


















































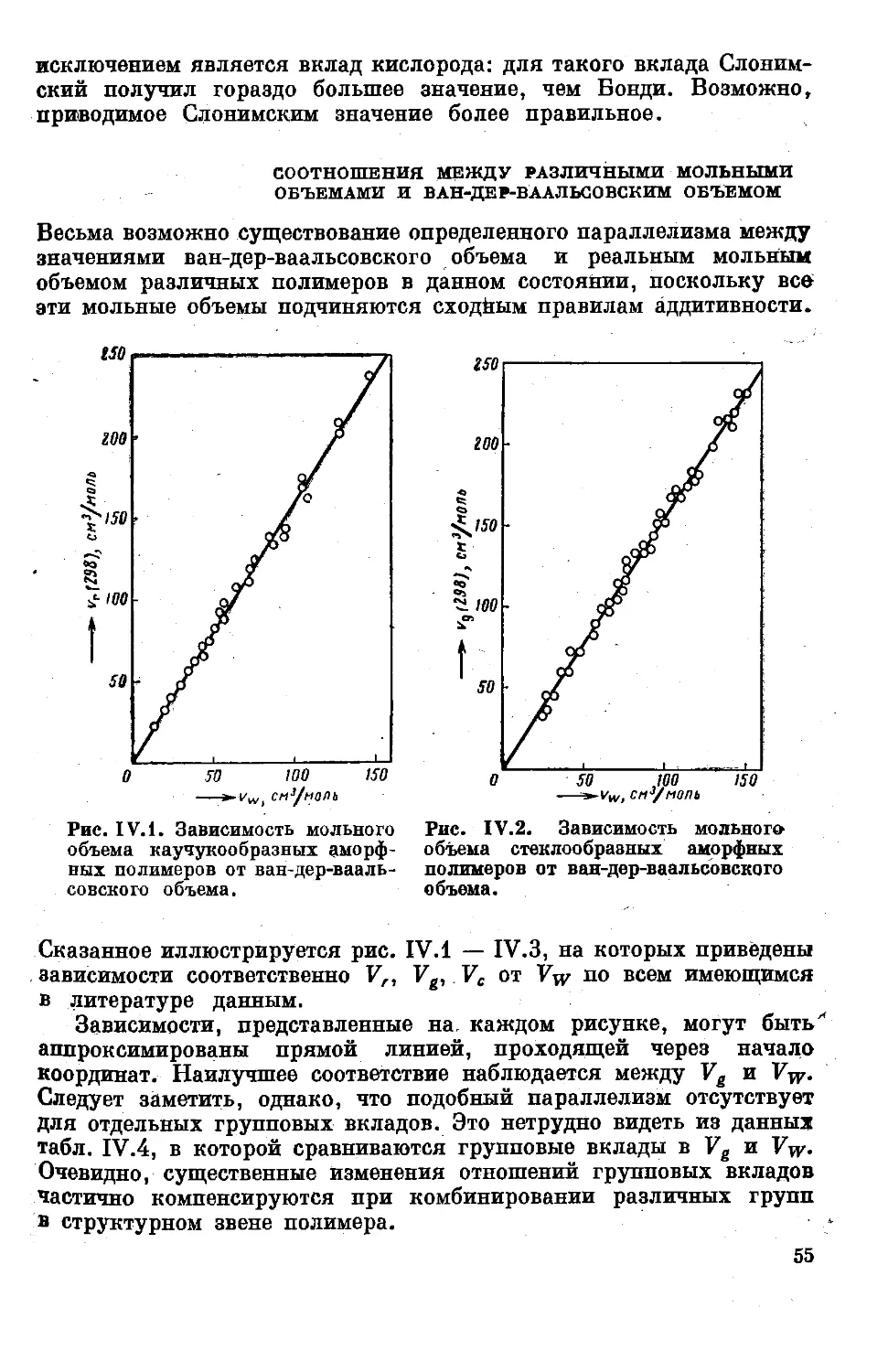






























































































































































































































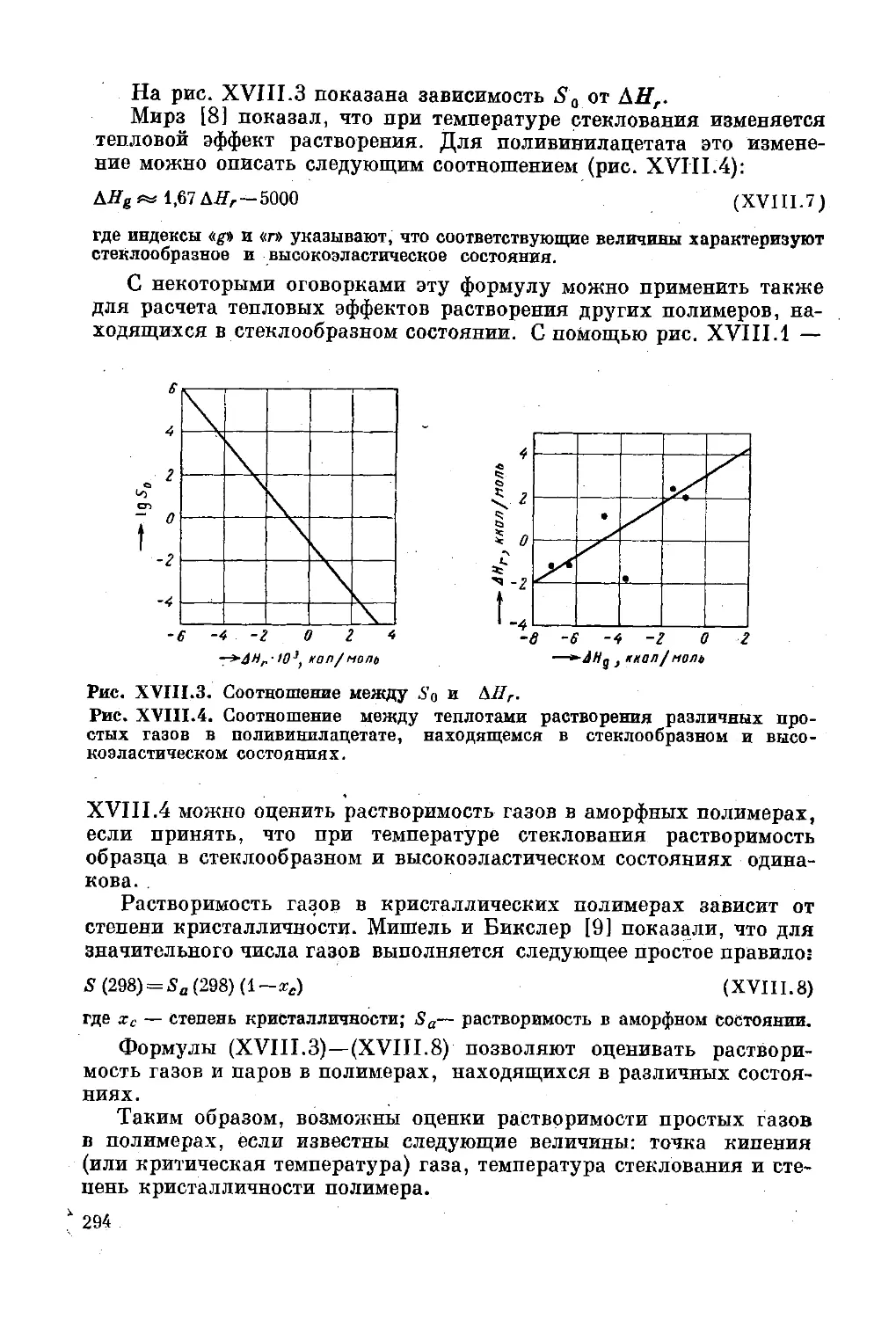





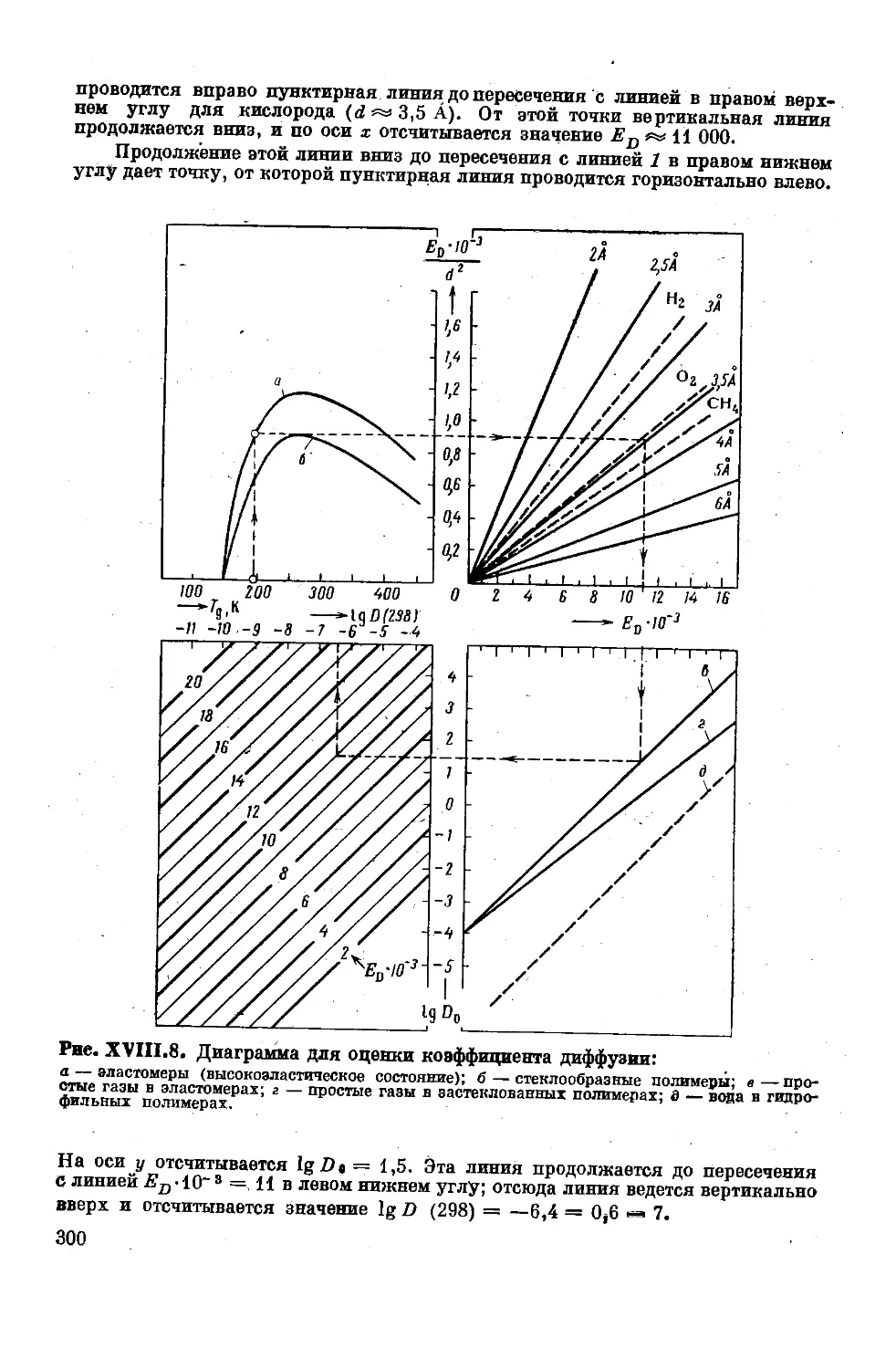
























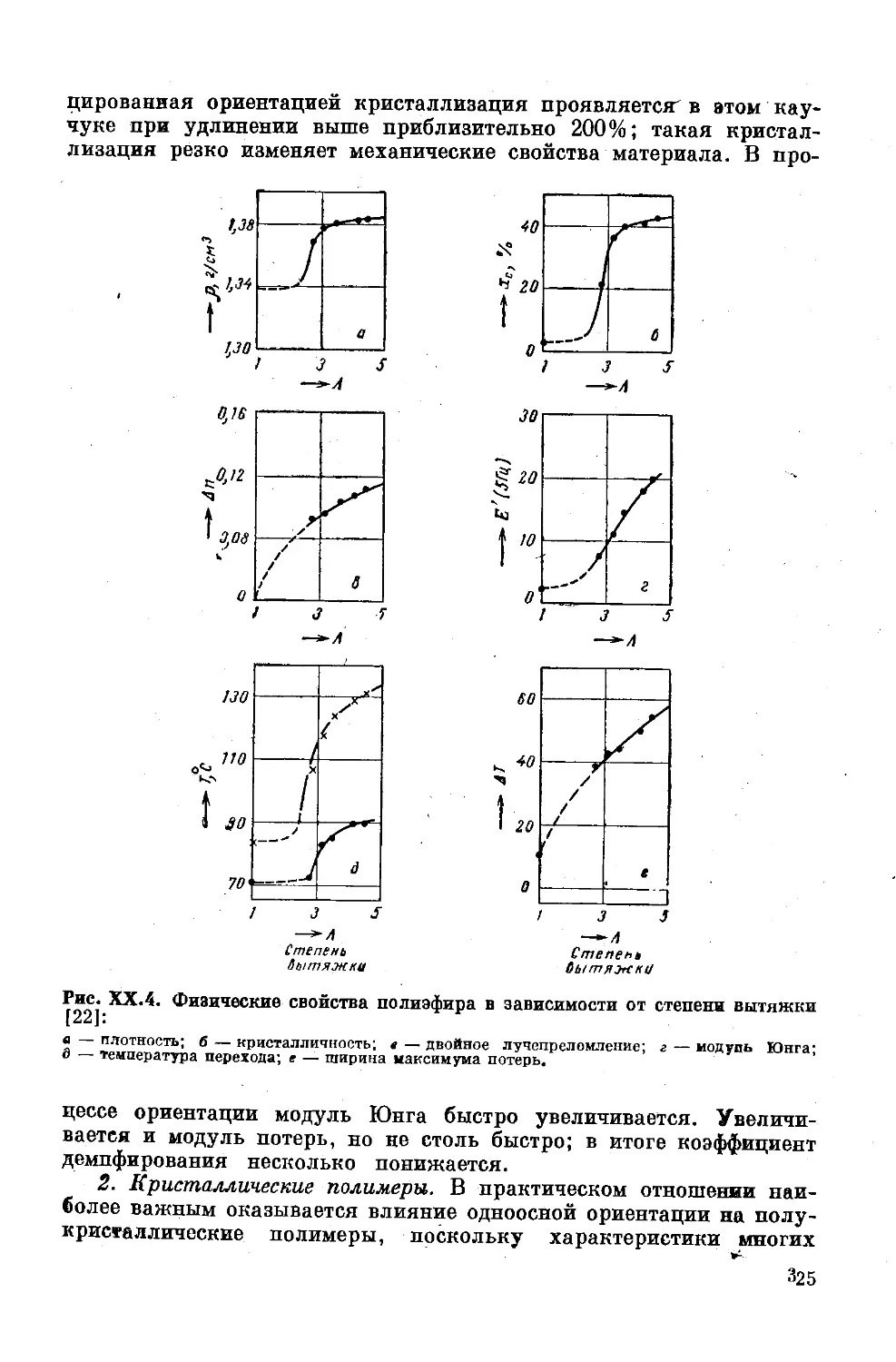





















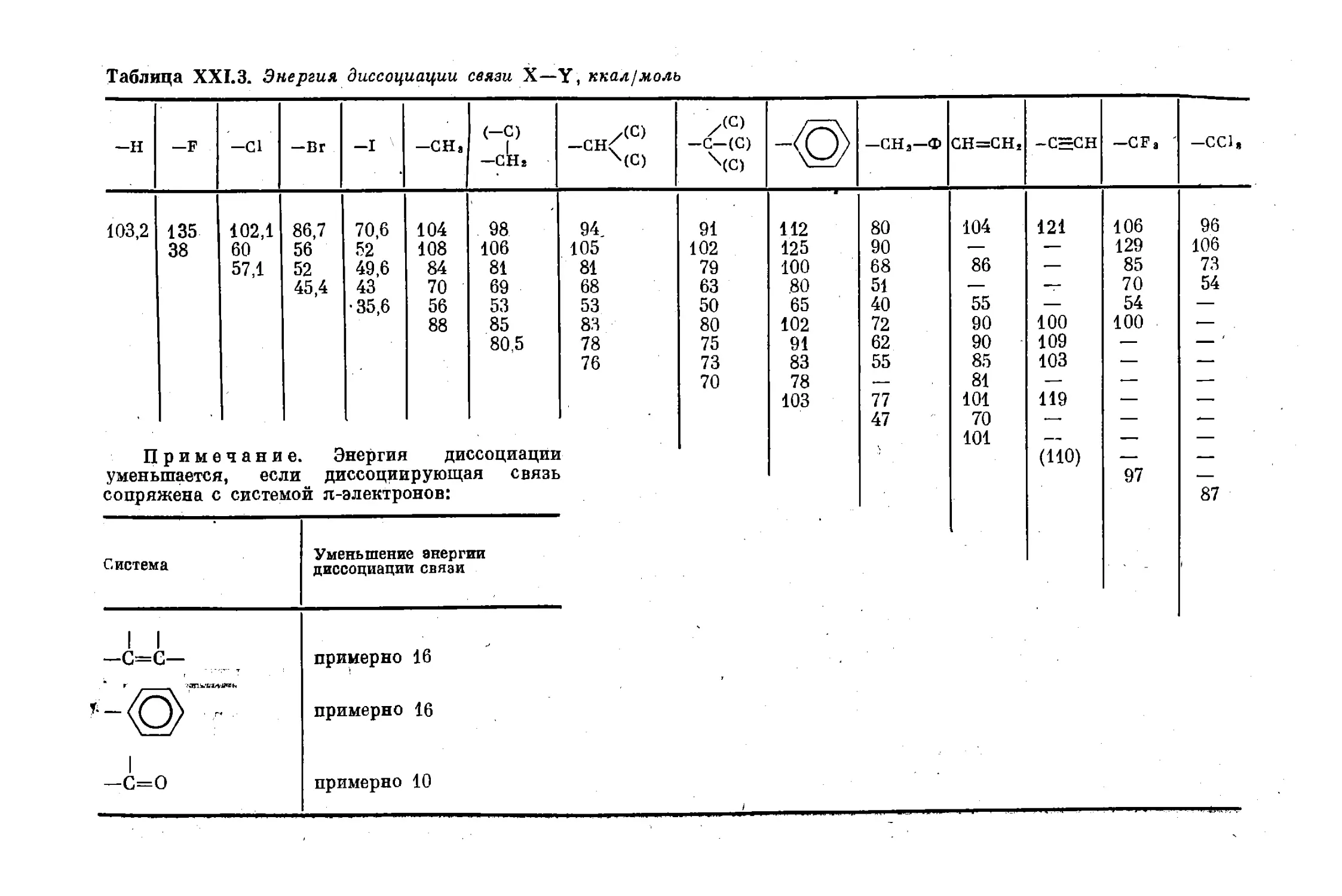



































































Текст
Д.ВВАН КРЕВЕЛЕН
Свойства
и химическое
строение
ПОЛИМЕРОВ
By D. W. VAN KREVELEN PR HPT?R TTl? Q
Professor at the University l llv/I UjLV 1 itlju
of Technology,
Delft, The Netherlands
and President
OF POLYMERS
of AKZO Research ,,/млпт^т * тхттттт
and Engineering N.V., CORRELATIONS WITH
Arnhem, The Netherlands
CHEMICAL STRUCTURE
With the collaboration of
P. J. HOFTYZER
Research Associate,
AKZO Research
and Engineering N. V.,
Arnhem, The Netherlands
elsevTer PUBLISHING
COMPANY AMSTERDAM-
LONDON—NEW YORK
1972
Д. В. ВАН КРЕВЕЛЕН QJ^QJ^Q^pgA
И ХИМИЧЕСКОЕ
СТРОЕНИЕ
ПОЛИМЕРОВ
Перевод с английского
канд. хим.' наук
Ф. Ф. ХОДЖЕВАНОВА
Под редакцией
доктора физ.-мат. наук
А. Я. МАЛКИНА
МОСКВА
ИЗДАТЕЛЬСТВО «ХИМИЯ» 19 75
УДК 678.6/.7 : 541,64/ 68
Ван Кревелен Д. В. ,
Свойства и химическое строение полимеров. Голландия,
1972. Пер. с англ. Под ред. А. Я. Малкина. М., «Химия»,
1976.
В книге подробно описаны термодинамические, механи-
ческие, оптические, электрические и магнитные свойства
полимеров, их растворов и расплавов; изложены методы
прогнозирования наиболее важных в технологическом отно-
шении показателей свойств полимерных материалов.
Следует особо отметить, что книга содержит обширный
и в ряде случаев уникальный справочный материал по фи-
зико-химическим характеристикам конкретных полимеров.
Книга предназначена для научных и инженерно-техни-
ческих работников, занимающихся исследованием и пере-
работкой полимеров. Она может представить интерес для
аспирантов и студентов старших курсов, изучающих химию
и физику полимеров.
416 с; 65 табл.; 136 рис.; список литературы 534 ссылок.
31410-049
050(01)-76
49-76
Перевод на русский язык.
<§) Издательство «Химия», 1976 г.
СОДЕРЖАНИЕ
ПРЕДИСЛОВИЕ РЕДАКТОРА ...................... 9
ПРЕДИСЛОВИЕ АВТОРА ......................... И
ЧАСТЬ ВВЕДЕНИЕ .................................. 13
ПЕРВАЯ
Глава I. Свойства полимеров................ 13
Подход .................................. 13
Понятие «свойства полимеров»...... 15
Литература ................................ 18
Глава II. Типология полимеров .............. 19
Строение полимеров ........................ 19
Молекулярный вес и молекулярно-весовое рас-
пределение ............................... 24
Переходы в полимерах. Классификация поли-
меров на основе механического поведения ... 26
Строение кристаллических полимеров .... 31
Литература ............... . 33
Глава III Типология количественпых характеристик ... 34
Безразмерные группы величин................ 34
Типы характеристик полимеров . ............ 37
Аддитивные мольные функции........ 37
Литература ................ 42
ЧАСТЬ ТЕПЛОФИЗИЧЕСКИЕ СВОЙСТВА
ВТОРАЯ ПОЛИМЕРОВ ................................. 44
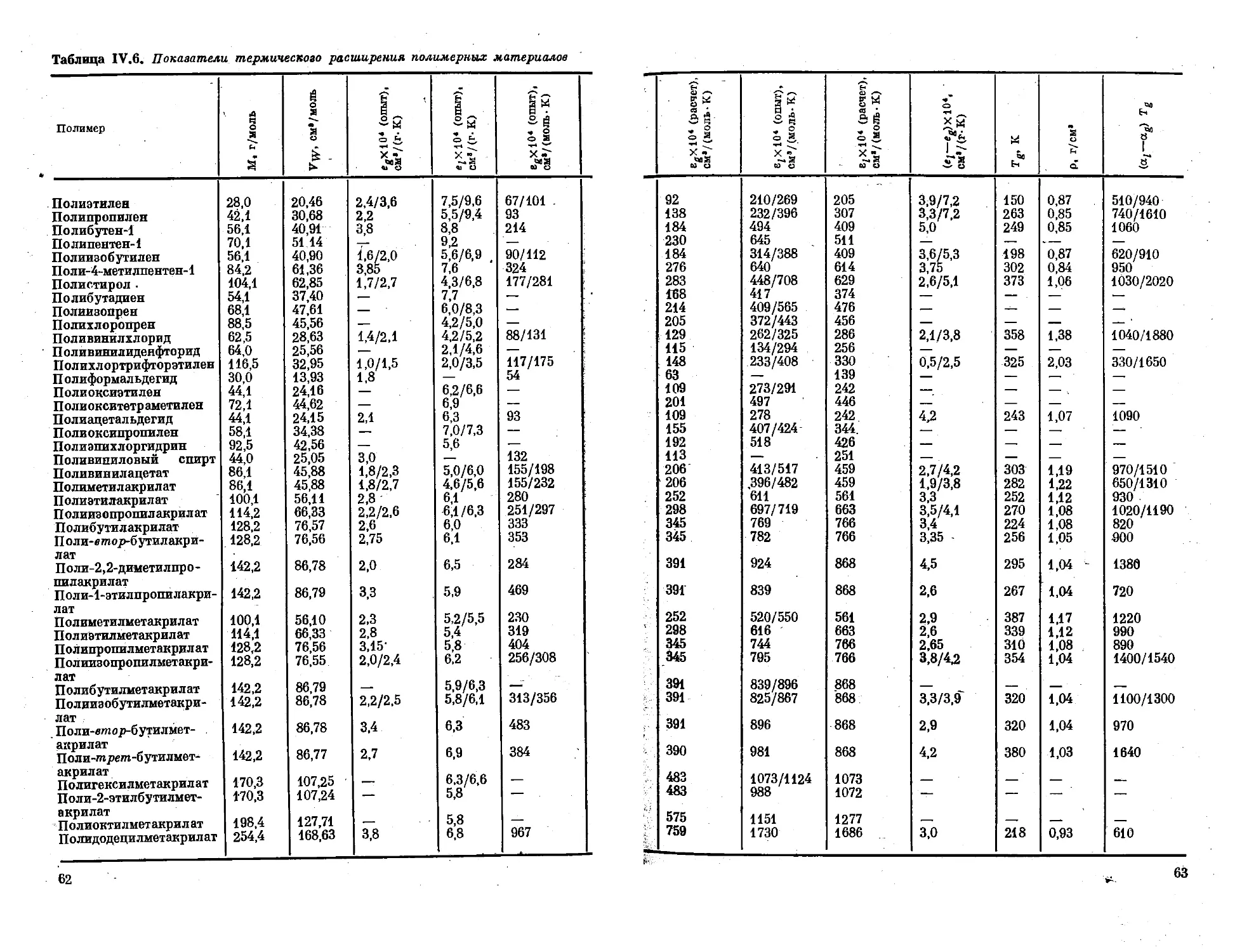
Глава IV. Объемные характеристики........... . 44
Объем и плотность ....................... 44
Тепловое расширение ....................... 57
Литература ................ . ... . 67
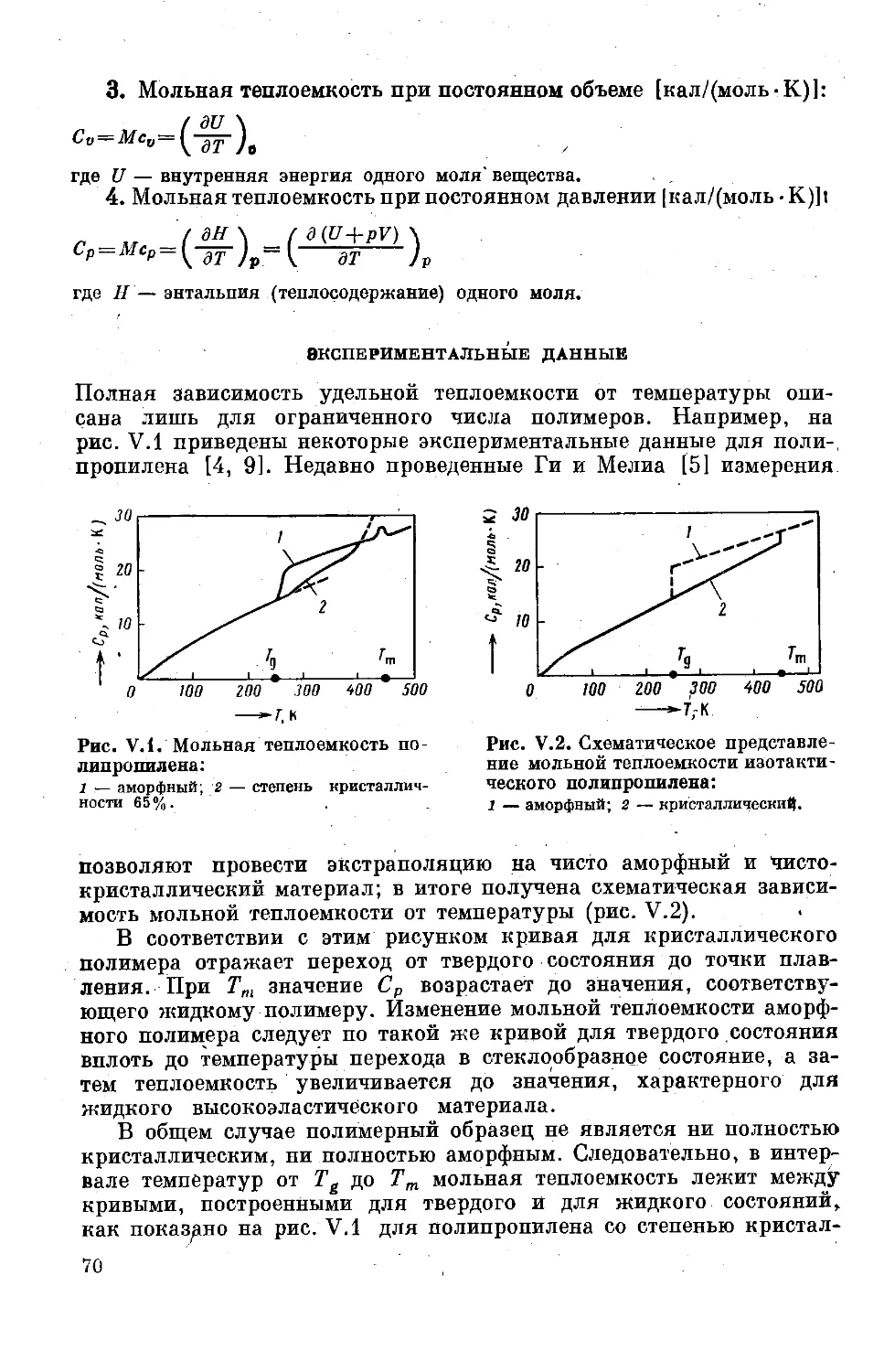
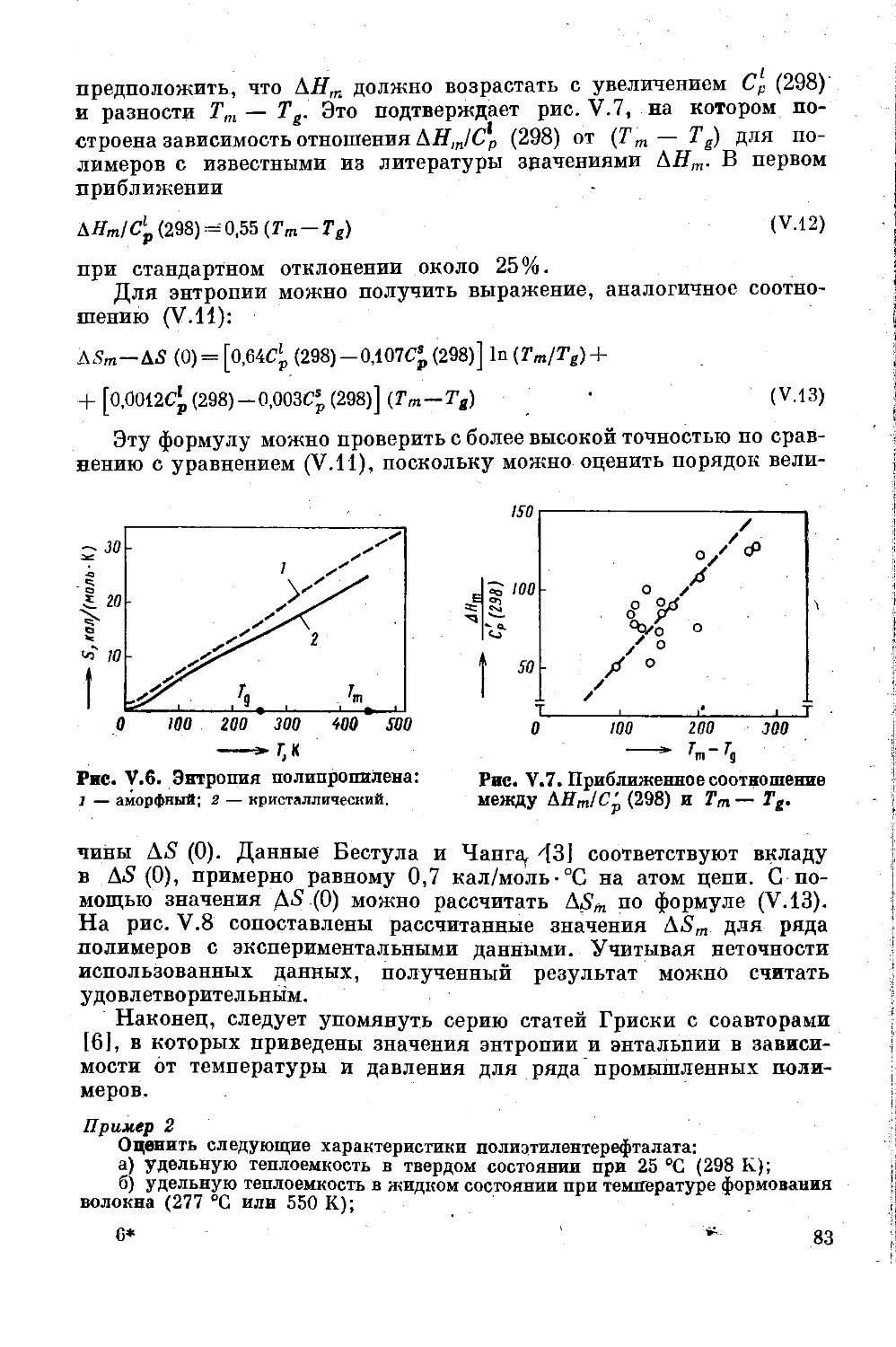
Глава V, Калориметрические характеристики.. 69
Теплоемкость .............................. 69
Теплота кристаллизации и плавления .... 78
Энтальпия и энтропия .......... . 81
Литература ................ 85
5
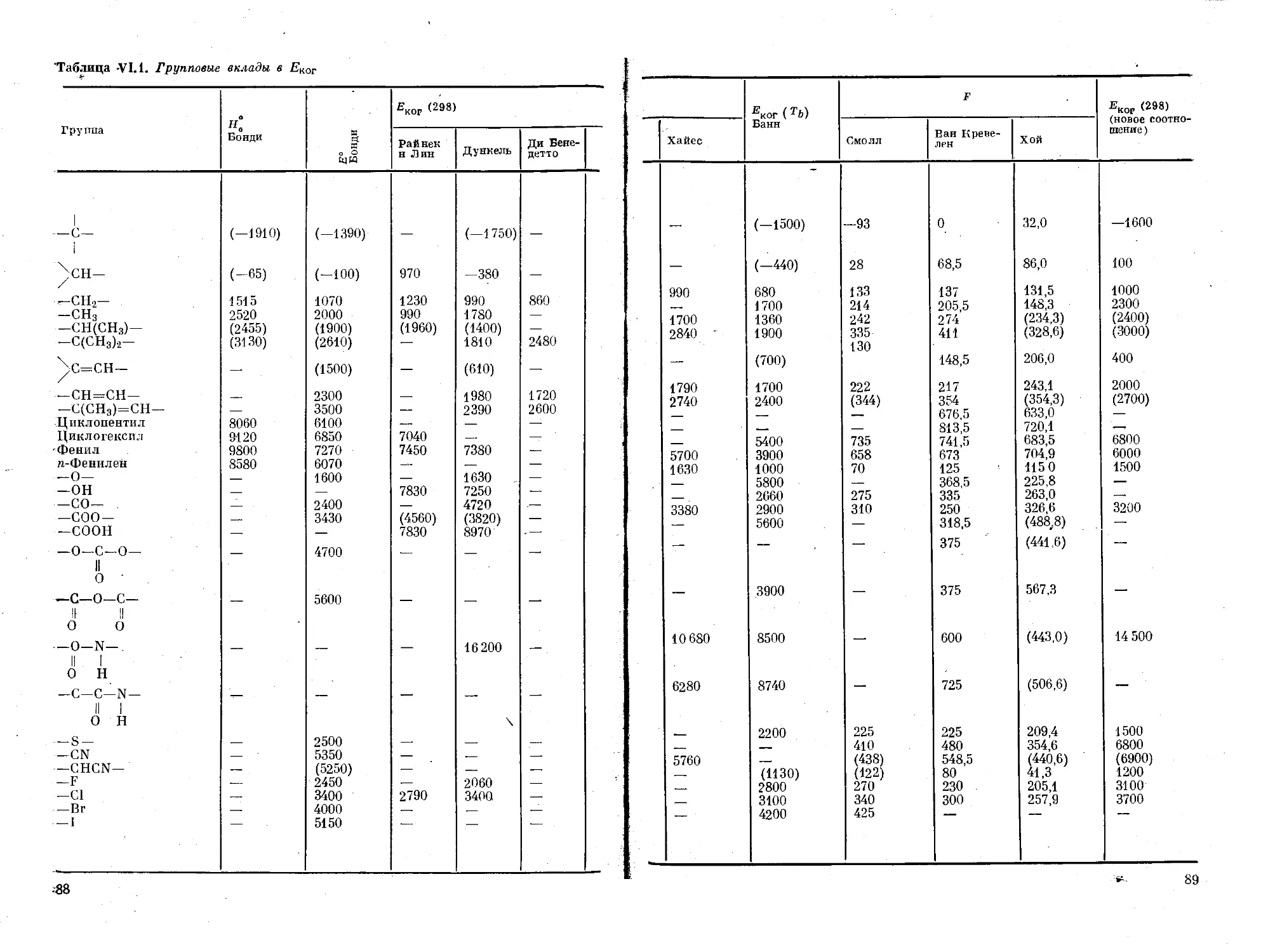
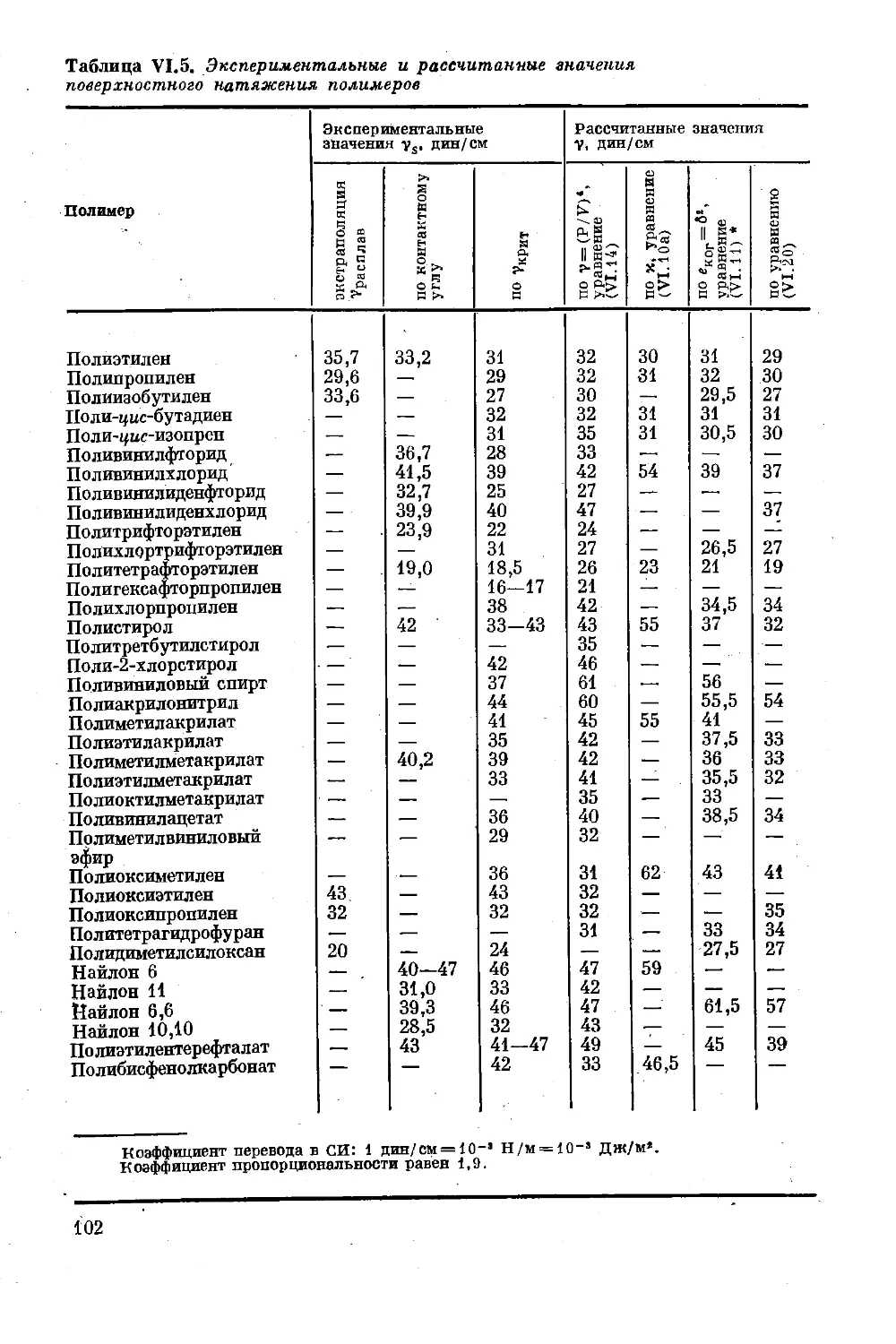
Глава VI. Характеристики когезии и адгезии............. 86
Мольная энергия когезии..................... 86
Внутреннее давление ........................ 92
Поверхностная энергия ...................... 95
Поверхностное натяжение жидкостей и расплавов 96
Межфазное натяжение на поверхности раздела
твердое тело — жидкость .................... 98
Поверхностная энергия нолииеров в твердом
состоянии ............................ 100
Обобщенная формула межфазного натяжения 103
Адгезия полимеров ......................... 105
Литература ................ 107
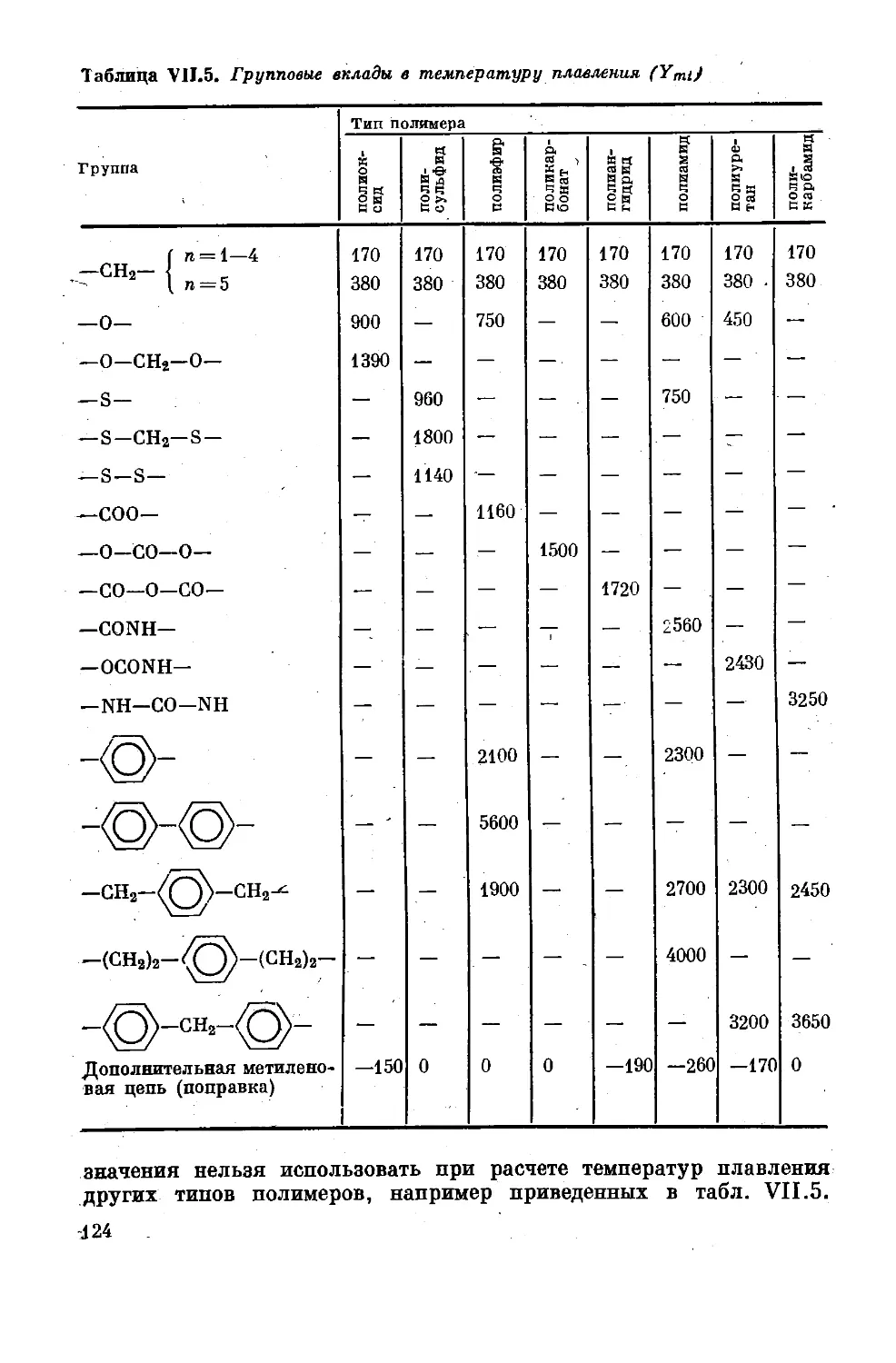
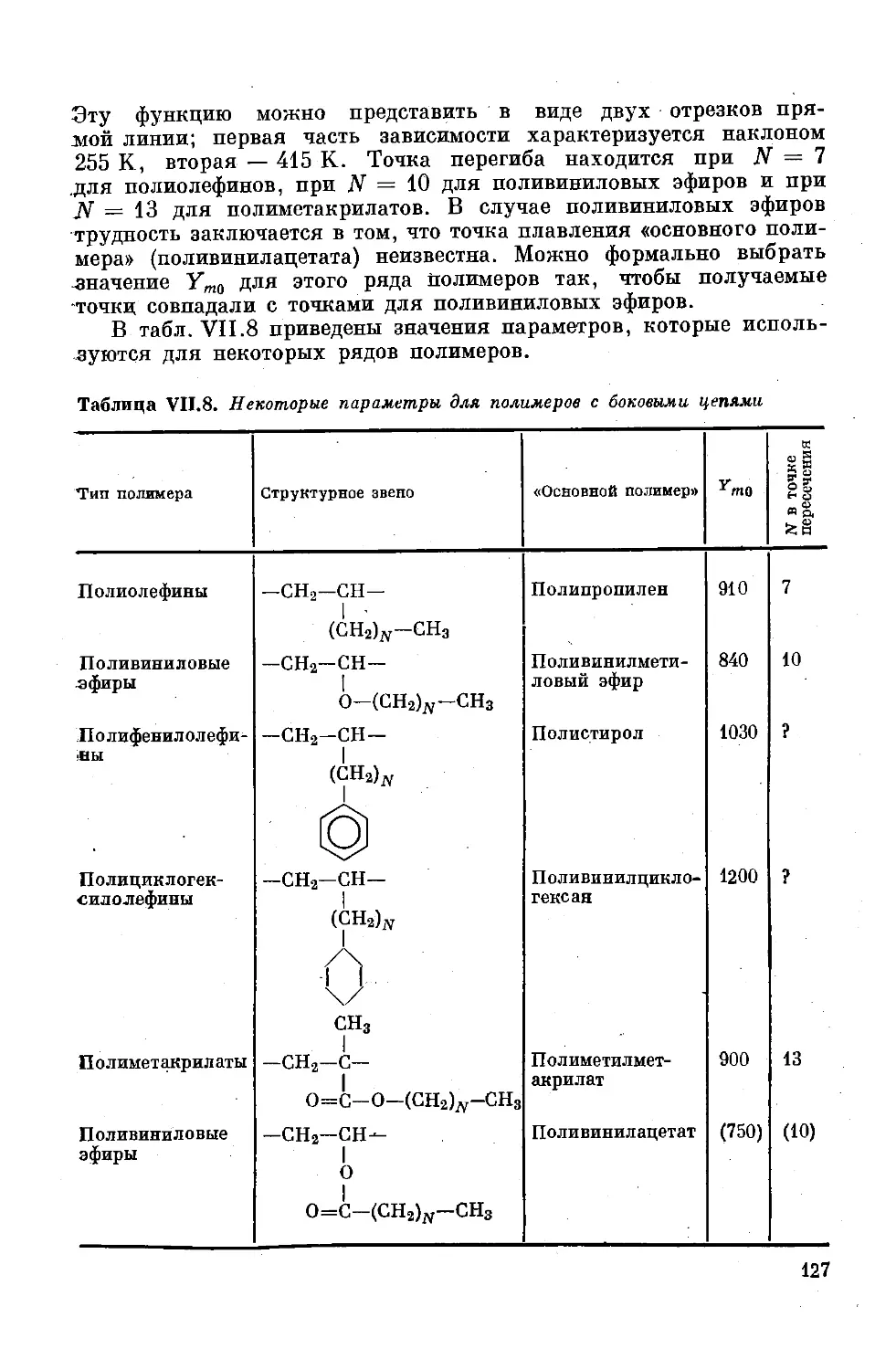
Глава VII. Температуры переходов ,..................... 108
Температура стеклования ............... 108
Точка плавления кристаллических полимеров 120
Соотношение между температурой стеклования
и температурой плавления полимеров ..... 129
Литература ................................ 131
Глава VIII. Растворимость............................. 133
Параметр растворимости .................... 133
Пределы растворимости...................... 141
Литература ................ 142
ЧАСТЬ СВОЙСТВА ПОЛИМЕРОВ
ТРЕТЬЯ В СИЛОВЫХ ПОЛЯХ . .......................... 143
Глава IX. Мехавгические (вязкоупругие) свойства .... 143
Упругие характеристики................... 143‘
Температурная зависимость модулей упругости 154
Высокоэластичность резин .................. 159
Вязкоупругость . .......................... 161
Динамические механические параметры . . . 162
Релаксация напряжений и ползучесть ..... 166
Модели вязкоупругого поведения ...... 172
Литература ................ 173
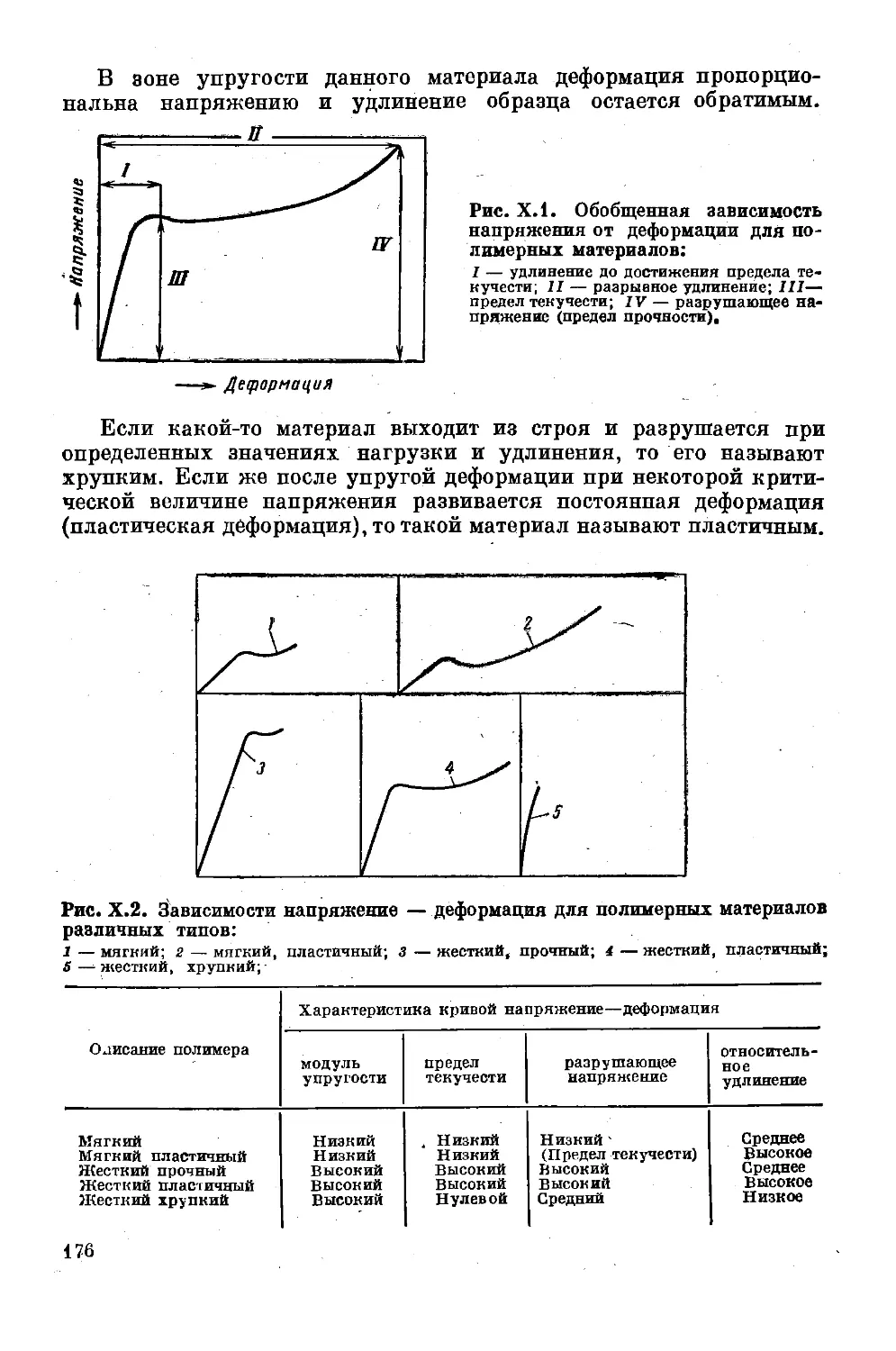
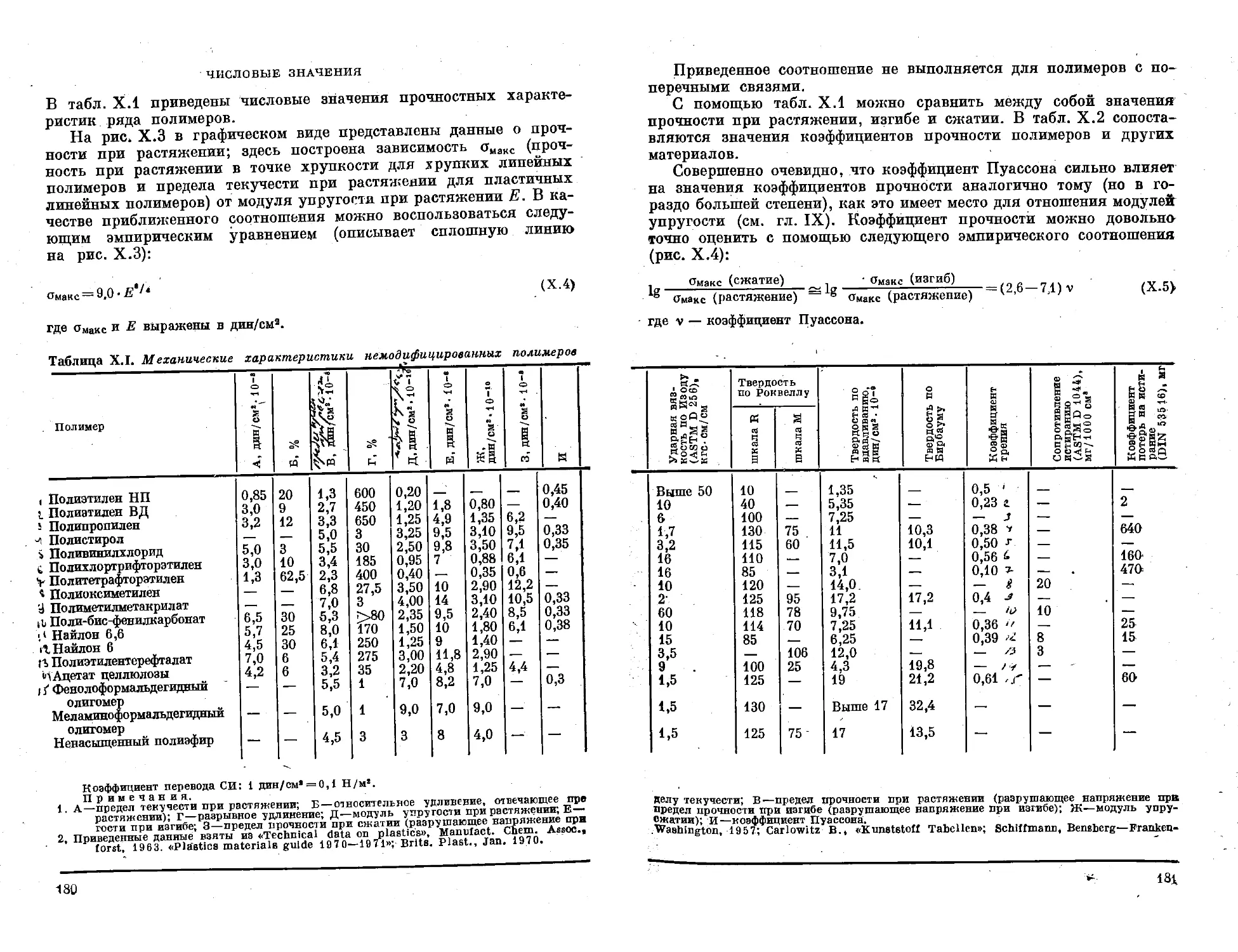
Глава X. Предельные механические характеристики . . . 175
Кривые напряжение — деформация для твердых
полимеров ................................ 175
Механические характеристики при длительных
воздействиях .............................. 186
Другие характеристики, связанные с механиче-
,ским разрушением ............. 189
Литература . .............................. 193
Глава XI. Оптические свойства ........................ 194
Поглощение света ....... 7"................ 195
Преломление света ......................... 199
Рассеяние света............................ 204
Отражение света .......................... 209
Литература ................................ 209
Глава XII. Электрические свойства ...................... 211
Диэлектрическая поляризация .............. 211
Статическая электризация и проводимость . . 219
Предельные электрические характеристики . . 220
Литература ................................. 221
Глава XIII. Магнитные свойства.......................... 222
Магнитная восприимчивость.............. 222
Магнитный резонанс ......................... 224
Ядерный магнитный резонанс............. 224
ЯМР-спектроскопия высокого разрешения . . . 226
ЯМР-спектроскопия широких линий........ 229
Электронный парамагнитный резонанс .... 230
Литература ................................. 232
ЧАСТЬ ЯВЛЕНИЯ ПЕРЕНОСА
ЧЕТВЕРТАЯ В ПОЛИМЕРАХ ............................... 233
Глава XIV. Перенос тепловой энергии................ . 233
Теплопроводность . . . ........................ 233
Литература ......... 239
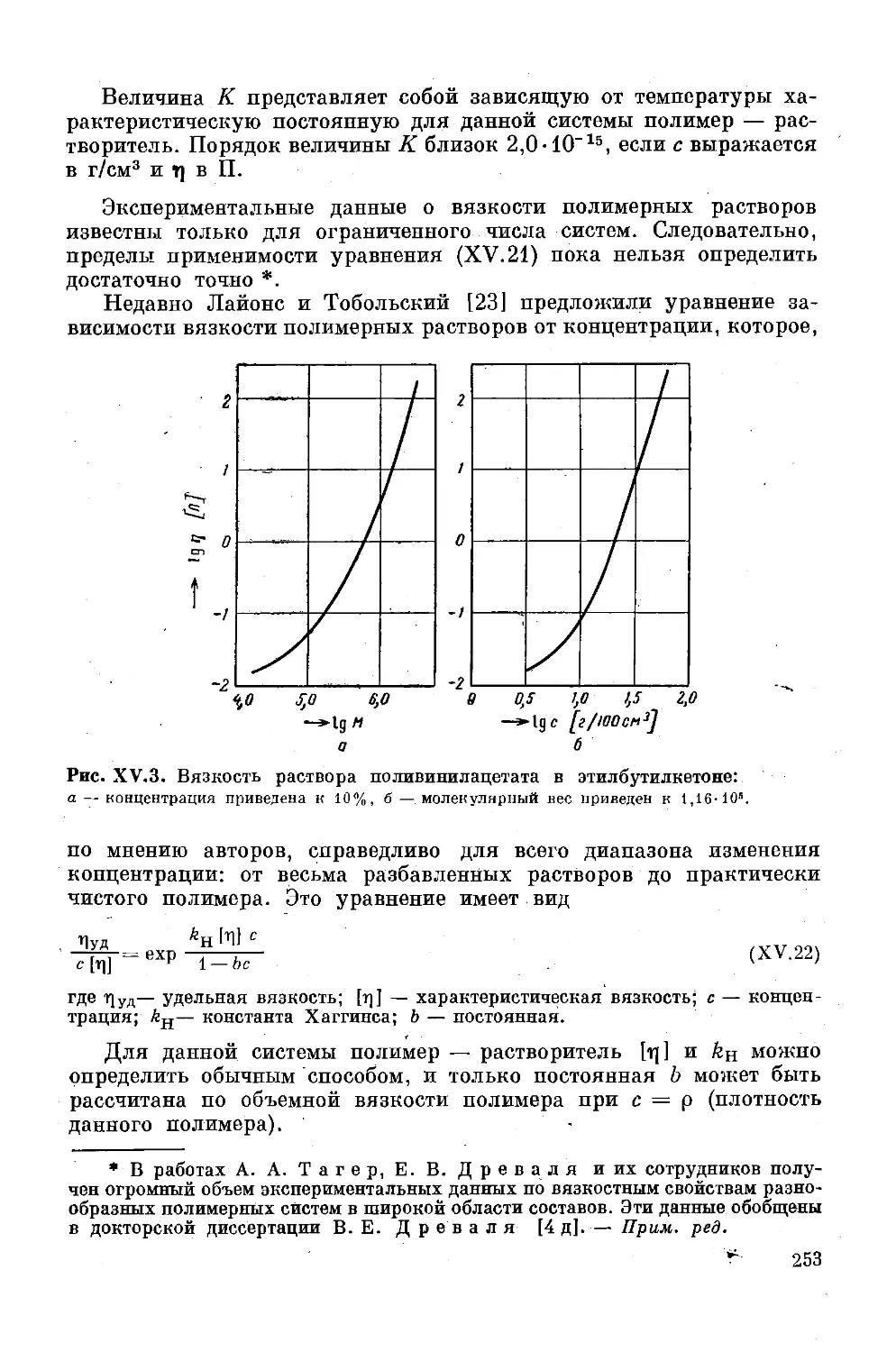
Глава XV. Вязкость растворов полимеров . . 240
Предельное число вязкости (характеристическая
вязкость) ..................................... 241
Вязкость растворов полимеров............ 250
Литература ....... 261
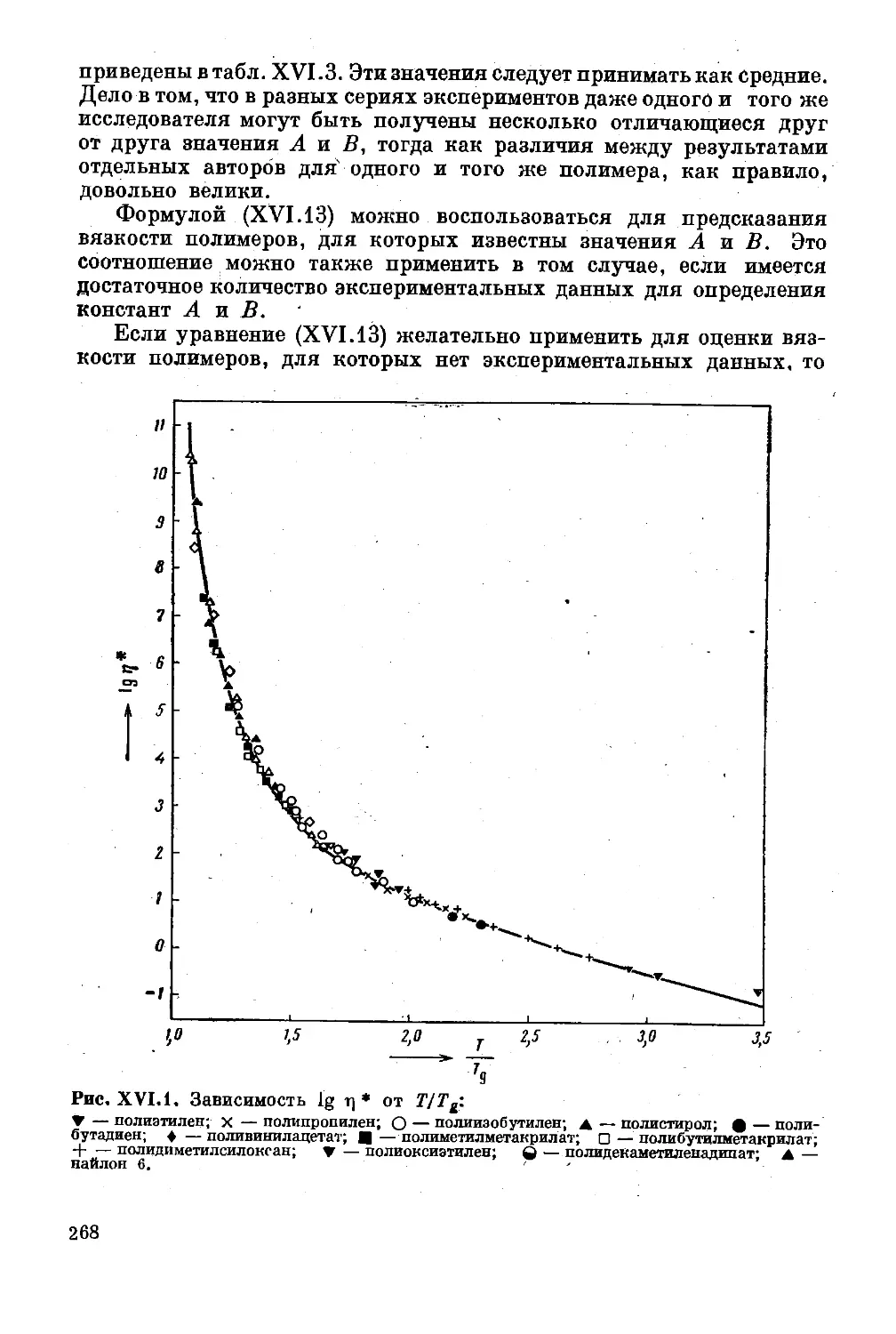
Глава XVI. Вязкость расплавов полимеров............ 262
Понятие о вязкости расплава............. 262
Влияние молекулярного веса на вязкость . . . 263
Влияние температуры на вязкость ...... 264
Влияние напряжения сдвигйг на вязкость . . . 270
Влияние гидростатического давления на вязкость 272
Упругость расплавов ................'. . . 273
Обобщенная «кривая течения» ........ 281
Литература ...... .......... 283
Глава XVII. Растворение и плавление полимеров........ 285
Растворение полимеров . . . ........... 285
Плавление полимеров..................... 289
Литература .................................... 289
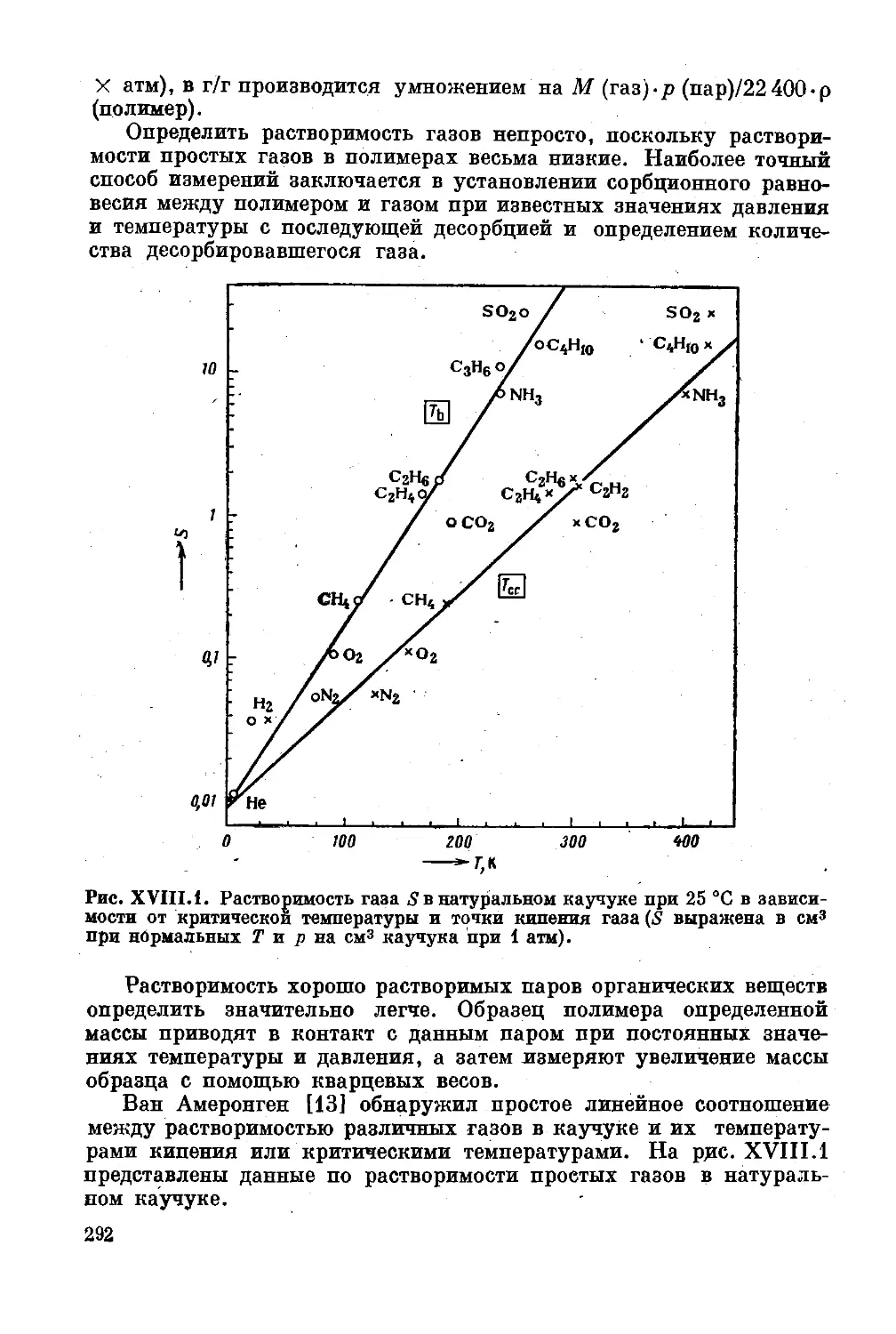
Глава XVIII. Проницаемость полимеров. Диффузионный пере*
нос газов, паров и жидкостей в полимерах . . . 291
Растворимость газов и паров в полимерах . . . 291
Коэффициент диффузии ......................... 295
Адсорбция и перенос влаги.............. 302
Литература ...... . ,....................... 307
7
ЧАСТЬ ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ
ПЯТАЯ. ПРЕВРАЩЕНИЯ В ПОЛИМЕРАХ .................... 308
Глава XIX. Кристаллизация ............................. 308
Кристалличность ............................ 308
Образование центров кристаллизации и рост кри-
сталлов .................................. 310
Суммарная скорость кристаллизации........... 313
Литература ................ 318
Глава XX. Ориентация ................................ 319
Степень ориентации ......................... 320
Одноосная ориентация ....................... 321
Двухосная ориентация ....................... 333
Литература ................ 333
Глава XXI. Термохимические характеристики ............. 335
Термодинамика и кинетика.................... 335
Расчеты изобарно-изотермического потенциала
реакции по групповым вкладам................ 337
Изобарно-изотермический потенциал реакций по-
лимеризации. Предельные температуры .... 343
Термодинамика свободных радикалов........... 345
Свободнорадикальная сополимеризация . . . 346
Литература ................................. 354
Глава XXII. Деструкция ................................. 355
Формы деструкции .......................... 355
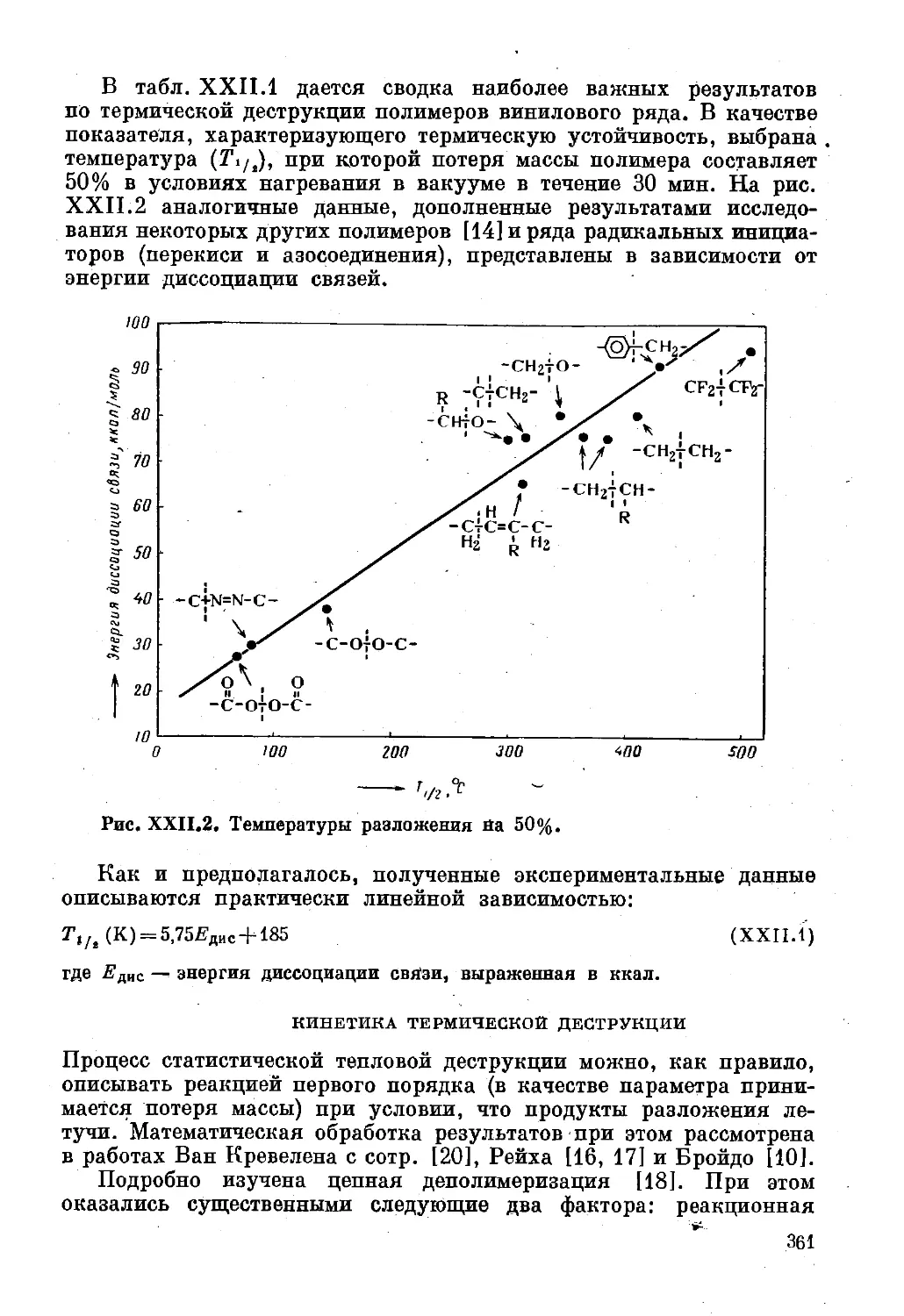
Количественное описание деструкции полимеров 360
Литература ................................ 364
ЧАСТЬ ОБСУЖДЕНИЕ ИЗЛОЖЕННЫХ
ШЕСТАЯ. ПРЕДСТАВЛЕНИЙ . . 366
Глава XXIII. Соотношения между характеристиками полимеров 366
Характеристические диаграммы инкрементов . . 366
Влияние функциональных групп на характери-
стики полимеров ........................... 371
Соотношения между физическими характеристи-
ками ...................................... 374
Заключение ................................. 381
Литература ................................. 381
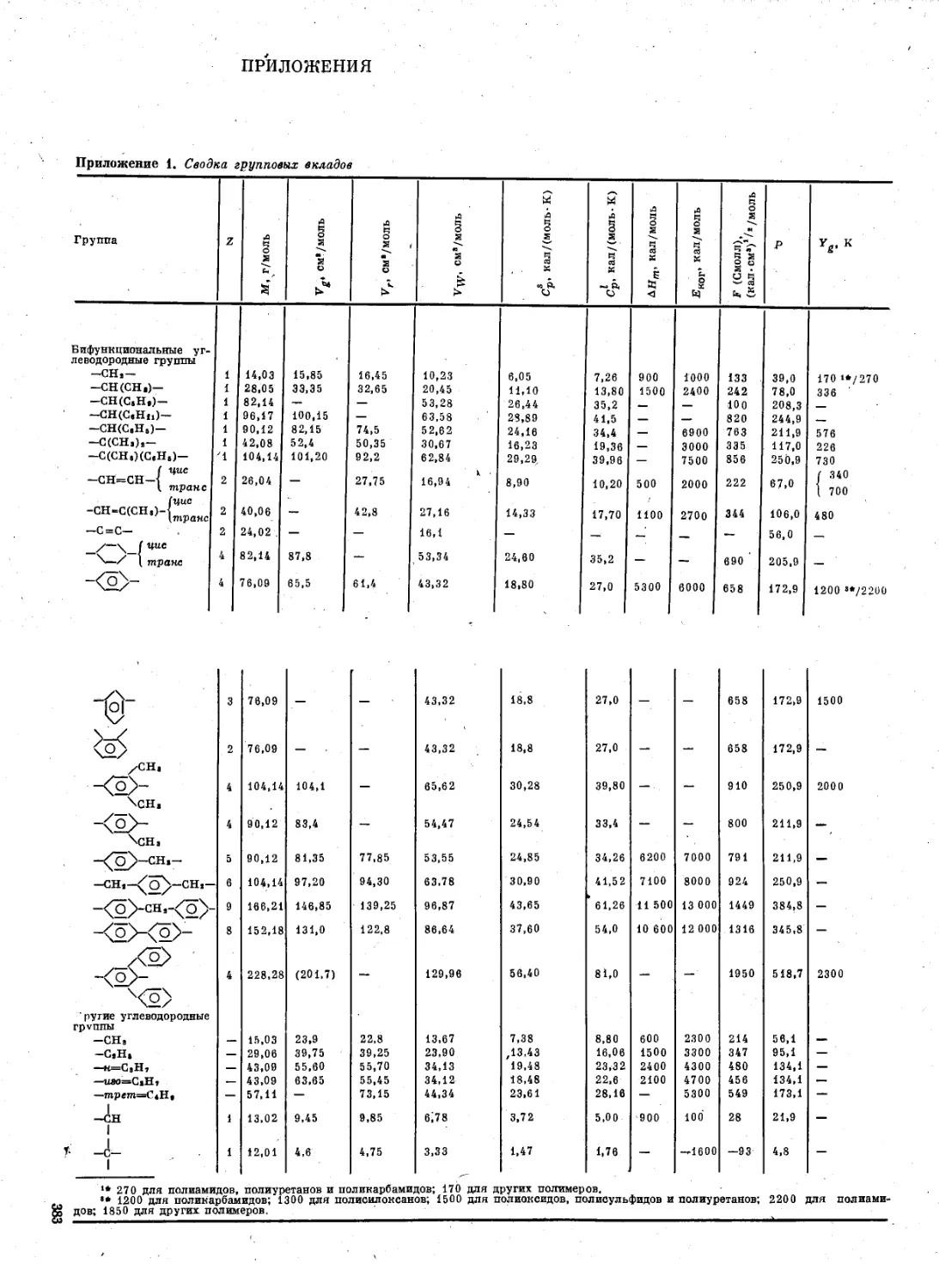
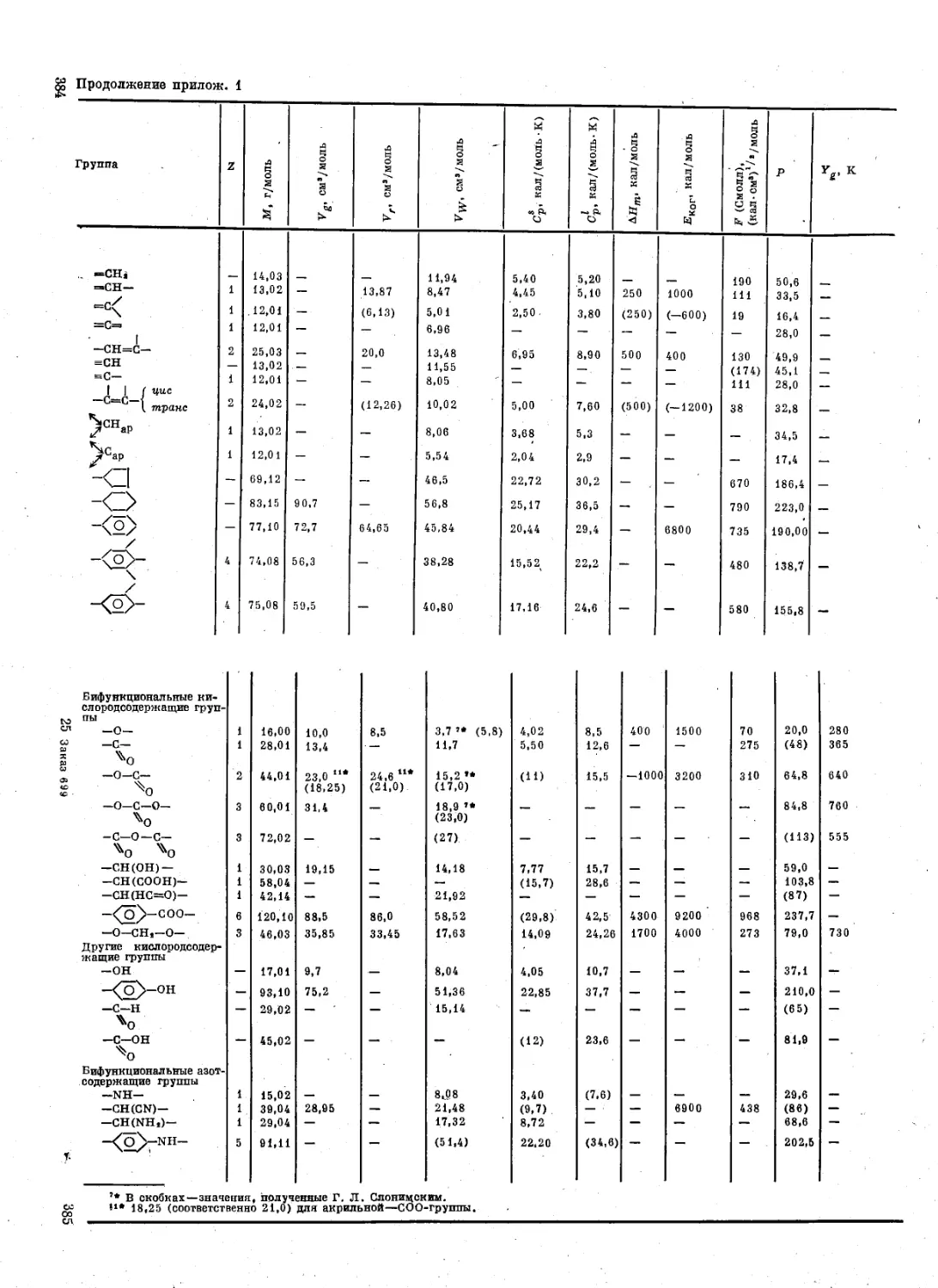
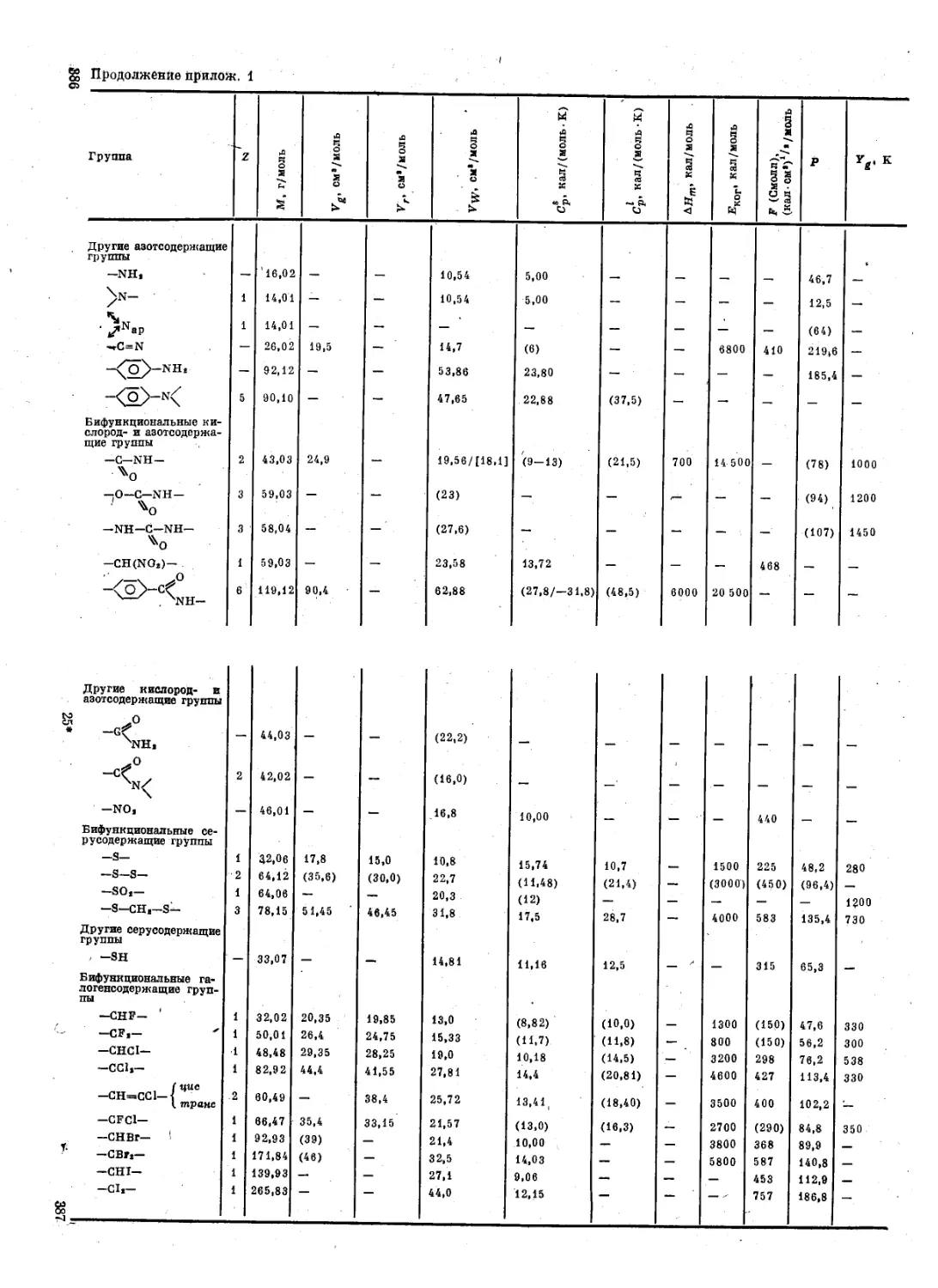
Приложения ................................. 382
Предметный указатель........................ 409
ПРЕДИСЛОВИЕ РЕДАКТОРА
Однажды было сказано, что в каждой науке столько истины, сколь?
ко в ней числа. Как и в каждом афоризме, здесь подчеркнута лишь
одна сторона дела, но, может быть, действительно, самая важная.
Накопление фактов и количественных закономерностей требует'
кропотливой и долголетней работы многих независимых исследова-
телей и продолжается неограниченно долго, пока конкретное науч-
ное направление представляет общественный интерес. Однако все-
гда наступает такой момент, когда созревает необходимость собрать
воедино разрозненные сведения, и тогда становится особенно ясно,
каков же фундамент реальных и объективных данных, на котором
основано представление о мире объектов, изучаемых нашей
наукой.
Сегодня такой момент наступил в науке о полимерах, и свидетель-
ством тому является книга известного голландского физико-химика
Ван Кревелена, перевод которой мы предлагаем советскому чита-
телю. Задача автора состояла в систематизации того поистине огром-
ного экспериментального материала, который скопился за десятиле-
тия (в основном все же за последние 8—10 лет) изучения свойств''
высокомолекулярных соединений и в установлении различных тео-
ретических и эмпирических корреляций между химическим строе-
нием и показателями всевозможных свойств полимеров. Появление )
такой книги беспрецедентно в области высокомолекулярных соеди-
нений, хотя подобные издания предпринимались для описания дру-
гих веществ, например газов, изучение свойств которых началось
гораздо раньше, чем полимеров. По своему характеру данную книгу
можно определить как справочник, но содержание ее несравненно
более глубоко и приближается к научной монографии. В книге
содержатся не только конкретные сведения о свойствах широкого
круга объектов. Более важным является обязательное обсуждение
вопроса о соответствии между химическим строением и свойствами.
Это позволяет распространить полученные результаты на великое
множество новых полимеров, даже не упоминаемых в книге. И еще:
существование многих эмпирических соответствий, подмеченных или
собранных автором, заставляет задуматься о природе описанных
закономерностей, и теоретикам здесь бросается недвусмысленный
вызов.
Несмотря на ограниченный объем, книга Ван Кревелена содер-
жит огромную информацию. Можно с очень большой долей уверен-
ности утверждать, что она дает ответ на любой вопрос, построенный
по такой схеме: чему равен показатель такого-то свойства полимера,
если задано его химическое строение. Точность выдаваемой инфор-
мации вполне отвечает существующим инженерным и во многих
случаях научным требованиям. Для облегчения пользования при-
водимым материалом немаловажное значение имеют многочисленные
примеры, иллюстрирующие предлагаемые расчетные схемы. Это
особенно удобно для специалиста, сталкивающегося со смежными
областями науки о полимерах, которые на первый взгляд кажутся
разделенными как бы непроницаемыми перегородками, но в действи-
тельности покоятся на общем фундаменте — химическом строении
цепи.
При подготовке русского перевода книги было решено сделать
некоторые дополнения, необходимость которых диктовалась двумя
обстоятельствами. Во-первых, тем, что автору, по-видимому, оста-
лись неизвестными некоторые работы советских специалистов,
имеющие непосредственное отношение к проблематике книги, и,
во-вторых, тем, что накопление фактов идет непрерывно и нельзя
не учитывать, что период времени от момента окончания работы
автора над рукописью книги до подписания в печать текста перевода
составляет более четырех лет. Эти примечания включены непосред-
ственно в авторский текст (в квадратных скобках).
Кому может понадобиться эта книга? Конечно, всем, кому по
роду работы нужны конкретные сведения о полимерных веществах,
т. е. специалистам практически всех прикладных областей. Общая
научная направленность книги заставляет думать, что она станет
настольной книгой исследователей — и теоретиков, и эксперимен-
таторов. Было бы также очень хорошо, если бы эта книга была
использована в процессе обучения студентов-старшекурсников, спе-
циализирующихся в различных областях полимерной науки, и
можно быть уверенным, что навык, приобретенный на студенческой
скамье, пригодится им в жизни. Думается, что книга Ван Кревелена
приобретет у советского читателя заслуженную популярность
ПРЕДИСЛОВИЕ АВТОРА
Книга предназначена для тех, кто работает над решением практиче-
ских задач в области полимеров и нуждается в оценочных данных
по свойствам полимеров. Книгат" адресована химикам-органикам,
которые занимаются синтезом новых полимеров и хотят знать,
будут ли реализованные ими на практике структуры обладать зара-
нее заданными свойствами. Книга предназначена для инженеров-
химиков, которые часто не располагают достаточными данными и не
могут получить числовые значения величин, соответствующих задан-
ным условиям определенного процесса. Книга призвана помочь
инженерам, занимающимся переработкой полимеров, которые пыта-
ются предугадать, как определённые физические параметры процесса
будут реагировать на изменение условий его проведения. Книга
адресуется технологам-полимерщикам, которые пытаются осмыс-
лить тесные взаимосвязи многих научных дисциплин в области тех-
нологии полимеров. Наконец, эту книгу можно рекомендовать всем
исследователям, которые заинтересованы в установлении связей
между химическим строением и физическими свойствами полимеров
и во взаимосвязях между этими свойствами.
Принимая за основу химическое строение полимеров,'мы ставим
задачу последовательно показать, что каждая структурная
группа в молекулярной цепи обладает функцией, которая отражается
на всех свойствах полимеров. Весьма широко использован тот
факт, что ряд величин и их комбинаций обладает свойствами адди-
тивности, конечно, н определенных пределах точности, Так что
эти величины могут быть рассчитаны простым способом с помощью
эмпирически полученных групповых вкладов или инкрементов.
Мы намеренно опустили теоретические выкладки, ва исключе-
нием тех случаев, когда пояснения совершенно необходимы для
правильного понимания смысла некоторых не столь широко распро-
страненных величин.
Данная книга писалась не для работающих в области полимер-
ной науки, особенно физиков и физико-химиков. Дело в т^м, что
построение книги слишком «эмпирично» для названной категорий
лиц и слишком направлено на практическую сторону проблемы.
Опытный исследователь не обнаружит в данной книге таких сведе-
ний, которые не были бы опубликованы в другрх источниках
11
Многие исследователи смогут даже представить серьезные возраже-
ния, зачастую вполне оправданные, против построения самой книги
и развиваемого в ней подхода.
К сожалению, разрыв между наукой и технологией полимеров
не только не сокращается, но постоянно растет. г Работа в области
полимерной науки становится все более сложной и противоречивой
как в экспериментальном, так и в теоретическом аспекте. Предла-
гаемая книга представляет собой попытку сократить этот разрыв.
Время покажет, насколько успешной окажется эта попытка.
Книга состоит из шести частей.
В первой части — введении — строение полимеров и их свойства
обсуждаются с позиций наиболее общего и широкого подхода. Цель
этого раздела — обеспечить основу для построения специфического
подхода, развиваемого в книге.
Во второй части — «Теплофизические свойства полимеров» —
рассматриваются основные физические свойства полимеров, в том
числе волюметрические и калориметрические, а также температуры
переходов и характеристики «взаимодействия».
Часть третья — «Свойства полимеров в силовых полях» — отве-
дена описанию поведения полимеров в полях механических и электро-
магнитных сил.
В четвертой части — «Явления переноса в полимерах» — дана
сводка величин, определяющих перенос тепла, момента количества
движения и вещества. Здесь рассмотрены особенно важные пока-
затели — теплопроводность, вязкость и коэффициенты диффузии.
В пятой части — «Физические и химические превращений в поли-
мерах» — рассмотрены все явления, которые обусловливают резкое
изменение характеристик полимеров: кристаллизация, ориентация
и деструкция. Сюда включена также глава о термохимических свой-
ствах полимеров.
В шестой части, ретроспективной, подводятся окончательные
итоги, которые можно рассматривать с общих позиций корреляции
и (.взаимосвязи.
Для повышения ценности книги и в надежде сделать ее справоч-
ным пособием мы приводим таблицы характеристик полимеров
и коэффициентов перевода единиц измерений в Международную
систему единиц (СИ).
Мы стремились следовать одной номенклатуре, однако, к сожа-
лению, нам не удалось избежать применения достаточно большого
количества символов, обладающих несколькими значениями. Такой
недостаток в значительной мере преодолен с помощью подстрочных
индексов, смысл которых понять весьма нетрудно.
ЧАСТЬ
ПЕРВАЯ
ВВЕДЕНИЕ
Глава i. СВОЙСТВА ПОЛИМЕРОВ
ПОДХОД
Интенсивное развитие современной промышленности сделало осо-
бенно важным получение информации р характеристиках ряда мате-
• риалов, среди которых много новых химических соединений, чьи
физические свойства ранее не были известны. Это прежде всего
относится к полимерным материалам. При конструировании техно-
логического и перерабатывающего оборудования необходимо хорошо
знать свойства перерабатываемых материалов. Сведения подобного
рода необходимы также для определения области использования
этих материалов.
В некоторых изданиях, например «Руководство по полимерам»
под редакцией Брэндрапа и Иммергата [1], «Физические константы
линейных гомополимеров» Льюиса [2] и др., можно найти часть
необходимых сведений. Однако во многих случаях нужную характе-
ристику материала нельзя получить из указанных источников.
Цель настоящей книги заключается в описании методов оценки
наиболее важных свойств твердых и расплавленных полимеров и их
растворов, особенно в тех случаях, когда отсутствуют эксперимен-
тально определенные характеристики этих соединений.
Предсказание каких-либо характеристик, как правило, основано
на сопоставлений уже имеющихся данных с результатами интерполя-
ции или экстраполяции некоторых известных зависимостей. Райд
и Шервуд [3] различают три типа таких зависимостей: чисто теоре-
тические зависимости (корреляции такого типа редко адекватно
отражают истинное положение); чисто эмпирические зависимости
(при экстраполяции подобного рода соотношений зачастую полу-
чают нереальные результаты и корреляции такого типа оказываются
бесполезными) и эмпирические зависимости, основанные на теорети-
ческих концепциях (подобные «полуэмпирические» корреляции наи-
более эффективны и дают достаточно надежные результаты в боль-
шинстве важных в практическом отношении случаев).
Довольно часто ученые пренебрежительно относятся к полу-
эмпирическим методам оценки характеристик материалов. Однако
такое отношение не находит никакого оправдания: существует боль-
шое количество формул для оценки различных характеристик мате-
риалов, которые получены не только теоретическим путем и в которых
13 '
весьма заинтересованы практики. Одним из огромных достижений
теоретической физики является современная кинетическая тео-
рия газов. На основании силовой функции, например силовой
функции Леннарда — Джонса, оказывается возможным получить
теоретические уравнения, описывающие все наиболее существенные
характеристики газов: соотношение давление — объем — темпера-
тура (р — v — Т-соотношение), вязкость, коэффициент диффузии,
теплопроводность и температуропроводность. Теоретически предска-
занные зависимости этих характеристик от температуры хорошо
согласуются с экспериментальными данными. Однако следует отчет-
ливо представлять, что эта столь плодотворная теоретическая раз-
работка основывается на чисто эмпирическом выражении для сило-
вой функции. За исключением наиболее простых случаев, не суще-
ствует достаточно строгой теории, способной описать взаимодействие
между молекулами. В то время как кинетическая теория газов пред-
ставляется относительно законченной, теория, описывающая свой-
ства твердых тел, остается менее разработанной, а теория жидкостей
развита в еще меньшей степени.
В относительно новой области полимерных материалов подобный
полуэмпирический подход представляется крайне необходимым,
а в ряде случаев — и единственно возможным.
Фундаментальная теория, как правило, слишком отдалена от
того явления, которое она призвана описывать. В действительности,
на практике, необходима формула, которая получена для непосред-
ственного описания конкретного явления и позволяет использовать
эксперимент. Описанный подход является прагматическим и важен
непосредственно для практических применений.
Для низкомолекулярных соединений Райд и Шервуд [3] решили
подобную задачу применительно к газам и жидкостям. Аналогичный
подход использовал Бонди [4] для твердых тел и жидкостей,
охватив при этом частично и область высокомолекулярных соеди-
нений.
Число публикаций, посвященных высокомолекулярным соедине-
ниям, уже сейчас чрезвычайно велико. Тем не менее очень часто
можно встретиться с такой проблемой, как отсутствие либо опреде-
ленных непосредственно в эксперименте характеристик, либо надеж-
ных методов для расчета этих характеристик. Именно это обстоя-
тельство оправдывает написание настоящей книги. Практическая
значимость методов оценки и сопоставления различных параметров
целиком и полностью определяется простотой применения этих
методов. Это положение является одним из основных руководящих
принципов для автора данной работы.
Совершенно очевидно, что надежно измеренные эксперименталь-
ные данные всегда пользуются преимуществом по сравнению с та-
кого же типа данными, но полученными путем применения тех или
иных оценок. В этом отношении все методы, описываемые в книге,
имею! ограниченную ценность.
14
ПОНЯТИЕ «СВОЙСТВА ПОЛИМЕРОВ»
Свойства вещества всегда можно разделить на три разные, хотя
и неотделимые друг от друга, категории: фундаментальные, или
характеристические, свойства, показатели, важные для перера-
ботки материала, и свойства готового продукта или изделия. На
рис. 1.1 схематически представлено понятие «свойства полимеров»
[51. Следует подчеркнуть, что
каждая из перечисленных кате-
горий свойств весьма тесно
связана с двумя другими. Но
в то время как фундаменталь-
ные или характеристические
свойства относятся всегда к са-
мому веществу, свойства про-
дукта относятся к целому изде-
лию. Такие характеристики за-
висят также от размеров и
формы изделия. Например,
можно говорить о проводимости
железа (характеристическое
свойство) и об электропровод-
ности железного провода опре-
деленного размера (свойство
изделия). Промежуточное поло-
жение занимают важные для
переработки свойства. Здесь
также может проявиться влия-
ние фактора формы.
Отличительное свойство по-
лимерных материалов заклю-
чается в том, что на конкретные
Рис. 1.1. Представление о характери-
стике материала.
характеристики полимеров может оказывать решающее влияние
метод получения и переработки: свойства полимеров оказываются
чувствительными к условиям процесса переработки и, в частности,
в существенной мере к ориентации вещества. Едва ли возможно при-
готовить образцы для определения характеристик материала без того,
чтобы при этом не наложился способ приготовления образца.
Из-за высокой вязкости расплавов полимеров все молекулярные
процессы в значительной степени замедленны. Это обусловливает
низкую теплопроводность и замедленную релаксацию.
Кроме того, упругость полимерного материала является при-
чиной такого его свойства, как «память» к предыстории деформи-
рования. В добавление к этому проявляется и определенная чувст-
вительность полимерных материалов к разложению (термическому,
механическому или химическому) в процессе переработки.
15
ФУНДАМЕНТАЛЬНЫЕ ХАРАКТЕРИСТИКИ
Фундаментальные характеристики материала связаны с химическим
и физическим строением вещества. Можно полагать, что химическое
строение не зависит от условий переработки, если не считаться
с возможностью разложения. Физическое строение всегда практи-
чески полностью определяется предысторией материала. Особенно
это относится к образцу, подвергаемому испытаниям в целях оценки
его механических характеристик, поскольку такой образец прохо-
дит через ряд стадий процесса переработки. Однако можно стандар-
тизовать способы приготовления образца и методы измерения таким
образом, чтобы получить характеристические свойства материала
в сопоставимой форме.
СВОЙСТВА ПРИ ПЕРЕРАБОТКЕ
Основные методы переработки полимерных материалов приведены
на схеме. Практически все полимеры перерабатываются из распла-
вов или довольно концентрированных растворов. В каждом методе
переработки можно выделить четыре стадии, которые зачастую
весьма тесно связаны друг с другом. К ним относятся:
транспортирование материала в формующее устройство перера-
батывающей машины (существенны характеристики переноса);
подготовка материала (главным образом путем нагревания)
к процессу формования (существенны теплофизические характери-
стики);
собственно формование (существенны реологические характе-
ристики);
фиксирование требуемой формы (существенны теплофизические
и реологические характеристики и особенно такие характеристики,
как теплопроводность, скорость кристаллизации и т. д.).
На каждой из перечисленных стадий в течение различных перио-
дов материал подвергается воздействию изменяющихся температур,
внутренних и внешних напряжений. Каждое из подобных воздейст-
вий вносит свой вклад во внутреннюю структуру материала. Именно
такой меняющийся характер условий процесса переработки и затруд-
няет выбор критериев для оценки технологических свойств
материала.
Для получения ответа на вопрос о способности материала к пере-
работке и для преодоления разрыва между данными научно-исследо-
вательских работ (ожидаемое поведение материала) и данными о по-
ведении материала на практике обычно проводят модельные экспе-
рименты, которые стремятся осуществлять в условиях, максимально
приближенных к практически достижимым.
ХАРАКТЕРИСТИКИ ПРОДУКТА ИЛИ ИЗДЕЛИЯ
Для продукта или изделия условие «неизменности» может считаться
наиболее важным. Это относится к требованиям неизменности формы
(стабильность размеров) и постоянству механических характеристик
16
Основные методы переработки полимера в изделие
2 Заказ 699 17
(ударная вязкость, разрушающее напряжение при растяжении и
усталостная прочность) под воздействием окружающей среды.
Характеристики свойств изделия или продукта можно разделить
на три основные подгруппы (рис. 1.2): эстетические характеристики,
эксплуатационные свойства.
технические показатели и
Рис. 1.2. Представление о харак-
теристиках изделия.
риала, неудовлетворительных
Большинство из них чрезвы-
чайно субъективны и определяются
(зачастую данные исследований от-
сутствуют) комбинациями фундамен-
тальных и дополнительных харак-
теристик. Практически все характе-
ристики изделия или продукта свя-
заны с определением свойств мате-
риала, находящегося в твердом со-
стоянии.
Поскольку все характеристики
изделия определяются выбором ма-
териала, способа переработки и пра7
вильностью области применения,
можно сказать, что не существует
«плохих» материалов самих по себе,
но есть только плохие изделия или
продукты. 1
Плохие изделия появляются в
результате ошибочного выбора мате-
условий переработки, неправиль-
ного применения и, зачастую, плохой конструкции изделия. По-
этому чрезвычайно нужны методы предсказания эксплуатационных
свойств на основе измерения фундаментальных характеристик мате-
риалов и параметров процесса переработки. Данная книга посвя-
щена главным образом фундаментальным, или характеристическим,
свойствам полимерных материалов.
ЛИТЕРАТУРА
1. Brandrup J. and Immergut Е. Н.
(Eds.), «Polymer Handbook», In-
terscience, New York, 1966.
2. Lewis, 0. Criffin, «Physical Con-
stants of Linear Homopolymers»,
Springer, Berlin — New York,
1968. “
3. Reid R. C. and Sherwood Th. K.,
«The Properties of Gases and
Liquids», McGraw-Hill, New York,
Its. ed^ 1958; 2nd ed. 1966.
4. Bondi A., «Physical Properties
of Molecular Crystals, Liquids and
Glasses», Wiley, New York, 1968.
5. Van Krevelen D. W., «Processing
Polymers to Products», Interna-
tional Congress 1966, p. 11—19.
t Raedthuys, Utrecht, 1967.
глава и. ТИПОЛОГИЯ ПОЛИМЕРОВ
Полимеры состоят из больших молекул, которые содержат повторя-
ющиеся химические единицы. Такие химические единицы соединены
в основном в линейные цепи, которые могут быть разветвленными
и взаимосвязанными и образовывать трехмерные сетчатые структуры.
В настоящей книге рассматриваются главным образом синтети-
ческие полимеры с линейными цепями. В принципе, такие полимеры
могут плавиться и растворяться в подходящем растворителе. Сетча-
тые полимеры не плавятся * и не растворяются. Следовательно,
некоторые характеристики полимеров, особенно те, которые отно-
сятся к жидкому состоянию и к растворам, применительно к сетча-
тым полимерам теряют смысл.
Полимеры, длина цепи которых конечна, содержат концевые
группы. Влияние концевых групп на физические характеристики '
полимеров, как правило, незначительно для тех степеней полимери-
зации, какие применяются на практике. »
СТРОЕНИЕ ПОЛИМЕРОВ
ФУНКЦИОНАЛЬНЫЕ ГРУППЫ
Каждую полимерную структуру можно рассматривать как совокуп-
ность функциональных групп. Функциональность какой-либо группы4
можно определить как количество свободных валентностей данной
группы. Длинная цепь может состоять в основном из бифункцио-/
нальных групп. Однако любая бифункциональная группа может
быть также замещена трехфункциональной или тетрафункциональ-
ной группой, которая в свою очередь содержит одну или две моно-
функциональные группы; в итоге снова образуется бифункциональ-
ная, но уже «составная» группа, например группа —СН2— может
- СНз
быть замещена группой —СН— или группой —С—.
I I
СН8 сн3
* Здесь под «плавлением» понимается переход в текучее состояние, так как
многие сетчатые полимеры могут кристаллизоваться, и при соответствующем
повышении температуры кристаллы «плавятся», хотя это и не приводит к те-
чению. — Прим. ред.
2* 19
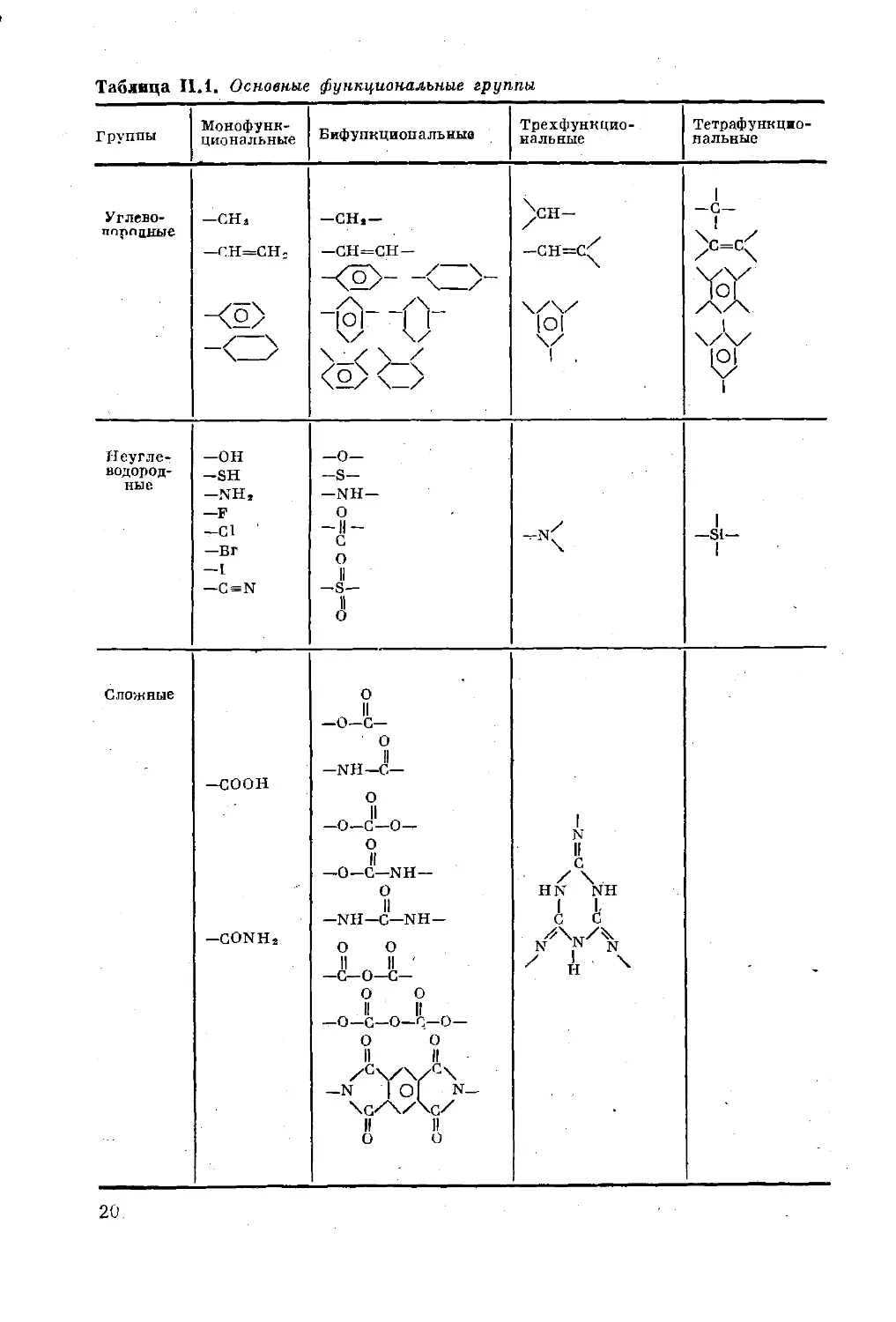
Таблица И.1. Основные функциональные группы
Группы Монофунк- циональные Бифункциональные Трехфункцио- нальные Тетрафункцжо- нальные
Углево- тгпрлцные —СНа —СН=СН2 -CHj- —СН=СН— 1 \/ о п И И II 1 А 1 “ —и— и 1 Z\
— । Y -С 1 Ж X Л Z
\ / 1 /\/ см ОС 1 . ¥ 1
Неугле- водород- ные —он -SH - NH, —F —С1 —В г —1 —G = N —о— -S- —NH— О -II- с о II —S— II о 1 —S1- 1
Сложные -соон —CONH2 О II -о-с- 0 II —NH—С— О II —О—С—О— О II —О—С—NH — О II —NH—С—NH— О О II II ' —С—О—С— о о II II —о—С—о—с—о— О О II II -N I of N- \С'/\/Хо/ II II О О 1 N II С HN \н Ь 'с /у и Х -
20
В табл. II. 1 приведены наиболее важные функциональные
группы.
В ряде случаев удобно рассматривать составную единицу струк-
туры как одну функциональную группу. В частности, иногда жела-
I I
тельно рассматривать группы С1—С—F и ОН—С=О как отдельные
функциональные группы, а не как комбинации —С—с—F,—С1
I
и ^>С=О с —ОН.
ЛИНЕЙНЫЕ ЦЕПНЫЕ ПОЛИМЕРЫ
Линейные цепные полимеры можно разделить на два основных
класса:
1) гомоцепные полимеры,, цепи главных валентностей которых
содержат только углеродные атомы; такие полимеры обычно полу-
чают путем цепной реакции полимеризации;
2) гетероцепные полимеры, в цепь главных валентностей кото-
рых входят различные атомы, первоначально присутствовавшие
в функциональных группах мономеров; такие полимеры обычно полу-
чают путем реакции поликонденсации или реакции ступенчатой поли-
меризации.
Большинство гомоцепных полимеров состоит из следующих
структурных звеньев:
СНз
—СИ— СН2— ИЛИ —i—СН2-
I I
R' R'
где R' — монофункциональная боковая группа, которая может содержать не—
с колько функциональных групп, например —С—О—(СН,)2—СН3.
II
О
Гетероцепные полимеры содержат следующие структурные звенья:
—АВ—R"—
где R’ — бифункциональная углеводородная группа, а —АВ-бифункцио-
нальная группа, образовавшаяся из исходных мономерных функциональных
групп, например — NH—С— из —>NH2 и НО—С—.
КОНФИГУРАЦИЯ И КОНФОРМАЦИЯ
Геометрическое расположение атомов в полимерной цепи может-
быть фиксированным и нефиксированным. Расположение, зафикси-
рованное химическими связями, называется конфигурацией.
21.
Конфигурация цепи не может изменяться без разрыва химиче-
ских связей. Примерами конфигураций могут служить цис- и транс-
изомеры или d- и Z-формы.
Поворот атомов относительно ординарной связи приводит к по-
явлению различных конформаций цепи. В разбавленных растворах
участки макромолекулы находятся в постоянном движении, и она
образует непрерывно изменяющиеся различные конформации; такое
состояние макромолекулы часто называют статистическим клубком.
В твердом состоянии многие полимеры принимают некоторые
типичные конформации, например складчатые цепи и спиральные
структуры. Было показано, что спиральные структуры, образуемые
полипептидами, состоят из двух или даже трех цепей.
КОНФИГУРАЦИИ ПОЛИМЕРНЫХ ЦЕПЕЙ
Регулярное строение поли-а-олефинов исследовал Натта [16], кото-
рый предложил принятую в настоящее время номенклатуру для
«описания стереорегулярных полимеров этого типа (Хаггинс с сотр.
[12]). Различные варианты стереорегулярных структур предста-
влены на рис. II.1
2Рис. II. 1. Плоскостное изображение полимеров винилового ряда (основная
углеродная цепочка представлена в полностью вытянутой конформации плоского
.зигзага; для простоты атомы водорода не показаны):
а„— синдиотактическая; б— изотактическая; в —- атактическая структура.
Рис. II.2. Плоскостное изображение полимеров с двойной углерод—углеродной
«связью:
-а — tfuc-1,4; б — траке-1,4; в — 1,2-изотактическая^ г — 1,2-синдиотактическая структура
22
Полидиены-1,3, содержащие после полимеризации одну двойную
связь в повторяющейся единице, могут быть построены из последо-
вательности звеньев с различными конфигурациями (рис. II.2).
Дополнительные сведения о стереорегулярных конфигурациях
можно найти в работе Коррадини [2].
Стереорегулярность играет весьма важную роль в структуре-
белков, нуклеиновых кислот и других соединений биологического
происхождения.
РАЗВЕТВЛЕННЫЕ ПОЛИМЕРЫ
При поликонденсации бифункциональных мономеров получаются <
линейные полимеры. При ступенчатой полимеризации полимеры/
могут иметь короткие или длинные ответвления, которые присоеди-1
йены случайным образом вдоль цепей. Особенно вероятно развет-'
вление цепей при радикальной полимеризации, а регулирование)
степени разветвленности в этом случае затруднительно. Разветвлен-)
ность изменяет характеристики полимера, находящегося в состоянии)
расплава и в растворе. Наиболее подходящими методами определе-
ния степени разветвленности оказались вискозиметрия, ЯДерный
магнитный резонанс и ИК-спектроскопия (см. также гл. XV).
СОПОЛИМЕРЫ
До сих пор рассматривались гомополимеры, т. е. такие полимеры,,
которые состоят из одинаковых повторяющихся звеньев. Однако-
существуют методы получения полимерных цепей с разными повторя-
Рис. 11,3. Типы сополимеров:
а статистические сополимеры (тип 1); б — чередующиеся сополимеры (тип 2); в — блоч-
ные сополимеры (тип 3); г — привитые сополимеры (тип 4).
ющимися звеньями и различной степенью упорядоченности или даже-
полимеров с цепью главных валентностей одного типа, а ветвями —
другого типа. Такие полимеры получили название сополимеров^
Сополимеры мджно разделить на четыре основные группы (рис. II.3),.
23
СЕТЧАТЫЕ ПОЛИМЕРЫ
Сетчатые полимеры образуются при полимеризации трех- или тетра-
функциональных мономеров.
Наиболее широкое применение нашли сетчатые фенолоформаль-
дегидные полимеры с повторяющейся группировкой
-CH2-R—СН2—
сн2
I
К сетчатым полимерам относятся также ненасыщенные полиэфиры,
полиэпоксиды, полиуретаны и вулканизированные каучуки.
МОЛЕКУЛЯРНЫЙ ВЕС
И МОЛЕКУЛЯРНО-ВЕСОВОЕ
РАСПРЕДЕЛЕНИЕ
Как правило, в полимере содержатся макромолекулы с различными
длинами цепей. Распределение длин цепей определяется некоторой
функцией, вид которой зависит от механизма процесса полимериза-
ции и условий его проведения.
За последние несколько лет стало очевидным, что многие харак-
теристики полимеров в процессе переработки и их свойства, связан-
ные с целевым применением изделий, определяются не только сред-
ним молекулярным весом, но шириной и формой молекулярно-весо-
вого распределения (МБР). Основная причина заключается в том,
что некоторые характеристики, в том числе разрушающее напряже-
ние при растяжении и стойкость к ударным нагрузкам, определяются
главным образом короткими молекулами; другие характеристики,
например вязкость растворов и расплавов при низких напряжениях
сдвига, зависят от макромолекул средней длины; наконец, ряд
характеристик, в том числе эластичность расплавов, резко зависит
-от содержания макромолекул с наиболее длинными цепями.
Можно считать, что молекулярно-весовое распределение охарак-
теризовано достаточно полно в том случае, если определены по мень-
шей мере три различных средних молекулярных веса: среднечисло-
вой (Мп), средневесовой (AfJ и z-средний (М2) молекулярные веса.
Эти молекулярные веса определяются по формулам
Ni= W/N
м» NiMi ='£wlMl
где Nt — число молекул с молекулярным весом Mr, N — общее число молекул;
u>i — весовая доля молекул с молекулярным весом Mr, W — общий вес.
:24
Рис. II.4. Распределение по
молекулярным весам для ти-
пичного полимера:
а — среднечисловой; б — средневяз-
костный; в — средневееовой; а—
z-средний.
фактором в определении
На рис. IL4 представлена кривая распределения по молекуляр-
ным весам. Характеристическими показателями являются Q =
= MJMn и Q' = Равенство Q = Q' = 1 соответствует
полностью однородному, или монодисперсному, полимеру. Большое
значение Q указывает на наличие низкомолекулярного полимера,
большое значение Q' свидетельствует о полимере с весьма большими
молекулярными весами. Значение Q может изменяться на практике
от 1,5—2,0 до 20—50 (меньшие значения Q соответствуют поли-
мерам, полученным поликонденсацией, более высокие — полимерам,
полученным радикальной полимеризацией).
Большинство термодинамических характеристик полимера опре-
деляется среднечисловым молекулярным весом. Многие такие харак-
теристики можно описать уравнением
типа
Х = ХОО-А/М„ . (II.1)
где X — рассматриваемая характеристика;
Хсо — асимптотическое значение этой харак-
теристики при очень больших молекулярных
весах; А — постоянная.
Многие характеристики подобного
рода, в том числе плотность, удельная
теплоемкость, показатель преломления
и т. п., достигают своих предельных
значенийХт уже при таких молекуляр-
ных весах, которые лежат ниже реаль-
ных значений молекулярных весов поли-
меров, Для таких характеристик кон-
фигурация только одного структур-
ного элемента оказывается решающим
свойства вещества.
Однако типичные механические характеристики, например раз
рушающее напряжение при растяжении, очень резко изменяются
ё зависимости от молекулярного веса в области значений молекуляр
ных весов реальных полимеров. Поскольку уравнение (II.1) приме
нимо к таким характеристикам и определяющим является средне
числовой молекулярный вес, основным фактором здесь оказывается
число концов цепей.
Характеристики, связанные с большими деформациями, напри-
мер вязкость расплава и раствора, полностью определяются средне
весовым молекулярным весом, т. е. той массой, которая должнг
быть перенесена. В этом случае ярко выраженное влияние на свой-
ства оказывают наличие разветвленности и степень поперечного
сшивания.
Типичные вязкоупругие характеристики, например высокоэла-
стичность расплава, определяются z-средним молекулярным весом.
•25
Ниже приведены наиболее важные методы определения средних
молекулярных весов:
Анализ концевых групп
Осмотическое давление
Понижение давления пара
Эбуллиометрия или криоскопия
Светорассеяние
Ультрацентрифугирование
Вязкость раствора
Хроматография на проницаемом геле
Мп
Мп
Мп
Мп
Ма>
мг
Мо Мш
Мп, Mnoi Мп
(полное распре-
деление по мо-
лекулярным
весам)
Более подробные сведения можно найти в специальных руковод-
ствах.
Три средних значения молекулярных весов характеризуют
распределение, но не дают более подробной информации. Полная
кривая молекулярно-весового распределения может быть получена
методом хроматографии на проницаемом геле.
Хроматография на проницаемом геле позволяет разделить моле-
кулы в соответствии с их размерами. Такой метод разделения осуще-
ствляется на хроматографической колонке, в которой в качестве
неподвижной фазы использован набухший в растворителе полимер-
ный гель с различными размерами пор; степень проницаемости
набухшего полимерного геля изменяется на много порядков. В про-
цессе прохождения жидкой фазы, содержащей полимер, сквозь
гель макромолекулы диффундируют внутрь тех частиц, которые не
создают механических препятствий диффузии молекул. Меньшие
молекулы проникают в гель более глубоко и удерживаются в порах
в течение более длительного времени по сравнению с более крупными
молекулами, которые проходят через колонку быстрее. Такой хро-
матограф калибруется по узкой фракции с известным молекуляр-
ным весом (молекулярный вес такой фракции определяется каким-
либо абсолютным методом).
ПЕРЕХОДЫ В ПОЛИМЕРАХ.
КЛАССИФИКАЦИЯ ПОЛИМЕРОВ НА ОСНОВЕ
МЕХАНИЧЕСКОГО ПОВЕДЕНИЯ
Невозможно понять природу характеристик полимеров без знания
тех типов переходов, которые осуществляются в полимерных мате-
риалах. Практически все характеристики полимеров определяются
этими переходами и температурами, при которых они осуществля-
ются.
26
ЛИНЕЙНЫЕ ТЕРМОПЛАСТИЧНЫЕ ПОЛИМЕРЫ
При охлаждении расплава полимера движение молекул затормажи-
вается. Если полимер не способен к кристаллизации, то при ком-
натной температуре он может стать либо твердым, жестким и хруп-
ким, либо мягким и гибким. Если же в процессе охлаждения рас-
плава происходит кристаллизация, то материал может стать твер-
дым, жестким и прочным.
Основным фактором, определяющим возможность кристаллиза-
ции полимера, является наличие последовательно расположенных
звеньев в цепи, конфигурация которых регулярна с геометрической
точки зрения. Если звенья цепи малы и идентичны друг другу, как,
например, в линейном полиэтилене, то кристаллизация происходит
быстро. Если же звенья цепи содержат объемные группы, например
полистирол, то полимер сможет закристаллизоваться лишь тогда,
когда такие группы расположатся в определенном геометрическом
порядке.
Характеристики аморфных полимеров в значительной мере
определяются степенью ограниченности молекулярного движения
их цепей. При охлаждении расплава полимер претерпевает переход,
напоминающий термодинамический переход второго рода *, так
называемый переход в стеклообразное состояние. Когда полимер
при комнатной температуре находится выше области перехода в сте-
клообразное состояние, то он ведет себя как мягкое и гибкое тело.
Если же полимер по температурной шкале находится ниже области
перехода в стеклообразное состояние, то он оказывается твердым,
и жестким и довольно часто — хрупким и прозрачным. Хрупкость
возникает в связи с ограниченностью молекулярного движения.
Высококристаллические полимеры, как правило, оказываются'
прочными и упругими. Полимеры с ограниченной степенью кристал-,
личности занимают промежуточное положение. Ниже температуры
стеклования аморфного материала эти полимеры — обычно твердые,
жесткие и довольно хрупкие вещества; в области между точкой
стеклования аморфного материала и точкой плавления кристаллов,
эти полимеры, как правило, упругие.
Измерить температуры переходов можно при помощи ряда экс-
периментальных методов.
Температуры стеклования обычно определяют по зависимости
удельного объема образца от температуры. В области перехода
в стеклообразное состояние наклон графика зависимости удельного
объема от температуры изменяется весьма резко. Аналогичные ре-
зультаты можно получить, если построить график температурной
зависимости теплосодержания или показателя преломления. В обла-
сти перехода в стеклообразное состояние также резко изменяются
теплоемкость, термический коэффициент линейного расширения
и модуль упругости.
* В действительности, это релаксационный переход, температура которого
определяется кинетическими факторами. — Прим. ред.
27
Точно таким же образом измеряют вторичные переходы в преде-
лах стеклообразного состояния. Влияние вторичных переходов на
изменение свойств полимера выражено значительно слабее, чем
влияние стеклования. В ряде случаев весьма полезными оказыва-
ются исследования механических или диэлектрических потерь *
и измерения ядерного магнитного резонанса в широком диапазоне
изменения температур. Основной переход в стеклообразное состоя-
ние происходит в том случае, когда сегменты цепи главных валент-
ностей макромолекул получают свободу движения; вторичные же
переходы осуществляются при температурах, допускающих свободу
движений или колебаний малых участков цепи или боковых ответ-
влений макромолекул. Отсюда очевидно, что температуры вторич-
ных переходов лежат ниже, чем температуры основного перехода.
Теоретически точка плавления кристаллического образца — это
максимальная температура, при которой могут существовать поли-
мерные кристаллы. Как правило, кристаллиты в полимерном образце
плавятся в определенном температурном интервале, а не при одной
температуре. Точку плавления можно измерить с помощью разных
методов. На кривой зависимости удельного объема от темпера-
туры наблюдается довольно резкое скачкообразное повышение
объема при температуре плавления. Следовательно, для определения
этой температуры удобно использовать дилатометрический метод.
Другие переходы осуществляются тогда, когда изменяется харак-
тер кристаллической структуры материала. Такие переходы, так же
как и плавление, являются термодинамическими переходами пер-
вого рода.
Из методов, позволяющих обнаружить переход в кристалличе-
’ ское состояние, можно отметить следующие: измерения дифракции
рентгеновских лучей, калориметрические измерения (в частности,
дифференциальный термический анализ) и измерения оптического
двойного лучепреломления в зависимости от температуры. В ряде
случаев точку плавления можно определить путем измерения меха-
нических характеристик.
’ Существуют вполне определенные соотношения между строением
полимера и температурами переходов. Наиболее существенным об-
стоятельством в этом отношении является гибкость полимерной
цепи, а также ее регулярность. В последующих главах книги будут
подробно рассмотрены соответствующие соотношения.
Температурная зависимость свойств линейных термопластичных
полимеров в общем случае полностью определяется положением
температур стеклования и кристаллизации. Вторичные переходы
могут играть существенную роль, несколько изменяя механические
* Измерение температурных зависимостей диэлектрических или механиче-
ских потерь при различных частотах является основным методом определения
положения температур вторичных переходов и их энергий активации. Темпера-
туре перехода отвечает максимум потерь. В то же время такие свойства, как тер-
мический коэффициент расширения, практически нечувствительны к этим пере-
ходам. — Прим. ред.
28
характеристики материала. Например, вторичный переход может •
оказаться ответственным за то, что жесткий материал становится
упругим, а не хрупким.
Рис. II.5. Изменение некоторых характеристик полимера при
температурах перехода.
На рис. II.5 схематически показано влияние температур основ-
ных переходов на некоторые физические величины.
ЛИНЕЙНЫЕ НЕТЕРМОПЛАСТИЧНЫЕ ПОЛИМЕРЫ
Некоторые полимеры, в частности целлюлоза, хоть и имеют линей-
ное строение, но обладают столь сильным межмолекулярным взаимо-
действием (главным образом за счет водородных связей и наличия
29
полярных групп), что не размягчаются и не плавятся. Следовательно,
температуры переходов имеют меньшее значение для полимеров
этого класса. Часто такие полимеры обладают высокой степенью
кристалличности, и точка плавления кристаллитов намного превы-
шает температуру разложения.
Физические характеристики подобных полимеров, да исключе-
нием плавления, соответствуют обычным параметрам кристалличе-
ских полимеров.
Некоторые полимерные материалы подвергаются пластификации
водой вследствие того, что вода оказывает сильное влияние на меж-
молекулярные взаимодействия. Эти полимеры, следовательно, можно
назвать гидропластами (в противоположность термопластам). При-
сутствие 'влаги в полимере может существенно понизить темпера-
туру перехода в стеклообразное состояние.
ПОЛИМЕРЫ С ПОПЕРЕЧНЫМИ СВЯЗЯМИ
Если в аморфном полимере образуются поперечные связи, то про-
исходит весьма существенное изменение основных его характеристик.
В некоторых отношениях поведение аморфного полимера с высокой
плотностью поперечных связей сходно с поведением полимера высо-
кой степени кристалличности. Кстати, кристаллизацию можно рас-
сматривать как способ образования физических поперечных связей.
Влияние температуры перехода в стеклообразное состояние на свой-
ства материала становится менее выраженным по мере увеличения
плотности поперечных связей.
Таблица П.2. Классификация полимеров по механическим показателям [J43]
Класс * Общее описание Рабочие температуры Степень кристаллич- ности Плотность поперечных связей
I. Расплавы Упруговязкие жид- кости Т>Т& 0 0
II. Эластомеры Мягкие и гибкие каучукообразные твердые тела т>тя 0 Низкая
III. Волокниты Жесткие твердые тела '1' <Z.'l m (Т>Те) 20-50 0
IV. Армированные эластомеры Твердые и гибкие тела T>Tg (T<Tm) 0 Промежу- точная
V. Реактопласты Прочные жесткие тела T<Tg 0 0
Прочные мягкие тела T<Tm От средней до высокой 0
VI. Реактоэлас- ты Твердые тела T<Tg 0 От средней до высокой
* Автор употребляет следующие Теркины, которые обычно не встречаются в техни-
ческой литературе: I — molllplaats; П —mollleiasts; III — fibroplasts; IV — libroplats;
V — duroplaflte; VI — duroplasts. — Дрим. ped.
30
КЛАССИФИКАЦИЯ ПОЛИМЕРОВ
ПО ИХ МЕХАНИЧЕСКИМ СВОЙСТВАМ
На основании общего описания поведения полимеров, рассмотрен-
ного в предыдущих разделах, можно предложить классификацию
для их практического применения. Такая классификация приве-
дена в табл. II.2. Здесь использована терминология, предложенная
Лейшем [14].
СТРОЕНИЕ КРИСТАЛЛИЧЕСКИХ
ПОЛИМЕРОВ ' - “
Одной из первых моделей, которая была применена для описания
характеристик частично кристаллических полимеров, является мо-
дель «бахромчатых мицелл». Согласно этой модели предполагалось,
что в образце сосуществуют зоны кристаллитов и аморфные участки.
Рис. II.6. Модель бахромчатых мицелл.
Рис. II.7. Модель паракристалли
ческого тела.
Это должно обеспечивать полностью упорядоченное состояние поли-
мерных цепей на расстояниях, соответствующих размерам кристал-
литов. Одновременно эти же самые полимерные цепи содержат раз-
упорйдоченные сегменты, принадлежащие аморфным областям
(рис. II.6).
С помощью модели бахромчатых мицелл и соотношения между
фракциями кристаллических и аморфных областей весьма просто
объяснить понятие степени кристалличности. На основании этой
модели были найдены многие удачные корреляции между строением
й свойствами полимерных тел.
Полученные в последнее время данные привели к необходимости
пересмотра концепции твердого состояния полимерных материалов.
Наиболее существенным было открытие и получение полимерных
монокристаллов (Шлезингер [18], Келлер [13]). Раньше считали,
что монокристаллы нельзя получить из растворов полимеров,
31
поскольку макромолекулы в растворах перепутаны. С 1953 г. были
описаны монокристаллы для столь большого числа полимеров, что
возможность образования монокристаллов, вероятно, имеет совер-
шенно общий характер. Такие монокристаллы представляют собой
пластины (ламелярные структуры) толщиной приблизительно 100 А,
в которых молекулы находятся в упорядоченном состоянии. Об этом
свидетельствуют данные дифракции электронов. Однако в таких
полимерных монокристаллах были обнаружены дислокации, совер-
шенно аналогичные дислокациям в металлах и кристаллах низко-
молекулярных веществ.
Вторым важным обстоятельством, приведшим к пересмотру поня-
тия о твердом состоянии полимеров, было создание Хоземанном
[10] теории, позволившей объяснить наблюдаемый характер диф-
ракции рентгеновских лучей. Сущность этой теории составило пред-
ставление о статистической разупорядоченности. Основой теории
является модель паракристаллического состояния (рис. II.7). По-
этому для объяснения характеристик полимеров уже не требуется
вводить представление об аморфной фазе. Различные явления,
например ползучесть, вторичная кристаллизация и прочностные
свойства образцов, лучше объясняются перемещениями дислокаций,
как обычно в физике твердого тела, а не моделью бахромчатых
мицелл.
Эти два новых понятия — ламелярность в сочетании со структур-
ной упорядоченностью, образуемой сложенными цепными молеку-
лами, а также паракристалличность — играют определяющую роль
в современных представлениях о морфологии кристаллических
полимерных материалов.
В большинстве твердых кристаллических полимеров с помощью
поляризационного микроскопа определяются по своему характер-
ному виду сферические структурные агрегаты — сферолиты. Элект-
ронная микроскопия поверхностей излома в сферолитах позволила
показать, что и здесь по всему телу сферолитов обнаруживаются
ламелярные структуры. Образование сферолитов, вероятно, явля-
ется нормальным следствием роста кристаллов из центра кристал-
лизации (довольно часто таким центром может быть инрродная
частица) в условиях избыточного содержания некристаллического
расплава.
В современной концепции строения кристаллических полимеров
остается лишь одна возможность применения модели бахромчатых
мицелл, а именно для полимеров с низкой степенью кристаллич-
ности. Для полимеров с промежуточной степенью кристалличности
наиболее вероятной представляется структура, содержащая «пара-
кристаллы» и дискретные аморфные зоны. Для полимеров высокой
степени кристалличности отсутствуют экспериментальные данные
относительно существования в них дискретных аморфных участков.
В этом случае модель бахромчатых мицелл должна быть исключена,
а для объяснения поведения таких образцов следует принять пара-
кристаллическую модель.
32
Сейчас становится все более очевидным, что полимерные моле-
кулы являются «нормальными» и только их цепное строение оказы-
вается «необычным» свойством и накладывает различные ограничения
на интерпретацию поведения макромолекул. Однако именно цепное
строение и обусловливает наличие новых характеристик полимер-
ных материалов.
В настоящей книге очень часто используется понятие «степень
кристалличности», хотя оно может быть достаточно точно определено
с помощью модели бахромчатых мицелл. Однако это понятие пред-
ставляет собой полезный показатель степени статистической упоря-
доченности также и в случае паракристалличности.
А
ЛИТЕРАТУРА
1. Billmeyer F. W., «Textbook of
Polymer Science», Interscience,
New York, 1962.
2. Corradini P., in «The Stereoche-
mistry of Macromolecules», A. D.
Ketley ed.; Marcel Dekker, New
York, volume 3, 1968.
3. Flory P. J., «Principles of Poly-
mer Chemistry», Cornell Univer-
sity Press, Ithaca, N. Y., 1953.
4. Holzmiiller W. and Altenburg K.,
«Physik der Kunststoffe», Akade-
mie Verlag, Berlin, 1961.
5. Ke B. (Ed.), «Newer Methods
of Polymer Characterization», In-
terscience, New York, 1964.
6. Miller M. L., «The Structure
of Polymers», Reinhold, New
York, 1966.
7. Rodriguez F., «Principles of Po-
lymer Systems», McGraw-Hill,
New York, 1970.
8. Staudinger H., «Die Hochmole-
kularen organischen Verbindun-
gen», Springer, Berlin, 1932. «Ar-
beitserinnerungen», Huttig Ver-
lag, Heidelberg, 1961.
9. Hermann K., Gerngross 0. and
Abitz W., Z. physik. Chem. BIO
(1930) 371.
10. Hosemann R., Z. Physik 128
(1950) 1, 465.
11. Hosemann R. and Bonart R.,
Kolloid-Z. 152 (1957) 53.
12. Huggins M. L., Natta G., Des-
reux V. and MarkH., J. Polymer
Sci. 56 (1962) 153.
13. Keller A., Phil. Mag. (8) 2 (1957)
1171.
14. Leuchs 0., «The Classifying of
High Polymers», Butterworth,
London, 1968.
15. McGrew F. C., J. Chem. Educa-
tion 35 (1958) 178.
16. Natta G. et al., J. Am. Chem.
Soc. 77 (1955) 1708.
17. Natta G. and Corradini P., J.
Polymer Sci. 20 (1956) 251; 39
(1959) 29.
18. Schlesinger W. and Leeper H. M.,
J. Polymer Sci. 11 (1953) 203.
3 Заказ 699
глава ш. ТИПОЛОГИЯ КОЛИЧЕСТВЕННЫХ
ХАРАКТЕРИСТИК
БЕЗРАЗМЕРНЫЕ ГРУППЫ ВЕЛИЧИН
Практическая значимость безразмерных групп величин была изве-
стна уже давно. Еще в 1873 г. Гельмгольц получил безразмерные
группы величин, которые теперь известны как числа Рейнольдса
и Фруда; однако первым дал названия этим числам Вебер в 1919 г.
127].
Наиболее существенными оказываются безразмерные группы
величин, которые связаны с переносом массы, энергии и количества
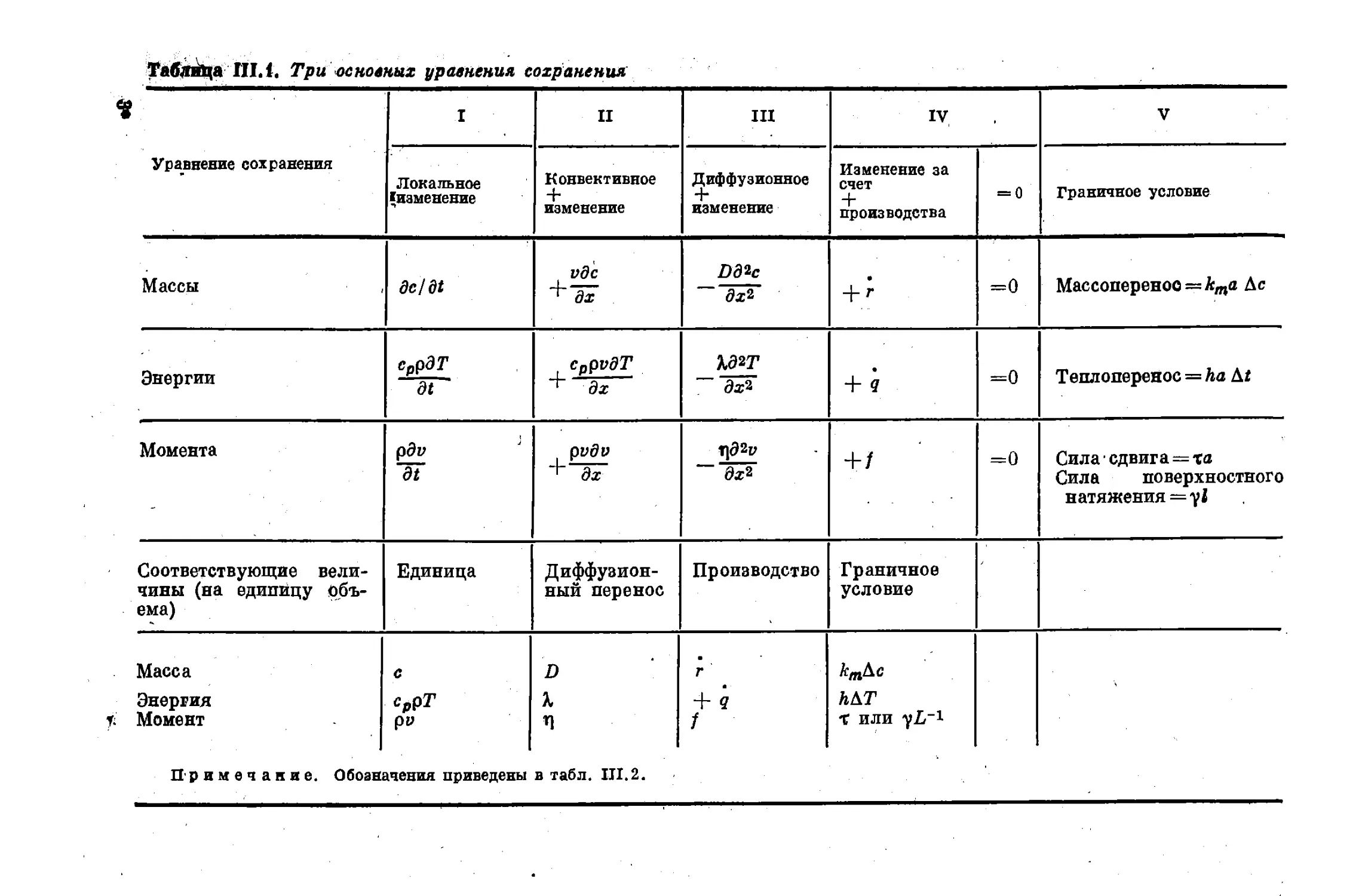
движения. В табл. III.1 приведены три основных уравнения сохра-
нения в их простейшей форме (одномерный случай). Полную систему
чисел можно получить путем образования отношений различных чле-
нов этих трех уравнений, как предположили Клинкенберг и Моой
[И]. Такая система чисел приведена в табл. III.2.
Из данной системы можно получить и другие числа, использовав
отношения чисел ряда массы и ряда энергии или количества движе-
ния в соответствующей колонке таблицы:
ЛГВр/ЛГРе = Х/сРР2? = ЛГЬе (Ее—Льюис)
Лгво/Лгне = 11/Рп = лгзс (Sc—Шмидт)
лгРе/ЛГНв==вР1,1Л=ЛгРг (Рс—Прандтль)
Другие числа можно^получить в виде отношений сил (/):
Сила тяжести
Градиент давления —?
Сила тяжести
Сила вязкого трения — Р®
Сила сдвига
— _-т / Е*
Сила упругости
Сила вязкого трения
Поверхностное натяжение =tlp/Y
Стандартное обозначение чисел — Nxy, где подстрочный] индекс
Ку представляет собой двухбуквенное сокращение фамилии того
ученого, именем которого названо данное число.
34
Таблица III Л. Три основных уравнения сохранения
I II III IV V
Уравнение сохранения Локальное {изменение Конвективное + изменение Диффузионное изменение Изменение за счет + производства = 0 Граничное условие
Массы de/dt идс + дх Рд^с ~ дх^ +/• =0 Массопереноо = кта &с
Энергии СррдТ dt , CppvdT г дх ~ дх* + я =0 Теилоиеренос = ha At
Момента pdv ~dt pvdv Г]Э2Р “ ~д^ +/ =0 Сила сдвига = та Сила поверхностного натяжения = у!
Соответствующие вели- чины (на единицу объ- ема) Единица Диффузион- ный перенос Производство Граничное условие
Масса С D Г /стДс
Энергия Момент СррТ рн П + 3 / ЛДТ X или
Примечание. Обозначения приведены в табл. III.2.
Таблица Ш.2. Система безразмерных групп («чисел»)
Отношение членов уравнений табл. Ш.1 III : I IV: I V : I II : III IV : II V : П IV : III V : III IV : V
Масса Dt £2 rt c kfni L vL D Bo rL I c Dall km V Me r£2 De Da II kmL D Sh rL k,nc
Энергия и cppL2 — qt cppT ht cppL CpPvL „ X — cppTV IPAlllI h St Cppv qL2 । xr lDaIV hL X Nu qL hT
Момент nt pL2 it pv ' xt pvL pvL n Re fL pp2 We T pp2 Fa M2 Y\U Po Bm fL T
Обозначения: а — поверхность единицы объема; с —^концентрация; Ср — удельная теплоемкость; D — коэффициент диффузии;
е — электрический заряд; Е — модуль упругости; /эл — электрическое поле единицы объема; g —ускорение силы тяжести; h — коэффи-
циент теплопереноса; h — константа скорости реакции; hm — коэффициент массопереноса; I— длина на единицу объема; L — характе-
ристическая длина; р—давление; t — время; Г — температура; о —скорость; а —линейная координата;
-у—поверхностное натяжение*, ц— вязкость; X— коэффициент теплопроводности; р—плотность; т — напряжение сдвига; <в — угловая
частота;
г—скорость реакции на единицу объема (для реакции первого порядка г = Хс; для реакции второго порядка г = кс2 ит. д.):
q — скорость образования тепла в единице объема; /—сила на единицу объема (сила тяжести / = gp; центробежная сила / = <о2Ер; градиент
давления f = ^p!L', упругая сила / = Е/Е; сила поверхностного натяжения /=у/Ь2; электрическая сила / = е/эл).
Числа: Вт — Бингама; Во — Боденштейна; Da — Дамкелера; Fa — Фаннигана; Fo — Фурье; Me — Меркеля; Nu — Нуссельта;
Ре — Пекле; Ро — Пуазейля; Бе — Рейнольдса; Sh — Шервуда; St — Стентона; We — Вебера.
ТИПЫ ХАРАКТЕРИСТИК ПОЛИМЕРОВ
С молекулярной точки зрения характеристики полимеров можно
разделить на три следующие категории:
1. Коллигативные характеристики. Эти характеристики имеют
одно и то же значение при расчете на грамм-молекулу вещества,
независимо от его строения. Следовательно, числовое значение дан-
ной величины, измеренное экспериментально, зависит от числа
грамм-молекул (молекул).
Истинно коллигативными характеристиками обладают только
идеальные газы и идеальные растворы. Примерами могут служить
осмотическое давление, понижение давления пара, повышение темпе-
ратуры кипения, понижение температуры замерзания, т. е. осмоти
ческие характеристики.
2. Аддитивные характеристики. При расчете на грамм-моле-
кулу эти характеристики имеют значение, которое в идеальном слу-
чае равно сумме значений подобных величин, составляющих моле-
кулу атомов. Строго аддитивным оказывается только молекуляр-
ный вес.
В известном приближении другие величины также являются
аддитивными, например мольный объем, мольная теплоемкость,
мольные теплота сгорания и ^теплота обра30вания7"мольная рефрак-
ция и т. д.
3. Конститутивные характеристики. Эти характеристики пол-
ностью определяются строением молекулы, причем не возникает
вопроса относительно коллигативности или аддитивности таких
характеристик. К типичным конститутивным характеристикам отно-
сятся избирательное поглощение света, магнитное резонансное погло-
щение и т. д. Довольно часто подобные характеристики оказываются
«визитной карточкой» данного соединения.
Межмолекулярные и внутримолекулярные взаимодействия ино-
гда весьма сильно влияют на коллигативность и аддитивность
свойств, а зачастую и подчеркивают конститутивные характеристики.
При последующем рассмотрении используются практически только
аддитивные характеристики, а также затрагивается промежуточная
область между аддитивными и конститутивными свойствами.
АДДИТИВНЫЕ МОЛЬНЫЕ ФУНКЦИИ
.Принцип аддитивности представляет собой весьма эффективное
средство в полуэмпирическом подходе к исследованию физических
характеристик вообще и характеристик полимеров в частности,.
Принцип аддитивности означает, что большое количество характе-
ристик, рассчитанных на моль вещества, можно вычислить путем
суммирования вкладов атомов, групп или связей:
F ~ У ntFi
? (III.1)
37
где F — мольная характеристика; щ — число компонентов типа I, вносящих
свой вклад в ату- характеристику; Ft —числовое значение вклада i-ro компо-
нента.
Полимеры оказываются идеальным материалом для применения
принципа аддитивности в связи с тем, что их структура образована
последовательностями простых групп. В общем случае концевые
группы играют незначительную роль. Следовательно, такие мольные
величины можно выражать в расчете на моль структурной единицы.
Понятие аддитивности оказалось чрезвычайно плодотворным
при изучении связи между химическим строением различных веществ
и физическими характеристиками этих веществ. Эффективность
понятия аддитивности распространяется как на индивидуальные
соединения, так и на их смеси, даже если такие смеси оказываются
весьма сложными, например смеси типа минеральных масел [23].
В ряде случаев заметное расхождение между числовыми значе-
ниями, рассчитанными с помощью принципа аддитивности, и значе-
ниями, полученными в эксперименте, оказывается чрезвычайно важ-
ным способом обнаружения эффектов, связанных с особенностями
строения вещества.
Основываясь на различиях природы структурных элементов,
можно указать три аддитивных метода.
7. Применение атомных вкладов. При условии полной аддитив-
ности данная характеристика молекулы может быть рассчитана из
вкладов атомов, составляющих эту молекулу. Такой наиболее про-
стой метод аддитивности обладает, однако, и ограниченной цен-
ностью. Точное сравнение мольных характеристик родственных
соединений позволило показать, что вклады одних и тех же атомов
могут быть до некоторой степени различными в зависимости от при-
роды окружения этих атомов. Это положение заставляет пользо-
ваться вторым методом аддитивности.
2. Применение групповых вкладов. В данном случае небольшие
изменения атомных вкладов, обусловленные природой окружения,
учитываются путем комбинирования таких атомов в наиболее часто
используемые молекулярные группы.
3. Применение связевых вкладов. Можно предложить систему
аддитивности; основанную на учете различных типов связей между
атомами. Подобный метод может быть применен на практике, но
здесь все же должны приниматься во внимание различные величины
вкладов одной и той же связи в зависимости от характера соседних
связей у данного атома.
Для практических целей следует отдать предпочтение методу
групповых вкладов. Дело в том, что метод атомных вкладов сильно
упрощает картину, а метод связевых вкладов приводит к неоправ-
данно большому количеству различных типов связей.
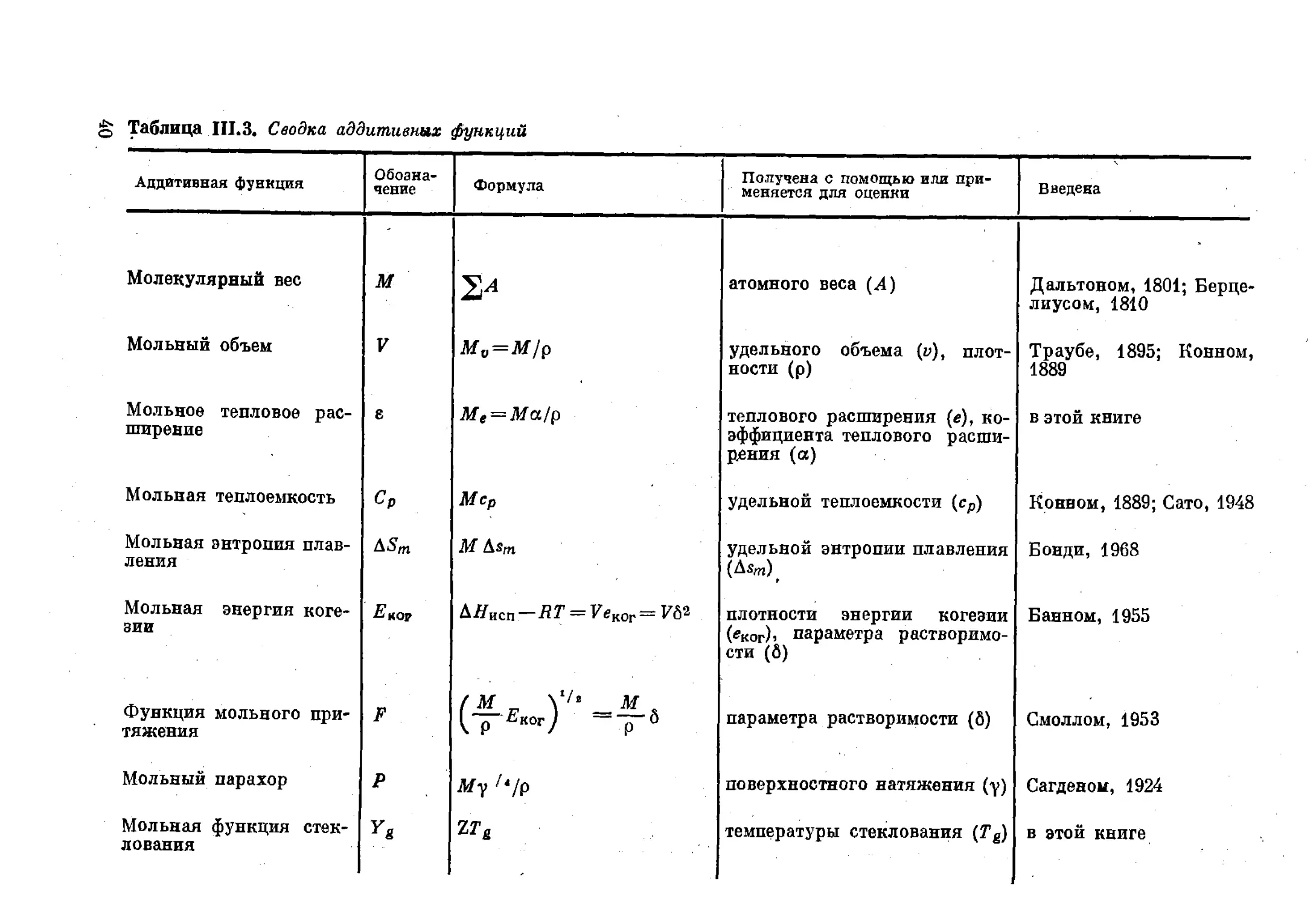
В табл. III.3 приведена сводка наиболее важных аддитивных
мольных функций и их обозначений.
г Если какая-либо мольная характеристика может быть рассчи-
тана на основании принципа аддитивности, то относящаяся к этой
38
характеристике физическая величина может быть найдена исключи-
тельно на основании данных о химическом строении молекулы.
Например, - поверхностное натяжение определяется соотношением
y=(P/V)4 (III.2)
Точность получения такого числового значения, конечно, ограни-
ченна , ПОСКОЛЬКУ аддитивность какого-либо мольного г.плйр.таа ттп-
когда не выполняется абсолютно точно. Вообще говоря, такая точ-
нбсть'достаточна^для практических целей.
Точность~этого расчёта можно "повысить двумя способами, а
именно путем применения «стандартной характеристики» или исполь-
зования «стандартного соединения».
1. Метод стандартных характеристик. Допустим, что необхо-
димая физическая характеристика какого-либо вещества, например
его поверхностное натяжение, неизвестна, но другая характери-
стика этого вещества, например показатель преломления, уже изме-
рена в эксперименте с высокой точностью. В таком случае можно
выбрать последнюю характеристику в качестве стандартной и вос-
пользоваться формулой
/ Р 1 м
V=(^ZT’ п2+2) (Ш.З)
Этот способ обладает двумя достоинствами. Во-первых, уравне-
ние (Ш.З) имеет то преимущество, что в нем нет абсолютного значе-
ния мольного объема V — величины наименее надежной из аддитив-
ных величин. Во-вторых, уравнение (Ш.З) можно легко преобразо-
вать в безразмерную группу
у1^4 Л г г
(д2—1)/(л2+2) P~ = i (III. 4)
что сразу же привносит все достоинства применения безразмерных
выражений.
2. Метод стандартных соединений. Такой метод можно приме-
нить, если неизвестна какая-либо физическая характеристика дан-
ного изучаемого вещества, но эта же самая характеристика уже
точно измерена для родственного соединения. В данном случае
можно принять это родственное соединение в качестве «модели»
жди «стандарта» (символ «О») и воспользоваться правилом:
Y । / Р Vo
<ш-5>
Это уравнение также оказывается безразмерным.
С учетом этих двух уточнений принцип аддитивности получает
еще бойее высокую практическую ценность и позволяет оценивать
физические величины с очень высокой точностью.
Рассмотренные в данной главе групповые методы аддитивности
можно считать частным случаем более общего метода, предложенного
39
g Таблица Ш.З. Сводка аддитивных функций
Аддитивная функция Обозна- чение Формула
Молекулярный вес м
Мольный объем V М„=М/р
Мольное тепловое рас- ширение е Ме = Ма/р
Мольная теплоемкость ср Мер
Мольная энтропия плав- ления Д5т М Esm
Мольная энергия коге- зии Ецог EH^n-RT = VeKOr=V^ (М Х*7’ М .
Функция мольного при- тяжения F 1 р Ек°г) р 6
Мольный парахор Р Му 7‘/р
Мольная функция стек- лования Yg zra
Получена с помощью или при- меняется для оценки Введена
атомного веса (4) Дальтоном, 1801; Берце- лиусом, 1810
удельного объема (v), плот- Траубе, 1895; Конном,
ности (р) 1889
теплового расширения (е), ко- эффициента теплового расши- рения (а) в этой книге
удельной теплоемкости (ср) Конном, 1889; Сато, 1948
удельной энтропии плавления (Д$/п) > Бонди, 1968
плотности энергии когезии («ког)> параметра растворимо- сти (6) Банном, 1955
параметра растворимости (6) Смоллом, 1953
поверхностного натяжения (у) Сагденом, 1924
температуры стеклования (Tg) в этой книге
Мольная функция плав- . ления Гт ZTm
Мольная рефракция Илл М га2 —1 р п2-[-2
Мольная рефракция
Мольная рефракция Мп
Мольная диэлектриче- ская поляризация р М е —1
Р е-|-2
Мольная магнитная восприимчивость X AfX
Мольная функция ско- рости звука (функция Рао) и / М \'/s / 1 + v
^Р“прод V3(l-v)>
Мольная функция ха- рактеристической вяз- кости б ( Me \,/а
К М'!< !
t Мольная свободная энер- гия образования АС/ A(AGz) = Z?Zln^paBH
температуры- плавления кри- сталлического вещества (Тт) в этой книге
показателя преломления (и) Лорентцом, 1880; Ло- ренцом, 1880
показателя преломления (п) Глэдстон-Дейлом, 1858
показателя преломления (и) Фогелем, 1950
диэлектрической проницаемо- сти (е) Дебаем, 1912
диамагнитной восприимчиво- Паскалем, 1923
сти (х)
продольной скорости звука (“прод) коэффициента Пуассона (v) Рао, 1940; Шуйером, 1958
характеристической вязкости ([*11), среднего молекулярного веса (М) Ван Кревеленом, Хоф- тицером, 1967
константы равновесия (Аравн) Франклином, 1949; Ван Кревеленом, Черминым, 1950
Хаггинсом [10]. Метод Хаггинса называется методом аддитив-
ного взаимодействия. При этом предполагается, что ряд характери-
стик жидкости или смеси равен сумме вкладов каждого взаимодей-
ствия между присутствующими в системе группами.
. В наиболее общей формулировке метод Хаггинса предполагает,
что характеристика F может быть рассчитана по соотношению
(П1.6)
1=1/-н
где wij — весовой коэффициент, учитывающий относительное значение контак-
тов между группами i и/; Рц — вклад в величину F, обусловленный контактом
между группами In/; п — число имеющихся в системе групп.
В теории Хаггинса предполагается, что весовые коэффициенты
Wq пропорциональны площади контактирования групп i и /.
Для системы, содержащей группы А и В, уравнение (Ш.6) при-
нимает вид
F = u^aa + u>W,F» + WabFab (III.7)
Если коэффициенты и?;;- выражены в долях числа молей пд и пв
nA ' nB 2иЛпВ
па+йв ; wbb- Пд+„в । и?аь-Пд+Ив
и при этом предполагается, что
11
РаЬ= ~2’Faa-{-~2'Fifb -
то уравнение (III.7) приводится к виду
F = njJ?aa + пЪ?ЬЬ (III.8)
Уравнение (II 1.8) представляет собой уравнение метода группо-
вой аддитивности для системы с двумя группами.
ЛИТЕРАТУРА
1. Bondi A., «Physical Properties
of Molecular Crystals, Liquids
and Glasses», Wiley, New York,
1968.
2. Bridgman P. IV., «Dimensional
Analysis», Yale University Press,
New Haven, 1931.
3. Catchpole J. P. and Fulford G.,
«Dimensional Groups», Ind. Eng.
Chem. 58 (1966) 46 and 60 (1968)
71.
4. Ехпет O., «Additive Physical
Properties», Collection Czecho-
slov. Chem. Commun. Vol. 31 and
32.
5. Langhaar H. L., «Dimensional
Analysis and Theory of Models»,
Wiley, New York, 1951.
6. Bunn C. W., J. Polymer Sci.
16 (1955) 323.
7. Debye P., Phys. Z. 13 (1912) 97,
8. Franklin J. L., Ind. Eng. Chem.
41 (1949) 1070.
9. Gladstone J. H. and Dale T. P.,
Trans. Roy. Soc. (London) A 148
(1858) 887; A 153 (1863) 317.
10. Huggins M. L., J. Paint Technol.
41 (1969) 509; J, Phys. Chem.
74 (1970) 371.
42
11. Klinkenberg A. and Mooy H. H.,
Ned T. Natuurk. 10 (1943) 29;
Chem. Eng. Progr. 44 (1948) 17.
12. Lorentz H. A., Wied; Aim. Phys.
9 (1880) 641.
13. Lorenz L, V., Wied. Ann. Phys.
11 (1880) 70.
14. Maxwell J. Cl., «А Treatise
on Electricity and Magnetism»,
Oxford, 1873.
15. Pascal P., Rev. Gen. Sci. 34
(1923) 388.
16. Rao R., Indian J. Phys. 14
[(1940) 109.
17. Satoh S., J. Sci. Research Inst.
(Tokyo) 43 (1948) 79.
18. Schuyer J., Nature 181 (1958)
' 1394; J. Polymer Sci. 36 (1959)
1475.
19. Small P. A., J. Appl. Chem. 3
(1953) 71.
20. Sugden S., J. Chem. Soc. 125.
(1924) 1177; «The Parachor and
Valency», London, 1930.
21. Traube J., Her. dtsch. Chem.
Ges. 28 (1895) 2722.
22. Van Krevelen D. W. and Cher-
min H. A. G., Ingenieur 38 (1950)
Ch. T. 1; Chem. Eng. Sci. 1
(1951) 66.
23. Van Krevelen D. W., «Coal»,
Elsevier, Amsterdam, 1961.
24. Van Krevelen. D. W. and Hofty-
zer P. J., J. Appl. Polymer Sci.
11 (1967) 1409.
25. Van Nes K. and Van Westen H. A.
«Aspects of the Constitution of
Mineral Oils»,' Elsevier, Amster-
dam, 1951.
26. Vogel A., Chem. and Ind. (1950)
358; (1951) 376; (1952) 514; (1953)
19; (1954) 1045.
27. Weber M., Jahrb. Schiffbautechn.
Ges. 20 (1919) 355.
ЧАСТЬ
ВТОРАЯ
ТЕПЛОФИЗИЧЕСКИЕ
СВОЙСТВА ПОЛИМЕРОВ
ГЛАВА IV.
ОБЪЕМНЫЕ ХАРАКТЕРИСТИКИ
Объемные характеристики чрезвычайно важны практически для
каждого явления или процесса. К основным объемным характери-
стикам относятся следующие:
1) удельные и мольные объемы и плотности — величины обрат-
ные удельным объемам; эти величины различны в стеклообразном,
высокоэластическом и кристаллическом состояниях;
2) удельный и мольный коэффициенты теплового расширения,
которые также зависят от физического состояния полимера;
3) удельное и мольное расширение при плавлении для кристал-
лических полимеров.
В настоящей главе показано, что мольные объемные характери-
стики можно рассчитывать с достаточной точностью на основании
аддитивных групповых вкладов. Кроме того, обнаруживается инте-
ресная корреляция этих величин с ван-дер-ваальсовским объемом.
ОБЪЕМ И ПЛОТНОСТЬ
Удельный объем и обратную ему величину — плотность — можно
рассматривать как наиболее существенные характеристики поли-
мерных материалов. Это важно как для практически применяемых
полимерных материалов, так и с теоретической точки зрения. Плот-
ность необходимо знать и для расчетов ряда других характеристик,
например термодинамических параметров. Наряду с этим, плотность
используется для оценки полимерных материалов в пределах данной
группы полимеров, например плотность теснейшим образом связана
со степенью кристалличности образца. Наконец, плотность полимера
можно легко определить экспериментально.
В связи со сказанным довольно удивительно, что нет точных и
надежных сводок экспериментальных данных о плотностях поли-
меров. В этой области есть лишь несколько литературных обзоров.
Следовательно, важен метод оценки плотности в зависимости от
строения полимеров. Такой метод был предложен Ван Кревеленом
и Хофтицером [33]. Сравнительные методы для предсказания плот-
ностей органических жидкостей были известны ранее. Эти методы
основаны на расчетах мольного объема путем суммирования груп-
повых вкладов структурных элементов.
44
ОПРЕДЕЛЕНИЯ
Ниже будут использованы следующие определения:
удельный объем (объем единицы массы)
г = 1/р (см3/г)
обычно в таблицах приводят плотности при определенных указан-
ных температурах, например при 20 или 25 °C;
мольный объем, представляющий собой произведение удельного
объема на массу 1 моль:
На практике применяют следующие мольные объемы:
мольный объем при определенной стандартной температуре,
например при комнатной V (298);
мольный объем при нулевой температуре или Vc0, который
представляет собой мольный объем наиболее стабильной конден-
сированной фазы (кристалла) при температуре 0 К;
ван-дер-ваальсовский объем Vw, ограниченный электронными
облаками молекул.
МОЛЬНЫЕ ОБЪЕМЫ ОРГАНИЧЕСКИХ ЖИДКОСТЕЙ
ПРИ КОМНАТНОЙ ТЕМПЕРАТУРЕ
Мольный объем при комнатной температуре — это одна из первых
физических величин, для которой был предложен метод групповых
вкладов. Методы атомных вкладов были развиты Ле Ба в 1915 г.
и Траубе [32]. Наиболее отчетливое различие между подходами этих
двух авторов заключается в том, что Траубе прибавлял к сумме
атомных вкладов для данного соединения дополнительную величину,
названную «остаточным объемом» (Й)
(298) =2 Vi (298)+ Q (IV.1)
i
Наличие остаточного объема подтвердили и другие исследователи.
Поскольку Ле Ба не учитывал этот эффект, его значения мольного
объема, рассчитанные но атомным вкладам, всегда больше по срав-
нению с найденными Траубе, и такие величины обладают незначи-
тельной практической ценностью. Однако до сих пор метод Ле Ба
описывается в руководствах.
Недавно Дэвис и Готлиб [12], а также Харрисон [15] усовер-
шенствовали метод Траубе, поскольку атомный вклад данного эле-
мента не- является постоянной величиной, а зависит от природы
окружающих атомов. Это обстоятельство приводит к существенному
увеличению количества величин «атомных» вкладов. Потому-то
45
и следует предпочесть метод групповых вкладов методу расчета по
атомным вкладам.
В последующих разделах будет применен только метод группо-
вых вкладов. Те литературные данные, Которые были найдены по
методу атомных вкладов, будут переведены в величины, соответ-
ствующие групповым вкладам.
Наиболее убедительные подтверждения аддитивности мольного
объема были получены при изучении гомологических рядов. Исследо-
вания нескольких рядов соединений с увеличивающимся числом
СЩ-групп позволило получить довольно точные значения вклада
втой группы в мольный объем. Ниже приведены данные, получен-
Таблица IV. 1. Групповые вклады в мольный объем органических жидкостей
при комнатной температуре (в смя/моль)
Группы Исследователи
Траубе, 1895 1 Куртц, 1941 Ли, 1956 Хаггинс, 1954 ДЭВИС, 1963 Экснер, 1967 Райнек, 1968
сн2- 16,1 16,3 16,5 16,5 16,6 16,6 16,5
-СН(СНв)- 32,2 32,6 — 32,3 33,2 33,5
- С(СН3)2- 48,3 48,8 z 47,6 49,8 49,7
—СН=СН- 24,3 26,4 26,5 25,0
—СН=С(СН3)- 40,4 42,6 — 40,3 40,6 —
—СвНю— 77,2 78,9 78,5 —-
- СН(СвНц) - 93,3 95,2 97,4 94,1 95,1 94,5
- СН(СБН9)- 77,2 82,0 82,6 80,3
—СН(СН=СН2)- 40,4 42,6 43,9 40,6 43,7 43,5-
—свн4— 58,6 60,3 — 61,8
-СН(СвН5)- 74,7 76,6 78,8 — 77,4 75,8 74,5
—О— 5,5 — —— 6,8 6,7
—со— 15,4 — — 10,2 8,5
—соо— 20,9 — — 23,9 15,5 19,5 16,5
—СН(ОН)— 18,4 — 15,8 14,5 11,4 8,2
—СН(СНО)- 31,5 — — 29,6 27,6 26 3 25,5
—СН(СООН)- 31,9 — 30,7 31,7 30,1 28,4 26,5
—S — 15,5 — 12,2 10,8
—CH(SH)- 31,6 — 31,7 30,0 27,0
—CONH— 20,0 . 17,5
—CH(CN)— 24,4 27,2 23,8
—CH(NH2)- — — —- 21,8 18,8 18,5
—CH(NO2)- — 30,1 25,7
-CHF— 18,5 — 17,8 16,3
—CHC1— 26,2 — 26,6 24,1 23,5
—CHBr— 26,2 — — 30,1 27,4
—CHI- 26,2 — ' — 37,3 — 34,1 —
* Коэффициент перевода в СИ: 1 см‘/моль = 10-« м’/кмоль.
46
ные рядом исследователей (среднее значение составляет 16,45 см3/моль
при стандартном отклонении 0,2 см3/моль):
Вклад группы СН3 в
Мольный объем,
Исследователи (см*/моль
Траубе, 1895 16,1
Курц и Липкин, 1941 16,3
Ван Нес и Ван Вестен, 1951 16,5
Сймха и Хадден, 1956 16,5
Ли, 1956 16,5
Хаггинс, 1958 16,5
Татевский, 1961 16,1
Дэвис и Готтлиб, 1963 16,6
Харрисон, 1965, 1966 16,4
Экснер, 1967 16,6
Райнек и- Лин, 1968 16,5
Для других групп вклады, полученные различными исследова-
телями, отличаются друг от друга значительно больше. Это следует
из данных табл. IV. 1, в которой приведены опубликованные значе-
ния вкладов для различных групп. Все результаты выражены в фор-
ме, соответствующей двухвалентным группам, поскольку ряд авто-
ров не делает различия между вкладами одновалентных концевых
групп и упомянутым выше остаточным объемом.
Вклады различных углеводородных групп известны достаточно
точно и могут быть предсказаны с ошибкой около 1 см3/моль. Для
других групп ошибка изменяется в диапазоне от 2 до 4 см3/моль.
Таким образом, следует принять, что мольный объем органических
жидкостей можно предсказать в лучшем случае с ошибкой до не-
скольких процентов.
- МОЛЬНЫЕ ОБЪЕМЫ ВЫСОКОЭЛАСТИЧЕСКИХ
АМОРФНЫХ ПОЛИМЕРОВ
Высокоэластическое состояние аморфных полимеров весьма сходно
с жидким состоянием органических соединений. Таким образом,
можно ожидать, что мольный объем полимеров, отнесенный к струк-
турному звену, в этом состоянии будет предсказываться на основа-
нии величин групповых вкладов (см. табл. IV.1):
F/ (298) = F/ (298) (IV. la)
' i
Молекулярный вес полимеров достаточно высок и, следовательно,
остаточным объемом Q [уравнение (IV.1)] можно пренебречь.
При комнатной температуре эти аморфные полимеры находятся
в высокоэластическом состоянии, температура стеклования которых
лежит^ ниже 25 °C. В табл. IV.2 приведены литературные данные
о плотностях полимеров этого класса. Для каждого полимера моль-
ный объем V, рассчитывался по измеренному значению его плот-
ности. Такие значения Vr сравнивались со значениями, рассчитанными
47
Таблица IV.2. Мольный объем каучукоподобных аморфных полимеров при 25 °C
Полимер Рг, г/см’ V . СМ*/МОЛЬ
опыт расчет
Полиэтилен 0,855 32,8 32,9
Полипропилен 0,85 49,5 49,1
Полибутилен 0,86 65,2 65,8
Полипентен 0,85 82,5 82,0
Полигексен 0,86 97,9 98,5
Полиизобутилен 0,86 66,8 66,8
Поли-5-фенилпентен 1,05 139,2 140,3
Поливинилиденхлорид 1,66 58,4 58,0
Политетрафторэтилен 2,00 50,0 49,5
Полиметилакрилат 1,22 70,6 70,1
Полиэтилакрилат 1,12 89,4 86,6
П олибутилметакрил ат 1,053 135,0 137,2
Полигексилметакрилат 1,007 169,1 170,1
Поли-2-этилбутилметакрилат 1,040 163,8 169,8
Полй-1-метилпентилметакрилат 1,013 168,1 169,8
Полиоктилметакрилат 0,971 204,2 203,0
Полидодецилметакрилат 0,929 273,8 268,8
Поливинилпропионат 1,02 98,1 90,2
Полиизопропилвиниловый эфир 0,924 93,2 90,3
Полибутилвиниловый эфир 0,927 108,0 107,0
Поли-втор-бутилвиниловый эфир 0,924 108,3 106,7
Полиизобутилвиниловый эфир 0,93 107,6 106,7
Полипентилвиниловый эфир 0,918 124,4 123,4
Полигексилвиниловый эфир 0,925 138,6 139,9
Полиоктилвиниловый эфир 0,914 171,0 172,8
Поли-2-этилгексилвиниловый эфир 0,904 172,9 172,5
Полидецилвиниловый эфир 0,883 208,7 205,7
Полидодецилвиниловый эфир 0,892 238,1 238,6
Полибутадиен 0,892 60,7 60,7
Поли-2-метилбутадиен 0,91 74,8 75,7
Полипентадиен 0,89 76,5 75,7
Полихлорбут адиен 1,243 71,2 71,3
Полиформальдегид 1.25 24,0 25,0
Полиоксиэтилен 1,13 38,9 41,4
Полиокситетраметилен 0,98 73,5 74,3
Полиацетальдегид 1,071 41,2 41,2
Полиоксипропилен 1,00 58,1 57,6
Поли-3,3-бис-хлорметилоксациклобутан 1,386 111,8 115,9
ПолиВинилметилсульфид 1,18 62,8 64,1
Поливинилбутилсульфид 0,98 118,6 113,5
по уравнению (IV. 1а) на основании групповых вкладов
(табл. IV.1). Погрешность подобного расчета, дающего, правда,
разумное соответствие между экспериментальными и рассчитанными
вначениями, все же слишком велика и не позволяет предсказать
плотность полимеров, имеющую какой-либо практический смысл.
Поэтому были получены новые значения групповых вкладов на
основании имеющихся данных по каучукоподобным аморфным поли-
мерам. Эти значения приведены ниже:
48
Труппы Vr, см’/моль V[, см’/моль
Двухвалентные
-СН2- 16,45 16,1-16,6
- СН(СН3) - 32,65 32,2—33,5
- С(СН3)2- 50,35 47,6—49,8
—СН=СН— 27,75 24,3—26,5
-СН=С(СН3)- 42,8 40,3-42.6
-свн4- 61,4- 58,6—61,8
-СН(СвН5)- 74,5 74,5-78,8
—О— 8,5 5,5—6,8
—СОО —(общего типа) 24,6 15,5—23,9
—СОО — (акриловая) 21,0 —
—S— 15,0 10,8—15,5
—CHF— 19,85 16,3-18,5
—СНС1— Ч етырехвалентная 1 28,25 23,5—26,6
1 —С— 1 4,75
1 Тр ехвалентные 1
—сн— 9,85
1 —сн=с— Одновалентные 20,0
- СНз 22,8
-свн5 64,65
—F 10,0
-С1 18,4
С* помощью этих значений были вычислены значения Vr (298),
представленные в табл. IV.2. Среднее расхождение между рассчи-
танными и экспериментальными значениями Vr составляет 1,5%.
В силу рассмотренных выше причин такое сравнение ограничи-
вается двухвалентными группами. Соответствие между обоими
рядами этих значений достаточно хорошее, кроме значений для
кислорода. В последнем случае вклад группы в величину Vr оказы-
вается много больше по сравнению с литературными данными. Это
заставляет применять две различные величины вклада группы СОО.
Меньшая величина используется для «акриловой» группы СОО,
т. е. группы, углеродный атом которой непосредственно присоеди-
нен к цепи главных валентностей.
Для удобства сложные двухвалентные группы, представленные
выше, были разбиты на ряд четырех-, трех- и одновалентных груп-
пировок. Однако такая классификация достаточно произвольна.
МОЛЬНЫЕ ОБЪЕМЫ СТЕКЛООБРАЗНЫХ
АМОРФНЫХ ПОЛИМЕРОВ
Аморфные полимеры, температура стеклования которых лежит
.выше 25 °C, находятся при комнатной температуре в стеклообразном
-Состоянии, В табл. IV. 3, приведены литературные данные о плотно-
стях таких полимеров. Из приведенных значений плотности могут
Заказ 699 49
Таблица IV.3, Мольный объем стеклообразных аморфных полимеров при 25 °C
Полимер р, г/см’ Vg, см1/моль
опыт расчет
Поли-4-метилпентен 0,84 100,2 98,4
Полистирол 1,05 99,0 98,0
Поли-а-метилстирол 1,065 111,0 117,1
Поли-п-метилстирол 1,04 113,7 114,7
П оли-п-трет-бутилстирол 0,95 168,7 167,1
П оливинилхлорид 1,385 45,1 45,2
Полихлортрифтбрэтилен 1,92 60,7 61,8
П оли-3,3,3-трифторпропилен 1,58 60,8 62,6
Прли-третп-бутилакрилат 1,00 128,2 119,9
Полиметилметакрилат 1,17 85,6 86,5
Полиэтилметакрилат 1,119 102,0 102,4
Полипропилметакрилат 1,08 118,7 118,2
Полииэопропилметакрилат 1,033 124,1 119,9
Поли-втпор-бутилметакрилат 1,052 135,2 135,7
Поли-третп-бутилметакрилат 1,022 139,1 138,9
Полииэопентилметакрилат 1,032 151,4 151,6
П оли-1 -метилбутилметакрил ат 1,030 151,7 151,6
Полинеопентилметакрилат 0,993 157,3 154,8
Поли-1,3-диметилбутилметакрил ат 1,005 169,5 169,1
Поли-3,3-диметилбутилметакрилат 1,001 170,1 170,6
Поли-1,2,2-тримети л пр опилметакри л ат 0,991 171,9 172,3 •
Полибензи лметакри л ат 1,179 149,5 152,0
Поли-1-фенилэтилметакрилат 1,129 168,5 168,6
Полидифенилметилметакрилат 1,168 216,0 217,5
Поли-1,2-дифенилэтилметакрилат 1,147 232,2 233,3
Поли-2-хлорэтилметакрилат 1,32 112,6 114,2
Поли-3,3,3-трифтор-1-метилэтилметакрил ат 1,34 134,4 133,3
Поливинилацетат 1,19 72,4 72,2
Найлон 6 1,084 104,4 104,2
Найлон 8 1,04 135,8 135,9
Найлон 11 1,01 181,5 183,4
Найлон 12 0,99 199,3 199,3
Найлон 6,6 1,07 211,5 208,3
Найлон 6,10 1,04 271,5 271,7
Полигликолевая кислота 1,60 36,3 38,9
Полиэтилентерефталат 1,33 144,5 143,2
Полиэтиленфтал ат 1,338 143,6 143,2
Полиэтиленизофталат 1,335 144,0 143,2
Политетраметиленизофталат 1,268 173,7 174,9
Поли-З-фенилпропен 1,046 113,0 113,9
Полифенилметакрилат 1,21 134,0 135,3
Полиэтилхлоракрилат 1,39 96,8 98,4
Полипропилхлоракрилат 1,30 114,3 114,2
Полиизопропилхлоракрилат 1,27 117,0 ' 115,9
Полибутилхлоракрилат 1,24 131,1 130,1
Поли-втор-бутилхлоракрилат 1,24 131,1 131,7
Поливинилхлорацетат. . 1,45 ' 83,1 84,1
Политриметилацетолактон 1,097 91,2 91,3
Поли-о-метилстирол 1,027 115,1 114,7
П оли-1-о-хлорфенилэтилметакрилат 1,269 177,1 181,4
Поли-л-трифтормети л стирол 1,32 130,5 128,1
Поли-п-циклогексилфенилмет акрилат 1,115 219,1 218,8
50
Продолжение табл. IV.3
Полимер р, г/см* У^, см"/моль
ОПЫТ расчет
Поливинилциклогексан 0,95 116,0 116,0
Полициклогексилметакрилат 1,10 152,9 153,3
Полициклогексилхлоракрилат 1,25 151,0 149,3
Полициклогексилендиметилентерефталат 1,19 230,5 231,0
Поли-4-фтор-2-трифторметилстирол 1,43 132,9 133,0
Полидиметилоксифенилен 1,07g 111,7 114,1
Полидифенилоксифенилен 1,14 214,3 211,7
Поли-4,4-метилендифениленкарбонат 1,24 182,4 178,3
Полй-4,4-изопропилендифениленкарбонат 1,20 211,9 214,8
Политиодифениленкарбонат 1,355 180,2 180,2
Поливиниловый спирт 1,26 35,0 35,0
Поливинилметилкетон 1,12 62,6 62,6
Полиакрилонитрил 1,184 44,8 44,8
Полиметакрйлонитрил 1,10 61,0 639
Полиметилцианоакрилат 1,304 82,5 82,1
быть рассчитаны мольные объемы каждого полимера, отнесенные
к одному структурному звену.
Как и предполагалось, мольные объемы стеклообразных полиме-
ров отличаются от значений, рассчитанных для высокоэластического
состояния по методу групповых вкладов (см. с. 52). Поэтому на осно-
вании литературных данных были рассчитаны групповые вклады
специально для стеклообразного состояния. В табл. IV.4 эти вели-
чины сравниваются с групповыми вкладами в Vr. Оба типа вкладов
для одной и той же группы оказываются всегда примерно одина-
ковыми, однако есть и существенные различия. Как и в случае кау-
чукоподобных полимеров, здесь оказалось необходимым пользо-
ваться специальной величиной вклада для «акриловой» СОО-группы.
В табл. IV.3 сравниваются экспериментальные знйчения~“ Vg
с величинами, рассчитанными на основании групповых вкладов
табл. IV.4. Среднее расхождение между экспериментальными и рас-
считанными значениями составляет^1,2%.
МОЛЬНЫЕ ОБЪЕМЫ ПОЛУКРИСТАЛЛИЧЕСКИХ
ПОЛИМЕРОВ
Для небольшой группы полимеров известны плотности как чисто
аморфного, так и чисто кристаллического состояний. Эти данные
собраны в. табл. IV.5. Отношение рс/ра изменяется довольно заметно;
среднее значение этого отношения составляет 1,13. Этих данных
явно недостаточно, чтобы провести разграничение между высоко-
эластическим и стеклообразным аморфным состоянием таких поли-
меров. Однако очевидно, что pc/pr > p£/pg.
4* 51
Таблица IV.4. Групповые вклады в Ve, Vr и Vw, см3/моль
Группы vi Vr vw vs/v«,
Двухвалентные
—СН2— 15,85 16,45 10,23 1,55
- СН(СН3) - 33,35 32,65 20,45 1,63
-С(СН3)2- 52,4 50,35 30,67 1,71
-С6Н4- 65,5 61,4 43,32 1,51
-СН(СвН5)- 82,15 74,5 52,62 1,56
- СвН3(СН3)- 83,4 — 54,47 1,53
—СзН2(СНз)2— 104,1 —. 65,62 1,59
—СбНю— 87,8 53,34 1,64
-СН(СвНц)- 100,15 — 63,58 1,58
—О— 10,0 8,5 3,7 (5,8) 2,70 (1,73)
—со- 13,4 — 11,7 1,14
—СОО— (общего типа) 23,0 24,6 15,2 (17,0) 1,52 (1,35)
—СОО— (акриловая) 18,25 21,0 15,2 1,20
—О—СО—О— 31,4 — 18,9 (23,0) 1,66(1,37)
—СН(ОН)- 19,15 —- 14,82 1,29
—CONH— 24,9 — 19,56 (18,1) 1,28(1,38)
—CH(CN)— 28,95 — 21,48 1,35
—S — 17,8 15,0 10,8 1,19
-CHF— 20,35 19,85 13,0 1,57
—СНС1— 29,35 28,25 19,0 1,54
Четырехвалентные
1 —С— 1 4,6 4,75 — —
1 1 х р и ZJ\ 56,3 — — —
Трехвалентные
1 —СН— 9,45 9,85 — /
59,5 — — —
Одновалентные
- СНз 23,9 22,8 — —
-с6н5 72,7 64,65 — —
- с6ни 90,7 — — —
-ОН 9,7 — —
—CN 19,5 . —- — —
—F 10,9 10,0 — —
—С1 19,9 18,4
52
Таблица 1V.5. Данные для кристаллических полимеров
Полимер рс, г/см* ра, г/см* Рс/Рд
Полиэтилен 1,00 0,85 1,18
Полипропилен 0,95 0,85 1,12
Полибутилен 0,95 0,86 1,10
Полиизобутилен 0,94 0,86 1,09
Полипентен 0,92 0,85 1,08
Полибутадиен 1,01 0,89 1,14
Полиизопрен (цис) 1,00 0,91 1,10
Полиизопрен (транс) 1,05 0,90 1,16
Полиацетилен 1,15 1,00 1,15
Полистирол 1,13 1,05 1,08
Поливинилхлорид 1,52 1,39 1,10
Поливинилиденфторид 2,00 1,74 1,15
Поливинилиденхлорид 1,95 1,66 1,17
Политрифторхлорэтилен 2,19 1,92 1,14
Политетрафторэтилен 2,35 2,00 1,17
Найлон 6 1,23 1,08 1,14
Найлон .6,6 1,24 1,07 1,16
Найлон4 6,10 1,19 1,04 1,14
Полиоксиметилен 1,54 1,25 1,25
Полиоксиэтилен 1,33 1,12 1,19
Полиоксипропилен 1,15 1,00 1,15
Полиокситетр аметилен 1,18 0,98 1,20
Полиэтилентерефталат 1,46 1,33 1,10
Полибисфенолкарбонат 1,31 1,20 1,09
Поливиниловый спирт 1,35 1,26 1,07
Полиметилметакрилат 1,23 1,17 1,05
Политриметилацетолактон 1,23 1,08 1,13
Среднее 1,13
Коэффициент перевода в СИ: 1 г/см’вЮ1 кг/м*. *
Для расчета плотности полукристаллических полимеров pslJ
можно воспользоваться следующим весьма приближенным соотно-
шением:
Р«с/Ра — Va/VSc l-f-O,13Xc
(IV.2)
где Хс — степень кристалличности.
ВАН-ДЕР-ВААЛЬСОВСКИЙ ОБЪЕМ
Ван-дер-ваальсовскйй объем молекулы можно определить как про-
странство, занимаемое этой молекулой, в которое не могут проник-
нуть другие молекулы с нормальной тепловой энергией (т. е. энер-
гией, соответствующей обычным температурам). Для возможности
сравнения с другими величинами, рассматриваемыми в этой главе,
йан-дер-ваальсовский объем будет выражаться в см3/моль структур-
ного звена.
53
Для приближенного' расчета принимается, что ван-дер-ваальсов-
ский объем ограничивается наружной поверхностью ряда взаимо-
проникающих сфер. За радиусы этих сфер принимаются (постоян-
ные) атомные радиусы элементов, входящих в данную молекулу,
а расстояния между центрами сфер представляют собой (постоянные)
длины связей.
Вклад данного атома радиуса гг в ван-дер-ваальсовский объем
определяется далее выражением
ДУиг=Аа Г4"яг1 —(г1—а^3)1
2 2 (IV.3)
. —h. Ti । г‘
»1— Т1 2 ~ 2Lt ' 2Lt
где Na — число Авогадро;' п — радиус i-того атома; £,• — длина связи между
рассматриваемым атомом и г-тым атомом.
Согласно такому определению, объемный вклад рассматри-
ваемого атома не является аддитивной величиной, поскольку зна-
чение этого вклада зависит от природы окружающих атомов. Можно
получить набор истинно аддитивных величин, если выразить их
через вклад двухвалентной группы, окруженной двумя СН^груп-
пами.
Ниже приведена сводка вкладов в величину Vw для двухвалент-
ных групп (в см®/моль) по данным Бонди [3, 8] . и Слонимского
с сотр. [26]:
Группа -сн2- Бонди [3, 8] 10,23 Слонимский с сотр. [26] 10,3
- СН(СН8) - 20,45 20,8
—С(СНз)2— 30,67 31,3
—сн=сн— 16,94 18,1
—СН=С(СНз)— 27,16 28,7
-свн4- 43,32 . .45,2
-CH(CeHs)- 52,62 55,7
—СвНю— 53,34 54,5
-СН(СвНц)- 63,58 64,8
—0- 3.7 5,8
—со— 11,7 ч 11,2
—соо— 15,2 17,0
—о—со-о— 18,9 23,0
—СН(ОН)— 14,82 14,8
—CONH- 19,56 18,1
—CH(CN)- 21,48 22,3
—S — 10,8 —
—CHF— 13,0 13,4
-СНС1— 19,0 19,2
-cf2- 15,3 16,6
-CC12- 27,8 28,2
В общем, величины вкладов, приводимые этими Двумя авторами,
удовлетворительно согласуются друг с другом. Наиболее важным
54
исключением является вклад кислорода: для такого вклада Слоним-
ский получил гораздо большее значение, чем Бонди. Возможно,
приводимое Слонимским значение более правильное.
СООТНОШЕНИЯ МЕЖДУ РАЗЛИЧНЫМИ мольными
ОБЪЕМАМИ И ВАН-ДЕР-ВААЛЬСОВСКИМ ОБЪЕМОМ
Весьма возможно существование определенного параллелизма между
значениями ван-дер-ваальсовского объема и реальным мольным
объемом различных полимеров в данном состоянии, поскольку все
эти мольные объемы подчиняются сходйым правилам аддитивности.
Рис. IV.1. Зависимость мольного
объема каучукообразных аморф-
ных полимеров от ван-дер-вааль-
совского объема.
Рис. IV.2. Зависимость мольног»
объема стеклообразных аморфных
полимеров от ван-дер-ваальсовского
объема.
Сказанное иллюстрируется рис. IV.1 — IV.3, на которых приведены
зависимости соответственно Vr, Vg, Vc от Vw по всем имеющимся
в литературе данным.
Зависимости, представленные на. каждом рисунке, могут быть
аппроксимированы прямой линией, проходящей через начало
координат. Наилучшее соответствие наблюдается между Vg и Vw.
Следует заметить, однако, что подобный параллелизм отсутствует
для отдельных групповых вкладов. Это нетрудно видеть из данных
табл. IV.4, в которой сравниваются групповые вклады в Vg и Vw
Очевидно, существенные изменения отношений групповых вкладов
частично компенсируются при комбинировании различных групп
в структурном звене полимера.
55
Были получены следующие средние значения отношения моль-
ых объемов:
V,(298)/7w=l,60; Va (298)/Vw = 1,55; Vc (298)/Vw=l,41 (IV.4)
Значение 1,41 для последнего отношения основано на данных,
приведенных в табл. IV.5. На рис. IV.3 представлены все имеющиеся
данные для кристаллических полимеров, собранные Льюисом [2].
Рис. IV.3. Зависимость мольного объема кристалличе-
ских полимеров От ван-дер-ваальсовского объема.
Этим данным соответствует среднее значение отношения
Vc (298)/Vw = 1,44; сюда же можно отнести и данные для некоторых
полукристаллических полимеров.
Среднее отклонение значений мольных объемов, рассчитанных
с помощью описанных выше отношений, при сравнении с экспери-
ментальными данными составляет примерно 3% для V, и Vs и около
6% для Vc.
56
МОЛЬНЫЙ ОБЪЕМ ПРИ НУЛЕВОЙ
ТЕМПЕРАТУРЕ У° (0)
Тиммеранс [29] и Мэтьюз [21] ввели понятие плотности при нулевой
температуре, основанное на экстраполяции значений плотности
для кристаллических и жидких веществ к температуре 0 К. Сагден
[28] и Бильтц [7] получили аддитивную систему для вычисления
величин V0 (0) по данным о химическом составе. Объем при нулевой
температуре тесно связан с ван-дер-ваальсовским объемом. Согласно
Бонди [9], хорошее приближение дает следующее соотношение]
v° (0)/Vw = Vc(0)/Vw^ 1,3 ' (lV.4a)
Пример 1
Рассчитать плотности аморфного и кристаллического полиэтил ентерефта-
латов.
Решение
Структурное звено полимера:
О О
II II
-O-C-/Q5-C—О—СН2-СН2
Молекулярный вес этого звена равен 192,2. При комнатной температуре аморф-
ный полиэтилентерефталат находится в стеклообразном состоянии. Из табл. IV.4
можно взять следующие групповые вклады:
Группы
2 (—СОО—)
2 (-СН2-)
Vg (298) Vw
65.5 43,32
46,0 30,40
31,7 20,46
143,2 94,18
Следовательно, pg (298) = 192,2/143,2 = 1,34. Эта величина хорошо коррели-
рует с экспериментальным значением pg (298) = 1,33. Отношение Vg (298)/ Vw =
= 143,2/94,18 = 1,52. Эта величина соответствует формуле (IV.4).
Применив соотношение (IV. 2), нетрудно найти, что
р с (298) = 1,13pf (298) = 1,52
Экспериментальное значение рс (298) изменяется в пределах от 1,46 до 1,50.
ТЕПЛОВОЕ РАСШИРЕНИЕ
ОПРЕДЕЛЕНИЯ
Для описания теплового расширения вещества используют различ-
ные обозначения:
1) удельный коэффициент теплового расширения (dvldT)p =
= е[см®/(г-К)];
2) температурный коэффициент плотности (др!дТ)р = д[г/(см3-К);
3) коэффициент теплового расширения ilv-{dvldT}p = а (К-1);
57
4) линейный коэффициент теплового расширения 1/L (дЫдТ)р =
= ₽ (К’1);
5) мольный коэффициент теплового расширения (dVldT)p =
— е [см3/(моль • К) ].
Эти величины связаны между собой следующими соотношениями:
< = —i>2g; е = ая = а/р; а = 30; q = —ep»; (IV 5)
q = — аР = — а/p; e, — Me = aV = aM/p
ФЕНОМЕНОЛОГИЯ
Экспериментальные данные позволяют предположить, что коэф-
фициент теплового расширения стекла имеет тот же порядок вели-
чины, что и коэффициент теплового расширения кристалла, и не
будет большой ошибкой принять, что
или eg<=siee (IV.6)
Строго говоря, коэффициент расширения полимера в твердом
ч состоянии зависит от температуры, постепенно повышаясь с ее ро-
стом (такой подъем заметен лишь при условии, что рассматриваемый
температурный интервал достаточно широк). Однако таким посте-
пенным повышением коэффициента можно пренебречь в сравнении-
с «го резким увеличением при прохождении через область стекло-
вания. Следовательно, зависимость объема от температуры можно
представить в виде двух прямых линий, пересекающихся в точке
перехода.
На практике принято приводить коэффициенты теплового расши-
рения непосредственно ниже (eg) и выше (ez) перехода в стеклооб-
разное состояние.
Эмпирическим путем было показано, что довольно часто вели-
чина q (= —ар= —ер2) практически не зависит от температуры
в весьма Широком диапазоне (например, [9]).
Коэффициент теплового расширения расплава или полимера
в каучукоподобном состоянии всегда выше, чем в стеклообразном
или кристаллическом полимере.
ЭМПИРИЧЕСКИЕ ПРАВИЛА
Ряд интересных соотношений приводится ниже *:
Правило Предложено
«/—5-10-4К-1 Тобольским, I960; Вики, 1962
ot/Tg «=> 0,16 Бойером и Спенсером, 1944
«сТт^О.И Бонди, 1968
(at—ag) 0,115 Симхой и Бойером, 1962
* Вопрос о соотношении разности коэффициентов термического расширения
выше и ниже температуры стеклования и величиной Tg недавно был заново пере-
смотрен Бойером (см. Boyer R. F., J. Macromol. Sci. Phys., 1973, v. B7,
№ 3, p. 487—501). Он нашел, что для 30 полимеров в среднем соблюдается эмпи-
58 /
ТЕОРИЯ
Расширение материала при нагревании определяется внутренними,
главным образом межмолекулярными, силами. Длины связей между
атомами не зависят от температуры. Точно такое же положение
справедливо и для длин связей между сегментами полимерных
цепей. Следовательно, коэффициенты теплового расширения поли-
мерных систем должны быть меньше по сравнению с наблюдаемыми
для низкомолекулярных жидкостей.
Ниже температуры перехода в стеклообразное состояние коэф-
фициент расширения уменьшается еще больше. При прохождении
точки стеклования исчезают структурные изменения, которые вносят
основной вклад в расширение жидкостей.
МОДЕЛЬ СИМХИ — БОЙЕРА
Одной из наиболее значимых, хотя и упрощенных, моделей, позво*
ляющих представить явление теплового расширения, является
мрдель, основанная на представлениях Симхи и Бойера (рис. IV.4).
При охлаждении жидкости ниже
температуры плавления вероятной
кристаллической структуры воз-
можны два случая: жидкость либо
кристаллизуется, либо переходит
в переохлажденное состояние.
Переохлажденное состояние воз-
никнет, если кристаллизация по-
давлена, например, из-за высокой
вязкости среды и отсутствия сим-
метрии строения молекул.
Переохлаждение жидкости мо-
жет происходить до тех пор, пока
не достигается такая темпера-
тура, при которой свободный объем
молекул оказывается слишком ма-
Лым и, следовательно, движения
всей молекулы в целом или ее
сегментов оказываются более не-
возможными. После этого дости-
гается стеклообразное состояние.
этому моменту, — это точка стеклования.
Vg[O
Ve(0
Стекло
1 ।
\Переахм&АЖидкаат
I денная [
^жидкость'
ve(T)
I Нилас та
'9е(П _________
Кристаллическое I кристал-
, piopdoe_merip_лиоации_
пло-
Оае рас-
ширение
расплава
Сбобр4-
ныи
абъец
v.
о
Г,
Рис. I V.4. Модель теплового расти -
рения полимеров, основанная на
представлениях Симхи и Бойера.
Температура, соответствующая
рическое правило ДаТй = 27,5 ккал/г. Однако гораздо-лучпше результаты полу-
чаются, если ввести несколько более сложное соотношение между Да и Те,,
а именно:
AaTfi =15 + 0,047' g
По-видимому, эту формулу следует считать наилучпшм сегодняшним достиже*-
кием в рассматриваемом вопросе. Тем не менее даже этой формуле отвечают
не все полимеры, исключения обычно связаны с особой ролью низкомолекуляр-
ных (вторичных) переходов в полимерах, для которых наблюдаются особенно
Сильные отклонения от приведенной формулы. — Прим. ред.
59
Предполагается, что мольные объемы переохлажденной жидкости
и кристаллического твердого тела при О К равны и, кроме того,
выполняется соотношение (IV.6), т. е. Vf0 = Vfg.
На основании рис. IV.4 для мольных теплоемкостей можно полу-
чить некоторые простые приближенные соотношения. Из рис. IV.4
видно, что
Vt(T)-Vc(0) V/(298)-Vc(0)
------Т------=-------298---~=Ё‘ <1УЛ1)
Аналогичным образом
Ид (Г)—Ид(0) ГС(Г)-УС(ОУ Ус (298) —Ус (0)
— Р Р — 298
Согласно формуле (IV.4), ,Vt (298) = Vr (298) = l,60Vw и
Vc (298) = 1,41 Vw. Если принять далее, что (см. [9]) Vc (0) = 1,3VW,
то получим
<=> 10-10-4yw (iv.lla)
«g «а 4 • 10-4yw (IV. 12a)
Записанные соотношения можно сравнить с экспериментальными
данными. На рис. IV.5 приводится зависимость величин ez и eg
от Vw. Несмотря на значительный разброс точек, соотношения между
указанными величинами можно аппроксимировать двумя пря-
мыми линиями; эти прямые описываются следующими уравнениями:
= 10,0-10-4 Vw (IV.116)
£g = 4,5 • 10-4 vw (IV.126)
Соответствие между формулами (IV.lla), (IV.116), (IV.12a) и
(IV.126) удовлетворительное.
В табл. IV.6 сопоставляются известные из литературы и рассчи-
танные с помощью формул (IV. 116) и (IV.126) величины ez и eg.
Среднее отклонение составляет 8% для ez и 15% для eg.
Простую модель, изображенную на рис. IV.4, можно использо-
вать также для получения соотношений, эквивалентных формулам
(IV.7)—(IV.10).
Если ez = 10,0-10-4Vw, eg = 4,5-10-4Fw, V, (298) = l,607w и
Vg (298) = l,55Vw, получается, что
.ai (298) >=« 10,0 • 10-4/1,60 6,3 • 10-4
ag (298) 4,5 • 10-4/1,55 2,9 • 10'4
и
ai (298) —ag(298)#=<3,4.10-4K-i (lV.7a)
Это соотношение соответствует формуле Тобольского — Вики, но
.с гораздо меньшей величиной коэффициента.
.60
Правило Симхи — Бойера можно получить в предположении, что
парциальный свободный объем Ф/ при Т& есть универсальная по-
•стоянная для всех полимеров. В этом случае
F/(Tg) = Ve(0) + e/T2
Поскольку при Tg величина Vt — Vg, то
Хе/ -е2) Те = Vg (0) - Vc (0) = Vf (0) = Vf (Tg) (I V.lOa)
(e; — eg) Tg _ e/fg____eg^g _ Vf (Tg) _
Vg (Tg) - Vi (Tg) Vg (Tg) - Vg (Tg) -
61
Таблица IV.6. Показатели термического расширения полимерных материалов
Полимер М, г/моль Уцг, см’/моль е-Х104 (опыт), см’/(г-К) ejXlO* (опыт), см’/(г-К) egxi0‘ (опыт), см’/(моль - К)
Полиэтилен 28,0 20,46 2,4/3,6 7,5/9,6 67/101 .
Полипропилен 42,1 30,68 2,2 5,5/9,4 93
Полибутен-1 56,1 40,91 3,8 8,8 214
Полипентен-1 70,1 51 14 —— 9,2 —
Полиизобутилен 56,1 40,90 1,6/2,0 5,6/6,9 90/112
Поли-4-метилпентен-1 84,2 61,36 3,85 7,6 324
Полистирол . 104,1 62,85 1,7/2,7 4,3/6,8 177/281
Полибутадиен 54,1 37,40 — 7,7 ——
Полиизопрен 68,1 47,61 — 6,0/8,3 —
Полихлоропрен 88,5 45,56 — 4,2/5,0 —
Поливинилхлорид 62,5 28,63 1,4/2,! 4,2/5,2 88/131
Поливинилиденфторид 64,0 25,56 — 2,1/4,6 —
Полихлортрифторэтилен 116,5 32,95 1,0/1,5 2,0/3,5 117/175
П олиформал ьдегид 30,0 13,93 1,8 — 54
Полиоксиэтилен 44,1 24,16 — 6,2/6,6 —
Полиокситетраметилен 72,1 44,62 — 6,9 —
Поли ацетальдегид 44,1 24,15 2,1 6,3 93
Полиоксипропилен 58,1 34,38 — 7,0/7,3 —
Полиэпихлоргидрин 92,5 42,56 — 5,6 —
Поливиниловый спирт 44,0 25,05 3,0 — 132
П оливинил ацетат 86,1 45,88 1.8/2.3 5,0/6,0 155/198
Полиметилакрилат 86,1 45,88 1,8/2,7 4,6/5,6 155/232
Полиэтилакри л ат 100,1 56,11 2,8 6,1 280
Полиизопропил акрил ат 1142 6633 2 2/2,6 6Д/63 251/297
Полибутилакрилат 128,2 76,57 2.6 6,0 333
П оли-втпо р-бутил акри- лат . 128,2 76,56 2,75 6,1 353
Поли-2,2-димети л пр о - пилакрилат 142,2 86,78 2,0 6,5 284
Поли-1-этилпропилакри- 142,2 86,79 з.з 5,9 469
Полиметилметакрилат 100,1 56,10 2,3 5,2/5,5 230
Полиэтилметакрилат 114,1 66,33 2,8 5,4 319
Полипропилметакрилат 128,2 76,56 3,15' 5,8 404
Полиизопропилметакри- лат 128,2 76,55 2,0/2,4 6,2 256/308
Полибутилметакрилат 142,2 86,79 — 5,9/6,3 —
Полиизобутилметакри- лат 142,2 86,78 2,2/2,5 5,8/6,1 31з/зьб
Поли-втор-бутилмет- 142,2 86,78 3.4 6,3 483
акрилат Поли-m ретп-бутилмет- 142,2 86,77 2,7 6,9 384
акрилат Полигексил метакрил ат 170,3 107,25 — 6,3/6,6 —
Поли-2-этилбутилмет- 1-70,3 107,24 — 5,8 —
акрилат Полиоктилмет акрил ат 198,4 127,71 — 5,8 —
Полидодецилметакрилат 254,4 168,63 3,8 6,8 967
62
BgXlO* (расчет), см’/(моль - К) BjXlO4 (опыт), см’/(моль- К) вгх10‘ (расчет), см’/(моль - К) о Sg м р, г/см’ йд в сГ
92 210/269 205 3,9/7,2 150 0,87 510/940
138 232/396 307 3,3/7,2 263 0,85 740/1610
184 494 409 5,0 249 0,85 1060
230 645 511 — — _
184 314/388 409 3,6/5,3 198 0,87 620/910
276 640 614 3,75 302 0,84 950
283 168 448/708 417 629 374 2,6/5,1 373 1,06 1030/2020
214 409/565 476 —
205 372/443 456 — — •
129 262/325 286 2,1/3,8 358 1,38 1040/1880
115 134/294 256 — — —
148 233/408 330 0,5/2,5 325 2,03 330/1650
63 — 139
109 273/291 242
201 497 446
109 278 242 4,2 243 1,07 1090
155 407/424- 344. —
192 518 426
113 —. 251
206 413/517 459 2,7/4,2 303 1,19 970/1510
206 ,396/482 459 1,9/3,8 282 1,22 650/1310
252 611 561 3,3 252 1,12 930
298 697/719 663 З/э/4,1 270 1,08 1020/1190
345 769 766 3,4 224 1,08 820
345 782 766 3,35 - 256 1,05 900
391 924 868 4,5 295 1,04 - 1380
391 839 868 2,6 267 1,04 720
252 520/550 561 2,9 387 1,17 1220
298 616 663 2,6 339 1,12 990
345 744 766 2,65 310 1,08 890
345 795 766 3,8/4.2 354 1,04 1400/1540
391 839/896 868
391 825/867 868 33/3,9 320 1,04 1100/1300
391 896 868 2,9 320 1,04 970
390 981 868 4,2 380 1,03 1640
483 1073/1124 1073
483 988 1072 —- — — —
575 1151 1277 __
759 1730 1686 3,0 218 0,93 610
63
Таблица IV.6. Показатели термического расширения полимерных материалов
Полимер • М, г/моль Й О Я Я о £ - е-Х10‘ (опыт), см4/(г. К) е^хЮ* (опыт), см’/(г-К) BgXlO4 (опыт), см’/(моль - К) BgXlO4 (расчет), см’/(моль - К) BjXlO4 (ОПЫТ), см’/(моль- К) в;х104 (расчет), см’/(моль - К) (Я--1)/«ио . ’»01Хгг—1») Е-. Я Tg
. Полиэтилен 28,0 20,46 2,4/3,6 7,5/9,6 67/101 . 92 210/269 205 3,9/7,2 150 0,87 510/940
Полипропилен 42,1 30,68 2,2 5,5/9.4 93 138 232/396 307 3,3/7,2 263 0,85 740/1610
Полибутен-1 56,1 40,91 3,8 8,8 214 184 494 409 5,0 249 0,85 1060
Полипентен-1 70,1 51 14 — 9,2 — 230 645 511
Полиизобутилен 56,1 40,90 1,6/2,0 5,6/6,9 90/112 184 314/388 409 3,6/5,3 198 0,87 620/910
Поли-4-метилпентен-1 84,2 61,36 3,85 7,6 324 276 640 614 3,75 302 0,84 950
Полистирол . 104,1 62,85 1,7/2,7 4,3/6,8 177/281 283 448/708 629 2,6/5,1 373 1,06 1030/2020
Полибутадиен 54,1 37,40 —— 7,7 —— 168 417 374 — —
Полиизопрен 68,1 47,61 — 6,0/8,3 — 214 409/565 476 — — — —.
Полихлоропрен 88,5 45,56 — 4,2/5,0 — 205 372/443 456 — — •
Поливинилхлорид 62,5 28,63 1,4/2,! 4,2/5,2 88/131 129 262/325 286 2,1/3,8 358 1,38 1040/1880
' Поливинилиденфторид 64,0 25,56 — 2,1/4,6 — 115 134/294 256 — —— —
Полихлортрифторэтилен 116,5 32,95 1,0/1,5 2,0/3,5 117/175 148 233/408 330 0,5/2,5 325 2,03 330/1650
П олиформал ьдегид 30,0 13,93 1,8 — 54 63 — 139 — — — —.
Полиоксиэтилен 44,1 24,16 — 6,2/6,6 — 109 273/291 242 — —. —.
Полиокситетраметилен 72,1 44,62 — 6,9 — 201 497 446 — — — —
Поли ацетальдегид 44,1 24,15 2,1 6,3 93 109 278 242 4,2 243 1,07 1090
Полиоксипропилен 58,1 34,38 — 7,0/7,3 — 155 407/424- 344. —. — —
Полиэпихлоргидрин 92,5 42,56 — 5,6 — 192 518 426 — —. —. —
Поливиниловый спирт 44,0 25,05 3,0 — 132 113 251 — ——
Поливинилацетат 86,1 45,88 1,8/2,3 5,0/6,0 155/198 206 413/517 459 2,7/4,2 303 1,19 970/1510
Полиметилакрилат 86,1 45,88 1,8/2,7 4,6/5,6 155/232 206 ,396/482 459 1,9/3,8 282 1,22 650/1310
Полиэтилакри л ат 100,1 56,11 2,8 6,1 280 252 611 561 3,3 252 1,12 930
Полиизопропил акрил ат 1142 6633 2 2/2,6 6Д/63 251/297 298 697/719 663 3,5/4,1 270 1,08 1020/1190
Полибутилакрилат 128,2 76,57 2.6 6,0 333 345 769 766 3,4 224 1,08 820
П оли-emo р-бутилакри- . 128,2 76,56 2,75 6,1 353 345 782 766 3,35 - 256 1,05 900
ЛАТ Поли-2,2-диметил про- 142,2 86,78 2,0 6,5 284 391 924 868 4,5 295 1,04 - 1380
пилакрилат Поли-1-этилпропилакри- 142,2 86,79 з.з 5,9 469 391 839 868 2,6 267 1,04 720
Полиметилметакрилат 100,1 56,10 2,3 5,2/5,5 230 252 520/550 561 2,9 387 1,17 1220
Полиэтилметакрйлат 114,1 66,33 2,8 5,4 319 298 616 663 2,6 339 1,12 990
Полипропилметакрилат 128,2 76,56 3,15- 5,8 404 345 744 766 2,65 310 1,08 890
Полиизопропилметакри- 128,2 76,55 2,0/2,4 6,2 256/308 345 795 766 3,8/4.2 354 1,04 1400/1540
лат Полибутилметакрилат 142,2 86,79 — 5,9/6,3 — 391 839/896 868 — —
Полиизобутилметакри- 142,2 86,78 2,2/2,5 5,8/6,1 313/356 - 391 825/867 '868 3.3/3,9 320 1,04 1100/1300
лат Поли-втор-бутилмет- , 142,2 86,78 3.4 6,3 483 391 896 868 2,9 320 1,04 970
акрилат Поли-m /?етп-бутилмет- 142,2 86,77 2,7 6,9 384 390 981 868 4,2 380 1,03 1640
акрилат Полигексил метакрил ат 170,3 107,25 — 6,3/6,6 — 483 1073/1124 1073 — -— ’ —
Поли-2-этилбутилмет- 170,3 107,24 — 5,8 — 483 988 1072 — — — —
акрилат Полиоктилмет акрил ат 198,4 127,71 — 5,8 — 575 1151 1277 — —.
Полидодецилметакрилат 254,4 168,63 3.8 6,8 967 759 1730 1686 • 3,0 218 0,93 610
62
63
Продолжение табл. IV.6
Полимер М, г/моль Vyp-, см’/моль egX104 (опыт), см*/(г- К) е^хЮ* (опыт), см’/(г-К.) BgXlO4 (опыт), см*/(моль. К)
Поли-2-метоксиэтилмет- 144,2 80,26 5,45
акрилат
Поли-2-пропоксиэтил- 172,2 100,72 — 6,1 —
метак рилат
Полициклогексилмет- 168,2 99,23 2,7 — 454
ПолиэтилеНтерефталат 192,2 94,18 2,2/2,4 6,0/7,4 423/461
Полидекаметилентере- 304,4 176,02 — 5,3 —
Полиэтилен фталат 192,2 94,18 1,7 5,9 327
Полиатиленизофталат 192,2 94,18 2,0 3,8/5,3 384
Полиэтилен-2,6-нафта- 242,2 119,86 — 4,9 —
лат
Полиэтилен-2,7-нафта- 242,2 119,86 —. 5,0 —
лат
По либисфено лкарб онат 254,3 135,71 2,4/2,9 4,8/5,9 610/737
Найлон 6 113,2 70,71 — 5,6 —
Найлон 7 127,2 80,94 3,5 — 445
Найлон 8 141,2 91,17 3,1 — 438
Найлон 9 155,2 101,40 3,6 — 559
Найлон 10 169,3 111,63 3,5 — 593
Найлон 11 183,3 121,86 3,6 — 660
Найлон 12 197,3 132,09 3,8 — 750
Найлон 10,9 324,5 213,03 — 6,6 —
Найлон 10,10 338,5 223,26 .— 6,7 —
Г Коэффициенты перевода в СИ:
1 г/моль = 1 кг/кмоль;
1 см*/моль = 10-’ м’/кмоль:
е^х10‘ (расчет), см’/(моль. К) EjXlO* (ОПЫТ), см’/(моль- К) ejX10‘ (расчет), см’/ (моль. К) О Л ь- u S 4«Z О я р, г/см’ «а Ь 8° L £
361 786 803 — — — —
453 1050 1007 — — — —
447 — 992 — — — —
424 1153/1422 942 3,6/5,2 342 1,33 1640/2360
792 1613 1760 — — — —
424 1134 942 4,2 290 1,34 1630
424 730/1019 942 1,8/3,3 324 1,34 780/1430
539 1187 1199 — .— — —
539 1211 1199 — — — —
611 1220/1500 1357 1,9/3,5 423 1,20 970/1780
318 634 707 — -—
364 — 809 —
410 — 912 — —
456 — 1014 — .—
502 — 1116 . —
548 — 1219 — —
594 — 1323 — .—.
959 2142 2130 i
1005 2268 2233 — — — —
Среднее
1100
1 см’/(г.К)= 10-’ м’/(кг-К); 1 см’/(моль.К)= 10-» м’/(кмоль-К); 1 г/см’ = 10’ кг/м’,
Следовательно, (az — ag) Tg — Ф[ — константа.
В табл. IV.6 данные, рассчитанные по формуле (IV. 10а), сопоста-
влены с имеющимися экспериментальными результатами. Среднее
значение Ф; равно 0,11 при среднем отклонении 17%. Это удовлет-
ворительно согласуется со значением Фр равным 0,115, предложен^
ным Симхой и Бойером *.
Теперь можно подставить в формулу (IV. 10) величину, получен-
ную выше для отношения az/ag = 2,17. Подстановка дает:
aj (1-1/2,17) Т£ = 0,И
arTg = 0,20 ' (!V.8a)
* См. примечание, касающееся недавней работы Бойера на с. 58. — Прим,
ред.
64
Аналогично ас (2,17—1) Tg = 0,11. Если принять, что Тт <=« 1,57^, то
ас7’„1^0.14 (IV. 9а)
Можно воспользоваться формулами (IV. 7а — IV. 10а) для оценки
величин az и ag. Следует предпочесть, однако, (IV. И б) и (IV. 126),
поскольку они обеспечивают меньшие отклонения от экспери-
ментальных данных, чем другие формулы.
РАСШИРЕНИЕ ПРИ ПЛАВЛЕНИИ
в практическом и теоретическом отношениях очень важно увеличе-
ние мольного объема в процессе плавления:
^Vm = vt(Tm)-Vc(Tm)
5 Заказ 699 65
Если принять, что картина, показанная на рис. IV.4, справед-
лива вплоть до точки плавления, то АУт можно рассчитать, поль-
зуясь данными для ра, рс, ez и ес. Если же одна из этих величин
неизвестна, то оценку интересующих величин можно провести по
методам, изложенным в данной главе.
.Для Tg < 298 К
Дvm = Va (298)-Vc (298) + (Тт-298) (gz-ес)
(IV. 13)
.Для Tg > 298 К
AVm = Va (298)-Vс (298) + (TOT-Te) (e/-eJ
(IV.13а)
Другой метод оценки А7т также основан на модели рис. IV.4.
Если принять, что Vt (298) = l,67w и 7С (298) = 1,417^, то •
Д7т=0,19 (Гт/298) Vw
(IV. 14)
В табл. IV.7 сравниваются значения Д7т, рассчитанные по
обоим методам, со значениями, полученными в эксперименте. Един-
ственный вывод, который можно сделать на основании такого срав-
нения, заключается в том, что рассчитанные значения Л7т пра-
вильны .по порядку величины. Расхождения между рассчитанными
и экспериментальными значениями А7т не превышают расхожде-
ний между данными различных авторов.
Таблица IV.7. Рассчитанные и экспериментальные значения Д7т
Полимер AVm, см’/моль Литератур- ный источ- ник
расчет (1) расчет (2) ОПЫТ
Полиэтилен 6,8 5,4 3,1/5,9 [23], [27]
Полипропилен 9,5 8,9 9,6 [6]
Полибутилен 9,4 9,9 8,4 [61
Полииз<?бутилен 8,5 7,2 6,7 [6]
Полистирол 11,5 21,0 11,4 [6]
Поли-4-метилпентен 16,3 20 9,8 [17]
П олитетрафторэтилен 11.8 12 7,3/14,5 [6], [27]
Полиформальдегид 4,0 4,2 3,5/5,1 [14], [27]
Полиоксиэтилен 6,8 5,2 5,3 [6]
Полиэтилентерефталат 25,8 33 11,5 [61
Найлон 6,6 33,8 48 24,8 [6]
Пример 2
Оценить коэффициент расширения расплава полиэтилентерефталата и плот-
ность его при температуре экструзии 277 ВС (550 К).
86
Решение
Применение формул (IV.11 б) и (IV.126) для мольных коэффициентов тепло-
вого расширения дает следующие результаты:
6g = Ёс = 4,5 - 1О’*Vw = 4,5 • 94,18.10-* = 4,2 - 10’2
в/= 10,0 • 10-*^^ = 10,0 • 94,18 • 10-* = 9,4 • 10-2
Таким образом, для = 343 К мольный объем расплава при 550 К составит:
Vi (550) = Vt (298) + eg (Tg -298) + et (550 — T g) =
= 143,2 + 4,2 -10-2.45 + 9,4 • 10’2.207 = 164,3
Это приводит к следующему значению плотности:
Р1 (550) = 192,2/164,3 = 1,17
Эта величина соответствует экспериментальному значению 1,16* полученному
в лаборатории автора. .
Вычисление коэффициентов теплового расширения дает для удельного
коэффициента теплового расширения:
е/= е//М = 9,4 • 10-2/192;2 = 4,8 • 10-*
и
1е = г,/М = 4,2.10-2/192,2 = 2,2-10-*
Средние величины по литературным данным составляют соответственно 6-10“*
и 2,3-10-*, что хорошо согласуется с рассчитанными значениями.
ЛИТЕРАТУРА
1. Brandrup J. and Immergut Е. Н.,
Eds., «Polymer Handbook», New
York, Interscience, 1966.
2. Lewis O., Griffin, «Physical Con-
stants of Linear Homopolymers»,
Springer, Berlin/New York,
1968.
3. Bondi A., «Physical Properties
of Molecular Crystals, Liquids
and Glasses», Wiley, New York,
1968.
4. Bueche F., «Physical Properties
of Polymers», Interscience, New
York, 1962.
5. Tobolsky A. V., «Structure and
Properties os Polymers», Wiley,
New York, 1966.
6. Allen <?., J. Appl.Chem. 14(1964) 1.
7. Biltz A., «Raumchemie der festen
Stoffe», Voss, Leipzig, 1934.
8. Bondi A., J. Phys. Chem. 68
(1964) 441.
9. Bondi A., see General references.
“ (1968a): Chapters 3 and 4; (1968b):
5»
Chapter 14; (1968c): p. 236?'
(1968d): p. 50.
10. Boyer R. F. and Spencer R. S.,
J. Appl. Phys. 15 (1944) 398.
11. Bueche F., see General references
(1962): Chapter 4.
12. Davis H. G. and Gottlieb S.,
Fuel. 42 (1963) 37.
13. Exner O., Collection Czech. Chem.
Comm. 32 (1967) 1.
14. Fortune L. R. and Malcom G. Ni,
J. Phys. Chem, 64 (i960) 934.
15. Harrison E. K., Fuel 44 (1965)
339; ibid. 45 (1966) 397.
16. Huggins M. L., J. Am. Chem.
Soc. 76 (1954) 843; «Physical
Chemistry of High Polymers»,
Wiley, New York, 1958.
17. Kirshenbaum I., J. Polymer Sci.
A3 (1965) 1869.
18. Krause S., Gormley J. J., Ro-
man N., Shetter J. A. ahd Wata-
nabe W. H., J. Polymer Sci.
A3 (1965) 3573.
*- 67
‘19. Kurtz S. S. and Lipkin M. R.,
Ind. Eng. Chem. 33 (1941) 779.
:20. Li K., Arnett R. L., Epstein M. B.,
Ries R. ВBitter L. P., Lynch J .M\
and Rossini F. D., J. Phys. Chem.
60 (1956) 1400.
.21. Mathews A. P., J. Phys. Chem.
20 (1916) 554.
22. Rheineck A. E. and Lin K. F.,
J. Paint Technol. 40 (1968) 611.
23. Robertson R. E., Macromolecules
2 (1969) 250.
:24. Simha R. and Boyer R. F., J.
Chem. Phys. 37 (1962) 1008.
25. Simha R. and Hadden S. T.,
J. Chem. Phys. 25 (1956) 702.
26. Слонимский Г. Л., Аскад-
скийА .А., Китайгородский А.Л.,
Высокомол. соед., 1970, т. 12,
с. 494.
27. Starkweather Н. W. and BoydB.H.,
J. Phys. Chem. 64 (1960) 410.
28. Sugden S., J. Chem. Soc. (1927)
1780 and 1786.
29. Timmermans J., Bull. Soc. Chim.
Belg. 26 (1913) 205.
30. Tatevskii V. M., Benderskii V. A.
and Yarovoi S. S., «Rules and
Methods for Calculating the Phy-
sico-chemical Properties of Paraf-
finic Hydrocarbons», Pergamon
Press, London, 1961.
31. Tobolsky A. V., (1960), cm.
ссылку 5.
32. Traube J., Ber. dtsch. Chem.
Ges. 28 (1895) 2722.
33. Van Krevelen D. W. and Hofty-
zer P. J., J. Appl. Polymer
Sci. 13 (1969) 871.
34. Van Nes K. and Van Westen H. A.,
«Aspects of the Constitution of
Mineral Oils», Elsevier, Amster-
dam, 1951.
ГЛАВА V.
КАЛОРИМЕТРИЧЕСКИЕ
ХАРАКТЕРИСТИКИ
К категории калориметрических характеристик относятся: удель-
ная и мольная теплоемкости, а также скрытые теплоты кристаллиза-
ции или плавления. В настоящей главе показано, что обе группы
этих характеристик могут рассчитываться как аддитивные мольные
величины. Кроме того, с помощью этих параметров можно оценить
мольные энтропию и энтальпию полимеров.
ТЕПЛОЕМКОСТЬ *
ОПРЕДЕЛЕНИЯ
Удельная теплоемкость — это количество теплоты, которое следует
сообщить одному грамму вещества для повышения его температуры
на один градус. Мольная теплоемкость есть удельная теплоемкость,
умноженная на молекулярный вес вещества или на молекулярный
вес структурного звена в полимерных соединениях. Удельные
и мольные теплоемкости можно определять при постоянном объеме
или при постоянном давлении. Сообщенная веществу теплота вызы-
вает изменение внутренней энергии и энтальпии (теплосодержания)
вещества.
Используются следующие величины.
1. Удельная теплоемкость при постоянном объеме (кал/(г-К)]:
° \дт Л
где Q — количество теплоты, сообщенное одному грамму вещества.
2. Удельная теплоемкость при постоянном давлении [кал/(г-К)Ь
* Обширный теоретический и экспериментальный материал по теплоемкости
полимерных материалов содержится в монографии: Вундерлих Б.,
Баур Г. Теплоемкость линейных полимеров. М., «Мир», 1972. 239 с.—
Прим, ред.
9
3. Мольная теплоемкость при постоянном объеме [кал/(моль-К)]:
/ ди \
Со—Мс„— ат )в
где U — внутренняя энергия одного моля' вещества.
4. Мольная теплоемкость при постоянном давлении [кал/(моль • К)]t
где И — энтальпия (теплосодержание) одного моля.
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
Полная зависимость удельной теплоемкости от температуры опи-
сана лишь для ограниченного числа полимеров. Например, на
рис. V.1 приведены некоторые экспериментальные данные для поли-
пропилена [4, 9]. Недавно проведенные Ги и Медиа [5] измерения
Рис. V.1, Мольная теплоемкость по-
липропилена:
1 аморфный; 2 — степень кристаллич-
ности 65%.
Рис. V.2. Схематическое представле-
ние мольной теплоемкости изотакти-
ческого полипропилена:
1 — аморфный; 2 — кристаллический.
позволяют провести экстраполяцию на чисто аморфный и Чисто-
кристаллический материал; в итоге получена схематическая зависи-
мость мольной теплоемкости от температуры (рис. V.2).
В соответствии с этим рисунком кривая для кристаллического
полимера отражает переход от твердого состояния до точки плав-
ления. При Тт значение Ср возрастает до значения, соответству-
ющего жидкому полимеру. Изменение мольной теплоемкости аморф-
ного полимера следует по такой же кривой для твердого состояния
вплоть до температуры перехода в стеклообразное состояние, а за-
тем теплоемкость увеличивается до значения, характерного для
жидкого высокоэластического материала.
В общем случае полимерный образец не является ни полностью
кристаллическим, ни полностью аморфным. Следовательно, в интер-
вале температур от Tg до Тт мольная теплоемкость лежит между
кривыми, построенными для твердого и для жидкого состояний,,
как показано на рис. V.1 для полипропилена со степенью кристал-
70
личности 65%. Это означает, что приведенные в литературе данные
об удельной теплоемкости полимеров следует принимать с осторож-
ностью. Надежные значения теплоемкости можно получить только
по кривой зависимости теплоемкости от температуры, найденной
для ряда образцов. -
Проверка имеющихся в литературе данных показала, что для
всех исследованных полимеров температурные зависимости мольной
теплоемкости для твердого и жидкого состояний можно аппроксими-
ровать прямыми линиями, за исключением твердого состояния при
температурах ниже 150 К. Таким образом, если тангенсы углов
наклона этих прямых известны, то теплоемкость при произвольной
температуре можно приближенно оценить на основании значения
теплоемкости при 298 К. Ниже приведены значения тангенсов
углов наклона температурных зависимостей теплоемкости, отне-
сенной к ее значению при 298 К, для ряда полимеров:
. dCs 1 . Р. ,п. 1 dCl р dT
с» (298) dT с£.(298)
Полиэтилен 3,04 1,03
Полибутилен 3,06 1,36
Полиметилпентен-4 2,98 —
Полиизобутилен 3,30 2,24
Полистирол 3,36 1,19
Полибутадиен 3,13 —
Полиизопрен 2,96 1,81
Поливинилхлорид 2,81 —
П олйвинилацетат 2,92 —
Полиметилметакрилат 2,95 1,47
Полиоксиэтилен 2,55 0,50
Полиокситетр аметилен 2.92 0,98
Полиоксипропилен 2,88 1,40
Полиоксифенилен 2,68 0,94
Полиэтиленсебацинат — 1,24
Полигёксаметиленадипамид 2,98 0,50
П олибисфенолкарбонат 3,18 1,37
Среднее значение тангенса угла наклона
кости твердых полимеров составляет:
прямых для теплоем-
I
с; (298) ’
dT
= 3 • 10-з
при среднем отклонении 5%.
Для жидких полимеров можно воспользоваться аналогичным
выражением, но при гораздо большей величине отклонения. В этом
случае
при среднем отклонении 30%. И все же, если экспериментальных
данных нет, температурную зависимость теплоемкости приходится
71
аппроксимировать с помощью этих средних величин. Следовательно
Ср (Г)
= 0.106+ 0.003Г (V.1)
с‘(Т)
- = 0,64 + 0,0012Г (V.2)
Ср (2Уо)
Константы 0,106 и 0,64 в уравнениях (V.1) и (V.2) не имеют
физического смысла, но они выбраны так, чтобы при Т — 298 К
Ср (Т) = Ср (298).
При помощи уравнений (V.1) и (V.2) можно приближенно найти
удельную теплоемкость в твердом и жидком состояниях при темпе-
ратурах, представляющих практический интерес, на основании зна-
чений теплоемкости при комнатной температуре. Абсолютные зна-
чения Ср (298) и Ср (298) будут обсуждены в следующем разделе.
МОЛЬНАЯ ТЕПЛОЕМКОСТЬ ТВЕРДЫХ
И ЖИДКИХ ПОЛИМЕРОВ ПРИ 25 »С
Надежные значения мольной теплоемкости для твердого и жидкого
состояний имеются только для ограниченного числа полимеров. Это
подчеркивает важность установления соотношений между Ср (298)
и Сj (298) и строением полимеров.
Для соединений низкого молекулярного веса известны соотно-
шения такого рода. Сато [И] предложил метод расчета Ср при 200,
300 и 400 К путем суммирования групповых вкладов. Таким же
методом воспользовался Шоу [12] для оценки С1Р (298), а также
Джонсон [7] для оценки С1Р (293). Однако неизвестно, можно ли
воспользоваться теми же значениями инкрементов для полимеров.
В табл. V.1 представлена сводка групповых вкладов в величину
С), (300) по Сато и в величину С1Р (298) по Шоу. Сато не приводит
значений вкладов наиболее важных групп —СОО—, —CONH—,
—SO2—, —F, тогда как Шоу опускает значения вкладов для —С1,
F и —CONH—. Наиболее вероятные значения вкладов для пере-
численных групп, соответствующие имеющимся экспериментальным
данным, даны в табл. V.1 в скобках. Значение вклада —CONH—
в Ср пока еще установлено весьма неточно.
z В табл. V.2 сравниваются имеющиеся экспериментальные данные
со значениями Ср (298), Ср (200) и С1Р (298), полученными расчет-
ным путем по методам Сато и Шоу. В общем, соответствие между
рассчитанными и экспериментальными значениями достаточно хоро-
шее. Среднее расхождение между экспериментальными и рассчитан-
ными величинами составляет 2% для Csp (298) и 3,5% для С1Р (298).
Значения С1Р, рассчитанные по методу Джонсона, отклоняются от
экспериментальных данных больше, чем рассчитанные по Шоу.
72
Таблица V.1. Групповые вклады в мольную теплоемкость
при 25 °C, кал/(моль • °C)
Группа Cs Ср Cp/R (на атом) Cp/R (на а том)
-СН3 7,38 8,80 0,92 1,10
-СН2- 6,05 7,26 1,01 1,21
1 —сн— 3,72 5,00 0,93 1,25
1 —с— 1,47 1,76 0,74 0,88
=сн2 5,40 5,20 0,90 0,87
=сн— 4,45 5,10 1,11 1,28
1 =с- 2,50 3,80 1,25 1,90
—СН2— (в пятичленном кольце) 4,75 6,3 0,79 1,05
—СН2— (в шестичленном кольце) 4,29 6,3 0,71 1,03
=СН— (ароматическая) 3,68 5,3 0,92 1,33
1 =С— (ароматическая) 2,04 2,9 1,02 1,45
—О- 4,02 8,5 2,01 <4,25
—S — 5,74 10,7 2,37 5,35
—F (5,1) (5,0) 2,55 2,50
-С1 6,46 (9,5) 3,23 4,75
—Вг 6,28 — 3,14 —
-I 5,34 — 2,67 —
—ОН 4,05 10,7 1,01 2,68
—SH 11,16 12,5 2,78 3,12
-nh2 5,00 — 0,83 —
—NH 3,40 (7,6) 0,85 1,90
/N— 4,08 (10,5) 2,04 5,25
—NO, 10,00 — 1,67 —
—CO— 5,so 12,6 1,38 315
—COO— (И) 15,5 1,83 2,58
—COOH (12) 23,6 1,50 2.95
-CONH— (9-13) (21,5) 1,12-1,63 2.68
-so2- (12) — 2,00
—SN (6) — 1,50 —
-© 20,44 29,4 0.94 1,35
18,80 27,0 0,95 1,36 -
15,52 22,2 0,98 1,40
73
Вундерлих и Джонс [15] опубликовали значения групповых вкладов
для расчетов Csp в интервале температур от 50 до 240 К. Если учесть
неточность экстраполяции этих данных к 300 К, то названные груп-
повые вклады соответствуют данным Сато.
Как вцдно из табл. V.2,’значение отношения г = Clp (298)/Ср (298)
в среднем отклоняется от усредненной величины г = 1,32 на 7%.
Эта величина уменьшается с цовышенией температуры, поскольку
Таблица V.2. Рассчитанные и определенные експерименталъно значения тепло
Полимер Твердое
Ср (298), кал/(г-К) Ср (298) (опыт), кал/(моль-К) Ср (298), кал/(моль- К) Ср (200) (опыт), кал/(моль-К) Ср (200), кал/ (моль • К)
Полиэтилен Полипропилен Полибутилен Полиизобутилен Лоли-4-метилпентен Полибутадиен Полиизопрен 4 Полистирол Полиоксиметилен Полиоксиэтилен Полиокситетраметилен Полиоксипропилен Поливинилхлорид Полихлортрифторэтилен Поливпнилиденхлорид Политетрафторэтилен Полихлоропрен Поливиниловый спирт Поливинилацетат - Полиметилметакрилат Полиакрилонитрил Найлон 6 Найлон 6,6 Найлон 6,10 Полиэтплентерефталат Полиэтпленсебацинат Полпбпсфенолкарбонат Полипропиленсульфон Полибутиленсульфон П олигексенсул ьфон Поли-2,6-диметил оксифенилен Алмаз Графит Сера , Кремний 0,37/0,42 0,39/0,42 0,37/0,42 0,40 0,40 0,39 0,38 0,29 0,34 0,30 0,38 0,34 0,23/0,26 0,22 0,23 0,31 0,35 0,33 0,30 035 0,35 0,38 0,27 028 0,28 0,28 0,33 0,30 10.4/11,8 16,5 >20,7 22,4 33,6 21,0 25,7 30,5 10,2 <16,7 28,2 19,7 1^4/16,2 25,0 23,0 13,6 30,2 33,0 15,9 391 79 107 52 72,4 29,4 29,4 49 35,4 1,46 2,06 5,75 . 4,9 12,1 17,15 23,2 22,3 34,3 21,0 26,4 30,2 10,1 16,1 28,2 21,2 16,2 (24,7) 20,4 (23,3) 25,5 13,8 (28,2) (33,3) (15,8) (39 2) (78,5) (102,7) (52,9) (82,5) (68,9) (29,2) (29,2) (47,3) 34,3 1.47 1,47 5,74 8,0 11,5 16,4 15,4 23,8 15 18,5 20,1 6,9 9,9 20,0/21,1 15,4 10,0/11,6 16,6 16,5 9,3 20,9 24 11,5 28,0 38 49,4 21,6 21,6 35,4 25,8 0,56 1,19 7,9 10,9 14,8 13,9 21,8 13,8 17,8 19.4 7,0 10,9 18,8 13,9 11,1 14,3 18,0 8,7 22,0
Коэффициенты перевода в СИ: 1 кал/г-К = 4,19-103 Дж/кг«К; 1 кал/моль«Кв4,19’
74
наклон кривой Csp больше, чем кривой С1Р. Для оценки С1Р и Ср в точ-
ке плавления можно воспользоваться линейными аппроксимациями
зависимости СР и С1Р от температуры [уравнения (V.l), (V.2)]. Отно-
шение CPICSP в точке плавления в среднем отклоняется на 6% от
среднего значения г = 1,12. Это нетрудно увидеть также из данных
табл. V.2.
емкостей полимеров
Жидкое
§ W 2" S'
~ Q да ~ ч • Л ~ ч оО 00 да
Oi Л см С- 05 я см 2-* [298 Цмо 05 см 05 СМ и °
о К -- А св О » -Art - А С oj & О ь* я •0 »св О К — Рч и — и «0 А и
0,54 15,1 14,5 1,28/1,46 410 15,9 16,8 1,06
0,51 21,6 21,1 1,31 450 24,3 26,3 1,08
0,51 28,6 28,3 — 400 301 32,6 1,08
0,47 26,4 26,6 1,18 320 23,9 26,6 1,11
— — 42,1 (1,26) 500/520 —-
0,45 24,3 24,7 1,16 370 25,6 27,5 1,08
0,46 31,3 32,2 1,22 309/340 29,0 31,7 1,09
0,41 42,6 41,7 1,40 513 50,2 54 1,07
0,50 15,0 15,8 1,47 460 - 14,1 17,9 1,27
0,49 21,6 23,0 >1,30 340 18,8 22,0 1.17
0,50 35,7 37,5 1,27 310 28,5 36,4 1,28
0,46 26,5 29,6 1,35 350 22,1 28,4 1,29
0,29 18,1 (21,8) — 485 25,3 -Н 29,1 1,15
— — (28,0) (1,12) 490 39,4 38
— — (28,0) (1,37) 463
0,23 23,0 (23,5) 1,00 300 23,5 23,5 1,00
— — (32,9) (1,29) 343 —
— 39,5 23,0 (1,69) 505/535 —.
0,46 36,6 1,31 —
0,43 43,5 42,1 1,32 433 46,4 50,5 1.09
— — — — 590 — ___
0,51/0,59 57,8 57,8 1,48 496 63,0 74,0 1,17
— —- 115,6 (1,47) 506
0,52 147 144,6 1,37 496 15,8 18,2 1.15
0,37 71 72,5 1,36 540 9,1 9,2 1,01
0,46 105,5 103,6 (1,28) 345
0,38 97,8 (94,9) 1,35 500 11,6 13,1 1.13
— — —• — 570 -
— — — — 570 — —
ч — —
0,42 50 5 483 143 535/750 60,1 64 1,07
— — — — —
— — — — — —.
— — — — — — — —
10* Дж/кмоль-К.
75
Вундерлих и Джонс [15] опубликовали значения групповых вкладов
для расчетов Csp в интервале температур от 50 до 240 К. Если учесть
неточность экстраполяции этих данных к 300 К, то названные груп-
повые вклады соответствуют данным Сато.
Как вцдно из табл. V.2,’значение отношения г = Clp (298)/Ср (298)
в среднем отклоняется от усредненной величины г = 1,32 на 7%.
Эта величина уменьшается с цовышенией температуры, поскольку
Таблица V.2. Рассчитанные и определенные експерименталъно значения тепло
Полимер Твердое
Ср (298), кал/(г-К) Ср (298) (опыт), кал/(моль-К) Ср (298), кал/(моль- К) Ср (200) (опыт), кал/(моль-К) Ср (200), кал/ (моль • К)
Полиэтилен Полипропилен Полибутилен Полиизобутилен Лоли-4-метилпентен Полибутадиен Полиизопрен 4 Полистирол Полиоксиметилен Полиоксиэтилен Полиокситетраметилен Полиоксипропилен Поливинилхлорид Полихлортрифторэтилен Поливпнилиденхлорид Политетрафторэтилен Полихлоропрен Поливиниловый спирт Поливинилацетат - Полиметилметакрилат Полиакрилонитрил Найлон 6 Найлон 6,6 Найлон 6,10 Полиэтплентерефталат Полиэтпленсебацинат Полпбпсфенолкарбонат Полипропиленсульфон Полибутиленсульфон П олигексенсул ьфон Поли-2,6-диметил оксифенилен Алмаз Графит Сера , Кремний 0,37/0,42 0,39/0,42 0,37/0,42 0,40 0,40 0,39 0,38 0,29 0,34 0,30 0,38 0,34 0,23/0,26 0,22 0,23 0,31 0,35 0,33 0,30 035 0,35 0,38 0,27 028 0,28 0,28 0,33 0,30 10.4/11,8 16,5 >20,7 22,4 33,6 21,0 25,7 30,5 10,2 <16,7 28,2 19,7 1^4/16,2 25,0 23,0 13,6 30,2 33,0 15,9 391 79 107 52 72,4 29,4 29,4 49 35,4 1,46 2,06 5,75 . 4,9 12,1 17,15 23,2 22,3 34,3 21,0 26,4 30,2 10,1 16,1 28,2 21,2 16,2 (24,7) 20,4 (23,3) 25,5 13,8 (28,2) (33,3) (15,8) (39 2) (78,5) (102,7) (52,9) (82,5) (68,9) (29,2) (29,2) (47,3) 34,3 1.47 1,47 5,74 8,0 11,5 16,4 15,4 23,8 15 18,5 20,1 6,9 9,9 20,0/21,1 15,4 10,0/11,6 16,6 16,5 9,3 20,9 24 11,5 28,0 38 49,4 21,6 21,6 35,4 25,8 0,56 1,19 7,9 10,9 14,8 13,9 21,8 13,8 17,8 19.4 7,0 10,9 18,8 13,9 11,1 14,3 18,0 8,7 22,0
Коэффициенты перевода в СИ: 1 кал/г-К = 4,19-103 Дж/кг«К; 1 кал/моль«Кв4,19’
74
наклон кривой Csp больше, чем кривой С1Р. Для оценки С1Р и Ср в точ-
ке плавления можно воспользоваться линейными аппроксимациями
зависимости СР и С1Р от температуры [уравнения (V.1), (V.2)]. Отно-
шение CPICSP в точке плавления в среднем отклоняется на 6% от
среднего значения г = 1,12. Это нетрудно увидеть также из данных
табл. V.2.
емкостей полимеров
Жидкое
§ W 2" S'
~ Q да ~ ч • Л ~ ч оО 00 да
Oi Л см С- 05 я см 2-* [298 Цмо 05 см 05 СМ и °
о К -- А св О » -Art - А С oj & О ь* я •0 »св О К — Рч и — и «0 А и
0,54 15,1 14,5 1,28/1,46 410 15,9 16,8 1,06
0,51 21,6 21,1 1,31 450 24,3 26,3 1,08
0,51 28,6 28,3 — 400 301 32,6 1,08
0,47 26,4 26,6 1,18 320 23,9 26,6 1,11
— — 42,1 (1,26) 500/520 —-
0,45 24,3 24,7 1,16 370 25,6 27,5 1,08
0,46 31,3 32,2 1,22 309/340 29,0 31,7 1,09
0,41 42,6 41,7 1,40 513 50,2 54 1,07
0,50 15,0 15,8 1,47 460 - 14,1 17,9 1,27
0,49 21,6 23,0 >1,30 340 18,8 22,0 1.17
0,50 35,7 37,5 1,27 310 28,5 36,4 1,28
0,46 26,5 29,6 1,35 350 22,1 28,4 1,29
0,29 18,1 (21,8) — 485 25,3 -Н 29,1 1,15
— — (28,0) (1,12) 490 39,4 38
— — (28,0) (1,37) 463
0,23 23,0 (23,5) 1,00 300 23,5 23,5 1,00
— — (32,9) (1,29) 343 —
— 39,5 23,0 (1,69) 505/535 —.
0,46 36,6 1,31 —
0,43 43,5 42,1 1,32 433 46,4 50,5 1.09
— — — — 590 — ___
0,51/0,59 57,8 57,8 1,48 496 63,0 74,0 1,17
— —- 115,6 (1,47) 506
0,52 147 144,6 1,37 496 15,8 18,2 1.15
0,37 71 72,5 1,36 540 9,1 9,2 1,01
0,46 105,5 103,6 (1,28) 345
0,38 97,8 (94,9) 1,35 500 11,6 13,1 1.13
— — —• — 570 -
— — — — 570 — —
ч — —
0,42 50 5 483 143 535/750 60,1 64 1,07
— — — — —
— — — — — —.
— — — — — — — —
10* Дж/кмоль-К.
75
Пример 1
Рассчитать теплоемкость полипропилена со степенью кристалличности 30%
при 25 °C.
Решение
Значения Ср (298) и С1р (298) рассчитываются суммированием групповых
вкладов (см. табл. V.1): С1 (298) р 7,26
(~СН2-) С» (298) 6,05
(—СН—) 3,72 5,00
(-СН3) 7,38 8,80
17,15 21,06
Для полимера с кристалличностью 30% оцененная мольная теплоемкость
Ср (298) = 0,3-17,15 + 0,7-21,06 = 19,9 кал/(моль-К). Удельная теплоемкость
tyM = 19,9/42,1 = 0,47 кал/(г- К).
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
Проведенное выше рассмотрение удельной теплоемкости полимеров
носило весьма эмпирический характер. В действительности, суще-
ствует несколько основных принципов, которые можно применить
для определения удельной теплоемкости. Так, при очень низких
температурах справедливы уравнения Дебая и Эйнштейна.
Если принять одинаковое распределение энергии молекулы по
степеням свободы, то максимальное значение мольной теплоемкости
должно соответствовать величине 37? на атом. В действительности,
часть степеней свободы всегда заморожена, что приводит к меньшей
величине мольной теплоемкости. Увеличение удельной тепло-
емкости с повышением температуры определяется возрастанием ко-
лебательных степеней свободы.
Эмпирически было показано, что мольная теплоемкость полиме-
ров при комнатной температуре имеет порядок величины R (на один
атом). Это нетрудно заметить из данных табл. V.1, в которой значе-
ния С*Р1М на атом были рассчитаны для груцповых вкладов в моль-
ную теплоемкость. Для углеводородных групп значения CPIR на
атом несколько меньше единицы; среднее значение ClplR близко
к единице. Группы, содержащие другие элементы, имеют большие
значения CP!R.
Интересно отметить, что для некоторых групп значения CPIR
на атом превышают максимальное значение, равное 3, которое соот-
ветствует всем колебательным степеням свободы данной группы. Это
означает, что наличие этих групп в молекуле влияет на свободу
движения соседних групп. Именно поэтому для таких групп пра-
вила линейной аддитивности не выполняются строго.
На основании теории жидкостей Вундерлих [14] пришел к вы-
воду, что разность между С1Р и Csp при температуре перехода в стекло-
образное состояние при расчете на структурную группу полимера
76
должна быть постоянной величиной для данного полимера. В этом?
случае структурная группа определяется как наименьший участок
молекулы, который может перемещаться как целое в процессе
внутреннего вращения.
Формулы (V.1) и (V.2) позволяют рассчитать примерные значения!
Clp (Tg) и Ср (7g) для ряда полимеров. Разность значений Ср, отне-
сенных на группу, при расчетах по этим уравнениям изменяется
в диапазоне 1,9—3,1 кал/моль-°С, что соответствует значению,,
полученному Вундерлихом (2,7 кал/моль • °C).
ОТНОШЕНИЯ Ср/С0
До сих пор рассматривались только ср и Ср — удельная и мольная
теплоемкости при постоянном давлении. Очевидно, что именно
с этими величинами приходится иметь дело при обычных изме-
рениях.
ю>____2__I______I______I______I______I______
‘ о,о 0,8 i,o 1,2 J4 1,6 I/
т/\
Рис. V.3. Зависимость Ср/С0от Т/Т„ для аморф-
ных
/,2
полимеров.
' 0,4 0,5 0,6 0,7 0,8 0,3 7,0
---* Т/Тт
Рис. V.4. Зависимость Ср/Сц от Т1Тт для полу-
кристаллических и кристаллических полимеров.
Существуют соотношения для расчета удельной теплоемкости
при постоянном объеме cv. Точный термодинамический расчет по-
зволяет получить следующее уравнение:
а2
cv=cP — Tv— (V.3)
где v — удельный объем; а — коэффициент расширения; и — сжимаемость.
Приближенные соотношения были получены для полимеров Уор-
филдом с соавторами [13]. Эти данные представлены на рис. V.3
и V.4.
77
ТЕПЛОТА КРИСТАЛЛИЗАЦИИ
И ПЛАВЛЕНИЯ
Теплота плавления (кристаллизации) или разность энтальпий
= (V.4)
представляет собой важную величину, используемую в расчетах
других термодинамических функций. Более того, знание &Нт необ-
ходимо для конструирования некоторых аппаратов для переработки
полимеров.
Однако экспериментальные значения \Нт известны только для
ограниченного числа полимеров. Это связано, вероятно, с трудностью
определения М1т. При непосредственных определениях следует
принимать в расчет степень кристалличности образца; при косвен-
ных определениях, например, исходя из характеристик растворов,
получаемое значение зависит от применимости термодинамической
формулы, которая используется для расчета. Поэтому и существует
большой разброс в опубликованных значениях \Нт. В общем слу-
чае наиболее вероятным является максимальное из известных зна-
чений &Нт для данного полимера.
В табл. V.3 дана сводка имеющихся литературных данных о кНт.
В связи с небольшим количеством полимеров, для которых опреде-
лено значение &Нт, крайне желательна разработка метода оценки
этой величины. Однако данных табл. V.3 недостаточно для получе-
ния надежного соотношения между &Нт и строением полимеров.
И все же ниже отмечен ряд групповых вкладов в \Нт, которые
согласуются с данными табл. V.3:
Группа
-СН3
-СН2
>СН-
—СН=СН—
^>С=СН-
-о-
-соо—
-CONH-
Групповой вклад,
кал/«оль
600
900
900
500
500
5300
400 '
—1000
700
Следует подчеркнуть, что значения ДЯт,. рассчитанные таким
шутем, оказываются весьма приближенными.
Маловероятно, что будет развит метод расчетов точных значений
ЛНт на основании простого -суммирования групповых вкладов.
Ведь подобный метод не может быть использован даже для низкомо-
78
Таблица V.3. Энтальпия и антропия плавления некоторых полимеров
Полимер ДНт, кал/моль к ASm. кал/(моль - К)
литературные данные расчет (см. с. 78)
Полиоксисоединения
полиформальдегид 1700 1300 460 3,7
полиоксиэтилен 2000—2200 2200 340 5,9—6,5
полиокситетраметилен 3000 4000 310 9,7
Полиэфиры
полиэфир 2,6 3800 3400 320 11,9
полиэфир 2,10 6100—7000 7000 345 17,7—20 3
полиэфир 10,6 10 200 10 600 343 29,8
полиэфир 9,9 10 300 12 400 338 30,5
полиэфир 10,9 10000 13 300 340 29,4
полиэфир 10,10 12 000 14 200 344 34,9
Политерефталаты
полиэтилентерефталат 5400—5800 5100 543 9,9—10,7
политетраметилентерефта- 7600 6900 505 15,0
лат
полигексаметилентерефта- 8300-8400 8700 434 19,1—19,4 .
лат
полидекаметилёнтерефта- 10400—11 600 12 300 411 25,2—28,2
лат
Полиамиды -
полиамид 6 5200—5600 5200 496 10,5-11,3
полиамид 11 9900 9700 463 21,4
полиамид 6,6 10 600—11 000 10 400 538 19,7—20.4
полиамид 6,10 13000—13 500 14000 496 26,2—27,2
Полиэтилен 1800—2000 1800 414 4,3—4,8
Полипропилен 2100—2600 2400 456 4,6-5,7
Полистирол 2000—2400 — 513 3,9-4,7
Полибутадиен 2200—2400 2300 421 5,2—5,7
Полпиэопрен 3000 2900 309-347 8,6—9,7
Поливинилхлорид 2700 — 558 4,8
Поливинилфторид 1800 — 473 3,8
Полихлортрифторэтилен 1200 — 491 2,4
Политетрафторэтилен 1400 — 600 2,3
Полихл оропрен 2000 — 316 6,3
Полиоксипропилен 2000 2800 348 5,7
Поливиниловый спирт 1600 — 531 3,0
Политетраметпленизофталат 10100 — 426 23,7
Поли-2,2-диметпламид 3100 — 546 5,7
Полибисфенолкарбонат 8800 — 5,40 16,3
пекулярных' соединений, для которых имеется большое количество
экспериментальных значений &Нт [1}.
Подобное положение соответствует ситуации, сложившейся и
в другом разделе термодинамики. Редлих, Дерр и Пьеротти [10}
попытались рассчитать энергию взаимодействия между молекулами
неэлектролитов в растворе как сумму вкладов, составляющих
79
молекулы групп. Вместо того чтобы приписать определенный вклад
каждой имеющейся в системе группе, эти авторы вынуждены были
прибавлять вклады, соответствующие каждой паре взаимодейству-
ющих групп. Это можно назвать правилом аддитивности второго
порядка, которое представляет единственно возможный путь оценки
теплоты растворения.
Использование этого способа оценки энтальпии гомологического
ряда органических соединений в жидком состоянии привело бы
к нелинейной зависимости энтальпии от числа метиленовых групп.
Именно такая зависимость обнаружена экспериментально для теп-
лоты плавления. Правило аддитивности второго порядка оказывается,
однако, слишком сложным для практического применения, если
при этом участвует большое число структурных групп. Это правило
в данном случае подразумевает сложение практически бесконечного
количества вкладов пар групп.
Как было установлено Бонди [1], энтропия плавления
обнаруживает гораздо большую регулярность зависимости от строе-
ния по сравнению с энтальпией плавлейия. При температуре пла-
вления Тт энтропию плавления можно рассчитать по соотношению
Д3т = 4^- - (V.5)
Имеющиеся экспериментальные значения кНт для ряда полиме-
ров, приведенные в табл. V.3, позволяют найти значения Д5,и для
этих полимеров. Данные для трех полиоксисоединений, шести поли-
эфиров, четырех политерефталатов и четырех полиамидов показы-
вают, что Д5т для этих образцов можно представить как сумму
ASm — fti \SL (V.6)
где щ — число групп типа г; Д8, — энтропийный вклад группы I.
Ниже приведены значения Д5,:
Группа ASp кал/(моль-К)
-сн2- 2,0
7,0
—О— 1.8
-соо- 0,0
—CONH— 0,0
Коэффициент перевода в СИ: = 4,9-10’ Дж/(кмоль-К). 1 кал/(моль К) =
Очевидно, что эфирные и амидные группы не дают вклада в энтро-
пию плавления. Вклад метиленовой группы в этих линейных поли-
мерах (2,0 кал/моль-К) гораздо меньше, чем в полиэтилене
{2,35 кал/моль-К). Последняя величина также отличается от боль-
ших значений вкладов метиленовых групп (около 2,65), наблюда-
80
емых в гомологическом ряду низкомолекулярных соединений [1].
Тем не менее для некоторых рядов низкомолекулярных соединений
было найдено значение вклада метиленовой группы, примерно
равное 2,35.
Аналогичный эффект наблюдали много лет назад Кинг и Гар-
нер [8] для теплоты плавления гомологических рядов низкомолеку-
лярных соединений. Было обнаружено, что длинные метиленовые
цепочки могли кристаллизоваться с образованием двух различных
кристаллических структур; значения теплоты плавления этих двух
структур существенно различались между собой.
Перечисленные факты позволяют сделать вывод о том, что для
предсказания теплот плавления полимеров не следует пользоваться
экспериментальными данными о низкомолекулярных соединениях.
Более того, вклад в термодинамические параметры метиленовой
группы в полиэтилене явно отличается от вклада метиленовой груп-
пы в ряду линейных полимеров, содержащих неуглеводородные
группы.
С помощью приведенного выше уравнения для ASm можно рас-
считать теплоту плавления линейных полимеров, содержащих мети-
леновые, n-фениленовые, кислородные, эфирные и амидные группы,
при условии, что известны их температуры плавления.
ЭНТАЛЬПИЯ И ЭНТРОПИЯ
При определении зависимости энтальпии и энтропии какого-либо
вещества от температуры, как правило, начинают с весьма точных
измерений удельной теплоемкости. Затем можно рассчитать энталь-
пию и энтропию путем интегрирования:
т
Я(Т) = Я(0) + J CpdT + ^bffi (V.7)
О
г
5(Г) = 5(0) + J Ср/Т dT + ^bSi (V.8)
о
где Н (0) и S (0) — энтальпия и энтропия при 0 К; ДЯ/ и Д5, — изменения
энтропии и энтальпии при фазовых переходах первого рода.
Если воспользоваться этим методом для расчета термодинами-
ческих параметров полимеров, то возникнут точно такие же затруд-
нения, что и при определении удельной теплоемкости, поскольку
большинство полимерных образцов частично кристаллизуется. Зна-
чения термодинамических величин лежат между значениями, най-
денными для чисто кристаллического и для совершенно аморфного
полимеров. Для получения данных, относящихся к этим двум идеа-
лизированным состояниям, необходимо выполнить очень много изме-
рений. Данные такого рода опубликованы только для ограниченного
числа полимеров.
В качестве примера на рис. V.5 и V.6 приведены зависимости
энтальпии и энтропии от температуры для полипропилена
6 Заказ 699 81
в соответствии с данными Ги и Мелия [5], Дэйнтона с сотр. [4|
и Пассаглиа с сотр. [9]. Соответствующие данные для других по-
лимеров можно найти в серии работ Дэйнтона с сотр. [4].
Как видно из рис. V.5, кривые изменения энтальпии для кристал-
лического и аморфного образцов полипропилена идут параллельно
друг другу вплоть до температуры стеклования. Расстояние между
этими кривыми равно кН (0) при 0 К и носит название энтальпии
аморфного полимера. Начиная
с точки стеклования, кривая для
аморфного полимера постепенно
приближается к кривой для рас-
плава, в то же время на кривой,
построенной для кристаллического
полимера, возникает разрыв при
температуре плавления. Расстоя-
ние между кривыми для кристал-
лического и жидкого состояний
в точке плавления представляет
собой скрытую теплоту плавле-
ния &Нт.
температурные зависимости энтро-
Рис. V.5. Энтальпия полипропилена:
J — аморфный; 2 — кристаллический.
Аналогичный характер имеют
пии для кристаллического и аморфного полимеров (рис. V.6).
Применяя уравнение (V.7) к полимеру, находящемуся в кри-
сталлическом и жидком (или высокоэластическом) состояниях, мож-
но записать, что
Т
HS(T) = HS(O)+ f CpdT (Т<Тт} (V.9)
о
Тт Т
tf;(T) = tfs(O)+y CspdT+ J ClvdT + bHm (V.10)
° I'm
В соответствии с данными рис. V.5 Ht (Tg) = Hs (Tg) + ДЯ (0).
Комбинация этих формул и подстановка в них выражений (V.1)
и (V.2) дает:
Д77т-Д7Г(0) = [0,64С], (298) —0,107Ср (298)] (Tm-Tg) +
+ [0,0006Ср (298) —0,0015Ср (298)] —Tg) (V.11)
Формулы (V.9) и (V.10) при очень низких температурах явно
становятся некорректными. Однако отклонения от правильных
значений энтальпии большей частью оказываются незначительными
за счет параллельности хода кривых Нг и Hs при низких темпера-
турах.
Применение формулы (V.11) к ряду полимеров позволяет полу-
чить правильный порядок разности кН,п — АН (0). Однако этим
выражением нельзя пользоваться для точного предсказания АНт
в связи с отсутствием данных для кН (0) и приближенным характе-
ром формул (V.9) и (V.10). Но на основании выражения (V.11) можно
82
предположить, что \Нт должно возрастать с увеличением СР (298)
и разности Тт — Tg. Это подтверждает рис. V.7, на котором по-
строена зависимость отношения \Н1П1С1Р (298) от (Т т— Тg) для по-
лимеров с известными из литературы значениями &Нт. В первом
приближении
ДЯт/eJ, (298)=-0,55 (Tm-Tg) (V.12)
при стандартном отклонении около 25%.
Для энтропии можно получить выражение, аналогичное соотно-
шению (V.11):
Д5т-Д5 (0) = [0,6467^ (298) —0,1076?’ (298)] In (Гт/Т£) +
+ [0,0012Ср (298) —О.ООЗСр (298)] (Тт—Тя) (V.13)
Эту формулу можно проверить с более высокой точностью по срав-
нению с уравнением (V.11), поскольку можно оценить порядок вели-
Рис. V.6. Энтропия полипропилена:
1 — аморфный; 2 — кристаллический.
Рис. V.7. Приближенное соотношение
между кИт/С? (298) и Тт — Тя.
чины AS (0). Данные Бестула и Чанга,^ 43] соответствуют вкладу
в Д5 (0), примерно равному 0,7 кал/моль-°С на атом цепи. С по-
мощью значения Д5.(0) можно рассчитать Д5т по формуле (V.13).
На рис. V.8 сопоставлены рассчитанные значения \Sm для ряда
полимеров с экспериментальными данными. Учитывая неточности
использованных данных, полученный результат можно считать
удовлетворительным.
Наконец, следует упомянуть серию статей Гриски с соавторами
[6], в которых приведены значения энтропии и энтальпии в зависи-
мости от температуры и давления для ряда промышленных поли-
меров.
Пример 2
Оценить следующие характеристики полиэтилентерефталата:
а) удельную теплоемкость в твердом состоянии при 25 °C (298 К);
б) удельную теплоемкость в жидком состоянии при температуре формования
волокна (277 °C или 550 К);
6* ' 83
в) теплоту плавления в точке плавления;
г) разность энтальпий твердого и высокоэластического состояний при тем-
пературе перехода в стеклообразное состояние.
Решение
Данные, приведенные в табл. V.1 н на с. 78, дают:
4СНаром
2Саром
2(—СН2—)
2(—СОО—)
Группа
Csp (298)
14,72
4,08
12,10
22
С^(298)
21,2 |
5,8 J
14,5
31,0
ДНт, ккал
5.3
1,8
-2,0
52,90 72,5 5,1
а. Удельная теплоемкость твердого полимера при 25 9С
4 (298) = 52,90/192,2 = 0,275 кал/К
что весьма удовлетворительно соответствует экспериментальному значению 0,27.
б. По формуле (V.2) мольная тепло-
емкость расплава
Рис. V.8. Рассчитанные и экспери-
ментальные значения ASm.
Ср (550) = С1р (298) [0,64 + 0,0012Т\ =94,3
Удельная теплоемкость с1р (550)=
=94,3/192,2 = 0,49; экспериментальное
значение 0,48.
в. Теплота плавления в точке плав-
ления составляет 5100 кал/моль, что
соответствует экспериментальному зна-
чению 5400 в разумных пределах.
г. По формуле (V.11)
ДЯ (Те) = ДЯ (0) = ДНт— [0,64Ср(298) -
-0,107^,(298)] {Tm-Tg)-
-[0,0006С],(298)-0,0015С* (298)]х
или
ДН (Те) = 5100 — 8200 + 6400 = 3300 кал/моль
Соответственно по формуле (V.13)
Д5 (Г£) = Д5т- [о,64Ср (298) —0,107Ср (298)] In (Tm/Tg)~
~ [О 0012Ср (298)—О.ООЗСр (298)] (Тт-Те)
следовательно
Д5т = ДЯт/7'т = 5100/543 = 9,4 кал/моль • К
Добавление групповых вкладов (см. с. 80) дает:
Д5т = 2 - 2,0 + 7,0 = 11,0 кал/моль К
84
Учет первого из этих значений Д5т дает:
AN (Те) = 9,4—(18,8—14,4) = 5,0 кал/моль К
Нетрудно видеть, что AN (Tg) <5 АН (Tg)/Tg, поскольку твердая и жидкая
формы не находятся в равновесии при Tg.
ЛИТЕРАТУРА
1. Bondi A., «Physical Properties
of Molecular Crystals, Liquids
and Glasses», Wiley, New York,
1968.
2. Reid R. C. and Sherwood Th. K.,
«The Properties of Gases and
Liquids», McGraw-Hill, New
York, 1958 (1st. ed.); 1966 (2nd
ed.).
3. Bestul A. B. and Chang S. S.,'
J. Chem. Phys. 40 (1964) 3731.
4. Dainton F. S., Evans D. M.,
Hoare F. E. and Melia T. P.,
Polymer 3 (1962) 286.
5. Gee D. R. and Melia T. P.,
Makromol. Chem. 132 (1970) 195.
6. Griskey R. G. et al., several
articles in Modern Plastics 43
(1966); 44 (1967).
7. Johnson A. J. and Huang C. J.,
Can. J. Technol. 33 (1955) 421.
8. King A. M. and Garner W. E.,
J. Chem. Soc. (1934) 1449.
9. Passaglia E. and Kevorkian R.r
J. Appl. Phys. 34(1936)90.
10. Redlich O., Derr E. L. and Pie-
rotti G. J., J. Am. Chem. Soc.
81 (1959) 2283.
11. Satoh S., J. Sci. Research Inst.
(Tokyo) 43 (1948) 79.
12. Shaw R., J. Chem. Eng. Data>
14 (1969) 461.
13. Warfield 7?. W., Pastine D. J.
and Petree M. C., U. S. Naval
Ordnance Lab., RPT NOLTR
gg______gg 1969.
14. Wunderlich B., ]. Phys. Chem.
64 (1960) 1052.
15. Wunderlich B. and Jones L. D.,
J. Macromol. Sci. Phys. Bi
(1969) 67.
глава vi. ХАРАКТЕРИСТИКИ КОГЕЗИИ
И АДГЕЗИИ
К наиболее важным характеристикам когезии и адгезии относятся
энергия когезии, внутреннее давление и поверхностная энергия.-
Для этой группы характеристик в принципе нельзя предпола-
гать простую аддитивность групповых вкладов в связи со сложными
межмолекулярными взаимодействиями, однако ниже будет показано,
что в качестве первого приближения применение метода аддитивных
групповых вкладов позволяет получить довольно приличные оценки
числовых значений рассматриваемых параметров.
МОЛЬНАЯ ЭНЕРГИЯ КОГЕЗИИ
Энергия когезии жидкости Еког определяется как энергия, необхо-
димая для разрушения всех межмолекулярных контактов и отнесен-
ная к одному молю этой жидкости. Эта величина, следовательно,
тесно связана с мольной теплотой испарения АНКСП:
/?Ког = ДЯисп— ЯТ
Непосредственно связаны с энергией когезии две другие' величины:
плотность энергии когезии <?КоГ = EKoTIV при 298 К, кал/см3; пара-
метр растворимости 6 = (ЕКоТ1У)'1* = еКог при 298 К, кал'/’/см3/2.
Для низкомолекулярных жидкостей Екот, а следовательно, еКоГ
и 6 можно рассчитать по теплоте испарения или по кривой зависи-
мости давления пара от температуры. Но полимер нельзя испарить,
поэтому для определения энергии когезии полимеров следует поль-
зоваться косвенными методами, например исходя из эксперимен-
тальных данных о набухании или растворении полимеров в жидко
-стях с известной плотностью энергии когезии.
АДДИТИВНЫЕ СВОЙСТВА Е„„ И Л
KOI KU1
Для низкомолекулярных соединений еще Дункель [10] рассматри
вал Елот в качестве аддитивной характеристики и получил группо-
вые вклады для энергии когезии жидкостей при комнатной темпера-
туре. Такой же метод применили к полимерам Хайес [16] и Ди Бене-
детто [9].
Райнек и Лин [24] показали, однако, что для гомологических
рядов низкомолекулярных жидкостей вклад метиленовой группы
«6
в энергию когезии не остается постоянной величиной, но зависит
от природы других структурных групп в данной молекуле.
Бенн [8] занимался исследованиями энергии когезии в точке
кипения; Бонди [7] изучал когезионные характеристики при О К
(т. е. Н°о) и при такой температуре, когда V/Vw = 1,7, т. е. вблизи
точки плавления (Е°).
Ранее Смолл [26] показал, что комплексная величина^
(2?KorV (298))'/= = F, названная им мольной константой притяжения, \
оказывается аддитивной величиной как для низкомолекулярных,?'
так и для высокомолекулярных соединений. <
Очень часто пользуются набором величин Смолла. Недавно Хой
[18] предложил значения групповых вкладов в F, которые незначи-
тельно отличаются от данных Смолла. Ван Кревелен [301 получил
набор атомных вкладов для расчета величины F. Более подробные
сведения можно найти в гл. VIII.
Зная F, можно получить косвенным путем значение Екот для
полимера.
В табл. VI. 1 приведена сводка вкладов в величину Еког наиболее
важных структурных групп. Значения, заключенные в скобки,
ранее не приводились в литературе.
Значения ЕКоТ у различных авторов в первом приближении согла-
суются друг с другом. Энергия когезии уменьшается при повышении
температуры, поэтому, как и следовало ожидать, в общем случае
выполняется следующее правило:
Я«>£8>Еког (298)>Ек0г (ТЬ)
В применении к низкомолекулярным соединениям приводимые
Банном данные позволяют наилучшим образом предсказать энер-
гию когезии. Однако эти значения можно применять к соединениям,
находящимся только при температуре кипения, потому к энергии
когезии полимеров эти данные не имеют прямого отношения. Удо-
влетворительная корреляция получается по методу Райнека и Лина,
но недостатком их подхода оказывается необходимость введения
большого количества поправок. К недостатку методов Ди Бене-
детто и Хайеса относится весьма ограниченное количество рас-
сматриваемых групп.
Работа Райнека и Лина показала, что принцип аддитивности но
может быть в строгом смысле применен для вычисления энергии
когезии при комнатной температуре; тем не менее с помощью этого
метода можно получить разумные значения энергии когезии поли-
меров. Но здесь должны использоваться величины, которые не обяза-
тельно идентичны аналогичным параметрам, применяемым для
низкомолекулярных соединений. Недавно Хофтицер и Ван Креве-
лен [17] показали, что по имеющимся данным для ЕКог полимеров
можно получить новый набор групповых вкладов, который дает
наилучшую возможную корреляцию со всеми приводимыми в лите-
ратуре данными. Эти новые значения групповых вкладов также
приведены в табл. VI.2. Для ряда из 30 полимеров среднее
87
Таблица -VI. 1. Групповые вклады в Еког
Группа Бонди Е° Бонди ЕКОР (298)
Райнек н Лин Дункель Ди Бене- детто
1 — С — 1 (-1910) (—1390) — (—1750) —
^СН- (-65) (-100) 970 -380 —
—сн2— 1.515 1070 1230 990 860
- СНз 2520 2000 990 1780 —
-СН(СН3)- (2455) (1900) (1960) (1400) —
—С(СН3)2— (3130) (2610) — 1810 2480
>с=сн— (1500) — (610) —
—сн=сн- 2300 — 1980 1720
—С(СН3)=СН— — 3500 — 2390 2600
Циклопентил 8060 6100 — — —.
Циклогекспл 9120 6850 7040 — —
-Фенил 9800 7270 7450 7380 —
га-Фенилен 8580 6070 — — —
—О— 1600 — 1630 . —
—ОН — — 7830 7250 —
-СО- — . 2400 — 4720 .—
—СОО — — 3430 (4560) (3820) —
—СООН — — 7830 8970 ,—
—О-С-О— II О ' — 4700 — — —.
—С—О—С— II II О О — 5600 — — —
—O-N—. II . 1 О н — — — 16 200 — .
—C-C-N- II 1 О н — — — \ —
— S — — 2500 —
—CN — 5350 — —
—CHCN— — (5250) — — —
—F — 2450 — 2060 —
—С1 — 3400 2790 3400
—Вг — 4000 — — —
— I — 5150 .— —
Ф8
^ког (Tb) Банн F ЕкоР (298) (новое соотно- шение )
Хайес Смолл Ван Крене- лен Хой
— (—1500) —93 0 32,0 —1600
— (-440) 28 68,5 86,0 100
990 680 133 137 131,5 1000
— 1700 214 205,5 148,3 2300
1700 1360 242 274 (234,3) (2400)
2840 ' 1900 335 411 (328,6) (3000)
130
— (700) 148,5 206,0 400
1790 1700 222 217 243,1 2000
2740 2400 (344) 354 (354,3) (2700)
— — — 676,5 633,0 —
— — — 813,5 720,1 —
— 5400 735 741,5 683,5 6800
5700 3900 658 673 704,9 6000
1630 1000 70 125 1150 1500
— 5800 — 368,5 225,8 —
— 2660 275 335 263,0
3380 2900 310 250 326,6 3200
— 5600 — 318,5 (488,8) —
— — — 375 (441,6) —
— 3900 — 375 567,3 —
10 680 8500 — 600 (443,0) 14 500
6280 8740 — 725 (506,6) —
2200 225 225 209,4 1500
— — 410 480 354,6 6800
5760 — (438) 548,5 (440,6) (6900)
— (ИЗО) (122) 80 41,3 1200
— 2800 270 230 205,1 3100
3100 340 300 257,9 3700
4200 425 — — —
89
Таблица -VI. 1. Групповые вклады в Еког
Группа Бонди Е° Бонди ЕКОР (298)
Райнек н Лин Дункель Ди Бене- детто
1 — С — 1 (-1910) (—1390) — (—1750) —
^СН- (-65) (-100) 970 -380 —
—сн2— 1.515 1070 1230 990 860
- СНз 2520 2000 990 1780 —
-СН(СН3)- (2455) (1900) (1960) (1400) —
—С(СН3)2— (3130) (2610) — 1810 2480
>с=сн— (1500) — (610) —
—сн=сн- 2300 — 1980 1720
—С(СН3)=СН— — 3500 — 2390 2600
Циклопентил 8060 6100 — — —.
Циклогекспл 9120 6850 7040 — —
-Фенил 9800 7270 7450 7380 —
га-Фенилен 8580 6070 — — —
—О— 1600 — 1630 . —
—ОН — — 7830 7250 —
-СО- — . 2400 — 4720 .—
—СОО — — 3430 (4560) (3820) —
—СООН — — 7830 8970 ,—
—О-С-О— II О ' — 4700 — — —.
—С—О—С— II II О О — 5600 — — —
—O-N—. II . 1 О н — — — 16 200 — .
—C-C-N- II 1 О н — — — \ —
— S — — 2500 —
—CN — 5350 — —
—CHCN— — (5250) — — —
—F — 2450 — 2060 —
—С1 — 3400 2790 3400
—Вг — 4000 — — —
— I — 5150 .— —
Ф8
^ког (Tb) Банн F ЕкоР (298) (новое соотно- шение )
Хайес Смолл Ван Крене- лен Хой
— (—1500) —93 0 32,0 —1600
— (-440) 28 68,5 86,0 100
990 680 133 137 131,5 1000
— 1700 214 205,5 148,3 2300
1700 1360 242 274 (234,3) (2400)
2840 ' 1900 335 411 (328,6) (3000)
130
— (700) 148,5 206,0 400
1790 1700 222 217 243,1 2000
2740 2400 (344) 354 (354,3) (2700)
— — — 676,5 633,0 —
— — — 813,5 720,1 —
— 5400 735 741,5 683,5 6800
5700 3900 658 673 704,9 6000
1630 1000 70 125 1150 1500
— 5800 — 368,5 225,8 —
— 2660 275 335 263,0
3380 2900 310 250 326,6 3200
— 5600 — 318,5 (488,8) —
— — — 375 (441,6) —
— 3900 — 375 567,3 —
10 680 8500 — 600 (443,0) 14 500
6280 8740 — 725 (506,6) —
2200 225 225 209,4 1500
— — 410 480 354,6 6800
5760 — (438) 548,5 (440,6) (6900)
— (ИЗО) (122) 80 41,3 1200
— 2800 270 230 205,1 3100
3100 340 300 257,9 3700
4200 425 — — —
89
Таблица VI.2. Энергия когезии полимеров
Полимер 6, кал /’/см /’ Л ч о 2 2 о Еког по б Я ког ( расчет) , юл/ ноль
ОТ до от до Райнек и. Лин Дункель Ди Бенедет- то Хайес Смоли Ван нреве- лен 43 О И новое соот- । ношение
Полиэтилен 7,7 8,35 32,9 1950 2290 2460 1980 1720 1980 2190 2330 2140 2000
Полипропилен 8,2 9,2 49,1 3300 4160 3190 2390 — 2690 2830 3400 2690 3400
Полиизоб утилен 7,8 8,1 66,8 4060 4380 .— 2880 3340 3830 3320 4560 3210 4000
Поливинилхлорид 9,4 10,8 45,2 3990 5270 4870 4010 4040 5170 4120 4200 4020 4200
Поливинилиденхлорид 9,9 12,2 58,0 5680 8630 — • 6040 — 3690 5930 6280 5930 5600
Поливинилбромид 9,5 — 53,8 4850 — — — — — 4670 4750 4200 4800
Политетрафторэтилен 6,2 — 50,0 1920 — 4740 —. 2300 1800 - 2050 1050 4600
Долихлортрифторэтилен 7,2 7,9 61,8 3200 3860 — 6080 —- — — 3820 2710 3500
Поливиниловый спирт 12,6 14,2 35,0 5560 7060 9870 7860 — — . — 9500 5670 —-
Поливинилацетат 9,35 11,05 72,2 6310 8820 7450 6210 6920 6070 6600 6140 6740 6600
Поливинилпропионат 8,8 — 90,2 6990 — 8580 7200 7780 7060 7670 7300 7780 7600
Полистирол 8,5 9,3 98,0 7080 8480 9210 7900 9800 8180 8250 9220 8350 7900
Полиметилакрилат 9,7 10,4 70,1 6600 7580 7450 6210 6920 6070 6600 6140 6740 6600
Полиэтилакрилат 9,2 9,4 86,6 7330 7650 8580 7200 7780 7060 7670 7300 7780 7600
Полипропилакрилат 9,05 — 103,0 8440 — 9710 8190 8640 8050 8750 • 8460 8830 8600
Полибутилакрилат 8,8 9,1 119,5 9260 9900 10 840 9180 9500 9040 9830 9610 9880 9600
Полиизобутилакрилат 8,7 11,0 114,2 9020 14 420 10 340 8600 — 8760 9300 9510 9270 10 000
Поли-2,2,3,3,4,4,4-гептафторбутилакри- 6,7 — 148,0 6640 — — 14 590 . — . — — 8860 7510 8900
Полиметилметакрилат 9,1 12,8 86,5 7160 14 170 — 6620 7210 6930 7290’ 7090 7200
Полиэтилметакрилат 8,9 9,15 102,4 8110 8570 — 7610 — 8200 8020 8450 8150 8200
Полибутилметакрилат 8,7 9,0 137,2 10 380 11 НО — 9590 — 10180 10 210 10 770 10 280 10 200
Полиизобутилметакрилат 8-,2 10,5 135,7 9120 14 960 — 9010 —. 9900 9700 10 670 9690 10 600
Поли-трет-бутилметакрилат 8,3 — 138,9 9570 — — 8430 — 10 050 9030 10 660 9070 10 200
Полиэтоксиэтилметакрилат 9,0 9,9 145,6 11 790 15 270 —. 11 220 — 11 810 10 680 42 220 11 540 11 700
Полибензилметакрилат 9,8 10,0 152,0 14 600 15 200 — 13 210 — — 13 580 14 330 13 990 12 700
Полиакрилонитрил 12,5 15,4 44,8 7000 10 620 '—. — — 6750 6490 9350 6520 7900 :
Полиметакрилонитрил 10,7 — 63,9 7320 — — — — — 6630 10180 6680 8500
Д о ли-а-циано метилакрилат 14,0 14,5 82,1 16 090 17 260 — — — — 10 790 13 080 И 220 11 700
Полибутадиен 8,1 8,6 60,7 3980 4490 — 3960 3440 3770 3930 3980 4110 4000
Полиизопрен 7,9 10,0 75,7 4730 7570 ' — 4370 4320 - 4720 4910 5210 4910 .4700
Полихлоропрен 8,2 9,25 71,3 4790 6100 5990 5000 4990 6230 5980 6310 5500
Полиэпихлоргидрин 9,4 — 69,7 6160 — — 6630 '— — 5840 7060 6500 6700
Полиэтилентерёфталат 9,7 10,7 143,2 13 470 16 390 1 — — 14 440 16 510 14'500 17 300 14 400
Лолигексаметиленадипамид 13,6 — 208,3 38 500 — —_ 42 300 — 31 260 -— 31 -750 23 290 39 000
Поли-8-аминокаприловая кислота 12,7 — 135,9 21 920 — —. 23 130 — 17 610 — 17 830 15 000 21 500
Полиформальдегид 10,2 11,0 25,6 2600 3030 — 2620 — 2620 1740 2890 2560 2500
Полиокситетраметилен 8,3 8,55 74,3 5120 5430 — 5590 — 5590 5020 6820 5700 5500
Полиэтиленсульфид 9,0 9,4 47,9 3880 4230 '— —- — — 5600 5790 5190 4100
Полиоксипропилен 7,5 9,9 57,6 3240 . 5650 —. — — 4320 3460 5010 4030 4900
Полистиролсульфид 9,3 — 115,8 10 020 — — — — — 11 630 12 710 И 410 10 000
П олидиметилсилоксан 7,3 7,6 75,6 4030 4300 — — — — 2800 — — —
'Коэффициснты’перевода в СИ: 1 кал’/г/см’/’ = 2,04-10* Дж^О/м’/*; 1 см’/моль =
10-* м*/кмоль! 1 кал/моль=4,19-10* Дж/кмоль.
90
91
Таблица VI.2. Энергия когезии полимеров
Полимер 6, кал ^/см Л ч Q 2 2 о £каг по 6 ЕКОг (расчет), кал/моль
ОТ до ОТ до Райнек и. Лин Дункель Ди Бенедет- то Хай ее Смолл Ван Креве- лен 43 О И новое соот- ношение
Полиэтилен 7,7 8,35 32,9 1950 2290 2460 1980 1720 1980 2190 2330 2140 2000
Полипропилен 8,2 9,2 49,1 3300 4160 3190 2390 — 2690 2830 3400 2690 3400
Полиизобутилен 7,8 8,1 66,8 4060 4380 .— 2880 3340 3830 3320 4560 3210 4000
Поливинилхлорид 9,4 10,8 45,2 3990 5270 4870 4010 4040 5170 4120 4200 4020 4200
Поливинилиденхлорид 9,9 12,2 58,0 5680 8630 — 6040 — 3690 5930 6280 5930 5600
Поливинилбромид 9,5 — 53,8 4850 — — — — 4670 4750 4200 4800
Политетрафторэтилен 6,2 — 50,0 1920 — 4740 —. 2300 1800 - 2050 1050 1600
Полихлортрифторэтилен 7,2 7,9 61,8 3200 3860 — 6080 .—- — — 3820 2710 3500
Поливиниловый спирт 12,6 14,2 35,0 5560 7060. 9870 7860 — 9500 5670 — '
П оливинилацетат 9,35 11,05 72,2 6310 8820 7450 6210 6920 6070 6600 6140 6740 6600
Поливинилпропионат 8,8 — 90,2 6990 — 8580 7200 7780 7060 7670 7300 7780 7600
Полистирол 8,5 9,3 98,0 7080 8480 9210 7900 9800 8180 8250 9220 8350 7900
Полиметилакрилат 9,7 10,4 70,1 6600 7580 7450 6210 6920 6070 6600 6140 6740 6600
Полиэтилакрилат 9,2 9,4 86,6 7330 7650 8580 7200 7780 7060 7670 7300 7780 7600
Полипропилакрилат 9,05 — 103,0 8440 — 9710 8190 8640 8050 8750 • • 8460 8830 8600
Полибутилакрилат 8,8 9,1 119,5 9260 9900 10 840 9180 9500 9040 9830 9610 9880 9600
Полиизобутилакрилат 8,7 11,0 114,2 9020 14 420 10 340 8600 8760 9300 9510 9270 10 000
Поли-2,2,3 >3,4,4,4-гептафторбутнл акри- лат 6,7 — 148,0 6640 — — 14 590 — . — — 8860 7510 8900
Полиметилметакрилат 9,1 12,8 86,5 7160 14 170 — 6620 — 7210 6930 7290 7090 7200
Полиэтилметакрилат 8,9 9,15 102,4 8110 8570 — 7610 — 8200 8020 8450 8150 8200
Полибутилметакрилат 8,7 9,0 137,2 10 380 11 НО — 9590 — 10180 10 210 10 770 10 280 10 200
Полиизобутилметакрилат 8,2 10,5 135,7 9120 14 960 9010 —. 9900 9700 10 670 9690 10 600
Поли-трет-бутилметакрилат 8,3 — 138,9 9570 — 8430 — 10 050 9030 10 660 9070 10 200
Полиэтоксиэтилметакрилат 9,0 9,9 145,6 И 790 15 270 — 11 220 — И 810 10 680 12 220 И 540 11 700
Полибензилметакрилат 9,8 10,0 152,0 14 600 15 200 — 13 210 — —. 13 580 14 330 13 990 12 700
Полиакрилонитрил 12,5 15,4 44,8 7000 10 620 —. — — 6750 6490 9350 6520 7900
Полиметакрилонитрил 10,7 — 63,9 7320 — — —, — 6630 10180 6680 8500
По ли-а-циано метилакрилат 14,0 14,5 82,1 16 090 17 260 — — 10 790 13 080 11 220 И 700
Полибутадиен 8,1 8,6 60,7 3980 4490 — 3960 3440 3770 3930 3980 4110 4000
Полиизопрен 7,9 10,0 75,7 4730 7570 ’ — 4370 4320 4720 4910 5210 4910 4700
Полихлоропрен 8,2 9,25 71,3 4790 6100 — 5990 5000 4990 6230 5980 6310 5500
Полиэпихлоргидрин 9,4 — 69,7 6160 — — 6630 .— 5840 7060 6500 6700
Полиэтилентерефталат 9,7 10,7 143,2 13 470 16 390 —. — — 14 440 16 510 14'500 17 300 14 400
Полигексаметиленадипамид 13,6 — 208,3 38 500 —. 42 300 — 31 260 — 31 -750 23 290 39 000
Поли-8-аминокаприловая кислота 12,7 — 135,9 21 920 —. 23 130 — 17 610 17 830 15 000 21 500
Полиформальдегид 10,2 11,0 25,0 2600 3030 — 2620 — 2620 1740 2890 2560 2500
Полиокситетраметилен 8,3 8,55 74,3 5120 5430 — 5590 — 5590 5020 6820 5700 5500
Полиэтиленсульфид 9,0 9,4 47,9 3880 4230 —. — — 5600 5790 5190 4100
Полиоксипропилен 7,5 9,9 57,6 3240 . 5650 .—. — — 4320 3460 5010 4030 4900
По листиро лсульфид 9,3 — 115,8 10 020 — — . 11 630 12 710 И 410 10 000
П олидиметилсилоксан • 7,3 7,6 75,6 4030 4300 — 1— — — 2800 — — —
'Коэффициснты’перевода в СИ: 1 кал'/г/см’Л = 2,04-10* Дж’/г/м’/»; 1 см’/моль =
10-* м’/кмоль) 1 кал/моль=4,19-10* Дж/кмоль.
•АО
91
отклонение групповых вкладов у Хайеса составляет 8%, а отклонение
Для новых значений вкладов у Хофтицера и Ван Кревелена не пре-
вышает 6%. Среднее отклонение значений ЕКоГ, полученных косвен-
ным методом расчета через F с групповыми вкладами Смолла, Хойя
или Ван Кревелена, составляет 7%. В табл. VI.2 даны величины
Еког для ряда из 41 полимера, рассчитанные разными методами,
и проводится сравнение их с экспериментальными значениями.
Пример 1
Оценить энергию когезии полибутилметакрилата
СН3
I
----с—сн2-----
О=с-О-(СН2)3—СНз
а) с помощью групповых вкладов, приведенных в этой главе;
б) по методу Смолла.
Решение
Сложение групповых вкладов, приведенных на С. ив табл. VI.2, приводит
к следующему результату:
Группы 2 Е/ 2Е/
4(—СН2—) 4000 532 65,80
2(—СНз) 4600 428 50,35
/ 1 \ 1 -С-1 \ 1 / —1600 -93
К-СОО-) 3200 310 21,00
Еког =10200 Е = 1177 Р = 137,15
а. Прямой метод дает Киог=10 200 кал/моль звеньев.
б. Метод Смолла дает £Kor=F2/y = 10 100 кал/моль.
Экспериментально определенные значения параметра растворимости 6
изменяются от 8,7 до 9,0. Это соответствует изменению значения Еког = 62V
от 10 380 до И ИОкал/моль.
ВНУТРЕННЕЕ ДАВЛЕНИЕ
Спенсер и Джилмор [28] показали, что р — v — ^-характеристика
полимерного расплава может быть представлена видоизмененным
уравнением состояния Ван-дер-Ваальса следующего вида:
(v-a)(p+ri) = RT/Ma (VI.1)
где р — давление;' v — удельный объем полимера; Мл — масса 1 моля «взаимо-
действующего сегмента».
Величины лио — это константы, которые должны определяться
экспериментально, как и параметр Ми. Величина л в этом уравне-
нии представляет собой внутреннее давление, которое не зависит
от удельного объема и, следовательно, от температуры и давления.
92
Спенсер и Джилмор оценили значения констант л и Ми по изме-
рениям серий значений р и v при постоянных температурах. Для
линейных синтетических полимеров Ма можно идентифицировать
с молекулярным весом структурного звена. В этом случае уравнение
состояния, поскольку Миа> = V (0), принимает вид
Г-У(0))(р+я) = /?7’ (VI.2)
В атмосферных условиях внутреннее давление л гораздо больше
внешнего давления р, следовательно, для жидкого состояния поли-
мера
RT
л = V (Т)_V (0) —(VI.3)
Такой же результат получается дифференцированием уравнения
состояния
/ dv \ R
к ОТ )р~ МиЛ
или
R R R
я - г w ~ Zl
\_ми ( aT )pJ
В табл. VI.3 сравниваются результаты расчета по уравнению
(VI.3) с экспериментальными данными. Полученные значения л
имеют правильный порядок величины.
Таблица V1.3. Постоянные в уравнении состояния
Полимер Опыт Расчет
л, атм <0. см8/г й Л*=—, fiZ * атм <й=У(0)/м = = i,3Vyp/M, см*/г
Полистирол 1850 ' 0,822 1440 0,785
Полиметилметакрилат 2150 0,734 1710 0,730
Полиэтилен 3250 0,875 3350 0,955
Коэффициенты перевода в СИ: 1 атм= 1,013 10* Н/м* = 1,013-10* Дж/м"; 1 см*/г=
= 10-’ м’/кг.
Недавно Смит [27] получил уравнение состояния для жидких
полимеров, исходя из дырочной теории жидкого состояния. При
повышенных температурах уравнение Смита можно привести к виду,
эквивалентному уравнению (VI. 1).
Следует отметить, что для производных целлюлозы (ацетата и
бутирата целлюлозы, этилцеллюлозы) значения Ми оказались го-
раздо меньше молекулярного веса структурных элементов.
93
Если в литературе нет данных для расплава полимера, можно
получить удобное выражение характеристики р— v — Тс помощью
уравнения (VI.2). В этом случае только значение Vw следует рас-
считывать по групповым вкладам, поскольку V (0} 1,3Vwh е/
10,3-10-47w.
Пример 2
Оценить удельный объем расплава полипропилена при: 200 °C, 1 атм;
250 °C, 1 атм и 200 °C, 600 атм.
Решение
Можно рассчитать и с помощью модифицированного уравнения (VI.1):
v = <о + R Т/Ми (р + я)
воспользовавшись следующими данными: R = 82,06 см3-атм/(моль-К); Ми =
= 42,1 г/моль = М\ <o=l,3VwIM = 1,3-30,68/42,1 = 0,948 см3/г; Vw=
= 30,68 см3/моль (табл. IV.4); л = Я/е; = 82,06/0,0396 = 2080 атм; е/ =
= 0,0396 см3/(моль • К) (табл. IV.6).
Расчет приводит к следующим результатам:
V, см*/г
Т, “С р, атм расчет опыт
200 1 1,39 1,34
250 1 1,44 1,39
200 600 . 1,29 1,28
Экспериментальные данные были получены Фостером, Вальдманом и Гриски
[11].
С помощью уравнения состояния можно получить уравнения
для коэффициента теплового расширения (а) и для сжимаемости (х).
Преобразование уравнения (VI.2) дает:
I г (0) + /?Т/(л + р) , (VI.4)
Отсюда вытекают следующие выражения в частных производных:
/ дУ \____R f дУ \ _ RT
\дт)р-р + п и \др)т~ (р + л)2 1 '
Подстановка приводит к уравнениям
“=~('ат’)р= г , V(0) (VL6)
__________1_________ (VI 7'
'V \др/т~ 7(0) О1-'/
(р + л) +-д?г-(р + я)2
Сжимаемость х представляет собой величину, обратную модулю
упругости при всестороннем сжатии или объемному модулю упру-
гости материала. Эта важная характеристика будет рассмотрена
в гл. IX.
94
Область применимости уравнения (VI.2) ограничена расплавами
полимеров. Для аморфных полимеров, находящихся ниже точки
плавления, внутреннее давление л можно определить так:
(VI.8)
где U — внутренняя энергия одного моля вещества; здесь л зависит от темпера-
туры и давления.
Значения внутреннего давления для некоторых полимеров при
комнатной температуре были приведены Алленом с соавторами [6].
Они оказались по порядку величины близкими к плотности энергии
когезии екоГ. Воке [31] теоретическим путем получил, что
Л 1 ,3бког
(VI.9)
Ниже сравниваются значения л и еког при 20 °C для ряда поли-
меров:
я, атм гког* а™
Полиэтилен 3200 2500-2900
Полиизобутилен 3300 2500—2700
Полистирол 4500 3000—3600
Полихлортрифторэтилен 3700 2200—2600
Поливинилацетат 4200 3600—5000
Полиэтилакрилат 4300 3500—3700
Полиметилметакрилат 3750 3400—6800
Полиоксипропилен 3700 2300—4100
П о лидимети л си локс ан 2350 2200—2400
Коэффициент перевода в СИ: 1 атм = 1,013 • 10* Н/м! = 1,013х
Х10» Дж/м'.
Отсюда видно, что уравнение (VI.9) действительно справедливо
в качестве первого приближения.
ПОВЕРХНОСТНАЯ ЭНЕРГИЯ
Поверхностная энергия — прямое проявление межмолекулярных
Взаимодействий. Молекулы, находящиеся на поверхности жидкости
или твердого тела, испытывают воздействие неуравновешенных
молекулярных сил, вследствие чего получают дополнительную
энергию по сравнению с молекулами, находящимися внутри
жидкости или твердого тела.
В жидкостях поверхностная энергия проявляет себя как сила,
стремящаяся уменьшить площадь поверхности до минимально воз-
можной величины. Поверхностная энергия измеряется в единицах
силы, отнесенной к единице длины, или в единицах энергии, отне-
сенной к единице площади.
Поверхность твердого тела подобно поверхности жидкости обла-
дает дополнительной свободной энергией. Но, поскольку подвиж-
ность молекул на поверхности твердых тел отсутствует, эта поверх-
ностная энергия не может непосредственно наблюдаться в виде
95
поверхностного натяжения. Измерять свободную энергию можно
лишь с помощью косвенных методов.
Дополнительная свободная энергия на поверхности раздела двух
конденсированных фаз известна под названием межфазного натя-
жения.
Поверхностное и межфазное натяжение оказываются важными
характеристиками потому, что они определяют ход ряда важных
[технологических процессов, например прядение волокон, адгезию
I полимеров, устойчивость дисперсий и набухание твердых полимеров
\ в жидкостях.
ПОВЕРХНОСТНОЕ НАТЯЖЕНИЕ
ЖИДКОСТЕЙ И РАСПЛАВОВ
ОПРЕДЕЛЕНИЯ
Поверхностное натяжение жидкости обозначается символом у/,
поверхностное натяжение твердого тела — ys, межфазное натяже-
ние на поверхности раздела жидкость — твердое тело — ysZ; по-
верхностное натяжение твердого тела, находящегося в равновесии
с насыщенным паром жидкости, обозначается ysa. Наконец, давле-
О ние. набухания в равновесных условиях
HpanH = Ys Yso
МЕТОДЫ ОПРЕДЕЛЕНИЯ ПОВЕРХНОСТНОГО
НАТЯЖЕНИЯ ЖИДКОСТЕЙ
Существует ряд независимых методов определения поверхностного
натяжения жидкостей. Один из первых использованных прямых
методов заключался в измерении силы, необходимой для отрыва,
например, металлического диска или кольца от поверхности жид-
кости.
Наиболее популярный квазистатический метод — определение
высоты подъема жидкости в капилляре. Наряду с этим существуют
два родственных метода (метод взвешивания капли и метод давления
в пузырьке) измерения поверхностного натяжения, связанные с опре-
делением избыточного давления под криволинейными поверхностями.
Наконец, существуют динамические методы, в которых измеряют
длину волны, образующейся на поверхности жидкости под действием
источника колебаний известной частоты, или измеряют период
колебаний вибрирующих капель.
ОЦЕНКА ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
ЖИДКОСТЕЙ из РОДСТВЕННЫХ ХАРАКТЕРИСТИК
Поверхностное натяжение есть проявление действия межмолекуляр-
ных сил, поэтому можно думать, что эта характеристика связана
с другими свойствами, в основе которых лежит действие межмоле-
кулярных сил. К таким характеристикам относятся, например,
внутреннее давление, сжимаемость и плотность энергии когезии.
96
Прежде всего, существуют соотношения между сжимаемостью
и поверхностным натяжением. Согласно МакГовену [20]
иу,/’ = 1,33-10"8 единиц СГС (VI.10)
или
у = 5,6 • 10-вх_,/* (VI.10а)
Другое интересное эмпирическое соотношение между поверх-
ностным натяжением и плотностью энергии когезии можно полу-
чить исходя из того, что сжимаемость обратно пропорциональна
плотности энергии когезии. Тогда с учетом формулы (VI.11) можйо
записать, что
Т-еУо’г-6'71 (у1-И)
где S — параметр растворимости. ,
Соотношение (VI. 12) показывает связь между поверхностным
натяжением, энергией когезии и мольным объемом. Гранберг и Нис-
сан [15] получили количественно правильное соотношение между
бтими величинами. Для низкомолекулярных соединений эти авторы
ввели понятие о работе когезии
И7Ког = 2у^д/аг’/' (VI.12)
где JVA — число Авогадро; поверхностное натяжение у следует выражать в кал/см2
для того, чтобы получить Иког в. кал/моль.
Отношение ^ког/Иког оказывается характеристической постоян-
ной для рассматриваемой жидкости. Значение этой постоянной
близко к 3,5 для неполярных жидкостей и заключается между 4 и
8 для жидкостей, в которых образуются водородные связи.
РАСЧЕТ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
ИЗ АДДИТИВНОЙ ФУНКЦИИ; ПАРАХОР
Мольный парахор является полезным параметром для оценки
значений поверхностного натяжения. Парахор представляет собой
следующую аддитивную величину:
p = Y,/4M/p = Y‘/4F . [VI.13)
Понятие парахора ввел Сагден [291; он же привел перечень атом-
ных постоянных. Позже атомные и групповые вклады несколько
видоизменили и усовершенствовали Мемфорд и Филлипс [21] и
Квейл [3].
Полученные различными исследователями групповые вклады
в парахор представлены в табл. VI.4. Если групповые вклады Р и V
известны, то у получают по формуле
Y.=(P/V)4 (VI,14)
7 Заказ 899 Q7
Таблица VI.4. Значения атомных и структурных вкладов в парахор,
полученные различными авторами *
Структурный элемент Сагден Мемфорд, Филлипс Фогель Квейл
сн2 39,0 40,0 40,0 40,0
с 4,8 9,2 8,6 9,0
н 17,1 15,4 15,7 15,5
О 20,0 20,0 19,8 19,8
О2 (в сложных эфирах) 60,0 60,0 54,8 54,8
N 12,5 17,5 —. 17,5
S 48,2 50,0 49,1 49,1
F 27,5 25,5 — 26,1
€1 54,3 55,0 55,2 55,2
Вг 68,0 69,0 68,8 68,0
I 91,0 90,0 90,3 90,3
Двойная связь 23,2 19,0 19,9 16,3-19,1
Тройная связь 46,4 38,0 40,6 40,6
Трехчленное кольцо 16,7 12,5 — 12,5
Четырехчленпое кольцо 11,6 6,0 — 6,0
Пятичленное кольцо 8,5 3,0 — 3,0
Шестичленное кольцо 6,1 0,8 — 0,8
Семичленное кольцо — 4,0 — 4,0
* Поверхностное натяжение дано в дин/см, мольный объем — в моль/см’.
Парахор практически не зависит от температуры, поэтому выра-
жение (VI. 14) можно также применять для расчета температурной
зависимости поверхностного натяжения.
МЕЖФАЗНОЕ НАТЯЖЕНИЕ НА ПОВЕРХНОСТИ
РАЗДЕЛА ТВЕРДОЕ ТЕЛО — ЖИДКОСТЬ
На рис. VI. 1 символ S изображает поверхность твердого тела,
находящегося в контакте с жидкостью. Здесь имеется в виду, что
система продолжается влево. Жидкость Lx смачивает твердое тело
и стремится растечься вправо по поверхности. Контактный угол
0 равен нулю. Во втором случае (жидкость L2) тенденция к растека-
нию по поверхности менее выражена, и значение контактного угла
лежит между 0 и л/2. В третьем примере (жидкость L3) жидкость
не смачивает поверхность, и контактный угол превышает л/2; жид-
кость стремится уменьшить площадь контакта с твердым телом.
Равновесные контактные углы между жидкостями и твердыми
телами, как правило, обсуждаются в терминах уравнения Юнга
Y1 •COS0=(Ys — Yst)— (Ys — Y<o) = (Ys — Ys/)— Лравнов Ys~Ys* (VI.15)
Величина cos 0 носит название адгезионного натяжения. Полное
смачивание возникает при cos 0 = 1 или 0 = 0°.
Очевидно, что смачиванию благоприятствуют относительно низ-
кая свободная энергия поверхности раздела, высокая поверхностная
98
энергия твердого тела и низкая свободная энергия поверхности
жидкости (поверхностное натяжение). К сожалению, для прямых
экспериментальных измерений можно выбрать только и fl. Однако
для того чтобы понять явление адгезии, важно знать значения и
ysl. Фокс и Зисман [13] сделали важный шаг на пути решения этой
проблемы. Эти авторы показали, что для гомологических рядов
жидкостей, помещаемых на одном и том же твердом теле, график
зависимости cos 0 от yt оказывается в общем случае прямой линией.
Рис. VI.1. Контактные углы, образованные жидко-
стями на поверхности твердого тела.
Зисман [5, 33] ввел понятие о критическом поверхностном натя-
жении смачивания (укрит)- Эта величина определяется как значение
У/ в точке пересечения графика зависимости cos 0 от yt с горизон-
тальной линией, соответствующей cos 0 = 1. Жидкость с yt меньше
Укрит растекается по поверхности твердого тела. Численно укрит
примерно равно уs.
Другой подход применили Джирифалко и Гуд [14], которые полу-
чили следующие важные соотношения между ys, уг и ysl:
Ys/ = Ys + y/—2Ф (YsYz)7’
где
4(WZ*
Комбинирование уравнений (VI.15) и (VI.16) дает
[Y/ (1 + COS 0) + Лравнов]2 (1-|-СО80)2
Ys = 4Ф2у/ —4Ф2
соз0 = 2Ф ^2фГ-^)‘Л-1
\ Y/ ' X У1 /
(VI.16)
(VI.17)
(VI.18)
(VI.19)
С помощью уравнения (VI. 18) можно рассчитать поверхностное
натяжение твердого тела из результатов измерений контактной
угла. Если ys известна, то уравнение (VI.16) позволяет рассчитан
межфазное* натяжение ysZ. Используя соотношения (VI. 19), можш
найти контактный угол в системах твердое тело — жидкость.
7* 99
ПОВЕРХНОСТНАЯ ЭНЕРГИЯ ПОЛИМЕРОВ
В ТВЕРДОМ СОСТОЯНИИ
МЕТОДЫ ОПРЕДЕЛЕНИЯ ПОВЕРХНОСТНОГО
НАТЯЖЕНИЯ ТВЕРДЫХ ТЕЛ
Существует три способа оценки поверхностного натяжения твер-
дого тела ys. Первый способ основан на измерении контактного
угла между твердым телом и различными жидкостями с последующим
„расчетом поверхностного натяжения по уравнению (VI. 18). Второй
способ заключается в определении укрит по Зисману [51, предпола-
гая, что ys ?крит- Третий способ представляет собой экстраполя-
цию данных по поверхностному натяжению расплавов полимеров
к комнатной температуре [25].
ОЦЕНКА ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
ТВЕРДЫХ ПОЛИМЕРОВ С ПОМОЩЬЮ ДРУГИХ ВЕЛИЧИН
Соотношения (VI. 10а) и (VI.И) также можно применить для
оценки поверхностного натяжения твердых полимеров, если изве-
Рис. VI.2. Соотношение между поверх-
ностным натяжением и энергией ко-
гезии.
рис. VI.2 можно описать уравнением
стно значение сжимаемости или
параметра растворимости.
Имеется и другая интерес-
ная корреляция между рабо-
той когезии 1см. формулу
(VI. 12)] и энергией когезии.
Если для неразветвленных по-
лимеров работа когезии и энер-
гия когезии оценены в расчете
на атом цепи главных валент-
ностей, то можно предположить
наличие корреляции между
EKor/Z и величиной у (VIZ)'1*.
Соответствующие данные пока-
заны на рис. VI.2 для тех по-
лимеров, для которых имеются
значения у и Еког. Кривую на
(VI.20)
Рис. VI.2 или формулу (VI.20) можно далее использовать для пред-
сказания у тех полимеров, для которых либо непосредственно,
либо по групповым вкладам известны значения Еког и V.
ОЦЕНКА ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
ТВЕРДЫХ ПОЛИМЕРОВ С ПОМОЩЬЮ ПАРАХОРА
Поверхностное натяжение твердых полимеров можно рассчитать
из величин парахора на структурное звено путем применения фор-
мулы (VI. 13).
100
ЧИСЛОВЫЕ ЗНАЧЕНИЯ И СРАВНЕНИЕ
РАЗЛИЧНЫХ МЕТОДОВ ОЦЕНКИ
ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
В табл. VI.5 сравниваются рассчитанные и экспериментальные зна-
чения поверхностного натяжения полимеров, полученные различ-
ными методами. Расчет выполнялся по парахору, а также по фор-
мулам (VI.10), (VI.И) и (VI.20). Сам парахор рассчитывался с по-
мощью группорых вкладов Сагдена, поскольку последние дают
наилучшее соответствие с экспериментом.
Расхождения между различными методами находятся в разумных
пределах. Рассчитанные значения, за немногим исключением, удо-
влетворительно соответствуют экспериментальным данным.
Таким образом, расчет с помощью групповых ^вкладов в парахор
дает весьма надежные результаты, особенно если отсутствуют экс-
периментальные данные. Еще более высокую точность можно полу-
чить, если воспользоваться методами «стандартных характеристик»
или «стандартных соединений», которые рассмотрены в гл. III.
Пример 3
Оценить поверхностное натяжение твердого полиметилметакрилата и кон-
тактный угол между ним и метилениодидом (у = 50,8).
Решение
Мономерное звено полиметилметакрилата:
СНз
—СН2—С-
O=(i—О—СНз
Из табл. VI.4 и IV.4 заимствуются следующие значения групповых вкладов
в_£парахор и мольный объем:
1(—СН2—) / 1 \ \ С/ 2(-СН3) 1(—СОО—) Следовательно у = (Р /У)4 = (220,8/86,5)4 = 2,554 - 42,5 Согласно уравнению (VI. 19) сов 0 2Ф (Yj/yz)'^ — 1 Ф = 4(Г|И1)‘/’/(гУЧ-^/,)а 39,0 15,85 4,8 4,6 112,2 47,8 64,8 18,25 220,8 86,5 101
Таблица VI.5. Экспериментальные и рассчитанные значения
поверхностного натяжения полимеров
Полимер Экспериментальные значения vs, дин/см Рассчитанные значения V, дин/см
экстраполяция ^расплав по контактному углу он по V=(P/V)‘, уравнение (VI. 14) по х, уравнение (VI. Юа) 110 еког=вг- уравнение (VI.И) * 1 по уравнению (VI.20)
Полиэтилен 35,7 33,2 31 32 30 31 29
Полипропилен 29,6 — 29 32 31 32 30
Полиизобутилен 33,6 — 27 30 —. 29,5 27
Поли-цис-бутадиен — — 32 32 31 31 31
Поли-^ис-изопрен — — 31 35 31 30,5 30
Поливинилфторид — 36,7 28 33 —. — —
Поливинилхлорид — 41,5 39 42 54 39 37
Поливинилиденфторид — 32,7 25 27 —_ — —
Поливинилиденхлорид — 39,9 40 47 — — 37
Политрифторэтилен — 23,9 22 24 — — —
Полихлортрифторэтилен — — 31 27 — 26,5 27
Политетрафторэтилен — 19,0 18,5 26 23 21 19
Полигексафторпропилен — —- 16—17 21 —. —
Полихлорпропилен — — 38 42 — 34,5 34
Полистирол — 42 33-43 43 55 37 32
Политретбутилстирол — — — 35 — — —
Поли-2-хлорстирол - —— — 42 46 —— — —
Поливиниловый спирт — — 37 61 — 56 —
Полиакрилонитрил — — 44 60 — 55,5 54
Полиметилакрилат — — 41 45 55 41 —
Полиэтилакрилат — — 35 42 — 37,5 33
Полиметилметакрилат — 40,2 39 42 — 36 33
Полиэтилметакрилат — — 33 41 — 35,5 32
Полиоктилметакрилат • —. — — 35 — 33 —
Поливинилацетат — — 36 40 —- 38,5 34
Прлиметилвиниловый эфир Полиоксиметилен — — 29 32 — — —
— — 36 31 62 43 41
Полиоксиэтилен 43 — 43 32 —- — —
Полиоксипропилен 32 — 32 32 —. — 35
Политетрагидрофуран — — — 31 — 33 34
Полидиметилсилоксан 20 — 24 — — 27,5 27
Найлон 6 —. 40-47 46 47 59 — —
Найлон 11 — 31,0 33 42 — — —
Найлон 6,6 ' — 39,3 46 47 — 61,5 57
Найлон 10,10 28,5 32 43 —— — —
Полиэтилентерефталат — 43 41-47 49 — 45 39
Полибисфенолкарбонат — — 42 33 46,5 — —
Коэффициент перевода в СИ: 1 дин/см = 10-’ Н/м = 10-а Дж/м!.
Коэффициент пропорциональности равен 1,9.
102
Выше было найдено, что Vs = 86,5. Для метилениодида М = 267,9 и р = 3,33;
следовательно, Vi = 80,6. Отсюда Ф = 1,00. Контактный угол для метилен-
иодида
cos0=«2(ys/yz)*/2—1 2 (42,5/50,8)‘л—1 = 0,83
так что 0 34°. Адгезионное натяжение метилениодида на полиметилметакри-
лате
у; сов 0 = 50,8 X 0,83 42 дин/см
Джарвис с соавторами [19] приводил экспериментальное значение 0 = 41’;
из уравнения (VI.19) следует, что Ys = 39 дин/см. Эта величина удовлетвори-
тельно согласуется с экспериментальными значениями укриг.
ОБОБЩЕННАЯ ФОРМУЛА МЕЖФАЗНОГО
НАТЯЖЕНИЯ
Выражение (VI. 16) можно переписать в обобщенном виде (поскольку
Ф ~ 1):
Y12 = Yi + Y2—2Ф (YiY2)‘/! (VI.21)
или
Y12*« (y1/2— yV*)2 (Vi.21а)
Однако полученное выражение справедливо лишь для соединений
без водородных связей, как это показал Фоуке [12].
Он, рассматривая теоретически действующие на поверхностях
разделов силы притяжения, предположил, что суммарная свободная
энергия поверхности образуется из вкладов различных межмолеку-
лярных сил, действующих на этой поверхности. Таким образом,
поверхностную свободную энергию можно записать в следующем
виде:
Y = y‘' + yA (VI.22)
где верхние индексы d и h указывают на дисперсионную составля-
ющую и составляющую, обусловленную образованием водородных
связей, соответственно. Исходя из этого предположения, Оуэнс
и Вендт [22] предложили следующую общую форму выражения для
межфазного натяжения:
Y12 = Yi,+ Y2-2(yiY2)‘/!-2(y?Y2),/‘ (VI.23)
или
Yi2 = [(YO,/!-(v0,/!]2 + [M),/2-(Y?)i/e]2 (VI.23а)
Вещества 1 и 2 могут быть либо в жидком, либо в твердом состояниях,
либо, наконец, могут представлять собой смесь твердого тела и
жидкости.
Если жидкости 1 и 2 не смешиваются, значения и известны
и, кроме того, одна из этих жидкостей неполярна (yft = 0), то
103
составляющие у для обеих жидкостей можно получить следующим
образом: измеряют межфазное натяжение одним из существующих
способов (у12) и решают уравнения (VI.23) и (VI.22). Таким способом
были исследованы, различные жидкости; значения yz, yf и у? приве-
дены в табл. VI.6.
Таблица V1.6. Составляющие силы, поверхностного натяжения
различных жидкостей по данным Фоукса [12]
и Оузнса и Вендта [22]
Жидкость ?z, дин/см d tl, дин/см Л VI , дин/см
н-Гексан 18,4 18,4 0
Диметилсилоксан 19,0 16,9 2,1
Циклогексан 25,5 25,5 0
Декалин 29,9 29,9 0
Трикрезилфосфат 40,9 39,2 ±4 «si
а-Бромнафталин 44,6 47±7 «ь0
Трихлордифенил 45,3 44±6 «з1.3
Метилениодид 50,8 , 49,5+Д «=<1,3
Формамид 58,2 39,5 ±7 «si 9
Глицерол 63,4 37,0±4 «s26
Вода 72,8 21.8+0.7 51
Коэффициент перевода в Си: 1 дин/см=10-“ Н/м = 10-* Дж/м*.
Пример 4
Оценить значения у'* и уА воды, если известно, что для воды у = 72,8 дин/см
при 20° С, межфазное натяжение на границе раздела циклогексан — вода соста-
вляет 50,2 дин/см при 20 °C, для циклогексана у = 25,5 дин/см.
Решение
Для циклогексана у* = 0, поэтому у^= уц = 25,5. Из уравнения (VI.23)
следует, что
Y12 = Yh«O, ц = 50,2 = 25,5+ 72.8 2 (25,5ygjO) I*
Отсюда 7н>о = 22,7 дин/см. Следовательно, Yh«o = 72,8 ~ 22>? = 50,1 дин/см.
Полученное значение хорошо согласуется с наиболее реальными значениями
величин YglO = 21,8 и Yhso= 51Д
Оуэнс и Вендт приводят также более общее выражение для (VI. 19):
1 + cos 0 «=! 2 ------------1-------------- (VI .24)
L П Y/ J
Это уравнение позволяет получить значения yd и yhs по данным изме-
рений контактных углов 0 двух жидкостей, если значения yz,
yf и у? обеих жидкостей известны.
104
Пример 5
Оценить ys и составляющие и для поливинилхлорида, если известно, что
для воды yHiO = 72,8, уд о= 21,8, удгО= 51,0; Для метилениодида уми= 50,8,
у„И = 49,5, у„и= 1,3; контактные углы для поливинилхлорида 0Н О=87а,
Оми = 36°.
Решение
Подстановка данных в уравнение (VI.24) дает:
для воды
1+0,052 = 2
(yf)*/’ 21,8*/’
72,8
(^)*/’51,0‘/« ’
72,8
для метилениодида
1 + 0,809 = 2 (y^)*/,49,51/» , (y^>Z,1.31/8 ~ 50,8 1 50,8
Совместное решение этих уравнений приводит к следующему результату:
yf = 40,0; у^=1,5; Ys = 41,5
Оуэнс и Вендт определили таким способом свободную поверхностную энергию
и составляющие свободной энергии для ряда твердых полимеров. Полученные
значения приведены в табл. VI.7.
Таблица VI.7. Составляющие поверхностной энергии
различных твердых полимеров (в дин/ем) по [22]
Полимер +рнт
Полиэтилен низкой плотности 33,2 0,0 33,2 31
Поливинилхлорид 40,0 1,5 41,5 39
Полив инилиденхлорид 42,0 3,0 45,0 40
Поливинилфторид 31,3 5,4 36,7 28
Поливинилиденфторид 23,2 7,1 30,3 25
Политрифторэтилен 19,9 4,0 23,9 22
Политетрафторэтилен . 18,6 0,5 19,1 18,5
Полиэтилентерефталат 43,2 4,1 47,3 43
Полиметилметакрилат 35,9 4,3 40,2 39
Найлон 6,6 40,8 6,2 47,0 46
Полистирол 41,4 0,6 •42,0 43
Графит 123
Коэффициент перевода в СИ: 1 эрг/см2 = 1 дин/см = 10-* Н/м=10-’ Дж/м2.
АДГЕЗИЯ ПОЛИМЕРОВ
Мерой взаимного притяжения двух твердых тел Sr и на их гра-
нице раздела является обратимая работа адгезии lTaar. Эта вели-
чина определяется соотношением Дюпре
^r = VSl + vSj-YSlSl^2[(Y/X)1/l + (Y^()1/!] (VI.25)
105
Величина Жадг представляет собой работу, которую необходимо
затратить для разделения и в результате такого разделения
создается единичная площадь поверхности тел Sx и S2 при одновре-
менном устранении единичной площади поверхности контакта тел
<$х и S2.
На практике важно знать, является ли в действительности адге-
зионный контакт устойчивым по отношению к жидкостям. В таком
случае работа адгезии
И^адг — YSiI + Ys,j — YS1s.
(VI.26)
Если поверхность контакта тел Sx и S2 погружена в жидкость
L, а работа адгезии Жадг, рассчитанная по соотношению (VI.26),
отрицательна, то такие условия благоприятствуют разделению 6’х
и 52, которое в этом случае произойдет самопроизвольно. Дело
в том, что разделение уменьшает свободную энергию всей системы.
Условие самопроизвольного разделения:
Ys^^Ys,! +YS1;
(VI.27)
Все межфазные натяжения можно рассчитать с помощью уравне-
ния (VI.23).
Пример 6 (Оуэнс [23])
Оценить возможность расслоения между покрытием и основой в следующем
случае: полипропиленовая пленка, обработанная газопламенным способом, по-
крыта сополимером винилиденхлорида с метакрилатом и погружена в 0,5% -нцй
раствор н-додецилсульфата натрия. Известны следующие характеристики поли-
мера, покрытия и жидкости:
+ у'
Полипропилен после обра- 33,5 4,1 37,3
ботки пламенем
Сополимер 38,9 14,7 53,6
Раствор н-додецилсульфата 29,0 8,2 37,2
натрия
Решение
Сначала рассчитываются значения межфазных натяжений (у12), т. е. yS1Sit
Ysii и Vs2p с помощью уравнения (VI.23):
YSiS2 = 37,6-|-53,6—2 (33,5-38,9)1/2-2(4,1.14,7)1л = 3,4 дин/см
YSli = 37,6+ 37,2—2 (33,5-29,0)!/2-2(4,1.8,2)l/i=0,9 дин/см .
Ys2j = 53,6+ 37,2-2 (38,9 29,0)V«-2 (14,7 • 8,2)V* =1,6 дин/см
Правило (VI.27) дает
ТГадг = 1,6 + 0,9 - 3,4 = -0,9
Расслоение произойдет, поскольку РГадг<гО.
106
ЛИТЕРАТУРА
1. Hildebrand J. H. and Scott R. L.,
«Solubility of Non-electrolytes»,
Reinhold, New York, 1950 (3rd
ed.).
2. Weiss Ph., (ed.), «Adhesion and
Cohesion», Elsevier, Amsterdam,
1962.
3. Quayle O. R., Chem. Revs. 53
(1953) 439.
4. Sugden S., «The Parachor and
Valency», London, 1930.
5. Zisman W. A., p. 1—51 in «Con-
tact Angle, Wettability and Adhe-
sion», Adv. in Chem. Ser. 43,
Am. Chem. Soc., 1964; p. 176—
208 in «Adhesion and Cohesion»,
ed. by P. Weiss, Elsevier, Amster-
dam, 1962.
6. Allen G., Gee G., Mangaraj D.,
SimsD. and Wilson G. J., Polymer
1 (1960) 467.
7. Bondi A., J. Chem. Eng. Data 8
(1963) 371; J. Polymer Sci. A2
(1964) 3159; «Physical Properties
of Molecular Crystals, Liquids
and Glases», Wiley, New York,
1968.
8. Bunn C. W., J. Polymer Sci. 16
(1955) 323.
9. Di Benedetto A. T., J. Polymer
Sci. Al (1963) 3459.
10. Dunkel M., Z. physik. Chem.
A138 (1928) 42.
11. Foster G. N., Waldman N. and
Griskey. R. G., Polymer Eng.
Sci. 6 (1966) 131.
12. Fowkes F. M., Ind. Eng. Chem.
56 (1964) 40.
13. Fox H. W. and Zisman W. A.,
J. Colloid. Sci. 7 (1952) 109
and 428.
14. Girifalco L. A. and Good R. J.,
J. Phys. Chem. 61 (1957) 904;
62 (1958) 1418 and 64 (1960)
561.
15. Grunberg L. and Nissan A. H.,
Trans. Faraday Soc. 45 (1949)
125.
16. Hayes R. A., J. Appl. Polymer
Sci. 5 (1961) 318.
17. Hoftyzer P. J. and Van Kreve-
len D. W., Paper (Nr. Illa—15)
presented at the International
Symposium on Macromolecules
of I UP AC, Leyden (1970).
18. Hoy K. L., J. Paint Tedinol.42
(1970) 76.
19. Jarvis N. L., Fox R. B. and
Zisman W. A., Advances in Che-
mistry 43 (1964) 317.
20. McGowan J. C., Polymer 8 (1967)
57.
21. Mumford S. A. and Phil-
lips J. W. C., J. Chem. Soc. 130
(1929) 2112.
22. Owens D. K. and Wendt R. C.,
J. Appl. Polymer Sci. 13 (1969)
1741.
23. Owens D. K., J. Appl. Polymer
Sci. 14 (1970) 1725.
24. Rheineck A. E. and Lin K. F., J.
Paint Technol. 40 (1968) 611.
25. Roe, Ryong-Joan, J. Phys. Chem.
69 (1965) 2809.
26. Small P. A., J. Appl. Chem. 3
(1953) 71.
27. Smith R. P., J. Polymer Sci. A2,
8 (1970) 1337.
28. Spencer R. S. and Gilmore G. D.,
J. Appl. Phys. 21 (1950) 523.
29. Sugden S., J. Chem. Soc. 125
(1924) 1177.
30. Van Krevelen D. W., Fuel. 44
(1965) 229.
31. Voeks J. F., J..Polymer Sci. A2
(1964) 5319.
32. Vogel A. I., J. Chem. Soc. (1948)
624, 1825.
33. Zisman W. A., Ind. Eng. Chem.
55 (1963) 19.
глава vii. ТЕМПЕРАТУРЫ ПЕРЕХОДОВ
В этой главе будет показано, что с химическим строением полимеров
можно связать температуры двух основных переходов: температуру
перехода из стеклообразного состояния в высокоэластическое и
температуру плавления кристаллического полимера. Подобного
рода корреляции получаются на основе метода групповых вкладов
и носят чисто эмпирический характер.
ВВЕДЕНИЕ
Как уже было показано в гл. II, невозможно понять природу харак-
теристик полимера, если не исследованы переходы, происходящие
в таких материалах, и не определены температуры этих переходов.
К основным переходам относятся переход между стеклообразным
и высокоэластическим состояниями и плавление кристаллического
материала.
Можно, однако, наблюдать и ряд других переходов, играющих
второстепенную роль. Для обозначения таких вторйчных переходов
не существует единого способа. Как правило, пользуются символами
Та, и т. д., но разные авторы обозначают одни и те же переходы
по-разному.
Вероятно, существуют по крайней мере три перехода в стекло-
образном состоянии ниже Tg: в областях от 0,57^ до 0,8Т£, от
0,357^ до 0,5Tg и при очень низких температурах от 4 до 40 К.
В высокоэластическом аморфном и в кристаллическом состояниях
могут наблюдаться переходы между Tg и Тт. Подробности читатель
найдет в обзорных „работах Бойера [2, 15], а также Шена и Айзен-
берга [9]. 4
ТЕМПЕРАТУРА СТЕКЛОВАНИЯ
Многие авторы предлагали различные соотношения между химиче-
ским строением и температурой перехода в стеклообразное состояние
полимеров. Используемые при этом методы, как правило, базиро-:
вались на предположении, что функциональные группы в повторя-
ющихся звеньях дают взвешенные аддитивные вклады в Tg. В слу-
чае идеальной аддитивности вклад данной группы не зависит от
природы соседних групп. Такой идеальный случай редко встречается
108
на практике, однако аддитивность зачастую можно аппроксимиро-
вать путем соответствующего выбора функциональных групп. Мы
еще вернемся к этому обстоятельству позже.
В общем виде соотношения для Ts имеют следующую форму:
Tg 2 — SSl?gl
(VII.1)
откуда
Tg—2 S(?,£<
i I i
(VII.2)
где Tgl- — характеристический вклад в Tg данной функциональной группы;
st — весовой коэффициент, приписываемый данной функциональной группе.
Хайес [19] принял, что У s^g,. идентично мольной энергии коге-
i
зии, и получил ряд правил для расчета отдельных значений
Бартон и Ли [10] предположили, что si идентична весовой или моль-
ной доле функциональных групп, и рассчитали значения характери-
стической постоянной Tgt для некоторых функциональных групп.
В более поздней работе Ли [23] сравнил четыре метода оценки
S{ для выбранной группы полимеров. Хорошую корреляцию уда-
лось получить в предположении, что значение Tgi данной функцио-
нальной группы зависит от природы соседних групп. Однако это
приводит к необходимости использовать большое количество вели-
чин Т61. Соотношение, полученное в предположении независимости
Tst от соседних групп, оказалось гораздо менее хорошим.
Вейланд, Хофтицер и Ван Кревелен [25] также исследовали раз-
личные методы оценки и нашли, что надежность получаемых соот-
ношений оказывается довольно нечувствительной к правилам, при-
меняемым при расчетах sz. Наилучшее соответствие получили в пред-
положении, что s. равно числу атомных расстояний в главной цепи
(т^е. пропорционально числу атомов Z, вдоль цепи главных валент-
ностей в пределах мономерного звена). Если в цепь главных валент-
ностей входит ароматическое кольцо, то Z оценивается следующим
образом:
о-фенилен, Z = 2; л-фенилен, Z = 3; n-фенилен, Z = 4
На основании этого можно рассчитать значения T&i для ряда важ-
ных функциональных групп.
В данной главе будет использован, в сущности, тот же метод.
Для упрощения записи аддитивную величину 2 ?i?gi обозначим
i
через Yg — мольную функцию перехода в стеклообразное состояние.
Уравнение (VII.2) примет вид
(VII.2а)
109
Применяемый корреляционный метод годен только для линейных
неразветвленных полимеров. В следующем разделе этот метод будет
Применен к таким линейным полимерам, которые могут содержать
боковые ответвления лишь из одной функциональной группы. При-
менение такого метода к полимерам с более длинными боковыми
цепями будет рассмотрено в другом разделе.
ТЕМПЕРАТУРА ПЕРЕХОДА В СТЕКЛООБРАЗНОЕ
СОСТОЯНИЕ ДЛЯ ПОЛИМЕРОВ БЕЗ БОКОВЫХ ЦЕПЕЙ
Как было установлено выше, на практике не выполняется принцип
идеальной аддитивности, т. е. независимости величин Ygl от при-
роды других имеющихся в структурной единице групп. Эффектив-
ный метод корреляции можно получить лишь при условии учета
влияния соседних групп на различных этапах применения метода.
Рис. VII.1. Зависимость Yg от п для Рис. VII.2. Зависимость Кйот п для
полиэфиров. полиамидов.
1. Выбор структурных единиц. В ряде случаев более хорошая
корреляция может быть получена, если две соседние группы рас-
сматривать как одну функциональную группу. Вклад в Yg эфирной
группы —С—О—, например, отличается от суммы вкладов групп
II
О
—С— и—О—. Другой пример — атом углерода с двумя метильными
II
О
группами, который при расчете рассматривается как одна бифунк-
циональная группа СН3—С—СН?.
110
2. Различные вклады метиленовых групп. Обычный вклад метиле-
новой группы равен 170. Для полиамидов, полиуретанов и поликарб-
амидов нужно, однако, пользоваться величиной 270. Это обстоя-
тельство может быть обусловлено действием водородных связей
между цепями названных полимеров.
В качестве иллюстрации на рис. VII.1 показана зависимость Yg
от числа метиленовых групп п для ряда полиэфиров общей формулы
[—(СОО)2—(СН2)П—J
На рис. VII.2 представлен соответствующий график для поли-
амидов. Тангенс угла наклона линии дает значение 170 для поли-
эфиров и 270 для полиамидов.
Таблица VII.1. Групповые вклады Ygt
Группа Ygl Группа
-СН2- { -СН(СНа)- —С(СН3)2— -СН(С6Н5)- - С(СНз)(Св сн полиамиды, поликарбамиды полиуретаны другие полимеры Н5)- полиамиды полиуретаны полиоксиды, полисульфи- ды полисилоксаны поликарбамиды другие полимеры j 270 170 336 226 576 730 2200 1500 1500 1300 1200 1850 1500 -СНС1- —cf2— -СС12- —CFC1 -О— >с=о -С-О- 11 О —О—С-О- 11 о -с-о-с- II II 0 0 -S- 538 300 330 350 280 365 640 780 555 280
-oz- 2000 o=s=o 1 ' 1200
СНз 1 o=s=o 1 450
-^1 2300 о —N—С— 1 II но —N—С—О 1 1! Н О —N—С—N— 1 II 1 НОН 1 1000 71200
—СН=СН— ц ис транс —СН=С(СН3)—уис транс —CHF— 340 700 480 480 330 1450
СН3—Si—СН3 1 20
111
Таблица VII.2. Экспериментальные и рассчитанные аначения Тг (К) полимеров
Л Полимер Tg (экспери- мент) тг (расчет) Полимер Tg (экспери- мент) гй (расчет)
Полипропилен 253 253 Поли-4,4' -метилендифениленкарбонат Поли-4,4'-изопропилидендифенилен- карбонат 393 388
Полистирол Полиизобутилен 373 198 373 198 414/423 392
Поливинилфторид Поливинилхлорид 253/313 356 250 354 Поли-1,4'-тетраметилендибензангид- рид 319 329
Поливинилиденфторид Поливинилиденхлорид 233/286 255/288 235 250 Поли-4,4'-метилендиоксидибензангид- рид 357 356
Поли-1,2-дифторэтилен 323/371 330 Полигексаметиленадипамид 318/330 335
Полихлортрифторатилен 318/325 325 Полидекаметиленсебацинамид 333 312
Поли-а -метилстирол 390/465 450 Полигептаметилентерефталамид 396 406
Дис-полибутадиен 171 170 Поли-га-фенилендиэтиленсебацинат- 378 372
Транс-полибутадиен 259 260 амид 303 319
Полиоксиметилен Полиоксиэтилен 190/243 206 225 207 Политетраметиленгексаметил ендиуре- тан
Полиокситриметилен Полиокситетраметилен 195 185/194 198 192 Полифенилендиметиленгексаметилен- диуретан 329 337
Поли-и-ксилилендисульфид Полидекаметилентетрасульфид 296 197 300 201 Полигексаметилендодекаметилен- димочевина 322 323
Полиэтиленадипинат Полиэтилендодеканат 203/223 202 230 208 По лиметилен-бис(оксиди-л-фенилен) - сульфон 453 467
Полидекаметиленадипинат 217 203 Полиокси-бис(оксиди-н-фенилен) кетон 423 431
Полиэтилентерефталат Полидекаметилентерефталат 343/350 268/298 347 268 Полиизопропилиденди-п-фенилен-л1- фенилендисульфонат 388 395
"Полпдиэтиленгликольмалонат 244 241 Поли-3,5-диметил-п-оксифенилен 453/515 456
Полидиэтиленгликольоктадекандионат Поли-л-фенилениаофталат 205 428 199 428 Полидиметилсилоксан 150 150 \
3. Различные вклады п-фениленовых групп. Нормальный группо-
вой вклад и-фенилена составляет 1850, но для некоторых типов
полимеров пользуются другими значениями.
В табл. VI 1.1 приведены те значения вкладов, которые следует
использовать для Yg. В табл. VII.2 сравниваются эксперименталь-
ные и рассчитанные значения Т&. В общей сложности для примерно
220 полимеров среднее значение разности между эксперименталь-
ными и рассчитанными значениями составляет 13°.
Следовало бы заметить, однако, что для некоторых комбинаций
структурных групп метод расчета по уравнению (VII.2а) ненаде-
жен. Так, иногда рассчитанные значения Tg оказываются слишком
большими, например для комбинации группы —СН(СН3)— с кисло-
родом или серой и для полиэфиров и полиамидов, содержащих
комбинацию ——СН2—.
Для других комбинаций групп рассчитанные значения Tg могут
оказаться слишком низкими,- например для полиэфиров, содержа-
щих две рядом расположенные n-фениленовые группы, и для поли-
амидов, содержащих только фениленовые группы.
ТЕМПЕРАТУРА ПЕРЕХОДА В СТЕКЛООБРАЗНОЕ
СОСТОЯНИЕ ДЛЯ ПОЛИМЕРОВ С БОКОВЫМИ ЦЕПЯМИ
В этом разделе рассматриваются полимеры общей формулы
Q
-Р-С-
R-(СНа)№ СНз
где Р, Q и R — произвольные структурные группы.
Такая структура может быть получена из «основного полимера»
Q
I
—Р—С—
R—СНз
путем добавления N метиленовых групп в боковую цепь.
Температуры перехода в стеклообразное состояние для этой
группы полимеров можно вычислить по формуле
У£ = 2’в(2о+^=У£о + Ь(ЛГ) (VII.3)
или
(VII А)
_ Удо+НУ)
Zo+ЛГ
где N — число метиленовых групп в боковой цепи (в расчете на структурную
единицу); h (V) — поправочный фактор для Уг, учитывающий боковую цепь
и зависящий от N-, ZQ и Уй0 относятся к строению «основного полимера».
8 Заказ 699 ' * 113
Поскольку
yMo=2’goZo (VI 1.5)
(где Tg0 — температура перехода «основного полимера» в стекло-
образное состояние), то функцию h (IV) можно рассчитать по формуле-
fe(V) = Tg (Zo + V)-TgoZo
(VII.6)
В табл. VII.3 дана сводка типов полимеров, для которых можно-
получить значения Tg описанным способом. На рис. VII.3 предста-
----
Рис. VII.3. Температуры переходов в стекло-
образное состояние для полимеров с боко-
выми цепями:
X — полиолефины; О — поли-п-стиролы; Д — поли-
акрилаты: Ф — полиметанрилаты; □ — поливинило-
вые эфиры.
влены полученные этим
способом значения h (N).
Из рисунка видно, что для
низких значений 2V отно-
шение h(N)/N близко к
190 К, т. е. примерно
равно величине Yg мети-
леновой группы в основ-
ной цепи. При более вы-
соких значениях N, од-
нако, наклон резко меня-
ется, доходя до 470 К.
Для полистиролов и поли-
метакрилатов это резкое
изменение происходит при
.V = 11, для полиолефи-
нов и полиакрилатов —
при V «= 8. Высокие значе-
ния коэффициента h (N)/N t
для полимеров с длин-
ными боковыми цепями
могут обусловливаться
кристаллизацией боковыхJ
ответвлений.
В табл. VII.4 сравни-
ваются экспериментальные
и рассчитанные значения
Tg для ряда полимеров с
боковыми цепями. В общей
сложности для 70 поли-
меров этого типа среднее
отклонение между рассчитанными и экспериментальными данными
по Тg составляло приблизительно 10 К.
Пример 1
ч Оценить температуру перехода в стеклообразное состояние для полизтилен-
терефталата.
114
Таблица VII.3. Типы полимеров с боковыми цепями
Тип полимера р Q
Полиолефины -сн2- —н
Поливиниловые эфиры -сн2- -н
Полиалкилстиролы —сн2— -н
Полиакрилаты -сн2- —н
Полиизоакрилаты —сн2— —II
Полиметакрилаты -сн2- -СНз
Полидиены -(СН2)2-СН= —
Полидиэтиленгликольалкилмалонаты —СО(СН2)2О(СН2)2ОС— II II О О —II
Тип полимера R Z,
Полиолефины — 2 506
Поливиниловые эфиры -0- 2 512
Полиалкилстиролы 2 746
Полиакрилаты —СО—0— 2 560
Полиизоакрилаты —СО—0—СН(СН3)— 2 568
Полиметакрилаты —СО—0— 2 756
Полидиены — 4 820
Лолидиэтиленгликольалкилмалонаты — 10 2440
Решение
Структурное звено полиэтилентерефталата:
о-с—О-С—О-(СН2)2
II
Значения групповых вкладов Zi и Ygt (табл. VI 1.1):
zi
2СН2 2 340
2СОО 4 1280
1СвН4 4 1850
И = 10 Уй = 3470
8*
115
Таблрца VI1.4. Экспериментальные и рассчитанные аначения Те
для ряда полимеров с боковыми цепями (К.)
Полимер тй (эксперимент) те (расчет)
Полипропилен 253 253
Полибутилен 228/249 232
Полипентен 221/233 222
Полиоктен 208/228 208
Полигексадецен. 246/313 287
Полиметилвиниловый эфир 252/260 256
Полипентилвиниловый эфир 207 212
Поли-4-метилстирол 366/379 373
Поли-4-этилстирол 300/351 312
Поли-4-нонилстирол 220 227
Поли-4-нонадецилстирол 305 303
Полиметилакрилат 279/282 280
ПолиэтилакриЛат 249/252 250
Полигептилакрилат 213 213
Полигексадецилакрилат 308 311
Полиизопропилакрилат 262/284 284
Полиизобутилакрилат 249/256 253
Полиметилметакрилат 378/399 378
Полиэтилметакрилат 285/338 315
Полигексилметакрилат 256/268 244
Полигексадецилметакрилат 288 274
Полиметилбутадиен 205/225 205
Полигексилбутадиен 190 197
Полидиэтиленгликольметилмалонат 244 244
ПолидиэтиленглиКольнонилмалонат 214 220
Следовательно, рассчитанное'значение составляет:
Тг = -^- = 3470/10 = 347 К
Экспериментальные значения изменяются от 343 до 350 К.
Пример 2
Оценить температуру перехода в стеклообразное состояние’ для полигекса-’
децилметакрилата.
Решение
Структурное звено полимера:
СН3
—СН2— С— .
О=(!:-О-(СН2)15-СН3
«Основной полимер» — полиметилметакрилат с = 378; Za = 2; следова-
тельно, Ye0 = 756. Для боковой цепи N = По рис. VII.3 h (N) = 3900;
С помощью формулы (VII.4) можно получить, что
Т6=(756 + 3900)/(2 +15) = 4656/17 = 274 К
Приводимое в Литературе значение составляет 288 К.
116
СООТНОШЕНИЕ МЕЖДУ ГРУППОВЫМИ ВКЛАДАМИ
В ВЕЛИЧИНУ Tg и УДЕЛЬНОЙ ТЕПЛОЕМКОСТЬЮ
С точки зрения теории выбор атомных расстояний в качестве основы
для оценки может оказаться довольно произвольным. Групповые
вклады в удельную теплоемкость, как это было обсуждено в гл. V»
теоретически могут быть более подходящей основой для оценки «(.
Для ряда групп, входящих в неразветвленный полимер, наблюдается
все же удивительное соответствие между этими двумя методами
оценки sit как это следует из данных, приведенных ниже:
-СН2--
л-СвНф—
—О—
—СОО—
—CONH—
sz-, атомные
расстояния (—^i)
1
4
1
2
2
удельная
теплоемкость
1
3,1
0,7
1.9
1,8
Расчеты с помощью соотношений, основанных на групповых
вкладах в удельную теплоемкость, позволяют получить такие зна-
чения Tg, которые практически совпадают с данными, вычислен-
ными с помощью описанного в этой главе метода. Следовательно,
более предпочтителен метод 'оценки на основании атомных рас-
стояний.
ТЕРМОДИНАМИКА ПЕРЕХОДА
СТЕКЛООБРАЗНОЕ — ВЫСОКОЭЛАСТИЧЕСКОЕ
СОСТОЯНИЕ
Переход стекло — каучук обнаруживает некоторое сходство с пере-
ходом второго рода. Если бы это был действительно переход второго-
рода, то должны были бы выполняться следующие соотношения:
dTg ' TgV (Т g) Да Дх
dp ~ Л.СР “Да
или
ДСрДх = Т4Р(Тг)(Да)2 (VII.8}
где Тg — температура перехода в стеклообразное состояние; р — давление;
V (7g) — мольный объем при Tg; Да = а/—ag — разность коэффициентов
теплового расширения при Tg; АСР= C£p(Tg) — Csp(Tg) — разность мольной
теплоемкости при Tg, Дх = х; — хй — разность сжимаемостей при‘Тг.
Ставерман [24] и Бройер и Рехаге [16] подробно рассмотрели
термодинамику перехода стекло — каучук. Они пришли к заключе-
нию, что это явление не является истинным переходом второго рода,
главным образом потому, что свойства материала в стеклообразном
состоянии не определяются однозначно термодинамическими пара-
метрами р, V, Т.
Было бы интересно проверить выполнение уравнений (VII.1)
и (VII.2) по прямым экспериментальным данным. К сожалению,,
имеющиеся результаты обнаруживают значительный разброс,
117
поэтому подобные расчеты позволяют получить всего лишь правиль-
ный порядок величины. Только измерения Бройера и Рехаге для
полистирола оказываются достаточно точными. Их данные позво-
ляют получить следующие результаты:
Да = 3,3 • 10~4 (К)-1; Ди = 1,65-10-11 см2/дин
ДСР = 6,4 кал/(моль • К); dTg/dp =2,5 • 10-8 см2-К/дин
Гй = 375 К; У (Tg) =1QO смз/моль .
~dp~^CP 2,5-10-8.6,4-41,3
TgV (Та) Да - 375 - 100 • 3,3 - 10~* - °’53
dTg Да 2,5-10-8-3,3-10-4
dp Дх ~ 1,65-10-ы ~0’50
______________1 07
TgV(Tg) (Да)2 -1>и'
•Очевидно, что уравнения (VI 1.7) и (VI 1.8) выполняются в пределах
точности имеющихся данных *.
Представленные значения несколько отличаются от опубликован-
ных ранее Джи [17]. Данные для полиизобутилена, поливинилаце-
тата, поливинилхлорида и полиметилметакрилата, приведенные
Бьянки [12] и Коваксом [21], имеют такой же порядок величин.
Следовательно, уравнения (VII.7) и (VII.8) можно применять
для оценки порядка одной из величин, если известны другие вели-
чины.
ВЛИЯНИЕ СТЕРЕОРЕГУЛЯРНОСТИ НА ТЕМПЕРАТУРУ
ПЕРЕХОДА В СТЕКЛООБРАЗНОЕ СОСТОЯНИЕ
Парад и МакНайт [20] привели сводку имеющихся данных по тем-
пературам перехода в стеклообразное состояние винильных поли-
меров общей формулы
Р
I
—сн2—С—
I
Q
•Эти авторы заметили, что стерическая конфигурация влияет на Tg
только в том случае, если Р Q ини Р, ни Q не являются атомами
водорода.
* Тем не менее, этот результат не является достаточным для трактовки стек-
лования как термодинамического перехода второго рода. Более того, значения
параметров, относящихся к стеклообразному состоянию, могут быть различными
в зависимости от предыстории образца, так что, например, нет какой-либо одно-
значной величины Да, отвечающей рассматриваемому переходу. Эксперименталь-
ные данные показывают, что обычно первая из формул (VII.7) выполняется
лучше, чем вторая, вследствие чего, как правило, dTg!dp <« Дх/Аа. Это следует
иметь в виду при оценке зависимости Tg (р), исходя из измерений термодинамиче-
ских свойств полимера выше и ниже температуры стеклования. — Прим. ред.
118
Теоретическое рассмотрение этого явления, основанное на тео-
рии Гиббса и Ди Марцио [18], позволяет заключить, что для ряда
полиалкилметакрилатов
Те (синдиотактический) — Tg (изотактический) = константа 112 °C
Значения Tg для чисто синдиотактического и чисто изотактиче-
ского полиалкилметакрилатов, полученные Карачем и МакНайтом,,
приведены ниже:
Полиметилметакрилат
Полиэтилметакрилат
Полииропропилметакрилат
П олибу ти лметакрилат
Полиизобутилметакрилат
Полициклогексилметакрилат
Гй(синдио), к
433
393
412
361
393
436
Tg (изо), К
316
281
300
249
281
324
[* Независимый метод расчета температуры стеклования Tg аморфных
полимеров, основанный на использовании принципа аддитивности, вкладов
элементов, образующих мономерные группы, был предложен в работах [1 д,
2 д]. Согласно работе [1 д]- Ts вычисляется как
, d V .
T'g — (1д)
где d — плотность; М — масса 1 моля повторяющегося звена полимера; А —
константа, зависящая от строения рассматриваемого ряда полимеров, Kt
характеризует вклад каждого элемента структуры звена, причем не только вхо-
дящих в него атомов, но и таких структурных особенностей, как двойные связи,
водородные связи, наличие ароматических и других циклов и т. д.
Экспериментальная проверка этой формулы подтвердила ее применимость
для большого числа ароматических полиэфиров (полиарилатов) и полиамидов
разнообразного строения.
Применение формулы (1д) для расчета Tg ограничено, однако, несколькими
рядами полимеров. Поэтому в работе [2д] была предложена более общая адди-
тивная схема расчета Tg, основанная на применении формулы
У1 К*
(2д)
где А К,— собственный мольный объем повторяющегося звена, складыва-
ющийся из объемов всех атомов, входящих в это звено. Сумма K*t—аддитив-
ная величина, связанная с коэффициентами упаковки и объемного термического-
расширения. Константа А имеет универсальное значение, равное 1,435. Методика
определения У, А У; описана в гл. IV настоящей книги. Значения инкрементов
Kt некоторых атомов, наиболее часто встречающихся в полимерных молекулах,
таковы [2 д]:
Углерод . 10,739 Азот 9,520
Кислород 3,925 Хлор 7,242
Водород —1,248 Кремний 10,401
Кроме того, при замене в полимерах винилового ряда атома водорода в основ-
ной цепи или боковом ответвлении радикалом любого типа, кроме фенильного,
11»
учитывается вклад «диполь-динольных взаимодействий», выражаемый коэффи-
циентом К*, равным 7,827. При замене двух атомов водорода в мономерном звене
одинаковыми радикалами вводится половина величины этого инкремента. До-
полнительный вклад, названный «коэффициентом симметрии» и равный 10,500,
вводится в расчет в том случае, если все ароматические ядра в основной цепи
замещены в пара-положениях.
Экспериментальная проверка изложенного метода расчета Tg была прове-
дена для 80 полимеров самого разнообразного химического строения и показала,
что он дает значения Tg, удовлетворительно согласующиеся с опытными данными.
Это позволяет рекомендовать формулу (2д) для оценки температур стеклования
вновь синтезируемых полимеров.*]— Ярил. ред.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1 д. Аскадский Л. Л., Высокомол. 2 д. Аскадскии А. А., Слоним-
соед., 1967, т. 9А, К» 2, с. 418— ский Г. Л., Высокомол. соед.,
432. 1971, т. 13А, №8, с. 1917—1919.
ТОЧКА ПЛАВЛЕНИЯ
КРИСТАЛЛИЧЕСКИХ ПОЛИМЕРОВ
Примечательно, что до сих пор в литературе практически отсут-
ствуют соотношения между химическим строением полимеров и их
температурой плавления, хотя экспериментальных данных для Тт
известно больше, нежели для Tg. Это обстоятельство может быть
связано с большим разбросом имеющихся данных по Тт.
Много лет тому назад уже отмечалась определенная корреляция
- между Т& и Тт для одного и того же полимера, что позволяет пред-
положить возможность применения метода оценки Tg для оценки Тт.
Эквивалентная формула имеет вид
(VII.9)
ii
( Однако существует принципиальное различие между Tg и Тт. Дело
I в том, что точка плавления есть температура истинного перехода
I первого рода, при которой свободные энергии каждой из двух нахо-
\ дящихся в равновесии фаз равны между собой. Следовательно
TmbSm = kHm (VII.10)
где ASm— энтропия плавления; &.Нт — энтальпия плавления.
Соответствие между (VII.9) и (VII.10) позволяет предположить,
что можно идентифицировать 2 s{ с Д5т и 2 siTml с N.Hm.
.1 I
Для полимеров количество данных по &Нт довольно ограничено.
В сводке, приведенной в гл. V, было показано, что теплота плавле-
ния ДН^ не подчиняется строгому правилу аддитивности, но энтро-
пию плавления Д6’т можно приближенно записать в виде формулы
= (VII.11)
420
Ниже приведены значения групповых вкладов Л5,:
ASZ 2ZZ Л81
—СН2— 2,0 2 —СОО— 0 4
7,0 8 -CONH— 0 4
—О— 1,8 2 -OCONH- (0) 6
Теперь можно воспользоваться уравнениями (VII.10) и (VII.11)
для расчетов &Нт полимеров с известными Тт.
Для данной группы полимеров значения &Нт, рассчитанные та-
ким способом, в общем случае не коррелируют строго с числом ме-
тиленовых групп п. Однако можно существенно улучшить такую
корреляцию, если опустить полимеры, содержащие цепи с нечетным
числом метиленовых групп или цепи с числом метиленовых групп
меньше 6. Для остальных полимеров с длинными метиленовыми
цепями обнаруживается практически линейное соотношение между
ЬНт и п. Это соотцошение можно записать в виде формулы
ЬНт = пхарЯхар + 770n
(VII.12)
где п — полное число метиленовых групп; raXap — число групп, характерное для
рассматриваемого ряда полимеров; ЯХар— групповой вклад специфической
группы.
Значения Яхар Для некоторых рядов полимеров приведены ниже:
Характеристическая
группа ихар
Полиэфиры
Полиамиды /
Полиуретаны
—СОО— —670
—CONH— 1650
—OCONH— 550
Из всего количества примерно 60 полимеров представляют собой
соединения, содержащие длинные цепи с четным числом метилено-
вых групп. Для рих среднее расхождение между эксперименталь-
ными значениями Тт и значениями Тт, рассчитанными по формулам
(VII.10)-^-(VII.12), составило приблизительно 4 °C.
Однако, если попытаться распространить этот метод на другие
типы полимеров, возникают определенные сложности. При наличии
коротких метиленовых цепей значения Тт для полиэфиров выше,
а для полиамидов ниже по сравнению с полученными описанным,
выше методом. Полимеры с другими дополнительными группами
(например, га-фениленовыми группами и атомом кислорода) обычно
содержат короткие метиленовые цепочки. Следовательно, нужно
ввести некоторую поправку для учета коротких метиленовых последо-
вательностей с тем, чтобы получить правильные формулы и для
таких полимеров. Кроме того, лишь для ограниченного числа типов
полимеров можно рассчитать А5„г. Таким образом, пока отсутствует
достаточное количество данных о &Нт, не нужно, вероятно, прово-
дить дальнейшую разработку этого метода.
121
Ввиду сказанного выше, расчеты Тт в этом разделе должны
основываться на уравнении (VII.9). Величина 2 st принимается рав-
ной числу атомов Z в цепи главных валентностей, как это было сде-
лано при получении формулы для расчета Т&. Нужно особо под-
черкнуть, что это приводит к исключительно эмпирическому методу
расчета, потому и не следует приписывать какой-либо физический
смысл абсолютным значениям групповых вкладов.
На стр. 121 групповые вклады в энтропию плавления А5(. срав-
ниваются с вкладами в число атомов Z, цепи главных валентностей.
Рис. VII.4. Вклады метиленовых групп в по-
лиэфирах.
Вклады эфирной и амид-
ной групп существенно
различаются, хотя и на-
блюдается определенная
пропорциональность для
вкладов метиленовых, фе-
ниленовых и кислородных
групп.
Формулу, эквивалент-
ную формуле (VII.2а),
можно применить к ряду
линейных полимеров без
боковых цепей:
у X Ymi
Tm =-£-= (VI 1.9а)
где Ym—аддитивная величина, которую можно назвать мольной1 функцией
перехода в расплавленное состояние.
Для некоторых полимеров, для которых имеются в литературе
значения Тт, с помощью соотношения (VII.9а) можно рассчитать
величину Ym. Была сделана попытка сопоставить эти величины
с групповыми вкладами Ymi. Это возможно, но лишь при условии,
если ограничить пределы применимости принципа аддитивности.
Эти ограничения сводятся к следующему.
1. Вклад метиленовой группы не является постоянной величи-
ной, а зависит от длины рассматриваемой метиленовой цепи. На
рис. VII.4 построена зависимость Ym от числа метиленовых групп
в цепи для ряда полиэфиров. Представлены данные для полимоно-
эфиров со структурным звеном
С«—О—(СНг)л
И для полидиэфиров со структурным звеном
^О-С-(СН2)п-С-О-(СН2)п-
122
В последнем случае откладывается величина 1/2Ym. Аналогичные
графики можно построить и для других типов полимеров.
Из этих графиков видно, что величина вклада Ymi метиленовой
группы постоянна только для достаточно длинных метиленовых
последовательностей.
Разумное приближение можно получить, если принять, что
первые четыре метиленовые группы в отдельной цепи дают вклад
Ymi = 170 К, а все остальные метиленовые группы — вклад Ymi —
= 380 К.
Для определенных типов полимеров, например полиамидов,
наблюдаются меньшие значения Тт, если метиленовые последова-
тельности состоят из нечетного числа метиленовых групп. Тогда
следует вводить поправку на нечетное число метиленовых групп
в цепи.
2. Вклады некоторых групп, а именно: n-фенилена, кислорода и
серы, определяются типом исследуемого полимера.
То же самое можно увидеть и для многих других групп, но в свя-
зи с отсутствием достаточных данных нельзя достоверно определить
их групповые вклады.
3. Вклады некоторых комбинаций групп отличаются от суммы
вкладов для каждой группы в этой комбинации. Например, вклад
комбинации
в полиэфирах составляет Yml = 1900, а сумма вкладов отдельных
групп этой комбинации составляет 2100 + 2-170 = 2440.
Принимая во внимание перечисленные выше ограничения, можно
теперь рассчитать ряд групповых вкладов. Полученные значения
представлены в табл. VII.5. В общей сложности для 500 полимеров
величины Тт, рассчитанные по групповым вкладам табл. VII.5,
отклоняются от экспериментальных значений в среднем на 14°.
Большие отклонения наблюдаются для полимеров, содержащих
короткие метиленовые последовательности. Отклонения от рассчи-
танных значений Тт для полимеров с асимметричной структурной
единицей, например содержащих комбинации —— или
—CL—, крайне велики. В табл. VII.6 сравниваются экспери-
ментальные и рассчитанные значения Тт для ряда полимеров.
В табл. VII.5 не приводятся значения групповых вкладов, необ-
ходимые для расчета Тт некоторых важных в промышленном отно-
шении винильных полимеров. Причина заключается в том, что рас-
сматриваемые группы дают совершенно различные вклады в зависи-
мости от того, в каком полимере они содержатся. Для полноты кар-
тины в табл. VII.7 приведены групповые вклады, рассчитанные по
точкам плавления рассматриваемых полимеров. К сожалению, эти
123
Таблица VII.5. Групповые вклады в температуру плавления (Тml)
Тип полимера
Группа полиок- сид поли- сульфид полиэфир поликар- бонат полиан- гидрид полиамид полиуре- тан поли- карбамид
( га = 1—4 —СН2— { 170 170 170 170 170 170 170 170
I п = 5 380 380 380 380 380 380 380 • 380
—О— 900 — 750 — — 600 450 —
—О—СН2—О— 1390 — — — • — — — ' —
—S— — 960 — — . — 750 — —
—S—СН2—S — — 1800 — — — . — — —
—S-S— — 1140 — — — — — —
—СОО— — — 1160 — — — — —
—О-СО—О— — — — 1500 — — — —
—СО—О—СО— — — — — 1720 — — —
—CONH— — — — — — 2560 — —
—OCONH- — ' — . — — — — 2430 —
—NH—CO-NH — — — — —• — — 3250
— — 2100 — — 2300 — —
— " — 5600 — — — — —
-СЩ-^-СН^ — — 1900 — 2700 2300 2450
-(СН2)2-<^)-(СН2)2- — — — — — 4000 — —
-0-^-0- 3200 3650
Дополнительная метилено- вая цепь (поправка) —150 0 0 0 —190 —260 -170 0
значения нельзя использовать при расчете температур плавления
других типов полимеров, например приведенных в табл. VII.5.
124
Таблица VH.6. Экспериментальное и рассчитанные значения Тт для ряда полимеров (К)
Полимер Тт (опыт) * tn (расчет) Полимер * tn (опыт) Л tn (расчет)
Полиоксиметилен 333/471 352/460 Полидекаметиленсебацинамид 469/489 467
Полиоксиэтилен 335/345 413 Поли-6-аминокапроновая кислота 487/506 480
Полиокситриметилен 308 315 Поли-11-аминоундекановая кислота 455/493 460
Полиокситетраметилен 308/333 316 Полинонаметиленазеламид 438/462 450
Политетраметиленацеталь 296 295 Полиэтилевтерефгаламид 728 776
Полидекаметиленацеталь 330 334 Полигексаметилентерефталамид 623/644 632
Полиэтил енсульфид 418/483 433 Полиоктадекаметилентерефталамид 528 516
Полидекаметиленсульфид 351/365 356 Поли-п-фенилендиметиленадипамид 606/613 607
Полиэтилендисульфид Полидекаметилендисульфид 386/403 318/332 370 341 Полигексаметилен-4,4'-оксидибутир- амид 460 450
Полиэтиленадипинат Полидекаметилеиадипинат 320/338 343/355 334 331 Политетраметиленгексаметилендиуре- тан 446/457 436
Полцдекаметиленсебацинат Полиэтилентерефталат 344/358 538/557 340 476 Полидекаметиленгексадекаметиленди - уретан 401 408
Полидекаметилентерефталат Поли-л-фенПлендиметиленадипинат 402/411 343/354 410 350 Поли-п-фенилендиметилентетрамети- лендиуретан 500 490
Политетраметиленангидрид Полигексадекаметиленангидрид 350/358 368 342 366 ,Полигексаметиленоктаметилендикарб - амид 498/526 507
Политетраметиленкарбонат Полидекаметиленкарбонат Полигексаметиленадипамид 332 328 523/545 311 343 517 Поли-п-фенилендиметиленгексамети- лендикарбамид 579 577
Таблица VII.7. Некоторые формальные значения групповых вкладов в Ут
(в других комбинациях не применяются)
Полимер Тт. к Характеристическая группа
Полиэтилен 414 -сн2- 414 '
Полипропилен 456 —СН(СН3)— 742
Полиизобутилен 275 —С(СН3)2— 380
Полистирол 513 -СН(СвН5)- 856
Поливиниловый спирт 531 —СП (ОН)— 892
Поливинилхлорид 558 -СНС1- 946
Полив инилиденхлорид 483 —СС12— 796
Поливинилфторид 473 —CHF— 776
Поливинилиденфторид 444 —cf2— 718
Поли-п-ксилилен 713 1 О т 3938
Формулу для расчета температур плавления полимеров с боко-
выми цепями можно получить таким же методом, как и соотношение
для температур стеклова-
ния этих полимеров. Для
этого следует воспользо-
ваться формулой
Ym0 + h'{N)
т~ Za + N
где Н — число метиленовых
групп в боковой цепи; h' (N) —
функция от N-, Ута — зна-
чение Ут для «основного поли-
мера», Zo — число атомов в
цепи главных валентностей
«основного полимера».
«Основной полимер» —
это первый член ряда,
т. е. полимер рассматри-
ваемого типа, для кото-
рого N = 0.
На рис. VII.5 построена
функция h' (N) для неко-
торых рядов полимеров.
Рис. VII.5. Температуры плав-
ления полимеров с боковыми
цепями:
X — полиолефины; ф — полимет-
акрилаты; □ — поливиниловые
эфиры (простые); + — поливини-
ловые эфиры (сложные).
126
Эту функцию можно представить в виде двух отрезков пря-
мой линии; первая часть зависимости характеризуется наклоном
255 К, вторая — 415 К. Точка перегиба находится при У = 7
.для полиолефинов, при У = 10 для поливиниловых эфиров и при
JV = 13 для полиметакрилатов. В случае поливиниловых эфиров
трудность заключается в том, что точка плавления «основного поли-
мера» (поливинилацетата) неизвестна. Можно формально выбрать
значение Ym0 для этого ряда Полимеров так, чтобы получаемые
точки совпадали с точками для поливиниловых эфиров.
В табл. VII.8 приведены значения параметров, которые исполь-
зуются для некоторых рядов полимеров.
Таблица VII.8. Некоторые параметры для полимеров с боковыми цепями
Тип полимера Структурное звено «Основной полимер» ^то в точке пересечения
Полиолефины —СН2—СН— Полипропилен 910 7
। (CH2)W-CH3
Поливиниловые эфиры —сн2—сн— 1 O-(CH2)N-CH3 Поливинилмети- ловый эфир 840 10
Полифенилолефи- ны —сн2-сн— (CH2)n Полистирол 1030 ?
Полициклогек- силолефины -сн2-сн— 1 (CH2)N Поливинилцикло- гексан 1200 ?
сн3
Полиметакрилаты 1 —сн2-с- О=С-О-(СН2)Л-СН3 Полиметилмет- акрилат 900 13
Поливиниловые эфиры -сн2-сн- 1 О Поливинилацетат (750) (10)
O=C-(CH2)N-CHg
127
Ниже сравниваются экспериментальные и рассчитанные значе-
ния Тт для некоторых полимеров с боковыми цепями:
- Т-т (эксперимент) Тт (расчет)
Полибутен 379/415 388
Полинонен 292 305
П олигексадецен 341 346
Поливинилэтиловый эфир 359 365
Поливинилдодециловый эфир 303 293
П о ли-3-фени лпр опен 458/513 428
Поли-З-циклогексилпропен 488/503 485
Политетрадецилметакрилат 264/271 282
По лидокозилметакрилат 328 328
П оливинилдодеканат 274 275
Поливинилдокозанат 330 338
В общей сложности для 37 полимеров с боковыми цепями средняя
величина разности между экспериментальными и рассчитанными
значениями Тт составила 10°.
Пример 3
Оценить температуру плавления полипарафенилендиметиленазеламида.
Решение
Структурное звено этого полимера:
—СН2
—СН2—N—С—(СН2)7—С—N—
По табл. VII.5 следует сложить групповые вклады:
zi Ymi
-ch2-(j2))-ch2- 6 2700
2—CONH— 4 5120
4—СН2— (170) 4 - 680
3—СН2— (380) 3 1140
Нечетная метиленовая цепь — —260
Z=17 Ym = 9380
Следовательно, Тт — 9380/17 = 552 К.
Пример 4
Оценить температуру плавления поливинил- 1-бутилового эфира.
Решение .
Структурное звено поливинил-1-бутилового эфира:
—СН2-СН-
0-(СН2)з-СНз
128
Согласно табл. VII.8, Ym» = 840. Поскольку N = 3, наклон линии зависи-
мости h' от N еще не изменился и h' (N) = 255А = 765. По уравнению (VII.13)
840+765 _
Zq + N ~ 2 + 3
В литературе приводится значение Тт = 337 К.
СООТНОШЕНИЕ МЕЖДУ ТЕМПЕРАТУРОЙ
СТЕКЛОВАНИЯ И ТЕМПЕРАТУРОЙ ПЛАВЛЕНИЯ
ПОЛИМЕРОВ
Бойер [13] первым отметил существование эмпирического линей-
ного соотношения между Tg и Тт для полимеров. Независимо от него
Биман [11] также обнаружил это соотношение. Позже Бойер [2, 14]
Рис. VII.6. Кривая интегрального распределения по Tg/Tm'
9 — симметричные полимеры; О — несимметричные полимеры.
рассмотрел эту проблему более полно и вывел следующие правила:
Тг ( 1/2 для симметричных полимеров
I'm I 2/3 для несимметричных нолимеров
0 Закаа 699
129
Несимметричными были названы полимеры, атом главной цепи
которых не содержит двух одинаковых заместителей; другие поли-
меры считаются симметричными.
Ли и Найт [22] проверили это соотношение для 138 полимеров
и обнаружили, что в действительности величина TgITm изменяется
в широких пределах.
На рис. VII.6 представлены результаты этих авторов. Кривые
интегрального распределения описывают зависимость числа поли-
меров ДГ, для которых значение TglTm меньше или равно заданному
значению, от величины отношения Т&1Тт. Приблизительно для
80% как симметричных, так и несимметричных полимеров величина
рассматриваемого отношения лежит в области 0,5—0,8 (при макси-
муме около 0,66); для остальных 20% полимеров это отношение
выходит за указанные пределы. Согласно выводам цитируемой ра-
боты, не существует реальной ос-
новы для разграничения между
симметричными и несимметричными
полимерами. Более того, авторы счи-'
тают, что с точки зрения термоди-
намики получение простого соотно-
шения между Tg и Тт маловероятно,
поскольку молекулярные механизмы
стеклования и плавления’ ~в прин-
ципе отличаются друг от друга. Как
при стекловании, так и при плавле-
нии состояние части полимерного
образца изменяется от относительно
жесткого твердого тела до жидкости,
однако твердые состояния, возника-
ющие в каждом из этих переходов,
обладают различными характеристи-
ками. Более того, соответствующие
жидкие состояния обладают суще-
ственно различными характеристиками при температурах Т&
и Тт, при которых эти состояния формируются.
Итак, окончательные выводы.
1. Полимеры с Т„!Тт ниже 0,5 оказываются высоко симметрич-
ными и обладают короткими повторяющимися звеньями из одного
или двух атомов цепи главных валентностей, при этом заместители
у этих атомов представляют собой только отдельные атомы (поли-
метилен; полиэтилен, политетрафторэтилен, полиоксиметилен). Эти
прдимеры . хя дактеризуются. - высокой. степенью к ристалличп ости?
2Г~Полимеры с Тg!Tm выше 0,76 оказываются несимметричными.
Эти полимеры также могут обладать высокой степенью кристаллич-
ности, если содержат длинные последовательности метиленовых
групп или обладают высокой степенью стереорегулярности. Строение
таких полимеров гораздо сложнее, чем полимеров, у которых TglTm
ниже 0,5.
Ьлоксополинеры борнильные У
/гомополи- У
f меры s
/ /
/ /
Е
fcmamucmusec-
’ кие сополимеры
/
— TS
Рис. VII.7. Схематическое пред-
ставление зависимости Тт Tg
для некоторых типов полимеров.
130
. 3. Для большинства полимеров отношение TJT,., лежит между
0,56 и 0,76 с максимумом вблизи 0,66. В этой группе есть как сим-
метричные, так и несимметричные полимеры.
Только что приведенные выводы можно считать модификацией
правил Бойера. Эти результаты полезны для практических оценок.
Совершенно другие значения.,7g/могут наблю1д^тьсяддя_хш1ш-
лимероВГ~В это^тшгая~следурт различать статистические сополимеры
и блок2соподджв.ы.
"Кристаллизация статистических сополимеров за счет нерегуляр-
ности их строения затруднена по сравнению с каждым из гомополи-
меров. В итоге точка плавления понижена, а температура перехода
в стеклообразное состояние может иметь нормальное значение, лежа-
щее между температурами стеклования гомополимеров. В резуль-
тате значения TJTm возрастают.
В блок-сополимерах, напротив, длинные последовательности
одинаковых структурных звеньев могут кристаллизоваться незави-
симо, так же, как и в случае гомополимеров. Можно получить ино-
гда такие блок-сополимеры, в которых сочетаются высокая темпе-
ратура плавления кристаллической части, соответствующая темпе-
ратуре плавления одного гомополимерного компонента, с низкой
температурой перехода в стеклообразное состояние, соответству-
ющей температуре стеклования другого гомополимера *. Все это
приводит к низкому значению отношения TJTm (рис. VII.7).
ЛИТЕРАТУРА
1. Alfrey Т. and Gurnee Е. F.,
«Organic Polymers», Ch. 3, Pren-
tice-Hall, Englewood Cliffis,N. J.,
1967.
2. Boyer R. F., «The Relation of
Transition Temperatures to Che-
mical Structure in High Poly-
mers», Rubber Chem. Technol.
36 (1963) 1303.
3. Boyer R. F. (ed.), «Transitions
and Relaxations in Polymers»,
Interscience, New York, 1967.
4. Brandrup J. and Immergut E. H.r
«Polymer Handbook», Wiley, New
York, 1966.
5. Bueche F., «Physical Properties
of Polymers», Ch. 4, 5, 11, Inter-
science, New York, 1962.
6. Bunn С. И7., «Melting and Second
Order Phenomena», Ch. 12 in
R. Hill (ed.), «Fibres from Synthe
tic Polymers», Elsevier, Amster
dam, 1953.
7. McCrum N. G., Read В. E. and
Williams G., «Anelastic and
Dielectric Effects in Polymer So-
lids», Wiley, New York, 1967.
8. Meares P., «Polymers: Structure
and Bulk Properties», Van Nost-
rand, Princeton, 1965.
9. Shen M. C. and Eisenberg A,,
«Glass Transitions in Polymers»,
Rubber Chem. Technol. 43 (1970)
95.
10. Barton J. M. and Lee W. A.,
Polymer 9 (1968) 602.
11. Beaman R. G., J. Polymer Sci. 9
(1953) 472.
12. Bianchi U., J. Phys. Chem. 69
(1965) 1497.
* В блок-сополимерах могут наблюдаться также раздельные температуры
стеклования каждого компонента, причем если длина последовательности звеньев
одного типа достаточно велика, то- соответствующая температура стеклования
не Отличается от 7’й такого же гомополимера. Типичным примером в этом отно-
шении являются блок-сополимеры бутадиена и стирола. — Прим. ред.
9* 131
13. Boyer R. F., 2nd Int. Conf.
Phys. Chem., Paris, June 6,
1952
14. Boyer R. F., J. Appl. Phys. 25
(1954) 825.
15. Boyer R. F., Am. Chem. Soc.
Preprints 30 (1970) nr. 2.
16. Breuer H. and Rehage G., Kol-
loid.-Z. 216/217 (1967) 159.
17. Gee G., Polymer 7 (1966) 177.
18. Gibbs J. H. and Di Marzio E. A.,
J. Chem. Phys. 28 (1958) 373.
19. Hayes R. A., J. Appl. Polymer
Sci. 5 (1961) 318.
20. Karasz F. E.and Mac Knight W. J.
Macromolecules 1 (1968) 537.
21. Kovacs A. J., Fortschr. Hochpol.
Forsch. 3 (1963) 394.
22. Lee W. A. and Knight G. J.,
Brit. Polymer J. 2 (1970) 73.
23. Lee W. A., J. Polymer. Sci.
A-2 8 (1970) 555.
24. Sta verman A. J., Rheologica Acta
5 (1966) 283.
25, Weyland H. G., Hoftyzer P. J.
and Van Krevelen D. W., Poly-
mer 11 (1970) 79.
ГЛАВА VIII.
РАСТВОРИМОСТЬ
Возможности количественной оценки растворимости полимеров
взаимодействий в растворах и пределов растворимости до сих по}
еще полностью не исследованы. Для молекул, не содержащих сильнс
полярные группы, параметр растворимости можно оценить с по
мощью правил мольной аддитивности; критические пределы рас
творимости коррелируют с разностью между параметрами раствори
мости Цолимера и растворителя.
ПАРАМЕТР РАСТВОРИМОСТИ
Соотношения растворимости в полимерных системах оказываются
более сложными по сравнению с правилами растворимости низко-
молекулярных соединений. Однако некоторые эмпирические законо-
мерности, полученные для малых молекул, можно с успехом приме-
нить и к полимерам.
Наиболее важным является то, что химическое и структурное
сходство облегчает растворение. Сродство между растворителем
и растворяемым веществом оказывается наибольшим, когда их моле-
кулы обладают сходной «полярностью».
Другое важное правило, являющееся основой фракционирования
полимеров по молекулярным весам, заключается в том, что раство-
римость уменьшается с увеличением молекулярного веса растворя-
емого вещества.
Кристаллические полимеры относительно плохо растворимы.
Они часто растворяются только при температурах, лишь немного
меньших их точки плавления.
Процесс растворения аморфного полимера в каком-либо раство-
рителе определяется известным уравнением для изменения изобарно-
изотермического потенциала смешения:
Ьвм = ьнм-Т bSM (VIII.1)
где AGM — изменение изобарно-изотермического потенциала; &НМ— теплота
смешения и Д<$м — энтропия смешения.
Растворение или смешение осуществляется лишь тогда,
когда изменение изобарно-изотермического потенциала смешения
133
отрицательно. Растворение полимера, как правило, связывают
с увеличением энтропии. Следовательно, знак и величина энталь-
пийного члена уравнения оказываются решающими факторами в
определении знака изменения свободной энергии.
Еще в 1916 г. Гильдебранд [И] заметил, что порядок раствори-
мости данного вещества в ряде растворителей определяется внутрен-
ним давлением растворителя. Позже Скетчард [13] ввел в теорию
Гильдебранда понятие «плотности энергии когезии» и приравнял
эту величину энергии испарения 1 см3 вещества. Наконец, Гильде-
бранд дал исчерпывающую разработку этого понятия [1] и предло-
жил применять в качестве параметра, определяющего характер
растворимости для данного растворителя, корень квадратный из
плотности энергии когезии. В 1950 г. Гильдебранд предложил на-
звание «параметр растворимости» и обозначил этот параметр сим-
волом 6:
б=(—(VIII.2)
где Д7?ц сп—энергия испарения при нулевом давлении, т. е. при бесконечном
удалении молекул друг от друга.
Согласно Гильдебранду, теплота смешения в эндотермическом
процессе пропорциональна (6Х — 62)2:
Д/гм=Ф1Ф2(б1-62)2 (VIII.3)
где &hM — теплота смешения на единицу объема; Ф — объемная доля компо-
нента 1 или 2.
Если теплота смешения должна быть не слишком большой, чтобы
не препятствовать смешению, то и величина (6Х — 62)2 должна
быть малой.
В предельном случае, когда бх — б2 = 0, растворение опреде-
ляется только энтропийным фактором. Если значения 6 двух ве-
ществ приблизительно равны друг другу, то эти вещества будут
смешиваться. Отсюда и название — «параметр растворимости».
Параметр растворимости определяет только теплоту смешения у
жидкостей или аморфных полимеров. Любой некристаллический 1
'полимер будет растворяться в растворителе с близким значением б. j
При получении соотношения для параметра растворимости было :
сделано предположение об отсутствии специфических сил взаимо-
действия. Однако наличие сильно полярных групп или водородных
связей в растворителе или полимере сильно усложняет картину.
При этом может возникать положительное взаимодействие между
полимером и растворителем, так что энтальпийный член станет отри-
цательным. Растворение будет осуществляться при этом даже тогда,
когда разность (6Х — 62) относительно велика.
134
ОЦЕНКА ПАРАМЕТРОВ РАСТВОРИМОСТИ
ПОЛИМЕРОВ
Параметр растворимости растворителя 6Р вполне поддается опреде-
лению. Параметр растворимости полимера 6П нельзя измерить непо-
средственно, поскольку полимеры не испаряются без разложения.
Тогда величину 6П можно определять как параметр растворимости
того растворителя, с которым полимер смешивается в любых отно-
шениях без теплового эффекта и изменения объема (и, конечно, без
химической реакции между полимером и растворителем).
Рис. VIII.1. Зависимость степени равновесного набухания от параметра раство-
римости растворителя для линейного и структурированного полистиролов:
2 — линейный полимер; 2 — линейный полимер, неограниченно смешивающийся с раствори-
телем; з — линейный полимер; 4 — сетчатый полимер.
Экспериментально параметр 6П можно определить по данным изме-
рений растворимости (средняя часть области растворимости в пре-
делах группы растворителей) или по степени набухания полимеров
с низкими плотностями поперечных сшивок (рис. VIII. 1).
Как уже отмечалось в гл. VI, Смолл [14] опубликовал таблицу
констант молекулярного притяжения, с помощью которых можно
оценить Параметр растворимости только по структурной формуле
данного соединения и по его плотности. Константы молекулярного
притяжения аддитивны и связаны с параметром растворимости сле-
дующим уравнением:
(VIII. 4)
135
Таким методом аддитивности не следует пользоваться при рас-
смотрении соединений с сильной склонностью к образованию водо-
родных связей, если удельный вес участвующих в образовании
водородных связей функциональных групп достаточно велик.
В табл. VIII.1 приведены константы, Смолла. Недавно Хой [12]
опубликовал некоторые значения групповых вкладов для . ра-
счета F, несколько отличающиеся от данных Смолла. Эти значения
также приведены в табл. VIII. 1.
Ван Кревелен [15] получил набор аналогичных атомных и груп-
повых вкладов для расчета V/7;
6-1
Ч Ьц
с о
Н 68,5
О простой эфир 125
сложный эфир 125<
кетон _ 335
спирт первичный 360
спирт вторичный 300
фенол 250
S тиоэфир 225
тиол (250)
F 80
С1 230
Вт 300
I 420
Двойная связь (неаро- 80
матическая)
Двойная связь (арома- 133
тическая)
Тройная связь 215
N алифатический первичный
амин
алифатический вторичный
амин
ароматический первичный
амин
гетероциклический
нитрильный
N+О в алифатической нитро-
группе
в ароматической нитро-
группе
в кислой амидной группе
Неароматическое кольцо
Разветвление в цепи
Сопряжение двойных свя-
зей
Две ОН-группы на приле-
жащем атоме С
g
к
S
ьГ
100
140
65
115
480
460
325
600
400
60
—20
25
—190
Разумеется, можно также рассчитать 6 непосредственно по дан-
ным энергии когезии (гл. VI). В табл. VIII.2 приведены для сравне-
ния некоторые экспериментальные и рассчитанные значения 6.
136
Таблица VJII.1. Константы, молекулярного притяжения при 25* С по Смоллу и Хойю
) Группа (кал-см’)1/1/моль Группа F, (кал-см1) '"/моль
Смолл Хой Смолл Хой
-СН, ^>СН, ^СН ° одинарной связью 1 -С- 1 =CHS «=сн- „ и с двойной связью ”=С\ % ^СН (ароматическая) % ^С— (ароматическая) Нѻї —С=С— Фенил Фенилен Нафтил пятичленное кольцо шестичлекиое кольцо Сопряжение —Н (в разных вариантах) —О— (простой эфир) 214 133 23 -93 (190 111 19 285 222 735 «53 1146 105—115 95—105 20—30 800—100 70 148,3 131,5 86,0 32,0 126,5 121,6 84,5 117,1 98,1 21,0 -23,4 23,3 115,0 j 1 —С=О (кетон) —C^Q (сложный эфир) —C-N —F ( (в среднем) I одиночный -С1{ \ /сс1« ' -СС1, —Вт одиночный —I одиночный )>СГ, 1 ? только фторуглероды —CF, j —S— (сульфиды) —SH (тиолы) —0N02 (нитраты) —NO, (алифатическая) 2Р°* —СО-О—СО— —NH— 275 310 410 260 270 260 250 340 425 150 274 225 315 440 440 500 263,0 326,6 354,6 41,3 207 258 209,4 567,3 180,0
Таблица VIII.2. Экспериментальные и рассчитанные значения 6 для различных
полимеров, кал Щсм !*
Полимер а (опыт) 0 (расчет)
Смолл Ван Креве- лен Хофти- цер. Ван Кревелен Хой
Полиэтилен 7,7-8,35 8,25 8,4 7,8 8,15
Полипропилен 8,2—9,2 7,55 8,3 8,3 7,35
Полиизобутилен 7,8—8,1 7,1 8,2 7,75 7,0
Полибутадиен 8,1-8,6 8,05 8,05 8,1 8,25
Полиизопрен 7,9—10,0 8,05 8,3 7,9 8,05
Полистирол 8,5-9,3 9,0 9,4 9,0 9,25
Поливинилхлорид 9,4-10,8 9,55 9,65 9,65 9,45
Поливинилиденхлорид 9,9—12,2 10,2 10,55 9,8 10,2
Поливинилбромид 9,5 9,3 9,4 9,45 8,85
Полихлортрифторэтилен 7,2-7,9 — 8,1 7,5 6,85
Политетрафторэтилен 6,2 6,0 6,4 3,65 4,6
Полихлоропрен 8,2-9,25 9,35 9,05 8,8 9,4
Поливинилацетат 9,35-11,05 9,65 9,3 9,55 9,75
Поливинилпропионат 8,8 9,4 9,1 9,2 9,45
Полиметилакрилат 9,7—10,4 9,6 9,3 9,7 9,75
Полиэтил акрилат 9,25—9,4 9,4 9,1 9,35 9,45
Полипропил акрилат 9,05 9,2 9,05 9,0 9,25
Цолибутилакрилат 8,8—9,1 9,1 8,95 8,95 9,1
Полиизобутилакрилат 8,7-11,0 8,8 8,9 9,15 8,75
П о лиметилметакрилат 9.1—12-8 8,9 9,15 9,1 9,0
Полиэтилметакрйлат 8,9-9,15 8,8 9,0 8,9 8,85
Полибутилметакрилат 8,7-9,0 8,75 8,9 8,6 8,7
Полиизобутилметакрилат 8,2—10,5 8,4 8,8 8,8 8,4
Полибензилметакрилат 9,8-10,0 9,5 9,7 9,15 9,6
Полиакрилонитрил 12,5—15,4 12,7 13,6 13,3 11,4
Полиметакрилонитрил 10,7 10,0 12,4 11,5 10,0
Полиоксиметилен 10,2—11,0 8,6 10,7 10,0 10,4
Полиокситетраметилен 8,55 8,35 9,35 8,6 8,9
Полиоксипропилен 7,5—9,9 7,8 9,35 9,2 8,4
Полиэтиленсульфид 9,0-9,5 10,9 11,0 9,25 10,95
Полидиметилсилоксан 7,3—7,6 6,1 — — —
Полиэпихлоргидрин 9,4 9,25 10,1 9,8 У, /
Полигексаметиленадипамид 13,6 — 12,4 13,7 10,55
Полиэтилентерефталат 9,7—10,7 10,7 10,4 10,0 10,95
Коэффициент перевода в СИ: 1 кал*I1 /см3/* = 2,04 -10* Дж /*/м /«•
Пример 1
Оценить параметр растворимости поли-н-бутилметакрилата.
Решение
С помощью табл. VIII.1 и IV.3 нетрудно получить:
Группа
4(—СН2—)
I । \
1 -с-
F (по Смоллу) V
532 65,8
—93 4,75
138
2 (—СНз) 428 45,6
1 (-С00-) 310 21,0
1177 137,15
8 = 1177/137,15 = 8,6
Точно так же, если пользоваться приведенными выше данными, можно получить;
Атом F (по Ван Кревелену)
8 (С) С
14 (Н) 959
2 (Оэфирн) 250
1209
6 = 1209/137,15 = 8,8
Экспериментальное значение лежит в области 8,7—9,0. Следовательно, соответ-
ствие вполне удовлетворительное.
ВОДОРОДНЫЕ связи
Образование водородных связей вносит в суммарную плотность
энергии когезии такой же существенный вклад, как и дисперсион-
ные взаимодействия. К сожалению, измерение энергии водородных
связей во всех случаях, исключая энергию водородных связей в про-
стейших молекулах, оказывается сложной задачей, и до сих пор не
получено приемлемое уравнение для теплоты смешения двух жид-
костей, образующих водородные связи.
Тем не менее Баррелл [6] предложил удобную на практике сис-
тему расчета путем произвольного разделения растворителей на
три класса в зависимости от их относительной способности образо-
вывать водородные связи. В тех случаях, когда использовалась эта
система, удалось избежать значительных расхождений с экспери-
ментальными данными.
На рис. VIII.2 приведены значения 6 для трех различных групп
растворителей в соответствии с их склонностью к образованию
водородных связей. Успешное применение понятия о параметре
растворимости определяется таким разделением растворителей на
группы.
УТОЧНЕНИЯ ПРЕДСТАВЛЕНИЙ
О ПАРАМЕТРЕ РАСТВОРИМОСТИ
Бланкс и Праузнитц [5] подошли к проблеме полярных взаимодей-
ствий путем расчетов параметров растворимости гомоморфных угле-
водородов. Эти авторы предположили, что суммарное значение
этого параметра, полученного по теплоте испарения, можно вычис-
лять как корень квадратный из суммы квадратов полярных и непо-
лярных компонентов. Авторы пришли к выводу, что подобные рас-
четы, хотя и вносят уточнения при наличии полярных групп, не при-
водят к получению надежных результатов, когда в системе имеются
водородные связи.
139
Хансен Ц01 и Кроули, Тидж и Лау 18] предложили «трехмер-
ные» параметры растворимости, с помощью которых они пытались
учесть три основных типа межмолекулярных взаимодействий, опре-
деляющих растворимость: дисперсионные, дипольные и водородные
Рис. VIII.2. Параметры растворимости различных растворителей по~Барреллу:
1 — нитрометан; 2 — ацетонитрил; з — нитрозтан; 4 — 1-бромнафталин; 5 — о-дихлор-
бензол; в — тетрагидронафталин; 7— толуол; 8— растворитель «Солвессо-150»; 9— мяг-
читель «Апко»; ю — н-гептан; 11 — н-пентав; 12 — зтиленкарбонат; 18 — пропиленкарбо-
нат; 14 — 2,3-бутиленкарбоиат; 15—диметилфталат; it—диоксан; 17—дибутилфталат;
it — метилпропионат; 19— н-бутилацетат; 20 — диизобутилкетон; 21 — диэтиловый эфир;
22 — метанол; 28 — этанол; 24 — н-пропанол; 25 — «-бутанол; 2в — н-пентанол; 21 —
2-этилбутапол; 21 — метилиэобутилкарбинол; 29 — 2-этилгексанол.
связи. Обе предложенных системы позволяют составить более пол-
ное представление о характере растворимости данного полимера и,
кроме того, позволяют, вероятно, учесть некоторые расхождения
между теорией и экспериментом для простых систем. Однако можно
сомневаться в том, что усилия, затраченные на получение характе-
ристик полимера таким путем, не окажутся много больше усилий,
необходимых для введения поправки экспериментальным путем.
Это позволило бы учесть несоответствия из-за сугубо качественного
разделения систем по склонности к образованию водородных свя-
зей (рис: VIII.2).
140
ВЛИЯНИЕ КРИСТАЛЛИЧНОСТИ
В самом начале главы отмечалось, что понятие о параметре раствори-
мости приложимо только к аморфным полимерам.
Для того чтобы приспособить этот метод к высококристалличе-
ским полимерам, надо ввести теплоту плавления \Нт в уравнение
для изобарно-изотермического потенцйала:
А^м = (AZZ^ + Т (AiSjjAiSm) (VIII.5)
Высококристаллические полимеры, например полиэтилен и поли-
тетрафторэтилен, при комнатной температуре не растворяются ни
в одном из известных растворителей. Однако эти полимеры подчи-
няются правилам, основанным на определении параметра раствори-
мости при Т 0,9Тт. Например, полиэтилен становится раство-
римым при температуре выше 80 °C. Более того, кристаллические
полимеры подчиняются правилам растворимости даже при комнатной
температуре в том смысле, в каком это относится к набуханию. Это
обстоятельство опять-таки показывает, что кристаллические обла-
сти играют роль (физических) поперечных сшивок.
Могут быть получены некоторые кристаллические полимеры
с группами, весьма активно участвующими в образовании водород-
ных связей, и способные растворяться при комнатной температуре.
Однако в подобных случаях между полимером и растворителем воз-
никают весьма специфические взаимодействия. Например, целлю-
лоза растворима в 70 %-ной серной кислоте и в водном растворе
тиоцианата аммония; найлон 6,6 растворим в феноле и в 15%-ном
метанольном растворе хлорида кальция.
ПРЕДЕЛЫ РАСТВОРИМОСТИ
Температура Флори (0Ф) определяется как температура, при которой
парциальный мольный изобарно-изотермический потенциал взаимо-
действия полимер — растворитель равен нулю. В этом случае сис-
тема полимер — растворитель находится в «идеальных» условиях.
Если Т = 0ф, то молекулы свободно могут проникать друг в друга.
При Т меньше 0Ф макромолекулы образуют ассоциаты. Наконец,
если температура много меньше 0Ф, происходит высаждение поли-
мера из раствора.
Термодинамическое рассмотрение позволяет получить следующее
уравнение для температуры, при которой начинается фазовое раз-
деление полимерных растворов:
---------- (VIII. 6)
(1+^й77;)
где С —, постоянная для данной системы полимер — растворитель.
Очевидно, что точка Флори есть критическая температура сме-
шения для предельного случая бесконечного молекулярного веса.
141
Фокс [9] получил соотношения между 0-точками систем поли-
мер — растворитель и параметром растворимости бр данного рас-
творителя. На рис. VIII.3 представлены графики зависимости 6Р
Рис. VIII.3. Соотношение между
параметрами растворимости и
0-температурами [16]:
а — полиметилметакрилат; б — поли-
стирол; в — полиизобутилен.
от 0Ф. С помощью этих графиков можно приблизительяо оценить
0ф различных систем полимер — растворитель:
6Ф 0,2256^ — 1 (бп —бР> при 6р < 6,1 (VIII.7а)
6Ф 0,22565п —1 <6Р~бп> при 5Р > 6п (VIII.76)
Линии, проведенные на рис. VIII.3, соответствуют этим соот-
ношениям.
Следует отметить, что построение графика зависимости 0Ф от 6Р
и последующая экстраполяция этой зависимости к О К составляют
ценный метод определения значений 6р.
ЛИТЕРАТУРА
1. Hildebrand J. Н. and Scott R. L.,
«The Solubility of Non-Electro-
lytes», Reinhold, New York, 1936
(2nd Ed.); 1959 (3rd Ed.).
2. Flory P. J., «Principles of Poly-
mer Chemistry», Cornell Univer-
sity Press, Ithaca N. Y., 1953.
3. Tanford C., «Physical Chemistry
of Macromolecules», Wiley, New
York, 1961.
4. Tompa H., «Polymer Solutions»,
Academic Press, New York, 1956.
5. Blanks R. F. and Prausnitz J. M.,
Ind. Eng. Chem. Fundamentals
3 (1964) 1.
6. Burrell H., Official Digest 27, .
Nr. 369 (1955) 726; 29, Nr. 394
(1957) 1069 and 1159.
7. Burrell H., in «Polymer Hand-
book», J. Brandrup and E. H. Im-
mergut, Eds., Part IV, p. 341,
Interscience, New York, 1966.
8. Crowley J, D., Teague G. S. and
Lowe J. W., J. Paint Technol.
38 (1966) 269; 39 (1967) 19.
9. Fox T. G., Polymer 3 (1962) 111.
10. Hansen С. M., J. Paint Technol.
39 (1967) 104 and 511.
11. Hildebrand J. H., J. Am. Chem.
Soc. 38 (1916) 1452.
12. Hoy K. L., J. Paint Technol. 42
(1970) 76.
13. Scatchard G., Chem. Revs. 8
(1931) 321.
14. Small P. A., J. Appl. Chem. 3
(1953) 71.
15. Van Krevelen D. W., Fuel 44
(1965) 236.
16. Van Krevelen D. W. and Hojty-
zer P. J., J. Appl. Polymer Sci. 11
(1967) 2189.
ЧАСТЬ
ТРЕТЬЯ
СВОЙСТВА ПОЛИМЕРОВ
В СИЛОВЫХ ПОЛЯХ
глава lx. МЕХАНИЧЕСКИЕ (ВЯЗКОУПРУГИЕ)
СВОЙСТВА
Единственная механическая характеристика, которую можно рас-
считать. с помощью аддитивных величин, — это объемный модуль
упругости или обратная ему величина — сжимаемость. С помощью
объемного модуля упругости можно оценить другие упругие посто-
янные материала. Их можно также найти с помощью эмпирического
соотношения между модулем упругости при сдвиге и температурами
переходов. Если известна температура стеклования, то можно также
оценить временную и температурную зависимости модуля.
ВВЕДЕНИЕ
Механические характеристики полимерных материалов пред ста- \
вляют интерес с точки зрения любых практических применений этих )
материалов в конструкциях. Механическое поведение материала — у
это деформация данного материала под действием прилюженныхГсил.
Простейшими оказываются"механические характеристики одно-
родного изотропного и чисто упругого материалов. Реакция таких
материалов на механическое воздействие может определяться всего
лишь двумя постоянными. Для описания механического поведения
анизотропных, ориентированных аморфных, кристаллических и
ориентированных кристаллических материалов требуется уже боль-
шее число постоянных.
УПРУГИЕ ХАРАКТЕРИСТИКИ
ОПРЕДЕЛЕНИЯ
Изотропные упругие материалы характеризуются тремя взаимосвя-
занными модулями, т. е. характеристическими отношениями между
напряжением и деформацией (рис. ТХ.1).
1. Объемный модуль (К, дин/см2)
Гидростатическое давление .
— ' Объемная деформация ~
__ Гидростатическое давление р ' _ pVo
Изменение объема, отнесенное к единице объема тела ДУ/Po АР
143
Рис. IX.1. Виды деформаций — гидростатическое давление (а), сдвиг (б), растя-
жение (в):
1 — нет сжатия; 2 — гидростатическое сжатие; 3 — иет сдвига; 4 — простой сдвиг; 5 — нет
нагрузки; в — растяжение.
Рис. IX.2. Соотношение между модулями упругости.
144
2. Модуль упругости при сдвиге, или жесткость (G, дин/см2)
Сдвиговое напряжение _
Деформация сдвига
______________Сила сдвига на единицу площади_____________
Величина сдвига, отнесенная к единице расстояния между
сдвигаемыми поверхностями
_ F/A _ F__________т_
S/D A tgy у
3. Модуль Юнга (Е, дин/см2)
Растягивающее напряжение ______________________
Деформация растяжения
_ Сила, отнесенная к единице площади F/А т
Удлинение, отнесенное к единице длины AL/Lo в
4. Когффициент Пуассона (у)
Изменение ширины на единицу ширины ______ Поперечное сжатие
* Изменение длины на единицу длины Продольная деформация
Перечисленные величины свя-
заны друг с другом (для упру-
гого тела) с помощью следую-
щего уравнения:
£ = 2G(l + v) = 3X(l —2v) (IX.1)
На рис. IX.2 и 1Х?3 это урав-
нение представлено в графическом
виде. Довольно часто пользуются
величинами, обратными модулям,
которые носят название податли-
востей:
1) упругая податливость (D —
= НЕ)-,
2) сдвиговая податливость
(J = i/G);
3) объемная податливость (х =
= НК), или сжимаемость.
Рис. IX.3. Зависимость отношения
модулей упругости при растяжении
и сдвиге от коэффициента Пуассона.
ЧИСЛОВЫЕ ЗНАЧЕНИЯ
В табл. IX. 1 приведены характерные значения модулей и коэф-
фициента Пуассона для ряда различных материалов: металлов,
керамики и полимеров.
Модули упругости полимеров изменяются в более широком диа-
пазоне значений по сравнению с модулями других материалов (от
10е дин/см2 для эластомеров до 1011 дин/см2 для жестких полимеров).
Это обстоятельство — одна из причин широкого применения поли-
мерных материалов. Абсолютные значения жесткости и прочности
10 Заказ 699 145
Таблица IX. 1. Механические характеристики различных материалов (модули выражены в дин/см2)
Материал V й ё К- 10-’» р oi-OI '(d/Л) 1 (С/р)- Ю-’»
Металлы чугун 0,27 90 35 66 7,5 12,0 4,7 8,8
мягкая сталь 0,28 220 86 166 7,8 28,0 11 21
алюминий 0,33 70 26 70 2,7 26,0 9,6 26
медь 0,35 120 44,5 134 8,9 13,5 4,5 15
свинец 0,43 15 5,3 36 11,0 13,6 4,8 33
ртуть 0,5 0 0 25 13,55 0 0 1,85
Неорганические твердые тела кварц 0,07 100 47 39 2,65 2,20 38,0 17,8 14,0 14,7
плавленый кварц 0,14 70 30,5 32,5 32,0 14,7
стекло 0,23 60 24,5 37 2,5 24,0 9,8 14,9
гранит 0,30 30 11,5 25 2,7 11,1 4,3 9,2
Полимеры полистирол 0,33 3,2 1,2 3,0 - 1,05 3,05 1,15 2,85
полиметилметакрилат 0,33 4,15 1,55 4,1 1,17 3,55 1,33 3,5
найлон 6,6 0,33 2,35 0,85 3,3 1,08 2,21 0,79 2,3
полиэтилен НД 0,45 1,0 0,35 3,33 0,91 1,1 0,385 3,7
эбонит 0,39 2,7 0,97 4,1 1,15 2,35 0,86 3,6
резина 0,49 0,002 . 0,0007 0,033 0,91 0,002 0,00075 0,04
Жидкости вода 0,5 0 0 2,0 1,0 0 0 2,0
органические жидкости 0,5 0 0 1,33 0,9 0 0 1,5
«Усы» (уискерсы) окиси алюминия 2000 1000 667 3,96 510 253 225
карборунда — 1000 500 333 3,15 315 160 106
графита — 1000 500 333 2,25 440 220 150
Коэффициент перевода в СИ: 1 дин/см2 = 0,1 Н/м2.
полимеров гораздо ниже, нежели металлов, однако, если сравнивать
такие материалы при одинаковом весе, то полимеры оказываются
предпочтительнее из-за их относительно низкой плотности. Наиболее
характерные значения модулей полимеров примерно в десять раз
меньше, чем модулей металлов.
Как уже отмечалось, объемный модуль упругости — единствен-
ная механическая характеристика, которая рассчитывается с по-
мощью аддитивных величин. Поэтому объемный модуль будет рас-
смотрен, в первую очередь, хотя другие модули более интересны
с точки зрения технических приложений и играют на практике
более существенную роль.
ОБЪЕМНЫЙ МОДУЛЬ УПРУГОСТИ (К)
Объемный модуль упругости — величина, обратная сжимаемости
х — важной термодинамической характеристики, уже упоминав-
шейся в гл. VI. Эту величину можно определить в экспериментах
при статическом сжатии, а также, что гораздо легче, путем измере-
ния скорости распространения продольных упругих волн, т. е.
звука.
В сплошной изотропной твердой среде возникают упругие волны двух неза-
висимых типов.
В случае волн сжатия твердое тело подвергается локализованным череду-
ющимся деформациям сжатия и растяжения. Отдельные молекулы или атомы
твердого тела, через которое проходит волна сжатия, перемещаются в направле-
ниях, перпендикулярных фронту падающей волны и параллельных распростра-
нению этой волны.
Второй тип упругой волны известен как деформационная волна. В непре-
рывной среде, через которую проходят деформационные волны, материал ло-
кально подвергается действию сил сдвига, и сама волна представляет собой рас-
пространение в твердом теле осциллирующего сдвигового движения. Такое
движение отдельных молекул осуществляется параллельно волновому фронту
и перпендикулярно направлению распространения волны (поперечная волна).
В средах, не обладающих упругостью формы, например в газах или низко-
молекулярных жидкостях, возможно распространейие только продольных волн.
В жестких, но несжимаемых средах могут распространяться только поперечные
волны.
Соотношение между объемным модулем и продольной скоростью
распространения звука в жидкостях имеет вид
“прод = “^ ' (IX.2)
Рама Рао [22] показал, что для органических жидкостей отно-
шение tP/’/p практически не зависит от температуры и что комбина-
ция
М
y = V“n/pofl <1Х-3)
обладает свойствами аддитивной мольной величины. Такая аддитив-
ная величина носит название функции Рао или мольной функции
скорости звука.
10* 147
(IX.4)
Для изотропных твердых тел функция Рао отличается от найден-
ной для тех же веществ, но находящихся в жидком состоянии.
Шуйер [28] показал, что для твердых тел начинать следует с
общего соотношения
, К 3(1—v)
I1л == — - - —
прод р 14-V
где v — коэффициент Пуассона.
Это означает, что обобщенное выражение для функции Рао
имеет вид
U=V^‘ Л
(IX.5)
ИЛИ
( и \3[-3(1—V) I1/.
“прод-у V ) |_ 1+v J
(IX.5а)
Ниже приведены групповые вклады в функцию Рао [24], опре-
деляемую по уравнению выражается в см/с): (IX.5) (для этих величин скорость звука
Группа и Группа и
- СН3 1361 —F (800)
-СН2- 895 —С1 1060
^>СН— 460 —Вг 1180
1 —с- 40 —I 1400
1 =сн2 1234 —О— 348
=сн— 745 /О - <©о— 1220
=с\ 255 /0 -О-С<£О- 1 1568
^СН (ароматическая) 758 -с=о 872
V— (ароматическая) 258 —он 628
-© 4045 —соон —C=N 1431 1308
3556 /О —C^NH— 1367
3500 /° —Ci2NH2 1839
—S — (550)
(3450) -SH 1050
-О 4874 —N\ 65
=сн“ 1108 -NH- 638
=с— 618 -nh2 967
148
Шуйер показал также, что для полиэтиленов функция Рао, опре-
деляемая по уравнению (IX.5), не изменяется с плотностью незави-
симо от причин варьирования самой плотности: за счет ли изменения
температуры или строения полимера. Величина v практически не
зависит от плотности, поэтому из формул (IX.5) или (IX.5а) можно
заключить, что продольная скорость звука приблизительно пропор-
циональна кубу плотности по-
лиэтилена. Это положение под-
тверждается данными рис. IX.4.
Совместное рассмотрение
формул (IX.4) и (IX.5) приво-
дит к соотношению
К fU\*
(1Х-6>
Это выражение позволяет рас-
считать объемный модуль уп-
ругости по аддитивным вели-
чинам U и V.
Рис. IX.4. Зависимость корня кубиче-
ского из продольной скорости звука от
плотности полиэтилена.
Пример 1
Оценить объемный модуль упругости аморфного поликарбоната
М = 254
"7 = 212
Решение
Из данных, приведенных выше, можно извлечь следующие значения вкла-
дов в функцию Рао:
/ । \
1 —С-|
\ I 7
2 (-СН3)
1 [-О(СО)О-]
7112
40
2 722
1 568
17 = 11442
Таким образом
17/7 = 11442/212 = 54
и
(17/7)8 = 2,5.10W = К/р
Кроме того
0=4/77 = 254/212 = 1.20
14S*
следовательно
К = 1.20 2,5 • 1010 = 3,0 • 10Ю дин/сма
Этот результат хорошо согласуется с экспериментальным значением, равным
2,4 • 1010, которое относится к модулю упругости при низких частотах. В этом слу-
чае значение объемного модуля упругости, как известно, несколько меньше по
Сравнению с данными измерений при звуковых частотах.
Объемный модуль упругости и сжимаемость связаны с другими
физическими характеристиками, поэтому числовые значения модуля
или сжимаемости можно рассчитать через другие аддитивные вели-
чины.
Представляют интерес следующие соотношения.
1. Поверхностное натяжение. МакГовен [18] показал, что
у жидкостей сжимаемость тесно свя&ана с поверхностным натяже-
нием:
х « 1,33.10-«у“‘/! (IX.7)
Но поверхностное натяжение можно рассчитать по мольному пара-
хору (см. гл. III и VI), поэтому получается такое соотношение:
/ Р V
К = 0,75-108 (IX.8)
2. Термические характеристики. Грюнайзен [14] получил полу-
эмпирическое соотношение между тепловым коэффициентом расши-
рения, удельной теплоемкостью и сжимаемостью твердых кристал-
лических веществ:
an aV еа
^Л=7^=7\Л=С0П81 (IX.9)
так что
С„
К~~г (IX.10)
Если К выражено в дин/см2, Cv (^Ср) — в кал/(моль -К) и ес —
в см3/(моль • К), то значение коэффициента пропорциональности
в уравнении (IX. 10) составляет примерно 3-106 дин-см/кал.
3. Плотность энергии когезии. Согласно Грюнайзену (см. также
Тобольский [8]), для простых молекулярных кристаллов справед-
ливо следующее соотношение:
К. 8,04еког (IX. 11)
Поскольку еког = £Ког/7, то получим
К — 3,5- 108£'КОГ/7 = 3,5-10863 (IX.lla)
Здесь коэффициент 3,5-10® обязан своим появлением переводу еког,
выраженной в кал/см3, в эрг/см® = дин/см2; 3,5-108 = 8,04-4,19-107.
В табл. IX.2 приведены результаты расчетов К этими четырьмя
независимыми методами, а также экспериментальные значения
150
Таблица IX.2. Экспериментальные и рассчитанные значения объемного модуля
(в дин/см2)
К-10- 10 (расчет), дии/см*
К- 10-1» О «Ol'S 3,5/10»
Полимеры р (опыт). со
дин/см2 р
в* со с;
£ й? о хэ
i ь О ьг
Аморфные, Tg < 298 К
полиэтилен (экстра- поляция) 0,85 1,8 2,2 1,6 — — —
полихлоропрен 1,24 2,3 3,0 2,3 — — —
Аморфные, Tg>> 298 К
полистирол 1,05 3,0 2,7 — 3,1 3,1 —
поливинилхлорид 1,41 4,1 4,1 — 3,8 3,3 —
полиметилметакрилат 1,17 4,0 3,75 — 3,9 2,9 —
поликарбонат 1,20 2,4 2,2 — — 2,4 —
полиэтилентерефта- 1,33 около 3 3,2 — 3,65 2,4 —
лат
Полукристаллические 2,3 *
полиэтилен НП 0,91 3,3 3,5 — 3,95 5,0 **
полиэтилен ВП 0,955 4,35 ' 4,95. — 3,95 2,4 * 5,0 **
полиэтилен ВП 0,97 5,0 5,5 — 3,95 2,5 * 5,0 **
полипропилен 0,905 около 3,5 3,6 — 3,7 2,55* 3,5 **
политрифторхлор- 2,15 2,0 2,9 — 4,7 1,05* 3,3 **
этилен полиоксиметилен 1,5 больше 4 8,7 4,9 4,4* I 6,4 **
найлон 6 1,1 3,3 4,4 —. 4,0 1 6,6 * 1 8,0 **
полиэтилентерефта- 1,4 около 4 4,75 — 3,65 2,4 * 5,3 **
лат
Полностью кристалличе- ские (экстраполяция)
полиэтилен 1,00 6 6,75 — 3,95 —. 5,0
полиоксиметилен 1,54 больше 4 10,4 — 4,8 — 6,4
поливинилиденхло- рид 1,95 больше 4 7,0 — 3,7 — 7,3
Коэффициент перевода в СИ: 1 дин/см* = 0,1 Н/м*.
* Для аморфного состояния.
** Для полностью кристаллического состояния.
К. Для частично кристаллических полимеров были использованы
два предельных значения энергии «когезии»: обычная энергия коге-
зии и теплота сублимирования (для полностью кристаллических
веществ).
Все эти методы позволяют получить правильный порядок вели-
чины и дают значения, расходящиеся друг с другом не более чем
в 2 раза, что следует считать хорошим соответствием. Можно ис-
пользовать среднее из четырех значений, получаемых из аддитивных
функций, если вообще нет никаких экспериментальных далных.
.151
МОДУЛЬ СДВИГА, ИЛИ ЖЕСТКОСТЬ G
Модуль сдвига можно определить путем измерения срлы, требуемой
для создания деформации кручения, или по скорости распростране-
ния поперечных волн:
ипопер= jj", (IX.2а)
Для изотропных тел G можно также оценить с помощью уравне-
ния (IX.1), если известны коэффициент Пуассона и объемный мо-
дуль упругости:
«-«(т-ттт) • . . (1х'1а)
Другой способ довольно точной оценки жесткости полимера при
комнатной температуре заключается в применении эмпирического
соотношения, полученного Ван Кревеленом и Хофтицером [35]:
G.(298) = Gfi (298)+^ [Ge (298)-Ge (298)] (IX.12)
(где^7?с — модуль упругости полностью кристаллического и пол-
ностью стеклообразного изотропных полимеров), причем
гр ___________298
Ge(298)~ - т100 - ЮМ (IX.13)
3
Gg (298) i-]_(600/Tg)'1010 (IX.14)
Если Tg меньше 298 К, то величиной Gg (298) следует пренебречь. Ниже приведены результаты, подученные с помощью этих фор-
мул для ряда полимеров, а также экспериментальные данные:
G-10-“ (опыт), G-10-1’ (расчет),
Аморфное состояние дин/см’ дин/см9
полистирол 1,1—1,2 1,15
поливинилкарбазо л 1,24 1,33
полиметилметакрилат 1,0—1,5 1,15
поливинилхлорид 1,1 . 1,10
поливинилацетат 1,0 1,0
поликарбонат 0,8-1,1 1,25
полиуретан 6,4 i Полукристаллическое состояние 0,9 1,0
полиэтилен (хс 0,8) 0,5—1,0 0,8
полипропилен (хс <=& 0,5) 0,7 0,75
полиоксиметилен (хс г=»1) 1,2—2,0 1,6
поливинилфторид (хс 0,25) 0,1 0,1
поливинилиденхлорид (хс «==* 0,75) 1,2 1,05
полиоксиэтилен (хс 1) 0,2 0,35
полиоксипропилен (хс 0,3) 0,05 (0,05)
найлон 6 (хс !==! 0,35) — 1 1.2
полиэтилентерефталат (хс «=< 0,3) — 1 1,1
политетраметилентерефталат (хс 0,2) ~1 1,0
Коэффициент перевода в систему СИ: 1 дин/см* = 0,1 Н/м».
Соответствие оказывается весьма удовлетворительным.
152
МОДУЛЬ ЮНГА Е
Модуль Юнга можно определить путем измерения напряжения
и отнесением полуденного значения к степени удлинения. Эту вели-
чину можно также рассчитать с помощью уравнения (IX.1), если
известны коэффициент Пуассона и либо К, либо G:
£’ = 3^(l-2v)^2G(l + v) ' (ГХ.16)
Значения Е определены для ряда монокристаллов линейных
неразветвленных полимеров. Это осуществлено прямыми измере-
ниями или расчетом по силовым постоянным для деформации валент-
ного угла и связей [13, 25, 26, 34].
Порядок величины Е для полимерных кристаллов соста-
вляет 1012 дин/см2. Это гораздо выше, чем значения, приведенные
в табл. IX. 1 и относящиеся к промышленным полимерам.
КОЭФФИЦИЕНТ ПУАССОНА V ' '
Коэффициент Пуассона изменяется от 0,5 для жидкостей до 0,1—0,2
для чисто упругих тел. На рис. IX.5 приведены экспериментальные
значения у, взятые из литературы, для многих материалов. Как пра-
Рис. IX.5. Коэффициенты Пуас-
сона для различных материалов:
1 — алюминий; 2 — медь; з — свинец;
4 — чугун; 5 — мягкая малоуглеро-
дистая сталь; < — ртуть; 7 — стекло;
i — двуокись кремния; 9 — плавле-
ный кварц; 10 — гранит; 11 — вода;
12 — полистирол; 13 — полиметилмет-
акрилат; 14— эбонит; 15—полностью
кристаллический полиэтилен (эстрапо-
ляция); 16 — органические жидкости;
а — стеклообразное состояние; б — мо-
лекулярные кристаллы и соединения с
поперечными связями; в — ковкие ме-
таллы.
вило, значение коэффициента Пуассона составляет около 0,33 для
стеклообразных полимеров, примерно 0,4 для полукристаллических
полимеров и 0,49—0,5 для эластомеров (каучуков).
Пример 2
Оценить модуль сдвига и модуль Юнга для поликарбоната.
Решение
В примере 1 было получено значение К = 2,4-1010. Для стеклообразных
полимеров коэффициент Пуассона составляет 1/3, поэтому для ^модуля сдвига
по уравнению (IX. 1) получается следующее значение:
3 «__оу
G= у X утру = 3/2-2,4-1010 • (1/3)/(4/3) = 0,9 • 1010 дин/см2
153
Это значение удовлетворительно согласуется с экспериментальным (0,8—1,1) X
Х10В * 10 дин/см2. Величину G можно также получить с помощью уравнения (IX. 14).
Поскольку Tg = 423 К, то
Gg (298) 3/(1 + 600/423)-1010=1,25- 1010 дин/см2
Далее, модуль ЮнгаДюлучается по формуле (IX.1):
Е = ЗК (1 — 2v) = 3 • 2,4 • 101° • 0,33 = 2,4 • 1010 дин/см2
ТЕМПЕРАТУРНАЯ ЗАВИСИМОСТЬ
МОДУЛЕЙ УПРУГОСТИ
Если^ построить зависимость модуля упругости от температуры,
то получится весьма характерная кривая, форма которой оказы-
вается различной для разных типов полимеров — линейных аморф-
ных, полукристаллических или эластомеров (поперечно сшитых
аморфных полимеров).
Рис. IX.6. Зависимость модуля от
температуры для изотактического
кристаллического полистирола, двух
линейных атактических образцов по-
листирола А и В с различными моле-
кулярными весами и атактического
полистирола с небольшой плотностью
поперечных связей [8):
1 — кристаллический; 2— с поперечными
связями.
В качестве типичного примера ниже рассматривается полистирол.
Этот ’полимер (обычно аморфный (атактический); в этом состоянии
в нем можно создать поперечные связи. Но полистирол может быть
и кристаллическим (изотактический полистирол). Зависимости мо-
дуля полистиролов различных типов от температуры представлены
на рис. ГХ.6.
На зависимости модуля упругости при сдвиге от температуры
для аморфного полимера обнаруживаются пять участков, характе-
ризующихся различными упругими свойствами (рис. IX.7): участок
стеклообразного состояния (Т Tg, G 10е’5—1010>6 дин/см2);
переходный («кожеподобный») участок (Т Tg; G изменяется от 1010
до 106 дин/см2); «плато» высокоэластического (каучукоподобного)
состояния (G сохраняет практически постоянное значение
<=* 10е’3 дин/см2); участок вязкоупругого течения (G изменяется от
10е’3 до IO4-6); участок течения жидкости (G ниже 10* дин/см2).
Протяженность плато высокоэластического состойния в очень
сильной степени зависит от длины молекулярных цепей, т. е. от
молекулярного веса. Взаимное перепутывание цепей есть причина
каучукоподобного поведения полимерной жидкости.
154
Плато можно «стабилизировать» путем поперечного сшивания
полимера. Это приводит к полному устранению областей течения,
поскольку химические связи закрепляют узлы сетки. Физические
поперечные связи возникают вследствие кристаллизации. Понятно,
что стеклообразное состояние более или менее «стабилизируется»
путем кристаллизации, так что переход становится менее выра-
женным.
Из рис. IX.6 видно, что при температуре перехода в стеклообраз-
ное состояние резко уменьшается жесткость аморфных полимеров.
Для полукристаллических полимеров также происходит уменьше-
ние жесткости, но эти полимеры остаются достаточно жесткими
Рис. IX.7. Области вязкоупругого поведения аморфных полимеров:
Механиче- ское поведение Стеклообраз- ное Кожеподоб- ное Высокоэла- стическое Вязкоупру- гое течение Вязкое течение
Молекуляр- ная под- вижность Колебания атомных групп Близкие диффузион- ные переме- щения сегментов цепей Быстрые короткие перемещения; замедленные дальние перемещения Проскальзы- вание зацеплений дальнего порядка дальнодейству- ющие изменения конформаций (движение молекулы в целом)
вплоть до точки плавления. В высококристаллических полимерах
практически отсутствует влияние перехода в стеклообразное состоя-
ние на свойства материала. Жесткость этих полимеров резко падает
при температуре плавления кристаллитов. Очевидно, что жесткость
стеклообразных полимеров сильно зависит от температуры стекло-
вания. В отличие от этого жесткость высококристаллических поли-
меров определяется положением точки плавления. Для полукри- -
сталлических полимеров существенную роль играют оба перехода.
Экспериментально полученные Ван Кревеленом и Хофтицером
[35] выражения (IX.12) —(IX.14) можно распространить и на зависи-
155.
мость жесткости полимеров от температуры. Для полностью кри-
сталлических полимеров при 100 <; Т <j Тт
Gc ^3-low=3- 10Ю(Гт/Г—1) '(IX.15)
Для полностью аморфных полимеров при Т << Те
3/2Те {Тй/Т)
1010 7ТТ7^=Э-1010 (tJt+2)
G& < 10’ при T > Тй
Для полукристаллических полимеров
GnojiynpHCT Gg (Ge Gs) (IX.17)
где xc — степень кристалличности.
На рис. IX.8 в качестве типичного примера приведены рассчи-
танные и экспериментальные данные для найлона 6.
[★ В настоящей главе лишь вскользь упоминается о важнейшем и специфи-
ческом для полимерного состояния вещества высокоэластическом поведении
и его количественной характеристике — плато высокоэластичности. В основном
тексте книги говорится только об одном параметре плато — его высоте, отвеча-
ющей значениям сдвигового модуля упругости, близким к 2 106 дин/см2, а также
указывается на факт сильной зависимости длины (протяженности) плато высоко-
зластического состояния от молекулярного веса полимера. Известны, однако,
методы, позволяющие дать количественную оценку длины плато и установить
корреляцию этой величины с молекулярными характеристиками полимера.
Протяженность плато высокоэластического состояния определяется как
область, в которой мбдуль упругости остается примерно постоянным в диапазоне
значений 10е—10’ дин/см2 при варьировании условий нагружения полимера.
Эта область может определяться по двум координатам — температуре и времени
(частоте) внешнего воздействия. В первом случае протяженность плато высоко-
эластического состояния характеризуется разностью температур (ДТ) между
температурой текучести и размягчения, во втором — разностью логарифмов
частот Д 1g <в, большая из которых отвечает переходу в стеклообразное состоя-
ние, а меньшая — в текучее состояние полимера. Разность Д Т находят в условиях
нормирования режима испытаний по временному (частотному) фактору, a A 1g со
определяется при изотермических условиях. В силу принципа температурно-
временной аналогии должна существовать связь между АТ и Д 1g со, а обе эти
величины могут быть сопоставлены с характеристиками структуры полимеров.
Общий подход к установлению связи между ДТ и Д 1g со и нахождению
корреляций этих величин с молекулярными параметрами полимера был предло-
жен в работах [1 д, 2 д}. В качестве основной характеристики полимерной цепи
в этих работах было предложено использовать безразмерное отношение х =
= М/Мпъ (где М — молекулярный вес данного полимера, а ЛГПв— такой моле-
кулярный вес в данном гомологическом ряду, начиная с которого полимер может
переходить в высокоэластическое состояние). Величина МПв — это мера жест-
кости макромолекулярной цепи, или величины кинетического сегмента. Суще-
ственно, что может быть установлена простая связь между 1ИПв и другими моле-
кулярными характеристиками гомологического ряда. Так, характеристикой
жесткости (или мерой длины кинетического сегмента) является величина Ме,
вычисляемая по значению динамического модуля в области плато высокоэластич-
ности Ge как Ме — pRT/Ge (где р — плотность полимера; R — универсальная
газовая постоянная; Т — абсолютное значение температуры). Другим молеку-
лярным параметром, связанным с химическим строением цепи, является вели-
156
чина Мс — критический молекулярный вес, определяемый по моменту резкого
изменения характера зависимости вязкости от молекулярного веса полимера,
которое, по существующим представлениям, обусловлено возникновением флук-
туационной сетки межмолекулярных «зацеплений» в расплаве по достижении М с-
Вопрос об определении Ме и его связи с некоторыми величинами, имеющими
общий смысл для многих полимерных систем, рассматривается в гл. XVI на-
стоящей книги.
Рис. IX.8. Зависимость модуля упругости при сдвиге от температуры:
X —данные Шмайдера и Вольфа [271; О — данные Хейджбоера [15].
Для общего подхода к оценке протяженности плато высокоэластического
состояния полимеров важным экспериментальным фактом является следующий
ряд соотношений между ЛГПВ, Ме и Мс, установленный в работах [2 д, 3 д]:
Мпй = 2Мс = Ше
Это позволяет найти для полимера заданного молекулярного веса параметр
х = М/Мпв= М12МС = М/4Ме, нормируя М по независимо найденным вели-
чинам Ме или Мс-
Зависимости ДТ и Д 1g <в от х для различных полимеров представляются
универсальными функциями, показанными соответственно на рис. 1, д и 2, д.
Первая из этих зависимостей может быть описана аналитическим выражением
вида
С 1g ж
где В 3,22 и С = 122 (очевидно, эта формула имеет смысл при lgz<^£).
157
Вторая зависимость описывается формулой
Д 1g <о = a lg z
где коэффициент а практически сопадает с показателем степени, равным 3,4,
в зависимости вязкости от молекулярного веса полимера в гомологическом ряду.
Этот результат связан с тем, что с ростом молекулярного веса (и величины х)
смещается длинновременная, или низкочастотная, граница плато высокоэластич-
ности, причем это смещение пропорционально изменению вязкости. В то же время
высокочастотная граница плато, отвечающая переходу в стеклообразное состоя-
ние, не зависит от длины всей цепи, так как ее положение определяется молеку-
лярными движениями, совершаемыми в пределах одного сегмента макромоле-
кулы.
Рис. 1, д. Зависимость протяженности плато высокоэластического состояния
по температурной шкале от относительной длины молекулярной цепи. Экспери-
ментальные точки относятся к полибутадиену (7), полистиролу (2) и полидиметил-
силоксану (3).
Рис. 2, д. Зависимость протяженности плато высокоэластичности по частотной
шкале от относительной длины молекулярной цепи. Экспериментальные точки
относятся к полибутадиенам (7), полистиролам (2) и поливинилацетатам (3).
Таким образом, приводимые рисунки и указанный метод нахождения аргу-
мента — величины х — по значениям Ме или Мс дают общую схему для вычисле-
ния протяженности плато высокоэластического состояния аморфных полимеров
по температурной и частотной шкалам.
Для одно компонентных полимерных систем высота плато высокоэластич-
ности не зависит от молекулярного веса и является характеристикой гомологи-
ческого ряда. Однако при введении в полимер пластификатора (растворителя)
изменяется не только низкочастотная граница плато из-за уменьшения вязкости
полимерной системы, но и снижается высота плато. Экспериментальных данных,
которые позволили бы дать общую количественную картину всех закономерно-
стей, отвечающих этому случаю, пока недостаточно. Однако ориентировочно
можно считать, что высота плато высокоэластичности (т. е. значение модуля
в области плато) снижается пропорционально уменьшению содержания полимера
в пластифицированном полимере, а сдвиг низкочастотной границы плато свя-
158
зан как с падением вязкости при пластификации, так и со смещением точки пере-
сечения плато с ветвью релаксационного спектра, отвечающей текучему состоя-
нию пластифицированного полимера [4 д].*1 — Прим. ред.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
д! Малкин А. Я., Даюра Е. А.,
Виноградов Г. В., ДАН СССР,
1969, т. 188, № 6, с. 1328—
г - 1331.
I 2 д.(Виноградов Г. В., Даюра Е. А.,
— *- Малкин А. Я., Инж.-физ. жур-
нал, 1970, т. 18, № 6, с. 965—
978.
3 д. Porter В. S., Johnaon J. F.,
Rheol. Acta, 1967, v. 7, № 4,
p. 332-334.
4 д. Блинова И. К. и др., «Механика
полимеров», 1973, № 1, с. 132—
137; Виноградов Г. В. и др.,
Europ. Polymer J., 1973, v. 9,
№ 11, р. 1231—1249.
ВЫСОКОЭЛАСТИЧНОСТЬ РЕЗИН
Особый случай представляют поперечно сшитые эластомеры. Из-за
наличия сетки поперечных связей этот класс полимеров практически
не обнаруживает течения. Кинетическая теория высокоэластичности
каучуков позволяет получить следующую формулу для модуля
Юнга при малых деформациях:
ВТа ВТ -
£ = 3 — =3z~?7—= ЗС0 (IX.18)
Мсегм V
где В— универсальная газовая постоянная, равная 8,316-10’ эрг/К-моль;
АГсегм— среднечисловой молекулярный вес сегментов между поперечными свя-
зями; р — плотность; z — среднее число поперечных связей на однв структур-
ное звено (M/Mcera)-,V — мольный объем структурного звена; Со = zBTIV.
Из соотношения (IX.18) видно, что модуль упругости резины
увеличивается с повышением температуры. Это резко отличается
от поведения других твердых тел. Причина заключается в том, что
высокоэластичность каучуков обладает энтропийной природой в от-
личие от «энергетической» упругости обычных твердых тел; модуль
возрастает с температурой потому, что увеличивается интенсивность
теплового, или броуновского, движения, вследствие чего усили-
вается нагрузка на молекулярные сегменты в точках их зацеплений,
поскольку возрастает тенденция к образованию более вероятной
клубкообразнои конформации цепи.
Из формулы (IX.18) видно также, что Е увеличивается с ростом z.
Обычно z имеет порядок 10“2, следовательно Е при 25 °C близко
к 107 дин/см2.
Теория упругости каучука позволяет получить также следу-
ющее соотношение между силой и деформацией для одноосного
растяжения и сжатия:
/ = Со(А —А-2) (IX.19)
где А = L/Lq — отношение длины образца в растянутом состоянии к его длине
до начала деформации.
159
Для простого сдвига соответствующее выражение имеет вид
/пс= Со tg у <=< Со (Л—Л-1) (IX.19а)
где у — угол наклона ребра элементарного кубика, вырезанного в теле, при
Сдвиге.
Равенство tg у = Л — Л"1 справедливо только при небольших
деформациях растяжения. Реальное поведение поперечно сшитых
каучуков при одноосном растяжении более или менее удовлетвори-
тельно описывается эмпирическим уравнением Муни — Ривлина
[19, 211:
7=(fi+-y-) (Л-Л-2) (IX.20)
где Сх и Сг — эмпирические постоянные.
При сжатии и при сдвиге поведение образца больше отвечает
модели идеальной упругости.
Уравнение (IX.20) лучше описывает экспериментальные данные
по одноосному растяжению резин по сравнению с уравнением (IX. 19),
однако с точки зрения предсказаний это уравнение обладает неболь-
шой ценностью, поскольку физический смысл постоянной С2 пробле-
матичен. Блоклэнд [10] разработал новую теорию высокоэластич-
ности резин. На основании данных изучения фотоупругости, рас-
сеяния света и электронной микроскопии он обнаружил некую
структуру в сетчатых системах, которая напоминает стержневид-
ные ассоциаты сегментов цепных молекул или «пачки». Эти струк-
турные элементы включают в себя около 5% цепных сегментов.
На основе предложенной модели Блоклэнд вывел соотношение
следующего типа:
/=С0[1-С3(Л)](Л-Л-2) (IX.21)
где С3 (Л) — функция поправок.
Эта формула позволяет сделать более ясным смысл параметра С2
в уравнении Муни — Ривлина.
Выражение (IX.20) можно переписать так:
/ = [(С1+С2)-(с2--у-)](Л-Л-2) (IX.20а)
или
с^Г2(1-4-)](Л-Л‘2) (IX.206)
В табл. IX.3 приведены значения (\ и С2 для эластомеров раз-
личных типов.
Если уравнение Муни — Ривлина переписать в виде:
/ = С0 [1-0,4 (1—J-)J(A-A-2) (1Х.20в)
(где kw €t 4- С2), то по форме оно станет аналогичным (IX.21).
160
Таблица IX.3. Константы уравнения Муни — Ривлина
(числовые значения взяты из [10\)
Каучук Ci-10-» (С, + С,)х хю-«
Натуральный 2,0 (0,9-3,8) 1,5 (0,9-2,0) 3,5 0,4 (0,25—0,6)
Бутиловый 2,6 (2,1—3,2) 1,5 (1,4-1,6) 4,1 0,4 (0,3-0,5)
Бутадиен-стирольный 1,8 (0,8-2,8) 1,1 (1,0—1,2) 2,9 0,4 (0,3—0,5)
Этилен-пропиленовый 2,6 (2,1-3,1) 2,5 (2,2—2,9) 5,1 0,5 (0,43—0,55)
Акриловый 1,2 (0,6-1,6) 2,8 (0,9—4,8) 3 0,5 (0,3—0,8)
Силиконовый 0,75 (0,3—1,2) 0,75 (0,3—1,1) 1,5 0,4 (0,25—0,5)
Уретановый 3(2,4-3,4) 2 (1,8-2,2) 5 0,4 (0,38—0,43)
В эластомерах, не содержащих поперечных связей, напряжение
монотонно уменьшается с течением времени. Чем больше молекуляр-
ный вес, тем медленнее проходит релаксация напряжения. По сво-
ему воздействию на свойства материала перепутывание цепей оказы-
вается сходным с воздействием не слишком многочисленных попереч-
ных связей. Пользуясь соотношением Флори
£~ЗС0(1-2^^
\ М„
(IX. 22)
(где Мп — среднечисловой молекулярный вес), можно оценить
порядок числа структурных звеньев, приходящихся на одну попереч-
ную связь в сетчатых структурах.
ВЯЗКОУПРУГОСТЬ
Полимеры не являются идеально упругими материалами. Наряду
с некоторыми свойствами упругих твердых тел полимеры обладают
также некоторыми характеристиками вязких жидкостей. Так, поли-
мерные материалы проявляют ползучесть под нагрузкой, и напря-
жения в деформированных образцах релаксируют. Следовательно,
полимеры представляют собой вязкоупругие среды.
Еще более важным оказывается динамическое механическое
поведение полимерных материалов в условиях изменяющихся по
гармоническому закону напряжений и деформаций, между которыми
возникает разность фаз. Динамические механические испытания
позволяют рассчитать модуль упругости и величину, ответствен-
ную за диссипацию энергии в виде тепла. Формально для описания
диссипативных характеристик механических свойств полимеров
можно ввести вязкость т|.
11 Заказ 699
161
ДИНАМИЧЕСКИЕ МЕХАНИЧЕСКИЕ
ПАРАМЕТРЫ
Идеально упругие материалы при деформировании запасают всю
работу внешних сил в виде потенциальной энергии. Вся энергия,
затрачиваемая на деформацию жидкостей, диссипирует в виде тепла.
Полимеры, обладая некоторыми характеристиками как жидкостей,
так и упругих твердых тел, в процессе деформации часть энергии
запасают, а часть рассеивают в виде тепла. Диссипированная энер-
гия проявляет себя как механическое демпфирование, или «потери».
Рис. IX.9. Области на логарифмической временной шкале, охватываемые раа-
личными экспериментальными методами, согласно Ставерману и Щварцлю [33]:
/ — распространение волн; II — вынужденные колебания; III — релаксация, ползучесть
и течение; IV — нерезонансные методы с дополнительной массой; V—свободные колеба-
ния с дополнительной массой; VI — отскок; VII — испытания на изгиб.
Для измерения динамических механических параметров пользу-
ются многими экспериментальными методами. Каждый конкретный
метод охватывает лишь некоторую часть всего диапазона частот.
Следовательно, необходимо пользоваться целым рядом различных
методов, дополняющих друг друга. Сводка существующих экспери-
ментальных методов приведена на рис. IX.9.
Измерение отклика материала в процессе деформирования под
действием периодически изменяющихся сил, например в процессе
вынужденных колебаний, показывает, что деформация отстает от
напряжения на фазовый угол б (угол механических потерь).
Если колебание осуществляется по синусоидальному закону
a = <Tosina>t
то деформация изменяется как
е = ео Мп (<о/— б)
В другой записи:
<r*=<wto(
е*=еое(<<»^)
Отсюда
°* Г.» °о /8 °о . - , . . х. По . , . ®0 . х
—г- = Е* = — —— (cos о 4-1 sin о) = — cosS-H — sin о
е* so ео ' 1 ' е0 е0
162
или
E* = E' + iE" (IX.23)
где Е* — комплексный модуль упругости; £' = оо/ео cos 6 — действительная
часть, или модуль накопления; Е" = Оо/ео sin 6 — мнимая часть, или модуль
потерь.
Точно так же можно записать в виде комплексной величины
и другие динамические модули. Для комплексного модуля остается
в силе основное соотношение (IX.1). Мнимые части комплексных
модулей представляют собой «демпфирующие» члены, которые опре-
деляют диссипацию энергии в процессе деформирования данного
материала; именно поэтому эти слагаемые носят название модулей
потерь. Действительные части комплексных модулей определяют
количество отдаваемой энергии, запасенной телом при деформиро-
вании, поэтому эти величины называют модулями накопления.
Комплексные модули связаны с комплексными вязкостями с по-
мощью следующих соотношений:
Е" = сот); = 2.4vat|J G” = (orj' = 2rtv(aT]' 1
£,' = cor|j = 2nv(ilr]( G' = <0T|" = 2n-voi']" j
где ш — частота, рад/с; уш — частота, Гц; гр — комплексная
вязкость при растяжении; ц* — комплексная динамическая
сдвиге.
(IX,24)
динамическая
вязкость при
Эти величины связаны между собой соотношением
^5-= g5- = 2(l+v) (IX.25)
Для несжимаемых жидкостей у = 1/2, следовательно r]f* = Зц*.
Выражения для абсолютных значений динамических механиче-
ских характеристик имеют вид.
I £*| = [(£')2 + (£”)2]’/‘
G‘| = [(G')2 + (G”)2]‘/«
| J*| = [(J')2+(J»)2],/!
ln*l = [(n')2+(n")2],/s и т. д.
(IX.26)
ТАНГЕНС УГЛА МЕХАНИЧЕСКИХ ПОТЕРЬ
Мерой интенсивности демпфирования является отношение энер-
гии, диссипируемой за цикл колебаний, к максимальной потенциаль-
ной энергии, запасенной на протяжении цикла. Это отношение
называется коэффициентом диссипации, или тангенсом угла механи-
ческих потерь (тангенс потерь):
Е" G" J" п'
£7 = tg6£; gr = 7r = ^r = tg6G; tgdG , (IX.27)
11* 163
Полиоксиметилен ГГ I
Лолиоксизтилен
Полиокситриметилен И
Лолиокситетраметилен HI тп
Полиэтилен НД 1 1 m
Политетрафторэтилен 1 1
Полиамиды 1 II и 1m
Полиуретан „6,4 ” 1 1
Поливинилфторид 1 ~|9
Поливинилхлорид ш
Политрирторхлорзтилен |0
Полипропилен 1 И 1 И 1 гл
Полистирол I В- Ж
Поливинил карбазол “1 Г" м
По ли метилмета крип ат 1 ! I] ж л
-зтил- я
-пропип- 1 О £1
-бутил- 91 1 W 1
-гексил- 4 я 1
-лаурил- 0
-стеарил- 1 я 1
-трет -бутил- f f ЙГ
-фенил- И
-цикпогексил~ Т 1 1 tl II 1
100 200 300 ^QQ SOO
—> T,K
Рис. IX.10. Области дисперсии (максимумы механических потерь) для некоторых полимеров:
g — переход в стеклообразное состояние; т — плавление.
Другими параметрами, по смыслу аналогичными tg 6, являются
логарифмический декремент Л и удельная демпфирующая способ-
ность, или внутреннее трение, ф. Соотношение между ними таково:
Д ib
<1Х-28>
Теплота, выделяемая в одном цикле единицей объема при деформа-
циях с постоянной амплитудой, есть
Q лЯ’е^ (IX.29)
где е0 — максимальная амплитуда деформации в цикле.
КОЛИЧЕСТВЕННЫЕ ДАННЫЕ
В каждой переходной области наблюдается изменение модуля и
прохождение тангенса потерь через максимум (рис. IX.11). Такое
явление носит название динами-
ческого перехода. Спектр подоб-
ных пиков демпфирования пред-
ставляет собой «визитную кар-
точку» полимера. Эта характе-
ристика показана на рис. IX.10
для целого ряда полимеров.
Максимумы механических
потерь в области между 130 и
170 К связаны с движением по-
следовательностей, состоящих
из более чем трех СН2-групп в
цепи. Дисперсионная область
вблизи 200 К обусловлена пере-
мещениями (может быть, вра-
щениями) цепных сегментов.
Главная дисперсионная область
связана с обычным переходом
из стеклообразного в высоко-
властическое состояние. Для
ряда метакрилатов отчетливо
видно, что длинные боковые
цепи действуют как пластифи-
каторы. В зоне между переходом
Рис. IX.11. Зависимости модуля упру-
гости при сдвиге G и тангенса угла ме-
ханических потерь tg 6 от температуры
для частично кристаллических поли-
меров .
в стеклообразное состояние и предплавлением в ряде случаев возни-
кают переходы в твердой фазе, которые приводят к появлению но-
вых максимумов потерь (для политетрафторэтилена, полиэтилена,
полиметилметакрилата).
Температура, при которой наблюдаются максимумы потерь,
не равна температуре, при которой наблюдается разрыв термодина-
мической функции. Максимум потерь всегда почти совпадает с точ-
кой перегиба кривой зависимости модуля от температуры, в то время
как температура перехода, по крайней мере при низкой частоте,
165
соответствует точке пересечения касательных к двум участкам
графика зависимости модуля от температуры. Для многих полимеров
значение такой разности Td — Т6№„лк„ч (см. рис. IX.11) может
составлять около 25 °C.
Весьма важная работа, посвященная исследованию влияния
химического строения на температуру максимума потерь, была
проведена Хейжбоером [15]. Особенно детально им была исследо-
вана роль строения эфирных групп в полиметакрилатах.
Рис. IX.12. Относительное уменьше-
ние модуля упругости в зависимости
от среднего значения тангенса угла
механических потерь tg б в интервале
температур от 20 до 60 °C для поли-
меров, находящихся при температу-
рах, существенно меньших Tg. Мо-
дуль определен при частоте 1 гц [15].
Хейжбоер обнаружил также интересное соотношение между
относительным уменьшением модуля с повышением температуры
и средним значением тангенса угла потерь в диапазоне от 20 до 60 °C.
Это соотношение показано на рис. IX. 12 и выполняется для жестких
стеклообразных полимеров преимущественно при температуре ни-
же Т„.
РЕЛАКСАЦИЯ НАПРЯЖЕНИЙ И ПОЛЗУЧЕСТЬ
РЕЛАКСАЦИЯ НАПРЯЖЕНИЙ
Релаксация напряжений представляет собой зависящее от времени
изменение напряжения при постоянной величине деформации и дан-
ной температуре.
Релаксационные характеристики полимеров чрезвычайно сильно
вависят от температуры, особенно в области температуры перехода
в стеклообразное состояние.
В переходной зоне зависимость логарифма функции релаксации
напряжений а (£)/е0 от логарифма времени описывается практически
прямой линией с отрицательным наклоном. При более высоких и
при более низких температурах такой наклон становится резче.
На рис. IX.13 представлены экспериментальные данные, иллюстри-
рующие поведение полиизобутилена (Tg = — 76 °C, Td = — 60 °C).
Такое поведение можно приблизительно описать эмпирической
формулой
= Er(t) = Kt~n (IX.30)
где X и п — постоянные.
166
Для аморфных полимеров значение постоянной п может коле-
баться между 0,5 и 1,0.
Величина п тесно связана с тангенсом угла механических потерь:
В качестве другого приближения для функции релаксации напря-
жений можно привести модель Максвелла
а (£)
-^2- = £,,(О = £'оехР(-//тг) (IX.32)
где — время релаксации, т. е. время] необходимое для уменьшения аве раз.
ПОЛЗУЧЕСТЬ
Постоянство размеров представляет собой одну из наиболее
существенных характеристик конструкционных материалов, но лишь
167
немногие твердые материалы оказываются совершенными с этой
точки зрения.
Ползучесть — это зависящая от времени относительная дефор-
мация при постоянной величине действующей силы (растяжение,
сдвиг или сжатие). Ползучесть представляет собой прямое проявле-
ние вязкоупругости среды.
Характеристики ползучести весьма сильно зависят от темпера-
туры. При температурах существенно ниже точки стеклования пол-
зучесть проявляется очень слабо даже на протяжении длительных
интервалов времени. При увеличении температуры скорость раз-
вития деформаций вследствие ползучести возрастает. В области
перехода в стеклообразное состояние характеристики ползучести
начинают необычайно резко зависеть от температуры. Для многих
полимеров скорость ползучести проходит через максимум вблизи
точки стеклования.
Гораздо большее удлинение наблюдается при температурах,
лежащих существенно выше точки перехода в стеклообразное состоя-
ние, т. е. податливость ползучести в этом случае возрастает.
Хорошо известно упрощенное уравнение для функции ползу-
чести при растяжении — формула Наттинга [20]:
=Dr(t) = K'tn (IX.33)
где К' и п — постоянные.
Величина п лежит в тех же пределах, что и константа, входящая
в формулу для релаксации напряжений. Если п л/2 много меньше
1, то ил/2 приближенно равно тангенсу угла механических потерь:
n-y«rftge (IX.34)
Другое приближенное выражение для функции ползучести при
растяжении получается из реологической модели Фойхта
= D,(t) = D0 [1-ехр(—t/т,)] (IX.35)
где т3 — время запаздывания.
Из формул (IX.32) и (IX.35) следует, что процессы релаксации
напряжений и развития ползучести в какой-то степени аналогичны
химической реакции первого порядка, поскольку константа скорости
такой реакции обладает тем же смыслом, что и обратное время релак-
сации или запаздывания.
Поведение кристаллического полимера может оказаться более
сложным. Характеристики ползучести кристаллических полимеров
не только быстро изменяются с температурой, но при данной темпе-
ратуре для кристаллического полимера ползучесть часто разви-
вается быстрее во времени по сравнению с ползучестью жестких
аморфных или поперечно-сшитых полимеров. Это определяется
степенью кристалличности, которая изменяется с температурой,
168
относительной удаленностью от точки стеклования, положением
температуры плавления и возможными явлениями перекристал-
лизации.
ПРИНЦИП ТЕМПЕРАТУРНО-ВРЕМЕННОЙ
СУПЕРПОЗИЦИИ
Выше температуры стеклования характеристики скорости релакса-
ции напряжений и ползучести аморфных полимеров подчиняются
принципу температурно-временной суперпозиции, или эквивалент-
ности. '
Лидерман [17] первым * предположил, что влияние временного
фактора и температуры на свойства вязко-упругих материалов
эквивалентно. Это означает, что данные, полученные при одной тем-
пературе, можно привести к данным, полученным при другой темпе-
ратуре, простым сдвигом кривых вдоль координатных осей. Вильямс,
Лэндел и Ферри [36], Тобольский [8] и Ферри [3] детально разрабо-
тали это предположение и показали действительную выполнимость
такого принципа. С помощью созданных этими ^авторами методов
можно свести данные о релаксации напряжений, полученные при
самых различных температурах, к одной кривой, охватывающей
много порядков времени при какой-то стандартной температуре.
Принцип эквивалентности можно использовать для релаксацион-
ного модуля а/е = Er (t), который определяется в зависимости от
времени и температуры. Пример такого подхода иллюстрируется
экспериментальными данными, приведенными на рис. IX.13, для
полиизобутилена. Эти кривые сначала корректируют, т. е. при-
водят к одной плотности и температуре. В качестве стандартной
выбирается произвольная температура То (К). Приведенные зна-
чения модуля рассчитывают по соотношению
[£г («)]пРивед=4г (IX.36)
Поправка берется согласно кинетической теории упругости
каучуков; эта поправка довольно мала, и в ряде случаев ею можно
пренебречь. После такого приведения экспериментальные кривые
снова строят, как показано на рис. IX.14 слева. Теперь эти приве-
денные кривые можно сдвигать вдоль временной шкалы по отноше-
нию к стандартной кривой (То = 25 °C) до тех пор, пока части кри-
вых не совместятся и не будет образована одна обобщенная кривая,
показанная на рис. IX.14 справа. Отрезок, на который следует
сдвинуть каждую исходную кривую вдоль оси логарифма времени
для получения обобщенной кривой, носит название фактора сдвига.
Его длина зависит от температуры (см. верхнюю правую часть
рис. IX.14).
* Впервые это было сделано в явной форме в работах А. П. Александрова
и Ю. С. Л азу р кина в 1939 г.; на эти работы, как на основополагающие в рассма-
триваемой области, ссылался и Лидерман. — Прим. ред.
169
Согласно Вильямсу, Лэнделу и Ферри [36], универсальная фор-
мула для фактора сдвига имеет вид:
, , —17,44(Т—Т«)
lgaT — lg [t/t (71g)]= 6-|- (71 —
(IX.37)
В этом соотношении в качестве стандартной температуры выбрана
точка стеклования *.
Рис. IX.14. Применение принципа температурно-временной суперпозиции для
обработки экспериментальных данных по релаксации напряжений в полиизобу-
тилене. Стандартная температура приведения 25 °C. Справа вверху показана
зависимость фактора сдвига от температуры [12]:
I — данные по релаксации напряжений; II — обобщенная кривая при 25 °C; III — фактор
температурного сдвига при стандартной температуре 25 °C.
Приведенное уравнение Вильямса — Лэндела — Ферри поз-
воляет рассчитать изменение частоты (времени) при постоянной тем-
пературе, которое приводит к тем же изменениям, что и эквивалент-
ное изменение температуры при постоянной частоте (времени).
На рис. IX.15 представлен график уравнения Вильямса — Лэн-
дела — Ферри.
При температурах, лежащих ниже точки стеклования, следует
ожидать отклонений от уравнения Вильямса — Лэндела — Ферри.
Это установили, в частности, Раш и Бек [23].
Фактор сдвига а т представляет собой отношение времени релак-
сации при температуре Т к времени релаксации при температуре 7'g,
если Т& принята за стандартную температуру. Но время релаксации
♦ «Универсальными» значениями констант в формуле (IX.37) следует поль-
зоваться с осторожностью, так как для различных материалов значения констант
в уравнении (IX.37) оказываются несколько различными (см. монографию
Ферри [3]), а результаты расчета ат весьма чувствительны к точным значениям
числовых постоянных. — Прим. ред.
170
связано с вязкостью. Отсюда и формула (IX.37) может быть пред-
ставлена в виде:
Ту________Г|УдР (^я)___________П
ат~тг (Те) - Г] (Tg) Тр (Т) ~ Г] (Tg)
(IX. 38)
Принцип температурно-временной эквивалентности аналогич-
ным образом можно применить к характеристикам1. ползучести.
И в этом случае указанный принцип приводит к факторам сдвига,
практически совпадающим со значе-
ниями, определяемыми для релаксации
напряжений;
Пример 3
Релаксационный модуль полиизобутилена
при 25 °C составляет 3-10® дин/см2 при вре-
мени измерений 1 ч. Оценить: а) модуль при
времени измерений 1 ч и температуре —80 °C
и б) температуру, при которой этот модуль
для времени измерений 10* • ч равен модулю
при температуре —80 °C и времени измере-
ния 1 ч.
Решение
а. По рис. IX. 13 логарифм фактора
Сдвига при —80 °C составляет около 12.
Обобщенная кривая при t>s=<10*12 ч дает
значение модуля примерно 1010 дин/см2. Зна-
чение модуля увеличивается в 1010/3-10® =
= 3000 раз.
Фактор сдвига при —80 5С можно также
получить с помощью формулы (IX.37). Для
Следовательно
—17,44 • 101
lgar(298)= 5^101 = -1760/152,6=-11,5
Рис. IX.15. Зависимость фак-
тора сдвига по Вильямсу —
Лэнделу — Ферри ат от тем-
пературы.
полиизобутилена Tg = 197 К.
17,44-4
lg а т (193)=-^-g—4 = 70/47,6=
1,5 .
аг(193)
ат(298)
1g
= 1,5+11,5=13,0
Обобщенная кривая при г«*10*13ч дает модуль около 2-1010 дин/см2.
б. Релаксационный модуль при —80 °C и времени измерений 1 ч составляет
1010 дин/см2. Для расчета изменения температуры, соответствующего фактору
Сдвига 10®, следует воспользоваться формулой (IX.37):
lgaT (193) = 1,5; lg [а (193)/в (Г)] = 6 = 1,5+4,5;
lg ат (Т) = -4,5 = -17,44 ДГ/(51,6 + ДТ)
Отсюда
ДТ = 18 или Т = 71Й + 18 = 215К = —58°С
Для кристаллических полимеров при температурах, лежащих
ниже точек плавления, уравнение Вильямса — Лэндела — Ферри
не выполняется. Зейтц и Балаш [32] показали, что соотношение
171
между ат и Т в этом случае имеет вид простой аррениусовской
зависимости:
t Кист / 1 1 \
lgaT-lg tQ - 2>ЗЯ V Т То)
(IX.39)
МОДЕЛИ ВЯЗКОУПРУГОГО ПОВЕДЕНИЯ
Для описания и моделирования вязкоупругих характеристик поли-
меров были предложены следующие реологические модели (рис.1Х.16).
1. Идеально упругий элемент представляется пружиной, дефор-
мации которой подчиняются закону Гука с определенной величиной
Числа элементов модели Модели
1 Гука Ньютона Т Ф'
2 Моксбелло Фойгто
J бюргерсо L J7’ ф”
Phc. IX.16. Модели вязкоупругих сред.
модуля упругости (элемент Гука). Упругая деформация разви-
вается мгновенно.
2. Идеально вязкий элемент можно представить поршнем, пере-
мещающимся в цилиндре, заполненном ньютоновской жидкостью;
деформация под действием приложенного напряжения лцнейно
изменяется во времени, и эффекты упругости восстановления совер-
172
шенно отсутствуют (элемент Ньютона). Напряжение точно.на 90е
отличается по фазе от деформации.
3. Элемент Максвелла (упругая деформация плюс течение) обра-
зуется соединенными последовательно пружиной и цилиндром с порш-
нем. Эта модель описывает свойство такого материала, который
упруго реагирует на нагрузку, но может также течь. В этой модели
два вклада в деформацию подчиняются закону аддитивности
8 = 8у пругая 4“ бвязкая
Деформация не совпадает по фазе с напряжением, а фазовый угол
изменяется от 0 до 90°.
4. Элемент Фойхта (задержанная упругая реакция) представляет
собой параллельно соединенные пружины и поршень с цилиндром.
Упругая реакция развивается не мгновенно, а задерживается вязким
сопротивлением. В этой модели правилу аддитивности подчиняются
два вклада в напряжение:
О = Супругов + ^вязкое
Помимо перечисленных, существует много различных моделей,
составленных из комбинаций пружин и поршней с цилиндрами.
Три компонента (4 элемента), которые имитируют простейшее пове-
дение реального полимерного образца в процессе ползучести, могут
представляться так называемой четырехпараметрической моделью,
т. е. последовательной комбинацией моделей Максвелла и Фойхта.
Наконец, существуют так называемые обобщенные модели, в ко-
торых осуществляются мгновенный упругий отклик, вязкое течение,
а также содержится большое число элементов Фойхта, каждый
из которых обладает своими собственными модулем упругости и
временем запаздывания. Такие модели правильно передают главные
характеристики вязкоупругого поведения полимеров и позволяют
получить спектр времен релаксации и запаздывания, однако они
имеют ограниченную ценность: они применимы только при малых
деформациях. Характер течения полимера, как правило, неньюто-
новский, и упругость полимера не описывается законом Гука. Однако
в качественном отношении описанные модели оказываются полез-
ными.
ЛИТЕРАТУРА
1. Alfrey Т., «Mechanical Behaviour
of High Polymers», Interscience,
New York, 1948.
2. Eirich F. R. (Ed.), «Rheology»,
Academic Press, New York, 1959—
1960.
3. Ferry J. D., «Visco-elastic Proper-
ties of Polymers», Wiley, New
York, 1970.
4. Flory P. J., «Principles of Poly-
mer Chemistry», Cornell Uni-
versity Press, Ithaca, N. Y., 1953.
5. McCrum N. G., Read R. E. and
Williams G., «Anelastic and Di-
electric Effects in Polymeric So-
lids», Wiley, New York, 1967,
6. Nielsen L. E., «Mechanical Pro-
perties of Polymers», Reinhold,
New York, 1962.
7. StuartH.A.,«DiePhysik derHoch-
polymeren», Springer, Berlin,1956.
8. Tobolsky A. V., «Structure and
Properties of Polymers», Wiley,
New York, 1966.
173
9. Treloar L. R. G,, «The Physics
of Rubber Elasticity», Clarendon
Press, Oxford, 1958.
10. Blokland R., «Elasticity and
Structure of Polyurethane Net-
works», Thesis Delft, University
of Technology, 1968.
11. Blokland R. and Prins W., J.
Polymer Sci. (A2) 7 (1969) 1595.
12. Catsif} E. and Tobolsky A. V.,
J. Colloid. Sci. 10 (1955) 375;
J. Polymer Sci. 19 (1956) 111.
13. Frensdorf} H. K., J. Polymer Sci.
A2 (1964) 333, 341.
14. Griineisen E., «Handbuch der
Physik», Vol. 10, Springer, Ber-
lin, 1926, p. 52.
15. Heijboer J., Kolloid-Z. 148 (1956)
36 and 171 (1960) 7; Makromol.
Chem. 35A (I960) 86; Proc.
Intern. Conf. Physics of Non-
crystalline Solids, Delft (1965)
p. 231; Brit. Polymer J. 1 (1969)
3; Plastica 10 (1957) 824; 11 (1958)
34; 12 (1959) 110, 598.
16. Heijboer J. and Schwarzl F., in
R. Nitsche and K. A. Wolf
(Eds.), «Kunststoffe», Springer,
Berlin, 1962.
17, Leaderman H., «Elastic and Creep
Properties of Filamentous Mate-
rials and other High Polymers»,
The Textile Foundation, Washing-
ton, D.C., 1943.
18, McGowan j. C., Polymer 8 (1967)
57.
19. Mooney M., J. Appl. Phys. 11
(1940) 582.
20, Nutting P., Proc. Am. Soc.
Testing Materials 21 (1921) 1162.
21, Rivlin R. S., Phil, Trans. Roy.
Soc. (London) A240 (1948) 459,
491, 509 and A241 (1948) 379.
22, Rama Rao, Indian J. Phys. 14
(1940) 109; J. Chem. Phys. 9
(1941) 682.
23. Rusch К. C. and Beck R. H.,
J. Macromol. Sci. B3 (1969) 365.
24. Sakiadis В. C. and Coates J,,
A. I. Ch. E. J. 1 (1955) 275.
25. Sakurada I., Nakushina Y. and
Ito T., J. Polymer Sci. 57 (1962)
651.
26. Sakurada I. and Keisuke K., J.
Polymer Sci. C31 (1970) 57.
27. Schmieder K. and Wolf K.,
Kolloid-Z. 134 (1953) 149.
28. Schuyer J., Nature 181 (1958)
1394; J. Polymer Sci. 36 (1959)
475.
29. Schwarzl F., Kolloid-Z. 165 (1959)
88.
30. Schwarzl F. and Staverman A. J.,
J. Appl. Phys. 23 (1952) 838.
31. Schwarzl F., Chapter 6 in «Che-
mie und Technologie der Kunst-
stoffe» (Houwink, Staverman,
Eds.), 4. Edition, Akademie Ver-
lagsgesellschaft, Leipzig, 1963.
32. Seitz J. T. and Balazs C, F.,
Polymer Eng. Sci. (1968) 151.
33. Staverman A. J. and Schwarzl F.,
Chapters 1, 2 and 3 in «Die
Physik der Hochpolymeren»,
Vol. 4 (H. A. Stuart, Ed.), Sprin-
ger, Berlin, 1956.
34. Treloar L. R. G., Polymer 1
(1960) 95, 279.
35. Van Krevelen D. W. and Hofty-
zer P. J. (1970) unpublished.
36. Williams M. L., Landel R. F. and
Ferry J. D., J. Am. Chem. Soc.
77 (1955) 3701.
глава х. ПРЕДЕЛЬНЫЕ МЕХАНИЧЕСКИЕ
ХАРАКТЕРИСТИКИ
Непосредственно из аддитивных величин нельзя оценить ни одну
из предельных механических характеристик. Существуют тем не
менее различные количественные соотношения, которые связывают
предельные механические характеристики с другими механическими
свойствами полимерных материалов.
ВВЕДЕНИЕ
Предельные механические свойства представляют собой параметры,
характеризующие механическое разрушение образца в условиях
эксплуатации. Грубо говоря, эти свойства можно разделить на две
категории:
а) прочностные характеристики, относящиеся к большим напря-
жениям и кратковременным воздействиям;
б) прочностные характеристики, относящиеся к малым напря-
жениям и длительным периодам воздействия.
К первой категории относятся результаты типичных испытаний
на прочность — кривые напряжение — деформация при различных
температурах и различных скоростях нагружения вплоть до точки
разрушения, ударная вязкость, определенная в разных условиях,
и т. д.
Ко второй категории относятся результаты таких испытаний,
как, например, разрушение при ползучести (напряжение действует
в течение длительного периода времени) и усталостное разрушение
(разрушение при циклическом деформировании).
Третья группа характеристик позволяет получить полезную
информацию относительно предельного поведения материалов: жест-
кости (сопротивления вдавливанию), сопротивления процарапыва-
нию (поверхностной жесткости), трения и теплостойкости.
КРИВЫЕ НАПРЯЖЕНИЕ — ДЕФОРМАЦИЯ
ДЛЯ ТВЕРДЫХ ПОЛИМЕРОВ
ДЕФОРМАЦИОННЫЕ ХАРАКТЕРИСТИКИ
Прочностные свойства твердых тел наиболее просто выявляются
с помощью диаграммы напряжение — деформация; такой диаграм-
мой описывают поведение однородного образца с постоянным
поперечным сечением в условиях одноосного растяжения (рис. Х.1).
175
В зоне упругости данного материала деформация пропорцио-
нальна напряжению и удлинение образца остается обратимым.
Рис. Х.1. Обобщенная зависимость
напряжения от деформации для по-
лимерных материалов:
1 — удлинение до достижения предела те-
кучести; II — разрывное удлинение; III—
предел текучести; IV — разрушающее на-
пряжение (предел прочности).
Если какой-то материал выходит из строя и разрушается при
определенных значениях нагрузки и удлинения, то его называют
хрупким. Если же после упругой деформации при некоторой крити-
ческой величине напряжения развивается постоянная деформация
(пластическая деформация), то такой материал называют пластичным.
Рис. Х.2. Зависимости напряжение — деформация для полимерных материалов
различных типов:
1 — мягкий; 2 — мягкий, пластичный; 3 — жесткий, прочный; 4 — жесткий, пластичный;
S—-жесткий, хрупкий;-
Описание полимера Характеристика кривой напряжение—деформация
модуль упругости предел текучести разрушающее напряжение относитель- ное удлинение
Мягкий Низкий . Низкий Низкий ' Среднее
Мягкий пластичный Низкий Низкий (Предел текучести) Высокое
Жесткий прочный Высокий Высокий Высокий Среднее
Жесткий пластичный Высокий Высокий Высокий Высокое
Жесткий хрупкий Высокий Нулевой Средний Низкое
176
b процессе пластической деформации в общем случае напряжение
возрастает с увеличением деформации; это явление известно как меха-
ническое упрочнение. Если напряжение снять в какой-то точке,
то такой материал упруго восстанавливается вдоль пути, практи-
чески параллельного линии упругих деформаций. После этого в об-
разце развивается остаточное пластическое удлинение.
Пластическое течение всегда связано с критическим сдвиговым
напряжением. Это обстоятельство подтверждается данными микро-
скопических исследований, которые показывают, что пластическая
деформация обычно сопровождается скольжением по кристаллогра-
фическим плоскостям за счет дислокаций. Продолжительная дефор-
мация вызывает «зацепления» дислокаций и все более затрудняет
их перемещения. Это приводит к механическому упрочнению в ре-
зультате деформации.
Существует принципиальное различие между разрушением об-
разца выше температуры перехода в стеклообразное состояние
(здесь цепи главных валентностей полимера получают возможность
изменять свои конформации до разрушения материала) и разруше-
нием при температуре существенно ниже Т& (здесь конформации
цепей главных валентностей практически «заморожены» на протя-
жении периода наблюдения и, следовательно, материалы разру-
шаются хрупко).
На рис. Х.2 представлены зависимости напряжение — деформа-
ция для различных типов полимерных материалов.
ПРЕДЕЛ ПРОЧНОСТИ ХРУПКИХ
МАТЕРИАЛОВ
Теоретическое значение предела прочности хрупкого материала
имеет порядок от 0,12?, где" Е — модуль Юнга.
Экспериментально наблюдаемая прочность обычно изменяется
в широких пределах, но всегда оказывается в 10—100 раз меньдпе
теоретического предела. И лишь для очень тонких волокон — «усов»,
полученных из некоторых материалов (например, двуокиси крем-
ния), предел прочности при растяжении достигает теоретического
значения. Причина подобного ослабления твердого тела заключается'
в присутствии в нем дефектов или трещин, особенно на поверхности
тела. Такие трещины действуют как концентраторы механического
напряжения.
Если в образце имеется трещина длиной L и радиусом при вер-
шине г, то величина растягивающего напряжения у края трещины
увеличивается в Ыг раз. Значение этого множителя может дости-
гать 10—100, но даже если радиус вершины трещины стремится
к нулю, материал все еще обладает определенной прочностью. Сле-
довательно, для объяснения таких фактов нужно привлекать допол-
нительные предположения. Гриффит [11] показал, что для роста
трещины недостаточно, чтобы напряжения в вершине трещины превы-
шали теоретическую прочность. Вместе с этим должно высвободиться
12 Заказ 699 177
достаточное количество упругой энергии, которое обеспечит образо-
вание избыточной поверхностной энергии и тем самым обусловит
развитие трещины.
Наименьшее напряжение Стгр, способное вызвать распростране-
ние трещины, имеет порядок:
оГр«^<тт V^a/L (ХМ)
где а — линейный размер в додели Гриффита; L — длина трещины. •
Поскольку поверхностная энергия должна равняться затрачен-
ной работе (затраченная работа ^Еа—у), то окончательный резуль-
тат имеет вид:
/ vE Х1/^
<^макс= jr J (Х.2)
где С — постоянная.
Величина у пропорциональна Ег/з, поэтому окончательный
результат записывается в форме:
Омаке ~£'e’8 (Х.З)
ПРЕДЕЛ ПРОЧНОСТИ ПЛАСТИЧНЫХ МАТЕРИАЛОВ
Если материал не подвергался заметному механическому упрочне-
нию после развития деформации в условиях нагружения, то его
предел текучести очень близок к максимальному напряжению,
выдерживаемому данным материалом перед разрушением, т. е.
его пределу прочности при растяжении, или разрушающему напря-
жению при растяжении.
Тейбор [14] показал, что предел текучести материала пропор-
ционален твердости, определенной по вдавливанию (см. ниже).
Последняя величина может быть представлена как степенная функ-
ция модуля упругости, поэтому предел текучести
°У — амажс ~ Еп
где п близко 0,75.
Если материал подвергается механическому упрочнению, что
характерно для многих кристаллических полимеров вследствие
ориентации в процессе пластической деформации, то предел проч-
ности этого материала, рассчитанный по исходному поперечному
сечению образца, конечно, оказывается выше предела текучести.
ПЕРЕХОД ОТ ПЛАСТИЧЕСКОГО РАЗРУШЕНИЯ
К ХРУПКОМУ
Дислокации оказываются одной из причин возникновения пластич-
ности. Однако если свободное перемещение дислокаций невозможно
из-за какого-либо барьера (за счет этого несколько дислокаций
могут накопиться в одном месте), то возможно формирование внутрен-
ней трещины и инициирование распространения трещины по мате-
178
риалу. Этим путем пластичное твердое тело может превратиться
в хрупкое.
Но и хрупкое твердое тело может стать пластичным, если к нему
прикладывается гидростатическое давление. Рассмотрим хрупкое
твердое тело, которое разрушается при растягивающем напряже-
нии о. Если приложено гидростатическое давление р, то необходи-
мое для разрушения растягивающее напряжение равно р -|- а.
Такое растягивающее напряжение создает напряжение сдвига,
равное х/2 (р + о). Если величина критического напряжения сдвига
меньше этого значения, то тело деформируется пластично, прежде
чем растягивающее напряжение станет достаточным для возникно-
вения хрупкого разрушения.
Явления, подобные описанному, хорошо известны на практике.
На больших глубинах залегания горные породы могут течь, хотя
в обычных условиях эти породы хрупкие. Даже в кварце может
наблюдаться пластическое течение при достаточно большом гидро-
статическом давлении.
В опытах по определению твердости процесс вдавливания зача-
стую может осуществляться путем развития пластических дефор-
маций даже в относительно хрупких материалах. Это обусловлено
торможением процесса хрупкого разрушения из-за большого зна-
чения гидростатической составляющей напряжений в таких опытах.
И значения твердости, полученные этим способом, представляют
собой меру именно пластических характеристик данного хрупкого
твердого тела.
Другим параметром, который резко влияет на характер разру-
шения, является ориентация. Если полимерный материал нагре-
вается выше температуры перехода в стеклообразное состояние,
растягивается в одном направлении на несколько сотен процентов
и, наконец, охлаждается до комнатной температуры, оставаясь под
действием напряжения, то Полимерные цепи сохраняют приданное
им не случайное распределение конформаций: более высокая сте-
пень ориентации наблюдается вдоль направления вытяжки. Такой
материал становится существенно анизотропным и оказывается
гораздо более прочным в направлении ориентации (соответственно
слабее в поперечном направлении). Этот эффект может выражаться
весьма резко. Например, если неориентированный стеклообразный
полимер должен был разрушаться хрупко при амакс = 0,003, то
после ориентации пластическое течение с разрушением происходит
только при омакс = 0,50. Площадь под кривой напряжение — дефор-
мация, являющаяся приближенной мерой прочности тела, может
оказаться почти в 20 раз больше для соответствующим образом
ориентированного образца по сравнению с неориентированным мате-
риалом (см. гл. XX).
Еще больше можно увеличить прочность, если кристаллизация
осуществляется одновременно с ориентацией. Наиболее прочными
полимерными материалами являются высокоориентированные кри-
сталлические полимеры. ,
179
ЧИСЛОВЫЕ ЗНАЧЕНИЯ
В табл. Х.1 приведены числовые значения прочностных характе-
ристик ряда полимеров.
На рис. Х.З в графическом виде представлены данные о проч-
ности при растяжении; здесь построена зависимость омакс (проч-
ность при растяжении в точке хрупкости для хрупких линейных
полимеров и предела текучести при растяжении для пластичных
линейных полимеров) от модуля упругости при растяжении Е. В ка-
честве приближенного соотношения можно воспользоваться следу-
ющим эмпирическим уравнением (описывает сплошную линию
на рис. Х.З):
омакс = 9.0 -еЧ* (Х.4)
где Омаке и Е выражены в дин/см2.
Таблица X.I. Механические характеристики немодифицированных полимеров
Полимер А, ДИн/см’.- 10-" Б, % /гми •’/-‘Г В, Дин/см» • 10-® г, % Д,дин/см’• 10-’’ —. —- Е, ДИн/СМ» • 10-’ 7 «а 2 Q Л К н 3, дин/см» • 10"’ н
i Полиэтилен НП 0,85 20 1,3 600 0,20 0,45
l Полиэтилен ВД 3,0 9 2,7 450 1,20 1,8 0,80 — 0,40
1 Полипропилен 3,2 12 3,3 650 1,25 4,9 1,35 6,2 —
“1 Полистирол — — 5,0 3 3,25 9,5 3,10 9,5 0,33
$ Поливинилхлорид 5,0 3 5,5 30 2,50 9,8 3,50 7,1 0,35
ь Полихлортрифторэтилен 3,0 10 3,4 185 0,95 7 0,88 6,1 —
V Политетрафторэтилен 1,3 62,5 2,3 400 0,40 — 0,35 0,6 —
Полиоксиметилен — — 6,8 27,5 3,50 10 2,90 12,2 —
9 Полиметилметакрилат — — 7,0 3 4,00 14 3,10 10,5 0,33
Поли-бис-фенилкарбонат 6,5 30 5,3 >80 2,35 9,5 2,40 8,5 0,33
Найлон 6,6 5,7 25 8,0 170 1,50 10 1,80 6,1 0,38
it Найлон 6 4,5 30 6,1 250 1,25 9 1,40 —— —
Г*> Полиэтилентерефталат 7,0 6 5,4 275 3,00 11,8 2,90 — —
И Ацетат целлюлозы 4,2 6 3,2 35 2,20 4,8 1,25 4,4 —
1 f Фенолоформальдегидный — — 5,5 1 7,0 8,2 7,0 — 0,3
олигомер Меламиноформальдегидный — — 5,0 1 9,0 7,0 9,0 — —
олигомер Ненасыщенный полиэфир — — 4,5 3 3 8 4,0 — —
Коэффициент перевода СИ: 1 дин/см* = 0,1 Н/м».
Примечания.
1. А—предел текучести при растяжении; Б — относительное удлинение, отвечающее пре
растяжении); Г—разрывное удлинение; Д—модуль упругости при растяжении; Е—
гости при изгибе; 3—предел прочности при сжатии (разрушающее напряжение при
2, Приведенные данные взяты из (Technical data on plastics», Manutact. Chem. Assoc.,
torst, 1963. «Plastics materials guide 1970—1971»; Brits. Plast., Jan. 1970.
180
Приведенное соотношение не выполняется для полимеров с по-
перечными связями.
С помощью табл. Х.1 можно сравнить между собой значения
прочности при растяжении, изгибе и сжатии. В табл. Х.2 сопоста-
вляются значения коэффициентов прочности полимеров и других
материалов.
Совершенно очевидно, что коэффициент Пуассона сильно влияет
на значения коэффициентов прочности аналогично тому (но в го-
раздо большей степени), как это имеет место для отношения модулей
упругости (см. гл. IX). Коэффициент прочности можно довольна
точно оценить с помощью следующего эмпирического соотношения
(рис. Х.4):
1 °МакС <Сжатие) ' Омаке (изгиб) /сети /у 51
Омаке (растяжение) ~ 8 Омаке (растяжение) ' ’ J ( • >
• где v — коэффициент Пуассона.
Ударная вяз- кость по Изоду (ASTM D 256), КГС- См/СМ Твердость по Роквеллу Твердость по вдавливанию, дин/см3 -10-® Твердость по Бирбауму Коэффициент трения Сопр отивление истиранию (ASTM D 1044), мг/lflOO см’ Коэффициент потерь на исти- рание (DIN 53516), мг
« л 5 X а . а «3 я к В
Выше 50 10 6 1,7 3,2 16 16 10 2' 60 10 15 3,5 9 1,5 1,5 1,5 10 40 100 130 115 110 85 120 125 118 114 85 100 125 130 125 75 60 95 78 70 106 25 75- 1,35 5,35 7,25 И 11,5 7,0 3,1 14,0. 17,2 9,75 7,25 6,25 12,0 4,3 19 Выше 17 17 10,3 10,1 17,2 11,1 19,8 21,2 32,4 13,5 0,5 1 0,23 г. — J 0,38 v 0,50 г 0,56 £ 0,10 > — g 0,4 — 'i> 0,36 ‘f 0,39 — Z3 — /У 0,61 20 10 8 3 2 640 160. 470 25 15 60
Делу текучести; В—предел прочности при растяжении (разрушающее напряжение при
предел прочности при изгибе (разрушающее напряжение при изгибе); Ж—модуль упру-
сжатии); И—коэффициент Пуассона.
Washington, 1957; Carlowitz В., «Kunststoff Tabellen»; Schiffmann, Bensberg—Franken-
181
ЧИСЛОВЫЕ ЗНАЧЕНИЯ
В табл. Х.1 приведены числовые значения прочностных характе-
ристик ряда полимеров.
На рис. Х.З в графическом виде представлены данные о проч-
ности при растяжении; здесь построена зависимость омакс (проч-
ность при растяжении в точке хрупкости для хрупких линейных
полимеров и предела текучести при растяжении для пластичных
линейных полимеров) от модуля упругости при растяжении Е. В ка-
честве приближенного соотношения можно воспользоваться следу-
ющим эмпирическим уравнением (описывает сплошную линию
на рис. Х.З):
Омакс — 9.0 ’Е
(Х.4)
где Смаке и Е выражены в дин/сма.
Таблица X.I. Механические характеристики ^модифицированных полимеров
Полимер А, ДИН/См'(. 10-' Б, % fc? г, % д,дин/см».10->3 7 о Я о д' я и н ж, дин/см» 10-1“ 7 о ^"Ч « Й о В rt п к
i Полиэтилен НП 0,85 20 1,3 600 0,20 0,45
l Полиэтилен ВД 3,0 9 2,7 450 1,20 1,8 0,80 — 0,40
s Полипропилен 3,2 12 3,3 650 1,25 4,9 1,35 6,2 —
Полистирол — — 5,0 3 3,25 9,5 3,10 9,5 0,33
$ Поливинилхлорид 5,0 3 5,5 30 2,50 9,8 3,50 7,1 0,35
Ь Полихлортрифторэтилен 3,0 10 3,4 185 0,95 7 0,88 6,1 —
у Политетрафторэтилен 1,3 62,5 2,3 400 0,40 — 0,35 0,6 —
1 Полиоксиметилен — 6,8 27,5 3,50 10 2,90 12,2 —
9 Полиметилметакрилат — — 7,0 3 4,00 14 3,10 10,5 0,33
Поли-бис-фенилкарбонат 6,5 30 5,3 >80 235 93 2,40 8Д ОДЗ
< Найлон 6,6 5,7 25 8,0 170 1,50 10 1,80 6,1 0,38
it Найлон 6 4,5 30 6,1 250 1,25 9 1,40 — —
t Ъ Полиэтилентерефталат 7,0 6 5,4 275 3,00 11,8 2,90 — —
i>i Ацетат целлюлозы 4,2 6 3,2 35 2,20 4,8 1,25 4,4 —
i ? фенолоформальдегидный — — 5,5 1 7,0 8,2 7,0 — 0,3
олигомер Меламиноформальдегидный — — 5,0 1 9,0 7,0 9,0 — —
олигомер Ненасыщенный полиэфир — — 4,5 3 3 8 4,0 —
Коэффициент перевода СИ: 1 дин/см* = 0,1 Н/м“.
Примечания.
1.А—предел текучести при растяжении; Б — относительное удлинение, отвечающее пре
растяжении); Г—разрывное удлинение; Д—модуль упругости при растяжении; Е—
гости при изгибе; 3—предел прочности при сжатии (разрушающее напряжение при
2, Приведенные данные взяты из «(Technical data он plastics», Manufact. Chem. Assoc.>
torst, 1963. «Plastics materials guide 1970—1971»; Brits. Plast., Jan. 1970.
180
Приведенное соотношение не выполняется для полимеров с по-
перечными связями.
С помощью табл. X 4 можно сравнить между собой значения
прочности при растяжении, изгибе и сжатии. В табл. Х.2 сопоста-
вляются значения коэффициентов прочности полимеров и других
материалов.
Совершенно очевидно, что коэффициент Пуассона сильно влияет
на значения коэффициентов прочности аналогично тому (но в го-
раздо большей степени), как это имеет место для отношения модулей
упругости (см. гл. IX). Коэффициент прочности можно довольна
точно оценить с помощью следующего эмпирического соотношения
(рис. Х.4):
1 Смаке (сжатие) ' Смаке (изгиб) _/2П--7И (Y 51
Омаке (растяжений 8 Омаке (растяжений ' > ' ' V ( • >
где v — коэффициент Пуассона.
Твердость й *
J, OcP 2 - по Роквеллу Mi О X И <и и в . 1 Я И S со
1рная j ГЬ ПО ] TM D • CM/CK И a ь « • Л 6- S ° В X Ef §|q о и« gn S
rt § «3 я g is 8 «во а® е § lisa eg»" eg gz
S о сд о Л X ' x X 3 X 3 ® я И "rtB Ь йй ® & я Н ьи sg. И ь О О t* и ° о Sq Meat
Выше 50 10 1,35 0,5 1
10 40 — 5,35 - 0,23 г. . 2
6 100 — 7,25 — — J —.
1,7 130 75 11 10,3 0,38 * 640
3,2 115 60 11,5 10,1 0,50 г -—
16 110 7,0 0,56 £ 160.
16 85 3,1 0,10 з- 470
10 120 14,0. — i 20 —
2- 125 95 17,2 17,2 0,4 — —
60 118 78 9,75 — — /V 10
10 114 70 7,25 11,1 0,36 ‘Г — 25
15 85 6,25 0,39 8 15
3,5 106 12,0 - — Z3 3
9 100 25 4,3 19,8 — /у —
125 — 19 21,2 0,61 ,г — 60
1,5 130 — Выше 17 32,4 — — —
1,5 125 75- 17 13,5 — — —
Делу текучести; В—предел прочности при растяжении (разрушающее напряжение при
предел прочности при изгибе (разрушающее напряжение при изгибе); Ж—модуль упру-
сжатии); И—коэффициент Пуассона.
Washington, 1957; Carlowitz В., «Kunststott Tabellen»; Schiffmann, Bensberg—Franken-
14
Таблица Х.2. Коэффициенты прочности различных материалов
Материал Коэффициент прочности (опыт) Коэффициент Пуассона
Сжатие /растяжение изгиб/растяжение
Плавленый кварц 30 0,14
Силикатное стекло 12,5 — 0,225
Фенолоформальдегидный 2,5 — 0,3
олигомер
Полистирол 1,9 1,9 0,33
Полиметилметакрилат 1,5 2,0 0,33
Поливинилхлорид 1,9 1,9 0,35
Полиэтилен НД — 0,6 0,40
Рис. Х.З. Соотношение между преде-
лом прочности при растяжении и
модулем упругости полимеров:
X — предел текучести (для пластичных
полимеров); О — предел прочности (для
хрупких полимеров).
Рис. Х.4. Коэффициенты прочности по-
лимеров:
1 — плавленый £кварц; 2 — стекло; з — фе-
нолоформальдегидная смола; 4 — полистирол;
5 — поливинилхлорид; в — полиметилмет-
акрилат; 7 — полиэтилен высокой плотности.
ЗАВИСИМОСТЬ ПРЕДЕЛА ПРОЧНОСТИ
(РАЗРУШАЮЩЕГО НАПРЯЖЕНИЯ)
ОТ СКОРОСТИ ДЕФОРМАЦИИ
При увеличении скорости удлинения возрастают прочность при
растяжении и модуль упругости, а разрывное удлинение в общем
случае уменьшается (исключая резины).
Обычно увеличение скорости испытания приводит к такому же
эффекту, как снижение температуры. Для каучуков с низкой плот-
ностью поперечных связей этот эффект может даже описываться
с помощью принципа температурно-временной эквивалентности (см.
ниже).
182
ПРЕДЕЛЬНЫЕ ХАРАКТЕРИСТИКИ ТИПА
НАПРЯЖЕНИЕ — ДЕФОРМАЦИЯ ДЛЯ АМОРФНЫХ
ЭЛАСТОМЕРОВ
Предельное механическое поведение эластомеров с низкой плот-
ностью поперечных связей оказывается достаточно простым. Смит
[13] показал, что предельные механические характеристики этого
класса полимерных материалов подчиняются принципу температур-
но-временной эквивалентности точно так же, как и адеструктивные
напряжения, возникающие вследствие вязкоупругости среды.
Рис. Х.5. Обобщенные предельные характеристики эластомеров [13]. Логариф-
мическая зависимость разрывного напряжения (а • 273/Т) от приведенной вели-
чины времени до разрушения («разр/яу) для вулканизованного витона В; стандарт-
ная температура составляет 313 К (40 °C):
Обозначения Температура, А X □ О А О • е
С 180 140 100 70 40 25 10 —5 —20
lg ау — -5,13 —4,40 3,29 2,02 0 1,53 3,70 7,00 12,70
Можно совместить зависимости напряжения, отнесенного к абсо-
лютной температуре, от логарифма времени до разрушения при
различных температурах путем введения зависящего от темпера-
туры фактора сдвига ат. Применение того же фактора сдвига поз-
воляет совместить зависимости коэффициента растяжения при раз-
рушении Ар от логарифма времени до разрушения при различных
температурах. Прямым следствием этого является то, что все значе-
ния предела прочности (разрушающего напряжения) при растяже-
нии, отнесенные к абсолютной температуре, в зависимости от
разрывного удлинения описываются одной обобщенной огибающей
разрушения независимо от температуры проведения каждого кон-
кретного испытания. На рис. Х.5 — Х.7 представлено поведении
эластомера витон В.
Если растяжение сопровождается кристаллизацией, то описан-
ный выше принцип простой температурно-временной эквивалент-
ности нарушается.
183
Тис. Х.6. Обобщенные предельные характеристики эластомеров [13]. Логариф-
мическая зависимость разрывного удлинения (АПр — 1) от приведенной величины
времени до разрушения (1р/ат) для вулканизованного витона В; стандартная
температура составляет 313 К. (40 °C):
Обозначения А X □ Q А О Я • в
Температура °C, 180 140 100 70 40 25 10 —5 —20
«1 аТ —5,13 —4,40 3,29 2,02 0 1,53 3,70 7,00 12, 70
Рис. Х.7. Обобщенные предельные характеристики эластомеров [13]. Общий
характер разрушения вулканизованного витона В: '
Обозначения А X □ ф А .0 3 • © Я
Температура, ®С <80 140 100 70 40 25 10 —5 —20
УДАРНАЯ ВЯЗКОСТЬ (МЕХАНИЧЕСКОЕ
ПОВЕДЕНИЕ ПРИ МАЛЫХ ДЛИТЕЛЬНОСТЯХ
ВОЗДЕЙСТВИЯ)
Ударная вязкость характеризует сопротивление разрушению в--
условиях высокой скорости воздействия. Этот показатель играет
весьма важную в практическом отношении роль, но в то же
время его чрезвычайно трудно определить с научной точки
зрения.
В большинстве случаев при ударных испытаниях измеряют энер-
гию, необходимую для разрушения стандартного образца в опреде—
ленных заданных условиях.
К наиболее распространенным
типам опытов относятся испы-
тание по Изоду (прибор маят-
никового типа, образец с над-
резом, удар наносится по не-
закрепленному концу образца),
испытание по Шарпи (прибор
маятникового типа, образец
опирается концами, удар нано-
сится по его средней части),
испытание падающим грузом
(стандартный шар падает на об-
разец с известной высоты) и вы-
сокоскоростное растяжение, да-
ющее кривую напряжение — де-
формация.
Ударная вязкость зависит
от температуры: вблизи области
стеклования ударная вязкость
—* Е, дин/см2
Рис. Х.8. Соотношение между ударной!
вязкостью и модулем Юнга:
1 — политетрафторэтилен; 2 — поликарбонат.
резко увеличивается с повышением
температуры. Вторичные переходы играют существенную роль;
полимер, у которого явно выражен низкотемпературный вторичный
переход в стеклообразном состоянии, почти всегда гораздо прочнее
полимера, не имеющего такого перехода.
Кристаллические полимеры обладают высокой ударной вяз-
костью, если их температура перехода в стеклообразное состояние-
лежит намного ниже температуры проведения испытания. При повы-
шении степени кристалличности и особенно с ростом размеров сферо-
литов ударная вязкость уменьшается.
Ударная вязкость термореактивных полимеров незначительно-
изменяется в зависимости от температуры в весьма широком темпе-
ратурном интервале.
В табл. Х.1 приведены значения ударной вязкости по Изоду
при комнатной температуре для ряда распространенных полимеров t
На рис. Х.8 представлено соотношение между ударной вязкостью
по Изоду и модулем упругости при комнатной температуре. Эти
результаты могут использоваться для получения приближенных
оценок.
185
Для некоторых полимеров наблюдается большой разброс вели-
чин ударной вязкости. Поликарбонат, например, очень хорошо
противостоит ударным нагрузкам, а политетрафторэтилен обладает
меньшей ударной вязкостью по сравнению с ожидаемой, по-
скольку образцы из этого материала получают методом спе-
кания.
Рис. Х.9. Соотношение между ударной вязкостью и удли-
нением, отвечающим достижению предела текучести.
Ударная вязкость коррелирует также с относительным уд-
линением, отвечающим пределу текучести, что видно из
рис. Х.9.
МЕХАНИЧЕСКИЕ ХАРАКТЕРИСТИКИ
ПРИ ДЛИТЕЛЬНЫХ ВОЗДЕЙСТВИЯХ
РАЗРУШЕНИЕ ПРИ ПОЛЗУЧЕСТИ
Испытания на ползучесть обычно проводят при малых нагрузках,
так что образец не разрушается. Если же величина нагрузки при-
ближается к пределу прочности, то разрушение произойдет спустя
некоторое время после приложения нагрузки. Теоретические и экс-
186
периментальные данные позволили получить * следующее соотно-
шение [9]:
In ip = Л 4- Е(Х.6>
где ip — время, которое материал выдерживает до разрушения при приложении
постоянного напряжения а; Е — энергия активации процесса разрушения»
А и В — постоянные.
Приведенная формула показывает, что приложение напряжения
снижает величину энергии активации от Е до Е — Вс.
[★ Большой объем данных по долговечности различных полимеров был полу- -
чей С. Н. Журковым и его школой. Обзор и обсуждение физического смысла
этих результатов см. в работах: Регель В. Р., Слуцкер А. И., Т ом а -
шевский Э. Е., УФН, 1972, т. 106, вып. 2, с. 193—228; Р е г е л ь В. Е.,
С л у ц к е р А. И,, Томашевский Э. Е. Кинетическая теория прочности
твердых тел, М., «Наука», 1974. 560 с.
Ниже приводятся только некоторые конкретные значения параметров,
входящих в уравнение для долговечности т, которое записывается в форме:
Для многих исследованных полимеров то принимает универсальное значение:
lgxo= 13 ±1, что связано с физическим смыслом то как среднего периода
тепловых колебаний атомов в конденсированных телах. Величина U« имеет
смысл энергии активации процесса разрушения (разрыва) связей в отсутствие
внешних напряжений. Она близка к энергии активации процесса термодеструк-
ции, поскольку элементарный акт разрушения твердых тел, согласно термофлук-
туационной теории прочности, связан с разрывом химических связей. Значения
U« определяются природой полимера и составляют для поликапроамида 45,
триацетата целлюлозы 49, полиэтилена 25, нитрата целлюлозы 38, полистирола
33, полиметилметакрилата 31 ккал/моль.
Величина у в формуле для долговечности отражает эффективность работы
внешних сил при разрушении тела, поскольку роль напряжения сводится к пони-
жению активационного барьера разрыва связей. Величина у зависит от физиче-
ской структуры материала, поскольку она определяет неоднозначность распре-
деления напряжений в теле. В зависимости от предыстории образца (его термо-
обработки, степени ориентации и т. п.) и условий его нагружения (в активной
среде, при облучении и т. д.) структурночувствительный коэффициент у изме-
няется в несколько раз. Например, для поликапроамида наблюдались изменения
у от 0,43 до 1,78 ккал мм2/моль • кг. Поэтому заранее трудно указать ожидаемые
значения у и дать количественные оценки долговечности того или иного изделия
в реальных условиях эксплуатации.
Таким образом, существующие в настоящее время представления о долго-
вечности полимерных материалов носят в основном качественный характер,
правильно указывая общие закономерности проявлений долговременной проч-
ности в зависимости от химической природы материала и условий нагружения,
но результаты соответствующих вычислений с инженерной точки зрения должны
рассматриваться как грубые прикидки ожидаемых значений т.*] — Прим. ред.
СОПРОТИВЛЕНИЕ УСТАЛОСТНОМУ РАЗРУШЕНИЮ
Разрушение образца и изменение его механических свойств в резуль-
тате повторяющегося приложения напряжения или деформиро-
вания известно как усталость материала. При этом долговечность
* С. Н. Журков предложил термофлуктуационную концепцию разрушения
твердых материалов и на ее основании получил и теоретически обосновал фор-
мулу (Х.6). См. ссылки й дополнении к этому разделу. — Прим. ред.
187
определяется как количество циклов деформирования, которое обра-
зец выдерживает до разрушения.
Для испытаний на сопротивление усталости получили распро-
странение различные экспериментальные схемы (изгибающаяся бал-
ка, вращающаяся балка, циклическое изменение напряжения или
деформаций с постоянной амплитудой, постоянная скорость увели-
чения амплитуды напряжения или деформаций и т. д.). Получаемые
при этом результаты представляют в виде зависимости числа циклов
до разрушения от величины прилагаемой нагрузки. Предельное
напряжение, ниже которого материал «никогда» не выйдет из строя,
называется пределом усталости, или пределом выносливости. Для
весьма многих полимеров этот предел выносливости составляет
приблизительно одну треть от предела прочности при растяжении
в условиях статической нагрузки. Следовательно, практически
важно рассчитывать конструкции таким образом, чтобы при вибра-
циях максимальные напряжения в этих конструкциях были ниже
лредела выносливости, а не ниже предела прочности при растяжении
в условиях статической нагрузки.
Для испытаний на усталость также существует определенный
тип температурно-временной эквивалентности: долговечность поли-
мера в общем случае уменьшается с повышением температуры.
Температурную зависимость усталостной долговечности £уст можно
выражать соотношением
В
1g 1уст = -АJ, (Х.7)
При определении долговечности важен эффект механического
демпфирования. Сильное демпфирование вблизи температуры пере-
хода может привести к усталостному разрушению из-за резкого
подъема температуры. С другой стороны, демпфирование может
оказаться благоприятным фактором, поскольку в отсутствие демпфи-
рования резонансные колебания часто приводят к разрушению.
(★ Тепловыделения при деформировании, обусловленные гистерезисными
потерями, могут привести к неконтролируемому повышению температуры, сни-
жению несущей способности материала и вследствие этого к разрушению изде-
лия. Это явление было подробно изучено С. Б. Ратнером, который показал, что
для каждого полимера существует критическая величина разогрева А Тк, прак-
тически не зависящая от внешних параметров (нагрузки, частоты, условий тепло-
отдачи), но зависящая от внутренних свойств материала (модуля упругости и угла
механических потерь) и режима нагружения. Верхняя предельно допустимая
температура экстраполяции 7’э> обеспечивающая безопасные условия работы
изделия, должна определяться из условия Тэ << Тр — Д Тк, где Тр — темпера-
тура размягчения (теплостойкость) полимера, по достижении которой наблю-
дается резкое падение модуля упругости и потеря деформационной стабильности
изделия. Проблема теплостойкости кратко рассмотрена в последнем разделе
настоящей главы. Ниже приводятся значения ДТкв "С (по С. Б. Ратнеру)
для ряда полимеров
Капролон
Капрон
Полиамид-68
(температура испытаний 20 °C):
при постоянной при постоянной при постоянной
амплитуде работе нагруже- амплитуде
напряжения ния за цикл деф ормации
15 17 19
15 16 18
11 13 15
188
39
28
Полипропилен 28 35
Фторопласт-3 26 32
Фторопластам 18 23
Характерные значения А Тк для некоторых других полимеров, определенные
при заданной амплитуде напряжения сто, таковы:
а0. кгс/см* со, цикл/мин Начальная температура, °C ДТК.
Полиформальдегид 425 2700 18 39
250 2700 62 36
Полиэтилен НД 90 2700 20 22
фторопласт-4 90 2700 22 26
100 2700 22 26
П олиметилметакри- 130 2700 70 и 75 20
лат
Разрушение полимерных материалов вследствие недопустимого повышения
температуры из-за гистерезисного разогрева по своей природе аналогично явле-
ниям теплового взрыва в экзотермической системе и теплового пробоя диэлек-
триков. — Прим. ред.
ДРУГИЕ ХАРАКТЕРИСТИКИ, СВЯЗАННЫЕ
С МЕХАНИЧЕСКИМ РАЗРУШЕНИЕМ
ТВЕРДОСТЬ
При испытании материалов довольно часто определяют твердость
по вдавливанию. В таких опытах в образец под действием приложен-
ной нагрузки вдавливается наконечник индентора той или иной гео-
метрической формы. При испытаниях по Бринеллю наконечник
представляет собой шарик из твердой стали, при испытаниях по
Виккерсу — алмазную пирамидку.
Механизм процесса вдавливания подробно исследовал Тэйбор
[14]. В процессе вдавливания шарика в поверхность твердого мате-
риала возникают упругие деформации. При повышении нагрузки
напряжения довольно скоро начинают превышать предел упругости,
и возникает пластическое течение. При дальнейшем повышении
нагрузки материал, находящийся непосредственно у вдавливаемого
наконечника, становится полностью пластичным. После снятия
нагрузки деформации в определенной степени восстанавливаются.
По теории почти две трети среднего давления в области вблизи
индентора составляет гидростатическая компонента, которая прак-
тически не принимает участия в создании пластического течения.
Таким образом, в приближенном виде давление ру, соответству-
ющее началу пластического течения, равно:
ру= W/A «=< Зоу (Х.8)
где W — приложенная нагрузка; А — площадь поверхности вдавливания; ау —
предел текучести при одноосном напряженном состоянии.
Таким способом по данным измерения твердости материала можно
определить предел текучести.
189
Впервые теоретические основы испытаний на твердость по Бри-
неллю разработал Герц [12], позднее Тимошенко [6] пересмотрел
теорию. Расчеты обоих авторов основаны на методах теории упру-
гости.
Исходя из концепций пластической деформации Тэйбора и анали-
зируя процесс упругого восстановления после снятия нагрузки,
Байер, Майер и Петерсон [8] получили следующую формулу:
W
(Х.9>
где W — приложенная нагрузка; Е — модуль Юнга; d1 — глубина вдавливания;
d± — d2 — упругое восстановление после деформации; D — диаметр наконеч-
ника индентора (шарик).
Рис. Х.10. Соотношение между твер-
достью (по вдавливанию шарика) и
модулем упругости.
наконечника индентора и
Измеряя исходную глубину
вдавливания и расстояние упру-
гого восстановления, можно опре-
делить модуль упругости Е.
Твердость находят следующим
образом:
W
Поскольку Dd — А (площадь по-
верхности вдавливания) и свя-
зана с модулем Юнга (о^ — Еп),
то, комбинируя уравнения (Х.8)
и (Х.10), можно записать следую-
щее соотношение (для стандартного
нагрузки):
стандартной
Hv~En (Х.11)
В табл. Х.1 приведены значения твердости для ряда исследо-
ванных полимеров. Данные рис. Х.10 показывают, что соотношение
(Х.11) приблизительно выполняется. Эмпирическое выражение
имеет вид
Яр^1,7£3/4 (Х.12)
здесь Яр и £ выражены в дин/см2.
УСТОЙЧИВОСТЬ К ПРОЦАРАПЫВАНИЮ
Хорошо известная шкала твердости основана на способности одного
материала процарапывать любой материал меньшей твердости.
Однако такое представление о твердости вовсе необязательно соот-
ветствует данным испытаний на твердость, проводимых по другому
методу.
190
Устойчивость к процарапыванию твердых полимеров связана
с истиранием. В общем случае для твердых полимеров устойчивость
к процарапыванию изменяется параллельно модулю упругости. Для
резины характерна высокая устойчивость к процарапыванию, кото-
рая обусловлена двумя факторами: легкостью деформирования
и совершенной упругостью.
В табл. Х.1 приведены величины устойчивости к процарапыва-
нию для ряда наиболее распространенных полимеров.
ТРЕНИЕ
Коэффициент трения р, определяется как отношение тангенциальной
силы F к нормальной нагрузке W в условиях, когда одна поверх-
ность материала перемещается по отношению к другой:
Р=4г (Х.13)
Коэффициент трения никоим образом не является постоянной
величиной, поскольку он зависит от нагрузки, площади контакта
трущихся поверхностей, структуры поверхности, скорости сколь-
жения, температуры и, конечно, от наличия и качества смазочных
материалов.
Из характеристических свойств полимеров наиболее важную
роль играют молекулярная адгезия, температура размягчения и
относительная твердость двух трущихся материалов. Трение ока-
зывается сильным, когда молекулярная адгезия высока, поэтому
трение между поверхностями одного и того же материала обычно
больше, чем трение разнородных материалов.
В соответствии с данными работ Боудена и Тэйбора [2J трение
оказывается суммарным эффектом, обусловленным действием раз-
личных факторов: внутренним трением, связанным с механическим
демпфированием; поверхностным сдвиговым трением, т. е. сдвигом
по поверхностям с разрушением их контакта; крайней формой такого
сдвига является вдавливание более твердого материала в менее
твердый.
1. Внутреннее трение. Этот тип трения играет наиболее важную
роль в периодических процессах, например при работе подшипников
качения и автомобильных шин. Механическое демпфирование и за-
держанное упругое восстановление обусловливают диссипацию энер-
гии. Следовательно, трение качения и механическое демпфирование
тесно связаны между собой. Для коэффициента трения при качении
твердого шарика по пластмассовой поверхности справедливо сле-
дующее выражение, полученное Фломом [10]:
(1 = 0,115 (ётД-ёТг) (Х.14)
W W — нагрузка, действующая на катящийся шарик радиуса г; G' — модуль
Упругости при сдвиге поверхности, по которой катится шарик; G"/G’ — коэффи-
циент диссипации, равный тангенсу угла механических потерь.
191
Выражение (Х.14) ясно показывает, что трение качения велико,
если велико отношение G"IG' = tg 6; следовательно, трение должно
быть особенно сильным в дисперсионных областях.
2. Поверхностное трение. Даже самые гладкие поверхности при
рассматривании в микроскоп оказываются шероховатыми. Контак-
тирующие поверхности соприкасаются друг с другом всего лишь
в отдельных точках. Скольжение одной поверхности по другой об-
условливает возникновение значительных сил, действующих в ме-
стах контактов. Во многих случаях трение связано с пластической
деформацией из-за того, что поверхности «привариваются» друг
к другу и сдвиг может происходить даже ниже уровня поверхности
более мягкого материала. Скольжение вызывает разрушение уме-
ренно прочных соединений трущихся поверхностей, образовав-
шихся при их контакте.
Если сдвиг оказывается основным фактором в процессе трения,
то коэффициент трения приближенно определяется выражением
т
Подвиг = ~ (Х.15)
где т — сдвиговая прочность более мягкого материала; ру— давление, отвеча-
ющее пределу текучести более мягкого материала.
Давление текучести ру связано с модулем упругости выражением
Ру~Еп (Х.16)
В случае, когда выступы более жесткого материала проникают
в более мягкий материал и проделывают в нем бороздки, наблюда-
ются сильное абразивное истирание и износ поверхности. Полимеры
представляют собой относительно мягкие материалы, поэтому вклад
втого эффекта в общую величину сопротивления трению может
оказаться весьма существенным в суммарном эффекте.
СКАЧКООБРАЗНОЕ ДВИЖЕНИЕ
Если коэффициент трения в статических условиях больше, чем в ди-
намических, то может возникнуть скачкообразное движение. В твер-
дых полимерных материалах динамический коэффициент трения
обычно ниже статического; для эластомеров справедливо обратное
соотношение. При высоких скоростях скольжения обычно довольно
трудно разделить воздействия скорости и температуры на процесс
трения.
ИСТИРАНИЕ
Истирание тесно связано с трением, особенно если существенную
роль играет составляющая силы трения, которая обусловлена «про-
никновением» одного материала в другой. И, конечно, истирание
связано также с устойчивостью к процарапыванию. Зепп [15] пока-
зал, что потери на истирание пропорциональны динамическому
192
коэффициенту трения и динамическому модулю упругости и обратно
пропорциональны прочности на растяжение.
В табл. Х.1 дана сводка коэффициентов трения для ряда поли-
мерных материалов, а также приведены данные об относительных
потерях на истирание.
ТЕПЛОСТОЙКОСТЬ
Испытание на теплостойкость аналогично испытанию на ползучесть,
за исключением того, что температура в первом случае повышается
с постоянной скоростью. При температуре размягчения или «тепло-
вого коробления» полимер начинает заметно деформироваться в уз-
ком температурном интервале.
Для аморфных полимеров температура размягчения лежит
вблизи точки перевода в стеклообразное состояние, а теплостойкость
высококристаллических полимеров близка к точке плавления.
Значение теплостойкости зависит от прилагаемого к образцу
напряжения. Чем больше прилагаемая нагрузка, тем ниже тепло-
стойкость полимера.
Исследование теплостойкости может использоваться как метод
определения «замороженных» напряжений, поскольку ориентирован-
ные полимеры дают быструю усадку при температуре выше точки
размягчения.
Усадка полимера происходит только в том случае, когда вели-
чина приложенного напряжения меньше величины «замороженного»
напряжения. Если же внешнее напряжение превышает внутреннее,
то усадки не происходит. Таким образом, исследование термостойко-
сти при различных приложенных напряжениях оказывается полез-
ным при изучении ориентированных полимерных образцов (напри-
мер, волокно) и влияния тепловой обработки на их свойства.
ЛИТЕРАТУРА
1. Andrews Е. Н., «Fracture in
Polymers», Elsevier, New York,
1968.
2. Bowden F. P. and Tabor D.,
«The Friction and Lubrication
of Solids», Clarendon Press, Ox-
ford, 1954.
3. Drucker D. C. and Gilman J. J.
(Eds.), «Fracture of Solids», In-
terscience, New York, 1963'.
4. Nielsen L. E., «Mechanical Proper-
ties-of Polymers», Reinhold, New
York, 1962.
5. Rosen В. (Ed.), «Fracture Proces-
ses in Polymeric Solids», Inter-
. science,. New York, 1964.
6. Timoshenko S., «Theory of Elasti-
city», McGraw-Hill, New York,
1934. ft
7. Winding С. C. and Hiatt G. D.,
13 Заказ 699
«Polymeric Materials», McGraw-
Hill, New York, 1961.
8. Baer E., Maier R. E. and Peter-
son R. N., SPE Journal 17
(1961) 1203.
9. Coleman B. D., J. Polymer Sci.
20 (1956) 447; Textile Research
J. 27 (1957) 393; 28 (1958) 393,
891.
10. Flom D. G., J. Appl. Phys. 32
. (1961) 1426.
11. Griffith A. A., Phil. Trans. Roy.
Soc. (London) A221 (1921) 163.
12. Hertz H., J. reine angew. Mathem.
92 (1881) 156.
13. Smith T. L., J. Appl. Phys. 35
(1964) 27.
14. Tabor D., Proc. Roy. Soc. (Lon-
don) 192 (1947) 247.
15. Zapp R. L., Rubber World 133
(1955) 59.
ГЛАВА XI.
ОПТИЧЕСКИЕ СВОЙСТВА
Показатель преломления и удельный инкремент показателя прело-
мления (важную в светорассеянии величину) можно оценить с по-
мощью аддитивных мольных характеристик. Но поглощение света
не является аддитивной величиной, а оказывается типичной внутрен-
ней характеристикой вещества. Другие оптические свойства, напри-
мер отражение света, зависят как от преломления, так и от поглоще-
ния света.
ВВЕДЕНИЕ
В общем случае взаимодействие между электромагнитным излуче-
нием (светом) и веществом определяется тремя характеристиками —
удельной электрической проводимостью а, электрической индуктив-
ностью в (обычно эту характеристику называют электрической по-
стоянной, или абсолютной диэлектрической проницаемостью) и маг-
нитной восприимчивостью р. (эту характеристику называют также
магнитной постоянной, или абсолютной магнитной проницаемостью).
Названные характеристики связаны с показателем преломления
и степенью поглощения света средой.
В ряде случаев полезно провести разграничение между проводя-
щей, или поглощающей (а >0),.и непроводящей (а = 0) средами.
В случае непроводящей среды скорость электромагнитной волны
обратно пропорциональна произведению (ре)1/’; точно так же ско-
рость распространения звуковой волны обратно пропорциональна
корню квадратному из сжимаемости.
Все материальные тела обладают набором собственных частот,
при которых излучение резонирует с некоторыми внутренними коле-
баниями данного тела. При таких критических частотах подобные
материальные тела оказываются сильными поглотителями излучения
даже в том случае, если эти тела проницаемы для излучения при
частотах выше или ниже критической.
К наиболее важным оптическим характеристикам относятся:
преломление, поглощение, отражение и рассеяние света. Три пер-
вые характеристики определяются усредненными оптическими свой-
ствами данной среды, рассеяние же определяется локальными флук-
туациями оптических свойств внутри среды.
194 ' -
В принципе все среды частично пропускают и частично отражают
падающий свет, однако для практических целей используются опи-
сательные понятия, позволяющие провести разграничение между
качественно различающимися случаями. Пропускание света каким-
либо материалом (отношение интенсивности проходящего света к
интенсивности падающего на образец света) определяется отраже-
нием, поглощением и рассеянием света. Если поглощением и рассея-
нием света можно пренебречь, то материал называют прозрачным
или проницаемым для излучения. Для непрозрачного материала
пропускание практически равно нулю в связи с высокой интенсив-
ностью рассеяния света. Материалы, которые практически не погло-
щают и пропускают свет с интенсивностью заметно больше нуля,
но меньше 90%, можно назвать полупрозрачными.
Полимерные материалы характеризуются глянцевитостью и мут-
ностью. Глянцевитость — это показатель отражающей способности
поверхности, связанный е внешним видом этой поверхности.
Мутность — это количество (в процентах) света, проходящего
сквозь образец, но отклоняющегося от направления падающего
пучка из-за рассеяния.
ПОГЛОЩЕНИЕ СВЕТА
Уменьшение интенсивности I светового пучка, проходящего через
среду путь длиной Z, описывается соотношением Ламберта — Бэра
/ = /оехр(^------J (XI.1)
В другом виде это соотношение записывается через коэффициент
экстинкции, который определяется уравнением
„ 1 , 1
ех=7Г1пТ7 (Х1Ла)
Показатель поглощения К есть характеристическая функция
длины волны. Большинство полимеров не имеют специфического
поглощения в видимой части спектра и, следовательно, оказываются
бесцветными.
' ИНФРАКРАСНОЕ ПОГЛОЩЕНИЕ
Все полимеры обладают полосами характеристического поглощения
в инфракрасной части спектра, поэтому определение спектров инфра-
красного поглощения — один из наиболее ценных методов анализа
полимеров. На рис. XI.1 представлены примерные длины волн неко-
торых полос инфракрасного поглощения, обусловленных колеба-
ниями функциональных групп и атомов. Некоторые полосы инфра-
красного поглощения изменяются в зависимости от конформации
полимерной цепи, поэтому метод ИК-спектроскопии позволяет
определить степень кристалличности. С помощью поляризованного
ИК-излучения можно раздельно определить ориентацию аморфной
и кристаллической частей в полукристаллическом полимере.
13* ” 195
Группы —Длина волны, мкм Обратное значение • 10* = волновое число, см~г J 4 5 6 7 в Э 10 11 12 13 14 15 10 17 18 19 20
снп Anutpamu- ческие СНГ с 1
CH3-N
СНг О
СНз-Si 1
-СН2 1
-сне
-С(СН3)з 1“
Насыщенное кольцо Циклопропан ~1 1 ,
Циклобутан
Цикпапентан
Ццклоеексан
с=с -сн=сн2 1
5"С=СНг
-сн=сн-
-сн=сн-
>с=сн-
-с=с=с-
Ароматические снаг 1
ьензольное кольцо
тнозомещенные
1,2-' 1,3- 7,4- Дилане щенные _ — — —
1,2,3- 1,3,5- 1,2Д- Тризоме ’ щенные — — —
с=с -с=сн 1
-C5C- Г л_
Рис. XI.l
Группы
» 5 в , 7 8 9 Ю П 12 13 14 15 16 П 18 19 20
-О- Простой э/pup. 1
Эпоксид 1
Пероксид ZE !!
Озонид II 1
-он Свободный 1
С внутренними связями *
Спиртовой первичный 1 1 1
вторичный
третичный zj I nJ
Фенольный
-ООН 1
-соон -соон i 1 1
-соо-
Ангидрид 7
-COCI -
1 о 1 Насыщенные, сложные эфиры
Ненасыщенные, сложные эфиры |
Ароматические, сложные Эфиры Г
>с«о Насыщенные кетоны 1
Ненасыщенные кетоны
Хинон
fl -лактон 1
Z а, ? ~ лактоны 1
-с*° С'Н Насыщенный альдегид I
Ненасыщенный альдегид n
< Серасадержацие группы 9. -SH 1
-S-
-S-S-
-c=s 1
- so2- 1 1
-SO3H • 1 1 1 I •
- OSOjO- 1 M
-so2nh2
-SCN ' ' " _L
Рис. XI.1
Продолзкение
»- мкм
3 4 5 6 7 в 9 Ю П 12 13 14 IS 16 П 18 19 20
Анина - группы -nh2 Свободная 1
Связанная г-
)NH Сводадная '
Связанная
-N< 1
nh4*
Аминный гал'огенводород ИВ»
Аминокислоты
Амидные группы '* - coNHa 1 1 п 1
-CONH- 1 1 1 1
-CON
-OCONH-
Кольца с азотом Пиридины г~
Триазины
Другие озатСоЗер- жащие группы -C=N 1
-N = C 1 *
-NCO
-NCS 1
-no2 1
-ONO п
-ono2 п 1
Галогенные группы -CF3
-F
-Cl
-Br
-I
Фоырорные группа! P-H
P-OH я t
p-o-c
P=O
P=S
Кремний- содержащие группы Si-H 1
Sl-C
Sl-O-C
Рис. XI. 1, Хаоактеоистические . полосы поглощения функциональных групп.
ПРЕЛОМЛЕНИЕ СВЕТА
Показатель преломления п представляет собой отношение скорости
света в вакууме к скорости света в рассматриваемом веществе.
Для непоглощающих сред аддитивной величиной, связывающей
оптическое преломление с химическим строением, является так назы-
ваемая мольная рефракция. В литературе предложены различные
определения мольной рефракции, например:
а) мольная рефракция по Лорентцу и Лоренцу [21, 22]
П2 — 1 М П2 — 1
Ядл = «24-2 ~р_= п2-Ь2 V (XI-2)
б) мольная рефракция по Гладстону и Дейлу [15]
Ягд=(п-1)у-=(«-1П (XL3)
в) мольная рефракция по Фогелю [25—28]
7?(1=пМ (XI. 4)
Формулы Гладстона и Дэйла, а также Фогеля — чисто эмпири-
ческие, но определение Лорентц — Лоренца основано на электро-
магнитной волновой теории света. В качестве стандартного обычно
используют показатель преломления света, измеренный для D-ли-
нии натрия (nD).
Некоторые исследователи рассчитали вклады атомных групп
или связей в мольную рефракцию [14, 18 , 24, 25 , 29 , 30]. Недавно
Годхарт [16] провел обширный анализ данных для приблизительно
тысячи органических жидкостей, содержащих 43 различные функцио-
нальные группы. С помощью полученных этим автором групповых
вкладов можно предсказать значение величины (л—1) при среднем
стандартном отклонении около 1%; такое стандартное отклонение
означает, что само значение п можно предсказать в среднем с точ-
ностью примерно 0,4%. Полученные Годхартом значения приведены
в табл. XI.1.
Бензольное кольцо довольно сильно влияет на другие группы,
поэтому Годхарт разграничил группы, непосредственно присоеди-
ненные к бензольному кольцу, и группы, отделенные от аромати-
ческого кольца одним или более атомом углерода. Групповой вклад
бензольного кольца с дополнительными заместителями был получёц
путем вычитания эквивалентного количества вкладов Наромат
из вклада фенильной или фениленовой групп.
Специальный инкремент — «стерическое препятствие» — был вве-
ден для преодоления трудности, связанной с множественным замеще-
нием на соседних атомах углерода. Инкремент стерического препят-
ствия должен использоваться, если в одной цепи на двух соседних
атомах углерода расположены поблизости друг от друга группы
типа СН3, CI или ОН. *
199
Таблица Х1.1. Групповые вклады в мольную рефракцию
при длине волны света 589 ммк [16]
Группы Характер или место присоединения группы Ялл ЛГД Нф
-СНз общего типа 5,644 8,82 17,66
присоединена к бензольному кольцу 5,47 8,13 15,4
—СН2- общего типа 4,649 7,831 20,64
присоединена к бензольному кольцу 4,50 7,26 18,7
^>СН- общего типа 3,616 6,80 23,49
] присоединена к бензольному кольцу 3,52 6,34 21,4
—с- общего типа 2,580 5,72 26,37
1 присоединена к бензольному кольцу 2,29 ( 4,96 25,1
\ ’ / циклогексил , 26,686 44,95 122,66
—^3/* фенил 25,52 44,63 123,51
о-фенилен 24,72 44,2 129,0
л<-фенилен 25,00 44,7 128,6
—— п-фенилен 25,03 44,8 128,6
Наромат. среднее значение 0,59 0,04 -5,2
-О- метиловые эфиры 1,587 2,96 23,85
высшие эфиры 1,641 2,81 23,18
присоединена к бензольному кольцу 1,77 2,84 22,6
ацетали 1,63 2,75 22,99
—он первичный спирт 2,551 4,13 24,08
вторичный спирт 2,458 3,95 23,95
третичный спирт 2,453 3,85 24,05
фенол 2,27 3,53 22,7
/>С=О метилкетон 4,787 8,42 43,01
высшие кетоны 4,533 7,91 43,03
присоединена к бензольному кольцу 5,09 8,82 41,9
0
II —с-н общего типа 5,83 9,63 40,69
—соон общего типа 7,212 11,99 64,26
-СОО- метиловые эфиры 6,237 10,76 65,32
этиловые эфиры 6,375 10,94 64,49
высшие эфиры 6,206 10,47 64,20
присоединена к бензольному кольцу 6,71 11,31 64,8
ацетаты 6,306 10,87 64,90
—осоо- метилкарбонаты 7,75 13,39 87,8
высшие карбонаты 7,74 13,12 86,8
—nh2 общего типа 4,355 7,25 22,64
присоединена к бензольному кольцу 4,89 8,40 23,7
200
Продолжение табл. XI. 1.
Группы Характер или место присоединения группы ВЛЛ «ГД Яф
>NH общего типа 3,585 6,29 24,30
присоединена к бензольному кольцу 4,53 8,68 26,9
^>N— общего типа 2,803 5,70 26,66
присоединена к бензольному кольцу 4,05 8,67 '30,7
—CONH- общего типа 7,23 15,15 69,75
присоединена к бензольному кольцу 8,5 18,1 73
—C^N 5,528 9,08 36,67
-no2 6,662 11,01 66,0
—SH первичная 8,845 15,22 50,61
вторичная 8,79 15,14 50,33
третичная 9,27 15,66 49,15
—s— метилсульфид 7,92 14,30 53,54
—ss- высшие сульфиды 8,07 14,44 53,33
16,17 29,27 107,63
—F моно 0,898 0,881 22,20
пер 0,898 0,702 20,92
-Cl первичный 6,045 10,07 51,23
вторичный 6,023 9,91 50,31
третичный 5,929 9,84 50,75
—Br присоединена к бензольному кольцу 5,60 8,82 48,4
первичный 8,897 15,15 118,5
вторичный 8,956 15,26 118,4
третичный 9,034 15,29 119,1
—I 13,90 25,0 —
Инкременты состава 0,41
Д «стерического затруднения» (соседство) —0,118 —0,18
Д изопропильной группы А этиленовой связи 0,068 0,05 -0,20
общего типа 1,65 1,90 -6,36
цис- 1,76 1,94 —5,56
транс- ( 1,94 2,09 ... —6,37
А циклического строения -0,18 —1,15 —5,06
циклопентан
циклогексан —0,13 —0,92 —4,44
тетрагидрофурил —0,12 -0,98 —4,36
фурил —0,086 —0,78 —4,53
пиперидил 1 —0,41 —1,94 ' —5,63
В табл. XI.2 сравниваются рассчитанные и полученные экспери-
ментально величины для ряда твердых аморфных полимеров. Ин-
тересно отметить, что очень простая формула Фогеля [25—281 R& =
пМ позволяет получить примерно такое же стандартное отклоне-
вие, как и более сложная теоретически обоснованная формула Ло-
рентц — Лоренца, а также формула Гладстона — Дейла.
Средний показатель преломления кристаллического полимера
всегда больше, чем такого же полимера в аморфном состоянии. Од-
нако, поскольку плотность кристаллического полимера также выше,
Мольная рефракция по Лорентц — Лоренцу и Гладстону — Дейлу
' 201
Таблица XL2. Экспериментальные и рассчитанные значения
показателей преломления
Полимер п (опыт) п рассчитан по (XI. 5)
ПО длл ПО лгд ПО Яф
Полибутадиен-1,3 1 1,516 1,511 1,513 1,514
Полиизопрен 1,520 1,514 1,514 1,504
Поли-2-трет-бутилбутадиен-1,3 1,506 1,517 Г,513 1,489
Поли-2-децилбутадиен-1,3 1,490 Г,487 1,489 <483
Полистирол 1,591 1,603 <600 1,590
Полиметилстирол 1,587 1^577 <585 <574
Полиизопропилстирол 1,554 1,562 <560 <555
Поливинилхлорид 1,544 <539 <543 <511
Полихлоропрен 1,558 1,560 <560 1,531
Поли-о-хлорстирол 1,610 1,612 1,607 1,583
Поли-о-метоксистирол 1,593 1,560 1,562 1,575
Поли-п-метоксистирол 1,597 1,565 1,566 1,572
Полиоксиметилен (1,510) 1,416 <426 1,453
Полиоксипропилен 1,457 1,451 1,455 1,463
Полиметилвиниловый эфир 1,467 1,450 1,457 <,474
Полиэтилвиниловый эфир 1,454 <457 1,460 1,465
Поли-и-бутилвиниловый эфир < 1,456 <463 1,465 <466
Полиизобутилвиниловый эфир 1,451 . 1,464 । <466 1,464
Поли-н-пентилвиниловый эфир 1,459 1,465 1,467 1,467
Полигексилвиниловый эфир 1,4601 1,4661 1,468 1,468
Полидецилвиниловый эфир 1,463 1,469 1^472 1,469
Поливинилацетат 1,467 <471 1,475 <472
Поливинилбензоат 1,578 <583 <583 <569
Полиметилакрилат .1,479 1^489 1,-188 <477
Полиэтилакрилат 1,469 1,487 1,488 1,468
Полибутилакрилат 1,466 1,481 1,480 <466
Полиметилметакрилат 1,490 1,484 1,484 1,475
Полиэтилметакрилат 1'485 1X88 1X88 1,467
Полиизопропилметакрилат 1,552 1,479 1,478 1,464
ПолИ'л-бутилметакрилат 1,483 1,475 1,475 1,466
Поли-трет-бутилметакрилат 1,464 1,467 1,472 1,464
Полиизобутилметакрилат 1,477 1,480 1,480 1,466
Полигексилметакрилат 1,481 1,476 1,475 <467
Полилаурилметакрилат 1,474 <476 1,475 <469
Полициклогексилметакрилат 1,507 1,515 <513 1,495
Полибензилметакрилат 1,568 1,563 1,560 1,539
Полиакрилонитрил 1,514 1,528 <529 1,523
Полиметилхлоракрилат 1,517 1,519 1,521 1,500
Полиэтилхлоракрилат 1,502 1,516 1,518 1,490
Поли-втор-бутилхлоракрилат 1,500 1,503 1,502 1,495
Полициклогексилхлоракрилат 1,532 1,529 1,528 1,508
Полигексаметиленадипамид 1,530 1,497 1,521 1,528
Полиэтилентерефталат - * 1,640 1,580 1,581 1,558
Поли-1,1-этан-бис (4-фенил) карбонат 1,594 1,600 <602 1,594
Поли-2,2-пропан-бис(4-фенил)карбонат Поли-1,1-бутан-бис(4-фенил)карбонат Поли-2,2-бутан-бис(4-фенил)карбонат 1,585 1,579 1,583 1,579 1,585 1,573 1,583 <587 1,577 1,590 1,582 1,584
Поли-1,1 -(2-метилпропан)б ис(4-фенил) карбонат 1,570 1,580 1,583 1,582
Полидифенилметан-бис(4-фенил)карбонат 1,654 <637 <630 1,628
Поли-1,1-циклопентан-бис(4-фенил)карбонат Поли-1,1-циклогексан-бис(4-фенил)карбонат 1,599 1,590 <589 1,583 <596 1,590 1,599 1,595
202
остается практически постоянной. Оценивать мольную рефракцию
кристаллических полимеров по формуле Фогеля нельзя, поскольку
в эту формулу не входит плотность полимера.
Из уравнений (XI.2)—(XI.4) можно легко получить следующие
выражения для показателя преломления:
/ 1 ч-2Ллл/^\*/.. дгд. Лф -
П^1-/?ЛЛ/Р у ’ n~1 + v > п~ м (Х1-5)
Совершенно ясно, что отношения 7?лл/К, 7?гд/К и R&IMхарактери-
зуют преломляющую способность данного полимера. То же самое
справедливо и для функциональных групп. Очевидно, ароматические
группы обладают высокой преломляющей способностью, а метиль-
ные группы и атомы фтора — очень низкой преломляющей способ-
ностью.
Пример 1
Оценить показатель преломления полиметилметакрилата.
Решение
Структурное звено полимера:
СН3
I
—СН2-С— М = 100,1
о=с—осн3
Из значений групповых вкладов, приведенных в табл. XI.1, следует, что
Лф = 1 (-СН2-)4-21(-СН3) + 1 (-СОО-) + 1
-С-
= 20,64+35,32+65,32+26,37 = 147,65
Следовательно
nD = 147,65/100,1 =1,475
Экспериментальное значение nD = 1,490.
ДВОЙНОЕ ЛУЧЕПРЕЛОМЛЕНИЕ
Если какое-либо вещество обладает различными оптическими харак-
теристиками в различных направлениях, то оно проявляет эффект
двойного лучепреломления. Двулучепреломление связано с измене-
нием показателя преломления в зависимости от направления рас-
пространения света в данном веществе.
- Кристаллические полимеры, и особенно ориентированные кри-
сталлические полимеры (см. гл. XX), обладают двойным лучепрелом-
лением, которое слагается из характеристического двойного лучепре-
ломления (вклад самих кристаллитов) и двойного лучепреломле-
ния формы, обусловленного формой кристаллитов или наличием
пустот.
203
Для полностью неориентированного материала вклады как кри-
сталлических, так и аморфных областей равны нулю. Причиной
двойного лучепреломления в твердых полимерах может оказаться
также и другой фактор, например ориентация веществ, вызывающих
набухание полимера, обусловленная самим строением цолимера^и,
что более существенно, внутренними напряжениями.
В полимерных растворах двойное лучепреломление возникает
в том случае, если под действием внешних сил происходит ориента-
ция молекул. Ориентация может возникнуть в текущем полимере
(двойное лучепреломление в потоке) под действием электрических
полей (эффект Керра) или магнитных полей (эффект Коттон — Му-
тона). Жанешитц-Кригл и Уолес [20] получили безразмерные
группы величин для обработки данных по двойному лучепреломле-
нию в потоке.
РАССЕЯНИЕ СВЕТА
Рассеяние света осуществляется оптическими неоднородностями
среды. Молекулы выступают в роли вторичных источников света,
как это было показано Редеем [23]. Если проинтегрировать интен-
сивность рассеянного единицей объема света iq по всем углам, то
получится величина мутности т:
п
т= У sin 0 dQ ' (XI.6)
о
В отсутствие поглощения мутность связана с интенсивностью свето-
вого пучка до и после прохождения среды толщиной I и вычисляется
по формуле '
/ = 70ехр(-т|) (XI.7)
В растворах рассеяние света частично обусловлено колебаниями по-
казателя преломления из-за флуктуаций состава. Именно на таких
представлениях основано хорошо известное уравнение рассеяния
света растворами:
I/ <Ш \
R^KRT.
(XI.8)
r2i0
где Re=7e(i + cOs2 0)
•я отношение Релея; г —* расстояние до рассеивающей
Свет молекулы;
2л2 Г_ dn "12
К'= X0Na Ln de J
(XI.9)
— длина волны свеп в вакууме; я — осмотическое давление; А^ — число
Авогадро.
204
Поскольку
1 f dn \ 1 ,
"йГ \~dT ) =~M ' 2АгС
где Аг — второй вириальный коэффициент; подстановка этого вы-
ражения в (XI.9) дает: ,
Кс 1
~п^ = ~м' + 242с ' (XI.IO)
Это уравнение представляет собой основу для определения молеку-
лярных весов полимеров методом рассеяния света — одним ив не-
многих абсолютных методов.
Однако соотношение (XI.10) справедливо только для оптически
изотропных частиц, которые много меньше по сравнению с длиной
волны падающего света. Если размер частицы превышает 1/20, как
это бывает в полимерных растворах, то волны рассеянного света
от различных частей одной и той же частицы интерферируют друг
с другом; это приводит к ослаблению интенсивности рассеянного
света в Р (9) раз. Функция Р (9) имеет вид
Р(0) = 1-уЛ2 </?£>+-. . (XI.11)
где Rg — радиус вращения частицы (молекулы полимера); h =4я (n/X«) sin 0/2.
Формула для избыточного рассеяния полимерным раствором по
сравнению’с рассеянием чистым растворителем теперь записывается
следующим образом:
Кс 1
^-й5чч+2Л*+--- <х,Л2>
При А -> 0 величина Р (9) -> 1, поэтому Зимм [31 ] предложил
строить график зависимости KcIRq от sin2 (9/2) + kc, где к — про-
извольно выбранная постоянная порядка 100. При таком построе-
нии можно получить сетку, с помощью которой осуществляется экс-
траполяция к с = 0 и 9 = 0 (рис. XI.2). Отсекаемый по оси ординат
отрезок дает AlMw, а по тангенсам двух углов наклона можно рассчи-
тать А 2 и <7?g>- С помощью описанного метода можно измерять
молекулярные веса полимеров в диапазоне от 104 до 10’. Для того
чтобы определить величину К, необходимо экспериментально изме-
рить показатель преломления раствора п и так называемый удельный
инкремент показателя преломления (dnldc). Но так как рассматри-
ваемый метод применяется к очень разбавленным растворам, то вели-
чину п можно заменить п0 — показателем преломления раство-
рителя.
Для таких разбавленных растворов инкремент показателя пре-
ломления является постоянной величиной для данной системы по-
лимер растворитель при выбранной температуре и его, как
205
правило, измеряют с помощью интерферометра или дифферен-
циального рефрактометра.
Как показал Годхарт [16], значения инкремента показателя пре-
ломления (dnldc) можно рассчитывать по групповым вкладам.
Рис. XI.2. График, показывающий метод экстраполяции данных рассеяния
света по Зимму:
• — экспериментальные точки; О — экстраполированные точки.
Наиболее приемлемые результаты были получены при помощи сле-
дующего простого уравнения:
dn
de
206
Пп — ПП V (Rv
Рп ~ м \ м
(XI.13)
где индексы «О» и «п» относятся к растворителю и полимеру соответ-
ственно. Значения п и р полимеров можно рассчитать с помощью
аддитивных мольных величин, что позволяет найти удельный инкре-
мент показателя преломления. Следовательно, для определения Мш,
<ZRqZ> и А 2 достаточно измерить только величины Rq.
В табл. XI.3 показаны экспериментальные и рассчитанные зна-
чения (dn/dc), которые удовлетворительно согласуются друг с дру-
гом.
Следует отметить, что применимость метода светорассеяния не
ограничивается растворами. Метод рассеяния света можно исполь-
зовать для получения информации относительно надмолекулярной
структуры твердых полимеров.
Пример 2
Оценить удельный инкремент показателя преломления dn/dc полистирола
в 1,4-диоксане (nD = 1,422).
Решение
Структурное звено полистирола:
—СН2-СН—
I
М = 104,1
Использование групповых вкладов, приведенных в табл. IV.4 и XI.1 дает:
1 <-СН2)
1(>сн-)
RVi
20,64 15,85
21,4 9,45
123,51 72,7
165,55 98,0
Из формулы (XI. 13) следует, что
dn V
(nV/M ~ п<>) = 98,0/104,1 • (165,55/104,1 -1,422) =
= 0,941 (1,590-1,422) = 0,158
Этот результат удовлетворительно согласуется с экспериментальными данными
(0,168).
207
Таблица XI.3. Экспериментальные и рассчитанные аначения dn/dc
растворов полимеров
Полимер Растворитель Опыт Расчет
dn/dc Т, °с X, ммк ПО Ярд ПО Пф
Полигексаме- ж-Крезол —0,016 25 546 -0,017 —0,011
тиленадипамид Дихлоруксусная кис- лота 0,098 25 546 0,051 0,057
Серная кислота, 95%-ная 0,082 25 546 0,076 0,083
.Полиметилмет- Ацетонитрил 0,137 25 546 0,120 0,112
акрилат Бромбензол 1 -Бромнафталин —0,058 —0,147 25 25 546 546 —0,065 -0,151 —0,073 -0,159
Бутил ацетат 0,097 — 546 0,078 0,069
Бутилхлорид 0,090 20 546 0,072 0,063
Хлорбензол -0,026 25 546 —0,035 —0,043
Изоамилацетат 0,091 20 546 0,072 0,063
Нитроэтан 0,094 25 546 0,082 0,074
1,1,2-Трихлорэтан 0,025 — 560 0,012 0,003
Ацетон 0,129 25 546 0,109 0,100
Ацетон 0,134 25 546 0,109 0,100
Бензол —0,010 25 546 —0,014 —0,022
2-Бутанон 0,109 — 546 0,090 0,081
2-Бутанон 0,114 25 546 0,090 0,081
Четыреххлористый углерод 0,023 25 546 0,019 0,010
Хлороформ 0,055 — 546 0,034 0,025
Хлороформ 0,063 — 546 0,034 0,025
Диоксан 0,071 — 546 0,055 0,046
Этилацетат 0,118 — 546 0,098 0,089
Тетрагидрофуран 0,087 — 546 0,070 0,061
Полиоксипро- Бензол —0,045 25 546 —0,046 —0,042
пилен Хлорбензол —0,064 25 546 —0,069 —0,065
Гексан 0,078 25 546 0,079 0,083
2-Метилгептан 0,066 35 546 0,061 0,065 ,
Метанол 0,118 25 546 0,123 0,127 0,084
Полистирол Бензол , 0,106 25 546 0,093
Бензол 0,110 25 546 0,093 0,084
Бромбензол 0,042 25 546 0,038 0,028
Бромнафталин -0,051 25 546 -0,056 —0,025
Четыреххлористый углерод 0,146 25 546 0,129 0,120
Хлорбензол 0,099 — 436 0,071 0,061
Хлороформ 0,195 20 436 0,145 0,136
Циклогексан 0,167 20 546 0,161 0,151
п-Хлортолуол 0,093 25 436 0,075 0,066
Декалин 0,128 20 546 0,122 0,113
Дихлорэтан 0,161 20 546 0,147 0,137
Диоксан 0,168 25 546 0,167 0,158
Тетрагидрофуран 0,189 25 546 0,184 0,175
Толуол 0,104 20 546 0,097 0,088
Полив инилаце- Ацетон 0,095 20 546 0,097 0,094
тат Ацетон 0,104 30 436 0,097 0,094
Ацетонитрил 0,104 — — 0,108 0,105
Бензол —0,026 25 560 —0,022 —0,025
Хлорбензол —0,040 25 560 -0,042 -0,045
208
Продолжение табл. X 1.3.
Полимер Растворитель Опыт Расчет
dn/dc Т, °C 1, ммк ПО Пр jy ПОПф
Диоксан 0,030 25 560 0,044 0,040
Этилформиат 0,095 — . ! 0,096 0,093
Метанол 0,131 25 546 0,121 0,117
2-Бутанон 0,080 25 546 0,079 0,075
Тетрагидрофуран 0,055 25 546 0,060 0,056
Толуол —0,020. 25 560 -0,018 —0,022
Поливинилхло- Ацетон 0,138 20 546 0,133 0,110
рид Циклогексанон 0,078 25 — 0,066 0,043
Диоксан 0,086 —- 546 0,087 0,064
Тетрагидрофуран 0,102 — — 0,100 0,077
ОТРАЖЕНИЕ СВЕТА
Отражение света на поверхности раздела двух непоглощающих сред
(например, полимер и воздух) зависит от показателей преломления
этих двух сред и угла падения. Для света, поляризованного в пло-
скости падения, доля отраженного на поверхности раздела света
определяется соотношением Френеля
r=tg2(e-oo)/tga(e+0o) (Xi.14)
где О — угол падения света на более-плотную среду; 0« — угол паденйя света
для второй среды.'
Для светового потока, падающего под прямым углом, это соотно-
шение принимает вид '
/ П—Пп \2 • __
(XI.15)
п — показатель преломления более плотной среды; п» — показатель преломле
ния второй среды.
Полное отражение происходит под углом 0В (угол Брюстера),
который определяется соотношением
tg0B = n/no
' ЛИТЕРАТУРА
1. Bellamy L. J., «Advances in Infra-
red Group Frequencies», Methuen,
London, 1968.
2. Conley R. T., «Infrared Spectro-
scopy», Allyn and Bacon, Boston,
‘ 1966.
3. Dodd R. E., «Chemical Spectro-
scopy», Elsevier, Amsterdam,
1962.
(XI. 16)
4. Flett M. St. C„ «Characteristic
Frequencies of Chemical Groups
in the Infrared», Elsevier, Am-
sterdam, 1963.
5. Freeman S. K. (Ed.), «Interpre-
tive Spectroscopy», Reinhold,
New York, Chapman, London,
1965.
14 Заказ 699
209
6. Hummel D. О., «Infrared Spectra
of Polymers: in the Medium and
Long Wavelength Regions», Inter-
science, New York, 1966. (Poly-
mer Reviews, ed. by Mark H. F.
and Immergut E. H., Vol. 14).
7. Oster G., Chem. Revs. 43 (1948)
319,
8. Stacey K. A., «Light Scattering
in Physical Chemistry», Butter-
worth, London, 1956.
9. Stuart H. A., «Die Physik der
Hochpolymeren», Bd. I., Sprin-
ger, Berlin, 1952.
10. Szymanski H. A., «Infrared Band
Handbook», Plenum Press, New
York, 1964.
11. Weissberger A. (Ed.), «Technique
of Organic Chemistry», Inter-
science, New York, 1961.
12. Zbinden R., «Infrared Spectro-
scopy of High Polymers», Acade-
mic Press, New York, 1964.
13'. Debye P., J. Appl. Phys. 15
(1944) 338; J. Phys, and Coll.
Chem. 51 (1947) 18.
14. Eisenlohr F., Z. physik. Chem.
75 (1911) 585; 79 (1912) 129.
15, Gladstone J. H. and Dale T. P.,
Trans. Roy Soc. (London) A 148
(1858) 887.
16. Goedhart D. J., Communication
Gel Permeation Chromatography
International Seminar, Monaco,
Oct. 12—15, 1969.
17. Heller W., Phys. Rev. 68 (1945)
5; J. Phys. Chem. 69 (1965) 1123;
J. Polymer Sci. A2—4 (1966)
209.
18. Huggins M., Bull. Chem. Soc.
Japan 29 (1956) 336.
19. Janeschitz-Kriegl H., Makromol.
Chem. 33 (1959) 55; 40 (1960)
140.
20. Janeschitz-Kriegl H. and Wa-
les J. L. S., Nature (1967) 1116.
21. Lorentz H. A., Wied. Ann. Phys.
9 (1880) 641.
22. Lorenz L. V., Wied. Ann. Phys.
11 (1880) 70.
23. Rayleigh J. W., Strutt, Lord,
Phil. Mag. (4) 41 (1871) 107,
224, 447.
24. Schoorl N., «Organische Analyse»
1 bd. 14, Centen, Amsterdam,
1920.
25. Vogel A., J. Chem. Soc. (1948)
1833.
26. Vogel A., Cresswell W. and Lei-
cester I., Chem. and. Ind. (1950)
358; (1953) 19.
27. Vogel A., Chem. and Ind. (1951)
376; (1952) 514.
28. Vogel A., Cresswell W. and Lei-
cester/., J. Phys. Chem. 58 (1954)
174.
29. Wibaut J. P., Hoog H., Langen-
dijk S. L., Overhoff J. and
Smittenberg J., Rec. Trav. Chim.
58 (1939) 329.
30. Young J. and Finn A., J. Rese-
arch Natl. Bur. Standards 24
(1940) 759.
31. Zimm R. H., «The Normal-Coor-
dinate Method for Polymer Chains
in Dilute Solution», Chapter 1 in
F. R. Eirich (Ed.), «Reology»,
Vol. 3, Academic Press, New
York, 1960.
ГЛАВА XII.
ЭЛЕКТРИЧЕСКИЕ СВОЙСТВА *
Представляют интерес две группы электрических свойств полимеров.
Первая группа обычно определяется по характеристикам полимера
в слабых электрических полях. К этой группе относятся диэлектри-
ческая проницаемость, коэффициент диэлектрических потерь, ста-
тическая электризация и электрическая проводимость.
Ко второй группе относятся характеристики, играющие суще-
ственную роль в очень сильных электрических полях. В частности,
к этим свойствам относятся напряжение пробоя и дугостойкость.
Названные свойства можно отнести к предельным электрическим
характеристикам материала.
Свойства первой группы непосредственно связаны с химическим .
строением данного полимера, а оценка свойств второй группы су-
щественно усложняется за счет дополнительного влияния самих
методов определения этих свойств.
С помощью аддитивных величин можно оценить только диэлект-
рическую проницаемость.
ДИЭЛЕКТРИЧЕСКАЯ ПОЛЯРИЗАЦИЯ
ДИЭЛЕКТРИЧЕСКАЯ ПРОНИЦАЕМОСТЬ
Диэлектрическая проницаемость (относительная) непроводящих ма-
териалов представляет собой отношение емкостей плоского конден-
сатора, измеренных при наличии и отсутствии данного диэлектрика
между пластинами конденсатора. Различие между емкостями в двух
указанных случаях обусловлено явленийи поляризации диэлектрика
в электрическом поле.
Диэлектрическую проницаемость е неполярного изолятора можно
выразить через показатель преломления п с помощью уравнения
Максвелла
е = «2 (XII.1)
* Обширные экспериментальные данные по электрическим свойствам
полимерных материалов содержится в кн.: Конструкционные свойства пласт-
масс. Составитель Э. Бэр. М., «Химия», 1967, с. 41 — 175. — Прим, ред-
14* w 211
Следует сравнивать значения этих двух величин, измеренных
при одной частоте. Однако диэлектрическую проницаемость обычно
измеряют при относительно низких частотах (102—106 Гц), тогда
как показатель преломления определяется в диапазоне частот ви-
димого света (5-1014—7-Ю14 Гц), как правило, на D-линии натрия.
Но даже простое сравнение величин е и nD уже дает интересную
информацию. Большое расхождение между е и п2 может служить
указанием на полупроводниковые свойства вещества, но гораздо
чаще подобное расхождение обусловлено присутствием постоянных
диполей в. данном диэлектрике.
МОЛЬНАЯ ПОЛЯРИЗАЦИЯ
Мольрая поляризация диэлектрика может определяться следующим
образом (сравните с /?лл):
р _ 1
*лл=7+2Г ' (XIL2>
или (сравните с 7?у)
Pr=e.'/‘M (ХП.З)
Групповые вклады в мольную поляризацию полимеров приве-
дены ниже:
— |<М 1* — |м
14- м 1 +
ttl ш л! и **03
- И II л II
в а, а, Л с а, ft,
в S. S. в S В р,
& Сн С й в* Сн К в
- СНз 5,64 17,66 - —соо- 15 95
—СН2- 4,65 20,64 —CONH— 30 125
^>сн— 3,62 235 —О—СОО— —F 22 (1.8) 125 (20)
1 -С1 (9,5) (60)
—с— 2,58 26,4 -C=N 11 (50)
1 —CFa— 6,25 70
25,5 123,5 -СС12- 17,7 145
—СНС1— 13,7 90
— S — 8 (60)
— 25,0 128,6 —ОН (спирт) (6) (30)
—ОН (фенол) — 20 — 100
—0=^ 52 (30)
/с=0 (10) (65)
С помощью формул (XII.2) и (ХП.З) можно рассчитать диэлек-
трическую проницаемость е, если известно строение полимера.
В табл. XII.1 приведены рассчитанные и экспериментально опре-
деленные значения е, а для сравнения значения п2. Согласие расчет-
ных и экспериментальных значений удовлетворительное.
212
Таблица XII.1. Относительная дивлектрическая проницаемость полимеров
Полимер 8 * (ОПЫТ) 8 рассчитана до (XII.2) е рассчитана по (XII.3)
Полиэтилен (экстраполяция на аморфное состояние) 2,2 (2,19) 2,20 2,20
Полипропилен (аморфная часть) 2,15 (2,19) 2,15 2,15.
Полистирол 2,5 2,53 2,55 2,60
Политетрафторэтилен (аморфный) 2,0 — 2,00 1,96
Поливинилхлорид 3,05 2,37 3,05 3,15
Поливинилиденхлорид (аморфный) 2,85 — 2,90 2,92
Поли-о-хлорстирол 2,63 2,60 2,82 2,82
Полиакрилонитрил 3,1 2,29 3,26 3,15
Полиоксиметилен 3,1 2,29 2,95 2,85
Поли-2,6-диметилоксифени леи 2,65 — 2,65 2,75
Полиметилметакрилат 3,15 2,22 2,94 3,15
Полиэтилметакрилат 2,9 2,20 2,80 3,00
Полиметил-а -хлоракрилат 3,4 2,30 3,45 3,32
Поляэтил-а -хлоракрилат 3,1 2,26 3,20 3,16
Пентон 3,0 — 2,95 2,80
Поливинилацетат 3,22 2,15 3,02 3,30
Полиэтилентерефталат (аморфный) 3,1 2,70 3,40 3,50
Полибисфенолкарбонат 3,05 2,50 3,00 3,05
Полигексаметиленадипамид 4,0 2,35 4,14 4,10
* Данные взяты из работы [8]; Kudststoff Taschenbuch, 1964; Polymer Handbook»
1966; Stoft-Hiitte, 1967.
ДИПОЛЬНЫЙ МОМЕНТ
В простейших случаях, например, когда диэлектрик представляет
собой чистое вещество и дипольный момент р. мал (р меньше 0,6 Д),
можно воспользоваться уравнением Дебая при 298 К:
Это уравнение показывает, что при наличии постоянных диполей е
больше и2. Например, вода обладает очень высокой диэлектрической
проницаемостью (81), тогда как п2 для нее составляет всего 1,77.
Для всех полимеров, обладающих полярными группами, е больше га2.
Для таких полймеров формула (XII.4) приводит к значениям
среднего дипольного момента мономерного звена от 0 для карбоцеп-
ных полимеров до примерно 1 Д для полиамидов. Измеренные зна-
чения оказываются ниже по сравнению с дипольными моментами
полярных групп в жидких средах.
Ниже приведены эффективные средние дипольные моменты в по-
лимерах, а также дипольные моменты, измеренные в жидких средах!
Группа
-С1
-СС12-
-cf2-
цЭфВ полимерах
0,45
0,40
0,25
ЦЭф В жидкости
2,0
213
Группа М-дф в полимерах Нэф в жидкости
—CsN 0,50 3,5
-О— 0,45 1,1
-с^о- X) 0,70 —
- C^-NH- ^0 1,00 —
Коэффициент перевода в СИ: 1 Д = 3,34.10-’“ К>м,
Пример 1
Оценить диэлектрическую проницаемость и средний дипольный момент
поликарбоната.
Решение
Структурное звено поликарбоната
ЛГ = 254
Г.(298)=215
С помощью групповых вкладов, приведенных на с. 212, можно получить:
Группы « s+2 ‘
2 (-©-) 50,0 257,2
1 (_(М \. 1 / 2,6 26,4
2 (-СН3) 11,3 35,3
1 (О-СОО-) 22 125
85,9 443,9
Отсюда е = 3,00 и е = 3,05 соответственно; результат удовлетворительно
согласуется с экспериментальным значением 8 = 3,05.
Средний дипольный момент оценивается с помощью мольной рефракции
Лорентц — Лоренца (см. табл. XI.1):
Группы 2(-©-) / 1 \ 1 —С— \ 1 / 2 (-СНз) 1 (-О-СОО-) Й1ЛЛ 50,06 2,29 11,29 7,74 71,38
.214
Подстановка этой величины в формулу (XII.4) дает:
20,6р.2=Р — Ялл = 85,9-71,4 = 14,5
или
р = (14,5/20,б)’1/’=0.84
СООТНОШЕНИЕ МЕЖДУ ДИЭЛЕКТРИЧЕСКОЙ
ПРОНИЦАЕМОСТЬЮ И ПАРАМЕТРОМ РАСТВОРИМОСТИ
Электрические силы, обусловленные поляризуемостью и дипольным
моментом, определяют энергию когезии, поэтому можно ожидать
наличия определенной корреляции между диэлектрической прони-
цаемостью и параметром растворимости. Дарби с сотр. [11] пред-
ложили метод сопоставления этих величин для органических соеди-
нений.
Оказывается, что для полимеров выполняется удивительно простое
соотношение
6^3,Зе (XII.5)
В табл. XII.2 сравниваются рассчитанные и экспериментальные
значения е.
Таблица XII.2. Относительная диэлектрическая проницаемость
и параметр растворимости
Полимер б (расчет) ♦ е (расчет) е (опыт)
Полиэтилен 7,9 2,4 2,2
Полипропилен 8,3 2,5 2,15
Полистирол 9,0 2,7 2,5
Поливинилхлорид 9,6 2,9 3,05
Поливинилиденхлорид 9,9 3,0 2,85
Политетрафторэтилен 5,7 1.7 2,0
Полиформальдегид 10,3 3,1 3,1
Поливинилацетат 9,6 2,9 3,2
Полиметилметакрилат 9,1 2,8 3,15
Полиэтилметакрилат 8,9 2,7 2,9
Полиакрилонитрил 12,5 3,8 3,1
Полиэтилентерефталат 10,0 3,0 3,1
Поликарбонат 9,9 ♦♦ 3,0 3,05
Найлон 6,6 13,7 4,1 4,0
Поли-о-хлорстирол 8,9 ♦♦ 2,7 2,6
Полихлортрифторэтилен 7,8 2,4 2,5
* Рассчитано по формулам, приведенным в гл. VI,
** Экспериментальное значение, j
215
КОЭФФИЦИЕНТ ДИЭЛЕКТРИЧЕСКИХ ПОТЕРЬ
Если электрическое поле изменяется во времени (например, если
создается переменный ток), то диэлектрическая поляризация также
изменяется во времени. Но в диэлектрике существует сопротивление
перемещению атомных групп, поэтому возникает разность фаз между
изменениями электрического поля и поляризации. Диэлектрическая
характеристика полимера в осциллирующем электрическом поле
Рис. XII. 1. Дисперсионные кривые диэлектрической проницаемости и тангенса
угла диэлектрических потерь для поливинилацетата [18].
-полностью аналогична динамической механической характеристике
при заданных переменных напряжениях. И в этом случае также за-
держка характеризуется величиной угла потерь (рис. XII.1).
Так называемый тангенс угла диэлектрических потерь tg S пред-
ставляет собой весьма полезный безразмерный параметр, являю-
щийся мерой отношения диссипированной электрической работы
к запасенной энергии в периодически изменяющемся электрическом
ноле. Произведение диэлектрической проницаемости на тангенс угла
потерь прямо пропорционально энергии, потерянной в диэлектрике,
например в высоковольтном кабеле.
По аналогии с величиной Е, используемой при характеристике
динамических механических свойств материала (см. гл. IX), е*
можно представить в комплексной форме:
е* = е' —ie" . (XII.6)
где е' — действительная часть (диэлектрическая проницаемость); е* — мнимая
часть (константа диэлектрического поглощения) диэлектрической проницаемости.
216
Значения е и tg 6 обычно измеряют в широком диапазоне частот—
от 50 Гц до нескольких МГц (см. рис. XII.1).
Комплексная диэлектрическая проницаемость тесно связана с оп-
тическими характеристиками вещества, например показателем пре-
ломления п и коэффициентом поглощения К-.
в* = («*)2
е* = е' — ге’;
(XII.7)
п* = п(1— 1К)
(XII.8)
е' = п2(1— R2)
е*=2л2К
e'/e' = tge = 2^/(l— К2)
(XII.9)
(XII.10)
(XII.11)
СООТНОШЕНИЕ МЕЖДУ ДИЭЛЕКТРИЧЕСКОЙ
ПОЛЯРИЗУЕМОСТЬЮ И ДИНАМИЧЕСКИМИ
МЕХАНИЧЕСКИМИ ХАРАКТЕРИСТИКАМИ
Поляризация полимера в электрическом поле осуществляется вслед-
ствие стремления связанных в единую цепь субъединиц разместиться
так, чтобы их дипольные моменты и сильно поляризованные связи
были ориентированы в направлении внешнего поля. Следовательно,
можно ожидать существование корреляции между динамическим ме-
ханическим поведением и электрическими характеристиками поли-
мера в переменном электрическом поле.
Одно время полагали, что более трудоемкие исследования меха-
нических характеристик можно заменить гораздо более легкими
электрическими. Существуют, вероятно, тесные аналогии между
общей формой температурных зависимостей механических и диэлек-
трических потерь. Однако количественная связь между этими явле-
ниями не столь проста, как предполагалось прежде. Электрические
измерения — полезное дополнение к исследованиям механических
свойств, но отнюдь не замена последних.
СВЯЗЬ МЕЖДУ ДИЭЛЕКТРИЧЕСКОЙ
ПОЛЯРИЗУЕМОСТЬЮ И ОПТИЧЕСКОЙ
ДИСПЕРСИЕЙ
Основные (конечно, упрощенные) закономерности изменения ди-
электрической поляризуемости в зависимости от частоты показаны
на рис. XII.2. При низких частотах суммарная поляризация про-
являет себя полностью. Однако ориентация полярных групп относи-
тельно слаба, и с увеличением частоты ориентация запаздывает.
Когда частота достигает значений порядка 1012 Гц, диполи оказы-
ваются не в состоянии следовать за изменениями поля (Рдип исчезает).
Сохраняется только статистическая ориентация, которая не вносит
вклада в результирующую поляризацию. Из суммарной поляриза-
ции остаются только атомная (Рат) и электронная (РЭп) поляризации.
217
При несколько более высокой частоте растяжение и изгибающие
деформации связей сильно затормаживаются и атомной поляризации
не происходит.
Частота возникновения подобного резонансного эффекта для
атомной поляризации имеет порядок 1013 Гц, таким образом в ин-
фракрасной части спектра возникает дисперсия, и могут наблю-
даться полосы ИК-поглощения. При частоте более 1014 Гц остается
только электронная поляризация.
Наконец, при частоте выше 10ls Гц, которая относится к оптиче-
скому диапазону, начинается запаздывание деформации электронных
Рис. XII.2. Дисперсионные кривые диэлектрической проницаемости и константы
диэлектрического поглощения в областях электронных, инфракрасных и оптиче-
ских частот:
1 — поглощение, обусловленное проводимостью; II — высокочастотная релаксация; III —
сверхвысокочастотная релаксация; IV — инфракрасная; V — оптическая; VI — ультра-
фиолетовая; VII — резонансная дисперсия.
оболочек атомов, вследствие этого появляется поглощение в оптиче-
ской части спектра.
Из рис. XII.2 видно, что поляризация (и показатель преломле-
ния) увеличивается с приближением к резонансной частоте и по-
степенно достигает «слишком» низкого значения (выше этой резо-
нансной частоты). Подобное явное и внезапное изменение поведения
когда-то посчитали аномалией свойств и назвали «аномальной» дис-
персией. С помощью электромагнитной волновой теории было по-
казано, что подобная «аномальная» дисперсия есть именно «нормаль-
ная» дисперсия, и она может объясняться как прямое следствие за-
кономерностей движения ядер и электронов.
218
СТАТИЧЕСКАЯ ЭЛЕКТРИЗАЦИЯ
И ПРОВОДИМОСТЬ
СТАТИЧЕСКАЯ ЭЛЕКТРИЗАЦИЯ
Если полимер сильно прижать к электрически нейтральной поверх-
ности и затем еще больше увеличить контакт, например путем тре-
ния, то полимер после отделения от поверхности окажется заря-
женным положительно или отрицательно. Если же два полимера
потереть друг о друга, а затем разделить, то один из них окажется
Рис. ХП.З. Трибоэлектрический ряд полимеров:
1 — найлон 6,6, (а также шерсть, шелк); 2 — целлюлоза; 3 — ацетат целлюлозы; 4 — поли-
метилметакрилат; 5 — полиацеталь; в — полиэтилентерефталат; 7 — полиакрилонитрил;
8 — поливинилхлорид; 9 — поли-бис-фенолкарбонат; 10 — полихлорзфир (пентон); ц —
поливинилиденхлорид; 12 — поли-2 6-диметилполифениленоксид; 13 — полистирол; 14 —
полиэтилен; 15— полипропилен; 16—политетрафторэтилен; 1—сухое состояние; II —
влажное состояние.
заряженным положительно (т. е. будет играть роль донора электро-
нов), а другой — отрицательно (т. е. будет играть роль акцептора
электронов). Расположение полимеров по порядку, соответствующему
219
их способности принимать или отдавать электроны, позволяет по-
лучить так называемый трибоэлектрический ряд.
Коэн [9] вывел общее правило: если два вещества заряжаются
вследствие тесного контакта друг с другом, то вещество с наиболь-
шей диэлектрической проницаемостью получит положительный за-
ряд, т. е. будет действовать как донор электронов. Данные рис. XII.3
показывают, что это правило выполняется в широких пределах:
последовательность расположения материалов в трибоэлектрическом
ряду оказывается точно такой, как и в ряду по величине их диэлек-
трической проницаемости. Диэлектрическая проницаемость гидро-
фильных полимеров сильно зависит от содержания влаги в связи
с большим влиянием высокой диэлектрической проницаемости воды.
Отсюда ясно, что’эти полимеры (например, шерсть) являются анти-
статиками во влажной среде, но способны накапливать статический
заряд в очень сухом состоянии. Наибольшие наблюдаемые в экспери-
менте заряды (500 эл,- ст. ед/см3) возникают тем не менее из-за пере-
носа относительно небольшого числа заряженных частиц; может ока-
заться достаточным перенос одного электрона на каждые 104 А2
площади, занимаемой сотнями атомов.
Более важно то, что сама электризация обусловлена скоростью
исчезновения заряда. Шашуа [16] показал, что для данной функ-
циональной группы заряд исчезает наиболее быстро, если эта группа
^играет роль заместителя в боковой цепи, а не в цепи главных валент-
ностей макромолекулы. Способность полимера «избирать» заряд,
вероятно, связана со способностью присоединения ионов к его поверх-
ности. Отсюда ясно, как небольшое количество добавки или включе-
ний может изменить способность к избирательности заряда данного
вещества (степень диссоциации ионных включений).
ЭЛЕКТРИЧЕСКАЯ ПРОВОДИМОСТЬ
Электрическое сопротивление большинства полимеров’ крайне вы-
соко, а проводимость, вероятно, обусловлена присутствием ионных
включений, подвижность которых ограничена высокой вязкостью
среды. Проводимость, а также энергия активации процесса проводи-
мости практически не зависят от степени кристалличности полимера.
При использовании полимеров в качестве изоляционных материалов
важными оказываются поверхностное и объемное удельные сопро-
тивления.
Наличие влаги существенно повышает проводимость полимеров.
ПРЕДЕЛЬНЫЕ ЭЛЕКТРИЧЕСКИЕ
ХАРАКТЕРИСТИКИ /
ДИЭЛЕКТРИЧЕСКАЯ ПРОЧНОСТЬ
В процессе увеличения напряжения электрического тока, приложен-
ного к изолятору, достигается такая точка, в которой физическое
разрушение данного диэлектрика обусловит чрезвычайно резкое па-
дение сопротивления. Такое напряжение носит название электриче-
ской прочности диэлектрика. Можно также построить зависимость
220
времени, которое проходит до разрушения, от приложенного напря-
жения. Такие кривые, как правило, состоят из двух различных
областей. Разрушение при малых временах обусловлено неспособ-
ностью электронов проводимости достаточно быстро диссипировать
эту энергию, которую Они получили от приложенного электрического
поля. Разрушение при больших временах обусловлено главным
образом воздействием коронного разряда.
Диэлектрическая прочность в сильной степени зависит от формы
образца, и этот фактор зачастую оказывается намного важнее, чем
изменение структуры молекул.
Между электрической и механической прочностью весьма много
общего. Это связано с тем обстоятельством, что при электрическом
прббое происходит физическая деструкция. В пределах одного тем-
пературного диапазона как модуль упругости, так и диэлектриче-
ская прочность существенно уменьшаются.
ДУГОСТОЙКОСТЬ ,
Поверхность некоторых полимеров, подвергаемых действию электри-
ческого разряда, может обуглиться и стать причиной появления
тока проводимости. Дугостойкость — мера такого свойства ма-
териала — оказывается весьма важным показателем при исполь-
зовании полимеров в качестве электроизоляционного материала, на-
пример в системах зажигания двигателей внутреннего сгорания. К со-
жалению, никакой корреляции этой характеристики с химическим
строением наити пока не удалось
ЛИТЕРАТУРА
1. Birks J. В. and Schulman J. Н.
(Eds.), «Progress in Dielectrics»,
Wiley, New York, 1959.
2. Bottcher C. J. F., «Theory of Elec-
tric Polarization», Elsevier, Am-
sterdam, 1952.
3. Frohlich H., «Theory of Dielec-
trics», Oxford Univ. Press, Ox-
ford, 1949.
4. McCrum N. G., Read В. E. and
Williams G., «Anelastic and Di-
electric Effects.in Polymeric So-
lids», Wiley, New York, 1967. .
5. Wiirstlin F. and Thum H.,
«Struktur und Electrische Eigen-
. schaften»j in H. A. Stuart (Ed.),
«Die Physik der Hochpolymeren»,
Vol. IV. Ch. 8, Springer, Berlin,
1956.
6. Gayler J., Wiggins R. E. and
Arthur J. B., «Static Electricity;
Generation, Measurement and its
Effects on Textiles», N. Carolina
State University, School of Tex-
tiles, Rayleigh, 1965.
7. LoebL.B.,«StaticElectrification»,
Springer, Berlin/New York, 1958.
8. Morton W. E. and HearleJ. W. S.,
«Physical Properties of Textile
Fibers», Chapter 21, The Textile
Institute, Butterworths, London,
1962.
9. Coehn A., Ann. Phys. 64 (1898)
217.
10. Cole R. H. and Cole K. S., J.
Chem. Phys. 9 (1941) 341.
11. Darby J. R., Touchette N. W.
and Sears K., Polymer Eng. Sci.
(1967) 295.
12. Debye P., Phys. Z. 13 (1912) 97.
13. Mosotti O. F., a Mem. Matem.
e Fisica Modema 24 (1850) 11.
14. Muller F. II. and Schmelzer Chr.,
Ergebn. exakt. Naturwiss. 25
(1951) 359.
15. Schmieder K. and Wolf K.,
Kolloid-Z. 127 (1952) 65.
16. Shashoua, J. Polymer Sci. A-l,
1 (1963) 169.
17. Seanor D. A., Adv. Polymer Sci.
4 (1965) 317.
18. Wiirstlin F., Kolloid-Z. 120 (1951)
102.
глава хш. МАГНИТНЫЕ СВОЙСТВА
Основными магнитными свойствами полимеров являются магнитная
восприимчивость и магнитный резонанс. Первое свойство харак-
теризует данный материал в целом, второе связано с особой конфигу-
рацией электронов и ядер внутри этого материала.
Аддитивными мольными свойствами обладают только магнитная
восприимчивость и второй момент ядерного магнитного резонанса.
МАГНИТНАЯ ВОСПРИИМЧИВОСТЬ
Магнитная восприимчивость % определяется как отношение интен-
сивности намагничивания к напряженности магнитного поля. Ве-
щество может быть диамагнитным, парамагнитным или ферромаг-
нитным. В табл. XIII.1 приведены критерии подобного разделения.
Таблица XIII.1. Диамагнитные, парамагнитные и ферромагнитные вещества
Класс Диапазон изме- нения х в ед. С Г СЕ Зависимость от темпера- туры Зависимость от напряженности поля
Диамагнитный Парамагнитный ~10-в 1а-в_ю-з Практически нет Обратная пропорцио- нальность абсолютной Практически нет То же
Ферромагнитный 10—105 температуре Сильная Достигает насы- щения
В органических полимерах ферромагнетизм проявляется только
при наличии включений и примесей. Последние могут полностью
маскировать истинный тип восприимчивости исследуемого вещества.
В дальнейшем будут рассматриваться вещества, не имеющие ферро-
магнетизма, поскольку сами полимеры не являются ферромагнит-
ными. /
Диамагнетизм есть универсальное свойство вещества. Парамаг-
нетизм возникает только в двух классах органических веществ —
в веществах, содержащих металлы переходных групп периодической
222
системы, и в соединениях, обладающих неспаренными электронами
или триплетными состояниями.
Величина %, экстраполированная к бесконечной температуре,
дает значение только диамагнитной части восприимчивости, по-
скольку
Хобщая — Хдиама г Хпарамаг
(XIII.1)
Паскаль исследовал диамагнитные восприимчивости большого числа
соединений и показал, что мольная восприимчивость
Х = Мх
(XIII.2)
может рассматриваться как аддитивная величина. Атомные и груп-
повые вклады в мольную диамагнитную восприимчивость приведены
ниже:
Группа —Xz-10*, ед. СГСЕ Группа — X f 10«, ед. СГСЕ
-СН3 14,04 \N- 5,48
-СН2— 11,36
>СН- 8,68 ** 12,4
| -teN И
—с— 6,0 —ОН 7,28
1 —О— 4,6
=сн2 8,61 . Л
=СН- 5,93 6,45
3,25 \н
//3=0 4,27
^““ароматическая 9,23 —соон 15,8
5,6 —СОО— 13,1
ароматическая о
"ч/тч > 9,7 -c^nh2 14,0
^('-'ароматическая о
=СН 8,30 —С<—NH— 11,9
—F 5,61 6,6 —C^n/ 9,2
—С1 —Вг 20 32 Структурные инкре- менты
—I —S — 44 15 сопряжение г 1 -0,8
—SH 17,68 1 О за группой—С— 1,6
-nh2 11,16 1
-NH— 8,48 О за группой >СН- - 1,3
Количество надежных данных о магнитной восприимчивости по-
лимеров крайне незначительно, поэ!ому невозможно сравнивать рас-
считанные и экспериментальные значения этого параметра.
223
МАГНИТНЫЙ РЕЗОНАНС
Магнитный резонанс возникает в том случае, когда помещенное в по-
стоянное магнитное поле вещество поглощает энергию переменного
магнитного поля вследствие присутствия в данном веществе неболь-
щих парамагнитных частиц. Подобное поглощение всегда обладает
резонансным характером, когда постоянное магнитное поле начинает
изменяться и частота осциллирующего поля поддерживается по-
стоянной. Два типа переходов могут обусловливать появление маг-
нитного резонанса:
а) переходы, связанные с переориентацией магнитного момента
электронов в стационарном магнитном поле; такой эффект известен
под названием электронного парамагнитного резонанса (ЭПР) или
электронного спинового резонанса;
б) переходы, обусловленные переориентацией магнитного мо-
мента ядер в стационарном магнитном поле; этот эффект известен
как ядерный магнитный резонанс (ЯМР).
В одинаковом магнитном поле электронный резонанс возникает
при гораздо более высокой частоте по сравнению с ядерным резонан-
сом, поскольку магнитный момент электрона примерно в 1000 раз
больше магнитного момента протона, хотя они и могут обладать оди-
наковым спиновым квантовым числом,- Электронный спиновый резо-
нанс наблюдается в микроволновом диапазоне частот (приблизи-
тельно 28 000 МГц), ядерный спиновой резонанс — при радиочасто-
тах (10—50 МГц).
При использовании метода обычной поглощательной спектроско-
пии наблюдают взаимодействие между осциллирующим электриче-
ским полем и веществом. Этот эффект приводит к возникновению
перехода между нормальными уровнями энергии системы электри-
чески заряженных диполей. Здесь более подошло бы название «элек-
трическая резонансная спектроскопия».
Магнитная резонансная спектроскопия связана с наблюдением
взаимодействия между осциллирующим магнитным полем и? веще-
ством. Такое взаимодействие приводит к переходу между энергети-
ческими уровнями магнитных диполей, а вырожденность этих уров-
ней обычно снимается путем наложения внешнего стационарного
магнитного поля.
Важное в практическом отношении различие между электриче-
ской и магнитной резонансной спектроскопией заключается в том,
что первый метод позволяет, как правило, наблюдать переходы
в отсутствие внешних полей. В магнитной резонансной спектроско-
пии это невозможно в принципе.
ЯДЕРНЫЙ МАГНИТНЫЙ РЕЗОНАНС
Ядра некоторых элементов обладают суммарным магнитным момен-
том. Квантово-механические расчеты показывают, что для такого
ядерного «магнита» существует ограниченное число устойчивых ориен-
таций во внешнем магнитном поле.
224
Для ядра атома водорода, или протона, существует две ориента-
ции, которые можно представить себе как параллельную и анти-
параллельную направлению приложенного поля. Такие ориентации
обладают различными энергиями, и, как уже говорилось, величина
этой энергии в ядерных переходах такова, что она попадает в радио-
частотную область электромагнитного спектра.
Исследование ЯМР, следовательно, представляет собой раздел
радиочастотной спектроскопии, и определение резонансного сигнала
можно осуществить при помощи телевизионной техники. Рассматри-
ваемое резонансное явление описывается так называемым ларморов-
ским уравнением:
v=KH (XIII.3)
где v — резонансная частота; Н — напряженность приложенного магнитного
ноля; К — постоянная, характерная для данного типа ядер.
Сама величина К определяется уравнением
K — gfi/h
где р — магнитный момент ядра (магнетон); h — постоянная Планка; g — опре-
деляемая экспериментально константа (Так называемый фактор расщепления).
Обычно практикуется изменение («качание») напряженности,
а не частоты магнитного поля. Однако самые последние и наиболее
мощные спектрометры основаны на методе изменения частоты.
Протон представляет собой наиболее важный в практическом
отношении тип магнитного ядра, поэтому подавляющая часть работ
в области ЯМР посвящена исследованиям протонного магнитного
резонанса. Другие методы спектроскопии основаны на использова-
нии свойств электронов, создающих химическую связь, и, следова-
тельно, на свойствах «скелета» молекулы. А протонный магнитный
резонанс имеет дело с «кожей» молекул, которая обычно содержит
атомы водорода.
Чувствительность измерений методом ЯМР такова, что с весьма
высокой точностью можно определить даже очень незначительные
изменения положения резонансной линии, обусловленные окруже-
нием ядер. Все сведения относительно химического и физического
строения, которые могут быть получены методом ЯМР, как раз
и определяются различиями в положениях резонансных линий.
Магнитное поле, действующее на ядра, несколько изменяется
из-за присутствия окружающих ядра электронов (химический сдвиг)
и вследствие взаимодействия между соседними ядрами (спин-спино-
вое сопряжение). В жидкостях, когда молекулы могут свободно
вращаться, эти факторы вызывают расщепление линии, которое на-
блюдается в условиях высокого разрешения (спектроскопия ЯМР
высокого разрешения).
В твердых телах, в которых все ядра занимают более или менее
фиксированные положения, наличие диполь-дипольного взаимодей-
ствия приводит к уширению линии резонансного поглощения (ЯМР-
спектроскопия широких линий).
15 Заказ 699 225
ЯМР-СПЕКТРОСКОПИЯ высокого
РАЗРЕШЕНИЯ
Главные области применения ЯМР в химии определяются тремя
эффектами: химическим сдвигом, спин-спиновым сопряжением и вре-
менной зависимостью измеряемых параметров. Эти явления можно
наблюдать только для образцов, находящихся в газообразном или
жидком состоянии или в растворе.
ХИМИЧЕСКИЙ сдвиг
Как уже отмечалось, химический сдвиг возникает вследствие различ-
ных положений электронов химических связей в непосредственной
близости к протонам в различных условиях химического окруже-
ния. Наиболее существенными являются различия, обусловленные
присутствием разных по электроотрицательности заместителей. Дру-
гие различия обусловлены эффектами перемещения электронов по
кратным связям под влиянием приложенного магнитного поля, пре-
вышающего их собственные поля. Кратные связи, например, обла-
- дают явно выраженной магнитной анизотропией; в ароматических
структурах, обладающих делокализованными электронами, на самом
деле имеются свидетельства возникновения «круговых токов» под
воздействием магнитного поля. Общее влияние химического окру-
жения можно описать с помощью константы экранирования о:
^уядра = ^о(1 ст) Ь4)
Величина «качания» Магнитного поля Д/Г составляет лишь очень
небольшую долю (10-5) полной напряженности поля. Следовательно,
положение резонансных линий различных протонов, определяемое
химическим сдвигом, нельзя регистрировать в абсолютных едини-
цах. Вместо этого в исследуемый образец добавляют Стандартное со-
единение и химический сдвиг определяют по уравнению
H—HR -
д = —ц-----Юв , (ХШ.5)
R
где Н — напряженность магнитного поля, соответствующая резонансной частоте
ядра в исследуемом соединении; HR — напряженность магнитного поля, соот-
ветствующая резонансной частоте того же самого ядра в стандартном соединении.
При исследованиях неводных растворов методом протонного маг-
нитного резонанса наиболее широко используют в качестве стан-
дартного соединения тетраметилсилан (CH3)4Si. Это соединение изо-
тропно как в магнитном, так и электрическом поле, химически
инертно и не образует ассоциатов ни с одним из наиболее распро-
страненных соединений. При использовании в качестве стандарта
тетраметил силана, поглощающего в сильном поле, большинство хи-
мических сдвигов, определенных уравнением (XIII.5), отрицательно.
На основании этого Тирс [17] предложил не приписывать резонанс-
ной линии тетраметилсилана значение 6 = 0 м. д., а прибавлять
226
Группа, содержащая протон Химический сдвиг, выраженный в '£ = 10+6
Группа присоеоинена к или содепжится 0 t" -*-J -2-1 О113456ТЙЗ 10
-сн3 ICHjfeSi
АЛКиЛ- г
- C=N
2С=О
5С = СС
Арал -
Галоген-
xSaO
-NC г
-он г
- о- ]
- NO2 и - NO3 1
-сн2- В цепи J
в кольце
>с=о
5 С=СС
Арил - [
Галоген- 1
-NC I
- О- 1
-NOz-
(с\ сн- ес/ Алкил- 1
Z с=о
5 С=СС
Арил- г
-N< J 1г
-О-
Галоген-
Я СНОд
HCS В нс=с- ]
НгС= - H;C=q<
нс = н-с=сн
н-с=Сн -
н-с=сс 1
Циклоокта тетраен т
(сн)„ бензол
Арал -
пуриоин
-NHj лнкипимины
Арипамины
Амиды
'nh Алкиламины
Арипамины г
Амиды
Алканоламины г
Циклические амины
-он Вддд
манигиарирооамные СЛиргЛЫ
ОалигидрироОанные спирты ]
Орто-замещенные фенолы
Арцгие фенолы
-соон Алкилкислоты
Арилкиспоты
’С'Н Алкилальдегиды г
Арилальдегиды
нсоо- Эфиры муравьиной кислоты
HCONC Формамиды
- SH Меркаптаны L
!йофенолы 1
zjShti СцпыроноОые кислоты а
Стандарты-
Рже. Х1П.1. Химические сдвиги основных функциональных групп.
15*
произвольно к этой величине -f-Ю м. д. и, тем самым, сделать боль-
шинство химических сдвигов положительными:
т = ЮЧ-СГ (м. д.)
На рис. XIII. 1 приводится сводка химических сдвигов основных
функциональных групп. Следует отметить, что величины химических
сдвигов до известной степени зависят от природы растворителя.
НЕПРЯМОЕ СПИН-СПИНОВОЕ
СОПРЯЖЕНИЕ
Резонансные сигналы довольно часто обладают тонкой структурой
с характерным типом расщепления, обусловленным спин-спиновыми
взаимодействиями ядра с соседними ядрами. Такое расщепление
соответствует несколько различным энергетическим уровням ядер,
спины которых параллельны и антипараллельны спинам соседних
ядер, и в ряде случаев может привести к существенному усложнению
спектра ЯМР. Такой тип спин-спинового сопряжения осуществляется
главным образом за счет электронов, образующих химические связи.
ЭФФЕКТЫ, ЗАВИСЯЩИЕ ОТ ВРЕМЕНИ
На характер сигналов ЯМР зачастую влияют эффекты, зависящие
от времени, например конформационные изменения. Скорость таких
изменений равна (или даже больше) обратной величине разности
между частотами переходов. Это означает, что методом ЯМР можно
исследовать и кинетические явления, особенно, если можно изменять
температуру исследуемого образца.
Можно регистрировать спектры ЯМР высокого разрешения рас-
творов полимеров с помощью широкого набора растворителей, при-
годных для исследования ЯМР, иногда и при повышенных темпера-
турах. Вязкость раствора не всегда влияет на ширину линии в спек-
тре ЯМР. Так, ЯМР-спектроскопию высокого разрешения можно
применить с успехом для определения химического строения, конфи-
гурации (тактичности) и конформации гомополимеров и сополимеров.
Интенсивность сигнала ЯМР пропорциональна числу поглоща-
ющих ядер, поэтому спектроскопию ЯМР высокого разрешения
можно применять как количественный метод, например при опреде-
лении функциональных групп в полимерах.
Точность анализов полимеров с помощью спектроскопии ЯМР
высокого разрешения резко возросла в связи с появлением спектро-
метров ЯМР на 100 и 220 МГц, снабженных электронно-вычислитель-
ными устройствами, которые проводят автоматическое усреднение
и выделение сигнала над уровнем шумов.
Обзорные работы по исследованиям полимеров методом спектро-
скопии ЯМР высокого разрешения опубликованы, например, Бовеем
[11, 12] — одним из создателей этой области исследований.
228
ЯМР-СПЕКТРОСКОПИЯ
ШИРОКИХ ЛИНИЙ
Твердые тела характеризуются спектрами с широкими линиями. Дело
в том, что локальные поля, возникающие при взаимодействиях ядер-
ных магнитных диполей, вносят существенный вклад в суммарное
поле, действующее на ядро в твердом веществе. Мерой такого непо-
средственного спин-спинового взаимодействия является время спин-
спиновой релаксации Т2. Это время гораздо меньше в твердых со-
единениях по сравнению с жидкостями, что и является причиной
появления более широких линий (шириной в несколько гаусс).
В этом случае информацию относительно взаимного положения со-
седних ядер дает огибающая линия поглощения.
Уширение резонансной полосы можно охарактеризовать шири-
ной линии на половине ее высоты (АЯ1/2) или вторым моментом
кривой (52). Второй момент определяется формулой
+J (Я—Я0)2/(Я) dH ,
= ------------ (ХШ.6)
f dH
— co
где Но — величина магнитного поля при резонансе; / (Я) — форма кривой в пере-
менном магнитном поле при постоянной частоте.
Упомянутое выше время спин-спиновой релаксации Т2 корре-
лирует с полушириной линйи:
11
<хш-7)
Наряду с описанным спин-спиновым взаимодействием существуют
взаимодействия между спинами и окружающей средой, т. е. так
называемое спин-решеточное взаимодействие. Специфической кон-
стантой перехода в этом случае служит время спин-решеточной
релаксации 7\. Величина Т2 практически не зависит от темпера-
туры, тогда как зависимость 7\ от температуры выражена достаточно
сильно. Это обстоятельство позволяет изучать два описанных про-
цесса раздельно.
Ричардс с соавторами [15] измерили ширину линий при низких
температурах (90 К) и показали, что второй момент S2 сильно зави-
сел от расстояния между атомами водорода. Но атомы водорода
в ароматическом кольце расположены дальше друг от друга, чем
в алифатических группах, поэтому такое изменение ширины линий
можно связать с изменением соотношения между ароматическими
и алифатическими компонентами в исследуемом веществе.
229
Ричардс получил аддитивные групповые вклады во второй момент
С помощью резонансных спектров ряда модельных соединений. Эти
вклады приведены ниже:
Вклад на атом
водорода во второй
Группа момент, Гс
СН ароматическая 9,7
(СН алифатическая) (10)
СНз алифатическая 27,5
СНз алифатическая 10,0
СНз пери-положение 22,4
Изменение ширины линии в зависимости от температуры может
дать полезные сведения относительно температур переходов в твер-
дом состоянии.
Разные типы переходов характеризуются различными энергиями
активации, поэтому ширина линии иногда уменьшается при измене-
нии температуры не непрерывным образом. При характеристических
температурах переходов время спин-решеточной релаксации TY до-
стигает минимума. Очень часто для понимания структурных или
конформационных особенностей весьма ценным оказывается сравни-
тельное исследование, проводимое, с. одной стороны, методом ЯМР
для измерения температур переходов и, с. другой стороны, методами
динамических механических и диэлектрических измерений.
Можно полагать, что возможности спектроскопии ЯМР широких
линий в отношении оценки молекулярных движений в полимерах
резко возрастут при распространении недавно разработанных усо-
вершенствований, например предложенных МакКоллом [13] (ядер-
' пая магнитная релаксация во вращающемся поле). Применение спек-
троскопии ЯМР широких линий для исследования полимеров рас-
смотрены в обзоре Шлихтера [161 — одного из пионеров этой об-
ласти исследований.
Другое важное применение спектроскопии ЯМР широких линий
заключается в определении степени кристалличности. Спектры не-
которых полимеров можно графическим способом разделить на ши-
рокие и узкие компоненты. Этим способом Уилсон и Пейк [18] опре-
делили кристалличность полиэтилена и политетрафторэтилена.
В итоге можно сказать, что измерение спектра ядерного магнит-
ного резонанса представляет собой чрезвычайно точный и весьма
полезный метод, позволяющий идентифицировать любое соединение
с магнитными ядрами.
ЭЛЕКТРОННЫЙ ПАРАМАГНИТНЫЙ
РЕЗОНАНС
Этот тип резонанса, часто называемый электронным спиновым резо-
нансом, обычно наблюдается в парамагнитных соединениях, и осо-
бенно в веществах с нечетным числом электронов. Энергия неспарен-
ного электрона зависит от того, как ориентирован его магнитный
230
момент по отношению к магнитному полю Н: параллельно или анти-
параллельно.
Разность энергий между этими двумя положениями определяется
формулой
= (ХШ.8)
где g — коэффициент спектроскопического расщепления (его значение равно
примерно 2); 0Б — магнетон Бора.
Вследствие того, что они обладают своим магнитным моментом,
неспаренные электроны могут взаимодействовать с электромагнит-
ным полем, если частота этого поля удовлетворяет условию резо^
нанса:
v = —— Н=К'Н . (XIII.9)
Результатом такого взаимодействия опять является суммарное по-
глощение электромагнитной энергии.
Обычно частоту поддерживают постоянной, а изменяют напря-
женность поля Н. Изменения поля обусловливают возникновение
полосы конечной ширины; полоса характеризуется вторым момен-
том S2 или шириной на половине высоты АЯ\/2. Одна из причин
возникновения линии конечной ширины — диполь-дипольное взаи-
модействие в данной системе спинов. Характеристика перехода
«порядок — беспорядок», обусловленного таким взаимодействием,
дается временем спин-спиновой релаксации Т2, которое связано
с шириной линии на половине высоты. Для линии лорентцевского
типа эта связь описывается соотношением
,h 1 1
7'2=лгрдя / х'дя/2 (хш.ю)
Наряду со спин-спиновым взаимодействием в этом случае имеется
также взаимодействие между спинами и-их окружением (так назы-
ваемое спин-решеточное взаимодействие). Характерная константа
перехода порядок — беспорядок для этого взаимодействия — это
время спин-решеточной релаксации Тх. Последняя величина сильно
зависит от температуры, тогда как Т2 практически от температуры
не зависит.
В растворе линия снова становится достаточно узкой и там не
наблюдается сверхтонкого расщепления, обусловленного взаимодей-
ствием неспаренного спина с магнитными ядрами радикала (например,
водорода, фтора, азота).
Таким образом, электронный парамагнитный резонанс является
важным средством определения и структурного анализа радикалов
в полимерных системах (в процессе их образования при окислении,
облучении, пиролизе и механическом разрушении образцов). На осно-
вании характеристик спектров ЭПР можно сделать заключение от-
носительно химической природы радикалов. Для исследования
короткоживущих радикалов созданы проточные системы спектромет-
ров ЭПР. В стеклообразных полимерах свободные радикалы могут
существовать весьма длительное время.
ЛИТЕРАТУРА
1. Andrew Е. Л., «Nuclear Magnetic
Resonance», Cambridge Univ.
Press, Cambridge, 1956.
2. Becker E. D,, «High Resolution
NMR; Theory and Chemical Ap-
plications», Academic Press, New
York, 1969.
3. Bhacca N. S., Johnson J. L.
and Shoolery J. N., «NMR Spectra
Catalog», Varian Associates, Palo
Alto, Cal. 1962.
4. Bible В. H., «Interpretation of
NMR Spectra; an Empirical Ap-
proach», Plenum Press, New York,
1965.
5. Bible R. H., «Guide to the NMR
Empirical Method, a Wor-
kbook», Plenum Press, New York,
1965.
6. Bovey F. A., «Nuclear Magnetic
Resonance Spectroscopy», Acade-
mic Press, New'York, 1969.
7. Emsley J. W., Feeney J. and.
Sutcliffe L. H., «High Resolution
Nuclear Magnetic Resonance
Spectroscopy», Vol. 1—11, Perga-
mon Press, Oxford, 1965.
8. Assenheim H. M., «Introduction
to Electron Spin Resonance»,
Hilger and Watts, London,
1966.
9. Ayscough P. B., «Electron Spin
Resonance in Chemistry», Met-
huen, London, 1967.
10. Ingram D. J. E., «Free Radicals
as Studied by Electron Spin
Resonance», Butterworth, Lon-
don, 1958.
11. Bovey F. A. and Tiers G. V. D.,
Fortschr. Hochpolym. Forsch. 3
(1963) 139.
12. Bovey F. A., Pure and Appl.
Chem. 12 (1966) 525.
13. McCall D. W., Douglass D. C.
and Falcone D. S., J. Phys. Chem.
71 (1967) 998.
14. Pascal P., Ann. Chem. et Phys.
(8) 16 (1909) 531; (8) 25 (1912)
289; (8) 29 (1913) 218; Bull.
Soc. Chim. France (4) 11 (1912)
636; Rev. Gen. Sci. 34 (1923) 388.
15. Richards R. E. et al: New-
man P. C., Pratt L. and Ric-
hard R. E., Nature 175 (1955)
645; Bell C. L. M., Richards R. E.
and Yorke R. W., Brennstoff
Chem. 39 (1958) 30.
16. Slichter W. P., Fortschr. Hochpo-
lym. Forsch. 1 (1958) 35; J.
Chem. Ed. 47 (1970) 193.
17. Tiers G. V. D., J. Phys. Chem.
62 (1958) 1151.
18. Wilson C. W. and Раке G. E.,
J. Polymer Sci. 10 (1953) 503,
ЧАСТЬ
ЧЕТВЕРТАЯ
ЯВЛЕНИЯ ПЕРЕНОСА
В ПОЛИМЕРАХ
глава xiv ПЕРЕНОС ТЕПЛОВОЙ ЭНЕРГИИ *
В данной главе показано, что с помощью аддитивных величин можно
рассчитать теплопроводность аморфных полимеров и полимерных
расплавов. Здесь аддитивными величинами являются функция Рао,
мольная теплоемкость и мольный объем. Кроме того, эмпирические
правила позволяют рассчитывать теплопроводность кристаллических
и полукристаллических полимеров.
Скорость переноса тепла в полимеры и через полимеры играет
весьма существенную роль. Теплопроводность хорошего теплоизоля-
ционного материала должна быть низкой. Но в процессах пере-
работки полимеров они должны нагреваться до температуры пере-
работки и охлаждаться до температуры окружающей среды в тече-
ние разумно короткого периода времени.
ТЕПЛОПРОВОДНОСТЬ
Не существует теории, которой можно было бы воспользоваться для
точного предсказания величин теплопроводности полимерных рас-
плавов или твердых полимеров. Подавляющее большинство пред-
ложенных теоретических или полуэмпирических выражений осно-
вано на схеме Дебая [3] для теплопроводности. Эта схема приводит
к выражению
X = AcopuA (XIV.1)
где cD — удельная теплоемкость; р — плотность; и — скорость прохождения
упругих волн (скорость звука); L — средняя длина свободного пробега; А —
постоянная порядка 1.
Кардос [11], а позже и Сакиадис с Коутсом [15] предложили
аналогичное уравнение ♦
X CppuL (XIV.2)
где L — расстояние между молекулами в «соседних изотермических слоях».
* Теплофизические свойства полимеров подробно рассмотрены в моногра-
фии; Новиченок Л. Н., Шульман 3. П. Теплофизические свойства
полимеров. Минск, «Наука и техника», 1971. 120 с. — Прим. ред.
233
Большинство теорий объясняют явление теплопроводности (в рас-
плавах и в аморфных твердых полимерах) на основе так называемой
«фононной» модели. Процесс теплопроводности, как предполагают,
связан с переносом энергии от слоя к слою квантованными порциями
со скоростью звука; при этом количество перенесенной энергии счи-
тают пропорциональным плотности и теплоемкости. Здесь не проис-
ходит переноса молекул.
В кристаллических твердых телах, а следовательно, и в высоко-
кристаллических твердых полимерах процесс теплопроводности уси-
ливается согласованным действием молекул.
АМОРФНЫЕ ПОЛИМЕРЫ И ПОЛИМЕРНЫЕ
РАСПЛАВЫ
На рис. XIV. 1 показана типичная зависимость теплопроводности от
температуры для аморфных полимеров и полимерных расплавов.
Кривая проходит через довольно плоский максимум при Tg, затем
постепенно и медленно понижается
в сторону, соответствующую жид-
кому состоянию. На этом же рисунке
показаны углы наклонов темпера-
турных зависимостей теплоемкости
при постоянных давлении, плот-
ности и скорости звука. Такие кри-
вые в соответствии с формулами
(XIV.1) и (XIV. 2) являются состав-
ными элементами кривой зависи-
мости теплоемкости от температуры.
Очевидно, что перемножение ср,
р и и позволяет получить ожидае-
мое изменение А. Если принять, что
A.L в (XIV.1) или L в (XIV.2) прак-
тически постоянны и не зависят от
температуры, можно ожидать пря-
мую пропорциональную зависимость
между коэффициентом температуро-
проводности (1/срр) и скоростью звука
и. Методы расчета и были рассмот-
рены в гл. IX. Тогда приняв, что
/ U \з г 3 (1—у) ТЛ
(XIV.3)
ИгСрод
Рис. XIV.1. Теплопроводность % можно получить:
аморфных полимеров и ее со-
ставляющие. к z, / V Г 3(1-у)
срр ~ \ V J L 14-V J
В табл. XIV.1 приведены соответствующие данные для комнат-
ной температуры, опубликованные Хеллвйджем [9], Айерманном
[5—8] и Кнаппе [12]. В определенных пределах (среднее отклонение
234
Таблица XIV. 1. Теплопроводности аморфных полимеров
Полимер и о 6 -2 •* о о •-, • «в «г к ср, кал/г - °C р, г/см* о «Р* ь Л/Л О 6
Полипропилен (атакти- ческий) 4,2 0,53 0,85 9,3 2 716 54,8 0,49 0,56
Полиизобутилен (3,0) 0,47 0,86 7,5 3 657 56,0 0,49 0,42*
Полиизопрен 3,7 0,45 0,91 9,0 4151 55,5 0,49 0,52
Полистирол 3,1 0,30 1,05 9,9 12,1 5 400 54,5 0,33 0,50
Поливинилхлорид 4,0 0,24 1,38 2 415 53,2 0,33 0,66
Полихлоропрен 4,6 0,38 1,24 9,8 3 850 53,9 0,49 0,62
Поливинилкарбазол 3,0 0,3 1.2 8,3 8 532 52,8 0,33 0,46
Полиметилметакрилат 4,6 0,33 1,17 11,9 4 877 57,0 0,33 0,52
Поливинилацетат 4,2 0,36 1,18 9,9 3 936 53,9 0,4 0,60
Полиоксиэтилен 4,9 0,48 1,13 1,335 9,0 2138 54,9 0,49 0,54
Полиэтилентерефталат 5,3 0,27 14,7 7 786 53,8 0,33 0,73
Полибисфенолкарбонат 5,7 0,28 1,20 16,9. 11 442 54,0 0,33 0,88
Полидиметилсилоксан 4,0 0,38 0,975 10,8 4180 вреднее 55,0 . / 0,49 0,64 0,60
Коэффициенты перевода в 1 кал/(г- °C) = 4,19- 10» Дж/(кг- К); * Значение неточное. СИ: 1 г/см3 1 кал/(с-см = 103 кг/м8. °С) = 4,19- 10* Дж/(с • м-К);
±0,08-10" 8) величина С сохраняет постоянное значение (0,60 • 10" 8).
На рис. XIV.2 представлена обобщенная температурная зависи-
мость ка-
[* Близкий по смыслу к способу, использованному в этой главе, способ
обобщения экспериментальных данных по температурной зависимости коэффи-
циента теплопроводности X различных полимеров был предложен Б. А. Арутю-
новым (см. «Механика полимеров», 1972, № 5, с. 912—913), который'построил
— г/г3
Рис. XIV.2. Обобщенная характеристика теплопроводности аморфных полимеров:
□ — силиконовый каучук; — полииэобутилен; О — натуральный каучук; 0 — поли-
пропилен; V — политрифторхлорэтилен; х — полиэтилентерефталат; ▲ — поливинил-
хлорид; • — полиметилметакрилат; А — полибисфенолкарбонат; о — поливинилкарба-
зол.
23 5
зависимость X/lg от 777g (1g — значение Л при температуре стеклования 7g)
, для той же области значений аргумента, что и на рис. XIV.2. Согласно цитиру-
емой работе, кроме полимеров, данные о которых собраны на рис. XIV.2, обоб-
щенной зависимостью 1/lg от 777g описываются также экспериментальные зави-
симости, определенные X (Т) для полистирола, полиэтилена и поливинилхлорида,
содержащего до 40% пластификатора. Однако достаточно хорошее соответствие
между данными, относящимися к различным полимерам, наблюдалось только
при T/Tg<< 1. В области, лежащей выше температуры стеклования, наблюдался
заметный разброс точек, полученных для разных полимеров. Причина такого
хода зависимостей 1/lg от T/Tgwpa T/Tg > 1 связана с различием относитель-
ного свободного объема / полимеров разной химической природы, т. е. отклоне-
ниями от универсального хода функции / (777g) для разных полимерных рядов
при Т > Tg. Если же в этой температурной области в качестве аргумента для
температурной зависимости l/lg принять величину/, нормированную по значению
относительного свбодного объема при температуре стеклования /g, то в этом
случае точки, относящиеся к различным полимерам, в координатах 1/1» — ///г
очень хорошо укладываются на одну общую кривую. Таким образом, обобщен-
ное представление зависимостей 1 (Т) достигается тогда, когда 1/lg рассматри-
вается при Т <• Tg в виде функции отношения T[Tg и при Т > Tg как функция
отношения — Прим. ред.
ВЫСОКОКРИСТАЛЛИЧЕСКИЕ ПОЛИМЕРЫ
Кристаллические полимеры обладают гораздо более высокой тепло-
проводностью. В качестве примера на рис. XIV.3 приведены экспе-
риментальные данные о теплопроводности полиэтилена, измеренные
Рис. XIV.3. Зависимость коэффициента теплопроводности полиэтилена от
степени кристалличности, по данным Айерманна [5].
236
Методом экстраполяции Айерманн 16, 7J получил следующее
соотношение для полимеров типа полиэтилена и полиоксиметилена
со «100%-ной» кристалличностью:
% «ь С/Т (кал/см) (XIV.5
где С — постоянная' порядка 0,5.
Таким образом, значение теплопроводности для этих высокоупо-
рядоченных полимеров оказалось равным примерно 17 X
X Ю“4 кал/см-К; для тех же полимеров в аморфном состоянии теп-
лоемкость составляет примерно 4-10’4 кал/см-К.
Для приблизительных оценок теплопроводности высокоупорядо-
ченных полимеров можно пользоваться соотношением
(-&) <XIVe>
С помощью этого соотношения можно вычислить теплопроводность
полностью кристаллических полимеров при комнатной температуре,
если известно отношение рс/ра.
Для обычных кристаллизующихся полимеров Айерманн [6] по-
лучил следующее соотношение:
— 1 = — 1) (XIV.7)
\ Ра J .
ЧАСТИЧНО КРИСТАЛЛИЧЕСКИЕ
ПОЛИМЕРЫ
Айерманн получил также формулы для теплопроводности частично
кристаллических полимеров, для которых известны степень кристал-
личности хс и отношение У.с!^а. Графики его формул представлены
на рис. XIV.4.
Пример 1
Оценить теплопроводность аморфного цолиметилмет акрилата
а) при комнатной температуре и б) при 200 еС.
Решение
а. Вычисления основываются на использовании формулы (XIV.4) при ср =
= 0,33 (гл. V), р = 1,17 (гл. IV), V = 0,33 (гл. IX) и М = 100,1.
Сначала рассчитывается функция Рао (гл. IX):
Vi
1 (-СН2-) 895
/ । А
1 —С— 40
\ I /
2 (—СН3) 2 722
1 (-СОО-) 1220
4 877
Следовательно
U /7=4877/(100,1/1,17) = 57,0
237
и
А = 0,6 • 0,33 • 1,17 • (57,0)3 (3.0,67/1,ЗЗ)1 /2 • 10“8 =
= 0,232 • 1,85 • 105.1,225 • Ю'» = 5,2 • 10'4 кал/(см • К)
Это значение соответствует данным Айерманна (4,6-10-4).
_____ _ Р- Рс
* J-С ~ -----------—
Pt “ Ра
Рис. XIV.4. Соотношение между коэффициентом теп-
лопроводности, степенью кристалличности и плот-
ностью.
б. При помощи данных рис. XVI.2 находится А при Tg. Поскольку Tg =
= 387 К, при комнатной температуре
Т/Тg = 298/387 = 0,77 и A (f)/A (Tg) =0,96
Следовательно
А (Те) = (5,2/0,96) • 10'4 = 5,4 • 10'4
Далее, при Т = 200 °C = 473 К
Т]Т& =473/387 = 1,22
и по данным рис. XIV. 2
А (473)/А (Г£) = 0,95 .
Следовательно, при 200 °C А = 5,1 «IO-4.
Пример 2
Теплопроводность аморфного полиэтилентерефталата при комнатной тем-
пературе составляет 5,3-10~4 йал/см-К. Рассчитать теплопроводность полукри-
сталлического полиэтилентерефталата со степенью кристалличности 0,40.
238
Решение
Поскольку ас = 1,465 и ра = 1,335, можно рассчитать с помощью фор-
мулы (XIV. 7):
= Ха И + 5,8 (рс/ра -1)1 = 5,3 • 10-4 [1 + 5,8 (1,465/1,335 -1)] =
= 5,3-1,58-10-4
По рис. XIV.4 находится, что А. (хс = 0,4). При Хс1Ха— 1>58 и хс = 0,4
Х-ка 1041-5,3
j--ji— (==> 0,36 или n nr—= 0,36
Лс — Лд о,иЭ — 0,0 -
X = (1,1+5,3) 10-4 = 6,4 • 10-4
Полученный результат удовлетворительно согласуется с экспериментальным
значением 6,5-10-4.
ЛИТЕРАТУРА
1. Bridgman Р. W., Proc. Am. Acad.
Arts and Sci. 59 (1923) 154.
2. Car slaw H. S. and Jaeger J. C.,
«Conduction of Heat in Solids»
(2nd Ed.), Clarendon Press, Ox-
ford, 1959.
3. Debye P., «Vortrage uber die
kinetische Theorie der Materie
und der Electrizitat», (Wolfskehl-
Vortrage), Berlin, 1954.
4. Stuart H. A., «Die Physik der
Hochpolymere», Vol. 1, Springer,
Berlin, 1952.
5. Eiermann K., Kolloid-Z. 180
(1962) 163; 198 (1964) 5, 96; 199
(1964) 63, 125; 201 (1965) 3.
6. Eiermann K., Kunststoffe 51
(1961) 512; 55 (1965) 335.
7. Eiermann K. and Hellwege К. H.,
J. Polymer Sci. 57 (1962) 99.
8. Eiermann K., Hellwege К. H.
and Knappe W., Kolloid-Z. 171
(1961) 134.
9. Hellwege К. H., Henning J. and
Knappe W., Kolloid-Z. 186 (1962) .
29; 188 (1963) 121.
10. Henning J., Kolloid-Z. 188 (1963)
159; 196 (1964) 136.
11. Kardos A., Forsch. Geb. Inge-
nieurw. 5B (1934) 14.
12. Knappe W., Z. angew. Physik,
12 (1960) 508; Kunststoffe 51(1961)
707; 55 (1965) 776.
13. Lohe P., Kolloid-Z. 203 (1965) 115;
204 (1965) 7; 205 (1965) 1.
14. Natta G. and Baccareda M.,
Makromol. Chem. 4 (1949) 135.
15. Sakiadis В. C. and Coates J.,
A. I. Ch. E. J. 1 (1955) 275;
2 (1956) 88.
ГЛАВА XV
ВЯЗКОСТЬ РАСТВОРОВ ПОЛИМЕРОВ
При постоянной температуре вязкость раствора полимера зависит
главным образом от химической природы, среднего молекулярного
веса и концентрации полимера, а также от природы растворителя.
На практике весьма полезным параметром оказывается так называе-
мое предельное число вязкости, или характеристическая вязкость.
Последнюю можно определить, если известны химическая структура
и средний молекулярный вес полимера, а также природа раствори-
теля.
ВВЕДЕНИЕ
Вязкость полимерного раствора зависит от линейных размеров поли-
мерной молекулы. Поэтому вязкость раствора линейных неразвет-
вленных полимеров эмпирически связана с молекулярным весом.
Следовательно, измерения вязкости составляют весьма ценный спо-
соб получения молекулярных характеристик полимеров.
Измерения вязкости раствора (т]) обычно осуществляют * путем
сравнения времени истечения t заданного объема раствора полимера
через капилляр с соответствующим временем истечения раствори-
теля t0 (вязкость растворителя т|0).
Вязкости растворов выражают с помощью различных величин.
Ниже приведены названия этих величин и уравнения, описывающие
их:
Принятое название
Относительная вязкость
Удельная вязкость
Приведенная вязкость
Собственная вязкость
Характеристическая
вязкость
Название, предложенное
Международным Союзом
чистой и прикладной химии
Коэффициент вязкости
Обозначение и определяю-
щее уравнение
Потн = П/По t/t0
Число вязкости
Логарифмическое число
вязкости
Предельное число вяз-
кости (показатель Штау-
дингера)
^УД — Лотн — 1 —
__ П —По _ t — to
По t0
т1пр = 'Пуд/с
'Псоб ~ 1и Цотн/^
[Ц] = Г^Д_) =
1 11 \_ с /с=.о
__ ( InTjoTH \
\ с А=0
Имеется в виду разбавленный раствор. — Прим. ред.
240
ПРЕДЕЛЬНОЕ ЧИСЛО ВЯЗКОСТИ
(ХАРАКТЕРИСТИЧЕСКАЯ ВЯЗКОСТЬ)
УРАВНЕНИЕ МАРКА — ХАУВИНКА
Вязкость раствора полимера зависит от природы полимера и раство-
рителя, концентрации полимера в растворе, среднего молекуляр-
ного веса полимера (молекулярно-весового распределения), темпера-
туры и, до некоторой степени, от скорости сдвига.
Влияние концентрации на результаты измерений можно исклю-
чить путем определения важной величины — предельного числа
вязкости, или характеристической вязкости:
, , Л Ло .. Луд 1ПЛОТН /VVM
(t|) = hm-— = lim—— = 11Ш -------- (XV.1)
С->0 Т|0с с C-+Q с
Величина [rj] определяется при комнатной температуре (если не
указана другая температура) и при «нулевой» скорости сдвига.
Концентрация с выражается в граммах растворенного вещества на
см3 раствора или, чаще, в граммах растворенного вещества на 100 см3
раствора. Предельное число вязкости, следовательно, выражается
в см3/г или (см3/г)-100.
Характеристическая вязкость [т] ] зависит только от природы
полимера и растворителя и от молекулярного веса полимера. Широко
применяемое на практике соотношение между характеристической
вязкостью и молекулярным весом М, полученное эмпирическим
путем, известно под названием уравнения Марка — Хаувинка:
[т\] = КМа (XV.2)
В этом уравнении эмпирические постоянные К и а зависят от
природы растворителя.
На практике молекулярный вес описывается каким-либо распре-
делением. Следовательно, оценка констант К и а должна основы-
ваться на измерениях [т| ] фракций полимера с узкими молекулярно-
весовыми распределениями. Если измерения проводят на нефрак-
ционированном образце полимера, то с помощью уравнения Марка—
Хаувинка получают так называемый средневязкостный молекуляр-
ный вес Ми.
Установлено, что для линейных гибких полимеров в 0-условиях
(т. е. при температуре Флори), когда парциальная мольная свобод-
ная энергия равна нулю, уравнение Марка — Хаувинка принимает
вид
{П]е = Х0М^2 (XV. 3)
где Х0 — константа, значение которой практически не зависит от растворителя.
Уравнение (XV. 3) можно получить теоретическим путем
для растворов полимерных молекул, принимающих конформации
1 fi Hawao ЙОО it.
статистических клубков при условии отсутствия взаимодействия
между растворителем и полимерными молекулами.
Уравнение Марка — Хаувинка (XV.2) нельзя получить с по-
мощью какой-либо простой модели раствора. Ряд теоретических ис-
следований позволяет сделать заключение, что для обычных поли-
мерных растворов показатель степени а больше значения 0,5, соответ-
ствующего 0-растворам. (Подробнее см. специальную литературу *.)
В литературе также приводится большое количество значений К
и а для различных систем полимер — растворитель. Зачастую для
одной и той же системы разные исследователи получают совер-
шенно другие комбинации значений К и а. Ё настоящее время нет
абсолютно надежного метода предсказания значений К и а для
данной системы полимер — растворитель. Однако некоторые- эмпи-
рические соотношения получены Ван Кревеленом и Хофтицером
[32]. Эти соотношения будут рассмотрены ниже, хотя и в несколько
видоизмененной форме по сравнению с описанной в оригинальных
работах.
Прежде всего следует заметить, что между константами К и а
существует определенная связь. Если для данного полимера срав-
нивают значения К и а, определенные в различных растворителях,
то обнаруживается отчетливая корреляция этих величин. Макси-
мальными значениями К всегда оказываются Кд, полученные длй
идеальных растворителей (в них а = 0,5), а с увеличением а значе-
ния К уменьшаются. Такое соотношение можно приблизительно
описать следующим уравнением:
-lg ~^ = С (а-0,5) (XV.4)
где С — постоянная, изменяющаяся от 3,0 до 4,5 в зависимости от природы
полимера.
Однако, если использовать значение С = 3,7, то это не приведет
к существенной ошибке в сравнении с разбросом имеющихся экспери-
ментальных данных. Сказанное иллюстрирует рис. XV.1, на кото-
ром приведены имеющиеся в литературе экспериментальные данные
по характеристической вязкости растворов полистирола в виде за-
висимости 1g (Кд/К) от а.
Величины К были рассчитаны по уравнению (XV.4) при С =3,7
для примерно 300 экспериментальных точек, исходя из приведенных
в табл. XV.2 экспериментальных значений а и Кд. Средняя раз-
ность между экспериментальными и рассчитанными значениями К
составила приблизительно 30%. В табл. XV.2 приведены экспери-
ментальные и рассчитанные значения К для ряда систем полимер —
растворитель.
* См. также монографию: Цветков В. Н., Эскин В. Е., Френ-
кель С. Я. Структура макромолекул в растворах. М., «Наука», 1964. 720 с.;
Маравец Г. Макромолекулы в растворе. М., «Мир», 1967. 398 с. — Прим,
ред.
242
Если формулу (XV.4) подставить в (XV.2), то можно избавиться
от постоянной К'.
[х\] = 10,1Кд{м1ЪООО)а (XV.5)
В полученном соотношении величина — постоянная для данного
полимера, а влияние природы растворителя на характеристическую
вязкость описывается только постоянной а.
----а
Рис. ХУЛ. Зависимость KJK от а для полистирола.
ОЦЕНКА ЗНАЧЕНИЙ Kg
Ван Кревелен и Хофтицер [32] предложили способ оценки Kq. Мето-
дами статистической механики полимерных цепей можно показать,
что К$ зависит от некоторых характеристических параметров цеп-
ных сегментов:
К е = р/* ]
0 L(Af/Z) /2 J
(XV.6)
где выражается в 100 см3/г; 6 = 2 61—аДДитивная величина (мольная функ-
I
ция характеристической вязкости), полученная по групповым вкладам; Z —
число атомов в цепи главных валентностей, отнесенное к одному мономерному
звену.
Под цепью главных валентностей понимают собственно поли-
мерную цепочку без боковых групп и ответвлений. Например, все
243
виниловые полимеры содержат два атома в каждом мономерном звене
в цепи главных валентностей. Вклад в величину Z п-фениленовой
или п-циклогексильной группы в основной цепи составляет 4. В ори-
гинальной работе был использован несколько отличный метод рас-
чета Z.
В табл. XV.1 приведены групповые вклады 6, для вычисления
2 6г- Значения К(), рассчитанные по уравнению (XV.6), оказываются,
L
Таблица XV. 1. Групповые вклады (ц
Бифункциональные группы вг10> Монофункцио- нальные группы •10» в главной или боковой цепи
в главной цепи в боковой цепи
1 ^с— 1 50 5 —н 0
-с- 65 10 -СН3 5
1!
О
1 1 *
—С=С—цис 75 \ х. ) ’ 35
Г—\ **
транс 100
) с 50
—— 250 30
***
—\ /—^ис (140) (20) —\О/ 75
©
транс (170) (25) —соон 10
-о— 35 5 -SO3H (40)
1 —N— 60 (Ю) —CN (40)
1 —Si— (70) —ОН (20)
1 -nh2 (40)
ci**** 20
вг**** (Ю)
* Также и другие моноциклические системы (приближенно).
** Также и другие бициклические системы (приближенно).
*** Также и другие трициклические системы (приближенно).
**** Только в главной цепи; при замещении в боковой цепи эффект незначителен.
244
Таблица XV.2. Расчет характеристической вязкости
Полимер Растворитель Кв- Ю‘, дл/г к- ie\ пл/г . а [д] для М = 2,5- 10s
опыт расчет | ОПЫТ - расчет | опыт расчет опыт | 1 расчет
Полииаобу- Диизобутилен 100 112 36 34 0,64 0,70 1,02 1,20
тилен Циклогексан — — 40 17 0,72 0,73 3,06 1,34
Четыреххлористый — — 29 24 0,68 0,70 1,35 1,20
углерод Толуол 87 67 0,56 0,65 0,91 0,99
Бензол — — 83 87 0,53 0,60 0,60 0,82
Полипропи- Циклогексан 150 150 16 12 0,80 0,74 3,29 1,87
лен (атак- Толуол — — 22 22 0,73 0,70 1,88 1,61
тический) Бензол — — 27 25 0,71 0,66 1,82 1,38
Полистирол Метилциклогексан 80 82 76 82 0,50 0,50 0,38 0,41 0,60
(атактиче- Бутилхлорид — — 15 21 0,66 0,60 0,55
ский) Циклогексан — — 82 82 0,50 0,62 0,41 0,64
Этилбензол — — 18 18 0,68 0,74 0,82 1,02
Толуол — — 12 13 0,72 0,75 0,90 1,06
Бензол — . — 11 12 0,73 0,73 0,98 0,98
Метил этилкетон — 29 36 0,60 0,72 0,50 0,95
Хлорбензол — — 7 10 0,75 0,71 0,82 0,91
Диоксан — — 15 16 0,69 0,66 0,79 0,76
Полибута- Циклогексан 170 169 И 16 0,75 0,67 1,24 1,26
диен (цис) Поливинил- Хлорбензол 125 124 71 58 0,59 0,72 1,09 1,43
Полихлоро- Толуол 115 113 50 42 0,62 0,67 1,11 1,08
Поливинил- Гептанон-3 90 77 82 77 0,50 0,50 0,41 0,38
ацетат Толуол — — 108 60 0,53 0,53 0,78 ‘ 0,43
Бензол . — 22 21 0,65 0,62 0,70 0,60
Этилформиат — — 32 21 0,65 0,67 1,03 0,73
Хлорбензол — — 94 46 0,56 0,58 0,99 0,51
Ацетон — — 17 14 0,70 0,73 1,04 0,92
Диоксан — —' 11 10 0,74 0,74 1,11 0,96
Ацетонитрил — — 16 13 0,71 0,62 1,09 0,60
Метанол — — 37 36 0,59 0,50 0,57 0,38
Полиметил- Бутилхлорид 65 69 51 69 0,50 0,50 0,25 0,34
метакрилат Гептанон-3 — — 63 69 0,50 0,50 0,32 0,34
(атактиче- Октанон-3 — — 58 (75) 0,49 0,50 0,26 0,34
ский) Гептанон-4 — — 59 (82) 0,48 0,56 0,23 0,43
Метилизобутират — — 10 16 0,67 0,60 0,41 0,50
Метилметакрилат — — 6,8 11 0,72 0,67 0,52 0,66
Толуол —— — 6,6 9,6 0,73 0,69 0,57 0,71
Бензол — — 6,3 7,6 0,76 0,73 0,79 0,83
Метилэтилкетбн — 6,8 11 0,72 0,74 0,52 0,86
Хлороформ — .— 4,9 4,5 0,82 0,74 1,29 0,86
Тетрахлорэтан — — 12,5 9,6 0,73 0,73 1,08 0,83
Этилендихлорид — — 17 15 0,68 0,73 0,79 0,83
Ацетон — — 7,5 13 0,70 0,72 0,45 0,80
Нитроэтан — — 5,7 8,9 0,74 0,60 0,56 0,60
Ацетонитрил — — 39 69 0,50 0,50 0,20 0,34
245
Продолжение табл. XV.2.
Полимер Растворитель хе-10*- дл/г К-10‘, дл/г а [т|1 для М=2,5- 10*
опыт расчет ОПЫТ расчет опыт расчет ОПЫТ расчет
Полиэтил- Метилэтилкетон 48 64 2,8 5,4 0,79 0,74 0,51 0,80
метакрилат Изопропанол — — 48 64 0,50 0,50 0,24 0,32
Полиоктил- Метилэтилкетон 30 52 4,5 10 0,69 0,65 0,24 0,46
метакрилат Полиакри- Диметилацетамид 225 251 29 27 0,76 0,63 3,6 2,05
лонитрил Диметилсульф- — — 32 30 0,75 0,75 3,55 3,26
ОКСИД Диметилформамид 18 23 0,78 0,75 0,73 2,81 3,26
Бутиролактон — — 34 35 0,73 2,97 3,02
Этиленкарбонат — — 30 42 0,71 0,56 0,61 2,25 1,90
Полиметак- Ацетон 220 196 , 96 118 0,68 1,00 1,94
рилонитрил Диметилформамид — — 306 196 0,50 0,67 1,53 1,87
Полидиме- Толуол 90 84 20 21 0,66 0,72 0,72 0,97
тилсилоксан Бензол — — 12 18 0,68 0,70 0,56 0,90
Метилэтилкетон — 81 84 0,50 0,70 0,41 0,90
Этилиодид — — 70 84 0,50 0,68 0,35 0,83
вообще говоря, в пределах точности имеющихся литературных
данных. В табл. XV.2 приведены некоторые рассчитанные значе-
ния Kq.
ОЦЕНКА ЗНАЧЕНИЙ а
Основная трудность при оценке характеристической вязкости для
данной системы полимер — растворитель заключается в предска-
зании значения показателя степени а. Ван Кревелен и Хофтицер
[32] предположили, что а зависит от параметров растворимости поли-
мера и растворителя. В графическом виде эта зависимость
представлена на рис. XV. 2.
Указанный путь довольно часто позволяет получить весьма удо-
влетворительное соответствие между рассчитанными и эксперимен-
тальными значениями а, однако остается ряд отклонений от точного
выполнения описанного соотношения. Как правило, эксперимен-
тальное значение а оказывается ниже предсказанного, если полимер
и растворитель обладают совершенно разным химическим строе-
нием. Очевидно, одних параметров растворимости недостаточно для
предсказания а. Можно добиться некоторого повышения точности
оценки а, если разделить параметр растворимости на ряд величин,
характеризующих ван-дер-ваальсовские силы, дипольные взаимодей-
ствия и образование водородных связей. Такой метод предложил
Хансен. Однако имеющихся данных явно недостаточно для прове-
дения подобного анализа.
246
В табл. XV.2 сравниваются экспериментальные и рассчитанные
значения а для ряда систем полимер — растворитель. Во многих
случаях соответствие оказывается вполне удовлетворительным, но
возникают и резкие расхождения. Предсказанные значения а исполь-
зовали для расчетов характеристической вязкости при М — 2,5-105
с помощью уравнения (XV. 5). В среднем различие между рассчитан-
ными и экспериментально определенными значениями [т] ] соста-
вляет приблизительно 40%.
Для разветвленных полимеров значения а меньше по сравнению
с той же величиной для линейных полимеров такого же химического
состава. Эти данные получили Зимм и Штокмайер [34], Зимм и Килб
[33].
Пример 1
Для характеристической вязкости полистирола в толуоле в литературе при-
водится более 20 разных комбинаций постоянных в уравнении Марка — Хау-
винка. Две из них таковы:
[т]]=4,4.10-Ш°*66 и [г)] = 1,15.10-Ш°-7>
Оценить, какое из этих двух уравнений надежнее.
Решение
а. Оценка К %.
Структурное звено полистирола
М = 104,1
Z=2
247
Из табл. XV. 1 получаются следующие значения групповых вкладов:
Группа -сн2— —СН— 1 Ю 50 50
-о 35
6=135-10-2
С помощью уравнения XV.6 можно найти, что
Кв = [(1,35/2)/(104,1/2)*/2]3 = 8,2 • Ю’4
Полученное значение удовлетворительно согласуется с экспериментальным
/от 6,7 -Ю’4 до 9,0-10-4).
б. Применение уравнения (XV.4).
Для первой комбинации К и а
—ig(K/Ke) = 3,7-0,15 = 0,555; Кв = К Ю0-535 =
= 3,6-4,4-10-4 = 15,8-IO'4
Для второй комбинации К и а
Х0= К-Ю°’814 = 6,5-1,15-10-4= 7,5-10-4
Последнее значение ближе к ожидаемому (Л0 = 8,2-Ю-4).
в. Оценка с помощью параметров растворимости.
Параметр растворимости полистирола 6П= 9,0, толуола 6р = 8,9. Согласно
рис. XV.2, а примерно равно 0,735. Сравнение обоих результатов показывает,
что второй набор значений К и а можно считать более надежным.
КОРРЕЛЯЦИЯ МЕЖДУ К0
И ПАРАМЕТРАМИ ПОЛИМЕРНОГО КЛУБКА
Теоретически К$ связана с некоторыми основными параметрами
растворенного полимерного клубка, что выражается формулой
А’0 = Ф(р/мо)>/ = (XV. 7)
где Р — длина статистически «эффективного» сегмента цепи; Мд — молекуляр-
ный вес сегмента; Ф — универсальная постоянная, равная приблизительно
2,5-1021.
Среди других широко используются следующие параметры поли-
мерного клубка:
радиус вращения в невозмущенном состоянии (2?Go), который
представляет собой среднеквадратичное расстояние от сегментов до
центра масс клубка в невозмущенном состоянии («о)1/з ;
среднеквадратичное расстояние между концами полимерного
клубка в невозмущенном состоянии
248
Перечисленные величины связаны между собой следующим соот-
ношением:
м
6
1 / 2
(XV.8)
Р2
Мо
Комбинация (XV. 7) и (XV.8) дает:
(XV.9)
Радиус вращения изменяется из-за межмолекулярных взаимодейст-
вий в пределах объема, занятого полимерной молекулой, например-
с другими полимерными молекулами и с молекулами растворителя.
Поэтому
BG = aRG0 (XV.10)
где а — так называемый коэффициент набухания клубка.
В качестве первого приближения для оценки а можно восполь-
зоваться соотношением
Подстановка (XV.4) в (XV.11) приводит к формуле
аз & (М/бООО)'’"*7'* (XV.12>
ГИДРОДИНАМИЧЕСКИЙ ОБЪЕМ ПОЛИМЕРОВ
Теория растворов полимеров позволяет получить выражение для
гидродинамического объема полимерного клубка; последний ока-
зывается пропорциональным произведению характеристической вяз-
кости на молекулярный вес. Согласно данным Бенуа с сотр., гидро-
динамический объем представляет собой основную характеристику
размеров макромолекулы, необходимую для получения универсаль-
ной калибровочной кривой при хроматографии на проницаемом геле
(см. гл. II), при использовании которой зависимости 1g (М [т]]) от
элюирующего объема для большого числа разных полимеров описы-
ваются одной кривой.
Отсюда вытекает, что для линейных и разветвленных макромо-
лекул с одинаковым гидродинамическим объемом илц элюирующим
объемом произведения характеристических вязкостей на молекуляр-
ные веса равны друг другу:
Л1лин Илин = -Мразв • [т)]разв (XV.13)
Уравнение (XV. 13) составляет основу метода определения молеку-
лярного веса разветвленных полимеров путем комбинирования дан-
ных вискозиметрии и хроматографии на проницаемом геле.
Методика подобных определений такова. Если для данной ко-
лонки с проницаемым гелем известна универсальная калибровочная
249
кривая (для линейных полимеров), то следующая стадия заклю-
чается в определении элюирующего объема и характеристической
вязкости фракции исследуемого разветвленного полимера. Далее
по универсальной калибровочной кривой отсчитывается произведе-
ние М [т|], соответствующее среднему элюирующему объему этой
фракции разветвленного полимера. Полученное значение следует
разделить на экспериментальную характеристическую вязкость, что
даст молекулярный вес исследуемой фракции разветвленного поли-
мера. С помощью полученного молекулярного веса можно рассчи-
тать характеристическую вязкость линейного полимера по уравне-
нию Марка — Хаувинка.
СООТНОШЕНИЕ МЕЖДУ ПАРАМЕТРАМИ
ДИФФУЗИОННОГО ПЕРЕНОСА
Характеристическая вязкость, коэффициент диффузии- (D) и
константа седиментации («сед=скорость седиментации/ускорение
центробежной силы) полимерного клубка тесно связаны друг
с другом. Все эти величины зависят от радиуса вращения молекулы,
а следовательно, и от ее молекулярного веса:
1^] = КМа; (fe2),/i = 7?G/6 = KhMb
(XV.14)
«сед = КседМ*; D = KDM'd
Показатели степеней связаны вледующими соотношениями:
« = ЗЬ —1=2—3«<=3d—1; b + c^d + c = l; b = d (XV.15) .
Приблизительную оценку постоянных Kh, Ксел и KD можно про-
вести по величине К с помощью следующих выражений:
К ^КГ) ЮИ^сед Ю’12^1 (XV.16)
ВЯЗКОСТЬ РАСТВОРОВ ПОЛИМЕРОВ
ВЯЗКОСТЬ РАЗБАВЛЕННЫХ РАСТВОРОВ
ПОЛИМЕРОВ
Вязкость разбавленных растворов полимеров удовлетворительно
описывается хорошо известным приближенным уравнением
Чуд — 1ч]с 4“ Г*!)2 с2
где А>н — константа Хаггинса.
250
(XV.17)
В литературе приводится большое количество значений Ан для
различных систем полимер — растворитель, но эти значения имеют
большой разброс. Какого-либо заметного влияния температуры или
самой характеристической вязкости на Ан определить не удалось.
Для одной и той же системы полимер — растворитель, вероятно,
существует корреляция между Ан и показателем степени а в урав-
нении Марка — Хаувинка.' Но в связи с сильным разбросом
Таблица XV.3. Константы Хаггинса и значения а
для систем полимер—растворитель
Полимер Растворитель йн а Лн+°
Полиметилметакрилат Толуол 0,43 0,73 1,16
Хлороформ 0,32 0,82 1,14
Бензол 0,35 0,76 1,11
Ацетон 0,48 0,70 1,18
Бутанол 0,40 0,72 1,12
Поливинилацетат Ацетон 0,37 0,70 1,07
Хлорбензол 0,41 0,56 0,97
Хлороформ 0,34 0,74 1,08
Диоксан 0,34 0,74 1,08
Метанол 0,47 0,59 1,06
Толуол 0,50 0,53 1,03
Бензол 0,37 0,65 1,02
Полистирол Толуол 0,37 0,72 1,09
Циклогексан 0,55 0,50 1,05
Бензол 0,36 0,73 1,09
Хлороформ 0,33 0,76 1,09
Бутанон 0,38 0,60 0,98
Этилбензол •- 0,23 0,68 0,91
Декалин 0,60 0,56 1,16
Полибутадиен Бензол 0,49 0,76 1,25
1 Изобутилацетат 1 0,64 , 0,50 1,14
Циклогексан 0,33 0,75 1,08
1 Толуол 1 0,33 0,70 1,03
Среднее _ -"1,08
значений Ан и а подобную корреляции нельзя установить однознач-
но. В общем случае для обычных полимерных растворов с а » 0,7
Константа Хаггинса составляет приблизительно 0,4. В идеальных рас-
творах, согласно данным Сакаи [29], константа Хаггинса изменяется
от 0,5 до 0,64. Но если показатель степени а приближается к единице
(например, растворы найлона 6,6 в муравьиной кислоте), константа
Хаггинса примерно равна 0,1.
Весьма приближенно соотношение между Ан и а можно выразить
следующим образом:
Лн«^1,1—Л (XV.18)
В табл. XV.3 приведены значения Ан и а для ряда систем полимер —
растворитель.
251
ВЯЗКОСТЬ КОНЦЕНТРИРОВАННЫХ ПОЛИМЕРНЫХ
РАСТВОРОВ (КОНЦЕНТРАЦИЯ ПОЛИМЕРА
ВЫШЕ 5 ВЕС. %)*
Характеристики течения концентрированных полимерных растворов
и полимерных расплавов близки. Оба случая важны в практическом
отношении; именно от таких показателей зависят способность к пере-
работке и возможность получения из полимеров разнообразных
изделий.
Для растворов, а также для расплавов полимеров зависимости
вязкости при нулевой скорости сдвига (т]) от молекулярного веса
полимера (М) в двойных логарифмических координатах зачастую
описываются двумя прямыми линиями. Как правило, существует
некая переходная зона, но тем не менее по точке взаимного пересе-
чения двух линейных участков кривой определяется так называе-
мый критический молекулярный вес Мкр. При М Мкр вязкость
примерно пропорциональна М, а при М > Л/Кр вязкость пропор-
циональна МЗА [14].
Оноги с сотр. [28] показали сходное влияние концентрации по-
лимера на вязкость. Кривые логарифмических зависимостей вяз-
кости от молекулярного веса и вязкости от концентрации для данной
системы полимер — растворитель могут быть совмещены путем
сдвига вдоль оси абсцисс; в итоге получаются «сглаженные» обоб-
щенные кривые (рис. XV.3).
Обобщенное уравнение Оноги для зависимости вязкости от мо-
лекулярного веса и концентрации полимера имеет следующий вид:
г] = КсаМ& (XV. 19)
где с — концентрация полимера; К, а и 0 — постоянные.
Для различных полимерных систем значение отношения 0/а из-
меняется от 0,54 до 0,72.
Вязкость при критическом молекулярном весе, по-видимому, не
зависит от концентрации, и соотношение между 7Икр и с принимает
вид
с • Мкра = постоянная 105) (XV.20)
где с выражена в 100 г/см3.
Для молекулярных весов, больших Л/кр
т] = К [С“/3-М]3-4 (XV.21)
где а/0 составляет примерно 1,5.
* Такая оценка области перехода от разбавленных растворов к концентри-
рованным весьма недостоверна, поскольку растворы высокомолекулярных поли-
меров проявляют все свойства концентрированных растворов уже при гораздо
меньшем содержании полимера в растворе. Правильнее условия перехода оцени-
вать по характерному значению безразмерного параметра с [ц] (с и ц должны
быть выражены в одинаковых единицах). Условию перехода от разбавленных
к концентрированным растворам отвечает величина этого параметра, близкая
к 5—6. — Прим. ред.
252
Величина К представляет собой зависящую от температуры ха-
рактеристическую постоянную для данной системы полимер — рас-
творитель. Порядок величины К близок 2,0 • 10-15, если с выражается
в г/см3 и 7] в П.
Экспериментальные данные о вязкости полимерных растворов
известны только для ограниченного числа систем. Следовательно,
пределы применимости уравнения (XV. 21) пока нельзя определить
достаточно точно *.
Недавно Лайонс и Тобольский [23] предложили уравнение за-
висимости вязкости полимерных растворов от концентрации, которое,
Рис. XV.3. Вязкость раствора поливинилацетата в этилбутилкетоне:
а — концентрация приведена к 10%, б — молекулярный вес приведен к 1,16* 10s.
по мнению авторов, справедливо для всего диапазона изменения
концентрации: от весьма разбавленных растворов до практически
чистого полимера. Это уравнение имеет вид
Луд *н [Ш '
с [л] ехР 1 — Ас
(XV.22)
где г]уд— удельная вязкость; [т]] — характеристическая вязкость; с — концен-
трация; Ад— константа Хаггинса; Ъ — постоянная.
Для данной системы полимер — растворитель [т]] и можно
определить обычным способом, и только постоянная Ъ может быть
рассчитана по объемной вязкости полимера при с = р (плотность
данного полимера).
* В работах А. А. Т а г е р, Е. В. Д р е в а л я и их сотрудников полу-
чен огромный объем экспериментальных данных по вязкостным свойствам разно-
образных полимерных систем в широкой области составов. Эти данные обобщены
в докторской диссертации В. Е. Д р е в а л я [4 д].— Прим. ред.
253
Лайонс и Тобольский с успехом применили уравнение (XV. 22)
для систем нолиоксипропилен — бензол и полиоксипропилен — ме-
тил циклогексан. Приложимость уравнения ограничивалась, однако
молекулярным весом 2000; этот молекулярный вес лежит ниже кри
тического значения Л/кр.
В литературе нет сведений о дальнейшей проверке применимости
этого уравнения *.
Пример 2
Оценить вязкость раствора полистирола в толуоле при 25 °C, если в растворе
содержится 0,1 вес. % полимера с молекулярным весом М = 3-10®.
Решение
Удельную вязкость т)удможно оценить с помощью уравнения (XV.17).
а. Рассчитать характеристическую вязкость [tjJ.
Воспользовавшись уравнением из примера 1
Гп] = 1,15 - 10-4ДГ0,72
можно получить при М = 3-10® [t]J = 5,3-100 см3/г.
б. Рассчитать концентрацию е.
Плотность разбавленного раствора полистирола в толуоле при 25 °C соста-
вляет 0,87 г/см3; концентрация 0,1 вес. % соответствует 0,1 г на 100 г раствора
или с = 0,087 г/100 см3. С помощью уравнения (XV.17) вычисляется т]уд:
Пуд = № + ГП]3 с2 = 5.3 0,087 + 0,40 • 5,32.0,0872 = 0,46 + 0,08 = 0,54
Вязкость толуола при 25 °C Т)е = 0,0056 П, тогда
г] = 1,54-0,0056 = 0,0086 П
Пример 3
Оценить вязкость раствора полистирола в толуоле при 40 °C, если в рас-
творе содержится 10 вес. % полимера с молекулярным весом М = З-'Ю®.
Решение
По уравнению (XV.21) •-
T] = 2,0-10-iSc5.iM3,4
Плотность 10%-ного раствора полистирола в толуоле при 40° С составляв
0,87 г/см3. Следовательно, с = 0,1-0,87 = 0,087 г/см3 и q = 2-10~15-4-10’6 X
X 1,1-1022= 90 П.
Этот результат согласуется с данными Оноги [28].
[* Константа Хаггинса &н, определяющая реологическую эффективность
появления межмолекулярных взаимодействий в полимерных растворах, может
'быть связана с термодинамическими характеристиками раствора и с химической
природой обоих компонентов — полимера и растворителя. Так, в работах Сакаи
[1 д] и Багданецкого [2 д] было показано существование корреляции между
Ади параметром разбухания макромолекулярного клубка а в выбранном раство-
рителе. Дальнейшее развитие этих представлений содержится в работах [3 д],
в которых вместо используется константа Мартина к^. Величина k'N предста-
вляет собой параметр, входящий в уравнение для концентрационной зависимости
* Так как область применимости этого уравнения для различных систем
полимер — растворитель действительно не установлена, вряд ли следует рассма-
тривать его как «универсальное». Примеры ограниченности его применимости
можно найти, например, в работе: Ганди К.,Уильямс М. В кн.: Вязко-
упругая релаксация в полимерах. Составитель М. Шен. Пер. в англ, под ред.
А. Я. Малкина. М., «Мир», 1974, с. 214—244. — Прим. ред.
254
вязкости при ее представлении экспоненциальной фэриулэй та па (
если принять, что в ней b = 0. Из этой же формулы видно, что Ам совпадает
с kH при малых концентрациях (когда с [т]] < 1). Ниже используется модифи-
цированная константа Мартина ZcM, равная А^/2,3.
Величина а, с которой сопоставляется ZcM (рис. 3, д), включает в себя как
энтропийные, так и энергетические эффекты межмолекулярных взаимодействий
в растворе. Влияние этих двух определяющих факторов можно разделить, что
позволяет найти общую форму зависимости /см от термодинамических характери-
стик растворов. Для этого, согласно [3 д], в качестве безразмернойхарактери-
Рис. 3, д. Соотношение между констан-
той Мартина и параметром разбухания
для полидиметилсилоксана (J), полии-
зобутилена (2), полистирола (<3) и поли-
винилацетата (4). Показаны также
экспериментальные данные Богданец-
кого для полистирола (5).
стики энергии взаимодействий в растворе используется параметр у, вычисляемый
через разность параметров растворимости полимера 62 и растворителя вх!
Х=Р1(б2-д1)2/ЯГ
где Vj — мольный объем растворителя; R — универсальная газовая постоянная;
Т — абсолютная температура.
Вопросу о вычислении 61Г § 2, их физическом смысле и о значении этих пара-
метров для получения растворов полимеров посвящена гл. XIII настоящей
книги. Использование содержащегося в ней материала позволяет без особых
трудностей находить значения %.
Рассмотрение обширных экспериментальных данных показало [3 д], что
для различных полимеров зависимость Ам от % оказывается гиперболической,
т. е. линейной в координатах — % (рис. 4, д). Различие в положениях линий
на этом рйсунке, отвечающих разным полимерам, связано с ролью энтропийного
фактора. Два параметра могут быть взяты из рис. 4, д: точка пересечения каждой
прямой с осью ординат (при % = 0) и тангенс угла наклона прямой. Первая
из этих величин, обозначаемая как отвечает значению константы Мартина
для раствора, в котором исключены энергетические взаимодействия,-поскольку
на оси ординат % = 0, или^ = б2. Вторая величина у = — dk^/dx характери-
зует степень влияния энергетических взаимодействий в растворе на рост кон-
станты А:м, т. е. на интенсивность повышения вязкости при возрастании концен-
трации полимера в растворе: чем выше /см (или &н), тем сильнее, при прочих
равных условиях, повышается вязкость по мере добавления полимера в раствор.
Величина 0 связана с так называемыми невозмущенными размерами
макромолекулярного клубка и сопоставляется с отношением (ЛдМ^)0'6, где
255
Л| — среднеквадратичное расстояние между концами цепи в тета-условиях,
а Ад — расстояние между концами свободно-сочлененной цени, т. е. отношение
(Ag/Ад)015 может трактоваться как мера внутренней жесткости макромолекулы,
обусловливающей возможность перехода между ее различными конформациями.
С увеличением (Л-§/Ад)0,5, т. е. с ростом скелетной жесткости цепи, 0 возра-
стает (Ajj 0 линейно убывает, как это показано на рис. 5, д). Зависимость Ам 0
от (Ag/Ад)0-5 описывает корреляцию структурного фактора и энтропийного
вклада в величину константы Мартина (или Хаггинса) при исключенных энер-
гетических эффектах.
Величина -у, отражающая вклад энергетических эффектов, кооррелирует
с плотностью когезионной энергии полимера 6а, а именно, с увеличением энер-
Рис. 4, д. Соотношение между константой Мартина и параметром 1 для ацетата
целлюлозы (Z), полистирола (2), полиизобутилена (<3) и полиметилсилоксана (4).
гии межмолекулярного взаимодействия полимера возрастает чувствительность
константы Ам к изменению / (рис. 6, д), причем зависимость у от б2 — линей-
ная, и у = 0 при S2 = 7,15.
Таким образом, приведенные выше экспериментальные данные свидетель-
ствуют о том, что константа Мартина, отражающая интенсивность возрастания
вязкости по мере увеличения концентрации полимера в растворе, зависит от сле-
дующих величин: разности параметров растворимости полимера и растворителя
и мольного объема растворителя (оба эти фактора суммируются величиной %),
скелетной жесткости цепи (Ag/Ад)0-6 и плотности когезионной энергии полимера
5 2. Этот результат может быть выражен следующим аналитическим выражением:
*м1 —W, o~YX
где
Ам, o =a-09Ao)o’BA
256
и
у = 7,15+Лд2
или (см. рис. 4, д):
Лм = ["-(М I ^о)0’8 Ь + (7.15*й2) х]"1
Здесь а, Ь, и к — постоянные коэффициенты, смысл и значения которых хорошо
видны из рис. 5, д и 6, д.
Последняя формула позволяет дать весьма надежные оценки величины
константы Лм(следовательно, и йн)по известным значениям61,62, ^и^д/Тф0'8.
Все эти величины могут быть найдены в соответствующих справочных руковод-
ствах или вычислены на основании материала, приводимого в настоящей книге.
Рис. 5гд. Связь между константой
к о и скелетной жесткостью
макромолекулярной цепи для по-
лидиметилсилоксана (7), полиизо-
бутилена (2), полистирола (3) и
ацетата* целлюлозы (4).
Рис. 6, д. Зависимость параметра у от
величины 62 Для ацетата целлюлозы (7),
полистирола (2), полиизобутилена (3) и
полидиметилсилоксана (4).
Для обобщенного представления экспериментальных данных о концентра-
ционной зависимости вязкости растворов принципиально важен метод «приве-
денных» (или нормированных) переменных, позволяющий использовать обобщен-
ные, или безразмерные, характеристики вязкости ц и объемной концентрации с.
Пусть характеристическая вязкость данного полимера в выбранном растворителе
Составляет [т|]. Тогда, согласно [3 д1, в качестве безразмерного показателя
вязкостных свойств раствора можно использовать отношение ц = Цр-ра/с [ц],
где т]р-ра—удельная вязкость раствора, выражаемая как (т)—Лр-ля)/Яр-ля
(Яр-ля — вязкость растворителя). Роль безразмерной концентрации может
играть произведение (с [т}J)- Согласно Дебаю, в области (с [rj) 1 эта величина
имеет физический смысл степени заполнения объема раствора полимером. В об-
ласти более высоких значений этого параметра такой ясный физический смысл
отсутствует, но величина (с Гл 1) остается характеристикой безразмерной кон-
центрации (подробное рассмотрение вопроса о корректности использования
17 Заказ 699 257
параметра (с [г] J), сопоставление его с другими мерами нормированной концентра-
ции, а также ссылки на оригинальные работы и их обсуждение см. в [3 д—5 д]).
Экспериментальные данные о вязкостных свойствах полимеров различных
молекулярных весов в координатах ц — с [г]J образуют единую кривую. При-
мер такой кривой показан на рис. 7, д для растворов полибутадиенов различных
молекулярных весов в трех различных растворителях [6 д]. Другие примеры
для различных полимеров можно найти в [3 д, 5 д), а в [3 д] также подробно
обсуждены ограничения рассмотренного метода обобщения экспериментальных
данных о вязкостных свойствах растворов полимеров.
Методика использования изложенного метода состоит в следующем: для
одного полимера любого молекулярного веса измеряется зависимость т] (с)
и представляется в координатах т) (с [т| ]), тогда для оценки вязкости раствора
полимера другого молекулярного веса любой концентрации достаточно измерить
величину ' [т] ] предельно разбавленного раствора; после этого для требуемого
значения с“величина rj находится по построенной обобщенной зависимости i)
от (с [Т]]).
Дальнейшее развитие метода построения обобщенных характеристик вяз-
костных свойств растворов некоторого полимера состоит в сведении зависимостей
т; (с [ц ]), отвечающих разным растворителям, в единую кривую. Согласно [3 д],
это достигается нормированием аргумента по константе /см, равной тангенсу
угла наклона начальной прямолинейной области зависимости 1g q от (с [ц]).
Существование такой области хорошо видно из рис. 7, д. Как уже говорилось,
величина &м равна Лщ/2,3, где — константа Мартина, отвечающая предста-
влению зависимости ц (с) в области низких концентраций в экспоненциальной
_ Ащс [В]
форме, а именно т) = е
Использование аргумента концентрационных зависимостей вязкости рас-
творов в-различных растворителях в форме (Аме [т)]) позволяет построить обоб-
щенную зависимость вязкостных свойств растворов данного полимера для всех
растворителей и широкой области молекулярных весов. Пример такой зависи-
мости показан на рис. 8, д [6 д].
Методика использования обобщенных зависимостей т; ([т]]) для оценки
вязкости раствора аналогична описанной выше (см. рис. 7, д). Однако положе-
ние здесь еще более упрощается по сравнению с рис. 7, д, поскольку использо-
вание рис. 8, д требует получения зависимости ц (с) во всем диапазоне составов
только для одной системы полимер — растворитель, а для любого другого рас-
твора, представляющего интерес, оказывается достаточным измерить зависимость
вязкости от концентрации в узкой области разбавленных растворов. Такие изме-
рения дают значения двух характерных параметров, определяющих свойства
растворов полимеров некоторого химического строения — [ц] и Ам. Дальней-
шие оценки вязкости растворов этого-полимера без труда выполняются по обоб-
щенной зависимости т] (Амс [г; ]), которая строится по экспериментальным точкам,
полученным для растворов образца с одним молекулярным весом в одном раство-
рителе.
Таким образом, изложенный метод построения обобщенных характеристик
концентрационной зависимости вязкости растворов полимеров в различных рас-
творителях основывается на измерении двух параметров ([rj ] и А:м), определяемых
для области разбавленных растворов. Первый из них связан со свойствами инди-
видуальной полимерной цепи, второй — отражает влияние на вязкость простей-
ших межсегментальных взаимодействий, приводящих к росту вязкости разба-
вленных растворов, в которых еще не образовалась сплошная трехмерная сетка
флуктуационных контактов (зацеплений), по мере увеличения концентрации.
При этом существенно, что возможен способ оценки Ам без прямых измерений,
как это описано в первой части этого раздела..
Несмотря на то, что указанные параметры характеризуют реологические
свойства разбавленных растворов, они сохраняют свое значение определяющих
258
факторов для всей области составов. Это обусловлено тем, что структура поли-
мерного раствора, от которой зависит его вязкость, закладывается еще в области
разбавленных растворов. При переходе к концентрированным растворам основ
ным структурным эффектом, по-видимому, является равномерное заполнени
Рис. 7, д. Безразмерные концентрационные зависимости вязкости растворов
полибутадиенов различных молекулярных весов в метилнафталине (7), толуоле
(2) и дигептйпфталате (3).
Рис. 8, д. Обобщенная характеристика вязкостных свойств растворов полибута'
диенов в трех различных растворителях. Обозначения те же, что на предыдущем
рисунке.
полимером всего объема раствора и переход от случайных (статистических)
контактов отдельных макромолекул к единой однородной структурной «сетке»,
в которую вовлекаются все макромолекулы. Но и в этом случае характеристики
отдельных цепей и их взаимодействий, которые измерялись в области разбавлен-
ных растворов, сохраняют свое значение.
Зависимости ц(А:ме [т] ]) индивидуальны для полимеров различной химиче-
ской природы. Сводка известных графиков функций такого рода представлена
17* " 259
на рис. 9, д (по [3 д]), который может использоваться для оценки вязкости рас-
творов ряда полимеров.
Форма зависимости г] (/смс [ц ]), по-видимому, чувствительна к тому, нахо-
дится ли полимер в области выше или ниже температуры стеклования Tg. Если
Т Тg, то во всем диапазоне составов система полимер — растворитель суще-
Рис. 9, д. Обобщенные кон-
центрационные зависимости
вязкости различных поли-
меров: полистирола (У), по-
ливинилацетата (2), ацетата
целлюлозы (<?), полиизобу-
тилена (4), полидиметилси-
локсана (5) и полибута-
диена (6).
ствует в виде вязкой жидкости. Этому отвечает выпуклая кривая на рис. 9, д,
охватывающая всю область составов. Если Т < Tg, то в области разбавленных
растворов система полимер — растворитель представляет собой вязкую жидкость,
а в области концентрированных растворов или пластифицированных полимеров
система полцмер — растворитель находится в стеклообразном состоянии.
Рис. 10, д. Схема, иллюстрирующая ха-
рактер концентрационной зависимости
температуры стеклования и переход рас-
твора по достижении концентрации с*
из текучего (/) в стеклообразное (II) со-
стояние при температуре эксперимента
Уэксп*
Поэтому в некоторой области концентраций имеет место переход из текучего
в стеклообразное состояние. Это схематично показано на рис. 10, д. В области
I при с < с* раствор существует в виде текучей жидкости, а в области II
при с > с* — в виде стекла; с* зависит от температуры эксперимента Тэксп-
При приближении состава к с* со стороны разбавленных растворов наблю-
дается очень резкий рост вязкости, завершающийся при с* стеклованием рас-
твора. На рис. 9, д этому отвечают вогнутые кривые со значением вязкости,
очень резко возрастающим в узком диапазоне значений аргумента.
Таким образом, форма кривой на рис. 9, д определяется соотношением
между температурой эксперимента и Тг полимера. Однако в настоящее время
260
8 тот вывод носит качественный характер, и каких-либо количественных корре-
ляций между формой зависимости t] (к^с [ц]) и природой полимерной цепи
пока не установлено.*] — Прим. ред.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1 д. Sakai Т., 3. Polymer Sci.,
1968, v. А-2, № 6, р. 1535.
2 Д. Bohdanecky, Coll. Czech. Chem.
Commun., 1970, v. 35, p. 1972.
3 Д. Древалъ В. E., Малкин А. Я.,
Ботвинник Г. О., J. Polymer
Sci., Polymer Phys. Ed., 1973,
v. 11, № 6, p. 1055—1076; Дре-
валъ В. E. и др., «Механика
ЛИТЕРАТУРА
1. Bueche F., «Physical Properties
of Polymers», Wiley, New York,
1962.
2. Casassa E. F., Ann. Rev. Chem.
11 (1960) 477.-
3. Eirich F. R., «Rheology», Vol. I,
Academic Press, New York, 1956.
4. Flory P. J., «Principles of Poly-
mer Chemistry», Cornell Univ.
Press, Ithaca N. Y., 1953.
5. Tanford C., «Physical Chemistry
of Macromolecules», Wiley, New
York, 1961.
6. Tampa H., «Polymer Solutions»,
Acabemfc Press, New York, 1956
7. Vollmert B., «Grundrisz der Mak-
romolecularen Chemie», Springer,
Berlin, 1962.
8. Budtov V. P., Int. Chem. Eng.
7 (1967) 684.
9. Bueche F., J. Chem. Phys. 40
(1964) 484.
10. Cooper W., Eaves D. E. and
Vaughan G., J. Polymer Sci. 59
(1962) 241.
11. Danusso F., Moraglio G. and
Gianotti G., J. Polymer. Sci.
51 (1961) 475.
12. Flory P. J. and Fox T. G., J.
Am. Chem. Soc. 73 (1951) 1904.
13. Flory R. J. and Krigbaum W. R.
J. Chem. Phys. 18 (1950) 1086.
14. Fox T. G., Gratch S. and Les-
ha# k S., in Eirich, «Rheology»,
Vol. I, Academic Press, New York,
1956, p. 446.
15. Fujlshige S., J. Polymer Sci, A3
(1965) 3219.
16. Gillespie T., J. Polymer Sci. C3
(1963) 31.
17. Houwink R., J. Pract. Chem.
157 (1941) 15.
полимеров», 1972, № 6,
c. 1110—1117.
4 д. Древалъ В. E. Докторская дис-
сертация. M., Физ.-хим. ин-т
им. Л. Я. Карпова, 1974.
5 д. Малкин А. Я., Rheol. Acta,
1973, v. 12, № 3/4, р. 486—495.
6 д. Виноградов Г. В. и др., Europ.
Polymer J., 1973, v. 9, № И,
p. 1231—1249.
18. Hoffmann M., Makromol. Chem.
24 (1957) 222.
19. Huggins M. L., J. Am. Chem.
Soc. 64 (1942) 2716.
20. Kapur S. L. and Gundiah S.,;J.
Polymer Sci. 26 (1957) 89.
21. Kurata M. and Stockmayer W. H.,
Fortschr. Hochpolymer. Forsch.
3 (1963) 196.
22. Kurata M. and Yamakawa H.,
J, Chem. Phys. 29 (1958) 311.
23. Lyons P. F. and Tobolsky A. V.,
Polymer Eng. Sci. 10 (1970) 1.
24. Matsuda H., Aonuna H. and
Kuroiwa S., J. Appl. Polymer
Sci. 14 (1970) 335. ,
25. Moore W. R. and Fort R. J.,
I. Polymer Sci. Al (1963) 929.
26. Moore W. R. and Murphy M.,
J. Polymer Sci. 56 (1962) 519.
27. Norisuje T., Kawahafe K. and
Fufita H., Polymer Letters 6
(1968) 849.
28. Onogi S., Kimura S., Kato T.,
Masuda T. and Miyanaga H.,
J. Polymer Sci C15 (1966) 381.
29. Sakai T., Macromolecules 3
(1970) 96.
30. Stockmayer W. H. and Fixman M.,
J. Polymer Sci CI (1963) 137.
31. Streeten D. J. and Boyer R. F.,
Ind. Eng. Chem. 43 (1951) 1790.
32. Van Krevelen D. W. and Hofty-
zer P. J., 3. Appl. Polymer Sci.
10 (1966) 1331; 11 (1967) 1409:
11 (1967) 2189.
33. Zimm В. H. and Kilb R. W.t
J. Polymer Sci. 37 (1959) 19.
34. Zimm В. H, and Stockmayer W. H
3. Chem. Phys. 17 (1949) 1301.
261
ГЛАВА XVI
ВЯЗКОСТЬ РАСПЛАВОВ ПОЛИМЕРОВ
Вязкость полимеров зависит от среднего молекулярного веса, тем-
пературы, напряжения или скорости сдвига и гидростатического да-
вления. Полуэмпирические соотношения для таких зависимостей
позволяют оценить вязкость расплава. Даже если ничего, кроме
строения полимера, не известно, все же можно получить разумное
первое приближение для вязкости его расплава.
ПОНЯТИЕ О ВЯЗКОСТИ РАСПЛАВА
Вязкость расплава tj определяется как отношение касательного на-
пряжения т к градиенту скорости (dv/dx) или к скорости сдвига у:
Эр
т—т] -^- = T1Y (XVI.1)
Если вязкость не зависит от скорости сдвига, то такая жидкость
называется ньютоновской или, как еще говорят, эта жидкость обла-'
дает «идеальным» характером течения. По своему поведению рас-
плавы полимеров в общем случае не являются ньютоновскими жидко-
стями и вязкость расплавов полимеров понижается при увеличении
скорости сдвига.
Совершенно ясно, что для получения истинного показателя вяз-
костных свойств величину т] необходимо измерять при весьма низ-
ких скоростях сдвига и проводить экстраполяцию эксперименталь-
ных данных к нулевой скорости сдвига.
Вязкость в сильной степени зависит от температуры и молекуляр-
ного веса, поэтому значение вязкости может изменяться от 10"1 до
более чём 1012 П. Столь широкий диапазон нельзя охватить каким-
либо одним методом измерений. Ниже указаны примерные диапа-
зоны методов измерения вязкости, получившие широкое применение:
Метод Диапазон
измерения, П
Стеклянный капилляр
Продавливание через капилляр
Параллельные пластинки (пластометр)
Вращающийся цилиндр
Вискозиметр типа «конус—плоскость»
Ползучесть при -растяжении
10-2—103
100-108
104—10»
100—1012
103—1012
105—>1012
262
ВЛИЯНИЕ МОЛЕКУЛЯРНОГО ВЕСА
НА ВЯЗКОСТЬ
Наиболее важным структурным параметром, определяющим вязкость,
является молекулярный вес или длина цепи. При больших молеку-
лярных весах вязкости расплавов полимеров при постоянной тем-
пературе могут описываться простым выражением
1g П=3,41g Ми,+А (XVI.2)
где А — эмпирическая постоянная, характеризующая данный полимер и не
зависящая от средневесового молекулярного веса.
Впервые это выражение получили Фокс и Флори [28]. Существо-
вание эмпирического соотношения столь общего характера позво-
ляет предположить, что существует и теоретическое соотношение
подобного характера. Последнее получил Вики [16] на основании
упрощенной, но принятой в настоящее время теории течения поли-
меров.
Уравнение (XVI.2) справедливо для области больших молекуляр-
ных весов. Для низких молекулярных весов это уравнение прини-
мает вид
1ёг\ = п1ёМш+В (XVI.3)
где п 1; В — постоянная.
Еще одно эмпирическое выражение для низких молекулярных
весов имеет вид
lgr\ = Ci(Mw)n+Ci (XVI.4)
где п V2; Ci и Сг — постоянные.
Первоначально предполагали, что существует довольно резкий
переход от (XVI.2) к (XVI.3) при некотором критическом значении
молекулярного веса Л/кр. Этот переход связывали с началом про-
цесса взаимного перепутывания цепей, молекулярный вес которых
выше Л/кр. Последующие исследования показали, что переход от
(XVI.3) и (XVI.2) осуществляется постепенно. Тем не менее вели-
чину Мкр можно определять как такой молекулярный вес, при кото-
ром пересекаются линейные участки двух предельных областей за-
висимости Т| (М).
Значение критического молекулярного веса сильно зависит от
строения полимера и может изменяться от 2 000 до 60 000. Фокс,
Грач и Лошек [29] показали, что критическое значение числа атомов
в цепи главных валентностей полимерной молекулы Хкр подвержено
гораздо меньшим изменениям. Однако диапазон изменения ZKp все
еще велик: от 300 до 800.
263
Фокс и Аллен . [27] связали Л/кр с молекулярными размерами
цепи, т. е. с безразмерной группой RGt /Mv, где RGts — радиус
вращения в невозмущенном состоянии (см. гл. XV); v — удельный
объем.
Еще лучшую корреляцию можно получить, однако, с величиной
Kq — коэффициентом в уравнении для характеристической вяз-
кости в идеальном растворителе (см. гл. XV). Приближенно такая
корреляция описывается соотношением
~0,14
(XVI.5)
В табл. XVI.1 приведены значения Мкр, ZKp и произведения К$ X
X М^р для ряда полимеров.
Таблица XVI. 1. Критические молекулярные веса некоторых полимеров
Полимер Мкр ZKP к0- Ю«
Полиэтилен 3500 250 2380 0,14
Полиизобутилен 16 000 570 . 1120 0,14
Полистирол 40 000 770 820 0,16
Полибутадиен 6000 330 2000 0,15
Полиизопрен 10 000 450 1600 0,16
Поливинилацетат 25 000 580 770 0,12
Полиметилметакрилат 30 000 600 690 0,12
Полиоксиэтилен 3400 240 1610 0,09
Полидекаметиленсукцинат 4000 250 1950 0,12
Полидекаметиленадипинат 4500 , 285 1990 0,13
Полидекаметиленсебацинат 4000 260 2070 0,13
Полинеопентилсукцинат 16 500 800 1500 0,19
Поли-е-капролактам 5000 310 2360' 0,17
Полидиметилсилоксан 30 000 810 840 0,15
Если вязкость полимера с критическим молекулярным весом обо-
значить т]кр, то формулы XVI.2 и XVI.3 можно переписать так:
18Г] = 1§т]кр + 3,418(Мш/МКр) при Mw> Мкр
lgn=lgnKp — lg(MKp/Mw) при Mw < Мкр
(XVI.6)
ВЛИЯНИЕ ТЕМПЕРАТУРЫ НА ВЯЗКОСТЬ
Влияние температуры на вязкость расплавов полимеров, не-
видимому, носит очень сложный характер. Поэтому в настоящее
время не существует адекватного математического описания влияния
температуры на вязкость в широком интервале температур. Для
температур, достаточно превышающих точку плавления данного
264
полимера, температурная зависимость вязкости описывается про-
стым экспоненциальным соотношением
г] = Л ехр(-ЕТ)/Я7’) (XVI.7)
где Е^ — энергия активации вязкого течения; А — константа.
Впервые это выражение получил Андраде [10]. Эйринг [23] ин-
терпретировал это уравнение с помощью «дырочной» теории жидко-
стей. Согласно этой теории, жидкость содержит незанятые места, или
«дырки», которые перемещаются случайным образом по всей жидко-
сти; перемещение происходит за счет заполнения их в одном месте
и возникновения в другом месте (дырки заполняются молекулами,
«перепрыгивающими» из одного места в другое). Каждый прыжок осу-
ществляется путем преодоления энергетического барьера высотой Еп.
Эта энергия активации связана для низкомолекулярных жидкостей
с теплотой испарения данной жидкости, поскольку удаление какой-то
молекулы из среды окружающих ее молекул можно рассматривать
или как процесс перескока, или как процесс испарения.
Однако в полимерах Еп принимает значение, не зависящее от мо-
лекулярного веса. Это означает, что в длинных цепях единица, уча-
ствующая в потоке, оказывается существенно меньше своей моле-
кулы. Вероятно, вязкое течение в полимерах осуществляется путем
последовательных прыжков каждого из сегментов до тех пор, пока
не переместится вся цепочка. у .
Более тщательные исследования показали, что вязкое течение не
является простым активированным процессом. Если построить за-
висимость логарифма вязкости от обратной абсолютной температуры
в широком диапазоне изменения Т, то даже для низкомолекулярных
жидкостей получаются кривые, приводящие к уменьшению кажу-
щейся величины Е^ с повышением Т. Подобное явление можно объяс-
нить тем, что вязкость зависит от избыточного свободного объема,
возникающего при тепловом расширении. Этот факт много лет на-
зад установил Бачинский [14], получивший соотношение
a z
(XVI.8)
которое, по крайней мере, не хуже уравнения Андраде описывает
поведение простых жидкостей. Бачинский показал, что v0 составляет
приблизительно одну треть критического объема и, следовательно,
близко к ван-дер-ваальсовскому объему для каждой жидкости.
Именно поэтому v — v0 есть эффективный свободный объем.
ВЯЗКОСТЬ АМОРФНЫХ ПОЛИМЕРОВ
ВЫШЕ ТЕМПЕРАТУРЫ СТЕКЛОВАНИЯ
Характеристики течения аморфных полимеров в состоянии расплава
(т. е. много выше температуры стеклования) весьма Сходны с харак-
теристиками расплавленных кристаллических полимеров. Для пол-
ностью аморфных полимеров нельзя наблюдать точку плавления.
265
Поэтому вязкость непрерывно изменяется в широком диапазоне
температур от точки стеклования и до температур, превышающих
точку плавления эквивалентных кристаллических полимеров.
Были предприняты попытки получить аналитическое выражение
для широкого температурного диапазона изменения вязкости поли-
мерных расплавов и аморфных полимеров. Дулитл 122] вновь рас-
смотрел понятие о свободном объеме при расчете вязкости и получил
уравнение
i) = A exp (B/cpf) или 1пт] = 1п А + В/ф/ (XVI.9)
где А и В — константы; ср/ — доля свободного объема.
Если принять, что cpf линейно возрастает с температурой, т. е.
<pf = <pg + а (Т — Те), то можно получить следующее уравнение:
, 1'1(7’) В / т — Tg \
ln i'l(T’g) ~ <рг ( ФгЛ+Г-Te) (XVI.10)
Это уравнение получили Вильямс, Лэндел и Ферри. В обобщенном
виде оно записывается так:
lgil(7’) = lgil(7’s)--C2+k(r_^) (XVI.11)
где Т8— стандартная температура.
Если за стандартную температуру принять точку стеклования,
то Сг = —17,44 и Са = 51,6. Если же стандартные температуры вы-
бираются произвольно с целью получить наиболее подходящую
универсальную функцию (см. табл. XVI.2, где приведены такие стан-
дартные температуры для различных полимеров), то «универсальные»
значения констант составят: Сг = —8,86, С2 = 101,6. Разность Ts — Tg
примерно равна 43 °C для весьма разнообразных полимеров. Ранее
уже говорилось, что уравнение Вильямса — Лэндела — Ферри иг-
рает важную роль в принципе температурно-временной суперпози-
ции процессов вязкоупругой релаксации (гл. IX).
Таблица XVI.2. Значения Tg и Т8 для различных полимеров
Полимер т8 те rs-Tg
Полиизобутилен 243 197 46
Полигексен-1 268 223 45
Полистирол 413 373 40
Поли-1/ис-изопрен 249 206 43
Поливинилацетат 350 305 45
Полиметилакрилат 324 282 42
Полиметилметакрилат 433 387 46
Поливинилхлорид 395 358 37
Полиуретан 283 238 45
Среднее 43
266
Формулы XVI. 10 и XVI. 11 позволяют достаточно точно описать
температурную зависимость вязкости в области от Т& до приблизи-
тельно Tg + 100 °C. При более высоких температурах удобнее поль-
зоваться следующим уравнением, предложенным Фоксом и Лошеком
[30]:
= (XVI.12)
где А, В и а постоянные; значение а обычно близко к 1.
Сходная формула была получена Корнельссеном и Уотерманом
[18] для различных масел, битумов, силиконов и стекол.
Таким образом, для описания температурной зависимости вяз-
кости в разных диапазонах температуры необходимо пользоваться
различными уравнениями:
а) формулой (XVI.10) или (XVI.11) при Tg < Т < (Т- + 100);
б) формулой (XVI.13) при + 50) < Т < 2Tg.
Каждое уравнение имеет собственный набор постоянных, которые
различаются для разных полимеров. Кроме того, если пользоваться
формулами (XVI.10) и (XVI.11), то необходимо знать вязкость при
температуре стеклования.
УНИВЕРСАЛЬНОЕ СООТНОШЕНИЕ ДЛЯ ВЯЗКОСТИ
РАСПЛАВЛЕННЫХ И КАУЧУКООБРАЗНЫХ АМОРФНЫХ
ПОЛИМЕРОВ
Магилл и Грит [35] попытались получить более универсальное соот-
ношение для описания влияния температуры на вязкость низкомо-
лекулярных жидкостей. Эти авторы построили зависимость лога-
рифма вязкости от отношения Тт1Т и получили семейство сходных
кривых. При высоких температурах эти кривые асимптотически при-
ближаются к прямой, соответствующей соотношению XVI.7. Однако
вблизи точки плавления связь между логарифмом вязкости и Тт1Т
была совершенно нелинейной.
Аналогичный метод можно применить к полимерам. Для поли-
меров наиболее характерной температурой является Tg. Следова-
тельно, в качестве безразмерного температурного параметра было
взято отношение TlTg. Для того чтобы учесть влияние молекуляр-
ного веса, рассматривались величины т]кр — значения вязкости при
критическом молекулярном весе. Построение зависимостей лога-
рифма T]Kg от TITg для ряда полимеров дает кривые совершенно
различной формы. Можно, однако, совместить эти кривые путем
следующего преобразования:
1йП*=-^^ + -В (XVI.13)
где А и В — постоянные, имеющие разные значения для каждого полимера;
1g т)* — универсальная функция от TlTg.
На рис. XVI.1 приведен график зависимости lg tj* от TlTg для
13 полимеров. Использованные здесь значения констант А п В
267
приведены в табл. XVI.3. Эти значения следует принимать как средние.
Дело в том, что в разных сериях экспериментов даже одного и того же
исследователя могут быть получены несколько отличающиеся друг
от друга значения А и В, тогда как различия между результатами
отдельных авторов для одного и того же полимера, как правило,
довольно велики.
Формулой (XVI. 13) можно воспользоваться для предсказания
вязкости полимеров, для которых известны значения А и В. Это
соотношение можно также применить в том случае, если имеется
достаточное количество экспериментальных данных для определения
констант А и В.
Если уравнение (XVI. 13) желательно применить для оценки вяз-
кости полимеров, для которых нет экспериментальных данных, то
Рис. XVI. 1. Зависимость 1g г] * от T/Tg'.
▼ — полиэтилен; х — полипропилен; О — полиизобутилен; А — полистирол; • — поли-
бутадиен; ф — поливинилацетат; — полиметилметакрилат; □ — полибутилметакрилат;
Н---полиднметилсилоксан; ▼ — полиоксиэтилен; Q — полидекаметиленадипат; А —
найлон 6. '
268
Таблица XVI.3. Средние аначения констант А и В в уравнении (XVI. 13)
Полимер А (опыт) В (опыт) о £ А (распет) В (расчет) 1
Полиэтилен 1,2 —0,5 150 1000 1,2 —0,5
Полипропилен 2,1 1,2 253 1700 0,7 0,5
Полиизобутилен 1,3 —0,5 198 2000 1,4 0
Полистирол 0,7 2,0 373 4350 0,8 1,7
Полибутадиен 1,0 0 165 1000 • 1,0 -0,3
Полиизопрен 0,7 0,3 220 1200 0,7 0,2
Поливинилацетат 1,0 1,0 301 3300 1,0 1,0
Полиметилметакрилат 0,8 -0,8 378 3600 0,7 1,8
Полибут илметакрилат 2,2 0,7 249 5100 2,2 0,5
Полиоксиэтилен 0,7 0,2 206 1150 0,7 0,1
Полидекаметиленадипинат 0,8 0,2 217 1150 0,7 . 0,2
Найлон 6 1,1 1,0 323 2800 0,7 1,2
Полидиметилсилоксан 0,6 -0,6 150 2000 2,4 -0,5
нужно иметь способ оценки констант А т& В. Для большинства поли-
меров, перечисленных в табл. XVI.3, значения 4 иВ можно оценить
с помощью следующих формул:
Л^27£,ког/Т’|г (XVI.14)
В ^7-^100-2 (XVI.15)
где Еког— энергия когезии, отнесенная к числу атомов в цепи главных валент-
ностей макромолекулы.
Значение молекулярной энергии когезии для некоторых полиме-
ров можно получить из табл. VI.2. Это значение следует разделить
на Z — число атомов в цепи главных валентностей, отнесенное к од-
ному структурному звену полимера. Наличие пропорциональности
между А и ЕКог весьма правдоподобно, поскольку А пропорцио-
нальна тангенсу угла наклона графика зависимости 1g ц от Т и, слет
дователйно, пропорциональна энергии активации вязкого течения-
В принципе вязкость данного полимера при любых значениях
молекулярного веса и температуры можно было бы предсказать
с помощью- формул (XVI.5), (XVI.6), (XVI. 13)-(XVI. 15) и
рис. XVI. 1. Однако точность предсказанной таким образом величины
будет довольйо низкой, поскольку неточности при оценках значе-
ний Tg, Еког и Кв, используемых в этих формулах, в конечном ре-
зультате суммируются. Следовательно,, рассчитанная с помощью
указанных выше соотношений величина должна рассматриваться
всего лишь как указание на порядок величины вязкости.
Пример 1
Рассчитать вязкость полиизобутилена с молекулярным весом 1,35-10*
при 25 °C.
269
Пусть известны следующие значения: Тг = 198 К: А = 1,3; В = —0,5MKD=
= 1,6-10*. .
Решение
При 25 °C T/Tg = 298/198 = 1,50. По рис. XVI.l 1g q* = 2,95. G помощью
уравнения (XVI.13) вычисляется rjKp:
1g т]кр = A (1g п* - В) = 1,3 (2,95 + 0,5) = 4,49
и по уравнению (XVI.6)
lg П = lg %р + 3,31g (У/1Икр)= 4,49 + 3,41g (1,35 • 106/1,6 • 10«) = 4,49 + 6,55 = 11,04;
т| = 1,1. юн П
Пусть известна только температура Стеклования Tg = 198 К.
Решение
Как и выше, Г/Гг = 1,50 и 1g ц* = 2,95. Энергия когезии полиизобутилена
Еког= 4000 кал/моль (см. табл. VI.2). Структурное звено содержит два атома
углерода в цепи главных валентностей, следовательно, E^or/Z — 2000. По урав-
нениям (XVI.14) и (XVI.15)
Л = 27-2000/1982 = 1,38 ц В = 198/100 — 2 = — 0,02
1g Пкр = A (1g т]* - В) = 1,38 (2,95 + 0,02) = 4,10
По табл. XV.2 Kq = 1,12 -10~3. С помощью уравнения (XVI.5) находится
значение AfKp:
Мкр = (0,14/Хе)2= 1,56-10*
В этом случае
lgr] = lgr]Kp + 3,41g(M/MKp) = 4,10 + 6,59 = 10,69 и г)=4,9-10ЮП
Экспериментальное значение, полученное Ферри [26], составляет: г] =
= 6,2-1010 П.
ВЛИЯНИЕ НАПРЯЖЕНИЯ СДВИГА
НА ВЯЗКОСТЬ
При низком напряжении сдвига (или небольшой скорости сдвига)
вязкость расплавов полимеров не зависит от условий деформирова-
ния. Полимер в этом случае ведет себя практически как ньютонов-
ская жидкость. С повышением напряжения (или скорости) сдвига
вязкость уменьшается довольно характерным образом. Для описа-
ния такого изменения вязкости был предложен целый ряд эмпириче-
ских соотношений. Наибольшую известность получили следующие
формулы. "
Оствальда-де Валле [37]:
т =к'уп или х\ = 4- = К'хп~1/п
У
Ферри [24]:
• Т / Т \ тпп
y=v(1+-g7) илич=Т+Т7ё7
270
(XVI. 16)
(XVI.17)
Спенсера и Диллона [39]:
У = т/д0 = ехр (т/6) или д = доежр ( —Т/Ь)
(XVI.18)
где До — предельное значение вязкости при «нулевой» скорости сдвига; К', га,
Gi и Ь — постоянные.
Более фундаментальный подход использовал Бики [1]. Исходя
из модели свободно вращающегося клубка при простом сдвиге, Бики
получил соотношение между коэффициентом ц/ц0 и безразмерной
скоростью сдвига Г . Послед-
няя величина определяется
формулой
Г=-л2'^т10 pRT (XVI. 19)
Результат обобщения экспе-
риментальных данных, пред-
ставляемых в координатах
т]/ц 0 — Г, показан на рис. XVI.2.
Аналогичный способ обра-
ботки экспериментальных дан-
ных предложили Виноградов
с соавторами [44], построив за-
висимость т]/т)о от произведе-
ния* уД0.
Мидлман [7] показал, что
значения вязкости, рассчитан-
ные с помощью формулы XVI.2,
Рис. XVI.2. Понижение вязкости рас-
плава в зависимости от безразмерной
скорости сдвига.
удовлетворительно соответствуют экспериментальным данным.
Расчеты Бики выполнены для моно дисперсного полимера. Однако
многие исследователи наблюдали влияние молекулярно-весового
распределения на неньютоновскую вязкость расплавов полимеров.
Ван дер Вегт [42] предложил эмпирический метод учета этого эф-
фекта. Он показал, что построение зависимости д/д0 от произведе-
ния iQ дает практически совпадающие кривые для полипропилена
с разными значениями средневесового , молекулярного веса и коэффи-
циента Q (Q = На рис. XVI.3 воспроизводится такая обоб-
щенная кривая.
Интересным следствием расчетов Бики оказывается влияние уйе-
личивающейся скорости сдвига на зависимость вязкости от молеку-
лярного веса. При низких скоростях сдвига т] = т] 0 и вязкость про-
порциональна Л/3 4. Однако в величину Г входит произведение
* В наиболее полной форме аргумент должен записываться как безразмер-
ное произведение скорости сдвига уо на характерное время релаксации (у0).
Однако во многих случаях, например для расплавов полимеров, исследуемых
при различных температурах, 0 с точностью до постоянной равно До, что оправ-
дывает запись аргумента в форме уд о- — Прим. ред.
271
вязкости при нулевой скорости сдвига на молекулярный вес, и Г
пропорциональна М*\ Но при постоянной скорости сдвига отноше-
ние rj/ij0 уменьшается с ростом М} и вязкость оказывается пропор-
циональной М в степени, меньшей чем 3,4. Совершенно аналогично
влияние температуры на вязкость должно уменьшаться с повыше-
Рис. XVI.3. Приведенная вязкостная
характеристика 33 марок полипропи-
лена при 210 °C (Q изменяется от 3,5
до 25) [42].
нием скорости сдвига *.
Пример 2
Оценить вязкость полистирола в
следующих условиях:
Л/=1,75 • 105 г/моль, 7=505 К,
у=1,65 • 105 с~1.
Решение
Для формулы (XVI.19) прини-
маются следующие величины: ц» =
= 2,0-104 П [20]; р = 0,885 г/см3
при 505 К (расчет по схеме гл. IV);
R = 8,32-10’дин • см/моль-К.
Таким образом
12 • 1,65 • 105 • 2,0 • 104 • 1,75 • 105_
Г ~ л20,885 - 8,32 • 10’ • 505
= 1,9 • 104
и
lgT = 4,28
По рис. XVI.2 1g ('П/'По) = —2,60 илип/ти = 2,5-10~3. Отсюда п = 2,5 X
X 10-з-2,0.104 = 50 П.
В заданных условиях величина вязкости, экспериментально опре-
деленная Кроссом [20], составила 40 П. •
ВЛИЯНИЕ ГИДРОСТАТИЧЕСКОГО
ДАВЛЕНИЯ НА ВЯЗКОСТЬ
Вязкость полимеров, как и низкомолекулярных жидкостей, увели-
чивается с ростом гидростатического давления. Влияние давления
на вязкость определяется коэффициентом
х=1/цР11/ад
Экспериментальных данных о значениях коэффициента % для
полимеров известно немного. Такие данные имеются лишь для трех
полимеров (табл. XVI.4). В принципе, для оценки коэффициента
можно воспользоваться следующим уравнением, полученным из
термодинамических соображений:
'/./А — х/а
* Это справедливо тогда, когда сравниваются значения вязкости, измерен-
ные при одной и той же скорости сдвига. Энергия активации, рассчитанная по
значениям вязкости, которые берутся при постоянном напряжении сдвига,
не зависит от напряжения. — Прим. ред.
272
где Л — температурный коэффициент вязкости ^равен
а — коэффициент теплового расширения.
х— сжимаемость,1
Однако, как правило, нет надежных термодинамических данных
для расчетов коэффициента х/а. По данным табл. XVI.4 можно за-
писать следующее эмпирическое соотношение:
Х/А ла константа
Таблица XVI.4. Коэффициент зависимости вязкости от давления
Полимер х-10’ см2/дин А, К"2 (-Х/А). 10» т, °C Литератур- ный источ- ник
Полиэтилен 1,4 -0,028 5,0 190 [46]
Полистирол , 4,3 —0,078 5,5 165 [32]
Полйдиметилсилоксан 0,73 —0,018 4,0 40 [33]
Коэффициент перевода в систему СИ; 1 см2/дин=10 м2/Н-
УПРУГОСТЬ РАСПЛАВОВ
Когда расплавленный полимер подвергается действию напряжения
сдвига, он не ведет себя подобно вязкой жидкости, как, например,
низкомолекулярные жидкости, а характеризуется частичной упру-
гостью. Это означает, что при стационарном течении полимерного
расплава возникает упругая деформация, от которой данный расплав
может «освободиться» при снятии напряжения сдвига. Такую упру-
гую деформацию можно выразить как = t/G, где G — модуль
упругости при сдвиге.
Упругая деформация увеличивается при повышении скорости
сдвига и оказывается тем больше, чем выше молекулярный вес
образца. Ширина молекулярно-весового распределения оказывает
определяющее влияние на упругие характеристики полимерных рас-
плавов. Согласно Ферри [3], упругая деформация связана с более
высокими средними молекулярными весами, чем средневесовой, на-
пример с z-средним молекулярным весом; здесь определяющей вели-
чиной оказывается M2-M2+1IMW. Высокая упругость расплава,
обусловленная широким распределением по молекулярным весам,
приводит к резко выраженным и важным практическим последствиям:
к длительному упругому последействию, высоким напряжениям при
формовании волокон, высокой, устойчивости пузырьков в пленке по-
лимера.
Одно из наиболее легко наблюдаемых последствий упругости
расплавов полимеров заключается в так называемом фильерном
расширении при экструзии (эффект Баруса). При выходе из
фильеры или литникового канала полимерный расплав, находившийся
под действием напряжения сдвига, сразу же испытывает упругое
18 Занаа 699 273
восстановление, и поперечное сечение струи возрастает. Фильерное
расширение определяется как отношение диаметра экструдата к диа-
метру канала. На практике это отношение изменяется от 1 до 4,5; сле-
довательно, поперечное сечение экструдата может увеличиться почти
в двадцать раз. Это одно из наиболее удивительных свойств термо-
пластов. Такой эффект проявляется во всех материалах, которые
обладают хоть сколько-нибудь заметной упругостью формы.
Другим следствием каучукоподобной упругости полимерных рас-
плавов оказывается ориентация. Если текущий расплав охлаждать
достаточно быстро (критерий «быстроты» — длительность охлажде-
ния должна быть меньше времени релаксации), то ориентация «за-
мораживается» и сохраняется в полученном изделии. Длительность
релаксации равна 0 = t\/G = TjJ, а податливость J (или 1/G) зависит
от молекулярно-весового распределения; следовательно, распределе-
ние по молекулярным весам сильно влияет на степень ориентации
экструдатов. Этот эффект также может иметь важное значение для
технологов, поскольку возможно искривление или нарушение формы
после изготовления изделия.
Известно еще одно следствие упругости расплавов — эффект
Вайссенберга, выражающийся в ряде специфических воздействий
расплава на вращающуюся в нем мешалку *.
КОМПЛЕКСНАЯ ВЯЗКОСТЬ
Реакцию полимера на воздействие переменного напряжения можно
описать комплексной вязкостью
Параметр^' называется динамической вязкостью; она зависит от
частоты со так же, как стационарная сдвиговая вязкость г| зависит
от скорости сдвига. Параметр т]" представляет собой меру упругой
реакции данного материала. Реакцию на переменное напряжение
можно с тем же успехом описать комплексным сдвиговым модулем,
как об этом говорилось в гл. IX:
G* = G' + iG"
где G' — упругая составляющая модуля сдвига; G‘>— мера диссипативных по-
тер ь.
Нетрудно показать, что
o’* = i<oT]*, G'=<ot]' и с”=сот]'
На практике важной задачей оказывается расчет стационарной
сдвиговой вязкости т] по результатам динамических измерений вяз-
кости. Кокс и Мерц [19] эмпирическим путем показали, что стацио-
нарная сдвиговая вязкость при заданной скорости сдвига практи-
* Обзор существующих теоретических представлений об упругости распла-
вов и растворов полимеров см. Малкин А. Я., «Механикаполимеров», 1975,
№ 1, с. 173—187— Прим. ред.
274
чесни равна абсолютному значению комплексной динамической
вязкости при частоте, численно равной этой скорости сдвига:
тГ(у) = |1]*1 (<>)
где
n*l=l(n')2+(n’)a],/1
Это положение подтвердили Оноги с сотр. [36] и Адамс с соавто-
рами [9] *.
[* Результаты многих экспериментальных исследований позволяют в на-
стоящее время предложить некоторые количественные оценки высокоэластиче-
ских свойств расплавов полимеров. Так, в области достаточно высоких молеку-
лярных весов, примерно в 10 раз превышающих критический молекулярный вес
-/Икр, модуль высокоэластичности монодисперсных полимеров G, вычисляемый
как отношение касательного напряжения т к обратимой деформации сдвига уе,
не зависит от их молекулярного веса. Характерные значения G, полученные
прямыми измерениями ув в области пропорциональности между т и уе, для неко-
торых монодисперсных полимеров, находящихся в текучем состоянии, ориенти-
ровочно таковы;
G-10-4, Литературный
дин/см* источник
Полибутадиены 6 1 д]
Полиизопрены 70 1 д, 2 д] 3 д]
Полистирол 60
Поли-а-метилстирол 140 3 д]
П олидиметилСилоке ан 100 4 д]
С увеличением напряжения (или скорости сдвига) модуль высокоэластич-
ности возрастает, т. е. имеет место эффект, обратный явлению аномалии вяз-
кости — снижению эффективной вязкости с ростом скорости сдвига.
Модуль высокоэластичности резко падает (т. е. обратная ему величина рав-
новесной сдвиговой податливости возрастает) с расширением молекулярно-
весового распределения (МБР). Мерой МБР в этом случае должно быть отношение
различных средних значений молекулярных весов, сводящееся к 1 при пере-
ходе к монодисперсным полимерам, так как для них G не зависит от молекуляр-
ного веса. Поэтому такой мерой МВР, определяющей величину модуля высоко-
эластичности, не может быть комплекс (М2-M2+i/Mw), предлагавшийся в ранних
источниках, хотя доминирующая роль высокомолекулярных фракций в про-
явлении высокоэластических свойств расплавов несомненна. В качестве характе-
ристик МВР, определяющих G, могут использоваться отношения типа (MwIMn),
(М2/Мш), (М2/Мп) ит. п. или некоторые степени и комбинации этих отношений.
Что касается эффекта Вейссенберга, лишь вскользь упомянутого в основном
тексте книги, то, резюмируя результаты многочисленных экспериментальных
исследований, выполненных на большом числе объектов, можно указать на следу-
ющие соответствия его с другими реологическими свойствами расплавов и рас-
творов полимеров (подробно см. обзор [5 д]). Первая разность нормальных напря-
жений а (возникающих при установившемся сдвиговом течении), представляю-
щая собой основную количественную характеристику эффекта Вейссенберга,
в широкой области режимов деформирования пропорциональна квадрату
* Подробное рассмотрение этого вопроса дается в статье: Виногра-
дов Г. В., М а л к и н А. Я. и др., «Механика полимеров», 1969, № 1, с. 164—
181; этой проблеме посвящен также обзор: Яновский Ю. Г., Мал-
ки н А. Я. В кн.: Успехи реологии полимеров. М., «Химия», 1970, с. 52—78. —
Прим. ред.
275
касательного напряжения: о ~ т2, причем коэффициентом пропорциональности
служит величина G-1, т. е.
O = T2/G
Поэтому для монодисперсных полимеров, когда G не зависит от молекуляр-
ного веса, а — Мт, где показатель степени т примерно равен 6,8—7,0, т. е.
удвоенной степени в зависимости наибольшей ньютоновской вязкости т)« от моле-
кулярного веса М (ср. с формулой XVI.2). Эта формула позволяет оценивать
величины нормальных напряжений, если известны более просто измеряемые
параметры цо и G; ойа также указывает на чрезвычайно сильный характер зави-
симости а от молекулярного веса. -
Расширение молекулярно-весового распределения приводит к уменьшению
G, и, как следствие этого, к росту нормальных напряжений а, т. е. интенсифика-
ции проявлений эффекта Вейссенберга при сопоставимых значениях касательных,
напряжений.
Записанная формула остается справедливой и для растворов полимеров.
Однако в этом случае для оценки величины напряжений в конкретных ситуациях
необходимо знать концентрационные зависимости вязкости и модуля высоко-
эластичноСти. Дальнейшее обсуждение этого вопроса, а также других количе-
ственных корреляций, связанных с вычислением нормальных напряжений и уста-
новлением соответствия этого эффекта с другими измеряемыми характеристи-
ками вязкоупругих свойств полимерных систем, интересующиеся могут найти
в обзоре [5 д].*] — Прим. _ред.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1 д. Малкин А. Я. и др., Еигор.
Polymer J . 1974, v. 10, № 5, р.
445—451.
2 д. Nemoto N. et al., «Macromole-
cula», 1971, v. 41, № 2, p. 215—
219.
3 Д. Odani H., Nemoto N., Kuvata M.
Bull. Inst. Chem. Res., Kyoto
Univ., 1972, v. 50, № 2, p. 117—
133.
4 д. Mills J., Europ. Polymer J.,
1969, v. 5, № 5, p. 675—695.
5 д. Малкин А. Я., «Механика по-
лимеров», 1975, № 1, с. 173—
187.
НЕУСТОЙЧИВОСТЬ ТЕЧЕНИЯ
ПОЛИМЕРНЫХ РАСПЛАВОВ
С увеличением скорости сдвига эффекты, связанные с упругостью
расплава, все в большей мере превосходят вязкую реакцию матери-
ала. Упругие деформации при этом возрастают пока не достигаются
такие условия, при которых расплав разрывается как упругое твер-
дое тело.
По Бенбоу и Лэмбу [15] для весьма разнообразных полимеров
расплав разрывается практически при одинаковом напряжении
сдвига, равном 1,25-106 дин/см2.
Виноградов с соавторами [44] для описания критических условий
течения ввели безразмерное число (так называемое «упругое» число
Рейнольдса) *.
NB — yQ = yi]/G = yx\J = T;/G = y (XVI.20)
где 0 — время релаксации, равное ц/G; G — модуль сдвига; / — податливость.
__________ <
* См. также Малкин А. Я.,Леонов А. И., ДАН СССР} 1963, т. 151,
№ 2, с. 380—383. — Прим. ред.
276
Если значение NB превышает примерно 7, то поток полимерного
расплава может разрушаться. В этих условиях в расплаве нака-
пливается максимальная упругая деформация, которую он может
выдержать. Величина критической упругой деформации опреде-
ляется главным образом разрывом очень длинных цепей и, следо-
вательно, сильно зависит от ширины распределения по молекуляр-
ным весам; предельная деформация до некоторой степени зависит
от наличия наиболее длинных цепей.
Барнетт [12] показал, что довольно точным критерием начала
разрушения раплава является «показатель разрушения расплава»:
(XVI.21)
где Q = MW/Mn.
Расплав разрывается при условии, когда значение VMF близко
к 1,0-107 дин/см2 (т]0 выражена в П; у — в с-1).
Бартош [13] предложил еще один критерий разрушения рас-
плава — критическое значение уменьшения вязкости
^mf/^o — 0.025
(XVI.22)
где т]о — ньютоновская вязкость.
Два эмпирических правила (XVI.21) и (XVI.22) будут удовлет-
ворены одновременно, если предположить, что начало разрушения
расплава происходит при критическом значении безразмерной ско-
рости сдвига Г [формула (XVI.19)], равном 350:
Гмр = 350 ' ~ . (XVI.23)
Имеющиеся экспериментальные данные обнаруживают, однако,
слишком большой разброс критических значений рассмотренных
параметров, поэтому нельзя оценить точность приведенных выше
критериев разрушения потока.
Задолго до собственно разрушения расплава могут проявляться
некоторые другие признаки неустойчивого течения: шероховатость
поверхности экструдата, плохая глянцевитость струи и потеря про-
зрачности. Влияние разрушения расплава можно существенно осла-
бить, если увеличивать скорость сдвига медленно. На практике это
осуществляется с помощью экструзионный головок с конусообразным
входом. Йри меньшей конусности критическая скорость сдвига выше.
Экспериментально это положение подтвердили Торделла -f41] и
Клегг [17].
Пример 3
Применить критерии (XVI.21)—(XVI.23) к образцу полистирола, свойства
которого рассматривались в примере 2.
277
Решение
а. Формула (XVI.23) дает:
Гмг = 350; уМР = Гшр • л2рЯ7’/12цоЛ/' =
= (350 • 3 • 142 • о,885 • 8,32 • 10? • 505)/(12 • 2,0 • 10< • 1,75 • 1Q5) = 3000 с-1
По рис. XVI.2:
т]мр/По = 0,023; т]мр = 0,023-2,0-101= 460 П;
?МР = 3000 • 460 = 1,38 • 10е дин/см2
Полученный результат по порядку величины соответствует эмпирическому
значению tmf = 1,25-10е дин/см2.
б, Формула (XVI.21) при Q = 6 дает:
•Ymf = -^мрО/По = Ю7.6/2 • 10< = 3000 с-1
в. Формула (XVI.22) дает:
т]МР = 0,025 • 2 • 10* = 500 П
Вопрос б причинах наступления неустойчивого режима течения, приво-
дящего к искажению поверхности изделия (экструдата), и критериях, позволя-
ющих оценить критические значения скорости сдвига и касательного напря-
жения т5, отвечающие этому моменту, чрезвычайно важен. Например, в техноло-
гии производства разнообразных полимерных изделий величина уs определяет
предельно допустимую степень интенсификации режима формования. Поэтому
неудивительно, что этой проблеме посвящено большое число разнообразных
исследований. Обзор ранних работ можно найти в статьях [1 д—3 д].
Общий результат ранних теоретических исследований сводится к введению
безразмерного критерия, представляющего собой отношение характерного вре-
мени релаксации 0 текущей полимерной системы (раствора или расплава) к ха-
рактерному временному масштабу процесса течения или же произведение 0
на некоторую скоростную характеристику потока. Такой характеристикой может
быть скорость сдвига у в установившемся течении. Введенный таким образом
критерий (у0), как об этом говорилось в основном тексте главы, может сводиться
к величине критической сдвиговой деформации уе, вытекающей либо из простей-
ших модельных соображений [4 д], либо следует из того, что для очень многих
полимерных систем существует универсальная связь между безразмерным
произведением (у0) и отвечающей ему величиной упругой деформации [5 д].
Хотя значения уе при этом получаются с довольно значительным разбросом,
все же критическое значение во многих случаях оказывается близким к 7 [1 д].
Роль скоростного параметра может играть также ускорение, или продоль-
ный градиент скорости [6 д], что приводит к введению качественно нового кри-
терия потери устойчивости, нежели величина (у0). Таким образом, хотя воз-
можны различные причины нарушения устойчивости течения вязкоупругой
жидкости, параметр такой структуры — произведение характерного времени
релаксации на масштаб скорости процесса деформирования — должен в любом
случае играть определяющую роль как критерий возникновения неустойчивости.
Общий характер такого критерия позволяет привести к нему многие другие
аналитические выражения различного строения, предлагавшиеся в литературе.
Применительно к критерию, предложенному О. Бартошем (см. формулу XVI.22),
выяснение его физического смысла, а также причин значительного разброса
экспериментальных точек, возникающего при его использовании, было дано
в работе [7 д]. Практически те же аргументы, позволившие связать критерий
(у0) с формулой XVI.22, использованы р основном тексте главы. Интересно
278
отметить, что критерий (XVI.21) также сводится к безразмерному произведению
(у'0). Этот результат следует из прямой связи, существующей между модулем
высокоэластичности G и шириной молекулярно-весового распределения поли-
мера, так что в первом приближении можно полагать, что G —(MwIMn)- Тогда
критерий Бернетта представляется в форме r\ay/Q — — VQ, где ц» — наи-
большая ньютоновская вязкость и 0 определяется как отношение (т) o/G).
Физический смысл критерия потери устойчивости, представляемого как
(у0), с общих позиций был рассмотрен в работах [8 д—10 д]. Принципиальный
подход, на котором основываются эти исследования, связан с трактовкой потери
возможности установившегося сдвигового течения как следствия перехода
из текучего в высокоэластическое состояние. Так, если представить модель поли-
мера в виде системы цепей, связанных временно существующими узлами флук-
туационной сетки с некоторым распределением времен жизни этих узлов, то,
очевидно, можно выбрать такую достаточно большую скорость, по отношению
к которой все узлы в сетке окажутся квазистабильными. Это означает, что при
такой (и те м более при большей) скорости деформации течение, которое должно
быть неизбежно связано с разрушением флуктуационных связей и проскальзы-
ванием цепей друг относительно друга в узлах, происходить не сможет, и поли-
мер будет вести себя подобно вулканизованному каучуку с его сеткой стабильных
химических связей. Из этой модели наглядно следует, что критические условия
потери текучести должны определяться некоторой характерной величиной
параметра (у0).
Наиболее отчетливо картина потери текучести с переходом полимера в вы-
сокоэластическое состояние проявляется для монодисперсных образцов (точнее,
для полимеров с очень узкими молекулярно-весовыми распределениями). Иссле-
дования таких полимеров привели к следующим выводам, важным для количе-
ственной оценки условий наступления неустойчивого режима деформирования
полимерных систем.
Во-первых, величина критического напряжения xs является характерным
параметром данного полимергомологического ряда и не зависит от молекуляр-
ного веса полимера и температуры, изменяясь при переходе от одного полимера
к другому в относительно узких пределах. Типичные примерные значения rs
таковы:
дин/см*
Полибутадиен 3,5
Полиэтилен линейный 3
Полиизопрен .1,5
Полистирол 1,0
По достижении xs течение становится невозможным. Поэтому, если полимер
все же понуждается внешней силой проходить через отверстие (канал, капилляр,
фильеру и т.п.), то это происходит по механизму пристенного скольжения.
Другими словами, достижение xs соответствует переходу от течения к скольже-
нию, или «срыву», когда из-за отделения полимера от стенки и падения гидравли-
ческого сопротивления канала скачкообразно возрастает объемный расход без
заметного увеличения давления (нагрузки). Различные интересные следствия
и возможные проявления этого эффекта, такие, как возникновение автоколеба-
ний, различные входовые эффекты, характер распределения напряжений в потоке
и др., детально описаны и обсуждены в работах [8 д—10 д].
Во-вторых, величина ys изменяется с изменением температуры и молекуляр-
ного веса полимера практически так же. как и наибольшая ньютоновская вяз-
кость т]е полимера, так что может вычисляться по простой формуле:
Т« = тв/т]о
поскольку явление аномалии вязкости не свойственно монодисперсным полиме-
рам в области температур, значительно отстоящих от температуры стеклования.
279
В-третьих, существует очень тесная корреляция между величинами ts и ys
динамическими (вязкоупругими, или релаксационными характеристиками поли-
мерных систем, так что xs может определяться из равенства
Ts Смаке 0,5Сй
где Смаке— значение модуля потерь в максимуме, отвечающем переходу из теку-
чего в высокоэластическое состояние; Се — значение модуля упругости в области
плато высокоэластического состояния.
Величину це при этом можно найти как отношение (G"l<o) при малых часто-
тах <о, когда модуль потерь пропорционален <о. Поэтому как rs, так и т]« может
находиться по результатам измерений динамических характеристик вязкоупру-
гих систем при малоамплитудных гармонических колебаниях.
При переходе к полидисперсному полимеру величина ts не изменяется,
так как она не зависит от молекулярно-весового распределения полимера.
Но неустойчивое течение развивается в более широком диапазоне скоростей,
чем в случае монодисперсного модельного образца с-тем же средним значением
среднего молекулярного веса и той же ньютоновской вязкостью. Поэтому вели-
чина ys, при которой неустойчивость становится критическим фактором, ограни-
чивающим возможность дальнейшего роста скорости течения, оказывается тем
выше, чем шире молекулярно-весовое распределение полимера.
При введении в высокомолекулярный полимер низкомолекулярного раство-
рителя величина rs понижается примерно пропорционально с1 2 3 4 (где с — объемная
доля полимера в растворе), так что критическое напряжение для раствора может
вычисляться как
Ts — Ts. i • С2
где ^s, 1 — значение ts при с = -1 (для непластифицированного полимера).
Так как при этом вязкость пропорциональна примерно с5 (см. гл. XV),
то критическая скорость сдвига, отвечающая прекращению установившегося
течения, резко возрастает по, мере разбавления полимера растворителем. При
этом усиливается эффект аномалии вязкости, что также способствует росту Ys-,
Эти соображения справедливы приблизительно при с > 0,3—0,5. При более'
низком содержании полимера в растворе аномалия вязкости становится чрез-
вычайно сильной и смазывает резкость перехода к неустойчивому режиму Дефор-
мирования, хотя возможность вязкоупругой неустойчивости сохраняется до еще
меньших концентраций, вплоть до таких, при которых еще существует флуктуа-
ционная сетка контактов макромолекул. Лишь при самых низких концентрациях
меняется характер проявлений вязкоупругости раствора [11 д], и неустойчи-
вость обсуждаемого типа становится невозможной.*] — Прим. ред.
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1 д, Малкин А. Я., Леонов А. И.
В кн.: Успехи реологии поли-
меров. Под ред. Г. В. Виногра-
дова.'М., «Химия», 1970, с. 98—
117.
2 д. Bialas G. A., White J. L.,
Rubb. Chem. Techn., 1969,
v. 42, № 3, p. 675-690.
3 д. Tordella J. P. in Rheology, New
York, Acad. Press, 1969.
4 д. Малкин А. Я., Леонов А. И.,
ДАН СССР, 1963, т. 151, № 2,
с. 380—383.
280
5 д. Исаев А. И., Бережная Г. В.
Малкин А. Я., Инж.-физ. ж.
1963, т. 24, № 1, с. 91— 96.
6 д. Еverage А. Е. Jr., Ballman Я. L.,
J. Appl. Polymer Sci., 1974,
v. 18, № 3, p. 933—937.
7 д. Малкин A. Я., Виноградов Г. В.,
J. Appl. Polymer Sci., 1966,
v. 10, № 5, p. 767-771.
8 д. Виноградов Г. В., Малкин А. Я.
и др., Высокомол. соед., 1972,
т. 14А, № 11, с. 2425—2442; J .
Polymer Sci., 1972, v. A2, № 6,
p. 1061—1084.
9 д. Виноградов Г. В., Высокомол.
соед., 1971, т. 13А, № 2, с. 294—
310; Rheol. Acta, 1973, v. 10,
№ 6, р. 357—373.
10 Д. Виноградов Г. В. и др., Poly-
mer Eng. Sci., 1972, v. 12, № 5,
р. 323—334.
11 д. Малкин А. Я., Бережная Г. В.,
Виноградов Г. В., J. Polymer
Sci., 1973, pt. 3, № 42, р. 1111 —
1115.
ОБОБЩЕННАЯ «КРИВАЯ ТЕЧЕНИЯ»
Ленк [34] описал обобщенную «кривую течения», которая анало-
гична полной кривой «напряжение — деформация», полимеров, на-
ходящихся в твердом состоянии. Эта кривая течения представлена на
рис. XVI.4 и XVI.5. Такая кривая суммирует возможные различные
Рис. XVI.4. Обобщенная кривая течения:
I — первая ньютоновская область; II — вторая ньютоновская область; III — область псев-
допластпчного поведения; IV—область дилатантного поведения; V — турбулентность
(разрыв потока расплава).
Рис. XVI.5. Полная кривая напряжение — деформация для жестких полимер-
ных материалов; условное ' напряжение проведено в истинное напряжение:
I — первая Гуковская область; II — Вторая Гуковская область (образование шейки, холод-
ная вытяжка); III — область деформационного размягчения; IV — область деформацион-
ного упрочнения; V — разрушение.
типы течения жидкости и описывает псевдопластическое, дилатант-
ное и другие типы поведения среды.
Псевдопластичность представляет собой наиболее общее реологи-
ческое свойство полимерных расплавов. Известно также существо-
вание области ньютоновского течения. Дилатантный тип течения
менее распространен среди полимерных материалов в обычных усло-
виях *.
Ленк объясняет переходы от одного типа течения к другому изме-
нениями структуры расплавов. Ньютоновское течение по Ленку —
* Кривые течения дилатантного типа характерны для пластизолей и неко-
торых растворов таких полимеров, в которых межмолекулярное взаимодействие
резко усиливается при ориентации. — Прим. ред.
281
это невозмущенный поток, в котором статистически сохраняется ис-
ходная структура материала. Псевдопластичное течение — это пере-
ход от основного исходного состояния к структуре, ориентированной
под действием сдвига. Вторая ньютоновская область должна была бы
соответствовать максимально возможной ориентации элементов среды
вдоль линий потока.
При очень высоких скоростях сегменты полимерных молекул,
ориентированные вдоль линий тока в максимально возможной сте-
пени, начнут взаимодействовать с соседними сегментами по меха-
низму трения. Форма движущихся частиц такова, что самому по-
току придается «беспорядочный» характер (дилатантная область).
Когда же такие центры «беспорядочности» потока (микровихри)
достигнут больших размеров, то течение окажется невозможным.
В итоге произойдет разрушение расплава.
ТЕЧЕНИЕ РАСПЛАВОВ ПОЛИМЕРОВ
ЧЕРЕЗ УЗКИЕ ТРУБКИ И КАПИЛЛЯРЫ
При течении ньютоновской жидкости через капилляр выполняется
закон Гагена — Пуазейля:
nr* dP
ф = -8г]_ dL
(XVI.24)
где Ф — объемная скорость потока; г» — радиус капилляра; dp/dL — градиент
давления.
Напряжение сдвига и скорость сдвига у стенки капилляра для
ньютоновской жидкости составляют:
го аР • ,л, ч
Тн = Т’5Ги =
(XVI.25)
Если расплав не обладает ньютоновскими свойствами, то необхо-;
димо ввести соответствующие поправки. Первая поправка учитывает
входовой и концевой эффекты; эту поправку предложил Бэгли [11].
Для исключения концевых эффектов необходимо пользоваться эф-
фективной длиной капилляра, вследствие чего выражение для на-
пряжения сдвига примет вид
г0 Ар _ Др
Xw~ 2_ £Эф 2(L/r0 + e)
(XVI.26)
где е — поправочный коэффициент, который можно определить, строя графин
зависимости Др от £/г« при различных постоянных значениях скорости сдвига j.
Вторая поправка учитывает неньютоновский характер течения.
Эту поправку предложил Рабинович [38]. Тогда формула для у
запишется следующим образом:
4Ф Зп + 1
лг® 4п
(XVI.27)
282
где в — определяется из соотношения
и = lim
L/r,->oo
dln Ст
dp \
dL J
d In
4Ф \
яго /
Постоянную n можно определить по графику зависимости
lg (0,5rQdpldL) от Ig(40/nro) при различных величинах L/r0 путем
экстраполяции к бесконечно большому значению Ыгй.
Действительное значение вязкости вычисляется как т| = тш/у.
Поправочный коэффициент е связан с постоянной лис упругой де-
формацией (величиной обратимого сдвига) соотношением
I . vw
e-n-f- 2 -п+ 2G
(XVI.28)
Модуль сдвига Gможно найти по графику зависимости е от xw.
Критическую величину напряжения сдвига нельзя превысить без
нарушения устойчивости потока (см. выше раздел о разрушении
расплава).
ЛИТЕРАТУРА
1. Bueche F., «Physical Properties
of. Polymers», Interscience, New
York, 1962.
2. Eirich F. R. (Ed.), «Rheology»,
Academic Press, New York,
1956.
3. Ferry J. D., «Viscoelastic Proper-
ties of Polymers», Wiley, New
York, 1961; 2nd ed. 1970. '
4. Glasstone S., Laidler K. J. and
Eyring H., «The Theory of Rate
Processes», McGraw-Hill, New
York, 1941.
5. Mason P. and Wookey N. (Eds.),
«The Rheology, of Elastomers»,
Pergamon Press, New York, 1957;
2nd ed. 1964.
6. McKelvey J. M., «Polymer Pro-
cessing», Wiley, New York,
1962.
7. Middleman S., «The Flow of
High Polymers», literscience, New
York, 1968.
8. Reiner M., «Deformation and
Flow», Lewis, London, 1948.
9. Adamse J. W. C., Janeschitz-
Kriegl H., den Otter J. L. and
Wales J. L. S., J. Polymer Sci.
A2-6 (1968) 871.
10. Andrade, E. N. da Costa, Nature
125 (1930) 309, 582.
11. Bagley E. B„ J. Appl. Phys. 28
(1957) 624; Trans. Soc. Rheol. 5
(1961) 355.
12. Barnett S. M., Polymer Eng. Sci.
7 (1967) 168.
13. Bartos 0., J. Appl. Phys. 35
(1964) 2767.
14. Batchinski A. J., Z. phys. Chem.
84 (1913) 643.
• 15. Benbow J. J. and Lamb P., SPE
Trans. 3 (1963) 1.
16. Bueche F., J. Chem. - Phys. 20
(1952) 1959; 22 (1954) 603; 25
(1956) 599.
17. Clegg P. L., in «Rheology of
Elastomers» (1958) see General
references; Brit. Plastics 39 (1966)
96.
18. Cornelissen J. and Water-
man H. J., Chem. Eng. Sci. 4
(1955) 238.
19. Cox W. P. and Merz E. H., J.
Polymer Sci. 28 (1958) 619.
20. Cross M. M., Europ. Polymer J.
2 (1966) 299.
21. De Waele A., J. Oil Col. Chem.
Assoc. 4 (1923) 33.
22. Doolittle A. K., J. Appl. Phys.
22 (1951) 1031, 1471; 23 (1952)
236.
23. Eyring ff., «The Theory of Rate
Processes», McGraw-Hill, New
York, 1941.
24. Ferry J. D., J. Am. Chem. Soc.
64 (1942) 1330.
25. Ferry J. D., (1961) see General
references.
283
26. Ferry J. D., Grandine L. D. and
Fitzgerald E. R., J. Appl. Phys.
24 (1953) 911.
27. Fox T. G. and Allen V. R., J.
Chem. Phys. 41 (1964) 344.
28. Fox T. G. and Flory P. J., I.
Am. Chem. Soc. 70 (1948) 2384;
J. Appl. Phys. 21 (1950) 581;
J. Polymer Sci. 14 (1954) 315.
29. Fox T. G., Gratch S. and Los-
haek S., in «Rheology» ed. by
F. R. Eirich, Academic Press,
New York, Vol. 1, 431 (1956).
30. Fox T. G. and Loshaek S„ J.
Polymer Sci. 15 (1955) 371.
31. GoppelJ. M and Vander Vegt A. K,
Proc. Intern. Plastics Congress
1966, 't Raedthuys, Utrecht 1967,
- 177.
32. Hellwege К. H., Knappe W.,
Paul P. and Semjonov V., Rheol.
Acta 6 (1967) 165.
33. Holzmuller W. and Dinter R.,
Exp. techn. Physik 8 (1960) 118.
34. Lenk R. S., J. Appl. Polymer
Sci. 11 (1967) 1033.
35. Magill J. H. and Greet R. J.,
Ind. Eng. Chem. Fundamentals
8 (1969) 701.
36. Onogi Masuda T. and Iba-
ragi T., Kolloid-Z. 222 (1968)
110.
37. Ostwald Wo., Kolloid-Z. 36 (1925)
99. '
38. RabinowitschB., Z. physik. Chem,
A145 (1929) 1.
39. Spencer R. S. and Dillon R. E.,
J. Colloid. Sci. 4 (1949) 241.
40. Spencer R. S., J. Polymer Sci.
5 (1950) 591; Proc. Sci. Int.
Congress Rheology, Oxford, 1953.
41. Tordella J. P., J. Appl. Phys.
27 (1956) 454; Trans. Soc. Pheol.
1 (1957) 203; Rheol. Acta 2/3
(1961) 216.
42. Van der Vegt A. K., Trans.
Plastics Inst. 32 (1964) 165.
43. Виноградов Г. В., Малкин АГ. Я.,
Прозоровская Н. В., Каргин В. А.,
ДАН СССР, 1966, т. 150, с. 767.
44. Виноградов Г. В., Малкин А. Я.,
Прозоровская Н. В., Каргин В. А.,
ДАН СССР, 1964, т. 154,
с. 1421.
45. Weisenberg К., Nature 159 (1947)
310.
46. Westover R. F., S. P. E. Trans.
1 (1961) 14.
47. White J. L., J. Appl. Polymer
Sci. 8 (1964) 2339; Rubber Chem.
Technol. 42 (1969) 675, 682, 691.
48. Williams M. L., Landel R. Ft
and Ferry J. D., I. Am. Chem(
Soc. 77 (1955) 3701.
глава xvn РАСТВОРЕНИЕ И ПЛАВЛЕНИЕ
ПОЛИМЕРОВ
При переработке весьма существенное значение имеют процессы
плавления и растворениц полимеров в различных жидкостях. В на-
стоящей главе рассматриваются основные физические величины,
определяющие скорости этих процессов.
РАСТВОРЕНИЕ ПОЛИМЕРОВ
Первая фаза процесса растворения полимеров заключается в про-
никновении молекул растворителя в полимер. Такое проникновение
обусловливает возникновение квазииндукционного периода, т. е.
периода времени, необходимого для образования поверхностного
набухшего слоя. Связь между таким «временем набухания» тнаб
и толщиной набухшего поверхностного слоя й описывается соотно-
шением
Тнаб = б2/б5 (XVII.1)
где D — средний коэффициент диффузии проникающих в полимер молекул рас-
творителя.
Вслед за таким квазииндукционным периодом может установиться
стационарное состояние. На протяжении периода стационарного со-
стояния объемные диффузионные потоки растворителя и полимера
будут равны друг другу. В этой стадии скорость растворения опре-
деляется выражением
. D
«=-§-ДФр ч (XVII.2)
где Фр — объемная доля растворителя; ДФР— полный градиент концентрации
растворителя (в объемных долях) между жидкостью и поверхностью полимера.
Если растворение происходит ц чистом растворителе, то ДФр ра-
вен единице и, следовательно, формула (XVII.2) принимает вид
's = D/i> (XVIL3)
ДИФФУЗИОННЫЙ слой
Согласно Юберейтеру [8], общий поверхностный слой в стеклообраз-
ных полимерах состоит из четырех областей:
— гидродинамический жидкий слой, который окружает каж-
дое твердое тело в движущейся жидкости;
285
ба — гелеобразный слой, который содержит набухший полимер-
ный материал в каучукоподобном состоянии;
б3 — твердый набухший слой, в котором полимер находится
в стеклообразном состоянии;
б4 — твердый инфильтрационный слой, т. е. каналы и отверстия
в полимере, заполненные молекулами растворителя.
На рис. XVII.1 приведены относительные размеры перечислен-
ных выше слоев и указаны концентрации полимера в этих слоях.
Рис. XVII. I. Зависимость концентрации полимера в поверхностном слое от
толщины слоя 6; 64 — инфильтрационный слой; 03 — твердый набухший слой;
+ 64— слой геля и жидкости; va, v&, vc— частоты перемешивания [8]).
В количественном отношении слои б4 и б2 оказываются наиболее
важными. Совершенно очевидно, что толщина полного поверхност-
ного слоя б зависит от характера течения жидкости, который опре-
деляется числом Рейнольдса (JVrc = уЛр/ц), поэтому можно предпо-
лагать наличие связи между числом Рейнольдса и толщиной всего
поверхностного слоя. Действительно, такая связь существует, как
это видно из данных рис. XVII.2. Ниже температуры перехода
в стеклообразное состояние влияние температуры на толщину всего
слоя б описывается соотношением
6 = boexp (-А/Т) (XVII.4)
В этих же пределах устойчивость гелеобразного слоя зависит'от
вязкости. Итак, толщина всего поверхностного слоя увеличивается
с повышением температуры. Величина А имеет порядок 1000 К.
В конечном счете величина б зависит, как и можно было ожидать
от молекулярного веса полимера. В области обычных молекулярных
весов эта зависимость описывается соотношением
& = км'/г (XVII.5)
286
При молекулярных весах выше 6 • 10в значение S быстро возрастает,
что можно объяснить усилением эффекта перепутывания макро-
молекул.
Общее выражение для толщины полного поверхностного слоя
в полимерах с «нормальными» молекулярными весами принимает вид
6~0.35.10-W1/. (XVIL6)
При обычных температурах и молекулярных весах толщина 6 имеет
порядок 10“2—10“1 см. Асмуссен и Юберейтер [5] показали, что
Рис. XVII.2. Зависимость толщины поверхностного слоя 6 от числа Рейнольдса
цифры на кривых — температуры растворения [7].
отношение толщины всего поверхностного слоя 6 к диаметру цолимер-
ной молекулы в клубкообразной конформации (Й2)1/* практически
постоянно и близко к 1,2-104 при комнатной температуре:
ss 1,2-10< (XVII.7)
Согласно данным гл. XV
(Л2)*,, = аМ1/г(Кв/Ф)'/’
и константа Ф близка к 2,5-1021.
' 'КОЭФФИЦИЕНТ ДИФФУЗИИ
В гл. XVIII, посвященной проницаемости, будет показано, что коэф-
фициент диффузии проникающих в полимер жидкостей, очевидно,
определяется вязкостью растворителя при комнатной температуре,
т. е. параметром, связанным с размером молекулы. Это заключение
подтверждается экспериментальными данными Юберейтера [7] по
диффузии различных пластификаторов; он одновременно измерил s
и б и смог рассчитать средний коэффициент диффузии по формуле
(XVII.3).
287
ТИПЫ РАСТВОРЕНИЯ
Юберейтер [8] показал, что состояние полимера в значительной сте-
пени влияет на характер растворений. Если аморфный полимер
растворяется при достаточно высокой температуре, например выше
температуры текучести, которая ограничивает сверху область высо-
коэластического состояния, то поверхностный слой образуется только
областью толщиной и процесс растворения сводится к простому
перемешиванию двух жидкостей.
Если полимер находится в высокоэластическом состоянии, то по-
верхностный слой состоит из областей толщиной и S2. Молекулы
растворителя могут быстрее проникнуть в полимерную матрицу, чем
полимерные молекулы смогут «распутаться» и перейти в раствор.
Большинство аморфных полимеров растворяется в стеклообраз-
ном твердом состоянии. В этом случае поверхностный слой разви-
вается полностью. Твердое состояние полимера позволяет существо-
вать всем четырем слоям. Наличие гелеобразного слоя очень важно
потому, что он «заживляет» трещины и отверстия, образовавшиеся
при удалении части растворяющихся макромолекул.
Иногда наблюдается растворение без образования гелеобразного
слоя, особенно при пониженных температурах. Вероятно, причина
этого явления заключается в растворении под действием напряже-
ния растрескивания. При этом наблюдаются трещины, идущие вглубь
полимерной матрицы, и образуются блоки полимерного вещества не-
больших размеров, которые целиком уходят с поверхности полимера.
Этот процесс, видимо, осуществляется за счет больших количеств
запасенной энергии деформации, замороженной при переходе в стек-
лообразное состояние и сконцентрированной вдоль широких кана-
лов и отверстий. В предельном случае сохраняется только один
слой Sv Здесь отсутствует индукционный период.
Растворение полукристаллических подимеров происходит не-
сколько по-иному, чем аморфных. Растворение кристаллов гораздо
более затруднено по сравнению с аморфными веществами, поскольку
энтальпия плавления должна компенсироваться в процессе растворе-
ния. Многие растворители, способные растворять стереорегулярные,
но застеклованные полимеры, не могут растворить эти же полимеры
в кристаллическом состоянии. Асмуссен с соавторами [4] показали,
что зависимость скорости растворения кристаллических полимеров
от температуры очень сходна с зависимостью скорости кристалли-
зации от температуры: полимеры, образовавшиеся при более высоких
скоростях роста кристаллов, растворяются также при более высо-
ких скоростях.
Пример 1
Оценить скорость растворения полистирола в толуоле при 35 °C (308 К):
а) При очень низком числе Рейнольдса 0), б) при числе Рейнольдса 1000.
Молекулярный вес полистирола 150 000; коэффициент диффузии толуола
в полистирол при 35 °C примерно равен 1,5-10~6 см2/с (оценка приведена
в гл. XVIII).
288
Решение '
Основные соотношения Даются формулой (XVI 1.6):
а) 6 = 0,35-10-2-(150 000)*^а-ехр (—1000/308) = 0,35-10-2.3,87-Ю2 X
X 3,88-Ю-2 = 5,25-10~2См; •
6)6 = 0,35-10-2 • 3,87-102-3,88-10~2/(1 + 0,35) = 3,9-10“ 2 см. Проверку
этой величины можно провести с помощью формулы (XVII.7). Из гл. XV
известно, что А0 для полистирола равна 80-IO-6, следовательно
(fia)V« = (K0/2,5 • 1021)’/а = а • 3,87 • 102 • (0,32 • 10"^)!/а = а • 2,65.10-е см
Поскольку а близок к 1,4, то 6 = 1,2-10* (ii2)^* =*li2-10*-l,4-2,65-10-e =
= 4,5-IO-2 см ири 25°С,'т. е. соответствие удовлетворительное.
Скорость растворения находится по. формуле
i =5/6 =1,5-10-8/5,25-10-2 = 3,0-10-6 см/с при АГКв₽а0
Юберейтер и Киршнер (7] экспериментально нашли, чт< « = 5 • 10* 8 см/с.
Соответствие можно считать хорошим.
ПЛАВЛЕНИЕ ПОЛИМЕРОВ
Плавление полимеров обычно осуществляют в пластицирующем ап-
парате, например в экструдере. Здесь можно рассматривать два раз-
личных процесса: первый — перенос тепла от нагретой поверхности
(цилиндр экструдера) к полимерным частицам, второй— превращение
механической энергии в тепло посредством деформации твердого
материала. Скорость первого процесса зависит от теплопроводности
и определяется, грубо говоря, площадью горячей поверхности. Ме-
ханизм второго процесса определяется количеством передаваемой
механической энергии.
Мэддок [6] подробно исследовал процесс плавления, который
начинается, когда контактирующие с поверхностью цилиндра поли-
мерные частицы неполностью расплавляются и эта поверхность по-
крывается пленкой расплавленного полимера (поверхностное пла-
вление). На этой стадии определяющим оказывается перенос тепла
через точки контакта полимера с горячей поверхностью. Как только
образуется достаточное количество расплава, твердые частицы сус-
пендируются в этом расплаве, расплав находится в движении, по-
этому интенсивность переноса тепла от цилиндра к жидкости высо-
кая. Далее процесс плавления происходит весьма быстро.
При переработке «жестких» полимеров, например полукристал-
лического полиэтилена, можно с помощью червяка подходящей
конструкции обеспечить выделение практически всей энергии, тре-
буемой для плавления полимера (в форме механической энергии);
ход процесса здесь почти совсем не зависит от внешнего обогрева.
289
ЛИТЕРАТУРА
1. Crank J. and Park G. S. (Eds.),
«Diffusion in Polymers», Acade-
mic Press, London/New York,
1968.
2. Ueberreiter K., «Kristallisieren,
Kristallzustand und Schmerzen»,
in «Struktur und physikalisches
Verhalten der Kunststolle»
(Nitsche und Wolf, Eds.) Vol. I,
Springer, Berlin, 1962.
3. Volmer.M., «Kinetik der Phasen-
bildung», Steinkopf, Dresden/Lei-
pzig, 1939.
4. Asmussen F., Ueberreiter K. and
Naumann H., in Diplomarbeit,
Fr. Univ. Berlin, 1965.
5. Asmussen F., Ueberreiter K., J.
Polymer Sci. 57 (1962) 199; Kol-
loid-Z. 185 (1962) 1.
6. Maddock В. H., S. P. E. Journal
15 (1959) 383.
7. Ueberreiter K., «Advances in Che-
mistry Series» 48 (1965) 35.
8. Ueberreiter K., «The Solution Pro-
cess», in «Diffusion in Polymers»,
1968.
ГЛАВА XVIII.
ПРОНИЦАЕМОСТЬ ПОЛИМЕРОВ.
ДИФФУЗИОННЫЙ ПЕРЕНОС ГАЗОВ,
ПАРОВ И ЖИДКОСТЕЙ В ПОЛИМЕРАХ
Степень проницаемости полимеров для небольших молекул опре-л
деляется раство римостью.и; диффузией Можно провести их приемле-
мую оценку, если имеются некоторые основные данные для прони-
кающих молекул (критическая температура, диаметр) и для полимера >
(строение, температура стеклования, степень кристалличности).'
Диффузия и проницаемость полимеров по отношению к различ-
ным газам и парам имеют большое практическое значение при по-
лучении и переработке полимерных материалов, и особенно при ис-
пользовании этих материалов в качестве защитных покрытий.
Проницаемость (Р) полимера для газа или пара образуется произ-
ведением двух величин — растворимости (5) данного газа или пара
в полимере и коэффициента диффузии (Д) газа или пара внутрь по-
лимера:
P = SD (XVIII.1)
Проницаемость можно измерить непосредственно как скорость
переноса газа или пара через полимерную пленку и выразить ее
путем отнесения полученного значения объемного потока к единице
площади, единице толщины и единице разности давлений по обе
стороны пленки.
РАСТВОРИМОСТЬ ГАЗОВ И ПАРОВ
В ПОЛИМЕРАХ
При достаточно малых значениях парциального давления газа или
пара выполняется закон Генри:
c = Sp (XVIII.2)
где с — равновесная концентрация растворенного вещества; р — парциальное
давление этого вещества.
В случае простых газов приведенное соотношение выполняется
для парциальных давлений, вплоть до 1 кгс/см2.
Растворимость простых газов при стандартных условиях обычно
выражается в см3/(см3-атм). Растворимость паров органических со-
единений обычно выражается в граммах на грамм полимера (при
давлении пара). Перевод растворимости, выраженной в см3(см3 X
291
X атм), в г/г производится умножением на М (газ)-р (пар)/22 400 • р
(полимер).
Определить растворимость газов непросто, поскольку раствори-
мости простых газов в полимерах весьма низкие. Наиболее точный
способ измерений заключается в установлении сорбционного равно-
весия между полимером и газом при известных значениях давления
и температуры с последующей десорбцией и определением количе-
ства десорбировавшегося газа.
Рис. XVIII. 1. Растворимость газа S в натуральном каучуке при 25 °C в зависи-
мости от критической температуры и точки кипения газа (S выражена в см3
при нормальных Т и р на см3 каучука при 1 атм).
Растворимость хорошо растворимых паров органических веществ
определить значительно легче. Образец полимера определенной
массы приводят в контакт с данным паром при постоянных значе-
ниях температуры и давления, а затем ^измеряют увеличение массы
образца с помощью кварцевых весов.
Ван Амеронген [13] обнаружил простое линейное соотношение
между растворимостью различных газов в каучуке и их температу-
рами кипения или критическими температурами. На рцс. XVIII. 1
представлены данные по растворимости простых газов в натураль-
ном каучуке.
292
Сплошные линии на этом рисунке можно описать следующими
выражениями [5 выражена в см3/(см3-атм)]:
lg S (298) = — 2,1 4- 0,00747’кр
lg S (298) = — 2,1 + 0,0123ТКИП
(XVIII.За)
(XVIII.36)
Phc.XVIII.2. Растворимость
и теплота растворения
газов в. эластомерах.
Црирода полимера влияет на растворимость и, вероятно, связана
.с параметром растворшдй^йПиолйЖраТ'Как о^ее’йрзпшатгчГ'кя-
честве первого приближения для аморфных эластомеров без сильно
полярных групп можно пользоваться
выражениями (XVIП. За) и (ХУТТТТЗб).
Ван Амеронген обнаружил отчетливо
выраженное избирательное влияние сте-
пени полярности полимера на раствори-
мость газа на примере бутадиен-акрило-
нитрильных сополимеров. С увеличением
содержания акрилонитрила в сополимере
быстро возрастает растворимость дву-
окиси углерода, в то время как раствори-
мость водорода, азота и кислорода
уменьшается.
Температурная зависимость раствори-
мости подчиняется уравнению Клапей-
рона — Клаузиуса:
d In 8
*H = -R d{i/T} (XVIII.4)
где А Я — теплота растворения.
Для самых маленьких молекул газов,
растворенных в эластомерах, теплота
растворения положительна (эндотермический эффект); для более
крупных молекул газов теплота растворения отрицательна (экзотер-
мический эффект). Процесс экзотермичен, если выделившаяся
энергия сорбции превышает энергию образования «дырки» молеку-
лярных размеров в полимере.
Ван Амеронген измерил тепловые эффекты для различных газов
и нескольких эластомеров; эти данные представлены на рис. XVIII.2,
и их можно описать с помощью уравнения
ДЯГ= — 2400 —20001g 8 (298) (XVII 1.5)
где индекс «г» обозначает величины; характеризующие высокоэластическое
состояние.
Таким образом, если известна критическая температура или точка
кипения газа или пара, то для таких эластомеров (71 g) можно
рассчитать не только S (298), но и AZT. Следовательно, обобщенное
выражение для растворимости газов в эластомерах выше Tg прини-
мает вид ’
8(Т) = 80ехр( —ДЯг/Л7’) = 8(298)ехр[(ДЯг/Л)(1/298-1/Т)] (XVIII.6)
293
На рис. XVIII.3 показана зависимость <S0 от \НГ.
Мирз [8] показал, что при температуре стеклования изменяется
тепловой эффект растворения. Для поливинилацетата это измене-
ние можно описать следующим соотношением (рис. XVIII.4):
ДЯв#« 1,67 ДЯГ—5000 (XVIII.7)
где индексы «g» и «г» указывают, что соответствующие величины характеризуют
стеклообразное и высокоэластическое состояния.
С некоторыми оговорками эту формулу можно применить также
для расчета тепловых эффектов растворения других полимеров, на-
ходящихся в стеклообразном состоянии. С помощью рис. XVIII.1 —
Рис. XVIII.3. Соотношение между и М1Г.
Рис. XVIII.4. Соотношение между теплотами растворения различных про-
стых газов в поливинилацетате, находящемся в стеклообразном и высо-
коэластическом состояниях.
XVIII.4 можно оценить растворимость газов в аморфных полимерах,
если принять, что при температуре стеклования растворимость
образца в стеклообразном и высокоэластическом состояниях одина-
кова.
Растворимость газов в кристаллических полимерах зависит от
степени кристалличности. Мишель и Бикслер [9] показали, что для
значительного числа газов выполняется следующее простое правило;
5 (298) = Sa (298) (1 —хс) (XVIII.8)
где хс — степень кристалличности; Sa— растворимость в аморфном состоянии.
Формулы (XVIII.3)—(XVIII.8) позволяют оценивать раствори-
мость газов и паров в полимерах, находящихся в различных состоя-
ниях.
Таким образом, возможны оценки растворимости простых газов
в полимерах, если известны следующие величины: точка кипения
(или критическая температура) газа, температура стеклования и сте-
пень кристалличности полимера.
294
Пример 1
Оценить растворимость кислорода (Ткип =90 К, ТкР= 155 К) в поливинил-
ацетате (7g = 303 К): а) при 316 К, т. е. выше температуры стеклования,
б) при 280 К, т. е. ниже температуры стеклования.
Решение
а. 'При температуре немного выше поливинилацетат находится в высоко-
эластическом состоянии. Это позволяет применить формулы (XVIII.За) и
(XVIII.36), что дает:
lg S (298) = —2,1 + 0,0074 • 155 = 0,045 — 1
lg S (298) =—2,1 + 0,0123 • 90 = 0,005 — 1
Два полученных значения S (298) составляют 0,11 и 0,10 или 0,105 в среднем.
С помощью (XVIII.5) получим:
Д Нг = -2400—2000 lg S (298) = —450
Следовательно
S (316) = 0,105 ехр [(—450/1,987) (1 /298 — 1 /316)] = 0,105 • 0,96 =
= 0,10 см3/см3-атм (при стандартных Т и р)
Экспериментальная величина (по Ван Амеронгену) составляет 0,06.
б. Сначала рассчитывается растворимость при = 303 К:
S (303) = 0,105 ехр [(-450/1,987) (1/298—1/303)] Q.105
Для стеклообразного поливинилацетата из формулы (XVIII.7) следует, что
ДЯг = 1,67 ДНГ —5000=—750—5000 =—5750 кал/моль
Экспериментальное значение (по Мирзу) составляет —6250 кал/моль.
Теперь можно рассчитать So для стеклообразного состояния:
S (7’g) = 0,105 = S0exp (—AZ7g/7?7’g) = Soexp (9,5)
ИЛИ
So = 0,105/1,35-IO* = 0,78 ИО-»
Следовательно
S (280) = So ехр (-ДЯй/2807?) = 0,78-10-5 ехр (10,35)
ИЛИ ;
S (280) = 0,78 • 10-а • 3,15 • 10* = 0,25
КОЭФФИЦИЕНТ ДИФФУЗИИ
ДИФФУЗИЯ ПРОСТЫХ ГАЗОВ
В общем случае диффузию газов можно рассматривать как терми-
чески активированный процесс, описываемый уравнением типа Ар-
рениуса:
Д = Оаехр(-£д/Я7л) (XVIII.9)
где 7)0 и Ed — постоянные для данного газа и данного полимера.
295
Стэннет [12] собрал все известные данные по диффузии газов
через различные полимеры.
Если построить зависимость lg Do от ED, то получится довольно
простое соотношение, как это показано на рис. XVIII.5 для многих
эластомеров. Для аморфных стеклообразных полимеров подобная
Рис. XVIII.6. Соотношение между Do и ED для стеклообразных полимеров:
1 — простые газы в эластомерах; 2 — простые газы в стеклообразиых полимерах; 3 —
водяной пар в гидрофильных полимерах.
корреляция менее точна, но характер зависимости такой же, как это
следует из рис. XVIII.6.
-Таким образом, если для определенной пары газ — полимер из-
вестна энергия активации E‘D, то можно рассчитать коэффициент
диффузии с помощью следующей формулы:
для эластомеров
lgDo = O,5(EBliQOO-8,O) (XVIII.10)
для стеклообразных полимеров
lg Do 0,4(#0/1000-10) (XVIII.ll)
296
Выражения (XVIII.10) и (XVIII.И) дают интересные примеры
так называемых компенсационных эффектов (частный случай —
повышение Ео с ростом Dn).
Комбинация (XVIII.9) с (XVIII.10) или с (XVIII.il) дает соотно-
шения:
для эластомеров
Ев —ЕП /1 1 \
lg D =lgD0— 2.3ЛТ = 2,ЗЯ \~Т 0?/ 4,0 (XVIII.12)
для стеклообразных полимеров
ed ed (1 1 \ л
lgP-lgPo-2^r=--2^-(T-—ej-4’0 (XVIII.13)
где
е1 = 2000/2.3Я = 435К и е2 = 2500/2,ЗЯ = 545К
Теперь задача заключается в том, чтобы найти соотношение
между Ео и характеристическими величинами для газа и полимера.
Энергия активации — это та энергия, которая обеспечивает
возможность перескока растворенной молекулы из одной «дырки»
в другую. Совершенно ясно, что для диффузии более крупных моле-
кул в полимере должны образоваться более крупные «дырки». Следо-
вательно, энергия активации диффузии более крупных молекул
больше, а коэффициент диффузии — меньше. По-видимому, это Спра-
ведливо во всех случаях.
Имеющиеся данные свидетельствуют о наличии довольно при-
ближенной корреляции между энергией активации диффузии и диа-
метром молекул газа; характер этой зависимости близок к степенной,
и Еъ пропорциональна диаметру молекулы проникающего газа в сте-
пени между первой и второй. Все же наилучшая корреляция полу-
чается, если принять, что ED пропорциональна квадрату диаметра
молекулы. В табл. XVIII.1 приведены значения диаметров молекул
газов; эти диаметры всегда выражаются в ангстремах. Если по-
строить зависимость Е^ от температуры стеклования данного по-
лимера, то получатся кривые, представленные на рис. XVIII.7 (дан-
ные Ван Кревелена). Из этого рисунка видно, что энергия активации
диффузии в стеклообразном состоянии меньше, чем в высокоэласти-
ческом {EDg 0,75EDr).
С помощью данных рис. XVIII.7 можно оценить ED, если известны
диаметр молекулы газа и температура стеклования полимера. По
известному теперь значению ED можно найти D с помощью формул
(XVIII.12) и (XVIII.13).
При кристаллизации полимера возникает тенденция к уменьше-
нию достижимого для диффузии объема аморфного материала ^кристал-
лические участки затрудняют перемещение молекул и обусловли-
вают повышение средней длины пути, проходимого молекулами.
297
Таблица XVIII.1. Диаметры простых молекул
Молекулы <газ) d, А d2. А2 Укр’ см* ТКИП’ К Гкр' К
Не 2,55 6,5 58 . 4,3 5,3
Н2О эф. 3,7 эф. 13,5 56 373 647
н2 2,82 7,9 65 20 33
Ne 2,82 7,9 (42) 27 44,5
NH3 2,90 8,4 72,5 240 406
О2 3,47 12,0 74 90 155
А 3,54 12,5 75 87,5 151
СН.ОН 3,63 13,2 118 338 513
Кг 3,66 13,4 92 121 209
СО 3,69 13,6 93 82 133
СН4 3,76 14,1 99,5 112 191
n2 3,80 14,4 90 77 126
со2 (3,8) (14,4) 94 195 .304
Хе" 4,05 16,4 119 164 290
so2 4,11 16,9 122 263 431
С2Н4 4,16 17,4 124 175 283
СН3С1 4,18 17,5 143 249 416
С2Н« 4,44 19,7 148 185 305
СН2С12 4,90 24,0 193 313 510
с3нв 5,12 26,2 200 231 370
СвНа 5,35 29,7 260 353 562
Рис. XVIII.7. Соотношение между энергией активации диффузии, размером
диффундирующих молекул и температурой перехода полимера в стеклообразное
состояние:
а — высокоэластическое состояние; б — стеклообразное состояние; к™,,,™» кямчтк-
а___яластомеры (1 — силиконовым каучук; 2 — полибуталиен, 3 оутиловыи каучук.
?-п=опрен; Ч -Теонрен; 6 - пЖиропилен атактический; 7 - поливиниладетат
каучукообразный; 8 — поливинилхлорид/ацетат каучукообразный 9 полиэтилен
терефталат каучукообразный); О — аморфные стеклообразные; полимер________пОЛИстипол-
винилацетат; 11 — полиэтилентерефталат; 12 — полиэтилметакрилат, 13 “°™СТ“Р° >
14 - поликарбонат); х — полукристаллические полимеры (15 — потигатилет плот-
ностью 0,91; 16 — полиэтилен плотностью 0,96 г/см", 17 —полиоксиметилен, is
политрифторхлорэтилен; 19 — политетрафторэтилен).
298
В качестве первого приближения для расчета коэффициента диф-
фузии через полукристаллические полимеры можно воспользоваться
следующим уравнением:
D = Da(i-xc) (XVIII.14)
где хс — степень кристалличности.
Экспериментально это уравнение подтвердили Мишель с соавто-
рами [10], исследовавшие диффузию различных газов через поли-
этилентерефталат.
Использованные в (XVIII.14) значения Da взяты из данных
рис. XVIII.7 и результатов расчетов по формулам (XVIII.10)—
(XVIII.13); от абсолютной величины Тя зависит, какой кривой на
рис. XVIII.7 следует воспользоваться для расчета Da — построен-
ной для эластомеров или для застеклованных полимеров.
Таким образом, коэффициент диффузии простых газов через по-
лимеры можно оценить при любой температуре, если известны диа-
метр молекулы газа, температура стеклования и степень кристал-
личности полимера. ь,
Обобщенная диаграмма показана на рис. XVIII.8.
Пример 2
Оценить коэффициент диффузии кислорода (d = 3,74 А) в высокоэластиче-
ский и стеклообразный поливинилацетат (Tg = 303 К) при комнатной темпера-
туре.
Решение
а. Высокоэластическое состояние. С помощью рис. XVIII.7 при Tg = 303 К
можно найти, что
£B/da=1170 или Ев=1170-3,472 = 14000
Экспериментальное значение (по Мирзу) 14 500 кал/моль. Подстановка этого
значения в (XVIII.12) дает:
1g 7) (298)=(—7?д/2,37?) (1/298—1/435) —4,0 = 3,33 — 4,0 = —7,33;
D (298) = 0,47 -IO-’
б. Стеклообразное состояние. С помощью рис. XVIII.7 для Tg = 303 К
в стеклообразном состоянии
£B/d2 = 900 или Вд =900-3,472 = 11 000
Следовательно
lg D (298) = (—Ed /2,37?) (1/298—1 /545) — 4,0 = —3.6—4,0 = —7,6;
D (298) = 0,25-10-7
Экспериментальное значение 0,51-10"’.
Пример 3
Оценить с помощью рис. XVIII.8 значения EN, D»nD (298) для кислорода,
диффундирующего через бутилкаучук (Ts= 200 К).
Решение
В верхнем левом углу рис. XVIII.8 при Tg = 200 К вертикальная пунктир-
ная кривая пересекает кривую «а» (эластомер). От этой точки пересечения
299
проводится вправо пунктирная линия до пересечения с линией в правом верх-
нем углу для кислорода (</«3,5А). От этой точки вертикальная линия
продолжается вниз, и по оси х отсчитывается значение «=» 11 000.
Продолжение этой линии вниз до пересечения с линией 1 в правом нижнем
углу дает точку, от которой пунктирная линия проводится горизонтально влево.
Рис. XVIII.8. Диаграмма для оценки коэффициента диффузии:
а — эластомеры (высокоэластическое состояние); б — стеклообразные полимеры; в — про-
стые газы в эластомерах; г — простые газы в застеклованных полимерах; S — вода в гидро-
фильных полимерах.
На оси у отсчитывается lgD« = 1,5. Эта линия продолжается до пересечения
С линией Ed • 10~ 3=,11 в левом нижнем углу; отсюда линия ведется вертикально
вверх и отсчитывается значение lg D (298) = —6,4 = 0,6 7.
300
В итоге получаются следующие результаты:
Эксперимент
Расчет (рис. XVIII.8) (Ван Амеронген [13])*
Ed 11000 11900
2>о. 31,5 43
D (298) 4-10-’ 0,81-10-’
Соответствие вполне удовлетворительное.
ДИФФУЗИЯ ПАРОВ
Коэффициенты диффузии некоторых паров через полимеры были
определены методом кинетики сорбции. Во всех случаях было най-
дено, что коэффициент диффузии сильно зависит от концентрации.
При высоких температурах 1g D линейно зависит от объемной доли
диффундирующего вещества фр; при меныпих температурах зависи-
мость 1g D от фд описывается кривой с выпуклостью в сторону оси
абсцисс.
Фуджита с сотр. [7] нашли следующее соотношение для диффу-
зии этилацетата -в полиметилакрилат;
Г я (Фо. л Т1
Llg D (0, Т)
0,1354
0,165 Т —То
Фо У’о
(XVIII.15)
где Т» — постоянная температура, близкая к точке стеклования (в данном слу-
чае 265 К).
Это уравнение справедливо для объемных долей фр, вплоть до
примерно 0,15. Такой характер влияния концентрации на коэффи-
циент диффузии получил теоретическое объяснение на основании
Рис. XVIII.9. Соотношение между энер-
гией активации диффузии в полистирол
и мольным объемом в диапазоне темпера-
тур Тй < Т-. •
1 — СН3ОН; ' 2 — С2Н„ОН; 3 — СН2С12; 4 —
С2Н5Вг; 5 •- СНС1,; в — C5HeN; 7 — С3Н-С1; 8—
С.Н.; 9 — С«Н„Е [15].
концепции свободного объема в предположении, что частота переско-
ков молекул диффундирующего вещества непосредственно зависит
от доли свободного объема в системе, как было показано Мирзом [8].
Для паров органических соединений нельзя пользоваться соот-
ношением Ed — d2, которое было получено для простых газов. Жур-
ков и Рыскин [15], а также Дуда и Врентас [6] получили соответ-
ствие между энергией активации ED и мольным объемом VD диффун-
дирующего вещества. Результаты этих авторов воспроизведены на
301
рис. XVIII.9; данные показывают существование линейного соот-
ношения между £j>h VD при очень низких концентрациях, когда
набухание полимера практически отсутствует.
В этом случае также выполняются общие соотношения (XVIII. 10)
и (XVIII.И); таким образом, если ED рассчитана по VD, то можно
рассчитать Do и D.
ДИФФУЗИЯ ЖИДКОСТЕЙ
Коэффициенты диффузии органических жидкостей в 'резину опреде-
лили Саутерн и Томас [11]; они измеряли скорость увеличения
массы резинового образца, погруженного в жидкость. На зависимости
увеличения массы от времени были обнаружены аномальные области,
которые образовывались за счет напряжений, возникающих в об-
разце в процессе набухания. Подобных аномалий на кривой уда-
валось избегать, если торцевые части образцов закрепляли соеди-
Рис. VIII.10. Соотношение между коэффициентом диффузии
D и вязкостью различных жидкостей ц при диффузии жид-
костей в натуральный каучук [11].
пением с металлическими пластинками, что обеспечивало постоян-
ство граничных условий в процессе набухания каучука. При иссле-
дованных концентрациях жидкостей (вплоть до объемной доли 0,8 для
наиболее активных агентов набухания) коэффициент диффузии, как
оказалось, зависел от вязкости жидкости, а не от совместимости
полимера с данной жидкостью. На рис. XVIII.10 показано полу-
ченное соотношение; эти результаты могут иметь общее значение.
АДСОРБЦИЯ И ПЕРЕНОС ВЛАГИ
>
Природа молекул воды заставляет выделить в особый случай харак-
теристики взаимодействия влаги с полимерами. Молекула воды от-
носительно невелика и обладает сильной тенденцией к образованию
водородных связей с окружающими молекулами воды в жидком
и твердом состояниях, а также с другими полярными группами.
В полярных полимерах равновесия как сорбции, так и диффузии силь-
302
но зависят от таких взаимодействий; кроме того, в менее полярных
полимерах могут возникать аномалии, например появиться ассо-
циаты сорбированных молекул воды (так называемые «кластеры»).
Равновесная сорбция воды (растворимость) описывается различ-
ными изотермами типа Брунауэра — Эммета — Теллера.
В большинстве гидрофильных полимеров, например целлюлозе
и белках, каждая полярная группа взаимодействует только с одной
молекулой воды. В гидрофобных полимерах, например полиолефи-
нах, закон Генри выполняется для всего диапазона относительных
давлений, но только для небольших количеств адсорбированной воды.
Чем больше полярных групп имеется в полимерной матрице,
тем выше сорбционное сродство полимера к воде. Однако доступ-
ность этих полярных групп, относительная прочность связей вода —
вода и вода — полимер, а также степень кристалличности полимер-
ной матрицы оказываются весьма существенными факторами, объ-
ясняющими отсутствие простой корреляции между числом полярных
групп и растворимостью. Например, высокоупорядоченные кристал-
литы недоступны для воды, но на их поверхности полярные группы
активно взаимодействуют с водой.
Бэрри [5] собрал все опубликованные данные по сорбции воды.
С помощью этих данных можно оценить влияние различных струк-
турных групп на сорбцию воды при различных степенях влажности.
В табл. -XVIII.2 представлена наилучшая возможная величина для
оценки сорбционной способности полимеров по отношению к воде:
количество воды, приходящееся на одну структурную группу в ус-
ловиях равновесия. По этим данным можно легко оценить раство-
римость (см3 водяного пара при стандартных температуре и давле-
нии на см3 полимера). Коэффициент пересчета составляет 22,4 X
X 103/V, где V — мольный объем структурного звена полимера.
Пример 4
Оценить содержание влаги в найлоне 6,6 при 25 °C и относительной влаж-
ности 0,7. Степень кристалличности равна 70%.
Решение
Структурное звено найлона 6,6:
—NHCO—(СН2)4—CONH—(СН2)в—
Из табл. XVIII.2 видно, что сорбционной способностью СН2-групп можно пре-
небречь. Следовательно, остаются две CONH-группы на структурное звено
с содержанием воды 2-0,75 =1,5 моль/звено при р/ро = 0,7. Молекулярный вес
звена 226,3; таким образом, на 226,3 г полимера адсорбировались 1,5-18 г воды,
или 12 г воды на 100 г полимера. Учет кристалличности полимера с помощью
формулы (XVIII.8) дает для растворимости этого кристаллического полимера
значение 0,3-12 = 3,6 г на 100 г полимера. Экспериментальное, значение соста-
вляет 4 г/100 г.
Коэффициент диффузии воды в полимеры также сильно зависит
от взаимодействия полимер — вода.
Если полимер содержит большое число групп, которые могут
участвовать в образовании водородных связей (целлюлоза, поливи-
ниловый спирт, белки и т. д., в меньшей степени — синтетические
303
Таблица XVIII.2. Мольное содержание води в полимерах
на одну полярную еруппу при различных относительных
е ложностях и температуре 25 ° С
Значения р/р0 (примерно равно относительной влажности)
0,3 0,5 0.7 0,9 1.0
-СН3
-сн2- (1,5-10-6) (2,5-10-5) (3,3-10-5) (4,5-10-5) (5-10-5)
—СН—
0,001 0,002 0,003 0,004 0,005
^>с=о 0,025 0,055 (0,11) (0,20) (0,3)
л
~с\ 0,025 0,05 0,075 0,14 0,2
хо-
—о— 0,006 0,001 0,02 0,06 0,1
—он 0,35 0,5 0,75 0,5 2
-nh2 0,35 0,5 0,75 (1.5) (2)
—NHJ — — 2,8 5,3 —
—соон 0,2 0,3 0,6 1,0 1.3
—СОО- 1,1 2,1 4,2 — —
-с<° 0,35 0,5 0,75 1.5 2
XNH-
—G1 0,003 0,006 0,015 ' 0,06 (0.1)
-CN 0,015 0,02 0,065 0,22 (0.3)
полиамиды), то коэффициент диффузии возрастает с увеличением со-
держания воды. Это объясняется сильной локализацией первона-
чально сорбированных молекул воды на ограниченном числе центров,
а при высоком содержании воды полимерная матрица набухает и под-
вижность сорбированной воды увеличивается. В качестве хорошего
приближения можно применить следующее соотношение:
lg D = lg Рг._0 + 0,08c ' (XVIII.16)
где с — содержание воды в г на 100 г полимера.
По сравнению с негидрофильными полимерами сам коэффициент
диффузии существенно понижается из-за сильных взаимодействий
в системе, и вместо (XVIII.10) и (XVIII.11) следует пользоваться
соотношением для расчета коэффициента диффузии воды в гидро-
фильных полимерах (пунктирная линия на рис. XVIII.6):
1g Ро = О,5(£в/1ООО-14) (XVIII.17)
304
Другой крайний случай имеет место для менее гидрофильных по-
лимеров, например полиэфиров и полиметакрилатов. Здесь коэффи-
циент диффузий заметно уменьшается с увеличением содержания
воды. Это объясняется усилением эффекта образования «кластеров»
молекул воды в полимере у полярных центров или в микрополостях,
поэтому часть молекул воды становится относительно неподвижной.
В подобной ситуации величину D можно оценить с помощью следу-
ющего выражения:
lgD = lgDc=0—0,08с (XVIII.18)
Кроме того, соотношение между Do и Ед оказывается точно та-
ким, как и для других простых газов.
Особо ведут себя истинно гидрофобные полимеры, например
полиолефины и некоторые полиэфиры. Здесь растворимость очень
низкая (термодинамически «идеальное» поведение), и коэффициент
диффузии не зависит от содержания воды. Диффузия водяного пара
осуществляется точно так, как и диффузия других простых газов.
Нетрудно представить себе, что диффузионный перенос (прони-
цаемость) воды в полимеры и через полимеры играет весьма важную
роль, в частности при изготовлении одежды из полимерных материа-
лов.
[ + Диффузия, как и вязкость расплавов и растворов полимеров, рпреде-
ляется сегментальной подвижностью. макромолекул- Однако зависимости коэф-
*фш(ЙентгГдиффузии“Т77~характеризующего подвижность низкомолекулярного
компонента в полимере, или скорость его переноса через выбранное сечение
в полимере, и вязкости ц от молекулярного веса и концентрации раствора прин-
ципиально различны. Прежде всего следует подчеркнуть очень сильную зависи-
мость ц и независимость D от молекулярного веса высокомолекулярных' образ-
цов. Затем следует отметить существенно различный характер концентрацион-
ных зависимостей D и ц. Материал, относящийся к вязкости полимерных систем,
изложен в гл. XV и XVI настоящей книги. Ниже обсуждаются некоторые сведе-
ния, касающиеся зависимости D от молекулярного веса и концентрации раствора,
дополняющие материал настоящей главы (в основном по [1 д1).
Значения D не зависят от молекулярного веса полимера в широкой области
концентраций, но не для разбавленных растворов. Если концентрация полимера
в растворе <р2 становится меньше некоторого предела, существование которого
связано с условиями образования трехмерной флуктуационной сетки зацеплений,
то коэффициент диффузии растворителя в области низких значений <р2 становится
тем выше, чем меньше молекулярный вес полимера. Такая же зависимость про-
является в области низких молекулярных весов, т. е. для низкомолекулярных
полимеров D также растет с понижением молекулярного веса полимера [1 д].
Зависимость D от концентрации раствора неоднозначна, и ее характер опре-
деляется термодинамическими свойствами системы полимер — растворитель.
Показательны в этом отношении детально исследованные [1 д] растворы полй-
бутадиенов в метилнафталине (I) и дигептилфталате (II). В обеих системах кон-
центрационная зависимость коэффициента самодиффузии D* растворителя,
характеризующая подвижность его молекул в отсутствие градиента концентра-
ции под действием броуновского движения, монотонна: подвижность уменьшается
•с ростом <р2. Иной оказывается концентрационная зависимость скорости пере-
носа, которая определяется свободным объемом системы и термодинамическим
взаимодействием полимера с растворителем. Так, при комнатной температуре
в системе I коэффициент взаимодиффузии изменяется по кривой с максимумом,
а в системе II — по кривой с минимумом. При этом существенно подчеркнуть,
305
что положение минимума на концентрационной зависимости D совпадает с рас-
положением максимума бинодали фазовой диаграммы системы II. При посте-
пенном повышении температуры качество дегиптилфталата как растворителя
для полибутадиена улучшается и это приводит к изменению формы концентра-
ционной зависимости коэффициента взаимодиффузии, которая постепенно при-
ближается к наблюдаемой для системы I.
Величина коэффициента самодиффузии D* представляет собой меру микро-
вязкости системы и составляет верхний предел значений коэффициента взаимо-
диффузии D. Связь между этими величинами выражается формулой
О = Л* £1 — (1—4") Ф1 —2ХФ1Ф2^
где — концентрация растворителя (ф!=1—<р2); х—отношение парциальных
объемов компонентов (для высокомолекулярного полимера х 1)‘, <Pj и <р2 —
объемные доли растворителя и полимера в системе; % — константа Хаггинса,
характеризующая энергетические взаимодействия в системе полимер — раство-
ритель.
В области концентрированных растворов при % 0 и D = D* (1 — фг) =
= />*ф2. В области разбавленных растворов значения D для самых разных
систем во всех случаях по порядку величины близки к 10-’ см2/с, и различия
значений коэффициентов диффузии для разных диффундирующих низкомолеку-
лярных растворителей аналогичны разнице значений их вязкостей.
Концентрационная зависимость Z)* определяется, прежде всего, относитель-
ным свободным объемом раствора и описывается формулой, следующей из мо-
дели свободного объема, а именно:
1 Д* д Рф1
ln Р (0) /(0) /(0) + рФ1
где / (0) доля свободного объема полимера; D (0) — значение D* при ф2 =
= 0; Р — доля свободного объема растворителя; В — эмпирическая постоянная,
для процесса диффузии близкая к 0,3.
Хотя записанная, формула правильно представляет концентрационную
зависимость D*, ее практическое применение затруднено необходимостью неза-
висимого определения ряда входящих в нее констант. Поэтому теоретические
оценки коэффициентов диффузии (величины D* и практически более интересной
величины D) весьма недостоверны и обычно позволяют указать лишь ожидаемый
порядок значений коэффициентов диффузии в различных системах. В' этой связи
особо важны экспериментальные значения коэффициентов диффузии В. Многие
конкретные сведения содержатся в обзоре [2 д], который может использоваться
как полезное дополнение к материалу настоящей главы.
Согласно [2 д], по характеру концентрационной зависимости коэффициента
диффузии полимерные системы условно делятся на три группы. К первой отно-
сятся гибкоцепные неполярные полимеры (такие, как натуральный каучук,
полибутадиен, полидиметилсилоксан и др.), коэффициент диффузии растворителя
в которых в диапазоне значений <р2 от 0,2 до 1,0 изменяется всего в 3—5 раз,
Вторую группу образуют полимеры с более высокой жесткостью цепи (полиизо-
бутилен, полиметилметакрилат) выше, температуры стеклования; коэффициент
диффузии в этих полимерах в том же самом диапазоне концентраций изменяется
уже в десятки (в 30—40) раз. Наконец, третья группа — это полярные стекло-
образные полимеры (поливинилацетат, полистирол, поливинилхлорид, полиами-
ды)^ которых изменение <р2 даже на 10% приводит к варьированию скорости диф-
фузии в сотни раз. Однако все эти цифры имеют характер сугубо ориентировоч-
ных оценок, поскольку точные значения коэффициентов диффузии зависят не
только от природы полимера и концентрации раствора, но и от природы диффун-
дирующего растворителя, температуры, гидростатического давления и т. д.*] —
Прим. ред.
306
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
1 д. Чалых А. Е. и др., Высокомол.
соед., 1974, т. 16А, № 8,
с. 1844—1851.
2 д. Чалых А. Е. В кн.: Рейтлин-
гер С. А. Проницаемость поли-
мерных материалов. М., «Хи-
мия», 1974, с. 11—41.
ЛИТЕРАТУРА
1. Barter R. М., «Diffusion in and
through Solids», Cambridge Uni-
versity Press, London, 1941 2nd
ed. 1951.
2. Crank-J. and Park G. S. (Eds.),
«Diffusion in Polymers», Acade-
mic Press, London/New York,
1968.
3. Crank J., «The Mathematics of
Diffusion», Oxford University
Press, London, 1956.
4. Glasstone S., Laidler K. J. and
Eyring H., «The Theory of Rate
Processes», McGraw-Hill, New
York, 1941.
5. Barrie J. A., Chepter 8 in «Diffu-
sion in Polymers» (1968).
6. Duda J. L. and Vrentas J. S.,
J. Polymer Sci. A2, 6 (1968)
675.
7. Fujita H., Kishimoto A. and
Matsumoto K., Trans. Faraday
Soc. 56 (1960) 424.
8. Meares P., J. Am. Chem. Soc. 76
(1954) 3415; Trans. Faraday Soc.
53 (1957) 101; Trans. Faraday
Soc. 54 (1958) 40.
9. Michaels A. S. and Bixler H. J.,
J. Polymer Sci. 50 (1961) 393
and 50 (1961) 413.
10. Michaels A. S., Vieth W.' R.
and Barrie J. A.,.J. Appl. Phys.
34 (1963) 1 and 13.
11. Southern E. and Thomas A. G.j
Trans. Faraday Soc. 63 (1967)
1913.
12. Stannett V., Chapter 2 in «Diffu-
sion in Polymers» (1968).
13. Van Amerongen G. J., J. Appl.
Phys. 17 (1946) 972; J. Polymer
Sci. 2 (1947) 381; 5 (1950) 307;
Rubber Chem. Technol. 37 (1964)
1065.
14. Van Krevelen D. W., (1970)
unpublished work.
15. Журков C.H.j Рыскин Г., ЖЭТФ,
1954, p. 24, с. 797.
ЧАСТЬ
ПЯТАЯ
ФИЗИЧЕСКИЕ И ХИМИЧЕСКИЕ
ПРЕВРАЩЕНИЯ В ПОЛИМЕРАХ
глава хи КРИСТАЛЛИЗАЦИЯ
Кристаллизация полимеров определяется вероятностями процессов
образования центров кристаллизации и роста кристаллов. На оба
эти процесса влияет регулярность строения полимера. Для оценки
скорости кристаллизации удалось получить довольно интересные
соотношения.
Большинство веществ в чистом виде обладают определенной тем-
пературой плавления, ниже которой происходит переход от статисти-
ческой структуры жидкости к высокоупорядоченной периодической
кристаллической структуре. Такой переход называется кристаллиза-
цией.
Для расплавов высокомолекулярных соединений характерна вы-
сокая вязкость, которая быстро возрастает при охлаждении. Это
затрудняет кристаллизацию и в ряде случаев до такой степени,
что при охлаждении образуется твердое тело с аморфной стекло-
образной структурой. И только полимеры, обладающие довольно ре-
гулярными и гибкими молекулярными цепями, способны кристалли-
зоватйся достаточно быстро, несмотря на высокую вязкость расплава.
КРИСТАЛЛИЧНОСТЬ
Полимеры не могут быть полностью кристаллическими, т. е. не мо-
гут обладать совершенно регулярной кристаллической решеткой,
поэтому было введено понятие степени кристалличности. Смысл
этого понятия до сих пор все еще обсуждается многими исследова-
телями (см. гл. II). В соответствии с первоначальной мицеллярной
теорией кристаллизации полимерный материал состоит из большого
числа небольших кристаллитов, т. е. упорядоченных участков ми-
целл; эти кристаллиты распределены в материале случайным обра-
зом и связаны друг с другом окружающими их аморфными участ-
ками. Сами же полимерные молекулы частично принадлежат кристал-
литам и частично — аморфным участкам.
Было показано, что многие твердые полимеры состоят в основ-
ном из складчатых ламелей, а уширение линий дифракции рент-
геновских лучей должно рассматриваться как результат либо
мозаичной структуры вещества, либо разупорядоченности внутри
ламели. Однако далеко не все экспериментальные данные соот-
ветствуют этим крайним точкам зрения.
308
Таблица XIX.1. Характеристические параметры кристаллизации
полимеров
Полимер макс *g йй Ь •й» Ь г’макс (опыт), мкм/мин Z 04 д и е 6 и макс* (рачет), мкм/мин
Полиэтилен 0,80 414 153 2000 2 2 0 5000
Полипропилен (изо- тактический) 0,63 456 264 — 20 2 1 1 20
Полибутен-1 (изотак- тический). 0,50 415 249 — 9 2 1 2 4
Полиизопрен (цис) 0,45 313 206 248 — 4 2 1 20
Полистирол (изотак- тический) 0,32 513 373 449 0,25 2 1 6 0,13
Полиоксиметилен —. 455 191 — 400 2 1 0 310
Полиоксиэтилен — 339 206 — 3 2 0 3000
Полиоксипропилен — 348 201 290 50 3 1 1 4
Полиэтиленсукцинат — 381 272 — 10 8 4 0 (310)
Полиэтилентерефта- лат (0,50) 543 342 459 7 10 2 0 (8)
Поликарбонат А * (0,30) 540 423 — 0,01 12 1 1 0,015
Поликарбонат F * — 513 393 — 1 12 1 0 0,2
Поликарбонат S * —. 513 383 — 1 12 1 0 0,2
Найлон 6 0,35 502 330 413 200 7 5 0 1300
Найлон 6,6 0,70 540 330 420 -1200 7 5 0 1300
Полихлортрифторэти- лен 0,70 491 325 — 27 2 1 1 20
О
—\ R—\~О/—О—С—О—, здесь поликарбонат А:
F: R = — СН2—, поликарбонат S: R = — S —.
R = — c(CHa)j —, поликарбонат-
Различные определения степени Шульцу перечислены ниже: кристалличности хс по Кавешу
Удельный объем (о) va — V Хс — с Vq — Vc
Удельная теплоемкость (ср) н J ej ЪР Чй 1 1 Г* о Ъ
Удельная энтальпия (h) Лд h Хс~ ha —hc
Удельная энтальпия плавления (Дйт) xc = ^hm/\hcm
Массовый коэффициент экстинк- ции в ИК-области спектра е для xc — ел/ех —1 „(а)
характеристического типа коле-
баний
309*
Интенсивность рассеяния рентге-
новских лучей (единица—пло-
щадь выбранного максимума)
1С ~ 1д
(^а)расплав
Ядерный магнитный резонанс
площадь широкой
хс ___ компоненты
1 —хс площадь узкой, ком-
поненты
Кавеш и Шульц [12] критически рассмотрели смысл и способы
измерений степени кристалличности в полимерах. Очевидно, что
различные способы определения степени кристалличности могут
приводить к довольно разным значениям для одного и того же по-
лимера.
В табл. XIX. 1 приведены максимальные степени кристаллич-
ности, полученные экспериментально для различных полимеров. Не-
трудно заметить, что степень кристалличности максимальна у тех
полимеров, которые обладают высокой степенью молекулярной упо-
рядоченности вдоль цепи и наиболее простым строением мономер-
ного звена.
ОБРАЗОВАНИЕ ЦЕНТРОВ
КРИСТАЛЛИЗАЦИИ И РОСТ КРИСТАЛЛОВ
В процессе кристаллизации можно выделить два основных явления:
•образование центров кристаллизации и рост кристаллов.
СКОРОСТЬ ЛИНЕЙНОГО РОСТА (с)
"Скорость линейного роста сферолитов исследовали для ряда поли-
меров. Более или менее обобщенная диаграмма, описывающая это
явление, представлена на рис. XIX.1 (верхняя часть, а). Этот рису-
нок можно характеризовать следующими показателями: максималь-
ной скоростью роста рмакс (Tk) ПРИ температуре Tk; температу-
рами Т\ и Tjj, получаемыми путем пересечения оси температур ка-
сательными к кинетической кривой.
Для скорости линейного роста было получено следующее теоре-
тическое выражение (Беккер [8], Хоффман [11], Горник и Хоффман
12]):
(ДУ'1* \ ( m \
kT / ехР у у (Т,ц Т) / (XI Х.1)
где ДЕ* — свободная энергия активации переноса через границу фазы; С3 —
постоянная, значение которой дается формулой
(XIX.2)
тде Ъ» — толщина цепных молекул; — свободная энергия поверхности раз-
дела, параллельной цепи; — свободная энергия поверхности раздела, перпен-
дикулярной цепи; \Нт — теплота плавления единицы объема.
Согласно Фишеру и Тёрнбуллу [15], AF * характеризует энерге-
тический барьер, который препятствует передвижению кристалли-
310
зующейся единицы к поверхности раздела. Величина этого барьера
связана с подвижностью сегмента полимерной цепочки и, следо-
вательно, с показателями вязкостных свойств материала. Именно
Гц
Рис. XIX. 1. Зависимость скорости образования центров кристаллизации и роста,
кристаллов от степени переохлаждения расплава (а) и влияние термической'
предыстории образца на число центров кристаллизации (б):
I — стекло; 11— переохлажденная жидкость; 111 — жидкость; IV — скорость гомоген-
ного процесса образования центров кристаллизации/; V—скорость роста кристаллов v;
VI — метастабильная зона переохлаждения; VII — число центров кристаллизации N (гисте-
резисная петля).
поэтому AF* приближенно описывается соотношением Вильямса —
Лэндела — Ферри:
SF* = Л7?вЛФ= (XIX.3}
где Cj примерно равно 4120; Са равно 51,6, если AF* выражено вг-моль.
311
Окончательное выражение для скорости роста принимает, следова-
тельно, вид
( А Г 4boV||Y_Lym
г-уоехр^ я(С2 + Т — Tg) \_h&.HmT(Tm—T)
(XIX.4)
В принципе это уравнение позволяет рассчитать скорость роста
сферолитов в кристаллизующемся полимере, однако остаются опре-
деленные трудности, поскольку значения v0, уц и неизвестны.
Оледовательно, можно воспользоваться эмпирическим уравнением
для зависимости v от температуры. Такое уравнение было предло-
жено Манделькерном с соавторами [141:
In v = 24,5
Ер
~ RT
263ТО
Т(Тй~Т)
(XIX.5)
-где Ев и То — величины, которые устанавливаются для данного полимера.
Эти величины по Манделькерну даны в табл. XIX.2.
Таблица XIX.2. Значения постоянных в формуле (XIX.5)
*Полимер к ^7П’ . К ed- 10-«, кал/моль гй’ к ED/Tg. кал/моль-К
Полидекаметиленсебацинат 356 358 3,0
Полиоксиэтилен 347 339 5,5 206 27
Полиэтилен 419 414- 7,0 153 46
Полиоксипропилен 354 348 9,7 201 47,5
Полиоксиметилен 456 456 9,8 191 51
Полибутилен (изотактический) 407 415 10,6 249 43
Полидекаметидентерефталат 418 411 11,1 268 43
Полипропилен 438 456 12,0 264 45,5
Найлон 6,10 516 499 12,8 323 39,5
Найлон 6 505 502 13,5 330 41
Найлон 9,6 529 515 13,6 .—
Полихлортрифторэтилен 499 491 14,2 325 43,5
Найлон 5,6 541 531 14,6 318 46
Найлон 6,6 553 540 15,4 330 46,5
Полистирол (изотактический) 527 513 20,2 373 54
Сред- 44
нее
Коэффициент перевода в СИ: 1
кал/мопь=4,19 X 10* Дж/кмопь.
Вообще говоря, температура То близка к температуре плавления
кристаллического образца Т т, но все же не может отличаться от
последней более чем на 10 °C. Параметр ED, вероятно, возрастает при
повышении температуры перехода в стеклообразное состояние. Од-
нако указанные правила выполняются с большим разбросом.
.312
МАКСИМАЛЬНАЯ СКОРОСТЬ
ЛИНЕЙНОГО РОСТА
Эмпирическое соотношение для максимальной скорости роста может
базироваться на том экспериментальном факте, что при высокой
регулярности молекулярной структуры скорость роста оказывается
также высокой. Ван Кревелен и Хофтицер показали, что довольно
Рие. XIX.2. Соотношение между
максимальной скоростью роста кри-
сталлов и максимальной степенью
кристалличности:
1 — полиэтилен; 2 — найлон 6,6; з —
найлон 6; 4 — полихлортрифторэтилен;
5 — полипропилен; "в — полибутадиен;
1 — полиэтилентерефталат; в — полисти-
рол; 9 — поликарбонат.
точное первое приближение можно получить с помощью следующего
правила:
(Лртх • 1
2 1 + Р
(Х1Х.6>
где Z — число атомов цепи в мономерном звене; псн. — число метильных,
или других эквивалентных групп в мономерном эвене; р — характеристика;
объема боковой группы, выраженная числом атомов углерода или эквивалентных
ему атомов в боковой группе.
В качестве групп, эквивалентных СН2, считаются группы —CF2—
и в еще большей степени — группы —СХ2— и'—S—, когда послед-
ние служат мостиками между ароматическими кольцами в цепочке..
Атом хлора считается эквивалентным группе СН3 в боковой цепи.
х В табл. XIX.1 приведены рассчитанные-^ измеренные на опыте
значения Умакс. Соответствие между ними удовлетворительное.
Другим интересным эмпирическим соотношением оказывается
связь между максимальной степенью кристалличности xCt иаке и
Гмакс- Как видно из рис.Х1Х.2, такая связь действительно существует.
313
Но это не является неожиданным, ибо весьма простое и регуляр-
ное строение мономерного звена обусловливает высокие степень
кристалличности и скорость роста кристаллов.
ТЕМПЕРАТУРА, СООТВЕТСТВУЮЩАЯ
МАКСИМАЛЬНОЙ СКОРОСТИ КРИСТАЛЛИЗАЦИИ
В табл. XIX.1 приведены температуры, соответствующие максималь-
ной скорости роста сферолитов (Tk), и для сравнения температуры
стеклования и плавления. Следующие соотношения позволяют по-
лучить довольно хорошее приближение:
П/Гй = 1,3; Tk/Tm 0,82; Tk/(Tg+Tm)=0,5 (XIX.7)
Пример 1
Для стереорегулярного полиоксипропилена оценить: а) температуру макси-
мальной скорости кристаллизации; б) скорость линейного роста при этой темпе-
ратуре; в) вероятную степень кристалличности.
Решение
а. С помощью уравнения (XIX.7) при 7’g=201K, Тт= 348К нахо-
дится Т*:
Tk = 0.5 + Тт) = 0,5 (201 + 348) = 275 К
Экспериментальное значение Tk — 290 К; соответствие вполне удовлетворитель-
ное.
6. Расчет основан на использовании уравнения Манделькерна (XIX.5).
Поскольку ED= 44Tg = 44-201 = 8800 кал/моль, Т» Тт= 348 К и Г = Tk =
= 275 К, то
2,31g омакс = 24,5—8800/(2 • 275)—263 -348/275 (348 —275) = 3,7; lg vMaKC = l,6;
^мякс === 40 мкм/мин
С помощью уравнения (XIX.6) находится vm-.
А’макс = 5000 (1/3 • 1/(1 + 1)а = 4 мкм/мин
Экспериментальное значение составляет 50 мкм/мин.
в. По рис. XIX.2 можно найти, что вероятная степень кристалличности
составляет 50—60% для значений рмакс между 4 и 40 мкм/мин.
I ★ Соотношение между равновесной температурой плавления Тт и ско-
ростью кристаллизации при различных степенях переохлаждения АТ было
детально проанализировано Ю. К. Годовским (см. Высокомол. соед., 1969,
т. II, А, № 10, с. 2129—2134) на примере 23 полимеров различного химического
строения. В качестве количественной характеристики кинетики кристаллизации
был выбран полупериод процесса то,5. Зависимость То,б от АГ носит экстремаль-
ный характер, и температура ТХакс. кр, при которой полупериод то,а-минимален,
имеет смысл температуры максимальной скорости кристаллизации. Обработка
экспериментальных данных показала, что для большого числа полимеров
Гмакс.кр = (0,82 — 0,83) Тт- Численный коэффициент в этой зависимости
совпадает с приводимым Ван Кревеленом.
Однако скорости кристаллизации полимеров разного строения при различ-
ных АГ, в том числе и при температуре максимальной скорости кристаллиза-
ции, различаются на десятичные порядки, что дает основание классифицировать
те или иные полимеры при сопоставимых условиях сравнения как «медленно»
или «быстро» кристаллизующиеся материалы.
При повышении Тт не только возрастает 7'макс. кр (при этом отнюдь не обя-
зательно, чтобы снижался To,s), но и расширяется температурный диапазон,
314
в котором возможна кристаллизация полимера. Действительно, этот диапазон;
определяется разностью между температурами плавления и стеклования
(Тт Tg), a Tg можно оценить с помощью эмпирического правила Tg
(V2 - 2/3) Тт (см. гл. VII). Отсюда следует, что разность (Тт— Tg)
(0,33 — 0,5) Тт и возрастает с повышением Тт-*] — Прим. ред.
ОБРАЗОВАНИЕ ЦЕНТРОВ КРИСТАЛЛИЗАЦИИ
Скорость гомогенного процесса образования зародышей J должна
быть весьма сходной со скоростью линейного роста, поскольку оба
процесса — это образование центров кристаллизации. Первый осу-
ществляется на трехмерном центре, второй — на двумерном центре
или на кристаллической поверхности. Температурная зависимость J
описывается следующим соотношением [15]:
J— Joexp R ) ехр
1
к (ЬНт)г Т(Тт-Т)2]
(XIX. 8)
где Jo — константа, пропорциональная числу эффективных молекулярных
сегментов в единице объема.
На рис. XIX.1 представлены графики температурных зависимо-
стей скоростей гомогенного образования зародышей кристаллизации
и роста кристаллов.
Для полимерных молекул температурный интервал ниже равно-
весной температуры плавления представляет собой метастабильную
зону, в которой скорость образования зародышей практически нуле-
вая, но при этом однажды образовавшиеся кристаллы могут расти.
Ниже этой метастабильной температурной зоны центры кристалли-
зации могут возникать спонтанно как по гомогенному, так и по гете-
рогенному механизму. Однако в процессе дальнейшего охлаждения
вещества достигается зона высокой вязкости, в которой снова ско-
рость образования центров кристаллизации падает и скорость роста
становится практически нулевой. Скорости образования центров
кристаллизации и роста кристаллов проходят через максимум, по-
скольку при более высоких температурах снижается движущая сила
процесса (перенасыщение), а при более низких температурах ско-
рость массопереноса резко падает из-за высокой вязкости среды.
Гомогенное образование центров кристаллизации, сопровождаю-
щееся ростом кристаллитов, может осуществляться только в том
температурном диапазоне, в котором обе кривые взаимно перекры-
ваются. Предполагается, что метастабильная зона переохлаждения
(перенасыщение) возникает вследствие более высокой растворимости
микроскопических зародышевых кристаллитов по сравнению с макро-
кристаллами й, следовательно, из-за более высокой энергии актива-
ции процесса образования зародышей кристаллизации по сравнению
с процессом роста кристаллов.
Очень трудно изучать гомогенный процесс образования центров,
кристаллизации, поскольку неоднородности, всегда имеющиеся в
315
полимерных расплавах, резко ускоряют гетерогенный процесс обра-
зования зародышей *.
Как было показано, для многих полимеров образование центров
кристаллизации связано с термической предысторией образца. Этот
процесс зависит от условий до начала кристаллизации, а также от
температуры плавления и длительности нахождения образца в со-
стоянии расплава. Очень маленькие участки с высокой степенью упо-
рядоченности, зачастую стабилизированные неоднородностями, мо-
гут существовать в расплаве длительное время и действовать как уже
образовавшиеся центры кристаллизации при дальнейшем охлажде-
нии. Количество и размеры центров кристаллизации, остающихся
в расплаве, определяются тремя факторами: температурой предвари-
тельной кристаллизации, температурой расплава и длительностью
изотермической выдержки расплава.
Для очень медленно кристаллизующегося полимера можно на-
блюдать интересные эффекты [9, 10]. При сильном переохлаждении
возникают «индуцированные» центры кристаллизации, которые
могут вырасти в эффективные центры кристаллизации при более
высоких температурах. Такая кристаллизация сильно переохлажден-
ного полимера полностью определяется этими индуцированными цен-
трами, количество которых превосходит число сохраняющихся в рас-
плаве зародышей на несколько порядков величины. Количество ин-
дуцированных центров можно уменьшить путем очистки полимера.
Если охлажденные полимеры нагревают до температур, лежащих
выше точки плавления, то индуцированные центры кристаллизации
при этом разрушаются и остаются сохраняющиеся в расплаве заро-
дыши, число которых невелико (нижняя часть рис. XIX.1).
СУММАРНАЯ СКОРОСТЬ КРИСТАЛЛИЗАЦИИ
“Суммарная скорость кристаллизации переохлажденной жидкости
определяется двумя уже перечисленными факторами: скоростью об-
разования зародышей размерами больше критических и скоростью
роста таких зародышей до размеров окончательных кристаллических
агрегатов.
Обычная методика изучения скорости кристаллизации заклю-
чается в быстром охлаждении полимерного образца от точки расплава
до температуры измерения и в последующем измерении скорости кри-
сталлизации при постоянной температуре (изотермическая кристал-
лизация).
Если в процессе кристаллизации образуются хорошо оформи-
вшиеся сферолиты, видимые в микроскоп, иногда удается одновре-
♦ Связанные с этим методические трудности были удачно преодолены в ра-
боте: Горник Ф.,Росс Дж., Фроули Л. В кн.: Физическая химия поли-
меров за рубежом. Пер. с англ. Под ред. 3. А. Роговина и А. Я. Малкина.
М., «Мир», 1970, с. 51—67 (по J. Polymer Sci., 1967, v. 18, р. 79—91). —
Прим. ред.
316
менно проследить за скоростью образования зародышей кристалли-
зации и за скоростью роста сферолитов (в ммкм/мин). Так как ра-
стущие сферолиты начинают мешать развитию друг друга, измерения
следует проводить на ранних стадиях кристаллизации.
Если образование центров кристаллизации и рост кристаллитов
нельзя изучать раздельно, то можно проследить за интегральным
эффектом превращения аморфного материала в кристаллический
с помощью какого-либо метода, позволяющего определить степень
кристалличности? Например, можно проводить измерения удельного
объема, Поместив кристаллизующийся образец в дилатометр. Как
правило, определяют скорость кристаллизации при заданной темпе-
ратуре как обратную величину времени, необходимого для достиже-
ния половины конечной степени кристалличности (io%)-
Согласно Аврами [7], развитие процесса кристаллизации при
постоянной температуре можно выразить следующим уравнением:
*(() = ! — exp (—Ktn) > (XIX.9)
где х (()— доля закристаллизовавшегося материала к моменту времени t;
X и п — постоянные.
В постоянную К входят параметры образования центров кри-
сталлизации и роста кристаллов; п — теоретически должно быть
целым числом, значение которого определяется механизмом образо-
вания зародышей и характером роста кристаллов. Численное значе-
ние К непосредственно связано с суммарной скоростью кристалли-
зации 17*5 формулой
7f = ln2(zoM" (XIX.10)
Теоретически вычисленные значения п и К приведены в табл. XIX.3.
Таблица XIX. 3. Значения постоянных п и К в уравнении Аврами
Форма кристаллов Тип образования центров кристаллизации
предопределенный (постоянное число центров в 1 см*) спонтанный (постоянная скорость)
п К п К
Сферолитная (шарики) 3 4 n/3i>3Jp*
Дискообразная (пластины) 2 nbv^Np* 3 -у- bv Vp*
Фибриллярная (палочки) 1 fvNp* 2 f Т * — Г/р*
Примечание. Ь — толщина пластин; / — поперечное сечение палочек; р* — отно-
сительная плотность рс/р; JV—число центров в единице объема; J—скорость образо-
вания центров кристаллизации в единице объема; о — скорость роста кристаллов.
317
Когда полимерный образец быстро охлаждается от температуры
расплава до температуры, при которой производятся измерения,
скорость кристаллизации сначала оказывается весьма небольшой.
После завершения индукционного периода скорость этого процесса
достигает максимальной величины, а затем снова уменьшается по
мере приближения структуры к конечному равновесному состоянию.
Большинство полимеров кристаллизуется с измеримой скоростью
в определенном диапазоне температур, который характерен для
каждого полимера. Этот диапазон может охватывать температуры
от точки примерно на 10 °C ниже температуры плавления до точки,
примерно на 30 °C превышающей температуру стеклования.
Скорость кристаллизации возрастает при уменьшении темпера-
туры ниже точкй плавления, достигает максимального значения
при Tk и снова понижается при последующем охлаждении.
Вряд ли существует какой-либо класс материалов, кроме поли-
меров, для которого объемные характеристики определяются кине-
тическими закономерностями. Вследствие самой природы процессов
образования центров кристаллизации и роста кристаллов даже после-
дующий длительный отжиг образцов не может полностью устранить
эффектов, связанных с предысторией образца.
ЛИТЕРАТУРА
1. Geil Р. Н., «Polymer Single Crys-
tals», Interscience, New York,
1963.
2. Gornick F. and Hoffman J. D.,
«Nucleation in Polymers», Ind.
Eng. Chem. 58 (1966) 41.
3. Keller A., «Growth and Perfec-
tion of Crystals», Wiley, New
York, 1958.
4. Mandelkern L., «Crystallization
of Polymers», McGraw-Hill, New
York, 1964.
5. Stuart H. A., «Die Physik der
Hochpolymeren», Vol. IV, Sprin-
ger, Berlin, 1956.
6. Uhlmann D. R. and Chalmers B.,
«Energetics of Nucleation», Ind.
Eng. Chem. 57 (1965) 19.
7. Avrami MJ. Chem. Phys. 7
(1939) 1103; 8 (1940) 212; 9
(1941) 177.
8. Becker R., Ann. Physik 32 (1938)
128.
9. Boon J., Thesis, Delft, 1966.
10. Boon J., Challa G. and Van Kreve-
len D. W., J. Polymer Sci. A2,
6 (1968) 1791, 1835.
11. Hoffman J. D., J. Chem. Phys.
28 (1958) 1192; SPE Trans. 4
(1964) 315.
12. Kavesh S. and Schultz J. M.,
Polymer Eng. Sci. 9 (1969) 5.
13. Magill J. H„ Polymer 3 (1962)
43; J. Appl. Phys. 35 (1964) 3249.
14. Mandelkern L., Jain N. L. and
Kim H„ J. Polymer Sci. A2, 6
(1968) 165.
15. Turnbull D. and Fisher J. C., :
J. Chem. Phys. 17 (1949) 71.
16. Van Krevelen D. W. and Hofty-
zer P. J. (1970) unpublished
work.
ГЛАВА XX.
ОРИЕНТАЦИЯ
Ориентация играет чрезвычайно важную роль в практических
приложениях, особенно при формовании из полимеров волокон и
пленок. Ориентация совершенно явно влияет на физические свой-
ства полимеров.' Получены некоторые интересные количественные
соотношения между коэффициентом удлинения, степенью ориента-
ции и механическими характеристиками полимерных материалов.
Однако, исходя из строения полимеров, пока не удается получить
значения констант, входящих в эти соотношения.
Если изотропный полимерный материал подвергается действию
направленного внешнего напряжения, то в образце осуществляется
перестройка структурных элементов, называемая ориентацией.
В аморфных полимерах ориентация заключается в простой пере-
группировке статистически свернутых в клубки цепных молекул
(молекулярная ориентация). В кристаллических полимерах явление
ориентации оказывается более сложным: кристаллиты могут пере-
ориентироваться или перегруппироваться, кроме того, под влиянием
приложенного напряжения может возникнуть направленная кри-
сталлизация. Подобные эффекты в кристаллических материалах
можно наблюдать с помощью методов рентгеновской дифракции.
Практически все полимерные изделия обладают некоторой ориен-
тацией. При образовании (формовании) данного образца молекулы
ориентируются в процессе вязкого течения, и ориентация частично
«замораживается» при охлаждении изделия. Однако этот тип ориен-
тации менее важен по сравнению с направленной ориентацией, воз-
никающей в процессах вытяжки.
Ориентация связана с деформацией полимера при температуре
стеклования или выше этой температуры. Закрепление развившейся
ориентации происходит тогда, когда растянутый полимер охлаждают
до температуры, лежащей ниже области стеклования, прежде чем
восстановится статистическая ориентация макромолекул. При нагре-
вании выше температуры стеклования ориентированный полимер
стремится сократиться; в аморфных полимерах сила, возникающая
при таком сокращении, является непосредственной мерой степени
достигнутой ориентации.
Ориентация резко влияет на физические характеристики поли-
меров. Характеристики ориентированных полимеров весьма сущё-
319
ственно различаются для разных направлений, т. е. полимеры ста-
новятся анизотропными. С помощью ориентации, например, можно
превратить хрупкие образцы (полистирол, полиметилметакрилат)
в пластичные, если напряжение прикладывается в направлении
ориентации.
Ориентированные полимерные материалы можно довольно грубо
разделить на два класса: материалы с одоосной ориентацией и мате-
риалы с двухосной ориентацией.
СТЕПЕНЬ ОРИЕНТАЦИИ
Ориентацию можно измерить с помощью ряда методов.
1. Двойное лучепреломление довольно часто оказывается наи-
более простым методом [8, 10, 20]. Двойное лучепреломление
(Аи) волокна определяется как разность показателей преломления
пучков света, распространяющихся параллельно и перпендикулярно
оси волокна (Аи = Иц — и^). В двойное лучепреломление вносят
вклад аморфные и кристаллические участки.
Усиление двойного лучепреломления вследствие ориентации
кристаллических полимеров обусловлено повышением средней сте-
пени ориентации поляризуемых цепных сегментов макромолекул.
В кристаллических участках эти сегменты вносят более существен-
ный вклад в суммарное двойное лучепреломление по сравнению
с менее упорядоченными участками. Кроме того, средняя ориента-
ция таких кристаллических участков, выступающих в роли струк-
турных единиц, может существенно отличаться от средней степени
ориентации сегментов.
2. Дифракция рентгеновских лучей в кристаллических полимерах.
Характер дифракции рентгеновских лучей для неориентированных
кристаллических полимеров сходен с наблюдаемым для порошко-
образных образцов низкомолекулярных кристаллических соедине-
ний; в обоих случаях дифрактограммы представляют собой набор
кольцевых рефлексов, а не отдельных точек. В результате ориента-
ции кольцевые рефлексы превращаются в дуги и точки. Степень
ориентации кристаллических участков можно рассчитать по азиму-
тальному распределению интенсивности рассеяния в этих дугах [16].
3. Дихроизм ИК-спектра. В ориентированных образцах степень
поглощения поляризованного ИК-излучения может сильно изме-
няться в зависимости от направления плоскости поляризации. Если
деформационные колебания определенных функциональных групп
осуществляются с изменением дипольного момента относительно
оси молекулы, то соответствующие полосы поглощения оказываются
более сильными для излучения, поляризованного перпендикулярно
оси цепочки, и более слабыми для излучения, поляризованного
параллельно этой оси. Иногда удается наблюдать раздельно погло-
щение для кристаллических и аморфных областей; тогда коэффи-
циент дихроизма такой полосы позволяет получить сведения отно-
сительно ориентации элементов в обоих участках.
320
4. Поляризация флуоресценции [17, 19]. Этот сравнительно не-
давно разработанный метод позволяет получать информацию не
только о степени ориентации в аморфных участках полимерных
образцов, но и относительно функции распределения макромолекул
по направлению ориентации. ВТ этом отношении названный метод
гораздо более полезен по сравнению с другими.
Количественные аспекты влияния ориентации на различные физи-
ческие характеристики полимеров будут рассмотрены позже.
ОДНООСНАЯ ОРИЕНТАЦИЯ
Одноосная ориентация сопровождает растяжение волокна, полоски
или прутка, осуществляемое в одном направлении. Обычно этот
процесс проводят при температуре, лежащей выше области стекло-
вания. Полимерные цепочки стремятся расположиться параллельно
направлению вытяжки, однако в действительности такая тенденция
реализуется только для небольшой части сегментов цепей, которые
ориентируются полностью.
Одноосная ориентация играет весьма большую роль в производ-
стве синтетических волокон, поскольку только путем вытяжки нить
приобретает стабильные размеры и теряет склонность к ползучести,
по крайней мере при комнатной температуре. Обычно для достиже-
ния ориентации, соответствующей оптимальным физическим харак-
теристикам, требуется отдельная стадия вытяжки. На практике
это осуществляется с помощью набора валков, вращающихся с раз-
ной скоростью.
Как уже отмечалось, ориентация существенно влияет на физиче-
ские свойства полимера. Ориентация повышает прочность при рас-
тяжении и жесткость. Конечно, при повышении степени ориентации
анизотропия свойств также усиливается. Ориентированные волокна
обладают прочностью в направлении оси вытяжки, но оказываются
довольно непрочными в перпендикулярном направлении.
Если процесс ориентации полукристаллических волокон про-
водят при температуре существенно ниже точки плавления поли-
мера, то волокно становится тоньше не постепенно, а скачкообразно
в узкой области из-за образования шейки. Следует заметить, что
образование шейки может наблюдаться и для аморфных полимеров.
Так называемая степень вытяжки (Л) представляет собой отно-
шение длины растянутого волокна к длине волокна до начала вы-
тяжки. Для многих полимеров величина степени вытяжки соста-
вляет примерно от 4 до 5. Однако границы этого диапазона дости-
гают, с одной стороны, 10 для линейных полиолефинов и, с другой
стороны, 2 для регенерированной целлюлозы.
В процессе вытяжки степень кристалличности резко не изменя-
ется, если до начала процесса образец уже обладал развитой кри-
сталличностью. Если же, напротив, образец до вытяжки не был
кристаллическим или был умеренно кристаллическим, то вытяжка
может индуцировать высокую степень кристалличности. Так
21 Заказ 699 v 321
называемая «холодная вытяжка», например найлона 6,6 и найлона 6,
йроводится при более или менее адиабатических условиях. Энергия
процесса вытяжки выделяется в виде тепла, за счет которого повы-
шается температура и снижается сопротивление деформированию.
Как только при растяжении полимерного волокна достигается пре-
дел текучести, процесс деформирования становится неустойчивым
в механическом отношении и образуется шейка.
На протяжении процесса вытяжки кристаллиты стремятся рас-
пасться на составные элементы. Возможно, это обусловлено рас-
прямлением, или деспирализацией цепей. На первой стадии процесса
вытяжки сферолиты стремятся сохранить свою целостность и пре-
вращаются в эллипсоиды. Разрушение волокна может осущест-
вляться по границам между сферолитами. Следовательно, появление
сферолитов в неориентированном волокне можно отнести к недостат-
кам структуры материала. После завершения первой стадии обрати-
мой деформации сферолитов может начаться вторая стадия, на про-
тяжении которой сферолиты
разрушаются, а отдельные спи-
ральные цепочки (например,
полиамидов) ориентируются па-
раллельно оси волокна.
ИЗМЕНЕНИЕ ХАРАКТЕРИСТИК
ПОЛИМЕРОВ ПРИ ОРИЕНТАЦИИ
1. Аморфные полимеры. Модуль
Юнга одноосно ориентирован-
ных аморфных полимеров ока-
зывается больше в направле-
нии, параллельном ориентации,
по сравнению с модулем в пер-
пендикулярном Направлении;
однако при малых степенях
ориентации этот эффект неве-
лик. Для того чтобы получить
значение модуля Юнга в напра-
влении, параллельном ориента-
ции, в 2 раза превышающее ве-
личину модуля неориентирован-
ного аморфного полимера, не-
обходимо реализовать весьма
высокие степени ориентации.
Гораздо сильнее влияет ори-
ентация на прочность при рас-
тяжении и на разрывное удли-
нение [е = (1 — & также
на ударную вязкость полимера.
На рис. ХХ.1 сказанное иллю-
Рис. ХХ.1. Влияние ориентации на
разрушающее напряжение при растяже-
нии (а), относительное удлинение при
разрыве (б) и ударную вязкость (в)
пленки полистирола [14].
322
Таблица XX. 1. Влияние степени вытяжки на свойства волокон
полизтилентерефталата [22]
Показатели Коэффициент вытяжки Л
1 2,77 3,08 3,56 4,09 4,49
Плотность р при 20 °C, г/см? 1,3383 1,3694 1,3775 1,3804 1,3813 1,3841
Степень кристалличности хс, 3 22 37 40 41 43
Двойное лучепреломление Ап при 20 °C 0,0068 , 0,1061 0,1126 0,1288 0,1368 0,1420
Разрывная прочность пмакс> гс/текс 11,8 23,5 32,1 43,0 51,6 64,5
Разрывное удлинение, % (450) 55 39 27 11,5 7,3
Модуль Юнга, дин/см2 •1О'1о Е' при 9370 Гц ... 8,3 12,3 17,4 20,2 22,9
Е' при 5 Гц 2,6 7,8 11,5 14,9 18,0 19,9
Коэффициент потерь tg б 4 0,20 0,165 0,155 0,135 0,12
Tg (динамич.), °C .... Т j (максимум демпфирова- ния), °C ......... 71’ 72 83 85 90 89
84 107 118 124 128 131
ATg (ширина максимума по- терь в области перехода) 10 39 43 45 50 55
Рис. XX.2. Зависимость динамического модуля Юнга для каучука от сте-
пени удлинения при частоте 1 кГц [18].
£23
стрируется данными, полученными при испытании полосок из ори-
ентированного аморфного полистирола [14]. Параллельно ориен-
тации прочность при растяжении (<тмакс) и разрывное удлинение
Рис. ХХ.З. Диаграмма напряжение — деформация не Ван-дер-Мееру. Цифры
на кривых — степень вытяжки [22]:
а — найлон 6; б — полиэтилентерефталат.
(®иакс) увеличиваются в 3 раза, в то же время ударная вязковть
может увеличиться в 8 раз.
Ориентация столь же сильно влияет на характеристики эласто-
мерных материалов, но в этом случае в процессе измерений поли-
мер должен находиться в растянутом состоянии. На рис. XX.2,
взятом из работы Мейсона [18], приведены результаты динамических
механических испытаний полосок из натурального каучука. Инду-
324
цированная ориентацией кристаллизация проявляется' в этом кау-
чуке при удлинении выше приблизительно 200%; такая кристал-
лизация резко изменяет механические свойства материала. В про-
—-4
Степени
Иитимки
Рис. XX.4. Физические свойства полиэфира в зависимости от степени вытяжки
[22]:
« — плотность; б — кристалличность; в — двойное лучепреломление; г — модуль Юнга;
д — температура перехода; е — ширина максимума потерь.
цессе ориентации модуль Юнга быстро увеличивается. Увеличи-
вается и модуль потерь, но не столь быстро; в итоге коэффициент
демпфирования несколько понижается.
2. Кристаллические полимеры. В практическом отношении наи-
более важным оказывается влияние одноосной ориентации на полу-
кристаллические полимеры, поскольку характеристики многих
325
синтетических волокон определяются именно ориентацией. Влияние
ориентации в этом случае можно изучать на образцах, подвергну-
тых разной степени вытяжки. На рис. XX.3 а и б представлены дан-
ные для полиэфирных волокон и волокон из найлона 6. Дополни-
тельные данные приведены в табл. ХХ.1 и на рис. ХХ.4 [22].
КОЛИЧЕСТВЕННЫЕ СООТНОШЕНИЯ
Ниже приводится сводка количественных соотношений между неко-
торыми физическими величинами после ориентации полимеров.
1. Плотность. Плотность изменяется лишь в том случае, когда
осуществляется индуцированная кристаллизация. При этом -урав-
нение (IV. 2) сохраняет свою силу:
Рполукрист/Раморф 1 -|-0,13хе (ХХ.1)
2. Тепловое расширение. Коэффициент объемного теплового рас-
ширения ориентированного полимера такой же, как у изотропного
материала. Для одноосной ориентации коэффициент объемного рас-
ширения (а) связан с коэффициентами линейного расширения в па-
раллельном и перпендикулярном оси направлениях (0ц и 0±) фор-
мулой
а = ЗРиво = Р || +2Рд_ (XX.2)
По данным измерений Тьядера и Протцмана [21], выполненных для
растянутого полиметилметакрилата, 0± и 0ц зависят от степени
вытяжки:
₽±/Рц-1 ~(А-1)/3 (ХХ.З)
Отсюда
3|| ~-3 2 <ХХ'4а)
1+-д-(А-1)
и
а i+4-(a-1)
Р± ~ 3 2 (XX.46)
1+т(А-1)
Для ориентированных кристаллических полимеров аналогичных
соотношений в литературе пока нет.
3. Теплопроводность “к. Айерманн [И] получил выражение,
эквивалентное (XX.2):
3 12
"Г---= _1— + "Г- (ХХ.5)
Лизо Л [| Л_[_ --
326
Кроме того, он показал, что для аморфных полимеров соотношение
между X и р имеет вид
^^2 4-0,8^ (XX.6)
Лорнент Ризо
где Рорнент— коэффициент линейного расширения ориентированного материала
(любая из двух, рп или величина).
Комбинация (XX.4), (XX.5) и (XX.7) позволяет рассчитать теп-
лопроводность ориентированных аморфных полимеров в зависимости
от степени вытяжки. Аналогичных соотношений для ориентирован-
ных кристаллических полимеров нет.
4. Двойное лучепреломление. Для показателя преломления спра-
ведлива формула, аналогичная (XX.2); формула получена Херман-
сом [13] в предположении, что плотность при ориентации не из-
меняется:
3«изо «ц+2«± (XX.7)
Как уже отмечалось, разность пц — = Ап называется двойным
лучепреломлением. В табл. XX.2 приведены некоторые данные по
двойному лучепреломлению волокон.
Таблица ХХ.2. Некоторые числовые данные по двойному
лучепреломлению волокон [4] \
Волокно "II п± Лп
Полиэтилентерефталат 1,725 1,537 . 0,188
Льняное волокно 1,596 1,528 0,068
Найлон 6,6 1,528 1,519 0,063
Найлон 6 1,580* 1,530* 0,050 *
Шелк 1,591 1,538 . 0,053
Хлопок 1,578 1,532 0,046
Полиэтилен 1,556 1,512 0,044
Вискозный шелк 1,539 1,519 0,020
Шерсть 1,553 1,542 0,010
Ацетат целлюлозы (вторичный) 1,539 1,519 0,020
Полиакрилонитрил 1,500 1,500 0,000
Стеклянное волокно 1,547 1,547 0,000
Триацетат целлюлозы 1,474 1,479 0,005
* Измерения проведены де-Фризом.
Общий характер соотношения между двойным лучепреломле-
нием и коэффициентом вытяжки (Л) для изотропного состояния поли-
меров получил де-Фриз [10]:
d (Дм)
<Ип"л" = т+^Дга
(ХХ.8)
где т к р — постоянные.
327
После интегрирования уравнение (XX.9) принимает вид (для
р¥=0):
(XX.9)
где d„30 nd— диаметры волокна в изотропном и ориентированном состояниях.
Это выражение позволяет оценить степень вытяжки волокна,
если значение Ап определено и т и р известны.
Де-Фриз [10] привел значения р и т для некоторых полимеров.
Эти значения даны в табл. XX.3.
Таблица ХХ.З. Значения постоянных р и т в уравнениях (XX.9) и (XX. 10)
Полимер Тип деформации р т Литера- турный источник
Регенерированная цел- Вытяжка в сухом воздухе 1,2 0,024 [10]
люлоза Вытяжка при намотке 1,0 0,012 [13]
Полистирол Горячая вытяжка 1,0 0,002 [9]
Полиэтилентерефталат Холодная вытяжка 0,5 0,094 [Ю]
Горячая вытяжка 0,5 0,083 [Ю],[15]
Найлон 6 Холодная вытяжка -0,5 0,050 [Ю]
Найлон 6,6 Холодная вытяжка -1,0 0,072 [Ю]
Линейный полиэтилен Горячая вытяжка -0,5 0,038 [Ю]
Важное следствие из этих данных заключается в том, что р поло-
жительно, если в полимерные цепи входят кольца, и отрицательно,
если полимерные цепи — чисто алифатические. Де-Фриз дал следу-
ющую качественную схему соотношений между значениями р и
химическим строением:
Одно повто- ряющееся звено Два чередую- щихся звена
Макромолекулы с али- Р = -1/2 р = — 1
фатическими звеньями
Макромолекулы с аро- р = +1 р = +1/2
магическими звеньями
В соответствии с (XX.9) т представляет собой тангенс угла наклона
начального участка кривой зависимости Дп от In Л.
5. Модуль упругости. Модуль упругости ориентированного поли-
мера всегда выше, чем неориентированного. Де-Фриз [10] вывел
общее правило для модуля упругости Е полукристаллических
полимеров после ориентации, измеренного в области звуковых
частот:
г 1 1 \
1пЛ = Сл(^-^) (ХХ’В * 10)
где Сл — константа, характерная для данного полимера.
В табл. XX.4 приведены значения Сд для некоторых полимеров.
При высоких степенях вытяжки могут возникать отклонения от
уравнения (ХХ.10), как это показано на рис. XX.5.
328
Таблица ХХ.4. Значения С\, Ееза, СЁ и Ео для некоторых полимеров
(данные [20] и неопубликованные ревулътаты измерений)
Полимер Вытяжка Удлинение
Сд Еизо Еизо/СД се Ео
Регенерированная целлюло- за (медноаммиачный процесс) — — — 9,0 20 2,2
Натуральный шелк — — — 8,5 17 2,0
Поливиниловый спирт — — — 7,3 34 4,7
Регенерированная целлюло- за (вискозный процесс) 8 5,0 0,63 5,5 10—40 1,8—7,3
Кератин (волосы) — — — 3,7 6,0 1,6 4,1
Полиакрилонитрил — — — 3,4 14
Полипропилен (изотактиче- ский) — — — 3,2 5,5 1,7
Политетрафторэтилен — — — 3,1 21 6,8
Полиэтилентерефталат 4 2,6 0,65 2,3 18,5 8,0
Природная целлюлоза (хло- пок) — — — 2,0 ♦* 16 8,0
Ацетат целлюлозы — — — 1,8 5 2,8
Полиэтилен (линейный) — — — 1,65 11,7 7,1
Поливинилхлорид — — — 1,6 7,0 4,4
Найлон 4 — — — 1,1 9,4 8,5
Найлон 6,6 — — — 0,9 0,65 7 7,8
Найлон 6 2,6 1,45 0,56 4,5 7,0
Найлон 11 — — — 0,4 5 12 5
* Типичные значения для волокон. Величины Сд,.Се, Еиз0 и Ео выражены
в io10 дин/см*. Модули упругости измерены при частоте приблизительно 9000 Гц.
** Измерено Вейландом [23].
Рис. XX.5. Влияние степени вытяжки на
модуль упругости Е' полиэфира:
Л - Е статич, Хервиг 12]; Q - ЕДннпри 5 Гц,
Ван-дер-Меер [22]; 4---Езвук при 9370 Гц,
Ван-дер-Меер [22].
Де-Фриз [10] получил сходное соотношение для удлинения 8
уже подвергнутого вытяжке волокна! .
In (1+8) = с. (^--40 (ХХ.Н)
где Ев — исходный модуль упругости волокна после вытяжки.
329
Константа Сг близка по порядку величины к С а, но не равна
последней.
Процессы вытяжки и растяжения ориентированного волокна
проводятся не при одинаковых условиях, поэтому имеет значение
различие между Сл и Се; отношение Сл/Се может, например, зави-
сеть от температуры.
В табл. XX.4 приведены значения Сг. Между Се и химическим
строением полимеров не удалось обнаружить какой-либо корреля-
ции. Низкое значение Се для хлопчатобумажных волокон обусло-
влено, вероятно, спиральным строением целлюлозы.
Аналогию между формулами (XX, 10) и (XX.И) нетрудно понять,
поскольку ориентационная вытяжка и растяжение ориентирован-
ного волокна в принципе очень сходны друг с другом.' если волокно,
растянутое в Ло раз, с начальным модулем упругости Ео (при таких
же условиях) было бы подвергнуто дальнейшему удлинению е,
то результирующая степень вытяжки стала бы:
Апо^кая = Ло (1 те) (XX.12)
Подстановка (XX.12) в (XX.10) приводит к следующему соотноше-
нию между модулем упругости и двойным лучепреломлением:
(ххлз)
г
Пример 1
Оценить модуль упругости в области звуковых частот волокна из найлона 6
при степени вытяжки 3,5. Для изотропного найлона 6 модуль упругости соста-
вляет 1,45 -IO10 дин/см2.
Решение
По табл. XX.4 СЛ для найлона 6 составляет 2,6-1О1о дин/см2. Использова-
ние выражения (XX. 11) дает:
!/£= 1/£изо-1нЛ/СЛ = (1/1,45—2,31g3,5/2,6).10-ю = (0,69-0,48)10-ю =
= 0,21-10-10
или
2? = 4,75 • 1010 дин/см2
Экспериментально измеренное значение Е составляет 4,4-101°.
Пример 2
Оценить двойное лучепреломление такого же волокна из найлона 6.
Решение
В табл. XX.3 приведены- следующие величины для найлона 6:
р = —0,5 и т = 0,05
Выражение (XX.10) позволяет получить:
Дя = (—0,05/0,5) (3,5-0.6 —1) = —0,1 (1/1,87 —1) = 0,047 •
330
о
о,os о,to nis
Такой же результат получится и с помощью выражения (XX.14);
2,3 lg (1 —10 Ди) = —0,5 2,6 • 10Ю (1 /1,45 — 1 /4,75) 10-ю
lg (1 -10 Дп) = -(0,5 • 2,6/2,3) (0,69 - 0,21) = -0,27
1 —10 Дп = 0,53
Дп = 0,47/10 = 0,047
В отличие от модуля Юнга мо-
дуль сдвига практически не зависит
от ориентации, однако предел проч-
ности при кручении уменьшается
с увеличением X.
6. Механическое демпфирование.
При одноосной ориентации максимум
потерь в области перехода стекло-
вания становится шире и ниже; и,
кроме того, он смещается в сторону
более высоких температур (см.
табл. XX. 1). Это означает, что пе-
реходная зона становится более ши-
рокой вследствие упорядоченности
молекул.
По данным Ван дер Меера можно
заключить, что для полукристалли-
ческих полимеров справедливо следу-
ющее правило (рис. XX.6):
ДТ = а + ЬДп (XX,14)
Td — Tg = a' + b' Дп (XX.15)
где ДУ — ширина максимума потерь в
точках перегиба; Td — температура, при
которой демпфирование максимально; а,
а', Ъ и Ь’ — постоянные.
За счет ориентации суммарная
ползучесть и релаксация напряже-
ний существенно уменьшаются, но
скорости ползучести и релаксации
часто увеличиваются.
7. Обобщенное соотношение между
для полимеров. Интересное соотношение обнаружил Хервиг [12]
для найлона 6. Для каждого волокна, характеризующегося своим
значением степени вытяжки Ло, существует собственная диаграмма
напряжение — деформация; в этой диаграмме напряжение а отно-
сится к поперечному сечению данного волокна и деформация е —
к первоначальной длине подвергнутого вытяжке волокна. Если,
однако, напряжение относится к поперечному сечению исходной
нити, а деформация — к первоначальной длине нити, то, очевидно,
все кривые после перехода через предел прочности выходят, на одну
331
---’ь-Дп
Рис. XX.6. Зависимости различ-
ных характеристик полиэфира от
степени ориентации [22].
напряжением и деформацией
и ту же огибающую, что показано на рис. XX.7. Эта суммарная
кривая есть не что иное, как зависимость напряжение — деформа-
ция для волокна, не подвергнутого вытяжке.
Для построения подобной кривой рассчитываются «приведен-
ные» напряжение и Деформация с помощью следующих соотношений:
Опривед— Оиабдюд/^0
1 + ЁпригеД — (1 + Ёнаблюд) ^0
(ХХ.16)
(ХХ.17)
где Л о — тот коэффициент вытяжки, при котором получено данное волокно.
Сравнение этих формул с (ХХ.13) показывает, что 1 + епривед =
= Лполная до тех пор, пока условия вытяжки и удлинения эквива-
лентны.
Из рис. XX.7 видно, что, если определена диаграмма напряже-
ние-деформация для не подвергавшегося вытяжке волокна, можно
оценить диаграммы напряжение — деформация для всех предвари-
тельно ориентированных волокон, полученных при определенном
значении степени вытяжки.
Рис. XX.7. Обобщенная диаграмма напряжение — деформация для найлона 6
J12] (1 текс= 1 г на 1000 м); после деления на плотность это значение становится
пропорциональным площади поперечного сечения пряжи:
а "• разрушающее напряжение при растяжении и относительное удлинение при разрыве.
Соотношения между кривыми напряжение — деформация ориен-
тированного и не подвергавшегося вытяжке волокна, определяемые
«производящей» кривой, еще раз подчеркивают основополагающую
роль степени вытяжки в определении физических характеристик
волокон; это уже было показано для двойного лучепреломления
(XX. 10) и модуля упругости в области звуковых частот (XX. 11)
и (ХХ.12).
332
ДВУХОСНАЯ ОРИЕНТАЦИЯ
Двухосная ориентация возникает в том случае, когда пленка поли-
мера растягивается в двух направлениях, перпендикулярных друг
другу. Сегменты полимерных цепочек стремятся расположиться
параллельно плоскости пленки, но в самой этой плоскости ориенти-
руются в более или менее случайных направлениях. В общем случае
пленки, подвергнутые двухосной вытяжке, все еще обладают некото-
рым направлением преимущественной ориентации, хотя степень
анизотропии пленки существенно меньше по сравнению с одноосно
вытянутым образцом.
Двухосная ориентация хрупких полимеров применяется для
того, чтобы улучшить прочностные показатели этих материалов при
одноосной ориентации в направлении, перпендикулярном оси одно-
осной ориентации. Описанный способ ориентации весьма важен при
получении пленок из кристаллических полимеров; такая ориента-
ция позволяет добиться во всех направлениях тех же показателей,
которые дает одноосно ориентированный материал (в направлении
расположения цепочек).
По сравнению с неориентированным полимером двухосно ориен-
тированные листы и пленки характеризуются более высокой проч-
ностью при растяжении и повышенной ударной вязкостью. Наи-
более типичные результаты, собранные Нильсеном [5], представлены
в табл. XX.5.
Таблица ХХ.5. Двухосная ориентация пленок
(сравнение с неориентированными материалами)
Полимер Ориентация Разрушаю- щее напря- жение при растяжении амакс* кгс/см? Относитель- ное удлине- ние при разрыве, % Ударная прочность
Полистирол Неориентированный 350—630 1-4 0,25-0,50
Двухосная 490-840 8—18 Выше 3
Полиметил- Неориентированный 525—700 5—10 4
метакрилат Двухосная 560—770 25—50 15
Де-Фриз [10] исследовал оптические характеристики двухосно
ориентированных пленок и влияние процесса вытяжки на двойное
лучепреломление в двух направлениях ориентации.
ЛИТЕРАТУРА
1. Bunn С. W., «Polymer Texture;
Orientation of Molecules and
Crystals in Polymer Specimens»,
Ch. 10 in R. Hill, Ed., «Fibres
from Synthetic Polymers», Else-
vier, Amsterdam, 1953.
2. Hermans P. H., «Contributions
to the Physics of Cellulose Fib-
res», Elsevier, Amsterdam, 1946.
3. Morgan L. B., «The Fibre-For-
ming Properties of Polymers»,
pp. 233—277 in J. C. Robb and
333
F. W. Peaker, Eds., «Progress
in High Polymers», Vol. I, Acade-
mic Press, New York, 1961.
4. Morton W. E. and Searle J. W. S.
«Physical Properties of Textile
Fibres», The Textile Institute
and Butterworths, London,
1962-
5. ' Nielsen L. E.t «Mechanical
Properties of Polymers», Ch. 10,
Reinhold, New York, 1962.
6. Stein R. S., «The Optical and
Mechanical Properties of High
Polymers», in Research Report
Nr. 14, High Polymer Series,
Chaps. 8 and 9, US Army Quar-
termaster Res. and Eng. Center,
Natick, Mass., 1960.
7. Stein R. S. and Onogi S. (Eds.), ,
US — Japan Seminar in Polymer
Physics (J. Polymer Sci., C. 15)
Interscience, New York, 1966.
8. Andrews R. D., J. Appl. Phys.
25 (1954) 1223.
.9 . Cleereman K. J., Karam H. J.,
and Williams J. L., Modern
Prastics 30 (1953) 119.
10. De Vries H., Thesis, Delft (1953);
Appl. Sci. Research A3 (1952)
111; Rayon Revue 10 (1956) 53;
J. Polymer Sci. 34 (1959) 761;
Angew. Chem. 74 (1962) 574;
Ned. Tijdschr. Natuuek. 31 (1965)
68.
11. Eiermann K., J. Polymer Sci.
C6 (1964) 157.
12. Herwig H. U., Internal Report
AKZO Research and Engineering
N. V. (1970).
13. Hermans P. H. and Vermaas D.,
Trans. Faraday Soc. 42B (1946)
155.
14, - Jackson G. B. and Ballman R. L.,
Soc. Plastics Eng. J. 16 (I960)
1147.
15. Kordes E., Gunther F., Buchs L.
and Golther IP., Kolloid-Z. 119
(1950) 23.
16 Kratky O., Z. physik. Chem.
B50 (1941) 255.
17. McGraw G. E., J. Polymer Sci.
A2, 8 (1970) 1323.
18. Mason P., J. Appl. Polymer Sci.
5 (1961) 428.
19. Nishijima Y. et al., Rept. Pro-
gress Polymer Phys., Japan 9
(1966) 457; J. Polymer Sci. Al,
5 (1967) 1021; J. Polymer Sci.
A2, 5 (1967) 23, 37.
20. Stein R. S. and Tobolsky A. V.,
Textile Research J. 18 (1948)
201, 302.
21. Tjader T. C. and Protzman T. F.,
J. Polymer Sci. 20 (1956) 591.
22. Van der Meer S. J., Thesis Delft,
1970.
23. Weyland H. G., Textile Research
J. 31 (1961) 629.
ГЛАВА XXI.
ТЕРМОХИМИЧЕСКИЕ ХАРАКТЕРИСТИКИ
В данной главе показано, что изобарно-изотермический потенциал
реакций можно рассчитать с помощью аддитивных групповых вкла-
дов. Этим способом можно оценить предельную температуру поли-
меризации. Получены интересные численные корреляции между
прочностями связей и отношениями реакционных способностей в про-
цессе сополимеризации.
ТЕРМОДИНАМИКА И К ИНЕТИКА
Все полимеры образуются и модифицируются в результате химиче-
ских реакций.
Наука о химических реакциях состоит из двух разделов: химиче-
ская термодинамика, которая имеет дело с равновесными состоя-
ниями, и химическая кинетика, которая изучает скорости реакций.
Термодинамические потенциалы представляют собой те движущие
силы, которые заставляют каждый процесс осуществляться в напра-
влении конечного состояния его равновесия. Термодинамика, следо-
вательно, определяет возможность или невозможность протекания
какой-либо реакции.
Реальность осуществления какой-либо реакции определяется
в действительности кинетическими факторами. Для того чтобы под-
держивалась практически любая реакция, необходимы определен-
ные количества энергии активации и энтропии активации. Поэтому,
если реакция термодинамически возможна, необходимо найти под-
ходящий катализатор, активность и селективность которого доста-
точны для проведения этой реакции. Классический пример — поли-
меризация этилена, которая проводится либо при высоком давлении
в присутствии радикальных инициаторов, либо при~низких давле-
ниях в присутствии катализаторов типа Циглера.
Термодинамика определяет возможность, а кинетика — реаль-
ность данного превращения.
Константа равновесия К связана с термодинамическими функ-
циями, в частности с энтальпией реакции АД0 и энтропией реак-
ции AS °:
—/?7’1пАраЕН = Д^°—7 A5O = AGO (XXI I)
где AG° — изменение стандартного изобарно-изотермического потенциала реак-
ции.
Выражение (XXI.1) можно переписать в хорошо известной форме
(уравнение Вант-Гоффа), которая описывает температурную зависи-
мость константы равновесия:
1пйГравн— — “Ь (XXI.2)
Аналогом уравнения Вант-Гоффа при равновесии является урав-
нение Аррениуса для константы скорости реакции:
1пА = -Е/ЯТ + 1пА (XXI.3)
где Е — энергия активации; А — постоянная.
Рис. XXI.1. Энергия активации и теп-
лота реакции в реакционной системе:
I — реагирующие компоненты; II — активиро*
ванный комплекс (переходное состояние); III —
продукты реакции.
Современные теории «переходного состояния» приводят к следу-
ющему виду, аналогичному (XXI.2):
ЛЯ* AS*
1п<! = --др +—+1п— (XXI.4)
где ЕН о — энтальпия активации; ASq — энтропия активации; k — константа
Больцмана; й — постоянная Планка.
На рис. XXL1 показана взаимосвязь между АН0 и ДЯр или Е.
Когда изучается одна и та же реакция в присутствии разных
катализаторов или сравниваются аналогичные реакции, то обычно
оказывается, что величины Е и А в уравнениях типа Аррениуса свя-
заны между собой. Увеличение Е компенсируется возрастанием А
согласно формуле
In А = -^0- + In В (XXI.5)
где 6 — характеристическая постоянная с размерностью температуры (так назы-
ваемая «изокинетическая температура»).
Подстановка этого выражения в (XXI.3) дает
Е / 1 1 \
1пй = — -----0-j+lnfi (XXI.6)
Эта формула представляет собой обобщенное уравнение Аррениуса
для семейства родственных реакций.
33S
Формула (XXI.5) математически выражает «компенсационный
эффект», отмеченный ранее при расчете констант диффузии.
Химическая термодинамика и химическая кинетика представляют
собой обширные разделы науки, поэтому здесь рассмотрены только
некоторые частные задачи:
расчет изобарно-изотермических потенциалов реакций по груп-
повым вкладам;
оценка предельной температуры в реакциях полимеризации;
рассмотрение термодинамики образования свободных радикалов;
определение термохимических факторов, определяющих состав
сополимеров.
РАСЧЕТЫ ИЗОБАРНО-ИЗОТЕРМИЧЕСКОГО
ПОТЕНЦИАЛА РЕАКЦИИ ПО ГРУППОВЫМ
ВКЛАДАМ
К сожалению, ценность соотношений химической термодинамики
зачастую ограничивается отсутствием достаточных данных. В таких
случаях важно иметь простой метод хотя бы для приближенных
расчетов этих данных.
Пусть имеет место следующая реакция образования соединения
С из составляющих это соединение элементов Ez:
2 с
i
Условия равновесия этой реакции при данной температуре описы-
ваются константой Kf:
А/=[С]/П[Е]П (XXI.7)
где П — это знак произведения.
Термодинамика устанавливает связь между константой равно-
весия и изобарно-изотермическим потенциалом образования АС®-.
AG® = — RTlaKf (XXI.8)
Из (XXI.7) следует, что равновесие смещается вправо, если Kf
больше 1, и влево, если Kf меньше 1. Отсюда вытекает, что согласно
(XXI.8) равновесие сдвинуто вправо при отрицательной величине
AG°f и влево при положительной величине Дб?’. Другими словами,
при данной температуре соединение устойчиво по сравнению с со-
ставляющими его элементами, если при этой температуре AGf <С 0.
Поэтому значение AGf может рассматриваться как количественная
мера устойчивости соединения сравнительно с его элементами.
Для любой произвольной химической реакции
У «аА — У пнВ
22 Заказ 699
337
Таблица XXI.1. Изобарно-изотермический потенциал образования
некоторых малых молекул и групповые вклады
в изобарно-изотермический потенциал образования больших молекул
Группа 6Gf (Т), кал/моль Группа 6Gf (T), кал/моль
СН4 —19 000+22,3 Г Н2О —58 000 + 11,5 T
-сн3 —11 500 + 22,9 Т -ОН -42 000 + 12,0 T
—сн2— -5 300+25,0 Т -О- —28000 + 14 T
Н2С=О —23 500+4,7 T
>сн- —750 + 30,0 Т —НС=О —29 000 + 5,5 T
X 3 000 + 36,5 Т \с=О —30000+9,5 T
=сн2 5 900 + 8,0 Т нсоон -87 000+21,6 T
=сн- 9000 + 9,0 Т -соон -94 500+25 T
=с\ 10 000 + 14,0 Т -соо- -80000 + 27 T
=с= 35 000-4 Т NH3 —11600 + 25,6 T
sCH 27 000—7,7 Т -nh2 -3800+2^1 T
=с— 27 000—6,0 Т —NH— 2 000 + 29,4 T
)}СНар 3100 + 6,1 Т /N- 6 000 + 37,2 T
^jCap 5 300 + 9,9 Т ^Nap 11500 + 11,1 T
^Сар*->(а/1Эо) 6 600+5,1 Т -no2 —9 000 + 37,0 T
18 600 + 36,6 Т H2S —5 000—10,0 T
— 20 800 + 40,4 Т -SH 4200-9,3 T
23000 + 44,2 Т -s- 10000—7,1 T
HF —64500-1,5 Т 1 p. co СЮ Z’S 14 500-14,4 T
—F —46000—2,2 Т -so- —15100 + 15,2 T
НС1 —22 000—22 Т -so2- -67 300 + 36,2 T
—С1 —11 700-2,2 Т co —26 600—21,2 T
НВг —8 700—13,6 Т co2 -94 000—0,6 T
—Вг —1000—13,3 Т ‘ cos -33 000-24 T
Вг2 (газ) 7400-22,2 Т cs2 27 500—40,3 T
HI 6200—19,7 Т COC12 —52 000—9,4 T
—I 16500—20 Т so2 —71 000—2,8 T
12 (газ) 15 000-34,4 Т so3 —94 500 + 19,9 T
HCN 31 200—8,3 Т NO 21500—3,0 T
—CN 29 000—7,2 Т no2 8000 + 14,81Т
(CN)2 72 000-10,6 Т S2 (газ) 31 000—39,2 T
338
Структурные поправки AGy (Т), кал/моль
Кольцевая замкнутая (неароматическая) 3 кольца 4 кольца 5 колец 6 колец Заместители в цепях 3 прилежащих группы /СН 25 000—30 Т 25 000—27 Т 5 000—25 Т —1000—16,5 Т 2100
1 2 прилежащих группы—С— 1 800
\ 1 ' прилежащие группы^СН— и —С— 2 600
в ароматических кольцах о положение ^-положение п-положение в неароматических кольцах одинарное замещение двойное 1,1-замещение 1,2; 1,3; 1,4-замещение, цис 1,2; 1,3; 1,4-замещение, транс Особые эффекты в ненасыщенных системах ^ис-транс-превращение сопряжение одной двойной связи с другой сопряжение с ароматическим кольцом сопряжение с ^>С=0-связью 1 000 350 0 -1 000 —2 000 —1 500 —3 000 —1400 + 1,7 Т -4000 —2 000 —1 000
равновесие при данной температуре определяется постоянной
П {В |" в
К = -- . (XXI.9)
П[А]а
Константа равновесия К связана с изменением изобарно-изотерми-
ческого потенциала реакции соотношением
AG0 = — RTiaK (ХХЫО).
Изменение изобарно-изотермического потенциала можно записать
и в виде разности этих потенциалов образования рассматриваемых
соединений:
ДС° = 2"вДС°В~1>АДС°А (XXI.И)
Следовательно, если известны изобарно-изотермические потенциалы
образования соединений, участвующих в какой-либо реакции, то
можно рассчитать условия равновесия этой реакции.
22*
339
На основании рассмотренного выше ясно, что для реакций поли-
меризации, деструкции и замещения весьма важно знать числовые
значения изобарно-изотермического потенциала образования.
Следует отметить, что термодинамические параметры получены
лишь для очень незначительного количества из огромного числа
известных органических соединений. Следовательно, чрезвычайно
важны методы расчетов подобного рода.
Теоретически термодинамические параметры можно рассчитать
методами статистической механики. Однако эти методы весьма тру-
доемки и, кроме того, они основаны на спектроскопических (эмпи-
рических) данных, которые обычно отсутствуют.
Метод групповых вкладов и здесь оказывается весьма эффектив-
ным. В этом направлении много сделали Андерсон, Бейер и Уотсон
[20], Бремнер и Томас [21], Саудерс, Мэтьюз и Хёрд [25] и Фран-
клин [23]. Наиболее полную и разработанную систему групповых
вкладов создали Ван Кревелен и Чермин [26]. Несколько упрощен-
ный вариант этой системы дан в табл. XXI. 1.
Приведенные здесь значения для соединений серы, брома и иода
отличаются от данных оригинальной работы [26]. За стандартные
состояния в табл. XXL1 приняты, как и обычно в термодинамике,
элементы в устойчивой форме при 25 °C и давлении 1 кгс/см2; в ори-
гинальной работе в качестве стандартных состояний были приняты
(газ), Вгг (газ) и 12 (газ).
ГРУППОВЫЕ ВКЛАДЫ
В системе Ван Кревелена и Чермина изобарно-изотермический по-
тенциал образования рассчитывается по групповым вкладам, и
вводятся поправки на особенности химического строения:
AG, = вклады составляющих групп 4-^ поправки на строение -\-RT In <т
(XXI.12)
где RT In а — поправка на степень "симметрии данной молекулы; о — число
симметрии, которое определяется количеством одинаковых положений молекулы
в пространстве, возможных путем простого вращения молекулы без деформации.
При этом предполагается, что групповые вклады линейно зависят
от температуры:
группы=4+ВГ (XXI.13)
Такое предположение основано на данных Шеффера [24]. Формула
(XXI.13) весьма похожа на общее термодинамическое выражение:
Дв = ДЯ—7Д5 (XXI.14)
При сравнении (XXI.13) и (XXI.14) нетрудно увидеть, что А обла-
дает размерностью теплоты образования, а В — энтропии образо-
вания. Согласно Улиху [27]:
А АН? (298) и В «й Д$°. (298) (XXI.15)
340
С помощью одного выражения (XXI. 13) нельзя точно описать тем-
пературную зависимость изобарно-изотермического потенциала
образования веществ в широком температурном-интервале. Однако
это выражение достаточно точно в интервале 300—600 К.
Все групповые вклады и структурные поправки рассчитаны на
основании опубликованных экспериментальных данных для органи-
ческих соединений.
Для углеводородов, для которых групповые вклады рассчиты-
вали по экспериментальным данным Россини с соавторами [6], оце-
ненные изобарно-изотермические потенциалы образования соответ-
ствовали опубликованным в литературе с ошибкой, не превышающей
0,8 ккал/моль. Для неуглеводородных соединений точность была
ниже, и максимальные отклонения достигали 3 ккал/моль.
Все значения изобарно-изотермического потенциала образования,
как правило, приведены к стандартным условиям для идеального
газа с летучестью 1 кгс/см2. Это также относится и к приведенным
групповым вкладам.
Пример 1
Оценить изобарно-изотермический потенциал образования газообразного
бутадиена-1,3 и газообразного (условно) полибутадиена.
Решение
Структурные звенья:
сн2=сн—сн=сн2 мономер Для мономера: • • -СН2—СН=СН—СН2 полимер
2(=СН2) 2(=СН—) R Т In а (а=2) сопряжение 11800 + 16,0 Т 18 000 + 18,0 Т 1,37 Т —4000
25 000+35,4 Т
При 300 К искомая величина равна 25 800 + 10 600 = 36 400 кал/моль; в лите-
ратуре приводится значение 36 490 кал/моль. При 600 К рассматриваемая вели-
чина равна 25 800 + 21 200 = 47 000 кал/моль; в литературе приводится зна-
чение 47 200 кал/моль.
Для газообразного (условно) полимерного звена:
2(—СН2—) 2(-СН=) —10 600 + 50,0 Т 18000+18,0 Т 7 400 + 68,0 Т
Мольный изобарно-изотермический потенциал полимеризации:
7400+ 68,0j71 — (25800 + 35,4 Т} = —18400 + 32,6 Т
Приведенное в литературе значение составляет —18 700 кал/моль.
344
ПОПРАВКИ, УЧИТЫВАЮЩИЕ ФИЗИЧЕСКОЕ
СОСТОЯНИЕ ВЕЩЕСТВА
Если реагирующие вещества или продукты реакции не находятся
в идеальном газообразном состоянии, но конденсированы (такое
состояние всегда наблюдается для полимеров), то необходимо ввести
поправки (см., например, Дайнтон, Ивин [22]).
Такие поправки представлены в виде диаграмм уровней энталь-
пии и энтропии; для реакций полимеризации соответствующие диа-
ДН реакции
Мономер/1) -----* Пааимер
AS реакции
Мономер)!)-—» Попимер
9<sl
—Т 9
------ (реакции)/-} I
А'Нат(+)
------ с
ЛН*др(~)
AlS°ap{+)
AS0Sg (реакции)/—}
'9
AS кар (“1
AS°m hl
a —-------------
I
с
Рис. XXI.2. Уровни энтальпии и энтропии мономера и полимера в различных
физических состояниях:
д — газообразное; в — жидкое; а — аморфное; е — кристаллическое; з — растворенное;
пар — испарение (конденсация); т — плавление (кристаллизация); 0 — стандартное состо-
яние (25 °C, 1 атм).
граммы приведены на рис. XXI.2. .Из этих диаграмм можно без
труда получить требуемые формулы. Ниже приведены поправки
ЛН° и А5° для негазообразного состояния:
Деполимеризация
ДЯг«а = ДЯг»г-ДЯ«сп
дя£к=дя°г—дя°сп—дя°л
ДЯ° = ДЯ° Ч-Д1Я° — дя°
дя» к = дя?г+д1^ - дя»сп -
-Д^л
ДЯ«к= ДЯ»Г+ Д1Я«сп-ДЯ»сп+
+ дя°л-дя“л
^^полимеризация
Д5»а = Д5?г-Д5«сп
Д5г°к = Д5гОг-Д5«сц-Д5»л
Д«жа= Д«?г + Д^исп-ДЗисп
ДДжк = Д^?г + Д1-5исп-Д^сп-
-ДД°л
Д5«к = Д5?г + Д14сп-Д^Сп+
+ Д^2л-Д^л
Примечание. дп’у — стандартный мольный тепловой эффект реакции, если мономер
в состоянии х превращается в полимер в состоянии у; AS^y — стандартное мольное изме-
нение энтропии, если мономер в состоянии х превращается в полимер в состоянии у.
Обозначения см. в подписи к рис, XXI.2.
342
При рассмотрении реакций полимеризации для оценки поправок
можно воспользоваться следующими эмпирическими правилами:
ДО“а^Д<?®г-2000+4 Т (XXI.16)
Д(3“к ^ДС°Г-4000+ 7,5 Т (XXI.17)
Дб^а *=» Дб®г+1000 — 12 Т (XXI.18)
ДС°К & ДѮà —7 7* (XXI.19)
ДС«К ДС®Г-9Т - (XXI.20)
где символ &.Gxy обозначает изменение мольного изобарно-изотермического по-
тенциала при переходе мономера из состояния х в полимер в состоянии у.
Пример 2
Оценить изобарно-изотермический потенциал полимеризации бутадиена-1,3,
в полибутадиен, если мономер находится в жидком состоянии, а полимер —
в аморфном твердом состоянии.
Решение
Из примера 1 следует, что
Дб“г (полим) = —18 400 + 32,6Т
Формула (XXI.18) дает:
ДС®а = ДС®г + 1000-12 Т
Следовательно
ДСжа=-17 400+20,6 Т
или ДЯ®я —17 400 и Д5® 20,6
Приведенные в литературе значения ДЯ^а = —17 400 и Д5^а =20,1—21,2.
ИЗОБАРНО-ИЗОТЕРМИЧЕСКИЙ
ПОТЕНЦИАЛ РЕАКЦИЙ ПОЛИМЕРИЗАЦИИ.
ПРЕДЕЛЬНЫЕ ТЕМПЕРАТУРЫ
В процессе полимеризации присоединением осуществляются сле-
дующие реакции (в качестве примера):
Н2С=СН2---► — Н2С—сн2-
II'
н2с=сн—> —н2с—сн—
I I
Н2С=С —► —Н2С—С—
I I
В табл. XXI.2 приведены экспериментальные и рассчитанные
с помощью соответствующих данных (см. с. 342) характеристики
этих реакций.
343
Таблица XXI.2. Сравнение вксперименгПальных и раесчитанных термодинамических величин
для некоторых реакций полимеризации (обозначения см. в подписи к рис. XXI.2)
Реакция полимеризации дв° ДН° да* гпр. к
гг гк или жа ОПЫТ расчет ОПЫТ расчет ОПЫТ расчет
Н2С=СН2 ► —СН2—СН2— -22400 + —26400+ —22,35 (гг) —22,4 (гг) -34,0 (гг) —34,0 (гг) 659 (гг) 658 (гг)
+ 34,0 7’ + 41,5 7’(гк) —25,5 (гк) —26,4 (гк) —41,5 (гк) —41,5 (гк) 615 (гк) 635 (гк)
(СН3) 1 1
Н2С=СН —> -сн2-сн- —20 950 + -19 950 + —20,7 (гг) —21,0 (гг) —39,9 (гг) —38,0 (гг) 520 (гг) 550 (гг)
+ 38,0 7’ +26,0 Т (жа) —20,1 (жа) —20,0 (жа) —27,5 (жа) —26,0 (жа) 707 (жа) 765 (жа)
(Ph) 1 1
Н2С=СН —> -сн2-сн- —18 950 + -17 950 + —17,8 (гг) -19,0 (гг) -35,5 (гг) -38,0 (гг) 550 (гг) 500 (гг)
+ 38,07’ +26,0 Т (жа) —16,7 (жа) —18,0 (жа) —25,0 (жа) —26,0 (жа) 670 (жа) 690 (жа)
(СНз)
1 1 Н2С=С —> -сн2—с— -18200 + -17 200+ —17,2 (га) —18,2 (гг) —41,0 (гг) —39,5 (гг) 419 (гг) 462 (гг)
1 1 (СНз) + 39,57’ +27,5.7’ (жа) —11,5 (жа) —17,2 (жа) —27,5 (жа) —27,5 (жа) 418 (жа) 625 (жа)
(СНз). 1
Н2С=С —> -сн2-с- —16 200 + -15200 + —8,4 (жа) —15,2 (жа) —27,5 (жа) 410 (гг)
1 Ph + 39,5 7’ +27,5 Т^жа) 550 (жа)
Н2С=О —> —СН2—О— -9800 + —15 200+ —9,8 (гг) —34,3 (гг) 275 (гг)
+ 34.3 71 +27,5 Т( гк) —13,0 (гк) —13,8 (гк) —41,7 (гк) —41,8 (гк) 312 (гк) 331 (гк)
(СН3)С=О —> -С-0- 250 + 38,5 Т 1250+ 26,5 7’ 0 (жа) 1,3 (жа)
1 1 (жа)
н н
В табл. XXI.2 приведены также рассчитанные и измеренные пре-
дельные температуры полимеризации. При такой температуре изме-
нение изобарно-изотермического потенциала равно нулю и, следо-
вательно
7\,Р = А/?полим/А5полим (XXI.21)
Если стандартное состояние относится к единичной концентра-
ции и мономер (М) ведет себя идеальным образом, т. е. AS = AS® +
+ R In [M], то
7’пР = ДЯ«олим<(Д5“олИМ+Я In [М]) (XXI.22)
Величина Т^р характеризует равновесие мономер — полимер неза-
висимо от того, чем вызвана полимеризация (свободными радикалами
или ионами). Следовательно, предельная температура — это такая
температура, выше которой полимеризация не может осущест-
виться; при такой температуре, кроме того, полимер становится
неустойчивым в отношении термической деструкции. На практике,
образовавшиеся полимеры, По-видимому, устойчивы при темпера-
турах выше предельной, но подобное состояние оказывается мета-
стабильным. Причина заключается в том, что для прохождения
процесса, деполимеризации образовавшегося полимера требуется
определенная энергия активации. Но полимеры, цепи которых не
закончили свой рост, могут деполимеризоваться спонтанно.
ТЕРМОДИНАМИКА СВОБОДНЫХ РАДИКАЛОВ
Многие реакции полимеризации и деструкции полимеров осущест-
вляются по радикальному механизму. Следовательно, важно знать
сравнительные термодинамические характеристики свободных ра-
дикалов и связанных групп.
Для ряда простых радикалов данные такого рода получены путем
исследования газопламенных реакций. Кроме того, методами термо-
химии определены энергии диссоциации различных химических
связей. В табл. XXI.3 представлена полная информация об энер-
гиях диссоциации связей.
Наконец, эмпирическим путем было установлено, что изменение
энтропии в реакциях разрыва простых связей составляет приблизи-
тельно 40 э. е. Следовательно, изменение энтропии на один образо-
вавшийся свободный радикал составляет примерно 20 э. е. (при рас-
чете на атом эта величина составит почти 10 э. е.).
При помощи этих эмпирических данных можно вычислить изо-
барно-изотермический потенциал свободных радикалов [81:
Радикал AG°, кал/моль
Н- 52 000—11,8 Т
F. 19 000—13,5 Т
С] • 2Й 940—12,8 Т
Вг- 26 800—23,7 Т
I • 25 500—29,0 Т
Радикал Дб°, кал/моль
• ОН) 9000—1,5 Т
• О— 8000—2 Т
О
II
• с— 5000—10 Т
345
Радикал Д<3°, кал/моль Радикал AG°, кал/моль
•сн3 32 090+2,1 Т z0
СН2— 34 000-1,0 Т • о-с^ —26000+10 Т
-СН<^ 36000+9 Т • NH- 4000+10 Т
38 000+15 Т •CN 110 000—15 Т
•0-0— 7500
Пример 3
В качестве примера рассматривается термодинамическая вероятность двух
типов реакций между радикалами: рекомбинация и диспропорционирование.
Решение
Эти реакции таковы: '
----СН2- +-СН2—СН2--------->------СН2—СН2—СН2------- (а)
или
----СН2-+-СН2-СН2---------->------СН3 + СН2=СН------ (б)
С помощью групповых вкладов можно рассчитать для реакции (а) AG° =
= —78 600 + 52,0 Т‘, для реакции (б) Д<?° = —59 300 + 16,9т. Следовательно^
разность изобарно-изометрических потенциалов для этих двух реакций состав-
ляет:
Д (Д<?°) = — 19 300 + 35,1Г = 0 при 550 К
Таким образом, при температуре выше 550 К, или 277 "С, преобладает диспропор-
ционирование, а ниже 550 К — рекомбинация.
СВОБОДНОРАДИКАЛЬНАЯ СОПОЛИМЕРИЗАЦИЯ
Все процессы полимеризации присоединением начинаются путем
активирования двойной связи. Реакционная способность двойной
связи в мономере в сильной степени зависит от природы заместите-
лей, которые обычно активируют двойную связь. Однако стерические
препятствия, связанные с наличием объемных заместителей, могут
привести к замедлению скорости реакции.
Заместители также оказывают сильное влияние на стабильность
свободного радикала; образовавшийся радикал может стабилизиро-
ваться за счет сопряжения с системой двойных связей в заместителе.
Подобная резонансная стабилизация может привести к подавлению
реакционной способности радикалов таким образом, что порядок
реакционных способностей радикалов станет обратным порядку
реакционных способностей мономеров.
Эффективность заместителей в повышенйи реакционных способ-
ностей мономеров приблизительно соответствует следующему ряду:
.О О о
II II II
—CH=CHS>— СвНБ>— с>— C=N>— С>— С1>—О— С> — OR> — Н
III
СН3 OR СН3
Влияние второго заместителя на этот же атом С обычно оказы-
вается аддитивным. Порядок резонансной стабилизации примерно
346
Таблица XXI.3. Энергия диссоциации связи X—Y, ккал/моль
(-С) /(С)
—н —F -С1 -вг -I -СНа 1 - СНа —сн< 4 (С)
103,2 135 102,1 86,7 70,6 104 98 94
38 60 56 52 108 106 105
57,1 52 49,6 84 81 81
45,4 43 70 69 68
•35,6 56 53 53
88 85 83
80,5 78
76
Примечание. Энергия диссоциации
уменьшается, если диссоциирующая связь
сопряжена с системой л-электронов:
Система Уменьшение анергии диссоциации связи
1 1 —с=с— примерно 16
“ г -•anvbiA-И’вь r._ примерно 16
1 —С=О примерно 10
/(С) —С—(С) \с> —СНа—Ф СН=СН, -С=СН - CFa ' -СС1,
91 112 80 104 121 106 96
102 125 90 — — 129 106
79 100 68 86 — 85 73
63 80 51 — — 70 54
50 65 40 55 — 54 —
80 102 72 90 100 100 —
75 91 62 90 109 — —
73 83 55 85 103 — —
70 78 — 81 — —— —
103 77 101 119 — —
47 70 — — —
101 — — —
(НО) — —
97 —
87
Продолжение табл. ХХ1.3
-он -О-(С) J. .0 \(С) ,О -о-с^ \() —nh2 (С) —NH —N< /С.) \С) —CN —SH -S-(С)
119 102 87 80 — 112 103 92 86 — 90 88 —н
— 59 '— — — — — — — — — — — F
— 49 — 82 — — — — — — — (65) —CI
— — — 07 51 - — ' — — — — — —Вг —I
91 80 75 82 — — 79 — — но — — - СН3
91 80 71 77 — . 78 — — — — — —СН2—(—С) АС)
92 81 - — 77 —сн< (С) /(С)
91 78 — — — — 77' —- — — — — —С—(С) \о
112 101 90 — — — 100 — — 130 — —
77 — 50 >5 — 65 — — 95 — — —CH2-Ph
— — — — — — — — — 121 — — сн=сн..
— — — - — — —с=сн
— — — — — — — — -CF3 -CC18V
51 — 100 102 — — — — — — — — —ОН
34 — — — — —О—(С)
60 60 59 — 90 — — — — — о я \/ о 1
60 — — 98 — — — — — /С С<\(С)
— — — — — — — \о-
-ю — /0 -о-с/ Х(С)
— — — — — — -nh2
(С)
37 — — — — 1 —NH
42 — — — АС) —N< \С)
145 — — —CN
(30) — SH
(63) —S—(С)
Продолжение табл. ХХ1.3
-он -О-(С) J. .0 \(С) \О— ,О -о-с^ „ \(С) —nh2 (С) —Ан —N< /С.) ''(С) —CN —SH —S—(С)
119 102 87 80 — 112 103 92 86 — 90 88 —н
— 59 .— — — — — — — — — — — F
— 49 — 82 — — — — — — — (65) —CI
— — — 07 - — — — — — — — —Вг
— — — 51 - — — — — — — —I
91 80 75 82 — — 79 — — но — — -СН3
91 80 71 77 — — . 78 — — — — — —СН2—(—С)
АС)
92 81 — — — , 77 —— — — — — —сн<
(С)
/(С)
91 78 — — — — 77 — — — — — —С—(С)
\о
112 101 90 — — — 100 — — 130 — —
77 — 50 >5 — 65 — — 95 — — —CH2-Ph
— — — — — — — — 121 — — сн=сн..
— — —= - — — —с=сн
— — — — — — — — — — — —CF з -CC18V
51 — 100 102 — — — — — — — — —ОН
34 — — — — —О—(С)
60 60 59 — 90 — — — — — о я \/ о 1
60 — — 98 — — — — /С \С) ZzO
— — Хо-
-ю — — — — — — /0 -о-с/ Х(С)
— — — — — — -nh2
(С)
37 — — — — 1 —NH
42 145 — — АС) —N< \(С) —CN
(30) — SH
(63) —S—(С)
такой же, как и указанный порядок реакционных способностей.
Влияние заместителя на подавление активности радикала выражено
гораздо сильнее по сравнению с влиянием на повышение реакцион-
ной способности мономера, поэтому влияние данного заместителя
на общую скорость реакции оказывается сложным. Например, реак-
ционная способность мономера стирола примерно в 50 раз выше по
сравнению с мономером винилацетата, но радикал стирола за счет
резонансной стабилизации примерно в 1000 раз менее реакционно-
способен, чем радикал винилацетата. В итоге полимеризация стирола
осуществляется примерно в 20 раз медленнее по сравнению с винил-
ацетатом.
Если радикал может взаимодействовать с двумя мономерами одно-
временно, но при этом проявляет преимущественное сродство к од-
ному из них, то последнее описывается отношением реакционных
способностей г. Когда активирование осуществляется в смеси двух
мономеров, радикалы, образовавшиеся из обоих мономеров, могут
реагировать с обоими мономерами.
Если скорость взаимодействия радикала с мономером выражается
константой реакции йху (где х обозначает радикал мономера X и у—
второго мономера Y), то отношение реакционных способностей опре-
деляется как гх = АхЛу Следовательно, для бинарных смесей
ri = ^и/^12 и r2 = k22/k21. Совершенно ясно, что для описания
процессов полимеризации чрезвычайно важно знать величины
Если каждый радикал реагирует преимущественно с другим мо-
номером, т. е. fj = г2 = 0, то образуется строго чередующийся сопо-
лимер. Если же каждый радикал не взаимодействует с другим моно-
мером, то сополимеризации не происходит и образуется смесь двух
полимеров. Наконец, если оба радикала не обладают преимущест-
венной склонностью к взаимодействию с любым из двух мономеров,
то образуется статистический сополимер. Условием этого является
соотношение = 1.
Между этими крайними случаями могут возникать промежуточ-
ные варианты. При этом, если известно соотношение реакционных
способностей, то появляется возможность рассчитать состав получа-
ющегося сополимера по составу исходной смеси мономеров.
Таким образом, очевидно, что крайне желательно иметь метод
оценки величин отношений реакционных способностей. Такой метод
был разработан Алфреем и Прайсом [19]. Они предположили, что
константы скорости реакций можно записать в следующем виде:
&ху = PxQy ехр (—fix^y) (XXI.23)
где Рх — реакционная способность радикала X; Qv— реакционная способность
мономера Y; ех и еу— «полярности» мономеров X и Y.
Отсюда
Г1 = А11/А!12=^1/<?2-еХР [——«2)]
r2 = *22/fe2i=<?2/<?i- exp [—e2(e2 —«1)] (XXI.24)
rjr2 = exp [—(й! —е2)2]
350
Условие статистической сополимеризации rxr2 « 1 по (XXI.4)
выполняется при ег = е2, т. е. если полярности обоих мономеров
приблизительно равны друг другу и имеют одинаковый знак.
Образование чередующегося сополимера происходит при =
= г2 >=» 0, т. е. если полярности мономеров резко различаются
•з
• 2
2
• 4
• 5
• 6
7
!
«
О
•8
II
9
10 •
• 12
• 17
,6*.!9
• 20
16
• 13
_ *19
• 15
• 21
22 23
• 24
• 26
• 25
• 28
-•29
33
• 27
30
• 31
*34 *3Z
•35
• >‘И
-/
-2
^001 0,01 0,1 1 10 100
----* Q
Рис. XXI.3. Диаграмма «е—<?»:
1 — тетрахлорэтилен; 2 — малеиновый ангидрид; 3 — винилиденцианид; 4 — трихлор-
этилен; 5 — винил фторид; 6 — тетрафторэтилен; 7 — акриламид; 8 — акрилонитрил; 9 —
итаконовый ангидрид; 20 — акриловая кислота; 11—акролеин; 12— метилацетат; 13—
метакриловая кислота; 14 — акриловый ангидрид; 15—метилметакрилат; 1в — винили-
денхлорид; 17 — аллиловый спирт; 18 — винилхлорид; 19 — аллил хлорид; 20 — винил-
сульфоновая кислота; 21 — хлоропрен; 22 — этилен; 23 — винилацетат; 24 — я-хлорстирол;
25 —пентен-1; 26 — пропилен; 27 — стирол; 28 — изобутилен; 29 — аллилацетат; 30 —
н-винилпирролидон; 31 — бутадиен; 32 — изопрен; 33 — из о бутил виниловый эфир; 34 —
а-метилстирол; 35 — винилкарбазол.
между собой, например ег имеет большое положительное значение,
а е2 — большое отрицательное. При любом процессе сополимериза-
ции благоприятное условие образования чередующегося сополимера
состоит в выполнении равенства Qr Q2.
В табл. XXI.4 приведены значения Q и е для наиболее распрост-
раненных заместителей у двойной связи. На рис. XXI.3 показана
так называемая е — (7-диаграмма для наиболее распространенных
мономеров при полимеризации присоединением.
351
Таблица XXI.4. Приближенные значения величин е и Q
для групповых заместителей
Группа сн2=снх СН2=С—X 1 СНз сн2=сх2 СХ2=СХ2 снх=снх
е Q е Q е Q е Q е Q
—Н -СН3 —СН—(С) /(С) -сн< \(С) -© -©© -©О© —сн=сн2 —CsCH —F —Cl —Вг -СН2С1 —сн2он - ОСН3 . /(С) -с-сн< \(С) -о-<° хн /0 —o-c<f Х(С) —соон /0 —с/ Х0—(С) -cf X(Q . //0 —с< хн —0,20 —0,8 —0,4 —1,0 -0,6 -0,8 —1,0 -1,6 -1,0 АО,4 1,3 0,2 —0,25 0,1 0,4 -1,2 -1,8 —0,8 -0,2 0,8 0,6 0,6 0,7 0,015 0,002 0,04 0,03 0,01 1,0 1,5 1,5 2,4 0,7 0,01 0,04 0,05 0,06 0,05 0,05 0,02 0,2 0,03 1,15 0,5 0,7 0,85 ° 1 ° -° 1 1 1 1 1 1 } 1 1 1 1 1 1 1 1111 g? 1 -Г * * .° _ _ со ьо со о 00 0,002 0 03 1,0 з,з 0,04 2,3 0,7 1,75 | । 1 . । । । 1 I 1 I 1 -2 । । L L о Ьо о , 0,015 0,03 0,2 <z> сч fill I I I I Г ”Si I I I I г ill । । 0,015 0,05 0,03 fill Г 1 1 1 1 -12 1 1 1 1 1 1 1 Й 5 1 1 0,015 0,01 0,4 0,2
352
Продолжение табл. XXI.4
Группа CH,=CHX CH,=C-X 1 CH, CH4=CX, cx,=cx, CHX=CHX
e Q e Q e Q e Q e Q .
-1оГПо| -1,4 0,4
Jo —с<^ -0,2 1,3 0,8 1,2 1,25 1,5
xnh2
_с/) '0 0,2
\NHR
-CN 1,2 0,6 0,8 1,1 2,6 2,0 — — 2,0 0,6
—SO3H 0 0,1 —
—SCH3 —1,5 0,3 —
-SO2CH3 1,3 0,1 —
Пример 4
Оценить отношение реакционных способностей для сополимеризации метил-
акрилата и стирола.
Решение
В табл. XXI.4 находим следующие значения:
I е Q
Метилакрилат 0,6 ч 0,5
Стирол —0,8 1,0 .
Следовательно
П = (0,5/1,0) ехр [—0,6 (0,6 + 0,8)] = 0,5 ехр (—0,84) = 0,215
г2 = (1,0/0,5) ехр [+0,8 (—0,8—0,6)] = 2 ехр (—1,12) = 0,655.
Экспериментально получены следующие величины:
т-i = 0,18 ±0,02-0,4 ±0,2; г2 = 0,7±0,1
Соответствие вполне удовлетворительное.
Состав полимера зависит от состава смеси мономеров и от соот-
ношения реакционных способностей. Для расчета состава полимера
по этим данным следует воспользоваться хорошо известным урав-
нением (см., например, [17]):
^1= (г + /1 ^2)/(Г!/1 Ч-гг/| + 2^1/2)
23 Заказ 699 353
где Fx — доля мономера 1 в полимере; /х — доля мономера 1 в смеси мономеров,
/2=1-/!-
Пусть /х = 2/3 и /2 = 1 — /1 = 1/3, тогда
Л = '(0,215-4/9-2/3 • 1/3)/(0,215 • 4/9 + 0,655• 1/9+2 • 2/9) =
= (0,0955 + 0,222)/(0,0955 + 0,0725 + 0,444) = 0,3175/0,612 «0,52
ЛИТЕРАТУРА
1. Jani G. J., «Estimation of Ther-
modynamic Properties of Organic
Compounds», Academic Press,
New York, 1958. ,
2. Lewis G. N. and Randall M.,
«Thermodynamics», 2nd Ed., Rev.
by Pitzer, K. S. and Brewer,
L., McGraw-Hill, New York,
1961.
3, Parks G. S. and Huffman H. M.,
«The Free Energies of some Orga-
nic Compounds», Chem. Cat. Co.,
New York, 1932.
4. Rossini F. D., «Chemical Thermo-
dynamics», Wiley, New York,
1950.
5. Rossini F. D. et al., «Selected
Values of Chemical Thermodyna-
mic Properties», Natl. Bur. Stan-
dards. Circ. 500, U. S. Printing,
Office, Washington, 1952.
6. Rossini F. D. et al., «Selected
Values of Physical and Thermody-
namic Properties of Hydrocar-
bons and Related Compounds»,
Carnegie Press, Pittsburgh, 1953.
7. Ivin K. J., «Heats and Entropies
of Polymerization, Ceiling Tempe-
ratures and Equilibrium Monomer
Concentrations», in «Polymer
Handbook» (Brandrup J. and
Immergut E. M., Eds.) Part II,
pp. 363—398, Interscience, New
York, 1966.
8. Sawada H., «Thermodynamics of
Polymerization», J. Macromol.
Sci., Revs. Macromol. Chem.
C3 (2) (1969) 313-396.
9. Bamford С. H. and Tipper C. F. H.
(Eds.), «Comprehensive Kinetics»,
Elsevier, Amsterdam, 1969.
10. Boudart M., «Kinetics of Chemi-
cal Processes», Prentice-Hall, En-
glewood, N. J., 1968.
11. Burnett G. M., «Mechanism of Po-
lymer Reactions», Interscience,
New York, 1954.
12. Glasstone S., Laidler K. J. and
Eyring H., «The Theory of Rate
Processes», McGraw-Hill, New
York, 1941.
13. Lefler J. E. and Grunwald E.,
«Rates and Equilibria of Chemi-
cal Reactions», Wiley, New York,
1963.
14. Mortimer С. T., «Reaction Heats
and Bond Strengths», Pergamon
Press, London/New York, 1962.
15. Franklin J. L., «Prediction of
Rates of Chemical Reactions Invol-
ving Free Radicals», Brit. Chem.
Eng. 7 (1962) 340.
16. Alfrey T., Bohrer J. J. and
Mark H., «Copolymerization», In-
terscience, New York, 1952.
17. Ham G. E., «Copolymerization»,
Interscience, New York, 1964.
18. Young L. J., «Transfer Constants
in Free Radical Polymerization;
Tabulation of Q—е-values», in
Polymer Handbook» (Brandrup
J. and Immergut E. M., Eds.),
Part II, pp. 77—362, Interscience,
New York, 1966.
19. Alfrey T. and Price С. C., J.
Polymer Sci. 2 (1947) 101.
20. Anderson J. W., Beyer G. H.
and Watson К. M., Natl. Petr.
News 36R (1944) 476.
21. BremnerJ.G. M. and ThomasG.D.,
Trans. Faraday Soc. 44 (1948)
230.
22. Dainton F. S. and 7 vin K. J.,
Trans. Faraday Soc. 46 (1950)
331.
23. Franklin J. L., Ind. Eng. Chem,
41 (1949) 1070.
24. Scheffer F. E. C., «De Toepassing
van de Thermodynamica op Che-
mische Processen», Waltman,
Delft, 1945.
25. Souders M., Matthews C. S.
and Hurd С. O., Ind. Eng. Chem.
41 (1949) 1037.
26. Van Krevelen D. W. and Cher-
min H. A. G., Chem. Eng. Sci.
1 (1951) 66; 1 (1952) 238.
27, Ulich H., «Chemisette Thermo-
dynamik», Dresden/Leipzig, 1930.
ГЛАВА XXII. ДЕСТРУКЦИЯ
Деструкция полимеров представляет собой типичное свойство поли-
мера, зависящее от его строения. В отношении деструкции получены
некоторые интересные количественные соотношения.
t
ФОРМЫ ДЕСТРУКЦИИ
Полимер может деструктировать, т. е. подвергаться химическим из-
менениям под действием тепла, излучения (типичным примером явг
ляется фотоокисление) или условий окружающей среды. Ниже каж-
дая из перечисленных возможностей рассмотрена более подробно.
ТЕРМИЧЕСКАЯ ДЕСТРУКЦИЯ
В АТМОСФЕРЕ ИНЕРТНОГО ГАЗА
Характер термической деструкции полимера в инертной атмосфере
определяется, с одной стороны, химическим строением самого поли-
мера и, с другой стороны, присутствием малых количеств неустойчи-
вых соединений (различных включений или добавок).
Термическая деструкция не начнется до тех пор, пока темпера-
тура не станет достаточной для разрушения первичных химических
связей. Большую работу в этой области провели Мадорски и Штраус
[3, 15]. Они показали, что при нагревании одни полимеры (полиме-
тилметакрилат, поли-а-метилстирол и политетрафторэтилен) рас-
падаются, главным образом, до своих мономеров, а.другие полимеры,
например типа полиэтилена, дают целую гамму продуктов разло-
жения, занимающих промежуточное положение между исходным
мономером и полимером. Эти два типа термической деструкции поли-
меров получили названия цепной деполимеризации и статистической
деструкции.
Цепная деполимеризация — это процесс последовательного от-
щепления мономерных звеньев от конца полимерной цепи или по
местам слабых связей; этот процесс, по сути дела, представляет
собой реакцию, обратную полимеризации. Деполимеризация начи-
нается в точке, соответствующей некоторой предельной температуре.
Статистическая деструкция осуществляется путем разрыва цепи по
случайно распределенным местам вдоль макромолекулы и приводит
23* 355
к образованию смеси фрагментов; длина последних обычно пре-
вышает размер мономерного звена. Два названных типа термической
деструкции могут осуществляться раздельно или в сочетании друг
с другом. Последний случай более распространен. Цепная деполи-
меризация зачастую определяет процесс деструкции полимеров
винилового ряда, в то время как деструкция конденсационных поли-
меров обусловлена в основном статистическим разрывом цепей.
ДЕСТРУКЦИЯ ПОД ДЕЙСТВИЕМ СВЕТА
Из всей электромагнитной энергии, излучаемой солнцем, только
небольшая часть достигает поверхности земли; этой частью яв-
ляется свет с длинами волн больше 290 ммкм. Рентгеновские лучи
поглощаются наиболее удаленным от поверхности земли слоем атмо-
сферы, а ультрафиолетовые лучи с длинами волн вплоть до 290 ммк—
содержащимся в атмосфере озоном. Суммарная интенсивность излу-
чения изменяется в весьма широких пределах в зависимости от атмо-
сферных и географических условий, однако «валовый» состав солнеч-
ного света остается практически постоянным.
В процессе фотохимической деструкции энергию активации
обеспечивает солнечный свет. Для обычных химических реакций
энергии активации изменяются от 15 до 65 ккал/моль. Это энерге-
тически эквивалентно излучению с длинами волн между 1900 и
440 ммкм. Значение энергии, необходимой для разрыва простой кова-
лентной связи, изменяется, за немногими исключениями, от 40 до
100 ккал/моль, что соответствует излучению с длинами волн от 710
до 290 ммкм (рис. XXII.1). Это означает, что энергия излучения
в ближней ультрафиолетовой части спектра (300—400 ммкм) доста-
точна для разрыва большинства простых ковалентных связей, за
исключением таких прочных связей, как С—Н и О—Н.
Химически активным может оказаться только часть всего излу-
чения, которое обычно поглощается данным материалом. Вследствие
высокой регулярности своей структуры большинство чистых орга-
нических полимеров, полученных синтетическим путем (полиэтилен,
полипропилен, поливинилхлорид, полистирол и т. д.), не поглощают
свет с длинами волн больше 300 ммкм и, следовательно, не подвер-
гаются действию солнечного света. Тот факт, что эти полимеры зача-
стую все же разрушаются под действием солнечного света, объяс-
няется присутствием небольших количеств включений или наличием
структурных дефектов, которые поглощают свет и оказываются
инициаторами деструкции. Подавляющее количество поглощенной
световой энергии, как правило, диссипирует путем либо безызлуча-
тельных процессов (вращения и колебания), либо вторичного излу-
чения (флуоресценция).
Природа включений или структурных дефектов, ответственных
за фоточувствительность, точно неизвестна, однако принято считать,
что подобные включения представляют собой различного типа карбо-
нильные группы (кетоны, альдегиды) и перекиси. Разрывы цепи
356
главных валентностей или образование свободных радикало в в
различных фотохимических превращениях часто сопровожда ются
возникновением хрупкости материала вследствие образования попе-
речных связей, а вторичные реакции, особенно в присутствии кис-
лорода, обусловливают дальнейшую деструкцию полимера. В подоб-
ных условиях могут резко изменяться механические характеристики
полимерных материалов, например прочность, разрывное удлинение
Рис. ХХП.1. Энергетическая эквивалентность световых волн:
I — поглощается земной атмосферой; II — ближняя УФ-часть спектра;
III — видимый свет; IV — ближняя ИК-часть спектра.
и ударная вязкость. Довольно часто наблюдается появление окра-
шенных продуктов деструкции. Кроме того, об индуцированной
ультрафиолетовым облучением деструкции может свидетельство-
вать поверхностное растрескивание образцов.
Одни полимеры подвергаются обесцвечийанию и обнаруживают
ухудшение механических показателей (например, ароматические
полиэфиры, ароматические полиамиды, поликарбонат, полиуретаны,
полиоксифенилен, полисульфон), другие проявляют только измене-
ние механических характеристик (полипропилен, хлопок) или всего
лишь изменяют окраску на желтую (шерсть, поливинилхлорид).
Деструкция подобного типа может выражаться слабее, если в поли-
мер включен поглотитель ультрафиолетового излучения. Роль по-
добных поглотителей УФ-излучения (обычно это о-оксибензофеноны
или о-оксифенилбензотриазолы) заключается в адсорбировании
357
излучения в диапазоне волн 300—400 ммкм и в диссипации энергии
этого излучения способом, безвредным для защищаемого материала.
В настоящее время в области защиты полимеров от воздействия
УФ-излучения применяют добавки (например, никелевые комплексо-
образователи), которые путем переноса энергии могут обеспечивать
гашение электронных возбужденных состояний в присутствующих
включениях (например, карбонильных групп в полипропилене).
ОКИСЛИТЕЛЬНАЯ ДЕСТРУКЦИЯ
При комнатной температуре полимеры обычно реагируют с кисло-
родом, но столь медленно, что окисление становится заметным
лишь спустя длительный период времени. Так, если полистирол
хранится на воздухе в темноте .в течение нескольких лет, то его
спектр поглощения в УФ-области практически не изменяется. В то
же время, если тот же полимер облучают УФ-светом в сходных
условиях в течение 12 суток, то в спектре поглощения полимера
возникают новые сильные полосы. Сказанное справедливо и в отно-
шении других полимеров, например полиэтилена и натурального
каучука.
Таким образом, проблема, по сути дела, сводится не к окисляе-
мости, как таковой, а к синергическому эффекту ряда факторов в про-
цессе окисления, например, электромагнитного излучения и тепло-
вой энергии. Под действием этих факторов в полимере образуются
свободные радикалы, которые вместе с кислородом выступают в роли
инициаторов цепной реакции. Следовательно, подавляющее болыпин-
ство^ реакций окисления обладает автокаталитической природой.
Если окисление индуцируется светом, то это явление называют
фотоокислением; если же окисление индуцируется исключительно
термическими факторами, то осуществляется термоокисление.
ФОТООКИСЛЕНИЕ
Наиболее подробно изучена окислительная деструкция натураль-
ного каучука. В 1943 г. Фармер и Сундралингхэм [11] показали,
что в процессе фотохимического окисления этого полимера обра-
зуется гидроперекись, а количество двойных связей в цепи остается
неизменным. Кислород, как выяснили эти авторы, действовал на
активированную метиленовую группу, но не по двойной связи, как
предполагали ранее.
Позже механизм окирления каучука широко изучали Болланд
с соавторами [9]. Исследования этих авторов проводились в основ-
ном на модельных соединениях. В своей первой работе, посвященной
указанной проблеме, Болланд предложил следующий механизм
реакции роста цепи:
Н-+О2 —► ROO-
ROO.-I-RH —► ROOH + R.
358
(а)
(б)
где RH — олефин; R — радикал, образовавшийся при отрыве атома водорода
в аллильной группе; ROO • — перекисный радикал, полученный присоединением
кислорода к первому радикалу.
Согласно Болланду, реакционные цепи обрываются путем реком-
бинации аллильного и перекисного радикалов, и длина основных
реакционных цепей составляет 50—100. Такой процесс можно ини-
циировать реакцией любого типа, идущей с образованием свободных
радикалов. Автокаталитический характер данной реакции обусло-
влен разложением гидроперекисей:
hv
ROOH —>- RO- + -OH (в)
Гидроперекиси оказываются также причиной вторичных реакций,
в которых образуются окрашенные смолообразные продукты (через
карбонильные соединения).
Стабилизировать полимер в отношении фотоокисления можно
при помощи подходящих поглотителей УФ-излучения в сочетании
(синергическое действие) с антиокислителями (АН), способными
предотвращать реакции (а) и (б):
R-+AH > RH + A- (г)
ROO-+AH ► ROOH + A- (д)
А- -—> неактивные продукты (е)
ТЕРМИЧЕСКОЕ ОКИСЛЕНИЕ
Многие полимеры, особенно при температуре выше комнатной,
разлагаются в атмосфере воздуха из-за окисления, которое не инду-
цировано световым воздействием (тепловое старение). Для ряда
полимеров ухудшение механических характеристик наблюдается
уже после нагревания в течение нескольких дней до температуры
порядка 100°С и даже до еще более низких температур (это харак-
терно, например, для полиэтилена, полипропилена, полиформаль-
дегида, по лиэти ленсу льфида).
Скорость окисления можно определить, измеряя объем поглощен-
ного кислорода при определенной температуре. Подобные измерения
показали, что интенсивность окисления полиэтилена низкой плот-
ности при 140°С увеличивается экспоненциально после индукцион-
ного периода длительностью 2 ч. На основании такого результата
можно заключить, что термическое окисление, подобно фотоокисле-
нию, обусловлено автокаталитическим процессом, и различие за-
ключается только в том, что образование радикала из гидропере-
киси активируется теплом.
Основной реакцией может оказаться прямое взаимодействие
с кислородом:
RH + O2 —R- + -OOH (ж)
Термическое окисление может ингибироваться антиокислителями
[уравнения (г), (д), (е)[.
359
ГИДРОЛИТИЧЕСКАЯ ДЕСТРУКЦИЯ
Гидролитическая деструкция играет существенную роль, если гид-
ролиз потенциально оказывается основной реакцией в разрыве свя-
зей, например в полиэфирах и поликарбонатах. Атака молекулой
воды может осуществиться быстро, если температура достаточно
высока; эффективность атаки кислотами зависит от активности
кислот и температуры. Деструкция под действием щелочных соеди-
нений определяется возможностью проникновения в полимер дан-
ного гидролитического агента: аммоний и амины могут приводить
к гораздо более интенсивному гидролизу по сравнению, например,
с каустической содой, поскольку последняя атакует главным обра-
зом поверхность соединения. Аморфные участки полимера подвер-
гаются атаке быстрее всего, но и кристаллические области тоже
подвергаются атаке гидролитических агентов.
РАСТРЕСКИВАНИЕ ПОД ДЕЙСТВИЕМ
ВНЕШНИХ НАПРЯЖЕНИЙ
Многие пластики подвергаются комбинированному воздействию дли-
тельных напряжений и жидкостей или паров. Наблюдаемый при
этом эффект ухудшения механических характеристик называют
растрескиванием под влиянием внешних напряжений. Напряжение
может быть статическим или периодически изменяющимся. Сущест-
венно то, что действующие на полимер жидкости или пары могут
не быть ни растворителями для данного полимера, ни даже просто
активными агентами набухания. Довольно часто растворы поверх-
ностно-активных веществ могут вызывать разрушение при низких
уровнях напряжения. Как правило, растрескивание под влиянием
внешних напряжений уменьшает длительность периода ползучести
до наступления разрушения.
КОЛИЧЕСТВЕННОЕ ОПИСАНИЕ
ДЕСТРУКЦИИ ПОЛИМЕРОВ
ТЕМПЕРАТУРА, ПРИ КОТОРОЙ
РАЗЛОЖЕНИЕ ПРОИСХОДИТ НА 50%
Термическая деструкция характеризуется прежде всего разрывом
наиболее слабой связи и, следовательно, определяется энергией
диссоциации такой связи.
Для практически всех реакций диссоциации изменение энтропии
близко по порядку величины, поэтому можно предположить, что
и энтропия активации должна быть примерно одинаковой. В прин-
ципе это означает, что такой процесс определяется энергией диссо-
циации связи. Следовательно, можно думать, что температура, при
которой достигается одинаковая степень конверсии, фактически
пропорциональна энергии диссоциации такой связи. В гл. XXI
приведены числовые значения энергии диссоциации связей (см.
табл. XXI.3).
360
В табл. XXII.1 дается сводка наиболее важных результатов
по термической деструкции полимеров винилового ряда. В качестве
показателя, характеризующего термическую устойчивость, выбрана
температура (71!/,), при которой потеря массы полимера составляет
50% в условиях нагревания в вакууме в течение 30 мин. На рис.
XXII.2 аналогичные данные, дополненные результатами исследо-
вания некоторых других полимеров [14] и ряда радикальных инициа-
торов (перекиси и азосоединения), представлены в зависимости от
энергии диссоциации связей.
Рис. XXII.2. Температуры разложения На 50%.
Как и предполагалось, полученные экспериментальные данные
описываются практически линейной зависимостью:
Г>/4(К) = 5,75Ядис+185 (XXII.1)
где Елис энергия диссоциации связи, выраженная в ккал.
КИНЕТИКА ТЕРМИЧЕСКОЙ ДЕСТРУКЦИИ
Процесс статистической тепловой деструкции можно, как правило,
описывать реакцией первого порядка (в качестве параметра прини-
мается потеря массы) при условии, что продукты разложения ле-
тучи. Математическая обработка результатов при этом рассмотрена
в работах Ван Кревелена с сотр. [20], Рейха [16, 17] и Бройдо [10].
Подробно изучена цепная деполимеризация [18]. При этом
оказались существенными следующие два фактора: реакционная
361
Таблица XXII.1. Термическая деструкция полимеров
Полимер T*/f °C k 350* % /мин Выход мономера. % Еакт* ккал/моль
Политетрафторэтилен 509 2Х10-в Выше 95 81
Поли-п-ксилилен 432 0,002 0 73
Поли-п-фениленметилен 430 0,006 0 50
Полиметилен 414 0,004 Ниже 0,1 72
Политрифторэтилен 412 0,017 Ниже 1 53
Полибут а диен 407 0,022 2 62
Полиэтилен (разветвленный) 404 0,008 Ниже 0,025 63
Полипропилен 387 0)069 Ниже 6,2 58
Полихлортрифторэтилсн 380 0,044 27 57
Поли-а-дейтеростирол 372 0,14 39 56
Поливинилциклогексан 369 0,45 0,1 49
Полистирол 364 0,24 40 55
Поли-Р-дейтеростирол 362 0,27 68 55
П оли-л-метилстирол 358 0,90 45 56
Полиизобутилен 348 2,7 20 49
Полиоксиэтилен 345 2,1 4 46
Поли-а, р, Р-трифторстирол 342 2,4 7,4 64
Полиметилакрилат 328 10 0 34
П олиметилметя крил ат 327 5.2 Выше 95 52
Полиоксипропилен (изотактический) 313 20 1 35
Полиоксипропилен (атактический) 295 5 1 20
П оли- а - мети лстир ол 286 228 Выше 95 55
П оливини лацетат 269 — 0 17
Поливиниловый спирт 268 0
Поливинилхлорид 260 170 о*** 32 ***
• Температура, при которой полимер теряет 50% своей массы при нагревании
в вакууме в течение 30 мин.
** Скорость выделения летучих продуктов (потеря массы) при 350 °C.
*** Определено по потерям НС1.
способность радикала, обрывающего цепь; наличие реакционноспо-
собного атома водорода, участвующего в передаче цепи.
Для всех полимеров, содержащих а-водородные атомы (например,
полиакрилаты, полиолефины и т. д.), наблюдаются низкие выходы
мономеров. Напротив, для полиметакрилатов и поли-а-метилстиро-
лов выходы мономеров высокие за счет блокирования реакции пере-
дачи цепи а-метильной группой. Выход мономера в случае политет-
рафторэтилена высок, поскольку прочные С—F-связи оказываются
устойчивыми в отношении реакций передачи цепей.
Такой тип деструкции можно также описать суммарной реакцией
квазипервого порядка, но кинетическая схема окажется более
сложной. G помощью кинетического анализа можно получить, на-
ряду с константой скорости реакции, два других параметра: кон-
станту передачи- цепи (£пер = вероятность передачи/вероятность
инициирования) и кинетическую длину цепи (Лкин = вероятность
362
роста цепи/суммарная вероятность обрыва цепи и передачи цепи).
Для полиэтилена ЛКИн близка к нулю (мономера не образуется);
для полиметилметакрилата Лкин составляет примерно 200 (образо-
вание мономера почти 100%).
РЕЛАКСАЦИЯ НАПРЯЖЕНИЙ
ЗА СЧЕТ ХИМИЧЕСКОЙ ДЕСТРУКЦИИ
Напряжение релаксирует в том случае, когда разрывается молеку-
лярная цепь, находящаяся под нагрузкой. Например, такие явления
наблюдаются в процессе окисления каучуков. Если цепной сегмент
растянут, то при разрыве он возвращается в недеформированное
состояние. Нагрузку несут только растянутые цепи, и нагрузка,
действовавшая на разорвавшейся цепочке в структуре сеткц, не
может передаваться ею на другие цепи. Можно предположить, что
скорость разрыва растянутых цепей в структуре сетки пропорцио-
нальна полному числу (п) цепей, несущих нагрузку:
—dn/dt = kn (XXII.2)
С помощью теории высокоэластичности каучука можно довольно
просто получить следующее соотношение:
о/а0 = сг (t)/a0 — exp ( — kt) (XXII.3)
Это выражение по форме весьма сходно с условием релаксации на-
пряжений в вязкоупругом материале (гл. IX). По аналогии вели-
чину i/k назвали «временем релаксации» т. Обычно температурные
зависимости скорости химических реакций удовлетворительно опи-
сываются уравнением Аррениуса, поэтому зависимость времени
релаксации от температуры можно выразить таким образом:
1пт = 1п1/А = 1пА+£,а/ЛТ (XXII.4)
где Еа — энергия активации данной химической реакции.
Наиболее характерное значение составляет 30 ккал/моль для
окислительной деструкции каучуков.
ДОЛГОВЕЧНОСТЬ ПОЛИМЕРНЫХ МАТЕРИАЛОВ
Ниже приведены факторы, влияющие на долговечность полимерных
материалов: , ’ .
Напряжение и деформа- Растворение
ция
Природа окружающей Размягчение
среды
Температура Растрескивание под дейст-
вием напряжения
Молекулярный вес Хрупкость
Молекулярная струк- Химическая деструкция
тура
Кристалличность Фотохимическая деструк-
ция
Ориентация Биологическая деструкция
363
Хаган и Томас [13] показали, каким образом можно применить
принцип температурно-временной аналогии для предсказания долго-
вечности полимерных материалов. Эти авторы задались постоянным
удлинением при разрушении (ползучестью) и воспользовались
принципом суперпозиции для ускоренного получения данных по
механическим свойствам полимеров. Однако такой метод связан
с известной опасностью, поскольку окружающие условия могут
чрезвычайно резко снижать долговечность и в ряде случаев на не-
сколько порядков величины.
Суезава с соавторами [19] показали, что долговечность полимеров
под нагрузкой можно описать обобщенной кривой; при этом, на-
ряду с напряжением, в качестве независимого параметра может
использоваться некоторая величина, характеризующая влияние
окружающей среды на долговечность. Для конкретного полимера
для каждой жидкой средь/ может быть получена постоянная вели-
чина «фактора сдвига», представляющего собой соотношение долго-
вечностей при испытании полимера в данной среде и в другой среде,
выбранной за эталонную. Это обстоятельство обнаружил Фалмер
[12]. Ниже приводятся значения некоторых факторов сдвига, полу-
ченные Бергеном [7], которые характеризуют влияние окружающей
среды на долговечность полимеров:
Фактор сдвига для
изопропилового
спирта
120
Сополимер акрилонитрила, 120
бутадиена и стирола
Поливинилхлорид 0,08
Поликарбонат 3
Сополимер стирола и акрило- 1500
нитрила
Другой способ обработки данных долговечности полимеров заклю-
чается в построении зависимости логарифма времени до разрушения
от действующей силы. Этот способ предложил Коулман * [3], кото-
рый использовал следующее уравнение:
In /разр — 1И &— bf
(ХХП.5)
где / — сила, приложенная к образцу; а и Ъ — постоянные, зависящие от окру-
жающей среды.
Постоянная а описывается выражением
а = ай exp (E1RT)
* См. Прим рев. к гл. X на С. 187.
364
ЛИТЕРАТУРА
1. Grassie N., «Chemistry of High
Polymer Degradation Processes»,
Interscience, New York, 1956.
2. Jellinek H‘. H. T., «Degradation
of Vinyl Polymers», Academic
Press, New York, 1955.
3. Madorsky S. L. and Straus S.,
«High Temperature Resistance
and Thermal Degradation of Po-
lymers», S. С. I. Monograph
13 (1961) 60—74.
4. Madorsky S. L., «Thermal Degra-
dation of Organic Polymers»,
Interscience, New York, 1964.
5. Neimann M. B. (Ed.), «Aging
and Stabilization of Polymers»,
Consulants Bureau, New York,
1965.
6. Voigt J., «Die Stabilisierung der
Kunststoffe gegen Licht und War-
me», Springer, Berlin, 1966.
7. Bergen R. Lt, S, P. E. Journal
20 (1964) 630.
8. Bolland J. L., Proc. Roy. Soc.
(London) A186 (1946) 218.
9. Bolland J. L. et al., Trans.
Faraday Soc. 42 (1946) 236, 244:
43 (1947) 201; 44 (1948) 669; 45,
(1949) 93; 46 (1950) 358.
10. Broido A., J. Polymer Sci. A2,
7 (1969) 1761.
11. Farmer E. H. and Sundraling-
ham A., J. Chem.- Soc. (1943)
125.
12. Fulmer G. E., Polymer Eng. Sci.
(1967) 280.
13. Hagan Rt S. and Thomas J. R.,
«Plastic Part Properties and their
Relationship to End-use Perfor-
mance», 67th Annual Meeting
ASTM, June 21—26 (1964).
14. Коршап B.B., Виноградова С. B.,
Усп. хим., 1968, т. 37, с. 11.
15. Madorsky S. L. and Straus S.,
J. Research Natl. Bur. Standards
53 (1954) 361; 55 (1955) 223; 63A
(1959) 261.
16. Reich L. and Levi D. W., Makro-
mol. Chem. 66 (1963) 102.
17. Reich L., Makromol. Chem., 105
(1967) 223.
18. Simha R. and Wall L. A., J.
Phys. Chem. 56 (1952) 707.
19. Suezawa Y., Hojo H., Ideda T.
and Okamura Y., Materials and
Research Standards 3 (1963) 550.
20. Van Krevelen D. W., Van Heer-
den C. and Huntjens F. J., Fuel
30 (1951) 253.
ЧАСТЬ
ШЕСТАЯ
ОБСУЖДЕНИЕ ИЗЛОЖЕННЫХ
ПРЕДСТАВЛЕНИЙ
глава ххш. СООТНОШЕНИЯ МЕЖДУ
ХАРАКТЕРИСТИКАМИ ПОЛИМЕРОВ
В предыдущих главах книги обсуждались аддитивные Величины,
приведенные в табл. Ш.З. Удалось показать, что с помощью этих
аддитивных величин можно провести оценку важных характеристик
полимерных материалов. Но всегда основное условие, обеспечива-
ющее подобную возможность, заключалось в том, что должны быть
известны некоторые эмпирические величины, связанные с группо-
выми вкладами или групповыми инкрементами, входящими в эти
аддитивные величины.
Кроме этого, в предыдущих главах был установлен ряд соотно-
шений между числовыми значениями характеристик.
В заключение будут обсуждены три следующих вопроса:
1. В какой мере групповые вклады характеризуют различные
аддитивные величины?
2. Сколь далеко распространяется влияние на них характеристик
полимерных материалов?
3. В какой мере эти соотношения могут быть объединены в одну
систему?
В Приложении 1 приводится сводка значений групповых вкладов
в аддитивные величины.
ХАРАКТЕРИСТИЧЕСКИЕ ДИАГРАММЫ
ИНКРЕМЕНТОВ
Характеристические диаграммы получаются путем построения зави-
симостей групповых вкладов в различные аддитивные величины
от молекулярных весов данных групп.
В диаграммах, представленных на рис. ХХШ.1—XXIII.12,
каждой группе соответствует определенная точка. Если соединить
эту точку с началом координат, то тангенс угла наклона прямой
будет мерой удельной величины, соответствующей данной группе,
например удельного объема, показателя преломления, парахора
и т. д.
На всех диаграммах проведены разделительные линии, облада-
ющие следующими свойствами:
а — линия проходит через начало координат и группу [—СН2—];
группа —СН2— принимается в качестве прототипа неполярных
групп;
366
Рис. XXIII.2. Мольный объем (стекло-
образное состояние).
Рис. XXIII.3. Ван-дер-ваальсовский
объеы-
Рис. XXIII. 1. Мольный объем (высо-
коэластическое состояние).
с$, кол/(моль°К)
О 20 60 80 /00 120 КО
Рис. ХХШ.4. Мольная теплоемкость
(твердое состояние).
Рис. ХХШ.6. Мольная функция при-
тяжения по Смоллу.
Рис. ХХ1П.5. Мольная теплоемкость
(жидкое состояние).
о го 40 во so юо /го мо
Рис. ХХ1П.7, Парахор.
Рис. ХХШ.9. Мольная рефракция.
Рис. XXIII.8. Мольная функция
скорости звука.
Мольная вееприим-
Рис. ХХП1.10. Мольная элек-
трическая поляризация.
Рис. XXIII.il.
чивость.
Рис. XXIII.12. Мольная функция ха-
рактеристической вязкости.
б — линия проходит через начало координат и группу [—СИ;
группа —С1 принимается за «нормальную» полярную группу;
в — линия соединяет четыре типичных алифатических углеводо-
родных группы [—СН3], [—СН2—], 1=СН—] и [—С—];
г — кривая соединяет типичные галогенный группы [—С1],
[—Вг] и [—I]; группа [—F] занимает особое положение.
Теперь можно сравнить между собой положения некоторых типич-
ных групп на разных диаграммах.
1. Положение четырех алифатических углеводородных групп (ли-
ния в). Эти группы фактически всегда располагаются регулярно и
во многих случаях почти на одинаковых расстояниях друг от друга.
2. Положение галогенных групп (линия г). Эта кривая резко
отличается от предыдущей линии. В одних случаях она вогнута,
в других выпукла. Иногда точки лежат почти на прямой. Однако
характерное свойство заключается в том, что экстраполированная
часть линий г пересекает линию в почти всегда в одном и том же
месте — между группами —СН3 и —СН2—.
Среди галогенных групп группа —F занимает уникальное поло-
жение — она всегда находится ниже линии г. Довольно часто — F
располагается на линии б или недалеко от линии б, но эта группа
лежит^между линиями а и б в диаграммах «Ср» — «М» и «U — М»'
и ниже линии б — в диаграммах «ВЛл — М», «Р — М» и «X — М».
3. Положения других типичных групп. В большинстве диаграмм
другие группы попадают между линиями а и б. Основные исключе-
ния: диаграмма «F — М», на которой группа [—CN] лежит выше
линии а, группа [—О—] — ниже линии би «X — М» диаграмма,
на которой несколько групп лежат ниже линии б.
Наиболее заметное свойство этих диаграмм заключается в том,
что они сходны друг с другом, но никоим образом не идентичны.
Другими словами, каждая аддитивная величина обладает индиви-
дуальностью. - '
ВЛИЯНИЕ ФУНКЦИОНАЛЬНЫХ ГРУПП
НА ХАРАКТЕРИСТИКИ ПОЛИМЕРОВ
Та’степень, в которой характеристики определяются специфическими
группами, лучше всего оценивается следующим образом. Все группы
можно скомбинировать так, чтобы получились только бифункцио-
нальные единицы, например
[_СН2~]
[-СВД +
I
СН—
I
= [СН(СН3)—], 2[—C1JH-
= [—СС12—] и т. д.
С помощью аддитивных величин для этих бифункциональных групп
можно рассчитать характеристики гипотетического полимера,
24*
371
& 1О'"Ч _71 7m
2,4 2,2 2/ 7/ 1,6 {4 7,2 1,0 О,в 0,6 о,о 0,2 -0 1<£г- ?50 22L 200 180 ISO МО по 10Q 80 60 Н3)- 60 20 0- 600 550 500 900 800 100 600 - 500 W0 306 гос 100 0 tCONM- t-s-- -CHCf-
|-со - н --С<0>-
11 --о-
‘ -CHF- -снег- ^-CCi2— --CHF-
-сг2- /л - -СО- -CONH-
Veer /и
- -о- - -СН(СНз)
--СОМ а : cr2“
ЧГНСг- -СНГ- $
ХЯ юо 250 гос 150 100 7C00-— =co-o-co
7 гСН^СНз
-сн(ск)- -conhJ ‘4-@-
$ п~ '-СОО- • -СР2 o-co-o-
- -С-© - -S-
|СН=С(СН, jrCH.CH- ^СН(СН,)- Ц’снг~1 У5 --CONH-* •ch(cn)! ^-s- уСН=С(СЕ -с(сн3)2 7C(ch3)2-
4 --снег-
--СС12- ж д ~СНС1~ —гСОО- r->CHF- —гСГ2“— S’CHfCH- тСН-СН
3 гСОО- 'ЧрСнГ-|
= СН(СН;)
тсСснЛ- $сн=сн
' L
1 1ЕЙН 1 т
- -о- ¥C(CH,W \-О- 5г -0-
0-
Ka (‘10<) (S <T 77D
30 25 20 15 Ю 5 -0 22 20 18 16 10 12 10 8 6 4 2 0 sfi 5,5 5,0 5,5 3,5 3,0 г,5 Zfl !,5 l,t 2,7 2,0 7,5 // 7,7 7/ 1,5 44 77 7,7 I,»
—ch(cn)-
- -co-
^LUNM ” -hC»L-l- ^0-
^COO-
^-CHfCN1)-
- -co- -c^0>
- -s- H ж _X*CO-
- -COO- --ch(cn)- -CC12-
--СО-ОтСО- _Xo>- — CH Cl- — ^-CHCr- ^CH ‘ CHt -ch(cnI?
= -CHCl-
--COO- ^CH (CH 3)-“ ^-CHCI - __hctiV ^H2-|- e сн- VCHF— \-ch(chJ- \-C(CHj)2- VCFj- -_-CC(2- -\H-chz-I ^v^ch(chj): JsSXchjV. ^=C00- - -CHF-
~^CCI2- — ^-(дУ H
- -C(CHj)2- - -O-
гсн(сн2)- - -CFj-
\-o- ^J-CH2~J
Рис. ХХШ.13. Влияние функциональных групп на различные характеристики полимеров.
полностью составленного из таких бифункциональных групп. Таким
путем определяются поверхностное натяжение, параметр раствори-
мости, показатель преломления и т. д. для этого гипотетического
полимера и, следовательно, для составляющей полимер бифункцио-
нальной группы. Тем самым определяется количественное влияние
данной группы на характеристики.
На рис. XXIII.13 и XXIII.14 описанный способ применен
к двум категориям, величин: категории (р, К, -у, Tg и 7\), дающей
информацию относительно механических и термодинамических
Рис. ХХШ.14. Характеристики, связанные с тепловым
расширением.
характеристик данного вещества, и категории (Кв, 8, е и nD), да-
ющей информацию относительно взаимодействий между полимерами и
взаимодействий полимера с электромагнитным полем. На обоих
рисунках отмечена группа —СН2—.
Если характеристики бифункциональных групп рассчитываются
по значениям групповых вкладов, то довольно часто привлекается
мольный объем V. В Приложении 1 приведены значения групповых
вкладов в мольный объем для высокоэластического и стеклообраз-
ного состояний. Для расчетов характеристик, отмеченных на
рис. XXIII.13 и ХХШ.14, применяются величины VH, если они
известны. В других случаях используются величины Vgi', такие дан-
ные показаны вертикальными стрелками.
Теперь следует кратко рассмотреть некоторые особенности этих
диаграмм.
1. В большинстве случаев углеводородные группы (насыщенные
и ненасыщенные) располагаются относительно близко друг к другу.
Исключение составляют характеристики Кд, Т&, Тм и nD. Величины
373
Kq, Т'&т1 Тя резко зависят от степени гибкости молекул; гибкость
весьма сильно уменьшается в случае ароматических цепей. При
наличии ароматического кольца показатель преломления резко
возрастает.
2. Группы [—[Tjj—] и [—СН—] обычно близки друг к другу,
но и здесь величина Kq составляет исключение; это понятно,
если учесть гибкость молекул. ’
3. Многие характеристики группы [—С—] имеют большие зна-
II
О
чения, но это не относится к величинам Ка, nD и Tg.
4. Группа [—CF2—] дает . чрезвычайно низкие значения 6, е
и и высокие значения р и К; для технологов это представляет
весьма большой интерес.
5. Группа [—CONH—] дает значения всех характеристик выше
средних; особенно это относится к величинам б, Те и Тм. Последние
зависят _ от тенденции к образованию водородных связей.
Можно утверждать, что рис. XXIII.13 и XXIII.14 представляют
собой «спектр» характеристик вещества: с помощью аддитивных
величин сложная картина («средняя величина») расщепляется как
бы на спектральные линии, т. е. система аддитивных величин играет
роль призмы^или дифракционной решетки.
СООТНОШЕНИЯ МЕЖДУ
ФИЗИЧЕСКИМИ ХАРАКТЕРИСТИКАМИ
На протяжении почти всей книги было неоднократно показано, как
определенные физические величины коррелируют друг с другом.
Теперь можно попытаться систематизировать все такие соотно-
шения. Для того чтобы получить подходящее расположение физиче-
ских величин в относительно простых схемах, их делят на следу-
ющие группы:
1) величины, описывающие тепловое расширение;
2) величины, определяющие когезию;
3) механические характеристики полимеров в твердом состоянии;
4) электромагнитные величины;
5) величины, определяющие свойства расплавов и растворов
полимеров;
6) термодинамические величины.
374
СООТНОШЕНИЯ МЕЖДУ ВЕЛИЧИНАМИ,
ОПИСЫВАЮЩИМИ ТЕПЛОВОЕ РАСШИРЕНИЕ
Такие соотношения были рассмотрены в гл. IV. Сводка их при-
ведена на рис. XXIII.15. Как и следовало ожидать, основу этой
схемы составляют все аддитивные мольные величины, определяющие
Основные аддитидные мольные функции
~ Уд ' Р
Рис. ХХШ.15.х Характеристики, связанные с энергией
когезии.'
тепловое расширение; сюда относятся мольные величины, описыва-
ющие постепенное расширение, и величины, определяющие темпе-
ратуры переходов ~
i . -
СООТНОШЕНИЯ МЕЖДУ ВЕЛИЧИНАМИ,
ОПРЕДЕЛЯЮЩИМИ КОГЕЗИЮ
В гл. VII и IX уже отмечались соотношения между плотностью
энергии когезии, поверхностным натяжением, внутренним давле-
нием, сжимаемостью и т. д. Все приведенные ниже величины имеют
размерность энергия/объем: еког, К, ср/е, RYg/ZV; кроме того, эти
величины связаны вполне определенными соотношениями с внутрен-
ним давлением я и поверхностным натяжением у. По аналогии
с плотностью энергии когезии объемный модуль упругости можно
представить как плотность упругой энергии, а коэффициент ср/е —
как плотность тепловой энергии.
На рис. XXIII.16 приведена сводка всех соотношений, получен-
ных в этой области.
375
Рис. ХХШ.16. Механические характеристики.
СООТНОШЕНИЯ МЕЖДУ МЕХАНИЧЕСКИМИ
ХАРАКТЕРИСТИКАМИ (РИС. ХХШ.17)
Механические характеристики рассматривались в гл. IX и X. Как
можно было ожидать, не только молярная функция скорости звука,
но и температуры переходов играют весьма существенную роль
Рис. XXIII. 17. Электромагнитные характеристики.
в проявлении механических свойств полимеров. Мольная функция
скорости звука связана с энергией когезии, поэтому можно считать,
что энергия когезии (Е^, и ДЯЖ) и вероятность переходов (Д5М)
376
определяют механические характеристики. Температура и время
связаны с температурами переходов с помощью фактора
сдвига (ат).
СООТНОШЕНИЯ МЕЖДУ
ЭЛЕКТРОМАГНИТНЫМИ ВЕЛИЧИНАМИ
Эти величины рассматривались в гл. XI—XIII. Соотношения между
ними представлены на рис. XXIII.18.
Рис. ХХП1.18. Характеристики расплавов и растворов.
СООТНОШЕНИЯ МЕЖДУ ВЕЛИЧИНАМИ,
ОПРЕДЕЛЯЮЩИМИ ХАРАКТЕРИСТИКИ РАСПЛАВОВ
И РАСТВОРОВ ПОЛИМЕРОВ
Эти величины рассматривались в гл. VI—VIII, XV и XVI.
Очень интересные и довольно новые соотношения были полу-
чены для величин, определяющих поведение полимеров в жидком
состоянии. Сказанное иллюстрирует рис. XXIII. 19. По-видимому,
решающими факторами здесь оказываются величины, определяющие
когезию и гибкость молекул. От этих факторов зависят не только
вязкости расплава и раствора, но также определяемая диффузией
скорость роста кристаллитов и константа диффузии растворенного
полимера.
На рис. XXIII.20 представлена диаграмма, в которой сравни-
ваются два упомянутых выше фактора: межмолекулярная когезия
(характеризуется величиной 6) и гибкость молекул (характеризуется
величиной Kq). Очевидно, что семейства полимеров представлены
на этой диаграмме характерными кривыми.
377
Рис. ХХП1.19. Диаграмма «К0— б»:
а— полиолефины и полидиены; Л — алифатические поли-
эфиры; в — полиакрилаты и полиметкрилаты; г — поли-
амиды; О — целлюлозы; 1 полиэтилен; 2 — полипропи-
лен; з — поли-н—бутен; 4 — полииаобутилен; 5 — поли-
бутадиен (троне); в — полибутадиен (цис); 7 — полиизо-
прен (тра-нс); 8 — полиизопрен (цис); 9 — полистирол;
10— полиоксиметилен; 11 — полиоксиэтилен, 12 — найлон
6; 13 — полиакрилонитрил; 14 — полиэтилентерефталат;
13 — поливинилацетат; 16 — полиметилакрилат; 17 — поли-
этйлакрилат; 18—полиакриловая кислота; 19—полиме-
тилметакрилат; 20 — полиэтилметакрилат; 21 — полимета-
акриловая кислота; 22 — целлюлоза; 23 — триацетат
целлюлозы.
40
30
го
10
0
-10
-го
-30
-40
•50
-so
-10
-80
-so
-40 -зо -го -10 О 10 20 30 40
-—в[(ъ -8S°(2S8)]
Рис. ХХШ-20. Графическое представление величин А и В
в уравнении (Г) = А + В Г:
а — тройные связи; б — сульфид; в — олефиновые и ароматические; г —
алкильные и аминные; & — карбонильные; е — окисные; ж ~ карбоксильные.
СООТНОШЕНИЯ МЕЖДУ
ТЕРМОДИНАМИЧЕСКИМИ ВЕЛИЧИНАМИ
В тл. XXI приведены аддитивные групповые вклады в свободную
энергию образования. Здесь также имеется ряд важных соотношений.
Прежде всего, на рис. XXIII.21 представлены величины А
ль АН} (298) и В «а —AS} (298), входящие в формулу AG} (298) =
= А + ВТ. Нетрудно видеть, что на этой диаграмме группы рас-
полагаются по семействам: углеводородные группы с одинарными,
двойными и тройными связями; галогенные группы (—F, —С1,
—Вг, —I);' окси-, амино- и сульфидные группы; карбоксильные
и карбонильные группы.
Более того, из рис. XXIII.21 видно, что отрыв атома водорода
от группы ХН с образованием новой группы X приводит к увели-
чению А и В. Подобное изменение AG} (Т), обозначенное Д (Дб/),
подчиняется определенным правилам.
Если величину А для группы ХН (Лхн) отложить в зависимости
от величины X (Лх), то получится универсальная функция, что
показано на рис. XXIII.22. Исключения немногочисленны, напри-
мер HI, Н2С=О, -НС=О,НСООН и -СООН.
Приближенно выполняется следующее правило:
Лх = 0,785А хн + 4000
379
Аналогичным способом величину В группы ХН (Вхн) можно срав-
нить с величиной В после отрыва атома водорода (Вх)- Если разность
Число атомоб боборода, оторбонных от
полностью гидрироооннои группы
Рис. ХХ1П.22. Влияние отрыва атома водорода на величину В.
Вх — Z?xh = A-Sx отложить как функцию числа оторванных атомов
водорода от полностью гидрированной группы, то получится диа-
грамма рис. XXIII.22. Примером являются следующие данные:
^сн4 = 22,3
в-сн. = 22,9
В—СН,— = 24,5
*1 СН— 1 = 29,0
= 36,5
дйх = 0,6
ДЯХ = 1,6
ДЯХ = 4,5
ДВХ = 7,5
Число оторванных от пол-
ностью гидрированной груп-
пы атомов Н
1
2
3
4
380
Здесь также выполняется более или менее очевидное правило,
несмотря на некоторые имеющиеся отклонения, которые по порядку
величины такие же, как и ошибки определения АВ.
Наличие упомянутых эмпирических, правил подтверждает един-
ство системы групповых вкладов.
ЗАКЛЮЧЕНИЕ
После завершения двадцати трех глав, составляющих данную
книгу, можно считать, что цель, сформулированная в предисловии,
достигнута, по крайней мере в общем плане.
Хотелось бы в заключение привести цитату из редакционной
статьи, опубликованной в журнале «Chemical and Enginering
News» от 1 марта 1971 г.: «Принцип групповой аддитивности может
стать столь же эффективным инструментом для химиков и техноло-
гов, каким стали бунзеновская горелка и логарифмическая линейка».
ПРИЛОЖЕНИЯ
Приложение 1. Сводка групповых вкладов
М м л А Л §
Л Л ч ч я
>0 t; Л 5 ч © § ф я Ф Я м
Группа Z Л ч о Я о Я t _Я S я. ч ч “Ч Р
ф я h Я ф /.«□ я о & § «1 ё. 1 р, кал X S X h о X i ?
£ и о < Ц о
Бифункциональные уг- леводородные группы
-сн,— 1 14,03 15,85 16,45 10,23 6,05 7,26 900 1000 133 39,0 170 ‘*/270
-СН(СН,)— 1 28,05 33,35 32,65 20,45 11,10 13,80 1500 2400 242 78,0 336 '
-СН(С,Н,)- 1 82,14 — 53,28 26,44 35,2 — — 10 0 208,3 —
- СН(С.Нц)- 1 96,17 100,15 — 63,58 28,89 41,5 — — 820 244,9 —
-CH(C.Hj)— 1 90,12 82,15 74,5 52,62 24,16 34,4 — 6900 763 211,9 576
-С(СН1)«- 1 42,08 52,4 50,35 30,67 16,23 19,36 — 3000 335 117,0 226
-С(СН.)(С,Н,)- '1 104,14 101,20 92,2 62,84 29,29. 39,96 — 7500 856 250,9 730
( цис -СН=СН-{ V транс 2 26,04 — 27,75 16,94 8,90 10,20 500 2000 222 67,0 Г 340 1 700
(цис -СН-С(СН«)-{ 2 40,06 42,8 27,16 14,33 17,70 1100 2700 344 106,0 480
' [транс 56,0
-с=с- 2 24,02 . — — 16,1 — —' — — — —
/—\ ( Цис 24,60
—/ 1 троне 4 82,14 87,8 — 53,34 35,2 — — 690 205,9 —
-<сГ>- 4 76,09 65,5 61,4 43,32 18,80 27,0 5300 6000 658 172,9 1200 ’*/2200
"joj 3 76,09 — — 43,32 18,8 27,0 — — 658 172,9 1500
\О/ 2 76,09 — — 43,32 18,8 27,0 — — 658 172,9 —
/СН, 910 2000
-<О>- 4 104,14 104,1 — 65,62 30,28 39,80 — - . — 250,9
\сн,
-\О>- 4 90,12 83,4 — 54,47 24,54 33,4 — — 800 211,9 —
\сн. 791
-<О>-СН.- —-CH t — 5 90,12 81,35 77.85 53,55 24,85 34,26 6200 7000 211,9 —
6 104,14 97,20 94,30 63,78 30,90 41,52 7100 8000 924 250,9 —
-<о><2>- 9 166,21 146,85 139,25 96,87 43,65 61,26 11 500 13 000 1449 384,8 —
8 152,18 131,0 122,8 86,64 37,60 54,0 10 600 12 000 1316 345,8 —
_/<о> 4 228,28 (201,7) — 129,96 56,40 81,0 — — 1950 518,7 2300
"<<О>
ругие углеводородные
группы -СН. -С.Н. 15,03 23,9 22,8 13,67 7,38 8,80 600 2300 214 56,1 —
29,06 39,75 39,25 23,90 ,13.43 16,06 1500 3300 347 95,1 —
43,09 55.60 55,70 34,13 19,48 23,32 2400 4300 480 134,1 —
43,09 63.65 55,45 34,12 18,48 22,6 2100 4700 456 134,1 —
— 57,11 — 73,15 44,34 23,61 28,16 — 5300 549 173,1
1 —сн 1 1 13,02 9,45 9,85 6i78 3,72 5,00 900 . 100 28 21,9 —
1 -с— 1 1 12,01 4,6 4,75 3,33 1,47 1,76 — —1600 —93 4,8 —
'** 270 пля полиамидов, полиуретанов и поликарбамидов; 170 для других полимеров. •• 12Оо”для поликарбамидов; 1300 для полисилоксанов; 1500 для полиоксидов, полисульфидов и полиуретанов. 2200 для полиами-
ПРИЛОЖЕНИЯ
Приложение 1. Сводка групповых вкладов
Группа Z 31, г/моль Vgt см’/моль Vr, См’/моль Уфу, см’/моль Ср, кал/(моль-К) 1 Ср, кал/(моль-К) ДНт, кал/моль Вког, кал/моль F (Смолл), (кал-см’)1/'/моль Р
Бифункциональные уг- леводородные группы - СН,— 1 14,03 15,85 16,45 10,23 6,05 7,26 900 1000 133 39,0 170 ‘*/270
-СН(СН,)— 1 28,05 33,35 32,65 20,45 11,10 13,80 1500 2400 242 78,0 336 '
-СН(С,Н,)- 1 82,14 —— — 53,28 26,44 35,2 — — 10 0 208,3 —
—СН(С«Нн)— 1 96,17 100,15 — 63,58 28,89 41,5 — — 820 244,9 —
-СН(С,Н,)— 1 90,12 82,15 74,5 52,62 24,16 34,4 — 6900 763 211,9 576
-С(СН,),- 1 42,08 52,4 50,35 30,67 16,23 19,36 — 3000 335 117,0 226
-С(СН,)(С,Н,)- '1 104,14 101,20 02,2 62,84 29,29. 39,96 — 7500 856 250,9 730
( цис -СН=СН~1 1 транс 2 26,04 — 27,75 16,94 8,90 10,20 500 2000 222 67,0 Г 340 t 700
(цис -СН-С(СН,)-^раис 2 40,06 — 42,8 27,16 14,33 17,70 1100 2700 344 106,0 480
-с=с- 2 24,02 . — — 16,1 — —' — — — 56,0 —
/—\ f 4UC 4 82,14 87,8 53,34 24,60 35,2 690 ' 205,9
1 транс -<О/- 4 76,0-9 65,5 61,4 43,32 18,80 27,0 5300 6000 658 172,9 1200 ’*/2200
"^oj 3 76,09 — — 43,32 18,8 27,0 — — 658 172,9 1500
2 76,09 — — 43,32 18,8 27,0 — — 658 172,9 —
/СН,
-<а>- 4 104,14 104,1 — 65,62 30,28 39,80 —- — 910 250,9 2000
\сн,
~\9У~ 4 90,12 83,4 — 54,47 24,54 33,4 — — 800 211,9 —
\сн,
-<7Г>-сн,- 5 00,12 81,35 77,85 53,55 24,85 34,26 6200 7000 791 211,9 —
—-СН 1 —6^>-СН i— 6 104,14 97,20 94,30 63,78 30,00 41,52 7100 8000 924 250,9 —
-<О>-сн,-<Т))>- 9 166,21 146,85 139,25 96,87 43,65 ’ 61,26 И 500 13 000 1449 384,8 —
-<2><2>- 8 152,18 131,0 122,8 86,64 37,60 54,0 10 600 12 000 1316 345,8 —
/<о> -<о>- 4 228,28 (201,7) — 129,96 56,40 81,0 — — 1950 518,7 2300
ч<2/
ругие углеводородные группы —СНа 15,03 23,9 22,8 13,67 7,38 8,80 600 2300 214 56,1 —
-с,н. - —СдН? 29,06 39,75 39,25 23,90 ,13,43 16,06 1500 3300 347 95,1 —
43,00 55,60 55,70 34,13 19,48 23,32 2400 4300 480 134,1 —
—U8O=C1H? 43,00 63.65 55,45 34,12 18,48 22,6 2100 4700 456 134,1 —
—mpem=C,H, — 57,11 — 73,15 44,34 23,61 28,16 — 5300 549 173,1 —
1 -CH | 1 13,02 9,45 9,85 6i78 3,72 5,00 900 100 28 21,9 —
1 -С— 1 1 12,01 4,6 4,75 3,33 1,47 1,76 — —1600 —93 4,8 —
1
** 270 для полиамидов, полиуретанов и поликарбамидов; 170 для других полимеров.
•• 1200 для поликарбамидов; 1300 для полисилоксанов; 1500 для полиоксидов, полисульфидов и полиуретанов; 2200 для полиами-
S дов; 1850 для других полимеров.
Продолжение прилож. 1
Группа г М, г/моль Vg, см’/моль Уг, см’/моль У|у, см8/моль 5 Ср, кал/(моль -К) г Ср, кал/(моль-К) 4Нт, кал/моль чггои/гген F (Смолл), (кал-см’)1/’/моль Р у£.к
.. =CHi 14,03 11,94 5,40 5,26 190 50,6
=яСН— 1 13,02 — 13,87 8,47 4,45 5,10 256 1000 111 33,5 —
=с< 1 . 12,01 — (6,13) 5,01 2,50- 3,80 (250) (-600) 19 16,4
=с= I 1 12,01 — — 6,96 — — — — — 28,0 —
-сн=с- 2 25,03 — 20,0 13,48 6,95 8,90 500 400 130 49,9
=сн — 13,02 .— — 11,55 — — — — (174) 45,1
«с— 1 1 ( цис 1 12,01 — — 8,05 — — — — 111 28,0 —
—С—С—1 1. транс %сн „ 2 24,02 — (12,26) 10,02 5,00 7,60 (500) (-1200) 38 32,8 —
ар 1 13,02 — — 8,06 3,68 5,3 — — — 34,5
Чс х'"‘аР 1 12,01 — — 5,54 2,04 2,9 — — — 17,4 —
-а — 69,12 — — 46,5 22,72 30,2 — — 670 186,4 —
—<\ у — 83,15 90,7 — 56,8 25,17 36,5 — — 790 223,0 —
-<о> — 77,10 72,7 64,65 45,84 20,44 29,4 — 6800 735 190,00 —
-\О>- 4 74,08 56,3 — 38,28 15,52 22,2 — — 480 138,7 —
-<сГ>- 4 75,08 5 9,5 — 40,80 17,16 24,6 — — 580 155,8 —
25 Заказ 699
Бифункциональные ки- слородсодержащие груп- пы -О- 1 16,00 10,0
—С— 1 28,01 13,4
%
1 О 1 О 1 2 44,01 23,0 “* (18,25)
—о—с—о— 3 60,01 31,4
ч
-С-0—с— 3 72,02
ч ч
—СН(ОН) — 1 30,03 19,15
—СН(СООН)— 1 58,04 —
-СН(НС=О)- 1 42,14 —
-<^сГ>-соо— 6 120,10 88,5
-о-сн,-о— 3 46,03 35,85
Другие кислородсодер- жащие группы
-ОН — 17,01 9,7
—<^О/-он — 93,10 75,2
—С-Н — 29,02 __
ч
—с—он — 45,02 —
%
Бифункциональные азот-
содержащие группы —NH— 1 15,02
—CH(CN)— 1 39,04 28,95
—CH(NHj)— 1 29,04 —
-<(O/-NH- 5 91,11
8,5 3,7 ’* (5,8) 11,7 4,02 5,50 8,5 12,6
24,6 “* (21,0) 15,2 ’* (17,0) (И) 15,5
— 18,9 ’* (23,0) — —
— (27) — —
14,18 7,77 15,7
— — (15,7) 28,6
— 21,92 — —
86,0 58,52 (29,8) 42,5
33,45 17,63 14,09 24,26
— 8,04 4,05 10,7
— 51,36 22,85 37,7
— 15,14 — —
— — (12) 23,6
8.08 3,40 (7.6)
21,48 (9,7) — •
— 17,32 8,72 —
— (51,4) 22,20 (34,6)
400 1 1500 70 275 20,0 (48) 280 365
—1000 3200 310 64,8 640
— — — 84,8 760
— — — (ИЗ) 555
59,0
— 103,8 —
— — — (87) —
4300 9200 ' 968 237,7 —
1700 4000 273 79,0 730
— — 37,1 —
— — — 210,0 —
— — — (65) —
— — — 81,9 —
29,6
— 6900 438 (86) —
— — — 68,6 —
— — — 202,5 —
’* В скобках—значения, полученные Г. Л. Слонимским.
Oj И* 18,25 (соответственно 21,0) для акрильной—СОО-группы.
СЛ —.....
g Продолжение прилож. 1
Группа Z М, г/мопь Vg, см’/моль Vr, см’/моль У|у, см8/моль 1 Ср, кал/(моль -К) Ср, кал/(моль- К) 4Нт, кал/моль ЕКог, кал/моль F (Смолл), (кал-см’)1/’/моль Р у£.к
.. =CHi 14,03 11,94 5,40 5,26 190 50,6
=яСН— 1 13,02 — 13,87 8,47 4,45 5,10 256 1000 111 33,5
=с< 1 . 12,01 — (6,13) 5,01 2,50 3,80 (250) (-600) 19 16,4
=с= 1 1 12,01 — — 6,96 — — — — — 28,0 —
-сн=с- 2 25,03 — 20,0 13,48 6,95 8,90 500 400 130 49,9
=сн — 13,02 — — 11,55 — — — — (174) 45,1
«с— 1 12,01 — — 8,05 — — — 111 28,0
1 1 ( цис —С=С—1 1. транс 2 24,02 — (12,26) 10,02 5,00 7,60 (500) (-1200) 38 32,8
УНаР 1 13,02 — 8,06 3,68 5,3 34,5
Уар 1 12,01 — — 5,54 2,04 2,9 — — 17,4
-а — 69,12 — — 46,5 22,72 30,2 — — 670 186,4 —
— 83,15 90,7 — 56,8 25,17 36,5 — — 790 223,0 —
-<о> — 77,10 72,7 64,65 45,84 20,44 29,4 — 6800 735 190,00 —
-\О>- 4 74,08 56,3 — 38,28 15,52 22,2 — — 480 138,7 —
4 75,08 5 9,5 — 40,80 17,16 24,6 — — 580 155,8 —
25 Заказ 699
Бифункциональные ки- слородсодержащие груп- пы -О- 1 16,00 10,0 8,5 3,7 ’* (5,8) 4,02 8,5 400 1 1500 70 20,0 280
—с— 1 28,01 13,4 11,7 5,50 12,6 — — 275 (48) 365
%
1 о 1 О 1 2 44.01 23,0 “* (18,25) 24,6 “* (21,0) 15,2 ’* (17,0) (И) 15,5 —1000 3200 310 64,8 640
1 о 1 о 1 о 1 3 60,01 31,4 — 18,9 ’* (23,0) — — — — — 84,8 760
-с-о—с— 3 72,02 — (27) — — — — (ИЗ) 555
ч ч
—СН(ОН) — 1 30,03 19,15 — 14,18 7,77 15,7 — — — 59,0 —
—СН(СООН)— 1 58,04 — — — (15,7) 28,6 — — — 103,8 —
-СН(НС=О)- 1 42,14 — — 21,92 — — — — — (87) —
-^о^-соо- 6 120,10 88,5 86,0 58,52 (29,8) 42,5 4300 9200 968 237,7 —
-о-сн,-о— Другие кислородсодер- 3 46,03 35,85 33,45 17,63 14,09 24,26 1700 4000 273 79,0 730
жащие группы 37,1
-он — 17,01 9,7 — 8,04 4,05 10,7 — — —
-<3>-он — 93,10 75,2 — 51,36 22,85 37,7 — — — 210,0 —
—с-н 29,02 15,14 — — — (65) —
%
—с—он % — 45,02 — — • — (12) 23,6 — - 81,9
Бифункциональные азот-
содержащие группы 29,6
—NH— 1 15,02 — — 8.08 3,40 (7.6) — — — ——
—CH(CN)— 1 39,04 28,95 — 21,48 (9,7) — • — 6900 438 (86) —
—CH(NHj)— 1 29,04 — — 17,32 8,72 — — — — 68,6 —
—^cT>-NH- 5 91,11 — — (51,4) 22,20 (34,6) — — — 202,5 —
’* В скобках—значения, полученные Г. Л. Слонимским.
•** 18,25 (соответственно 21,0) для акрильной—СОО-группы.
00
Продолжение при лож. 1
I
а
Группа Z М, г/моль Vg, СМ’/МОЛЬ Vr, см’/моль Ууу, елЧчопъ Ср, кал/(моль - К) Ср, кал/(моль-К) ДНт, кал/моль Яког, кал/моль F (Смолл), (кал-см’)‘/’/моль Р V К
Другие азотсодержащие группы —NH, ^N— ' ^*NaP —C=N —<^О>—NH« Бифункциональные ки- слород- и азотсодержа- щие группы —С—NH— % O-C-NH- % -NH-C-NH- \\0 —CH(NGa)— ' /—к -<О>-сГ 4— . XNH- 1 1 5 2 3 3 1 6 '16,02 14,01 14,01 26,02 92,12 90,10 43,03 59,03 58,04 59,03 119,12 19,5 24,9 90,4 1 1 1 1 1 1 1 1 1 1 1 10,54 10,54 14,7 53,86 47,65 19,56/[18,1] (23) (27,6) 23,58 62,88 5,00 5,00 (6) 23,80 22,88 (9-13) 13,72 (27,8/—31,8) (37,5) (21,5) (48,5) 700 г— 6000 6800 14 500 20 500 410 468 46,7 12,5 (64) 219*6 185,4 (78) (94) (107) 1000 1200 1450
ьо сл Другие кислород- и азотсодержащие группы XNH, -сС° 2 44,03 42,02 — — (22,2) (16,0) — — — — — — —
—NO, — 46,01 — — 16,8 10,00 — — 440 —
Бифункциональные се- русодержащие группы
—S— 1 32,06 17,8 15,0 10,8 15,74 10,7 — 1500 225 48,2 280
-S-S- 2 64,12 (35,6) (30,0) 22,7 (11,48) (21,4) — (3000) (450) (96,4) —
-so,- 1 64,06 — — 20,3 (12) — — — — — 1?00
—S-СН,—S- 3 78,15 51,45 46,45 31,8 17,5 28,7 — 4000 583 135,4 730
Другие серусодержащие группы
, —SH — 33,07 — — 14,81 11,16 12,5 — ? — 315 65,3 —
Бифункциональные га- логенсодержащие груп- пы
-CHF- ' 1 32,02 20,35 19,85 13,0 (8,82) (Ю.О) — 1300 (150) 47,6 330
— —CF,— ' 1 50,01 26,4 24,75 15,33 (11,7) (11,8) — 800 (150) 56,2 300
-CHCI- 1 48,48 29,35 28,25 19,0 10,18 (14,5) — 3200 298 76,2 538
—СС1,- 1 82,92 44,4 41,55 27,81 14,4 (20,81) — 4600 427 113,4 330
( цис -CH^CCl— < 1 транс 2 60,49 — 38,4 25,72 13,41, (18,40) — 3500 400 102,2 *—
—CFC1— 1 66,47 35,4 33,15 21,57 (13,0) (16,3) — 2700 (290) 84,8 350
-СНВг— ' 1 92,93 (39) — 21,4 10,00 — — 3800 368 89,9 —
у; -СВГ,— 1 171,84 (46) — 32,5 14,03 — — 5800 587 140,8 —
-CHI- 1 139,93 — — 27,1 9,06 — — 453 112,9 —
-С1,- 1 265,83 — — 44,0 12,15 — — — 757 186,8 —
8?
Продолжение прилож. 1
Группа Z М, г/моль V сМ’/МОЛь сМ’/МОЛь Vw< См’/МОль W л ч о £ - 5 и « ft О Ср, кал/(моль К) 4 Ч О S § £ ЙЗ < л § Я ч к о ьГ Ч1гож/1у1(,иэ-гген) ‘(irirowo) Я р к
Другие азотсодержащие
группы
—NHi — '16,02 — — 10,54 5,00 — — — — 46,7 —
1 14,01 — — 10,54 5,00 — — — — 12,5 —
An 1 14,01 — (64) —
J"wap
-C=N — 26,02 19,5 — 14,7 (6) — — 6800 410 219,6 —
—<j5^>—NHj — 92,12 — — 53,86 23,80 — ' — — — 185,4 —
5 90,10 — 47,65 22,88 (37,5) — — — — —
Бифункциональные ни-
слород- и азотсодержа-
щие группы (9—13)
—С—NH— 2 43,03 24,9 — 19,56/(18,1] (21,5) 700 14 500 — (78) 1000
%
—О—С—NH— 3 59,03 — — (23) — — г— — — (94) 1200
ч
-NH-C-NH- 3 58,04 — — (27 6) — — — — — (107) 1450
\\0
-CH(NG2)- 1 59,03 — — 23,58 13,72 — — — 468 — —
О -<0>-сг 6 119,12 90,4 — 62,88 (27,8/—31,8) (48,5) 6000 20 500 — — —
4 ' . XNH-
Другие кислород- и азотсодержащие группы -<0 XNH, .0 —с€ 1 2 44,03 42,02 — (22,2) (16,0) — — ' — 1 1 — —
—NO, — 46,01 — — 16,8 10,00 — а— — 440 — —
Бифункциональные се- русодержащие группы —S— 1 32,06 17,8 15,0 10,8 15,74 10,7 1500 225 48,2 280
-S-S- 2 64,12 (35,6) (30,0) 22,7 (11,48) (21,4) — (3000) (450) (96,4) —
—SO,— 1 64,06 — — 20,3 (12) — — — — — 1?оо
—S-CH,-S- 3 78,15 51,45 46,45 31,8 17,5 28,7 — 4000 583 135,4 730
Другие серусодержащие группы , -SH — 33,07 — 14,81 11,16 12,5 / 315 65,3
Бифункциональные га- логенсодержащие груп- пы -CHF- ' 1 32,02 20,35 19,85 13,0 (8,82) (10,0) 1300 (150) 47,6 330
—CFi— ' 1 50,01 26,4 24,75 15,33 (11,7) (И.8) — 800 (150) 56,2 300
-CHCI- 1 48,48 29,35 28,25 19,0 10,18 (14,5) — 3200 298 76,2 538
—СС1,— 1 82,92 44,4 41,55 27,81 14,4 (20,81) — 4600 427 113,4 330
(цис -СН«СС1—< 2 60,49 38,4 25,72 13,41, (13,0) (18,40) 3500 400 102,2
1 транс —GFCI— 1 66,47 35,4 33,15 21,57 (16,3) — 2700 (290) 84,8 350
-СНЕГ— ' I 92,93 (39) — 21,4 10,00 — — 3800 368 89,9 —
-СВ»,— 1 171,84 (46) — 32,5 14,03 — — 5800 587 140,8 —
—CHI- 1 139,93 — — 27,1 9,06 — — — 453 112,9 —
-С1,- 1 265,83 — — 44,0 12,15 — — — 757 186,8 —
со Продолжение прилож. 1
Группа 2 М, г/моль Vg, см’/моль Vr, см’/моль У И’, см’/моль £ л § а, i и « а о Ср, кал/(моль-К) ДНт, кал/моль Еког, кал/моль F (Смолл), (кал-см’)*/»/моль Р Ye, К
Другие галогенсодержа- щие группы —F 19,00 10,9 10,0 6,0 (5,1) (5,0) 1200 (122) 25,7
—СР» 69,01 37,3 34,75 21,33 (16,77) (16,76) — 2000 (274) 81,9 —
-CHF, — 51,02 31,25 29,85 18,8 (13,92) (15,0) — 2500 (272) 73,3 —
—CH»F — 33,03 26,75 26,45 16,2 (11,15) (12,26) — 2200 (255) 64,7 —
—Cl — 35,46 19,9 18,4 12,2 6,46 (9,5) — 3100 270 54,3 —
—СС1, — 118,38 64,3 59,95 (40) 20,85 (30,3) — 7700 657 167,7 —
—СНС12 — 83,93 49,25 46,65 31,3 16,64 (24,0) — 6300 548 130,5
—СН,С1 — 49,48 35,75 34,85 22,5 12,51 (16,76) — 4100 403 93,3 —
-<o>-G1 — 111,55 .85,4 79,8 55,3 25,26 ' (36,5) — 9100 828 227,2 —
—Вг — 79,2 — — 14,6 6,28 — — 3700 340 68,0
—СВг» — 251,76 — — (47,1) 20,31 — — 9500 (927) 208,8
—СНВГи — 172,85 — — 36,0 16,28 — — 7500 (708) 157,9
—СНЕВг — 93,94 — — 24,8 12,33 — — 4700 473 107,0
—I — 126,91 — — 20,4 5,34 — — — 425 91,0
—CI, — 392,74 — — (64,4) 17,49 — — — (1182) 277,8 —
-CHI, — 266,84 -г- — 47,5 14,40 — — — (878) 203,9 —
—СН»1 — 140,94 30,6 11,39 — — — 558 130,0 —
Продолжение прилож. 1
Группа] V Ядд, см’/моль Ярд, см’/моль Яф, см’/моль Рдд, см’/моль -X-10% ед. СГС а- Ю’ ДСу, кал/моль
главная цепь боковая цепь
Бифункциональные уг- леводородные группы —СН,- 170’*/380 895 465 7,83 20,64 4,65 11,36 50 5 — 5300-|-25,0Т
-СН(СН»)- (742) 1821 9,26 15,62 41,14 9,26 22,72 55 10 — 12 250+52,97
-СН(С»Н,)- — — 25,6 43,77 124,48 — — — — — 17 700+135,07
-СЩС.Н,,)- — 5334 30,30 51,75 146,15 — — — — — 29 000+168,57
-СН(С.Н„)- (856) 4505 29,03 50,97 144,91 29,12 60,43 85 40 20 050+70,47
-С(СН»)»- (380) 2762 13,87 23,36 61,69 13,86 34,08 60 15 — 20 000+82,37
-СОЗНаНС.Н»)- — 5446 33,44 58,41 166,27 33,72 71,79 90 45 12 300+99,87
( цис -СН=СН-{ „ _ V транс — 1490 8,88 15,50 40,62 — 11,86 | 75 1 100 — 18 000 + 18,07
f цис -СН-С(СНа)-{ „ (транс — 2361 . 13.49 23,24 61,16 — 23,22 < 80 1 105 — 7500+45,97
—С=С- — 1236 — — — — 11,22 — 54 000—12,07
/—\ ( чис — 25,70 44,0 125,10 ( 140 < 20 —23 700+143,57
\—/. 1 транс -\О>- 2100’*/2300 3556 25,03 44,8 128,6 25,0 48,12 1 170 250 1 25 30 23 000+44,27
_(о)~ — 3500 25,00 44,7 128,6 — 48,12 — — 23 350+44,2Т
— (3450) 24,72 , 44,2 129,0 — 48,12 — — 24 000+44,27
й* 170 для n = 1—4; 380 для п == 5.
** 2100 для полиэфиров; 2300 для полиамидов.
s
Продолжение прилож. 1
Группа М, г/моль Vg, см’/моль Vr, см’/моль Vyy, см’/моль £ § i к « а и Ср, кал/(моль-К) ЛНт, кал/моль Еког, кал/моль F (Смолл), (кал-см’)*/’/моль Р Ye, К
Другие галогенсодержа- щие группы —F 19,00 10,9 10,0 6,0 (5,1) (5,0) 1200 (122) 25,7
—СР» —. 69,01 37,3 34,75 21,33 (16,77) (16,76) — 2000 (274) 81,9 —
—CHF» — 51,02 31,25 29,85 18,8 (13,92) (15,0) — 2500 (272) 73,3 —
—CH»F — 33,03 26,75 2б,45 16,2 (11,15) (12,26) —* 2200 (255) 64,7 —
—d — 35,46 19,9 18,4 12,2 6,46 (9,5) — 3100 270 54,3 —
—СС1» — 118,38 64,3 59,95 (40) 20,85 (30,3) — 7700 657 167,7 —
—снсь — 83,93 49,25 46,65 31,3 16,64 (24,0) — 6300 548 130,5 —
—СН.С1 — 49,48 35,75 34,85 22,5 12,51 (16,76) — 4100 403 93,3 —
-<o>-cl — 111,55 85,4 79,8 55,3 25,26 (36,5) — 9100 828 227,2 —
—Вг — 79,2 — — 14,6 6,28 — — 3700 340 68,0
—СВг» — 251,76 — — (47,1) 20,31 — — 9500 (927) 208,8
—СНВГи — 172,85 — — 36,0 16,28 — — 7500 (708) 157,9
—СНЕВг — 93,94 — — 24,8 12,33 — — 4700 473 107,0
—I — 126,91 — — 20,4 5,34 — — — 425 91,0
—CI, — 392,74 — — (64,4) 17,49 — — — (1182) 277,8 —
-CHI, — 266,84 -7- — 47,5 14,40 — — — (878) 203,9 —
—СН»1 — 140,94 — — 30,6 11,39 — — — 558 130,0 —
Продолжение прилож. 1
Группа] и 4) R О ^8 Я о Й ч й5 Ярд, см’/моль Яф, см’/моль РдЛ1 см’/моль —X-10% ед. СГС О- Ю’ AGy, кал/моль
главная цепь боковая цепь
Бифункциональные уг- леводородные группы —GH3— 170 ’*/380 895 465 7,83 20,64 4,65 11,36 50 5 —5300 + 25,ОТ
—СН(СН»)— (742) 1821 9,26 15,62 41,14 9,26 22,72 55 10 — 12 250+52,9Т
-СН(С.Н,)- — — 25,6 43,77 124,48 — — — — — 17 700+135,07
—СН(СаНц)— — 5334 30,30 51,75 146,15 — — — — — 29 000+168,5Т
—СН(С,Н,)- (856) 4505 29,03 50,97 144,91 29,12 60,43 85 40 20 050+70,4Т
-С(СНа)«- (380) 2762 13,87 23,36 61,69 13,86 34,08 60 15 —20 000+82.3Т
-C(CH»)(C,HS)- — 5446 33,44 58,41 166,27 33,72 71,79 90 45 12 300+99,87
( цис —СН=СН—{ 1 транс — 1490 8,88 15,50 40,62 — 11,86 ( 75 1 100 — 18 000 + 18,07
f цис -СН-С(СЦа)-{ „ „ ( транс — 2361 . 13.49 23,24 61,16 — 23,22 < 80 1 105 — 7500+45,97
—С=С- — 1236 — — — — 11,22 — 54 000—12,07
/—\ I uuc — — ' 25,70 44,0 125,10 Г 140 Г 20 —23 700+143,57
\—/. | транс -<о>- 2100’*/2300 3556 25,03 44,8 128,6 25,0 48,12 1 170 250 1 25 30 23 000+44,27
“(ор — 3500 25,00 44,7 128,6 — 48,12 — — 23 350+44,2Т
— (3450) 24,72 44,2 129,0 — 48,12 — — 24 000+44,2Т
•* 170 для п = 1—4; 380 для п = 5.
оо ** 2100 для полиэфиров; 2300 для полиамидов.
СО Продолжение прилож. 1
Группа Д |ШЛ и ЛЛЛ> см’/моль Лрд, см’/моль Лф, см’/моль Л § я 3 ч Ч а, —X-10’, ед. СГС б-10’ AGy, кал/моль
главная цепь боковая цепь
zCH, -<о5- 5270 34,8 61,0 16.9,8 68,94 (6400 + 97,67)
\сн, 4409 29,9 52,19 149,2 — 58,53 — — (14 700+70,97)
\сна -\О/-СН,- — 4451 29,53 52,06 147,3 29,65 59,48 300 — 17 700+69,2Т
1900 ‘*/2700 5346 34,03 59,32 166,0 34,3 70,84 350 — 12 400+94,2Г
3200 «*/3650 8007 54,56 96,9 275,9 54,65 107,60 550 — 40 700+113,4Т
(5600) 7112 50,06 89,6 275,2 50,0 96,24 500 — 46 000+88,4Т
9, Yo) 1 10 638 74,8 - 134,0 386,0 — 144,36 — — (71 000+132,6ТУ
\о> Другие углеводородные группы —СН, 1361 5,64 8,82 17,66 5,64 14,04 5 —11 500+22,97
2256 10,29 16,65 38,30 10,29 25,40 — 10 —16 800+47,9Т
—и=С,Н? 3151 14,94 24,48 58,94 1'4,94 36,76 — 15 —22 100+72,9Т
—иао=СаН? 3182 14,90 24,44 58,81 14,90 36,76 — 15 —23 750+75,87
* —тпрет=СфНв — 4123 19,51 32,18 79,35 19,51 48,12 20 —31 500+105,27
1 —CH 1 — 460 3,62 6,80 23,49 3.62 8,68 50 5 —750+30,07
|_ — 40 2,58 5,72 26,37 2.58 6,0 50 5 3000 + 36,57
1 =сн, 1234 5,47 8,78 17,46 8,61 — 5900+8,07
=сн- — 745 4,44 7,75 20,31 — 5,93 — — 9000+9,07
=с< — 255 3,40 6,67 23,19 — 3,25 — — 10 000 + 14,07
=с= — 4,23 7,62 20,01 — — 35 000—47
-сн=*с- , 1000 7,85 14,42 43,50 — 9,18 — — 19 000+23,0?
=сн — 1108 — — — — 8,30 — — 27 000—7,7?
ЕС — 1 1 (цис — 618 — — — . — 5,61 / 75 — 27 000—6,0?
—с=с— { ( транс — 510 6,81 . 13,34 46,38 /— 6,50 t 100 — 20 000 + 28,0?
^СНар — 758 — . — — — 9,23 — — 3100+6,1?
^»СаР— — 258 — — — — 5,6 — — 5300+9,97
-CI — — 22,0 36,97 100,99 — — — — —16 950+105,0?
— 4874 26,69 44,95 122,66 — — • — 35 —28 250+138,57
— — 4045 25,51 44,63 123,51 25.5 51,75 — 35 20 800+40,4?
-<о>- — 2548 23,8 44,72 139,0 — 40,86 — — 27 400+51,8?
-<о^- Бифункциональные ки- слородсодержащие груп- пы — 3048 24,4 44,76 133.8 — 44,49 — — 25 200+48,0?
—О— 450 «*/900 348 1,59 <*/1,77 2,75 ’*/2,96 22,6 ’*/23,85 5.2 4,6 35 5 —28 000+14?
•* 1900 для полиэфиров; 2300 для полиуретанов; 2450 для поликарбамидов; 2700 для полиамидов.
' •* 3200 для полиуретанов; 3650 для поликарбамидов.
"* 450 для полиуретанов; 600 для полиамидов; 750 для полиэфиров; 900 для полиоксидов.
’* 1,59, 2,96 и 23,85 для метиловых эфиров; 1,63, 2,75 и 22,99 для ацеталей; 1,64, 2,81 и 23,18 для высших эфиров; 1,77, 2,84 и
eg 22,6 для присоединения к бензольному кольцу. __________________________________________________________
Продолжение прилож. 1
Группа я ‘шл и ЛЛЛ> см’/моль Врд, см’/моль Лф, см’/моль Л § я i ч £ —X-10’, ед. СГС б-10’ AGy, кал/моль
главная цепь боковая цепь
хСН, -<о$- } 11 5270 34,8 61,0 16.9,8 68,94 (6400 + 97,67)
\сн, 4409 29,9 52,19 149,2 — 58,53 — — (14 700+70,97)
\сна
— 4451 29,53 52,06 147,3 29,65 59,48 300 — 17 700+69,2Т
1900 ‘*/2700 5346 34,03 59,32 166,0 34,3 70,84 350 — 12 400+94,2Т
3200 «*/3650 8007 54,56 96,9 275,9 54,65 107,60 550 — 40 700+113,4Т
(5600) 7112 50,06 89,6 275,2 50,0 96,24 500 — 46 000+88,4Т
1 Ю|\ 10 638 74,8 - 134,0 386,0 — 144,36 — — (71 000+132,67У
\о>
другие углеводородные группы — 11 500+22,97
-сн, — 1361 5,64 8,82 17,66 5,64 14,04 — 5
2256 10,29 16,65 38,30 10,29 25,40 — 10 —16 800+47,9Т
—и=С,Н? 3151 14,94 24,48 58,94 1'4,94 36,76 — 15 —22 100+72,9Т
—иао=СаН? 3182 14,90 24,44 58,81 14,90 36,76 — 15 —23 750+75,87
* —тпрет=СфНв — 4123 19,51 32,18 79,35 19,51 48,12 20 —31 500+105,27
1 —CH 1 — 460 3,62 6,80 23,49 3,62 8,68 50 5 —750+30,07
|_ — 40 2,58 5,72 26,37 2,58 6,0 50 5 3000 + 36,57
=сн, 1234 5,47 8,78 17,46 8,61 — 5900+8,07
=сн- — 745 4,44 7,75 20,31 5,93 — 9000+9,07
=с< — 255 3,40 6,67 23,19 — 3,25 — — 10 000 + 14,07
=с= — 4,23 7,62 20,01 — -F- — 35 000—47
-сн=*с- . 1000 7,85 14,42 43,50 — 9,18 — 19 000+23,07
=сн — 1108 — — — — 8,30 — — 27 000—7,77
ЕС — 1 1 (цис — 618 — — — 5,61 ( 75 — 27 000—6,07
—С=С—{ ( транс — 510 6,81 . 13,34 46,38 6,50 1 100 — 20 000 + 28,07
^!СНар — 758 — . — — 9,23 — — 3100+6,17
^СаР— — 258 — — —. — 5,6 — — 5300+9,97
-CI — — 22,0 36,97 100,99 — — — — —16 950+105,07
— 4874 26,69 44,95 122,66 — — • — 35 —28 250+138,57
— — 4045 25,51 44,63 123,51 25,5 51,75 — 35 20 800+40,47
— 2548 23,8 44,72 139,0 — 40,86 — — 27 400+51,87
Бифункциональные ки- слородсодержащие груп- пы — 3048 24,4 44,76 133.8 — 44,49 — — 25 200+48,07
т. -О- 450 «*/900 348 1,59 ’*/1,77 2,75 ’*/2,96 22,6 ’*/23,85 5,2 4,6 35 5 —28 000+147
•* 1900 для полиэфиров; 2300 для полиуретанов; 2450 для поликарбамидов; 2700 для полиамидов.
' •* 3200 для полиуретанов; 3650 для поликарбамидов.
"* 450 для полиуретанов; 600 для полиамидов; 750 для полиэфиров; 900 для полиоксидов.
в* 1,59, 2,96 и 23,85 для метиловых эфиров; 1,63, 2,75 и 22,99 для ацеталей; 1,64, 2,81 и 23,18 для высших эфиров; 1,77, 2,84 и
g 22,6 для присоединения к бензольному кольцу. __________________________________________________________
gj Продолжение прилож. 1
Группа м g и ЛЛЛ« см’/моль Лрд, см’/моль Вф, см’/моль РдЛ' см’/моль -X-10’, ед. СГС 6-10’ AGft кал/моль
главная цепь боковая цепь
—С— 872 4,53 «“*/5,09 7,91 ”*/8,82 41,9 ”*/43,0 (Ю) 4,27 65 10 —30 000+9,5Т
1 1 1160 1220 6,21 ”*/6,71 10,47 ”«/11,31 64,20 ”*/65,32 15 13,1 100 15 —80 000+27?
Ч -о-с-о— 1500 1568 7,75 *’* 13,12 ”*/13.39 86,8 ”*/87,8 22 135 20 -
Ч
-с-о-с— 1720 2092 (10,8) (18.4) (107) (25) 165 25 __
Ч Ч
—СН(ОН)— (892) 1088 6,07 10,75 47,4 (Ю) 15,96 (70) (25) —42 750+42.07
—СН(СООН)— —- 1891 10,83 18,79 87,75 — 24,48 60 15 —95 250+55?
-СН(НС=О)— — • — 9,45 16,43 64,2 — 15,13 — — — 29 750+35.57
—<^р\-СОО— — 4776 31,74 56,1 193,4 40 61,2 350 — 57 000+71,27
—О—СН,—О— (1390) 1591 7,93 13,45 . 67,0 15,05 20,6 120 —61 300+53?
Другие кислородсодер-
жащие группы —ОН — ' 628 2,45 S‘*/2,55 3,85 ”*/4,13 23,95 ”*/24,08 (6) 7,28 (20) —42 000+12,07
—\0/-он — 4184 27,30 48,33 151,3 (45) 55,4 — 19 000+56,27
-с-н ч — — 5,83 9,63 40,69 — 6,45 — — —29 000+5,5 7
-с-он ч — 1431 7,21 11,99 64,26 — 15,8 — 10 —94 000+25Т
Бифункциональные азот-
содержащие группы —NH— — 638 3,59 6,29 24,30 — 8,48 60 — 2000 + 29,47 *
-CH(CN)- —CH(NHi)— — ,1768 1427 9^,14 7,97 15,88 14,05 60,16 46,13 14,6 19.7 19,84 (90) (90) — 28 250+22,8Т 4550+57,17
NH— — 4194 29,56 53,48 115,5 — 56,6 — — 25 000+73,67
Другие азотсодержащие группы —NH, 967 4,36 7,25 22,64 — 11,16 — (40) —3800+27,17
\n— —. 65 2,00 5,70 26,66 — 5,48 60 (Ю) 6000+37,27
g. й J“ III и l — 1308 5,53 9,08 36,67 11 12,4 11 — (40) И 500+11,17 29 000—7,2?
-^O^-NHj — 4523 29,9 2 53,2 152,3 — 59,3 — — 19 200+71,3?
— 3621 29,08 53,5 159,3 — 53,6 — — 29 000+81,4?
Бифункциональные ки- слород- и азотсодержа- щие группы —С—NH— ч —О—С—NH— ч 2560 2400 1367 (1715) 7,23 15,5 69,75 30 11,9 125 160 (20) (25) (-28 000+ +38,9?)
—NH-C-NH— ч —CH(NO,)— 3250 {(2005) 10,28 17,81 89,49 — — 185 — —9750 + 67,0?
.0 -<о >-СГ т 4—' лян— *> 4923 33,5 62,9 201,6 55 60 .— — —
41.9 для присоединения к
и
бензол*-
эфиров;
4,79, 8,42 и 43,01 для метилкетонов; 4,5К3( 7(91
10,76 и 65,32 для метиловых эфиров; 6,38.
7«75. 13*39 й 87.8 для метилкарбонатов; 7,74» 13*12 и 86.8 для высших карбонатов.
1,1 и, у ____о ta о ПК -П ОО ак тттгст nTnnmitn.TY ПГГГТПТЛ
и 43.03 для высших кетонов; 5,09, 8(82
ному1>*>^24.' 10,76 и 65,32 для метиловых эфиров; 6,38, 10,94 и 64,49 для этиловых эфиров; 6,21, 10,47 и 64,20 для высших
6 71, 11,31 и 64,8 для присоединения к бензольному кольцу; 6,31, 10,87 и 64,90 для ацетатов.
’ 1:11: Ю88 s™ °^,о5 для этичных спиртов.
393
g? Продолжение прилож. 1
Группа М g и ЛЛЛ« см’/моль Лрд, см’/моль Вф, см’/моль РЛЛ' см’/моль -X-10’, ед. СГС 6-10’ AGp кал/моль
главная цепь боковая цепь
—С— ч -о-с- ч -о-с-о— ч -с-о-с— \\0 ч —СН(ОН)— —СН(СООН)— -СН(НС=О)— —<(сГ)-соо— —О—СН,—О— Другие кислородсодер- жащие группы —ОН -\О/~0Н -С-Н ч -с-он ч Бифункциональные азот- содержащие группы —NH— -CH(CN)- —CH(NH,)— -^Q^>—NH— Другие азотсодержащие группы —NH, yN— ^Nap —C = N -<^>-NH, Бифункциональные ки- слород- и азотсодержа- щие группы -C-NH— ч —О—С—NH— ч —NH-C-NH— ч -CH(NO,)- О -<о >-СГ 4 ' ''NH— 1160 1500 1720 (892) (1390) 2560 2400 3250 872 1220 1568 2092 1088 1891 4776 1591 ' 628 4184 1431 638 ,1768 1427 4194 967 65 1308 4523 3621 1367 (1715) 1(2005) 4923 4,53 «“*/5,09 6,21 >’*/6,71 7,75 >’* (10,8) 6,07 10,83 9,45 31,74 7,93 2,45 >‘*/2,55 27,30 5,83 7,21 3,59 9^,14 7,97 29,56 4,36 2,00 5,53 29,9 2 29,08 7,23 10,28 33,5 7,91 >«*/8,82 10,47 ”*/11,31 13,12 >’*/13.39 (18.4) 10,75 18,79 16,43 56,1 13,45 3,85 >‘*/4,13 48,33 9,63 11,99 6,29 15,88 14,05 53,48 7,25 5,70 9,08 53,2 53,5 15,5 17,81 62,9 41,9 >«*/43,0 64,20 >’*/65,32 86,8 >’*/87,8 (107) 47,4 87,75 64,2 193,4 . 67,0 23,95 >‘*/24,08 151,3 40,69 64,26 24,30 60,16 46,13 115,5 22,64 26,66 36,67 152,3 159,3 69,75 89,49 201,6 (Ю) 15 22 (25) (Ю) 40 15,05 (6) (45) 14,6 И 30 55 4,27 13,1 15,96 24,48 15,13 61,2 20,6 7,28 55,4 6,45 15,8 8,48 19.7 19,84 56,6 11,16 5,48 12,4 11 59,3 53,6 11,9 60 65 100 135 165 (70) 60 350 120 60 (90) (90) 60 125 160 185 10 15 20 25 (25) 15 (20) 10 (40) (Ю) (40) (20) (25) —30 000+9,57 —80 000+277 —42 750+42,07 —95 250+557 — 29 750+35.57 — 57 000+71,27 —61 300+537 -42 000+12,07 — 19 000+56,27 —29 000+5,5 7 —94 000+257 2000 + 29,47 * 28 250+22,87 4550+57,17 25 000+73,67 —3800+27,17 6000+37,2Т 11 500+11,17 29 000—7,27 , 19 200+71,37 29 000+81,47 (-28 000+ +38,97) —9750 + 67,07
41,9 для присоединения к беизоль-
и
и 43*03 для высших кетонов; 5*09, 8*82
10,94 и 64,49 для этиловых эфиров; 6*21, 10*47 и 64*20 для высших эфиров;
•»* 4,79, 8,42 и 43,01 для метилкетонов*, 4,5К3, 7,91
ному^льцу. 0 и 65 32 _ля метиловых эфиров; 6,38, хи.и» и о»,»» wy* оч-ч^», ----------.
6 71. 11*31 и 64,8 для присоединения к бензольному кольцу; 6*31 * 10*87 и 64*90 для ацетатов.
’ 2>45. з,85 a2W для тре^х спиртов.
393
Продолжение прилож. 1
Группа ут< К и ЙЛЛ> СМ*/моль Ярд, см’/моль ' Нф, см’/моль «а § я а i а, -X-10*. ед. СГС в- 10* ' Дб/, кал/моль
главная цепь боковая цепь|
Другие кислород- и -
азотсодержащие группы
-О
“с\ 'NHj — 1839 — — — — 14,0 — — —
,О
-C<N< — 937 — — — — 9,2 — —
-NO, — — 6,66 11,1 66,0 —9 00 0+37, ОТ
Бифункциональные се-
русодержащие группы
-S— 750,<’*/960 (550) 8,07 14,44 53,33 8 15 10 000-7,1Т
_ g—s— (1140) (1100) 16,17 29,27 107,63 16 . (30) (20 000—14,2т)
—SO1— — — — — —67 300+36,2Т
—S—СН,—S— (1800) (2000) 20,08 36,71 127,3 21 41 14 700+10,8Г
Другие серусодержащие
группы
—8Н — 1050 8,75 ‘•*/9,27 15,14 «••/15,66 50,61 <•* 17,68 4200—9,ЗТ
Бифункциональные га-
яогенсодержащие груп-
пы
—CHF— (776) (1260) 4,51 7,68 45,7 5,42 15,28 —46 750+27,8Г
—CF.— (718) (1640) 4,38 7,12 68,2 6,25 19,2 —89 000+32,1Т
—СНС1— (946) 1520 9,64 —16,71 73,8 13,7 28,7 70 — 12 450 + 27,8Т
—GClj— j (796) 2160 14,63 25,54 127,0 17,7 46,0 90 — —20 400+32,1Т
- ( 95
—СН=СС1— ’ tfue 2060 13,87 24,33 93,8 — 29,2 { 120 — 7300 + 20,8T
транс
-CFC1— (1900) 9,50 16,3 97,6 13,9 32,6 — — —54 700+32,IT
—СНВг— 1640 12,57 22,06 141,9 — 40,7 (60) — -.1750+16,7T
—СВГ1— 2400 20,49 36,24 263,2 — 70,0 (70) — 1000+9,9T
—CHI— 1860 17,52 31,80 — — 52,7 — — 15 750+Ю,0T
—CIf— — 2840 30,38 55,72 — — 94,0 — — 36 000+3,5T
другие галогенсодержа-
щие группы — F (800) 0,90 <’* 0,70 *’*/0,88 20,92 <’*/22,20 1,8 6,6 — — —46 000+2,2T
—СЕ, (2440) 5,27 7,83 89,1 — 25,8 — — -135 000+29,9T
—CHF, (2060) 5,41 8,20 65,3 — 21,9 — — —92 750+25,6T
—CH.F (1695) 5,55 8,71 42,84 6,45 18,0 — — —51 300+22.8T
—С1 1060 5,93 <’*/6,02 9,84 *’*/10,07 50,31 *’*/51,23 9,5 20 20 — —11 700+2,2T
—СС1. 3220 20,7 35,93 180,1 — 66, — — —32 100+29,9 Г
-СНС1. > 2580 15,7 26,94 125,9 — 48,7 — — -24 150+25,6T
—СН.С1 ' +- 1955 10,7 17,90 71,9 14,15 31,4 — — — 17 000+22,81
-<О>-С1 4616 20,63 53,62 1 177,0 34,5 68 — — 11 300+42,ОТ
^7 1180 8,9 0 *’*/9,03 15,15 *•*/15,29 118,4 ***/119,1 — 32 (10) — —1000+13,ЗТ
—СВг. 3580 23,30 51,2 381,9 — 102 — 0—3,4T
—СНВГ. 2820 21,4 37,10 260,5 — 72,7 — —2750+3.4Т
—CHiBr 2075 13,5 22,98 139,1 — 43,4 — — —6300 + 11,71
—I 1400 13,90 25,0 / — — 44 — — 16 500—20Т
—CI. 4240 44,3 80,7 — — 138 — — 52 500—23,5Т
-CHIi — 3260 31,4 56,8 — — 96,7 — — 32 250—ЮТ
—CH.I — 2295 18,5 32,83 — — 55,4 — — 11 200+5,ОТ
11* 750 для полиамидов; 960 для полисульфидов. „„ Л „„ „„„ „„„„„
>•* 8,85, 15,22 и 50,61 для первичных тиоспиртов; 8,79, 15,14 и 50,33 для вторичных тиоспиртов, 9,27, 15,66 и 49,15 для третич-
вых тиоспйртов.
0,91)» 0,88 и 22,20—моно: 0,90, 0,70 и 20,92—пер. , __
it* 6 05. 10,07 и 51,23—первичный; 6,02> 9,91 и 50,31—вторичный; 5,93 , 9,84 и 50, 75—третичный.
оэ it* в 90, 15,15 и 118,5—первичный; 8,96, 15,26 и 118,4—вторичный; 9,03, 15,29 и 119,1—третичный.
со _________•----------------------------------------------------—---------.-
сл — ч" .
Продолжение прилож. 1
Группа к и ДЛЛ> см’/моль Лгд, см’/моль ’ Лф, см’/моль РЛЛ« см’/моль -X-10’, ед. СГС в-10’ ' Дб/, кал/моль
главная цепь боковая цепь|
Другие кислород- и -
азотсодержащие группы
-О
“с\ 'NH. 1839 — — — — 14,0 — — —
,О
— 937 — — — — 9,2 — —
-NO, — — 6,66 11,1 66,0 —9 00 0+37, ОТ
Бифункциональные се-
русодержащие группы
-S— 750,»’*/960 (550) 8,07 14,44 53,33 8 15 10 000-7,1Т
—8—S— (1140) (1100) 16,17 29,27 107,63 16 . (30) (20 000—14.2Г)
—so*— — — — « — — —67 300+36,2Т
-S-СН,—S— (1800) (2000) 20,08 36,71 127,3 21 41 14 700+10,8Т
Другие серусодержащие
группы
—8Н — 1050 8,75 >«*/9,27 15,14 >«*/15,66 50,61 >•* 17,68 4200—9,31
Бифункциональные га-
логенсодер жащие груп-
пы
—CHF— (776) (1260) 4,51 7,68 45,7 5,42 15,28 —46 750+27,8Т
—CF,— (718) (1640) 4,38 7,12 68,2 6,25 19,2 —89 000+32.1Т
—СНС1— (946) 1520 9,64 — 16,71 73,8 13,7 28,7 70 — 12 450 + 27,8Т
—СС1г— j (796) 2160 14,63 25,54 127,0 17,7 46,0 90 — —20 400+32,1Т
- Г 95
-СН=СС1- tpie тпракс — 2060 13,87 24,33 93,8 — 29,2 { 120 — 7300 + 20,8Т
—CFC1— (1900) 9,50 16,3 97,6 13,9 32,6 — — —54 700+32,1Т
—СНВГ— 1640 12,57 22,06 141,9 — 40,7 (60) — —.1750+16,7 Т
—СВГ1— 2400 20,49 36,24 263,2 — 70,0 (70) — 1000+9,91
—CHI— 1860 17,52 31,80 — — 52,7 — — 15 750+10.0Т
—CIj— — 2840 30,38 55,72 — — 94,0 — — 36 000+3,5Т
другие галогенсодержа-
щие группы
— F (800) 0,90 >’* 0,70 >’*/0,88 20,92 >’*/22,20 1.8 6,6 — — —46 000+2,2Т
—CF, (2440) 5,27 7,83 89,1 — 25,8 — — -135 000+29,9Т
—CHF, (2060) 5,41 8,20 65,3 — 21,9 — — —92 750+25,6Т
—CH.F (1695) 5,55 8,71 42,84 6,45 18,0 — — —51 300+22.8Т
—С1 1060 5,9 3 >’*/6,02 9,84 »•*/10,07 50,31 >«*/51,23 9,5 20 20 — —11 70 0+2,2Т
—СС11 3220 20,7 35,93 180,1 — 66. — — —32 100+29,9 Т
-CHCli > 2580 15,7 26,94 125,9 — 48,7 — — -24 150+25,6Т
—СН,С1 +_ 1955 10,7 17,90 71,9 14,15 31,4 — — — 17 000+22,8F
-<О>-С1 4616 20,63 53,62 1 177,0 34,5 68 — — И 300+42,ОТ
1180 8,9 0 >’*/9,03 15,15 >«*/15,29 118,4 И*/119,1 — 32 (Ю) — —1000+13,31
—СВГа 3580 23,30 51,2 381,9 — 102 — 0—3,4Т
—СНВГ. 2820 21,4 37,10 260,5 — 72,7 — —2750+3.4Т
—СНаВГ , 2075 13,5 22,98 139,1 — 43,4 — — —6300 + 11,71
—I 1400 13,90 25,0 / — — 44 — — 16 500—20Т
—CI1 4240 44,3 80,7 — — 138 — — 52 500—23,5Т
-CHIi — 3260 31,4 56,8 — — 96,7 — — 32 250—ЮТ
—CH,I — 2295 18,5 32,83 — — 55,4 — 9— 11 200+5,ОТ
15,66 и 49,15 для третич-
со
сл
я* 750 для полиамидов; 960 для полисульфидов. Л ___ А __
1е* 8,85, 15,22 и 50,61 для первичных тиоспиртов; 8»79* 15*14 и 50,33 для вторичных тиоспиртов; 9,27*
тиоспйртов.
»’* 0,90, 0,88 и 22,20—моно; 0,90, 0,70 и 20,92—пер. .
»»* 6 05. 10,07 и 51,23—первичный; 6,02, 9,91 и 50,31—вторичный; 5,93 , 9,84 и 50, 75—третичный.
«•* 8 90, 15,15 и 118,5—первичный; 8,96, 15,26 и 118,4—вторичный; 9,03, 15,29 и 119,1—третичный.
g Приложение 2. Физические характеристики полимеров
[Полимер. М, г/моль Ра, Г/СМ" Я о и h я о о г» и о £ Я Q О •чч Ср, кал/(г.°С) Ср, кал/(г. °C) кал/моль
Полиолефины
полиэтилен полипропилен 28,1 42,1 0,85* 0,85 0,86 < 0,90 1,00 0,95 0,95 0,93 0,92 0,91 0,84 0,95 0,94 >0,96 0,963 2,4/3,6 2,2 7,5/9,6 0,37/0,42 0,54 1820/2000
поли-1-бутен 58,1 5,5/9,4 0,368/0,425 0,51 0,509 650/2600
поли-З-метил-1 -бутен 70,1 3,8 8,8 0,37/0,42 970/1710
поли-1-пентен поли-4-метил-1 -пентен поли-1-гексен 70,1 84,2 84,2 0,85 0,838 0,86 3,83 9,2 7,61 0,400 2850
поли-5 -метил-1 -гексен 98,2 — ——
поли-1-октадецен 252,5 0,86 0,86 — мм
полиизобутилен 1,2-поли-1,3-бутадиен(изо) 56,1 54,1 1,6/2,0 5,6/6,9 0,40 0,467 —
1,2-поли-1,3-бутадиен(синдио) Полистиролы 54,1 <0,92 — — — — —
полистирол * поли-а-метилстирол 104,1 118,2 1,05 1,065 1,027 1,13 1,7/2,6 4,3/6,5 0,293 0,41 2000/2400
поли-2-метилстирол 118,2 1,07 —— —
поли-4-метилстирол 118,2 1,04 -— —
поли-4-метоксистирол 134,2 >1,12 —— —
поли-4-фенилстирол 180,2 —
поли-3-фенил-1 -пропен 118,2 1,046 > 1,052 ——
поли-2-хлорстирол 138,6 < 1,25 —
поли-4-хлорстирол 138,6
Полигалогенолефины — — —
поливинилфторид 46,0 < 1,37 1,44 1800 600/2700
поливинилхлорид поливинилбромид 62,5 107,0 1,385 П52 1,1/2,1 1,4/5,2 0,226 0.29
поливинилиденфторид 64,0 1,74 2,00 2,1/4,6 —
поливинилиденхлорид 97,0 1,66 1 95
политетрафторэтилен 100,2 2,00 2,35 3,0 4,77 2,0/3,5 0,23 0,22 0,23 330 13 70 1200/2100
полихлортрифторэтилен Поливинилы 116,5 1,92 2,19 1,0/1,5
поливинилциклопентан 96,2 <0,965 0,986 — — — — —
поливинилциклогексан
поли-а-винилнафталин
поливиниловый спирт
поливинилметиловый эфир
поливинилэтиловый эфир
поливинилпропиловый эфир
поливинилизопропиловый эфир
поливинилбутиловый эфир
поливинилизобутиловый эфир
полив инил-ешор-бутиловый
эфир
поливинил-тиретп-бутиловый
эфир
поливинилгексиловый эфир
поливинилоктиловый эфир
полив инилметилнетон *
полиметилизопропенилкетон
п оливинил формиат
поливинилацетат
поливинилпропионат
поливинилхлорацетат
поливинилтрифторацетат
поливинилбенэоат
поли-2-винилпиридин
поливинилпирролидон
полив инилкарбазол
Полиакрилаты
полиакриловая кислота
полиметилметакрилат
полиэтилакрилат '
полипропилакрилат
полиизопропилакрилат
полибутилакрилат
полиизобутил акрилат
поли-етор-бутилакрилат
поли-тпрет-бутилакрилат
Полиметакрилаты
полиметакриловая кислота
полиметилметакрилат
полиэтилметакрилат
полипропилметакрилат
полиизопропилметакрнпат
г полибутилметакрилат
110,2 154,2 0,95 0,982 1,12 — —
44,1 1,26 1,35 3
58,1 < 1,03 1,175 — —
72,1 0,94 >0,97 —— —м
86,1 <0,94 —— мм ——
86,1 0,924 >0,93 —— —•
100,2 < 0,92 0,944 мм
100,2 0,93 0,94 •—
100,2 0,92 — — —
100,2 — 0,978 — —
128,2 0,925 >0,925 —
156,3 0,914 >0,91 —
70,1 1,12 1,216 —— —
84,1 1,12/1,15 1,15/1,17 — —
72,1 < 1,35 1,49 — —
86,1 1,19 >1,194 1,8/2,4 5,0/6,0
100,1 1,02 ___ — —
120,5 1,45 1,3 3,4
140,1 — — —— ——
148,2 — — — —
105,1 ‘ — мм
111,1 1,25 —— —— ——
193,2 < 1,19/1,2 — —
72,1 . —
86,1 1,22 1.8/2,7 4,6/5,6
100,1 1,12 —— 2,8 6,1
114,1 < 1.08 >1,18 — ““
114,1 1,08/1,18 2,2/2,6 6,1/6,3
128,2 1,00/< 1,09 —— 2,6 6,0
128,2 < 1,05 1,24 —— “
128,2 <1,05 1,06 2,75 6,1
128,2 1,00 1,04/> 1,08 — —
86,1 _ .
100,1 1,17 1.23 1, 2/2, 3 2,3/5,4
114,1 1,119 2,75 5,40
128,2 1,08 __ 3,2 5,7
128,2 1,033 2,0/2,4 6,2
142,2 1,055 6,1
0,35
0,31
а 328
. -
— 1640
— ——
— мм
— —
——
—— ——
—
— —
— —
__ —
—— ——
—— —
__
0,47 —
^м ——
— 1800
—— ——
— ——
——
— —
—
——
—— —
— 1400
мм ——
—
— —
0,43 —
— —--
— ——
——
* Л =2,62‘1.0"4 кал/(см-с-°С).
g Приложение 2. Физические характеристики полимеров
О 3
h £ и §
[Полимер. л Я я з я
о Я Q §
Af, г/i . . о О •чч § к 5 й №
аа г» «а, — О- *3
Полиолефины
полиэтилен полипропилен 28,1 42,1 0,85* 0,85 0,86 < 0,90 1,00 0,95 0,95 0,93 0,92 0,91 0,84 0,95 0,94 >0,96 0,963 2,4/3,6 2,2 7,5/9,6 0,37/0,42 0,54 1820/2000
поли-1-бутен 58,1 5,5/9,4 0,368/0,425 0,51 650/2600
поли-З-метил-1 -бутен 70,1 3,8 8,8 0,37/0,42 0,509 970/1710
поли-1-пентен поли-4-метил-1 -пентен поли-1-гексен 70,1 84,2 84,2 0,85 0,838 0,86 3,83 9,2 7,61 0,400 2850
поли-5 -метил-1 -гексен 98,2 ——
поли-1-октадецен 252,5 0,86 0,86 — — —
полиизобутилен 1,2-поли-1,3-бутадиен(изо) 56,1 54,1 1,6/2,0 5,6/6,9 0,40 0,467 —
1,2-поли-1,3-бутадиен(синдио) Полистиролы 54,1 <0,92 — — — — —
полистирол * поли-а-метилстирол 104,1 118,2 1,05 1,065 1,13 1,7/2,6 4,3/6,5 0,293 0,41 2000/2400
поли-2-метилстирол 118,2 1,027 1,07
поли-4-метилстирол 118,2 1,04 — —
поли-4-метоксистирол 134,2 >1,12 —
поли-4-фенилстирол 180,2 — , —
поли-З-фенил-1-пропен 118,2 1,046 > 1,052 ——
поли-2-хлорстирол 138,6 < 1,25
поли-4-хлорстирол ’ 138,6 —
Полигалогенолефины —— —
поливин ИЛ фторид 46,0 < 1,37 1,44 1800 600/2700
поливинилхлорид полив инилбромид 62,5 107,0 1,385 1,52 1,1/2,1 1,4/5,2 0,226 0.29
поливинилиденфторид 64,0 1,74 2,00 2,1/4,6 —
долив инилиденхлорид 97,0 1,66 1 95
политетрафторэтилен 100,2 2,00 2,35 3,0 4,77 2,0/3,5 0,23 0,22 0,23 330 1370 1200/2100
полихлортрифторэтилен Поливинилы 116,5 1,92 2,19 1,0/1,5
поливинилциклопентан 96,2 <0,965 0,986 — — — — —
поливинилциклогексан
поли-а-винилнафталин
поливиниловый спирт
поливинилметиловый эфир
поливинилэтиловый эфир
поливинилпропиловый эфир
поливинилизопропиловый эфир
поливинилбутиловый эфир
поливинилизобутиловый эфир
полив инил-етнор-бутиловый
эфир
поливинил-трети-бутиловый
эфир
поливинилгексиловый эфир
поливинилоктиловый эфир
полив инилметилкетон *
полиметилизопропенилкетон
п оливинил формиат
поливинилацетат
поливинилпропионат
поливинилхлорацетат
поливинилтрифторацетат
поливинилбенэоат
поли-2-винилпиридин
поливинилпирролидон
полив инилкарбазол
Полиакрилаты
полиакриловая кислота
полиметилметакрилат
полиэтилакрилат '
полипропилакрилат
полиизопропилакрилат
полибутилакрилат
полиизобутил акрилат
поли-етор-бутилакрилат
иоли-тпретп-бутилакрилат
Полиметакрилаты
полиметакриловая кислота
полиметилметакрилат
полиэтилметакрилат
полипропилметакрилат
полиизопропилметакрилат
полибутилметакрилат
110,2 154,2 0,95 0,982 1,12 — —
44,1 1,26 1,35 3
58,1 < 1,03 1,175 — —
72,1 0,94 >0,97 —— —«
86,1 <0,94 —— ——
86,1 0,924 >0,93 — —•
100,2 < 0,92 0,944
100,2 0,93 0,94 •—
100,2 0,92 — — —
100,2 — 0,978 — —
128,2 0,925 >0,925 —
156,3 0,914 >0,91 —
70,1 1,12 1,216 — —
84,1 1,12/1,15 1,15/1,17 — —
72,1 < 1,35 1,49 — —
86,1 1,19 >1,194 1,8/2,4 5,0/6,0
100,1 1,02 ___ — —
120,5 1,45 1,3 3,4
140,1 — — —— ——
148,2 — — — —
105,1 ‘ —
111,1 1,25 —— —— ——
193,2 < 1,19/1,2 — —
72,1 . —
86,1 1,22 1.8/2,7 4,6/5,6
100,1 1,12 —— 2,8 6,1
114,1 < 1.08 >1,18 — ““
114,1 1,08/1,18 2,2/2,6 6,1/6,3
128,2 1,00/< 1,09 2,6 6,0
128,2 < 1,05 1,24 —— “
128,2 <1,05 1,06 2,75 6,1
128,2 1,00 1,04/> 1,08 — —
86,1 _ .
100,1 1,17 1.23 1, 2/2, 3 2,3/5,4
114,1 1,119 2,75 5,40
128,2 1,08 __ 3,2 5,7
128,2 1,033 2,0/2,4 6,2
142,2 1,055 — — 6,1
0,31
а 328
-
__
— 1640
—— —
— —*
— —
——
—— —
«а ——
— —
— —
— —
— ——
—— —-
——
0,47 —
——
— 1800
—— ——
— ——
——
— —
—
—-
—— —
— 1400
——
——
— —
0,43 —
— —--
—— ——
, ——
* %О=2,62-Ю"4 кал/(см-с-°С).
g Продолжение прилож. 2
00
Иолимер М, г/моль Ро, г/см* Рс, г/см* G я о о ez-10‘, см*/(г-’С) |4~ и о . § к - S. о АНт, кал/моль
Полииетакрилаты
полиизобутилметакрилат 142,2 1,045 2,4 ‘ 6,0
поли-втор-бутилметакрилат 142,2 1,052 —— 3,3 6,3
поли-трет-бутилметакрилат 142,2 1,03 2,7 7,0 -
поли-2-етилбутилметакрилат 170,2 1,040 5,76 __ . -
полигексилметакрилат 170,2 1,01 —— 6,3/6,6 ' 1
полиоктилметакрилат 198,3 0,971 —— — 5,8 __
попидодецилмет акрил ат 254,4 0,929 —— 3,8 6,8 __ -
полиоктадецилметакрилат 338,6 •— >0,97 —
полифенилметакрилат 162,2 1,21 — 1,3 4,4 —
полибензилметакрилат 176,2 1,179' — 1,45 4,2
полициклогексилыетакрилат - 168,2 1,098 2,7 __ __ - -
Другие полиакрилы
полиметилхлоракрилат 120,5 1,45/1,49 —— — — __
полиакрилонитрил 53,1 1,184 1,27/1,54 1,4 2,9 0,30 1160/1250
полиметакрилонитрил 67,1 1,10 — ——
полиакриламид 71,1 1,302 —— —— —— __ __
поли-И-изопропилакриламид 113,2 1,03/1,07 1,18
Полидиены
поли-1,з-бутадиен (цис) 54,1 1,01 —- 2000/2200
поли-1,3-бутадиен (транс) 54,1 —— 1,02 —— —— __ 2400/3300
поли-1,3-бутадиен (смесь) 54,1 0,892 2,0 7,7 0,395
поли-1,3-пентадиен (транс) 68,1 0,89- 0,98 —
поли-2-метил-1, 3-бутадиен 68,1 0,908 1,00 2,0 6,0/7,4 - — — 1050
поли-2-метил-1,3-бутадиен 68,1 0,904 1,05 8,3 3050
(транс)
поли-2-метил-1,3-бутадиен 68,1 — — —— — 0,38 0,457
(смесь)
поли-2-трет-бутил-1,3-бут ади- 110,2 <0,88 0,906
ен (цис)
поли-2-хлор-1,3-бутадиен 88,5 —— 1,09/1,66 —— __ 2000
(транс)
поли- 2-хлор-1,3 -бутадиен 88,5 1,243 1,356 5,0 —
(смесь) ч /
Полиоксиды
полиоксиметилен
полиоксиэтилен
политетраоксиметилен
полиэтиленформаль
поЛитетраметиленформаль
полиацетальдегид
полиоксипропилен
полиоксигексен
полиоксиоктен
поли-транс-2-оксибутея
полиоксистирол
поли-3-метоксиоксипропилен
поли-3-бутоксиоксипропилен
поли-З-гексоксиорсипропилен
поли-З-феноксиоксипропилен
поли- 3 -хлороксипр опилен
поли-2,2-бис(хлорметил)триме-
тилен-З-оксид(пентон)
поли- 2,6 -диметил-1,4 -фенил ей-
оксид
Полисульфиды
полипропиленсульфид
полифениленсульфид
Полиэфиры
полигликолевая кислота
полиэтиленсукцинат
полиэтиленадипинат
политетр аметиленадипинат
полиэтиленазелаинат
полиатиленсебацинат
полидекаметиленадипинат
полидекаметиленсебацинат
поли-ct,а-диметилпропиолактон
полиэтиленизофталат
полиэтилентерефталат *♦
политетр аметиленизофтал ат
политетр аметилентерефталат
полигексаметилентерефталат
полидекаметилентерефгалат
поли-1,4-циклогександимети-
лентерефталат (транс)
Т полиэтилен-1,5 -нафтал ат
30,0 44,1 1,25 1,125 1,54 1,33 1,8 6,4 0,34 0,30 0,50 0,49 890/1780 1750/2080
72,1 0,98 1,18 __ 6,9 0,394 0495 3000
74,1 - - 0,308 0,439 4000
102,1 — 0,339 0,453 3350/3500
44,1 1,071 1,14 2,1 6,3 —— —— ——
58,1 1,00 1,10/1,21 7,2 0,34 0,458 2000
100,2 <0,92 >0,97 — — — —
128,2 <0,94 >0,97 —— — — —
72,1 <1,01 1,099 — —— —— — ““
120,1 1,15 >1,18 — —— — —— —
88,1 1,095 —— — — — —— —
130,2 <0,982 — —— — —
158,2 <0,966 — —— —— — — —
150,2 < 1,21 1,035 — — —— ——
92,5 155,0 1,37 1,39 1,461 1,47 — 5,6 3,18 0,23 — 54 9Й/5 74 0
120,1 < 1,06 — — — 0,294 0,42 1400
74,1 < 1,10 >1,12 — — х
108,2 <1,34 — — — — —
58,0 1,60 1,70 — — — —
144,1 1,175 1,358 3,16 4,04 —• —— ——
172,2 < 1,183 1,25/1,45 — — — — 3800
200,2 < 1,019 — — —— —
214,3 228,3 284,4 340,5 1,04/1,11- 1,17/1,22 1,12/1,21 1,96 3,58 7,34 7,50 т 0,46 3300/6950 3800/10 200 7200/12 300
100,1 1,097 1,23 — — — — —
192,2 192,2 1,34 1,335 >1,38 1,50 2,0 1,4/2,4 3,8/5,3 6,0/7,4 0,27 7,37 2200/5820 10 100 7600 8000/8500 10 400/11 600
220,2 1,288 1,309 — — ——
220,0 248,3 304,4 1,131 1,012 5,3
274,3 149 1,265 — г- —
242,2 <1,37 — 1,56 3,43 __ 1
** А.^6,8* Ю“4 кал/(см*с**С).
Продолжение прилож. 2
Полимер М, г/моль Ро, г/см* "а о с? S' я о о •W4 Ъо а о о тч О) Ср, кал/(г-°С) Ср, кал/(г-’С) ДНт, кал/моль
Полиметакрилаты
полиизобутилметакрилат 142,2 1,045 2,4 ‘ 6,0 __
поли-втор-бутилметакрилат 142,2 1,052 3,3 6,3 ——
поли-трет-бутилметакрилат 142,2 1.03 2,7 7,0 -
поли-2-этилбутилметакрилат 170,2 1,040 5,76
полигексилметакрилат 170,2 1,01 — 6,3/6,6 ’ —V ' — ——
полиоктилметакрилат 198,3 0,971 —— — 5,8
полидодецилметакрилат 254,4 0,929 —— 3,8 6,8
полиоктадецилметакрилат 338,6 >0,97 — -
полифенилметакрилат 162,2 1,21 1,3 4,4 —
полибензилметакрилат 176,2 1,179' —— 1,45 4,2
полициклогексилыетакрилат - 168,2 1,098 2,7 — — —
Другие полиакрилы
полиметилхлоракрилат 120,5 1,45/1,49 — —— — ——
полиакрилонитрил 53,1 1,184 1,27/1,54 1,4 2,9 0,30 1160/1250
полиметакрилонитрил 67,1 1,10 —— —— —
полиакриламид 71,1 1,302 — —— —— — — —
поли-N-изопропилакриламид 113,2 1,03/1,07 1,18 —— —— ——
П олидиены
поли-1,з-бутадиен (цис) 54,1 1,01 — —_ — 2000/2200
поли-1,3-бутадиен (транс) 54,1 — 1,02 — —— —— —— 2400/3300
поли-1,3-бутадиен (смесь) 54,1 0,892 2,0 7,7 0,395 —
поли-1,3-пентадиен (траке) 68,1 0,89- 0,98 — —— — —— __
поли-2-метил-1,з-бутадиен 68,1 0,908 1,00 2,0 6,0/7 ,4 - — — 1050
(t(UC) поли-2-метил-1, 3-бутадиен 68,1 0,904 1,05 ' — 8,3 3050
(тррнс)
поли-2-метил-1,3-бутадиен 68,1 — — — _ 0,38 0/,57
(смесь)
поли-2-трвш-бутил-1,3-бут ади- 110,2 <0,88 0,906 — — — ——
ен (цис)
поли-2-хлор-1, з-бутадиен 88,5 —— 1,09/1,66 •— —— — 2000
(транс)
поли-2-хлор-1,3 -бутадиен 88,5 1,243 1,356 •— 5,0 —
(смесь) ч /
Полиоксиды
полиоксиметилен
полиоксиэтилен
политетраоксиметилен
полиэтиленформаль
поЛитетраметиленформаль
полиацеталь де гид
полиоксипропилен
полиоксигексен
полиоксиоктен
поли-транс-2-оксибутен
полиоксистирол
попи-3-метоксиоксипропилен
поли-3-бутоксиоксипропилен
поли-З-гексоксиодсипропилен
поли-3-феноксиоксипроиилен
поли- 3 -х л ороксипр опилен
поли-2,2-бис(хлорметил)триме-
тилен-З-оксид(пентон)
поли- 2,6-диметил-1,4-фенилеН-
оксид
Полисульфиды
полипропиленсульфид
нолифениленсульфид
Полиэфиры
полигликолевая кислота
полиэтил енсукцинат
полиэтиленадипинат
политетр аметиленадипинат
полиэтиленазелаинат
полиэтиленсебацинат
полидекаметиленадипинат
долидекаметиленсебацинат
цоли-а.а-диметилпропиолактон
полиэтиленизофталат
полиэтилентерефталат **
политетраметиленизофталат
политетр аметилентерефталат
полигексаметилентерефталат
полидекаметилентерефталат
поли-1,4-циклогександимети-
леитерефталат (траке)
Т полиэтилен-1,5 -нафталат
30,0 1,25 1,54 1.8 0,34 0,50 890/1780
44,1 1,125 1,33 6,4 0,30 0,49 1750/2680
72,1 74,1 0,98 1,18 — 6,9 0,394 0,308 0,495 0,439 3000 4000
102,1 . 0,339 0,453 3350/3500
44,1 1,071 1,14 2,1 6,3 —— — —
58,1 1,00 1,10/1,21 — 7,2 0,34 0,458 2000
100,2 <0,92 >0,97 —— — — —
128,2 <0,94 >0,97 — — —
72,1 <1,01 1,099 —— — — — —
120,1 1,15 >1,18 —— — ——
88,1 1,095 — — — — — —
130,2 <0,982 — — — — ——
158,2 <0,966 —— — —• —
150,2 < 1,21 1,035 —— — —— — —
92,5 155,0 1,37 1,39 1,461 1,47 5,6 3,18 0,23 — 549Й /5740
120,1 . < 1,06 — — — 0,294 0,42 1400
74,1 <1,10 >1,12 — — — — X
108,2 <1,34 — — — —
58,0 1,60 1,70 — — — — —
144,1 1,175 1,358 3,16 4,04 — 3800
172,2 < 1,183 1,25/1,45 — — —
200,2 < 1,019 — — —— —
214,3 228,3 284,4 340,5 1,04/1,11- 1,17/1,22 1,12/1,21 1,96 3,58 7 34 7,50 1 и । 0,46 3300/6950 3800/10 200 7200/12 300
100,1 1,097 1,23 — — —
192,2 192,2 1,34 1,335 >1,38 1,50 2,0 1,4/2,4 3,8/5,3 6,0/7,4 0,27 0,37 2200 /5820 10 100 7600 8000/8500 10 400/11 600
220,2 1,288 1,309 — — —
220,0 248,3 304 Д 1,131 1 012 5 3 — —
274,3 7,19 1,265 — —
242,2 <1,37 — 1,56 3,43 __ \
•* X—6,8-10-1 кал/(см-с-*С).
ш ___
Продолжение прилож. 2
“^00 26 Заказ 699
Полимер М, г/моль рд1 г/см» Я о 10», см»/(г.°С) и о Я о о о и о се й «о Ji, cj,, кал/(г-°С) ЬНт, кал/моль
Полиафиры < 1,33
полиэтилен- 2,6-нафталат 242,2 >1,35 1,41 А.Я6
Полиамиды 113,2 1,084 У
поли-6-аминогексановая кисло- та (найлон 6) 1,23 — 5,6 .0,35 0,51/0,59 4200/5500
поли-7-аминогептановая кис- 127,2 <1,095 1,21 3,5 ">0 АО
лота (найлон 7) 141,2
поли-8-аминооктановая кислота 1,04 1,04/1,18 3,1
(найлон 8) поли-9-аминононаяовая кислота 155,2
< 1,052 > 1,066 3,6
(найлон 9)
поли-10-аминодекановая кис- лота (найлон 10) 169,3 < 1,032 1,019 3,5 — — — —
поли-11-аминоундекановая кис- 183,3 1,01 1,23/1,12 3,6 — 9900
лота (найлон И) *
поли-12-аминододекановая кис- лота (найлон 12) 197,3 226,3 0,99 1,106 3,8 — 0,17 — 4000
полигексаметипенадипамид (найлон 6,6) 1,07 1,24 — — 0,35 — 10 600/11 200
254,4
полигептаметиленпимеламид ? (найлон 7,7) < 1,06 1,108 — — — — —
282,4
полиоктаметиленсуберамид (найлон 8,8) < 1,09 — — — — — —
полигексаметиленсебацамид 282,4 1,04 1,19 __ 0,38 0,52 13 000/13 500
(найлон 6,10) 310,5 < 1,043
полипонаметиленазеламид (най- лон 9,9) ' — — — — — —
324,5 < 1,044
полидекаметиленазеламид (пай- —— 6,6 __ 8750
лон 9,10) полидекаметиленсебацамид 338,5 < 1,032 > 1,063 7800/12 200
6,7
(найлон 10,10) 246,3 < 1,22 1,22/1,251
ноли-л-ксилиленадипамид __ __
поли-п-ксилиленсебацамид 302,4 < 1,14 1,169 —— — —
поли-2,2,4-триметилгексамети- 288,4 1,12 — — — 0,35 — —
лентерефталамид
полипиперазинсебацамид
Поликарбонаты
полиметан-бис(4-фенил)карбо-
нат
поли-1,1 -этан-бис(4- фенил) кар-
бона.'
поли-2,2-пропан-бис(4-фенил)
карбонат
поли-1,1-буган-бис(4-фенил)
карбонат
п оли-1,1 - (2 -метилпропан) бис
(4-фенил)-карбонат
поли-2,2-бутан-бис(4-фенил)
карбонат
поли-2,2-пентан-бис(4-фенил)
карбонат
поли-4,4-гецтан-бис(4-фенил)
карбонат
поли-1,1- (1 -фенилетан)бис (4 -фе-
нилкарбонат)
полидифенилметан-бис(4-фенил)
карбонат
поли-1,1-цикл опентан-бис(4-фе-
нил)карбонат
поли-1,1 -циклогексан-бис (4 -фе-
нил)карбонат
политио-бис(4-фенил)карбонат
поли-2,2-пропан-бис[4-(2-ме-
тилфенил) ]карбонат
поли-2,2-пропан-бис[4-(2-хлор-
фенил)]карбонат
поли-2,2-пропан-бис[4-(2,6-
дихлорфенил)]карбонат
поли-2,2-пропан-бис[4-(2,6-ди-
бромфенил)]карбонат
поли-1,1-циклогексан-бис[4-
(2,6-дих л орфенил) ]карбонат
Другие полимеры
поли-п-ксилилен
полихлор-п-кеилилен
поли-а,а,а',а'-тетрафтор-п-кси-
лилен
У- поли-4,4»-тетраметилендиокси-
дибензойный ангидрид
полидиметилсилоксан
252,4
226,2
240,3
254,3
268,3
268,3
268,3
282,3
310,4
316,3
378,4
280,3
294,3
244,3
282,3
323,2
392,1
569,9
432,1
104,1
138,6
176,1
312,3
74,1
1,24
1,20
< 1,18
1.27
< 1,10
< 1.28
< 1,506
< 1,266
0,98
— 5,9
1,303 — — —
>1,22 — — —
131 2.4/2,9 4,8/5,9 0,285
> 1,17 — / —
> 1,18 — — —
— — —
>1,13 — — —
> 1,16 — — —
> 1,21 — — —
— — —
> 1,21 — — —
> 1,20 — — —
1,500
> 1,22 — — —
> 1,32 — — —
> 1,42 — — —
>1,91 — — —
1,38 — — —
1,084/1,248 — —— х
> 1,597 — — —
>1,301 — — —
1,07 2,7/3,2 9/14 —
0,385
6300
8800
8
продолжение прилож. 2
Полимер М, г/моль Я ^3 Я о и 8 о и о а о и о S й cj,, кал/(г-°С) Affm, кал/моль
Полиэфиры < 1,33
полиэтилен- 2,6-нафталат 242,2 >1,35 1,41 А.Я6
Полиамиды 113,2 1,084 У
поли-6-аминогексановая кисло- та (найлон 6) 1,23 — 5,6 0,35 0,51/0,59 4200/5500
поли-7-аминогептановая кис- 127,2 <1,095 1,21 3,5 >0 АО
лота (найлон 7) 141,2
поли-8-аминооктановая кислота 1,04 1,04/1,18 3,1
(найлон 8) иоли-9-аминононаяовая кислота 155,2
< 1,052 > 1,066 • 3,6
(найлон 9) 169,3
поли-10-аминодекановая кис- лота (найлон 10) < 1,032 1,019 3,5 — — — —
поли-11-аминоундекановая кис- 183,3 1,01 1,23/1,12 3,6 — 9900
лота (найлон И) *
поли-12-аминододекановая кис- лота (найлон 12) 197,3 226,3 0,99 1,106 3,8 — 0,17 — 4000
полигексаметиленадипамид (найлон 6,6) 1,07 1,24 — — 0,35 — 10 600/11 200
254,4
полигептаметиленпимеламид ? (найлон 7.7) < 1,06 1,108 — — — — —
282,4
полиоктаметиленсуберамид (найлон 8,8) < 1,09 — — — — — —
полигексаметиленсебацамид 282,4 1,04 1,19 __ 0,38 0,52 13 000/13 500
(найлон 6,10) 310,5 < 1,043
полидонаметиленазеламид (най- —— —— __ __ — __
лон 9,9) z полидекаметиленазеламид (най- 324,5 < 1,044 ; 6,6 8750
лон 9,10) полидекаметиленсебацамид 338,5 < 1,032 > 1,063 7800/12 200
6,7
(найлон 10,10) 246,3 < 1,22 1,22/1,251
поли-лс-ксилиленадипамид __
поли-п-ксилиленсебацамид 302,4 < 1,14 1,169 —— — —
поли-2,2,4-триметилгексамети- 288,4 1,12 — — 0,35 — —
26 Заказ 699
лентерефталамид полипиперазинсебацамид Поликарбонаты полиметан-бис(4-фенил)карбо- нат поли-1,1 -»тан-бис(4- фенил) кар- бона.' поли-2,2-пропан-бис(4-фенил) карбонат поли-1,1-буган-бис(4-фенил) карбонат 252,4 226,2 240,3 254,3 268,3 1,24 1,20 1,303 > 1,22 131 > 1,17 2.4/2,9 5,9 4,8/5,9 / 0,285 0,385 6300 8800
п оли-1,1 - (2 -метилпропан) бис (4-фенил)-карбонат 268,3 268,3 < 1,18 > 1,18 — — — — —
поли-2,2-бутан-бис(4-фенил) карбонат >1,13 — — — —
282,3
поли-2,2-пентан-бис(4-фенил) карбонат — — — —
310,4 > 1,16
поли-4,4-гецтан-бис(4-фенил) карбонат — — — — —
> 1,21
поли-1,1- (1 -фенилетан)бис (4 -фе- нилкарбонат) полидифенилметан-бис(4-фенил) 316,3 — — — — — —
378,4 < 1.27 — — ' —
карбонат поли-1,1-цикл он ентан-бис(4-фе- 280,3 > 1,21
нил)карбонат поли-1,1 -циклогексан-бис (4 -фе- 294,3 > 1,20 — —
нил)карбонат политио-бис(4-фенил)карбонат 244,3 1.355 1,500 — — —
поли-2,2-пропан-бис[4-(2-ме- тилфеиил) ]карбонат 282,3 323,2 > 1,22 > 1,32 — — — -- — '
поли-2,2-пропан-бис[4-(2-хлор- фенил)]карбонат — — —
392,1 > 1,42
поли-2,2-пропан-бис[4-(2,6- дихлорфевил)]карбонат — — ““
569,9 >1,91
поли-2,2-иропан-бис[4-(2,6-ди- 5ромфенил)]карбонат — — — —
432,1 1,38
поли-1,1 -циклогексан-бис[4- (2,6-дих л орфенил) ]карбонат
Другие полимеры поли-п-ксилилен 104,1 <1,10 1,084/1,248 — —— х — —
полихлор-п-ксилилен 138,6 < 1,28 —— —— — — —
поли-ос,а,сь*,а*-тетрафтор-п-кси- лилен 176,1 < 1,506 <1,266 > 1,597 >1,301 -- —
7- поли-4,4*-тетраметилендиокси- 312,3 — — — —
дибензойный ангидрид полидиметилсилоксан 74,1 0,98 1,07 2,7/3,2 9/14 — — , —
ю
Продолжение прилож. 2
S * - 1
«Hi и о
Полимер я Я "я 6
й М § X о - Я 3 О '
- и 4е « © X Й м << X ф
Полиолефины полиэтилен 31/36 150/188/253 368/414 7,7/8.35 2,0/3,0 1,49/1,52 2,3 9,5/11,5 230/250
полипропилен 29,4 253/299 438/462 8,1/9,2 3 1,49 2,2 2,1/5,3 120/182
поли-1-бутен 34 228/249 379/415 — — — — — 1U5/123
попи-З-метил-1-бутен — <323 573/583 — — ——
поли-1-пентен —— 223/287 384/403 — —— — — —— 113/120
поли-4 -метил-1 -пентен — 295/315 501/523 — — — 2,12 — —
поли-1 -гексен —— 223 218 — — —
поли-5-метил-1-гексен < 259 383/403 — — —— —
поли-1-октадецен <328 341/353 — — 1,471/1,507 — —— —-
полиизобутилен 27/33,6 198 275 7,9 6,5 1,508 — 2,95 91/107
1,2-поли-1,3-бутадиен(изо) — 208 398 —т- —— — —— — —
1,2-поли-1.3-бутадиен(синдио) — — 428 — — — —« —
Полистиролы 1,591 2,55 3,13 67/98
полистирол * 33/43 373 498/523 8,5/9,3 3,3
поли-а-метилстирол 36 443/465 —— — — — — —— 70/77
поли-2-метил стирол -— 409 633 . — — 1,5874 —— — —
' поли-4-метилстирол — 366/379 — —— — — —— —— 66
поли-4-метоксистирол — 362 511 — —— 1,5967 —• — —-
поли-4-фенилстирол —— 434 — — —— — — —— —
поли-з-фенил-1-пропен —— 333 503/513 — —— — —— —— —-
поли-2-хлорстирол 42 392 — 8,9 — 1,6098 2,63 2,59 — ——
поли-4-хлорстирол Полигалогенолефины — 383/399 473 — — — 2,78 50
поливинилфторид 28/37 253/313 —— — — — —
поливинилхлорид 39/42 356 558 9,4/10,8 2,4 1,539 3,9 125/156
поливинилбромид —— 373 —— 9,6 — — —— 40
поливинилиденфторид 25/32,7 233/286 410/445 — —— 1,42 8/13 —— —
поливинилиденхлорид 40 225 463/483 9,9/12,2 —— 1,60 2,85 — —
политетрафторэтилен 18,5 160/400 303/600 6,21 —— 1,35 2,1 5,9 /
полихлортрифторэтилен 31 318/325 483/495 7,2/7,9 5,0 — 2,3/2,8 3,3 —
Поливинилы <348 565
поливинилциклопентан
поливинилциклогексан поли-а-винилнафталин — <363 408/435 575/656 633 — — 1,6818 — — —
поливиниловый спирт 37 343/372 531/538 12,6/14,2 1,5 1,467 8/12 160/220
оз поливинилметиловый эфир 29 252/260 417/423 —
* ПОЛИВИНИЛ8ТИЛОВ ЫЙ Э Змр — 231/254 359 — —— 1,4540 __
поливинилпропиловый афир — — 349 — __
поливинилизопропиловый афир — 270 464 — — — __
поливинилбутиловый эфир 220 337 —— — 1,4563 __
поливинилизобутиловый афир — 246/255 443 — 1,4507 __
поливинил-атор-бутиловый эфир поливинил-трвт-бутиловый — 253 443 — — 1,4740 — — —
— 1 361 533 — — — — — —
t поливинилгексиловый эфир 196 — 1,4591
поливинилоктиловый эфир —— 194 —— — —- 1,4613
поливинилметилкетон — 443 — — 1,50 — ——
полиметилизопропенилкетон 353/387 473/513 •— 1,5200 1,4757 — —— ' —.
пол ивинилф ормиат — 304/310 —— — —- — —
поливинилацетат 36 301 — 8,8/11,5 2,0 1,467 3,25 — 80/101
поливинилпропионат — 283 — 8,85 — 1,4665 —> — —
поливинилхлорацетат — 304 — — —— 1,513 — — ' •
поливинилтрифторацетат 319 448 —- —— 1,375 — ——
поливинилбензоат 341 — — 1,5775 — — 62
поли-2-винилпиридин 377 488 — — — —— 82
полив инилпирролидон — 418/448 —— — 1,53 —— — 74/90
поливинилкарбазол —— 473/48Г1 —— —- 1,683 —м 3,02 74
Полиакрилаты *
полиакриловая кислота 379 — — —- __ —— 76
полиметилметакрилат 41 281 9,7/10,4 — 1,479 4,4/5,5 — 54/81
полиэтилакрилат 35 251 ——. 9,4 9,00 -5,0 1,4685 — 90
полипропилакрилат ' •— 229 388/435 —— — — 1 — —
полиизопропилакрилат 262/284 389/453 — —— —. — — —
полибутилакрилат 221 320 8,9 —. 1,466 — —
полиизобутилакрилат —— 249/256 354 9,0 — — — — —
поли-втор-бутилакрилат поли -трет-оутилакрилат 250/256 313/316 403 466/473
Полиметакрилаты
полиметакриловая кислота —— — — —1 — 66
полиметилметакрилат 33/44 378/399 433/473 9,1/12,9 2,5 1,490 2,6/3,7 3,5/4,8 42/90
полиэтилметакрилат 33 285/338 —— 8,95 — 1,485 2,7/3,4 47
полипропилметакрилат 308/316 —— — —— 1,484 3,1 — —
полиизопропялметакрилат 300/354 —. 1,552 3,0 2,5/3,1 —— —
полибутилметакрилат — 249/300 — 8,8 .— 1,483 — 30/38
Т
* lc = 2,62.10-‘ кал/(см- с- °C).
g Продолжение прилож. 2
ю
< й * а
«Hi и О
Полимер g ж о "s о
й 5 X о * Я £ а О '
- W. 4е « о X Й м << X ф
Полиолефины полиэтилен 31/36 150/188/253 368/414 7,7/8.35 2,0/3,0 1,49/1,52 2,3 9,5/11,5 230/250
полипропилен 29,4 253/299 438/462 8,1/9,2 3 1,49 2,2 2,1/5,3 120/182
поли-1-бутен 34 228/249 379/415 — —— — — — 105/123
поли-3-метил-1-бутен — <323 573/583 —- —— —— ——
поли-1-пентен 223/287 384/403 — — —— —— —“ 113/120
поли-4-метил-1-пентен —— 295/315 501/523 — —— — 2,12 —
поли-1 -гексен —— 223 218 — — — ““
поли-5-метил-1-гексен < 259 383/403 _| ——• 1,471/1,507 — — —
поли-1-октадецен <328 341/353 — — — — ——
полиизобутилен 27/33,6 198 275 7,9 6,5 1,508 — 2,95 91/107
1,2-поли-1,3-бутадиен(изо) — 208 398 —1- — — — —
1,2-поли-! ,3-бутадиен(синдио) — — 428 — — —- —
Полистиролы полистирол * 33/43 373 498/523 8,5/9,3 3,3 1,591 2,55 3,13 67/98 70/77
поли-а-метилстирол 36 443/465 —— — — —
поли-2-метил стирол — 409 633 — 1,5874 — — 66
' поли-4-метилстирол — 366/379 — — — — —
поли-4-метоксистирол — 362 511 — —— 1,5967 —- - —
поли-4-фенилстирол —— 434 —- — — — — —
поли-З-фенил-1-пропеи — 333 503/513 — — — — -— -—
поли-2-хлорстирол 42 392 — 8,9 —— 1,6098 2,63 То
поли-4-хлорстирол Полигалогенолефины — 383/399 253/313 356 473 558 — — — 2,59 2,78
поливинилфторид поливинилхлорид 28/37 39/42 9,4/10,8 2,4 1,539 2,8/3,05 3,9 125/156
поливинилбромид —— 373 —— 9,6 — — — — 40
поливинилиденфторид 25/32,7 233/286 410/445 —— —— 1,42 8/13 — —
поливинилиденхлорид 40 225 463/483 9,9/12,2 — 1,60 2,85 —
политетрафторйтилен 18,5 160/400 303/600 6,21 —— 1,35 2,1 5,9 , —
полихлортрифторетилен 31 318/325 483/495 7,2/7,9 5,0 — 2,3/2,8 3,3 —
Поливинилы <348 565
поливинилциклопентан —
поливинилциклогексан поли-а-винилнафталин 1 <363 408/435 575/656 633 — — 1,6818 — — —
поливиниловый спирт 37 343/372 531/538 12,6/14,2 —_ 1,5 8/12 __ 160/220
оз поливинилметиловый эфир 29 252/260 417/423 — 1,467 __
* поливинилэтиловый эфир 231/254 359 — — 1,4540 —
поливинилпропиловый афир —— — 349 —— — — ——
поливинилизопропиловый афир — 270 464 — —— — ш— —
поливинилбутиповый эфир 220 337 —— — 1,4563 — __
поливинилизобутиловый афир — 246/255 443 — 1,4507 __ —
поливинил-втор-бутиловый — 253 443 — —- 1,4740 — — —
поливинил-трвт-бутиловый — 1 361 533 — — — — — —
t поливинилгексиловый афир 196 — — — 1,4591 — — —
поливинилоктиловый эфир —— 194 —— — — 1,4613
поливинилметилкетон —— - 443 — 1,50 — —
полиметилизопропенилкетон 353/387 473/513 — —- 1,5200 — —— ' —
пол ивинилф ормиат — 304/310 —— — 1,4757 — — —
поливинилацетат 36 301 —— 8,8/11,5 2,0 1,467 3,25 — 80/101
полив инилпропионат — 283 — 8,85 —— 1,4665 — — —
поливинилхлорацетат — 304 — — —в 1,513 — — —
поливинилтрифторацетат 319 448 —— —- 1,375 — — •
поливинилбензоат __ 341 — — —— 1,5775 — — 62
поли-2-винилпиридин —_ 377 488 — — —— —— 82
полив инилпирролидон 418/448 — —— 1,53 —— 74/90
поливинилкарбазол — 473/48Г1 —— — — 1,683 3,02 74
Полиакрилаты
полиакриловая кислота 379 —— — —> —— 76
полиметилметакрилат 41 281 —, 9,7/10,4 — 1,479 4,4/5,5 — •>4/81
полиэтилакрилат 35 251 — 9,4 .5,0 1,4685 — 90
пол ипропил акрил ат ___ 229 388/435 9,00 — 1 — —
полиизопропилакрилат 262/284 389/453 — — —. — —— —
п о л иб у тил ак рил ат 221 320 8,9 1,466 —
полиизобутилакрилат — 249/256 354 9,0 —- — — — —
поли-втор-бутилакрилат поли-пгрещ-бутилакрилат 250/256 313/316 403 466/473 — — —
Полиметакрилаты
полиметакриловая кислота —_ —— — —— — —- — 66
полиметилметакрилат 33/44 378/399 433/473 9,1/12,9 2,5 1,490 2,6/3,7 3,5/4,8 42/90
полиэтилметакрилат 33 285/338 —— 8,95 1,485 2,7/3,4 - 47
полипропилметакрилат ' — 308/316 —— —— 1,484 3,1 — —
полиизопропилметакрилат —— 300/354 1,552 3,0 — —
пояибутилметакрилат — 249/300 — 8,8 ' 1,483 2,5/3,1 — 30/38
Т /
* 1С = 2,62.10“* кал/(см- с- °C).
Продолжение прилож. 2
I
Полимер V, дин/см Я ,3J. м о, (кал/см’) /* Н- 10U, см'/дин й 6) о о- si • R вя «< X Кд-10», ([т1], дл/г)
Полиметакрил аты
полиизобутилметакрилат ж— 281/326 8,65 1,477
поли-ипор-бутилметакрилат поли-трет-оутилметакрилат 333 ' 380 377/438 8,3 1,4638 — —
поли-2-втилбутилметакрилат — 284 - 35
полигексилметакрилат —•— 256/268 -- 1,4813 42
полиоктилметакрилат 253 __ 30
полидодецилметакрилат 208/218 239 __ 1,4740 34
полиоктадецилметакрилат __ 309 __ __
полифенилметакрилат полибензилметакрилат — 378/393 327 93 — 1,7515 1,5679 — —
нолициклогексилметакрилат жж 324/377 __ —ж 1,5 065
Другие полиакрилы
полиметилхлоракрилат 416 —ж ___ 1,517 3,4 жж
полиакрилонитрил 44 353/378 591 12,5/15,4 1,514 3,1/4,2 __ 225
полиметакрилонитрил 393 523 10,7 — 220
полиакриламид 35/40 438 жж 260
поли-ы-изопропилакриламид 358/403 473 10,7 -
полидиены
поли-1,3-бутадиен (цис) 32 171 277 8,6 __ 1,516 __ 145/185
поли-1,3-бутадиен (транс) 31 255/263 421 жж — __ 200
поли-1,3-бутадиен (смесь) 188/215 8,3 __ ___
поли-1,3-пентадиен (транс) 213 368 жж ж—
поли-2-метил-1,3-бутадиен (цис) поли-2-метил-1,3-бутадиен 31 203 287/309 7,9/8,4 — 1,520 — • — 119/130
30 205/220 347 ж- —ж 230
(транс')
поли-2-метил-1, 3-бутадиен (смесь) — 225 — — — — 2.4 — —
поли-2-треш-бутил-1,3-бутади- ен ("цис) —- 298 379 — — 1,506 — — —
поли-2-хлор-1,3-бутадиен (транс) поли-2-хлор-1,3-бутадиен — 225 353/388 — — — — — —
38 228 316 8,2/9,3 4,3 1,558 4.6 115
(смесь)
Полиоксиды
полиоксиметилен
полиоксиэтилен
политетраоксиметилён
полиатиленформаль
политетраметиленформаль
полиацетальдегид
полиоксипропилен
полиоксигексен
полиоксиоктен
поли-транс-2-оксибутен
полиоксистирол
поли- 3 -метоксиоксипропилен
поли-3-бутоксиоксипропилен
поли-3-гексоксиоксипропилен
поли- 3-феноксиоксипропилен
ноли-3-хлороксипропилен
поли-2,2-бис(хлорметил)триме-
тилен-З-оксид(пентон)
поли-2, в-диметил-1,4-фенилен-
оксид
Полисульфиды
полипропиленсульфид
полифениленсульфид
Полиэфиры
полигликолевая кислота
полиетиленсукцинат
поливтиленадипинат
политетраметиленадипинат
полиэтиленаэелаинат
полйэтиленсебацинат
полидек аметиленадипинат
полидекамётиленсебацинат
поли-а,а-диметилпропиолактон
полиэтиленизофталат
иолиэтилентерефталат **
политетраметиленизофталат
политетраметилентерефталат
полигексаметилентерефталат
полидекам етилентере фталат
поли-1,4-циклогександимети-
лентерефталат (транс)
полиэтилен-1, 5-нафталат
36 190/243 333/471
43 206 335/345
32 185/194 308/333
— 209 328/347
189. 296
— 243 438
32 200/212 333/348
— 204 345
203 360
— 277 387
— 312 413/452
— 211 330
— 194 300
жж 188 317
— 315 485
—. — 390/408
281 353/459
— 453/515 543/548
— 221/236 313/326
— 423 527/563
— 311/368 496/533
— 272 379
— 203/223 320/338
205 327
— • 228 319
— 243 351
217 343/355
—— — 344/358
— 258/315 513 •
— 324 416/513
43 343/350 538/557
—— — 426
— 295/353 505
— 264/318 427/434
— 268/298 402/411
— 365 591
344 503
10.2/11,0 2,5 5 1,510
8,3/8,55 —
— ЖЖ
—— — —
— —ж
7,5/9,9 6 1,450/1,457
жж —
— — 1,469
^ж — ЖЖ
—ж _ж
— ж- 1, 463
— 1,458
— — 1,459
—— —— Ж_
9,4 — жж
— — —
— — —
— — 1,596
— .— —
— —
жж _ж ЖЖ
9,5 ЖЖ —Ж
—— ——
— — —
— — ^ж
— —
—ж • —
— — жж
_ж
9,7/10,7 3 1,64
—— —
— —•
— — ——
— — —
-Г- —
3,1/3,6 9,65/10,1 1 130/380
4,5 — 100/230
— — 180/270
—— ЖЖ ж—
— — —
ЖЖ —ж
— ж— 115
ж— —ж
— —— жж
—— •ж— жж
—— — ^ж
— — —
ж— жж —.
— — жж
— —
——> ЖЖ
3,0 — —
2,6 — —
3,1 6,84 —
жж
5,0/5,5 жж
5,2
3,1 —ж
3,95 ЖЖ жж
4,1 жж жж
жж —ж
3,35 жж 220
жж ЖЖ —
— ЖЖ -
2,9/3,2 5,2 175
ж_ жж ЖЖ
— жж »
^ж жж
—
— — —
— —
%с = 6,8-10_‘ кал/(см-с-°С).
сл
so?
^0?
д
•иэ)/1ГВ!1 ,_(П -8'9
Д
_ о
яаасипЕипвпквпавиайййа н^вкЕииоиаиаииаиаива
§Sgggggggggggggggg“gg.5hg§ggggggggggggggggg|
" “aSiSffiiaSSSSaSSo
IS й К Д Дс <В Л Д Д й Д Д м
ФОЛНдНччОДЛЮОл
нччпЗйинощ нЬч{ 2
m * гп А А А 1О* Я И И ХД Я А М *й
ifiigsggdOmOs
I§glf«sp,gg'§ss§4S|
2 йй s
?5и2
и
р'
о И
е*№
в?
Л
л
я
л
л
g i
® е
л ®
я Я -о -о д х
^§§§§
“§§§§
Л<оя 2
Л Я Я и
S
л
е*™ Я
О О' И
« &
к я
Л '
Я
д
о
й
Продолжение прилож.
V. джн/см
377/438 239 309 591 523 473 277 421 368 287/309 347 379 353/388 316 Тт< к -
8,65 8,3 9^9 12,5/15,4 10,7 10,7 8,6 8,3 7,9/8,4 8,2/9,3 а, (кал/см’)*/«
f 1 1 1 1 1 1 1 1 1 1 II 1 1 1 1 1 1 1 1 1 1 1 1 1 СО X- 10U, см'/дия
1,477 1,4638 1,4813 1,4740 1,7515 1,5679 1,5065 1,517 1,514 1,516 1,520 1,506 1,558 п
1 1 i м 1 1 1 1 1 1 111 ^ссс 1 1 1 1 1 1 1 1 1 1 1 &
5-1 | 1 | 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 Q5 Ха.10‘, кал/(см-о.“С)
35 42 30 34 225 220 260 145/185 200 119/131' 230 115 К0-10», ([л], дл/г)
901
Продолжение прилож. 2
Полимер V, дин/см «о М е! -о а ч св X, . ъ X. 1011, СМ1/ДИН • и о О - о Ч Q Св «-г х Кв- 10*. ([!)], дл/г)
Полиефиры
полиэтилен-2,6-нафталат — 386/453 533/541 — —— — — —
Полиамиды
поли-6-аминогексановая кис- 42 323/348 487/506 — 3,0 — 4,2/4,5 — 190
лота (найлон 6)
поли-7-аминогептановая кис- — 325/335 490/506 — — — — — —
лота (найлон 7)
поли-8-аминооктановая кисло- — 324 458/482 12,7 — — — —• —
та (найлон 8)
поли-9-аминононановая кисло- — 324 467/482 —— — — —— —*
та (найлон 9)
поли-10-аминодекановая кис- — 316 450/465 — — — — — —
лота (найлон 10)
поли-11-аминоундекановая кдс- 33 319 455/493 — — — —
лота (найлон 11)
поли-12-аминодОдёкановая кис- — 310 452 — — — 2,8/3,6 5)8/6,3 —
лота (найлон 12) полигексаметиленадипамид 46 318/330 523/545 13,6 3,0 1,475/1,580 3,8/4,3 — 190/250
(найлон 6,6)
полигептаметиленпимеламид 43 —- 469/461 — —— — — —
(найлон 7,7)
полиоктаметнленсуберамид 34 — 478/498 — — — — —
(найлон 8,8) полигексаметиленсебацамид 303/323 488/506 — 6 1,475/1,565 3,5 — —
(найлон 6,10)
полинонаметиленааеламид (най- 36 — 450 — — — —
лон 9,9)
полидекаметиленазеламид (най- —— — 487 — — — — —
лон 9,10) г
полидекам ет ил енсебацамид 32 333 469/489 — — — 3,4/3,8 —
(найлон 10,10)
поли-л€-ксилиленадипамид — 363 518 — 1 — —•
поли-п-ксилиленсебацамид — 388 541/573 — — — —
поли-2,2,4-триметилгексамети- — 1)566 3) 1 / 3)5 5
пентерефталамид
полипиперазинсебацамид Поликарбонаты полиметан-бис(4-фенил)карбо- — 355. 455 — — — —
— 393 513/573
нат
поли-1,1 -этан-бис(4- фенил кар- бонат — 403 468 — — 1,5937 2,9 — —
поли-2,2-пропан-бис(4-фенил) карбонат 42 ’ 414/423 498/540 9,9 4,2 1,5850 2,6/3,0 5,7 180/210
поли-1 Л-бутан-бис(4-фенил) карбонат — 396 443 — — 1,5792 3,3 —
поли-1,1-(2-метилпропан)бис (4-фенил) карбонат — 422 453 — — 1,5702 2,3 — —
поли-2,2-бутан-бис(4-фенил) карбонат — 407 - 495 — — 1,5827 * 3,1 — —
поли-2,2-пентан-бис(4-фенил) 1 карбонат — 410 493 — — 1,5745 — — —
поли-4,4-гептан-бис(4-фенил) — 421 473 1,5602
карбонат поли-1,1-(1-фенилвтан)бис(4- фенилкарбонат) — 449/463 503 — — — — —
1,6130
полидифенилметан-бис(4-фенил) карбонат поли-1,1 -циклопентан-бис(4-фе- — 394 503 — — 1,6539 — — —
440 523 1,5993
нил)карбонат
поли-1,1 -циклогексан-бис(4-фе- нил)карбонат — 446 533 1 — — 1,5900 2,6 — —
политио-бис(4-фенил)карбонат — 383 513 .
по ли-2,2-пропан-бис [4-(2-ме- тилфенил)]карбонат — 368/418 443 — . — 1,5783 — — —
поли-2,2-пропан-бис[4-(2-хлор- фешш)]карбонат — 420 483 — — 1,5900 — — —
поли-2,2-пропан-бис[4-(2,в-ди- хлОрфенил)]карбонат •— 453/493 533 — — 1,6056 — — —
поли-2,2-пропан-бис[4-(2,6-ди- бромфенил)]карбонат — 430 533 — — 1,6147 — — —
поли-1,1-циклогексан-бис[4- (2,6-дихлорфенил)]карбонат —- 446 543 — — 1,5858 — — —
Другие полимеры
поли-п-ксилилен —— 333/533 648/713 __ 1,669 2,65
полихлор-п-ксилилен —— 353/373 552/572 —— 1,629 3»0
поли-а,а,а',а'-тетрафтор-п- ксилилён — 363 773 — — 2,35 — —
поли-4,4 '-тетраметияендиокси- '• дибенаойный ангидрид 20/24 348 477 — — — — — —
полидиметилсилоксан 150 234/244 7,4 и 1,4036 — — 75/90
§ '
901
Продолжение прилож. 2
Полимер V, дин/см «о М е! -о а ч св X, . ъ X. 1011, СМ1/ДИН • и о О - о Ч Q Св «-г х Кв- 10*. ([!)], дл/г)
Полиефиры
полиэтилен-2,6-нафталат — 386/453 533/541 — —— — — —
Полиамиды
поли-6-аминогексановая кис- 42 323/348 487/506 — 3,0 — 4,2/4,5 — 190
лота (найлон 6)
поли-7-аминогептановая кис- — 325/335 490/506 — — — — — —
лота (найлон 7)
поли-8-аминооктановая кисло- — 324 458/482 12,7 — — — —• —
та (найлон 8)
поли-9-аминононановая кисло- — 324 467/482 —— — — —— —*
та (найлон 9)
поли-10-аминодекановая кис- — 316 450/465 — — — — — —
лота (найлон 10)
поли-11-аминоуидекановая кдс- 33 319 455/493 — — — —
лота (найлон 11)
поли-12-аминодОдёкановая кис- — 310 452 — — — 2,8/3,6 5)8/6,3 —
лота (найлон 12) полигексаметиленадипамид 46 318/330 523/545 13,6 3,0 1,475/1,580 3,8/4,3 — 190/250
(найлон 6,6)
полигептаметиленпимеламид 43 —- 469/461 — —— — — —
(найлон 7,7)
полиоктаметиленсуберамид 34 — 478/498 — — — — —
(найлон 8,8) полигексаметиленсебацамид 303/323 488/506 — 6 1,475/1,565 3,5 — —
(найлон 6,10)
полинонаметиленааеламид (най- 36 — 450 — — — —
лон 9,9)
полидекаметиленазеламид (най- —— — 487 — — — — —
лон 9,10) г
полидекам ет ил енсебацамид 32 333 469/489 — — — 3,4/3,8 —
(найлон 10,10)
поли-л€-ксилиленадипамид — 363 518 — 1 — —•
поли-п-ксилиленсебацамид — 388 541/573 — — — —
поли-2,2,4-триметилгексамети- — 1)566 3) 1 / 3)5 5
пентерефталамид полипиперазинсебацамид Поликарбонаты полиметан-бис(4-фенил)карбо- нат поли-1,1 -этан-бис(4- фенил кар- бонат — 355. 393 403 455 513/573 468 — — 1,5937 2,9 — —
поли-2,2-пропан-бис(4-фенил) карбонат i2 ’ 414/423 498/540 9,9 4,2 1,5850 2,6/3,0 5,7 180/210
поли-1,1-бутан-бис(4-фенил) карбонат поли-1,1-(2-метилпропан)бис (4-фенил) карбонат — 396 422 443 , 453 — — 1,5792 1,5702 3,3 2,3 — —
поли-2,2-бутаи-бис(4-фенил) карбонат поли-2,2-пентан-бис(4-фенил) 407 410 495 493 — — 1,5827 * 1,5745 3,1 — —
1 карбонат
поли-4,4-гептан-бис(4-фенил) карбонат — 421 473 — — 1,5602 — — —
поли-1,1-(1-фенилвтан)бис(4- фенилкарбонат) — 449/463 503 — — 1,6130 — — —
полидифенилметан-бис(4-фенил) карбонат поли-1,1 -циклопентан-бис(4-фе- нил)карбонат — 394 503 — — 1,6539 — — —
— 440 523 — — 1,5993 — — —
поли-1,1 -циклогексан-бис(4-фе- нил)карбонат — 446 533 1 — — 1,5900 2,6 — —
политио-бис(4-фенил)карбонат — 383 513 .
по ли-2,2-пропан-бис [4-(2-ме- тилфенил)]карбонат — 368/418 443 — . — 1,5783 — — —
поли-2,2-пропан-бис[4-(2-хлор- фешш)]карбонат — 420 483 — — 1,5900 — — —
поли-2,2-пропан-бис[4-(2,в-ди- хлОрфенил)]карбонат •— 453/493 533 — — 1,6056 — — —
поли-2,2-пропан-бис[4-(2,6-ди- бромфенил)]карбонат — 430 533 — — 1,6147 — — —
поли-1,1-циклогексан-бис[4- (2,6-дихлорфенил)]карбонат —- 446 543 — — 1,5858 — — —
Другие полимеры
поли-п-ксилилен —— 333/533 648/713 __ 1,669 2,65
полихлор-п-ксилилен —— 353/373 552/572 —— 1,629 3»0
поли-а,а,а',а'-тетрафтор-п- ксилилён — 363 773 — — 2,35 — —
поли-4,4 '-тетраметияендиокси- '• дибенаойный ангидрид —• 348 477 — — — — — —
полидиметилсилоксан § ' 20/24 150 234/244 7,4 и 1,4036 75/90
СПИСОК КНИГ, УПОМЯНУТЫХ
Д.';В. ВАН КРЕВЕЛЕНОМ В МОНОГРАФИИ
И ПЕРЕВЕДЕННЫХ НА РУССКИЙ ЯЗЫК
А л ф р е й Т. Механические свойства высоклполимеров. Пер.
с англ. Под ред. М. В. Волькенштейна. М., Издатинлит, 1952. 620 с.
Гл>есстон С., Л ей д л ер К., Эйринг Г. Теория
абсолютных скоростей реакций. Пер. о англ. Под ред. А. А. Балан-
дина и Н. Д. Соколова. М.. Издатинлит, 1948. 584 с.
Г р а с с и Н. Химия процессов де^грукции^полимеров. Пер.
с англ. Под ред Ю. М. Малинского. М., Издатинлит, 1959. 252 с.
Д ж е й л Ф. X. Полимерные монокристаллы. Пер, с англ.
Под ред. С. Я. Френкеля.*Л., «Химия», 1968. 552 с.
М и д л м а н. С. Течение полимеров. Пер. с англ. Под ред.
А. Я. Малкина. М-, «Мир», 1971. 260 с.
Новейшие методы исследования полимеров. Пер. с англ. Под
ред. В. А. Каргина и Н. А. Плата. М., «Мир», 1966. 572 с.
Переходы и релаксационные явления в полимерах. Составитель
Р. Бойер. Пер. с англ. Под ред. А. Я. Малкина. М., «Мир», 1968.
384 с.
Реология. Теория и приложения. Под ред. Ф. Эйриха. Пер.
с англ. Под ред. Ю. Н. Работнова и П. А. Ребиндера. М., Издатин-
лит, 1962. 824 с.
Тобольский. А. Свойства и структура полимеров. Пер.
с англ. Под ред. Г. Л. Слонимского и Г. М. БартеневаЛМ., «Химия»,
1964. 322 с.
Ферри Дж. Вязкоупругие свойства полимеров. Пер. с англ
Под ред. В. Е. Гуля. М., Издатинлит, 1963. 536 с.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Аврами уравнение 317
Адгезионные характеристики 86 сл.^З
Адгезия полимеров 105 сл.
Аддитивные мольные функции 37 сл.
Аддитивные характеристики 37, 86
Адсорбция 302 сл.
Аморфные полимеры 27, 29, 234,
265, 322
Армированные эластомеры 30
Аррениуса уравнение 336
Атактическая структура 22
Атомные вклады
в константы молекулярного при-
тяжения 136
в парахор 98
Баруса эффект 273
Бахромчатые мицеллы 31
Безразмерные jpynntj величин 34 сл.
Блочные сополимеры 23
Брюстера угол 209
Бюргерса модель 172
Вайссенберга эффект 274, 275
Ван-дер-ваальсовский объем 45,
53, 367
Бант-Гоффа уравнение 336 сл.
Вильямса — Ландела — Ферри урав-
нение 170, 171, 266, 311
Вискозиметрия 23
Внутреннее давление 86, 92
Внутреннее трение 191
Водородные связи 139
Волокниты 30
Высокоэластические свойства рас-
плавов 275
Высокоэластичность 155, 156
резин 159
Вязкость 161
аморфных полимеров 265, 267
и гидростатическое давление 272:
комплексная 274
и молекулярный вес 263
и напряжение сдвига 270
расплавов 262 сл., 267
растворов 240 сл., 250 сл., 252
и температура 264
Вязкоупругие свойства 143 сл., 161,.
172
Гагена — Пуазейля закон 282
Генри закон 291
Гетероцепные полимеры 21
Гидродинамический объем 249
Гидролитическая деструкция 360 ’
Гидропласты 30
Гидростатическое давление 262, 272
Глянцевитость 195
Гомоцепные полимеры 21, 23
Групповые вклады 38, 111, 230 , 382
в изобарно-изотермический по-
тенциал 337 сл., 340
в константы молекулярного при-
тяжения 136
в мольный объем 46, 52
в мольную полярйзацию 212
в мольную рефракцию 200
в мольную теплоемкость 73
в параметр растворимости 244
409>
Групповые вклады
в парахор 98
в температуру плавления 124
в температуру стеклования 117
в функцию Рао 148
в энергию когезии 86
в энтальпию 81
в энтропию 80, 121
Гука модель 172
Двойное лучепреломление 203, 320'
327
Двухосная ориентация 333
Дебая, уравнение 213
Демпфирование 331
Деполимеризация 355
Деструкция 355 сл.
гидролитическая 360
окислительная 358
термическая 359
фотохимическая 356, 358
Деформационные характеристики 175
Диаграммы
. e-Q 351
инкрементов 366 сл.
Диамагнитные вещества 222
Динамические механические харак-
теристики 162 сл.
и диэлектрическая поляри-
зуемость 217
Динамический переход 165
Дипольный момент 213
Дифракция рентгеновских лучей
320
Диффузионный перенос 250, 291
Диффузия 291
газов 295 сл.
жидкостей 302
паров 301 сл.
Диффузионный слой 285
Дихроизм ИК-спектра 321
Диэлектрическая поляризация
211 сл.
Диэлектрическая поляризуемость
и динамические механические ха-
рактеристики 217
и оптическая дисперсия 217
Диэлектрическая проницаемость
194, 211 сл., 219
и параметр растворимости 215
Диэлектрическая прочность 220
Диэлектрические потери 28, -216
Долговечность 187, 363
Дугостойкость 211, 221
«Дырочная» теория жидкостей 265
е—((-диаграмма 351
Жесткость 152
Изобарно-изотермический потен-
циал 337 сл., 343 сл., 345
«Изокинетическая температура» 336
Изотактическая структура 22
Инфракрасная спектроскопия 23
Инфракрасное поглощение 195
Истирание 192
Калориметрические характеристи-
ки 69 сл.
Кинетика
деструкции 361
полимеризации 335 сл.
Клапейрона — Клаузиуса уравнение
293
Когезионные характеристики 86 сл.,
150
Коллигативные характеристики 37
Компенсационные эффекты 297
Комплексная вязкость 274
Константа
Мартина 254, 255, 256
молекулярного притяжения 135,
137
седиментации 250
Хаггинса 250, 254
Конститутивные характеристики 37
Контактный угол 98, 99
Конфигурация 21, 22
Конформация 21, 22
Коэффициент
вязкости 240, 272
диссипации 163
диффузии 250, 285, 287, 291,
295 сл., 301
диэлектрических потерь 216
410
Коэффициент
набухания клубка 249
прочности 181
Пуассона 145, 153
самодиффузии 306
теплового расширения 57, 94
Кривые
напряжение — деформация 175 сл.
течения 281
Кристаллизация 308 сл.
образование центров 310 сл.,
315 сл.
полимера 27
рост кристаллов 310 сл., 313 сл.
суммарная скорость 316 сл.
Кристаллические полимеры 29, 236
325
Кристалличность 308 сл.
Ламберта — Вара уравнение 195
Ламелярные структуры 32, 308
Лармора уравнение 225
Линейные полимеры 21, 27, 29
Линейный коэффициент теплового
расширения 58
Логарифмическое число вязкости 240
Магнитная восприимчивость 194,
222 сл., 370
Магнитные свойства 222 сл.
Магнитный резонанс 222, 224
Максвелла' модель 172, 173
Максвелла уравнение 211
Марка — Хаувинка уравнение 241
Мартина константа 254, 255, 256
Межфазное натяжение 96, 98 сл.,
103 сл.
Механические потери 28, 163
Механические свойства полимеров
31, 143 сл., 175 сл., 185,
186, 376
Механическое демпфирование 331
Механическое поведение полимеров
26 ,сл.
Модели
вязкоупругого поведения 172
кристаллических полимеров 31
Модуль
высокоэластичности 275
сдвига 152 .
упругости 29, 145, 154, 328
Юнга 145, 153
Молекулярно-весовое распределение
24 сл.
Молекулярный вес 24 сл.
Мольная магнитная восприимчивость
370 .
Мольная поляризация 212
Мольная рефракция 199, 369
Мольная теплоемкость 69, 72 сл.,
233, 368
Мольная функция
притяжения 368
скорости звука 147, 148, 369
характеристической вязкости
370
Мольная электрическая поляриза-
ция 370
Мольная энергия когезии 86
вольные объемы 45, 233, 367
высокоэластических аморфных по-
лимеров 47
органических жидкостей 45-
полукристаллических полимеров
51
при нулевой температуре 57
стеклообразных аморфных по-
лимеров 48
Мольный коэффициент теплового рас-
ширения 58
Монодисперсный полимер 25
Монокристаллы 31
Муни — Ривлина , уравнение 160,
161
Мутность 195, 204
Напряжение
и деформация 331
пробоя 211
сдвига 262
Наттинга уравнение 168
Ньютона модель 172, 173
Объем 29, 233, 249
411
Объемная податливость 145
Объемные характеристики 44 сл.
Объемный- модуль упругости 143,
147 сл., 150
Одноосная ориентация 321 сл,
Однородный полимер 25
Окислительная деструкция 358
Оптическая дисперсия 217
Оптические свойства 194 сл.
Ориентация 274, 319 сл,
двухосная 333
и прочность 322
«Остаточный объем» 45
Относительная вязкость 240
Отражение света 194, 209
Паракристаллическое тело 31
Парамагнитные вещества 222
Параметр растворимости 133 сл,,
139, 293
и диэлектрическая проницае-
мость 215
и кристалличность полимеров 141
Парахор 97, 369
Переносы в полимерах 233 сл., 302 сл.
Переработка полимеров 17
Переходы в полимерах 26 сл,
165
Плавление полимеров 19, 285, 289
Пластическое разрушение 178 сл.
Пластичные полимеры 176
Плотность 44, 326
энергии когезии 134, 150
Поверхностная энергия 86,95,100 сл.
Поверхностное натяжение 150
жидкостей и расплавов 96 сл,
твердых полимеров 100 сл.
Поверхностное трение 192
Поглощение света 194, 195 сл.
Податливость 145
Показатель преломления 194
Ползучесть 166, 186
Полимеры с поперечными связями
30
Полукристаллические полимеры 29,
237
Поляризация флуоресценции 321
412
Предел
прочности 177, 178, 182, 183
текучести 189
Предельное число вязкости 240,
241 сл., 250
Предельные механические харак-
теристики 175 сл.
Предельные температуры 343 сл.
Предельные электрические харак-
теристики 220
Преломление света. 194, 199 сл.
Приведенная вязкость 240
Привитые сополимеры 23
Притяжение 368
Пробой 211
Проводимость 219
Проницаемость полимеров 291 сл.
Процарапывание 190
Прочностные характеристики 175
Псевдопластичность расплавов
281
Пуассона коэффициент 145, 153
Разветвленные полимеры 22
Разрушающее напряжение 177, 178,
182
Разрушение при ползучести 186 сл.
Рао функция 147, 148, 233
Расплавы полимеров 30, 377
неустойчивость течения 276
течение 282
Рассеяние света 194, 204 сл,
Растворение полимеров 285,
288
Растворимость 133 сл., 141, 291
газов и паров 291 сл.
и диэлектрическая проницае-
мость 215
Растворы 377
Растрескивание 360
Расширение при плавлении 65
Реактопласты и реактоэласты 30
Реакционная способность 350
Релаксация напряжений 166, 363
Релея уравнение 204
Рефракция 369
Самодиффузия 306
Свободнорадикальная сополимери-
зация 346 сл.
Свойства полимеров 13, 366 сл.
механические 31
при переработке 15, 16
в силовых полях 143 сл.
и изделий 15, 16, 18
Связевые вклады 38
Сдвиговая податливость 145
Сетчатые полимеры 24
Сжимаемость 94, 143, 145, 150
Силовые функции 14
Симхи — Бойера' модель 59
Синдиотактическая структура 22
Складчатые цепи 22
Скорость
звука 369
сдвига 262.
Собственная вязкость 240
Сополимеры 23, 346 сл.
Спин-спиновое сопряжение 228
Спиральные структуры 22
Средневековой молекулярный вес
24, 25
Среднечисловой молекулярный вес
24, 25
z-Средний молекулярный вес 24, 25
«Стандартные характеристики» и
«стандартные соединения» 39
Статистическая [разупорядоченность
32
Статистические сополимеры 23, 350
Статистический клубок 22
Статическая электризация 219
Стеклообразное состояние 27
Степень
вытяжки 321
кристалличности 33, 308
ориентации 320
Стереорегулярные полимеры 22
Строение полимеров 19 сле
кристаллических 31
Структурная упорядоченность 32
Сферолиты 32
Тангенс угла диэлектрических по-
терь 216
Тангенс угла механических потерь
163
Твердость 189
Текучесть 189
Температурно-временная суперпози-
ция 169
Температурный коэффициент плот-
ности 57
Температуры
деструкции 360
кристаллизации 28, 314
переходов 29, 108 сл., 117,118 сл.
плавления 28, 108, 120, 124,
125, 129
стеклования 27, 28, 108 сл.,
НО, 112, 113, 129
Флори 141
Тепловое расширение 57, 326, 373
375
Теплоемкость 69, 233, 368
Теплопроводность 29, 233, 326
аморфных полимеров 234
расплавов 234
Теплостойкость 193
Теплота плавления и кристаллиза-
ции 78
Теплофизические свойства полиме-
ров 44 сл.
Термическая деструкция 355
Термические характеристики 150
Термический коэффициент расшире-
ния 29, 150
Термическое окисление 359
Термодинамика
переходов 117 сл.
полимеризации 335 сл.
свободных радикалов 345 сл.
Термодинамические характеристики
379
Термохимические свойства 335 сл.
Технические характеристики (свой-
ства) 18
Типология
количественных характеристик
34 сл.
полимеров 19 сл.
Транспортирование материала 16
Трение 191
413
Ударная вязкость 185 сл.
Удельная вязкость 240
Удельная теплоемкость 29, 69, 117,
*150
Удельный инкремент показателя пре-
ломления 194, 205
Удельный коэффициент теплового
расширения 57
Удельный объем 44, 45
Упругая податливость 145
Упругие характеристики 143
«Упругое» число Рейнольдса 276
Упругость расплавов 273
Усталостное разрушение 187 сл.
Усталость материала 187
Устойчивость к процарапыванию 190
Уравнение
Авраами 317
Аррениуса 336
Вант-Гоффа 336 сл.
Вильямса — Лэндела — Ферри
170, 171, 266, 311
Дебая 213
Клапейрона — Клаузиуса 293
Ламберта — Бэра 195
Лармора 225
Максвелла 211
Марка — Хаувинкд 241
Муни — Ривлина 160, 161
Наттинга 168
Релея 204
Френеля 209
Ферромагнитные вещества 222
Физические превращения 308 сл.
Флори температура 141 <
Фойхта модель 172, 173
«Фононная» модель 234
Формование полимера 16
Фотоокисление 358
Фотохимическая деструкция 356
Френеля уравнение 209
Фундаментальные свойства 15, 16
Функциональные группы 19, 20,
110, 371 сл.
Функция Рао 147, 148
Хаггинса константа 250, 254
Характеристическая вязкость 240,
241 сл., 250, 370
Характеристические диаграммы
инкрементов 366 сл.
Характеристические свойства 15
Химическая деструкция 363
Химические превращения 308 сл.
Химический сдвиг 226
Хромотография на проницаемом ге-
ле 26
Хрупкие полимеры 176
Хрупкое разрушение 178 сл.
Чередующиеся сополимеры 23, 350,
351
Число вязкости 240
Штаудингера показатель 240
Эластомеры 30
Электрическая индуктивность 194
Электрическая поляризация 370
Электрическая проводимость 194,
220
Электрические свойства 211 сл.
Электромагнитные свойства 376,
377
Эксплуатационные свойства 18
Энергия
активации диффузии 297, 298
диссоциации связей 347
когезии 86, 90, 375
Энтальпия 78, 79, 81
Энтропия 80, 81 1
ЭПР 224, 230 сл.
Эстетические характеристики (свой-
ства) 18
Юнга модуль 145, 153
ЯМР 23, 224
ЯМР-спектроскопия 226 сл., 229 сл.
Д. В. ВАН-КРЕВЕЛЕН
СВОЙСТВА
И ХИМИЧЕСКОЕ
СТРОЕНИЕ
ПОЛИМЕРОВ
Редактор
А. А. РОГАЙЛИНА
Технический редактор
А. С. КОЧЕТОВА
Художник
В. М. КАЗИНЦЕВ
Художественный редактор
н. в. носов
Корректоры
Л. В. ЛАЗУТКИНА,
М. С. ХРИПУНОВА
Сдало в наб. 15/ХП 1975 г.
Поди, к печ. 24/111 1976 г. Формат бумаги 60x90‘/i«.
Бумага тин. 2. Уел. печ. л. 26.
Уч.-изд. л. 25,83. Тираж 7400 экз.
Зак. J4 699. Изд. № 931.
Цена 1 р. 94 к.
Издательство «X ими я»,
107076, Москва, Стромынка, 13.
Ленинградская типография Л5 6 Союзполиграфпрома
Государственного комитета Совета Министров СССР
пр делам издательств, полиграфии и книжной торговли.
196006, Ленинград, Московский пр., 91.