ~1% при 1500 К и ~2% при 2000 К.
Погрешности табулированных значений Ф° (Т) при 298,15, 1000 и 2000 К оцениваются в 0,15»
Ю,4 и 0,8 Дж-К^-моль.
Значения Ф°(Т), приведенные во втором издании настоящего справочника [337] (до 2300 К),
таблицах JANAF [2834] (до 2000 К), книге Шпильрайна и др. [1179] (до 1800 К) и справочнике
Халтгрена и др. [2752] (до 1700 К) согласуются с данными табл. 1104 в пределах 0,4, 0,2 и
0,3 Дж«К.моль соответственно. Расхождения обусловлены различием^ выборе энтальпии
Na(K) при 298,15 К и тем, что в настоящем издании принято более быстрое возрастание C°(Na, ж)
при Т > 1000 К. .
В настоящем справочнике в качестве стандартного состояния натрия принято кристаллическое
•состояние и в соответствии с этим
Ь,Н0(Ил, к) = 0.
Давление пара натрия в реакции Na(K, ж)=Ыа(г) вычислено с использованием значения
Ari/°@)=107,763+0,7 кДж-моль, соответствующего принятой в справочнике энтальпии
сублимации
A,#°(Na, к, 298,15 К) = 107,5 + 0,7 кДж • моль.
* Данные [3408] и [844] по теплоемкости Ш(ж) были проинтегрированы и использовались в виде значений
энтальпий Н° (Т)—Н° (Тт) в указанных интервалах температур A0 значений для [3408],и 12 значений для[844]).
311
Na(K, ж)
Глава 46
Таблица 46.2. Результаты определения A,2T°(Na, к, 298,15 К) (в кДжмоль)
Авторы
Метод
As#°(Na, к, 298,15 К)!
II закон
III закон
Руфф, Йохансен, 1905 [4091]
Хейкок, Ламплуг, 1912 [2635]
Ладенбург, Минковский, 1921
[3158]
Хабер, Циш, 1922 [2538]
Точек кипения, 1150,65 К, 1 атм, 1 точка
То же, 1156 К, 1 атм, 1 точка
Магнито-оптический, 509—695 К, 1 -10~в—
0,9-10 атм, 21 точка
Протока, 746—838 К, 3 -Ю—2 -10 атм,
4 точки
Эффузионный, 455—534К, 6-Ю-8—3-Ю-6 атм, 107,2±2,7 108,40+0,15
5 точек
Статический, 787—870 К, 7 -Ю—3 -Ю атм, 109,1 ±2,6 107,37 ±0,15
5 точек
Эдмондсон, Эджертон, 1927 [2144] Эффузионный, 496—571 К, 6-10-'—1-Ю атм, 107,7±4,2 108,48+0,16
4 точки
Магнито-оптический, 519,5—590,5 К, 1-Ю^6— 109,8±3,8 108,12±0,14
4 -10~6 атм, 10 точек
Статический, 924—1118 К, 6 -Ю—6 -10 атм, 109,2 ±2,9
11 точек
То же, 536—670 К, 4 -10-0—4 -10"* атм, 28точек 108,2±1,0
Родбуш, Деврие, 1925 [4023]
Вейлер, 1929 [4682]
Родбуш, Вальтере, 1930 [4025]
— 107,1
— 107,5
108,2 ±2,3 108,17 ±0,17
108,31 ±0,86 108,62 ±0,01
108,48 ±0,16
Родбуш, Генри, 1930 [4024]
Ладенбург, Тиль, 1930 [3159]
Льюис, 1931 [3248]
Тиль, 1932 [4505]
108,38 ±0,06
Протока, 615—771 К, 8-Ю—5-Ю"8 атм, 108,0±1,1 108,11+0,08
6 точек
Эффузионный, 630—763 К, 1-10-4-4-Ю-3 атм, 108,46±0,21 107,97 ±0,02
12 точек
Протока, 649-701 К, 2-Ю"*—9-Ю"* атм, 109,24±0,58 107,99±0,06
16 точек
Кистяковский, 1941 [3021]
Макензи и др., 1955 [3357]
Кириллов, Грачев, 1959 [512]
Точек кипе/ния, 1155,15 К, 1 атм, 1 точка — 107,4
То же, 894—1408 К, 5 -Ю—6,5 атм, 55 точек 108,93 ±0,95 107,2±0,1 б
Статический, 1173—1573 К, 1,2-14,9 атм, 107,0±2,1 108,07 ±0,17 б
9 точек
Атомно-адсорбционный, 393—408 К, 9 -Ю-10— 111 ±12 108,11 ±0,14
3 -Ю-9 атм, 35 точек
Шпильрайн, Зверева, 1963 [1164] Статический, 921—1136 К, 6-Ю—8-Ю атм, 110,2±1,1 107,27±0,10 б
16 точек
Видаль, 1960 [4612]
Сова, 1963 [4351]
Бук, Паули, 1965 [1709]
Точек кипения,Ш73—1663 К, 1,16—24,8 атм, 110,3±1,9 107,60+0,19"
16 точек '¦
106+11 107,81 ±0,71
Ионизационный, 400—453 К, 1 -Ю-*—
1 • 10~' атм, 11 точек
Статический, 1072—2154 К, 0,5—120,0 атм, 108,97±0,91 107,29±0,11 б
49 точек
То же, 1157—1533, 1,02—12,96 атм, 59 точек 107,6±0,2 107,68±0,07 б
Тоже, 1196—1666 К, 1,42—23,82атм, 27точек 108,95±0,49 107,68+0,07 б
То же, 864—1160 К, 2 -Ю—1,0 атм, 18 точек 108,02±0,66 107,63 ±0,15 б
Точек кипения, 883—1229 К, 4-Ю—1,94 атм, 110,5+1,1 Ю7,85±0,15б
33 точки
Статический, 795—1080 К, данные представ- 108,72±0,26 107,66±0,11
лены в виде уравнения
Точек кипения, 1116—1390 К, 0,7—5,8 атм, 108,4±3,1 107,47 ±0,37 б
4 точки
Герасимов и др., 1969 [273, 2386] Эффузионный, 339—538 К, 3-Ю2—5-Ю атм, 110,45±0,94 108,49+0,13
49 точек
Мурадова, Фишман, 1970 [704] Атомно-адсорбционный, 391—439 К, 6-Ю0— 103,9±5,1 108,83+0,18
2 -Ю-8 атм, 23 точки
Шине и др., 1971 [4184] Точек кипения, 1116—1403 К, 0,67—5,8 атм, 105,5±1,3 107,36+0,11 б
42 точки
Байс, Бонилла, 1977 [1551] Статический, 1255—2500 К, 2,3—251,0 атм, 109,1 ±1,4 107,73+0,08б
94 точки
а Приведенные величины рассчитаны с учетом образования в насыщенном паре дииеррых молекул, б Введены
поправки на отклонение от идеальности. Учтены только измерения, выполненные npi Т < 17С0 К.
Боулс, Розенблюм, 1965 [1639,
1640]
Стоун и др., 1965 [4400]
Стоун и др., 1966 [4402]
Виноградов, Воляк, 1966 [217]
Аченер, Ютас, 1966 [1214]
Фишер, 1966 [2259]
Боданский, Шине, 1967 [1604]
Натрий и его соединения
Na(r>
Принятая величина основана на представленных в табл. 46.2 результатах измерений давления
насыщенного пара натрия. Приведенные в таблице погрешности отражают лишь
воспроизводимость измерений. Расчет энтальпии сублимации проводился для модели идеальной смеси
химически реагирующих атомарной и молекулярной компонент. При высоких температурах учитывалась
поправка на неидеальность в модели диссоциирующих идеальных газов по варианту метода смеси t
разработанному Фокиным [1060]. Такие расчеты могли быть проведены для измерений при
температурах до 1700 К. Метод расчета изложен в Приложении 8.
В результате обработки данных, приведенных в табл. 46.2, для большинства работ получены
хорошо согласующиеся значения энтальпий сублимации. Рекомендованное значение вычислено как
средневзвешенное из наиболее надежных данных [2635, 3021, 4184, 1604], которые были получены
методом точек кипения, и данных A164, 4023, 4400, 4402], где использовались различные варианты
статического метода (всего 176 точек в интервале 787—1666 К). Погрешность принятой величины
обусловлена в основном погрешностями термодинамических функций натрия в конденсированном
состоянии, которые приводят к погрешностям в энтальпии сублимации от 0,2 кДж-моль для
измерений при 700 К до 1,2 кДж-моль для измерений при 1700 К. Принятое значение
энтальпии сублимации натрия совпадает с рекомендацией КО ДАТА—МСНС [1887]. Ряд спектральных
работ [1917, 1918, 2214, 2780, 2846], в которых определялись величины, пропорциональные
давлению пара, при выборе энтальпии сублимации не учитывались. Менее точные измерения давления
пара были приведены в работах [1108, 2383, 2541, 2542, 2563, 2996, 3735].
Na(r). Термодинамические свойства газообразного натрия в стандартном состоянии в
интервале температур 100—10 000 К приведены в табл. 1105.
"Уровни энергии атома Na, использованные при расчете термодинамических функций,
приведены в табл. 46.3, в которой даны валентные состояния Na, соответствующие электронным
конфигурациям . . .3s—3d. Приведенные в этой таблице
значения основаны на результатах измерений
Рисберга [4006], которые практически
совпадают с рекомендацией Мур [3550]. При этом для
состояния 2D принято значение, усредненное
для двух подсостояний.
Термодинамические функции Na(r)
вычислены по уравнениям A. 3)—A.6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммировани-
Таблица 46.3.
Уровни энергии атомов Na и Na+,
принятые для расчета
термодинамических функций
Атом
Na
Na+
Электронная
конфигурация
Is22pe3s
Is22p63p
№2рШ
Ь22р«
Состояние
2S
¦>P>ll
*D
T<
CM"
0
16 956,172
16 973,368
29 172,86
0
Pi
2
2
4
10
ем по уровням энергии. Погрешности
вычисленных значений термодинамических функций при
Т <[ 5000 К определяются только неточностью
фундаментальных постоянных и не превышают
0,02 Дж-К^-моль в значениях Ф°(Т) и S°(T).
При более высоких температурах погрешности
возрастают из-за пренебрежения ридберговски-
ми состояниями и при Г=6000 и 10 000 К могут достигать соответственно 0,05 и 0,5 Дж- К~1-моль~1
в значениях S°{Т).
Ранее таблицы термодинамических функций Na(r) вычислялись в разных работах, в том числе
в предыдущих изданиях настоящего справочника [337, 340], в таблицах JANAF [2834] и Мак-
Брайд и др. [3439] до 6000 К, а также в работах Гурвича и др. [335], Хилзенрата и др. [2652] и
Рейтера [3973] до 10 000 К, Кольского и др. [3067] до 8000 К и Эванса и др. [2195] до 2500 К.
Расхождения результатов этих расчетов при Т < 4000 К и данных настоящего справочника, как
правило, не превышают 0,01—0,02 Дж-К^-моль" в значениях Ф°(Т). Расхождения с данными
[3067, 3973] достигают 0,06 Дж-К^-моль, по-видимому, вследствие ошибок в этих расчетах.
При Т > 4000 К термодинамические функции Na(r), вычисленные в других работах, возрастают
с температурой быстрее, чем в настоящем справочнике. Максимальные расхождения имеют место'
с расчетами Гурвича и др. [335] и Рейтера [3973], "
в Ф°A0 000 К) (см. Li(r».
Энтальпия образования
A,#°(Na, г, 0) = 107,763 + 0,70 кДж • моль
соответствует принятой энтальпии сублимации натрия.
где они достигают 7—8 Дж-К 1-моль
313
Na+(r), Na2(r) Глава 46
Na+(r). Термодинамические свойства газообразного положительного иона натрия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 1106.
В табл. 46.3 приведено только основное состояние Na+, относящееся к типу 1>S'; возбужденные
состояния Na+ являются ридберговскими.
Термодинамические функции Na+(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10) при условии, что In $вн=0. Погрешности вычисленных значений термодинамических
функций во всем интервале температур обусловлены только неточностью фундаментальных
постоянных и не превышают 0,01—0,02 Дж-К^-моль.
Ранее термодинамические функции Na+(r) вычислялись в предыдущих изданиях настоящего
^справочника [337, 340], в таблицах JANAF [2834] до 6000 К, а также работах Хилзенрата и др.
[2652] до 10 000 К и Грина и др. [2473] до 50 000 К. Результаты всех расчетов совпадают с
данными табл. 1106 в пределах 0,01 Дж-К-моль~1 в значениях Ф°(Г) и S°(T).
Константа равновесия реакции Na+(r)-j-e(r)=Na(r) вычислена с использованием значения
Arff°@)=^-495,845 +0,001 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Na)=41 449,44+0,10 см-*=495,845 +0,001 кДж-моль.
Это значение рекомендовано Мур [3549] с погрешностью 0,03 см на основании результатов
измерений ридберговских серий в спектре Na, выполненных Рисбергом [4006]. С учетом
результатов других работ (см. [3550]) погрешность принятой в справочнике величины увеличина.
Принятому значению /0(Na) соответствует
AfH°(Na.+, г, 0) = 603,608 ± 0,70 кДж • моль.
Na2(r). Термодинамические свойства газообразного динатрия в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 1107.
В табл. 46.4 приведены молекулярные постоянные Na2, принятые на основании результатов
спектральных исследований и квантовомеханических расчетов. В электронном спектре Na2
наблюдались пять систем полос: А1^ ** Х1^ [1761, 2326, 2327, 2328, 2916, 3149,' 4566, 4761, 4762,
4763, 4764], В1Пи*ъХ1Ъ+г [2039, 2040, 2328, 3151, 3293, 3294, 3300, 3301, 4324, 4616, 4761,
4762, 4763, 4764], #41.4* Х1^ [1407, 3007, 3555, 4298, 4299, 4302, 4646, 4688, 4764], ЯП.**
^ХгЦ [1407, 3007, 3555,4298,4646, 4764], G4I, ** Х1!* [3555] и •Ejr*b»?+ [5064]. Согласно
теоретическим расчетам [1392, 4012,4998] молекула Na2 помимо экспериментально наблюдаемых
имеет также стабильные состояния а3Пи, C1Hff и D1^, диссоциирующие на атомы Na в
состояниях 2S-\-2P x. Молекулярные состояния, коррелирующие с более высокими атомными уровнями
(в том числе и экспериментально изученные /г^П,,, F1^ и €Р-Пш), имеют энергии возбуждения
30 000 см и выше и в настоящем справочнике не рассматриваются.
Молекулярные постоянные состояний ХЧ1* и ^П,, приняты по результатам анализа системы
В1Пи -> X1!,* в спектре лазерной флуоресценции, выполненного Кушем и Хесселом [3151]. Из
трех наборов постоянных, полученных авторами [3151], выбран второй, соответствующий
обработке спектральных переходов между всеми наблюдаемыми колебательными уровнями (v' ^ 45
v" ^ 29) и невысокими вращательными уровнями. Постоянные состояния А1!,* получены
Каминским [2917] из анализа системы А1!,* -> Х1!^ (у' <[ 44) в спектре лазерной флуоресценции.
Поскольку интеркомбинационные переходы в спектре Na2 не наблюдались, энергия состояния
?3ПК принята на основании сопоставления результатов квантовомеханических расчетов Роача
[4012], Бардслея и др. [1392], Коновалова и др. [4998] и приближенного анализа возмущений
в системе Л.12* -* Х1!^, проведенного Кэрролом [1761]. Постоянные ш, и В, в состоянии а3П„,
а также постоянные Г„ (о# и Ве в состояниях &3??, СШд и D1!,*, приведенные в табл. 46.4,
приняты по работам [1392, 4012, 4998]. Погрешности принятых значений Те могут достигать 1000 см,
значений ю, и Ве — 10—15%.
Термодинамические функции Na2(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн и ее производные ^вычислялись по соотношениям A.90)—
JS
Поскольку квантовомеханический расчет [4998] показал, что ван-дер-ваальсово состояние 3SJ- имеет очень
плоский минимум (D,= 180 см, г„— 5,206 А), оно не учитывается при расчете термодинамических функций.
314
Натрий в его соединения
Na2(r)
Таблиц
Молекула
Naa
NaO
NaH
NaF
Na^Cl
Na81Br
Nal
Na'Li
a 48.4.
Состояние
а3Пи
A^i
ЪЗЧ
cm,
D14
xm
A2 S+
Xl2+
Л12+
а3П
x1^
X!2+
X1 2+
X1^
о3П
632+
541
?12+
Иолекулярные постоянные Na2,
0
14 000
14 680,581
16 800
20 319,166
21500
21 500
0a
1800
0
22 719,1
31800
0
0
0
0
0
14 700 б
14 700 б
20 000 б
20 226,01 г
20 400 б
ше
шехе
159,109 39 0,721419
160
0,714
117,323 22 0,357 583
150
124,442
60
80
504
519
1172,2
310,6
41ft
563,10
364,60
298,49
259,20
256,8
—
—
—
0,912
76 0,759 934
0,613
" 1,09
5,0
3,83
19,72 а
—5,41 г
50
3,83
1,755
1,16
0,964
1,612 а
—
—
.
—
NaO, NaH, NaF,
Be
CM
62 a 0,154 740 08
0,150
46 r 0,110 784 07
0,125
91 ж 0,125 886 89
0,071
0,080
0,425
0,470
4,898
1,728
3,53
0,436 901
0,218 063
0,151 253
0,117 806
0,376 б
—
—
0,307 г
Na85Cl, Na81Br,
a, • 10'
0,868 268 26 б
i.o
0,548 836 74 Д
1,1
0,935 927 27 3
0,98
1,3
4,2
5,6
132 б
—78,3 Д
850
4,557
1,625
0,941
0,6478
3,0 б
—
_
Nal, Na'Li
De ¦ 10е
0,589 72142
0,53
0,388 160 28
0,35
0,518 669 61
0,40
0,32
1,2 6
1,5 6
340 B
235 e
800 ж
1,161
0,312
0,1553
0,097 34
3,2 B
__
_
re
A
B 3,078 5
3,13
e 3,638 37
3,42
и 3,413 15
4,54
4,29
2,05
1,95
1,888
3,179
2,22
1,926 03
2,360 90
2,502 01
2,711 43
2,888 б
_
.
3,196 г
Примечание. Ниже все постоянные даны в см.
№ь,: а шеуе=-0Л53 543 61 -Ю, meze=0,23u 758 18 -Ю"*, ыЛ,=-0,330 534 55 -Ю, а>А=—0,127 203 83 -Ю"8;
(„^=—0,457 455 48 -Ю-10; б а2=—0,428 553 22 -10, а3=0,147 844 88 -Ю, а4=0,198 892 57 -ЮЛ
а5=0,817 444 20 -Ю0, а„=0,122 515 48 -Ю1, а,=0,155 754 06 -Ю3; в §,=—0,390 680 31 -Ю"8,
p0911 879 28 Ю10 C3=0,246 104 38 -Ю'11, В4=0,463 816 55 -Ю3, %=—0,164 220 15 -10-",
&!==—0,360 550 56 -Ю-13, А2=0,342 212 15 -Ю5, й3=0,115 329 57 -Ю5,
S1=—0,102 747 22-Ю-17, ЛГЙ=О,562 317 76 -101; г а),уе=5,167 111 3 -Ю"8,
и>ед,=—1,455 958 3 -Ю; Д а2=1,625 161 1 -Ю"8, а3=—3,164 514 3 -Ю"8,
е Я,=1,129 249 8 -Ю2; ж швг/в=0,401 891 61 -Ю, coeze=0,419 756 70 -Ю,
0463 645 93 Ю6 0806 101 60 Ю8 0998 511 98 Ю10
5, ,
pg=0,911 879 28 -Ю-10,
#„=0,280 165 53 -Ю-11,
Le=0,443 441 81-10-",
@^=—9,276 970 7 -Ю-6,
<х4=—9,204 607 6 -Ю0;
0157 363 33 Ю4
4,, ; вг/в, , ee,
<oeg,=0,157 363 33 -Ю, (oeS(,=0,463 645 93 -Ю^6, м?,ге=0,806 101 60 -Ю"8, <oeit,=0,998 511 98 -Ю0;
з а2=— 0,532 871 50 -Ю, а3=0,371 658 54 -Ю, а4=—0,197 691 36 -Ю, а5=—0,300 052 23 -Ю"8,
а6=— 0,177 280 42 -Ю-9, а,=—0,460 286 77 -Ю1, as=—0,501 018 54 -Ю3; и ^=—0,379 955 26 -Ю"8,
|32=0,422 230 71 -Ю"9, |33=0,301 289 91 -Ю0, р4=0,221 71 743 -Ю1, S6=0,977 153 27 -Ю3,
36=0,241811 33 -Ю-14, #„=0,198 620 77 -Ю1, /гг=—0,108 62019 -Ю3, Л2==0,209 072 57 -Ю4, Л3=0,271 651 73 -10-",
Л4=—0,294 370 61 -Ю6, ?,,=0,329 035 23 Ю6, 81=—0,229 148 77 -Ю7, 1Ге=0,343 813 41 -Ю1.
NaO: a А=100, оценка; & вычислено по соотношению A. 68).
NaH: a u>eye=ifi Л0~\ o)eze=5 -Ю-3; б а2=2,2-Ю; в 8,=3 -
<o,ge=1 ,Ъ -10-5; Д а2=-9,3э Л0-3, а3=-3,<38 ЛО, а4=-6,
ж приведено значение Do.
NaF: а <х2=2,281 58 -Ю; 6 ^=7,005 -Ю-10, #,=—4,105 -Ю3.
NaCl: а <х3=5,136 89-10-'; б ^=8,339-Ю0, #,=—3,402-Ю4.
NaBr: a а2=2,432 35 -Ю"»; б ^=4,97 -Ю0.
Nal: a os=l,430 99 -Ю"8; б ^=4,6699 -Ю"".
NaLi: а ш,уе=—7,5 -10~3; б оцэяка; в ватаелэно по соотношению A. 68);г приведено значение для
неизвестного уровня и'.
г
¦10-«;
">Л=-1,97 -10-1, »А=7,3 -Ю-*,
6 ^=4,1 -10-е, ^2=-4 -10-', ^,=-2 -Ю-8;
315
Na2(r)
Глава 46
A. 92) с учетом шести возбужденных электронных состояний в предположении, что для состояний
с Те > 20 000 см (?кол.вр.=Р<(?кол.вр- Колебательно-вращательная статистическая сумма по
состояниям X1!,*, а3Пи, А1^, Ь3Е* и ее производные находились непосредственным суммированием
по колебательным и вращательным уровням. В расчете учтены все уровни этих состояний,
ограниченные предельной кривой диссоциации для состояния X1¦?*, а для состояний а3Ии, А1!,*
и Ь3Щ — уровни имеющие значения J ^ JmSlXtV (последнее находилось по формуле A.82)).
Уровни энергии вычислялись по уравнениям A. 65), A. 63), A. 64а) с коэффициентами Ykl,
приведенными вместе со значениями ушах, /1Im и коэффициентами а( в табл. 46.5. Для обеспечения
Таблица 46.5а.
Значения коэффициентов в уравнениях, описывающих уровни энергии (в ем),
а также значения г;шах и i/lim, принятые для расчета термодинамических функций
Na2 и NaO
Коэффициенты
Na2
NaO
Те -10-*
Ущ-Ю-2
Но
У31-10'
У41-10«
У.! -Ю12
У71-1014
У°2-10»
Угг'
у«-
у62 -io«
У0з-10^
У13-10«
У23-10«
Узя -10"
(ao=De) -Ю-з
о2 -10е
а4 -10»
' О
1,591 094
—7,214 196
—1,535 436
—2,357 582
—3,305 346
0,127 204
-0,457 456
1,547 400
-0,868 268
—4,285 531
—1,478 449
1,988 926
-8,174 442
1,225 155
—1,557 540
—5,897 214
—3,906 803
—9,118 793
2,461 044
—4,638 165
—1,642 202
2,801 655
3,605 505
3,422 122
—1,153 296
—1,339 417
5,988 941
1,356 460
—2,227 466
1,445 205
60
275
1,40
1,60
-7,140
1,50
-1,0
—5,30
0,832 371
8,963 587
0,446 877
-0,519 700
0,255 834
111
331
1,468 058 1,680
1,173 293 1,50
-3,597 909 —9,12
0,315 214 —
-1,205 689 —
6,376142 —
-1,535 672 —
1,464 310 —
-4,927 853 —
1,107 841 1,25
-0,548 837 —1,1
0,016 252 —
0,316 451 —
-0,920 461 —
0,348 584 —
—3,881 603 —3,5
1,129 250 -0,364 01
3,401410
114
383
81
296
О
5,008 508
—38,986 06
10,000 92
0,18
5,139 927
—20,440 49
1,514 42
4,25
-4,2
4,957 26
4,70
-5,6
16,720 96
—12,0
-15,0
—9,485 725
0,579 205
115
314
150
419
Na2: a Состояние
ВЩЯ
Те, см-1 20 319,166
21500
СШ5
21500
NaO: a А=100 смт
316
Натрий и его соединения
Na2(r)
Таблица 46.56. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения fmax и Jiim, принятые для расчета термодинамических функций
NaH, NaF, NaCl, NaBr, Nal и NaLi
Коэффициенты
те -ю-4
У10 -10
Уао-Ю-1
У40 -102
у50 -103
у60 -105
у01
Уц -Ю1
у21 -ю»
У31-Ю*
У51-108
у02 -10*
У12Юв
У22-Ю'
У32 -108
¦* 03 и
(ао=Т>е)
а2 -103 "
«з-Ю6
а4 -108
JUm
NaF: a
NaCl: a
NaBr: a
Nal: a
NaLi: a
Х1^
0
1,172 925
-2,032 465
3,701 124
—3,961 830
2,664 850
—7,631 467
4,898
—1,32
0,22
—
.—
—
-3,40
3,0
—
—
142,071
—
•10-* 1,574143
0,259 194
-0,005 145
0,000 452
20
78
NaH
А1^
. 2,271 910
0,310 560
0,543 976
—2,055 281
0,027 976
0,036 362
—0,061 562
1,728
0,783
—9,35
3,98
-6,80
1,767 059
—2,35
4,10
4,0
-2,0
369,419
—
0,999 532
0,011 173
—0,000 090
0,000 003
36
126
коэффициенты потенциальной
коэффициенты потенциальной
коэффициенты потенциальной
коэффициенты потенциальной
Состояние А12+ а3П
Те, см-1 14
700 14 700
а"п
3,180
0,419
—4,025
—
—
—
—
3,533
—8,530
—
—
—
—
-8,0
—
—
—
-7318,394
—
0,097
9,679
—2,265
1,654
3
27
функции
функции
функции
функции
б3 2+
20 000
NaF»
Х*2+
0
390 0,53610
—0,383
—
—
—
—
0,436 901
—0,045 571
0,022 8
—
—
—
—0,011 607
0,000 700
—
—
—0,004 105
—3,74
480 —
199 —
222 —
735 —
222
571
2О=164 456 см-1,
io=152 402 см,
20=147 259 см,
io=142 575 см,
NaCl1
0
0,363 61
-0,174 5
—
—
—
—
0,216 882
NaBra
Х1!*
0
0,298 07
-0,115 7
—
—-
—
—
0,150 832
-0,016116 —0,009 370
0,005 08
—
—
—
—0,003 087
0,000 82
—
—
—0,000 335
—
—
—
— -
—
196
686
d1=3,133 13,
dx=3,076 33,
^=3,04612,
d1=3,016 40,
ВЧ1 С12+
20 226,01 20 400
0,002 42
—
—
—
-0,001 545
0,000 49
—
—
—
—
—
—
183
769
d2=6,426.
da=6,466.
da=6,485.
Aj=5,918.
Nala
0
0,259 20
—0,096 4
—
—
—
0,117 806
—0,006 478
0,001 43
—
—
—
—0,000 973
0,000 047
—
—
164
756
NaLia
X1^
0
0,258 927
-0,199 712
0,291 535
-0,072 374
—
0,379 113
—0,030 373
—0,032 532
—
0,166 242
43
193
условий A. 59) для состояния X1?,* было изменено значение У04| а для обеспечения условий
A. 59) и A. 50) для состояния А1!,* добавлены коэффициенты У04, У60 и У~70. Мультиплетность
и вырождение состояний а3Пм и Ь3И* учитывались статистическими весами 6 и 3 соответственно.
Погрешности рассчитанных термодинамических функций Na2(r) при температурах до 1000 К
обусловлены главным образом неточностью фундаментальных пострянных. При более высоких
температурах основной вклад в погрешность вносит неопределенность энергий возбуждения три-
плетных состояний. Общие погрешности в значениях Ф°(Т) оцениваются в 0,02, 0,05 и 0,4 Дж-К~хХ
Xмоль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции Na2(r) ранее вычислялись в таблицах JANAF [2834],
справочнике Мак-Брайд и др. [3439], а также в работах Фебера и Херрика [2228], Эванса и др. [2195]
(Т < 1500 К), Артыма [46] и Рейтера [3973] (Т < 10 000 К) и Шнейдера [4197а] (<?вн при Т <
^ 2400 К). В расчетах [2834] и [3439] применение приближенных методик и пренебрежение
возбужденными электронными состояниями привели к частичной компенсации ошибок,
расхождения этих данных с табл. 1107 не превышают 4 и 8 Дж-К-^моль в значениях Ф°(Т) и S°(T)
317
NaO(r) Глава Ш
соответственно. Результаты расчета Артыма [46] согласуются с табл. 1107 в пределах 0,4 и 1 ДжХ
хК-моль в значениях Ф°(Т) и S°(T) соответственно. Несколько больше расхождения (до
2 Дж-К^-моль в значениях 5°) с данными Рейтера [3973] из-за использования разных методик
ограничения (?вр. Фебер и Херрик [2228] не учитывали триплетные состояния Na2, и расхождения
с данными табл. 1107 доетигают| 2 и 10 Дж-К^-моль в значениях Ф°F000 К) и ?°F000 К)
соответственно.
Константа равновесия реакции Na2(r)=2Na(r) вычислена с использованием значения
"""" |ДгЯ°@) = D0(Net) = 5908 ± 20 см« 70,60 + 0,25 кДж . моль.
Это значение получено Кушем и Хесселом [3151] короткой (~560 см) экстраполяцией
колебательных уровней основного состояния X1 2* молекулы Na2 по уравнению 7-й степени. Принято©
значение согласуется с менее точными спектральными данными Демтредера и Стока [2040] E820 +
+30 gm), Лумиса и Нусбаума [3300] F154+200 см), а также с результатами
термодинамических исследований равновесия реакции Na2(r)=2Na(r), полученных Льюисом [3248] E887 +
+80 см'1) и Воляком [236] E912+30 см).
Принятой энергии диссоциации соответствует
Na,, г, 0) = 144,856±1,4 кДж-моль.
NaO(r). Термодинамические свойства газообразного оксида натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1108.
Экспериментальные данные о молекулярных постоянных NaO отсутствуют; значения,
приведенные в табл. 46.4, приняты на основании результатов квантовомеханических расчетов.
Неэмпирические расчеты в приближении Хартри—Фока [3702, 4339] показали, что молекула NaO имеет
основное состояние Х2П, (электронная конфигурация . . .а2л3) и одно стабильное низколежащее
состояние А2И + (конфигурация . . .ал4). Принятая симметрия основного электронного состояния
качественно согласуется с экспериментами по отклонению молекулярного пучка NaO в
электрическом и магнитном полях в работе [2616].
Молекулярные постоянные NaO в состояниях Х2Пф. и Л2Е+ приняты по результатам
неэмпирического расчета [3702], проведенного с расширенным базисом. Погрешности в значениях Тег
(в, и Ве можно оценить в 300, 50 и 0,04 см соответственно. Значение шехе основного состояния
изменено по сравнению с данными [3702] на основании сопоставления с результатами расчета
молекулы LiO [4803]. Постоянная спин-орбитального взаимодействия для состояния X4li оценена
по данным для молекулы LiO с погрешностью 50 см.
Термодинамические функции NaO(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Qm и ее производные вычислялись по соотношениям A.90)—
A. 92) с учетом состояний X2IIf и А*Ъ+. При этом компонента 4L/t рассматривалась как
возбужденное состояние с энергией Т,=А и QVOi вр BП1/;)=()КОЛ вр (Х21Ц). Статистическая сумма по
колебательно-вращательным уровням состояний Х2Пз/г и А2И + и ее производные находились
непосредственным суммированием но колебательным уровням и интегрированием по / с помощью
соотношений типа A. 81). В расчете учитывались уровни со значениями / ^ JmaXtt, последние
находились по формуле A. 82). Уровни энергии вычислялись по уравнениям A. 65) и A. 64а)
с коэффициентами Ykl, приведенными вместе со значениями i>max и /lim в табл. 46.5. Коэффициенты
Y30 и У03 получены из условий A.50) и A.59). Вырождение и мультиплетность состояний Х2Пз„,
21Ь/г и IaS+ учитывались статистическим весом 2.
Погрешности рассчитанных термодинамических функций NaO(r) обусловлены главным
образом неточностью молекулярных постоянных основного электронного состояния Х2П,- и энергии
возбуждения состояния A2Z+. Общие погрешности в значениях Ф°(Т) могут составить 0,5, 1
и 2 Дж-К'моль~1 при 298,15, 3000 и 6000 К соответственно. Погрешность в значении #сB98,15)—
—Н°@) может достигать 0,6 кДж-моль.
Термодинамические функции NaO(r) ранее вычислялись в таблицах JANAF [2834],
справочнике Мак-Брайд и др. [3439], в работах Брюэра и Розенблатта [1666] (Ф°(Т) при Т < 3000 К)
и О'Харе и Уола [3702] (Т ^ 1500 К). Расхождения с данными [2834] не превышают 2 и 4 ДжХ
хК~1-моль в значениях Ф°(Т) и S°(T) и объясняются главным образом тем, что авторы [28341
пренебрегли состоянием А2Л+, не учли расщепление состояния Х2П,- и приняли другое значение
ше. Существенно большие расхождения с данными [1666, 3439,, 3702] (до 10—12 Дж-К^-моль
318
Натрий и его соединения NaO2(K, ж)
в значениях Ф°(Г)) объясняются ошибкой в выборе типа основного состояния ([3439]) и иной
оценкой молекулярных постоянных ([1666, 3702]).
Константа равновесия реакции NaO(r)=Na(r)+O(r) вычислена с использованием принятого'
в справочнике значения
Дг#°@) = D0(NaO) = 247 ± 20 кДж • моль я» 20 600 ± 1700 см.
Принятая величина основана на исследовании равновесия реакции Na2O(K)=Na(r)+NaO(r),
выполненного масс-спектрометрическим методом в работе Хйлденбранда и Мурада [2648] A056—
1100 К, 5 точек). Погрешность оценена авторами работы [2648].
Принятой энергии диссоциации соответствует
A//°(NaO, г, 0)= 107,546 + 20 кДж . моль.
NaO2(K, ж). Термодинамические свойства кристаллического и жидкого диоксида натрия
в стандартном состоянии при температурах 100—2000 К приведены в табл. 1109.
Значения постоянных, принятые для расчета термодинамических функций, приведены в табл.
46.1. В справочнике за стандартное состояние NaO2(K) в интервале 0—196,2 К принята
ромбическая модификация (структурный тип марказита), в интервале 196,2—223,3 К — кубическая
модификация (структурный тип пирита) и в интервале 223,3—770 К — кубическая модификация^
(структурный тип NaCl) [4820].
При Т ^ 298,15 К термодинамические функции NaO2(K) вычислены по результатам измерений
теплоемкости, выполненных Тоддом [4519] E2—296 К) на образце, содержавшем в качестве
примесей 6% Na2O2 и 1,5% Na2CO3 (поправка на примеси вводилась автором работы [4519]).
Изменения энтальпии NaO2(K) вблизи точек фазовых переходов в интервалах 185—208 К и 208—235 К
были определены автором [4519] в специальной серии опытов и составляют 3,12+0,06 и 3,50-f
+0,07 кДж-моль соответственно. Экстраполяция теплоемкости для Т <[ 51 К по уравнению
С°(Г)=1>A75/Г)+2ЯC13/Г) привела к значению 5°E1 К)=9,33 Дж-К'^моль. Погрешности
принятых значений ?°B98,15 К) и #°B98,15 К)—#°@) составляют 1,5 Дж-К-моль и 0,1 кДжХ
X моль ~1 соответственно.
При Т > 298,15 К единственное экспериментальное исследование термодинамических свойств
NaO2(K) (измерение энтальпии) проведено Веденеевым и Скуратовым [199] в интервале
температур 292—373 К. Авторы [199] исследовали образец, содержавший ~89% NaO2 (примеси: Na2O2 —
2%, Na2CO3 — 1,1%, NaOH — 7,1%, Н2О — 0,65%) и получили значение средней теплоемкости
71,7 Дж-К~1-моль (при 332 К). Это значение меньше найденного Тоддом [4519] при 298,15 К
G2,13 Дж-К-моль~1)х. Поэтому данные [199] в справочнике не учитываются и для теплоемкости
в интервале 298,15—770 К принято уравнение (см. табл. 46.1), выведенное по значениям
теплоемкости при 298,15 и 770 К. Последнее (88+4 Дж-К~1-моль~1) оценено на основании сравнения
экспериментальных данных для Na2O и NaatJ, теплоемкость которых в точках фазовых
превращений составляет —ЗОге Дж-К-моль, где га — число атомов в молекуле.
Температура плавления NaO2 (800+50 К) оценена с учетом экспериментальных данных для
КО2(к) и RbO2(K). Принятому значению не противоречит величина, приведенная в справочнике
JANAF [2834] (825 К), основанная на неопубликованных данных Маргрейва. Значение
Amff°(NaO2)=20,5+4 кДж-моль оценено на основании предположения о равенстве энтропии
плавления NaO2 и К02. Теплоемкость жидкого диоксида натрия A00+10 Дж-К~1-моль)
оценена равной 33га Дж-К^-моль".
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 1, 8
и 16 Дж-К^-моль соответственно.
Термодинамические функции NaO2(K), вычисленные ранее в справочниках [1395], [2834],
незначительно отличаются от приведенных в табл. 1109 @,5—0,7 Дж-К-моль~1 в значении Ф°(Г)
при Т <^ 800 К). Термодинамические функции NaO2(ffl) публикуются впервые.
Константа равновесия реакции NaO2(K, ж)=^(г)+2О(г) вычислена с использованием
значения Дг#°@)=865,487+3,1 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
к, 298,15 К) = —261 ±3 кДж • моль.
1 Согласно данным [4519] теплоемкость NaO2 (к) при температурах выше 223 К почти постоянна до ~280 К„
а затем начинает увеличиваться.
319
Na2O(K, ж) Глава 46
Эта величина основана на измеренной Гиллесом и Маргрейвом [2403] энтальпии реакции:
2NaO2(K)+12 001 H2O(>K)=2NaOH(p-p, 6000 Н2О)+3/2О2(г), ДГЯ°B98,15 К)=-133+6 кДжХ
Хмоль. В работе было использовано 6 образцов NaO2, содержавших от 92,5 до 94,0% основного
вещества. Главным источником погрешности является низкая чистота образцов и их недостаточная
устойчивость.
Na2O(K, ж). Термодинамические свойства кристаллического и жидкого оксида динатрия
в стандартном состоянии при температурах 100—3500 К, приведены в табл. 1110.
Значения постоянных, принятые для расчета термодинамических функций, приведены в табл.
46.1. В справочнике за стандартное состояние Na2O(K) в интервале 0—1023 К принята кубическая
модификация y-Na2O (структурный тип флюорита, CaF2 [4824]), в интервале 1023—1243 К —
Р-модификация Na2O и в интервале 1243—1405 К — а-модификация; данные о кристаллической
структуре двух последних модификаций в литературе отсутствуют.
При Т <; 298,15 К термодинамические функции Na2O(K) основаны на результатах измерений
теплоемкости, выполненных Фурукава [2349] A5—380 К) на образце, содержавшем 2,33% Na2CO3
и 0,91% Na2O2. Авторы справочника JANAF [2834] ввели поправки к данным работы [2349]
на содержание Na2CO3 (соответствующие поправки на примесь Na2O2 несущественны).
Экстраполяция теплоемкости привела к значению 5°A5 К)=0,051 Дж-К^-моль. Значения
термодинамических функций при 100, 200 и 298,15К приняты по [2834]; погрешности значений iSr0B98,15K)
и #°B98,15К)—#°@) (см. табл. 46.1) оцениваются в 0,4 Дж-К^-моль и 0,06 кДж-моль
соответственно.
Уравнение для теплоемкости Na2O(K) в интервале 298,15—1023 К (см. табл. 46.1) выведено
совместной обработкой результатов измерений энтальпии в работах Гримли и Маргрейва [2494]
F53—1170 К, чистота образца 96,8%) и Фредриксона и Часанова [2323] E00—1300 К). При
обработке погрешности этих данных оценивались в 2,5 и 1,5% соответственно. Рассчитанные по
этому уравнению значения теплоемкости в области 298—380 К практически совпадают с
измеренными Фурукава [2349]. Температуры полиморфных превращений A023+5 и 1243+10 К) и
соответствующие им энтальпии превращений A,3+0,3 и 9,0+0,9 кДж-моль) приняты по
термографическим данным Буазиза и др. [1629]. Существование этих переходов подтверждается резким
ростом энтальпии в интервале 1025—1825 К (от 60+1 до 89,8+1,5 кДж-моль), отмеченным в
работе [2323]. Теплоемкость C- и a-Na2O принята равной A00+10 Дж-К-моль); эта величина
не противоречит данным по измерению энтальпии Na2O(K), полученным в работе [2323].
Значения Гт=1405+2 К и Д ^°=36,0+4,0 кДж-моль, приведенные в справочнике,
основаны на согласующихся между собой термографических измерениях, выполненных в работах
[1629] и [3762]. Более ранние измерения Тт привели к заниженным значениям A193, 1190 К)
и не учитывались.
Теплоемкость жидкого оксида динатрия A00+10 Дж-К^-моль) оценена на основании
соответствующих данных для 1л20(ж) и принятого в справочнике эмпирического соотношения (см.
т. 1, кн. 1, с. 79).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
2, 6 и 12 Дж-К-моль~1 соответственно. Расхождения термодинамических функций,
приведенных в табл.,1110 и справочнике [2834], составляют 1—1,5 Дж-К~1-моль при ? ^ 1000 К и
достигают ~10 Дж-К~1-моль~1 при 3000 К, что обусловлено различием принятых значений
теплоемкости и энтальпии плавления.
В справочнике принята энтальпия образования
/±fH°(Na2O, к, 298,15 К) = — 414,57 + 0,30 кДж • моль.
Эта величина основана на приведенных в табл. 46.6 результатах измерений энтальпии
растворения Na2O. Наиболее точные измерения с тщательно очищенным образцом были проведены
О'Харой [3698], и принятая в справочнике энтальпия образования вычислена по данным этой
работы. Результаты более ранних измерений менее точны, в первую очередь из-за наличия
неучтенных примесей в образцах. Приближенное значение —421 кДж-моль может быть
вычислено из измерений энтальпии растворения Na2O в соляной кислоте, проведенных Гроссом и
Вильсоном [2503].'Исследования равновесия реакции Na2O(K)=2Na(r)+1/2O2(r) приводят при расчете
по уравнению III закона к значениям —417+6 кДж-моль (масс-спектрометрический метод,;
987—1114 К, 10 точек) [2648] и —412+6 кДж-моль (торзионный метод, 1144—1304 К, 29 то-
320
Натрий и его соединения
Na2O(r)
Таблица 43.6. Результаты измерений Aa?jff°(Na2O, к->а>Н2О, 298,15 К) (в кДж - моль)
Авторы
Бекетов, 1879 [1454]
Ренгад, 1907 [3975, 3976]
Мацуи, Ока, 1929 [3424]
Мацуи, Наката, 1929 [3422]
Рот, Кауле, 1947 [4060, 4067]
О'Харе, 1972 [3698]
а Измерялась энтальпия реакции
Конечная концентрация, температура
измерений
1600 Н2О, 292,4 К, 5 измерений
4600 Н2О, 298,15 К, 4 измерения
7890 Н2О, 298,15 К, 4 измерения
825 Н2О, 295,55 К, 6 измерений
298,15 К, 9 измерений а
Na2O(K)+5,263(NaOH -555,27 H2O)=7,263(NaOH
ДагЯ°(сх>Н2О, 298,15 К)
—232
-238,2
—237,54+0,90
—237,17+0,70
—235,56+0,85
—240,25+0,20
• 402,23 Н2О).
чек) [3832], а исследование ЭДС реакции 4Na(>K)+Oa(r)=2NaaO(K) G14—934 К) [1284] — к
значению —404+5 кДж-моль. Эти измерения уступают по точности калориметрическим данным.
Давление "пара в реакции Na2O(K, }K)=Na2O(r) вычислено с использованием принятой в
справочнике энтальпии сублимации
\Н°{0) = As#°(Na30, к, 0) = 396 ± 10 кДж • моль.
Принятое значение основано на измерениях давления насыщенного пара Na2O(K) в работе
Хилденбранда и Мурада [2648] (масс-спектрометрический метод, 1033—1114 К, 7 точек). Расчет
по уравнениям II и III законов приводит к значениям 294+25 кДж-моль и 396,1+2,5 кДжХ
Хмоль соответственно. При оценке погрешности принятой величины помимо воспроизводимости
измерений учтена неточность термодинамических функций и сечений ионизации.
Na2O(r). Термодинамические свойства газообразного оксида динатрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1111.
Молекулярные постоянные, использованные для расчета термодинамических функций Na2O,
приведены в табл. 46.7. Экспериментальные данные о структуре и частотах колебаний Na2O в
литературе отсутствуют. В настоящем справочнике по аналогии с молекулой Li2O принимается,
что молекула Na2O в основном электронном состоянии X1 S* имеет линейную симметричную
структуру (симметрия D^b)- Приведенное в табл. 46.7 значение момента инерции Na2O соответствует
Таб
тина 46 7. Молекулярные постоянные, а также о и р(, принятые для расчета термодин,
* функций Na2O, Na2O+, Na2O2, NaOH, Na2O2H2, Na2SO4, NaNO2, NaNO3, NaBO.
термодинамических
O NBO
Молекула
Na2O
Na2O+
Na,O2
a a
NaOH
Na2O2H2
Na2SO4
NaNO2
NaNOs
NaBO3
Состояние
^2
v4
CM
Х^-Ц 320
I2 SJ 300
Р4г 850
Х^-Ц 431
ХЫд 3700
ХЫ 961
lUi 1330
fr-At 1484
X^-A 1974
Na2O: a приведено значение I •
NaaO+: a приведено значение /•
Na2O2H2: a v
Na2SO4: a %
NaNO3: a %
60B)
55B)
450
337 B)
450
550
790
1023
1089
1039 г-см2.
1038 г-см2.
7=190, v8=300, v9=300, vlo=32O, v
7=360, v8B)=H00, v9(
7=200, v8=825, v9=7E
600
550
350
3700
230
280
307
730
580
u=3700,
2)=610, vioB)=295, v
) CM
__
— —
80 524
— —
310 300
470 1137
1224 206
300 1283
577 364
v12=390 см-1.
uB)=75 cM-i.
—
254
—
300 a
650 a
70
730 a
109
г3 ¦ см6
2,9 -10 a
3,4 -10 a
23 -102
6,677 898
7-103
7,64-10*
13 -10a
5-103
2 -103
2
2
4
1
4
4
2
2
1
Pi
1
4
1
1
1
1
1
1
1
321
Na2O+(r) Глава 46
межатомному расстоянию r(Na—О) = 1,95+0,05 А, оцененному на основании межатомного
расстояния r(Na—О) в молекуле гидроксида натрия (см. ниже). Погрешность величины / составляет
1 • 10~39 г-см2. Приведенные в табл. 46.7 частоты колебаний Na2O рассчитаны по значениям
силовых постоянных, которые были оценены в предположении, что отношение силовых постоянных
связей Na—О в двухатомной и трехатомной молекулах равно аналогичному отношению для
молекул LiO и Li2O, а отношение силовой постоянной связи к деформационной силовой постоянной
сохраняется таким же, как в Li2O *. Погрешности принятых значений частот оцениваются в 20—
25%. Сведения о возбужденных электронных состояниях Na2O в литературе отсутствуют.
Термодинамические функции Na2O(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 126),
A. 129). Погрешности рассчитанных термодинамических функций обусловлены неточностью
принятых постоянных, в том числе структуры молекулы Na2O, тем, что не учитываются
возбужденные электронные состояния, которые могут оказаться стабильными по аналогии с найденным
для 1ляО [2507], а также приближенным характером расчета и составляют 6, 10 и 18 Дж-К-1Х
Хмоль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций Na2O(r) публикуется впервые.
Константа равновесия реакции Na2O(r)=2Na(r)-|-O(r) вычислена с использованием значения
Дгй\0) = 476,018 + 10 кДж . моль,
соответствующего принятым в справочнике энтальпиям образования и сублимации NaaO(K).
Этим величинам также соответствует
AfH°(Na2O, г, 0) = —13,709 ±10 кДж • моль.
Na2O+(r). Термодинамические свойства газообразного положительного иона оксида динатрия
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 1112.
Молекулярные постоянные, использованные для расчета термодинамических функций Na2O+,
приведены в табл. 46.7. Экспериментальные данные о структуре и частотах колебаний Na2O +
в литературе отсутствуют. По аналогии с молекулой Na2O принимается, что ион Na2O + в основном
электронном состоянии Z2EJ имеет линейную симметричную структуру (симметрия D^,).
Приведенные в табл. 46.7 постоянные были оценены по принятым постоянным молекул Na2O, Н2О и Н2О +
в предположении, что соотношение между постоянными Na2O и Na2O + такое же, как у Н2О и Н2О +.
Момент инерции Na2O+ рассчитан со значением r(Na—O)=2,l-j-0,l А, его логрешность
оценивается в 5-109 г-см2. Погрешности в значениях v1( v2 и v3 составляют 70, 15 и 150 см. Молекула
Na2O+, так же как молекула NaO,. должна иметь электронные состояния с низкими энергиями
возбуждения. Однако обоснованная оценка их энергий невозможна, и в связи с невозможностью
корректной оценки постоянных Na2O+ даже в основном состоянии возбужденные состояния этой
молекулы в справочнике не рассматриваются.
Термодинамические функции Na2O+(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A.10), A. 122)—A. 124), A.126),
A. 129) без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены грубой оценкой молекулярных постоянных Na2O+,
пренебрежением возбужденными состояниями, а также приближенным характером расчета; они составляют
10, 15 и 20 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций Na2.O+(r) публикуется впервые.
Константа равновесия реакции Na2O+(r)-f-e(r)=2Na(r)+O(r) вычислена с использованием
значения ДгЯ°@) =—54,982+51 кДж-моль, соответствующего принятому в справочнике
потенциалу ионизации
/0(Na2O)=531+50 кДж-моль.
Это значение основано на измерениях Хилденбранда и Мурада [2648], выполненных методом
электронного удара. Близкое значение 550 кДж-моль было получено в неопубликованной работе
Нормана (см. [2648]). Принятому потенциалу ионизации соответствует
A//°(Na2O+, г, 0) = 517,291 + 51 кДж • моль. '
1 /г=1,40, /а/г2= 0,023 (в dO5 дпк-см).
322
Натрий и его соединения Na2O2(K, ж)
Na2O2(K, ж). Термодинамические свойства кристаллического и жидкого диоксида динатрия
в стандартном состоянии при температурах 100—2500 К приведены в табл. 1113.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние Na2O2(K) в интервале 0—785 К принята
тетрагональная ^-модификация [5059], в интервале 785—948 К а-модификация; данные о
кристаллической структуре этой модификации в литературе отсутствуют.
При Т ^ 298,15 К термодинамические функции Na2O2(K) вычислены по измерениям
теплоемкости, выполненным Тоддом [4519] E0—298,15 К) на образце, содержавшем в качестве примесей
2,4% Na2CO3 и 3,6% Na2O. Экстраполяция теплоемкости к Г=0 по уравнению C°p(T)=DB52/T)+
+3Z?C99/7') [4519] приводит к значению S°E1 К)=4,44 Дж-К~1-моль~1. Погрешности принятых
значений ?"B98,15 К) и Я°B98,15 К)—Я0 @) составляют 1,5 Дж-К^-моль-1 и 0,12 кДж-моль
соответственно.
Для теплоемкости Na2O2(K) в интервале 298,15—785 К принято уравнение (см. табл. 46.1),
полученное обработкой результатов измерений энтальпии, выполненных Чандрасекхарайя и др.
[1806] C75,4—869,2 К) на образце, содержавшем в качестве примесей 1,5% Na2CO3 и 0,2% Na2O.
При обработке погрешность этих данных оценивалась в 1 %. Измерения энтальпии образца Na2O2t
содержавшего 5,7% NaOH и 1,15% Na2GO3, выполненные Веденеевым и Скуратовым [199] B92—
373 К), лежат ниже принятых данных на 3% и не учитывались.
Температура полиморфного превращения G85+2 К) принята по измерениям Толмана и Мар-
грейва [4465] G85 К) и Роде и Гольдера [881] G83 К). Данные [2287] G58 К) не надежные и не
учитывались. Энтальпия полиморфного превращения E,36+0,06 кДж-моль) найдена в работе
[1806]. Теплоемкость [3-Na2O2 в интервале 785—948 К принята постоянной A13+5 Дж-КХ
Хмоль) на основании результатов измерения энтальпии [1806].
Принятое значение температуры плавления Na2O2 (948+5 К) получено Бунцелем и Кольмейе-
ром [1721 ]. Энтальпия плавления A7+2 кДж-моль) и теплоемкость Ка2О2(ж) A34+15 Дж-К~хХ
Хмоль) оценены при подготовке справочника в соответствии с принятыми методиками (см. т. 1,
кн. 1, гл. 2).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000 и 2000 К оцениваются в 1, 3
и 12 Дж-К-моль~1. Термодинамические функции Na2O2(K), приведенные в таблицах JANAF
(Т ^ 2500 К) [2834] и настоящем справочнике, согласуются в пределах 0,08 Дж-К^-моль.
Термодинамические функции Na2O2^) публикуются впервые.
В справочнике принята энтальпия образования
A^°(Na2O2, к, 298,15 К) = —512 + 5 кДж.моль.
В табл. 46.8 приведены результаты измерений этой величины. Принятое значение основано
на наиболее надежных измерениях, выполненных в работе Гиллеса и Маргрейва [2403];
совпадающее в пределах погрешности значение было вычислено по данным работы Рота и Кауле [4060,
4067].
Давление пара в реакции Na2O2(K, H{)=Na2O2(r) вычислено с использованием значения ДгЯ°@)=
= k.sH°{N&2O2, к, 0)=388,184+30 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования Na2O2 (к) и атомизации Na2O2(r).
Таблица 46.8. Результаты определений Ayjy°(Na2O2, к, 298,15 К) (в кДж-моль)
Авторы
Метод
AfH°(NazO2, к, 298,15 К)
Форкран, 1898 [2289] Измерение энтальпии растворения Na2O2 в соля- —498
ной кислоте
Центнершвер, Блюменталь, 1931 Исследование равновесия реакции 2№2О2(к)= —495+50 (II)
[1589, 1774] =2Na2O(K)+O2(r) динамическим методом, 741— —489+6 (III)
922 К, 60 точек
Рот, Кауле, 1947 [4060, 4067] Измерение энтальпии растворения Na2O2 в воде, —509,6
1200 Н2О; 291,65 К; 8 измерений
Гиллес, Маргрейв, 1956 [2403} То же, 3800 Н2О; 298,15 К; 3 измерения —512,3±5,0
323
Na2Oii(r), NaH(K, ж) Глава 46
Na2O2(r). Термодинамические свойства газообразного диоксида динатрия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1114.
Молекулярные постоянные, использованные для расчета термодинамических функций NaaO2,
приведены в табл. 46.7. Структура Na2O2 экспериментально не исследовалась. На основании
теоретического расчета, проведенного' Алавеном и др. [4915], а также результатов исследования
ИК-спектров молекулы Na2O2 [1327] в справочнике принимается, что в основном электронном
состоянии ХХА молекула Na2O2 имеет плоскую ромбическую структуру (симметрия D2h) со связью
кислород—кислород в цикле. Приведенное в табл. 46.7 произведение моментов инерции
рассчитано по оцененным структурным параметрам r(Na—О)=2,0+0,1 А (из Na2F2, NaF и NaO) и г(О—
—О)=1,5+0,1 А (найдено при рентгеноструктурном исследовании Na2O, [4316]) *. Эти значения
в пределах погрешностей согласуются с результатами расчета [4915]. Погрешность 1А1в1с
составляет 1-Ю14 г3-см6.
Приведенные в табл. 46.7 значения частот v5 и ve найдены Эндрюсом [1327] в ИК-спектре
молекул Na2O2, изолированных в матрице из Аг. С принятым значением v5 согласуются результаты
измерений [4316, 4366], частота ve в других работах не наблюдалась. Приведенные в табл. 46.7
значения частот vx—v4 оценены при подготовке справочника на основании данных для молекулы
диоксида дилития. Значение валентной частоты связи О—O(vx) принято таким же, как в Li2O2,
в предположении, что оно мало изменяется в ряду молекул М2О2, где М — щелочной металл 2.
Эндрюс [1327] предположительно отнес к частоте внеплоскостного колебания v4 слабую полосу
в ИК-спектре в области 239 см. Однако так же, как для Li2O2, в настоящем справочнике
значение этой частоты принято по оценке (см. 1Л2О2). Погрешности экспериментально измеренных
частот колебаний с учетом матричного сдвига составляют 20 см, а оцененных — 20—25%.
Сведения о возбужденных электронных состояниях Na2O2 в литературе отсутствуют. По
аналогии с 1Л2О2 можно ожидать, что энергии возбужденных состояний превышают 25 000 см.
Термодинамические функции Na2O2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Их погрешности обусловлены главным образом неточностью принятых молекулярных
постоянных, а также приближенным характером расчета, они оцениваются в 5, 10, 15 Дж-КХ
Хмоль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно (в том числе 3—4 Дж-К~хХ
Хмоль из-за неточности молекулярных постоянных).
Таблица термодинамических функций Na2O2(r) публикуется впервые.
Константа равновесия реакции Na2O2(r)=2Na(r)+2O(r) вычислена на основании принятой
в справочнике энтальпии атомизации
Дгй °@) = 827 + 30 кДж • моль.
Эта величина была оценена сопоставлением энтальпий атомизации в рядах М2О2(г), М2О(г)
и МОН(г), где M=Li, Na, К, Rb, Cs. Принятой энтальпии атомизации соответствует
Д/И^Оа, г, 0) = —117,908 ± 30 кДж • моль.
NaH(K, ж). Термодинамические свойства кристаллического и жидкого гидрида натрия в
стандартном состоянии при температурах 100—1000 К приведены в табл. 1115.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние NaH(K) в интервале 0—911 К принята
кубическая модификация (структурный тип NaCl).
При Т <^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости NaH(K), проведенным Веструмом и др.3 в интервале 10—350 К. Экстраполяция
теплоемкости ниже 10 К приводит к значению 5°A0 К)=0,033 Дж-К-моль-1. Погрешности значений
5°B98,15 К) и #"B98,15 К)—Я°(О) оцениваются в 0,4 Дж-К^-моль и 0,06 кДж-моль
соответственно. С данными Веструма и др. в пределах 1—2% согласуются результаты измерений
теплоемкости NaH, проведенных Сейром и Вивером [4164] в интервале температур 60—92 К.
1 Эндрюс [13271 также принял значение г (О—О), полученное для кристаллического Na2O2 A,50 А), а для г (Na—
О) рекомендовал 1,90 А.
2 Частота колебания иона Oj2 в кристаллах М2О2 меняется в ряду Li2O2, Na2O2, K2O.,, Rb2O2 от 790 до 760 см
[2209], а частота колебаний иона О2 в МО2 увеличивается от 1097 см в LiO2 до 1116 см в CsO2 [1329].
s Данные Веструма и др. не опубликованы и известны по таблицам JANAF [2834].
324
Натрий и его соединения
NaH(K, ж)
Для теплоемкости NaH(K) при Т >¦ 298,15 К принято уравнение (см. табл. 46.1), выведенное
29815 К С°E00 КL7 Д^
), д
по значению теплоемкости при 298,15 К и значениям С°E00 К)=47,1 Дж-К^-моль
и С^(911 К)=63 Дж-К~1-моль~1, полученным сопоставлением экспериментальных данных по
теплоемкости гидридов лития и натрия (для NaH(K) — по данным Веструма и др. до 350 К).
Погрешности оцененных значений теплоемкости NaH составляют 3% при 500 К и 8 — 10% до 1000 К.
Температура плавления (911+5 К) при равновесном давлении водорода 107,3 бар принята
по результатам термографического исследования, выполненного Скуратовым и др. [940];
воспроизводимость ее определения оценена авторами в 2°. Исследованный образец гидрида натрия
содержал не менее 99,4% NaH. Принятое значение энтальпии плавления B6+3 кДж-моль)
вычислено Скуратовым и др. [940] на основании данных по давлению диссоциации NaH,
имеющихся в литературе и полученных в работе [940].
Значение теплоемкости жидкого NaH E6+8 Дж-К^-моль) оценено с учетом
экспериментальных данных по теплоемкости 1ЛН(ж).
Погрешности вычисленных значений Ф°(Т) при 298,15, 500 и 1000 К оцениваются в 0,3, 0,7
и 3 Дж-К~1-моль~1 соответственно. Термодинамические функции NaH(K), вычисленные
авторами справочника JANAF [2834], согласуются с приведенными в табл. 1115 в пределах 0,1 ДжХ
ХК~1>моль~1 в значениях Ф°(Т). Таблица термодинамических функций NaH^) публикуется
впервые.
Принятая в справочнике энтальпия образования
Ay#°(NaH, к, 298,15 К) = —56,38 + 0,20 кДж • моль
основана на приведенных в табл. 46.9 результатах калориметрических измерений энтальпий
растворения NaH(K) и Na(K) в воде и исследований равновесия диссоциации 2NaH(K, JK)=2Na(jK)+
+Н2(г). В тех калориметрических работах, где были измерены энтальпии растворения NaH(K)
Таблица 46.9. Результаты определений A/ff°(NaH, к, 298,15 К) (в кДж • моль)
Авторы
Форкран, 1905 [2293]
Хаген, Зивертс, 1929 [2547]
Мессер и др., 1955 [3488]
Ганн, Грин, 1958 [2525, 2527]
Труст, Отфей, 1874 [4552]
Кейс, 1912 [2979]
Жукова, 1927 [426]
Соллерс, Креншоу, 1937 [4343]
Херолд, 1951 [2619, 2620]
Аддисоя и др., 1964 [1254]
Скуратов и др., 1976 [940]
Дымова и др., 1974 [394]
Банус и др., 1955 [1383]
Метод
Калориметрический, измерение энтальпии
растворения NaH(K) в воде
A/#°(NaH,
II закон
к, 298,15 К)
III закон
—76 A-й путь)
Калориметрический, измерение энтальпии рас- —53,6
творения NaH(K) и Na(K) в воде
То же, 10 и 6 измерений
То же, 7 и 5 измерений
Статический, 2NaH(K, ж)=2№(ж)+Н2(г), 603—
703 К, 11 точек
То же, 540—688 К, 42 точки
То же, 558—698 К, 5 точек
То же, 583—633 К, 9 точек, расчет по уравнению
То же, 561—687 К, 6 точек
То же, 373—523 К, 10 точек
То же, 936—1032 К, 10 точек
ДТА, NaH(K), 700—850 К, расчет по уравнению
Точек кипения, NaH(K), 773—873 К, 5 точек
-56,3+2,1
—56,9+2,6
—56,31+0,22
-56,44+0,21
—63+6
—60+2
-54+8
-58
—56+2
—53+10
-62
—51
—58+2
B-й путь)
A-й путь)
B-й путь)
A-й путь)
B-й путь) ¦
—56,3+1,0
—56,3+0,8
—56,6+1,0
-56,5+0,6
—55,9+1,0
—55,7+1,3
—55,4+3,0
-56,6+1,7
—55,7+2,0
и Na(K) в воде, расчет энтальпии образования NaH(K) проводился двумя путями: по энтальпии
растворения NaH в воде A-й путь) и по разности энтальпий растворения NaH и Na в воде
B-й путь). Наиболее точные измерения с хорошо охарактеризованными образцами были выполнены
Ганном и Грином [2525, 2527], и рекомендуемая в справочнике величина принята по результатам
этих измерений. Совпадающая в пределах погрешностей величина была получена по данным
работы Мессера и др. [3488]. Значения энтальпии образования NaH(K), вычисленные по
результатам исследований равновесия реакции диссоциации, находятся в хорошем соответствии с резуль-
325
NaH(r) Глава 46
татами калориметрических измерений. Приведенные в таблице погрешности включают
воспроизводимость измерений и погрешности использованных в расчетах термохимических величин. Для
величин, рассчитанных по измерениям констант равновесия, учтена также неточность
термодинамических функций, приводящая к погрешности в энтальпии образования от 0,8 (измерения при
600 К) до 3,4 кДж-моль (измерения при 1000 К).
Давление пара гидрида натрия в реакции NaH(K, at)=NaH(r) вычислено с использованием
значения ДгЯ°@)= hsH°(NaR, к, 0)=194,742+5,0 кДж-моль, соответствующего принятым
в справочнике значениям энтальпии образования NaH(K) и энергии диссоциации NaH(r).
NaH(r). Термодинамические свойства газообразного гидрида натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1116.
В табл. 46.4 приведены молекулярные постоянные, принятые на основании результатов
исследований спектров и квантовомеханических расчетов NaH. В спектре NaH проанализирована только
одна система полос А1Л+^±Х1Л + [2706, 2707, 2868, 3717, 3727, 3751, 3752], однако теоретический
анализ [3687, 4112, 4112а] показал, что эта молекула имеет три связанных валентных состояния
ХЧ;^. . .4о2), АгЛ+(. . .4<з5а) и а3П(. . .4о2п), которые коррелируют с атомами NaB5)-fHB5)
и NaBP)+HBS).
Колебательные постоянные NaH в состояниях X1L+ и А1Е+, приведенные в табл. 46.4,
вычислены Панкхурстом [3752] по началам полос системы А1 Л* -> X1t,+ на основании его
собственных измерений B5 полос с 1 ^ i>' ^ 8 и 3 ^ ;;" ^ 8) и данных Хори D1 полоса с 3 ^ v' ^ 20
и v" ^ 2) [2706, 2707]. При этом Панкхурст учел уточнение нумерации полос, проведенное Ол-
соном [3717] на основании сопоставления спектров молекул NaH и NaD. В связи с аномальным
характером состояния А1!,* (см. LiH), принятые постоянные только приближенно описывают
энергию колебательных уровней этого состояния при у'=20; погрешность достигает 6 см.
Вращательные постоянные состояний ХХЕ+ и А1 Л*, приведенные в табл. 46.4, получены при подготовке
справочника совместной обработкой значений Bs и Dv, найденных в работах [2707, 3717, 3752].
Молекулярные постоянные в состоянии а3П приняты по расчету Сакса и др. [4112а], погрешность
значения Ге(а3П), по-видимому, не превышает 400 см.
Термодинамические функции NaH(K) были рассчитаны по уравнениям A.3)—A.6), A.9),
A. 10), A. 93)—A. 95). Значения QBS и ее производные вычислялись по уравнениям A. 90)—
A. 92)' с учетом двух возбужденных состояний. Величины (?^,вр для состояния Х1?+, Аг1, + и а3П
и их производные находились по уравнениям A. 70)—A. 75) непосредственным суммированием
по колебательно-вращательным уровням энергии. В расчете учитывались все уровни,
ограниченные предельными кривыми диссоциации указанных состояний. Энергия
колебательно-вращательных уровней вычислялась по уравнениям A. 65) и A. 64а). Значения коэффициентов Ykl в этих
уравнениях приведены в табл. 46.5 вместе со значениями ушах, /Иш и коэффициентами а{,
аппроксимирующими предельные кривые диссоциации. Коэффициенты УБ0, У60 и У03 для состояния X1 ? +,
Yeo и Yos для состояния А1Т, + ш Ym для состояния а3П получены из условий A. 50) и A. 59). Муль-
типлетность и вырождение состояния а3Л учитывались статистическим весом 6.
Погрешности вычисленных термодинамических функций при Т ^ 3000 К обусловлены в
основном неточностью фундаментальных постоянных и не превышают 0,03 Дж-К~1-моль в
значениях Ф°{Т). При Т 5> 3000 К становится существенной неточность аппроксимации высоких
колебательно-вращательных уровней состояния X1 ? + уравнением с принятыми значениями
постоянных. Погрешность значения Ф°F000 К) может достигать 0,2—0,3 Дж-К~1-моль.
Ранее термодинамические функции NaH(r) вычислялись в таблицах JANAF [2834],
справочниках Мак-Брайд и др. [3439] и Хара и др. [2537], а также в работах Горбова .и др. [306] (Т <J
< 6000 К) и Шнейдера [4197а] (Т < 4000 К). Расхождения данных [2834] и [3439] с
приведенными в табл. 1116 достигают 2, 3 и 8 Дж-К^-моль в значениях Ф°F000 К), 5°F000 К) и
6ГF000 К) соответственно и объясняются главным образом различием методов расчета. Данные
[306] и табл. 1116 согласуются ъ пределах 0,08 и 0,6 Дж-К^-моль в значениях Ф°(Т) и S°(T).
Константа равновесия реакции NaH(r)=Na(r)+H(r) вычислена с использованием принятой
в справочнике энергии диссоциации
D0(NaH)=15 160+400 см « 181+5 кДж-моль.
Эта величина была получена Мейером и Росмусом [3495] в результате квантовомеханического
расчета, выполненного с учетом электронной корреляции в приближении парного взаимодействия.
326
Натрий и его соединения " NaOH(K, ж)
С принятым значением хорошо согласуется интерпретация континиума в спектре испускания NaH
в области 3000 А как перехода из отталкивательного состояния ВЩ в основное состояние этой
молекулы [2707, 2709, 2710].
Принятому значению энергии диссоциации соответствует
г, 0) = 142,797+ 5,0 кДж • моль.
МаОЩк, ж). Термодинамические свойства кристаллического и жидкого гидроксида натрия
в стандартном состоянии при температурах 100—3000 К приведены в табл. 1117.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние NaOH(K) в интервале 0—572 К принята
ромбическая а-модификация, а в интервале 572—596 К — моноклинная модификация p-NaOH.
При Т ^ 298,15 К термодинамические функции NaOH(K) вычислены по результатам измерений
теплоемкости, проведенных Марчем и Джиоком [3592] A2—320 К) на образце, содержавшем в
качестве примесей 0,01 мол.% Na2CO3 и 0,04 мол.% И2О. Экстраполяция теплоемкости к Т=0 по
закону Дебая привела к значению 5°A5 К)=0,12 Дж-К~1-моль~1. Погрешности принятых
значений 5°B98,15К) и #°B98,15 К)—Н°@) оцениваются в 0,2 Дж-К^-моль и 0,04 кДж-моль
соответственно.
Для теплоемкости a-NaOH в интервале 298,15—572 К принято уравнение (см. табл. 46.1),
выведенное обработкой результатов измерений энтальпии, проведенных Дугласом и Девером
[2106] B73—973 К; чистота образца 99,05%). По данным [2106] рассчитана также теплоемкость
p-NaOH(K) (86+4 Дж-К^-моль). При обработке погрешность данных [2106] оценивалась в 1%.
Измерения энтальпии NaOH(K) в работе Попова и Гинзбурга [807] B98,15—1500 К; чистота
образца 98,79%), представленные в виде уравнений для С°Р(Т), не учитывались, так как расчет
теплоемкости NaOH(K) по полученному в этой работе уравнению приводит к значительно более низким
значениям (вблизи точки фазового перехода на 13%), чем это следует из данных [2106].
Температура полиморфного превращения E72+3 К) принята по термографическим данным Решетникова
и Баранской [866], хорошо согласующимся с результатами работ [116, 742, 869, 872, 873, 874,
1073, 1074, 1629, 1895, 1898, 2553, 2782, 2836, 3323, 3765, 4039] \ Данные менее точных работ
[247, 807, 949, 1335, 2106, 2208, 3559. 4332] E56—578 К) не учитывались. Энтальпия полиморфного
превращения E,85+0,60 кДж-моль) принята по работе Дугласа и Девера [2106] F,36 кДжХ
Хмоль) с учетом уравнений, выведенных для энтальпии а- и (З-NaOH. Значения Ь.иД,
полученные в работах [807] (8,08 кДж-моль) и [866] G,2+0,7 кДж-моль), ненадежны.
Температура плавления E96+2 К) определена из термографических данных [866] и хорошо
согласуется с измерениями [113,~380, 807, 869, 872, 873, 874, 1073, 1074, 1335, 1895, 2836, 3116,
3323, 3765, 4039, 4079]. В работах [116, 247, 742, 949, 1072, 1898, 2106, 2553, 2782, 3559, 4241,
4332] получены менее точные результаты E92—603 К), которые не учитывались. Энтальпия
плавления G,8+1,5 кДж-моль) получена из уравнений, принятых для энтальпии P-NaOH и NaOH(m),
и согласуется в пределах погрешности сданными работ [2106] F,36 кДж-моль), [807] F.82 кДжХ
Хмоль-!), [866] F,59 кДж-моль) и [3116] (8,37 кДж-моль).
Для теплоемкости NaOH(Ht) принято уравнение (см. табл. 46.1), выведенное при совместной
обработке результатов измерений энтальпии, проведенных авторами работы [2106] и Пауэрсом
и Блейлоком E92—1000 К) 2. В совместную обработку вводились два значения энтальпии
#°A500 К)—Я°E96 К)=76,8 кДж-моль и #"B000 К)-#°E96 К) = 119,6 кДж-моль,
полученные экстраполяцией теплоемкости к Г=2000 К. Погрешность экстраполированных значений при
обработке оценивалась в 2%, погрешность данных [2106] и Пауэрса и Блейлока — в 1,5%.
Уравнение для теплоемкости КаОН(ж), выведенное в работе [807], приводит к более низким значениям,
чем это следует из согласующихся между собой данных работ [2106] и Пауэрса и Блейлока (при
1000 К на 7,5%), поэтому оно не принималось во внимание.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
1,5 и 8 Дж • К -моль соответственно. Термодинамические функции NaOH(K, ж), приведенные в
справочнике и в таблицах JANAF [2834], близки между собой при Т <^ 1000 К; при более высоких
температурах расхождения, обусловленные различием принятых значений C°(NaOH, ж),
увеличиваются (до 3,5 Дж-К-моль-1 в значении Ф°C000 К)).
1 Данные о полиморфном превращении при Г=513 К, полученные Папеном и Буази [3765], не подтвердились
в других исследованиях и в справочнике не учитывались.
- Результаты работы Пауэрса и Блейлока известны по справочнику Гмелина [2422].
327
NaOH(r) Глава 46
Принятая в справочнике энтальпия образования
, к, 298,15 К) =—425,88 + 0,15 кДж • моль
основана на приведенных в табл. 46.10 измерениях энтальпии растворения NaOH(K) в воде.
Наиболее точные измерения были выполнены Марчем и Джиоком [3592] и Решетниковым [864]. В
справочнике для последующих расчетов принимается
\qH°(oD Н2О; 298,15 К) = —44,45 + 0,12 кДж • моль.
Давление пара гидроксида натрия в реакции NaOH(K, ?K)=NaOH(r) вычислено с
использованием значения ДгЯ°@) = As-flro(NaOH, к, 0)=239,335+10 кДж-моль, соответствующего принятым
в справочнике энтальпиям образования NaOH в кристаллическом и газообразном состояниях.
Таблица 46.10. Результаты измерений Aa.JEr°(NaOH, к-> соНаО, 298,15К) (в кДж-моль)
Авторы
Конечная концентрация, температура
измерений
Д Я°(соН3О, 298,15 К)
Бертло, 1875 [1531] 154Н2О, 283,65 К, 2 измерения -43,3+2,5
Томсен, 1883 [4511] 163—19Ш2О, 290,47 К, 4 измерения —43,12
Форкран, 1901 [2292] 160Н2О, 294,65 К, 1 измерение —44,09
160Н2О, 294,65 К, 11 измерений —45,15+0,75 а
Рот и др., 1942 [4069] 5,8 и 424Н2О, 292,95 К, 2 измерения —43,4
Бобтельский, Лэрш, 1950 [1592] 228Н2О, 296,15 К, 1 измерение —42,64
Решетников, 1961 [864] 400НаО, 298,15 К, 5 измерений —44,16+0,25
Марч, Джиок, 1962 [3592] 182Н2О, 298,15 К, 5 измерений —44,52±0,12
Хартшорн и др., 1972 [2577] 437Н2О, 298,15 К, 2 измерения —43,52±1,5
Фучс, Хаган, 1973 [2342] 55 500—111000 НаО, 298,15 К —43,93±0,85
а Величина получена экстраполяцией энтальпии растворения кристаллогидратов различного состава к безводной
соли.
NaOH (г). Термодинамические свойства газообразного гидроксида натрия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1118.
Молекулярные постоянные, использованные для расчета термодинамических функций NaOH,
приведены в табл. 46.7. Исследования микроволнового [3140, 3792] и инфракрасного [1246]
спектров, а также квантовомеханический расчет [451] показывают, что в основном электронном
состоянии X1!!,* молекула NaOH имеет линейную структуру (симметрия Cmh). Приведенный
в табл. 46.7 момент инерции / вычислен по вращательной постоянной i?00°0=0,419 191 8+3-10~7 см,
найденной при исследовании микроволнового спектра Пирсоном и Трубладом [3792] х. Этому
значению Вт°0 в предположении, что г„(О—Н)=0,92+0,02 А, соответствует ro(Na—О) = 1,954 +
+0,002 А. Приведенные в табл. 46.7 значения частот vx и v2 измерены Аквиста и Абрамовицем
[1246] в инфракрасном спектре молекул NaOH, изолированных в Ar-матрице. С принятым
значением v2 в пределах погрешности согласуется частота 437+10 см, наблюдавшаяся Спинаром
и Маргрейвом [4367] в ИК-спектре паров NaOH при 1100 К. Ненаблюдавшаяся в спектре частота v3
принята приближенно равной соответствующей частоте молекулы CsOH. Погрешности частот
vl5 v2 и v3 оцениваются в 10, 20 и 100 см. По аналогии с изоэлёктронной молекулой NaF энергии
возбужденных электронных состояний NaOH должны превышать 20 000 см. Согласно
неэмпирическому расчету [451 ] энергия изомера HNaO превышает энергию NaOH на 28 000 см.
Термодинамические функции NaOH(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126), A. 129) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены неточностью принятых значений молекулярных постоянных, а также приближенным
характером расчета и составляют 1, 4 и 6 Дж-К~1-моль в значениях Ф°(Т) при 298,15, 3000 и
6000 К (в том числе 1—2 Дж-К~1-моль~1 из-за неточности молекулярных постоянных).
1 В работе [3792] определено также значение Z?oo°o= (9,58+0,2)-10~7 см, а в работе f3140J —значения Bwo—
328
Натрий и его соединения
Na2O2H2(r)
Ранее термодинамические функции NaOH(r) вычислялись в таблицах JANAF [2835 ] и
справочнике Мак-Брайд и др. [3439]. Данные [2835] и табл. 1118 согласуются в пределах 0,1—0,2 Дж-Кх
Хмоль. В справочнике [3439] принята нелинейная структура молекулы NaOH и значение v2 =
=950 см, соответствующие расхождения с табл. 1118 достигают 12 Дж-К~1-моль~1 в значениях
Ф°(Г).
Константа равновесия реакции NaOH(r) = Na(r)+O(r)+H(r) вычислена с использованием
значения Дг#°@) = 752,580+10 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
bfH°(Na0R, г, 0)= —182 ± 10 кДж • моль.
Принятая величина основана на приведенных в табл. 46.11 результатах исследований
равновесия реакций с участием NaOH(r) в работах [2855, 2965, 3935]. Расчеты, основанные на
исследованиях испарения гидроксида натрия [4656], привели к завышенной стабильности NaOH(r), так как
Таблица 46.11. Результаты определений A^H"°(NaOH, г, 0) (в кДж • моль)
Авторы
Вартенберг, Альбрехт, 1921 [4656]
Скунмейкер, Портер, 1958 [4209]
Рао, Скунмейкер, 1960 [3935]
Енсен, Педли, 1966 [2855]
Коттон, Дженкинс, 1969 [1941]
Келли, Педли, 1971 [2965]
Метод
Точек кипения, Ш0Н(ж)=№0Н(г), 1283—1681 К,
19 точек
Масс-спектрометрический, NaOH^)=NaOH(r),
625—694 К
Эффузионный с селектором скоростей молекул,
NaOHEK)=NaOH(r), 936 К, 1 точка
Спектрофотометрическое измерение p(Na) в пламенах
H2+O2+N2, Na(r)+H20(r)=Na0H(r)+H(r), 2475 К,
1 точка
То же, 2030 К, 6 измерений
То же, пламена Н2+О2+СО2,1950—2750 К, 33 точки,
данные представлены в виде уравнения
A/ff°(NaOH, г, 0)
II закон
-246
-267
—
—182+8
III закон
-214
—
-181+10
—180+12
—196+15
-186+7
в них не было учтено образование в парах димерных молекул. Измерения [4209J позволяют провести
расчет только по уравнению II закона и не достаточно точны. В работе Коттона и Дженкинса
[1941] отсутствуют многие данные, необходимые для оценки надежности этих измерений.
Na2O2H2(r). Термодинамические свойства газообразного димера гидроксида натрия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 1119.
Молекулярные постоянные, использованные для расчета термодинамических функций Na2O2H2,
приведены в табл. 46.7. Спектр и структура молекулы Na2O2H2 экспериментально не исследовались,
поэтому ее молекулярные постоянные оценены на основании сравнения соответствующих
постоянных молекул К2О2Н2, Rb2O2H2 и Gs2O2H2 и постоянных димеров фторидов щелочных металлов
M2F2, принятых в настоящем справочнике. Произведениемоментов инерции IaIbIc вычислено
в предположении, что в основном электронном состоянии Х1Ад молекула Na2O2H2 имеет плоскую
ромбическую структуру со связями ОН, расположенными на одной прямой (симметрия D2h) х,
и структурные параметры 2} О—Na—0=100+5°, r(Na—О)=2,12+0,05 А, г(О—Н)=0,97+0,03 А.
310114 36 П й
Погрешность
))
оценивается в 3-10~114 г3-см6. Погрешности принятых значений частот
2
р р 1
и vu оцениваются в 100 см, всех остальных — 20% 2. Сведения о возбужденных электронных
состояниях Na2O2H2 в литературе отсутствуют.
Термодинамические функции Naa02H2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор». Погрешности рассчитанных термодинамических функций обусловлены неточностью
принятых значений молекулярных постоянных, а также приближенным характером расчета и со-
1 С ромбической структурой фрагмента NaONaO согласуются результаты исследования отклонения
молекулярного пучка Na2O2H2 в электрическом поле [1707].
2 Отнесение частот к различным типам колебаний см. в тексте по К2О2Н2 (г).
329
NaF(K, ж)
Глава 46
ставляют 5, 12 и 20 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К (в том числе 2—
5 Дж-К~1-моль~1 из-за неточности молекулярных постоянных).
Ранее термодинамические функции Na2O2H2(r) вычислялись в таблицах JANAF [2835] и
справочнике Мак-Брайд и др. [3439]. Расчеты были выполнены в предположении, что молекула
Na2O2H2 имеет симметрию C2h (с атомами водорода, находящимися в трансположении относительно
плоскости ONaONa) и иные значения частот; соответствующие расхождения с табл. 1119
достигают в значениях Ф°(Г) 32 и 23 Дж-К-моль' соответственно.
Константа равновесия реакции NaaO2H2(r)=2Na(r)+2O(r)+2H(r) вычислена с использованием
значения Дг#°@) = 1721,160+25 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A//°(Na2O2H2, г, 0) = —580 + 25 кДж • моль.
Принятая величина является средней из приведенных в табл. 46.12. Указанные в таблице
погрешности учитывают воспроизводимость измерений и погрешности термодинамических
функций и термохимических величин, использованных в расчетах. При оценке погрешности
рекомендованного значения был также учтен разброс результатов измерений в разных работах.
Таблица 46.12. Результаты определения Aj:II°(Na2O2B.2, г, 0) (в кДж ¦ моль)
Авторы
Метод
A^Na-AHa, г, 0)
II закон
III закон
Скунмейкер, Портер, 1958 [4209] Масс-спектрометрический, 2NaOH^)=Na2O2H2(r), —679+15 —
625—695 К, 8 точек
Тоже, Na2O2H2(r)=2NaOH(r), 625—695 К, 8 точек —592+30 —
Портер, Скунмейкер, 1958 [3871] Масс-спектрометрический, Na2O2H2(r)=2NaOH(r), — —562+20
666 К, 1 точка
Скунмейкер, Портер, 1959 [4210] Масс-спектрометрический, 2NaOH(r)+K2O2H2(r)= — —570+20
=2KOH(r)+Na3O2H2(r), 883 К, 4 измерения
Рао, Скунмейкер, 1960 [3935] Эффузионный с селектором скоростей молекул, — —599+15
2NaOH()K)=Na2O2H2(r), 936 К, 1 точка
NaF(K, ж). Термодинамические свойства кристаллического и жидкого фторида натрия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1120.
Значения постоянных, использованные для расчета термодинамических функций,
представлены в табл. 46.1. В справочнике за стандартное состояние NaF(K) в интервале 0—1269 К
принимается кубическая модификация (структурный тип NaCl) 1.
При Т ^ 298,15 К термодинамические функции вычислены для Т ^ 20 К по результатам
измерений теплоемкости в работах Харрисона и др. [2574] @,05—15 К) и Берча и др. [1563]
B,3—20 К), в интервале 20—50 К — по данным Киркхема и Йейтса [3019] B5—300 К) и в
интервале 50—298,15 К — по согласующимся между собой данным Кинга [3009] E4—296 К)
и работы [3019]. Результаты старых измерений тенлоемкости Корефом [3072] (82—273 К) и Брен-
стедом [1683] B73—293 К) не учитывались. Принятые значения ?°B98,15 К) и Я°B98Д5 К)—
Н°@), приведенные в табл. 46.1, имеют погрешности 0,15 Дж-К~1-моль~1 и 0,02 кДж-моль
соответственно.
При Т > 298,15 К для теплоемкости NaF(K) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии NaF в работах"О'Брайена и Келли [3692] D06—1746 К),
Маклеода [3337] E26—1268 К) и Ускоковича и Ристича [4581] C73—1073 К). При этом данные
О'Брайена и Келли были скорректированы в связи с тем, что Франк [2315] обнаружил
допущенную ими ошибку при измерениях температуры. Скорректированные данные [3692] практически
совпадают с результатами измерений [4581] и гораздо лучше согласуются с данными Маклеода
[3337]. При обработке погрешность данных [3692] и [4581] оценивалась в 1%, данных [3337] —
в 2%. Измерения энтальпии NaF, выполненные ранее Ляшенко [637] B93—1287 К) и Крестовни-
ковым и Каретниковым [586] B98—1073 К), не принимались во внимание ввиду их существенных
(до 5%) отклонений от результатов более точных работ [3337, 3692, 4581].
* Кубическая фаза высокого давления (структурный тип CsCl) стабильна при 298 К и р > 16,8 кбар [3851].
330
Натрий и его соединения NaF(n, ж)
Температура плавления NaF A269+1 К) основывается на многочисленных измерениях [1415,
1654, 1740, 3084, 3173, 3337, 3806, 4030, 4079, 4234, 4886], выполненных на хорошем уровне и на
весьма чистых образцах; менее точные значения были получены в работах [82, 668, 1701, 1867,
2267, 2315, 2691, 3063, 3827, 3868]. Для энтальпии плавления принята величина C4,25 +
+0,40 кДж-моль), полученная в результате обработки данных по энтальпии кристаллического
и жидкого NaF. В калориметрических измерениях получены значения 32,9 [637], 33,6 [3692]
и 33,5 [3337] кДж-моль соответственно. Энтальпия плавления определялась также методом
высокотемпературной криоскопии [3806] C3,0 кДж-моль) и по диаграммам плавкости бинарных
систем [1701, 3857] C3,6 и 32,7 кДж-моль соответственно).
Теплоемкость NaF(}K) G0,1+1,5 Дж-К~1-моль~1) принята по результатам совместной
обработки данных [3692] A247—1761 К; 7 измерений) и [3337] A284—1576 К; 6 измерений). В более
ранней работе [637] было получено значение 73,6 Дж-К~1-моль.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
0,6, 2 и 5 Дж-К~1-моль~1 соответственно.
Расхождения между термодинамическими функциями, табулированными в настоящем издании
и в справочниках [337] (Т < 3600 К), [1395] (Г < 1900 К), [2834] (Т < 3500 К), [2960] (Т <
< 1900 К), достигают 1,5 Дж-К^-моль в значениях Ф°(Г) для NaF(K) и 2,8 Дж-К^-моль-*
для NaF(ffl) и обусловлены существенным уточнением теплоемкости в работах [3337] и [4581].
В справочнике принята энтальпия образования
&fH°(NaF, к, 298,15 К) = —576,6+ 0,7 кДж • моль.
Это значение основано на измерениях энтальпии растворения NaF(ic) в воде, представленных
в табл. 46.13, и измерениях энтальпии реакции NaCl(K)-)-0,5F2(r)=NaF(K)+0,5Cl2(r) в работах,
Таблица 46.13. Результаты
Авторы
Гунц, 1884 [2530]
Латимер, Джолли, 1953 [3199]
Хеплер и др., 1953 [2614]
Девис, Бенсон, 1965 [1999]
Цветков, Рабинович, 1969 [1097]
Кук и др., 1972 [1916]
Паттал и др., 1978 [3688]
Гермейн и др., 1979 [2390]
измерений AaqH°(NaF, к-*соН2О, 298,15 К) (в
Конечная концентрация, температура
измерений
400 Н2О, 235,15 К, 1 измерение
1390 и 2880 Н2О, 298,15 К, 2 измерения
1390 Н2О, 298,15 К, 1 измерение
220 Н2О, 298,15 К, 1 измерение .
240 Н2О, 298,15 К, 1 измерение
620—2780 И2О, 298,15 К, 10 измерений
420—3470 Н3О, 298,15 К, 10 измерений
820 Н2О, 298,15 К, 8 измерений
кДж
• МОЛЬ)
ДагЯ°(ооН20, 298,15 К)
0,44
0,96
0,91
0,99
0,88
0,948+0,017
0,937+0,042
0,808+0,042
Вартенберга, Фитцнера [4659] и Шмитца и Шумахера [4194], а также реакции NaCl(K)+HF(p-p,
5,7 H2O)=NaF(K)+HCl(p-p, 12,7 Н2О) в работе Кофлина [1943]. Наиболее полные и точные
измерения энтальпии растворения были проведены авторами работ [1916, 2390, 3688]. Принятая
в справочнике знтальп'я образования вычислена с использованием значения ДЯ2#°(ооНгО,
298,15 К)=0,898+0,050 кДж-моль, среднего из этих данных. По результатам исследований
[4194, 4659] и [1943] были вычислены значения энтальпии образования, равные —576,3 +
+1,0 кДж-моль и —576,7+0,7 кДж-моль соответственно, которые практически совпадают
с принятым. Основным источником погрешности принятой величины является неточность
используемого в расчетах значения энтальпии образования иона F~(aq).
Давление пара в реакции NaF(K, ж)=КаР(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = &sH°(NaF, к, 0) = 280,7 + 4,0 кДж • моль.
Эта величина основана на измерениях давления и состава насыщенного пара NaF(K, ж). В
состав насыщенного пара NaF при Гт=1269 К по принятым в настоящем справочнике данным
входят 28,3 мол.% димерных и 0,7 мол.% тримерных молекул. Методика выбора термохимических
величин для компонентов насыщенного пара галогенидов щелочных металлов изложена в
Приложении 8.
331
NaF(r)
Глава 46
Результаты расчетов энтальпии сублимации NaF в виде мономерных молекул представлены
в табл. 46.14. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятое значение является средневзвешенным из приведенных в таблице величин. При
Таблица 46.14. Результаты определения AsH"°(NaF, к, 0) (в кДж-моль)
Авторы
Вартенберг, Шульц, 1921 [4663]
Руфф и др., 1922 [4096]
Нива, 1938 [3665]
Нарышкин, 1939 [707]
Сенс и др., 1957 [4234]
Новоселова и др., 195.8 [738]
Айзенштадт и др., 1958 [2166]
Пью, Барроу, 1958 [3896]
Миллер, Куш, 1957 [3504]
Энтнер, Иекель, 1967 [2182]
Ветюков и др., 1959 [211]
Алиханян и др., 1972 [28]
Лукашенко и др., 1973 [636]
Метод
Точек кипения, 1669—1830 К, 5 точек
То же, 1699—1974 К, 14 точек
Эффузионный, 1053—1112 К, 5 точек
Протока, 1188—1273 К, 4 точки
То же, 1208—1348 К, 16 точек
То же, 1344—1470 К, 6 точек
Эффузионный, 1113—1149 К, 6 точек
Торзионный, 1023—-1165 К, данные
представлены в виде уравнения
Эффузионный, 1146 К, 1 точка
Торзионный, 1016—1236 К, данные представлены
в виде уравнения
Точек кипения, 1480—1600 К, 3 точки
Масс-спектрометрический, 1136 К, 1 точка
Эффузионный, 1073—1223 К, 4 точки
As#°(NaF, к, 0)
II закон
315+80
299+6
262+14
170
310+6
284+20
280+60
283+4
—
285
277 '
—
271
III закон
276,8+2,7
274,3+0,7
275,1 +0,4
277+11
280,9+0,2
284,4 ±0,6
293,7+1,1
281,7+0,2
286,5
280,0+1,2
275,3 ±0,7
279,6
279,6+3,8
вычислении весов учитывалось отклонение результатов данной работы от среднего. Погрешность
принятой величины учитывает воспроизводимость измерений, расхождения результатов измерений
в разных работах, неточности определения состава насыщенного пара и термодинамических
функций веществ.
NaF(r). Термодинамические свойства газообразного фторида натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1121.
Приведенные в табл. 46.4 молекулярные постоянные NaF приняты на основании анализа
результатов исследований микроволнового [1432, 4595], ПК-[61, 4007] и УФ-[1406] спектров этой
молекулы. В работе [2685] было изучено поведение молекулярного пучка NaF в неоднородном
электрическом поле; в работах [169, 558, 566, 567, 1497, 2700, 4009] выполнены полуэмпирические
оценки постоянных NaF. По аналогии с молекулой LiF и на основании исследования электронного
спектра [1406] принимается, что валентные возбужденные электронные состояния имеют
энергии, превышающие 40000 см"; они не рассматриваются в справочнике. Приведенные
в табл. 46.4 колебательные и вращательные постоянные NaF в состоянии X1'L+ получены Визе
и Горди [4595] при обработке результатов измерений микроволновых переходов / -»¦ /+1 (/ «^ 9)
на колебательных уровнях с v=0, 1 и 2. Соответствующие постоянные ше и шехе рассчитаны с
помощью соотношений Данхема [2126]. Они хорошо согласуются с результатами исследований ИК-
спектра [4007] (ше=536+2, шА=3,4+0,25 cm) и [61] (а>е=537+4, (оА==3,3 см).
Термодинамические функции NaF(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QSB и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dt и d2 приведены в табл. 46.5 и получены по принятым
молекулярным постоянным NaF.
Погрешности рассчитанных термодинамических функций NaF(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах дополнительный вклад в погрешность вносит неточность метода экстраполяции
потенциальной функции. Погрешности в значениях Ф°(Т) составляют 0,02, 0,05 и 0,2 Дж-К~1Х
Хмель при 298,15, 3000 и 6000 К соответственно.
332
Натрий и его соединения
Na2F2(r)
Термодинамические функции NaF(r) ранее вычислялись в предыдущих изданиях настоящего
справочника [337, 340], в таблицах JANAF [2834], справочнике Мак-Брайд и др. [3439], а также
в работе Брюэра и Брэкета [1660] (Ф°(Л> т < 2000 к)- Расхождения данных [337] и табл. 1121
достигают 3 и 7,5 Дж-К~1-моль~1 в значениях Ф°F000 К) и 5°F000 К) соответственно и
объясняются главным образом различием принятых молекулярных постоянных. Расхождения данных
[2834] и табл. 1121 не превышают 0,9 и 3,2 Дж-К^-моль в значениях Ф°(Г) и S°(T) и
объясняются тем, что авторы [2834] использовали приближенный метод расчета.
Константа равновесия реакции NaF(r)=Na(r)+F(r) вычислена с использованием значения
Arff°@)=D0(NaF) =478,548 +4,2 кДж-моль^^До 000+350 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации NaF(K). Этим величинам также соответствует
г, 0) = —293,510 + 4,1 кДж • моль.
Близкие значения E06+30 и 485+8 кДж-моль) были найдены Булевич и др. [1719] и Гур-
вичем и Рябовой [336] при изучении реакции образования NaF в водородно-воздушных пламенах.
Существенно более высокое значение E15+13 кДж-моль) получил Хам [2556] по хемилюминес-
ценции Na, образующегося по реакции Na2+F.
Na2F2(r). Термодинамические свойства газообразного дифторида динатрия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1122.
Молекулярные постоянные, использованные для расчета термодинамических функций Na2F2,
приведены в табл. 46.15. На основании расчетов для ионной модели молекулы [961, 4689], а также
Таблица 46.15. Молекулярные постоянные, а также с и р%, принятые для расчета термодинамических
функций Na2F2, Na3F3, Na3Cl2, Na3Cl3, Na2Br2, Na2I2, NaAlF4
Молекула
Na2F2
Na3F3
Na2Cl2
Na3Cl3
Na2Br2
Na2I2
NaAlF4
NaAlF4: a v
Состояние
J?3A 0 406
X^At 261
ХЫд 263
fiij 186
X1Ag 207
txAg 178
XUX 808
,=300, vio=893, vn
229
233
165
149
125
96
613
=100,
v3
271
443
186
301
160
143
350
V4
176
157
116
95
106
98
220
v12=304 CM~
v5
CM
350
419 B)
228
289 B)
186
152
378
l
371
361 B)
274
217 B)
230
199
200
_
140 B)
—
99B)
—.
—
669
_
95B)
—-
55B)
—
—
329 a
We-"»"'
Г3 • CM6
67 • 102
10 • 104
55 • 103
15 • 105
35 • 104
13 • 105
48 • 103
a
4
6
4
6
4
4
2
Pi
1
1
1
1
1
1
1
квантовомеханического расчета [4100] в справочнике принимается, что молекула Na2F2 в основном
электронном состоянии XxAff имеет плоское ромбическое строение (симметрия D2k).
Приведенное в табл. 46.15 произведение моментов инерции соответствует структурным параметрам r(Na—
—F)=2,08+0,04 А и ^ F—Na—F=95+5°, найденным Соломоником и Красновым [961, 963]1.
Погрешность значения /д/д/с составляет 1-Ю"14 г3-смв. Приведенные в табл. 46.15 значения
v4, vs. ve найдены Измаилом и др. [2793] при исследовании ИК-спектра молекул Na2F2,
изолированных в матрице из Аг, они хорошо согласуются со значениями, полученными в работах [1977,
2721, 4190]. Исключение составляет работа Мартина и Шабера [3410], в которой на основании
исследования ИК-спектра молекул Na2F2 в Ar-матрице предложено другое отнесение частот,
которое представляется ошибочным. Для ненаблюдавшихся экспериментально частот vx, v2 и v3
приняты значения, рассчитанные Соломоником и Красновым [961, 963]. Погрешности принятых
значений v4, v5( v6, оцениваются в 15 см, остальных — в 40—80 см.
Термодинамические функции Na2F2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), "A. 9), A. 10), A. 122)—A. 124), A. 128),
1 Данные Акшдина и Рамбиди [1277] для Na2F2 и других димерных молекул Ме3Х3 (Me=Na, К, Rb, Cs, X=F,
Cl, Br, I) не принимались во внимание, так как в этой работе при расшифровке электронограмм не учитывалось
содержание в паре мономерных молекул [961].
333
Na3F3(r) Глава 46
A. 130) без учета возбужденных электронных состояний. Их погрешности обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета и
составляют 4, 8 и 12 Дж-К-моль~1 в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно
(в том числе 2—4 Дж-К~1-моль~1 из-за неточности молекулярных постоянных).
Ранее термодинамические функции Na2F2(r) рассчитывались в таблицах JANAF [2834],
справочнике Мак-Брайд и др. [3439], а также в работах Сивина и др. [1975] (до 2000 К) и Краснова и др.
[569] (до 5000 К). Данные [2834] и табл. 1122 отличаются на 6—13 Дж-К~1-моль~1, так как авторы
этой работы использовали другие значения основных частот. Результаты расчетов [3439] и [569]
согласуются с табл. 1122 в пределах 3 и 1 Дж-К~1-моль~1 в значениях Ф°(Г). В работе [1975],
по-видимому, допущена ошибка при вычислении Ф°(Т), хотя значения S°(T) согласуются с
данными настоящего справочника в пределах 2 Дж-К~1-моль.
Константа равновесия реакции Na2F2(r)=2Na(r)+2F(r) вычислена с использованием значения
&rH°(Q)=1199,076 +9,1 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A///°(Na2F2, г, 0) = —829 + 9 кДж • моль.
Принятая величина основана на результатах исследований равновесия реакции NaF(K)+
-j-NaF(r)=Na2F2(r), выполненных эффузионным методом Мюллером и Кушем [3504] A146 К,
1 точка, ДД @)=50,9 кДж-моль (III закон)) и Эйзенштадтом и др. [2166] A113—1149 К, 6
точек, Ari/°@)=44-f-40 кДж-моль (II закон) и 42,5+0,6 кДж-моль (III закон)), а также масс-
спектрометрическим методом Алиханяном и др. [28] A136 К, 1 точка, ДД°@)=35,6 кДж-моль
(III закон)). Для дальнейших расчетов принимается значение Дг//°@)=39+8 кДж-моль,
являющееся средним из работ [28] и [2166]. Его погрешность оценена с учетом расхождения
результатов измерений в этих работах и неточности термодинамических функций. Расчеты по
температурной зависимости интенсивности ионных токов, измеренных в работах [2166, 3504], приводят
к значениям ДгЯ°@) равным 97 и 46 кДж-моль (II закон) соответственно.
Na3F3(r).iТермодинамические свойства газообразного трифторида тринатрия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1123.
Молекулярные постоянные, использованные для расчета термодинамических функций
Na3F3, приведены в табл. 46.15. Структура молекулы Na3F3 экспериментально не исследовалась.
В справочнике для молекулы Na3F3 в основном электронном состоянии Х1А1 на основании
результатов расчетов по ионной модели [4689] принята плоская гексагональная конфигурация
симметрии D%k. Приведенное в табл. 46.15 произведение моментов инерции соответствует значению
r(Na—F)=l,88+0,1 А, найденному Уэлчем и др. [4689]. Погрешность значения IaIbIc
составляет 5-Ю13 г3-см6. Приведенные в табл. 46.15 значения частот v4, v5, ve, v7 найдены Измаилом
и др. [2793] при исследовании ИК-спектра молекул Na8F3, изолированных в матрице из Аг х.
Для частот колебаний, не активных в ИК-спектре, в табл. 46.15 приведены значения,
рассчитанные Соломоником и Даниловой [961, 962] по оцененным значениям силовых постоянных.
Погрешности принятых частот v4—v7 оцениваются в 15—20 см, остальных — в 20% от их величин.
Термодинамические функции Na3F3(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Их погрешности обусловлены неточностью принятых молекулярных постоянных, а также
приближенным характером расчета и составляют 6, 18, 25 Дж-К~1-моль в значениях Ф°(Т)
при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Na3F3(r) рассчитывались Соломоником и Даниловой [962]
(до 5000 К). Расхождения результатов этого расчета и данных табл. 1123 не превышают
3,5 Дж-К~1-моль~1 в значениях Ф°(Г) и объясняются различием в межатомном расстоянии
r(Na—F), принятом в [962] по величине r(Na—F) в молекуле Na2F2.
Константа равновесия реакции Na3F3(r)=3Na(r)+3F(r) вычислена с использованием значения
ДД°@) = 1896,114+12 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д,Я °(Na,F8, г, 0) = -1341 ± 12 кДж • моль.
1 Расчет частот колебаний димера Na—F—Na—F с линейной структурой, выполненный Соломоником [961], но
подтвердил сделанное авторами [2793] отнесение полос при 419, 361, 157 и 140 см к такому димеру и позволил
отнести эти полосы к молекуле Na3F3.
334
Натрий и его соединения N аСЦк, ж)
Эта величина основана на масс-спектрометрическом исследовании равновесия реакции 2NaF(K)+
+NaF(r)=Na3F3(r), выполненном Алиханяном и др. [28] A136 К, 1 точка, Дг#°@)=101+ 12кДжХ
Хмоль). Основным источником ее погрешности является неточность термодинамических
функций. Менее точное значение энтальпии этой реакции G9 кДж-моль) было получено Миллером
и Кушем [3504] эффузионным методом с селектором скоростей A146 К, 1 точка). Температурной
зависимости ионных токов, измеренных в работе [3504], соответствует значение ДгЯ°@) =
=40 кДж-моль.
NaCl(K, ж). Термодинамические свойства кристаллического и жидкого хлорида натрия
в стандартном состоянии при температурах 100—3400 К приведены в табл. 1124.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние NaCl(K) в интервале 0—1074 К принимается
кубическая гранецентрированная модификация х.
Теплоемкость NaCl при Т < 298,15 К измерена в работах Корефа [3073] (85—273 К), Нерн-
ста [3642] B5—84 К), Бренстеда [1683] B73—293 К), Мак-Грау [3457] (95—245 К), Клузиуса и др.
[1883] A1—277 К), Мартина [3400] B-30 К), Моррисона и сотр. [3568, 3569] B,5—20 и 12—
270 К) и Баррона и др. [1404] C—267 К). Наиболее достоверными являются измерения Баррона
и др. [1404]. С данными [1404] хорошо согласуются результаты измерений Мартина [3400] и
Моррисона и др. [3568, 3569]. Результаты измерений Клузиуса и др. [1883] в интервале 16—
210 К на 3% выше данных Баррона и др. [1404], далее кривые теплоемкости пересекаются, и
С° B98,15 К) по данным [1883] становится на 1,4% ниже величины, полученной в работе [1404].
Результаты определений теплоемкости в старых работах [1683, 3073, 3642] получены с большим
разбросом и имеют низкую точность. Термодинамические функции NaCl(K) при Т <^ 298,15 К
рассчитаны по данным [1404]. Погрешности принятых значений ?°B98,15 К) и Я°B98,15 К)—
—Н°@), приведенных в табл. 46.1, оцениваются в 0,2 Дж-К~1-моль~1 и 0,02 кДж-моль
соответственно.
При Т > 298,15 К для теплоемкости NaCl(K) принято уравнение, выведенное совместной
обработкой результатов измерений теплоемкости и энтальпии NaCl в работах Доусона и др. [2007]
F72—1128 К), Лидбеттера и Сеттатри [3211] C04-773 К) и Ляшенко [637] D55—1268 К). При
обработке погрешности данных [2007] и [3211] оценивались в 0,5%, данных [637] — в 2%. В
старых работах Плато [3856] B93—1048 и 1112—1205 К), Магнуса [3340] B90—372, 291-541, 291—
829 и 291—1036 К), Рота и Бертрама [4064] B93—1098 К) приведены результаты единичных
определений средней теплоемкости NaCl в различных температурных интервалах, их точность
невелика.
Температура плавления NaCl A074+1 К) принята по согласующимся между собой
измерениям Филлипса и др. [3827] A074+0,5 К), Доусона и др. [2007] A073,6+0,8 К), Костенски
и Малиновского [3084] A073,8+1,0 К), Таллона и Робинсона [4466] A074+0,1 К); менее точные
значения получены в работах [939, 1567, 1599, 1654, 2409, 2545, 2566, 3371, 4663] A072—1077 К).
Энтальпия плавления B8,2+0,2 кДж-моль) принята по согласующимся между собой данным
[637] B8,37 кДж-моль-1), [2007] B8,28+0,13 кДж-моль) и [2137] B7,99 кДж-моль).
Измерения [4064] приводят к завышенному значению C1,0 кДж-моль), а результаты криоскопи-
ческих определений энтальпии плавления [2573] B7,45 кДж-моль) и [3982] B7,41 кДж-моль)
явно занижены.
Теплоемкость NaCl^) F7,9+1,0 Дж-К-моль) получена усреднением хорошо
согласующихся между собой данных [637] F8,1 Дж-К^-моль, 1123-1268 К), [3856] F6,6 Дж-К^Х
Хмоль, 1112—1205 К) и [3594] F9,0 Дж-К^-моль, в стоградусном интервале вблизи
температуры плавления). Данные [2007], по-видимому, ошибочны, так как приводят к резкому
возрастанию теплоемкости в интервале 1078—1279 К (от 73 до 87 Дж-К^-моль).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
0,6, 2 и 5 Дж-К~1-моль~1. Расхождение между термодинамическими функциями NaCl(K, ж),
приведенными в табл. 1124 и в справочниках [337] (Т < 3400 К), [1395] (Т < 1700 К), [2834]
(Т < 3500 К), [2960] (Т < 1700 К), не превышают 0,5 Дж-К^-моль-1 в значениях Ф°(Г) во всем
интервале температур.
1 Прп высоких давлениях известны кубическая модификация (структурный тип CsCl), стабильная при 298 К
и р > 290 кбар [3840], а также металлическая фаза NaCl, образующаяся при р > 1000 кбар [203].
335
NaCl(K, ж)
Глава 46
Принятая в справочнике энтальпия образования
A//°(NaCl, к, 298,15 К) = —411,26 + 0,12 кДж • моль
основана на измерениях энтальпии растворения NaCl(K) в воде, представленных в табл. 46.16.
В таблице приведены только наиболее полные и точные измерения, выполненные при 298,15 К.
Таблица 46.16. Результаты измерений \aqS°(NaCl, к->ооН2О, 298,15 К) (в кДж ¦ моль)
Авторы
Рандал, Биссон, 1920 [3920]
Вгоот, Ланге, 1925 [4787]
Липсет и др., 1927 [3278]
Коэн, Кой, 1928 [1892]
Бенсон, Бенсон, 1955 [1473]
Бенсон и др., 1956 [1474]
Крисе, Кобл, 1961 [1964]
Базлова и др., 1965 [59]
Цветков, Рабинович, 1969 [1097]
Штерн, Куллук, 1969 [4392]
Сергеева и др., 1970 [923]
Дьяконова и др., 1970 [393]
Штерн и др., 1974 [4393]
Абросимов и др., 1977 [2, 3]
Дэдгер, Тахериан, 1977 [1983]
Штерн и др., 1979 [4391]
Воробьев и др., 1980 [245]
Таневска-Осинска, 1980 [4472]
Конечная концентрация
107—430 Н2О, 7 измерений
10—100 Н2О, 14 измерений
10—800 Н2О, 20 измерений
100 Н2О, 4 измерения
43—1110 Н2О, 16 измерений
111 Н2О; 3 измерения
2775—55510 Н2О, 17 измерений
50—1600 Н2О, 6 измерений
250 Н2О, 1 измерение
3700—7930 Н2О, 6 измерений
631 Н2О
1590 Н20, 10 измерений
3114 Н2О, 10 измерений
48 и 278 Н2О, 2 измерения
1850—5550 Н2О, 5 измерений
1110—2780 Н2О, 8 измерений
—
111—5551 Н2О, 14 измерений
ДагЯ°(аоН2О, 298,15 К)
3,887+0,021
3,895+0,042
3,862+0,021
3,828 ±0,021
3,883+0,013
3,879 ±0,042
3,879 ±0,042
3,923 ±0,040
3,868 ±0,080
3,954 ±0,063
3,895+0,040
3,770 ±0,060
3,866 ±0,040
3,951 ±0,040
3,831+0,040
3,912 ±0,042
3,883 ±0,043
3,789 ±0,040
Помимо них, в литературе известны измерения [24, 252, 482, 747, 899, 900, 902, 1347, 1354, 1358,
1450, 1534, 1907, 2253, 2699, 2885, 3176, 3179, 3280, 3732, 3838, 4228, 4313, 4511, 4516, 4742], многие
из которых выполнены на достаточно высоком уровне, но все же уступающие по точности
приведенным в таблице работам. Менее точные измерения были выполнены в работах [498, 689, 1297,
1528, 1532, 1680, 1838, 2224, 2453, 2699, 2889, 3059, 3226, 3732, 3995, 4136, 4206, 4373, 4374, 4380,
4391, 4392, 4471, 4472, 4590, 4592, 4819]. Анализ исследований, выполненных до 1965 г., проведен
в обзорной работе Паркер [3771].
Результаты приведенных в таблице исследований согласуются в пределах приписанных им
погрешностей. В справочнике для последующих расчетов выбрано средневзвешенное значение
Дй2Я°(соН20; 298,15 К)=3,878+0,020 кДж-моль. Его погрешность оценена с учетом
воспроизводимости измерений и расхождений измерений в разных работах.
Давление пара в реакции NaCl(K, ж)=КаС1(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Лг#°@) = As#°(NaCl, к, 0) = 230,7 + 3,0 кДж • моль.
Эта величина основана на измерениях давления и состава насыщенного пара NaCl(K, ж). В
состав насыщенного пара NaCl при 7тот=1074 К по принятым в настоящем справочнике данным
входит примерно 32 мол.% димерных и 0,5 мол.% тримерных молекул. Методика выбора
термохимических величин для компонентов насыщенного пара галогенидов щелочных металлов
изложена в Приложении 8.
Результаты расчетов энтальпии сублимации NaCl в виде мономерных молекул представлены
в табл. 46.17. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятое значение является средневзвешенным из приведенных в таблице данных, за
исключением выпадающих работ [1416, 2482, 2919, 3155, 3352, 3504]. При вычислении весов
учитывалось отклонение результата данной работы от среднего. Погрешность принятой величины
336
Натрий и его соединения
NaCl(r)
Таблица 48.17. Результаты определений As?T°(NaCl, к, 0) (в кДж-моль)
Авторы
Вартенберг, Альбрехт, 1921 [4656]
Руфф, Мугдан, 1921 [4094]
Майер, 1925 [3352]
Фиок, Родебуш, 1926 [2258]
Хоряба, Баба, 1928 [2711]
Грейнер, Елинек, 1933 [2482]
Руфф, Буше, 1934 [4092]
Мейер, Винтнер, 1938 [3433]
Кангро, Викинг, 1938 [2919]
Нива, 1938 [3665]
Нарышкин, 1939 [707]
Цимм, Мейер, 1944 [4823]
Бартон, Блюм, 1956 [1416]
Несмеянов, Сазонов, 1957 [718]
Миллер, Куш, 1957 [3504]
Бартон, Блюм, 1959 [1417]
Милн, Клейн, 1960 [3510]
Акишин и др., 1959 [8], Горохов,
1972 [309]
Мургулеску, Топор, 1966 [3595]
Глазов, Нарышкин, 1968 [287]
Лэствр, Саморджей, 1968 [3234]
Фэтер, Сирей, 1971 [2226]
Эвинг, Штерн, 1974 [2199]
Эвинг, Штерн, 1975 [2200]
Эмонс и др., 1976 [2173]
Шатова и др., 1979 [1145] .
Вагнер, Шефер, 1979 [5067]
Кванде и др., 1979 [3155]
Метод
Точек кипения, 1429—1703 К, 12 точек
То же, 1433—1723 К, 8 точек
Статический, 1020—1420 К, 11 точек
То же, 1250—1429 К, 8 точек
То же, 1073—1514 К, 10 точек
Протока, 1453 К, 2 точки
Точек кипения, 1485—1652 К, 5 точек
Эффузионный, 890—944 К, 6 точек
Протока, 1133—1263 К, 3 точки
Эффузионный, 883—973 К, 10 точек
Протока, 1028—1188 К, 5 точек
Эффузионный, 760—897 К, 11 точек
Точек кипения, 1330—1538 К а
Эффузионный, 743—948 К, 11 точек
То же, 920 К, 1 точка
Точек кипения, 1290—1416 К, 3 точки
Масс-спектрометрический, Тср=930 К а
То же, Гср=1000 К а
Статический, 1323—1442 К, 8 точек
Эффузионный, 1073—1173 К а
Испарение с поверхности, 821—919 К, 13 точек
Масс-спектрометрический, Гср=800 К а
То же, Гор=937 К а
Эффузионный, 860—966 К, 28 точек
Испарение с поверхности, 7'0р=873 К а
Эффузионный, 1073—1243 К а
Точек кипения, 1173—1408 К, 6 точек
Масс-спектрометрический, 742—916 К а
Истечение через капилляр, 1285—1378 К, 9 точек
а Данные представлены в виде уравнения.
As#°(NaCl, к, 0)
II закон
257 ±15
252+20
227 ±10
238 ±2
206 ±4
—
252 ±10
208 ±14
214+80
220 ±3
199 ±70
232+4
246 ±18
222 ±7
—
234 ±10
230
231 ±5
242 ±6
232 ±6
201 ±40
240 ±8
229 ±7
236 ±4
234
232
207 ±40
234
228 ±17
III закон
228,8 ±1,6
228,9 ±1,6
223,8 ±0,3
231,4 ±0,3
225,5 ±1,6
222,2 ±1,1
229,2 ±27
230,9 ±0,6
225,8+2,0
231,3 ±0,3
228,7 ±4,1
231,0 ±0,5
239,1 ±1,3
230,9 ±0,6
227,4
230,6 ±0,5
—
230,3 ±0,3
230,7+0,5
231,2+1,2
—
—
230,7 ±0,1
—
231,2 ±1,0
233,8 ±2,7
—
236,2 + 0,4
учитывает воспроизводимость измерений, расхождения измерений в разных работах, неточность
термодинамических функций и определения состава насыщенного пара.
NaCl(r). Термодинамические свойства газообразного хлорида натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1125.
Приведенные в табл. 46.4 молекулярные постоянные NaCl приняты на основании анализа
результатов исследований микроволнового [1874, 1875, 1876, 2700, 4396] и УФ- [3239] спектров
этой молекулы. Результаты исследования электронного спектра [3239] и аналогия с молекулой
LiF свидетельствуют, что валентные возбужденные электронные состояния NaCl имеют энергию
свыше 34 000 см и могут не рассматриваться в справочнике.
Приведенные в табл. 46.4 колебательные и вращательные постоянные NaCl в состоянии X1!,*
получены Клаузером и Горди [1876] обработкой результатов измерений микроволновых
переходов / -> /+1 (/ ^ 24) на колебательных.уровнях у=0,1 и 2. Соответствующие постоянные со„
и и>ехе были рассчитаны с помощью соотношений Данхема. Они удовлетворительно согласуются
с результатами исследования ИК-спектра [3984] (а>е=366+4, соехе=2,05 см).
Термодинамические функции NaGl(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A.10), A.93)—A.95). Значения QBB и ее производные вычислялись без учета возбужденных состояний
337
Na2Cl2(r) • Глава 46
по методике, изложенной в Приложении 7. Использованные в расчете коэффициенты
потенциальной функции d0, dx и d2 приведены в табл. 46.5 и получены по принятым молекулярным
постоянным NaCl. Расчет выполнен для природной изотопической смеси изотопов хлора.
Погрешности рассчитанных термодинамических функций NaCl(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопического состава хлорида натрия. При более высоких температурах
дополнительный вклад в погрешность вносит неточность экстраполяции потенциальной функции, а также
пренебрежение возбужденными электронными состояниями. Погрешности в значениях Ф°(Т),
составляют 0,02, 0,05 и 0,3 Дж-К-^моль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции NaCl(r) ранее вычислялись в предыдущем издании настоящего-
справочника [337], в таблицах JANAF [2834], справочнике Мак-Брайд и др. [3439], а также
в работах Брюэра и Брэкхет [1660] (Ф°(Т), Т < 2000 К), Уилкинса [4728] (Т < 6000 К), Раиса
и Клемперера [3985] (Т < 2000 К). Расхождения данных [337], [2834], [3439], [4728] и табл. 1125
не превышают 0,8—2,2 Дж-К~1-моль~1 в значениях Ф°(Г) и объясняются тем, что в этих работах
использовались приближенные методы расчета.
Константа равновесия реакции NaCl(r)=Na(r)+Cl(r) вычислена с использованием значения
&r#°@)=D0(NaCl)=407,493+3,l кДж• моль-1 « 34 100+260 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации NaCl(K). Этим величинам также
соответствует
t\fH°{Na.Cl, г, 0) = —180,110 ±3,0 кДж • моль.
Практически совпадающие, но менее точные значения энергии диссоциации D12+13 и 408 +
+8 кДж-моль) были получены в спектрофотометрических исследованиях Гурвича и Вейц [332,
333] и Булевич и др. [1719], а также в работе Су и Рейли [4427] методом фотофрагментарной
спектроскопии D08+8 кДж-моль).'
Na2Cl2(r). Термодинамические свойства газообразного дихлорида динатрия в стандартном^со-
стоянии в интервале температур 100—6000 К приведены в табл. 1126.
Молекулярные постоянные, использованные для расчета термодинамических функций Na2Cla,
приведены в табл. 46.15. На основании электронографического исследования Мики и др. [35011
для молекулы Na2Cla в основном электронном состоянии ХгАд принята плоская ромбическая
структура (симметрия D2h). Приведенное в табл. 46.15 произведение моментов инерции
соответствует структурным параметрам r(Na—С1)=2,515 +0,017 А и -4С1—Na—01=101,4+0,8°,.
найденным в работе [3501]. Погрешность величины IaIbIc оценивается в 5-10~114г3-см6.
Приведенные в табл. 46.15 частоты v4—ve найдены Измаилом и др. [2794] при исследовании ИК-спектра
молекул Na2Gl2, изолированных в матрице из Аг, их величины хорошо согласуются со значениями,
полученными Мартином и Шабером [3410]. Для не наблюдавшихся экспериментально частот
Vl—v3 приняты значения, рассчитанные Соломоником и Красновым [961, 963] с использованием
ионной модели молекулы. Погрешности принятых значений v4—v6 оцениваются в 15-см,
остальных частот — до 20% от их величин.
Термодинамические функции NaaCl2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—1. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Их погрешности при низких температурах
обусловлены в основном неточностью принятых молекулярных постоянных, а при высоких —
приближенным характером расчета и составляют 4, 10, 15 Дж-К-моль~1 в значениях Ф°(Т) при 298,15,.
3000 и 6000 К соответственно.
Ранее термодинамические функции Na2Cl2(r) рассчитывались в таблицах JANAF [2834],
справочнике Мак-Брайд и др. [3439], а также в работе Краснова и др. [569] (до 3000 К). Все
расчеты выполнены по оцененным значениям молекулярных постоянных. Расхождения в
значениях Ф°(Т), рассчитанных в работах [569, 2834, 3439] и приведенных в табл. 1126, не превышают
3,5 Дж-К~1«моль и объясняются различием использованных в расчетах постоянных Na2Cl2.
Константа равновесия реакции Na2Cl2(r)=2Na(r)+2Cl(r) вычислена с использованием
значения Дг^Г°@)=1015,766+7,1 кДж-моль'1, соответствующего принятой в справочнике энтальпик
образования
bfH°(Na2Cl2, г, 0) = —561 + 7 кДж • моль.
338
Натрий и его соединения
Na3CI8(r)
Эта величина основана на результатах исследований равновесия реакции NaCl(K, ж)+
+NaCl(r)=Na2Cl2(r), приведенных в табл. 46.18. Для дальнейших расчетов использовалось зна-
Таблица 46.18. Результаты определений энтальпии реакции NaCI(K, ж) -f NaCl(r) = Na2CI2(r
(в кД;к • моль)
Авторы
Метод
ДГЯ°(О) ]
II закон
III закон !
Миллер, Куш, 1957 [3504] Эффузионный с селектором скоростей, 920 К, — 29,2
1 точка ;
Эйзенштадт, Родберг и др., 1959 [2165, То же, 932—972 К а 26+2 32,0+0,2 '.
4073] I
Берковиц, Чапка, 1958 [1501] Маес-спектрометрический, ~930 К, 1 точка — 30,5+3,0
Чапка, 1959 [1851] То же, Гср=800 Ка 47 ±13 —
Бартон, Блум, 1959 [1417] Метод протока и точки кипения, 1290—1416 К, 21 ±30 20,1 ±0,3
3 точки
Милн, Клейн, 1960 [3510] Масс-спектрометрический, Гср=930 Ка 39 —
Лестер, Саморджей, 1968 [3234] То же, Гср=800 К а 39 ±8 —
Мургулеску, Топор, 1966 [3595] Метод протока и статический, 1373—1473 К, —53 ±95 19,6 ±3,0
5 точек
Фетер, Сирей, 1971 [2226] Масс-спектрометрический, 968 К, 1 точка — 29,1
Акишин и др., 1959 [8]; Горохов, Масс-спектрометрический с двойной эффузион- 21+8 31,1
1972 [309] ной камерой, 834—903 К а
Вагнер, Шефер, 1979 [5067] Масс-спектрометрический, 742—916 Ка 28±5 29,3
а Данные представлены в виде уравнения.
чение ДгЯ°@)=30±6 кДж-моль, среднее из приведенных в таблице данных, за исключением
выпадающих измерений [1417, 3595]. Его погрешность определяется в первую очередь
неточностью термодинамических функций Na2Cla(r) (около 6 кДж-моль для измерений при 1000 К), i
В работах [309, 1417, 1851, 1995, 2165, 3234, 3504, 3510, 5067] приведены данные, позволяю-*
щие найти для реакции Na2Cl2(r)=2NaCl(r) средние значения энтальпии 209+20 кДж-моль
(II закон) и 213+10 кДж-моль (III закон). В пределах погрешностей^эти данные согласуются
со значением 199+7 кДж-моль, вычисленным на основании принятых в|[справочнике величин.
¦ ..?
Na3Cl3(r). Термодинамические свойства газообразного трихлоридаГтринатрия в-'стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1127. <• ' Г"^'
Молекулярные постоянные, использованные для расчета термодинамических функций Na3Cl3,
представлены в табл. 46.15. Структура и спектр молекулы Na3Cl3 экспериментально не
исследовались. По аналогии с Li3F3 для молекулы NasCl3 в основном состоянии XxAlt принята плоская
гексагональная конфигурация (симметрия D3h). Приведенное в табл. 46.15 произведение моментов
инерции соответствует r(Na—С1)=2,51+0,1 А, рассчитанному Уэлчем и др. [4689] с
использованием ионной модели молекулы. Погрешность значения /аЛУс оценивается в 5-1012 г3«см*.
Для основных частот молекулы Na3Cl3 приняты значения, рассчитанные Соломоником и
Даниловой [961, 962] по оцененным силовым постоянным. Погрешности принятых частот могут'дости-
гать 20—25% от их величины. |"те
Термодинамические функции Na3Cl3(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A.10), A. 122)—A. 124)/A. 128),
A. 130) без учета возбужденных состояний. Их погрешности обусловлены неточностью принятых
молекулярных постоянных, а также приближенным характером расчета и составляют 10, 20,
30 Дж-К^-моль-1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно (в том числе
~10 Дж-К-моль из-за неточности молекулярных постоянных).
Ранее термодинамические функции Na3Cl3(r) рассчитывались Соломоником и Даниловой
[962] (до 5000 К). Результаты расчета [962] согласуются с данными табл. 1127 в пределах
0,5 Дж-К^-моль в значениях Ф°(Г).
339
NaBr(K, ж)
Глава 46
Константа равновесия реакции Na3Gl3(r)=3Na(r)+3Cl(r) вычислена с использованием
значения Дг#°@)=1591,149+16 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
ЬУН°(Ш3С13, г, 0) = —909 ± 16 кДж • моль.
Эта величина основана на приведенных в табл. 46,19 результатах исследований равновесия
реакции 2NaCl(K, ffl)+NaCl(r)=Na3Cl3(r). Во всех работах измерения абсолютных давлений
выполнялись только при одной температуре. Для дальнейших расчетов принимается среднее
значение Дг?Г°@)=93+16 кДж-моль. Основным источником его погрешности является
неточность термодинамических функций Na3Gl3(r).
Таблица 46.19. Результаты определения энтальпии реакции 2NaCI(K, ж) -j- NaCl(r) = NasCI3(r)
(в кДж-моль)
Авторы
Метод
ДгЯ°@)
II закон
III закон
Берковиц, Чапка, 1958 [1501] Масс-спектрометрический, 930 К, 1 точка — 91,7
Акишин и др., 1959 [8], Горохов, То же, 834—1000 Ка 83+12 98,2
1972 [309]
Фетер, Сирей, 1971 [2226] То же, Гср=937 Ка 60 ±20 93,8
Вагнер, Шефер, 1979 [5067] То же, 742-916 Ка 53 86,5
а Данные представлены в виде уравнения.
NaBr(K, ж). Термодинамические свойства кристаллического и жидкого бромида натрия
в стандартном состоянии при температурах 100—3400 К приведены в табл. 1128.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние NaBr(K) в интервале 0—1020 К
принимается кубическая модификация (структурный тип NaCl) 1.
При Т ^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работах Гарднера и Тейлора [2362] G—302 К) и Бёрча и др. [1563] B,7—19,4 К).
При Г<^7К использованы данные [1563], в интервале 7—20 К значения теплоемкости получены
усреднением значений [2362] и [1563], а при Т > 20 К использованы данные [2362]. Значения
?°B98,15 К) и #°B98,15 К)—#°@), приведенные в табл. 46.1, имеют погрешности 0,2 Дж-К"хХ
Хмоль и 0,02 кДж-моль соответственно. Единичные измерения теплоемкости NaBr в работах
Корефа [3073] (82—273 К) и Брёнстеда [1683] B73—293 К) выполнены с недостаточной
точностью и не учитывались.
При Т > 298,15 К для теплоемкости NaBr(K) принято уравнение, полученное обработкой
данных по энтальпии, приведенных в работе Гарднера и Тейлора [2363] C74—1195 К).
Погрешность этих данных оценивается в 0,3%. Результаты измерений энтальпии в интервалах 290—372
и 290—543 К, выполненных Магнусом [3340], на 1% выше данных [2363].
Температура плавления NaBr A020+2 К) принята по согласующимся между собой
измерениям Бредига и др. [1654], A020+0,5 К), Гарднера и Тешщра [2363] A020 К), Маргеритиса
и др. [3386] A020 К); менее точные значения получены в работах [5, 461, 1577, 1711, 1867, 3371,
4196, 4663, 47911. Энтальпия плавления B6,23+0,20 нДж-моль) найдена в работе [2363].
С принятым значением прекрасно согласуется величина B6,11 кДж-моль), полученная Двор-
киным и Бредигом [2137].
Теплоемкость NaBr^) F8,0+1,5 Дж-К~1-моль~1) принята по результатам девяти измерений
энтальпии, проведенных Гарднером и Тейлором [2363] A020—1195 К). Мало отличающееся
значение F7,2 Дж-К~1>моль~1) получено в работе Мургулеску и Теле [3594], выполненной
в узком температурном интервале вблизи точки плавления.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,2,
0,4, 2 и 5 Дж-К~1-моль~1 соответственно. Различие между термодинамическими функциями,
Кубическая фаза высокого давления (структурный тип CsCl) стабильна при 448 К и р > 11,5 кбар [3S48].
340
Натрий и его соединения
NaBr(K, ж)
приведенными в табл. 1128 и справочнике JANAF [2834] (Т <[ 2500 К), достигают 1,4 Дж-К~1Х
Хмоль в значениях Ф°(Г) и обусловлены тем, что в настоящем справочнике учтены
экспериментальные данные [2363]. Расхождения с рекомендациями [1395] (Т <^ 1600 К) и [2960] (Г ^
^ 550 К) не превышают 0,6 Дж-К^-моль.
Принятая в справочнике энтальпия образования
Д^^КаВг, к, 298,15 К) = —361,16 + 0,20 кДж • моль
основана на измерениях энтальпии растворения NaBr(K) в воде, представленных в табл. 46.20.
Таблица 46.20. Результаты измерений Aavfl"°(NaBr, к-> ооНаО, 298,15К) (в кДж-моль)
Авторы
Томсен, 1883 [4511]
Вюст, Ланге, 1925 [4787]
Ланге, Дюрр, 1926 [3176]
Чипман и др., 1929 [1838]
Аскю и др., 1934 [1354]
Сланский, 1940 [4313]
Уоллес, 1949 [4642]
Фучс, Хаган, 1973 [2342]
Джоли, Перашон, 1978 [2885]
Воробьев и др., 1980_[245]
Конечная концентрация, температура
измерений
200 НаО, 290 и 293 К
6,4-54,4 Н2О, 298,15 К, 15 измерений
100 Н2О, 298,15 К, 4 измерения
40—500 Н2О, 298,55 К, 15 измерений
5550 Н2О, 293,15 К, 1 измерение
298,15 К
925—1850 Н2О, 298,15 К, 6 измерений
55 510-111000 Н2О, 298,15 К, 6 измерений
2920 Н2О, 298,15 К, 1 измерение
298,15 К
ДагЯ°(гаН2О, 298,15 К)
—0,33
-0,623+0,060
—1,04
—0,19+0,80
—0,75
-0,17
—0,602+0,060
-0,552+0,040
-0,662 ±0,040
—0,640+0,035
Помимо перечисленных в таблице работ, измерения энтальпии растворения NaBr(K) проводились
в ныне устаревших работах [1530, 2225, 4592]. В наиболее совершенных работах [245, 2342,
2885, 4642] получены достаточно хорошо согласующиеся значения. Придавая больший вес
работам Воробьева и др. [245] и Джоли и Перашона [2885], для дальнейших расчетов принимается
значение AaqH°(coH20; 298,15 К) = — 0,640+0,040 нДж-моль.
Давление пара в реакции NaBr(K, ж)=^Вг(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дгй°@) = A,#°(NaBr, к, 0) = 217,0 + 3,0 кДж • моль.
Эта величина основана на измерениях давления и состава насыщенного пара NaBr(K, ж).
В состав насыщенного пара NaBr при Гт=1020 К по принятым в настоящем справочнике данным
входит около 28 мол.% димера. Методика выбора термохимических величин для компонентов
насыщенного пара галогенидов щелочных металлов изложена в Приложении 8.
Таблица 46.21. Результаты определений A,-Hro(NaBr, к, 0) (в кДж-моль)
Авторы
Вартенберг, Альбрехт, 1921 [4656]
Руфф, Мугдан, 1921 [4094]
Мейер, Винтнер, 1938 [3433]
Нива, 1938 [3665]
Коджин, Кимбелл, 1948 [1890]
Блум и др., 1958 [1584]
Мургулеску, Топор, 1966 [3595]
Метод
Точек кипения, 1411—1667 К, 9 точек
Точек кипения, 1338—1683 К, 8 точек
Эффузионный, 891—930 К, 6 точек
То же, 833—933 К, 11 точек
То же, 758—911 К, данные представлены в виде
уравнения
Точек кипения, 1143—1403 К, данные
представлены в виде уравнения
Статический, 1273—1444 К, 10 точек ,;
Д,Я°(МаВг, к, 0)
II закон
226 ±5
225 ±13
223+70
213 ±2
236 ±4
254
223 ±3
III закон
215,6 ±0,5
215,8+0,9
219,5 ±0,9
217,0 ±0,1
221,6 ±3,0
216,2 ±4,4
216,0+0,3
341
NaBr(r), Ка2Вг2(г) Глава 46
Результаты расчетов энтальпии сублимации NaBr в виде мономерных молекул представлены
в табл. 46.21. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятое значение является средневзвешенным из приведенных в таблице данных. При
вычислении весов учитывалось отклонение результата измерений в данной работе от среднего.
Погрешность принятой величины учитывает воспроизводимость измерений, расхождения
результатов измерений в разных работах, неточности термодинамических функций и определения
состава насыщенного пара.
NaBr(r). Термодинамические свойства газообразного бромида натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1129.
Приведенные в табл. 46.4 молекулярные постоянные NaBr приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3984] и УФ- [3239] спектров этой
молекулы. По аналогии с молекулой LiF и на основании исследования электронного спектра
[3239] принимается, что валентные возбужденные электронные состояния NaBr имеют энергии,
превышающие 30 000 см'1 и могут не рассматриваться в справочнике.
Приведенные в табл. 46.4 колебательные и вращательные постоянные NaBr в состоянии ХгЕ +
получены Раском и Горди [4101] обработкой результатов измерений микроволновых переходов
/ -> /+1 (/ ^ 21) на колебательных уровнях у=0,1 и 2. Постоянные и>е и шехе были получены
не непосредственно по экспериментальным данным, а рассчитаны с помощью соотношений Дан-
хема. Они удовлетворительно согласуются с результатами исследования ИК-спектра [3984]
(сов=302,1+4, 0)^=1,50 см).
Термодинамические функции NaBr(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QBB и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 46.5 и получены по принятым
молекулярным постоянным Na79Br. Расчет выполнен для природной изотопической смеси изотопов
брома.
Погрешности рассчитанных термодинамических функций NaBr(r) при температурах до 3000 К
обусловлены главным образом неточностью физических постоянных. При более высоких
температурах дополнительный вклад в погрешность вносят неточность способа экстраполяции
потенциальной функции, а также пренебрежение возбужденными электронными состояниями.
Погрешности в значениях Ф°(Т) составляют 0,02, 0,05 и 0,4 Дж-К~1-моль~1 при 298,15, 3000 и
и 6000 К соответственно.
Термодинамические функции NaBr(r) ранее вычислялись в таблицах JANAF [2834], а также
в работах Уилкинса [4728] (Т < 6000 К), Раиса и Клемперера [3985] (Т < 2000 К), Брюэра
и Брэкета [1660] (Ф°(Г), Т < 2000 К). Расхождения данных [2834], [4728] и табл. 1129 не
превышают 0,5 и 1,5 Дж-К^-моль в значениях Ф°(Г) и S°(T) соответственной объясняются тем,
что в справочнике [2834] и работе [4728] использовался приближенный метод расчета.
Константа равновесия реакции NaBr(r)=Na(r)+Br(r) вычислена с использованием значения
Ar#°@)=D0(NaBr)=362,716+3,1 кДж• моль~х «* 30 320+260 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации NaBr(K). Этим величинам также
соответствует
A^^NaBr, г, 0) = —137,030 + 3,0 кДж • моль.
Практически совпадающие значения C63,8+2,0 и 359+4 кДж-моль) были получены
методом фрагментарной спектроскопии в работах Ван Веена и др. [4589] и Су и Райли [4427]. Бойтлер
и Леви [1539] при исследовании флуктуационных полос молекулы NaBr в поглощении и
испускании нашли близкое значение C56+8 кДж-моль). Булевич и др. [1719] получили значение
371+8 кДж-моль на основании спектрофотометрического изучения равновесия реакции
образования NaBr в пламенах.
Na2Br2(r). Термодинамические свойства газообразного дибромида динатрия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. ИЗО.
Молекулярные постоянные, использованные для расчета термодинамических функций Na2Br2,
приведены в табл. 46.15. На основании теоретических расчетов [961, 4689], а также по аналогии
с молекулой Na2Gl2 в справочнике принимается, что молекула Na2Br2 в основном состоянии ^Av
имеет плоскую ромбическую структуру (симметрия D2&). Приведенное в табл. 46.15 произведе-
342
Натрий и его соединения NaI(R, ж)
ние моментов инерции соответствует структурным параметрам r(Na—Вг)=2,69 +0,05 А и
-$Br—Na—Вг=108+5°, найденным Соломоником и Красновым в результате расчета с
использованием ионной модели молекулы [961, 963 ]. Погрешность значения IAIBI<; оценивается в 1 • 10 ~112 г3 • см6.
Приведенные в табл; 46.15 значения частот v4—ve найдены Мартином и Шабером [3410] в
ИК-спектре молекул Na2Br2, изолированных в матрице из Аг. Для не наблюдавшихся
экспериментально частот vx—v3 приняты значения, рассчитанные Соломоником и Красновым [961,
963]. Погрешности принятых значений v4—v6 оцениваются в 15 см, остальных частот — в 20%
от их величины.
Термодинамические функции Na2Br2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Их погрешности обусловлены неточностью принятых
молекулярных постоянных, а также приближенным характером расчета и составляют 5, 10,
15 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно (в том числе 4—
6 Дж-К-моль~1 из-за неточности молекулярных постоянных).
Ранее термодинамические функции Na2Br2(r) рассчитывались в таблицах JANAF [2834] и
в работе Краснова и др. [569] (до 3000 К). Расхождения в значениях, приведенных в этих работах
и в табл. ИЗО, объясняются различием молекулярных постоянных и не превышают 3 Дж-К~хХ
Хмоль [569] и 2 Дж-К^-моль-1 [2834] в значениях Ф°(Т).
Константа равновесия реакции Na2Br2(r)=2Na(r)+2Br(r) вычислена с использованием
значения ДгЯ°@)=914,372+7,1 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(Na2BT2, т, 0) — — 463 + 7 нДж-моль.
Принятая величина основана на результатах исследований равновесия реакции NaBr(K, ж)+
H~NaBr(r)=Na2Br2(r), выполненных масс-спектрометрическим методом Берковицем и Чапкой
11501] (890 К, 1 точка,, ДгЯ°@)=28+6 кДж-моль). Меньшее значение (ДгЯ°@)=18+11 кДжХ
Хмель) было получено Топором и Мургулеску [3595, 4524] комбинацией измерений методом
протока и статическим методом A373—1473 К, 5 точек).
Измерения плотности ненасыщенного пара NaBr^), выполненные Датцем и др. [1995]
статическим методом D293—1434 К, 4 точки), приводят для реакции Na2Br2(r)=2NaBr(r) к
значению ДгЯ°@)=199+12 кДж-моль, которому соответствует А^Н°(№а2Вт2, г, 0) = —473+13 кДжХ
Хмоль -, согласующееся в пределах -.^грешности с принятым в справочнике.
NaI(K, ж). Термодинамические свойства кристаллического и жидкого иодида натрия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1131.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние Nal(K) в интервале 0—934 К принимается
кубическая модификация (структурный тип NaCl) x.
Теплоемкость Nal при Т < 298,15 К измерялась в работах Корефа [3073] (82—192 и 196 —
273 К), Брёнстеда [1683] B73-293 К), Берга и Моррисоца [1492] B,8—20 и 12-270 К), Клэй-
тора и Маршалла [1869] B,3—7,9 К), Гарднера и Тейлора [2362] E6—301 К) и Мартина [3407]
•@,5—1,5 К). При расчете термодинамических функций в температурном интервале 0—20 К ис-
яользованы хорошо согласующиеся между собой результаты измерений [1492, 1869, 3407],
а в интервале 20—50 К — измерения [1492]. В интервале 50—298,15 К значения теплоемкости
получены усреднением данных [1492] и [2362]. Значения ?°B98,15 К) и Я°B98,15 К)—Я°@),
приведенные в табл. 46.1, имеют погрешности 0,3 Дж-К~1-моль~1 и 0,03 кДж-моль
соответственно.
При Т > 298,15 К для теплоемкости Nal(K) принято уравнение, полученное обработкой
результатов измерений энтальпии в работе Гарднера и Тейлора [2362] C57—1074 К). При обра-
(ботке погрешность данных [2362] оценивалась в 0,4%. Температура плавления Nal(K) (934 +
+1 К) принята по измерениям Бредига и др. [1654] (934+0,5 К) и Гарднера и Тейлора [2362]
<934+1 К); менее точные значения получены в работах [1637, 1867, 3386, 4196, 4791] (924—
937 К). Энтальпия плавления B3,68+0,20 кДж-моль) принята по согласующимся между собой
данным Гарднера и Тейлора [2362] B3,77 нДж-моль) и Дворкина и Бредига [2137] B3,60 кДжХ
1 Кубическая фаза высокого давления (структурный тип CsCl) стабильна при 437 К и р > 10,2 кбар [3848].
343
NaI(K, ж)
Глава 46
Хмель). Близкие значения были найдены авторами [1577] B2,18+0,88 кДж-моль) и [1636]
B5,10+0,84 кДж-моль) менее точными методами.
Теплоемкость Nal(jK) F7,0+1,0 Дж-К~1-моль) принята по результатам шести измерений
энтальпии в интервале 935—1074 К в работе [2362]. Мургулеску и Теле [3594] измерили
теплоемкость Ка1(ж) вблизи температуры плавления; найденная ими величина F5,6 Дж-К>моль~1)
в пределах погрешности согласуется с принятой.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,2,
0,5, 3 и 8 Дж-К~1-моль~1. Расхождения между термодинамическими функциями, приведенными
в табл. 1131 и в справочниках JANAF [2834] (Т < 3000 К) и Барина и Кнакке [1395 ] (Т < 1500 К),
достигают 2,3 Дж-К-моль~1 в значениях Ф°(Т); они обусловлены тем, что в настоящем
издании для Т > 298,15 К использованы данные [2362], в то время как расчеты [1395, 2834] были
основаны на оценках.
Принятая в сп равочнике энтальпия образования
Ay#°(NaI, к, 298,15 К) = —289,63 + 0,12 кДж-моль
основана на измерениях энтальпии растворения Nal(K) в воде, представленных в табл. 46.22.
Использованное в дальнейших расчетах значение A^#°(ooH20; 298,15 К)=—7,570+0,030 кДжХ
Хмоль является средневзвешенным из наиболее точных измерений в работах [245, 583, 687,
778, 923, 1097, 2885, 4472, 4787].
Таблица 46.22. Результаты измерений AaJ5T°(NaI, к-*-соН2О, 298,15К) (в кДж• моль)
Авторы
Бертло, 1875 [1530]
Томсен, 1882 [4511]
Пиккеринг, 1888 [3839]
Варэ, 1896 [4592]
Мознер, 1897 [3570]
Бюст, Ланге, 1925 [4787]
Аскю и др., 1934 [1354]
Мищенко, Сухотин, 1954 [687]
Цветков, Рабинович, 1969 [1097]
Сергеева и др., 1970 [923]
Крестов и др., 1972 [583]
Фучс, Хаган, 1973 [2342]
Джоли, Перашон, 1977 [2885]
Перелыгин и др., 1980 [245, 778]
Таневска-Осинска, 1980 [4472]
Конечная концентрация, температура
измерений
500 Н2О, 284,2 К, 1 измерение
200 Н2О, 293,3 К, 2 измерения
200 Н2О, 291,2 К, 1 измерение
111 Н2О, 289,2 К, 1 измерение
800 Н2О, 288,2 К, 1 измерение
5,7—88 Н2О, 298,15 К, 14 измерений
2780 Н2О, 293,15 К, 2 измерения
555—2780 Н2О, 298,15 К, 5 измерений
500 Н2О, 298,15 К, 1 измерение
555—1790 Н2О, 291,2—305,2 К, 17 измерений
1110-5550 Н2О, 298,15 К
111 000—555 000 Н2О, 298,15 К, 12 измерений
3100 Н2О, 298,15 К, 1 измерение
2780 Н2О, 298,15 К
111—5551 Н2О, 298,15 К, 12 измерений
ДвгЯ°(юН2О, 298,15 К)
-7,53+0,85
—5,88 ±0,45
-6,91 +0,85
—6,55+0,85
—7,20+0,85
—7,565+0,085
—7,45+0,15
—7,48+0,10
—7,67+0,15
—7,636+0,040
—7,573+0,035 .
—7,531 +0,045
—7,464+0,045
—7,581+0,030
—7,536+0,040
Таблица 46.23. Результаты определений AiSro(NaI, к, 0) (в кДж-моль)
Авторы
Вартенберг, Альбрехт, 1921 [4656]
Руфф, Мугдан, 1921 [4094]
Грейнер, Елинек, 1933 [2482]
Коджин, Кимбелл, 1948 [1890]
Мургулеску, Топор, 1970 [3598]
Уорк, 1981 [4765]
Метод
Точек кипения, 1336—1570 К, 5 точек
Точек кипения, 1339—1568 К, 6 точек
Протока, 1453 К, 2 точки
Эффузионный, 736—903 К, данные представлены
в виде уравнения
Статический, 1272—1377 К, 11 точек
Протока, 890—1004 К, 33 точки
As.ff°(NaI, к, 0)
II закон
220 ±6
223 ±12
—
235
207,3 ±1,3
219+10
III закон
200,9 ±1,7
201,0+1,6
192,4 ±3,3
198,7 ±9,3
201,0 ±0,2
202,5 ±0,4
344
Натрий и его соединения Nal(r>
Давление пара в реакции NaI(K, 5K)=NaI(r) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = A,#°(NaI, к, 0) = 201K±3,0кДж-моль-1.
Принятая величина основана на измерениях давления и состава насыщенного пара Nal(к, ж).
В состав насыщенного пара Nal при jTm=934 К по данным настоящего справочника входит
27 мол.% димерных молекул. Методика выбора термохимических величин для компонентов
насыщенного пара галогенидов щелочных металлов изложена в Приложении 8.
Результаты расчета энтальпии сублимации Nal в виде мономерных молекул представлены
в табл. 46.23. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая величина является средневзвешенной из приведенных в таблице данных, за
исключением выпадающей работы [2482]. При вычислении весов учитывалось отклонение
результатов каждой работы от среднего. Погрешность принятой величины учитывает
воспроизводимость измерений, расхождения результатов измерений в разных работах, неточность
термодинамических функций и определения состава насыщенного пара.
Nal(r). Термодинамические свойства газообразного иодида натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1132.
Приведенные в табл. 46.4 молекулярные постоянные Nal(r) приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3984] и УФ- [3239] спектров этой
молекулы. По аналогии с молекулой LiF и на основании исследования электронного спектра
[3239] принимается, что валентные возбужденные электронные состояния имеют энергии,
превышающие 25 000 см.
Приведенные в табл. 46.4 колебательные и вращательные постоянные Nal в состоянии X1!,*
получены Раском и Горди [4101 ] обработкой результатов измерений микроволновых переходов
/ -» /+1 (/ ^ 27) на колебательных уровнях у=0,1 и 2. Колебательные постоянные и>с и шехе
были получены не непосредственно по экспериментальным данным, а рассчитаны с помощью
соотношений Данхема. Они удовлетворительно согласуются с результатами исследования
ИК-спектра [3984] (юе=258+4, (вех(,=1,08 см).
Термодинамические функции Nal(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A., 10),
A. 93)—A. 95). Значения Qm и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 46.5 и получены по принятым
молекулярным постоянным Nal.
Погрешности рассчитанных термодинамических функций Nal(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах дополнительный вклад в погрешность вносит неточность способа экстраполяции
потенциальной функции, а также пренебрежение возбужденными электронными состояниями.
Погрешности в значениях Ф°(Г) составляют 0,02, 0,05 и 0,5 Дж-К^-моль при 298,15, 3000
и 6000 К соответственно.
Термодинамические функции Nal(r) ранее вычислялись в работах Уилкинса [4728] (Т <^
< 6000 К), Раиса и Клемперера [3985] (Т < 2000 К), Брюэра и Брэкета [1660] (Ф°(Г), Г<
<J 2000 К). Расхождения данных работы [4728] и табл. 1132 не превышают 0,5 и 1,2 Дж-К~хХ
Хмоль в значениях Ф°(Г) и S°(T) соответственно и объясняются тем, что в этой работе
использовался приближенный метод расчета.
Константа равновесия реакции NaI(r)=Na(r)+I(r) вычислена с использованием значения
Ar#°@)=D0(NaI)=302,458+3,l кДж-моль я» 25 280+260 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации Nal(K). Этим величинам также
соответствует
г, 0) = —87,532 + 3,0 кДж • моль.
Близкие значения энергии диссоциации были получены Су и Райли [4427] и Ван Вееном и др.
[4589] методом фотофрагментарной спектроскопии C00,4+2,1 и 306,8+0,9 кДж-моль), Бойт-
лером и Леви [1539] по границе обрыва флуктуационных полос в спектрах испускания и
поглощения Nal B97+4 кДж-моль), Булевич и др. [1719] при спектрофотометрическом изучении
равновесия реакции образования Nal в пламенах C02+8 кДж-моль), Более низкое значение?
345
J4a5jI2(r), Na2SO4(K, ж) Глава 46
B89 кДж-моль) было получено Берковицем и Чапка [1502] в результате фотоионизационных
измерений. Берг и Скьюс [4925] на основании анализа дискретной структуры электронного спектра
поглощения Nal нашли более высокое значение C15+3 кДж-молъ), однако их
экспериментальные данные допускают другую интерпретацию, приводящую к меньшей величине (см. [1523]).
Na2I2(r). Термодинамические свойства газообразного дииодида динатрия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1133.
Молекулярные постоянные, использованные для расчета термодинамических функций Na2I2,
приведены в табл. 46.15. На основании теоретических расчетов J961, 4689], а также по аналогии
с молекулой Na2Gl2 для молекулы Na2I2 в основном состоянии ХХА^ принимается плоская
ромбическая структура (симметрия D2h). Приведенное в табл. 46.15 произведение моментов инерции
соответствует структурным параметрам r(Na—1)=2,91+0,08 А и -$I—Na—1 = 111+5°,
найденным Соломоником и Красновым в результате расчета с использованием ионной модели [961, 963].
Погрешность IaIbIc оценивается в 5-1012 г3-см6. Приведенные в табл. 46.15 значения частот
v4—v6 получены Мартином и Шабером [3410] при изучении ИК-спектра молекул Na2I2,
изолированных в матрице из Аг. Для не наблюдавшихся экспериментально частот vx—v3 приняты
значения, рекомендуемые Соломоником и Красновым [961, 963]. Погрешности принятых значений
v4—v6 оцениваются в 15 см, остальных частот — в 20—25% от их величины.
Термодинамические функции Na2I2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных состояний. Их погрешности обусловлены неточностью принятых
молекулярных постоянных, а также приближенным характером расчета и составляют 6, 12,
20 Дж-К~1-моль в значениях Ф°(Г) при 298,15 , 3000 и 6000 К соответственно (в том числе 5—
8 Дж-К-моль~1 из-за неточности молекулярных постоянных).
Ранее таблицы термодинамических функций Na2I2(r) вычислялись в работах Краснова и др.
[569] (до 3000 К) и Фрурипа и Бландера [2340] (до 5000 К). Расхождения в значениях Ф°(Т),
приведенных в табл. 1133 и в работе [569], не превышают 3 Дж-К-моль~1 и объясняются
некоторым отличием принятых молекулярных постоянных. Фрурип и Бландер [2340] провели
расчет термодинамических функций ряда димерных галогенидов щелочных металлов по
эмпирическим соотношениям. Для Na2I2 данные [2340] и табл. 1133 согласуются в пределах
1,5 Дж-К^-моль в значениях Ф°(Т).
Константа равновесия реакции Na2I2(r)=2Na(r)+2I(r) вычислена с использованием
значения Дг/7°@)=780,852+7,1 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д^°(Na2T2, г, 0) = —351 + 7 кДж • моль.
Принятое значение является средним из полученных по результатам исследований
равновесия реакции NaI(K)+NaI(r)=Na2I2(r) в работах Миллера и Куша [3504] (эффузионный метод
с селектором скоростей, 817 К, 1 точка, Дг#°@)=23,8 кДж-моль) и Берковица и Чапки [1501]
(масс-спектрометрический метод, 816 К, 1 точка, Дг#°@)=26,9 кДж-моль). Для дальнейших
расчетов было принято значение ДгЯ°@)=25+6 кДж-моль. Основным источником его
погрешности является неточность термодинамических функций. Мургулеску и Топор [3598]
статическим методом и методом протока A272—1377 К, 11 точек) нашли Дг#°@)=9+30 кДж-моль
(II закон) и 8,8+0,9 кДж-моль (III закон). Эти величины, вычисленные комбинированием
•измерений двумя методами, представляются менее надежными по сравнению с приведенными
выше. Погрешность термодинамических функций при температурах измерений в работе [3598]
приводит к погрешности в энтальпии реакции около 12 кДж-моль.
Измерения плотности ненасыщенного пара Nal(ffl), выполненные Датцем и др. [1995]
статическим методом A212—1337 К, 4 точки), приводят к значению Дг#°@)=195+12 кДж-моль
для реакции Na2I2(r)=2NaI(r), которому соответствует AfH°(Na.2I2, г, 0) = —370+13 кДж-моль,
согласующееся в пределах погрешностей с принятым.
Na2SO4(K, ж). Термодинамические свойства кристаллического и жидкого сульфата динатрия
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1134.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние Na2SO4(K) в интервале 0—458 К принята
Натрий и его соединения Na2SO4 (к, ж>
ромбическая модификация (kV) [3675], в интервале 458—514 К — ромбическая модификация
^kIV) [2055] и в интервале 514—1157 К гексагональная модификация (к1) [936] *.
При Т ^ 298,15 К термодинамические функции Na2SO4(K) рассчитаны по результатам
измерений теплоемкости, выполненных Питцером и Коултером [3852] A4—300 К) и обработанных
в работе Бродейла и Джиока [1676]. Образец был приготовлен дегидратацией Na2SO4-10H2O
в вакууме при температуре —350 К и содержал не менее 0,1 % следов исходного
кристаллогидрата. Экстраполяция теплоемкости по кубическому закону привела к значению ?°A5 К) =
=0,31Дж К^-моль. Погрешности вычисленных значений S°B98,15 К) и #"B98,15 К)—ff°@)
оцениваются в 0,4 Дж-К^-моль и 0,08 кДж-моль соответственно.
Уравнение для теплоемкости Na2SO4(K) в интервале 298,15—514 К (см. табл. 46.1) получено
на основании измерений теплоемкости модификации kV в работах Шмидт и Соколова [1155] C08—
1123 К), Шмидт [1153] B98—770 К) и Попова и Гальченко [805] C73—973 К); результаты
измерений [805] хорошо согласуются с данными первых двух работ только в интервале 300—400 К,
а затем на кривой теплоемкости, полученной Поповым и Гальченко, наблюдается аномальный
рост теплоемкости (Х-кривая) в интервале 460—550 К, поэтому данные [805] учитывались только
до 400 К 2.
Температуры полиморфных превращений D58+2 и 514+2 К) и соответствующие энтальпии
@,3+0,1 и 10,9+0,6 кДж-моль) приняты по данным Бродейла и Джиока [1676]. Принятое
значение &trH для перехода kIV -> kI в пределах погрешности совпадает с данными Шмидт и
Соколова [1155] A0,9 кДж-моль), Попова и Гальченко [805] A0,8-кДж-моль) и Девиса и Стэнд-
¦форда [2055] A0,8 кДж-моль). С принятым значением Tir согласуются данные [96, 157, 286,
530, 1075, 1597, 2262, 3040, 3098].
Уравнение для теплоемкости Na2SO4(K) в интервале 514—1157 К (см. табл. 46.1) выведено
по хорошо согласующимся результатам измерений теплоемкости в работах [805, 1153, 1155] 3.
При выводе уравнений для теплоемкости Na2SO4(V) и NaaS04(I) не учитывались данные по
энтальпии, полученные в работах Попова и Гинзбурга [278, 807] B93—1373 К), Дениелоу и др.
[2041, 2043, 2044] D00—1523 К) и Кофлина [1942] B98—1850 К), поскольку имеющиеся у Na2SO4
метастабильные переходы могли существенно исказить результаты этих измерений.
Температура плавления A157+1 К) основана на совпадающих измерениях [17, 19, 21, 62,
36, 105, 108, 115, 264, 372, 374, 379, 396, 679, 863, 892, 974, 1076, 1371, 1942, 2825, 3040,
3116, 3615, 3779, 3805, 3810, 3849, 4017]. Энтальпия плавления 23,5+0,6 нДж-моль принята
по результатам калориметрических определений Дениелоу и др. [2041, 2043, 2044] и согласуется
в пределах погрешности с данными [1942] B3,7 кДж-моль) и [807] B4,1 кДж-моль). Менее
точные измерения [3805] B1,9 кДж-моль) и [3116] B4,3 кДж-моль) не учитывались.
Теплоемкость жидкого сульфата натрия B04+2 Дж-К-моль~1) рассчитана из данных по
энтальпии Na2SO4^), полученных Дениелоу и др. [2041, 2043, 2044] A157—1523 К). Измерения
Кофлина [1942] A157—1850 К) приводят к более низкому значению 197 Дж-К^-моль и не
учитывались. По менее точным данным Гинзбурга [278, 807] A157,2—1500 К) теплоемкость резко
падает с ростом температуры от 219 Дж-К~1-моль~1 при 1157 К до 115 Дж-К^-моль при 1500 К.
Погрешности вычисленных значений Ф°{Т) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,3, 1,5, 10 и 20 Дж-К~1-моль соответственно.
Термодинамические функции, приведенные в табл. 1134, отличаются от данных справочника
JANAF [2835] (Т < 3000 К) не более чем на 0,8 Дж-К^-моль в значениях Ф°(Т) при Г<
<^ 1000 К, с ростом температуры расхождения увеличиваются до 4 Дж-К~1-моль из-за
различия принятых значений C°(Na2SO4, ж).
Принятая в справочнике энтальпия образования
A/F°(Na2SO4, к, 298,15 К)= —1387,9 + 0,4 кДж • моль
1 Сульфат динатрия имеет ряд метастабильных модификаций и переходов. По данным работы Бродейла и Джиока
;. [1676] метастабильной в интервале 0—517 К является модификация кШ, а также модификации kV, в интер-
" вале 514—516 К и к1 — в интерчале 509—514 К; метастабильные переходы кШ ->• к1, kV ->• к1 п kV-> кШ
происходят при 509, 512 и 517 К соответственно. В работе [1676] дана также схема стабильных переходов,
принятая в настоящем справочнике. Термодинамические свойства Na2SO4 (кШ) рассмотрены ниже.
2 В связи с отсутствием данных о свойствах модификации kIV, стабильной в области 458—514 К, в справочнике
принято одно уравнение для теплоемкости модификаций kV и kIV.
3 Сведения о существовании полиморфного превращения у Na2SO4 (I -> 8), происходящего по данным [1075, 1076]
при 843 К, а по данным [1155] при 963—993 К, противоречивы и в справочнике не учитывались.
347
Na2SO4(K, ж)
Глава 46
Таблица 46.24. Результаты измерений Д.й„Ж"о^а28О4, к-»<х>Н2О, 298,15 К) (в кДж-моль)
Авторы
Конечная концентрация, температура
измерений
Дв,Д°(ооНаО, 298,15 К)
Пягцер, Коултер, 1938 [3852] 1100 Н2О, 298,15 К, 2 измерения
Воскресенская, Пономарева, 1944 [251] 300 Н2О, 298,15 К
Кофлян, 1955 [1942] 1029 Н2О, 298,15 К, 5 измерений
Ныоман, 1958 [3649] 1000 Н2О, 298,15 К, 5 измерений
Риднер, Кобл, 1969 [3952] 298,15 К
Грицус и др., 1969 [329] 278 Н2О, 298,15 К, 1 измерение
Фролов и др., 1976 [1063] 68—2220 Н2О, 298,15 К, 18 измерений
—2,343+0,042
—3,025
—2,272 ±0,15
—2,238+0,040
-2,372+0,10
-2,824
-2,343 ±0,15
основана на приведенных в табл. 46.24 результатах измерений энтальпии растворения Na2SO4(K)-
в воде. Придавая больший вес наиболее детальным и надежным измерениям [1942, 3649, 3852],
для дальнейших расчетов принимается значение Д^//°(ооНаО, 298,15 К)=—2,30+0,05 кДжХ
Хмоль, которому соответствует принятая энтальпия образования Na2SO4(K). В табл. 46.24 не-
включены менее точные измерения энтальпии растворения и измерения, выполненные при
нестандартной температуре в работах [252, 330, 1530, 1532, 1922, 2225, 2451, 3799, 3837, 4511, 4517,
4590]. Калориметрические измерения энтальпий реакций с участием Na2SO4 в кристаллическом
состоянии или в растворе [1333, 1526, 1527, 2223, 2224, 2524, 2526, 2528, 2622, 3415, 3519, 3761,
3955, 3993, 4088, 4511, 4513] и исследования равновесия реакций с участием Na2SO4(K, ж) [2247,
3081, 3367, 3743, 4262, 4499] приводят к менее точным значениям энтальпии образования по
сравнению с рассмотренными измерениями энтальпии растворения.
Давление пара в реакции NaaS04(K, }K)=Na2SO4(r) вычислено с использованием принятой:
в справочнике энтальпии сублимации
\Н*{0) = A,ff e(Ne,SO4, к, 0) = 350 ± 20 кДж • моль.
Эта величина основана на результатах измерений давления пара над NaaSO4(jK), приведенных
в табл. 46.25. Испарение NaaS04 происходит с частичным разложением и в состав пара, помимо
Na2SO4, входят также Na, SO2 и О2. Приведенные в таблице результаты исследований, в которых
измерялось общее давление, были пересчитаны с использованием принятых в справочнике значе-
Таблица 46.25. Результаты
Авторы
определений Asffo(Na2SO4, к, 0) (в кДж-моль)
Метод
Кубмчотги, Кенеши, 1972 [1970] Протока, газ-носитель N2, 1400—1624 К, 8 точек
Фрикселл и др., 1973 [2341]
Коль и др., 1975 [3061]
Яганнатан, Уайтт, 1978 [2825]
Воннел, Хасти, 1979 [1615]
Халле, Штерн, 1980 [2554]
Хилденбранд, 1982 [2645]
а Данные представлены в виде
То же, газ-носитель SO2+O2, 1395—1663 К,
12 точек
То же, газ-носитель воздух, 1339—1477 К,
20 точек
Масс-спектроыетрический, 1196—1349 К а
Протока, газ-носитель SO2+O2, 1289—1412 К,
6 точек
Эффузионный, 1217—1404 К, 14 точек
Масс-спектрометрический, 1069—1281 К а
Эффузионный и масс-спектрометрический-, 1173—
1473 К а
Торзионный, 1200 К, 1 точка
Масс-спектрометрический, Тср=1200 К а
уравнения.
As//°(Na2SO4, к, 0)
11 закон
416 ±40
392+20
361 ±50
358
379+23
445 ±70
339+15
401
408
III закон
346,7 ±3,9
350,8 ±1,9
358,4+1,9
359,7 ±2,0
338,6±1,6
339,6 ±4,0
—.
360,5 ±5,0
345,5
—
348
Натрий и его соединения Na2SO4 (к, ромб.), Na2SO4(r)
лий термодинамических свойств компонентов насыщенного пара, что позволило вычислить
парциальные давления Na2SO4. Приведенные в таблице величины относятся к испарению сульфата
натрия в виде Na2SO4(r). Результаты расчетов расходятся больше, чем это можно объяснить их
воспроизводимостью, что связано со сложностью изучаемого процесса. Принятая величина
является средневзвешенной из приведенных в таблице величин. Она практически совпадает с данными
Хилденбранда [2645] и Кубиччотти и Кенеши [1970], представляющимися наиболее
достоверными. Приведенные в таблице погрешности учитывают только воспроизводимость измерений.
Основным источником погрешности принятого значения является неточность термодинамических
функций.
Na2SO4(K, ромб.). Термодинамические свойства ромбической модификации Na3SO4(KlII) при
температурах 100—509 К приведены в табл. Д.67.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 46.1. Метастабильная ромбическая модификация NaaSO4(KlII) образуется в результате мета-
стабильных переходов Na2SO4(KV) -> Na2SO4(KlI.I) при 517 К и Na2S04(Kl, метастаб.) -> Na2SO4(KlV)
при 509 К [1676].
При Т ^ 298,15 К термодинамические функции Na2SO4(III) вычислены по результатам
измерений теплоемкости, выполненных в работе Бродейла и Джиока [1676] A3—298 К), в которой
термодинамические свойства сульфата динатрия изучались очень тщательно и подробно.
Измерения проводились в адиабатическом калориметре на образце, который не содержал Na2SO4(KV).
•Экстраполяция теплоемкости по кубическому закону Дебая привела к значению S°A5 K) =
=0,52 Дж-К-моль~1. Данные по теплоемкости Na2SO4(KlII), полученные Пауковым и
Лаврентьевой [763] A4—296 К; адиабатический вакуумный калориметр, образец с содержанием
~0,008% воды и следов Na2SO4(KV)), ниже результатов работы [1676] при 20—30 К на 15—18%,
при 100 К это расхождение уменьшается до 1 %, при 200 К оно составляет 0,5 % ж при комнатных
температурах не превышают 0,1%. Погрешности вычисленных значений ?°B98,15 К) и
jfiT°B98,15 К)—Н° @) оцениваются в 0,4 Дж-К^-моль и 0,08 кДж-моль соответственно.
Уравнение для теплоемкости Na2SO4(KlII) в интервале 298,15—509 К (см. табл. 46.1) выведено
по результатам измерений теплоемкости, выполненных Шмидт и Соколовым [1155] C18—521,65 К)
с погрешностью не более 0,5% \
Погрешности вычисленных значений Ф°(Г) при 298,15 и 500 К оцениваются в 0,3 и 0,5 ДжХ
XК-моль соответственно. Термодинамические функции, приведенные в табл. Д.67,
отличаются от данных таблиц JANAF [2834] не более чем на 0,08 Дж-К~1-моль~1 в значениях Ф°(Г).
Принятая в справочнике энтальпия образования ромбической кШ модификации Na2SO4
A/ff°(NaaSO4, кШ, ромб., 298,15 К) = —1384,9 ± 0,4 кДж.моль
основана на измерениях энтальпии перехода модификации kV, которая равновесна при комнатной
температуре, в модификацию кШ. Бродейл и Джиок [1676] измерили энтальпию растворения
в воде модификации кШ и, используя результаты аналогичных измерений Питцера и Коултера
[3852] для модификации kV, получили Д,г#°B98,15 К)=2,983+0,040 кДж-моль. Кофлин [1942]
измерил энтальпии растворения в воде двух упомянутых выше модификаций Na2S04 и нашел
(см. исправления в работе [1676]) значение 2,996+0,017 кДж-моль в хорошем согласии с
данными [3852 ].|
Na3SO4(r). Термодинамические свойства газообразного сульфата натрия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 1135.
Молекулярные постоянные Na2SO4, принятые для расчета термодинамических функций,
приведены в табл. 46.7. Молекула Na2SO4 имеет четное число электронов, поэтому в справочнике
ее основное электронное состояние принимается синглетным (ХХА). Сведения о возбужденных
электронных состояниях Na2SO4 в литературе отсутствуют. На основании исследований ИК-
и КР-спектров молекул Na2SO4, изолированных в матрицах инертных газов и азота [360, 1356],
а также по аналогии с молекулами Cs2SO4 и K2SO4 (см. ниже) в справочнике для молекулы Na2SO4
принимается «бициклическая» равновесная геометрическая конфигурация (симметрия D2d), т. е.
1 Данные по энтальпии NaaSO4 (кШ), полученные в работах Верма и др. [4607] C74,87—497 К) и Кофлина [1942]
B98—514 К), не учитывались, поскольку имеющиеся у Na2SO4 метастабильные превращения могли существенно
исказить результаты измерений.
349
NaNO2(K, ж) Глава 4g
структура, содержащая тетраэдрический ион SO|~ и расположенные симметрично относительно
него два атома Na, связанные мостиками с атомами кислорода. Структурные параметры молекулы
Na2SO4 экспериментально не определялись. Произведение моментов инерции Na2SO4,
приведенное в табл. 46.7, вычислено по структурным параметрам: r(Na—O)=2,2+0,l A, r(S—О)=1,47 +
+0,03 А и J$O—S—О = 109,5 +10°. Значение r(Na—О) было оценено по величине r(Cs—О) и г(К—О)
в молекулах Cs2SO4 и K2S04 и закономерностям в изменении расстояний металл—кислород в
молекулах МОН и кристаллах M2SO4. Для группы SO4 приняты те же параметры, как в молекулах
K2SO4 и Cs2SO4. Погрешность значения IaIbIc оценивается в 2-1013 г3-см6. Приведенные в табл.
46.7 основные частоты vlt v5, v6, v8 и v9, отвечающие колебаниям группы SO4, наблюдались в ИК-
спектрах и спектрах КР молекул Na2SO4, изолированных в матрице из Аг, в работах [360, 1356] 1..
Частоты v2 и v4, также принадлежащие колебаниям группы SO4, но не наблюдаемые
экспериментально, рассчитаны по ионной модели [360]. Частоты колебаний связей Na—О приняты на
основании результатов исследования ИК-спектра молекул Na2SO4, изолированных в матрице из Аг
(v7, v10) и расчета по ионной модели (v3, vn) [360]. Погрешности принятых значений оцениваются
в 15 см для v6, v9 и vu, в 20 см — для vx, v6 и v8, в 50 см — для v3, v4, v, и v10 и в 75 см —
для v2, они учитывают возможный матричный сдвиг, ненадежность отнесения некоторых
наблюдавшихся частот, а также модельных расчетов. jfe.
Термодинамические функции Na2SO4(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128), A. 130) без учета возбужденных состояний. Погрешности рассчитанных
значений обусловлены приближенным характером расчета и неточностью принятых
молекулярных постоянных, они составляют 8, 12 и 20 Дж-К-моль~1 в значениях Ф°(Т) при 298,15, 300О
и 6000 К.
Ранее термодинамические функции Na2SO4(r) вычислялись в таблицах JANAF [2835] и работе
Кубиччотти и Кенеши [1970] (Г=1000 и 1700 К). Расхождения данных табл. 1135 и [2835] не
превышают 2 Дж-К-моль~1 в значениях Ф°(Т). Расхождения с данными [1970] достигают 18 ДжХ
XК-моль в значениях ФсA700 К) и обусловлены тем, что в этой работе для Na2SO4 принята
структура симметрии C2v и другие значения основных частот.
Константа равновесия реакции Na2SO4(r)=2Na(r)+S(r)-(-4O(r) вычислена с использованием
значения Дг#°@)=2503,851+20 кДж-моль,^соответствующего принятым|в справочнике
энтальпиям образования и сублимации Na2SO4(K). t "~*
Этим величинам также соответствует 98 ЕйШ •;
A^°(Na2SO4,fr,|0) = —1026,408 ± 20^кДж • моль.
NaNO2(K, ж). Термодинамические свойства кристаллического и жидкого нитрита натрия
в стандартном состоянии при температурах 298,15—2500 К приведены в табл. 1136.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние NaNO2(K) в интервале 0—557 К принята
ромбическая модификация [2943] 2. Для NaNO2(K) характерны два фазовых превращения,
обусловленных переходами ферроэлектрик =?* антиферроэлектрик при 436,7 К (точка Кюри) и антиферро-
электрик ** параэлектрик при 437,9 К (точка Нее ля) 3.
При Т < 298,15 К измерения теплоемкости NaNO2 выполнены в работе Вилляра и др. [46151
B—40 К). Значение С°р B98,15 К) принято на основании измерений теплоемкости в*интервале
275—468 К, проведенных Сакиямой и др. [4117] методом теплового потока. Рекомендуемое
значение iSr0B98,15 К) получено усреднением оценок, выполненных методами Келли [3771 ], Лати-
мера [614] и Цагарейшвили [1094]. Значение ЯсB98,15 К)—#°@) оценено по методу Карапетьянца
[485] на основании сопоставления экспериментальных данных в рядах нитриты—метабораты
щелочных металлов. Погрешности принятых значений 5°B98,15 К) и #°B98,15 К)—#°@)
(см. табл. 46.1) могут достигать 10 Дж-К-моль и 0,8 кДж-моль "соответственно.
Для Vj приведено значение, наблюдаемое в спектре КР ^молекул Na2SO4, изолированных в матрице из N2 [1356].
В аргоновой матрице частота vt не наблюдалась.
2 При высоких давлениях у NaNO2 стабильны еще три фазы с неизученной структурой [1009].
8 При расчете термодинамических функций промежуточная фаза (NaNO2—антиферроэлектрик) не
рассматривалась как самостоятельная, поскольку она существует в очень узком температурном интервале. Температура
I перехода антиферроэлектрик—параэлектрик получена в работах [209, 3412, 3936, 4117].
350
Натрий и его соединения NaNO2(K, яф
Имеющиеся в литературе данные по теплоемкости [2585, 2586, 2716, 3673, 4117] и энтальпии
[1765, 2915] NaNO2 при Т > 298,15 К крайне противоречивы. Так, значения Н°(Т)—#°B98,15 К)
в интервале 452—468 К, общем для работ [4117] и [1765] отличаются на 14—20%. Результаты
измерений теплоемкости в работах Номуры [3673] B93—483 К), Хошино [2716] (в области
фазовых превращений), Хатты и Икушимы [2585, 2586] C02—470 К) представлены только на
графиках. Расчет термодинамических функций при Т > 298,15 К выполнен по данным Сакиямы и др.
[4117] B75—468 К) и Кейсиса [1765] D52—695 К, погрешность измерений не больше 1%).
Данные [1765] хорошо согласуются с измерениями [2915] B98—573 К), результаты которых
представлены только на графике. Для согласования данных [1765, 4117] в справочнике была
принята следующая схема расчета: ниже T(r—4SQ,7 К использованы данные Сакиямы и др. [4117],
аппроксимированные двумя уравнениями, первое из которых описывает плавный рост
теплоемкости в интервале 298,15—400 К, а второе — восходящую ветвь Х-кривой теплоемкости в
интервале 400—436,7 К. Для теплоемкости высокотемпературной модификации в интервале 436,7—
557 К принимается постоянное значение С°(Г)=133 Дж-К~1-моль [1765]. Расхождение между
величинами Н°(Т)—#°B98,15 К) в точке перехода D36,7 К) по данным [4117] и [1765] отнесено
за счет скрытой энтальпии превращения.
Температура перехода ферроэлектрик «* антиферроэлектрик D36,7+1,0 К) принята по
данным [2172, 3412, 3936, 4117]; с принятым значением Ttr согласуются результаты измерений [454,,
705, 824, 1535, 2559, 2804, 2826, 4118, 4148, 4445] D35—437 К). Энтальпия перехода C,2 кДжХ
Хмоль) получена на основании принятых уравнений для энтальпии низко- и
высокотемпературной модификаций NaNO2(K). Погрешность этого значения может достигать 50%. Энтальпии
превращения, приведенные в работах [4117] A,82 кДж-моль) и [3673] A,19 кДж-моль), получены
интегрированием аномальной части кривой теплоемкости и, по-видимому, не включают
изотермическую часть энтальпии перехода. В работах [209, 1253, 3928], выполненных методами ДТА,
ДСК и термографии, получены более низкие значения A,05, 1,25 и 1,05 кДж-моль). *-,
Температура плавления E57 +2 К) принята по согласующимся между собой измерениям
Беруль, Бергмана и Никоновой [98, 133, 3673] и Проценко и др. [820, 826, 1152]; менее точные
результаты получены в работах [254, 841, 869, 1870, 3936, 4177] E43—561 К). Энтальпия
плавления A4,94+0,80 кДж-моль) и теплоемкость КаЖ>2(ж) A00+3 Дж>К'Мрль~1) приняты
на основании измерений энтальпии в работе Кейсиса [1765]; для жидкой фазы автором выполнена
16 измерений в температурном интервале 557—695 К. В работах [2915] E52—573 К) и [5018]
E65—583 К) для теплоемкости NaNO2^) получены значения 114 и 121 Дж-К~1-моль~1 на
основании измерений, выполненных в узких интервалах температур.
Значение энтальпии плавления, полученное в работе [2915] A5,3 кДж-моль), согласуется
с принятым в пределах погрешностей. Величина &тН°, рассчитанная Верхотуровым и др. [209]
на основании термографических определений A3,0 кДж-моль), представляется заниженной.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000 и 2000 К оцениваются в 7, 12
и 20 Дж-К-моль~1 соответственно. Таблица термодинамических функций NaNO2(K, ж)
публикуется впервые.
Принятая в справочнике энтальпия образования
A^o(NaN02, к, 298,15 К) = —354,6 + 0,8 кДж.моль
основана на приведенных в табл. 46.26 результатах измерений энтальпии растворения NaNOa(K)>
в воде. Наиболее точные измерения были выполнены Кейсисом и др. [1766] и принятая для
дальнейших расчетов величина Дяд#°(ооН20; 298,15 К)=14,005 +0,015 кДж-моль основана на этой
работе. Основную погрешность в энтальпию образования NaNO2(K) вносит неточность
использованного в расчетах значения энтальпии образования иона NOj (см. Приложение 5). Приведенные
в таблице величины были пересчитаны к стандартному состоянию с использованием энтальпий
разведения и теплоемкости растворов NaNO2, измеренных в работе [1766]. В работе [4873] было
получено значение, заметно отличающееся от данных других авторов.
Доде [2088, 2089] измерил энтальпию реакции нейтрализации NaOH(p-p)-fHNO2(p-p)=
=NaNO2(p-p)+H2O(p-p), Дг#°=—44,1 кДж-моль (температура измерений и концентрации
не указаны), ей соответствует значение —353,9 нДж-моль", хорошо согласующееся с принятой
величиной Д^77О. Использованное в расчете значение энтальпии образования HNO2(p-p) принято*
по данным справочников [1009] и [4626] (см. Приложение 5).
351
NaNO2(r) Глава 46
Таблица 46.26. Результаты измерений Aa?JH"°(NaNO2, к-»¦ со Н20, 298,15 К) (в кДж-моль)
Авторы
Конечная концентрация, температура
измерений
Дй//°(со Н2О, 298,15 К)
Свентоолавский, 1909 [912] 350 Н2О, 291 К, 1 измерение 13,8
Матиньон, Маршал, 1920 [3419] 350 Н2О, 293 К, 3 измерения 13,9
Бюро, 1937 [1724] 400 Н2О, 293 К, 1 измерение 14,09
Доде, 1937 [2089] 430 Н2О, 285 К, 2 измерения 13,18
Перре, 1941 [3800] . 250—400 Н2О, 287 К, 4 измерения 13,82+0,50
Решетников, 1961 [864] 330 Н2О, 298,15 К, 5 измерений8 13,81 ±0,10
By, Хеплер, 1962 [4777] 1270-7430 НаО, 298,15 К, 8 измерений6 13,65
Кейсис и др., 1977 [1766] 280—1110 Н2О, 298,15 К, 24 измерения 14,005+0,015
Андреева и др., 1979 [34] 4,4—111 Н20, 298,15 К, 29 измерений 13,79+0,20
а Измерялась энтальпия растворения механической смеси кристаллических NaNO2 и NaOH (мольное отношение 1:1).
б Измерялась энтальпия растворения в разбавленных растворах NaOH.
Менее точные значения были получены при исследованиях равновесия реакции NaN03(m) =
=NaNO2^)+V2O2(r), которые проводились статическим методом Центнершвером [1773] (см.
также [3123] G40—806 К, —412 кДж-моль (II закон), —393 кДж-моль (III закон)), Фрима-
ном [2329] (873—973 К, 3 точки, — 360 кДж-моль (II закон), —374+10 кДж-моль-1 (III закон))
и Сироткиным [938] (823—973 К, 4 точки, —356+15 кДж-моль (II закон), — 358+10 кДжХ
Хмоль (III закон)). Приведенные погрешности обусловлены главным образом неточностью
термодинамических функций; кроме того, в работе [1773] не были учтены активности компонентов
расплава.
Давление пара в реакции NaNO2(K, ffl)=NaNO2(r) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = A.,#°(NaNOa, к, 0) = 190 + 8 кДж • моль.
Это значение основано на масс-спектрометрическом исследовании Верхотурова и др. [205, 207],
в котором использовалась двойная эффузионная камера, что позволило показать, что основным
компонентом пара является мономер (количество димера составляет 3,4%) Ч При 694 К авторы
[205, 207] методом полного изотермического испарения нашли давление мономера в парах, что
позволило вычислить по уравнению III закона принятое в справочнике значение энтальпии
сублимации. Его погрешность обусловлена в основном неточностью термодинамических функций
NaNO2(K, ж). Измерениям интенсивностей ионных токов, проведенным в интервале 616—680 К,
соответствует значение 172 кДж-моль (II закон).
NaNO2(r). Термодинамические свойства газообразного натрия в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 1137.
Молекулярные постоянные, использованные для расчета термодинамических функций NaNO2,
приведены в табл. 46.7. Структура NaNO2 не исследовалась. В согласии с результатами
исследования ИК-спектра молекул NaNO2 в матрице из Аг, проведенного Беляевой и др. [88], в
справочнике принимается, что эта молекула имеет плоское циклическое строение (симметрия С2„).
Произведение моментов инерции IaIbIc в состоянии Х1А1 вычислено по структурным параметрам
r(Na—О)=2,2±0,1 A, r(N-O)=1,25+0,05 Аи4 О—N—0=115+5°. Величина r(Na—О) оценена
по значениям г(М—О) в молекулах GsNO2 и RbNO2 с учетом ее изменения в соединениях МОН,
МВО2, MN03, МР03, М2Сг04, M2SO4, М2МоО4 и M2W04, где М — щелочной металл. Остальные
структурные параметры были оценены по данным для RbNO2 и CsNO2. Погрешность значения
IaIbIc оценивается в 5-Ю15 г3-см6. Принятые значения частот v3=vNa_0, v4=vN_0 и vB=vNa_0_N
наблюдали Беляева и др. [88] в ИК-спектре молекул NaNO2, изолированных в матрице из Аг.
1 Авторы работы [1706] пришли к ошибочному заключению о преобладании в паре димерных молекул; это было
обусловлено тем, что в работе не было детально проанализировано происхождение основного иона в масс-спектре
(Na+).
352
Натрий и его соединения NaNO3(K, ж)
Близкое значение для v4 получили Миллиган и Джекокс [3508]. Частота неплоского колебания v6
принята равной соответствующей частоте молекулы Na2SO4 (см. текст). Поскольку частоты N0^
в нитритах во всех фазовых состояниях и в свободном ионе N0^ характеристичны, частота va=
= vo_N_o принята такой же, как в CsNOa, a v1=vN_o принята равной соответствующей частоте
свободного иона N0^. Близкие значения частот NOi" наблюдались в спектрах кристаллов и
расплавов NaN02 [1251, 2476, 2681, 2830, 4536]. Погрешность принятого значения ve оценивается
в 15 см, остальных частотах — в 10—20 см. Сведений о возбужденных электронных
состояниях NaNO2 в литературе нет.
Термодинамические функции NaNO2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета и составляют 4, 8 и 12 Дж-К~1-моль в значениях Ф°(Т)
при 298,15, 3000 и 6000 К (в том числе ~2 Дж-К~1-моль~1 из-за неточности молекулярных
постоянных). ¦
Таблица термодинамических функций NaNO2(r) публикуется впервые.
Константа равновесия реакции NaNO2(r)=Na(r)+N(r)+2O(r) вычислена с использованием
значения ДгЯ°@)=1233,771+8,1 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации NaNO2(K). Этим величинам также соответствует
Д f?P(NaNO2, r,t.O) = —161,624 + 8,0 кДж • моль.
NaNO3(K, ж). Термодинамические свойства кристаллического и жидкого нитрата натрия
в стандартном состоянии при температурах 100—2000 К приведены в табл. 1138.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние NaNO3(K) в интервале 0—549 К
принимается гексагональная а-модификация (структурный тип кальцита), а в интервале 549—579,6 К —
высокотемпературная разупорядоченная ^-модификация (группа симметрии R3m) [3937]) г.
При Т ^ 298,15 К термодинамические функций вычислены по результатам измерений
теплоемкости, выполненных Саузардом и Нелсоном [4350] A6—287 К). Величины ?°A6 К)=0,58 ДжХ
хК-моль и С°B98,15 К)=93,05+0,5 Дж-К^-моль получены в результате соответствующей
экстраполяции данных [4350]. В пределах погрешности с принятым значением согласуется
величина, полученная Рао и др. [3927] при измерении теплоемкости NaNO3(K) в интервале 173—373 К
с помощью дифференциального сканирующего калориметра (92,3 -^-2 Дж-К~1-моль~1).
Приведенные в табл. 46.1 значения S°B98,15 К) и #°B98,15 К)—Н°@) имеют погрешности 0,3 Дж-К^Х
Хмоль и 0,08 кДж-моль соответственно.
При Т > 298,15 К расчет термодинамических функций NaNO3(K) основан на результатах
измерений теплоемкости в работах Миекк-Ойи [3500] C20—574 К), Соколова и Шмидт [959] E45—
661 К), Мустайоки [3603] C34—624 К), Рейнсборо и Уэтмора [3966] D23—580 К) и измерений
энтальпии в работе Янца и др. [2842] D84—747 К). Кубичар и Мариани [3128, 3129] измерили
теплоемкость в интервале 323—573 К, однако соответствующие данные представлены только
на графике. Данные по энтальпии, полученные в работе Нгуен-Дюи и Даней [3652] D50—650 К)„
отличаются от результатов измерений теплоемкости на ~6% и не учитывались при выводе
уравнения для теплоемкости.
Фазовый переход у NaNO3(K) сопровождается А^-аномалией кривой теплоемкости с максимумом
при 549 К. Поэтому теплоемкость в интервале 298,15—579,6 К была аппроксимирована двумя
уравнениями для а-фазы и постоянной величиной для Р-фазы. Первое уравнение, описывающее
медленное возрастание теплоемкости в интервале 298,15—500К, основано на значениях С°B98,15 К)
(см. табл. 46.1), С;D00К)=11.3 Дж-К^-моль-1, С;D50 К) = 126 Дж-К^-моль-1 и С°р(ЪОО К)=
=152 Дж-К~1-моль, из которых три последних получены в результате усреднения данных
[959, 3500, 3603]. При выводе уравнения для интервала 500—549 К, аппроксимирующего
восходящую ветвь Х-кривой, были использованы значения СрE00 К)=152 Дж-К^-моль, С°E25 К)=
=183 Дж-К^-моль и Я°E49 К)—Я°E00 К)=9,53 кДж-моль; два первых значения получены
усреднением данных [959, 3500, 3603], а последнее — усреднением данных [959, 2842, 3500, 3603„
1 Фаза высокого давления NaNO3 стабильна при р > 42 кбар (Г=733 К) [1009].
353
NaNO3(K, ж)
Глава 46
3966]. В интервале 549—579,6 К теплоемкость принята постоянной A40 Дж-К~1-моль~1) [959,
3500, 3603].
Температура фазового перехода E49+1 К) основана на результатах калориметрических
измерений [2842, 3500, 3603, 3966]. С принятым значением хорошо согласуются результаты работ
[550, 846, 986, 1952, 2141, 3027, 3028, 3093].
Температура плавления E79,6+0,5 К) и энтальпия плавления A5,05+0,10 кДж-моль)
приняты по измерениям [959], (образец ©'содержанием примесей меньше 0,01%). С этими данными
совпадают результаты определений Тт в работах [2319, 3603, 4217, 4303]; менее точные данные
получены в работах [112, 984, 1893, 2005, 2842, 3652, 3739, 4177] E77-582 К). С принятым
значением Ат Н хорошо согласуются измерения [2728, 3051, 3319, 3375, 4815] A4,94, 15,06, 15,13,
15,06, 15,00 кДж-моль соответственно). Энтальпия фазового перехода II рода определялась
в работах [846, 2842, 3500, 3603, 3652, 4217] и изменяется в пределах 0,7—4,8 кДж-моль.
Теплоемкость NaN03(m) A41+3 Дж-К-моль) принята на основании данных [959] E83—
661 К; 23 измерения). В работах [3603] E80—624 К; 13 измерений) и [2842] E98—747 К)
получены значения ~153 и 131 Дж-К~1-моль~1 соответственно. Значение С°(ж)=212 Дж-К^-моль",
приведенное в работе [3652], представляется ошибочным. По данным [2065] C°(NaNO3, ж) вблизи
температуры плавления составляет 142 Дж-К~1-моль~1 и уменьшается с повышением температуры.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 0,2, 3
и 10 Дж-К^-моль-1.
Расхождение между термодинамическими функциями NaNO3(K, ж), приведенными в табл. 1138
и в справочниках Барина и Кнакке [1395] (Т < 700 К) и Келли [2960] (Т < 700 К), не
превышают 0,03 Дж-К~1-моль в значениях Ф°(Г).
В справочнике принята энтальпия образования
A//o(NaN03, к, 298,15 К) = —467,7 ± 0,5 кДж • моль,
основанная на результатах измерений энтальпии растворения NaNO3(K) в воде, представленных
в табл. 46.27. Принятая величина ДадЯ°(соН2О; 298,15 К)=20,59+0,20 кДж-моль^1, является
Таблица 46.27. Результаты измерений Aa2i7o(NaN03, к->-соН2О, 298,15 К) (в кДж-моль4)
Авторы
Конечная концентрация, температура
измерений
&aqH°(co H2O, 298,15 К
Воскресенская, Пономарева, 1946 [252]
Ланге, Мартин, 1937 [3179]
Киселева, 1958 [514]
Ван Тассель, Вендландт, 1959 [4588]
Мищенко, Шпигель, 1967 [688]
Хренова и др., 1970 [1083]
Ояри, 1971 [746]
300 Н2О, 298,15 К
460 Н2О, 298,15 К, 1 измерение
132 Н2О
200 Н2О, 298,15 К, 5 измерений
93—1111 Н2О, 298,15 К, 7 измерений
5—6,5 Н2О, 298,15 К, 5 измерений
1000 Н2О
20,35
20,56
21,05
21,09 ±0,42
20,36+0,13
20,42
20,42+0,30
средней из данных [252, 688, 3179, 4588]. Публикации [514, 746] не содержат ряда сведений,
необходимых для оценки их надежности, а в работе [1083] измерения проводились при очень высоких
концентрациях, что приводит к значительным погрешностям при пересчете к бесконечному
разбавлению. В литературе известен еще ряд работ, где измерялась энтальпия растворения NaNO3(K)
в воде [581, 584, 741, 980, 1530, 2548, 3540, 3541, 4206, 4374, 4511, 4517, 4590, 4742, 4814]. Часть
их относится не к стандартной температуре или их основной целью являлось измерение энтальпии
растворения в смешанных растворителях. Эти работы приводят к менее точным значениям
стандартной энтальпии растворения.
Близкие, но менее точные значения энтальпии образования NaNO3(K) могут быть вычислены
по измерениям энтальпии нейтрализации растворов NaOH и HNO3 [1333, 1348, 1526, 2222, 2224,
2623, 3415, 3994, 4511]. Наиболее точное из этих исследований [3994] приводит к величина
—467,3 кДж-моль (см. [1555, 3771]), совпадающей с принятой. Исследования ЭДС реакции
KGl(K)+NaNO3(K)=NaCl(K)+KNO3(K), выполненное Бренстедом [1682] B95—355 К, данные
354
Натрий и его соединения NaNO3(r)
представлены в виде уравнения), приводят к значению —466,8 кДж-моль, также близкому к
принятому.
Давление пара в реакции NaNO3(K, ;-K) = NaNO3(r) вычислено с использованием принятой .
в справочнике энтальпии сублимации
Дг//°@) = As/7°(NaNO3, к, 0) = 184 + 5 кДж • моль.
Принятая величина основана на масс-спектрометрическом исследовании, выполненном Бага-
ратьян и Никитиным [56, 58]. Опыты с двойной эффузионной ячейкой показали, что основным
компонентом пара является мономер (количество димера при 700 К составляет 6%). При 693,4 К
методом изотермического испарения было найдено парциальное давление мономера, которому
соответствует вычисленное по уравнению III закона и принятое в справочнике значение. Его
погрешность обусловлена в основном неточностью термодинамических функций NaNO3(r).
Измерения интенсивностей ионных токов, проведенные в интервале 647—709 К, приводят к значению
208 кДж-моль~х (II закон).
В работе этих же авторов [4851 ] методом электронного удара была найдена энергия связи
D0(Na—NO3)=410+9 кДж-моль, которой соответствует значение Д8#°@)=236+25кДж-моль~1,
значительно отличающееся от принятого в справочнике. Это свидетельствует о неточности
измерений энергии связи D0(Na—NO3).
NaNO3(r). Термодинамические свойства газообразного нитрата натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1139.
Молекулярные постоянные, использованные для расчета термодинамических функций NaNO3,
приведены в табл. 46.7. Произведение моментов инерции IaIbIo в состоянии ХхАх вычислено для
плоской циклической бидентатной конфигурации молекулы NaNO3 (симметрия С2о) с ^Оц—N—
ц = 115±5°, »\N-O1()=1,29+J?5 A, r(N-Oraem) = l:21+°'5jA, r(Na-O4) = 2,2 +0,1 A;
погрешность значения LJeI;- составляет 1-10~114 г3-см6. Конфигурация молекулы NaNO3
и значения геометрических параметров группы NO3 приняты такими же,, как в молекулах
LiNO3, RbNOs и CsNO3. Значение r(Na—Оц) принято таким же, как в молекуле NaPO3, для
которой оно было определено в здектронографическом эксперименте [786]. Результаты выполненного
Ходченковым и др. [10S0] визуального электронографического исследования паров NaNO3, в
котором была найдена монодентатная конфигурация этой молекулы (симметрия Cs) с ^. Na—О—N=?
= 105°, ^ O=N=O = 134°, r(N=O) = l,22 A, r(N-O) = l,40 A, r(Na-O) = 1,90 А, представляются
ошибочными, поскольку Бюхлер и Штауфер [1706] при масс-спектральном исследовании NaNO3
показали, что в паре присутствуют примерно в одинаковых количествах мономерные и димерные
молекулы вместе с заметным количеством продуктов разложения. Принятые значения частот
валентных колебаний v1( v2, v5 и неплоского колебания v8 иона NO" измерены Смитом и др. [4319]
в ИК-спектре молекул NaNO3, изолированных в матрице из аргона х. Для деформационных
колебаний иона NOg в плоскости, v3 и ve, приняты значения, промежуточные между наблюдавшимися
в матрице из аргона для LiNO3 (v3 и v6) [4333] и KNO3 (v6) [1444]. Близкие значения частот NO"
наблюдались при исследованиях спектров кристаллов, расплавов и растворов NaNO3 [607, 803
1252, 1546, 1547, 1685, 1714, 1717, 2052, 2100, 2143, 2476, 2515, 2628, 2634, 2784, 2829, 2841*
2982, 3091, 3216, 3231, 3420, 3625, 3639, 3914, 3997, 4163, 4214, 4638, 4733, 4738]. Для частот
v4, v7 и v9, связанных с колебаниями иона Na+ относительно иона NOg, приняты значения
соответствующих частот NaNO2 (см. текст NaNO2). Близкая величина v4 B87 см) наблюдалась в ИК-
спектре молекул NaPO3 в Аг-матрице [2852]. Погрешности частот vlt v2, v5, v8 и ve оцениваются
в 10—15 см, v3, v4, v6, v7 — в 30 см". Сведения о возбужденных электронных состояниях NaNO3
в литературе отсутствуют.
Термодинамические функции NaNO3(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. ДО),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор — гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены приближенным характером расчета и неточностью принятых значе-
1 В соответствии с данными Битти и др. [1444] в справочнике принято отнесение частот у1 л v5, противоположное
сделанному в работе [4319].
355
Na2CO3(K, ж) Глава 46
ний молекулярных постоянных, они составляют 4, 8 и 12 Дж-К~1-моль~1 в значениях Ф°(Т) при
298,15, 3000 и 6000 К.
Таблица термодинамических функций NaN03(r) публикуется впервые.
Константа равновесия реакции NaNO3(r)=Na(r)+N(r)+3O(r) вычислена с использованием
значения Д,.#о@)=1596,043+5,1 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации NaNO3(K). Этим величинам также соответствует
t\fHo(NeiN03, г, 0) == —277,113 + 5,0 кДж • моль.
Na2CO3(K, ж). Термодинамические свойства кристаллического и жидкого карбоната динатрия
в стандартном состоянии при температурах 100—3000 К приведены в табл. 1140.
Значения постоянных, принятые для расчета термодинамических функций, приведены в табл.
46.1. За стандартное состояние Na3CO3(K) в интервале 0—623 К принята моноклинная (а)
модификация, в интервале 623—752 К — моноклинная неупорядоченная (Р) модификация и в
интервале 752—1131 К — гексагональная (у) модификация [3763].
При Т ^ 298,15 К термодинамические функции Na2CO3(K) вычислены по результатам
измерений теплоемкости, выполненных Уотерфилдом и др. [4671] (9—300 К, образец прокален при
583 К для устранения примеси высокотемпературной фазы). Экстраполяция теплоемкости по
закону Дебая привела к значению 5°A0 К)=0,19 Дж-К~1-моль. Погрешности принятых значений
5°{298,15 К) и Я°B98;15 К)—#°@) (см. табл. 46.1) оцениваются в 0,4 Дж-К^-моль и 0,06 кДжХ
Хмоль. Данные Андерсона [1315] E5—300 К) при Т < 250 К лежат ниже, а при Т > 250 К —
выше измерений [4671], и различие между ними составляет ~1%.
При Т > 298,15 К энтальпия Na2CO3(K) измерялась в работах Попова и Гинзбурга [807]
B98—1373 К), Янца и др. [2845] E48—1248 К), Ролина и Рекапе [4035] E73—1273 К);
теплоемкость измерена Поповым и Гальченко [805] D73—873). Обработка экспериментальных данных
проводилась с учетом двух полиморфных превращений Na2CO3(K), которые, согласно [805],
приводят к широким максимумам теплоемкости в интервалах 550—630 и 650—752 К. Уравнение
для теплоемкости a-Na2GO3(K) (см. табл. 46.1) получено по данным [805] без учета аномалии С'р
при Т > 550 К. Это же уравнение принято для C-Na2CO3(K), причем аномальное изменение
теплоемкости учитывалось в виде энтальпий полиморфных превращений при 623 и 752 К (см. ниже).
Данные [278, 807] в интервале 298—723 К лежат ниже принятого уравнения на 2%.
Температура первого полиморфного превращения F23+5 К) и соответствующая ему энтальпия
0,80+0,08 кДж-моль приняты по работе [805]. Принятое значение Ttr согласуется в пределах
погрешности с данным [1560, 1628, 3733, 3764, 3968]; менее точные измерения [659, 807, 1014,
1077, 1687] E93—643 К) не учитывались. Принятое значение &trH согласуется в пределах
погрешности с данными [3733] @,88 кДж-моль).
Температура второго полиморфного превращения G52+2 К) х принята по согласующимся
между собой результатам термографических [1014, 1077, 1628, 3764, 3968], рентгенографических
[1560] и калориметрических измерений [805, 807]; соответствующая этому превращению
энтальпия B,1+0,2 кДж-моль) вычислена по калориметрическим данным [4035] и согласуется в
пределах поГрешности с рекомендациями [805] A,9 кДж-моль) и [3733] B,0 кДж-моль).
Результаты менее точных определений Ttr [659, 1687, 3733, 4035] G18—762 К) не учитывались.
Уравнение для теплоемкости y-Na2CO3 (см. табл. 46.1) найдено совместной обработкой данных по
энтальпии, полученных в работах [2845] G07-1127 К), [4035] G35-1118 К) и [278, 807] G23-
1123 К).
Температура плавления A131+1 К) принята по хорошо согласующимся между собой данным
термографических исследований [229, 1628, 2167, 2843, 3733, 3764] и калориметрических работ
[2845, 4035]. Результаты менее точных определений Тт [278, 807, 1014, 1160, 3658, 3968] A073—
1127 К) не учитывались. Энтальпия плавления B8+2 кДж-моль) вычислена по уравнениям,
принятым для изменения энтальпии у-модификации и жидкого карбоната натрия, она совпадает
с величиной, найденной Янцем и др. [2845], и отличается от значений, полученных в работах
[4035] C3,5 кДж-моль), [807] C0,5 кДж-моль) и [3733] C2,2 кДж-моль).
Теплоемкость Na2CO3^) A95+8 Дж-К~1-моль~1) основывается на результатах измерений
энтальпии в работе Янца и др. [2845] A127—1210 К). Это значение не противоречит величинам,
1 Данные о наличии у Na2CO3 (к) третьего полиморфного превращения, полученные в работах [659] (891 К)
и [3968] (866 К), не подтвердились в последующих исследованиях.
356
Натрий и его соединения NaBO3(K, ж)
полученным для теплоемкости жидких карбонатов лития и калия. Данные [278, 807] и [4035]
не учитывались, поскольку их обработка привела для [4035] к резко заниженному значению
теплоемкости A62,5 Дж-К~1-моль~1), а для [278, 807] — к уравнению с максимумом при 1300 К.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,2,
2, 10 и 20 Дж-К~1-моль соответственно. Расхождения между термодинамическими функциями
Na2CO3(K, ж), приведенными в справочнике JANAF [2834] (Т ^ 2500 К) и в табл. 1140,
составляют 2—3,5 Дж-К-моль и обусловлены различием принятых уравнений для теплоемкости
и энтальпий фазовых переходов. ¦ .
Константа равновесия реакции Na2CO3(K, ж)=2^(г)+С(г)+ЗО(г) вычислена с
использованием значения Дг.йг°@)=2790,007+1,5 кДж-моль". соответствующего принятой в справочнике
энтальпии образования
A//°(Na2GO3, к, 298,15 К) = —1129,19 ± 0,26 кДж • моль.
Эта величина основана на выполненных Бергом и Вандерзее [1490] прецизионных
измерениях энтальпии реакций GO,(r)+2NaOH(ag)=Na2CO3(ag')+HaO(ffl), ДГЯ°B98,15 К)=—107,51 ±
+0,17 кДж-моль и Na2C03(K)+coH20=2Na+H)+C032M, Даг#°(соН20; 298,15 К) = -26,65±
+0,13 кДж-моль. Использованный в работе [1490] путь определения энтальпии образования
Ка2СО3(к) приводит к более точному значению по сравнению с расчетом, основанным на
измерениях энтальпии только второй из указанных реакций, так как при этом из расчета исключается
энтальпия образования СОд(ад), имеющая значительную погрешность @,25 кДж-моль).
Полученная в работе [1490] энтальпия растворения Na2CO3(K) подтверждается данными [4099]
(-26,61 кДж-моль-1), [4671] (-26,26 нДж-моль), [4528] (-26,36 кДж-моль), [3421, 3423]
(—26,35 кДж-моль) и [4446] (—27,1 кДж-моль). В перечисленных работах измерения
проводились при высокой степени разведения конечного раствора (моляльность менее 0,1). Их пересчет
к стандартному состоянию проводился с учетом гидролиза по методике, изложенной в работе
[4099]. Измерения при более высоких конечных концентрациях [1530, 1532, 2095, 3732, 3838,
4511, 4517] приводят к менее точным значениям стандартной энтальпии растворения Na2CO3(K).
К менее точным значениям энтальпии образования Na2CO3(K) приводят также измерения
энтальпии ряда реакций с участием растворов Na2GO3 [1526, 3582, 4511] и Na2CO3 в кристаллическом
состоянии [3518] (см. [1555]), а также исследования равновесия реакций с участием Na2CO3 12722,
2844, 2883, 2961, 3081, 3112, 3116, 3214, 3571, 3743].
КаВО2(к, ж). Термодинамические свойства кристаллического и жидкого метабората
натрия в стандартном состоянии при температурах 100—3000 К приведены в табл. 1141.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 46.1. В справочнике за стандартное состояние NaBO2(K) в интервале 0—1239 К принимается
гексагональная модификация [3383].
При Т <; 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работе Гренье и Веструма [2484] E—346 К, образец содержал 47,11+0,20% Na2O
и 52,91+0,13% В2О3 в хорошем соответствии с теоретическим составом: 47,09 и 52,91%
соответственно; содержание воды в образце не превышало 0,1%). Погрешность измерений оценена
авторами [2484] в 5% при Т < 14 К, 1% — в интервале 14—25 К и 0,2% при Т > 25 К.
Погрешности принятых значений S "B98,15 К) и #°B98,15 К)—#°@) (см. табл. 46.1) оцениваются
в 0,2 Дж-К-моль и 0,04 кДж-моль соответственно.
При Т ^> 298,15 К для теплоемкости NaBO2(K) принято уравнение, полученное при обработке
результатов измерений энтальпии в работе Панкраца и Ферранте [3753] D04—1200 К). При
обработке погрешность данных [3753] оценивалась в 1%.
Температура плавления A239+2 К) принята по согласующимся между собой данным Морп
и Мервина [3561] A239 К),' Пети и Егера [3807] A239 К), Бергмана и др. [99, 110, 111, 145, 1671
A239 К), Лесных и др. [627] A239 К); менее точные результаты получены в работах [106, 876,
1900, 2850, 3509, 3753] A237—1247 К). Энтальпия плавления C3,5+2,0 кДж-моль) основана
на согласующихся результатах расчетов из данных по диаграммам состояния двойных систем,
проведенных авторами [2835] (использованы работы [99, 110, 627, 3039]) и Пети и Егером [3807].
Теплоемкость КаВО2(ж) A45+15 Дж-К~1-моль) оценена с учетом найденной
экспериментально величины для метабората лития.
357
NaBO2(r)
Глава 46
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,1, 1, 5 и 10 Дж-К~1-моль. Термодинамические функции NaBO2(K, ж), приведенные
в табл. 1141, незначительно (в пределах 0,2 Дж-К^-моль в значениях Ф°(Г)) отличаются от
вычисленных в таблицах JANAF [2835] (Г <! 2500 К).
Принятая в справочнике энтальпия образования
bfH°(NaBO2, к, 298,15 К) =—976,5 + 2,5 кДж • моль
основана на измерениях Шульца и др. [1181 ] и Адами и Джо [1248]. Шульц и др. [1181 ] измерили
энтальпии растворения NaBO2(K) и В2О3(стекл.) в 2N азотной кислоте и нашли ДГЯ°B98,15 К)
равное —53,1+0,8 и —38,5+0,8 кДж-моль соответственно. Вместе с вычисленной по данным
настоящего справочника энтальпией растворения Na2O(K) в 2ЛГ азотной кислоте (Ari7°B98,15 K) =
=—351,9+0,5 кДж-моль) это дает AfH°(NaBO2, к, 298,15 К) =—976,8+2,4 кДж-моль.
Совпадающее значение : энтальпии растворения NaBO2(K) в 2N азотной кислоте было вычислено
(см. [2834]) по данным работы Шертсиса и Каппса, [4252] (см. также работы [2495] и [2902]).
Адами и Джо [1248] измерили энтальпии растворения NaBO2(K) и В2О3(к) в НСЫ2,731Н2О,
откуда для реакции V2Na2O(K)+1/2B2O3(K) = NaBO2(K) было получено ДГЯ°B98,15 К) =—131,8 +
+ 3,3 кДж-моль. Этой величине соответствует AjH°(NaBO2, к, 298,15 К) = —975,8+3,4 кДжХ
Хмоль, что находится в хорошем соответствии с результатами [1181].
Давление пара метабората натрия в реакции NaBO2(K, ж) = КаВО2(г) вычислено с
использованием принятой в справочнике энтальпии сублимации
ДгЯ°@) = Ag#°(NaBO2, к, 0) = 341+6 кДж • моль.
Результаты определений энтальпии сублимации на основании измерения давления пара и
равновесия реакций представлены в табл. 46.28. Приведенные в таблице погрешности учитывают
Таблица 46.28. Результаты определений A,H"°(NaBO2, к, 0) (в кДж • моль)
Авторы
Коул, Тейлор, 1935 [1901]
Бюхлер, Берковяц-Маттук, 1963
[1703]
Крстер, Зсрштрём, 1965 [3115]
Горохов и др., 1971 [311, 642]
Иенсен, 1969 [2853]
Метод
Протока, 1423—1623 К, 5 точек
Масс-спектрометрпческпй, 1070 К; 1 точка
То же, 990—1140 К
Эффузпоныый, 1110—1316 К, 21 точка
То же, 1270 К, 1 точка
Маес-спектрометрическтш, 1080—1230 К, 7 серий
Спектрофотометрпя в пламенах H2+O2+N2,
Na(r)+HBOa(r) = NaBO2(r)+H(r), 2020—2560 К,
данные представлены в виде уравнения
Asff°(NaBO2, к, 0)
II закон
III закон
471+45 336+10
- 353
320 ±13 —
386+12 341+6
— 338,3
305+10 —
310 323+20
воспроизводимость измерений и неточность использованных в расчетах термодинамических
функций. Принятая величина основана на результатах эффузионных измерений, выполненных Крёге-
ром и Зёрштрёмом [3115]. В пределах погрешностей эта величина согласуется с результатами
измерений [311,642,1901]. Основным источником погрешности принятой величины является
неточность термодинамических "функций.
NaBO2(r). Термодинамические свойства газообразного метабората натрия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1142.
Молекулярные постоянные, использованные для расчета термодинамических функций NaBO2,
приведены в табл. 46.7. Электронографические исследования [15, 16, 402], исследование ИК-
спектра молекул NaBO2, изолированных в Аг- и Кг-матрицах [4237 ], и расчет функции
потенциальной энергии на основании ионной модели показали, что молекула NaBO2B электронном состоянии
ХХА' имеет угловую конфигурацию с линейным фрагментом О—В = О (симметрия Cs). При этом
в электронографическом исследовании [402] для -^Na—О—В получено эффективное значение
358
Натрий и его соединения NaAlF4(r)
106+5°, в спектральной работе [4237] найдено значение 90°, а минимуму функции потенциальной
энергии NaBO2 соответствует равновесный «$Na—О—В=95+5° [400]. Расчет [400] показал
также, что энергии линейной и циклической конфигурации NaBO2 превышают энергию угловой
конфигурации на 900+100 и 6000+300 см соответственно. Учитывая заметные отличия
равновесного и эффективного значений -^М—О—В в метаборатах щелочных металлов, а также
ограниченные возможности электронографического метода в определении -^tNa—-О—В в NaBO2, в
настоящем справочнике для него принято эффективное значение 130+30°, равное соответствующему
углу в CsBO2, где оно определено с наибольшей точностью по сравнению с метаборатами других
щелочных металлов. Приведенное в табл. 46.7 произведение моментов инерции рассчитано по
структурным параметрам r(Na—О) = 2,14 + 0,03 А, г(В—О) =1,30 +Ц'^А и г(В=О) = 1,20+^ А
|4.02]. Погрешность значения IaIbIc составляет 1 • 10~114 г3 • см6.
Основные частоты NaBO2 приняты по работе Сешадри и др. [4237] после пересчета их на
природную смесь изотопов бора 1. В ИК-спектре высокотемпературного пара над 1ЧаВО2(к)
наблюдались частоты vx, v3 и v4 [1705] и vx—v6 [648], значения которых хорошо согласуются между собой,;
но на 15—70 см отличаются от данных [4237] 2. С учетом результатов исследований спектра
высокотемпературного пара [648, 1705] и возможных матричных сдвигов погрешности принятых
частот vx—ve оцениваются равными 40, 60, 20, 20, 20 и 10 см".
Термодинамические функции NaBO2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), <1. 9), A. 10), A. 122)—A. 124), A. 128) и A. 130)
без учета возбужденных электронных состояний. Погрешности вычисленных значений
определяются неточностью принятых молекулярных постоянных, а также приближенным характером
расчета и составляют 5, 8 и 12 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К.
Ранее термодинамические функции NaBO2(r) рассчитывались в справочнике JANAF [2835].
Расхождения данных табл. 1142 и [2835] не превышают 1 Дж-К-моль~1 в значениях Ф°(Т).
Константа равновесия реакции NaBO2(r)=Na(r)+B(r)+2O(r) вычислена с использованием
значения ArZT°@) = 1792,097+8,2 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации NaBO2 (к).
Этим величинам также соответствует
Д,Я°(NaBO2, г, 0) = —630,768 + 6,5 кДж • моль.
NaAlF4(r). Термодинамические свойства газообразного тетрафторалюмината натрия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 1143.
Молекулярные постоянные, использованные для расчета термодинамических функций NaAlF4,
приведены в табл. 46.15. На основании электронографического [968] и спектроскопических [1978,
2750] исследований в справочнике принимается, что молекулы NaAlF4 в основном электронном
состоянии Х^А^ имеет бидентатную структуру (симметрия С2ь). Приведенное в табл. 46.15
произведение моментов инерции вычислено для структурных параметров: r(Na—F)=2,ll +0,02 А,
r(Al-FBHemn)=r(Al-F)K00I=1,69+0,04 A, -$FBBema-Al-FMemH=-*FMM-Al-FMOT=109,5+10°,
найденных в электронографическом исследовании Спиридонова и Ерохина [968] 8. Погрешность
IaIbIg составляет 1-1013 г3-см6.
Основные частоты NaAlF4 приняты на основании результатов изучения ИК-спектров молекул
NaAlF4, изолированных в матрице из Ne (Сивин и др. [1978]) и в матрицах из Аг и N2 (Хаглен
и др. [2750]). Частоты колебаний, не наблюдавшиеся экспериментально (v3, v4, v6 и v9), были
рассчитаны авторами [1978] и [2750] по силовым постоянным. Спектры, полученные в работах [1978,
2750], хорошо согласуются между собой, за исключением двух частот в области 379 и 620 см.
Частота 379 см, отнесенная в работе [1978] к молекуле NaAlF4, не была обнаружена в Аг- и
К2-матрицах в спектрах NaAlF4 и других фторалюминатов щелочных металлов [2750]. Хаглен
и др. [2750] объяснили ее появление как результат матричного расщепления частоты 372 см
1 Шаповалов [1141] исследовал ИК-спектр молекул NaBO2 в Аг-, Кг- и Хе-матрицах в области 400—30 см 1, но
идентифицировал одну полосу vf>=365 см в матрице из Аг.
^Частота v5=290 см, наблюдавшаяся в работе [648], по-видимому, не принадлежит NaBO2.
3 Авторы [968] отмечают возможность небольшого отклонения цикла AlF2Na от плоской структуры.
359
NaAlF4 (г) Глава 4С
(наблюдавшейся в матрице из Ne [1978] и идентичной с частотой 364 см в матрице из Аг [2750])
Различие в интерпретации частот в области 610—630 см (см. LiAlF4).
В табл. 46.15 для всех частот, кроме v3, v4, ve и v9, приняты значения, рекомендованные авторам!
[2750] для свободных молекул NaAlF4 на основании данных, полученных ими при исследования?
в матрицах. Значения v4 и ve были оценены при подготовке справочника на основании соответ
ствующих частот A1F" (см. т. 3) с учетом изменения частот колебаний в молекулах фторалюминато!
щелочных металлов. Частоты валентных колебаний Na—F (v3 и v9) были оценены на основанш
постоянных молекул Na2F2, Li2F2 и LiAlF4. Расчет этих частот в работе [2750] представляете}
'ошибочным. Погрешности в значениях v3, v4, v6 и v9 оцениваются в 20—25% от их величины, осталь
ных частот — в 10—15 см.
Термодинамические функции NaAlF4(r) вычислены в приближении «жесткий ротатор—гармо
нический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128)
A. 130) без учета возбужденных состояний. Их погрешности обусловлены приближенным харак
тером расчета и неточностью принятых молекулярных постоянных, они составляют 5, i5, 25 Дж><
хК^Хмоль в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции NaAlF4(r) рассчитывались в справочнике JANAF [2834]
а также в работах Снелсена и Сивина [4336] (до 2000 К), Морозова и др. [696] (до 5000 К) и Хаг
лена и др. [2750] (до 2000 К). Расхождения в функциях, приведенных в табл. 1143 и вычисленный
в [4336], [2750] и [2834], составляют 1,5, 3 и 5 Дж-К^-моль в значениях Ф°(Г) и обусловлень
различием в принятых частотах колебаний. Соответствующие расхождения для работы [696
достигают 7 Дж-К-моль~1.
Константа равновесия реакции NaAlF4(r)=Na(r)+Al(r)+4F(r) вычислена с использование!
значения ДгЯ°@)=2594,209+16 кДж-моль, соответствующего принятой в справочнике энталь
пии образования
A/F°(NaAlF4, г, 0) = —1850 ± 15 кДж • моль
Результаты определений этой величины представлены в табл. 46.29. Приведенные в таблиц
погрешности учитывают воспроизводимость измерений, неточность термодинамических функцш
Таблица 46.29 Результаты
Авторы
Колосов и др., 1974 [536]
Гриотейм и др., 1974 [2496]
Никитин, Сидоров, 1980 [3659]
определений A^_ff°(NaAlF4, г, 0) (в кДж-моль)
Метод
Масс-спектрометрический, 1/2Na5Al3F14(K)=
=1/2Na3AlFe(K)+NaAlF4(r), 910 К, 1 точка
То же, Na3AlF6(K)=2NaF(K)+NaAlF4(r), 910 К,
1 точка
То же, NaAlF4(r)=NaF(r)+AlF3(r), 910 К, 1 точка
То же, V5Na6Al3F14(K)-p/5AlF3(K)=NaAlF4(r),
910 К, 1 точка
Эффузионный, 1/5Na5Al3F14(K)+2/5AlF3(K)=NaAlF4(i
910 К, 1 точка
Электронный удар, D0(NaF—A1F3)—D0(LiF—A1F3)=
=60,0+2,5
To же, NaAlF4(r)+e(r)=Na+(r)+AlF3(r)+F(r)+2e(r
ДгЯ°@)=1297±25
III закон
—1842+15
—1860+15
—1845+15
—1843+15
), —1845+15
-1862+15
-1823+25
и термохимических величин, использованных в расчетах. Полученные величины совпадаю
в пределах их погрешностей. Принятая величина является средневзвешенной. (См. также работ]
[211, 536, 675, 724, 932, 933, 934, 1159, 2496, 2694, 2749, 4279, 4525], где приводятся дополнител!
ные экспериментальные данные и проводится их обсуждение.) Погрешность принятого значени
из-за неточности использованных термодинамических функций составляет ~10 кДж-моль"]
360
Натрий и его соединения Na3AIF6{n, ж)
Na3AlF6(K, ж). Термодинамические свойства кристаллического и жидкого гексафторалюмината
тринатрия (криолита) в стандартном состоянии при температурах 100—4000 К приведены
в табл. 1144.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 46.1. За стандартное состояние Na3AlF6(K) в интервале 0—838 К принята моноклинная
а-модификация, а в интервале 838—1286 К кубическая В-модификация.
При Т <J 298,15 К термодинамические функции вычислены по измерениям теплоемкости,
выполненным Кингом [3009] E1—298,15 К, образец содержал 32,76% Na и 13,04% А1 при
теоретическом составе 32,85 Na и 12,85% А1; примеси —0,3% К и 0,007% Li). Полученные данные
скорректированы на 0,1—0,2% для приведения к стехиометрическому составу'. Экстраполяция
теплоемкости по уравнению D (Ш/Т)+4Е B36/Т)+5ЕE68/Т) привела к значению ?°E1 К)=17,3 ДжХ
хК'^моль. Погрешности принятых значений <S°B98,15 К) и Я°B98,15 К)—Я°@) оцениваются
в 1,7 Дж-К-моль~1 и 0,17 кДж-моль соответственно.
При Т > 298,15 К энтальпия Na3AlFe(K) измерена Ротом и Бертрамом [4064] B93—1371 К),
Лященко [637] D40—1217 К) и Крестовниковым и Каретниковым [586] E73—1273 К); все эти
данные различаются между собой в пределах 6—8% и не согласуются с данными [3009] для Т ^
< 298,15 К.
Уравнения для теплоемкости a-Na3AlF6 B98,15—838 К) и |3-Na3AlF6 (838—1286 К)
(см. табл. 46.1) получены при обработке результатов измерений энтальпии, проведенных О'Брай-
еном и Келли [3692] на образце, использованном в работе [3009]. Для кристаллического криолита
данные [3692] скорректированы на 0,01—0,06%, так же как измерения [3009] при низких
температурах. Погрешность данных [3692] оценивается в 0,3% х.
Температура полиморфного превращения (838+3 К) и температура плавления A286+2 К)
принята по данным Ландона и Убеллоде [3173] 2. С принятым значением Ttr в пределах
погрешности согласуются результаты измерений [637, 1742, 2410, 4064, 4605], а с принятым значением
Тт согласуются данные [2311, 2688, 2693]. Значения Ttr и Тт, найденные в работах [1279, 2693,
3692, 3827, 4031, 4064], менее точны и не учитывались. Энтальпия полиморфного превращения
(9,15+1,7 кДж-моль) рассчитана из уравнений, принятых для изменения энтальпий а- и В-
модификаций. Энтальпия плавления A14,4+2,0 кДж-моль) рассчитана из уравнений, принятых
для изменения энтальпии Na3AlFe(K, ж) по данным [3692], и согласуется с результатами работы
Холма и Гронвольда [2688] A14,2+2,0 кДж-моль).
Теплоемкость Na3AlF6^) C91+8 Дж-К-моль) рассчитана из данных по энтальпии
жидкого криолита, полученных О'Брайеном и Келли [3692] A301,1 —1370,5 К) 3, и согласуется
в пределах погрешности с измерениями Холма и Гронвольда [2688] A290—1340 К), которые
приводят к значению 397,1 Дне-К-моль.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 3000 и 4000 К оцениваются в 1,
3, 40 и 55 Дж-К-моль~1 соответственно. Термодинамические функции криолита ранее
вычислялись в таблицах JANAF [2834] (Т < 3000 К). Расхождения в значениях Ф°(Г) при Т < 2000 К
не превышают 1 Дж-К~1-моль и увеличиваются до 2 Дж-К-моль~1 при 3000 К.
Константа равновесия реакции Na3AlF6(K)=3Na(r)-|-Al(r)+6F(r) вычислена с использованием
значения Дг#о@)=4424,335+6,3 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
/^"(NagAlFg, к, 298,15 К) = —3322,4 + 4,0 кДж • моль.
Результаты определений этой величины приведены в табл. 46.30. Указанные в таблице
погрешности учитывают воспроизводимость измерений, неточность термодинамических функций и
энтальпий образования, использованных в расчетах. За исключением работы [1431], все величины
совпадают в пределах их погрешностей. Принятая в справочнике величина является
средневзвешенной, причем основной вес придавался наиболее точным измерениям, проведенным в работах
[1943, 2064].
С принятым уравнением для |3-Na3AlF6 в пределах 1—2% согласуются данные по энтальпии, полученные Холмом
и Гронвольдом [2688] в узком интервале температур A070—1175 К).
Данные о существовании полиморфного превращения при 1154 К, полученные авторами работы [3173], не
подтвердились результатами других исследований.
Результаты измерений для жидкого криолита скорректированы на 0,1% в соответствии со стехиометрическим
составом.
361
Na5Al3Fu(K, ж)
Глава 46
Таблица 46.30. Результаты определений AyjH"°(Na3AlFe, к, 298,15 К) (в кДж •
Авторы
Метод
моль 2)
A/l°(Na3AlF6, к, 298,15 К)
II закон
III закон
Машовец, Юдин, 1962 [675] Протока, ^NagAlF^+HaCKr^VsAlaOatK^HFtrH 3301+12 —3322+7
+2NaF(K), 1101—1315 К, 9 точек
Оно и др., 1965 [3719] ДТА, 2NaF(K)+-1/3A^)=Na(r)+1/8Na3AlF6(K), 1063- -3188 -3326+11
1153 К, данные приведены в виде уравнения
Дьюпнг, 1970 [2063] Протока, 2NaF(K)+1/3Al(K, ж)=Ка(г)+-1^а3А1Р6(к.), —3329+7 —3326+8
908—1089 К, 13 точек
Дьюинг, 1970 [2064] ЭДС, 3NaF(K)+AlF3(K)=Na3AlFe(K), 839—943 К, —3316+5 —3321,9+4,0
9 точек
Бо, 1904 [1431] Калориметрический, растворение в плавиковой кис- —3257
лоте, 3NaF(K)+AlF3(K)=Na3AlF6(K), Дг#°B98,15 К)=
=-170,3
Кофлин, 1958 [1943] Калориметрический, растворение в соляной и плави- —3323,4+4,0
ковой кислоте, 303 К, Al(K)+2,96NaCl(K)+5,96HF (р-р,
5,716 Н2О)+3,617 H2O(ж)=Na2 96A1F5 96(к)+
+2,96НС1 (р-р, 12,736Н2О)+3/2Н2(г), Д,#°B98,15К)=
=-641,1+0,6
Гросс и др., 1961 [2502] Калориметрический, 3/2PbF2(K)+Al(K)+3NaF(K)= —3322,5+6,5
=3/2Pb(K)+Na3AlFe(K), ДГЯ°B98,15 К)=-578,7+0,5
Хонг, Клеппа, 1978 [2698] Калориметрический, двойной высокотемпературный —3308+10
калориметр, 3NaF(K)+AlF3(K)=Na3AlF6(K),
Д,Я°A320 К)=— 44,8+2,0
Na5Al3F14(K, ж). Термодинамические свойства кристаллического и жидкого хиолита (фторалю-
мината натрия состава Na5Al3F14) в стандартном состоянии при температурах 100—3000 К
приведены в табл. 1145.
Значения постоянных, использованные для расчета термодинамических функций,
представлены в табл. 46.1. За стандартное состояние хиолита в интервале 0—1010 К принята
тетрагональная модификация [4981а].
При Т ^ 298,15 К термодинамические функции хиолита вычислены по результатам измерений
теплоемкости в работе Стьюва и Ферранте [5054а] 1 E,8—304 К, образец содержал
~99,9% Na5Al3F14, включая Na — 24,80%, А1 — 17,54%, F — 57,58%). Экстраполяция
теплоемкости ниже 5,8 К привела к значению 5°E,8 К)=0,14 Дж-К~1-моль~1 [5054а]. Погрешности
значений S°B98,15 К) и Я°B98,15К)—#°@), приведенных в табл. 46.1, оцениваются в 1 Дж-К^Х
Хмоль и 0,2 кДж-моль соответственно.
Данные по энтальпии Na5Al3F14(K) при Т > 298,15 К, полученные в работах Стьюва и Ферранте
• [5054а] D03—992 К, 8 измерений) и Холма [2694] F67—932 К, 8 измерений, образец содержал
0,2% примесей), систематически расходятся между собой (данные [2694] выше в среднем на —3%).
Причины расхождений неясны; в справочнике отдано предпочтение измерениям [5054а], поскольку
они выполнены в более широком интервале температур и хорошо согласуются с измерениями
теплоемкости, проведенными в той же работе. Уравнение для теплоемкости хиолита в интервале 298,15—
1010 К (см. табл. 46.1) выведено совместной обработкой данных [5054а] и [2694]; погрешности
этих данных оценены в 0,5 и 3% соответственно.
Температура плавления хиолита A010+10 К) принята по согласующимся между собой данным
Холма [2694] и Дьюинга [2064]. Хиолит плавится инконгруэнтно (с образованием расплава
и небольшого количества Na3AlFe(K)), однако двухфазная область очень узка A010—1040 К),
ввиду чего при расчете термодинамических функций Na5Al3F14^) принимался конгруэнтный
характер его плавления.
1 В работе [5054а] установлена слабая аномалия («выпуклость») на кривой теплоемкости хиолита в интервале
140—175 К, которая была учтена в расчетах. При использовании данных [5054а] по теплоемкости хиолита при
Т < 304 К учитывались исправления, которые авторы провели после публикации работы.
362
Натрий и его соединения NaLi(r)
Энтальпия плавления B29+8 кДж-моль) вычислена по уравнениям для энтальпии
кристаллического и жидкого хиолита, найденным в работе Холма [2694], и отнесена к 1010 К.
Использование этого значения LmH вместе с принятым в справочнике уравнением для теплоемкости
NagAl3F14(K)'равнозначно введению поправки (—8 кДж-моль) в данные Холма [2694] по
энтальпии жидкого хиолита. Принятое значение теплоемкости жидкого хиолита (974+30 Дж-К~1-моль~1)
также основано на работе Холма [2694], в которой энтальпия Na5Al3F14^) была измерена в
интервале 1047—1173 К (9 точек). Погрешности значений Ф°(Г) для Na6Al3F14(K, ж) при 298,15, 1000,
2000 и 3000 К оцениваются в 0,7, 3, 35 и 87 Дж-К^-молъ соответственно. В справочных
изданиях термодинамические функции Na5Al3F14(K, ж) ранее не публиковались.
Константа равновесия реакции Na5Al3F14(K)=5Na(r)+3Al(r)+14F(r) вычислена с
использованием значения АгЯ°@) = 10 130,763+17 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^i^NagAlgF^, к, 298,15 К) = —7555 + 10 кДж • моль.
Принятая величина была вычислена по уравнению III закона на основании результатов
исследования реакции 5NaF(K)+3AlF3(K) = Na5Al3F14(K) (844—996 К, 20 точек) методом ЭДС в
работе Дьюинга [2064]. Расчет по уравнению II закона приводит к значению —7563+10 кДж-моль.
NaLi(r). Термодинамические свойства газообразного натрия—лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1146.
Молекулярные постоянные Na7Li, приведенные в табл. 46.4, приняты на основании анализа
результатов спектральных исследований [2624], квантовомеханических расчетов [1533, 4012,
4050] и оценок. В спектре молекулы Na'Li изучена лишь одна система полос В1!! -> ХгЕ+ [2624].
Квантовомеханический расчет методом псевдопотенциала, проведенный Роачем [4012], показал,
что трем первым диссоциационным пределам LiB6')+NaB>S), LiBP)+NaB5) и LiB5)+NaBP)
соответствуют основное Х~Ч,+ и пять возбужденных связанных состояний а3П,Л1Е+, 63Е+, ВШш
Колебательные постоянные NaLi в состоянии ZXE+, приведенные в табл. 46.4, были определены
Хесселом [2624] при анализе системы 541 -*¦ ХгТ,+{и" ^ 8) в спектре лазерной флуоресценции.
Полученные в работе [2624] вращательные постоянные E„=0,396 см, ах=3,6-10~3 см, ге =
=2,814 А) вызывают сомнение нз-за их существенного отличия от результатов прецизионного
неэмпирического квантовомеханического расчета Росмуса и Мейера [4050] E^=0,383 см, а1 =
=3,2-10 см, ге=2,873 А) и оценки по принятым постоянным молекул Li2 и Na2 [1772]
{ге = 2,875 А). Поэтому в табл. 46.4 приведены значения Ве, а1 и ге, оцененные при подготовке
справочника по интерполяционной схеме Кавальере и др. [1772] с использованием
молекулярных постоянных гомоядерных двухатомных соединений щелочных металлов. Погрешность
значения Ве не превышает 0,01 см. Молекулярные постоянные в состоянии В1!! приняты по данным
Хессела [2624] и относятся к уровню с неизвестным значением колебательного квантового числа и,
¦с которого наблюдалась флуоресценция. Энергии возбуждения экспериментально не
наблюдавшихся состояний оценены по соотношению A. 89) и данным для молекулы KNa, поскольку такая
оценка для состояния .б1!! (Те=20 600 см) оказалась более близкой к экспериментальному
значению, чем результат расчета Роача [4012] (Гс = 21 400 см). Погрешность оцененных
значений Те оценивается в 1000—2000 см.
Термодинамические функции NaLi(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн и ее производные вычислялись по A. 90)—A. 92) с
учетом пяти возбужденных электронных состояний, в предположении, что <?Й,.еР.=.Р»<2к5.бр-
Статистическая сумма по колебательно-вращательным уровням энергии основного состояния X1 S +
и ее производные находились по уравнениям A. 73)—A. 75) непосредственным суммированием
по колебательным уровням и интегрированием по / с помощью соотношений типа A. 82). В
расчете учитывались уровни с / <С /шах „, последние находились по формуле A. 81). Уровни энергии
вычислялись по уравнениям A. 62), A. 65) с коэффициентами Ykl, приведенными вместе со
значениями утах и /lim в табл. 46.5. Величины Ykl были получены при помощи соотношений A. 66) по
постоянным молекулы Na'Li из табл. 46.4 без учета состава природной смеси изотопов. Для
обеспечения условий A. 50) и A. 59) были введены коэффициенты Y30 и 1^03 соответственно.
363
NaLi(r) Глава 46
Погрешности вычисленных термодинамических функций NaLi(r) обусловлены неточностью
вращательной постоянной в состоянии X1 S+, неточностью энергии диссоциации и энергии
возбужденных электронных состояний. Общие погрешности в значениях Ф°(Т) при 298,15, 3000 и
6000 К соответствуют 0,1, 1 и 4 Дж-К~1-моль соответственно.
Термодинамические функции NaLi(r) ранее вычислялись в работе Змбова и др. [4826] (Ф°(Т)
при Г=400-^-800 К); расхождения данных [4826] и табл. 1146 достигают 0,5—1,5 Дж-К^-моль
в значениях Ф°(Т) и объясняются различием принятых вращательных постоянных и методов
расчета.
Константа равновесия реакции NaLi(r) = Li(r)+Na(r) вычислена с использованием принятого
в справочнике значения A7.^°@) = D0(NaLi)=85,8+2,0 кДж-моль~хг«7170 + 170 см.
Принятое значение было получено Змбовым и др. [4826] при масс-спектроскопическом
исследовании равновесия реакций: Na2(r)-|-Li(r)=NaLi(r)+Na(r) и NaLi(r)=Na(r)+Li(r). Оно
удовлетворительно согласуется с результатами квантовомеханических расчетов 80,7 [1533], 85 [4012]
и 82,3 кДж-моль-1 [4050].
Принятой энергии диссоциации соответствует
i\fH°{NaLi, г, 0) = 179,693 + 2,3 кДж • моль.
Глава 47
КАЛИЙ И ЕГО СОЕДИНЕНИЯ
2(к, ж), К2О2, КН(к, ж),
, К2С12, КВг(к, ж), КВг,
O2, KNO3(k, ж), KNO3,
КАЮЛк ж) KA1F
В настоящей главе рассматриваются калий и его соединения с кислородом, водородом и галогенами,
а также некоторые соли калия и кислородсодержащих кислот, фторалюминаты и фторуранат
калия и его соединения с литием и натрием — всего 33 вещества. Для 19 веществ представлены
сведения о свойствах в конденсированном состоянии, для 28 — в газообразном состоянии, причем
для К и К+ — при температурах до 10 000 К.
Термодинамические свойства всех веществ рассчитаны для природной смеси изотопов калия
(атомный вес 39,0983) и других элементов. Однако различие молекулярных постоянных разных
изотопомеров учитывалось только для некоторых двухатомных соединений (см. тексты).
В Приложении 3.10 приведены сведения о критических постоянных элементарного калия,
а также постоянные, позволяющие учитывать отклонения термодинамических свойств К и КС1
от их свойств в стандартном состоянии. В справочнике не рассматриваются некоторые
соединения калия, образующиеся при высоких температурах (К+, K2F+, KF2 и др.).
К(к, ж). Термодинамические свойства кристаллического и жидкого калия в стандартном
состоянии при температурах 100—2200 К 1 приведены в табл. 1147.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние К(к) в интервале 0—336,86 К принята
кубическая модификация (структурный тип Na).
При Т ^ 298,15 К термодинамические функции вычислены по измерениям теплоемкости калия
в работах Крира и др. [3107] A2—320 К), Филби и Мартина [2251] @,4—26 К) и Дофинэ и др.
[1997] C0—330 К), с учетом данных Лина и Филлипса [3258, 3259] @,17—4,1 К) и Робертса
[4020] A,5-20 К). Измерения [2142] F9-102 К) и [4296] A5-277 К) менее точны и не
учитывались. Погрешности принятых значений 5°B98,15 К) и Я°B98,15 К)—Н°@), которые совпадают
с рекомендованными КО ДАТА—MGHG [1887], оцениваются в 0,2 Дж-К^-моль и 0,02 кДжХ
Хмоль соответственно.
В интервале 298,15—336,86 К для теплоемкости твердого калия принято уравнение,
выведенное по данным Дугласа и др. [2105] B98—332 К) и Крира и др. [3107]; их погрешность в
указанном интервале оценивается в 0,5%.
Температура плавления C36,86+0,02 К) принята по работе Отта и др. [2428, 3735], где она
была определена для образца калия чистотой 99,9%. Близкие величины были получены Карпен-
тером и Стюардом [1759] C36,56 К), Дугласом и др. [2105] C36,35 К) и Стоксом [4398] C36,86 К).
Энтальпия плавления калия B,321+0,020 кДж-моль) принята по результатам измерений
энтальпии, проведенным Дугласом и др. [2105]; в работах [1759, 3358, 3978] были получены
значения 2,377, 2,314 и 2,39 кДж-моль соответственно. Уравнение для теплоемкости жидкого калия
выведено совместной обработкой результатов измерений энтальпии К(ж), выполненных Дугласом
и др. [2105] C73—1070 К), Шпильрайном и Каганом [1167] E19—1316 К), Леммоном и др. C73—
1173 К) 2 и Пчелкиным [844] 3 C73—1473 К). Погрешность данных [2105] оценена в 0,7%,
данных [1167] и [1179] — в 3% и данных [844] — в 5%. Приведенному в табл. 47.1 уравнению
соответствует уменьшение теплоемкости от 6*^C36,86 К)=32,13 Дж-К~1-моль~1 до СG50 К) =
=29,8 Дж-К~1-моль~1 (минимум теплоемкости), а затем ее ускоренное возрастание до 54 Дж-К~*Х
1 Гст(К)=2280 + 50 К.
2 В связи с отсутствием оригинальной работы данные Леммона и др. заимствованы из книги Шпильрапна и др.
[1179].
3 Данные [844] по теплоемкости К (ж) были проинтегрированы и вводились в обработку в виде 12 значений эн-
тальпни На(Т)—Н°(Тт) в интервале 373—1473 К.
365
К (к, ж)
Глава 47
Таблица 47.1. Принятые значения термодинамических величин для калия и его соединений
в кристаллическом и жидком состояниях
Вещество
К
ко2
К2О
к2о2
кн
кон
KF
КС1
КВг
KI
K2SO4
KNO2
KNO3
к2со3
K2Si03
K2Si205
кво2
КА102
Состояние
к, куб.
ж
к11, тетр.(р)
к1, куб.(с)
ж
кШ, куб(ч)
к11,куб.C)
к1, гекс. (а)
ж
к, ромб,
ж
к, куб.
ж
к11, монокл.
к1, куб.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
к11, ромб.
к1, гекс.
ж
к11, гекс.
к1, куб.
ж
КИ, ромб.(а)
к1, гекс. ф)
ж
1
ю
^ч , ,
оо" S
Qi 0
estu
?3 I
кДжХ
X моль
7,088
16,82
13,7
17
7,8
12,15
10,00
11,37
12,21
12,70
25,435
22,94
18,75
к11, монокл.(а) 22,665
к1, гекс. (C) —
/К
к, ромб,
ж
кШ, монокл.
кН, (р)
к1(т)
ж
к, гекс.
ж
кП, куб.
к1, куб.
21,86
(а) 28,82
12,114
13,395
ОС
аз
2L
о
со
ОС
аг
CNJ
о R.
Дж ¦ К X
X моль J
64,68
125,4
96
117
51,3
78,87
66,50
82,57
95,92
106,05
175,56
152,1
132,9
155,50
146
190,58
80,00
%Ъ,ЪЪ
29,60
77,53
72
• 91
37,9
64,90
48,99
51,30
52,47
52,80
131,46
107,4
95,06
114,43
117,57
160,95
67,03
16,12,
Коэффициенты в уравнении
для с;(Г)
а
82,924
39,288
52,325
88
100
51,057
100
100
100
76,088
134
34,031
56
42,596
79
83
48,273
70,3
44,008
72,0
43,241
69,5
43,538
70,3
96,928
-287,152
200
3321,454
81,714
102
21,540
-418,441
141
67,798
108,922
205,5
116,485
190
191,852
158,005
224,225
275
81,993
145
66,206
6-Ю3
_394,997
—24,334
84,538
70,242
50,014
33,233
74,808
12,996
33,165
21,117
22,396
131,099
339,765
—16355,917
16,736
207,936
1215,640
156,403
68,852
52,007
36,585
90,826
4,418
22,7?й
36,42.7
30,975
с • 10*
°а «
0,865 б
—
—
5,369
2,807
0,002 а
-2,607
-2,2?8
4,049
—1399,339
а
-10,244
—267,060 а
—
12,819
37,166
9,987
19,342
13,006
—68,200
Интервал
температур
или
тт
К
298,15-336,86
336,86—2200
298,15—422
422—808
808—2000
298,15—590
590-645
645-1013
1013-3000
298,15—818
818—2000
298,15—892
892—1200
298,15—520
520—678
678-3000
298.15—1131
1131—3400
298,15-1044
1044—3400
298,15-1007
1007—3400
2ГР.1?-'"Г/(
954—3300
298.15—857
857—1342
1342—3500
298,15—314,7
314,7—711
711—2500
298,15—402
402—607,7
607,7—2500
298.15—693
693—1173
1173—3000
298,15—1249
1249—4000
298,15-510
510—867
867—1318
1318—4000
298.15—1220
1220—2500
298,15—810
810—1986
336,86
422
808
590
645
1013
818
892
520
678
1131
1044
1С07
954
857
1342
314,7
711
402
607,7
693
1173
1249
510
867
1318
1220
810
1986
или
кДгк X
X моль"
2,321
0
20,6
0,7
4
27
20,5
21
6,45
9,4
28
26,32
25,52
24,02
• 8,0
36,3
0,75
18,7
5,04
9,86
2,1
30,5
20
1,2
1,6
35,2
33,5
1,295 !
82 [
366
Калий и его соединения
К(к, ж)
Таблица 47.1 (окончание)
Вещество
K3A1F6
Состояние
К)-
Ю
СО "~
loff
кДж
X
X моль^1
ж
к11, тетр. 42,
к1, куб.
ж
Примечание. C°p(T)=a-\-b ¦
К: а
КС1: а
й=724,957 -Ю-6; б
d=—35,991 -Ю-6,
5,15
см
о
Дж-
сг
ю
со
о~а.
О
К X
X моль ^
— —
6 285
_
—
Коэффициенты в уравнении
а
- 130
223 152,870
— 240,540
- 393
T—c-T-2+d-T2+e-Ts (в Дж
й=15,863
е=22,850
К)"8. KN02:
Ю-9. KNOS:
ДЛЯ Up(l )
Ъ ¦ 103
с ¦ 105
—
235,216 —
55,770 —
— —
•К -моль-1).
а ^=0, «=62 725,964-10-
а d=—678,412 -Ю-6.
Интервал
температур
К
1986—4000
298,15—600
600—1273
1273—4000
или
Т
-
600
1273
—
AtrH°
или
кДж X
X моль-1
6,5
122,6
—
Хмоль'1 при 2000 К. Точность описания энтальпии жидкого калия этим уравнением оценивается
в 0,7% при 1000 К, 1,5% при 1500 К и 3% при 2000 К. Погрешности вычисленных значений
Ф°(Т) при 298,15, 1000, 1500 и 2000 К составляют 0,15, 0,5, 0,8 и 1,5 Дж-К^-моль-1
соответственно.
Значения Ф°(Т), приведенные во втором издании настоящего справочника [337] (Т ^ 2300 К),
таблицах JANAF [2834] (Т < 2000 К), справочнике Халтгрена и др. [2752] (Т < 1600 К) и книге
Шпильрайна и др. [1179] (Т <С 1500 К), согласуются с данными табл. 1147 в пределах 0,04 ДжХ
хК~1-моль~1 при Т ^ 1000 К и 1 Дж-К~1-моль~1 при более высоких температурах. Расхождения
обусловлены в основном тем, что в настоящем справочнике принято более быстрое возрастание
теплоемкости К (ж) при Т > 1000 К.
В справочнике в качестве стандартного состояния калия принято кристаллическое состояние
и в соответствии с этим
bfH°(K, к) = 0.
Давление пара калия в реакции К (к, ж)=К (г) вычислено с использованием значения Ari7°@) =
= 89,891+0,5 кДж-моль, соответствующего принятой в справочнике энтальпии сублимации
\Н°(К, к, 298,15 К) = 89,0+ 0,5 кДж ¦ моль.
Принятая величина основана на представленных в табл. 47.2 результатах измерений давления
насыщенного пара калия. Приведенные в таблице погрешности учитывают только
воспроизводимость измерений. Расчет энтальпии сублимации проводился для модели идеальной смеси химически
реагирующих атомарной и молекулярной компонент. При высоких температурах учитывалась
поправка на неидеальность с использованием уравнения состояния для калия. Метод расчета
изложен в Приложении 8.
Большинство из приведенных в табл. 47.2 работ приводят к хорошо согласующимся между
собой значениям энтальпии сублимации. Рекомендованное значение вычислено как
средневзвешенное из наиболее надежных данных [1171, 1209, 3356, 4091, 4184, 4635] (метод точек кипения)
и данных [1176, 1609, 1638, 3999, 4402] (различные варианты статического метода). Всего для
расчета средневзвешенного было использовано 360 точек, охватывающих интервал 718—1644 К.
Погрешность принятой величины обусловлена главным образом неточностью термодинамических
функций калия в конденсированном состоянии, приводящей к погрешности в энтальпии
сублимации от 0,3 кДж-моль для измерений при 700 К до 1,5 кДж-моль^1 для измерений при 1700 К.
Принятое значение энтальпии сублимации совпадает со значением, рекомендованным КОДАТА—
MCHG [1887].
Данные работ [1108, 2563, 2635, 2997, 4682] менее точны и не принимались во внимание.
367
К(г)
Глава 47
Таблица 47.2. Результаты определений Л,Д"°(К, к, 298,15 К) (в кДж ¦ моль)
Авторы
Метод
Д,Я°(К, к, 298,15 К) а
II закон
III закон
Руфф, Иохаязен, 1905 [4091]
Хакдшиль, 1913 [2542]
Точек кипения, 1030,65 К, 1 атм, 1 точка
Статический, 537—673 К, 1-Ю—6-Ю"8
8 точек
Кронер, 1913 [3119] То же, 523—672 К, 7 -Ю-5—5 -Ю атм, 17 точек
Фиок, Родбуш, 1926 [2258] То же, 679—1033 К, 6 Л0~3—1 атм, 10 точек
Эдмондсон, Эджертон, 1927 [2144] Эффузионный, 373—474 К, 2 -Ю"8—9 -Ю атм,
10 точек
атм, 87,9+12,0
89,0
88,04+0,68
86,9±6,8 89,Е5±0,61
90,84 ±0,16 89,41 ±0,24
89,92±0,55 89,71 ±0,04
Льюис, 1931 [3248]
Майер, 1931 [3432]
Нейман, Волькер, 1932 [3647]
Макензи и др., 1956 [3356]
Редер и Моравнц, 1956 [4026]
Грачев, Кириллов, 1960 [316]
Бонилла, 1961 [1609]
Воллинг, 1963 [4635, 4636]
Ригней и др., 1965 [3999]
Аченер, 1965 [1209]
Бук, Паули, 1965 [1709]
Стоун и др., 1966 [4402]
Виноградов, Воляк, 1966 [217]
Шпильрайн, и др., 1968 [1176]
Боулс, 1968 [1638]
То же, 581—714 К, 5 -10-*—1 -Ю атм, 7 точек
2-Ю-8—6-10-' атм,
85,9 ±5,0
90,2±2,5
90,0±0,4
88,58 ±0,ЕЗ
89,56±0,02
88,73+0,04 б
92,0±1,2 89,59+0,11
92,0
90,39 ±0,63 б
Торзионный, 332—416 К,
30 точек
То же, 418—472 К, 5 -Ю—9 -Ю атм, 28 точек 89,64 ±0,55
Точек кипения, 860—1281 К, 2-Ю—6,5 атм, 88,51 ±0,5
26 точек
Торзионный, 443—543 К, 2-Ю—2-10 атм
11 точек
Статический, 825—1553 К, 5-Ю—23,1 атм в
То же, 1029—1107 К, 1,02—1,97 атм, 20 точек
Точек кипения, 758—1431 К, 3-Ю—153 атм, 89,61 ±0,62 88,66 ±0,04 б
51 точка
Статический, 1065—1495 К, 1,3—19,2 атм, 45 точек 92,21 ±0,19
Точек кипения, 798—1316 К, 6-Ю—7,8 атм в 89,45±0,19
Ионизационный, 338—398 К, 1 -Ю"8—1-10~7 атм, 92,0±3,0
9 точек
Статический, 925—1585 К, 0,3—27,3 атм, 32 точки 90,76±0,22
То же, 725—1037 К, 1 -Ю-2—1 атм, 20 точек 90,8±2,4
То же, 794—929 К, 6 -Ю—4 -Ю атм, 10 точек
То же, 945—2169 К, 0,4—132 атм, 51 точка
88,73±0,06 88,45±0,01б
88,77 ±0,07 б
88,92±0,07 б
89,7±0,2
Герасимов и др., 1968 [273, 2386] Эффузионный, 343—471 К, 1-Ю"8—1-Ю атм в
Кирияненко, 1970 [513]
Шпильрайн, Никаноров, 1970
[1171]
Шине и др., 1971 [4184]
Кондратьев, Парфентьева, 1971
[539]
Фрейланд, Гензель, 1972 [2334]
Чернеева, Проскурин, 1972 [1110]
Точек кипения, 1061—1400 К, 1,4—12,35 атм,
9 точек
89,2+1,1
93,04 ±0,31
90,68
91,7+1,3
88,81 ±0,05 б
89,3 ±0,2
88,94 ±0,06
88,5 ±0,08 б
89,43 ±0,15
88,51 ±0,39 б
То же, 769—1554 К, 3 -10-2_24,4 атм, 108 точек 89,16+0,15 88,70±0,03 б
То же, 718—1006 К, 2-Ю-2—0,8 атм, 22 точки 89,98±0,8 88,94±0,09
То же, 731—1033, 2-Ю-2—1,4 атм, 24 точки 88,10±0,38 88,41 ±0,24
То же, 1262—1726 К, 5,2—45 атм, 6 точек 96,6+2,3 88,75±0,5б
То же, 1017—1833 К, 0,9—63,6 атм, 195 точек 91,28±0,12 90,13±0,07б
а Приведенные величины рассчитаны с учетом образования в насыщенном паре димерных молекул; б введены
поправки на отклонение от идеальности. Учтены точки, лежащие в интервале 1033 г$ Т sS 1644 К; в данные
представлены в видеуравнения.
К(г). Термодинамические свойства газообразного калия в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 1148.
Уровни энергии атома К, использованные при расчете термодинамических функций, приведены
в табл. 47.3, в которой даны валентные состояния К, соответствующие пяти валентным
электронным конфигурациям. Приведенные в этой таблице значения основаны на результатах спектральных
измерений Ридберга [4006], которые практически совпадают с рекомендацией Корлисса и Шугара
[1935].
Термодинамические функции К(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
значений при Т <С 5000 К определяются главным образом неточностью фундаментальных по-
368
Калий и его соединения
К+(г)
стоянных и не превышают 0,02 Дж«-К-иоль~1 в значениях Ф°(Т) и S°(T). При более высоких
температурах погрешности возрастают из-за пренебрежения ридберговскими состояниями и
при 6000 и 10 000 К могут достигать соответственно 0,05 и 1 Дж>К~1'Моль~1 в значениях
Ф°(Т) и 0,3 и 5 Дж-К-^моль в значениях S°(T).
Ранее термодинамические функции К (г) вычислялись в предыдущих изданиях настоящего
справочника [337, 340], таблицах JANAF [2834] и Мак-Брайд и др. [3439] до 6000 К, а также
в работах Гурвича и др. [335], Хилзенрата и др. [2652] и Рейтера [3973] до 10 000 К,
Кольского и др. [3067] до 8000 К и Эванса и др. [2195] до 2500 К. Расхождения результатов этих
расчетов и данных табл. 1148 при Т <4000 К, как правило, не превышают 0,03 Дж-К~1-моль
в значениях Ф°(Т). Расхождение сданными [3067,3973], по-видимому, вследствие ошибок в этих
расчетах, достигают 0,07 Дж-К~1-моль. При Т > 4000 К термодинамические функции К(г),
вычисленные в других работах, возрастают с температурой быстрее, чем в настоящем
справочнике. Максимальные расхождения имеют место с расчетами Гурвича и др. [335 ] и Рейтера [3973];
они достигают 10—11 Дж-К^-моль в Ф°A0 000 К) (см. Li(r)).
Энтальпия образования
Д,Я0(К, г, 0) = 89,891 ±0,5 кДж . моль
соответствует принятой энтальпии сублимации калия.
К+(г). Термодинамические свойства газообразного положительного иона калия в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 1149.
В табл. 47.3 приведено только основное электронное состояние XS иона К+, относящееся к
конфигурации . . .Зрв; состояния, относящиеся к конфигурации . . .3p63d, имеют энергии выше
160 000 см [1935], а другие возбужденные состояния К+ являются ридберговскими.
Термодинамические функции К+(г) были вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
для условия In (?вя=0. Погрешности вычисленных значений во всем интервале температур
обусловлены только неточностью фундаментальных постоянных и не превышают 0,02 Дж-К~1-моль.
Ранее термодинамические функции К+(г) вычислялись в предыдущих изданиях настоящего
справочника [337, 340] и в таблицах JANAF [2834] до 6000 К, а также в работах Хилзенрата и др.
[2652] до 10 000 К и Грина и др. [2473] до 50 000 К. Результаты этих расчетов совпадают с
данными табл. 1149 в пределах 0,01 Дж-К-моль~1, за исключением значений Ф°(Т) и S°(T) при
10 000 К в расчете [2652], где для этих
величин, по-видимому, допущена ошибка в 0,2
Дж-К~1-моль~1.
Константа равновесия реакции К+(г)+
-)-е(г)=К(г) вычислена на основании
значения ДГЯ°(О)=—418,810+0,001 кДж-моль,
соответствующего принятому в справочнике
потенциалу ионизации
/О(К)=35 009,77 ±0,10 см
=418,810+0,001 кДж-моль.
Это значение рекомендовано Мур [3549]
с погрешностью 0,02 см на основании
измерений ридберговских серий, выполненных
Рисбергом [4006]. С учетом результатов
других работ (см. [3550]) в настоящем
справочнике погрешность принятой величины
несколько увеличена. Принятому значению
соответствует •,
Таблица 47.3. Уровни энергии атомов К и К+,
принятые для расчета
термодинамических функций
AfH°{K+, г, 0) = 508,701 ±0,5 кДж • моль
-1
Атом
К
К+
Электронная
конфигура-
. ция
"¦ ¦ . . .4s
..Ар
, . .3d
. .Ad
• -...4/
' .. .зР«
Состояние
Г.
СМ
25 0
2i»Vi 12 985,17
2А/а' , . 13 042,88
*D.,t 21534,70
2Д8/а 21537,00
Ю,/г 27 397,10
2D3/j 27 398,14
aF 28 127,85
*Я 0
2
2
4
6
4
6
4
14
1
369
Ка(г)
К2(г). Термодинамические свойства газообразного дикалия в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 1150.
Молекулярные постоянные К2, принятые на основании анализа результатов исследований
электронного спектра и квантовомеханических расчетов, приведены в табл. 47.4. В
электронном спектре К2 наблюдались семь систем полос А1!,*—Х1^ [1761, 1955, 3463, 4008], СП,—
ХХЕ+ [1955, 2328, 3299, 4469], ЕШи—ХгЦ [4646, 4688, 4982а, 5047а], РШи—ХгЦ, G—Х1^:
[5047а, 50476], Н—Х1!^ и I—Х1!^ [5070а, 50706]. Согласно расчету методом псевдопотенциала
[4012], молекула К2 помимо экспериментально наблюдаемых имеет еще ряд стабильных три-
плетных и синглетных состояний (asHu, &32J, В1!,*, D1!!^, диссоциирующих на атомы в
состояниях 2?+2.Р 1. Молекулярные состояния, коррелирующие с более высокими атомными
уровнями, как и в случае Li2 и Na2, в справочнике не рассматриваются.
Молекулярные постоянные К2 в состоянии X1!,* и константы Те, Ве и De в состоянии СШ^
приняты по результатам анализа структуры полос с v'^.2 и v" ^ 20 системы СЩи—X1!,*
, в спектре лазерной флюоресценции, выполненного Танго и др. [4469]. Колебательные
постоянные, а также постоянные <хг и |3Х состояния СуП.в приняты по данным Лумиса и Нусбаума [3299],
которые получили их обработкой кантов полос с v' <^ 27 системы СЧ1и—X1!,*. Колебательные
постоянные и значение Те состояния -4*2* были найдены Крейном и Кристи [1955] при анализе
полос с v' ^ 32 и v" ^ 10 системы А1!^—Х1!^. Поскольку анализ тонкой структуры полос
этой системы не проводился, значение Ве в состоянии А1^ было оценено по соответствующим
постоянным молекулы Na2. Молекулярные постоянные в состояниях с3Пя, Ь3Е*, В1^ и ZI^
оценены на основании сопоставления результатов квантовомеханического расчета Роача [4012 f.
для молекул К2 и Na2 с величинами, принятыми в справочнике для Na2. Погрешности значений Те.
могут достигать 1000—2000 см, постоянных В„ и шв — 10—15%.
Термодинамические функции К2(г) были вычислены по уравнениям A.3)—A.6), A.9)г
A.10), A.93)—A.95). Значения QBS и ее производные вычислялись по соотношениям A.90)—
A.92) с учетом шести возбужденных электронных состояний. Колебательно-вращательные
статистические суммы всех состояний и их производные находились непосредственным
суммированием по колебательным уровням и интегрированием по значениям / с помощью 'соотношений
типа A. 81). В расчете учтены уровни, имеющие значения / <I «/max, p» последние находились
по формуле A.82). Уровни энергии вычислялись по уравнениям A. 64а) и A.65) с
коэффициентами Ykl, полученными для молекулы К2, соответствующей природной смеси изотопов калия,
по соотношениям A.66) и данным табл. 47.4. Для обеспечения условий A.50) для состояния Х1^,
добавлены коэффициенты Y80, У40, а для выполнения условия A.59) для всех состояний добавлен:
коэффициент Y03. Постоянные Ykl вместе со значениями vm% и /,,ш, приведены в табл. 47.5. Муль-
типлетность и вырождение состояний а3Пи, Ь3?*г С1Пи и Вги.д учитывались статистическими
весами 6, 3, 2 и 2 соответственно.
Погрешности рассчитанных термодинамических функций К2(г) при температурах до 1000 К
обусловлены главным образом неточностью энергии диссоциации молекулы К2 и связанной с ней
неопределенностью экстраполяции наблюдаемых колебательно-вращательных уровней. При более
высоких температурах основной вклад в погрешность вносит неточность энергий возбуждения
триплетных состояний. Погрешности в значениях Ф°(Г) оцениваются в 0,05, 0„5 и 2 Дж-К~1Х
Хмоль" при 298,15„ 3000 и 6000 К соответственно.
Термодинамические функции К2(г) ранее вычислялись в таблицах JANAF [2834], а также-
в работах Эванса и др. [2195], Гриффела [2486], Фебера и Херрика [2228], Артыма [45] и
Рейтера 13973] (Т < 10 000 К) и Шнейдера [4197а] (<?вн при Т < 2400 К). Применение
приближенной методики и неучет возбужденных электронных состояний в таблицах JANAF [2834] привели
к частичной компенсации ошибок, и расхождения этих данных и табл. 1150 не превышают 3 и
4 Дж-К-моль~1 в значениях Ф°(Г) и S°(T) соответственно. Результаты расчета Артыма [45]
согласуются с табл. 1150 в пределах 0,4 и 0,7 Дж-К'^моль в значениях Ф°(Т) и S°(T)
соответственно. Расхождения с данными Рейтера [3973], достигающие 3 Дж-К-моль в значении
Баскар и др. [1548] наблюдали в ИК-спектре паров К2 поглощение в области 1,1—1,6 мкм, связанное с
переходом из нестабильного состояния 3 2J в состояние Ь3 SJ.
370
Калии и его соединения
К,(г)
Т а б л и ц а 47.4. Молекулярные постоянные К2, Кf, КО, КН, KF, KC1, КВг, КГ, KLi, KNa
Молекула
Состояние
D, ¦ 10е
А
39к,
А12+
38Kt
ко
39КН
MKF
39К81Вг
3SKI
S9K7Li
38KNa
cm
СТЛ
от
о
9 600в
11 683,58
13 000 а
15 100 в
15 379,44
15 900 в
О
18 300
О
0а
0
19 064,8
О
0
0
0
0а
17 572
0
5 065,0
12 000 е
12 139,65
16 500 е
16 800 »
16 993,83
20 089,594
20 200
92,025
90
69,09
70 в
40 в
72,870
20 в
72,5
30
420 а
440
983,37
219,4
426,04
279,80
219,17
186,53
201
130
124,028 72
22,816
131 е
79,852
122 е
125 е
71,33
82,782 393*
64
0,282 9 а
0,267 г
0,153
0,294 г
0,193 г
-0,2535 3
0,079 г
0,2
0,2
3,8 а
2,5 а
14,32
^7,78 б
2,43
1,167
0,758
0,574
0,79 б
0,056 743
0,056 8 в
0,041 8 ж
0,047 2 в
0,022 1 в
0,048 763
0,0222 в
0,0428
0,024
0,274 б
0,302 6
3,412 3
1,271
0,279 939
0,128 635
0,081221
0,060 874 7
0,244 б
0,165 б
0,251 Д
0,138 Л
0,286 Д.
0,143Д
0,235
0,131 Д
0,18
0,22
2,9 в
2,3 в
8,17
-3,72 в
2,335 а
0,789 9 а
0,404 8 а
0,267 8 а
1,1 б
0,0863
0,090 е
0,061 е
0,086 е
0,027 е
0,080 6 и
0,11 е
0,060
0,062
0,47 г
0,57 г
1,53 а
0,95 а
0,483 4 6
0,109 6
0,044 62 б
0,025 93 6
1,4 в
3,905
3,90 в
4,55 ж
4,82 в
6,26 в
4,212
6,24 в
4,5
6,0
2,330 б
2,217 б
2,242
3,67
2,171 55
2,667 72
2,820 75
3,047 81
3,41 б
0,495 899 08 а 0,095 218 624
0,591 6 г 0,039 15
0,087 2 ж —
0,451 179 30 б 0,223 516 76 в 3,499 23
1,236 з
0,442 736 51 н
0,057 44
0,067 727 296
1,09 Д
0,078 и
0,450 477 81°
0,43
3,21 к' л
О,КЗ 508 29"
5,457
3,63 е
4,10 е
4,49 е
4,24 е
4,505
4,149 08
4,19
Примечание. Ниже все величины даны в см.
К2: а ч>еуе=—2,055 -10~3; б аа=—7,2-10"*, а3——1,5 -10; в квантовомеханическвй расчет;
A 67) Д A 69) е (
еычислсео по
оценка;
еуе, ; а,, 3, ; й р;
соотношению A. 67); Д вычислено по соотношению A. 69); е вычислено по соотнсшснвю A. 68);
з а)ег//!=_о,О69 16, u>eze=—0,003 347 2, ше9е=—0,598 6 -10-*; и рх=—0,74 Ю"8.
КО: а оценка; б квантовомеханический расчет; в вычислено по ссотнсшенЕЮ A.69); Г вычислено по
соотношению A. 68).
КН: а графическая оценка DexzD,; б <й^„=—3,64 -10, <»eze=—7,53 -Ю"8, ш,д,=—7,56 Ю; в аа=—2,74 -Ю-",
а3=-4 10-8.
KF: a аа=6,9 -10-»; б Р!=2,33 -Ю0.
КС1: а а,=1,в -Ю-«; б pl==8,3 -Ю"».
КВг: a а2=7,7-10-'; 6 р^г.О-Ю-и.
KI: a а2=3,88 -Ю-7; б ^=Afi -КИ1.
KLi: a АгЪ+, Те—12 500, а3П, Ге=12 500, 63S+, Ге=16 600, В1^, 7^=17 300; б опенка, см.текст; в вычислено
по соотношению A. 68).
KNa: а шеу.=—0,726 658 05 -Ю'3, шА=0,215 849 53 -Ю"*, ше9(==—0,116 201 37 -Ю"*, ша-=—0,255 751 64 -Ю,
Ше(в=_0,482 825 67 -Ю-10; б а2=—0,168 532 27 -Ю-5, а5=0,108 010 02 -Ю, а4=0,212 762 76 -Ю"8, а6=
=0,333 898 37 -Ю-10; в Р,=—0,159 638 63 -Ю"8, Р2=О,278 115 11 -Ю0, рь=0,173 4S6 03 -Ю1, Р4=
=0,578 658 82-Ю-13, #,=0,518 299 07 -Ю2, ^=—0,766 268 06 Ю4, Л2==—0,915 643 87 -10-», L,—
=0,374 985 17-10-", 81=0,974 337 11 -10-"; г ш^е=—0,346-Ю, <оА=—0,47-Ю; Д а2=—0,33-10'*;
е квантовомеханический расчет; ж «>еуе=—0,3887 -10'2; 3 шеуе=8,8 Ю, m,z4=2,23 -10"*; и а2=
=—2,3-10-*; к р1==—7,62-10~7; ра=1,6-10-»; д см. текст; м оценка, см. текст;11 шеуе=— 0,291 644 81 -Ю,
(o,z,=0,lll 061 66 -Ю-3; ° аа=0,190 519 00 -Ю, а8==0,434 076 80 -10-»; И ^=0,428 829 59 -Ю"8, Р2=
=0,436 432 45 -Ю-9, Я,=0,237 160 96 -Ю2, Лх=0,128 647 94 -Ю2.
371
Таблица 47.5а. Коэффициенты в уравнениях, описывающих уровни энергии (в см), а также значения «шах и
принятые для расчета термодинамических функций К2, KJ, КО
со
-^
to
Коэффициенты
Т. -10-*
Но-ю-1
Но-ю1
Но-ю2
По-Ю» -
Но-Ю»
Hi-ю3
Hi-юв
Hi-10'
У„-10»
Hi-ioi
Нз-юи
"max
КО: а 422+,
Х1Ч
0
9,165 977
—2,240 258
-0,744 155
0,187 960
-0,184 679
0,565 477
—0,164 149
—7,150 517
1,487 125
—2,890 174
—0,857 069
—
0,756 438
66
401
Г,=0.
«зп„ .
0,96
8,984 497
—2,660 810
—
—
0,566 045
-0,249 705
—
—
-0,893 815
0,600 391
168
496
АЩ-
1,168 358
6,887 924
-1,353 187
-0,104 469
0,023 832
—0,015 819
0,416 561
-0,137 288
—0,605 808
—
0,627 110
115
508
к2
ъщ
1,3
6,987 943
-2,929 880
—
0,470 375
-0,284 525
. .
-0,854 090
—
-0,056 324
118
397
1,51
3,993 НО
-1,923 357
—
—¦
.—
0,220 239
-0,142 262
-0,268 144
—
-0,278 051
. 103
407
стя
1,537 944
7,274 447
2,526 274
-6,880 322
3,324 19е
-5,934 621
0,848 951
-0,233 786
—
-0,800 461
—7,336 483
-2,082 255
31
276
D4Jg
1,59
1,996 555
-0,787 281
0,221 23€
5 —0,130 32^
—
—
—1,092 440
4,271 115
126
333
Х*Ц
0
7,237 511
-1,993 116
—
—
—
> 0,426 527
t -0,179 071
—
_
—
-0,595 877
—
0,388 859
181
533
1,83
2,994 832
—1,993 116
—
—
—
0,239 174
—0,218 865
—
—
—
-0,615 739
—
—1,065 776
74
288
КО
0
41,606 58
-24,038 73
0,416 157
—
—
2,74 ,
-2,9
6,363 01
—
—
-4,7
—
-21,467 5
130
410
Таблица 47.56. Коэффициенты в уравнениях, описывающих уровни энергии (в см), а также значения г>шах и >Тцш,
принятые для расчета термодинамических фу нкций КН, KF, KCI, KBr<, KI, KLi, KNa
Я
Коэффициенты
КН
ОТ
Л*2+
KF
XrE+ a
КС1
от-
КВг
ОТ-
KI
ХЧГ"
KLi
ОТ-
KNa
Х>2+
а»2+ »
т. -ю-*
УИ.-10-8
у20
*V«2
У«Ю*
По -10*
у°1о»
Ум-Ю"
Уог-10*
Уи-10»
Уи-10*
У,.10«
У41-10»
У.х-10»
Уи-Ю«
У1а-10»
У2а-10"
о
0,983 826
-14,651 59
70,246 4
-36,044 2
34,123
-81,7
Уоз -10"
У„ -10»
K=D.)-10-*
а2 -10*
а,-10»
а4 -Ю12
-153,0
205,742 3
1,538 826
1,332 739
—1,895 600
1,184925
27
92
1,906 480
' 0,219 400
7,78
-36,4
75,3
—7,56
12,71
37,2
-27,4
40,0
-95,0
О
0,425 8
-2,427 258
О
0,278 563
-1,156 701
0 0 0
0,218 47 0,186 284 0,200 982
-0,753163 —0,572 488 —0,484 208
_ _ —1,953 067
0,952 156
0,038 213
-0,019 898
0,004 621
41
156
2,796 215
—2,330 95
0,068 84
-0,482 34
0,229
-0,009 624
-6,54
324
773
1,274 996
-0,779 477
0,015 72
-0,106 83
0,08118
-0,002 025
-0,318 5
359
971
0,807 027
-0,400 941
0,007 602
0,607 144
-0,266 703
0,003 856
2,461 062
—1,114 274
-0,044 05 -0,025 80 -1,424 274
0,001 97 -0,042 7 —
—0,000 556 —0,000 268 0,649 845
376
1093
307
1085
50
236
О
0,123 980
-0,513 664
0,307 024
-3,698 520
1,613 210
-3,971 073
4,853 768
-2,334 534
0,950 970
-0,450 315
-0,016 810
-0,107 665
2,119 484
-3,324 081
-0,222946
-1,591 293
-2,770 508
1,727 216
-5,757 079
0,051 632
7,928 136
-9,109 747
-0,012 525
0,526 770
0,006 545
-0,000 763
0,000 034
66
331
0,5065
0,022 801
-0,590 844
-0,345 337
0,468 800
0,391 00
-1,087 912
-0,329 158
-0,428 902
-1,711 572
0,020 397
0,037 382
-0,004 895
0,024 715
16
100 :
Примечание. Ниже все величины даны в см, sa исключением коэффициентов потенциальной функции 4 и 4, являющихся безразмерными,
KF: a коэффициенты потенциальной функции 4=162 099, 4==3,115 66, 4=6,347.
KG1: а коэффициенты потенциальной функции 4=152 152, 4=3,22617, 4=6,962.
КВг: а коэффициенты потенциальной функции du=iVI 855, 4=3,24153, 4=6,913. .
KI: a коэффициенты потенциальной функции 4=142889, 4=3,246 31, 4=6,837.
KLi: i
KNa:
8 Состояние
т.
а Состояние
12 500
ьт
12 000
12 500
а1?.*:
12 139,65
1'6 9ОО
с3 2+
16 500
?12+
17 300
?12+
16 800
cm
17 572
cm
16 993,83
Dm d8n
20 089,59 20 2C0
К?(г) ^^^ ; Глава 47
5"F000 К), обусловлены использованием разных методик ограничения вращательной
статистической суммы. Фебер и Херрик [2228] не учитывали триплетные состояния К2, в связи с чем
расхождения с данными табл. 1150 достигают 8 и 18 Дж-К-моль~1 в значения Ф°(Т)
и S°(T) соответственно.
Константа равновесия реакции К2(г)=2К(г) вычислена с использованием принятого в
справочнике значения
D0(K2)=3146+200 см=49,8+2 кДж-моль,
полученного Лумисом и Нусбаумом [3299] короткой (~100 см) экстраполяцией колебательных
уровней состояния СШи. Принятое значение заметно отличается от результата далекой
экстраполяции колебательных уровней основного состояния [4469] D516 см) и данных по
исследованию равновесия К2(г)=2К(г), полученных Льюисом [3248] E4,0+3,6 кДж-моль) и Воляком
[236] E3,8+0,5 кДж-моль). Причины этих расхождений остаются не установленными.
Принятой энергии диссоциации соответствует
Д^Ка, г, 0) = 129,982 ± 2,2 кДж • моль.
К?(г). Термодинамические свойства газообразного положительного иона дикалия в
стандартном состоянии при температурах 298,16—6000 К приведены в табл. 1151.
|В табл. 47.4 приведены молекулярные постоянные 39Щ, принятые на основании исследования
фотоионизационного спектра, результатов квантовомеханических расчетов и приближенных
оценок. Полуэмпирические расчеты молекулы KJ показали, что двум первым диссоциационным
пределам К+A5)+КB5) и К+A5Г)+КBР) соответствуют два стабильных электронных состояния
Z8?* и АЧ1и [4923а,|5023а, 5063а]. Колебательные постоянные в состоянии Х2Е+ приняты по
данным Лейтвилера и др. [5006]г, полученным на основании детального анализа
фотоионизационных переходов на колебательные уровни этого состояния молекул 39Kj и39> 41К? (v=0-~8).
Величина re(KJ) была оценена на основании сопоставления соответствующих данных для ридбергов-
ских состояний молекул К2 и Li2 и основного состояния LiJ. Погрешность оцененной величины
не превышает 0,1 А. Молекулярные постоянные К* в состоянии А2Ии были оценены на основании
постоянных^ Lij, погрешность принятой энергии этого состояния не превышает 1000 см.
Термодинамические функции KJ(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения (?вн и ее производных рассчитывались по уравнениям A. 90)—A. 92)
с учетом одного возбужденного состояния. Статистические суммы по колебательно-вращательным
уровням энергии состояний Х2%* и А2Ии и их производные вычислялись по соотношениям
A.|73)—A. 75) непосредственным суммированием по колебательным уровням и интегрированием
по вращательным уровням с помощью уравнений типа A. 82). В расчете учитывались все уровни
со значениями / <j /ша1> в, где /ша1 „ определялись из A. 81). Колебательно-вращательные уровни
вычислялись по уравнениям A. 65), A. 63а) со значениями коэффициентов Ykl, приведенными
вместе с величинами vmxx и /Ит в табл. 47.5. Для соблюдения условия A. 59) добавлен
коэффициент Y03. В расчетах использовались значения Ykl для изотопомера KJ, соответствующего
природной смеси изотопов калия, которые были получены по уравнениям A. 66) и постоянным
молекулы 39К? из. табл. 47.4. Мультиплетность и вырождение состояний Х2?* и А2Пи учитывались
статистическими весами 2 и 4 соответственно.
Погрешности вычисленных термодинамических функций при низких температурах
обусловлены главным образом неточностью принятых молекулярных постоянных, при высоких
температурах дополнительный вклад вносит пренебрежение всеми возбужденными состояниями, кроме
состояния А2Ла; при 298,15,: 3000 и 6000 К погрешности в значениях ФО(Г) составляют 0,5, 1
и 4 Дж-К~1-моль.
Таблица термодинамических функций К?(г) публикуется впервые.
Константа равновесия реакции Кз(г)+е(г)=2К(г) вычислена с использованием значения
ДгЯ°@)=-^-341,997+210 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(К2)=391,797+0,015 кДж-моль « 32 751,7+1,3 см.
Это значение было получено Лейтвилером и др. [5006, 5007] методом двухфотонной лазерной
фотоионизации и хорошо согласуется с результатами менее точных фотоионизационных измере-
374
Калий и его соединения КО(г), КО2(к, ж)
ний [4973а] C91,6+0,1 кДж-моль) и [2312] C86+10 кДж-моль), но заметно отличается
от найденного при масоспектрометричееком исследовании равновесия реакций К*(г) в работе
1592] C76+10 кДж-моль"). Принятому значению соответствуют также
D0(KJ) = 76,813 ±2,2 кДж.моль-1«!б420 +180 ем,
Ь,Н°(К$, г, 0) = 521,779 + 2,2 кДж • моль.
КО(г). Термодинамические свойства газообразного оксида калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1152.
Молекулярные постоянные КО, принятые на основании экспериментальных и теоретических
данных, приведены в табл. 47.4. Надежные сведения о типе основного электронного
состояния КО отсутствуют. На основании результатов исследования спектра ЭПР молекул КО,
изолированных в матрицах инертных газов [3265], в настоящем справочнике принимается, что
основным электронным состоянием КО является состояние Х2П. Поскольку приближенный квантово-
механический расчет [4339] и эксперименты по отклонению молекулярного пучка в
электрическом и магнитном полях [2616] свидетельствуют в пользу того, что основное состояние имеет
симметрию 22+, энергия возбуждения последнего принимается равной нулю с погрешностью
500 см.
Частоты колебаний ше для состояний Х2П и42Е+ приняты на основании сопоставления
экстраполированного на газовую фазу значения частоты, зарегистрированной Спикером и Эндрюсом
14365] в матрице C83,6 см), с найденными в расчете Со и Ричардса [4339]. Постоянные Ве
приняты по расчету [4339], постоянные шехе оценены по соответствующим данным для молекул LiO
и NaO. Погрешность значений ше и Ве оценивается в 40 и 0,03 см соответственно.
Термодинамические функции КО(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения (?вн и ее производные рассчитывались по соотношениям A. 90)—A. 92)
с учетом одного возбужденного состояния. Статистические суммы по
колебательно-вращательным уровням состояний Х2П и Л2Е+ и их производные находились непосредственным
суммированием по колебательным уровням и интегрированием по / с помощью соотношений типа A.81).
В расчете учитывались уровни со значениями / <^ /Шах, ») последние находились по формуле
A. 82). Уровни энергии вычислялись по уравнениям A. 64а) и A. 65) с коэффициентами Ykl,
полученными без учета различия постоянных разных изотопомеров молекулы КО; эти
коэффициенты приведены вместе со значениями ушах и /1Ш в табл. 47.5. Для обеспечения условий A. 50)
и A. 59) были введены коэффициенты Y30 и YQ3. Мультиплетность и вырождение состояний Х2П
ш A2L+ учитывались статистическими весами 4 и 2 соответственно.
Погрешности рассчитанных термодинамических функций КО(г) обусловлены главным
образом отсутствием данных об энергии нижних электронных состояний этой молекулы; они
составляют 4—6 Дж-К-^моль в значениях Ф°(Г) во всем интервале температур.
Термодинамические функции КО(г) ранее вычислялись в таблицах JANAF [2834] и работе
Брюэра и Розенблатта [1666] (Ф°(Т) ^ 3000 К). Расхождения с данными [2834] составляют
около 4 Дж-К-моль-1 при всех температурах и обусловлены тем, что в [2834] не учитывалось
состояние Л2Е+.
Константа равновесия реакции КО(г)=К(г)+О(г) вычислена с использованием принятого
в справочнике значения
ДгЯ°@) = D0(KO) = 270 + 20 кДж • моль я» 22 600 ± 1700 см.
Принятая величина основана на масс-спектрометрическом исследовании равновесия реакции
К0(г)=К(г)+1/2О2(г), выполненного Элертом [2160] A163 К, 1 точка). Это равновесие
исследовалось в парах над карбонатом калия.
Принятой энергии диссоциации соответствует
г, 0) = 66,674 + 20 кДж
КО2(к, ж). Термодинамические свойства кристаллического и жидкого диоксида калия в
стандартном состоянии при температурах 100—2000 К приведены в табл. 1153.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние КО2(к) в интервале 0—7,1 К принята три-
375
КО8(к, ж)
Глава 47
клинная модификация, в интервале 7,1—12,3 К — модификация с неизученной структурой,
в интервале 12,3 — 193,5 К — моноклинная В-модификация, в интервале 193,5—229,7 К —
ромбическая у-модификация, в интервале 229,7—422 К — тетрагональная ^-модификация и в
интервале 422—808 К — кубическая а-модификация (структурный тип NaCl) [4858].
При Т < 298,15 К теплоемкость КО2(к) измерена Тоддом [4519] E0 — 298,15 К) и Зумсте-
гом и др. [4828] B—20 К). В работе [4519] использовался образец, содержащий 3,5% Na202
и 4,1% К2СО3. В интервалах 180—200 К и 217—243 К автор [4519] в специальной серии опытов
определил энтальпии полиморфных превращений B,31+0,04 кДж-моль при 193,5 К и 2,00 +
+0,04 кДж-моль при 229,7 К). Результаты измерений теплоемкости [4828] представлены
в виде мелкомасштабных графиков и в расчете термодинамических функций КО2(к) не
использовались. Термодинамические функции КО2(к) при Т <J 298,15 К вычислены по данным работы
[4519] с учетом энтальпий фазовых переходов при 7,1 и 12,3 К, определенных в работе [48281
{0,016 и 0,07 кДж-моль). Экстраполяция теплоемкости ниже 51 К по уравнению Z>A39/T)+
+2?B60/Т) [4519] с учетом данных [4828] приводит к значению 5°E1 К)=22,8 Дик-К^-моль.
Погрешности принятых значений ?°B98,15 К) и #°B98,15 К)—Я°@) (см. табл. 47.1)
оцениваются в 3 Дж-К-моль~1 и 0,3 кДж-моль соответственно.
При Т > 298,15 К единственное экспериментальное исследование свойств КО2(к) проведено
Веденеевым и Скуратовым [199] (измерение энтальпии, 292—373 К, образец содержал ~94%
КО2, примеси: К2О2 — 2,8%, К2С2 — 1%, КОН -± 1,2%, Н2О — 0,93%), которые нашли
значение средней теплоемкости &C32 К)=81,1 +7 Дж-К-моль~1. Приведенное в табл. 47.1
уравнение теплоемкости КО2(к) в интервале 298,15—422 К выведено по двум значениям теплоемкости
С;B98,15 К) (см. табл. 47.1) и С°D22 К)=88+4 Дж-К^-моль", последнее оценено так же, как
для NaO2(K) (см. выше). В пределах 1% это уравнение согласуется с данными [199].
Температура полиморфного превращения D22+5 К) принята по результатам наиболее
надежных измерений [3090, 4151]. Энтальпия этого превращения принята равной нулю, поскольку
ее величина должна быть небольшой. Теплоемкость КО2(к) в интервале 422—808 К (88 +
+4 Дж>К~1-моль) оценена на основании экспериментальных данных для a-Na2O2 и a-NaO2.
Температура плавления (808+10 К) принята по термографическим данным [1059, 1102, 4151].
Энтальпия плавления B0,6+0,5 кДж-моль) рассчитана Джонгом и Броерсом [2900] на
основании изучения фазовой диаграммы системы KNO3—КО2. Теплоемкость К02(ж) оценена равно!
100+10 Дж-К^-моль (см. т. 1, кн. 1, с. 79).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 2, 1С
и 18 Дж-К-моль~1 соответственно. Расхождения в значениях Ф°(Г), приведенных в табл. 1153
и в справочнике JANAF [2835] (Т <^ 800 К), составляют ~2-f-3 Дж-К~1-моль и обусловлены
различием принятых значений ?°B98,15 К), а также разной оценкой величины С°р(КО2, к).
Константа равновесия реакции КО2(к, ж)=К (г)+20 (г) вычислена с использованием значения
AfJT°@)=868,107+3,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования КО2(к)
bfH°(KO2, к, 298,15 К) = —283,6 + 3,0 кДж • моль.
Таблица 47.6 Результаты определений A^J2"°(KO2, к, 298,15 К) (в кДж • моль)
Авторы
Форкран, 1914 [2303]
Казарновская, Казарновский, 1951
1471]
Гиллес, Маргрейв, 1956 [2403]
Д'Орацио, Вуд, 1965 [2096]
Метод
Измерение энтальпии растворения КО8(к) в
серной кислоте
То же, и энтальпия растворения K3SO4(k) в
серной кислоте, 295,2 К 13 измерений
Измерение энтальпии растворения КО2(к) в воде,
разведение КОН 10 000 Н2О, 298,15 К, 5 изме-
То же, разведение КОН -18 000 Н4О, 298,15 К,
5 измерений
Д/Я°(КОЯ, к, 298,15 К)
—286
—280,6±5,0
—283,9 ±3,5
-284,9 ±3,5
376
Калий и его соединения К2О(к, ж), К2О(г>
Эта величина основана на приведенных в табл. 47.6. результатах калориметрических
исследований. В справочнике принято средневзвешенное значение из данных [471, 2096, 2403],
которые хорошо согласуются между собой. Основным источником его погрешности являются примеси*
в исходных образцах и их нестабильность.
К2О(к, ж). Термодинамические свойства кристаллического и жидкого оксида дикалия в
стандартном состоянии при температурах 298,15—3000 К приведены в табл. 1154.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние К2О(к) в интервале 0—590 К принята
кубическая ^-модификация (структурный тип флюорита, CaF2), в интервале 590—645 К —
кубическая C-модификация и в интервале 645—1013 К — гексагональная а-модификация [3635»
4534]. В литературе отсутствуют какие-либо сведения о термодинамических свойствах К2О(к, ж).
Значения термодинамических функций при 298,15 К, приведенные в табл. 47.1, оценены при
подготовке справочника с помощью различных эмпирических методик [1094, 1741, 3127, 3198]
и с учетом экспериментальных данных о свойствах окислов Li, Na и Cs. Погрешности значений
5°B98,15 К) и #°B98,15 К)—Н°@) оцениваются в 6 Дж-К^-моль и 0,8 кДж-моль
соответственно. Принятое значение ?°B98,15 К) удовлетворительно согласуется с приведенными в
справочниках [2834] и [3124] (94±6 Дж-К-^моль) и (98,3+10,5 Дж-К^-моль).
Для теплоемкости К2О в интервале 298,15—590 К принято уравнение (см. табл. 47.1),
выведенное по значениям теплоемкости при 298,15 К (см. табл. 47.1) и 590 К (92,5+6 Дж-К~1-моль);
последнее оценено так же, как и в случае В.Ь2О(к) (см. ниже). Температуры полиморфных
превращений E90+5 К и 645+10 К) приняты по результатам термографических исследований [3635,,
4534]. Соответствующие этим превращениям энтальпии @,7+0,2 и 4+1 кДж-моль) оценены
так же, как для В.Ь2О(к). Теплоемкость |3- и ос-модификаций A00+10 Дж»К-моль) оценена
на основании предположения о равенстве теплоемкости высокотемпературных фаз К2О и Na2O.
Температура плавления A013+10 К) принята по термографическим данным Козака и^др. [3090],
полученным для образца чистотой 98%. Данные работ [3635, 4534] (958 К) менее точные и не
учитывались. Энтальпия плавления К2О B7+3 кДж-моль) оценена на основании
предположения о равенстве энтропии плавления К2О и Na2O. Теплоемкость К20(ж) A00+10 Дж-К~1-моль)<
оценена так же, как и в случае Na2O^).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 4,
13, 23 и 30 Дж-К~1-моль~1 соответственно. Расхождения термодинамических функций,
приведенных в табл. 1154 и таблицах JANAF [2834] (Т ^ 1100 К), составляют ~2 Дж-К-моль
и обусловлены разной оценкой значений С° (К2О, к), 5°B98,15 К) и Тт. Термодинамические
функции, вычисленные в работе Спенсера и др. [5050а], отличаются от приведенных в табл. 1154 на
2—3 Дж-К-моль~1 при Т ^ 1100 К; при 2000 К расхождение достигает 17 Дж-К-мольг
что обусловлено разной оценкой значений С°(К2О, к, ж). -
Принятая в справочнике энтальпия образования
Д^КгО, к, 298,15 К) = —361,7 ± 4,0 кДж • моль
основана на измерениях энтальпии растворения К20(к) в воде, выполненных Ренгардом [3975,
3976] (разведение КОН• 2300Н2О; 291,2 К; 5 измерений). Измерения Бекетова [1455] были
проведены на образце, содержавшем большое количество примесей, и соответствуют величине
—393 кДж.моль [1555].
Давление пара в реакции К2О(к, ж)=К20(г) вычислено с использованием значения ДгЯ°@)=
= Д,^Г°(К2О, к, 0)=287,448+16 кДж-моль, соответствующего принятым в справочнике
энтальпиям атомизации КгО(г) и образования К2О(к).
К2О(г). Термодинамические свойства газообразного оксида дикалия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1155.
Молекулярные постоянные, использованные для расчета термодинамических функций К2О(г)г,
приведены в табл. 47.7. Спикер и Эндрюс [4365] на основании изотопического расщепления
частоты v3 в ИК-спектре молекул К2О, изолированных в матрице из N2, пришли к выводу, что
наиболее вероятное значение угла К—О—К лежит в интервале 160—180°. В настоящем
справочнике на основании этих данных для молекулы К2О в основном электронном состоянии XxAt при-
377
Ж2О(г)
Глава 47
нята угловая структура (симметрия C2v) *. Приведенное в табл. 47.7 произведение моментов
инерции соответствует структурным параметрам г(К—О)=2,21+0,03 А (оценка по величине г(К—О)
в молекуле КОН) (см. ниже) и Д К—О—К=160+10° (рекомендован в работе Спикера и Эндрюса
[4365]). Погрешность значения 1А1в1с составляет 1-10~ш г3-смв. Приведенное в табл. 7.7
значение частоты v3 принято по работе Спикера и Эндрюса. Частоты vx и v2 рассчитаны по
соотношениям простого поля валентных сил 2. Погрешности принятых значений v1? v2 и v3 оцениваются
в 25-30 см.
Таблица 47.7. Молекулярные постоянные, а также а и р%, принятые для расчета
термодинамических функций КЛ), К2О+, К2О2, КОН, К2О2Н2, ,K2F2, K2C12, ¦
К2Вг2, K2I2, K2SO4, KNO2, KNO3, K2CO3, KBO2, KA1F4, KUF6
Молекула
К2О
к2о+
к2о2
кон
К2О2Н2
K2F2
КаС12
К2Вг2
К212
K2SO4
KNOa
KNO3
касо3
кво2
KA1F4
KUFa
КОН:
К2О2Н2:
K2SO4:
ТС С*С\ *
2 3-
Состояние
хч,
%2АХ
ХЧд
Х1^
КЧд
хчд
ХЧд
ХЧд
ХЧд
хч-i
хчг
1чг
ХЧ±
ХЧ'
хч,
?чг
v2
Ve
СМ
230
200
850
408
3700
323
212
157
131
962
1330
1460
1450
1966
805
650
85
75
400
300 B)
360
162
111
87
69
520
790
1031
1050
1083
618
600 C)
а приведено значение /
а vlo=27O
, vn=3700,
« v10 B)=230,
a v 4/л
v10— i-iu
502
450
300
3700
165
220
145
118
104
200
227
720
720
585
270
530
¦1039 г
_
—
70
.—
300
110
84
54
45
460
1214
230
270
581
210
200 B]
•CM2.
x12=294 см-1.
vuB)=62 см-1.
7ПА \
_
—
433
—
250
275
187
145
125
1126
160
1293
260
265
351
180
_
— —
200 —
— —
290 120
305 —
193 —
156 —
139 -г
640 262
60 —
721 ч 160
880 200
88 —
200 695
150 B) 120 B)
KA1F4
KUF5
_
— —
_ —
— —
295 290a
—
— —
— —
—
1099 B) 609 B) a
— —
827 60
230 230a
. .
317 250a
270 250a
TIT 10117
'a'b'c'1U
Г3 ¦ CM6
G
1,2 -103 2
2 -103 2
12 -103 4
10,23458 a 1
34 -103 4
34 -103 4
25 -10* 4
13 -105 4
¦48-105 4
26 -10* 4
4 -103 2
12 -103 2
15 -10* 2
5 103 1
12-10* 2
4 -105 2
a vio=877, vn=90, v12=303 см.
PS
1
2
1
1
1
1
1
1
1
1
1
1
1
1
1
1
a vio—90 см х. Возбужденные электронные
состояния:
Т„, см 500
Pi 4
3000 8000 12 000 20 000
9 25 20
31
Сведения о возбужденных электронных состояниях К2О в литературе отсутствуют.
Термодинамические функции К2О(г) вычислены в приближении «жтесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены приближенным характером расчета, неточностью оценки молекулярных
постоянных, в частности величины ДК—О—К, а также тем, что в расчете не учитывались
возбужденные электронные состояния, которые по аналогии с молекулой Li2O могут иметь низкие энергии;
они оцениваются в 6, 20 и 30 Дж-К-^моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К
соответственно.
Таблица термодинамических функций К2О(г) публикуется впервые.
См. примечание к тексту по Cs2O (г).
/г=1.01-106 (рассчитано по принятому значению v,), /а/г2=0,017-105 дин-см ( оценено в предположении, что
отношение /г//а сохраняется таким же, как в Ы2О п КОН).
378
Калий и его соединения К2О+(г), К2О2(к, ж)
Константа равновесия реакции К2О(г)=2К(г)+О(г) вычислена с использованием принятого
з справочнике значения энтальпии атомизации
ДГЯ°(О) = 496 + 15 кДж • моль.
Эта величина 9снована на результатах масс-спектрометрического исследования равновесия
реакции К2О(г)=2К(г)+1/2О2(г), выполненного Гусаровым и Гороховым [309, 343, 344] A066—
1223 К, 9 точек, 496+15 кДж-моль). Аналогичное, но менее детальное и точное исследование
было выполнено Элертом и др. [2160, 4297] A164 К, 1 точка, 547 кДж-моль). Клемм и Шарф
[3031] исследовали испарение К2О с открытой поверхности (детали эксперимента не сообщаются).
-Эти измерения были обработаны авторами [3031] в предположении, что единственным продуктом
испарения является К2О(г). Из этих данных следует, что энтальпия атомизации меньше
-580 кДж-моль.
Принятой энтальпии атомизации соответствует
bfH°(K3O, г, 0) = — 69,435 + 15 кДж • моль.
КаО+(г). Термодинамические свойства газообразного положительного иона оксида дикалия
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 1156.
Молекулярные постоянные, использованные для расчета термодинамических функций К2О+,
приведены в табл. 47.7. Экспериментальные данные о структуре и частотах колебаний К2О+ в
литературе отсутствуют. По аналогии с молекулой К2О в справочнике принимается, что К2О+ в
основном электронном состоянии %2Аг имеет угловую структуру (симметрия C2t). Приведенные
в табл. 47.7 постоянные были оценены по принятым постоянным молекул К2О, Н2О и Н2О+ (см.
табл. 5.4 и 5.7) в предположении, что соотношение между постоянными К2О и К2О+ такое же,
как у Н2О и Н2О+. Произведение моментов инерции было рассчитано по структурным параметрам
г(К—О)=2,3+0,1 А и ДК—О—К=160+15°, его погрешность оценивается в 1,5-10'114 г3-см6.
Погрешности в значениях vx, v2 и v3 составляют 50, 25 и 100 см. Молекула К2О+, так же как
молекула КО, должна иметь электронные состояния с низкими энергиями возбуждения.
Обоснованная оценка энергии этих состояний невозможна, и, учитывая нетрчность принятых
постоянных К2О+ в основном состоянии, они не рассматриваются в справочнике.
Термодинамические функции К2О+(г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130).
Погрешности рассчитанных термодинамических функций обусловлены грубой оценкой
молекулярных постоянных К2О+„ в частности, структуры этой молекулы, пренебрежением ее возбужден-
лыми состояниями, а также приближенным характером расчета; они составляют 10, 15
и 20 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций К2О+(г) публикуется впервые.
Константа равновесия реакции K2O+(r) +e(r)=2K(r)-f-0(r) вычислена с использованием
значения Дг#°@)=60+21 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(К2О)=436+15 кДж-моль.
Принятое значение основано на результатах масс-спектрометрического исследования
равновесия реакции К2О+(г)+К(г)=К+(г)+К2О(г), выполненного Кудиным и др. [592] A060 К,
1 точка). Согласующиеся в пределах погрешности значения 482+50 кДж-моль и 444 +
+30 кДж-моль были получены методом электронного удара Гусаровым и Гороховым [344]
и Гороховым и др. [312]. В работе Элерта и др. [2160, 4297] методом электронного удара было
получено значение 733+10 кДж-моль. Столь большое различие с величинами, полученными
в других работах, объясняется, возможно, тем, что зарегистрированные в этой работе ионы К2О +
являлись продуктом реакции диссоциативной ионизации КаО2Н2(г)+е(г)=К2О+(г)+Н2О(г)+
-f-2e(r), а не реакции К2О(г)+е(г)=К2О+(г)+2е(г), как предполагали авторы [2160, 4297].
Принятому потенциалу ионизации соответствует
К2О2(к, ж). Термодинамические свойства кристаллического и жидкого диоксида дикалия
m стандартном состоянии при температурах 298,15—2000 К приведены в табл. 1157.
379
К2О2(к, ж)
Глава 47
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике эа стандартное состояние К2О2(к) в интервале 0—818 К принята
ромбическая модификация [4958]. Какие-либо сведения о термодинамических свойствах К2О2(к, ж)
в литературе отсутствуют. Значения термодинамических функций при 298,15 К, приведенные
в табл. 47.1, были оценены при подготовке справочника с помощью эмпирических методик [1094,
1741, 3127, 3198], а также на основании экспериментальных данных для соединений щелочных
металлов типа Ме2О, Ме2О2 и МеО2. Погрешности принятых значений ?°B98,15 К)
и Н°B98,15 К)—Н°@) составляют 12 Дж-К~1-моль и 0,8 кДж-моль соответственно. Близкое
значение S°B98,15 К) принято в таблицах JANAF [2834] A13+6 Дж-К-^моль).
Для теплоемкости К2О2(к) в интервале 298,15—818 К принято уравнение (см. табл. 47.1),
полученное по Значениям теплоемкости при 298,15 К (см. табл. 47.1), и 818 К A17+12 Дж-К~*Х
Хмоль); последнее оценено на основании экспериментальных данных для Na2O2(K),
теплоемкость последнего в точке фазового перехода составляет примерно ЗОп, где п — число атомов
в молекуле. Принятое значение температуры плавления К2О2 (818+10 К) получено Козаком
и др. [3090] при термографических измерениях. Результаты менее точных работ [774, 1774] G63,
733 К) не учитывались. Энтальпия плавления B0,5+4,0 кДж-моль) оценена так же, как для
Na2O2 (см. выше). Теплоемкость К2О2(ж) A34+15 Дж-К-моль) оценена при подготовке
справочника (см. гл. 2.5).
Погрешности вычисленных значений Ф°(Т) при температурах 298,15, 1000 и 2000 К
оцениваются в 6, 20 и 30 Дж-К~1-моль~1. Различия в термодинамических функциях, приведенных
в табл. 1157 и справочнике JANAF [2834] (Г <С 700 К), составляют ~4 Дж-К~1-моль при
298,15 К, с ростом температуры расхождение уменьшается и при 700 К не превышает 1 Дж-КХ
Хмоль.
В справочнике принята энтальпия образования
Ь,Н°(К2О2, к, 298,15 К) = — 443 + 20 кДж-моль.
Результаты исследований равновесия реакций с участием К2О2(к) представлены в табл. 47.8.
Указанные погрешности учитывают воспроизводимость измерений, неточность
термодинамических функций и термохимических величин, использованных в расчетах. Принятая величина
основана на результатах исследования равновесия реакции 2К2О2(к, ж)=2К2О(к, ж)+02(г).
Таблица 47.8. Результаты определений А/?ГО(К2О2, к, 298,15К) (в кДжмоль)
Авторы
Центнершвер, Блюменталь, 1932
[1591, 1774]
Казарновский, Райхштейн, 1947 [472
Лефлер, Видерхорн, 1964 [3223]
Метод
Компенсационный, 2К»Оо(ж)=2К203(к, ж)+0»(г),
719-912 К, 57 точек
То же, 2К2О2(к, ж)=2К2О(к, ж)+02(г), 785—
882 К, 43 точки
] Статический, 2КО2(к)=К2О2(к)+О2(г), 573—643 К,
3 точки
То же, 2К2О2(к)=2К2О(к)+О2(г), 633 К, 4
измерения
Статический, 2КО2(к, ж)=К2О2(к, ж)+02(г),
573-873 К, 5 точек
То же, 2К2О2(к, ж)=2К2О(ж)+Оа(г), 773-923 К,
33 точки
AfH°(K2O2,
II закон
—473 ±35
—421 ±25
—485+40
—
—484 ±15
—430 ±35
к, 298,15 К)
III закон
—493 ±30
—442 ±20
—495 ±12
—438 ±20
—497±15
-448 ±20
Исследования этой реакции во всех работах привели к близким величинам. Однако результаты
исследований реакции образования К2О2 из высших окислов очень противоречивы. Центнершвер
и Блюменталь [1591, 1774] обнаружили фазу К2О3(к), существование которой опровергается
Казарновский и Райхштейном [472] и в ряде других работ (см. [3101]). В табл. 47.8 приведены
результаты обработки измерений [1591, 1774], при которой были использованы оцененные
термодинамические функции К2О3(к) и энтальпия образования (—529+5 кДж-моль), полученная
Форкраном [2304]. Расхождение данных [1591, 1774] для двух реакций достигают 50 кДж-моль»
380
Калий и его соединения К2О2(г), КН(к, ж)
что можно было бы считать дополнительным доводом, опровергающим существование фазы КаО3(к).
¦Однако обработка измерений [472, 3223 ] в предположении, что в равновесии с К2О2(к) находится
КО2(к), также приводит к плохо согласующимся данным — расхождения и в этом случае
достигают 50 кДж-моль. Таким образом, исследования равновесий реакции окисления К2О2
противоречивы и не использовались для выбора энтальпии образования К202(к).
Основным источником погрешности принятой величины является неточность
термодинамических функций.
Давление пара диоксида дикалия в реакции К2О2(к, ж)=К2О2(г) вычислено с использованием
значения Дг#°@)=Д,#°(К2О2, к, 0)=252,142+28 кДж-моль, соответствующего принятым
в справочнике энтальпиям образования К2О2 в кристаллическом и газообразном состояниях.
К2О2(г). Термодинамические свойства газообразного диоксида дикалия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1158.
Молекулярные постоянные, использованные для расчета термодинамических функций К2О2,
приведены в табл. 47.7. Структура КаОя экспериментально не исследовалась. На основании
теоретического расчета, проведенного Аллавена и др. [4915], в справочнике принимается, что в
основном электронном состоянии %}-Ад молекула К2О2 имеет плоскую ромбическую структуру
(симметрия D2h) со связью кислород—кислород в цикле. Приведенное в табл. 47.7 произведение
моментов инерции рассчитано по оцененным структурным параметрам г(К—О)=2,3+0,1 А (из
K2F2, KF и КО) и г(О—О)=1,50+0,1 А (из Li2O2). Погрешность величины 1А1в1с составляет
5.1О-и4 гз.смб<
Значение частоты v5 найдено Эндрюсом и др. [1328, 4366] при исследовании ИК-спектра
молекул К2О2, изолированных в матрице из Аг. Частоты v4, ve в ИК-спектре К2О2 (Rb2O2, Cs2O2)
не наблюдались, так как их величины, вероятно, ниже 200 см — нижнего предела
применявшейся спектральной аппаратуры. Приведенные в табл. 47.7 значения частот v4, ve, а также va
и v3 были оценены на основании частот, принятых для 1Л2О2, и с учетом закономерного изменения
частот колебаний в ряду молекул М2О2, где М — щелочной металл. Значение валентной частоты
колебания связи О—О (vx) принято таким же, как в молекуле Li2Oa (см. Na2O2).
Погрешность принятого значения v5 оценивается в 15—25 см, остальных частот — в 20—
25 % от их величины.
. Сведения о возбужденных состояниях К2О2 в литературе отсутствуют.
Термодинамические функции К2О2(г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A-9), A.10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета и
составляют 6, 12 и 20 Дж-К -моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций К2О2(г) публикуется впервые.
Константа равновесия реакции К2О2(г)=2К(г)+.2О(г) вычислена с использованием значения
^О)=858,348+20 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A^°(K2O2, г, 0) = —185+ 20 кДж-моль.
Принятая величина основана на масс-спектрометрическом исследовании равновесия реакции
К2О2(г)=2К(г)+О2(г) A060 и 1146 К, 5 измерений), выполненном Гусаровым и Гороховым [309,
343, 344]. Ее погрешность обусловлена в основном неточностью термодинамических функций
и сечений ионизации.
КН(к, ж). Термодинамические свойства кристаллического и жидкого гидрида калия в
стандартном состоянии при температурах 298,15—1200 К приведены в табл. 1159.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние КН(к) в интервале 0—892 К принята
кубическая модификация (структурный тип NaCl) 1. В литературе отсутствуют экспериментальные
данные по теплоемкости и энтальпии КН(к, ж).
1 Башкин и др. [1419] установили, что при высоких давлениях гидрид калия имеет фазовый переход из
кубической модификации типа NaCl в кубическую модификацию типа CsCl. При 298 К это превращение происходит при
4,5 -104 атм при повышении давления и при 2,5 -10* атм при понижении давления; с ростом температур величина
гистерезиса уменьшается. . '
381
КН(к, ж) Глава i'i
Термодинамические функции КН(к) при 298,15 К оценены на основании
экспериментальных данных для 1ЛН(к) и NaH(K), имеющих тот же тип кристаллической структуры. Для энтропии
КН(к) при 298,15 К принято значение, которое приводит к совпадению энтальпии образования
КН(к), вычисленной из данных по равновесию диссоциации КН(к, ж) в наиболее надежных
исследованиях, с точным калориметрическим значением этой величины (см. ниже). Погрешности
принятых значений <S°B98,15 К) и #°B98,15 К) — Я°@), приведенных в табл. 47.1, оцениваются
в 2 Дж-К^'моль и 0,6 кДж-моль соответственно.
Теплоемкость КН(к) при Т > 298,15 К была оценена на основании соответствующих данных
для гидридов лития и натрия и фторидов щелочных металлов г. Приведенное в табл. 47.1
уравнение для теплоемкости КН(к) получено по значениям теплоемкости при 298,15 К (см. табл. 47.1),
при 500 К D8,5 Дж-К'моль~1) и при температуре плавления F3 Дж>К-моль). Погрешность
оцененных значений теплоемкости составляет 8—10%. %»¦ $?Щ.
Температура плавления (892+5 К) при равновесном давлении водорода F7,3 бар) принята,
по результатам термографического исследования, выполненного Скуратовым и др. [940];
воспроизводимость ее определения оценена авторами в 2°. Исследованный образец гидрида калия
содержал не менее 98,9% КН. Принятое значение энтальпии плавления B1+3 кДж'Моль)
вычислено Скуратовым и др. [940] на основании данных по давлению диссоциации КН, имеющихся
в литературе и полученных в работе [940]. Значение теплоемкости жидкого КН E6+6 Дж'К-1Х
Хмоль) оценено с учетом экспериментальных данных по теплоемкости 1лН(ж).
Погрешности рассчитанных Ф°(Т) для КН(к, ж) при 298,15, 500 и 1000 К оцениваются в 2,
4 и 6 Дш-К-моль~1 соответственно.
Термодинамические функции КН(к), приведенные в справочнике JANAF [2834] (Т <; 800 K)s
и табл. 1159, согласуются в пределах 0,3 Дж'К^маль в значениях Ф°(Т).
Таблица термодинамических функций КН(ж) публикуется впервые.
Принятое в справочнике значение энтальпии образования
к, 298«15К) = — 57,82 + 0,15 кДж • моль
основано на калориметрических измерениях энтальпии растворения К(к) и КН(к) в воде, выпол*-
ненных Ганном и Грином [2527] и скорректированных в более поздней работе Ганна [2525].
Аналогичные измерения, выполненные Мессером и др. [3488]; привели к менее точному значению»
_63,4+1,8 кДж-моль, что, видимо, связано с недостаточной чистотой образца гидрида калия.
Результаты расчетов энтальпии образования КН(к) на основании исследований равновесие
реакции 2КН(к, ж)=2К(ж)+Н2(г) представлены в табл. 47.9. Приведенные в таблице
погрешности включают вклады, обусловленные неточностью термодинамических функций, которые
составляют 2 кДж'МОЛь для измерений при 600 К и 8 кДж-моль для измерений при 1000 К..
Таблица 47.9. Результаты
равновесия
Авторы
Труст, Отфей, 1874 [4552]
Кейс, 1912 [2979]
Соллерс, Креншоу, 1937 [4342]
Херолд, 1947 [2618]
Херолд, 1947 [2617]
Херолд, 1949 [2619, 2620]
Дымова и др., 1974 [394]
Скуратов и др., 1976 [940]
а Данные представлены в виде
определений А^ДГ°(КН, к, 298,15 К) на основании
реакции 2КН(к, ж) = 2К(ж) + Н2(г) (в кДжмоль
Метод
Статический, 603—703 К, 11 точек
То же, 488—688 К, 66 точек
То же, 543—653 К, 49 точек8
Статический, 587—663 К, 7 точек
Динамический, 675—754 К, 24 точки
Статический, 562—689 К, 6 точек
ДТА, 714—847 К а
Статический, 932—1015 К, 9 точек
уравнения.
исследований
)
bfH°(KE, j
II закон
—59±6
-60±3
—57
—54 ±6
—55 ±5
—58+3
—65
-60+10
к, 298,15 К)
III закон
—57,1+2,5
—58,0 ±2,0
—58,2+2,0
—58,1 ±2,2
—59,5±3,5
-58,5 ±2,6
-58,2+3,0
—57,9±6,0
1 Аналогичные оценки проведены для теплоемкости RbH(K> и СзН(к), поскольку все эти соединения относятся!
к одному и тому же структурному типу.
382
Калий и его соединения КН(г)-
Давление пара гидрида калия в реакции КН(к, зк)=КН(г) вычислено с использованием значения
ДгЯ°@) = Д8Я°(КН, к, 0)=182,223+18 кДж-моль, соответствующего принятым в справочнике-
энтальпии образования КН(к) и энергии диссоциации КН(г).
КН(г). Термодинамические свойства газообразного гидрида калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1160.
Молекулярные постоянные, принятые в справочнике на основании анализа результатов
исследований электронного спектра и теоретических расчетов, приведены в табл. 47.4.
В спектре КН исследована только одна дискретная система полос А1 Л*—Х1И + [1299, 1300,
1413, 2708, 2772, 2773]. Кроме того, в спектре испускания в области 3000—4000 А наблюдался
континуум [1299, 2710], предположительно отнесенный к переходу из отталкивательного
состояния В1!! в основное состояние молекулы. В согласии с экспериментальными данными
теоретический расчет, выполненный Намричем и Трухларом [3687], показал, что только два валентных
состояния (ZXS + и ^4XS+), связанные с пределами КB?)+НB?) и КB.Р)+НB?), являются
стабильными. Довольно глубокий минимум имеет также состояние 12+, которое коррелирует с атомами
KEs2iS)+HB5r) и должно рассматриваться как ридберговское состояние; поэтому оно не включено-
в табл. 47.4.
Колебательные постоянные 39КН в ХХЕ+ и А1!,* состояниях, принятые в справочнике,
вычислены по началам полос системы А11,+—Х11,+ F2 полосы, 2 <[ v' <^ 26 и v" <^ 4),
полученным Бартки [1413] в результате анализа экспериментальных данных Хори [2708].
Вращательные постоянные в основном состоянии взяты из работы Бартки [1413]. Вращательные постоянные
в Аг1,+ состоянии рассчитаны по значениям Bt, приведенным в этой работе для v <J[ 261.
Термодинамические функции КН(г) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QBM и ее производные вычислялись по уравнениям A. 90)—A. 92)<
с учетом одного возбужденного состояния. Статистические суммы по колебательно-вращательным
уровням энергии состояний Х1?+иЛ1Е+и их производные находились по уравнениям A. 70)—
A. 75) непосредственным суммированием по уровням энергии. В расчете учитывались все уровни,
ограниченные предельными кривыми диссоциации указанных состояний.
Колебательно-вращательные уровни вычислялись по уравнениям A. 65), A. 64а) и A. 62). Значения коэффициентов
Ykl в этих уравнениях приведены в табл. 47.5 вместе со значениями i;max, /1№ и коэффициентами air
аппроксимирующими предельные кривые диссоциации; они не учитывают различия постоянных:
разных изотопомеров этой молекулы. Для соблюдения условий A. 50) и A. 59) добавлены
коэффициенты Г30, Yi0 и Y03 Для состояния Хг1,+.
Погрешности вычисленных термодинамических функций при Т ^ 3000 К обусловлены
в основном неточностью фундаментальных постоянных и не превышают 0,02 Дж-К-моль
в значениях Ф°(Г). При Т > 3000 К становится существенной неточность энергий диссоциации
и экстраполяции колебательно-вращательных уровней. Общая погрешность в значении Ф9F000 К)
может достигать 0,3—0,4 Дж-К-моль~1.
Ранее термодинамические функции КН(г) вычислялись в справочниках JANAF [2834] и Хара
и др. [2537], а также в работе Шнейдера [4197а] (QIB для 1000 < Т < 3000 К). Расхождения
данных [2834] и [2537] с приведенными в табл. 1460 достигают 0,8, 1,2 и 6 Дж-К~1«моль в
значениях Ф°(Т), S°(T) и С°(Т) и объясняются главным образом различием методов расчета.
Константа равновесия реакции КН(г)=К(г)+Н(г) вычислена с использованием принятого
в справочнике значения
ДгЯ°@) = D0(KH) = 178 ± 18 кДж • моль «* 14 900 + 1500 см.
Эта величина основана на сопоставлении результатов экстраполяции колебательных уровней
основного состояния и оценки энергии последнего наблюдаемого колебательно-вращательного
уровня в состоянии Л12 + .по методике Алми и Бейлера [1299]. Принятое значение удовлетво-
Состояние А1 2+, как и у всех гидридов щелочных металлов, аномально: кривые Дб (»+1/») и В, имеют максимум
при р=10 и г=7 соответственно. Значения постоянных существенно зависят от степени полинома, выбранного
для аппроксимации энергии колебательных уровней и величин Вс, а также от числа использованных данных.
В работе Бартки [1413] колебательные и вращательные постоянные были получены для v < 12. Расчет по этим
постоянным энергии уровня »=26, /=0 приводит к расхождению с экспериментальными данными примерно>
на 300 см, а рассчитанное значение Bie @,79 см) меньше экспериментального на 0,3 см. Постоянные КН
в обоих состояниях, полученные Янгом и др. [4794], близки к приведенным в табл. 47.4.
383
КОН(К) ж) = ===««==»===„ : Глава 47
рительно согласуется с интерпретацией континуума в области 3920—3530 А. [2709, 2710] как
перехода из отталкивательного состояния В1!! в основное состояние молекулы, а также с
рекомендациями Гейдона [2377] и Хьюбера и Герцберга [2736].
Принятой энергии диссоциации соответствует
ДуЯ°(КН, г, 0) = 127,925 + 18 кДж • моль.
КОН(к, ж). Термодинамические свойства кристаллического и жидкого гидроксида калия
в стандартном состоянии при температурах 100—3000 К приведены в табл. 1161.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние КОН(к) в интервале 0—520 К принята
' моноклинная модификация, а в интервале 520—678 К — кубическая модификация.
При Т ^ 298,15 К термодинамические функции КОН(к) вычислены по результатам
измерений теплоемкости, выполненных Сталлом и др. [4420] A5—298,15 К) на образце, содержавшем
в качестве примеси 2,2% К2СО3. При расчетах учитывалась аномалия в ходе теплоемкости
(>.-кривая) в интервале 210—240 К с максимумом С°р{Т) при 227,4 К. Экстраполяция теплоемкости
по закону Дебая привела к значению ?°A5 К) =0,225 Дж>К-моль~1. Погрешности принятых
значений 5°B98,15 К) и #"B98,15 К)—Н°@) оцениваются в 0,4 Дж-К^-моль и 0,08 кДжХ
X моль -1 соответственно.
Для теплоемкости КОН(к) в интервале 298,15—520 К принято уравнение (см. табл. 47.1),
полученное обработкой результатов измерений энтальпии, проведенных Гинзбургом и др. [280]
B98—1073 К, в работе даны уравнения, аппроксимирующие значения С°), и результатов
измерений теплоемкости при низких температурах [4420]. Погрешность значений теплоемкости,
вычисленных по принятому уравнению, оценивается в 1 %. Температура полиморфного превращения
E20+3 К) принята в результате усреднения данных работ [280, 872, 874, 1102, 1446, 3323, 3497,
3498, 3877, 4079, 4080, 4242]. Данные работ [114, 370, 866, 1039, 1073, 1074, 1894, 1895, 3496,
4039] E13—531 К) не учитывались. Энтальпия полиморфного превращения F,45+0,50 кДжХ
Хмоль) принята по согласующимся между собой калориметрическим данным Пауэрса и Блей-
лока [3877] х и термографическим данным Решетникова и Баранской [866]. Данные [280]
E,15 кДж-моль) менее точны и не учитывались.
Теплоемкость КОН(к) в интервале 520—678 К G9+1 Дж-К'1-моль~1) принята на основании
данных по энтальпии, полученных в работе [3877]. Результаты измерений [280] не учитывались,
так как приведенная в ней зависимость С°р(Т) для высокотемпературной фазы имеет слишком
крутой ход (от 75+2 Дж-К^-моль при 520 К до 104+3 Дж-К^-моль при 677 К).
Температура плавлений F78+2 К) принята по хорошо согласующимся между собой
измерениям [114, 116, 117, 370, 380, 866, 868, 870, 874, 1072, 1073, 1074, 1446, 1895, 3496, 3497, 3498,
3877, 4039, 4079, 4080]. Энтальпия плавления (9,4+1,2 кДж-моль) принята по результатам
калориметрических измерений [3877] и согласуется в пределах погрешности с
термографическими измерениями в работах [866] (8,3 кДж-моль) и [3116] (8,4 кДж-моль). Менее надежные
величины, полученные в работах [280, 4242] G,3—7,7 кДж'Моль), не учитывались.
Теплоемкость КОН(ж) (83+4 Дж-К^-моль) принята по результатам исследования
энтальпии в работе [3877] и согласуется в пределах погрешности с данными работы [280] (87 Дж-К-1Х
Хмоль").
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,3,
1, 3 и 7 Дж-К^-моль соответственно.
Термодинамические функции КОН(к, ж), приведенные в табл. 1161, согласуются с принятыми
в справочнике JANAF [2834] (Т < 2500 К) в пределах 1 Дж-К^-моль в значениях Ф°(Т).
Принятая в справочнике энтальпия образования
bfH°{KOH, к, 298,15 К) = —424,58 + 0,25 кДж.моль
основана на измерениях энтальпии растворения КОН в воде, представленных в табл. 47.10.
Наиболее точные измерения с хорошо охарактеризованным образцом были выполнены
Решетниковым [864], и на них основано принятое значение. Более ранние измерения [252, 1530, 2291,
4511] проводились с недостаточно чистыми образцами.
1 Ввиду недоступности работы {3877] результаты обработки полученных в ней данных заимствованы из таблиц
JANAF [2834].
384
Калий и его соединения КОН(г)
Таблица 47.10.Результаты определений АаяН°(КОЯ, к-*соН2О, 298,15К) (в кДжмоль-1)
Авторы
Конечная концентрация, температура
измерений
ДагД°(ооН20, 298,15 К)
Бертло, 1875 [1530, 1531] 260 Н2О, 284,55 К, 1 измерение —55,52
Томсен, 1883 [4511] 260 Н2О, 290,42 К, 1 измерений —57,52
Форкран, 1901 [2291] 150—190 Н2О, 294,65 К, 7 измерений —50,2ч—55,5
Решетников, 1961 [864] 650 НаО, 298,15 К, 6 измерений -57,62±0,20
Давление пара гидроксида калия в реакции КОН(к, ж)=КОН(г) вычислено с использованием
значения ArH°@)=AtH°(KOH, к, 0) = 197,067+8 кДж-моль, соответствующего принятом
в справочнике энтальпиям образования КОН в кристаллическом и газообразном состояниях.
КОН(г). Термодинамические свойства газообразного гидроксида калия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1162.
Молекулярные постоянные, использованные для расчета термодинамических функций KOJJ,
приведены в табл. 47.7. Исследования микроволнового [3133, 3139, 3793, 3794] и ИК-спектров
[87, 89, 4367], а также результаты квантовомеханического расчета [2178] показывают, что в
основном электронном состоянии Х1^* молекула КОН имеет линейную структуру (симметрия Сю1).
Приведенный в табл. 47.7 момент инерции для изотопомера, соответствующего «природной
смеси изотопов», вычислен по вращательным постоянным Z?00O0C9K16OH)=0,273 812 1 [3793] и
?00О0D1К16ОН)=0,269 817 0 см [3794]. Микроволновым данным [3793, 3794] соответствуют
го(К—О)=2,2115 А, г0(О—Н)=0,9120 А. Данные, полученные Пирсоном и др. [3793, 3794], были
подтверждены в работе Куйперса и др. [3139] х. Измерения Кучковским и др. [3133] менее точны.
Приведенное в табл. 47.7 значение частоты vx=vK_0 измерено Спинаром и Маргрейвом 143671
в ИК-спектре газообразного КОН при температуре 1100—1200 К. Практически то же значение
рекомендовано в работе [89]. Принятое значение частоты Деформационного колебания v2
получено Беляевой и др. [89] в ИК-спектре молекул КОН, изолированных в Аг-матрице. Не
наблюдавшаяся экспериментально частота v3=v0_h принята приближенно равной соответствующей
частоте молекулы CsOH. Погрешности частот vx, vj и v3 не превышают 20, 20 и 100 см
соответственно. Сравнение с изоэлектронной молекулой KF свидетельствует, что энергии возбужденных
электронных состояний КОН должны превышать 20 000 см. ; ,
Термодинамические функции КОН(г) вычислены по уравнениям A. 3)—A. 6), A. 9), A. ДО),
A. 122)—A. 124), A. 126), A. 129) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены неточностью принятых значений молекулярных постоянных, а
также приближенным характером расчета и составляют ,1, 4 и 8 Дж-К~1-моль~1 в значениях
Ф°(Г) при 298,15, 3000 и 6000 К. • \ ''
Ранее термодинамические функции КОН(г) вычислялись в справочнике JANAF [2835];
расхождения данных [2835] и табл. 1162 не превышают 2 Дж-К^-моль й значениях Ф°(Т).
Константа равновесия реакции КОН(г)=К(г)+О(г)+Н(г) вычислена с использованием
значения ДГ^Г°(О)=776,708+8 кДж-моль, соответствующего принятой в справочнике энтальпии
образования :
Д^^КОН, г, 0) = —224 + 8 кДж • моль.
Принятая величина основана на представленных в табл. 47.11 данных. Приведенные в таблице
погрешности включают воспроизводимость измерений, неточность термодинамических функций
и термохимических величин. При пересчетах данных работ, в которых давление пара над КОН
измерялось методом точек кипения [2805, 4656] и протока [2121], было приближенно учтено
образование в парах димерных молекул К2О2Н2 (поправки составляют 4—5 кДж-моль). За ис-
1 В работах [3139, 3793, 3794] определены также значения некоторых постоянных колебательно-вращательного
взаимодействия (ait y<,), постоянная центробежного растяжения ?>00оо=4,07-10~7 см и одна постоянная
ангармоничности. '
385
Глава 47"
Таблица 47.11. Результаты определений Л^-Н"°(КОН, г, 0) (в кДж-моль)
Авторы
Метод
ДуД^КОН, г, 0)
II закон
III закон
Вартенберг, Альбрехт, 1921 [4656] Точек кипения, КОН(ж)=КОН(г), 1443—1600 К, —237±10 —223+8
9 точек
Джексон, Морган, 1921 [2805] То же, КОН(ж)=КОН(г), 1068 К, 1 точка — —232±12
Енсен, Педли, 1966 [2855] Спектрофотометрия в пламенах H2+O2+N~, К(г)+ — —210+12
+Н2О(г)=КОН(г)+Н(г), 2475 К, 1 точка
Гусаров, Горохов, 1968 [345], см. Масс-спектрометрический, КОН(к)=КОН(г), —224+8 —
[755] 548-698 К, 16 точек
Дюбуа, Милле, 1969 [2121] Протока, КОН(ж)=КОН(г), 925—1300 К, —237 —227+12
64 точки, данные представлены в виде уравнения
Коттон^ Дженкинс, 1969 [1941] Спектрофотометрия в пламенах H2+O2+N2, К(г)+ — —227+15
+Н2О(г)=КОН(г)+Н(г), 1800 К, 33 измерения
Келли, Педли, 1971 [2965] То же, в пламенах Н2+СО+О2, К(г)+Н2О(г)= —222+10 —221+13
=КОН(г)+Н(г), 1950—2750 К, 32 точки
Фбжье, 1972 [2246] То же, детали эксперимента не приведены — —189
Горохов и др., 1976 [312, 755] Электронный удар,'КОН(г)+е(г)=К+(г)+ОН(гН 216+15 —
' +2е(т)
Кудин, 1974 [591] : Масс-спектрометрический, КОН(к)=КОН(г), 760— —223±80 —227+8
844 К, 6 точек
ключением работы [2246], оценить надежность которой не представляется возможным,
результаты всех измерений совпадают в пределах их погрешностей. Принятое значение является
средневзвешенным из приведенных в таблице величин, включая величины, вычисленные по уравнению
II закона [345, 2965].
К2О2Н2(г). Термодинамические свойства газообразного димера гидроксида калия в стандартном,
состоянии в интервале температур 100—6000 К приведены в табл. 1163.
Молекулярные постоянные, использованные для расчета термодинамических функций К2О2Н2Г.
приведены в табл. 47.7. Произведение моментов инерции 1д1в1с в основном электронном
состоянии %хАд вычислено для плоской ромбической структуры со связями ОН, расположенными на*,
одной прямой (симметрия D2k) с ДО—К—0=94+10°, г(К—О)=2,38+0,05 А, г(О—Н)=0,97+-
4-0,03 А. Эти структурные параметры рассчитаны Беляевой [87] с использованием и*нной
модели. Погрешность IjJbIo составляет 5 «10 U г3-см8 х. Частоты vl5 v5, v8 активны в спектре КРГ.
частоты ve, v7, ve—vls — в ИК-^пектре. Валентным колебаниям кольца соответствуют частоты
v2(>4f), v6{Blg), vlo(jB2e), v12(fiSl(), плоской деформации кольца — Vg^), неплоской деформацию
кольца— vi(Blu), валентным колебаниям внешних связей ОН — vi(Ag), vn(B3ll), плоской
деформации ОН — v4(Z?1?), v9E2m), неплоской деформации ОН — ve(Z?la), vs(B2g). Значения всех частот-
приняты по работе Беляевой [87]. Частоты ve, v9, v10, v12 измерены в ИК-спектре молекул КаО2Н2,
изолированных в матрице из Аг, vx и vn оценены равными 3700 см, остальные частоты
вычислены с использованием ионной модели. Погрешности значений ve, v9, v10, v12 оцениваются в 10 см,
vi и vn B 150 см, всех остальных частот в 15—20% от их величины. Сведения о возбужденных
электронных состояниях К2О2Н2 в литературе отсутствуют.
Термодинамические функции К2О2Н2(г) вычислены по уравнениям A. 3)—A. 6), A. 9)—A. 10),
A 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены неточностью принятых значений молекулярных постоянных и приближенным
характером расчета, они могут составлять 8, 15 и 25 Дж-К~1-моль в значениях Ф°G') при 298,15,
3000 и 6000 К.
1 Близкие значения структурных параметров (г (К—О)=2,43+0,02 А, Д О—К—0=85°) получены в электроно-
графическом исследовании Гиричова и др. [4861], опубликованном после завершения подготовки этой главы.,
386
Калий и его соединения
KF(k, ж)
Ранее термодинамические функции К2О2Н2(г) вычислялись в справочнике JANAF [2835]
для молекулы симметрии Си (с атомами водорода, находящимися в трансположении
относительно плоскости ОКОК). Расхождения данных [2835] и табл. 1163 достигают 35 Дж-К^-моль
в значениях Ф°(Г).
Константа равновесия реакции К2О2Н2(г)=2К(г)+2О(г)+2Н(г) вычислена с использованием
значения Дг/Г°@) = 1735,416+15 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
КаОаНа, г, 0) = —630 + 15 кДж • моль.
Принятая величина является средневзвешенной из приведенных в табл. 47.12 величин,,
исключая выпадающие значения, вычисленные по данным [3870] и по уравнению II закона по
данным [3871]. Указанные в таблице погрешности включают воспроизводимость измерений,
неточность использованных в расчетах термодинамических функций, энтальпий образования
и сечений ионизации.
Таблица 47.12. Результаты определении Ду.Ыо(К202Н2, г,
Портер,
Портер,
Гусаров
Авторы
Скунмейкер, 1958 [3870]
Скунмейкер, 1958 [3871]
, Горохов, 1968 [345]
Метод
0) (в кДж ¦ моль)
Масс-спектрометрический, 2КОН(к)=КаО2Н2(г),
626 К, 1 точка
То же, КаО2Н2(г)=2КОН(г)
То же, 2КОН(к)=К2О2Н2(г)
То же, К2О2Н2(г)=2КОН(г),
То же, КОН(к)+КОН(г)=К
(комбинация с измерениями
641 и 666 К, 2 точки
, 598—653 К, 13 точек
602—777 К, 14 точек
гО2Н2(г), 646 К,_ 1 точка
интенсивностей ионных
токов при 598—653 К, см. выше)
Д/Я°(К2С
II закон
_
—499
-631 ±10
—628 ±20
—630±15
>2Н2, г, 0)
III закон
-667
—619 ±30
—
—
-630±15
KF(k, ж). Термодинамические свойства кристаллического и «жидкого фторида калия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1164.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние KF(k) в интервале 0—1131 К принимается
кубическая модификация (структурный тип NaCl)x.
При Т <^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работе Веструма и Питцера [4715] A6—323 К). С учетом величины S°A6 К)=
=0,155 Дж'К-моль, полученной в результате экстраполяции экспериментальных данных
[4715], приняты приведенные в табл. 47.1 значения ?°B98,15 К) и #"B98,15 К)—#°@); их
погрешности оцениваются в 0,2 Дж-К-моль~1 и 0,025 кДж-моль соответственно 2.
При Т > 298,15 К для теплоемкости KF(k) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии KF, проведенных Веструмом и Питцером [4715] C25—
530 К), Маклеодом [3337] E05—1482 К) и Ляшенко [637] D29—1187 К). При обработке
погрешность данных [4715] оценивалась в 0,3%, данных [3337] — в 0,5% и данных [637] — в 3%.
Температура плавления A131 +1 К) принята по согласующимся между собой измерениям
[163, 668, 1653, 2691, 2796, 2879, 3868]. Теплоемкость KF(hs) G0,3±1,0 Дж-К^.моль)
получена в результате совместной обработки данных по энтальпии жидкого KF, полученных в работах
[637] A148—1187 К; 3 измерения) и [3337] A148-1482 К; 6 измерений).
Энтальпия плавления B8,0+0,4 кДж-моль) вычислена как разность изменений энтальпии KF
в жидком и твердом состояниях при Гт=1131 К, принятых в настоящем справочнике. В
работах [637] и [3337] приводятся более высокие значения B8,7 и 29,5 кДж-моль). Расчеты из
данных пв диаграммам состояния двойных систем [1361, 3806, 4990] приводят к значениям &тН3
в пределах 27,2—28,0 кДж-моль.
1 Кубическая фаза высокого давления (структурный тип CsCl) стабильна при р > 40 кбар (Г=298 К) [2031].
8 Измерения средней теплоемкости в работе Бренстеда [1683] B73—293 К) привели к значению, которое на 0,3%
ниже данных [4715].
387
KF(k, ж)
Глава 47
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
0,5, 2 и 5 Дж-К-моль. Расхождения между термодинамическими функциями KF(k, ж),
приведенными в табл. 1164 и в справочниках [2834] (Т < 3000 К), [2960] (Г < 1800 К), [337] (Т <
^ 3200 К), составляют 0,6, 0,7 и 1,7 Дж-К-моль в значениях Ф°(Т) соответственно и
обусловлены тем, что в настоящем издании учтены экспериментальные данные [3337].
Принятая в справочнике энтальпия образования
к, 298,15 К)=: — 569,9 + 0,7 кДж • моль"-1
основана на измерениях энтальпии растворения KF(k) в воде, результаты которых приведены
в табл. 47.13. Для дальнейших расчетов принимается значение Aa^°(ooH20; 298,15 К)=
=—17,65+0,10 кДж-моль, основанное на хорошо согласующихся данных [580, 1097, 2389,
2390, 3178].
Таблица 47.13. Результаты определений Aa?ff°(KF, к-»соН2О, 298,15 К) (в кДжмоль4)
Авторы
Конечная концентрация, температура
измерений
Да2Я°(ооН20, 298,15 К)
Гунц, 1884 [2530]
Форкран, 1911 [2300, 2302]
Ланге, Эйхлер, 1927 [3178]
Ланге, Мартин, 1937 [3179]
Цветков, Рабинович, 1969 [1097]
Крестов и др., 1974 [580]
Гермейн и др., 1979 [2389, 2390]
200 Н2О, 291,15 К, 1 измерение
110 Н2О, 288,15 К, 1 измерение
18—540 НаО, 298,15 К, 8 измерений
180 Н2О, 298,15 К, 1 измерение
500 Н2О, 298,15 К, 1 измерение
560—11000 Н2О, 298,15 К
1160 Н2О, 298,15 К, 8 измерений
—16,76
—19,38
-17,73+0,17
—16,77+0,84
—17,62+0,42
—17,70 ±0,21
—17,56 ±0,19
Давление пара в реакции KF(k, ж)=КГ(г) вычислено с использованием принятого в
справочнике значения энтальпии сублимации
0) = bsH°(KF, к, 0) = 242,0 ±4,0 кДж • моль.
Принятая величина основана на измерениях давления и состава насыщенного пара KF(k, ж).
В насыщенном паре над фторидом калия присутствуют молекулы KF и K2F2. Согласно данным
[28, 2166] и принятым в справочнике термодинамическим свойствам их содержание при Tm(KF)=
=1131 К составляет 81 и 19 мол.%. Методика выбора термохимических величин для
компонентов насыщенного пара галогенидов щелочных металлов изложена в Приложении 8.
Результаты расчетов энтальпии сублимации KF(k) в виде мономерных молекул представлены
в табл. 47.14. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая величина является средневзвешенной из приведенных в таблице данных. При
Таблица 47.14. Результаты определений ASH"°(KF, к, 0) (в кДж ¦ моль)
Авторы
Вартенберг, Шульц, 1921 [4663]
Руфф и др., 1922 [4096]
Нива, 1938 [3665]
Нарышкин, 1939 [707]
Пью, Барроу, 1958 [3896]
Эйзеяштадт и др., 1958 [2166]
Алиханян и др., 1972 [28]
Топор, 1976 [4527]
Метод
Точек кипения, 1624—1776 К, 5 точек
То же, 1551—1773 К, 6 точек
Эффузионный, 913—973 К, 6 точек
Протока, 1023—1233 К, 4 точки
Торзионный, 902—1023 К, данные представлены
в виде уравнения
Эффузионный, 974—1052 К, 6 точек
Масс-спектрометрический, 1027 К, 1 точка
Статический, 1262—1448 К, 38 точек
ASH°(KF, к, 0)
II закон
III закон
241 + 10 239,4+0,3
257+30 239,3 + 1,5
238+3 232,1 + 0,2
228 + 60 239,2+4,4
244 242,6+1,2
248+5 248,9+0,1
- 244,7
243+3 242,4+0,1
388
Калий и его соединения KF(r), K2F.,(r)
вычислении весов учитывалось отклонение результата данной работы от среднего значения.
Погрешность принятой величины учитывает воспроизводимость измерений, расхождения
измерений в разных работах, неточности термодинамических функций и определения состава
насыщенного пара.
KF(r). Термодинамические свойства газообразного фторида калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1165.
Приведенные в табл. 47.4 молекулярные постоянные KF приняты на основании анализа
результатов исследований микроволнового [4595], ИК- [61, 4007} и УФ- [1406] спектров этой
молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406]
принимается, что валентные возбужденные электронные состояния KF имеют энергии больше
41 000 см и не рассматриваются в справочнике. Полуэмпирические оценки постоянных KF
проводились в работах [169, 558, 567, 1497, 2700, 3786, 4009].
Приведенные в табл. 47.4 колебательные и вращательные постоянные KF в состоянии X1!!*
получены Визе и Горди [4595] обработкой результатов измерений микроволновых переходов
/ -> /+1 (/ ^ 13) на колебательных уровнях с v=0, 1 и 2. Соответствующие постоянные ше
и и>ехе были получены не непосредственно по экспериментальным данным, а рассчитаны с
помощью соотношений Данхема. Они хорошо согласуются с результатами исследования ИК-спектра
[4007] (со, =429+3, (оЛ=2,4±0,25 см) и [61] (сое=426+4, <оЛ=2,4±0,2 см).
Термодинамические функции KF(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции dg, d± и d2 приведены в табл. 47.5 и получены по принятым
молекулярным постоянным KF для изотопомера, соответствующего природной смеси изотопов
калия.
Погрешности рассчитанных термодинамических функций KF(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава фторида калия. При более высоких температурах дополнительный вклад
в погрешность дает неточность принятого способа экстраполяции потенциальной функции.
Погрешности в значениях Ф°(Т) составляют 0,02, 0,05 и 0,3 Дж-К-моль~1 при 298,15, 3000 и
и 6000 К соответственно.
Термодинамические функции KF(r) ранее вычислялись в предыдущем издании настоящего
справочника [337], в таблицах JANAF [2834], а также в работе Брюэра и Бреккетт [1660]
(Ф°(Г), Т <; 2000 К). Расхождения данных [337] и табл. 1165 достигают величин 3,0 и
7,7 Дж-К^-моль" в значениях Ф°F000 К) и 5°F000 К) соответственно и объясняются
различием принятых молекулярных постоянных и метода расчета. Расхождения данных [2834]
и табл. 1165 не превышают 1 и 3,5 Дж-К~1-моль~1 в значениях Ф°(Т) и S°(T) и объясняются тем,
что авторы [2834] использовали приближенный метод расчета.
Константа равновесия реакции KF(r)=K(r)+F(r) вычислена с использованием значения
Ar#°@)=D0(KF)=493,566 +4,1 кДж• моль & 41 260+340 см,' соответствующего принятым
в справочнике энтальпиям образования и сублимации KF(k).
Булевич и др. [1719] на основании спектрофотометрического изучения равновесия реакции
K(r)-fHF(r)=KF(r)+H(r) (пламена H2+O2+N2 с добавками солей калия и G7F14, 1500—2600 К)
нашли значение D0(KF)=490+13 кДж-моль, согласующееся в пределах погрешности с
принятым в справочнике.
Принятым в справочнике энтальпиям образования и сублимации KF(k) соответствует
bfH°(KF, г, 0) = — 326,400 ±4,1 нДж-моль.
K2F2(r). Термодинамические свойства газообразного дифторида дикалия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1166.
Молекулярные постоянные, использованные для расчета термодинамических функций K2F2,
приведены в табл. 47.7. На основании расчета для ионной модели [961, 963, 4689] в
справочнике принимается, что молекула K2F2 в основном электронном состоянии %.хАд имеет плоское
ромбическое строение (симметрия Du). Приведенное в табл. 47.7 произведение моментов инер-
389
КС1(к, ж) Глава 47
ции соответствует структурным параметрам г(К—F)=2,35+0,05 А и Д F—К—F=88+5°,
найденным Соломоником в результате расчета с использованием ионной модели [961, 963] х.
Погрешность значения IaIbIc оценивается в 5-Ю14 г3-см8. Приведенные в табл. 47.7 значения
частот v4, v5 и v6 найдены Шабером и Мартином [4167] при исследовании ИК-спектра молекул
K2F2, изолированных в матрице из Аг; их величины хорошо согласуются со значениями,
полученными другими авторами [1362, 2721, 2793]. Для не наблюдавшихся экспериментально частот vlr
v2, v3 приняты значения, рассчитанные Соломоником [961, 963]. Погрешности принятых значений
v4— v6 оцениваются в 15—20 см, частот vx— v3 — в 30—60 см.
Термодинамические функции K2F2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета, они
составляют 4, 10 и 15 Дж-К~1-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно
(в том числе 2—4 Дж>К~1-моль~1 из-за неточности молекулярных постоянных).
Ранее термодинамические функции K2F2(r) рассчитывались в справочнике JANAF [2834],
а также в работе Краснова и др. [569] (до 5000 К). Расхождения данных этих расчетов и табл. 1166
не превышают 2 Дж-К~1-моль в значениях Ф°(Г) и объясняются некоторым отличием принятых
значений молекулярных постоянных. \
Константа равновесия реакции K2F2(r)=2K(r)+2F(r) вычислена с использованием значения
Дг#°@)=1189,332+7,1 кДж'Моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(KaF2, г, 0) = —855 + 7 кДж • моль.
Принятая величина основана на результатах исследований равновесия реакции KF(K)+KF(r)=
=K2F2(r), выполненных Эйзенштадтом и др. [2166] (эффузионный метод с селектором скоростей,
974—1052 К, 6 точек, Дг#°@)=36+14 кДж-моль (II закон) и 41,4+0,3 кДж-моль (III
закон)) и Алиханяном и др. [28] (масс-спектрометрический метод, 1027 К, 1 точка, ДгЯ°@)=
=39,0+0,5 кДж-моль). Для дальнейших расчетов принимается средняя величина АГН @)=
=40+7 кДж-моль, основным источником ее погрешности является неточность
термодинамических функций.
Рыков [4898] масс-спектрометрическим методом исследовал равновесие реакции K2F2(r)=
=2KF(r) A058 К, 9 измерений). Его данным соответствует значение Д^°(К2Р2, г, 0) = —843 +
±15 кДж-моль, совпадающее с принятым в пределах погрешности.
КС1(к, ж). Термодинамические свойства кристаллического и жидкого хлорида калия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1167.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние КС1(к) в интервале 0—1044 К принимается
кубическая модификация (структурный тип NaCl) 2.
Теплоемкость КС1 при Т < 298,15 К измерена в работах Нернста [3642] B3—86 К), Корефа
[3073] (81-192 и 196-273 К), Брёнстеда [1683] B73-293 К), Саузарда и Нелсона [4350] A7-
285 К), Кеезома и Кларка [2952] B,3-4,3 и 4,6—17,1 К), Феодосьева [1051] (82-298 К), Клу-
зиуса и др. [1883] (И—268 К), Сейра и Бивера [4164] F9—80 К), Кеезома и Пёрлмана [2950]
A,4—4,3 К), Стрелкова и др. [990] A2-298 К), Уэбба и Уилкса [4676] A,5-4,0 К), Берга
и Моррисона [1492] B,8—20 и 12—270 К), Соколова и Шарпатой [958] (88—299 К), Киркхема
и Йейтса [3018] B0—300 К), Теле и др. [4492] F4—274 К). Анализ перечисленных
экспериментальных данных показал, что ниже 90 К результаты всех работ согласуются очень хорошо. Выше
90 К экспериментальные точки группируются с обеих сторон относительно данных Берга и
Моррисона [1492] и хорошо согласующихся с ними данных Саузарда и Нелсона [4350]. Поэтому
при расчете термодинамических функций КС1(к) за основу были приняты измерения Берга и
Моррисона [1492]. Погрешности принятых здачений 5°B98,15 К) и Я°B98,15 К)—Я°@),
приведенных в табл. 47.1, оцениваются в 0,17 Дж-К-моль~1 и 0,02 кДж-моль соответственно.
1 Результаты электронографического исследования структуры молекул М2Х3 (M=Na, К, Rb, Cs, X=F, Cl, Br, I)
в работе [14] не принимались во внимание, так как в этой работе не учитывался сложный состав пара и имелись
систематические ошибки.
2 Кубическая фаза высокого давления (структурный тип CsCl) стабильна при р > 19,5 кбар G"=298 К) [628].
390
Калий и его соединения
КСЦщ at).
При Т > 298,15 К для теплоемкости КС1(к) принято уравнение, полученноегсовместной
обработкой результатов измерений теплоемкости и энтальпии в работах Ляшенко [6371 D09—1127 К)
Попова и др. [812] F73--923 К), Скуратова и Лапушкина [941] F73—933 К), Лидбеттера и Сёт-
татри [3211] C14—641 К), Марчидана и Чопеца [3368] C87—714 К), Томпсона и Фленгаса [4509]
C75—1306 К), Марчидана и Панделе [3372] E13—1141 К) и Гомельского и др. [4864] F73—
"970 К). При совместной обработке погрешность данных [3211] и [4864] оценивалась в 0,5%,
.данных [812, 941, 3372] — в 1%, данных [637, 4509] — в 2% и данных [3368] — в 3%.
Температура плавления KG1 A044+1 К) принята по согласующимся между собой
измерениям [190, 436, 497, 1361, 1599, 1867, 2545, 2879, 3371, 3386, 3513, 4196, 4466, 4758, 4864] A043—
1045 К); менее точные значения были получены в работах [20, 103, 523, 634, 909,, 2566, 3828,,
4017] A042—1048 К). Энтальпия плавления B6,32+0,20 кДж-моль) принятд но
согласующимся между собой данным [637, 2137, 2834, 3372, 4509] B6,32, 26,53, 26,28, 26,36, 2645 «ДжХ
Хмоль соответственно). В работе [4864] получено более высокое значение B6,6 кДж'Моль~*),;
включающее энтальпию предплавления. Менее точные результаты получены в
калориметрических работах [656] B5,4 кДж-моль) и [1361J B6,8+0,8 нДж-моль).
Теплоемкость КС1(ж) G2,0+1,5 Дж-К-моль) принята на основании измерений энтальпии
в интервале 1050—1141 К [3372], выполненных с высокой точностью. С принятым значением
¦в пределах погрешности удовлетворительно согласуются данные Дугласа [цитируется по 2834]
<73,6 Дж-К-^моль; 1053-1173 К), [656] G4,4 Дж-К^-мвль; 1043-1173 К) и [3594]
«^73,2 Дж-К~1'Моль~1; в стоградусном интервале вблизи температуры плавления).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются
« 0,15, 0,5, 2 и 5 Дж-К-моль соответственно. Расхождения между термодинамическими
функциями КС1(к, ж), приведенными в табл. 1167 ив справочниках [337] (Г <; 3100 К), [2834 J
{Т < 2000 К), [2960] (Т < 1500 К) достигают 3, 0,7 и 1 Дж-К-^моль в значениях Ф°(Г)
соответственно. Эти различия обусловлены тем, что в настоящем издании учтены новые
экспериментальные данные.
В справочнике принята энтальпия образования
, к, 298,15 К) = —436,49+.0,13 кДж • моль, ':?"
«основанная на результатах измерений энтальпии растворения КС1(к) в воде, приведенных
да табл. 47.15. В таблице представлены только наиболее полные и точные измерения при 298,15 К.
Ломимо них в литературе известны многочисленные другие измерения энтальпии растворения
Таблица 47.15. Результаты определений Aarff°(KCl, к-> га НаО, 298,15К) (в кДж-моль)
Авторы
Мищенко, Каганович, 1949 [683]
Сомсен и др., 1963 [43461
Ганн, 1965 [2524]
Васильев, Лобанов, 1966 [172]
Абросимов, Крестов, 1967 [4]
Палкин и др., 1969 [751]
Гуревич, Соколов, 1972 [341]
Олофссон и др., 1973 [3714]
Уагман, Килди, 1973 [4627]
Медведев, Ефимов, 1975 [677]
Привалова и др., 1976 [816]
Блахо и др., 1976 [1574]
Рыхлы, Пекарек, 1977 [4107]
Джонсон, Гейер, 1979 [2871]
Килди, 1980 [2995]
Килди, 1980 [2994]
1 ¦ — "
Конечная концентрация,
температура измерений
200 НаО, 6 измерений
2800—8000 Н4О, 10 измерений
200 Н2О, 12 измерений
450 HjjO, 7 измерений
879—2684 Н8О, 13 измерений
2540—3200 Н2О, 8 измерений
370 Н2О, 8 измерений
450 НаО, 5 измерений
561—945 Н2О, 3 измерения
1600 Н2О, 8 измерений
35000-45500 Н2О, 7 измерений
13500—25000 Н2О, 8 измерений
1720—2050 Н2О, 7 измерений
6 измерений
600 Н2О, 5 измерений
500 Н2О, 53 измерения
ДвгЯ°(ооН2О, 298,15 К)
17,200 ±0,042
17,192+0,021
17,236 ±0,088
17,203+0,070
17,196±0,040
17,205 ±0,052
17,158±0,021
17,236 ±0,020
17,218±0,095
17,187 ±0,033
17,217 ±0,063
17,220±0,029
17,278 ±0,030
17,234 ±0,021
17,234 ±0,033
17,241 ±0,018 .
391
ж)
Глава 47
КС1(к) в воде, в том числе проведенные на высоком экспериментальном уровне в работах [3, 171,
407, 444, 499, 552, 553, 672, 776, 777, 864, 1342, 1378, 1857, 2286, 2523, 2638, 2640, 2760, 2885,
3182, 3514, 3547, 3650, 3867, 4066, 4359, 4442]. В работах [683, 684, 685, 1951, 2524, 2994, 3547,
3771, 4346] были проанализированы имевшиеся к моменту их подготовки данные и
рекомендованы наиболее надежные значения.
Результаты приведенных в табл. 47.15 исследований в основном согласуются в пределах их
погрешностей. Во многих этих работах были выполнены детальные исследования влияния таких
факторов, как степень измельчения и гомогенность образца, полнота его осушки, особенности
калориметрической аппаратуры. В справочнике для последующих расчетов принимается
значение Даг#°(ооН20, 298,15 К)=17,241 +0,018 нДж-моль, полученное Килди [2994] в
результате наиболее детальных и'точных измерений.
Давление пира в реакции KGl(k, ж)—КС1(г) вычислено с использованием принятой в
справочнике йнтйльпии сублимации
' ' ДгЯ°@) = Д8Яй(КС|1, к, 0) = 223,4+ 3,0 кДж-моль.
¦ (Принятая величина основана на измерениях давления и состава насыщенного пара КС1(к, ж),
В насыщенном паре хлорида калия содержатся молекулы КС1 и К2С12. Согласно данным [341,
393] и принятым в справочнике термодинамическим свойствам при Гт(КС1)=1044 К их
содержание равно 78 и 22 мол. % соответственно. Методика выбора термохимических величин для
компонентов насыщенного пара галогенидов щелочных металлов изложена в Приложении 8.
Таблица; 47.16. Результаты определений ASJ2"°(KC1, к, 0) (в,«Дж-моль)
...,„. Авторы .
Вартенберг, Альбрехт, 1921 [4656]
Руфф, Мугдан, 1921 [4094]
Джексон, Морган, 1921 [2805] ~;;
Фиок, Родебуш, 192§,{2258] ,
Хрриба, Баба, 1928 [2711] ,?
Грейнер, Еллинек, 1933 [2482]
Дейц, 1936 [2023]
Кангро, Викинг, 1938 12919]
Мейер, Винтнер, 1938 13433]
Нива, 1938 [3665]
Нарышкин, 1939 [707]
Цимм, Мейер, 1944 [4823]
Тредуэлл, Вернер, 1953 [4538]
Брэдли, Волане, 1953 [1642]
Несмеянов, Сазонов, 1957 [719]
Кнаке и др., 1957 [3046]
Пью, Барроу, 1958 i[3896]
Бартон, Блум, 1959 [1417]
Чалка, 1959 [1851]
Милн, Клейн, 1960 [3510]
Шриер, Кларк, 1963 [4212]
Мургулеску, Марта, 1966 [3593]
Лукашенко и др., 1970 [635]
Глазов, Нарышкин, 1970 [288]
Эмонс и др., 1976 [2173]
Вагнер, Шефер^ 1979 [4629]
Метод
Точек, кипения, 1389—1691 К, 16 точек
То же, Т393—1688 К, 7 точек
Протока, 1074-^1317 К, 3 точки-
Статический, 1250—1428 К, 8 точек
. То же, 973—1524 К, -ДЗ точек
Протока, 1453 К, 2 точки
Торзионный, 847—936 К, 9 точек
Эффузионный, 913—932 К, 6 точек
Протока, 1133—1263 К, .3 точки
Эффузионный, 899—935 К, 5 точек
То же, 853—955 К, 11 точек
Протока, 1023—1188 К, 3 точки
Эффузионный, 624—945 К, 21 точка
Протока, 859—1024 К, 13 точек
Эффузионный, 713—870 К, 5 точек
То же, 753—897 К, 7 точек
То же, 750—870 К, 9 точек
Торзионный, 819-^945 К а
Точек кипения, 1250—1473 К,~4 точки
Масс-спектрометрический, 7^=800 К а
То же, Гср=900 К
Точек кипения,,1189—1418 К а
Статический, 1373—1473 К, 5 точек
Эффузионный, 1023—1123 К, 4 точки
То же, 1043—1143 К а
То же, 1063—1243 К а
Масс-спектрометрический, 821—882 К а
а Данные представлены в виде уравнения.
Д,Я°(КС1, к, 0)
II закон
224+6
238 ±8
181 ±75
236±4
177+19
—
223+4
221+30
187+60
236+25
211 + 2
222 ±80
221 + 2
218+2
224±4
217 ±8
211+3
225
225 ±2
225
221
228
228 ±30
228+40
224
235
224
III закон
222,1 ±0,4
223,1 + 1,0
222,5 + 10
229,0±0,4
219,0 ±3,0
225,3 + 0,2
224,8±0,2
225,4±0,3
218,2+4,5
223,5±0,3
224,1 ±0,3
221,6+1,0
222,8+0,4
224,9+0,3
223,4 + 0,3
223,9+0,6
221,3+0,6
223,2+1,5
222,5+0,4
—
—
223,1 + 1,0
222,7+0,6
226,6+0,7
224,2±1,0
223,1 ±2,5
—
392
Калии и его соединения КС1(г), К2С12(г)'
Результаты расчетов энтальпии сублимации КС1(к) в виде мономерных молекул представлены
в табл. 47.16. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая-величина является средневзвешенной из приведенных в таблице величин.
Ее погрешность учитывает воспроизводимость измерений, расхождения измерений в разных
работах, неточность термодинамических функций и неточность определения состава
насыщенного пара.
КС1(г). Термодинамические свойства газообразного хлорида калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1168.
Приведенные в табл. 47.4 молекулярные постоянные КС1 приняты на основании анализа
результатов исследований микроволнового [1874, 1875, 1876, 2700, 4479], ИК- [3984] и УФ- [1406}
3239] спектров этой молекулы. По аналогии с молекулой LiF и на основании исследования
электронного спектра [1406, 3239] принимается, что валентные возбужденные электронные состояния
КС1 имеют энергии больше 35 000 см.
Приведенные в табл. 47.4 колебательные и вращательные постоянные КС1 в состоянии X1!!4"
получены Клаузером и Гбрди [1876] обработкой результатов измерений микроволновых
переходов / -> /+1 (/ ^ 25) на колебательных уровнях с i?=0, 1 и 2. Соответствующие
постоянные о>е и шехе были получены не непосредственно по экспериментальным данным, а рассчитаны
с помощью соотношений Данхема. Они удовлетворительно согласуются с результатами
исследования ИК-спектра [3984] (и,=281+6, wexe=l,3 см).
Термодинамические функции КС1(г) были вычислены по уравнениям A. 3)—A. 6), A.9),,
A. 10), A. 93)—A. 95). Значения Qm и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dt и d2 приведены в табл. 47.5 и получены по принятым
молекулярным постоянным 39К36С1 для изотопомера, соответствующего природной смеси
изотопов калия и хлора.
Погрешности рассчитанных термодинамических функций КС1(г) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава хлорида калия. При более высоких температурах дополнительный вклад
в погрешность вносят неточность принятого способа экстраполяции потенциальной функции,
а также пренебрежение возбужденными электронными состояниями. Погрешности в значениях
Ф°(Т) составляют 0,02, 0,05 и 0,5 Дж-К^-моль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции КС1(г) ранее вычислялись в предыдущем издании настоящего
справочника [337], в таблицах JANAF [2834], а также в работах Уилкинса [4728] (Г<6000К)г
Брюэра и Брэккетт [1660] (Ф°(Г), Т < 2000 К), Раиса и Клемперера [3985] (Т < 2000 К).
Расхождения данных [337, 2834, 4728] и табл. 1168 не превышают 1,1 и 4,0 Дж-К-моль в
значениях Ф°(Т) и S°(T) соответственно и объясняются тем, что в этих справочниках и работе
использовались приближенные методы расчета.
Константа равновесия реакции КС1(г)=К(г)+С1(г) вычислена с использованием значения
Ar#°@)=D0(KCl, г)=422,293+3,0 кДж• моль ри 35300+250 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации КС1(к).
Близкие значения энергии диссоциации D17+8 кДж-моль" и 429+8 кДж-моль) нашли
Булевич и др. [1719] на основании спектрофотометрического исследования равновесия реакции
К(г)+НС1(г)=КС1(г)+Н(г) (пламена H2+O2+N2, 1500-2600 К) и Су и Райли [4427] методом
фотодиссоциативной спектроскопии. Бойтлер и Леви [1539] по коротковолновой границе флук-
туационных полос в спектре поглощения КС1 получили более высокое значение D35+8 кДжХ
Хмоль).
Принятым в справочнике энтальпиям образования и сублимации КС1(к) соответствует
' г, 0) = —212,782 ±3,0 кДж • моль.
К2С12(г). Термодинамические свойства газообразного дихлорида дикалия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1169.
Молекулярные постоянные, использованные для расчета термодинамических функций
К2С12(г), приведены в табл. 47.7. На основании теоретических расчетов [961, 963, 4689] для
молекулы К2С12 в основном электронном состоянии XхА д принимается плоская ромбическая
структура (симметрия D2h). Приведенное в табл. 47.7 произведение моментов инерции соответствует
393
КВг(к, ж)
Глава 47
структурным параметрам г(К-^-С1)=2,86+0,05 А и Д С1—К—С1=98+5°, найденным Соломо-
ником в результате расчета с использованием ионной модели [961, 963]. Погрешность значения
IaIbIc составляет 5• 10~118 г3-см6. Приведенные в табл. 47.7 значения частот v4—ve найдены
Шабером и Мартином [4167] при исследовании ИК-спектра молекул К2С12, изолированных в матрице
из Агг их величины хорошо согласуются с данными, полученными Измаилом и др. [2794]. Для
не наблюдавшихся экспериментально частот vx— v3 приняты значения, рассчитанные Соломоником
[961, 963]. Погрешности принятых значений v4— v6 составляют с учетом возможного матричного
сдвига 15—20 см, а частот va—v3 30—40 см.
Термодинамические функции К2С12(г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A.10), A.122)—A. 124), A.128),
A. 130) без учета возбужденных электронных состояний. Погрешности рассчитанных величин
при низких температурах обусловлены в основном неточностью принятых молекулярных
постоянных, а при высоких температурах — приближенным характером расчета; они составляют 5Л
10 и 15 Дж-К^-моль в значениях Ф°(Т) при.298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции К2С12(г) рассчитывались в справочнике JANAF [2834]
и в работе Краснова и др. [569] (до 3000 К). Результаты расчетов согласуются с данными
табл. 1169 в пределах 3 Дж-К~1-моль и 0,5 Дж-К моль в значениях Ф°(Г) для [2834]
и [569] соответственно.
Константа равновесия реакции КаС12(г)=2К(г)-{-2С1(г) вычислена с использованием значения
ДгЯ°@)=1031,022+7,0 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
AfH°(K2Gl2, г, 0) = —612 + 7 кДж • моль.
Эта величина основана на приведенных в табл. 47.17 результатах исследований равновесия
реакции КС1(к, ж)+КС1(г)=К2С12(г). Для дальнейших расчетов использовалось значение
Ar-ff°@)=37+8 кДж-моль, среднее из приведенных в таблице величин. Его погрешность
определяется в первую очередь неточностью термодинамических функций.
Таблица 47.17. Результаты определений энтальпии реакции КС1(к, ж) + КС1(г) = К2С12(г) (в кДж • моль)
Авторы
Дейц, 1936 [2023]
Миллер, Куш, 1957 [3504)
Берковиц, Чапка, 1958 [1501]
Бартон, Блюм, 1959 [1417]
Шриер, Кларк, 1963 [4212]
Мургулеску, Марта, 1966 [3593]
Чапка, 1959 [1851]
Милн, Клейн, 1960 [3510]
Вагнер, Шефер, 1979 [4629]
Метод
Эффузионный и торзионный, 913—932 К,
Эффузнонный с селектором скоростей, 897
Масс-спектрометрический, 915 К, 1 точка
Протока и точек кипения, 1250—1473 К,
Протока и точек кипения, 1153—1352 К,
Протока и статический, 1373—1473 К,
Масс-спектрометрический, 7|ср=800 К а
То же, Гср=900 К а
То же, 821—882 К а
а Данные представлены в виде уравнения. '•
2
7
10
22
точки
точка
точек
точек
точки
Д,.Яо@)
II закон
—
—
45+25
39 ±13
13 ±55
48 ±13
48
37,0
III закон
44,1 ±24
39,5
39,3
31,1 ±1,4
32,6 ±0,6
33,0±1,3
—
—
38,2
В работах [1417, 1851, 1995, 2023, 3504, 3593] было исследовано равновесие реакции К2С12(г)=
=2КС1(г). Эти измерения приводят к значениям Д/Я°(К2С12, г, 0)=—615+10 кДж-моль (III
закон) и —625+20 кДж-моль (II закон), которые совпадают с принятым в пределах погрешностей.'
КВг(к, ж). Термодинамические свойства кристаллического и жидкого бромида калия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1170.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние КВг(к) в интервале 0—1007 К
принимается кубическая модификация (структурный тип NaCl) Ч
1 Кубическая фаза высокого давления (структурный тип CsCl) стабильна при р > 17 кбар (Г=298 К) [4584].
394
Калий и его соединения
КВг(к,
При Т <^ 270 К термодинамические функции вычислены по результатам согласующихся
между собой измерений теплоемкости в работах Клузиуса и др. [3043] A1—270 К), Берга и Мор-
рисона [1492] B,8-20 и 12—270 К), О'Нила и Филлипса [3718] (Т < 1 К) и Мартина [3407]
@,5—1,5 К). В интервале 270—298,15 К были использованы интерполированные значения
теплоемкости между данными работы [1492] и результатами измерений при Т > 298,15 К (см. ниже).
Данные по теплоемкости, полученные в работе Теле и др. [4492] F4—273 К), практически
совпадают с принятыми в интервале 160—220 К, лежат выше них при Т <С 160 К и ниже при Т >
> 220 Kj при этом максимальное расхождение достигает 1,5%. Результаты единичных
измерений средней теплоемкости в старых работах Корефа [3073] (82—192 и 196—273 К), Нернста
[3642] G9—в9 К) и Брёнстеда [1683] B73—293 К) имеют большой разброс и не учитывались.
Приведенные в табл. 47.1 значения <S°B98,15 К) и fT°B98,15 К)—Н°@) имеют погрешности
•0,2 Дж-К~1-моль и 0,025 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости КВг(к) принято уравнение, полученное при совместной
^обработке результатов измерений теплоемкости и энтальпий, проведенных Скуратовым и Лапуш-
киным [941] F73—933 К), Купером [1919] E70—993 К), Лидбеттером и Сеттатри [3211] C05—
762 К), Бибасом и Леонарди [1554] C00—1070 К). Погрешность данных [941] оценивалась
в 0,5%, данных [1919] и [3211] — в 1% и данных [1554] — в 2%. Результаты отдельных
измерений энтальпии в работах [249, 812, 3340] и теплоемкости в работе [3602] менее точны и не
учитывались.
Температура плавления A007+1 К) принята по прецизионным измерениям Джонсона и Бре-
дига [2879], с которыми в указанных пределах погрешности хорошо согласуются данные [1361,
2409, 3828, 4196, 4791 ] A006—1008 К); библиографию менее точных определений этой величины
•см. [1009]. Энтальпия плавления B5,52+0,40 кДж-моль) принята по данным Дворкина и Бре-
дига [2137]. С принятым значением хорошо согласуется величина теплоты плавления, получен-
яая в работе [1554] B5,15 кДж.моль). Измерения [1361] B8,0 кДж-моль), [1577] B4,8 кДжХ
Хмоль) и [3433] B2,2 кДж-моль) приводят к менее достоверным величинам. Теплоемкость
КВг(ж) F9,5+1,5 Дж-К~1-моль) принята по согласующимся между собой данным Дворкина
[2135] F9,9 Дж-К~1-моль в интервале 1010—1100 К; цитируется по справочнику JANAF)
и Мургулеску и Теле [3594] F9,2 Дж-К~1-моль~1). Величина теплоемкости КВг(ж), полученная
в работе [1554] D5,8 Дж-К~1-моль~1), явно занижена.
Таблица 47.18. Результаты определений Ae?-ff°(KBr, к->соНаО, 298,15К) (в кДж-моль)
Авторы
Томсен, 1883 [4511]
Брёнстед, 1906 [1680]
Вальден, 1907 [4640]
Вюст, Ланге, 1925 [4787]
Чипман и др., 1929 [1838]
Попов и др., 1930 [3866]
Фёдоров, Сильченко, 1937 [1042]
Ланге, Мартин, 1937 [3179]
Попов и др., 1940 [813]
Хиетала, 1960 [2638]
Крестов, Абросимов, 1967 [579]
Цветков, Рабинович, 1969 [1097]
Уагман, Килди, 1973 [4627]
Джоли и др., 1973 [2885, 2886]
Олафссон, 1977 (см. [2157])
Перелыгин и др., 1978 [245, 777]
Ефимов и др., 1979 [2157]
Конечная концентрация, температура
измерений
200 Н2О, 292,8 К, 2 измерения
700 Н2О, 293,2 К, 8 измерений
200 Н2О, 289,8 К, 2 измерения
10,6—150 Н2О, 298,15 К, 31 измерение
130—600 Н2О, 298—300 К, 30 измерений
300 Н2О, 273 К, 7 измерений
100—400 Н2О, 296,6 К, 3 измерения
430 Н2О, 1 измерение
460 Н2О, 273, 6 К
200 Н2О, 298,15 К, 3 измерения
1320—6170 Н2О, 283—373 К
500 Н2О, 298,15 К, 1 измерение
838 и 426 Н2О, 298,15 К, 2 измерения
3190 Н2О, 298,15 К, 1 измерение
800—1110 Н2О, 298,15 К, 7 измерений
2780 Н2О, 298,15 К
555—7000 Н2О, 298,15 К, 11 измерений
1260 Н2О, 298,15 К, 3 измерения
1320—3700 Н2О, 298,15 К, 7 измерений
ДвдЯ°(ю Н2О, 298,15 К)
19,95+0,40
19,90+0,20
19,78+0,20
19,99+0,20
19,89 ±0,40
19,60+0,25
20,46+0,50
20,01 ±0,30
19,00+0,40
19,828+0,085
20,43±0,10
19,80+0,20
19,772+0,050
19,60+0,09
19,813±0,044
19,809+0,035
19,851 ±0,027
19,845+0,010
19,709+0,044
395
КВг(г)
Глава 47"
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000,2000 и 3000 К оцениваются в 0,15,.
0,6, 4 и 9 Дж-К-моль~1 соответственно. Значения термодинамических функций КВг(к, ж)».
рассчитанные в справочниках [2834] (Г < 2500 К) и [2960] (Т < 1000 К), согласуются с
данными табл. 1170 в пределах 0,4 и 1 Дж-К-моль в значениях Ф°(Т).
Принятая в справочнике энтальпия образования
bfH°(KBr, к, 298,15 К) = — 393,45 + 0,20 кДж • моль
основана на результатах измерений энтальпии растворения КВг(к) в воде, представленных
в табл. 47.18. Принятое значение Дй?#°(соН20; 298,15 К)=19,78+0,08 кДж-моль основано
на наиболее точных данных [245, 777, 2157, 2638, 4627] и близко к среднему из всех приведенных
в таблице величин. Менее точные измерения энтальпии растворения КВг(к) в воде были
выполнены в работах [54, 519, 520, 921, 3859].
Давление пара в реакции КВг(к, ж)=КВг(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
0) = Д,Я°(КВг, к, 0)^216,3 + 4,0 кДж • моль.
Эта величина основана на измерениях давления и состава насыщенного пара КВг(к, ж).
Насыщенные пары бромида калия состоят из молекул КВг и К2Вг2; согласно данным [2546] и
термодинамическим свойствам соединений калия, принятым в справочнике, содержание этих молекул
при Гт=1007 К составляет 74 и 26 мол.% соответственно. Методика расчета термохимических
величин для компонентов насыщенного пара галогенидов щелочных металлов изложена в
Приложении 8.
Результаты расчетов энтальпии сублимации КВг (к) в виде мономерных молекул представлены
в табл. 47.19. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая величина является средневзвешенной из приведенных в таблице данных.
Погрешность рекомендованного значения учитывает воспроизводимость измерений, расхождение
результатов измерений в разных работах, неточность термодинамических функций и определения
состава насыщенного пара.
Таблица 47.19. Результаты определений AgJ2"°(KBr, к, 0) (в кДж • моль)
Авторы
Вартенберг, Альбрехт, 1921 [4656]
Руфф, Мугдан, 1921 [4094]
Фиок, Родебуш, 1926 [2258]
Нива, 1938 [3665]
Мейер, Винтнер, 1938 [3433]
Цимм, Мейер, 1944 [4823]
Чапка, 1959 [1851]
Хагемарк и др., 1966 [4969а]
Мургулеску, Марта, 1966 [3593]
Эвинг, Штерн, 1975 [2200]
Метод
Точек кипения, 1368—1654 К, 14 точек
То же, 1361—1668 К, 10 точек
Статический, 1173—1336 К, 10 точек
Эффузионный, 823—923 К, 6 точек
То же, 885—929 К, 6 точек
То же, 637—900 К, 15 точек
Масс-спектрометрический, Тор=800 К а
Статический, 1143—1408 К а
То же, 1373—1473 К, 5 точек
Испарение с поверхности, Гср=753 К,
измеренные давления не указаны
а Данные представлены в виде уравнения.
А8Н°(КВт, к, 0)
II закон
III закон
221+5 215,2 ±0,3
224+7 217,1 ±0,5
215 ±6 216,0 ±0,2
204+1 218,3+0,7
237 ±40 217,6+0,7
215 ±2 213,8 ±0,2
219 ±2 —
217 216,2+1,1
218±3 215,7+0,1
209 —
КВг(г). Термодинамические свойства газообразного бромида калия в стандартном состоянии»
в интервале температур 100—6000 К приведены в табл. 1171.
Приведенные в табл. 47.4 молекулярные постоянные КВг приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3984] и УФ- [1406, 3239] спектров
этой молекулы. По аналогии с молекулой LiF и на основании исследования электронного спектра
[1406, 3239] в справочнике принимается, что валентные возбужденные электронные состояния
КВг имеют энергии больше 32 000 см.
396
Калий в его соединения К2Вг2(г)
Приведенные в табл. 47.4 колебательные и вращательные постоянные КВг в состоянии
получены Раском иТорди [4101] обработкой результатов измерений микроволновых переходов
J -*¦ /+1 (/ ^ 40) на колебательных уровнях с v=0, 1 и 2. Соответствующие постоянные <о,
и шехе были получены не непосредственно по экспериментальным данным, а рассчитаны с
помощью соотношений Данхема. Они удовлетворительно согласуются с результатами
исследования ИК-спектра [3984] («„=213+6, <оЛ=0,8 см).
Термодинамические функции КВг(г) были вычислены по уравнениям A. 3)—A. 6), A. 9),
•A. 10), A. 93)—A. 95). Значения QBB и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 47.5 и получены по принятым
постоянным молекулы 39К81Вг для изотопомера, соответствующего природной смеси изотопов
калия и брома.
Погрешности рассчитанных термодинамических функций КВг(г) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах дополнительный вклад в погрешность вносят неточность принятого способа
экстраполяции потенциальной функции, а также пренебрежение возбужденными электронными
состояниями. Погрешности в значениях Ф°(Т) составляют 0,02, 0,05 и 0,6 Дж-К~1-моль при
298,15, 3000 и 6000 К соответственно.
Термодинамические функции КВг(г) ранее вычислялись в таблицах JANAF [2834], а также
ъ работах Уилкинса [4728] (Т < 6000 К), Брюэра и Брэкет [1660] (Ф°(Т), Т < 2000 К).
Расхождения данных [2834, 4728] и табл. 1171 не превышают 1,3 и 4,1 Дж-К-моль в значениях
Ф°(Г) и S°(T) соответственно и объясняются тем, что авторы [2834, 4728] использовали
приближенный метод расчета.
Константа равновесия реакции КВг(г)=К(г)+Вг(г) вычислена с использованием значения
дгЯ°@)=Б0(КВг)=377,826+4,0 кДж• моль я» 31 580+330 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации КВг(к).
Близкие значения энергии диссоциации КВг были получены методом фотофрагментарной
спектроскопии в работах Ван Вина и др. [4589] C76,3+2,0 кДж-моль) и Су и Райли [5056]
{375,3+4,2 кДж-моль), а также при спектрофотометрическом изучении равновесия реакции
К(г)+НВг(г)=КВг(г)+Н(г) в пламенах в работе Булевич и др. [1719] A500—2600 К, 379 +
+8 кДж-моль). Исследования коротковолнового предела флуктуационных полос в спектре
.поглощения [4345] и в спектрах испускания и поглощения КВг [1539] привели к значениям
>366 и 385+8 кДж-моль-1.
Принятым в справочнике энтальпиям образования и сублимации КВг(к) также соответствует
г, 0) = —170,012 ±4,0 кДж • моль.
К2Вг2(г). Термодинамические свойства газообразного дибромида дикалия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1172.
Молекулярные постоянные, использованные для расчета термодинамических4 функций К2Вг2,
приведены в табл. 47.7. На основании теоретических расчетов [961, 4689] для молекулы К2Вг2
в основном электронном состоянии ХхАд принята плоская ромбическая структура-(симметрии/Jд).
Приведенное в табл. 47.7 произведение моментов инерции соответствует структурным параметрам
,г(К—Вг)=3,02+0,05 А и Д Вг—К—Вг=101+10°, рассчитанным Соломоником в результате
использования ионной модели [961, 963]. Погрешность значения IaIbIc оценивается
в 3-Ю"12 г3-см6. Приведенные в табл. 47.7 значения v5, ve найдены Шабером и Мартином [4167]
в ИК-спектре молекул К2Вг2, изолированных в матрице из Аг. Для не наблюдавшихся
экспериментально частот приняты значения, рассчитанные Соломоником [961, 963]. Погрешности
принятых значений частот v5, ve оцениваются с учетом матричного сдвига в 15 см, а частот vx—v4
в 20—25% от их величины.
Термодинамические функции К2Вг2(г) вычислялись в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124),
A. 128), A. 130) без учета возбужденных состояний. Погрешности рассчитанных величин
обусловлены неточностью принятых молекулярных постоянных, а также приближенным характером
расчета, они составляют 8, 12, 16 Дж-К-^моль'1 в значениях Ф°(Т) при 298,15, 3000 и 6000 К
соответственно.
397
К1(к, ж) Глава 47
Ранее термодинамические функции К2Вг2(г) рассчитывались в справочнике JANAF [2834 F
и в работе Краснова и др. [569] (до 3000 К). Результаты этих расчетов согласуются с данными
табл. 1172 в пределах 0,8 и 2 Дж-К-моль~1 в значениях ФО(Г).
Константа равновесия реакции К2Вг2(г)=2К(г)+2Вг(г) вычислена с использованием
значения Дгйг°@)=936,628+10 кДж-моль", соответствующего принятой в справочнике энтальпии;
образования
Д/&°(КяВг2, г, 0) = —521 + 10 кДж-моль.
Эта величина основана на исследованиях равновесия реакции КВг(к, ж)+КВг(г)=К2Вг2(г)^
представленных в табл. 47.20. Приведенные в таблице погрешности учитывают только
воспроизводимость измерений. Для дальнейших расчетов принимается среднее значение АгН°@) =
=35+10 кДж-моль, Его погрешность обусловлена в основном неточностью термодинамических
функций.
Таблица 47.20. Результаты измерений энтальпии реакции КВг(к, ж) + КВг(г) = К2Вг2(г) (в кДж • моль).
Авторы
Берковиц, Чапка, 1958 [1501]
Чапка, 1959 [1851]
Хагемарк, Хенгстенберг, 1966 [4969а]
Мургулеску, Марта, 1966 [3593]
Метод
Масс-спектрометрический, 890 К, 1 точка
То же, Гср=800 К
Статический и измерение плотности пара,
1143—1408 К, 2 точки
Статический и протока, 1373—1473 К,
22 точки
ДгЯ°@)
II закон
III закон
- 39,8
44+13 —
38 ±4 35,2 ±3,0
19+50 30,6±1,3
В работах [1851, 4969а, 35931 было исследовано равновесие реакции К2Вг2(г)=2КВг(г). Этим,
данным соответствует значение Д/Я°(К2Вг2, г, 0)=—524+15 кДж-моль (III и II законы),
согласующееся в пределах погрешности с принятым в справочнике.
К1(к, ж). Термодинамические свойства кристаллического и жидкого иодида калия в
стандартном состоянии при температурах 100—3300 К приведены в табл. 1173.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние К1(к) в интервале 0—954 К принимается
кубическая модификация K.I (структурный тип NaCl) *.
Теплоемкость KI при Т <^ 298,15 К измерялась в работах Клузиуса и др. [1883] A0—270 К),
Берга и Моррисона [1492] B,8—20 и 12-270 К), Скейлса [4165] B—7 К), Мартина [3407!1
@,5—1,5 К), Теле и др. [4492] F5—273 К). С учетом точности эксперимента, совершенства
методик и чистоты исследованного образца термодинамические функции KI при Т ^ 298,15 К
вычислены по данным Берга и Моррисона [1492], с которыми хорошо согласуются измерения
Скейлса [4165]. В старых работах Корефа [3073] (82—193 и 196—273 К) и Брёнстеда [1683]*
B73—293 К) приведены результаты измерений средней теплоемкости, точность которых невелика.
Принятые значения ?°B98,15 К) и Я°B98,15 К)—#°@), приведенные в табл. 47.1, имеют
погрешности 0,2 Дж-К-моль и 0,025 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости К1(к) принято уравнение, полученное совместной
обработкой измерений энтальпии иодида калия, проведенных Скуратовым и Лапушкиным [941}
F23—933 К), Купером [1919] E70—973 К) и Дворкиным [2135] (854—954 К; цитируется по-
справочнику [2834]). При обработке погрешность данных [941] и [2135] оценивалась в 0,5%,
а данных [1919] — в 1%.
Температура плавления (954+1 К) принята по измерениям Джонсона и Бредига [28791
(953,7+0,5 К), проведенным для весьма чистого образца. С выбранным значением
удовлетворительно согласуются результаты определений в работах [1307, 1576, 3828, 3949, 4054]; менее
точные величины были найдены в работах [1644, 3041, 3386, 39411 (951—959 К).
1 Кубическая фаза высокого давления (структурный тип CsCI) стабильна при р > 17,5 кбар (Т=29& К) [2969]~
398
Калий и его соединения
К1(к,
Энтальпия плавления B4,02+0,40 кДж-моль"*) принята по результатам калориметрических:
измерений, проведенных Дворкиным и Бредигом [2137]. В литературе известны также значения,
полученные методом высокотемпературной криометрии [1576] .B5,1+1,0 кДж-моль),
калориметрическим методом [1636] B8,9+0,8 кДж-моль), а также рассчитанное по диаграммам
плавкости двойных систем [4990] A7,2 кДж-моль).
Теплоемкость К1(ж) G0,3+2,0 Дж-К~1-моль) получена усреднением данных [2135]
G2,4 Дж-К^-моль) и [3594] F8,2 Дж-К-моль). В обеих работах измерения энтальпии
жидкого KI проводились в узком интервале температур вблизи точки плавления.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются;
в 0,15, 0,7, 4 и 10 Дж-К~1-моль. Расхождение между термодинамическими функциями К1(к, ж),,
приведенными в табл. 1172 и в справочнике [2834] (Г ^ 2500 К), не превышают 1 Дж-К-1Х
Хмель в значениях Ф°(Г). Расхождения с данными [2960] (Т <; 950 К) составляют 2 Дж-КХ
Хмоль в значении 5°B98,15 К) и объясняются уточнением теплоемкости К1(к) в работе [1492].
Принятая в справочнике энтальпия образования
bfH°{Kl, к, 298,15 К) = —329,30 + 0,17 кДж • моль
основана на результатах измерений энтальпии растворения К1(к) в воде, представленных
в табл. 47.21. Принятое в справочнике значение ДйдЯс(ооН20;, 298,15 К)=20,23+0,10 кДжХ
Хмоль является средним из результатов наиболее точных последних работ [2, 3, 245, 777, 2157],
удовлетворительно согласующихся между собой, и близко к среднему из всех данных
приведенных в таблице. Менее точные измерения энтальпии растворения KI в воде были выполнены в
работах [2030, 3895].
Таблица 47.21. Результаты измерений ДЙ4Й"С(К1, к->оэН2О, 298,15 К) ( в кДж •
Авторы
Томсен, 1883 [4511]
Бюст, Ланге, 1925 [4787]
Ланге, Мартин, 1937 [3179]
Попов и др., 1940 [813]
Бобтельский, Лэрш, 1950 [1592]
Капустинский, Дракин, 1952 [482]
Цветков, Рабинович, 1969 [1097]
Крестов и др., 1972 [583]
Джоли и др., 1973 [2885, 28$В]
Крестов и др., 1974 [5801
Абросимов и др., 1977 [2, 3]
Перелыгин и др., 1978 [245, 777]
Ефимов и др., 1979 [2157] !
Конечная концентрация, температура
измерений
200 HjjO, 292 К, 2 измерения
7—190 Н2О, 298,15 К, 24 измерения
500 Н2О, 298,15 К, 1 измерение
620 Н3О, 293,6 К, 1 измерение
185 Н2О, 297,6 К, 1 измерение
70000 Н2О, 298,15 К, 1 измерение
500 Н2О, 298,15 К, 1 измерение
1100—5500 Н2О, 298,15 К
3000 Н2О, 298,15 К, 1 измерение
555—11100 Н2О, 298,15 К
50—760 НаО, 298,15 К, 4 измерения
2780 Н2О, 298,15 К
?00—9250 Н2О, 298,15 К, 11 измерений
моль)
ДягЯо(соН20, 298,15 К)
20,29 ±0,40
20,49 ±0,25
20,39 ±0,25
20,10 ±0,40
21,00 ±0,80
20,21 ±0,30
20,18 ±0,25
20,50 ±0,10
20,56 ±0,21
20,41 ±0,21
20,294 ±0,060
20,254 ±0,030
20,145 ±0,024
Давление пара в реакции К1(к, ж)=К1(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = Д,Я°(К1, к, 0) = 203,3 + 4,0 кДж ¦ моль.
Эта величина основана на измерениях давления и состава насыщенного пара К1(к, ж).
Насыщенный пар иодида калия состоит из молекул KI и К212; согласно данным [1501, 3504] и
принятым в справочнике термодинамическим свойствам при Гт(К1)=954 К их содержание
составляет 87 и 13 мол.%. Методика выбора термохимических величин для компонентов насыщенного
пара галогенидов щелочных металлов изложена в Приложении 8.
Результаты расчета энтальпии сублимации К1(к) в виде мономерных молекул представлены
в табл. 47.22. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая величина является средневзвешенной из приведенных в таблице данных. По-
399
К1(г) Глава 47
Таблица 47.22. Результаты определений А,Я"°(К1, к, 0) (в кДж-моль)
Авторы
Вартенберг, Альбрехт, 1921 [4656]
Руфф, Мугдан, 1921 [4094]
Фиок, Родебуш, 1926 [2258]
Грейнер, Еллинек, 1933 [2482]
Нива, 1938 [3665]
Цимм, Мейер, 1944 [4823]
Кодяшн, Кимбелл, 1948 [1890]
Кнаке и др., 1957 [3046]
Мургулеску, Топор, 1970 [3598]
Метод
Точек кипения, 1336—1606 К, 9 точек
То же, 1319—1590 К, 12 точек
Статический, 1116—1302 К, 9 точек
Протока, 1453 К, 2 точки
Эффузионный, 803—873 К, 8 точек
То же", 618—893 К, 10 точек
То же, 721—898 К, 29 точек
То же, 700—800 К, 9 точек
Статический, 1273—1357 К, 9 точек
Д,Я°(К1, к, 0)
II закон
III закон
214+5 204,1 ±0,5
220 ±10 203,3+0,8
205 ±2 203,4 ±0,1
— . 205,5
200±3 203,1 ±0,1
209 ±3 204,3 ±0,7
211 ±4 200,9 ±0,4
— 203,5 ±0,5
198+3 201,8 ±0,1
грешность рекомендованного значения учитывает воспроизводимость измерении, расхождение
результатов измерений в разных работах, неточность термодинамических функций и определения
состава насыщенного пара.
К1(г). Термодинамические свойства газообразного иодида калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1174.
Приведенные в табл. 47.4 молекулярные постоянные KI приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3984] и УФ- [1406, 1539, 3239]
спектров этой молекулы. По аналогии с молекулой LiF и на основании исследования
электронного спектра [1406, 1539, 3239] в справочнике принимается, что валентные возбужденные
электронные состояния имеют энергии больше 27 000 см.
Приведенные в табл. 47.4 колебательные и вращательные постоянные KI в состоянии Z1S +
получены Раском и Горди [4101] обработкой результатов измерений микроволновых переходов
/ -> /+1 (J ^ 50) на колебательных уровнях с v=0, 1 и 2. Соответствующие постоянные <о,
и шехе были получены не непосредственно по экспериментальным данным, а рассчитаны с
помощью соотношений Данхема.
Термодинамические функции К1(г) были вычислены по уравнениям A. 3)—A. 6), A. 9),;
A. 10), A.93)—A.95). Значения (?вн и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dt и d2 приведены в табл. 47.5 и получены по принятым
постоянным молекулы 39К1 для изотопомера, соответствующего природной смеси изотопов калия.
Погрешности рассчитанных термодинамических функций К1(г) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава иодида калия. При более высоких температурах дополнительный
вклад в погрешность вносят неточность принятого способа экстраполяции потенциальной
функции, а также пренебрежение возбужденными электронными состояниями. Погрешности в
значениях Ф°(Г) составляют 0,02, 0,05 и 0,7 Дж-К^-моль-1 при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции К1(г) ранее вычислялись в таблицах JANAF [2834], а также в
работе Брюэра и Брэкет [1660] (Ф°(Г), Т < 2000 К). Расхождения данных [2834] и табл. 1174 не
превышают 1,2 и 3,1 Дж-К-моль в значениях Ф°(Г) и S°(T) соответственно и объясняются
тем, что в справочнике JANAF использовался приближенный метод расчета.
Константа равновесия реакции К1(г)=К(г)+1(г) вычислена с использованием значения
дг#°@)=Б0(К1)=322,068 +4,0 кДж• моль ^ 26 920+330 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации К1(к).
Исследования энергии диссоциации KI методом фотофрагментарной спектроскопии в работах
Ван Вина и др. [4589] и Су и Рейли [5055] привели к значениям 321+1 и 319+2 кДж-моль,
которые согласуются с принятым в пределах погрешностей. К близким значениям привели также
исследования коротковолновой границы флуктуационных полос KI в спектрах поглощения и
испускания, выполненные Зоммермайером [4345] C20 кДж'Моль) и Бойтлером и Леви [1539]
C18+4 кДж-моль). Более высокая и существенно менее точная величина C36+13 кДж-моль)
400
Калин и его соединения К212(г)
была получена Булевич и др. [1719] при спектрофотометрическом изучении равновесия реакции
К(г)+Ш(г)=К1(г)+Н(г) в пламенах A500-2600 К).
Принятым в справочнике значениям энтальпии образования и сублимации К1(к) соответствует
bfH°(Kl, г, 0) = —125,014 ±4,0 кДж-моль.
К212(г). Термодинамические свойства газообразного дииодида дикалия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1175.
Молекулярные постоянные, использованные для расчета термодинамических функций К212,
приведены в табл. 47.7. На основании теоретических расчетов [961, 4689], а также по аналогии
с молекулами Li2X2 (X=F, C1, Вг, I) для молекулы К212 в основном электронном состоянии %}-Ад
принята плоская ромбическая структура (симметрия D2h). Приведенное в табл. 47.7 произведение
моментов инерции соответствует структурным параметрам г(К—I)=3,26 +0,08 А и Д I—К—1 =
=104+10°, найденным Соломоником в результате расчета с использованием ионной модели [961,
963]. Погрешность значения IaIbIc составляет 1-Ю11 г3-см6. Приведенные в табл. 47.7
значения частот v5, v6 найдены Шабером и Мартином [4167] в ИК-спектре молекул К212,
изолированных в матрице из Аг. Для не наблюдавшихся экспериментально частот vt—v4 приняты значения,
рассчитанные Соломоником [961, 963]. Погрешности принятых значений частот v5, v6
оцениваются в 15 см, частот vt— v4 — в 20—25% от их величин.
Термодинамические функции К212(г) вычислялись в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью оценки молекулярных постоянных, а также приближенным характером расчета,
они составляют 10, 15 и 20 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции К212(г) рассчитывались в справочнике JANAF [2834]
и в работе Краснова и др. [569] (до 3000 К). Результаты этих расчетов согласуются с данными
табл. 1175 в пределах 2 и 1 Дж-К^-моль в значениях Ф°(Т) для [2834] и [569].
Константа равновесия реакции Ка12(г)=2К(г)+21(г) вычислена с использованием значения
ДгЯ°@)=807,108+10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
^Н°{К212, г, 0) = — 413 + 10 кДж'.
'Р Эта величина основана на исследованиях равновесия реакции К1(к, ж)+К1(г)=К212(г),
приведенных в табл. 47.23. Приведенные в таблице погрешности учитывают только воспроизводимость
Таблица 47.23. Результаты измерений энтальпии реакции К1(к, ж)-(-К1(г) = К21а(г) (в кДжмоль-1)
Авторы
Метод
*
ДгЯ°@)
II закон
III закон
Миллер, Куш, 1957 [3504] Эффузионный с селектором скоростей, 823 К, — 41,5
1 точка
Берковиц, Чапка, 1958 [1501] Масс^спектрометрический, 830 К, 1 точка — 40,6
Мургулеску, Топор, 1970 [3598] Статический и протока, 1273—1357 К, —86+100 38,4+4,0
9 точек
измерений. Для дальнейших расчетов принимается среднее значение ДгЯ°@)=40+15 кДжХ
Хмоль. Его погрешность обусловлена в основном неточностью термодинамических функций.
Исследования равновесия реакции К212(г)=2К1(г) [1995, 3504, 3598] приводят к значениям
A^°(K2I2, г, 0)=—422+20 кДж-моль (II закон) и — 415+10 кДж-моль (III закон), которые
согласуются с принятым в пределах их погрешностей.
401
K2SO«(k, ж) Глава 47
K2SO4(k, ж). Термодинамические свойства кристаллического и жидкого сульфата дикалия
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1176.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние K2SO4(k) в интервале 0—857 К принята
ромбическая модификация, а в интервале 857—1342 К — гексагональная модификация [3529].
При Т <С 298,15 К термодинамические функции K2SO4(k) вычислены по результатам
измерений теплоемкости в работе Паукова [756] A2,5—299,5 К, образец чистотой —99,9%). Среднее
отклонение экспериментальных точек от сглаженной кривой не превышает 0,7% в интервале
12—20 К и 0,12% — в интервале 50—300 К. Графическая экстраполяция теплоемкости
привела к значению 5°A2 К)=0,3 Дж-К^-моль. Данные Мура и Келли [3551] E2,7—295,4 К,
образец чистотой 99,7%) лежат ниже полученных в работе [756] на 0,2—0,4% во всем интервале
температур вплоть до комнатных, для которых расхождение увеличивается до 0,7—0,9%. При
расчете термодинамических функций K2SO4(k) данные [3551 ] не учитывались. Погрешности
принятых значений 5°B98,15 К) и #°B98,15 К)—Я°@) (см. табл. 47.1) оцениваются в 0,6 Дж-К^Х
Хмоль и 0,08 кДж-моль соответственно.
Уравнение для теплоемкости K2SO4 в интервале 298,15—857 К (см. табл. 47.1) выведено
совместной обработкой результатов измерений теплоемкости, выполненных Шмидт [1153], B98,15—
770 К) и энтальпии, выполненных Рубинчик и др. [887] G50—1345 К), Дворкиным и Бредигом
[2138] D30—980 К), Шомейтом и Нейлором [4273] D60—1700 К) и Дениелоу [2042, 2043, 2044]
D00—1540 К). При обработке погрешность данных [1153] оценивалась в 0,5%, [2042, 2043,
2044] и [887] — в 1%, а [2138] и [4273], имеющих наибольший разброс, — в 1,5%. Результаты
измерений энтальпии в работе Ферланда и Крог-Мо [2306] B98—1123 К) при выводе уравнений
не учитывались, поскольку они приведены только в виде графической зависимости Н(Т)—
—#B98 К) от Т.
Температура полиморфного превращения (857+2 К) принята по согласующимся между собой
данным [530, 890, 1518, 1519, 3354, 3438, 3484, 3557, 3756, 4017, 4736] (термический анализ),
[20, 97] (визуально-политермический метод), [1517, 2262, 3757] (высокотемпературная
рентгенография) и [756, 887, 1397, 2000, 2042, 2043, 2138, 2457, 4273] (калориметрические измерения).
Энтальпия полиморфного превращения (8,0+0,9 кДж-моль) рассчитана из данных по энтальпии
высокотемпературной модификации, полученных Рубинчик и др. [887] с учетом уравнения,
выведенного для энтальпии низкотемпературной модификации сульфата калия. Принятое значение
в пределах погрешности согласуется с приведенными в работах [2138] (8,45 кДж-моль), [2042,
2043, 2044] (8,9 кДж-моль) и [3528] (8,1 кДж-моль).
Уравнение для теплоемкости в интервале 857—1342 К (см. табл. 47.1) получено обработкой
наиболее точных измерений энтальпии высокотемпературной модификации сульфата калия в
работе Рубинчик и др. [887] D1 точка). При этом погрешность измеренных величин [887]
оценивалась в 1%. Принятое уравнение хорошо описывает данные Дениелоу [2042, 2043, 2044], за
исключением области, близкой к Ttr. Результаты измерений энтальпии высокотемпературной
модификации K2SO4 в работах Дворкина и Бредига [2138] и Шомейта и Нейлора [4273] не
учитывались, так как в работе [2138] исследовался .узкий интервал (869—980 К) и соответствующие
данные имели разброс в 5—10%, а данные [4273] выше измерений [887] на ~4%.
Температура плавления A342+2 К) принята по согласующимся между собой данным [17,
18, 20, 21, 22, 81, 83, 84, 373, 374, 530, 887, 1158, 1391, 2042, 2043, 2044, 2454, 2555, 3116,
3241, 3805, 4017, 4076]. Результаты менее точных измерений (см. [1009]) не учитывались.
Энтальпия плавления C6,3+0,8 кДж-моль) вычислена из данных по энтальпии жидкого сульфата
калия [887] с учетом уравнений, принятых для энтальпии K2SO4(k) и величины b-trH°. Принятое
значение согласуется в пределах погрешности с данными Дениелоу [2042, 2043, 2044] C6,8 кДжХ
Хмоль). Результаты измерений [3805] C4,8 кДж-моль) и [3116] C3,9 кДж-моль) не
учитывались. Значение C°(K2SO4, ж)=200+4 Дж-К~1-моль~1 рассчитано из данных по энтальпии
К28О4(ж), полученных Дениелоу [2042, 2043, 2044] A370—1540 К), Шомейтом и Нейлором [4273]
A359-1698 К).
Погрешности вычисленных значений Ф°(Г) при 298,15 К, 1000, 2000 и 3000 К оцениваются
в 0,4, 1,6, 6 и 15 Дж-К-моль~1 соответственно.
Различия в термодинамических функциях, приведенных в табл. 1176 и справочнике JANAF
[2835] (Т < 3000 К), при Т < 1000 К не превышают 0,1 Дж-К^-моль, с повышением
температуры расхождения увеличиваются и при 3000 К достигают 1,5 Дж-К~1-моль~1 в значении
402
Калий и его соединения
TC2SO4(k,
Ф°(Т). Расхождения с данными справочника Барина и Кнаке [1395] (Т ^ 2000 К) составляют
~0,8-И Дж-К^-моль-1.
Принятая в справочнике энтальпия образования
A^KaSO^ к, 298,15 К) = —1437,7 ± 0,5 кДж • моль
основана на результатах измерений энтальпии растворения K2SO4(k) в воде, представленных
в табл. 47.24. Приведенные в таблице величины находятся в хорошем соответствии друг с другом.
Наиболее детальные измерения были выполнены Мищенко и Прониной [686], и принятое для
дальнейших расчетов значение Дйг/7°(соН20; 298,15 К)=23,75+0,05 кДж-моль основывается
на этой работе. Измерения энтальпии растворения в работах [1532, 1681, 1743, 3541, 3836, 3837»
3838, 4206, 4511, 4517, 4590] менее точны или относятся к температурам, сильно отличающимся
от стандартной.
Таблиц а 47.24. Результаты определений Аа .ff°(K2SO4, к->соН2О, 298,15 К) (в кДж • моль)
Авторы
Конечная концентрация, температура
измерений
Аа Я°(соН2О, 298,15 К) \
Коэн, Кой, 1928 [1892]
Мищенко, Пронина, 1936 [686]
Цветков, Рабинович, 1969 [1097]
Грицус и др., 1971 [330]
400 Н2О, 293 К, 4 измерения
83—800 Н2О, 298,15 К, 12 измерений
500 Н2О, 298,15 К
59—3703 Н2О, 298,15 К, 49 измерений
23,680 ±0,040
23,748 ±0,040
23,82
23^88
Расчеты энтальпии образования K2SO4(k) на основании измерений энтальпий ряда реакций
с участием K2SO4(p-p) [1333, 1526, 1527, 2223, 2224, 2622, 3583, 4511] и исследования равновесия
реакций с участием K2SO4(k, ж) [418, 1682, 2247, 2497, 3082, 3116, 3203, 3743, 4262] приводят
к менее точным значениям по сравнению с расчетами, основанными на измерениях энтальпии
растворения.
Давление пара в реакции K2SO4(k, ж)=К28О4(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = As#°(K2SO4, к, 0) = 345 + 15 кДж • моль.
Эта величина основана на результатах измерений давления пара над K2SO4(k, ж),
приведенных в табл. 47.25. Испарение K2SO4 происходит с частичным разложением, и в состав
насыщенного пара помимо K2SO4(r) входят также К, SO2 и О2 (по данным работы [3203] количество K2SO4
в насыщенном паре составляет около 63%). G использованием принятых в настоящем справочнике
Та блица 47.25. Результаты определений A4JEr°(K2SO4, к, 0) (в кДж-моль)
Авторы
Метод
4827°(K2SO4, к, 0)
II закон
III закон
Косуги, 1967 [3080] Эффузионный, 1409—1658 К, 23 точки 353+45 342,8±1,1
-Дюбуа, Милле, 1968 [2120] Протока, газ—носитель N2, 1342—1600 К а 358 316,4+9,2
Фикалора и др., 1968 [2247] Масс-спектрометрический, 1080—1230 К, 351 ±8 —
14 точек
Холстед, 1970 [2555] Эффузионный, 1180—1342 Ка 333 326,6±2,0
Протока, газ-носитель N2 и SO2+O2,1342— 347 322,5±3,0
Ефимова, Горохов, 1978 [418] Масс-спектрометрический, 1171—1331 Ка 367 346,5±3,3
Лау и др., 1979 [3203] Торзионный, 1180—1300 К, 7 точек 374±6 345,2±0,9
Масс-спектрометрический, 1170—1300 К, 366 ±6 — ;¦
19 точек
а Данные представлены в виде уравнения \
403
KaSO4(r), KNO2(k, ж) Глава 47
термодинамических свойств продуктов разложения были вычислены парциальные давления К,
SOa и 02 в насыщенном паре, и в результаты работ, где измерялось общее давление, были внесены
соответствующие поправки. Приведенные в таблице величины относятся к испарению сульфата
калия в виде молекул K2SO4(r). Наиболее надежные измерения были проведены в работах [418,
3080, 3203] и принятая величина является средней из них. Ее погрешность определяется в
первую очередь неточностью термодинамических функций.
|K2SO4(r). Термодинамические свойства газообразного сульфата калия в стандартном состоянии
ири температурах 100—6000 К приведены в табл. 1177.
Молекулярные постоянные K2SO4, принятые для расчета термодинамических функций,
приведены в табл. 47.7. Электронографическое исследование структуры молекулы K2SO4 в паре
fflti ], а также исследование ИК-спектров и спектров КР молекул K2SO4, изолированных в мат-
рщах инертных газов и азота [91, 92, 93, 360, 1356], показывают, что в основном электронном
состоянии XXAX молекула K2SO4 имеет бициклическую равновесную геометрическую
конфигурацию симметрии D2d, т. е. структуру, содержащую тетраэдрический ион SOf- и расположенные
симметрично относительно него два атома К, связанные мостиками с атомами кислорода.
Приведенное в табл. 47.7 произведение моментов инерции соответствует структурным параметрам
|<К—О)=2,45+0,03 A, r(S—О)=1,47±0,01 А, Д О—S—0=109,5+5°, найденным
Спиридоновым и Лутошкиным [971 ] при электронографическом исследовании паров K2SO2. Погрешность
значения IaIbIc составляет 5-1018 г3-см6. Приведенные в табл. 47.7 основные частоты,
отвечающие колебаниям группы SO4 (vx. v5, v6, v8 и v9), найдены в результате исследований ИК-спектра
в спектра КР молекул K2SO4, изолированных в матрице из Аг [360, 1356] г, частоты v2 и v4
рассчитаны по ионной модели в работе [360]. Частоты колебаний связей К—О приняты на основании
результатов исследования ИК-спектра молекул K2SO4, изолированных в аргоновой матрице
К» vioi vu)i и результатов расчета по ионной модели (v3) [91, 92, 93, 360]. Погрешности принятых
частот оцениваются в 10—15 см'1 для v7, v10 и vu, в 15—20 см для v6 и v9, в 20 см для vr, v5
ж v8, в 50 см для VjiB 100 см для v2 и v4. Сведения о возбужденных электронных состояниях
K2SO4 в литературе отсутствуют.
Термодинамические функции K2SO4(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A, 128),
A. 130). Погрешности рассчитанных значений обусловлены приближенным характером расчета
и неточностью принятых молекулярных постоянных и составляют 8, 16 и 25 Дж-К-моль~1
в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции K2SO4(r) вычислялись в справочнике JANAF [2835]
Сйо 6000 К) и работе Гурвича и др. [2534] (до 3000 К). Расхождения данных [2835], [2534] и
табл. 1177 не превышают 1,3 и 0,5 Дж«К'Моль~1 в значениях Ф°(Г).
Константа равновесия реакции K3SO4(r)=2K(r)-f-S(r)+4O(r) вычислена с использованием
значения ДгЯ°@)=2523,882+15 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации K2SO4(k).
Этим величинам также соответствует
^#°(K2SO4, г, 0) = —1082,183 ± 15 кДж • моль.
KNO2(k, ж). Термодинамические свойства кристаллического и жидкого нитрита калия в
стандартном состоянии при температурах 100—2500 К приведены в табл. 1178.
«f'f Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние KNO2(k) в интервале 0—264,1 К
принимается моноклинная модификация (кШ), в интервале 264,1—314,7 К — гексагональная
модификация (к11) и в интервале 314,7—711 К —кубическая модификация (к1) [4341, 4477]2.
При Т ^ 298,15 К термодинамические функции вычислены по данным Мроу и др. [3573],
выполнившим измерения теплоемкости KNO2(k) в интервале 10—476 К с погрешностью 0,1% от 86
до 210 К и 0,3% от 330 до 476 К. Исследуемый образец подвергался тщательной очистке, однако
* Для vx приведено значение, полученное в спектре КР молекул K2SO4, изолированных в матрице из N2 [1358].
В аргоновой матрице частота vt не наблюдалась.
*• При высоких давлениях у KNO2 стабильны еще три модификации: ромбическая (kIV), кубическая, структурный
CsCl (kV) и тетрагональная (kVI) [1009].
404
Калий и его соединения KNO2(k, ж)
данные анализа не приведены. Экстраполяция теплоемкости ниже 10 К по уравнению Дебая
дает S°A0 К)=0,28 Дж-К^-моль. Принятые значения ?°B98,15 К) и Я°B98,15 К)-Я°@),
приведенные в табл. 47.1, имеют погрешности 0,8 Дж-К^-моль и 0,1 кДж-моль
соответственно.
При Т > 298,15 К расчет термодинамических функций KNO2(k) также основан на результатах
измерений теплоемкости в работе [3573]. Теплоемкость KNO2(k) в интервале 298,15—711 К была
аппроксимирована двумя уравнениями: первое для kII B98,15—314,7 К) описывает резкий рост
теплоемкости, а второе для к1 — ее медленный рост выше 314,7 К.
Температуры фазовых превращений B64,1+0,5 К) и C14,7+0,5 К) приняты по работе Мроу
и др. [3573]. Менее точные значения были получены в работах [133, 827, 828, 1863, 3850, 3948,
4341] B53-271 К) и [133, 134, 209, 822, 824, 827, 828, 834, 837, 1253, 1871, 2598, 2983, 3776,
3850, 3928, 3947, 4341] C12-338 К) соответственно.
Температура плавления G11 +2 К) принята по согласующимся между собой данным [133,
134, 209, 822, 827, 828, 834]; значения, полученные в работах [824, 832, 837, 1409, 2088, 2598,
3232, 4177], лежат в пределах 692—714 К х. Энтальпия полиморфного превращения -KNO2(k
III) -> KNO2(kII) измерена в работах [3573] E,05 кДж-моль) и [3948] E,02 нДж-моль);
в старой работе [1671] получена заниженная величина (—4 кДж-моль), что, по-видимому,
связано с плохим качеством образца. Принятая энтальпия фазового превращения KNO2(kII) -*¦
KNO2(kI) @,75 + 0,08 кДж-моль) представляет собой изотермический вклад, выделенный
сравнением величин Я°D20 К)-#°B98,15 К)=11,92 нДж-моль [3573] и #°D20 К)-#°B98,15 К)**
=11,17 кДж-моль (рассчитано пр принятым аппроксимационным уравнениям). Суммарная
энтальпия фазового превращения, включающая также вклад, полученный интегрированием
аномальной части кривой теплоемкости, по данным [3573] составляет 2,11 кДж-моль~х. В
работах [209, 1253, 3928, 3936], выполненных менее точными методами, получены значения 0,50,
0,83, 1,67, 2,09 кДж-моль соответственно. Энтальпия плавления A6,7+2 кДж-моль'1)
принята по данным Верхотурова и др. [209], полученным термографическим методом.
Теплоемкость 1Ш02(ж) A02+3 Дж-К~1-моль~1) принята на основании измерений энтальпии
жидкой соли, выполненных Бартоломью в интервале 701—780 К [1409].
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 оцениваются в 0,5, 3!и
12 Дж-К^-моль. ;
Таблица термодинамических функций KNO2(k, ж) публикуется впервые.
Принятая в справочнике энтальпия образования ?_'*"<•
,15 K) = —365,9 ± 1,0 кДж.моль
основана на результатах измерений энтальпии растворения KNO2(k) в воде,, приведенных
в табл. 47.26. Пересчет к стандартной температуре проводился с использованием значения ДС° =
=—39,3 Дж-К^-моль" для реакции KN02(K)+ooH20=K+(ag)+NO^(ag). Результаты
измерений оказались в хорошем соответствии и дальнейшие расчеты основаны на величине Дя„#°(оэНаО;
298,15 К)=13,45+0,40 кДж-моль, которой соответствует принятое значение энтальпии обрай-
зования KNO2(k). Основным источником погрешности принятой величины является неточность
энтальпии образования NO^ag) (см. Приложение 6).
р ^g) ( )
Таблица 47.26. Результаты определений AajB'o(KN02, к-»ооН2О, 298,15К) (в кДж• моль')
Авторы
Конечная концентрация, температура
измерений
ДагЯ°(юН20, 298,15 К)
Доде, 1937 [2089] 400 НаО, 285 К, 5 измерений 13,46+0,50
Бюро, 1937 [1724] 400 Н2О, 293 К, 1 измерение 13,61
Решетников, 1961 [864] 330 Н2О, 298,15 К, 3 измерения 13,41 ±0,40
Андреева и др., 1979 [35] 1,5—111 Н2О, 298,15 К, 40 измерений ' 13,38+0,20
1 Значительный разброс экспериментальных данных обусловлен, по-видимому (см. [134]), способностью солж
к образованию твердых растворов со многими веществами (в силу повышенной пластичности), что затруднжв
ее очистку.
405
KNO2(r) Глава 47
Доде [2088, 2089] измерил энтальпию реакции нейтрализации КОН (p-p)+HNO2(p-p) =
=KNO2(p-p)-j-H2O(p-p), Дг#°=—45,0 кДж-моль (температура измерений и концентрации
не указаны), которой соответствует величина A^T°(KNO2, к, 298,15 К) =—365,8 кДж-моль",
подтверждающая принятое значение.
Таблица 47.27. Результаты определений AyJ3"°(KNO2, к, 298,15К) на основании исследований
равновесия реакции KNO3(jk) = КРТО2(ж) -f- 1/2О2(г) (в кДж • моль)
Авторы
: Цетнершвер, 1930 [1773]
.(см. также [3123])
:Фриман, 1956 [2329]
1 Сироткин, 1959 [938]
^Бартоломью, 1966 [1409]
Метод
Компенсационный, 753—811 К, 5 точек
Статический, 923—1023 К, 6 точек
Определение отношения KNO3/KNO2
в расплавах, 873—973 К, 3 точки
Статический и определение отношения
KNO8/KNO2 в расплавах, 921—1023 К,
11 точек
Д/Я°(КМО
II закон
—450
-335
—370
-367 ±20
2, к, 298,15 К)
III закон
—404
—373,6+3,7
-375,6+3,3
—370,4 ±3,2
В табл. 47.27 представлены результаты исследований равновесия реакции 1Ш03(ж) =
^KNOj^+VsjO^r). Приведенные в таблице погрешности включают воспроизводимость
измерений, неточность термодинамических функций и энтальпии образования KNO3(k). Результаты
этих работ приводят к более отрицательным значениям.
Давление пара»в реакции KNO2(k, ж)=КЖJ(г) вычислено с использованием принятой
s справочнике энтальпии сублимации
ДгЯ°@) = \H°(KNOa, к, 0) = 181 ± 5 кДж • моль.
Эта величина основана на масс-спектрометрическом исследовании, выполненном Верхотуро-
вым и др. [205, 206]. Опыты с двойной эффузионной камерой показали, что основным
компонентом пара является мономер (количество димера составляет 1,8%). При 675 К методом полного
изотермического испарения было определено парциальное давление мономера, что позволило
вычислить по уравнению III закона принятое в справочнике значение. Его погрешность
обусловлена в основном неточностью термодинамических функций KNO2(r) и воспроизводимостью
измерений. Измерения интенсивностей ионных токов, выполненные в интервале 607—675 К, приводят
к значению 183 кДж-моль (II закон).
KNO2(r). Термодинамические свойства газообразного нитрита калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1179.
Молекулярные постоянные, использованные для расчета термодинамических функций KNO2,
приведены в табл. 47.7. Структурные исследования KNO2 не проводились. Результаты
исследования ИК-спектра молекул KNO2, изолированных в матрице из Аг, выполненного Беляевой
ндр. [88], показали, что в основном электронном состоянии Х1А1 молекула KNO2 имеет плоское
циклическое строение (группа симметрии С2„). Приведенное в табл. 47.7 произведение моментов
инерции вычислено для структурных параметров r(K—O)=2,5+0,l A, r(N—O)=l,25+0,05 A
¦ ДО—N—0=115+5°. Величина г(К—О) оценена по экспериментальным значениям г(М—О)
в молекулах RbNO2 и CsNO2 с учетом изменения величины г(М—О) в соединениях МОН, МВ02,
MN03, МРО3, M2Cr04, M2SO4, М2МоО4 и M2WO4, где М — щелочной металл. Остальные
структурные параметры KNO2 оценены по экспериментальным данным для RbNO2 и CsNO2. Погрешность
значения IaIbIc составляет 1-Ю14 г3-смв. Для частот v3(^x) и v4(.81) приняты значения, найден-
иые Беляевой и др. [88| и Миллиганом и Джекокс [3508] в ИК-спектре молекул KNO2 в Аг-матрице.
Частота v8Ex) оценена на основании сравнения экспериментального значения v6 у молекулы
NaNO2 и соответствующих частот сульфатов щелочных металлов. Частота неплоского колебания
цикла ve(B2) принята равной соответствующей частоте K2SO4 (vn), частота валентного колебания
N—0(v1D1)) — равной аналогичной частоте свободного иона NO^. Беляева и др. [88] наблюдали
406
Калий и его соединения КРЮа(к, ж)
в спектре KNO2, изолированного в Ar-матрице, частоту va(^41)=805 см. Однако в связи с тем,
что частоты v4 нитритов щелочных металлов в матрице из Аг на 5—20 см выше, чем в паре, в
настоящем справочнике для v2 принято значение этой частоты, наблюдавшейся в спектре
газообразного CsNO2 (см. ниже). Погрешности принятых значений v3, v4 и ve оцениваются в 15—20 см,
частот vx, v2 и v6 — в 30—40 см. Сведения о возбужденных электронных состояниях KNO2 в
литературе отсутствуют.
Термодинамические функции KNO2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор^-гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены приближенным характером расчета и неточностью принятых значений
молекулярных постоянных, они составляют 4. 8 и 12 Дж-К~1-моль~1 в значениях Ф°(Т) при 298,15, 3000
и 6000 К (в том числе 2—3 Дж-К~1-моль~1 из-за неточности молекулярных постоянных).
Таблица термодинамических функций KNO3(r) публикуется впервые.
Константа равновесия реакции KNO2(r) =K(r)+N(r)+2O(r) вычислена с использованием
значения Дг#°@)=1242,011 +5,1 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации KNO2(k).
Этим величинам также соответствует
г, 0) = —187,736 ±5,1 кДж • моль.
KNO3(k, ж). Термодинамические свойства кристаллического и жидкого нитрата калия в
стандартном состоянии при температурах 100—2500 К приведены в табл. 1180.
Значения постоянных, использованные для расчета термодинамических функций,
представлены в табл. 47.1. В справочнике за стандартное состояние KNO3(k) в интервале 0—402 К
принимается ромбическая а-модификация, а в интервале 402—607,7 К — гексагональная
^-модификация [3662] г> 2.
При Т <[ 298,15 К термодинамические функции KNO3(k) вычислены по результатам
измерений теплоемкости в работе Саузарда и Нелсона [4350 ] A6—293 К) с учетом данных Шмидт
и Максимова [1154] B94—641 К). В целях согласования результатов низкотемпературных и
высокотемпературных измерений теплоемкости была проведена интерполяция между данными
[4350] и [1154], в результате которой получено значение С°B98,15 К)=95,06 Дж-К^-моль1
Экстраполяция теплоемкости ниже 16 К по уравнению Дебая приводит к величине ?°A6 К) =
0,98 Дж-К^-моль. Принятые значения 5°B98,15 К) и #"B98,15 К)—#°@), приведенные
в табл. 47.1, имеют погрешности 0,4 Дж-К~1-моль~1 и 0,06 кДж-моль соответственно.
При Т > 298,15 К расчет термодинамических функций основан на результатах измерений
теплоемкости в прецизионной работе [1154] B94—641 К; погрешности измерений 0,5%).
Теплоемкость KNO3(k) в интервале 298,15—607,7 К аппроксимирована двумя уравнениями,
выведенными по данным [1154] с использованием следующих величин: для a-KNO3 С°B98,15 К)=95,06,
С;C50 К) = 102,68 и СрC90 К) = 109,37 Дж-К^-моль, и для P-KNO3 С; D10 К)=124,8
С;D50 К) = 123,1, С;E00 К)=126,6 и С;E80 К)=137,8 Дж-К^-моль-1. С выбранными значениями
теплоемкости в пределах 1—1,5% согласуются данные Миекк-Ойи [3500] C21—573 К) и
Соколова и Шмидт [960] C05—667 К). Кларк [1865], измеривший энтальпию в интервале 512—663 К,
принял для теплоемкости KNO3 постоянную величину A31,8+5,3 Дж-К^-моль). В работе
Гэнта [2360] измерена энтальпия KNO3 в интервале 353—698 К и для ^-модификации также
принято постоянное значение A25 Дж-К~1-моль). Энтальпию KNO3(k) измерили также Нгуен-
Дюи и Даней [3652] E00—675 К); расхождение этих данных и полученных в работе [1154]
составляет —1%.
Температура фазового превращения D02,0 + 0,5 К) принята по согласующимся измерениям
11154, 2163, 2366, 3500, 3606, 4462, 4680]. Энтальпия фазового превращения E,04+0,08 кДжХ
Хмоль) принята по работе [1154]. С принятым значением в пределах погрешностей согласуются
1 При охлаждении ниже 402 К гексагональная {3-модификация не переходит непосредственно в а-ромбическую,
я переохлаждается и при 397 К претерпевает энантиотропное превращение (Д/г?Г°=2,8+0,2 кДж-моль)
в ферроэлектрическую у-фазу. При дальнейшем охлаждении у-фазы при 385 К наблюдается ее монотропяый
переход в ромбическую a-фазу ,(Д;г#°=2,34+0,20 кДж-моль).
2 При высоких давлениях у KNO3 стабильны еще пять модификаций: кШ, гекс, kIV, кУ, kVI и kVII [1009].
407
KNO,(k, ж) Глава 47
результаты определений в работах [95, 960, 1338, 2053, 2360, 2457] E,1, 5,08, 4,9, 4,98, 4,98,
5,0 кДж-моль соответственно). Величины энтальпии фазового превращения, приведенные
в [1103, 1189, 3376, 3500, 3606, 3741, 3742, 3983, 4648], больше принятого значения на 0,4—
0,7 кДж-моль и, по-видимому, представляют собой сумму энтальпии изотермического
превращения и величины, полученной интегрированием аномальной части кривой теплоемкости.
Температура F07,7+0,2 К) и энтальпия плавления (9,80+0,08 кДж-моль) приняты по
результатам измерений Шмидт и др. [960, 1154] (образец с содержанием примесей не более 0,01 %).
Менее точные данные по температуре плавления получены авторами [112, 1189, 1409, 1579, 1865,
2005, 2098, 2163, 2330, 2679, 3376, 3742, 4183, 4303] F06-610 К). С принятым значением АтН°
согласуются определения авторов [960, 1189, 1865, 2098, 2163, 3373, 3375, 3376, 3983] (9,6—
10,0 кДж-моль).
Теплоемкость 1Ш0з(ж) A41+2 Дж-К-моль) принята по измерениям [1154] F12—641 К,
5 измерений), с которыми согласуются данные [960] A40 Дж-К~1-моль~1, 611—667 К, 11
измерений) и [2442] A41 Дж-К-моль~1). Более низкие величины получены в работах [1865]
A36 Дж-К^-моль-1, 621—663 К) и [2360] A37 Дж-К^-моль, 630—698 К); к явно заниженным
результатам привели измерения [1409] A23 Дж-К«моль~1). Значение теплоемкости,
полученное в работе [3652] B33 Дж-К'^моль, 611—675 К), представляется ошибочным. По данным
[2065] C°(KNO3, ж, 608 К)=142 Дж-К-моль~1, но с повышением температуры теплоемкость
жидкого KNO3 заметно падает.
Погрешность вычисленных значений Ф°(Т) при 298,15, 1000 и 2000 К оцениваются в 0,2, 2
и 10 Дж«К~1-моль. Расхождения между термодинамическими функциями KNO3(k, ж),
приведенными в табл. 1180 и в справочниках [1395] (Т < 700 К) и [2960] (Т < 700 К), составляют
0,03 и 0,6 Дж-К-моль~1 в значениях Ф°(Г) и обусловлены тем, что в настоящем издании учтены
экспериментальные данные [1154] по теплоемкости KNO3(k, ж).
В справочнике принята энтальпия образования
bfH°(KNOs, к, 298,15 К) = —494,0 ±0,5 кДж.моль,
основанная на приведенных в табл. 47.28 наиболее точных измерениях энтальпии растворения
KNO3(k) в воде. Принятая величина Aas#°(ooHaO; 298,15 К)=34,97+0,15 кДж-моль
является средней из этих измерений. Менее точные измерения энтальпии растворения KNO3(k) в воде
Таблица 47.28. Результаты измерений b.aqH°(KNO3, к-»ооН2О, 298,15К) (в кДж.моль)
Авторы
Беренгер-Кальве, 1927 [1480] (см. [1555])
Рот, Эйман, 1943 [4059, 4066]
Коэн, Кой, 1928 [1892] (ом. [1555])
Ланге, Монхейм, 1930 [3181]
Хюккель и др., 1940 [2744]
Воскресенская, Пономарева, 1946 [251,
252]
Веотрум, Эйринг, 1952 [4712]
Билле, Коттон, 1960 [1561]
Мищенко, Шпигель, 1967 [688]
Цветков, Рабинович, 1969 [1097]
Таневска-Осинска, Логвиненко, 1974
[4470]
Конечная концентрация, температура
измерений
23—617 Н2О, 287—289 К, 30 измерений
350 Н2О, 294 К, 4 измерения
200—350 Н2О, 293,4 К, 6 измерений
800—1111 Н2О, 298,15 К, 5 измерений
460 Н2О, 291,7 К, 3 измерения
300 Н2О, 298,15 К, 1 измерение
5Е60 НаО, 298,15 К, 4 измерения
400 Н2О, 298,15 К, 3 измерения
111—600 Н2О, 298,15 К, 9 измерений
500 Н2О, 298,15 К, 1 измерение
111—1750 НаО, 298,15 К, 6 измерений
Д^Я°(ооН20, 298,15 К)
34,96+0,17
35,04
35,03+0,08
35,05+0,17
35,21
34,66+0,17
34,83+0,08
35,14+0,17
35,08+0,17
34,93
34,76 ±0,17
были проведены в ряде работ. В некоторых из этих работ измерения проводились не при
стандартной температуре, при высоких концентрациях конечного раствора или не сообщаются
данные, необходимые для оценки их надежности. Значительная их часть проведена с недостаточно
хорошо охарактеризованными образцами и на примитивной калориметрической аппаратуре
(см. также обзорные работы [1555, 3771]).
408
Калий и его соединения KNO3(r)
Измерения энтальпии нейтрализации растворов КОН и HNOS [1331, 1332, 1333, 1526, 2222,
2224, 2453, 2623, 3994] приводят к значению A^°(KNO8, к, 298,15 К) = —493,9 кДж-моль,
которое совпадает с вычисленным по измерениям энтальпии растворения, но уступает ему по
точности.
Давление пара в реакции KNO3(k, ж)=К]>Ю3(г) вычислено с использованием принятой
в справочнике энтальпии сублимации
ArH°@) = A,H°(KNOs, к, 0) = 181 +5 кДж • моль.
Принятая величина основана на масс-спектрометрическом исследовании, выполненном
Багаратьян и др. [56, 57]. Опыты с двойной эффузионной ячейкой показали, что основным
компонентом пара является мономер (количество димера при 680 К составляет 3%). При 649,2 К
методом изотермического испарения было найдено парциальное давление мономера, что позволило
вычислить по уравнению III закона принятое в справочнике значение. Его погрешность
обусловлена в основном неточностью термодинамических функций KNO3(r). Измерения интенсивно-
стей ионных токов в интервале 649—688 К, приводят к значению 208 кДж-моль (II закон).
В работе этих же авторов [4851] методом электронного удара была найдена энергия связи
D0(K—NO3)=438 + 13 кДж-моль, которой соответствует значение ^#"@)=182+25 кДжХ
Хмоль, совпадающее в пределах погрешности с принятым.
KNO3(r). Термодинамические свойства газообразного нитрата калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1181.
Молекулярные постоянные, использованные для расчета термодинамических функций KNO3,
приведены в табл. 47.7. На основании исследования ИК-спектра молекул KNO3, изолированных
в матрице из Аг, выполненного Битти и др. [1444], в справочнике принимается, что в основном
электронном состоянии ХгАх молекула KNO3 имеет циклическую бидентатную структуру
(симметрия С2е). Приведенное в табл. 47.7 произведение моментов инерции вычислено по
структурным параметрам r(N-Oa) = 1,29+0,04 A, |V(N-OBHem)=l,21+0,04 А, ДОц-К-Оц=115+10°
и г(К—Ов)=2,5+0,1 А. Структурные параметры группы NO3 приняты такими же, как в
молекулах LiNO3, RbNO3 и CsNO3. Значение г(К—Оц) оценено по величине г(М—О) в молекулах
LiNO3, RbNO3 и CsNO3 с учетом ее изменения в рядах МОН и МВО2 (М — щелочной металл).
Погрешность значений IaIbIc может достигать 4-10~114 г3>см6. Приведенные в табл. 47.7 частоты
валентных (vx, v2, v5) и деформационных (ve, v8) колебаний иона NOg найдены Битти и др. [1444].
С принятыми значениями удовлетворительно согласуются результаты исследований ИК-спектра
KNO3 в различных матрицах [3864, 4319, 4333] х. Для деформационного колебания v3 иона N03
принято то же значение, что для частоты v6, поскольку значения v3 и ve в молекуле LiNO3 близки.
Частоты v4, v7 и v9, характеризующие колебания иона К+ относительно иона NOg, приняты
равными соответствующим частотам KN02 (см. текст по KNO2). Погрешности в значениях vx,
V2> V5. V6) V8 и V9 оцениваются в 15—20 см, частот v3, v4 и v7 — в 30—40 см. Сведения о
возбужденных электронных состояниях KNO3 в литературе отсутствуют.
Термодинамические функции KN03(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены приближенным характером расчета и неточностью принятых значений
молекулярных постоянных, они составляют 6, 12 и 18 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000
и 6000 К.
Таблица термодинамических функций KNO3(r) публикуется впервые.
Константа равновесия реакции KNO3(r)=K(r)+N(r)+3O(r) вычислена с использованием
значения ДД°@)=1608,363+5,0 нДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации KNO3(k).
Этим величинам также соответствует
AjH°{KNOa, г, 0) = —307,305 + 5,0 кДж • моль.
Близкие значения дляШОз наблюдались в спектрах кристаллического и расплавленного KNOS и в спектрах
его растворов [706, 803, 1685, 1717, 2052, 2059, 2100, 2240, 2321, 2476, 2628, 2841, 3625, 4638].
409
К2СО3(к, ж) Глава 47
К2СО3(к, ж). Термодинамические свойства кристаллического и жидкого карбоната дикалия
в стандартном состоянии при температурах 100—3000 К приведены в табл. 1182.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 47.1. За стандартное состояние К2СО3 в интервале 0—693 К принята моноклинная (а)
модификация1, а в интервале 693—1173 К — гексагональная (р) модификация [4268].
При Т <С 298,15 К термодинамические функции К2СО3(к) вычислены по измерениям
теплоемкости, выполненным Сталлом и др. [4420] A5—298,15 К, образец чистотой 99,9%).
Отклонение экспериментальных точек от сглаженной кривой составляет ~0,5% в интервале 100—
298,15 К, 1% в интервале 50—100 К и —10% при 15 К. Экстраполяция теплоемкости по закону
Дебая приводит к значению S°AQ K)=0,65 Дж-К-моль~1. Погрешности принятых значений
5го B98, 15 К) и Я°B98,15 К)—Я°@) (см. табл. 47.1) оцениваются в 0,8 Дж-К^-моль и
0,1 кДж-моль соответственно.
При Т > 298,15 К энтальпия К2СО3(к, ж) измерялась Гинзбургом [279, 280] B98—1273 К),
Янцем и др. [2845] E48—1248 К) и Ролиным, Рекапе [4035] G53—1306 К). Данные, полученные
в работе [2845] в области 970—1170 К, лежат ниже результатов измерений Ролина и Рекапе
[4035] на 2—2,5%, при Т <^ 970 К расхождения резко возрастают. В интервале 298,15—693 К
для теплоемкости К2СО3(к) принято уравнение, полученное Гинзбургом [279, 280] для
температур 298,15—523 К. Температура полиморфного превращения F93+10 К) принята по
результатам наиболее надежных измерений, проведенных Шнейдером и Левиным [4268] (чистота образца
выше 99%). Принятое значение согласуется в пределах погрешности с данными [280, 646, 2824,
3763] 2. Менее надежные определения в работах [1306] F78 К) и [1777] G12 К) не учитывались.
Энтальпия полиморфного превращения B,1+0,3 кДж-моль) рассчитана из уравнений,
принятых для а- и р-модификаций, она хорошо согласуется с суммарной энтальпией полиморфного
превращения, найденной Гинзбургом [279, 280] B,16 кДж'.моль).
Уравнение для теплоемкости Р-К2СО3 (см. табл. 47.1) получено усреднением данных Янца
и др. [2845], Ролина и Рекапе [4035] и Гинзбурга [279, 280] по энтальпии.
Температура плавления A173+2 К) принята как среднее значение между согласующимися
результатами калориметрических [280, 2845, 4035] и термографических исследований [646, 1391,
2843, 3498, 3968]. Результаты менее точных работ [78, 229, 850, 1306] A164—1169 К) не
учитывались. Энтальпия плавления C0,5+3,0 кДж-моль) вычислена из уравнений, принятых для
изменения энтальпии р-модификации К2СО3(к) и К2С03(ж) и согласуется в пределах погрешности
с данными [2845] B7,6 кДж-моль) и [4035] B7,2 кДж-моль).
Теплоемкость К2СО3(ж) B05+10 Дж-К-моль~1) принята по измерениям энтальпии в
работе [4035] A178—1306 К) и хорошо согласуется с рекомендацией [2845] B08,7 Дж-К-моль~1).
Данные [279, 280] приводят к более низкому значению A81 Дж-К^-моль).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,5,
3, 10 и 25 Дж-К-моль~1 соответственно.
Термодинамические функции, приведенные в справочнике JANAF [2834] (Т ^ 2500 К), при
Т ^ 500 К согласуются с данными табл. 1182; при повышении температуры расхождения
увеличиваются до 4 Дж-К~1-моль~1 в значениях Ф°B500 К).
Принятая в справочнике энтальпия образования
Д/Я°(КаСО3, к, 298,15 К) = —1151,5 ± 2,0 кДж • моль
основана на значении энтальпии растворения К2СО3(к) в воде, найденном Бенджамином [1471].
В результаты этой работы (разведение 1 моль К2СО3 на 1750, 5000 и 7000 Н2О) были внесены
поправки, учитывающие гидролиз (см. [4099]). Полученные величины экстраполированы к
бесконечному разведению, что дало для реакции К2С03(к)+соН20(ж)=2К+(а9г)+СОз2(ад') значение
^«5-^c(c°H2O; 298,15 К) =—28,0+2,0 кДж-моль, которому соответствует принятая энтальпия
образования К2СО3(к).
Измерения энтальпии растворения К2СО3(к) в воде при более высоких конечных
концентрациях [1530, 1532, 2723, 4511], так же как измерения энтальпии ряда реакций с образованием
1 По данным Папена [37631 моноклинная модификация при 613 К претерпевает фазовый переход
порядок—беспорядок. Этот переход не был обнаружен при измерениях энтальпии и поэтому не учитывался в справочнике
при расчете термодинамических функций.
2 Существование полиморфных превращений К2СО3(к) при 523 и 895 К, отмеченное в работах [280, 646],
опровергнуто термографическими и рентгенографическими исследованиями [1777, 3763, 3968, 4268].
410
Калий и его соединения К2СОа(г)
растворов К2СО3 [1526, 3582], приводят к менее точным величинам (см. [1555]). Исследования
равновесий с участием К2СО3 [1106, 2120, 2497, 2723, 2961, 3082, 3116, 3214, 4297] не позволяют
получить надежных значений энтальпии образования К2СО3 вследствие сложного состава
продуктов реакции и отсутствия данных по коэффициентам активности их компонентов.
Давление пара карбоната калия в реакции К2СО3(к, ж)=К2СО3(г) вычислено с использованием
принятой в справочнике энтальпии сублимации
ДгЯ°@) = Д8#°(К2СО3, к, 0) = 343 + 20 кДж • моль.
В табл. 47.29 приведены результаты расчетов энтальпии сублимации К2СО3(к) на основании
измерений давления насыщенного пара над К2СО3(к, ж). Приведенные в таблице погрешности
учитывают только воспроизводимость измерений.
Таблица 47.29. Результаты определений Ав?Го(К2СО3, к, 0) (в кДж • моль)
Авторы
Крёгер, Штратманн, 1961 [3116]
Дюбуа, Милле, 1968 [2120]
•
Симмонс и др., 1977 [4297]
Метод
Эффузионный, 1072—1208 К, 8 точек
Протока, газ-носитель СО2, 1250—1550 К,
данные представлены в виде уравнения
Масс-спектрометрический, 1037—1170 К,
,13 точек
Д8Я°(К2
II закон
209 ±14
338
351 ±17
СО3, к, 0)
III закон
311,3 ±3,5
287,3 ±6,0
342,8±0,5
Принятая величина основана на данных работы [4297], где расчет парциальных давлений
производился по интенсивности ионного тока K2COJ. Предполагалось, что ионы К2О+ образуются
лз молекул К2О. Однако достаточно высокий потенциал появления этого иона G24+20 кДжХ
Хмель') и низкая равцовесная концентрация К2О(г) в насыщенных парах дают основание
связать его образование с диссоциативной ионизацией К2СО3, поэтому приведенная в табл. 47.29
величина была пересчитана в предположении, что ионы K2GOJ и К2О+ образуются из К2СО3.
Ее погрешность оценена с учетом неточности термодинамических функций и сечений ионизации.
Величина, вычисленная по данным [3116], оказалась несколько заниженной, вероятно,
вследствие конденсации на мишени не только молекул К2СО3, но и входящих в состав насыщенного
пара продуктов диссоциации (К, СО2, О2). В работе [2120] получено дакже заниженное значение,
как и в некоторых других работах этих же авторов (см. текст по энтальпии сублимации K2SO4).
К2СО3(г). Термодинамические свойства газообразного карбоната дикалия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 1183.
Молекулярные постоянные К2СО3, принятые для расчета термодинамических функций,
приведены в табл. 47.7. Экспериментальные данные о строении и спектрах газообразного К2СО3 в
литературе отсутствуют. На основании сравнения с газообразными молекулами сульфатов, нитратов
л нитритов щелочных металлов в справочнике принимается, что молекула К2СО3 в основном
электронном состоянии XrAx имеет плоскую структуру (симметрия C2v), причем группа СО3 имеет
конфигурацию симметрии DSh, а один атом металла связан Костиковыми связями с двумя атомами
кислорода, образуя цикл, тогда как другой соединен с третьим атомом кислорода. Приведенное
в табл. 47.7 произведение моментов инерции соответствует структурным параметрам г(К—О) =
=2,45+0,1 -А, г(С—О)=1,30+0,05 А и ДО—С—О=120±10°. Близость расстояний металл-
кислород в солях различных кислородсодержащих кислот позволяет принять расстояние К—О
таким же, как в молекуле K2SO4. Для группы СО3 приняты параметры, отвечающие средним
значениям соответствующих величин в кристаллическом К2СО3 [2372, 2757]. Этот выбор основан
на том, что для молекул сульфатов и нитратов щелочных металлов структурные параметры групп
S04 и NO3 в газе и кристалле близки. Погрешность вычисленного значения I&IbIg оценивается
•в 5-Ю"13 г3-см8. Приведенные в табл. 47.7 частоты vx, v2, v3, ve, v10 и vu, отвечающие колебаниям
группы СО3, приняты близкими соответствующим значениям частот свободного иона COj2 [1447,
2807, 4010] и кристаллических карбонатов щелочных металлов [1157, 1447, 1716, 4218]. Частоты
колебаний связей К—О оценены на основании сравнения с частотами колебаний связей металл—-
411
K2Si03(K, ж) Глава 47
кислород, принятыми для молекул K2SO4, KNO3, KNO2 и КВО2. Погрешности принятых
значений v1? v2, ve и v10 оцениваются в 100—150 см, частот v3, v4 и vix — в 75 см, частот v6, v7, vs
и v9 — в 50 см, частоты v12 — в 20—25 см.
Термодинамические функции К2СО3(г) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130),
без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью молекулярных постоянных и приближенным характером расчета, они могут достигать 10,
20 и 30 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций К2СО3(г) публикуется впервые.
Константа равновесия реакции K2CO3(r)=2K(r)-j-C(r)-f-3O(r) вычислена с использованием
значения АгН°@)=2434,232 + 20 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации К2СО3(к).
Этим величинам соответствует также
kfH°(K2CO3, г, 0) =—802,916+ 20 кДж.моль.
K2Si03(K, ж). Термодинамические свойства кристаллического и жидкого силиката дикалия
в стандартном состоянии при температурах 100—4000 К приведены в табл. 1184.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние K2SKK(k) принимается ромбическая
модификация, устойчивая в интервале 0—1249 К. .
При Т <J 298,15 К термодинамические функции вычислены по данным Сталла и др. [4420],
которые исследовали теплоемкость K2Si03(K) A5—298 К, погрешность измерений ~3% ниже
20 К, 1,0—0,5% в интервале 20—80 К и 0,3% при Т >»80 К). Чистота исследованного образца
вызывает сомнения, ввиду чего принятые по [4420] значения 5°B98,15 К) и #°B98,15 К)—Н°@)
(см. табл. 47.1) могут иметь погрешности 0,8 Дж-К-моль и 0,1 кДж-моль соответственно х.
При Т >¦ 298,15 К для теплоемкости принято уравнение (см. табл. 47.1), основанное на
результатах измерений энтальпии в работе Бейера и др. [1545] D03—1195 К, образец чистотой
99,94%, погрешность измерений 0,3%) 2. Температура плавления A249+2 К) принята по
согласующимся между собой измерениям Мори и Боуэна [3558] и Крачека и др. [3094, 3097]. В
литературе отсутствуют измерения энтальпии К2ЭЮ3(ж), что затрудняет выбор величины ДШЯ°. Оценка
этой величины, проведенная авторами справочника JANAF [2835] сравнением данных для
других силикатов, привела, по-видимому, к резко завышенному значению 50+12 кДж-моль.
Выполненные Такахаши и Йошио [4460] измерения энтальпий растворения кристаллического
и стеклообразного K2SiO3 в растворах плавиковой кислоты дали для энтальпии перехода стекло—
кристалл при 298,15 К значение 9,2 кДж-моль; пересчет этой величины к температуре
плавления с использованием данных [2835] дает значение энтальпии плавления 11 кДж-моль. Эта
величина, возможно, занижена вследствие частичной кристаллизации стеклообразного K2SiQ3.
Аналогичные данные для K2Si205 [44611 показывают, что ошибка может достигать 10 кДж-моль,
ввиду чего в справочнике принимается энтальпия плавления K2SiO3 равная 20 + 10 кДж-моль.
Теплоемкость жидкого K2SiO3 A90+20 Дж-К-моль) оценена" как округленная разность
значений теплоемкости К2812О5(ж) B75 Дж-К~1-моль~1) и ЭЮ2(ж) (83,5 Дж-К-моль).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 4000 К оцениваются в 0,6,
1, 7 и 26 Дж-К-моль~1 соответственно. Расхождения между термодинамическими функциями
K2Si03(K, ж), приведенными в справочнике JANAF [2835] (Т ^ 3000 К) и табл. 1184, растут
с температурой и составляют 4 и 15 Дж-К-моль~1 в значениях Ф°(Г) при 1000 и 3000 К
соответственно, они обусловлены тем, что в настоящем издании использованы данные [1545] и
отличающиеся значения Am#°(K2Si03) и C°(K2Si03, ж).
Константа равновесия реакции K2Si03(k, ж)=2К(г)+81(г)+ЗО(г) вычислена с использованием
значения Дг#°@)=2900,242+ 13 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
&.fH°(K2Si03, к, 298,15 К) = —1543 + 10 кДж • моль.
1 Для согласования данных [4420] с результатами измерений энтальпий Н°(Т)—Н° B98,15 К) [1545] (интервал
298,15—1195 К) значение С°р B98,15 К)=118,4 Дж• К • моль" [4420] было уменьшено на 0,7% (см. табл. 47.1).
2 Для аппроксимации данных [1545] использовались 8 точек D03—1103 К), измерение при 1195 К отйлоняетСя
от кривой на —2% и не принималось во внимание.
412
Калий и его соединения K2Si205(K, ж)
Имеющиеся в литературе калориметрические измерения энтальпии образования K2Si03(K)
противоречивы. Такахаши и Йошио [4460] провели измерения энтальпии реакций растворения
ряда веществ в плавиковой кислоте при 298,15 К, откуда для реакции К28Ю3(к)+8Ю2(кварц)=
=K2Si205(K) было получено Дг#°B98,15 К) =—7,5+2,5 кДж-моль. Этой величине соответствует
AfH°(K2SiOa, к, 298,15 К) =—1586,8+6,0 кДж-моль. Хаттон и др. [2587] также из измерений
энтальпии реакций растворения в плавиковой кислоте нашли для реакции 2КОН(к)+8Ю2(кварц) =
=К28Ю3(к)+Н2О(ж) значение Дг#°B98,15 К)=—73,2+2,0 кДж-моль, которому соответствует
AfH°(K2SiO3, к, 298,15 К)=—1547,2+5,0 кДж-моль-1.
Расхождение величин, полученных в работах [4460] и [2587], намного превышает их
оцененные погрешности. Приведенные в этих публикациях сведения очень ограничены (данные [2587]
известны только по справочнику [2834]), что не позволяет решить, какая из этих работ более
надежна. Однако измерения Крёгера и Фингаса [3113] (см. также [3114]), где статическим
методом было исследовано равновесие реакции К2СО3(кL-8Ю2(кварц)=К28Ю3(к)+СО2(г) G48—
848 К, 11 точек), дают основание считать работу [2587] более надежной. По данным [3113] были
получены значения &fH°(K2SiOs, к, 298,15 К)=—!539+5 кДж-моль (III закон) и —1530 +
+12 кДж-моль (II закон), причем найденная в этой же работе энтальпия образования
iT2Si205(K) оказалась в хорошем согласии с имеющимися калориметрическими измерениями (см.
ниже). Принятое в справочнике значение является средним из величин, полученных в работах
[2587] и [3113]. Его погрешность увеличена с учетом противоречивости имеющихся данных.
В литературе известны оценки энтальпии образования K2SiO3(K), которые привели к значениям
—1606 кДж-моль [3095] (оценка по зависимости энтальпии растворения силикатов натрия
и калия в плавиковой кислоте) и —1544 кДж-моль [1040] (оценка с использованием цикла
Борна—Габера)
K2Si205(K, ж). Термодинамические свойства кристаллического и жидкого дисиликата дикалия
в стандартном состоянии при температурах 100—4000 К приведены в табл. 1185.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние K2Si205(k) в интервале 0—510 К
принимается моноклинная модификация (а), а в интервалах 510—867 К и 867—1318 К — соответственно
р- и у-модификации с неисследованными структурами.
При Г^ 298,15 К термодинамические функции вычислены по данным Бейера и др. [1545],
которые измерили теплоемкость K2Si205(K) D,8—308 К, образец с содержанием примеси
металлов в 0,01%, молярным отношением К2О : SiO2, равным 1 : 2,004, погрешность измерений 1%
при Т < 25 К, 0,5% — в интервале 25—50 К и 0,2% — при Т > 50 К). Экстраполяция
теплоемкости ниже 5 К приводит к значению S°E K)=0,02 Дж-К~1-моль. Принятые по [1545]
значения ?°B98,15 К) и Я°B98,15 К)—#°@), приведенные в табл. 47.1, имеют погрешности
0,4 Дж-К^-моль и 0,05 кДж-моль соответственно.
При Т > 298,15 К уравнения для теплоемкости а-, [3- и у-модификаций приняты по данным
Бейера и др. [1545], которые измерили энтальпии этих модификаций в интервалах 402—474,
518—803 и 873—1258 К соответственно (погрешность измерений по оценке авторов 0,3%). Менее
точные данные по энтальпии K2Si208 (погрешность до 3%) получены Такахаши и Йошио [4461]
B98—1309 К), они завышены на 2% и не учитывались.
Температуры полиморфных превращений E10+5 К и 867 +5 К) и их энтальпии A,2+0,2
и 1,6+0,3 кДж-моль) приняты по работе Бейера и др. [1545]. Крачек и др. [3097] методом
ДТА liaimra для (р— -^-превращения Д/г#°=1,1+0,4 кДж-моль при 867 К.
Температура плавления A318+5 К) принята по данным Крачека и др. [3097], менее точные
значения получены в работах [3560] A314 К), [3094] A309 К) и [3657] A288 К). Энтальпия
плавления C5,2+1,0 кДж-моль) принята по работе Бейера и др. [1545]. Такахаши и Йошио [4461]
с использованием своих данных по энтальпиям растворения кристаллического и стеклообразного
K2Si205, а также по изменению энтальпии K2Si2O5, нашли значение 31,8 кДж-моль, которое
представляется менее точным.
Теплоемкость жидкого K2Si206 B75+10 Дж-К^-моль) принята на основании результатов
трех измерений энтальпии в интервале 1363—1454 К [1545].
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000, 3000 и 4000 К
оцениваются в 0,3, 1, 8, 20 и 40 Дж-К~1-моль~1 соответственно. В справочных изданиях таблицы
термодинамических функций K2Si205(K, ж) не публиковались. Термодинамические функции
413
КВО2(к, ж) Глава 47
K2Si20B(K, ж), приведенные в работе Бейера и др. [1545] (Т ^ 1600 К), практически совпадают
с данными табл. 1185. -
Константа равновесия реакции K2Si205(K, >K)=2K(r)+2Si(r)+5O(r) вычислена с
использованием значения Дг//°@) =4796,536 +17 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
!±fHo{K?,\20b, к, 298,15 К) = —2505 + 5 кДж • моль.
Принятая величина является средневзвешенной из совпадающих в пределах их погрешностей
величин, полученных в калориметрических измерениях Крачека [3095] и Бэрани [1386] и в
результате исследований равновесия реакций, выполненных Крёгером и Фингасом [3113]. В
работах [3095] и [1386] были проведены измерения энтальпий реакций растворения ряда веществ
в 20% плавиковой кислоте, для реакции 2КС1(к)+28Ю2(кварц)+Н2О(ж)=К2812О6(к)+
+2НС1(р-р 12,73Ш2О) они дают ДДСB98,15 К)=147,1+4,0 кДж-моль. Этой энтальпии
реакции соответствует A^°(K2Si205, к, 298,15 К) = — 2509+5 кДж-моль. '
Крбгер и Фингас [3113] статическим методом исследовали равновесие реакций в системе
К2СО3—SiO2—СО2. Совместная обработка данных для двух реакций, исследованных в этой
работе G48—1222 К, 44 точки), приводит для реакции К2812О5(к)+СО2(г)=К2СОз(к)+28Ю2(кварц}
к значениям ДГЯСB98,15 К)=—85 + 8 кДж-моль (III закон) и —90+25 кДж-моль (II закон).
Этим величинам соответствуют энтальпии образования K2Si205(K), равные —2495+8 кДжХ
Хмоль и —2490+25 кДж-моль.
КВО2(к, ж). Термодинамические свойства кристаллического и жидкого метабората калия
в стандартном состоянии при температурах 100—2500 К приведены в табл. 1186.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47Л. В справочнике за стандартное состояние КВО2(к) в интервале 0—1220 К
принимается гексагональная модификация [4200] х.
При Т ^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работе Паукова и др. [771] A2—312 К, образец содержал Si (~0,03%), Mg (~0,003%),
Fe (~0,001%), Na (~0,1%) и Са (—0,001 %), следы воды не обнаружены). Среднее отклонение
экспериментальных точек от сглаженной кривой не превышало 0,34% в температурном интервале
12—25 К и 0,1 % при всех остальных температурах. Экстраполяция теплоемкости ниже 12 К
по закону Дебая приводит к величине ?°A2 К) =0,14 Дж-К~1-моль~1. Погрешности принятых
значений S°B98,15 К) и #°B98,15 К)—#°@) (см. табл. 47.1) оцениваются в 0,2 Дж-К^-моль
и 0,03 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости КВО2(к) принято уравнение, полученное на основании
значений С»B98,15 К)=67,03 [771], С°вG00 К)=94 и С»A200 К)=108 Дж-К^-моль.
Последние две величины оценены на основании зависимости от температуры теплоемкоетей метаборатов
лития и натрия, для которых известны экспериментальные данные.
Температура плавления A220+3 К) принята по результатам измерений Бергмана и др. [101,
102, 104, 107]; менее точные данные получены в работах [106, 626, 651, 875, 876, 4037] A213—
1223 К). Энтальпия плавления C3,5+4,0 кДж-моль) оценена по энтропии плавления, которая
принята равной этой величине B7 Дж-К-моль~1) для метабората натрия. Теплоемкость КВО2(ж)
A45+15 Дж-К-моль~1) принята равной теплоемкости жидкого метабората лития.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 0,1, 5
и 14 Дж-К-моль. Расхождения между термодинамическими функциями КВО2(к, ж),
приведенными в табл. 1186 и в справочнике [2835] (Т <; 2000 К), не превышают 0,7 Дж-К'^моль
в значениях Ф°(Т) и объясняются несколько различной оценкой теплоемкости КВО2(к) при Т >
> 298,15 К и энтальпии плавления.
Принятая в справочнике энтальпия образования
к, 298,15 К) = —983 + 5 кДж • моль
Рза-Заде и Мамедова [875] методом ДТА наблюдали два полиморфных превращения КБО2 при 713 и 1053 К.
Однако тепловой эффект на термограмме при 713 К очень мал и, возможно, вызван примесями, а температура,
при которой наблюдался второй тепловой эффект A053 К), очень близка к температурам эвтектик системы КВО2
с К4В2О5 и с К,В4О7 ( 1033 и 1053 К). Поскольку авторы [875] не приводят доказательств однофазности
образцов КВО2, этот тепловой эффект может быть вызван плавлением эвтектических смесей. Поэтому до появления
дополнительных исследований возможность полиморфизма у КВО2 не следует принимать во внимание.
414
Калий и его соединения КВО2(г)?
основана на измерениях энтальпий растворения в 2N азотной кислоте КВО2(к) (Шартсис, Каппе
L4252J, ДГЯ°B98,15К)=—бЭ+бкДж-моль-1) \ в В2О3(стекл.) (Шульцидр. [1181J, АгНаB98,15 К) =
= —38,5+0,8 кДж-моль) и энтальпии растворения К2О(к) в 2N азотной кислоте, вычисленной
по принятым в настоящем справочнике данным (Дг#°B98,15 К)=—428,5+4,0 кДж-моль).
Давление пара метабората калия в реакции КВО3(к, ж)=КВО2(г) вычислено с
использованием принятой в справочнике энтальпии сублимации
Дг#°@) = Д,Д°(КВО2, к, 0) = 313+ 15 кДж-моль-1.
Эта величина основана на измерениях давления пара и равновесия реакций, представленных
в табл. 47.30. Приведенные в таблице погрешности включают воспроизводимость измерений и
неточность использованных в расчетах термодинамических функций. Наиболее точные измерения
проведены эффузионным методом Крёгером и Зёрштрёмом [3115] и Гороховым и др. [311, 642],
и на них основывается рекомендованное значение. Основным источником его погрешности
является неточность термодинамических функций, в первую очередь КВО2 в конденсированном
состоянии.
Таблица 47.30. Результаты определений AejEro(KB02, к, 0) (в кДж-моль)
Авторы
Метод
Дв#°(КВО2, к, 0)
II закон
III закон
Крёгер, Зёрштрём, 1965 [3115] ! Эффузионный, 1087—1232 К, 17 точек 344±32 311+10
Горохов и др., 1971 [311, 642] То же, 1053—1124 К, 5 точек 291+100 315+10
Масс-спектрометрический, 1050—1147 К, 9 се- 315 ±5 —
рий
Енсен, 1969 [2853] Спектрофотометрия в пламенах H2+O2+N2, 294 295+20
К(г)+НВО2(г)=КВО2(г)+Н(г), 2020—2560 К,
данные представлены в виде уравнения
КВО2(г). Термодинамические свойства газообразного метабората калия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1187.
Молекулярные постоянные, использованные для расчета термодинамических функций
КВО2(г), приведены в табл. 47.7. Электронографическое исследование пара КВО2 [402],
изучение ИК-спектра различных изотопомеров молекул КВО2, изолированных в Аг-, Кг- и Хе-матри-
цах [1141, 1143, 1144, 4237], и расчет функций потенциальной энергии на основании ионной
модели [400] показывают, что молекула КВО2 в основном электронном состоянии ХХА' имеет угловую
конфигурацию с линейным фрагментом О—В=О (симметрия Cs). При этом в электронографиче-
ском исследовании [402] для ДК—О—В получено эффективное значение 100°, в работе [4237]
на основании измеренного изотопного расщепления найдено значение 90°, а минимуму функции
потенциальной энергии КВО2 соответствует равновесный ДК—О—В=95° [400]. Расчет [400]
показал также, что энергии линейной и циклической конфигурации КВО2 превышают энергию
угловой конфигурации на 800+100 и 5500+300 см соответственно, и, следовательно, ион К+
может почти свободно перемещаться вокруг иона ВО^. Учитывая заметные отличия
равновесного и эффективного значений ДМ—О—В в молекулах других метаборатов щелочных металлов,
для угла принято эффективное значение 130+30°, такое же, как в молекуле CsBO2, для которой
оно определено с наибольшей точностью по сравнению с остальными метаборатами.
Произведение моментов инерции, приведенное в табл. 47.7, рассчитано по структурным
параметрам г(К-О)=2,40 +0,05 А, г(В-О)=1,31+0,05 А и г(В=О)=1,21+0,05 А (см. [397, 402]).
Погрешность значения IaIbIc составляет 3-Ю14 г3-см6. Основные частоты КВО2 приняты по
работе Сешадри и др. [4237] после пересчета их на природную смесь изотопов бора. Близкие
значения частот v1( v2, v4, v5 были получены Шаповаловым и др. [1141, 1143, 1144], но частоты v3 и v6
этими авторами не наблюдались. Полосы vx—v8l которые наблюдались Мальцевым [648] в ИК-
1 Эта величина найдена линейной экстраполяцией полученных в [4252] энтальпий растворения соединений яК2Ох.
ХВ2О3 к составу КВО2 (к).
415
КАЮ2(к, ж) Глава 47
спектре высокотемпературного пара над КВО2, имеют частоты, отличающиеся от данных [4237]
на 10—50 см. С учетом данных [648] и возможных матричных сдвигов, погрешности принятых
значений v3, v4, v5 и ve оцениваются в 10—20 см, частот vx и v2 — в 40—50 см.
Термодинамические функции КВО2(г) рассчитаны в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128) и A. 130)
без учета возбужденных состояний и возможных изомеров. Погрешности вычисленных значений
определяются приближенным характером расчета, а также неточностью принятых
молекулярных постоянных; они составляют 8, 12 и 16 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000
и 6000 К.
Ранее термодинамические функции КВО2(г) вычислялись в справочнике JANAF [2835];
расхождения данных [2835] и табл. 1187 не превышают 1 Дж-К~1-моль~1 в значениях Ф°(Г).
Константа равновесия реакции КВО2(г)=К(г)+В(г)+2О(г) вычислена с использованием
значения АгН°@)=1808,579+17 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации КВО2(к).
Этим величинам соответствует также
Д/Я°(КВОа, г, 0) = —665,122 + 16 кДж • моль.
КА1О2(к, ж). Термодинамические свойства кристаллического и жидкого алюмината калия
в стандартном состоянии при температурах 100—4000 К приведены в табл. 1188.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние КАЮ2(к) в интервалах 0—810 К и 810—
1986 К принимаются две кубические модификации, из которых первая имеет упорядоченную
гранецентрированную решетку, а вторая — разупорядоченную решетку с статистическим
распределением ионов калия и алюминия [155].
При Т ^ 298,15 К термодинамические функции вычислены по данным Бейера и др. [1544],
которые измерили теплоемкость КАЮ2 в интервале 5—303 К (образец с содержанием примесей
~0,05% и с молярным отношением К2О : А12О3, равным 1 : 1,005). Погрешность измерений
составляла 1% при Т <[ 25 К, 0,5% в интервале 25—50 К и 0,2% при Т > 50 К; экстраполяция
теплоемкости ниже 5 К приводит к значению S°E K)=0,004 Дж-К-моль. Принятые по [1544]
значения 5°B98,15 К) и #"B98,15 К)—#°@) (см. табл. 47.1) имеют погрешности 0,3 Дж-К^Х
Хмоль и 0,04 кДж-моль соответственно.
При Т > 298,15 К уравнения для теплоемкости двух модификаций КАЮ2 (см. табл. 47.1)
основаны на данных Бейера и др. [1544], которые измерили энтальпии этих модификаций в
интервалах 406—742 К и 818—1198 К с погрешностью 0,3%. Температура превращения (810+5 К)
принята по согласующимся между собой данным [1544] (810 К), [155] (813 К) и [3734] (808 К),
энтальпия превращения A,295+0,100 кДж-моль) — по данным [1544]. Температура
плавления A986 К) и оцененные значения энтальпии плавления (82+20 кДж-моль) и теплоемкости
жидкого КАЮ2 A36+20 Дж>К~1-моль~1) приняты по рекомендации Спенсера и др. [5050а] (см.
также [2168]).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000, 3000 и 4000 К
оцениваются в 0,2, 0,6, 4, 12 и 18 Дж-К~1-моль~1 соответственно. В справочных изданиях
термодинамические функции КАЮ2(к, ж) не публиковались. Термодинамические функции КАЮ2(к),
вычисленные в работе Бейера и др. [1544] (Т ^ 1400 К), согласуются с данными табл. 1188 в пределах
0,1 Дж-К^-моль-1 в значениях Ф°(Г).
Константа равновесия реакции КАЮ2(к, ж)=К(г)+А1(г)+2О(г) вычислена с использованием
значения АгН°@)=2034,423 +5,0 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A/ff°(KAlOI, к, 298,15 К) = —1130,6 ± 3,0 кДж • моль.
Принятая величина основана на измерениях энтальпий растворения К2СО3(к), А12О3(к)
и КАЮ2(к) в расплаве окислов бора и свинца, выполненных Зыганом и др. [449].
Комбинирование этих измерений дает для реакции К2СО3(к)+А12О3(к)=2КАЮ2(к)+СО2(г) значение
ДГЯ°(975 К)=156,4+5,0 кДж-моль, которому соответствует принятая в справочнике
энтальпия образования КАЮ2(к).
416
Калий и его соединения KAlF4(r)
KA1F4(p). Термодинамические свойства газообразного твтрафторалюмината калия в стайдарт-
ном состоянии в интервале темШратур 100—6000 К приведены в табл. 1189.
Молекулярные постоянные, использованные для расчета термодинамических функций KA1F4,
приведены в табл. 47.7. На основании электронографического [4586] и спектроскопического
[2750] исследований для молекулы KA1F4 в основном электронном состоянии ХгАх принята би-
дентатная структура симметрии C2t. Приведенное в табл. 47.7 произведение моментов инерции
вычислено по структурным параметрам r(Al—F)BHeiI11I=1,692+0,008 А, г(А1—Р)йост=1,692 +
±0,010 A, r(K~FH2,513±0,014 А, Д FBHeniH-Al-FBHenIH=102,9+l,l°, Д FMOCT-A1-FMOOT=
=117,7+0,8°, найденным Вайда и др. при электронографическом исследовании KAlF4 [4585] *.
Погрешность значения IaIbIc оценивается в 2-10~11а г8-см*.
Основные частоты, приведенные в таблице, основаны на результатах исследования ИК-спектра
молекул KA1F4, изолированных в матрицах из Аг и N2, в работе Хаглена и др. [2750]. Авторы
[2750] наблюдали в спектре 8 основных частот и рекомендовали их значения для газовой фазы,
принятые в справочнике частоты v3, v4, ve и v9 не были зарегистрированы в спектре, и Хаглен
и др. [2750] оценили их значения в результате расчета по уравнениям поля валентных сил.
Найденные таким образом значения v3 и v9 A49 и 175 см) представляются заниженными для
колебаний кольца KFfcAl, а частоты v4 и ve B84 и 269 см) завышенными для иона AlF^. Приведенные
в таблице значения этих частот были оценены по частотам соответствующих колебаний молекул
K2F2 и AlF^ (ем. NaAlF4). Погрешности оцененных значений vs, v4, ve и v9 могут достигать 50—
60 см, остальных частот — 15—20 см.
Термодинамические функции KAlF4(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A.6), A.9), A.10), A. 122)—A. 124), A.128),
A. 130) без учета возбужденных электронных состояний и возможности существования изомеров
у этой молекулы. Погрешности рассчитанных величин обусловлены приближенным характером
расчета и неточностью принятых молекулярных постоянных; они могут достигать 10, 15
и 20 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции KAlF4(r) рассчитывались в работах Морозова и др. [696]
(до 5000 К) и Хаглена и др. [2750] (до 2000 К). Расхождения данных [696], [2750] и табл. 1189
достигают 5 и 2 Дж-К~1-моль~1 в значениях Ф°(Т) соответственно.
Константа равновесия реакции KAlF4(r)=K(r)+Al(r)+4F(r) вычислена с использованием
значения Дг#°@)=2626,337±20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(KA1F4, г, 0) = —1900 + 20 кДж • моль'1.
Результаты определений этой величины представлены в табл. 47.31. Указанные погрешности
учитывают воспроизводимость измерений, неточность термодинамических функций и термохими-
Таблица 47.31. Результаты определений Ay-H"°(KA1F4, г, 0) (в кДж-моль)
Авторы
Метод
bfH°{KA.l?it г, 0)
^III закон
Колосов и др., 1974 [535] Масс-спектрометрический, K3AlFe(K)=2KF(K)+ —1920+25
+KAlF4(r), 1020 К, 1 точка, конденсированная
фаза — K3AlFe
То же, конденсированная фаза K3A1F6+KF —1898 ±25
Масс-спектрометрический, K3AlFe(K)=2KF(r)+ —1902+25
+KAlF4(r), 1020 К, 1 точка, конденсированная
фаза — K3A1F6
То же, конденсированная фаза K3AlFe+KF —1902+25
Никитин, Сидоров, 1980 [3659] Электронный удар, D0(KF—A1F3)— D0(NaF— —1897+18
A1F3)=15,6+15
Авторы [4585] пришли к выводу, что цикл A1F2K должен быть неплоским с выходом атома К из плоскости A1F2
на угол 25,9 ±25°. Большая погрешность определения величины этого угла, а также большая амплитуда
колебаний не позволяют определить однозначно равновесную структуру этой молекулы. В справочнике
произведение моментов инерции рассчитано для плоского кольца A1FKF.
27 Заказ № 1590 417
K,A1F,,(k, ж), KUF5(r) Глава 47
ческих величин, использованных в расчетах. Приведенные в таблице:величины совпадают в
пределах их погрешностей. Принятая величина является средневзвешенной. См. также работы [551,
724, 1063, 1159, 2749, 4279, 4510, 4882], где приводятся дополнительные экспериментальные
данные и дано их обсуждение.
K3A1F6(k, ж). Термодинамические свойства кристаллического и жидкого гексафторалюмината
трикалия (калиевого криолита) при температурах 298,15—4000 К приведены в табл. 1190.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 47.1. В справочнике за стандартное состояние K3A1F6(k) приняты в интервале 0—600 К
тетрагональная модификация, а в интервале 600—1273 К — кубическая модификация. Ввиду
отсутствия данных по теплоемкости K3A1F6 при низких температурах значения iSl0B98,15 К),,
#°B98,15 К)—Н°@) и С°B98,15 К), приведенные в табл. 47.1, были оценены сравнением
соответствующих экспериментальных данных для A1F3(k), MF(k) и M3A1F6(k), где M=Li, Na и К.
Погрешности принятых значений 5°B98,15 К) и /Г°B98,15 К)^Н°@) оцениваются в 10 ДжХ
ХК~1-моль~1 и 1 кДж-моль соответственно. Единственные измерения энтальпии кристаллического
и жидкого KgAlFg выполнены Холмом и Гронволдом [2688] в очень узких интервалах температур
A126—1190К и 1290—1317 К) с целью определения энтальпии плавления K3A1F6. По этим данным
{2688]приняты энтальпия плавления A22,6+2,0 кДж-моль) и теплоемкость К3А1Г6(ж) C93 +
+20 Дж-К-моль~1). Температура полиморфного превращения F00+5 К) определена Холмом
[2690]; энтальпия этого превращения F,5+2,0 кДж-моль) оценена в предположении, что
энтропия перехода у K3A1F6(k) такая же, как у Na3AlFe(K) (Д)В<?=11 Дж-К~1-моль~1). Уравнения для
теплоемкости тетрагональной и кубической модификаций (см. табл. 47.1) получены на основании
найденного в работе [2688] значения #°A273 К)—#°B98,15 К)=285,6 кДж-моль и величины
AtrH°FO0 К).
Температура плавления A273+20 К) получена усреднением значений, найденных в работах
Холла и Инсли [2551] A293 К), Филлипса и др. [3814] A258 К) и Мальцева и Бухаловой [6501
A259 К); она совпадает со значением, рекомендованным в работе [2688].
Погрешности значений Ф°(Т) для K3A1F6(k, ж) при 298,15, 1000, 2000, 3000 и 4000 К
оцениваются в 7, 15, 30, 50 и 70 Дж-К~1-моль~1. Значения термодинамических функций K3A1F6(k),
вычисленные в справочнике JANAF [2834] (Т ^ 1200 К), согласуются с приведенными
в табл. 1190 в пределах 5 Дж-К~1-моль~1 в значениях Ф°(Т); имеющиеся расхождения
обусловлены тем, что в настоящем справочнике использованы данные [2688]. Термодинамические функ-.
ции K3AlF6(ffl) публикуются впервые.
Константа равновесия реакции K3A1F6(k, ж)=ЗК(г)+А1(г)+6Р(г) вычислена с использованием
/Значения Дг?Г°@)=4397,965+20 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(K3A1F6, к, 298,15 К) = —3347 + 20 кДж.моль.
Эта величина была найдена Хонгом и Клеппа [2698] методом высокотемпературной
калориметрии. Ее погрешность определяется в основном неточностью значения энтальпии K3A1F6(jk)
при 1320 К, которое было использовано для пересчета результатов измерений к стандартной
температуре. В справочнике [2834] по аналогии с Na3AlFe(K), Na3AlCl6(K) и К3А1С1в(к) было
оценено значение Дг#°B98,15 К)=—126 кДж-моль для реакции 3KF(k)+A1F3(k)=K3A1F6(k),
которому соответствует А^Н°(К3А1?в, к, 298,15 К)=—3346 кДж-моль.
KUF6(r). Термодинамические свойства газообразного пентафторураната калия в стандартном;
состоянии в интервале температур 100—6000 К приведены в табл. 1191.
Молекулярные постоянные, использованные для расчета термодинамических функций KUF5).
приведены в табл. 47.7. Экспериментальные данные о строении и основных частотах молекулы
KUF5 в литературе отсутствуют. В настоящем справочнике молекулярные постоянные KUF6
оценены на основании принятых выше постоянных молекул KA1F4, K2F2 и UFJ.
Для молекулы KUF5 в основном состоянии Х1А1 принята структура с циклом KF2U,
плоскость которого перпендикулярна плоскости равностороннего треугольника, образованного
остальными тремя атомами фтора, и проходит через биссектрису этого треугольника (симметрия'
С2с). Произведение моментов инерции, приведенное в табл. 47.7, рассчитано по структурным
параметрам r(U-F)=2,0+0,l А (как в молекуле UF"), r(K-F)=2,5+0,l Аи Д F—K-F=105+1001
418
Калий и его соединения К Li (г)
(как в KA1F4), 6=60° (угол между связями r(U—FK0H1I) и осью, проходящей через атомы К и U.
Погрешность значения IaIbIc оценивается в 2-10~112 г3-см6. Приведенные в таблице значения
основных частот KUFB основаны на основных частотах UF" (средних для конфигураций с
симметрией С4„ и D3h) и частотах К—F в KA1F4. Близкие по величине частоты объединены, и им
приписан суммарный статистический вес. Погрешности приведенных значений v1; v2 оцениваются
в 150 см, v3 — в 100 см, v4, v5, v8, v9, v10 — в 50 см, v6 и v7 — в 30—40 см.
Энергии возбужденных состояний KUF5 приняты такими же, как у UFJ. Уровни с близкими
энергиями объединены и приведены с суммарным статистическим весом, погрешности этих
величин оцениваются в 10—30%.
Термодинамические функции KUF6(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
с учетом пяти возбужденных состояний по уравнениям A. 168)—A. 170). Погрешности
рассчитанных значений определяются главным образом неточностью молекулярных постоянных, а также
приближенным характером расчета; они составляют 20, 30 и 40 Дж-К-моль в значениях
Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций KUF6(r) публикуется впервые.
Константа равновесия реакции KUF5(r)=K(r)+U(r)-(-5F(r) вычислена с использованием
значения ДгЯ°@)=3231,131 +31 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
Д/Я°(КиРБ, г, 0) = —2220 + 30 кДж.. моль.
Принятая ведичина основана на масс-спектрометрическом исследовании равновесия реакции
KUF6(r)=KF(r)-fUF4(r) A087 К, 1 точка), выполненном Барковым [4857] (см. также [4280, 5047]).
KLi(r). Термодинамические свойства газообразного калия—лития в стандартном состоянии
в интервале температур 100—С000 К приведены в табл. 1192.
Молекулярные постоянные 39K7Li, приведенные в табл. 47.4, приняты на основании анализа
результатов спектральных исследований [4646, 4688], квантовомеханического расчета [4012]
и приближенных оценок. В спектре поглощения KLi исследована одна система полос в области
5520—5840 А [4646, 4688], которая по аналогии с молекулой KNa может быть отнесена к
переходу СХП <- X12+. Теоретический расчет методом псевдопотенциала [4012] показал, что
молекула KLi должна иметь и другие стабильные электронные состояния, коррелирующие с атомами
KBP)+LiBS) (а3П, А1^, b3L+) и KBS)+LiBP) (В1!,*).
Приближенный анализ полос системы СХП <— Х1Л+, выполненный в работе Вейцеля и Кульпа
[4688], позволил определить только ше ;« 207 см. Поэтому молекулярные постоянные KLi в
основном состоянии ХХЕ+, приведенные в табл. 47.4, оценены на основании сравнения
постоянных гомоядерных и гетероядерных молекул щелочных металлов [1772]. Погрешности оцененных
значений ше и Ве составляют 10 и 0,01 см соответственно. Энергия состояния СЧ1 принята по
работе [4688]. Энергии остальных возбужденных состояний оценены по соотношению A. 89) и
данным для молекулы KNa, их погрешности могут достигать 1000—2000 см.
Термодинамические функции KLi(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Qm и ее производные вычислялись по соотношениям A. 90)—
A. 92) с учетом пяти возбужденных электронных состояний, если принять Qfflj.^—PiQ^l вр-
Статистическая сумма по колебательно-вращательным уровням энергии основного состояния
Х11,+ и ее производные находились по уравнениям A. 73)—A. 75) непосредственным
суммированием по колебательным уровням и интегрированием по / с помощью соотношений типа A. 82).
В расчете учтены все уровни с / ^ Jm^, «i последние находились по формуле A. 81). Уровни
энергии вычислялись по уравнениям A. 64а), A. 65) с коэффициентами Ykl, приведенными вместе
со значениями i;mMt и /Ип, в табл. 47.5. Величины Ykl были получены для изотопомера,
соответствующего природной смеси изотопов, при помощи соотношений A. 66) и постоянных молекулы
39K7Li (см. табл. 47.4). Для обеспечения условий A. 50) и A. 59) добавлены коэффициенты Ym
и Y03 соответственно.
Погрешности вычисленных термодинамических функций KLi(r) обусловлены неточностью
молекулярных постоянных, энергии диссоциации и энергий возбужденных электронных
состояний. Общие погрешности в значениях ФС(Г) при 298,15, 3000 и 6000К составляют 0,5, 1 и 2 ДжХ
К1 1
419
KNa(r) Глава 47
Термодинамические функции KLi(r) ранее вычислялись в работе Змбова и др. [4826] (Ф°(Г)
при r=400-f-800 К) в приближении «жесткий ротатор—гармонический осциллятор».
Расхождения данных [4826] и табл. 1192 составляют до 3 Дж-К~1-моль в значениях Ф°(Т) и
объясняются различием принятых постоянных и методов расчета.
Константа равновесия реакции К1л(г)=К(г)+1л(г) вычислена с использованием принятого
в справочнике значения
D0(KLi) = 75,4 + 2 кДж • моль «* 6300 ± 170 см.
Эта величина была получена Змбовым и др. [4826] при масс-спектрометрическом исследовании
равновесия реакции K2(r)+Li2(r)=2KLi(r). Принятое значение D0(KLi) удовлетворительно
согласуется с результатами квантовомеханических расчетов [4012, 4139] F855 и 6353 см).
Принятой энергии диссоциации соответствует
bfH°(KLi, г, 0)= 172,226 ±2,3 кДж^моль.
KNa(r). Термодинамические свойства газообразного калия-натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1193.
Молекулярные постоянные 39KNa, приведенные в табл. 47.4, приняты на основании анализа
результатов исследований электронного спектра и квантовомеханических расчетов KNa. В
спектре этой молекулы изучено 6 систем полос: ^l1S+«-X1E+ [3295, 3296], C1n**Z1S+ [1291,
1402, 3648, 3888, 3889, 4008, 4301], Д1П<*Х12+ [2164, 3296, 3722, 4529, 4565, 4688,49736],
ЕЧ1+*ьХЧ1+ [3788, 4301], Z>4I->a3I+ [1655, 1656, 1840, 3446] и dm -* a32+ [2549].
Неэмпирический квантовомеханический расчет [2839] и расчет методом псевдопотенциала [4012]
показали, что помимо экспериментально исследованных имеется еще ряд триплетных и синглет-
ных электронных состояний, коррелирующих с атомами в состояниях NaC2iS)+KDa.P) (Ь3П,с3?+)
и NaC2P)+KD2S) EXS+).
Молекулярные постоянные в состояниях X1!,* и D41 приняты по работе Хессела [49736),
который наблюдал спектр флуоресценции и проанализировал в системе ?IП -> Хх? + полосы
с v' ^ 23, v" ^ 44. Колебательные постоянные в состоянии Лх? + были определены Лумисом
и Эрвином [3296] по кантам полос системы Л1Е+«-Х1Е+ (v" ^ 10, v' ^ 26). Колебательные
и вращательные постоянные в состоянии СШ были получены Аллегрини и др. [1291 ] при
совместной обработке данных Л умиса и Эрвина [3296] по кантам полос с v" ^ 8 и v' ^ 26
системы СХП ±j X1!]* и результатов собственного анализа этой|системы в спектре флуоресценции
(г/ < 22I.
Постоянные слабосвязанного состояния а3? + приняты по данным Брефорда и Энгелке [1656],
которые провели анализ системы D1!! -*¦ а3? + (у" ^ 14), наблюдаемой в спектре лазерной
флуоресценции. Энергия состояния Б1!!* принята по оценке Эйзеля и др. [2164], сделанной на
основании интерпретации некоторых особенностей спектра флуоресценции, ее погрешность
оценивается в 700 см. Молекулярные постоянные состояния d3U приняты по результатам
приближенного анализа перехода д?Л -> а3? + в спектре лазерной флуоресценции [2549]. Энергии
возбуждения (с погрешностью 1000—2000 см), значения юе, Ве и ге состояний Ь3П !и c3S+, а также
значения Ве и ге состояния AXL+ приняты по расчету Яношека и Ли [2839].
Термодинамические функции KNa(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения (?вн и ее производные вычислялись по соотношениям A. 90)—
A. 92) с учетом восьми возбужденных электронных состояний, принимая для состояний сГе>
> 12 000 см @?ол.вр=.Р»(??5.вр- Статистические суммы по колебательно-вращательным уровням
энергии состояний Х11,+ и a3S+ и их производные находились по уравнениям A. 73)—A. 75)
непосредственным суммированием по колебательным и вращательным уровням, ограниченным
предельной кривой диссоциации. Уровни энергии вычислялись по уравнениям A. 63), A. 64а)
и A. 65) с коэффициентами Ykl, приведенными вместе со значениями ишах, /1Ш и а{ в табл. 47.5.
Величины Ykl были получены при помощи соотношений A. 66) для изотопомера,
соответствующего природной смеси изотопов К на основании постоянных за KNa из табл. 47.4. Для обеспе-
1 Анализ вращательной структуры, выполненный Аллегрини и др. [1291], вызывает сомнение, так как полученное
в этой работе значение постоянной De почти в 20 раз превышает величину, найденную по соотношению A.68).
420
Калий н его соединения KNa(r)
чения условий A. 50) и A. 59) в состоянии Х*Е+ добавлен коэффициент Y80 и исправлен
коэффициент F04, а для состояния а3?+ добавлены коэффициенты Yb0 и Y03. Мультиплетность уровней
состояния а3?+ учитывалась статистическим весом 3.
Погрешности рассчитанных термодинамических функций при Т ^ 1000 К обусловлены
главным образом неточностью фундаментальных постоянных. При более высоких температурах
основной вклад в погрешность вносит неточность энергий возбуждения триплетных состояний. Общие
погрешности в значениях Ф°(Т) могут составить 0,02, 0,05 и 1 Дж-К-моль при 298,15, 3000
и 6000 К соответственно.
Термодинамические функции KNa(r) ранее вычислялись в работах Тоцкого и Шпильрайна
[1025] (Г <! 1500 К) и Змбова и др. [4826] (Т ^ 800 К) приближенными методами по оцененным
молекулярным постоянным. Расхождения данных [1025] и [4826] с приведенными в табл. 1193
не превышают 0,4 и 0,8 Дж-К~1-моль~1 в значениях Ф°(Г) соответственно.
Константа равновесия реакции KNa(r)=K(r)+Na(r) вычислена с использованием принятого
в справочнике значения
ДгЯ°@) = Do(KNa) = 5205,8 + 0,8 см = 62,275 ± 0,010 кДж • моль.
Эта величина была получена Брефордом и Энгелке [1656] короткой экстраполяцией (на
несколько См) колебательных уровней состояния а3Л+ с учетом поведения ван"-дер-ваальсового
потенциала на больших расстояниях. Принятое значение хорошо согласуется с менее точными
величинами, полученными ранее при исследованиях спектров [2164, 3296, 4529^ 4826].
Принятой энергии диссоциации соответствует
г, 0) = 135,379 ±0,86 кДж . моль.
Глава 48
РУБИДИЙ И ЕГО СОЕДИНЕНИЯ
Ш)(к, ж), Rb, Rb+, Rb2, RbO, RbO2(K, ж), Rb2O{K, ж), Rb2O, Rb2O2(K, ж), Rb2O2, RbH(u, ж),
RbH, RbOH(K, ж), RbOH, Rb2O2H2, RbF(K, ж), RbF, Rb2F2, RbCl(K, ж), RbCl, Rb2Cl2, RbBr(it, ж),
RbBr, Rb2Br2, Rbl(u, ж), КЫ, Rb2I2, Rb2SO4(K, ж), RboSO4, RbNO2(K, ж), RbNO2, RbNO3(K, ж),
RbNO3, Rb2CO3(K, ж), RbBO2(K, ж), RbBO2, RbLi, RbNa, RbK
В настоящей главе рассматриваются рубидий и его соединения с кислородом, водородом,
галогенами, литием, натрием и калием, а также соли рубидия и некоторых кислородсодержащих кислот —
всего 26 веществ. Для 15 веществ приведены термодинамические свойства в конденсированном
состоянии, для 24 веществ — в газообразном состоянии. Термодинамические свойства двух газов
(Rb, Rb+) рассчитаны до 10 000 К, остальных — до 6000 К. Термодинамические свойства всех
веществ приведены для природной смеси изотопов рубидия (атомный вес 85,4678) и других
элементов, однако различие молекулярных постоянных разных изотопомеров учитывалось только
для некоторых двухатомных газов ввиду того, что для остальных соединений соответствующие
эффекты существенно меньше погрешностей рассчитанных термодинамических свойств.
В Приложении 3.10 для элементарного рубидия приведены сведения о критических постоянных,
а также данные, необходимые для учета отклонения термодинамических 1,ойств Rb(r) при
высоких давлениях от свойств в стандартном состоянии.
Rb(K, ж). Термодинамические свойства кристаллического и жидкого рубидия в стандартном
состоянии при температурах 100—2100 К приведены в табл. 1194.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние Rb(K) в интервале 0—312,47 К принята
кубическая модификация (структурный тип Na).
При Т ^ 298,15 К термодинамические функции вычислены по измерениям теплоемкости,
проведенным на образцах рубидия чистотой лучше 9Э,9% Филби и Мартином [2251] @,4—320 К)
и Мартином [3409] @,4—3 К). Данные по теплоемкости [1997, 2061, 3259, 3363, 3444] менее точны
и не учитывались. Принятые значения 5°B98,15 К) и #°B98,15 К)—Я°@) совпадают с
рекомендованными КО ДАТА—MGHC [1887 ], их погрешности оцениваются в 0,3 Дж -К -моль и 0,02 кДж X
Хмоль соответственно.
В интервале 298,15—312,47 К для теплоемкости рубидия принято уравнение, выведенное
по данным Филби и Мартина [2251]. Температура плавления C12,47 +0,03 К) принята по
результатам измерений Мартина [3409]; принятое значение согласуется в пределах погрешности с
данными Гоутса и др. [2427, 2428] C12,45 +0,02 К). Данные Филби и Мартина [2251] C12,65 К)
лежат несколько выше принятого значения, а данные [3978] C12,15 К) и [1997] C11,7+0,5 К) —
менее точны и не учитывались. Энтальпия плавления B,192 +0,005 кДж -моль) принята по
согласующимся между собой определениям Мартина [3409! и Филби и Мартина [2251]. Данные [1997,
3358, 3978, 4292] менее точны, но в пределах их погрешностей согласуются с принятым значением.
¦Я Уравнение для теплоемкости жидкого рубидия выведено совместной обработкой результатов
измерений энтальпии (или теплоемкости) Rb^) в работах Шпильрайна и Кагана [1165] D05—
1334 К), Новикова и др. [735] C76-1234 К), Теппера и др. E04—1268 К) 1 и Пчёлкина [845] 2
C73—1073 К); при этом погрешность измерений [11651 оценена в 1,5%, данных [735] — в 3%,
а данных Теппера и др. и [845] — в 5%. Учитывая ускоренный рост теплоемкости жидких калия
и цезия (см. гл. 47 и 49), при выводе уравнения для теплоемкости жидкого рубидия были
использованы дополнительно три значения энтальпии при 1400, 1600 и 1800 К, оцененные по
экспериментальным данным для К и Gs. Полученному в результате такой обработки уравнению для
теплоемкости Rb^K) (см. табл. 48.1) соответствует уменьшение теплоемкости от С°C12,47 К)=31,8 ДжХ
XК-моль до С°F00 К)=30,4 Дж-К~1-моль (минимум теплоемкости), а затем ее возрастание
1 Результаты измерений Теппера и др. цитируются по справочнику Шшшьрайна и др. [1179].
3 Данные [845] по теплоемкости Rb (ж) были пересчитаны и вводились в обработку в впде восьми значений эн-
тальшш в интервале 373—1073 К.
422
Рубидий и его соединения
Ш>(к, ж>
Таблица 48.1. Принятые значения термодинамических величин для рубидия и его соединений
в кристаллическом и жидком состояниях
Вещество
Rb
RbO2
RbaO
Rb2O2
RbH
RbOH
RbF
RbCl
RbBr
Rbl
Rb3SO4
RbNOa
RbNO3
Rb2CO3
RbBO2
Состояние
к, куб.
ж
кП, тетр.(р)
к1, куб. (а)
ж
кШ, куб.(т)
кП, куб.(C)
к1, гекс.(а)
ж
кП, ромб.(Р)
Kl, куб.(а) :
ж
к, куб.
ж
кИ
Kl
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
к, куб.
ж
кИ, ромб.(а)
КИ, ромб.(а)
к1, гекс. (8)
ж
к1, куб.
ж
kIV, гекс.
кШ, куб.
кП, гекс.
к1, куб.
ж
кП, монокл.(а)
к1, гекс.ф)
ж
кП, (|3)
Kl, (а)
Ж
Примечание. Cp{T)=a+l
Rb:
Rb,SO4:
RbN03:
a d=— 23,180 -10-
a d=350,531 -Ю-6
Ю
. -o
oo ^Z"
О} о
«ч I
кДжХ
Хмоль
7,489
—
16,76
—
15,6
—
19,7
—
9,1
14,5
—
10,9
—
12,16
—
13,06
—
13,34
—
27,014 '
—
—
—
23,4 5
—
19,64
—
—
—
24,48
—.
—
13,31
—
—
,15 K)
CO
СЭ
CM
,15 K)
t, oo
O3
CNJ
Дж - К Х
X моль
76,78
—
130,1
—
125
—
160
—
63,7
—
92
—
77,7
95,23
—
110,1
—
118,8
—
197,5
—
162
—
144,9
—
181,32
—
94,32
—
31,06
—
77,57
—
74,0
—¦<
93,0
—
39,3
—
69,0
—
50,63
—
52,26
¦ —¦
52,76
—
52,59
—
134,055
—
84,0
—
99,0
—
117,6
—
, 73,39
—
тТ—c-T-2+d-T*+e-T3 (i
6; e=10,
• б с=0,
a c=o, d=522,854 -Ю"8; б
824 -10-
Коэффициенты в уравне-
а
3,53
23,082
52,66с
88
100
51,473
100
100
100
21,336
117
134
35,446
56
51,240
80
83
42,343
71
48,515
73,4
48,629
72,8
41,527
72,1
236,935
2880,717
92,071
206,5
75,736
104
120,354
75,833
896,8Г
146
146
НИИ ДЛЯ С'р(Т)
b-i03
с ¦ 10
92,337 —
20,330 —4,198
! 83,540 —
_ —
75,556 —
'
240,361 —
— —
32,955 5,308
_ ' _
59,566 —
,
— —
26,024 —0,469
— —
12,560 —
— —
13,856 —
— —
24,862 —3,245
— —
—297,629 40,270
—5056,942б —
102,324 —
— —
27,718 —
— —
—227,512а —
136,667 —
7 —2346,157 s —
— . _
70,968 156,403 —
112,716
205
78,362
108
145
) Дж -К-1
<Z=0, e=2699,000-10-«.
c=0, d=0, e=3261,123-
68,852 —
— —
32,042 12,912
— —
•МОЛЬ).
Ю"9.
Интервал
температур
или
Т
1 m
К
298,15—312,47 312,47
а 312,47—2100 —
298,15—423 423
423—813 813
813—2000 —
298,15—543 543
543—613 613
613—778 778
778—2500 —
298,15—398 398'
398—843 843
843—2000 —
298,15—858 858
858—1200 —
298,15—508 508
508-658 658
658-3000 —
298,15—1068 1068
1068—3400 —
298,15—997 997
997—3400 —
298,15-965 965
965-3500 —
298,15—929 929
929—3400 —
а 298,15—800 —
800—931 931
931-1343 1343
1343-3500 —
298,15—695 695
695—2500 —
298,15-437 437
437—493 493
493—556 556
556—583 583
583—2500 —
298,15—576 576
576—1146 1146
1146-3500 —
298,15—968 968
968-1133 1133
1133—3000 —
ИЛИ
АтН
кДжХ^
2,192
—
21
—
0,7
4
20
—
0
21
—
22
—
5,4
8,9
—
25,82
—
24,4
—
23,3
—
22,05
—
—
0
37,3
—
12,1
-1
3,9
3,2
0,97
4,6
—
1,3
30
—
0
31
—
423
ИЬ(к, ж)
Гдана 48
до значения С°B000 К)=57,7 Дж-К-моль. Это уравнение позволяет описать данные по
энтальпии жидкого рубидия с погрешностью в 1 % при 1000 К, 2% при 1500 К и 3% при 2000 К.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 1500 и 2000 К оцениваются в 0,2, 0,7, 1,2
и 2 Дж-К-моль соответственно. Значения термодинамических функций НЬ(к, ж), рассчитанные
во втором издании настоящего справочника [337] (Г ^ 2300 К), в справочниках Шпильрайна и др.
[1179] (Т < 1500 К) и Халтгрена и др. [2752] (Т < 1300 К) согласуются с приведенными
в табл. 1194 в пределах 1 Дж -К -моль в значениях Ф°(Т); эти расхождения обусловлены разным
выбором значения 1S(Rb, к, 298,15 К) и тем, что в данном издании принято ускоренное возрастали»
теплоемкости ИЬ(ж) при Т > 1000 К.
В настоящем справочнике в качестве стандартного состояния рубидия принято кристаллическое
состояние и в соответствии с этим
Д^^ЛЬ, к) = 0.
Давление пара в реакции Rb(K, ж)=Ш>(г) вычислено с использованием значения ДгЯ°@)=
=82,192 +0,8 кДж -моль, соответствующего принятой в справочнике энтальпии сублимации
Д,Я°(Ш>, к, 298,15 К) = 80,9 ±0,8 кДж • моль.
Принятая величина основана на представленных в табл. 48.2 результатах измерений давления
насыщенного пара рубидия. Расчет энтальпии сублимации проводился для модели идеальной смеси
химически реагирующих атомарной и молекулярной компонент. При высоких температурах
учитывалась поправка на неидеальность к модели диссоциирующих идеальных газов по варианту
метода смеси, разработанному Фокиным (см. Приложение 8); эти расчеты могли быть проведены
только для измерений, выполненных при Т ^ 1673 К.
В результате проведенной обработки данных для большинства указанных в табл. 48.2 работ
Табл
иц а
48.2. Результаты
Автор
определения
"(Kb,
к, 298,15 К)
Метод
II
,#°(Rb,
закон
к, 298
III
,15К)а
закон
Руфф, Йохансен, 1905 [4091]
Хакшпиль, 1913 [2542]
Скотт, 1924 [4221]
Киллиан, 1926 [2997]
Бонилла и др., 1962 [1611], см.
также [4277]
Везерфорд и др., 1963 [4674]
Теппер и др., 1963 [4493]
Аченер, 1964 [1208]
Бук, Паули, 1965 [1709]
Боданский, Шине, 1967 [1604]
60,5 ±6,2
80,27
79,46+0,18
Точек кипения, 994 К, 1 точка
Статический, 523—640 К, 8 -Ю—8 -Ю атм,
11 точек
То же, 364—400 К, 7-10-8-8-10-'атм, 8 точек 85,3+6,7 81,48+0,20
То же, 313—376 К, 2 -Ю"9—4 -Ю атм, 10 точек 84,2 ±2,5 79,73 ±0,21
Точек кипения, 718—1222 К, 0,04—7,38 атм, 83,80 ±0,12 80,93 ±0,02б
32 точки
То же, 958—1101 К, 0,9—3,2 атм, 11 точек
То же, 717—1352 К, 0,04—14,6 атм, 56 точек
То же, 745—1260 К, 0,06—9,0 атм, 14 точек
Ионизационный, 307—363 К, 1 -Ю"9—1 -Ю"' атм,
3 точки
Точек кипения, 750—1153 К, 0,07—4,8 атм, 7 точек 84,75 ±1,06 80,81 ±0,10 б
Герасимов и др., 1968 [273, 2386] Эффузионный, 326—396 К, 1-Ю—1-Ю атм, 81,37 ±0,93 80,55 ±0,07
5 точек
Статический, 593—960 К, 1 -Ю—1,0 атм, 49 точек 85,85 ±0,80 80,92 ±0,04 б
Точек кипения, 771-1153 К, 0,09—4,8 атм, 85,04 ±0,53 80,81 ±0,07б
34 точки
То же, 707—1541 К, 0,04—33 атм, 61 точка 83,45 ±0,05 80,83 ±0,01 6
81,5 ±1,6 80,89 ±0,12 б
81,97 ±0,19 80,97 ±0,02 б
83,06 ±0,34 81,03 ±0,01 б
83,4 ±33,4 81,25 ±0,60
Воляк и др., 1968 [240]
Шине и др., 1971 [4184]
Шпильрайн, Никаноров, 1971
[11721
Чернеева, Проскурин, 1972 [1111] То же, 1074—1370 К, 2,7—16,2 атм, 75 точек
Байс, Бонилла, 1973 [1550] То же, 1048—2073 К, 2,0—126 атм, 17 точек
Пьяченте и др., 1973 [3831] Масс-спектрометрический, 402—551 К, 3,5-10-'—
1,3 -Ю-3 атм, 66 точек
а Приведенные величины рассчитаны с учетом образования в насыщенном паре димерных молекул.
б Введены поправки на отклонение от идеальности. Учтены только измерения, выполненные при Т < 1673 К-
86,18 ±0,19 80,84 ±0,03 б
86,3 ±1,6 81,35 ±0,2 б
82,5 ±1,5 82,72 ±0,10
424
Рубидий и его соединения
И»(г)
получены хорошо согласующиеся между собой значения энтальпий сублимации. Приведенные
в таблице погрешности отражают лишь воспроизводимость измерений. Рекомендованное значение
вычислено по наиболее надежным данным [240, 1172, 1208, 1611, 4184, 4277, 4493], полученным
различными вариантами метода точек кипения (всего 246 измерений в интервале 707—1541 К).
Погрешность принятой величины обусловлена в основном погрешностями термодинамических
функций рубидия в конденсированном состоянии, которые приводят к погрешностям в энтальпии
сублимации от 0,4 кДж-моль для измерений при 700 К до 1,6 кДж-моль для измерений при
1400 К. Принятое значение энтальпии сублимации совпадает с рекомендованным КО ДАТА—
МСНС [1887]. Менее точные измерения давления пара рубидия были проведены в работах [273,
1111, 1550, 1604, 1709, 2386, 2542, 2997, 3831, 4091, 4221, 4674].
Rb(r). Термодинамические свойства газообразного рубидия в стандартном состоянии
в интервале температур 100—10 000 К приведены в табл. 1195.
Уровни энергии атома Rb, использованные в расчете термодинамических функций,
представлены в табл. 48.3, в которой даны валентные состояния Rb, соответствующие электронным
конфигурациям . . .4p65s — . . Ap*5g, . . Ape4d и . . Apeif. Принятые значения основаны на результатах
изучения спектра Rb в работе Иоханссона [2862 ], которые практически совпадают с рекомендацией
Мур [3550]. При этом для состояний . . Ad(*D), . . AfBF) и . . .5fBF) приняты усредненные
значения для двух подсостояний, основанные на данных [2862].
Таблица 48.3.
Уровни энергии атомов Rb и Rb+, принятые для расчета термодинамических
функций
Атом
Электронная
конфигурация
Rb . . .5s
. . .Ър
. . Ad
Состояние
Ti
CM
2S 0
2i>Vj 12 578,950
*P,l 12 816,545
Ю * 19 355,38
Ю,и 25700,536
Ю,'г 25 703,498
Pi
2
2
4
10
4
6
Атом
Rb
Rb+
Электронная
конфигурация
...if
...5/
...bg
¦ ¦ Ap*
Состояние
Ti
CM
*F 26 792,10
*F 29 277,78
2G 29 297,6
JS 0
Pi
14
14
18
1
Термодинамические функции Rb(r) вычислены по формулам A. 3)—A. 6), A. 9), A. 10), A. 23)—
A. 25) непосредственным суммированием по уровням энергии. Погрешности вычисленных
термодинамических функций при Т <С 4000 К определяются главным образом неточностью
фундаментальных постоянных и не превышают 0,02—0,03 Дж-К-моль~1 в значениях Ф°(Т) и S°(T).
При более высоких температурах погрешности возрастают из-за того, что в расчете не учитывались
ридберговские состояния, при 6000 и 10 000 К они могут достигать 0,1 и 2 Дж -К -моль в
значениях Ф°(Г) и 1 и 10 Дж-К-моль в значениях S°(T) соответственно.
Ранее термодинамические функции Rb(r) вычислялись неоднократно, в том числе в
предыдущем издании настоящего справочника [337] (Т ^ 6000 К), в работах Гурвича и др. [335], Хилзен-
рата и др. 12652] и Рейтера [3973] (Г < 10 000 К), Кольского и др. [3067] (Т < 8000 К) и Эванса
и др. [2195] (Т < 2500 К), Результаты расчетов [335, 337] при Т < 4000 К, а также расчета
[2652] во всем интервале температур удовлетворительно согласуются с данными табл. 1195;
расхождения в значениях Ф°(Т) не превышают 0,01—0,02 Дж-К'1-моль.Данные [3067, 3973]
при Т < 4000 К отличаются от табл. 1195 на 0,06—0,07 Дж -К -моль. При Т > 4000 К
результаты расчетов [335, 337, 3067, 3973] возрастают с температурой быстрее, чем данные настоящего
справочника и при 10 000 К расхождения достигают 10 Дж>К-моль~1 в значениях Ф°(Г)
(см. Li(r)).
Энтальпия образования
bfH°(Rb, г, 0) = 82,192 ±0,80 кДж • моль
соответствует принятой в справочнике энтальпии сублимации рубидия.
425
Rb+(r), Rfo2 (г) Глава 48
Rb+(r). Термодинамические свойства газообразного положительного иона рубидия в
стандартном состоянии в интервале температур 298,15—10 000 К приведены в табл. 1196.
В табл. 48.3 приведено только основное электронное состояние XS иона Rb+, относящееся
к конфигурации . . Лре; состояния, принадлежащие конфигурациям . . АрьЫ и . . .4р54/, имеют
энергии свыше 125 000 см [3550], а другие возбужденные состояния Rb+ являются ридбергов-
скими.
Термодинамические функции Rb+(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
при условии, что In <2BH=0. Погрешности вычисленных значений термодинамических функций
во всем интервале температур обусловлены только неточностью фундаментальных постоянных
и не превышают 0,02 Дж>К-моль.
Ранее термодинамические функции Rb+(r) вычислялись в предыдущем издании настоящего
справочника [337] (Т < 6000 К), а также в работах Хилзенрата и др. [2652] (Г < 10 000 К)
и Грина и др. [2473] (Т ^ 50 000 К). Результаты этих расчетов совпадают с данными табл. 1196
в пределах 0,01 Дж-К~1-моль, за исключением значений Ф°(Г) и S°(T) при 10 000 К в расчете
[2652], где, по-видимому, допущена ошибка в 0,2 Дж-К~1-моль.
Константа равновесия реакции Rb+(r)+e(r)=Rb(r) вычислена с использованием значения
АгН°@) = —403,032+0,002 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Rb)=33 690,81+0,2 см=403,032+0,002 кДж-моль.
Это значение рекомендовано Мур [3549] с погрешностью 0,01 см на основании результатов
измерений ридберговских серий Rb в работе Йоханссона [2862]. Погрешность принятой величины
несколько увеличена с учетом данных других работ (см. [3550], а также [2129, 2579]).
Принятому потенциалу ионизации соответствует
&fH\Rb+, г, 0) = 485,224 +^0,80 кДж • моль.
Ш>2(г). Термодинамические свойства газообразного дирубидия в стандартном состоянии в
интервале температур 100—6000 К приведены в табл. 1197.
Молекулярные постоянные, приведенные в табл. 48.4, приняты на основании спектральных ¦,
исследований и оценок. В спектрах Rb2 наблюдались четыре системы полос, связанные с
переходами АхЪ+и *- Хх?; [3427, 3428], ВхПи^ Xх Z+ [1540, 1737, 1764, 3087, 3427, 3428, 3463, 4646],
ЕхПи *- XхЦ [3427, 3428] и FXUU <- Z1E+-[3427, 3428, 3689]. По аналогии с двухатомными
молекулами других щелочных металлов у Rb2 следует ожидать существование низколежащих
электронных состояний «3ПИ, Ь3?*, Сх1у и DlTlg, диссоциирующих на атомы в состояниях 2S и 2Р. Кроме
того, можно предполагать существование состояний с энергией около 20 000 см,
диссоциирующих на атомы RbBS) и Rb^D).
Принятые значения колебательных постоянных в состоянии X12* получены Котник-Каруза
и Видалом [3087] по кантам полос с v" ^ 29 системы В1Пи <- Xх Щ, наблюдавшихся в спектре
магнитного вращения. Вращательные постоянные состояния Xх 2^ и все постоянные состояния
ВхПи получены Колдвеллом и др. [1737] в результате анализа той же системы в спектре лазерной
флуоресценции (г/ <J 17, v" ^ 2). Постоянные состояния ЕхИи получены Тзи-зе и Тзин-Сан-
Тзингом [3689] по кантам полос системы ЕхИи <~ Xх ?^ (v' ^ 18).
Поскольку в работах [3427, 3428] анализ системы Ах?*-*- Хх^+ не проводился, энергия
состояния А11*1 оценена по^(l. 89) и данным для молекул Na2 и К2. Аналогично были оценены
энергии остальных триплетных и синглетных состояний, погрешность этих величин достигает
1000—2000 см. Состояние Я-Пя (Те—22 777,5 см) соответствует ридберговским состояниям
атомов и не рассматривается в справочнике.
Термодинамические функции Rb2(r) были вычислены по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения QBa и ее производные вычислялись по соотношениям A. 90)—A. 92) с учетом семи
возбужденных состояний в предположении, что Q^J0\ Bf=PiQSl. вр- Статистическая сумма
по колебательно-вращательным уровням состояния Xх Е1" и ее производные находились
непосредственным суммированием по колебательным уровням и интегрированием по / с помощью
соотношений типа A. 81). В расчете учитывались все уровни со значениями / ^ /шах> „, последние
находились по формуле A. 82). Уровни энергии рассчитывались по уравнениям A. 64а) и A. 65) с коэф-,
426
Рубидий и его соединения
Rb2(r)
Таблица
Молекула
48.4. Молск
Состояние
уляриые 1
Те
85Rb2 Х*2+а о
not
70ЯННЫ8 Rt
57,7743
Вт* 14 665,447 47,431
Ета 20 835,1
85Rb0 Хг 2+ 0
АШ 600
85RbH ХХ2+ 0
Л12+ 18 219,8
85RbF X12+ 0
S5RbCl X1^ 0
85RbBr X12+ 0
85RbI X12+ 0
8iRb7Li X12+ 0a
Cm 17 550
85RbNa JPE+ 0
СЩ 16 421,8
S5RbK X12+ 0a
Примечание. Ниже все величины
36,46
390 а
370 а
936,94
211,74
373,27
233,34
169,46
138,51
184 б
81
106,64
в 61,49
76 б
даны в см.
Rb2: a оцененные электронные состояния:
Состояние а3Пм А
12+ ь»2 +
Те 9000 11 000 12 500
6 (о,г/е=2,806 -Ю-*. в о)ег/е=
RbO: а оценка; б вгиислеяо по
RbH: а шеу,=8ЛЪ -Ю; 6 а2=3
=0,59874 -Ю-2.
соотношению A.
Ю-*; в 3,, = 1,2-
Д а2=—7,08-Ю-3, а3=—3,42-10-4,'а4=—7
RbF: a а2=3,3 -10; б ^=3,7 ¦
RbCl: a а2=7,0-10-'; б ^=2,0-
RbBr: a а2=2,28-10-'.
Ю-10.
Rbl: a а2=1,18-10-'; 6 j31=5,27 -10-".
RbLi: a Состояние А12+, а3П
Te 12 000
б оценка; в вычислено по
RbNa: a оценка; б вычислено по
Состояние А1 2+, а3П
Те 11 800
RbK: a oi
(енеяные электронные с
б оценка; в вычислено по
Ь3 2+ В12+
16 300 16 600
соотношению A.
соотношению A.
• б3 2+ В1 2+
16 000 16 300
ос
RbO, RbH
ыехе
см
0,13 885 б
0,15 326 в
0,124
2а
За
14,205 а
-6,47 г
1,8
0,856^
0,463
0,335
0,68 б
—
0,455
0,945
0,21 б
С!2+, Dm
15 000
, RbF
Bt
-i
0,022
, RbCl, RbBr, Rbl, RbLi, RbNa, RbK
0! • 103
?,-10°
78 0,047 402 0,015 06
0,19 938 0,070 029 0,014 034
0,236
0,217
3,020
1,103
0,210
0,087
0,047
0,032
0,207
0,071
0,037
—
a 1,6б
a 2,1 6
72 6
6 — 60 Д
665 1,523 a
640 5 0,453 6 a
527 9 0,186 0 a
832 9 0,109 5 a
6 0,85 б
—
7a 0,28a
—
76 0,10б
69); в вычислено по соотношению
,0-10; е
68).
—
0,35 B
0,30 B
122,4 B
123,4 e
0,268 4 б
0,049 47 e
0,014 95
0,007 38 б
1,0 B
—
0,13 б
—
0,037 B
A. 68).
„=—2,53 -Ю-1, (o,ze=—3,04 -10;
,3-ЮЛ pa=2,12-10-'.
68); в оцененные электронные состояния:
гоянпя: Состояние А11
т
соотношению
A-
10
68).
+, a3ll Ь32+ В1^
400
14 100 14 400
cm
14 500
re
A
4,175
4,457
—
2,30 a
2,40 a
2,367
3,916
2,270 450
2,786 865
2,944 71
3,176 84
3,54 б
—
3,61 a
—
4,09 s
фициентами Уы,вычисленными по формулам A. 66), и постоянным из табл.48.4 для изотопомера,
соответствующего природной смеси изотопов. Эти коэффициенты приведены вместе со значениями
vm% и /Иш в табл. 48.5. Для обеспечения условий A. 50) и A. 59) добавлены коэффициенты Yi0,
Y50 и У0з-
Погрешность рассчитанных термодинамических функций Rb2(r) при низких температурах
обусловлена главным образом неточностью принятых значений фундаментальных постоянных
427
Таблица 48.5. Коэффициенты в уравнениях, описывающих уровни энергии (в ем), а также значения «шах и Jiim,
принятые для расчета термодинамических функций Rb2, RbO, RbH, RbF, RbCl, RbBr, Rbl, RbLi, RbNa, RbK
Коэффициенты
Rb,a
Г„ • 10-' 0
У10 • 10-1 0,574 209
Yia ¦ 10-' -0,009 853
У* • 10' -0,024 218
У<0 • 103 0,070 104
Уи ¦ 105 -0,055 336
У„ ¦ 101 0,226 318
У„ • 10' -0,046 94
У31 • 10* -
У„ • 10' -
У„ • 10« -
У„ ¦ 10»
У„ • 10е -0,014 S65
Y» ¦ 10»
ую • ю7
У* • 10" 0,000 714
Ум • Ю15
(а„ = De) X —
xio-'
а2 • 10< -
а3 • 10» -
а, ¦ Ю" —
«шах 90
Jlim 604
RbO
X'Z+
АШ
RbH
Х'Ъ+
0 0,060 0 0
3,881309 3,669 593 9,368 345
-0,133 547 -0,192 051 -1,422 298
0,013 653 0,030 053 1,083 421
_ _ -3,798 215
_
2,36 2,17 30,200
—1,6 —2,1 —72,0
0,0241 0,042 5 3,0
— — —
_ — —
_ — —
-0,35 -0,30 -122Д
— - 120,0
_ — —
—0,020 7 —0,109 163,021
— —
_ - 1,516 488
_ — 1,083 177
— _ —1,464 514
_ - 8,451 231
218 144 28
584 451 96
А'Ъ+
1,821980
2,116 600
0,652 650
-2,654 417
4,100 429
-3,079 525
11,036
60,00
-70,6
3,42
-7,0
5,1
-123,4
630,0
-2,12
1558,703
—
0,976 107
0,030 118
-0,013 617
0,027 443
47
168
RbFa
RbCla
X'S+
0 0
3,730 48 2,319 86
-0,179 8 -0,084 61
— _
— —
—
2,104134 0,866 264
-1,520 071 -0,445 764
0,032 944 0,006 844
— —
— _
— —
-0,267 748 -0,048 329
0,036 583 -0,002 27
— —
-0,004 59 -0,000 799
-0Д36 -0,010 5
'— _
_ _
— —
— -
312 559
833 1288
Примечание. Ниже все величины даны в см-1, за исключением коэффициентов d,, d2, являющихся
Rb,: а Состояние а
П„ A Vt b'st В 41,, С'it.
D'na Е'Пи
Те 9000 11000 12 500 14 665,447 15 000 20 835,1
RbF: a коэффициенты потенциальной функции: do = 165 347,
RbCl: а коэффициенты потенциальной функции: d0 = 155 315,
RbBr: a коэффициенты потенциальной функции: d0 — 151052,
Rbl: а коэффициенты потенциальной функции: d0 = 146 083,
d,== 3,134 67,
^ = 3,296 76,
d, — 3,325 52,
d, = 3,344 09,
RbLi: a
d, = 6,586. RbNa: a
d, = 7,074.
d, -- 7,329.
dj = 7,176.
RbK: a
RbBra
X'Z+
0
1,686 51
-0,045 86
—
—
_
0,470 752
-0,183 344
0,002 235
—
—
_
-0,014 671
—
—
-0,000 149
—
—
_
—
-
514
1447
Rbla
Х'Ъ+
0
1,382 41
—0,033 37
—
—
—
0,327 050
—0,108 822
0,001 168
—
—
—
-0,007 322
-0,000 522
—
—0,000 056
—
—
—
—
—
482
1499
безразмерными.
Состояние
Te
Состояние
т.
Состояние
Те
A 'S+, asa
12 000
A'S+, а3П
11800
А '?+, а"П
10 400
RbLia
X't+
0
1,857 546
-0,091176
-0,075 643
—
—
2,089 633
—0,?62 122
—
—
—
—
-1,019 059
—
—
0,355 0
—
—
—
—
—
53
253
Ь3Е+
16 300
16 000
b3S+
14 100
RbNa*
0
1,066 511
-0,048 968
0,041 135
—0,075 254
—
0,716
-0 279 4
—
—
—
—
—0,13
—
—
0,008 0
—
—
—
—
—
68
359
В'Е+ С
RbKa
0
0,755 00
-0,010 78
-0,025 465
—
—
0,375 34
-0,099 34
—
—
—
—
-0,036 7
—
—
0,001 549
—
—
—
—
—
85
482
П
16 600 17550
B'S+ С
¦п
16 300 16 421,8
В 'Z+ С
14 400 14
¦п
500
о?
Рубидий и его соединения RbO(r)
и неточностью аппроксимации колебательно-вращательных уровней уравнением с принятыми
постоянными. При температурах выше 3000 К к ним добавляется погрешность из-за неточности
принятых энергий ряда низколежащих состояний и пренебрежения рядом состояний с энергией
^ 20 000 см. Общие погрешности в значениях Ф°(Г) при 298,15, ЗОШ и 6000 К могут составить
0,05, 1,0 и 5 Дж-К-моль соответственно.
Термодинамические функции Rb2(r) ранее вычислялись в работах Рейтера [3973]
(Т <; 10 000 К) и Эванса и др. [2195] (Т < 1000 К). Расхождения с данными [3973] достигают
4 Дж -К -моль в значениях Ф°F000 К) и объясняются различием в способе учета возбужденных
электронных состояний.
Константа равновесия реакции Rb2(r)=2Rb(r) вычислена с использованием принятого в
справочнике значения
D0(Rb2)=3910±10 cm-x=46,77 ±0,12 кДж-моль,
полученного Брефордом и Энгелке [1657] в результате исследования предиссоциации в состоянии
.РПЯ, наблюдаемой в спектре флуоресценции. Принятое значение хорошо согласуется с величиной,
полученной экстраполяцией колебательных уровней в основном и возбужденных состояниях
{47 кДж-моль) [3297, 3689], но существенно отличается от результата масс-спектрального
исследования [3831] D1,8+3,6 кДж-моль).
Принятой энергии диссоциации соответствует
A,?r(Rba г, 0) = 117,614 + 1,6 кДж .моль.
RbO(r). Термодинамические свойства газообразного оксида рубидия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1198.
Экспериментальные данные о молекулярных постоянных RbO отсутствуют. Значения
постоянных, приведенные в табл. 48.4, приняты на основании оценок и квантовомеханических расчетов.
Исследование спектра ЭПР молекул RbO, изолированных в матрицах инертных газов в работе
Линдсея и др. [3265], показало, что основным электронным состоянием молекулы является
состояние Х22+. Приближенный квантовомеханический расчет, выполненный Со и Ричардсом
[4339], привел к выводу, что у молекулы RbO должно быть стабильным возбужденное состояние
А2И с энергией 600 см, погрешность определения которой можно оценить в 500 см.
Частоты колебаний ш„ для состояний Х22+ и А2П оценены на основании сопоставления
соответствующих величин в молекулах КО, CsO, KF, RbF, CsF и результатов квантовомеханического
расчета [4339]. Погрешность этих величин может достигать 40 см. Значения шех„ были оценены
по соответствующим значениям, принятым для молекул NaO и КО. Приведенные вращательные
постоянные соответствуют межатомным расстояниям r,(Rb—О)=2,30дА (ХаЕ+) и re(Rb—О) =
=2,40 А (А2Щ, оцененным по данным для молекулы RbOH и результатам расчета [4339], их
погрешность составляет 0,03 см.
Термодинамические функции RbO(r) были вычислены по уравнениям A. 3)—A. 10), A. 93)—
{1. 95). Значения QBS и ее производные вычислялись по соотношениям A. 90)—A. 92) с учетом
одного возбужденного состояния. Статистическая сумма по колебательно-вращательным уровням
состояний X2 ?+ и А2Н и ее производные находились непосредственным суммированием по
колебательным уровням и интегрированием по значениям / с помощью соотношений типа A. 81). В
расчете учитывались уровни со значениями /^ Jm&%, „. последние находились по формуле A. 82).
Уровни энергии вычислялись по уравнениям A. 64а) и A. 65) с коэффициентами Ykl, приведенными
вместе со значениями ушах и /Иш в табл. 48.5. Для обеспечения условий A. 50) и A.59) были введены
коэффициенты Y30 и Y03. Мультиплетность и вырождение уровней состояний Х2? + и А2П
учитывались статистическими весами 2 и 4 соответственно. '
Погрешности рассчитанных термодинамических функций RbO(r) обусловлены главным образом
неточностью оценки молекулярных постоянных, в том числе энергии состояния А2Л, а. при
высоких температурах — отсутствием данных о других возбужденных состояниях; они составляют
3, 5 и 8 Дж -К -моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Термодинамические функции RbO(r) ранее вычисляли Брюэр и Розенблатт [1666] (Ф°(Т),
Т < 3000 К). Расхождения данных [1666] и табл. 1198 достигают 4 Дж-К-моль при 3000 К.
429
КЬО2(к, ж) Глаза 48
Константа равновесия реакции RbO(r)-— Rb(r)+O(r) вычислена с использованием принятого'
в справочнике значения
Дг#°@) = Do(RbO) = 275 + 25 кДж • моль «* 23 000 + 2000 см.
Принятая величина была оценена сопоставлением энергии атомизации в рядах МОН(г), МпО(г).
и МО(г), где M=Li, Na, К, Rb, Cs. Совпадающее значение было получено на основании расчета
с использованием ионной модели [346] после корректировки с учетом разности результатов
аналогичных расчетов для LiO, NaO, КО и имеющихся для этих молекул экспериментальных данных.
Принятой энергии диссоциации соответствует
г, 0) = 53,975+ 25 кДж • моль.
Ш>О3(к, ж). Термодинамические свойства кристаллического и жидкого диоксида рубидия
в стандартном состоянии при температурах 100—2000 К приведены в табл. 1199.
Значения постоянных, принятые для расчета термодинамических функций, приведены в табл.
48.1. В справочнике за стандартное состояние RbO2(K) в интервале 0—15 К принята моноклинная
Т '-модификация, в интервале 15—178 К — ромбическая у-модификация, в интервале 178—423- К —
тетрагональная ^-модификация (структурный тип СаС2) и в интервале 423—813 К — кубическая
а-модификация (структурный тип NaCl) [4049, 4325].
При Т < 298,15 К термодинамические функции RbO2(K) вычислены по результатам измерений
теплоемкости, выполненных Пауковым и др. [766, 767] A2,9—297,8 К) на образце чистотой 99,8%.
Термографический анализ, проведенный авторами [766], показал, что фазовые переходы в области
15 и 178 К не имеют скрытой энтальпии и являются переходами второго рода. Экстраполяция
теплоемкости проводилась графически и привела к значению S°A2 K)=3,0 Дж-К~1-моль.
Погрешности принятых значений >S°B98,15 К) и #°B98,15 К)—Н°@) оцениваются в 0,6 Дж-К-1Х
Хмоль и 0,05 кДж-моль соответственно.
Экспериментальные данные по термодинамическим свойствам RbO2(K) при Т ]>.298,15 К
в литературе отсутствуют. Для теплоемкости p-RbO2 в интервале 298,15—423 К принято уравнение
(см. табл. 48.1), выведенное по значению теплоемкости при 298,15 К (см. табл. 48.1) и значению
С°D23 К)=88 +4 Дж«К-моль; последнее оценено так же, как для NaO2(K). Температура
полиморфного превращения D23 +5 К) принята по термографическим данным Ценципер и
Добролюбовой [1101] г. Энтальпия этого превращения принята равной нулю, поскольку оценить ее не
представляется возможным. Теплоемкость a-RbO2 (88+5 Дж-К~1-моль) оценена так же, как
для <х-КО2 (см. выше).
Температура плавления (813 + 10 К) принята по термографическим данным, полученным в
работе [1101 ]. Измерения Центнершвера и Блюменталя [1774] привели, как и для других соединений,
к заниженной величине F85 К) и не учитывались. Энтальпия плавления B1 +4 кДж -моль'1)
оценена на основании предположения о равенстве энтропии плавления RbO2 и КО2. Значение
C°(RbO2, ж)=100+10 Дж-К-моль~1 оценено по принятой методике (см. т. 1, гл. 2).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 0,4, 8
и 16 Дж-К~1-моль.
Таблица термодинамических функций RbO2(K, ж) публикуется впервые.
Константа равновесия реакции RbO2(K, ж) = КЬ(г)+2 О (г) вычислена с использованием
значения Дг#°@)=855,447 +2,2 кДж-моль, соответствующего принятой в справочнике^энтальпии
образования , ','t.J
1 A^RbOj, к, 298,15 К) = —279,1+ 2,0 кДж-моль-1..:
Принятая величина основана на измерениях Д'Орацио и Вуда [2096] энтальпии растворения
RbO2(K)+20 000,5 Н,О(ж)=11ЪОН(р-р, 20 000 НаО)+% О2(г); Aff?#°B98,15 K)=-59,0±
+1,8 кДж'Моль, шесть измерений с двумя образцами. Основным источником погрешности
являются примеси в образцах RbO2 (до 5% RbHCO3).
В работе [1101] методом Д ТА у RbO2 обнаружено еще два небольших термически эффекта — экзотермический
при 628 К и эндотермический при 723 К; природа этих эффектов не установлена. В справочнике эти переходы
не учитываются.
430
Рубидий и его соединения ШJО(к, ж), Rb2O(r)
Rb2O(K, ж). Термодинамические свойства кристаллического и жидкого оксида дирубидия
в стандартном состоянии при температурах 298,15—2500 К приведены в табл. 1200.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние Rb2O(K) в интервале 0—543 К принята
кубическая у-модификация (структурный тип флюорита, CaF2), в интервале 543—613 К — кубическая
Р-модификация и в интервале 613—778 К — гексагональная а-модификация [4535 ]. В литературе
отсутствуют какие-либо экспериментальные данные о термодинамических свойствах В.Ь2О(к, ж).
Значения термодинамических функций при 298,15 К, приведенные в табл. 48.1, оценены с помощью
эмпирических методик [1094, 1741, 3127, 3198] с учетом экспериментальных данных по
теплоемкости оксидов Li, Na, Cs. Погрешности принятых значений 5°B98,15 К) и #"B98,15 К)—Н°@)
оцениваются в 6 Дж-К-моль и 0,8 кДж-моль соответственно.
Для теплоемкости Y-Rb2O в интервале 298,15—543 К принято уравнение (см. табл. 48.1),
выведенное по значениям теплоемкости С°B98,15 К)=74+4 Дж-К-моль и С°>E43 К)=92,5 +
+6 Дж-К~1-моль; последнее оценено на основании предположения о равенстве теплоемкостей
T-Rb2O и *f-Na2O в точках фазовых переходов. Температуры полиморфных превращений E43+5 К)
и F13 +10 К) приняты по термографическим измерениям Тузена и Калле [4535]. Соответствующие
этим превращениям энтальпии @,7 +0,3 кДж-моль) и D +2 кДж-моль) оценены на основании
предположения о равенстве энтропии превращения Rb2O(K) и Ка2О(к). Теплоемкость р- и a-Rb3O
в интервалах 543—613 К и 613—778 К A00 +10 Дж-К~1-моль~1) оценена так же, как для К2О(к).
Температура плавления G78 +5 К) найдена в результате термографического исследования
Тузена и Калле[4535]. Энтальпия плавления B0+2 кДж-моль) оценена на основании
предположения о равенстве энтропии плавления Rb2O и Na2O. Теплоемкость КЬ2О(ж) A00+10 Дж-К"хх
Хмоль) оценена так же, как для Ка2О(ж) (см. выше).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 4, 12 и
20 Дж •К-моль соответственно. Расхождения между термодинамическими функциями,
приведенными в табл. 1200 и в справочнике Барина и Кнакке [1395] (Г ^ 900 К), достигают 10 —
18 Дж-К~1-моль и обусловлены различием оценки значений 5°B98,15 К) и Cl°(Rb20, к).
Принятая в справочнике энтальпия образования
Ayff°(Rb20, к, 298,15 К) = — 338 + 8 кДж • моль
основана на энтальпии реакции Rb2O(K)+6451 HaO(jK)=2RbOH(p-p, 3225 Н2О), Дв4#°B91,4 К) =
=—335,0 кДж-моль, измеренной Ренгадом [3975, 3976]. Пересчет этой величины к 298,15 К
и бесконечному разведению дает AagiT°(oo H2O, 298,15 К) =—338,2 кДж -моль, чему соответствует
принятое значение энтальпии образования Rb2O(K). Его погрешность оценена на основании
расхождения других величин, измеренных в работах [3975, 3976], с современными данными.
Энтальпия растворения Rb2O в воде была также измерена Бекетовым [14561. Найденная по данным этой
работы энтальпия образования (—383 кДж -моль) неточна вследствие наличия больших
количеств неучтенных примесей в образцах.
Давление пара в реакции Rb2O(K, ж)=Rb2O(г) вычислено с использованием значения \гН°@) =
= As#°(Rb20, к)=230,448+22 кДж-моль, соответствующего принятым в справочнике
энтальпии образования Rb2O(K) и энтальпии атомизации Rb2O(r).
Rb2O(r). Термодинамические свойства газообразного оксида дирубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1201.
Молекулярные постоянные, использованные для расчета термодинамических функций Rb2O(r),.
приведены в табл. 48.6. Спикер и Эндрюс [4365] на основании величины изотопического
расщепления частоты колебания v3 нашли, что наиболее вероятное значение угла Rb—О—Rb лежит
в интервале 160—180°. Другие сведения о структуре Rb2O в литературе отсутствуют. В
справочнике на основании данных [4365], а также аналогичных данных для молекул К2О и Cs2O,
принимается, что молекула Rb2O в основном электронном состоянии Х1А1 имеет угловую структуру
(симметрия С2A) (см. примечание к тексту по Cs2O (г)). Приведенное в табл. 48.6 произведение
моментов инерции рассчитано по структурным параметрам r(Rb—О)=2,32+0,03 А (оценка
по величине r(Rb—О) в молекуле RbOH) и 4Rb—О—Rb=160+10° (рекомендован в
работе [4365]). Погрешность значения IJbIc оценивается 1 -Ю13 г3-см6. Приведенное в табл. 48.6
значение v3 найдено Спикером и Эндрюсом [4365] при исследовании ИК-спектра молекул Rb2Or
изолированных в матрице из Аг. Частота vx рассчитана по величине v3 и соотношениям простого
431
Rb2O(r)
Глава 48
Таблица 48.6. Молекулярные постоянные, а также о (рх = 1), принятые для расчета
термодинамических функций Rb2O, Rb2O2, RbOH, Rb2O2Ha, Rb2Fa, RbeCl2, Rb2Br2,
Rbal2, Rb2S04, RbNO2, RbNO3, RbBOa
Молекула
Состояние
Rb2O ТЫг
Rb2O2 ХхАд
RbOH Xi 2+
Rb2OaH2 XхАд
Rb2F2 fr-A9
Rb2Cl2 X>Ag
RbaBr2 XхA g
Rb2I2 .?M,
Rb2SO4 -?44
RbNO2 XXAX
RbNO3 1^!
RbBO2 XiA'
Примечание. Ниже все
vx
170
850
354
3700
287
185
127
101
960
1330
1456
1965
v2
75 473
350 250
309 B) 3700
306 111
115 195
77 120
65 89
55 74
510 130
790 183
1033 720
1082 586
постоянные приведень
RbOH: a приведено значение
Rb2O3H2: a vlo=244, vu
Rb2SO4: a vle=195 B),
=3700
vu=4C
/ -1039 т -см2.
v12=253.
B).
V4
_
60
—
290
99
54
39
31
460
1213
180
581
t В CM"
CM~
_
389
—
218
270
160
120
98
1125
130
1293
225
i.
v7
-8
v9
i
_^_ ^_ _j_
180 — — —
— — __ —
278 100 296 284a
233 — — —
160 — — —
125 — — —
103 — — —
636 215 1098B) 608 B) a
40 — — —
720 130 830 40
70 — — —
VbVio1"
r3 • cm6
9 -103
70 -103
13,36 a
18-10*
18 -10*
11-106
49 -105
16-10е
1,5 -106
85 -102
27 -103
10 -103
a
2
4
1
4
4
4
4
4
4
2
2
1
поля валентных сил. Для частоты v2 принято среднее из значений, рассчитанных в предположении,
что отношение силовой постоянной связи Rb—О и деформационной силовой постоянной
сохраняется таким же, как в молекулах Ы2О и RbOH г. Погрешности принятых частот оцениваются
в 30 см для vj и в 20 см для v2 и v3.
Сведения о возбужденных электронных состояниях Rb2O в литературе отсутствуют.
Термодинамические функции 'ПЬ2О(г) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Погрешности рассчитанных термодинамических
функций обусловлены неточностью оценки молекулярных постоянных, в частности, структуры этой
молекулы, пренебрежением возбужденными электронными состояниями, а также приближенным
характером расчета; они составляют 10, 20 и 30 Дж-К-моль в значениях Ф°(Г) при 298,15,
3000 и 6000 К соответственно.
Таблица термодинамических функций Rb2O(r) рассчитана впервые.
Константа равновесия реакции Rb2O(r)=2Rb(r)+O(r) вычислена с использованием принятой
в справочнике энтальпии атомизации
ДгЯ°@) = 515 + 20 кДж • моль.
Принятая величина была оценена сопоставлением энтальпий атомизации в рядах М2О(г) и
МОН(г), где M=Li, Na, К, Rb, Gs. В работе Нормана и Стейли [3680] масс-спектрометрическим
методом было исследовано равновесие реакции Rb2O(r)=2Rb(r)+1/2O2(r) A010—1160К, данные
представлены в виде уравнения). По данным этой работы были вычислены значения энтальпии
атомизации, равные 468 кДж -моль (III закон) и 459 кДж -моль (II закон). Эти величины плохо
согласуются с имеющимися данными для молекул М2О. В работе Гусарова [343] высказывается
предположение, что в работе [3680] могли быть допущены неточности в определении парциального
давления кислорода, что должно привести к заниженному значению вычисляемой энтальпии
атомизации.
Принятой энтальпии атомизации соответствует
bfH°(Rh20, г, 0) = —103,833 + 20 кДж • моль.
1 /r=0,99-105, fa/r2=0,016-106 (из Ы2О) и 0,018-106 (из RbOH), в дин-см~1.
432
Рубидий и его соединения
КЬ2О2(к, ж)
Rb2O2(K, ж). Термодинамические свойства кристаллического и жидкого диоксида дирубидия
в стандартном состоянии при температурах 298,15—2000 К приведены в табл. 1202.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние ШJО2(к) в интервале 0—398 К принята
ромбическая ^-модификация, а в интервале 398—843 К — кубическая а-модификация [1845].
Какие-либо сведения о термодинамических свойствах Rb2O2(K, ж) в литературе отсутствуют.
Значения термодинамических функций при 298,15 К, приведенные в табл. 48.1, оценены так же,
как для К2О2(к) (см. выше). Погрешности принятых значений ?°B98,15 К) и #°B98,15 К)—#°@)
оцениваются в 17 Дж-К-моль и 1,5 кДж-моль~х соответственно.
Для теплоемкости |3-Rb2O2 в интервале 298,15—398 К принято уравнение (см. табл. 48.1),
выведенное по значениям теплоемкости С°B98,15 К)=93 +6 Дж-К-моль~1 и С°C98 К)=117 +
+12 Дж-К-моль; последнее оценено так же, как для К2О2(к). Температура полиморфного
превращения C98 ±5 К) принята по термографическим данным, полученным Шретьеном и Алла-
маньи [1845]. Энтальпия полиморфного превращения принята равной нулю, поскольку оценить
ее значение не представляется возможным. Теплоемкость a-Rb2O2 A17+15 Дж-К~1-моль~1)
оценена на основании экспериментальных данных по энтальпии a-Na2O2(K). Температура
плавления (843+50 К) найдена в работе Центнершвера и Блюменталя [1774], а энтальпия плавления
B1+4 кДж-моль) оценена так же, как для Na2O2. Теплоемкость В.Ъ2Ог(ж) A34+15 Дж>К~1Х
Хмоль") принята на основании оценки (см. т. 1, гл. 2).
Погрешности вычисленных значений Ф°(Т) при температурах 298,15, 1000 и 2000 К
оцениваются в 12, 20 и 30 Дж-К~1-моль соответственно.
Таблица термодинамических функций Rb2O2(K, ж) публикуется впервые.
В справочнике принята энтальпия образования
^fH°(Rb2O3, к, 298,15 К) = —410 + 20 кДж.моль',
основанная на приведенных в табл. 48.7 исследованиях равновесия реакций с участием Rb2O
2O2.
Таблица 48.7. Результаты определений A^Br°(Rb202, к, 298,15К) (в кДж-'моль)
Авторы
Центнершвер, Блюменталь, 1933
[1590, 17741
Краус, Петрочелли, 1962 [3101]
Метод
Компенсационный и динамический, 211Ь2О3(ж)=
=2Rb2Oa(}K)+O2(r), 853—1058 К, 15 точек
Динамический, 2КЬ2О2(ж)=2Rb2OEK)+O2(r),
1069—1156 К, 4 точки
Статический, 2RbO2(K)=Rb2O2(K)+O2(r), 553—
633 К, 30 точек
То же, 2Rb2Oa(K)=2RbaO(K)+O2(r), 573—633 К,
16 точек
II закон
—451 ±40
—458+30
—474+13
—369 ±15
, к, 298,15 К)
III закон
—462 ±35
—428+30
-461 ±10
-401 ±20
Предполагаемый в работах [1590,1774] механизм окисления Rb2O2(K) включает фазу Rb2O3(K).
Существование этой фазы оспаривается в более поздней работе [3101], где она не была обнаружена
и было высказано предположение, что окисление Rb2O2(K) идет до RbO2(K). Точка зрения авторов
работы [3101] представляется более убедительной (см. текст по энтальпии образования К2О2(к)).
Однако расчет энтальпии образования Rb2O2 по двум реакциям, исследованным в работе [3101],
приводит к значениям, отличающимся на 60 кДж-моль, что свидетельствует о наличии
систематических ошибок в работе [3101].
Результаты исследования реакции восстановления 2Rb202(K)=2Rb20(K)+02(r) в работах
{1590, 1774, 3101] оказались в хорошем соответствии, и принятое значение основывается на
величинах, полученных этим путем. Погрешность принятого значения определяется в основном
неточностью использованных в расчетах термодинамических функций.
Давление пара в реакции Rb2O2(K, ж)=Rb2O2(г) вычислено с использованием значения
Arffo@)=As#°(Rb2O2, к, 0)=196,990+36 кДж'Моль", соответствующего принятым в
справочнике энтальпиям образования Rb2O2(K) и атомизации Rb2O2(r).
433
Rb2O2(p), Ш>Н(к, ж) Глава 48
Rb2O2(r). Термодинамические свойства газообразного диоксида дирубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1203.
Молекулярные постоянные, использованные для расчета термодинамических функций Rb2O2(r),
приведены в табл. 48.6. Структура молекулы Rb2O2 экспериментально не исследовалась. По
аналогии с молекулой Li2O2 для Rb2O2 в основном состоянии Х1Ад принято плоское ромбическое
строение (симметрия Z?2A) со связью кислород—кислород в цикле. Приведенное в табл. 48.6
произведение моментов инерции рассчитано по оцененным структурным параметрам r(Rb—О)=2,4+0,1 А
(из RbaF2, RbF и RbO) и г(О—О)=1,50+0,1 А (из Li2O2). Погрешность величины 1А1в1с
составляет 3-1013 г3-см6.
Частота \ найдена Эндрюсом [1328] при исследовании ИК-спектра молекул Rb2O2,
изолированных в матрице из Аг. Остальные частоты Rb2O2 в спектрах не наблюдались, и их величины
оценены в справочнике так же, как для К2О2. Погрешности приведенных в табл. 48.6 значений
частот могут достигать 100 см для vx, 50—60 см для v2, v3 и v6, 20 см для v4 и v6.
Сведения о возбужденных электронных состояниях Rb2O2 в литературе отсутствуют.
Термодинамические функции Rb2O2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A.10), A. 122)—A. 124), A. 128)г
A. 130) без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным характером расчета,
они составляют 8, 15, 20 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К
соответственно.
Таблица термодинамических функций Rb2O2(r) публикуется впервые.
Константа равновесия реакции Rb2O2(r)=2Rb(r)-f-2O(r) вычислена с использованием принятой
в справочнике энтальпии атомизации
Дг#°@) = 867 ± 30 кДж • моль.
Принятая величина была оценена сопоставлением энтальпий атомизации в рядах М2О2(г)г
МаО(г) и МОН(г), где M=Li, Na, К, Rb и Cs.
Принятой энтальпии атомизации соответствует
" a, г, 0) =—209,050 ± 30 кДж • моль.
RbH(K, ж). Термодинамические свойства кристаллического и жидкого гидрида рубидия в
стандартном состоянии при температурах 298,15—1200 К приведены в табл. 1204.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние RbH(K) в интервале 0—858 К принята
кубическая модификация (структурный тип NaCl). В литературе отсутствуют какие-либо данные о
теплоемкости и энтальпии RbH(K, ж), ввиду чего термодинамические функции были вычислены на
основании оценок, выполненных по соответствующим постоянным фторидов щелочных металлов,
1ЛН(к, ж) и NaH(K, ж), для которых имеются экспериментальные данные; все эти вещества
относятся к одному и тому же структурному типу (NaCl). Значение <S°B98,15 К) было оценено на осно:
вании сопоставления принятого значения A^i/o(RbH, к) и значения, найденного по методу III
закона из данных [940, 2619, 2620] по давлению диссоциации RbH(K, ж). Погрешности значений
5°B98,15 К) и #°B98,15 К)—Н°@), приведенных в табл. 48.1, оцениваются в 3 Дж-К-^моль'1
и 1 кДж-моль соответственно.
Уравнение для теплоемкости RbH(K), приведенное в табл. 48.1, было вычислено п© значениям
С;B98,15 К)=39,3 Дж-К^-моль, С;E00 К)=49,8 Дж-К^-моль и С;(858 К)=63 Дж-К^Х
Хмоль, оцененным таким же способом, как и в случае КЩк). Точность аппроксимации
теплоемкости этим уравнением составляет примерно 10%.
Температура плавления (858 +5 К) при равновесном давлении водорода A48 атм) принята го
результатам термографического исследования Скуратова и др. [940] (воспроизводимость
определения оценена авторами в 2°). Исследованный образец был получен при 860—890 К и давлениях
Н2 в 150—200 атм из металла чистотой 99,9% и водорода, содержавшего N2 и О2 в количествах
не более 10~4%; образец содержал не менее 99,5% RbH. Энтальпия плавления B2+3 кДж-моль)
принята по расчетам Скуратова и др. [940], основанных на данных по давлению диссоциации
RbH, полученных авторами [940] и имеющихся в литературе. Значение теплоемкости жидкого
434
Рубидий и его соединения RbH(r)
RbH E6+8 Дж-К^-моль) оценено с учетом экспериментальных данных по теплоемкости
1ЛН(ж).
Погрешности вычисленных значений Ф°(Т) для БЬН(к, ж) при 298,15, 500 и 1000 К оцениваются
в 2, 4 и 8 Дж-К-моль~1 соответственно.
Таблица термодинамических функций RbH (к, ж) публикуется впервые.
Принятое в справочнике значение энтальпии образования
bfH°(RbR, к, 298,15 К) =—52,30 ± 0,15 кДж • моль
основано на выполненных Ганном [2525] калориметрических измерениях энтальпии растворения
Б.Ь(к) и RbH(K) в воде. Принятое значение является разностью этих величин.
Таблица 48.8. Результаты определений A/-ff°(RbH, к, 298,15К) по исследованиям равновесия
2НЬН(к, ж) = 2ИЬ(ж) + Н2(г) (в кДж • моль)
Авторы
Хакшпиль, Барокко, 1938 [1401,
2543]
Херолд, 1951 [2619, 2620]
Скуратов и др., 1976 [940]
Метод
Статический, RbH(K), 463—593 К, 14 точек
То же, RbH(K), 497—623 К, 6 точек
То же, НЬН(ж), 870—973 К, 8 точек
A/i?o(RbH, к, 298,15 К)
II закон
-40 ±7
—52+2
-57+6
III закон
-54,1 +1,0
-52,4 ±0,1
-52,4+0,1
В табл. 48.8 приведены значения энтальпии образования ИЬН(к), вычисленные по результатам
исследований равновесия реакции 2ИЬН(к, ж)=2КЬ(ж)+Н2(г). Приведенные в таблице
погрешности отражают только воспроизводимость измерений. Неточность термодинамических функций
приводит к дополнительным погрешностям, составляющим от 1,5 кДж-моль для измерений при
500 К до 7 кДж-моль для измерений при 1000 К. Как видно из таблицы, результаты расчетов
равновесия диссоциации гидрида рубидия и калориметрических измерений находятся в
удовлетворительном согласии.
Давление пара гидрида рубидия в реакции ИЬН(к, ж)=КЬН(г) вычислено с использованием
значения &rH°@)= AaH°(Rbli, к, 0)=171,903+4,0 кДж-моль, соответствующего принятым
в справочнике энтальпиям образования RbH(K) и энергии диссоциации RbH(r).
RbH(r). Термодинамические свойства газообразного гидрида рубидия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1205.
В табл. 48.4 представлены молекулярные постоянные RbH, принятые на основании анализа
результатов исследований электронного спектра этой молекулы. В электронном спектре RbH
(а также RbD) наблюдалась только одна система полос ^41E+<±:Z1E+ [1411, 2379, 2378, 2726].
Комбинирующие состояния коррелируют с пределами RbBP)+HBJSl) и RbB5f)+HB<SI)
соответственно. По аналогии с молекулами NaH, KH и CsH в справочнике принимается, что другие
состояния RbH, связанные с этими состояниями, являются отталкивательными. Валентные состояния
а'Д и В1^*, коррелирующие с атомами RbBZ>)+HB<S), так же как у молекулы CsH, могут быть
связанными, имея энергии возбуждения 32 000—34 000 см. Эти состояния, по-видимому, должны
быть мало стабильными и поэтому не включены в табл. 48.4. s|
Вращательные постоянные RbH в состоянии ZXE+, приведенные в табл. 48.4, получены Гей-
доном и Пирсом [2379] в результате анализа вращательной структуры 34 полос системы АгЛ+—
—ХХЕ + C <С v' <I 14, v" ^ 5). Колебательные постоянные в состояниях ХХЕ+ и АгЛ+ вычислены
Бартки [1411] по началам полос, найденным в работе [2379], после того как автор [1411] смог
однозначно установить нумерацию полос на основании сравнения изученного им спектра RbD
(полосы 11—1 и 10—1) с известным спектром RbH1. Постоянные, рассчитанные автором [1411]
для уровней v' t^L 9, хорошо описывают начала всех наблюдавшихся полос. Вращательные по-
.стоянные в состоянии Л1?+, приведенные в табл. 48.4, получены при подготовке справочника
1 Исследование спектра лазе рной флуоресценции RbH в работе Хсиеха и др. [2726] (t>" < 12) привело к близким
значениям колебательных постоянных в состоянии X1 2+.
435
НЬОН(к, ж) Глава 48
обработкой данных [2379] с учетом колебательной нумерации полос, установленной в работе
A411]. Состояние -Ах?+ у молекулы RbH, так же как у молекул гидридов других щелочных
металлов, имеет аномальный характер. Кривые зависимости AG(v+V2) и Bv от v проходят через
максимум при и=11 и v=7 соответственно.
Термодинамические функции RbH(r) были рассчитаны по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения @ви и ее производные вычислялись по уравнениям A. 90)—A. 92) с учетом одного
возбужденного состояния. Статистические суммы по колебательно-вращательным уровням
состояний ХХЕ+ и 41?+ и их производные находились по уравнениям A. 70)—A. 75)
непосредственным суммированием по уровням энергии. В расчете учитывались все уровни, ограниченные
предельными кривыми диссоциации этих состояний. Колебательно-вращательные уровни вычислялись
по уравнениям A.65) и A.64а). Значения коэффициентов YkI в этих уравнениях приведены
в табл. 48.5 вместе со значениями vmta, Jlim и коэффициентами aif аппроксимирующими
предельные кривые диссоциации. Для соблюдения условий A. 50) и A. 59) добавлены коэффициенты
Yi0 и Уоа для состояния Хх?+ и коэффициенты Y&a и У03 для состояния А1!,*.
jflorp ности вычисленных термодинамических функций при Т ^ 3000 К обусловлены
главным образом неточностью фундаментальных постоянных и не превышают 0,03 «Дж-К~1-моль.
При Т>3000К становится существенной неточность: экстраполяции колебательно-вращательных
уровней и погрешность в значении Ф°F000 К) может составить 0,2—0,3 Дж«К-моль.
Пренебрежение состояниями а3 А и 5ХЕ+, а также различием постоянных разных изотопомеров молекулы
RbH практически не влияет на точность расчета.
1Ранее термодинамические функции RbH(r) вычислялись Харом и др. [2537] (Т <[ 5000 К)
и в работе Шнейдера [4197а] (Qm для 1000 ^ Т 4^ 3000 К). Расхождения данных [2537] и приведенных
в табл. 1205 достигают 0,4, 0,8 и 4 Дж-К^-моль в значениях Ф°(Т), S°(T) и С°р(Т) и объясняются
главным образом различием методов расчета.
Константа равновесия реакции RbH(r)=Rb(r)+H(r) вычислена с ^использованием значения
D0(RbH)=176±4 кДж.моль-1;»^ 700+300 см,
принятого на основании сопоставления результатов линейной экстраполяции колебательных
уровней состояния Х*?+ молекулы RbH A4 980 см) и молекул гидридов других щелочных
металлов с принятыми значениями их энергий диссоциации. Близкое значение получается при
рассмотрении зависимости величин D0(MH) и D0(M2) в ряду Li—Na—К—Rb—Cs.
Принятой энергии диссоциации соответствует
AfH°(RhE, г, 0) = 122,226 ± 4,0 кДж • моль.
НЬОН(к, ж). Термодинамические свойства кристаллического'и жидкого гидроксида рубидия
в'стандартном состоянии при температурах 298,15—3000 К приведены в табл. 1206.
m'j Значения постоянных, принятые для расчета термодинамических функций, приведены
в^табл. 48.1. За стандартное состояние RbOH(K) приняты модификации, устойчивые в интервалах
температур 0—508 и 508—658 К; данные об их кристаллических структурах в литературе
отсутствуют. В литературе отсутствуют также какие-либо сведения о термодинамических свойствах
RbOH(K, ж). Принятые значения ?°B98,15 К), #°B98,15 К)-#°@) и С;B98,15 К) (см. табл. 48.1)
оценены с помощью эмпирических методик [1094, 1741, 3127, 3198], а также на основании
сравнения соответствующих данных для гидроксидов Li, Na и К. Погрешности принятых значений
?°B98,15К) и Я°B98,15 К)—Я°@) оцениваются в 8 Дж-К-^моль и 1 кДж-моль
соответственно. Приведенные в таблице значения С°B98,15 К) и #°B98,15 К)—#°@) согласуются с
оценками Краснова и Филиппенко [570] G1 Дж-К-моль~1 и 15,8 кДж-моль).
Для теплоемкости в интервале 298,15—508 К принято уравнение (ем. табл. 48.1) полученное
по значениям С°рB98,15 К)=69+4 Дж-К'^моль и СрE08К)=81,5+5 Дж-К-^моль; последнее
принято равным экспериментальному значению теплоемкости КОН в точке фазового перехода.
Температура полиморфного превращения E08 +2 К) принята по согласующимся между собой
данным [1895, 2795, 3765, 4041, 4531]. Результаты измерений [744, 866, 1101, 2631, 2632] D78—
518 К) менее надежны. Энтальпия полиморфного превращения E,4+0,5 кДж-моль) принята
по термографическим данным Решетникова и Баранской [866]. В работах [2631, 2632] было
получено более высокое значение G,1 кДж-моль). Теплоемкость в интервале 508—658 К принята
равной теплоемкости высокотемпературной фазы КОН(к) (80+6 Дж«К~1-моль~1). Температура
436
Рубидий и его соединения RbOH(r)
плавления F58+3 К) и энтальпия плавления (8,9+0,8 нДж-моль) были определ..ны на основании
термографических измерений [866]. Принятое значение Тт согласуется в пределах погрешности
с результатами работ [64, 1101, 1600, 1895, 3765, 4041, 4531]. Близкое значение АтН было
получено в работах [2631, 2632] F,8 кДж-моль). Теплоемкость жидкого гидроксида рубидия (83 +
+8 Дж-К-моль) принята равной теплоемкости КОН(ж).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 5,
11, 20 и 25 Дж-К-моль~1 соответственно.
Таблица термодинамических функций ШЮН(к, ж) публикуется впервые.
Принятая в справочнике энтальпия образования
-1
AfH°(RbOH, к, 298,15 К) = —418,8 + 4,0 кДж • моль
основана на измерении энтальпии растворения RbOH(K) в воде, выполненном Форкраном [2294J
(разведение 111Н2О, 288,15 К, одно измерение, —59,7 кДж-моль). Пересчет этой величины
к стандартным условиям дает Аа1Н°(соН20, 298,15 К)=—62,3 кДж-моль. Ее погрешность была
оценена сопоставлением измерений, выполненных Форкраном с гидроксидами других щелочных
металлов, с результатами более современных работ (см. тексты по выбору энтальпии образования
ЫОН(к), NaOH(K) и КОН(к)).
Давление пара гидроксида рубидия в реакции ШЮН(к, ж)=11ЬОН(г) вычислено с
использованием значения ArH°@)—AsH (RbOH., к, 0)=180,236+15 кДж-моль, соответствующего
принятым в справочнике энтальпиям образования RbOH в кристаллическом и газообразном
состояниях.
RbOH(r). Термодинамические свойства газообразного гидроксида рубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1207.
Молекулярные постоянные, использованные для расчета термодинамических функций
RbOH(r), приведены в табл. 48.6. Исследования микроволнового [3255, 3425] и инфракрасного
[87, 1246] спектров показали, что в основном электронном состоянии Х1!,* молекула RbOH
имеет линейную структуру (симметрия Cmk). Приведенный в таблице момент инерции вычислен
по постоянным ?00O0(86RbOH)=0,209 817 и 500O0(87RbOH)=0,209 023 см, полученным Мацу-
мура и Лайдом [3425]. Погрешность значения /0 составляет 3-10~41 г «см2. Микроволновым
данным соответствуют структурные параметры ro(Rb—О)=2,316 А и г0(О—Н)=0,913 А. Принятые
значения vx и v2 найдены Аккуиста и Абрамовицем [1246] и Беляевой [87] в ИК-спектре
молекул RbOH, изолированных в матрице из Аг х, а не наблюдавшаяся в спектре частота v3 принята
приближенно равной соответствующей частоте GsOH. Спинар и Маргрейв [4367] в
инфракрасном спектре газообразного RbOH при 1100—1200 К получили для vx значение 365+20 см,
которое в пределах погрешности согласуется с принятой величиной. Погрешности частот vx, v2 и v3
оцениваются в 20, 20 и 100 см. По аналогии с молекулами RbF, LiOH и NaOH можно ожидать,
что изомер HRbO, а также возбужденные электронные состояния RbOH имеют энергии выше
20 000 см.
Термодинамические функции RbOH(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),;
A. 122)—A. 124), A. 126), A. 129) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных электронных состояний. Погрешности вычисленных
термодинамических функций обусловлены приближенным характером расчета, а также неточностью
принятых значений молекулярных постоянных и составляют 1, 4 и 8 Дж>К~1-моль~1 в значениях
Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций RbOH(r) публикуется впервые.
Константа равновесия реакции RbOH(r)=Rb(r)+O(r)+H(r) вычислена с использованием
значения Дг.й"°@)=782,009+15 кДж-моль, основанного на принятом в справочнике значении
энтальпии образования
, г, 0) = —237 + 15 кДж . моль.
Значения частот vj и v2) наблюдавшихся Беляевой в спектрах молекул RbOH, изолированных в матрицах из Кг
и Хе, близки к принятым.
437
Rb2O2H2(r), RbF(K, ж)
Глава 48
Принятая величина является средней из представленных в табл. 48.9 результатов
исследований. Приведенные в таблице погрешности включают воспроизводимость измерений и
погрешности использованных в расчетах термодинамических функций и энтальпий образования.
Таблица 48.9. Результаты определений Ayfi"°(RbOH, г, 0) (в кДж • моль)
Авторы
Енсен, Педли, 1966 [2855]
Коттон, Дженкинс, 1969 [1941]
Келли, Педли, 1971 [2965]
1 Панченков, 1976 [755]
Метод
Спектрофотометрия в пламенах H2+O«+N2,
Rb(r)+H2O(r)=RbOH(r)+H(r), 2475 К, 1 точка
То же, 1570 К, 39 измерений
То же, пламена Н2+О2+СО2, 1950—2750 К,
32 точки
Масс-спектрометрический, Rb (r)+CsOH (г)=
=Cs(r)+RbOH(r)
A^°(RbOH, г, 0)
II закон
III закон
— —228 ±15
— —248 +15
—237 —239 +15
_ _235 +15
Rb2O2H2(r). Термодинамические свойства газообразного дигидроксида дирубидия в
стандартном состоянии в интервале температур 100—6000 К приведены в табл. 1208.
Молекулярные постоянные, использованные для расчета термодинамических функций
Rb2O2H2(r), приведены в табл. 48.6. Произведение моментов инерции в основном электронном
состоянии Х^Ад вычислено для плоской ромбической структуры со связями ОН,
расположенными на одной прямой (симметрия D2h), и с Д О—Rb—0=93+10°, r(Rb—О)=2,50+0,10 А,
г(О—Н)=0,97+0,03 А. Структурные параметры рассчитаны Беляевой [87] с использованием
ионной модели молекулы. Погрешность значения IaIbIc составляет 5-Ю13 г3-см6. Частоты
v±—v5, v8 активны в спектре КР, частоты ve, v7,
в ИК-спектре. Валентным колебаниям
кольца соответствуют частоты v2D?), \{Blg), \0(В2и), v12(jB3m), плоской деформации кольца —
v3(Ag), неплоской деформации кольца — vi(Blu), валентным колебаниям внешних связей ОН—
\(Ад), Vu(B3l), плоской деформации ОН — vi(Blff), v9B?2a), неплоской деформации ОН — ^вE1а),
vs(B2g). Значения всех частот приняты по работе Беляевой [87]. Частоты v6, v7, v9, v10, v12
наблюдались в ИК-спектре молекул Rb2O2H2, изолированных в матрице из Ar, vx и vn оценены как
характеристические колебания ОН, а остальные частоты вычислены с использованием ионной
модели молекулы. Погрешности в значениях частот v± и vn оцениваются в 150 см, частот v2,
V4; V5 и ve — в 50—60 см, частоты v3 — 30 см, остальных частот в 20 см. Сведения о
возбужденных электронных состояниях Rb2O2H2 в литературе отсутствуют.
Термодинамические функции Rb2O2H2 вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний. Погрешности рассчитанных термодинамических
функций обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета и составляют 8, 15 и 20 Дж-К~1-моль в значениях Ф°(Г) при
298,15, 3000 и 6000 К.
Таблица термодинамических функций Rb2O2H2(r) публикуется впервые.
Константа равновесия реакции Rb2O2H2(r) =2Rb(r)+2O(r)+2H(r) вычислена с
использованием значения ДгЯ°@)=1735,018+30 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
kfH°(Rb202H2, г, 0)== —645 ± 30 кДж • моль.
Принятая величина основана на масс-спектрометрическом исследовании равновесия реакций
2NaOH(r)+Rb2O2H2(r)=2RbOH(r)+Na2O2H2(r) (823 К, 3 измерения, A^RbAH,, г, 0)=
= -646+40 кДж-моль) и 2KOH(r)+Rb2O2H2(r)=2RbOH(r)+K2O2H2(r) G28 К, 3 измерения,
A/ff°(Rb202H2, г, 0)=—643+30 кДж-моль), выполненном Скунмейкером и Портером [4210].
RbF(K, ж). Термодинамические свойства кристаллического и жидкого фторида рубидия
в стандартном состоянии при температурах 298,15—3400 К приведены в табл. 1209.
438
Рубидий и его соединения RbF(K, ж)
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние RbF(K) в интервале 0—1068 К принимается
кубическая модификация (структурный тип NaCl) x.
Экспериментальные данные по теплоемкости RbF при Т <^ 298,15 в литературе отсутствуют,
за исключением работы Бренстеда [1683], который определил среднюю теплоемкость RbF в
интервале 273—293 К. Принятое значение С°B98,15 К) основано на этих измерениях. Значение
^°B98,15 К) принято по оценке Ощерина и Федотовой [745], выполненной по зависимости
энтропии фторидов щелочных металлов от главного квантового числа валентных электронов.
Результаты других оценок [1088, 2963, 3125] G5—82 Дж-К-моль~1) менее надежны, хотя и приводят
к близким величинам. Значение #°B98,15 К)—Н°@) получено интерполяцией между принятыми
в справочнике на основании экспериментальных данных значениями для KF(k) и GsF(k).
Принятые значения 5ГОB98,15 К) и Я°B98,15 К)—#°@) приведены в табл. 48.1, их погрешности
оцениваются в 2 Дщ-К~1-моль~1 и 0,3 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости RbF(K) принято уравнение, полученное совместной
обработкой результатов измерений энтальпии, проведенных Кейлором и др. [2944, 4321] C27—
1199 К) и Маклеодом [3337] E07—1376 К). При обработке погрешность данных [3337]
оценивалась в 1%, данных [2944, 4321] —в 3%.
Температура плавления A068 +3 К) принята по измерениям Шмитц-Дюмонта и Шмитца
[4196] A067 К), Сенса и Стоуна [4235] A071 К), Холма [2691] A066 К) и Бизота [4929] A069 К);
менее точные данные получены в работах [82, 364, 2808, 4663]. Энтальпия плавления B5,82 +
+0,40 кДж-моль) основана на согласующихся данных Маклеода [3337] B5,86 кДж-моль)
и Дворкина и Бредига [2137] B5,73 кДж-моль). Измерения Кейлора и др. [2944] приводят
к более низкому значению B3,0 кДж-моль).
Теплоемкость RbF^) G1,0+1,5 Дж«К-моль~1) принята на основании экспериментальных
данных по энтальпии жидкого RbF [3337] A078—1376 К). Близкое значение G0,6 Дж-К^Х
Хмоль) получается из данных [2944] A076—1199 К).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 1,
1,5, 5 и 10 Дж-К-моль соответственно. Расхождения между термодинамическими функциями
RbF(K, ж), приведенными в табл. 1209 и в справочнике Барина и Кнакке [1395] (Г < 1600 К),
достигают 3 Дж-К'^моль в значениях S°(T) и Ф°(Т). Различия обусловлены уточнением оценки
.5ОB98,15 К) в работе [745], а также тем, что в настоящем справочнике учтены
экспериментальные данные [3337] по энтальпии RbF(K, ж). Расхождения с термодинамическими функциями,
табулированными в работе [3337] (Г < 2000 К), не превышают 1 Дж-К^-моль" в значениях
Ф°(Т).
В справочнике принята энтальпия образования
^fH°(RhF, к, 298, 15 К) = —559,7 + 0,7 кДж • моль.
Эта величина основана на измерениях энтальпии растворения RbF(K) в воде, выполненных
Ефимовым и Леонидовым [415, 416] (разведение 2060—2520Н2О, 298,15 К, 6 измерений,
ДйгЯ°(ооН20, 298,15 К) =—26,769+0,030 кДж-моль). Близкое значение, —26,6 кДж-моль,
было получено Форкраном [2300, 2301] (разведение 110Н2О, 288,15 К, одно измерение).
Давление пара в реакции RbF(K, ж)=RbF(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДД°@) = Ae#°(RbF, к, 0) = 227,5 + 4,0 кДж • моль.
Принятая величина основана на измерениях давления и состава насыщенного пара RbF(K, ж).
Насыщенный пар фторида рубидия состоит из молекул RbF и Rb2F2. Содержание этих молекул
при 77m(RbF) = 1068 К согласно данным [2166] и принятым в справочнике термодинамическим
свойствам составляет 85 и 15 мол. % соответственно. Методика выбора термохимических величин
для компонентов насыщенного пара галогенидов щелочных металлов изложена в Приложении 8.
Результаты расчетов энтальпии сублимации RbF(K) в виде мономерных молекул
представлены в табл. 48.10. Приведенные в таблице погрешности учитывают только
воспроизводимость измерений. Принятая величина является средневзвешенной из приведенных в
таблице данных. Она совпадает с наиболее надежными измерениями [3896, 4235, 4526]. При
1 Кубическая фаза высокого давления (структурный тип CsC]) стабильна при р > 30 кбар B98 К) [2031].
439
RbF(r) Глава 48
Таблица 48.10. Результаты определений A,S"°(RbF, к, 0) (в кДжмоль)
Авторы
Метод
Вартенберг, Шульц, 1921 [4663] Точек кипения, 1436—1683 К, 11 точек
Руфф и др., 1922 [4096] То же, 1415—1673 К, 6 точек
Пью, Барроу, 1958 [3896] Торзионный, 853—860 К а
Сенс, Стоун, 1958 [4235] Протока, 862—1071 К а
То же, 1071—1332 К а
Эйзенштадт и др., 1958 [2166] Эффузионный, 951—1026 К, 6 точек
Топор, 1974 [4526] .Статический, 1189—1387 К, 46 точек
а Данные представлены в виде уравнения.
A,#°(RbF, к, 0)
II закон
233 ±7
245 ±10
232
225 ±1
235 ±2
228+4
236 ±2
III закон
224,1 ±0,4
225,3 ±1,5
227,6 ±3,2
226,8 +2,2
226,0 ±1,2
235,1 ±0,2
226,9 ±1,0
вычислении веса учитывалось отклонение данной величины от средней. Погрешность
рекомендованного значения оценивалась с учетом воспроизводимости измерений, расхождения результатов
измерений в разных работах, неточности термодинамических функций и определения состава
насыщенного пара.
RbF(r). Термодинамические свойства газообразного фторида рубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1210.
Приведенные в табл. 48.4 молекулярные постоянные RbF приняты на основании анализа
результатов исследований микроволнового [4595], ИК- [61] и УФ- [1406, 1771] спектров этой
молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406, 1771}
в справочнике принимается, что валентные возбужденные электронные состояния молекулы RbF
имеют энергии больше 41 000 см.
Приведенные в табл. 48.4 колебательные и вращательные постоянные RbF в состоянии ХХ2+
получены Визе и Горди [4595] обработкой результатов измерений микроволновых переходов
/ -* /+1 (/ ^ 23) на колебательных уровнях с v=0, 1 и 2. Соответствующие постоянные ше
и шехв были получены не непосредственно по экспериментальным данным, а рассчитаны с
помощью соотношений Данхема. Они хорошо согласуются с результатами исследования ИК-спектра
[61] ((о,=374+4, ауг,=1,9 см)-
Термодинамические функции RbF(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения @вн и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, d± и ds приведены в табл. 48.5 и получены по принятым
постоянным молекулы 85RbF для изотопомера, соответствующего природной смеси изотопов
рубидия.
Погрешности рассчитанных термодинамических функций RbF(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава фторида рубидия. При более высоких температурах дополнительный
вклад в погрешность дает неточность принятого способа экстраполяции потенциальной функции.
Погрешности в значениях Ф°(Г) составляют 0,02, 0,05 и 0,5 Дж-К-моль при 298,15, 3000
и 6000 К соответственно.
Термодинамические функции RbF(r) ранее вычислялись в справочнике Галкина и др. [4860]
(Г <; 5000 К), а также в работе Брюэра и Брэкет [1660]. Расхождения данных [4860] и табл. 1210
достигают 3 Дж-К-моль~1 в значениях Ф°(Г) и S°(T) и, по-видимому, обусловлены ошибкой
в расчете [4860]. •
Константа равновесия реакции RbF(r)=Rb(r)+F(r) вычислена с использованием значения
Ar#°@)=D0(RbF)=490,666+4,2 кДж-моль «* 41 010+350 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации RbF(K). Этим величинам также
соответствует
A/Te(RbF, г, 0) = —331,199 + 4,1 кДж ¦ моль.
440
Рубидий и его соединения Rb2F2(r), RbCl(n, ж>
Булевич и др. [1719] в результате спектрофотометрического изучения равновесия реакции
Rb(r)+HF(r)=RbF(rL-H(r) (пламена H2+O2+N2, добавки солей рубидия и C7Fn, 1500—
2600 К) нашли значение D0(RbF)=502+30 кДж-моль, которое совпадает с принятым в
пределах своей погрешности.
Rb2F2(r). Термодинамические свойства газообразного дифторида дирубидия в стандартном,
состоянии в интервале температур 100—6000 К приведены в табл. 1211.
Молекулярные постоянные, использованные для расчета термодинамических функций Rb2F2(r)f
приведены в табл. 48.6. На основании теоретических расчетов [961, 4689] для молекулы Rb2F2
в основном электронном состоянии XхА д принимается плоская ромбическая структура
(симметрия D2h). Приведенное в табл. 48.6 о произведение моментов инерции соответствует
структурным параметрам r(Rb—F)=2,45+0,07 Аи Д F—Rb—F=84+10°, найденным Соломоником в
результате расчета для ионной модели [961, 963]. Погрешность значения IaIbIc составляет
5-Ю13 г3-см6. Приведенные в табл. 48.6 значения частот v4, v5, v6 найдены Шабером и Мартином
[4167] в ИК-спектре молекул Rb2F2, изолированных в матрице из Аг, их величины хорошо
согласуются с данными, полученными в работе Олта и Эндрюса [1362] *. Для частот vx—v3 приняты
значения, рассчитанные Соломоником [961, 963]. Погрешность принятого значения частоты v2
оценивается в 60 см, частоты v3 — в 40 см, остальных частот — в 15—30 см.
Термодинамические функции Rb2F2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130>
без учета возбужденных электронных состояний. Погрешности рассчитанных величин
обусловлены неточностью принятых молекулярных постоянных, а также приближенным методом
расчета; они составляют 8, 12, 16 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции Rb2F2(r) рассчитывались в работах Краснова и др. [569],
Фрурипа и Бландера [2340] (до 5000 К). Расхождения данных [569] и [2340] сданными табл. 1211
не превышают 3 Дж-К~1-моль~1 в значениях Ф°(Г).
Константа равновесия реакции Rb2F2(r)=2Rb(r)+2F(r) вычислена с использованием
значения Ari7o@) = 1168,934+15 кДж-моль,, соответствующего принятой в справочнике энтальпии
образования
, г, 0) = —850 ± 15 кДж ¦ моль.
Принятая величина основана на исследованиях равновесия реакции RbF(K)+RbF(r)=Rb2F2(r),.
проведенных Эйзенштадтом и др. [2166] эффузионным методом с селектором скоростей (951—
1026 К, 6 точек), ДгЯ°@)=40+13 кДж-моль (II и III законы). В работе [2166] было также
изучено равновесие реакции Rb2F2(r)=2RbF(r), что приводит к значению AfH0(Rh2F2, г, 0) =
=—854+15 кДж-моль (III закон). Масс-спектрометрическое исследование этой же реакции,,
выполненное Рыковым [4898] A058 К, 9 измерений), приводит к значению A/^°(Rb2F2, г, 0)==
=—851+15 кДж-моль. Эти величины подтверждают принятое значение.
RbCl(K, ж). Термодинамические свойства кристаллического и жидкого хлорида рубидия
в стандартном состоянии при температурах 100—3400 К приведены в табл. 1212.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние RbCl(K) в интервале 0—997 К принимается
кубическая модификация (структурный тип NaGl) 2.
Термодинамические функции ЙЬС1(к) для. Т <С 298,15 К в интервале 0—14 К вычислены по
значениям теплоемкости, полученным в результате усреднения данных Роллефсона и Перессини
[4036] @,1—13 К), Хигашигаки и Чихары [2639] C—300 К), Уоткинса и Фенишеля [5069] @,8—
16 К), в интервале 14—55 К — по данным Паукова и Хриплович [769] A4—314 К), а при Т >
> 55 К — по усредненным значениям теплоемкости, полученным в работах [769, 2639}
и Киркхема и Иейтса [3018] B0—300 К); при усреднении экспериментальным данным
[769, 2639, 3018] была приписана погрешность в 0,3, 0,5 и 1% соответственно. Измерения
1 Говард и Эндрюс [2721] отнесли наблюдаемую ими полосу~266,1 см х к колебанию v2 молекулы Rb+FjT, отметив;
возможность отнесения, принятого в справочнике (к колебанию vB молекулы RbaF2).
2 Кубическая фаза высокого давления (структурный тип GsCl) стабильна при р > 5,2 кбар B98 К) [4584].
441
«ЬС1(к, ж) Глава 48
-средней теплоемкости ИЬСЦк) в ранней работе Бренстеда [1683] B73—293 К) не учитывались.
Значения 5°B98,15 К) и Я°B98,15 К)—Я°@), приведенные в табл. 48.1, имеют погрешности
•0,4 Дж-К~1-моль~1 и 0,04 кДж-моль соответственно.
При Т > 298,15 К данные по энтальпиям и теплоемкости RbCl(K) в литературе отсутствуют.
Известна работа Маркова и др. [657], в которой была измерена энтальпия жидкого RbCl в
интервале 1017—1316 К и определена энтальпия плавления. На основании этих данных было
рассчитано значение (ЯТт — Я298Д5)крист = (ЯТиг —Я29816)ж— ДтЯ = 40,27 кДж • моль, которое было
использовано для вывода уравнения для теплоемкости ИЬСЦк) (см. табл. 48.1).
Температура плавления (997+3 К) принята по данным [139, 743, 1140, 1368, 1478, 1599, 3371,
3513, 4791] (995—999 К). Энтальпия плавления B4,4+0,4 кДж-моль) принята по
согласующимся между собой измерениям Маркова и др. [657] B4,9 кДж-моль), Дворкина и Бредига
[2137] B3,7 кДж-моль), Марчидана и Гамбино [3371] B4,6 кДж-моль). Более низкая энталь-
лия плавления получена в работах [4819] A9,2 кДж-моль), [4663] B0,9 кДж-моль), [3433]
{19,2 кДж-моль-1) и [4538] B0,1 кДж-моль).
Теплоемкость ЙЬС1(ж) G3,4+1,5 Дж-К~1-моль~1) принята по результатам измерений
энтальпии [657] A017—1316 К). С принятым значением хорошо согласуются данные Мургулеску и Теле
[3594] G3,5 Дж-К^-моль).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,3,
1, 5 и 10 Дж-К~1-моль~1. Расхождения между термодинамическими функциями RbCl(K, ж),
приведенными в табл. 1212 и в справочнике Барина и Кнакке [1395] (Т ^ 1600 К), достигают
7 Дж-К-моль~1 в значениях Ф°(Г), они обусловлены существенным уточнением величины
.S°(RbCl, к, 298,15 К) в работах [769, 2639, 3018] и энтальпии плавления — в работах [657, 2137,
3371].
В справочнике принята энтальпия образования
l, к, 298,15 К) =—435,22 + 0,16 кДж • моль,
основанная на измерениях энтальпии растворения RbCl(K) в воде, приведенных в табл. 48.11.
Результаты этих измерений находятся в хорошем соответствии. Для дальнейших расчетов
принимается средневзвешенное ДягЯ°(соН20, 298,15 К)=17,01+0,03 кДж-моль. Близкие, но
менее точные значения энтальпии растворения RbCl в воде были получены в работах [2, 3, 27,
.244, 257, 580, 901, 902, 2295, 2300, 2301, 2548, 4514, 4781, 4819].
Таблица 48.11. Результаты определений Аа„Н°(КЬС1, к-»соН2О, 298,15К) (в кДж-моль^1)
Авторы
Цветков, Рабинович, 1969 [1097]
Сергеева и др., 1970 [922]
Де Виссер и др., 1976 [2057]
Дэдгер, Тахериан, 1977 [1983]
Воробьев и др., 1978 [245, 778]
Монтгомери и др., 1978 [3548]
Джонсон, Гейер, 1979 [2871]
Конечная концентрация, температура
измерений
500 Н2О, 298,15 К, 1 измерение
530 Н2О, 295—305 К, 5 измерений
2780—11100 Н2О, 298,15 К, 3 измерения
1850—5550 H2Q, 298,15 К, 5 измерений
2780 Н2О, 298,15 К
1113—1843 Н2О, 298,15 К, 6 измерений
1700 Н2О, 298,15 К, 5 измерений
Да(гЯ°(соН2О, 298,15 К)
17,03 + 0,20
17,45 + 0,20
17,05+0,10
17,07 + 0,20
17,04±0,12
16,99+0,10
17,002 + 0,030
Давление пара в реакции RbCl(K, ж)=RbCl(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = A8#°(RbCl, к, 0) = 214 + 4 кДж • моль".
Принятая величина основана на измерениях давления и состава насыщенного пара над
RbCl(K, ж). Насыщенный пар хлорида рубидия состоит из молекул RbCl и Rb2Cl2. Согласно
измерениям [1501, 3504] и термодинамическим свойствам, принятым в справочнике, при Tm(RbCl) =
=997 К их содержание составляет 90 и 10 мол. % соответственно. Методика выбора
термохимических величин для компонентов насыщенного пара галогенидов щелочных металлов изложена
в Приложении 8.
442
Рубидий и его соединения
RbCl(r)
Таблица 48.12. Результаты определения AsJ3"°(RbCI, к, 0) (в кДж • моль)
; Авторы
Вартенберг, Шульц, 1921 [4663]
' Руфф, Мугдан, 1921 [4094]
Кангро, Викинг, 1938 [2919]
Мейер, Винтнер, 1938 [3433]
i Нива, 1938 [3665]
: Тредуэлл, Вернер, 1953 [4538]
- Несмеянов, Сазонов, 1957 [718]
Чапка, 1959 [1851]
Милн, Клейн, 1960 [3510]
Мургулеску, Топор, 1967 [3596]
Вдовенко и др., 1967 [196]
Мургулеску, Топор, 1968 [3597]
Хасти, Свинглер, 1969 [2583]
Эмонс и др., 1976 [2173]
Вагнер, Шефер, 1979 [4630]
Метод
Точек кипения, 1434—1657 К, 8 точек
То же, 1415—1668 К, 6 точек
Протока, 1133—1263 К, 3 точки
Эффузионный, 885—926 К, 5 точек
То же, 833—964 К, 11 точек
Протока, 831—944 К, 11 точек
Эффузионный, 676—868 К, 13 точек
Масс-спектрометрический, Гср=800 К а
То же, Гср=800 К а
Статический, 1213—1372 К, 8 точек
Эффузионный, 673—873 К а
Статический, 1275—1429 К, 6 точек
Масс-спектрометрический, 890—980 К
Эффузионный, 993—1223 К а
Масс-спектрометрический, 785—860 К а
: а Данные представлены в виде уравнения.
А,Н°(ЕЪС1, к, 0)
II закон
220+6
262+40
197 + 25
219+15
206+2
199+6
204+6
214
207
199+4
204
200+5
214+4
196
207
III закон
214,3 + 0,3
217,4±3,8
219,4+3,0
214,2+0,2
219,5+0,5
214,5+0,5.
211,1 + 0,6
•—
_
212,6 + 0,5
212,2+1,0
213,1 + 0,5
¦—
214,2+2,0
—
Результаты расчетов энтальпии сублимации RbCl(K) в виде мономерных молекул представлены
в табл. 48.12. Приведенные в таблице погрешности отражают только воспроизводимость
измерений. Принятая величина является средневзвешенной из приведенных в таблице величин.
При вычислении веса учитывалось отклонение данной величины от средней. Погрешность
рекомендуемого значения оценена с учетом воспроизводимости измерений, расхождения результатов
измерений в разных работах, неточности термодинамических функций и определения состава
насыщенного пара.
RbCl(r). Термодинамические свойства газообразного хлорида рубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1213.
Приведенные в табл. 48.4 молекулярные постоянные RbCl приняты на основании анализа
результатов исследований микроволнового [1874, 1876], ИК- [3984] и УФ- [1406] спектров этой
молекулы,
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406]
в справочнике принимается, что валентные возбужденные электронные состояния молекулы RbCl
имеют энергии больше 35 000 см.
Приведенные в табл. 48.4 колебательные и вращательные постоянные RbCl в состоянии
Хх? + получены Клаузером и Горди [1876] обработкой результатов измерений микроволновых
переходов / ->/+1 (J ^48) на колебательных уровнях с v=0, 1, 2 и 3. Соответствующие
постоянные (ое и шехе были получены не непосредственно по экспериментальным данным, а
рассчитаны с помощью соотношений Данхема. Они хорошо согласуются с результатами исследования
]
щ Д
ИК-спектра [3984] (ш,=228+6,
см).
р (, а )
Термодинамические функции RbCl(r) были вычислены по уравнениям A. 3)—A. 6), A.9),
A. 10), A. 93)—A. 95). Значения QBa и ее производных вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 48.5 и получены по принятым
постоянным молекулы 85Rb35Cl. Расчет был выполнен для изотопомера, соответствующего
природной смеси изотопов рубидия и хлора.
Погрешности рассчитанных термодинамических функций RbCl(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава хлорида рубидия. При более высоких температурах дополнительный
вклад в погрешность вносят неточность принятого способа экстраполяции потенциальной функции.
443
Rb2Cl2(r) Глава 48
а также пренебрежение возбужденными электронными состояниями. Погрешности в значениях
Ф°(Г) составляют 0,02, 0,05 и 0,7 Дж-К^-моль при 298,15, 3000. и 6000 К соответственно..
Термодинамические функции RbCl(r) ранее вычислялись в работе Уилкинса [4728] (Г ^
< 6000 К), Раиса и Клемперера [3985] (Г < 2000 К), Брюэра и Брэкет [1660] (Ф°(Г), Т <
^ 2000 К). Расхождения данных [4728] и табл. 1213 не превышают 1,2 и 6,4 Дж-К"х-моль"*
в значениях Ф°(Т) и S°(T) соответственно и объясняются тем, что в этой работе использовался
приближенный метод расчета.
Константа равновесия реакции RbCl(r)=Rb(r)+Cl(r) вычислена с использованием значения
Ar#°@)=D0(RbCl)=423,113+4,l кДж-моль « 35 370+340 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации RbCl(K). Этим величинам также
соответствует
Д/HRbCl, г, 0) = —221,301 ± 4,0 кДж • моль.
Практически совпадающие значения энергии диссоциации были получены Су и Райли [44271
D24+8 кДж-моль) методом фотофрагментарной спектроскопии и Булевич и др. [1719] D25 +
+8 кДж-моль) на основании спектрофотометрического изучения равновесия реакции Rb(r)+
+HCl(r)=RbGl(r)+H(r) в пламенах A500—2600 К). Более высокое значение D36+3 кДж-моль)
было найдено Берковицем [1499] при фотоионизационных измерениях, однако учет теплового-
возбуждения и кинетической энергии в условиях этого эксперимента приводит, как показали
Дронин и Горохов [389]t к величине 424+4 кДж-моль.
Rb2Cl2(r). Термодинамические свойства газообразного дихлорида дирубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1214.
Молекулярные постоянные, использованные для расчета термодинамических функций
Rb2Cl2(r), приведены в табл. 48.6. На основании теоретических расчетов [961, 4689] для
молекулы Rb2Gl2 в основном электронном состоянии ХгАд принимается плоская ромбическая
структура (симметрия D2h). Приведенное в табл. 48.6 произведение моментов инерции соответствует
структурным Параметрам r(Rb-Cl)=2,98+0,08 А и Д С1—Rb—С1=95+10°, найденным Соло-
моником в результате расчета с использованием ионной модели [961, 963]. Погрешность
значения IaIbIc составляет 3-1012 г3>см6. Приведенные в табл. 48.6 значения частот v5 и v6 найдены
Шабером и Мартином [4167] в ИК-спектре молекул Rb2Cl2, изолированных в матрице из Аг. Для
частот vx—v4 приняты значения, рассчитанные Соломоником [961, 963]. Погрешность значения мг
оценивается в 40 см, частоты v3 — в 30 см, остальных частот — в 20 см.
Термодинамические функции Rb2Cl2(r) вычислялись в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128)г
A. 130) без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью принятых молекулярных постоянных, а также приближенным методом расчета;
они составляют 8, 15 и 20 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции Rb2Cl2(r) рассчитывались в работах Краснова и др. [569]
(до 3000 К) и Фрурипа и Бландера [2340] (до 5000 К). Расхождения результатов этих расчетов
и табл. 1214 не превышают 2 и 4 Дж-К~1-моль~1 соответственно.
Константа равновесия реакции Rb2Cl2(r)=2Rb(r)+2Cl(r) вычислена с использованием
значения Дг-/7°@)=1018,624+15 кДж-моль, соответствующего "принятой в справочнике энтальпии
образования
bfH°(Rb2Cl2, г, 0) = —615+ 15 кДж-моль.
Принятая величина основана на исследованиях равновесия реакции RbCl(K, ж)+RbCl(г)=
=Rb2Gl2(r), представленных в табл. 48.13. Приведенные в таблице погрешности учитывают только
воспроизводимость измерений. Для дальнейших расчетов принимается среднее значение АгН°@)=
=41+15 кДж-моль. Его погрешность обусловлена в основном неточностью
термодинамических функций.
В работах [391, 1995, 3504, 3597] было исследовано равновесие реакции Rb2Cl2(r)=
=2RbCl(r). По этим данным были вычислены значения A^°(Rb2Gl2, г, 0)=—624+18 кДжХ
Хмоль (III закон) и —678+50 кДж«моль"х (II закон), совпадающие в пределах погрешностей
с принятым.
444
Рубидий и его соединения RbBr(u, ж)
Таблица 48.13. Результаты определений энтальпии реакции RbCl(is) + RbCI(r) = Rb2CI2(r)
(в кДж-моль)
Авторы
Миллер, Куш, 1957 [3504]
Берковиц, Чапка, 1958 [1501]
Милн, Клейн, 1960 [3510]
Мургулеску, Топор, 1968 [3597]
Вагнер, Шефер, 1979 [4630]
Метод
Эффузионный с селектором скоростей, 866 К,
1 точка
Масс-спектрометрический, 870 К, 1 точка
То же, Гор=870 К
Статический и протока, 1275—1429 К, 12 точек
Масс-спектрометрический, 785—860 К, данные
представлены в виде уравнения
ДгД°@)
II закон
—
49+8
99+30
33
III закон
41,1
41,1
—
39,9 ±2,1
—
ЛЬВг(к, ж). Термодинамические свойства кристаллического и жидкого бромида рубидия
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1215.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние RbBr(K) в интервале 0—965 К принимается
кубическая модификация (структурный тип NaCl) 1.
При Т <С 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работе Клузиуса и др. [3043] A1—273 К). Экстраполяция экспериментальных данных
[3043] дает 5°A1 К)=0,92 Дж-К^-моль. Приведенные в табл. 48.1 значения 5°B98,15 К)
и #°B98,15 К)—Н°@) имеют погрешности 0,8 Дж-К'моль и 0,08 кДж-моль соответственно.
Измеренная Брёнстедом [1683] средняя теплоемкость ИЬВг(к) B73—293 К) на 2% ниже данных
[3043].
Экспериментальные данные по теплоемкости или энтальпии RbBr при высоких температурах
в литературе отсутствуют. Для теплоемкости В_ЬВг(к) при Т > 298,15 К принято линейное
уравнение, полученное на основании значений СрB98,15 К)=52,76 Дж-К»моль~1 и 6*^(965 К)=
=62,0 Дне-К-моль. Первая величина получена экстраполяцией данных [3043], вторая —
оценкой по данным о теплоемкости бромидов щелочных металлов в точках плавления.
Температура плавления (965 +2 К) принята по согласующимся между собой измерениям
Иоффе и Артсдалена [4791] (965+2 К), Марчидана и Гамбино [3371] (966 К), Товмасьяна и др.
[1016] (963 К) и Маргеритиса и Флора [3386] (967 К). В работах [2808, 4663, 4758] получены
более низкие значения (954—962 К). Энтальпия плавления B3,3+0,4 кДж-моль) принята
по измерениям Дворкина и Бредига [2137], выполненным для чистого образца. Несколько
большая величина получена в работе Марчидана и Гамбино [3371] B4,2 кДж-моль). Теплоемкость
ИЬВг(ж) G2,8+2,0 Дж-К~1-моль~1) принята по данным Мургулеску и Теле [3594], полученным
в узком интервале температур вблизи точки плавления.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,5,
2, 6 и 11 Дж-К~1-моль~1. Расхождения между термодинамическими функциями RbBr(K, ж),
приведенными в табл. 1215 и в справочнике Барина и Кнакке [1395] (Т ^ 1600 К) достигают
14 Дж-К-моль в значениях Ф°(Т) при Т > Тт. Столь значительные расхождения обусловлены
использованием в настоящем справочнике данных [2137, 3371] по энтальпии плавления RbBr.
В справочнике принята энтальпия образования
A/ffe(RbBr, к, 298,15 К) = —394,77 + 0,25 кДж • моль,
соответствующая энтальпии растворения RbBr(K) в воде Дад#°(соН20, 298,15 К)=22,15 +
+0,15 кДж-моль. Эта величина является средневзвешенной из результатов работ [245] и
[4514], приведенных в табл. 48.14.
Давление пара в реакции RbBr(K, ж)=ВЬВг(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) == \Н°(ПЪВг, к, 0) = 206 + 3 кДж • моль.
1 Кубическая фаза высокого давления (структурный тип CsCl) стабильна при р > 4,2 кбар B98 К) [4584].
445
RbBr(r)
Глава 48
Таблица 48.14. Результаты определений Aa9ir0(RbBr, к->ооНаО, 298,15 К) (в кДж-моль)
Авторы
Конечная концентрация, температура
измерений
Аа Я°(соН20, 298,15 К)
Форкран, 1911 [2300, 2301]
Ланге, Мартин, 1937 [3179]
Воробьев и др., 1980 [245]
Тоури, Перашон, 1980 [4514]
110 Н2О, 288,15 К, 1 измерение
500 Н2О, 298,15 К, 1 измерение
2780 Н2О, 298,15 К, 3 измерения
55500 Н2О, 298,15 К, 1 измерение
23,37
21,88
22,13+0,12
22,36 +0,30
Принятая величина основана на измерениях давления и состава насыщенного пара RbBr(K, ж).
Насыщенный пар бромида рубидия состоит из молекул RbBr и Rb2Br2. Согласно данным [35971
и термодинамическим свойствам, принятым в настоящем справочнике, при Tm(RbBr)=965 К
их содержание в насыщенном паре составляет 85 и 15 мол.%. Методика выбора термохимических
величин для компонентов, насыщенного пара галогенидов щелочных металлов изложена в
Приложении 8.
Результаты расчетов энтальпии сублимации RbBr(K) в виде мономерных молекул представлены
в табл. 48.15. Приведенные в таблице погрешности учитывают только воспроизводимость изме-
Таблица 48.15. Результаты определений A,.ff°(RbBr, к, 0) (в кДж-моль")
Авторы
Вартенберг, Шульц, 1921 [4663]
Руфф, Мугдан, 1921 [4094]
Мейер, Винтнер, 1938 [3433]
Мургулеску, Топор, 1967 [3596]
Мургулеску, Топор, 1968 [3597]
Макаров, Ступин, 1968 [645]
Ефимова, 1981 [4171
Метод
Точек кипения, 1369—1613 К, 10 точек
То же, 1323—1638 К, 7 точек
Эффузионный, 856—911 К, 5 точек
Статический, 1213—1396 К, 8 точек
То же, 1263—1379 К, 5 точек
Эффузионный, 650—853 К а
Масс-спектрометрический, 853—950 К а
а Данные представлены в виде уравнения.
A,#°(RbBr, к, 0)
II закон
216+7
218+17
219±18
201 ±6
204,5+6
204+1
201 + 6
III закон
206,2±0,6
205,8±1,2;
208,6+0,5
204,9 ±0,3*
204,6+0,3
205,7 ±2,4
—
рений. Результаты измерений находятся в хорошем согласии и принятое значение является
средневзвешенным из приведенных в таблице величин. При вычислении среднего взвешенного
учитывалось отклонение результата данной работы от среднего. Погрешность принятой величины
учитывает воспроизводимость измерений, расхождения измерений в разных работах, неточность
термодинамических функций и определения состава насыщенного пара.
RbBr(r); Термодинамические свойства газообразного бромида рубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1216.
Приведенные в табл. 48.4 молекулярные постоянные RbBr приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3984] и УФ- [1406] спектров этой
молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406]
в справочнике принимается, что валентные возбужденные электронные состояния молекулы имеют
энергии больше 32 000 см.
Приведенные в табл. 48.4 колебательные и вращательные постоянные RbBr в состоянии Z1?+
получены Раском и Горди [4101] обработкой результатов измерений микроволновых переходов
/ -> /+1 (/ <С 69) на колебательных уровнях с v=0, 1 и 2. Соответствующие постоянные we
и и>ехе были получены не непосредственно по экспериментальным данным, а рассчитаны с
помощью соотношений Данхема.
Термодинамические функции RbBr(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Q3U и ее производные вычислялись без учета возбужденных элек-
446
Рубидий и его соединения Rb2Br2(r)i
тронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 48.5 и получены по принятым
постоянным молекулы 86Rb79Br. Расчет был выполнен для изотопомера, соответствующего
природной смеси изотопов рубидия и брома.
Погрешности рассчитанных термодинамических функций RbBr(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава бромида рубидия. При более высоких температурах дополнительный,
вклад в погрешность вносят неточность использованного способа экстраполяции потенциальной
функции, а также пренебрежение возбужденными электронными состояниями. Погрешности
в значениях Ф°(Г) составляют 0,02, 0,05 и 0,8 Дж -К -моль при 298,15, 3000 и 6000 К
соответственно.
Термодинамические функции RbBr(r) ранее вычислялись в работе Брюэра и Брэкет [1660]
(Ф°(Т), Т <; 2000 К). Расчеты в работе [1660] выполнены по молекулярным постоянным,
близким к принятым в настоящем издании; соответствующие расхождения с данными табл. 1216 не
превышают 0,7 Дж-К"г-моль~1 в значениях Ф°(Т) и объясняются главным образом различием
метода расчета <?вв. Щ ШШ
Константа равновесия реакции RbBr(r)=Rb(r)+Br(r) вычислена с использованием значения
A^#°@)=D0(RbBr)=382,196+3?l кДж-моль я» 31 900 ±260 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации RbBr(K). Этим величинам также
соответствует ffiibi! И
A/ff°(RbBr, г, 0) = —182,081 + 3,0 кДж • моль.
Практически совпадающее значение энергии диссоциации C84 +8 кДж -моль) было
получено Булевич и др. [1719] на основании спектрофотометрического изучения равновесия реакции
Rb(r)+HBr(r)=RbBr(r)+H(r) в пламенах A500—2600 К). Берковиц [1499] в результате
фотоионизационных измерений нашел D0(RbBr)=380+.3 кДж-моль; учет теплового возбуждения
и кинетической энергии осколков в условиях этого эксперимента, выполненный в работе [389]г
уменьшил это значение до 377 +4 кДж -моль. Определение этой величины методом
фотофрагментарной спектроскопии в работе Су и Райли [5055 ] также привело к значению 377 +4 кДж X
Хмоль. §|
Rb2Br2(r). Термодинамические свойства газообразного дибромида дирубидия в стандартном*
состоянии в интервале температур 100—6000 К приведены в табл. 1217.
Молекулярные постоянные, использованные для расчета термодинамических функций
Rb2Br2(r), приведены в табл. 48.6. На основании теоретических расчетов [961, 4689] для
молекулы Rb2Br2 в основном электронном состоянии Х1Ад в справочнике принимается плоская
ромбическая структура (симметрия Du). Приведенное в табл. 48.6 произведение моментов инерции
соответствует структурным параметрам r(Rb—Вг)=3,15+0,10 А и Д Вг—Rb—Вг=98+10°,
найденным Соломоником в результате расчета с использованием ионной модели молекулы [961,.
963]. Погрешность значения IaIbIc оценивается в 1-Ю11 г3-см6. Приведенные в табл. 48.6
значения частот v5, ve найдены Шабером и Мартином [4167 ] в ИК-спектре молекул Rb2Br2,
изолированных в матрице из Аг. Для частот vx—v4 приняты значения, рассчитанные Соломоником [961 т
963]. Погрешность частоты vx оценивается в 30 см, остальных частот — в 15—20 см.
Термодинамические функции Rb2Br2(r) вычислялись в приближении «жесткий ротатор—
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128)г
A. 130) без учета возбужденных электронных состояний. Погрешности рассчитанных величин
обусловлены неточностью принятых молекулярных постоянных, а также приближенным
характером расчета; они составляют 10, 15 и 20 Дж-К^-моль в значениях Ф°(Т) при 298,15, 300Q
и 6000 К соответственно.
Ранее термодинамические функции Rb2Br2(r) рассчитывались в работах Краснова и др. [569]
(до 3000 К) и Фрурипа и Бландера [2340] (до 5000 К). Расхождения данных [569] и [2340] с
данными табл. 1217 не превышают 1 и 4 Дж-К-моль в значениях Ф°(Т) соответственно.
Константа равновесия реакции Rb2Br2(r)=2Rb(r)+2Br(r) вычислена с использованием
значения Ду#°@)=934,230 +14 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
t'a, г, Of =—534 + 14 кДж • моль.
447
ВЫ(к, ж) Глава 48
Принятая величина основана на результатах исследований равновесия реакции ЫЬВг(к, ж)+
+RbBr(r)=Rb2Br2(r), выполненных Мургулеску и Топор [3597] (статический метод и метод
протока, 1263—1379 К, 9 точек, Дг#°@)=34,1 +2,5 кДж-моль (III закон) и 116+18 кДжХ
Хмоль (II закон)) и Ефимовой [417] (масс-спектрометрический метод, 853—950 К, данные
представлены в виде уравнения, АгН°@)=40,0 ±2,0 кДж-моль (III закон) и 31+6 кДж-моль
(II закон)). В качестве погрешностей указаны воспроизводимости измерений. Неточность
термодинамических функций увеличивает погрешность величин, вычисленных по уравнению III
закона, до 18 кДж-моль [3597] и 12 кДж-моль [417]. Результаты измерений в работах [3597]
и [417] совпадают в пределах указанных погрешностей. Для дальнейших расчетов принимается
среднее Дг.й"°@)=37±12 кДж «моль. Хагемарк и Хенгстенберг [2546] исследовали
статическим методом плотность ненасыщенных паров RbBr, 1310—1421 К, откуда была получена
энергия димеризации, которой для приведенной выше реакции соответствует Д,.Яо@)=33 ±23 кДжХ
Хмоль (II закон)|и 30 ±15 кДж-моль (III закон), что подтверждает выбранное значение.
RbI(K, ж). Термодинамические свойства кристаллического и жидкого иодида рубидия в
стандартном состоянии лри температурах 100—3400 К приведены в табл. 1218.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние Rbl(K) в интервале 0—929 К принимается
кубическая модификация (структурный тип NaCl) x.
При Т <С 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости Rbl в работе Клузиуса и др. [1883] A1—277 К). Экстраполяция экспериментальных
данных [1883] дает S°(ll K)=l,77 Дж-К~1-моль~1. Погрешности приведенных в табл. 48.1
значений 5°B98,15 К) и #°B98,15 К)—#°@) оцениваются в 1 Дж-К^-моль и ОД кДж-моль
соответственно. Измерения средней теплоемкости в работе Бренстеда [1683] B73—293 К)
приводят if величине, которая на 1% ниже данных [1883].
Экспериментальные данные по термодинамическим свойствам Rbl (к) при высоких температурах
в литературе отсутствуют. При Т > 298,15 К для теплоемкости Rbl(K) принято уравнение,
полученное на основании значений СрB98,15 К)=52,59 Дж-К^-моль, СрD00 К)=53,56 Дж-К-1Х
Хмель" и СД929 К)=65,70 Дж-К~1-моль. Первые две величины получены экстраполяцией
данных [1883], третья — оценкой по принятым значениям теплоемкости других галогенидов
щелочных металлов в точках плавления.
Температура плавления (929 ±2 К) принята по согласующимся между собой измерениям
Роштози и Кубиччотти [4054] (929 К) и Маргеритиса и др. [3386] (929 К). Результаты измерений
[86, 911, 4523, 4791] имеют большой разброс (от 915 до 945 К), что, по-видимому, обусловлено
недостаточной чистотой исследованных образцов. Энтальпия плавления B2,05 ±0,40 кДжХ
Хмоль) основана на данных Дворкина и Бредига [2137]. Менее надежная величина приведена
в работе [1636] B4,6 кДж-моль, погрешность 3%). Теплоемкость НЫ(ж) G2,1 ±2,0 Дж-К"хХ
Хмоль) принята vo измерениям Мургулеску и Теле [3594], проведенным в узком температурном
интервале вблизи точки плавления.
Погрешности вычисленных значений Ф0(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,5,
2, 5 и 10 Дж-К~1-моль. Расхождения между термодинамическими функциями RbI(K, ж),
приведенными в табл. 1218 и в справочнике Барина и Кнакке [1395] (Т ^ 1500 К), достигают
6 Дж-К~1-моль~1 в значениях Ф°(Т) при Т~^>Тт. Имеющиеся расхождения обусловлены
существенным уточнением энтальпии плавления в работе [2137].
В справочнике принята энтальпия образования
bfH°(B.M, к, 298,15 К) =—333,60 ± 0,25 кДж'-моль,
основанная на приведенных в табл. 48.16 измерениях энтальпии растворения Rbl(K) в воде.
Используемая в последующих расчетах величина Дя?#°(ооН2О, 298,15 К)=25,58 ±0,15 кДжХ
Хмоль является средневзвешенной из данных [245, 778, 954, 4514].
Давление пара в реакции Rbl (к, ж)=Rbl (г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = A,#°(RbI, к, 0) = 198,0 ±3,0 кДж • моль.
Кубическая фаза высокого давления (структурный тип CsCl) стабильна при р > 3,6 кбар B98 К) [1989].
448
Рубидий и его соединения
Rbl(r)
Таблица 48.16. Результаты
Авторы
Мознер, 1897 [3570]
Форкран, 1911 [2300, 2301]
Крестов и др., 1972 [583]
Новоселов и др., 1975 [736]
Амиго и др., 1975 [1312]
Смирнова и др., 1978 [954]
Воробьев и др., 1978 [245, 778]
Тоури, Перашон, 1980 [4514]
определений b.aqH°(BbI, к-к» Н2О, 298,15 К) (в
Конечная концентрация, температура
измерений
925 Н2О, 288,15 К, 1 измерение
110 Н2О, 288,15 К, 1 измерение
1110—5550 НаО, 298,15 К
298,15 К
28 000—185 000 Н2О, 293,15 К, 21 измерение
5780—12 620 Н2О, 298,15 К, 5 измерений
2780 Н2О, 298,15 К
55500 Н2О, 298,15 К, 1 измерение
кДж • моль)
ДйгЯ°(ооН20, 298,15 К)
23,74
26,04
25,44
25,65
24,09
25,77 ±0,30
25,60 ±0,12
25,28 ±0,30
Принятая величина основана на измерениях давления и состава насыщенного пара RbI(K, ж).
Насыщенный пар иодида рубидия состоит из молекул Rbl и Rb2I2. Содержание этих молекул
при 21m(RbI)=929 К согласно данным [3598] и принятым в справочнике термодинамическим
свойствам составляет 90 и 10 мол.% соответственно. Методика выбора термохимических
величин для компонентов насыщенного пара галогенидов щелочных металлов изложена в
Приложении 8.
Результаты расчетов энтальпии сублимации Rbl(K) в виде мономерных молекул представлены
в табл. 48.17. Приведенные в таблице погрешности учитывают только воспроизводимость изме-
Таблица 48.17. Результаты определений KsH°(Rbl, к, 0) (в кДж • моль)
Авторы
Вартенберг, Шульц, 1921 [4663]
Руфф, Мугдан, 1921 [4094]
Нива, 1938 [3665]
Мургулеску, Топор, 1967 [3596]
Макаров, Ступин, 1968 [645]
Мургулеску, Топор, 1970 [3598]
Бифельд, 1978 [1557]
Ефимова, 1981 [417]
Метод
Точек кипения, 1308—1581, 10 точек
То же, 1348—1598 К, 6 точек
Эффузионный, 779—873 К, 9 точек
Статический, 1235—1395 К, 8 точек
Эффузионный, 650—850 К а
Статический, 1269—1324 К, 7 точек
Эффузионный, 753—936 К, 29 точек
Масс-спектрометрический, 771—910 К, 46 точек а
а Данные представлены в виде уравнения.
Д„Я°AШ, к, 0)
II закон
III закон
210+12 198,9+0,9
210 ±28 198,7 ±1,4
189+4 198,3+0,6
201 ±3 197,7 ±0,2
200 194,2 ±9,0
200 ±3 197,8 ±0,1
195 ±4 198,2+0,5
196 ±4 —
рений. Результаты измерений согласуются в пределах их воспроизводимости. Принятое значение
является средневзвешенным из приведенных в таблице величин, кроме данных [645]. При
вычислении средневзвешенного учитывалось отклонение результата данной работы от среднего.
Погрешность принятой величины учитывает воспроизводимость измерений, расхождения
результатов измерений в разных работах, неточность термодинамических функций и неточность
определения состава насыщенного пара.
Rbl(r). Термодинамические свойства газообразного иодида рубидия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1219.
Приведенные в табл. 48.4 молекулярные постоянные Rbl приняты на основании анализа
результатов исследований микроволнового [2700, 4101], ИК- [3984] и УФ- [1406] спектров этой
молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406]
в справочнике принимается, что валентные возбужденные электронные состояния молекулы Rbl
имеют энергии больше 27 000 см.
29 Заказ № 1590
449
Rb2I2(r) Глава 48
Приведенные в табл. 48.4 колебательные и вращательные постоянные Rbl в состоянии Хх? +
получены Раском и Горди [4101] обработкой результатов измерений микроволновых переходов
/ -> /+1 (/ <^ 100) на колебательных уровнях с и=0, 1, 2 и 3. Соответствующие постоянные we
и и>вхв были получены не непосредственно по экспериментальным данным, а рассчитаны с
помощью соотношений Данхема.
Термодинамические функции*; Rbl (г) были вычислены по уравнениям A. 3)—A. 6), A.9),
A. 10), A. 93)—A. 95). Значения Qbn и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 48.5 и получены по принятым
постоянным молекулы 85RbI. Расчет был выполнен для природной смеси изотопов рубидия.
Погрешности рассчитанных термодинамических функций Rbl (г) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным
учетом изотопного состава иодида рубидия. При более высоких температурах дополнительный
вклад в погрешность вносят неточность использованного способа экстраполяции потенциальной
функции, а также пренебрежение возбужденными электронными состояниями. Погрешности
в значениях Ф°(Т) составляют 0,02, 0,05 и ЗДж-К^-моль при 298,15, 3000^и 6000 К
соответственно.
Термодинамические функции Rbl(r) ранее вычислялись в работе Брюэра и Брэкет [1660]
(Ф°(Г), Т ^ 2000 К). Расчеты в работе [1660] выполнены по молекулярным постоянным, близким
к принятым в настоящем издании; соответствующие расхождения с данными табл. 1219 не
превышают 0,4 Дж-К~1-моль в значениях Ф°(Г) и объясняются главным образом различием
метода расчета <?вн.
Константа равновесия реакции RbI(r)=Rb(r)+I(r) вычислена с использованием значения
Artfo@)=D0(RbI)=324,208+3,l кДж -моль'1 ^ 27 100 +260 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации Rbl (к). Этим величинам также^соответ-
ствует ^
l, г, |0) = —134,853)±|3,0 1кДж[. моль.
Практически совпадающее значение энергии диссоциации было получено|Берковицем [1499]
C23 ±4 кДж -моль) на основании фотоионизационных измерений. Учет теплового возбуждения
и кинетической энергии осколков в условиях этого эксперимента, выполненный Дрониным и
Гороховым [389], уменьшает для Rbl полученную величину только на 1 кДж-моль. Более низкое
значение было найдено методом фотофрагментарной спектроскопии в работе Су и Райли [50551
C15 +2 кДж -моль); оно противоречит не только перечисленным выше данным, но и
определению нижней границы возможных значений энергии диссоциации, которая была определена Зом-
мермайером [4345] по коротковолновому пределу флуктуационных полос в спектре поглощения
Rbl C20 кДж-моль). Спектрофотометрическое изучение равновесия реакции Rb(r)+HI(r)=
=RbI(r)+H(r) в пламенах в работе Булевич и др. [1719] A500—2600 К) привело к значению
D0(RbI)=340+8 кДж'МОЛь, по-видимому, из-за_ неустановленной ошибки^ в интерпретации
данных. |
Rb2I2(r)- Термодинамические свойства газообразного дииодида дирубидия|
в^стандартном"состоянии в интервале температур 100—6000 К приведены в табл. 1220.
Молекулярные постоянные, использованные для расчета термодинамических функций Rb2I2,
приведены в табл. 48.6. На основании теоретических расчетов [961, 4689] дляРмолекулы Rb2I3
в основном электронном состоянии XMJb справочнике?принимаетсярллоская|ромбическая
структура (симметрия Dih). Приведенное в'табл.Ц^З.бЦпроизведение моментов|инерции соответствует
структурным параметрам r(Rb—1)=3,39 ±0,10 А и Д l—Rb—1=101+10°, найденным Соло-
моником в результате расчета по ионной модели [961, 963]. Погрешность значения IAIBh
составляет 5-1011 г3-см6. Приведенные в табл. 48.6 значения частот v5, ve найдены Шабером и
Мартином [4167] в ИК-спектре молекул Rb2I2, изолированных в матрице из Аг. Для частот vj—v4
приняты значения, рассчитанные Соломоником [961, 963]. Погрешности принятых частот
оцениваются в 25 см для частоты v1? 15 см — для частоты v4, 20 см — для остальных частот.
Термодинамические функции Rb2I2(r) вычислялись в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям. A.^3)—A. 6), A.9), A.10), A. 122)—A. 124), A.128),
A. 130) без учета возбужденных состояний. Погрешности рассчитанных величин обусловлен»
450
Рубидий и его соединения
Rb2SO4(K, ж)
неточностью принятых молекулярных постоянных, а также приближенным характером расчета;
они составляют 10, 15 и 20 Дж-К-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции Rb2I2(r) рассчитывались в работах Краснова и др. [569]
(до 3000 К), Бифелда [1557] (до 1300 К) и Фрурипа и Бландера [2340] (до 5000 К). Результаты
этих расчетов согласуются с данными табл. 1220 в пределах 2 Дж-К-моль для [569, 1557]
и 5 Дж-К~1-моль для [2340] в значениях Ф°(Г).
Константа равновесия реакции Rb2I2(r)=2Rb(r)+2I(r) вычислена с использованием значения
дг#°@)=805,710+12 кДж-моль", соответствующего принятой в справочнике энтальпии
образования!
A/&°(Rb2I2): г,|0) = — 427|±|122[кДж|.(моль-1.
Принятая величина основана на результатах исследований равновесия реакции RbI(K, ж)+
+RbI(r)=Rb2I2(r), приведенных в табл. 48.18. Приведенные в таблице погрешности учитывают
Таблица 48.18. Результаты определений энтальпии реакции Rbl(u, ж) + КЫ(г)=КЬ21г(г)
Авторы
Метод
ДЛ«@)
II закон
III закон
Мургулеску, Топор, 1970 [3598] Статический и протока, 1269—1324 К, 7 точек —54±100 39,4±2,8
Бифельд, 1978 [1557] Масс-спектрометрический, 720—846 К, 6 точек 42 ±4 40,3 ±2,0
Ефимова, 1981 [417] То же, 771—910 К, 7 точек 35 ±6 43,9 ±1,7
только воспроизводимость измерений. Для дальнейших расчетов принимается среднее значение
Дг#°@)=41 +10 кДж-моль. Погрешность его оценена с учетом неточности термодинамических
функций, которым соответствует погрешность в А,Л°@), равная 16 кДж-моль для измерений
при 1300 К и 10 кДж -моль для измерений при 900 К.
Rb2SO4(K, ж). Термодинамические свойства кристаллического и жидкого сульфата дирубидия
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1221.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние ЙЬ2ЭО4(к) в интервале 0—931 К принята
ромбическая а-модификация, а в интервале 931—1343 К — гексагональная ^-модификация
[44561 l. &ZT 2
При Т < 298,15 К термодинамические функции Rb2SO4(K) вычислены по измерениям
теплоемкости, выполненным Пауковым и Лаврентьевой [762]| A2—303 К, образец чистотой 99,5%).
Среднее отклонение экспериментальных точек от сглаженной кривой не превышает 0,5% в
интервале 12—20 К и 0,13% при Т > 20 К. Графическая экстраполяция теплоемкости привела к
значению 5°A2 К)=0,82 Дж-К'1-моль.! Погрешности значений 5°B98,15 К) и Я°B98,15 К)—
—Я°@), приведенных в табл. 48.1, оцениваются в 0,4 Дж •К-моль и 0,08 кДж-моль
соответственно.
Анализ экспериментальных данных поТэнтальпии Rb2SO4, полученныхгв работах Соколова
и др. [957] G40—1334 К) и Дениелоу [2041, 2043, 2044] D00—1523 К), показал, что полиморфное
превращение при 931 К не имеет изотермической энтальпии перехода. Поэтому теплоемкость
а-модификации Rb2SO4 в интервале 298,15—931 К описана двумя уравнениями. Первое
уравнение описывает медленное возрастание теплоемкости в интервале 298,15—800 К и получено
совместной обработкой результатов измерений теплоемкости, выполненных Шмидт [1153] B98—
770 К), и данных по энтальпии, полученных в работах [957] и [2041, 2043, 2044]; при обработке
их погрешности оценивались в 0,5, 0,5 и 1% соответственно. Второе уравнение в интервале 800—
1 В работах [80, 1516] и [4456J прп помощи рентгенографического и дилатометрического методов и метода ДТА
были получены указания о существовании у RboSO,, (к) фазовых переходов при 573 и 723 К [1516], 471 и 556 К
[80], 548, 748 и 848 К [4456]. Согласно [4456] эти переходы протекают без изменения структуры ромбической
модификации с малыми энтальпиями переходов. Ввиду противоречивости приведенных данных, а также в связи
с тем, что эти переходы не были обнаружены при измерениях теплоемкости, в справочнике они не учитывались.
451
Rb2SO4(r) Глава 48
931 К аппроксимирует восходящую ветвь Х-кривой и выведено усреднением согласующихся между
собой данных по энтальпии [957, 2041, 2043, 2044]. Результаты измерений Полищука и др. [800]
(819—1300 К) не учитывались, так как они лежат выше данных других работ на 5—8%,
по-видимому, из-за систематической ошибки измерений.
Температура полиморфного превращения (931 +3 К) принята по согласующимся данным
[1516, 2764, 2778] (метод термического анализа), [2262,4456] (высокотемпературная
рентгенография) и [800, 2041, 2043, 2044] (калориметрические измерения). Поскольку полиморфное
превращение при 931 К не имеет изотермической энтальпии перехода (см. выше), имеющиеся в литературе
данные по энтальпии этого перехода [800, 2041, 2043, 2044, 2778] B,2—8,8 кДж-моль) не
учитывались; эти определения относятся к интервалу аномальной части теплоемкости <x-Rb2SO4
вблизи температуры превращения.
Уравнение для теплоемкости в интервале 931—1343 К (см. табл. 48.1) получено усреднением
данных по энтальпии, найденных Соколовым и др. [957] и Дениелоу [2041, 2043, 2044].
Температура плавления A343 +4 К) принята после усреднения измерений, проведенных в работах
[373, 374, 379, 794, 795, 1057, 1136, 2041, 2043, 2764, 2808, 4213]. Менее точные данные [80, 83,
84, 159, 852, 853, 3581, 4195] A163—1333 К) не учитывались. Энтальпия плавления 37,3 +
+0,8 кДж -моль рассчитана из данных по энтальпии жидкого сульфата рубидия, полученных
Дениелоу [2041, 2043, 2044], и значению #°A343 К)-#°B98,15 К) для Rb2SO4(K), полученному
по принятым выше уравнениям.
Теплоемкость жидкого сульфата рубидия B06,5+6 Дж-К"-моль~1) рассчитана из данных
по энтальпии Rb2SO4(ffl), полученных в работах [2041, 2043, 2044] A353—1532 К).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,3,
2, 6 и 15 Дж-К~1-моль~1 соответственно.
Таблица термодинамических функций Rb2SO4(K, ж) публикуется впервые.
Принятая в справочнике энтальпия образования
к, 298,15 К) = —1435,9 ± 0,6 кДж • моль
основана на результатах измерений энтальпии растворения Rb2SO4(K) в воде, выполненных
Воробьевым и др. [244] и Кузнецовой и др. [598]. В этих работах были получены значения
ДядЯ°(соН20, 298,15 К)=23,69+0,15 кДж-моль [244] A300-5200 Н2О, 298,15 К) и 24,42 +
+0,15 кДж-моль [598] C3—11 111 Н2О, 298,15 К). Для дальнейших расчетов принимается
среднее значение 24,06+0,30 кДж-моль, которому соответствует принятая энтальпия
образования Rb2SO4(K). Близкое значение 23,6 кДж-моль было вычислено по результатам измерений
Форкрана [2295].
Давление пара в реакции Rb2SO4(K, ffi)=Rb2SO4(r) вычислено с использованием принятой
в справочнике энтальпии сублимации
Дг#°@) = \#°(Rb2SO4, к, 0) = 343 + 20 кДж • моль.
Принятая величина является средней из найденных по результатам эффузионных
исследований Кубиччотти [1969] A090—1295 К, 19 точек, 376+10 кДж-моль (II закон) и 345,8 +
+0,8 кДж-моль (III закон)) и Яганнатана, Уайята [2825] A159—1337 К, 15 точек, 349 +
+30 кДж-моль (II закон) и 339, 9+1,4 кДж-моль (III закон)). Испарение Rb2SO4
сопровождается частичной диссоциацией с образованием Rb(r), SO2(r) и О2(г). С использованием принятых
в настоящем справочнике термодинамических свойств продуктов разложения были вычислены
парциальные давления Rb, SO2 и О2 в насыщенном паре ив результаты измерений [1969] и [2825]
внесены соответствующие поправки. Приведенные выше величины относятся к испарению
сульфата рубидия в виде молекул Rb2SO4(r). Погрешность принятой величины определяется в
основном неточностью термодинамических функций.
По результатам масс-спектрометрического исследования, выполненного Фикалорой и др.
[2247] A010—1230 К, 20 точек), по уравнению II закона была найдена близкая величина 339 +
+10 кДж-моль.
Rb2SO4(r). Термодинамические свойства газообразного сульфата дирубидия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 1222.
Молекулярные постоянные Rb2SO4, принятые для расчета термодинамических функций,
приведены в табл. 48.6. Молекула Rb2SO4 имеет четное число электронов, поэтому в справочнике
452
Рубидий и его соединения RbNO2(K, ж)
ее основное электронное состояние рассматривается как синглетное (Х^-А). Сведения о
возбужденных электронных состояниях Rb2SO4 в литературе отсутствуют. На основании результатов
исследований ИК-спектров молекул Rb2SO4, изолированных в матрицах инертных газов [91,
92, 93, 360], а также результатов электронографического изучения структуры молекул Cs2SO4
и K2SO4, в справочнике для молекулы Rb2SO4 принимается «бициклическая» равновесная
геометрическая конфигурация (симметрия D2d), содержащая тетраэдрическую группу SO4 и
расположенные симметрично относительно нее два атома Rb, связанные с двумя атомами кислорода
каждый. Произведение моментов инерции, приведенное в табл. 48.6, вычислено по структурным
параметрам r(Rb—О)=2,65+0,10 А (оценка по величинам г(К—О), r(Rb—О) и r(Cs—О) в
молекулах K2SO4, Cs2SO4, МОН и RbNO3), r(S—O)=l,47+0,02 А и Д O-S-O=109,5+5° (оценка
по K2SO4 и Cs2SO4). Погрешность значения IaIbIc составляет 4-10"12 г3-см6. Приведенные
в табл. 48.6 частоты v5, v6, vg и v9, отвечающие колебаниям группы SO4, найдены Дворкиным [360]
в ИК-спектре молекул Rb2SO4, изолированных в матрице из Аг. Частота vx группы S04 принята
равной ее величине в молекулах Na2SO4 и K2SO4, для которых она наблюдалась в спектре КР
в матрице из N2 [1356]. Частоты v2 и v4 этой группы приняты по расчету для ионной модели
Rb2SO4 [360]. Частоты колебаний связей Rb—О приняты по результатам исследования ИК-спектра
молекул Rb2SO4, изолированных в аргоновой матрице (v7 и v10), и результатам расчета по ионной
модели (v3 и vn) в работах Беляевой и др. [91, 92, 93, 360]. Погрешности принятых частот
оцениваются в 10—15 см для ve, v7, v9, v10 и vn, в 20—25 см для v3, v5 и v8, в 40 см для vx
и в 100 см для v2 и v4.
Термодинамические функции Rb2SO4(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Погрешности рассчитанных значений обусловлены
приближенным характером расчета и неточностью принятых молекулярных постоянных; они
оцениваются в значениях Ф (Т) при 298,15, 3000 и 6000 К в 10, 17 и 25 Дж-К~1-моль
соответственно.
Ранее термодинамические функции Rb2SO4(r) вычислялись в работе Кубиччотти [1969] (до
1300 К). Расх'ождения данных [1969] и табл. 1222 достигают 22 Дж-К-моль~1 в значении
Ф°A300 К) и обусловлены тем, что Кубиччотти принял другую структуру этой молекулы и
существенно отличающиеся значения постоянных.
Константа равновесия реакции Rb2SO4(r)=2Rb(r)+S(r)+4O(r) вычислена с использованием
значения АгН°@)=2509,461 +20 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации Rb2SO4(K).
Этим величинам также соответствует
O4, г, 0) = —1083,160 +20 кДж . моль.
RbNO2(K, ж). Термодинамические свойства кристаллического и жидкого нитрита рубидия
в стандартном состоянии при температурах 298,15—2500 К приведены в табл. 1223.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние RbNO2(K) в интервале 0—261 К
принимается моноклинная модификация, а в интервале 261—695 К — кубическая модификация [3996] *¦ 2.
Экспериментальные данные по теплоемкости RbNO2(K) при Т <^ 298,15 К отсутствуют,
поэтому приведенные в табл. 48.1 значения ?ОB98,15 К) и Я°B98,15 К)—#°@) были получены
оценкой по методу Карапетьянца [485 ] на основании соответствующих данных в рядах нитриты—
матабораты щелочных металлов. Погрешности этих величин составляют 8 Дж-К~1-моль~1 и
0,8 кДж-моль соответственно.
При Т ]> 298,15 К для теплоемкости RbNO2(K) принято уравнение, полученное на основании
значений С B98,15 К)=84 Дж-К-моль~1 и СрF95 К)=95 Дж-К~1-моль, которые были
оценены по теплоемкости нитритов других щелочных металлов. Температура плавления F95+2 К)
принята по измерениям Верхотурова и др. [209] и Проценко и др. [822, 827, 828, 834, 839]; ме-
1 Температура фазового превращения B61+2 К) измерена в работах [827, 828, 3996]. В работе [3633] при изучении
свойств RbNO3 (к) методами ДТА и электропроводности был отмечен эндоэффект при 359 К, который был
приписан неизвестному ранее превращению. Однако данные [3633] не могут рассматриваться как достаточно
надежные, поскольку исследуемый образец содержал —1,2% воды.
2 При высоких давлениях у RbNO2 стабильны еще две фазы с неизученной структурой [1009].
453
RbNO2(r) Глава 48
нее точные данные получены в работах [824, 830, 832, 837]. Энтальпия плавления A2,1 +
+1,2 кДж-моль) найдена Верхотуровым и др. [209] в результате термографических измерений.
Теплоемкость RbNO2(>K) A04+10 Дж-К~1-моль~1) оценена по данным о теплоемкости жидких
нитритов натрия и калия.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 2500 К оцениваются в 5,
15, 25 и 30 Дж-К^-моль.
Таблица термодинамических функций RbNOa(K, ж) публикуется впервые.
Принятая в справочнике энтальпия образования
A,ffe(RbNO,, к, |298,15 К) = —367 + 5 кДж • моль
была''оценена сопоставлением энтальпий образования и энтальпий растворения в рядах MNO2(k)
и MNO3(k), где M=Li, Na, К, Rb, Cs. Исследования равновесия реакции 2RbNO3^)=
=2RbNO2^)-j-O2(r) компенсационным методом, выполненные Центнершвером [1773] (см. также
[3123]) (802—825 К, 5 точек, —384 кДж-моль (II закон) и —399 кДж-моль (III закон)), а
также измерения энергии связи D0(Rb—NO2)=375 кДж-моль, выполненные Верхотуровым
и;!др. [208] методом электронного yflapaJ(A^°(RbNO,, к, 298,15 К) = —440 кДж-моль),
приводят |к менее точным'зн i ¦: :.,иям (см. тексты по LiNO2(K), NaNO2(K), KNO2(k) и CsNO2(k)).
t|| Давление пара в реакции RbNO2(K, ж)=RbNO2(г) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯф@) = \H°(RbNOa, к, 0) = 187|±|10 кДж
Принятая величина основана на масс-спектрометрическом исследовании, выполненном
Верхотуровым и др. [205, 206]. Опыты с двойной эффузионной камерой показали, что основным
компонентом пара является мономер (количество димера составляет 1,5 мол.%). Парциальное давление
мономера при 686 К было определено методом полного изотермического испарения; принятое
значение рассчитано из этих данных по уравнению III закона. Его погрешность обусловлена
в основном неточностью термодинамических функций RbNO2(K) и RbNO2(r). Измерения интен-
сивностей ионных токов в интервале^623—686ЛК, выполненные в^этих^аботах^привели к
значению 177 кДж-моль
RbNO2(r). Термодинамические свойства газообразного нитрита рубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1224.
Молекулярные постоянные, использованные для расчета термодинамических функций
RbNO2(r), приведены в табл. 48.6. произведение моментов инерции в состоянии ХгАх вычислено
пля плоской циклической молекулы (симметрия С2р) по структурным параметрам r(Rb—О)=
=2,64+0,02 A, :r(N—О)=1,252+0,005 А и Д О—N—0=116+3°, наиденным1Куликовым и др.
[606] при электронографическом исследовании паров RbNO2 (~570 °С). ИК-спектры молекул
RbNO2, изолированных в матрице из Аг, изученные Беляевой и др. [88], и ИК-спектры паров
над RbNO2 при 600—900 °С, наблюдавшиеся Шаповаловым и др. [1141, 1144], также были
интерпретированы в предположении о плоском циклическом строении этой молекулы. Погрешность
значения IaIbIo оценивается в 1-1014 г3-см6.
Принятые значения частот v3 (колебание связи Rb—О) и v4 (колебание связи N—О) найдены
Беляевой и др. [88] в ИК-спектре молекул RbNOa в матрице из Аг. Близкие значения v4
получены Миллиганом и Джекокс [3508] в матрице из Аг и Шаповаловым и др. [1141, 1144] в ИК-
спектре паров. Частота чх (колебание связи N—О) принята такой же, как в NO2, поскольку она
практически характеристична в спектрах нитритов 1, а частота v2 (колебание О—N—О) —
такой же, как в молекуле CsNO2 (см. ниже). Приведенные в табл. 48.6 значения v5 (колебание Rb—
О—N) и ve (неплоское колебание цикла) были оценены, первое — по величине этой частоты в
молекуле NaNO2 и изменению частоты аналогичного колебания в ряду молекул M2SO4, второе —
по величине аналогичного колебания в молекуле Rb2SO4 (vlx). Погрешности принятых значений
v3, v4 и ve оцениваются в 20 см, частот v2 и v5 — в 40 см, частоты vx — в 50 см. Сведения
о возбужденных электронных состояниях молекулы RbNO2 в литературе отсутствуют.
1 Шаповалов и др. [1141, 1144] отнесли к частоте ч1 полосу в ИК-спектре паров над RbNO2 в области 1460 см;
это отнесение, так же как сделанные в этпх работах оценки частот v3, v5 и ve, представляется ошибочным.
454
Рубидий и его соединения RbNO3(K, ж)
Термодинамические функции RbNO2(r) вычислены по уравнениям A. 3)—A.'6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний и возможных изомеров этой молекулы. Погрешности
рассчитанных термодинамических функций обусловлены неточностью принятых значений
молекулярных постоянных, а также применением приближенного метода расчета; они составляют
6, 10 и 15 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000К.
Таблица термодинамических функций RbNO2(r) публикуется впервые.
Константа равновесия реакции RbNO2(r)=Rb(r)+N(r)+2O(r) вычислена с использованием
значения Дг.?Го@)=1229,471+11 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации RbNO2(K).
Этим величинам также соответствует
^ г, 0) = —182,895 + И кДж • моль.
RbNO3(K, ж). Термодинамические свойства кристаллического и жидкого нитрата рубидия
в стандартном состоянии при температурах 100—2500 К приведены в табл. 1225.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние RbNO3(K) в интервале 0—437 К
принимается гексагональная модификация (kIV), в интервале 437—493 К — кубическая модификация
(структурный тип GsGl, кШ), в интервале 493—556 К — гексагональная, модификация (к11)
и в интервале 556—583 К — кубическая (структурный тип NaCl, к1) [3740] х> 2.
При Т <^ 298,15 К термодинамические функции RbNO3(K) вычислены по результатам
измерений теплоемкости, выполненным Березовским и Пауковым [131] (8—319 К, содержание
примесей менее 0,02 мае. %, содержание воды менее ~1-10~3 мас.%, среднее отклонение
экспериментальных значений теплоемкости от сглаженной кривой ~0,25% в интервале 8—20 К и 0,04% —
от 20 до 319 К). По оценке авторов [131] 5°(8 К)=0,14 Дж-К^-моль. Приведенные в табл. 48.1
значения 5°B98,15 К) и #°B98,15 К)—#°@) имеют погрешности 0,3 Дж-К^-моль и
0,04 кДж-моль" соответственно.
При Т > 298,15 К для теплоемкости четырех кристаллических модификаций RbNO3(K)
приняты уравнения, полученные на основании результатов измерений теплоемкости в работе
Аристовой и др. [4849] C00—690 К). Измерения выполнены на образце, использованном в работе
Березовского и Паукова [131], их погрешности оцениваются в 0,6%. При выводе уравнений были
использованы следующие значения: для RbNO3(KlV) — С;B98,15 К)=99, С°C80 К)=109,4
и С;D30 К)=119,2 Дж-К^-моль; для RbNO3(KIII) - <?;D60 К) = 138,7 и С;D90 К)=
= 142,8 Дж-К^-моль; для RbNO3(KlI) — С;E05 К)=132, СрE35 К)=141 и С;E50 К)=
= 149 Дж-К~1-моль. Для теплоемкости RbNO3(Kl) из-за ограниченности данных [4849] в
интервале 556—583 К принято постоянное значение A46 Дж-К-моль), равное теплоемкости
RbNO3^). Результаты измерений теплоемкости в работе Мустайоки [3605] C24—632 К) на
образце неизвестной чистоты, по-видимому, ненадежны (систематически завышены) и не
учитывались.
Температура фазового превращения RbNO3(KlV) -> RbNO3(KlII) D37+2 К) принята по
работам [77, 359, 491, 1272, 2077, 2237, 2346, 2974, 4849, 4914, 5044]; в работах [358, 834, 1692,
3605, 3740, 3880, 3983] были получены значения в интервале 433—440 К. Температура фазового
превращения RbN03(KlII) -> RbNO3(KlI) D93 +2 К) принята по данным [358, 839, 1340, 1692,
1871, 1872, 2346, 2798, 4849, 4914]. Температура фазового превращения RbNO3(KlI) ->
->RbNO3(Kl) E56+2 К) принята по измерениям [1872, 2235, 2970, 2972, 3605, 3738, 3983, 4122,
4849, 4914]; в работах [793, 839, 1692, 1871, 2237, 2330, 3690] получены более высокие значения
E59—564 К). Температура плавления RbNO3 E83+2 К) определена в работах [839, 1272, 2237,
3605, 3739, 3983, 4177, 4914]. Энтальпии фазовых переходов C,9+0,2 кДж-моль, 3,2+0,2 кДжХ
Хмоль, 0,97+0,20 кДж-моль) приняты по результатам согласующихся измерений [1340,
3376, 3605, 3983, 4849]. Энтальпия плавления D,6+0,2 кДж-моль'1) определена авторами [3037,
1 Фазовое превращение RbNO8 (к) при 22S К, о котором сообщают авторы [2235, 2236], пе было обнаружено в
работах других исследователей.
2 Ромбическая фаза высокого давления RbNO3 стабильна при р > 24 кбар (при 608 К) [1009].
455
RbNO3(r)
Глава 48
3374, 3605, 3983, 4849]; менее точные значения были получены в работах 1209] E,4 кДт-моль~г)
и [3376] D,3 кДж-моль).
Теплоемкость RbNO3(ffl) A46+3 Дж-К^-моль-1) принята по данным [4849] E90—690 К,
19 измерений). В работе [3605] E88—632 К, 13 измерений) получено более высокое значение
A62 Дж-К-^моль).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 2500 К оцениваются в 0,2,
2, 10 и 15 Дж-К^-моль-1.
Таблица термодинамических функций RbNO3(K, ж) публикуется впервые.
В справочнике принята энтальпия образования
i\fH°(RbNOs, к, 298,15 К) = —494,7 + 0,5 кДж • моль,
основанная на результатах измерений энтальпии растворения RbNO3(K) в воде, приведенных
в табл. 48.19. Наиболее точные и детальные измерения были проведены Воробьевым и др. [244];
Таблица 48.19. Результаты определений AajBro(RbN03, к-»ооН20, 298,15 К) (в кДжмоль)
Авторы
Хейг, 1912 [2548]
Воробьев и др., 1966 [244]
Иоффе и др., 1974 [464]
Ояри, 1971 [746]
Крестов, Егорова, 1967 [582]
Крестов, Харитонов, 1974 [585]
Конечная концентрация, температура
измерений
400 Н20, 294,2 К, 3 измерения
720 Н2О, 298,15 К, 4 измерения
1430 Н2О, 298,15 К, 2 измерения
1760 Н2О, 298,15 К, 5 измерений
2860 Н2О, 298,15 К, 2 измерения
12,9—17362 Н2О, 298,15 К, 26 измерений
1000 Н2О
298,15 К
1111—9260 Н2О, 298,15 К
ДвгЯ°(ооН2О, 298,15 К)
36,65 ±0,50
36,74+0,25
36,69 ±0,25
36,69+0,25
36,74 ±0,25
36,7
37,3+0,6
36,3
36,6
на основании этих измерений в дальнейших расчетах используется Aa3H°(coH.20, 298,15 К)=
=36,71 +0,25 кДж-моль. Результаты остальных работ совпадают с этой величиной в
пределах их погрешностей.
Давление пара в реакции RbNO3(K, ж)=RbNOз(г) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = A,#°(RbNO3, к, 0) = 183 + 8 кДж • моль.
Принятая величина основана на масс-спектрометрическом исследовании, выполненном Ба-
гарятьян и др. [56, 57]. Опыты с двойной эффузионной камерой показали, что основным
компонентом пара является мономер (количество димера при 680 К составляет 2 мол.%). Парциальное
давление мономера при 639,4 К было найдено методом изотермического испарения; принятое
в справочнике значение вычислено по этим данным и уравнению III закона. Его погрешность
обусловлена в основном неточностью термодинамических функций RbNO3(r). Измерения интен-
сивностей ионных токов, выполненные авторами этих работ F38—679 К), приводят к значению
198+8 кДж-моль.
В работе [4851] методом электронного удара была также определена энергия связи RbNO3r
равная 457+14 кДж-моль'1; ей соответствует значение AsH°@)=191 +25 кДж-моль,
совпадающее в пределах погрешности с принятым в справочнике.
RbNO3(r). Термодинамические свойства газообразного нитрата рубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1226.
Молекулярные постоянные, использованные для расчета термодинамических функций
RbNO3(r), приведены в табл. 48.6. Куликов и др. [605] при высокотемпературном электроногра-
фическом исследовании нашли, что молекула RbNO3 имеет плоскую бидентатную конфигурацию
(симметрия С2г) с Д Оц—N—Оц=115+5°, средним значением r(N—О)=1,252 +0,003 А и r(Rb—
Оц)=2,65+0,03 А. Приведенное в табл. 48.6 произведение моментов инерции в состоянии Х1А1
вычислено по данным работы [605], однако если учесть, что электронографический эксперимент
456
Рубидий и его соединения Ш>2СО3(к, ж)
не чувствителен в пределах 0,1 А к возможной неравноценности длин связей N—О, в справочнике
приняты r(N—О„)=1,29+0,02 А и r(N—Овнеш)=1,21 +0,02 А. Погрешность значения 1л1в1с
оценивается в 3-1014 г3-смв. Принятые значения частот валентных колебаний vt, v2, v5 и
неплоского колебания v8 иона NO.s найдены Смитом и др. [4319] в ИК-спектре молекул RbNO3,
изолированных в матрице из Ат1. Шаповалов [1141] наблюдал в ИК-спектре поглощения паров над
RbNO3(K) полосы 1630, 1460, 1270 и 1020 см и отнес их к валентным колебаниям группы NOJ,
выдвинув гипотезу о присутствии в паре двух изомеров — тридентатного (симметрия СЪг)) и би-
дентатного (симметрия C2v), приняв за более устойчивый первый из них. Однако исследования
спектров молекул нитратов щелочных металлов, изолированных в матрицах, а также результаты
неэмпирических расчетов LiNO3 ставят под сомнение эту гипотезу и отнесение полосы 1630 см
к RbNO3. Ранее Шаповалов и др. [1144] дали для молекулы RbNO3 некорректные рекомендации
значений всех (кроме v1=1460 см) частот. Принятые в справочнике значения частот
деформационных колебаний иона NOg в плоскости (v3 и ve) перенесены из молекулы KNO3, поскольку
для расплавов нитратов щелочных металлов наблюдается весьма незначительное уменьшение
этих частот в ряду Li—Na—К—Rb—Cs (см., например, [4638]). Близкие к принятым значения
частот колебаний иона NO3 измерены в спектрах кристаллов и расплавов RbNO3 [706, 803, 1685,
2058, 2476, 2828, 2841, 4638]. Для частот, характеризующих колебания иона Rb+ относительно
иона NO3 (v4, v7 и ve), приняты значения соответствующих частот молекулы RbNO2 (см. выше).
Погрешности значений частот v1; v2, v5, v8 и v9 оцениваются в 15—20 см, остальных частот—
в 40—50 см. Сведения о возбужденных электронных состояниях RbNO3 в литературе
отсутствуют.
Термодинамические функции RbNO3(r) вычислены по уравнениям A. 3)—A. 6), A. 9)г
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний и возможной изомерии молекулы RbNO3. Погрешности
рассчитанных термодинамических функций обусловлены неточностью принятых значений
молекулярных постоянных, а также применением приближенного метода расчета, они составляют
8, 15 и 20 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К (в том числе 3—
3,5 Дж-К-моль из-за неточности молекулярных постоянных).
Таблица термодинамических функций RbNO3(r) публикуется впервые.
Константа равновесия реакции RbNO3(r)=Rb(r)+N(r)+3O(r) вычислена с использованием
значения Дг#°@)=1599,853+8,0 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации R1jNO3(k). Этим величинам соответствует также
AfH°(RbNOz, г, 0) = —306,494 ± 8,0 кДж • моль.
Ш>2СО3(к, ж). Термодинамические свойства кристаллического и жидкого карбоната дирубидия
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1227.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 48.1. За стандартное состояние Rb2CO3 в интервале 0—576 К принята моноклинная (а)
модификация, а в интервале 576—1146 К — гексагональная (Р) модификация [3029].
При Т <^ 298,15 К термодинамические функции Rb2CO3(K) вычислены по результатам
измерений теплоемкости, выполненных Пауковым и др. [773] A4—316 К, образец с суммарным
содержанием примесей ~0,1%). Среднее отклонение экспериментальных значений от сглаженной
кривой не превышает 0,15%. Экстраполяция данных [773] по закону Дебая дает 5°A4 К) =
= 1,5 Дж-К^-моль. Погрешности значений 5°B98,15 К) и #°B98,15 К)—Я°@), приведенных
в табл. 48.1, оцениваются в 0,4 Дж-К~1-моль~1 и 0,04 кДж-моль соответственно.
В литературе отсутствуют какие-либо сведения о термодинамических свойствах Rb2CO3(K, ж)
при Т ^> 298,15 К. Уравнение для теплоемкости a-Rb2CO3 в интервале 298,15'—576 К (см.
табл. 48.1) оценено на основании сопоставления данных по теплоемкости a-модификаций Na2CO3
и К2СО3. Температура полиморфного превращения E76+3 К) принята по результатам
термографического исследования, проведенного Рейсманом [3968] в атмосфере СО2 на образце чистотой
99,9%, и согласуется в пределах погрешности с данными Клемента и Коэна [3029] E73 К),
полученными при обычном давлении. Значение Г^=935 К, найденное Жженых и Кащеевым [422]
1 В соответствии с рекомендацией Битти и др. [1444] в справочнике дано отнесение ч1 hv5, обратное приведенному
в работе [4319].
457
RbBO2(K, ж) Глава 48
при исследовании двойных систем Rb2GO3—Rb2SO4, представляется ошибочным и не учитывалось.
Энтальпия полиморфного превращения A,3+0,6 кДж-моль) оценена из сопоставления
термограмм, полученных в работах [3968] для К2СО3 и Rb2GO3. Уравнение для теплоемкости j3-Rb2CO3
(см. табл. 48.1) получено на основании сравнения экспериментальных данных по энтальпии и
теплоемкости высокотемпературных модификаций карбонатов натрия и калия. Температура
плавления A146+5 К) принята по данным Рейсмана [3968]. Менее надежные измерения [376,
378, 422] A001—1138 К) не учитывались. Энтальпия плавления C0+6 кДж-моль) оценена
на основании предположения о равенстве суммы энтропии превращения и плавления карбонатов
щелочных металлов. Теплоемкость Rb2CO3^) B05+20 Дж-К-моль~1) оценена на основании
соответствующих данных для К2СО3(ж) и соотношения, приведенного в т. 1, разд. 2.5.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,2,
6, 20 и 35 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
Rb2CO3(K), приведенными в справочнике Барина и Кнакке [1395] (до 1113 К) и в табл. 1227,
составляют 6—8 Дж-К~1-моль~1 и обусловлены в основном различием принятых значений
5°B98,15 К).
Термодинамические функции Rb2CO3^) публикуются впервые.
Константа равновесия реакции ЙЬ2СО3(к, ж)=2Rb(г)^-C(г)+ЗO(г) вычислена с
использованием значения Дг-?Г°@)=2743,947+4,3 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
bfH°(RbaCO3, к, 298,15 К) = —1132,6 + 4,0 кДж • моль.
Принятая величина основана на измеренных Форкраном энтальпиях реакций 2RbOH(p-p)+
+CO2(p-p)=Rb2CO3(p-p)+H2O(p-p), Дг#°B88 К) = — 86,1 кДж-моль [2297] и Rb2CO3(K)+
+2000H2O=Rb2CO3(p-p, 2000H2O), ДГЯB88 К)= — 38,0 кДж-моль [2298]. Эти данные вместе
с данными [1009] для реакции растворения СО2 в воде приводят к значению ДГДГB88 К)=
=—67,8 кДж-моль для реакции 2RbOH(p-p)+CO2(r)=Rb2CO3(K)+H2O^). Приближенный
пересчет этой величины к стандартной температуре дает для энтальпии реакции Дг//°B98,15 К) =
= —63,6 кДж-моль, которой соответствует значение &/H°(Rb2GO3, к, 298,15 К)=
=—1132,7 кДж-моль.
Пересчет энтальпии растворения Rb2GO3(K) в воде, найденной в работе [2298], к стандартной
температуре приводит к значению Дг#°B98,15 К)=—41,1 кДж-моль (поправка для
бесконечного разбавления была оценена в предположении, что энтальпия разбавления раствора Rb2GO3
такая же, как для Rb2SO4 [3184]). С учетом поправки на гидролиз (—2,6 кДж-моль, см. [4099])
для реакции Rb2C03(K)+coH20=2Rb+(a?)+COj2(ag) было найдено Дад#°(соН20, 298,15 К) =
= —44,8 кДж-моль, чему соответствует AjH°(Rh2GO3, к, 298,15 К) =—1132,6 кДж-моль.
Результаты расчетов по приведенным двум схемам совпадают. Косвенным подтверждением
надежности измерений в работах Форкрана является также то, что в аналогичных измерениях
с 1л2СО3(к) и Cs2CO3(k) (cm. соответствующие тексты) этим автором были получены величины,
совпадающие с более точными современными данными в пределах 1,5 кДж-моль и 4,0 кДжХ
Хмоль соответственно.
В работе Раффелини и Май Ле Вана [3910] сообщается об измерении стандартной энтальпии
растворения Rb2GO3(K) в воде, Дад#°(оэН20, 298,15 К) =—26,3+2,6 кДж-моль". В этой
работе не приводится непосредственно измеренная величина, концентрация конечного раствора,
метод учета поправки на гидролиз и на разведение до бесконечного разбавления, что затрудняет
оценку надежности полученной величины и ее использование для расчета энтальпии образования
Rjd2CO3(k). Расхождение результатов измерений [3910] и [2298] намного превышает их
оцененные погрешности. Учитывая отсутствие первичных данных в публикации [3910], значительное
расхождение полученной в ней энтальпии растворения Gs2CO3 с другими данными (см. ниже),
в справочнике отдано предпочтение более старым измерениям Форкрана [2297,Ц2298].
Ш)ВО2(к, ж). Термодинамические свойства кристаллического и жидкого метабората рубидия
в стандартном состоянии при температурах 100—3000 К приведены в табл. 1228.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 48.1. В справочнике за стандартное состояние RbBO2(K) приняты в интервале 0—968 К
^-модификация, а в интервале 968—1133 К а-модификация с неизученными структурами.
298,15|К термодинамические функции вычислены по результатам измерений тепло-
458
Рубидий и его соединения ' RbBO2(r)
емкости, выполненных Пауковым и др. [772] A2—313 К, примеси Na, К, Cs и Са в количествах
до 3-1СГ2 мае. % каждого элемента, среднее отклонение экспериментальных точек от сглаженной
кривой не превышало 0,18%). Экстраполяция теплоемкости ниже 12 К по закону Дебая
приводит к величине <5°A2 K)=0,59 Дж-К~1-моль~1. Погрешности значений 5°B98,15 К) и
#°B98,15 К)—#°@), приведенных в табл. 48.1, оцениваются в 0,25 Дж-К^-моль и 0,03 кДжХ
Хмоль соответственно.
При Т > 298,15 К для теплоемкости C-RbBO2 принято уравнение, полученное на основании
значений С;B98,15 К)=73,39 [772], С;F00 К)=94 и Ср(968 К) = 108 Дж-К^-моль.
Последние две величины оценены из данных для теплоемкости метаборатов лития и натрия. Для
теплоемкости <x-RbBQ2 принято постоянное значение A08 Дж-К-моль~1).
Температуры полиморфного превращения (968+5 К) и плавления A133+10) найдены Кохером
и Ролле [3058, 4042] методом ДТА. Оценка температуры плавления по диаграмме состояния
системы Rb2O—В2О3, полученной Димитровым и др. [2081], приводит к близкому, но более
высокому значению A151 К). Сведения об энтальпии полиморфного превращения в литературе
отсутствуют, в справочнике она принята равной нулю. Энтальпия плавления C1+6 кДж-моль)
оценена на основании предположения, что энтропии плавления метаборатов рубидия и натрия
равны.
Теплоемкость КЬВО2(ж) A45+15 Дж-К~1-моль") оценена равной теплоемкости жидкого мета-
бората лития. Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К
оцениваются в 0,2, 6, 15 и 23 Дж-К^-моль.
Таблица термодинамических функций RbBO2(K, ж) публикуется впервые.
Принятая в справочнике энтальпия образования
AfH°(RbBO2, к, 298,15 К) = —975 + 20 кДж ¦ моль
оценена методом, предложенным Карапетьянцем, в результате сопоставления энтальпий
образования в рядах соединений МВО2(к) и МОН(к), где М — щелочной металл.
Давление пара метабората рубидия в реакции RbBO2(K, ж)=RbBO2(г) вычислено с
использованием принятой, в справочнике энтальпии сублимации
ДД°@) = A,#°(RbBO2, к, 0) = 295 ± 20 кДж • моль.
Принятая величина основана на измерениях давления пара, представленных в табл. 48.20,
где в качестве погрешностей приведены воспроизводимости измерений. Приведенные в таблице
Таблица 48.20. Результаты определений AsJ5r°(RbBO2, к, 0) (в кДж-моль")
Авторы
Метод
Д,#°(ЙЫЮ2, к, 0)
II закон
III закон
Адаме, Квон, 1966 [1250] Протока, 1151—1272 К, 5 точека 296 294,1
Горохов и др., 1971 [ЗЦ, 642] Эффузионный, 1028—1110 К, 5 точек 256+60 296,0+2,0
Масс-спектрометрический, 1008—1084 К, 6 серий 296 +10 —
а Данные представлены в виде уравнения.
величины совпадают в пределах указанных погрешностей. Принятая величина основана на
обработке по уравнению III закона результатов измерений, выполненных методом протока [1250]
и эффузионным методом [311 ¦, 642]. Погрешность принятой величины определяется в основном
неточностью термодинамических функций.
RbBO2(r). Термодинамические свойства газообразного метабората рубидия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1229.
Молекулярные постоянные, использованные для расчета термодинамических функций
RbBO2(r), приведены в табл. 48.6. Электронографические исследования паров RbBO2 [401, 402,
2211], исследование ИК-спектра различных изотопических модификаций молекул RbBO2, изоли-
459
RbLi(r) Глава 48
рованных в матрицах из Аг и Кг [4237], и расчет функции потенциальной энергии на основании
ионной модели [400] показали, что молекула RbBO2 в основном электронном состоянии Х1А'
имеет угловую конфигурацию с линейным фрагментом О—В=О (симметрия Cs). В электроногра-
фических исследованиях [401, 2211] для Д Rb—О—В получено эффективное значение 112+5°,
в работе [4237] на основании изотопического сдвига частот было найдено значение 90°, а минимуму
потенциальной энергии RbBO2 в расчете [400] соответствует равновесный угол в 105+5°. Расчет
[400] показал также, что энергии линейного и циклического изомеров этой молекулы превышают
энергию нелинейного изомера на 800 +100 и 4600 +300 см соответственно, и, по-видимому,
ион Rb+ может относительно свободно перемещаться вокруг иона ВОг.
Приведенное в табл. 48.6 произведение моментов инерции рассчитано по структурным
параметрам Д Rb—О—В = 130+30° (эффективное значение, см. ниже GsBO2, для которого электроно-
графический эксперимент дал наиболее надежное значение), r(Rb—О)=2,55+0,05 А, г(В—О) =
= 1,31+0,03 А и г(В = О)=1,23+0,02 А (средние из данных [402, 2211 ]).Погрешность значения
IaIbIc оценивается в 7-Ю14 г3-см6. Значения частот чг—v5, принятые в справочнике, рассчитаны
для природной смеси изотопов бора на основании результатов изучения ИК-спектра молекул
RbBO2, изолированных в матрицах, в работе Сешедри и др. [4237]. Полосы vx—v4, наблюдавшиеся
в работах Мальцева [648] и Квона и Адамса [3904] в ИК-спектре паров над RbBO2(K), согласуются
с данными [4237] в пределах 5—50 см. Частота v6, приведенная в табл. 48.6, оценена на
основании значений этой частоты у молекул LiBO2, NaBO2 и КВО2. Погрешности принятых значений
v1 и v2 оцениваются в 40 см, остальных частот — в 20—25 см.
Термодинамические функции RbBO2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A.10), A. 122)—A. 124), A.128)
и A. 130) без учета возбужденных состояний и изомерии молекул RbBO2. Погрешности
рассчитанных значений определяются неточностью принятых молекулярных постоянных и применением
приближенного метода расчета; они составляют 10, 15 и 20 Дж-К-моль~1 в значениях Ф°(Т)
при 298,15, 3000 и 6000 К.
Таблица термодинамических функций RbBO2(r) публикуется впервые.
Константа равновесия реакции RbBO2(r) = Rb(r)+B(r)-(-2O(r) вычислена с использованием
значения &гН°@)=1811,675 +28 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации RbBO2(K). Этим величинам также соответствует
A/ff°(RbBO2, г, 0) == —675,917 + 28 кДж • моль.
RbLi(r). Термодинамические свойства газообразного рубидия-лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1230.
В табл. 48.4 приведены молекулярные постоянные RbLi, принятые на основании результатов
спектральных исследований и приближенных оценок. В электронном спектре RbLi исследована
система полос в области 5680—5800 А [4646, 4688], которую по аналогии с молекулой KNa можно
отнести к переходу СШ <- X1S+. Как и другие гетероядерные молекулы щелочных металлов,
молекула RbLi должна иметь ряд стабильных низколежащих состояний, коррелирующих с
атомами RbBP)+LiBS) (а3П,^11Е+, &32+) и RbB?')+LiBP) (В1!,*), а также состояния с энергией около
23 000 см, коррелирующие с атомами RbBZ))-|-LiB5).
Молекулярные постоянные RbLi в состоянии ХХ2+ приняты по оценке, сделанной по данным
для гомоядерных молекул [1772]. Принятое значение све хорошо согласуется с найденным в
результате приближенного анализа системы полос СХП <— Х1!!", выполненного Вейцелем и Кульпом
[4688] (сое?«185 см). Погрешности принятых значений ше и Ве оцениваются в 10 и 0,01 см
соответственно. Постоянные RbLi в состоянии СХП приняты по данным [4688]. Энергии остальных
возбужденных состояний оценены по соотношению A. 89) и данным для молекулы KNa, их
погрешность составляет 1000—2000 см.
Термодинамические функции RbLi(r) были вычислены по уравнениям A. 3)—A. 10), A. 93)—
A. 95). Значения <?ън и ее производные вычислялись по соотношениям A. 90)—A. 92) с учетом
пяти возбужденных электронных состояний, если принять ОД2.БР =PiQw.*r Статистическая
сумма по колебательно-вращательным уровням энергии состояния X1 ?+ и ее производные
находились по уравнениям A. 73)—A. 75) непосредственным суммированием по колебательным уровням
и интегрированием по / с помощью соотношений типа A. 82). В расчете учитывались все уровни
с / <^ Лпах,»! последние находились по формуле A. 81). Уровни энергии вычислялись по уравне-
460
Рубидий и его соединения Rb Na(r)
ниям A. 64а) и A. 65) с коэффициентами YM, приведенными вместе со значениями итах и /цт
в табл. 48.5. Величины Ykl для изотопомера, соответствующего природной смеси изотопов, были
получены по соотношениям A. 66) и постоянным молекулы 86Rb7Li из табл. 48.4. Для обеспечения
условий A. 50) и A. 59) дополнительно введены коэффициенты Y30 и Y03 соответственно.
Погрешности вычисленных термодинамических функций RbLi(r) обусловлены главным образом
неточностью молекулярных постоянных, значения энергии диссоциации и энергий возбужденных
электронных состояний, а также пренебрежением состояниями с Те ^ 23 000 см; погрешности
в значениях Ф°(Г) при 298,15, 3000 и 6000 К составляют 0,5, 1 и 4 Дж -К -моль соответственно.
Таблица термодинамических функций RbLi(r) публикуется впервые.
Константа равновесия реакции RbLi(r) = Rb(r) + Li(r) вычислена с использованием принятого
в справочнике значения
ДгЯ @) = D0(RbLi) — 73,9 + 2,0 кДж ¦ моль я« 6180 ± 170 см.
Эта величина была оценена как среднее между энергиями диссоциации молекул Rb2 и Li2.
Принятое значение существенно отличается от теоретической оценки [4139] E5 кДж-моль).
Принятой энергии диссоциации соответствует
A/ff°(RbLi, г, 0) = 166,027 + 2,4 кДж • моль.
RbNa(r). Термодинамические свойства газообразного рубидия-натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1231.
Молекулярные постоянные RbNa, принятые на основании результатов спектральных
исследований и приближенных оценок, приведены в табл. 48.4. В спектре RbNa исследована одна система
полос СХП <- X12+ [3147, 4646, 4688]. По аналогии с другими двухатомными гетероядерными
соединениями щелочных металлов молекула RbNa должна иметь и другие стабильные триплетные
и синглетные состояния, коррелирующие с валентными состояниями атомов RbBP)+NaB>S)
(а3П, 4Х2+, Ь3?+) и RbBS)+Na(aP) {BXZ+), а также состояния с энергией ~20 000 см и
соответствующие пределу RbBZ))+NaB<S).
Приведенные в табл. 48.4 колебательные постоянные RbNa в состояниях X1 Е+ и C4Q получены
Кушем [3147] по кантам полос системы СХП <- ХХ2 + (и" ^ 6, и' ^ 5) в спектре магнитного
вращения. Вращательные постоянные в состоянии Z1S+ оценены по постоянным молекул Rb2 и Na2,
погрешность принятого значения Ве составляет 0,004 см. Энергии возбуждения состояний
41!!", а3И, Ь3Е+ и S1!!" были оценены по соотношению A. 89) и данным для молекулы KNa, их
погрешности составляют 1000—2000 см.
Термодинамические функции RbNa(r) вычислены по уравнениям A. 3)—A. 10) и A. 93)—
A. 95). Величина Qsn и ее производные рассчитывались по уравнениям A. 90)—A. 92) с учетом пяти
возбужденных состояний в предположении, что <?koLbp=.P.(?k5.bp- Статистическая сумма по
колебательно-вращательным уровням состояния Z1S+ и ее производные вычислялись
непосредственным суммированием по колебательным уровням и интегрированием по / с помощью
уравнений типа A. 82). В расчете учитывались все уровни этого состояния со значениями / ^ /шм „,
последние находились по формуле A. 81). Энергия уровней рассчитывалась по уравнениям A. 64а)
и A. 65) с коэффициентами Ykl, приведенными в табл. 48.5 вместе со значениями vmaj% и /lim.
Значения Y1H для изотопомера RbNa, соответствующего природной смеси изотопов рубидия и натрия,
были получены по формулам A. 66) и постоянным молекулы 85RbNa из табл. 48.4. Для
соблюдения условий A. 50) и A. 59) были дополнительно введены коэффициенты У8о и ^оз-
Погрешности вычисленных термодинамических функций обусловлены неточностью постоянной
Ве в состоянии Х12+, энергии диссоциации RbNa и энергий низколежащих электронных
состояний, а также пренебрежением рядом состояний с энергией ^> 20 000 см. Погрешности в
значениях Ф°(Т) при 298,15, 3000 и 6000 К могут достигать 0,3, 0,6 и 4,0 Дж-К-^моль.
Таблица термодинамических функций RbNa(r) публикуется впервые.
Константа равновесия реакции RbNa(r)=Rb(r)+Na(r) вычислена с использованием принятого
в справочнике значения
Дг#°@) = D0(RbNa) = 55,2 + 4,0 кДж • моль «* 4610|+ 330 см.
Эта величина была рекомендована Гейдоном [2377 ] на основании скорректированного значения,
полученного линейной экстраполяцией колебательных уровней с помощью постоянных,
найденных Кушем [3147].
461
RbK(r) Глава 48
Принятой энергии диссоциации соответствует
г, 0) = 134,755 ±4,1 кДж ¦ моль.
RbK(r). Термодинамические свойства газообразного рубидия-калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1232.
Экспериментальные данные по молекулярным постоянным RbK отсутствуют; приведенные
в табл. 48.4 постоянные RbK приняты на основании приближенных оценок. Колебательные и
вращательные постоянные в состоянии X1 Е+ были оценены по постоянным молекул Rb2 и К2.
Погрешность принятых значений ше и Ве не превышает 4 и 0,002 см соответственно. Энергии
низколежащих электронных состояний RbK оценены по соотношению A. 89) и данным для
молекулы KNa, их погрешность может достигать 1000—2000 см.
Термодинамические функции RbK(r) вычислены по уравнениям A. 3)—A. 10) и A. 93)—A. 95).
Величина QBB и ее производные рассчитывались по уравнениям A. 90)—A. 92) с учетом пяти
возбужденных состояний в предположении, что i Qi2.T.s=PiQifl.Bi!' Статистическая сумма ,по
колебательно-вращательным уровням энергии состояния X1 Е + и ее производные вычислялись
непосредственным суммированием по колебательным уровням и интегрированием по / с помощью
уравнений типа A. 82). В расчете учитывались все уровни этого состояния со значениями / ^ /Ш|„
последние находились по формуле A. 81). Энергия уровней рассчитывалась по уравнениям A. 64а)
и A. 65) с коэффициентами Ykl, приведенными в табл. 48.5 вместе со значениями уши и /нт.
Значения Yы для изотопомера, соответствующего природной смеси изотопов рубидия и калия,
вычислялись по формулам A. 66) и постоянным 86RbK из табл. 48.4. Для соблюдения условий A. 50)
и A. 59) были добавлены коэффициенты Yso и У03.
Погрешности вычисленных термодинамических функций обусловлены неточностью оценки
молекулярных постоянных и энергии возбужденных электронных состояний, а также
пренебрежением рядом состояний с энергией больше 20 000 см. Погрешности в значениях ФС(Г) при 298,15,
3000 и 6000 К могут достигать 0,5, 1 и 5 Дж-К-моль соответственно.
Таблица термодинамических функций RbK (г) публикуется впервые.
Константа равновесия реакции RbK(r)=Rb(r)+K(r) вычислена с использованием принятого
в справочнике значения
|ДгЯ°@) = D0(RbK) = 48,3|±B,0 кДнф моль-^ЩШО ±|1
Эта величина была оценена как среднее между энергиями диссоциации молекул Rb2 и К2.
Принятое значение находится в удовлетворительном согласии с теоретическими оценками [4139,
4844] D4 и 47 кДж-моль).
Принятой энергии диссоциации соответствует
г, 0) = 123,783 + 2,2 кДж • моль.
Глава 49
ЦЕЗИЙ И ЕГО СОЕДИНЕНИЯ
Св(к?яс), Се, Cs+, Cs2, CsO, CsO2(k, ж), Cs2O(k, ж), Cs2O, Cs2O+, Cs2O2(k, ж), Cs2O2, CsH(k, ж),
CsH, CsOH(k, ж), CsOH, Cs2O2H2, CsF(k, ж), CsF, Cs2F2, CsC1(k, ж), CsCl, Cs2Cl2, CsBr(K, ж), CsBr,
Cs2Br2,|.CsI(K, ж), Csl, Cs2I2, Cs2SO4(k, ж), Cs2SO4, CsNO2(k, ж), CsNO2, CsNO3(k, aic),|CeNO,t
Cs2CO3(k, ж), Cs2CO3, CsBO2(k, ж), CsBO2, CsLi, CsNa, CsK,CsRb
В настоящей главе рассматриваются цезий и его соединения с кислородом, водородом,' галогенами,
другими щелочными металлами, а также соли цезия и некоторых кислородсодержащих^ кислот —
всего 28 веществ. Для 15 веществ в справочнике представлены сведения об их свойствах в
конденсированном состоянии, для 27 веществ — в газообразном состоянии. Термодинамические свойства
Cs(r) и Cs+(r) рассчитаны до 10 000 К, остальных газов — до 6000 К. В Приложении 3.10 для
Cs приведены сведения о критических постоянных, а также данные, ^позволяющие учесть
отклонения свойств Cs(r) от его свойств в стандартном состоянии.
В справочнике не рассматриваются такие ионизированные соединения цезия, как CsJ, CsJ,
Cs2F+, CsFi, его комплексные соединения типа CsMXM и некоторые другие. Эти соединения могут
присутствовать в некоторых системах, но ограниченность данных затрудняет расчеты их свойств.
Приближенные модельные оценки влияния некоторых из этих соединений на свойства
соответствующих систем могут быть выполнены на основании данных, приведенных в^справочнике для
соединений лития и калия.
Cs(k, ж). Термодинамические свойства кристаллического ш[ жидкого цезия в стандартном
состоянии при температурах 100—2000 К приведены в табл. 1233.
Значения постоянных, использованных для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние Cs(k) в интервале 0—301,59 К принята
кубическая модификация (структурный тип Na). Термодинамические функции твердого цезия
вычислены по измерениям теплоемкости в работах Филби и Мартина [2251 ] @,4—301 К) и
Мартина [3409] @,4—3 К), проведенных на образцах цезия чистотой лучше 99,9%. Данные [1997,
3259, 3399, 3444] менее точны и в расчетах не учитывались. Погрешности принятых значений
?°B98,15 К) и #°B98,15 К)—#°@), которые совпадают с рекомендованными КОДАТА—МСНС
[1887], оцениваются в 0,4 Дж -К -моль и 0,02 кДж -моль соответственно. Принятая в
справочнике температура плавления C01,59+0,02 К) определена в работах Гоутса и др. [2428, 3736]
для образца чистотой 99,9%. Измерения [639, 1886, 2016, 2251, 3311, 3409, 4005, 4487] менее точны
и не учитывались. Энтальпия плавления B,096 ±0,005 кДж-моль) принята по совпадающим
данным Мартина [3409] и Филби и Мартина [2251]; значения, полученные в работах [639, 1886,
3358], менее точны, хотя и не противоречат принятому значению.
Уравнение для теплоемкости жидкого цезия (см. табл. 49.1) получено совместной обработкой
результатов измерений энтальпии в работе Шпильрайна и Кагана [1165] D72—1330 К) и значений
энтальпии, вычисленных по измерениям теплоемкости в работе Благонравова и Филиппова [138]
A200—1900 К) г. При обработке погрешность данных [1165] была оценена в 3%, а данных [138] —
в 5%. Полученному уравнению для теплоемкости Св(ж) соответствует уменьшение теплоемкости
от С° C01,59 К)=32,6 Дж-К-моль до С°р(800 К)=29,1 Дж «К'1-моль (минимум теплоемкости),
а затем возрастание до значения С°B000 К)=62,7 Дж-К»моль. Погрешности аппроксимации
энтальпии жидкого цезия можно оценить в 1% при 1000 К, 1,5% при 1500 К и 2% при 2000 К.
Погрешности табулированных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 0,3,
0,7 и 1,5 Дж -К"-моль. Значения термодинамических функций Cs(k, ж), приведенные во втором
издании настоящего справочника [337] {Т < 2400 К), в таблицах JANAF [2834] (Т <; 1500 К),
книге Шпильрайна и др. [1179] (Т ^ 1500 К) и в справочнике Халтгрена и др. [2752]' (Г <^
<; 1400 К), согласуются с данными табл. 1233 в пределах 1—2 Дж-К^-моль в значениях Ф°B");
имеющиеся расхождения обусловлены различием выбора значения S°(Cs, к, 298,15 К) и тем, что
в настоящем справочнике учтены измерения теплоемкости в работе [138].
1 Данные [138] были пересчитаны к давлению 1 атм и вводились в обработку в виде восьми значений энтальпии
в интервале 1200—1900 К.
463
Таблица 49.1. Принятые значения термодинамических величин для цезия и его соединений в кристаллическом
и жидком состояниях
Вещество
Состояние
Ю
GO ,—^
кДж ¦ моль
и"
оо"
оэ
о
Дж • К
Cs к, куб. 7,711 85,23
ж — _
CsO2 kII, тетр. (а) 18 142
к1, куб. (г) — _
ж —
Cs2O к, гекс. 17,68 146,9
ж — __
Cs2Oa к, ромб. 21,3 180
ж — _
CsH к, куб. 10,5 73
ж — _
CsOH kII, ромб. 16 102
к1, куб. — _
ж _
CsF к, куб. 11,76 92,96
ж — _
CsCl kII, куб. (а) 12,45 101,17
к1, куб. (Р)
ж _ _
CsBr к, куб. 13,135 112,94
Ж —
Csl к, куб. 13,47 122,20
ж — _
Cs2SO4 Kill, ромб, (а) 27,74 211,91
KlI, (р) _ _
к1, гекс. (у) .
ж _ _
CsNO2 к1, куб. 23,42 174,0
ж _
CsNOa к11г гекс. (а) 20,05 153,83
к1, куб. (р) —
ж _ _
Gs2CO3 к 25,727 204,47
ж _
CsBO2 к, куб. 14,368 104,35
ж
Примечание. С°р{Т)=а+Ъ -Т—с -T^+d Г2+е -Т3 (в Дж
ю
00
СП
о"»,
СО
• МОЛЬ
32,21
—
79,0
—
76,00
.
95,0
40,6
.
73,0
—
51,09
52,47
—
52,93
—
52,47
134,893
—
91,34
,
96,14
—
—
123,85
80,58
—
К -моль-i).
Cs: а <г= 24,449-Ю-6. Cs2SO4: a d=24,449-Ю, е=205 089-Ю"9
Csl: a d=54,926 10-е. CsNO2: a d=49,317 -Ю-".
Коэффициенты в уравнении
а
4,922
46,727
53,408
88
100
66,024
100
83,469
134
34,443
56
60,255
80
83
42,306
72
55,095
65,2
73,5
48,908
75
61,097
71
142,364
-195,000
84,942
207
95,725
106
47,608
183,872
150
122,928
205
84,329
145
для Ср(Т)
6-Ю3
91,525
—40,865
85,837
.
33,461
38,675
36,467
. .
42,746
29,462
3,527
13,489
-39,968
80,837
442,708
117,414
—29,412
162,777
-38,214
71,895
29,630
—.
с • 10~5
3,630 а
.
4,192
3,268
1,416 а
21,480 а
, ,
0а
, ,
72,131
18,235
11,186
Интервал
температур
Ttr
или Тт
К
298,15—301,59
301,59-2000
298,15-403
403-723
723—2000
298,15-768
768—2500
298,15—867
867—2000
298,15-801
801—1200
298,15-497
497—616
616—3000
298,15—976
976—3400
298,15—743
743—919
919—3400
298,15-910
910—3400
298,15—905
905—3400
298,15—920
920-1000
1000—1288
1288—3500
298,15—679
679—2500
298,15-427
427—682
682—2500
298,15—1066
1066—3500
298,15-1005
1005—3000
301,59
403
723
768
.
867
801
497
616
976
743
919
910
.
905
920
1000
1288
679
427
682
1066
1005
.—
LtrH°
или \пН°
кДжХх
X моль
2,096
.
0
18,4
20
22
15
7Д
7,3
21,7
2,93
20,38
23,6
25,65
0
0
35,1
10,9
3,6
13,8
31
27
—
Цезий и его соединения
Cs(k, ж)
В справочнике в качестве стандартного состояния цезия принято кристаллическое состояние
и в соответствии с этим
Давление пара цезия в реакции Cs(k, ?K)=Cs(r) было вычислено с использованием значения
Дг#°@)=78,014 +1,0 кДж -моль, соответствующего принятой в справочнике энтальпии сублимации
As#°(Cs, к, 298,15 К) = Д/Я°(С8, г, 298,15 К) = 76,5±1,0 кДж • моль.
Принятая величина основана на представленных в табл. 49.2 результатах измерений давления
насыщенного пара цезия. Расчет энтальпии сублимации проводился для модели идеальной смеси
химически реагирующих атомарной и молекулярной компонент. При высоких температурах
учитывалась поправка на отклонение от идеальности с использованием уравнения состояния,
полученного Эвингом. Эти расчеты могли быть проведены для интервала 980—1672 К. Метод
расчета изложен в Приложении 8.
В результате проведенных расчетов для большинства приведенных в табл. 49.2 работ получены
хорошо согласующиеся значения энтальпии сублимации. Приведенные в таблице погрешности
Таблица 49.2. Результаты определения AsH°(Cs, к, 298,15 К) (в кДж-моль)
Авторы
Метод
As#°(Cs, к, 298,15 К)
II закон III закон
Руфф, Йохансен, 1905 [4091]
Хакшпиль, 1913 [2541, 2542]
Крёнер, 1913 [3119]
Скотт, 1924 [4221]
Тейлор, Лангмюр, 1937 [4487]
Бонилла и др., 1962 [1611]
Теппер и др., 1963 [4493, 4494]
Аченер, 1964 [1208]
Бук, Паули, 1965 [1709]
Эвинг и др., 1966 [2204, 2206]
Стоун и др., 1966 [44021
Боданский, Шине, 1967 [1603,
1604]
Шпильрайн, Белова, 1967 11162]
Воляк и др., 1968 [239]
Новиков, Рощупкин, 1969 [733]
Эвинг и др., 1970 [2207]
Шпильрайн и др., 1970 [1175]
Шине и др., 1971 [4184]
атм, 76,9 ±8,0
76,06
77,45 ±0,68J
77,67 ±0,26
73,73 ±0,15
76,71 ±0,04
80,8 ±1,0 76,20 ±0,05 б
96,8 ±2,1
78,7 ±2,0
75,8 ±1,6
Точек кипения, 943,15 К, 1 атм, 1 точка
Статический, 503—670 К, 3 -Ю-4—1-10
10 точек
То же, 523—629 К, 4 -Ю—9 -10 атм, 12 точек 81,1 ±4,4
То же, 321—387 К, 5-Ю"8—4-Ю"8 атм, 8 точек 74,6 ±2,8
Ионизационный, 239—346 К, 8 -Ю-13—1 -10-' атм, 76,19 ±0,73
|14 точек
Точек кипения, 674—1200 К, 0,03—7,15 атм,
41 точка
То же, 729—1334 К, 0,07—13,6 атм, 22 точки
То же, 752—1144 К, 0,1—4,8 атм, 14 точек
Ионизационный, 293—348 К, 1 -Ю"9—1 -10"' атм,
9 точек
Статический, 943—1545 К, 1,0—32,1 атм, 47 точек 79,65 ±0,20 76,41 ±0,03 б
То же, 921—1475 К, 0,8—24,7 атм, 66 точек 77,95 ±0,12 76,52 ±0,02 б
гТочек кипения, 723—1137 К, 0,06—4,8 атм, 78,86 ±5,93 76,10 ±0,24б
17 точек
Статический, 823—1198 К, 0,3—6,6 атм, 121 точка 78,3 ±2,5
То же, 563—946 К, 2-Ю-3—1,04 атм, 45 точек 79,7 ±1,3
То же, 500—1400 К, 4 -Ю"*—18 атм, данные пред- 79,12 ±0,34
ставлены в виде уравнения
То же, 1118—1730 К, 4,2—56,1 атм, 19 точек 80,2 ±0,6
То же, 732—1348 К, 0,08—14,78 атм, 46 точек 79,1 ±0,20 76,50 ±0,02 б
Точек кипения, 723—1139 К, 0,07—4,8 атм, 79,13 ±0,31 76,34 ±0,06б
59 точек
Чернеева, Проскурин, 1972 [1111] То же, 1028—1872 К, 2,1—80,5|атм, 162 точки 80,79 ±0,11
78,1 ±1,1 б
76,64 ±0,05 б
76,71 ±0,12
76,6 ±0,1 б
76,48 ±0,20
76,57 ±0,07 б
76,44 ±0,11 б
б
Шпильрайн,
[1173]
Воляк и др., 1975 [243]
Гущин и др., 1975 [351]
Беренс и др., 1977 [1452]
76,13+0,41 б
Никоноров, 1972 То же, 675—1499 К, 0,03—27,4 атм, 56 точек 79,19 ±0,15 76,46 ±0,05 б
То же, 954—1637 К, 1,15—42,6 атм, 15 точек 79,5 ±1,1 76,86 ±0,4б
То же, 483—642 К, 2 -10"*—1 -Ю атм, 69 точек 77,21 ±0,26 76,87 ±0,01
Эффузионный, 327—448 К, 4 -Ю"8—3 -Ю атм, 77,5 ±2,7 77,23 ±0,27
34 точки
а1Прявэдены величины, рассчитанные с учетом образования в насыщенном паре димерных молекул.
6 Введены поправки на отклонение от идеальности. Учтены только измерения, выполненные при 980 < Т < 1672 К.
465
Cs(r)
Глава 49
отражают только воспроизводимость измерений. Рекомендованное значение вычислено как
средневзвешенное нз наиболее надежных данных [4487], полученных ионизационным методом, измерений
[1173, 1208, 1611, 4184], выполненных методом точек кипения, и данных работ [239, 733, 1162,
1175, 2204, 2206, 2207, 4402], в которых применялись различные варианты статического метода.
Всего при вычислении средневзвешенного использовано 416 точек, лежащих в интервале 238—
1672 К. Погрешность принятой величины обусловлена главным образом погрешностью
термодинамических функций цезия в конденсированном состоянии, которым соответствуют погрешности
в энтальпии сублимации от 0,1 кДж-моль для измерений, выполненных при 300 К, до 2,2 кДжХ
Хмоль для измерений при 1700 К. Принятое значение энтальпии сублимации цезия совпадает
со значением, рекомендованным КОДАТА—MGHC [1887]. Работы [495, 620, 2343, 3191, 3192,
3193, 3511, 3881], где измерялись величины, пропорциональные давлению пара, при выборе
энтальпии сублимации цезия не учитывались. Работа [4288], в которой измерения были выполнены
в основном при Т > 1672 К, также|не учитывалась при выборе энтальпии сублимации.
Cs(r). Термодинамические свойства газообразного цезия в стандартном состоянии в интервале
температур 100—10 000 К приведены в табл. 1234.
Уровни энергии атома Cs, использованные в расчете термодинамических функций, приведены
в табл. 49.3, в которой даны валентные состояния, соответствующие электронным конфигурациям
Т а б|л и ц а 49.3. Уровни энергии атомов Cs и
Электронная
ция
Cs . . .6s
..Ар
. . .bd
. . Ad
Состояние
Ti
см
25 0
2Р1А 11 178,2697
2Р8/ 11 732,3079
2Z)8/ 14 499,2568
Ю% 14 596,8452
2?>з/з 22 588,793
Ю% 22 631,658
Cs+, принятые
Pi
2
2
4
4
6
4
6
Cs
Cs+
цля расчета термодинамических функций
Электронная
конфигурация
...4/
...5/
...5*
...6/
• • Ag
. . Ah
. . .5р«
Состояние
2G
2F
2G
2tf
*5
CM"
24 472,12
26 971,22
27 010
28 329,45
28 347
28 356
0
Pi
14
14
18
14
18
22
1
. . .5p66s—. . .5p6?>h, . . .5p65d—. . .5pe5g и . . .5pe4/. Принятые в этой таблице значения основаны
на результатах спектральных исследований Клеймана [3024], Эрикссона и Венакера [2187],
Йоханссона [2862 ], а также рекомендаций Мур [3550 ]. Данные различных авторов несущественно
отличаются друг от друга. Для состояний . . .6рBР), . . AfBF), . . .5fBF), . . .6/BF) приняты
значения, усредненные по данным [2187 ] для двух соответствующих подсостояний.
Термодинамические функции Cs(r) вычислены по уравнениям A.3)—A.-6), A. 9), A. 10),
A. 23)—A. 25) непосредственным суммированием по уровням энергии. Погрешность вычисленных
значений термодинамических функций при Т <^ 4000 К определяется главным образом неточностью
фундаментальных постоянных и не превышает 0,02—0,03 Дж-К-моль~1 в значениях Ф°(Г)
и S (Т). При более высоких температурах погрешности возрастают из-за пренебрежения ридбер-
говскими состояниями и при 6000 К и 10 000 К могут достигать соответственно 0,3 и 3 Дж-КХ
Хмоль в значениях S°(T).
Ранее термодинамические функции Cs(r) вычислялись неоднократно, в том числе в предыдущем
издании настоящего справочника [337], таблицах JANAF [2834] до 6000 К, а также в работах
Гурвича и др. [335], Хилзенрата и др. [2652] и Рейтера [3973] до 10 000 К, Кольского и др. [3067]
до 8000 К и Эванса и др. [2195] до 2500 К. Результаты расчетов [337, 2834] при Т < 3000 К,
а также расчета [2652] во всем интервале температур совпадают с данными табл. 1234 в пределах
0,01—0,02 Дж -К -моль. При Т > 3000 К результаты расчетов [335, 337, 3067, 3973] возрастают
с температурой быстрее, чем в настоящем справочнике: при 10 000 К расхождения в значениях
Ф°(Т) для расчетов [335, 3973] достигают приблизительно 9—10 Дж-К-моль (см. Li(r)).
466
Цезий и его соединения Cs+(r), Cs2(r)
Энтальпия образования
&fH°(Cs, г, 0) = 78,014 + 1,0 кДж-моль
соответствует принятой энтальпии сублимации цезия.
Cs+(r). Термодинамические свойства газообразного положительного иона цезия в стандартном
состоянии в интервале температур 298,15—10 000 К приведены в табл. 1235.
В табл. 49.3 приведено только основное электронное состояние *S иона Cs+, относящееся к
конфигурации . . .5s25p6; состояния, относящиеся к конфигурациям . . .5р6Ы—. . .5p55g, имеют
энергии выше 100 000 см [3550], а другие возбужденные состояния Cs+ являются ридберговскими.
Термодинамические функции Cs+(r) были вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
при условии, что In (?BB=0. Погрешности вычисленных значений термодинамических функций
во всем интервале температур обусловлены только неточностью фундаментальных постоянных
и не превышают 0,02 Дж-К-моль.
Ранее термодинамические функции Cs+(r) вычислялись в предыдущем издании настоящего
справочника [337] и в таблицах JANAF [2834] (Т ^ 6000 К), а также в работах Хилзенрата
и др. [2652] (Т < 10 0С0 К) и Грина и др. [2473] (Г < 50 000 К). Результаты всех расчетов
совпадают с данными табл. 1235 в пределах 0,01—0,02 Дж-К^-моль.
Константа равновесия реакции Cs+(r)-f-e(r)=Cs(r) вычислена с использованием значения
ДгЛ°@) ——375,705+0,001 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Cs)=31 406,45+0,02 см=375,705 +0,001 кДж.моль.
Это значение выбрано на основании хорошо согласующихся между собой результатов измерений
ридберговских серий в работах Йоханссона [2862] C1406,47+0,01 см), Клеймана [3024]
C1 406,450+0,030 см), Эрикссона и др. [2186] C1 406,474 см), [2187] C1 406,454+0,005 см)
и Лоренцена с сотрудниками [3308] C1 406,46+0,03 см), [3309] C1 406,468+0,006 см).
Рекомендованное Мур [3549] значение C1 406,432+0,010 см)|совпадает с принятым в пределах
указанной погрешности.
Принятому значению соответствует
&fH°(Cs+, г, 0) = 453,719 + 1,0 кДж • моль.
Cs2(r). Термодинамические свойства газообразного дицезия в стандг,ртном состоянии в
интервале температур 100—6000 К приведены в табл. 1236.
В табл. 49.4 приведены молекулярные постоянные Cs2, принятые на основании анализа
результатов исследований электронного спектра и приближенных оценок. В спектре Cs2 наблюдалось
шесть систем полос: ЛХ1+ ^ Х*Г+ [2255, 2702, 3297, 3427], ВШ, *» Х1^ [2626, 2702, 3104,
3138, 3297, 3427], ЕЩи ^ X1!* [2625, 3053, 3147, 3297, 3427, 3907,4044], ЛП„ ?* Х1!* [3147],
G1!!,, ч* Х1!^ [3297] и а3Пя ^ Х1!^ [1468]. По аналогии с другими двухатомными молекулами
щелочных металлов у Cs2 следует ожидать существование еще ряда связанных низколежащих
состояний (i>3?J, СЩд и D1^), диссоциирующих на; атомы в состояниях ZS и 2Р. Особенностью
атома Cs по сравнению с атомами других щелочных металлов является то, что уровень 2Z> лежит
ниже первого ридберговского состояния 2S. Поетому можно предполагать, что состояния Е и F
коррелируют с состояниями атомов Cs B»S)-)-CsBD). Более высокие состояния, в том'числе
наблюдавшееся в спектре состояние С!1П11, в справочнике не рассматриваются. ;;
Молекулярные постоянные Cs2 в состоянии Х1!'^ приняты по результатам анализа большого
числа линий переходов А1^ -> Z]S+ и В1^ -» ХЩ в спектре флуоресценции (у" < 65),
выполненного Хонигом и др. [2702]. Колебательные постоянные для состояний ЯЧ!,,, 2?гПм и Рх11и
найдены Хесселом и Кушем [2625, 2626] и Кушем [3147] по кантам полос соответствующих систем.
Вращательные постоянные в состоянии Е1Ии получены Раабом и др. [3907] из анализа полосы
0—0 системы ^ХПЯ — ХгЩ в спектре лазерной флуоресценции. Молекулярные постоянные Cs2
в состояниях а3Пи и Аг^% приняты по данным Бенедикта и др. [1468], которые провели полуколи-
честЕенвый анализ систем а3П„ <- Х1^ и A1 2J. *- Х1!^. Погрешность соответствующих значений
Те оценивается в 500 см. Энергии состояний ЪЪЩ, D1^ и СхИд оценены путем сопоставления
соответствующих величин для молекул Cs2 и Na3. Их погрешность может достигать 1000—2000 см.
467
Таблица 49.4. Молекулярные постоянные Cs2, CsO, CsH, CsF, CsCl, CsBr, Csl, CsLi, CsNa, CsK, CsRb
Состояние
ХЩ
а3П„
A *S+
Ь32+
вти
стд
ff
Fma
Z2S+
А2П
A 12+
а3 А
Z12+
X12+
сш
.041
СШ
Г,
<°А
<*! • 103
De ¦ 107
СМ
0
7 850
9 450
11000
13 043,88
13 500
13 500
15 948
16 175,80
0
1 000 a
0
17 840,7
27 174
28 370 ж
0
0
0
0.
0а
16 477
0а
18 250
0а
0а
13 747,21
42,020 596
50
34
34,329 3
—
29,703 0
27,34
360
340 а
891,0
168,1
50
350
352,56
214,165
149,662
119,18
168 б
74
97 б
65
67 й
49,41 б
38,46
0,081 957 932 а
—
—
—
0,079 962 г
—
—
0,057 56 Д
0,073 3
1,5 а
2*4 а
12,93 а
—7,36 в
-0,39
37
1,62
0,731
3,740
2,505.
0,56 б
0,32 б
0,16 б
0,11 б
—
0,011 733 025
0,013 7
0,009 20
—
—
—
0,012 315 049 6
-—
0,207 а
0,190 а
2,709
1,056
0,8
1,64
0,184 37
0,072 091 5
0,036 069 3
0,023 627 4
0,18 б
0,060 5 б
0,029 4 б
0,016 3 б
—
0,022 422 338 6
—
—
—
—
—
0,035 7 е
—
1,2 б
1,6 б
57,9
—30 г
6,9
280 3
1,176 а
0,337 6 а
0,124 0 а
0,068 26 а
0,62 б
0,20 б
0,065 б
0,034 б
—
0,033 575 23 в
—
—
_
—
0,080 130 56 ж
—.
2,7 в :у :¦
2,4 в
1000 б
850 Д
300 е
300 б
2,017 б
0,326 7 б
0,083 8 б
0,037 15 б
8,3 в
0,94 в
0,23 в
0,069 в
—
А
4,646
4,3
5,25
—
—
—
—
4,532
—
2,39 а
2,49 а
2,494
3,995
4,6
3,21
2,345 46
2,906 26
3,072 26
3,315 15
3,745 б
3,77 б
4,36
4,46 б
—.
133Cs,
CsO
i33CsH
133CsF
133Cs35Cl
133Cs79Br
133CsI
133CsLi
CsNa
CsK
CsRb
Примечание. Ниже все величины даны в см.
Cs2: a (oei/e=—0,972 400 88-Ю-4, шА=0,223 394 2 -Ю, a>ege=-0,882 590 24-10-*; б а2=-0,620 290 69 -10, <х3=0,106 079 52 -10"8; в 81 =
=—0,154 815 77 -Ю0, ра=0,269 482 85 -Ю2, Не = —0,331 853 367 -Ю4; г юег/„=—0,1511 -Ю'3, о>,г, = 0,2860 -Ю-6; Д <оег/е=0,1608Х
ХЮ-2, o>eze = 0,130 12 -Ю; е вычислено по соотношению A.69); ж w = —0,100 295 1-Ю3, Le = — 0,717 023 5 -Ю8, М„ =
= -0,583 140 7 -10-2в.
CsO: а оценка; б вычислено по соотношению A. 69); в вычислено по соотношению A. 68).
~-- - - ¦-- .--.--. - - -- . - г а2=_2L-10-3,
«)вгв=— 3,22-Ю;
8 а„=0,04.
а3=—4,4-Ю; Д приведено значение
CsH: a а.Л=0,105, швге=1,78-10-3; " De^D,; B i»,y.=-0,268,
для у=3; е приведено значение для у=15; ж 40=14,65;
CsF: a a2=l,2-10-«; 6 81=3,1-Ю-10.
CsCl: а а2=3,4 -Ю-7: 6 е^З.8 -Ю-".
CsBr: а а2=1,02 -10; б В,=6,0 -Ю3.
Csl: а а2=4,893 38-10-8, ос3=—1,14412-Ю-10; б 81=—2,298 25-Ю2, Я,=—2,5043-Ю"", Le=-8,876 -Ю3.
CsLi: а а3П, Г„==11 300; А12+, Te—ll 300; Ь3Е+, Гв=15 400; В12+, Гв=15 700; 6 оценка, см. текст; в вычислено по соотношению A. 68).
CsNa: а а3П, Ге=11 300, А 12+, Гв=11 300, Ь3Е+, Гв=15 300, BW, Гв=15 600, Cm, Те=15 800; б оценка, см. текст; в вычислено по
соотношению A. 68).
CsK: а а3П, Г„=9700, А 12+, Ге=9700, 63S+, Ге=13 200, В W, Ге=13 500, СШ, ?\,= 13 600; б
A. 68).
CsRb: a а3П, Г,=9500,
Ге=9500, Ь32+, Те=12 900,
оценка, см. текст; в вычислено по соотношению
7",=13 200; б оценка, см. текст; в вычислено по соотношению A. 68).
м
Цезий и его соединения
Cs2(r)
Термодинамические функции Gs2(r) были вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 93)—A. 95). Значения Qm и ее производные вычислялись по соотношениям A. 90)—A. 92)
с учетом восьми возбужденных состояний, приведенных в табл. 49.4, в предположении, что <?^.вр=
=pi(??).Bp- Статистическая сумма по колебательно-вращательным уровням состояния X1 Е? и ее
производные находились непосредственным суммированием по колебательным уровням и
интегрированием по /с помощью соотношений типа A. 81). В расчете учитывались все уровни со
значениями / ^/шаХ; „> последние находились по формуле A.82). Уровни энергии вычислялись по
уравнениям A. 64а) и A. 65) с коэффициентами Ykl, приведенными вместе со значениями vma% и
/нш в табл. 49.5. Для обеспечения условия A. 59) был добавлен коэффициент YOi.
Погрешности рассчитанных термодинамических функций Gs2(r) при Т ^ 1000 К обусловлены
главным образом неточностью экстраполяции колебательно-вращательных уровней основного
состояния. При более высоких температурах основной вклад в погрешность вносит неточность
принятых энергий низколежащих состояний а3Ии и A1 Li. Общие погрешности в значениях Ф°(Т)
при 298,15, 3000 и 6000 К могут составить 0,05, 0,8 и 3 Дж-К-моль соответственно.
Термодинамические функции Cs2(r) ранее вычислялись в справочнике JANAF [2834], а также
в работах Рейтера [3973] (Т < 6000 К), Беренса и др. [1452] (Т < 2000 К) и Эванса и др. [2195]
(Т ^ 1500 К). В справочнике JANAF [2834] расчет выполнен приближенным методом и без учета
триплетных состояний, расхождения данных [2834] и табл. 1236 достигают 11 и 23 Дж -К -моль
Таблица 49.5а. Значения коэффициентов в уравнениях, описывающих уровни энергии (в см),
а также значения vm&x и </iim, принятые для расчета термодинамических
функций Cs2, CsO и CsH
Коэффициенты
те -ю-4
У10 -Ю-2
Уго
V -1П4
ув0 -1о6
¦у
У31-104
У31-Юв
У41-107
Ухг-Ю»
У22-1013
Уоз "Ю11
УоИО"»
(ao=De) -10-*
а2-103
а3 -10е
at-109
утах
Jllm
Примечание.
Cs2: а а3Пи
7850
Cs2
X '2+ а
0
0,420 206
—0,081 958
—0,009 724
—0,000 223
—0,000 883
—
0,011 733
—0,022 422
-0,000 620
-0,001 061
—
—0,003 358
—1,548 158
—2,694 829
-0,000 332
1,474 361
—
—
—
—
134
794
CsO
0
3,592 255
—1,226 685
0,139 498
—
—
—
0,207
-1,20
0,016 741
—
—
—0,27
—
—
—0,010 719
—
—
—
—
—
283
600
Ниже все величины даны
А *^"*"щ
Ь32+ В1
АЧ
0,1
3,376
—1,582
0,221
—
—
—
0,190
—1,60
0,026
—
—
—0,24
—
—
—0,067
—
—
—
—
—
160
492
в см.
9450 11 000 13 043,88
[
974
473
129
722
268
0
8,911
—13,073
15,368
—91,254
49,984
-12,524
¦ 2,709
-57,90
—
—
—
—100,0
—
—
155,03
—
1,504
0,089
—0,011
0,000
27
101
ОХ2+
13 500 13 500
h
579
05
77
92
63
81
233
370
148
593
Е
1?
A JS+
1,784 07
1,679 302
7,441 01
—28,195 79
41,894 66
—2,082 75
—0,166
1,056
30,0
—24,0
44,0
—3,74
—85,0
—
—
.—
—
0,893 363
0,001 805
—0,000 071
0,000 001
46
180
CsH
В 12+
2,717
0,5
0,39
10,0
—30,0
—
—
0,80
—6,90
-1,0
—
—
—30,0
—
-614,4
—
0,213
0,017
—0,001
0,000
30
142
948 16175,80
4
413
117
623
043
аЧ
2,837
3,5
—37,0
—
—
—
—
1,64
-280,0
400,0
—
—
-30,0
—
—1692,0
—
0,082
2,461
—3,061
1,248
4
35
770
739
838
190
469
Глава 49
Таблица 49.56. Значения коэффициентов в уравнениях, описывающих уровни энергии (в сиг1),
а также значения vmst и J],., принятые для расчета термодинамических функций
CsF, CsCl, CsBr, Csl, CsLi, CsNa, CsK и CsRb
Коэффициенты
CsF
CsCl
Х11.+ л
JV«-* 0 0
У10 -10-2 3,525 56 2,130 045
Уао —1,615 —0,722 9
Ум-Ю» -
Yi0 -10* -
Уо1-10 1,843 697 0,713 123
Уи-103 —1,175 623 —0,332 099
У21-106 1,179 8 0,334 55
У31-108 1,775 0,172
У02-10' —2,016 8 —0,319 72
У1а-1010 —3,1 —0,373
^оз^О15 —7,1 —4,563 21
Yoi -1020 —1,79 —0,581 6
Vmt* 467 856
JUm 1014 1704
CsBr
Csl
Xi2+a
0 0
1,490 815 -1,191776
—0,371 1 —0,250 5
— —
0,357 900 0,236 274
—0,122 574 -0,068 263
0,1004 0,048 93
0,0317 0,011 4
—0,082 508 —0,037 146
—0,062 5 —0,022 98
—0,718 33 —0,250 43
—0,040 48 —0,008 876
890 626
1979 1829
Примечание. Ниже все величины даны в см за исключением
являющихся безразмерными.
CsF: a коэффициенты потенциальной
CsCl: a коэффициенты потенциальной
CsBr: a коэффициенты потенциальной
Csl: a коэффициенты потенциальной
CsLi: a а3П А *2+ Ь*2+
11 300 11 300 15 400
CsNa: а а3П A1!* 632+
11 300 11 300 15 300
CsK: a а3П А12+ Ь32+
9700 9700 13 200
CsRb: a а3П A1S+ Ь32+
9500 9500 12 900
[ функции: do=168 541,
функции: do=159 057,
функции: do=155 248,
функции: do=150 284, с
?!s+ cm
15 700 16 477
CsLi
XW a
0
1,688 210
—0,542 213
—3,532 691
—1,082 376
1,818 476
—0,629 570
—
—
—8,471 264
—
3732,627
—
55
270
CsNa
0
0,970 380
-0,344 230
4,439 512
—0,779 885
0,605
-0,2
—
—
—0,94
—
71,805
—
71
401
CsK
XI 2+a
0
0,669 063
—0,156 864
—0,419 139
—0,086 947
0,293 217
—0,064 741
—
—
—0,229
—
10,223
—
91
540
CsRb
XX2+»
0
0,493 182
—0,113 603
0,719 564
—0,086 875
0,162 353
—0,033 798
¦ —
—
—0,068 5
—
0,053 9
113
691
коэффициентов потенциальной функции d1 и d2,
^=3,032 2
ij=3,31833,
J1=3,377 65,
!х=3,428 82,
ВХЪ+ C4L D}U.
15 600 15 800 18 250
в^+ cm
13 500 13 600
5XS+ С 41
13 200 13747,21
<Z2=5,653.
d2=7,006.
<22=7,348.
da=7,628.
в значениях Ф°F000 К) и ?°F000 К); расхождения в значениях СР(Т) достигают при 6000 К почти
50% из-за неприменимости приближенных методов расчета термодинамических функций Cs2(r)
в этой области температур. Расхождения с данными [1452] и [3973] составляют 2 и 6 ДжХ
ХК^-моль в значениях Ф°B000 К) и 5°B000 К) и 1 и 4 Дж-К-^моль в значениях Ф°F000 К)
и 5°F000 К) соответственно.
Константа]равновесия реакции Cs2(r)=2Cs(r) вычислена^с]использованием принятой в
справочнике энергии диссоциации
D0(Cs2)=3530±20 см~1=42,23 ±0,24 кДж -моль.
^^Принятая величина получена Хонигом и др. [2702] на основании экстраполяции
колебательных уровней основного состояния и приближенной интерпретации флуоресцентного перехода
из возбужденного в слабосвязанное состояние x3S«, коррелирующее с основными состояниями
атомов Gs. Принятое значение существенно отличается от полученного ранее Кушем и Хесселом
470
Цезий и его соединения CsO(r)
13148] C176+80 см) и от результатов исследования диссоциации молекул Cs2 чри
столкновениях [2414] B900-3130 см).
Принятой энергии диссоциации соответствует
bfH°{Cs2, г, 0) = 113,798 ± 2,0 кДж • моль.
CsO(r). Термодинамические свойства газообразного оксида цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1237.
Молекулярные постоянные CsO, принятые на основании анализа экспериментальных данных
и приближенных оценок, приведены в табл. 49.4. На основании исследования спектра ЭПР молекул
CsO, изолированных в матрицах инертных газов, в работе Линдсея и др. [3265] и экспериментов
по отклонению молекулярного пучка в неоднородных электрическом и магнитном полях [2616]
в справочнике принимается, что основным электронным состоянием СзО является состояние X SS*.
По аналогии с молекулой RbO молекула CsO должна иметь возбужденное состояние 42Пс низкой
энергией. Энергия этого состояния была оценена на основании тенденции к ее увеличению при
переходе от КО к RbO. Погрешность принятого значения может достигать 500 см. Приведенная
в табл. 49.4 частота колебания в состоянии Х2?+ оценена Спикером и Энлрюсом [4365] на
основании результатов исследования ИК-спектра молекул CsO, изолированных в матрице из ^ И*/.-
=314 см). Погрешность этой величины может достигать 30 ем'1. Значение fre(Gs—О)=2,4±
+0,1 А оценено по величине r(Cs—О) в молекуле CsOH. Постоянная шехе в состоянии Х2Е+ и все
молекулярные постоянные в состоянии А 2П оценены из сопоставления с данными для оксидов
других щелочных металлов1.
Термодинамические функции CsO(r) были вычислены по уравнениям A. 3)—A. 6), A. 9)^
A. 10), A. 93)—A. 95). Значения QBa и ее производные вычислялись по соотношениям A. 90)—
A. 92) с учетом одного возбужденного состояния. Статистические суммы по
колебательно-вращательным уровням состояний Х2?+ и А2П и их производные находились непосредственным
суммированием по колебательным уровням и интегрированием по / с помощью соотношений типа A. 81).
В расчете учитывались все уровни со значениями / <^ /шах „, последние находились по формуле
A. 82). Уровни энергии вычислялись по уравнениям A. 64а) и A. 65) с коэффициентами Yklm
приведенными вместе со значениями ушах и /цт в табл. 49.5. Для обеспечения условий A.50)
и A. 59) были введены коэффициенты Y30 и Уоз. Мультиплетность и вырождение состояний Х2?+
и А 2П учитывались статистическими весами 2 и 4 соответственно.
Погрешности рассчитанных термодинамических функций CsO(r) при Т ^ 3000 К обусловлены
главным образом отсутствием экспериментальных данных о вращательной постоянной в состоянии
X2S+ и об энергии состояния А 2П. При более высоких температурах становятся существенными
погрешности из-за отсутствия данных о других возбужденных состояниях CsO. Погрешности
в значениях Ф°(Г) при 298,15, 3000 и 6000 К оцениваются в 2, 3 и 5 Дж-К^-моль соответственно.
Термодинамические функции CsO(r) ранее вычислялись в справочнике JANAF [2834] и работе
Брюэра и Розенблатта [1666] (Ф°(Г), Т < 3000 К). Расхождения данных [2834] и табл. 1237
наиболее значительны при низких температурах (около 7 Дж-К-моль в значении Ф°B98 К))
и обусловлены тем, что авторы [2834] приняли для основного состояния статистический вес 4.
Расхождения с данными [1666] достигают 4 Дж-К-моль.
Константа равновесия реакции CsO(r)=Cs(r)+O(r) вычислена с использованием принятой
в справочнике величины |
Дг#°@) = D0(CsO) = 285 + 25 кДж • моль« 23800 + 2000 см.
тягтая величина была ощенена сопоставлением анергии атпмиаашиш в ргтдах МОШ(г), М„О (г)
о nononi DODonnwc ноннод мололи [346] посше KODDeKIHDOBKI О X2gI0M ВШ9?1В В§Ш?Ш§§
с использованием ионной модели Io4b J, после корректировки с учетом разности результатов
аналогичных расчетов для LiO, NaO, КО и имеющихся для этих молекул экспериментальных данных.
Принятой энергии диссоциации соответствует <
AfH°(CsO, г, 0) = 39,797 ± 25 кДж • моль.
1 Для CsO оценка значения ч>,хе по соотношению A.67) некорректна ввиду большой полярности этой молекулы.
471
CsO2(k, ж), Cs2O(k, ж) Глава 49
CsO2{k, ж). Термодинамические свойства кристаллического и жидкого диоксида цезия в
стандартном состоянии при температурах 298,15—2000 К приведены в табл. 1238.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние CsO2(k) в интервале 0—9,5 К принята
ромбическая §-модификация, в интервале 9,5—190 К ромбическая ^-модификация с другими параметрами
кристаллической решетки, в интервале 190—403 К тетрагональная а-модификация и в интервале
403—723 К кубическая у-модификация [4858]. В литературе отсутствуют какие-либо сведения
о термодинамических свойствах CsO2(k, ж). Термодинамические функции CsO2(k) при 298,15 К,
приведенные в табл. 49.1, оценены при подготовке справочника с помощью эмпирических методик
[1094, 1741, 3127, 3198], а также на основании принятых данных для соединений щелочных
металлов типа Ме2О(к), Ме2О2(к) и МеО2(к). Принятое значение 5°B98,15 К) согласуется в пределах
погрешности с оценкой, выполненной в справочнике [1395] A46 Дж-К~1-моль~1). Погрешности
принятых значений ?°B98,15 К) и #"B98,15 К)—#°@) оцениваются в 8 Дж-К^-моль
и 1 кДж-моль соответственно.
Для теплоемкости CsO2(k) в интервале 298,15—403 К принято уравнение (см. табл. 49.1),
основанное на значении теплоемкости при 298,15 К (см. табл. 49.1) и величине С?D03 К)=88 +
+5 Дж-К-моль~1; последняя была оценена так же, как для NaO2 (см. выше). Температура
полиморфного превращения D03+10 К) принята по данным [390, 4858]. Энтальпия полиморфного
превращения принята равной нулю, поскольку оценить ее значение не представляется возможным.
Теплоемкость y-GsO2 (88+5 Дж-К~1-моль~1) принята равной теплоемкости <x-CsO2 в точке
фазового превращения. Приведенная в табл. 49.1 температура плавления G23+10 К) найдена Вольно-
вым и Матвеевым [4859]. Энтальпия плавления A8,4+4,0 кДж-моль) оценена на основании
предположения о равенстве энтропии плавления CsOa и КО2, а теплоемкость СзО2(ж) A00 +
+10 Дж-К~1'Моль~1) оценена так же, как для диоксидов других щелочных металлов.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000 и 2000 К оцениваются в 5, 14
и 21 Дж-К~1-моль~1 соответственно. Расхождения в значениях Ф°(Г), приведенных в табл. 1238
и справочнике [1395] (Т <J 700 К), достигают 5 Дж-К~1-моль~1 и обусловлены разной оценкой
теплоемкости и энтропии этого соединения.
Константа равновесия реакции CsO2(k, ж)=Сз(г)+2О(г) вычислена с использованием значения
Дг/Г°@)=859,287+2,2 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
-1
AfH°(CsO2, к, 298,15 К) = —286,1 + 2,0 кДж • моль-
Принятая величина основана на энтальпии реакции GsO2(k)+20 000,5 Н20(ж)=Сз0Н (р-р;
20 000 Н2О)+3/4О2(г), даг#°B98,15К)=—59,0+1,7 кДж-моль, измеренной Д'Орацио и Вудом
[2096]. Авторы провели 6 измерений с двумя образцами. Основным источником погрешности
являются примеси в образцах, составляющие 3,4 и 6,5% Gs2GO3. Менее точная величина
(—293 кДж-моль) соответствует энтальпии растворения CsO2 в серной кислоте, измеренной
Форкраном [2299].
Cs2O(k, ж). Термодинамические свойства кристаллического и жидкого оксида дицезия в
стандартном состоянии при температурах 100—2500 К приведены в табл. 1239.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние Cs2O(k) в интервале 0—768 К принята
гексагональная модификация (структурный тип CdCl2).
При Т ^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости, выполненных Флотовым и Осборном [2273] E—350 К, образец чистотой 98,72%).
Погрешность измерений [2273] оценивается в 5% при 5 К, 1% при 15 К и 0,3% выше 20 К.
Экстраполяция теплоемкости по закону Дебая приводит к значению S°E K)=l,14 Дж-К~1-моль~1.
Погрешности значений ?°B98,15 К) и Я°B98,15 К)—#°@), приведенных в табл. 49.1, оцениваются
в 0,4 Дж-К~1-моль~1 и 0,05 кДж-моль соответственно.
В литературе отсутствуют сведения об измерениях энтальпии или теплоемкости Cs2O выше
350 К. Для теплоемкости в интервале 298,15—768 К было принято уравнение, полученное
авторами [2273] по результатам измерений в интервале 298,15—350 К. Температура плавления
G68+5 К) принята по термографическим данным, полученным Тузеном [4533]; в пределах
погрешности это значение согласуется с результатами измерений в работах [3974, 3977] G63+10 К).
472
Цезий и его соединения
Энтальпия плавления B0+2 кДж-моль) оценена на основании предположения о равенстве
энтропии плавления Gs2O и Ы2О. Теплоемкость жидкой окиси цезия A00+10 Дж-К"-моль)
оценена так же, как для Na2O^).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 2500 К оцениваются в 0,25,
4, 12 и 15 Дж-К~1-моль~1 соответственно. Расхождения в значениях термодинамических функций,
приведенных в табл. 1239 и справочнике Барина и Кнаке [1395] (Т <; 768 К), составляют
19 Дж-К~1-моль~1 и обусловлены неверной оценкой значения <S°B98,15 К) авторами [1395].
Принятая в справочнике энтальпия образования
A//°(Gs2O, к, 298,15 К) =—346,4 + 1,2 кДж • моль
основана на измеренной Сеттлом и др. [4240] энтальпии реакции Cs2O(k)+0,740(GsOHx
X5555,5 H2Oj=2,740 (CsOff-1500 Н2О), AaqE~°B98,15 K)=-343,80+l,2 кДж.моль. Пересчет
к бесконечному разбавлению дает Дйг#(соН20; 298,15 К) = — 343,80+1,2 кДж-моль. Ренгад
[3975, 3976] измерил энтальпию реакции Сй2О(к)+7001Н2О(ж)=2СзОН(р-р, 3500 Н2О),
Дад#B90,9 К) = —348,5 кДж-моль. Пересчет к стандартным условиям приводит к значению
—352+8 кДж-моль, которому соответствует энтальпия образования —338+8 кДж-моль.
Измерениям энтальпии растворения Cs2O(k) в воде, выполненным Бекетовым [1457], соответствует
неверное значение энтальпии образования —387 кДж-моль из-за присутствия большого
количества примесей в использованном образце.
Давление пара в реакции Cs2O(k, ж)=Сз2О(г) вычислено с использованием значения &гН°@)=
= AsH°(Cs20, к, 0)=207,128+15 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования Cs2O(k) и атомизации Cs2O(r).
Cs2O(r). Термодинамические свойства газообразного оксида дицезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1240.
Молекулярные постоянные, использованные для расчета термодинамических функций Cs2O,
приведены в табл. 49.6. На основании исследования инфракрасного спектра [4365, 4366], а также
отклонения молекулярного пучка в неоднородном электрическом поле [1707] в справочнике для
молекулы Cs3O в основном электронном состоянии Х1А1 принята угловая структура
(симметрия C2v). Приведенное в табл. 49.6 произведение моментов инерции Cs2O соответствует структур-
Таблица 49.6. Значения молекулярных постоянных, а также а (рх = 1), принятые для расчета
термодинамических функций Cs2O, Cs2O+, Cs2t>2, CsOH, Cs2<?2i5f2, Cs2F2, Cs2C/2, C's2Br2,
Cs2I2, Cs2SO4, CsNO2, CsNO3, Cs2CO3 и CsBO2
Молекула
Состояние
Csa0 ХЫг
Cs2O+ iMj
Cs2O2 ХЫд
CsOH I12+
CjS2O2ri2 •"- A g
Cg2cJ ?i?
Cs2Br2 Xi A g
Gs2I2 %ikg
Gs2SO4 ^i
CsNO2 ^Ax
CsNO3 XXAX
Cs2CO3 ХЫг
CsBO2 frA'
Примечание. Ниже все
250
200
850
336
3700
268
170
113
89
960
1330
1450
1450
1960
V3
45 457 —
40 400 —
300 200 50
306 B) 3705 —
275 86 275
97 184 76
62 109 74
52 76 32
44 62 25
500 100 460
790 170 1210
1035 720 170
1050 720 210
1078 585 580
постоянные приведены в см
GsOH: a Приведено значение
Gs2O2H2: a vlo=227,
/-1039 г- см2.
vu=3700, v12=235.
va
см-1
,
— —
357 160
— —
185 272
251 205
161 144
104 106
82 90
1125 633
НО 30
1295 720
200 880
206 60
—
—
—
113
—
.—
—
—
196
—
110
100
—
CsaSO4: a vlo=16O
. —
— —
_ _
— —
281 278a
— —
— —
— —
— —
1095 B) 607 B) a
_ _
830 30
160 160a
— —
B), vu=30B).
Cs2CO3: a vlo=14OO, vu=700, v
Til 4ПИ7
1axb'c•iU
Г3 ¦ CM6
1,1-105
1,0 -105
1,9 -105
1,52555 -10 a
5,2-105
5,4 -105
3,3 -10е
1,3-107
3,9-10'
4,1 -106
1,37 -104
4,3 -104
2,44 -106
1,4-10*
12=30.
2
2
4
1
4
4
4
4
4
4
2
2
2
1
473
Cs2O+(r) Глава 49
ным параметрам r(Cs—О)=2,40+0,03 А (оценка по величине r(Gs—О) в молекуле CsOH) и Д Cs—
О—Cs=130+20° (найден по изотопическому сдвигу частоты v3 в работах Спикера и Эндрюса
[4365, 4366]). Погрешность величины 1д1в1с оценивается в 6-Ю13 г3-см6. Приведенная
в табл. 49.6 частота v3 найдена Спикером и Эндрюсом [4366] в результате исследования ИК-спектра
молекул Cs2O, изолированных в матрице из Аг. С принятой величиной хорошо согласуется
значение, полученное в ИК-спектре молекул Cs2O, изолированных в матрице из N2 D55 см'1).
Значение vx рассчитано по соотношениям поля валентных сил и принятой величине v8; для v2
принято среднее из значений, рассчитанных в предположении, что отношение силовой постоянной
связи и деформационной силовой постоянной в Cs2O сохраняется таким же, как в молекулах
Li2O и CsOH x. Погрешности приведенных значений v1? v2 и v3 оцениваются в 50, 20 и 30 см.
Сведения о возбужденных электронных состояниях Csa0 в литературе отсутствуют.
Термодинамические функции Cs2O(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены приближенным характером расчета, неточностью принятых
молекулярных постоянных, в особенности структурных параметров, а также тем, что в расчете не
учитывались возбужденные электронные состояния; погрешности могут достигать 10, 20 и 30 ДжХ
ХК^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Cs2O(r) рассчитывались авторами справочника JANAF
[2834]. Расхождения в термодинамических функциях, приведенных в табл. 1240 и в справочнике
[2834], не превышают 6 Дж-К-моль в значениях Ф°(Т).
Константа равновесия реакции Cs2O(r)=2Cs(r)+O(r) вычислена с использованием принятой
в справочнике энтальпии атомизации
ДгЯ°@) = 540 + 15 кДж- моль.
Принятая величина основана на масс-спектрометрических исследованиях равновесия реакции
Cs2O(r)=2Cs(r)+V2O2(r), выполненных Норманом и Стейли [3680] A434—1600 К, 6 точек,
данные представлены в виде графика) и Панченковым и др. [309, 343, 347, 755] A118 К, 6
измерений). Обработка результатов этих измерений по методу III закона привела к близким значениям
энтальпии атомизации 545+20 к Дж-моль и 537+13 кДж-моль, соответственно.!
Принятой энтальпии атомизации соответствует
AfH°(Gs2O, г, 0) = —137,189 + 15 кДж • моль.
Cs2O+(r). Термодинамические свойства газообразного положительного иона оксида дицезия
в стандартном состоянии в интервале температур 298,15—6000 К приведены в табл. 1241.
Молекулярные постоянные, использованные для расчета термодинамических функций
Cs2O+(r), приведены в табл. 49.6. Экспериментальные данные о структуре и основных частотах
молекулы Cs2O+ в литературе отсутствуют. По аналогии с молекулой Cs2O в справочнике
принимается, что Cs2O+ в основном электронном состоянии Х2АХ имеет угловую структуру (симметрия
Civ), см. примечание к тексту по Cs2O. Приведенные в табл. 49.6 постоянные были оценены по
принятым постоянным молекулы Cs2O, а также молекул Н2О и Н2О+. Сравнение последних
свидетельствует, что ионизация Н2О сопровождается уменьшением частот и увеличением r(O—H)
и Д Н—О—Н. То же изменение было принято при оценке постоянных Cs2O+. Приведенное
в табл. 49.6 произведение моментов инерции Csa0+ рассчитано по структурным параметрам r(Cs—
—О)=2,5+0,1 А и Д Cs—О—Cs=140+30°, его погрешность оценивается в 8-10"13 г3-см6.
Погрешности принятых значений vlt v2 и v3 оцениваются в 50, 20 и 100 см. Молекула Cs2O+
так же, как молекула CsO, должна иметь электронные состояния^ низкими энергиями возбужде-
1 Можно предполагать, что в равновесном состоянии молекула Cs2O (а также молекулы К2О и ВЬ2О) имеет
линейную структуру, так как принятым структурным параметрам соответствует г (Gs—Cs)~4,3+0,3 А, что меньше
величины re (Gs2). Эффекты, наблюдавшиеся в работах [1707, 4365, 4366], могут быть обусловлены низкой
частотой деформационного колебания, и им соответствует некоторая эффективная структура. Расчет
термодинамических функций газов в приближении гармонического осциллятора дает для «эффективной угловой модели»
молекулы XY2 лучшее согласие с расчетом, учитывающим ангармоничность потенциальной функции деформационного
колебания линейной молекулы, чем расчет в гармоническом приближении для линейной модели этой молекулы.
Яоэтому в справочнике отдано предпочтение структуре с ДСв—Q— Cs ф 180°.
474
Цезий и его соединения Cs2O2(k, ж)
ния. Однако обоснованная оценка их энергии невозможна, и в связи с невозможностью
корректной оценки постоянных Gs2O+ даже в основном состоянии возбужденные состояния этой молекулы
в справочнике не рассматриваются.
Термодинамические функции Gs2O+(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных электронных состояний. Погрешности рассчитанных
термодинамических функций обусловлены грубой оценкой молекулярных постоянных Gs2O+,
пренебрежением возбужденными состояниями, а также приближенным характером расчета, они составляют
10, 15 и 20 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций Cs2O+(r) публикуется впервые.
Константа равновесия реакции Cs2O+(r)+e(r)=2Cs(r)+O(r) вычислена с использованием
значения ДгД"°@)==120+25 кДж-моль, соответствующего принятому в справочнике потенциалу
ионизации
/0(Cs2O)=420+20 кДж-моль.
Принятая величина является средней из совпадающих в пределах погрешностей результатов
измерений, выполненных Емельяновым и др. [410] методом электронного удара D29+20 кДжХ
Хмоль) и Кудиным и др. [592] в масс-спектрометрическом исследовании равновесия реакции
Cs2O+(r)+Cs(r)=Cs2O(r)+Cs+(r) D16+16 кДж-моль, 990 К, 1 точка). Несколько большее
значение D40+50 кДж-моль) было получено методом электронного удара Норманом и Стейли
[3680].
Принятому потенциалу ионизации соответствует
A^°(Cs2O+, г, 0) = 282,811 +Ц25 кДж • моль.
Cs2O2(k, ж). Термодинамические свойства кристаллического и жидкого диоксида дицезия
в стандартном состоянии при температурах 298,15—2000 К приведены в табл. 1242.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние Gs2O2(k) в интервале 0—867 К принята
ромбическая модификация [4858]. Какие-либо сведения о термодинамических свойствах
Cs2O2(k, ж) в литературе отсутствуют. Стандартные значения термодинамических функций,
приведенные в табл. 49.1, оценены так же, как для К2О2(к). Погрешности принятых значений
5°B98,15К) и Я°B98,15 К)—Я°@) оцениваются в 17 Дж-К^-моль и 1,5 кДж-моль
соответственно. Для теплоемкости Gs2O2(k) в интервале 298,15—867 К принято уравнение (см.
табл. 49.1), выведенное по значению теплоемкости при 298,15 К (см. табл. 49.1) и значению
СД867|К)=117+12 Дж-К-моль; ^последнее оценено так же, как для К2О2(к). (Температура
плавления (867+50 К) принята по данным Центнершвера, Блюменталя [1774]. Энтальпия
плавления B2+5 кДж-моль'1) оценена по величине энтропии плавления так же, как для Na2O2(K).
Теплоемкость Сз2О2(ж) A34+15 Дж-К~1-моль~1) принята на основании оценки, выполненной
по методике, изложенной в главе 2, раздел 5.]
Погрешности вычисленных значений ФО(Г) при 298,15, 1000 и 2000 К оцениваются в 12, 25
ж 35 Дж-К~1-моль~1 соответственно.
Таблица термодинамических функций Gs2O2(k, ж) публикуется впервые.;
Принятая в справочнике энтальпия образования
A^CsA, к, 298,15 К) = —440 + 25 кДж • моль
основана на исследованиях равновесия реакции 2СзаО2(ж)=2Сз2О(ж)+О2(г), выполненных Цент-
нершвером и Блюменталем [1590, 1774]. Измерения проводились динамическим методом (ПОЗ—
1187 К, 4 точки). По этим данным для энтальпии образования Gs2O2(k) было получено —449 +
+35 кДж-моль (II закон) и —440+25 кДж-моль (III закон). В этих же работах [1590, 1774]
была исследована реакция окисления Cs2O2, для которой принималось уравнение 2Сз2О3(ж) =
=2Сз2О2(ж)+О2(г), однако существование фазы Cs2O3 вызывает сомнение (см. тексты по
В.Ь2О2(к, ж) и К2О2(к, ж)), и эти измерения не были использованы для выбора энтальпии
образования Gs2O2.j
475
Cs2O2(r), CsH(k, ж) Глава 49
Давление пара в реакции Gs2O2(k, jk)=Cs2O2(:t) вычислено с использованием значения Дг-йГ°(О) =
= AsH°(Cs202, к, 0) = 196,790+32 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования Gs2O2(k) и атомизации Gs2O2(r).
Cs2O2(г).Термодинамические свойства газообразного диоксида дицезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1243.
Молекулярные постоянные, использованные для расчета термодинамических функций
Cs2O2(r), приведены в табл. 49.6. Структура молекулы Gs2O2 экспериментально не исследовалась.
По аналогии с молекулой Li2O2 для CsaO2 в основном электронном состоянии ХгАд принято
плоское ромбическое строение (симметрия D2k) со связью кислород—кислород в цикле. С такой
структурой согласуются результаты исследования отклонения молекулярного пучка Cs2O2 в
неоднородном электрическом поле [1707]. Приведенное в табл. 49.6 произведение моментов
инерции рассчитано по оцененным структурным параметрам r(Cs—О)=2,5+0,1 А (по принятым
постоянным молекул CsF, Gs2F2 и CsO) и г(О—О) = 1,50+0,1 А (см. Li2O2). Погрешность величины
III 6Ю113 36
Принятое значение v5 найдено Эндрюсом и др. [1330, 4366] при исследовании ИК-спектра
молекул Cs2O2, изолированных в матрице из Аг. Остальные частоты Gs2O2 в спектрах не
наблюдались, и их величины были оценены в справочнике так же, как для молекулы К2О2.
Погрешность принятого значения va оценивается в 100 см, частот v2, v3 и v6 — в 50—60 см и частот \
и v5 — в 20 см.
Сведения о возбужденных электронных состояниях Cs2O2 в литературе отсутствуют.
Термодинамические функции Cs2O2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
неточностью оценки молекулярных постоянных, а также приближенным характером расчета
и составляют 8, 15 и 25 Дж^К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К
соответственно.
Таблица термодинамических функций Cs2O2(r) публикуется впервые.
Константа равновесия реакции Cs2O2(r)=2Cs(r)-f2O(r) вычислена с использованием
принятой в справочнике энтальпии атомизации
ДгЯ°@) = 890 ± 20 кДж • моль.
Принятая величина основана на масс-спектрометрических исследованиях равновесия реакции
Cs2O2(r)=Cs2O(r)+1/2O2(r), выполненных Норманом и Стейли [3680] G70—960 К, данные
представлены в виде уравнения, 884+25 кДж-моль (II закон) и 885+20 кДж-моль (III закон)),
и равновесия реакции Cs2O2(r)=2Cs(r)+O2(r), выполненных Гусаровым и др. [347] A118 К,
892+18 кДж-моль).
Принятой энтальпии атомизации соответствует
i\fH°(Gs202, г, 0) = —240,406 + 20 кДж • моль.
CsH(k, ж). Термодинамические свойства кристаллического и жидкого гидрида цезия
в стандартном состоянии при температурах 298,15—1200 К приведены в табл. 1244.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние CsH(k) в интервале 0—801 К принята
кубическая модификация (структурный тип NaCI). В литературе отсутствуют какие-либо данные
по теплоемкости и энтальпии CsH(k, ж), ввиду чего термодинамические функции этого
соединения так же, как КН(к, ж) и Ш)Н(к, ж), вычислены по постоянным, которые были оценены на
основании принятых данных для фторидов щелочных металлов, ЫН(к, ж) и NaH(K, ж). Все эти
вещества имеют один и тот же структурный тип (NaCI). Значение 5°B98,15 К) было оценено на
основании сопоставления принятого значения AyH°(Csii, к) с величинами, рассчитанными по
методу III закона из данных [2619, 2620] по давлению диссоциации GsH(k, ж). Погрешности
значений ?°B98,15 К) и #°B98,15 К)—#°@), приведенных в табл. 49.1, оцениваются в 4 Дж-К*
Хмоль и 1 кДж-моль соответственно. Уравнение для теплоемкости в интервале 298,15—
801 К, приведенное в табл. 49.1, получено на основании значений СрB98,15 К)=40,6 Дж-К~*Х
Хмель, С°E00К)=51 Дж-К-^моль и С°(801 К)=63 Дж-К^-моль, которые были оценены
476
Цезий и его соединения CsH(r)
так же, как для КН(к) и RbH(K). Предполагается, что теплоемкость CsH(k) аппроксимируется
этим уравнением с погрешностью, не превышающей 10%.
Температура плавления (801+5 К) при равновесном давлении водорода 31,5 бар принята по
результатам термографического исследования, выполненного Скуратовым и др. [940];
воспроизводимость определения температуры плавления была оценена авторами работы в 2°
(исследованный образец был получен при 803—833 К и давлении водорода 33—60 атм из металла чистотой
99,9% и водорода, содержащего не больше 10~4% N2 и О2, химический анализ показал, что
образец содержал не менее 99,5% CsH). Энтальпия плавления A5+3 кДж-моль) принята по
расчету Скуратова и др. [940], основанному на данных по давлению диссоциации CsH(k, ж),
имеющихся в литературе и полученных авторами [940]. Значение теплоемкости жидкого CsH E6 +
+8 Дж-К~1-моль~1) оценено с учетом экспериментальных данных по теплоемкости 1лН(ж).
Погрешности рассчитанных значений Ф°(Т) оцениваются в 3, 5 и 8 Дж-К-моль~1 при 298,15,
500 и 1000 К соответственно.
Таблица термодинамических функций CsH(k, ж) публикуется впервые.!
Принятое в справочнике значение энтальпии образования
&fH°(CsE, к, 298,15 К) = —54,04 + 0,15 кДж • моль
основано на выполненных Ганном [2525] калориметрических измерениях энтальпии растворения
Cs(k) и CsH(k) в воде. Разность этих величин соответствует приведенному выше значению
энтальпии образования CsH(k). Смит и Басе [4327] измерили энтальпии растворения CsC1(k) и CsH(k)
в соляной кислоте, откуда была найдена энтальпия образования —59,7 кДж-моль.
Расхождение, по-видимому, объясняется недостаточной чистотой образца CsH (98,7% по Cs и 94,7% по
водороду), использованного в этой работе.
В табл. 49.7 представлены энтальпии образования CsH(k), вычисленные по результатам
исследований равновесия реакций 2CsH(k, ж)=2Сз(ж)-(-Н2(г). В качестве погрешностей указана
Таблица 49.7. Результаты определения A/fi"o(CsH, к, 298,15 К) по исследованиям
равновесия 2CsH(k, ж) = 2С8(ж) + Н2(г) (в кДж • моль)
Авторы
Метод
AjH°(CsR, к, 298,15 К)
II закон
III закон
Хашшгаль, Борокко, 1938 [1401, 2543] Статический, CsH(k), 473—593 К, 13 точек —37,1+4,0 —53,0±0,6
Херолд, 1949 [2619, 2620J То же, CsH(k), 497—651 К, 7 точек —54,6 ±2,5 —54,0 ±0,1
Скуратов и др., 1976 [940] То же, СэЩж), 813—969 К, 14 точек —59,5+5,0 —53,1+0,2
воспроизводимость измерений. Неточность термодинамических функций CsH в конденсированном
состоянии увеличивает погрешность на 3 кДж-моль для измерений при 600 К и на 6 кДжХ
Хмоль для измерений при 900 К. Как видно из приведенных в таблице данных, результаты
калориметрических измерений и исследований равновесия реакции диссоциации находятся
в удовлетворительном согласии.
Давление пара гидрида цезия в реакции CsH(k, ж)=СзН(г) вычислено с использованием
значения ArH°@) = AsH°(GsH., к, 0) = 171,643 +4,0 кДж-моль, соответствующего принятым в
справочнике энтальпии образования CsH(k) и энергии диссоциации CsH(r).
CsH(r). Термодинамические свойства газообразного гидрида цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1245.
В табл. 49.4 приведены молекулярные постоянные, принятые на основании анализа
результатов спектральных исследований. В электронном спектре CsH изучены две системы полос,
связанных с переходами между тремя электронными состояниями: А1!,* ^а Х1Е+ [1302, 1412, 1967,
1968, 4004, 4467] и В1!,* <- ХХЕ+ [4004]. Анализ возмущений в системе В1!,* *- X1!,* показал
существование еще одного стабильного состояния а3Д. Основное состояние ХХЕ+ коррелирует
с атомами CsB/Sr)+HB5), состояние А1!,* — с атомами CsBP)+RBS) и состояния В1?,* и а3Д —
с атомами GsBD)-{~llBS). По аналогии с гидридами калия и рубидия следует ожидать, что все
остальные состояния, соответствующие указанным диссоциационным пределам, нестабильны.
4 77
CsOH(k, ж) Глава 49
Принятые молекулярные постоянные CsH в состоянии Z1E+ получены Тамом и Хэпером
[4467] в результате анализа флуоресцентных переходов из состояния А1!,* на уровни с v" ^ 14
состояния X1!,*. Они хорошо согласуются с данными Алми и Рассвайлера [1302], полученными
для уровней с v" ^ 2. Молекулярные постоянные в состоянии А1!,* были вычислены в работе
Хсиеха и др. [2725] на основании данных Алми и Рассвайлера [1302] для системы Аг1,+—ХХЕ+
C ^ г/ ^ 20) и уточненной нумерации полос, предложенной в работе Бартки [1412]. При
расчете постоянных учитывались результаты исследования системы АгЛ+—ХгЛ+ молекулы CsD
в работах [1412, 1968]. Состояние А1!,* у молекулы CsH, как и у молекул гидридов других
щелочных металлов, аномально. Кривые зависимости ДС(у+1/2) и Bv от v проходят через максимум
при у=13 и v=l~B> соответственно.
Молекулярные постоянные в состоянии BXZ+ были рассчитаны при подготовке настоящега
справочника по данным Рингстрема [4004] для одиннадцати полос прогрессии v"—0 системы
-В1Е+ <— Х12+, если принять нумерацию и значения те и Ве, оцененные автором [4004] по
интенсивности полос и форме, рассчитанной по РКР-потенциальной кривой. Постоянные в состоянии
а3Д также рассчитаны по данным Рингстрема [4004] в предположении, что нижний
обнаруженный по возмущениям колебательный уровень соответствует квантовому числу v—0.
Термодинамические функции CsH(r) были рассчитаны по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QBS и ее производные вычислялись по уравнениям A. 90)—
A. 92) с учетом трех возбужденных состояний. Статистические суммы по
колебательно-вращательным уровням состояний ХХЕ+, АХ1,+, В1!,* и а3Д и их производные находились по уравнениям
A. 70)—A. 75) непосредственным суммированием по уровням энергии. В расчете учитывались
все уровни, ограниченные предельными кривыми диссоциации указанных состояний.
Колебательно-вращательные уровни вычислялись по соотношениям A. 65), A. 62) и A. 64а). Значения
коэффициентов Ykl в этих уравнениях приведены в табл. 49.5 вместе со значениями vm&x, Jum
и коэффициентами а, в уравнениях, аппроксимирующих предельные кривые диссоциации. Для
соблюдения условий A. 50) и A. 59) дополнительно введены для состояния ХХЕ+ коэффициенты
^50* ^во и ^оз; Для состояния А1 Е+ — коэффициенты Y50 и У60 и для состояний 51Е+ и а3Д —
коэффициент YQS.
Погрешности вычисленных термодинамических функций при Т ^ 3000 К обусловлены в
основном неточностью физических постоянных и не превышают 0,03 Дж-К-моль в значениях Ф°(Т).
При Т ~^> 3000 К становятся существенными неточность экстраполяции колебательно-вращ атель-
ных уровней и пренебрежение ридберговскими состояниями; погрешность в значении Ф°F000 К)
может достигать 0,3—0,5 Дж-К~1-моль~1.
Ранее термодинамические функции CsH(r) вычислялись в справочнике Хара и др. [2537}
(Т < 5000 К) и в работе Шнейдера [4197а] (<?вн для 1000 < Т < 3000 К). Расхождения данных
[2537] с приведенными в табл. 1245 достигают 0,4, 0,8 и 1 Дж-К-моль в значениях Ф°(Т),
S°(T) и С°р(Т) и объясняются различием использованных методов расчета и тем, что авторы [2537]
не учитывали возбужденные состояния CsH (ошибки частично компенсируют друг друга).
Константа равновесия реакции CsH(r)=Cs(r)+H(r) вычислена с использованием принятой
в справочнике энергии диссоциации
ДгЯ°@) = Do(CsH) = 14 600 + 400 см = 175 + 4 кДж • моль.
Принятая в еличина получена на основании экстраполяции колебательных уровней состояния
X1!,* примерно на 6000 см A4 900 см), а также энергии последнего наблюдавшегося уровня
состояния 51!" B8 800 см), которой соответствует_ D0(CsH)\> 14 250 см [4004].
Принятой энергии диссоциации соответствует
г, 0)=419,048+|4,0 кДж^моль.
CsOH(k, ж). Термодинамические свойства кристаллического и жидкого гидроксида цезия
в стандартном состоянии при температурах 298,15—3000 К приведены в табл. 1246.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 49.1. За стандартное состояние CsOH(k) в справочнике в интервале 0—497 К принята
ромбическая модификация (структурный тип анти-a-NaOH), а в интервале 493—616 К —
кубическая модификация (структурный тип NaCl) [2806]. В литературе отсутствуют какие-либо
сведения о теплоемкости CsOH(k, ж). Принятые значения ?°B98,15 К), Я°B98,15 К)—Н°ф) и
С°B98,15 К) оценены так же, как для В.ЬОН(к), погрешности значений ?"B98,15 К) и
478
Цезий и его соединения CsOH(r)
#°B98,15 К)—Я°@) оцениваются в 8 Дж-К~1-моль~1 и 1 кДж-моль соответственно.
Выполненные оценки согласуются в пределах погрешностей с оценками других авторов, за исключением
величины С° B98,15 К), приведенной в справочнике JANAF [2834] и составляющей 67,8 ДжХ
X К-моль.
Для теплоемкости CsOH(k) в интервале 298,15—493 К принято уравнение (см. табл. 49.1L
основанное на значениях С° B98,15 К)=73+4 Дж-К^-моль и С°р(№1 К)=88+5 Дж-К"хХ
Хмоль; последнее оценено так же, как для ЙЬОН(к).
Температура полиморфного превращения D97+1 К) и энтальпия превращения G,1 +
+0,5 кДж-моль) приняты по измерениям Якобса и Харбрехта [49816], выполненным методом
ДСК на образце, полученном по реакции CsOH-H2O+CsNH2=2CsOH+NH3 в среде аммиака
(по химическому анализу образец CsOH содержал более 98,6% CsOH). Результаты других работ
относятся, по-видимому, к менее чистым образцам CsOH, химический анализ которых, как
правило, не проводился. В работах [865, 2632] получены близкие значения температуры
превращения D96—497 К), в ряде других работ [65, 814, 866, 1896, 1897, 4040, 4532] — более низкие
значения в пределах 488—493 К. Величина ^irH° определена также в работах Хевеси [2632]
G,4 кДж-моль) и Решетникова и Баранской [866] F, 1 кДж-моль") термографическим методом1.
Теплоемкость кубической модификации CsOH в интервале 497—616 К (80+8 кДж-моль)
принята так же, как и для RbOH(i<), равной теплоемкости высокотемпературной фазы КОН(к).
Температура плавления F16+5 К) основана на усреднении данных [814, 1896, 1897,4040,4532,
49816]. В работах [65, 865, 866, 2544, 2632] были получены существенно более низкие значения
E46—589 К). Энтальпия плавления G,3+0,5 кДж-моль) принята по данным Якобса и
Харбрехта [49816], полученным методом ДСК (см., выше). Термографические измерения дали менее
точные значения 6,7 кДж-моль [2632] и 4,6 кДж-моль [866]. Теплоемкость жидкого гидро-
ксида цезия (83+8 Дж-К-моль) принята равной теплоемкости КОН(ж).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 6,
15, 20 и 25 Дж-К-моль~1 соответственно.
Расхождения в термодинамических функциях CsOH(k, ж), приведенных в табл. 1246 и
справочнике JANAF [2834] (Т <С 2000 К), составляют ~3 Дж-К-моль~1 и обусловлены главным,
образом разницей в оценке значения 5°B98,15 К).
Принятая в справочнике энтальпия образования
A^CsOH/Ik, 298,15 К) = —416,6,+|4,0|кДж • моль
основана на измерениях энтальпии растворения CsOH(k) в воде, выполненных Форкраном [2294]
(разведение 111 Н20; 298,15 К; одно измерение, —68,7 кДж-моль, Дад#°(озН20; 298,15 К) =
= —71,5 кДж-моль) и подтвержденных более ранними измерениями Бекетова [1453]
(разведение 325 Н2О; 289,15 К; одно измерение, —66,4 кДж-моль, Да}#°(соН20; 298,15 К) =
=—69,0 кДж-моль). Погрешность оценена на основании сопоставления измерений,
выполненных Форкраном с гидроксидами лития, натрия и калия, с измерениями, проведенными в
последующих исследованиях (см. соответствующие тексты).
Давление пара гидроксида цезия в реакции CsOH(k, ж)=СэОН(г) вычислено с использованием
значения Дг#°@)= Д*Я°(СзОН, к, 0) = 156,314+10 кДж-моль, соответствующего принятым
в справочнике^энтальпиям образования CsOH вv кристаллическом и газообразном состояниях.
CsOH(r). Термодинамические свойства газообразного гидроксида цезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1247.
Молекулярные постоянные, использованные для расчета термодинамических функций
CsOH(r), приведены в табл. 49.6. Исследования микроволнового [3133, 3254, 3255] и
инфракрасного [87, 1247] спектров показывают, что в основном электронном состоянии Xх 1? молекула
CsOH имеет линейную структуру (симметрия Cms). Приведенный в таблице момент инерции
вычислен по вращательной постоянной Дзо°о=0,183 496+3 -10~6 см, найденной Лайдом и Куч-
ковски [3254], этой величине также соответствуют структурные параметры ro(Cs—О)=2,403 А,
г0(О—Н)=0,920 А. Принятые значения vx и v2 найдены Беляевой [87] и Аккуиста и др. [1247],
1 Сведения о полиморфном превращении при 410 К с энтальпией 1,4 кДж-моль ', приведенные в работе
Решетникова и Баранской [865, 8fi6], не подтвердились в последующих исследованиях.
479
Cs2O2H2(r)
Глава 49
а частоты v3 — Беляевой [87] в ИК-спектре молекул CsOH, изолированных в матрице из Аг.
Близкие значения частот vx и v2 были зарегистрированы Беляевой [87] в ИК-спектре в матрице
из Хе C28 и 322 см). С учетом матричных сдвигов частот vx и v2 и различий в значениях частоты\
у молекул NaOH, КОН и RbOH в газовой фазе и в матрицах, погрешности принятых значений
частот оцениваются в 20 см. По аналогии с молекулами LiOH, NaOH и GsF можно ожидать, что
изомер HCsO и возбужденные электронные состояния CsOH имеют энергии свыше 20 000 см.
Термодинамические функции CsOH(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 126), A. 129) в приближении «жесткий ротатор—гармонический осциллятор»
без учета возбужденных электронных состояний. Погрешности рассчитанных термодинамических
функций обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета и составляют 1, 4 и 8 Дж-К-моль~1 в значениях Ф°(Т) при
298,15, 3000 и 6000 К (в том числе 1—2 Дж-К-моль из-за неточности молекулярных
постоянных).
Ранее термодинамические функции CsOH(r) вычислялись в справочнике JANAF [2835],
результаты обоих расчетов согласуются в пределах 0,1 Дж-К~1-моль в значениях Ф°(Т).
Константа равновесия реакции CsOH(r)=Cs(r)+H(r)+O(r) вычислена с использованием
значения АгН°@)=800,831 +10 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
bfH°(CsOH, г, 0) = —260 + 10 кДж • моль.
Принятая величина является средневзвешенной из представленных в табл. 49.8 значений.
Приведенные в таблице погрешности включают воспроизводимость измерений, неточность
термодинамических функций и термохимических величин, использованных в расчетах. ,.;
Таблица 49.8. Результаты определений Ayff°(CsOH, г, 0) (в кДж • моль)
Авторы
Метод
AjH°(CsOtt, г, 0)
II закон
III закон
Смит, Сагден, 1953 [4323[ Спектрофотометрия в пламенах Н,+воздух, CsOH(r)= — —264
=Cs(r)+OH(r), 2095—2290 К, 7 точек
Енсен, Педли, 1966 [2855] То же, в пламенах H2+O2+N2, Cs(r)-f-H2O(r)= — —265+13
=CsOH(r)+H(r), 2475 К, 1 точка
Коттоя, Дженкинс, 1969 [1941] То же, в пламенах H2+O2+N2, Cs(r)+H2O(r)= — —258+15
=CsOH(r)+H(r), 1570 К, 37 измерений
Емельянов и др., 1967 [410] Электронный удар, CsOH(r)+e(r)=Cs+(r)+OH(r)+ —227 +30
+2е(г)
Панченков и др., 1970 [312, 755] То же, CsOH(r)+e(r)=Cs+(r)+OH(r)+2e(r) —252+15
То же, D0(Cs-OH)—D0(K-OH)=24±5 —260+10
Масс-спектрометрический, CsOH(r)+K(r)=KOH(r)+ —242 —268+10
+Cs(r), 795—1044 К, 12 точек
Cs2O2H2(r). Термодинамические свойства газообразного дигидроксида дицезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1248.
Молекулярные постоянные, использованные для расчета термодинамических функций
Cs2O2H2(r), приведены в табл. 49.6. Произведение моментов в основном электронном состоянии
Х*Ад вычислено для плоской ромбической структуры со связями О—Н, расположенными на одной
прямой (симметрия D2h) х и Д О—Cs—0=92+10°, r(Cs—O)=2,61 +0,10 А, г(О—Н)=0,97 +
+0,03 А. Структурные параметры были рассчитаны Беляевой [87] с использованием ионной
модели молекулы. Погрешность значения IaIbIc оценивается в 2-Ю12 г3-см6. Частоты \—v6,
б
\6,
v8 активны в спектре КР, частоты v6, v7, v9—v12 — в ИК-спектре. Валентным колебаниям кольца
соответствуют частоты \{Ад), v5(Z?ly), vlo(B2tt), vlz(B3u), плоской деформации кольца— ^(
не-
1 С ромбической структурой фрагмента CsOCsO согласуются результаты исследования отклонения
молекулярного пучка Cs2O2H2 в электрическом поле [1707].
480
Цезий и его соединения CsF(k, ж)
плоской деформации кольца — ^(В1и), валентным колебаниям внешних связей ОН — ^i(Ag),
vn(BSu), плоской деформации ОН — \{В1д), ve(i?2e), неплоской деформации ОН — ve(Blu), v8(#2ir).
Значения всех частот приняты по работе Беляевой [87]. Частоты ve, v7, v9, v10, v12 наблюдались
в ИК-спектре молекул Gs2OaH2, изолированных в матрице из Ar, vx и vn оценены как
характеристические колебания ОН, а остальные частоты были рассчитаны по ионной модели. Погрешности
в значениях частот vx и vn оцениваются в 150 см, частот v2, v4, v5 и vg — в 50 см, остальных
шести частот — в 20—25 см. Сведения о возбужденных электронных состояниях Cs2O2H2 в
литературе отсутствуют.
Термодинамические функции Gs2O2H2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),;
{1. 122)—A. 124), A. 128) и A.130) в приближении «жесткий ротатор—гармонический осциллятор»
я без учета возбужденных состояний. Погрешности рассчитанных термодинамических функций
обусловлены неточностью принятых значений молекулярных постоянных, а также
приближенным характером расчета; они составляют 10, 15 и 20 Дж-К~1-моль в значениях Ф°(Т) при
298,15, 3000 и 6000 К.
Ранее термодинамические функции Cs2O2H2(r) вычислялись в справочнике JANAF [2835].
Расхождения данных [2835] и табл. 1248 достигают 40 Дж-К~1-моль~1 в значениях Ф°(Г), они
обусловлены тем, что авторы [2835] приняли другую структуру и отличающиеся значения
постоянных молекулы Cs2O2H2.
Константа равновесия реакции Cs2O2H2(r)=2Cs(r)+2O(r)+2H(r) вычислена с использованием
значения АгН°@) = 1756,662 +30 кДж-моль, соответствующего принятой в справочнике
энтальпии образования
A^°(Cs2O2H2, г, 0) = —675 + 30 кДж • моль.
Принятая величина основана на масс-спектрометрическом исследовании равновесия реакций
2KOH(r)+Cs2O2H2(r)=2CsOH(r)+K2O2H2(r) F92 К, 4 измерения, AfH°(Cs202H2, г, 0)=
= _676+30 кДж-мдль) и 2RbOH(r)+Cs2O2H2(r)=2GsOH(r)+Rb2O2H2(r), 673 К, 3 измерения,,
AfH°(Gs202H2, г, 0)=—673+45 кДж-моль), выполненном Скунмейкером и Портером [4210].
Приведенные погрешности учитывают воспроизводимость измерений, неточность
термодинамических функций и термохимических величин, использованных в расчетах.
CsF(k, ж). Термодинамические свойства кристаллического и жидкого фторида цезия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1249.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние GsF(k) в интервале 0—976 К принимается
кубическая модификация (структурный тип NaCl) *.
При Т <^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости CsF в работе Паукова и Рахменкулова [765] A3—301 К). С учетом значения S°A3 К)=
=1,05 Дж-К~1-моль~1, найденного экстраполяцией данных [765] к Т=0, получены
приведенные в табл. 49.1 величины 5°B98,15 К) и Я°B98,15 К)—#°@), погрешности которых
оцениваются в 0,17 Дж-К"х-мольг1 и 0,02 кДж-моль соответственно. Данные Брбнстеда [1683] по
средней теплоемкости CsF(k) в интервале 273—293 К не учитывались.
При Т > 298 К энтальпия CsF(k) измерена Шпильрайном и Каганом [1166] F24—1275 К)
и Маклеодом [3337] E05—1375 К). Известна также величина Я°(976 К)-Я°B98 К)=39,33 кДжХ
Хмоль, полученная Дворкиным [2135] (цитируется по справочнику [2834]), который провел
измерения энтальпии в узком интервале вблизи температуры плавления. Совместная обработка
данных [1166] и [3337] приводит к необъяснимо высоким значениям теплоемкости (до 95 ДжХ
хК-моль~1 вблизи Тт), что противоречит данным [2135]. В работе [1166] для CsF(k) было
получено всего 4 значения энтальпии с большим разбросом, по-видимому, из-за недостаточной
точности измерений. В работе [3337] измерения проводились на образце, исходная чистота которого
составляла 98%; возможно, что аномалия теплоемкости, обнаруженная автором [3337] и
обусловившая низкое значение энтальпии плавления CsF, объясняется наличием примесей. Ввиду
этого при Т >¦ 298,15 К для теплоемкости CsF(k) принято уравнение, полученное с
использованием значения С;B98Д5 К) (см. табл. 49.1) и значения #°(976 К)—#°B98,15 К)=41,4 кДжХ
1 Кубическая фаза высокого давления (структурный тип CsCl) стабильна при р > 19,7 кбар B98 К) [3851].
31 Заказ N» 1590 481
CsF(k, ж) Глава 4Я
Хмоль. Последняя величина получена усреднением данных [2135] и [3337] для энтальпии
СэР(ж, Тт), из которых вычиталось значение энтальпии плавления.
Температура плавления (976+1 К) принята по измерениям Бредига и др. [1653] (976+1 К),
Роббинса и др. [4015] (976+2,5 К) и Бизота [4929] (977 К); менее точные результаты получены
в работах [150, 1897, 4196, 4663]. Энтальпия плавления B1,7+0,4 кДж-моль) принята
поданным Дворкина и Бредига [2137], полученным для весьма чистого образца. В работе [11661
получено значение 23 кДж-моль. Измерения [3337] приводят к очень низкому значению
теплоты плавления A4 кДж-моль). Теплоемкость СэР(ж) G2+_2 Дж-К^-моль) получена
усреднением данных по энтальпии жидкого CsF, найденных в работах [1166] (999—1275 К; 9 измерений)
и [3337] (983—1375 К; 8 измерений).
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,1,
2, 5 и 10 Дж-К-моль~1. Расхождения между термодинамическими функциями CsF(k, ж),
приведенными в табл. 1249 и в справочнике [2834] (Т ^ 3000 К), достигают 6 Дж-К-моль в
значениях S°(T) и Ф°(Т) и обусловлены существенным уточнением «S^CsF, к, 298,15 К) в работе
[765]. Термодинамические функции, вычисленные Маклеодом [3337] (Т ^ 2000 К) завышены
на 7 и 19 Дж-К-моль в значениях Ф°A000 К) и Ф°B000 К) из-за аномального роста
теплоемкости при Т > 780 К по данным [3337].
В справочнике принята энтальпия образования
^"(CsF, к, 298,15 К) = —557,1 +0,7 кДж • моль,
основанная на приведенных в табл. 49.9 результатах измерений энтальпии растворения CsF(k)
в воде. Результаты измерений в работах [4347, 4871а] оказались в хорошем согласии и на них
Таблица 49.9. Результаты измерений haiJH° (CsF, к-»-ооН2О, 298,15 К) (в кДж • моль)
Авторы
Конечная концентрация, температура
измерений
4„Я°(оо Н2О, 298,15 К)
Форкран, 1911 [2300, 2301] 110 Н2О, 288,15 К, 1 измерение —36,86
Сомсен,"Веда, 1964 [4347] 13 200—46 260 Н2О, 298,15 К, 8 измерений —36,07+0,30
Цветков, Рабинович, 1969 [1097] 500 Н2О, 298,15 К, 1 измерение —37,35+0,47
Тоури и др., 1980 [4515] 2840 Н2О, 298,15 К —40,54+0,97
Ефимов и др., 1981 [4871aJ 4000—6400 Н2О, 298,15 К, 6 измерений —36,33+0,10
основано средневзвешенное значение Ar#°og(CsF, к -* ооН20; 298,15 К)=—36,30+0,10 кДжХ
Хмоль, принимаемое для дальнейших расчетов. Близкие значения были получены также в
работах [1097] и [2300, 2301]. Величина, полученная в работе [4515], представляется ошибочной.
Эта работа опубликована в очень краткой форме, не позволяющей оценить ее надежность и
провести обоснованное сопоставление с данными других работ.
Давление пара в реакции CsF(k, ж)=СбГ(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = \Н°(Св?, к, 0) = 195 ± 4 кДж • моль.
Принятая величина основана на измерениях давления и состава насыщенного пара над
CsF(k, ж). В насыщенном паре фторида цезия присутствуют молекулы CsF и Cs2F2. По данным
работ [9, 309, 2166] и термодинамическим свойствам, принятым в справочнике, при Tm(CsF)=
= 976 К в насыщенном паре содержится 85 и 15 мол.% этих соединений. Методика выбора
термохимических величин для компонентов насыщенного пара галогенидов щелочных металлов
изложена в Приложении 8.
Результаты расчетов энтальпии сублимации CsF(k) в виде мономерных молекул представлены
в табл. 49.10. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая величина является средневзвешенной из всех приведенных величин. При вычислении
средневзвешенного принималось во внимание отклонение результатов данной работы от среднего.
482
Цезий и его соединения
CsF(r)
Таблица 49.10. Результаты определений As-ff°(CsF, к, 0) (в кДж • моль)
Авторы
Метод
Вартенберг, Шульц, 1921 [4663] Точек кипения, 1228—1554 К, 8 точек
Руфф и др., 1922 [4096] То же, 1306—1528 К, 7 точек
Пыо, Барроу, 1958 [3896] Торзионный, 753—856 К а
Эйзенштадт и др., 1958 ]2166] Эффузионный, 838—919 К, 7 точек
Шир, Файн, 1962 [4178] То же, 677—878 К, 32 точки
Горохов, 1972 [309] Масс-спектрометрический, 745—803 К а
Сонин, Поляченок, 1973 [966] Точек кипения, 1071—1409 К, 19 точек
Топор, 1974 |4526] Статический, 1108—1288 К а
а Данные представлены в виде уравнения.
AsH°(CsF, к, 0)
II закон
200 ±5
203 + 20
206
201 + 2
203+3
195
202 + 5
197+4
III закон
192,3 + 0,4
192,2+1,0
197,2+1,4
206,4+0,2
194,1 ±0,3
—
194,5+0,4
195,0±0,4
Погрешность принятой величины учитывает воспроизводимость измерений, расхождения изме»
рений в разных работах, неточности термодинамических функций и определения состава насы*
щенного пара.
CsF(r). Термодинамические свойства газообразного фторида цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1250.
Приведенные в табл. 49.4 молекулярные постоянные CsF приняты на основании анализа
результатов исследований микроволнового [2697, 2700, 2701, 4595], ИК- [61] и УФ-[1406, 1771]
спектров этой молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406, 1771]
в справочнике принимается, что валентные возбужденные электронные состояния CsF имеют
энергии больше 42 000 см и не рассматриваются в справочнике.
Приведенные в табл. 49.4 колебательные и вращательные постоянные CsF в состоянии X1!!*
получены Хонэрьягером и Тишером [2697] обработкой результатов измерений микроволновых
переходов / ->/+1 (/^ 2) на колебательных уровнях с ь>=0, 1, . . ., 9. Соответствующие по-
стоянные сое и шехе были получены не непосредственно по экспериментальным данным, а рассчи~
таны с помощью соотношений Данхема. Они хорошо согласуются с результатами исследования
ИК-спектра [61] (а>е=353+4, шехе = \,1 см).
Термодинамические функции CsF(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения Qm и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dx и d2 приведены в табл. 49.5 и получены по
принятым молекулярным постоянным CsF.
Погрешности рассчитанных термодинамических функций CsF(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах дополнительный вклад в погрешность дает неточность принятого способа
экстраполяции потенциальной функции. Погрешности в значениях Ф°(Г) составляют 0,02, 0,05 и
0,5 Дж-К^-моль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции CsF(r) ранее вычислялись в таблицах JANAF [2834], а также
в работе Брюэра и Брэкет [1660] (Ф°(Г), Т < 2000 К). Расхождения данных [2834] и табл. 1250
не превышают 1,2 и 4 Дж-К-моль в значениях Ф°(Т) и S (Т) соответственно и объясняются
тем, что в этом справочнике использовался приближенный метод расчета.
Константа равновесия реакции CsF(r)=Cs(r)+F(r) вычислена с использованием значения'
Ar#°@)=D0(CsF)=517,026+4,2 кДж • моль « 43 220+350 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации CsF(k). Этим величинам также соответствует
Д/nCsF, г, 0) = —361,737+ 4,1 кДж • моль.
Булевич и др. [1719] на основании спектрофотометрического исследования равновесия
реакции Cs(r)+HF(r)=CsF(r)+H(r) (пламена H2+O2+N2 с добавками солей цезия и C7F14, 1500—
483
Cs2F2(r) Глава 49
2600 К) получили значение D0(CsF)=515+30 кДж-моль. Значительно более низкое значение
D69+10 кДж-моль) было получено Берковицем [1498] фотоионизационным методом. Как
отмечает автор [1498], форма полученной кривой эффективности ионизации оказалась аномальной,
поэтому найденное в этой работе значение не может считаться надежным.
Cs2F2(r). Термодинамические свойства газообразного дифторида дицезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1251.
Молекулярные постоянные, использованные для расчета термодинамических функций Cs2F2,
приведены в табл. 49.6. На основании теоретических расчетов [961, 4689] для молекулы Cs2F2
в основном электронном состоянии ХгАд принимается плоское ромбическое строение (симметрия
Du). Приведенное в табл. 49.6 произведение моментов инерции соответствует структурным
параметрам r(Cs—F)=2,56+0,05 А и Д F—Cs—F=79+5°, рассчитанным Соломоником для ионной
модели [961, 963]. Погрешность значения IaIbIc составляет 1-1012 г3-см6. Приведенные
в табл. 49.6 значения частот v4— ve найдены Мартином и Шабером [3410] в ИК-спектре молекул
Cs2F2, изолированных в матрице из Аг, их величины хорошо согласуются со значениями,
полученными в работе Олта и Эндрюса [1362] г. Для частот vx—v3 приняты значения, рассчитанные
Соломоником [961, 963]. Погрешности принятых значений частот vx и v3 оцениваются в 40—
60 см, остальных частот — в 20—25 см.
Термодинамические функции Cs2F2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128), A. 130)
без учета возбужденных электронных состояний. Погрешности рассчитанных величин при
низких температурах обусловлены в основном неточностью принятых молекулярных постоянных,
а при высоких — приближенным характером расчета; они составляют 8, 12 и 16 Дж-К~1-моль~1
в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Gs2F2(r) рассчитывались в справочнике JANAF [2834]
и в работе Краснова и др. [569] (до 5000 К). Расхождения данных [569] и [2834] и табл. 1251
достигают 5 и 6 Дж-К-моль в значениях Ф°(Т) соответственно, они объясняются тем, что
в обоих расчетах использовались оцененные значения всех постоянных.
Таблица 49.11. Результаты определений энтальпии реакции CsF(k) + CsF(r) = Cs2F2{r) (в кДж • моль)
Авторы
Метод
ДгЯ°@)
II закон
III закон
Эйзенштадт и др., 1958 [2166] Эффузионный с селектором скоростей, 838—919 К, 31+14 34,1+0,4
7 точек
Акишин и др., 1960 [9]; Горохов, Масс-спектрометрический с двойной эффузионной ка- 42 29,9+3,5
1972 [309] мерой, 793—860 К, 3 точки
Масс-спектрометрический, 704—860 К 36+1 35,4
Константа равновесия реакции Cs2F2(r)=2Cs(r)-|-2F(r) вычислена с использованием значения'
Дг/?°@)=1197,578+12 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
A/Z/°(Cs2F2, г, 0) = —887 ± 12 кДж • моль.
Принятая величина основана на результатах исследования равновесия реакции CsF(k)+
+CsF(r)=Cs2F2(r), представленных в табл. 49.11. Приведенные в таблице погрешности
учитывают только воспроизводимость измерений. Для дальнейших расчетов принимается среднее
значение Дг#°@)=32+10 кДж-моль. Погрешность его оценена с учетом неточности
термодинамических функций. Близкие значения AfH°(Cs2F2, г, 0)=—892+15 кДж-моль (II закон) и
—897+15 кДж-моль" (III закон) были получены в работах [9, 309, 2166] при исследовании
равновесия реакции Cs2F2(r)=2CsF(r).
Говард и Эндрюс [2721] отнесли наблюденную ими частоту 248,3 см J к v2 колебанию молекулы Cs+F2, однако
авторы не исключали возможность принятого в настоящем справочнике отнесения (к v3(Cs2F2)).
484
Цезий и его соединения CsC1(k, ж)
CsC1(k, ж). Термодинамические свойства кристаллического и жидкого хлорида цезия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1252.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние CsC1(k) в интервале 0—743 К принимается
кубическая объемноцентрированная модификация (а, структурный тип GsGl), a в интервале 743—
919 К — кубическая гранецентрированная (|3, структурный тип NaCl).
При Т ^ 298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости в работе Тейлора и др. [4486] G—299 К). С учетом значения S°{1 К)=0,13 Дж-К^Х
Хмоль, полученного экстраполяцией данных [4486], в справочнике принимаются
приведенные в табл. 49.1 величины ?°B98,15 К) и #°B98,15 К)—#°@), погрешности которых оцениваются
в 0,2 Дж-К~1-моль~1 и 0,03 кДж-моль соответственно. Данные Брёнстеда [1683], который
измерил среднюю теплоемкость CsC1(k) в интервале 273—293 К, не учитывались.
Для теплоемкости a-CsCl в интервале 298,15—743 К принято уравнение, полученное на
основании значения С;B98,15 К) (см. табл. 49.1) и значений #°E00 К)-#°B98,15 К)=10,96 кДжХ
Хмоль и #°G00 К)—#°B98,15 К)=22,22 кДж-моль. Две последние величины приняты по
результатам измерений энтальпии в работах Кейлора и др. [2945] C85—905 К) и Смита и др.
[4321] B98,15—918 К). Для теплоемкости |3-CsCl принято постоянное значение 65,2 Дж-КХ
Хмоль, найденное совместной обработкой данных Кейлора и др. [2945, 4321] и Маркова и др.
[658] G63—913 К). Данные Марчидана и Панделе [3372] G65—905 К) не учитывались,
поскольку приводят к резко завышенному значению теплоемкости {3-CsCl.
Температура полиморфного превращения G43 +2 К) принята по согласующимся между собой
результатам измерений [644, 1339, 2570, 2571, 2704, 2878, 2945, 3481, 3565, 3878, 3879, 4681,
4760]; данные [204, 945, 946, 3195, 3209, 3355, 3566, 4225, 4539] имеют большой разброс G31—
753 К). Температура плавления (919+1 К) определена в работах [160, 162, 204, 331, 654, 658,
669, 797, 946, 1050, 4225]; менее точные данные получены авторами [195, 365, 384, 386, 459, 849,
1140, 1599, 1653, 1867] (913—922 К). Энтальпия полиморфного превращения B,93+0,40 кДжХ
Хмоль) найдена Арелом и др. [1339], с этим значением хорошо согласуются измерения Кларка
[1867] B,93+0,40 кДж-моль), Пойхонена и Мансикка [3878] B,89+0,12 кДж-моль), Уэйма
и Арендса [4681] C,14+0,33 кДж-моль). В работах [2945] и [4321] получены величины 2,4
и 3,8 кДж-моль соответственно. Энтальпия плавления B0,38+0,4 кДж-моль) принята по
данным Кейлора и др. [2945] B0,8 кДж-моль), Смита и др. [4321] B0,04 кДж-моль), Маркова
и др. [658] B0,33+0,40 кДж-моль), Марчидана и Панделе [3372] B0,5 кДж-моль) и Двор-
кина и Бредига [2137] B0,25 кДж-моль); заниженное значение этой величины получено в
работе Йококава и др. [4799] A9,5+0,5 кДж-моль-1). Теплоемкость СбСЦж) G3,5+1,5 Дж-К^Х
Хмоль) получена в результате совместной обработки данных по энтальпии расплавленного
CsCl [2945] (924-1168 К, 8 измерений), [658] (948-1222 К, 6 измерений) и [3372] (925-1080 К,
7 измерений). Мургулеску и Теле [3594] измерили теплоемкость СэС1(ж) в узком температурном
интервале вблизи точки плавления и получили значение 75,9 Дж-К~1-моль~1.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,15,0,7, 4 и 10 Дж^-К-моль соответственно. Расхождения между термодинамическими
функциями CsC1(k, ж), приведенными в табл. 1252 и в справочнике [2834] (Т <^ 3000 К), не
превышают 0,3 Дж-К-моль в значениях Ф°(Т) во всем интервале температур.
В справочнике принята энтальпия образования
к, 298,15§К) = —442,31 +0,16 кДж ¦ моль,
основанная на измерениях энтальпии растворения CsC1(k) в воде, представленных в табл. 49.12.
Для дальнейших расчетов принимается средневзвешенное значение ДадДг°(соН20; 298,15 К)=
= 17,19+0,03 кДж-моль. Помимо представленных в табл. 49.12 наиболее детальных работ,
энтальпия растворения CsC1(k) была измерена также в работах [24, 257, 747, 885, 901, 902, 1097,
2295, 2300, 2301, 2548, 4781], результаты которых приводят к величинам, совпадающим с принятой
в пределах их погрешностей.
Давление пара в реакции CsC1(k, ж)=СзС1(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = A,#°(CsCl, к, 0) = 202,4 + 4,0 кДж • моль.
485
CsC1(k, ж)
Глава 49
Таблица 49.12. Результаты
Авторы
Воробьев и др., 1966 [244]
Сергеева и др., 1970 [922]
Дэдгер, Тахериан, 1977 [1983]
Воробьев и др., 1978 [245, 779]
Монтгомери и др., 1978 [3548]
Джонсон, Гайер, 1979 [2871]
Тоури, Перашон, 1980 [4514]
определений Aajfl"°(CsCI, к-^-соН2О, 298,15 К) (в
Конечная концентрация, температура
измерений
820—3280 Н2О, 288,15 К, 9 измерений
720 Н2О, 291—305 К, 4 измерения
1790 Н2О, 293—305 К, 7 измерений
1850—5550 Н2О, 298,15 К, 5 измерений
2780 Н2О, 298,15 К
774—922 Н3О, 298,15 К, 6 измерений
724—1022 Н2О, 298,15 К, 6 измерений
600 Н2О, 298,15 К, 7 измерений
600 Н2О, 298,15 К, 6 измерений
55 500 НаО, 298,15 К, 1 измерение
кДж • моль х)
Дяг//°(со Н2О, 298,15 К)
17,37 +0,17
17,28+0,20
17,18+0,20
17,41 +0,20
17,35 ±0,12
17,205+0,050
17,200+0,050-
17,189+0,030
17,155+0,030
17,39+0,30
Принятая величина основана на измерениях давления и состава насыщенного пара над
CsC1(k, ж). В насыщенном паре над CsG1(k, ж) присутствуют молекулы GsCl и Gs2Cl2. Согласно
данным [9, 309] и термодинамическим свойствам, принятым в справочнике, в паре при Tm(GsCl)=
=919 К содержание этих соединений составляет 93 и 7 мол.% соответственно. Методика выбора
термохимических величин для компонентов насыщенного пара галогенидов щелочных металлов
изложена в Приложении 8.
Результаты расчетов энтальпии сублимации CsC1(k) в виде мономерных молекул представлены
в табл. 49.13. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая величина является средневзвешенной из данных табл. 49.13. При вычислении
средневзвешенного принималось во внимание отклонение результата данной работы от среднего.
Таблица 49.13. Результаты определений Asfl"°(CsCl, к, 0) (в кДж ¦ моль)
Авторы
Вартенберг, Шульц, 1921 [4663]
Руфф, Мугдан, 1921 [4094]
Фиок, Родебуш, 1926 [2258]
Нива, 1938 [3665]
Кангро, Викинг, 1938 [2919]
Коджин, Кимбелл, 1948 [1890]
Тредуэлл, Вернер, 1953 [4538]
Несмеянов, Сазонов, 1960 [721]
Милн, Клейн, 1960 [3510]
Шир, Файн, 1962 [4178]
Шриер, Кларк, 1963, [4212]
Вдовенко и др., 1967 [196]
Мургулеску, Топор, 1967 [3596]
Мургулеску, Топор, 1968 [3597]
Дудчик и др., 1970 [391]
Колесников, 1971 [532]
Акишин и др., 1960 [9]; Горохов,
1972 13041
Шнып, Новиков, 1972 [1156]
Бурылев, Миронов, 1976 [158]
Шефер, Вагнер, 1979 [4176]
Метод
Точек кипения, 1335—1577 К, 9 точек
То же, 1259—1568 К, 7 точек
Статический, 1098—1293 К, 9 точек
Эффузионный, 793—893 К, 11 точек
Протока, 1133—1263 К, 3 точки
Эффузионный, 690—891 К, 29 точек
Протока, 784—905 К, 10 точек
Эффузионный, 605—851 К, 16 точек
Масс-спектрометрический, Гор=5;815 К а
Эффузионный, 675—892 К, 21 точка
Точек кипения, 1102—1384 К а
Эффузионный, 673—873 К а
Статический, 1174—1353 К, 10 точек
То же, 1328—1436 К, 4 точки
Статический и точек кипения, 1103—1493 К а
Масс-спектрометрический, 532—565 К а
То же, 745—847 К а
Точек кипения, 1200—1500 К а
То же, 1202—1421 К а
Масс-спектрометрический, 747—839 К а
а Данные представлены в виде уравнения.
A,fl°(CsCl, к, 0)
11 закон
213+5
225+16
208+3
200 +28
210 ±90
207 ±3
202 ±2
193+3
189
208 ±3
206
209
208+2
213 ±2
210
205+7
199+8
212
218
204
III закон
201,8+0,6
201,2 ±1,7
202,7+0,2
208,4 +0,3
202,9 ±1,5
200,3 ±0,3
204,4 +0,2
204,1 ±0,6
—
201,1+0,3
202,4 ±4,8
199,4 ±15
202,8 ±0,2
202,0 ±0,6
200,4 ±3,1
—
—
201,2 ±2,9
205,0 ±2,6
—
486
Дезий и его соединения CsCl(r), Cs2Cl2(r)
Оценка погрешности принятой величины производилась с учетом воспроизводимости измерений,
расхождения результатов измерений в разных работах, неточности термодинамических функций
и определения состава насыщенного пара.
CsCl(r). Термодинамические свойства газообразного хлорида цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1253.
Приведенные в табл. 49.4 молекулярные постоянные CsCl приняты на основании анализа
результатов исследований микроволнового [1874, 1875, 1876, 2697, 2700, 2701, 4396], ИК- [3984]
и УФ- [1406] спектров этой молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406] в
справочнике принимается, что валентные возбужденные электронные состояния CsCl имеют энергии
больше 36 000 см.
Приведенные в табл. 49.4 колебательные и вращательные постоянные CsCl в состоянии Х1Е +
получены Хонэрьягером и Тишером [2697] обработкой результатов измерений микроволновых
переходов / -» /+1 (/ ^ 5) на колебательных уровнях с v=0, 1, . . ., 13, соответствующие
постоянные ше и шехе были получены не непосредственно по экспериментальным данным, а
рассчитаны с помощью соотношений Данхема. Они хорошо согласуются с результатами исследования
ИК-спектра, [3984] (сое=209+6, шЛ=0,75 см).
Термодинамические функции CsCl(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QBB и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, dt и d2 приведены в табл. 49.5 и получены по принятым
молекулярным постоянным 133Cs3SCl для изотопомера, соответствующего природной смеси изотопов
хлора.
Погрешности рассчитанных термодинамических функций CsCl(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным учетом
изотопного состава хлорида цезия. При более высоких температурах дополнительный вклад в
погрешность вносят неточность принятого способа экстраполяции потенциальной функции, а также
пренебрежение возбужденными электронными состояниями. Погрешности в значениях Ф°(Г)
составляют 0,02, 0,05 и 1,0 Дж-К^-моль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции CsCl(r) ранее вычислялись в таблицах JANAF [2834], а также
в работах Уилкинса [4728] (Т < 6000 К), Раиса и Клемперера [3985] (Т < 2000 К), Брюэра
и Брэкет [1660] (Ф°(Г), Т < 2000 К). Расхождения данных [2834], [4728] и табл. 1253 достигают
1,7 и 7,0 Дж-К-моль~1 в значениях Ф°(Т) и S°(T) соответственно и объясняются тем, что авторы
[2834] и [4728] использовали приближенный метод расчета.
Константа равновесия реакции CsCl(r)=Cs(r)+Cl(r) вычислена с использованием значения
Ar#°@)=D0(CsCl)=437,693+4,l кДж-моль~1я^36 590+340 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации CsC1(k). Этим величинам также соответствует
A/ff°(CsCl, г, 0) = —240,059 + 4,0 кДж • моль. '
Близкие значения энергии диссоциации D42+7 кДж-моль и 437+8 кДж-моль) были
получены Берковицем [1498] (фотоионизационный метод) и Су и Райли [4427] (метод
фотофрагментарной спектроскопии) соответственно. Булевич и др. [1719] на основании спектрофотометрического
изучения реакции Cs(r)+HCl(r)=CsCl(r)+H(r) (пламена H2+O2+N2 с добавками солей цезия
и СНС13 или СС14, 1500—2600 К) нашли D0(CsCl)=449+13 кДж-моль, согласующееся с принятым
в пределах погрешности.
Cs2Cl2(r). Термодинамические свойства газообразного дихлорида дицезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1254.
Молекулярные постоянные, использованные для расчета термодинамических функций Cs2Cl2(r),
приведены в табл. 49.6. На основании теоретических расчетов [961, 4689] для молекулы Cs2Cl2
в основном электронном состоянии ХхАд в справочнике принимается плоская ромбическая
структура (симметрия D2h). Приведенное в табл. 49.6 произведение моментов инерции соответствует
структурным параметрам r(Cs—С1)=3,11+0,05 А и Д С1—Cs—0=91+10°, рассчитанным Со-
ломоником и Красновым по ионной модели [961, 963]. Погрешность значения IaJbIq составляет
1-Ю11 г3-см6. Приведенные в табл. 49.6 значения частот v4—ve найдены Мартином и Шабером
C410] в ИК-спектре молекул Cs2Cl2, изолированных в матрице из Аг. Для частот v1—v3 приняты
487
CsBr(K, ж)
Глава 49
значения, рассчитанные Соломоником и Красновым [961, 963]. Погрешность принятого значения Vj.
оценивается в 40 см, остальных частот — в 20 см.
Термодинамические функции Cs2Cl2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных состояний. Погрешности рассчитанных величин обусловлены
главным образом неточностью принятых молекулярных постоянных, а также приближенным
характером расчета; они составляют 10, 15 и 20 Дж-К~1-моль~1 в значениях Ф°(Г) при 298,15Т
3000 и 6000 К соответственно.
Ранее термодинамические функции Cs2Gl2(r) рассчитывались в справочнике JANAF [28341
и работе Краснова и др. [569] (до 3000 К). Результаты этих расчетов согласуются с данными
табл. 1254 в пределах 2 Дж-К-моль~1 в значениях Ф°(Г).
Константа равновесия реакции Cs2Cl2(r)=2Cs(r)+2Cl(r) вычислена с использованием значения
Дг-йгс@)=1036,268+15 кДж-моль", соответствующего принятой в справочнике энтальпии
образования
bfH°(Csfil2, г, 0) = —641 + 15 кДж-моль-1.
Принятая величина основана на результатах исследований равновесия реакции CsC1(k, ж)-|-
+GsCl(r)=Cs2Gl2(r), представленных в табл. 49.14. Приведенные в таблице погрешности учиты-
Таблица 49.14. Результаты определений энтальпии реакции CsCI(k, ж) -f- CsCl(r) = Cs2CI2(r) (в кДж • моль)
Авторы
Берковиц, Чапка, 1958 [1501 ]
Милн, Клейн, 1960 [3510]
Шриер, Кларк, 1963 [4212]
Мургулеску, Топор, 1968 [3597]
Колесников, 1971 [532]
Акишин и др., 1960 [9]; Горохов,
1972 [309]
Шефер, Вагнер, 1979 [4176]
Метод
Масс-спектрометрический, —845 К, 1 точка
То же, Гср=815 К а
Протока и точек кипения, 1209—1350 К, 5 точек
Протока й статический, 1328—1436 К, 6 точек
Масс-спектрометрический, испарение с
поверхности, 569—617 К а
Масс-спектрометрический с двойной эффузион-
ной камерой, 745—847 К а
Эффузионный, 812—828 К, 3 точки
Масс-спектрометрический, 747—839 К а
а Данные представлены в виде уравнения.
ДгЯ°@)
II закон
39,3
70 ±25
48 ±100
48 ±13
49 ±4
79 ±2
32
III закон
43,4
—
24,5 ±2,7
24,3 ±2,7
—
—
40,4 ±5,0
—
вают только воспроизводимость измерений. Для дальнейших расчетов [принимается значение-
Дг-НР°@)=42+15 кДж-моль, среднее из работ Горохова, Акишина и др. [9, 309] и Берковица
и Чапки [1501]. Значения, полученные в работах [3597, 4212] комбинацией измерений двумя
методами, представляются менее надежными. Неточность термодинамических функций вносит
погрешность в АгН°@), составляющую 10 кДж-моль (для измерений при 900 К, работы [9, 309,,
1501]) и 16 кДж-моль (при 1400 К, работы [3597, 4212]).
В работах [9, 309, 391, 532, 1995, 3597] было исследовано равновесие реакции Cs2Cl2(r)=
=2CsGl(r). Средние значения из этих исследований приводят к величинам A^°(Gs2Cl2, г, 0)=
="—658+25 кДж-моль (II закон) и —659+15 кДж-моль (III закон), которые в пределах
погрешностей согласуются с принятой величиной.
СзВг(к, ж). Термодинамические свойства кристаллического и жидкого бромида цезия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1255.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние СэВг(к) в интервале 0—910 К принимается
кубическая модификация (структурный тип CsCl)г.
1 В работах [1812, 2878, 4368, 4681], выполненных различными методами, ршеется указание на структурный переход
в бромиде цезия из кубической модификации типа CsCl в кубическую модификацию типа NaCl, который
осуществляется вблизи точки плавления, ввиду чего высокотемпературная модификация не имеет области
термодинамической стабильности.
488
Цезий и его соединения CsBr(K, ж}
При Т ^ 298,15 К термодинамические функции кристаллического бромида цезия' вычислены
по результатам измерений теплоемкости в работах Паукова и др. [764] A2—307 К), Сораи и др.
[4348] B,8—21 и 15—281 К), Роббинса и Маршалла [4014] A,1—5,6 К). Данные Киркхема и
Иейтса [3019] B5—300 К) лежат ниже принятых значений теплоемкости: в интервале 30—
70 К — на 2—4% и в интервале 180—300 К — на 0,5—1,5%. Измерения средней теплоемкости
в работе Брёнстеда [1683] B73—293 К) не учитывались. Приведенные в табл. 49.1 значения
?°B98,15 К) и Я°B98,15 К)—#°@) имеют погрешности 0,4 Дж-К-^моль и 0,04 кДж-моль
соответственно.
При Т > 298,15 К для теплоемкости СвВг(к) принято линейное уравнение, полученное на
основании значений С;B98,15 К)=52,93 Дж-К^-моль [764] и Я°G50 К)-Я°B98,15 К) =
= 25,29 кДж-моль. Последняя величина вычислена по уравнению для энтальпии, приведенному
в работе Бибаса и Леонарди [1553] F23—909 К).
Температура плавления (910 +2 К) принята по согласующимся между собой измерениям Спинд-
лера [4368] (911 ±2 К), Диогенова и Буковой [371] (909 К), Маргеритиса и др. [3386] (910 К),
выполненным для весьма чистых образцов; менее точные результаты получены в работах [85, 457,.
1015, 1016, 1812, 4663] (900—914 К). Энтальпия плавления B3,6+0,4 кДж-моль) принята по
данным Дворкина и Бредига [2137]. Измерения Бибаса и Леонарди [1553] приводят к несколько
меньшему значению B1,6+0,9 кДж-моль). Теплоемкость СэВг(ж) G5+4 Дж-К~1-моль~1)
оценена на основании имеющихся данных о теплоемкости других жидких галогенидов цезия,
а также расплавленных галогенидов других щелочных металлов. Экспериментальные данные
[1553] F4г4 Дж-К~1'моль~1) и [3594] G7,9 Дж-К^-моль) ненадежны, поскольку в работе
[1553] не указана чистота исследованного образца, а данные [3594] представляются несколько
завышенными.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,3,.
1,5, 5 и 10 Дж-К-моль~1. Расхождения между термодинамическими функциями СэВг(к, ж),
приведенными в справочнике [1395] (Т <^ 1500 К) и в табл. 1255, не превышают 0,4 Дж-К~1-моль~1
в значениях Ф°(Г) во всем интервале температур.
В справочнике принята энтальпия образования
r, к, 298,15 К) =—405,60 + 0,25 кДж • моль,
основанная на измерениях энтальпии растворения СэВг(к) в воде, представленных в табл. 49.15.
Как видно из этой таблицы, результаты измерений, за исключением работы [2300, 2301], находятся
Таблица 49.15. Результаты
Авторы
Форкран, 1911 12300, 2301]
Ланге, Мартин, 1937 [3179]
Воробьев и др., 1966 [244]
Де Виссер, Сомсен, 1973 [2056]
Крестов и др., 1974 [2, 3, 580]
Монтгомери и др., 1978 [3548]
Воробьев и др., 1978 [245, 779]
Тоури, Перашон, 1980 [4514]
измерений Aarff°(CsBr, к->соН20, 298,15 К) (в
Конечная концентрация, температура
измерений
110 Н2О, 288,15 К, 1 измерение
500 Н2О, 298,15 К, 1 измерение
1040 Н2О, 298,15 К, 4 измерения
2050 Н2О, 298,15 К, 2 измерения
5550—55 500 Н2О, 298,15 К, 3 измерения
14—11100 Н2О, 298,15 К, 2 измерения
864—955 Н2О, 298,15 К, 4 измерения
2780 НаО, 298,15 К
55500 Н2О, 298,15 К, 1 измерение
кДж • моль)
ДагЯ°(ооН2О, 298,15 К)
26,66
25,98 +0,30
26,20 ±0,20
26,17 ±0,20
26,18+0,10
25,99 ±0,25
25,95 ±0,10
26,02 ±0,12
26,10 ±0,30
в хорошем соответствии. Для дальнейших расчетов принимается средневзвешенное значение
Дв(гЯ°(соЯ20; 298,15 К)=26,07+0,10 кДж-моль, близкое к результатам наиболее точных
измерений в работах [245, 779, 2056, 3548].
Давление пара в реакции CsBr(K, ж)=СэВг(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
ДгЯ°@) = A,ff°(CsBr, к, 0) = 201,5 ± 4,0 кДж • моль.
489
CsBr(r)
Глава 49
Принятая величина основана на измерениях давления и состава насыщенного пара над
CsBr(K, ж). В насыщенном паре бромида цезия присутствуют молекулы CsBr и Cs2Br2. По данным
работ [9, 309, 4073] и принятым в справочнике термодинамическим свойствам при Tm(CsBr)=
=910 К содержание этих компонентов составляет 94 и 6 мол. % соответственно. Методика выбора
термохимических величин для компонентов насыщенного пара галогенидов щелочных металлов
изложена в Приложении 8.
Результаты расчетов энтальпии сублимации CsBr(K) в виде мономерных молекул представлены
в табл. 49.16. Приведенные в таблице погрешности учитывают только воспроизводимость измере-
Таблица 49.16. Результаты определений AsH°(CsBr, к, 0) (в кДж ¦ моль)
Авторы
Метод
As#°(CsBr, к, 0)
II закон
III закон
Вартенберг, Шульц, 1921 [4663] Точек кипения, 1325—1574 К, 8 точек 211+4 202,6+0,5
Руфф, Мугдан, 1921 [4094] То же, 1251—1578 К, 7 точек 211+17 202,0+1,2
Нива, 1938 [3665] Эффузионный, 783—853 К, 8 точек 196+6 201,5+0,2
Коджин, Кимбелл, 1948 [1890] То же, 775—900 К, 46 точек 215+4 201,5+0,2
Шир, Файн, 1962 [4178] То же, 715—900 К, 28 точек 205+2 199,7 ±0,2
Макаров, Ступин, 1968 [645] То же, 650—850 К, данные представлены в виде 203 197,9+10
уравнения
Мургулеску, Топор, 1967 [3596] Статический, 1250—1363 К, 12 точек 206+3 202,5+0,2
Мургулеску, Топор, 1968 [3597] То же, 1250—1363 К, 5 точек 180+30 201,6+1,2
Колесников, 1971 [532] Масс-спектрометрический, испарение с поверхно- 202+8 —
сти, 524—570 К
Акишин и др., 1960 [9]; Горохов, Масс-спектрометрический, 786—821 К 199+12 —
1972 [309]
ний. Принятая величина является средневзвешенной из приведенных в таблице величин, за
исключением данных [645]. При вычислении средневзвешенного принималось во внимание отклонение
результатов данной работы от среднего. Оценка погрешности принятой величины производилась
с учетом воспроизводимости измерений, расхождения результатов измерений в разных работах,
неточности термодинамических функций и определения состава насыщенного пара.
CsBr(r). Термодинамические свойства газообразного бромида цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1256.
Приведенные в табл. 49.4 молекулярные постоянные CsBr приняты на основании анализа
результатов исследований микроволнового [2697, 2700, 2701, 4101], ИК- [3984] и УФ- [1406]
спектров этой молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406] в
справочнике принимается, что валентные возбужденные электронные состояния CsBr имеют энергии
больше 34 000 см.
Приведенные в табл. 49.4 колебательные и вращательные постоянные CsBr в состоянии ХХЕ+
получены Хонэрьягером и Тишером [2697] обработкой результатов измерений микроволновых
переходов / -> /+1 (/ ^ 11) на колебательных уровнях с г;=0,1, . . ., 17, соответствующие
постоянные (о„ и шехе были получены не непосредственно по экспериментальным данным, а
рассчитаны с помощью соотношений Данхема.
Термодинамические функции CsBr(r) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 93)—A. 95). Значения QBa и ее производные вычислялись без учета возбужденных
электронных состояний по методике, изложенной в Приложении 7. Использованные в расчете
коэффициенты потенциальной функции d0, d± и d2 приведены в табл. 49.5 и получены по принятым
постоянным молекулы 133Cs79Br для изотопомера, соответствующего природной смеси изотопов брома.
Погрешности рассчитанных термодинамических функций CsBr(r) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных и приближенным учетом
изотопного состава бромида цезия. При более высоких температурах дополнительный вклад в по-
490
Цезий и его соединения Cs2Br2(r)
грешность вносят неточность принятого способа экстраполяции потенциальной функции, а также
пренебрежение возбужденными электронными состояниями. Погрешности в значениях Ф°(Т)
составляют 0,02, 0,05 и 1,0 Дж-К^-моль при 298,15, 3000 и 6000 К соответственно.
Термодинамические функции CsBr(r) ранее вычислялись в работе Брюэра и Брэкет [1660]
(Ф°(Т), Т ^ 2000 К). Расчеты в работе [1660] выполнены по молекулярным постоянным, близким
к принятым в настоящем издании, соответствующие расхождения с данными табл. 1256 не
превышают 0,4 Дж-К-моль в значениях Ф°(Г) и объясняются главным образом различием метода
расчета.
Константа равновесия реакции CsBr(r)=Cs(r)+Br(r) вычислена с использованием значения
Ar#°@)=D0(CsBr)=393,201+4,l кДж-моль-хя»32 870+340 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации СэВг(к). Этим величинам также соответствует
A^^CsBr, г, 0) = —197,264+ 4,0 кДж • моль.
Нижняя граница возможных значений D0(CsBr) была определена Зоммермайером [4345]
{379 кДж-моль) по коротковолновой границе флуктуационных полос в спектре поглощения
CsBr. Существенно более высокое значение D16+13 кДж-моль) было получено Булевич и др.
[1719] при спектрофотометрическом исследовании равновесия реакции Cs(r)+HBr(r)=CsBr(r)+
+Н(г) в пламенах H2+O2+N2 A500—2600 К). Берковиц [1498] в результате фотоионизационных
измерений нашел D0(CsBr)=402+5 кДж-моль. Обработка данных [1498] с учетом теплового
возбуждения и кинетической энергии осколков, выполненная Дрониным и Гороховым [389],
привела к более низкой величине, 392^5 кДж-моль, практически совпадающей с принятой.
Еще более низкое значение C85+4 кДж-моль) было получено методом фотофрагментарной
спектроскопии в работе Су и Райли [5056].
Cs2Br2(r). Термодинамические свойства газообразного дибромида дицезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1257.
Молекулярные постоянные, использованные для расчета термодинамических функций Cs2Br2(r),
приведены в табл. 49.6. На основании теоретических расчетов [961, 4689] для молекулы Cs2Br2
в основном электронном состоянии ХгАд принимается плоская ромбическая структура (симметрия
ZJA). Приведенное в табл. 49.6 произведение моментов инерции соответствует структурным
параметрам r(Cs—Вг)=3,29 +0,08 А и ДВг—Cs—Вг=94+10°, рассчитанным Соломоником по ионной
модели [961, 963]. Погрешность значения IaIbIc составляет 4-10~т г3-см6. Приведенные
в табл. 49.6 значения частот v6, v6 найдены Мартином и Шабером [3410] в ИК-спектре молекул
Cs2Br2, изолированных в матрице из Аг. Для частот vx—v4 приняты значения, рассчитанные
Соломоником [961, 963] для ионной модели молекулы. Погрешность принятого значения \
оценивается в 25 см, остальных частот — в 10—15 см.
Термодинамические функции Cs2Br2(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130). Их погрешности обусловлены неточностью принятых молекулярных постоянных и
приближенным характером расчета, они составляют 12, 18, 25 Дж-К~1-моль~1 в значениях Ф°G|)
при 298,15, 3000 и 6000 К соответственно.
Ранее термодинамические функции Cs2Br2(r) рассчитывались Красновым и др. [569] (до 3000 К)
и Фрурипом и Бландером [2340] (до 5000 К). Расхождения данных [569, 2340] и табл. 1257
достигают 3 и 5 Дж-К~1-моль~1 в значениях Ф°(Г) соответственно.
Константа равновесия реакции Cs2Br2(r)=2Cs(r)+2Br(r) вычислена с использованием значения
Дг#°@)=939,874+15 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
t\fH°(Cs2Bi-2, г, 0) = —548 + 15 кДж • моль.
Принятая величина основана на результатах исследований равновесия реакции CsBr(K, ж)+
+CsBr(r)=Cs2Br2(r), представленных в табл. 49.17. Приведенные в таблице погрешности
учитывают только воспроизводимость измерений. Для дальнейших расчетов принимается значение
ДгЯ°@)=48+12 кДж-моль, среднее из величин, полученных в работах Горохова, Акишина и др.
[9, 309] и Родберга и др. [4073]. Менее точной представляется величина, полученная
комбинированием результатов измерений статическим методом и методом протока [3597]. Неточность термо-
491
CsI(k, ж)
Глава 49
Таблица 49.17. Результаты определений энтальпии реакции CsBt(k, ж) + CsBr(r) = Cs2Br2(r)
(в кДж • моль)
Авторы
Ротберг и др., 1959
Акишин и др., 1960
1972 [309]
Мургулеску, Топор,
[4073]
[9]; Горохов
1968 [3597]
Колесников, 1971 [532]
Метод
Испарение с поверхности с селектором скоростей,
785-800 К, 3 точки
Масс-спектрометрический с двойной эффузионной
камерой, 786—830 К, данные представлены в виде
уравнения
Статический и протока, 1250—1363 К, 7 точек
Масс-спектрометрический, испарение с поверхности,
524-570 К
АГВ
II закон
—208 ±180
58
20 ±30
44+8
°@)
III
закон
47,4+2,0
48
31
3±4,0
7+1,0
—
динамических функций вносит погрешность в значение АгН°@), равную 10 кДж-моль для
измерений при 800 К [9, 309, 4073] и 20 кДж-моль для измерений при 1300 К [3597].
В работах [309, 532, 2546,3597] было исследовано равновесие реакции Cs2Br2(r)=2CsBr(r).
Средним из этих данных соответствуют AfH°(Cs2BT2, г, 0) =—549+25 кДж-моль (II закон) и —561 +
+15 кДж-моль (III закон), совпадающие с принятым в пределах погрешностей.
CsI(k, ж). Термодинамические свойства кристаллического и жидкого иодида цезия в
стандартном состоянии при температурах 100—3400 К приведены в табл. 1258.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние GsI(k) в интервале 0—905 К принимается
кубическая модификация (структурный тип GsGl).
При Т ^ 298,15 К расчет термодинамических функций выполнен по результатам исследований
теплоемкости в работах Маршалла и Кункеля [3396] B—10 К), Сораи и др. [4348] A,5—21 и
15—300 К), Тейлора и др. [4486] A3—298 К). Данным [3396] и [4348] соответствует значение
5°A3К)=2,26 Дж-К^-моль-1. Погрешности значений ?°B98,15 К) и #°B98,15 К)-#°@),
приведенных в табл. 49.1, оцениваются в 0,6 Дж-К~1-моль~1 и 0,06 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости CsI(k) принято уравнение, полученное обработкой данных
по энтальпии, найденных Кейлором и др. [2945, 4321] для температур 484—904 К B4 точки,
содержание примесей в образце ~0,2%, погрешность измерений 2%I.
Температура плавления Csl (905+2 К) принята по работе Спиндлера [4368]. Близкие значения
были получены в работах [4791] (903+2 К) и [906] (907 К). Энтальпия плавления B5,65 +
+0,40 кДж-моль) основана на принятых в справочнике уравнениях для энтальпии
кристаллического и жидкого иодида цезия. Значения, полученные экспериментально B5,02 [2945], 25,82
[4321], 23,60 [2137] и 27,61 кДж-моль [1636]), имеют большой разброс, и отдать предпочтение
одной из работ не представляется возможным. Теплоемкость Сэ1(ж) G1+2 Дж-К~1-моль~1)
принята по результатам обработки данных по энтальпии, полученных Кейлором и др. [2945, 43211
в интервале 908—1172 К A0 измерений). Мургулеску и Теле [3594] измерили теплоемкость
Сэ1(ж) вблизи температуры плавления и нашли значение С°(ж)=74,3 Дж-К~1-моль~1.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,4,
2, 5 и 10 Дж-К~1-моль~1 соответственно. Расхождения между термодинамическими функциями
Csl (к, ж), приведенными в справочнике [1395] (Т <^ 1500 К) и в табл. 1258, составляют 3 ДжХ
ХК~1-моль в значениях Ф°(Т) в связи с тем, что авторы [1395] принимали оцененное значение
для ?°B98,15 К).
В справочнике принята энтальпия образования]
AfH°{CsI, к, 298,15 К) =—348,10 + 0,18 кДж • моль,
1 Авторами [2945, 4321] измерения были выполнены в области 369—1172 К, однако сравнение этих данных и
данных [4348, 4486] позволяют предполагать, что для Т= 369+484 К A0 точек) Кейлор с соавторами получили
заниженные значения энтальпии.
492
Цезий и его соединения
CsI(k, ж)
«снованная на измерениях энтальпии растворения CsI(k) в воде, представленных в табл. 49.18.
Для дальнейших расчетов принимается значение Даг#°(ооН20; 298,15 К)=33,16+0,05 кДж-моль,
которое является средневзвешенным из величин, приведенных в таблице, за исключением
недостаточно точных и выпадающих данных [455, 1458, 2300, 2301, 3759].
Таблица 49.18. Результаты
Авторы
Бекетов, Бекетов, 1904 [1458]
Форкран, 1911 [2300, 2301]
Митчелл, Кобл, 1964 [3515]
Паолетти и др., 1965 [3759]
Иванова, Котлярова, 1968 [455]
Воробьев и др., 1966 [244]
Цветков, Рабинович, 1969 [1097]
Крестов и др., 1972 [583]
Новосёлов и др., 1975 [736]
Амиго и др., 1975 [1312]
Воробьев и др., 1978 [245, 779]
Монтгомери и др., 1978 [3548]
Тоури, Перашон, 1980 [4514]
определений Aajff°(CsI, к->оо Н2О, 298,15 К) (в
Конечная концентрация, температура
измерений
350—460 Н2О, 291,15 К, 3 измерения
110 Н2О, 288,15 К, 1 измерение
4440—23 320 Н2О, 298,15 К, 10 измерений
298,15 К, 2 измерения
298,15 К
1270 Н2О, 298,15 К, 5 измерений
2590 Н2О, 298,15 К, 2 измерения
500 Н2О, 298,15 К, 1 измерение
1110—5550 Н2О, 298,15 К
298, 15 К
28 000—185 000 Н2О, 293,15 К, 23 измерения
2780 Н2О, 298,15 К
1258—1672 Н2О, 298,15 К, 6 измерений
55 500 Н2О, 298,15 К, 1 измерение
кДж • моль)
ДагЯ°(ооНаО, 298,15 К)
33,25 ±0,65
33,28+0,50
33,150 ±0,050
32,55 ±0,20
32,68+0,70
33,29+0,13
33,26 ±0,13
33,10 ±0,30
33,01 +0,30
33,10+0,30
31,53+0,50
33,18 ±0,12
33,06 ±0,10
33,31 ±0,30
Давление пара в реакции CsI(k, ж)=Св1(г) вычислено с использованием принятой в
справочнике энтальпии сублимации
Дг#°@) = As#°(CsI, к, 0) = 198,7 + 3,0 кДж • моль.
Принятая величина основана на измерениях давления и состава насыщенного пара над
CsI(k, ж). В насыщенном паре иодида цезия присутствуют молекулы Csl и Gs2I2. Согласно
измерениям [9, 309, 2023] и принятым в справочнике термодинамическим свойствам при rm(CsI)=905 К
•содержание этих компонент составляет 93 и 7 мол. % соответственно. Методика выбора термохими-
Таблица 49.19. Результаты определений Asff°(CsI, к, 0) (в кДж-моль)
Авторы
Вартенберг, Шульц, 1921 [4663]
Руфф, Мугдан, 1921 [4094]
Дейц, 1936 [2023]
• Коджин, Кимбелл, 1948 [1890]
Шир, Файн, 1962 [4178]
Мургулеску, Топор, 1967 [3596]
Макаров, Ступин, 1968 [645]
Мургулеску, Топор, 1970 [3598]
Колесников, 1971 [532]
Акишин и др., 1960 [9]; Горохов,
1972 [309]
Метод
Точек кипения, 1296—1553 К, 5 точек'
То же, 1325—1553 К, 9 точек
Эффузионный, 789—815 К, 6 точек
Торзионный, 767—847 К, 9 точек
Эффузионный, 696—895 К, 54 твчки
То же, 671—876 К, 22 точки
Статический, 1157—1375 К, 12 точек
Эффузионный, 650—850 К а
Статический, 1276—1376 К, 4 точки
Масс-спектрометрический, испарение с
поверхности, 534—554 К а
Масс-спектрометрический, 716—815 К а
| а Данные представлены в виде уравнения.
A,H°(Csl, к, 0)
II закон
208 ±12
255 ±15
197 +18
207 ±4
206 ±13
203 ±3
200 ±3
198
—
197 ±8
190 ±8
III закон
198,1 ±1,2
198,8 ±2,2
200,1 +0,2
199,7+0,2
198,2 ±0,9
195,7 ±0,3
197,6+0,1
191,4 ±11
197,7 ±0,3
—
—
493
Csl(r) Глава 49
ческих величин для компонентов насыщенного пара галогенидов щелочных металлов изложена
в Приложении 8.
Результаты расчетов энтальпии сублимации CsI(k) в виде мономерных молекул представлены
в табл. 49.19. Приведенные в таблице погрешности учитывают только воспроизводимость
измерений. Принятая величина является средневзвешенной из приведенных в таблице величин, за
исключением данных [645]. При вычислении средневзвешенного принималось во внимание отклонение
результата каждой работы от среднего. Оценка погрешности принятой величины производилась
с учетом воспроизводимости измерений, расхождения измерений в разных работах, неточности
термодинамических функций и определения состава насыщенного пара.
Csl(r). Термодинамические свойства газообразного иодида цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1259.
Приведенные в табл. 49.4 молекулярные постоянные Csl приняты на основании анализа
результатов исследований микроволнового [2697, 2700, 4101], ИК- [3984] и УФ- [1406] спектров этой
молекулы.
По аналогии с молекулой LiF и на основании исследования электронного спектра [1406] в
справочнике принимается, что валентные возбужденные электронные состояния Csl имеют энергии
больше 28 000 см.
Приведенные в табл. 49.4 колебательные и вращательные постоянные Csl в состоянии Х*?+
получены Хонэрьягером и Тишером [2697] обработкой результатов измерений микроволновых
переходов / ->• /+1 (/ <; 19) на колебательных уровнях с v=0, 1, . . ., 21, 23, 24.
Соответствующие постоянные ш„ и шехе были получены не непосредственно по экспериментальным данным,
а рассчитаны с помощью соотношений Данхема.
Термодинамические функции Csl(r) были вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)г
A. 93)—A. 95). Значения QBa и ее производные вычислялись без учета возбужденных электронных
состояний по методике, изложенной в Приложении 7. Использованные в расчете коэффициенты
потенциальной функции d0, dt и d2 приведены в табл. 49.5 и получены по принятым постоянным
молекулы 133CsI.
Погрешности рассчитанных термодинамических функций Csl (г) при температурах до 3000 К
обусловлены главным образом неточностью фундаментальных постоянных. При более высоких
температурах дополнительный вклад в погрешность вносят неточность принятого способа
экстраполяции потенциальной функции, а также пренебрежение возбужденными электронными
состояниями. Погрешности в значениях Ф°(Т) составляют 0,02, 0,05 и 1,0 Дж-К~1-моль при 298,15,
3000 и 6000 К соответственно.
Термодинамические функции Csl(r) ранее вычислялись в работе Брюэра и Брэкет [1660J
(Ф°G1), Т <1 2000 К). Расчеты в работе [1660] выполнены по молекулярным постоянным, близким
к принятым в настоящем издании, соответствующие расхождения с данными табл. 1259 не
превышают 1 Дж-К-моль~1 в значениях Ф°(Т) и объясняются главным образом различием метода
расчета <?вн.<
Константа равновесия реакции CsI(r)=Cs(r)+I(r) вычислена с использованием значения
Ar#°@)=D0(CsI)=333,738+3,2 нДж-моль;^? 900+270 см, соответствующего принятым
в справочнике энтальпиям образования и сублимации CsI(k). Этим величинам также соответствует
Ь,Н°(Св1, г, 0) = —148,561 ±3,0 кДж • моль.
Практически совпадающее значение энергии диссоциации C34+2 кДж-моль) было получено
Су и Райли [5055] методом фотофрагментарной спектроскопии. Берковиц [1498] на основании
фотоионизационных измерений нашел более высокое значение C44+5 кДж-моль). Однако учет
теплового возбуждения и кинетической энергии осколков в экспериментах [1498] приводит, как
показали Дронин и Горохов [389], к величине 332+5 кДж-моль, практически совпадающей
с принятой.
Спектрофотометрическое исследование равновесия реакции Cs(r)+HI(r)=CsI(r)+H(r),
выполненное Булевич и др. [1719] (пламена Ha+O2+N2, 1500—2600 К), привело к менее точному
значению C50+17 кДж-моль), а старые исследования электронного спектра в работах Зоммер-
майера [4345] (граница флуктуационных полос) и Буткова и Теренина [1735] (атомная
флуоресценция) — к значениям 322 и 310+17 кДж-моль.
494
Цезий и его соединения
Cs2I2(r)
Cs2I2(r). Термодинамические свойства газообразного дииодида дицезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1260.
Молекулярные постоянные, использованные для расчета термодинамических функций Cs2I2(r),
приведены в табл. 49.6. На основании теоретических расчетов [961, 4689] для молекулы Cs2I2
в основном электронном состоянии Х1Ад принимается плоская ромбическая структура (симметрия
ZJA). Приведенное в табл. 49.6 произведение моментов инерции соответствует структурным
параметрам r(Gs—1)=3,54+0,10 А и Д1—Cs—1=98+10°, рассчитанным Соломоником по ионной
модели [961, 963]. Погрешность значения IaIbIc оценивается в 1 -10~110 г3-см6. ИК-спектр молекул
Cs2I2, изолированных в матрице из Аг, наблюдался в работе Мартина .и Шабера [3410]. Авторам
[3410] удалось определить только частоту ve, значение которой согласуется с рассчитанным в
работах [961, 963] (89 см). В справочнике значения всех основных частот приняты по расчету
[961], их погрешности оцениваются в 25 см для vx, v5, ve и в 10—15 см для остальных частот.
Термодинамические функции Cs2I2(r) вычислялись в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A. 130) без учета возбужденных электронных состояний. Погрешности рассчитанных величин
обусловлены неточностью принятых молекулярных постоянных и приближенным характером
расчета; они составляют 15, 20, 25 Дж-К^-моль в значениях Ф°(Г) при 298,15, 3000 и 6000 К
соответственно.
Ранее термодинамические функции Cs2I2(r) рассчитывались Красновым и др. [569] (до 3000 К)
и Фрурипом и Бландером [2340] (до 5000 К). Расхождения данных [569, 2340] и табл. 1260 не
превышают 2 и 6 Дж-К~1-моль в значениях Ф°(Т) соответственно.
Константа равновесия реакции Cs2I2(r)=2Cs(r)-b2I(r) вычислена с использованием значения
&гН°@)=818,354 +20 кДж-моль, соответствующего принятой в справочнике энтальпии
образования
i\fHo(Cs2Ji2, г, 0) = —448 + 20 кДж • моль-.
Принятая величина основана на результатах исследования равновесия реакции CsI(k, ж)+
+CsI(r)=Cs2I2(r), представленных в табл. 49.20. Приведенные в таблице погрешности учитывают
Таблица 49.20. Результаты определений энтальпии реакции CsI(k, ж) + Csl(r) = Cs2I2(r) (в кДж • моль)
Авторы
Дейц, 1936 [2023 J
Ротберг и др., 1959 [4073]
Мургулеску, Топор, 1970 [3598]
Колесников, 1971 [532]
Акишин и др., 1960 [9J; Горохов
1972 [309]
Метод
Эффузионный и торзионный, 789 К, 2 точки
Испарение с поверхности с селектором скоростей,
757-772 К, 2 точки
Статический и протока, 1276—1376 К, 12 точек
Масс-спектрометрический, испарение с поверхности
554—596 К а
4J4J х V %J \J Л- ч_
, Масс-спектрометрический с двойной эффузионной
камерой, 787—820 К, 2 точки
Масс-спектрометрический, 716—815 К а
а Данные представлены в виде уравнения.
АГН
II закон
—
82 ±30
48+10
67
57
"@)
III закон
47+8
59+7
32,5+1,2
—
48,4 ±3,0
50,9
только воспроизводимость измерений. Для дальнейших расчетов принимается Дг-/У°@)=48 +
+15 кДж-моль, среднее из приведенных в таблице величин. Его погрешность оценена с учетом
неточности термодинамических функций, которая вносит в значение Дг#°@) погрешность,
равную 14 кДж-моль для измерений при 800 К [9, 309, 2023, 4073] и 20 кДж-моль для измерений
при 1300 К [532].
В работах [9, 309, 532, 3598] было исследовано равновесие реакции Cs2Ia(r)=2CsI(r). Средние
значения из этих работ приводят к величинам AfH°(Cs2I2, г, 0) = —426+25 кДж-моль (II закон)
и —465+15 кДж-моль (III закон), которые в пределах погрешности совпадают с значением,
принятым в справочнике.
495
€s2SO4(k, ж) Глава 49
Cs2SO4(k, ж). Термодинамические свойства кристаллического и жидкого сульфата дицезия
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1261.
Значения постоянных, принятые для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние Cs2SO4 в интервале 0—920 К принята
ромбическая а-модификация, в интервале 920—1000 К — ^-модификация с неисследованной структурой
и в интервале 1000—1288 К — гексагональная ^-модификация [4455] *.
При Т < 298,15 К термодинамические функции Cs2SO4(k) вычислены по измерениям
теплоемкости, выполненным Пауковым и др. [770] A2,82—307,72 К, образец чистотой 99,9%). Среднее
отклонение экспериментальных точек от сглаженной кривой не превышает 0,1%. Графическая
экстраполяция теплоемкости привела к значению 5°A2 К)=1,3 Дж-К~1-моль~1. Погрешности
значений ?°B98,15 К) и #°B98,15 К)—#°@), приведенных в табл. 49.1, оцениваются в 0,4 ДжХ
хК~1-моль~1 и 0,08 кДж-моль соответственно.
Уравнение для теплоемкости a-Gs2SO4 в интервале 298,15—920 К (см. табл. 49.1) получено
совместной обработкой хорошо согласующихся между собой результатов измерения теплоемкости
в работе Шмидт [1153] B98—770 К) и измерения энтальпии в работах Соколова и др. [957]
F72,84—1288,07 К) и Дениелоу [2042, 2043, 2044] D00—1540 К). При обработке погрешности
всех измерений оценивались в 0,5%. Измерения Полищука и др. [800] (800—1380 К) так же, как
и в случае Rb2SO4(K), превышают данные других авторов на 6—7% и при выводе уравнений для
теплоемкости Cs2SO4(k) не учитывались.
Согласно результатам тщательных измерений энтальпии в работе Соколова и др. [957] F8
измерений; разброс не более 0,5%) в интервале 900—1000 К у Cs2SO4(k) имеются два фазовых
превращения II рода. Многочисленные измерения температуры полиморфного превращения сульфата
цезия не противоречат этим данным. В работах [156, 795, 1359, 2262, 3581] приведены значения
923—933 К, соответствующие первому переходу, а в работах [84, 363, 795, 853, 1136, 2185, 2778,
3241, 4454] получены значения 983—1055 К, относящиеся, по-видимому, ко второму переходу
(в работе [795] найдены оба перехода). Поскольку литературные данные о полиморфизме Cs2SO4
достаточно противоречивы, температуры фазовых переходов II рода 920+10 и 1000+10 К приняты
по наиболее надежным данным [957]. Результаты калориметрических определений [800, 2042,
2043, 2044], в которых определена температура полиморфного превращения (940 К) и энтальпии
превращения B,5 кДж-моль" [2042, 2043, 2044] и 1,3 кДж-моль [800]), не учитывались.
Уравнения для теплоемкости P-Cs2SO4 в интервале 920—1000 К и y-Gs2SO4 в интервале 1000—
1288 К найдены по результатам измерений энтальпии в работе Соколова и др. [957]; для у-моди-
фикации принятое уравнение практически совпадает с данными Дениелоу [2042, 2043, 2044].
Температура плавления A288+5 К) принята по результатам наиболее надежных работ [22,
79, 83, 84, 156, 363, 374, 795, 853, 861, 892, 967, 2764, 2808, 2825, 4213]. Энтальпия плавления
C5,1+0,8 кДж-моль) рассчитана из данных по энтальпии Сэ2ЗО4(ж), полученных Дениелоу
[2042, 2043, 2044] и значения для Cs2SO4(k) при Тт, полученного с помощью уравнений, принятых
для энтальпии Cs2SO4(k).
Теплоемкость жидкого сульфата цезия B07+2 Дж-К-моль~1) также рассчитана из данных
[2042, 2043, 2044] A300—1540 К).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0,3,
2, 12 и 24 Дж-К-моль соответственно.
Термодинамические функции, приведенные в справочнике Барина и Кнаке [1395] (Т ^ 1800 К),
отличаются от данных табл. 1261 на 4—10 Дж-К-моль в значениях Ф°(Г) из-за различия
принятых значений 5'°B98,15 К) и C°(Cs2SO4, к, ж).
Принятая в справочнике энтальпия образования
A^CsaSO^,, к, 298,15 К) = —1442,9 + 0,5 кДж • моль
основана на измерениях энтальпии растворения Cs2SO4(k) в воде, представленных в табл. 49.21.
Принятое для дальнейших расчетов значение ДагйГ°(соН20; 298,15 К)=17,19+0,10 кДж-моль
основано на наиболее точных измерениях в работах [244, 1063]. Эта величина относится к реакции
1 В работах [1359] и [4455] найдено еще два фазовых перехода — при 593 и 733 К по данным [1359] и в интервалах
473—523 и 723—773 К по данным [4455]; исследования проводились рентгенографическим, дилатометрическим
и ДТА-методами и показали, что переходы не сопровождаются изменением структуры ромбической модификации
Cs2SO4 (к). Эти переходы не были обнаружены при измерениях теплоемкости Cs2SO4 и не учитывались в
справочнике.
496
Цезий и его соединения Cs2SO4(r)
Таблица 49.21. Результаты измерений AOj?r°(Cs2S04, к-> с» Н20, 298,15 К) (в кДж • моль)
Авторы
Конечная концентрация, температура
измерений
Д Я°(оо Н2О, 298,15 К)
Форкран, 1906 [2295] 288 К 16,3
Воробьев и др., 1966 [244] 1760—5030 Н2О, 298,15 К, 15 измерений 17,21 ±0,08
Цветков, Рабинович, 1969 [1097] 500 Н2О, 298,15 К 17,1 а
Кузнецова и др., 1974 [597] 346 и 8410 Н2О, 298,15 К, 2 измерения 16,98±0,18
Фролов и др., 1976 [1063] 28—18503 Н2О, 298,15 К, 22 измерения 17,16±0,08
а В работе [1097], видимо, ошибочно указана величина 25,9 кДж-моль.
Cs2S04(K)+coH20=2Gs+(agr)+S042(a9r). Результаты работ [597, 1097, 2295] совпадают в пределах
их погрешностей с принятым значением.
Исследование растворимости Cs2SO4 в воде [2891] приводит к значению —1433 кДж-моль
(II закон).
Давление пара в реакции Cs.2SO4(k, jK)=Cs2SO4(r) вычислено с использованием принятой в
справочнике энтальпии сублимации
Ar#e@)=A,#o(CsaSO4 к, 0) = 329 + 20 кДж . моль.
Принятая величина является средней из найденных по результатам эффузионных
исследований Кубиччотти [1969] A092—1262 К, 22 точки, 346 +9 кДж-моль (II закон) и 329,9 + 0,4кДжХ
Хмель (III закон)) и Яганнатана, Уайтта [2825] A102—1270 К, 15 точек, 394+30 кДж-моль
(II закон) и 327,3+2,4 кДж-моль" (III закон)). Сублимация Cs2SO4 сопровождается частичной
диссоциацией с образованием Cs(r), SO2(r) и О2(г). С использованием принятых в справочнике
термодинамических свойств были вычислены парциальные давления Cs, SO2 и О2 в насыщенном
паре и в результаты измерений [1969] и [2825] внесены соответствующие поправки. Приведенные
выше величины относятся к испарению сульфата цезия в виде молекул Cs2SO4(r). Погрешность
принятой величины определяется в основном неточностью термодинамических функций.
По результатам масс-спектрометрического исследования, выполненного Фикалора и др. [2247]
(960—1200 К, 14 точек), по уравнению II закона была найдена величина 312+20 кДж-моль,
совпадающая с принятой в пределах погрешности.
Cs2SO4(r). Термодинамические свойства газообразного сульфата цезия в стандартном состоянии
при температурах 100—6000 К приведены в табл. 1262.
Молекулярные постоянные Cs2SO4, принятые для расчета термодинамических функций,
приведены в табл. 49.6. Молекула Cs2SO4 имеет четное число электронов, поэтому в справочнике
принимается, что ее основное электронное состояние является синглетным (XXA). Сведения о
возбужденных электронных состояниях Cs2SO4 в литературе отсутствуют. Элёктронографические
исследования структуры молекулы Cs2SO4 [603, 971, 4573], изучение поведения молекулярного
пучка в неоднородном электрическом поле [1707], а также исследование ИК-спектра молекул
Cs2SO4, изолированных в матрицах инертных газов и азота [90—93,360], свидетельствуют, что эта
молекула имеет «бициклическую» равновесную геометрическую конфигурацию (симметрия D2d)
с тетраэдрической группой S04 и расположенными симметрично двумя атомами Сз,связанными
с двумя атомами кислорода каждый. Приведенное в табл. 49.6 произведение моментов инерции
рассчитано для структурных параметров r(Cs—O)=2,80+0,05 A, r(S—О) = 1,47 +0,01 А (найдены
в электронографическом исследовании Куликова и др. [603] г) и *$ О—S—0=109,5 +5° 2.
Погрешность значения IaIbIo оценивается в 1-10~ш г3-смв. Приведенные в табл. 49.6 основные частоты v6,
v6, v8 и v9, отвечающие колебаниям группы SO4, приняты равными значениям, наблюдаемым в ИК-
1 Ранее в работах f971, 4573] было найдено г (Cs—О)=2,60 А,однако авторы [603] показали, что это значение
является неверным.
2 В работе [603] в качестве независимых параметров рассматривалась вся совокупность межъядерных
расстояний. Найденным значениям r(S—О) и г (О ... О) отвечает Д О—S—О=110,7±3°. В пределах погрешности
эта величина совпадает с тетраэдрическим углом.
32 Заказ N1 1590 497
CsNO2(k, ж) Глава 49
спектре молекул Cs2SO4, изолированных в матрице из Аг [90, 360]. Из трех остальных частот
группы S04 одна (vx) принята равной значению этой частоты в спектре КР молекул Na2SO4 и K2SO4,
изолированных в матрице из N2 [1356], а две (v2 и v4) приняты по расчету для ионной модели этой
молекулы [360]. Частоты колебаний связей Cs—О приняты по результатам исследования ИК-
спектра молекул Gs2SO4, изолированных в аргоновой матрице (v7 и v10), и результатам расчета
по ионной модели (v3 и vn) [91, 92, 93, 360]. Погрешности принятых значений оцениваются в 10—
15 см для v7, v10 и vn — в 20 см для v3, v5, ve, vg и v9 — в 40 см для vx и в 100 см для v2 и \,
они отражают возможный матричный сдвиг для наблюдавшихся экспериментально величин и
неточность рассчитанных значений.
Термодинамические функции Cs2SO4(r) вычислены в приближении «жесткий ротатор —
гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
A.130) без учета возбужденных состояний и возможных изомеров этой молекулы. Основные
погрешности вычисленных функций при низких температурах обусловлены неточностью
молекулярных постоянных, а при высоких температурах — приближенным характером расчета;
погрешности в Ф°(Т) при 298,15, 3000 и 6000 К составляют 10, 20 и 30 Дж-К^-моль соответственно.
Ранее термодинамические функции Cs2SO4(r) вычислялись в работах Кубиччотти [1969] (до
1300 К) и Гурвича и др. [2534] (до 3000 К). Расхождения данных [1969] и табл. 1262 достигают
9 Дж-К-моль~1 в значениях Ф°(Г), они обусловлены тем, что Кубиччотти принял для
молекулы Cs2SO4 структуру с симметрией Съ. Расхождения с данными [2534] достигают 4 Дж-КХ
Хмоль и обусловлены уточнением ряда постоянных Cs2SO4 за последние годы.
Константа равновесия реакции Cs2SO4(r)=2Cs(r)+S(r)+4O(r) вычислена с использованием
значения ДгЯ°@)=2522,387+20 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации Gs2SO4(r).
Этим величинам также соответствует
A/ff°(CsaSO4, г, 0) = —1104,442 + 20 кДж • моль.
CsNO2(k, ж). Термодинамические свойства кристаллического и жидкого нитрита цезия в
стандартном состоянии при температурах 100—2500 К приведены в табл. 1263.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние CsNO2(k) в интервале 0—209 К принимается
гексагональная модификация, а в интервале 209—679 К — кубическая модификация [1399,
3996] i. .
При Т <С 298,15 К термодинамические функции CsNO2(k) вычислены по согласующимся
результатам измерений теплоемкости в работах Мроу и Стейвли [3574] (83—479 К) и Мория и др.
[5020] A3—350 К) (измерения выполнены на хорошо аттестованных образцах, содержание Rb,
Na и К не превышало 0,01—0,02%, погрешность измерений оценена авторами [3574] в 0,1%).
Данные [5020] представлены в графическом виде, поэтому при Т > 83 К использованы результаты
измерений [3574], а для Т ^ 83 К — значения теплоемкости по работе [5020]. Экстраполяция
теплоемкости ниже 13 К дала S°A3 К)=2,32 Дж-К-моль~1. Приведенные в табл. 49.1
значения S°B98,15 К) и #°B98,15 К)—Я°@), имеют погрешности 3 Дж-К^-моль и 0,2 кДж-моль.
При Т ~^> 298,15 К для теплоемкости GsNO2(k) принято уравнение, полученное на основании
данных Мроу и Стейвли [3574]; в расчете были использованы значения С°B98,15 К)=91,34,
С°C80 К)=91,67 и С°D80 К)=92,97 Дж-К'^моль. Температура плавления F79+3 К) определена
Проценко и др. [831, 839, 840, 1824]; менее точные значения получены в работах [209, 822, 834,
837, 838] F73—692 К). Энтальпия плавления A0,9+1,2 кДж-моль) принята по измерениям
Верхотурова и др. [209], выполненным термографическим методом. Теплоемкость CsNO2(h{)
A06+10 Дж-К~1-моль~1) оценена на основании данных для жидких нитритов натрия и калия.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 2500 К оцениваются в 2,
7, 15 и 20 Дж-К-моль соответственно.
1 Сведения о фазовых превращениях CsNO2 противоречивы, по-видимому, вследствие наличия примесей в
исследованных образцах (см. [3574]). Для температуры фазового перехода в работах [827, 828, 833, 1399, 3574, 3633,
3996, 5020] получены значения в интервале 175—209 К; последнее по данным [3574, 5020] соответствует
максимуму Х-кривой теплоемкости B08,85+0,10 К и 209, 16+0,10 К) и принимается в справочнике. В работе [5020]
отмечена небольшая аномалия теплоемкости при —41 К, объясненная эффектом стеклования; в работах [3633J
и[3574] вайдены термические эффекты при 365—368 К и 408,4 К, но нашедшие подтверждения в других
исследованиях, поэтому они не учитывались в справочнике.
498
Цезий и его соединения CsNO2(r)
Таблица термодинамических функций CsNO2(k, ж) публикуется впервые.
Принятая в справочнике энтальпия образования
A^°(CsNO2, к, |298,15 К) = —379,9 + 2,0 кДж ¦ моль
основана на измерениях растворимости CsNO2 в воде [821, 825, ИЗО] и коэффициентов
активности CsNO2 в растворе [ИЗО]. Из этих данных для реакции CsN02(K)+ooH20=Cs+(ag')+NOi(ag)
было найдено значение Дй26?°(ооН20; 298,15 К)=—10,0+0,3 кДж-моль. При расчете энтальпии
образования использовано значение Ay?°(NC>2, ооН20; 298,15 К)=—37,1+1,0 кДж-моль^
рекомендованное в справочнике [1009].
Исследования равновесия реакции 2CsNO3^)=2CsNO2^)+O2(r), выполненные Центнер-
швером [1773] (см. также [3123]) (811—865 К, 6 точек, —412 кДж-моль (II закон), —404 кДжХ
Хмоль (III закон)) приводят к неточным значениям, так же как для нитритов других
щелочных металлов. Измерения энергии связи D0(Cs—NO2)=325 кДж-моль, выполненные Верхо-
туровым и др. [208] методом электронного удара, в комбинации с данными по энтальпии
сублимации CsNO2 также приводят к более высокой величине (—429 кДж-моль).
Давление пара в реакции CsNO2(k, }K)=CsNO2(r) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = As#°(CsNO2, к, 0) = 177 ± 10 кДж-моль.
Принятая величина основана на масс-спектрометрическом исследовании, выполненном Верхо-
туровым и др. [205, 206]. Опыты с двойной эффузионной камерой показали, что основным
компонентом пара является мономер (количество димера составляло 1,4%). Парциальное давление
мономера при 728 К было определено методом полного изотермического испарения; принятое
в справочнике значение рассчитано из этих данных по уравнению III закона. Его погрешность
обусловлена неточностью термодинамических функций CsNO2^, г) и воспроизводимостью
измерений. Измерения интенсивности ионных токов, выполненные в интервале 576—660 К, приводят
к значению 178 кДж-моль.
CsNO2(r). Термодинамические свойства газообразного нитрита цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1264.
Молекулярные постоянные, использованные для расчета термодинамических функций
CsNO2(r), приведены в табл. 49.6. Электронографические исследования паров над CsNO2 [606,
1035], исследования ИК-спектра паров над CsNO2 при 870—1170К [1141, 1142, 1144] и ИК-спектра
молекул CsNO2, изолированных в матрицах благородных,газов [88, 1141, 3508], показали, что
молекула GsNO2 имеет циклическую структуру (симметрия С2е). Значение IaIbIc в состоянии
Х1А1 вычислено для плоской структуры с параметрами Д О—N—0=118+3°, rg (N—O) =
= 1,256+0,005 А и rg (Cs—O)=2,79+0,02 А, найденными Куликовым и др. [606]. Тусеев и др.
[1035] в результате электронографических исследований, пришли к выводу, что молекула CsNO2
имеет неплоскую структуру (Д <х=36+4°) с параметрами Д О—N—0=100,6°, r(Cs—О)=2,814 +
+0,009 А и r(N—О)=1,380+0,004 А. Эти данные противоречат результатам исследований
структуры молекул нитритов других металлов и представляются сомнительными. Погрешность
значения IaIbIg оценивается в 2-Ю14 г3-см6.
В спектрах молекул CsNO2, изолированных в матрицах благородных газов, однозначно
идентифицирована только одна частота v4 (колебание связи N—О), ее значение принято на основании
согласующихся измерений Беляевой [88], Шаповалова и др. [1141, 1142, 1144] и Миллигана
и Джекокс [3508]. Для v2 (колебание О—iN—О) принято значение частоты, наблюдавшейся
в ИК-спектре паров над CsNO2(k) в работах [1141, 1142, 1144]. Для vx (колебание N—О) принято
значение частоты соответствующего колебаниям иона NOj, поскольку частоты колебаний этого
иона характеристичны в спектрах изученных нитратов металлов. Отнесение к этому колебанию
полосы 1460 см в ИК-спектре паров CsNO2, сделанное в работах [1141, 1142, 1144],
представляется ошибочным. Приведенные в табл. 49.6 значения v3 и \ (колебания Cs—О и Cs—О—N)
оценены по значениям соответствующих частот в молекулах нитритов и сульфатов щелочных
металлов. Частота внеплоскостного колебания ve была оценена по частоте соответствующего колебания
молекулы Cs2SO4 (vn). Погрешности приведенных в табл. 49.6 частот \ и ve оцениваются в 15—
20 см, частот v2, v3 и v5 — в 30 см, частоты v2 — 50 см. Сведения о возбужденных
электронных состояниях GsNO2 в литературе отсутствуют.
499
CsNO3(k, ж) Глава 49
Термодинамические функции CsNO2(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденных состояний этой молекулы и ее возможных изомеров. Погрешности
рассчитанных термодинамических функций обусловлены неточностью принятых молекулярных
постоянных, в том числе ее структуры, а также приближенным характером расчета и составляют 6,
10 и 15 Дж-К'^моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К соответственно.
Таблица термодинамических функций GsNO2(r) публикуется впервые.
Константа равновесия реакции CsNO2(r)=Cs(r)+N(r)-j-2O(r) вычислена с использованием
значения Дг#°@)=1247,991+10 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации CsNO2(k). Этим величинам также соответствует
bfH9(CsN02, г, 0) = —205,593 ± 10 кДж • моль.
CsNO3(k, ж). Термодинамические свойства кристаллического и жидкого нитрата цезия в
стандартном состоянии при температурах 100—2500 К приведены в табл. 1265.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние CsNO3(k) в интервале 0—427 К
принимается гексагональная а-модификация, а в интервале 427—682 К — кубическая ^-модификация
[3740] ь 2.
При Т ^ 298,15 К термодинамические функции вычислены по согласующимся между собой
результатам измерений теплоемкости в работах Флотова и др. [2270] E—350 К) и Паукова и др.
[761] E—302 К), выполненных на хорошо аттестованных образцах. Погрешность измерений
не превышала 5% вблизи 6 К, 1% при 14 К и 0,2% — выше 25 К. Приведенные в табл. 49.1
значения ?"B98,15 К) и Я°B98,15 К)—Я°@) имеют погрешности 0,3 Дж-К^-моль'1 и 0,04 кДжХ
Хмоль соответственно.
При Т > 298,15 К для теплоемкости а- и ^-модификаций GsNO3(k) приняты уравнения,
полученные на основании результатов измерений теплоемкости в работах Флотова и др. [2270] (до
350 К) и Аристовой и др. [4850] C00—675 К). Измерения [4850] выполнены на образце, который
использовался в работе Паукова и др. [761], их погрешность составляет 0,6%. При выводе
уравнения в интервале 298,15—427 К были использованы значения С°B98,15 К)=96,14 Дж-КХ
Хмель и С°C50 К)=104,58 Дж-К^-моль-1, а в интервале 427—682 К — С°D40 К)=129,8,
С;E40К)=138,5 и с;F70 К)=142,2 Дж-К^-моль [4850]. Данные Мустайоки [3604] C28-
719 К), полученные на образце неизвестной чистоты, систематически завышены (как и в случае
RbNO3) и не учитывались.
Температура фазового превращения D27+1 К) определена в работах [491, 793, 834, 2719,
2974, 4850]. Энтальпия фазового превращения C,6+0,4 кДж-моль) принята по
согласующимся в пределах погрешности измерениям [1399, 2719, 3604, 3983, 4850] C,3, 4,0, 3,7, 3,7,
3,3 кДж-моль соответственно). В работе [3376] получено более высокое значение энтальпии
перехода E,0 кДж-моль). Температура плавления F82+2 К) принята по данным [820, 839,
984, 985, 2185, 3740, 4850]; значения Тт, определенные в работах [793, 865, 1565, 2330, 2447,
3037, 3604, 3983, 4177], лежат в пределах 675—690 К, что, вероятно, обусловлено недостаточной
чистотой образцов и точностью измерений. Энтальпия плавления A3,8+0,4 кДж-моль)
получена усреднением согласующихся измерений [209, 3037, 3374, 3376, 3604, 3983] A3,4, 13,4, 14,1,
13,8, 14,1, 14,1 кДж'Моль соответственно); более низкое значение получено в работе [4850]
A2,9 кДж-моль).
Теплоемкость CsNO3^) измерялась в работе Мустайоки [3604] F85—719 К, 11 измерений),
который получил необоснованно высокое значение — 175 Дж-К-моль~1. В справочнике
принято значение A50+15 Дж-К-моль), оцененное на основании известных данных по тепло-
емкостям жидких нитратов других щелочных металлов.
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 2500 К оцениваются в 0,2,
2, 10 и 15 Дж-К~1-моль соответственно. Расхождения между термодинамическими функциями
1 Фазовое превращение CsNO3 (к) при 238 К, отмеченное в работах [2235, 2236J, не обнаружено другими
исследователями. По мнению авторов [3738], аномалия диэлектрической постоянной при 238 К, обнаруженная в
работах [2235, 2236] свидетельствует скорее о способе приготовления образца, чем об аномалии в кристаллической
решетке CsNO3.
" Ромбическая фаза высокого давления CsNO3 стабильна при р > 27 кбар (при 608 К) [1009].
500
Цезий и его соединения
CsNO3(r>
GsNO3(k, ж), приведенными в табл. 1265 и в работе Флотова и др. [2270] (Т <^ 725 К), достигают
1,3 Дж-К-^моль в значениях Ф°(Т).
В справочнике принята энтальпия образования
&fH°(GsNOs, к, 298,15 К) = —505,0 + 0,5 кДж • моль,
основанная на измерениях энтальпии растворения CsNOs(k) в воде, приведенных в табл. 49.22.
Наиболее точные и детальные исследования были выполнены О'Харе и Боэрио [3699], на
основании этой работы в последующих расчетах в справочнике используется значение Д Н°(соШ20;
298,15 К)=40,11 ±0,05 кДж-моль.
Таблица 49.22. Результаты определений A(j?J3r0(CsN08, к-»соН2О, 298,15 К) (в кДж • моль)
Авторы
Хейг, 1912 [2548]
Носова, Самойлов, 1964 [741]
Крестов, Егорова, 1967 [581, 582]
Ояри, 1971 [746]
Крестов, Куракина, 1974 [584]
Кузнецова и др., 1975 [599]
Крестов, Харитонов, 1974 [585]
О'Харе, Боэрио, 1975 [3699]
Конечная концентрация, температура
измерений
400 Н2О, 294,2 К, 4 измерения
~790 Н2О, 298,15 К
298,15 К
1000 Н2О
30—5560 Н2О, 13 измерений8
50—2780 Н2О, 298,15 К, 15 измерений3
1111—9260 Н2О, 298,15 К б
1089 Н2О, 298,15 К, 6 измерений
10727 Н2О, 298,15 К, 7 измерений
а Данные представлены в виде графика, б Данные представлены в виде уравнения.
44J>Hj0, 298,15 К)
39,6+1,3
40,25 ±0,80
39,6
39,2 ±0,6
40,7 ±0,6
40,3 ±0,3
40,4
40,10 ±0,04
40,18 ±0,18
Давление пара в реакции CsNO3(k, ж)=Сз]ЧО3(г) вычислено с использованием принятой
в справочнике энтальпии сублимации
ДгЯ°@) = Д,Я°(С8ГЮ„ к, 0) = 190 + 8 кДж • моль.
Принятая величина основана на приведенных в табл. 49.23 результатах измерений давления
пара. Как было показано Багаратьян и др. [56, 57], пары CsNO3 при 680 К состоят в основном
Таблица 49.23. Результаты определений As?T°(CsNO3, к, 0) (в кДж-моль)
Лаудер и щ
Эймс и др.,
Багаратьян
Багаратьян,
Авторы
)., 1971 [3206]
1973 [1311]
и др., 1978 [56,
Никитин, 1978
57]
[4851]
Метод
Масс-спектрометрический, 620—646 К
То же, средняя температура 770 К
То же, 626—670 К, 22 точки
То же, 666,9 К, 1 точка
Электронного удара, D0(Cs—NO3)=451 +6
A,#°(CsNO3,
II закон
III
226
149
221
204 ±20
к, 0)
закон
202
186
из мономерных молекул (количество димера составляет 2%). Принятая в справочнике величина
основана на наиболее детальном исследовании, выполненном Багаратьян и др. [56, 57], в котором
методом полного изотермического испарения при 666,9 К было найдено парциальное давление
мономерных молекул. Основным источником погрешности принятого значения является
неточность термодинамических функций CsNO3(r).|
CsNO3(r). Термодинамические свойства газообразного нитрата цезия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1266.
Молекулярные постоянные, использованные для расчета термодинамических функций
CsNO3(r), приведены в табл. 49.6. Куликов и др. [605а] при высокотемпературном электроно-
501
Cs2CO3(k, ж) Глава 49
графическом исследовании нашли, что молекула CsNO3 имеет бидентатную конфигурацию
(симметрия С2с) со ДО,,—N—Оц=117+5°, средним значением r(N—О) = 1,252 +0,004 А и r(Cs—Оц)=
—2,80+0,02 А. Приведенное в табл. 49.6 произведение моментов инерции GsNO3 в состоянии
ХгА± вычислено по данным [605а], однако, поскольку электронографический эксперимент в
пределах 0,1 А нечувствителен к возможной неравноценности длин связей N—О, в справочнике
+0 01 ° '+0 04 °
приняты r(N—Оц) = 1,29 __q'04 ^ и r(N—Оювш) = 1,21 g'g^ А. Погрешность значения 1А1в1о
оценивается в 1-1013 г3-см6. Принятые частоты валентных колебаний vx, v2, v5 и неплоского
колебания v8 иона NO3 получены экстраполяцией значений этих частот в молекулах LiNO3s
NaNO3, KNO3, RbNO3 (см. выше). Значения частот деформационных колебаний иона NOj в
плоскости (v3 и ve) перенесены из молекулы KNO3, поскольку для расплавов нитратов щелочных
металлов наблюдается лишь незначительное уменьшение этих частот в ряду Li—Na—К—Rb—Cs.
Близкие к принятым значения частот иона NOj наблюдались в спектрах кристаллов и расплавов
CsNO3 [803, 1685, 2237, 2476, 2841, 3091]. Для частот v4, v7 и v9, характеризующих колебания
иона Gs+ относительно иона NO3, приняты значения соответствующих частот молекулы CsNO2.
Погрешность приведенной в табл. 49.6 частоты v9 оценивается в 15 см, остальных частот —
в 40—50 см. Шаповалов и др. [1141, 1142, 1144] в ИК-спектре поглощения паров над CsNO3(k)
наблюдали 4 полосы A630, 1460, 1270 и 1020 см) и отнесли их к валентным колебаниям NOg
труппы, выдвинув гипотезу о существовании двух изомеров GsNO3 — тридентатного
(симметрия C3v) и бидентатного (симметрия C2v), из которых более устойчив тридентатный [1141]. Однако
исследования спектров молекул других нитратов щелочных металлов и результаты
неэмпирических расчетов молекулы LiNO3 (см. выше) ставят под сомнение и эту гипотезу и отнесение полосы
1630 см к CsNO3. Ранее Шаповалов и др. [1142, 1144] дали некорректные рекомендации
значений всех частот молекулы CsNO3 (кроме vx). Сведения о возбужденные электронные состояниях
CsNO3 в литературе отсутствуют.
Термодинамические функции CsNO3(r) вычислены по уравнениям A. 3)—(!. 6), A. 9), A. 10),
A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический сцилля-
тор» без учета возбужденных состояний и возможной изомерии этой молекулы. Погрешности
рассчитанных термодинамических функций обусловлены грубой оценкой молекулярных
постоянных, а также применением приближенной методики расчета; они составляют 8, 14 и 20 Дж-К-1Х
Хмоль в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций CsNO3(r) публикуется впервые.
Константа равновесия реакции CsNO3(r)=Cs(r)-bN(r)-|-3O(r) вычислена с использованием
значения Дг^Г°@)=1599,163+8,1 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации GsNO3(k). Этим величинам также соответствует
Д/ИСбГЮз, г, 0) = —309,982 + 8,0 кДж • моль.
Cs2CO3(k, ж). Термодинамические свойства кристаллического и жидкого карбоната дицезия
в стандартном состоянии при температурах 100—3500 К приведены в табл. 1267.
Значения постоянных, принятые для расчета термодинамических функций, приведена
в табл. 49.1. За стандартное состояние в интервале 0—1066 К принята кристаллическая
модификация Gs2GO3, структура которой еще не исследована.
При Т ^ 298,15 К термодинамические функции Сз2СО3(к) вычислены по результатам
измерений теплоемкости, выполненных Пауковым и др. [768] A3—307 К) на образце с суммарным
содержанием примесей ~0,02 мае. %. Отклонение экспериментальных точек от сглаженной кривой
не превышало 0,5% в интервале 13—25 К и 0,15% в интервале 100—300 К. Экстраполяция этих
экспериментальных данных дает S°A3 К)=3,37 Дж-К-^моль. Погрешности значений
iS°B98,15 К) и iPB98,15 К)—Н°@), приведенных в табл. 49.1, оцениваются в 0,6 Дж-К'^моль
и 0,08 кДж-моль соответственно.
В литературе отсутствуют сведения о термодинамических свойствах Gs2GO3(k, ж) при Т >
> 298,15 К. Уравнение для теплоемкости Gs2GO3(k) в интервале 298,15—1066 К (см. табл. 49.1)
получено по значениям Тр{298,15 К)=1 3,85+0,4 Дж-К^-моль [768], С;F00К) = ю1 ДяГх
ХК«моль~1 и G^A000 К)=193 Дж-К-моль~1; последние две величины оценены (с
погрешностью ~5%) на основании данных о теплоёмкости К2СО3(к) и Na2GO3(K) вблизи точек фазовых
502
Цезий и его соединения Св2СО3(г)
переходов и температур плавления. Температура плавления A066 +1 К)' принята по
согласующимся между собой данным термографических измерений [443, 1896, 3968], полученным для
¦чистых образцов Gs2CO3 (99,9%). Результаты менее надежных исследований [375, 377, 422, 423]
A003 К) не учитывались. Энтальп :я плавления C1+6 кДж-моль) оценена на основании
предположения о равенстве энтропии плавления карбонатов щелочных металлов. Теплоемкость
Сз2СО3(ж) B05+20 Дж-К-моль) оценена так же, как и в случае ЫЬ2СО3(ж) (см. выше).
Погрешности вычисленных значений Ф°(Т) при 298,15, 1000, 2000 и 3000 К оцениваются в 0^4,,
7, 20 и 40 Дж-К-моль соответственно.
Термодинамические функции Gs2GO3(k), приведенные в справочнике Барина и Кнаке [1395]
(Т ^ 1065 К) и табл. 1267, хорошо согласуются до 500 К; при повышении температуры
расхождения увеличиваются (до 2,1 Дж-К~1>моль~1 в значениях Ф°A000 К)).
Термодинамические функции Сэ2СО3(ж) публикуются впервые.
Принятая в справочнике энтальпия образования
A^°(Cs2COs, к, 298,15 К) == —1134,9 ± 0,7 кДж • моль
основана на прецизионных измерениях энтальпии реакций растворения CsaCO3(K) в растворе
CsOH и в воде в работе Джонсона и Гейера [2872]. Эти измерения дали для реакции Cs2GO3(k)
+оэИ20(ж)=2С&+(ад)+СО;2(ад) значения A^°(ooH20; 298,15 K)=—56,38+0,52 кДж-моль
и —56,7 кДж-моль соответственно.
В работе Раффелини и Май Ле Вана [3910] сообщается об измерении Да4Я°(ооН20; 298,15 К)=
=—46,8+2,6 кДж-моль. В работе [3910] не приводятся детали экспериментов и расчетов,
что не позволяет оценить ее надежность (см. также текст по выбору энтальпии образования
Rb2CO3(K)), а найденное значение существенно отличается от полученного в более надежных
измерениях [2872].
Форкран [2297] измерил энтальпии реакций взаимодействия растворов CsOH и СО2 и энталь
пию растворения Cs2CO3'(k) в воде. Результаты этих измерений приводят к значению AfH°(CsfiO,m
к, 298,15 К)=—1139,2 кДж-моль. Расчет, основанный на энтальпии растворения Cs2CO3(k)
в воде [2297], дает значение ДадЯ°(соН20; 298,15 К)=—52,8 кДж-моль"^ чему соответствует
^fH°(Gs2GO3, к, 298,15 К) = —1138,5 кДж • моль.
Исследования равновесия диссоциации Gs2GO3(k) [3214] не позволяют получить надежного
значения его энтальпии образования вследствие сложного состава продуктов реакции-и отсутствия
данных по коэффициентам активности их компонентов.
Давление пара карбоната дицезия в реакции Cs2CO3(k, ж)=Cs2CO3(r) вычислено с
использованием принятой в справочнике энтальпии сублимации
-1
Дг#°@) = &sH°(Cs2CO3, к, 0) = 333 + 20 кДж • моль-
Принятая величина была найдена по результатам масс-спектрометрических измерений
зависимости ионного тока Cs2CO3: от температуры в работе Гусарова и др. [348 ]> (928—1086 К, 13
точек, расчет по уравнению II закона).
Cs2CO3(r). Термодинамические свойства газообразного карбоната дицезия в стандартном
состоянии при температурах 100—6000 К приведены в табл. 1268.
Молекулярные постоянные Gs2GO3, принятые для расчета термодинамических функций,
приведены в табл. 49.6. Экспериментальные данные о строении и спектрах газообразного Cs2GO3
в литературе отсутствуют. Из сравнения с газообразными молекулами сульфатов, нитратов и
нитритов мелочных металлов в справочнике принято, что в основном электронном состоянии
Х1А1 м лекула Gs2CO3 имеет структуру (симметрия C2v), в которой группа СО3 имеет
конфигурацию симметрии DSh, один атом Cs связан мостиковыми связями с двумя атомами кислорода,
образуя цикл, а другой соединен с третьим атомом кислорода. Приведенное в табл. 49.6
произведение моментов инерции рассчитано по структурным параметрам r(Gs—О)=2,8+0,1 Аж г(С—О) =
+0°
= 1,30+0,05 А и ДО—С—О=120_10о. Расстояние Cs—О принято таким жея как в молекулах
503
CsBO2(k, ж) Глава 49
Gs2SO4, CsNO2 и GsNO3, а структурные параметры группы С03 — такими же, как в молекуле
К2СО3 х. Погрешность вычисленного значения 1А1в1с составляет 6-Ю12 г3-см6.
Приведенные в табл. 49.6 основные частоты молекулы Gs2CO3 оценены так же, как для
молекулы К2СО3. Погрешности в значениях частот v1; v2, v3, v6, v10, vlx оцениваются в 100 см, частот
V4' V5> V7—V9 — в 30—40 см, частоты v12 — в 10—15 см. Сведения об электронных состояниях
молекулы Cs2CO3 в литературе отсутствуют.
Термодинамические функции Gs2CO3(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A. 9), A. 10), A. 122)—A. 124), A. 128),
(Г. 130) без учета возбужденных состояний. Погрешности вычисленных величин обусловлены
грубой оценкой молекулярных постоянных, в частности пренебрежением возможной изомерией
Gs2GO3; они составляют 12, 20 и 30 Дж-К^-моль в значениях Ф°(Т) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций Cs2CO3(r) публикуется впервые.
Константа равновесия реакции Cs2CO3(r)=2Cs(r)+C(r)-f-3O(r) вычислена с использованием
значения ДгЯ°@)=2405,694+20 кДж-моль, соответствующего, принятым в справочнике
энтальпиям образования и сублимации Cs2CO3(k). Этим величинам также соответствует
Д_,Л °(Cs2CO3,>, 0) = —798,132 ± 20 кДж • моль.
CsBO2(k, ж). Термодинамические свойства кристаллического и жидкого метабората цезия
в стандартном состоянии при температурах 100—3000 К приведены в табл. 1269.
Значения постоянных, использованные для расчета термодинамических функций, приведены
в табл. 49.1. В справочнике за стандартное состояние CsBO2(k) в интервале 0—1005 К
принимается кубическая модификация [3058].
При Т ^298,15 К термодинамические функции вычислены по результатам измерений
теплоемкости, выполненных Хриплович и др. [1091] E—307 К, образец содержал Na, К и Rb в
количестве 7-Ю, 9-10 и 3,8-10 мас.% соответственно, среднее отклонение экспериментальных
точек от сглаженной кривой составляло 1,2% для Г=5—20 К и 0,08% при Т > 20 К).
Экстраполяция теплоемкости ниже 5 К по закону Дебая дает S°E К)=0,09 Дж-К-моль.
Погрешности значений ?°B98,15 К) и Я°B98,15 К)—#°@), приведенных в табл. 49.1, оцениваются
в 0,2 Дж-К~1-моль~1 и 0,02 кДж-моль соответственно.
При Т > 298,15 К для теплоемкости CsBO2(k) принято уравнение, полученное на основании
величин С;B98,15 К)=80,58 [1091], СД600 К)=99 и С;A005 К)=113 Дж-К-^моль.
Последние две величины оценены на основании данных о теплоемкости метаборатов лития и натрия
(см. выше).
Температура плавления A005+3 К) принята по результатам измерений Кохера [3058]
A005 К) и Мукерджи [3576] A005 К) (исследуемые образцы подвергались тщательной очистке,
но анализ не проводился). В работах [100, 625, 626, 627, 671] получены более низкие значения
(973—982 К), по-видимому, из-за недостаточной чистоты образцов. Энтальпия плавления B7 +
+6 кДж-моль) оценена в предположении, что энтропии плавления ме'таборатов цезия и натрия
равны B7 Дж-К-моль). Теплоемкость ОзВ02(ж) A45+15 Дж-К-моль) оценена по
теплоемкости жидкого метабората лития.
Погрешности вычисленных значений Ф°(Г) при 298,15, 1000, 2000 и 3000 К оцениваются
в 0,15, 6, 16 и 24 Дж-К^-моль.
Таблица термодинамических функций CsBO2(k, ж) публикуется впервые.
Принятая в справочнике энтальпия образования
A^°(GsBO2, к, 298,15 К) = —962 + 20 кДж • моль
оценена по методике, предложенной Карапетьянцем, на основании сопоставления энтальпий
образования в рядах соединений МВО2(к) и МОН(к), где М — щелочной металл.
Давление пара метабората цезия в реакции CsBO2(k, ж)=СзВО2(г) вычислено с
использованием принятой в справочнике энтальпии сублимации
Дг#°@) = A,#°(CsBO2, к, 0) = 275 + 15 кДж • моль.
1 Неравноценность связей Cs—О в молекуле Cs2CO8, а также возможность существования других ее изомеро»
в справочнике не учитывались.
504
Цезий и его соединения CsBO2(r), CsLi(r)
Принятая величина основана на измерениях давления пара, выполненных Гороховым и др.
[311, 642] эффузионным методом (968—971 К, 4 точки, 271,0+3,0 кДж-моль (III закон)) и масс-
спектрометрическим методом (886—977 К, 6 серий, 286+10 кДж-моль (II закон)) и Бисвасом
и Мукерджи [1564, 3576] методом протока A038—1361 К, 6 точек, 233+16 кДж-моль (II
закон) и 281,5+5,0 кДж-моль (III закон)). Основным источником погрешности принятой
величины является неточность термодинамических функций CsBO2(k, ж) и CsBO2(r).
CsBO2(r). Термодинамические свойства газообразного метабората цезия в стандартном
состоянии в интервале температур 100—6000 К приведены в табл. 1270.
Молекулярные постоянные, использованные для расчета термодинамических функций
CsBO2(r), приведены в табл. 49.6. Электронографические исследования [538, 604, 2211],
изучение ИК-спектра различных изотопических модификаций молекул CsBO2, изолированных в Аг-
н Кг-матрицах [4237], и расчет функций потенциальной энергии на основании ионной модели
[400] показа ;и, что молекула GsBO2 в электронном состоянии Х1А' имеет угловую
конфигурацию с линейным фрагментом О—В=О (симметрия Cs).
Для эффективной величины Д В—О—Cs в условиях высокотемпературных электронографи-
ческих экспериментов в работах [538, 604, 2211] были получены значения от 129+6° до 143+10°.
Изотопический сдвиг частот в ИК-спектре молекул CsBO2, изолированных в матрицах, привел
к существенно более низкому и менее точному значению угла Д В—О—Gs=90° [4237], а
минимуму потенциальной энергии, рассчитанной для ионной модели этой молекулы, соответствует
угол 110+10° [400]. Расчет Ежова и Виноградова [400] показал также, что энергии линейного
и циклического изомера CsBO2 превышают энергию изомера с Д Cs—О—В=110+10° всего на
550+100 и 2200+300 см соответственно. По-видимому, потенциальная поверхность этой
молекулы такова, что ион Cs+ почти свободно перемещается относительно иона ВОа, а значение Д Cs—
О—В, определяемое в высокотемпературных электронографических экспериментах существенно
отличается от равновесного.
Приведенное в табл. 49.6 произведение моментов инерции рассчитано по структурным
параметрам Д Cs—О—В = 130+30° (эффективное значение, среднее из данных [538, 604, 2211]),
r(Cs—О)=2,65+0,05 А, г(В—О) = 1,27+0,02 А и г(В=О)=1,23+0,02 А (средние величины из
данных. [604, 2211]). Погрешность принятого значения IAIBIc оценивается в 1 • 10~113 г3-см6.
Приведенные в табл. 49.6 значения основных частот vx—v6 основаны на результатах
исследования Сешадри и др. [4237] ИК-спектра молекул CsBO2, изолированных в матрицах
благородных газов, которые были пересчитаны для изотопомера, соответствующего природной смеси
изотопов бора. Частоты полос vx, v3 и v4, наблюдавшихся в ИК-спектре паров над CsBO2(k) в работе
Бюхлера и Маррана [1705], согласуются с данными [4237] в пределах 15—30 см. Частота v6
экспериментально не наблюдалась, принятое значение было оценено по соответствующим
частотам молекул LiBO2, NaBO2 и КВО2. Погрешности принятых частот оцениваются для vx и v2
в 30 см, для остальных частот — в 20 см.
Термодинамические функции CsBO2(r) вычислены в приближении «жесткий
ротатор—гармонический осциллятор» по уравнениям A. 3)—A. 6), A.9), A.10), A. 122)—A. 124), A.128)
и A. 130) без учета возбужденных состояний и возможной изомерии молекулы CsBO2.
Погрешности табулированных величин обусловлены неточностью принятых молекулярных
постоянных, а также применением приближенного метода расчета, они составляют 10, 15 и 20 Дж-КХ
Хмоль в значениях Ф°(Г) при 298,15, 3000 и 6000 К.
Таблица термодинамических функций CsBO2(r) публикуется впервые.
Константа равновесия реакции CsBO2(r)=Cs(r)+B(r)+2O(r) вычислена с использованием
значения ДгЯ°@) = 1815,333+25 кДж-моль, соответствующего принятым в справочнике
энтальпиям образования и сублимации CsBO2(k). Этим величинам также соответствует
A^^CsBO,,, г, 0) = —683,753 +25 кДж • моль.
CsLi(r). Термодинамические свойства газообразного цезия-лития в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1271.
В табл. 49.4 приведены молекулярные постоянные CsLi, принятые на основании результатов
спектральных исследований и приближенных оценок. В электронном спектре CsLi исследована
система полос в области 6050—6200 А [4646, 4688], которая по аналогии с молекулой KNa
отнесена к переходу СЧ1 <- Х1Е+. Так же как и другие двухатомные гетероядерные соединения,
505
CsNa(r) " Глава 4?
щелочных металлов, молекула CsLi должна иметь также стабильные состояния с низкими
энергиями, коррелирующие с валентными состояниями атомов CsBP)+LiB5) (а3П, А1^*, Ь3Е+) и
CsBi5)+LiBi>) EXS+). Кроме того, в области 23 000 см молекула CsLi должна иметь
состояния, связанные с пределом GsBD)+LiB5').
Молекулярные постоянные CsLi в состоянии ХХ2+ оценены по постоянным молекул Cs2 и Li2
с помощью соотношений, полученных в работе [1772]. Приведенное в табл. 49.4 значение шв
хорошо согласуется с результатами приближенного анализа системы СШ «- ХХ2+, проведенного
Вейцелем и Кульпом ((!>„?« 167 см). Погрешность принятых значений шв и Ве оценивается в 2
и 0,01 см соответственно. Энергия состояния СХП принята по работе [4688]. Энергии четырех
других возбужденных состояний оценены по соотношению A. 89) и данным для молекулы KNa,
погрешности этих величин составляют 1000—2000 см.
Термодинамические функции CsLi(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
и A. 93)—A. 95). Величина Q3B и ее производные рассчитывались по уравнениям A. 90)—A. 92)
с учетом пяти возбужденных состояний в предположении, что (?йг.вр==.Р*(?1ет>.вр* Статистическая
сумма по колебательно-вращательным уровням энергии состояния X1 2+ и ее производные
вычислялись непосредственным суммированием по колебательным уровням и интегрированием
по/с помощью уравнений типа A. 82). В расчете учитывались все уровни этого состояния со
значениями / <J /шазс, v, последние находились по формуле A.81). Энергия уровней
рассчитывалась по уравнениям A. 64а) и A. 65) с коэффициентами Ykl, приведенными в табл. 49.5 вместе
со значениями vmm и /цт. Значения Ykl для изотопомера CsLi, соответствующего природной смеси
изотопов лития, были получены по формулам A. 66) и постоянным Cs'Li из табл. 49.4. Для
соблюдения условий;A. 50) и A. 59) были введены коэффициенты Y30 и Уоз соответственно.
Погрешности вычисленных термодинамических функций обусловлены неточностью оценки
молекулярных постоянных CsLi в состоянии X12 +, энергии диссоциации этой молекулы и энергий
низколежащих электронных состояний, а также пренебрежением состояниями с энергией больше
20 000 см. Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К могут достигать 0,3, 0,5
и 2 Дж •К~1-моль~1 соответственно.
Таблица термодинамических функций CsLi(r) публикуется впервые.
Константа равновесия реакции GsLi(r)=Cs(r)+Li(r) вычислена с использованием принятого
в справочнике значения
= 71,6 + 2,0 кДж моль «5990 ± 170 ом'1.
Эта величина была оценена как средняя между энергиями диссоциации молекул Cs2 и Li2.
Она существенно отличается от теоретических оценок [4139, 4844] E0 и 52 кДж •моль).
Принятой энергии диссоциации соответствует
bjH°(CeLi, г, 0) == 164,149 + 2,4 кДж • моль.
CsNa(r). Термодинамические свойства газообразного цезия-натрия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1272.
Молекулярные постоянные CsNa, приведенные в табл. 49.4, приняты на основании
результатов спектральных исследований и приближенных оценок. В спектре поглошения CsNa
исследованы три системы полос в области 5480—5733, 4054—4243 и 3930—4034 А [4646,4688]. Первая
система по аналогии с молекулой Cs2 отнесена к переходу 1L1 <— Z1E+, верхнее состояние
которого коррелирует с атомами Cs^J+Na^^). Две другие системы, по-видимому, обусловлены
переходами в состояния, связанные с ридберговскими уровнями атомов. По аналогии с другими
двухатомными гетероядерными соединениями щелочных металлов следует ожидать, что
молекула CsNa имеет ряд триплетных и синглетных состояний, коррелирующих с атомами СзBР)+
+NaBS) (а3П, ЛХ2+, Ь32+) и CsB5)+NaBP) (В1?,*, СЧТ), а также группу состояний в области
20 000 см, связанных с пределом CsfZO+Na^S).
Молекулярные постоянные CsNa в состоянии ХХ2+ были оценены по постоянным молекул Cs2
и Na2 [1772]. Приведенное в табл. 49.4 значение ш, хорошо согласуется с результатами
приближенного анализа трех систем полос, выполненного Вейцелем и Кульпом [4688] (96, 97 и 99 см).
Погрешность значений <о<, и В, оценивается в 3 и 0,003 см соответственно.
Для состояния Z)xn принята энергия возбуждения, полученная в работе [4688]. Энергии
остальных возбужденных состояний оценены по соотношению A. 89) и данным для молекулы
KNa, погрешности принятых значений составляют 1000—2000 см.
506
Цезий и его соединения CsK(r)
Термодинамические функции CsNa(r) вычислены по уравнениям A. 3)—A. 6), A.9), A.10)
и A. 93)—A. 95). Величина Qm и ее производные рассчитывались по уравнениям A. 90)—A.92)
с учетом шести возбужденных состояний в предположении, что <?й.вр=Р»<?юл).вг> Статистическая
сумма по колебательно-вращательным уровням энергии состояния Х1?** и ее производные
вычислялись непосредственным суммированием по колебательным уровням и интегрированием по /
с помощью уравнений типа A. 82). В расчете учитывались все уровни этого состояния со
значениями / ^/шах, „, последние находились по формуле A.81). Энергия уровней рассчитыва-.
лась по уравнениям A. 64а) и A. 65) с коэффициентами Ykl, приведенными в табл. 49.5 вместе
со значениями утах и Уцт. Для соблюдения условий A. 50) и A. 59) дополнительно введены
коэффициенты Y30 и Y03 соответственно.
Погрешности вычисленных термодинамических функций обусловлены неточностью оценки
молекулярных постоянных CsNa в состоянии Хх?+, энергии диссоциации и энергии возбуждения
низколежащих электронных состояний, а также пренебрежением состояниями с энергией больше
20 000 см. Погрешности значений Ф°(Т) при 298,15, 3000 и 6000 К могут достигать 0,4, 1 и
3 Дж-К-моль соответственно.
Таблица термодинамических функций CsNa (г) публикуется впервые.
Константа равновесия реакции CsNa(r)=Cs(r)+Na(r) вычислена с использованием принятого
в справочнике значения
ДгЯ°@) = Do(CsNa) = 56,4 + 2,0 кДж • моль «* 4710 + 170 см.
Эта величина была оценена как средняя между энергиями диссоциации молекул Cs2 и Na2.
Она находится в удовлетворительном согласии с теоретической оценкой [4844] E1 кДж-моль).
Принятой энергии диссоциации соответствует
A/fiTo(GsNa, г, 0) = 129,377 ± 2,3 кДж • моль.
CsK(r). Термодинамические свойства газообразного цезия-калия в стандартном состоянии
в интервале температур 100—6000 К приведены в табл. 1273.
Экспериментальные данные по молекулярным постоянным CsK отсутствуют. Приведенные
в табл. 49.4 молекулярные постоянные CsK приняты на основании приближенных оценок.
Колебательные и вращательные постоянные CsK в состоянии X12+ оценены по постоянным молекул
Cs2 и К2. Погрешность принятых значений ше и Ве составляет 7 и s0,003 см соответственно.
Энергии пяти возбужденных электронных состояний оценены по соотношению A. 89) и данным
для молекулы KNa, их погрешности могут достигать 1000—2000 см. Молекула CsK должна
также иметь валентные состояния с энергией —20 000 см, связанные с пределом CsBZ))+KBS).
Термодинамические функции CsK(r) вычислены по уравнениям A. 3)—A. 6), A. 9), A. 10)
и A. 93)—A. 95). Величина QBH и ее производные рассчитывались по уравнениям A. 90)—A. 92)
с учетом пяти возбужденных состояний в предположении, что Qia\.m~PiQml.B»- Статистическая
сумма по колебательно-вращательным уровням энергии состояния ХХ2+ и ее производные
вычислялись непосредственным суммированием по колебательным уровням и интегрированием по /
с помощью уравнений типа A. 82). В расчете учитывались все уровни этого состояния со
значениями / <i /шах_ „, последние находились по формуле A. 81). Энергия уровней рассчитывалась
по уравнениям A.' 64а) и A. 65) с коэффициентами Ykl, приведенными в табл. 49.5 вместе со
значениями vmgx и /iim. Для соблюдения условий A. 50) и A. 59) были добавлены коэффициенты Y30
и Y03 соответственно.
Погрешности вычисленных термодинамических функций обусловлены неточностью оценки
молекулярных постоянных CsK в состоянии Z12+, энергии диссоциации этой молекулы и энергии
возбуждения ее низколежащих электронных состояний, а также пренебрежением состояниями
с энергией около 20 000 см. Погрешности значений Ф°(Г) при 298,15, 3000 и 6000 К могут
достигать 0,5, 2 и 4 Дж-К-моль соответственно.
Таблица термодинамических функций CsK(r) публикуется впервые.
Константа равновесия реакции GsK(r)=Cs(r)+K(r) вычислена с использованием принятого
в справочнике значения
Д,#°@) = D0(CsK>= 46,0 ± 2,0 кДж • моль ж 3850 ± 170 см.
507
CsRb(r) Глава 49
Эта величина была оценена как средняя между энергиями диссоциации молекул Cs2 и К2. Онг
находится в удовлетворительном согласии с теоретической оценкой [4844] D5 кДж-моль).
Принятой энергии диссоциации соответствует
bfH°(CsK, г, 0) = 121,905 ±2,3 кДж • моль.
CsRb(r). Термодинамические свойства газообразного цезия-рубидия в стандартном состояши
в интервале температур 100—6000 К приведены в табл. 1274.
В табл. 49.4 приведены молекулярные постоянные CsRb, принятые на основании результатеt
спектральных исследований и приближенных оценок. В электронном спектре CsRb наблюдалис!
две системы полос в области 7230—7400 А [3147] и 5640 А [4646]. Первую из них можно отнестк
к переходу В1!! <- Х1!!", поскольку она была отмечена Лумисом и Кушем [3297] в спектре
магнитного вращения. Отнесение второй системы не известно. По аналогии с другими гетероядернымг
молекулами щелочных металлов молекула CsRb должна иметь ряд стабильных низколежащиз
состояний, коррелирующих с атомами RbBP)+CsBS) (Сх?+) и RbB5)+CsBP) (а3П, А^+, Ь32+),
Кроме того, в области 20 000 см у нее, по-видимому, существуют стабильные состояния,
связанные с диссоциационными пределами Rb^Z^+Cs^/S) и Rb^S^+Cs^Z)).
Значения частот колебаний CsRb в состояниях X1S+ и ВЩ и энергия возбуждения последнегс
приняты по работе [3147], в которой был выполнен анализ 10 полос системы ВЩ <- Х1^
(v" ^ 5, v' <C 3). Остальные молекулярные постоянные в основном состоянии оценены по
постоянным молекул Rb2 и Cs2. Погрешность принятого значения Ве оценивается в 0,001 см.
Энергии возбужденных состояний были оценены по соотношению A. 89) и данным для
молекулы LiNa, погрешности этих величин составляют 1000—2000 см.
Термодинамические функции CsRb(r) были вычислены по уравнениям A. 3)—A. 6), A. 9).
A. 10), A. 93)—A. 95). Значения (?„„ и ее производных вычислялись по уравнениям A. 90)—
A. 92) с учетом пяти возбужденных состояний в предположении, что Q^ol.-BV=PiQmLnr
Статистическая сумма по колебательно-вращательным уровням энергии состояния Xх ?+ и ее
производные вычислялись непосредственным суммированием по колебательным уровням и
интегрированием по / с помощью уравнений типа A. 82). В расчете учитывались все уровни этого
состояния с значениями / <^/шаХ)(,, последние находились по формуле A. 81). Энергия уровней
рассчитывалась по уравнениям' A. 64а) и A. 65) с коэффициентами Ykl, приведенными в табл. 49.?
вместе со значениями i>max и /нт- Значения Ykl изотопомера CsRb, соответствующего природной
смеси изотопов рубидия, вычислялись по формулам A. 66) и постоянным молекулы 133Cs85Rb
из табл. 49.4. Для соблюдения условий A. 50) и A. 59) были введены коэффициенты Y30 и У03.
Погрешности рассчитанных термодинамических функций CsRb(r) при низких температурах
обусловлены главным образом неточностью оценки вращательной постоянной в состоянии Х1Б+,
а при Т ^> 3000 К — неточностью оценки энергии диссоциации и энергий возбужденных
состояний, а также пренебрежением рядом состояний в области 20 000 см. Погрешности в значениях
Ф°(Г) при 298,15, 3000 и 6000 К оцениваются в 0,5, 2 и 4 Дж -К -моль соответственно.
Таблица термодинамических функций CsRb(r) публикуется впервые.
Константа равновесия реакции CsRb(r)=Rb(r)+Cs(r) вычислена с использованием принятого
в справочнике значения
ДгЯ°@) = Do(CsRb) = 44,5 + 2,0 кДж • моль «13720 ± 170 см.
Эта величина была оценена как средняя между энергиями диссоциации молекул Cs2 и Rb2. Она
находится в удовлетворительном согласии с теоретической оценкой [4844] D5 кДж-моль).
Принятой энергии диссоциации соответствует
г, 0) = 115,706+ 2,4 кДж • моль.
ПРИЛОЖЕНИЯ
Приложение 3*
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ГАЗОВ
ПРИ ПОВЫШЕННОМ ДАВЛЕНИИ
П3.10. Обзор данных для веществ IV тома
Критические постоянные металлов. До недавнего времени экспериментальные данные по
критическим постоянным металлов были известны только для щелочных металлов и ртути [290, 5028].
Для приближенной оценки этих постоянных обычно применяются полуэмпирические методы.
Краткий обзор таких методов дан в Приложении П3.9 настоящего справочника (см. т. 3, кн. 1,
с. 421). Для металлов, рассмотренных в третьем томе, были приняты оценки, сделанные
Фортовым и др. [4907] по методу, предложенному этими авторами. (
В последние годы опубликован ряд экспериментальных работ, посвященных исследованию
металлов при высоких температурах и давлениях [4846а, 4856а, 4888, 4962а, 5044а]
динамическими методами (подробный обзор см. [4906а]). В рамках определенных модельных
представлений авторы этих работ определили критические постоянные некоторых металлов. В частности
в работах [4846а, 5044а] проведено экспериментальное изучение ударного сжатия и построены
уравнения состояния для ряда металлов в широком диапазоне параметров. Полученные данные
по критическим постоянным меди и свинца находятся в хорошем согласии с оценками по
методу [4907].
В настоящем справочнике в табл. Д.68 приведены значения критических постоянных Мо
и W, определенные в работах Фуке и Сейделя [4962а, 5044а] методом электрического
импульсного нагрева. Этот метод позволяет определить температуру на спинодали как функцию давления
и с помощью представлений, развитых Скриповым [4901], вычислить критические параметры.
Погрешности полученных таким образом величин составляют около 10% для" Гкр и 25% для РкГ
Результаты других экспериментальных исследований, по-видимому, менее надежны из-за
сильной зависимости от ряда других параметров. Поэтому для Cr, V, Nb, Та, Ti, Zr, Hf, Sc, Y, La,
Th и U в табл. Д.68 приняты значения критических постоянных, оцененные Фортовым и др.
[4907 ]. Для плутония в этой работе нет данных, и критические постоянные Ри приведены
в табл. Д. 68 по оценке, сделанной в обзоре Озе и Типпельскирха [5028] с использованием
принципа соответственных состояний г.
UO2. Диоксид урана — одно из немногих тугоплавких веществ, для которого имеются данные
по уравнению состояния при температурах, значительно превышающих точку плавления. В
основном это экспериментальные данные, полученные методами динамического импульсного
нагрева [4931а,4937а,4956, 4965а, 5017а, 5025—5027] (в работе Озе и др. [5026] дан подробный
обзор этих данных, а также физических моделей, использованных при их обработке). При
построении уравнения состояния и оценке критических постоянных UO2 используются главным
образом две модели: принцип соответственных состояний (теория термодинамического подобия)
[4931а, 4937а, 5017а] и теория характеристических структур Эйринга [4937а, 4956, 4965а].
Оценка по принципу соответственных состояний дает большой разброс результатов. Это
обусловлено различием как методов обработки, так и исходных данных по давлению пара, по
изменению объема в точке плавления и по коэффициенту объемного расширения жидкости. Кроме
того, серьезные сомнения вызывает применение принципа соответственных состояний в случае
UO2, поскольку этот принцип сформулирован для низкокипящих жидкостей и не учитывает
ионный характер связи. Поэтому рядом авторов в качестве альтернативной модели рекомендуется
теория Эйринга [4955а], которая достаточно успешно применялась к ионным расплавам (типа
галогенидов щелочных металлов) и в то же время хорошо приспособлена к учету различных
усложняющих факторов. Исходная информация, используемая в теории Эйринга, включает
данные по давлению пара, по наличию дефектов и вакансий кристаллической решетки, по
молекулярным постоянным и др. Молекулярные постоянные UO2, наиболее близкие к принятым в на-
* Окончание. Начало Приложения 3 см. в т. 1,?кн. 1, с. 397; в т. 2, кн. 1, с. 352 и в т. 3, кн. 1, с. 420.
1 Выбор критических постоянных для щелочных металлов обсуждается ниже.'
510
Термодинамические свойства газов при повышенном давлении
стоящем справочнике для расчета термодинамических функций, использованы в работе Фишера
и др. [4956], в которой, в частности, также принята линейная структура молекулы и учтен вклад
возбужденных электронных состояний. Поэтому в настоящем справочнике для критических
постоянных UO2 в табл. Д.68 приняты значения, рекомендованные в работе [4956].
Согласно расчетам [4956, 4989] согласование данных по фазовому равновесию при обработке
по II и III законам термодинамики вплоть до 5000 К обеспечивается при использовании
термодинамических функций UO2(r) в стандартном состоянии без учета поправок на неидеальность.
Поэтому применение теории Эйринга для оценки неидеальности UO2(r) оправдано только в
околокритической области.
UF6. Обзор работ по теплофизическим|свойствам UF6, опубликованных до 1974 г.,'выполнен
Беляниным [4853а]. Основной массив экспериментальных /?УГ-данных для температур 364—
592 К и давлений до 24,2 МПа получен Малышевым [48876]. Этим же автором получено
уравнение состояния для температур выше 463,3 К. Второй вириальный коэффициент был измерен также
Моризо и др. [5021] C28,15—463,15 К) и Хейнцом и др. [4972] C20-470 К).
Недавно в работе [5076] была выполнена тщательная обработка />7Г-данных для UF6 с
использованием метода «идеальных кривых», разработанного Недоступом и др. [4889а, 48896],
и построена система уравнений состояния, а также вычислены таблицы свойств. Согласно [4889а,
48896] уравнение состояния для газовой фазы может быть записано в виде
где Б — второй вириальный коэффициент, являющийся функцией аргумента 77A—р/р0), и р0 —
характерный для данного вещества параметр, по порядку величины близкий к плотности
кристалла при Т=0 и р=0.
Выведенное в [5076] уравнение для газовой фазы охватывает область температур от тройной
точки 337,2 К [4853] до точки Бойля 1175 К [5076] при Ро=15,23 моль .дм. Для ОТв(ж) при
р > 1,5рвр было составлено отдельное уравнение состояния, основанное на предложенной
в [4879а] ячеечной модели жидкости.
Таблиц
Т
К
500
600
700
800
900
1000
а ПЗ
V
3,83
4,79
5,69
6,56
7,43
8,28
Примечание.
18. Термодинамически*
оЯ
-1140
-756,8
—473,1
—336,5
-311,7
—281,8
V даны в
а
bS
—1,64
—0,94
-0,49
—0,31
--0,28
-0,25
дм3 -моль
i свойства UFe при повышенных
Р = 5МПа
V
ЬН
0,79 —4275,9
1,04 —2426,6
1,25 —1748,3
1,45 -1600,6
1,64 -1368,1
bS
_
-5,44
-2,57
-1,65
-1,47
—1,23
~1, Ш — в Дж -моль,
давлениях
р=10МПа
V
_
0,28
0,46
0,59
0,71
0,82
bS — I
ЬН
_
—9749,8
—5030,4
-3657,9
-3224,2
-2538,4
! ДЖ -К
55
_
—12,78
—5,39
—3,53
—3,02
-2,3
•МОЛЬ.
р = 20МПа
V
0,17
0,23
0,30
0,36
0,41
ЬН
—13880,7
—9267,7
-7046,4
-5740,4
—3702,4
bS
-17,21
—10,06
-7,06
-5,52
—3,39
Таблица термодинамических свойств UFe (табл. П3.18) рассчитана х по данным [5076].
Из этой же работы принято аппроксимационное уравнение для второго вириального
коэффициента, константы которого приведены в табл. Д.67. Критические постоянные UFe (табл. Д.68)
приняты по данным Малышева [48876].
РиО2. В работе [4937а] приведены значения критических |констант РиО2 (Гкр=7000 К,
р =111,5 МПа, FKp=0,131 дм3-моль и ZKp=0,25), оцененные с использованием принципа
соответственных состояний. Как указано выше (см. UO2), такой подход мало оправдан для подобных
веществ, и поэтому в настоящем справочнике оценка критических постоянных РиО2 выполнена
1 Термодинамические свойства UFf), приведенные в табл.
В. И. Недоступом и М. Б. Беккером.
511
П3.18, рассчитаны для настоящего справочника
Приложение 3
заново на основании сопоставления оценок для UO2, сделанных по принципу соответственных
состояний и по теории характеристических структур Эйринга. Полученные таким образом
значения критических постоянных РиО2 приведены в табл. Д.68.
Щелочные металлы. Пары щелочных металлов в интервале от тройной точки до критической
характеризуются разнообразием состояний — от идеального одноатомного газа до плотной
плазмы. Экспериментальные данные о свойствах щелочных металлов в состоянии плотной плазмы
ограничены измерениями давления паров Na, К, Rb и Cs, плотности насыщенных паров Rb [2201]
и Cs [1994], а также /^Г-зависимости для Cs [556, 557]. Эти данные недостаточны для
проведения полного анализа и согласованной обработки, и в настоящем Приложении
рассматриваются пары щелочных металлов только в состоянии диэлектрика, для которого известны
экспериментальные данные о свойствах при температурах ниже 1500—1700 К и давлениях менее
2,5-3 МПа.
Для описания свойств паров щелочных металлов в диэлектрическом состоянии используются,
как правило, три типа уравнения состояния.
Во-первых, это уравнение состояния в вириальной форме [2203]
Z = l-f
к=2
где
Вк (Т) = (_1)*« exp [qO?T~* -f qi*>T+ + <rf*> + g{*> In T] (П3.52)
и ?-2> <7-i> 9o и 4i — параметры, определяемые из анализа экспериментальных данных.
Во-вторых, это уравнение состояния смеси реагирующих идеальных газов [2205], в которой
кроме мономеров и димеров учитываются также тримеры и тетрамеры. Соответствующие
константы равновесия находятся из экспериментальных /^Г-данных.
В-третьих, это модифицированное уравнение состояния диссоциирующего газа в виде [1060]
11' (ПЗ-53)
где a=[l+4p/Kl(T)]"I/j — степень диссоциации Ма=2М, К°2(Т) — константа равновесия этой
реакции, а &В=Ь0+Ь2/Т2, С=со+с2/Т2 и D=d2/T2 — полуэмпирические поправки на парные
несвязанные, а также тройные и четверные взаимодействия. Уравнение (П3.53) содержит 6
параметров (включая энергию диссоциации Do).
Таблицы термодинамических свойств паров щелочных металлов приведены в справочнике
Шпильрайна и др. [1179]. Соответствующие уравнения состояния для Na, К, Rb и Cs основаны
на модели диссоциирующих идеальных газов с поправкой на «физическую» часть второго ви-
риального коэффициента, найденную из анализа данных по давлению насыщенных паров рвы
по уравнению
Рвас
(«L)*$ (П3.54)
При этом экспериментальные рУГ-данные в справочнике [1179] не учитывались.
Приведенные в справочнике Варгафтика [165] таблицы рассчитаны для модели
диссоциирующих идеальных газов. Как отмечено в [241], в этих таблицах введена поправка на
экспериментальные pVT-данные для Na, К, Rb и Cs только к удельным объемам.
Не совсем последовательная комбинация уравнения состояния в вириальной форме и
уравнения состояния смеси была использована в работе Ли и др. [3218] при вычислении свойств
паров щелочных металлов.
Для лития отсутствуют экспериментальные данные по рТ^Г-свойствам, поэтому расчеты для
него, в отличие от других щелочных металлов, проводились различными авторами только по
схеме диссоциирующих идеальных газов. Соответствующие таблицы термодинамических свойств
512
Термодинамические свойства газов при повышенном давлении
приведены в справочнике [1179] до 2000 К и 0,9 МПа, а также в работах [165, 237] до 3000 К
и 1 МПа. При давлениях менее 1 Па в [165, 237] учитывалась ионизация.
С|При подготовке настоящего справочника критические постоянные лития (табл. Д.68)
оценены по аналогии с величинами для остальных щелочных металлов, а также по данным о давлении
пара. Погрешности оцененных величин достигают 300 К для Гкр, 1,5 МПа для рк и 0,02 дм3Х
Хмоль для 7кр. Принятым значениям соответствует довольно низкое значение Zx =0,11 ±
±0,03, что находится в согласии с оценками Ликальтера [4887а] для щелочных металлов.
Для натрия экспериментальные р7Г-данные были получены Стоуном и др. [4401] методом
пьезометра постоянного объема для интервала температур 1233—1690 К и давлений 0 19—
2,53 МПа. Таблицы термодинамических свойств по этим данным рассчитаны Эвингом и др. [22031
и Аченером и др. [1215] на основе вириального уравнения (П3.51), а также в справочных изданиях
[2254, 4340] на основе уравнения состояния смеси. Недостатком всех этих таблиц является
использование для давлений пара уравнений не согласованных с помощью III закона
термодинамики с термодинамическими функциями конденсированной фазы и уравнением состояния пара.
Таблицы термодинамических свойств паров натрия (а также калия) на основании уравнения
состояния с групповыми коэффициентами составлены в работе [4895а]. При этом наряду с
экспериментальными pVT-данными частично использовались сведешия о потенциалах взаимодействия
атомов в молекуле.
Корреляция термодинамических свойств натрия и таблицы свойств от 3 МПа до критической
области выполнена в работах [2254, 3744] на основании теории термодинамического подобия.
Оценка свойств натрия в сверхкритической области проведена в работах Барнеса [1400] и
Браунинга [4836а].
При подготовке настоящего справочника параметры уравнения состояния (П3.51) для Na
были пересчитаны заново в результате критического анализа исходных данных [4401] и
аппроксимации их с помощью нелинейного метода наименьших квадратов с весами. Соответствующие
параметры уравнения состояния приведены в табл. П3.19. Термодинамические свойства натрия,
приведенные в табл. П3.20, рассчитаны по этим параметрам.
Таблица П3.19. Параметры уравнения состояния в вириальной форме (П3.51)—(П3.52) для Na, К и Cs»
Вещество
Na
К
Cs
а Вк - в (дм3
Температурный
интервал
К
к
1000—1700 2
3
4
900—1700 2б
3
4
900—1700 2
3
4
5
•моль-1)*-1. 6 Для калия В2C
9-з
9-1
9о
- 8 940,7 -12,3133
- 15 799,1 -7 8501
- 23 589,2 -11,8388
___
— 8168,7 —4,1141
- 10 060 -4,8170
— 5116,9 —10,5213
7,693-10» -6 820,1 2 1779
- 1 125,7 i;3165
— — 2,210
г)=-6,8117-10-Ч-1,4628-10-*Х Ш-ехр F256,2/Г)j.
9,
i
1
__
^^
1
—¦*
В процессе расчета параметров|вириального уравнения (ПЗ. 51) матрица ошибок D(q) была
найдена в предположении, что ошибки в исходных р7Г-данных имеют случайный характер. Это
позволило оценить погрешности термодинамических функций Y
(П3.55)
Результаты этих" оценок для "натрия в виде 28 приведены в табл. П3.21.
Значения критических постоянных натрия (табл. Д.68) рекомендованы по данным [1551
1552] (прямое измерение критического давления) с учетом корреляции величины Zx для щелоч'
513
Приложение 3
Таблица П3.20. Мольные объемы н поправки на неидеальность
Т
К
1000
1100
1200
1300
1400
1500
1600
1700
900
1000
1100
1200
1300
1400
1500
1600
1700
800
900
1000
1100
1200
1300
1400
1500
р = 0,01 МПа
V
799,2
898,3
989,6
1075,2
1160,3
1244,5
1328,4
1412,0
734,4
823,8
909,8
994,5
1078,5
1162,1
1245,7
1329,1
1412,4
648,9
740,0
826,5
911,6
996,3
1079,6
1163,8
1246,7
ЬН
bS
—2893 —2,560
—1339 —1,069
—682 —0,488
—392 —0,266
—241 —0,151
—159 —0,091
—122 —0,053
-85 -0,050
—996 —0,941
-494 -0,394
-305 —0,225
—194 -0,118
—122 —0,061
-89 -0,04
—65 —0,03
—51 —0,02
-40 -0,01
—1168 —1,051
—557 —0,440
—302 —0,240
—182 —0,107
—120 —0,082
—84 —0,077
—59 —0,04
—48 —0,02
Примечание. У даны в дм3«моль-1, ЪН — в
V
Na
__
91,77
102,83
112,81
122,17
131,13
139,89
к
—
86,72
96,53
105,72
114,60
123,28
131,84
140,32
Cs
78,22
88,06
97,30
106,21
114,92
123,52
Дж-моль-i,
р = 0,1 МПа
ЬН
__
—5923
-3611
—2287
—1515
—1064
—775
—
—2729
—1680
—1139
—832
—604
-493
—382
—2607
—1697
—1156
—830
—624
—487
bS — в Дн
цля Na, К, <
bS
Р
V
_
—4,218 —
—2,364 —
—1,376 —
—0,856 10,49
—0,54 11,70
-0,50 12,83
— —
—2,037 —
—1,148 —
-0,700 —
—0,443 10,03
—0,313 11,15
—0,214 12,19
—0,183 13,17
—1,658 —
—0,848 —
—0,531 —
—0,334 9,15
—0,281 10,28
—0,123 11,33
t-K «моль.
= 1 МПа
ЬН
'_
_
—11424
—8743
—6752
—
—7244
—5491
—4324
—3471
—7103
—5555
—4461
IS
_
—6,104
—4,364
—3,165
—
-3,936
—2,751
—1,987
—1,447
—2,72
—1,87
-1,45
Таблица П3.21. Оценка погрешности 2Э(У) термодинамических свойств Y(p, T)паров натрия
Т
К
1000
1200
У
V
н
S
V
н
S
Примечание.
Р ^^
= 0,01 МПа
3
5-Ю2
0,2
0,8
МО2
0,07
= 0,1 МПа
0,3
4-Ю2
0,3
Погрешности для V даны в дм3
= 1 МПа
—
Т
К
1400
1600
•моль-1, для Н — в Дж
У
V
н
S
V
н
S
= 0,01 МПа
=
0,2
2-101
0,015
0,07
6
0,002
•моль и S — в Дж -К
Р =
0,1 МПа
0,14
2-Ю2
0,10
0,07
6-101
0,04
•моль-1.
р '—'
= 1МПа
0,02
4-Ю2
0,3
ных металлов [290]. Погрешности рекомендуемых значений могут составлять 50 К для Гкр,
1,5 МПа для р.ъ и 0,02 дм3-моль для V . Критическим постоянным натрия соответствует ?,„=
=0,13 ±0,03.
Для калия основной массив экспериментальных pVT-данных получен в работах Эвинга с сотр.
[2201, 4401] A170-1660 К, 0,24-2,8 МПа), Аченера и др. [1213] A150-1480 К, 0,25-1,4 МПа)
в Варгафтика и др. [166] A430—2075 К, 1,25—7,24 МПа). Известны также немногочисленные
514
Термодинамические свойства газов при повышенном давлении
экспериментальные данные по теплоемкости и скорости звука при давлении около 0,1 МПа НОЮ
1011, 1012], а также по энтальпии испарения [1209, 1210]. Результаты работ [2201, 4401]
использованы при составлении таблиц термодинамических свойств в справочнике [4340 ] по модели
смеси мономеров, димеров и тетрамеров и в работах Эвинга и др. [2202] и Хеймела [2602]
по уравнению состояния в вириальной форме, причем в [2602] оно было экстраполировано до
2100 К. Аченер и др. [1205] также рассчитали таблицы термодинамических свойств калия на
основании уравнения в вириальной форме, выведенного по собственным экспериментальным данным
[1213]. Наиболее полная обработка экспериментальных данных по свойствам паров калия была
выполнена Теряевым [1010], который методом наименьших квадратов с учетом весов провел
совместную обработку рУГ-данных [1213, 4401] и данных по давлению насыщенного пара калия
при Т < 1600 К и вычислил параметры вириального уравнения состояния (ПЗ. 51). Для
функции В2(Т) в работе [1010] была использована комбинированная форма модели диссоциирующих
газов и потенциала прямоугольной ямы. Для остальных функций Вк{Т) сохранена форма (ПЗ. 52),
Найденные в работе [1010] параметры приведены в табл. П3.19. Значения термодинамических
свойств, приведенные в табл. П3.20, также вычислены по этим данным.
Критические постоянные калия, приведенные в табл. Д.68, выбраны в результате аналдза
экспериментальных данных по рК9 [2857], корреляции данных по давлению насыщенного пара
и оценки критической плотности по правилу прямолинейного диаметра с учетом данных 117.8 Ь
Погрешности выбранных величин составляют Q для Гкр, 1 МПа для ркр и 0,03 дм3-моль для Ух ,
Для рубидия Аченер с сотр. [1207, 1215] составили уравнение состояния в вириальной форме
(ПЗ. 51) и рассчитали таблицы термодинамических свойств на основании экспериментальных
рУГ-данных [1207] в интервале температур 1067—1475 К и давлений 0,21—1,5 МПа. Результаты
обработки [1207, 1215] не представляются достаточно надежными. Известна также работа Стоуна
и др. [4399а], в которой получено уравнение состояния для рубидия в вириальной форме как
комбинация уравнений для калия [2202] и цезия [2204]. Имеются немногочисленные
экспериментальные данные по скорости звука [4905а] и энтальпии испарения [1208] рубидия. При
подготовке настоящего справочника была проведена совместная обработка рУГ-данных [1207] и
давления насыщенного пара при Т < 1673 К и по методу [1060] определены параметры уравнения
состояния (ПЗ. 53): Ьо=—1,447 дм3-моль, &2=0,412-106 дм3-К2-моль и с2=0,477 дм3-КаХ
X Па-моль. Постоянные с0 и d2 оказались незначимыми. Для энергии диссоциации Rb3 было
использовано принятое в настоящем справочнике значение.
Значения критических постоянных рубидия, приведенные в табл. Д.68, выбраны на основании
экспериментальных данных для рЕр [1551, 1552] и ортобарических плотностей [2201], а также
данных работы [4961а]. Погрешности принятых значений могут составлять 20 К'для Т
1,5 МПа для ркр и 0,025 дм3-моль для Укр. кр'
Для цезия исследование рУГ-свойств проводилось в работах Аченера и др. [1207] A068—
1490 К, 0,21-1,3 МПа), Эвинга с сотр. [2206, 4401] (980-1685 К, 0,11-3,32 МПа)
Коршунова и др. [556, 557] (870-2470 К, 2-30,5 МПа), Воляка и др. [242, 243, 4593] A366-1940 К
1,5-5,2 МПа), Гурьяновой [342] A645-2117 К, 3,2-8,9 МПа). В работах [174, 817, 818, 1027]
была измерена также скорость звука в парах цезия до 1260 К.
. В работе Эвинга и др. [2206] на основании рУГ-данных [2206, 4401] было получено уравнение
состояния для смеси мономеров, димеров и тетрамеров, справедливое в области до 2 9 МПа
и уравнение состояния в вириальной форме (ПЗ. 51). Параметры последнего уравнения приве^
дены в табл. П3.19. Эти параметры были использованы для расчета термодинамических свойств
цезия, приведенных в табл. П3.20. Уравнение в вириальной формуле получено также Аченером
и др. [1204, 1215] по собственным измерениям рУГ-данных, Трелиным и др. [1026] при
совместной обработке рУТ-данных [1204, 2206, 4401 ] и данных по скорости звука, а также Воляком и др.
[241] с учетом дополнительных рУГ-данных, полученных в работах [242, 243, 342, 2198, 4593],'
Таблицы термодинамических свойств цезия рассчитаны в [241, 1215, 2204] на основании
уравнения состояния в вириальной форме, причем в [241] расчеты доведены до 7=2050 К и р=10МПа.
Но результаты последнего расчета для энтропии должны быть скорректированы на Д In Z.
Значения критических постоянных, приведенные в табл. Д.68 для цезия, получены на
основании анализа высокотемпературных рУГ-данных [556, 557, 2198], результатов измерения орто*-
барических плотностей [1994] и критического давления [4288]. Погрешности принятых значений
могут достигать 15 К для Т^, 0,3 МПа для ркр и 0,03 дм3-моль для Укр.
515
Приложение 3
Для оценки второго вириального коэффициента для Na, К и Cs в области, где имеются
экспериментальные pFT-данные, могут быть использованы величины В2(Т) из (ПЗ. 51),
вычисленные по параметрам, приведенным в табл. П3.19. Для рубидия оценка второго вириального
коэффициента может быть проведена по соотношению В=—RTУК%(Т)-\- Д5.
кроме величин К°(Т)=р(М) для щелочных металлов, приведенных в основных таблицах
книги 2 настоящего тома и представляющих собой парциальное давление соответствующего
одноатомного газа в реакции М(к, ж)=М(г), определенный практический интерес имеет также полное
давление насыщенного пара этих металлов. В работе Покрасина и др. [798] для этого было
предложено интерполяционное уравнение, описывающее полное давление насыщенного пара
щелочных металлов от тройной точки до критической (для Li — до 2500 К) в виде
к
In ршл = с In -с 4- 2 а»х'. t=r»10". (ПЗ. 56)
Параметры выражения (ПЗ. 56) были определены методом наименьших квадратов по
экспериментальным данным о полном давлении насыщенного пара с учетом весов. Для согласования
этих данных в интервале температур от тройной точки до 1000 К для Li и до 700 К для Na, К,
Rb и Cs вместо экспериментальных значений давления насыщенных паров были использованы
значения р(М), приведенные в соответствующих основных таблицах термодинамических свойств.
Параметры уравнения (ПЗ. 56) для щелочных металлов приведены в табл. П3.22. *jj
Таблица П3.22
Параметры
С
a-i
а0
а1
аг
а3
Ч
J'mini К
-ь шах» -Ь-
. Параметры
Li
—2,0532
—19,4268
9,4993
0,7530
—
453,69
2500
выражения (П3.5) для
Na
—2,4946
—13,2905
7,8441
1,7093
—0,1716
—0,0088
—0,0091
0,0029
371,01
2503
щелочных металлов
К
-0,9875
—10,8427
8,9156
—1,5573
1,1129
—0,1124
—0,1276
0,0324
336,86
2280
(давление в МПа)
Rb
—0,9148
—9,8420
8,5965
—1,6227
1,2416
—0,2943
—0,0055
0,0042
312,47
2106
Cs
—0,7063
—9,3205
8,7226
—2,4528
1,2463
0,4933
—0,5969
0,1350
301,59
2043
NaCl, KC1. Критические температуры галогенидов щелочных металлов лежат в области, не
доступной для прямых экспериментальных исследований. Киршенбаум и др. [4992] по данным
о плотности NaCl и КС1 в жидком и газообразном состояниях построили диаграмму ортобариче-
ских плотностей и линию криволинейного диаметра, что позволило определить приближенно
Гкр и Ущ для этих веществ. Величины ркр, оцененные авторами [4992] экстраполяцией данных
Бартона и Блума [4922] по давлению паров, по-видимому, несколько завышены C5,5 МПа для
NaCl и 22,3 МПа для КС1). Рекомендованные в [4992] величины имеют погрешность 200 К для
Г^р и около 30% для рщ и Укр.
Расчет критических постоянных по методу Эйринга в работе [4941] привел к близким
значениям Т^ и Укр, но существенно меньшему значению р^.
Оценки, выполненные для ряда галогенидов щелочных металлов в работах [5045, 5072],
представляются менее надежными, так как они основаны на обобщенном уравнении Ван-дер-
Ваальса, мало пригодном для ионных жидкостей, и сильно расходятся с опытными данными.
В табл. Д.68 для NaCl и КС1 приведены значения Ткр и 7кр из работы [4992] и pKV из работы
[4941], скорректированные в пределах их погрешности для согласования с результатами
расчетов по III закону термодинамики.
В работе [4992] оценены также критические постоянные других щелочных галогенидов с
помощью полуэмпирического метода, основанного на предположении об универсальной зависимости
энтропии испарения от приведенной температуры. Результаты этих оценок, имеющих
погрешность около 500 К в Гкр, приведены в справочнике [1009].
516
Приложение 5*
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ВОДНЫХ ИОНОВ
В СТАНДАРТНОМ СОСТОЯНИИ
В дополнение к ионам, рассмотренным в Приложении 5 в третьем томе, для вычисления
энтальпий образования нитритов щелочных металлов, рассмотренных в настоящем четвертом томе,
необходима энтальпия образования иона NO^ в стандартном состоянии водного раствора.
В справочных изданиях [406, 4626] приняты совпадающие значения
bfH°(NO-, p-pcoH20, 298,15 К) = —105 кДж • моль
без указания погрешности. Эта рекомендация основана на измерениях, выполненных в
основном еще в прошлом веке (см., например, [1555]).
Ефимов и др. [48716] измерили энтальпию окисления NaNO2 сернокислым раствором К3Сг207.
Из этих измерений было вычислено
Ь,Н°(№-, р-рсоИаО, 298,15 К) = —100,3 ±0,8 кДж • моль.
Эта величина принята в настоящем справочнике для расчета энтальпий образования нитритов
щелочных металлов.
* Окончание. Начало Приложения 5 см. в т. 3, кн. 1, с. 423.
Приложение 6
ПРИБЛИЖЕННАЯ ОЦЕНКА ЭНЕРГИИ
ЭЛЕКТРОННЫХ СОСТОЯНИЙ АТОМОВ И ИОНОВ
Для большинства рассматриваемых в настоящем справочнике атомов и ионов элементов с
достраивающимися электронными d-оболочками отсутствуют экспериментальные данные о части
валентных электронных состояний, преимущественно высокорасположенных. Для оценки
энергии этих состояний в справочнике использовалась известная формула Ридберга—Рида (см. т. 1,
кн. 1, раздел 1.3)
где Тп f — энергия состояния, соответствующего электронной конфигурации с квантовыми
числами гаи/, /<"' ') — ионизационный предел данной ридберговской серии, Дм>, — квантовый
см
постоянО
1
2
3
4
5
2,6 ±0,2
2,1 ±0,5
1,1+0,2
0,03 ±0,02
0,003 ±0,002
0
3,6 ±0,1
3,2+0,2
2,1 +0,2
0,03 ±0,02
0,003 ±0,002
0
4,6+0,2
4,3 ±0,3
3,0 ±0,1
1,0 ±0,1
0,03 ±0,02
0,003 ±0,002
дефект и #га=109 737,317 7
ная Ридберга.
Величина Тп1 для мультиплетных состояний
определяется в этом случае как центр тяжести
мультиплета по формуле
21.,, = 2B7< + 1)Г^2B7< + 1),
где суммирование проводится по всем
компонентам мультиплета.
Величина /<"• г> определяется как сумма
первого потенциала ионизации атома (иона) и
энергии того электронного состояния однократно (двукратно) заряженного иона, к которому сходится
данная ридберговская серия.
Величины квантовых дефектов д t были рассчитаны при подготовке настоящего справочника
на основании имеющихся спектральных данных для ридберговских серий атомов и ионов 4-го,
5-го и 6-го периодов. Найденные таким образом средние значения Ди г приведены в таблице.
Погрешность оценки Тп , определяется неточностью значений /("> г> и д г.
Рассмотрим в качестве примера оценку энергии ненаблюдавшихся электронных состояний
2S, 2P, 2D, 2F и 2G атома Sc с суммарным статистическим весом 2 B74 -)-1) = 50, соответствую-
i
щих электронной конфигурации . . .3daAZ)Ld. В этом случае ге=4 и 1=2. Первый потенциал
ионизации атома Sc равен 52 922,0 ±0,5 см, а энергия электронного состояния . . .3d? *D
иона Sc+ равна 10 944,56 см, откуда /?,*>'>=63 866,56 см. Из таблицы находим ДЯ1^
=1,1 ±0,1. Тогда Гя i=50 818,14 см. Погрешность оценки Тп>, в данном случае определяется
преимущественно неточностью Дя п и для расчета термодинамических функций принимается
окончательно Тщ 2=51 000 ±2000 см.
Приложение 7
ВЫЧИСЛЕНИЕ Q,
ЧЕРЕЗ ПОТЕНЦИАЛ МЕЖАТОМНОГО ВЗАИМОДЕЙСТВИЯ
юи. вр
Методы экстраполяции колебательно-вращательных уровней энергии двухатомных молекул
при вычислении QK0Ii вр, изложенные в разделе 1.4 настоящего справочника (см. т. 1, кн. 1,
с. 35—52), могут привести к заметным ошибкам в случае ионных молекул, к которым, в частности,
относятся галогениды щелочных металлов, рассматриваемые в главах 45—49 настоящего тома.
В этом случае оказывается целесообразным использовать метод расчета QK0Xm вр через потенциал
межатомного взаимодействия, развитый в работах Иориша и др. [4875, 4876, 4877, 4979, 4980].
Согласно этому методу
J
<?*ол Bi = exp[-pG(O)] 2 B/ + l)<?Jexp[-pFJ(riain)],
j=o
где
n
dr{pj(r, r; P) + X-Xexp[—pFj(r)](erfZj —1)}. (П7.2)
• 0
S
Здесь $=hc/kT; гя и гь — корни уравнения Vj (r)=*Wj; Wj — энергия максимума на
эффективной потенциальной кривой Vj(r); pj(r, г; р) — диагональный элемент матрицы плотности,
соответствующей свободным атомам, связанным и квазисвязанным состояниям: X =
j
2 <¦
длина волны Де Бройля; Х3- = {|3[ТГ/—Vj (г)]}1/' и erf Xj — -jz= i dtexp(—t7) — интеграл
вероятности. *% о
При вычислении матричных элементов р(г, г'; Р) используется итерационная схема Сторера
[4994, 5053, 5054], в соответствии с которой задается начальное так называемое осцилляторное
приближение [4875 ]:
где
) D
Здесь ге — температурный множитель, и интегрирование проводится на пути от га до г, с шагом Д,
зависящим от свойств потенциала и температуры. В результате перемножения матриц находятся
элементы матрицы плотности
Р<,(*Д, /Д; 2-<«'р)= 2 Р5(»д. ^; 2-р)
и
Pj(iA, /Д; 2~(Я~2)Р) = 2 Pj (*A Ад." 2"(Я~1)Р) р^ (ЛД, /Д; 2"Ся
ж
519
Приложение 7
В большинстве случаев с достаточной точностью можно ограничиться только диагональными
элементами матрицы плотности в виде
Расчет по этому уравнению проводился для иодидов и бромидов Na, К, Rb и Gs от
комнатной температуры и выше; для Lil, CsF, RbF, хлоридов Na, К, Rb и Cs от 500 К и выше; для
LiBr и LiCl от 1000 К и выше и для фторидов Li, Na и К от 1500 К и выше.
Для получения производных (?ЕМ вр по температуре при расчете термодинамических функций
использовалось численное дифференцирование по пяти точкам. Оптимальный шаг определялся
на основе численного эксперимента.
В качестве потенциальной функции был использован так называемый паде-аппроксимант
ПА[2, 2], предложенный Иорданом и др. [4986]:
где |=(г—re)/re; dOi dy и d2 — первые коэффициенты ряда Данхэма [2126]
которые выражаются через постоянные в уравнении для уровней энергии двухатомных молекул
(см. т. 1, кн. 1, уравнение A. 65)):
Так как паде-аппроксиманты для галогенидов щелочных металлов сходятся к ионному диссо-
циационному пределу, а реальные потенциалы имеют своим пределом атомные состояния
галогена и щелочного металла, то в расчетах использовался потенциал вида
где ге — межатомное расстояние, соответствующее точке пересечения ионного потенциала с
ковал ентным диссоциационным пределом.
Необходимая для расчета суммы по состояниям величина G @) выражается через параметры
потенциала
Значения Ykl и коэффициенты d0, dx и d2 для двухатомных молекул галогенидов щелочных
металлов приводятся в таблицах соответствующих глав.
Приложение 8
ВЫЧИСЛЕНИЕ ЭНТАЛЬПИИ СУБЛИМАЦИИ
ПРИ СЛОЖНОМ СОСТАВЕ ПАРА
П8.1. Щелочные металлы
Давление насыщенных паров щелочных металлов измерялось в широком интервале температур,
включая область, где пар нельзя рассматривать как одноатомный идеальный газ. Это приводит
к усложнению расчета энтальпии реакции М(к)=М(г) по данным о давлении пара.
Возможны три случая:
1. Пар состоит из атомов, образующих идеальный газ. В этом случае расчет Д8Я°@)
проводился, как обычно, по уравнению C0) (см. т. 1, кн. 1, с. 27).
2. Пар состоит из атомов и двухатомных молекул, образующих химически реагирующую
смесь идеальных газов. Зная энергию диссоциации и термодинамические функции одноатомного
и двухатомного газов, можно выделить парциальное давление атомов р(Ш). При этом следует
учесть, что измеряемое эффективное давление связано с величинами р{М) и р(М2) неодинаково
в зависимости от метода измерения (см. ниже, раздел П8.2).
После выделения р(М) расчет ДвЯ°@) проводился, как и в первом случае, по уравнению C0).
3. Пар представляет собой реальный газ. В этом случае расчет &еН°@) проводился по
уравнению (ПЗ. 54) (см. раздел П3.10), в котором интегральный член включает в себя уравнение
состояния.
Для натрия, калия и цезия использовалось уравнение состояния в вириальной форме (ПЗ. 51).
Соответственно в этом случае
= ГДГФ°(Г) - RT Ы Рт - RT [ЩР- +i^p.+i%gl+i%gl _ in i^.]. (П8.1)
Величины Вк(Т) находились на основании параметров, приведенных в табл. П3.19.
Для рубидия использовалось модифицированное уравнение состояния (ПЗ. 53), и тогда
Д8Я°@) = ТАГФ°(Т) _ ЯГ In р,ае - ЯГ In т|^-4 *BPme - | Cplc -A Dp^. (П8.2)
Коэффициенты Д#, С и D этого уравнения вычислялись по параметрам, приведенным в
разделе П3.10. Уравнение такого вида, наряду с уравнением в вириальной форме, было
использовано также для натрия и получены совпадающие результаты. ~~
П8.2.[ |Галогениды^щелочньгаГ металлов^.
При вычислении энтальпии реакции МХ(к)=МХ(г) из данных по давлению насыщенных паров
галогенидов щелочных металлов необходимо, как и в случае щелочных металлов, учитывать
сложный состав пара, в котором кроме мономеров MX могут в заметных количествах
присутствовать димеры М2Х2, а иногда и тримеры М3Х3.
Результаты экспериментальных исследований состава пара галогенидов щелочных металлов
обычно удобно выражать для реакций
MX (к, ж) + М X (г) = МаХ,'(г)> (Пв!)
1 (П8.4)
На основании экспериментально определенных значении энтальпии этих реакции и принятых
в справочнике термодинамических функций вычислялись константы равновесия соответствую-
521
Приложение 8
щих реакций, т. е. отношения парциальных давлений димера или тримера и мономера (pJPi
или p3/Pi)-
Наиболее точные измерения давления насыщенного пара щелочных галогенидов проведены
методами, позволяющими найти только суммарное давление компонентов пара. При этом авторы
работ обычно проводили обработку полученных данных в предположении о существовании в паре
только мономеров. Получаемое таким образом эффективное давление р* связано с
парциальными давлениями компонентов пара соотношениями, вид которых зависит от метода измерения.
В случае измерений статическим методом, торзионным методом и методом точки кипения
Р* = А + Л + А = ft (*к+ Pal Pi + Pal Pi)-
В случае измерений эффузионным методом и методом испарения с открытой поверхности
Р* = Pi + V2 Ра + V3 PS =. А A + V2 А/А + \/3 а/а).
В случае измерений методом протока
р* = А + 2а + Зр3 = А A + 2А/А + За/А).
Эти уравнения действительны для случая, когда вещество испаряется конгруэнтно.
Эти соотношения были использованы для вычисления парциальных давлений мономеров по
результатам измерений давления насыщенных паров галогенидов щелочных металлов.
Необходимые для этих расчетов отношения pJPi и p3lpi вычислялись, как указано выше, из констант
равновесия реакций (П8.3) и (П8.4). Такие расчеты проводились для каждой экспериментальной
точки. По найденным таким образом парциальным давлениям мономеров были вычислены
энтальпии сублимации АвН°(МХ, к, 0). На основании этой величины, а также принятых значений
энтальпии реакции (П8.3) и (П8.4) определялись значения энтальпии образования М2Х2(г) и
М3Хз(г).
ЛИТЕРАТУРА
1. Абашкин Ю. Г., Дементьев А. И. — В кн.: Кон- 27.
ференция по теории атомов и молекул, Вильнюс,
1979: (Тез. докл.). Вильнюс, 1979, ч. 2, с. 75. 28.
2. Абросимов В. К., Ионов А. В., Крестов Г. А. —
Изв. вузов. Хим. и хим. технол., 1977, 20, с. 1555. 29.
3. Абросимов В. К., Ионов А. В., Крестов Г. А. —
Радиохимия, 1977, 19, с. 862.
4. Абросимов В. К., Крестов Г. А. — Журн. физ. 30.
хим., 1967, 41, с. 3150.
5. Агеев А. И. — Уч. зап. Азерб. Гос. ун-та. Сер.
физ.-мат. и хим. наук, 1961, № 2, с. 61.
6. Агте К., Вацек И. — В кн.: Вольфрам и молибден. 31.
М.; Л.: Энергия, 1964, с. 283.
7. Айвазов М- Л., Домашнее И. А., Саркисян А. Г., 32.
Гуров С. В. — Изв. АН СССР. Неорган, матер.,
1972, 8, с. 1069. 33.
8. Акишин П. А., Горохов Л. Н., Сидоров Л. Н. —
Вестн. МГУ. Сер. хим., 1959, № 6, с. 194. 34.
9. Акишин П. А., Горохов Л. Д., Сидоров Л. Н. —
Докл. АН СССР, 1960, 135, с. ИЗ. 35.
10. Акишин П. А., Наумов В. А. — Журн. структур,
хим., 1961, 2, с. 3.
11. Акишин П. А., Наумов В. А., Татевский В. М. — 36.
Вестн. МГУ. Сер. мат., мех., астрон., физ., хим.,
1959, 1, с. 229. ' 37.
12. Акишин П. А., Наумов В. А., Татевский В. М. —
Кристаллография, 1959, 4, с. 174. 38.
13. Акишин П. А., Рамбиди Н. Г. — Докл. АН СССР,
1958, 118, с. 973. 39.
14. Акишин П. А., Рамбиди Н. Г. — Журн. неорган,
хим., 1960, 5, с. 23. 40.
15. Акишин П. А., Спиридонов В. П. — Журн."^
структур, хим., 1961, 2, с. 63. 41.
16. Акишин П. А., Спиридонов В. П. — Журн.
структур, хим., 1962, 3, с. 267. 42.
17. Акопов Е. К. — Журн. неорган, хим., 1961, 6,,
с. 1211. 43.
18. Акопов Е. К. — Укр. хим. журн., 1967, 33, с. 446.
19. Акопов Е. К. — Журн. неорган, хим., 1968, 13, 44.
с. 1941.
20. Акопов Е. К., Бергман А. Г. — Изв. сектора 45.
физ.-хим. анализа ИОНХ АН СССР, 1954, 25, с. 255. 46.
21. Акопов Е. К., Коробка Е. И. — Журн. неорган. 47.
хим., 1968, 13, с. 2312.
22. Акопов Е. К., Моисеенко Ж- Г., Паниева А. А. — 48.
Журн. неорган, хим., 1973, 18, с. 226.
23. Алексеев В. А., Овчаренко В. Г., Рыжков Ю. Р., 49.
Сенченков А. Л. — Письма в ЖЭТФ, 1970, 12,
с. 306. 50.
24. Алешко-Ожевская Л. А., Ильин Л/. К.,
Макаров А. В., Никитин О. Т. — Вестн. МГУ. Сер.
хим., 1978, 19, с. 564. 51.
25. Алешко-Ожевская Л. А., Ильин' М- К.,
Макаров А. В., Никитин О. Т. — Вестн. МГУ. Сер.
хим., 1978, 19, с. 681.
26. Алешко-Ожевская Л. А., Ильин М- К., Макаров 52.
А. В., Никитин О. Т. — Вестн. МГУ. Сер. хим.,
1980, 21, с. 248.
Алешко-Ожевский Ю. П. — Журн. физ. хим., 1979,
53, с. 1369.
Алиханян А. С, Шольц В. В., Сидоров Л. Н. —
Вестн. МГУ. Сер. хим., 1972, 13, с. 639.
Алямовский С. И., Зайнулин Ю. Г., Швейкин Г. П.
Оксикарбиды и оксинитриды металлов IVA и VA
подгруппы. М.: Наука, 1981.
Амоненко В. М., Иванов В. Е., Ковтун Г. П.,
Павлов В. С, Круглых А. А. — В кн.: Экспер.
и теор. методы высокотемлер. измерений. Ин-т
металлургии АН СССР. М.: Наука, 1966, с. 85.
Амоненко В. М., Павлов В. С. — Изв. АН СССР.
Металлы, 1968, № 5, с. 212.
Андреева Г. Т., Келлер Е. К. — Журн. неорган,
хим., 1964, 9, с. 633.
Андреева Т. А., Бондаренко Н. Б. —
ОНИИТЭХИМ. Деп. № 3129-79. Черкассы, 1979.
Андреева Т. А., Бондаренко Н. В., Богма К. К. —
ОНИИТЭХИМ. Деп. № 3131-79. Черкассы, 1979.
Андреева Т. А., Голованова Т. Г.,
Бондаренко Н. Б. — ОНИИТЭХИМ. Деп. № 3130-79.
Черкассы, 1979.
Андрианов Г. О., Дричко И. Л., Лайхтман Б. Д. —
Журн. эксперим. и теор. физ., 1978, 75, с. 2246.
Андрианов Г. О., Жданова В. В., Сергеев В. П. —
Физ. твердого тела, 1980, 22, с. 1249.
Анисимов В. М., Воляк Л. Д. — Теплофизика
высоких температур, 1969, 7, с. 317.
Ария С. М., Морозова М. Л. — Журн. общ. хим.,
1962, 32, с. 2081.
Ария С. М., Морозова М. П., Вольф 9. — Вестн.
ЛГУ. Сер. физ. и хим., 1956, № 22, с. 91.
Ария С. М., Морозова М- П., Вольф 9. — Журн.
неорган, хим., 1957, 2, с. 13.
Ария С. М., Щукарев С. А., Глушкова В. Б. —
Журн. общ. хим., 1953, 23, с. 1241.
Ария С. М., Щукарев С. А., Глушкова В. Б. —
Журн. общ. хим., 1953, 23, с. 2063.
Артым Р. И. — Теплофизика высоких температур,
1968, 6, с. 1010.
Артым Р. И Журн. физ. хим., 1969, 43, с. 990.
Артым Р. И Журн. физ. хим., 1970, 44, с. 2380.
Арутюнов А. В. — Автореф. дис. . . .канд. техн.
наук. М.: МГУ, 1970.
Арутюнов А. В., Банчила С. И., Филиппов Л. П. —
Теплофизика высоких температур, 1971, 9, с. 535.
Арутюнов А. В., Банчила С. Н., Филиппов Л. П. —
Теплофизика высоких температур, 1972, 10, с. 425.
Арутюнов А. В., Макаренко Л. Н., Труханова Л. Н.,
Филиппов Л. П. — Вестн. МГУ. Сер. физ., астрон.,
1970, И, с. 340.
Арутюнов А. В., Макаренко И. Н.,
Филиппов Л. П. — В кн.: Теплофизические свойства
веществ и материалов. М.: Изд-во стандартов, 1972,
вып. 5, с. 105.
Арутюнов А. В., Филиппов Л. П. — В кн.: Тепло-
физические свойства веществ и материалов, вып. 5.
М.: Изд-во стандартов, 1972, с. 97.
523
Литература
53. Астафьева М- Л., Ветренко Е. А., Микулинский
А. С, Фришберг И. В. — Журн. физ. хим., 1964,
38, с. 523. ^
54. Афанасьев В. А., Клопов В. И., Крестов Г. А. —
Журн. физ. хим., 1979, 53, с. 267.
55. Бабаджан А. А. — Труды ин-та металлургии
Уральского фил. АН СССР, 1957, 1, с. 74.
56. Багаратьян Л. В. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1978.
57. Багаратьян Л. В., Ильин М- К., Никитин О. Т. —
Вестн. МГУ. Сер. хим., 1977, 18, с. 17.
58. Багаратьян Л. В., Никитин О. Т. — Вестн. МГУ.
Сер. хим., 1977, 18, с. 387.
59. Базалова И. В., Стаханова Af. С, Карапетъ-
янц М. X., Власенко К. К. — Журн. физ. хим.,
1965, 39, с. 1245.
60. Байков В. И. — Автореф. дис. . . . канд. физ.-мат.
наук. Л.: ГОИ, 1974.
61. Байков В. И., Василевский К. П. — Оптика и
спектроскопия, 1967, 22, с. 364.
62. Баку мекая Е. П., Бергман А. Г. — Журн. неорган,
хим., 1956, 1, с. 1629.
63. Банчила С. Н., Филиппов Л. П. — Инж.-физ.
журн., 1974, 27, с. 68.
64. Баранская Е. В., Решетников Н. А. — В кн.:
Физико-химические исследования взаимодействия
солей щелочных металлов в расплавах и продуктов
деструкции сопропелитов. Иркутск: Иркутский
политехи, ин-т, 1972, с. 122.
65. Баранская Е. В., Решетников Л. А. — В кн.:
Физико-химические исследования взаимодействия
солей щелочных металлов в расплавах и продуктов
деструкции сопропелитов. Иркутск: Иркутский
политехи, ин-т, 1972, с. 134.
66. Бархатов Л. С, Каган Д. Л., Цыцаркин А. Ф.,
Шпилърайн 9. 9., Якимович К. А. — Теплофизика
высоких температур, 1973, 11, с. 1188.
67. Баталов В. С. — Журн. физ. хим., 1971, 45, с. 708.
68. Баташов В. И., Морозов А. А., Резниченко В. А. —
В кн.: Металлургия вольфрама, молибдена, ниобия.
М.: Наука, 1967, с. 133.
69. Бацанов С. С, Киселев Ю. М., Копанева Л. И. —
Журн. неорган, хим., 1980, 25, с. 1987.
70. Безус Е. В., Соколовская Е. М., Григорьев А. Т.,
Павлов В. П., Переладова Л. А., Соколова И. Г. —
Вестн. МГУ. Сер. хим., 1966, № 5, с. 74.
71. Бекетов А. Р., Власов В. Г. — Журн. прикл.
хим., 1965, 38, с. 2103.
72. Бекетов А. Р., Власов В. Г., Стрекаловский В. Н.
— Журн. неорган, хим., 1965, 10, с. 737.
73. Бекетов А. Р. Стрекаловский В. Н.,
Власов В. Г. — Журн. структур, хим., 1965, 6, с. 75.
74. Белов А. Л., Лопатин С. И., Семенов Г. А. —
Журн. физ. хим., 1981, 55, с. 932.
75. Белов А. Н., Семенов Г. А. — Журн. физ. хим.,
1979, 53, с. 3050.
76. Белов А. Л., Семенов Г. А. — Журн. физ. хим.,
1980, 54, с. 1537.
77. Беломестных В. Л., Поздеева 9. В., Толмачева
Л. Д. — В кн.: Молодые ученые и специалисты
Томской области в 9-й пятилетке. Секция физ.
твердого тела. Томск: Томский Гос. ун-т, 1975,
с. 213.
78. Белых П. Д., Захвалинский М- Л. — В кн.:
Взаимодействие хлоридов и сульфатов щелочных и
щелочноземельных металлов в расплавах. Иркутск:
Иркутский Гос. ун-т, 1972, с. 176.
79. Беляев И. Л., Казанбеков Р. Г. — Журн. неорган,
хим., 1969, 14, с. 1365.
80. Беляев И. Л., Казанбеков Р. Г. — Журн. неорган,
хим., 1969, 14, с. 2553.
81. Беляев И. Л., Нестерова А. К. — Докл. АН СССР,
1952, 86, с. 949.
82. Беляев И. Л., Сигида Л. П Журн. общ. хим.,
1956, 26, с. 1553.
83. Беляев И. Л., Чикова Н. Л. — В кн.: Физико-
химический анализ солевых систем. Ростов-на-
Дону: Ростовский Гос. ун-т, 1962, с. 69.
84. Беляев И. Н., Чикова Н. Н. — Журн. неорган.
хим., 1963, 8, с. 1442.
85. Беляев И. Н., Шургинов Е. А. — Журн. неорган.
хим., 1970, 15, с. 883.
86. Беляев И.Н., Шургинов Е.А., Кудряшов Л. С. ¦—
Журн. неорган, хим., 1972, 17, с. 2812.
87. Беляева А. А. — Автореф. дис. . . . канд. физ.-мат.
наук. Л.: ЛГУ, 1978.
88. Беляева А. А., Бухмарина В. Н., Предтечен-
ский Ю. Б., Щерба Л. Д. (в печати).
89. Беляева А. А., Дворкин М-И., Щерба Л. Д. —
Оптика и спектроскопия, 1971, 31, с. 392.
90. Беляева А. А., Дворкин М. И., Щерба Л. Д. —
Оптика и спектроскопия, 1971, 31, с. 585.
91. Беляева А. А., Дворкин М. И., Щерба Л. Д. —
Оптика и спектроскопия, 1975, 3 8, с. 308.
92. Беляева А. А., Дворкин М. И., Щерба Л. Д. —
Оптика и спектроскопия, 1975, 38, с. 516.
93. Беляева А. А., Дворкин М. И., Щерба Л. Д. —
Оптика и спектроскопия, 1977, 43, с. 198.
94. Белякова П.Е. — Шв. АН СССР. Металлы, 1975,
№ 4, с. 78.
95. Берг Л. Г., Ясникова Т. Е. — Журн. неорган.
хим., 1966, 11, с. 886.
96. Бергман А. Г., Акопов Е. К. — Изв. сектора физ.-
хим. анализа ИОНХ АН СССР, 1953, 23, с. 222.
97. Бергман А. Г., Бакумская Е. Л. — Журн. общ.
хим., 1956, 26, с. 629.
98. Бергман А. Г., Берулъ С. И., Никонова И. Н. —
Изв. сектора физ.-хим. анализа ИОНХ АН СССР,
1953, 23, с. 183.
99. Бергман А. Г., Бондарева Д. И. — Журн. неорган,
хим., 1969, 14, с. 1118.
100. Бергман А. Г., Бондарева Д. И. — Журн. неорган.
хим., 1971, 16, с. 2286.
101. Бергман А. Г., Вартбаронов О. Р. — Журн.
неорган, хим., 1957, 2, с. 642.
102. Бергман А. Г., Вартбаронов О. Р. — Журн.
неорган, хим., 1957, 2, с. 2641.
103. Бергман А. Г., Кислова А. И., Посыпайко В. И. —
Журн. общ. хим., 1954, 24, с. 1935.
104. Бергман А. Г., Кислова А. И., Посыпайко В. И. —¦
Журн. общ. хим., 1955, 25, с. 1890.
105. Бергман А. Г., Коробка Е. И. — Журн. неорган.
хим., 1959, 4, с. НО.
106. Бергман А. Г., Литовская Н. А. — Журн. неорган.
хим., 1969, 14, с. 1672.
107. Бергман А. Г., Матросова В. А.—Журн.
неорган, хим., 1969, 14, с. 1354.
108. Бергман А. Г., Матросова В. А., Казаченко
Е. Л. — Журн. неорган, хим., 1968, 13, с. 1712.
109. Бергман А. Г., Михалкович Л. Н. — Журн.
неорган, хим., 1969, 14, с. 3163.
110. Бергман А. Г., Михалкович Л. Н.— Журн.
неорган, хим., 1970, 15, с. 1390.
111. Бергман А. Г., Михалкович Л. Л. — Журн.
неорган, хим., 1970, 15, с. 2270.
112. Бергман А. Г., Ногоев К. — Журн. неорган,
хим., 1964, 9, с. 1423.
113. Бергман А. Г., Решетников Л. А. — Изв. сектора
524
Литература
физ.-хим. анализа ИОНХ АН СССР, 1954, 25,
с. 208.
114. Бергман А. Г., Решетников Н. А. — Изв. физ.-
хим. научно-иссл. ин-та при Иркутском Гос.
ун-те, 1959, 4, с. 9.
115. Бергман А. Г., Санжаров А. С. — Журн. неорган.
хим., 1970, 15, с. 583.
116. Бергман А. Г., Хитрое В. А. — Изв. сектора
физ.-хим. анализа ИОНХ АН СССР, 1952, 21,
с. 199.
117. Бердичевский Я. И., Морачевский А. Г. —Журн.
прикл. хим., 1966, 39, с. 791.
118. Березин Б. Я. — Автореф. дис. . . . канд. техн.
наук. М.: ИВТАН, 1973.
119. Березин Б. Я., Каи С. А., Кенисарин М. М-,
Чеховской В. Я. — Теплофизика высоких
температур, 1974, 12, с. 524.
120. Березин Б. Я., Каи С. А., Чеховской В. Я. —
Теплофизика высоких температур, 1976, 14, с. 497.
121. Березин Б. Я., Кенисарин М- М-, Чеховской
В. Я. — Теплофизика высоких температур, 1972,
10, с. 1214.
122. Березин Б. Я., Чеховской В. Я. — Изв. АН СССР.
Металлы, 1977, № 3, с. 63.
123. Березин Б. Я., Чеховской В. Я. — Теплофизика
высоких температур, 1977, 15, с. 772.
124. Березин Б. Я., Чеховской В. Я., ШейндлинА, Е. —
Докл. АН СССР, 1971, 201, с. 583.
125. Березовский Г. А. — Автореф. дис. . . . канд.
хим. наук. Новосибирск: ИНХ СО АН СССР, 1976.
126. Березовский Г. А., ИкарскийВ. Я., Пауков И. Е. —
ВИНИТИ. Деп. № 2111-74. М., 1974.
127. Березовский Г. А., Лукащук Е. И. — Журн. физ.
хим. (в печати).
128. Березовский Г. А., Пауков И. Е. — ВИНИТИ.
Деп. № 1303-74. М., 1974.
129. Березовский Г. А., Пауков И. Е. — Журн. физ.
хим., 1974, 48, с. 2138.
130. Березовский Г. А., Пауков И. Е. — В кн.:
Фазовые переходы металл—диэлектрик: Краткое
содержание докладов представл. на II Всесоюз.
конф. по фазовым переходам металл—диэлектрик.
М.; Львов: Львовский Гос. ун-т, 1977, с. 103.
131. Березовский Г. А., Пауков И. Е. — Журн. физ.
хим., 1982, 56, с. 1541.
132. Березовский Г. А., Пауков И. Е. — Изв. СО АН
СССР. Сер. хим. наук, 1981, вып. 2, № 4, с. 19.
133. Беруль С. И. — Журн. неорган, хим., 1958, 3,
с. 2427. ,
134. Берулъ С. И., Бергман А. Г. — Изв. сектора
физ.-хим. анализа ИОНХ АН СССР, 1952, 21,
с. 172.
435. Бессонов А. Ф. — Журн. неорган, хим., 1971,
16, с. 3.
136. Бессонов А. Ф. — Изв. вузов. Цвет, мет., 1971,
№ 1, с. 102.
137. Бессонов А. Ф., Стрекаловский В. Я., Устъян-
цев В. М. — Кристаллография, 1965, 10, с. 570.
138. Благонравов Л. А. — Автореф. дис. . . . канд.
физ.-мат. наук. М.: МГУ, 1978.
139. БлидинВ. П. — Журн. общ. хим., 1941, 11, с. 891.
140. Бобыренко Ю. Я., Жолнин А. Б., Коновалов
В. К. — Журн. физ. хим., 1972, 46, с. 1305.
141. Боганов А. Г., Руденко В. С, Макаров Л. П. —
Докл. АН СССР, 1965, 160, с. 1065.
142. Бодиско А. — Журн. Рус. физ.-хим. об-ва, 1888,
20, с. 503.
143. Бодиско А. — Журн. Рус. физ.-хим. об-ва, 1889,
21, с. 7.
144. Болдырев А. И., Соломонип В. Г., Закжевский В. Г.,
Чаркин О. Л. — Журн. неорган, хим., 1980, 25,
с. 2307.
145. Бондарева Д. И., Бергман А. Г. — Журн.
неорган, хим., 1970, 15, с. 2302.
146. Борисоглебский Ю. В., Ветюков М- М., Шатова
3. Ю., Козьмова О. Я. — Изв. вузов. Цвет, мет.,
1980, № 4, с. 20.
147. Боярский С. В., Новиков И. И. — Теплофизика
высоких температур, 1978, 16, с. 534.
148. Боярский С. В., Новиков И. И. — Теплофизика
высоких температур, 1981, 19, с. 201.
149. Брезгин Ю. А. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1972.
150. Бреусов О. П., Новоселова А. В., Симанов Ю. П. —
Докл. АН СССР, 1958, 118, с. 935.
151. Броунштейн Б. П., Юрков Г. Я. — Журн. физ.-
хим., 1959, 33, с. 1289.
152. Будников П. П., Тресвятский С. Г., Кушаков-
ский В. И. — Докл. АН СССР, 1959, 128, с. 85.
153. Бурлаков В. Д. — Физ. металлов и
металловедение, 1957, 5, с. 91.
154. Бурлакова В. М., Бухалова Г. А. — Изв. вузов.
Хим. и хим. технол., 1968, 11, с. 259.
155. Бурмакин Е. И., Буров Г. В., Розанов И. Г.,
Шехтман Г. Ш. — Журн. неорган, хим., 1978,
23, с. 3366.
156. Бурмистрова Я. П., Воложанина 9. Г. — Журн.
физ. хим., 1968, 42, с. 2795.
157. Бурмистрова И. П., Воложанина 9. Г.,
Маркова Л. Я. — Журн. неорган, хим., 1967, 12,
с. 784.
158. Бурылев Б. П., Миронов В. Л. — Изв. Сев.-
Кавказ. научного центра высш. школы. Сер.
естеств. наук, 1976, 4, с. 61.
159. Бухалова Г. А., Казаченко Е. Л., Семенцова Д. В.,
Керопян В. В. — Журн. неорган, хим., 1975, 20,
с. 1378.
160. Бухалова Г. А., МарЭиросова И. В . Журн.
неорган, хим., 1967, 12, с. 2199.
161. Бухалова Г. А., Семенцова Д. В. — Журн.
неорган, хим., 1965, 10, с. 1027.
162. Бухалова Г. А., Семенцова Д. В. — Журн.
неорган, хим., 1965, 10, с. 1886.
163. Бушуев Ю. Г., Дьяконов Я. И., Балакир 9. А.,
Рыбаков И. И. — Изв. СО АН СССР. Сер. хим.
наук, 1968, 1, с. 138.
164. Валиев К. А., Копаев Ю. В., Мокеров В. Г.,
Раков А. В. — Журн. эксперим. и теор. физ., 1971,
60, с. 2175.
165. Варгафтик П. Б. — В кн.: Справочник по тепло-
физическим свойствам газов и жидкостей. М.:
Наука, 1972, с. 86.
166. Варгафтик Я. Б., Воляк Л. Д., Анисимов В. М.,
Степанов В. Г., Кожевников В. Ф., Челебаев
А. К. — Инж.-физ. журн., 1980, 39, с. 986.
167. Вартбаронов О. Р., Бергман А. Г. — Изв. вузов.
Хим. и хим. технол., 1967, 10, с. 491.
168. Василев X., Николов Т., Чимбулев М. — Год.
Висш. Хим.-технол. институт. София, 1967 (опубл.
1971), 14, с. 321.
169. Василевский К. П., Байков В. И. — Тезисы
XI Всесоюзной конференции по спектроскопии.
Л., 1960.
170. Василевский К. П., Байков В. И. — Оптика
и спектроскопия, 1961, 11, с. 41.
171. Васильев В. П., Козловский Е. В. — Журн. физ.
хим., 1972, 46, с. 2442.
172. Васильев В.П., Лобанов Г. А. — Журн. неорган,
хим., 1966, 11, с. 703.
525
Литература
173. Васильев В. П., Орлова Г. Д., Кочергина Л. А. — 204.
Труды Ивановен, хим.-технол. ин-та, 1975,
вып. 18, с. 122.
174. Васильев И. Н., Трелин Ю. С, Романов А. А. —
Теплофизика высоких температур, 1971, 9, с. 59. 205.
175. ВасильеваИ. А., Герасимов Я. И., Кушнир Э. Ю.—
Докл. АН СССР, 1977, 234, с. 1362. 206.
176. Васильева И. А., Герасимов Я. И., Кушнир Э. Ю.—
Докл. АН СССР, 1977, 235, с. 839.
177. Васильева И. А., Герасимов Я. И., Огородова 207.
Л. П. — Докл. АН СССР, 1977, 232, с. 600.
178. Васильева И. А., Герасимов Я. И., Огородова 208.
Л. П. — Докл. АН СССР, 1977, 232, с. 835.
179. Васильева И. А., Герасимов Я. И., Симонов Ю. П.—
Журн. физ. хим., 1957, 31, с. 682. 209.
180. Васильева И. А., Герасимов Я. И., Симонов
Ю. П. — Журн. физ. хим., 1960, 34, с. 1811. 210.
181. Васильева И. А., Грановская Ж. В. — Журн. физ.
хим., 1974, 48, с. 1536. 211.
182. Васильева И. А., Грановская Ж. В. — Журн. физ.
хим., 1977, 51, с. 2100. 212.
183. Васильева И. А., Грановская Ж- В., Серегин
А. Я. — Журн. физ. хим., 1977, 51, с. 2102. 213.
184. Васильева И. А., Мудрецова С. Я., Сулейма-
нова Г. С. — Вести. МГУ. Сер. хим., 1981, 22,
с. 403. . 214.
185. Васильева И. А., Огородова Л. П., Стпепанец
Л. И. — Вести. МГУ. Сер. хим., 1976, 17, с. 47. 215.
186. Васильева И. А., Пожилъцова Л. И. — Вестн.
МГУ. Сер. хим., 1978, 19, с. 141.
187. Васильева И. А., Стпепанец Л. И. — Вестн. МГУ. 216.
Сер. хим., 1977, 18, с. 33.
188. Васильева И. А., Сухушина И. С, Грановская 217.
Ж. В., Балабаева Р. Ф., Майорова А. Ф. — Журн.
физ. хим., 1975, 49, с. 2169. 218.
189. Васильева И. А., Шаулова д. Ю- — Журн. физ.
хим., 1969, 43, с. 3047. 219.
190. ВасильковаИ. В., Ефимов А. И., РязанцеваЛ. В. —
Вестн. ЛГУ. Сер. физ. и хим., 1976, вып. 2, № 10, 220.
с. 144.
191. Васютинский Б. М., Картмазое Г. М-, Фин- ?21.
келъ В. О. — Укр. физ. журн., 1962, 7, с. 661.
192. Васютинский Б. М-, Картмазое Г. М-, Фин- 222.
келъ В. О. — Физ. металлов и металловедение,
1963, 16, с. 771. 223
193. Ватолин Н. А., Моисеев Г. К. — ВИНИТИ.
Дел. № 4027-76. М., 1976. 224.
194. Ващенко В. А., Кашпорое Л. Я., Фролов Ю. В. —
Институт хим. физики АН СССР, Черноголовка. 225.
Препринт, 1974. 21 с.
195. Вдовенко В. М-, Волков В. А., Кожина И. И., 226
Суглобова И. Г. — Радиохимия, 1972, 14, с. 482.
196. Вдовенко В. М., Макаров Л. Л., Стукин Д. Ю. — 227
Вестн. ЛГУ. Сер. физ. и хим., 1967, вып. 2,
№ 10, с. 124. 228.
197. Вдовенко В. М., Маширов Л. Г., Блохина В. К.,
Суглобова И. Г. — Радиохимия, 1963, 5, с. 80. 229.
198. Вдовенко В. М., Романов Г. А., Щербаков В. А. —
Радиохимия, 1963, 5, с. 581. 9ЧП
199. Веденеев А. В., Скуратов С. М. — Журн. физ. zou'
хим., 1951, 25, с. 837.
200. Величко А. В., Зайцева Л. Л. —Журн. неорган. 231.
хим., 1972, 17, с. 2799.
201. Вейц И. В., Гуреич Л. В. — Докл. АН СССР,
1967,173, с. 1325.
202. Вейц И. В., Гуреич Л. В., Кобъиянский А. И., 232-
Смирнов А. Д., Суслов А. А. — J. Quant. Spectrosc.
and Radiat. Transfer, 1974, 14, p. 221.
203. Верещагин Л. Ф., Яковлев Е. Н., Виноградов Б. В., 233.
Сакун В. П. — Письма в ЖЭТФ, 1974, 20, с. 540.
Вержбицкий Ф, Р., Василевская М. М., Буров Г. В.,
Смирнов М. В. — Труды ин-та электрохимии,,
Уральск, научн. центр АН СССР, 1971,вып. 17г
с. 7.
Верхотуров Е. Н. — Автореф. дис. . . . канд.
хим. наук. М.: МГУ, 1978.
Верхотуров Е. Н.„ Макаров А. В.,
Никитин О. Т. — Вестн. МГУ. Сер. хим., 1976, 17,
с. 299.
Верхотуров Е. Н., Макаров А. В.', Никитин
О. Т. — Вестн. МГУ. Сер. хим., 1977, 18, с. 392.
Верхотуров Е. Н., Макаров А. В.,
Никитин О. Т. — Вестн. МГУ. Сер. хим., 1979, 20,
с. 535.
Верхотуров Е. Н., Один И. Н., Шер А. А. —
Изв. АН СССР. Неорган, матер., 1980, 16, с. 1688.
Ветренко Е. А., Микулинский А. С, Фриш-
берг И. В. — Журн. физ. хим., 1962, 36, с. 1585.
Ветлокое М. М., Блюштейн М. Л., Ноддымо»
В. П. — Изв. вузов. Цвет, мет., 1959, № 6, с. 126.
Видавский Л. М., Бяхова Н. И., Ипполитова
Е. А. — Журн. неорган, хим., 1965, 10, с. 1746.
Видавский Л. М., Бяхова Н. И., Ковальчук В. Ю.,
Ипполитова Е. А. — Вестн. МГУ. Сер. хим.,
1964, № 4, с. 33.
Видавский Л. М., Ипполитова Е. А. —
Радиохимия, 1971, 13, с. 299.
Видавский Л. Ж., Лавут Э. Г., Ковба Л. М.,
Ипполитова Е. А. — Докл. АН СССР, 1964»
154, с. 1371.
Вилков Л. В., Спиридонов В. П., Харгиттаи И. —
Вестн. МГУ. Сер. хим., 1977, 18, с. 635.
Виноградов Ю. К., Воляк Л. Д. — Теплофизика
высоких температур, 1966, 4, с. 50.
Винтайкин Е. 3. — Докл. АН СССР, 1958, 118,
с. 977.
Винтайкин Е. 3. — Докл. АН СССР, 1959, 129,
с. 1368.
Винтайкин Е. 3. — Физ. металлов и
металловедение, 1962, 13, с. 157.
Власов В. Г., Бекетов А. Р. — Изв. АН СССР.
Металлургия и горное дело, 1963, № 6, с. 110.
Власов В. Г., Лебедев А. Г. — Журн. прикл. хим.,
1961, 34, с. 1739.
Власов В. Г., Лебедев А. Г. — Изв. вузов. Черн.
мет., 1960, № 7, с. 5.
Власов В. Г., Лисняк С. С. — Изв. вузов. Черн.
мет., 1958, № 7, с. 45.
Власов В. Г., Семавин Ю. Н. — Журн. прикл.
хим., 1966, И, с. 1210.
Волк В. И., Зайцева Л. Л. — Журн. неорган.
хим., 1971, 16, с. 2845.
Волков А. Н., Неуймин А. Д. — Заводская
лаборатория, 1980, 46, с. 916.
Волков В. А., Суглобова И. /\, Чиркст Д. Э. —
Координационная химия, 1980, 6, с. 417.
Волкова Л. Ф. — Изв. СО АН СССР. Сер. хим.
наук, 1958, № 7, с. 33.
Волкова Н. М., Гельд П. В. — Изв. вузов. Цвет,
мет., 1963, № 5, с. 89.
Володкович Л. М., Вечер Р. А., Вечер А. А. —
В кн.: V Всесоюз. симпозиум по химии неорган.
фторидов. Днепропетровск, 1978. Тезисы
докладов. М.: Наука, 1978, с. 74.
Володкович Л. М., Петров Г. С, Вечер Р. А.,
Вечер А. А. — Вестн. БГУ. Сер. 2, 1981, № 1,
с. 20.
Вольф Э. — Автореф. дис. . . . канд. хим. наук.
Л.: ЛГУ, 1958.
526
Литература
234. Волъф д., Ария С. М. — Журн. общ. хим., 1959, 260.
29, с. 2470.
235. Волъф 9., Толкачев С. С, Кожин И. И. — Вести.
ЛГУ. Сер. физ. и хим., 1959, вып. 2, № 10, с. 87. 261.
236. Воляк Л. Д. — В кн.: Теплофизические свойства
твердых тел при высоких температурах. Труды 262.
всесоюз. конференции 1966: Сборник/Под ред.
И. И. Новикова и А. Н. Гордова. М.: Комитет
стандартов, мер и измерит, приборов при Совете 263.
Министров СССР, 1966, с. 232.
237. Воляк Л. Д. — В кн.: Теплофизические свойства
веществ и материалов, вып. 1. М.: Изд-во стан- 264.
дартов, 1968, с. 49.
238. Воляк Л. Д. Автореф. дис. . . . докт. техн. наук. 265.
М.: МАИ, 1969.
239. Воляк Л. Д., Виноградов Ю. К., АнисимовВ. М. — 266.
Теплофизика высоких температур, 1968, 6, с. 545.
240. ВолякЛ. Д., Виноградов Ю.К., АнисимовВ. М.— 267.
Теплофизика высоких температур, 1968, 6, с. 754.
241. Воляк Л. Д., Фокин Л. Р., Челебаев А. К. — 268.
В кн.: Исследование термодинамических и
переносных свойств нейтральных и ионизированных
газов: Тематический сборник научных трудов ин- 269.
ститута. М.: МАИ, 1979, вып. 498, с. 80.
242. Воляк Л. Д., Челебаев А. К. — Теплофизика вы- 270.
соких температур, 1976, 14, с. 913.
243. Воляк Л. Д., Челебаев А. К., Гурьянова Л. П. —
В кн.: Теплофизические свойства рабочих тел 271.
и теплоносителей новой техники: Тематический
сборник научных трудов института. М.: МАИ,
1975, вып. 319, с. 102. 272.
244. Воробьев А. Ф., Набилъ Ахмед Ибрагим,
Скуратов С. М. — Журя, неорган, хим., 1966, 11, с. 25.
245. Воробьев А. Ф., Перелягин Б. Г., Бывальцев 273.
JO. А. В кн.: Термодинамические свойства
растворов: Труды Моск. хим.-технол. ин-та им. 274.
Менделеева. Межвузовский сборник № 1, 1980,
111, с. 3.
246. Воробьев А. Ф., Умярова Р. С, Урусов В. С. — 275.
Журн. общ. хим., 1974, 44, с. 979.
247. Воронин Л. К., МеркулъевА. П., Неймарк Б. Е. —
Теплофизика высоких температур, 1970, 8, с. 780. 276.
248. Воронов Н. М., Данилин А. С, Ковалев И. Т. —
In: Thermodynamics of Nuclear Materials. Proc. 277.
Symp. Vienna, 1962. Vienna: IAEA, 1963, p. 789.
249. Воскресенская Н. К., Бонашек Е. И. — Жгв. 278.
сектора физ.-хим. анализа ИОНХ АН СССР, 1954,
25, с. 150. 279.
250. Воскресенская Н. К., Пацукова И. Н. — Докл.
АН СССР, 1952, 87, с. 219. 280.
251. Воскресенская Н. К., Пономарева К. С. — Докл.
АН СССР, 1944, 45, с. 200. 281.
252. Воскресенская П. К., Пономарева К. С. — Журн.
физ. хим., 1946, 20, с. 433.
Й53. Воскресенская Н. К., СрколовВ. А., БанашекЕ. И., 282.
Шмидт Н. Е. — Изв. сектора физ.-хим. анализа
ИОНХ АН СССР, 1956, 27, с. 233.
254. Воскресенская Н. К., Янковская Г. Н., Аносов
В. Я Журн. прикл. хим., 1948, 21, с. 18. 283.
255. Гавриш А. М., Сухаревский Б. Я., Криворучко
П. П. — Докл. АН СССР, 1967, 177, с. 886.
256. Гагаринский Ю. В., Ханаев Е. И. — Журн. 284.
неорган, хим., 1967, 12, с. 111.
257. Гайдукова Т. И., Шадский С. В. — ВИНИТИ.
Деп. № 5306-73. М., 1973. 285.
258. Галицкий Н. В. — Журн. неорган, хим., 1968,
13, с. 3120. 286.
259. Галицкий И. В., Минина К. П. — В кн.: Общая
и прикл. химия: Сборник. Минск: Высш. школа, 287.
1969, вып. 1, с. 35.
Галкин Н. П., Судариков Б. И., Зайцев В. А. —
Труды Моск. хим.-технол. ин-та им. Д. И.
Менделеева, 1963, 43, с. 64.
Галъбрайх 9. И., Ария С. М. — Журн. физ. хим.,
1970, 44, с. 2773.
Гальченко Г. Л., Гагаринский Ю. В.,
Попов М. М. — Журн. неорган, хим., 1960, 5,
с. 1631.
Гарбер Р. П., Харитонова Ж. Ф., Ажижа П. М.,
Въюгов П. Н., Великодная О. А. — Физ. металлов
и металловедение, 1973, 35, с. 863.
Гасаналиее А. М., Мальцев В. Т. —'Жури,
неорган, хим., 1970, 15, с. 1688.
Гвелесиани Г. Г., Яшвили Т. С. — Журн. нёорган.
хим., 1967, 12, с. 3233.
Гельд П. В., Алямовский С. И. — Физ. металлов
и металловедение, 1960, 9, с. 315.
Гельд П. В., Алямовский С. И., Матвеенко И. И.—
Журн. структур, хим., 1961, 2, с. 301.
Гельд П. В., Кусенко Ф. Г. — Изв. АН СССР.
Отд. техн. наук. Металлургия и топливо, 1960,
№ 2, с. 79.
Гельд П. В., Путинцев Ю- В. — Труды Уральск,
политехи, ин-та, 1970, № 186, с. 199.
Герасимов Я. И., Васильева И. А., Грановская
Ж. В., Майорова А. Ф. — Докл. АН СССР, 1973,
210, с. 1347.
Герасимов Я. И., Васильева И. А., Чусова Т. П.,
Гейдерих В. А., Тимофеева М- А. — Докл. АН
СССР, 1960, 134, с. 1350.
Герасимов Я. И., Васильева И. А., Чусова Т. П.,
Гейдерих В. А., Тимофеева М. А. — Жури. физ.
хим., 1962, 36, с. 358.
Герасимов Я. И., Воронин Г. Ф., Шиу Н. Т. —
Журн. физ. хим., 1968, 41, с. 1468.
Герасимов Я. И., Глазов В. М., Лазарев В. Б.,
Жаров В. В., Пашинкин А. С. — Докл. АН СССР,
1977, 235, с. 846.
Герцберг Г. Колебательные и вращательные
спектры многоатомных молекул. М.: Изд-во иностр.
лит., 1949.
Герцберг Г. Электронные спектры и строение
многоатомных молекул. М.: Мир, 1969.
Герцикен С. Д., Слюсарь Б. П. — Укр. физ.
журн., 1962, 7, с. 439.
Гинзбург Д. М. — Журн. общ. хим., 1956, 26,
с. 968.
Гинзбург Д. М. — ВИНИТИ. Деп. № 3304-71.
М., 1971.
Гинзбург Д. М., Губа Н. И., Кочколда В. Е. —
Журн. физ. хим., 1971, 45, с. 2939.
Гиричев Г. В., Данилова Т. Г., Гиричева П. И.,
Краснов К. С, Засорин Е. 3. — Изв. вузов. Хим.
и хим. технол., 1977, 20, с. 1233.
Гиричев Г. В., Данилова Т. Г., Гиричева Н. И.,
Краснов К. С, Петров В. М., Уткин А. Н.,
Засорин Е. 3. — Изв. вузов. Хим. и хим. технол.,
1978, 21, с. 627.
Гиричева П. И., Бобкова В. А., Миронова Е. В.,
Краснов К. С, Данилова Т. Г. — ВИНИТИ.
Деп. № 515-74. М., 1974.
Гиричева Н. И., Засорин Е. 3., Гиричев Г. В.,
Краснов К. С, Спиридонов В. П. — Журн.
структур, хим., 1976, 17, с. 797.
Гладущенко В. А., Бергман А. Г. — Труды
Новочеркасского политехи, ин-та, 1956, 27/41, с. 49.
Гладущенко В. А., Бергман А. Г. — Труды
Новочеркасского политехи, ин-та, 1959, 65, с. 67.
Глазов В. И., Нарышкин И. И. — Журн. прикл.
хим., 1968,-41, с. 2297.
527
Литература
288. Глазов В. Я., Нарышкин И. И. — Журн. прикл.
хим., 1970, 43, с. 2728.
289. Глыбин В. П., Добротин Р. Б., Акулова Г. В. —
Журн. неорган, хим., 1971, 16, с. 2640.
290. Гоголева В. В., Фокин Л. Р. Оценка критических
параметров лития и франция: препринт ИВ ТАН
№ 1-061. М., 1981, с. 44.
291. Годун И. В. — ЦНИИЭИцветмет, 5 окт. 1976.
Деп. № 245, М., 1976.
292. Голубцов И. В. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1966.
293. Голубцов И. В. — В кн.: Теплофизические
свойства твердых веществ: Сборник/Под ред.
Г. В. Самсонова. М.: Наука, 1971, с. 154.
294. Голубцов И. В. — В кн.: Современные проблемы
физической химии. М.: МГУ, 1972, т. 6, с. 343.
flF:
295.f[Голубцов И. В., Лапицкий А. В., Ширяев В. К. —
Изв. вузов. Хим. и хим. технол., 1962, 5, с. 691.
296. Голубцов И. В., Микульская Г. Ю. — В кн.:
Термодинамические свойства металлических
сплавов. Баку: Элм, 1975, с. 62.
297. Голубцов И. В., Микульская Г. Ю-,
Павлова Л. М. — Вестн. МГУ. Сер. хим., 1972, 13,
с. 206.
298. Голубцов И. В., Несмеянов Ан. Н. — Вестн. МГУ.
Сер. хим., 1965, № 5, с. 31.
299. Голутвин Ю. М. — Журн. физ. хим., 1959, 33,
с. 1798.
300. Голутвин Ю- М., Козловская Т. М. _ Журн.
физ. хим., 1960, 34, с. 2350.
301. Голутвин Ю. М., Козловская Т. М. — Журн.
физ. хим., 1962, 36, с. 362.
302. Голутвин Ю. М., Масленникова Э. Г. — Изв.
AHfCCCP. Металлы, 1970, № 5, с. 174.
303. Голутвин Ю. М., Цзинь-куй Л. — Журн. физ.
хим., 1961, 35, с. 129.
304. Горбатый Н.А., ШуппеГ.Н. — Изв. АН Узб. ССР .
Сер. физ.-мат. наук, 1957, № 3, с. 39.
305. Горбатый Н. А., Шуппе Г. Я. — Докл. АН
Узб. ССР, 1957, № 12, с. 13.
306. Горбов С. И., Гурвич Л. В., Иориш В. С, Юнз-
ман В. С. — Теплофизика высоких температур,
1975, 13, с. 503.
307. Горелик В. С, Желудев И. С, Сущинский М. М. —
Кристаллография, 1966, 11, с. 604.
308. Горохов Л. Н. — Докл. АН СССР, 1962,142, с. 113.
309. Горохов Л. Н. — Автореф. дис. . . . докт. хим.
наук. М.: МГУ, 1972.
310. ГороховШ. Н., Гусаров А. В., Емельянов А. М. —
В кн.: Восьмая Всесоюзная конференция по
калориметрии и химической термодинамике, Иваново,
1979. Тезисы #докладов. Иваново: Ивановский
хим.-технол. ин-т, 1979, с. 329.
311. Горохов Л. Н., Гусаров А. В., Макаров А. В.,
Никитин, О. Т. Теплофизика высоких
температур, 1971, 9, с. 1173.
312. Горохов Л. Я., Гусаров А. В., Панченков И. Г. —
Журн. физ. хим., 1970, 44, с. 269.
313. Горохов Л. Н., Емельянов А. М., Ходеев Ю. С. —
Теплофизика высоких температур, 1974,12, с. 1307.
314. Горячева В. П., Бергман А. Г., Кислова А. Г. —
Журн. неорган, хим., 1959, 4, с. 2744.
315. Госик М. И., Емлин Б. Я., Хитрик С. И. —
Изв. вузов. Черн. мет., 1970, № 3, с. 59.
316. Грачев Я. С, Кириллов П. Л. — Инж.-физ.
журн., 1960, 3, с. 62.
317. Григорович В. К. — Изв. АН СССР. Отд. техн.
наук. Металлургия и топливо, 1960, № 6, с. 93.
318. Григорьев А. И., Орлова Ю. В., Сипачев В. А.,
Новоселова А. В. —Докл. АН СССР, 1963, 152,
с. 134.
319. Григорьев А. Т., Гусева Л. Я., Соколовская Е. М.,
Максимова М. В. — Журн. неорган, хим., 1959,
4, с. 2168.
320. Григорьев А. Т., Е Юй-Пу, Соколовская Е. М.
Журн. неорган, хим., 1960, 5, с. 1642.
321. Григорьев А. Т., Соколовская Е. М. — Вестн.
МГУ. Сер. хим., 1961, № 6, с. 3.
322. Григорьев А. Т., Соколовская Е. М., Максимова
М. В., Соколова И. Г., Недумов Н. А. — Журн.
неорган, хим., 1960, 5, с. 2640.
323. Григорьев А. Т., СоколовскаяЕ. М., Недумов Н. А.,
Максимова М. В., Соколова И. Г., Е Юй-Пу —
Журн. неорган, хим., 1961, 6, с. 1248.
324. Григорьев А. Т., Соколовская Е. М., Нефедов А. П.,
Максимова М. В.'—Вестн. МГУ. Сер. хим.,
1961, № 2, с. 19.
325. Григорьев А. Т., Соколовская Е. М., Симанов
Ю. П., Соколова И. Г., Максимова М. В.,
Пятигорский Л. И. — Журн. неорган, хим., 1960, 5,
с. 2136.
326. Григорьев А. Т., Соколовская Е. М., Симанов
Ю. П., Соколова И. Г., Павлов В. Я.,
Максимова М. В. — Вестн. МГУ. Сер. хим., 1960, № 4,
с. 23.
327. Григорян А. Л. — Научные труды н.-и. горно-
метал. ин-та Арм.ССР, 1961, 2, с. 113.
328. Григорян А. Л., Энфиаджан М. А. Сб. научн.
тр. Ереванского политехи, ин-та. Хим. и хим.
технол., 1957, № 16, с. 131.
329. Грицус Б. В., Ахумов Е. И., Жилина Л. П. —
Журн. прикл. хим., 1969, 42, с. 208.
330. Грицус Б. В., Ахумов Е. П., Жилина Л. П. —
Журн. прикл. хим., 1971, 44, с. 186.
331. Громаков С. Д. — Журн. физ. хим., 1950, 24,
с. 641.
332. Гурвич Л. В., Вейц И. В. — Докл. АН СССР,
1957, 116, с. 811.
333. Гурвич Л. В., Вейц И. В. — Изв. АН СССР. Сер.
физ., 1958, 22, с. 673.
334. Гурвич Л. В., Карачевцев Г. В., Кондратьев В. Н.,
Лебедев Ю. А., Медведев В. А., Потапов В. К.,
Ходеев Ю- С. Энергия разрыва химических связей.
Потенциалы ионизации и сродство к электрону.
М.: Наука, 1974.
335. Гурвич Л. В., Квливидзе В. А., Ртищева Н. П. —
Журн. физ. хим., 1962, 36, с. 219.
336. Гурвич Л. В., Рябова В. Г. — Теплофизика
высоких температур, 1964, 2, с. 401.
337. Гурвич Л. В., Хачкурувов Г. А., Медведев В. А.,
Вейц И. В., Бергман Г. А., Юнгман В. С,
Ртищева Н. П., Куратова Л. Ф., Юрков Г. М.,
Кане А. А., Юдин Б. Ф., Броунштейн Б. Я.,
Байбуз В. Ф., Квливидзе В. А., Прозоровский Е.А.,
Воробьев Б. А. Термодинамические свойства
индивидуальных веществ / Под ред. В. П. Глушко.
М.: Изд-во АН СССР, 1962, т. 1—2.
338. Гурвич Л. В., Юнгман В. С, Дорофеева О. В.,
Горохов Л. Я., Мунвез С. С. —
Термодинамические свойства газообразной системы U—F.
Препринт № 1-0018. М.: ИВТАН, 1977.
339. Гурвич Л, В., Юнгман В. С, Дорофеева О. В.,
Горохов Л. Н., Мунвез С. С. —
Термодинамические свойства газообразной системы W—F.
Препринт № 1-0019. М.: ИВТАН, 1977.
340. Гурвич Л. В., Юнгман, В. С, Хачкурузов Г. А.,
Бергман Г. А., Медведев В. А., Ртищева Я. П.,
Броунштейн Б. И., Вейц И. В., Коробов В. В.,
Куратова Л. Ф. Термодинамические свойства
528
Литература
компонентов продуктов сгорания / Под ред. 368.
В. П. Глушко. М.: Изд-во АН СССР, 1956, т. 1—3.
341. Гуревич В. М., Соколов В. А. — Журн. физ. хим.,
1942, 46, с. 1868.
342. Гурьянова Л. Д. — В кн.: Тематический сборник 369.
научных трудов института. М.: МАИ, 1977, вып.
420, с. 52. 370.
343. Гусаров А. В. — Автореф. дис. . . . канд. хим. 371.
наук. М.: МГУ, 1968.
344. Гусаров А. В., Горохов Л. Н. — Теплофизика вы- 372.
соких температур, 1966, 4, с. 590.
345. Гусаров А. В., Горохов Л. Н. — Журн. физ. хим., 373.
1968, 42, е. 860,
346. Гусаров А. В., Горохов Л. Я. — Теплофизика вы- 374.
соких температур, 1971, 9, с. 505.
347. Гусаров А. В., Горохов Л. Я., Ефимова А. Г. — 375.
Теплофизика высоких температур, 1967, 5, с. 584.
348. Гусаров А. В., Горохов Л. Я., Ефимова А. Г. — 376.
Теплофизика высоких температур, 1967, 5, с. 783.
349. Гусаров А. В., Пятенко А. Т., Сидорова И. В., 377.
Горохов Л. Я. — В кн.: Восьмая конференция
по калориметрии и хим. термодинамике, Иваново,
1979. Тезисы докладов. Иваново: Ивановский
хим.-технол. ин-т, 1979, с. 314. 378.
350. Гусева Е. А., Болгар А. С, Гордиенко С. П.,
Горбатюк В. А. —Теплофизика высоких темпе- 379.
ратур, 1966, 4, с. 649.
351. Гущин Г. И., СубботинВ.А., Хачатуров 9. X. — 380.
Теплофизика высоких температур, 1975,13, с. 747.
352. Гшнейдер К. А. — В кн.: Свойства и применение 381.
редкоземельных металлов: Материалы
конференции по редкоземельным металлам, г. Чикаго, 382.
ноябрь 1959. М.: Изд-во иностр. лит., 1960, с. 7.
353. Гшнейдер К. А. — В кн.: Проблемы современной 383.
металлургии: Сборник статей. М.: Изд-во иностр.
лит., 1960, № 2, с. 53.
354. Давыдов К. Я., Гельд П. В., Серебренников Я. И. 384.
— В кн.: Физико-химические основы производства
стали. М.: Изд-во АН СССР, 1957, с. 350. 385.
355. Данилов С. В., Суриков В. И., Миллер И. И.,
Суриков Вал. И. — Физика твердого тела, 1980, 386.
22, с. 2511.
356. Данилова Т. Г., Гиричев Г. В., Гиричева Н. И.,
Краснов К. С, Засорин Е. 3. — Изв. вузов. 387.
Хим. и хим. технол., 1977, 20, с. 1069.
357. Данилова Т. Г., Гиричев Г. В., Гиричева Я. И., 388.
Краснов К. С, Засорин Е. 3. — Изв. вузов. Хим.
и хим. технол., 1979, 22, с. 101. 389.
358. Данцигер А. Я., Фесенко Е. Г. —
Кристаллография, 1963, 8, с. 894. 390.
359. Данцигер А. Я., Фесенко Е. Г. —
Кристаллография, 1965, 10, с. 338. 391.
360. Дворкин М. И. — Автореф. дис. . . . канд. физ.-
мат. наук. Л.: ЛГУ, 1977.
361. Дегтярева В. Ф. — Физика твердого тела, 1973,
15, с. 3436. 392.
362. Делимарский Ю. К., Кириллов С. А. —Журн.
теор. физ., 1974, 10, с. 201. 393
363. Дергунов Е. Л. — Журн. физ. хим., 1951, 25,
с. 584.
364. Дергунов- Е. П., Бергман А. Г. — Журн. физ. 394
хим., 1948, 22, с. 625.
365. Дергунов Е. П., Бергман А. Г. — Дркл. АН СССР, 395
1950, 75, с. 815.
366. Дерибас А. А., Ручкин Е. Д., Филаткина В. С,
Хрипин Л. А. —• Физ. горения и взрыва, 1966, 396
№ 4, с. 143.
367. Десятник В. Я., Дубинин Г. В. — Изв. вузов. 397
Цвет, мет., 1973, № 6, с. 84.
Десятник В. Я., Курбатов Я. Я., Стрелов В. А. —
В кн.: V Всесоюзный симпозиум по химии неорган,
фторидов, Днепропетровск, 1978. Тезисы
докладов. М.: Наука, 1978, с. 99.
Десятник В. Я., Распопин С. Я., Трифонов
Я. И. — Изв. вузов. Цвет, мет., 1972, № 1, с. 100.
Диогенов Г. Г. — Докл. АН СССР, 1951, 78, с. 697.
Диогенов Г. Г., Букова Т.Е. — Журн. неорган,
хим., 1970, 15, с. 2555.
Диогенов Г. Г., Ермачков В. И. — Журн. неорган,
хим., 1966, 11, с. 1220.
Диогенов Г. Г., Ермачков В. И. — Труды
Иркутского политехи, ин-та, 1966, вып. 27, с. 64.
Диогенов Г. Г., Ермачков В. Я. — Журн. неорган,
химии, 1967, 12, с. 575.
Диогенов Г. Г., Жжёных В. Я. — Труды
Иркутского политехи, ин-та, 1970, вып. 44, с. 134.
Диогенов Г. Г., Иванова В. Ф. — Труды
Иркутского политехи, ин-та, 1971, вып. 66, с. 168.
Диогенов Г. Г., Калинченко Ф. И. — В кн.:
Некоторые вопросы химии распл. солей и продуктов
деструкции сопропелитов. Иркутск: Иркутский
политехи, ин-т, 1974, с. 69.
Диогенов Г. Г., Кириллова В. Ф. — Журн.
неорган, хим., 1973, 18, с. 2830.
Диогенов Г. Г., Мавридис Р. Я. — Журн. неорган,
хим., 1968,-13, с. 2014.
Диогенов Г. Г., Решетников Я. А. — Труды
Иркутского политехи, ин-та, 1971, вып. 66, с. 37.
Дихтер Я. Я., Лебедев С. В. — Теплофизика
высоких температур, 1970, 8, с. 55.
Дихтер И. Я., Лебедев С. В. — Теплофизика
высоких температур, 1971, 9, с. 929.
Домбровская Я. С, Колоскова 3. А. — Изв.
сектора физ.-хим. анализа ИОНХ АН СССР, 1953,
22, с. 178.
Домбровская О. С. — Журн. общ. хим., 1933, 3,
с. 1017.
Дракин С. И., Чжан Ю-Мин. — Журн. фнз. хим.,
1964, 38, с. 2800.
Дробот А. В., Аникина Г. П., Дуринина Л. В.,
Коршунов Б. Г. —¦ Журн. неорган, хим., 1965,
10, с. 562.
Дробышев В. Я., Резухина Т. Я. — Журн. физ.
хим., 1965, 39, с. 151.
Дробышев В. Я., Резухина Т. Я., Тарасова Л. А. —
Журн. физ. хим., 1965, 39, с. 141.
Дронин А. А., Горохов Л. Я. — ВИНИТИ. Деп.
№ 2977-74. М., 1974.
Дударёв В. Я., Ценципер А. Б., Добролюбова
М. С. — Кристаллография, 1973, 18, с. 759.
Дудчик Г. Л., Комшилова О. Я., Новиков Г. И.,
Лоляченок О. Г. —-В кн.: Термодинамические
и термохимические константы/ Отв. ред. К. В.
Астахов. М.: Наука, 1970, с. 39.
Дьяков И. Г., Швец А. Д. — Журн. эксперим.
и теор. физ., 1965, 49, с. 1091.
Дьяконова М. Е., Дракин С. И., Карапетянц
М. X. — Труды Моск. хим. техн. ин-та им.
Д. И. Менделеева, 1970, вып. 67, с. 12.
Дымова Т. Я., Бакунин С. М., Мирсаидов У. —
Докл. АН СССР, 1974, 216, с. 87.
Евсеев А. М., Пожарская Г. В., Несмеянов А. Я.,
Герасимов Я. М. — Журн. неорган, хим., 1959,
4, с. 2196.
Евсеева Я. Я. — Изв. сектора физ.-хим. анализа
ИОНХ АН СССР, 1953, 22, с. 162.
, Ежов Ю. С. — Журн. структур, хим., 1977, 18,
с. 89.
34 Заказ JVS 1590
529
Литература
398. Ежов Ю. С, Акишин П. А., Рамбиди П. Г. —
Журн. структур, хим., 1969, 10, с. 571.
399. Ежов Ю. С, Акишин П. А., Рамбиди Н. Г. —
Журн. структур, хим., 1969, 10, с. 763.
400. Ежов Ю. С, Виноградов В. С. — Журн. структур.
хим., 1978, 19, с. 600.
401. Ежов Ю. С., Комаров С. А. — Журн. структур,
хим., 1975, 16, с. 662.
402. Ежов Ю. С, Толмачев С. М., Рамбиди Н. Г. —
Журн. структур, хим., 1972, 13, с. 972.
403. Ежов Ю. С, Толмачев С. М., Спиридонов В. П.,
Рамбиди Н. Г. — Теплофизика высоких
температур, 1968, 6, с. 68.
404. Елисеев А. А., Ярембаш Е. И., Вигилева Е. С,
Антонова Л. И., Заготская А. В. — Журн.
неорган, хим., 1964, 9, с. 1032.
405. Елютин В. П.', Маурах М. А., Мишук В. Я.,
Свердлов Г. М. — Изв. вузов. Цвет, мет., 1967,
№ 1, с. 60.
406. Елютин В. П., Маурах М. А., Свердлов Г. М. —
Изв. вузов. Цвет, мет., 1967, № 10, с. 87.
407. Емелин В. П., Кесслер Ю. М. — В кн.: Пятая
Всесоюз. конф. по калориметрии, 21—25 июня
1971: (расширенные тезисы докл.). М.: МГУ,
1971, с. 443.
408. Емелин В. П., Кесслер Ю- М. — Журн. структур,
хим., 1972, 13, с. 323.
409. Емельянов А. М., Гусаров А. В., Горохов Л. Н. —
В кн.: Седьмая Всесоюз. конф. по калориметрии,
31 янв.—3 февраля 1977. Москва: (расширенные
тезисы докл.). Черноголовка, 1977, с. 325.
410. Емельянов А. М., Гусаров А. В., Горохов Л. Н.,
Садовникова Н. А. — Теор. и эксперим. хим.,
1967, 3, с. 226.
411. Емельянов А. М., Дередвиеина В. А., Горохов
Л. И. — Теплофизика высоких температур, 1971,
9, с. 190.
412. Еременко Л. Т., Колесов Ю. Р., Кустова Л. В. —
Журн. физ. хим., 1964, 38, с. 1259.
413. Ерофеева М. С, Лукиных И. Л., Ария С. М. —
Журн. физ. хим., 1961, 35, с. 772.
414. Есин О. А., Захаров И. И. — Изв. вузов. Черн.
мет., 1959, № 10, с. 9.
415. Ефимов М. Е., Леонидов В. Я. — В кн.: V Всесоюз.
симпоз. по химии неорганических фторидов,
Днепропетровск, 1978. Тезисы докладов. М.:
Наука, 1978, с. 109.
416. Ефимов М. Е., Леонидов В. Я. — Журн. физ.
хим., 1980, 54, с. 1323.
417. Ефимова А.Г.—В кн.: III Всесоюз. конф. по масс-
спектрометрии, Ленинград, 1981, Тезисы
докладов. Л.: СКВ АН, 1981, с. 169.
418. Ефимова А. Г., Горохов Л. И. — Теплофизика
высоких температур, 1978, 16, с. 1195.
419. Ефремова Р. И., Матизен д. В. — Теплофизика
высоких температур, 1982, 20, с. 265.
420. Жагрина Г. А., Фокин Л. Р., Чеховской В. Я. —
В кн.: Теплофизические свойства веществ при
высоких температурах: Сборник научных трудов.
М.: ИВТАН, 1978, с. 15.
421. Жданов Г. С, Русаков А. А. — Труды ин-та крж-
сталлографии АН СССР, 1954, вып. 9, с. 165.
422. Жжёных В. #., Кащеев Г, Н. — Труды
Иркутского политехи, ин-та, 1971, вып. 66, с. 5.
423. Жжёных В. Н., Кащеев Г. И. —Труды
Иркутского политехи, ин-та, 1971, вып. 66, с. 26.
424. ЖилинскийВ.И., МакушинЮ- С, Уленико*(Э. II.,
Чеглоков А. Е. — Журн. структур, хим., 1980,
21, с. 3.
425. Жмакин Л. И. — Автореф. дис. . . . канд. техн.
наук. М.: ИВТАН, 1977.
426. Жукова И. И. — Изв. сектора физ.-хим. анализа
ИОНХ АН СССР, 1927, 3, с. 600.
427. Ж-уковский В. М., Ткаченко Е. В., Власов В. Г. —
Физ. металлов и металловедение, 1963, 15, с. 210.
428. Забей ворота И. С.,ЛыкасовА. А., Михайлов Г. Г.,
Шахин Е. Л'..—'Изв. АН СССР. Неорган, матер.,.
1977, 13, с. 388.
429. Заборенко К. В., Оеькина, Т. Е., Чернова Н. А. —
Радиохимия, 1974, 16, с. 625.
430. Зайнулин Ю. Г., Алямовский СИ., Швейкин Г. П.,.
Попова С. В. — Докл. АН СССР, 1977, 234, с. 358.
431. Зайнулин Ю. Г., Алямовский С. И., Швейкин Г. П.,
Попова С. В. — Журн. неорган, хим., 1978, 23,
с. 1155.
432. Зайцева Г. Г., Крафтмахер Ю. А. — Журн.
прикл. мех. и техн. физ., 1965, 3, с. 117.
433. Закжевский В. Г. — Автореф. дис. . . , канд. хим.
наук. М.: МФТИ, 1979.
434. Закжевский В. Г., Болдырев А. И., Чаркин О. П.,
Боженко К. В., Клименко Н. М. — Журн.
неорган, хим., 1979, 24, с. 3171.
435. Зандберг Э. Я., Ионов Н. П., Тонтегоде Н. Я. —
Журн. техн. физ., 1965, 35, с. 1504.
436. Запольский С. В., Стерлядкина 3. К., Ми-
хеева В. И. — Журн. неорган, хим., 1970, 15,
с. 404.
437. Зарецкий Е. Б., Пелецкий В. Э. — Теплофизика
высоких температур, 1980, 18, с. 84.
438. Зарецкий Е. Б., Пелецкий В. 9. — Теплофизика
высоких температур, 1982 (в печати).
439. Засорин Е. 3. — Автореф. дис. . . . канд. физ.-мат.
наук. М.: МГУ, 1965.
440. Захарова Б. С, Решетникова Л. П.,
Новоселова А. В. — Вестн. МГУ. Сер. хим., 1967, 22^
с. 102.
441. Захарченко М- А., Гладущгнко В. А.,
Асланов С. М- — Журн. неорган, хим., 1966, И,
с. 2408.
442. Захвалинский М. Н., Белых П. Д. — В кн.: Вза1-
имодействие хлоридов, сульфатов щелочных и
щелочноземельных металлов в расплавах. Иркутск,:
Иркутский Гос. ун-т, 1972, с. 169.
443. Захвалинский М. П., Белых П. Д. — В кн.:
Взаимодействие хлоридов, сульфатов щелочных
и щелочноземельных металлов в расплавах.
Иркутск: Иркутский Гос. ун-т, 1972, с. 172.
444. Зверев В. А., Крестов Г. А. — Журн. физ. хим.,
1968, 42, с. 540.
445. Зеликман А. П., Белявская А. В. — Журн. прикл.
хим., 1954, 27, с. 1151.
446. Зеликман А. П., Горовиц Н. П., Просенкова Т. Е.—•
Журн."неорган, хим., 1956, 1, с. 632.
447. Зенков И. Д. — В кн.: Восьмая Всесоюз. конф.
по калориметрии и хим. термодинамике, Иваново,
1979. Тезисы докладов. Иваново: Ивановский
хим.-технол. ин-т, 1979, с. 470.
448. Зиновьев О. Ш., Лебедев С. В. — Теплофизика
высоких температур, 1976, 14, с. 83.
449. Зыган В. Н., Кеслер Я. А., Гордеев И. В.,
Третьяков Ю. Д. — Изв. АН СССР. Неорган, матер.,
1978, 14, с. 1087.
450. Зюбина Т. С, Чаркин О. П. — Журн. неорган,
хим., 1979, 24, с. 2529. ,
451. Зюбина, Т. С, Чаркин О. П. — Журн. неорган,
хим., 1980, 25, с. 609.
452. Иванов А. А. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1976.
453. Иванов В. Е., Круглых А. А., Павлов В. С, Ко*-
530
Литература
тун Г. П., Амоненко В. М- — In:
Thermodynamics of Nuclear Materials. Proc. Symp. Vienna,
1962. Vienna: IAEA, 1963, p. 735.
454. Иванова Е. А., Числер 9. В. — Физика твердого
тела, 1975, 17, с. 2873.
455. Иванова Е. Ф., Котлярова Г. П. — В кн.: Труды
1-ой конференции по аналитической химии
неводных растворов и их физ.-хим. свойствам. М., 1968,
ч. 2, с. 11.
456. Игнатьев Д. В., Лебедев Ю. И. — В кн.: Физико-
химические основы производства стали. М.: Изд-во
АН СССР, 1961, с. 305.
457. Ильясов И. И. — Журн. неорган, хим., 1968, 13,
с. 1659.
458. Ильясов И. И., Бергман А. Г.— Укр. хим.
журнал, 1965, 31, с. 772.
459. Ильясов И. И., Волчанская В. В., Лагу-
това Р. П. — Журн. неорган, хим., 1970, 15,
с. 1429.
460. Ильясов И. И., Давранов М., Искандаров К. И. —¦
Журн. неорган, хим., 1974, 19, с. 1919.
461. Ильясов И. И., Чаурский И. И. — Журн. неорган,
хим., 1966, 11, с. 2171.
462. И играм М., Дроварт Д. — В кн.: Исследования
при высоких температурах: Сборник. М.: Изд-во
иностр. лит., 1962, с. 274.
463. Институт высоких температур АН СССР.
Важнейшие результаты научно-исследовательских
работ за 1977 г. М.: Наука, 1978, с. 27.
464. Иоффе О. В., Кузнецова Г. П., Ловецкая Г. А.,
Преснякова В. М-, Степан Б. Д. — ВИНИТИ.
Деп. № 2116-74. М., 1974.
465. Ипполитова Е. А., Симанов Ю. П., Ефремова
К. М., Ивацпий В. М- — В кн.: Исследования
в области химии урана. М.: МГУ, 1961, с. 29.
466. Ищенко А. А., Будников С. С, Берсукер И. Б.,
Спиридонов В. П. — Теор. и эксперим. хим.,
1975, 11, с. 740.
467. Кабанкова Н. Н., Москвитина Е. Н., Кузяков
Ю. Я. — Вестн. МГУ. Сер. хим., 1975, 16, с. 485.
468. Кабанкова Н. Н., Москвитина Е. Н., Кузяков
Ю. Я. — Вестн. МГУ. Сер. хим., 1975, 16, с. 620.
469. Каганович Ю. Я., Мищенко К. П. — Журн. общ.
хим., 1951, 21, с. 28.
470. Каганович Ю. Я., Мищенко К. П. — Докл. АН
СССР, 1952, 87, с. 89.
471. Казарновская Л. И., Казарновский Ю- А. —
Журн. физ. хим., 1951, 25, с. 293.
472. Казарновский И. А., Райтсштейн С. И. — Журн.
физ. хим., 1947, 21, с. 245.
473. Казенас Е. К. — Автореф. дис. . . . канд. техн.
наук. М.: Институт металлург, им. А. А. Бай-
кова АН СССР, 1968.
474. Казенас Е. К., Цветков Ю. В. — ЗКурн. физ. хим.,
1967, 41, с. 3112.
475. Казенас Е. К., Цветков Ю. В. — Журн. неорган,
хим., 1969, 14, с. 11.
476. Казенас Е. К., Чижиков Д. М. — В кн.: Давление
. и состав пара над окислами химических элементов.
М,: Наука, 1976, с. 201.
477. Казенас Е. К., Чижиков Д. М. — В кн.: Давление
и состав пара над окислами химических элементов.
М.: Наука, 1976, с. 209.
478. Казенас Е. К., Чижиков Д. М-, Цветков Ю. В. —
В кн.: Исследование процессов в металлургии
цветных и редких металлов. М.: Наука, 1969, с. 19.
479. Калишевич Г. И., Гельд П. В., Кренцис Р. П
Журн. физ. хим., 1965, 39, с. 2999.
480. Капель О. М., Крафтмахер Я. Л. —Физика
твердого тела, 1966, 8, с. 283.
481. Кантор П. Б., Фомичев Е. Н. — В кн.: Тепло*
физические свойства твердых тел при высоких
температурах. М.: Комитет станд. мер и измер.
приборов при Сов. Мин. СССР, 1969, с. 406.
482. Капустинский А. Ф., Дракин С. И. — Журн. физ,
хим., 1952, 26, с. 581.
483. Капустинский А. Ф., Майер А. И., Дракин
С. И. — Труды Моск. хим.-технол. ин-та им.
Д. И. Менделеева, 1962, вып. 38, с. 10.
484. Капустинский А. Ф., Стаханова М. С. — Труды
Моск. хим.-технол. ин-та им. Д. И. Менделеева,
1956, вып. 22, с. 21.
485. Карапетянц М. X. — Методы сравнительного
расчета физико-химических свойств. М.: Наука,
1965.
486. Карасев Р. А., Поляков А. Ю., Самарин А. М. —
Докл. АН СССР, 1952, 85, с. 1313.
487. Карелин В. В., Несмеянов Ан. П., Приселков
Ю. А. — Докл. АН СССР, 1962, 144, с. 352.
488. Карелин В. В., Несмеянов Ан. Н., Приселков
Ю. А. — Изв. АН СССР. Отд. техн. наук.
Металлургия и топливо, 1962, № 5, с. 117.
489. Карелин В. В., Несмеянов Ан. П., Приселков Ю. А.,
' Чжоу-Кунъ-ин. — Вестн. МГУ. Сер. хим., 1962,
№ 2, с. 40.
490. Карнаухова И. М., Каражанова Г. И., Лужниц-
кий В. А., Маслов И. И., Терентъева 3. П. —
В кн.: Методы и применение нейтроноактивирую-
щего анализа. Рига: Зинатне, 1969, с. 167.
491. Карпов С. В., Шултин А. А. — Физика твердого
тела, 1975, 17, с. 2868.
492. Кац Дж., Рабинович Е. Химия урана. М.: Изд-во
иностр. лит., ч. 1, 1954.
493. Кац С. А. — Автореф. дис. . . . канд. техн. наук.
М.: ИВТАН, 1978.
494. Кац С. А., Чеховской В. Я. — Журн. физ. хим.,
1980, 72, с. 768.
495. Кватер Г. С, Мейстер Т. Г. — Вестн. ЛГУ.
Сер. физ. и хим. 1952, вып. 7, № 9, с. 137.
496. Канисарин М. М. — Автореф. дис. . . . канд. техн
наук. М.: ИВТАН, 1974.
497. Керопян В. В., Казаченко Е. П., Бергман А. Г. —
Журн. неорган, хим., 1970, 15, с. 3320.
498. Кесслер Ю. М., Горбанев А. И. — Изв. Вост.
филиала АН СССР, 1957, с. 75.
499. Кесслер Ю. М., Емелин В. П. — ВИНИТИ.
Деп. № 3079-71. М., 1971.
500. Киреев В. А. Методы практических расчетов в
термодинамике химических реакций. 2-е изд. М.:
Химия, 1975.
501. Кириллин В. А., Шейндлин А. Е., Чеховской
В. Я. — Докл. АН СССР, 1961, 139, с. 645.
502. Кириллин В. А., Шейндлин А. Е., Чеховской
В. Я. — Докл. АН СССР, 1962, 142, с. 1323.
503. Кириллин В. А., Шейндлин А. Е., Чеховской
В. Я. — Теплоэнергетика, 1962, 9, с. 63.
504. Кириллин В. А., Шейндлин А. Е., Чеховской В. Я.
— В кн.: Исследования при высоких
температурах/ Пер. с англ. Под ред. В. А. Кириллина
и А. Е. Шейндлина. М.: Наука, 1967, с. 258.
505. Кириллин В. А., Шейндлин А. Е., Чеховской
В. Я., Жукова И. А. — Теплофизика высоких
температур, 1965, 3, с. 395.
506. Кириллин В. А., Шейндлин А. Е., Чеховской В. Я.,
Жукова И. А. — Теплофизика высоких
температур, 1965, 3, с. 860.
507. Кириллин В. А., Шейндлин А. Е., Чеховской В. Я.,
Жукова И. А. — Теплофизика высоких
температур, 1967, 5, с. 1124.
531
Литература
508. Кириллин В. А., ШейндлинА. Е., Чеховской В. Я.,
Жукова И. А., Тарасов В. Д. — Теплофизика
высоких температур, 1966, 4, с. 878.
509. КириллинВ. А., ШейндлинА. Е., Чеховской В. Я.,
Петров В. А. — Докл. АН СССР, 1962, 144, с. 390.
510. КириллинВ. А., ШейндлинА. Е., Чеховской В. Я.,
Петров В. А. — Журн. физ. хим., 1963, 37,
с. 2249.
511. КириллинВ. А., ШейндлинА. Е., Чеховской В. Я.,
Петров В. А. — В кн.: Труды III Всесоюз. научно-
техн. конф. по термодинамике, Ленинград, 1968:
Сборник докл. секции «Теплофизические свойства
веществ». Л.: Наука, 1969, с. 80.
512. Кириллов П. А., Грачев И. С. — Инж.-физ. журн.,
1959, 2, с. 3.
513. Кирияненко А. А. Исследование теплофизических
свойств. Новосибирск: Наука, 1970.
514. Кисилева Е. В. — Научн. докл. высш. школы.
Хим. и хим. технол., 1958, № 3, с- 405.
515. Китайгородский А. И., Попова Т. А., Ботвин-
кин О. К. — Журн. физ. хим., 1933, 4, с. 380.
516. Китайгородский А. И., Попова Т. А., Ботвин-
кин О. К. — Изв. сектора физ.-хим. анализа
ИОНХ АН СССР, 1933, 6, с. 135.
517. Клименко Ю. А., Чеховской В. Я., Устинов С. В. —
В кн.: Восьмая Всесоюз. конф. по калориметрии
и хим. термодинамике, Иваново, 1979. Тезисы
докладов. Иваново: Ивановский хим.-технол. ин-т,
1979, с. 400.
518. Климов В. Л., Кочина Е. А., Краснов К. С,
Морозов Е. В., Данилова Т. Г. — Изв. вузов. Хим.
и хим. технол., 1970, 13, с. 1104.
519. Клопов В. И., Афанасьев В. А., Крестов Г. А. —
Труды Ивановск. хим.-технол. ин-та, 1973, вып. 16,
с. 98.
520. Клопов В. И., Афанасьев В. А., Пирогов А. И.,
Крестов Г. А. — Изв. вузов. Хим. и хим. технол.,
1973, 16, с. 1181.
521. Клопов В. И., Пирогов А. И., Крестов Г. А. —
Журн. структур, хим., 1972, 13, с. 141.
522. Клочко М. А. — Журн. общ. хим., 1933, 3, с. 1026.
523. Ковалёв Ф. В., Иоффе В. М., Карцев В. Е. —
Журн. неорган, хим., 1970, 15, с. 1966.
524. Ковба Л. М., Видавский Л.} М., Лавут Э. Г. —
Журн. структур, хим., 1963, 4, с. 627.
525. Ковтун Г. П., Круглых А. А. — Изв. АН СССР.
Металлы, 1967, № 2, с. 222.
526. Ковтун Г. П., Круглых А. А., Павлов В. С. —
Укр. физ. журн., 1962, 7, с. 336.
527. Кожевников И. Г. — Теплофизика высоких
температур, 1974, 12, с. 896.
528. Кожина И. И., Быков В. А., Малинии Р. В.,
Ядринцев В. Б. — Вестн. ЛГУ. Сер. физ. и хим.,
1973, вып. 2, № 10, с. 73.
529. Кожина И. И., Ширяева Л. В. — Радиохимия,
1974, 16, с. 360.
530. Кокот И. Ф., Мохосоев М. В., Кисель Н. Г. —
Изв. вузов. Цвет, мет., 1970, № 1, с. 87.
531. Колесник В. В., Жиров А. И., Дунаев К. М., Спи-
цын В. И. — Журн. неорган, хим., 1980, 25,
с. 1329.
532. Колесников Б. Я. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1971.
533. Колесов В. Я., Скуратов С. М. — Журн. неорган,
хим., 1961, 6, с. 1741.
534. Колесов В. П., Скуратов С. М., Зайкин И. Д. —
Журн. неорган, хим., 1959, 4, с. 1237.
535. Колосов Е. Я., Туваева Т. Я., Сидоров Л. Я. —
ВИНИТИ. Деп. № 2941-74. М., 1974.
536. Колосов Е. Н., Шольц В. Б., Сидоров Л. Н. —
Журн. физ. хим., 1974, 48, с. 2199.
537. Колчин О. П., Сумароков Н. В. — Атомная
энергия, 1961, 10, с. 168.
538. Комаров С. А., Ежов Ю. С. — Журн. структур.
хим., 1975, 16, с. 899.
539. Кондратьев Я. С, Парфентьева И. Ф. — В кн.:
Тепло-массоперенос в одно—двухфазных средах.
М.: Наука, 1971, с. 19.
540. Копытин Л. М., Гагаринский Ю. В. — Атомная
энергия, 1961,' 10, с. 238.
541. Коренев Ю. М., Новоселова А. В., Украинский
Ю. М. — Журн. неорган, хим., 1970, 15, с. 1970.
542. Корнилов А. Н. — Журн. физ. хим., 1967, 41,
с. 3096.
5-53. Корнилов А. Я., Леонидов В. Я., Скуратов С. М.
— Докл. АН СССР, 1962, 144, с. 355.
544. Корнилов А. Н., Леонидов В. Я., Скуратов
С. М. — Журн. физ. хим., 1964, 38, с. 2008.
545. Корнилов А. Я., Леонидов В. Я., Скуратов
С. М. — Журн. физ. хим., 1964, 38, с. 2013.
546. Корнилов А. Я., Ушакова И. М. —¦ Докл.
АН СССР, 1971, 200, с. 1382.
547. Корнилов А. Н., Ушакова И. М. — Журн. физ.
хим., 1976, 50, с. 2951.
548. Корнилов А. Я., Ушакова И. М., Скуратов
С. М. — Журн. физ. хим., 1967,41, с. 200.
549. Корнилов И. П., Глазова В. В. — Изв. АН СССР.
Металлы, 1965, № 1, с. 189.
549а. Корнилов И. И., Глазова В. В. Взаимодействие
тугоплавких металлов переходных групп с
кислородом. М.: Наука, 1967.
550. Корнфельд М. Я., Чудинов А. А. —¦ Журн. экспе-
рим. и теор. физ., 1957, 33, с. 33.
551. Коробов М. В., Сидоров Л. П. — ВИНИТИ.
Деп. № 916-78. М., 1978.
552. Королев В. П., Егоров Г. И., Кухаренко В. А.,
Крестов Г. И. — Журн. физ. хим., 1980, 54,
с. 248.
553. Королев В. П., Колкер А. М., Крестов Г. А. —
Журн. физ. хим., 1977, 51, с. 751.
554. Королева Т. И., Афанасьев Ю. А., Дурасов
В. Б. — Изв. АН СССР. Сер. хим., 1971, № 3,
с. 480.
555. Коротаева Л. Т., Ремизов В. Г., Дударева
А. Г. — Журн. неорган, хим., 1975, 20, с. 2197.
556. Коршунов Ю. С, Ветчинин С. П., Сенченков А. П.,
Асиновский д. И. — Теплофизика высоких
температур, 1975, 13, с. 517.
557. Коршунов Ю. С., Сенченков А. Я., Асиновский
Э. П., Купавин А. —¦ Теплофизика высоких
температур, 1970, 8, с. 1288.
558. Коряжкин В. А., Татевский В. М., Харитонов
Ю. А. — Вестн. МГУ. Сер. хим., 1961, № 1, с. 48.
559. Костиков В. И., МаурахМ. А., Митин Б. С,
Пеньков И. А., Свердлов П. М. — В кн.:
Высокотемпературные материалы: Сб. 49.
Московский ин-т стали и сплавов. М.: Металлургия, 1968
с. 106.
560. Кострюков В. П. — Журн. физ. хим., 1961, 35,
с. 1759.
561. Костюк Б. Г., Воробьев А. Ф. — Журн. физ. хим.,
1972, 46, с. 2445.
562. Котляр А. А., Андреева Р. Т. — В кн.: Сборник
материалов по вакуумной технике. М.; Л.: Гос-
энергоиздат, 1960, вып. 23, с. 51.
563. Кочержинский Ю. А., Кобзенко Г. Ф., Пан В. М.,
Свириденко В. К., Юпко Л. М. — В кн.: Вопросы
физики металлов и металловедения: Сборник.
Киев: Наукова думка, 1963, вып. 17, с. 209.
532
Литература
564. Кочубей Л. А., Маргулис Е. В., Шокорёв М. М., 595.
Портеелова Я. В. — Журн. неорган, хим., 1978,
23, с. 2275. 596.
565. Краев О. А. — Теплофизика высоких температур,
1967, 5, с. 817. 597.
566. Краснов К. С. — Журн. неорган, хим., 1957, 2,
с. 1725.
567. Краснов К. С. — Журн. неорган, хим., 1959, 4, 598.
с. 530.
568. Краснов К. С, Климов В. Л., Морозов Е. В.,
Кочина 9. А. — Изв. вузов. Хим. и хим. технол., 599.
1966, 9, с. 205.
569. Краснов К. С, Соломоник В. Г., Морозов Е. В. — 600.
Теплофизика высоких температур, 1972, 10,
с. 760. 601.
570. Краснов К. С, Филиппенко Н. В. — Изв. вузов.
Хим. и хим. технол., 1971, 14, с. 1435. 602.
571. Крафтмахер Я. А. — Журн. прикл. мех. и техн.
физ., 1962, с. 176.
572. Крафтмахер Я. А. — Журн. прикл. мех. и техн. 603.
физ., 1963, с. 158.
573. Крафтмахер Я. А. — Физика твердого тела, 1963, 604.
5, с. 950.
574. Крафтмахер Я. А. — Физика твердого тела, 1964, 605.
6, с. 503.
575. Крафтмахер Я. А. — В кн.: Исследования при 605а
высоких температурах: Сборник. Новосибирск:
Наука, 1966, с. 5. 606.
576. Крафтмахер Я. А., Стрелков П. Г. — Физика
твердого тела, 1962, 4, с. 2271. 607.
577. Кренцис Р. П., Гельд П. В., Серебренников
Н. Н. — Изв. вузов. Черн. мет., 1960, № 12, с. 5. 608.
578. Крестов Г. А. Термохимия соединений
редкоземельных и актинидных элементов. М.: Атомиздат, 609.
1972.
579. Крестов Г. А., Абросимов В. К. — Изв. вузов. 610.
Хим. и хим. технол., 1967, 10, с. 1005.
580. Крестов Г. А., Абросимов В. К., Макаров Г. Н. —¦ 611.
Изв. вузов. Хим. и хим. технол., 1974, 17, с. 1867.
581. Крестов Г. А., Егорова И. В. — Изв. вузов. Хим.
и хим. технол., 1967, 10, с. 750. 612.
582. Крестов Г. А., Егорова И. В. — Теор. и экспер.
хим., 1967, 3, с. 128. 613.
583. Крестов Г. А., Зверев В. А., Кротов В. С-—
Изв. вузов. Хим. и хим. технол., 1972, 15, 1414. 614.
584. Крестов Г. А., Куракина Г. И. — Журн. неорган,
хим., 1974, 19, с. 886.
585. Крестов Г. А., Харитонов Е. Б. — ВИНИТИ. 615.
Деп. № 3081-74. М., 1974.
586. Крестовников А. Н., Каретников Г. А. — Легкие
металлы, 1934, 3, № 4, с. 29.
587. Круглое А. Я., Панарина О. М., Тринова Л. В. — 616.
ОНИИТЭХИМ. Деп. № 3253-79. Черкассы,
1979. 617.
588. Круглых А. А., Лавлов В. С. — Изв. АН СССР.
Металлы, 1966, № 1, с. 178. 618.
589. Круглых А. А., Павлов В. С, Тихвинский Г. П. —
Укр. физ. журн., 1964, 9, с. 214. 619.
590. Ксенофонтова Р. Ф., Васильева И. А.,
Герасимов Я. И. — Докл. АН СССР, 1962, 143, с. 1105.
591. КудинЛ. С. — Автореф. дис. . . . канд. хим. наук. 620.
Иваново: Ивановский хим.-технол. ин-т, 1974.
592. Кудин Л. С, Гусаров А. В., Горохов Л. Я., Крас- 621.
нов К. С. — Теплофизика высоких температур,
1977, 15, с. 505. 622.
593. Кудрявцев Н. С, Иванов Л. И. — Приборы и
техника эксперимента, 1968, № 4, с. 163.
594. Кудрявцев Я. С, Иванов Л. И. — Заводская лабо- 623.
ратория, 1969, 35, с. 368.
Кузнецов А. К., Келер Э. К., Фанъ Фу-Кан. —
Журн. прикл. хим., 1965, 38, с. 233.
Кузнецов Л. М., Цвигунов А. Я. — Радиохимия,
1976, 18, с. 414.
Кузнецова Г. П., Ловецкая Г. А., Преснякова В. М.,
Степин Б. Д. — Журн. физ. хим., 1974, 48,
с. 2141.
Кузнецова Г. П., Ловецкая Г. А., Преснякова В. М.,
Степин Б. Д., Смирнова Т. Г. — Журн. неорган,
хим., 1975, 20, с. 2546.
Кузнецова Г. П., Ловецкая Г. А., Степин Б. Д. —
ВИНИТИ. Деп. № 584-75. М., 1975.
Кузьмин А. А., Моисеева Т. Г. — Физ. металлов
и металловедение, 1964, 18, с. 925,
Кузьмин А. А., Палатник Л. С. — Физ. металлов
и металловедение, 1962, 14, с. 795.
Кулагин В. И., Борисов М. И., Антонов А. А.,
Маслов П. Г., Степанов А. С. — ВИНИТИ.
Деп. № 1677-70. М., 1970.
Куликов В. А., Угаров В. В., Рамбиди Я. Г. —
Журн. структур, хим., 1982, 23, с. 184.
Куликов В. А., Угаров В. В., Рамбиди Я. Г. —
Журн. структур, хим., 1982, 23, с. 182.
Куликов В. А., Угаров В. В., Рамбиди Я. Г. —
Журн. структур, хим., 1981, 22, с. 196.
. Куликов В. А., Угаров В. В., Рамбиди Я. Г. —
Журн. структурн. хим., 1981, 22, с. 168.
Куликов В. А., Угаров В. В., Рамбиди Я. Г. —
Журн. структур, хим., 1981, 22, с. 183.
Кулюпин Ю. А., Яценко А. Ф. — Физика
твердого тела, 1965, 5, с. 2756.
Кусенко Ф. Г., Гельд Л. В. — Труды Уральск,
политехи, ин-та, 1959, 92, с. 121.
Кусенко Ф. Г., Гельд Л. В. — Журн. общ. хим.,
1960, 30, с. 3847.
Кусенко Ф. Г., Гельд П. В. — Изв. СО АН СССР,
1960, № 2, с. 46.
Кусенко Ф. Г., Гельд Л. В. — В кн.: Физ.-хим.
основы производства стали: Сборник статей. М.:
Изд-во АН СССР, 1961, с. 41.
Лаврентьев В. И., Герасимов Я. И., Резу хина
Т. Я. — Докл. АН СССР, 1961, 136, с. 1372.
Лазарева Л.С, Кантор П. В., Кандыба В. В. —
Физ. металлов и металловедение, 1961, 11, с. 628.
Латимер В. М. Окислительные состояния
элементов и их потенциалы в водных растворах. М.:
Изд-во иностр. лит., 1954.
Латыев Л. Я., Петров В. А., Чеховской В. Я.,
Шестаков Е. Я. Излучательные свойства твердых
материалов: Справочник / Под ред. А. Е. Шейнд-
лина. М.: Энергия, 1974.
Лащенко П. Н., Компанский Д. И. — Журн.
прикл. хим., 1935, 8, с. 628.
Лебедев С. В. — Журн. эксперим. и теор. физ.,
1957, 32, с. 199.
Лебедев С. В., Можаров Г. И. — Теплофизика
высоких температур, 1977, 15, с. 53.
Лебедев С. В., Савватимский А. И., Смирнов
Ю. Б. — Теплофизика высоких температур, 1971,
9, с. 635.
Лебедев С. Я., Стависский Ю. Я. — Приборы и
техн. эксперим., 1962, № 1, с. 142.
Левальт-Езерский М. — Журн. Рус. физ.-хим.
об-ва. Химия, 1912, 44, с. 665.
Левина М. Е., Новоселова А. В., Симонов Ю. П. —
Вестн. МГУ. Сер. мат., мех., астрон., физ., хим.,
1956, № 1, с. 239.
Левицкий В. А., Ченцов В. Я., Сколис 10. Я.,
Клименко А. Я., Марин В. П., Меньшенин
533
Литература
Ю. В. — Изв. АН СССР. Неорган, матер., 1980,
16, с. 949.
624. Лесных Д. С, Бергман А. Г. — Журн. общ. хим.,
1953, 23, с. 894.
625. Лесных Д. С, Гарматина Е. П., Шилина Е. В. —
Журн. неорган, хим., 1968, 13, с. 3130.
626. Лесных Д.' С, Черняховская С. А. -— Журн.
неорган, хим., 1967, 12, с. 3178.
627. Лесных Д. С, Эйхенбаум И. Г., Черняховская
С. А. — Журн. неорган, хим., 1970, 15, с. 824.
628. Лившиц Л. Д., Рябинин Ю. Н., Ларионов Л. В. —•
Журн. техн. физ., 1969, 39, с. 1115.
629. Линтон Е. А. Сверхпроводимость / Пер. с англ.
2-е изд., исправ. и доп. М.: Мир, 1971. 262 с.
630. Липилина И. И., Самойлов О. Я. — Докл.
АН СССР, 1954, 98, с. 99.
631. ЛитвиненкоВ. Ф., Муратов В. Б., Момот Г. Г. —
В кн.: Структурные исследования новых
материалов. Киев: Ин-т проблем металловедения, 1979,
с. 173.
632. Лифшиц Г. М. — Журн. общ. хим., 1956, 26,
с. 20.
633. Лопато Л. М., Шевченко А. В., Кучевский А. Е.,
Тресвятский С. Г. — Изв. АН СССР. Неорган,
матер., 1974, 10, с. 1481.
634. Лужная Н. П., Верещетина И. П. — Журн.
прикл.|хим., 1949, 22, с. 952.
635. Лукашенко 9. Е., Коробейников А. П., Хомайко
И. А.— Журн. физ. хим., 1970, 44, с. 341.
636. Лукашенко Э. Е., Шаповалов В. С, Кислое В. Е. —
ВИНИТИ. Деп. № 6995-73. М., 1973.
637. Ляшенко В. С. —Металлургия, 1935, 10, № И,
с. 85.
638. Макаренко И. Н. — Автореф. дис. . . . канд.
физ.-мат. наук. М.: Ин-т кристаллографии
АН СССР, 1971.
639. Макаренко И. #., Иванов В. А., Стишов С. М. —
Письма в ЖЭТФ, 1973, 18, с. 320.
640. Макаренко И. Д., Труханова Л. Д., Филиппов
Л. П. — Теплофизика высоких температур, 1970,
8, с. 445.
641. Макаренко И. Н., Труханова Л. Н., Филиппов
Л. П. — Теплофизика высоких температур, 1970,
8,?с. 667.
642. Макаров А. В. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1972.
643. Макаров Г. Н., Абросимов В. К., Крестов Г. А. —
Журн. физ. хим., 1974, 48, с. 2157.
644. Макаров Л. Л., Алой А. С, Ступин Д. Ю. —
Радиохимия, 1969, 11, с. 116.
645. Макаров Л. Л.,\Ступин Д. Ю. — Журн. физ. хим.,
1968, 42, с.4508.
646. Макаров С. 3., Шульгина М. П. — Изв. АН СССР.
Отд. хим. наук, 1940, № 5, с. 691.
647. Малышеве.В. — Докл. АН СССР, 1971,197, с. 73.
648. Мальцев А. А. — Автореф. дис. . . . докт. хим.
наук. М.: МГУ, 1965.
649. МалъцевУВ. А., Гагаринский Ю. В., Попов М. М. —
Журн. неорган, хим., 1960, 5, с. 228.
650. Мальцев В. Г., Бухалова Г. А. — Изв. вузов.
Хим. и хим. технол., 1966, 9, с. 151.
651. Мамедова\3. Д., Самедов Ф. Р., Рза-Заде П. Ф. —
Азерб. хим. журн., 1969, № 3, с. 82.
652. Мардыкин И. П. — В кн.: Физико-механические
и теплофизические свойства металлов. М.: Наука,
1976, с. 105.
653. Мардыкин И. П., Кашин В. И., Сбитнев П. Л. —
Изв. АН СССР. Металлы, 1973, № 6, с. 77.
654. Марков Б. Ф., Панченко И. Д. —¦ Журн. общ. хим.,
1955, 25, с. 2038.
655. Марков Б. Ф., Панченко И. Д., Костенко Т. Г. —
Укр. хим. журн., 1956, 22, с. 287.
656. Марков Б. Ф., Тишура Т. А. — Укр. хим. журн.,
1973, 39, с. 757.
657. Марков Б. Ф., Тишура Т. А., Бударина А. Н. —¦
Укр, хим. журн., 1972, 38, с. 823.
658. Марков Б. Ф., Тишура Т. А., Бударина А. Н. —
Укр. хим. журн., 1974, 40, с. 242.
659. Марков С. 3., Шульгина М. П. — Изв. АН СССР.
Отд. хим. наук., 1940, № 5, с. 691.
660. Мармер 9. Н., Жуков В. В., Стуканов А. Ф. —
Теплофизика высоких температур, 1965, 3, с. 771.
661. Мартынкевич Г. М., Молярова Г. В. — Журн.
физ. хим., 1964, 38, с. 1283.
662. Мартынюп М. М. — Журн. физ. хим., 1977, 51,
с. 1199.
663. Мартынюк М. М., Каримходжаев И. — Журн.
физ. хим., 1974, 48, с. 1243.
664. Мартынюк М. М., Каримходжаев И., Цапков
В. И. — Журн. техн. физ., 1974, 44, с. 2367.
665. Мартынюк М. М., Цапков В. И. — Журн. физ.
хим., 1973, 47, е. 1308.
666. Мартынюк М. М., Цапков В. И. — Изв. АН СССР.
Металлы, 1974, № 2, с. 181.
667. Мартынюк М. М., Цапков В. И., Пантелейчук
О. Г., Каримходжаев И. — В кн.: Исследование
физических свойств металлов методом
импульсного нагрева. М.: Ун-т дружбы народов им. П. Лу-
мумбы, каф. физ., 1972.
668. Матейко 3. А., Бухалова Г. А. — Журн. неорган.
хим., 1959, 4, с. 1649.
669. Матейко 3. А., Ягубьян Е. С, Бухалова Г. А. —
Журн. неорган, хим., 1966, 11, с. 2405.
670. Матизен 9. В., Ефремова Р. И. — Теплофизика
высоких температур, 1982 (в печати).
671. Матросова В. А., Бергман А. Г. — Журн.
неорган, хим., 1969, 14, с. 2279.
672. Матюшин Ю. Д., Конькова Т. С,
Мирошниченко Е. А., Ларионов Б. П., Воробьев А. Б.,
Лебедев Ю. А. — В кн.: Седьмая Всесоюз. конф.
по калориметрии, 31 янв.—-3 февр. 1977, Москва,
(расширенные тезисы докл.). Черноголовка, 1977,
с. 503.
673. May pax M. А., Митин Б. С. — Жидкие
тугоплавкие окислы. М.: Металлургия, 1979.
674. Маурах М. А., Мишук В. Я., Свердлов Г. М. —
Заводская лаборатория, 1967, № 4, с. 515.
675. Машовец В. П., Юдин Б. Ф. — Изв. вузов. Цвет.
мет., 1962, № 5, с. 95.
676. Медведев Б. С, Проценко П. И. — Журн.
неорган, хим., 1964, 9, с. 2017.
677. Медведев В. А., Ефимов М. Е. — Журн. физ. хим.,
1975, 49, с. 1324.
678. Микульская Г. Ю. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1972.
679. Михайленко И. Е., Спицин В. И. — Докл.
АН СССР, 1963, 148, с. 613.
680. Миллер И. И., Суриков В. П., Ярош 9. М.,
Данилов С. В., Калистратова Л. Ф. — ВИНИТИ.
Деп. № 682-80. М., 1980.
681. Миллер И. И., Суриков В. И., Ярош Э. М.,
Данилов С. В., Калистратова Л. Ф. — Инж.-физ.
журн., 1980, 39, с. 1115.
682. Митькина Е. А. — Атомная энергия, 1959, 7,
с. 163.
683. Мищенко К. П., Каганович Ю. Я. — Журн.
прикл. хим., 1949, 22, с. 1078.
684. Мищенко К. П., Полторацкий Г. М. — В кн.:
Вопросы термодинамики и строения водн. и невод-
534
Литература
ных растворов электролитов. Л.: Химия, 1968,
с. 35.
^685. Мищенко К. П., Полторацкий Г. М. — В кн.:
Термодинамика и строение водн. и неводных
растворов электролитов. 2-е изд., перераб. и доп. Л.:
Химия, 1976, с. 47.
686. Мищенко К. П., Пронина М. 3. — Журн. общ.
хим., 1936, 6, с. 85.
687. Мищенко К. П., Сухотин А. М. —Докл.
АН СССР, 1954, 98, с. 103.
-688. Мищенко К. П., Шпигель Л. П. —Журн. общ.
хим., 1967, 37, с. 2145.
689. Мищенко К. П., Яковлев И. Ф. — Журн. общ.
хим., 1959, 29, с. 1761.
690. Моисеев Г. К., Ватолин Н. 4. —ВИНИТИ.
Дел. № 596-77. М., 1977.
€91. Моисеев Г. К., Ватолин Н. А. — Изв. АН СССР.
Неорган, матер., 1978, 14, с. 1462.
€92. Мокеров В. Г., Бегишев А. Р., Игнатьев А. С. —
Физика твердого тела, 1979, 21, с. 1482.
693. Мордовии О. А., Тимофеева Н. И., Дроздова
Л. Н. — Изв. АН СССР. Неорган, матер., 1967,
3, с. 187.
694. Морозов А. И., Морозов И. С. — Журн. неорган,
хим., 1973, 18, с. 989.
<695. Морозов А. И., Морозов И. С. — В кн.: Химия
парообразных неорганических соединений и
процессов парообразования. Материалы Всесоюзной
конференции. Минск. 24—26 мая 1973. Минск,
1973, с. 121.
<696. Морозов Е. В., Курникова Л. А., Гиричева Н. И.,
Краснов К. С, Данилова Т. Г. — ВИНИТИ.
Деп. № 4544-72. М., 1972.
697. Морозова М. П., Гецкина Л. Л. — Вестн. ЛГУ.
Сер. физ. и хим., 1959, вып. 4, № 22, с. 128.
¦698. Морозова М. П., Гецкина Л. Л. — Журн. общ.
хим., 1959, 29, с. 1049.
699. Морозова М. П., Егер Г. — Журн. общ. хим.,
1960, 30, с. 3514.
700. Морозова М. П., Конопелька М. В., Пинчук
И. В. — Вестн. ЛГУ. Сер. физ. и хим., 1964,
вып. 4, № 22, с. 109.
701. Морозова М. П., Столярова Т. А. — Журн. общ.
хим., 1960, 30, с. 3848.
702. Морозова Н. Ю., Селиванова Н. М., Хожаннова
Т. И. — Журн. неорган, хим., 1978, 23, с. 341.
703. Мочаладзе Т. Е., Чичанидзе Г. Д., Пирцхалава
Р. Н. — Сообщ. АН ГрузССР, 1973, 71, № 1,
с. 109.
704. Мурадова О. Н., Фишман И. С. — Журн. прикл.
спектроскопии, 1970, 12, с. 971.
705. Мясникова Т. П., Евсеева Р. Я. — В кн.:
Кристаллизация и фазовые превращения. Минск: Наука
и техника, 1971, с. 41.
706. Мясникова Т. П., Яценко А. Ф. — Физика
твердого тела, 1966, 8, с. 2814.
707. Нарышкин И. И. — Журн. физ. хим., 1939, 13,
с. 528.
708. Наумкин О. П., Терехова В. Ф., Савицкий Е. М. —
В кн.: Вопросы теории и применения
редкоземельных металлов. М.: Наука, 1964, с. 71.
709. Наумов Г. В., Рыженко Б. Н., Ходаковский И- Л.
Справочник термодинамических величин для
геологов. М.: Атомиздат, 1971.
710. Недумов Н. А., Григорович В. К. — В кн.:
Высокотемпературные неорганические соединения:
Сборник ин-та проблем металловедения АН СССР.
Киев: Наукова думка, 1965, с. 25.
711. Неймарк Б. Е., Беляева П. Е,, Бродский Б. Р.,
Воронин Л. К., Корытииа С. Ф., Меркулъев
A. Н. — В кн.: Теплофизические свойства твердых
веществ: Сборник / Под ред. Г. В. Самсонова. М.:
Наука, 1971, с. 63.
712. Некрасова Н. П., Обломов Е. Н., Голованова В. П.,
Безносиков А. В.—Атомная энергия, 1967, 22,
с. 293.
713. Немухин А. В., Степанов Н. Ф. — Журн.
структур, хим., 1978, 19, с. 771.
714. Несмеянов Ан. Н. — Давление пара химических
элементов. М.: Изд-во АН СССР, 1961.
715. Несмеянов Ан. Н., Белых Л. П. — Журн. физ.
хим., 1960, 34, с. 841.
716. Несмеянов Ан. Н.,ДёДык Май — Докл. АН СССР.
1960, 131, с. 1383.
717. Несмеянов Ан. Н., Приселков Ю. А., Карелин-
B. В. — In: Thermodynamics of Nuclear Materials.
Proc. Symp. Vienna, 1962. Vienna: IAEA, 1963,
p. 667.
718. Несмеянов Ан. Н., Сазонов Л. А. — Журн.
неорган, хим., 1957, 2, с. 946.
719. Несмеянов Ан. Н., Сазонов Л. А. — Журн.
неорган, хим., 1957, 2, с. 1183.
720. Несмеянов Ан. Н., Сазонов Л. А. — Журн.
неорган, хим., 1959, 4, с. 231.
721. Несмеянов Ан. Н., Сазонов Л. А. — Журн.
неорган, хим., 1960, 5, с. 519.
722. Никитин В. С, Мальцев А. А. — Вестн. МГУ.
Сер. хим., 1966, 7, с. 28.
723. Никитин В. С, Мальцев А. А. — Вестн. МГУ.
Сер. хим., 1969, 10, с. 109.
724. Никитин М. И., Сидоров Л. Н. — В кн.: V Все-
союз. симпозиум по химии неорганических
фторидов, Днепропетровск, 1978. Тезисы докладов.
М.: Наука, 1978, с. 199.
725. Никитин М. И., Скокан Е. В., Сорокин И. Д.,
Сидоров Л. Н. — Докл. АН СССР, 1979, 247,
с. 151.
726. Николаев Г. И., Немец A.M. — Журн. прикл.
спектроскопии, 1970, 13, с. 695.
727. Новиков Г. И., Орехова С. Е. — В кн.: Химия
и хим. технология: Сборник. Минск: Высшая
школа, 1974, вып. 7, с. 12.
728. Новиков И. И., Александров В. В., Борзяк А. Н.,
Боярский С. В., Гончаров В. Г., Орлова Т. И.,
Рощупкин В. В., Семашко Н. А. — В кн.:
Конструкционные и жаропрочные материалы для
новой техники. М.: Наука, 1978, с. 166.
729. Новиков И. И., Груздев В. А., Краев О. А.,
Одинцов А. А., Рощупкин В. В. — Теплофизика
высоких температур, 1969, 7, с. 71.
730. Новиков И. И., Костюков В. И., Филиппов
Л. П. — Изв. АН СССР. Металлы, 1978, 4, с. 89.
731. Новиков И. П., Мардыкин И. П. — Атомная
энергия, 1974, 37, с. 348.
732. Новиков И. П., Мардыкин И. П. — Теплофизика
высоких температур, 1975, 13, с. 318.
733. Новиков И. И., Рощупкин В. В. — В кн.: Труды
III Всесоюз. научно-техн. конференции по
термодинамике, Ленинград, 1968: Сборник докладов
секции «Теплофизические свойства веществ»'. Л.:
Наука, 1969, с. 87.
734. Новиков И. И., Рощупкин В. В., Семашко П. А.,
Фордеева Л. К. — Инж.-физ. журн., 1980, 39,
с. 1013.
735. Новиков И. И., Рощупкин В. В., Фордеева Л. К. —
Теплофизика высоких температур, 1976, 14, с. 75.
736. Новоселов Н. П., Рябченко О. И., Мищенко К. П.,
Клюева М. Л. — Журн. прикл. хим., 1975, 48.
с. 2296.
535
Литература
737. Новоселова А. В., Коренев Ю.М., Симанов Ю. П. — 768.
Докл. АН СССР, 1961, 139, с. 892.
738. Новоселова А. В., Муратов Ф. Ш., Решетникова 769.
Л. П., Гордеев И. В. — Вестн. МГУ. Сер. хим.,
1958, № 6, с. 181. 770.
739. Новоселова А. В., Симанов Ю. П., Ярембаш Е. И. —
Журн. физ. хим., 1952, 26, с. 1244. 771.
740. Новохатский И. А., Ленев Л. М. — Журн.
неорган, хим., 1966, 11, с. 2014. 772.
741. Носова Т. А., Самойлов О. Я. — Журн. структур,
хим., 1964, 5, с. 363. 773.
742. Овечкин Е. К., Шевцова Л. Н. — Журн. неорган,
хим., 1973, 18, с. 1084. 774.
742а. Основные свойства неорганических фторидов.
Справочник / Под ред. Н. П. Галкина. М.: Атом-
издат, 1976.
743. ОстриковаН. В., Мирошник 3. А., Кулешова В. Д.,
Дудкина Н. П., Илющенко А. И. — Журн. не- 775.
орган, хим., 1969, 14, с. 2229.
744. Островитянова С. 9., Иткина Л. С. — Журн. 776.
неорган, хим., 1969, 14, с. 577.
745. Ощерин Б. Н., Федотова Е. Ю. — ВИНИТИ. 777.
Деп. № 2891-79. М., 1979.
746. Ояри П. У. — Журн. структур, хим., 1971, 12, 778.
с. 913.
747. Ояри Л. У., Самойлов О. Я. — Журн. структур. 779.
хим., 1971, 12, с. 915.
748. Падунова И. Д. — Автореф. дис. . . . канд. хим. 780.
наук. М.: МГУ, 1975.
749. Пак Т. А. — Журн. физ. хим., 1972, 46, с. 2121. 781.
750. Палкин А. П. — Журн. Рус. физ. хим. об-ва,
1928, 60, с. 317.
751. Палкин В. А., Горбунов В. Е., Капитонова 782.
Т. А. — Журн. физ. хим., 1969, 43, с. 1627.
752. Пан В. М. — Сборник научных трудов ин-та ме- 783.
. таллофизики АН УССР, 1964, № 20, с. 130.
753. Пан В. М., Прохоров В. Г., Шевченко А. Д., Довго- 784.
пол В. П. — Физика низких температур, 1977, 3,
с. 1266. 785.
754. Панова Т. П., Исупова Е. П., Келлер Е. К. —
Изв. АН СССР. Неорган, матер., 1978, 14, с. 781.
755. Панченков И. Г. — Автореф. дис. . . . канд. хим. 786.
наук. М.: МГУ, 1976.
756. Пауков И. Е. — Журн. физ. хим., 1969, 43, 787.
с. 2021.
757. Пауков И. Е., Анисимов М. П., Лукьянова
И. Г. — ВИНИТИ. Деп. № 524-74. М., 1974. 788.
758. Пауков И. Е., Березовский Г. А. — Журн. физ.
хим., 1972, 46, с. 2683.
759. Пауков И. Е., Березовский Г. А. — ВИНИТИ. 789.
Деп. № 4502-72. М., 1972.
760. Пауков И. Е., Березовский Г. А., Суховей К. С. — 790.
Изв. СО АН СССР. Сер. хим. наук, 1981, вып. 2,
№ 4, с. 11. 791.
761. Пауков И. Е., Березовский Г. А., Фролова Г. П.,
Румянцева Л. П., Лаврентьев М. Н. — Журн. 792.
физ. хим., 1982, (в печати).
762. Пауков И. Е., Лаврентьева М. Н. — Журн. физ. 793.
хим., 1968, 42, с. 1842.
763. Пауков И. Е., Лаврентьева М. Н. — Журн. физ. 794.
хим., 1969, 43, с. 2116.
764. Пауков И. Е., Лаврентьева М. Н., Анисимов 795.
М. П. — Журн. физ. хим., 1969, 43, с. 2120.
765. Пауков И. Е., Рахменкулов Ф. С. — Журн. физ. 796.
хим., 1969, 43, с. 788.
766. Пауков И. Е., Рахменкулов Ф. С. — Физика
твердого тела, 1971, 13, с. 2198. 797.
767. Пауков И. Е., Рахменкулов Ф. С, Добролюбова
М. С, Ценципер А. Б. —Изв. АН СССР. Сер. 798.
хим., 1970, № 9, с. 2135.
Пауков И. Е., Рахменкулов Ф. С, Лукьянова
И. Г. — Журн. физ. хим., 1970, 44, с. 256.
Пауков И. Е., Хриплович Л. М. —• Журн. физ,
хим., 1969, 43, с. 2678.
Пауков И. Е., Хриплович Л. М., Коротких
А. М. — Журн. физ. хим., 1968, 42, с. 1267.
Пауков И. Е., Хриплович Л. М., Попов А. П. —
Журн. физ. хим., 1970, 44, с. 547.
Пауков И. Е., Хриплович Л. М., Попов А. П. —
Журн. физ. хим., 1971, 45, с. 1295.
Пауков И. Е., Хриплович Л. М., Попов А. П. —
Журн. физ. хим., 1971, 45, с. 2451.
Пейко Д., Брюнере В., Сауко Ж. — В кн.: Все-
союз. совещ. по химии неорганических перекисных
соединений. Тез. докл. / Ред. И. И. Вольнов,
А. Я. Блюм. Рига: Рижский политехи, ин-т, 1973,
с. 37.
Пелецкий В. 9., Дружинин В. П. — Теплофизика
высоких температур, 1971, 9, с. 539.
Перелигин Б. Г., Бывальцев Ю. А., Воробьев
А. Ф. — Журн. физ. хим., 1977, 51, с. 2398.
Перелигин Б. Г., Бывальцев Ю. А., Воробьев
А. Ф. — Журн. физ. хим., 1978, 52, с. 484.
Перелигин Б. Г., Бывальцев Ю. А., Воробьев
А. Ф. — Журн. физ. хим., 1978, 52, с. 1558.
Перелигин Б. Г., Бивалъцев Ю. А., Воробьев-
А. Ф. — Журн. физ. хим., 1978, 52, с. 1836.
Перельман Ф. М., Зворикин А. Я. — Молибден
и вольфрам. М.: Наука, 1968.
Переляев В. А., Миллер В. П., Переляева Л. А.,
Гребенщиков Н. Е., Швейкин Г. П., Чирков
А. К. — Журн. неорган, хим., 1977, 22, с. 2344.
Перов П. А., Новиков В. Н., Мальцев А. А. —
Вестн. МГУ. Сер. хим., 1972, 13, с. 89.
Перфильева О. Г., Решетников Я. А. — Журн.
неорган, хим., 1964, 9, с. 2602.
Петров В. М. —• Автореф. дис. . . . канд. хим.
наук. Иваново: Ивановский хим.-техн. ин-т, 1978.
Петров К. П., Куликов В. А., Угаров В. В., Рам-
биди Н. Г. — Журн. структур, хим., 1980, 21,
с. 71.
Петров К. П., Угаров В. В., Рамбиди Н. Г. —
Журн. структур, хим., 1981, 22, с. 158.
Петрова И. И. — В кн.: Теплофизические
свойства веществ при высоких температурах: Сб. науч.
работ ИВТАН. М.: ИВТАН, 1978, с. 158.
Пигалъская Л. А., Юрчак Р. П., Макаренко И. П.,
Филиппов Л. П. — Теплофизика высоких
температур, 1966, 4, с. 144.
Пилоян Г. О., Евсеев А. М., Герасимов Я. И. —
Журн. физ. хим., 1960, 34, с. 1768.
Пирс Р., Гейдон А. Отождествление
молекулярных спектров. М.: Изд-во иностр. лит., 1949.
Плотников В. А., Якубсон С. И. — Журн. физ,
хим., 1938, 12, с. 113.
Плющев В. Е., Амосов В. М. — Изв. АН СССР.
Неорган, матер., 1965, 1, с. 1155.
Плющев В. Е., Маркина И. Б., Шкловер Л. П. —
Докл. АН СССР, 1956, 108, с. 645.
Плющев В. Е., Марковская Н. Ф. — Журн. общ.
хим., 1954, 24, с. 1302.
Плющев В. Е., Самусеева Р. Г., Полетаев И. Ф. —
Журн. неорган, хим., 1962, 7, с. 860.
Плющев В. Е., Стёпина С. Б., Зимина Г. В.г
Молчанова О. П., Савельева Л. В., Яшков Д. А. —
Изв. вузов. Цвет, мет., 1970, № 1, с. 65.
Плющев В. Е., Шахно И. С, Пожиткова С. А. —
Журн. общ. хим., 1955, 25, с. 1072.
Покрасин М. А., Рощупкин В. В., Фокин Л. Р.,
Хандомирова П. Э. — В кн.: Теплофизические
536
Литература
свойства веществ и материалов. М.: Изд-во стан- 830.
дартов, 1982, вып. 20.
799. Полищук А. Ф., Тишура Т. А., Бударина А. П. — 831.
В кн.: Шестая Всесоюз. конф. по калориметрии,
1973 (расширенные тезисы докл.). Тбилиси: Meif- 832.
ниереба, 1973, с. 433.
800. Полищук А. Ф., Тишура Т. А., Бударина А. Н. — 833.
Укр. хим. журн., 1974, 40, с. 120.
801. Половое В. М. — Журн. эксперим. итеор. физ., 834.
1974, 66, с. 2164.
802. Поляков А. Ю. — Журн. физ. хим., 1946, 20,
с. 1021. 835.
803. Понятепко М. А., Радченко И. В. — Укр. физ.
журн., 1966, 11, с. 317. 836.
804. Попов И. А., Ширяева Н. В. — Журн. неорган,
хим., 1961, 6, с. 2334. 837.
805. Попов М. М., Гальченко Г. Л. — Журн. общ.
хим., 1951, 21, с. 2220. 838.
806. Попов М. М., Галъченко Г. Л., Сенин М. Д. —
Журн. неорган, хим., 1958, 3, с. 1734.
807. Попов М. М., Гинзбург Д. М. — Журн. общ. 839.
хим., 1956, 26, с. 971.
808. Попов М. М., Иванов М. И. — Атомная энергия,
1957, 7, с. 360.
809. Попов М. М., Костылев Ф. А., Зубова Н. В. — 840.
Журн. неорган, хим., 1958, 4, с. 1708.
810. Попов М. М., Костылев Ф. А., Зубова П. В. — 841.
Журн. неорган, хим., 1959, 4, с. 770.
811. Попов М. М., Костылев Ф. А., Карпова Т. Ф.— 842.
Журн. неорган, хим., 1957, 2, с. 9.
812. Попов М. М., Скуратов С. М., Никонова И. И. — 843.
Журн. общ. хим., 1940, 10, с. 2017.
813. Попов М. М., Скуратов С. М., Стрельцова 844.
М. М. — Журн. общ. хим., 1940, 10, с. 2023.
814. Портнова С. М., Иткина Л. С. — Журн. неорган. 845.
хим., 1974, 19, с. 1624.
815. Портной К. П., Мордвинов О. А., Тимофеева
Н. И. — Изв. АН СССР. Неорган, матер., 1968,
4, с. 1368. 846.
816. Привалова Н. М., Смирнова Л. С, Воробьев
А. Ф. — Журн. физ. хим., 1976, 50, с. 1334. 847.
817. Проскурин В. Б. — Автореф. дис. . . . канд. техн.
наук. М.: МИФИ, 1968. 848.
818. Проскурин В. Б., Трелин Ю. С, Баранов В. М.,
Морозов Л. Г. — Измерительная техника, 1968, 849.
№ 7, с. 91.
819. Проценко А. В., Проценко П. И. — Изв. вузов. 850.
Цвет, мет., 1964, № 5, с. 34. 851.
820. Проценко А. В., Проценко П. И., Децен Л. П. —•
Изв. вузов. Хим. и хим. технол., 1971, 14, с. 868. 852.
821. Проценко П. П., Андреева Т. А. — Журн.
неорган, хим., 1964, 9, с. 1441. 853.
822. Проценко П. П., Бордюшкова Е. А., Венеров-
ская Л. Н. — Укр. хим. журн., 1965, 31, с. 1200. 854.
823. Проценко П. И., Брыкова Н. А. — Укр. хим.
журн., 1964, 30, с. 448. 855.
824. Проценко П. П., Брыкова Н. А. — Журн. неорган,
хим., 1965, 10, с. 1220. 856.
825. Проценко П. И., Брыкова Н. А., Иванова Е. М. —.
Журн. неорган, хим., 1965, 10, с. 1477. 857.
826. Проценко П. И., Жилин Г. С. — Изв. вузов. Хим.
и хим. технол., 1967, 10, с. 377.
827. Проценко П. И., Коломин Л. Г. — Изв. вузов. 858.
Сер, физ., 1971, 7, с. 105.
828. Проценко П. И., Коломин Л. Г., Паниева Л. А.,
Венеровская Л. Н. — Изв. вузов. Хим. и хим. 859.
технол., 1969, 12, с. 3.
829. Проценко П. И., Медведев Б. С. — Журн. неорган. 860.
хим., 1963, 8, с. 2737.
Проценко П. И., Медведев Б. С. — Журн.
неорган, хим., 1964, 9, с. 2438.
Проценко П. И., Медведев Б. С. — Журн.
неорган, хим., 1965, 10, с. 1906.
Проценко П. И., Медведев Б. С. — Укр. хим.
журн., 1966, 32, с. 690.
Проценко П. И., Савенкова М. А. — Изв. вузов.
Хим. и хим. технол., 1974, 17, с. 802.
Проценко П. И., Ходаков А. А., Мирская Е. 3.,
Венеровская Л. Я. — В кн.: Сегнетоэлектрики.
Ростов-на-Дону: Ростов. Гос. ун-т, 1961, с. 21.
Проценко П. И., Шишолина Р. Н. — Журн.
неорган, хим., 1963, 8, с. 2744.
Проценко П. И., Шишолина Р. П. — Изв. вузов.
Хим. и хим. технол., 1964, 7, с. 887.
Проценко П. И., Шишолина Р. П. — Укр. хим.
журн., 1964, 30, с. 912.
Проценко П. И., Шишолина Р. П., Иванова
Е. М. — Изв. вузов. Хим. и хим. технол., 1964,
7, с. 180.
Проценко П. И., Шмелькова Г. Ф. — В кн.:
Гетерогенные равновесия в нитритных и нитратных
системах / Отв. ред. А. Г. Бергман. Элиста:
Калмыцкое кн. изд-во, 1970, с. 66.
Проценко П. И., Шмелькова Г. Ф. —• Журн.
неорган, хим., 1972, 17, с. 867.
Проценко П. И., Шокина О. Н. — Журн. неорган,
хим., 1959, 4, с. 2554.
Прусов А. Н., Захаров А. Г., Крестов Г. А. —
Изв. вузов. Хим. и хим. технол., 1979, 22, с. 1523,
Прымова Л. А., Селиванова Н. М., Орлова В. Т. —•
Изв. вузов. Хим. и хим. технол., 1972, 15, с. 3.
Пчелкин И. М. — Автореф. дис. . . . канд. техн.
наук. М.: ЭНИН, 1962.
Пчелкин И. М. — В кн.: Теплообмен,
гидродинамические и теплофизические свойства веществ:
Сборник / Под ред. И. Т. Аладьева. М.: Наука,
1968, с. 258.
Равич Г. Б., Егоров Б. П. — Журн. неорган, хим.,
1960, 5, с. 2603.
Равич М. И., Воровать Ф. Е. — Изв. сектора физ.-
хим. анализа ИОНХ АН СССР, 1954, 24, с. 280.
Равич М. И., Ястребова Л. Ф. — Журн. неорган,
хим., 1963, 8, с. 202.
Радищев В. П. — Журн. Рус. физ.-хим. об-ва.
Химия, 1930, 62, с. 1063.
Радищев В. П. — Журн. общ. хим., 1933, 3, с. 852.
Рассонская И. С, Семендяева Н. К. — Журн.
неорган, хим., 1963, 8, с. 1419.
Рассонская И. С, Семендяева Н. К. — Журн.
неорган, хим., 1966, И, с. 1134.
Рассонская И. С, Семендяева Н. К. — Журн.
неорган, хим., 1970, 15, с. 52.
Рахменкулов Ф. С, Пауков И. Е. — Журн. физ.
хим., 1982, 56, с. 2283.
Рачев В. В., Ковба Л. М., Ипполитова Е. А. —
Докл. АН СССР, 1964, 159, с. 1371.
Рачев В. В., Ковба Л. М., Ипполитова Е. А. —
Журн. неорган, хим., 1965, 10, с. 573.
Рачев В. В., Смурова В. С, Ковба Л. М.,
Ипполитова Е. А. — Журн. неорган, хим., 1965, 10,
с. 2796.
Регниченко В. А., Холимое Ф. Б., Уколова Т. П. —
В кн.: Титан и его сплавы. М.: Изд-во АН СССР,
1963, вып. 9, с. 42.
Резухина Т. Н., Голованова Ю. Г. — Изв.
АН СССР. Неорган, матер., 1967, 3, с. 867.
Резухина Т. П., Трошина 3. В. — Журн. физ.
хим., 1962, 36, с. 637.
537
Литература
861. Ремизов В. Г., Коротаева Л. Г., Дударева А. Г., 894
Иванов-Эмин Б. Л. — Журн. прикл. хим., 1971,
44, с. 2319. 895,
862. Ремизов В. Г., Коротаева Л. Г., Дударева А. Г.,
Иванов-Эмин Б. Л. — Изв. вузов. Хим. и хим.
технол., 1971, 14, с. 461. 896,
563. Ремизов В. Г., Коротаева Л. Г., Дударева А. Г.,
Иванов-Эмин В. И., Ежов А. И. — Журн. не- 897,
орган, хим., 1971, 16, с. 2308.
$64. Решетников Н. А. — Журн. неорган, хим., 1961, 898.
6, с. 682.
865. Решетников Н. А., Баранская Е. В. — Журн. 899.
неорган, хим., 1965, 10, с. 183.
866. Решетников Л. А., Баранская Е. В. — Изв. вузов. 900.
Хим. и хим. технол., 1967, 10, с. 496.
867. Решетников Н. А., Баранская Е. В. — Научные 901.
труды Иркутск, мед. ин-та, 1969, вып. 95, с. 135.
Решетников И. А., Бергман А. Г. — Изв. физ.- 902.
хим. НИИ при Иркутском ун-те, 1959, 4, с. 9.
Решетников Н. А., Вилутис Н. И. — Журн.
неорган, хим., 1958, 3, с. 366. 903.
870. Решетников Н. А., Диогенов Г. Г. — Докл.
АН СССР, 1952, 85, с. 819.
871. Решетников Н. А., Перфильева О. Г. — Журн. 904.
неорган, хим., 1968, 13, с. 1662.
872. Решетников Н. А., Романова Е. В. — Журн. не- 905.
орган, хим., 1961, 6, с. 1381.
873. Решетников И. А., Унжаков Г. М. — Журн. не- 906.
орган, хим., 1958, 3, с. 1433.
874. Решетников Н. А., Унжаков Г. М. — Изв. физ.- 907.
хим. НИИ при Иркутском ун-те, 1959, 4, с. 41.
875. Рза-Заде П. Ф., Мамедова 3. Д. ~ В кн.: Исследо- 908.
вания в области неорганической и физической
химии: Сборник работ аспирантов и соискателей. 909.
Баку: Изд-во АН АзербССР, 1971, с. 118.
876. Рза-Заде П. Ф., Рустамов П. Г. — Труды ин-та 910.
химии АН АзербССР, 1960, 18, с. 38.
877. Роддатис Л. М. — Автореф. дис. . . . канд. хим. 911.
наук. М.: МГУ, 1975.
878. Роде Е. Я., Лысанова Г. В. — Докл. АН СССР, 912.
1962, 145, с. 351.
S79. Роде Т. В. — Докл. АН СССР, 1953, 90, с. 1075. 913.
880. Роде Т. В. — Докл. АН СССР, 1953, 91, с. 313.
881. Роде Т. В., Гольдер Г. А. — Изв. АН СССР. Отд. 914.
хим. наук, 1956, № 3, с. 299.
882. Роде Т. В., Добрынина Т. А., Гольдер Г. А. — 915.
Изв. АН СССР. Отд. хим. наук, 1955, № 4, с. 611.
883. Родигина Э. Н., Гомельский К. 3. — Журн. физ. 916.
хим., 1961, 35, с. 1828.
884. Родигина Э. И., Гомельский К. 3., Лугинина
В. Ф. — Журн. неорган, хим., 1959, 4, с. 975.
885. Родникова М. Н. — Журн. неорган, хим.,. 1958,
3, с. 2417. 917.
886. Романов О. А., Шевчук В. Г. — Журн. неорган.
хим., 1974, 19, с. 2990.
887. Рубинчик С. М., Банашек Е. И., Соколов 918.
B. А. — Журн. физ. хим., 1971, 45, с. 1069.
888. Руда Г. И., Корнилов И. И., Вавилова В. В. — 919.
Изв. АН СССР. Металлы, 1975, № 5, с. 203.
889. Румянцев А. В., Макаренко И. Н., Банчила 920.
C. Н. — Инж.-физ. журн., 1979, 36, с. 581.
890. Рунов Н. Н. — Уч. зап. Яросл. пед. ин-та, 1969,
№ 66, с. 147. 921.
891. Рухляда Н. Я., Трофимов А. Г., Шишкин В. Б. —
Вестн. МГУ. Сер. физ., астрон., 1979, 20, с. 70. 922.
892. Рютина Н. М., Максина Н. В., Финкельштейн
Н. А. — Журн. неорган, хим., 1973, 18, с. 1101. 923.
893. Савватимский А. И. — Теплофизика высоких
температур, 1973, 11, с. 1182.
Савватимский А. И. — Автореф. дис. . . . канд.
техн. наук. М.: ИВТАН, 1975.
Савицкий Е. М., Вурханов Г. С. — В кн.: Титан
и его сплавы. М.: Изд-во АН СССР, 1962, вып. 7,
с. 51.
Савицкий Е. М., Наумкин О. П., Ефимов Ю. В. —
Изв. АН СССР. Металлы, 1971, № 2, с. 178.
Савицкий Е. М., Терехова В. Ф., Наумкин О. П. —
Усп. физ. наук, 1963, 79, с. 263.
Савицкий Е. М., Тылкина М. А., Поварова К. В. —
Атомная энергия, 1959, 7, с. 470.
Самойлов О. Я. — Докл. АН СССР, 1951, 81,
с. 641.
Самойлов О. Я. — Изв. АН СССР. Отд. хим. наук,
1952, с. 398.
Самойлов О. Я. — Изв. АН СССР. Отд. хим. наук,
1956, с. 1415.
Самойлов О. Я., Буслаева М. Л. — В кн.:
Строение вещества и спектроскопия. М.: Изд-во
АН СССР, 1960, с. 102.
Самойлова А. Л., Ефремов Ю. М., Журавлев Д. А.,
Гурвич Л. В. — Химия высоких энергий, 1974,
8, с. 229.
Самсонов Г. В. — Укр. хим. журн., 1957, 23,
с. 287.
Самусева Р. Г., Плющев В. Е. — Журн. неорган,
хим., 1961, 6, с. 2139.
Самусева Р. Г., Плющев В. Е. — Журн. неорган,
хим., 1964, 9, с. 2433.
Сандлер Р. А., Яскеляйнен Э. И., Василъкова
И. В. — ВИНИТИ. Деп. № 4566-72. М., 1972.
Санталова Н. А., ВидавскийЛ. М., Дунаева К. М.,
Ипполитова Е. А. — Радиохимия, 1971, 13, с. 592.
Сафонов В. В., Коршунов Б. Г., Яровой А. А. —
Журн. неорган, хим., 1966, 11, с. 1720.
Сафонов В. В., Лемешко О. В. — Журн. неорган,
хим., 1973, 18, с. 1957.
Сафонов В. В., Лемешко О. В., Коршунов Б. Г. —•
Журн. неорган, хим., 1971, 16, с. 2283.
Свентославский В. — Журн. Рус. физ.-хим. об-ва,
1909, 41, с. 587.
Свечников В. Л. — Физ. металлов и
металловедение, 1963, 15, с. 616.
Свечников В. Л., Кобзенко Г. Ф. — Докл.
АН СССР, 1964, 155, с. 611.
Севастьянова Т. Л., Карпенко Н. В. — Журн.
неорган, хим., 1969, 14, с. 3130.
Семашко Л. Л., Новиков И. И., Рощупкин В. В.,
Гончаров В. Г., Мозговой А. Г. — В кн.: Восьмая
Всесоюз. конф. по калориметрии и хим.
термодинамике. Иваново, 1979: Тезисы докладов. Иваново:
Ивановский хим.-техн. ин-т, 1979, с. 399.
Семенов Г. А. — В кн.: Силикаты и окислы в
химии высоких температур. М.: Изд-во АН СССР,
1963, с. 228.
Семенов Г. А. — Журн. неорган, хим., 1965, 10,
с. 2390.
Семенов Г. А. — Изв. АН СССР. Неорган, матер.,
1969, 5, с. 67.
Семенов Г. А., Францева К. Е., Шалкова Е. К. —
Вестн. ЛГУ. Сер. физ. и хим., 1970, вып. 3, № 16,
с. 83.
Семин А. В., Клопов В. И. — Изв. вузов. Хим.
и хим. технол., 1979, 22, с. 248.
Сергеева Р. И., Дракин С. И., Карапетъянц
М. X, — Журн. физ. хим., 1970, 44, с. 2611.
Сергеева Р. И., Дракин С. П., Карапетъянц
М. X. ¦— Труды Моск. хим.-технол. ин-та
им. Д. И. Менделеева, 1970, вып. 67, с. 18.
538
Литература
S24. Серебренников Л. В. — Автореф. дис. . . . канд.
хим. наук. М., МГУ, 1975.
925. Серебренников Л. В., Мальцев А. А. — Вестн.
МГУ. Сер. хим., 1975, 16, с. 486.
926. Серебренников Л. В., Мальцев А. А. — ВИНИТИ.
Деп. № 27—75. М., 1975.
927. Серебренников Л. В., Мальцев А. А. — Вестн.
МГУ. Сер. хим., 1976, 17, с. 38.
928. Серебренников Л. В., Мальцев А. А. — Вестн.
МГУ. Сер. хим., 1980, 21, с. 148.
929. Серебренников Л. В., Мальцев А. А. — ВИНИТИ.
Деп. № 54—80. М., 1980.
330. Серебренников Н. Л., Гелъд П. В. — Труды
Уральск, политехи, ин-та, 1954, № 7, с. 583.
¦931. Серебренников Л. Л., Гельд П. В. — Изв. вузов.
Цвет, мет., 1961, № 4, с. 80.
¦932. Сидоров Л. Л., Ерохин Е. В., Акишин П. А.,
Колосов Е. Л. — Докл. АН СССР, 1967,173, с. 370.
933. Сидоров Л. Л.; Колосов Е. Л. — Журн. физ. хим.,
1968, 42, с. 2617.
934. Сидоров Л. Л., Колосов Е. Л., Шолъц В. Б. —
Журн. физ. хим., 1968, 42, с. 2620.
935. Сидоров Л. #., Колосов Е. Л., Давыдов В. А.,
Шолъц В. Б. — Вестн. МГУ. Сер. хим., 1973,
14, с. 35.
936. Симанов Ю. П., Киркина Д. Ф. — Журн. неорган,
хим., 1957, 2, с. 699.
«37. Сирота Л. Д., Куделъко Ж. М. — Докл. АН СССР,
1973, 209, с. 1068.
938. Сироткин Г. Д. — Журн. неорган, хим., 1959,
4, с. 2558.
939. Скляренко С. И., Краузе И. д. — Журн. физ. хим.,
1937, 10, с. 449.
S40. Скуратов О. А., Павлов О. Л., Данилкин В. И.,
Волков И. В. — Журн. неорган, хим., 1976, 21,
с. 2910.
941. Скуратов С. М., Лапушкин С. А. — Журн. общ.
хим., 1951, 21, с. 2217.
942. Слюсарь Н. П., Криворотенко А. Д., Фомичев
Е. Л., Калашник А. А., Бондаренко В. П. —
ВИНИТИ. Деп. № 6306-73. М., 1973.
943. Слюсарь Н. П., Криворотенко А. Д., Фомичев
Е. #., Калашник А. А., Бондаренко В. П. —
Журн. физ. хим., 1973, 47, с. 2706.
944. Слюсарь Л. П., Криворотенко А. Д., Фомичев
Е. Л., Калашник А. А., Бондаренко В. П. —
Теплофизика высоких температур, 1973, 11, с. 213.
•945. Смирнов М. В., Василевская М. М., Буров Г. В.,
Антонов Б. Д. — Труды ин-та электрохимии,
Уральск, научн. центр АН СССР, 1971, 17, с. 3.
946. Смирнов М. В., Василевская М. М., Буров Г. В.,
Вержбицкий Ф. Р., Антонов Б. Д. — В кн.:
Физ. химия и электрохимия расплав, солей и
шлаков. Киев: Наукова думка, 1969, ч. 1, с. 12.
947. Смирнов М. В., Любимцева И. Я. — Труды ин-та
электрохимии Уральского фил. АН СССР, 1970,
вып. 16, с. 82.
948. Смирнов М.В., Любимцева И. #., Циовкина Л. Л.,
Краснов Ю. Л. — Журн. неорган, хим., 1971, 16,
с. 251.
949. Смирнов М. П., Стрельникова Л. М. — Сборник
науч. трудов Моск. ин-та цвет. мет. и золота
и ВНИТО цвет, мет., 1965, № 23, с. 67.
950. Смирнов Ю. Л., Финкелъ В. А. — Физ. металлов
и металловедение, 1963, 16, с. 637.
951. Смирнов Ю. Л., ФинкелъВ. А. — Журн. эксперпм.
и теор. физ., 1965, 49, с. 1077.
952. Смирнова В- И., ОрмонтБ. Ф. — Докл. АН СССР,
1955, 100, с. 127.
953. Смирнова В. И., Ормонт Б. Ф. — Журн. физ.
хим., 1956, 30, с. 1327.
954. Смирнова Л. С, Привалова Л. М., Воробьев
A. Ф. — Вестн. МГУ. Сер. хим., 1978, 19, с. 579.
955. Соболева Л. В., Косырбасова М. Г., Огаджанова
B. В. — Изв. АН СССР. Неорган, матер., 1972,
8, с. 533.
956. Соколов В. А. — Журн. техн. физ., 1948, 18, с. 813.
957. Соколов В. А., Банашек Е. И., Рубинчик С. М. —
Жури» физ. хим., 1982, 55 (в печати).
958. Соколов В. А., Шарпатая Г. А. — Журн. неорган,
хим., 1964, 9, с. 1542.
959. Соколов В. А., Шмидт Л. Е. — Изв. сектора физ.-
хим. анализа ИОНХ АН СССР, 1955, 26, с. 123.
960. Соколов В. А., Шмидт Л. Е. — Изв. сектора физ.-
хим. анализа ИОНХ АН СССР, 1956, 27, с. 219.
961. Соломоник В. Г. — Автореф. дис. . . . канд. хим.
наук. Иваново: Ивановский хим.-техн. ин-т, 1977.
962. Соломоник В. Г., Данилова Т. Г. — Журн. физ.
хим., 1973, 47, с. 1063.
963. Соломоник В. Г., Краснов К. С. — Журн. физ.
хим., 1979, 53, с. 284.
964. Соломоник В. Г., Краснов К. С, Гиричев Г. В.,
Засорин Е. 3. — Журн. структур, хим., 1979, 20,
с. 427.
965. Соломоник В. Г., Озерова В. Л., Болдырев Л. И. —
Журн. неорган, хим., 1982 (в печати).
966. Сонин В. П., Лоляченок О. Г. — ВИНИТИ.
Деп. № 5539-73. М., 1973.
967. Сосновская Л. К., Финкелъштейн Л. А. — Журн.
неорган, хим., 1974, 19, с. 221.
968. Спиридонов В. П., Ерохин Е. В. — Журн. неорган.
хим., 1969, 14, с. 636.
969. Спиридонов В. П., Ерохин Е. В., Брезгин Ю. А. —
Журн. структур, хим., 1972, 13, с. 321.
970. Спиридонов В. П., Ерохин Е. В., Лутошкин
Б. И. — Вестн. МГУ. Сер. хим., 1971, 12, с. 296.
971. Спиридонов В. П., Лутошкин Б. И. — Вестн.
МГУ. Сер. хим., 1970, И, с. 509.
972. Спиридонов В. П., Ходченков А. Л., Акишин
П. А. — Журн. структур, хим., 1965, 6, с. 633.
973. Спицын В. И., Кулешов И. М. — Журн. общ.
хим., 1951, 21, с. 1365.
974. Спицын В. И., Михайленко И. Е., Воронин
Ю. В. — Радиохимия, 1971, 13, с. 866.
975. Справочник по плавкости солевых систем /
Под ред. Н. К. Воскресенской. М.: Изд-во
АН СССР, 1961.
976. Старостина Т. С, Сидоров Л. П., Акишин П. А.,
Карасев Л. М. — Изв. АН СССР. Неорган, матер.,
1967, 3, с. 727.
977. Стегний А. П., Шевченко А. В., Лопато Л. М.,
Рубан А. К., Дверняков В. С, Пасичный В. В. —
Докл. АН УССР, 1979, № 6, с. 484.
978. Степанов Л. Ф., Немухин А. В. — Оптика и
спектроскопия, 1978, 45, с. 1193.
979. Степанов Л. И., Кабанкова И. Л., Москвитина
Е. Л., Кузяков Ю. Я. — Вестн. МГУ. Сер. хим.,
1973, 14, с. 645.
980. Степин Б. Д., Ловецкая Г. А., Иоффе О. В. —
Журн. неорган, хим., 1978, 23, с. 2247.
981. Стеценко П. Л., Авксентьев Ю. И. — Журн.
эксперим. и теор. физ., 1964, 47, № 9, с. 806.
982. Стеценко П. Л., Авксентьев Ю. И. — Приборы
и техника эксперимента, 1965, 10, с. 178.
983. Стороженко В. Л. — Журн. физ. хим., 1974, 48,
с. 1709.
984. Сторонпин А. В., Василъкова И. В., Козина
Т. М. — Вестн. ЛГУ. Сер. физ. и хим., 1976,
вып. 4, № 22, с. 90.
539
Литература
985. Сторонкин А. В., Василькова И. В., Менде- 1013.
леееаС. В. — Журн. физ. хим., 1973, 47, с. 2031.
986. Сторонкин А. В., Василькова И. В., Шамко 1014.
В. И. — Вестн. ЛГУ. Сер. физ. и хим., 1973,
вып. 2, № 10, с. 67. 1015.
987. Стрекаловский В. Л., Бекетов А. Р., Власов
В. Г. — Журн. неорган, хим., 1964, 9, с. 2496. 1016.
988. Стрекаловский В. Л., Бекетов А. Р., Власов
B. Г. — Труды ин-та электрохимии Уральского 1017.
фил. АН СССР, 1965, вып. 6, с. 131.
989. Стрекаловский В. Н., Зубанков В. Н., Пальгуев 1018.
C. Ф, — Труды ин-та электрохимии Уральского
фил. АН СССР, 1969, вып. 12, с. 120. 1019.
990. Стрелков П. Г., Ицкевич Е. С, Кострюков В. Н.,
Мирская Г. Г. — Журн. физ. хим., 1954,28, с. 645. 1020.
991. Суганеев Ю. С, Таубин М. Л., Якутпович М. В. —
Изв. АН СССР. Металлы, 1970, № 6, с. 215. 1021.
992. Суглобова И. Г., Чиркст Д. 9. — Радиохимия,
1978, 20, с. 211. 1022.
993. Суглобова И. Г., Чиркст Д. 9. — Радиохимия,
1980, 22, с. 779. 1023.
994. Сумарокова Т. Н., Модестова Т. П. — Изв.
АН КазССР. Сер. хим. наук., 1964, 14, с. 26.
995. Сумин В. В., Пейзулаев Ж. И. — Журн. физ. хим.,
1973, 47, с. 1604. 1024.
996. Супоницкий Ю. Л., Бодров В. Г., Карапетъ-
янц М. X. — Труды Моск. хим.-технол. ин-та им. 1025.
Д. И. Менделеева, 1973, вып. 75, с. 11.
997. Супоницкий Ю. Л., Цветков А. А., Селезнев В. П., 1026.
Карапетъянц М. X., Судариков Б. Н.,
Громов Б. В.—Журн. физ. хим., 1971, 45, с. 995. 1027.
998. Суриков Bad. И., Данилов СВ., Суриков Вал. И.,
Штольц А. К. —Изв. вузов. Сер. физ., 1981,
24, с. 112. 1028.
999. Суриков Bad. И., Миллер И. И., Ярош 9. М.,
Суриков Вал. И., Кондратьева Н. Е. — Физика 1029.
твердого тела, 1979, 21, с. 1863.
1000. Сурков Ю. С. — Изв. вузов. Хим. и хим. технол.,
1968, 11, с. 1190. 1030.
1001. Сухаревский Б. Я. — Докл. АН СССР, 1966, 167,
с. 1046. 1031.
1002. Сухаревский Б. Я., Вишневский И. И. — Докл.
АН СССР, 1962, 147, с. 882. 1032.
1003. Сухаревский Б. Я., Вишневский И. И., Гав-
риш А. М. — Докл. АН СССР, 1961, 140, с. 884. 1033.
1004. Сухушина И. С. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1973.
1005. Суховей К. С, Анишин В. Ф., Березовский Г. А., 1034.
Пауков И. Е. — ВИНИТИ. Деп. № 2893-75.
М., 1975. 1035.
1006. Суховей К. С, Анишин В. Ф., Березовский Г. А.,
Пауков И. Е. — Журн. физ. хим., 1976, 50, с. 802. 1036.
1007. Талакин О. Г., Аханщикова Л. А., Сосновский
Е. Н., Панкратов А. В., Зерченинов А. Н. ¦— 1037.
Журн. физ. хим., 1962, 36, с. 1065.
1008. Терехов Г. И., Синяков С. И., Иванов О. С. — 1038.
В кн.: Физико-химия сплавов и тугоплавких
соединений с торием и ураном: Сборник статей.
М.: Наука, 1968, с. 9. 1039.
1009. Термические константы веществ: Справочник/
Отв. ред. В. П. Глушко. М.: ВИНИТИ, 1965— 1040.
1981, вып. 1—10. 1041.
1010. Теряев В. В. — Автореф. дис. . . . канд. техн.
наук. М.: МИФИ, 1981.
1011. Теряев В. В., Трелин Ю. С. — В кн.: Доклады
IX Всесоюзной акустической конференции. Сек- 1042.
ция Г. М.: Акустический институт, 1977, с. 59.
1012. Теряев В. В., Трелин Ю. С. — В кн.: Вопросы 1043.
теплофизики ядерных реакторов. М.: Атомиздат,
1978, с. 44. 1044.
Тимошинин В. С, Краснов К. С. — Журн.
структур, хим., 1969, 10, с. 161.
Ткачев К. В., Ремпелъ П. С, Стержнева И. И. —•
Журн. неорган, хим., 1968, 13, с. 3179.
Товмасъян И. К., РадинаВ. П. — Журн. неорган,
хим., 1968, 13, с. 2268.
Товмасъян И. К., Пушба Т. М., Бергман А. Г. —
Журн. неорган, хим., 1970, 15, с. 2268.
Товмасъян И. К., Шолохова В. И. — Укр. хим.
журн., 1973, 39, с. 576.
Токарева М. В. — Науч. зап. Ворошиловоград-
ского пед. ин-та, 1956, вып. 6, с. 3.
Токарева М. В., Бергман А. Г. — Журн.
неорган, хим., 1956, 1, с. 2570.
Толмачев С. М., Засорин Е. 3., Рамбиди Н. Г. —
Журн. структур, хим., 1969, 10, с. 541.
Тонкое Е. Ю. Фазовые диаграммы элементов
при высоком давлении. М.: Наука, 1979.
Тополь И. А., Дементьев А. И. — Журн.
структур, хим., 1980, 21, с. 13.
Торопов Н. А., Барзаковский В. П. и др.
Диаграммы состояния силикатных систем:
Справочник. Л.: Наука, 1969, вып. 1; 1970, вып. 2;
1972, вып. 3; 1974, вып. 4.
Торопов Н. А., Васильева В. А. — Журн.
неорган, хим., 1962, 7, с. 1938.
Тоцкий Е. Е., Шпилърайн 9. 9. — Теплофизика
высоких температур, 1963, 1, с. 456.
Трелин Ю. С, Теряев В. В., Фокин Л. Р. —
Теплофизика высоких температур, 1974,12, с. 998.
Трелин Ю. С, Теряев В. В., Фокин Л. В. —
Обзоры по теплофизическим свойствам веществ.
М.: ИВТАН, 1981, № 1 B7).
Тресвятский С. Г., Кушаковский В. И. —
Атомная энергия, 1960, 8, с. 56.
Тресвятский С. Г., Павликов В. Н., Лопа-
то Л. М. — Изв. АН СССР. Неорган, матер.,
1966, 2, с. 269.
Третъяков Ю. Д. — Химия нестехиометрических
окислов. М.: МГУ, 1974.
Труханова Л. Н. — Автореф. дис. . . . канд.
физ.-мат. наук. М.: МГУ, 1970.
Турдакин В. А., Тарасов В. В. — Журн. физ.
хим., 1968, 42, с. 2787.
Турчанин А. Г., Бабенко С. А., Екимов С. Е. —
Теплофизика высоких температур, 1980, 18,
с. 995.
Турчанин А. Г., Орданъян С. С, Фесенко В. В. —
Порошковая металлургия, 1967, с. 23.
Тусеев Н. П., Засорин Е. 3., Спиридонов В. П. —¦
Журн. структур, хим., 1979, 20, с. 587.
Тылкина М. А., Поворова К. Б., Савицкий
Е. М. — Журн. неорган, хим., 1960, 5, с. 1907
Тылкина М. А., Цыганова И. А., Савицкий
Е. М. — Журн. неорган, хим., 1960, 5, с. 1906.
Уикс К. Е., Блок Ф. Е. Термодинамические
свойства 65 элементов, их окислов, галогенидов,
карбидов и нитридов. М.: Металлургия, 1965.
Унжаков Г. М. — Докл. АН СССР, 1952, 87,
с. 791.
Урусов В. С. — Геохимия, 1965, 55, с. 551.
Ушакова И. М., Корнилов А. Н. — В кн.:
Шестая Всесоюз. конф. по калориметрии, 1973
(расширенные тезисы докл.). Тбилиси: Мецниереба,
1973, с. 57.
Федоров А. С, Силъченко Г. Ф.— Укр. хим.
журн., 1937, 12, с. 53.
Федоров В. Б. — Теплофизика высоких
температур, 1975, 13, с. 666.
Федоров Г. Б. — В кн.: Металлургия и металло-
540
Литература
ведение чистых металлов. М.: Атомиздат, 1960, 1070.
вып. 2, с. 141.
4045. Федоров Г. Б., Калинин П. И., Смирнов Е. А., 1071.
Иванов К. В. — Журн. физ. хим., 1971, 45,
с. 1218. 1072.
1046. Федоров Г. Б., Смирнов Е. А. — В кн.:
Металлургия и металловедение чистых металлов. М.: 1073.
Атомиздат, 1961, вып. 3, с. 34.
1047. Федоров Г. Б., Смирнов Е. А. — Атомная энер- 1074.
гия, 1968, 25, с. 54.
1048. Федоров Г. Б., Смирнов Е. А., Жомов Ф. И. — 1075.
В кн.: Металлургия и металловедение чистых
металлов. М.: Атомиздат, 1963, вып. 4, с. 110. 1076.
1049. Федоров Г. Б., Смирнов Е. А., Михлин Н. А.,
Калинин П. Н. — Журн. физ. хим., 1968, 42, 1077.
с. 3.
1050. Федоров Н. Я., Петров Е. С. — В кн.: Редкие 1078.
щелочные элементы: Сборник докл. 2-го Всесоюз.
совещ., Новосибирск, 1964/Под ред. В. Е. Плю- 1079.
щева. Новосибирск: Наука, 1967.
1051. Феодосьев Н. — Журн. физ. хим., 1938, 12, с. 291. 1080.
1052. Филатов С. К., Чинь-Тхи Ле Тхы. — Журн.
неорган, хим., 1972, 17, с. 1547. 1081.
1053. Филиппов Л. П., Арутюнов А. В., Макаренко
И. Н., Мардыкин И. П., Труханова Л. Д., Ху-
саинова Б. И. — Труды III Всесоюз. научно- 1082.
техн. конф. по термодинамике, Ленинград, 1968:
Сборник докл. секции «Теплофизические свойства 1083.
веществ». Л.: Наука, 1970, с. 242.
1054. Филиппов Л. П., Арутюнов А. В., Макаренко 1084.
И. Н., Мардыкин И. П., Труханова Л. Н.,
Хусаинова Б. #., Юрчап Р. П. — В кн.:
Физические параметры тепло- и массопереноса. Минск: 1085.
Наука и техника, 1968, т. 7, с. 583.
1055. Филиппов Л. П., Юрчак Р. П. — Инж.-физ.
журн., 1971, 21, с. 561. 1086.
1056. Финкель В. А., Гламазда В. И., Ковтун Г. П. —
Журн. эксперим. и теор. физ., 1969, 57, с. 1065. 1087.
1057. Финкельшщейн Н. А., Сосновская Л. К. — Жу?н.
неорган, хим., 1976, 21, с. 1901. 1088.
1058. Финкельштейн Н. А., Сосновская Л. К. — Журн.
неорган, хим., 1976, 21, с. 2586.
1059. Фирсова Т. П., МолодкинаА. Н., Морозова Т. Г., 1089.
Аксенова И. В. — Изв. АН СССР. Сер. хим.
наук, 1965, № 9, с. 1678. 1090.
1060. Фокин Л. Р. — Автореф. дис. . . . канд. техн.
наук. М.: МЭИ, 1969. 1091.
1061. Францева К. Е. — Автореф. дис. . . . канд. хим.
наук. Л.: ЛГУ, 1968. 1092.
1062; Францева К. Е., Семенов Г. А, — Теплофизика
высоких температур, 1969, 7, с. 55.
1063. Фролов Ю. Г., Жигунова Л. К., Винецкая Т. Н.,
Насонова Г. И. — ВИНИТИ. Деп. № 2641-76.
М., 1976. 1093.
1064. Фугас Дж., Браун Д. — Радиохимия, 1980, 22,
с. 617. 1094.
1065. Хаит Ю. Г., Хачкурузов Г. А. — ОНИИТЭХИМ.
Деп. № —82. Черкассы, 1982 (в печати).
1066. Хаит Ю. Г., Юрков Г. П., Хачкурузов Г. А. —
В кн.: Термодинамика и кинетика химических 1095.
процессов: Сборник научных трудов ГИПХ.
Л., 1981, с. 3.
1067. Ханаев Е. И. — Изв. СО АН СССР. Сер. хим. 1096.
наук, 1968, 9, с. 123.
1068. Ханаев Е. И., Хрипин Л. А. — Радиохимия,
1970, 12, с. 178. 1097.
1069. Хансен М., Камен Е., Кесслер Г., Макфер-
сон Д. — В кн.: Редкие металлы. Титан. М.: 1098.
Изд-во иностр. лит., 1954, ч. 2, с. 195.
Харитонов Е. Б., Крестов Г. А. — Изв. вузов.
Хим. и хим. технол., 1971, 14, с. 1446.
Харитонов Ю. Я., Гержа Т. В. — ВИНИТИ.
Деп. № 3575-77. М., 1977.
Хитрое В. А. — Изв. Воронежского пед. ин-та,
1948, 10, с. 49.
Хитрое В. А. — Изв. сектора физ.-хим. анализа
ИОНХ АН СССР, 1954, 25, с. 236.
Хитрое В. А., Хитрова Н. П., Хмельков В. Ф. —
Журн. общ. хим., 1953, 85, с. 1630.
Хлапова А. Н. — Докл. АН СССР, 1955, 105,
с. 500.
Хлапова А. Н. — Журн. неорган, хим., 1956,
1, с. 2561.
Хлапова А. Н. — Докл. АН СССР, 1957, 116,
с. 979.
Хлюстов В. Г., Борухович А. С, Переляев
В. А. — Физика твердого тела, 1972, 14, с. 2142.
Ходеев Ю. С. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1964.
Ходченков А. Н., Спиридонов В. П., Акишин
П. А. — Журн. структур, хим., 1965, 6, с. 765.
Хомяков К. Г., Спицын В. И., Жванко С. А. —
В кн.: Исследования в области химии урана.
М.: МГУ, 1961, с. 141.
Хренова Т. Л., Ахумов Е. И. — Журн. прикл.
хим., 1969, 42, с. 2597.
Хренова Т. Л., Ахумов Е. И., Жилина Л. П. —
Журн. прикл. хим., 1970, 43, с. 1599.
Хрипин Л. А., Гагаринский Ю. В., Заднепров-
ский Г. М., Лукьянова Л. А. — Атомная энергия,
1965, 19, с. 437.
Хрипин Л. А., Гагаринский Ю. В., Лукьянова
Л. А. — Изв. СО АН СССР. Сер. хим. наук,
1965, № 3, с. 14.
Хрипин Л. А'., Лукьянова Л. А. — Изв. СО АН
СССР. Сер. хим. наук, 1966, № 3, с. 131.
Хрипин Л. А., Подузова С. А., Заднепровский
Г. М. — Журн. неорган, хим., 1968, 13, с. 2796.
Хриплович Л. М. — В кн.: Химическая
термодинамика и термохимия: Сборник статей/
Отв. ред. В. А. Тулупов. М.: Наука, 1979, с. 38.
Хриплович Л. М., Лукьянова И. Г., Пауков
И. Е. — ВИНИТИ. Деп. № 2978-74. М., 1974.
Хриплович Л. М., Пауков И. Е. — Журн. физ.
хим., 1982 (в печати).
Хриплович Л. М., Попов А. П., Пауков И. Е. —
Журн. физ. хим., 1976, 50, с. 567.
Хукстра Г., Зигелъ С. — В кн.: Материалы
Международной конференции по мирному
использованию атомной энергии, Женева, 8—
20 авг. 1955, М.: Гос. научно-техн. изд-во химич.
литературы, 1958, т. 7, с. 482.
Хусаинова Б. П., Филиппов Л. П. —
Теплофизика высоких температур, 1968, 6, с. 929.
Цагарейшвили Д. Ш. Методы расчета
термических и упругих свойств кристаллических
неорганических веществ. Тбилиси: Мецниереба,
1977.
Цагарейшвили Д. Ш., Гвелесиани Г. Г. — Труды
Грузинского ин-та металлургии, 1964 (опубл.
1965), вып. 14, с. 187.
Цагарейшвили Д. Ш., Яшвили Т. С,
Гвелесиани Г. Г. — Сообщ. АН Груз. ССР, 1968, 49,
№ 1, с. 175.
Цветков В. Г., Рабинович И. Б. — Журн. физ.
хим., 1969, 43, с. 1213.
Цвигунов А. Н., Ковба Л. М. — Вестн. МГУ,
Сер. хим., 1970, 11, с. 59.
541
Литература
1099. Цвигунов А. И., Ковба Л. М., Зубова Е. В.,
Савранский В. В., Опарников Г. Л. — Докл.
АН СССР, 1973, 210, с. 365.
1100. Цвигунов А. П., Кузнецов Л. М. — Раджохямия,
1979, 21, с. 747.
1101. Ценципер А. В., Добролюбова М. С. — В кн.:
Редкие щелочные элементы. Пермь: Пермский
политехи, ин-т, 1969, с. 147.
1102. Ценципер А. Б., Рогожникова Т. И., Баку липа
В. М. — Изв. АН СССР. Сер. хим. наук, 1967,
№ 9, с. 2073.
1103. Цой Г. К., Топор Н. Д., Санникова А. И., Це-
ханская Ю. В. — Журн. физ. хим., 1975, 49,
с. 2960.
1104. Чаркин О. П. — Итоги науки и техники. М.:
ВИНИТИ, т. 4, 1976. (Серия: Строение молекул
и химическая связь).
1105. Чаркин О. П., Зюбина Т. С. — Журн. неорган,
хим., 1979, 24, с. 2877.
1106. Чахалъян О. X., Данкова И. Н., Пучков Л. В. —
Журн. общ. хим., 1980, 50, с. 2393.
1107. Челебаев А. К. — Автореф. дис. . . . канд. техн.
наук. М.: МАИ, 1977.
1108. Чернеев Е. И. — В кн.: Теплофизические
свойства рабочих тел энергетических установок:
Сборник трудов. М.: ЭНИН, 1977, вып. 61, с. 49.
1109. Чернеев Е. И. — В кн.: Теплофизические
свойства рабочих тел энергетических установок:
Сборник трудов. М.: ЭНИН, вып. 61, с. 58.
1110. Чернеева Л. И., Проскурин В. Н. —
Теплофизика высоких температур, 1972, 10, с. 674.
1111. Чернеева Л. И., Проскурин В. Н'. —
Теплофизика высоких температур, 1972, 10, с. 765.
1112. Черниховский А. — Теплофизика высоких
температур, 1968, 6, с. 809.
1113. Черняев В. С, Летун С. М., Швейкин Г. П. —
Труды Уральск, политехи, ин-та, 1970, № 186,
с. 167.
1114. Черняев В. С, Швейкин Г. П., Гелъд П. В. —
Труды Уральск, политехи, ин-та, 1970, № 186,
с. 205.
1115. Черняев В. С, Швейкин Г. П., Гелъд П. В. —
Изв. АН СССР. Неорган, матер., 1972, 8, с. 459.
1116. Черняев В. С, Щетников Е. Н., Швейкин Г. П.,
Гельд П. В. — Изв. АН СССР. Неорган, матер.,
1968, 4, с. 2117.
1117. Черняев И. И., Соколов В. А., Палкин В. А. —
Изв. сектора платины и других благородных
металлов ИОНХ АН СССР, 1954, 28, с. 142.
1118. Чеховской В. Я. — Инж.-физ. журн., 1962, 5,
с. 43.
1119. Чеховской В. Я. — Обзоры по теплофизическмм
свойствам веществ. М.: ИВТАН, 1979, № 6 B0).
1120. Чеховской В. Я., Березин Б. Я. — Инж.-физ.
журн., 1971, 21, с. 309.
1121. Чеховской В. Я., Березин Б. Я. — В кн.: Тепло-
физич. св-ва твердых веществ. Материалы 4-й
Всесоюз. теплофизич. конф. по свойствам
веществ при высоких температурах/ Под ред.
Г. В. Самсонова. М.: Наука, 1973, с. 128.
1122. Чеховской В. Я., Жукова И. А. — Изв. АН СССР.
Металлы, 1980, № 3, с. 67.
1123. Чеховской В. Я., Жукова И. А., Тарасов В. Д. —
Теплофизика высоких температур, 1979, 17,
с. 754.
1124. Чеховской В. Я., Калинкина Р. Г. —
Теплофизика высоких температур, 1973, 11, с. 885.
1125. Чеховской В. Я., Калинкина Р. Г. — Журн.
физ. хим., 1975, 49, с. 740.
1126. Чеховской В. Я., Кац С. А., Коваленко М. Д. —
Теплофизика высоких температур, 1982 (в
печати).
1127. Чеховской В. Я., Петров В. А. — Теплофизика,
высоких температур, 1968, 6, с. 752.
1128. Чеховской В. Я., Петров В. А. — Физика
твердого тела, 1970, 12, с. 3056.
1129. Чеховской В. Я., ШумяцкийБ. Я., ЯкимовичК. А.
— Инж.-физ. журн., 1962, 5, с. 13.
ИЗО. Чехунова Н. П., Проценко П. И., Венеровскал
Л. Н. — Журн. физ. хим., 1969, 43, с. 2070.
1131. Чижиков Д. М., Казенас Е. К., Ермилова И. О. —
В кн.: Химия парообразных неорганических
соединений и процессов парообразования:
Материалы Всесоюз. конф., Минск, 24—26/V-1973.
Минск, 1973, с. 217.
1132. Чижиков Д. М., Казенас Е. К., Ермилова И. О. —
Журн. физ. хим., 1976, 50, с. 1454.
1133. Чижиков Д. М., Казенас Е. К., Ермилова И. О. —
Журн. физ. хим., 1976, 50, с. 3130.
1134. Чижиков Д. М., Павлов Ю. А., Цветков Ю. В.,
Казенас Е. К., Нестеренко П. А. — Изв. вузов.
Черн. мет., 1970, № 7, с. 5.
1135. Чижиков Д. М., Цветков Ю. В., Казенас Е. Л".,
Тагиров В. К. — Журн. физ. хим., 1972, 46,
с. 806.
1136. Чикова П. Н. — В кн.: Сборник материалов
четвертой научной конференции аспирантов. Ростов-
на-Дону: Ростовский Гос. ун-т, 1962, с. 103.
1137. Числер 9. В. — Физика твердого тела, 1969,
11, с. 1272.
1138. Числер 9. В., Гончарик И. Н. — Физика
твердого тела, 1971, 13, с. 1384.
1139. Чудинов 9. Г., Чопоров Д. Я. — Журн. физ.
хим., 1970, 44, с. 1955.
1140. Шапкин П. С, Василъкова И. В., Сусарев М. П. —
Журн. неорган, хим., 1967, 12, с. 2838.
1141. Шаповалов А. М. — Автореф. дис. . . . канд.
хим. наук. М.: МГУ, 1980.
1142. Шаповалов А. М., Шевелъков В. Ф., Мальцев А. А*
— Вестн. МГУ. Сер. хим., 1973, 14, с. 151.
1143. Шаповалов А. М., Шевелъков В.Ф., Мальцев А. А.
— Вестн. МГУ. Сер. хим., 1975, 16, с. 153.
1144. Шаповалов А. М., Шевелъков В. Ф., Мальцев'
А. А. — В кн.: XI Менделеевский съезд по
общей и прикладной химии: Рефераты докл. и
сообщений. М.: Наука, 1975, № 1, с. 12.
1145. Шатова 3. Ю., Борисоглебский Ю. В., Ветю-
ков М. М., Ширпановский В. В. — Журн. прикл.
хим., 1979, 52, с. 542.
1146. Шахно И. В., Плющев В. Е. — Журн. неорган.
¦ хим., 1960, 5, с. 1172.
1147. Шевченко В. Г., Кононенко В. И., Сухман А. Л. —
Журн. физ. хим., 1978, 52, с. 1867.
1148. Шевченко А. В., Лопато Л. М., Майстер И. М.г
Зайцева 3. А. — Изв. АН СССР. Неорган,
матер., 1980, 16, с. 1450.
1149. Шевченко А. В., Лопато Л. М., Рубан А. К. —
Допов. АН УРСР. Сер. Б: геол., хим., биол.
наук, 1976, № 10, с. 922.
1150. Шестопал В. Д. — Физика твердого тела, 1965,
7, с. 3461.
1151. Широков II. И. — Изв. АН СССР. Металлы,
1973, № 2, с. 102.
1152. Шишолина Р. П., Проценко П. И. — Журн.
неорган, хим., 1963, 8, с. 2741.
1153. Шмидт Н. Е. — Журн. неорган, хим., 1967,
12, с. 1766.
1154. Шмидт Н. Е., Максимов Д. П. — Журн. физ»
хим., 1979, 53, с. 1895.
542
Литература
1155. Шмидт Н. Е., Соколов В. А. — Журн. неорган,
хим., 1961, 6, с. 2613.
1156. Шнип В. Л., Новиков Г. И. — ВИНИТИ. Деп. 1183.
№ 4528-72. М., 1972.
1157. Шоканов А. К., Бакеев М. И. — Труды хим.- 1184.
металлург, ин-та АН Каз. ССР, 1967, 3, с. 102.
1158. Шолохович М. Л., Крамаров О. П., Берберова 1185.
Л. М. — В кн.: Совещание по физ. химии рас-
плавл. солей и шлаков: Труды совещания, Киев, 1186.
1963. Киев: Изд-во АН УССР, 1969, с. 73.
1159. Шолъц В. В., Сидоров Л. Н. — Вестн. МГУ. 1187.
Сер. хим., 1972, 13, с. 371.
1160. Шорина Л. Л., Полищук А. В., Мондин Л. Я. — 1188.
Изв. АН СССР. Неорган, матер., 1975, 11, с. 1715.
1161. Шпакова С. Г., Дракин С. И., Карапетъянц
М. X. — Труды Моск. хим.-технол. ин-та им. 1189.
Д. И. Менделеева, 1973, вып. 75, с. 3.
1162. Шпильрайн 9. 9., Белова A.M. — Теплофизика 1190.
высоких температур, 1967, 5, с. 531.
1163. Шпильрайн, 9. 9., Белова A.M. — Теплофизика
высоких температур, 1968, 6, с. 342.
1164. Шпильрайн Э. 9., Зверева А. М. —Инж.-физ.
журн., 1963, 6, с. 74. 1191.
1165. Шпильрайн 9. 9., Каган Д. Н. — Теплофизика
высоких температур, 1969, 7, с. 362. 1192.
1166. Шпильрайн 9. 9., Каган Д.Н. — Теплофизика
высоких температур, 1969, 7, с. 577. 1193.
1167. Шпильрайн 9. 9., Каган Д. Н. —Теплофизика
высоких температур, 1970, 8, с. 1. 1194.
1168. Шпильрайн 9. 9., Каган Д. Н., Бархатов Л. С,
Жмапин Л. И., Королева В. В. — Инж.-физ. 1195.
журн., 1980, 38, с. 581.
1169. Шпильрайн 9. 9., Каган Д. Н., Кречетова Г. А.— 1196.
Теплофизика высоких температур, 1971, 9, с. 1301.
1170. Шпильрайн 9. 9., Ковердяев Б. Н. Цит. по: 1197.
[1179, с. 51].
1171. Шпильрайн 9. 9., Никоноров 9. В. — В кн.:
Теплофизические свойства газов. М.: Наука, 1198.
1970, с. 141.
1172. Шпильрайн 9. 9., Никоноров Э. В. —
Теплофизика высоких температур, 1971, 9, с. 434. 1199.
1173. Шпильрайн 9. 9., Никоноров 9. В. —
Теплофизика высоких температур, 1972, 10, с. 297. 1200.
1174. Шпильрайн 9. 9., Солдатенко Ю. А., Якимо-
вич К. А., Фомин В. А., Савченко В. А., Бе- 1201.
лова В. А., Каган Д. Н., Крайнева И. Ф. —
Теплофизика высоких температур, 1965, 3, с. 930. 1202.
1175. Шпилърайн 9. д., Тоцкий Е. Е., Кар'мышин
Ю. В. — Труды Моск. энергетич. ин-та, 1970, 1203.
вып. 75, с. 62.
1176. Шпильрайн Э. Э., Тоцкий Е. Е., Шерешевский 1204.
В. А. — Теплофизика высоких температур, 1968,
6, с. 924. 1204а
1177. Шпильрайн 9. 9., Якимович К. А. Гидрид лития.
Физико-химические и теплофизические
свойства. М.: Изд-во стандартов, 1972.
1178. Шпилърайн 9. 9., Якимович К. А., Мозговой
А. Г. — Теплофизика высоких температур, 1977,
15, с. 1104.
1179. Шпильрайн 9. 9., Якимович К. А., Тоцкий Е. Е.,
Тимрот Д. Л., Фомин В. А. Теплофизические 1205.
свойства щелочных металлов / Под. ред. В. А.
Кириллина. М.: Изд-во стандартов, 1970. 1206.
1180. Шпильрайн 9. 9., Якимович К. А.,
Шерешевский В. А. — Теплофизика высоких температур, 1207.
1977, 15, с. 661.
1181. Шульц М. М., Борисова Н. В., Ведищева Н. М., 1208.
Пивоваров М. М. — Физика и химия стекла,
1979, 5, с. 36. 1209.
1182. Щукарев С. А., Семенов Г. А. — В кн.: Иссле-
543
дования в области химии силикатов и окислов
М.; Л.: Наука, 1965, с. 208.
Щукарев С. А., Семенов Г. А., Францева К. Е. —
Журн. неорган, хим., 1959, 4, с. 11.
Щукарев С. А-, Семенов Г. А., Францева К. Е. —
Журн. неорган, хим., 1959, 4, с. 2638.
Щукарев С. А., Семенов Г. А., Францева К. Е. —
Докл. АН СССР, 1962, 145, с. 119.
Щукарев С. А., Семенов Г. А., Францева К. Е. —
Изв. вузов. Хим. и хим. технол., 1962, 5, с. 691.
Щукарев С. А., Семенов Г. А., Францева К. Е. —
Журн. неорган, хим., 1966, 11, с. 233.
Юдин,Б. Ф., МогилевскийВ.И., ПолонскийЮ. А.г
Лапшин С. А. — Журн. прикл. хим., 1976, 49,
с. 776.
Ягфаров М. Ш. — Журн. физ. хим., 1969, 43,
с. 1620.
Якимович К. А., Мельникова Т. Н. —
Термодинамические свойства гидрида лития и его
изотопических модификаций в твердой фазе: Обзоры
по теплофизическим свойствам веществ. М.:
ИВТАН, 1979, № 5/A9).
Яковлева М. С, Красилова 3. Л. — Вестн. ЛГУ.
Сер. физ. н хим., 1961, вып. 3, № 16, с. 136.
Ямпольский В. И. — Автореф. дис. . . . канд.
хим. наук. М.: МГУ, 1973.
Ямпольский В. И., Мальцев А. А. — Журн.
неорган, хим., 1970, 15, с. 1998..
Ямпольский В. И., Мальцев А. А. ¦— Журн.
неорган, хим., 1973, 18, с. 262.
Ярым-Агаев Н. А. — Журн. физ. хим., 1964,
38, с. 2579.
Яцимирский К. Б., Графова 3. М. — Журв.
общ. хим., 1953, 23, с. 717.
Яшвили Т. С, Цагарейшвили Д. Ш., Гвеле-
сиани Г. Г. — Сообщ. АН Груз.ССР, 1967, 46,
с. 409.
Яшвили Т. С, Цагарейшвили Д. Ш., Гвелеси-
ани Г. Г. — Теплофизика высоких температур,
1968, 6, с. 817.
Abrahams E., Grant N. — J. Metals, 1956, 8,
p. 975.
Abramowitz S. — U. S. NTIS, AD-A Rept N 008438.
1975.
Abramowitz S., Acquista N. — J. Phys. Chem.,
1972, 76, p. 648.
Abramowitz S., Acquista N. — J. Res. NBS, 1974,
A78, p. 421.
Abramowitz S., Acquista N., Levin I. W. — J. Res.
NBS, 1968, A72., p. 487.
Abramowitz S., Acquista N., Thompson K. R. —
J. Phys. Chem., 1971, 75, p. 2283.
. Abramowitz S., Armstrong G. Т., Beckett C. W.,
Churney K. L., Dibeler V. H., Douglas Т. В.,
Herron J. Т., Krause R. F., Jr., McCulloh К. Е.,.
Reilly M. L., Rosenstock H. M., Tsang W. —
New Ideal-Gas Thermochemical Tables (In the
Format of the JANAF thermochemical tables).
AFOSR Sci. Rept AFOSR-TR-72-2004. NBS Rept
10904. Wash., 1972.
Abramson R. — In: Metal Plutonium. Chicago
Univ. Press, 1961, p. 123.
Accao G., Ioshida Т., Ando R. — J. Phys. Soc.
Japan, 1962, 17, p. 442.
Achener P. Y. — U. S. Aerojet General Corp.
Rept AGN-8192, vol. 1. San Ramon (Calif.), 1964.
Achener P. Y. — U. S. Aerojet General Corp..
Rept AGN-8090, vol. 1, San Ramon (Calif.), 1964.
Achener P. Y. — U. S. Aerojet General Corp.-
Rept AGN-8141. San Ramon (Calif.), 1965.
Литература
1210. Achener P. У. — In: Alkali metals. Intern. Symp.
Held in Natingham, July, 1966. Chem. Soc. Spec.
Publ. N 22, L., 1967, p. 75.
1211. Achener P. Y., Fisher D. L. — U. S. Aerojet
General Corp. Rept AGN-8191, vol. 2. San Ramon
(Calif.), 1967.
1212. Achener P. Y., Fisher D. L. — U. S. Aerojet
General Corp. Rept AGN-8191, vol. 6. San Ramon
(Calif.), 1967.
1213. Achener P. Y., et al. — U. S. Aerojet General
Corp. Rept AGN-8494, vol. 2. San Ramon (Calif.),
1967.
1214. Achener P. Y., Jouthas J. T. — U. S. Aerojet
General Corp. Rept AGN-8191, vol. 1. San Ramon
(Calif.), 1966.
1215. Achener P. Y., Mackewichz W. F., Fisher D. L.,
Camp D. C. — U. S. Aerojet General Corp. Rept
AGN-8195, vol. 1. San Ramon (Calif.), 1968.
1216. Ackermann R. J., Faircloth R. L., Rand M. H. —
J. Phys. Chem., 1966, 70, p. 3698.
1217. Ackermann R. /., Garg S. P., Rauh E. G. — J.
Amer. Ceram. Soc, 1977, 60, p. 341.
1218. Ackermann R. J., Gilles P. W., Thorn R. J. —
J. Chem. Phys., 1956, 25, p. 1089.
1219. Ackermann R. J., Rauh E. G. — J. Chem. Phys.,
1962, 36, p. 448.
1220. Ackermann R. J., Rauh E. G. — J. Phys. Chem.,
1963, 67, p. 2596.
1221. Ackermann R. J., Rauh E. G. — J. Phys. Chem.,
1969, 73, p. 769.
1222. Ackermann R. J., Rauh E.G. — J. Chem. Thermo-
dyn., 1971, 3, p. 445.
1223. Ackermann R. J., Rauh E. G. — J. Chem. Thermo-
dyn., 1971, 3, p. 609.
1224. Ackermann R. J., Rauh E. G. — High Temp.
Sci., 1972, 4, p. 272.
1225. Ackermann R. /., Rauh E. G. — High Temp. Sci.,
1972, 4, p. 496.
1226. Ackermann R. J., Rauh E. G. — J. Chem. Thermo-
dyn., 1972, 4, p. 521.
1227. Ackermann R. J., Rauh E. G. — High Temp.
Sci., 1973, 5, p. 463.
1228. Ackermann R. J., Rauh E. G. — 3. Chem. Thermo-
dyn., 1973, 5, p. 331.
1229. Ackermann R. J., Rauh E. G. — J. Chem. Phys.,
1974, 60, p. 2261.
1230. Ackermann R. J., Rauh E. G. — J. Chem. Phys.,
1974, 60, p. 2266.
1231. Ackermann R. J., Rauh E. G. — J. Chem. Ther-
modyn., 1975, 7, p. 211.
1232. Ackermann R. J.,Rauh E. G., Rand M.H.— Rev.
int. hautes temp, et refract., 1978, 15, p. 259.
1233. Ackermann R. J., Rauh E. G. — In:
Thermodynamics Nuclear Materials. Proc. Symp.. Jiilich,
29 Jan. — 2 Feb. 1979. Vienna: IAEA, 1980,
vol. I, p. 11.
1234. Ackermann R. J., Rauh E. G., Alexander С. А. —
High Temp. Sci., 1975, 7, p. 304.
1235. Ackermann R. J., Rauh E. G., Chandrasekhara-
iah M. S. — J. Phys. Chem., 1969, 73. p. 762.
1236. Ackermann R. J., Rauh E. G., Thorn R. J. —
J. Chem. Phys., 1964, 40, p. 883.
1237. Ackermann R. J., Rauh E. G., Thorn R. J. —
J. Chem. Phys., 1976, 65, p. 1027.
1238. Ackermann R. J., Rauh E. G., Thorn R. J.,
Cannon M. C. — J. Pbys. Chem., 1963, 67, p. 762.
1239. Ackermann R. J., Rauh E. G., Walters R. R. —
J. Chem. Thermodyn., 1970, 2, p. 139.
1240. Ackermann R. J., Sorrell Ch. A. — High Temp.
Sci., 1970, 2, p. 119.
1241. Ackermann R. J., Thorn R. J. — In: XVI Congr.
intern, de chimie pure and applique, Paris, 1957:
Mem. sect. chim. minerale, P., 1958, p. 667.
1242. Ackermann R. J., Thorn R. J. — In: Proc. 2nd
Intern. Peaceful Uses Atom. Energy, Geneva,
1958. Geneva: United Nations, 1958, vol. 28,
p. 180.
1243. Ackermann R. J., Thorn R. J. — In: Progress
in ceramic science/Ed. J. E. Burke. Oxford etc.,
1961, vol. 1, p. 39, p. 81.
1244. Ackermann R. J., Thorn R. J. — In: Thermodyn.
Nucl. Mater. Proc. Symp. Vienna, 1965. Vienna;
IAEA, 1966, vol. I, p. 243.
1245. Ackermann R. J., Thorn R. J., Alexander C,
Tetenbaum M. — J. Phys. Chem., 1960, 64, p. 350.
1246. Acquista N., Abramowitz S. — J. Chem. Phys.,
1969, 51, p. 2911.
1247. Acquista N., Abramowitz S., bide D. R. — J.
Chem. Phys., 1968, 49, p. 780.
1248. Adami L. #., Joe C. J. — U. S. Bur. Mines. Rept
Invest., 1968, N 7167.
1249. Adams A., Klemperer W., Dunn T.—Canad.
J. Phys., 1968, 46, p. 2213.
1250. Adams С E., Quan J. T. — J. Phys. Chem.,
1966, 70, p. 331.
1251. Adams D. M., Sharma Sh. K. — Chem. Phys.
Lett., 1975, 36, p. 407.
1252. Adams D. M., Sharma S. K. — J. Chem. Soc.
Faraday Trans. II, 1976, 72, p. 2069.
1253. Adams M. /., House J. E. — Trans. Illinois
State Acad. Sci., 1970, 63, N 1, p. 83.
1254. Addison C. C, Pulham R. J., Roy R. J. — 1.
Chem. Soc, 1964, p. 4895.
1255. Adenstedt H. K. — Trans. Amer. Soc. Metals,
1952, 44, p. 949.
1256. Adenstedt H. K., Pequignot J. R., Raymer J. M. —
Trans. Amer. Soc. Metals, 1952, 44, p. 990.
1257. Adler D., Feinleib J. — Phys. Rev. Lett., 1964,
12, p. 700.
1258. Adler S. F., Selwood P. W. — J. Amer. Chem.
Soc, 1954, 76, p. 346.
1259. Adlhart W., Fritsch G., Luscher E. ~ Phys. Lett.,
1974, A48, p. 239.
1260. Ado G. — These doct. Fac. Sci. Paris Univ. P.,
1971, p. 72.
1261. Afaf M. — Proc. Phys. Soc. London, 1950, A63,
p. 674.
1262. Afaf M. — Proc. Phys. Soc. London, 1950, A63,
p. 1156.
1263. Affortit C. — Rapport CEA-R 3287. Paris, 1967.
1264. Affortit C. — High Temp.-High Pressur., 1969,
I, p. 27.
1265. Affortit C, Lallement R. — Rev. int. hautes temp,
et refract., 1968, 5, p. 19.
1266. Affortit C., Marcon J. P. —¦ Rev. int. hautes temp,
et refract., 1970, 7, p. 236.
1267. Agarwal K. L., Retterton J. O. — In: Proc. Intern.
Conf. Low Temp. Phys., 13th 1972 (Publ. 1974)/
Ed. K. D. Timmermans et al., N. Y., 1974, vol. 4,
p. 597.
1268. Agarwal K. L., Betterton J. O. — J. Low Temp.
Phys., 1974, 17, p. 515.
1269. Agostini P. — Atti Accad. naz. Lincei Rend. Cl.
sci. fis., mat. e natur., 1928, 71, p. 1030.
1270. Agron P. A. — Office of Technical Information
Extension. U. S. AEC Rept 5290. Oak Ridge
(Tennessee), 1948, vol. 2, p. 610.
1271. Agte C, Alterthum H. — Z. tech. Phys., 1920,
II, S. 182.
544
Литература
1272. Ahtee M., Hewat A. W. — Phys. status solidi,
1980, A58, p. 525.
1273. Akerlind L. —Ark. fys., 1957, 11, s. 395.
1274. Akerlind L. — Ark. fys., 1962, 22, s. 41.
1275. Akerlind L. —Ark. fys., 1962, 22, s. 65.
1276. Akimov A. I., Kraftmakher Y. A. — Phys. status
solidi, 1970, 42, p. K41.
1277. Akishin P. A., Rambidi N, G. — Z. phys. Chem.
Leipzig, 1960, 213, S. 111.
1278. Albrecht Д., Hdusler H., Mobius R. — Z. anorg.
und allg. Chem., 1970, 377, S. 310.
1279. Albright D. M. — Ph. D. Thesis. Carnegie Inst.
Technol., 1956.
1280. Albright J. G. — Z. Kristallogr., 1932, 84, S. 150.
1281. Albritton G. L., Harrop W. /., Schmeltekopf A. L.,
Zare R. N., Grow E. E. — J. Mol. Spectrosc,
1973, 46, p. 67.
1282. Alcock С. В., Grievson P. — J. Inst. Metals, 1962,
90, p. 304.
1283. Alcock С. В., Peleg M. — Trans. Brit. Ceram.
Soc, 1967, 66, p. 217.
1284. Alcock С. В., Stavropoulos G. P. — Canad. Met.
Quart., 1971, 10, p. 257.
1285. Alcock С. В., Zador S., Steele В. С H. — Proc.
Brit. Ceram. Soc, 1967, p. 231.
1286. Alekseevski N. E., Migunov L. — J. Phys. (USSR),
1947, 11, p. 95.
1287. Alessandrini V. A., Cracknell A. P., Przystawa
J. A. — Commun. Phys., 1976, 1, p. 51.
1288. Alexander D. <?., Carlson O. N. —Met. Trans.,
1971, 2, p. 2805.
1289. Alexander L. E., Beattie J. R., Bukovszky A.,
Jones P. J., Marsden C. J., Van Schalkwyk G. J. —
J. Chem. Soc. Dalton Trans., 1974, p. 81.
Alfred A. M., Myles K. M. — Trans. Met. Soc.
AIME, 1964, 230, p. 736.
Allegrini M., Moi L., Arimondo E. — Chem.
1290.
1291.
1292. Allen Z. C, Russell
Phys. Lett., 1977, 45, p. 245.
Allen L. C, Russell J. D. — J.
1967, 46, p. 1029.
Chem. Phys.,
1293. Allen R. D. — Nature, 1962, 193, p. 769.
1294. Allen R. P., Arrowsmith H. W. — U. S. Rept
BNWL-SA-5328, 1975, p. 12.
1295. AllendorferA. — Z. Naturforsch., 1950, 59, S. 234.
1296. Alles A., Westrum E. F. — Fifth Intern. Conf.
on Chem. Thermodyn., Aug., 23—26, 1977. Ron-
neby Sweden. Abstr. of Poster Papers, p. 22.
1297. Allmand A. /., Polack W. G. — J. Chem. Soc,
1919, 115, p. 1020.
1298. Almldf J., Ishenko A. A. — Chem. Phys. Lett.,
1979, 61, p. 79.
1299. Almy G. M., Better A. C. — Phys. Rev., 1942,
61, p. 476.
1300. Almy G. M., Наше С. D. —Phys. Rev., 1932,
42, p. 242.
1301. Almy G. M., Irwin G. R. — Phys. Rev., 1936,
49, p. 72.
1302. Almy G. M., Rassweiler M. — Phys. Rev., 1938,
53, p. 890.
1303. Altman R. L. — J. Chem. Phys., 1959, 31, p. 1035.
1304. Amadori M. — Atti Accad. naz. Lincei Mem. Cl.
sci. fis., mat. e nat. Sez. II, 1912, 21, p. 467.
1305. Amadori M. — Atti Accad. naz. Lincei Mem.
Cl. sci. fis., mat. e nat. Sez. II, 1912, 21, p. 688.
1306. Amadori M. — Atti Accad. naz. Lincei Mem. Cl.
sci. fis., mat. e nat. Sez. II, 1912, 21, p. 1166.
1307. Amadori M., Pampanini G. — Atti Accad. naz.
Lincei Mem. Cl. sci. fis., mat. e nat. Sez. II, 1911,
20, p. 473.
1308. Amaya K., Keesom P. #., Wenger L. E. — Phys.
Rev., 1977, B16, p. 1042.
1309. Ambrosetti R., Angelone R., Colligiani A., Riga-
monti A. — Phys. Rev., 1977, B15, p. 4318.
1310. Ames L. L., Walsh P. TV., White D. — J. Phys.
Chem., 1967, 71, p. 2707.
1311. Ames L. L., Wang J. L.-F., Margrave J. L. —
Inorg. and Nucl. Chem. Lett., 1973, 9, p. 1243.
1312. Amigo J. M., Curreras G., Cesari E., Navarro J.,
Reventos M. M., Rulio A., Torra V. — In: Quan-
trinie conf. intern, de thermodynam. chim. Mont-
pellier, 1975, vol. 1, p. 209.
1313. Amirthalingam V. — i. Nucl. Mater., 1966, 20,
p. 193.
1314. Amphlett С. В., Mullinger L. W., Thomas L. F. —
Trans. Faraday Soc, 1948, 44, p. 927.
1315. Anderson С. Т. — J. Amer. Chem. Soc, 1933
55, p. 3621.
1316. Anderson С. Т. — J. Amer. Chem. Soc, 1936,
58, p. 564.
1317. Anderson С. Т. — J. Amer. Chem. Soc, 1937
59, p. 488.
1318. Anderson E., Noyes H. A. — J. Phys. Chem.,
1913, 17, p. 249.
1319. Anderson J. S. — Bull. Soc chim. France, 1953,
N 9, p. 781.
1320. Anderson J. S., Sawyer J. O., Worner H. W.,
Willis G. M., Bannister M. J. — Nature, 1960,
185, p. 915.
1321. Anderson J. W., Gilmor R. R., Marman W. J.—
In: Plutonium 1965. Proc. 3d Intern. Conf. London,
1965 / Ed. A. E. Kay, M. B. Walderon. L.:
Chapman and Hall, 1967, p. 39.
1322. Andersson G. — Acta chem. scand., 1963, 8, p. 413.
1323. Andersson S. — Ark. Kemi, 1960, 15, s. 247.
1324. Andersson S., Collen В., Kuylenstierna U., Mag-
neli A. —Acta chem. scand., 1957, 11, p. 1641.
1325. Andres K. — Phys. Rev., 1970, B2, p. 3768.
1326. Andrews L. — J. Chem. Phys., 1969, 50, p. 4288.
1327. Andrews L. — J. Phys. Chem., 1969, 73, p. 3922.
1328. Andrews L. — J. Chem. Phys., 1971, 54, p. 4935.
1329. Andrews L. — Appl. Spectrosc. Rev., 1976, 11,
p. 125.
1330. Andrews L., Hwang J. Т., Trindle C. — J. Phys.
Chem., 1973, 77, p. 1065.
1331. Andrews T. —Ann. chim. phys., 1845, 14, p. 68.
1332. Andrews T. — Brit. Assoc. Adv. Sci. Rept., 1849,
19, p. 63.
1333. Andrews T. — J. Chem. Soc, 1870, 23, p. 432.
1334. Anthony A. M., Kiyoura R., Sata T. — C. r. Acad.
sci., 1962, 255, p. 1606.
1335. Antropoff A. V., Sommer W. — Z. phys. Chem.
Leipzig, 1926, 123, S. 161.
1336. Aoyama S., Kanda E. — J. Chem. Soc. Jap.,
1934, 55, p. 1174.
1337. Aral Т., Sakanoto M. — J. Chem. Phys., 1958,
28, p. 32.
1338. Arell A. —Ann. Acad. sci. Fennicae, Ser. AVI,
1962, N 101.
1339. Arell A., Roiha M., Aaltonen M. — Phys. kondens.
Mater., 1967, 6, S. 140.
1340. Arell A., Varteva M. — Ann. Acad. sci. Fennicae,
Ser. AVI, 1961, N 88.
1341. Aring K., Sievers A. J. — J. Appl. Phys., 1967,
38, p. 1496.
1342. Argue G. R., Mercer E. E., Cobble J. W. — J. Phys.
Chem., 1961, 65, p. 2041.
1343. ArkelA. E., Flood E. A., Bright N. F.H. — Canad.
J. Chem., 1953, 31, p. 1009.
545
Литература
1344. Armentrout Р. В., Beauchamp J. L. — Chem.
Phys., 1980, 50, p. 21.
1345. Armentrout P. В., Beauchamp J. L. — Chem
Phys., 1980, 50, p. 27.
1346. Armstrong L. D., Grayson-Smith H. — Canad.
J. Res., 1950, A28, p. 51.
1347. Arnett E. M., Campion J. J. — 3. Amer. Chem.
Soc, 1970, 92, p. 7097.
1348. Arrhenius S. — Z. phys. Chem. Leipzig, 1889, 4,
S. 96.
1349. Arrott A., Goldmann J. E. — Phys. Rev., 1955,
99, p. 1641.
1350. Arrott A., Goldman J. E. — Phys. Rev., 1957,
108, p. 948.
1351. Arthur J. S. — J. Appl. Phys., 1950, 21, p. 732.
1352. Asbrink G., Asbrink S., Magneli A., Okinaka H.,
Kosuge K., Kachi S. — Acta chem. scand., 1971,
25, p. 3889.
1353. Asbrink S., Magneli A. — Acta crystallogr.,
1959, 12, p. 575.
1354. AskewF.A.,BullockE., SmithH. Т., Tinkler R. K.,
Gatty O., Wolfenden J. W. — J. Chem. Soc,
1934, p. 1368.
1355. Athenour Ch., Bacis R., Femenias J. L., Strin-
gat R. — С. г. Acad. sei., 1970, B271, p. 567.
1356. Atkins R. M., Gingerich K. A. — Chem. Phys.
Lett., 1978, 53, p. 347.
1357. Atoji M., McDermott M. J. —Acta crystallogr.,
1970, B26, p. 1540.
1358. Attree R. W., Cushing R. L., Ladd J. A., Pie-
гот J. J. — Rev. Sci. Instrum., 1958, 29, p. 491.
1359. Auby R., Bernard M. J., Massaux M, — С. г.
Acad. sci., 1968, 266, p. 425.
1360. Audrieth L. F., Mills J. R., Netherton L. E. —
J. Phys. Chem., 1954, 58, p. 482.
1361. Aukrust E., Bjorge В., Flood #., Forland T. —
Ann. N. Y. Acad. Sci., 1960, 79, p. 830.
1362. Ault B. S., Andrews L. — J. Amer. Chem. Soc,
1976, 98, p. 1591.
1363. Austin I. G. — Philos. Mag., 1962, 7, p. 961.
1364. Austin J. M., Mair A. D. — J. Phys. Chem.,
1962, 66, p. 519.
1365. Avert M., Graig R. S., Waite T. R., Wallace
W. E. — Phys. Rev., 1956, 102, p. 1263.
1366. Babeliowsky T. P. J. H. — Physica, 1962, 28,
p. 1160.
1367. Babeliowsky T. P. J. H., Boerbom A. J. H. —
In: Advances Mass Spectrometry: Proc. Conf.
2nd. Oxford, 1961. Oxford etc.: Pergamon Press,
1963, vol. 2, p. 135.
1368. Baboian R., Flengas S. N. — Canad. J. Chem.,
1967, 45, p. 813.
1369. Bacis R., Collomb R., Bessis N. — Phys. Rev.,
1973, AS, p. 2255.
/. - J.
189, p. 124.
1370. Backhurst
Iron and Steel Inst., 1958,
1371. Badr Y. A., El-Kaffany F., Tosson M. — Phys.
status solidi, 1979, A53, p. K51.
1372. Bagnall K. W., Wakerley M. W. — J. Inorg.
and Nucl. Chem., 1975, 37, p. 329.
1373. Balfour W. /., Lindgren B. — Phys. scr., 1980,
22, s. 36.
1374. Balfour W. J., Tatum J. B. — J. Mol. Spectrosc,
1973, 48, p. 313.
1375. Balducci G., De Maria <?., Guido M., Piacente V. —
J. Chem. Phys., 1972, 56, p. 3422.
1376. Balducci <?., Gigli G., Guido M. — High Temp.
Sci., 1977, 9, p. 149.
1377. Balducci G., Gigli G., Guido M. — l. Chem. Phys.,
1977, 67, p. 147.
1378. Balk P., Benson G. С — J. Phys. Chem., 1959r
63, p. 1009. m
1379. Bando Y., Nagasawa K., Kato Y., Takada T. —
J. Appl. Phys. Jap., 1969, 8, p. 633.
1380. Bannister M. J. — J. Nucl. Mater., 1967, 24,.
p. 340.
1381. Bannister G. #., Thomson J. R. — J. Nucl. Mater.,.
1964, 12, p. 16.
1382. Banus M. D. — Mater. Res. Bull., 1968, 3, p. 723..
1383. Banus M. D., McSharry J. J., Sullivan E. A. —
J. Amer. Chem. Soc, 1955, 77, p. 2007.
1384. Banus M. D., Reed Т. В. — In: The chemistry
of extended defects in non-metallic solids/ Ed-
L. R. Eyring, M. O'Keeffe. Amsterdam; Londonr
North-Holl. Publ. Co., 1970, p. 488.
1385. Banus M. D., Reed Т. В., Strauss A. J. — Phys..
Rev., 1972, B5, p. 2775.
1386. Barany R. — U. S. Bur. Mines. Rept. Invest.,. .
1964, N 6356.
1387. Barany R., Adami L. H. — U. S. Bur. Mines..
Rept Invest., 1966, N 6873.
1388. Barbi G. B. — J. Phys. Chem., 1964, 68, p. 1025.
1389. Barbi G. B. — J. Less-Common Metals, 1970,.
22, p. 487.
1390. Barbi G. B. — Z. Naturforsch., 1970, A25, S. 1515.
1391. Barde R., Heuzi A., Dubois J., Millet J. — С. г..
Acad. sci., 1967, C265, p. 1257.
1392. Bardsley J. N., Junker B. R., Norcross D. W. —
Chem. Phys. Lett., 1976, 37, p. 502.
1393. Barfknecht А. Т., Keer H. V., Honig J. M. —
Phys. Rev., 1977, B15, p. 2406.
1394. Barieau R. E., Giauque W. F. — J. Amer. Chem..
Soc, 1950, 72, p. 5676.
1395. Barin /., Knacke O. Thermochemical properties
of inorganic substances. B. etc: Springer-Verl.,
1973.
1396. Barin I., Knacke O., Kubaschewski O.
Thermochemical properties of inorganic substances:
Supplement. B. etc: Springer-Verl., 1977.
1397. Barner J. O., Fulrath R. M. — Rept N 10706,.
Univ. Calif. Lawrence Radiat. Lab., 1963, p. 147.
1398. Barnes G. — Colloq. intern. Centre nat. rech. sci.,.
1972, N 205, p. 327.
1399. Barnes J. C, Sesay L. J. — J. Less-Common,
Metals, 1979, 63, p. 301.
1400. Barnes J. F. — In: Thermodynamics of Nuclear
Materials. Vienna: IAEA, 1975, vol. 1, p. 327.
1401. Barocco A. — С. г. Acad. sci., 1938, 206, p. 1117..
1402. Barrat S. — Proc. Roy. Soc. London, 1924,
A105, p. 221.
1403. Barri G. B. — Z. Naturforsch., 1968, A23, S. 800..
1404. Barren T. H. K., LeadbetterA. J., Morrison J. A.—
Proc. Roy. Soc. London, 1964, A279, p. 62.
1405. Barros H. L., Chandrashekhar G. V., Chi T. C.r
Honig J. M., Sladek R. J. — Phys. Rev., 1973,
B7, p. 5147.
1406. Barrow R. F., Caunt A. D. — Proc. Roy. Soc.
London, 1953, A219, p. 120.
1407. Barrow R. F., Travis N., Wright C. V. — Nature,
1960, 187, p. 141.
1408. Barta C, Petry F., Nojek B. — Naturwissen-
schaften, 1958, 45, S. 36.
1409. Bartholomew R. F. — J. Phys. Chem., 1966, 70,
p. 3442.
1410. Bartholomew R. F., Frankl D. R. — Phys. Rev.,.
1969, 187, p. 828.
1411. Bartky I. R. — J. Mol. Spectrosc, 1966, 21, p. 1.
1412. Bartky I. R. — J. Mol. Spectrosc, 1966, 21, p. 25.
1413. Bartky I. R.— J. Mol. Spectrosc, 1966, 20,
p. 299.
546
Литература
1414. Barton С. J., Grimes W. Д., Insley H., Moore R. E.,
Thoma R. E. — J. Phys. Chem., 1958, 62, p. 665.
1415. Barton С J., Redman J. D., Strehlow R. A. —
J. Inorg. and Nucl. Chem., 1961, 20, p. 45.
1416. Barton J. L., Bloom H. — J. Phys. Chem., 1956,
60, p. 1413.
1417. Barton J. L., Bloom H. — Z. Phys. Chem., 1959,
63, p. 1785.
1418. Bar-ZivE., Freiberg M., Weiss S. — Spectrochim.
acta, 1972, A28, p. 2025.
1419. Bashkin I. 0., Dymova T. N., Ponyatovskii E. G. ¦—
Phys. status solidi, 1980, B100, p. 87.
1420. Basili R., El-Sharkawy A., Atalla S. — Rev.
int. hautes temp, et refract., 1979, 16, p. 331.
1421. Baskin С P., Bender С F., Kollman F. A. —
J. Amer. Chem. Soc, 1973, 95, p. 5868.
1422. Baskin Y. — J. Amer. Ceram. Soc, 1965, 48,
p. 652.
1423. Bates G. N., Barnes G. — Appl. Phys. Lett.,
1967, 11, p. 75.
1424. Bates J. K., Dunn Т. М. — Canad. J. Phys.,
1976, 54, p. 1216.
1425. Bates J. K., Gruen D. M. — J. Chem. Phys.,
1979, 70, p. 4428.
1426. Bates J. K., Gruen D. M. — J. Mol. Spectrosc,
1979, 78, p. 284.
1427. Bates J. L. — In: Thermodyn. Nucl. Mater. Proc.
Syrup. Vienna, 1965. Vienna: IAEA, 1966, p. 73.
1428. Bates J. L. — J. Amer. Ceram. Soc, 1966, 49,
p. 395.
1429. Batt R. H. — Ph. D. Diss. Univ. of California.
Berkeley, 1964; Diss. Abstrs, 1965, 26, p. 2491.
1430. Battles J. E. — Ph. D. Diss. Ohio State Univ.,
1964; Diss. Abstrs, 1965, 25, p. 6509.
1431. Baud E. — Ann. chim. phys., 1904, 1, p. 8.
1432. Bauer R. K., Lew H. — Canad. J. Phys., 1963,
41, p. 1461.
1433. Bauer S. H., Ino Т., Porter R. F. — J. Chem.
Phys., 1960, 33, p. 685.
1434. Bauer T. W., Johnston H. L., Kerr E. С — J.
Amer. Chem. Soc, 1950, 72, p. 5174.
1435. Baun W. L. — Science, 1963, 140, p. 1330.
1436. Baur E., Brunner R. — Helv. chim. acta, 1934,
17, s. 958.
1437. Beatte I. R., Ogden J. S., Price D. D. — Inorg.
Chem., 1978, 17, p. 3296.
1438. Beauchamp J. L. — J. Chem. Phys., 1976, 64,
p. 718.
1439. Beauchamp J. L. — J. Chem. Phys., 1976, 64,
p. 929.
1440. Beaudry B. J. — J. Less-Common Metals, 1968,
14, p. 370.
1441. Beaudry B. J., Daane A. H. — Trans. Met. Soc.
AIME, 1962, 224, p. 770.
1442. Beaudry B. J., Daane A. H. — Trans. Met. Soc.
AIME, 1963, 227, p. 867.
1443. Beattie I. R., Marsden С J., Ogden J. S. — J.
Chem. Soc. Dalton Trans., 1980, p. 535.
1444. Beattie I. R., Ogden J. S., Price D. D. — J. Chem.
Soc. Dalton Trans., 1979, p. 1460.
1445. Beaumont R. H., Chihara H., Morrison J. A. —
Philos. Mag., 1960, 5, p. 188.
1446. Bee C, Counioux J. J., Papin G., Sebaoun A. —
С. г. Acad. sci., 1974, C278, p. 1193.
1447. Becher H. J., Friedrich F., Willner H. — Z. anorg.
und allg. Chem., 1976, 426, S. 15.
1448. Beck G. — Z. anorg. und allg. Chem., 1930, 174,
S 31
1449. Becker G., Roth W. A. — Z. phys. Chem. Leipzig,
1933, A167, S. 16.
1450. Becker G., Roth W. A. — Z. phys. Chem. Leipzig,
1935, A174, S. 104.
1451. Bedford R. G. — J. Appl. Phys., 1965, 36, p. 113.
1452. Behrens R. G., Woodrow H. O., Aronson S. —
J. Chem. Thermodyn., 1977, 9, p. 1035.
1453. Beketoff N. — Bull. Acad. Sci. Russ., 1842, 34,
p. 169. '
1454. Beketoff N. — Ber. Dtsch. chem. Ges., 1879, 12
S. 856.
1455. Beketoff N. — Bull. Acad. Sci. Russ., 1888, 32,
p. 186.
1456. Beketoff N. — Bull. Acad. Sci. Russ., 1890, 33,
p. 173.
1457. Beketoff N. — Bull. Acad. Sci. Russ., 1894, 35,
p. 541.
1458. Beketov N., Beketov W. — Z. anorg. und. allg.
Chem., 1904, 40, S. 355.
1459. Belbeoch В., Boivineau J. C. — Bull. Soc. frang.
mineral, et cristallogr., 1967, 90, p. 558.
1460. Belbeoch В., Boivineau J. C, Perio P. — J. Phys.
and Chem. Solids, 1967, 28, p. 1267.
1461. Belbeoch В., Kleinberger R., Roulliay M. — J.
Phys. and Chem. Solids, 1978, 39, p. 1007.
1462. Belbeoch В., Kleinberger R., Roulliay M. — Solid
State Communs, 1978, 25, p. 1043.
1463. Belbeoch В., Piekarski C, Perio P. — Bull. Soc.
franQ. mineral, et cristallogr., 1960, 83, p. 206.
1464. Belbeoch В., Piekarski C, Perio P. — Acta cry-
stallogr., 1961, 14, p. 837.
1465. Bell J. Т., Friedman H. A., Billings M. R. —
J. Inorg. and Nucl. Chem., 1974, 36, .p. 2563.
1466. Belton G. R., Jordan A. S. — J. Phys. Chem.,
1965, 69, p. 2065.
1467. Bender С F., Davidson E. R. — J. Chem. Phys
1968, 49, p. 4222.
1468. Benedict R. P., Drummond D. L., Schlie L. A. —
J. Chem. Phys., 1977, 66, p. 4600.
1469. Benediktsson G., Astrom H. U., Rao K. V. — J.
Phys. F: Metal. Phys., 1975, 5, p. 1966.
1470. Benezech G., Foex M. — С. г. Acad. sci., 1969,
C268, p. 2315.
1471. Benjamin L. — J. Chem. and Eng. Data, 1962
7, p. 239.
1472. Bennett S. L., Lin Sin-Shong, Gilles P. W. —
J. Phys. Chem., 1974, 78, p. 266.
1473. Benson G. C, Benson G. W. — Rev. Sci. Instrum
1955, 26, p. 477.
1474. Benson G. C, Goddard E. D., Hoeve С A. — Rev.
Sci. Instrum., 1956, 27, p. 725.
1475. Benton A. — J. Chem. and Eng. Data, 1964, 9
p. 198.
1476. Benz R. — J. Nucl. Mater., 1969, 29, p. 43.
1477. Benz R. — J. Nucl. Mater., 1970, 34, p. 86.
1478. Benz R., Douglass R. M. — J. Phys. Chem., 1961
65, p. 1461.
1479. Benz R., Hoffman С G., Rupert G. N. — J. Amer.
Chem. Soc, 1967, 89, p. 191.
1480. Berenger-Calvet. ¦— J. chim. phys. et phys.-chim.
biol., 1927, 24, p. 325.
1481. Berak J. M., Sienko M. J. — J. Solid State Chem.,
1970, 2, p. 109.
1482. Berezin B. Ya., Chekhovskoy V. Ya., Sheindlin
A. E. -r High Temp.-High Pressur., 1971, 3,
p. 287.
1483. Berezin B. Ya., Chekhovskoy V. Ya.,
Sheindlin A. E. — High. Temp. Sci., 1972, 4, p. 478.
1484. Berezin B. Ya., Chekhovskoy V. Ya., Kats S. A. —
In: Quatrieme conf. intern, de thermodynam.
chim., Montpellier, 1975, vol. 9, p. 77.
547
Литература
1485. Berezin В. Ya., Chekhovskoy V. Ya., Kats S. A., 1517.
Kenisarin M. M. — In: Proc. 6th Symp. Thermo-
dyn. Prop., Aug., 6—8, 1973/Ed. P. E. Liley. 1518.
N. Y.: ASME, 1973, p. 263.
1486. Berg G. J. van den, Klinnerberg P. F. A., Bosch 1519.
/. С van den. — Physica, 1952, 18, p. 221.
1487. Berg J. R. — Diss. Abstrs, 1962, 22, p. 2599. 1520.
1488. Berg J. R., Speeding F. H., Daane A. H. — U. S.
AEC Iowa State Univ. Rept IS-327. Iowa State
Univ., 1961, p. 35. 1521.
1489. Berg R. A., Wharton L., Klemperer W., Buchler A.,
Stauffer J. L. — J. Chem. Phys., 1965, 43,
p. 2416. 1522.
1490. Berg R. L., Vanderzee C. E. — J. Chem. Thermo-
dyn., 1978, 10, p. 1113. 1523.
1491. Berg W. T. — Phys. Rev., 1976, B13, p. 2641.
1492. Berg W. Т., Morrison J. A. — Proc. Roy. Soc.
London, 1957, A242, p. 467. 1524.
1493. Berggren G., Forsyth R. S. — In: Plutonium 1965.
Proc. 3rd Intern. Conf. London, 1965/ Ed. A. E. 1525.
Kay, M. B. Walderon. London: Chapman and Hall,
1967, p. 828. 1526.
1494. Berglund С TV., Guggenheim H. J. — Phys. Rev., 1527.
1969, 185, p. 1022. 1528.
1495. Berglund S., Kierkegaard P. — Acta chem. scand., 1529.
1969, 23, s. 329. 1530.
1496. BergsteinA., Spitz /., Didier R. — Czech. J. Phys., 1531.
1968, B18, s. 538. 1532.
1497. Berkowitz J. — J. Chem. Phys., 1958, 29, p. 1386.
1498. Berkowitz J. — J. Chem. Phys., 1969, 50, p. 3503. 1533.
1499. Berkowitz J. — In: Advances in high temperature
chemistry/ Ed. L. Eyring. N. Y.; L.: Acad. Press, 1534.
1971, vol. 3, p. 123.
1500. Berkowitz /., Batson C. II., Goodman G. L. — 1535.
J. chim. phys. et phys.-chim. biol., 1980, 77, 1536.
p. 631.
1501. Berkowitz J., Chupka W. A. — J. Chem. Phys., 1537.
1958, 29, p. 653.
1502. Berkowitz /., Chupka W. A. — J. Chem. Phys., 1538.
1966, 45, p. 1287.
1503. Berkowitz J., Chupka W. A., Blue G. D., Mar- 1539.
grave J. L. — J. Phys. Chem., 1959, 63, p. 644.
1504. Berkowitz J., Chupka W. A., Inghram M. G. — 1540.
J. Chem. Phys., 1957, 27, p. 85.
1505. Berkowitz /., Chupka W., Inghram M. G. — 1541.
J. Chem. Phys., 1957, 27, p. 87.
1506. Berkowitz J., Chupka W., Inghram M. — J. Phys. 1542.
Chem., 1957, 61, p. 1569.
1507. Berkowitz J., Inghram M. G., Chupka W. A. — 1543.
J. Chem. Phys., 1957, 26, p. 842.
1508. Berkowitz J., Meschi D. J., Chupka W. A. — 1544.
J. Chem. Phys., 1960, 33, p. 533.
1509. Berkowitz J., Tasman H. A., Chupka W. A. — 1545.
J. Chem. Phys., 1962, 36, p. 2170.
1510. Berkowitz-Mattuck J. В., Buchler A. — J. Phys.
Chem., 1963, 67, p. 1386. 1546.
1511. Berkowitz-Mattuck J. В., Buchler A., Engelke J. L.,
Goldstein S. N. — J. Chem. Phys., 1963, 39, 1547.
p. 2722.
1512. Berman A., Zemansky M. W., Boorse H. A. — 1548.
Phys. Rev., 1958, 109, p. 70.
1513. Bernard A., Bacis JR., Luc P. — Astrophys. J., 1549.
1979, 227, Part 1, p. 338.
1514. Bernard A., Gravina R.—Astrophys. J. Suppl. 1550.
Ser., 1980, 44, p. 223.
1515. Bernard A., Sibai A. M. — Z. Naturforsch., 1981,
A35, S. 1313. 1551.
1516. Bernard M., Chaume P., Massaux M. — С. г.
Acad. sci., 1967, C265, p. 1136.
Bernard M., Hocart R. — Bull. Soc. franc, mineral,
et crystallogr., 1961, 84, p. 396.
Bernard M., Jaffray J. — C. r. Acad. sci., 1955,
240, p. 1078.
Bernard M., Jaffray J. — C. r. Acad. sci., 1956,
242, p. 2512.
Bernheim R. A., Gold L. P., Kelly P. В., Kitt-
rell C, Veirs D. K. — Phys. Rev. Lett., 1979,
43, p. 123.
Bernheim R. A., Gold L. P., Kelly P. В., Kitt-
rell C, Veirs D. K. — Chem. Phys. Lett., 1980,
70, p. 104.
Bernstein L. S., Abramowitz S., Levin I. W. —
J, Chem. Phys., 1976, 64, p. 3228.
Berry R. S. — In: Alkali halide vapors, spectra
and reaction dynamics/ Ed. P. Davidovits, D. L.
McFadden. N. Y.: Acad. Press, 1979, p. 77.
Berry R. C, Klemperer N. — Bull. Amer. Phys.
Soc. Ser. II, 1956, 1, p. 284.
Berry R. C, Klemperer TV. — J. Chem. Phys.,
1957, 26, p. 724.
Berthelot M. — Ann. chim. phys., 1873, 29, p. 433.
Berthelot M. — Ann. chim. phys., 1873, 30, p. 456.
Berthelot M. — Ann. chim. phys., 1875, 4, p. 5.
Berthelot. M. — Ann. chim. phys., 1875, 4, p. 21.
Berthelot M. — Ann. chim. phys., 1875, 4, p. 74.
Berthelot M. — Ann. chim. phys., 1875, 4, p. 513.
Berthelot M., Ilosvay J. N. — Ann. chim. phys.,
1883, 29, p. 295.
Bertoncini P. /., Das <?., Wahl A. C. — J. Chem.
Phys., 1970, 52, p. 5112.
Bertrand G. L., Milero F. J., Wu Ching-hsier,
Hepler L. G. — J. Phys. Chem., 1966, 70, p. 699.
Betsujaku H. — J. Phys. Soc. Jap., 1966, 21, p. 187.
Betz G., Frohberg M. G. — High Temp.-High Pres-
sur., 1980, 12, p. 169.
Betz G., Frohberg M. G. — Z. Metallk., 1980, 71,
S. 451.
Betz G., Frohberg M. G. — Metall (W. Berlin),
1981, 35, S. 299.
Beutler #., Levi H. — Z. Elektrochem., 1932, 38,
S. 589.
Bevan R. V. — Proc. Roy. Soc. London, 1911,
85, p. 61.
Bevolo A. J., Shanks H. R., Sidles P. #., Daniel-
son G. С — Phys. Rev., 1974, B9, p. 3220.
Beyer R. P. — Bull. Chem. Thermodyn., 1978,
N 21, p. 307.
Beyer R. P., Daut G. E. — J. Chem. and Eng.
Data, 1979, 24, p. 170.
Beyer R. P., Ferrante M. /., Brown R. R. —
J. Chem. Thermodyn., 1980, 12, p. 985.
Beyer R. P., Ferrante M. J., Brown R. R., Daut
G. E. — U. S. Bur. Mines. Rept Invest., 1980,
N 8410.
Bhagavantam S. — Proc. Indian Acad. Sci., 1941,
A13, p. 543.
Bhagavantam S., Venkatarayudu T. — Proc. Indian
Acad. Sci., 1939, A9, p. 224.
Bhaskar TV. D., Zouboulis E., McClelland Т., Нар-
per W. — Phys. Rev. Lett., 1979, 42, p. 640.
Bhatnagar O. TV., Campbell A. TV.—Chem.
Instrum., 1973, 4, p. 179.
Bhise V. S., Bonilla C. F. — In: Proc. 6th Symp.
Thermophys. Properties, Atlanta, 1973. N. Y.:
ASME, 1973, p. 362,
Bhise V. S., Bonilla С F. — In: Proc. Intern.
Conf. Liquid Metal Technol. in Energy Production,
Bristol, 1977, vol. 2, p. 657.
548
Литература
1552. Bhise V. S., Bonilla С. F. — In: Proc. 7th Symp.
Thermophys. Properties. N. Y.: ASME, 1977,
p. 910.
1553. Bibas /., Leonardl J. — C. r. Acad. sci., 1968,
C266, p. 1319.
1554. Bibas J., Leonardi J. — C. r. Acad. sci., 1969,
C268, p. 877.
1555. Bichowsky F. R., Rossini F. D. — The
Thermochemistry of the Chemical Substances. N. Y.:
Reinhold Publ. Corp., 1936.
1556. Bickerdike R. L., Hughes G. — J. Less-Common
Metals, 1959, 1, p. 42.
1557. Biefeld R. M. — J. Chem. Thermodyn., 1978, 10,
p. 907.
1558. Bieganski Z., Stalinski B. — Bull. Acad. polon.
sci. Ser. sci. chim., 1961, 9, s. 367.
1559. Bigeleisen J., Mayer M. G., Stevenson P. C, Tur-
kevich J. — J. Chem. Phys., 1948, 16, p. 442.
1560. Billhardt H. W. — Glastechn. Ber., 1969, 42,
S. 272.
1561. Bills J. L., Cotton F. A. — J. Phys. Chem., 1960,
64, p. 1477.
1562. Biltz W., Mailer H. — Z. anorg. und allg. chem.,
1927, 163, S. 258.
1563. Birch J. A., Collins J. G., White G. K. — Aust.
J. Phys., 1979, 32, p. 463.
1564. Biswas S. R., Mukerji J. — J. Chem. and Eng.
Data, 1971, 16, p. 336.
1565. Bizouard M., Cerisier P., Pantaloni J. — C. r.
Acad. sci., 1967, C264, p. 144.
1566. Bjorge В., Jenssen B. — Acta chem. scand., 1968,
22, p. 1347.
1567. Bjorklund С W., Reavis J. G., Leary J. A.,
Walsh K. A. — J. Phys. Chem., 1959, 63, p. 1774.
1568. Black W. C, Johnson R. Т., Wheatley J. C. —
J. Low Temp. Phys., 1969, 1, p. 641.
1569. Blackburn M. В., Mermet J. M., Winefordner
J. D. — Spectrochim. acta, 1978, A34, p. 847.
1570. Blackburn P. E. — Diss. Abstrs, 1960, 20, p. 2519.
1571. Blackburn P. E. — In: Chemical and thermodyn.
properties at high temperatures: Symp. XVIIIth
Intern. Congr. of Pure and Appl. Chemistry,
Montreal, Aug. 6—12, 1961, p. 105.
1572. Blackburn P. E. Thermodynamic and kinetic
studies for a refractory materials, program/ Ed. L. A.
McClaine. Air Force Mater Lab., Res. and Techn.
Div., Air Force Systems Comm., Wright-Patterson
Air Force Base, Ohio, Rept ASD-TDR-62-204,
Pt III, April 1964. Ohio, 1964.
1573. Blackburn P. E., Hoch M., Johnston H. L. —
J. Phys. Chem., 1958, 62, p. 769.
1574. Blaho D., Abbrent M., Pekarek V. — Chem. zvesti,
1976, 30, s. 621.
1575. Blanc M. — C. r. Acad. sci., 1958, 246, p. 570.
1576. Blanc M. — С. г. Acad. sci., 1959, 249, p. 2012.
1577. Blanc M. — Ann. phys. (Fr.), 1960, p. 615.
1578. Blaugher R. D., Hulm J. K., Rayne J. A.,
Veal B. W., Hein R. E. — In: Proc. Intern. Gonf.
Low Temp. Phys., 8th. L.: Butterworths, 1963,
p. 147.
1579. Block R. — Sitzungsber. Osterr. Akad. Wiss.
Math.-naturwiss. Kl. Ha, 1953, 162, S. 99.
1580. Blocher J. M., Campbell I. E. — J. Amer. Chem.
Soc, 1949, 71, p. 4040.
1581. Blomeke J. O., Ziegler W. T. — J. Amer. Chem.
Soc, 1951, 73, p. 5099.
1582. Bloom D. S., Grant N. J. — Trans. AIME, 1951,
191, p. 1009.
1583. Bloom D. S., Putman J. W., Grant N. J. —
J. Metals, 1952, 4, p. 626.
1584. Bloom H., Bockris J., Richards N., Taylor R. —
J. Amer. Chem. Soc, 1958, 80, p. 2044.
1585. Blum P. L., Guinet P., Vaugoyeau H. — С. Г.
Acad. sci., 1963, 257, p. 3401.
1586. Blume M. — Phys. Rev., 1966, 141, p. 517.
1587. Blumenthal B. — J. Nucl. Mater., 1960, 2, p. 23.
1588. Blumenthal В., Baumriicker J. E., Lloyd L. Т.—
U. S. Argonne Nat. Lab., Rept ANL-N 5957, 1959.
1589. Blumental M. — Rocz. chem., 1931, 11, s. 865.
1590. Blumental M. — Rocz. chem., 1934, 14, s. 233.
1591. Blumental M. — Rocz. chem., 1932, 12, s. 119.
1592. Bobtelsky M., Lairsch R. D. — J. Chem. Soc,
1950, p. 3612.
1593. Bockstahltr L. — Phys. Rev., 1925, 25, p. 677.
1594. Bodisco A. — Berichte, 1888, 21, S. 586.
1595. Bodisco A. — Z. phys. Chem. Leipzig, 1889, 4,
S. 482.
1596. Вое G. H., Morrice E. — Acta chem. scand., 1974,
A28, p. 286.
1597. Boeke H. E. — Z. anorg. und allg. Chem., 1906,
50, S. 355.
1598. Boerboom A. J. M. — In: Mass Spectrometry /
Ed. R. J. Reed. N. Y.; L.: Acad. Press, 1965,
p. 251.
1599. Bogacz A., Trzebiatowski W. — Rocz. chem., 1964,
38, s. 729.
1600. Bogart D. — J. Phys. Chem., 1954, 58, p. 1168.
1601. Bogros A. — C. r. Acad. sci., 1930, 191, p. 322.
1602. Bogros A. — Ann. phys., 1932, ser. 10, 17, p. 199.
1603. Bohdansky /., Schins H. E. J. — J. Appl. Phys.,
1965, 36, p. 3683.
1604. Bohdansky J., Schins H. E. J. — J. Phys. Chem.,
1967, 71, p. 215.
1605. Boldyrev A. I., Solomonik V. G., Zakzhevskii V. G.,
Charkin O. P. — Chem. Phys. Lett., 1980, 73,
p. 58.
1606. Bolton W., von. — Z. Elektrochem., 1907, 13,
S. 145.
1607. Bommer H., Hohmann E. — Z. anorg. und allg.
Chem., 1941, 248, S. 357.
1608. Bondarenko V. P., Fomichev E. N., Kandyba
V. V. — High Temp.-High Pressur., 1973, 5, p. 5.
1609. Bonilla C. F. — U. S. AEC Rept ORNL-3605.
Oak Ridge, 1961, vol. 1, p. 286.
1610. Bonilla C. F. — High Temperature Properties
of Nuclear Reactor Coolants: Rept COQ-3027-30,
Univ. Columbia, 1977.
1611. Bonilla C. F., Sawhney D. L., Makansi M. M. —
Trans. Amer. Soc. Metals, 1962, 55, p. 877.
1612. Bonnefoi J. — С. г. Acad. sci., 1897, 124, p. 771.
1613. Bonnefoi J. — С. г. Acad. sci., 1900, 130, p. 1394.
1614. Bonnefoi J. — Ann. chim. phys., 1901, 23, p. 317.
1615. BonnellD. W., Hastie J. W. — In: Proc. 10th
Materials Res. Sympos. Gaithersburg, Maryland,
Sept. 18—22, 1978 / Ed. J. W. Hastie. Wash.:
NBS, 1979, p. 357. >
1616. Bonnell D. W., Valerga A. J., Margrawe J. L. —
Abstracts. Amer. Ghem. Soc. Meeting. Wash., 1971.
1617. Bonnell D. W., Valerga A. J., Margrave J. L.
Radiation and conduction loss corrections to-free
drop calorimetry data: Preprint. Houston: Rice
Univ., 1972. «
1618. Boorse H. A., HirshfeldA. Т., Leupold H, — Phys.
Rev. Lett., 1960, 5, p. 246.
1619. Boorse H. A., Leupold H. — In: Problems in Low-
Temperature Physics and Thermodynamics.
¦ Oxford etc.: Pergamon Press, 1962, vol. 3, p. 153.
1620. Boring M., ffecht H. G. — J. Chem. Phys., 1978,
69, p. 112.
549
Литература
1621. Boring М., Moskowitz J. W. — Chem. Phys. Lett.,
1976, 38, p. 185.
1622. Boring M., Wood J. #., Moskowitz J. W. —
J. Chem. Phys., 1974, 61, p. 3800.
1623. Bornstedt A., Edvinsson G. — Phys. scr., 1970, 2,
.s. 205.
1624. Bornstedt A., Edvinsson G., Lagerquist A., Ren-
horn I. — Phys. scr., 1979, 20, s. 599.
1625. Bosworth Y. M., Clark R. J. H., Rippon D. M. —
J. Mol. Spectrosc, 1973, 46, p. 240.
1626. Better B. J., Kooter J. A., Mulder J. J. С —
Chem. Phys. Lett., 1975, 33, p. 532.
1627. Bouaziz R., Maraine C. H. — С. г. Acad. sci.,
1972, C274, p. 390.
1628. Bouaziz R., Papin G. — C. r. Acad. sci., 1968,
C266, p. 1530.
1629. Bouaziz R., Papin G., Rollet A. P. — С. г. Acad.
sci., 1966, C262, p. 1051.
1630. Bounzel E. G., Kohlmeyer E. J. — Z. anorg. und
allg. Chem., 1974, 254, S. 1.
1631. Boureau G., Gerdanian P. — High Temp.-High
Pressur., 1970, 2, p. 681.
1632. Bousquet J., David J.-C, Diot M., Gullon A. —
Bull. Soc. chim. France, 1968, N 6, p. 2390.
1633. Bousquet J., Pirachon G. — С. г. Acad. sci., 1963,
256, p. 694.
1684. Bousquet J., Pirachon G. — C. r. Acad. sci., 1964,
258, p. 934.
1635. Bousquet J., Pirachon G. — C. r. Acad. sci., 1964,
258, p. 3869.
1636. Bousquet J., Perachon G., Remy J.-C. — Bull. Soc.
chim. France, 1967, N 1, p. 238.
1637. Bousquet J., Remy J.-C. — С. г. Acad. sci., 1962,
254, p. 691.
1638. Bowles K. J. — NASA Techn. Note D-4535.
Cleveland (Ohio), 1968.
1639. Bowles K. J., Rosenblum L. — J. Chem. and Eng.
Data, 1965, 10, p. 321.
1640. Bowles K. J., Rosenblum L. — NASA Techn.
Note D-2849. Clevelandf(Ohio), 1965.
1641. Bradbury M. H., Ohse R. W. — J. Chem. Phys.,
1979, 70, p. 2310.
1642. Bradley R. S., Volans P. — Proc. Roy. Soc.
London, 1953, A217, p. 508.
1643. Bradshaw F. F., Pearson S. — Proc. Phys. Soc.
London, 1956, B69, p. 441.
1644. Brand H. — Zentr. Mineral. Geol., 1912, S. 26.
1645. Brandt O. G., Walker С. Т. —Phys. Rev. Lett.,
1967, 18, p. 11.
1646. Brauer G. — Z. anorg. und allg. Chem., 1941, 248,
S. 1.
1647. Brauer G., Littke W. — J. Inorg. and Nucl. Chem.,
1960, 16, p. 67.
1648. Brauer G., Marawietz H. — Z. anorg. und allg.
Chem., 1962, 317, S. 13.
1649. Braun M., Kohlhaas R., Vollmer O. — Z. angew.
Phys., 1968, 25, S. 365.
1650. Braun H., Rudy E. — Z. Metallk., 1960, 51, S. 360.
1651. Braunstein R., Trischka J. W. — Phys. Rev., 1955,
98, p. 1092.
1652. Bredig M. A. — J. Phys. Chem., 1942, 46, p. 747.
1653. Bredig M. A., Bronstein H. R., Smith W. T. —
J. Amer. Chem. Soc, 1955, 77, p. 1454.
1654. Bredig M. A., Johnson J. W., Smith W. T. —
J. Amer. Chem. Soc, 1955, 77, p. 307.
1655. Breford E. J., Engelke F. — Chem. Phys. Lett.,
1978, 53, p. 282.
1656. Breford E. /., Engelke F. — J. Chem. Phys., 1979,
71, p. 1994.
1657. Breford E. J., Engelke F. — Chem. Phys. Lett.,
1980, 75, p. 132.
1658. BreivogelF. W., HebertA. J., Street K. — J. Chem.
Phys., 1965, 42, p. 1555.
1659. Brewer L. — Chem. Rev., 1953, 52, p. 1.
1660. Brewer L., Brackett E. —Chem. Rev., 1961, 61,
p. 425.
1661. Brewer L., Green D. W. — High Temp. Sci., 1969,
1, p. 26.
1662. Brewer L., Lamoreaux R. H. — In: Atomic Energy
Review. Special Issue N 7. Vienna: IAEA, 1980,
p. 11.
1663. Brewer L., Margrave J. L. — J. Phys. Chem.,
1955, 59, p. 421.
1664. Brewer L., Mastick D. F. — J. Amer. Chem. Soc,
1951, 73, p. 2045.
1665. Brewer L., Rosenblatt G. M. — Chem. Rev., 1961,
61, p. 257.
1666. Brewer L., Rosenblatt G. M. — In: Advances
in high temperature chemistry / Ed. L. Eyring.
N. Y.; L.: Acad. Press, 1969, vol. 2, p. 1.
1667. Brewer L., Searcy A. —Ann. Rev. Phys. Chem.,
1956, 7, p. 259.
1668. Brice J. C. — In: The growth of crystals from
liquids. Amsterdam etc.: North-Holl. Publ. Co.,
1973, p. 64.
1669. Brickwedde F. G., Hoge H. J., Scott R. B. —
J. Chem. Phys., 1948, 16, p. 429.
1670. Bridgman P. W. — Phys. Rev., 1914, 3, p. 153.
1671. Bridgman P. W. — Proc. Amer. Acad. Sci., 1915,
51, p. 55.
1672. BriggsA. G., Kemp R. J. — J. Chem. Soc. Dalton
Trans., 1972, 12, p. 1223.
1673. Briggs G. G. — U. S. Natl. Lead Corp. Rept
NLCO-720. Ohio, Cincinnati, 1960. Цит. по: [2344].
1674. Briscoe H. V. A., Evans C, Robinson P. L. —
3. Chem. Soc, 1932, p. 1101.
1675. Brittain R., Powell D., Kreglewski M., Vala M. —
Chem. Phys., 1980, 54, p. 71.
1676. Brodale G. E., Giauque W. F. — J. Phys. Chem.,
1972, 76, p. 737.
1677. Brom J. M., Broida H. P. — J. Chem. Phys.,
1975, 63, p. 3718.
iblS. Brom J. M., Durham С. Н., Weltner W. —
J. Chem. Phys., 1974, 61, p. 970.
1679. Bronson H. L., Chisholm H. M., Dockerty S. M. —
Canad. J. Res., 1933, 8, p. 282.
1680. Bronsted J. N. — Z. phys. Chem. Leipzig, 1906,
56, S. 645.
1681. Bronsted J. N. — Z. phys. Chem. Leipzig, 1911,
77, S. 315.
1682. Bronsted J. N. — Z. phys. Chem. Leipzig, 1912,
80, S. 206.
1683. Bronsted J. N. — Z. Elektrochem., 1914, 20,
S. 554.
1684. Brooker M. #., Bredig M. A. — J. Chem. Phys.,
1973, 58, p. 5319.
1685. Brooker M. H., Irish D. E. — Canad. J. Chem.,
1970, 48, p. 1183.
1686. Brooker M. #., Quist A. S., Boyd G. E. — Chem
Phys. Lett., 1971, 9, p. 242.
1687. Brouns E., Visser J. W. — Acta crystallogr., 1964,
17, p. 614.
1688. Brown A., Zemansky M. W., Boorse H. A. —
Phys. Rev., 1952, 86, p. 134.
1689. Brown A., Zemansky M. W., Boorse H. A. —
In: Proc. NBS Semicentennial Symposium low-
Temp. Phys. U. S. NBS, 1952, circ. 519, p. 99.
1690. Brown A., Zemansky M. W., Boorse H. A. —
Phys. Rev., 1953, 92, p. 52.
550
Литература
4691. Brown О. L. I., Latimer W. M. — J. Amer. Chem.
Soc, 1936, 58, p. 2228.
4692. Brown R. TV., McLaren A. C. — Acta crystallogr.,
1962, 15, p. 974.
.1693. Brown S. D., Gard G. L., Loehr T. M. — J. Chem.
Phys., 1976, 64, p. 1291.
,1694. Bruce R. #., Cannell D. S. — Phys. Rev., 1977,
B15, p. 4451.
1695. Bruckner W., Moldenhauer W., Wich H., Woef E.,
Oppermann #., Gerlach U., Reichelt W. — Phys.
status solidi, 1975, A29, p. 63.
1696. Brumer P., Kurplus M. — J. Chem. Phys., 1976,
64, p. 5165.
1697. Brunton G. — Acta crystallogr., 1968, B24, p. 1703.
•1698. Brunton G. — Acta crystallogr., 1969, B25, p. 2161.
1699. Bryant С A., Keesom P. H. — J. Chem. Phys.,
1961, 35, p. 1149.
1700. Brynestadt J., Grjotheim K., Gronvold F., Holm
J. L.y Urness S. — Disc. Faraday Soc, 1961,
N 32, p. 90.
11 $1. Brynestadt J., Grjotheim K., Urness S. — Met.
ital., 1960, 52, p. 495.
1762. Bucher E., Heiniger F., Muller J. — In: Proc.
9th Intern. Conf. Low Temp. Phys. Columbus
(Ohio), 1964, (B), p. 1059.
1703. Buchler A., Berkowitz-Mattuck J. B. — J. Chem.
Phys., 1963, 39, p. 286.
4704. Buchler A., Berkowitz-Mattuck J. — In: Advances
in high temperature chemistry / Ed. L. Eyring.
N. Y.; L.: Acad. Press, 1967, vol. 1, p. 130.
1795. Buchler A., Marram E. P. — J. Chem. Phys.,
1963, 39, p. 292.
17§6. Buchler A., Stauffer J. L. — J. Phys. Chem.,
1966, 70, p. 4092.
1797. Buchler A., Stauffer J. L., Klemperer W.—
J. Chem. Phys., 1967, 46, p. 605.
1708. Buchler A., Stauffer J. L., Klemperer W., Whar-
ton L. — J. Chem. Phys., 1963, 39, p. 2299.
1709. Buck U., Pauly H. — Z. phys. Chem. Leipzig,
1965, 44, S. 345.
1710. Bucle E. R., Ubbelohda A. R. — Proc. Roy. Soc.
London, 1960, A259, p. 61.
1711. Bucle E. Д., Ubbelohde A. R. — Proc. Roy. Soc.
London, 1960, A259, p. 335.
1712. Budreick J. I. — Hiys. ftev., l%0, 119, p. 1ЭТ8.
1713. Buenker R. L., Peyerimhoff S. D. — J. Chem.
Phys., 1966, 45, p. 3682.
1714. Bite* W. — Angew. Chem., 1954, 66, S. 336.
1715. Bues W. — Z. phys. Chem. (BRD), 1957, 10, S. 1.
1716. Buijs K., Schutte С J. H. — Spectrochim. acta,
1961, 17, p. 927.
1717. Buijs K., Schutte C. J. H. — Spectrochim. acta,
1962, 18, p. 307.
1718. Bulewicz E. M., Padley P. J. — Proc. Roy. Soc.
London, 1971, A323, p. 377.
1719. Bulewicz E. M., Phillips L. F., Sugden Т. М. —
Trans. Faraday Soc, 1961, 57, p. 921.
1720. Bumps E. S., Kessler H. D., Hansen M. — Trans.
Amer. Soc. Metals, 1953, 45, p. 1008.
1721» Bunzel E. G., Kohlmeyer E. J. — Z. anorg. und
allg. Chem., 1947, 254, S. 1.
1722. Burdese A. — Ann. chim. (Ital.), 1957, 47, p. 785.
1723. Burdese A., Abbattista F. — Ric. sci., 1958, 28,
p. 1634.
1724. Bureau J. — Ann. chim. (Fr.), 1937, 8, p. 1.
.1725. Burgers W. G., Jacobs F. M. — Z. Kristallogr.,
1936, 94, S. 299.
,4726. Burgess G. K., Wallenberg P. G. — Z. anorg. und
allg. Chem., 1913, 82, S. 361.
1727. Burk D. L., Estemann /., Friedberg S. A. —
Z. phys. Chem. (BRD), 1958, 16, S. 183.
1728. Burke T. G., SmithD. F., Nielsen A. H. — J. Chem.
Phys., 1952, 20, p. 447.
1729. Burmistrova N. P., Lisov N. I., Sabadash N. G. —
In: Proc. 1st Europ. Symp. Therm. Anal., Salford,
1976. L., 1976, p. 225.
1730. Burns J. #., Osborne D. W., Westrum E. F. —
J. Chem. Phys., 1960, 33, p. 387.
1731. Burns J. H., Tennissen A. C, Brunton G. D. —
Acta crystallogr., 1968, B24, p. 225.
1732. Burns R. P., DeMaria G. D., Drowart J., Grim-
ley R. T. — J. Chem. Phys., 1960, 32, p. 1363.
1733. Busey R. H., Bevan R. B. — Low-Temperature
Heat Capacity of 7Li Hydride and 7Li Deuteride.
U. S. Oak Ridge Natl. Lab. Rept ORNL—4706.
Oak Ridge (Tennesse), 1971.
1734. Busey R. #., Dearman H. H., Bevan R. B. —
J. Phys. Chem., 1962, 66, p. 82.
1735. Butkov K., TereninA. — Z. Phys., 1928, 49, S. 865.
1736. Buzzard R. W., Liss R. В., Fickle D. P. — J. Res.
NBS, 1953, 50, p. 209.
1737. Caldwell C. D., Engelke F., Hage H. — Chem.
Phys., 1980, 54, p. 21.
1738. Campbell I. E., Jaffee R. I., Blocher J. M., Gur-
land /., Gonser B. W. — J. Electrochem. Soc.,
1948, 93, p. 271.
1739. Caanelli G., Cannelli G. B. -~ J. Appl. Phys.,
1976, 47, p. 17.
1740. Cantor S. — J. Phys. Chem., 1961, 65, p. 2208.
1741. Cantor S. — Inorg. and Nucl. Chem. Lett., 1973,
9, p. 1275.
1742. Cantor S., HitchB. F., HeatherlyD. E. — In: Proc.
Intern. Symp. Molten Salts /Ed. J. P. Pemsler,
J. Braunstein, K. Noble. Princeton: Electrochem.
Soc, 1976, p. 417.
1743. Cappellina F., Napolitano G. — Ann. chim. (Ital.),
1967, 57, p. 1087.
1744. Carette P., Blondeau J. M. — С. г. Acad. sci.,
1969, B268, p. 1743.
1745. Carette P., Houdart R. — С. г. Acad. sci., 1970,
B271, p. 110.
1746. Carette P., Houdart R. — С. г. Acad. sci., 1971,
B272, p. 595.
1141. CarlUe S. J., Christian J. W., Hume-Rothery W. —
J. Inst. Metals, 1949, 76, p. 169.
1748. Carlson K. D., Ludena E., Moser С — J. Chem.
Phys., 1965, 43, p. 2408.
1749. Carlson K. D., Moser С — J. Chem. Phys., 1966,
44, p. 3259.
1750. Carlson K. D., Moser C. — J. Chem. Phys., 1967,
46, p. 35.
1751. Carlson O. N., Dickenson J. M. —Trans. Met.
Soc. AIME, 1954, 200, p. 915.
1752. Carlson O. N., Owen C. V. — J. Electrochem.
Soc, 1961, 108, p. 88.
1753. Carlson O. N., Schmidt F. A., Sever J. С — Met.
Trans., 1973, 4, p. 2407.
1754. Carlson O. N., Schmidt F. A., Wilhelm H. A. —
J. Electrochem. Soc, 1957, 104, p. 51.
1755. Carnall W. Т., WybourneB. G. — J. Chem. Phys.,
1964, 40, p. 3428.
1756. Carnelly T. — J. Chem. Soc, 1878, 33, p. 273.
1757. Caro P., Derouet J., Beaury L., Teste de Sagey G.,
Chaminade J. P., Aride J., Pouchard M. —
3. Chem. Phys., 1981, 74, p. 2698.
1758. Carpenter L. G., Mair W. N. — Proc. Phys. Soc.
London, 1951, B64, p. 57.
1759. Carpenter L. G., Steward T. F. — Philos. Mag.,
1939, 27, p. 551.
551
Литература
1760. Carr Р. #., Foner S. J. — J. Appl. Phys., 1960,
31, p. 344.
1761. Carrol T. — Phys. Rev., 1937, 52, p. 822.
1762. Carsky P., Zahradnik R., Kozak I. — Chem. Phys.
Lett., 1976, 41, p. 165.
1763. CarstensD. H. W., GruenD. M., Kozlowski J. F. —
High Temp. Sci., 1972, 4, p. 436.
1764. Carter T. S. — Phys. Unserer Zeit., 1910, 11,
S. 632.
1765. Cases J. С — Rev. chim. miner., 1973, 10, p. 577.
1766. Cases J. C, Parker V. В., Kilday M. V. — J. Res.
NBS, 1977, 82, p. 19.
1767. Catalan M. A., Rico F. R. — An. Real. soc. esp.
fis. у quim., 1952, A48, p. 328.
1768. Catalan M. A., Velasco R. —An. fis. Real. Soc.
esp. fis у quim., 1952, A48, p. 247.
1769. Cater E. D., Plante E. R., Gilles P. W. — J. Chem.
Phys., 1960, 32, p. 1269.
1770. Catlow С R. A., LidiardA. B. — In: Thermodyn.
Nuclear Mater., pt II, Vienna, 1974. Vienna: IAEA,
1975, p. 27.
1771. Caunt A. D., Barrow R. F. — Nature, 1949, 164,
p. 753.
1772. Cavaliere P., Ferrante G., Lo Cascio L. — J. Chem.
Phys., 1975, 62, p. 4753.
1773. Centnerszwer M. — J. chim. phys. et phys.-chim.
biol., 1930, 27, p. 9.
1774. Centnerszwer M., Blumenthal M. — Bull. int.
Acad. polon. cl. sci. math, nat., A, 1933, p. 499.
1775. Centnerszwer M.t Blumenthal M. — Bull. int.
Acad. polon. cl. sci. math, nat., A, 1936, p. 81.
1776. Cerisier P. — J. chim. phys. et phys.-chim. biol.,
1969, 66, p. 1593.
1777. Cerisier P., Poux F. — Solid. State Communs,
1978, 26, p. 661.
1778. Cesbron F., Vachey H. — Bull. Soc. franc, miner,
et cristallogr., 1970, 93, p. 235.
1779. Cezairliyan A. — In: 3rd Symp. Thermophys.
Properties Papers, Lafaeltte, 1965. N. Y., 1965, p. 253.
1780. Cezairliyan A. — U. S. NBS Rept 10326, chap. 7,
8. Wash., 1970.
1781. Cezairliyan A. — J. Res. NBS, 1971, A75, p. 565.
1782. Cezairliyan A. — High Temp.-High Pressur., 1972,
4, p. 453.
1783. Cezairliyan A. — High Temp. Sci., 1972, 4, p. 248.
1784. Cezairliyan A. — U. S. NBS Rept 10904. Wash.,
1972.
1785. Cezairliyan A. — In: 8th Thermophys. Conf. Palm
Springs, California, July, 1973, Paper N 73—743.
Calif.: AIAA, 1973.
1786. Cezairliyan A. — Faraday Symp. Chem. Soc, 1974,
N 8, p. 7.
1787. Cezairliyan A., McClure J. L. — J. Res. NBS,
1971, A75, p. 283.
1788. Cezairliyan A., McClure J. L. — J. Res. NBS,
1975, A79, p. 431.
1789. Cezairliyan A., McClure J. L. — High Temp.-
High Pressur., 1976, 8, p. 461.
1790. Cezairliyan A., McClure J. L. — В кн.: V Европ.
конф. по теплофиз. свойствам твердых веществ,
Москва, 18—21/V.1976. М., 1976, с. 7.
1791. Cezairliyan A., McClure J. L., Beckett С. W. —
J. Res. NBS, 1971, A75, p. 1.
1792. Cezairliyan A., Miiller A. P.— High Temp.-High
Pressur., 1977, 9, p. 319.
1793. Cezairliyan A., Miiller A. P. — J. Res. NBS, 1977,
A82, p. 119.
1794. Cezairliyan A., Miiller A. P.— J. Res. NBS, 1978,
83, p. 127.
1795. Cezairliyan A., Miiller A. P.— Intern. J.
Thermophys., 1980, 1, p. 195.
1796. Cezairliyan A., Morse M. S., Beckett С W. —
Rev. int. hautes temp, et refract., 1970, 7, p. 382.
1797. Cezairliyan A., Morse M. S., Bermann H. A.,
Beckett C. W. — J. Res. NBS, 1970, A74, p. 65.
1798. Cezairliyan A., Righini F. — J. Res. NBS, 1974,
A78, p. 509.
1799. Cezairliyan A., Righini F. — J. Res. NBS, 1975,.
A79, p. 81.
1800. Cezairliyan A., Righini F. — Rev. int. hautes
temp, et refract., 1975, 12, p. 180.
1801. Cezairliyan A., Righini F. — Rev. int. hautes
temp, et refract., 1975, 12, p. 201.
1802. Cezairliyan A., Righini F., McClure J. L. —
J. Res. NBS, 1974, A78, p. 143.
1803. Chalek С L., Gole J. L. — J. Chem. Phys., 1976,
65, p. 2845.
1804. Chalek С L., Gole J. L. — Chem. Phys., 1977,
19, p. 59.
1805. Chamberland B. L. — Mater. Res. Bull., 1967, 2r
p. 827.
1806. Chandrasekharaiah M. C, Grimley R. Т.,
Margrave J. L. — J. Phys. Chem., 1959, 63, p. 1505.
1807. Chandrashekhar G. V., Barros H. L. C, Honig
J. M. — Mater. Res. Bull., 1973, 8, p. 369.
1808. Chang C. #., Margrave J. L. — Mater. Res. Bull.,
1967, 2, p. 929.
1809. Chang C. #., Margrave J. L. — J. Amer. Chem.
Soc, 1968, 90, p. 2020.
1810. Chang L. L. Y., Scroger M. G., Phillips B. —
J. Amer. Ceram. Soc, 1966, 49, p. 385.
1811. Chang Y. A. — J. Less-Common Metals, 1969, 17,
p. 325.
1812. Chanh N. В., De Luzy de Pelissac. — Bull. Soc
frang. mineral, et cristallogr., 1968, 91, 13.
1813. Chantry G. W., Gebbie H. A., Hamlin A. G.,
Lomas B. — Infrared Phys., 1970, 10, p. 95.
1814. Charlesworth J. H. — Techn. Rept AFML-TR-70-
137. Ohio, Air Force Base Dayton, Oct., 1970.
1815. Charlu T. V., Kleppa O. J. — J. Chem.
Thermodyn., 1971, 3, p. 697.
1816. Charlu T. V., Kleppa O. J. — High Temp. Sci.,
1973, 5, p. 260.
1817. Charlu T. V., Kleppa O. J. — J. Chem.
Thermodyn., 1973, 5, p. 325.
1818. Charlu T. V., Kleppa O. J., Reed Т. В. — J. Chem.
Thermodyn., 1974, 6, p. 1065.
1819. Chaudron M. G. — Ann. chim. (Fr.), 1921, 16,
p. 221.
1820. Chaudron M. G. — С. г. Acad. sci., 1920, 170,
p. 1056.
1821. Chauvenet E. — Ann. chim. phys., 1911, 23, p. 425.
1822. Cheetham С /., Barrow R. F. — In: Advances
in high temperature chemistry. / Ed. L. Eyring.
N. Y.; L.: Acad. Press, 1967, vol. 1, p. 7.
1823. Cheetham C. J., Barrow R. F. — Trans. Faraday
Soc, 1967, 63, p. 1835.
1824. Chekhovskoi V. Ya., Petrov V. A. — In: 1st
Intern. Conf. Calorim. and Thermodyn., Warsaw,
Aug. 31—Sep. 4. Warsaw, 1959, L—1.
1825. Chekhovskoi V. Ya., Sheindlin A. E., Berezin
B. Ya. — High Temp-High Pressur., 1970, 2,
p. 301.
1826. Chen H. H., Chipman J. — Trans. Met. Soc. AIMEr
1947, 38, p. 70.
1827. Chen H.-L. S., Sladek R. J. — Phys. Rev., 1978r
B18, p. 6824.
1828. Cheng С #., Gupta K. P., van Reuth E. C.r
Beck P. A. — Phys. Rev., 1962, 126, p. 2030.
552
Литература
1829. Chikalla Т. D. — U. S. AEC, Rept HW-69832. 1863.
Richland, Wash., 1961. 23 p.
1830. Chikalla T. D. — J. Amer. Geram. Soc, 1963, 1864.
46, p. 323.
1831. Chikalla T. D., McNeilly C. E., Bates J. L., 1865.
Rasmussen J. J. — Colloq. intern. Centre nat. Rech.
sci., P., 1972, N 205, p. 351. 1866.
1832. Chikalla T. D., McNeilly C. E., Skavdahl R. E. —
U. S. General Electric Co. Rept HW-74802. Han- 1867.
ford Atomic Products Operation. Richland. Wash., 1868.
1962, Sept. 30 p.
1833. Chikalla T. D., McNeilly C. E., Skavdahl R. E. — 1869.
J. Nucl. Mater., 1964, 12, p. 131.
1834. Chiotti P. — J. Electrochem. Soc, 1954, 101, 1870.
p. 567.
1835. Chiotti P. — Rev. Sci. Instrum., 1954, 25, p. 876. 1871.
1836. Chiotti P., Dooley G. J. — J. Nucl. Mater., 1966,
23, p. 45. 1872.
1837. Chiotti P., Gill K. J. — Trans. Met. Soc. AIME,
1961, 221, p. 573. 1873.
1838. Chipman H. Д., Johnson F. M. G., Maass O. —
Proc. Trans. Nova Scotian Inst. Sci., 1929, 17, 1874.
p. 149. 1875.
1839. Chiswik H. H. — Metallography, 1959, 76, p. 242.
1840. *Chin Chi-Lian, Chang Hua. — Chem. Phys. Lett., 1876.
1980, 73, p. 167.
1841. Choain Ch., Marion F. — C. r. Acad. sci., 1961, 1877.
252, p. 3258.
1842. Chodura В., Maly J. — In: Proc. 2nd Intern. Conf. 1878.
Peaceful Uses of Atomic Energy, Geneva, 1958.
Geneva, 1958, p. 223. 1879.
1843. Chou C, White D., Johnston H. L. — Phys. Rev.,
1958, 109, p. 788. 1880.
1844. Choudary U. V., Gingerich K. A., Kingcade J. E. —
J. Less-Common. Metals, 1975, 42, p. 111. 1881.
1845. Chretien A., Allamagny P. — С. г. Acad. sci.,
1965, 260, p. 1425. 1882.
1846. Christensen J. A. — U. S. AEC Rept HW-69234.
Richland, Wash., 1962. 1883.
1847. Christensen J. A., Allio R. /., Biancheria A. —
U. S. AEC. Rept WCAP-6065. Pittsburgh, 1965. 1884.
1848. Christy A. — Phys. Rev., 1929, 33, p. 701.
1849. Christy A. — Astrophys. J., 1929, 70, p. 1. 1885.
1850. Chung J. W., Bonilla С F. — In: Proc. 6th Symp.
Thermophys. Properties. N. Y.: ASME, 1973, 1886.
p. 397.
1851. Chupka W. — J. Chem. Phys., 1959, 30, p. 458. 1887.
1852. Chupka W.A., Berkowitz J., Giese C.F. — J. Chem.
Phys., 1959, 30, p. 827.
1853. Chupka W. A., Berkowitz J., Inghram M. G. —
J. Chem. Phys., 1957, 26, p. 1207.
1854. Chupka W. A., Berkowitz /., Inghram M. G. —
U. S. Argonne Nat. Lab. Rept OOK-AD-107062. 1888.
1957. 12 p. ' 1889.
1855. Chupka W. A., Berkowitz J., Meschi D. J., Tas-
man H. A. — In: Adv. Mass Spectrometry. Oxford
etc.: Pergamon Press, 1963, vol. II, p. 99. 1890.
1856. Chupka W. A., Inghram M. G., Porter R. F. —
J. Chem. Phys., 1956, 24, p. 792. 1891.
1857. CiampoliniM.,PaolettiP. — J. Phys. Chem., 1961,
65, p. 1224. 1892.
1858. Cingolani A., Berchiesi M. A., Pianloni G., Leo-
nesi D. — Z. Naturforsch., 1972, A27, S. 159. 1893.
1859. Cini R., Cocchi M. — Ric. sci., 1957, 27, p. 2187.
1860. Cizeron G., Lacombe P. —Mem. sci. Rev. met., 1894.
1960, 57, p. 179.
1861. Claassen H. #., Goodman G. L., Holloway J. #., 1895.
Selig H. — J. Chem. Phys., 1970, 53, p. 341.
1862. Claassen H. H., Weinstock В., Malm J. G. — 1896.
J. Chem., Phys., 1956, 25, p. 426.
Clark J. В., Rapoport E. — J. Chem. Phys., 1968,
49, p. 2453.
Clark M. J., Lynton H. — Canad. J. Chem., 1969,.
47, p. 2579.
Clark R. P. — J. Chem. and Eng. Data, 1973,
18, p. 67.
Clark' R. P. — J. Chem. and Eng. Data, 1975r
20, p. 17.
Clark S. P. — J. Chem. Phys., 1959, 31, p. 1526.
Clausing P. — Z. anorg. und allg. Chem., 1932,
204, S. 33.
Claytor R. N., Marshall B. J. — Phys. Rev., 1960,
120, p. 332.
Cleaver В., Rhodes E., Ubbelohde A. R. — Proc.
Roy. Soc. London, 1963, A276, p. 437.
Cleaver В., Rhodes E., Ubbelohde A. R. — Proc-
Roy. Soc. London, 1963, A276, p. 453.
Cleaver В., Williams J. F. — J. Phys. Chem.
Solids, 1968, 29, p. 877.
Cloud W. H., Schreiber D. S., Babcock K. R. —
J. Appl. Phys., 1962, 33, p. 1193.
Clouser P. L. — Diss. Abstrs, 1964, 25, p. 3051.
Clouser P. L., Gordy W. — Bull. Amer. Phys.
Soc, 1963, 8, p. 326.
Clouser P. L., Gordy W. — Phys. Rev., 1964, A134,
p. 863.
Clusius K., Eichenauer W. — Z. Naturforsch., 1956,
All, S. 715.
Clusius K., Franzosini P. — Z. phys. Chem. (BRD),
1958, 16, S. 194.
Clusius K., Franzosini P. — Z. Naturforsch., 1959,
A14, S. 99.
Clusius K., Franzosini P. — Z. Naturforsch., 1962,
A17, S. 522.
Clusius K., Franzosini P. — Gazz. chim. ital.,
1963, 93, p. 221.
Clusius K., Franzosini P., Piesbergen U. — Z.
Naturforsch., 1960, A15, S. 728.
Clusius K., Goldmann J., Perlick A. — Z.
Naturforsch., 1949, A4, S. 424.
Clusius K., Losa С G. — Z. Naturforsch., 1955,
A10, S. 939.
Clusius K., Piesbergen U. —¦ Helv. phys. acta,
1958, 31, p. 302.
Clusius K., Stern II. — Z. angew. Phys., 1954,
6, S. 194.
CODATA Recommended Key Values for
Thermodynamics: CODATA Bull., 1971, N 5; CODATA
Bull., 1973, N 10; CODATA Bull., 1975, N 17;
CODATA Bull., 1976, N 22; CODATA Bull., 1977,
N 28; CODATA Tentative Set of Key Values
for Thermodynamics. Part VIII, April 1980.
Coey J. M. D. — Physica (B+C), 1977, 91, p. 59.
Coey J. M. D., Roux-Buisson H., Schlenker C,
Lakkis S., Dumas J. — Rev. gen. thermiq., 1976,
15, p. 1013.
Cogin G. E., Kimball G. E. — J. Chem. Phys.,
1948, 16, p. 1035.
Cohen E., Helderman W. D., Moesveld A. L. T. —
Z. phys. Chem. Leipzig, 1920, 96, S. 259.
Cohen E., Kooy J. — Z. phys. Chem. Leipzig,.
1928, A139, S. 273.
Cohen L. H., Klement W. J. — J. Chem. and Eng.
Data, 1974, 19, p. 210.
Cohen-Adad R., Michaud M. — С. г. Acad. sci.,
1956, 242, p. 2569.
Cohen-Adad R., Michaud M., Said J., Rollet
A. P. — Bull. Soc. chim. France, 1961, N 2, p. 356.
Cohen-Adad R., Ruby C. — C. r. Acad. sci., 1964r
C258, p. 6163.
553
Литература
1897. Cohen-Adad R., Ruby С — С. г. Acad. sci., 1966, 1932.
C263, p. 1014.
1898. Cohen-Adad R., Ruby C, Pichon M. J. — C. r. 1933.
Acad. sci., 1965, 260, p. 2200.
1899. Coheur F. P. — Bull. soc. roy. sci. Liege, 1943, 1934,
12, p. 98. 1935.
1900. Cole S. S., Scholes S. R., Amberg C. R. — J. Amer.
Ceram. Soc, 1935, 18, p. 58. 1936.
1901. Cole S. S., Taylor N. W. — J. Amer. Ceram.
Soc, 1935, 18, p. 53. 1937.
1902. Collings E. W., Ho J. С — Phys. Rev., 1970,
B2, p. 235. 1938.
1903. Collings E. W., Ho J. C. — Phys. Rev., 1971,
B4, p. 349. 1939.
1904. Collins J. G. — J. Phys.: Atom, and Mol. Phys.,
1975, 8, p. 304. 1940.
1905. Collongues R., Revcolevschi A., Foex M., Traverse
J. P., Rouanet A. — Colloq. intern. Centre nat. 1941.
rech. sci., 1972, N 205, p. 241.
1906. Colomina M. Цит. по: [3771]. 1942.
1907. Colson A. — С. г. Acad. sei., 1915, 161, p. 414.
1908. Colson A. — С. г. Acad. sci., 1915, 161, p. 458. 1943.
1909. Compton R. N. — J. Chem. Phys., 1977, 66,
p. 4478. 1944.
4910. Conard B. R., Bennett J. E., Gilles P. W. —
J. Chem. Phys., 1975, 63, p. 5502. 1945.
,1911. Conway J. В., Hein R. A. — J. Nucl. Mater., 1946.
1965, 15, p. 149.
1912. Conway J. В., Hein R. A. — In: Advances in ther-
mophysical properties at extreme temperature and 1947.
pressures / Ed. S. Gratch. N. Y.: ASME, 1965,
p. 131. 1948.
1913. Conway J. В., Hein R. A., Fincel R. M., Losekamp
A. C. — U. S. AEC General Electric Co. Rept 1949.
GE-TM-64-2-8. Cincinnati (Ohio), 1965.
1914. Cook D. В., Zemansky M. W., Boorse H. A. — 1950.
Phys. Rev., 1950, 80, p. 737.
1915. Cook O. A. — S. Amer. Chem. Soc, 1947, 69, 1951.
p. 331.
1916. Cook R. O., Davies A., Staveley L. A. K. — J. 1952.
Chem. Soc. Faraday Trans. I, 1972, p. 1384.
1917. CoolenF. С M.,BaghiusL. С. J., HagedoornH. L., 1953.
Heide J. A. van der. — J. Opt. Soc. Amer., 1974,
64, p. 482.
1918. Coolen F. С M., Hagedoorn H. L. — J. Opt.
Soc. Amer., 1975, 65, p, 952. 1954.
1919. Cooper С. В. — J. Chem. Phys., 1953, 21, p. 777.
1920. Cooper D., Longstroth G. O. — Phys. Rev., 1929, 1955.
33, p. 243.
1921. Coops J., Balk A. N., Tolk M. W. — Recueil 1956.
trav. chim., 1956, 75, p. 75.
1922. Coppellina F., Napolitano G. — Ann. chim. (Ital.), 1957
1967, 57, p. 1087.
1923. Coppens P., Smoes S., Drowart J. — Trans. Fara- 195»
day Soc, 1967, 63, p. 2140.
1924. Coppens P., Smoes S., Drowart J. — Trans. Fara- ,nl-o
day Soc, 1968, 64, p. 630. 1959"
1925. Corak W. S., Goodman В. В., Satterthwaite С. В.,
Wexler A. — Phys. Rev., 1956, 102, p. 656. I960.
1926. Corbino О. М. — Phys. Z., 1912, 13, S. 375.
1927. Cordfunke E. H. P. — I. Inorg. and Nucl. Chem., 1961.
1961, 23, p. 285.
1928. Cordfunke E. H. P. — J. Phys. Chem., 1964, 68, 1962.
p. 3353.
1929. Cordfunke E. H. P., Aling P. — Trans. Faraday 1863.
Soc, 1965, 61, p. 50.
1930. Cordfunke E. H. P., Giessen A. A. — J. Inorg. 1864.
and Nucl. Chem., 1963, 25, p. 553.
1931. Cordfunke E. H. P., Muis R. P., Prins G. — 1965.
J. Chem. Thermodyn., 1979, 11, p. 819.
Cordfunke E. H. P., Ouweltjes W. — J. Ghea.
Thermodyn., 1977, 9, p. 71.
Cordfunke E. H. P., Ouweltjes W. — J. Chem.
Thermodyn., 1981, 13, p. 193.
Corliss Ch. — J. Res. NBS, 1969, A73, p. 277.
Corliss Ch., Sugar J. — J. Phys. and Chem. Ref.
Data, 1979, 8, p. 1109.
Cormier M., Claisse F. — J. Less-Common Metals,
1974, 34, p. 181.
Cornman W. R. — U. S. AEC. Du Pont de
Nemours Co. Rept DP-749. Wilmington, 1962.
Cornman W. Д.— U. S. AEC. Du Pont de Nemours
Co. Rept DP-750. Wilmington, 1962.
Corsan J. M., Cook A. J. — Phys. Lett., 1969,
A28, p. 500.
Cosgrove L. A., Snyder P. E. — J. Amer. Chem.
Soc, 1953, 75, p. 1227.
Cotton D. H., Jenkins D. R. — Trans. Faraday
Soc, 1969, 65, p. 1537.
Coughlin J. P. — J. Amer. Chem. Soc, 1955,
77, p. 868.
Coughlin J. P. — J. Amer. Chem. Soc, 1958,
80, p. 1802.
Coughlin J. P., King E. G. — J. Amer. Chem.
Soc, 1950, 72, p. 2262.
Coutures J. P. These. Paris, Universitat, 1971.
Coutures J. P. Melting point of refractory oxides:
Lanthanides sesquioxides: Preprint. Lab. Ultra-
Refract., CNRS, Odeillo-Fontromeu (Fr.), 198©.
Coutures J. P., Verges R., Foex M. — Rev. int.
hautes temp, et refract., 1975, 12, p. 181.
Cowley R. A., Dolling G. — Phys. Rev., 1968,
167, p. 464.
Cox D. M., Elliott J. — Spectrosc Lett., 1979,
12, p. 275.
Cox J. D., Harrop D. — Trans. Faraday Soc,
1965, 61, p. 1328.
Cox J. D., Herington E. F. — Pure and Appl.
Chem., 1974, 40, p. 399.
Craft W. Z., Slutsky L. Z. — J. Chem. Phys.,
1968, 49, p. 638.
Cramer E. M., Hawes L. L., Miner W. N., Schon-
feld F. W. — In: Metal Plutonium/ Ed. A. S. Cof-
finberry, W. N. Miner. Chicago; Univ. Press, 1961,
p. 112.
Cranderna A. W., Rao С N. R., Honig J. M. —
Trans. Faraday Soc, 1958, 54, p. 1069.
Crane W. O., Christy A. — Phys. Rev., 1930, 36,
p. 421.
Crangle J., Smith T. F. — Phys. Rev. Lett., 1962,
9, p. 86.
Crangle J., Temporal J. — J. Phys.: Metal Phys.,
1973, F3, p. 1097.
Crawford F. H., Jorgensen T. — Phys. Rev., 1935,
47, p. 358.
Crawford F. H., Jorgensen T. — Phys. Rev., 1935,
47, p. 932.
Crawford F. H., Jorgensen T. — Phys. Rev., 1936,
49, p. 745.
Crawford J. A., Montgomery D. J. — Bull. Amer.
Phys. Soc, 1957, 2, p. 299.
Creutz E. С — Rept CP-255, MP Chicago 17,
Sept. 15, 1942, Цит. по: [492, с 129].
Creutz E. С — Rept A-95, MP Chicago 16. Цит.
no: [492, с 129].
Criss С M., Cobble J. W. — J. Amer. Chem.
Soc, 1961, 83, p. 3223.
Cristenscu S., Simon F. — Z. phys. Chem. Leipzig,
1933, B25, S. 273.
554
Литература
1966. Crowder В. L., Sienko H. J. — Inorg. Chem., 1965,
4, p. 73.
1967. Csaszar L., Koczkas E. — Acta phys. Acad. sci. 1999.
hung., 1967, 23, p. 211.
1968. Csaszar L., Koczkas E., Matrai T. — Kozp. fiz. 2000.
kut. intez. kozl., 1964, 12, dd. 135.
1969. Cubicciotti D. — High Temp. Sci., 1971, 3, p. 349. 2001.
1970. Cubiociotti D., Keneshea F. J. — High Temp. Sci.,
1972, 4, p. 32. 2002.
1971. Cunningham P. Т., Johnson S. A., Cairns E. J. — 2003.
J. Electrochem. Soc, 1972, 119, p. 1448.
1972. Curtis C. E., Donney L. M., Johnson J. R. — 2004.
J. Amer. Ceram. Soc, 1954, 37, p. 458.
1973. Curtiss L. A.— Chem. Phys. Lett., 1979, 68, 2005.
p. 225.
1974. Cypres R., Wollast R., Raucq J. — Ber. Dtsch. 2006.
keram. Ges., 1963, 40, S. 527.
1975. Cyvin B. N., Cyvin S. J., Rao D. В., Snelson A. — 2007.
Acta chem. scand., 1971, 25, p. 470.
1976. Cyvin S. J., Cyvin B. N., Bhogeswara Rao D. В., 2008.
Snelson A. — Z. anorg. und allg. Chem., 1971,
380, S. 212.
1977. Cyvin S. J., Cyvin B. N., Snelson A. — J. Phys. 2009.
Chem., 1970, 74, p. 4338.
1978. Cyvin S. J., Cyvin B. N., Snelson A. — J. Phys. 2010.
Chem., 1971, 75, p. 2609.
1979. Cyvin B. N., Hargittai M., Cyvin S. J., Hargit- 2011.
tai I. — Acta chim. Acad. sci. hung., 1975, 84,
p. 55. 2012.
1980. Daane A. H. — Iowa State Coll. J. Sci., 1955,
29, p. 400. 2013.
1981. Daane A. H., Spedding F. H. — J. Electrochem.
Soc, 1953, 100, p. 442. 2014.
1982. Dabringhaus #., Meyer H. J. — J. Cryst. Growth,
1977, 40, p. 139. 2015.
1983. Dadgar A., Taherian M. R. — J. Chem. Thermo- 2016.
dyn., 1977, 9, p. 711.
1984. Dahl A. /., Cleaves H. E. — J. Res. NBS, 1949, 2017.
43, p. 513.
1985. Dahl A. J., Van Dusen M. S. — J. Res. NBS, 2018.
1947, 39, p. 53.
1986. Dangley W. J., Mulay L. N. — Mater. Res. Bull., 2019.
1972, 7, p. 739.
1987. Dao N. Q., Knidiri M. — Spectrochim. acta, 1976, 2020.
A32, p. 1113.
1988. Darnell A. J., McCollum W. A. — U. S. North 2021.
American Aviation Inc., Rept NAA-SR-6498.
Downey, Calif., 1961. 18 p.
1989. Darnell A. J., McCollum W. A. — J. Phys. Chem. 2022.
Solids, 1970, 31, p. 805.
1990. Darnell A. J., McColum W. A., Milne T. A. — 2023.
J. Phys. Chem., 1960, 64, p. 341. 2024.
1991. Da Silva J. F., Burgemeister E. A., Dokoupil Z. —
Phys. Lett., 1967, A25, p. 354. 2025.
1992. Da Silva J. F., Burgemeister E. A., Dokoupil Z. —
Physica, 41, p. 409. 2026.
1993. Da Silva J. F., Scheffer J., Duykeren N. W. J.
van, Dokoupil Z. — Phys. Lett., 1964, 12, p. 166. 2027.
1994. Das Gupta S., Bhise V., Stuteville D., Chung J. W.,
Bonilla С F. — In: Proc. Sixth Symp. Thermo- 2028.
phys. Properties. N. Y.: ASME, 1973, p. 387.
1995. Datz S., Smith W., Taylor E. — J. Chem. Phys., 2029.
1961, 34, p. 538.
1996. Daunt J. G., Mendelssohn K. — Proc. Roy. Soc. 2030.
London, 1937, A160, p. 127.
1997. Dauphinee T. M., Martin D. L., Preston-Tho- 2031.
mas H. — Proc. Roy. Soc London, 1955, A233,
p. 214. 2032.
1998. Dauphinee Т. М., McDonald D. К. С, Preston-
Thomas H. — Proc. Roy. Soc. London, 1954, A221,
p. 267.
Davies D. H., Benson G. C. — Canad. J. Chem.,
1965, 43, p. 1912.
Davies J. E. D., Sandford W. F.— J. Chem. Soc.
Dalton Trans., 1975, p. 1912.
Davies Т., Singer S. S., Stavely L. A. K. —
J. Chem. Soc, 1954, p. 2304.
Davis D. N. — Astrophys. J., 1947, 106, p. 28.
Davis D. N., Keenan P. C. — Publ. Astron. Soc.
Pacif., 1969, 81, p. 230.
Davis D. S., Andrew K. L. — J. Opt. Soc. Amer.,
1978, 68, p. 235.
Davis W. J., Rogers S. E., Ubbelohde A. R. —
Proc. Roy. Soc. London, 1953, A220, p. 14.
Dawihl W., Shroter K. — Z. anorg. und allg.
Chem., 1937, 233, S. 178.
Dawson R., Brackett E. В., Brackett Т. Е. —
J. Phys. Chem., 1963, 67, p. 1669.
Dawton V. M., Wilkinson K. L. — Gr. Brit. Atom.
Energ. Res. Estab. Rept GPR-1906. Gr. Brit,
1955, p. 6.
Deadmore D. L. — J. Amer. Ceram. Soc, 1965,
48, p. 357.
Dean D. J., Kay A. E., Loasby R. G. — J. Inst.
Met., 1958, 86, p. 464.
Deardorff D. K., Hayes E. T. — J. Metals, 1956,
8, p. 509.
Deardorff D. K., Kato H. — Trans. Met. Sec.
AIME, 1959, 215, p. 876.
Deardorff D. K., Kato H. — Trans. Met. Sec.
AIME, 1963, 227, p. 264.
Debets P. С -~ J. Inorg. and Nucl. Chem., 1964,
26, p. 1468.
Debets P. С — Acta crystallogr., 1966, 21, p. 589.
De Boer J. ff., Broos J., Emmens H. — Z. anorg.
und allg. Chem., 1930, 191, S. 113.
De Boer J. H., Burgers W. G., Fast J. D. — Proc.
Koninkl. nederl. akad. Wet., 1936, 39, p. 515.
De Boer J. #., Fast J. D. — Z. anorg. und allg.
Chem., 1930, 187, S. 193.
De Boer J. H., Fast J. D., Clausing P. — Recueil
trav. chim., 1936, 55, p. 450.
Defacqz E., Gluichard M. — Ann. chim. phys.,
1901, 24, p. 139.
De Fre.'s D. /., Levi B. A., Pollack S. K., Hehre
W. J., Binkley J. S., Pople J. A. — J. Amer.
Chem. Soc, 1979, 101, p. 4085.
Degreve F., Drowart J. — Rev. А. Т. В. Met.
(Belgium), 1966, 6, p. 204.
Deitz V. — J. Chem. Phys., 1936, 4, p. 575.
Dekker H. — Bull. Chem. Thermodyn., 1961, 4,
p. 22.
De Коек С W., Wesley R. D., Radtke D.D. —
High Temp. Sci., 1972, 4, p. 41.
Del Bene J. E. — Chem. Phys. Lett., 1979, 64,
p. 227.
Delepine M. M. — С. г. Acad. sci., 1900, 131,
p. 184.
Delepine M. M. — Bull. Soc. chim. France, 1903,
29, p. 1166.
Delepine M. M., Hallopeau L. A. — C. r. Acad.
sci., 1899, 129, p. 600.
Delesalle G., Dervainne P., Heubel J. — С. г. Acad.
sci., 1969, C268, p. 553.
Demarest H. H., Cassell C. R., Jamieson J. C. —
J. Phys. Chem. Solids, 1978, 39, p. 1211.
De Maria G., Burns R. P., Drowart J., Inghram
M. G. — J. Chem. Phys., 1960, 32, p. 1373.
Литература
2033. De Maria G., Drowart J., Inghram M. G. — 2069.
J. Chem. Phys., 1959, 30, p. 318.
2034. De Maria G., Gigli R., Malaspina L. — SIPS 2070.
(soc. ital. progr. sci.), sci. tec, 1963, 7, p. 26.
2035. De Maria G., Malaspina L., Piacente V. — SIPS
(soc. ital. progr. sci.), sci. tec, 1963, 7, p. 33. 2071.
2036. Dementjev A., Kracht D. —Chem. Phys. Lett.,
1975, 35, p. 243. 2072.
2037. Demidov A. V., Ivanov A. A., Ivashkevich L. S.,
Ischenko A. A., Spiridonov V. P., Almlof J., 2073.
Strand T. G. — Chem. Phys. Lett., 1979, 64,
p. 528. 2074.
2038. Dempesy C. W., Gordon J. E., Romer R. H. —
Phys. Rev. Lett., 1963, 11, p. 547. 2075.
2039. Demtroder W., McClintock M., Zare R. N. —
J. Chem. Phys., 1969, 51, p. 5495. 2076.
2040. Demtroder W., Stock M. — J. Mol. Spectrosc,
1975, 55, p. 476. 2077.
2041. Denielou L., Fournier Y., Petitet J. P., Tequi С —
С. r. Acad. sci., 1969, C269, p. 1577. 2078.
2042. Denielou L., Fournier Y., Petitet J. P., Tequi C. —
C. r. Acad. sci., 1970, C270, p. 1854. 2079.
2043. Denielou L., Fourier Y., Petitet J. P., Tequi С —
Rev. int. hautes temp, et refract., 1971, 8, p. 119. 2080.
2044. Denielou L., Petitet J. P., Tequi С — Thermo-
chim. acta, 1974, 9, p. 135. 2081.
2045. Denker S. P. — J. Appl. Phys., 1966, 37, p. 142.
2046. Denning R. G., Snellgrove T. R., Woodwork 2082.
D. R. —Mol. Phys., 1976, 32, p. 419.
2047. Denning R. G., Snellgrove T. R., Woodwork 2083.
D. R. — Mol. Phys., 1979, 37, p. 1109.
2048. Dennison D. H., Gschneidner K. A. J., Daane
A. H. — J. Chem. Phys., 1966, 44, p. 4273. 2084.
2049. De Poorter G. L., Rofer-De Poorter С. К. —
Spectrosc. Lett., 1975, 8, p. 521. 2085.
2050. De Poorter G. L., Rofer-De Poorter С. К. — Inorg.
and Nucl. Chem. Lett., 1977, 13, p. 101. 2086.
2051. Derge G., Cefola M. — Rept CT-2277. Chicago,
Oct. 26, 1944. Цит. по: [492, с 129]. 2087.
2052. Derkosch /., Preisinger A., Steffan L. — Micro-
chim. acta, 1975, 2, S. 477. 2088.
2053. Deshpande V. V., Karkhanavala M. D., Rao 2089.
U. R. K. — J. Therm. Anal., 1974, 6, p. 613.
2054. De Sorbo W. — J. Phys. Chem., 1958, 62, p. 965. 2090.
2055. Devis J. E. D., Standford W. F. — J. Chem. Soc. 2091.
Dalton Trans., 1975, p. 1912.
2056. DeVisser C, Somsen G. — J. Chem. Soc. Faraday 2092.
Trans. I, 1973, 69, p. 1440.
2057. DeVisser C, Van Netten E., Somsen G. — Electro- 2093.
chim. acta, 1976, 21, S. 97.
2058. Devlin J. P., James D. W. — Chem. Phys. Lett., 2094,
1970, 7, p. 237. 2095.
2059. Devlin J. P., Li P. C, Pollard G. — J. Chem.
Phys., 1970, 52, p. 2267. 2096.
2060. DeVries R. C, Roy R. — Bull. Amer. Ceram. Soc,
1954, 33, p. 370. 2097.
2061. Dewar J. ~ Proc. Roy. Soc. London, 1913, A89,
p. 158. 2098.
2062. Dewing E. — J. Amer. Chem. Soc, 1955, 77,
p. 2639. 2099.
2063. Dewing E. W. —Met. Trans., 1970, 1, p. 1691.
2064. Dewing E. W. —Met. Trans., 1970, 1, p. 2211. 2100.
2065. Dewing E. W. — J. Chem. and Eng. Data, 1975,
20, p. 221. 2101.
2066. Dewing E. W. — J. Electrochem. Soc, 1976, 123,
p. 1289.
2067. Dewing E. W. — Met. Trans., 1980, Bll, p. 245. 2102.
2068. D'Eye R. W. M., Martin F. S. — J. Chem. Soc,
1957, p. 1847. 2103,
Dharwadkar S. R., Chandrasekharaiah M. S. —
High Temp. Sci., 1973, 5, p. 5.
Dharwadkar S. R., Chandrasekharaiah M. S.r
Karkhanavaba M. D. — J. Therm. Anal., 1975,,
7, p. 219.
Diamond J. J., Schneider S. J. — J. Amer. Ceram.
Soc, 1960, 43, p. 1.
Dichtl H. J. — Radex Rundschau, 1967, N 3/4,
S. 608.
Dickson D. S., Myers J. R., Pool M. J., Saxer
R. K. — Trans. Met. Soc. AIME, 1969, 245, p. 175.
DicksonD. S., Myers J. R., Saxer R. K. — J. Phys.
Chem., 1965, 69, p. 4044.
Dietzel A., Tober H. — Ber. Dtsch. keram. Ges.r
1953, 30, S. 47.
Dietzel A., Tober H. — Berl. Dtsch. keram. Ges.,
1953, 30, S. 71.
Dikant /., Mariani E., Jonova A., Dobrzanskii
G. F. — Acta phys. Slov., 1977, 27, s. 266.
Dichter I. Ya., Lebedev S. V. — High. Temp.-
High Pressur., 1970, 2, p. 55.
Dill J. D., Schleyer P. R., Einkley J. S., Pople
J. A. — J. Amer. Chem. Soc, 1977, 9, p. 6159.
Dillon J. G., Swanson B. S., Nelson P. — J. Chem.
Phys., 1966, 44, p. 4229.
Dimitriev J. В., Marinov M. R., Stavrakeva
D. A. — С. г. Acad. Bulg. Sci., 1966, 19, p. 1055.
DitmarsD. A. — High Temp.-High Pressur., 1979,
11, p. 615.
Ditmars D. A., Cerzairliyan A., Ishihara S.,
Douglas T. B. — U. S. NBS Spec. Publ. 260—55.
Wash., 1977. 80 p.
Ditmars W. E., Johnston H. L. — J. Amer. Chem.
Soc, 1953, 75, p. 1830.
Dixit L., Kapoor S. K., Singh J. D., Gupta P. L. —
Indian J. Phys., 1977, B51, p. 116.
Docken K. K., Hinze J. — J. Chem. Phys., 1972r
57, p. 4928.
Docken K. K., Hinze J. — J. Chem. Phys., 1972,.
57, p. 4936.
Dode M. — С. г. Acad. sci., 1936, 203, p. 365.
Dode M. — Bull. Soc. chim. France, 1937, 4,
p. 2093.
Doi H. — l. Appl. Phys., 1969, 40, p. 3080.
Domagala R. F., McPerson D. J. — Trans. Met.
Soc, AIME, 1954, 200, p. 238.
Domagala R. F., Ruh R. — Trans. Amer. Soc.
Metals, 1965, 58, p. 164.
Domange L., Wohlhuter M. — C. r. Acad. sci.,,
1949, 228, p. 1591.
Donath J. — Ber. chem. Ges., 1879, 12, S. 742.
Donnan F. G., Hope G. D. — Trans. Faraday Soc,
1909, 5, p. 244.
D'Orazio L. A., Wood R. H. — J. Phys. Chem.,
1965, 69, p. 2550.
Doucet Y., Le Due J. A. — С. г. Acad. sci., 1953,
C237, p. 52.
Doucet Y., Vallet C. — C. r. Acad. sci., 1964,
C259, p. 1517.
Doucet Y., Vallet С — С. г. Acad. sci., 1968,
C267, p. 200.
Doucet У., Vallier J. — С. г. Acad. sci., 1962,
C255, p. 2935.
Dougherty G. /., Dunn M. R., McEwan M. J.,
Phillips L. F. — Chem. Phys. Lett., 1971, 11,
p. 124.
Douglas A. E., Veillette P. M.— J. Chem. Phys.,
1980, 72, p. 5378.
Douglas Т. В. — J. Res. NBS, 1954, 52, p. 223,
556
Литература
2104. Douglas Т. В. Private communication. — U. S. 2139.
NBS, 1961, May 23. [JANAF].
2105. Douglas Т. В., Ball A. F., Ginnings D. C, Da-
vies W. D. — J. Amer. Chem. Soc, 1952, 74, 2140.
p. 2472.
2106. Douglas Т. В., Dever J. L. — J. Res. NBS, 1954, 2141.
53, p. 81.
2107. Douglas Т. В., Dever J. L. — J. Amer. Chem. 2142.
Soc, 1954, 76, p. 4826.
2108. Douglas Т. В., Dever J. L., Harman A. W. — 2143.
U. S. NBS. Rept N 6297, Wash., 1960.
.2109. Douglas Т. В., Ditmars D. A. — V. S. NBS 2144.
Rept 73-280, Wash., 1973.
2110. Douglas Т. В., Epstein L. F., Dever J. L., How- 2145.
land W. H. — J. Amer. Chem. Soc, 1955, 77, 2146.
p. 2144.
.2111. Douglas Т. В., Victor A. C. — Z. Res. NBS, 2147.
1958, 61, p. 13.
2112. Drowart J. — Thesis. Fac. sci. Univ. Brussels, 2148.
Belgium, 1959.
2113. Drowart J. — In: Proc. Intern. Symp. on Conden- 2149.
sation and Evaporation of Solids, Dayton, Ohio,
Sept. 12—14, 1962. N. Y.; L.: Gordon and Breach, 2150.
1964, p. 255. 2151.
2114. Drowart /., Coppens P., Smoes S. — J. Chem.
Phys., 1969, 50, p. 1046. 2152.
2115. Drowart J., DeMaria G., Burns B. P., Inghram
M. G. — J. Chem, Phys., 1960, 32, p. 1366. 2153.
2116. Drowart /., Exsteen G., Verhaegen G. — Trans.
Faraday Soc, 1964, 60, p. 1920. 2154.
2117. Drowart J., Pattoret A., Smoes S.~ Proc. Brit.
Ceram. Soc, 1967, N 8, p. 67. 2155.
2118. Drowart J., Pattoret A., Smoes S., Degreve F.,
Detry D. — In: Advances in Mass Spectrometry/ 2156.
Ed. W. L. Mead. London, 1966, vol. 3, p. 931.
2119. Drucker С — Arkiv kemi, mineral., geol., 1935, 2157.
All, N 18, 27 p.
2120. Dubois /., Millet J. — С. г. Acad. sci., 1968,
C266, p. 852. 2158.
2121. Dubois /., Millet J. — C. r. Acad. sci., 1969,
C269, p. 1336.
2122. Dubois L. H., Gole J. L. — J. Chem. Phys., 1977, 2159.
66, p. 779.
2123. Dubrovin I. TV., Evseev A. K. — Kernenergie, 1960,
3, S. 577. 2160.
2124. Dugdall J. S., Morrison J. A., Patterson D. — 2161.
Proc. Roy. Soc. London, 1954, A224, p. 228.
.2125. Dummer G. Z. — Z. Phys., 1965, 186, S. 249. 2162.
2126. Dunham J. L. — Phys. Rev., 1932, 41, p. 721. 2163.
2127. Dunn Т. М. ~ Mol. Spectrosc. Modern Res., 1972,
p. 231.
2128. Dunn T. M,, Hanson L. K., Bubinson K. A. — 2164.
Canad. J. Phys., 1970, 48, p. 1657.
2129. Duong Hong Tua, Liberman S., Pinand J. — 2165.
Opt. Communs, 1976, 18, p. 533.
2130. Duquesnoy A. — Rev. int. hautes temp, et refract., 2166.
1965, 2, p. 201.
2131. Duquesnoy A., Marion F. — С. г. Acad. sci., 1964, 2167.
258, p. 5657.
2132. Duwez P. — J. Appl. Phys., 1951, 22, p. 1174. 2168.
2133. Duwez P. — Trans. AIME, 1951, 3, p. 765.
2134. Duwez P. — J. Appl. Phys., 1953, 24, p. 152. 2169.
2135. Dworkin A. S. Private communication. — Oak
Ridge Nat. Lab., 1964, Dec. 1 (JANAF).
2136. Dworkin A. S. — J. Inorg. and Nucl. Chem., 1972, 2170.
34, p. 135.
2137. Dworkin A. S., Bredig M. A. — J. Phys. Chem., 2171.
1960, 64, p. 269.
2138. Dworkin A. S., Bredig M. A, — J. Phys. Chem., 2172.
1970, 74, p. 3403.
Dyke J. M., FayadN. K., Morris A., Trickle I. B.,
Allen G. С — J. Chem. Phys., 1980, 72,
p. 3822.
Each D. Т., Carlson O. N. — Trans. ASME, 1960,
52, p. 1097.
Eades B. G., Hughes D. G., Andrew E. B. — Proc.
Phys. Soc. London, 1958, 71, p. 1019.
Eastman E. D., Bodebusch W. H. — J. Amer. Chem.
Soc, 1918, 40, p. 489.
Eckhardt В., Eggers ?>., Slutsky L. J. — Spectro-
chim. acta, 1970, A26, p. 2033.
Edmondson W., Egerton A. C. — Proc. Roy. Soc
London, 1927, A113, p. 520.
Edvinsson G. — Thesis. Stockholm, 1971.
Edvinsson G., Bornstedt A., Nylen P. — Akr. fys.,
1968, 38, s. 193.
Edvinsson G., Nylen Ch. — Phys. scr., 1971, 3,
s. 261.
Edvinsson G., Selin L. E. — Phys. Lett., 1964,
9, p. 238.
Edvinsson G., Selin L. E., Aslund N. —¦ Ark. fys.,
1965, 30, s. 283.
Edwards J. G. — J. Chem. Phys., 1971, 54, p. 545.
Edwards J. W., Johnston H. L., Blackburn P. E. —¦
J. Amer. Chem. Soc, 1951, 73, p. 172.
Edwards J. W., Johnston H. L., Blackburn P. E. —
J. Amer. Chem. Soc, 1951, 73, p. 4727.
Edwards J. W., Johnston H. L., Blackburn P. E. —¦
J. Amer. Chem. Soc, 1952, 74, p. 1539.
Edwards J. W., Johnston H. L., Ditmars W. E. —
J. Amer. Chem. Soc, 1953, 75, p. 2467.
Edwards J. W., Speiser В., Johnston H. L. —
J. Appl. Phys., 1951, 22, p. 424.
Effenberger H., Zemann J. — Z. Kristallogr., 1979,
150, S. 133.
Efimov M. E., Klevaichuk G. N., Medvedev V. A.,
Kilday M. V. — J. Res. NBS, 1979, 84,
p. 273.
Efremov Yu. M., Gurvich L. V., Savchenko A. N.,
Sviridenkov E. A. — Chem. Phys. Lett., 1979, 61,
p. 179.
Egorova N. M., Bambidi N. G. — In: Molecular
structure and vibrations/ Ed. S. J. Cyvin.
Amsterdam: North-Holl. Publ. Co, 1972, p. 212.
Ehlert Т. С — High Temp. Sci., 1977, 9, p. 237.
Ehlert T. C, Margrave J. L. — J. Amer. Ceram.
Soc, 1958, 41, p. 330.
Eichler A., Gey W. — Z. Phys., 1972, 251, S. 321.
Einhorn P. A. — In: Therm. Anal.: Proc.
2nd Intern. Conf. 1968. N. Y.: Acad. Press, 1969,
vol. 1, p. 149.
Eisel D., Zevgolis D., Demtroder W. — J. Chem.
Phys., 1979, 71, p. 2005.
Eisenstadt M., Bao V. S., Hothberg G. M. —
J. Chem. Phys., 1959, 30, p. 604.
Eisenstadt M., Bothberg G., Kusch P. — J. Chem.
Phys., 1958, 29, p. 797.
Eitel W., Skaliks W. — Z. anorg. und allg. Chem.,
1929, 183, S. 263.
Eliezer I., Howald B. A. — High Temp. Sci., 1978,
10, p. 1.
Eller P. G., Larson A. C, Peterson J. В., Ensor
D. D., Young J. P. — Inorg. chim. acta, 1979,
37, p. 129.
Elliott B. P. — U. S. At. Energy Comm. ARF-
2120, 1960, p. 74.
Elliott B. P. — Trans. Amer. Soc. Metals, 1960,
52, p. 990.
Ema K., Hamano K., Hatta I. — J. Phys. Soc.
Jap., 1975, 39, p. 726.
557
Литература
2173. Emons Н. П., Brautigam G., Thomas R. — Chem.
zvesti, 1976, 30, s. 773.
2174. Engel T. K. — J. Nucl. Mater., 1967, 23, p. 25. 2208
2175. Engel T. K. — U, S. Mount Lab. Rept MLM-1347. 2209,
Miamisburg (Ohio), 1967.
2176. Engel T. K. — J. Nucl. Mater., 1969, 31, p. 211. 2210.
2177. Engel T. K., Jordan K. C, Scott D. M., Otto
G. W. — U. S. AEC. Division Technical
Information Exstension. Rept TID-7641, 1962. 2211.
2178. England W. B. — J. Chem. Phys., 1978, 68,
p. 4896. 2212.
2179. Engmann R., Wolff P. M. — Acta crystallogr.,
1963, 16, p. 993.
2180. Ennen G. — Ber. Max-Planck Inst. Stromungs- 2213.
lorsch., 1976, 15, S. 1.
2181. Ennen G., Ottinger C. — Chem. Phys. Lett., 1975, 2214.
36, p. 16.
2182. Entner P., Neekel A. — Monatsh. Chem., 1967, 2215.
98, S. 1083.
2183. Epstein L. F. — J. Nucl. Mater., 1967, 22, p. 340. 2216.
2184. Epstein L. F., Howland W. H. — J. Amer. Ceram.
Soc, 1953, 36, p. 334. 2217.
2185. Erdey Z., Liptay G., Gal S. — Talanta, 1965, 12,
p. 257. 2218.
2186. Eriksson К. В., Johansson I., Norben G. — Ark.
fys., 1964, 28, s. 233. 2219.
2187. Eriksson К. В., Wenaker I. — Phys. scr., 1970,
1, s. 21. 2220.
2188. Erlich P. — Z. Electrochem., 1939, 45, S. 362. 2221.
2189. Erlich P. — Z. anorg. und allg. Chem., 1941, 247, 2222
S. 53. 2223.
2190. Ermisher W., Hauser O., Schenk M. — J. Nucl. 2224.
Mater., 1965, 16, p. 341.
2191. ErokhinE.V.,Prikhod'koA.Ya.,SpiridonovV.P., 2225
Kiselev Yu. M. — High Temp. Sci., 1978, 10,
p. 269. 2226.
2192. Esperon J. F., Almandos J. R., Suares С. В. —
Spectrosc. Lett., 1978, 11, p. 411. 2227
2193. Estermann /., Friedberg S. A., Goldman J. E. —
Phys. Rev., 1952, 87, p. 582.
2194. Evans J. С — Chem. Communs, 1969, p. 682.
2195. Evans W. H., Jacobson R., Munson T. R., Wag- 2228
man D. D. — J. Res. NBS, 1955, 55, p. 83.
2196. Evans D. F., Richards R. E. — J. Chem. Soc,
1952, p. 3932.
2197. Ewing С. Г., Chang D., Stone J. P. et al. — 2229
J. Chem. and Eng. Data, 1969, 14, p. 210.
2198. Ewing С. Т., Spann J. R., Miller R. R. — 2^30
J. Chem. and Eng. Data, 1971, 16, p. 27.
2199. Ewing C, Stern K. — J. Phys. Chem., 1974, 78, 2231
p. 1998.
2200. Ewing С. Т., Stern К. Н. — J. Phys. Chem., 1975, 2232
79, p. 2007. 2233'
2201. Ewing С. Т., Stone J. P. et al. — U. S. Naval
Research Lab. Rept NRL-6233. Wash., 1965.
2202. Ewing С. Т., Stone J. P., Spann J. R., Mil- 2234.
ler R. R. — J. Chem. and Eng. Data, 1966, 11, 2235.
p. 460.
2203. Ewing С. Т., Stone J. R., Spann J. R., Miller 2236.
R. R. — J. Chem. and Eng. Data, 1966,11, p. 468.
2204. Ewing С. Т., Stone J. P., Spann J. R., Mil- 2237.
ler R. R. — J. Chem. and Eng. Data, 1966, 11,
p. 473. 2238.
2205. Ewing С. Т., Stone J. R., Spann J. R.,
Miller R. R. — J. Phys. Chem., 1967, 71, p. 473. 2239.
2206. Ewing С. Т., Stone J. P., Spann J. R., Stein-
kuller E. W., Williams D. D., Miller R. R. —
U. S. Naval Research Lab. Rept NRL-6246. Wash., 2240.
1965. 2241.
2207. Ewing С. Т., Stone J. P., Spann J. R., Stein-
kuller E. W., Williams D. D., Miller R. R. —
J. Chem. and Eng. Data, 1970, 15, p. 508.
Ewles I. — Philos. Mag., 1923, 45, p. 957.
Eysel H. H., Thym S.— Z. anorg. und allg. Chem.,.
1975, 411, S. 97.
Eysel W. — In: Therm. Anal.: Proc. 3rd Intern.
Conf.: 23—28 Aug. 1971, Davos (Sz), 1971, vol. 2,
p. 179.
Ezhov Yu. S., Komarov S. A. — J. Mol. Struct.,.
1978, 50, p. 305.
Faber J., Lander G. #., Cooper B. R. — AIP Conf.
Proc, 1976, 29. (Magn. Magn. Mater., Annu. Conf.
21st, 1975), p. 379.
Fabricand B. P.. Carlson R. O., Lee C. A., Rabi-
J. J. — Phys. Rev., 1953, 91, p. 1403.
Fairbank W. M., Hdnsch T. W., Schawlow A. L. —
Opt. Soc. Amer., 1975, 65, p. 199.
Farber M., Srivastava R. D. — J. Chem. Soc.
Faraday Trans. I, 1973, 69, p. 390.
Farber M., Uy O. M., Srivastava R. D.— J. Chem..
Phys., 1972, 56, p. 5312.
Farr J. D., Huber E. J., Head E. L., Holleij
С. Е. — J. Phys. Chem., 1959, 63, p. 1455.
Farrar P., Margolin H. — Trans. Met. Soc. AIME,.
1955, 206, p. 101.
Fast J. D. — Z. anorg. und allg. Chem., 1939,,
241, S. 42.
Fast J.D. — Recueil trav. chim., 1939, 58, p. 973.
Fast J. D. — J. Appl. Phys., 1952, 23, p. 350.
Favre P. A. — C. r. Acad. sci., 1871, 73, p. 767.
Favre P. A. — С. т. Acad. sci., 1871, 71, p. 772.
Favre P. A., Silberman J. T. — Ann. chim. phys.,
1853, 37, p. 406.
Favre P. A., Valson С A. — С. г. Acad. sci.,
1873, 77, p. 577.
Feather D. H., Searcy A. W. — High Temp. Sci.,.
1971, 3, p. 155.
Feber R. C., Harrick С. С. Ideal gas thermodyna-
mic functions of lantanide and actinide elements:
Rept LA-3184, UC-4. Los Alamos Sci. Lab., Univ.
California, 1965.
Feber R. C, Herrick С. С. An improved calculation
of the ideal gas thermodynamic functions of
selected diatomic molecules. — U. S. Rept LA-3597.
Los Alamos (Calif.), 1966/1967.
Feber R. C, Herrick С. С. — J. Chem. and Eng.
Data, 1967, 12, p. 85.
Fehrenbacher L. L., Jacobson L. A. — J. Amer.
Ceram. Soc.,.1965, 48, p. 157.
Feinleib J., Paul W. — Phys. Rev., 1967, 155,
p. 841.
Feiser /. — Metall und Erz, 1931, 28, S. 297.
Feldhause I. В., Lang J. I. — U. S. Wright Air
Development Div. Rept WADD-TR-60-904. Wright-
Petterson AFB, Ohio, 1961.
Ferguson W. F. C. — J. Res. NBS, 1932, 8, p. 381.
Fermor J. H., Kjekshus A. — Acta chem. scand.,.
1972, 26, p. 2645.
Fermor J. H., Kjekshus A. — Acta chem. scand.,.
1973, 27, p. 915.
Fernandes J. R., Ganguly S., Rao С N. R. —
Spectrochim. acta, 1979, A35, p. 1013.
Fernando I., Krishnamurthy S. G. — Curr. Sci.,
1949, 18, p. 371.
FerranteR. F., Wilkerson J. L., Graham W. R. M.r
Weltner J. W. — J. Chem. Phys., 1977, 67r.
p. 5904.
Ferraro J. R. — J. Mol. Spectrosc, 1960, 4, p. 99.
Ferris L. M., Baird F. G. — J. Electrochem. Soc.t.
1960, 107, p. 305.
558
Литература
2242. Ferris L. M., Gabbard E. F. Canada. Defence Res.
Northern Lab. Rept DRNL-2401. Fort Churchill,
Monitoba, 1958. 22 p.
2243. Ferroni E., Cocchi M. — Z. Kristallogr., 1959, 111,
S. 154.
2244. Ferroni E., Sabatini A., Orioli P. — Ric. sci.,
1957, 27, p. 1557.
2245. Fert A. — Bull. Soc. franc mineral, et cristallogr.,
1962, 85, p. 267.
2246. Feugier A. — These doct. sci. phys. Fac. sci. Univ.
Paris, 1972.
2247. Ficalora P. J., Uy 0. M., Muenow D. W.,
Margrave J. L. — J. Amer. Ceram. Soc, 1968, 51,
p. 574.
2248. Fiegel L. J., Mohanty G. P., Healy J. H. —
J. Chem. and Eng. Data, 1964, 9, p. 365.
2249. Fieldhouse I. В., Long J. I. — U. S. Wright Air
Devel. Div. Tech. Rept WADD-60-904. Wrigh-
Petterson AFB, Ohio, July, 1961.
2250. Filby J. D., Martin D. L. — Proc. Roy. Soc.
London, 1963, A276, p. 187.
2251. Filby J. D., Martin D. L. — Proc. Roy. Soc.
London, 1965, A284, p. 1396.
2252. Finch A., Gardner P. J., Steadman С J. — Canad.
J. Chem., 1968, 46, p. 3447.
2253. Fineman M. A., Wallace W. E. — J. Amer. Chem.
Soc, 1948, 70, p. 4165.
2254. Fink J. K., Leibowitz L. — U. S. AEC Argonne
Natl. Lab. Rept ANL-CEN-RSD—79-1, Lemont
ail.), 1979.
2255. Finkelnburg W. von, Hahn O. Th. — Z. Phys.,
1938, 39, S. 98.
2256. Finnemore D. K., Johnson D. L., Ostenson J. E.,
Spedding F. #., Beaudry B. J. — Phys. Rev.,
1965, A137, p. 550.
2257. Finnemore D. K., Stromberg T. F., SwensonC.A. —
Phys. Rev., 1966, 149, p. 231.
2258. Fiock E. F., Rodebush W. H. — J. Amer. Chem.
Soc, 1926, 48, p. 2522.
2259. Fischer D. R. — Rev. Sci. Instrum., 1966, 37,
p. 717.
2260. Fischer W. .— Angew. Chem., 1949, 61, S. 336.
2261. Fischmeister H. F. — J. Inorg. and Nucl. Chem.,
1956, 3, p. 182.
2262. Fischmeister H. F. — Monatsch. Chem., 1962, 93,
S. 420.
2263. Fisher E. S., Renken C. J. — Phys. Rev., 1964,
A135, p. 482.
2264. Fiske M. D. — Phys. Rev., 1942, 61, p. 513.
2265. Fitzgibbon G. C, Holley C. E., Wadso I. —
J. Phys. Chem., 1965, 69, p. 2464.
2266. Fitzgibbon G. C, Pavone D., Holley С. Е. —
J. Chem. and Eng. Data, 1967, 12, p. 122.
2267. Flood H., Fykse V., Urnes S. — Z. Elektrochem.,
1955, 59, S. 364.
2268. Flood #., Kleppa O. J. — J. Amer. Chem. Soc,
1947. 69, p. 998.
2269. Flotow H. E., Lohr H. R. — J. Phys. Chem.,
1960, 64, p. 904.
2270. Flotow H. E., O'Hare P. A. G., Boerio-Goates J.
— J. Chem. Thermodyn., 1981, 13, p. 477.
2271. Flotow H. E., OsborneD. W. — Phys. Rev., 1966,
151, p. 564.
2272. Flotow H. E., OsborneD. W. — Phys. Rev., 1967,
160, p. 467.
2273. Flotow H. E., OsborneD. W. — J. Chem.
Thermodyn., 1974, 6, p. 135.
2274. Flotow H. E., OsborneD. W., Fried S. M., Malm
I. G..— J. Chem. Phys., 1976, 65, p. 1124.
2275.
2276.
2277.
.2278.
2279.
2280.
2281.
2282.
2283.
2284.
2285.
2286.
2287.
2288.
2289.
2290.
2291.
2292.
2293.
2294.
2295.
2296.
2297.
2298.
2299.
2300.
2301.
2302.
2303.
2304.
2305.
2306.
2307.
2308.
2309.
2310.
2311.
2312.
2313.
2314.
2315.
2316.
2317.
2318.
2319.
Flotow H. E., Osborne D. W., Westrum E. F. —
J. Chem. Soc, 1968, 49, p. 2438.
Flotow H. E., Tetenbaum M. — J. Chem. Phys.,.
1981, 74, p. 5269.
Foex M. — J. rech. Centre nat. rech. sci., 1958.,
N 42, p. 1.
Foex M. — Sol. Energy, 1965, 9, p. 61.
Foex M. — Rev. intern, hautes temp, et refract.,.
1966, 3, p. 309.
Foex M. — High Temp.-High Pressur., 1977, 9,,
p. 269.
Foex M. <?., Goldsztaub S., Wey R., Jaffray J.,
Lyand R., Wueher J. — J. rech. Centre nat. rech.
sci., 1952, N 21, p. 237.
Foex M., Traverse J. P. — С. г. Acad. sci., 1965,.
261, p. 2490.
Foex M., Traverse J. P. — Rev. int. hautes temp.
et refract., 1966, 3, p. 429.
Foex M., Wyart J. — Bull. Soc. fran$. miner..
et cristallogr., 1953, 76, p. 102.
Fonda G. R. — Phys. Rev., 1923, 21, p. 343..
Fontell N. — Soc. sci. Fennicae Common Phylos.-
Math., 1938, 10, s. 18.
Foppl H. — Z. anorg. und allg. Chem., 1957, 291,
S. 12.
Foppl H. — Z. anorg. und allg. Chem., 1962, 314,
S. 46.
Forcrand R. — С. г. Acad. sci., 1898, 127, p. 514.
ForcrandR. — С. г. Acad. sci., 1900, 130, p. 1465..
Forcrand R. — С. г. Acad. sci., 1901, 133, p. 157.
Forcrand R. — С. г. Acad. sci., 1901, 133, p. 223.
Forcrand R. — С. г. Acad. sci., 1905, 140, p. 990.
Forcrand R. — С. г. Acad. sci., 1906, 142, p. 1252.
Forcrand R. — G. r. Acad. sci., 1906, 143, p. 98.
ForcrandR. — С. г. Acad. sci., 1907, 144, p. 1402-
Forcrand R. — С. г. Acad. sci., 1908, 146, p. 511
Forcrand R. — С. г. Acad. sci., 1909, 149, p. 97.
Forcrand R. — С. г. Acad. sci., 1910, 150, p. 1399.
Forcrand R. — Ann. chim. phys., 1911, 24, p. 256.
Forcrand R. — С. г. Acad. sci., 1911, 152, p. 27..
ForcrandR. — С. г. Acad. sci., 1911, 152, p. 1073.
ForcrandR. — С. г. Acad. sci., 1914, 158, p. 843.
Forcrand R. — С. г. Acad. sci., 1914, 158, p. 991.
Fo'rland 2\, Krogh-Moe J. — Acta chem. scand..,.
1957, 11, p. 565.
Forland Т., Krogh-Moe J. — Acta chem. scand.,
1959, 13, p. 1051.
Forneris R., Tavares-Forneris Y. — J. Mol. Struct.,.
1974, 23, p. 241.
Forsythe W. E. — Astrophys. J., 1911, 34, p. 353.
Forsythe W. E., Adams E. Q. — J. Sci. Lab. Deni-
son Univ., USA, 1937, April.
Forsythe W. E., Worthing A. G. — Astrophys. J..
1925, 61, p. 146.
Foster P. A. — J. Amer. Chem. Soc, 1968, 51,.
p. 107.
Forster P. /., Leckenby R. E., Robbins E. J. —
J. Phys: Atom, and Mol. Phys., 1969, 2, p. 478.
Foz O. R., Colomina M. — An. Real. soc. exp...
fis. у quim., 1946, 42, p. 129.
Fraga S., Ransil B. J. — J. Chem. Phys., 1961.
35, p. 669.
Frank W. B. — J. Phys. Chem., 1961, 65, p. 2081. .
Franklin J. E., Stickney R. E. — High Temp.
Sci., 1971, 3, p. 401.
Franz G., Freyland W., Hensel F. — J. phys. (Fr.). .
1980, 41, p. C8-70.
Franzen H. F. — Diss. Abstrs, 1963, 23, p. 2713.
Franzosini P., Sinistre C. — Ric. sci., 1963, АУ,..
p. 411.
559
Литература
2320. FrazerB. С, Shirane G., Cox D. E. — Phys. Rev.,
1965, A140, p. 1448.
2321. Freeh R., Devlin V. P. — Chem. Phys. Lett., 1976,
38, p. 79.
2322. Fredrickson D. R., Chasanov M. G. — J. Chem.
Thermodyn., 1970, 2, p. 623.
2323. Fredrickson D. R., Chasanov M. G. — J. Chem.
Thermodyn., 1973, 5, p. 485.
2324. Fredrickson D. R., Chasanov M. G. — J. Chem.
Thermodyn., 1974, 6, p. 629.
2325. Fredrickson D. R., Kleb R., Nuttall R. L., Hub-
bard W. N. — Rev. Sci. Instrum., 1969, 40,
p. 1022.
2326. Fredrickson W. R. — Phys. Rev., 1928, 31,
p. 1130.
2327. Fredrickson W. R., Stannard С R. — Phys. Rev.,
1933, 44, p. 632.
2328. Fredrickson F. W., Watson W. W. — Phys. Rev.,
1927, 30, p. 429.
2329. FreemanE. S. — J. Phys. Chem., 1956, 60, p. 1487.
2330. Freeman E. S., Anderson D. A. — Nature, 1963,
199, p. 63.
2331. Freiberg M., Ron A., Schnepp 0. — J. Phys. Chem.,
1968, 72, p. 3526.
2332. French R. A. — Cryogenics, 1968, 8, p. 301.
2333. Freund S. M., Herbst E., Marietta R. P., Jr.,
Klemperer W. — J. Chem. Phys., 1972, 56,
p. 1467.
2334. Freyland W. F., Hensel F. — Ber. Bunsenges.
phys. Chem., 1972, 76, S. 16.
2335. Fried S., Davidson N. R. — U. S. Chicago Univ.
Repts CN-2689, March 3, 1945; CN-3053, June 28,
1945.
2336. Friederich E., Sittig L. — Z. anorg. und allg.
Chem., 1925, 145, S. 127.
2337. Friedman L. — J. Chem. Phys., 1955, 23, p. 477.
2338. Fries R. J. — U. S. AEC Accesion N 13029
Rept N LA-3423. N. Мех., 1965. 22 p.
2339. Fries В., Claassen H. H. — J. Chem. Phys., 1967,
46, p. 4603.
2340. Frurip D. J., Blander M. — J. Chem. Phys., 1980,
73, p. 509.
2341. Fryxell R. E., Frythall С A., Perkins R. J. —
Corrosion, 1973, 29, p. 423.
2342. Fuchs R., Hagan С P. — J. Phys. Chem., 1973,
77, p. 1797.
2343. Fuchtbauer C, Bartels H. — Z. Phys., 1921, 4,
S. 337.
2344. Fuger J., Parker V. В., Hubbard W. N., Oetting
F. L. The chemical thermodynamics of actinide
elements and compounds. Part 8. The actinide
halides. Vienna: IAEA, 1982 (in press).
2345. Fuji K., Miyake C, Imoto S. — J. Nucl. Sci.
Technol., 1979, 16, p. 207.
2346. Fujimoto S., Yasuda N., Shimizu #., Tsuboi S.,
Kawabe K., Takagi Y., Midorikawa M. — J. Phys.
Soc. Jap., 1977, 42, p. 911.
2347. Funaki K., Asada K. — J. Electrochem. Soc. Jap.,
1950, 18, p. 250.
2348. Furry W. H. — Phys. Rev., 1932, 39, p. 1015.
2349. Furukawa G. T. — U. S. NBS, Preliminary Data.
Wash., 1967.
2350. Furukawa G. Т., Reilly M. L. — U. S. NBS,
Rept-7093, Wash., 1961.
2351. Furukawa G. Т., Saba W. G., FordJ. С — J. Res.
NBS, 1970, A74, p. 631.
2352. Gabelnick S. D., Reedy G. Т., Chasanov M. G. —
Chem. Phys. Lett., 1973, 19, p. 90.
2353. Gabelnick S. D., Reedy G. Т., Chasanov M. G. —
J. Chem. Phys., 1973, 58, p. 4468.
2354. Gabelnick S. D., Reedy G. Т., Chasanov M. G. —
J. Chem. Phys., 1973, 59, p. 6397.
2355. Gabelnick S. D., Reedy G. Т., Chasanov M. G. —
J. Chem. Phys., 1974, 60, p. 1167.
2356. Gado P. — Hiradastechn. ipari kut. intez. kozl.,
1968, 8, dd. 103.
2357. Gaehr P. F. — Phys. Rev., 1918, 12, p. 396.
2358. Gallagher P. K., King E. L. — J. Amer. Chem.
Soc, 1960, 82, p. 3510.
2359. Gallaher T. N., DeVore Т. С — High Temp. Sci.,
1979, 11, p. 123.
2360. Gant F. A. — Dissertat. Univ. Alabama, 1967.
2361. Gardner E. R., Markin T. L., Street R. S. —
J. Inorg. and Nucl. Chem., 1965, 27, p. 541.
2362. Gardner T. E., Taylor A. R. — U. S. Bur. Mines.
Rept Invest., 1964, N 6435.
2363. Gardner T. E., Taylor A. R. — U. S. Bur. Mines.
Rept Invest., 1967, N 7040.
2364. Gam P. D. — Anal. Chem., 1969, 41, p. 447.
2365. Garn P. D. — Anal. Calorim., 1974, 3, p. 787.
2366. Garn P. D., Diamondstone B. I., Menis O. — J.
Therm. Anal., 1974, 6, p. 623.
2367. Garner CD., Mather R., Dove M. F.A. — J. Chem.
Soc. Chem. Communs, 1973, p. 633.
2368. Garton G., Wanklyn В. М. — J. Inorg. and Nucl.
Chem., 1965, 27, p. 2466.
2369. Garton W. R. S., Reeves E. M. — Proc. Roy.
Soc. London, 1973, A 333, p. 17.
2370. Garton W. R. S., Reeves E. M., Tomkins F. S.
Ercoli B. — Proc. Roy. Soc. London, 1973, A333,
p. 1.
2371. Gasic M. C, Boskovic S. В., Mikijelj V. D., Ri-
stic H. H. — In: Thermodyn. Nuclear Mater.
Proc. Symp. Vienna. 1967. Vienna, 1968, p. 537.
Discus., p. 542.
2372. Gatehouse B. M., Lloyd D. J. — J. Chem. Soc.
Dalton Trans., 1973, p. 70.
2373. Gathers G. R., Shaner J. W., Hixson R. S.,
Young D. A. — High Temp.-High Pressur., 1979,
11, p. 653.
2374. Gatterer A., Junkes J., Sulpeter E., Rosen B. —¦
In: Molecular spectra of metallic oxides/Ed. Spe-
cola Vaticana. Vatican City: Vatican Press, 1957,
p. 80.
2375. Gatterer A., Krishnamurthy S. G. — Nature, 1952,
169, p. 543.
2376. Gaut J. — Trans. Faraday Soc, 1953, 49, p. 1122.
2377. Gaydon A. G. Dissociation energies and spectra
of diatomic molecules. 3 ed. L.: Chapman and
Hall, 1968.
2378. Gaydon A. G., Pearse R. W. B. — Nature, 1938,
142, p. 291.
2379. Gaydon A. <?., Pearse R. W. B. — Proc. Roy.
Soc. London, 1939, A 173, p. 28.
2380. Geach G. A., Harper M. E. — Metallurgia, 1953,
47, p. 269.
2381. Gearhart R. C, Beck J. D., Wood R. H. — Inorg.
Chem., 1975, 14, p. 2413.
2382. Geballe T. H., Matthias В. Т., Corenzwit E.,
Hull G. W. — Phys. Rev. Lett., 1962, 8, p. 313.
2383. Gebhardt A. — Berichte, 1905, 38, S. 184.
2384. Gebhardt E., Seghezzi H., Keil H. — Z. Metallk.,
1962, 53, S. 524.
2385. George A. M., Karkhanavala M. D. — J. Phys.
Chem. Solids, 1963, 24, p. 1207.
2385a. George P. M., Beauchamp J. L. — Chem. Phys.,
1979, 36, p. 345.
2386. Gerasimov Ya. I., Voronin G. F., Shiu N. T. —
J. Chem. Thermodyn., 1969, 1, p. 425.
560
Литература
2387. Gerdanian Р., Матисса F., Bode M.— С. г. Acad.
sci., 1963, 256, p. 2591.
2388. Gerding P., Leden I., Sunner S. — Acta chem.
scand., 1963, 17, p. 2190.
2389. Germain P., Perachon G., Thourey J. — J. Fluor.
Chem., 1978, 11, p. 555.
2390. Germain P., Perachon G., Thourey J. — J. Fluor.
Chem., 1979, 13, p. 141.
2391. GershikovA. G., Spiridonov V. P., ButayevB. S. —
Chem. Phys. Lett., 1978, 55, p. 599.
2392. Gershikov A. G., Spiridonov V. P., Prikhod'ko
A. Ya., Erokhin E. V. — High Temp. Sci., 1981,
14, p. 17.
2393. Gerstein B. C, Taylor W. A., Schickell W. D.,
Spedding F. H. — J. Chem. Phys., 1971, 54,
p. 4723.
2394. Ghose J., Greenwood N. N., Hallam G. C, Read
D. A. — J. Solid State Chem., 1976, 19, p. 365.
2395. Ghosh С — Z. Phys., 1932, 78, S. 521.
2396. Gibson E. D., Loomis B. A., Carlson O. N. —
Trans. Amer. Soc. Metals, 1958, 50, p. 348.
2397. Gidon E., Uzy E. — J. Appl. Phys., 1966, 37,
p. 4633.
2398. Giessen B. C, Rump I., Grant N. /. — Trans.
Met. Soc. AIME, 1962, 224, p. 60.
2399. Giessen В. С, Rump I., Grant N. /. — Trans.
Met. Soc. AIME, 1963, 227, p. 265.
2400. Gilchrist K. E., Preston S. D. — High Temp.-
High Pressur., 1979, 11, p. 643.
2401. Gilles P. W. — J. Chem. Phys., 1967, 46, p. 4987.
2402 Gilles P. W., Carlson K. D., Franzen H. F., Wahl-
beck P. G. — J. Chem. Phys., 1967, 46, p. 2461.
2403. Gilles P. W., Margrave J. L. — J. Phys. Chem.,
1956, 60, p. 1333.
2404 Gilles P. W., Rinehart G. H., Sheldon R. I. —
J. Chem. Phys., 1977, 66, p. 2229.
2405. Gilles P. W., Sheldon R. I. — Rev. int. hautes
temp, et refract., 1978, 15, p. 315.
2406. Gilles P. W., Wheatley Q. L. — J. Chem. Phys.,
1951, 19, p. 129.
2407. Ginnings D. C, Corruccini R. J. — J. Res. NBS,
1947, 39, p. 309.
2408. Ginnings D. C, Douglas Т. В., Ball A. F. — J.
Res. NBS, 1950, 45, p. 23.
2409. Ginnings D. C, Phtpps Т. Е. — J. Amer. Chem.
Soc, 1930, 52, p. 1340.
2410. Ginsberg H., Wefers K. — Z. Erz. und Metall.,
1967, 20, S. 156.
2411. Gioia L. G., Cingolani A. —Ann. chim. (Ital.),
1975, 65, p. 361.
2412 Girdhar H. L., Westrum E. F. — J. Chem. and
Eng. Data, 1968, 13, p. 531.
2413. Gire G. — Ann. chim. (Fr.), 1925, 4, p. 183.
2414. Glas H. J., Weber H. G. — Z. Phys., 1978, A284,
S. 253.
2415. Glassner A. Thermochemical properties of the
oxides, fluorides and chlorides to 2500 K. Argonne
Nat. Lab., ANL-5750, 1957, p. 70.
2416. Glauber R., Schomaker V. — Phys. Rev., 1953,
89, p. 667.
2417. Gleditsh E. — Naturwissenschaften, 1940, 28, S. 127
2418. Gleiser M., Chipman J. — J. Phys. Chem., 1962,
66,-p. 1539.
2419. Glemser O., Haeseler R. — Z. anorg. und allg.
Chem., 1962, 316, S. 168.
2420 Glushkova V. В., Kaehler E. K. — Mater. Res.
Bull., 1967, 2, p. 503.
2421 Gmelins Handbuch der anorganischen Chemie.
В.: Verl. Chemie, I960, N 20.
2422. Gmelins Handbuch der anorganischen Chemie.
B: Verl. Chemie, 1960, N 21.
2423. Gmelins Handbuch der anorganischen Chemie.
В.: Verl. Chemie, 1951, N 41.
2424. Gmelins Handbuch der anorganischen Chemie
В.: Verl. Chemie, 1967, N 4.
2425. Gmelins Handbuch der anorganischen Chemie.
В.: Verl. Chemie, 1970, N 49.
2426. Gmelins Handbuch der anorganischen Chemie.
В.: Verl. Chemie, 1962, N 52.
2427. Goates J. R., Ott J. В., Hsu С. С. — Trans.
Faraday Soc, 1970, 66, p. 25.
2428. Goates J. R., Ott J. В., Delawarde E. — Trans.
Faraday Soc, 1971, 67, p. 1612.
2429. Gode H. — Latv. PSR Zinatnu Akad. vsstis, 1949.
N 3, p. 91.
2430. Gohel V. B. — Indian J. Pure and Appl. Phys.
1974, 12, p. 367. '
2431. Gokcen N. A. — Trans. Met. Soc. AIME, 1953
197, p. 1019.
2432. Goldberg A., Massalski Т. В. — In: Plutonium:
Proc. 4th Intern. Conf. Plutonium and Other
Actinides, Santa Fe, N. M., 1970. N. Y.: Met.
Soc. and Amer. Inst. Min. Met., Petrol. Eng
Inc., 1970, vol. 2, p. 875.
2433. Golden G. H., Tokar J. V., Miller D. — TJ.S.
Argonne Nat. Lab. Rept ANL-7323. Lemont
A11.), 1967.
2434. Golden G. #., Tokar J. V., Miller D. — Reactor
and Fuel-Process. Technol., 1967/68, 11, p. 27
(перевод экспресс-информ.: Теплофизика. М.:
ВИНИТИ, 1969, № 4, с. 1).
2435. Goldsmith A., Watermann Т. Е., HirschhornH. /.—
In: Handbook of Thermophys. Properties of Solid
Materials. Oxford etc.: Pergamon Press, 1961,
vol. 1, Elements (Melting Temp, above 1000 °F)
p. 235.
2436. Goldstein H. W., Neilson E. F., Walsh P. N.t
White D. — J. Phys. Chem., 1959, 63, p. 1445.
2437 Goldstein H. W., Walsh P. N., White D. — J.
Phys. Chem., 1961, 65, p. 1400.
2438. Goldwater D. L., Danforth W. E. — J. Franklin
Inst., 1960, 270, p. 317.
2439. Gole J. L. — Evidence for on Inflected D2 2+
State in LaO. Частное сообщение.
2440. Gole J. L., Chalek С L. — J. Chem. Phys., 1976
65, p. 4384.
2441. Goller S., Romo P., Remeika J. P. — Z. Kristal-
logr., 1967, 124, S. 136.
2442. Goodwin H. M., Kalmus J. H. — Phys. Rev
1909, 28, p. 1.
2443. Gordon J. E., Hall R. O. A., Lee J. A., Martl-
mer M. J. — Proc. Roy. Soc. London, 1976, A351,
p. 179.
2444. Gordon J. E., Montgomery H., Noer R. /., Pic-
kett G. R., Tabon R. — Phys. Rev., 1966, 152,
p. 432.
2445. Gordon J. S. — Amer. Rocket Soc. J., 1959, 29
p. 455.
2446. Gordon J. S., Robinson R. — In: Kinetics
equilibria and performance of high-temperature systems:
Proc. of the 2nd Conf., Apr. 1962 / Ed. G. S. Bahn.
N. Y.; L., 1963, p. 39.
2447. Gordon S., Campbell C. — Anal. Chem., 1955
27, p. 1102.
2448. Gotoo K., Naito K. — J. Phys. and Chem. Solids
1965, 26, p. 1673.
2449. Goubeau J., Kolb H., Krall H. G. — Z. anorg.
und allg. Chem., 1938, 236, S. 45.
36 Заказ № 1590
561
Литератур*
2450. Grabner L., Hughes V. — Phys. Rev., 1950, 79, 2490.
p. 819.
2451. Graham O. — Ann. chim. phys., 1845, 13, p. 188. 2491.
2452. Graham T. — Philos. Mag., 1843, 22, p. 329.
2453. Graham T. — Philos. Mag., 1844, 24, p. 401. 2492.
2454. Grahmann W. — Z. anorg. und allg. Chem., 1913,
81, S. 257. 2493.
2455. Grain C. F., Garvie R. С — U. S. Bur. Mines.
Rept Invest., 1965, N 6619. 2494.
2456. Grassman P. — Z. Phys., 1932, 77, S. 616.
2457. Gray A. P. — In: Therm. Anal.: Proc. 4th Intern. 2495.
Conf. Therm. Anal., Budapest, 1974. Bp., 1975,
vol. 3, p. 991. 2496.
2458. Greaves C, Fender В. E. F. — Acta crystallogr.,
1972, B28, p. 3609.
2459. Green D. W. — J. Mol. Spectrosc, 1971, 38, p. 155. 2497.
2460. Green D. W. — J. Mol. Spectrosc, 1971, 40, p. 501.
2461. Green D. W. — Canad. J. Phys., 1971, 49, p. 2552. 2498.
2462. Green D. W. — J. Chem. Phys., 1971, 75, p. 3103.
2463. Green D. W. Argonne Nat. Lab. Rept ANL-CEN- 2499.
RSD-80-1. 1980, March.
2464. Green D. W. — Int. J. Thermophys., 1980, 1, p. 61. 2500.
2465. Green D. W. — J. Nucl. Mater., 1980, 88, p. 51.
2466. Green D. W., Gabelnick S. D., Reedy G. T. — J. 2501.
Chem. Phys., 1976, 64, p. 1697.
2467. Green D. W., Gruen D. M., Korfmaeher W. — 2502.
J. Chem. Phys., 1973, 58, p. 404.
2468. Green D. W., Lew H. — Canad. J. Phys., 1960, 2503.
38, p. 482.
2469. Green D. W., Reedy G. T. — J. Chem. Phys., 1978, 2504.
69, p. 544.
2470. Green D. W., Reedy G. Т., Gabelnick S. D. — J. 2505.
Nucl. Mater., 1977, 66, p. 200.
2471. Green D. W., Reedy G. Т., Gabelnick S. D. — J. 2506.
Chem. Phys., 1980, 73, p. 4207.
2472. Green D. W., Reedy G. Т., Kay J. G. — J. Mol. 2507.
Spectrosc, 1979, 78, p. 257.
2473. Green J. W., Poland D. E., Margrave J. L. — 2508.
J. Chem. Phys., 1960, 33, p. 35.
2474. Green P.D., Gross P., Haymari С — Trans. Faraday 2509.
Soc, 1968, 64, p. 633.
2475. Greenaway H. Т., Johnstone S. Т. M., McQuil- 2510.
Ian M. K. — J. Inst. Metals, 1951, 80, p. 109.
2476. Greenberg J., Hallgren L. J. — J. Chem. Phys., 2511.
1960, 33, p. 900.
2477. Greene P. D., Gross P., Heyman С — Trans. 2512.
Faraday Soc, 1968, 64, p. 633.
2478. Greenwood H. C. — Proc. Roy. Soc. London, 1909, 2513.
A82; p. 396.
2479. Greenwood H. С — Proc. Roy. Soc. London, 2514.
1910, A83, p. 483.
2480. Greenwood N. N., Prince D. — J. Ghem. Soc. A,
1969, p. 2876.
2481. Gregory N. W., Mohr R. H. — J. Amer. Chem. 2515.
Soc, 1955, 77, p. 2142.
2482. Greiner В., Jellinek K. — Z. phys. Chem. Leipzig, 2516.
¦'¦ 1933, A165, S. 97.
2483. Greiner E. S., Ellis W. C. — Trans. AIME, 1949, 2517.
180, p. 657.
2484. Greiner <?., Westrum E. F. — J. Amer. Chem. 2518.
Soc, 1956, 78, p. 6226.
2485. Greutz E. С — Цит. по: [492, с. 129]. 2519.
2486. Griffel M. — J. Chem. Phys., 1953, 21, p. 1908.
2487. Griffel M., Skochdopole R. E. — J. Amer. Chem. 2520.
Soc, 1953, 75, p. 5250.
2488. Griffis R. C. — J. Electrochem. Soc, 1958, 105, 2521.
p. 398.
2489. Griffis R. С — J. Electrochem. Soc, 1959, 106, 2522.
p. 418.
Griffith G. H., Eastwood H. K.—J. Appl. Phys.,.
1974, 45, p. 2201.
Griffiths E. — Proo. Roy. Soc. London, 1914, A89,.
p. 561.
Grimley R. Т., Burns R. P., Inghram M. G. —
J. Chem. Phys., 1961, 34, p. 664.
Grimley R. Т., Forsman J. A., Grindstaff O. G. —
J. Phys. Chem., 1978, 82, p. 632.
Grimley R. Г., Margrave J. L. — J. Phys. Chem.,.
1960, 64, p. 1763.
Grinier G., White D. — J. Phys. Chem., 1957, 61,
p. 1681.
Grjotheim K., Motzfeldt K., Rao D. В.— In: Proc.
Symp. 100th AIME. Annual Meeting, New York Г
Ed. T. G. Edgeworth. N. Y., 1974, March 1—4.
Gromb S. — J. chim. phys. et phys.-chim. biol.,
1960, 57, p. 101.
Gr0nvold F. — J. Inorg. Nucl. Chem., 1955, 1,.
p. 357.
Gr0nvold F., Kveseth N. /., Sveen A., Tichy J. —
J. Chem. Thermodyn., 1970, 2, p. 665.
Groschuff E. — Z. anorg. und allg. Chem., 1908r
58, S. 113.
Gross B. — Berlin Inst. Kerntechn. Techn. Univ.
Rept TUBIK-4. В., Okt. 1966.
Gross P., Heyman C, Levi D. L. — Metallurgy
Soc. Conf., 1961, 8, p. 903.
Gross P., Wilson G. L. — J. Chem. Soc. A, 1970,-
p. 1913.
Grossman L. N., Kaznoff A.I. — J. Amer. Ceram.
Soc, 1968, 51, p. 59.
Grossmann G., Proskurenko O. W., Aria S. M. —¦
Z. anorg. und allg. Chem., 1960, 305, S. 121.
Groves W. O., Hoch M., Johnston H. L. — J. Phys.
Chem., 1955, 59, p. 127.
Grow D. Т., Pitzer R. M. — J. Chem. Phys., 1977,
67, p. 4019.
Grube G., Flad M. — Z. Elektrochem., 1939. 45,.
S. 835.
Grube G., Flad M. — Z. Elektrochem., 1942, 48,
S. 377.
Grube G., Knabe R. — Z. Elektrochem., 1936,,
42, S. 793.
Grube G., Kubaschewski O., Zwiauer K. — Z.
Elektrochem., 1939, 45, p. 885.
Gruber J. В., Hecht H. G. — J. Chem. Phys.,
1973, 59, p. 1713.
Gschneidner K. A. J. — 8th Annual Conf. on Va-
kuum Metallurgy. N. Y., 1965.
Gschneidner K. A. J., Tsang T. W. E., Queen J.,
Ledvold S., Schmidt F. A. — In: Rare Earths
and Actinides, 1977: Int. Conf. Durham, 1977.
Bristol; London, 1978, p. 23.
Guerchais J. E. — C. r. Acad. sci., 1964, 259,
p. 2850.
Guerreschi L., Romita JR. — Ric. sci. Rend. Ser.
A, 1963, 3, p. 513.
Gugenheimer К. М. — Proc. Phys. Soc. London^
1946, 58, p. 456.
Guha J. P. — Trans. Indian Ceram. Soc, 1969,,
28, p. 97.
Gulbransen E. A., Andrew K. F. — J.
Electrochem. Soc, 1952, 99, p. 402.
Gulbransen E. A., Andrew K. F. — Trans. Met.
Soc. AIME, 1961, 221, p. 1247.
Gulbransen E. A., Andrew K. F., Brassart F. A. —
J. Electrochem. Soc, 1963, 110, p. 242.
Gundersen G., Hedberg K. — J. Chem. Phys.,.
1969, 51, p. 2500.
562
Литература
2523. Gunn S. R. — Rev. Sci. Instrum., 1958, 29,
p. 377.
2524. Gunn S. R. — J. Phys. Chem., 1965, 69, p. 2902.
2525. Gunn S. R. — J. Phys. Chem., 1967, 71, p. 1386.
2526. Gunn S. R. — J. Chem. Thermodyn., 1970, 2,
p. 535.
2527. Gunn S. R., Green L. G. — J. Amer. Chem. Soc,
1958, 30, p. 4782.
2528. Gunn S. R., Watson J. A., MachleH., Gundry И. A.,
Head A. J., M&nsson M,, Sunner S. — J. Chem.
Thermodyn., 1970, 2, p. 549.
2529. Gunther P. — Ann. Phys., 1.920, 63, S. 21.
2530. Guntz A. — Ann. chim. phys. (Fr.), 1884, 3, p. 5.
2531. Guntz A. — С. г. Acad. sci., 1896, 122, p. 244.
2532. Guntz А. —С. г. Acad. sci., 1896, 123, p. 694.
2533. Gupta S. K., Gingerich K. A. — 1. Chem. Phys.,
1979, 70, p. 5350,
2534. Gurvich L. V., Dorofeeva O. V., Yungman V. S. —
In: Conf. High Temp. Sci. Open-Cycle, Coal-
Fired MHD Syst., Argonne Nat. Lab. Rept ANL-
77-21. Argonne A11.), 1977, p. 241. "
2535. Gurvich L. V., Yungman V. S. — In:
Thermodynamics: Proc. Symp. Vienna, 1965. Vienna: IAEA,
1966, vol. 2, p. 613.
2536. Gurvich L. V., Yungman V. S., Dorofeeva O. V.,
Gorokhoy L. N., Munvez S. S. — In: Thermo-
dynamic properties of gaseous W—F and U—F
systems: Proc. 7th Symp. Thermophys. Prop.,
Gaithersburg (Md), 1977, N. Y., 1977, p. 615.
2537. Haar L., Friedman A. S., Beckett Ch. W., Ha-
ber F., Zisch W. — Ideal gas thermodynamic
functions and isotope exchange functions for the
diatomic hydrides, deuterides and tritides. Wash.:
NBS, 1961.
2538. Haber F., Zisch W. — Z. Phys., 1922, 9, S. 302.
2539. Habermann С E., Daane A. H. — J. Less-Common
Metals, 1963, 5, p. 134.
2540. Habermann С E., Daane A. H. — J. Chem. Phys.,
1964, 41, p. 2818.
2541. Hackspill M. L. — С. г. Acad. sci., 1912, 154,
p. 877.
2542. Hackspill M. L. — Ann. chim. phys., 1913, 28,
p. 613.
2543. Hackspill L., Barocco A. — Bull. Soc. chim.
France, 1939, 6, p. 91.
2544. Hackspill L., Thomas G. — C. r. Acad. sci., 1948,
227, p. 797,
2545. Haendler H. M., Sennett P. S., Wheeler С. М. —
J. Electrochem. Soc, 1959, 106, p. 264.
2546. Hagemark K., Hengstenberg D. — J. Phys. Chem.,
1967, 71, p. 3337.
2547. Hagen H., Sieverts A. — Z. anorg. und allg.
Chem., 1929, 185, S. 254.
2548. Haigh F. L. — J. Amer. Chem. Soc, 1912, 34,
p. 1137.
2549. Hajime Kato, Chifuru Noda. — J. Chem. Phys.,
1980, 73, p. 4940.
2550. Hake R. R., Cape J. A. — Phys. Rev., 1964,
A135, p. 1151.
2551. Hall F. P., Insley H. — J. Amer. Ceram. Soc,
1938, 21, p. 113.
2552. Hall R. O. A., Mortimer M. J. — J. Low Temp.
Phys., 1977, 27, p. 313.
2553. Halla F., Tompa II. — Z. anorg. und allg. Chem.,
1934 219 S. 321.
2554. Halle J.' C., Stem K. II. — J. Phys. Chem.,
1980, 84, p. 1699.
2555. Halstead W. D. — Trans. Faraday Soc, 1970, 66,
p. 1966.
2556. Ham D. O. — J. Chem. Phys., 1974, 60, p. 1802.
2557. Hamano K. — Proc. Phys. Soc. Jap., 1964, 19
p. 208. '
2558. Hamano K. — Bussei Kenkyu (Study of Property
of Mater.) (Jap.), 1976, 25, p. B3-B4.
2559. Hamano K., Ema K. — J. Phys. Soc. Jap., 1978
45, p. 923.
2560. Hambourger P. D., Marcus J. A. — Phvs. Rev
1967, 157, p. 438.
2561. Hammer P. D., Davis S. P. — Astrophys. J.
1980, 237, p. L 51.
2562. Hampson P. J., Gilles P. W. — J. Chem. Phvs
1971, 55, p. 3712. ''
2563. Hansen J. — Berichte, 1909, 42, S. 210.
2564. Hansen M., Kamen E. L., Kessler D. J., Pher-
son D. J. —¦ J. Metals, 1951, 3, p. 881.
2565. Harding J. H., Masri P., Stoneham A. M.~-
J. Nucl. Mater., 1980, 92, p. 73.
2566. Hare A. — Philos. Mag., 1924, 48, p. 412.
2567. Hargittai I., Hargittai M., Spiridonov V. P
Erokhin E. V. — J. Mol. Struct., 1971, 8, p. 3l!
2568. Hargittai M., Tremmel J., Hargittai I. — J
Chem. Soc. Dalton Trans., 1980, N 1, p. 87
2569. Harmelin M., Lehr P.'~ С. г. Acad.';sci., 1975
С 280, p. 1011.
2570. Harpur W. W., Ubbelohde A. R. — Nature 1952
170, p. 975. ' '
2571. Harpur W. W.t Ubbelohde A. R. — Proc. Roy
Soc. London, 1955, A232, p. 310.
2572. Harrington J. A., Seel R. M., Hebert G. R., Ni-
cholls R. W. — Identification Atlas of Molecular
Spectra, York University 1967/72, 1970.
2573. Harrison J. — С. г. Acad. sci., 1955, 241, p. :298.
2574. Harrison J. P., Lombardo Y., Peressini P p
J. Phys. Chem. Solids, 1968, 29, p. 557.
2575. Hartmann H., Schneider RA— Z. anorg.'und allp
Chem., 1929, 180, S. 275! g>
2576. Hartmanshenn O., Barral J.-C. — C. r.rAcad sci
1971, C272, p. 2139. ' ' M
2577. Hartshorn S. R., Moodie R. В., Stead К — J
Chem. Soc. Perkin Trans. 11, 1972, N 2, p. 127.
2578. Harvey A., Jenkins F. A.~ Phys. Rev., 1932
35, p. 789.
2579. Harvey K. C, Stoicheff B. P.1—[Phys.'Rev. Lett.,
1977, 38, p. 537.
2580. Hasapis A. A., Melveger A.*J., Parish M. B.
Reif L., Rosen C. L. — U. S. AEC. Rept. WADD-
TR-60-463, pt 2, 1960.
2581. Hastie J. W., Hauge R. II., Margrave J. L —
High Temp. Sci., 1971, 3, p. 56.
2582. Hastie J. W., Hauge R. H., Margrave J. L. —
J. Less-Common Metals, 1975, 39, p 309
2583. Hastie J. W., SwinglerD. LA—IHigh Temp. Sci
1969, 1, p. 46. '
2584. Hatta I. — J. Phys. Soc. Jap., 1970, 28, p. 1266.
2585. Hatta I., Ikushima A. — Phys.'Lett., 1971, A37
p. 207. '
2586. Hatta I., Ikushima A. — J. Phys. Chem. Solids
1973, 34, p. 57.
2587. Hatton W. E., Hildenbrand G. C, Sinke G. C,
Stall D. R. — Unpublished data of the Dow
Chem. Co., Midland (Mich.), [-1959. Пит по-
[2835].
2588. Hauge R. H., Hastie J. W., Margrave J. L. —
J. Less-Common Metals, 1971, 23, p. 359
2589. Hausner H. H. — J. Nucl. Mater., 1965, 15, p. 179.
2590. Hausner H. II., Mills R. G. — [JNTucleonics, 1957,
15, p. 94.
2591. Hautecler S., Rosen B. — Bull. cl. sci. Acad.
roy. Belgique, 1959, 45, p. 790.
563
Литература
2592. Hawkings В., Onillon M., Orr R. L. — J. Chem. 2628.
and Eng. Data, 1963, 8, p. 628.
2593. Hawkins D., Orr R. L. — J. Chem. and Eng. 2629.
Data, 1964, 9, p. 505.
2594. Haworts С W., Hume-Rothery W. — J. Inst.
Metals, 1959, 87, p. 265. 2630.
2595. Hay P. J., Wadt W. R., Raffenetti R. C,
Phillips D. H. — J. Chem. Phys., 1979, 71, p. 1767. 2631.
2596. Hayes E. E.^Kaufmann A. R. — In: Zirconium
and Zirconium Alloys. A Symp. on Zirconium 2632.
and Zirconium Alloys, Los Angeles, March 23—27,
1953, Amer. Soc. Metals. N. Y., 1953, p. 241. 2633.
2597. Hayman C. — In: Thermodyn. Symp. in
Heidelberg: Sess. 1/ Ed. K. L. Schafer. 1967, Pap. N 7. 2634.
2598. HazelwoodiF. J., Rhodes E., Ubbelohde A. R. — 2635.
Trans. Faraday Soc, 1963, 59, p. 2612.
2599. Hebert A. J., Rreivogel F. W., Street K. — J. 2636.
Chem. Phys., 1964, 41, p. 2368.
2600. Hebert A. J., Story T. L., Breivogel F. W., Hoi- 2637.
lowell С D., Street K. — UCRL — 11828, p. 193
(Цит. по: NSA, 1965, N 19, p. 11). 2638.
2601. Hegedus%A. J., Millner Т., Neugebauer J., Sas-
vari K. — Z. anorg. und allg. Chem., 1955, 281, 2639.
S. 64.
2602. Heimel S. — *U. S. National Advisary and Space 2640.
Administration. Rept NASA TND-4165, 1967.
2603. Hein R. A., Flagella P. N. — U. S. AEC General 2641.
Electric Co. Rept GEMP-578. Cincinnati (Ohio),
1968. 2642.
2604. Hein R. A., Flagella P. N., Conway J. B. — J.
Amer. Ceram. Soc, 1968, 51, p. 291. 2643.
2605. Hein R. A., Gibson J. M., Pablo M. R., Blang-
her R. D. — Phys. Rev., 1963, 129, p. 136.
2606. Hein R. A., Sjodahl L., Szwarc R. — J. Nucl. 2644.
Mater., 1968, 25, p. 99.
2607. Heiniger F., Mailer J. — Phys. Rev., 1964, A134, 2645.
p. 1407.Ц
2608. Helms A., Klemm W. — Z. anorg. und allg. 2646.
Chem., 1939, 241, S. 97.
2609. Henning gF. — Naturwissenschaften, 1925, 13, 2647.
S. 661.
2610. Henning F. — Temperaturmessung, Leipzig, 1951. 2648.
2611. Henning F., Heuse W. — Z. Phys., 1923, 16,
S. 63. 2649.
2612. Henry W. E. — Phys. Rev., 1958, 109, p. 1976.
2613. Henshaw D. <?., Brockhouse B. N. — Bull. Amer. 2650.
Phys. Soc, 1957, 2, p. 9.
2614. Hepler L. G., Jolly W. L., Latimer W. M. — J.
Amer. Chem. Soc, 1953, 75, p. 2809.
2615. HerakJR., Jovanovic B. — Inorg. and Nucl. Chem. 2651.
Lett., 1969, 5, p. 693.
2616. Herm R. R., Herschbach D. R. — J. Chem. Phys., 2652.
1970, 52, p. = 5783.
2617. Herold A. — С. г. Acad. sci., 1947, 225, p. 249.
2618. Herold А. —С. г. Acad. sci., 1947, 224, p. 1826.
2619. Herold A.—C. r. Acad. sci., 1949, 228, p. 686. 2653.
2620. Herold A. — Ann. chim. (Fr.), 1951, 6, p. 537.
2621. Herzberg G. — Molecular spectra and molecular 2654.
structure. I. Spectra of diatomic molecules. 2 ed.
Toronto etc., 1950. 2655.
2622. Hess G. I. — Ann. Phys., 1840, 50, S. 3884.
2623. Hess H. — Ann. Phys., 1841, 52, S. 97. 2656.
2624. Hessel M. M. — Phys. Rev. Lett., 1971, 26,
p. 215. 2657.
2625. Hessel M. M., Kuseh P. — J. Mol. Spectrosc,
1968, 25, p. 205. 2658.
2626. Hessel M. M., Kusch P. — J. Mol. Spectrosc,
1969, 32, p. 181. 2659.
2627. Hessel M. M., Vidal С R. — J. Chem. Phys.,
, 1979, 70, p. 4439. 2660.
Hester R. E., Plane R. A. — Inorg. Chem., 1964,
3, p. 769.
Heumann F. K., Salmon O. N. — U. S. AEC.
Knolls Atomic Power Lab. Rept KAPL-1667.
Schenectady (N. Y.), 1956, Dec. 1.
Heus R. J., Egan J. J. — Z. phys. Chem. (BRD),
1966, 49, S. 38.
Hevesy G. von. — Z. anorg. und allg. Chem., 1910,
67, S. 242.
Hevesy G. von. — Z. phys. Chem. Leipzig, 1910,
73, S. 667.
Hewett W. D., Newton J. H., Weltner W. — J.
Phys. Chem., 1975, 79, p. 2640.
Hexter R. M. — Spectrochim. acta, 1958,10, p. 291.
Heycock С. Т., Lamplough F. E. — Proc Chem.
Soc. London, 1912, 28, p. 3.
Hieber W., Feder E. E. — Z. Elektrochem., 1938,
44, S. 881.
Hieber W., Miihlbauer F. — Z. anorg. und allg.
Chem., 1930, 186, S. 97.
Hietala T. — Ann. Acad. Sci. Fennicae, Ser. AVI,
1960, N 63, p. 57.
Higashigaki Y., Chihara H. — Bull. Chem. Soc.
Jap., 1976, 49, p. 2089.
Higgins T. L., Westrum E. F. — J. Phys. Chem.,
1961, 65, p. 830.
Hildenbrand D. L. — J. Chem. Phys., 1972, 57,
p. 4556.
Hildenbrand D. L. — Chem. Phys. Lett., 1976,
44, p. 281.
Hildenbrand D. L. — U. S. NTIS AD-Rept 1976,
AD—AO25661. From Gov. Rept Announce Index
(U. S.), 1976, 76, p. 81.
Hildenbrand D. L. — J. Chem. Phys., 1977, 66,
p. 4788.
Hildenbrand D. L. — J. Chem. Phys., 1982 (in
press).
Hildenbrand D. L., Hall W. E., Potter N. D. —
J. Chem. Phys., 1963, 39, p. 296.
Hildenbrand D. L., Hall W. F., Ju F., Potter
N. D. — J. Chem. Phys., 1964, 40, p. 2882.
Hildenbrand D. L., Murad E. — J. Chem. Phys.,
1970, 53, p. 3403.
Hildenbrand D. L., Murad E. — J. Chem. Phys.,
1974, 61, p. 1232.
Hildenbrand D. L., Theard L. P., Hall W. F.,
Ju F., LaViola F. S., Potter N. D. — Aeronutronic
Publication NU-2055, March 15, 1963. Цит. по:
[2834].
Hill J. D. — J. Less-Common Metals, 1962, 4,
p. 376.
Hilsenrath J., Messina С G., Evans W. H.
Tables of ideal gas thermodynamic functions for
73 atoms and their first and second ions to 10000 K.
AD-606163. Wash.: NBS, 1964.
Hilti E., Laves F. — Naturwissenschaften, 1968,
55, S. 130.
Hilti E., Laves F. — Naturwissenschaften, 1968,
55, S. 131.
Hinz I., Dietzel A. — Ber. Dtsch. keram. Ges.,
1962, 39, S. 489.
Hiraoka Т., Sano N., Matsuhita Y. — Tetsu to
Hagane, 1969, 55, p. 470.
Hiraoka Т., Sano N., Matsuhita Y. — Trans.
Iron Steel Inst. Jap., 1971, 11, p. 102.
Hirshfeld А. Т., Leupold H. A., Boorse H. A. —
Phys. Rev., 1962, 127, p. 1501.
Ho J. C, Phillips N. E. — Inorg. Mater. Res.
Division Ann. Rept UCRL-17330, 1966, p. 25.
Ho W.-H. — Proc. Nat. Sci. Counc Pt 1 (Taiwan),
564
Литература
1977, 10, p. 175. (Chem. Abstrs, 1978, 88, N 6,
42307 с).
2661. Hobbs W. E. — J. Chem. Phys., 1958, 28, p. 1220.
2662. Hoch M. — J. Phys. Chem. Solids, 1963, 24, p. 157.
2663. Hoch M. — High Temp.-High Pressur., 1969, 1,
p. 531.
2664. Hoch M., Iyer A. S., Nelken J. — J. Phys. Chem.
Solids, 1962, 23, p. 1463.
2665. Hoch M., Johnston H. L. — J. Amer. Chem. Soc,
1954, 76, p. 4833.
2666. Hoch M., Johnston H. L. — J. Phys. Chem.,
1961, 65, p. 1184.
2667. Hoch M., Johnston H. L. — J. Phys. Chem., 1961,
65, p. 855.
2668. Hoch M., Nakata M., Johnston H. L. — J. Amer.
Chem. Soc, 1954, 76, p. 2651.
2669. Hocking W. H., Gerry M. C. L., Merer A. J. —
Canad. J. Phys., 1979, 57, p. 54.
2670. Hocking W. H., Merer A. J., Milton D. J.,
Jones W- E., Krishnamurty G. — Canad. J. Phys.,
1980, 58, p. 516.
2671. Hodge N. — Adv. Fluorine Chem., 1961, 2, p. 138.
2672. Hoekstra H. R., Siegel S. — J. Inorg. and Nucl.
Chem., 1961, 18, p. 154.
2673. Hoekstra H. R., Siegel S., FuchsL. H., KatzJ. J. —
J. Phys. Chem., 1955, 59, p. 136.
2674. Hoekstra H., Siegel S., Gebert E. — J. Inorg.
and Nucl. Chem., 1966, 28, p. 2065.
2675. Hoenig С L. — J. Amer. Ceram. Soc, 1971, 54,
p. 391.
2676. Hoermann F. — Z. anorg. und allg. Chem., 1929,
177, S. 145.
2677. Hoerster H., Kaner E., Kettel F. — Colloq. intern.
Centre nat. Rech. sci., 1972, N 205, p. 39.
2678. Hoeven B. I. C, van der Keesom P. H. — Phys.
Rev., 1964, A134, p. 1320.
2679. Hogan V. D., Gordon S. — J. Phys. Chem., 1960,
64, p. 172.
2680. Hohne E., Kutschabsky L. — Z. anorg. und allg.
Chem., 1966, 344, S. 279.
2681. Holah G. D., Happ H. — J. Phys.: Solid State
Phys., 1970, C3, p. 1807.
2682. Holland L. R. — J. Appl. Phys., 1963, 34, p. 2350.
2683. Hollander J. M., Perlman J., Seaborg G. T. —
Revs Mod. Phys., 1953, 25, p. 469.
2684. Holley С E., Mulford R. N. R., Huber E. J.,
Head E. L., Ellinger F. H., Bjorklund C. W. ¦—
In: Proc. 2nd Intern. Conf. Peaceful. Uses Atom.
Energy, Geneva, 1958. Geneva, 1958, vol. 6,
p. 215.
2685. Hollowell С D., Hebert A. J., Street K. S. —
J. Chem. Phys., 1964, 41, p. 3540.
2686. Holluta J., Werner H. — Z. phys. Chem. Leipzig,
1927, 129, S. 262.
2687. Holm B. J. — In: Proc. 1st Europ. Symp. Therm.
Anal., Salford, 1976. London etc., 1976, p. 111.
2688. Holm B. J., Gronvold F. — Acta chem. scand.,
1973, 27, p. 2043.
2689. Holm B. J., Holm J. L. — Thermochim. acta,
1973, 5, с 273.
2690. Holm J. L. — Acta chem. scand., 1965, 19, p. 261.
2691. Holm J. L. — Acta chem. scand., 1965, 19, p. 638.
2692. Holm J. L. — Acta chem. scand., 1966, 20, p. 1167.
2693. HolmJ. L. — Acta rhem. scand., 1968, 22, p. 1004-
2694. Holm J. L. — High Temp. Sci., 1974, 6, p. 16'
2695. Holtzberg F., Reisman A. — J. Phys. Chem., 1961'
65, p. 1192.
2696. Holtzberg F., Reisman A., Berry M-, Berken-
blit M. — J. Amer. Chem. Soc, 1957, 79, p. 2039.
2697. Honerjager R., Tischer R. — Z. Naturforsch.,
1974, A29, S. 819.
2698. Hong K. C, Kleppa O. — J. Phys. Chem., 1978,
82, p. 176.
2699. HonickeD. — Electrochim. acta, 1970, 15, p. 757.
2700. Honig A., MandelM., StitchM. L., Townes С. Н. —
Phys. Rev., 1954, 96, p. 629.
2701. Honig A., Stitch M- L., Mandel M. — Phys.
Rev., 1953, 92, p. 901.
2702. Honig G., Czajkowski M., Stock M., Demtro-
der W. — J. Chem. Phys., 1979, 71, p. 2138.
2703. Honig J. M., Reed Т. В. — Phys. Rev,, 1968,
174, p. 1020.
2704. Hoodless I. M., Morrison I. N. — J. Phys. Chem.,
1962, 66, p. 577.
2705. Horbe V. R., Knacke O., Prescher К. Е. — Metall
und Erz, 1961, 14, S. 232.
2706. Hori T. — Z. Phys., 1930, 62, S. 352.
2707. Hori T. — Z. Phys., 1931, 71, S. 478.
2708. Hori T. — Mem. Ryojun Coll. Eng., 1933, 6, p. 1.
2709. Hori T. — Mem. Ryojun Coll. Eng., 1933, 6,
p. 115.
2710. Hori T. — Jap. J. Phys., 1933, 8, p. 151.
2711. Horiba S., Baba H. — Bull. Chem. Soc. Jap.,
1928, 3, p. 11.
2712. Horiuchi H., Morimoto N., Takanami M. — J.
Solid State Chem., 1976, 17, p. 407.
2713. Horowitz M., Daunt J. G. — Phys. Rev., 1953,
91, p. 379.
2714. Horowitz M., Daunt J. G. — Phys. Rev., 1953,
91, p. 1099.
2715. Horowitz N. H., Bohm H. V. — Phys. Rev. Lett.,
1962, 9, p. 313.
2716. Hoshino S. — J. Phys. Soc. Jap., 1964, 19, p. 140.
2717. Hoshino S., Shibuja I. — J. Phys. Soc. Jap.,
1961, 16, p. 1254.
2718. Jougne J. T. — Canad. J. Phys., 1962, 40, p. 598.
2719. Hovi V., Arell A., Varteva M. — Ann. Acad. sci.
Fennicae, Ser. AVI, N 39.
2720. Howard J. C, Conway J. G. — J. Chem. Phys.,
1965, 43, p. 3055.
2721. Howard W. F., Jr., Andrews L. — Inorg. Chem.,
1975, 14, p. 409.
2722. Howarth J. Т., Turner W. E. S. — J. Soc. Glass
Tech., 1930, 14, p. 394.
2723. Howarth J. Т., Turner W. E. S. — J. Soc. Glass
Tech., 1931, 15, p. 360.
2724. Hewlett D. L., Lester J. E., Samorjai G. A. —
J. Phys. Chem., 1971, 75, p. 4049.
2725. Hsieh Y. K., Yang S. C, Tam A. C, Stawlley
W. С — J. Chem. Phys., 1978, 68, p. 1448.
2726. Hsieh Y. K., Yang S. С, Тат А. С, Verma К. К.,
Stawlley W. С — J. Mol. Spectrosc, 1980, 83,
p. 311.
2727. Ни R. H., Ni Xi.-R., Dai Mu.-Zh., Liu K. W.,
Ни C.-Q., Shi D.-J., Zhang M., Huang W. —
Sci. sinica, 1980, 23, p. 1386.
2728. Ни Т., Ко Н. С, Hepler L. G. — J. Phys. Chem.,
1964, 68, p. 387.
2729. Hubberstey P., Pulham R. J., Thunder A. E. —
J. Chem. Soc. Faraday Trans. I, 1976, 72, p. 431.
2730. Huber E. J., Fitzgibbon G. C, Head E. L.,
Holley С. Е. — 1. Phys. Chem., 1963, 67, p. 1731.
2731. Huber E. /., Head E. L., Holley С. Е. — J. Phys.
Chem., 1957, 61, p. 497.
2732. Huber E. J., Head E. L., Holley С. Е. — J. Phys.
Chem., 1963, 67, p. 1730.
2733. Huber E. /., Head E. L., Holley С. Е. — J. Phys.
Chem., 1964, 68, p. 3040.
565
Литература
2734. НиЪег Е. /., Head E. L., Holley С. Е., Bowman
A. L. — J. Phys. Chem., 1963, 67, p. 793.
2735. НиЪет Е. J., Head E. L., Holley С. Е., Storms E. К.,
Krikorian N. Н. — J. Phys. Chem., 1961, 65,
p. 1846.
2736. НиЪег К. R., Herzberg G. — Molecular Spectra
and Molecular Structure. IV. Constants of
Diatomic Molecules. N. Y. etc: Van Nostrand Reinhold
Co., 1979.
2737. Huber E. J., Holley С. Е. — J. Amer. Chem.
Soc, 1953, 75, p. 3594.
2738. Humber E. J., Holley С. Е. — J. Chem. and Eng.
Data, 1968, 13, p. 252.
2739. Huber E. J., Holley C. E. — J. Chem. Thermodyn.,
1969, 1, p. 267.
2740. Huber E. J., Holley С. Е. — J. Chem. Thermodyn.,
1969, 1, p. 301.
2741. Huber E. J., Holley С. Е. — В кн.: Шестая
Всесоюз. конф. по калориметрии, 1973: (расшир.
тез. докл.). Тбилиси: Мецниереба, 1973, с. 52.
2742. Huber Е. /., Holley С. Е., Meierkord Е. Н. —
J. Amer. Chem. Soc, 1952, 74, p. 3406.
2743. Hubert H., Ozin G. A. — J. Mol. Spectrosc, 1972,
41, p. 595.
2744. Huckel W., Datow J., Simmersbach E. — Z. phys.
Chem.'Leipzig, 1940, A186, S. 129.
2745. Hudson R. — J. Chem. Phys., 1965, 43, p. 1790.
2746. Huff G., Squitieri E., Shyder P. E. — J. Amer.
Chem. Soc, 1948, 70, p. 3380.
2747. Hughes H. "K. — Phys. Rev., 1947, 72, p. 614.
2748. Hughes V., Grabner L. — Phys. Rev., 1950, 79,
p. 314.
2749. Huglen R. — Gaseous aluminium and iron fluo-
ridesfcomplexes: A matrix isolation study. Inst.
for Uorganisk Kjemi Norges Tekniske Hogskoll.
Univ. Trondheim. 1976, Sept. Abh. N 32.
2750. Huglen R., Cyvin S. /., Oye H. A. — Z. Natur-
forsch., 1979, A34, S. 1118.
2751. Hulm J. K., Jones С. К., Hein R. A., Gibson
J. W. — l. Low Temp. Phys., 1972, 7, p. 291.
2752. Hultgren R., Desai R. D., Hawkins D. Т., Glei-
ser M., Kelley K. K., Wagman D. D. — Selected
values of the thermodynamic properties of the
elements. Ohio: ASM, 1973.
2753. Hultgren R., Land С — Trans. Met. Soc. AIME,
1959, 215, p. 165.
2754. Humphrey G. L. — J. Amer.
75, p. 1587.
2755. Humphrey G. L. — J. Amer. Chem. Soc, 1953,
75, p. 2806.
2756. Humphrey G. L. — J. Amer. Chem. Soc, 1954,
76, p. 978.
2757. Hunter F. D., Jaffrey G. A. — J. Chem. Phys.,
1967, 47, p. 3297.
2758. Huntzicker J. J., Westrum E. F. — J. Chem.
Thermodyn., 1971, 3, p. 61.
2759. Hurd С. В., Moor С. А. — J. Amer. Chem. Soc,
1935 57 p 332
2760. Hutchinson E., Manchester К. Е., Winslow L. —
J. Phys. Chem., 1954, 58, p. 1124.
2761. Hutchinson W. P., White A. G. — J. Sci. Instrum.,
1955, 32, p. 309.
2762. Huttig G. F., Magierkiewicz S., Fichmann J. —
Z. phys. Chem. Leipzig, 1929, A141, S. 1.
2763. Huttig G. F., Wehling H. — Kolloidchem. Bei-
hefte, 1927, 23, S. 354.
2764. Huttner K., Tammann G. — Z. anorg. und allg.
Chem., 1905, 43, S. 215.
2765. lafrate G. J., Leupold H., Breslin J. Г., Roth-
Chem. Soc, 1951,
warf F. — Bull. Amer. Phys. Soc, 1976, 21,
p. 302.
2766. Ibrahim M., Bright N. F., Rowland J. F. — J.
Amer. Ceram. Soc, 1962, 45, p. 329.
2767. Igrtatowicz S., Davies M. W J. Less-Common
Metals, 1968, 15, p. 100.
2768. Ihle H. R., Wu С H. — J. Inorg. and Nucl.
Chem., 1974, 36, p. 2167.
2769. Ihle H. R., Wu С. Н. — J. Chem. Phys., 1975,
63, p. 1605.
2770. Iijima K. — Bull. Chem. Soc. Japan, 1977, 50,
p. 373.
2771. Ikeda Y., Ito H., Matsumoto G., Nasu S. — Shit-
suryo Bunseki, 1979, 27, p. 263.
2772. Imanishi S. — Nature, 1932, 143, p. 165.
2773. Imanishi S. — Sci. Pap. Inst. Phys. and Chem
Res. (Tokyo), 1942, 39, p. 45.
2774. Inaba H., Naito K. — J. Nucl. Mater., 1973, 49,
p. 181.
2775. Inaba H., Shimizu H., Naito K. — J. Nucl.
Mater., 1977, 64, p. 66.
2776. Inghram M. G., Chupka W. A., Berkowitz J. —
J. Chem. Phys., 1957, 27, p. 269.
2777. Inghram M. G., Chupka W. A., Berkowitz J. —
Mem. Soc. roy. sci. Liege, 1957, 18, p. 513.
2778. Ingraham T. R., Marier P. — Canad. Met. Quart.
1965, 4, p. 169.
2779. International Practical Temperature Scale of 1968
(IPTS). Amended Edition of 1975. — Metrologia,
1976, 12, p. 7.
2780. loli N., Strumia F. — J. Opt. Soc. Amer., 1971,
61, p. 1251.
2781. Iorns T. V., Stafford F. E. — J. Amer. Chem
Soc, 1966, 88, p. 4819.
2782. Ioshizawa S., Watanabe N., O'Hare Y. — Denki
Kagaku, 1964, 32, p. 162.
2783. Irish D. E., Nelson D. L., Brooker M. H. — J.
Chem. Phys., 1971, 54, p. 654.
2784. Irish D. E., Tang S.-Y., Talts H., Petrucci S -~
J. Phys. Chem., 1979, 83, p. 3268.
2785. Irving A. J., Wadso I. — Acta chem. scand.,
1964, 18, p. 195.
2786. Ischenko A. A., Strand T. G., Demidov A. V.,
Spiridonov V. P. — J. Mol. Struct., 1978, 43
p. 227.
2787. Ishigama M-, Sakurai T. — J. Amer. Ceram
Soc, 1977, 60, p. 367.
2788. Ishihara S., Douglas Т. В. — NBS Rept 10481
(OFOSR-TR-71-2584). Wash.: NBS, 1971, p. 59.
2789. Ishu Т., Naito K., Oshima K. — J. Nucl. Mater
1970, 35, p. 335.
2790. Ishu Г., Naito K., Oshima K. — Solid State
Communs, 1970, 8, p. 677.
2791. Ishu Т., Naito K., Oshima K., Hamaguchi Y. —
J. Phys. and Chem. Solids, 1971, 32, p. 235.
2792. Ishikawa M., Toth L. E. — Phys. Rev., 1971,
Bl, p. 1856.
2793. Ismail Z. K., Hauge R. II., Margrave J. L. —
J. Inorg. and Nucl. Chem., 1973, 35, p. 3201.
2794. Ismail Z. K., Hauge R. H., Margrave J. L.—
J. Mol. Spectrosc, 1975, 54, p. 402.
2795. Itkina L. S., Portnova S. M., Ostrovitianova S. E. —
In: Therm. Anal. Proc. 4th Intern. Conf. Therm.
Anal., Budapest, 1974, Bp., 1975, vol. 1, p. 603.
2796. Iuchi Т., Ono K. — Sci. Repts Res. Inst. Tohoky
Univ., 1961, A13, p. 456.
2797. Ivanov A. A., Demidov A. V., Popenko N. I.,
Zasorin E. Z., Spiridonov V. P., Hargittai I.
J. Mol. Struct., 1980, 63, p. 121.
566
-Литература
.2798. Iversen A. J., Kennedy S. W. — Acta crystallogr.,
1973, B29, p. 1554.
2799. Iwai T. — J. Phys. Soc. Jap., 1960, 15, p. 1596. 2834,
2800. Iwai Т., Sakai K., Watanabe T. — Solid State
Phys., 1973, 8, p. 505.
2801. Iwasaki H., Bright N. F. H. — J. Less-Common 2835.
Metals, 1970, 21, p. 353.
2802. Iwasaki #., Bright N. F. H., Rowland J. F. — 2836.
J. Less-Common Metals, 1969, 17, p. 99.
.2803. Iwase M., Yasuda M., Mori T. — Electrochim. 2837.
acta, 1979, 24, p. 261.
2804. Iwata Y., Koyano N., Shibuya I. — Ann. Repts 2838.
Res. React. Inst. Kyoto Univ., 1978, 11, p. 129.
2805. Jackson D. D., Morgan J. J. — Ind. and Eng. 2839.
Chem., 1921, 13, p. 110.
2806. Jacobs H., Harbrecht В. — Ъ. Kristallogr., 1981, 2840.
156, S. 59.
2807. Jacox M. E., MilliganD. E. — J. Mol. Spectrosc, 2841.
1974, 52, p. 363.
2808. Jaeger F. M Z. anorg. und allg. Chem., 1917. 2842.
101, S. 1.
2809. Jaeger F. M., Bottema J. A., Rosenbohm E. — 2843.
Proc. Acad. Sci. Amsterdam, 1936, 39, p. 921.
2810. Jaeger F. M., Germs H. С — Z. anorg. und allg. 2844.
Chem., 1921, 119, S. 145.
,2811. Jaeger F. M., Rosenbohm E. — Proc. Acad. Sci. 2845.
Amsterdam, 1927, 30, p. 1069.
2812. Jaeger F. M., Rosenbohm E. — Recueil trav. 2846.
chim., 1928, 47, p. 513.
2813. Jaeger F. M., Rosenbohm E. — Proc. Acad. Sci. 2847.
Amsterdam, 1930, 33, p. 457.
.2814. Jaeger F. M., Rosenbohm E. — Recueil trav. 2848.
chim., 1932, 52, p. 1.
2815. Jaeger F. M., Rosenbohm E. — Proc. Acad. Sci. 2849.
Amsterdam, 1934, 37, p. 489.
2816. Jaeger F. M., Rosenbohm E., Fonteine R. — Proc. 2850.
Acad. Sci. Amsterdam, 1936, 39, p. 442.
.2817. Jaeger F. M., Rosenbohm E., Fonteine R. — Re- 2851.
cueil trav. chim., 1936, 55, p. 615.
.2818. Jaeger F. M., Veenstra W. A. — Proc. Acad. Sci. 2852.
Amsterdam, 1934, 37, p. 61.
2819. Jaeger F. M., Veenstra W. A. — Proc. Koninkl. 2853.
nederl. akad. Wet., 1934, 37, p. 327.
2820. Jaeger F. M., Veenstra W. A. — Recueil trav. 2854.
chim., 1934, 53, p. 677. 2855.
2821. Jaeger F. M., Veenstra W. A. — Recueil trav.
chim., 1934, 53, p. 917. 2856.
2822. Jaffray J., Dumas A. — J. rech. Centre nat. rech.
sci., 1954, N 27, p. 360.
2823. Jaffray J., Lyand R. — C. r. Acad. sci., 1951, 2857.
233, p. 133.
2824. Jaffray J., Martin P. — J. phys. et radium, 1953,
14, p. 553. 2858.
2825. Jagannathan V., Wyatt P. — J. Chem. Res. 2859.
Synop., 1978, N 6, p. 203.
2826. Jain A. K., Upreti G. C. — Proc. Nucl. Phys. 2860.
and Solid State Phys. Symp., 1973, C16, p. 161.
2827. Jain Y. S. — Indian J. Phys., 1976, 50, p. 206. 2861.
2828. James D. W., Leong W. H. — J. Chem. Phys., 2862.
1968, 48, p. 5089. 2863.
2829. James D. W., Leong W. H. — J. Chem. Phys.,
1969, 51, p. 640.
2830. James D. W., Leong W. H. — Aust. J. Chem., 2864.
1970, 23, p. 1087.
2831. James T. C, Norrish W. G., Klemperer W. — J. 2865.
Chem. Phys., 1960, 32, p. 728.
2832. Jamieson J. C, Olinger B. — Science, 1968, 161, 2866.
p. 893. 2867.
2833. JANAF thermochemical tables PB-168370: / Ed.
D. R. Stull, 1965. First Addendum, PR-168370-1, 2868.
1966; Second Addendum PB-168370-2, 1967;
Third Addendum PB-168370-3, 1968.
JANAF thermochemical tables. 2 ed./ Ed. D. R.
Stull, H. Prophet. Wash.: NSRDS—NBS, 1971,
N 37.
JANAF thermochemical tables. Supplements.
Wash., 1974—1978.
Janecke E. — Z. anorg. und allg. Chem., 1920,
188, S. 72.
Janke D., Fischer W. A. — Arch. Eisenhuttenw.,
1975, 46, S. 755.
Janninck A. E., Whitmore D. H. — J. Phys.
Chem. Solids, 1966, 27, p. 1183.
Janoschek R., Lee H. U. — Chem. Phys. Lett.,
1978, 58, p. 47.
Jansson В., Lunden A. — Z. Naturforsch., 1970,
A25, S. 697.
Jam G. /., James D. W. — J. Chem. Phys., 1961,
35, p. 739.
Janz G. J., Kelley F. J., Perano J. L. — J. Chem.
and Eng. Data, 1964, 9, p. 133.
Janz G. J., Lorenz M. R. — J. Chem. and Eng.
Data, 1961, 6, p. 321.
Janz G. J., Lorenz M. R. — J. Chem. and Eng.
Data, 1964, 9, p. 94.
Janz G. J., Neuenschwander E., Kelley F. J. —
Trans. Faraday Soc, 1963, 59, p. 841.
Jarett S. M., Franken P.A. — J. Opt. Soc. Amer.,
1965, 55, p. 1603.
Jasinski J. P., Holt S. L., Wood J. H., Asprey
L. B. — J. Chem. Phys., 1975, 63, p. 757.
Jayaraman A., Klement W., Kennedy G. С —•
Phys. Rev., 1963, 131, p. 644.
Jeannin Y., Mannerskantz C, Richardson F. D. -—
Trans. Met. Soc. AIME, 1963, 227, p. 300.
Jenckel E. — Z. anorg. und allg. Chem., 1936, 227,
S. 214.
Jennings L. D., Miller R. E., Spedding F. H. —
J. Chem. Phys., 1960, 33, p. 1849.
Jenny S. N., Ogden J. S. — J. Chem. Soc. Dalton
Trans., 1979, p. 1465.
Jensen D. E. — Trans. Faraday Soc, 1969, 65,
p. 2123.
Jensen D. E. — J. Phys. Chem., 1970, 74, p. 207.
Jensen D. E., Padley P. J. — Trans. Faraday Soc,
1966, 62, p. 2132.
Jensen D. E., Miller W. J. — In: 13th Symp.
Intern, on Combustion, 1970. Pittsburg: The
Combustion Inst., 1971, p. 363.
Jerez W., Bhise V., Das Gupta S., Bonilla С F. —
In: Proc. Sixth Symp. Thermophys. Properties.
N. Y.: ASME, 1973, p. 353.
Jette E. R. — J. Chem'. Phys., 1955, 23, p. 365.
Jevons W. — Proc. Phys. Soc. London, 1929, 41,
p. 520.
Jevons W. — Report on Band-Spectra of Diatomic
Molecules. L., 1932.
Johansson I. — Ark. fys., 1959, 15, s. 169.
Johansson I. — Ark. fys., 1961, 20, s. 135.
JohnsI. В., Walsh K.A. — V.S. AEC Los Alamos
Lab. Rept LA-381. Los Alamos (N. Мех.), 1945,
Aug. 30.
Johnson С E., Heinrich R. R., Crouthamel С. Е. —
J. Phys. Chem., 1966, 70, p. 242.
Johnson D. L. — U. S: Ames Lab. Iowa State
Univ. Rept IS-T-124. Ames (Iowa), 1966.
Johnson D.L. — Diss. Abstrs, 1967, B27, p. 4087.
Johnson D. L., Finnemore D. K. — Phys. Rev.,
1967, 158, p. 376.
Johnson E. H. — Phys. Rev., 1927, 29, p. 85.
567
Литература
2869. Johnson G. К. — J. Chem. Thermodyn., 1979,
11, p. 483.
2870. Johnson G. K., Deventer E. #., Kruger 0. L.,
Hubbard W. H. — J. Chem. Thermodyn., 1969,
1, p. 89.
2871. Johnson G. K., Gayer К. Н. — J. Chem.
Thermodyn., 1979, 11, p. 41.
2872. Johnson G. K., Gayer К. Н. — J. Chem.
Thermodyn., 1980, 12, p. 705.
2873. Johnson G. K., Grow R. Т., Hubbard W. N. —
J. Chem. Thermodyn., 1975, 7, p. 781.
2874. Johnson G. K., O'Hare P. A. G. — Z. Chem.
Thermodyn., 1978, 10, p. 577.
2875. Johnson G. K., Steele W. V. — J. Chem.
Thermodyn., 1981, 13, p. 717.
2876. Johnson I., Johansen R. Г. — Proc. Okla Acad.
Sci., 1951, 32, p. 63.
2877. Johnson J. S., Kraus K. A. — J. Amer. Chem.
Soc, 1952, 74, p. 4436.
2878. Johnson J. W., Agron P. A., Bredig M. A. —
J. Amer. Chem. Soc, 1955, 77, p. 2734.
2879. Johnson J. W-, Bredig M. A. — J. Phys. Chem.,
1958, 62, p. 604.
2880. Johnson L. W., Johnson R. С — Proc. Roy. Soc.
London, 1931, A133, p. 207.
2881. Johnson R. Т., Vilches O. E., Wheatley J. C,
Gydax S. — Phys. Rev. Lett., 1966, 16, p. 101.
2882. Johnston H. L., Bauer T. W. — J. Amer. Chem.
Soc, 1951, 73, p. 1119.
2883. Johnston J Z. phys. Chem. Leipzig, 1908, 62,
S. 330.
2884. Joly R. D., Perachon G. — In: 4th Conf. intern,
thermodyn. chim., Montpellier, 1975, vol. 1, p. 143.
2885. Joly R. D., Perachon G. — Thermochim. acta,
1977 21 d 333
2886. Joly'R.' Thourey J., Perachon G. — С. г. Acad.
sci., 1973, C277, p. 1179.
2887. Jones D. A., Jones R. V. — Proc. Phys. Soc.
London, 1962, 79, p. 351.
2888. Jones D. M. — Diss. Ph. D. The Univ. Wisconsin,
1965. 186 p.
2889. Jones E. D., Burgess D. S., Amis E. S. — Z. phys.
Chem. Leipzig, 1955, 4, S. 220.
2890. Jones E. S., Mosher С J. F., Speiser R., Spret-
nak J. W. — Corrosion, 1958, 14, p. 20.
2891. Jones E. V., Lietzke M. H., Marshall W. L. —
J. Amer. Chem. Soc, 1957, 79, p. 267.
2892. Jones F. O., Knapton A. <?., Savill J. — J. Less-
Common Metals, 1959, 1, p. 80.
2893. Jones G. O., Martin D. L. — Philos. Mag.4, 1954,
45, p. 649.
2894. Jones H. A., Langmuir I., Mackay G. M. J. —
Phys. Rev., 1927, 30, p. 201.
2895. Jones L. H., Ekberg S. — J. Chem. Phys., 1977,
67, p. 2591.
2896. Jones L. #., Ekberg S. A. — J. Chem. Phys.,
1979, 71, p. 4765.
2897. Jones L. V., Etter D. E., Hudgens С R.,
Huffman A. A., Rhinehammer Т. В., Rogers N. E.,
Tucker P. A., Wittenberg L. J. — J. Amer. Ceram.
Soc, 1962, 45, p. 79.
2898. Jones R. W., Gole J. L. — J. Chem. Phys., 1976,
65, p. 3800.
2899. Jones W. M., Gordon /., Long E. A. — J. Chem.
Soc, 1952, 20, p. 695.
2900. Jong J. M., Broers G. H. J. — J. Chem.
Thermodyn., 1976, 8, p. 367.
2901. Jongejan A., Wilkins A. L. — J. Less-Common
Metals, 1969, 19, p. 185.
2902. Joshio Т., Takahashi K. — J. Ceram. Soc. Jap.r
1976, 84, p. 62.
2903. Jostosons A., Jenkins A. E. — J. Phys. Chem.,
1969, 73, p. 749.
2904. Jostsons A., McDougall P. — In: Sci. Technol.
Appl. Titanium: Proc. Intern. Conf. 1968. N. Y.:
Pergamon Press, 1970, p. 745.
2905. Joubert P., Bougon R., Gaudreau B. — Canad.
J. Chem., 1978, 56, p. 1874.
2906. Juneja J. M., Bhatt Y. J., Garg S. P. — J,
Less-Common Metals, 1980, 69, p. 313.
2907. Jungermann A. — Spectrochim. acta, 1977, A33,,
p. 725.
2908. Junker E. — Z. anqrg. und allg. Chem., 1936, 228,
S. 97.
2909. Justice B. H., Westrum E. F. — J. Phys. Chem.,.
1963, 67, p. 339.
2910. Kachi S., Kosuge K., Okinaka H. — J. Solid
State Chem., 1973, 6, p. 258.
2911. Kachi S., Takada Т., Kosuge K. — J. Phys. Soc,
Jap., 1963, 18, p. 1839.
2912. Kahn L. R., Hay P. J., Shavitt I. — J. Chem.
Phys., 1974, 61, p. 3530.
2913. Kaiser E. W., Falconer W. E., Klemperer W. —
J. Chem. Phys., 1972, 56, p. 5392.
2914. Kajzar F., Parette G. — Solid State Communs,.
1978, 25, p. 535.
2915. Kamimoto M. — Thermochim. acta, 1980, 41,.
p. 361.
2916. Kaminsky M. E. — Thesis. Rept N 2531. Stanford
Univ., 1976.
2917. Kaminsky M. E. — J. Chem. Phys., 1977, 66,.
p. 4951.
2918. Капа'an A. S., Hauge R. H., Margrave J. L. —
J. Chem. Soc. Faraday Trans. II, 1976, 72, p. 1991.
2919. Kangro W., Wieking H. W. — Z. phys. Chem.
Leipzig, 1938, A183, S. 199.
2920. Kant A., Lin S.-S. — J. Chem. Phys., 1969, 51,
p. 1644.
2921. Kant A., Strauss B. — J. Chem. Phys., 1964, 41,
p. 3806.
2922. Kant A., Strauss B. — In: Proc. Army Sci. Conf.
West. Point, New York, 1964, Pt II. Wash., 1965,
p. 55.
2923. Kantola M., Vilhonen E. — Ann. Acad. sci.
Fennicae, Ser. AVI, 1960, N 54.
2924. Kao J. — J. Mol. Street., 1979, 59, p. 147.
2925. Karkhanovala M. D., George A. M. — J. Nucl..
Mater., 1966, 19, p. 267.
2926. Karpov S. V., Shultin A. A. — J. Phys. Chem.
Solids, 1968, 29, p. 475.
2927. Kasai P. H. — J. Chem. Phys., 1968, 49, p. 4979.
2928. Kasai P. H., Weltner W. — J. Chem. Phys., 1965,
43, p. 2553.
2929. Kasai P. H., Weltner W. — J. Chem. Phys., 1967,
46, p. 3172.
2930. Katayama I., Kozuka Z. — Techn. Repts Osaka
Univ., 1973, 23, p. 411.
2931. Kats S. A., Chekhovskoi V. Ya. — High Temp.-
High Pressur., 1979, 11, p. 629.
2932. Katsuda H., Ishigai Т., Furukawa K. — Nucl.
Technol., 1977, 32, p. 297.
2933. Katsura Т., Hasegava M. — Bull. Chem. Soc.
Jap., 1967, 40, p. 561.
2934. Katz T. J., Margrave J. L. — J. Chem. Phys.,.
1955, 23, p. 983.
2935. Kaufman M-, Muenter J., Klemperer W. — J.
Chem. Phys., 1967, 47, p. 3365.
2936. Kaufmann L. — Semi-annual Report N 2, Manlabs^
Inc. Cambridge, 1963, Apr.
568
Литература
2937. Kaufmann L., Clougerty E. V. — U. S. Air Force
Mater. Lab. Rept RTD-TDR-63-4096. Wright-
Patterson, AFB (Ohio), 1963.
2938. Kawabe V., Yanagi Т., Sawada S. — J. Phys.
Soc. Jap., 1965, 20, p. 2059.
2939. Kawakubo Т., Nakagawa T. — J. Phys. Soc.
Jap., 1964, 19, p. 517.
2940. Kawano S., Kosuge K., Kachi S. — J. Phys. Soc.
Jap., 1966, 21, p. 2744.
2941. Kay A. E. — In: Metal Plutonium/Ed. A. S.
Coffinberry, W. N. Miner. Chicago: Univ. Press.,
1961, p. 183.
2942. Kay A. E., Loasby R. G. — Philos. Mag., 1964, 9,
p. 37.
2943. Kay M. I., Frazer В. С — Acta crystallogr.,
1961, 14, p. 56.
2944. Kaylor C. E., Walden G. E., Smith D. F. — J.
Amer. Chem. Soc, 1959, 81, p. 4172.
2945. Kaylor С E., Walden G. E., Smith D. F. — J.
Phys. Chem., 1960, 64, p. 276.
2946. Kaznoff A. I., Grossman L. N. — In:
Thermodynamics nuclear materials: Proc. Symp. Vienna,
1967, Vienna: IAEA, 1968, p. 25.
2947. Keenan P. С — Publs. Astron. Soc. Pacif., 1957,
69, p. 5.
2948. Keenan P. C, Schroeder L. W- — Astrophys. J.,
1952, 115, p. 82.
2949. Keenan T. K., Asprey L. B. — Inorg. Chem., 1969,
8, p. 235.
2950. Keesom P. H., Pearlman N. — Phys. Rev., 1953,
91, p. 1354.
2951. Keesom P. H., Pearlman N. — Phys. Rev., 1958,
112, p. 800.
2952. Keesom W. H., Clark C. W. — Physica, 1935, 2,
p. 698.
2953. Keesom W. H., Desirant M. — Physica, 1941, 8,
p. 273.
2954. Kehl W. L., Hay R. G., Wahl D. — J. Appl.
Phys., 1952, 23, p. 212.
2955. Keller D. V., Kanday. A., King A. Z. — J. Phys.
Chem., 1958, 62, p. 732.
2956. Kelley K. K. — J. Amer. Chem. Soc, 1940, 62,
p. 818.
2957. Kelley K. K. — J. Chem. Phys., 1940, 8, p. 316.
2958. Kelley К. К. — Ind. and Eng. Chem., 1944, 36,
p. 377.
2959. Kelley К. К. — Ind. and Eng. Chem., 1944, 36,
p. 865.
2960. Kelley К. К. — Contributions to the data on
theoretical metallurgy. XIII High temperature heat-
content, heat capacity and entropy data for the
elements and inorganic compounds. — Bull. Bur.
Mines (USA), 1960, N 584.
2961. Kelley К. К., Anderson С. Т. — Bull. Bur. Mines
(USA), 1935, N 384.
2962. Kelley K. K., Boericke F. S., Moore G. E.,
Huffman E. H., Bangert W. M. — U. S. Bur. Mines.
Techn. Paper, 1944, N 662.
2963. Kelley K. K., King E. G. — Contributions to the
data on theoretical metallurgy. XIV. Entropies
of the elements and inorganic compounds. — Bull.
Bur. Mines (USA), 1961, N 592.
2964. Kelley K. K., Mah A. — U. S. Bur. Mines. Rept
Invest., 1959, N5490.
2965. Kelley R., Padley P. J. — Trans. Faraday Soc,
1971, 67, p. 740.
2966. Kemori N., Katayama I., Kozuka Z. — J. Chem.
Thermodyn., 1979, 11, p. 215.
2967. Kendall W. B. — J. Chem. and Eng. Data, 1962,
7, p. 540.
2968. Kenisarin М- М-, Beresin B. Ya., Chekhovskoi
V. Ya. — High. Temp.-High Pressur., 1972, 4,
p. 707.
2969. Kennedy G. S., LaMori P. N. — J. Geophys. Res.,
1962, 67, p. 851.
2970. Kennedy S. W. — Phys. status solidi, 1970, A2r
p. 415.
2971. Kennedy S. W. — J. Cryst. Growth, 1972, 16,
p. 274.
2972. Kennedy S. W-, Kriven W. M. — J. Mater. Sci.,.
1976, 11, p. 1767.
2973. Kennedy S. W., Odlyha M. — Austral. J. Chem.,.
1974, 27, p. 1121.
2974. Kennedy S. W., Taylor G. F., Patterson J. H. —
Phys. status solidi, 1966, 16, p. 175.
2975. Kennedy S. W., Ubbelohde F. K. S. — Proc. Roy.
Soc. London, 1953, A219, p. 303.
2976. Kent R. A. — High Temp. Sci., 1969, 1, p. 169.
2977. Kerr H. V., Barros H. L., Dickerson D. L., Barf-
knecht А. Т., Honig J. M. — Mater. Res. Bull.,
1977, 12, p. 137.
2978. Kerr H. V., Dickerson D. L., Kuwamoto H.,
Barros H. L., Honig J. M. — J. Solid State Chem.,.
1976, 19, p. 95.
2979. Keyes F.G. — J. Amer. Chem. Soc, 1912, 34, p. 779..
2980. Keys L. K., Mulay L. N. — Appl. Phys. Lett.,
1966, 9, p. 248.
2981. Keys L. K., Mulay L. N. — J. Appl. Phys., 1967,
38, p. 1466.
2982. Khanna R. K. — Proc. Nat. Inst. Sci. India,
1961, A27, p. 489.
2983. Kher V. C, Hete V. N., Deshphande D. A. —
Indian J. Pure and Appl. Phys., 1975, 13, p. 653.
2984. Kibler G. M., Lyon T. F., Linevsky M. /., De-
Santis V. f. — WADD Techn. Rept TR-60-646,.
1964.
2985. Kieffer R., Benesovsky F. — Planseeber. Pulver-
met., 1957, 5, S. 56.
2986. Kieffer R., Benesovsky F., Schmid H. — Z.
Metallic., 1956, 47, S. 247.
2987. Kiess С. С. — Publs Astron. Soc. Pacific, 1948,
60, p. 252.
2988. Kiess С. С — J. Res. NBS, 1953, 51, p. 247.
2989. Kiess С. С — J. Res. NBS, 1962, A66, p. 111.
2990. Kiess С. С, Kiess H. K. — J. Res. NBS, 1931,
6, p. 621.
2991. Kiess С. С, Stowell E. L. — J. Res. NBS, 1934,
12, p. 459.
2992. Kigoshi K. — Bull. Chem. Soc. Jap., 1950, 23,
p. 67.
2993. Kigoshi K. — J. Chem. Soc. Jap. Pure Chem.
Sec, 1951, 72, p. 57.
2994. Kilday M. V. — J. Res. NBS, 1980, 85, p. 467.
2995. Kilday M. V. — J. Res. NBS, 1980, 85, p. 449.
2996. Killackey J. J. — U. S. AEC. Brookhaven Nat.
Lab. Rept BNL-756 (c-35): TID-4500, 18th Ed.
Upton (N. Y.), 1962, p. 155.
2997. Killian T. I. — Phys. Rev., 1926, 27, p. 578.
2998. Killingbeck S. — Diss. Abstrs, 1972, B34, p. 138.
2999. Kilner S. B. — J. Amer. Chem. Soc, 1952, 74,
p. 5221.
3000. Kilpatrick M., Lott S. K. — J. Less-Common
Metals, 1965, 8, p. 299.
3001. Kim K. C, Fleming R., Seitz D. — Chem. Phys.
Lett., 1979, 63, p. 471.
3002. Kim K. C, Fleming R., Seitz D., Reisfeld M. —
Chem. Phys. Lett., 1979, 62, p. 61.
3003. Kim K. C, Reisfeld M. J., Person W. B. —
J. Mol. Struct., 1980, 60, p. 205.
569
Литература
3004. Kimizuka N., Saeki M., Nakamura M. — Mater.
Res. Bull., 1970, 5, p. 403.
3005. Kimura H., Asano M., Kubo K. — J. Nucl. Mater.,
1980, 92, p. 221.
3006. Kimura M., Schomaker V., Smith D. W., Wein-
^.^stock B. — J. Chem. Phys., 1968, 48, p. 4001.
'MQlT Kimura M., Uchida J. — Sci. Papers Inst. Phys.
and Chem. Res. Tokyo, 1932, 18, p. 109.
3008. King E. G. — J. Amer. Chem. Soc, 1954, 76,
p. 3289.
3009. King E. G. — J. Amer. Chem. Soc, 1957, 79,
p. 2056.
3010. King E. G. — J. Amer. Chem. Soc, 1958, 80,
p. 1799.
3011. King E. G., Christensen A. U. — U. S. Bur. Mines.
Rept Invest., 1961, N 5709.
3012. King E. G., Christensen A. U. — U. S. Bur. Mines.
Rept Invest., 1961, N 5789.
3013. King E. G., Weller W. W., Christensen A. U. —
U. S. Bur. Mines. Rept Invest., 1960, N 5664.
3014. King E. G., Weller W. W., Pankratz L. B. —
U. S. Bur. Mines. Rept Invest., 1961, N 5857.
3015. Kirillin V. A., Sheindlin A.E., Chekhovskoi V. Ya. —
Intern. J. Heat and Mass Transfer, 1962, 5, p. 1.
3016. Kirillin V. A., Sheindlin A. E.,
Chekhovskoi V. Ya. — In: Proc Intern. Sympos. on High
Temp. Technol. Wash., 1974, p. 471.
3017. Kirkerud Т., Klaeboe P., Oye H. A. — J. Inorg.
and Nucl. Chem., 1979, 41, p. 183.
3018. Kirkham A. J., Yates B. — Cryogenics, 1968, 8,
p. 381.
3019. Kirkham A. J., Yates B. — J. Phys.: Solid State
Phys., 1968, Cl, p. 1162.
3020. Kiss A. B. — Acta chim. Acad. sci. hung., 1975,
84, p. 393.
3021. Kistiakowsky G. B. — Naval Res. Lab. Rept
N 2958. Wash., 1941.
3022. Kitahiro I., Watanabe A., Sasaki H. — J. Phys.
Soc Jap., 1966, 21, p. 196.
3023. Kiukkola K. — Acta chem. scand., 1962, 16,
p. 327.
3024. KleimanH. — J. Opt. Soc. Amer., 1962, 52, p. 441.
3025. Klein A. H., Danielson G. C. — In: Proc Black
Hills Summer Conf. on Transport Phenom., Aug.
1962, Rapid City (South Dakota). Pap. A 4, 1962,
p. 47.
3026. Kleman В., Liljeqvist B. — Arkiv fys., 1955, 9,
p. 377.
3027. Klement W. J. — J. Phys. Chem., 1970, 74, p. 2751.
3028. Klement W. J. — J. Inorg. and Nucl. Chem.,
1974, 36, p. 1916.
3029. Klement W. J., Cohen L. H. — Ber. Bunsenges.
phys. Chem., 1975, 79, S. 327.
3030. Klemm K., Pirchner P. — Optik, 1948, 3, S. 75.
3031. Klemm L., Scharf H. J.—Z. anorg. und allg.
Chem., 1960, 303, S. 263.
3032. Klemm W-, Grimm L. — Naturwissenschaften,
1939, 27, S. 787.
3033. Klemperer W. — J. Chem. Phys., 1955, 23, p. 2452.
3034. Klemperer W., N orris W. G. — J. Chem. Phys.,
1961, 34, p. 1071.
3035. Klemperer W., N orris W. G., Buchler A. — J.
Chem. Phys., 1960, 33, p. 1534.
3036. Klemperer W., Rice S. A. — J. Chem. Phys.,
1957, 26, p. 619.
3037. Kleppa O. J., McCarty F. E. — J. Chem. and
Eng. Data, 1963, 8, p. 331.
3038. Kleykamp H., Supawan A. — J. Less-Common
Metals, 1979, '63, p. 237.
3039. Klooster H. S Z. anorg. und allg. Chem.,
1911, 69, S. 122.
3040. Klooster H. S. — Z. anorg. und allg. Chem.,
1914, 85, S. 49.
3041. Klooster H. S., Stearns E. I. — J. Amer. Chem.
Soc, 1933, 55, p. 4121.
3042. Klopp W. D., Maykuth D. J., Ogden H. R., Jaf-
fee R. I. — Trans. Met. Soc. AIME, 1960, 218,
p. 971.
3043. Klusius K., Goldmann J., Perlick A. — Z. Natur-
" forsch., 1949, A4, S. 424.
3044. Knacke O., Lossmann G., Mailer F. — Z. anorg.
und allg. Chem., 1969, 370, S. 91.
3045. Knacke O., Lossmann G., Muller F. — Z. anorg.
und allg. Chem., 1969, 371, S. 32.
3046. Knacke O., Schmolke Д., Stranski I. N. — Z. Kri-
stallogr., 1957, 109, S. 184.
3047. Knapton A. G., Savill J., Siddall G. — J. Less-
Common Metals, 1960, 2, p. 357.
3048. Kneip G. D., Betterton J. O. — J. Electrochem.
Soc. 1956, 103, p. 684.
3049. Kneip G. D., Betterton J. O., Scarbrough J. O. —
Phys. Rev., 1963, 130, p. 1687.
3050. Knidiri M., Benarafa L. — C. r. Acad. sci., 1978,
C287, p. 55.
3051. Ко Н. С, Ни Т., Spencer J. G., Huang C. Y. —
J. Chem. and Eng. Data, 1963, 8, p. 364.
3052. Kobayashi M. — Bull. Chem. Soc. Jap., 1933, 8,
p. 231.
3053. Kobyliansky A. I., KulikovA. H., Gurvich L. V. —
Chem. Phys. Lett., 1979, 62, p. 198.
3054. Koch R. K., Anable W. E. — U. S. Bur. Mines.
Rept Invest., 1968, N 7063.
3055. Koch R. K., Anable W. E., Beall R. A. Vapor
pressures of liquid Nb B740—3140 K.) and liquid
Hf B500—2810 K). — U. S. Bur. Mines. Rept
BM-RI-7125, May 1968.
3056. Koch R. K., Anable W. E., Calvert E. D., Be-
all R. A. — In: Trans. Vac. Met. Conf. 19th.
N. Y., 1966, p. 1.
3057. Koch R. K., Calvert E. D., Thomas C. R., Be-
all R. A. — U. S. Bur. Mines. Rept Invest.,
1969, N 7271.
3058. Kocher J. — Bull. Soc. chim. France, 1968, 3,
p. 919.
3059. Koehler M. F. — U. S. Bur. Mines. Rept Invest.,
1960, N 5700.
3060. Koelling D. D., Ellis D. E., Bartlett R. J. — J.
Chem. Phys., 1976, 65, p. 3331.
3061. Kohl F. J., Stearns С A., Fryburg G. С — In:
Met.-slag-gas react, processes: Pap. Intern. Symp. /
Ed. Z. A. Foroulis, W. W. Smeltzer. Princenton
(N. Y.): Electrochem. Soc. Inc., 1975, p. 649.
3062. Kohlhaas R., Braun M., Vollmer O. — Z. Natur-
forsch., 1965, A20, S. 1077.
3063. Kojima H., Whiteway S. G., Masson C. R. — Ca-
nad. J. Chem., 1968, 46, p. 2968.
3064. Kolitsch W-, Muller U. — Z. anorg. and allg.
Chem., 1975, 418, S. 235.
3065. Kolsky H. G., Gilles P. W. — J. Chem. Phys.,
1954, 22, p. 232.
3066. Kolsky H. G., Gilles P. W. — J. Chem. Phys.,
1956, 24, p. 828.
3067. Kolsky H. G., Gilmer R. M., Gilles P. W. — J.
Chem. Phys., 1957, 27, p. 494.
3068. Komarek K. L., Silver M. — In: Proc. Intern.
Symp. on Thermodyn. Nucl. Materials, 1962.
Vienna: IAEA, 1963, p. 749.
3069. Konishi Y. — J. Chem. Soc. Jap. Ind. Chem.
Sec, 1936, 39, p. 209 B.
570
Литература
3070. Konobeevsky S. Т., Zaimcfusky A. S., Levitsky В. М.,
Sokursky Y. N., Chebotarev N. Т., Bobkov Y. V.,
Egorov P. P., Ntkolaev G. N., Ivanov A. A. —
In: Proc. 2nd Intern. Conf. Peaceful Uses Atom.
Energy, Geneva, 1958. Geneva: UN, 1958, p. 194.
3071. Konowalow D. D., Olson M. L. — J. Chem. Phys.,
1977, 67, p. 590.
3072. Korbitz F. W. — Thesis. Iowa State Univ. Sci.
and Techn., 1964.
3073. Koref F. — Ann. Phys., 1911, 36, S. 49.
3074. Korhonen U. — Ann. Acad. sci. Fennicae, Ser. Al,
1953, N 150, 16 p.
3075. Kornilov A. N., Ushakova I. M., Huber E. J.,
Holley C. E. — J. Chem. Thermodyn., 1975, 7,
p. 21.
3076. Korreng E. — Neues Jahrb. Mineral. Geol. Bei-
lage Bd., 1919, 37, S. 51.
3077. Kosuge K. — J. Phys. Chem. Solids, 1967, 28,
p. 1613.
3078. Kosuge K. — J. Phys. Soc. Jap., 1967, 22, p. 551.
3079. Kosuge K., Takada Т., Kachi S. _ J. Phys.
Soc. Jap., 1963, 18, p. 318.
3080. Kosuge T. — J. Chem. Soc. Jap. Ind. Chem. Sec,
1967, 70, p. 2089.
3081. Kosugi T. — J. Chem. Soc. Jap. Ind. Chem. Sec,
1970, 73, p. 1087. '
3082. Kosugi T. — Bull. Chem. Soc. Jap., 1972, 45, p. 15.
,3083. Kostenskd I., Corba J., Malinovsky M. — Che.
zvesti, 1974, 28, s. 546.
3084. KoHenskd /., Malinovsky M.— Cbem. zvesti, 1974,
28, s. 553.
3085. Kothen C. W. — Diss. Ohio State Univ., 1952
(Diss. Abstrs, 1957, 17, p. 2842).
3086. Kothen С W-, Johnston H. L. — J. Amer. Chem.
Soc, 1953, 75, p. 3101.
3fl87. Kotnik-Karuza D., Vidal C. R. — Chem. Phys.,
1979, 40, p. 25.
3088. Kouchkovsky R., Lecomte M. — C. r. Aead. sci.,
1968, B267, p. 260.
3089. Kovacs I. Rotational structure of the spectra of
diatomic molecules. L., 1969.
3090. Kozak A., Bardin J. C, Erb A. — Rev. chim.
miner., 1976, 13, p. 190.
3091. Kozlowski T. R. — Appl. Opt., 1968, 7, p. 795.
3092. Kracek F. С — J. Phys. Chem., 1930, 34, p. 22i5.
3093. Kracek F. С — J. Amer. Chem. Soc, 1931, 53,
p. 2609.
3094. Kracek F. С — J. Phys. Chem., 1932, 36, p. 2529.
3095. Kracek F. С — Ann. Rept Director of Geoph'ys.
Lab. N 1215. Wash.: Carnegie Inst., 1952/1953,
p. 69.
3096. Kracek F. C, Barth T. E. W., Ksanda С J. —
Phys. Rev., 1932, 40, p. 1034.
3097. Kracek F. C, Bowen N. L., Morey G. W. — J.
Phys. Chem., 1937, 41, p. 1183.
3098. Kracek F. C, Ksanda С J. — J. Phys. Chem.,
1930, 34, p. 1741.
3099. Kracek F. C, Posnjak E., Hendricks S. B. —
J. Amer. Chem. Soc, 1931, 53, p. 3339.
3100. Kraftmacher Ya. A. — High Temp.-High Pressur.,
1973, 5, p..433.
3101. Kraus D. L., Petrocelli A. W. — J. Phys. Chem.,
1962, 66,'p. 1225.
3102. Krauss F. — Z. Metallk., 1958, 49, S. 386.
3103. KrebsB., МШегА., FadiniA J. Mol. Spectrosc,
1967, 24, p. 198.
3104. Krefft H. — Z. Phys., 1932, 77, S. 752.
.3105. Kreig G.,'Genter R. В., Gross A. V. — Inorg. and
Nucl. Chem. Lett., 1969, 5, p. 819.
3106. Kremers H. C, Stevens K. G. — J. Amer. Chem.
Soc, 1923, 45, p. 614.
3107. Krier С A., Craig R. S., Wallace W. E. — J. Phys.
Chem., 1957, 61, p. 522.
3108. Krikorian N. H., Wallace T. C. — J. Electrochem.
Soc, 1964, 111, p. 1431.
3109. Krikorian О. Н. — J. Phys. Chem., 1963, 67,
p. 1586.
3110. Krikorian O. #., Carpenter J. H. — J. Phys.
Chem., 1965, 69, p. 4399.
3111. Krikorian O. #., Carpenter J. H., Newbury R. S
High Temp. Sci., 1969, 1, p. 313.
3112. Kroger C, Fingas E. — Z. anorg. und allg. Chem.,
1933, 212, S. 269.
3113. Kroger C, Fingas E. — Z. anorg. und allg. Chem.,
1933, 213, S. 12.
3114. Kroger C, Fingas E. — Z. anorg. und allg. Chem.,
1935 223 S 257
3115. Kroger C.\ Sorstrom L. _ Glastechn. Ber., 1965,
38, S. 313.
3116. Kroger C, Stratmann J Glastechn. Ber., 1961,
34, S. 311.
3117. Krohn B. J., Person W. В., Overend J. — J. Chem.
Phys., 1976, 65, p. 969.
3118. Kroll W. J., Schlhten A. W. — Metals Technol.,
1947, 14, p. 2179.
3119. Kroner A. — Ann. Phys., 1913, 40, S. 438.
3120. Kruger O. L., Savage H. — J. Chem. Phys., 1968,
49, p. 4540.
3121. Krupka M. С High temp, vapor behaviour and
thermodynamic properties of HfB2. Los Alamos
(N. Мех.): Los Alamos Seint Lab. of Univ. of
Calif., 1962.
3122. Kruss G., Nilsbn L. F. — Z. phys. Chem. Leipzig,
1887, 1, S. 391.
3123. Krustinsons J. — Latv. Univ. Raksti Kim. Fak.
Ser. 1, 1929,1pp. 29.
3124. Kubaschewski O., Alcock С. В. Metallurgical
thermochemistry. 5th ed. Oxford etc.: Pergamon
Press, 1979.
3125. Kubaschewski O., Evans E. L., Alcock С. В.
Metallurgical thermochemistry. 4th ed. Oxford:
Pergamon Press, 1967.
3126. Kubaschewski O., Heymer G. — Acta met., 1960,
8, p. 416.
3127. Kubaschewski O., Vnal H. — High Temp-High
Pressur., 1977, 9, p. 361.
3128. Kubicar L. — Fys. Casop., 1968, 18, p. 58.
3129. Kubicar L., Mariani E. — Fys. Casop., 1968, 18,
p. 251.
3130. Kucharczyk D., Niklewski T. — Appl. Crystal-
logr., 1979, 12, p. 370.
3131. Kucharczyk D., Pietraszko A. — Acta Univ. Wra-
ticl., 1977, N 341, s. 61.
3132. Kucirek F., Papousek D. — Collect. Czechosl.
Chem. Communs., 1961, 26, s. 1458.
3133. Kuczkowski R. L., Lide D. R., Jr., Krisher L. С —
J. Chem. Phys., 1966, 44, p. 3131.
3134. Kudo #., Wu С. И., Ihle H. R. — J. Nucl. Mater.,
1978, 78, p. 380.
3135. Kudrak D. R., SienkoiM. J. — Inorg. Chem.,
1967, 6, p. 880.
3136. Kugel R., Williams C, Fred M., Malm J. G.,
Carnell W. Т., Hindman J. C, Childs W. /.,
Goodman L. С — J. Chem. Phys., 1976, 65, p. 3486.
3137. Kuhlman С W. — MCW, 1948, p. 118. Цит. по:
[2344].
3138. Kuhn P. — Z. Phys., 1932, 76, S. 782.
3139. Kuijpers P., Toning Т., Dymanus A. — Z. Na-
turforsch., 1975, A30, S. 1256.
571
Литература
3140. Kuijpers P., Torring Т., Dynamus A. — Chem. 3175.
Phys., 1976, 15, p. 457.
3141. Kunze K. R., Hauge R. #., Hamill D.,
Margrave J. L. — J. Chem. Phys., 1976, 65, p. 2026.
3142. Kunze K. R., Hauge R. #., Hamill D., Mar- 3176.
grave J. L. — J. Phys. Chem., 1977, 81, p. 1644.
3143. Kuroda N., Fan H. Y. — Phys. Rev., 1977, B16, 3177.
p. 5003.
3144. Kuroda Т., Suzuki T. — Denki Shikenjo Iho, 3178.
1958, 22, p. 509.
3145. Kurosawa Т., Hasegawa R., Yagihashi T. — 3179.
Trans. Jap. Inst. Metals, 1965, 6, p. 229.
3146. Kurti N., Simon F. — Proc. Roy. Soc. London, 3180.
1935, A151, p. 610.
3147. Kusch P. — Phys. Rev., 1936, 49, p. 217. 3181.
3148. Kusch P., Hessel M. M. — J. Mol. Spectrosc,
1969, 32, p. 181. 3182.
3149. Kusch P., Hessel M. M. — J. Chem. Phys., 1975,
63, p. 4087. 3183.
3150. Kusch P., Hessel M. M. — J. Chem. Phys., 1977,
67, p. 586. 3184.
3151. Kusch P., Hessel M. M. — J. Chem. Phys., 1978,
68, p. 2591. 3185.
3152. Kutscher J., Schneider A. — Z. anorg. und allg.
Chem., 1971, 386, S. 38. 3186.
3153. Kutzelnigg W-, Staemmler V., Gelus M. — Chem.
Phys. Lett., 1972, 13, p. 496. 3187.
3154. Kuwamoto H., Otsuka N., Sato H. — J. Solid
State Chem., 1981, 36, p. 133. 3188.
3155. Kvande H., Linga H., Motzfeldt K., Wahl-
beck P. G. — Acta chem. scand., 1979, A33, p. 281. 3189.
3156. Kybett B. D., Margrave J. L. — Tech. Rept 3190.
AFML-TR-65-123. Rice Univ., Aug. 1965. 3191.
3157. Lacombe P., Cizeron G. — Mem. sci. Rev. met.,
1960, 57, p. 179. 3192.
3158. Ladenburg R., Minkowski R. — Z. Phys., 1921,
6, S. 153. 3193.
3159. Ladenburg R., Thiele E. — Z. phys. Chem.
Leipzig, 1930, B7, S. 161. 3194.
3160. Laemmel R. — Ann. Phys., 1905, 16, S. 551.
3161. Lagergren S., Magneli A. — Acta chem. scand., 3195.
1952, 6, p. 444.
3162. Lagerqvist A. — Mem. Soc. roy. sci. Liege, 1957, 3196.
18, p. 550. 3197.
3163. Lagerqvist A., Huldt L. — Ark. fys., 1957, 12,
s. 491. 3198.
3164. Lagerqvist A., Selin L. E. — Naturwissenschaften,
1955, 42, S. 65. 3199.
3165. Lagerqvist A., Selin L. E. — Ark. fys., 1957, 11,
s. 429. 3200.
3166. Lagerqvist A., Selin L. E. — Ark. fys., 1957, 12,
s. 553. 3201.
3167. Lagerqvist A., Uhler U., Barrow R. F. —Ark. fys.,
1954, 8, s. 281. 3202.
3168. LaGrandel L. D., Dykslra L. J., Dixon J. M-,
Merten U. — J. Phys. Chem., 1959, 63, p. 2035. 3203.
3169. Lakkis S., Schlenker C, Chakraverty В. К., Bu-
der R., Marezio M. — Phys. Rev., 1976, B14, 3204.
p. 1429.
3170. Lambertson W. A., Handwerk J. H. — US AEC 3205.
Argonne Nat. Lab. Rept ANL-5053. Lemont
A11.), 1956. 3206.
3171. Lambertson W- A., Mueller M. H. — J. Amer.
Ceram. Soc, 1953, 36, p. 329. 3207.
3172. Lambertson W. A., Mueller M. H., Gunzel F. H. — 3208.
J. Amer. Ceram. Soc, 1953, 36, p. 397.
3173. Landon G. J., Ubbelohde A. R. — Proc. Roy. 3209.
Soc. London, 1957, A240, p. 160.
3174. Landspersky H., Sedldkova, L., Jakes D. — J. Appl. 3210.
Chem., 1964, 14, p. 559.
Lang J. I. — In: Thermodynamic and transport
properties of gases, liquids and solids: Symp. on
Thermal Properties. N. Y.: McGraw-Hill, 1959,
p. 405.
Lange E., Durr F. — Z. Elektrochem., 1926, 32,
S. 85.
Lange E., Dwr F. — Z. phys. Chem. Leipzig,
1926, 121, S. 361.
Lange E., Eichler A. — Z. phys. Chem. Leipzig,
1927, 129, S. 285.
Lange E., Martin W. — Z. phys. Chem.,
Leipzig, 1937, A180, S. 233.
Lange E., Monheim J. — Z. Elektrochem., 1929,
35, S. 29. /
Lange E., Monheim J. — Z. phys. Chem.
Leipzig, 1930, A150, S. 349.
Lange E., Rounsefell E. O. — Unpublished paper
(см.: Z. Elektrochem., 1930, 36, S. 777).
Lange E., Schwartz G. — Z. phys. Chem. Leipzig,
1928, 133, S. 129.
Lange E., Streeck H. — Z. phys. Chem. Leipzig,
1931, A157, S. 1.
Lange F. — Z. phys. Chem. Leipzig, 1924, 110,
S. 343.
Langer S., Blankeship F. F. — J. Inorg. and Nucl.
Chem., 1960, 14, p. 26.
Langmuir D. В., Matter L. — Phys. Rev., 1939,
55, p. 748.
Langmuir D. В., Matter L. — Phys. Rev., 1939,
55, p. 1138.
Langmuir /. — Phys. Rev., 1913, 2, p. 329.
Langmuir I. _ Phys. Rev., 1915, 6, p. 138.
Langmuir I., Kingdon К. Н. — Phys. Rev., 1923,
21, p. 80.
Langmuir I., Kingdon К. Н. — Proc. Roy. Soc.
London, 1925, A107, p. 61.
Langmuir I., Kingdon К. Н. — Phys. Rev., 1929,
34, p. 129.
Langmuir I., Mackay G. M. J. — Phys. Rev.,
1914, 4, p. 377.
Lantelme F., Pauly L. — С. г. Acad. sci., 1959,
249, p. 677.
Laporte O., Mack J. E— Phys. Rev., 1943, 63, p. 246.
Larson A. C, Roof R. В., Cromer Don T. — Act»
crystallogr., 1964, 17, p. 555.
Latimer W. M. — J- Amer. Chem. Soc, 1951,
73, p. 1480.
Latimer W. M-, Jolly W. L. — J. Amer. Chem.
Soc, 1953, 75, p. 1548.
Latta R. E., Duderstadt E. C, Fryxell R. С —
J. Nucl. Mater., 1970, 35, p. 345.
Latta R. E., Fryxell R. E. — Trans. Amer. Nucl.
Soc, 1965, 8, p. 376.
Latta R. E., Fryxell R. E. — J. Nucl. Mater.,
1970, 35, p. 195.
Lau K. H., Cubicciotti D., Hildenbrand D. L. —
J. Electrochem. Soc, 1979, 126, p. 490.
Lauchlan L. J., Brom J. M-, Jr., Broida H. P. —
J. Chem. Phys., 1976, 65, p. 2672.
Laud В. В., Kalsulkar D. R. — Indian J. Phys.,
1968, 42, p. 61.
Lauder J., Towler C, Wuth T. — Adv. Mass
Spectrom., 1971, 5, p. 379.
Laun D. D. — J. Res. NRS, 1964, A68, p. 207.
Laun D. D., Corliss С. Н. — J. Res. NBS, 1968,
A72, p. 609.
Laurent J. F., Bernard J. — Phys. and Chem.
Solids, 1957, 3, p. 7.
Laveissiere J. — Bull. Soc. frang. mineral, et cri-
stallogr., 1967, 90, p. 304.
572
Литература
3211. Leadbetter A. /., Settatree G. R. — Proc. Phys. 3248.
Soc. London. (Solid State Phys.), 1969, 2, p. 385. 3249.
3212.- Leary H. /., Jr., Rooney T. A., Cater E. D., Fried-
rich H. B. — High Temp. Sci., 1971, 3, p. 433.
3213. Leask J. F., Roberts L. E. J., Walter A. /., 3250.
Wolf W. P. — J. Chem. Soc, 1963, p. 4788.
3214. Lebeau P. — Ann. chim. phys., 1905, 6, p. 422. 3251.
3215. Lechner F. — Wien Berichte, 1932, 141, S. 291.
3216. Lecomte J. — Boll. sci. Fac. chim. ind. Bologna, 3252.
1963, 21, p. 51.
3217. Lee С A., Fabricand B. P., Carlson R. 0., 3253.
Rabi J. J. — Phys. Rev., 1953, 91, p. 1395.
3218. Lee С S., Lee D. J., Bonilla С F. — Nucl. Eng. 3254.
and Des., 1969, 10, p. 83.
3219. Lee J. A., Mardon P. G. — In: Metal Plutonium / 3255.
Ed. A. S. Coffinberry, W. N. Miner. Chicago:
Chicago Univ. Press, 1961, p. 133. 3256.
3220. Lee J. A., Mendelssohn K., Sutcliff P. W. — Proc.
Roy. Soc. London, 1970, A317, p. 303. 3257.
3221. Lee J. A ., Sutcliff P. W., Mendelssohn K. — Phys.
Lett., 1969, A30, p. 106. 3258.
3222. Lee V., Mohan В. Н. —"J. Chem. Phys., 1965,
42, p. 2893. 3259.
3223. Leffler A. J., Wiederhorn N. M J. Phys. Chem.,
1964, 68, p. 2882. 3260.
3224. Lefkowitz /., Dowell M. В., Shields M.A. — J.
Solid State Chem., 1975, 15, p. 24. 3261.
3225. Lehrman A., Breslau D. S. — J. Amer. Chem.
Soc, 1938, 60, p. 873.
3226. Lehtonen L. — Soc. sci. Fennicae, Comment. 3262.
Phys.-Math., 1922, 1, p. 1.
3227. Leibowitz L., Chasanov M. G., Mishler L. W. — 3263.
Trans. Met. Soc. AIME, 1969, 245, p. 981. 3264.
3228. Leibowitz L., Chasanov M- G., Mishler L. W., 3265.
Fischer D. F. — J. Nucl. Mater., 1971, 39, p. 115.
3229. Leibowitz L., Mishler L. W., Chasanov M. G. — 3266.
J.Nucl. Mater., 1969, 29, p. 356. 3267.
3230. Leiser D. В., Whittemore O. J. — J. Amer. Ceram.
Soc, 1967, 50, p. 60.
3231. Lemley А. Т., Lagowski J. J. — J. Phys. Chem.,
1974, 68, p. 708. 3268.
3232. Lengyel B. — Naturwissenschaften, 1933, 21, S. 848.
3233. Lesourd J. В., Vallet C, Doucet I. — J. Electro-
anal. Chem. Interfacial Electrochem., 1973, 48 A), 3269.
p. 99.
3234. Lester J. E., Samorjai G. A. — J. Chem. Phys., 3270.
1968, 49, p. 2940.
3235. Leston В. В., Taylor A. R. — U. S. Bur. Mines. 3271.
„ Rept Invest., 1965, N 6583. 3272.
3236. Leupold H. A., Boorse H. A. — Phys. Rev., 1964, 3273.
A134, p. 1322. 3274.
3237. Leupold H. A., lafrate G. J., Rothwarf F., Bres-
lin J. Т., Edmiston D., Аи Coin T. R. — J. Low 3275.
Temp. Phys., 1977, 28, p. 241.
3238. Leute V. — Z. phys. Chem. (BRD), 1966, 48, S. 307. 3276.
3239. Levi H. — Diss. В., 1934.
3240. Levin E. M. — J. Amer. Ceram. Soc, 1965, 48, 3277.
p. 491.
3241. Levin E. M., Benedict J. Т., Sciarello J. P., Mon- 3278.
sour S. — J. Amer. Ceram. Soc, 1973, 56, p. 427.
3242. LevinsonL. S. — J. Chem. Phys., 1964, 40, p. 3584. 3279.
3243. Levinson L. S. — J. Nucl. Mater., 1966, 19, p. 50.
3244. Levitski V. A., Chentson V. N., Scolis Yu. Ya., 3280.
Gerasimov Ya. I. — Rev. int. hautes temp, et
refract., 1980, 17, p. 82. 3281.
3245. Levy J. A., Taylor J. C, Welson P. W. — J.
Inorg. and Nucl. Chem., 1977, 39, p. 1989. 3282.
3246. Lew H. — Canad. J. Phys., 1964, 42, p. 1004.
3247. Lew H., Morris D., Geiger F. E., Eisinger J. T. — 3283.
Canad. J. Phys., 1958, 36, p. 171.
Lewis L. - Z. Phys., 1931, 69, S. 786.
Lewis W. В., Asprey L. В., Jones L.H.,
McDowell D. S., Rabideau S. W., Zeltmann A. #.,
Paine P. T. — J. Chem. Phys., 1976, 65, p. 2707.
Li K. C, Stwalley W. C. — J. Mol. Spectrosc,
1978, 69, p. 294.
Li P. C, Devlin J. P. — J. Chem. Phys., 1968,
49, p. 1441.
Libby W. F. Rept A—1228. Aug. 29, 1944. Пит.
no: [492, с 301].
LideD.R., Cahill P., Gold J. P. — J. Chem. Phys.,
1964, 40, p. 156.
Lide D. R., Jr., Kuczkowski R. L. — J. Chem.
Phys., 1967, 46, p. 4768.
LideD. R., Jr., Matsumura Ch. — J. Chem. Phys.,
1969, 50, p. 3080.
Liempt J. A. M- — Z. anorg, und allg. Chem.,
1923, 129, S. 263.
Liempt J. A. M. — Recueil trav. chim., 1931,
50, p. 343.
Lien W. #., Phillips N. E. — Phys. Rev., 1960,
118, p. 958.
Lien W. #., Phillips N. E. — Phys. Rev., 1964,
A133, p. 1370.
Lierde W-, Pelsmaekers /., Lecocq-Robert A. —
J. Nucl. Mater., 1970, 37, p. 276.
Lietz J. — In: Hamburger Beitrage zur Ange-
wandten Mineralogie und Kristallphysik. В.:
Rose-Festschr., 1956, S. 229.
Linde R. K., DeCarli P. S. — J. Chem. Phys.,
1969, 50, p. 319.
Lindgren B. — J. Mol. Spectrosc, 1972, 43, p. 474.
Lindgren B. — J. Mol. Spectrosc, 1973, 48, p. 322.
Lindsay D. M-, Herschbach D. R., Kwiram A. —
J. Chem. Phys., 1974, 60, p. 315.
Linevsky M.J. — i. Chem. Phys., 1963, 38, p. 658.
Linevsky M- J. Spectroscopic studies of the
vaporization of refractory materials: Final Rept
AD609121. Philadelphia: Gen. Electr. Co, 1964,
Nov., 61 p.
Linevsky M- J. Spectroscopic studies of the
vaporization of refractory materials: Final Rept
AFML-TR-64-420, 1965.
Linevsky M. J. — NASA Accession N 65-31315.
Rept AD-467028, 1965.
Linga #., Motzfeldt K., Oye H. A. — Ber. Bunsen-
ges. phys. Chem., 1978, 82, S. 568.
Linton С — Canad. J. Phys., 1972, 50, p. 312.
Linton C. — J. Mol. Spectrosc., 1974, 50, p. 235.
Linton C. — J. Mol. Spectrosc., 1978, 69, p. 351.
Linton C, Broida H. P. — J. Mol. Spectrosc,
1977, 64, p. 382.
Linton C., Broida ff. P. — J, Mol. Spectrosc,
1977, 64, p. 389.
Linton C, Nicholls R. W. — J. Phys.: Atom, and
Mol. Phys., 1969, B2, p. 490.
Linton C, Singhal S. R. — J. Mol. Spectrosc,
1974, 51, p. 194.
Lipsett S. G., Johnson F. M. G., Maass O. — J.
Amer. Chem. Soc, 1927, 49, p. 925.
Lipsett S. G., Johnson F. M. G., Maass O. — J.
Amer. Chem. Soc, 1927, 49, p. 1940.
Lister M. W., Meyers N. F. — J. Phys. Chem.,
1958, 62, p. 145.
Litaka J. — Sci. Rept Tohoku Imp. Univ., 1919,
8, p. 99.
Litton F. B. — J. Electrochem. Soc, 1951,
98, p. 488.
Liu С H., Lieto L. R. — J. Chem. and Eng. Data,
1969, 14, p. 83.
573
Литература
3284. Liu H. P. D., Verhaegen G. — Bull. Soc. chim.
Belg., 1972, 81, p. 109.
3285. Liu Ming Biann, Wahlbeck P. G. — High Temp.
Sci., 1974, 6, p. 179.
3286. Liu Ming Biann, Wahlbeck P. G. — J. Chem.
Phys., 1975, 63, p. 1694.
3287. Livey D. Т. — J. Less-Common Metals, 1959, 1,
p. 145.
3288. Llewellyn D. R. — J. Chem. Soc, 1953, p. 28.
3289. Lloyd L., Wyatt P. A. H. — J. Chem. Soc, 1957,
p. 4262.
3290. Loasby R. — In: Plutonium I960: Proc. 2nd
Intern. Conf. Plutonium Metallurgy. Grenoble,
1960, p. 98.
3291. Lockwood С W. — Astrophys. J., 1969, 157,
p. 275.
3292. Long F. A., Davidson N. R. Цит. по: [3919].
3293. Loomis F. W. — Phys. Rev., 1927, 29, p. 607.
3294. Loomis F. W. — Phys. Rev., 1928, 31, p. 323.
3295. Loomis F. W., Arvin M. J. — Phys. Rev., 1933,
44, p. 126.
3296. Loomis F. W., Arvin M. J. — Phys. Rev., 1934,
46, p. 286.
3297. Loomis F. W., Kusch P. — Phys. Rev., 1934, 46,
p. 292.
3298. Loomis F. W., Nusbaum R. E. — Phys. Rev.,
1931, 38, p. 1447.
3299. Loomis F. W., Nusbaum R. E. — Phys. Rev.,
1932, 39, p. 89.
3300. Loomis F. W., Nusbaum R. E. — Phys. Rev.,
1932, 40, p. 380.
3301. Loomis F. W., Wood R. W. — Phys. Rev., 1928,
32, p. 223.
3302. LoopstraB. O. — Actacrystallogr., 1964,17, p. 651.
3303. Loopstra B. O. — J. Phys., 1964, 25, p. 429.
3304. LoopstraB. O. — J. Nucl. Mater., 1969, 29, p. 354.
3305. Loopstra В. О. — 3. Appl. Crystallogr., 1970, 3,
p. 94.
3306. Loopstra B. O., Cordfunke E. H. P. — Recueil
trav. chim., 1966, 85, p. 135.
3307. Loopstra B. O., Taylor J. C, Waugh A. B. — J.
Solid State Chem., 1977, 20, p. 9.
3308. Lorenzen C. /., Niemax K. — J. Quant. Spectrosc.
and Radiat. Transfer, 1979, 22, p. 247.
3309. Lorenzen С /., Weber K. #., Niemax K. — Opt.
Communs, 1980, 33, p. 271.
3310. Lorenzelli V., Gesmundo F. — Rend. sci. fis. mat:
e nat. Lincei, 1964, 37, p. 70.
3311. Losano L. — Gazz. chim. ital., 1935, 65, p. 855.
3312. Lounasmaa O. V., Sundstrdm L. J. — Phys. Rev.,
1967, 158, p. 591.
3313. Lovas F. J. — J. Phys. and Chem. Ref. Data,
1978, 7, p. 1445.
3314. Lovas F. J., Tiemann E. — J. Phys. and Chem.
Ref. Data, 1974, 3, p. 609.
3315. Lowater F. «— Proc. Phys. Soc. London, 1929,
41, p. 557.
3316. Lowater F. — Proc. Phys. Soc. London, 1932,
44, p. 51.
3317. Lowater F. — Philos. Trans. Roy. Soc. London,
1935, A234, p. 355.
3318. Lowenthal G. С — Austral. J. Phys., 1963, 16,
p. 47.
3319. Lowings M. G., McCurdy K. G., Hepler L. G. —
Thermochim. acta, 1978, 23, p. 365.
3320. Luce R. <?., Trischka J. W. — Phys. Rev., 1951,
83, p. 851.
3321. Luce R. G., Trischka J. W. — J. Chem. Phys.,
1953, 21, p. 105.
3322. Lumme P., Lahermo P., Tummavuori J. — Acta
chem. scand., 1965, 19, p. 2175.
3323. Lux #., Brandl F. — Z. anorg. und allg. CheiH.,
1963, 326, S. 25.
3324. Lyhamn L. E., Cyvin S. J. — Z. Naturforsch.,
1979, A34, S. 867.
3325. Lyhamn L.E., Cyvin S.J., Cyvin B.N., Brun-
voll J. — Z. Naturforsch., 1976, A31, S. 1589.
3326. Lynam P., Scurlock R. G., Wray E. M. — In:
Proc. IXth Intern. Conf., Columbus, Ohio, 1964,.
pt B. N. Y.: Plenum Press, 1965, p. 905.
3327. Lynch С. Т., Vahldiem F. W., Robinson L. B. —
J. Amer. Ceram. Soc, 1961, 44, p. 147.
3328. Lynch E.D., Handwerk J.H., Hoenig С L. —
J. Amer. Ceram. Soc, 1960, 43, p. 520.
3329. Lyon T. F. Condensation Evaporation Solids.
Proc. Intern. Symp. Ohio, 1962, Publ. 1964r
p. 435.
3330. Lyon W. L., Baily W. E. — U. S. General
Electric Co. Rept GEAP—4640, 1964.
3331. Lyon W. L., Baily W. E. — U. S. General
Electric. Co. Rept. GEAP—4878, 1965.
3332. Lyon W. L., Baily W. E. — Trans. Amer. Nucl.
Soc, 1965, 8, p. 376.
3333. Lyon W. L., Baily W. E. — J. Nucl. Mater.,
1967 22 v 332
3334. McChesney J. В., Guggenheim N. J. — J. Phys.
and Chem. Solids, 1969, 30, p. 225.
3335. Mclnnes D.A., Cattow C. R. A. — J. Nucl.
Mater., 1980, 89, p. 354.
3336. Maclead A.C. — J. Chem. Thermodyn., 1972,
4, p. 699.
3337. Maclead A. C. — J. Chem. Soc. Faraday Trans. I,
1973, 69, p. 2026.
3338. Mader C. L. Ideal gas thermodynamic properties
of detonation products. — U. S. Atom. Energy
Commis. Rept AECU-4508, 1959.
3339. Maglic K., Herak R. — Rev. int. hautes temp.
et refract., 1970, 7, p. 247.
3340. Magnus A. — Phys. Z., 1913, 14, S. 5.
3341. Magnus A., Danz H. — Ann. Phys., 1926, 81,
5. 407.
3342. Magnus A., Holzmann H. — Ann. Phys., 1929,,
3, S. 585.
3343. Mah A. D. — J. Amer. Chem. Soc, 1954, 76,
p. 3363.
3344. Mah A. D. — J. Phys. Chem., 1957, 61, p. 1572.
3345. Mah A.D. — J. Amer. Chem. Soc, 1958, 80,
p. 3872.
3346. Mah A.D. — J. Amer. Chem. Soc, 1959, 8lr
p. 1582.
3347. Mah A.D. — U. S. Bur. Mines. Rept Invest.,
1962, N 5965.
3348. Mah A.D., Kelley K. K. — U. S. Bur. Mines..
Rept Invest., 1961, N 5858.
3349. Mah A. D., Kelley К. К., Gellert N. ?.,.
King E. G., O'Brien C. J. — U. S. Bur. Mines.
Rept Invest., 1957, N 5316.
3350. Mahanti P. С — Proc. Phys. Soc. London,
1935, 47, p. 433.
3351. Maier С G. — Bull Bur. Mines (USA), 1942,
N 436.
3352. Maier С G. — U. S. Mines. Techn. Paper, 1925,
N 360.
3353. Maier O. Diss. Deutsche Universitat, Prag, 1927.
3354. Majumdar A., Roy R. — J. Phys. Chem., 1965,
69, p. 1684.
3355. Majundar A. J., Rustum R.—'J. Inorg. and
Nucl. Chem., 1965, 27, p. 1961.
574
Литература
3356. Makansi M. M., Madsen M., Selke W. Н., Во-
nilla С. F. — J. Phys. Chem., 1956, 60, p. 128.
3357. Makansi M. M., Muendel С. #., Selke W. H. —
J. Phys. Chem., 1955, 59, p. 40.
3358. Malaspina L., Gigli R., Piacente V. — Gazz.
chim. ital., 1971, 101, p. 197.
3359. Malinovsky M. — Chem. zvesti, 1967, 21, s. 783.
3360. Malinovsky M., Kostenska I. — Chem. zvesti, 1974,
28, s. 509.
3361. Malter L., Langmuir D. B. — Phys. Rev., 1939,
55, p. 743.
3362. Mamantov G., Marassi R., Poulsen F. W.,
Springer S. E., Wiaux I. P., Huglen R.,
Smyrl N. R. — J. Inorg. and Nuel. Chem., 1979,
41, p. 260.
3363. Manchester F. D. — Canad. J. Phys., 1959, 37,
p. 525.
3364. Manneback С — Physica, 1963, 29, p. 769.
3365. Many A. — Phys. Rev., 1946, 70, p. 511.
3366. Maraine—Giroux C, Beuaziz R., Perez G. —
Rev. chim. miner., 1972, 9, p. 779.
3367. Marchal G. — ВиЦ. Soc. chim. France, 1929,
45, p. 88.
3368. Marchidan D. I., Ciopec M. — Rev. roum. chim.,
1970, 15, N 8, p. 1177.
3369. Marchidan D. I., Ciopec M. — Res. roum. chim.,
1970, 15, p. 1287.
3370. Marchidan D. I., Ciopec M. — In: 4me Conf.
intern, thermodyn. chim. Montpellier, 1975,
vol. 3, p. 55.
3371. Marchidan D. I., Gambino M. — Bull. Soc. chim.
France, 1966, N 6, p. 1954.
3372. Marchidan D. I., Pandele L. — Rev. roum. chim.,
1975, 20, p. 299.
3373. Marchidan D.I., Telea С— Rev. roum. chim.,
1968, 13, p. 1291.
3374. Marchidan D. I., Telea C. — Rev. roum. chim.,
1969, 14, p. 1361.
3375. Marchidan D. I., Vasu L. — Rev. roum. chim.,
1972, 17, p. 1649.
3376. Marchidan D. I., Vasu L. — Rev. roum. chim.,
1973, 18, p. 1295.
3377. Marden J. W., Rentschler H. С — Ind. and Eng.
Chem., 1927, 19, p. 97.
3378. Mardon P. G., Nichols J. L., Pearce J. H.,
Poole D. M. — Nature, 1961, 189, p. 566.
3379. Marezio M., Dernier P. D. — J. Solid State
Chem., 1971, 3, p. 340.
3380. Marezio M., Dernier P. D., McWhan D. В.,
Remeika J. P. — Mater. Res. Bull., 1970, 5,
p. 1015.
3381. Marezio M., McWhan D.B., Dernier P.D.,
Remeika J. P. — Phys. Rev. Lett., 1972, 28,
p. 1390.
3382. Marezio M., McWhan D.B., Derner P.D.
Remeika J. P. — J. Solid State Chem., 1973, 6,
p. 213.
3383. Marezio M., Plettinger H. A., Zachariasen W. H. —
Acta crystallogr., 1963, 16, p. 594.
3384. Marezio M., Remeika J. P. — J. Phys. Chem.
Solids, 1965, 26, p. 2083.
3385. Marezio M., Remeika J. P. — J. Chem. Phys.,
1966, 44, p. 3348.
3386. Margheritis C, Flor G., Sinistri С — Z. Natur-
forsch., 1973, A28, S. 1329.
3387* Margrave J. L. — High Temp.-High Pressur.,
1970, 2, p. 583.
3388. Mariani E., Dikant J., Jonova A. — Phys.
status solidi, 1975, A31, p. 749.
3389. Marinder B. O. — Ark. kemi, 1962, 19, s. 435.
3390. Marion F., Choain-Maourin C. — Chim. et ind.r
1962, 88, p. 483.
3391. Mark S. D. — J. Amer. Ceram. Soc, 1959, 42,
p. 208.
3392. Markin T. L., Bones R. J., Wheeler V. J. —
Proc. Brit. Ceram. Soc., 1967, N 8, p. 51.
3393. Markin T. L., Roberta L. E., Walter A. — In:
Thermodynamics Nuclear Materials Proc. Symp.-
Vienna, 1962. Vienna: IAEA, 1963, p. 693.
3394. Marple D.T.F., Triichka J. W. — Phys. Rev.,
1956, 103, p. 597.
3395. Mttrples J. A. C. — In: Plutonium 1975 and
other Aetinides. Proc. 5th Intern. Conf., Baden-
Baden, 1975. Amsterdam etc., 1976, p. 353.
3396. Marshall B. J., Kunkel J. R. — 1. Appl. Phys.,
1969, 40, p. 5191.
3397. Marshall W. L., Loprest F. L., Secoy С. Н. —
J. Amer. Chem. Soc, 1958, 80, p. 5646.
3398. Martin A. E., Edwards R. K. — J. Phys. Chem.,^
1965, 69, p. 1788.
3399. Martin B.D., Zych D.A., Heer С V. — Phys.
Rev., 1964, 135, p. 671.
3400. Martin D. L. — Philos. Mag., 1955, 46, p. 751.
3401. Martin D. L. — Physica, 1959, 25, p. 1193.
3402. Martin D. L. — Proc. Roy. Soc. London, I960,.
A254, p. 433.
3403. Martin D. L. — Proc. Roy. Soc. London, I960,
A254, p. 444.
3404. Martin D. L. — Phys. Rev., 1961, 124, p. 438.
3405. Martin D. L. — Proc. Roy. Soc. London, 1961,
A263, p. 378.
3406. Martin D. L. — Canad. J. Phys., 1962, 40,
p. 1166.
3407. Martin D. L. — Proc. Phys. Soc. London, 1964,
83, p. 99.
3408. Martin D. L. — Phys. Rev., 1967, 154, p. 571.
3409. Martin D. L. — Canad. J. Phys., 1970, 48, p. 1327.
3410. Martin T. P., Schaber H. — J. Chem. Phys.,
1978, 68, p. 4299.
3411. Martin W.C., Zalubas R., Hagan L. Atomic
energy levels. The rare-earth elements. — U. S.
NBS NSRDS-60. Wash., 1978.
3412. Marujama N., Sawada S. — J. Phys. Soc. Jap.,
1965, 20, p. 811.
3413. Masaki N. — J. Appl. Crystallogr., 1974, 7,
p. 247.
3414. Masi J. F. — J. Chem. Phys., 1949, 17, p. 755.
3415. Mathews J. H., Germann A. F. O. — J. Phys.
Chem., 1911, 15, p. 73.
3416. Mathewson С. Я., Spire E., Semans С. Н. —
Trans. Amer. Soc. Steel Treating, 1932, 20,
p. 357.
3417. Mathur B. P., Rothe E. W. — J. Chem. Phys.,
1977, 67, p. 377.
3418. Matignon С — Ann. chim. phys., 1906, 8, p. 433.
3419. Matignon C, Marshal G. — C. r. Acad. sci.,
1920, 170, p. 232.
3420. Matossi F., Hohler V. — Z. Naturforsch., 1967,
A22, S. 1525.
3421. Matsui M., Kambara S., Miyamura K. — J. Soc.
Chem. Ind. Jap., 1932, 35, p. 227.
3422. Matsui M., Nakata S. — J. Chem. Soc, Jap..
Ind., 1929, 32, p. 79.
3423. Matsui M., Nakata S., Akayama K., Bito K. —
J. Chem. Soc. Jap. Ind., 1928, 31, p. 53.
3424. Matsui M., Oka S. — J. Chem. Soc. Jap. Ind.,
1929, 32, p. 85.
3425. Matsumura Chi, Lide D. R., Jr. — J. Chem.
Phys., 1969, 50, p. 71.
575
Литература
3426. Matsushima T. — Denki Kagaku Oyobi Kogyo 3464.
Butsuri Kagaku, 1969, 37, p. 778.
3427. Matsuyama E. — Nature, 1934, 133, p. 567. 3465.
3428. Matsuyama E. — Sci. Repts Tohoku Univ., 1934,
23, p. 208. 3466.
3429. Mattias В. Т., Geballe T. #., Corenzwit E., 3467.
Hull G. W. — Phys. Rev., 1963, 129, p. 1025.
3430. Maucherat M. — С. г. Acad. sci., 1939, 208, p. 499. 3468.
3431. Maucherat M. — J. phys. et radium, 1939, 10,
p. 441. 3469.
3432. Mayer H. — Z. Phys., 1931, 67, S. 240.
3433. Mayer J. E., Wintrier I. H. — J. Chem. Phys.,
1938, 6, p. 301. 3470.
3434. Maykuth D.J., Ogden H. R., Jaffe R. I. —
J. Metals, 1953, 5, p. 225.
3435. Maylotte D. H., Peters R. L. St., Messmer R. P. — 3471.
Chem. Phys. Lett., 1976, 38, p. 181. 3472.
3436. Mazandarany F. N., Pehlke R. D. — J. Electro-
chem. Soc, 1974, 121, p. 711.
3437. Mazieres С — Ann. chim. France, 1961, 6, p. 575. 3473.
3438. McAdie H. G. — J. Therm. Anal., 1971, 3, p. 79.
3439. McBride B. J., Heimal S., Ehlers J. G., Gor- 3474.
don S. Thermodynamic properties to 6000 К for
210 substances involving the first 18 elements, 3475.
NASA-SP-3001. Wash., 1963.
3440. McCabe С L., Hudson R. G., Paxton H. W. — 3476.
Trans. Met. Soc. AIME, 1958, 221, p. 102.
3441. McCaldin J. O., Duwer P. — J. Metals, 1954, 3477.
6, p. 619.
3442. McCart В., Lander G. H., AldredA. T. — J. Chem. 3478.
Phys., 1981, 74, p. 5263.
3443. McClaine L.A. — XJ.S. Wright Air Develop. 3479.
Div. Rept WADD-TR-ASD-TDR-62-204. Part III.
Wright-Patterson AFB, Ohio, 1964.
3444. McCollum D.C., Jr., Silsbee H. B. — Phys. 3480.
Rev., 1962, 127, p. 119.
3445. McConville Т., Serin B. — Revs Mod. Phys.,
1964, 36, p. 112. 3481.
3446. McCormack J., McCaffery A. J. — Chem. Phys.
Lett., 1979, 64, p. 98. 3482.
3447. McCory L. D., Paule R. C, Margrave J. L. — 3483.
J. Phys. Chem., 1963, 67, p. 1087. 3484.
3448. McDiarmid R. — J. Chem. Phys., 1976, 65, p. 168.
3449. McDonald H. J., Seltz H. — J. Amer. Chem. 3485.
Soc, 1939, 61, p. 2405.
3450. McDonald J. D., Margrave J. L. — J. Inorg. 3486.
and Nucl. Chem., 1968, 30, p. 665.
3451. McDowell R. S., Asprey L.B. — l. Mol. Spec-
trosc, 1973, 48, p. 254. 3487.
3452. McDowell R. S., Asprey L. В., Paine R. T. —
J. Chem. Phys., 1974, 61, p. 3571. 3488.
3453. McEwan M. J., Phillips L. F. — Combust, and
Flame, 1967, 11, p. 63. 3489.
3454. McGinnety J. A.— Acta crystallogr., 1972, B28,
p. 2845. 3490.
3455. McGlynn S. P., Smith J. K. — J. Mol. Spectrosc,
1961, 6, p. 188. 3491.
3456. McGlynn S. P., Smith J. K. — J. Mol. Spectrosc,
1961, 6, p. 164. 3492.
3457. McGraw J. — J. Amer. Chem. Soc, 1931, 53,
p. 3683.
3458. Mclntyre N. S., Thompson K. R., Weltner W. — 3493.
J. Phys. Chem., 1971, 75, p. 3243.
3459. McKellar A. — Phys. Rev., 1933, 44, p. 155. 3494.
3460. McLaren A. C. — Rev. Pure and Appl. Chem.,
1962, 12, p. 54. 3495.
3461. McLean A. D. — 1. Chem. Phys., 1963, 39, p. 2653.
3462. McLean A. D. — J. Chem. Phys., 1964, 40, p. 243.
3463. McLennan J. C, Ainslie D. S. — Proc. Roy.
Soc. London, 1923, 103, p. 304. 3496.
McMaster O. D., Larsen W. L. — J. Less-Common
Metals, 1961, 3, p. 312.
McPherson D. J. — Trans. Amer. Soc. Metals,
1952, 44, p. 1029.
McQuillan A.D. —Nature, 1949, 164, p. 24.
McQuillan A.D. — J. Inst. Metals, 1950, 78,
p. 249.
McQuillan A.D.—Proc. Roy. Soc. London,
1950, A204, p. 309.
McWhan D. В., Remeika J. P., Bader S. D.,
Triplett В. В., Phillips N. E. — Phys. Rev.,
1973, B7, p. 3079.
McWhan D.B., Remeika J. P., Rice T.M.,
Brinkman W. F., Malta J. P., Menth A. — Phys.
Rev. Lett., 1971, 27, p. 941.
Mees R. — Цит. по: [1891], S. 259.
Meggers W. F., Moore Ch. E. The First Spectrum
of Hafnium (Hfl). — U. S. NBS Monograph 153,
Wash., 1976.
Meggers W. F., Russel H. N. — J. Res. NBS,
1936, 17, p. 125.
Meggers W. F,, Wheeler J.A. — J. Res. NBS,
1931, 6, p. 239.
Meggers W. F., Wheeler J. A. — Proc. Roy. Soc.
London, 1931, A133, p. 207.
Mehrotra P. K., Hoffman R. — Theor. chim.
acta, 1978, 48, p. 301.
Meissner W., Franz H. — Z. Phys., 1930, 63,
S. 558.
Meissner W., Franz H., Westerhoff F. — Ann.
Phys., 1933, 17, S. 593.
Mellor A.M., Glassman I. — Western States
Sect. Combust. Inst. Paper WSS-cI N 66—33,
1966, 25 p.
Mellor J. W. Comprehensive treatise on inorganic
and theoretical chemistry. L.; Longman, Green
and Co., 1929, vol. 9.
Menary J. W., Vbbelohde A. R., Woodward J. —
Proc. Roy. Soc London, 1951, A208, p. 158.
Mendelssohn K. — Nature, 1941, 148, p. 316.
Mendiola J. — Rev. cienc. apl., 1966, 20, p. 487.
Menis O., Sterling J. T. — U. S. NBS Spec.
Publ., N 338, 1973, p. 61.
Meredith R. E., Jenney J. A. — J. Chem. Phys.,
1963, 39, p. 3127.
Merritt R. R., Hyde B. G., Bursill L. A.,
Philp D. K. — Philos. Trans. Roy. Soc. London,
1973, A274, p. 627.
Mesnard G., Uzan R., Cabaud B. — Rev. phys.
appl. France, 1966, 1, p. 123.
Messer C. E., Fasolino L. G., Thalmayer С. Е. —
J. Amer. Chem. Soc, 1955, 77, p. 4524.
Messer C. E., Levy I. S. — Inorg. Chem., 1964,
4, p. 543.
Messer C. E., Mellor J., Krol J. A., Levy I. S. —
J. Chem. and Eng. Data, 1961, 6, p. 328.
Messier D. R. — J. Amer. Ceram. Soc, 1968,
51, p. 710.
Metcalf R. P. — U. S. AEC. Oak Ridge Nat.
Lab. Rept ORNL-1375. Oak Ridge (Tennesse),
1952, p. 90.
Meyer G., Oosterong J. F., Roo J. L. — Recueil
trav. chim., 1959, 78, p. 412.
Meyer W., Rosmus P. — J. Chem. Phys., 1975,
63, p. 2356.
Mezaki R., Tilleux E. W., Jambois T. F.,
Margrave J. L. Advances in thermophys. properties
at extreme temperature and pressures Ed/
5. Gratch. N. Y. : ASME, 1965, p. 138.
Michaud M. — С. г. Acad. sci., 1961, 253, p. 1947.
576
Литература
3497. Michaud M. — С. г. Acad. sci., 1967, С264,
p. 1939.
3498. Michaud M. — Rev. chim. miner., 1968, 5,
p. 89.
3499. Michaud M., Erb A., Ado G. — С. г. Acad. sci.,
1971, C272, p. 842.
,3500. Miekk-Oja H. — Ann. Acad. sci. Fennicae, Ser.
AI, 1941, N 7, 65p.
3501. Miki H., Kakumoto K., I no Т., Kodera S., Kaki-
noki J. — Acta crystallogr., 1980, A36, p. 96.
3502. Miller J. C, Allison S. W., Andrews L. — J. Chem.
Phys., 1979, 70, p. 3524.
3503. Miller R. C, Kusch P. — J. Chem. Phys., 1956,
25, p. 860.
3504. Miller R. C, Kusch P. — J. Chem. Phys., 1957,
27, p. 981.
3505. Miller R. E., Getty R. R., Treuil K. L.,
Leroi G. E. — J. Chem. Phys., 1969, 51, p. 1385.
3506. Miller W. J. — J. Chem. Phys., 1972, 57, p. 2354.
3507. Miller W. J. — In: NBS Spec. Publ. 561. Wash.,
1979, p. 433.
3508. Milligan D. E., Jacox M. E. — J. Chem. Phys.,
1971, 55, p. 3404.
3509. Milman Т., Bouaziz R. — Ann. chim. France,
1968, 3, p. 311.
3510. Milne Th., Klein Л.М. — 1; Chem. Phys.,
1960, 33, p. 1628.
3511. Minkowski R., Mulenbruch W. — Z. Phys., 1930,
63, S. 198.
3512. Minomura S., Nagasaki H. — J. Phys. Soc. Jap.,
1964, 19, p. 131.
3513. Mirabel P. — С r. Acad. sci., 1971, C272, p. 534.
3514. Mishchenko K. P. — Z. Elektrochem., 1930, 36,
S. 777.
3515. Mitchell R. E., Cobble J. W. — J. Amer. Chem.
Soc, 1964, 86, p. 5401.
3516. Mitsuhashi Т., Kleppa O. J. — J. Amer. Ceram.
Soc, 1979, 62, p. 356.
3517. Mitsuhashi Т., Takahashi Y. — J. Ceram. Soc.
Jap., 1980, 88, p. 305.
3518. Mixter W. G. — Amer. J. Sci., 1907, 24, p. 130.
3519. Mixter W. G. - Amer. J. Sci. """ "
3520. Mixter W. G. — Amer. J. Sci.
3521. Mixter W. G. — Amer. J. Sci.
3522. Mixter W. G. — Amer. J. Sci.
3523. Mixter W. G. — Z. anorg. und allg. Chem., 1912,
74, S. 122.
3524. Mixter W. G. — Z. anorg. und allg. Chem., 1912,
78, S. 221.
3525. Mixter W. G. — Z. anorg. und allg. Chem., 1912,
78, S. 222.
3526. Mixter W. G. — Amer. J. Sci., 1914, 37, p. 526.
3527. Mixter W. G., Dana E. S. — Justus Liebigs Ann.
Chem., 1873, 169, p. 388.
3528. Miyake'M., Minato I., Iwai S. — In: Proc. 5th
Intern. Conf. Therm. Anal./Ed. H. H. Chihara.
L., 1977, p. 170.
3529. Miyake M., Morikawa #., Iwai S. — Acta
crystallogr., 1980, B36, p. 532.
3530. Moers K. — Z. anorg. und allg. Chem., 1920,
117, S. 179.
3531. Mohamad N. S. — Indian J. Pure and Appl.
Phys., 1978, 16, p. 646.
3532. Mohamad N. S. — Intern. J. Quantum Chem.,
1979, 15, p. 769.
3533. Mohan S. — Indian J. Pure and Appl. Phys.,
1977, 15, p. 738.
3534. Mohanty G. P., Fiegel L. J., Healy J. H. —
J. Phys. Chem., 1964, 68, p. 208.
1908, 26, p. 125.
1909, 27, p. 393.
1910, 29, p. 488.
1912, 34, p. 141.
3535. Mohanty S. R., Upadhyay S. R. — Indian J.
Chem., 1965, 3, p. 285.
3536. Moles E., Perez-Vitoria A. -»¦ An. quim. Real.
soc. esp. fis. у quim., 1932, 30, p. 99.
3537. Momin A. C, Deshpande V. V., Karkhana-
vala M. D. — In: Proc. Chem. Symp., Aligarch.,
1972, vol. 2, p. 341.
3538. Momin A. C, Deshpande V. V., Karkhana-
vala M.D. — J. Nucl. Mater., 1973, 49, p. 98.
3539. Momin A. C, Mathews M. D., Karkhana-
vala M.D. — Indian J. Chem., 1971, 9, p. 582.
3540. Mondain-Monval P. — С. г. Acad. sci., 1923,
176, p. 301.
3541. Mondain-Monval P. — С. г. Acad. sci., 1923,
176, p. 889.
3542. Mondain-Monval P. — Ann. chim. (Fr.), 1925,
3, p. 72.
3543. Monnier G. — Bull. Soc. chim. France, 1957,
N 6, p. 766.
3544. Montero S., Schmolz R., Haussiihl S. — J. Raman
Spectrosc, 1974, 2, p. 101.
3545. Montgomery H., Pells G. P. — Proc. Phys. Soc.
London, 1961, 78, p. 622.
3546. Montgomery R. L., Hubert T. D. — U. S. Bur.
Mines. Rept Invest., 1960, N 5659.
3547. Montgomery R. L., Melaugh R. A., Lau Ch.,
Meier G. H., Chan H. H., Rossini F. D. — J. Chem.
Thermodyn., 1977, 9, p. 915.
3548. Montgomery R. L., Melaugh R.A., Lau Ch.,
Meier G. #., Grow R. Т., Rossini F. D. — J. Chem.
and Eng, Data, 1978, 23, p. 245.
3549. Moore Ch. E. Ionization potentials and ionization
limits derived from the analyses of optical
spectra. NSRDS-NBS-34. Wash., 1970.
3550. Moore Ch. E. Atomic energy levels. NSRDS-NBS
35. Wash., 1971, vol.1—3.
3551. Moore G. E., Kelley K. K. — J. Amer. Chem.
Soc, 1942, 64, p. 2949.
3552. Moore G. E., Kelley К. К. — J. Amer. Chem.
Soc, 1947, 69, p. 2105.
3553. Moore J. C, Devlin J. P. — J. Chem. Phys.,
1978, 68, p. 826.
3554. Moose J. E., Parr S. W. — 1. Amer. Chem. Soc,
1924, 46, p. 2656.
3555. Morales V. — An. quim Real., soc. esp. fis.
у quim., 1963, A59, p. 3.
3556. Moran T. J., Trischka J.W. — J. Chem. Phys.,
1961, 34, p. 923.
3557. Moreau R. — Bull. soc. roy. sci. Liege, 1963, 32,
p. 252.
3558. Morey G. W., Bowen N. L. — J. Phys., Chem.,
1924, 28, p. 1167.
3559. Morey G. W., Burlew J. S. — J. Phys. Chem.,
1964, 68, p. 1706.
3560. Morey G. W., Fenner С N. — J. Amer. Chem.
Soc, 1971, 39, p. 1173.
3561. Morey G. W., Merwin H. E. — J. Amer. Chem.
Soc, 1936, 58, p. 2248.
3562. Morin F. J. — Phys. Rev. Lett., 1959, 3, p. 34.
3563. Morin F. /., Maita J. P. — Phys. Rev., 1963,
129, p. 1115.
3564. Morizur G., Radenac A., Cretenet J. С — High
Temp.-High Pressur., 1976, 8, p. 113.
3565. Morlin Z. — Acta phys Acad. sci. hung., 1966,
21, p. 137.
3566. Morlin Z., Tremmel J. — Acta phys. Acad. sci.
hung., 1966, 21, p. 129.
3567. Morrison J. — In: Nat. Symp. Vacuum Technol.
Trans. L.: Pergamon Press, 1960, p. 291.
577
Литература
3568. Morrison J. A., Patterson D. — Trans. Faraday
Soc, 1956, 52, p. 764.
3569. Morrison J.A., Patterson D., Dugdale J.S.—
Canad. J. Chem., 1955, 33, p. 375.
3570. Mosnier A. — Ann. chim. phys., 1897, 12, p. 374.
3571. Motzfeldt K. — J. Phys. Chem., 1955, 59, p. 139.
3572. Moyer D. F. — J. Phys. Chem. Solids., 1965,
26, p. 1459.
3573. Mraw S. C, Book R. J., Staveley L. A. K. —
J. Chem. Thermodyn., 1978, 10, p. 359.
3574. Mraw S. C, Staveley L. A. K. — J. Chem.
Thermodyn., 1976, 8, p. 1001.
3575. Mukaibo Т., Naito K., Sato K., Uchijma T. —
In: Thermodynamics Nuclear Materials: Proc.
Symp., Vienna, 1962. Vienna: IAEA, 1963, p. 723.
3576. Mukerji J. — Trans. Indian Ceram. Soc, 1971,
30, p. 57.
3577. Mukhtar A., Winand R. — C. r. Acad. sci., 1965,
260, p. 3674.
3578. Mulay L. N., Danley W. J. — J. Appl. Phys.,
1970, 41, p. 877.
3579. Mulford R. N. R.— In: Thermodynamics: Proc.
Symp. Vienna, 1965. Vienna: IAEA, 1966, vol.
1, p. 231.
3580. Mulford R. N. R., Lamar L. E. — In: Plutonium,
1960/ Ed. E. Grivson, W. B. H. Lord, R. D.
Fowler. L.: Cleaver-Hume Press Ltd, 1961, p. 411.
3581. Muller H. — Z. Kristallogr., 1914, 53, S. 511.
3582. Muller J. A. — Ann. chim. phys., 1888, 15,
p. 517.
3583. Muller J. A. — Bull. Soc. chim. France, 1918,
23, p. 8.
3584. Muller W., Schafer K., Schuller D. — Chem.-
Ingr-Techn., 1971, 43, p. 619.
3585. Muller W., Schuller D. — Ber. Bunsenges. phys.,
Chem., 1971, 75, S. 79.
3586. Mulliken R. S. — Phys. Rev., 1930, 36, p. 1440.
3587. Mulliken R. S. — Rev. Mod. Phys., 1932, 4, p. 1.
3588. Mulliken R. S. — Phys. Rev., 1936, 50, p. 1017.
3589. Mumpton F. A., Roy R. — J. Amer. Ceram. Soc,
1960, 43, p. 234.
3590 Murabayashi M., Tanaka S., Takahashi Y. —
J. Nucl. Sci. and Technol., 1975, 12, p. 661.
3591 Murad E., Hildenbrand D. L. — J. Chem. Phys.,
1975, 63, p. 1133.
3592. Murch L. E., Giauque W. F. — J. Phys. Chem.,
1962, 66, p. 2052.
3593. Murgulescu I. G., Marta L. — In:
Thermodynamics: Proc. Symp. Vienna, 1965. Vienna: IAEA,
1966, vol. 1, p. 345.
3594. Murgulescu I. G., Telea С. ~ Rev. roum. chim.,
1977, 22, p. 683.
3595. Murgulescu I. G., Topor L. — Rev. roum. chim.,
1966, 11, p. 1353.
3596. Murgulescu I. G., Topor L. — Rev. roum. chim.,
1967, 12, p. 1077.
3597. Murgulescu I. G., Topor L. — Rev. roum. chim.,
1968, 13, p. 1109.
3598. Murgulescu I. G., Topor L. — Rev. roum. chim.,
1970, 15, p. 997.
3599. Murray P., Allianson E. B. — Trans. Brit.
Ceram. Soc, 1954, 53, p. 335.
3600. Murty P. S. — Astrophys. and Space Sci., 1980,
68, p. 513.
3601. Murty P. S. — Astrophys. J., 1980, 240, p. 585.
3602. Mustajoki A. — Ann. Acad. sci. Fennicae, Ser.
AI, 1951, N 98, 7 p.
3603. Mustajoki A. — Ann. Acad. sci. Fennicae, Ser.
AVI, 1957, N 5. 17 p.
3604. Mustajoki A. — Ann. Acad. sci. Fennicae, Ser.
AVI, 1957, N 7. 12 p.
3605. Mustajoki A. — Ann. Acad. sci. Fennicae, Ser.
AVI, 1958, N 9. 17 p.
3606. Mystajoki A. — Suomalais tiedeakat toimituks,
Ser. AVI, 1962, N 99. 11 p.
3607. Muthmann W., Weiss L. ^~ Justus Liebigs Ann.
Chem., 1904, 331, S. 1.
3608. Muthmann W., Weiss L., Metzger J. — Justus
Liebigs Ann. Chem., 1907, 355, S. 137.
3609. Muthmann W., Weiss L., Ridelbauch R. — Justus
Liebigs Ann. Chem., 1907, 355, S. 58.
3610. Muthmann W., Weiss L., Ridelbauch R. — Justus
Liebigs Ann. Chem., 1907, 355, S. 67.
3611. Muthmann W., Weiss L., Ridelbauch R. — Justus
Liebigs Ann. Chem., 1907, 355, S. 82.
3612. Myers C. E., Norman L. J., Loew L. M. — Inorg.
Chem., 1978, 17, p. 1581.
3613. Myklust R. L., Daane A. H. — Trans. Met. Soc.
AIME, 1962, 224, p. 354.
3614. Nacken R. — Neues Jahrb. Mineral. Geol. und
Palaontol. Beilage, 1907, 24, S. 1.
3615. Nacken R. — Nachr. Ges. wiss. Gottingen. math.-
phys. Kl., 1907, S. 602.
3616. Nacken R. — Zement, 1930, 19, S. 818.
3617. Nagarajan G. — Bull. Soc. chim. Belg., 1962,
71, p. 77.
3618. Nagarajan G. — Austral. J. Chem., 1963, 16,
p. 908.
3619. Nagarajan G. — Z. phys. Chem. Leipzig, 1967,
234, S. 406.
3620. Magarajan <?., Brinkley D.C. — Ъ. Naturforsch.,
1971, A26, S. 1658.
3621. Naito K. — J. Nucl. Mater., 1974, 51, p. 126.
3622. Naito K., Ishii Т., Hamaguchi Y., Oshima K. —
Solid State Communs, 1967, 5, p. 349.
3623. Naito K., Kamegashira N., Sasaki N. ~ J. Solid.,
State Chem., 1980, 35, p. 305.
3624. Naito K., Tsuju Т., Matsui T. — J. Nucl. Mater.,
1973, 48, p. 58.
3625. Nakagawa I., Walter J. L. — J. Chem. Phys.,
1969, 51, p. 1389.
3626. Nakahira M., Horiuchi S., Ooshima H. — J. Appl.
Phys., 1970, 41, p. 836.
3627. Nakamura G. — Z. Phys., 1929, 59, S. 218.
3628. Nakamura G. — Jap. J. Phys., 1931, 7, p. 33.
3629. Nakamura J., Takahashi Y., Izumi S., Kanno M:—
J. Nucl. Mater., 1980, 88, p. 64.
3630. Nakata M. M., McKisson R. L., Pollock B. D. —
U. S. North American Aviation Inc. Rept NAA-
SR-6095. Downey (Calif.), 1961.
3631. Narasimham K. V. — In: Development of
applied spectroscopy. Chicago: Spectrosc. Soc,
1963, vol. 2, p. 142.
3632. Narasimhamurty H. V. L., Iyer A. S., Hoch M. —
J. Phys. Chem., 1965, 69, p. 1420.
3633. Natarajan M., Novi V. — Suomalais. tiedeakat.
toimituks. Ser. AVI, 1972, N 400, 9 s.
3634. Nater K. A. — In: Recent developments in mass
spectroscopy. Proc. Intern. Conf. Mass Spectrosc.,
Kyoto, 1969 Ed. K. Ogata, T. Hayakawa. Tokyo:
Univ. Tokyo Press, 1970, p. 1136.
3635. Natola F., Touzain P. — Canad. J. Chem., 1970,
48, p. 1955.
3636. Navrotsky A., Kleppa 0. J. — J. Amer. Ceram.
Soc, 1967, 50, p. 626.
3637. Naylor B. F. — J. Amer. Chem. Soc, 1946, 68,
p. 1077.
3638. Nedeljkovic Lj.—Z. anorg. und allg. Chem.,
1968, 357, S. 103.
578
Литература
3639. Nedungadi Т. М. К. — Proc. Indian Acad. Sci.,
1939, А10, p. 197.
3640. Neel D. S., Pears С D. — In: Progress in
international research on thermodynamic and
transport properties/ Ed. J. F. Masi, D. H. Tsai.
N. Y.; L.: Acad. Press, 1962, p. 500.
3641. Nemukhin A. V., Stepanov N. F. — Intern. J.
Quant. Chem., 1979, 15, p. 49.
3642. Nernst W. — Ann. Phys., 1911, 36, S. 395.
3643. Neubert A., Zmbov K. F. — High Temp. Sci.,
1974, 6, p. 303.
3644. Neuman E. W. — J. Chem. Phys., 1935, 3, p. 243.
3645. Neuman В., Kroger C, Kurtz H. — Z. anorg.
und allg. Chem., 1934, 218, S. 379.
3646. Neumann В., Kroger C, Kunz H. — Z. anorg.
und allg. Chem., 1934, 218, S. 381.
3647. Neumann K., Volker E. — Z. phys. Chem.
Leipzig, 1932, A161, S. 33.
3648. Newman B. — Philos. Mag., 1924, 48, p. 159.
3649. Newman E. S. — J. Res. NBS, 1958, 61, p. 75.
3650. Newman E. S. — J. Res. NBS, 1962, A66, p. 381.
3651. Newrirk H. W., Bates J. L. — U. S. Hanford
Atomic Products Operation. Rept HW-59468.
Richland, Wash., 1959.
3652. Nguyen-Duy P., Dancy E. A. — Thermochim.
acta, 1980, 39, p. 95.
3653. Nguyen-Long-Den, Be Saint Simon M. — In: Adv.
Mass. Spectrom. L.: Heyden and Son Ltd., 1971,
vol. 5, p. 413.
3654. Nichols R. W. — Nucl. Eng., 1958, 3, p. 327.
3655. Nicolosi S. L., Tang I., Munkelwitz H. High
temperature mass spectrom. study of Cs2O and
Rb2O, NUREG/Cr-0867, BNL-NUREG-51030, R-8.
N. Y.: Brookhaven, Nat. Lab. Upton, 1979.
3656. Niebuhr J. — J. Less-Common Metals, 1966, 11,
p. 191.
3657. Niggli P. — J. Amer. Chem. Soc, 1913, 35,
p. 1693.
3658. Niggli P. — Z. anorg. und allg. Chem., 1919,
106, S. 126.
3659. Nikitin M. /., Sidorov L. N. — Intern. J. Mass
Spectrom. and Ion Phys., 1980, 35, p. 101.
3660. Nilson L. F., Petersson O. — Z. phys. Chem.
Leipzig, 1887, 1, S. 27.
3661. Nimmo J. K., Lucas B. W. — Nature Phys. Sci.,
1972, 237, p. 61.'
3662. Nimmo J. K., Lucas B. W. — Acta crystallogr.,
1976, B32, p. 1968.
3663. Ninomiya M.—3. Phys. Soc. Jap., 1955, 10,
p. 829.
3664. Nishimura H., Kimura H. — J. Jap. Inst. Metals,
1956, 20, p. 524.
3665. Niwa K. — J. Chem. Soc. Jap., 1938, 59, p. 637.
3666. Noguchi T. — In: Advances in High Temperature
Chemistry/Ed. L. Eyring. N. Y.; L.: Acad. Press,
1969, vol. 2, p. 235.
3667. Noguchi Т., Kozuka T. — Solar Energy, 1966,
10, p. 125.
3668. Noguchi Т., Kozuka T. — Solar Energy, 1966,
10, p. 203.
3669. Noguchi Т., Mizuno M. — Solar Energy, 1967,
11, p. 90.
3670. Noguchi Т., Mizuno M., Kozuka T. — J. Chem.
Soc. Jap. Ind. Chem. Sec, 1966, 69, p. 1705.
3671. Nolte G., Kordes E. — Z. anorg. und allg. Chem.,
1969, 371, S. 149.
3672. Nolting H. I., Simon С R., Klingenberg I. I. —
J^ Inorg. and Nucl. Chem., 1960, 14, p. 208.
3673. Nomura S. — J. Phys. Soc, Jap., 1961, 16,
p. 1352.
3674. Nomura S., Kawakubo Т., Yanagi T. — J. Phys.
Soc. Jap., 1961, 16, p. 706.
3675. NordA. G. — Acta chem. scand., 1973, 27, p. 814.
3676. Nord A. G. — Acta crystallogr., 1974, B30,
p. 1640.
3677. Nord A. G. — Acta chem. scand., 1976, A30,
p. 199.
3678. NordA. G. — Acta crystallogr., 1976, B32, p. 982.
3679. Norman J. #., Staley H. G. — J. Chem. Phys.,
1965, 43, p. 3804.
3680. Norman J. H., Staley H. G. — U. S. General
Atomic Division General Dynamics Corp. Rept
GA-7247. San Diego (Calif.), 1966.
3681. Norris L., Worthing A. G. — Phys. Rev., 1933,
44, p. 323.
3682. Notz I. I., Mendel M. G. — J. Inorg. and Nucl.
Chem., 1960, 14, p. 55.
3683. Notz K. J., Bach R. O. — Chimia, 1963, 17,
S. 158.
3684. Notz K. J., Huntington C. W., Burkhardt W. —
Ind. Eng. Chem. Process Design and Developm.,
1962, l,.p. 213.
3685. Novikov'l. I., Lepeshkin Y. ?>., Bojarski S. V.,
Roschupkin V. V., Semashko N. A., Forde-
jeva L. K. — Rev. int. hautes temp, et refract.,
1979, 16, p. 413.
3686. Novotny V., Meincke P. P. M. — J. Low Temp.
Phys., 1975, 18, p. 147.
3687. Numrich R. W., Truhlar D. G. — J. Phys. Chem.,
1975, 79, p. 2745.
3688. Nuttal R. L., Churney K. L., Kilday M. V. — J.
Res. NBS, 1978, A83, p. 335.
3689. Ny Tsi-Ze, Tsien-San-Tsiang. — Phys. Rev., 1937,
52, p. 91.
3690. Nygren M., Magneli A. — Ark. kemi, 1968, 28,
s. 217.
3691. Oberhammer #., Boggs J. E. — J. Mol. Struct.,
1979, 56, p. 107.
3692. O'Brien C. J., Kelley K. K. — J. Amer. Chem.
Soc, 1957, 79, p. 5616.
3693. Ochs L. — Z. Naturforsch., 1957, B12, S. 215.
3694. Oetting F. L., Navratil J. D. — J. Chem. and
Eng. Data, 1972, 17, p. 230.
3695. Oetting F. L., Rand M. H., Ackermann R. J.
The chemical Thermodynamics of actinide
elements and compounds. Pt 1: The actinide elements.
Vienna: IAEA, 1976.
3696. Ogden H. R., Maykuth D. J., Finlay W. L.,
Jaffe R. I. — J. Metals, 1951, 3, p. 1150.
3697. Ograd A. E., Leary J. A. — In: Thermodynamics
Nucl. Mater. Proc Symp. Vienna, 1967. Vienna:
IAEA, 1968, p. 651.
3698. O'Hare P. A. G. — J. Chem. Phys., 1972, 56,
p. 4513.
3699. O'Hare P. A. G., Boerio J. — J. Chem. Thermo-
dyn., 1975, 7, p. 937.
3700. O'Hare P. A. G.,JohnsonG. K., Cordjunke E. H. P.,
Wijbenga G., Halstead G. B. — In: 6th Intern.
Conf. on Thermodyn.: Abstracts of Poster Paper.
Merseburg, GRD, Aug. 26—29, 1980, p. 22.
3701. O'Hare P. A. G., Settle J. L., Feder H. M., Hub-
bard W. N. — In: Thermodynamics Nucl. Mater.
Proc. Symp. Vienna, 1967. Vienna: IAEA, 1968,
p. 265.
3702. O'Hare P. A. G., WahlA. С — J. Chem.
Thermodyn., 1972, 56, p. 4516.
579
Литература
3703. Ohnysty В., Rose F. К. — J. Amer. Ceram. Soc, 3738.
1964, 47, p. 398.
3704. Ohse R. W. — J. Chem. Phys., 1966, 44, p. 1375. 3739.
3705. Ohta Т., Sata T. — J. Ceram. Soc. Jap., 1974, 3740.
82, N 947, p. 347.
3706. Ohtsuka Т., Kimura Y. — Physica, 1971, 55, 3741.
p. 562.
3707. Ohwada K. — Spectrochim. acta, 1968, A24, 3742.
p. 595.
3708. Ohwada K. — J. Inorg. und Nucl. Chem., 1971, 3743.
33, p. 1615.
3709. Ohwada K. — J. Inorg. and Nucl. Chem., 1972, 3744.
34, p. 2357.
3710. Okada S., Kokubo S., Matsuo K. — J. Soc. Chem. 3745.
Ind. Jap., 1943, 46, p. 324.
3711. Okaz A. M., Keesom P. H. — Phys. Rev., 1975, 3746.
B12, p. 4917.
3712. O'Keefe M., Valigi M. — J. Chem. Phys., 1969, 3747.
50, p. 1490.
3713. Oliver G. D., Milton H. Т., Grisard J. W. — 3748.
J. Amer. Chem. Soc, 1953, 75, p. 2827.
3714. Olofsgon G., Sunner S., Efimov M., Laynez J. —
J. Chem. Thermodyn., 1973, 5, p. 199. 3749.
3715. Olson M. L., Konowalow D. D. — Chem. Phys., 3750.
1977, 21, p. 393.
3716. Olson M. L., Konowalow D. D. —Chem. Phys., 3751.
1977, 22, p. 29. 3752.
3717. Olsson E. — Z. Phys., 1935, 93, S. 206.
3718. O'Neal R. #., Phillips E. N. — U. S. Univ. 3753.
Calif., Lawrence Radiat Lab. Rept UCRL-10706,
1963. ¦ 3754.
3719. Ono K., Matsushita Т., Ito T. — J. Jap. Inst.
Metals, 1965, 29, p. 501. 3755.
3720. Oppermann H., Reichelt W., Gerlach U., Wolf E.,
Bruckner W., Moldenhauer W., Wich H. — Phys. 3756.
status solidi, 1975, A28, p. 439.
3721. Oriani R. A., Jones T. S. — Rev. Sci. Instrum., 3757.
1954, 25, p. 248.
3722. Orr R. D. — Diss. Abstrs, 1972, B32, p. 4138. 3758.
3723. Orr R. L. — J. Amer. Chem. Soc, 1953, 75,
p. 1231. 3759.
3724. Orr R. L. — J. Amer. Chem. Soc, 1953, 75,
p. 2808. 3760.
3725. Orr R. L. — J. Amer. Chem. Soc, 1954, 76,
p. 857. 3761.
3726. Orth F. В., Stwalley W. C. — J. Mol. Spectrosc,
1979, 76, p. 17. 3762.
3727. Orth F. В., Stwalley W. C, Yang S. C, Hsieh 3763.
Y. K. — J. Mol. Spectrosc, 1980, 79, p. 314. 3764.
3728. Osbome D. W., Westrum E. F. — J. Chem. Phys., 3765.
1953, 21, p. 1884.
3729. Osborne D. W., Westrum E. F., Lohr H. R. — 3766.
J. Amer. Chem. Soc, 1955, 77, p. 2737.
3730. Osborne D. W., Westum E. F., Lohr H. R. — J. 3767.
Amer. Chem. Soc, 1957, 79, p. 529.
3731. Oster G. F., Bonilla С F. — In: Proc. Fifth Symp. 3768.
Thermophysical Properties. N. Y.: ASME, 1970,
p. 468. 3769.
3732. Ostwald W. — J. prakt. Chem., 1882, 25, S. 1.
3733. Otsubo Y., Yamaguchi K. — J. Chem. Soc. Jap. 3770.
Pure Chem. Sec, 1961, 82, p. 557.
3734. Otsubo Y., Yamaguchi K., Kawamura Y. — Nip- 3771.
pon Kagaku Zasshi, 1962, 83, p. 352.
3735. Ott J. В., Goates J. R., Anderson D. R., Hall
H. T. — Trans. Faraday Soc, 1969, 65, p. 2870. 3772.
3736. Ott J. В., Goates J. R., Oyler D. E. — Trans.
Faraday Soc, 1971, 67, p. 31.
3737. Ouensanga A. — J. Less-Common Metals, 1981, 3773.
77, p. 151.
Owen W. R., Kennard С H. L. — Austral. J.
Chem., 1971, 24, p. 1295.
Owens В. В. — J. Chem. Phys., 1965, 42, p. 2259.
Owens F. /.—Chem. Phys. Lett., 1979, 64,
p. 116.
Ozawa Т., Isozaki H., Negishi A. — Thermochim.
acta, 1970, 1, p. 545.
Ozawa Т., Momota M., Isozaki H. — Bull. Chem.
Soc. Jap., 1967, 40, p. 1583.
Pacault A., Gromb S.—C. r. Acad. sci.,
1958, 247, p. 451.
Padilla A. — In: Proc. 7th Symp. on Thermophys.
Properties. Wash.: ASME, 1977, p. 887.
Page F. M., Sugden Т. М. — Nature, 1959,
183, p. 1672.
Paine R. Т., McDowell R. S., Asprey L. В.,
Jones L. H. — J. Chem. Phys., 1976, 64, p. 3081.
Palmer H. В., Hsu C. J. — J. Mol. Spectrosc,
1972, 43, p. 320.
Pan P. H., Finnemore D. K., Bevolo A. J., Shanks
H. R., Beaudry B. J., Schmidt F. A., Denielson
G. C. — Phys. Rev., 1980, 21, p. 2809.
Pandey J. D. — Indian J. Chem., 1969, 7, p. 251.
Panish M. В., Reif L. — J. Chem. Phys., 1963,
38, p. 253.
Pankhurst R. С — Nature, 1941, 147, p. 643.
Pankhurst R. C. — Proc. Phys. Soc. London,
1949, 462, p. 191.
Pankratz L. В., Ferrante M. J. — U. S. Bur.
Mines. Rept Invest., 1971, N 7551.
Pankratz L. В., Kelly K. K. — U. S. Bur. Mines.
Rept Invest., 1963, N 6198.
Pankratz L. В., King E. <?., Kelly K. K. — U. S.
Bur. Mines. Rept Invest., 1962, N 6033.
Pannetier G., Gaultier M. — Bull. Soc. chim.
France, 1966, N 1, p. 188.
Pannetier G., Gaultier M. — Bull. Soc. chim.
France, 1966, N 3, p. 1069.
Paolettt P. — Trans. Faraday. Soc, 1965, 61,
p. 219.
Paoletti P., Sabatini A., Vacca A. — Trans.
Faraday Soc, 1965, 61, p. 2417.
Papadopoulos M. N., Giauque W. F. — J. Amer.
Chem. Soc, 1955, 77, p. 2740.
Papee H. M., Canady W. J., Laidler K. J. —
Canad. J. Chem., 1956, 34, p. 1677.
Papin G. — С r. Acad. sci., 1973, C277, p. 497.
Papin G. — С r. Acad. sci., 1973, C277, p. 691.
Papin G. — These doct. sci. Univ. Paris, 1973.
Papin G., Bouaziz R. — C. r. Acad. sci., 1973,
C277, p. 771.
Papin <?., Michaud M., Bouaziz R. — С. г. Acad.
sci., 1969, C268, p. 1691.
Papousek D. — Trans. Faraday Soc, 1961, 57,
p. 884.
Papousek D., Valentova M. — Collect. Czechosl.
Chem. Communs, 1961, 26, p. 3157.
Pardue W. M., Keller D. L. — J. Amer. Ceram.
Soc, 1964, 47, p. 610.
Parker R. — Trans. Met. Soc. AIME, 1965, 233,
p. 1545.
Parker V. B. Thermal properties of aqueous uni-
univalent electrolytes. Wash.: NSRDS-NBS-2,
1965.
Parker V. B. An analysis and interpretation of
ДЯf (UFe c). Memorandum for IAEA, Authors,
October 15, 1976. Цит. по: [3773].
Parker V. B. The thermochemical properties
of the uranium-halogen containing compounds.
580
Литература
NBSIR-80-2029: Interium rept preparated for
IAEA and OSRD, NBS. Wash., 1980, July.
3774. Parkinson D. H., Simon F. E., Spedding F. H. —
Proc. Roy. Soc. London, 1951, A207, p. 137.
3775. Parkinson W., Nichols R. W. Shock excitation
of powdered solids: Sci. Rept N 1, Contract AF
19/604—4560, 1959.
3776. Parry G. S., Schuyff A., Vbbelhde A. R. —Proc.
Roy. Soc. London, 1965, A285, p. 360.
3777. Partington J. R., Soper W. E. — Philos. Mag.,
1929, 7, p. 20.
3778. Paseard R. — Acta met., 1959, 7, p. 305.
3779. РаЫ A. R., Koshy J. — J. Cryst. Growth, 1968,
2, p. 128.
3780. Patel M. M., Gohel V. B. — Indian J. Pure and
Appl. Phys., 1973\ 11, p. 60.
3781. Pathak C. M., Palmer H. B. — J. Mol. Spectrosc,
1970, 33, p. 317.
3782. Patil R. N., Subbarao E. C. — Acta crystallogr.,
1970, A26, p. 535.
3783. Pattoret A., Drowart J., Smoes S. — In: Thermo-
dyn. Nucl. Mater. Proc. Symp. Vienna, 1967.
Vienna: IAEA, 1968, p. 613.
3784. Pattoret A., Drowart J., Smoes S. — Trans.
Faraday Soc, 1969, 65, p. 98.
3785. Paul W. — Mater. Res. Bull., 1970, 5, p. 691.
3786. Pauling L. — Proc. Nat. Acad. Sci. India, 1956,
A25, p. 1.
3787. Pears С D., Osment D. The thermal properties
of twenty six solid materials to 5000 °F or their
destruction temperatures: Rept ASD-TDR 62-765.
Southern Res. Inst. Birmingam (Alabama), 1963,
Jan.
3788. Pears R. W. В., Sinha S. P. — Nature, 1947,
160, p. 159.
3789. Pearson A. D. — J. Phys. Chem. Solids, 1958,
5, p. 316.
3790. Pearson E. F., Gordy W. — Phys. Rev., 1969,
177, p. 52.
3791. Pearson E. F., Gordy W. — Phys. Rev., 1969,
177, p. 59.
3792. Pearson E. F., Trueblood M. B. — Astrophys.
J., 1973, 179, pt 2, p. L. 145.
3793. Pearson E. F., Trueblood M. B. — J. Chem.
Phys., 1973, 58, p. 826.
3794. Pearson E. F., Winnewisser B. P., Trueblood
M. B. — Z. Naturforsch., 1976, A31, S. 1259.
3795. Peery B. F., Beebe R. F. — Astrophys. J., 1970,
160, p. 619.
3796. Peletskii V. E., Druzhinin V. P., Sobol Ya. G. —
High Temp.-High Pressur., 1971, 3, p. 153.
3797. Pell E. M. — J. Phys. Chem. Solids, 1957, 3,
p. 77.
3798. Pemsler J. P. — J. Elektrochem. Soc, 1961, 108,
p. 744.
3799. Perreu J. — C. r. Acad. sci., 1937, 204, p. 1330.
3800. Perreu J. — С. г. Acad. sci., 1941, 212, p. 442.
3801. Person С. С — Ann. chim. phys., 1851, 33,
p. 448.
3802. Peslak J., Jr. — J. Mol. Struct., 1972, 12, p. 235.
3803. PessallN., Gupta K. P., Cheng С. H., Beck P.A.—
J. Phys. Chem. Solids, 1964, 25, p. 993.
3804. Petersen D. Т., Beernstein D. J. — Trans. Amer.
Soc. Metals, 1960, 52, p. 763.
3805. Petit G., Bourlange С — С. г. Acad. sci., 1957,
245, p. 1788.
3806. Petit G., Cremieu A. — С. г. Acad. sci., 1956,
243, p. 360.
3807.
3808.
3809.
3810.
. 3811.
3812.
3813.
3814.
3815.
3816.
3817.
3818.
3819.
3820.
3821.
3822.
3823.
3824.
3825.
3826.
3827.
3828.
3829.
3830.
3831.
3832.
3833.
3834.
3835.
3836.
3837.
3838.
3839.
3840.
3841.
3842.
Petit G., Jaeger M. — С. г. Acad. sci., 1957,
244, p. 1734.
Petterson A. V. — Ark. fys., 1959, 16, p. 185.
Petterson A. V., Lindgren B. — Ark. fys., 1962,
22, p. 491.
Phedorov P. /., Chzan Chi-Yun. — Acta chim.
sin., 1957, 23, p. 9.
Phedorov G. В., Smirnov E. A. — In: Thermodyn.
Nucl. Mater. Proc. Symp. Vienna, 1962. Vienna:
IAEA, 1963, p. 285.
Phillips В., Chang L. L. Y. — Trans. AIME,
1964, 230, p. 1203.
Phillips В., Chang L. L. Y. — Trans. AIME,
1965, 233, p. 1433.
Phillips В., Warshaw СМ., Mockrin I. — J.
Amer. Ceram. Soc, 1966,
Phillips J. G. — Astrophys. J.
Phillips J. G. — Astrophys. J.
Phillips J. G. — Astrophys. J.
Phillips J. G. — Astrophys. J.
Phillips J. G. — Astrophys. J
p. 631.
1950, 111, p. 314.
1951, 114, p. 152.
1969, 157, p. 449.
1971, 169, p. 185.
Suppl., 1973, 26,
p. 313.
Phillips J. G., Davis S. P. — Astrophys. J., 1971,
167, p. 209.
Phillips J. G., Davis S. P. — Astrophys. J.,
1972, 175, p. 583.
Phillips J. G., Davis S. P. — Astrophys. J.,
1976, 206, p. 632.
Phillips J. G., Davis S. P. — Astrophys. J. Suppl.,
1976, 32, p. 537.
Phillips J. G., Davis S. P. — Astrophys. J., 1976,
229, p. 867.
Phillips J. G., Davis S. P. — Astrophys. J.,
1979, 234, p. 393.
Phillips J. G.,DavisS. P., GalehouseD. С —
Astrophys. J., 1979, 234, p. 401.
Phillips N. W. F., Singleton R. H., Holling-
shead E. A. — J. Elektrochem. Soc, 1955, 102,
p. 690.
Phipps T. E., Partridje E. G. — J. Amer. Chem.
Soc, 1929, 51, p. 1331.
Phipps T. E., Sears G. W., Seifert K. L.,
Simpson О. С — In: Proc. Intern. Conf. Peaceful
Uses Atomic Energy. Geneva, 1955. N. Y.: U. N.,
1956, vol. 5, p. 382.
Phipps Т. Е., Sears G. W., Simpson О. С — J.
Chem. Phys., 1950, 18, p. 724.
Piacente V., Bardi G., Malaspina L. — J. Ghem.
Thermodyn., 1973, 5, p, 219.
Piacente V., Desideri A., Bardi G. — J.
Elektrochem. Soc, 1972, 119, p. 75.
Picard C, Gerdanian P. — J. Solid State Chem.,
1975, 14, p. 66.
Pickardi G. — Atti Accad. naz. Lincei, 1933, 17,
p. 654.
Pickard G. L., Simon F. E. — Proc. Phys. Soc.
London, 1948, A61, p. 1.
Pickering S. U. — J. Chem. Soc, 1885, 47, p. 98.
Pickering S. U. — J. Chem. Soc, 1886, 49, p. 260.
Pickering S. U. — J. Chem. Soc, 1887, 51, p. 290.
Pickering S. U. — J. Chem. Soc, 1888, 53, p. 865.
Piermarini G. J., Block S. — Rev. Sci. Inst.,
1975, 46, p. 973.
Pierre G. R., St., Ebihara W. Т., Pool M. J.,
Speiser R. — Trans. Met. Soc. AIME, 1862, 224,
p. 259.
Pijanowski S. W., DeLuca L. S. — U. S. Knolls
Atomic Power Lab., Rept KAPL-1957. Schene-
ctady (N. Y.), 1960.
581
Литература
3843. Pinchemel В., Schamps J. — Chem. Phys., 1976, 3876.
18, p. 481.
3844. Pintchovski F., Glaunsinger W. S., Navratsky A. —
J. Phys. Chem. Solids, 1978, 39, p. 941. 3877.
3845. Pirani M. — Verh. Deutsch. Phys. Ges., 1912,
14, S. 1037.
3846. Pirani M., Alterthum H. — Z. Elektrochem., 3878.
1923 29 S 5
3847. Pirani M., Meyer A. R. — Z. Elektrochem., 3879.
1911, 17, S. 908.
3848. Pistorius С W. F. T. — Nature, 1964, 204, p. 467. 3880.
3849. Pistorius С W. F. T. — J. Chem. Phys., 1965,
43, p. 2895. 3881.
3850. Pistorius С W. F. Т., Richter P. W. — Z. anorg.
und allg. Chem., 1972, 389, S. 315. 3882.
3851. Pistorius С W. F. Т., Snyman H. С — Z. phys.
Chem. (BRD), 1964, 43, S. 1.
3852. Pitzer K. S., Coulter L. V. — J. Amer. Chem.
Soc, 1938, 60, p. 1310.
3853. Plante E. R. — Ph. D. Thesis. Univ. Kansas, 3883.
1960. 3884.
3854. Plante E. R. — Diss. Abstrs, 1960, 21, p. 1081.
3855. Plante E. Д., Sessams A. B. — J. Res. NBS, 3885.
1973, A77, p. 237. 3886.
3856. Plato W. — Z. phys. Chem. Leipzig, 1906, 55,
S. 721. 3887.
3857. Plato W. — Z. phys. Chem. Leipzig, 1907, 58,
S. 350.
3858. Plies V., Geuehn R. — J. Less-Common Metals, 3888.
1975, 42, p. 77. 3889.
3859. Pointud Y., Juillard J. — J. Chem. Soc. Faraday
Trans. I, 1977, 73, p. 1907. 3890.
3860. Poko Z., Fodor M., Szabo E. — Acta chim. Acad.
sci. hung., 1968, 56, p. 357. 3891.
3861. Polaczkowa E., Stoch L. — Bull. acad. pol. sci.
Ser. sci. chim., 1968, 16, p. 317. 3892.
3862. Poland D. E., Green J. W., Margrave J. L. —
J. Chem. and Eng. Data, 1962, 7, p. 389. 3893.
3863. Pollard A. J. — J. Amer. Ceram. Soc, 1961, 44,
p. 630. 3894.
3864. Pollard G., Smyrl N., Devlin J. P. — J. Phys.
Chem., 1972, 76, p. 1826. 3895.
3865. Pompe R. — Thermochim. acta, 1977, 20, p. 229.
3866. Popov M. M., Bundel A., Choller V. — Z. Phys. 3896.
Chem. Leipzig, 1930, A147, S. 302.
3867. Popov M. M., Khomyakov K. (?., Feodosev N_. N., 3897.
Schirokich P. K. — Z. phys. Chem. Leipzig,
1933, A167, S. 29. 3898.
3868. Porter В., Brown E. A. — J. Amer. Ceram. Soc,
1962, 45, p. 49. 3899.
3869. Porter N. D., Boyer M. H., Ju F., Hildenbrand
D. L., Murad E., Hall W. F. — U. S. Aeronutro- 3900.
nic Division PHILCO Final Techn. Rept
AD-715567, N-U-4859, 31 Aug., 1970. 3901.
3870. Porter R. F., Schoonmaker R. С — J. Phys.
Chem., 1958, 62, p. 234. 3902.
3871. Porter R. F., Schoonmaker R. C. — J. Phys.
Chem., 1958, 62, p. 486. 3903.
3872. Porter R. F., Zeller E. — J. Chem. Phys., 1960,
33, p. 85. 3904.
3873. Potts A. W., Lempka H. /., Streets D. G., Price
W. С — Philos. Trans. Roy. Soc. London, 1970, 3905.
A268, p. 59.
3874. Pouget J. P., Launois H., Rice Т. М., Dernier
P., Gossard A., Villeneuve G., Hagenmuller P. — 3906.
Phys. Rev., 1974, B10, p. 1801.
3875. Powers H., Welch F., Trice J. B. — In: U. S. 3907
Fairchild Engine and Airplane Corp. NEPA.
Div. Oak Ridge (Tennesse), 1961, July; (Nucl. 3908,
Sci. Absts, 1962, 16, N 5, Absts 5304).
Powers R. M., Cavallaro Y., Mathern J. P. —
U. S. Sylvania-Corning Nuclear Corp. Rept
SCNC-317, Bayside (N. Y.), 1960.
Powers W. D., Blalock G. C. — U. S. AEC Oak
Ridge Nat. Lab. Rept ORNR-1653. Oak Ridge
(Tennesee), 1954. 48 p.
Poyhdnen J., Mansikka K. — Phys. kondens.
Mater., 1965, 3, S. 218.
Poyhdnen J., Ruuskanen A. — Suomalais. tiedea-
kat. toimituks. Sar. AVI, 1964, N 146.
Poyhdnen /., Sivonen T. — Ann. Acad. sci.
Fennicae, Ser. AVI, 1964, N 170.
Pradel P., Roussel F., Spiess G. — Rev. Sci. Inst-
rum., 1974, 45, p. 45.
Prasad R., Nagarajan K., Singh Z., Bhupathy M.,
Venugopal V., Sood D. D. — In: Thermodyn.
of nucl. mater. Proc. Intern. Symp., Vienna,
29 Jan. — 2 Febr. 1979. Vienna: IAEA, 1979,
vol. 1, p. 551
Premaswarup D, — Current Sci., 1955, 24, p. 45.
Premaswarup D. — Indian J. Phys., 1955, 29,
p. 109.
Premaswarup D. — Nature; 1955, 175, p. 1003.
Premaswarup D., Barrow R. F. — Natute, 1957,
180, p. 602.
Prince A. T. Wilkins A. L. Canad. Dep. Energy
Mines Res. Mines Branch, Rept MS-64-63.
Ottawa, 1964.
Pringsheim P. — Z. Phys., 1926, 38, S. 161.
Prinsheim P., Rosen B. — Z. Phys., 1927, 43,
S. 519.
Prodan A., Marinkovic V., Prosek M. —Mater.
Res. Bull., 1974, 9, p. 121.
Prodan A., Marinkovic V., Prosek M. —Mater.
Res. Bull., 1974, 9, p. 551.
Proks I., Eliasova M., Pack L., Zlatovsky I. —
Chem. zvesti, 1967, 21, s. 908.
Prunier C, Radenac A., Roux C, Rapin M. —
С. г. Acad. sci., 1969, C268, p. 1999.
Prunier C, Radenac A., Roux C, Rapin M. —
Ser. met., 1969, 3, p. 381.
Prunier C, Roux C, Hochid В., Rapin M. —
High Temp.-High Pressur., 1970, 2, p. 183.
Pugh A., Barrow R. F. —Trans. Faraday Soc,
1958, 54, p. 671.
Pugliese L. A., Fitterer G. R. — Met. Trans.,
1970, 1, p. 1997.
Рига В., Przedmojski J. — Phys. status solidi,
1975, 69, p. K37.
Pushin N. A., Radoicic M. — Z. anorg. und allg.
Chem., 1937, 233, S. 41.
Putman J. W., Potter R. D., Grant N. J. —
Trans. Amer. Soc. Metals, 1951, 43, p. 824.
Pynn R., Axe J. D. — J. Phys. C: Solid State
Phys., 1976, 9, L199.
Pynn R., Axe J. D., Raccah P. M. — Phys. Rev.,
1978, B17, p. 2196.
Pynn Д., Axe J. D., Thomas R. — Phys. Rev.,
1976, B13, p. 2965.
Quan J. Т., Adams С. Е. — J. Phys. Chem.,
1966, 70, p. 340.
Quarterly Summary Res. Rept ISC—531 in
Metallurgy for April, May, June. Iowa State College,
1954.
Quist A. S., Bates J. В., Boyd G. E. — J. Phys.
Chem., 1972, 76, p. 78.
Raab M., Honig G., Castell R., Demtroder W. —
Chem. Phys. Lett., 1979, 66, p. 307.
Radebauh i?., Keesom P. H. — Phys. Rev., 1966,
149, p. 209.
582
Литература
3909. Radziemski L. J., Mack J. M. — Physica, 1980,
B102, p. 35.
3910. Raffelini F., My Le Van. — С. г. Acad. sci.,
1971, C273, p. 92.
3911. Raftery J., Scott P. R., Richards W. G. — J.
Phys.: Atom, and Mol. Phys., 1972, 5, p. 1293.
3912. Rahmel A.—Z. phys. Chem. (BRD), 1971, 74,
S. 206.
3913. Rahmel A., Schorr M. — Z. phys. Chem. (BRD),
1972, 77, S. 331.
3914. Ramdas A. K. — Proc. Indian Acad. Sci., 1953,
A37, p. 441.
3915. Ramsey J. TV., Caplan D., Burr A. A. — J. Elec-
trochem. Soc, 1956, 103, p. 135
3916. Rand M. H. — In: Thorium. Physico-chem.
properties of its compounds and alloys: Atomic Energy
Review Spec. Issue N 5. Vienna: IAEA, 1975,
p. 7.
3917. Rand M. H. — In: Thermodyn. of Nucl. Mater.
Proc. Intern. Symp., Vienna, 1979. Vienna:
IAEA, 1980, vol. 1, p. 197.
3918. Rand M. H., Ackermann R. J., Gr0nvold F.,
Oetting F. L., Pattoret A. — Rev. int. hautes
temper, et refract., 1978, 15, p. 355.
3919. Rand M. H., Kubaschewski O. The thermochemical
properties of uranium compounds. N. Y.: Intern.
Sci. Publ., 1963.
?920. Randall M., Bisson С S. — J. Amer. Chem.
Soc, 1920, 42, p. 347.
3921. Rao C. N. R. — Canad. J. Chem., 1961, 39,
p. 498.
3922. Rao С N. R., Natarajan M., Rao G. v. S., Loeh-
man R. E. — J. Phys. and Chem. Solids, 1971,
32, p. 1147.
3923. Rao С N. Д., Ramdas S., Loehman R. E., Ho-
nig J. M. — J. Solid State Chem., 1971, 3, p. 83.
3924. Rao С N. R., Yoganarasimhan S. R., FoethP.A. —
Trans. Faraday Soc, 1961, 57, p. 504.
3925. Rao C. R. M., Mehrotra P. N. — Thermochim.
acta, 1979, 29, s. 180.
3926. Rao D. B. — High Temp. Sci., 1970, 2, p. 381.
3927. Rao K. /., Helphrey D. В., Angell C. A. — Phys.
and Chem. Glass., 1973, 14, p. 26.
3928. Rao K. /., Rao C. N. R. — Brit. J. Appl. Phys.,
1966, 17, p. 1653.
3929. Rao K. S. — Nature, 1952, 170, p. 670.
3930. Rao K. S. — Nature, 1954, 173, p. 1240.
3931. Rao K. S. — Proc. Nat. Inst. Sci. India, 1955,
A2i, p. 188.
3932. Rao K. S. — Proc. Nat. Inst. Sci. India, 1955,
A21, p. 219.
3933. Rao V. R. — Indian J. Phys., 1950, 24, p. 35.
3934. Rao V. R., Premaswarup D. — Indian J. Phys.,
1953, 27, p. 399.
3935. Rao V. S., Schoonmaker R. С — J. Chem. Phys.,
1960, 33, p. 1718.
3936. Rapoport E. — J. Chem. Phys., 1966, 45, p. 2721.
3937. Rapoport E. — J. Phys. Chem. Solids, 1966, 27,
p. 1349.
3938. Rapp R. A. — Trans. Met. Soc. AIME, 1963,
227, p. 371.
3939. Rasor N. S., McClelland J. D. — J. Phys. Chem.
' Solids, 1960, 15, p. 17.
3940. Rasser В., Remy M. — Intern. J. Mass Spectrom.
and Ion Phys., 1976, 21, p. 159.
3941. Rassow H. — Z. anorg. und allg. Chem., 1920,
114, S. 117.
3942. Rauh E. G. — Canad. Met. Quart., 1975, 14,
p. 205. " Hi
3943. Rauh E. G., Ackermann R. J. — J. Chem. Phys.,
1974, 60, p. 1396.
3944. Rauh E. G., Ackermann R. J. — J. Chem. Phys.,
1979, 70, p. 1004.
3945. Rauh E. G., Garg S. P. — J. Amer. Ceram. Soc,
1980, 63, p. 239.
3946. Rauh E. G., Thorn R. I. — J. Chem. Phys.,
1954, 22, p. 1414.
3947. Ray J. D. — J. Inorg. and Nucl. Chem., 1960,
15, p. 290.
3948. Ray J. D., Ogg R. A. — J. Phys. Chem., 1956,
60, p. 1599.
3949. Ray R. C, Dayal V. — Trans. Faraday Soc,
1936, 32, p. 741.
3950. Rayne J. — Phys. Rev., 1954, 95, p. 1428.
3951. Rayne J. A., Kemp W. R. G. — Philos. Mag.,
1956, 1, p. 918.
3952. Readnour J. M., Cobble J. W. — Inorg. Chem.,
1969, 8, p. 2174.
3953. Rechenberg С — J. prakt. Chem., 1879, 19, S.
143.
3954. Rechenberg C. — J. prakt. Chem., 1880, 22, S. 1.
3955. Recoura A. — Ann. chim. phys., 1895, 4, p. 494.
3956. Redington R. L. — J. Chem. Phys., 1966, 44,
p. 1238.
3957. Redmond R. F., Lones J. — U. S. AEC. Oak
Ridge Nat. Lab. Rept ORNL-1342. Oak Ridge
(Tennessee), 1952. 20 p.
3958. Regnault V. — Ann. chim. phys., 1840, 73, p. 5.
3959. Regnault V. — Ann. chim. phys., 1840, 73, p. 50.
3960. Reichelt W., Terukov E. I. — Z. Chem., 1977,
17 S 309.
3961. Reid A. F., Rinwood A. E. — J. Geophys. Res.,
1969, 74, p. 3238.
3962. Reid A. F., Wilmhurst R. E., Wylie A. W. —
J. Electrochem. Soc, 1963, 110, p. 429.
3963. Reiman A. L. — Philos. Mag., 1938, 25, p. 834.
3964. Reiman A. L., Grant С. К. — Philos. Mag., 1936,
22, p. 34.
3965. Reinders W., Vervloet A. W. — Recueil trav.
chim., 1923, 42, p. 625.
3966. Reinsborough V. C, Wetmore F. E. W. —
Austral. J. Chem., 1967, 20, p. 1.
3967. Reisfeld M. J., Crosby G. A. — Inorg. Chem.,
1965, 4, p. 65.
3968. Reisman A. — J. Amer. Chem. Soc, 1958, 80,
p. 3558.
3969. Reisman A., Holtzberg F. — J. Amer. Chem.
Soc, 1955, 77, p. 2115.
3970. Reisman A., Holtzberg F. — J. Amer. Chem.
Soc, 1959, 81, p. 3182.
3971. Reisman A., Holtzberg F. — In: High temperature
oxides. Pt II/Ed. A. M. Alper. N. Y.; L.: Acad.
Press, 1970, p. 217.
3972. Reisman A., Holtzberg F., Berkenblit M., Berry
M. — J. Amer. Chem. Soc, 1956, 78, p. 4514.
3973. Reiter F. W. — Z. Naturforsch., 1973, A28,
S. 1676.
3974. Rengade E. — Ann. chim. phys., 1907, 11, p. 348.
3975. Rengade E. — C. r. Acad. sci., 1907, 145, p. 236.
3976. Rengade E. — Ann. chim. phys., 1908, 14, p. 540.
3977. Rengade E. — Bull. Soc. chim. France, 1909,
5, p. 994.
3978. Rengade E. — С. г. Acad. sci., 1913, 156, p. 1897.
3979. Renkert H., Hensel F., Frank E. — Ber. Bun-
senges. phys. Chem., 1971, 75, S. 507.
3980. Revcolevschi A. — Rev. int. hautes temp et
refract., 1970, 7, p. 73.
3981. Rezakhina T. N., Kravchenko L. I. — J. Слгщ,
Thermodyn., 1972, 4, p. 655.
583
Литература
3982. Riccardi R., Benaglta С. — Gazz. chim. ital.,
1961, 91, p. 315.
3983. Riccardi R., Sinistri C. — Ric. sci. rend., 1965,
A8, p. 1026.
3984. Rice S. A., Klemperer W. — J. Chem. Phys., 1957,
27, p. 573.
3985. Rice S. A., Klemperer W. — J. Chem. Phys.,
1957, 27, p. 643. '
3986. Rice W. W., Wampler F. В., Oldenborg R. C,
Lewis W. В., Tiee J. J., Pack R. T. — J. Chem.
Phys., 1980, V2, p. 2948.
3987. Richards D. R. — Ph. D. Thesis. Oxford, 1969.
3988. Richards D. R., Barrow R. F. — Nature, 1968,
217, p. 842.
3989. Richards D. R., Barrow R. F. — Nature, 1968,
219, p. 1244.
3990. Richards G. W., Wool} A. A. — Z. Chem. Soc. A,
1968, p. 470.
3991. Richards T. W., Buell W. — J. Amer. Chem. Soc,
1917, 39, p. 1816.
3992. Richards T. W., Jackson F. G. — Z. phys. Chem.
Leipzig, 1910, 70, S. 414.
3993. Richards T. W., Rowe A. W. — Proc. Amer.
Acad. Arts. Sci., 1913, 49, p. 173.
3994. Richards T. W., Rowe A. W. — J. Amer. Chem.
Soc, 1922, 44, p. 684.
3995. Richardson L. Т., Wells R. С — J. Wash. Acad.
Sci., 1931, 21, p. 243.
3996. Richter P. W., Pistorius C. W. F. T. — J. Solid
State Chem., 1972, 5, p. 276.
3997. RiddellJ.D.,LockwoodD. J.,IrishD. E. — Canad.
J. Chem., 1972, 50, p. 2951.
3998. Rieck G. D. — Recueil trav. chim., 1943, 62,
p. 427.
3999. Rigney D. V., Kapelner S. M., Cleary R. E. —
U. S, Pratt and Whitney Aircraft Div. Rept.
TIM-810. Hardford (Connect.), 1965.
4000. Rigney D. V., Kapelner S. M., Cleary R. E. —
U. S. Pratt and Whitney Aircraft Div. Rept
TIM-844. Hardford (Connect.), 1965.
4001. Riley B. — J. Sci. Instrum., 1964, 41, p. 504.
4002. Riley B. — Rev. int. hautes temp, et refract.,
1966, 3, p. 327.
4003. Riley B. — Rev. int. hautes temp, et refract.,
1966, 3, p. 343.
4004. RingstrSm U. — J. Mol. Spectrosc, 1970,36, p. 232.
4005. Rink E. — C. r. Acad. sci., 1934, 199, p. 1217.
4006. Risberg P. — Ark. fys., 1956, 10, p. 583.
4007. Ritchie R. K., Lew H. — Canad. J. Phys., 1964,
42, p. 43.
4008. RitshelR., VillarsD. — Naturwissenschaften, 1928,
16, S. 219.
4009. Rittner E. S. — J. Chem. Phys., 1951, 19, p. 1030.
4010. Rives-Arnau V., Munuera G., Criado J. M. —
Spectrosc. Lett., 1979, 12, p. 733.
4011. Rizzo F. E., Bidwell L. R., Frank D. F. — Trans.
AIME, 1967, 239, p. 1901.
4012. Roach A. C. — J. Mol. Spectrosc, 1972, 42, p. 27.
4013. Roake W. E. — J. Elektrochem. Soc, 1957, 104,
p. 662.
4014. Robbins B. A., Marshall B. J. — J. Appl. Phys.,
1971, 42, p. 2562.
4015. Robbins G. D., Thomo R. E., Insley H. — J. Inorg.
and Nucl. Chem., 1965, 27, p. 559.
4016. Roberts B. W., Tittman В., Hamilton D., Jayara-
man A. — J. Appl. Phys., 1963, 35, p. 723.
4017. Roberts H. S. — Phys. Rev., 1924, 23, p. 386.
4018. Roberts J. E., Laubengayer A. W. — J. Amer.
Chem. Soc, 1957, 79, p. 5895.
4019. Roberts L. E., Walter A. J. — J. Inorg. und Nucl.
Chem., 1961, 22, p. 213.
4020. Roberts L. M. — Proc. Roy. Soc. London, 1957,
B70, p. 744.
4021. Robie R. A., Waldbaum D. R. — U. S. Geol.
Surv. Bull., 1968, N 1259, p. 146.
4022. Robiette A. G., Hedberg K., Hedberg L. — J. Mol.
Struct., 1977, 37, p. 105.
4023. Rodebush W. H., DeVries T. — J. Amer. Chem.
Soc, 1925, 47, p. 2488.
4024. Rodebush W. #., Henry W. F. — J. Amer. Chem.
Soc, 1930, 52, p. 3159.
4025. Rodebush W. #., Walters E.G. — J. Amer. Chem.
Soc, 1930, 52, p. 2654.
4026. RoederA., Morawitz W. — Z. Elektrochem., 1956,
60, S. 431.
4027. Rogers B. A., Atkins D. E. — J. Metals, 1955, 7,
p. 1034.
4028. Rogers D. В., Shannon R. D., Sleight A. W.,
Gillson J. L. — Inorg. Chem., 1969, 8, p. 841.
4029. Rogers L. J. — J. Chem. Thermodyn., 1980, 12,
p. 51.
4030. Rolin M. — Bull. Soc. chim. France, 1960, N 4,
p. 671.
4031. Rolin M., Bernard M. — Bull. Soc. chim. France,
1962, N 3, p. 423.
4032. Rolin M., Gallay J. J. — Elektrochim. acta, 1962,
7, p. 153.
4033. Rolin M., Latreille H., Pham H. — Bull. Soc.
chim. France, 1969, N 7, p. 2271.
4034. Rolin M., Muhletholer R. — Bull. Soc. chim.
France, 1964, N 10, p. 2593.
4035. Rolin M., Recapet J. M. — Bull. Soc. chim.
France, 1964, N 10, p. 2504.
4036. Rollefson R. J., Peressini P. P. — J. Appl. Phys.,
1972, 43, p. 727.
4037. RolletA. P. — С. г. Acad. sci., 1935, 200, p. 1763.
4038. Rollet A. P., Bouaziz R. — С. г. Acad. sci., 1955,
240, p. 2417.
4039. Rollet A. P., Cohen-Adad R., Choucroun J. —
Bull. Soc. chim. France, 1959, N 1, p. 146.
4040. Rollet A. P., Cohen-Adad R., Ferlin С — С. г.
Acad. sci., 1963, 256, p. 5580.
4041. Rollet A. P., Cohen-Adad R., Michaud M., Tran-
quard A. — С. г. Acad. sci., 1958, 246, p. 3249.
4042. Rollet A. P., Kocher J. — С. г. Acad. sci., 1964,
259, p. 4692.
4043. Romans P. A., Paasche Q. G., Kabo H. — J. Less-
Common Metals, 1965, 8, p. 213.
4044. Rompe R. — Z. Phys., 1932, 74, S. 175.
4045. Rorer D. С — Diss. Ph. D. Duke Univ., 1964.
124 p. (Diss. Abstrs, 1964, 25, p. 1287).
4046. Rorer D. C, Onn D. G., Meyer H. — Bull. Amer.
Phys. Soc, 1964, 9, p. 30.
4047. Rorer D. C, OnnD. G., Meyer H. — Phys. Rev.,
1965, A138, p. 1661.
4048. Rosen A., Fricke B. — Chem. Phys. Lett., 1979,
61, p. 75.
4049. Rosenfeld M., Ziegler M., Konzig W. — Helv.
phys. acta, 1978, 51, p. 298.
4050. Rosmus P., Meyer W. — J. Chem. Phys., 1976,
65, p. 492.
4051. Ross R. J., Hume-Rothery W. — J. Less-Common
Metals, 1963, 5, p. 258.
4052. Rossini F. D., Wagman D. D., Evans W. H.,
Levine S., Jaffe J. Selected values of chemical
thermodynamic properties: Circ. 500. Wash.:
NBS., 1952.
4053. Rostoker W., Yamamoto A. S. — Trans. Amer.
Soc. Metals, 1954, 46, p. 1136.
584
Литература
4054. Rosztoczy F. E., Cubicciotti D. — J. Phys. Chem.,
1965, 69, p. 1687.
4055. Roth R. S., Waring J. L. — J. Res. NBS, 1961,
A65, p. 337.
4056. Roth R. S., Waring J. L. — J. Res. NBS, 1962,
A66, p. 451.
4057. Roth W. A. — Z. phys. Chem. Leipzig, 1927, 130,
S. 539.
4058. Roth W. A. — Naturwissenschaften, 1931, 19,
S. 860.
4059. Roth W. A. — Z. Elektrochem., 1943, 49, S. 322.
4060. Roth W. A. — Z. anorg. und allg. Chem., 1948,
255, S. 324.
4061. Roth W. A., Becker G. — Z. phys. Chem., Leipzig,
1929, A145, S. 481.
4062. Roth W. A., Becker G. — Z. phys. Chem. Leipzig,
1931, Badenstein-Festband, S. 55.
4063. Roth W. A., Becker G. — Z. phys. Chem. Leipzig,
1932, A159, S. 1.
4064. Roth W. A., Bertram W. — Z. Elektrochem., 1929,
35 S 297
4065. Rath W. A., Borger E., Siemonson H. — Z. anorg.
und allg. Chem., 1938, 239, S. 321.
4066. Roth W. A., Eymann С — Z. phys. Chem. Leipzig,
1929, A143, S. 321.
4067. Roth W. A., Kaule H. L. — Z. anorg. und allg.
Chem., 1947, 253, S. 352.
4068. Roth W. A., Muller F. Unpublished measurements.
Цит. по: [4057].
4069. Roth W. A., Wirths G., Berendt H. — Z.
Elektrochem., 1942, 48, S. 264.
4070. Roth W. A., Wolf U. — Recueil trav. chim.,
1940, 59, p. 511.
4071. Roth W. A., Wolf U. — Z. Elektrochem., 1940,
46, S. 45.
4072. Roth W. A., Wolf V., Fritz O. — Z. Elektrochem.,
1940, 46, S. 42.
4073. Rothberg G. M., Eisenstadt M., Kusch P. — J.
Chem. Phys., 1959, 30, p. 517.
4074. Rothe E. W. — U. S. Chicago operations office.
Rept COO-2850-1, 1976.
4075. Rothe E. W. — Energy Res. Abstrs, 1977, 2 B1),
N 52659.
4076. Rowe J. H., Morey С W., Harisen I. D. — J. Inorg.
and Nucl. Chem., 1965, 27, p. 53.
4077. Rubbens A., Barbier P., Mairesse G., Wallart F.,
Wignacourt J. P. — Canad. J. Spectrosc, 1977,
22, p. 39.
4078. Rubbens A., Wallart F., Barbier P., Mairesse G.,
Wignacouart J. P. — J. Raman Spectrosc, 1978,
7, p. 249.
4079. Ruby С — С. r. Acad. sci., 1967, C264, p. 1172.
4080. Ruby C, Sebaoun A., Vouillon J. C. — С r.
Acad. sci., 1968, C267, p. 1043.
4081. Rudkin R. L., Parker W. J., Jenkins R. J.
Temperature, its measurement and control in science
and industry. Pt. 2 / Ed. A. I. Dahl. N. Y.:
Reinhold Publ. Corp., 1962, vol. 3.
4082. Rudorff W., Walter G., Stadler J. — Z. anorg.
und allg. Chem., 1958, 297, S. 1.
4083. Rudy E., Barman D. P. — U. S. Air Force Mater.
Lab. Techn. Rept AFML-TR-65-2. Pt 1. Ohio, 1965.
4084. Rudy E., Progulski J. — Planseeber. Pulvermet.,
1967, 15, S. 13.
4085. Rudy E., Stecher P. — J. Less-Common Metals,
1963, 5, p. 78.
4086. Ruff O., Ebert F. — Z. anorg. und allg. Chem.,
1929, 180, S. 19.
4087. Ruff O., Ebert F., Woitinek H. — Z. anorg. und
allg. Chert)., 1929, 180, S. 252.
4088. RuffO., FriedrichL. — Z. anorg. und allg. Chem..
1914, 89, S. 279.
4089. Ruff O., Goecke O. — Z. angew. Chem., 1911, 24,
S. 1459.
4090. Ruff O., Heinzelmann A. — Z. anorg. und allg.
Chem., 1911, 72, S. 63.
4091. RuffO., lohansenO. — Berichte, 1905, 38, S. 3601.
4092. RuffO., LeBoucherL. —Z. anorg. und allg. Chem.,
1934, 219, S. 376.
4093. Ruff O., Martin W. — Z. angew. Chem., 1912,
25, S. 49.
4094. Ruff O., Mugdan S. — Z. anorg. und allg. Chem..
1921, 117, S. 147.
4095. Ruff O., Plato W. — Ber. Dtsch. chem. Ges.r
1903, 36, S. 2357.
4096. Ruff O., Schmidt G., Mugdan S. — Z. anorg. und
allg. Chem., 1922, 123, S. 83.
4097. Ruff O., Seiferheld H., Suda J. — Z. anorg. und
allg. Chem., 1913, 82, S. 373.
4098. Ruh R., Garriett H. J., Domagala R. F., Tal-
lanN. M. — J. Amer. Ceram. Soc, 1968, 51, p. 23.
4099. Rupert J. P., Hopkins H. P., Wulff C. A. —I.
Phys. Chem., 1965, 69, p. 3059.
4100. Rupp M.. Ahlrichs R. — Theor. chim. acta, 1977,
46, p. 117.
4101. Rusk J. R., Gordy W. — Phys. Rev., 1962, 127,
p. 817.
4102. Russel L. E. Plutonium 1960 /Ed. E. Grison,
W. Lord, R. Fowler. L.: Cleaver Hume Ltd, 1961,
p. 492.
4103. Russel R. B. — J. Appl. Phys., 1953, 24, p. 232.
4104. Russell A. S. — Z. Phys., 1912, 13, S. 59.
4105. Russell H. N. — Astrophys. J., 1927, 66, p. 347.
4106. Ryan R. R., Penneman R. A., Asprey L. В.,
Paine R. T. — Actacrystallogr., 1976, B32, p. 3311.
4107. Rychly R., Pekarek V. — J. Chem. Thermodyn.,
1977, 9, p. 391.
4108. Ryoitiro S. — Nature, 1966, 188, p. 222.
4109. Rytter E., Oye H. A., Cyvin S. J., Cyvin B. N.,
Klaeboe P. — J. Inorg. and Nucl. Chem., 1973r
35, p. 1185.
4110. Sacconi L., Paoletti P., Ciampolini M. •— Ric.
sci., 1959, 29, p. 2412.
4111. Sacconi L., Paoletti P., Ciampolini M. — J. Amer.
Chem. Soc, 1960, 82, p. 3828.
4112. Sachs E. S., Hinze J., Sabelli N. H. — J. Chem.
Phys., 1975, 62, p. 3367.
4112a. Sachs E. S., Hinze J., Sabelli N. H. — J. Chem..
Phys., 1975, 62, p. 3377.
4113. Sakata K. — J. Phys. Soc. Jap., 1969, 26, p. 582.
4114. Sakata K. — J. Phys. Soc. Jap., 1969, 26, p. 867.
4415. Sakata K. — Acta crystallogr., 1979, B35, p. 2836.
4116. Sakata Т., Sakata K., Nishida I. — Phys. status-
solidi, 1967, 20, p. К 155.
4117. Sakiyama M., Kimoto A., Seki S. — J. Phys.
Soc. Jap., 1965, 20, p. 2180.
4118. Sakurai J., Cowley R. A., Dolling G. — J, Phys.
Soc. Jap., 1970, 28, p. 1426.
4119. Salomon M. В., Gamier P. R., Golding В., Ви-
ehler E. — J. Phys. Chem. Solids, 1974, 35, p. 851.
4120. Salomon M. В., Simons D. S., Gamier P. R. —
Solid State Communs, 1969, 7, p. 1035.
4121. Sale F. R. — Thermochim. acta, 1979, 30, p. 163.
4122. Salhotra P. P., Subbarao E. C, Venkateswarlu P. —
J. Phys. Soc. Jap., 1969, 27, p. 621.
4123. Salje E. — Ferroelectrics, 1976, 12, p. 215.
4124. Salje E. — Acta crystallogr., 1978, B34, p. 1105.
4125. Salje E., Genig R., Vismanathan K. — J. Solid1
State Chem., 1978, 25, p. 239.
585
Литература
4126. Salje E., Hoppmann G. — High Temp.-High
Pressur., 1980, 12, p. 213.
4127. Salje E., Vistvanathan K. — Acta crystallogr.,
1975, A31, p. 356.
4128. Salmon O. N., Ahman D. H. — J. Phys. Chem.,
1956, 60, p. 13.
4129. SamoilovaA. N., Efremov Yu. M., GurvichL. V. —
J. Mol. Spectrosc, 1981, 86, p. 1.
4130. Sandenaw T. A. — J. Phys. Chem. Solids, 1962,
23, p. 1241.
4131. Sandenaw T. A. — J. Nucl. Mater., 1963, 10,
p. 165.
4132. Sandenaw T. A., Gibney R. B. — J. Chem. Ther-
modyn., 1971, 3, p. 85.
4133. Sandenaw T. A., Harbur D. R. — J. Phys. Chem.
Solids, 1974, 35, p. 795.
4134. Sandenaw T. A., Olsen С E., Gibney R. B. —
In: Plutonium 1960/Ed. E. Grison, W. Lord,
R. Fowler. L.: Cleaver Hume Press, 1961, p. 66.
4135. Sandin T. R., Keesom P. H. — Phys. Rev., 1969,
117, p. 1370.
4136. Sandonnini C, Gerosa G. — Gazz. chim. ital.,
1925, 55, p. 916.
4137. Sandonnini C, Gerosa G. — Gazz. chim. ital.,
1925, 55, p. 938.
4138. Sandonnini C, Scarpa G. — Atti. Accad. naz.
Lincei Mem. Cl. sci. fis., mat. e nat. Sez. II, 1913,
22, p. 517.
4139. Sannigrahi А. В., Mohammad S. N. — Mol.
Phys., 1974, 27, p. 269.
4140. Sara R. V. — J. Amer. Ceram. Soc, 1965, 48,
p. 243.
4141. Sara R. V. — Trans. Met. Soc. AIME, 1965, 233,
p. 1683.
4142. Sasaki H., Watanabe A. — ]. Phys. Soc. Jap.,
1964, 19, p. 1748.
4143. Sasaki N., Kubo K., Asano M. — Bull. Eng. Res.
Inst. Kyoto Univ., 1970, 38, p. 38.
4144. Sasaki N., Kubo K., Asano M. — Mass Spectrosc.
(Jap.), 1970, 18, p. 1189.
4145. Sastry B. S. R., Hummel F.A. — J. Amer. Ceram.
Soc, 1959, 42, p. 216.
4146. Sata T. — Bull. Tokyo Inst. Technol., 1963, 56,
p. 21.
4147. Sata Т., KiyouraR. — Bull. Tokyo Inst. Technol.,
1963, 53, p. 39.
4148. Sato Y., Gesi K., Takagi Y. — J. Phys. Soc. Jap.,
1961, 16, p. 2172.
4149. Sato Y., Gesi K., Takagi Y. — J. Phys. Soc.
Jap., 1964, 19, p. 449.
4150. Satyamurthy H. В., Srinivasan S., Sivarama-
Krishnan V. — Indian J. Pure and Appl. Phys.,
1974, 12, p. 71.
4151. Sauka J., Bruners V. — Latvijas PSR Zinatnu
Akad. Vestis, Kim. Ser. 1966, N 1, lpp. 18.
4152. Savage A. W., Browne J. C. — J. Amer. Chem.
Soc, 1960, 82, p. 4817.
4153. Sawada S. — Phys. Rev., 1953, 91, p. 1010.
4154. Sawada S. — J. Phys. Soc. Jap., 1956, 11, p. 1237.
4155. Sawada S. — J. Phys. Soc. Jap., 1956, 11, p. 1246.
4156. Sawada S., Ando K., Nomura S. — Phys. Rev.,
1951, 84, p. 1054.
4157. Sawada S., Danielson G. С — Phys. Rev., 1959,
113, p. 803.
4158. Sawada S., Danielson G. С — Phys. Rev., 1959,
113, p. 1005.
4159. Sawada S., Nomura S. — Phys. Rev. Lett., 1958,
1, p. 320.
4160. Sawada S., Nomura S., Asao /..— J. Phys. Soc.
Jap., 1961, 16, p. 2207.
4161. Saxer R. K. Ph. D. Diss. Ohio State Univ., 1962.
4162. Sayama Y. — J. Phys. Soc. Jap., 1946, 1, p. 13.
4163. Sayre E. V. — J. Chem. Phys., 1959, 31, p. 73.
4164. Sayre E. V., Beaver J. J. — J. Chem. Phys., 1950,
18, p. 584.
4165. Scales W. W. — Phys. Rev., 1958, 112, p. 49.
4166. Scarpa G. — Atti. Accad. naz. Lincei Mem. Cl.
sci. fis., mat. e nat. Sez. I, 1915, 24, p. 476.
4167. Schaber H., Martin T. P. — J. Chem. Phys.,
1979, 70, p. 2029.
4168. Schadee A., Davis D. N. — Astro phys. J., 1968,
152, p. 169.
4169. Schdfer H. — Z. anorg. und allg. Chem., 1975,
414, S. 151.
4170. Schdfer H., Berger D., Gruehn R. — Z. anorg. und
allg. Chem., 1969, 365, S. 31.
4171. Schdfer #., Breil G. — Z. anorg. und allg. Chem.,
1952 267 S 265
4172. Schdfer Hi, Campbell R. /., Wedyk P., BextenL. —
Z. anorg. und allg. Chem., 1970, 376, S. 11.
4173. Schafer H., Gruehn R., Schulte F. — Z. angew.
Chem., 1966, 1, S. 28.
4174. Schdfer H., Liedmeier F. — Z. anorg. und allg.
Chem., 1964, 329, S. 225.
4175. Schdfer H., Wagner K. — Z. anorg. und allg.
Chem., 1979, 450, S. 88.
4176. Schafer H., Wagner K. — Z. anorg. und allg.
Chem., 1979, 451, S. 61.
4177. Schamm R., Toedheide K. — High Temp.-High
Pressur., 1976, 8, p. 65.
4178. Scheer M. D., Fine J. — J. Chem. Phys., 1962,
36, p. 1647.
4179. Scheer M. D., Fine J. — J. Chem. Phys., 1965,
42, p. 3645.
4180. Scheibe H. — Keram. Z., 1963, 15, S. 199.
4181. Schick H. L. Thermodynamics of certain refractory
compounds. N. Y.; L. : Acad. Press, 1966, vol. 1, 2.
4182. Schimpff H. — Z. phys. Chem. Leipzig, 1910, 71,
S. 257.
4183. Schinke H., Sauerwald F. — Z. anorg. und allg.
Chem., 1960, 304, S. 25.
4184. Schins H. E., Van Wijk R. W. M., Dorpema B. —
Z. Metallk., 1971, 62, S. 330.
4185. Schissel P. O., Trulson О. С — J. Chem. Phys.,
1965, 43, p. 737.
4186. Schlechter M. — J. Nucl. Mater.; 1970, 37, p. 82.
4187. Schlenker C, Ahmed S., Buder R., Gourmala M. —
J. Phys.: Solid State Phys., 1979, C12, p. 3503.
4188. Schlenker C, Buder R., Schlenker M.,
Houlihan J. F., Mulay L. N. — Phys. status solidi,
1972, B54, p. 347.
4189. Schlenker C., Lakkis S., Coey J. M. D. — Phys.
Rev., Lett., 1974, 32, p. 1318.
4190. Schlick S., Schnepp O. — J. Chem. Phys., 1964,
41, p. 463.
4191. Schmid E., Fritsch <?., Luescher E. — Z. angew.
Phys., 1969, 27, S. 35.
4192. Schmidt H. G., Wolf G. — Solid State Communs,
1975, 16, p. 1085.
4193. Schmidt U., Vollmer O., Kohlhaas R. — Z. Natur-
forsch., 1970, Л25, S. 1258.
4194. Schmitz H., Shumacher H.lJ. — Z. Naturforsch.,
1947, A2, S. 362.
4195. Schmitz-Dumont O., Heckmann I. — Z. anorg.
und allg. Chem., 1949, 260, S. 49.
4196. Schmitz-Dumont O., Schmitz E. — Z. anorg. und
allg. Chem., 1944, 252, S. 329.
4197. Schneider A., Hilmer O. — Z. anorg. und allg.
Chem., 1956, Л286, S. 97.
586
Литература
4197а. Schneider J. — Z. Phys. Chem. (DRD), 1974,
255, S. 986.
4198. Schneider S. J. — In: Computation on the melting
point of the metal oxides: NBS Monograph 68.
Wash., 1963.
4199. Schneider S. J., Waring J. L. — J. Res. NBS,
1963, АЪ7, р. 19.
4200. Schneider W., Carpenter G. B. — Acta crystallogr.,
1970, B26, p. 1189.
4201. Schofield Т. Н. — Proc. Phys. Soc. London, 1954,
B67, p. 845.
4202. Schofield Т.Н. — J. Inst. Metals, 1956, 85, p. 68.
4203. Schofield Т. Н. — J. Inst. Metals, 1957, 85, p. 372.
4204. Schofield T. H., Bacon A. E. — J. Inst. Metals,
1953, 82, p. 167.
4205. Schofield T. H., Bacon A. E. — J. Inst. Metals,
1955, 84, p. 47.
4206. Scholz R. — Ann. Phys., 1892, 45, S. 193.
4207. Schomaker V., Glauber R. — Nature, 1952, 170,
p. 290.
4208. SchonA. #., Szwarc M. — Ann. Rev. Phys. Chem.,
1957, 8, p. 439.
4209. Schoonmaker R. C, Porter R. F. — J. Chem. Phys.,
1958, 28, p. 454.
4210. Schoonmaker R. C, Porter R. F. — J. Chem. Phys.,
1959, 31, p. 830.
4211. Schoonveld L., Sundaram S. — Astrophys. J.
Suppl., 1974, 27, N 246, p. 307.
4212. Schrier E. E., Clark H. M. — J. Phys. Chem.,
1963, 67, p. 1259.
4213. Schroeder K., Kwist A., Zfungmark H. — Z. Na-
turforsch., 1972, Л27, S. 1252.
4214. Schroeder R. A., Weir С E., Lippincott E. R. —
J. Chem. Phys., 1963, 36, p. 2803.
4215. Schubel P. — Z. anorg. und allg. Chem., 1914,
87, S. 81.
4216. SchUlz B. — Rev. int. hautes temp, et refract.,
1975, 12, p. 132.
4217. Schiirmann E., Nedeljkovic L. J. — Ber. Bunsen-
ges: phys. Chem., 1970, 74, S. 462.
4218. Schutte C. J. H., Buijs K. — Spectrochim. acta,
1961, 17, p. 921.
4219. Schwerdtfeger K. — Trans. Metallurg. Soc. AIME,
1967, 239, p. 1276.
4220. Schwiete H. E., Ziegler G. — Ber. Dtsch. keram.
Ges., 1958, 35, S. 193.
4221. Scott D. H. — Philos. Mag., 1924, 47, p. 32.
4222. Scott J. L. — U. S. AEC Oak Ridge Nat. Lab.
Rept ORNL-2328. Oak Ridge (Tennessee), 1957.
4223. Scott P. R., Richards W. G. — Chem. Phys. Lett.,
1974, 28, p. 101.
4224. Seifert H. /., Klatyk K. — Naturwissenschaften,
1962, 49, S. 539.
4225. Seifert H. J., Landenbach U. — Z. anorg. und allg.
Chem., 1969, 386, S. 39.
4226. Seip H. M. — Acta chem. scand., 1965,19, p. 1955.
4227. Seip H. M., Seip R. — Acta chem. scand., 1966,
20, p. 2698.
4228. Seki S., Suzuki K. — Bull. Chem. Soc. Jap.,
1953, 26, p. 63.
4229. Sekula S. Т., Kernohan R. H. — Phys. Rev.,
1972, B5, p. 904.
4230. Selbin J., Ballhausen С J.,DurrettD. G. — Inorg.
Chem., 1972, 11, p. 510.
4231. Selbin J., Orteso J. D. — Chem. Rev., 1969, 69,
p. 657.
4232. Seltz tf., DunkerleyF. J., Dewitl B.J. — J. Amer.
Chem. Soc, 1943, 65, p. 600.
4233. Senozan N. M. — Diss. Abstrs, 1966, 26, p. 3623.
4234. Sense K. A., Alexander С A., Bowman R. E.,
Stone R. W., Filbert R. B. — J. Phys. Chem.,
1957, 61, p. 384.
4235. Sense K. A., Stone R. W. — 3. Phys. Chem., 1958,
62, p. 1411.
4236. Sergienko V. I., Davidovich R. L. — Spectrosc.
Lett., 1970, 3, p. 35.
4237. Seshadri K. S., Nimon L. A., White D. — J. MoK
Spectrosc, 1969, 30, p. 128.
4238. Seshadri K. S., WhiteD., MannD. E. — J. Chem.
Phys., 1966, 45, p. 4697.
4239. Settle J. L., Feder H. M., Hubbard W. N. — J.
Phys. Chem., 1963, 67, p. 1892.
4240. Settle J. L., Johnson G. K., Hubbard W. N. —
J. Chem. Thermodyn., 1974, 6, p. 263.
4241. Seward R. P. — J. Amer. Chem. Soc, 1942, 64,
p. 1053.
4242. Seward R. P., Martin К. Е. — J. Amer. Chem.
Soc, 1949, 71, p. 3564.
4243. Seydel U., Banhof H., Fucke W., Wadle H. —
High Temp.-High Pressur., 1979, 11, p. 635.
4244. Seydel U., Fischer U. — J. Phys. F: Metal. Phys.,
1978, 8, p. 1397.
4245. Seydel U., Kitzel W. — J. Phys. F: Metal. Phys.,
1979, 9, p. L153.
4246. Shafer M. W., Roy R. — Z. Kristallogr., 1958,
110, S. 241.
4247. Shaner J. W., Gathers G. Д., Hosson W. M. —
In: Proc. 7th Symp. Thermophysical Properties,
Gaithersburg (Md.), 1977. N. Y., 1977, p. 896.
4248. Shaner J. W., Gathers G. Д., Minichino С —
High. Temp.-High Pressur., 1977, 9, p. 331.
4249. Shapiro E. — J. Amer. Chem. Soc, 1952, 74,
p. 5233.
4250. Shapiro S. M., Axe J. D., Shirane G., Rac-
cah P. M. — Solid State Communs, 1974,15, p. 377.
4251. Sharan B. — Phys. status solidi, 1961, 1, p. 243.
4252. Shartsis L., Capps W. — J. Amer. Ceram. Soc,
1954, 37, p. 27.
4253. Sheer M. D., Fine J. — J. Chem. Phys., 1965, 42,
p. 3645.
4254. Sheindlin A. E., Berezin B. Ya., Chekhov-
skoi V. Ya. — High Temp.-High. Pressur., 1972,
4, p. 611.
4255. Sheindlin A. E., Berezin В. Ya., Chekhovskoi V. Ya.
Third Europ. Conf. on Thermophys. Properties
at High Temperatures. Italy, Turin, 20—23 June,
1972.
4256. Sheldon R. I., Gilles P. W. — J. Chem. Phys.,
1977, 66, p. 3705.
4257. Shen Y. L. — Ph. D. Thesis Univ. Calif. Berkeley,
1965; Diss. Abstrs, 1966, 27, p. 118.
4258. Shen Y. L., Senozan N. M., Phillips N. E. — Phys.
Rev. Lett., 1965, 14, p. 1025.
4259. Shenyavskaya E. A., Gurvich L. V. — J. Mol.
Spectrosc, 1980, 81, p. 152.
4260. Shenyavskaya E. A., Ryabov B. S. — J. Mol.
Spectrosc, 1976, 63, p. 23.
4261. Shibasaki Y., Kanamaru F., Koizumi M.,
Ките S. — J. Amer. Ceram. Soc, 1973, 56, p. 248.
4262. Shibata E., Oda S., Furukawa S. — J. Sci.
Hiroshima Univ., 1933, A3, p. 227.
4263. Shibata Z. — Techn. Repts Tohoku Imp. Univ.,
1929, 8, p. 255.
4264. Shibata Z., Tenasaki Y. — Nippon Kagaku Zasshi,
1936, 57, p. 1208.
4265. Shibuyal. — J. Phys. Soc. Jap., 1961, 16, p. 490.
4266. Shin J. В., Nicholls R. W. — Spectrosc. Lett.,
1977, 10, p. 923.
4267. Shirley D. A. — J. Amer. Chem. Soc, 1960, 82,
p. 3841.
587
Литература
4268. Shneider S. J., Levin E. M. — J. Amer. Ceram.
Soc, 1973, 56, p. 218.
4269. Shut V. #., Singh P. I., Kivel В., Bressel E. R. —
AIAA Journal, 1979, 17, p. 1178.
4270. Shomate С H. — J. Amer. Chem. Soc, 1946, 68,
p. 310.
4271. Shomate С. Н. — J. Amer. Chem. Soc, 1947, 69,
p. 218.
4272. Shomate С #., Cohen A. J. — J. Amer. Chem.
Soc, 1955, 77, p. 285.
4273. Shomate С H., Naylor B. F.— J. Amer. Chem.
Soc, 1945, 67, p. 72.
4274. ShpiVrainE. E., KaganD. N., Barkhatov L. S. —
High Temp.-High Pressur., 1972, 4, p. 605.
4275. ShpiVrain E. E., Kagan D. N., Barkhatov L. S.,
Koroleva V. V. — High Temp.-High Pressur., 1976,
8, p. 183.
4276. ShpiVrain E. E., Kagan D. N., Barkhatov L. S.,
Zhmakin L. I., Koroleva V. V. — High Temp.-
High Pressur., 1979, 11, p. 539.
4277. Shuiman G. Thesies Univ. Columbia School of
Engineer and Appl. Sci. Ohio, 1963.
4278. Sidorov L. N., Nikitin M. I., Skokan E. V., So-
rokin I. D. — In: Adv. Mass Spectrom.: Proc.
8th Intern. Conf. Oslo, 1979, L., 1980, vol. 8, p. 462.
4279. Sidorov L. N., ShoVts V. B. — Intern. J. Mass.
Spectrom. and Ion Phys., 1972, 8, p. 437.
4280. Sidorov L. N., Skonan E. V., Nikitin M. I., So-
rokin I. D.,—Int. J. Mass Spectrom. and Ion.
Phys., 1980, 35, p. 215.
4281. Siegel S. — Acta crystallogr., 1955, 8, p. 617.
4282. Siegel S., Hoekstra #., Sherry E. — Acta crystal
logr., 1966, 20, p. 292.
4283. Siemens R. E., Babitzke H. Д., Koto H. — U. S.
Bur. Mines. Rept Invest., 1964, N 6492.
4284. Siemonsen #., Ulich И. — Z. Elektrochem., 1940,
46, S. 141.
4285. Sieverts A., Gotta A. — Z. anorg. und allg. Chem.,
1931, 199, S. 384.
4286. Sieverts A., Gotta A., Halberstadt S. — Z. anorg.
und allg. Chem., 1930, 187, S. 155.
4287. Sieverts A., Gotta A., Halberstadt S. — Z. anorg.
und allg. Chem., 1930, 187, S. 159.
4288. Silver I. L., Bonilla C. F. — In: Proc. Fifth Symp.
on Thermophysical Properties / Ed. С F. Bonilla.
N. Y.: ASME, 1970, p. 461.
4289. Silvidi A. A., Daunt J. G. — Phys. Rev., 1950,
77, p. 125.
4290. Simeon V., IviSicN., Tkalcec M. — Z. phys. Chem.
(BRD), 1972, 78, S. 1.
4291. Simnad M., SpilnersA. — Trans. Met. Soc AIME,
1955, 203, p. 1011.
4292. Simon A. — Z. anorg. und allg. Chem., 1973,
395, S. 301.
4293. Simon F. E. Rept BR-280, Aug. 16, 1943.
(British). Цит. по: [492 с 129].
4294. Simon F. E., Ruhemann M. — Z. phys. Chem.
Leipzig, 1927, A129, S. 321.
4295. Simon F. E., Swain R. С — Z. phys. Chem.
Leipzig, 1935, B28, S. 189.
4296. Simon F. E., Zeidler W. — Z. phys. Chem.
Leipzig, 1926, 123, S. 383.
4297. Simons L. L., Lowden L. F., Ehlert Т. С. — 1.
Phys. Chem., 1977, 81, p. 706.
4298. Sinha S. P. — Proc. Phys. Soc. London, 1947,
59, p. 610.
4299. Sinha S. P. — Indian J. Phys., 1948, 22, p. 401.
4300. Sinha S. P. — Proc. i nys. Soc. London, 1948,
A60, p. 443.
4301. Sinha S. P. — Proc. Phys. Soc. London, 1948,
A60, p. 447.
4302. Sinha S. P. — Proc. Phys. Soc. London, 1949,
A62, p. 124.
4303. Sinistri C, Franzosini P. — Ric sci. rend. Ser. All,
1963, 3, p. 419.
4304. Sjoblom C. A. — High Temp.-High Pressur.,
1976, 8, p. 499.
4305. Sjostrand M. E., Keesom P. H. — Phys. Rev.
Lett., 1971, 27, p. 1434.
4306. Sjostrand M. E., Keesom P. H. — Phys. Lett.,
1972, A39, p. 147.
4307. Skaggs S. R. — High Temp. Sci., 1977, 9, p. 197.
4308. Skinner G. B. Ph. D. Diss. Ohio State Univ.,
1951; Diss. Abstrs, 1958, 18, p. 1646.
4309. Skinner G. В., Beckett C. W., Johnston H. L. —
U. S. Ohio State Univ. Techn. Rept 102—AC49/2—
100—3. Ohio, 1950.
4310. Skinner G. В., Edwards J. W., Johnston H. L. —
J. Amer. Chem. Soc, 1951, 73, p. 174.
4311. Skinner G. В., Johnston H. L. — J. Amer. Chem.
Soc, 1951, 73, p. 4549.
4312. Skinner G. В., Johnston H. L. — J. Chem. Phys.,
1953, 21, p. 1383.
4313. Slansky С. М. — J. Amer. Chem. Soc, 1940, 62,
p. 2430.
4314. Slonim C, Huttig G. F. — Z. anorg. und allg.
Chem., 1929, 181, S. 55.
4315. Slowinski E., Elliot N. — Acta crystallogr., 1952,
5, p. 768.
4316. Smardzewski R. R., Andrews L. — J. Chem. Phys.,
1972, 57, p. 1327.
4317. Smeets C. — Naturw. Tijdschr. Belg., 1933, 15,
S. 105.
4318. SmiSek M., Krestanova V. — Chem. listy, 1974,
68, s. 738.
4319. Smith D., James D. W., Devlin J. P. — J. Chem.
Phys., 1971, 54, p. 4437.
4320. SmithD. F.,BrownD.,DworkinA.S., SasmorD.J..
Van Artsdalen E- R- — J. Amer. Chem. Soc,
1956, 78, p. 1533.
4321. SmithD. F., KaylorC. E., Walden G. E., Taylor
A. R., Gayle J. B. — U. S. Bur. Mines. Rept
Invest., 1961, N 5832.
4322. SmithD. K., Cline С F. — J. Amer. Ceram. Soc,
1962, 45, p. 249.
4323. Smith #., Sugden Т. М. — Proc. Roy. Soc.
London, 1953, A219, p. 204.
4324. Smith H. G. — Proc. Roy. Soc. London, 1924,
A106, p. 400.
4325. Smith H. G., Nicklow P. M., Raubenheimer L. /.,
Wilkinson M. K. — Acta crystallogr., 1966, A21,
p. 210.
4326. Smith K. K., Bigler P. W. — Phys. Rev., 1922,
19, p. 268.
4327. Smith M. В., Bass G. E. — J. Chem. and Eng.
Data, 1963, .8, p. 342.
4128. Smith P. L., WolcottN. M. — Suppl. Bull. Intern.
Inst. Refrig. Annexe, 1955, p. 283.
4329. Smith T. S., Daunt J. G. — Phys. Rev., 1952,
88, p. 1172. J4
4330. Smoes S., Drowart J., Myers С. Е. — J. Chem.
Thermodyn., 1976, 8, p. 225.
4331. Smoes S., Drowart J., Verhaegen G. — J. Chem.
Phys., 1965, 43, p. 732.
4332. Smothers W. J., Kruh R. F., Carlton J. K. —
J. Appl. Chem., 1954, 4, p. 268.
4333. Smyrl N., Devlin J. P. — J. Phys. Chem., 1973,
77, p. 3067.
588
Литература
4334. Smyrl N. Д., Mamantov G., McCurry L. E. —
J. Inorg. and Nucl. Chem., 1978, 40, p. 1489.
4335. Snelson A. — J. Chem. Phys., 1967, 46, p. 3652.
4336. Snelson A., Cyvin S. J. — Ъ. anorg. und allg.
Chem., 1972, 390, S. 316.
4337. Snelson A., Cyvin B. N., Cyvin S. J. — J. Mol.
Struct., 1975, 24, p. 165.
4338. Snelson A., Pitzer K. S. — J. Phys. Chem., 1963,
67, p. 882.
4339. So S. P., Richards W. G. — Chem. Phys. Lett.,
1975, 32, p. 227.
4340. Sodium — NaK. Engineering Handbook / Ed.
0. J. Foust. N. Y.: Gordon and Breach. Vol. 1.
Sodium chemistry and physical properties, 1972.
4341. Solbakk J. K., Stromme K. 0. — Acta chem.
scand., 1960, 23, p. 300.
4342. Sellers E. F., Crenshaw J. L. — J. Amer. Chem.
Soc, 1937, 59, p. 2015.
4343. Sollers E. F., Crenshaw J. L. — J. Amer. Chem.
Soc, 1937, 59, p. 2724.
4344. Sommer R. G., Cater E.D. — J. Electrochem. Soc,
1975, 122, p. 1391.
4345. Sommermeyer K. — Z. Phys., 1929, 56, S. 548.
4346. Somsen G., Coops J., Tolk M. W. — Recueil trav.
chim., 1963, 82, p. 231.
4347. Somsen G., Weeda L. — Recueil trav. chim., 1964,
83, p. 810.
4348. Sorai M., Suga #., Seki S. — Bull. Chem. Soc.
Jap., 1968, 41, p. 312.
4349. Southard J. C. — J. Amer. Chem. Soc, 1941, 63,
p. 3142.
4350. Southard J. C, Nelson R. A. — J. Amer. Chem.
Soc, 1933, 55, p. 4865.
4351. Sowa E. S. — Nucleonics, 1963, 21, p. 76.
4352. Spedding F. H. — U. S. Quarterly Summary
Research Report in Metallurgy. Iowa State College.
Rept ISC-531. Ames, 1955.
4353. Spedding F. H., Daane A. H. — J. Metal, 1954,
6, p. 504.
4354. Spedding F. #., Daane A. H. — Met. Rev., 1960,
5, p. 297.
4355. Spedding F. #., Daane A. H. — Trans. Met. Soc.
AIME, 1957, 209, p. 895.
4356. Spedding F. #., Daane A. #., Wakefield G.. Den-
nison D. H. — Trans. Met. Soc. AIME, 1960, 218,
p. 608.
4357. Spedding F. #., Flynn J. P. — J. Amer. Chem.
Soc, 1954, 76, p. 1474.
4358. Spedding F. #., Gschneidner K. A., Daane A. H. —
Trans. Met. Soc. AIME, 1959, 215, p. 192.
4359. Spedding F. H., Miller С F. — J. Amer. Chem.
Soc, 1952, 74, p. 3158.
4360. Speiser R., Blackburn P., Johnston H. L. —
J. Electrochem. Soc, 1959, 106, p. 52.
4361. Speiser R., Johnston H. L., Blackburn P. —
J. Amer. Chem. Soc, 1950, 72, p. 4142.
4362. Spencer H. M., Justice J. L. — J. Amer. Chem.
Soc, 1934, 56, p. 2308.
4363. Speros D. M., Woodhouse R. L. — J. Phys. Chem.,
1963, 67, p. 2164.
4364. Spiker R. C, Andrews L. — J. Chem. Phys., 1973,
58, p. 702.
4365. Spiker R. C, Andrews L. — J. Chem. Phys., 1973,
58, p. 713.
4366. Spiker R. C, Andrews L. — J. Chem. Phys., 1973,
59, p. 1851.
4367. Spinar L. #., Margrave J. L. — Spectrochim. acta,
1958, 12, p. 244.
4368. Spindler H. — Naturwissenschaften, 1965, 52,
S. 184.
4369. Spriet B. — Thes. Doct. Sci. Fac. Sci. Univ.
Paris, 1966, 88 p.
4370. Squire С F., Kaufmann A. R. — J. Chem. Phys.,
1941, 9, p. 673.
4371. Sridhar R., Johnson С E., Cairns E. J. — J. Chem.
and Eng. Data, 1970, 15, p. 244.
4372. Srivastava S. K., Cartwright D. C, Trajmar S.,
Chutjian A., Williams W. X. — J. Chem. Phys.,
1976, 65, p. 208.
4373. Stackelberg E. — Z. phys. Chem. Leipzig, 1896,
20, S. 159.
4374. Stackelberg E. — Z. phys. Chem. Leipzig, 1898,
26, S. 533.
4375. Stacy D. W., Johnstone J. K., Wilder D. R. —
J. Amer. Ceram. Soc, 1972, 55, p. 482.
4376. Stalinski В., Bieganski Z. — Rocz. chem., 1961,
35, s. 273.
4377. Stammreich H., Kawai K., Tavares Y. —
Spectrochim. acta, 1959, 15, p. 438.
4378. Stanfield O. M. — J. Amer. Ceram. Soc, 1965,
48, p. 436.
4379. Staskiewicz B. A., Tucker J. R., Snyder P. E. —
J. Amer. Chem. Soc, 1955, 77, p. 2987.
4380. Staub G. — Ann. Phys. Beibl., 1890, 14, S. 493.
4381. Steeb S., Brucklacher D. — J. Less-Common
Metals, 1966, 11, p. 263.
4382. Steel W. С — U. S. Air Force Materials Lab.
Rept AFML-TR-65-343, Pt 3. Wright-Petterson
AFB, Ohio, 1968, Apr.
4383. Steele В. С H., Alcock С. В. — Trans. Met. Soc.
AIME. 1965, 233, p. 1359.
4384. Stein C, Grant N. — J. Metals, 1955, 7, p. 127.
4385. Stephens H. P. — High Temp. Sci., 1974, 6, p. 156.
4386. Stephenson C. C, Abojan P. G., Provost R.,
Wulff С — J. Chem. and Eng. Data, 1968, 13,
p. 191.
4387. Stephenson C. C, Hopkins H. P., Wulff С A.—
J. Phys. Chem., 1964, 68, p. 1427.
4388. Stephenson N. C, Roth R. S. — Acta crystallogr.,
1971, B27, p. 1037.
4389. Stephenson N. C, Roth R. S. — J. Solid State
Chem., 1971, 3, p. 145.
4390. Stern J. #., Anderson С W. — J. Phys. Chem.,
1964, 68, p. 2528.
4391. Stern J. H., Goeders B. L., Withers G. L., Wujs
S. L. — J. Chem. and Eng. Data, 1979, 24, p. 314.
4392. Stern J. H., Kulluk J. D. — J. Phys. Chem.,
1989, 73, p. 2795.
4393. Stern J. #., Lowe E., O'Connor M. E. — J. Solut.
Chem., 1974, 3, p. 823.
4394. Stern Т. Е. — Phys. Rev., 1928, 32, p. 298.
4395. Sterrett K. F., Wallace W- E. — J. Amer. Chem.
Soc, 1958, 80, p. 3176.
4396. Stitch M. L., Honig A., Townes С. Н. — Phys.
Rev., 1952, 86, p. 813.
4397. Stockdale J. A. P., Compton R. N., Schweinler
11. С — J. Chem. Phys., 1970, 53, p. 1502.
4398. Stokes R. H. — J. Phys. Chem. Solids, 1986, 27,
p. 51.
4399. Stone J. Д., Ewing С. Т., Karp R. L., Spann J. R.,
Miller R. R. — J. Chem. and Eng. Data, 1967,
12, p. 352.
4400. Stone J. P., Ewing С. Т., Spann J. R., Stein-
kuller E. W., Williams D. D., Miller R. R. —
U. S. Naval Res. Lab. Rept NRL-6241. Wash.,
1965.
4401. Stone J. P., Ewing С. Т., Spann J. R., Steinkul-
lerE. W., WilliamsD.D., Miller R. R. — J. Chem.
and Eng. Data, 1966, 11, p. 309.
589
Литература
4402. Stone J. P., Ewing С. Т., Spann J. R., Steinkul-
ler E. W-, Williams D. D., Miller R. R. —
J. Chem. and Eng. Data, 1966, 11, p. 315.
4403. Storms E., Calkin В., Yencha A. — High'Temp.
Sci., 1969, 1, p. 430.
4404. Storms E. K., Goffin J. — High Temp. Sci., 1973,
5, p. 291.
4405. Storms E. K., Huber E. J. — J. Nucl. Mater.,
1967, 23, p. 19.
4406. Storms E. K., Krikorian N. H. — J. Phys. Chem.,
1960, 64, p. 1471.
4407. Storms E. K., Lowe А., Baca Е., Griffin J. —
High Temp. Sci., 1973, 5, p. 276.
4408. Storms E. K., McNeal R. J. — J. Phys. Chem.,
1962, 66, p. 1401.
4409. Strassmair #., Stark D. — Z. angew. Phys., 1967,
23, S. 40.
4410. Straumanis M. E., Li H. W. — Z. anorg. und
allg. Chem., 1960, 305, S. 143.
4411. Stretz L. A., Bautista R. G. — Met. Trans., 1974,
5, p. 921.
4412. Stretz L. A., Bautista R. G. — J. Chem. Thermo-
dyn., 1975, 7, p. 83.
4413. Stringat R., Athenour Ch., Femenias J. L. — Ca-
nad. J. Phys., 1972, 50, p. 395.
4414. Stringer J. — J. Less-Common Metals, 1965, 8, p. 1.
4415. Stromberg T. F., Swenson С. А. — Phys. Rev.
Lett., 1962, 9, p. 370.
4416. Stroud J. E., Tripp W. C, Wimmer J. M. —
J. Amer. Ceram. Soc, 1974, 57, p. 172.
4417. Simpler N., Morette A. — С. г. Acad. sci., 1965,
260, p. 1971.
4418. Stubican V. S., Hink R. C, Ray S. P. — J. Amer.
Ceram. Soc, 1978, 61, p. 17.
4419. Stacker N. — Sitzungsber. math.-natur. Wiss. Kl.
Kaiserlichen Akad. Wiss. Wien, 1905, 114, S. 657.
4420. Stull D. R., Hildenbrand D. L., Oetting F. L.,
Sinke G. C. — J. Chem. and Eng. Data, 1970,
15, p. 52.
4421. Stull D. R., Sinke G. C. — Thermodynamic
properties of the elements: Advances in chemistry.
Wash.: Amer. Chem. Soc, 1956, Ser. N 18.
4422. Stwalley W. C. — Chem. Phys. Lett., 1970, 7,
p. 600.
4423. Stwalley W. C, Way K. R., Velasco R. —
J. Chem. Phys., 1974, 60, p. 3611.
4424. Stwalley W. C, Yang S.-C, Hsieh Y.-K., Orth
F. В., Li К. С — J. Chem. Phys., 1978, 69,
p. 1791.
4425. Stwalley W. C, Zemke W. Т., Way K. R., Li К. С,
Proctor T. R. — J. Chem. Phys., 1977, 67, p. 4785.
4426. Stwalley W- C, Zemke W. Т., Way K.R.,LiK.C,
Proctor T. R.—J. Chem. Phys., 1977, 66, p. 5412.
4427. Su T.-M. R., Riley S. J. — J. Chem. Phys., 1980,
72, p. 6632.
4428. Suares С. В. — J. Phys.: Atom, and Mol. Phys.,
1970, B3, p. 729.
4429. Suares С. В. — Chem. Phys. Lett., 1972, 16, p. 515.
4430. Subbarao E. С — J. Amer. Ceram. Soc, 1961,
44, p. 92.
4431. Sudo K., Kigoshi A. — Bull. Res. Inst. Mineral.
Dress, and Metallurgy. Tohoku Univ., 1959, 15,
. p. 31.
4432. Sudo K., Kigoshi A. — Sci. Repts Res. Inst. To-
hoku Univ. Ser. A, 1961, 13, p. 31.
4433. Sudo K., Kigoshi A. — Sci. Repts Res. Inst.
Tohoku Univ. Ser. A, 1961, 13, p. 448.
4434. Sue P. — J. chim. phys. et phys.-chim. biol.,
1939, 36, p. 280.
4435. Sue P. — С r. Acad. sci., 1939, 208, p. 1088.
4436. Sugar J. — J. Chem. Phys., 1973, 59, p. 788.
4437. Sugar /., Corliss Ch. — J. Phys. and Chem. Ref.
Data, 1977, 6, p. 317.
4438. Sugar /., Corliss Ch. — J. Phys. and Chem. Ref.
Data, 1978, 7, p. 1191.
4439. Sugar /., Corliss Ch. — J. Phys. and Chem. Ref.
Data, 1979, 8, p. 1.
4440. Sugar J., Corliss C. — J. Phys. and Chem. Ref.
Data, 1980, 9, p. 473.
4441. Sundaram S. — Z. phys. Chem. (BRD), 1962, 34,
S. 225.
4442. Sunner S., Wadso I. — Acta chem. scand., 1959,
13, p. 97.
4443. Surana S. S. L. — Indian J. Pure and AppL
Phys., 1975, 13, p. 480.
4444. Suzuki K., Sambongi K. — Tetsu to Hagane, 1972,
58, p. 1579.
4445. Suzuki S., Takagi M. — J. Phys. Soc. Jap., 1966,
21, p. 554.
4446. Swallow J. C, Alty b. — J. Chem. Soc, 1931,.
p. 3062.
4447. Swaminathan S., Srinivasan S. — Acta crystal-
logr., 1975, A31, p. 628.
4448. Swaminathan T. W., Krishnamurthy S. G. —
Current Sci., 1954, 23, p. 258.
4449. Swepston P. N., Sellers H. L., Schafer L. —
J. Chem. Phys., 1981, 74, p. 2372.
4450. Szczytowski A., Zyrnicki W- — Pr. nauk. Inst*
chem. nieorgan. i metalurg. pierwiast. rzadkich
Politech. Wroclaw, 1975, N 24, s. 105.
4451. Szwarc R. — J. Phys. Chem. Solids, 1969, 30,.
p. 705.
4452. Szwarc R., Plante E. R., Diamond J. J. — J. Res.
NBS, 1965, A69, p. 417.
4453. Tables internationales de constantes selectionnees-
17. Donnees spectroscopiques relatives aux
molecules diatomiques / Ed. B. Rosen. Oxford etc.:
Pergamon Press, 1970.
4454. Tabrizi D., Gaultier M. — Bull. Soc. chim. France,
1968, N 3, p. 935.
4455. Tabrizi D., Pannetier G. — Bull. Soc. chim.
France, 1969, N 12, p. 4280.
4456. Tabrizi D., Pannetier G. — Bull. Soc. frang.
mineral et cristallogr., 1970, 93, p. 437.
4457. Tacchini G. — Gazz. chim. ital., 1924, 54, p. 777.
4458. Takagi Y., Gesi K. — J. Phys. Soc. Jap., 1964,
19, p. 142.
4459. Takahashi #., Meshitsuka S., Hagasi K. —
Spectrochim. acta, 1975, A31, p. 1617.
4460. Takahashi K., Yoshio T. — J. Ceram. Soc. Jap.,
1970, 78, p. 29.
4461. Takahashi K., Yoshio T. — J. Ceram. Soc. Jap.,
1973, 81, p. 524.
4462. Takahashi Y., Ozawa T. — J. Therm. Anal., 1975,
8, p. 125.
4463. Takei #., Koide S. — J. Phys. Soc. Jap., 1966,
21, p. 1010.
4464. Takeuchi S., Houma Т., Satow Т., Hirei T. —
Trans. Jap. Inst. Met., 1966, 7, p. 59.
4465. Tallman R. L., Margrave J. L. — J. Inorg. and
Nucl. Chem., 1961, 21, p. 40.
4466. Tallon J. L., Robinson W. H. — J. Phys. Chem.,
1978, 82, p. 1277.
4467. Tarn A. C, Happer W. — J. Chem. Phys., 1976,
64, p. 2456.
4468. Tanaka Т., Itorie T. — Proc. Phys. Soc. Jap.,
1941, 23, p. 464.
4469. Tango W- /., Link J. K., Zare R. N. — J. Chem.
Phys., 1968, 49, p. 4264.
590
Литература
4470. Taniewska-Osinska S., Logwinienko R. — Prace 4504.
Lodzkie towarz. nauk. Wydz. Ill, 1974, 18, s. 17.
4471. Taniewska-Osinska S., Logwinienko R. — Acta
Univ. Lodz., Ser. 2, 1976, 6, s. 69. 4505.
4472. Taniewska-Osinska S., Logwinienko R. — Canad. J. 4506.
Chem., 1980, 58, p. 1584.
4473. Tanifuji Т., Shirozawa K., Nasu S. — J. Nucl. 4507.
Mater., 1978, 78, p. 422.
4474. Tanisaki S. — J. Phys. Soc. Jap., 1959,14, p. 680. 4508.
4475. Tanisaki S. — J. Phys. Soc. Jap., 1960, 15, p. 566.
4476. Tanisaki S. — J. Phys. Soc. Jap., 1963, 18,
p. 1181. 4509.
4477. Tanisaki S., Ichimatsu T. — J. Phys. Soc. Jap.,
1965, 20, p. 1277. 4510.
4478. Tashiro M. — Glass. Ind., 1956, 37, p. 549.
4479. Tate P. A., Strandberg M. — J. Chem. Phys., 4511.
1954, 22, p. 1380.
4480. Tatum J. В., Balfour W- J. — I. Mol. Spectrosc, 4512.
1973, 48, p. 292.
4481. Taylor A., Doyle N. J. — J. Less-Common Metals, 4513.
1964, 7, p. 37.
4482. Taylor A., Doyle N. J. — High Temp.-High Pres- 4514.
sur., 1969, 1, p. 679.
4483. Taylor A., Doyle N. J. — In: The chemistry 4515.
of extended defects in non-metallic solids /
Ed. L. R. Eyring, M. O'Keeffe. Amsterdam; Lon- 4516.
don: North-Holland Publ Co., 1970, p. 523. 4517.
4484. Taylor A., Doyle N. J., Kagle B. J. — J. Less-
Common Metals, 1961, 3, p. 265. 4518.
4485. Taylor A., Kagle B. J., Doyle N. J. — J. Less-
Common Metals, 1963, 5, p. 26. 4519.
4486. Taylor A. R., Gardner T. E., Smith D. F. — U. S.
Bur. Mines. Rept Invest., 1963, N 6157. 4520.
4487. Taylor J. В., Langmuir I. — Phys. Rev., 1937,
51, p. 753. 4521.
4488. Taylor J. C, Loasby R. G., Dean D. /., Lin-
ford P. E. — In: Plutonium 1965. Proc. 3d Intern. 4522.
Conf., London, 1965 / Ed. A. E. Kay, M. B. Wal-
deron. L.: Chapman and Hall, 1967, p. 162. 4523.
4489. Taylor J. C, Wilson P. W. — Acta crystallogr.
1973, B29, p. 1073. 4524.
4490. Taylor R. E., Finch R. A. — U.S. North American 4525.
Aviation, Inc. Rept NAA-SR-6034. Downey (Ca- 4526.
lif.), 1961, p. 28. 4527.
4491. Taylor R. E., Finch R. A. — J. Less-Common 4528.
Metals, 1964, 6, p. 283.
4492. Telea C, Ferloni P., Franzosini P. — Gazz. chim. 4529.
ital., 1972, 102, p. 546. 4530.
4493. Tepper F., Murchison A., Zelenak /., Roelich F. — 4531.
U. S. /Air Force Materials Lab. Rept RTD-TDR- 4532.
63-4018, pt 1. Wright-Petterson AFV, Ohio, 1963. 4533.
4494. Tepper F., Murchison A., Zelenak J., Roelich F. — 4534.
U. S. AEC Oak Ridge Nat. Lab. Rept ORNL-3605.
Oak Ridge (Tennessee), 1963 (Publ. 1964), p. 26. 4535.
4495. Terao N. — Jap. J. Appl. Phys., 1967, 6, p. 21.
4496. Tereuchi #., Cohen J. B. — J. Phys. Chem. So- 4536.
lids, 1978, 39, p. 681. 4537.
4497. Terauchi H., Cohen J. В., Reed Т. В. — Acta
crystallogr., 1978, A34, p. 556.
4498. Terenin A. — Z. Phys., 1927, 44, S. 713. 4538.
4499. Terres E. — Brennstoff-Chem., 1954, 35, S. 225.
4500. Tetenbaum M., Hunt P. D. — J. Nucl. Mater., 4539.
1970, 34, p. 86.
4501. Thayer W- L., Robbins J. L. — In: Plutonium 4540.
1975 and other actinides. Proc 5th Intern. Conf.,
Baden-Baden, 1975. Amsterdam etc., 1976, p. 85. 4541.
4502. The Chemistry and Metallurgy of Miscellaneous
Materials. Thermodynamics / Ed. L. L. Quill. 4542.
N. Y. etc., 1950, Pap. 3—7.
4503. Theimer O. — Monatsh. Chem., 1950, 81, S. 424.
Thermophysical properties of high temperature
Solid Materials / Ed. Y. S. Touloukian. N. Y.;
L.: Macmillan, 1967, vol. 1.
Thiele E. — Ann. Phys., 1932, 14, S. 937.
Thilo E., Schroder H. — Z. phys. Chem. Leipzig,
1951 197 S 39
Thoma R.'E., Insley H., Hobert G. M. — J. Amer.
Ceram. Soc, 1963, 46, p. 37.
Thoma R. E., Insley H., Landau В. С, Friedman
H. A., Grimes W. R. — J. Amer. Ceram. Soc,
1959, 42, p. 21.
Thompson W. Т., Flengas S. N. — Canad. J. Chem.,
1971, 49, p. 1550.
Thompson W. Т., Good D. G. W. — Canad. J.
Chem., 1976, 54, p. 3342.
Thomsen J. Thermochemische Untersuchungen.
Leipzig: Verl. von J. A. Barth, 1882—1886.
Thorn R. J., Winslaw G. #., Ziomek J. S. —
J. Nucl. Mater., 1979, 84, p. 416.
Thorvaldson Т., Brown W. G., Peaker C. R. —
J. Amer. Chem. Soc, 1929, 51, p. 2678.
Thourey J., Perachon G. — Thermochim. acta,
1980, 39, p. 243.
Thourey J., Perachon G., Germain P. — J. Fluor.
Chem., 1980, 15, p. 315.
Tichelaar G. W. Цит. по: [3771].
Tilden W- A. — Proc. Roy. Soc. London, 1885,
38, p. 401.
Todd S. S. — J. Amer. Chem. Soc, 1950, 72,
p. 2914.
Todd S. S. — J. Amer. Chem. Soc, 1953, 75,
p. 1229.
Todd S. S. — J. Amer. Chem. Soc, 1953, 75,
p. 3035.
Todd S. S., Bonnickson K. R. — J. Amer. Chem..
Soc, 1951, 73, p. 3894.
Tonosaki K. — Bull. Inst. Phys. Chem. Res.
Tokyo, 1940, 19, p. 126.
Topol L. E., Owens В. В. — J. Phys. Chem., 1968,
72, p. 2106.
Topor L. — J. Chem. Thermodyn., 1972, 4, p. 739.
Topor L. — Stud, cercet. chim., 1972, 20, p. 1269.
Topor L. — Rev. roum chim., 1974, 14, p. 1569.
Topor L. — J. Chem. Thermodyn., 1976, 8, p. 777.
Torgeson D. R. — Ind. and Eng. Chem., 1948,
40, p. 1152.
Toueg J. — Diss. Abstrs. 1975, B36, p. 2874.
Touzain P. _ Canad. J. Chem., 1969, 47, p. 2639.
Touzain P. — С. г. Acad. sci., 1973, C276, p. 1583.
Touzain P. — С. г. Acad. sci., 1974, C279, p. 41.
Touzain P. — J. Therm. Anal., 1976, 9, p. 441.
Touzain P., Brisse F. — Canad. J. Chem., 1970,
48, p. 3358.
Touzain P., Caillet M. — Rev. chim. miner., 1971,
8, p. 277.
Tramer A. — C. r. Acad. sci., 1959, 248, p. 3546.
Trarieux H., Bernier J. C. — In: Influence chan-
gements phase prop. phys. Corpusculars solides.
P.: Masson et c*e, 1970, p. 101.
Treadwell W- D., Werner W- — Helv. chim. acta,
1953, 36, p. 1436.
Tremmel J., Morlin Z. — Radex Rundsch., 1967,
N 2, S. 449.
Treswjatskij S. G., et al. — Colloq. int. Centre
nat. rech. sci., 1971, N 205, p. 247.
Tretjakow J. D., Schmalzried H. — Ber. Bunsen-
ges. phys. Chem., 1965, 69, S. 396.
Treverton J. A., Margrave J. L. — In: Proc.
5th Symp. on Thermophys. Properties / Ed. С F.
Bonilla. N. Y-: ASME, 1970, p. 489.
591
Литература
4543. Treverton J. A., Margrave J. L. — l. Ghem. 4582.
Thermodyn., 1971, 3, p. 473.
4544. Triplett В. В., Phillips N. E., Thorp T. L., Shir- 4583.
ley D. A., Brewer W- D. — J. Low Temp. Phys.,
1979, 12, p. 499. 4584.
4545. Trischka J. W. — Phys. Rev., 1948, 74, p. 718.
4546. Trischka J. W. — Phys. Rev., 1949, 76, p. 1365. 4585.
4547. Trischka J.W- — J. Chem. Phys., 1952,20, p. 1811.
4548. Trischka J. W-, Braunstein R. — Phys. Rev., 1954, 4586.
96, p. 968.
4549. Trombe F., Foex M., La Blanchetais С. Н. — 4587.
С. г. Acad. sci., 1946, 223, p. 317.
4550. Trombe F., Foex M., La Blanchetais С. H. — Ann. 4588.
chim., 1947, 2, p. 385.
4551. Trombe M. F. — С. г. Acad. sci., 1932,194, p. 1653. 4589.
4552. Troost L., Hautefeuille P. — C. r. Acad. sci., 1874,
78, p. 807.
4553. Truchot M. — C. r. Acad. sci., 1884, 98, p. 1330. 4590.
4554. Trugman S., Gordon R. G J. Ghem. Phys., 4591.
1976, 64, p. 4625.
4555. Trulson 0. C, Goldstein H. W. Mass spectrometric 4592.
study of zirconium diboride: Techn. Rept N C-25, 4593.
Union Carbide Research Institute. Wash.: 1964,
Oct.
4556. Trulson O. C, Goldstein H. W. — J. Phys. Chem.,
1965, 69, p. 2531. 4594.
4557. Trzebiatowski W., Horyn R. — Bull. Acad. polon.
sci. Ser. sci. chem., 1965, 13, p. 303. 4595.
4558. Trzebiatowski W-, Horyn R. — Bull. Acad. polon.
Sci. Ser. sci. chim., 1965, 13, p. 311. 4596.
4559. Tsang T. W. E., Gschneidner K. A. J., Schmids
F. A. — In: Proc. Rare Earth Res. Conf. 12th.
1976, vol. 2, p. 847.
4560. Tsuboi M., Terada M., Shimanouchi T J. Chem. 4597.
Phys., 1962, 36, p. 1301. 4598.
4561. Tummavuori /., Lumme P. — Acta chem. scand., 4599.
fcf 1968, 22, p. 2003.
4562.. TurkdoganiE. T. — J. Iron Steel Inst. London, 4600.
1954, 178, p. 278.
4563. Turnbull A. G. — J. Phys. Ghem., 1961, 65, 4601.
p. 1652.
4564. Туе R. P., Desjarlais A. O., Bourne J. G. — 4602.
In: Proc. 7th Symp. on Thermophys. Properties / 4603.
Г3 Ed. A. Cezairliyan. N. Y.: ASME, 1977, p. 189. 4604.
4565. Uchida Y. — Jap. J. Phys., 1929, 5, p. 145.
4566. Uchida Y. — Jap. J. Phys., 1932, 8, p. 25. 4605.
4567. Uchijima Т., Naito K., Mukaibo T. — J. Atomic
Energy Soc. Jap., 1962, 4, p. 111. 4606.
4568. Veda Y. — Nippon Kagaku Zasshi, 1931, 52, p. 740.
4569. Veda Y. — Sci. Repts Tohoku Imp. Univ. Ser. I,
1933, 22, p. 448. 4607.
4570. Veda Y. — Sci. Repts Tohoku Imp. Univ. Ser. I,
1933, 22, p. 879. 4608.
4571. Veda Y., Kosuge K., Kachi S. — J. Solid State
Chem., 1980, 31, p. 171. 4609.
4572. Veno K. — J. Chem. Soc. Jap., 1941, 62, p. 990. 4610.
4573. Ugarov V. V., Ezhov Yu. S., Rambidi N. G. —
J. Mol. Struct., 1975, 25, p. 357. 4611.
4574. Vhler V. — Ark. fys., 1954, 8, s. 265.
4575. Vhler U. — Ark. fys., 1954, 8, s. 295. 4612.
4576. Vhler V., Akerlind L. — Ark. fys., 1956, 10, s. 431.
4577. Vhler V., Akerlind L. — Naturwissenschaften, 4613.
1959, 46, S. 488. 4614.
4578. Vhler V., Akerlind L. — Ark. fys., 1961, 19, s. 1.
4579. Vmino S. — Sci. Repts Tohoku Imp. Univ. Ser. I, 4615.
1926, 15, p. 597.
4580. Vrey H. С — U. S. Rept A-330, Oct. 20, 1942. 4616.
Цит. по: [492, с 301].
4581. Vskokovic D. P., Ristic M. M. — Inst. Tech. Sci. 4617.
Serb. Acad. Sci., Belgrade, Yugoslavia, 1976, s. 97.
Vy О. М., Drowart J. — High. Temp. Sci., 1970,
2, p. 293.
Vahldiek F. W. — J. Less-Common Metals, 1966,
11, p. 99.
Vaiday S. N., Kennedy G. С J. Phys. Chem.
Solids, 1971, 32, p. 951.
Vajda E., Hargittai I., Trommel J. — Inorg. chim.
acta, 1977, 25, p. 143.
Vance J. E., Huffman J. K. — Phys. Rev., 1935,
47, p. 215.
Van Leirsburg D. A., DeKock C. W. — J. Phys.
Chem., 1974, 78, p. 134.
Van Tassel J. H., Wendlandt W. W. — J. Amer.
Ghem. Soc, 1959, 81, p. 813.
Van Veen N. J. A., DeVries M. S., Sokol J. D.,
Bailer Т., DeVries A. E. — Chem. Phys., 1981,
56, p. 81.
Varali-Thevenet A. — Nuovo cim., 1902, 4, p. 186.
Vardia M. S. — Astron. and Astrophys., 1970,
5, p. 162.
Varet R. — Ann. chim. phys., 1896, 8, p. 240.
Vargafiic N. В., Volyak L. G., Stepanov V. G.,
Chelebayev A. K., Kozhevnikov V. F., Alekseev
V. A., Ryzhkov Yu. F. — High Temp.-High Pres-
sur., 1976, 8, p. 659.
Vasilev C, Nikolov Т., Chimbulev M. — Trans.
Inst. Mining and Met., 1968, C77, p. 36.
Veazey S. E., Gordy W- — Phys. Rev, 1965, A138,
p. 1303.
Vedernikov M. V., Terehov G. I., Ioffe A. F. —
In: Proc. Intern. Conf. Electron. Struct. Actini-
des, 2nd. 1976. Wroclaw: Pol. Akad. Nauk, 1977,
p. 263.
Velasco R. — Canad. J. Phys., 1957, 35, p. 1204.
Velasco R. — Opt. pura у apl., 1974, 7, p. 14.
Velasco Д., Ottinger Ch., Zare R. N. — J. Chem.
Phys., 1969, 51, p. 5522.
Velasco R., Rivero F. — Opt. pura у apl., 1974,
7, p. 45.
Velasco R., Ruano O., Rico F. R. — Opt. pura
y. apl., 1970, 3, p. 159.
Veleckis E. — J. Phys. Chem., 1977, 81, p. 526.
Veleckis E. — J. Nucl. Mater., 1979, 79, p. 20.
Veleckis E., Van Deventer E. H., Blander M- —
J. Phys. Chem., 1974, 78, p. 1933.
Verdan M-, Monnier R. — Rev. int. hautes temp.
et refract., 1972, 9, p. 205.
Verhaegen G., Smoes S., Drowart J. — U. S. Wright
Air Development Div. Rept. WADD-TR-30-782,
part XXVIII. Wright-Petterson AFB, Ohio, 1965.
Verma A ., Johnson K. E., Sherman E. O. — Canad.
J. Chem. Eng., 1976, 54, p. 285.
Vernay Anne-Marie. —. These doct. Univ. Claude
Bernard-Lyon, 1972, 87 p.
Veseth L. — Phys. ser., 1975, 12, p. 125.
Victor A. C, Douglas Т. В. — J. Res. NBS, 1961,
465, p. 105.
Vidal R., Li K. C, Orth F. В., Stwalley W- C. —
to be publ.
Vidale G. L. — U. S. General Electric Company,
Space Sci. Lab. TIS. Rept R 60SD, 1980.
Vidale G. L. — J. Phys. Chem., 1950, 64, p. 314.
Viechnicki D., Stubican V. S. —- J. Amer. Caram.
Soc, 1965, 48, p. 292.
Villar R., Gmelin E., Vieira S. — Solid State Com-
muns, 1981, 38, p. 807.
VillarsD. S. — Proc. Nat. Acad. Sci. U. S., 1928,
14, p. 508.
Vittalachar V. V., Krishianurthy S. G. —• Current
Sci., 1954, 23, p. 357.
592
Литература
4618.
4619.
4620.
4621.
4622.
4623.
4624.
4625.
4626.
4627.
4628.
4629.
4630.
4631.
4632.
4633.
4634.
4635.
4636.
4637.
4638.
4639.
4640.
4641.
4642.
4643.
4644.
4645.
4646.
4647.
4648.
4649.
4650.
4651.
Vogel К., Топп W. — Z. anorg. und allg. Chem.; 4652.
1931 201 S 293
Volger /.'—Nature, 1952, 170, p. 1027. 4653.
Vollmer O., Braun M., Kohlhaas K. — Z. Natur-
forsch., 1967, A22, S. 833.
Vozzella P. A., Miller A. D., DeCrescente A. —
J. Chem. Phys., 1964, 41, p. 589.
Vrzhesnevskii I. B. — Z. anorg. Chem., 1912, 74,
S 95
Wach'i F. M-, Gilmartin D. E. — High Temp. Sci.,
1972, 4, p. 423.
WackeriP. F., Cheney R. K. — l. Res. NBS,
1947, 39, p. 317.
Wadt W. R., Hay P. J. — J. Amer. Chem. Soc,
1979, 101, p. 5198.
Wagman D. D., Evans W. #., Parker V. В.,
Hallow J., Bailey S. M., Schumm R. H. — In:
Selected values chemical thermodynamic properties.
NBS, Techn. Notes, 1965, N 270-1; 1966, N 270-2,
1968, N 270-3; 1969, N 270-4; 1971, N 270-5;
1971, N 270-6; 1973, N 270-7; 1981, N 270-8.
Wagman D. D., Kilday M. V. — l. Res. NBS,
1973, A77, p. 569.
Wagner E. L Theor. chim. acta, 1974, 32,
p. 295.
Wagner K., Schafer H. — Z. anorg. und allg.
Chem., 1979, 450, S. 115.
Wagner K., ScMfer H. — Z. anorg. und allg.
Chem., 1979, 451, S. 57.
Wagner O. G. — Z. phys. Chem. Leipzig, 1928,
131, S. 409.
Wahlbeck P. G., Gilles P.'W. — J. Amer. Ceram.
Soc, 1966, 49, p. 180.
Wahlbeck P. G., Gilles P. W.— J. Chem. Phys.,
1967, 46, p. 2465.
Wahlin H. В., Whitney L. V. — Phys. Rev., 1936,
50, p. 735.
Wailling J. F. — J. Phys. Chem., 1963, 67,
p. 1380.
Wailling J. F., Nuzum H. K., Lemmon A. W. —
NASA. Rept CR-52425, 1963.
Wait E. — J. Inorg. and Nucl. Chem., 1955, 1,
;p. 309.
Wait S. C, Ward А. Т., Jam G. J. — J. Chem.
Phys., 1966, 45, p. 133.
Waite T. R., Craig R. S., Wallace W. E. — Phys.
Rev., 1956, 104, p. 1240.
Walden P. T. — Z. phys. Chem. Leipzig, 1907,
58, S. 479.
Wallace D. C. — Phys. Rev., 1960, 120, p. 84.
Wallace W- F. ~ 1. Amer. Chem. Soc, 1949, 71,
p. 2485.
Walsh P. N., Dever D. F., White D. — J. Phys.
Chem., 1961, 65, p. 1410.
Walsh P. N., Goldstein H. W., White D. —
J. Amer. Ceram. Soc, 1960, 43, p. 229.
Walsh P. N., White D. — U. S. Ohio State Univ.
Techn. Note N 2, 1958, p. 22 (Nucl. Sci. Abstrs,
1958, 12, p. 113).
Walter J. M-, Barrat S. — Proc. Roy. Soc.
London, 1928, A119, p. 257.
Wang С. С, Grant N. J. — 1. Metals, 1956, 8,
p. 184.
Wang E. Y. — 1. Electrochem. Soc. 1976, 123,
p. 435.
Warekois E. P. — J. Appl. Phys., 1960, 31, p. 346.
Waring J. L., Roth R. S. — J. Res. NBS, 1968,
A72, p. 175.
Waring J. L., Roth R. S., Parker H. S. — J. Res.
NBS, 1973, A77, p. 705.
4654.
4655.
4656.
4657.
4658.
4659.
4660.
4661.
4662.
4663.
4664.
4665.
4666.
4667.
4668.
4669.
4670.
4671.
4672.
4673.
4674.
4675.
4676.
4677.
4678.
4679.
4680.
4681.
4682.
4683.
Warren W. W., Gossard A. C. — J. Appl. Phys..
1970, 41, p. 881.
Wartenberg H. — Z. Elektrochem., 1909, 15,
S. 866.
Wartenberg H. — Z. Electrochem., 1914, 20,
Wartenberg H. — Z. anorg. und allg. Chem., 1959,
299, S. 227.
Wartenberg H., Albrecht P. — Z. Elektrochem..
1921, 27, S. 162.
Wartenberg H., Aoyama S. — Z. Elektrochem.,
1927, 33, S. 144.
Wartenberg H., Eckhardt K. — Z. anorg. und allg.
Chem., 1937, 232, S. 179.
Wartenberg H., Fitzner O. — Z. anorg. und allg.
Chem., 1926, 151, S. 313.
Wartenberg H., Prophet E. — Z. anorg. und allg.
Chem., 1932, 208, S. 369.
Wartenberg H., Reusch H. J. — Z. anorg. und
allg. Chem., 1932, 207, S. 1.
Wartenberg H., Reusch H. J. — Z. anorg. und
allg. Chem., 1932, 208, S. 369.
Wartenberg H., Schulz H.—Z. Elektrochem.,
1921, 27, S. 568.
Washburn C. A. — Diss. Abstrs, 1969, B30,
p. 2215.
Wassermann A. —Z. phys. Chem. Leipzig, 1930,
A146, S. 409.
Watanabe D., Castles J. R., Jostsons A., Malin
A. S. — Nature, 1966, 210, p. 934.
Watanabe D., Castles J. R., Jostsons A., Malin
A. S. — Acta crystallogr., 1967, 23, p. 307.
Watanabe D., Terasaki O., Jostsons A., Castles
J. R. — In: Chemistry of extended defects non-
metallic solids. Amsterdam: North-Holland Publ.
Co, 1970, p. 238.
Watanabe N Bull. Electrotechn. Lab. (Jap.),
1966, 30, p. 923.
Watanabe Т., Sakai K., Iwai S. — Bull. Tokyo
Inst. Technol., 1973, N 177, p. 13.
Waterfield C. G., Linford R. G., Goalby В. В.,
Bates T. R., Elyard С A., Staveley L. A. K. —
Trans. Faraday Soc, 1968, 64, p. 868.
Watson D. K., Cerjan С /., Guberman S., Dal-
garno A. —Chem. Phys. Lett., 1977, 50, p. 181.
Way К. Д., Stwalley W. С — J. Chem. Phys.,
1973, 59, p. 5298.
Weatherford W. D., Johnston R. K., Valtierra
M. L., Roades J. V. — U. S. Aeronautical Systems
Div. Rept ASD-TDR-63-413. Ohio:
Wright—Patterson AFB, 1963.
Weaver С F., Thoma R. E., Friedman H. A.,
Hebert G. M. — J. Amer. Ceram. Soc, 1961, 44,
p. 146.
Webb F. J., Wilks J. — Proc. Roy. Soc. London,
1955, A230, p. 549.
Weber R., Street R. — J. Phys. F.: Metal. Phys.,
1972, 2, p. 873.
Wedekind E., Lewis S. J. — Proc. Amer. Chem.
Soc, 1908, 24, p. 170.
Wegdam G. H., Elsken J. — Solid State Communs,
1971, 9, p. 1867.
Weidenthaler P. — J. Phys. and Chem. Solids,
1964, 25, p. 1491.
Weijma H., Arends J. — Phys. status solidi, 1969,
35, p. 205.
Weiler J. — Ann. Phys., 1929, 1, S. 361.
Weinstock В., Crist R. H. — J. Chem. Phys.,
1948, 16, p. 436.
593
Литератур»
4684. Weinstock В., Goodman G. L. — Advan. Chem.
Phys., 1965, 9, p. 169.
4685. Weiss L., Kaiser H. — Z. anorg. und allg. Chem.,
1910, 65, S. 345.
4686. Weiss L., Martin A., Stimmelmayr A. — Z. anorg
und allg. Chem., 1910, 65, S. 279.
4687. Weiss L., Neumann E. — Z. anorg. und allg.
*¦"" Chem., 1910, 65, S. 248.
46.88. Weizel W., Kulp M. — Ann. Phys., 1930, 4,
"~yr S. 971.
46Д9. Welch D. O., Lazareth O. W., Dienes G. /.,
Hatcher R. D. — J. Chem. Phys., 1976, 64, p. 835.
4690. Welch F. H. Lithium hydride properties. Rept
DC-61-3-73. Aricraft Nuclear Propulsion
Department, 1962, March.
4691. Welch F. H., Northrup H. W., Mink W. #.,
Lemmon A. W. — U. S. AEC. Rept TID-12738,
1961. 17 p.
4692. Weller W. W., Kelley K. K. — U. S. Bur. Mines.
Rept Invest., 1962, N 5984.
4693. Weller W. W., King E. G. — U. S. Bur. Mines.
Rept Invest., 1963, N 6245.
4694. Wells J. C, Jr, Gruber J. B. — Chem. Phys.,
1977, 24, p. 391.
4695. Wells P., Lanchester P. C, Joney D. W., Jordan
R. G.— J. Phys. F: Metal. Phys., 1976, 6, p. 11.
4696. Weltner W. — In: Advances in High Temperature
Chemistry/Ed. L. Eyring. N. Y., L.: Acad. Press,
1969, vol. 2, p. 85.
4697. Weltner W., McLeod D. — J. Chem. Phys., 1965,
42, p. 882.
4698. Weltner W., McLeod D. — J. Phys. Chem., 1965,
69, p. 3488.
4699. Weltner W., McLeod D. — J. Mol. Spectrosc,
1965, 17, p. 276.
4700. Weltner W., McLeod D., Kasai P. H. — J.
Chem. Phys., 1967, 46, p. 3172.
4701. Welty J. R., Wicks С E., Boren H. O. — U. S.
Bur. Mines. Rept Invest., 1963, N 6155.
4702. Wenger L. E., Keesom P. H. — Phys. Rev.,
1975, B12, p. 5288.
4703. Wenger L. E., Keesom P. H. — Phys. Rev.,
1977, B15, p. 5953.
4704. Wentink Т., Spindler R. J. — J. Quant. Spectrosc.
and Radiat. Transfer, 1972, 12, p. 1569.
4705. Weppner W., Huggins R. A. — Solid State Ionics,
1980, 1, p. 3.
4706. Wesley R. D., De Коек С W. — J. Chem. Phys.,
1971, 55, p. 3866.
4707. Wesley R. D., De Коек С W. — J. Phys. Chem.,
1973, 77, p. 466.
4708. West С D. — Z. Kristallogr., 1934, 88, S. 94.
4709. West E. D., Ishihara S. — U. S. NBS, Rept 9803.
Wash., 1968.
4710. Westman S. — Acta chem. scand., 1961, 15, p. 217.
4711. Westrum E. F. — In: Thermodyn. Nucl. Mater.
Proc. Symp. Vienna, 1965. Vienna: IAEA, 1966,
p. 497.
4712. Westrum E. F., Eyring L. — J. Amer. Chem. Soc,
1952, 74, p. 2045.
4713. Westrum E. F., Gr0nvold F. — J. Amer. Chem.
Soc, 1959, 81, p. 1777.
4714. Westrum E. F., Grvnvold F. — In: Thermodyn.
Nucl. Mater. Proc. Symp. Vienna, 1962. Vienna:
IAEA, 1963, p. 3.
4715. Westrum E. F., Pitzer K. S. — J. Amer. Chem.
Soc, 1949, 71, p. 1940.
4716. Westrum E. F., Takahashi Y., Gr0nvold F. —
J. Phys. Chem., 1965, 69, p. 3192.
4717. Wharton L., Klemperer W., Gold L. P., Strauch R.,
Gollagher J. /., Derr V. E. — J. Chem. Phys.,.
1963, 38, p. 1203.
4718. Wheatley Q. D., Sheldon R. /., Gilles P. W. —
J. Chem. Phys., 1977, 66, p. 3712.
4719. Wheeler V. J., Dell R. M., Wait E. — J. Inorg.
and Nucl. Chem., 1964, 26, p. 1829.
4720. White D., Chou Chien, Johnston H. L. — Phys.
Rev., 1958, 109, p. 797.
4721. White D., Sechadri K. S., Dever D., Mann D. E.,
Linevsky M. J. — J. Chem. Phys., 1963, 39,
p. 2463.
4722. White G. K., Sheard F. W. — J. Low Temp.
Phys., 1374, 14, p. 445.
4723. White W. В., Roy R. — Amer. Ceram. Soc. Bull.,
1963, 42, p. 203.
4724. Wigner E., Witmer E. E. — Z. Phys., 1929, 51,.
S. 859.
4725. Wijbenga G., Cordfunke E. H. P., Johnson G. K. —
В печати, 1982. Цит. по: [1932].
4726. Wilhelm H. A., Carlson О. N., Dickinson J. M. —
Trans. Met. Soc. AIME, 1954, 200, p. 915.
4727. Wilhelmi K. A., Jonsson O. — Acta chem. scand.T
1958, 12, p. 1532.
4728. Wilkins R. L. — J. Chem. and Eng. Data, I960,,
5, p. 337.
4729. Wilkins R. L., Loduig R. M., Greene S. A. — In:
8th Symp. Combustion. Pasadena (Calif.), 1960.
Publ. 1962, p. 375.
4730. Williams D. E., Pechin W. H. — Trans. Amer..
Soc. Metals, 1958, 50, p. 1081.
4731. Williams I. S., Gopal E. S. R., Street R. — J.
Phys. F.: Met. Phys., 1979, 9, p. 431.
4732. Williams J. T. — Trans. Met. Soc. AIME, 1955,.
203, p. 345.
4733. Williamson K., Li P., Delvin J. P. — J. Chem.
Phys., 1968, 48, p. 3891.
4734. Willis В. Т. М. — J. phys. (Fr.), 1964, 25, p. 431.
4735. Willis B. T. M., Taylor R. J. — Phys. Lett.,
1965, 17, p. 188.
4736. Willmann G. — Z. anal. Chem., 1973, 266, S. 21.
4737. Wilmshurst J. K. — J. Chem. Phys., 1963, 39,
p. 1779.
4738. Wilmshurst J. K., Senderoff S. — J. Chem. Phys.,
1961, 35, p. 1078.
4739. Wilson H. W., Beam K. W., Cooper W. I. —
U. S. Aeronautical Systems Division. Rept
ASD-TR-61-187. Ohio: Wright-Petterson AFB,
June 30, 1961.
4740. Wilson W. B. — J. Inorg. and Nucl. Chem.,
1961, 19, p. 212.
4741. Wilson. W. В., Austin A. E., Schwarts С. М. —
Trans. Met. Soc. AIME, 1958, 212, p. 52.
4742. Winkelmann A. — Ann. Phys., 1973, 149, S. 1.
4743. Wisnyi L. G., Pijanawski S. W. — U. S. AEC
Knolle Atomic Power Lab. Rept KAPL-1702.
Schenectady (N. Y.), 1957.
4744. Woemer R. F., Wakefield G. F. — Rev. ScL
Instrum., 1962, 33, p. 1456.
4745. Wdhler L. — Z. anorg. und allg. Chem., 1907,
120 S 199.
4746. Wohler L., Balz O. — Z. Elektrochem., 1921,
27, S. 406.
4747. Wdhler L., Gunther R. — Z. Elektrochem., 1923,
29, S. 276.
4748. Wohler L., Prager W. — Z. Elektrochem., 1917,
23, S. 199.
4749. Wdhler L., Shibata Z., Kunst R. — Z.
Elektrochem., 1932, 38, S. 808.
4750. Wolcott N. M. — In: Proc. conf. phys. de basses-
594
Литература
temp., Paris, 1955. P.: Inst. intern, froid, 1955,
p. 286.
4751. Wolcott N. M. — Philos. Mag., 1957, 2, p. 1246.
4752. Wolcott N. M., Hein R. A. — Philos. Mag.,
1958, 3, p. 591.
4753. Wolf A. S., Posey J. C, Rapp K. E. — Inorg.
Chem., 1965, 4, p. 751.
4754. Wolff P. M. — Acta crystallogr., 1961, 14, p. 322.
4755. Wolten G. M. — J. Amer. Ceram. Soc, 1963, 46,
p. 418.
4756. Wood G. J., Bursill L. A. — Proc. Roy. Soc.
London, 1981, A375, p. 105.
4757. Wood J. H., Boring M., Woodruff S. B. — J.
Chem. Phys., 1981, 74, p. 5225.
4758. Wood L. J., Breithaupt L. J. — J. Amer. Chem.
Soc, 1952, 74, p. 2355.
4759. Wood L. J., Secunda W., McBrige С. Н..~ J.
Amer. Chem. Soc, 1958, 80, p. 307.
4760. Wood L. J., Sweeney C, Derbes S. M. T. — J.
Amer. Chem. Soc, 1959, 81, p. 6148.
4761. Wood R. W. — Proc. Amer. Acad. Arts and Sci.,
1906, 42, p. 235.
4762. Wood R. W. — Philos. Mag., 1908, 15, p. 581.
4763. Wood R. W., Gait R. H. — Astrophys. J., 1911,
33, p. 72.
4764. Wood R. W., Hackett F. E. — Astrophys. J.,
1909, 30, p. 1339.
4765. WorkD. — J. Chem. Thermodyn., 1981,13, p. 491.
4766. Worley R. D., Zemansky M. W., Boorse H. A. —
Phys. Rev., 1955, 99, p. 447.
4767. Worner H. W. — Austral. J. Sci. Res., 1951,
A4, p. 62.
4768. Worrell W. L. — J. Phys. Chem., 1964, 68,
p. 952.
4769. Worrell W. L. Measurements of the thermodynamic
stabilities of the Niobium and Tantalum oxides
using a high temper, galvanic cell. Univ. of
California, UCRL-11957 Paper SM-66166. Berkley
(Calif.), 1965.
4770. Worthing A. G. — Phys. Rev., 1917, 10, p. 377.
4771. Worthing A. G. — Phys. Rev., 1918, 12, p. 199.
4772. Worthing A. G. — Z. Phys., 1924, 22, S. 9.
4773. Worthing A. G. — Phys. Rev., 1925, 25, p. 846.
4774. Wouch G., Gray E. L., Frost R. Т., Lord A. E. —
High Temp. Sci., 1978, 10, p. 241.
4775. Wu С. H. — J. Chem. Phys., 1976, 65, p. 2040.
4776. Wu С. Н. — J. Chem. Phys., 1976, 65, p. 3181.
4777. Wu С #., Hepler L. G. — J. Chem. and Eng.
Data, 1962, 7, p. 536.
4778. Wu C. H., Kudo H., Ihle H. R. — J. Chem.
Phys., 1979, 70, p. 1815.
4779. Wu Hung Yuan, Wahlbeck P. G. — High Temp.
Sci., 1971, 3, p. 469.
4780. Wu Hung Yuan, Wahlbeck P. G. — J. Chem.
Phys., 1972, 56, p. 4534.
4781. Wu Y.-Ch., Friedman H. L. — J. Phys. Chem.,
1966, 70, p. 501.
4782. Wurm K. — Naturwissenschaften, 1928, 16,
S. 1028.
4783. Wurm K. — Z. Phys., 1929, 58, S. 562.
4784. Wurm K. — Z. Phys., 1929, 59, S. 35.
4785. Wurm K.—Z. Phys., 1936, 13, S. 199.
4786. Wust F., Meuthen A., Durrer R. — Forsch. Arb.
Ver. Deutsch. Ing., 1918, Heft N 204.
4787. Wust J., Lange E. — Z. phys. Chem. Leipzig,
1925, 116, S. 161.
4788. Wyart J. F. — Phys. scr., 1978, 18, p. 87.
4789. Wyart J. F., Kaufman V., Sugar J. — Phys.
scr., 1980, 22, p. 389.
4790. Wyckoff S., Clegg R. E. S. — Mon. Not. Roy.
Astron. Soc, 1978, 184, p. 127.
4791. Yaffe I. S., Van Artsdalen E. R. — J. Phys.
Chem., 1956, 60, p. 1125.
4792. Yamada Т., Mizuno M., Sekiya Т., Noguchi T. —
Rept Govern. Industr. Res., Inst., Nagoya, 1971,
20, p. 172.
4793. Yamada Y., Shibuya I., Hoshino S. — J. Phys.
Soc. Jap., 1963, 18, p. 1594.
4794. Yang S. C., Hsieh Y. K., Verma К. К., Stwal-
ley W. C. — J. Mol. Spectrosc, 1980, 83, p. 304.
4795. Yates В., Wostenholm G. H., Bingham J. L. ~ J.
Phys.: Solid State Phys., 1974, C7, p. 1769.
4796. Yates J. H., Pitzer R. M. — J. Chem. Phys.,
1977, 66, p. 3592.
4797. Yeh H. J., Radle J. L. — J. Phys. Chem., 1968,
72, p. 3688.
4798. Yeranos W. A.—Z. phys. Chem. (BRD), 1965,
45, S. 72.
4799. Yokokawa H., Kleppa O. J., Nachtrieb N. Й. —
J. Chem. Phys., 1979, 71, p. 4099.
4800. Yoshida I., Sawada S. — J. Phys. Soc. Jap.,
1961, 16, p. 2467.
4801. Yoshida I. V. P., Hase J. — Appl. Spectrosc,
1978, 32, p. 590.
4802. Yoshida, Y., Matsushima T. — Keikinzoku, 1969,
19, p. 488.
4803. Yoshimine M. — J. Chem. Phys., 1972, 57, p. 1108.
4804. Zachariasen W. H. — Acta crystallogr., 1948, 1,
p. 265.
4805. Zachariasen W. H. — Acta crystallogr., 1948, 1,
p. 277.
4806. Zachariasen W. H. — Acta crystallogr., 1949, 2,
p. 296.
4807. Zachariasen W. H. — Acta crystallogr., 1949, 2,
p. 388.
4808. Zachariasen W. H. — Acta crystallogr., 1964, 17,
p. 749.
4809. Zakzhevskii V. G., Boldyrev A. I., Charkin O. P. —
Chem. Phys. Lett., 1980, 70, p. 147.
4810. Zalabak С F. NASA Techn. Rept TN-D-761,
Wash., 1961.
4811. Zalubas R., Corliss Ch. H. — J. Res. NBS, 1974,
A78, p. 163.
4812. Zarzyski G. — Colloq. intern. Centre nat. rech.
sci., 1952, N 39. Electrolyse, p. C34.
4813. Zarzyski G. — Ann. phys. (Fr.), 1953, 8, p. 392.
4814. Zawidzki J., Schagger A. — Kosmos, 1910, 35,
p. 498.
4815. Zeeb K. G., bowings M. G., McCurdy K. G.,
Hepler L. G. — Thennochim. acta, 1980, 40, p. 245.
4816. Zeegers P. J. Т., Alkemade С. Т. J. — Combust.
and Flame, 1965, 9, p. 247.
4817. Zeegers P. J. Т., Alkemade С. Т. J. — Combust,
and Flame, 1970, 15, p. 193.
4818. Zeldes H. Report by Miles F. T. in Division D.
Monthly Rept M-2589 (DE), Jan. 5, 1946. Пит.
no: [492, с 302].
4819. Zemczuzny S., Rambach F. — Z. anorg. und
allg. Chem., 1910, 65, S. 403.
4820. Ziegler M., Rosenfeld M., Kanzig W., Fischer
P. — Helv. phys. acta, 1976, 49, p. 57.
4821. Ziegler W. Т., Bruksch W. F., Hickman J. W. —
Phys. Rev., 1942, 62, p. 354.
4822. Ziger H. G., Bolef D. — Phys. Rev., 1952, 85,
p. 788.
4823. Zimm В., Mayer J. — J. Chem. Phys., 1944, 12,
p. 362.
4824. Zintl E., Baumboch H. H. — Z. anorg. und allg.
Chem., 1931, 198, S. 88.
595
Литература
4825. Zmbov К. F. — In: Proc. first, intern, conf. on
calorimetry and thermodynamics, Warsaw, 31
Aug. — 4 Sept. Pol. sci. publ., 1969, p. 423.
4826. Zmbov K. F., Wu C. H., Ihle H. R. — J. Chem.
Phys., 1977, 67, p. 4603.
4827J-Zukowsky G. J. — Z. anorg. und allg. Chem.,
1911, 71, S. 403.
4828. Zumsteg A., Ziegler M., Bosch M., Kanzig W. —
Helv. phys. acta, 1973, 46, p. 15.
4829. Zwicker С — Physica, 1925, 5, p. 249.
4830. Zwicker C. — Physica, 1926, 6, p. 361.
4831. Zwicker C. — Physica, 1927, 7, p. 71.
4832. Zwicker C. — Physica, 1928, 8, p. 240.
4833. Zwicker С — Physica, 1928, 8, p. 241.
4834. Zwicker C. — Z. Phys., 1928, 52, S. 668.
4835.iZyrnieki W. — Rocz. chem., 1971, 45, s. 1151.
4836. Zyrnicki W. — Rocz. chem., 1973, 47, s. 23.
4837. Zyrnicki W. — Rocz. chem., 1974, 48, s. 1159.
4838. Zyrnicki W. — J. Quant. Spectrosc. and Radiat.
Transfer, 1975, 15, p. 575.
4839. Zyrnicki W. — Pol. J. Chem., 1978, 52, p. 465.
4840. Zyrnicki W. — Pr. nauk. inst. chem. neorgan.
i metalurg. pierwiast. rzadkieh PWr, 1978, N 39,
s. 1.
4841. Zyrnicki W., Czernichowski A. — Pr. nauk inst.
chem. neorgan. i metalurg. pierwiast. rzadkieh
PWr, 1970, N 2, s. 57.
4842. Zyrnicki W., Czernichowski A. — Acta phys. pol.,
1971, A39, p. 429.
4843. Zyrnicki W., Palmer H. B. — Physica, 1976,
C84, p. 152.
Дополнительная литература'
4844. Абраменков А. В. — Автореф. дне. . . . канд.
хим. наук. М.: МГУ, 1975.
4845. Акишин П. А., Ходеев Ю. С. —ЗКурн. физ.
хим., 1961, 35, с. 574.
4846. Александров В. В., Борзук А. Н.,
Новиков И. И. — В кн.: Диэлектрические и
конструкционные материалы для криогенных ЛЭП.
М.: ЭНИН, 1979, с. 113.
4846а. Алыпшулер Л. В., Бушман А. В., Жернокле-
тов М. В., Зубарев В. И., Леонтьев А. А.,
Фортов В. Е. — Журн. эксперим. и теор. физ.
1980, 78, с. 741.
4847. Алыиин Б. И., Астров Д. Н. — Журн.
эксперим. и теор. физ., 1963, 44, с. 1195.
4848. Ангелов С. А., Механджиев Д. Р. — Докл.
Българск. акад. наук, 1973, 26, с. 1213.
4849. Аристова Н. М., Деменский Г. К., Теплое О. А.,
Петров Л. А. — Журн. физ. хим., 1983 (в
печати).
4850. Аристова Н. М., Деменский Г. К., Теплое О. А.,
Петров Л. А. — Изв. АН СССР. Неорган, матер.,
1983 (в печати).
4851. Багаратъян Н. В., Никитин. ОТ.— Вестн.
МГУ. Сер. хим., 1978, № 3, с. 253.
4852. Бакулина И. Н., Ионов Н. И. — Журн.
эксперим. и теор. физ., 1959, 9, с. 709.
4853. Белянин В. С. — В кн.: Обзоры по теплофизи-
ческим свойствам веществ. М.: ИВТАН, 1976,
№ 1, сб.
4854. Березовский Г. А., Пауков И. Е. — Журн. физ.
хим., 1982, 56, с. 1541.
4855. Ворщевский А. Я., Рудный Е. Б. — ВИНИТИ:
Деп. № 690-80. М., 1980.
4855а. БорщевскийА. Я., Рудный Е. Б., БуцкийВ. Д.,
Сидоров Л. Н. — В кн.: IV Всесоюзный
симпозиум по химии неорганических фторидов.
21—23 июля 1981 г.: Тез. докл. Новосибирск,
1981, с. 170.
4856. Бучельникова Н. С. — Усп. физ. наук, 1958,
65, с. 351.
4856а. Бушман А. В., Фортов В. Е. —
Широкодиапазонные уравнения состояния металлов.
Препринт. Отделение ИХФ АН СССР,
Черноголовка, 1982 (в печати).
4857. Варкое М. В. — Автореф. дпс. . . . канд. хим.
наук. М.: МГУ, 1978.
4858. Вольное И. И. Перекисные соединения щелочных
металлов М.: Наука, 1980.
4859. Вольное И. И., Матвеев В. В. — В кн.: Редкие
щелочные элементы. Новосибирск: Наука, 1967,
с. 31.
4860. Галкин, Н. П., Туманов Ю. Я., Бутылкин. Ю. П.
Термодинамические свойства неорганических
фторидов: Справочник. М.: Атомиздат, 1972.
4861. Гиричев Г. В., Васильева С. Б., Краснов К. С. —
В кн.: Тез. докл. 14-го Всесоюз. Чугаевского
совещания по химии комплексных соединений,
Иваново, 1981. Иваново, 1981, ч. 2, с. 511.
4862. Голубев М. С, Бергман А. Г. — Журн. общ.
хим., 1954, 24, с. 1940.
4863. Голубев М. С, Медведев Б. С. — Журн.
неорган, хим., 1962, 7, с. 2600.
4864. Гомельский К. 3., Лугиника В. Ф., Сенни-
кова В. Н. —^Измерительная техника, 1981,
№ 1, с. 26.
4865. Горбань Ю. А., Павлинов Л. В., Быков В. Н. —
Атомная энергия, 1967, 22, с. 465.
4866. Горохов Л. И., Смирнов В. К. — Журн. физ.
хим., 1983 (в печатя).
4867. Горохов Л. Н., Смирнов В. К., Ходеев Ю. С. —
Докл. АН СССР, 1981, 261, с. 397.
4868. Горохов Л. Н., Смирнов В. К., Ходеев Ю. С, —
Журн. физ. хим., 1983 (в печати).
4868а. Гуреич Л. В., — Труды IX Всесоюз. конф.
по калориметрии и химической термодинамике,
сент. 1982 г.: Тез. докл. Тбилиси, 1982, с. 5.
4869. Днепрова В. Г. — Автореф. дне. . . . канд. хпм.
наук. М.: МГУ, 1968.
4870. Днепрова В. if., Резухина Т. Н.,
Герасимов Я. И. — Журн. физ. хим., 1968, 42, с. 1530.
4870а. Ежов Ю. С, Комаров С. А. — ЖСХ, 1982 (в
печати).
4871. Емельянов А. М., Ходеев Ю. С, Горохов Л. Н. —
Теплофизика высоких температур, 1970, 8,
с. 296.
4871а. Ефимов М. Е., Киселев Ю. М.,
Леонидов В. Я. — В кн.: VI Всесоюз. симпозиум по
химии неорганических фторидов. Новосибирск,
21—23 июля 1981 г.: Тез. докл. Новосибирск,
1981, с. 167.
48716. Ефимов М. Е., Смоляр М. И., Хренова И. В.,
Медведев В. А. — В кя.: IX Всесоюз. конф. по
химической термодинамике и калориметрии.
Тбилиси, 22—24 сент. 1982 г.: Тез. докл., 1982, с. 29.
4872. ЗасоринЕ. 3., ГершиковА. Г., Спиридонов В. П.,
Иванов А. А. — ЖСХ, 1983 (в печати).
4872а. Засорин Е. 3. — ЖСХ, 1983 (в печати).
4873. Иванова Е. Ф., Керн А. П., Довгопол И. В. —
Журн. физ. хим., 1981, 55, с. 1603.
596
Литература
4874. Ионов Н. И., Митцев М. А. -~ Журн. эксперим.
и теор. физ., 1961, 40, с. 741.
4875. Иориш В. С. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1976.
4876. Иориш В. С, Щербак Н. Б. — ВИНИТИ. Деп.
№ 1127-80. М., 1980.
4877. Иориш В. С, Щербак Н. Б. — Теплофизика
высоких температур, 1980, 18, с. 1322.
4878. Каледин Л. А. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1982.
4878а. Каталъников С. Г., Андреев Б. М. — Атомная
энергия, 1961, 11, с. 240. «' %;
4879. Кенисарин М. М., Чеховской В. Я. Исследования
температур плавления тугоплавких веществ:
Обзоры по теплофизическим свойствам веществ.
М.: ИВТАН, 1976, № 4.
4879а. Кессельмаи П. М., Онуфриев И. В. — Инж.-
физ. журн., 1979, 37, с. 316.
4880. Кобылянский А. И., Куликов А. Н., Гур-
еич Л. В. — Онтика и спектроскопия, 1982 (в
печати).
4881. Колонцова Е. В., Корнеев А. Е., Луценко В. П. —
Кристаллография, 1978, 23, с. 656.
4882. Колосов Е. Н., Туваев Т. Н., Сидоров Л. Н. —
Журн. физ. хим., 1975, 49, с. 805.
4883. Коренев Ю. М., Рыков А. Н., Новоселова А. В. —
Журн. неорган, хим., 1979, 24, с. 2201.
4884. Коренев Ю. М., Скокан Е. В., Карасев И. М.,
Акишин Я. Л. —ВИНИТИ. Деп. № 2196-79.
М., 1979.
4885. Кофстад П. Отклонение от стехиометрии,
диффузия и электропроводность в простых окислах
металлов. М.: Мир, 1975.
4886. Кувакин М.А. — В кн.: Взаимодействие
хлоридов калия, магния, аммония с их нитритами
и фосфатами. Саранск: Мордовский Гос. ун-т,
1977, с. 35.
4887. Лавренко В. А. — В кн.: Вопросы порошковой
металлургии и прочности материалов. Киев:
Изд-во АН УССР, 1959, вып. 7, с. 25.
4887а. Ликалътер А. А. — Докл. АН СССР, 1981,
259, с. 96.
48876. Малышев В. В. — В кн.: Теплофизические
свойства газов. М.: Наука, 1973, с. 142.
4887в. Малюсов В. А., Орлов В. Ю., Малафеев И. А.,
Умник Н. Н., Жаворонков Н. М. — Атомная
энергия, 1961, И, с. 435.
4888. Мартынюк М. М. — Физика горения и взрыва,
1977, 13, с. 213.
4888а. Международная практическая температурная
шкала 1968 г.: Редакция 1975 г. М.: Изд-во
стандартов, 1976.
4889. Можаров Г. И., Савеатимский А. И. —
Теплофизика высоких температур, 1981, 19, с. 954.
4889а. Недоступ В. И., Беккер И. В. — Теплофизика
высоких температур, 1980, 18, с. 1168.
48896. Недоступ В. И., Галькееич Е. П. — Инж.-физ.
журн., 1980, 38, с. 702.
4890. Новиков И. И., Рощупкин В. В., Мозговой А. Г.,
Семашко Н. А. — Теплофизика высоких
температур, 1981, 19, с. 958.
4891. Пятенко А. Т. — Автореф. дис. . . . канд. физ.-
мат. наук. М.: МФТИ, 1981.
4892. Пятенко А. Т., Гусаров А. В., Горохов Л. Н. —
Теплофизика высоких температур, 1980, 18,
с. 1154.
4893. Пятенко А. Т., Гусаров А. В., Горохов Л. П. —
Теплофизика высоких температур, 1981, 19,
с. 329.
4894. Пятенко А. Т., Гусаров А. В., Горохов Л. Н. —
Журн. физ. хим., 1983 (в печати).
4895. Рассонская И. С, Семендяева Н. К. — Журн.
неорган, хим., 1967, 12, с. 900.
4895а. Рева Т. Д., Семенов А. М. — В кн.:
Диссоциирующие газы как теплоносители и рабочие
тела АЭС: Тез. докл. V Всесоюз. конф. Февр.
1981. Минск: Ин-т ядерной энергетики, . 1981,
с. 26.
4896. Рогацкий А. Л., Томберг С. Э. — Журн. физ.
хим., 1983 (в печати).
4897. Романов Г. В., Спиридонов В. П. — Журн.
структур, хим., 1966, 7, с. 882.
4898. Рыков А. Н. — Автореф. дис. . . . канд. хим.
наук. М.: МГУ, 1974.
4899. Савинцев П. А., Исаков Л. А., Зилъбер-
ман П. Ф. — Укр. хим. журн., 1980, 46, с. 716.
4900. Семенов Г. А., Белов Н. В., Байдин В.Н.,
Иванаускас П. И., Вишняускас В. В., Маяу-
скас Ю. С, Караулов А. Г., Тарануха Н. В. —
Труды Лит. АН ССР, 1977, В5, с. 115.
4900а. Скрипов В. П. — Метастабилвная жидкость.
М.: Наука, 1972.
4901. Смирнов М. В., Ивановский Г. Е. — Журн.
неорган, хим., 1957, 2, с. 238.
4902. Смирнов Ю. Я., Фиикель В. А. — Журн.
эксперим. и теор. физ., 1964, 47, с. 476.
4903. Соломоник В. Г., Погребная Т. П. — Журн.
структур, хим., 1982, (в печати).
4904. Стриганов А. Р. Атомный спектр плутония и
его классификация. Препринт ИАЭ-2965. М.:
Ин-т атомной энергии им. И. В. Курчатов;),
1978.
4905. Тарасов В. Д., Иргашов X., Чеховской В. Я. —
Теплофизика высоких температур, 1982 (в
печати).
4905а. Трелин Ю. С, Теряев В. В., Егоров Ю. В. —
В кн.: Вопросы теплофизики ядерных
реакторов. М.: Атомиздат, 1974, вып. 4, с. 54.
4906. Филиппов Л. П., Арутюнов А. В.,
Макаренко И. Н., Мардыкина И. П., Труханова Л. Н.,
Хусаинова Б. Н. — В кн.: Труды 3-й Всесоюз.
научно-техн. конф. по термодинамике,
Ленинград, 1968: Сборник докладов секции «Тепло-
физические свойства веществ». Л.: Наука, 1969,
с, 116.
4906а. Фомичев Е. Н., Семинъко И. В., Криворо-
тпенко А. Д. — Огнеупоры, 1982 (в печати).
49066. Фортов В. Е. — Усп. физ. наук, 1982 (в
печати).
4907. Фортов В. Е., Дрёмин А. И., Леонтьев А. А. ~
Теплофизика высоких температур, 1975, 13,
с. 1072.
4908. Халдояниди К. А.г Гранкина З.А. — Шв. СО
АН СССР. Сер. хим. наук, 1976, вып. 1, № 2.
с. 47.
4909. Ходеее Ю. С, Дружерученко А. Г.,
Смирное В. К. — В кн.: Третья Всесоюз. конф. ш>
масс-спектрометрии, Ленинград, 1981: Тез.
докл. Л.: СКВ АН, 1981, с. 120.
4910. Четвериков А. 'П., Габескирия В. Я., Пуш-
ков В. В. — Журн. техн. !физ., 1981, 51, с. 130.
4910а. Шпилърайн Э. Э., Якимович К. А. — Таблицы
рекомендуемых справочных данных
«Калорические свойства гидрида, дейтерида и тритида
597
Литература
лития в жидком состоянии». ВНИИКИ, Деп.
Р 11-81. М.: 1981.
49106. Якимович К. А. — Автореф. дис. . . . докт.
техн. наук. М.: ИВТАН, 1980.
4910в. Якимович К. А., Мельникова Т. Н., Цир-
лина Е. А. — Таблицы рекомендуемых
справочных данных «Калорические свойства гидрида,
дейтерида и тритида лития в твердом состоянии».
ВНИИКИ, Деп. Р 12-81. М.: 1981.
4911. Ackermann R. J., Chandrasekharaiah M. S. —
In: Thermodynamics Nuclear Materials. Proc.
Symp. Vienna, 1974. Vienna: IAEA, 1975, vol, II,
p. 3.
4912. Ackermann R. J., Garg S. P., Rauh E. G. —
High Temp. Sci., 1979, 11, p. 199.
4913. Ackermann R. J., Rauh E. G., Rand M. H. —
In: Thermodynamics of Nuclear Materials. Proc.
Symp. Jiilich, 29 Jan. — 2. Feb. 1979. Vienna:
IAEA, 1980, vol. I, p. 11.
4913a. Ackermann R. J., Tetenbam M. — In:
Thermodynamics of Nuclear Materials. Proc. Symp.
Jiilich, 29 Jan. — 2 Feb. 1979. Vienna: IAEA,
1980, vol. I, p. 29.
4914. Ahree M., Hewat A. W. — Phys. status solidi,
1980, A58, p. 525.
4915. Allavena M., Blaisten-Barojas E., Silvi B. —
J. Chem. Phys., 1981, 75, p. 787.
4916. Almenningen A., Samdal S., ChristenD. — J. Mol.
Struct., 1978, 48, p. 69.
4916a. Ames L. L., Barrow R. F. — Proc. Phys. Soc,
1967, 90, p. 869.
4917. Amini M., Fincham D., Hockney R. W. —
J. Phys., 1979, C12, p. 4707.
4918. Ando R., Sawada S. — Rept. Inst. Sci. Technol.
Univ. Tokyo, 1950, 4, p. 223.
4919. Arrott A., Werner S. A., Kendrick H. — Phys.
Rev., 1967, 155, p. 528.
4920. Avni R., Klein F. S. — Spectrochim. acta, 1973,
B28, p. 331.
4921. Balducci G., Gigli G., Guido M. — J. Chem. Soc.
Faraday Trans. II, 1981, 77, p. 1107.
4921a. Barrow R. F., Clements R. M., Harris S. M.,
Jenson P. P. — Astrophys. J., 1979, 229 A),
Part I, p. 439.
4922. Barton J. L., Bloom H. — J. Phys. Chem., 1956,
60, p. 1413.
4923. Bauche /., Blaise J., Fred M. — J. Opt. Soc.
Amer., 1964, 54, p. 565.
4923a. Bellmonte L., Cavaliere P., Ferrante G. — J. Chem.
Phys., 1974, 61, p. 3225.
4924. Belton G. R., McCarron R. L. — J. Phys. Chem.,
1964, 68, p. 1852.
4925. Berg R.A., Skewes G.W. — l. Chem. Phys.,
1969, 51, p. 5430.
4926. Berglund S., Sahle W. — J. Solid State Chem.,
1981, 36, p. 66.
4927. Berthelot С — J. phys. et radium, 1962, 23, p. 447.
4928. Betz G., Frohberg M. G. — Scr. MetalL, 1981,
15, p. 269.
4929. Bizot D. — J. Fluor. Chem., 1978, 11, p. 497.
4930. Blaise J., Radziemski L. J. ¦— J. Opt. Soc. Amer.,
1976, 66, p. 644.
4931. Bohmhammel K., Wolf G., Gross G., Madge H. —
J. Low Temp. Phys., 1981, 43, p. 521.
4931a. Booth D. L. — TRG, Report 1759 (R/X)
UKAEA, London 1968, 1974.
4932. Boyd R. /., Gupta A., Langler R. F., Lownie S.,
Pincock J. A. — Canad. J. Chem., 1980, 58,
p. 331.
4933. Braekken H. — J. Appl. Phys., 1966, 37, p. 3635.
4934. Braekken H. — Kgl. norske vid. selskabs For-
handl., 1969, 42, s. 25.
4935. Brewer L. — J. Opt. Soc. Amer., 1971, 61,
p. 1101.
4936. Brewer L. — J. Opt. Soc. Amer., 1971, 61, p. 1666.
4936a. Browning P. — UK Atomic Energy Authority
Rt. AERE-R. 9847. — Harwell, 1981.
4937. Browning P. — J. Nucl. Mater., 1981, 98, p. 345.
4937a. Browning P., Gillan M. J., Potter P. E. — Rev.
int. hautes temp, et Refract. 1978, 15, p. 333.
4938. Bursill L. A. — Acta crystallogr., 1972, A28,
p. 187.
4939. Caers J., Casieels F. G., Brabers M. J. — High
Temp. — High Pressur., 1980, 12, p. 411.
4940. Carling R. W. — J. Electrochem. Soc, 1980,
127, p. 411.
4941. Carlson СМ., Eyring H., Ree T. — Proc. Nat.
Acad. Sci., 1960, 46, p. 333.
4942. Carlson K.D., Nesbet R. K. — J. Chem. Phys.,
1964, 41, p. 1051.
4943. CarnahanN. F., Starling K. E. — J. Chem. Phys.,
1969, 51, p. 635.
4944. CarnahanN. F., Starling К. E. — J. Chem. Phys.,
1970, 53, p. 472.
4945. CarnahanN. F., Starling К. E. — J. Chem. Phys.,
1970, 53, p. 600.
4945a. Cash W. M., Brooks С R. — J. Chem. Ther-
modyn., 1982, 14, s. 351.
4946. Chereau P., Dean G., De Franco M., Gardanian P.—
J. Chem. Thermodyn., 1977, 9, p. 211.
4946a. Christensen J. A. — J. Amer. Ceram. Soc, 1963,
46, p. 607.
4947. Christian P. A., Cardies J. N., Disalvo F. /.,
Waszczak J. W. — J. Electrochem. Soc, 1980,
127, p. 2315.
4948. Clark R. J. H., Mitchell P. D. — J. Chem. Soc.
Dalton Trans., 1972, p. 2429.
4949. Copper D. L., Richards G. W. — J. Chem. Phys.,
1980, 73, p. 3515.
4950. Dharwadkar S. R., Tripathi S. N., Karkhana-
vala M. D., Chandrasekharaiah M. S. — In:
Thermodynamics Nuclear Materials, Proc. Symp.
Vienna, 1974. Vienna: IAEA, 1975, vol. II,
p. 455.
4951. Dulick M., Field R. W., Beaufils J. С — J. Mol.
Spectrosc, 1981, 87, p. 268.
4952. Dulick M., Field R. W., Beaufils J. C, Schamps J .—
J. Mol. Spectrosc, 1981, 87, p. 278.
4953. Ekstrom Т., Nygren M. — Acta chem. scand.,
1972, 26, p. 1827.
4954. Ellison F. O., Delle Donne M. J. — J. Chem.
Phys., 1973, 59, p. 6179.
4955. Endo Y., Saito S., Hirota E. — J. Chem. Phys.,
1981, 74, p. 1568.
4955a. Eyring H., Jhon M. S. — Significant liquid
structure. Wiley, N. Y., 1969.
49556. Fisher E. A. — In: Thermodynamics Nuclear
Materials. Proc. Symp. Julich 1979. Vienna.
IAEA, 1979, vol. I, p. 115.
4956. Fisher E. A., Kinsman P. R., Ohse R. W. —
J. Nucl. Mater., 1976, 59, p. 125.
4957. Foex M. — С. г. Acad. sci., 1945, 220, p. 917.
4958. Foppl H. — Z. anorg. und allg. Chem., 1957,
291 S 772
4959. Foster J. S.' — Diss. Abstrs, 1965, 25, p. 7180.
4960. Fox R.E., Hickam W. H., Grove D.J., Kjel-
598
Литература
daas Т., Jr. — Rev. Sci. Instrum., 1955, 26,
p. 1101.
4961. Franck E. U. — Pure and Appl. Chem., 1974,
38, p. 4.
4962. Fred M. — частное сообщение.
4962a. Fucke W., Seydel U. — High Temperat. — High
Pressures, 1980, 12, p. 419.
4963. Gamier P. R., Salamon M. B. — Phys. Rev.
Lett., 1971, 27, p. 1523.
4964. GarstensD. H. W.,DruenD. M., KozlowskiJ. P.—
High Temp. Sci., 1972, 4, p. 436.
4965. Garton W. R. S., Wilson M. — Astrophys. J.,
1966, 145, p. 333.
4965a. Gillan M. J. — In: Thermodynamics Nuclear
Materials. Proc. Symp. Vienna, 1974. Vienna,
IAEA, 1975, vol. I, p. 269.
4966. Giroud M., Nedelec 0. — J. Chem. Phys., 1980,
73, p. 4151.
4967. Gole J. L., Childs R. H., Dixon D. A., Ea-
des R. A. — J. Chem. Phys., 1980, 72, p. 6368.
4968. Green D.W., Ervin K. M. — J. Mol. Spectrosc,
1981, 89, p. 145.
4968a. Green D. W., Reedy G. Т., Leibowitz L. — Proc.
7th Symp. Thermophys. Prop., Gaithersburg,
Md., 1977. N. Y., 1977, p. 379.
•4969. Gruendler W., Friedemann R., Nguyen Huu Thong,
Shimpf J. — Z. anorg. und allg. Chem., 1974,
410, p. 287.
4969a. Hagemark K., Blander M., Luchsinger E. B. —
J. Phys. Chem. 1966, 70, p. 276.
4970. Hammer P. D., Davis S. P., ZookA. С — J. Chem.
Phys., 1981, 74, p. 5320.
4971. Heideman S. A., Reed Т. В., Gilles P. W. —
High Temp. Sci., 1980, 13, p. 79.
4972. Heintz A., Meisinger E., Lichtenthaler R. — Ber.
Bunsenges. Phys. Chem., 1976, 8, p. 163.
4973. Henderson G. A., Zemke W. Т., Wahl A. C. —
J. Chem. Phys., 1973, 58, p, 2654.
4973a. Herrmann A., Schumacher E., Woste L. — J.
Chem. Phys., 1978, 68, p. 2327.
49736. Hessel M. M., Cotton S. G. — J. Mol. Spectrosc.
(in press).
4974. Hildenbrand D. L. — J. Chem. Phys., 1968, 48,
p. 2457.
4975. Hoekstra H. R. — Inorg. Chem., 1966, 5, p. 754.
4975a. Holm J. L., Jenssen B. — Acta chem. scand.,
1969, 23, p. 1065.
4976. Honda K., Sone T. — Sci. Repts Tohoku Univ.,
1914, 3, p. 139.
4977. Huber M. C, Sandeman R. J., Tubbs E. F. —
Proc. Roy. Soc. London, 1975, A342, p. 431.
4978. Huff V. N., Gordon S., Morrell V. E. General
method and thermodynamic tables for
computation of equilibrium composition and temperature
of chemical reactions: Rept 1037, Cleveland
(Ohio): NASA, 1951.
4979. lorish V. S., Baibuz V. F., Zitserman V. Yu.,
Zavalnyi A. A. — Chem. Phys., 1976, 18, p. 257.
4980. lorish V. S., Zitserman V. Yu. — Chem. Phys.
Lett., 1975, 34, p. 378.
4981. Izumi F., Kodama H. — J. Less-Common Metals,
1979, 63, p. 305.
4981a. Jacoboni C, Leble A., Rousseau J. J. — J. Solid
State Chem., 1981, 36, p. 297.
49816. Jacobs H., Harbrecht B. — Z. Naturforsch.,
1981, B36, S. 270.
4982. Jaffray J., Viloteau J. — C. r. Acad. sci., 1948,
226, p. 1701.
4982a. Jamamoto H. — Jap. J. Phys., 1929, 5, p. 153.
4983. Janes G. S., Itzkan I., Pike С. Т., Levy R. H.,
Levin L. — IEEE J. Quant. Electron., 1976,
QE-12, p. 111.
4984. Jensen D.E., Miller W.J. — J. Chem. Phys.,
1970, 53, p. 3287.
4985. Joliet J. F. — С. г. Acad. sci., 1961, 252, p. 719.
4986. Jordan K. D., Kinsey J. L., Silbey R. — J. Chem.
Phys., 1974, 61, p. 911.
4987. J0rgensen С К. — Mol. Phys., 1964, 7, p. 417.
4988. Kanno M., Takahashi Y., Yamauchi S., Iwato S.,
ffakamura J. — Sago Shikensho Nenpo (Tokyo
Daigaku kogakubu), 1980, 39, p. 161.
4989. Karow H. W. — Rev. intern, hautes temp, et
refract., 1978, 16, p. 347.
4990. Kelley К. К. Contributions to the Data on
Theoretical Metallurgy. V. Evalution of the Heats of
Fusion of Metals from Freezing Point Lowering
(Equilibrium Diagrams). — Bull. Bur. Mines
(USA), 1936, N 393.
4991. Kim Y.-W., Belton G. R. — Metall. Trans., 1974,
5, p. 1811.
4992. KirshenbaumA. D., CahileJ. A., McConigalP. J.,
Grosse A. V. — J. Inorg. and Nucl. Chem., 1962,
24, p. 1287.
4993. Kleinschmidt P. D., Hildenbrand D. L. —
J. Chem. Phys., 1979, 71, p. 196.
4993a. Klement W. — Z. phys. Chem. (BRD) 1976,
103, p. 65.
4994. Klemm A. D., Storer R. G. — Austral. J. Phys.,
1973, 26, p. 43.
4995. Kofstad P. — J. Electrochem. Soc, 1962, 109,
p. 776.
4996. Kofstad P. — Corrosion, 1968, 24, p. 379.
4997. Konowalow D. D., Rosenkrantz M. E. — Chem.
Phys. Lett., 1979, 61, p. 489.
4998. Konowalow D. D., Rosenkrantz M. E.,
Olson M. L. — J. Chem. Phys., 1980, 72, p. 2612.
4999. Kordes E., Zigler G. — Z. Elektrochem., 1954,
58, p. 168.
5000. Kordis /., Gingerich K.A.—J. Chem. Phys.,
1977, 66, p. 483.
5001. Kubaschewski O., Hopkins В. Е. — J.
Less-Common Metals, 1960, 2, p. 172.
5002. Kushawaha V.S. — J. Phys. Chem., 1973, -77,
p. 2885.
5003. Lagerqvist A., Huldt L. — Z. Naturforsch., 1953,
A8, S. 493.
5003a. Lau K. H., HildenbrandD. L. — J. Chem. Phys.,
1982, (in press).
5004. Lefebvre /., Currat R., Fouret R., More M. —
J. Phys., 1980, C13, p. 4449. ; .:'
5005. Leitnaker J. M. — High Temp. Sci., ?980, 12,
p. 289.
5006. Leutwyler S., Herrman A., Waste L.,
Schumacher E. - Chem. Phys., 1980, 48, p. 253.
5007. Leutwyler S., Hofmann M., Harri H. P.,
Schumacher E. — Chem. Phys. Lett., 1981, 77, p. 257.
5008. Levason W., Ogden J. S., Rest A. J. — J. Cheiftl.
Soc. Dalton Trans., 1980, p. 419.
5009. Linton C, Dulick M., Field R. W., Carette P.,
Barrow R. F. — J. Chem. Phys., 1981, 74, p. 189.
5010. Manivannan G., Sengodan V., Srinivasa-
charya K. G. — Z. Naturforsch., 1981, A36,
S. 975.
5011. Mann J. B. - J. Chem. Phys., 1964, 40, p. 1632.
5012. Markin T. L., Rand M. H. — In:
Thermodynamics. Proc. Symp. Vienna, 1965. Vienna: IAEA,
1966, vol. I, p. 145.
599
Литература
5013. Mathur В. P., Rothe E. W., Reck G. P., Light-
man A. J. — Chem. Phys. Lett., 1978, 56, p. 336.
5014. Matsumoto Т., Mitsui T. —J. Phys. Soc. Jap.,
1969, 27, p. 786.
5015. McGuire T. R., Scott E. J., Grannis F. H. —
Phys. Rev., 1956, 102, p. 1000.
5016. Mebed M. M., Yurchak R. P., Filippov L. P. —
High Temp. — High Pressur., 1973, 5, p. 253.
5017. Meggers W. F., Corliss С. Л., Scribner В. F. —
U. S. NBS Monogr. 32, part I. Wash., 1961.
5017a. MenziesD. C. — TRG Report 1119 (D), UKAEA.
L., 1966.
5018. Mikami M., Odawara O., Kawamura K. — J. Chem.
and Eng. Data, 1981, 26, p. 411.
5019. Miron E., DavidR., Erez G., Lavi S., LevinL. A.—
3. Opt. Soc. Amer., 1979, 69, p. 256.
5020. Moriya K., Matsuo Т., Suga H. — Chem. Phys.
Lett., 1981, 82, p. 581.
5021. Morizot P., Ostorero /., Plurien P. — J. chim.
phys. et phys.-chim. biol., 1973, 70, p. 1582.
5022. Muller W., Jungen M. — Chem. Phys. Lett., 1976,
40, p. 199.
5023. Nagarajan K., Bhupathy M., Prasad R. — J.
Chem. Thermodyn., 1980, 12, p. 329.
5023a. Nemukhin A. V., Stepanov N. F. — Chem. Phys.
Lett., 1979, 60, p. 421.
5024. Ohlendorf D., Wicke E. — J. Phys. Chem. Solids,
1979, 40, p. 721.
5025. OhseR. W.,Babelot J. F., FrezzottiA., Long K. A.,
Magill J. — High Temp. — High Pressur., 1980,
12, p. 537.
5026. OhseR. W.,BabelotJ. F., FrezzottiA., Long K. A.,
Magill J. — High Temp. Sci., 1980, 13, p. 35.
5027. Ohse R. W., Kinsman P. R. — High Temp. —
High Pressur., 1976, 8, p. 209.
5028. Ohse R. W., Von Tippelskirch H. — High Temp.—
High Pressur., 1977, 9, p. 367.
5029. Perri J. A., Banks E., Post B. — J. Appl. Phys.,
1957, 28, p. 1272.
5030. Pfeiffer G. V., Ellison F. O. — J. Chem. Phys.,
1965, 43, p. 3405.
5031. Plutonium: Physico-Chemical Properties of its
Compounds and Alloys., vol. 4, Special Issue
N I/Ed. O. Kubaschewski. Vienna: IAEA, 1966.
5032. Polla L., Piccardi G. — Atti Accad. naz. Lincei,
Rend. cl. sci. fis. mat. e natur., 1927, 5, p. 546.
5032a. Rand M. H. — In: Plutonium. Physico-chem.
Properties of its Compounds and Alloys. Atomic
Energy Review, vol. 4, Special Jssue N 1,
Vienna: IAEA, 1966, p. 7.
5033. Rauh E. G., Garg S. P. — High Temp. Sci.,
1981, 14, p. 121.
5034. Reedy G. Т., Chasanov M. G. — J. Nucl. Mater.,
1972, 42, p. 341.
5035. Rehn J., Cefola R. — U. S. Rept CK-1240, Jan.
19, 1944. Цит. по: [492, с 217].
5036. Richards E. W. Т., Ridgeley A. — Spectrochim.
acta, 1965, 21, p. 1449.
5036a. Robertson E. W., Barrow R. F. — Proc. Chem.
Soc, 1961, p. 329.
5036b. Rolin M., Recapet J. M. — Bull. Soc. chim.
France, 1964, 9, p. 2104.
5037. Rollon C. E., Gallegos G. F. — J. Therm. Anal.,
1981, 21, p. 159.
5038. Rosenstock H. M., Draxl K., Steiner B. W., Her-
ron J. T. — J. Phys. and Chem. Ref. Data, 1977,
6, p. 1.
5038a. Sandenaw T. A., Gibney R. B. — J. Phys. Chem.
Solids, 1958, 6, p. 81.
5039. Sawada S., Ando R. — Repts Inst. Sci. and
Technol. Univ. Tokyo, 1951, 5, p. 143.
5040. Sawada S., Ando Д., Nomura S. — Phys. Rev.,
1951, 82, p. 952.
5041. Schdfer #., Durkop A., Jori M. — Z. anorg.
und allg. Chem., 1954, 275, S. 289.
5042. Schimanouchi T. — J. Phys. and Chem. Ref.
Data, 1977, 6, p. 993.
5042a. Schoonmaker R. C, Porter R. F. — J. Chem.
Phys., 1959, 30, p. 283.
5043. Schung K. P., Wagner H. G. — Z. phys. Chem.
(BRD), 1977, 108, S. 173.
5044. Segel S. L. — J. Chem. Phys., 1980, 73, p. 4146.
5044a. Sevdel U., Fucke W. — J. Phys. F: Metal. Phys.,
1978, 8, p. L157.
5045. Sharma R. V., Sharma К. С — Indian J. Phys.,
1978, B52, p. 340.
5046. Shenyavskaya E. A., Egorova I. V., Lupa-
nov V. N. — J. Mol. Spectrosc, 1973, 47, p. 355.
5047. Sidorov L. N. — Intern. J. Mass Spectrom. and
Ion. Phys., 1981, 38, p. 49.
5047a. Sinha S. P. — Proc. Phys. Soc. London, 1948,
60, p. 4361.
50476. Sinha S. P. — Proc. Phys. Soc. London, 1950,
A63, p. 952.
5048. Smith D. H., Hertel G. R. — J. Chem. Phys.,
1969, 51, p. 3105.
5049. Smith D. #., Webb R. F. — U. S. Rept Y-2095.
Oak Ridge, Tennessee, Y-12 Plant, Dec. 2, 1977.
550. Solarz R. W., May С A., Carlson L. R., Wor-
den E. F., Johnson S. A., Palsner J. A. — Phys.
Rev., 1976, A14, p. 1129.
5050a. Spencer F. E., Hendrie J. C, Bienstock ?>. —
Proceedings of the 6th Intern. Conf. on MHD-
Electric Power Generation. Wach., 1975, vol. II,
p. 181.
5051. Sleiger R. P., Cater D. E. — High Temp. Sci.,
1975, 7, p. 288.
5052. Steinhaus D. W., Radziemski L. J., Cowan R. D.,
Blaise J., Guelachvili (?., Osman Z. В., Verges J. —
U. S. Rept LA-4501. Los Alamos Sci. Lab. Los
Alamos N. Мех., 1971, 243pp.
5053. Storer R. G. — J. Math, and Phys., 1968, »,
p. 964.
5054. Storer R. G. — Phys. Rev., 1968, 176, p. 326.
5054a. Stuve J. M., Ferrante M. J. — U. S. Bur. Mines.
Rept Invest., 1980, N 8442.
5055. Su T.-M. R., Riley S. J. — J. Chem. Phys.,
1979, 71, p. 3194.
5056. Su T.-M. R., Riley S. J. — J. Chem. Phys.,
1980, 72, p. 1614.
5057. Sugar J. — J. Chem. Phys., 1974, 60, p. 4103.
5058. Sze N. #., Meaden G. T. — Phys. Lett., 1971T
A35, p. 329.
5059. Tallman R. L. — Diss. Abstrs, 1960, 20, p. 4293.
5059a. Taylor J. C, Linford P. F. F., Dean D. J. —
J. Inst. Met., 1968, 96, p. 178.
5060. Trounson E. P., Bleil D. F., Wangsness R. K.,
Maxwell L. R. — Phys. Rev., 1950, 79, p. 542.
5061. Tsai K. K., Harris PA M., Lassetre E. N, —
J. Phys. Chem., 1956, 60, p. 338.
5062. Veda R., Ichinokawa T. — Phys. Rev., 1951,
82, p. 563. . тг
5063. Ueda R., Ichinokawa T. — Busseiron Kenkyu
(Res. on Chem. Phys.), 1951, N 36, p. 64.
5063a. Valance A. — J. Chem. Phys., 1978, 69, p. 355.
5064. Vasilakis A., Bhaskar N. D., Happer W. — J.
Chem. Phys., 1980, 73, p. 1490.
600
Литература
5065. Verma К. K.,VuT.H., Stwalley W.C. — J. Mol. 50706.
Spectrosc, 1981, 85, p. 131.
5066. Veza D., Rukavina J., Movre M., Vujnovic V., 5070b.
Pichler G. — Opt. Commun., 1980, 34, p. 77.
5067. Wagner K., Schdfer H. — Z. anorg. und allg. 5071.
Chem., 1979, 450, S. 107.
5068. Wang K. C, Dreger L. #., В adapt V. V., Mar- 5072.
grave J. L. — J. Amer. Ceram. Soc, I960, 43,
p. 509. 5073.
5069. Watkins G. M., Fenichel H. — Phys. Lett., 1979, 5074.
A73, p. 445.
5070. WeUs W. D., Kohlhaas R. — Z. Naturforsch., 5075.
1964, A19, S. 1631.
5070a. Yoshinaga H. — Proc. Phys. Math. Soc, Japan, 5076.
1937, 19, p. 847.
Yoshinaga H. — Proc. Phys. Math. Soc, Japan,-
1937, 19, p. 1073.
Westrum E. F. — J. Chem. Thermodynam., 1982
(in press).
Younes C., Nguyen L. D., Pattoret A. — High
Temp.—High Pressur., 1981, 13, p. 105.
Young D.A., Alder B. J. — Phys. Rev., 1971,
A3, p. 364.
Zalubas R. — J. Res. NBS, 1976, A80, p. 223.
Zasorin E. Z., Rambidi N. G., Akishin P. A. —
Acta crystallogr., Suppl., 1963, 16, p. A130.
Zeegers P. J. Т., Townsend W. P., Wineford~
ner J. D. — Spectrochim. aeta, 1969, B24, p. 243-
O'Hare P., Malm J. G. — l. Chem.
Thermodynam., 1982, 14, p. 323.
УТОЧНЕНИЕ ВЫБОРА ПОСТОЯННЫХ В I и III ТОМАХ
Приложение 9
НОВЫЙ РАСЧЕТ ТАБЛИЦ
ТЕРМОДИНАМИЧЕСКИХ СВОЙСТВ
НО2)
, DO2) DO;, HOC1, SF5, SF*, BC1
Ниже приводятся тексты, объясняющие новый выбор молекулярных и термохимических
постоянных и расчеты термодинамических функций и констант равновесия перечисленных
веществ, который обусловлен появлением в литературе в течение 1977—1981 гг. новых данных.
Для работ, цитированных ранее в томах I—III, сохранены прежние номера библиографических
ссылок; список новых работ приведен в конце этого Приложения и им присвоены номера 1п—13п.
Рассчитанные таблицы термодинамических свойств приведены в Дополнении 2 второй книги
этого тома, для них сохранены те же номера, которые были использованы в предыдущих томах,
яо с добавлением буквы «а».
Том I, книга 1, с. 109.
HJ (г). Термодинамические свойства газообразного ионизированного триводорода в стандартном
состоянии в интервале температур от 298,15 до 20 000 К приведены в табл. 14а.
Молекулярные постоянные, использованные для расчета термодинамических функций Нд (г),
приведены в табл. 5.4а. Теоретические расчеты [705, 961, 1038, 1123, 2980, 3363, 1п], изучение
Таблица 5.4а Значения молекулярных постоянных, Р{ и о,
принятые для расчета термодинамических
функций HJ
Состояние
х^
То
v2
v3
см-
0
3185
iaibic-io™
Г3 • СМ6
2521,56B) — 5,58064.10
6
Pi
1
спектров энергетических потерь при рассеянии атомов Ne на молекулах HJ [3001 ] и
доследование инфракрасного спектра [2п] показали, что молекула Щ в основном состоянии Х1А1 имеет
структуру равностороннего треугольника (симметрия D3ll). Согласно квантовомеханическим
расчетам [Зп, 4п] молекула имеет малостабильное возбужденное состояние а32„ с линейной структурой
и энергией —40 000 см. Значения основных частот в основном состоянии, приведенные в
таблице, найдены квантовомеханическим расчетом (vx) Карни и Портером [1п] и в результате
анализа тонкой структуры полосы v2 в работе Ока [2п]. Произведение моментов инерции вычислено
на основании значений вращательных постоянных Во=43,568 +0,048 см и С0=20,708 +
+0,048 см, определенных Ока; им соответствует также значение г (Н—Н)=0,8837 А.
Термодинамические функции Нд(г) были вычислены по уравнениям A. 3)—A. 6), A. 9),
A. 10), A. 122)—A. 124), A. 128) и A. 130) в приближении «жесткий ротатор—гармонический
осциллятор» без учета возбужденного электронного состояния. Основные погрешности
вычисленных термодинамических функций Щ(г) обусловлены проведением расчета в приближении
«жесткий ротатор—гармонический осциллятор» и составляют 0,1, 0,5, 1 и 2 Дж-К~1-моль в
значениях Ф°(Г) при 298,15, 3000, 6000 и 20 000 К соответственно.
Использование новых экспериментальных данных по молекулярным постоянным позволило
уточнить термодинамические функции
ур
(г) на 0,15—2 Дж-К-моль в значениях Ф°(Г).
602
Уточнение выбора постоянных в I и III томах Щ (г), НО2 (г), HOJ (г), D02 (г), DOJ (г)
Том I, книга 1, с. 116
НО2 (г). Константа равновесия реакции НО2(г)=2О(г)-|-Н(г), приведенная в табл. 18а,
вычислена с использованием значения Дг#° @)=697,0 +3,0 кДж -моль, основанного на принятой
в справочнике энтальпии образования
г, 0) = 12,6 + 3,0 кДж -моль-1.
Это значение вычислено по уравнению III закона по результатам исследования константы
равновесия реакции НО2 (r)-j-NO (г)=ОН (r)+NO2 (г), выполненного Говардом [5п] (измерения
скорости прямой и обратной реакции, 425—1115 К). В работах Кочубея и Моина [133, 134] было
получено бодее низкое значение E + 4 кДж-моль), основанное на предположении, что энергии
активации исследованных авторами реакций НХ (г)+О2 (г)=НО2 (г)+Х (г), где Х=С1, Вг, I,
равны энтальпиям этих реакций. Более ранние попытки определения величины Do (Н—О2) на
основании измерений потенциала появления иона НО? из Н2О2 и потенциала ионизации НО2
[1502, 1506, 3212] привели к менее надежным результатам, что, видимо, связано с избыточной
кинетической энергией и с энергией возбуждения продуктов диссоциативной ионизации.
Том I, книга 1, с. 116
НО~(г). Константа равновесия реакции НО^ (г)=2О (г)+Н (г)+е (г), приведенная в табл. 19а,
вычислена с использованием значения &ГН° @)=867,505+ 3,0 кДж-моль, соответствующего
принятой в справочнике энтальпии образования
Д^^НО", г, 298,15 К) = —167 ±15 кДж • моль.
Принятая величина основана на оцененной Бенсоном и Нангия [6п] энтальпии растворения
НО" (г) в воде (8,5 + 11 кДж-моль) и энтальпии образования h.fH° (НО~, ооН20, 298,15 К)=
=—159,0+1,7 кДж-моль, принятой по рекомендации справочника [1009]. Менее точное
значение AfH° (НО;, г, 298,15 К)=—132 + 40 кДж -моль было получено Эвансом и Юри [1424, 1425]
из термохимического цикла, включающего реакции диссоциации и ионизации Н2О (ж) и Н2О2 (ж),
причем в расчетах был использован ряд оцененных величин.
Принятой энтальпии образования соответствует
= —170,505 + 15 кДж-моль.
Том I, книга 1, с. 131
DO2 (г). Константа равновесия реакции DO2 (r)=2O (r)+D (г), приведенная в табл. 28а,
вычислена с использованием принятого в справочнике значения
ДгЯ° @) = D0(DO2) = 704 + 3 кДж . моль,
которое было рассчитано по соотношению C. 7). В этом расчете были использованы значения
G @, 0, 0; НО2)=2948 см и G @, 0, 0; DO2)=2337 см. Принятой энергии атомизации
соответствует
Д/Н° (DO2, г, 0) = 9,373 + 3,0 кДж • моль.
Том I, книга 1, с. 131
(г). Константа равновесия реакции DO^ (r)=D (г)+2О (г)+е (г), приведенная в табл. 29а,
вычислена с использованием принятого в справочнике значения
ДгЯ° @) = 875 + 15 кДж • моль.
Это значение было рассчитано по соотношению C. 7). В расчете были использованы величины
G @, 0, 0; НО")=2955 см и G @, 0, 0; DO7)=2340 см. Принятой энергии атомизации
соответствуют „
bfH°(DO~, г, 0)==—161,627 ±15 кДж-моль,
a) = —171 + 15 кДж • моль-1.
603
Н0С1 (г), SF6 (г)
Приложение 9
Том I, книга 1, с. 173
НОС1 (г). Константа равновесия реакции НОС1 (г)=Н (г)+О (г)+С1 (г), приведенная в табл. 59а,
вычислена с использованием значения ДгЯ°@)=655,237 + 5,0 кДж-моль, соответствующего
принятой в справочнике энтальпии образования
Ь,Н° (НОС1, г, 0) = —72,8 + 5,0 кДж • моль.
Эта величина основана на исследовании равновесия реакции Н2О (г)+С12О (г)=2НОС1 (г),
выполненном Ники и др. [7п] методом ИК-спектроскопии B95 К, 1 точка). Измерения энтальпии
ионизации НОС1 в водном растворе и энтальпии восстановления иона СЮ"(ад) перекисью
водорода, выполненные Флис и др. [270, 271], вместе с оцененной энтальпией растворения НОС1(г)-
в воде [1587, 2095] приводят к менее точной величине —90 + 15 кДж-моль.
Том I, книга 1, с. 275
SF5 (г). Константа равновесия реакции SFB (r)=S (r)-{~5F (г), приведенная в табл. 136а,
вычислена с использованием значения Д,//0 @)=1556,16+ 10 кДж-моль, соответствующего
принятой в справочнике энтальпии образования
bfH°(SFb, г, 0) = —895 + 10 кДж • моль.
Эта величина основана на результатах измерений энергии связи SF5—F, представленных
в табл. 12.14а. Принятое значение является средним из полученных в работах [8п—12п], хорошо»
Таблица 12.14а Результаты определений энергии разрыва связи SF5—F и энтальпии
образования SF5 (г) (в кДж-моль")
Авторы
Метод
D0(SF6-F)
г. 0)
Курран, 1961 [1132]
Кей и Пейдж, 1964 [2204]
Ботт и Джекобе, 1969 [710]
Харланд и Тинне, 1969 [1843]
(см. [1936])
Фрост и др., 1968 [1552]; Фезен-
фельд, 1971 [1457] (см. [1936])
Модика, 1973 [2686]
Лифпшц и др.. 1973 [2432]
Лаймэн, 1977 [12п]
Бенсон, 1978 [9п]
Кьянг и др., 1979 [Юп]
Кьянг и Заре, 1980 [Ип]
Бэбкок и Стрейт, 1981 [8п]
Масс-спектрометрическое определение порога
реакции SF, (г)+е (r)=SF5 (r)+F~ (г) и кинетической
энергии продуктов реакции
Измерение магнетронным методом
Исследование кинетики термической диссоциации
SFe в ударных волнах, 1650—2050 К
Масс-спектрометрическое определение порога
реакции SFe (r)+e (r)=SF5 (r)+F"(r)
Масс-спектрометрические измерения А Р (SF5+) из SFe
и /e(SF6)
Исследование разложения SFe, SF5C1 в ударных
волнах при 1700—2300 К
Масс-спектрометрический, измерение порогов ряда
ионно-обменных реакций
Пересчет кинетических данных работы [710]
То же
Исследование спектров хемилюминесценпии при
реакции молекул SFe с атомами Са (SP) и Sr CP)
То же, с определением поступательной энергии ато-
тов Са CР) и Sr CP)
Определение АР (SF5+) из SFe и Io (SF5) по
результатам исследований ионно-молекулярных реакций SF6
в проточном реакторе при 298,15 К
309 + 15 —974+15
360+15 -
318+10» ¦
или
334+10 а -
<328
>318 ¦
273
385
389±12
376 ±14
386±12б
396+13
-923+15
-S65+10»
-949±10а
<—955
>—965
—1010
<—936
-898
-894+12
-907 + 14
-901±12
-887 + 13
а В работе [710] были получены два ряда значений Do (SF5—F) и кинетических параметров реакции, одинэксео
хорошо соответствующих результатам измерений.
б Приведено значение, соответствующее исследованию реакции SF6 со Sr (SP) и принятому в справочнике значению
Do (SrF)=540±6 кДж -моль-i.
604
Уточнение выбора постоянных в I и III томах
(г), ВС1 (г)
«согласующихся между собой. В более ранних работах были получены заниженные значения
энергии связи.
Том I, книга 1, с. 275
SFT (г). Константа равновесия реакции SF~ (r)=S (r)+5F (г)+е (г), приведенная в табл. 137а,
«вычислена с использованием значения Д^0 @)=1852,16±14 кДж-моль. . . (далее текст
сохраняется таким, как это приведено в томе I).
Принятому значению Ао (SF6), равному —296 ±10 кДж-моль, соответствует
bfH°(SF-, г, 0) = —1191 ± 14 кДж-моль-1.
Изменение этих величин связано с изменением используемого при их вычислении значения
j, г, 0) (см. выше).
Том III, книга 1, с. 61
ВС1 (г). Константа- равновесия реакции ВС1 (г)=В (г)-(-С1 (г), приведенная в табл. 572а,
вычислена с использованием принятого в справочнике значения
ДгЯ° @) = Do (BC1) = 506 + 12 кДж • моль« 42 300 + 1000 см.
Принятая величина основана на результатах измерений, приведенных в табл. П9.1. Наиболее
точные измерения были выполнены Хилденбрандом [13п] и принятое значение является
средневзвешенным из величин, полученных при обработке этих данных. Сравнение принятого значения
с найденным Барроу при экстраполяции уровней состояния Л 41 позволяет предполагать наличие
максимума на потенциальной кривой этого состояния высотой —2000 см.
, Принятой энергии диссоциации соответствует
HfH° (BC1, г, 0) = 173,620 + 13 кДж • моль.
Таблица П9.1 Результаты определений Do (BC1, г) (в кДж-моль-1)
Авторы
Барроу, 1960 [589]
Кренев и др., 1969
Хилденбранд, 1969
[199-201]
[13п]
Метод
Экстраполяция колебательных уровней состояния Л1!!
Протока, 2В(к)+ВС13(г)=ЗВС1(г), 1373—1573 К,
8 точек
Масс-спектрометрический, MgCL (г)+ВС 1 (г)=Mg(r)+
+ВС13(г), 1348-1593 К, 9 точек
То же, BCl(r)+Sr(r)=SrCl(r)+B(r), 1763-1828 К,
5 точек
То же, 2MgCl(r)+BCl(r)=2Mg(r)+BCl3(r)
DO(BG1)
II закон
III закон
533 ±25
567 ±18
511 ±20
—
464 ±45
571 ±10
509 ±12
511 ±12
492 ±15
Литература
In. Carney G. D., Porter R. N. — J. Chem. Phys.,
1976, 65, p. 3547.
2п. Oka Т. — Phys. Rev. Lett., 1980, 45, p. 531.
3n. Ahlrichs R., Votava C, Zirz С — J. Chem. Phys.,
1977, 66, p. 2771.
4n. Schaad L. J., Hicks W. V. — J. Chem. Phys., 1974,
61, p. 1934.
5n. Howard C. J. — J. Amer. Chem. Soc, 1980, 102,
p. 6937.
6n. Benson S. W., Nangia P. S. — J. Amer. Chem.
Soc, 1980, 102 p. 2843.
7n. Niki H., Maker P. D., Savage С. М., Breiten-
bach L. P. — Chem. Phys. Lett., 1979, 66, p. 325.
8n. Babcock L. M., Strelt G. E. — J. Chem. Phys.,
1981, 74, p. 5700.
9n. Benson S. W. — Chem. Rev., 1978, 78, p. 23.
Юп. Kiang Т., Estler R. C, Zare R. N. — J. Chem.
Phys., 1979, 70, p. 5925.
lln. Kiang Т., Zare R. N. — J. Amer. Chem. Soc,
1980, 102, p. 4024.
12n. Lyman J. L. — l. Chem. Phys., 1977, 67, p. 1868.
13n. Hildenbrand D. — Private commun., 1981.
ФОРМУЛЬНЫЙ УКАЗАТЕЛЬ ВЕЩЕСТВ
Формульный указатель веществ, рассматриваемых в I, II, III и IV томах настоящего
справочника, составлен в соответствии с принятым в справочнике порядком расположения текстов к
таблиц термодинамических свойств элементов (рисунок) и их соединений (см. также т. I, кн. 1,
с. 17). Для каждого вещества указаны том и книга (например, I—1 следует читать: том I,,
книга 1 и т. п.).
Li 1
Rb
Cs
Fr
Be i
Mg
Ca
Sr
Ba
Ra
Al
Sc
La
Ac
Hf
Nb
Та
Cr
Mo
- W
-
Mn "I
Tc
L- Re
Ru
-
Rh
-
Pd
Os
-
Ir
-
Pt
Cu
Ag
Zn п Ga
Cd
L An L Hg L Tl
In
С п
Ge
Sn
Pb
Sb
1
?
Se
Те
Po
h
-
He ¦
Ne
Ar
Кг
Xe
Rn
T
F
1
Cl
!
Br
1
|
¦ At
Ce
Pr
Nd — Pm
Sm
Eu
— Gd —
Tb
Dy _
Ho
Er
Tm
U
Th — Pa — U — Np - Pu - Am — Cm — Bk — Cf — Es — Fm — Md — No i-j [03
Последовательность расположения элементов
606
Формульный указатель веществ
е(г) 1-1, 90; 1-2, 10, 11 ТО(ОТ)(г) 1-1, 129, 140; 1-2, 70
О(г) 1-1, 90, 91; 1-2, 12, 13 ТаО(г) 1-1, 132, 140, 141; 1-2, 71
О+(г) 1-1, 90, 91; 1-2, 14, 15 НТ(г) 1-1, 127, 141, 142, 401, 415; 1-2, 72, 73,
О-(г) 1-1, 90-92; 1-2, 16, 17 324-326
О2(г) 1-1, 92-96, 399, 409, 410; 1-2, 18, 19, 324- НТО(г) 1-1, 132, 142, 143; 1-2, 74
326 DT(r) I—I, 127, 143, 401, 415; 1-2, 75, 76, 324-326
О?(г) 1-1, 93, 94, 96-98; 1-2, 20, 21 DTO(r) 1-1, 132, 143, 144; 1-2, 77
O;f(r) I—1, 93, 94, 98, 99; 1-2, 22, 23
О3(г) 1-1, 99-101, 399, 409; 1-2, 24, 324-326 F(r) 1-1, 145; 1-2, 78, 79
F"(r) I—1, 145, 146; IV—1, 621; 1-2, 80, 81
H(r) I—1, 102; 1—2, 25, 26 F2(r) I—1, 146—149, 201, 399, 410, 415; I—2, 82,
H+(r) 1-1, 102, 103; 1-2, 27, 28; IV—2, 558 83, 324—326
H-(r) 1-1, 103; 1-2, 29, 30; IV-2, 558 FO(r) I—1, 147, 149, 150; 1-2, 84
H2(r) I—1, 103-107, 401, 409, 410, 413; 1-2, 31, F2O(r) I—1, 150, 151, 415; 1-2, 85, 324-326
32, 324—326 HF(r) I—1, 147, 151, 152, 410, 416, 417; 1—2, 86,.
o-H2(r) I—1, 107; 1—2, 309 87, 324—326
и-Н2(г) I—1, 107, 413; 1-2, 309, 326 H2F2(r) I—1, 152-155; 1-2, 314; IV-2, 543
Hf(r) I—1, 105, 107—109; 1—2, 33, 34 H3F3(r) I—1, 153—155; 1—2, 314
Щ(г) I—1, 109, 110; IV—1, 602; 1-2, 35, 36; H4F4(r) I—1, 153-155, 156; 1-2, 315
IV-2, 536, 537 H5F6(r) I—1, 153, 154, 156, 157; 1-2, 315
OH(r) 1-1, 105, 110-113; 1-2, 37; 38; IV-2, 558 H6Fe(r) I—1, 154, 157, 158; IV—1. 621; 1-2, 31&
OH+(r) I—1, 105, 112—114; 1—2, 39, 40 H7F7(r) I—1, 153, 154, 158, 159; 1—2, 316
OH-(r) I—1, 105, 112, 114, 115; 1—2, 41, 42 HOF(r) I—1, 153, 159; 1—2, 88
HO2(r) I—1, 110, 115, 116; IV—1, 603, 1-2, 43; DF(r) I—1, 147, 159, 160; 1-2, 89, 90
IV—2, 538 TF(r) I—1, 147, 160, 161; 1—2, 91, 92
HOj(r) I—1, 110, 116; IV—1, 603; 1—2, 44;
IV-2, 539 Cl(r) I—1, 162; 1-2, 93, 94
Н2О(к, ж) I—1, 117, 118; 1-2, 310, 326 Cl~(r) I—1, 162, 163; 1-2, 95, 96
H2O(r) I—1, 118—120, 410, 413, 414; 1—2, 45, 46, Cl2(r) I—1, 163—166, 201, 399, 411, 416, 418;
' 324—326 1—2, 97, 98, 324—326
H2O+(r) I—1, 110, 120, 121; 1—2,-47, 48 ClO(r) I—1, 163, 165—168; 1—2, 99; IV—2, 558
Н2О2(к, ж) 1-1, 117, 121; 1-2, 311, 326 ClO2(r) I—1, 168, 169; 1-2, 100
H,O2(r) I—1, 121-124, 414; 1-2, 49, 324-326 Cl2O(r) I—1, 169-171, 418; 1-2, 101, 324-326
H3O+(r) I—1, 110, 124, 125; 1-2, 50, 51 HCl(r) I—1, 163, 165, 171, 172, 411, 418; 1-2, 102,
D(r) I—1, 126; 1-2, 52, 53 324—326
D2(r) I—1, 126-128. 401, 414; 1-2, 54, 55, 324-326 HOCl(r) I—1, 170, 172, 173; IV—1, 604; 1-2, 103;
o-D2(r) I—1, 128; 1-2, 312 IV—2, 544
n-D2(r) I—1, 128; 1-2, 312; IV—2, 558 DCl(r) I—1, 163, 165, 173, 174; 1-2, 104
DO(OD)(r) I—1, 127-130; 1—2, 56 TCl(r) I—1, 163, 165, 174, 175; 1-2, 105
DO-(OD-)(r) I—1, 129, 130; 1-2, 57; IV-2, 540 ClF(r) I—1, 163, 165, 175—177, 191; 1—2, 106
DO2(r) I—1, 130, 131; IV—1, 603; 1-2, 58; IV— ClFs(r) I—1, 178, 179, 418; 1-2, 107, 324-326
2, 541 GlF6(r) I—1, 178-180, 419; 1-2, 108, 324-326
DOa(r) I—1, 130, 131; IV—1, 603; 1—2, 59; IV—
2, 542 Br(r) I—1, 181; 1—2, 109, 110
D2O(r) 1-1, 131-133, 410, 414, 415; 1-2, 60, 324— Br~(r) I—1, 181, 182; 1—2, 111, 112
326 Вг2(к, ш) I—1, 182, 183; IV—1, 621; 1-2, 317, 326;
D2O2(r) I—1, 133—135, 415; 1-2, 61, 324-326 IV-2, 558
HD(r) I—1, 127, 135, 136, 401, 415; 1—2, 62, 63, Br2(r) I—1, 183—187, 201, 399, 411, 419; IV—1, 621;
324—326 1-2, 113, 324-326
HDO(r) I—1, 132, 136, 137; 1-2, 64 BrO(r) I—1, 184, 187, 188; 1—2, 114
HDO2(r) I—1, 134, 137, 138; 1-2, 65 HBr(r) I—1, 184, 188, 189, 411, 419; 1-2, 115,
T(r) I—1, 138, 139; 1—2, 66, 67 . 324—326
T2(r) I—1, 127, 139, 401, 415; 1—2, 68, 69, 324— DBr(r) 1-1, 184, 188—190; 1-2, 116
326 TBr(r) I—1, 184, 188, 190, 191; 1-2, 117
o-T2(r) I—1, 140; 1-2, 313 BrF(r) 1-1, 184, 186, 191-193; 1-2, 118
n-T2(r) I—1, 140; 1—2, 313 BrF3(r) I—1, 193, 194; 1—2, 119
607
Формульный указатель веществ
BrF5(r) I—I, 194—196; 1—2, 120
BrCl(r) I—i, 184, 186, 191, 196, 197; 1—2, 121
l(r) I—1, 198; 1—2, 122. 123
Г(г) I—1, 198, 199; IV—1, 621; 1—2, 124, 125
12(к,ж) 1-1, 199, 200; IV—1, 621; 1—2, 317, 318, 326
12(г) 1-1, 200-205, 399, 411, 419; 1-2, 126, 324-
326
IO(r) I—1, 202, 204—206; 1—2, 127
HI(r) I—1, 202, 204, 206, 207, 411, 419; 1-2, 128,
324—326; II—2, 343
DI(r) I-i, 202, 204, 207, 208; 1-2, 129
TI(r) I—1, 202, 204, 208; 1—2, 130
IF(r) I—1, 191, 209—211; 1—2, 131
IF5(r) I—i, 212, 213; 1—2, 132
IF7(r) I—1, 212—214; 1—2, 133
ICl(r) I—1, 191, 209, 211, 214—216; 1—2, 134
IBr(r) I—1, 191, 209, 211, 216, 217; 1—2, 135
He(r) I—1, 218-220, 401, 419, 420; 1—2, 136, 137,
324-326
He+(r) I—1, 218—220; 1—2, 138, 139
Ne(r) I—1, 218—220, 401, 412, 420; 1—2, 140, 141,
324—326
Ne+(r) I—1, 219—221; 1—2, 142, 143
Ar(r) I—1, 219, 221, 412, 420; 1-2, 144, 145, 324—
326; IV—2, 558
Ar4(r) I—1, 219, 221, 222; 1—2, 146, 147
Kr(r) I—1, 219, 222, 412, 420, 421; 1—2, 148, 149,
324—326
Kr+(r) I—1, 219, 222; 1—2, 150, 151
KrF2(r) I—1, 223; 1—2, 152, IV—2, 558
Xo(r) I—I, 219, 223, 224, 412, 421; 1—2, 153, 154,
324—326
Xe+(r) I—1, 220, 224; 1—2, 155, 156
Xe2(r) I—1, 224, 225; 1—2, 318; IV—2, 545
XeO3(r) I—1, 225, 226; 1—2, 157
XeO4(r) I—1, 226, 227; 1—2, 158
XeF(r) I—1, 224, 227, 228; IV—1, 621; 1—2, 319
XeF2(K, ж) I—1, 228, 229; 1—2, 319
XeFa(r) I—1, 223, 229, 230; 1—2, 159
XeF4(K, ж) I—1, 229, 230, 231; 1—2, 320
XeF4(r) I—1, 226, 231, 232; 1—2, 160
XeF6(K, ж) I—i, 229, 232, 233; 1-2, 320, IV—2, 558
XeF6(r) I—1, 226, 233, 234; IV—1, 621; 1-2, 161
XeO2F2(r) I—1, 226, 234, 235; 1—2, 162
XeO3F2(r) I—1, 226, 235, 236; 1—2, 163
XeOF4(r) I—1, 226, 236; 1—2, 164
Rn(r) I—1, 220, 237; 1—2, 165, 166; IV—2, 558
Rn+(r) I—1, 220, 237; 1—2, 167, 168; IV-2, 558
S(k, ж) I—1, 238—240; 1—2, 169, 326
S(r) I—1, 240, 241, 421, 4221; 1-2, 170, 171, 324*,
326 l
Данные для пара серы (Sn, я < 8).
S'(r) I—1, 240, 241; 1-2, 172, 173
S2(r) I—1, 241—246; 1—2, 174
Sjfr) I—1, 242,244, 246, 247; 1—2, 175
S3(r) I—1, 247—250; IV—1, 621; 1—2, 176
S4(r) I—1, 248, 250, 251; 1-2, 177
Se(r) I—1, 248, 251; 1-2, 178
Se(r) I—1, 248, 251, 252; IV-1, 621; 1—2, 179
S,(r) I—1, 248, 252, 253; 1—2, 180; IV—2, 558
S8(r) I—1, 248, 253, 254; 1-2, 181
SO(r) I—1, 242, 244, 255, 256; 1-2, 182
SO-(r) I—I, 242, 244, 256, 257; IV-1, 621; 1—2, 183
SO2(r) I—I, 257—259, 422; IV—1, 621;
1—2, 184, 324—326
SOj(r) I—1, 259, 260; 1-2, 185
SO3(r) I—1, 259-262, 399, 422; 1-2, 186, 324—326
S2O(r) I—1, 259, 262, 263; 1—2, 187
SH(r) I—1, 242, 244, 263, 264; IV—1, 621;
1—2, 188; IV—2, 546
SH-(r) I—1, 242, 244, 264, 265; IV-1, 621; 1-2,
189; IV—2, 547
H2S(r) I—1, 258, 265, 266, 399, 422; 1—2, 190, 324—
326
H2SO4(r) I—1, 248, 266-268; 1-2, 191
SF(r) I—1, 242, 244, 268, 269; 1—2, 192
SF-(r) I—1, 242, 244, 269, 270; 1—2, 193; IV—2,
548
SF2(r) I—1, 259, 270, 271; IV—1, 621; 1—2, 194
SF3(r) I—1, 259, 271, 272; 1—2, 195
SFs(r) 1-1, 259, 272; IV-1, 621; 1-2, 196
SF4(r) I—1, 259, 273; 1-2, 197
SF5(r) I—1, 259, 273, 274; IV—1, 604, 605; 1—2,
198; IV—2, 549
SFg-(r) I—1, 259, 274, 275; IV-1, 605; 1—2. 199;
IV—2, 550
SFe(r» I—1, 259, 275, 276, 399, 412, 422, 423;
IV-1, 621; 1—2, 200, 324-326
SOF2(r) I—I, 259, 276, 277; 1—2, 201
SO2F2(r) I—1, 259, 277, 278; 1-2, 202
N(r) 1-1, 279, 280; 1—2, 203, 204
N+(r) I—1, 279, 280; 1—2, 205, 206
N2(r) 1-1, 280—284, 399, 412, 423; 1-2, 207, 208,
324—326
N?(r) I—1, 281, 283-286; 1-2, 209, 210
N3(r) I—1, 286, 287; 1—2, 211
NO(r) I—1, 287—290, 399, 423; 1—2, 212, 213, 324-
326; IV—2, 558
NO+(r) I—1, 288-293; 1—2, 214, 215
NO2(r) I—1, 292—294; 1-2, 216
NO^r) I—1, 286, 294—296; 1-2, 217
NO3(r) I—1, 286, 296; 1—2, 218
N2O(r) I—1,|292, 297-299, 399, 423; 1—2, 219, 324—
326
N2O3(r) I—1, 298—300; 1—2, 220
N2O4(r) I—1, 298, 300, 301, 423, 424; 1-2, 221,
324—326
608
Формульный указатель веществ
N2O5(r) 1-1, 298, 301—303; 1—2, 222
NH(r) I—1, 303—306; 1—2, 223, 224
NH+(r) I—I, 303, 305-307; 1—2, 225, 226; IV~2, 558
NH2(r) 1-1, 307—309; 1—2, 227
NH3(r) I—1, 308—311, 413, 425; 1—2, 228, 324—
326
NHf(r) I—1, 286, 311, 312; 1—2, 229
N2H2(r) I—1, 312-314; 1—2, 230; IV—2, 558
mpattc-N2H2(r) I—1, 312—314; 1—2, 231
jfMc-N2H2(r) I—1, 312, 314, 315; 1—2, 232
l,l-N2Hg(r) I—1, 312, 315; 1—2, 233
N2H4(r) I—1, 298, 315—317, 413, 425; I~2, 234,
324-326; IV-2, 558
HN3(r) I—1, 307, 317; 1-2, 235
HNO(r) 1-1, 307, 317, 318; 1-2, 236
HNO2(r) I—1, 312, 319, 320; 1—2, 237
mpaHc-HNO2(r) I—1, 312, 320, 321; 1—2, 238
ifKc-HNO2(r) I—1, 312, 321; 1—2, 239
HNO3(r) I—1, 298, 321, 322; 1—2, 240; IV—2, 558
NH20H(r) I—1, 298, 322, 323; 1—2, 241
NH2NO2(r) I—1, 298, 323, 324; 1—2, 242
NH4NO3(k, ж) 1-1, 324, 325; IV-1, 621; 1-2, 321
NF(r) I—1, 325—327; 1—2, 243;lIV-2; 558
NF2(r) I—1, 307, 327, 328; 1—2, 244
NF3(r) I—1, 309, 329, 330, 413, 425; [1—2, 245,
324—326
N2F2(r) I—1, 312, 330, 331; 1—2, 246
mpaHc-N2F2{T) I—1, 312, 332, 425, 426; 1—2, 248,
324, 326
4«e-NaF2(r) I—I, 312, 331, 332, 425, 426; 1—2, 247,
324, 326
N2F4(r) I—1, 312, 332, 333, 426; 1-2, 249, 324, 326
mpeKc-N2F4(r) I—1, 312, 333; 1—2, 250; IV—2, 558
eoiu-N2F4(r) I—1, 312, 333, 334; 1—2, 251
FNO(r) I—I, 292, 334, 335, 426; 1—2, 252, 324, 326
FNO2(r)I—1,307, 335, 336, 426; 1—2, 253, 324, 326;
II-2, 343; IV-2, 558
FNO3(r) I—1, 298, 336, 337; 1—2, 254; IV—2, 558
F3NO(r) I—1, 286, 337, 338, 426; 1—2, 255, 324,
326
NHF(r) I—1, 286, 338; 1—2, 256
NH2F(r) I—1, 286, 338, 339; 1—2, 257
NH4F(k, ж) I—1, 325, 339; 1—2, 322
NHF,(r) I—1, 286, 339, 340, 426; 1—2, 258, 324, 326
ClNO(r) I—1, 292, 340, 341; 1—2, 259
ClNO2(r) I—1, 307, 341—343; 1—2, 260
NS(r) I—I, 326, 343, 344; 1—2, 261
Р(к, белый, ж) I—1, 345, 346; 1—2, 323; IV—2, 558
P(r) I—1, 346, 347, 426, 427»; 1—2, 262, 263, 324,
326 i
P2(r) I—1, 347—350; 1—2, 264; IV—1, 621; IV-2,
558
P,(r) 1-1, 350-352; 1-2, 265
P4 (r) I—1, 351-353; I—2, 266
PO(r) I—1, 347, 349, 353, 354; 1—2, 267
PO-(r) 1-1, 347, 349, 355; 1-2, 268
PO2(r) I—1, 355, 356; 1-2, 269
POifr) I—1, 356, 357; 1-2, 270
P2O3(r) 1-1, 351, 357, 358; 1-2, 271
P2O4(r) I—1, 351, 358; 1-2, 272
P2O6(r) I—1, 351, 358, 359; 1-2, 273
P3O,(r) I—1, 351, 359, 360; 1—2, 274
P4Oe(r) I—1, 351, 360, 361; 1-2, 275
P4O7(r) I—1, 351, 361; 1-2, 276
P4O8(r) I—1, 351, 361, 362; 1-2, 277
P4O9(r) I—1, 351, 363; 1—2, 278
Р4О10(к, ж) I—1, 345, 363—365; 1-2, 279
P4O10(r) I—1, 351, 366; 1—2, 280; IV-2, 558
PH(r) I—1, 349, 366—368; 1—2, 281
PH2(r) I—1, 356, 368, 369; 1-2, 282
РНг(г) I—1, 356, 369; 1-2, 283
HPO(r) I—i, 356, 369, 370; 1-2, 284
PF(r) I—1, 349, 367, 370, 371; 1-2, 285; IV~2, 558
PF2(r) 1-1, 356, 371, 372; 1-2, 286; IV-2, 558
PFa(r) I—1, 356, 372; 1—2, 287
PF3(r) 1-1, 372-374, 427; 1-2, 288, 324, 326
PF5(r) I—1, 373, 374; 1—2, 289
POF3(r) I—1, 373—375; 1—2, 290
PCl(r) I—1, 349, 367, 375, 376; 1-2, 291
PCl2(r) I—1, 356, 376, 377; 1-2, 292
PCli-(r) I—1, 356, 377; 1—2, 293
РС13(г) I—1, 373, 377, 378, 427; 1—2, 294, 324, 326;
IV—2, 558
PCl5(rf I—1, 373, 378, 379; 1-2, 295
РОС13(г) I—1, 373, 379—381; 1—2, 296
PFCl(r) 1-1, 356, 381; 1-2, 297
PFCl-(r) I—1, 356, 381, 382; 1—2, 298
PF2Cl(r) I—1, 373, 382, 427; 1—2, 299, 324, 326
PF4Cl(r) I—1, 373, 382, 383; 1—2, 300; IV-2, 558
PFCl2(r) I—1, 373, 383, 427; 1-2, 301, 324, 326
PF3Cl2(r) I—1, 373, 383, 384; 1—2, 302
PF2Cl3(r) I—1, 373, 384; 1—2, 303
PFCl4(r) I—1, 373, 385; 1—2, 304
POFaGl(r) I—I, 373, 385, 386; 1-2, 305
POFCl2(r) I—1, 373, 386; 1—2, 306
PS(r) I—1, 349, 367, 386, 387; 1—2, 307
PN(r) I—1, 349, 367, 387, 388; 1-2, 308
С(к, ж) II—1, 10-12; II-2, 10, 11, 340
С(к, алмаз) II—1, 10, 12, 13; II—2, 332
C(r) II—1, 13,^14, 3521; II-2, 12, 13, 338-340»
C+(r) II—1, 13, 14; II-2, 14, 15
C-(r) II—1, 13—15; II—2, 16
C2(r) II—1, 15-19; II-2, 17, 18
СЦт) II—1, 16, 18—20; II-2, 19, 20
C2 (r) II—1, 16, 18, 20, 21; II—2, 21, 22;
IV—2, 559
Данные для пара фосфора {Р„, п—ъ, ¦*).
39 Заказ № 1590
l» д.ля пара
609
Формульный указатель веществ
Сз(г) II—1, 21—23; Н-2, 23, 24
С4(г) II—1, 21, 23, 24; П-2, 25, 26
С^г) П-1, 21, 25; И-2, 27, 28
СО(г) И—1, 25—30, 252, 352, 353; II—2, 29, 30,
338—340
СО+(г) II—1, 26, 28, 30, 31; II—2, 31, 32
СО2(г) II—1, 31—33, 352, 353; II—2, 33, 34, 338—
340
СО?(г) II—1, 33—35; II—2, 35, 36; IV—2, 559
С2О(г) П-1, 34, 35; И-2, 37
С3О2(г) II—1, 21, 35—37; II—2, 38
СН(г) 1—1, 37—40; И—2, 39, 40
СН+(г) П-1, 38-41; И-2, 41, 42; IV-2, 559
СН2(г) II—1, 41—44; И—2, 43
СН3(г) II—1, 44—46; II—2, 44
СН4(г) II—1, 46—48, 353, 354; II—2, 45, 338—340
С2Н(г) П-1, 42, 48, 49; П-2, 46, 47
С2Н2(г) II—1, 49, 50, 353—355; И—2, 48, 49, 338—
340
С2Н3 (г) П-1, 50—52; II—2, 50
С2Н4(г) П-1, 52, 53, 353, 355; П-2, 51, 338-340
СаН5 (г) П-1, 53-55; IV-1, 622; И-2, 52;
p.. IV-2, 559
С2Нв (г) II—1, 53, 55-57, 353, 355; П-2, 53, 338-
340
НСО(г) II—1, 34, 57, 58; IV—1, 622; И—2, 54
СООН(г) II—1, 58, 59; II—2, 55
Н2СО(г) II—1, 59, 60; II—2, 56
НСООН(г) II—1, 51, 60-62; П-2, 57
транс-ЯСООЩт) П-1, 51, 63; Н-2, 59
Чмс-НСООН(г) II—1, 51, 62, 63; II—2, 58
СН3О(г) II—1, 51, 63, 64; II—2, 60
СН2ОН(г) П-1, 51, 64, 65; П-2, 61
С2Н3Г(г) II—1, 53, 92, 93, 358; Н-2, 83, 338—340
G2H2F2(r) II-1, 93; И-2, 84
mpaHC-C2R2F2(T) II—1, 94, 96; II—2, 87
^uc-C2H2F2(r) II—1, 94-96; II—2, 86
l,l-C2H2F2(r) II—1, 93-95, 358; И—2, 85, 338—340
C2HF3(r) II—1, 53, 96, 97; И-2, 88
HFCO(r) II—1, 59, 97; И—2, 89
СС1(г) II—1, 38, 39, 98, 99, 230; IV—1, 622; II—2, 90
СС12(г) II—1, 42, 99, 100; II—2, 91
СС18(г) II—1, 44, 100—102; II—2, 92
СС14(г) II—1, 46, 101—103, 358; II—2Д93, 338—340
С2С1(г) II—1, 42, 102, 103; II—2, 94
С2С12(г) II—1, 44, 104; Н-2, 95
С2С13(г) II—1, 51, 104, 105; II—2, 96
С2С14(г) II—1, 53, 105, 358; II—2, 97Д338—340
С2С15(г) И-1, 54, 105, 106; П-2, 98"
С2С1„(г) II—1, 53, 106, 107; II—2, 99
С1СО(г) II—1, 34, 107, 108; II—2, 100
С12С0(г) II—1, 59, 108, 109, 359; И-2, 101, 338-
340
СНС1(г) II—1, 42, 109. НО; II—2, 102
СН2С1(г) II—1, 44, 110, 111; И-2, 103
СН3С1(г) П-1, 87, 112, 113, 359; Н-2, 104, 338-
340
СНС12(г) Н-1, 44, 112-114; Н-2, 105
СН2С12(г) П-1, 90, 113-115, 359; И-2, 106, 338-
340
СНС13(г) Н-1, 87, 115, 116, 359; Н-2, 107, 338-
340
С2НС1(г) П-1, 44, 116, 117; Н-2, 108
С2Н3С1(г) П-1, 53, 117, 118, 359; Н-2, 109, 338-
340
С2Н2С12(г) II—1, 118; Н-2, НО; IV—2, 559
CH3OH(r) II—1, 54, 66—68, 353, 356; IV—1, 622; га^амс-С2Н2С12(г) II—1, 94, 120, 360; П-2, ИЗ,
II—2, 338—340 338—340
С2Н5ОН(г) П-1, 54, 68, 69, 356, 357т IV-1, 622; ifKc-C2H2Cl2(r) II-l, 94, 119, 120, 360; П-2, 112,
II—2, 63, 338—340 338—340
CF(r) II—1,38, 39, 69—71, 230; IV—1, 622; И—2, 64 1,1-С2Н2С12(г) П-1, 94, 118, 119, 359; П-2, 111,
л 1-1 / \ ж* м /о гчл г*а. Т? п DC QOOO/A
CF2(r) П-1, 42, 71-73; Н-2, 65
CF3(r) П-1, 44, 72-74; П-2, 66
CF4(r) II—1, 46, 74—76, 357; И—2, 67, 338-340
C2F(r) П-1, 42, 75, 76; Н-2, 68
C2F,(r) II—1, 44, 76, 77; II—2, 69
C2Fs(r) II-l, 51, 77, 78; П-2, 70
C2F4(r) II—1, 53, 78, 79, 357; Н-2, 71, 338-340
C2F6 (г) Н-1, 54, 79, 80; Н-2, 72
C2F6(r) П-1, 53, 80, 81; П-2, 73, 338-340
FCO(r) II—1, 34, 81, 82; Н-2, 74
F2CO(r) П-1, 59, 82, 83; П-2, 75
CHF(r) II—1, 42, 83-85; П-2, 76
CH2F(r) П-1, 44, 85, 86; П-2, 77
CH3F(r) II—1, 86—88, 357; И—2, 78, 338—340
CHF2(r) П-1, 44, 88, 89; И-2, 79
CH2F2(r) II-l, 89, 90, 357; П-2, 80, 338—340
CHF3(r) II—1,87,90,91,354,358; Н-2, 81, 338-340
C2HF(r) П-1, 44, 91, 92; Н-2, 82
338-340
С2НС13(г) II—1, 53, 120, 121, 360; IV—1, 622;
II—2, 114, 338—340
НСЮО(г) II—1, 59, 121, 122; И-2, 115
CFCl(r) II—1, 42, 122, 123; П-2, 116
CF2Cl(r) II—1, 44, 123; II—2, 117
CF3Cl(r) II-l, 87, 123, 124, 354, 360; II—2, 118,
338-340
CFCl2(r) II—1, 44, 124, 125; И—2, 119
CF2C]2(r) II—1, 90, 125, 126, 360, 361; И-2, 120,
338-340
CFCl3(r) II-l, 87, 126, 127, 361; И-2, 121, 338-
340
C2FCl(r) II—1, 44, 127; H-2, 122
C2F3Cl(r) II-l, 53, 128, 361, 362; H-2, 123, 338-
340
C2F2Cl2(r) II-l, 128, 129; H-2, 124; IV-2, 551
mpaKC-C2F2Cl2(r) II-l, 94, 130; H-2, 127
610
Формульный указатель веществ
4Mc-C2F2Cl2(r) II—1, 94, 129, 130; Н-2, 126
l,l-C2F2Cl2(r) II—1, 94, 129; Н-2, 125
C2FCl3(r) II—1, 53, 130, 131; II—2, 128
FClCO(r) II—1. 59, 131, 132; II—2, 129
CHFCl(r) II—1, 44, 132; Н-2, 130
CH2FCl(r) II—1, 90, 132, 133; II—2, 131
CHFaCl(r)II—1, 133, 134, 354, 362; П-2, 132, 338—
340
CHFCl2(r) II—1, 133-135, 362; II-2, 133, 338-340
C2H2FCl(r) II—1, 135; H-2, 134
mpaKc-C2H2FCl(r) II—1, 94, 136, 137; II—2, 137
ijwc-C2H2FC](r) II—1, 94, 136; II—2, 136
l,l-C2H2FCl(r) II-l, 94, 135, 136; II—2, 135
C2HF2Cl(r) II—1, 137, 362; II—2, 138, 338-340
uipaBc-CjjHFjjCUi) II—1, 94, 138, 139; II—2, 141
i^uc-C2HF2Cl(r) II—1, 94, 138; II—2, 140
l,l-C2HF2Cl(r) II—1, 94, 137, 138; II—2, 139
C2HFCl,(r) II—1, 139; II-2, 142
mpoKc-C2HFCl2(r) II-l, 94, 139, 140; H-2, 143
4uc-C2HFCl2(r) II—1, 94, 140; H-2, 144
l,l-C2HFCl2(r) II—1, 94, 140, 141; H-2, 145
CBr(r) II—1, 38, 39, 141, 142, 230; IV—1, 622;
II—2, 146
СВг2(г) II—1, 42, 142, 143; H-2, 147
CBr3(r) II-l, 44, 143, 144; II—2, 148
СВг4(г) II-l, 46, 144; II—2, 149
СН3Вг(г) II-l, 87, 145, 146, 363; H-2, 150, 338—
340
CH2Br2(r) II—1, 90, 146, 147; И-2,„151
СНВгд(г) II—1, 87, 147; H-2, 152
CF3Br(r) II—1, 87, 147, 148, 363; II—2, 153, 338—
340
CF2Br2(r) II—1, 90, 148; II—2, 154
CFBrs(r) II—1, 87, 148, 149; II—2, 155
CH2FBr(r) II-l, 90, 149; H-2, 156
CHF2Br(r) II—1, 133, 150; H-2, 157
CHFBr2(r) II-l, 133, 150, 151; H-2, 158
CCl3Br(r) II—1, 87, 151; II—2, 159
CCl2Br2(r) II-l, 90, 152; II—2, 160
СС1Вг3(г) II—1, 87, 152, 153; II-2, 161
СН2С1Вг(г) II—1, 90, 153; II—2, 162
CHCl2Br(r) II-l, 133, 153, 154; II—2, 163
СНС1Вг2(г) II—1, 133, 154; II—2, 164
CF2ClBr(r) II—1, 154, 155, 363; H-2, 165, 338-340
CFCl2Br(r) II—1, 155, 156; II—2, 166
CFClBr2(r) II-l, 155, 156; II—2, 167
CHFClBr(r) II—1, 133, 157; II—2, 168
CI(r) II—1, 38, 39, 157, 158; 230; II—2, 169
CI2(r) II-l, 42, 158, 159; H-2, 170
CI3(r) II—1, 44, 159; H-2, 171
CI4(r) II—1, 46, 159, 160; II—2, 172
CH3I(r) II—1, 87, 160, 161, 363; II—2, 173, 338-
340
CH2I2(r) II—1, 90, 161, 162; II—2, 174
CHI3(r) II—1, 87, 162, 163; II—2, 175
CF3I(r) II—1, 87, 163, 164; II—2, 176
CF2I2(r) II—1, 90, 164; II—2, 177
CFI3(r) II—1, 87, 164, 165; II—2, 178
CH2FI(r) II—1, 90, 165; II—2, 179
CHF2I(r) II-l, 133, 165, 166; II—2, 180
CHFI2(r) II—1, 133, 166, 167; II—2, 181
CCl3I(r) II-l, 87, 167; II-2, 182
CCl2I2(r) II—1, 90, 167, 168; II—2, 183
CClI3(r) II—1, 87, 168; II—2, 184
CH2ClI(r) II—1, 90, 168, 169; II—2, 185
CHCl2I(r) II—1, 133, 169; II—2, 186
CHCH2(r) II-l, 133, 169, 170; П-2, 187
CF2CH(r) II-l, 155, 170; II—2, 188
CFCl2I(r) II-l, 155, 171; H-2, 189; IV-2, 559
CFClI2(r) II—1, 155, 171, 172; II—2, 190
CHFClI(r) II-l, 133, 172; H-2, 191
CBr3I(r) II-l, 87, 172, 173; H-2, 192
CBr2I2(r) II—1, 90, 173; II—2, 193
CBrI3(r) II—1, 87, 173, 174; II—2, 194'
CH2BrI(r) II—1, 90, 174; II—2, 195
CHBr2I(r) II—1, 133, 175; II—2, 196
CHBrI2(r) II—1, 133, 175, 176; H-2, 197
CF2BrI(r) II-l, 155, 176; II—2, 198
CFBr2I(r) II-l, 155, 176, 177; II—2, 199
CFBri2(r) II—1, 155, 177; II—2, 200
CCl2BrI(r) II—1, 155, 177, 178; II—2, 201
CClBr2I(r) II—1, 155, 178; II—2, 202
CClBrI2(r) II—1, 155, 178, 179; II—2, 203
CHFBrl(r) II—1, 133, 179; II—2, 204
CHClBrl(r) II—1, 133, 180; II—2, 205
CFClBrl(r) II-l, 155, 180, 181; II-2, 206
CS(r) II-l, 26, 181-183, 252; IV-1, 622;] II—2, 207
CS2(r) II—1, 32, 184, 185, 363; II—2, 208, 338—340
COS(r) II—1, 32, 185, 186, 363; H-2, 209, 338-340
CN(r) II—1, 182, 187—190; II—2, 210, 211
CN+(r) II—1, 182, 187, 190, 191; H-2, 212, 213
CN-(r) II—1, 182, 187, 191, 192; П-2, 214, 215
NCN(r) II—1, 34, 192, 193; II—2, 216
CNN(r) II—1, 34, 193, 194; II—2, 217
CNC(r) II—1, 34, 194, 195; II—2, 218
CCN(r) II-l, 34, 195; IV-1, 622; II—2, 219
C2N2(r) II—1, 49, 195, 196, 363, 364; H-2, 220,
338—340
NCO(r) II-l, 34, 196, 197; II—2, 221
HCN(r) II—1, 32, 197—199, 364; II—2, 222, 223.
338—340
HNC(r) II—1, 34, 199; H-2, 224
HC2N(r) II—1, 44, 199, 200; II—2, 225
FCN(r) II—1, 32, 200, 201; II—2, 226
ClCN(r) II—1, 32, 201, 202; II—2, 227
CP(r) II—1, 182, 187, 202, 203; II—2, 228
Si(it, ж) II—1, .204—207; II—2, 229, 230
Si(r) II—1, 206—208; II—2, 230, 231
Si+(r) II—1, 207, 208; II—2, 232, 233
Si2(r) II—1, 208—213; II—2, 234
Sis(r) II-l, 213-215; II—2, 235
611
Формульный указатель веществ
SiO(r) II—1, 209—211, 214—217, 252; II—2, 236;
IV—2, 552, 553
SiO2(n, ж)|Н—1, 205, 217—219; Н-2, 237;
IV—2, 554
SiO2(K, кварц) II—1, 205, 219; II—2, 332
SiO2(K, кристобалит) II—1, 2Q5, 220; II—2, 333
8Ю2(к,*тридимит) II—1, 205, 220, 221; И-2, 333
8Ю2(стекл.) II—1, 205, 221, 222; И—2, 334
SiO2(r) II—1, 213, 222, 223; П—2, 238
SiH(r) II—1, 210, 211, 223—225; II—2, 239
SiH2(r) II—1, 225, 226; И—2, 240
вШз(г){П—1, 226—228; И-2, 241
SiH4(r) II—1, 227—229, 364; II—2, 242, 338-340
SiF(r)|.II—1, 210, 211, 224, 229—231; II—2, 243
SiF2(r) II—1, 226, 231, 232; II—2, 244
SiFs(r)lII—1, 227, 233, 234; II—2, 245
SiF4(r) II—1, 227, 234, 364; II—2, 246, 338—340
SfflF(r) II—1, 226, 234, 235; II—2, 247JJ
SiCl(r) II—1, 210, 211, 224, 230, 235—237;
П-2, 248
SiCl2(r) П—1, 226, 237, 238; II—2, 249
SiCla(r) II—1, 227, 238, 239; II—2, 250
SiCl4(r) II—1, 227, 239-241, 364; II-2, 251, 338-
340
SiHCl(r) II—1, 226, 240, 241; II-2, 252
SiFCl(r) II—1, 226, 242; II—2, 253
SiBr(r) II—1, 210, 211, 224, 230, 242, 243;
II—2, 254
SiBr2(r) II—1, 226, 244, 245; II—2, 255
SiBr3(r) II—1, 227, 245; II—2, 256
SiBr4(r) II—1, 227, 245, 246, 364, 365; II-2, 257,
338Л340
Sil(r) II—1, 210, 211, 224, 230, 246-248; II—2, 258
SiI2(r) II—1, 226, 248, 249; II—2, 259
SiI3(r) II—1, 227, 248, 249; II-2, 260
SiI4(r) II—.1, 227, 249—251, 364, 365; II—2, 261,
338, 340
SiS(K, ж) II—1, 205, 251; IV—1, 622; И—2, 262
SiS(r);II—1, 209—211, 251—254; II—2, 263
SiS2(K, ж)р1—1, 205, 254, 255; II—2, 264
SiS2(r) II—1, 213, 255, 256; II—2, 265
SiN(r) II—1, 209-211, 256, 257; II—2, 266;
IV— 2Д559]
Si3N4(K)|II—1, 205, 257—259; II—2, 267
SiO(K, ж);Ш—1, 205, 258-260; IV—1, 622; (
II—2, 268; IV—2, 555 Щ
SiC(n, куб.) II—1, 205, 259, ,260; II—2, 334
SiC(r) II—1, 209—211, 261, 262; II—2, 269
SiC2(r) II—1, 213, 262, 263; II—2, 270
Si2C(r)lH-l, 213, 263-265; II—2, 271
Ge(K, ж)III—1, 266—268, 365; II—2, 272, 340
Ge(r) II—1, 268, 269, 365; II—2, 273, 274
Ge+(r) II—1, 269, 271; II—2, 275, 276
Ge2(r) II—1, 270—273; II—2, 277
GeO(r) II—1, 252, 270, 272—276; II—2, 278
GeO2(K, ж) II—1, 266, 276, 277; II—2, 279
GeO2(K, гекс.) II—i, 266, 277, 278; II—2, 335
GeO2(cTeroi.) II—1, 266, 278, 279; II—2, 335
GeO2(r) II—1, 279, 280; II—2, 280
GeF(r) II—1, 230, 270, 272, 280, 281; II—2, 281
GeF2(r) II—1, 279, 281, 282; II—2, 282
GeF,(r) II—1, 279, 282, 283; II—2, 283
GeF4(r) II—1, 279, 283; II—2, 284
GeCl(») II—1, 230, 270, 272, 283-285; II—2, 285
GeCl2(r) II—1, 279, 285, 286; II-2, 286
GeClg(r) II—1, 279, 286, 287; II—2, 287
GeCl4(r) II—1, 279, 287, 288, 365; П-2, 288, 338,
340
GeS(K, ж) II—1, 266, 288, 289; IV—1, 622;
II—2, 289
GeS(r) II—1, 252, 270, 272, 289, 290; II-2, 290;
IV—2, 559
GeS2(K, ж) II—1, 266, 291, 292; II—2, 291
GeS2(r) II—1, 279, 292, 293; II—2, 292
Sn(K, ж) II—1, 294—296; IV—1, 622; II—2, 293, 340
Sn(r) II—1, 296, 297, 365; II—2, 294, 295, 340
Sn+(r) II—1, 297; II—2, 296, 297
Sn2(r) II—1, 298—300; II—2, 298
SnO(K, ж) II—1, 294, 301, 302; II—2, 299
SnO(r) II—1, 252, 298, 300, 302—304; II—2, 300
SnO2(K, ж) II—1, 294, 304—306; II—2, 301
SnO2(r) II—1, 306; II-2, 302
SnF(r) II—1, 230, 298, 300, 307, 308; IV—1, 622;
II—2, 303
SnF2(K, ж) II—1, 294, 308, 309; IV—1, 622;
II—2, 304
SnF2(r) II—1, 306, 309, 310; II—2, 305
SnCl(r) II—1, 230, 298, 300, 310, 311; II—2, 306
SnCl2(K, ж) II—1, 294, 311, 312; IV—1, 622;
II—2, 307
SnCl2(r) II—1, 306, 313; II—2, 308
SnCl4(r) II—1, 365; II—2, 338, 340; IV-2, 559
SnS(K, ж) II—1, 294, 313—315; II—2, 309
SnS(r) II—1, 252, 298, 300, 315—317; II—2, 310
SnS2(K) II—1, 294, 317, 318; II—2, 311
SnS2(r) II—1, 306, 318, 319; II—2, 312
РЬ(к, ж) II—1, 320—322, 365; II—2, 313, 340
Pb(r) II—1, 323, 365; II—2, 314, 315; IV—2, 559
Pb+(r) II—1, 323, 324; II—2, 316, 317
Pb2(r) II—1, 324—328; II—2, 318
РЬО(к, ж) II—1, 321, 328—331; IV—I, 622;
II—2, 319,
РЬО(к, ромб., желт.) II—1, 321, 329, 331;
II—2, 336|
РЬО(г) II—1, 252, 325, 327, 331—333; II—2, 320
РЬО2(к) II—1, 321, 333; II—2, 336
РЬО2(г) И—1, 333—335; II—2, 321
РЬ2О3(к) II—1, 321, 335; И—2, 337
612
Формульный указатель веществ
РЬ3О4(к) II—1, 321, 335, 336; И—2, 337
PbF(r) II—1, 230, 325, 327, 336-338; IV—1, 622;
II—2, 322
PbF2(K, ж) II—1, 321, 338—340; IV—1, 622;
II—2, 323
PbF2(r) II—1, 334, 340, 341; II—2, 324
РЬС1(г) II—1, 230, 325, 327, 341, 342; II—2, 325
РЬС12(к, ж) II—1, 321, 342-345; П—2, 326
РЬС12(г) И-1, 334, 344, 345; II—2, 327
PbS(K, ж) II—1, 321, 346-348; IV—1, 622;
И-2, 328j
PbS(r) II—1, 252, 325, 327, 348, 349; II—2, 329
PbS2(r) II—1, 334, 350^ И-2, 330
В(к, ж) III—1, 8—10, 422; III—2, 12, 13, 395
В(аморф.) III—1, 9—11; III—2, 389
В(г) III—1, 11, 12, 422; III—2, 14, 15, 395
В+(г) III—1, 12, 13; III—2, 16, 17
В2(г) III—1, 13—15; III—2, 18
ВО(г) III—1, 13, 15—17; Ш-2, 19, 20
ВО-(г) III—1, 13, 15, 17, 18; Ш-2, 21
ВО2(г) III—1, 17—19; III—2, 22, 23
BOj(r) III—1, 20, 21; Ш-2, 24
В2О(г) III—1, 20, 21; III—2, 25
В2О2(г) Ш-1, 20—22; III—2, 26
В2О3(к, ж) III—1, 9, 22—24; III—2, 27
В2О3(стекл.) Ш-1, 9, 24, 25; Ш-2, 389
В2О3(г) III—1, 25—27; III—2, 28
ВН(г) III—1, 27—31; Ш-2, 29
ВН2(г) III—1, 31, 32; Ш-2, 30
ВН3(г) III—1, 32—34; III—2, 31
В2Нв (г) III—1, 34—36, 420; Ш-2, 32, 395
НВО(г) III—1, 20, 36, 37; III—2, 33
ВОН(г) III—1, 20, 37; Ш-2, 34
НВО2(к, ж) III—1, 9, 37, 38; III—2, 389
НВО2(г) Ш-1, 38, 39; III—2, 35
НВОН(г) III—1, 38-40; III—2, 36
В(ОНJ(г) III—1, 25, 40, 41; III—2, 37
Н2ВОН(г) Ш-1, 25, 41, 42; III—2, 38
НВ(ОНJ(г) Ш-1, 42, 43; III—2, 39
Н3ВО3(к, ж) III—1, 9, 43, 44; Ш-2, 389
Н3ВО3(г) III—1, 42, 44, 45; III—2, 40
В2(ОНL(г) III—1, 42, 45, 46; III—2, 41
Н3В3О3(г) III—1, 42, 46, 47; Ш-2, 42
Н3В3Ов (г) Ш-1, 35, 47, 48; Ш-2, 43
BF(r) III—1, 28, 29, 48, 49; III—2, 44
BFa(r) Ш-1, 31, 49, 50; Ш-2, 45
BFa(r) Ш-1, 31, 50, 51; III—2, 46
BF3(r) III—1, 32, 51, 52; 420, III—2, 47, 395
BFj(r) III—1, 32, 52, 53; III-2, 48
B2F4(r) III—1, 42, 53; III—2, 49
FBO(r) III—1, 20, 54; III—2, 50
F2BO(r) III—1, 38, 54, 55; III—2, 51
F3B3O3(r) III—1, 42, 55, 56; III-2, 52
BHF(r) III—1, 31, 56, 57; HI—2, 53
BH2F(r) III—1, 32, 57; III—2, 54
BHF2(r) III—1, 32, 57, 58; III—2, 55
FBOH(r) III—1, 38, 58, 59; III-2, 56
FB(OHJ(r) III—1, 42, 59; III—2, 57
F2BOH(r) III—1, 25, 59, 60; III—2, 58
BCl(r) III—1, 28, 29, 60, 61; IV—1, 605; III—2, 59;
IV-2, 556
ВС12(г) III—1, 31, 61, 62; Ш-2, 60
BCl3(r) III—1, 32, 62-64, 420, 421; III—2, 61, 395
B2Cl4(r) HI—1, 25, 64; III—2, 62
ClBO(r) III—1, 20, 64, 65; -III—2, 63
Cl2BO(r),III—1, 38, 65; HI—2, 64
Cl3B3O3(r) III—1, 42, 66; III—2, 65
BHCl(r) III—1, 31, 66, 67; III—2, 66
BH2Cl(r) HI—1, 32, 67; III—2, 67
ВНС12(г) III—1, 32,' 67—69; III—2, 68
ClBOH(r) III—1, 38, 69; HI—2, 69
ClB(OHJ(r) III—1, 42, 69, 70; III—2, 70
Cl2BOH(r) III—1, 25, 70; III—2, 71
BFCl(r) III—1, 31, 70, 71; III—2, 72
BF2Cl(r) III—1, 32, 71, 72; HI—2, 73
BFCla(r) HI—1, 32, 72; HI—2, 74
F2ClB3O3(r) III—1, 35, 72, 73; III—2, 75
FCl2B3O3(r) III—1, 35, 73; III—2, 76
BHFCl(r) III—1, 32, 74; III—2, 77
BBr(r) III—1, 28,.29, 74, 75; III-2, 78
BBr2(r) III—1, 31, 75; III—2, 79
BBr3(r) HI—1, 32, 75, 76, 420, 421; HI—2, 80, 395
BI(r) III—1, 28, 29, 76, 77; III—2, 81
BI2(r) III—1, 31, 77, 78; III—2, 82
BI3(r) III—1, 32, 78, 79, 420, 421; III—2, 83, 395
BS(r) III—i, 13, 15, 79, 80; III—2, 84
BS2(r) III—1, 20, 80, 81; HI—2, 85
B2S(r) III—1, 20, 81, 82; HI—2, 86
B2S2(r) III—1, 20, 82; III—2, 87
B2S3(k, ж) III—1, 9, 82, 83; III—2, 390
B2S3(r). Ill—1, 25, 83, 84; III—2, 88
BN(k, ж) HI—1, 9, 84, 85; III—2, 89
BN(r) III—1, 13, 15, 85, 86; III—2, 90
BH3NH3(r) III—1, 42, 86, 87;f III-2, 91
B3N3He(r) III—1, 35, 87, 88;$ III—2, 92
BC(r) III—1, 13, 15, 88, 89; "ill—2, 93
BC2(r) III—1, 20, 8§, 90; Ш-2, 94
B2C(r) HI—1, 20, 90; III—2, 95
B4C(k) III—1, 9, 90, 91; III—2, 96
А1(к, ж) III—i, 92—94, 422; III—2, 97,1395
Al(r) III—1, 94, 95, 422; IH-2, 98, 99, 395
Al+(r) III—1, 95; HI—2, 100, 101
Al2(r) III—1, 96-98; III—2, 102
AlO(r) HI—1, 96-101; III—2, 103, 104
AlO-(r) III—1, 96, 97, 101, 102; Ш-2, 105
АЮг(г) III—1, 102-104; III-2, 106
A10j(r) III—1, 102, 104; III—2, 107
Al2O(r) HI—1, 102, 104—106; III—2, 108
Al2O2(r) III—1, 106-108; III—2, 109
А12О3(к, ж) III—1, 93, 108, 109; IH-2, 110, 111
613
Формульный указатель веществ
7-А1,О3(к) Ш—1, 93, 109, 110; Ш-2, 390
А120(г) III—1, НО, 111; Ш-2, 112
А1Н(г) III—1, 111—114; III—2, 113
А1Н2(г) III—1, 114, 115; III—2, 114
А1Н3(к) III—1, 93, 116; Ш-2, 390
А1Н3^г) III—1, 116, 117; III—2, 115
АЮН(г) III—1, 102, 117, 118; III—2, 116
НАЮ(г) III—1, 102, 118, 119; III—2, 117 '.':,
НАЮа(г) III—1, 107, 119, 120; III—2, 118
А1(ОН)а(г) III—1, 110, 120; III—2, 119
А1(ОНK(к) III—1, 93, 120, 121; III—2, 390
А1(ОНK(г) III—1, НО, 121, 122; IV—1, 622;
III—2, 120
AlF(r) Ш—1, 112, 113, 122, 123; III «2, 121
AlF,(r) III—1, 115,1123—125; Ш-2, 122
AlF2(r) III—1, 115, 124,125; Ш-2, 123;
IV—2, 557
AIFjj(k) III—1, 93, 125—127; III—2, 124
AlF3(r) III—1, 117, 127, 128; III—2, 125
AlFi(r) III—1, 117, 128, 129; IV—1, 622; III-2, 126
Al2Fe(r) III—1, 129, 130; III—2, 127
FAIO(r) III—1, 102, 130, 131; III—2, 128
F2A10(r) III—1, 107, 131; III—-2, 129
AlHF(r) III—1, 115, 132; III-2, 130 '
AlH2F(r) III—1, 117, 132, 133; Ш-2, 131
AlHF2(r) III—1, 117, 133; III—2, 132
FAlOH(r) III—1, 107, 133, 134; III—2| 133
FAl(OHJ(r) III—1, 110, 134; III—2, 134
F2A10H(r) III—1, 110, 134, 135; III—2, 135
AlCl(r) III-l, 112, 113, 135—137; III—2, 136
А1С12(г) III—1, 115, 137, 138; III—2, 137
А1С13(к, ж) III—1, 93, 138, 139, 421; III—2, 390, 395
А1С13(г) III—1, 117, 139, 140, 421; III—2, 138, 395
AlaCle(r) III-l, 129, 140, 141, 421; III-2, 139, 395
ClAlO(r) III—1, 102, 141, 142; III—2, 140
Cl2A10(r) III-l, 107, 142, 143; III—2, 141 ]• i
AlHCl(r) III-l, 115, 143, 144; III—2, 142 ','
AlH2Cl(r) III—1, 117, 144; III-2, 143
AlHCl2(r) III—1, 117, 144, 145; III—2, 144
ClAlOH(r) III—1, 107, 145; III—2, 145 *?}
ClAl(OHJ(r) III—1, 110, 145, 146; Ш-2, 146
Cl2A10H(r) III—1, 110, 146; III—2, 147
AlFCl(r) III-l, 115, 146, 147; III—2, 148
AlF2Cl(r) III—1, 117, 147; III—2, 149
AlFCl2(r) III—1, 117, 147, 148; III—2, 150
AlHFCl(r) III—1, 117, 148; HI—2, 151
AlBr(r) III—1, 112, 113, 148—150; III—2, 152
А1Вг2(г) III—1, 115, 150, 151; III—2, 153
А1Вг3(к, ж) HI—1, 93, 151, 152, 421; III—2, 391, 395
AlBr3(r) III—1, 117, 151—153, 421; III—2, 154, 395
Al2Br6(r) III—1, 129, 153, 154, 421; III—2, 155, 395
AH(r) III-l, 112, 113, 154, 155; III-2, 156
All2(r) III-l, 115, 156; fill—2, 157 < g iff
АИ3(к, ж) III—1, 93, 156, 157, 421; III—2, 391 ^
AlI3(r) III—1, 117, 157, 158, 421; III—2, 158, 395
Al2I6(r) III—1, 129, 158, 159, 421; III—2, 159, 395
AlS(r) III—1, 96, 97, 159, 160; III—2, 160
AlS2(r) III—1, 102, 160, 161; III—2, 161
Al2S(r) III—1, 102, 161, 162; III—2, 162
Al2S2(r) III-l, 107, 162, 163; III—2, 163
A12S3(k, ж) III—1, 93, 163; III—2, 164
АШ(к, ж) III-l, 93, 164, 165; Ш-2, 165
AlN(r) III—1, 96, 97, 165, 166; III—2, 166
AlC(r) III—i, 96, 97, 166; III-2, 167
AlC2(r) III—1, 102, 166, 167; III-2, 168
Al2C2(r) III—1, 107, 167; Ш-2, 169
А14С3(к) III—1, 93, 167, 168; III-2, 170
Ga(K, ж) III—1, 169-171, 422; III—2, 171, 395
Ga(r) III—1, 171, 422; III—2, 172, 173
Ga+(r) III—1, 171, 172; III—2, 174, 175
GaO(r) HI—1, 172—174; III—2, 176
Ga2O(r) III-l, 174-176; Ш—2, 177
Ga2O3(K, ж) III—1, 169, 176, 177; III—2, 178
GaH(r) III—1, 173, 174, 177—179; III—2, 179
GaOH(r) III—1, 175, 179, 180; III—2, 180
GaF(r) III—1, 173, 174, 180, 181; HI—2, 181
GaF2(r) III—1, 175, 181; III—2, 182
GaF3(K) III—1, 169, 181, 182; III—2, 183
GaF3(r) III-l, 175, 182, 183; III—2, 184
GaCl(r) III—1, 173, 174, 183—185; HI—2, 185
GaCl2(r) III-l, 175, 185; III—2, 186
GaCl3(K, ж) III-l, 169, 186, 187; III—2, 391
GaCl3(r) HI—1, 175, 187; HI—2, 187
Ga2Cle(r) III-l, 175, 187, 188,' III—2, 188
Iii(k, ж) III-l, 189-191, 422; III—2, 189, 395
In(r) III-l, 191, 192, 422; III—2, 190, 191
In+(r) III—1, 192; III-2, 192, 193
InO(r) III—1, 193—195; III—2, 194
In2O(r) III—1, 195—197; HI—2, 195
1п2О3(к, ж) III—1, 189, 197, 198; III—2, 196
InH(r) III—1, 193, 194, 198—200; III—2, 197
InOH(r) III-l, 195, 200, 201; III—2, 198
InF(r) III—1, 193, 194, 201, 202; III—2, 199
InF2(r) III—1, 195, 202; III—2, 200
InF3(K, ж) III-l, 189, 202, 203; III—2, 201
InF3(r) III-l, 195, 204; III—2, 202
1пС1(к, ж) III-l, 189, 204, 205; HI—2, 203
InCl(r) III-l, 193, 194, 205—207; III—2, 204
InCl2(r) HI—1, 195, 207; III—2, 205
1пС13(к, ж) III—1, 189, 207—209; III—2, 391
InCl3(r) III-l, 195, 209; III—2, 206
In2Cl8(r) III—1, 195, 209, 210; III—2, 207
Т1(к, ж) III-l, 211—213, 422; III—2, 208, 395
Tl(r) III-l, 213, 214, 422; HI—2, 209, 210
Tl+(r) HI—1, 213, 214; III—2, 211, 212
TlO(r) III—1, 214-216; III—2, 213
ТЮ2(к, ж) III-l, 211, 216, 217; III—2, 214
Tl2O(r) III-l, 217—219; III—2, 215
Т12О3(к, ж) III-l, 211, 219, 220; III—-2, 216
614
Формульный указатель веществ
ТШ(г) III—1, 215, 216, 220, 221; III—2, 217
ТЮН(г) III—1, 217, 221, 222; Ш-2, 218
T1F (к, ж) III—1, 211, 222, 223; III—2, 219
TlF(r) III—1, 215, 216, 223, 224; HI—2, 220
Tl2F2(r) III—1, 217, 224, 225; Ш-2, 221
Т1С1(к, ж) III—1, 211, 225, 226; III—2, 222
TlCl(r) III—1, 215, 216, 226—228; III—2, 223
Tl2Cl2(r) III—1, 217, 228; 229; III—2, 224
Сг(к, ж) IV—1, 9—12; IV—2, 12, 534
Сг(г) IV-1, 12; IV-2, 13, 14
Cr+(r) IV—1, 12, 13; IV-2, 15, 16
CrO(r) IV—1, 13—16; IV—2, 17, 18
CrO2(r) IV—1, 15-17; IV-2, 19
CrO3(r) IV—1, 15-18; IV-2, 20
СгОз(г) IV—1, 16, 18; IV—2, 21
Сг2О3(к, ж) IV—1, 9, 18-20; IV—2, 22, 23
Мо(к, ж) IV—1,121—24; IV—2, 24, 25, 534
Мо(г) IV—1, 24; IV—2, 26, 27
Мо+(г) IV—1, 24, 25; IV-2, 28, 29
МоО(г) IV—1, 25—27; IV—2, 30, 31
МоО2(к) IV—1, 21, 27, 28; IV-2, 32
МоО2(г) IV—1, 28—30; IV—2, 33
МоО3(к, ж) IV—1, 21, 30, 31; IV—2, 34
МоО3(г) IV—1, 29, 30-32; IV—2, 35
MoOg(r) IV—1, 29, 32, 33; IV—2, 36
Мо2Ов(г) IV—1, 29, 33, 34; IV-2, 37
Мо3О9(г) IV—1, 29, 34-36; IV—2, 38
Мо4ОХ2(г) IV—1, 29, 36-38; IV—2, 39
МоБО1В(г) IV—1, 29, 37, 38; IV—2, 40
W(k, ж) IV—1, 39-42; IV—2, 41, 42, 534
W(r) IV-1, 42; IV—2, 43, 44
W+(r) IV—1, 42, 43; IV—2, 45, 46
W0(r) IV—1, 43—46; IV—2, 47, 48
W02(k, ж) IV—1, 39, 46, 47; IV—2, 49
WO2(r) IV—1, 47-49; IV—2, 50
W03(k, ж) IV-1, 39, 49, 50; IV—2, 51 '
W03(r) IV—1, 47, 48, 50, 51; IV—2, 52
WOs(r) IV—1, 47, 51, 52; IV—2, 53
W2O6(r) IV—1, 47, 52, 53; IV—2, 54
W3O9(r) IV-1, 47, 53, 54; IV—2, 55
W4O12(r) IV—1, 47, 54, 55; IV—2, 56
WsO16(r) IV-1, 47, 56; IV-2, 57
V(k, ж) IV-1, 57-59, 6lf IV-2, 58, 534
V(r) IV—1, 59, 60; | IV—2, 59, 60
V+ (r) IV—1, 60; IV—2, 61, 62
VO(k, ж) IV—1, 58, 60-62; IV—2, 63
VO(r) IV—1, 62-64; IV—2, 64, 65
VO2(r) IV—1, 64, 65; IV—2, 66
V2O3(k, ж) IV—1, 58, 61, 65-67; IV—2, 67
V2O4(k, ж) IV—1, 58, 67, 68; IV—2, 68
V2O5(k, ж) IV—1, 58, 68, 69; IV—2, 69
V4O10(r)'IV—1, 64, 69, 70; IV-2, 70
Nb(K, ж) IV-1, 71—74; IV—2, 71, 72, 534
Nb(r) IV—1, 74; IV—2, 73, 74
Nb+(r) IV-1, 74, 75; IV-2, 75, 76
NbO(K, ж) IV—1, 71, 75, 76; IV—2, 77
NbO(r) IV—1, 76—78; IV—2, 78, 79
NbO2(K, ж) IV—1, 71, 78-80; IV—2, 80
NbO2(r) IV—1, 80, 81; IV-2, 81
Nb2O6(K. ж) IV-1, 71, 81, 82; IV-2, 82
Ta(K,^) IV—1, 83-85; IV-2, 83, 84, 534
Ta(r)'lV-l, 85, 86; IV-2, 85, 86
Ta+(r) IV—1, 86; IV—2, 87, 88
TaO(r) IV—1, 86-89; IV—2, 89, 90
TaO2(r) IV-1, 89-91; IV-2, 91
Та2Об(к, ж) IV-1, 83, 91, 92; IV-2, 92
Ti(K, ж) IV-1, 93-96; IV—2, 93, 94, 534
Ti(r) IV—1, 96, 97; IV—2, 95, 96
Ti+(r) IV—1, 97; IV—2, 97, 98
ТЮ(к, ж) IV—1, 94, 98, 99; IV—2, 99, 100
TiO(r) IV-1, 99-104; IV-2, 101, 102
TiO+(r) IV-1, 100, 102, 104, 105; IV—2, 103, 104
ТЮ2(к, ж) IV-1, 94, 105-107; IV-2, 105
TiOa(K, анатаз) IV—1, 94, 107, 108; IV—2, 106
TiO,(r) IV-1, 108, 109; IV—2, 107
Ti2O3(K, ж) IV-1, 94, 109-111; IV-2, 108, 109
Ti3O5(K, ж) IV-1, 94, 111, 112; IV-2, 110
Т14О?(к, ж) IV-1, 94, 112, 113; IV-2, 111
Zr(K, ж) IV—1, 114—117;- IV-2, 112, 113, 534
Zr(r) IV—1, 117; IV—2, 114, 115
Zr+(r) IV-1, 117, 118; IV-2, 116, 117
ZrO(r) IV-1, 101, 118-122; IV-2, 118, 119
ZrO+(r) IV-1, 119, 120, 122, 123; IV-2, 120, 121
ZrO2(K, ж) IV—1, 114, 123-125; IV—2, 122, 123
ZrO2(r) IV-1, 125, 126; IV-2, 124
Hf(K, ж) IV—1, 127—129; IV-2, 125, 126. 534
Hf(r) IV—1, 129, 130; IV-2, 127, 128
Hf+(r) IV—1, 130, 131; IV—2, 129, 130
HfO(r) IV-1, 101, 131-133; IV-2, 131, 132
HfO+(r) IV-1, 131—134; IV—2, 133, 134
НЮ2(к, ж) IV-1, 127, 134-136; IV-2, 135, 136
HfOa(r) IV—1, 136; IV-2, 137
Sc(k, ж) IV—1, 137—139; IV-2, 138, 534
Sc(r) IV—1, 139; IV—2, 139, 140
Sc+(r) IV—1, 139, 140; IV-2, 141, 142
ScO(r) IV—1, 140—142; IV—2, 143, 144
ScO+(r) IV-1, 140—143; IV-2, 145, 146
ScO2(r);iV-l, 143, 144; IV-2, 147
Sc2O(r) IV-1, 144, 145; IV-2, 148
Sc2O2(r) IV—1, 144, 145; IV—2, 149
ScaO8(K,^) iv—1, 137, 145, 146; IV-2, 150, 151
615
Формульный указатель веществ
Y(k, ж) IV—1, 147—149;$ IV—2, 152, 534
У (г) IV—1, 149; IV—2, 153, 154
Y+(r) IV—1, 149, 150; IV—2, 155, 156
УО(г) IV-1, 150-153;^ IV-2, 157, 158
УО+(г) IV—1, 150, 151, 153, 154; IV—2, 159,-160
УО2(г) IV—1, 154, 155; IV—2, 161
У2О(г) IV—1, 154, 155; IV—2, 162
У2О2(г) IV—1, 154—156;5» IV—2, 163
У2О3(к, ж) IV—1, 147, 156, 157; IV—2, 164, 165
La(K, ж) IV—1, 158—160; IV—2, 166, 167, 534
La(r) IV—1, 160; IV—2, 168, 169
La+(r) IV—1, 160, 161; IV-2, 170, 171
LaO(r) IV—1, 161—164; IV—2, 172, 173
LaO+(r) IV—1, 161, 163—165; IV—2, 174, 175
LaO2(r) IV—1, 165; IV—2, 176
La2O(r) IV—1, 165, 166; IV—2, 177
La2O2(r) IV—1, 165—167; IV—2, 178
La2O3(K, ж) IV—1, 158, 167, 168; IV-2, 179, 180
ТЬ(к, ж) IV—1, 169—171; IV—2, 181, 182, 534
Th(r) IV—1, 171; IV—2.-183, 184
Th+(r) IV—1, 171, 172; IV—2, 185, 186
ThO(r) IY-1, 172—175; IV—2, 187, 188
ThO+(r) IV—1, 173—176; IV—2, 189, 190
ТЬО2(к, ж) IV—1, 169,176—178; IV—2, 191, 192
ThO2(r) IV—1, 178, 179; IV—2, 193, 194
ThO2+(r) IV—1, 178—180; IV—2, 195, 196
ThO2-(r) IV—1, 178, 180; IV—2, 197, 198
U(k, ж) IV-1, 181-184; IV-2, 199, 200, 534
U(r) IV—1, 184Д185;! IV—2, 201,|202
U+(r) IV—1, 185, 186; I IV—2, 203, 204]
UO(r) IV—1, 186—189;" IV~2,s205, |206
UO+(r) IV—1, 187—190; IV—2, 207*, 208
UO2(k, k)!IV-1, 182, 190—192, 510, 511;
IV—2, 209, 210, 534
UO2(r) IV-1, 193-194; IV-2, 211, 212
UOJ(r) IV—1, 193—195; IV—2, 213, 214
UOj(r) IV—1, 193, 195; IV—2, 215, 216
UOs(k) IV—1, 182, 195—197, 228; IV—2, 217
UOs(r) IV—1, 193, 197, 198; IV—2, 218, 219
UOa(r) IV—1, 193, 198; IV—2, 220, 221
U8O8(k, ж) IV—1, 182, 199, 200; IV—2, 222
U4O,(k) IV—1, 182, 201, 202; IV—2, 223
UF(r) IV—1, 187, 188, 202, 203; | IV—2, 224, 225
UF+(r) IV—1, 187, 188, 203, 204;! IV—2, 226, 227
UF-(r) IV—1, 187, 188, 204, 205;* IV-2, 228, 229
UF2(r) IV—1, 205—207; i IV—2, 230, 231
UF^r) IV—1, 206—208; IV—2, 232, 233
UFi(r) IV—1, 206, 208; IV—2, 234, 235
UF8(k, ж) IV—1, 182, 209, 210; IV—2, 236
UF3(r) IV—1, 206, 210,5211; IV—2, 237, 238
UFJ(r) IV—1, 206, 211,* 212; IV—2, 239, 240
UFa(r) IV—1, 206, 212;iIV—2, 241, 242
UF<(k, ж) IV-1, 182, 212-215; IV-2, 243
UF4(r) IV—1, 206, 215—217; IV—2, 244, 245
UFf(r) IV—1, 206, 216, 217;? IV—2, 246, 247
UF4(r) IV—1, 206, 217; IV—2, 248, 249
UF6(k, ж) IV—1, 182, 217—219; IV—2, 250
UFB(r) IV—1, 206, 219, 220; IV—2, 251, 252
UFJ(r) IV—1, 206, 220, 221; IV—2, 253, 254
UF?(r) IV—1, 206, 221; IV-2, 255, 256
UFe(K, ж) IV—1, 182, 222, 223, 228, 511;
IV—2, 531, 534
UF,(r) IV—1, 206, 223, 224; IV—2, 257, 534
UFe(r) IV—1, 206, 224, 225; IV—2, 258
UOF(r) IV—1, 225, 226; IV—2, 259
UO2F(r) IV—1, 225, 226; IV—2, 260
UOF2(r) IV—1, 225—227; IV—2, 261
UO2F2(k) IV—1, 182, 227, 228; IV—2, 262
UO2F2(r) IV—1, 225, 228, 229; IV—2, 263
UOF3(r) IV—1, 225, 229, 230; IV—2, 264
UOF4(r) IV—1, 225, 230; IV—2, 265
Pu(k, ж) IV—1, 231—234; IV—2, 266, 267, 534
Pu(r) IV—1, 234, 235; IV—2, 268, 269
Pu+(r) IV—1, 235, 236; IV—2, 270, 271
PuO(r) IV—1, 236—238; IV—2, 272, 273
PuO+(r) IV—1, 236—239; IV—2, 274, 275
РиО2(к, ж) IV—1, 231, 239, 240, 511, 512;
IV—2, 276, 277, 534
PuO2(r) IV—1, 240, 241; IV—2, 278, 279
PuOf(r) IV—1, 240, 241; IV—2, 280, 281
РиОг(г) IV—1, 240—242; IV—2, 282, 283
Pu2O3(k, ж) IV—1, 231, 242, 243; IV-2, 284, 285
Ве(к, ж) III—1, 230—232, 422; III—2, 225, 395
¦Be(r) III—1, 232, 233, 422; III—2, 226, 227
Be+(r) III—1, 233, 234; III—2, 228, 229
ВеО(к, ж) HI—1, 231, 234, 235; III—2, 230, 231
BeO(r) III—1, 235—238; III—2, 232, 233
Be2O(r) III—1, 239; HI—2, 234
Be2O2(r) III—1, 239, 240; III—2, 235
Be3O3(r) III—1, 240, 241; HI—2, 236
Be4O4(r) III—1, 240—242; III—2, 237
BeH(r) III—1, 236, 237, 242, 243; III—2, 238
ВеН2(аморф.) Ill—1, 231, 243; Ш-2, 391
BeOH(r) III—1, 239, 243, 244; III—2, 239
Ве(ОНJ(к) III—1, 231, 244, 245; Hf—2, 391
Be(OHJ(r) III—1, 240, 245-247; HI—2, 240
BeF(r) III—1, 236, 237, 247, 248; HI-2, 241
BeF2(K, ж) III—1, 231, 248—250; III—2, 242
BeF2(r) HI—1, 239, 250, 251; III—2, 243
Be2F4(r) III—1, 240, 251, 252; III—2, 244
BeCl(r) III—1, 236, 237, 252, 253; III—2, 245
ВеС12(к, ж) III—1, 231, 253, 254; HI-2, 246
р-ВеС12(к) III—1, 231, 254, 255; III—2, 392
ВеС12(г) III—1, 239, 255; III—2, 247
Be2Cl4(r) III—1, 240, 255, 256; HI—2, 248
BeBr(r) III—1, 236, 237, 256, 257; III—2, 249
ВеВг2(к, ж) III—1, 231, 257, 258; III—2, 250
616
Формульный указатель веществ
ВеВг2(г) III—1, 239, 258, 259; III—2, 251
Bel(r) III—I, 236 237, 259, 260; Ш—2, 252
Ве12(к, ж) III—1, 231, 260, 261; III—2, 253
BeI2(r) III—1, 239, 261; III—2, 254
BeS(K) III—1, 231, 261, 262; III—2, 255
BeS(r) III—1, 236, 237, 262, 263; III—2, 256
BeSO4(K, ж) III—1, 231, 263, 264; III—2, 257
Be8N2(K, ж) III—1, 231, 264, 265; IH-2, 258
BeCO3(K) III—1, 231, 265; HI—2, 392
Mg(K, ж) III—1, 266—268, 422; III—2, 259, 395
Mg(r) III—i, 268, 269, 422; HI—2, 260, 261
Mg+(r) III—1, 268, 269; III—2, 262, 263
Mg2(r) III—1, 269-272; III—2, 392
MgO(K, ж) III—1, 267, 271, 273; HI—2, 264
MgO(r) III—1, 270, 272^276; IH-2, 265, 266
MgH(r) Ш—i, 270, 272, 275-277; IH-2, 267
MgH2(K, ж) HI—i, 267, 277, 278; III—2, 393
MgOH(r) III—i, 278, 279; III—2, 268
Mg(OHJ(K) III—1, 267, 279, 280; III—2, 393
Mg(OHJ(r) III—1, 279-281; IH-2, 269
MgF(r) III—1, 270, 272, 281, 282; HI—2, 270
MgF2(K, ж) III—1, 267, 283, 284; HI—2, 271
MgF2(r) III—1, 279, 284, 285; III—2, 272
MgCl(r) III—1, 270, 272J285, 286; III—2, 273
MgCl2(K, ж) III—1, 267, 286—288; III—2, 274
MgCl2(r) III—1, 279, 288, 289; III—2, 275
MgBr(r) III—1, 270, 272, 289, 290; III—2, 276
MgBr2(K, ж) III—1, 267, 290, 291; HI—2, 277
MgBr2(r) III—1, 279, 291; IH-2, 278
Mgl(r) III—1, 270, 272, 291, 292; HI—2, 279
MgI2(K, ж) HI—1, 267, 292, 293; HI—2, 280
Mgljs(r) III—1, 279, 293, 294; III—2, 281
MgS(K, ж) III—1, 267, 294, 295; III—2, 282
MgS(r) III—1, 270, 272, 295, 296; IH-2, 283
MgS04(K, ж) III—1, 267, 296, 297; III—2, 284
Mg3N2(K) III—1, 267, 297—299; III—2, 285
MgCO3(K, ж) HI—1, 267, 299, 300; III—2, 286
Са(к, ж) III—1, 301—303, 422; IH-2, 287, 395
Ca(r) III—1, 303, 304, 422; III—2, 288, 289
Ca+(r) HI—1, 304, 305; III—2, 290, 291
Ca2(r) III—1, 305-307; III—2, 393
СаО(к, ж) III—1, 302, 307, 308; III—2, 292
CaO(r) III—1, 305, 306, 308—311; III—2, 293, 294
CaO+(r) HI—1, 305, 306, 311; III—2, 295, 296
CaH(r) III—1, 305, 306, 312, 313; III—2, 297
СаН2(к, ж) III—1, 302, 313, 314; III—2, 298
CaOH(r) III—1, 314, 315; III—2, 299
CaOH+(r) III—1, 315, 316;? Ill—2, 300
Са(ОНJ(к, ж) III—1, 302, 316, 317; III—2, 301
Ca(OHJ(r) III—1, 315, 317, 318; Ш-2, 302
CaF(r) III—1, 318—321; III—2, 303
CaF+(r) III—i, 319-322; Ш-2, 304
CaF2(K, ж) III—i, 302, 322, 323; III—2, 305
CaF2(r) III—1, 315, 323, 324; ^ III—2, 306
CaCl(r) III—1, 319, 320, 324—326; III—2, 307
CaCl+(r) III—1, 319, 320Д326; Ш-2, 308
СаС12(к, ж) III—1, 302, 326-328; IV—1, 622;
III—2, '309
СаС12(г) III—1, 315, 328, 329; Ш-2, 310
CaBr(r) III—1, 319, 320, 329, 330; III-2, 311 1
СаВг2(к, ж) III—1, 302, 330, 331; III—2, 312
CaBr2(r) III—1, 315, 331, 332; III—2, 313
Cal(r) III—1,' 319, 320, 332, 333; 1 III—2, 314
Са12(к, ж) III—1, 302, 333, 334; III—2, 315
CaI2(r) III—1, 315, 334; III—2, 316
CaS(K, ж) III—1, 302, 334, 335; III—2, 317
CaS(r) III—1, 305, 306, 336; III—2, 318
CaSO4(K, ж) Ш—1, 302, 336-338; III—2, 319
СаСО3(к, ж) III—1, 302, 338-340; 111-2,1320
Sr(K, ж) III—1, 341—344, 422; III—2, 321, 395
Sr(r) III—1, 344, 345; 422; HI—2, 322, 323 '
Sr+(r)jIH-l, 344, 345; III-2, 324, 325
Sr2(r) JII—1, 345—347; HI—2, 394
SrO(K, ж) III—1, 342, 347, 348; III—2,1326
SrO(r) III—1, 346—351; HI—2, 327, 328
SrO+(r) III—1, 346, 347, 351; III—2, 329, 330j.
SrH(r) III—1, 346, 347, 351—353; Ш-2, 331 j"
SrH2(K, ж) III—1, 342, 353; IH-2, 332
SrOH(r) III—1, 353, 354; III—2, 333
SrOH+(r) HI-1, 354, 355; IH-2, 334
Sr(OHJ(K, ж) III—1, 342, 355, 356; III—2, 335
Sr(OHJ(r) HI—1, 354, 356, 357; III—2, 336
SrF(r) III—1, 357—360; III—2, 337
SrF+(r) III—1, 358, 359—361; HI—2, 338
SrF2(K, ж) III—1, 342, 361, 362; 111—2,1339
SrF2(r) Ш—I, 354, 362, 363; III—2, 340
SrCl(r) III—i, 358, 359, 363, 364; III—2, 341
SrCl+(r) III—1, 358, 359, 364, 365; III—2, 342
SrCl2(K, ж) III—1, 342, 365-367; III—2, 343
SrCl2(r) III—1, 354, 367, 368; III—2, 344
SrBr(r) III—1, 358, 359, 368, 369; III—2, 345
SrBr2(K, ж) III—1, 342, 369, 370; III—2, 346
SrBr2(r) III—1, 354, 370, 371; III—2, 347
Srl(r) III—1, 358, 359, 371, 372; IH-2, 348
SrI2(K, ж) III—1, 342, 372; III—2, 349
SrI2(r) HI—1, 354, 372, 373; III—2, 350
SrS(K, ж) III—1, 342, 373, 374; III—2, 351
SrS(r) III—1, 346, 347, 374—376; III—2, 352
SrSO4(K, ж) III—1, 342, 376, 377; III—2, 353
SrCO3(K, ж) III—1, 342, 377, 378; III—2, 354
Ва(к, ж) III—1, 379-382, 422; III—2, 355, 395
Ba(r) HI—1, 381—383, 422; HI—2, 356, 357
Ba+(r) III—1, 382—385; III—2, 358, 359
Ba2(r) III—1, 384-386; III—2, 394
ВаО(к, ж) HI—1, 380, 386, 387; HI-2, 360
BaO(r) III—1, 384, 385, 387—390; III—2, 361, 362
BaO+(r) III—1, 384, 385, 390; Ш-2, 363, 364
BaH(r) III—1, 384, 385, 390—392; HI—2, 365
617
Формульный указатель веществ
ВаН2(к, ж) III—1, 380, 392; III—2, 366
ВаОН(г) III—I, 393, 394; III—2, 367
ВаОН+(г) III—1, 393—395; III—2, 368
Ва(ОНJ(к, ж) III—1, 380, 395; III—2, 369
Ва(ОНJ(г) III—1, 393, 395—397; III—2, 370
BaF(r) III—I, 397—400; III—2, 371
BaF+(r) III—1, 398-401; III—2, 372
BaF2(K, ж) HI—1, 380, 401, 402; III-2, 373
BaF2(r) III—1, 393, 402, 403; III—2, 374
BaCl(r) III—1, 398, 399, 403—405; III—2, 375
BaCl+(r) III—1, 398, 399, 404, 405; III—2, 376
ВаС12(к, ж) III—1, 380, 406, 407; III—2, 377
ВаС12(г) HI—1, 393, 407; III—2, 378
BaBr(r) III—1, 398, 399, 407-409; III—2, 379
ВаВг2(к, ж) III—1, 380, 408—410; III—2, 380
BaBr2(r) HI—I, 393, 410; III—2, 381
Bal(r) HI—I, 398, 399, 410—412; HI—2, 382
Ва12(к, ж) III—1, 380, 412; III—2, 383
BaI2(r) III—1, 393, 412, 413; HI—2, 384
BaS(K, ж) III—1, 380, 413, 414; III—2, 385
BaS(r) HI—1, 384, 385, 414—416; III—2, 386
BaSO4(K, ж) HI—1, 380, 416, 417; HI—2, 387
ВаСО3(к, ж) HI—1, 380, 417, 418; III—2, 388
Ы(к, ж) IV—1, 244—247; IV—2, 286, 534
Li(r) IV—1, 247, 512—516; IV-2, 287, 288
Li+(r) IV—1, 247, 248; IV—2, 289, 290
Li2(r) IV—1, 248—250, 252; IV—2, 291
LiJ(r) IV—1, 249—252; IV —2, 292
Щ(т) IV—1, 251, 256, 260; IV—2, 293
LiO(r) IV—1, 250, 252, 256, 257; IV—2, 294
Ы20(к, ж) IV—1, 245, 257—259; IV—2, 295
Li2O(r) IV—1, 259, 260; IV—2, 296
Li2O+(r) IV—1, 260, 261; IV—2, 297
Ы2О2(к) IV—1, 245, 261, 262; IV—2, 298
Li2O2(r) IV—1, 260, 262, 263; IV—2, 299
LiH(K, ж) IV—1, 245, 263—265; IV—2, 300 .
LiH(r) IV—1, 253, 265—267; IV—2, 301
LiD(K, ж) IV—1, 245, 267, 268; IV—2, 302
LiD(r) IV—1, 254, 268, 269; IV—2, 303
ЫТ(к, ж) IV—1, 245, 269, 270; IV-2, 304
LiT(r) IV—1, 255, 270; IV—2, 305
LiOH(K, ж) IV—1, 245, 270, 271; IV-2, 306
LiOH(r) IV—1, 260, 272, 273; IV—2, 307
Li2O2H2(r) IV—1, 260, 273; IV—2, 308
LiF(K, ж) IV—1, 245, 273-275; IV-2, 309
LiF(r) IV—1, 250, 252, 275, 276; IV—2, 310
Li2F2(r) IV—1, 276—278; IV—2, 311
Li3F3(r) IV—1, 276, 278, 279; IV-2, 312
ЫС1(к, ж) IV—1, 245, 279-281; IV—2, 313
LiCl(r) IV—1, 250, 252, 281, 282; IV—2, 314
Li2Cl2(r) IV—1, 276, 282, 283; IV—2, 315
Li3Cl3(r) IV—1, 276, 283, 284; IV—2, 316
LiBr(K, ж) IV-1, 245, 284, 285; IV—2, 317
LiBr(r) IV—1, 250, 252, 285, 286; IV—2, 318
LiaBra(r) IV—1, 276, 286, 287; IV—2, 319
Li3Br3(r) IV—1Д276, 287; IV—2, 320
LiI(K, ж) IV—1, 245, 287—289; IV—2, 321
Lil(r) IV—I, 250, 252, 289, 290; IV—2, 322
Li2I2(r) IV—1, 276, 290; IV-2, 323
Li3I3(r) IV—1, 276, 290, 291; IV-2, 324
Li2SO4(K, ж) IV—1, 245, 291, 292; IV—2, 325
LiNO2(K, ж) IV—1, 245, 292—294; IV—2, 326
LiNO2(r) IV—1, 260, 294; IV—2, 327
LiNO3(K, ж) IV—1, 245, 294—296; IV-2, 328
LiNOg(r) IV—1, 260, 296, 297; IV—2, 329
1Л2СО3(к, ж) IV—1, 245, 297, 298; IV-2, 330
LiBO2(K, ж) IV—1, 245, 298, 299; IV—2, 331
LiBO2(r) IV—1, 260, 299, 300; IV-2, 332
LiAlF4(r) IV—1, 260, 300, 301; IV—2, 333
Li3AlF6(K, ж) IV—1, 245, 301—303; IV—2, 334
?Li(K, ж) IV—1, 245, 303; IV—2, 335
SLi(r) IV—1, 247, 304; IV—2, 336, 337
SLir) IV—1, 304; IV—2, 338, 339
ШН(к, »)IIV—1, 245, 304, 305; IV—2, 340
fiLiH(r) IV-1, 250, 251, 253, 305; IV-2, 341
!LiD(K, ж) IV—1, 245, 305, 306; IV-2, 342
SLiD(r) IV—1, 250, 251, 254, 306; IV—2, 343
eLiT(r) IV—1, 255, 306; IV—2, 344
*Li(r) IV—1, 247, 306, 307; IV—2, 345, 346
*Li+(r) IV—1, 307; IV—2, 347, 348
?LiH(r) IV—1, 250, 251, 253, 307; IV-2, 349
*LiD(r) IV—1, 250, 251, 254, 307, 308; IV—2, 350
?LiT(r) IV—1, 255, 308; IV—2, 351
Na(K, ж) ".IV—1, 309—313; IV—2, 352, 534
Na(r) IV—1, 313, 512-516; IV—2, 353, 354, 534
Na+(r) IV—1, 313, 314; IV—2, 355, 356
Na2(r) IV—1, 314—318; IV-2, 357
NaO(r) IV—1, 315, 316, 318, 319; IV—2, 358
NaO2(K, ж) IV—1, 310, 319, 320; IV—2, 359]
Na2O(K, ж) IV—1, 310, 320, 321; IV—2, 360
Na2O(r) IV—1, 321, 322; IV—2, 361
Na2O+(r) IV—1, 321, 322; IV—2, 362]
Na2O2(u, ж) IV—1, 310, 323; IV—2, 363
Na2O2(r) IV—1, 321, 324; IV—2, 364
NaH(K, ж) IV—1, 310, 324—326; IV—2, 365J
NaH(r) IV—1, 315, 317, 326, 327; IV-2, 366
NaOH(K, ж) IV—1, 310, 327, 328; IV—2, 367
NaOH(r) IV—1, 321, 328, 329; IV—2, 368
Na2O2H2(r) IV—1, 321, 329, 330; IV-2, 369
NaF(K, ж) IV—1, 310, 311, 330—332; IV—2, 370
NaF(r) IV—1, 315, 317, 332, 333; IV—2, 371
Na2F2(r) IV-1, 333, 334; IV—2, 372
Na3F3(r) IV—1, 333—335; IV-2, 373
NaCl(K, ж) IV—1, 310, 311, 335—337; IV—2, 374,
534
NaCl(r) IV—1, 315, 317, 337, 338; IV—2, 375
Na2Gl2(r) IV—1, 333, 338, 339; IV-2, 376
Na3Gl3(r) IV—1, 333, 339, 340; IV-2, 377
NaBr(K, ж) IV—1, 310, 340—342; IV—2, 378
NaBr(r) IV—1, 315, 317, 342; IV—2, 379
618
Формульный указатель веществ
Na2Br2(r) IV—1, 333, 342, 343; IV-2, 380
NaI(K, ж) IV—1, 310, 343—345; IV—2, 381
Nal(r) IV—1, 315, 317, 345, 346; IV-2, 382
Na2I2(r) IV—1, 333,|346; IV—2, 383
Na-jSO^K, ж) IV—1, 310, 311, 346—349; IV—2, 384
Na2SO4(K, ромб.) IV—1, 310, 349; IV—2, 533
Na2SO4(r) IV—1, 321, 349, 350; IV-2, 385
NaNO2(K, ж) IV—1, 310, 311, 350—352; IV-2, 386
NaNO2(r) IV—1, 321, 352, 353; IV—2, 387
NaNO3(K, ж) IV—1, 310, 311, 353—355; IV—2, 388
NaNO3(r) IV—1, 321, 355, 356; IV—2, 389
Na2CO3(K, ж) IV—i, 310, 356, 357; IV—2, 39C
NaBO2(K, ж) IV—1, 310, 311, 357, 358; IV—2, 391
NaBO2(r) IV—1, 321, 358, 359; IV—2, 392
NaAlF4(r) IV—1, 333, 359, 360; IV—2, 393
Na3AlFe(K, ж) IV—1, 311, 361, 362; IV—2, 394
Na5Al3FM(K, ж) IV—1, 311, 362, 363; IV—2, 395
NaLi(r) IV—1, 315, 317, 363, 364; IV—2, 396
К(к, ж) IV—1, 365-368; IV-2, 397, 534
K(r) IV—1, 368, 369, 512—516; IV—2, 398, 399, 534
K+(r) IV—1, 369; IV—2, 400, 401
K2(r) IV—1, 370—372, 374; IV-2, 402
KJ(r) IV—1, 371, 372, 374, 375; IV—2, 403
KO(r) IV—1, 371, 372, 375; IV-2, 404
КО2(к, ж) IV—1, 366, 375—377; IV—2, 405
К2О(к, ж) IV—1, 366, 377; IV—2, 406
K2O(r) IV—1, 377—379; IV—2, 407
K2O+(r) IV—1, 378, 379; IV-2, 408
К2О2(к, ж) IV—1, 366, 379—381; IV—2, 409 .
K2O2(r) IV—1, 378, 381; IV—2, 410
КН(к, ж) IV—1, 366, 381—383; IV—2, 411
КН(г) IV—1, 371, 373, 383, 384; IV—2, 412
КОН(к, ж) IV—1, 366, 384, 385; IV—2, 413
КОН(г) IV—1, 378, 385, 386; IV—2, 414
К2О2Н2(г) IV—1, 378, 386, 387; IV—2, 415
KF(k, ж) IV—1, 366, 387—389; IV—2, 416
KF(r) IV—1, 371, 373, 389; IV—2, 417
K3F2(r) IV—1, 378, 389, 390; IV—2, 418
КС1(к, ж) IV—1, 366, 367, 390-393; IV—2, 419,
534
КС1(г) IV—1, 371, 373, 393; IV—2, 420
К2С1а(г) IV—1, 378, 393, 394; IV—2, 421 '
КВг(к, ж) IV—1, 366, 394—396; IV—2, 422
КВг(г) IV—1, 371, 373, 396, 397; IV—2, 423
К2Вг2(г) IV—1, 378, 397, 398; IV—2, 424«
К1(к, ж) IV-1, 366, 398-400; IV-2, 425?
К1(г) IV—1, 371, 373, 400, 401; IV—2, 426
Й^12(г) IV—1, 378, 401; IV—2, 427
K2SO4(k, ж) IV—1, 366, 402—404; IV—2, 4282
K2SO4(r) IV—1, 378, 404; IV—2, 429
KNO2(k, ж) IV—1, 366, 367, 404—406; IV—2, 430 j
KNO2(r) IV—1, 378, 406, 407; IV—2, 431
KNO3(k, ж) IV—1, 366, 367, 407—409; IV—2, 432
KNO3(r) IV—1, 378, 409; IV—2, 433
К2СО8(к, ж) IV—1, 366, 410, 411; IV-2, 434
К2СО3(г) IV—1, 378, 411, 412; IV—2, 435
K2Si08(K, ж) IV—1, 366, 412, 413; IV—2, 436
K2Si205(K, ж) IV—1, 366, 413, 414; IV—2, 437
КВО2(к, ж) IV—1, 366, 414, 415; IV—2, 438
КВО2(г) IV—1, 378, 415, 416; IV—2, 439
КА1О2(к, ж) IV—1, 366, 367, 416; IV—2, 440
KAlF4(r) IV—1, 378, 417, 418; IV—2, 441
K3A1F6(k, ж) IV—1, 367, 418; IV—2, 442
KUF6(r) IV—1, 378, 418, 419; IV—2, 443
KLi(r) IV—1, 371, 373, 419, 420; IV—2, 444
KNa(r) IV—1, 371, 373, 420, 421; IV—2, 445
Rb(K, ж) IV—1, 422—425; IV-2, 446, 534
Rb(r) IV—1, 425, 512—516; IV—2, 447, 448, 534
Rb+(r) IV—1, 425, 426; IV—2, 449, 450
Rb2(r) IV—1, 426—429; IV—2, 451
RbO(r) IV—1, 427—430; IV—2, 452
RbO2(K, ж) IV—1, 423, 430; IV—2, 453
Rb2O(K, ж) IV—1, 423, 431; IV—2, 454
Rb2O(r) IV—1, 431, 432; IV-2, 455
Rb2O2(K, ж) IV—1, 423, 33, 434; IV-2, 456
Rb2O2(r) IV—1, 432, 434; IV-2, 457
RbH(K, ж) IV—1, 423, 434, 435; IV—2, 458
RbH(r) IV—1, 427, 428, 435, 436; IV—2, 459
RbOH(K, ж) IV-1, 423, 436, 437; IV—2, 460
RbOH(r) IV—1, 432, 437, 438; IV—2, 461
Rb2O2H2(r) IV—1, 432, 438; IV—2, 462
RbF(K, ж) IV—1, 423, 438—440; IV—2, 463
RbF(r) IV—1, 427, 428, 440, 441; IV-2, 464
Rb2F2(r) IV—1, 432, 441; IV-2, 465
RbCl(K, ж) IV—1, 423, 441—443; IV—2, 466
RbCl(r) IV—1, 427, 428, 443, 444; IV—2, 467
Rb2Cl2(r) IV—1, 432, 444, 445; IV—2, 468
RbBr(K, ж) IV—1, 423, 445, 446; IV—2, 469
RbBr(r) IV—1, 427, 428, 446, 447; IV-2, 470
Rb2Br2(r) IV—1, 432, 447, 448; IV—2, 471
RbI(K, ж) IV—1, 423, 448, 449; IV-2, 472
Rbl(r) IV—1, 427, 428, 449, 450; IV—2, 473
Rb2I2(r) IV—1, 432, 450, 451; IV—2, 474
Rb2SO4(K, ж) IV—1, 423, 451, 452; IV—2, 475
Rb2SO4(r) IV-1, 432, 452, 453; IV-2, 476
RbNO2(K, ж) IV-1, 423, 453, 454; IV—2, 477
RbNO2(r) IV—1, 432, 454, 455; IV—2, 478
RbNO3(K, ж) IV—1, 423, 455, 456; IV—2, 479
RbNO3(r) IV—1, 432, 456, 457; IV—2, 480
Rb2CO3(K, ж) IV—1, 423, 457, 458; IV-2, 481
RbBO2(K, ж) IV—1, 423, 458, 459; IV—2, 482
RbBO2(r) IV—1, 432, 459, 460; IV—2, 483
RbLi(r) IV—1, 427, 428, 460, 461; IV—2, 484
RbNa(r) IV—1, 427, 428, 461, 462; IV—2, 485
RbK(r) IV—1, 427, 428, 462; IV-2, 486
Cs(k, ж) IV—1, 463—466; IV—2, 487, 534
Gs(r) IV—1, 466, 467, 512—516; IV—2, 488, 489, 534
Gs+(r) IV—1, 466, 467; IV—2, 490, 491 "
Cs2(r) IV—1, 467—471; IV—2, 492
619
Формульный указатель веществ
CsO(r) IV—1, 468, 469, 471; IV—2, 493
CsO2(k, ж) IV—1, 464, 472; IV—2, 494
Cs2O(k, ж) IV—1, 464, 472, 473; IV—2, 495
Cs2O(r) IV—1, 473, 474; IV—2, 496
Cs2O+(r) IV—1, 473—475; IV—2, 497
Cs2O2(k, ж) IV-1, 464, 475, 476; IV-2, 498
Cs2O2(r) IV—1, 473, 476; IV—2, 499
CsH(k, ж) IV—1, 464, 476, 477; IV—2, 500
CsH(r) IV—1, 468, 469, 477, 478; IV—2, 501
CsOH(k, ж) IV—1, 464, 478, 479; IV—2, 502
CsOH(r) IV—1, 473, 479, 480; IV-2, 503
Cs2O2H2(r) IV—1, 473, 480, 481; IV-2, 504
CsF(k, ж) IV—1, 464, 481—483; IV—2, 505
CsF(r) IV—1, 468, 470, 483, 484; IV—2, 506
Cs2F2(r) IV—1, 473, 484; IV-2, 507
CsC1(k, ж) IV—1, 464, 485—487; IV—2, 508
CsCl(r) IV—1, 468, 470, 487; IV-2, 509
Cs2Cl2(r) IV—1, 473, 487, 488; IV—2, 510
CsBr(K, ж) IV—1, 464, 488—490; IV—2, 511
CsBr(r) IV—1, 468, 470, 490, 491; IV—2, 512
Cs2Br2(r) IV—1, 473, 491, 492; IV—2, 513
CsI(k, ж) IV—1, 464, 492-494; IV—2, 514
Csl(r) IV—1, 468, 470, 494; IV-2, 515
Cs2I2(r) IV—1, 473, 495; IV—2, 516
Cs2SO4(k, ж) IV—1, 464, 496, 497; IV-2, 517
Cs2SO4(r) IV—1, 473, 497, 498; IV-2, 518
CsNO8(k, ж) IV—1, 464, 498, 499; IV—2, 519
CsNO2(r) IV—1, 473, 499, 500; IV—2, 520
CsNO3(k, ж) IV—1, 464, 500, 501; IV—2, 521
CsNO3(r) IV—1, 473, 501, 502; IV-2, 522
Cs2CO3(k, ж) IV—1, 464, 502, 503; IV-2, 523
Cs2CO3(r) IV—1, 473, 503, 504; IV-2, 524
CsBO2(k, ж) IV—1, 464, 504, 505; IV-2, 525
CsBO2(r) IV—1, 473, 505; IV—2, 526
CsLi(r) IV—1, 468, 470, 505, 506; IV—2, 527
CsNa(r) IV—1, 468, 470, 506, 507; IV—2, 528
CsK(r) IV—1, 468, 470, 507, 508; IV—2'. 529
CsRb(r) IV—1, 468, 470, 508; IV-2, 530 j
Страница
12
16
34
34
37
37
37
38
41
41
55
56
56
56
57
60
61
61
70
П f.
/4
74
75
75
Строка
22 св.
20 св.
5 св.
22 св.
Табл. 1.2,
заголовок
3 св.
9 св.
23 и 24 св.
5 св.
22 гя
21 сн.
Табл. 1.7
5 стб.
12 сн.
И сн.
9 сн.
2 св.
И св.
7 св.
4 сн.
11 сн.
8 св.
18 св.
19 сн.
14—17 сн.
СПИСОК ИСПРАВЛЕНИЙ
Напечатано
Том I, кн.1
[437а]
Np
дТ2
807
в Дж-К^-моль
(Т/2)R
Bv BД + 1)
У (У + 4)
B2/+1)
Л •*
1
— а?
а1
и„ехр
«5
+0,36380
+ In (
Щ
^2
2k
d[H°{T) — H°{T0)
dT
а + F/2)Г2 +
+ сГ-1 + d
B.23)—B.27)
f=D — 2gTlt
« = G-2/24-
— ЗгГ2
._ -1'
6 = В — 2e7\ —
— 3/Г| — 4#Г|
Р .
+ 4—ЗеГ|—
— 4/Г? — 5gT\
а == — ю \ 1/ ~~
, ^у2 2еТ34
+ 3/rj + igl !
Должно быт»
[473a]
Cm
J*2 вн Л_
-./
""" дТ
870
^о g Дж • К~5 • г~^
и i° — в кДж • г
(Т/2)(Д + 1)
В„ BJ? + 3)
У (У-4)
2B7 + 1)
г А
а '
ю«
п 2 (BeDe)'l*
1
— ~aF
dnu,,exp
dnu\
—0,36380
,
+ -~-ln (
Щ
2-n.k
12
&[H °{T) — H° G1,)]
dT
af+ (Ь/2)Г2 +
_j_ cf-i + g.
B.23), B.25)-B.27)
я = Л — 2ВГ,. +
+ CT\ + C^Tj),
Ь = 2 B? — 2CjTj +
+ DTf),
c ^y2
d = 3(C — 2DT1 +
+ ?,Jj),
- e = i(D ~2ETi),
f = bE,
— C°p {Ti) Ti
Страница
79
145
145
146
57
182
183
199
199
199
199
227
232
242
244
247
248
256
259
259
270
275
312
325
348
391
396
451
К ТОМАМ I—III
Строка
25 СН.
8 СВ.
5 сн.
18 св.
Табл. 7.8
4 стб.
Табл. 9.2
5 стб.
8 стб.
3 св.
6 св.
8 св.
11 сн.
18 св.
Табл. 10.2
5 стб.
6 стб.
7 стб.
8 стб.
17 сн.
17 сн.
Табл. 12.3
5 стб.
8 сн.
примечание
Табл. 12.4
6 стб.
7 стб.
9 стб. .
8 сн.
Табл. 12.6
17 стб.
14 сн.
10 св.
Табл. 12.10
12 стб.
21 сн.
12 сн.
Табл. 13.18
3 стб.
16 стб.
16 стб.
17 стб., 2 сн
17 стб., 1 сн
18 стб., 2сн
18 стб., 1 сн
Табл. 13.22
7 стб.
2 сн.
Табл. Ш.2
5 стб.
Табл. П2.4
1 стб., 2 ев
Прав. стб.
Напечатано
31 п
полиморфных
от 100
F
322
126,5802
298,15
298,15
295,59
—295,59
298
298,15
—133,0993
423,24
—5,4481
298,15
Хе2
12
11,5
so-
740
780
—1,15
35
296
кДж -моль
4,80 -102
100
296,81
2,770 -103
838 268,0
127
0 D38)
1,16-10*
1,5-10*
1
1
2
2
1470,0
В работе
10,129 385
0
Должно быть
31
полимерных
, от 298,15
F"
3322
126,971
310
310
—295,60
—295,60
320
320
—6,976
154,055
—1,3351
320
XeF
1,2
8,1
SO
74
78
—0,81
50
267
Дж -К -моль
4,08 -102
298,15
—296,81
2,670 -103
898 268,0
138
0 D70)
1,38-10*
1,18-10*
2
2 '
1
1
—1470,0
2 В работе * ЗЕ
10,012 938 5 Ц: §
—0
1108а. CoxonJ.A.,
Wickramaaratchi
М. A. — «J. Mol.
Spectrosc.», i 1977,
68, 372.
621
Окончание
а
в
g.
So
О з-
7
34
53
55
58
66
69
183
222
228
230
232
236
237
251
253
258
259
273
289
294
Строка
24 сн.
Табл. 15.13
4 стб.
Табл. 15.24
6 стб.
14 св.
Табл. 15.28
2 стб.
3 стб.
6 св.
15 и 16 св.
18 св.
19 св.
22 св.
1 св.
1 сн.
20 сн.
18 сн.
Табл. 16.17
3 стб.
4 стб.
5 стб.
6 стб.
7 стб.
Табл. 16.18
1 стб.
16 св.
24 св.
7 св.
9 св.
26 св.
7 сн.
9 св.
15 сн.
Табл. 17.13
4 стб.
Табл. 18.1
3 стб.
Напечатано
Том II, кн. 1
Фирсовой
325 B)
859
2383,786
462+15
52±15
4673а
1088
Должно быть
Фирсановой
309,3 B)
850
2384,172
489+15
78±15
4673
1775
Лисом и Джерри Джерри и др.
[1088]
1088
Лизом
452а
6,37
в возбужденных
4303
22 939,35
23 010,4
20 937,6
21 204,9
29 886,01
23 920
20 289,7
42 694,2
43 386
34 642,45
34,192,3
33 572,7
32 380,3
49 402,1
36 003,2
33 160
39 520,41
35 617,85
34 000
42 800
39 400
1977
16.4
16.12
100
Бредохля
6000
802,005
. . . <з2л3я3
GeS, г
74,8
17,0
[1775]
1775
Лисом
425а
276,37
и возбужденных
4115
22 858,4
22 906,8
20 728,6
20 879,9
29 805,06
23 710,5
19 964,4
42 655,65
43 318,5
34 561,5
34 007
33 363,2
32 055,3
49 363,56
35 939,3
32 990
39 439,46
35 529,3
42 900
39 460
1978
16.12
16.13
298,15
Бредохля и др.
5800
802,884
... О2 Я3 711 "
GeS, к
—74,8
17,33
ipanu-
Н
295
308
321
328
336
338
346
365
369
375
391
404
404
423
435
9
121
125
125
125
129
129
196
326
328
380
424
455
Строка
4 стб.
5 стб.
6 стб.
7 стб.
14 сн.
18 св.
Табл. 19.1
8 стб.
9 стб.
13 св.
5 сн.
28 сн.
2 св.
7 сн.
Лев. стб.,
2 сн.
Прав, стб.,
16 и 20 сн.
15 и 19 сн.
17 сн.
Прав, стб.,
3 св.
Лев. стб.,
38 св.
Прав, стб.,
11 сн.
Прав, стб.,
22 св.
Прав, стб.,
14 сн.
Табл. 20.1
8 стб.
17 сн.
19 сн.
17 сн.
8 сн.
5 св.
11 св.
колонтитул
4 сн.
Табл. 27.16
2 стб.
Табл. 29.1
2 сн.
примечание
Прав, стб.,
5 св.
Прав, стб.,
18 сн.
Напечатано
Должно выть
134,0 134,1
82,0 178,05
71,25 62,005
36,06 53,817
303,308 301,308
100 298,15
0,533 0,553
—1,134а —1,134
6537,505б 6537,505а
—2923,933 —2923,933е
2500 3000
2000 3200
19.2 19.4
100 298,15
2500 3000
SnCl2 SnCl4
1683 1083
Сычев В. М. Сычев В. В.
Цымарны/й В. Ф. Цымарный В. А.
1979 1981
Domande Domange
282 2821
А51 &
1972 1974
4122 4422
Том III, кн. 1
31,38 —31,38
большими меньшими
1289,896 1366,896
—138 —215
—808 —885
_1780 —1857
—1954 —2031
In2O3 In2O
7—30 7—300
1142—1281 1111—1281
2,083 -Ю-8 —2,083 -Ю"»
хим. наук физ.-мат. наук
1929 1925
622
ТЕРМОДИНАМИЧЕСКИЕ СВОЙСТВА ИНДИВИДУАЛЬНЫХ ВЕЩЕСТВ
Том IV. Книга 1
Утверждено к печати Институтом высоких температур Академии наук СССР
Редактор издательства В. П. Сироткина. Художник В. Г, Виноградов
Художественный редактор Т. П. Поленова. Технический редактор Э. Л. Кунина
Корректоры Н. Б. Габасова, Л. В. Письман
ИБ № 27023
Сдано в набор 09.07.82. Подписано к печати 23.12.82. Т-22818. Формат 84Х1081/1в. Бумага типографская № 1.
Гарнитура обыкновенная. Печать высокая. Усл. печ. л. 65,52. Усл.-кр. отт. 67,2. Уч.-изд. л. 76,2. Тираж 7100 экз.
Тип. зак. 1590. Цена IV т. (кн. 1) 6 руб.
Издательство «Наука». 117864, ГСП-7, Москва, В-485, Профсоюзная ул., 90
Ордена Трудового Красного Знамени Первая типография издательства «Наука»
199034, Ленинград, В-34, 9-я линия, 12
Банк данных
ИВТАНТЕРМО
В Институте высоких температур АН СССР функционирует автоматизированная
система данных по термодинамическим свойствам веществ, ИВТАНТЕРМО,
предназначенная для обеспечения расчетов высокотемпературных процессов.
Главная особенность ИВТАНТЕРМО, отличающая его от существующих
зарубежных и отечественных банков данных, состоит в том, что все накапливаемые в нем
термодинамические величины не являются компиляцией данных, рекомендованных
в различных справочниках, а генерируются с помощью комплекса специальных
программ на основании постоянных, отбираемых в результате критического анализа
и обработки всей первичной литературы. Этот экспертный анализ и обработка,
а также расчеты таблиц термодинамических свойств проводятся группой
высококвалифицированных специалистов, создавших под руководством академика
В. П. Г л ушко фундаментальные справочные издания АН СССР «Термодинамические
свойства индивидуальных веществ» и «Термические константы веществ».
Банк данных ИВТАНТЕРМО, состоящий из ряда информационных баз данных
и комплекса расчетных и сервисных программ, работает в диалоговом режиме и
выдает потребителям следующую информацию!
Каталог веществ, представленных т ИВТАНТЕРМО на день запроса;
Полную таблицу термодинамических свойств выбранного вещества по формату
справочника «Термодинамические свойства индивидуальных веществ»;
Выборку из полной таблицы, например: сведения о свойствах в заданном
интервале температур; уравнения, аппроксимирующие термодинамические функции
в'широком интервале температур вместе с термохимическими величинами и
характеристикой точности этих данных;
Значения констант равновесия любой реакции с участием веществ,
представленных в ИВТАНТЕРМО.
Для всех вариантов запросов обеспечивается выдача информации на различных
носителях (экран дисплея, АЦПУ, магнитная лента, магнитная кассета, перфолента).
Выдача на магнитную ленту'ироизводится в формате, читаемом на ЭВМ|ЕС.
Предполагается открыть доступ к ИВТАНТЕРМО по телефонным линиям связи.
ВрШТАНТЕРМО'на 1 ноября 1982 г. содержатся данные для всех соединений,
рассмотренных в четырех томах справочника «Термодинамические свойства
индивидуальных веществ», причем ряд таблиц уточнен на основании новейших исходных
данных. К концу 1983 г. в ИВТАНТЕРМО будут рассчитаны термодинамические
свойства соединений железа, кобальта, никеля, меди и селена, а также галоидных
соединений "титана, циркония, гафния, ванадия, ниобия и тантала.
При разработке программы развития ИВТАНТЕРМО, а также необходимых
для этого теоретических и экспериментальных исследований, будут учитываться
запросы заинтересованных организаций.
Запросы следует направлять по адресу:
127412, MIOC'KIBA, И-412,
Коровинское шоссе, ИВТАН,
Отдел химической
термодинамики.