









































































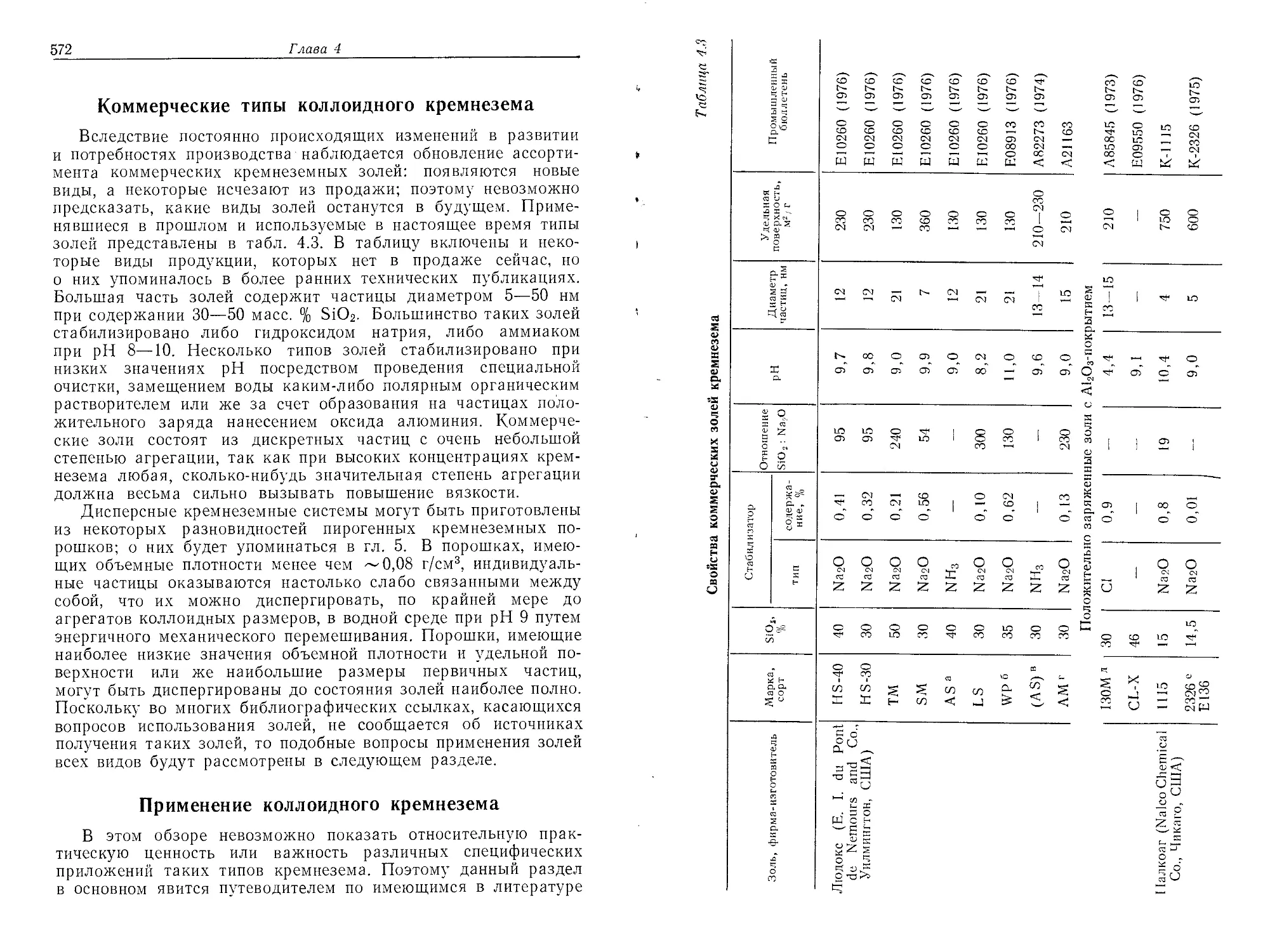



















































































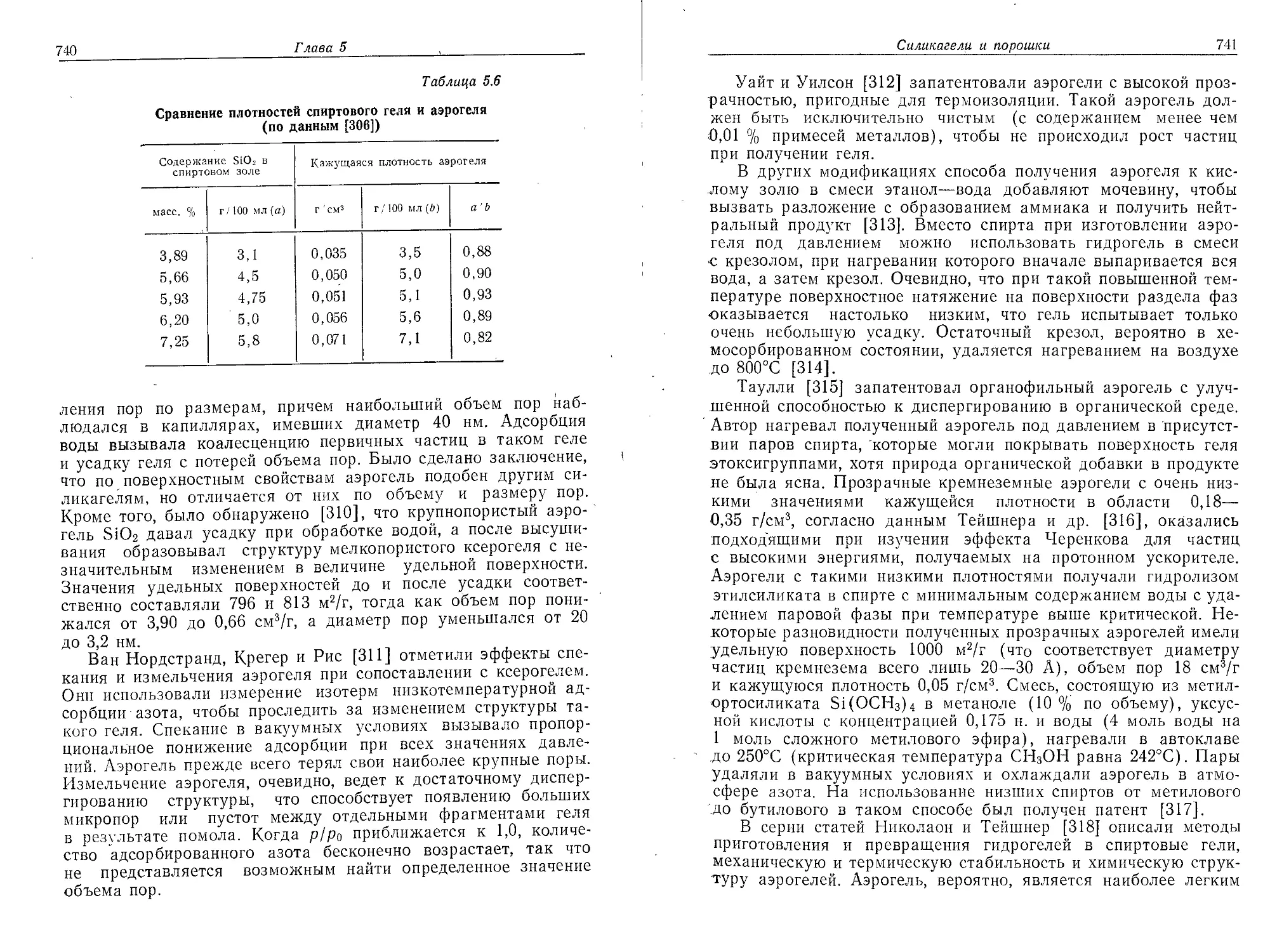




































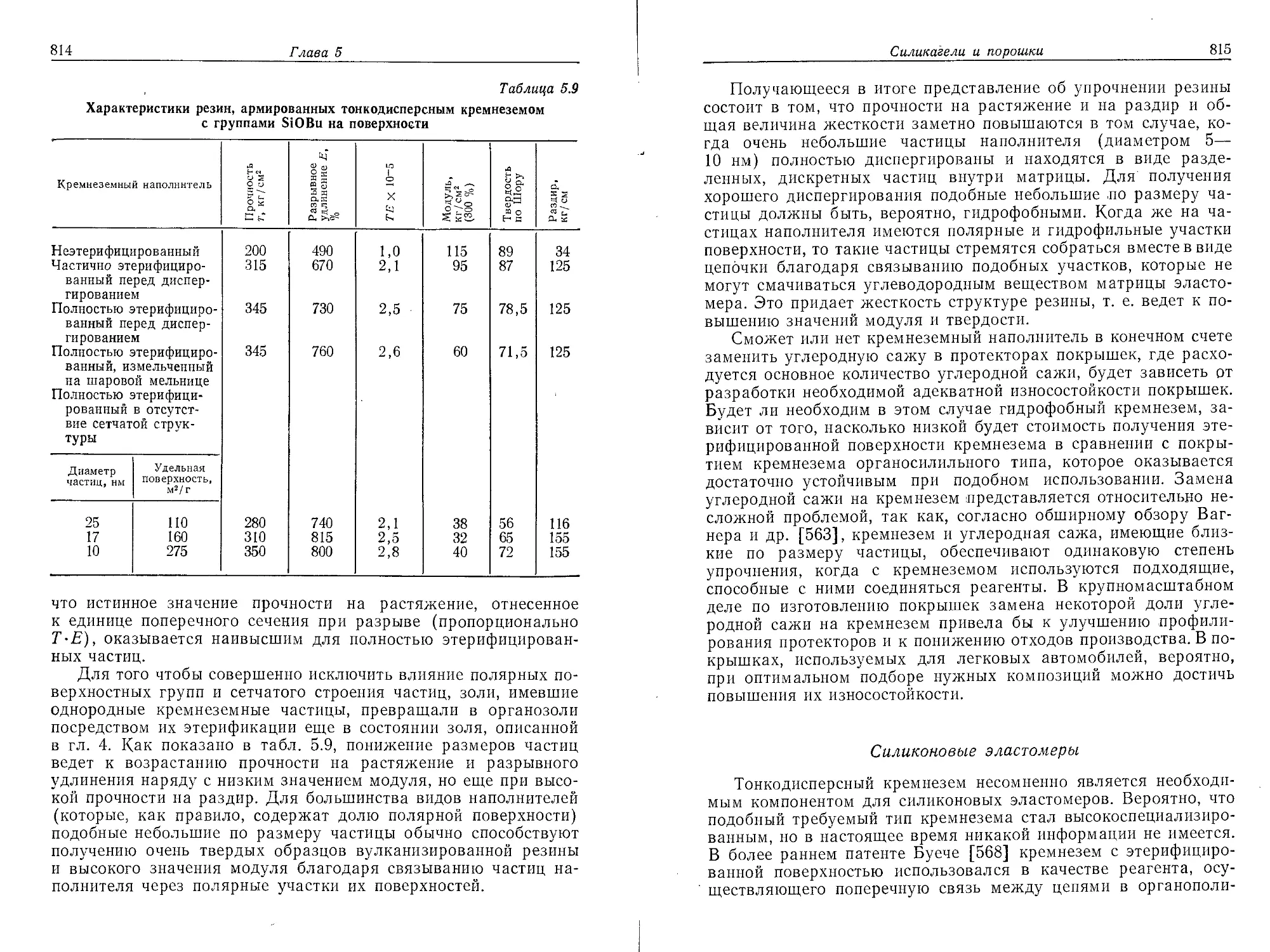


















































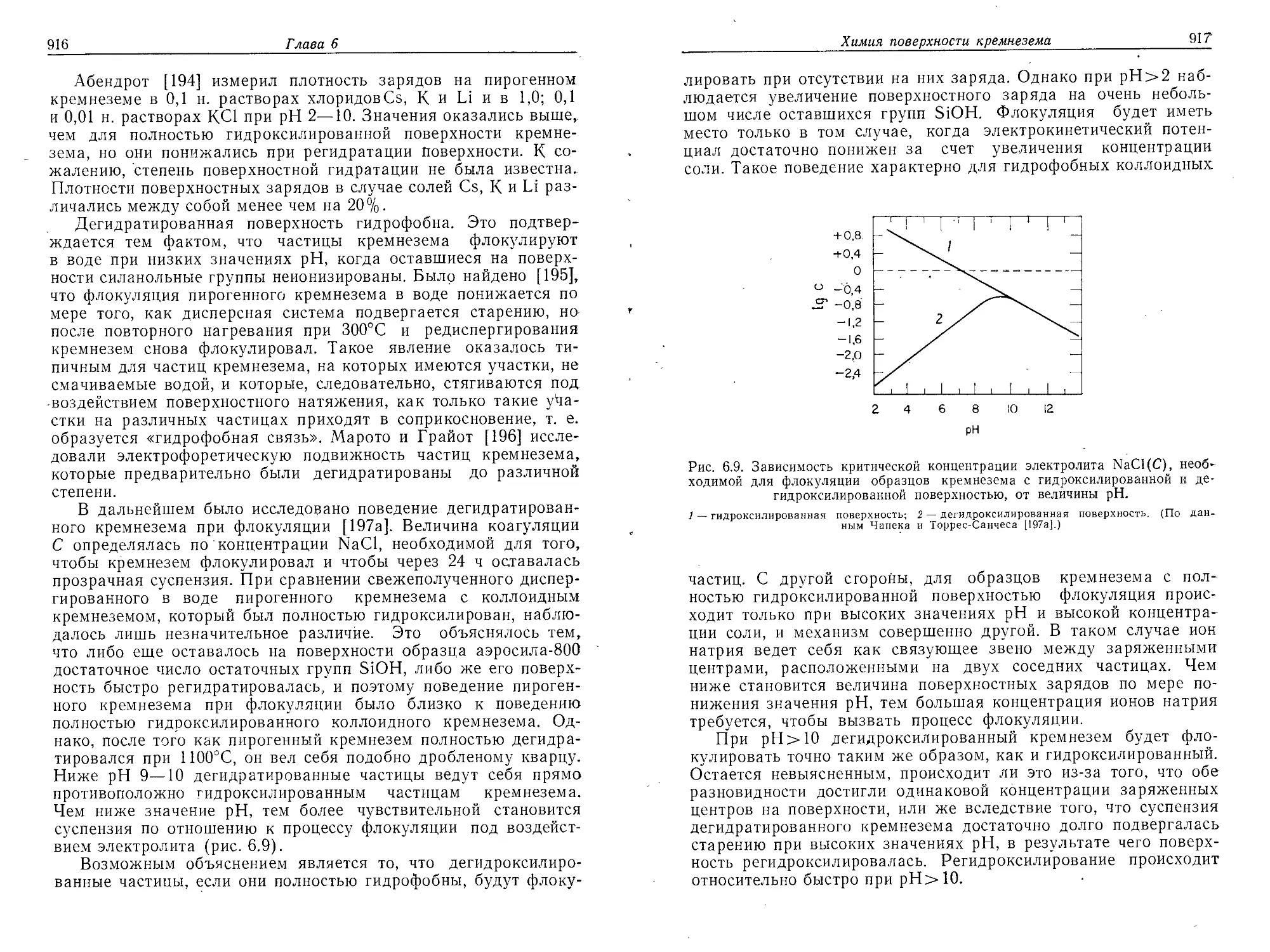













































































































Текст
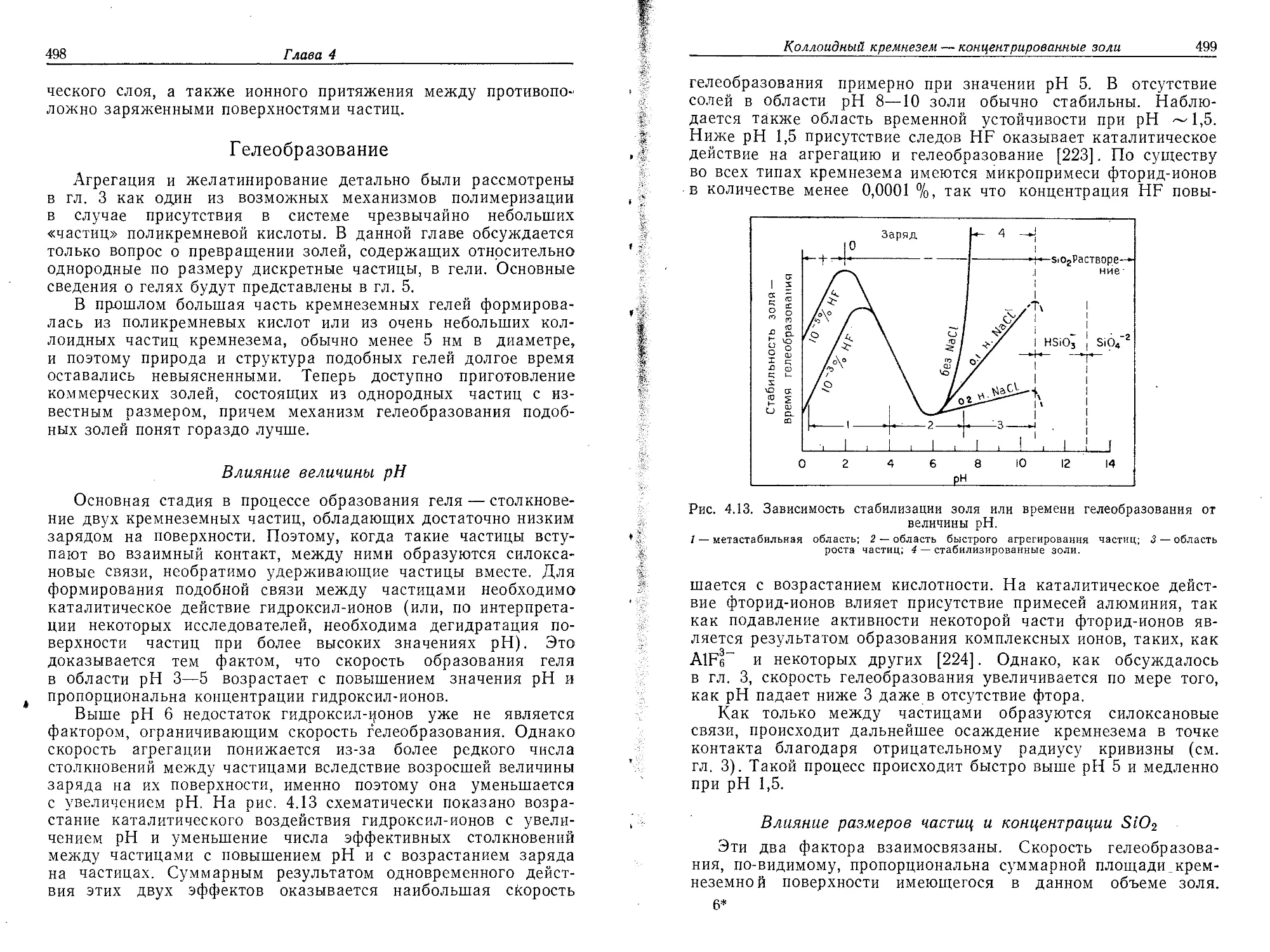
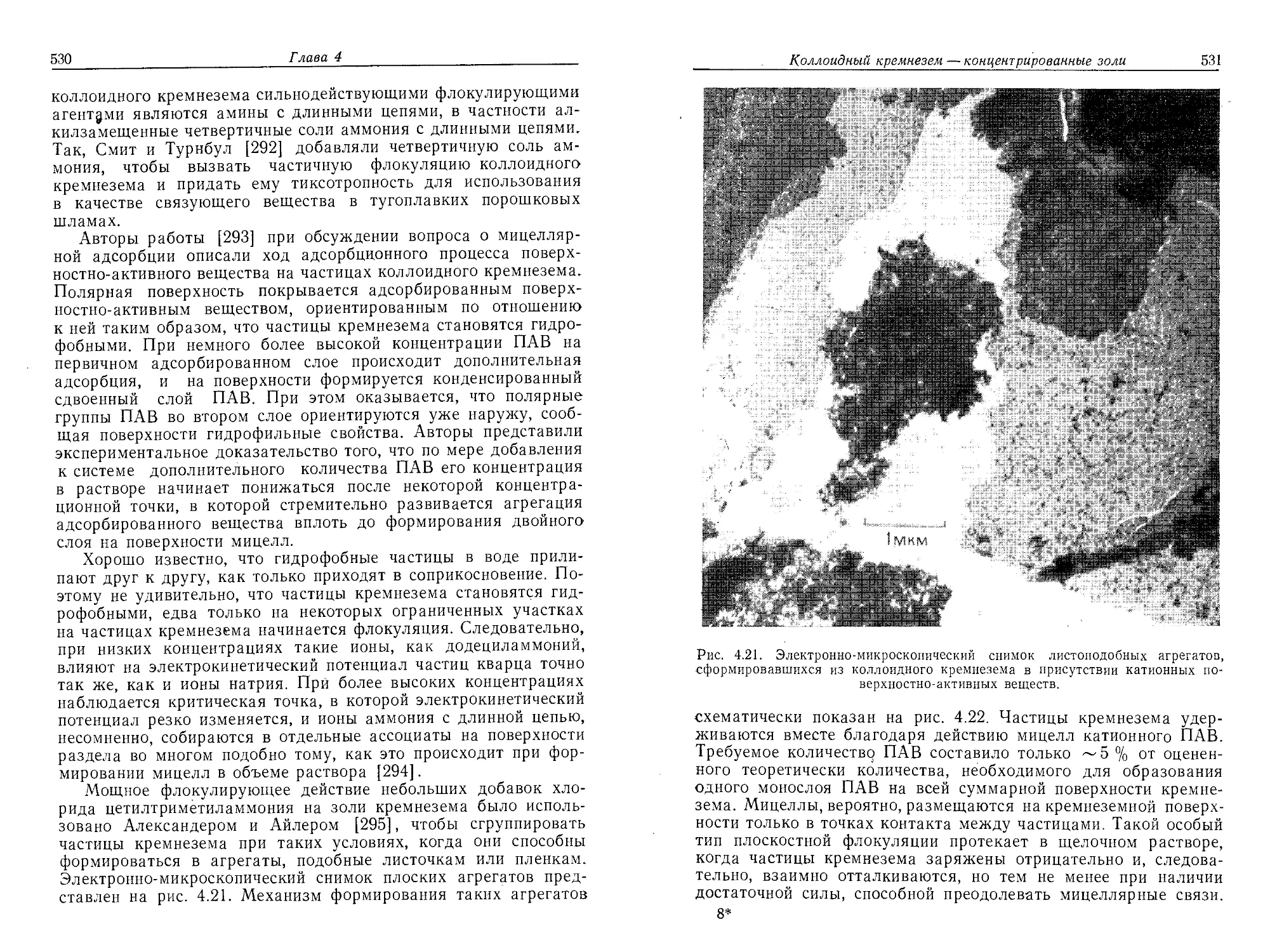
РДИЛЕР
ХИМИЯ
КРЕМНЕЗЕМА
THE CHEMISTRY
OF SILICA:
Solubility, Polymerization,
Colloid ana Surface Properties
and Biochemistry of Silica
Ralph K. Iler
A Wiley-lnterscience Publication
'New York-Chichester-Bnsbano-Toronto
РАЙДЕР
ХИМИЯ
КРЕМНЕЗЕМА
Р а створи моешь, п олимеризация,
коллоидные и поверхностные свойства
биохимия
В 2 частях
1
'Перевод с английского
канд. физ.-мат. наук
Л.Т.Журавлева
под редакцией
д-ра техн, наук
проф. В.П.Прянишникова
Москва
«Мир» 1982
ББК 24.124
А 36
УДК 546.28
Айлер Р.
А 36 Химия кремнезема: Пер. с англ.— М.: Мир, 1982.
Ч. 2,—712 с„ ил.
Фундаментальный труд американского физикохимика, охватывающий широ-
кий круг вопросов, связанных с изучением и применением чистого кремне-
зема. В русском переводе выходит в 2-х частях.
Часть 2 посвящена золям, гелям кремнезема и кремнеземным порош-
кам. Рассматриваются способы их получения, очистки и обогащения, вопросы
дегидратации и регидратации, поверхностная энергия силанольной и сило-
ксановой поверхностей и др. Приводятся сведения по адсорбции различных
соединений на кремнеземе и модифицированию его поверхности. Дается об-
зор работ по биохимической роли кремнезема в фауне и флоре.
Предназначена для научных и инженерно-технических работников в об-
ласти химии, биологии, медицины, почвоведения, металлургии, горнорудной
и строительной промышленности.
1802000000-106
А 041(01)—82
65—82,
ч.
ББК 24.124
540
1
Редакция литературы по химии
© 1979 by John Wiley & Sons, Inc. All rights reserved. Authorized
translation from English language edition published by John Wiley
& Sons, Inc.
© Перевод на русский язык, «Мир», 1982
Глава 4
КОЛЛОИДНЫЙ КРЕМНЕЗЕМ —КОНЦЕНТРИРОВАННЫЕ
ЗОЛИ
Определение коллоидного кремнезема.
Историческая справка
Термин «коллоидный кремнезем» в данной монографии отно-
сится к стабильным дисперсиям или золям, состоящим из диск-
ретных частиц аморфного кремнезема. Такое определение иск-
лючает растворы поликремневых кислот, в которых полимерные
молекулы или частицы настолько малы, что оказываются не-
.стабильными. ^Растворы поликремневых кислот, обычно полу-
чаемые подкислением растворов силиката натрия, гидролизом ;
сложных эфиров или галоидных соединений кремния при обыч- i
ных температурах, были рассмотрены в гл. 3 как стадия, пред- j
шествующая образованию коллоидных кремнеземных частиц.)
Стабильные концентрированные золи кремнезема, отличаю-
щиеся от геля и не выпадающие в осадок в течение по крайней
мере нескольких лет, стали доступными с 1940 г., после того,
как был найден способ приготовления однородных коллоидных
частиц, больших по размеру, чем ~5 нм, и стабилизированных
при оптимальном количестве основания.
Когда Вейл [1] в 1925 г. и Тредвелл и Виланд [2] в 1930 г.
рассматривали проблему коллоидного кремнезема, золи кремне-
зема с содержанием более 10 % SiOs были редкостью, причем
подобные золи не были устойчивы по отношению к процессу
гелеобразования. В 1933 г. Гриссбах [3] в своем обзоре сооб-
щил о том, что 10 %-ный золь, стабилизированный аммиаком,
был приготовлен фирмой ИГ Фарбениндустри АГ. В 1941 г.
Бёрд [4] запатентовал способ удаления щелочи из разбавлен-
ного раствора силиката натрия. Использовалась ионообменная
смола в водородной форме. Для стабилизации кремнезема пов-
торно добавлялось небольшое количество щелочи и проводилось
концентрирование системы выпариванием воды при нагревании.
Теперь мы знаем, что при подобных условиях частицы кремне-
зема могут вырастать до 5—10 нм. В 1945 г. Уайт [5] запатен-
товал способ вымывания солей из кремнеземного геля, приго-
товленного подкислением раствора силиката натрия. Гель про-
питывали щелочным раствором и нагревали до такой степени,
чтобы большая его часть пептизировала в золь. Обычно такими,
способами получали золи с содержанием 15—20 % SiOs, по
крайней мере временно стабилизированные против гелеобразо-
422
Глава 4
вания или выпадения в осадок, В 1951 г. Бечтольд и Снайдер
[6] впервые разработали способ приготовления однородных и
регулируемых по размеру коллоидных частиц кремнезема. Рул
[7] определил оптимальные концентрации щелочи, требуемые
для стабилизации системы при ограниченном содержании при-
месей электролитов. В книге, опубликованной в 1954 г., Айлер
[8] привел исторический обзор развития данной области химии
кремнезема и обобщил достигнутые к тому времени успехи. Даль-
нейшие усовершенствования, предложенные Александером [9]
в отношении контролирования размеров частиц, степени агреги-
рования, чистоты и оптимальной концентрации стабилизирующей
щелочи, дали возможность получать устойчивые зблй с размером
частиц ~8 нм, с содержанием кремнезема более 30 %. В рабо-
тах [10, И] были запатентованы способы приготовления ста-
бильных прозрачных водных золей, содержащих до 50 масс.%
SiO2 и имеющих размер частиц 20—25 нм. Для стабилизации си-
стемы вводили оптимальное количество щелочи, а для пони-
жения вязкости (без нарушения устойчивости) добавляли до-
статочное количество соли.
Золи с размером дискретных частиц 300 нм и более, обра-
зующие осадок при стоянии, приготовляли автоклавной обработ-
кой смеси влажного геля кремнезема с основанием при давле-
нии выше атмосферного с последующим помолом незначительно
агрегированных частиц в коллоидной мельнице [12а].
Таким образом, за прошедшие 30 лет были разработаны спо-
собы приготовления дискретных стабилизированных частиц
кремнезема во всей области коллоидных размеров, что позво-
лило организовать выпуск концентрированных технических
золей.
Нэпер и Хантер [126] написали обширный обзор по спосо-
бам приготовления и свойствам «гидрозолей».
Рост и стабилизация дискретных частиц
Как было показано в гл. 3 (см. рис. 3.1), кремневая кислота
полимеризуется в области значений pH 7—10 с образованием
дискретных частиц. Никакой агрегации частиц не происходит,
если концентрация электролита составляет менее 0,1—0,2 н.
в зависимости от концентрации кремнезема..
Вследствие неоднородного распределения частиц по разме-
рам, особенно когда их диаметр меньше 10 нм, наблюдается
самопроизвольный рост частиц. Размер, которого могут достиг-
нуть частицы, зависит в сильной степени от температуры си-
стемы. Как было представлено в табл. 3.5, при температуре
50—100°С диаметр частиц составляет 4—8 йм, тогда как в ав-
токлаве при температуре 350°С они могут вырастать до 150 нм.
Коллоидный кремнезем — концентрированные золи 423
Такой самопроизвольный рост оказался относительно незави-
симым от концентрации кремнезема. При автоклавной обра-
ботке водных золей размер получаемых частиц ограничен в об-
ласти высоких температур превращением аморфного кремнезема
в кристаллический кварц. Файр и Мак-Кей [13] показали, что
скорость превращения в автоклаве при 330°С и соответствующем
давлении пропорциональна квадрату концентрации гидроксил-
ионов. Олер [14] сообщил, что при автоклавной обработке
(150°С и 2000 бар) продолжительностью 4 недели образовыва-
лись микросферы из кристаллического тридимита диаметром
50 мкм.
Увеличение размера частиц при добавлении
«активного» кремнезема
Стабильные концентрированные золи не могли быть полу-
чены из-за того, что коллоидный кремнезем с частицами не-
большого размера приготовляли при комнатной температуре.
Поскольку было известно, что при более высокой температуре
процесс гелеобразования ускоряется, то казалось вполне логич-
ным проводить приготовление золей именно при условии под-
держания комнатной температуры. Следовательно, предельный
размер частиц в таких золях редко превышал 2—3 нм. Когда
же значение pH для золя данного вида повышали до 8—10 и,
затем проводили концентрирование кремнезема в вакууме при
20—30°С, то при достижении концентрации SiO2~ 10 % наблю-
дался процесс гелеобразования. В то время еще не было изве-
стно, что подобную кремнеземную систему можно нагреть до
100°С и выпаривать при такой температуре до получения устой-
чивого значения концентрации SiOz, равной 10—20 %, поскольку
при более высокой температуре размер частиц повышается до
4—6 нм.
Однако для получения еще более концентрированных золей
необходимы большие по размеру частицы. Впервые это было
доказано путем дальнейшего осаждения растворимого кремне- f
зема на частицах. Добавляли кремневую кислоту с частицами, ’
размер которых был меньше 5 нм (в основном менее 2 нм), или ;
даже представляющих собой еще более мелкие полимерные об-
разования. Такие частицы и называются «активным» кремнезе-
мом, поскольку они оказываются более растворимыми в при-
сутствии частиц большего размера или «зародышей», на кото- „
рых осаждается кремнезем.
Такой способ был разработан Бечтольдом и Снайдером [6],
которые первыми получили стабильные концентрированные
золи коллоидного кремнезема с любым желаемым размером
424
Глава 4
частиц от 10 до 130 нм. Прежде всего приготовляют 3,5 %-ный
раствор кремневой кислоты, пропуская раствор силиката натрия
через слой ионообменной смолы в водородной форме, что не-
обходимо для удаления натрия. После этого добавляют доста-
точное количество щелочи, чтобы получить pH>7, используя
для этой цели кремнезем с нанесенным на него NasO в количе-
стве около 1 масс. %. Эта часть процесса заимствована из спо-
соба, предложенного Бёдом [4]. Однако, далее, вместо прямого
упаривания всего раствора проводят нагревание некоторой его
части до 100°С, в результате чего в данной порции образуются
частицы кремнезема диаметром по крайней мере 4—6 нм. За-
тем остальная часть подщелоченного раствора поликремневой
кислоты, в которой размер частиц не превышает 3—4 нм, по-
степенно добавляется к указанному горячему золю по мере по-
вышения его концентрации в процессе выпаривания. Выполняя
такую процедуру достаточно медленно, добиваются того, чтобы
весь поступающий кремнезем осаждался на исходных частицах,
которые таким образом растут с постоянной скоростью. Обычно
при этом одновременно происходит испарение воды, так что при
сохранении суммарного числа частиц в единице объема системы
частицы будут увеличиваться в размере по мере повышения
концентрации золя. В патентной литературе США первона-
чально подщелоченный золь с частицами кремнезема, которые
способны действовать в качестве зародышей, получил наиме-
нование «пяты»*, понимаемое в том смысле, что такой золь
представляет собой «осадок» в частично заполненном сосуде,
к которому может добавляться раствор с меньшей вязкостью.
Модифицировав процесс, предложенный Бечтольдом и Снай-
дером, Рул [7] в своем способе начал с аналогичного подщело-
ченного основного золя. Но затем к такому золю он добавлял
раствор поликремневой кислоты, полученный ионным обменом,
причем перед этой процедурой никакой щелочи в систему не
вводилось. Таким образом, частицы кремнезема вырастали
в среде, имевшей постоянную концентрацию щелочи, необходи-
мую для стабилизирования подобных систем, что обеспечивало
получение стабильных концентрированных золей при минималь-
ном ее содержании. Альбрехт [15] запатентовал способ добав-
ления поликремневой кислоты с оптимальной скоростью в выше-
указанном процессе Рула с целью получения частиц кремнезема
размером 45—100 нм. Аналогичным способом, но применяя дав-
ления выше атмосферного, удавалось получать частицы разме-
ром вплоть до 150 нм [16].
* В советской научно-технической литературе такой золь носит название
«основного золя», а добавляемый золь называется «золем-питателем».— Прим,
ред.
Коллоидный кремнезем — концентрированные золи 425
Частицы диаметром более чем 100 нм приготовлялись Мин-
диком и Воссосом [17] путем добавления кремнезема со средней
молекулярной массой ниже 9р000 к основному золю, имевшему
pH 7—11 и содержавшему ~0,1 масс. % относительно одно-
родных сферических частиц кремнезема со средним диаметром,
равным по крайней мере 30 нм. Скорость подачи питательного
раствора поддерживалась ниже максимальной, определяемой
по соотношению Ft — kStCt, где Ft— максимальная скорость
в момент времени t, выраженная числом граммов кремнезема,
добавляемого к 1 мл смеси за 1 ч; k — заранее определяемая
константа скорости реакции, равная 0,005 для процесса, про-
текающего при 100°С и постоянном объеме системы; St — удель-
ная поверхность частиц в момент времени t, м2/г SiO2; Ct —
концентрация кремнезема в смеси в момент времени t, г/мл.
Подобный прием увеличения размеров частиц кремнезема
иногда называют «способом наращивания». Коллоидные ча-
стицы, в которых только лишь поверхность состоит из кремне-
зема, а внутренняя часть содержит какое-либо другое нераст-
воримое вещество, могут быть приготовлены «способом наращи-
вания», если выбрать в качестве исходных частиц пригодные
коллоидные зародыши, отличающиеся по составу от кремнезема.
Таким образом, создается возможность получения золей, имею-
щих дисперсность и поверхностные характеристики коллоидного
кремнезема, но состоящих из пластинчатых или волокнистых
частиц.
Айлер [18] разработал способ кремнеземного покрытия,
благодаря которому кремнезем может быть нанесен на исход-
ные коллоидные дисперсные материалы, представляющие со-
бой пластинчатые или волокнистые силикатные минералы, ок-
сиды, металлы или какие-либо другие материалы в коллоид-
ном состоянии. В процессе увеличения размеров частиц по этому
способу добавление «активного» кремнезема к системе не дол-
жно производиться быстрее, чем его может поглотить имею-
щаяся поверхность кремнезема. В противном случае раствор
становится настолько пересыщенным, что начинают формиро-
ваться новые небольшие зародыши, и, следовательно, оконча-
тельно образовавшийся золь не будет состоять из однородных
по размеру частиц. Как было показано в гл. 1, максимальная
скорость добавления кремнезема, при которой не происходит
образования зародышей, составляет при 90°С около 10 г актив-
ного SiO2 в расчете на 1000 м2 площади поверхности кремнезема
За 1 ч. Теоретически увеличение размеров частиц зависит от
так называемого «отношения наращивания» Вг:
426
Глава 4
Wa— количество «активного» кремнезема, добавляемого
к частицам; Wn — масса кремнезема, первоначально находяще-
гося в системе в виде зародышей. Очевидно, что диаметр ча-
стицы будет увеличиваться от начального размера di до конеч-
ного df в соответствии со следующим уравнением:
( df У Wn + Wa ( df У , , Г,
~ wn или Uz)— + Br
Айлер и Уолтер [19] разработали способ, с помощью кото-
рого 15 %-ный золь можно приготовить непосредственно ион-
ным обменом. По этому способу основной золь в воде или в раз-
бавленном водном растворе силиката натрия нагревают и пере-
мешивают. Затем к нему одновременно добавляют увлажнен-
ную, отстоявшуюся, регенерированную ионообменную смолу
(предпочтительно слабокислотного типа) и относительно кон-
центрированный раствор силиката натрия. Скорость добавления
регулируется так, чтобы поддерживать pH около 9, и, кроме
того, она зависит от температуры и относительного количестйа
и размера частиц основного золя. Это способствует увеличению
размера частиц кремнезема и предотвращает процесс образо-
вания зародышей. В способе с применением’колонны или псевдо-
ожиженного слоя смола непрерывно добавляется в верхнюю
часть колонны и перемещается вниз противотоком по отноше-
нию к движению золя кремнезема [20].
С целью изучения роста частиц кремнезема, происходящего
за счет добавления мономерного кремнезема к дисперсионной
среде из предварительно сформированных зародышей, было
исследовано образование зародышевых частиц при pH 8 в про-
цессе старения кремневой кислоты с низкой молекулярной мас-
сой, приготовленной из силиката натрия ионным обменом [21].
Очень чистый силикат натрия был получен из этилового эфира
ортокремневой кислоты. В этом случае 2,5 %-ный золь, подще-
лоченный до нужного значения pH, сохраняли при температуре
4°С, чтобы свести к минимуму самопроизвольный рост частиц и
промотировать формирование микрогеля, чему способствует вы-
сокая концентрация частиц небольшого размера. При нагрева-
нии агрегаты такого микрогеля конденсировались, и после
усадки из них формировались более или менее плотные сфери-
ческие частицы диаметром около 100 А. Однако следует отме-
тить, что зародыши получались гораздо более однородными по
размеру при медленном ступенчатом нагревании от 4 до 80°С
(в течение 2 ч), чем в случае быстрого нагревания (в течение
10 мин), как это было показано большей однородностью золей,
полученных на конечной стадии в результате добавления све-
жеприготовленного раствора кремневой кислоты, предвари-
тельно также отрегулированной до pH 8.
Коллоидный кремнезем — концентрированные золи 427
Одновременно с ростом небольших частиц (менее 20 нм) по
указанному процессу наблюдается и их некоторой самопроиз-
вольный рост. Это важно иметь в виду при приготовлении ча-
стиц с размером, меньшим чем 15 нм. Действительно, даже и
без добавления какой-либо формы «активного» кремнезема дол-
жен происходить некоторый рост зародышевых частиц за период
в несколько часов, требуемых обычно для такого процесса.
Приведенные выше замечания применимы к системам, когда
SiOa добавляется в виде раствора «активного» кремнезема, при-
готовленного, как правило, ионным обменом. Более быстрый
рост частиц наблюдается в том случае, когда раствор силиката
натрия непосредственно прибавляется к «основному золю», со-
стоящему из зародышевых частиц, из которого непрерывно уда-
ляются ионы натрия за счет использования ионообменной смолы
в водородной форме, что будет рассмотрено ниже. В указанном
примере кремнезем первоначально находился в системе в виде
мономера и олигомеров. Следовательно, не требовалось затрат
времени на процесс деполимеризации более высокомолекуляр-
ных поликремневых кислот, которые постоянно присутствуют,
если кремнезем.вначале приготовляется в виде отдельного рас-
твора «активного» кремнезема.
Другие разновидности способа «наращивания» упоминаются
также в работах ряда исследователей. Альбрехт [22] определил
максимальную концентрацию, до которой исходные золи с раз-
личающимися по размеру частицами могут быть сконцентриро-
ваны путем процесса осаждения, когда размер частиц увеличи-
вается до 100 нм. По данным Клосака [23], по способу «нара-
щивания» при температуре выше 100°С и давлениях пара 0,35—
7 кг/см2 получаются золи с концентрацией до 55 % SiOa- По
этому способу должны получаться частицы большего размера,
чем те, которые формируются только при 100°С. Был исследо-
ван [21] рост частиц в подщелоченных растворах чистой крем-
невой кислоты с последующим измерением размеров кремне-
земных частиц сферической формы методом рассеяния света.
Если необходимо повысить концентрацию золя без дальнейшего
увеличения размера частиц, то можно выпаривать золи с содер-
жанием 28—38 % S1O2 (в виде частиц диаметром 13—50 нм)
с одновременным добавлением золя точно такого же состава до
тех пор, пока не будет достигнута концентрация 52 % SiOg [24].
Ирани [25] предложил приготовлять способом «наращива-
ния» частицы с очень однородным распределением по размерам.
Исходным раствором служит или чистая вода, или «основной
золь» с однородными маленькими частицами, к которому при
60- 150°С добавляют силикат натрия и ионообменную смолу
в водородной форме при установленных заранее условиях.
В другой модификации данного способа [26] предусматри-
428
Глава 4
вается 'использование золя «активного» кремнезема, образую-
щегося непосредственно в результате ионного обмена при pH
2,5—5,5, а не приготовленного заранее в щелочной среде. Этот
золь-питатель затем добавляют к «основному золю» в процессе
выпаривания. Однако, вероятно, что отношение SiO2:Na2O дол-
жно непрерывно повышаться во время такого процесса. Сиппел
[27] утверждает, что, работая по приведенному выше способу,
можно получать золи низкой плотности, если к «основному
золю» с частицами размером 8—20 нм добавлять кремневую
кислоту и поддерживать при этом pH 8,5—9,5 за счет введения
раствора силиката натрия, что обеспечивает постоянство отно-
шения SiO2:Na2O. Другой способ приготовления предложили
Уэлдес, Бойль и Бобб [28], которые добавляли к чистой воде
«активный» кремнезем. Каммингс [29] указывает необходимые
специфические условия для получения частиц диаметром свыше
50 нм, формируемых из зародышей при добавлении кремневой
кислоты. Эгбрегт [30] в своей работе предложил использовать
циркулирование разбавленного раствора силиката натрия через
колонну, наполненную ионообменной смолой в водородной
форме, при введении с определенной скоростью еще некоторого
количества силиката натрия.
Как отмечалось в гл. 1, кремнезем может осаждаться на
многих подложках различного рода. Как только поверхность
какой-либо частицы покрывается монослоем SiO2, то в после-
дующем во всех случаях происходит отложение кремнезема на
кремнеземе. Так, например, непроницаемым кремнеземным
слоем могут покрываться частицы хромата свинца [31]. Уже
упоминалось покрытие кремнеземом пигмента TiO2, нашедшее
широкое применение.
Способы приготовления частиц размером
менее 10 нм
Первоначально технические золи приготовлялись с размером
частиц, превышающим 8 нм, и, следовательно, могли быть скон-
центрированы по крайней мере до 30 % SiO2. Такие золи вполне
транспортабельны, так как чрезвычайно стабильны против даль-
нейшего роста частиц при обычной температуре (см. рис. 3.52).
Впоследствии выяснилось, что для некоторых случаев должно
быть предпочтительнее использование частиц еще меньшего раз-
мера, и поэтому были разработаны золи с более низкими кон-
центрациями кремнезема. Однако при этом возникает проблема,
заключающаяся в том, что частицы размером меньше 8 нм, и
особенно меньше 5 нм, начинают самопроизвольно расти в кон-
тейнере, в котором они находятся, что’сопровождается соответ-
ствующими изменениями в их свойствах. Более того, поскольку
такие золи в общем случае можно сконцентрировать по крайней
Коллоидный кремнезем — концентрированные золи
429
мере до 15 %, частицы золя диаметром меньше 5 нм могут при
хранении частично агрегировать с определенными изменениями
свойств золя. К вопросу получения и стабилизации золей дан-
ного типа имеет отношение целый ряд патентов.
Александер [9] впервые приготовил очищенные от солей золи
с размером частиц 5—8 нм, для которых определил предельные
концентрации кремнезема и щелочи, требуемой для их стабили-
зации, в зависимости от размера частиц. Диаметр частиц свя-
зан следующей зависимостью со средним значением удельной
поверхности кремнезема:
Удельная
поверхность,
м2'/г
600
350
Диаметр, нм
4,6
7,9
Концентрация
SiO2, %
15-20
15-34
Отношение
SiO2: Na3O
20-200
34-340
Айлер [32] разработал способ приготовления золя с содер-
жанием 10 % SiOs и размером частиц -~3 нм. По этому спо-
собу предусматривалось добавлять к золю, размер частиц кото-
рого менее 3 нм, силикат натрия в таком количестве, чтобы вве-
сти не более 4 % SiO2 и чтобы концентрация ионов натрия не
превышала 0,4 н. Золь подвергали старению в течение 10 мин,
затем пропускали через колонну, заполненную сильнокислотной
ионообменной смолой в водородной форме, чтобы снова полу-
чить золь с pH 3,5. После этого повторяли весь процесс, под-
держивая комнатную температуру.
Для того чтобы повысить концентрацию кремнезема, но не
допустить роста частиц более 5—10 нм, раствор кремневой кис-
лоты подщелачивают силикатом натрия, а затем добавляют до-
полнительное количество кремневой кислоты, в то время как
воду удаляют вакуумной дистилляцией. При данных условиях
использование низкой температуры позволяет избежать чрез-
мерного роста частиц, несмотря на добавление к системе «актив-
ного» кремнезема [33]. По более ранней методике необходимо
было сформировать частицы до желаемого размера в разбавлен-
ном растворе, а затем сконцентрировать кремнезем за счет вы-
паривания воды и добавления еще некоторого количества раз-
бавленного золя. Так как добавляемые частицы кремнезема
имеют тот же самый размер, что и частицы, находящиеся в ис-
парителе, то никакого «наращивания» не происходит [34].
Использование аммиака в качестве стабилизирующего сред-
ства, несомненно, сводит к минимуму рост частиц. Уэлдес и
Деролф [35] утверждают, что золи с размерами частиц 2—3 нм,
содержащие вплоть до 12 % SiO2, получаются, когда система
стабилизируется смесью NaOH и NH4OH при отношениях
(NH4)2O:Na2O в интервале 25—150 и SiO2: (NH4)2O в интер-
430
Глава 4
вале 2—8. Вероятно, в этом случае адсорбция NH3 на поверх-
ности кремнезема понижает скорость растворения кремнезема
и, следовательно, замедляет рост частиц. Беркхеймер [36]
также считает, что золь стабилизируется в избытке аммиака, и
затем этот избыток удаляется в процессе выпаривания.
^Мак-Нелли и Розенберг [37] методом деионизации раствора
силиката натрия слабокислотной ионообменной смолой в водо-
родной форме (предварительно нейтрализованной в виде суспен-
зии при pH 6—7) приготовляли частицы кремнезема, размер
которых оценивался в 1—3 нм. Вероятно, благодаря обработке
смолы при pH 6—7 предотвращаются образование зародышей
кремнезема и рост частиц до большого размера, которые могли
бы сформироваться при более низком значении pH суспензии
в процессе предварительной нейтрализации смолы. Отношение
SiO2: Na2O этих золей составляло от 4: 1 до 40 : 1 при концент-
рациях 15—25 % SiO2. Возможно, что в отсутствие каких-либо
зародышей большего размера рассматриваемый золь стано-
вится необычно устойчивым по отношению к самопроизвольному
росту частиц, хотя высокая концентрация, достигающая 25 %,
наводит на мысль, что в последнем случае размер частиц дол-
жен превышать 3 нм.
Воссос [38] сделал авторскую заявку на золи с частицами
менее 5 нм, стабилизированные при pH 9—11 и содержащие до
25 % SiO2. Для того чтобы свести к минимуму рост частиц, ав-
тор использовал подщелоченный разбавленный золь, который
концентрировался в вакууме при температуре ниже 65°С.
Реутер и Ревен [39] приготовили 15 %-ный золь с части-
цами диаметром 7,5 нм из золя с частицами величиной 1—3 нм
посредством нагревания при pH 8,7 и проведения «наращива-
ния» при температуре 75°С.
Маротта [40] получил частицы размером 5—40 нм добавле-
нием раствора кремневой кислоты к разбавленному раствору
силиката натрия, нагревая смесь в течение нескольких часов
при 25—50°С, затем более длительно при 50—90°С и, наконец,
в течение еще более продолжительного периода при 70—100°С
с одновременным выпариванием воды. Очевидно, что такой спо-
соб включает в себя сочетание процессов дополнительного об-
разования зародышей и «наращивания» частиц.
Другие способы приготовления золей с частицами диамет-
ром менее 5 нм и стабилизирование таких золей рассматрива-
лись в разделе, посвященном полисиликатам. Золи с такими
небольшими по размеру частицами стабилизировались относи-
тельно большими количествами основания; при этом допуска-
лось, что характерные свойства растворимых силикатов или
полисиликатов преобладали над соответствующими свойствами
коллоидного кремнезема.
Коллоидный кремнезем, — концентрированные золи
431
Стабилизация системы для предотвращения
роста частиц (теория Пауля Йетса)
Как было показано в гл. 3, полисиликаты или золи с очень
небольшими по размеру частицами стабилизировались за счет
введения в систему достаточного количества щелочи. Йетс [41]
предположил, что стабилизированные щелочью золи устойчивы
не только по отношению к гелеобразованию, но также и в тер-
модинамическом понимании. Он указывал, что существуют тер-
модинамические факторы, предотвращающие самопроизвольный
рост частиц или их агрегирование и стабилизирующие высоко-
развитую поверхность раздела твердое тело—жидкость в случае
системы кремнезем—вода. Главным таким фактором, который
противодействует изменению свободной энергии, происходящему
при уменьшении площади поверхности в системе кремнезем—
вода, является сильная адсорбция жидкой фазы, стабилизирую-
щих противоионов или существование каких-либо иных адсорби-
рованных разновидностей на поверхности дисперсной фазы,
изменяющая таким образом значение свободной энергии поверх-
ности раздела.
Такой адсорбционный эффект можно легко понять при рас-
смотрении изменений свободной энергии, происходящих в трех-
компонентной системе SiO2— Н2О — NaOH на следующих гипо-
тетических обратимых изотермических стадиях, начиная со ста-
билизированного щелочью золя:
1. Десорбция щелочи NaOH с поверхности коллоидных частиц,
AFj.
2. Десорбция воды с поверхности коллоидных частиц, AF2 (сво-
бодная энергия смачивания поверхности).
3. Уменьшение поверхности коллоидных частиц до минимально
возможной площади, допускаемой плотностью коллоидной фазы,
AF3 (свободная энергия твердых коллоидных образований).
4. Возврат растворителя (воды) к поверхности коллоидных ча-
стиц, сократившихся до минимально возможной площади по-
верхности, AF4.
5. Возврат щелочи NaOH в рассматриваемую систему, AF3.
По определению
AF] + AF5— изменение свободной энергии в результате про-
исшедших адсорбционных процессов для системы NaOH — кол-
лоидные частицы;
AF2 + АГз + AF4— изменение свободной энергии поверхности раз-
дела коллоидных частиц для бинарной системы SiO2 — Н2О.
Если
AFj + AF5 = AF2 + AF3 + AF4 = О
432
Глава 4
то исходная коллоидная система, состоящая из трех компонен-
тов, находится в термодинамическом равновесии. Другими сло-
вами, золь оказывается термодинамически стабильным, если сво-
бодная энергия адсорбции гидроксида натрия на поверхности
коллоидных частиц равна свободной поверхностной энергии си-
стемы в отсутствие NaOH.
До сих пор все подобные золям коллоидные системы рас-
сматривались как термодинамически нестабильные.
В золях кремнезема свободная энергия поверхности раздела
аморфный кремнезем—вода оказывается равной ~50 эрг/см2
(см. рис. 3.32). Понижение свободной поверхностной энергии,
сопровождающее уменьшение величины поверхности, доходит
до 900 кал/моль поверхностных атомов кремния, если считать,
что на 1 нм2 приходится приблизительно 8 атомов кремния.
Если коллоидные частицы стабилизированы термодинамически,
то равновесная величина свободной энергии должна быть при-
мерно такого же порядка. Эта стабилизирующая энергия появ-
ляется в результате адсорбции ионов ОН ~ и противоионов Na+
на поверхности коллоидных частиц.
Йетс подсчитал количество щелочи, необходимой для стаби-
лизации частиц кремнезема (предотвращения их роста). Со-
гласно его расчетам, изменение свободной энергии, связанное
с ионизацией групп на поверхности кремнезема, зависит от кон-
станты кислотной диссоциации, на которую в свою очередь
влияют изменения концентрации соли и степени ионизации по-
верхностных групп. Изменение свободной энергии при постоян-
ных условиях записывается как
ДГ = а(-/?Пп^) (1)
где а — доля ионизированных поверхностных групп и К' — зна-
чение константы диссоциации в данный момент времени при дан-
ном значении а и при других фиксированных условиях в пред-
положении, что на поверхности содержится один моль силаноль-
ных групп.
Йетс вывел также формулу, раскрывающую функциональ-
ную зависимость константы К' от pH, концентрации ионов #Na+
и степени диссоциации поверхностных групп а (которая будет
подробно рассмотрена ниже в связи с обсуждением величины
заряда на поверхности частиц):
pH = р7( —/г 1g—0,74 lg (A-AW) (2)
где К — суммарная константа диссоциации и (А-Агха+)—про-
изведение активности ионов натрия на их нормальность.
Из данных по титрованию при фиксированной величине кон-
центрации ионов Na+ вычерчивается графически зависимость
Коллоидный кремнезем — концентрированные золи 433
—lg[(l— а)/а] от pH, из которой по наклону линии опреде-
ляется величина п, а из пересечения линии с осью ординат —
величина р/С Полученное из экспериментальных данных значе-
ние п оказалось равным 3,47.
Соответствующее выражение, которое включает в себя ве-
личину К', записывается в виде
pH = p/C'-Ig-*zJL (з)
где
РК' = р/< - (п - 1) 1g -----0,74 1g (Л • А\а+) (4)
Из уравнений (1) и (4) получается
AF = 2,3а7?7’ [-1g К - (п - 1) 1g -ЦтД - 0,74 1g (Л • AW)] (5)
Можно показать, что
где г — массовое отношение SiO2:'Na2O в рассматриваемой
системе и 5 — удельная поверхность кремнезема, м2/г. Йетс при-
нимал плотность кремнезема равной 2,3 г/см3.
Подставив числовые значения К, а и п, получим
AF = 3,38- 106 г 12,08 _ 2,47 rS-2430 _ 0)74 lg (Л . ^+)-1
I | J
(7)
Это уравнение выражает изменение свободной энергии в за-
висимости от числа молей поверхностных групп при любом
уровне ионизации или содержания соли в растворе.
Для того чтобы понизить удельную поверхностную свободную
энергию золя кремнезема путем увеличения размеров частиц или
степени их агрегации, необходимо изменить ионизацию поверх-
ностных групп на противоположную по знаку и возвратить ад-
сорбированные ионы в межмицеллярную жидкую фазу. При
этом формируется золь с более низкой величиной поверхности.
Согласно Йетсу, изменение- свободной энергии равно yS, где
у — величина удельной свободной энергии поверхности раздела
между кремнеземом и водой. Эта величина является функцией
размера частиц и, следовательно, удельной поверхности S.
Пусть изменение свободной энергии, происходящее в том
случае, когда частицы с диаметром D уменьшают свою поверх-
ность на величину AS в см2, составляет
AFo==_YoAS (8)
2 Заказ № 250
434
Глава 4
Аналогично для плоской кремнеземной поверхности
AFo=-YoAS (9)
и
A.FD-^F0=(y0-yD)^S (10)
Последнее выражение представляет собой разность между сво-
бодными энергиями коллоидных частиц диаметром D и той же
самой коллоидной фазы, но с плоской поверхностью. Эта раз-
ность должна равняться — /?7’1п(5л/5о), где SD и So — величины
растворимости кремнезема с искривленной и с плоской поверх-
ностями соответственно.
Йетс использовал уравнение, опубликованное Айлером
в 1955 г. [8]:
где D выражено в миллимикронах. Указанное уравнение было
основано на значении энергии поверхности раздела, равной
80 эрг/см2. Однако, как было показано в гл. 1 (см. также
рис. 1.10а и 3.32), более правильно принять значение равным
около 50 эрг/см2. Следовательно, уравнение (11) должно быть,
вероятно, записано в виде
Тогда
\FD-\F0=-RT-^- (12)
Из уравнений (8) и (10) имеем
(Yo - yD) AS = -2,4-^- (13)
IO/1 RT Yd —Yo + 2,4 daS (14)
AFd = -y'0AS-2,4-^- (15)
Однако диаметр D может быть выражен через удельную поверх-
ность S, учитывая плотность кремнезема, которую Йетс принял
равной 2,3 г/см3:
d==2^4 (16)
Коллоидный кремнезем — концентрированные золи
435
Тогда
-bFD = yobS + ^^S (17)
Так как при 25°С 7=298 К, /? = 1,987 кал/(град-моль), то
-\Fd = yo AS + 0.56S (18)
При yo = 5O эрг/см2 AS равно площади, занимаемой 6• 1023 ато-
мов кремния на поверхности кремнезема. Если допустить, что
на 1 нм2 приходится 8 атомов кремния, то значение S составит
7,5-108 см2. Если выразить значение энергии не в эргах, а в ка-
лориях, то можно записать
- kFD = 50 (2,39 • 10~8) (7,5 • 108) + 0.56S (19)
или
- \Fd = 896 + 0,56S
В таком случае величина —AFD, полученная из уравнения (19),
должна быть равна величине А/7, определяемой из уравнения
(7), а именно:
896 4- 0,56S = 3’3-8е1°6 [ 12*08 — 2,47 1g rS ~ff-30 —
1 rS L ’ » 2430
-0,74 1g (Л-AW)] (20)
Откуда г равно
3,38 • 106 £12,08 - 2,47 Ig[(rS - 2430)/2430] - 0,74 1g (А . lVNa+)}
Г = 5 (896 + 0,565)
(21)
При отсутствии посторонних солей концентрация ионов Na+
легко подсчитывается из значений концентрации кремнезема С3
и силикатного отношения г, так как
[SiO2]
[NazO] — Г
или
[Na20] =
Поскольку концентрация ионов натрия в стабилизированных зо-
лях редко превышает 0,1 н., то можно принять, что активность
приблизительно равна единице:
tNa+] = 2£_
2*
436
Глава 4
где С — молярная концентрация SiO2 в рассматриваемой си-
стеме:
3,38 • 106 {12,08 _ 2,47 lg [(rS - 2430)/2430] - 0,74 lg (2C/r)}
r~ 5 ( 896 + 0,565)
Для конкретных примеров при определенных значениях S и
С из последнего уравнения были подсчитаны значения отноше-
ний г. Для 15 %-ного золя с размером частиц 4,5 нм (600 м2/г)
отношение SiO2:Na2O составляет 47:1, а для 39 %-ного золя
с частицами диаметром 13,4 нм (200 м2/г) такое отношение
равно 170:1. (Ранее подсчитанные Йетсом соотношения
SiO2: Na2O были равны 32 : 1 и 115:1.)
В коммерческих золях с приведенными размерами частиц
фактически используются отношения, соответственно, равные
примерно 25: 1 и 100 : 1. Как указывалось в гл. 2, частицы диа-
метром ~16 нм, стабилизированные при отношении 100:1,
очень мало увеличились в размере за 20 лет. Однако частицы
размером 4—5 нм в некоторых случаях незначительно увеличи-
ваются в диаметре (на 10—20%) в течение примерно года, не-
смотря на то что они были стабилизированы при отношении
25:1. Тем не менее может иметь место множество отклонений
от теоретически расчитываемых соотношений SiO2:Na2O, ве-
роятно, из-за разнообразия распределения исходных частиц по
размерам.
Выше приведенные формулы скорее всего не применимы
к частицам, имеющим диаметр значительно ниже 7—8 нм, по-
скольку их повышенная растворимость приводит к появлению
заметных количеств силикат-ионов, что необходимо принимать
во внимание при рассмотрении подобных стабилизированных
систем. Вероятно, для таких случаев потребовалось бы боль-
шее количество стабилизирующей щелочи, чем это следует из
указанных уравнений, т. е. более низкие значения г.
Стабилизация для предотвращения агрегации
Концентрированные кремнеземные золи стабилизируют
! с целью предупреждения образования силоксановых связей
между частицами. Этого можно достичь, во-первых, за счет
образования ионных зарядов на частицах, что обеспечивает
удерживание таких частиц порознь из-за сил отталкивания, и,
во-вторых, путем адсорбции, в общем случае мономолекулярного
слоя инертного вещества, что позволяет отделить поверхности
кремнеземных частиц друг от друга настолько, чтобы предупре-
дить возможность прямого контакта силанольных групп между
собой. Последний случай называется «стерической» стабилиза-
цией.
Коллоидный кремнезем — концентрированные золи 437
Для значительных по своему размеру частиц, особенно при/
низких значениях pH, когда образование силоксановых связей!
протекает медленно, самопроизвольное связывание не харак-j
терно. Так, для частиц размером более 100 нм эти связи, по-
видимому, не возникают даже в концентрированных золях в пре-
делах всей области pH, если только золь не высушивают. При
таких больших размерах частиц, несмотря на возможное обра-
зование незначительного числа силоксановых связей в точках
контакта, указанные связи, вероятно, недостаточны, чтобы про-
тивостоять механическим напряжениям, имеющим место тогда,
когда пара подобных частиц вступает в столкновение с третьей
частицей при броуновском движении.
Седиментация, возникающая за счет силы тяжести, является
другой формой нестабильности при хранении золей. Благодаря
этому на дне контейнера формируется очень концентрированный
вязкий слой из частиц золей большого размера. В некоторых
случаях появляется отчетливо различимый граничный слой
между двумя слоями жидкости, что является подтверждением
формирования концентрированного коацервата. Наблюдения,
проведенные в течение 20 лет на серии 30 °/о-ных золей, стаби-
лизированных при отношениях SiOz:Na2O примерно в области
100—200, показали, что седиментация была явно выражена
только в том случае, когда диаметр частиц превышал 70 нм.
Золи с частицами меныцего размера, вероятно, оставались в ос-
новном в гомогенном состоянии вследствие конвекционных по-
токов, возникавших в сосуде, так как при хранении температура
колебалась в пределах 20—30°С. Роль конвекции при формиро-
вании расслаивающихся слоев в суспензиях кремнезема была
рассмотрена в работе [42].
Лишь один тип нестабильности не встречается в данной си-
стеме, а именно процесс кристаллизации. Как отмечал Уолтон
[43], чем выше степень пересыщения, тем меньше размер кри-
тического зародыша кристаллизации. Такой зародыш может
быть настолько малым по размеру, что не соответствует какой-
либо отдельной кристаллической фазе. Эта твердая фаза фор-
мируется затем структурно неупорядоченно. Когда кристаллизу-
ются простые соединения, имеется возможность быстрой перест-
ройки ионов, или молекул в системе для того, чтобы
удовлетворить требованиям минимума энергии. В случае же
кремнезема энергетическое различие между аморфным и кри-
сталлическим состояниями системы слишком мало. Более того,
для разрыва силоксановых связей в системе и ее перестройки
требуется высокое значение энергии активации. Поэтому не об-
наруживалось никаких кристаллических форм в золях или
гелях кремнезема даже после 25 лет старения при комнатной
температуре.
438
Глава 4
Стабилизация путем образования ионных зарядов
Основным механизмом стабилизации коммерческих золей
служит возникновение ионных зарядов на поверхности частиц
в присутствии щелочи. Однако полностью удовлетворительной
теории стабилизации золей до сих пор не создано. Основные
принципы стабилизации посредством возникающего вокруг ча-
стиц двойного ионного слоя были развиты Дерягиным и Лан-
дау [44] и Фервей и Овербеком [45]. Соответствующая теория
получила название теории «ДЛФО», и она применима, в част-
ности, к частицам сферической формы [46а]. Оттевил [466]
представил превосходное обобщение всех сил, оказывающих воз-
действие на стабильность дисперсных систем.
Неппер [47] суммировал условия стабильности коллоидных
систем, включая принципы и электростатической, и «стериче-
ской» стабилизации. Фундаментальное изучение вандерваальсо-
вых сил, возникающих между поверхностями аморфного кремне-
зема, было проведено Роулером [48]. Он измерил в условиях
высокого вакуума силы притяжения между двумя поверхностями
плавленого кремнезема, причем поверхности были покрыты тон-
кими пленками металлического хрома. Однако трудно перене-
сти полученные им результаты на водные системы кремнезема.
В обзоре Виссера [49] представлены константы Гамакера для
многих коллоидных систем, включая систему SiO2— Н2О.
Как указывал Китченер [50], по крайней мере при некото-
рых условиях, стабильность системы кремнеземных золей нахо-
дится в полном противоречии с теорией «ДЛФО». Так, при
pH 2, когда заряд на поверхности частиц кремнезема равен
нулю, частицы агрегируют наименее быстро и такой золь имеет
самую высокую стабильность во времени. Однако только в ще-
лочном растворе, когда заряд частиц наиболее высок, золи ока-
зываются постоянно устойчивыми. В последнем случае теория
двойного слоя применима.
Матиевич [51] обсудил возможность применения теории
«ДЛФО» к различным неорганическим золям. Для кремнезем-
ных золей наиболее важным фактором является природа элек-
тролита. Процесс адсорбции и образования стабильных комплек-
сов, на поверхности кремнезема настолько сильно влияет на ка-
тионы, что упомянутая теория в данном случае имеет неболь-
шое практическое значение. К тем же самым выводам пришли
авторы работы [52] в отношении коллоидной системы, содер-
жащей частицы ТЮ2.
Максимальная концентрация золей. Наибольшую трудность
представляет получение устойчивых высококонцентрированных
золей кремнезема, в которых возникает необходимость с точки
Коллоидный кремнезем — концентрированные золи
439
зрения экономической выгоды при продаже и транспортировке.
При этом никаких заметных изменений в свойствах золей не
должно происходить, по крайней мере, в течение года при обыч-
ных условиях хранения.
Коммерческие золи, по-видимому, стабилизируют при опти-
мальных значениях pH и концентрируют до максимального со-
держания, насколько это допустимо при выбранных размерах
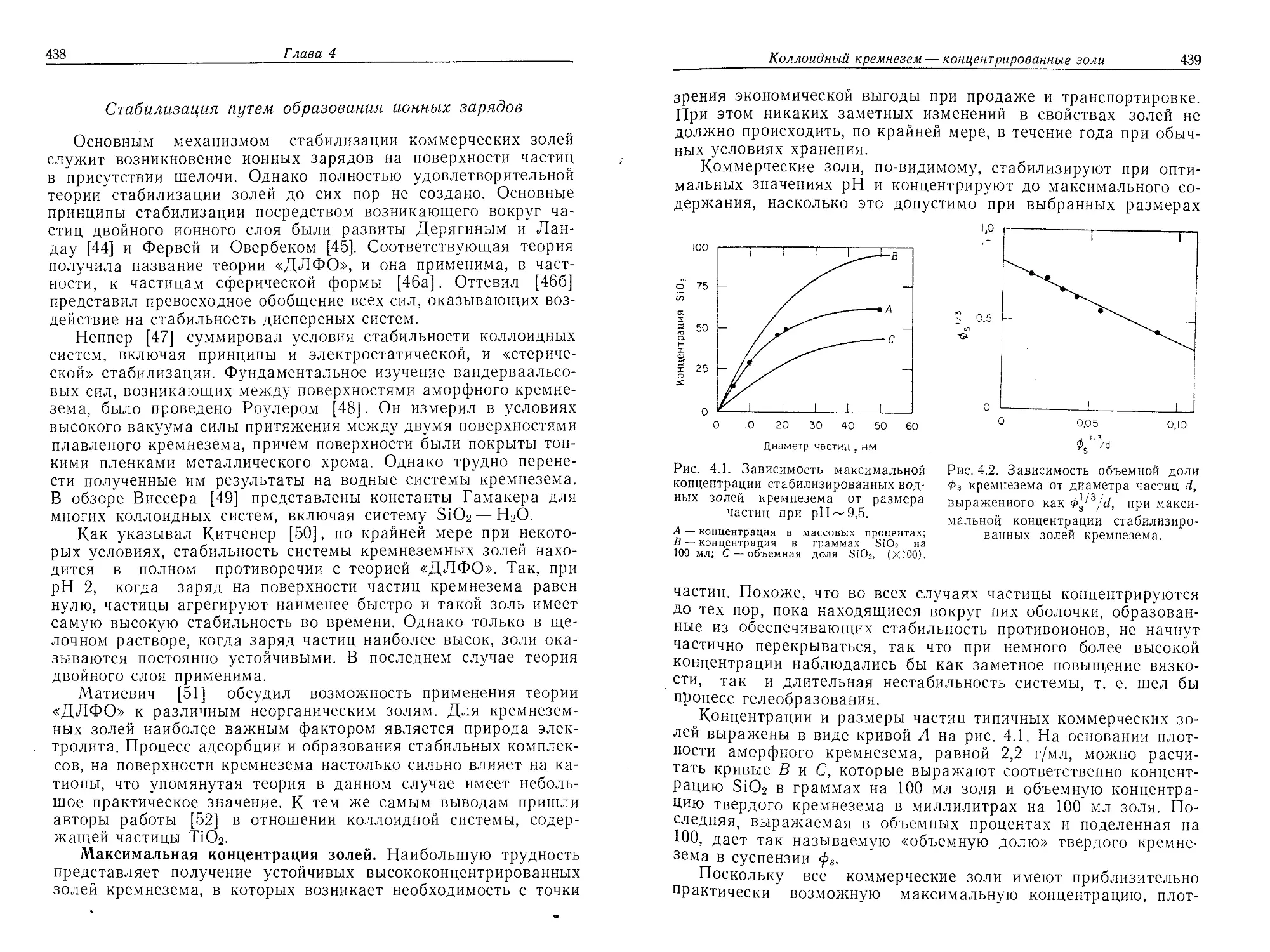
Рис. 4.2. Зависимость объемной доли
0s кремнезема от диаметра частиц it,
выраженного как при макси-
мальной концентрации стабилизиро-
ванных золей кремнезема.
Диаметр частиц, нм
Рис. 4.1. Зависимость максимальной
концентрации стабилизированных вод-
ных золей кремнезема от размера
частиц при pH ~ 9,5.
А — концентрация в массовых процентах;
В — концентрация в граммах SiO2 на
100 мл; С — объемная доля SiO2, (Х100).
частиц. Похоже, что во всех случаях частицы концентрируются
До тех пор, пока находящиеся вокруг них оболочки, образован-
ные из обеспечивающих стабильность противоионов, не начнут
частично перекрываться, так что при немного более высокой
концентрации наблюдались бы как заметное повышение вязко-
сти, так и длительная нестабильность системы, т. е. шел бы
процесс гелеобразования.
Концентрации и размеры частиц типичных коммерческих зо-
лей выражены в виде кривой А на рис. 4.1. На основании плот-
ности аморфного кремнезема, равной 2,2 г/мл, можно расчи-
тать кривые В и С, которые выражают соответственно концент-
рацию SiOa в граммах на 100 мл золя и объемную концентра-
цию твердого кремнезема в миллилитрах на 100 мл золя. По-
следняя, выражаемая в объемных процентах и поделенная на
100, дает так называемую «объемную долю» твердого кремне-
зема в суспензии </>«.
Поскольку все коммерческие золи имеют приблизительно
практически возможную максимальную концентрацию, плот-
440
Глава 4
ность упаковки частиц золя с их оболочками из окружающих
ионов, вероятно, во всех случаях примерно одинакова. Такая
максимальная плотность упаковки пропорциональна объемной
доле дисперсной фазы </>s, когда дисперсной фазой считается
частица с ее ионной оболочкой и диаметр частицы равен d:
Ь+2^)
где К — константа и da — толщина ионной оболочки или тол-
щина двойного слоя Гуи, образуемого вокруг частицы. Приве-
дение этого соотношения дает
^-(W13 + №)1/3 - 1 = 0
Отложив на осях величины^'и взятые изданных для
кривой С (рис. 4.1.), получаем линейную зависимость, показан-
ную на рис. 4.2. Из этого линейного графика получаем da =
= 2,43 нм и К= 1,68.
Для частиц больших размеров, когда толщина ионного слоя
много меньше диаметра частицы d, получаем ф8 = 1/К = 0,6. Это
показывает, что частицы в таких золях упакованы несколько
более плотно, чем это имеет место при произвольной упаковке.
Толщина da (в ангстремах) двойного слоя Гуи для данной
системы зависит только от ионной силы i [53]:
da = 3,05i°’s
Поскольку в коммерческих золях величина da в среднем со-
ставляет около 24 А, то ионная сила, рассчитанная из дан-
ного уравнения имеет среднее значение примерно 0,016 н. для
любых золей. Фактически в случае типичного стабилизирован-
ного золя с размером частиц 15 нм и содержанием '-—500 г
ЗЮ2/л обычное значение силикатного отношения SiO2: Na2O
составляет около 100:1, а содержание Na2O равно 5 г/л или
примерно 0,16 н. В подобных концентрированных золях боль-
шая часть ионов натрия должна находиться очень близко к по-
верхности (в так называемом слое Штерна), образуя концент-
рацию во внешнем двойном слое порядка 0,016 н.
В общем случае количество щелочи, необходимое для ста-
билизации системы, пропорционально площади поверхности
кремнеземных частиц в золе. Это количество составляет прибли-
зительно 1,0—1,5 молекул NaOH на 1 нм2 для большинства
золей, в которых допускается содержание примесей солей около
10~2 н. Однако если подобный золь почти очищен от соли (со-
держание соли менее 10-3 н.), то концентрация NaOH, необхо-
Коллоидный кремнезем — концентрированные золи 441
димая для стабилизирования системы, может приближаться
к теоретическому значению 0,016 н., а поверхностная концент-
рация при этом равняется 0,3 молекулы NaOH/нм2.
Добавление соли для понижения вязкости
Золи с размером частиц, большим чем 10 нм в диаметре, ко-
торые могут быть сконцентрированы до 500—700 г SiO2M, имеют
высокую вязкость. Понизить ее можно незначительным умень-
шением толщины двойного слоя при добавлении небольшого
количества соли, но не допуская при этом нарушения стабиль-
ности золя. Впервые этот способ был применен Аткинсом [54].
Он показал, что добавление таких солей, как Na2SO4 или NaCl,
к стабилизированным щелочью и очищенным от солей золям
с размером частиц в области 15—40 нм позволяет получать
концентрации 0,01—0,04 н. Такой способ дает возможность кон-
центрировать золи до значительно более высоких содержаний
кремнезема без чрезмерного увеличения вязкости, что имело бы
место, если бы соль не добавлялась. Позднее Воссос и Миндик
[55] в своей работе добавляли очищенную от металла соль
аммония или органического основания с анионами очень сла-
бой кислоты в количестве по крайней мере 0,003 мае. %, в пе-
ресчете на кремнезем. Так, добавление NH4HCO3 понижает
вязкость концентрированного золя без введения каких-либо
примесей, которые могли бы оказаться нелетучими при высу-
шивании кремнезема. Проводя исследования с целью понижения
вязкости концентрированного золя, Маротта [56] показал, что
при комбинировании 0,055—0,095 н. Na2SO4 с достаточным ко-
личеством силиката натрия для получения pH 8,8—9,9 форми-
руется стабильный золь, но если для установления необходи-
мого значения pH применяется NaOH, то происходит гелеоб-
разование.
Стерическая стабилизация
Вопросы теории стерической стабилизации золей рассмат-
ривались в работах [57, 58]. Были получены частично водные
золи кремнезема, которые затем стабилизировались исключи-
тельно за счет адсорбции неионных молекул. Типичным при-
мером служит 35 %-ный золь, приготовленный Лувиси [59].
Этот золь был свободен от соли и имел pH 3—4,5 при содер-
жании 30—90 % одноатомного спирта, например изопропило-
вого. При pH 4—4,5 добавление глиоксаля, очевидно, оказывало
стабилизирующее воздействие на золи кремневой кислоты и од-
новременно способствовало переведению в нерастворимую форму
таких водорастворимых полимеров, как желатин или поливини-
442
Глава 4
ловый спирт [60]. Хорошо известно, что золи кремневой кис-
лоты, освобожденные от соли, при низких значениях pH оказы-
ваются более стабильными в присутствии веществ,- способных
к образованию водородных связей, таких, например, как смеси
низших спиртов с полиэфирами. Без сомнения, и более кон-
центрированные золи могут быть стабилизированы подобным
образом. Однако максимальная устойчивость достигается лишь
при удалении всей воды. Такие золи, обычно называемые орга-
нозолями, будут рассмотрены позже в настоящей главе. В ти-
пичном процессе предусматривается полное удаление катионов
и анионов из кремнеземного золя при поддержании значения
pH в интервале 1—7, а затем частичное или же полное заме-
щение воды на спирт, простой эфир или кетон [61].
Стерическая стабилизация была применена и к частицам
очень небольшого размера в качестве дополнения к ионной
стабилизации. Так, Йетс [62] стабилизировал золи с очень ма-
лыми по размеру частицами, комбинируя неорганическое или
же органическое основание сводорастворимым неароматическим
полиокси- или неполным эфиром многоатомного спирта, напри-
мер поливинилового спирта. Некоторая стерическая стабили-
зация имеет место также, вероятно, и тогда, когда присутствует
катион органического основания, такой, как (CH3)4N+, по-
скольку, по данным Уолтера [63], золь кремнезема этого типа
можно выпаривать до получения сухого порошка, способного
повторно самопроизвольно диспергировать в воде. Такие золи
можно также подвергать повторному диспергированию после
их замораживания.
Стерическая стабилизация за счет нанесения монослоя при-
соединенных водородными связями молекул воды [64] должна
иметь место в случае золей, которые оказываются устойчивыми
по крайней мере в течение нескольких месяцев при низких зна-
чениях pH. Существенным фактором является чрезвычайно низ-
кая концентрация ионных примесей, не считая кислоты, которая
необходима для поддержания низких значений pH. Щелочь
имеет тенденцию к внедрению в частицы и, медленно выделяясь
из них, повышает pH и вызывает тем самым гелеобразование,
если только не принимаются специальные меры предосторож-
ности. Миндик и Ревен [65] неоднократно подвергали золь про-
цессам деионизации, старения и повторной деионизации путем
воздействия смесью, состоящей из катионита и анионита, вплоть
до тех пор, пока значение pH не поднималось выше 3 во время
хранения. Необходимо отметить, что во всех коммерческих сили-
катах присутствуют следы алюминия, которые могут находиться
на поверхности кремнезема в виде SiA10PH+ в достаточном ко-
личестве, чтобы способствовать стабильности системы при pH 3.
Коллоидный кремнезем — концентрированные золи
443
Пористые частицы
Первичные частицы коллоидного кремнезема обычно не по-
ристы, если они сформированы или выращены в щелочном раст-
воре и в особенности если они образованы при повышенной тем-
пературе (либо в водном растворе выше 60°С, либо сконденси-
рованы из газовой фазы при очень высокой температуре). Плав-
леный кремнезем (стекло) имеет плотность около 2,20 г/см3.
При измерениях плотности порошков различных типов коллоид-
ного аморфного кремнезема методом погружения в ксилол
с учетом поправки на небольшое содержание поверхностных
ОН-групп были получены следующие значения плотностей (счи-
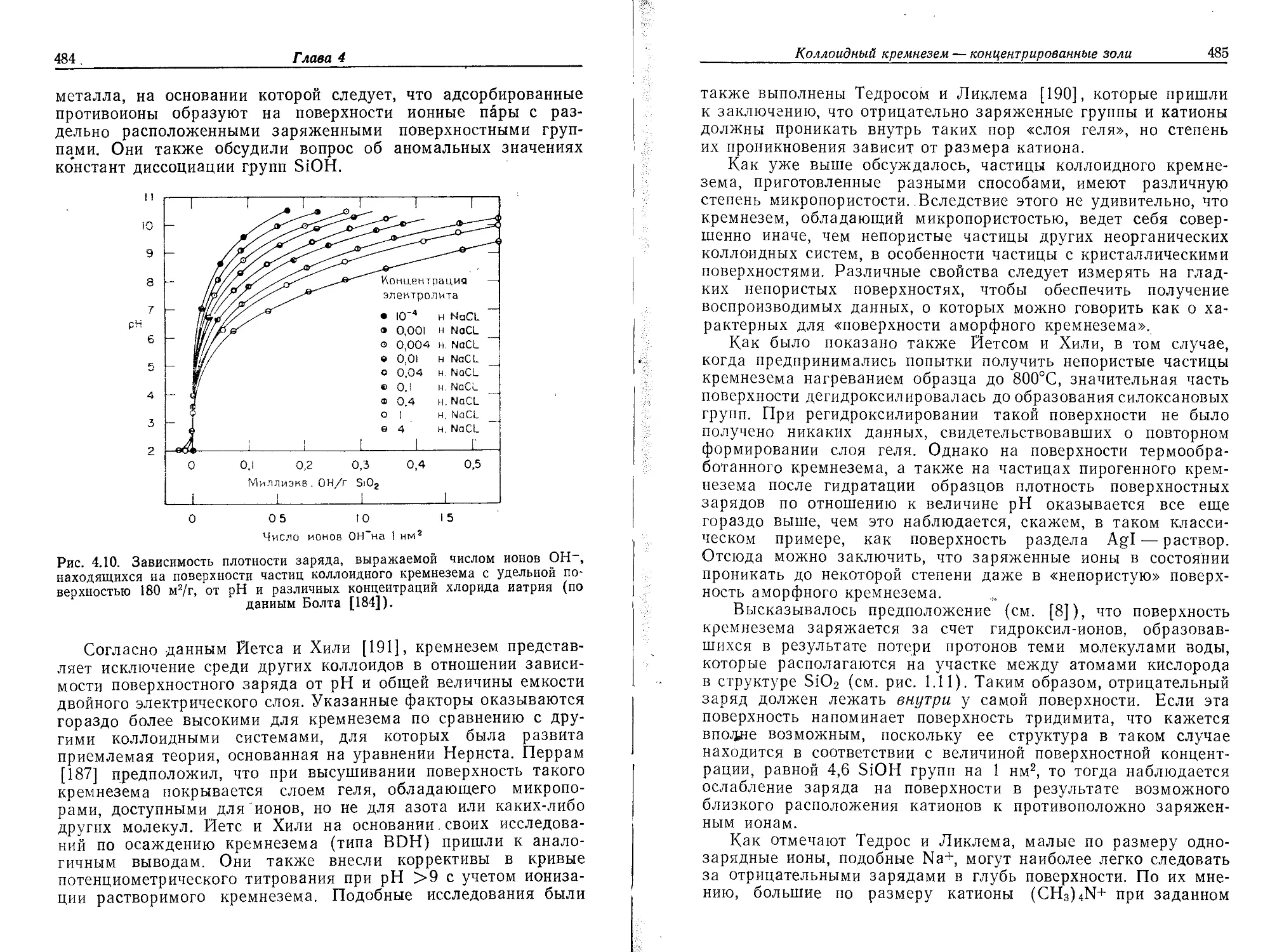
тается, что эквивалентная ОН-группам вода имеет плотность
1,0 г/см3):
Тип кремнезема • Плотность, г/см3
Коммерческие золи 2,2—2,3
Коммерческий силикагель (крупиопо- 2,22
ристый)
Конденсированный из паровой фазы 2,16
Осажденный из горячего раствора 2,0
Для всех таких порошков величина поверхности, определяемая
по адсорбции азота, примерно соответствует значениям, подсчи-
танным из распределения частиц по размерам, если плотность
кремнезема принимается равной 2,2 г/см3, а кривая распреде-
ления строится по данным, полученным методом электронной
микроскопии [66]. Из этих результатов можно заключить, что
частицы рассмотренного типа имеют пористость (по отношению
к молекулам азота) менее чем 5—10 объемн. %.
С другой стороны, могут быть приготовлены золи кремне-
зема, в которых дискретные частицы оказываются довольно
пористыми. Обычно такие частицы формируются при комнат-
ной температуре в воде в результате равномерного агрегирова-
ния гораздо меньших по размеру первичных частиц диаметром
не более 5 нм. Радчевский и Рихтер [67] получили путем гид-
ролиза SiCU с последующим старением нейтрализованного раст-
вора пористые частицы диаметром 200 нм. Штобер, Финк и
Бон [68] приготовили аналогичные частицы гидролизом этил-
силиката в смеси аммиака, воды и спирта. Наиболее детально
выполненные исследования проведены на пористых частицах
кремнезема, которые приготовлялись растворением измельчен-
ного кремния в воде. Балтис [69] обнаружил, что тонкоизмель-
ченный порошок кремния, промытый разбавленным раствором
HF для удаления оксидной пленки, вступает в реакцию с водой
с выделением водорода в присутствии катализатора — органиче-
ского основания. Это позволяет получать прозрачные разбав-
ленные золи с диаметром частиц 8—15 нм, которые можно
444
Глава 4
затем концентрировать выпариванием. Такие частицы оказались
непористыми. Балтис затем обнаружил, что в присутствии ам-
миака (вместо органического основания) происходит формиро-
вание частиц большего размера вплоть до 35 нм. К тому же
было показано, что такие частицы являлись пористыми. Эти
частицы деполимеризовались или растворялись в разбавленной
щелочи с более высокой скоростью, чем непористые частицы
такого же диаметра. Суммарная поверхность пористых частиц,
измеренная по адсорбции азота (методом БЭТ), оказалась в два
или три раза больше по сравнению с их наружной поверхностью,
измеренной путем адсорбции красителя метилового красного из
раствора бензола.
В дальнейшем Мак-Миллан [70] детально исследовал эту ре-
акцию и приготовил частицы диаметром более 100 нм, которые
оказались высокопористыми. Меняя условия проведения реак-
ции, можно было изменять как пористость во внутренней и в на-
ружной областях подобных частиц, так и их размер в интервале
100—500 нм и получать частицы сферической формы с почти
равными диаметрами.
Изучение структуры таких частиц, выполненное измерением
скорости деполимеризации (растворения) в щелочи, показало,
что их составной частью были первичные частицы диаметром
4 нм.
Свежеприготовленные частицы, получаемые в результате про-
ведения реакции при 25—30°С, оказывались высокопористыми
по отношению к азоту, а их плотность составляла около 1,3 г/см3,
что соответствовало пористости 40 % по объему. Сферические
частицы диаметром 200 нм с величиной наружной удельной по-
верхности, равной 15 м2/г, получались в результате высушива-
ния золя до состояния порошка. Однако внутренняя площадь по-
верхности, измеренная перед высушиванием по адсорбции ионов
ОН~, составила 745 м2/г, что соответствовало размеру предельно
малых, или первичных, частиц около 4 нм в диаметре.
Когда золь, имеющий такие пористые частицы, нагревали
при 90°С и pH 9,8, то поверхность, определяемая по адсорбции
азота, понижалась до 22 м2/г. Это указывает на то, что диаметр
внутренних пор уменьшался настолько, что молекулы азота уже
не могли проникать в них, и, следовательно, эти молекулы ад-
сорбировались только лишь на наружной поверхности. Тем не
менее микропористость все еще существовала, поскольку пло-
щадь, определяемая по адсорбции ионов ОН , оказалась равной
707 м2/г. При нагревании золя в течение более продолжитель-
ного времени при pH 10 тонкие поры могли в дальнейшем за-
крываться, так что поверхность частиц становилась совершенно
непроницаемой. Без сомнения, некоторое количество воды и ще-
лочи удерживалось внутри частиц, как в ловушке.
Коллоидный кремнезем — концентрированные золи
445
Коллоидные, по-видимому, пористые частицы были приготов-
лены Каммингсом [29] в виде золя молочного цвета добавле-
нием раствора кремневой кислоты в горячий раствор силиката
натрия, содержавшей 0,35—0,4 н. ионов натрия. По мере того
как концентрация ионов натрия понижалась в результате до-
бавления кремневой кислоты, последняя частично агрегировала
за счет окружения частиц кремнезема ионами натрия. Таким
образом, при осаждении кремнезема на подобных небольших
агрегатах образуется золь с размерами частиц 20—250 нм.
Айлер наблюдал образование золя с пористыми коллоид-
ными частицами в том случае, когда раствор силиката натрия
с отношением SiO2 : Na2O = 3,25 : 1,0, разбавленный до 4 % SiO2,
нагревали до 90°С и затем медленно нейтрализовали 60 %-ную
щелочь добавлением кислоты H2SO4. Требовалась более высо-
кая температура, так как при проведении нейтрализации при
60°С получался пластичный прозрачный гель.
Механизм этого процесса, по-видимому, можно представить
следующим образом. В исходном растворе силиката натрия с от-
ношением 3,25 около одной трети кремнезема находится в виде
коллоидных частиц диаметром 1—2 нм, а остальная часть —
в виде ионов HSiO-T. При нейтрализации части щелочи кремне-
зем, образующийся из ионов HSiOT, осаждается на коллоидных
частицах, которые при высокой температуре увеличиваются
в размере до 3—4 нм. В то же самое время концентрация суль-
фата натрия составляет 0,3—0,4 н., что превышает критическое
значение коагуляции. Вследствие этого, несмотря на весьма вы-
сокий заряд на поверхности, частицы агрегируют в пористые
сферические образования диаметром 15—20 нм. Тот факт, что
эти сферы образовывались из плотных кремнеземных частиц
размером до 4—5 нм, подтверждался величиной удельной по-
верхности, равной 600 м2/г, измеренной по адсорбции ионов ОН~
еще до высушивания таких сфер. Но после высушивания удель-
ная поверхность, измеренная по адсорбции азота, составила
только 160 м2/г, что соответствовало плотным сферическим обра-
зованиям диаметром 17 нм. Следовательно, исходные сфериче-
ские агрегаты дают усадку, составляющие их первичные частицы
плотно упаковываются и внутренняя пористость исчезает.
В том случае, когда сформированные в самом начале влаж-
ные частицы подкисляются до pH 2 и промываются пропанолом
перед их высушиванием, внутренняя пористость должна сохра-
няться в значительной степени.
Процесс агрегации небольших частиц кремнезема происходит
в несколько стадий. Получившиеся большие частицы в свою оче-
редь превращаются в еще большие по размеру образования, ко-
торые были описаны Гриром [71]. Он показал наличие стадий
446
Глава 4
на частицах размером 1,5, 10, 60 и 331 нм и вывел следующее
уравнение:
d — диаметр сферической частицы на стадии роста п, а — диа-
метр таких частиц на первоначальной стадии роста иг — по-
стоянная, равная 6.
Если предположить, что а=1,5 нм, то тогда размеры сфери-
ческих образований на разных стадиях должны иметь значения
9, 54 и 324 нм. Условия, вызывающие формирование таких
агрегатов, не были раскрыты автором.
Микропористость в коллоидных частицах в некоторых слу-
чаях может быть продемонстрирована методом малоугловой
дифракции рентгеновских лучей. Когда определяемый таким
методом размер частиц оказывается значительно меньшим, чем
размер, подсчитанный из величины удельной поверхности, ко-
торая измерялась по адсорбции азота или наблюдалась по элек-
тронно-микроскопическим снимкам, то это означает, что подоб-
ные частицы составлены из еще меньших дискретных единич-
ных образований, их упаковка так плотна и получающиеся при
этом поры настолько малы, что молекулы азота в них не про-
никают [72]. Большая часть гелей состоит из первичных частиц,
пронизанных порами, доступными молекулам азота. Однако
Ледерер, Шурц и Янцон [73] сообщили, что, по-видимому, в по-
лученных ими определенных разновидностях гелей кремнезема
наблюдалась некоторая «внутренняя» поверхность, поскольку
соответствующие высокие значения гидратации для таких гелей,
равные 0,15—0,26 г НДт SiOa, должны означать наличие высо-
кой пористости.
Удлиненные частицы
Огино и Куронума [74] сообщили о получении необычных
частиц кремнезема удлиненной или веретенообразной формы.
Эти авторы первыми проследили за формированием раздельных
частиц кремнезема коллоидных размеров с формой, отличаю-
щейся от сферической. Такие частицы получались при медлен-
ном добавлении ионообменной смолы в Н+-форме к разбавлен-
ному раствору силиката натрия (SiCh: Na2O = 3,52) при 40°С
до тех пор, пока значение pH не падало до 3, с последующим
добавлением аммиака для повышения pH до 8—9 и нагреванием
образца при 80°С в течение 1 ч.
Коллоидный кремнезем — концентрированные золи
447
Частицы с некремнеземной сердцевиной
До тех пор пока поверхность коллоидных частиц состоит из
кремнезема, такие частицы обладают теми же самыми свойст-
вами, что и частицы, состоящие сплошь из кремнезема. Как
было показано Дилером [18] и рассмотрено в гл. 1, кремнезем
можно осаждать на разнообразных поверхностях. Для того
чтобы покрыть кремнеземом частицы золя, несущие на поверх-
ности положительный заряд и подобные частицы БегОз или
AI2O3, необходимо прежде всего изменить знак заряда на обрат-
ный посредством добавления разбавленного золя в разбавлен-
ный (10 %-ный) раствор силиката натрия при интенсивном
перемешивании. Возможно также изменение знака заряда на
обратный путем введения перед смешиванием с раствором си-
ликата натрия вещества, способного вызывать образование хе-
латных соединений, такого, например, как цитрат. При этом по-
верхность покрывается отрицательно заряженным молекуляр-
ным слоем адсорбированного силиката, к которому уже может
присоединяться слой SiO2. Так были приготовлены [75] частицы
оксида тория с нанесенным слоем кремнезема. Стабилизирован-
ные золи оксида тория с частицами размером свыше 50 нм со-
держали вплоть до 60 % твердого оксида, а кремнеземное по-
крытие составляло около 50 масс. % от массы сердцевины из
оксида тория. Фитч, Санчез и Веник [76] заявили о получении
частиц оксида тория и оксида урана, покрытых кремнеземом.
Кинетика адсорбции кремнезема на поверхности частиц из
оксида тория изучалась в автоклавных условиях при 100—
200°С [77].
Методы приготовления золей
Было предложено и в настоящее время применено множество
процессов приготовления коллоидного кремнезема из имеющих
низкую стоимость растворов силиката натрия. Золи также изго-
товляются из способных гидролизоваться соединений, таких, на-
пример, как этилсиликат или тетрахлорид кремния. Чтобы по-
лучить золи, устойчивые при достаточно высокой концентрации,
необходимо вырастить частицы до некоторого определенного
размера в щелочных условиях, при которых такие частицы остаГ
ются отрицательно заряженными и, следовательно, не будут флок-
кулировать или образовывать гель. К тому же эти частицы нё,
должны быть пористыми.
Коллоидный кремнезем формируется в природных условиях,
когда вода насыщается кварцем при высоких температуре и
давлении, а затем такой раствор отделяется и охлаждается.
448
Глава 4
Подобные золи содержат всего лишь несколько десятых долей
процента SiC>2, но их можно легко сконцентрировать ультра-
фильтрацией. Получение коллоидного кремнезема этим способом
изучалось Китахара и Ошима [78]. Кроме того, они исследовали
скорость растворения частиц кремнезема при разбавлении золя.
Нейтрализация растворимых силикатов кислотами
Когда разбавленный раствор силиката натрия частично ней-
трализуется кислотой до pH 8—9, то получается не гель, а золь
кремнезема, если концентрация образуемой соли натрия оказы-
вается менее чем ~ 0,3 н., и процесс нейтрализации выпол-
няется при повышенной температуре, так что частицы начинают
расти, как только их размер достигнет нескольких миллими-
крон. Таким образом, 3 %-ный золь кремнезема можно приго-
товлять путем частичной нейтрализации разбавленного раствора
коммерческого силиката натрия кислотой в соответствии с услц-
виями, запатентованными Александером, Айлером и Уолтером
[79]. Из золя, состоящего из кремнеземных зародышей, вначале
удаляют ионы натрия, пропуская через ионообменную смолу
раствор силиката натрия с содержанием 2,2 % SiC>2 вплоть до
момента, когда массовое отношение SiO2:Na2O становится рав-
ным 85:1. Такой разбавленный золь затем нагревается при
100°С примерно в течение 10 мин с целью формирования заро-
дышей желаемого размера . После этого в золь одновременно
вливают разбавленные растворы силиката натрия и серной кис-
лоты при энергичном перемешивании смеси в течение 8 ч, под-
держивая рН-~9 и температуру 95°С. Концентрация ионов нат-
рия не должна превышать 0,3 н., так как в противном случае
будет происходить агрегация частиц. При указанных условиях
на кремнеземных зародышах осаждается кремневая кислота,
образующаяся при добавлении силиката натрия и серной кис-
лоты. При этом происходит формирование частиц размером, на-
пример, 37 нм.
В другом патенте Александер и Айлер [80] описали выде-
ление частиц, сформированных по рассмотренному выше про-
цессу, путем их коагуляции с ионами металла (например, каль-
ция), промывания осадка для удаления соли натрия и после-
дующей пептизации продукта до получения более концентриро-
ванного золя кремнезема посредством удаления ионов кальция
ионным обменом.
При приготовлении золя нейтрализацией разбавленного раст-
вора силиката кислотой существенным фактором является необ-
ходимость его быстрого перемешивания, чтобы образовавшаяся
смесь не находилась бы при pH 5—6 в течение заметного про-
Коллоидный кремнезем — концентрированные золи 449
межутка времени, поскольку при таком pH почти мгновенно про-
исходит формирование гелей кремневой кислоты. Поэтому тре-
буется, чтобы серная кислота и силикат натрия интенсивно пе-
ремешивались, причем, чтобы предотвратить локальное геле-
образование, необходим избыток серной кислоты или силиката
натрия. Даже в наиболее критичной области pH —7 способ пе-
ремешивания, запатентованный Армстронгом и Каммингсом
[81], позволяет получать однородный раствор кремневой кис-
лоты с концентрациями вплоть до 20 % SiO2. Такой раствор
даже при наиболее неблагоприятных pH не переходит в гель
по крайней мере в течение нескольких минут.
Другим способом, применяемым при приготовлении коллоид-
ного кремнезема по реакции силиката натрия с серной кисло-
той, является формирование кислого золя и осаждение соли
натрия в сильнокислой среде. Поликремневая кислота временно
устойчива при pH 2, и если натриевая соль кислоты, исполь-
зуемой для нейтрализации силиката натрия, оказывается в до-
статочной мере нерастворимой, то ее можно осадить и отделить.
Как только соль натрия отделяется от кислого золя, то появ-
ляется возможность подщелачивания поликремневой кислоты,
необходимого для роста коллоидных частиц и стабилизации по-
лучаемого продукта, или же, по-видимому, золь можно исполь-
зовать в других процессах выращивания часТиц кремнезема до
желаемого размера. Так, Тейчер [82] проводил нейтрализацию
силиката натрия кислотой с целью получения кислого золя, со-
держащего способную смешиваться с ним органическую жид-
кость, такую, например, как спирт, вызывающий осаждение
соли. В более раннем способе, предложенном Уайтом [83], суль-
фат натрия осаждается из золя, который был приготовлен из си-
ликата натрия и серной кислоты с добавлением ацетона. Мар-
чегет и Гендон [84] вместо этого использовали в своей работе
вещества, позволяющие формировать чрезвычайно мало рас-
творимую соль натрия, такие, например, как продукт реакции
между сульфит-ионами и глиоксалем, причем сульфит в этом
случае действует как кислота при нейтрализации щелочи в си-
ликате. В результате реакции силиката натрия с щавелевой кис-
лотой получается кислый золь, из которого осаждается гидроок-
салат натрия, с сохранением в растворе концентрации ионов
натрия приблизительно 0,13 н. при 15°С [85]. Оставшуюся соль
можно затем удалить ионным обменом [86].
Электродиализ
Коллоидный кремнезем приготовляется различными спосо-
бами, в том числе электродиализом, посредством которого ионы
натрия удаляются из раствора силиката натрия и образуется
3 Заказ № 250
450
Глава 4
золь. Такие способы были рассмотрены в обзоре Айлера [8],
но ни в одном случае не получался устойчивый продукт. Санчез
[87] и Айлер [88] запатентовали способы элёктродиализа раст-
вора силиката щелочного металла с непрерывным удалением
катионов вплоть до получения золя.
Электродиализ может в конечном счете заменять ионный об-
мен при приготовлении коммерческих' золей, так как щелочь,
кислород и водород можно регенерировать, и, следовательно,
необходимо удалять гораздо меньшее количество отработанной
воды.
В способе, предложенном Айлером, используются три камеры.
Раствор серной кислоты циркулирует вокруг свинцового анода
в анодной камере. В катодной камере, в которой производится
гидроксид натрия, щелочь циркулирует вокруг катода, сделан-
ного из стали. Камеры располагаются с противоположных сто-
рон двух параллельных, близко расположенных катионообмен-
ных мембран, между которыми раствор, используемый в дан-
ном способе, быстро циркулирует при температуре 60—90°С.
Такой раствор представляет собой золь кремнезема, содержа-
щий приблизительно 0,05 н. Na2SO4, добавляемого в качестве
проводящего или дополнительного электролита. Раствор сили-
ката натрия добавляется во входящий поток с целью повыше-
ния pH до 9,5. Плотность потока и скорость его течения регу-
лируются таким образом, чтобы на выходе потока из ячейки
значение pH не было ниже 8. Выделяемый в свободном состоя-,
нии кремнезем осаждается на частицах кремнезема, которые та-
ким образом растут до желаемого размера. С помощью такого
способа можно непосредственно приготовлять 25 %-ный'золь
кремнезема с частицами размером 15 нм. После этого электро-
лит удаляется ионным обменом, устанавливается значение pH,
необходимое для оптимальной устойчивости золя, и золь кон-
центрируется до содержания 30—50 % SiO2.
При таком способе кислота по существу не расходуется, за
исключением небольшого ее количества, требуемого в начале
каждой очередной загрузки для нейтрализации разбавленного
раствора силиката натрия (0,5 % SiO2) до pH 9 при температуре
60—90°С и получения зародышей кремнезема для инициирова-
ния процесса. Чтобы свести к минимуму расход энергии и избе-
жать отложений кремнезема между мембранами, зазор между
ними должен быть небольшим и равномерным. Вода добав-
ляется в анодную камеру, после чего она медленно подается
в катодную камеру, из которой постоянно отбирается раствор
гидроксида натрия. Анолит и католит циркулируют от соответ-
ствующей электродной камеры по направлению к разделяющим
перегородкам для удаления таких газов, как кислород и во-
дород.
Коллоидный кремнезем — концентрированные золи
451
Был запатентован способ [89], в котором для удаления нат-
рия из силиката натрия применяется ртутный катод. Никакого
дополнительного электролита в этом случае не используется, так
что, по мере того как щелочь расходуется, вследствие высокого
электрического сопротивления золя требуется повышение затра-
чиваемой мощности. Было предложено [90] трехкамерное уст-
ройство с использованием ионообменных мембран для приготов-
ления золей методом электродиализа. Здесь также никакого
дополнительного электролита не использовалось. Конечный золь
имел pH 2—3.
В разновидности электролитического процесса, запатентован-
ной Триппом [91], используется растворение кремниевого анода
в спирте, в котором содержится катализатор — соль металла
(такая, например, как сульфат меди), необходимая для получе-
ния кремнеземного органозоля.
При исследовании переноса кремнезема через мембраны
в процессе электродиализа Боари и др. [92] обнаружили, что
никакого переноса или осаждения кремнезема не происходит
до тех пор, пока pH соответствует значениям, при которых мо-
гут существовать ионы HSiOT-Это согласуется с наблюдениями
[88], указывающими на то, что электродиализ необходимо вы-
полнять при рН<9,5 для того, чтобы не осаждать кремнезем на
мембране.
Ионный обмен
Работа Бёрда [4], в которой натрий удаляется из силиката
натрия ионным обменом с последующим концентрированием
золя выпариванием при атмосферном давлении, ставшая пионер-
ской в этой области, привела к получению стабилизированных
золей кремнезема, содержащих приблизительно 20 % SiOs.
В дальнейшем [6] удалось осуществить контроль размера фор-
мируемых частиц. Другие усовершенствования, введенные Алек-
сандером [9] и Эткинсом [10] в отношении величин допусти-
мой концентрации соли и оптимального содержания щелочи,
дали возможность получить целый ряд концентрированных зо-
лей кремнезема, охватывающий широкую область размеров
частиц. В указанных работах золи кремнезема приготовляли
пропусканием относительно разбавленного раствора силиката
натрия через слой ионообменной смолы. Получали достаточно
очищенный от натрия кислый золь кремнезема, который затем
стабилизировали, и выращивали частицы до желаемого размера.
Второй способ, разработанный Уолтером и Айлером [93], за-
ключался в том, что ионообменную смолу в водородной форме
и силикат натрия добавляли к слабощелочной водной среде при
3*
452
Глава 4
pH около 9 и повышенной температуре. При этих условиях,
обеспечивающих отсутствие агрегации, частицы кремнезема ро-
сли непрерывно и непосредственно получались сравнительно
концентрированные золи с содержанием' 10—15 % кремнезема.
Миндик и Ревен [94] получили патент на еще один способ при-
готовления относительно концентрированного 12 %-ного кислого
золя поликремневой кислоты. По этому способу охлажденный
раствор силиката натрия пропускали через ионит; следова-
тельно, концентрированный золь формировался при ^сниженной
температуре, что позволило избежать гелеобразования.
Было предложено множество вариантов получения золя ионо-
обменным способом. Дирнбергер [95] показал, что более кон-
центрированные золи можно приготовить пропусканием раствора
силиката натрия снизу вверх через слой суспензии ионообмен-
ной смолы; тем самым удается избежать гелеобразования. Дру-
гие варианты ионообменного способа представлены в ряде па-
тентов [96].
Согласно Миндику и Ревену [65], оставшиеся следы электро-
литов можно удалять из золя кремнезема, полученного каким-
либо из стандартных способов, нагревая золь и повторно про-
пуская его через ионообменную смолу в водородной форме
с целью извлечения щелочи, высвобождаемой из частиц.
Использование катионита в NHi1 -форме дает возможность
удалять натрий из раствора силиката натрия без понижения pH
в какой-либо части раствора для предотвращений агрегации
частиц. Уолтер обнаружил, что при отношении SiO2:Na2O
3,25: 1 раствор силиката натрия, содержащий вплоть до 6 %
SiO2, не превращался в гель при пропускании его через колонну
со смолой в МН^-форме. Дополнительные количества силиката
натрия можно прибавлять к вытекающему щелочному потоку,
а затем образующийся раствор вновь обрабатывать тем же
катионитом [97].
Для получения золей с очень небольшим размером частиц
Шеннон [98] добавлял силикат натрия к кислой суспензии, при-
готовленной из ионообменной смолы, до тех пор, пока концент-
рация кремнезема не достигала 8%. Затем извлеченный золь
подщелачивался прибавлением NaOH и NH3 для того, чтобы
на частицах' SiO2 оказались 1 % Na и 3 % NH3.
При приготовлении кремневой кислоты пропусканием раст-
вора силиката натрия с содержанием более 3—4 % SiO2 через
слой ионообменной смолы в водородной форме внутри пор ис-
пользуемой смолы образуется гель кремнезема. Это приводит
не только к потере кремнезема и к необходимости проводить
очистку слоя ионита, но также вызывает раздробление гранул
ионообменной смолы. По данным Гофмана [99] элюент крем-
Коллоидный кремнезем — концентрированные золи 453
левой кислоты, содержащий до 6 % SiOs, может быть получен
без подобных затруднений, если использовать макросетчатый
катионит (Амерлит IR-200), имеющий поры диаметром ~10 нм,
пористость 32 % и удельную поверхность, измеренную по ад-
сорбции азота, равную 42 м2/г. Такую смолу нужно обрабаты-
вать раствором каустика для того, чтобы удалять небольшие
количества кремнезема после каждого применения смолы и пе-
ред последующей ее регенерацией.
Согласно Матейка [100], ионообменные смолы можно реге-
нерировать электролитическим способом.
Для удаления натрия из раствора силиката натрия в сер-
ной кислоте вместо обычной ионообменной смолы можно исполь-
зовать катионообменную мембрану [101]. Горячий золь, состоя-
щий из зародышевых частиц, быстро циркулирует по трубо-
проводу, заполненному ионообменным полимером, находящимся
в разбавленной серной кислоте. Силикат натрия прибавляют
к золю с такой скоростью, чтобы поддерживать pH около 8—10.
При этом выделяющийся кремнезем осаждается на частицах
золя, увеличивая тем самым их размер. Некоторое количество
сульфат-ионов проникает через мембрану, и, следовательно, со
временем в золе медленно возрастает концентрация сульфата
натрия. Золь можно очищать и концентрировать методом ульт-
рафильтрации. Однако концентрацию золя следует поддержи-
вать на таком уровне, чтобы нормальность соли натрия не пре-
вышала величины N = 0,26—0,005 С—0,0012 (Т—40), где С —
содержание SiO2, г/100 мл, Т — температура, °C.
Пептизация гелей
Еще в 1864 г. Томас Грэм сообщил, что гель кремнезема
может быть разжижен за счет добавления очень небольших
количеств щелочи. Такой процесс автор назвал «пептизацией
студня». В 1922 г. Преториус и Вульф [102] получили золь крем-
незема из геля посредством его нагревания в воде при повы-
шенной температуре и давлении. Ньюндлингер [103] пригото-
вил золи с содержанием около 10 % кремнезема обработкой
геля вначале аммиаком, а затем нагреванием системы без вы-
паривания воды вплоть до образования золя. Уайт [5] и Трейл
[104] разработали аналогичные, но улучшенные способы.
Модификация способа получения золя из геля описана Аль-
бертом и Симпсоном [105], которые проводили формирование
геля в щелочных условиях путем неполной нейтрализации ще-
лочи в растворе силиката натрия. Кислоты для нейтрализации
брали меньше, чем эквивалентно количеству силиката натрия.
Затем из геля вымывали соли и влажный гель нагревали, чтобы
вызывать его пептизацию. В этом случае гораздо большее ко-
454
Глава 4
личество геля превращается в золь, чем когда вначале приго-
товляется кислый гель. Характеристики золей, приготовленных
этим способом, не были представлены, но, вероятно, получались
зол^ с частицами диаметром 15—45 нм. Подобный способ был
описан Легалем [106]. Согласно патенту Мерца [107], превра-
щение геля следует осуществлять в автоклаве, при этом обра-
зуется 30 %-ный золь, который стабилизируют аммиаком. Бубы-
рева и Биндас [108] изучали действие ультразвукового диспер-
гирования на гель кремнезема. .
Гидролиз соединений кремния
Радчевский и Рихтер [67] сообщили, что очень чистые золи
кремнезема были получены гидролизом тетрахлорида кремния
с удалением образующейся кислоты для достижения pH 6,8.
Сформированные по этому способу сферические частицы дости-
гали размера 200 нм и, как оказалось при их электронно-мик-
роскопическом исследовании, представляли собой образования,
подобные губке. Позже Штобер и Финк [68] обнаружили, что
гидролиз низшего алкилсиликата в спиртовой среде, содержащей
необходимые количества воды и аммиака, приводит к возникно-
вению очень однородных сферических частиц кремнезема почти
любого желаемого размера вплоть до 1 мкм. Впоследствии был
применен метод меченых атомов [109, ПО]: в систему удалось
ввести определенное количество радиоактивных изотопов, не
оказывавших воздействия на рост частиц кремнезема во время
их формирования; однако не было уверенности в том, что такие
изотопы равномерно распределялись по всему объему таких
сферических частиц. Были получены частицы размером от 50
до 900 нм. Похоже, что крупные сферические частицы представ-
ляют собой в действительности агрегаты, состоящие из боль-
шого числа малых частиц размером 10 нм или менее, как это
описывал Радчевский.
По данным Бринсмида и Брауна [111], «золь кремневой кис-
лоты» с содержанием 43 % SiC>2 был приготовлен дефлегмацией
смеси, состоящей из этилсиликата и изопропилового спирта, в ко-
торую медленно добавляли разбавленный водный раствор кис-
лоты так, чтобы обеспечить стехиометрическое количество воды,
необходимое для гидролиза. Не были представлены данные по
полноте смешиваемости с водой, которые свидетельствовали бы
о том, что гидролиз протекал полностью. Отсутствовали также
какие-либо указания на формирование в данном случае дискрет-
ных частиц кремнезема.
Силикат натрия гидролизуется, если он достаточно разбав-
лен. Такой разбавленный раствор, имеющий высокое отношение
SiO2:Na2O, может гидролизоваться в автоклаве с образованием
Коллоидный кремнезем — концентрированные золи
455
коллоидных частиц, которые коагулируют и формируют оса-
док, если концентрация ионов натрия достаточно высока. Шнюрх
и Костер [112] сообщили, что при разбавлении раствора сили-
ката натрия с массовым соотношением SiO2:Na2O 3,89:1 до
концентрации 20 г SiO2M и последующем выдерживании при
150°С в течение 1,5 ч 38 % кремнезема осаждается в виде тон-
кодисперсных частиц SiO2.
Растворение элементарного кремния
Если порошкообразный кремний вначале обработать плави-
ковой кислотой, чтобы удалить оксидную пленку с поверхности,
то он приобретает способность быстро вступать в реакцию с во-
дой в щелочной среде, особенно при добавлении аммиака, обра-
зуя коллоидный кремнезем, который стабилизируется щелочью.
Этот способ был запатентован Балтисом [69]. Процесс уско-
ряется, если реакция проводится во время помола кремния [113].
Размер частиц золей, приготовляемых из элементарного
кремния, меняется в интервале 8—35 нм, а при некоторых усло-
виях может достигать 150 нм. Способ приготовления и харак-
тер получаемого вещества обсуждались в разделе «Пористые
частицы».
Для приготовления золей с содержанием до 50 % SiO2 по-
средством растворения кремния Бобб [114] предложил исполь-
зовать водный раствор неорганического основания (NaOH,
КОН), который катализирует растворение кремния при 50—
100°С и стабилизирует получающийся золь. Примечательно то,
что золи, приготовленные при 90—95°С, оказывались очень вяз-
кими, но имели обычную низкую вязкость, если готовились при
98—100°С. Частицы имели размеры 15—45 нм. К тому же золи
оказались необычными в том отношении, что при подкислении
не образовывалась жесткая сетка геля, а формировались лишь
пластичные коацерваты. Такое поведение системы необъяснимо.
В том случае, когда кремний используется в качестве анода и
растворяется под действием электролиза в водно-спиртовой
смеси, наблюдается образование алкозоля. Согласно данным
Триппа [91] и Чилтона [115], добавление кислоты или соли ме-
талла обеспечивает электропроводность золя.
Диспергирование пирогенного кремнезема
Испарение кремнезема происходит только при очень высокой
температуре — приблизительно при 2000°С. Однако в присутст-
вии восстановителя, способствующего образованию моноксида
кремния S1O, температура сублимации понижается до 1700°С.
По мере того как моноксид кремния испаряется в окислитель-
456
Глава 4
ной атмосфере, происходит конденсация диоксида кремния
в чрезвычайно тонкодисперсной форме. Окислению может под-
вергаться этилсиликат; образующиеся пары SiO2 затем конден-
сируются. В наиболее широко распространенном способе предус-
матривается сгорание тетрахлорида кремния в смеси с природ-
ным газом, при этом выделяются хлористый водород и пары
диоксида кремния, которые конденсируются в виде очень рых-
лого, занимающего большой объем, порошка. При контролиро-
вании условий сжигания примерно так, как это делается при
получении углеродной сажи, можно приготовлять вещества
с различными размерами первичных частиц и разными степе-
нями коалесценции частиц. В другом способе предусматривается
испарение __ кремнезема в электрической дуге с конденсацией
образующихся паров. Порошки такого типа рассматриваются
здесь только по той причине, что из некоторых их разновид-
ностей могут приготовляться коллоидные дисперсии. Соответ-
ствующие способы получения и свойства формируемых на осно-
вании этих способов порошков кремнезема будут рассматри-
ваться в гл. 5.
Диспергирование кремнезема в золь, состоящего из дискрет-
ных первичных частиц, представляет определенные трудности,
поскольку слипание частиц меняется в значительной степени.
Кроме того, во многих случаях поверхность частиц оказывается
почти безводной — всего лишь с несколькими гидрофильными
силанольными группами. По этим причинам свойства золей
такого типа обычно отличаются от свойств золей, приготовлен-
ных в водном растворе. Золи, получаемые сжиганием, не обра-
зуют прочных гелей и находят небольшое применение только
в качестве неорганических связующих веществ.
Анализ патентной литературы наводит на мысль, что для до-
стижения приемлемого диспергирования в воде или в полярных
органических жидкостях необходимы значительные механиче-
ские силы независимо от того, идет ли речь о сухих рыхлых
объемистых порошках или образующихся из них водных суспен-
зиях. В водных растворах для смачивания гидрофобной силокса-
новой поверности частиц применяются смачивающие реагенты,
а для промотирования поверхностной гидратации и диспергиро-
вания частиц добавляют щелочь [116, 117]. .
Более устойчивые дисперсии получаются при добавлении
к тетрахлориду кремния некоторых количеств хлорида титана
или хлорида алюминия, для того чтобы полученный кремнезем
содержал немного примесей оксида металла. В этом случае об-
разуются очень стабильные концентрированные золи, содержа-
щие 40—60 % твердого кремнезема [118]. Кремнезем фирмы
Degussa получаемый гидролизом в пламени, например, с со-
держанием 1,3 % оксида алюминия, нанесенного на поверхность
Коллоидный кремнезем — концентрированные золи 457
частиц кремнезема, аэросил-МОХ, особенно пригоден для при-
готовления концентрированных водных дисперсий с первичными
частицами размером 20—40 нм, а также более мелкими [119].
Кремнезем Cab-0-Sil, образуемый гидролизом в пламени,
имеющий величину удельной поверхности 200—400 м2/г, диспер-
гирует в воде при pH 9 за счет добавления, например, аммиака.
Это дает возможность получать золи с содержанием до
30 масс. % кремнезема. Такой золь пропускают через гомогени-
затор, чтобы разрушить на отдельные части трехмерную сетку
кремнезема, состоящую из первичных частиц. Образующиеся
в результате гомогенизации отдельные образования состоят
большей частью из цепеобразных агрегатов, которые повышают
вязкость системы [120].
В общем случае способ, называемый «дымовым» или «гидро-
лизом в пламене», не дает возможности получать кремнезем,
диспергирующий в воде с образованием золей, состоящих из
дискретных частиц и имеющих низкую вязкость при высокой
концентрации кремнезема, т. е. не дает золей с типичными ха-
рактеристиками, которые приготовляются полимеризацией
в водных средах. Тем не менее соответствующей обработкой по-
добный пирогенный кремнезем, содержащий первичные частицы
размером 10—25 нм, можно дезагрегировать и диспергировать
и получить из него водные золи с содержанием вплоть до 40 %
кремнезема, если применять подходящую механическую обра-
ботку и диспергаторы.
Согласно данным Спенсера, Смита и Космана [121], можно
приготовить чрезвычайно тонкодисперсный кремнезем, состоя-
щий из частиц с размером всего лишь 25—50 А и имеющий
удельную поверхность, достигающую 1000 м2/г. Такой кремнезем
получается обработкой влажной углеродной сажи диметил-
дихлорсиланом с последующим выжиганием органического ве-
щества на воздухе при 500°С. Образующийся в количестве 4 % от
исходной массы углеродной сажи кремнезем представлял собой
опалесцирующий порошок в форме небольших сферических кру-
пинок около 1 мм в диаметре, т. е. примерно того же самого
размера, что и гранулы углеродной сажи. Удельная поверхность
такого кремнезема была 1094 м2/г, что указывает на размер
первичных частиц ~2,5 нм. Подобное вещество легко дисперги-
рует в воде с формированием частиц в виде цепочек длиной
До 20 мкм. Это дает возможность предполагать, что частицы
были частично гидрофобными.
Коллоидное измельчение пирогенного кремнезема в воде
в присутствии борной кислоты или щелочного бората показано
в работе [122]. По этому способу можно приготовлять 30 %-ный
золь. Некоторые дополнительные патенты, относящиеся к полу-
чению золей из пирогенного кремнезема, большей частью пре-
458
Глава 4
дусматривают щелочную стабилизацию, для чего применяются
силикат натрия, гидроксид натрия, гидразин, гидроксиламин
или их смеси с пирогенными оксидами металлов [123—125].
Очистка, обогащение, стабилизация дисперсий
Приготовляемые посредством некоторых процессов золи со-
держат соли или другие вещества, количество которых должно
быть уменьшено или которые вообще необходимо удалять перед,
тем, как золь окончательно концентрируется.
Ионный обмен
Согласно данным Рула [7], чтобы получить золь максималь-
ной чистоты, требуются специальные методики очистки для
удаления солей из готовых концентрированных золей. Сюда
обычно включается обработка ионообменными смолами для
удаления растворимых солей с последующей стабилизацией зо-
лей минимальным количеством основания, в том числе аммиака.
Этот метод стал настолько обычным, что не требуется его даль-
нейшего обсуждения. После того как частицы уже сформиро-
ваны, удалить натрий довольно трудно, поэтому Шефер и Га-
маге использовали в своей работе алканоламин в качестве ос-
нования в процессе роста частиц [126а].
Диализ и электродиализ
Диализ представляет собой давно известный способ очистки,,
применяемый для удаления растворимых примесей из золей. Как
только были разработаны способы получения разбавленных зо-
лей кремнезема по реакции между кислотой и силикатом или
гидролизом подходящего вещества, такого, например, как тет-
рахлорид кремния, то сразу же было признано, что для удале-
ния электролита требовалась очистка. Еще Грэм [1266], один ид
самых первых исследователей золей кремнезема, в 1861 г. ис-
пользовал диализ для удаления электролитов из кремнеземной
системы и, таким образом, приготовлял относительно чистый
коллоидный кремнезем. Поскольку процесс диализа протекает
относительно медленно, он не находит широкого применения
в промышленных масштабах. Поэтому был предложен более
быстрый способ, не требующий использования плоских мембран.
В нем предусматривается пропускание золя через колонну или
слой, заполненный набухшим полимерным гелем с такой тонко-
пористой структурой, что через него способны проникать только
растворимые соли, но не коллоидные частицы. Полимерный гель
может представлять собой способные к регенерации целлюлозу
или желатин, поперечное связывание у которых осуществляется
Коллоидный кремнезем — концентрированные золи
459
введенным формальдегидом. Слой с полимерным гелем реге-
нерируется вымыванием из него солей [127].
Более быстрая очистка может выполняться с помощью элек-
тродиализа, при котором ионы переносятся к мембранам за
счет наложения электрического поля. Предложены различные
усовершенствования аппаратуры [128], особенно предназначен-
ной для обессоливания морской воды. Однако в отношении
очистки золей кремнезема этот способ не заменяет более деше-
вый ионный обмен.
Промывание
Методы, в которых концентрируются частицы золя, но не ра-
створимые компоненты, могут быть использованы для очистки
золя. Концентрированный золь разбавляется чистой водой, и
весь процесс повторяется еще раз.
Седиментационный способ, основанный на действии силы тя-
жести, в общем случае слишком длителен, но применение цент-
рифугирования позволяет довольно быстро концентрировать
частицы кремнезема размером больше 30—50 нм. В другом ва-
рианте кремнезем подвергается флокуляции за счет взаимодей-
ствия с двухзарядными ионами металла; образовавшийся осадок
сначала очищается от растворимых солей промыванием, а затем
пептизируется удалением флокулирующих ионов. Александер и
Айлер [80] применяли такие ионы, как Mg2+, Са2+ и Ва2+, и
удаляли их ионным обменом или в случае Ва2+ — осаждением
в виде нерастворимого сульфата.
В методе промывки можно также применять концентриро-
вание путем ультрафильтрации или электродекантации.
Обогащение
Пригодными являются многие методы обогащения золей, но
все же выпаривание воды остается наиболее общепринятым
промышленным способом. Однако в связи с ростом стоимости
энергии и водяного пара, безусловно, представляется целесооб-
разным рассмотреть и другие известные методы.
Выпаривание воды
Для получения стабилизированных золей, таких, как золи
коллоидного кремнезема, обычно используются выпарные ап-
параты с принудительной циркуляцией. Необходимо соблюдать
меры предосторожности, чтобы не допустить слишком высокого
концентрирования золя или высокой степени высушивания на
каких-либо участках оборудования, и особенно на теплообмен-
460
Глава 4
ных поверхностях. Если же это происходит, то на стенках нара-
щивается слой прочно связанного с поверхностью твердого
кремнезема. Такая проблема становится особенно острой, когда
содержание кремнезема приближается к конечной высокой кон-
центрации и одновременно повышается вязкость золя.
Выпаривание имеет то преимущество, что высокая темпера-
тура часто играет важную роль при затвердевании структуры
отчасти пористых частиц кремнезема, образованных при более
низкой температуре, а также способствует дальнейшему уве-
личению размеров частиц.
Центрифугирование
Обычно этот способ используется для удаления остатков ор-
ганических веществ и для осветления золей кремнезема, но не
для их концентрирования. До тех пор пока размер частиц не
превышает ~30 нм, требуется применять очень высокие ско-
рости центрифугирования. Следовательно, такой метод не прак-
тичен для большинства коммерческих золей.
У льтрафильтрация
Значительные успехи были достигнуты в течение последних
20 лет в развитии оборудования и мембран, используемых в ме-
тоде ультрафильтрации. Схема этого метода показана на
рис. 4.3. По существу такой способ позволяет удалять воду, не-
большие по размеру ионы и растворенные вещества из золя
кремнезема или коллоидной суспензии, которые, таким образом,
концентрируются без образования осадка или отложения на
мембранном фильтре.
Этот метод становится все более важным для проведения
очистки и концентрирования коллоидов с минимальным потреб-
лением энергии. Так, способ, описанный Айлером [129], дает
возможность приготовлять коллоидный кремнезем посредством
частичной нейтрализации горячего раствора силиката натрия
кислотой при таком разбавлении, что образующиеся частицы
не коагулируют под действием соли натрия. Золь (2—3 % SiO2)
охлаждается до 50°С и обогащается способом ультрафильтра-
ции, тогда как соль в то же время вымывается водой. Для из-
бежания агрегации частиц или формирования микрогеля необ-
ходимо добавлять воду с такой скоростью, чтобы поддерживать
концентрацию соли ниже некоторого значения нормальности А,
рассматриваемой как
N = 0,26 — 0,005С — 0,002 (Т — 40)
где С — концентрация SiO2 г/100 мл; Т — температура, °C.
Коллоидный кремнезем — концентрированные золи
461
Температура постепенно повышается до 75°С, в то время как
нормальность уменьшается до 0,15 н. и ниже, а С увеличивается
до 10 г/100 мл. Процедуры фильтрации через ультрафильтр и
вымывания солей продолжаются вплоть до получения устойчи-
вого стабилизированного
30—40 %-ного золя. Размер
частиц золей не должен пре-
вышать 10 нм, а уровень со-
держания соли поддержива-
ется несколько ниже по срав-
нению с уровнем для частиц
большего размера.
Чилтон [130] предложил
понижать значение pH золя
кремнезема до 2—4 перед
тем, как проводить его обога-
щение ультрафильтрацией.
Однако такие золи оказыва-
ются неустойчивыми при
высокой концентрации, и
особенно при повышенной
температуре, когда процесс
ультрафильтрации протекает
гораздо быстрее.
Новые успехи ультра-
фильтрации были достигну-
ты благодаря улучшенным
вариантам мембран. Наибо-
лее тонкие сорта фильтро-
вальной бумаги имеют поры,
достигающие в диаметре
1000 нм (1 мкм), тогда как
мембраны для ультрафиль-
трации могут изготовляться с
Рис. 4.3. Схемы фильтрации и ультра-
фильтрации.
Фильтр задерживает большие по размеру
частицы в виде слоя отфильтрованного осадка,
ио пропускает коллоидные частицы (тем-
ные кружочки) и растворенные соли. Ультра-
фильтр задерживает коллоидные частицы
в виде слоя концентрированного золя, ио про-
пускает растворенные соли.
порами диаметром от 1000 до 2—
3 нм. В течение многих лет использовались «целлофановые» или
свежеобразованные пленки из коллоидного материала (нитро-
целлюлозы). Но в настоящее время многие фирмы снабжают
оборудование прочными, гибкими и долговечными мембранами
с высокой степенью однородности пор по размерам, сохраняю-
щими, однако, высокое значение пористости, что позволяет воде
достаточно быстро протекать через такие мембраны. Были раз-
работаны мембраны из пористого стекла, а также из пористого
угля. Мембраны из пористой керамики с микропористым по-
кровным слоем обеспечивают высокую стойкость ультрафильт-
ров по отношению к воздействию повышенных температур и
химических веществ.
462
Глава 4
Михаэле [131] в своем обзоре осветил развитие мембранной
ультрафильтрации, изложил ее основные принципы, виды обо-
рудования и области применения в химической промышленности.
Портер и Михаэле [132] провели сравнение областей размеров
молекул и частиц, доступных процессам разделения. Они опи-
сали применяемые мембраны с однородными по размеру порами,
Рис. 4.4. Схема ультрафильтрации, «концентрационной поляризации»
частиц и образования микрогеля.
По мере циркуляции над мембраной разбавленный золь будет концентрироваться у ее
поверхности, однако его частицы имеют возможность диффундировать обратно в об-
ласть разбавленного золя. Частицы микрогеля не диффундируют подобным образом,
оставаясь около поверхности мембраны.
имеющими в диаметре 1—20 нм (10—200 А). Была составлена
библиография по применению мембран [133]. Оборудование и
необходимую информацию можно получить в ряде фирм, выпу-
скающих широкий ассортимент разработанной аппаратуры и
мембранных устройств.
Разработке гораздо более эффективной аппаратуры и рас-
ширению ее применения способствует лучшее понимание основ-
ных принципов ультрафильтрации. Теория ультрафильтрации
была подробно изложена Портером [134], который в особенно-
сти имел дело с проблемой «концентрационной поляризации».
Как представлено на рис. 4.4, движение частиц по направлению
к мембране приводит к формированию концентрированного слоя
золя с высокой вязкостью. Такой слой может замедлить поток
или скорость фильтрации до небольшой доли от скорости жид-
кой среды в отсутствие коллоидных частиц. Сопротивление по-
току— следствие не только закупоривания пор или даже фак-
тически образовавшегося сплошного слоя (геля) плотно упако-
ванных коллоидных частиц. Айлер наблюдал, что оно представ-
Коллоидный кремнезем — концентрированные золи 463
ляет собой прямую функцию от высокой вязкости концентриро-
ванного слоя золя. При пропускании струи воды в верхней ча-
сти системы частицы кремнезема начинают диффундировать
в направлении от граничного слоя, и скорость воды не меняется.
В ходе этого процесса распределение коллоидных частиц стано-
вится «поляризованным» в том смысле, что их концентрация
оказывается более высокой вблизи поверхности ультрафильтра и
значительно меньшей в отдалении от него. Такая «поляризация»
полностью обратима.
Для поддержания высокой скорости потока необходимо,
чтобы толщина и концентрация коллоидного слоя вблизи по-
верхности фильтра сохранялись минимальными за счет созда-
ния значительного смещения слоев жидкости около мембраны.
Это достигается увеличением линейной скорости потока золя,
проходящего над поверхностью фильтра, и особенно его тур-
булентным режимом. В небольших лабораторных ультрафильт-
рах с горизонтально расположенной мембраной интенсивный
турбулентный режим поддерживается механической мешалкой,
находящейся очень близко к мембране. При поддержании неиз-
менными других условий скорость потока быстро уменьшается
с возрастанием концентрации золя. Обычно увеличение давления
вблизи поверхности ультрафильтра не играет такой роли, как
достижение минимума концентрационной поляризации.
С ростом температуры наблюдается быстрое увеличение по-
тока. Следовательно, там, где это возможно, ультрафильтрацию
растворов необходимо проводить при максимально высокой тем-
пературе.
Ограничения, возникающие при удалении солей методом
ультрафильтрации. Ультрафильтрация с непрерывным добавле-
нием воды достаточно эффективна для удаления солей вплоть
до их остаточного содержания 0,03 н. даже из концентрирован-
ного золя кремнезема. При таких низких уровнях содержания
для солей характерна тенденция оставаться в ассоциирован-
ном состоянии с заряженной поверхностью коллоидных частиц.
Вполне возможно, что до сих пор это не было известно, по
крайней мере в химии коллоидного кремнезема, но на такое яв-
ление необходимо обращать внимание при удалении солей из
других ионных коллоидных систем. Высказывается предполо-
жение, что в разбавленных золях, когда заряженные частицы от-
далены друг от друга и в то же время концентрация противо-
ионов натрия вокруг частиц достаточно высока, сульфат-ионы
будут стремиться концентрироваться в виде вторичного слоя
вблизи слоя ионов натрия. Как показано на рис. 4.5, вокруг
каждой частицы кремнезема существует граничный слой с пре-
обладающим содержанием ионов Na+. Снаружи от него в не-
посредственной близости должен находиться вторичный слой,
464
Глава 4
в котором содержится больше отрицательно заряженных суль-
фат-ионов, чем катионов натрия. Таким образом, наблюдается
тенденция повышения концентрации сульфат-ионов вблизи ча-
стиц кремнезема, а не в толще слоя воды. Следовательно,
в фильтрате будет находиться относительно меньшее содержа-
ние сульфат-ионов.
Рис. 4.5. Схема концентрирования соли около коллоидных частиц кремнезема.
Вокруг отрицательно заряженной частицы кремнезема находится слой противоположно
заряженных катионов натрия. К нему примыкает слой, в котором сульфат.анионы скон-
центрированы в большей степени, чем в объеме раствора.
Такой эффект становится заметен в золях, состоящих из не-
больших частиц и содержащих менее 20 % кремнезема при
pH 9. Путем измерения концентрации ионов SO^b прошедшем
через ультрафильтр фильтрате, а также в золе прослеживались
типичные результаты наблюдений.
К 6,5 л 9 %-ного золя SiO2 с частицами размером 6 нм при
pH 9 и содержании 0,04 н. сульфата натрия добавляли 10 л
воды для промывания, но, несмотря на то что фильтрат уда-
ляли, объем золя оставался постоянным. Подсчеты показали,
что содержание соли при таком разбавлении должно пони-
зиться до 0,009 н., но фактически оно оказалось равным 0,022 н.
Одновременно выполненный анализ подтвердил, что концентра-
ция сульфат-ионов во время этого процесса в фильтрате оказы-
валась меньше, чем в золе. В окончательно получаемом золе
на 1 нм2 поверхности кремнезема приходилось 0,15 сульфат-
иона и 0,8 противоиона натрия. Можно, конечно, осуществить
Коллоидный кремнезем — концентрированные золи 465
дальнейшее понижение содержания сульфат-ионов посредством
длительного промывания водой, но эта процедура вскоре стано-
вится неэффективной.
Действие, оказываемое микрогелем на золь кремнезема. Наи-
более важным фактором при использовании метода ультрафильт-
рации для концентрирования золей кремнезема оказывается
присутствие микрогеля или агрегатов из частиц кремнезема раз-
мером порядка полмикрона или более. Эти большие по размеру
агрегаты диффундируют настолько медленно по сравнению
с одиночными частицами, что переносятся потоком только до
поверхности мембраны, где необратимо осаждаются в виде геля,
уменьшая тем самым поток воды и ионов через мембрану.
В серии контрольных опытов по ультрафильтрации, выполнен-
ных с 4 %-ным золем SiO2, поток понижался на 50 %, если всего
лишь 0,5 % кремнезема присутствовало в виде микрогеля, и на
80 % в присутствии 1,5 % микрогеля. Таким образом, при со-
держании кремнезема в суспензии, достигающем 0,02 масс.%,
сразу же наблюдается резкое уменьшение потока.
Электродекантация
При прохождении электрического тока через разбавленный
золь кремнезема с низким содержанием соли будет происходить
Рис. 4.6. Схема аппарата для электродекантации.
Анод помещен в разбавленную кислоту. Три пористых мембраны нли барьера типа уль-
трафильтра размещаются в емкости таким образом, что сверху и снизу имеются зазоры.
Катод находится в разбавленной щелочи. Ионообменные мембраны, пропускающие ка-
тионы, но не пропускающие анионы, располагаются вблизи каждого электрода.
электрофорез, и отрицательно заряженные частицы будут дви-
гаться по направлению к аноду. Посредством размещения между
ионообменными мембранами ряда барьеров в виде мембран для
Ультрафильтрации, как это схематично показано на рис. 4.6
4 Заказ № £50
466
Глава 4
[135], с целью выделения золя из электродных камер, запол-
ненных электролитом, представляется возможным сконцентри-
ровать золь. Частицы концентрируются на сторонах мембран,
обращенных к катоду, образуя густую жидкость, которая опу-
скается на дно резервуара, и затем отделяется. Золь должен
иметь низкое содержание растворимых солей для того, чтобы
электрический ток главным образом переносился заряженными
частицами и соответствующими противоионами, тогда будет
обеспечиваться наиболыйая эффективность этого способа.
Стабилизация дисперсий
Поразительным фактом оказывается то, что в концентриро-
ванных золях кремнезема часто вырастают микроорганизмы.
Они могут появляться в виде плавающих вязких масс, взвешен-
ных в суспензии частиц зеленого, желтого, красного или корич-
невого цвета или в виде беловатых волокнистых агрегатов. При-
сутствие бактерий часто обнаруживается по запаху сероводорода
и темному окрашиванию вследствие осаждения следов тяжелых
металлов. Могут появляться и морские водоросли в виде зеле-
ных волокнистых масс или коричневатых хлопьев.
В качестве стабилизаторов дисперсий часто добавляются
такие вещества, как формальдегид [136], или многоатомный
спирт вместе с пентахлорфеноловым эфиром [137]. Такой мно-
гоатомный спирт, как этиленгликоль, стабилизирует золь в от-
ношении замерзания. Другими патентованными стабилизато-
рами являются 3,5-диметилтетрагидро-1,3,5-2Н-тиопиримидин-
2-тиокетон совместно с формальдегидом [138] или 0,0025 —
0,02 %-ный хлорит натрия NaClOz [139].
По некоторой, еще неизвестной причине золи, стабилизиро-
ванные аммиаком, оказываются не способными поддерживать
рост микроорганизмов, даже если они стабилизированы при том
же самом значении pH, как и в случае стабилизации гидрокси-
дом натрия.
Характеристики золей
Лишь после того, как были разработаны методы, позволяю-
щие прослеживать полимеризацию кремнезема, измерять раз-
меры частиц и определять степень их агрегирования, что вполне
достаточно для изучения поведения коллоидного кремнезема,
появилась возможность контролировать размер частиц и пред-
сказывать их поведение во время какой-либо обработки. Для
того чтобы получать стабилизированные системы, т. е. выращи-
вать частицы до определенного размера без их агрегации, не-
обходимо привлечение сведений по химии поверхности кремне-
Коллоидный кремнезем — концентрированные золи
467
зема и понимание природы взаимодействия кремнезема с водой
в пределах широкой области значений pH.
Наиболее важными характеристиками золя кремнезема яв-
ляются следующие.
Химический состав. Кремнезем, стабилизирующее основание,
углерод, включая диоксид углерода и углерод в органическом
соединении, растворимые соли щелочных металлов. Химическим
.анализом определяется общее содержание твердых веществ, со-
держание золы без включений кремнезема, металлов, в том
числе алюминия и железа.
Характеристики частиц. Размер частиц и их распределение
по размерам, пористость, степень агрегации, удельная поверх-
ность, скорость растворения.
Физические характеристики. Значения pH, плотность, вяз-
кость, мутность, показатель преломления, рассеяние света, ско-
рость седиментации при ультрацентрифугировании.
Химический анализ
Содержание кремнезема может быть оценено приблизительно
по плотности и показателю преломления или же определено ка-
ким-либо стандартным аналитическим способом (см. гл. 1).
Количество стабилизирующего основания определяется титрова-
нием до pH ~3,5 или 2,0 с введением поправки на присутствие
10-2 н. кислоты. Может быть также использован традиционный
метод определения «бескремнеземной золы». По этому способу
высушивают и нагревают образец в платиновой чашке, взве-
шивают оставшийся образец, удаляют кремнезем действием
смеси дымящих кислот HF+H2SO4 и окончательно взвешивают.
В остатке легко можно определить другие элементы методом
атомно-абсорбционного анализа. Вся потеря в массе относится
к кремнезему.
Такой металл, как алюминий, представляет- особый интерес,
поскольку его присутствие показывает, что поверхность образца
может быть модифицирована за счет присутствия на ней алю-
мосиликатных групп, которые оказывают воздействие на пове-
дение коллоидных частиц. Алюминий на поверхности кремнезема
может быть определен экстракцией высушенного кремнезема
хлористым водородом с последующим анализом экстракта.
Карбонаты и органические вещества оказываются важными
в том смысле, что карбонат действует как дополнительный элек-
тролит, и может потребоваться коррекция при титровании осно-
вания, а органические вещества (обычно получаемые экстрак-
цией низкомолекулярного полимера из ионообменной смолы)
могут оказывать воздействие на поведение кремнезема в неко-
4*
468
Глава 4
торых областях применения. Анализ на суммарный азот и угле-
род показывает возможное присутствие органических оснований.
Характеристики частиц иногда измеряются непосредственно,
а в других случаях выводятся из таких физических свойств золя,
как вязкость, мутность, рассеяние света и скорость седимен-
тации.
Измерение pH
Значение pH золя кремнезема — одна из наиболее важных
его характеристик. Поведение золя может сильно меняться при
незначительном изменении pH. Например, возрастание pH всего
на 0,5 приведет к удвоению скорости процесса гелеобразования
при pH 4 или превратит золь в раствор силиката натрия при
pH 10,5.
Обычно измерения pH выполняются со стандартными стек-
лянными, устойчивыми к щелочи электродами. Такие элек-
троды, предназначенные для растворов с высоким содержанием
ионов натрия, должны использоваться, когда измерения pH про-
водятся в присутствии соли при определении удельной поверх-
ности методом титрования.
Болт [140] отмечал, что для получения наибольшей точно-
сти при проведении измерений pH, в частности в концентриро-
ванных золях, необходимо использовать «измерительный мост
для изучения процесса гелеобразования» для того, чтобы прео-
долеть «эффект концентрирования золя» на электроде сравнения.
Концентрация электролита
Для определения солевой концентрации в золе с достаточ-
ной точностью, не прибегая к химическому анализу, следует
проводить измерения электропроводности приблизительно при
pH 4,5. При таком значении pH заряд на поверхности частиц
кремнезема незначителен, и, следовательно, электропроводность
обуславливается главным образом присутствием соли. Что
касается золя, стабилизированного щелочью, то он титруется
обычной стандартной кислотой до pH 4,5, и получаемая в ре-
зультате этого концентрация соли натрия вычитается из общей
концентрации соли, подсчитанной из величины электропровод-
ности. Александер [9] дает эмпирическое уравнение
N = 8(L— 4,3- ИГ5)
где L — удельная электропроводность золя, разбавленного до
4 % SiO2, при 25°С и N — нормальность соли Na2SO4.
Коллоидный кремнезем — концентрированные золи
469
Характеристики частиц
Размер частиц
При содержании в золе более 10—15 % кремнезема о по-
рядке размера частиц можно судить визуально по степени
помутнения жидкости. Если диаметр частиц меньше чем ~7 нм,
то золь оказывается почти таким же прозрачным, как вода.
При размерах 10—30 нм наблюдается характерная опалесцен-
ция и золь просвечивается на некоторую глубину. Если диа-
метр частиц превышает ~50 нм, золь представляется белым
или молочным по цвету. Наконец, когда размер частиц превы-
шает 100 нм, при стоянии золя в течение нескольких суток или
недель просматривается прозрачный верхний слой.
Кун [141] представил обзор методов, пригодных для полу-
чения характеристик тонкодисперсных частиц.
Электронная микроскопия. Электронный микроскоп дает
возможность прямым методом определять размеры коллоидных
частиц (рис. 4.7). При применении усовершенствованных моде-
лей приборов можно различать отдельные частицы вплоть до
таких, размеры которых составляют всего лишь 1—2 нм.
Однако измерение частиц величиной меньше 5 нм оказывается
затруднительным. Александер и Айлер [142] впервые продемон-
стрировали, что размеры коллоидных частиц кремнезема, изме-
ренные с помощью электронного микроскопа, коррелируют
с соответствующими размерами, определенными методом рас-
Элекгрошю-микроскоппческий снимок частиц коллоидного кремне-
зема диаметром ~20 нм.
Рис. 4.7.
470
Глава 4
сеяния света в растворе, а также с размерами частиц, подсчи-
танными из величины удельной поверхности высушенного по-
рошка кремнезема.
Полностью монодисперсных золей практически не наблю-
дается, так как диаметры частиц золей обычно различаются по
крайней мере на несколько миллимикрон. Принято несколько
способов выражения средних размеров частиц. Например,
среднечисленный диаметр частиц dn представляет собой просто
среднее значение диаметров большого числа частиц. С другой
стороны, размер можно выражать диаметром частиц, которые
имеют массу, равную средней массе частиц. В этом случае
имеем среднемассовый диаметр частиц dw. Размер может вы-
ражаться также как диаметр ds частиц, которые имеют поверх-
ность, равную среднему значению поверхностей всех рассмат-
риваемых частиц.
Метод измерения и расчета величин dn и ds в общих чертах
был описан в патентах, опубликованных Балтисом [69] вслед
за описанием самого метода, предложенного Уотсоном [143].
Для того чтобы проводить различие между дискретными
частицами и возможными остаточными агрегатами частиц,
золь нужно разбавлять настолько сильно, чтобы при его высу-
шивании на сетке не наблюдалось агрегирование частиц. Грин
и др. [144] описывают использование положительно заряжен-
ного слоя цитохрома С на сетке с напыленным углеродным
слоем, что позволяет адсорбировать на такой подложке отри-
цательно заряженные частицы.
Для адсорбции кремнезема может быть использован свежий
0,1 %-ный раствор бычьего альбумина при pH 3, дающий воз-
можность получать тонкую высушенную пленку на экране с на-
пыленным углеродом. Капля образца, содержащая 0,1 % SiO2,
наносится и сразу же смывается. В результате этого на экране
остается характерная заселенность адсорбированных частиц
и агрегатов. Затем экран высушивается на воздухе для после-
дующего исследования.
Контрастность кремнеземных частиц может быть повышена
путем использования 1 %-ного раствора ацетата уранила при
pH 4,6 и смывания избыточного количества кремнезема [145].
Удельная поверхность Sc может быть подсчитана из вели-
чины ds, если принять плотность кремнезема равной 2,2 г/см3,
что соответствует плотности аморфного безводного непористого
кремнезема
4 6 • 103 2720
d плотность . ds ds
Диаметр выражается в миллимикронах, а удельная поверхность
имеет размерность м2/г. Удельная поверхность, определяемая
Коллоидный кремнезем — концентрированные зо^и
471
расчетным путем из известной величины ds при допущении, что
частицы кремнезема представляют собой плотные образования,
обозначается символом Ad-
Бейли, Бетти и Буз [146] в своей работе всесторонне
обсудили вопросы определения размеров частиц. Позже Ундер-
вуд [147] дал еще более широкое рассмотрение размеров и
форм частиц.
Электронно-микроскопическим методом было получено рас-
пределение частиц по размерам в коммерческом коллоидном
кремнеземе людокс [148]. Например, диаметр dw, представляю-
щий среднемассовую величину, определяемую из электронно-
микроскопической кривой распределения частиц по размерам,
оказался равным 20,0 нм, что в пределах 5%-ной эксперимен-
тальной ошибки находится в согласии со значениями диамет-
ров частиц, высчитанных из данных по рассеянию света.
Показатель однородности частиц, представляющий собой
отношение среднечисленного диаметра частиц к среднемассо-
вому диаметру, может определяться методами, описанными
Бейли, Бетти и Вузом [146].
Рассеяние света. Методом рассеяния света Александер и
Айлер [142] охарактеризовали ряд золей, полученных Бечтоль-
дом и Снайдером [6]. Эти данные находились в согласии с раз-
мерами частиц, определяемыми из электронно-микроскопиче-
ских снимков. Если допустить, что кремнезем состоит из одно-
родных частиц сферической формы с плотностью 2,2 г/см3,
имеющих среднечисленный диаметр dn, то тогда теоретически
вычисляемая молекулярная масса М (в миллионах) дается
в виде соотношения
lgd„ = 0,3331gM + 1,054
Диаметр выражен в нанометрах. Однако график, показываю-
щий зависимость получаемой величины lg dw от 1gМ, лучше
описывается уравнением
1g <4, = 0,27 1g МД- 1,15
Некоторое расхождение между этими уравнениями может быть
обусловлено различиями в соответствующих кривых распреде-
ления частиц по размерам, -так как для подобных образцов
кремнезема отношение среднемассового диаметра частиц
к среднечисленному изменялось от 1,13 до 1,35.
Коммерческий коллоидный кремнезем людокс, поставляемый
в виде золя с частицами размером 15 нм, имеющими молеку-
лярную массу около 2,5-106, был использован Мароном и Лоу
[149] в качестве стандартного образца для калибровки фото-
метров, работающих на принципе рассеяния света. Авторы пред-
ложили усовершенствованный способ калибровки. Примерно
472
Глава 4
в тот же период Трэи и Германе [150] применили людокс для
калибровки нефелометрической аппаратуры, измеряя абсолют-
ную величину помутнения в исследованиях по пропусканию
света. Позднее Горинг и др. [151] сообщили о некоторых труд-
ностях, возникавших в отношении получения воспроизводимых
результатов при использовании в качестве эталонов различав-
шихся между собой образцов коллоидного кремнезема. Они
также нашли, что при разбавлении образцов дистиллированной
водой, а не разбавленным раствором соли, данные оказывались
ошибочными. В дальнейшем упомянутое явление исследовали
Дежелич и Кратохвил [148, 152, 153] и отметили точно такое
же поведение системы в процессе наблюдения за изменением
второго вириального коэффициента в зависимости от концент-
рации электролита. Они подтвердили, что результаты, получен-
ные по методу рассеяния света, оказывались гораздо более
воспроизводимыми в присутствии по крайней мере 0,025 М
раствора NaCl. Другие авторы [154] рекомендовали проводить
измерения светорассеяния в солевом растворе с ионной силой,
равной 0,1, с целью подавления влияния поверхностных за-
рядов.
Дженнингс и Жерард [155] провели более детальное
сравнение размеров частиц двух видов коммерческих золей
кремнезема методами рассеяния света, электронной микроско-
пии и ультрацентрифугирования.
Было изучено [156] влияние на величину рассеяния света
изменяющегося показателя преломления жидкой фазы в кол-
лоидном кремнеземе и обнаружено, что собственно частицы
коллоидного кремнезема не поглощают свет при изменении
показателя преломления жидкой фазы, а избыточное помут-
нение и рассеяние света при этом падают до нуля.
Классон и Охман [157] описали прибор с автоматизирован-
ной регистрацией светорассеяния, калибровка которого произ-
водилась по коллоидному кремнезему.
Мутность, поглощение. Интенсивность светового потока
уменьшается по мере того, как он проходит через золь. Про-
пускание и поглощение связываются уравнением
где А — поглощение и Т — процент светопропускания, выра-
жаемый соотношением
Т= Ш10/1
где /о — интенсивность падающего света и I — интенсивность
проходящего света. Как показано на рис. 4.8а, поглощение
света пропорционально концентрации кремнезема (при низких
концентрациях) и возрастает с увеличением размеров частиц.
Коллоидный кремнезем — концентрированные золи
473
Рис. 4.8а. Зависимость поглощения света кремнеземными золями о*г концен-
трации кремнезема при длине волны 400 нм и различных размерах частиц.
Поглощение света при длине
волны 400 нм, как это представ-
лено на рис. 4.8а, не эффективно
для определения приблизитель-
ного размера частиц диаметром
менее 20 нм. При более корот-
ких длинах волн достигается луч-
шая чувствительность. Поглоще-
ние света представляет собой
удобный способ наблюдения из-
менений, происходящих в веще-
стве, в том числе размеров ча-
стиц, и определения присутствия
в системе агрегатов, частиц боль-
ших размеров или остатков ор-
Рис. 4.86. Зависимость поглощения
света кремнеземными золями при
длине волны 400 нм от концентра-
ции S1O2. Максимальное поглоще-
ние наблюдается в концентриро-
ванных зо-лях.
[ убывает, как это показано-
кажется удивительным, что
ганических веществ.
Линейное соотношение между
величиной поглощения и концент-
рацией кремнезема сохраняется
только при концентрациях менее
5—10 % SiO2. При увеличении
концентрации мутность золей
проходит через максимум, а зате
на рис. 4.86. На первый взгляд
почти прозрачный концентрированный золь становится более
мутным по мере разбавления. Поглощение и рассеяние света
взаимосвязаны между собой; рассеяние оказывается функцией от
среднего расстояния между поверхностями раздела кремнезем—
474
Глава 4
вода. Пока частицы находятся друг от друга на расстоянии,
в несколько раз превышающем их диаметр, критическим рас-
стоянием будет диаметр частиц. При концентрации ~7 объ-
емн. % (или ~ 15 масс. %) расстояние между поверхностями
двух частиц равно величине диаметра одной частицы. Следова-
тельно, при более высоких концентрациях расстояние между
двумя поверхностями раздела становится меньше критического,
и рассеянние и поглощение света снижаются. В очень концент-
рированном золе свет в действительности проходит через массу
кремнезема, в которой находятся лишь небольшие области воды,
которые и вызывают рассеяние и поглощение.
Малоугловое рассеяние рентгеновских лучей. Брилл, Вейл
и Шмидт [158] продемонстрировали возможность использова-
ния малоуглового рассеяния рентгеновских лучей для измере-
ния распределения частиц по размерам в относительно разбав-
ленных золях. Образцы коллоидного кремнезема с номиналь-
ным размером частиц около 15 и 10 нм (лю'докс-HS и
людокс-SM.) разбавляли примерно до 1 % SiO2. Диаметры
частиц измеряли с помощью электронного микроскопа при
увеличении вплоть до 32000 с погрешностью всего ~1 нм.
Было сделано заключение, что данные по распределениям частиц
по их диаметрам, полученные с помощью малоуглового рас-
сеяния рентгеновских лучей и электронной микроскопии, для
таких образцов кремнезема находятся в хорошем согласии
между собой в пределах экспериментальной погрешности.
Аналогичное исследование кремнезема людокс-HS приводится
в работе [159]: в растворах, разбавленных до 0,5%, обнару-
жены частицы диаметром около 18 нм.
По методу малоуглового рассеяния рентгеновских лучей
можно определять как диаметры частиц, так и средние рас-
стояния между частицами, произвольным образом находящи-
мися в исследуемом пространстве. Используя частицы разме-
рами 10—50 000 нм, Драгсдоре [160] показал, что к мень-
шим по размеру частицам применима теория дифракции,
тогда как к большим целесообразнее применять теорию пре-
ломления и отражения, основанную на законах геометрической
оптики.
Метод использовался для определения размеров частиц
золей кремнезема наряду с методами электронной микроскопии
и адсорбции азота. Ледерер [73] сообщил о том, что отмеча-
лось почти полное соответствие между данными, полученными
разными методами, если принималось во внимание распределе-
ние частиц по размерам. Однако вывод о том, что частицы
содержали «внутреннюю гидратную воду» в количестве 15—
26 масс. % по отношению к БЮг, вне всякого сомнения был
ошибочным, если частицы золя приготовлялись, как это утвер-
Коллоидный кремнезем — концентрированные золи
475
ждалось, способом, которым готовится коллоидный кремнезем
людокс [161].
Ультрацентрифугирование. Методом ультрацентрифугирова-
ния коллоидного кремнезема людокс Германе и Рюке [162]
получили распределение частиц по размерам, согласующееся
с электронно-микроскопическими данными Александера и
Айлера [142]. Дженингс и Жерард [155] измерили размер
частиц кремнезема марок людокс-HS и сайтон-2Х, используя
аналитическую ультрацентрифугу, а также электронный микро-
скоп и метод светорассеяния. Они пришли к заключению, что
оба золя могли быть использованы в качестве калибровочного
вещества:
Диаметр частиц (нм), определенный следующими методами
Золь Ультрацентри- фугирование Электронная микроскопия Рассеяние света
Людокс-HS 17,4 19,9 19,5
Сайтон-2Х 14,2 14,5 14,2
Некоторые различия в данных, полученных для кремнезема
людокс, были приписаны более широкому распределению
частиц по размерам. Пертофт и др. [145] исследовали распре-
деление частиц по размерам в золях людокс-HS и людокс-SM.
Было обнаружено, что некоторые частицы имели предельные
для этих золей размеры 8—25 и 5—15 нм соответственно.
Разделение частиц по размерам. Центрифугирование пред-
ставляет собой классический метод разделения частиц. Для
частиц размером меньше ~-30 нм необходимы очень высокие
силы гравитации, соответствующие скоростям вращения ротора,
превышающим 10000 оборотов в минуту. Были изучены раз-
личные методы разделения, требующие менее дорогостоящего
оборудования. Некоторый прогресс был достигнут при исполь-
зовании хроматографической аппаратуры, которая хотя и до-
статочно дорога, но в общем более доступна.
Бернс и Чилтон [163] получили патент на способ, основан-
ный на гель-хроматографии. Декстрановый гель с размером
пор 40—120 мкм использовался для заполнения колонок.
Кремнезем со средней молекулярной массой 6 млн. (т. е. со
средним размером частиц 20 нм) разделялся по размерам во
всей области молекулярных масс. Другие авторы [164] исполь-
зовали шарики из пористого стекла для заполнения колонок
с целью их применения в эксклюзионной хроматографии кол-
лоидов. Применительно к условиям разделения коллоидного
кремнезема толщина двойного слоя на поверхности частиц при
pH 9,5 составляла 6 нм. Поэтому эффективный диаметр частиц
оказался на 12 нм больше, чем истинный, а эффективный диа-
476
Глава 4
метр пор в шариках из пористого стекла — на 12 нм меньше,
чем их фактический размер.
Гидродинамическая хроматография, в которой частицы-золя
с различными размерами распределяются по-разному вдоль
стенок в направлении потока, была применена для разделения
коллоидных частиц. Смолл [165] описал такой способ и запа-
тентовал его [166] для разделения частиц субмикронных раз-
меров. Однако, по всей вероятности, этот способ не может быть
применен для разделения коллоидных частиц очень небольшого
размера. Применительно к этому методу была предложена
[167а] математическая модель. Дальнейшее усовершенствова-
ние в области седиментационного фракционирования в потоке
было описано Гиддингсом и соавторами [1676, 167в].
Новый метод разделения латексных частиц посредством
электрофореза непрерывного действия может оказаться полез-
ным для разделения коллоидного кремнезема. Мак-Канн и др.
[168] обнаружили, что при выполнении определенных условий
в отношении ионного заряда и ионной силы различающиеся по
размеру частицы будут в процессе электрофореза перемещаться
с различными скоростями. Авторы разработали устройство для
разделения с учетом преимуществ такого явления.
Уже давно известен способ разделения различающихся по
молекулярным массам органических полимеров путем селектив-
ной коагуляции и осаждения, но, очевидно, этот способ никогда
не применялся для разделения частиц коллоидного кремнезема.
Айлер [169] доказал, что для флокуляции частиц кремнезема,
различающихся по размерам, требуются разные по значению
критические концентрации ионов кальция. Было продемонстри-
ровано, что в узкой области pH, примерно 8—9, частицы раз-
мером 10—24 нм могут быть разделены путем селективного
осаждения наибольших по размеру частиц..
Разновидности кремнезема субколлоидного размера. На ха-
рактеристики золя кремнезема может в заметной степени
влиять присутствие относительно небольших количеств так
называемых «активных» разновидностей кремнезема, таких, как
мономер Si (ОН) 4, низкомолекулярные поликремневые кислоты
или же силикат-ионы в щелочных растворах. Все эти разно-
видности поддаются обнаружению по реакции золя с молибде-
новой кислотой и регистрированию скорости образования и
количества желтого кремнемолибденового комплекса. Некото-
рые авторы достаточно произвольно подразделяют кремнезем
на тип А, который вступает в реакцию с молибденовой кисло-
той в течение 3—5 мин, и тип В, который реагирует с кислотой
гораздо медленнее.. Типичная кривая развития интенсивности
окрашивания, полученная Гото и Окура [170] для золя с ча-
стицами небольшого размера, представлена на рис. 4.9. Такой
Коллоидный кремнезем — концентрированные золи 477
золь перед добавлением к молибдатному реактиву разбавляли
приблизительно в десять раз. В исходном золе 0,01 —
0,02 масс. % мономерного кремнезема должно было бы нахо-
диться в состоянии равновесной растворимости с частицами
золя. При разбавлении золя в кислой среде мгновенного даль-
нейшего растворения не происходит, поскольку первоначально
в системе находится только примерно 0,002 % мономерного
Рис. 4.9. Зависимость количества кремнезема, вступившего в реакцию с молиб-
деновой кислотой, от времени (по данным Гото и Окура [170]).
кремнезема, способного быстро вступать в реакцию с молибда-
том. Коллоидная или полимерная фракция кремнезема (тип В)
продолжает деполимеризоваться с фиксированной скоростью,
давая на графике постоянный наклон линии. Согласно авторам,
экстраполяция такой линии к нулевому моменту времени дает
возможность находить первоначальную концентрацию присут-
ствовавшего мономера. Скорость образования окраски для
кремнезема типа В, которая представляет собой скорость де-
полимеризации, зависит от условий, при которых приготовляют
полимер или частицы. В другой работе Гото [171] подтвер-
ждает, что деполимеризация ускоряется в присутствии молиб-
деновой кислоты и пропорциональна суммарной величине
поверхности частиц коллоидного кремнезема. В аналогичных
исследованиях Бауман [172] останавливал реакцию образова-
ния комплекса желтого цвета путем добавления цитрата, а за-
тем определял содержание кремнезема путем превращения
желтого комплекса в молибденовую синь, что позволяет про-
водить более точные измерения концентрации кремнезема.
Растворы, содержащие коллоидный кремнезем, частицы ко-
торого достаточно малы и вследствие этого более растворимы
478
Глава 4
по сравнению с образцами массивного аморфного кремнезема,
в общем случае содержат 0,011—0,015 масс. % SiO2 в виде
мономера, а если система стабилизирована при pH ~9, то
также и ионы HSiOF-
Возникает вопрос: будет ли существовать равновесие при
pH 9—10, когда наряду с ионами НБЮГ и мономером Si(OH)4
присутствуют и полисиликатные ионы? Айлер обнаружил, что
в золе, подвергшемся старению, скорость реакции частиц крем-
незема с молибденовой кислотой не изменяется при удалении
коллоидных частиц центрифугированием и повторным разбав-
лением водой. Это указывает на отсутствие полисиликатов
в системе при данных условиях.
Удельная поверхность
В золях, в которых частицы относительно однородны и
имеют размер более 5 нм в диаметре, а также нормальное рас-
пределение по размерам по отношению к среднему значению,
определение удельной поверхности представляет собой быстрый
и надежный метод для оценки среднего размера частиц. Если,
с другой стороны, золи содержат частицы, размер которых
меняется в широкой области, как, например, в случае, золей,
приготовленных из пирогенного кремнезема или других по-
вторно диспергированных порошков, то результаты могут вво-
дить в заблуждение.
Соотношение между размером частиц, определяемым из
электронно-микроскопических снимков, и удельной поверх-
ностью кремнезема, определяемой по низкотемпературной ад-
сорбции азота (метод БЭТ), как уже ранее обсуждалось, было
впервые получено Александером и Айлером [66].
Обычно диаметр частиц, высчитываемый из значения удель-
ной поверхности, несколько меньше диаметра, о котором можно
судить на основании электронно-микроскопических снимков,
поскольку наименьшие по размеру частицы могут не наблю-
даться на таких снимках, но, однако, давать значительный
вклад в величину удельной поверхности. Так, например, на
электронно-микроскопическом снимке частиц кремнезема раз-
мером 100 нм добавление 0,1 % кремнезема, состоящего из
частиц диаметром 5 нм, вероятно, совсем не отразится и по-
этому не будет особенно важным. Но в то же время такие
небольшие частицы повышают кажущуюся величину удельной
поверхности больших по размеру частиц на 25—30 м2/г. К тому
же частицы кремнезема могут иметь некоторую шероховатость
поверхности, не различимую под электронным микроскопом.
Для определения удельной поверхности следует подкислить
золь примерно до нейтральной точки, добавить равный объем
Коллоидный кремнезем. — концентрированные золи 479
трет-бутилового или «-пропилового спирта и превратить золь
в гель, который затем высушить на воздухе. При таких усло-
виях • частицы кремнезема формируются в гель с открытой
сеткой и минимальным числом точек контакта между части-
цами; следовательно, потери поверхности при высушивании
будут минимальны. Высушенный таким образом порошок на-
гревают затем до 150°С, чтобы удалить физически адсорбиро-
ванную воду и спирт, и удельную поверхность определяют
одним из адсорбционных методов.
Адсорбция азота. Метод БЭТ [173] представляется наибо-
лее надежным и точным и в дальнейшем будет подробно рас-
смотрен в гл. 5. В методах быстрого определения удельной
поверхности порошков по адсорбции азота используется аппа-
ратура с непрерывным потоком газа, основанная на принципах
газовой хроматографии [174]. Эберли [175] изучил пригод-
ность подобного метода к образцам кремнезема и сообщил
о хорошем согласии с данными, полученными обычным методом
равновесных изотерм. Этр и Циплински [176] представили
обзор по газохроматографическим методам определения удель-
ной поверхности и их приложениям.
Адсорбция из растворов. Альтернативой адсорбции азота
является адсорбция красителя метилового красного из органи-
ческого растворителя. Однако проведение такой адсорбции из
раствора требует большего времени [177, 178]. Краситель
адсорбируется только на полностью гидроксилированной по-
верхности кремнезема, т. е. в присутствии воды, и высушивается
при умеренной температуре. Каутски и Михель [179] исполь-
зовали адсорбцию флуоресцентного красителя, такого, как ро-
дамин В. При адсорбции в виде катионов краситель дает
розово-красную флуоресценцию, но если краситель находился
не в ионизированном состоянии, то окраска была голубовато-
красной. Подобный метод полезен для определения удельной
поверхности полностью гидроксилированного золя кремнезема,
поскольку краситель может быть адсорбирован из воды.
Унгер и Выдра [180] использовали адсорбцию из водного
раствора Zn(NH3)4+ или Zn(en)2+ на порошках кремнезема
с целью определения удельной поверхности. Равновесие дости-
гается после непрерывного встряхивания порошка в растворе
в течение 5 ч. Удельная поверхность определяется по урав-
нению
А = 480с + 6,6
где А — удельная поверхность, м2/г; с — количество адсорби-
рованного Zn(en)2+ , ммоль/г SiOs- Авторы приводят следую-
щие состав раствора и условия опыта: 0,1 М Zn; 2,0 М NH4NO3;
0,4 М NH2C2H4NH2; pH 8,2; температура 20°С. 1 г SiO2 встря-
480
Глава 4
хивают с 25 мл раствора в течение 5 ч/ после чего отфильтро-
ванный раствор анализируют на содержание Zn, которое
понижается в ходе опыта.
Метод с использованием радиоактивного гафния, вероятно,
мог бы быть разработан на основании работы Штрикера и
Матиевича [181].
Метод титрования. Адсорбция основания на поверхности
кремнеземных частиц золя дает возможность быстро оценивать
поверхность раздела кремнезем—вода. В методе титрования
Сирса [182], т. е. титрования щелочью поверхности кремнезема
в крепком растворе соли в интервале pH 4,0—9,0, вместо вы-
сушенного порошка используется золь кремнезема. Как уже
обсуждалось в гл. 3, такой метод дает возможность наиболее
просто следить за изменением величины удельной поверхности,
а следовательно, и за изменением размеров частиц в том
случае, когда размер частиц кремнезема меньше 5—10 нм.
Однако метод оказывается равным образом полезным и для
частиц большего размера, вплоть до 1 мкм и более в диаметре,
если только представляется возможным доказать отсутствие
микропористости у частиц путем сравнения полученных резуль-
татов с величиной удельной поверхности, определяемой по
адсорбции азота или паров воды или же из рассмотрения элек-
тронно-микроскопических снимков.
Этот метод представляет интерес главным образом для
сравнения относительных значений удельных поверхностей, за-
висящих от размеров частиц в заданной системе, которая может
быть стандартизирована. Полученные при таких условиях ре-
зультаты воспроизводимы в пределах +5 %. В тех случаях,
когда имеются вариации в типах изучаемого кремнезема, раз-
личие значений удельных поверхностей от соответствующих
значений, получаемых методом БЭТ, может доходить до ±10 %.
Образец золя, содержащий 1,50 г SiOa, разбавляют очищен-
ной от СО2 водой при 25°С до концентрации 2—3 % S1O2,
подкисляют кислотой НС1 до pH ~3 и разбавляют до объема
135 мл. Затем добавляют 30 г чистой кристаллической соли
NaCl и смесь быстро перемешивают. Сразу же после растворе-
ния соли pH доводится до 4,00 за счет добавления 0,1 н. NaOH.
Для измерения pH используется стеклянный электрод для
«высокого содержания натрия» (прибор «Бекман», тип Е или
равноценный). Смесь быстро титруют до pH 9,00, добавляя
0,1 н. NaOH и отмечая объем после того, как значение pH
9,00 ± 0,05 сохраняется постоянным в течение примерно 1 мин.
(Если частицы не пористы, то конечная точка титрования ока-
зывается устойчивой.)
Хотя раньше наблюдалось некоторое различие в уравне-
ниях, связывающих объем титранта с величиной удельной
Коллоидный кремнезем ~ концентрированные золи
481
поверхности кремнезема, серия контрольных опытов, выполнен-
ных Айлером на пяти подвергавшихся в достаточной степени
старению золях (от 50 до 420 м2/г) и приготовлявшихся перво-
начально при повышенной температуре, дала возможность
получить усредненное значение, соответствующее уравнению
A = 26,4(yt-Vb)
А — удельная поверхность, определяемая методом БЭТ, м2/г;
Vt — объем NaOH, требуемый для титрования 1,50 г SiOa, мл;
Уь — объем титранта в холостом опыте при отсутствии кремне-
зема, обычно составляющий около 0,3 мл (см. для сравнения
уравнение на стр. 280 в гл. 3).
Вполне вероятно, что частицы золя кремнезема могут отли-
чаться по степени шероховатости поверхности. К тому же зна-
чения удельных поверхностей, определяемые по адсорбции
азота, могут зависеть отчасти от способа предварительного
высушивания золя. Упоминавшийся выше метод определения
удельной поверхности по адсорбции азота включал процессы
деионизации золя обработкой его смесью анионо- и катионо-
обменных смол, регулирования значения pH до 2,0 и выпарива-
ния золя при 25—40°С до тех пор, пока не произойдет геле-
образование. Гидрогель подвергали диспергированию по край-
ней мере в десятикратном избыточном по массе количестве
н-пропилового спирта и высушивали на воздухе. Полученные
образцы затем нагревали на воздухе при температурах 150
и 350°С в течение 16 ч, после чего определяли значения удель-
ной поверхности этих образцов по адсорбции азота с использо-
ванием стандартного метода БЭТ. Значения удельных поверх-
ностей, определяемых на двух идентичных образцах, высушен-
ных при указанных выше температурах, отклонялись между
собой менее чем на ±2 %. В том случае, когда размер частиц
оказывался меньше 7—8 нм, то даже при очень осторожном
проведении процедуры гелеобразования и высушивания крем-
незема не удалось предотвратить некоторую потерю величины
удельной поверхности из-за возникновения контактов между
частицами.
Скорость растворения частиц. Взаимозависимость между
размерами частиц и скоростью растворения кремнезема по-
дробно рассматривалась в гл. 1.
Вполне логично предположить, что скорость деполимериза-
ции или растворения частиц коллоидного кремнезема должна
быть пропорциональна величине удельной поверхности. Следо-
вательно, различные растворители, такие, например, как раз-
бавленная плавиковая кислота, молибденовая кислота или же
разбавленная щелочь, т. е. растворители, способные превращать
мономерный кремнезем в другие соединения кремния, могут
5 Заказ № 250
482
Глава 4
применяться для измерения удельной поверхности. Трудность,
однако, заключается в том, *йто типы изучаемых частиц крем-
незема настолько сильно отличаются друг от друга и плохо
идентифицируются, что такое приближение вплоть до послед-
него времени не представляло какой-либо ценности. И только
недавно этот подход стал оправданным, когда появилась воз-
можность получать золи с однородными по размеру дискрет-
ными твердыми частицами. Но даже и при выполнении послед-
него условия значение метода сомнительно, если только нельзя
быть уверенным в том, что все исследуемые образцы кремне-
зема имеют, одинаковые состав и структуру. Переменными
величинами при определении скорости растворения, отнесенной
к единице площади поверхности, будут следующие:
1. Степень пористости или величина внутренней гидратации
в виде несконденсированных силанольных групп.
2. Количество примесей, особенно алюминия, когда частицы
растворяются в щелочи.
3. Размер частиц, так как растворимость возрастает с умень-
шением размера, особенно когда частицы менее 5 нм в диа-
метре.
4. Неоднородность частиц по размерам: меньшие по раз-
меру частицы растворяются более быстро.
Ионный заряд на поверхности частиц
Ионизация силанольных групп на поверхности раздела крем-
незем—вода рассматривалась, в гл. 3 в связи с процессом
полимеризации кремнезема. Изоэлектрическая точка и точка
нулевого заряда кремнезема располагаются при pH ~2.
Скорость конденсации силанольных групп SiOH, приводящей
к образованию силоксановых связей Si—О—Si, как и скорость
обратной реакции гидролиза, будет наименьшей при этом зна-
чении pH, так как в обеих реакциях участвует ион = Si~:
=SiO- + HOSia= = =SiOSi^ + он-
' Таким образом, несмотря на то что при pH 2 частицы крем-
-незема несут на поверхности лишь очень небольшой заряд или
же он вообще отсутствует, вследствие чего частицы могут
сталкиваться, образование связей Si—О—Si между частицами
„и формирование геля происходят медленно.
Однако только при значениях pH выше ~7, т. е. когда на
поверхности частиц образуется достаточно высокая концентра-
ция отрицательных зарядов, способных вызывать взаимное
отталкивание частиц, система стабилизируется настолько, что
гелеобразование не будет происходить. При промежуточных
значениях pH гелеобразование протекает наиболее быстро.
Коллоидный кремнезем — концентрированные золи
483
Природа ионного заряда
Как будет обсуждено ниже в гл. 6, отрицательный заряд на
поверхности аморфного кремнезема может быть интерпретиро-
ван несколькими способами. Одна из точек зрения заключается
в том, что структура кремнезема представляется достаточно
открытой, и гидроксил-ион способен занимать некоторый уча-
сток на поверхности. Поэтому координационное число каждого
соседнего с гидроксил-ионом атома кремния повышается на
некоторую дробную величину (см. рис. 1.11). Другая трактовка
предусматривает возможность ионизации поверхностных сила-
нольных групп:
=SiOH = =SiO- + Н+
Помимо исследований, упоминаемых в гл. 3 в связи с иони-
зацией моно- и поликремневых кислот, имеется гораздо больше
данных, относящихся к золям и гелям. Брайент [183] изучал
влияние размера частиц и концентрации электролита на кри-
вую кислотно-основного титрования золей кремнезема и интер-
претировал полученные данные в понятиях теории полимерных
электролитов, принимая во внимание преимущественную ад-
сорбцию катионов, а также тот факт, что при высоких значе-
ниях pH кремнеземные частицы растворяются с образованием
силикатов. В области pH 3,5—10,5 гидроксил-ионы адсорби-
руются на поверхности кремнезема, как показано Болтом [184],
в количествах, возрастающих с увеличением pH (рис. 4.10).
Концентрация анионных зарядов может выражаться., как число
миллиэквивалентов основания, приходящихся,, на 1 м2 площади
поверхности, или как число гидроксил-ионов, или зарядов,
в расчете на 1 нм2. Болт использовал в своих измерениях ком-
мерческий золь кремнезема с диаметром частиц 15 нм, имею-
щий удельную поверхность 180 м2/г. Автор нашел, что одина-
ковые значения заряда на поверхности наблюдаются при
концентрациях золя 3—30 % SiO2. Хестон, Айлер и Сирс [185]
показали, что при данном значении pH число зарядов, прихо-
дящихся на единицу поверхности кремнезема, не зависит от
размера частиц. Фундаментальные исследования ионных заря-
дов на поверхности раздела вода—оксиды металлов и вода—
кремнезем были выполнены Грэмом [186], Перрамом [187],
Парксом [188], Алленом, Матиевичем и Местесом [189], Тед-
росом и Ликлема [190], Йетсом и Хили [191] и другими.
Высоцкий и Стражеско [192] обратили особое внимание на
природу катионообмена или заряженных мест на поверхности.
Йетс, Левин и. Хили [193] представили модель связанных
между собой участков на поверхности раздела вода—оксид
5*
484 .
Глава 4
металла, на основании которой следует, что адсорбированные
противоионы образуют на поверхности ионные пары с раз-
дельно расположенными заряженными поверхностными груп-
пами. Они также обсудили вопрос об аномальных значениях
констант диссоциации групп S1OH.
О 0.1 0,2 0,3 0,4 0,5
Миллмэнв. ОН/г Si02
J____________I___________1_________-J______
Число ионов ОН'на 1 нмг
Рис. 4.10. Зависимость плотности заряда, выражаемой числом ионов ОН-,
находящихся на поверхности частиц коллоидного кремнезема с удельной по-
верхностью 180 м2/г, от pH и различных концентраций хлорида натрия (по
данным Болта [184]).
Согласно данным Йетса и Хили [191], кремнезем представ-
ляет исключение среди других коллоидов в отношении зависи-
мости поверхностного заряда от pH и общей величины емкости
двойного электрического слоя. Указанные факторы оказываются
гораздо более высокими для кремнезема по сравнению с дру-
гими коллоидными системами, для которых была развита
приемлемая теория, основанная на уравнении Нернста. Перрам
[187] предположил, что при высушивании поверхность такого
кремнезема покрывается слоем геля, обладающего микропо-
рами, доступными для ионов, но не для азота или каких-либо
других молекул. Йетс и Хили на основании. своих исследова-
ний по осаждению кремнезема (типа BDH) пришли к анало-
гичным выводам. Они также внесли коррективы в кривые
потенциометрического титрования при pH >9 с учетом иониза-
ции растворимого кремнезема. Подобные исследования были
Коллоидный кремнезем — концентрированные золи
485
также выполнены Тедросом и Ликлема [190], которые пришли
к заключению, что отрицательно заряженные группы и катионы
должны проникать внутрь таких пор «слоя геля», но степень
их проникновения зависит от размера катиона.
Как уже выше обсуждалось, частицы коллоидного кремне-
зема, приготовленные разными способами, имеют различную
степень микропористости. Вследствие этого не удивительно, что
кремнезем, обладающий микропористостью, ведет себя совер-
шенно иначе, чем непористые частицы других неорганических
коллоидных систем, в особенности частицы с кристаллическими
поверхностями. Различные свойства следует измерять на глад-
ких непористых поверхностях, чтобы обеспечить получение
воспроизводимых данных, о которых можно говорить как о ха-
рактерных для «поверхности аморфного кремнезема».
Как было показано также Йетсом и Хили, в том случае,
когда предпринимались попытки получить непористые частицы
кремнезема нагреванием образца до 800°С, значительная часть
поверхности дегидроксилировалась до образования силоксановых
групп. При регидроксилировании такой поверхности не было
получено никаких данных, свидетельствовавших о повторном
формировании слоя геля. Однако на поверхности термообра-
ботанного кремнезема, а также на частицах пирогенного крем-
незема после гидратации образцов плотность поверхностных
зарядов по отношению к величине pH оказывается все еще
гораздо выше, чем это наблюдается, скажем, в таком класси-
ческом примере, как поверхность раздела Agl — раствор.
Отсюда можно заключить, что заряженные ионы в состоянии
проникать до некоторой степени даже в «непористую» поверх-
ность аморфного кремнезема.
Высказывалось предположение (см. [8]), что поверхность
кремнезема заряжается за счет гидроксил-ионов, образовав-
шихся в результате потери протонов теми молекулами воды,
которые располагаются на участке между атомами кислорода
в структуре SiO2 (см. рис. 1,11). Таким образом, отрицательный
заряд должен лежать внутри у самой поверхности. Если эта
поверхность напоминает поверхность тридимита, что кажется
вподае возможным, поскольку ее структура в таком случае
находится в соответствии с величиной поверхностной концент-
рации, равной 4,6 S1OH групп на 1 нм2, то тогда наблюдается
ослабление заряда на поверхности в результате возможного
близкого расположения катионов к противоположно заряжен-
ным ионам.
Как отмечают Тедрос и Ликлема, малые по размеру одно-
зарядные ионы, подобные Na+, могут наиболее легко следовать
за отрицательными зарядами в глубь поверхности. По их мне-
нию, большие по размеру катионы (CH3)4N+ при заданном
486
Глава 4
значении pH способствуют образованию гораздо меньшего
поверхностного заряда. К такому же заключению пришел Айлер
• [169], который показал, что специфическое коагулирующее
действие ионов кальция отличается от поведения ионов натрия
из-за большего расстояния, на которое удалены ионы кальция
от внешнего силанольного слоя по сравнению с ионами натрия.
Подобный геометрический фактор приводит к предпочтительной
коагуляции больших по размеру коллоидных частиц кремне-
зема в присутствии меньших частиц. Этого не происходит
в случае ионов натрия.
Характерная особенность аморфного кремнезема, возможно,
состоит в, том, что отрицательный заряд на поверхности раздела
твердое тело—вода может образовываться скорее отчасти
внутри у поверхности, чем на такой поверхности, и поэтому
относительно большие количества кремнезема и меньшие воды
учитываются в экспериментально определяемом среднем зна-
чении диэлектрической проницаемости.
Так как настоящая глава относится главным образом к рас-
смотрению частиц коллоидного кремнезема, которые приготов-
ляются по способам, предназначенным для формирования
плотных непористых частиц, то интересно сравнить плотность
поверхностного заряда, получаемую при заданных условиях,
с плотностью заряда на изученных кремнеземных порошках.
В табл. 4.1 представлены условия опытов, одним из которых
является 0,1 н. концентрация электролитов NaCl, КС1 или
KNO3, дающих однозарядные ионы при pH 9,0.
Таблица 4.1
Сравнение величины плотности заряда на поверхности кремнезема
прн фиксированных условиях н pH 9
Авторы Кремнезем Удельная поверхность, мг2/г Электролит (0,1 н.) Плотность заряда, мкКл/см2 Число зарядов $ на 1 нм2
Йетс, Хили BDH 73 KNO3 40,0 2,51
(500 °C) 64 KNO3 11,4 0,716
(800 °C) 36 KNO3 8,8 0,552
Болт Людокс-HS 200 NaCl — 0,68
Хестон и др. Золь 318 Na+a — 0,63
а Присутствуют только противоионы, нет никаких других небольших по размеру
анионов, кроме HSiO£" . Данные в незначительной степени экстраполированы.
б 1 мкКл/см2—0,0628 заряд/нм2.
Коллоидный кремнезем — концентрированные золи 487
Результаты, полученные Болтом [184] (см. рис. 4.10),
а также Хестоном, Дилером и Сирсом [185], на непористых
частицах кремнезема сравниваются с результатами Йетса и
Хили, полученными на осажденном кремнеземе (BDH), пори-
стом до тех пор, пока его термически не обработают при высо-
кой температуре. Вполне очевидно, что порядок величины
плотности поверхностного заряда на частицах золя будет тем
же самым, что и на осажденном порошке после его термообра-
ботки. Частицы золя никогда не нагревались выше 100°С и тем
не менее оказывались плотными, поскольку были приготовлены
путем медленного осаждения молекулярного кремнезема. Если
же частицы формируются иным путем, то они получаются
пористыми, что проявляется в более высоком значении плот-
ности заряда. Однако кажется, что даже рассматриваемые
частицы золя еще не в такой степени плотные, как частицы,
прошедшие термообработку до 800°С. Это доказывается не-
сколько более высокой плотностью заряда, равной в первом
случае 0,63—0,68, по сравнению с величиной 0,55 для термо-
обработанных частиц кремнезема (см. табл. 4.1).
Противоионы и двойной электрический слой
Ионы натрия и другие катионы, окружающие в водной среде
заряженные частицы кремнезема, не удаляются полностью
в процессе фильтрации или центрифугирования и поэтому
остаются на поверхности кремнезема после его высушивания.
Если частицы кремнезема не находились в постоянном «броу-
новском» движении, то в водных растворах противоионы, такие,
например, как ионы натрия, должны образовывать сплошной
слой вблизи адсорбированных на поверхности кремнезема гид-
роксил-ионов: Однако прй термическом возбуждении частиц
кремнезема из большей части указанных противоионов вокруг
частиц формируется диффузное облако, называемое «слоем Гуи».
Оставшаяся часть противоионов вблизи поверхности рассмат-
ривается как «слой Штерна». Толщина диффузного слоя Гуи
определяется расстоянием от поверхности частицы до точки,
в которой потенциал составляет только 1/е или 0,37 значения
потенциала в бесконечности. «Дзета-потенциал» измеряется
посредством электрофореза и рассматривается как потенциал
между «плоскостью скольжения» на наружной границе слоя
Штерна, которая перемещается вместе с движущейся частицей,
и бесконечно удаленной точкой дисперсионной среды.
Адсорбированные на поверхности анионы и сопровождаю-
щее частицу заряженное облако, состоящее из катионов, обра-
зуют «двойной слой». Распределение ионов, толщина слоя и
плотность зарядов четко рассмотрены и объяснены Майселсом
488
Глава 4
[194] и подробно представлены в обзоре Грэма [195]. Плот-
ность заряда может быть выражена числом единичных зарядов
(например, числом адсорбированных на поверхности кремне-
зема гидроксил-ионов), приходящимся на единицу поверхности.
Заполненный монослой, состоящий из ионов средних размеров,
должен обеспечивать плотность, приблизительно равную пяти
единичным зарядам на 1 нм2. Однако такая плотность зарядов
не достигается из-за взаимного отталкивания ионов. Даже
значение 1 заряд/нм2 представляет собой очень высокую плот-
ность заряда для стабилизированной коллоидной системы.
Ликлема [196] в своем обзоре рассмотрел образование
зарядов на поверхности частиц и ряд других факторов, обуслов-
ливающих устойчивость коллоидных систем, а также относя-
щиеся к данной теме теоретические вопросы. Общая теория
была также рассмотрена Нэппером [197]. Превосходную
библиографию содержат книги Шелудько [198] и Круита [199].
Лонг и Росс [200] исследовали толщину двойного диффуз-
ного слоя, окружающего частицы кремнезема, которые имели на
поверхности постоянные отрицательно заряженые анионные
центры вследствие присоединения четырехкоординированного
оксида алюминия. В своей работе авторы использовали ком-
мерческий коллоидный кремнезем типа людокс-АМ. Измеряя
электрофоретическую подвижность при постоянном значении pH
и содержание электролита в пределах ряда выбранных концент-
раций кремнезема, Лонг и Росс изучили перекрывание двойных
диффузных слоев в довольно концентрированных золях'. Хотя
повышение концентрации электролита приводило к уменьшению
толщины двойного электрического слоя, оно также вызывало
возрастание адсорбции гидроксил-ионов и вследствие этого уве-
личение поверхностного потенциала. Таким образом, полная
величина электрокинетического потенциала, определяющая
электрофоретическую подвижность, оставалась относительно
неизменной.
Необычный метод изучения заряда на поверхности частиц
кремнезема был применен в работе [201]. Коллоидный кремне-
зем типа людокс, частицы которого имели диаметр 15 нм, под-
вергали ультразвуковой обработке. Помещая электроды в узло-
вых точках и в пучностях волн, авторы замеряли потенциал
такой коллоидной системы, который повышался с возрастанием
концентрации кремнезема в интервале от 0 до ~4 % SiO2.
Потенциал, по-видимому, возникает благодаря движению заря-
женных частиц по отношению к фиксированному положению
электрода.
Янгер и др. [202] измеряли скорость звуковых колебаний
в концентрированных золях кремнезема в пределах некоторой
области температур. Действие температуры на «гидратную обо-
Коллоидный кремнезем — концентрированные золи 489
лочку», образуемую вокруг частицы кремнезема, оказывается
важным фактором, но, как сообщалось в других исследованиях
систем кварц—вода, не было обнаружено каких-либо термиче-
ских аномалий.
Типы противоионов. В большей части патентов, относящихся
к коллоидному кремнезему, в качестве стабилизирующего осно-
вания вводятся гидроксиды калия, лития, натрия и аммония.
Золи наиболее часто стабилизируются гидроксидом натрия.
Если его необходимо заместить другими основаниями, золи
деионизируют в слое, состоящем из смеси анионита и катионита,
и повторно стабилизируют выбранным основанием. Когда жела-
тельно получить чистый кремнезем, употребляются золи, стаби-
лизированные аммиаком. Вместо аммиака могут быть использо-
ваны низшие амины, но обычно их избегают из-за неприятного
запаха [203].
Литий, согласно данным Рула [7], единственный в своем
роде по стабилизирующему действию среди других катионов
щелочных металлов. При использовании лития в отличие от
натрия к золю можно добавлять смешиваемые с водой спирты,
не опасаясь превращения золя в гель; кроме того, образую-
щиеся золи оказываются морозостойкими.
Айлер [204] обнаружил, что гидроксид лития непохож на
гидроксид натрия и в том отношении, что может быть добавлен
к золю в качестве стабилизатора в любом количестве, не
вызывая процесса коагуляции. Таким образом, золи, имеющие
соотношения SiO2: ЫгО в интервале от 4:1 до 25:1, оказы-
ваются устойчивыми, совместимыми с органическими жидко-
стями, смешивающимися с водой, и при высушивании образуют
водостойкие пленки (см. гл. 2, разд. «Полисиликаты лития»).
Как уже было рассмотрено в гл. 2 в связи с вопросом
о силикатах и полисиликатах лития, ионы лития исключительны
в том отношении, что способны предпочтительно адсорбиро-
ваться на поверхности кремнезема, формируя непроницаемый
слой, который замедляет процесс растворения кремнезема при
высоких значениях pH [206].
Уолтер [205] нашел, что ионы низших алифатических соеди-
нений четвертичного аммониевого основания, в частности
тетраметиламмоний, за счет образования адсорбированного
слоя из ионов вокруг поверхности частицы кремнезема спо-
собны обеспечивать такую степень стабилизации, что получен-
ный золь можно высушивать до порошка, который при
погружении в воду будет самопроизвольно диспергировать,
образуя водный золь. Подобные золи также очень морозо-
стойки и совместимы с органическими растворителями, способ-
ными смешиваться с водой, например с ацетоном и спиртом.
Амины с длинными цепями или ионы четвертичного аммоние-
490
Глава 4
вого основания также с длинной цепью оказывают, с другой
стороны, сильное флокулирующее действие на коллоидный
кремнезем и поэтому не могут быть использованы в качестве
стабилизаторов дисперсии в водных системах, но пригодны для
изменения заряда на поверхности частиц на противоположный.
Влияние концентрации противоионов на поверхностную
плотность зарядов. Хорошо известно, что в присутствии такого
электролита, как хлорид натрия, при заданном значении pH
достигается более высокая плотность заряда на поверхности.
Гораздо менее очевидно то, что в относительно концентриро-
ванных, стабилизированных щелочью золях концентрация
противоионов натрия должна учитываться как «электролит»
во взаимозависимости между поверхностной плотностью заряда
и величиной pH.
Болт [184] выполнил измерения в присутствии введенной
в систему соли NaCl, а Хестон, Айлер и Сирс [185] провели
аналогичные измерения в ее отсутствие, но с учетом концент-
рации противоионов Na+ в системе. В последней работе поверх-
ностный заряд адсорбированных ионов ОН- выражался
в значениях относительной величины f — доли от максимально
возможной плотности заряда, равной 3,5±0,3 ОЬГ /нм2. Данные
Болта, которые можно выразить таким же способом при pH 9,
а также данные Хестона и др. представлены ниже:
Концентрация Поверхностный заряд, число ионов ОН~ на 1 нм2
Противоионов Na+, и. NaCl, и. По Хестону и др. По Болту
0,001 — 0,28 —
— 0,001 — 0,29
0,01 — 0,42 —
— 0,01 .— 0,44
0,1 — 0,63 —
— 0.,1 — 0,68
Чтобы зарядить поверхность кремнезема в концентрированных
золях, состоящих из очень небольших частиц, требуется отно-
сительно высокое количественное отношение натрия к кремне-
зему, при этом концентрация ионов натрия может оказаться
настолько высокой, что достигает значения «критической кон-
центрации коагуляции» при данном pH, и поэтому такой золь
будет неустойчивым. Это накладывает другое ограничение на
концентрацию электролита для золей подобного типа. (Кон-
центрированные золи, содержащие очень малые по размеру
частицы и стабилизированные в области отношений SiO2: Na2O
6—20, становятся нестабильными и подвергаются гелеобразо-
ванию.) Такова характерная особенность ионов натрия, но не
ионов лития или калия. Для последних типов ионов растворы,
Коллоидный кремнезем — концентрированные золи 491
состоящие из концентрированных золей, могут приготовляться
независимо от размеров частиц кремнезема: от коллоидных до
высокомолекулярных полисиликатов. Ионы лития, как отмеча-
лось, ведут себя необычным образом [206].
Вязкость
Как отмечал Круит [207], вязкость золя зависит от объем-
ной доли «дисперсной фазы» согласно уравнению Эйнштейна
In ты — 1 +2,5<£
где T]rez — относительная вязкость или отношение вязкости дис-
персной системы к вязкости дисперсной среды; ф— доля
объема, занимаемая дипергированными, однородными по раз-
меру сферическими частицами. Муни [208] распространил
уравнение Эйнштейна на суспензии с ограниченным значением
концентрации:
. 2,5^
In — !_114з^
Был выведен целый ряд других уравнений, по которым можно
предсказывать даже с большей точностью вязкость более вы-
соко концентрированных суспензий, состоящих из сферических
частиц, но эти уравнения в основном не были применены
к золям. Симха [209], Вэнд [210] и Форд [211] использовали
такие уравнения в своих исследованиях.
В 1965 г. Томас [212] опубликовал обзор, в котором рас-
смотрел большой объем экспериментальных данных по значе-
ниям относительной вязкости суспензии с однородными по
размеру сферическими частицами и связал эти данные с урав-
нениями, полученными из предварительного теоретического
анализа. Он пришел к заключению, что к указанным экспери-
ментальным данным в пределах всей области концентраций
лучше всего подходит следующее уравнение (при значениях
концентраций <0,25 экспоненциальный член в уравнении
можно опустить):
Лгег = 1 + 2,5ф + 10,05ф2 + 0,002 73 ехр (16,6ф)
Льюис и Нильсен [213] распространили эту теорию на
вязкость непрерывно образующихся агрегатов из сферических
частиц, начиная от агрегатов, состоящих из двух или трех
частиц, и вплоть до кластеров, в которых удерживается вместе
большое число таких сферических частиц. Можно ожидать, что
эта теория более правильно описывает не те золи кремнезема,
в которых все частицы представляют собой индивидуальные
сферы, а те, в которых уже начинает формироваться фаза геля,
492
Глава 4
стабилизирующаяся впоследствии. Теорию можно также было
бы применить к золям, содержащим агрегаты, подобные тем,
которые образуются в кремнеземах пирогенного типа.
Федоре [214] принял во внимание максимальную плотность
упаковки сферических частиц в суспензии, что позволило ему
получить уравнение для очень высоких концентраций золей.
Объемная доля упакованных произвольным образом, но плотно,
сферических частиц составляет 0,63.
Другие исследования вязкости сферических частиц, находя-
щихся в суспензии, были выполнены Менли и Мейсоном [215]
и Хеппелом [216], а реология суспензий кремнезема (некол-
лоидного типа) описана Пивинским [217].
Как показано на рис. 4.11, при заданной массовой концент-
рации кремнезема в золе объемная доля дисперсной фазы изме-
няется в зависимости от объема ионной оболочки, окружающей
каждую частицу кремнезема в виде двойного слоя, и от по-
ристости или степени гидратации частиц. Если вязкость изме-
ряется при pH 2, когда отсутствует заряд на поверхности
частиц, то величина объемной доли может быть использована
для установления того, сколько воды связано с поверхностью
твердых частиц или удерживается внутри частиц с губчатой
структурой и агрегатов, поскольку эта вода может рассматри-
ваться как часть дисперсной фазы.
В серии измерений вязкости, выполненных при pH 2 Айде-
ром и Дальтоном [64] на ряде образцов золей, состоящих из
плотных частиц с диаметром менее 8 нм, было показано, что
такая связанная вода соответствует слою толщиной в одну
молекулу воды. Если диаметр частиц свыше 20 нм, подобный
фиксированный гидратный слой воды толщиной всего лишь
около 0,3 нм оказывает небольшое воздействие на вязкость,
но, когда частицы кремнезема имеют размер только 5 нм, раз-
личие в 0,6 нм повышает эффективный диаметр частиц до
5,6 нм и, следовательно, увеличивает эффективный объем
«дисперсной фазы» на 40 %.
Что же касается пористых частиц, которые рассматривались
выше, то вязкость оказывается не очень чувствительным пока-
зателем для оценки пористости, особенно если объем пор состав-
ляет менее 10—20%. С другой стороны, как упоминалось
в гл. 3, при pH 2 вязкость представляет собой чувствительный
критерий агрегирования частиц или присутствия в системе
микрогеля. Для частиц, в диамётре меньших ~20 нм, необхо-
димо принимать во внимание влияние гидратного слоя.
Из известной величины удельной поверхности, которая не
сильно изменяется по мере формирования микрогеля, можно
оценить средний размер частиц. В таком случае оказывается
возможным подсчитать из размера частиц вязкость золя, со-
Коллоидный кремнезем — концентрированные золи
493
" 08000^
°о О°оо Ог
>грооо°оос
О?) U
Рис. 4.11. Схема влияния различных факторов на вязкость золей.
При одинаковых концентрациях кремнезема высоко заряженные частицы (а) способст-
вуют более высокому значению вязкости золей по сравнению со слабо заряженными ча-
стицами (б). Вязкость золей с плотными частицами (в) более низка, чем в случае пори-
стых агрегатов, состоящих из меныпих по размеру частиц (г). Не образующие агрегатов
небольшие частицы (д) также дают более низкую вязкость, чем агрегированные ча-
стицы (г).
стоящего из неагрегированных частиц, и его концентрацию.
Окончательно величина относительной вязкости вследствие
присутствия в системе микрогеля берется как отношение факти-
ческой вязкости золя к ее значению, подсчитанному для золя,
состоящего из неагрегированных частиц.
Зависимость вязкости концентрированных золей от тол-
щины двойного ионного слоя следует рассматривать с точки
зрения наибольших достижимых на практике концентраций, до
которых могут быть сконцентрированы золи с различающи-
мися по размеру частицами. Для того чтобы охарактеризовать
494
Глава 4
концентрированные коммерческие золи, представляется полез-
ным ввести такую величину, как «выраженная в масс. % доля,
занимаемая кремнеземом в дисперсной фазе». Если такая
дисперсная фаза состоит из больших твердых сферических
частиц кремнезема, когда связанная с поверхностью вода
составляет незначительную часть по сравнению с массой самой
частицы, то масс. % SiO2 близок к 100 %. Для меньших по
размеру частиц, когда связанная с поверхностью вода и сила-
нольные группы на поверхности частицы составляют значи-
тельную долю от ее массы, масс. % SiO2 оказывается менее
100%. Однако, когда частицы кремнезема агрегируют и обра-
зуют суспендированную в системе массу микрогеля, подобные
агрегаты составляют дисперсную фазу, которая также вклю-
чает в себя и воду. В последнем примере масс. % SiO2 значи-
тельно понижен. (Для обозначения концентрации кремнезема
в дисперсной фазе Александер использовал символ S, но в дан-
ной монографии такая концентрация будет обозначаться
символом Cd.) Величина вязкости дает возможность измерять
содержание геля кремнезема, полученного из золя, при усло-
вии, что измерение выполняется при pH 2. Александер [9]
подробно описал этот способ измерений. Золь кремнезема
деионизируют и значение pH доводят до 2 добавлением силь-
ной кислоты. Наиболее удобна для проведения эксперимента
концентрация ~4% SiO2, которая должна быть в точности
известна. Измерения вязкости выполняют с образцами объемом
в 10 мл при 30°С в пипетках Оствальда № 100. Величина
— объемная доля дисперсной фазы — высчитывается из урав-
нения Муни [208], a Cd — доля (в масс. %), занимаемая
в дисперсной фазе кремнеземом,— определяется по уравнению
Р _______________Ct___________
d + Ф (1 -0,00566Cz)
где Ct — суммарное содержание кремнезема в золе, масс. %.
Плотность аморфного кремнезема при этом принимается равной
2,2 г/см3. Золи, не содержащие сколько-нибудь заметного
количества фазы геля, имеют Cd менее 100 %, обычно 70—80 %,
благодаря наличию воды, связанной с поверхностью частиц [64].
Если золь подвергается старению при низком значении pH,
а вязкость повышается вследствие начинающегося процесса
гелеобразования, то доля Cd падает ниже 50 %.
В области значений рН*3—4, когда поверхность кремнезема
заряжается очень слабо, в коммерческих кремнеземных золях
на поверхности может обнаруживаться заметно больший отри-
цательный заряд, определяемый присутствием немногочислен-
ных алюмосиликатных центров, так как алюминий почти всегда
имеется на поверхности в качестве загрязнения. При pH 2
Коллоидный кремнезем — концентрированные золи 495
такие центры подавляются. Авторы работы [218] считают, что
данные по вязкости дают основание предполагать существова-
ние на поверхности частиц кремнеземных золей при pH 2,8
гидратной оболочки толщиной 15 А, приблизительно соответ-
ствующей пяти мономолекулярным слоям воды. Толщина слоя
представляет собой среднее расстояние от поверхности кремне-
зема до граничной поверхности скольжения при условии, что
подобная поверхность частично заряжена. При высоких значе-
ниях pH гидратный слой вокруг частицы, содержащий проти-
воионы, становится во много раз толще, чем это имеет место
в разбавленных золях. Но в концентрированных, стабилизиро-
ванных щелочью золях окружающая частицу оболочка из
противоионов может быть сжата до такой степени, что ее тол-
щина подсчитанная из вязкости, не составляет более 20—30 А.
Как уже обсуждалось выше в связи с вопросом о получаемых
практически наиболее высоких концентрациях коммерческих
золей, содержащих различные по размеру частицы, было сде-
лано заключение о том, что среднее значение минимальной
толщины двойного слоя составляет примерно 2,4 нм, или 24 А.
Можно отметить, что для достижения наиболее высокой
возможной концентрации коллоидного кремнезема с целью
определенного его применения необходимо понижать значение
pH до 2 для исключения электрического двойного слоя, а также
использовать смесь, состоящую из двух или трех разновидностей
золей, в значительной степени различающихся по размерам
частиц [219]. Высушенный гель, полученный в виде смеси,
состоящей из 10 массовых частей частиц размером 100 нм и
6,24 массовых частей частиц размером 7 нм, имел пористость,
составляющую только 20 объемн. %.
Агрегация частиц
Определения
Некоторые исследователи считают, что не существует каких-
либо принципиальных различий между гелеобразованием,
с одной стороны, и коагуляцией или флокуляцией — с другой.
Оба процесса включают в себя как совместное связывание
коллоидных частиц, так и формирование трехмерных сеток.
Однако в том случае, когда золь превращается в гель, сначала
повышается вязкость системы, а затем происходит ее затвер-
девание. С другой стороны, при коагуляции или флокуляции
золя формируется осадок. В концентрированных золях такой
осадок может оказаться слишком объемистым и трудноотдели-
мым и будет сохраняться в виде тиксотропной массы. В раз-
496
Глава 4
ООоОС
6
Рис. 4.12. Схемы гелеобразования (б) и флокуляции и осаждения (в) золя (а).
бавленных золях подобный осадок оседает на дно. На рис. 4.12
показано различие между рассматриваемыми системами.
Впоследствии Ламер и Хили [220] предложили различать
понятия «коагуляция» и «флокуляция». Термин «флокуляция»
они отнесли к особому случаю коагуляции, когда окончательно
сформировавшаяся структура удерживается мостиками, состоя-
щими из органических молекул или из каких-либо коллоидный
неорганических частиц. При этом образуется свободная трехмер-
ная пористая сетка, обладающая свойствами фильтрации. Таким
образом, авторы сохраняют выражение «флокуляция» в его пер-
воначальном значении как процесса формирования свободной
открытой структуры и получения флокулированного осадка по-
добного пучку шерсти. В отличие от этого термин «коагуляция»
происходит от латинского выражения, означающего «переме-
щаться вместе», и, следовательно, используется для тех случаев,
когда первичные частицы переносятся вместе с образованием
относительно компактных агрегатов, или кластеров, способных
к плотному осаждению. Таким образом, по сравнению с фло-
куляцией подобный плотный осадок с трудом удаляется по-
Коллоидный кремнезем — концентрированные золи 497
средством фильтрации. Вполне очевидно, что не существует
сильного различия между отмеченными двумя терминами.
Понятие «агрегация» всегда используется в тех случаях,
когда коллоидные частицы связываются вместе. Таким обра-
зом, агрегация включает в себя следующие процессы:
1. Желатинирование. Связанные вместе частицы образуют
разветвленные цепочки, которые целиком заполняют объем
золя. Поэтому не наблюдается повышение концентрации крем-
незема в любой выбранной макроскопической области в среде
системы. Вместо этого вся среда становится вязкой, а затем
затвердевает в состоящую из связанных частиц сетку. Эта
сетка благодаря своему капиллярному строению может удер-
живать жидкость.
2. Коагуляция. Частицы составляют вместе сравнительно
плотные комочки, в которых кремнезем более сконцентрирован,
чем в исходном золе. Следовательно, такие коагуляты осаж-
даются в виде относительно плотного осадка.
3. Флокуляция. Частицы связываются вместе мостиками,
состоящими из агентов, способствующих подобному образова-
нию. Эти флокулирующие агенты имеют достаточную длину,
поэтому агрегированная структура остается открытой, занимая
значительный объем системы.
Очевидно, что указанные различия будут особенно заметны
в разбавленных золях, содержащих всего лишь несколько про-
центов кремнезема. В концентрированных золях можно отли-
чать гель, после того как он затвердеет, но нельзя провести
различие между коагулятом и флокулированным осадком.
4. Коацервация. Кремнеземные частицы окружены слоем
какого-либо адсорбированного вещества, понижающего гидро-
фильность частиц, но неспособствующего образованию мости-
ковых связей между ними. Такие частицы агрегируют в виде
концентрированной жидкой фазы, несмешивающейся с водной
фазой.
Грубер и Нелл [221] дали описание метода анализа золя,
содержащего агрегаты, который включал в себя измерение
рассеяния света и вязкости системы. По этому методу мо-
жно подсчитать размеры и массовую долю, занимаемую агре-
гатами.
В агрегацию включается и адгезия, возникающая между
коллоидными частицами. Виссер [222] в своей работе, в кото-
рой представлены данные, выбранные из 295 источников, под-
робно рассмотрел возникающее притяжение и образование свя-
зей между частицами. Особое внимание в его работе уделено по-
груженным в жидкую среду коллоидным системам, в которых
рассматриваются силы взаимодействия Лондона — Ван-дер-Ва-
альса и отталкивания за счет образования двойного электри-
6 Заказ № 250
498
Глава 4
ческого слоя, а также ионного притяжения между противопо-
ложно заряженными поверхностями частиц.
Гелеобразование
Агрегация и желатинирование детально были рассмотрены
в гл. 3 как один из возможных механизмов полимеризации
в случае присутствия в системе чрезвычайно небольших
«частиц» поликремневой кислоты. В данной главе обсуждается
только вопрос о превращении золей, содержащих относительно
однородные по размеру дискретные частицы, в гели. Основные
сведения о гелях будут представлены в гл. 5.
В прошлом большая часть кремнеземных гелей формирова-
лась из поликремневых кислот или из очень небольших кол-
лоидных частиц кремнезема, обычно менее 5 нм в диаметре,
и поэтому природа и структура подобных гелей долгое время
оставались невыясненными. Теперь доступно приготовление
коммерческих золей, состоящих из однородных частиц с из-
вестным размером, причем механизм гелеобразования подоб-
ных золей понят гораздо лучше.
Влияние величины pH
Основная стадия в процессе образования геля — столкнове-
ние двух кремнеземных частиц, обладающих достаточно низким
зарядом на поверхности. Поэтому, когда такие частицы всту-
пают во взаимный контакт, между ними образуются силокса-
новые связи, необратимо удерживающие частицы вместе. Для
формирования подобной связи между частицами необходимо
каталитическое действие гидроксил-ионов (или, по интерпрета-
ции некоторых исследователей, необходима дегидратация по-
верхности частиц при более высоких значениях pH). Это
доказывается тем фактом, что скорость образования геля
в области pH 3—5 возрастает с повышением значения pH и
пропорциональна концентрации гидроксил-ионов.
Выше pH 6 недостаток гидроксил-цонов уже не является
фактором, ограничивающим скорость гелеобразования. Однако
скорость агрегации понижается из-за более редкого числа
столкновений между частицами вследствие возросшей величины
заряда на их поверхности, именно поэтому она уменьшается
с увеличением pH. На рис. 4.13 схематически показано возра-
стание каталитического воздействия гидроксил-ионов с увели-
чением pH и уменьшение числа эффективных столкновений
между частицами с повышением pH и с возрастанием заряда
на частицах. Суммарным результатом одновременного дейст-
вия этих двух эффектов оказывается наибольшая скорость
Коллоидный кремнезем — концентрированные золи 499
гелеобразования примерно при значении pH 5. В отсутствие
солей в области pH 8—10 золи обычно стабильны. Наблю-
дается также область временной устойчивости при pH ~1,5.
Ниже pH 1,5 присутствие следов HF оказывает каталитическое
действие на агрегацию и гелеобразование [223]. По существу
во всех типах кремнезема имеются микропримеси фторид-ионов
в количестве менее 0,0001 %, так что концентрация HF повы-
Рис. 4.13. Зависимость стабилизации золя или времени гелеобразования от
величины pH.
1 — метастабильная область; 2 — область быстрого агрегирования частиц; 3 — область
роста частиц; 4 — стабилизированные золи.
шается с возрастанием кислотности. На каталитическое дейст-
вие фторид-ионов влияет присутствие примесей алюминия, так
как подавление активности некоторой части фторид-ионов яв-
ляется результатом образования комплексных ионов, таких, как
AlFe- и некоторых других [224]. Однако, как обсуждалось
в гл. 3, скорость гелеобразования увеличивается по мере того,
как pH падает ниже 3 даже в отсутствие фтора.
Как только между частицами образуются силоксановые
связи, происходит дальнейшее осаждение кремнезема в точке
контакта благодаря отрицательному радиусу кривизны (см.
гл. 3). Такой процесс происходит быстро выше pH 5 и медленно
при pH 1,5.
Влияние размеров частиц и концентрации SiO2
Эти два фактора взаимосвязаны. Скорость гелеобразова-
ния, по-видимому, пропорциональна суммарной площади,крем-
неземной поверхности имеющегося в данном объеме золя.
6*
500
Глава 4
Рис. 4.14. Зависимость времени гелеоб-
разования от концентрации кремнезема
для частиц диаметром 14 нм при различ-
ных значениях pH.
Поскольку удельная поверх-
ность кремнезема, как было
показано выше, изменяется
обратно пропорционально
диаметру частиц, то, следо-
вательно, золи, имеющие
одно и то же отношение
концентрации кремнезема
к диаметру частиц, будут
подвергаться гелеобразова-
нию приблизительно с оди-
наковой скоростью. Таким
образом, при эквивалентных
условиях можно ожидать,
что 10 %-ный золь с части-
цами диаметром 10 нм бу-
дет переходить в гель с той
же самой скоростью, что и
20 %-ный золь с частицами
размером 20 нм.
Для частиц заданного
размера (14 нм) при 25°С
в присутствии 0,1 н. NaCl
соотношение между време-
нем гелеобразования и кон-
центрацией кремнезема ока-
зывается непростым. Как
видно из рис. 4.14, влияние
концентрации кремнезема на время гелеобразования неодина-
ково при разных значениях pH.
Электролиты и органические жидкости
Ниже pH 3,5 присутствие солей слабо влияет на скорость
гелеобразования, тогда как смешиваемые с водой органические
жидкости, подобные спирту, замедляют этот процесс. Выше
pH 3,5, когда коммерческие золи кремнезема начинают нести
на поверхности частиц отрицательный заряд, добавление соли
понижает суммарный результирующий эффект отталкивания
частиц, и, как это было показано Бакстером и Брайантом [225],
коагуляция и желатинирование значительно ускоряются.
Добавление соли к разбавленному золю вызывает во всей си-
стеме коагуляцию и осаждение, тогда как такое добавление
к концентрированному золю приводит к тому, что коагуляция
протекает быстро и не отличима от гелеобразования. Таким
образом, при добавлении соли к концентрированному золю
Коллоидный кремнезем — концентрированные золи 501
кремнезема при pH 9 первоначально образующиеся гель или
осадок .могут повторно пептизировать в золь, если сразу же
провести разбавление или подкисление, но даже в течение не-
скольких секунд может наступить необратимый процесс геле-
образования.
Смешиваемые с водой органические жидкости оказывают до
некоторой степени примерно такое же дестабилизирующее
влияние на золи, стабилизированные щелочью, как и добавле-
ние электролита, поскольку, вероятно, при этом диэлектриче-
ская постоянная среды понижается.
Температура
Поскольку можно говорить о кинетике процесса агрегации,
то уместно ожидать, что скорость гелеобразования возрастает
с повышением температуры. Это не всегда имеет место в про-
цессе флокуляции, когда образуются водородные связи с орга-
ническими молекулами, но при гелеобразовании наблюдается
заметный температурный коэффициент скорости формирования
силоксановых мостиковых связей между частицами. Была
измерена энергия активации процесса гелеобразования в поли-
кремневой кислоте, но, однако, имеется мало данных по пре-
вращению золей с известными размерами частиц в гели. Ниже
pH 2, когда скорость реакции пропорциональна концентрации
водородных ионов (HF оказывает каталитическое действие),
энергия активации составляет около 9,5 ккал/моль. Согласно
данным Пеннера [226], при pH 4,5, когда реакция катализи-
руется гидроксил-ионами, энергия равна 16,1 ккал/моль. Броде,
Браун и Хофф [227] определили близкое значение, равное
15,5 ккал/моль, при pH 5,5. При pH 8,5 они обнаружили более
низкое значение 9,6. ккал/моль, тогда как при pH 10,5 в при-
сутствии хлорида калия энергия активации составила
14,6 ккал/моль. Очевидно, что температурный коэффициент
скорости гелеобразования должен также зависеть от изменяю-
щихся констант диссоциации рассматриваемых веществ по мере
возрастания температуры. Ранее выполненные исследования по
механизму гелеобразования были обобщены Айлером [8].
Во многих исследованиях гелеобразования поликремневой
кислоты, состоящей из очень небольших частиц, интерпретация
результатов измерения осложнялась тем фактом, что с ростом
температуры частицы увеличивались в размере. Существенные
данные, относящиеся к энергии активации процесса агрегации
частиц, могут быть получены только в том случае, когда частицы
уже закончили свой рост и стабилизированы при более высокой
температуре, чем предусматривалось в проводимых эксперимен-
тах. Кроме того, энергия активации зависит не только от вели-
502
Глава 4
чины pH, определяющей заряд на частицах, но также и от
изменения концентраций кремнезема и электролита. Следова-
тельно, бессмысленно приводить данные для энергии активации
гелеобразования, если только подобные переменные величины
точно не определены.
Для деионизированного золя с частицами размером 14 нм
(значение pH устанавливалось добавлением NaOH) энергия
активации подсчитывалась из времени гелеобразования при 23
и 60°С. Ее значения оказались следующими:
Концентрация S1O2, % pH
Энергия активации,
ккал/моль
20 5,5 10,7
30 5,5 7,6
20 3,0 16,4
30 3,0 11,9
Очевидно, что рассматриваемые факторы оказываются слож-
ными.
Теория прочности гелей
Коллоидный кремнезем находит многочисленные применения
благодаря тому, что после высушивания он необратимо превра-
щается в нерастворимый кремнезем. Его пригодность к исполь-
зованию в качестве загустителя и связующего для неоргани-
ческих волокон и порошков зависит от прочности геля, форми-
рующегося около точек контакта между макроскопическими
частицами веществ, подлежащих связыванию.
Настоящее обсуждение вопроса ограничивается гелями, полу-
ченными из дискретных частиц коллоидного кремнезема разме-
ром более чем 5 нм в диаметре. Не-сделано никаких попыток
рассмотрения огромной по объему совокупности литературных
данных, относящихся к гелям, полученным из поликремневых
кислот посредством подкисления растворимых силикатов ще-
лочных металлов. В том случае, когда в процессе участвуют
поликремневые кислоты или частицы диаметром менее 5 нм,
процесс гелеобразования оказывается гораздо более сложным
и предусматривает конденсацию силанольных групп внутри
частиц и циклизацию с образованием трехмерных силоксановых
колец, что приводит к формированию зародышевых безводных
частиц SiO2.
Томас и Мак-Коркл [228] сформулировали основные поло-
жения теории образования геля из коллоидных частиц. Они
показали, что теория Фервея—Овербека, описывающая взаимо-
действие двойных электрических слоев, окружающих две сосед-
ние сферические коллоидные частицы, приводит к объяснению
Коллоидный кремнезем — концентрированные золи 503
явления изотропной флокуляции. Новые коллоидные частицы
могут более легко присоединяться к концам цепеобразного
флокулированного осадка, т. е. в тех местах, где минимален
энергетический барьер, обусловленный силами отталкивания.
Это как раз тот тип агрегации, благодаря которому золь пре-
вращается в гель только в определенных точках контакта
частиц путем формирования бесконечной сетки из цепочек
коллоидных частиц, распространяющейся по всему объему золя
(см. также гл. 3).
Превращение золя в высушенный гель происходит по сле-
дующим стадиям:
а) затвердевание золя вплоть до образования трехмерной
сетки геля, состоящей из разветвленных цепочек кремнезема,
в которой жидкость удерживается капиллярными силами;
б) упрочнение связей между соседними частицами в точках
контакта за счет коалесценции частиц;
в) усадка трехмерной кремнеземной сетки по мере испаре-
ния воды;
г) развитие напряжений в кремнеземной сетке в процессе
объемной усадки;
д) появление трещин в высушенном геле кремнезема и об-
разование из него отдельных кусочков.
Как показано на рис. 4.15, усадка кремнеземного геля
легко просматривается в том случае, когда тонкий слой золя
высушивается на гладкой поверхности. На первом этапе фор-
мируется прозрачная, связанная с поверхностью пленка геля,
а затем по мере высушивания пленка сжимается с разрывами,
образующимися в направлении высушивания, так что полу-
чается непрочный волокнистый или похожий на волосы остаток
кремнеземного геля. Частицы размером 100 нм могут образо-
вывать ленточки геля шириной вплоть до 1 мм, а при исполь-
зовании частиц еще большего размера остаются относительно
большие тонкие пленки. Золь, состоящий из малых частиц,
превращается в гель при более низкой концентрации кремне-
зема, и, следовательно, образовавшийся по мере испарения воды
из пленки золя с данной концентрацией кремнезема гель сжи-
мается более сильно и растрескивается на небольшие, относи-
тельно более твердые кусочки геля. Таким образом, получается
некоторая определенная небольшая по толщине пленка геля,
которая полностью покрывает поверхность сплошным слоем.
Хотя сам по себе золь кремнезема при высушивании может
и не образовывать сплошной пленки, тем не менее он способен
вести себя подобно прочному гелю и заполнять промежутки
между большими по размеру частицами или волокнами. Подоб-
ным образом при нанесении на испытуемую подложку, напри-
мер на поверхность стекла, золь с заданной концентрацией БЮг
504
Глава 4
а
о°роо &о2ооо°о°§
Рис. 4.15. Схема превращения испаряющейся пленки золя кремнезема в гель
и высушивания геля (показано поперечное сечение системы).
а — золь; б—начало агрегации частиц в концентрированном золе; в — сжатие геля под
действием поверхностного натяжения; г — растрескивание геля в результате усадки; д —
высушенные несвязанные между собой кусочки геля, w — поверхность воды, s — твердая
подложка.
может образовывать при высушивании хрупкое, не прилипаю-
щее к поверхности отложение относительно большой толщины,
однако при высушивании очень тонкого слоя такого золя на
чистой гидрофильной поверхности может получиться сплошная
сцепленная с подложкой пленка, которая остается растянутой
по поверхности в процессе ее высушивания. Если эта пленка
достаточно тонка, например толщиной всего 1 мкм, то в том
случае, когда золь, образующий пленку, был сконцентрирован
почти до точки гелеобразования, окончательное высушивание
пленки протекает настолько быстро, что частицы кремнезема
оказываются в плотно упакованном состоянии, прежде чем
успевает образоваться гель, и поэтому не происходит никакой
Коллоидный кремнезем — концентрированные золи
505
усадки или растрескивания; кремнеземная пленка при данных
условиях будет прочно сцеплена с поверхностью.
В щелочных золях небольшие добавки солей, таких, напри-
мер, как сульфат натрия, ускоряют желатинирование при вы-
сушивании золя. Так как для получения более прочной струк-
туры геля золь должен высушиваться вплоть до достижения
возможно более высокой концентрации кремнезема, то очевидно,
что в стабилизированных щелочью золях необходимо избегать
присутствия электролитов. Так, например, Рейтер [229] утвер-
ждает, что при использовании в качестве связующего золя
кремнезема, имеющего небольшие по размеру частицы при pH
в пределах 8,5—9 и минимальном содержании электролита,
электропроводность золя обусловливается только присутствием
коллоидных частиц кремнезема и стабилизирующих систему
противоионов, но не примесями электролита.
Виссер [222] рассмотрел роль поверхностного натяжения
воды и шероховатости поверхности подложки при адгезии кол-
лоидных частиц на гладких поверхностях.
Очевидно, трудно получить плотную прочно сцепленную
с поверхностью пленку кремнезема, если использовать золь,
состоящий из однородных по размеру частиц. Айлер [219]
исходил в своем патенте из того факта, что когда золь с части-
цами диаметром 100 нм высушивается в виде тонкой пленки на
поверхности, то пленка хотя и остается размягченной, но сжи-
мается и растрескивается гораздо в меньшей степени по сравне-
нию с пленкой, полученной из золя, состоящего из меньших по
размеру частиц. Добавлением серий постепенно уменьшаю-
щихся по размеру частиц кремнезема получали смесь частиц
разного размера, которая при высыхании превращалась в проч-
ную и гладкую сплошную пленку. Использование только одного
золя с частицами размером 100 нм приводило к формированию
непрочной пленки, в которой относительный объем, занимае-
мый кремнеземом, составлял примерно 70 %. Вместе с тем
смесь, состоящая из 74 % золя с частицами размером 100 нм,
11,9 % золя с частицами 22 нм, 2,3 % золя с частицами 10 нм
и 2,8 % золя с частицами размером 7 нм, давала прочную
пленку, в которой относительный объем, занимаемый кремнезе-
мом, был равен 80,4 %. В такой пленке не появлялись волосные
трещины и не происходило ее растрескивания при высушива-
нии, поскольку промежутки между частицами большего размера
заполнялись частицами меньшего размера, и, следовательно,
масса пленки не могла сжиматься в значительной степени под
действием сил поверхностного натяжения (см. рис. 4.16).
Декстер и Таннер [230] теоретически рассмотрели упаковку
сферических частиц трех различных размеров.
506
Глава 4
Однородные по размеру коллоидные частицы полезно исполь-
зовать при приготовлении гелей с однородными большими
порами, но оказалось, что чем больше по размеру исходные
коллоидные частицы, тем заметно менее прочными получаются
гели. Однако Иетс [231] обнаружил, что добавлением относи-
тельно небольшого количества растворимого силиката для
образования кремневой кислоты удается усилить связи между
Рис. 4.16. Схема формирования прочного плотного геля из смеси больших и
малых частиц кремнезема.
частицами и получить гораздо более прочные гели. Подобным
же образом Сиппел [232] нашел, что прочный гель с большей
пористостью можно приготовлять из золя, содержащего частицы
двух различающихся размеров: 40 % частиц, больших по раз-
меру, диаметром D и 60 % частиц диаметром 0,4—0,8.0. Отно-
сительно низкая прочность гелей, приготовляемых из частиц
большего размера, частично компенсируется тем, что большие
частицы упаковываются более плотно. Белоцерковский с соав-
торами [233] наблюдал, что в ксерогелях эффективный диаметр
пор имеет тенденцию сохраняться постоянным, тогда как раз-
мер частиц в исходном золе изменяется. Малые частицы могут
стягивать пленку, сохраняя при этом поры вплоть до величины
равной диаметру частиц или даже более, а большие частицы
(превышающие 50—100 нм) становятся более плотно упако-
ванными с сохранением пор размером меньше диаметра частиц.
Как было предложено Шоупом (см. лит. к гл. 2 [97, 96]),
очень прочные покрытия могут, вероятно, приготовляться за
счет использования жидкой смеси, способной формировать гель,
состоящий из силиката калия, коллоидного кремнезема и схва-
тывающего агента.
Коллоидный кремнезем — концентрированные золи
507
Коагуляция
Хотя некоторые одинаковые факторы имеют место как при
коагуляции, так и в процессе гелеобразования, эти два явле-
ния во многом и различны. При гелеобразовании, по мере того,
как повышается его вязкость, золь, по-видимому, остается одно-
родным и в основном прозрачным. С другой стороны, при
коагуляции по определению частицы образуют агрегаты, кото-,
рые имеют более высокий показатель преломления по сравне-
нию со средой. Следовательно, наблюдать за коагуляцией
можно, регистрируя возрастание мутности системы или же по-
нижение светопропускания.
В работе [234] было исследовано различие в помутнении
вследствие формирования дублетов из одиночных частиц.
Помимо этого агрегация частиц прослеживалась под ультра-
микроскопом в потоке [235].
Коагуляция была также предметом исследования Ван Ол-
фена и Миселса [236].
Механизм коагуляции
Коллоиды подразделяются на «лиофильные» и «лиофобные»
или же для случая водных золей на «гидрофильные» и «гидро-
фобные». Стабильность гидрофобных золей зависит большей
частью от заряда на частицах, тогда как для гидрофильных
золей это обстоятельство менее важно, поскольку частицы
стабилизируются посредством «сольватации» или «гидратации».
Коллоидный кремнезем относится к гидрофильным, хотя
при некоторых условиях, когда коагуляция частиц вызывается
солями, кремнезем отчасти и гидрофобен. Эти термины не столь
важны по отношению к кремнезему, за исключением случая,
когда предусматривается использование органических флоку-
лирующих агентов и адсорбция этих веществ на поверхности
Кремнезема превращает ее в поверхность с выраженными гид-
рофобными свойствами.
Коагуляция рассматривается как результат воздействия
вандерваальсовых сил притяжения, которые стягивают вместе
две частицы в момент их столкновения, если только этому при-
тяжению не противостоит гидратный барьерный слой или силы
электростатического отталкивания между одинаково заряжен-
ными частицами, или же обе эти причины. Следовательно,
существуют два фактора, которые замедляют коагуляцию крем-
незема:
1. Гидратация поверхности частиц посредством образования
слоя молекул воды, связанных водородной связью с поверх-
ностными группами SiOH.
508
Глава 4
2. Отрицательный ионный заряд, возникающий на частицах
выше pH ~3,5, и окружающее частицу положительно заряжен-
ное облако, состоящее из противоионов — катионов, таких,
как Na+, что ведет к образованию «двойного электрического
слоя».
Для большинства разбавленных кремнеземных золей при
рН~2, когда на частице находится лишь слабый ионный
заряд, при воздействии на систему электролитом никакой
коагуляции не будет наблюдаться, по-видимому, из-за гидрат-
ного слоя. Однако Хардинг [237] обратил внимание на тот
факт, что относительно большие по размеру коллоидные ча-
стицы кремнезема, диаметром 50—100 нм или более, начинают
флокулировать при низких значениях pH, тогда как небольшие
частицы еще не подвержены этому процессу. Остается опреде-
лить, обусловливается ли флокуляция вандерваальсовыми си-
лами притяжения между частицами или же образованием
многократных водородных связей между покрытыми силаноль-
ными группами поверхностями в пределах области контакта
частиц при их столкновении.
В интервале значений ‘pH 7—10 золи кремнезема стабильны,
если концентрация электролита незначительна, но начинают
коагулировать при добавлении солей. Таким образом, следует
рассмотреть два механизма агрегации:
1. Притяжение частицы к частице за счет действия сил
Ван-дер-Ваальса, математическое описание которых выполнено
Лондоном и распространено впоследствии на коллоидные ча-
стицы Гамакером [238].
2. «Связывание» мостиковой связью двух частиц под дей-
ствием флокулирующего или коагулирующего агента.
В первом случае притяжение ведет к коагуляции, когда
силы отталкивания между частицами, несущими сходные ион-
ные заряды, понижаются посредством добавления солевого
коагулянта при критической концентрации (к. к. к.). Большая
часть добавленной соли остается в растворе, поэтому концент-
рация по существу сохраняется неизменной, в то время как
флокуляция развивается. Лишь очень небольшая доля ионов
прибавляемой соли адсорбируется на поверхности флокулирую-
щих частиц, и потерей этих ионов в самом растворе обычно
пренебрегают. В рассматриваемом случае внимание, как пра-
вило, в первую очередь концентрируется на свойствах диспер-
сионной среды, а не на возможной адсорбции ионов на флоку-
лирующих частицах.
С другой стороны, когда агентом, вызывающим агрегацию,
является полимерная молекула или же другая коллоидная
частица, большая доля такого агента адсорбируется на.части-
цах до тех пор, пока в системе не будет достигнута точка
Коллоидный кремнезем — концентрированные золи
509
к. к. к., в которой только небольшая часть флокулирующего
агента может в действительности находиться в растворе. В та-
ком случае требуемое количество агента непосредственно свя-
зывается с поверхностью флокулирующих частиц. Несомненно,
имеются также промежуточные ситуации, в частности, когда
агент представляет собой многозарядный ион или поликатион
небольших размеров.
По-видимому, различие состоит в том, что если флокули-
рующим агентом оказывается большая молекула или частица
и если этот агент преимущественно адсорбируется на изучае-
мых частицах, когда в системе имеется критическая концент-
рация коагулянта, то тогда можно наглядно выявить количе-
ство агента, адсорбированного на флокулированных частицах.
Однако если такой агент представляет собой простую соль,
как, например, хлорид натрия, который требуется в большом
избытке в растворе в точке к. к. к., то тогда адсорбцию этого
агента на коагулянте не так легко измерить, а также трудно
продемонстрировать возможное образование мостиковых связей
между частицами посредством ионов соли. Принимается, что
вандерваальсовы силы являются источником притяжения
частиц; в действительности может происходить удерживание
вместе частиц кремнезема в точках их контакта посредством
адсорбированных ионов, таких, как Na+.
Вандерваальсовы силы часто называют «дисперсионными
силами», что не совсем правильно, так как это силы притяже-
ния, а не дисперсионные в физическом понимании. Роувелер и
Овербек [239] исследовали силы, возникающие между кремне-
земными поверхностями. Подобным же образом Табор и Уин-
тертон [240] измерили силы между поверхностями пластинок
слюды и пришли к заключению, что присутствие воды снижает
притяжение примерно в 10 раз.
Вполне вероятно, что дисперсионные силы играют незначи-
тельную роль или вовсе не имеют никакого значения в рас-
сматриваемой системе кремнезема. Как отметили Дюмон и
Уотиллон [241], кремнезем представляет собой наиболее слож-
ную из всех известных оксидную систему, так как она прояв-
ляет максимальную устойчивость в точке нулевого заряда.
В данной точке очень низка константа Гамакера, и поэтому
дисперсионные силы оказываются незначительными, так что
по крайней мере в некоторых областях значений pH агрегация
частиц включает образование химической связи. В более де-
тальном исследовании, выполненном Депассе и Уотиллоном
[242], отмечалось, что одного монослоя воды достаточно,
чтобы экранировать дисперсионные силы между небольшими
коллоидными частицами кремнезема (меньшими 25 нм в диа-
метре).
510
Глава 4
Таким образом, дисперсионные силы, вероятно, не играют
никакой роли, за исключением, может быть, золей с частицами
по размеру, большими 100 нм.
Различие между кремнеземом и другими оксидами, согласно-
данным Визе и Хили [243], подчеркивается тем фактом, что
оксиды, подобные TiO2 и А12О3, подвержены флокуляции под
действием солей и величины pH, удовлетворительно объясняе-
мой до известной степени теорией ДЛФО, всякий раз, когда
электрокинетический потенциал падает ниже 14 + 4 мВ.
Коагуляция под действием электролитов
Эта проблема исследовалась в течение последних 50 лет,
однако механизм подобного действия еще полностью не понят.
Понижение электрокинетического потенциала при добавлении
электролитов поддерживало представление о том, что частицы
сближаются, поскольку величина заряда на них понижается до
некоторого значения, когда предполагаемое «вандерваальс’ово
притяжение» может преодолеть ионное отталкивание. Авторы
работы [53] суммировали теории и расчеты, относящиеся
к двойному электрическому слою, предложенные Гуи в 1910 г.,
Дебаем и Хюккелем в 1923 г., Бартеном [244], Гамакером
[238], Дерягиным [245], Фервем и Овербеком [246, 247]. В слу-
чае лиофобных коллоидов, но не кремнезема расчеты находятся
в согласии с экспериментальными данными [248]. Теории
механизмов, посредством которых катионы адсорбируются на
поверхности кремнезема, подробно рассматриваются в гл. 7.
Аллен и Матиевич [249—251] исследовали коагуляцию
коллоидного кремнезема в области pH 6—И и нашли, что
механизм в данном случае иной, чем для лиофобных коллоидов.
Критические концентрации коагулянта для различных солей не
коррелируют с изменениями электрофоретической подвижности
или с изменением электрокинетического потенциала. Хардинг
[237] провел аналогичные наблюдения, хотя и сообщил, что
кремнеземные частицы большего размера, т. е. диаметром
50 мм (вместо 12 нм), ведут себя в соответствии с теорией
двойного электрического слоя. Поведение же небольших частиц
кремнезема вследствие сказанного не может быть объяснено
общепринятой теорией. Аллен и Матиевич [249] обнаружили,
что катион коагулирующей соли вступает в обмен с протоном
силанольной группы на поверхности. Коагулирующий эффект,
вызываемый целым рядом катионов, определяется скорее числом
ион-эквивалентов вступающих в обмен ионов, чем валентностью
иона. Авторы предположили, что после адсорбции очередного
катиона поверхность кремнезема теряет одну силанольную
группу, способную образовывать водородную связь с водой,
Коллоидный кремнезем — концентрированные золи
511
и в этом смысле поверхность оказывается «дегидратирован-
ной». Подобное усиление «лиофобного» характера поверхности
превращает золь кремнезема в систему, более чувствительную
к коагуляции под действием электролита.
Депассе и Уотиллон [242] предполагают, что прямое «свя-
зывание частиц между собой», а не «дегидратация» поверх-
ности может рассматриваться как механизм коагуляции крем-
незема. Они высказывают мнение, что при pH 7—11, а также
выше некоторых значений критических концентраций солей
всех щелочных металлов происходит коагуляция, поскольку
вначале частицы соединяются в первую очередь кислотно-
основными связями, но не связями' Si—О—Si и поскольку
свежеобразованный коагулят пептизируют при понижении зна-
чения pH:
I I
-Si-OH...O-Si-
I I
(После того как силоксановые связи Si—О—Si образуются,
они уже не разрываются при действии кислотой.) Однако если
такая связь появляется, как это утверждают авторы, то трудно
понять, почему процесс конденсации не происходит сразу же
после того, как появляются ионы ОН~ — катализаторы
процесса.
Было обнаружено, что выше pH 11, когда поверхность
покрывается основными группами, соли натрия и лития про-
должают вызывать коагуляцию, но соли калия, рубидия и
цезия не оказывают такого действия. Авторы предполагают,
что поскольку частицы, полностью покрытые основными груп-
пами при данном значении pH, не в состоянии образовывать
кислотно-основные связи, то, следовательно, ионы натрия или
лития должны представлять собой агент, связывающий частицы,
или «агент, обеспечивающий мостиковую связь». Заметное
различие в коагулирующем действии, оказываемом разными
однозарядными ионами, было показано Уотиллоном [252],
который отметил, что ниже pH 7 ионы Na+, К+, Rb+ и Cs+
имеют одно и тоже значение к. к. к., но выше pH 10 флокули-
рующий эффект отсутствовал в случае использования ионов
К+, Rb+ и Cs+, тогда как ионы Na+ и Li+ еще продолжали
воздействовать.
Однозарядные катионы как агенты, формирующие
мостиковую связь
Айлер предложил несколько отличающуюся гипотезу, в ко-
торой утверждается, что во всех случаях флокуляция обуслов-
ливается связыванием частиц между собой посредством катио-
512
Глава 4
нов, но при значениях pH выше 11 большие по своему размеру
ионы калия, рубидия и цезия при высоких концентрациях
образуют вокруг каждой частицы полный двойной слой доста-
точной толщины, способный вызывать диспергирование частиц
и предотвращать их агрегацию. Золи, стабилизированные избы-
точным количеством ионов тетраметиламмония, имеющих еще
больший размер, могут даже высушиваться до порошкообраз-
ного состояния без флокуляции [63].
Теория двойного электрического слоя не была использована
для объяснения рассматриваемого ниже явления, когда адсор-
бированные катионы способной вызывать коагуляцию простой
соли могут оказаться «фактором образования мостиковой
связи» при коагуляции кремнезема аналогично частично боль-
шим по своим размерам изополикатионам, таким, например,
как катионы основных солей железа или алюминия. Если про-
исходит формирование мостиковой связи, то количество (в рас-
чете на единицу поверхности кремнезема) адсорбированных
или «способных вступать в ионный обмен» катионов, вызываю-
щих флокуляцию, должно уменьшаться с возрастанием разме-
ров частиц. Не существует очевидного* способа подтверждения
того, что в процессе флокуляции подобные адсорбированные
катионы будут концентрироваться вблизи мест контакта частиц
между собой, как это следовало бы ожидать, если бы катионы
образовывали мостиковые связи.
Если катионы натрия действуют как ионы, формирующие
мостиковую связь, то следует принимать во внимание их спо-
собность к гидратации. В водном растворе ион натрия в резуль-
тате гидратации окружается шестью атомами кислорода, при-
надлежащих молекулам воды. Это подтверждается тем, что
при адсорбции иона натрия на поверхности кремнеземной
частицы один или несколько атомов кислорода молекул гидрат-
ной воды могут замещаться атомами кислорода, принадлежа-
щими поверхностным силанольным группам SiOH, так что
последние оказываются непосредственно связанными с атомом
натрия. Следовательно, положительный заряд иона натрия
способен нейтрализовать отрицательный заряд адсорбирован-
ного вблизи него гидроксил-иона, который как раз определяет
поверхностный заряд частицы. В результате на поверхности
образуется нейтральный адсорбционный комплекс. Если это
так, то при высоких концентрациях ионов натрия (выше точки
«критической концентрации коагулянта») ничто не препятствует
замещению одной или более молекул воды, которые до тех пор
были связаны с ионом натрия с внешней стороны, а не у по-
верхности кремнеземной частицы, на поверхностные силаноль-
ные группы, принадлежащие второй вступающей в столкновение
частицы (рис. 4.17а, а, б, в). Таким образом, ион натрия может
Коллоидный кремнезем — концентрированные золи
513
Рис. 4.17а. Схема образования связи между частицами кремнезема посредст-
вом координации вызывающими флокуляцию катионами металла.
а—‘ гидроксил-ионы передают отрицательные заряды слою молекул воды, связанных
водородной связью с силанольными группами, находящимися иа поверхности частицы
кремнезема; такой процесс эквивалентен адсорбции гидроксил-ионов на поверхности;
б — гидратированные ионы натрия адсорбируются иа отрицательно заряженных участках
поверхности, образуя нейтральные комплексы; в — при столкновении с незаряженными
участками поверхности второй частицы ион натрия координирует кислородные атомы
силанольных групп и кислородные атомы молекул воды, связанных с поверхностью, об-
разуя координационную связь между частицами. Размер коллоидных частиц кремнезема г
по отношению к размерам атомов на самом деле гораздо больше, чем это показано иа
рисунке, поэтому между двумя частицами может сформироваться целый ряд мостиковых
связей, образующихся посредством иона натрия.
действовать как мостиковая связь между двумя частицами
кремнезема.
Если такая теория корректна, то коагулят может формиро-
ваться, как только на поверхности каждой частицы кремнезема
будет находиться достаточное число ионов натрия или других
катионов, координационно связанных с атомами кислорода,
способными к ионному обмену, вследствие чего каждая частица
кремнезема обеспечивается возможностью контактировать
с другими частицами более чем в двух точках. Контакт в двух
точках допускал бы формирование из частиц только лишь
7 Заказ № 250
514
Глава 4
цепочек, но не агрегатов. С возрастанием числа ионов натрия
на поверхности каждая частица могла бы соприкасаться
с окружающими ее другими частицами в трех и более (вплоть
до шести) точках, что приводило бы к образованию агрегатов
со все возрастающей плотностью.
Необходимо также иметь в виду, что выше pH 8,5—9,0
в растворе образуются силикат-ионы, сопровождаемые своими
катионами, которые также должны учитываться, как если бы
они были добавляемой к системе солью или другим электроли-
том. Этот вопрос обсуждался Алленом и Матиевичем [249].
Как отметили Депассе и Уотиллон [242], при pH >11
только Na+ и Li+ будут продолжать образовывать мостиковые
связи; большие по размеру ионы К+, Rb+ и Cs+- не ведут себя
подобным образом. По-видимому, в последнем случае частицы
кремнезема полностью покрываются слоем таких адсорбиро-
ванных катионов, снимающих в свою очередь соответствующие
отрицательные заряды на кремнеземной поверхности.
Депассе и Уорлус [253] сообщили, что при pH 7,5 ионы
тетраметиламмония оказываются сильными коагулирующими
агентами по сравнению с ионами гуанидина, аммония и натрия.
Соответствующие значения точек к. к. к. оказались равными
для этих ионов примерно 0,01, 0,32, 1,0 и 2,2 М. Ион (СН3)4Ы+
адсорбируется сильно и поэтому вызывает процесс коагуляции
уже при малых концентрациях, но свободное основание может
при pH 9—10 воздействовать как стабилизирующий агент,
поскольку монослой адсорбированных катионов обеспечивает
появление «стерической» стабилизации [205]. Тот факт, что
ион натрия может быть координированным с должным образом
расположенными в пространстве атомами кислорода в дикето-
нах, тогда как большие по размеру ионы не могут координиро-
вать подобным образом, отмечал в своей работе Гарнер [254].
Он высказал предположение, что наблюдаемое между ионами
натрия и калия различие в способности адсорбироваться на
поверхности кремнезема могло быть обусловлено присутствием
поверхностных углублений, в которые способны проникать
ионы натрия, но не калия.
Айлер измерил коагуляцию коммерческих стабилизирован-
ных щелочью золей кремнезема, имевших различные по раз-
меру частицы, при действии на них ионов Na+. Частицы крем-
незема были непористыми. За развитием процесса агрегации
наблюдали следующим образом. Исследуемые образцы цент-
рифугировали при скоростях вращения, достаточных только,
чтобы вызвать отделение дискретных частиц размером при-
мерно 1 мм. Затем измеряли количество кремнезема в агреги-
рованной форме, седиментированного из слоя золя толщиной
7,5 см. Полученные данные находились в соответствии с вели-
Коллоидный кремнезем — концентрированные золи
515
чиной, выраженной процентом светопропускания при длине
волны 400 нм и определенных условиях опыта. Частицы разме-
ром 8, 15 и 25 нм в 0,24 и 0,30 н. растворах Na2SO4 при кон-
центрации 12 % SiO2 агрегировали соответственно при 25, 40
и 55°С. Значение pH составляло ~9. Температурный коэффи-
циент флокуляции оказался гораздо более низким в интервале
25—40°С по сравнению с интервалом 40—55°С. Соответствую-
щие подсчитанные значения энергий активации составляли 4,7
и 10,6 ккал/моль. Во всех изученных случаях частицы меньшего
размера проявляли повышенную чувствительность к изменениям
в концентрации электролита и к температурным изменениям,
чем большие. При равных концентрациях кремнезема и темпе-
ратуре 55°С скорость агрегации была прямо пропорциональна
величине площади поверхности в единице объема золя и, сле-
довательно, обратно пропорциональна диаметру частиц.
В общем случае золи с обычными размерами частиц (5—
25 нм) не могут подвергаться какой-либо обработке без того,
чтобы не вызвать ту или иную степень агрегации, если только
концентрация соли натрия не превосходит некоторого опреде-
ленного уровня. По этой причине концентрация кремнезема,
необходимая для получения дискретных частиц путем нейтра-
лизации раствора силиката натрия с отношением SiO2:Na2O
3,25 в горячем состоянии до значения pH 9, не может
быть более чем 1—2%. Айлер [101] дает эмпирическую фор-
мулу, связывающую максимальное значение нормальности
ионов натрия N с концентрацией кремнезема С, выраженной
числом граммов SiO2 в 100 мл,, и с температурой Т (в градусах
Цельсия), по которой можно вычислять необходимую для
практических процессов нормальность, когда агрегация частиц
не происходит по крайней мере в течение нескольких часов:
N = 0,26 — 0,005С — 0,0012 (Т — 40)
Коагуляция под действием двухзарядных
катионов металлов
Характерной особенностью двухзарядного катиона является
то, что при его адсорбции на поверхности аморфного кремне-
зема, по крайней мере на первом этапе, происходит нейтрали-
зация лишь одного отрицательного заряда, т. е. освобождается
только один ион водорода.
При pH ~9 двухзарядный ион адсорбируется и проявляет
себя подобно положительно заряженному центру на поверх-
ности. Конечно, коагуляция происходит задолго до того, как
поверхность кремнезема полностью насыщается двухзарядными
ионами, поскольку они могут действовать подобно мостикам
благодаря реакции с двумя частицами кремнезема в точках их
у*
516
Глава 4
контакта. Например, Тедрос и Ликлема [255] изучили адсорб-
цию ионов кальция при различных значениях pH и показали,
что в области pH 8—9 на поверхности кремнезема на каждый
адсорбированный ион Са2+ приходился только 1 ± 0,05 ионов
ОН-. Ионы кальция, по-видимому, не образуют полимерных
катионных разновидностей в отличие от трехзарядных ионов
металлов, таких, например, как А13+.
Айлер [169] проверял подобное поведение ионов кальция
при исследовании флокуляции частиц кремнезема различных
размеров. Хили, Джеймс и Купер [256а] сообщили о похожем
поведении двухзарядных ионов кобальта. Они обнаружили, что
заряд на частицах кварца становился обратным по знаку в при-
сутствии 10-4 М ионов Со2+ при pH 7.
Были и другие исследования, в которых рассматривалась
коагуляция под действием двухзарядных катионов, но вне
зависимости от размеров частиц. Так, Мак-Фейдин и Матиевич
[2566] нашли, что при pH 5,2 коллоидные частицы кремнезема
могут коагулировать при введении всего лишь К)—3 моль/л
сульфата меди (II), но при немного более высоком значении pH
содержащий кристаллизационную воду гидроксид меди и кол-
лоидный кремнезем будут осаждаться совместно. Это подтвер-
ждает, что многоосновные катионы металлов будут вызывать
коагуляцию и осаждение коллоидного кремнезема при pH, только
немного меньшем, чем то, при котором происходит осаждение
водного оксида или гидроксида металла. Если значение pH
оказывается выше подобной критической точки, зависящей от
характера самого иона металла, то коллоидный кремнезем
будет коагулировать наряду с гидроксидом металла.
Коагуляция под действием многозарядных катионов
основных солей металла
По-видимому, имеется заметное различие между коагуля-
цией, вызываемой двухзарядным ионом Са2+ и действием трех-
зарядных ионов А13+ или La3+ и многозарядных основных поли-
катионов трехвалентных металлов. В последнем случае пред-
полагается присутствие очень небольших по размеру положи-
тельно заряженных коллоидных частиц — поликатионов метал-
лов со своими характерными свойствами, которые способны
нести многократные заряды и адсорбироваться на поверхности
частиц кремнезема из таких чрезвычайно разбавленных раство-
ров, что процесс адсорбции оказывается по существу необра-
тимым.
Агрегация при воздействии простого электролита или соли
обычно обозначается термином «коагуляция», тогда как агре-
гация, происходящая за счет добавления основных солей поли-
Коллоидный кремнезем — концентрированные золи 517
валентных металлов,— термином «флокуляция». Фактически же
в разбавленных растворах солей, содержащих ионы А13+ и Fe3+,
присутствуют как мономерные катионы, так и полимерные
многозарядные катионы, причем последние часто оказываются
настолько большими по размеру, что отождествляются как
положительно заряженные коллоидные частицы. По этой при-
чине коагулирующее действие как солей поливалентных метал-
лов, так и соответствующих коллоидных частиц этих солей
рассматривается как один процесс.
Вероятно, что коагуляция кремнезема под действием моно-
мерных или одиночных катионов, поликатионов или положи-
тельно заряженных коллоидных частиц происходит посредством
мостикового механизма, понимаемого в том смысле, что ука-
занные положительно заряженные единичные образования
служат как для нейтрализации отрицательных зарядов на
кремнеземных частицах в точках их контакта, так и для
образования осадка. Однако относительная эффективность мно-
гозарядных катионов зависит от того, какая доля этих агентов
коагуляции адсорбируется на.частицах кремнезема при крити-
ческой концентрации коагуляции в растворе. Поскольку чем
больше коагулянт по своему размеру и по числу положитель-
ных зарядов, тем выше при равновесии его адсорбированная
доля, и оказывается, что полимерные разновидности проявляют
гораздо большую эффективность по сравнению с мономерными.
Так, О’Мелиа и Стамм [257] отметили, что полимерные гидро-
ксокомплексы железа(III) адсорбируются значительно силь-
нее, чем мономерный трехзарядный ион металла. Такие ком-
плексы действуют в качестве коагулянтов при гораздо более
низкой суммарной критической концентрации коагуляции
(к. к. к.), чем это имеет место для простых ионов Fe3+, по-
скольку комплексы большей частью находятся в адсорбирован-
ном состоянии на кремнеземных частицах и лишь очень незна-
чительная их доля остается в растворе. В случае же ионов Fe3+
только часть их адсорбируется на кремнеземе в точке к. к. к.
Когда большая часть сложного коагулянта в рассматриваемой
системе адсорбирована на частицах кремнезема, то соотноше-
ние между точкой к. к. к. и суммарной величиной поверхности
кремнезема в системе становится более очевидным. Авторы
пришли к заключению, что адсорбированные разновидности
поликатионов железа (III) вызывают агрегацию частиц крем-
незема посредством формирования мостиков.
Те же авторы провели необычное наблюдение, которое
показало, что кремнезем способен коагулировать при воздейст-
вии железа, даже если последнее находится в системе выше
своей изоэлектрической точки, и, таким образом, оказывается
отрицательно заряженным, как и частицы кремнезема. Они
518
Глава 4
пришли к заключению, что «специфические химические силы
взаимодействия» должны иметь возможность преодолевать элек-
тростатические силы отталкивания. Адсорбция таких разновид-
ностей железа на кремнеземе при одинаковом по знаку заряде
должна включать образование связей Si—О—Fe. Такое поло-
жение согласуется с идеей о том, что образование химических
связей между частицами кремнезема посредством включения
в связь промежуточного мостикового атома или же промежу-
точной частицы фактически может представлять собой «силы
притяжения» вместо предполагаемых «вандерваальсовых сил»,
по крайней мере для случая кремнезема. Согласно некоторым
авторам [220, 258], положительно заряженные коллоидные
частицы — поликатионы действуют подобно мостикам между
отрицательно заряженными частицами кремнезема, формируя
таким образом трехмерную сетку. В таком случае коагулянт
представляет собой часть осадка. Хан и Стамм [259, 260]
выявили стадию, определяющую скорость процесса коагуляции
частиц кремнезема при использовании гидролизованных ионов
алюминия. Они постулируют три различающиеся стадии:
а) образование коагулянта в виде поликатионной разновидности
посредством гидролиза и полимеризации алюминия (III); б) де-
стабилизацию дисперсии в результате специфической адсорб-
ции изополикатионов, которая понижает потенциал поверхности
коллоидных частиц кремнезема; эта стадия обозначается как
«адсорбционная коагуляция»; в) перенос коллоидных частиц
за счет броуновского движения или же существования гра-
диента скоростей. Стадии а) и б) протекают быстро, тогда как
стадия в) оказывается медленной, т. е. этапом, определяющим
скорость всего процесса. Скорость коагуляции была получена
как произведение значения частоты столкновений частиц на
фактор эффективности таких столкновений. Авторы провели
различие между «адсорбционной коагуляцией» в том случае,
когда имеется скопление коллоидных частиц с гидролизован-
ными ионами металла, способными сильно адсорбироваться на
поверхности коллоидного кремнезема, и «дестабилизацией»
в случае существования негидролизованных ионов металла,
когда адсорбция указанных ионов оказывается значительной
относительно общего количества ионов, присутствующих в ра-
створе.
Однако, как отмечалось выше, оценить число адсорбирован-
ных «мостиковых ионов» довольно трудно, если только очень
небольшая доля от общего количества флокулирующих ионов
в системе оказывается адсорбированной на коагуляте, т. е.
когда такие ионы образуют мостиковые связи. Их наблюдения
подтверждают, что имеется большое различие в эффективности
коагуляции между мономерными разновидностями, но не исклю-
Коллоидный кремнезем — концентрированные золи
519
чается и возможность того, что катионную мостиковую связь
могут образовывать даже мономерные катионы.
Хан и Стамм [259] отмечали, что только некоторая доля от
общего числа наблюдаемых соударений между частицами
приводит к образованию устойчивых скоплений частиц. Вполне
возможно, что, когда две частицы, несущие заряды только лишь
на определенных участках своей поверхности, ориентированы
друг относительно друга таким образом, что участок с адсор-
бированным ионом на первой частице обращен к ионному
участку на поверхности второй частицы, отталкивание между
соответствующими ионами препятствует тесному сближению
частиц, и поэтому никакого соединения этих частиц не проис-
ходит. Но в том случае, когда частицы ориентированы так, что
заряженный участок одной частицы приближается к незаря-
женному участку другой, могут иметь место столкновение и
объединение частиц [251]. Стамм и О’Мелиа [261] выдвигают
важный вопрос о том, что флокуляция представляет собой
•стехиометрическую реакцию, и рассматривают данное явление
с этих позиций. Между вызывающими флокуляцию ионами и
флокулируемыми разновидностями имеют место не только сте-
хиометрические соотношения, но также и специфические хими-
ческие взаимодействия. Стамм, Хуанг и Дженкинс [262]
пришли к заключению, что теория Дерягина—Ландау—Фер-
вея—Овербека (ДЛФО) не дает объяснений роли специфических
взаимодействий между коагулирующим ионом и поверхностью
коллоидной частицы.
Некоторые координационные комплексы металлов с акво-
лигандами способны сильно связываться с поверхностью крем-
незема, поскольку аквогруппа может обмениваться на сила-
нольную. К тому же поверхностная группа O3SiCH может
замещать С1~ в таком комплексе, как транс-Со [ (en^CU] +•
Подобные адсорбированные комплексные ионы оказываются
гораздо более устойчивыми по отношению к. гидролизу, чем
когда они существуют в растворе [263].
Во многих случаях в первую очередь необходимо рассмат-
ривать не просто ионное притяжение, а специфическую реакцию,
ведущую к образованию связей металл—кислород—кремний.
Сходное образование связей РЬ—О—А1 происходит тогда,
когда положительно заряженный ион РЬ2+ сильно адсорби-
руется на у-оксиде алюминия ниже точки нулевого заряда при
условии, что оксид алюминия также заряжен положи-
тельно [264].
Согласно данным Гото [265], удаление коллоидного и
растворимого кремнезема путем флокуляции и адсорбции на
оксиде алюминия оказывалось оптимальным для коллоидной
формы при pH 4,5, а для растворимой формы при pH 9.
520
Глава 4
Джеймс и Хили [266а] выполнили обширные исследования
по адсорбции многозарядных ионов металлов на кремнеземе.
Ионы Fe3+, Сг3+, Са2+, Со2+, La3+ и Th4+ адсорбировались все
более и более сильно с увеличением pH, прежде всего нейтра-
лизуя заряд на поверхности кремнезема и далее образуя заро-
дыши в виде гидроксидов металлов на поверхности кремнезема.
Окончательный результат — покрытие поверхности кремнезема
гидроксидами металлов.
В общем случае наблюдалось, что в смеси раствора соли
поливалентного металла и поликремневой кислоты или кол-
лоидного кремнезема при низком значении pH происходит их
соосаждение по мере того, как pH повышается до значения,
лишь немного не достигающего pH начала осаждения гидро-
ксида металла из раствора соли металла в отсутствие кремне-
зема в системе. Шиндлер и др. [2666] попытались установить
взаимосвязь параметров в рассматриваемом явлении на коли-
чественной основе. Они обнаружили, что имеется зависимость
между константами равновесия адсорбции ионов металлов на
поверхности кремнезема и константами равновесия образования
гидроксидов (см. более подробно в гл. 6).
В работе [267] было показано, что коагуляция галогенида
серебра при воздействии гидролизованных солей алюминия за-
медляется добавлением фторид-ионов, что приводит к образо-
ванию комплексных ионов с алюминием. Подобное понижение
эффективности коагуляции в случае других катионов металлов,
таких, как Fe3+ и Th4+, при их воздействии на коллоидный
кремнезем можно предвидеть заранее.
Согласно данным Портёра и Вебера [268], ионы железа(III)
и ионы уранила адсорбируются на кремнеземе даже при низком
значении pH (4,3-10~2 М НСЮ4). Оксид алюминия адсорби-
руется на кремнеземе, вероятно, в виде многоосновных ионов.
Это способствует присоединению к поверхности жирных кислот,
так что частицы кремнезема становятся в достаточной степени
гидрофобными по мере их выделения с помощью «микрофло-
тации» [269, 270].
Гетерокоагуляция коллоидных частиц, несущих противо-
положные заряды, представляет собой очень сложное явление,
поскольку поведение системы зависит от относительных разме-
ров и концентрации частиц, способа приготовления смеси и
некоторых операций, выполняемых после смешивания. Как
было показано Хили и Др. [271], при смешивании коллоидных
кремнезема и оксида алюминия “Происходит коагуляция. В том
случае, когда коагулят диспергируется путем перемешивания,
то он формируется снова, но уже более медленно, пока коагу-
ляция не прекратится. Или один, или оба оксида растворяются
и осаждаются одновременно до тех пор, пока все поверхности
Коллоидный кремнезем — концентрированные золи
521
частиц не станут одинаковыми и коагуляция после этого пре-
кратится. Айлер [272] отмечал такое взаимное воздействие
друг на друга при исследовании растворимости кремнезема.
Вначале растворимость подавлялась вследствие того, что неко-
торая доля поверхности кремнезема вступала в реакцию
с ионами АЮГ’ но затем поверхность с адсорбированными на
ней ионами покрывалась осажденным кремнеземом.
Хардинг [273] изучил ряд факторов, влияющих на процесс
гете'рокоагуляции образцов коллоидного кремнезема с коллоид-
ным оксидом алюминия. Относительное число частиц, требуемое
для оптимального протекания коагуляции, зависит от их относи-
тельных размеров. Высокая концентрация в системе нейтраль-
ного электролита затормаживает взаимную коагуляцию. Хогг
и др. [274] рассмотрели влияние, оказываемое на коагуляцию
размером частиц. О подобном эффекте сообщалось Чернобереж-
ским, Голиковой и Гирфановой [275], которые наблюдали, что
в смеси золей SiO2 и Fe2O3 при pH 3 коагуляция не происхо-
дила, когда концентрация КС1 превышала 10~3 моль/л. Такая
система также изучалась Мади и др. [276].
Влияние концентрации кремнезема' и других факторов
Поскольку одним из этапов процесса флокуляции является
столкновение частиц, то очевидно, что скорость флокуляции
зависит в большей степени от числа частиц в единице объема
раствора, чем от массовой концентрации кремнезема. Таким
образом, при концентрации кремнезема 1 % в золе с размером
частиц 10 нм в единице объема содержится в 1000 раз больше
частиц, чем в золе с частицами 100 нм в диаметре. К тому же
частицы меньшего размера перемещаются с большими скоро-
стями (броуновское движение), что влияет на силу соударения
частиц, причем такая сила должна быть достаточно большой,
чтобы преодолеть ионный барьер.
До сих пор не было проведено никаких существенных работ
по измерению относительных скоростей коагуляции под дейст-
вием электролитов в зависимости от размеров частиц и кон-
центрации кремнезема. К тому же не было выполнено деталь-
ных исследований, касающихся количественного содержания
коагулянта в коагуляте.
Коагуляция имеет место и в случае замораживания золя.
Это происходит потому, что частицы кремнезема концентри-
руются между растущими кристаллами льда до тех пор, пока
они не сдавливаются вместе, после чего происходит связывание
частиц. Хотя представляется возможным добавлять какой-либо
антифриз, например гликоль или этиловый спирт, это не столь
легко выполнимо в случае коммерческих золей. Также может
522
Глава 4
происходить и повторное диспергирование частиц, если в си-
стеме присутствуют достаточно большие по размеру противо-
ионы — катионы, способные покрыть поверхность кремнеземных
частиц в такой степени, что последние не смогут более вступать
в прямой контакт [63]. Вузах и Рорзетцер [277] отметили, что-
уплотнение частиц путем замораживания аналогично их кон-
центрированию за счет удаления воды через мембранный
ультрафильтр [278]. Возможность повторного диспергирования
частиц кремнезема зависит от природы пленки, образующейся
между уплотненными частицами.
Влияние размера частиц
Согласно теории ДЛФО, критическая концентрация коагули-
рующего иона не должна зависеть от размера коллоидной ча-
стицы, но Визе и Хили [279] показали, что при дальнейшем
уточнении теории, принимая во внимание и первый, и второй
минимумы, можно объяснить, что устойчивость коллоидной
системы возрастает с увеличением размера частиц. Стабиль-
ность достигает максимума, а затем убывает. Подобное пове-
дение системы никогда не регистрировалось в случае коллоид-
ного кремнезема с размерами частиц в общепринятой области
5—100 нм.
Оттевил и Шоу [280] сообщили о некоторых наблюдаемых
колебаниях в значениях к.к.к. ионов Ва2+ в зависимости от раз-
меров частиц полистирольного латекса (30—212 нм), но не
были представлены достаточно веские аргументы, доказываю-
щие реальность этого эффекта [281]. Однако авторы работы
[282] описали аналогичный эффект, в котором точка к.к.к. до-
стигала максимального значения при использовании частиц раз-
мером 758 нм. По-видимому, 'полученные в этом случае резуль-
таты объясняются воздействием второго минимума, предусмат-
риваемого теорией ДЛФО, который имеет отношение только
к относительно большим по размерам частицам.
Френс [283] отметил, что золи, содержащие металлические
частицы, фактически можно фракционировать в соответствии
с их размерами путем коагуляции электролитами различной
концентрации. Полученные результаты объясняются понижен-
ным значением вандерваальсового притяжения между неболь-
шими частицами металла. Хардинг [237] в дальнейшем провел
исследование этого явления в суспензиях, получаемых из пиро-
генного кремнезема.
Что касается коллоидного кремнезема, то существуют опре-
деленные условия, когда флокуляция непосредственно зависит
от размеров частиц. Айлер [169] сообщил, что при флокуляции
частиц коллоидного кремнезема диаметром 4—130 нм под дей-
« Коллоидный кремнезем — концентрированные золи 523
ствием ионов Са2+ в области pH 8—9,5 наблюдаются заметные
изменения значений к.к.к. с увеличением размеров частиц. Од-
нако, чтобы наблюдать этот эффект, необходима определенная
величина к.к.к., представляющая собой количество ионов каль-
ция, еще находящихся в растворе, без учета этих ионов, адсор-
бированных на поверхности кремнезема.
При тщательном контролировании условий эксперимента
большие по размеру частицы кремнезема могут быть скоагули-
рованы и отделены от меньших частиц. Необходимое для коа-
гуляции критическое число ионов кальция определяется как ко-
личество таких ионов, адсорбированных на одном квадратном
нанометре поверхности кремнезема независимо от размеров его
частиц; но чтобы достичь такой величины адсорбции в том слу-
чае, когда частицы становятся меньшими по размеру, в раст-
воре должна поддерживаться более высокая концентрация ионов
кальция. На поверхности кремнезема, которая уже несет от-
рицательные заряды, каждый адсорбированный ион кальция
освобождает только один ион водорода, образуя при этом один
дополнительный отрицательный заряд на кремнеземной поверх-
ности. Таким образом, каждый адсорбированный ион кальция
сохраняет за собой один единичный положительный заряд (см.
также [255]). Для частиц меньших размеров вследствие их бо-
лее сильно искривленной поверхности каждый ион кальция, ад-
сорбированный на внешней стороне поверхности частицы, будет
отталкиваться от своих соседей по поверхности, причем резуль-
тирующая сила будет направлена от поверхности кремнезема,
поэтому для поддержания критической концентрации коагуля-
ции адсорбированных ионов кальция требуется их более вы-
сокая концентрация в растворе. Коагуляция, вероятно, обуслов-
лена силами притяжения между поверхностями частиц крем-
незема, несущими на себе мозаично расположенные положи-
тельно и отрицательно заряженные участки.
Такое поведение, вероятно, характерно для больших по раз-
меру двухзарядных и в любом случае многозарядных катионов,
так как для ионов натрия нет никаких доказательств того, что
концентрация отрицательных зарядов на поверхности частиц
при заданных значениях pH и концентрации ионов натрия ме-
няется в зависимости от размеров частиц. Другие большие по
размеру катионы не были исследованы таким образом. Крити-
ческие концентрации коагуляции для различающихся по разме-
рам частиц показаны на рис. 4.176. Флокуляция под действием
ионов Са2+, очевидно, не ведет к образованию постоянных си-
локсановых связей между частицами кремнезема. Ион кальция
отличается от иона натрия тем, что первый из них достаточно
большой по своему размеру; это позволяет предотвращать пря-
мой контакт между частицами кремнезема, и непосредственные
524
Глава 4
связи Si—О—Si между частицами не возникают. Так, Алексан-
дер и Айлер [80] обнаружили, что частицы кремнезема могут
коагулировать из разбавленного раствора, но затем коагулят
Рис. 4.176. Зависимость критических концентраций коагуляции ионов кальция
от величины pH для различающихся по размерам частиц коллоидного крем-
незема:
Кривая
Удельная поверхность, м2/г
Диаметр частиц а, нм
А 540 5
В 404 7
С 215 13
114 24
Еб 0 оо
Л1в 210 13
а Значения диаметров подсчитаны из удельной поверхности.
б Экстраполированная кривая.
в По данным Аллена и Матиевича.
может повторно диспергироваться в воде при удалении ионов
кальция.
Это явление, показывающее, что ион кальция нейтрализует
только один заряд на поверхности кремнезема и сохраняет тем
самым один свой положительный заряд, отмечалось также Бо-
эмом и Шнайдером [284]. Гудмен [285] предложил соответствую-
Коллоидный кремнезем — концентрированные золи
525
щую теорию, которая описывает возможность притяжения частиц
кремнезема друг к другу под действием мозаично размещенных
на поверхности каждой частицы положительно и отрицательно
заряженных участков. Присутствие катионных зарядов, возни-
кающих на поверхности кремнезема вследствие адсорбции ионов
кальция, наглядно доказывается тем фактом, что подобная по-
верхность способна затем адсорбировать анионы жирных кислот,
что приводит к ее гидрофобизации. Такое явление используется
в процессе флотации с целью удаления кремнезема из металли-
ческих руд [286].
Частично дегидратированная поверхность
По-видимому, существует значительное различие между ча-
стицами кремнезема, приготовленными в водной среде, и полу-
ченными при высокой температуре, т. е. пирогенным способом,
когда частицы сразу после их приготовления подвергаются дис-
пергированию в воде. Какая-то часть негидроксилированной по-
верхности пирогенного кремнезема в течение некоторого вре-
мени может сохраняться в неизменном виде, поэтому только
часть поверхности будет покрыта способными ионизоваться
группами SiOH. Такое различие было ясно продемонстрировано
Чапеком и Торресом Санчесом [287], которые показали, что
дегидратированный кремнезем ведет себя так, как если бы его
поверхность была гидрофобной. При низком значении pH, когда
на поверхности имеется очень незначительный заряд, пироген-
ный кремнезем способен флокулировать под действием следов
соли в системе.
По этой причине диспергирование золей, приготовляемых пу-
тем суспендирования пирогенного кремнезема в воде, затруд-
нено и становится возможным тогда, когда поверхность гидра-
тируется за счет каталитического действия щелочи.
В качестве дальнейшего доказательства этих фактов Рубио
и Гольдфарб [288] представили данные, показывающие, что
в водных дисперсиях пирогенного кремнезема, такого, напри-
мер, как аэросил, необходимое для коагуляции количество ионов
четвертичного аммониевого основания оказывается гораздо
меньшим, чем в случае полностью гидратированного кремнезема.
Кроме того, рассматриваемый золь повторно стабилизируется
заметно меньшим избыточным количеством органических ка-
тионов.
Флокуляция
Флокуляция золя кремнезема происходит при добавлении ка-
кой-либо полимерной или коллоидной мицеллы или частицы, спо-
собной одновременно адсорбироваться на поверхностях двух раз-
526
Глава 4
личных частиц кремнезема, связывая их таким образом вместе.
Эта адсорбция имеет место в присутствии катионного агента
либо способного притягиваться к отрицательно заряженной по-
верхности кремнезема, либо содержащего электронно-донорную
группу (например, атом кислорода в полиэфире), что позволяет
удерживать его на поверхности кремнезема за счет образования
водородной связи. Последнее из отмеченных явлений не наблю-
дается при высоких значениях pH, когда поверхность кремнезема
сильно заряжена, но имеет место при низких pH, когда поверх-
ность Покрыта нейтральными силанольными группами. Таким
образом, органическими флокулирующими агентами могут быть:
а) катионные поверхностно-активные вещества, которые обра-
зуют мицеллы; б) катионные органические полимеры и в) не-
ионные водорастворимые соединения или полимеры, содержащие
электронно-донорные группы (например, эфирные, гидроксиль-
ные или амидогруппы). Эти данные суммированы в таблице 4.2,
Таблица 4.2
Разновидности флокулирующих агентов
Тип агента Пример Механизм действия pH
Катионное ПАВ Низкомолекулярная Бромид октадецилтри- масса Ионное притяже- 4-10,5
Неионное ПАВ метиламмония Неионные моющие ве- ние, образова- ние мицеллы Водородная связь, <3
Основная соль щества Основной хлорид алю- образование ми- целлы Ионное притяже- (плюс соль) <7
металла Катионный поли- миния В ысо ко мо леку л я р н а Четвертичные аммони- ние I масса Ионное притяже- 3-9
мер Неионный поли- евые замещенные по- лиакрилаты Полиэтиленоксид ние Водородная связь <5
Катионное неор- Коллоидный оксид алю- Ионное притяже- 3-7
ганическое кол- лоидное веще- ство МИНИЯ ние
а механизмы флокуляции схематически изображены на
рис. 4.18—4.20.
Механизм, благодаря которому частицы кремнезема удержи-
ваются вместе, зависит от типа флокулирующего агента.
1. Когда подобный агент адсорбируется на поверхности ча-
стицы и ориентируется таким образом, что образуются гидрофоб-
Рис. 4.18. Схема флокуляции частиц кремнезема.
Образование мостиковых связей между двумя частицами посредством положительно за-
ряженных поверхностно-активных мицелл, например ионов цетилтриметиламмония (/);
трехмерных полимерных молекул, например крахмала (2); линейных полимеров, напри-
мер молекул полиэтиленимина (3).
Рис. 4.19. Схема флокуляции кремнеземных частиц под действием других кол-
лоидных частиц с противоположным ионным зарядом.
528
Глава 4
ные участки, то частицы вступают в контакт с формированием
мицеллы именно благодаря этим участкам, расположенным на
разных частицах. Частицы кремнезема удерживаются вместе
благодаря наличию мощных сил поверхностного натяжения, со-
средоточенных в зоне контакта частиц около искривлений по-
верхности с отрицательными радиусами кривизны. Это так на-
зываемая гидрофобная связь. Представляется возможным также
Рис. 4.20. Схема флокуляции и коацервации кремнеземных частиц, вызванных
образованием водородных связей с эфирами, спиртами и амидами при низких
значениях pH.
рассматривать такой тип флокуляции, как адсорбцию двух крем-
неземных частиц на противоположных сторонах одной мицеллы
поверхностно-активного вещества в растворе. Однако флокуля-
ция может иметь место и тогда, когда концентрация катионного
ПАВ оказывается ниже критической концентрации, необходимой
для образования мицелл в отсутствие кремнеземных частиц.
Тем самым выдвигается предположение, что мицелла стабилизи-
руется именно вследствие существования флокулирующего
агента вблизи точки контакта между твердыми частицами.
2. Второй, более широко признаваемый механизм заклю-
чается в том, что полимерная частица органического вещества
или коллоидная неорганическая частица, отличная от кремне-
зема, присоединяясь к двум различным кремнеземным частицам,
способна действовать как мостиковая связь.
Для какого-либо конкретного золя при заданных условиях
постепенное добавление в систему флокулирующего агента ведет
вначале к флокуляции только лишь некоторой доли имеющихся
частиц. Дальнейшее добавление агента будет приводить к воз-
растанию количества флокулированного осадка до тех пор, пока
Коллоидный кремнезем — концентрированные золи
529
в растворе полностью не исчезнут одиночные коллоидные ча-
стицы. Полная флокуляция имеет место только тогда, когда
в системе присутствует достаточное количество адсорбирован-
ного на частицах флокулянта, способного создавать мостики
между частицами в среднем в трех точках контакта в расчете
на одну частицу, так что формируется трехмерная сетка. После
этого флокулянт, находящийся в системе в избытке, адсорби-
руется на осадке. Адсорбция продолжается до тех пор, пока вся
поверхность осадка не будет покрыта. В зависимости от при-
роды флокулирующего агента дальнейшее его добавление мо-
жет вызвать повторное диспергирование кремнеземных частиц,
причем каждая из них будет теперь окружена адсорбирован-
ным слоем «флокулянта», а частицы будут иметь положитель-
ный заряд на поверхности.
Отсюда следует, что термином «флокуляция» характеризуется
действие агента, присутствующего в системе в количестве, соот-
ветствующем критическому содержанию, поскольку при дальней-
шем добавлении флокулянт уже становится стабилизирующим
агентом.
Для предотвращения пептизации в присутствии избыточного
количества флокулирующего агента могут быть использованы
наблюдения Руэрвейна и Уорда [258]. Если перед введением
в систему органического полимера коллоидный кремнезем скоа-
гулирован так, что его частицы находятся в контакте друг
с другом, то после введения полимера происходит его адсорбция
вокруг точек контакта, упрочняющая мостики между частицами.
Однако полимер не помещается между частицами, и поэтому не
вызывает повторного диспергирования частиц. С другой сто-
роны, если кремнеземные частицы разделены во время добав-
ления флокулирующего агента, то все поверхности частиц могут
быть окружены таким агентом, и при добавлении избыточного
количества агента образуется золь кремнезема.
Была предложена [289] общая модель структуры флокулиро-
ванного осадка, основанная на том, что коагуляция происходит,
когда в системе одиночные частицы добавляются к дублетам,
триплетам и к агрегатам, состоящим из большего числа частиц,
которые также могут сталкиваться между собой и сцепляться.
Смелли и Ламер [290] развили теорию флокуляционной плот-
ности и фильтрации.
Флокуляция под действием катионных
поверхностно-активных веществ
Взаимодействие гидрофобных групп, адсорбированных на по-
верхности кремнезема, влечет за собой образование «гидрофоб-
ной связи». Тэнфорд [291] подробно описал этот эффект. Для
3 Заказ № 250
530
Глава 4
коллоидного кремнезема сильнодействующими флокулирующими
агентами являются амины с длинными цепями, в частности ал-
килзамещенные четвертичные соли аммония с длинными цепями.
Так, Смит и Турнбул [292] добавляли четвертичную соль ам-
мония, чтобы вызвать частичную флокуляцию коллоидного
кремнезема и придать ему тиксотропность для использования
в качестве связующего вещества в тугоплавких порошковых
шламах.
Авторы работы [293] при обсуждении вопроса о мицелляр-
ной адсорбции описали ход адсорбционного процесса поверх-
ностно-активного вещества на частицах коллоидного кремнезема.
Полярная поверхность покрывается адсорбированным поверх-
ностно-активным веществом, ориентированным по отношению
к ней таким образом, что частицы кремнезема становятся гидро-
фобными. При немного более высокой концентрации ПАВ на
первичном адсорбированном слое происходит дополнительная
адсорбция, и на поверхности формируется конденсированный
сдвоенный слой ПАВ. При этом оказывается, что полярные
группы ПАВ во втором слое ориентируются уже наружу, сооб-
щая поверхности гидрофильные свойства. Авторы представили
экспериментальное доказательство того, что по мере добавления
к системе дополнительного количества ПАВ его концентрация
в растворе начинает понижаться после некоторой концентра-
ционной точки, в которой стремительно развивается агрегация
адсорбированного вещества вплоть до формирования двойного
слоя на поверхности мицелл.
Хорошо известно, что гидрофобные частицы в воде прили-
пают друг к другу, как только приходят в соприкосновение. По-
этому не удивительно, что частицы кремнезема становятся гид-
рофобными, едва только на некоторых ограниченных участках
на частицах кремнезема начинается флокуляция. Следовательно,
при низких концентрациях такие ионы, как додециламмоний,
влияют на электрокинетический потенциал частиц кварца точно
так же, как и ионы натрия. При более высоких концентрациях
наблюдается критическая точка, в которой электрокинетический
потенциал резко изменяется, и ионы аммония с длинной цепью,
несомненно, собираются в отдельные ассоциаты на поверхности
раздела во многом подобно тому, как это происходит при фор-
мировании мицелл в объеме раствора [294].
Мощное флокулирующее действие небольших добавок хло-
рида цетилтриметиламмония на золи кремнезема было исполь-
зовано Александером и Айлером [295], чтобы сгруппировать
частицы кремнезема при таких условиях, когда они способны
формироваться в агрегаты, подобные листочкам или пленкам.
Электронно-микроскопический снимок плоских агрегатов пред-
ставлен на рис. 4.21. Механизм формирования таких агрегатов
Коллоидный кремнезем — концентрированные золи
531
Рис. 4.21. Электронно-микроскопический снимок листоподобных агрегатов,
сформировавшихся из коллоидного кремнезема в присутствии катионных по-
верхностно-активных веществ.
схематически показан на рис. 4.22. Частицы кремнезема удер-
живаются вместе благодаря действию мицелл катионного ПАВ.
Требуемое количество ПАВ составило только ~5 % от оценен-
ного теоретически количества, необходимого для образования
одного монослоя ПАВ на всей суммарной поверхности кремне-
зема. Мицеллы, вероятно, размещаются на кремнеземной поверх-
ности только в точках контакта между частицами. Такой особый
тип плоскостной флокуляции протекает в щелочном растворе,
когда частицы кремнезема заряжены отрицательно и, следова-
тельно, взаимно отталкиваются, но тем не менее при наличии
достаточной силы, способной преодолевать мицеллярные связи.
8*
532
Глава 4
При этих условиях агрегаты будут расти в тонком слое, подобно
листочку толщиной всего лишь в одну кремнеземную частицу.
Так происходит потому, что частицы кремнезема способны сбли-
жаться и присоединяться друг к другу только по краям расту-
щего листочка, поскольку'
в этой области оказывается
наименьшим ионное оттал-
кивание. Как только фор-
мирование подобных лис-
точков или «плоскостных
агрегатов» заканчивается,,
на них начинает осаждаться
избыточный кремнезем. Это
приводит к прочному связы-
ванию ’Тастиц вместе и за-
полнению пустот между ни-
ми, так что в результате по-
лучается непроницаемая
слоистая частица кремнезе-
ма, имеющая толщину час-
тицы коллоидного размера.
О’Коннор и Сандерс
[296] исследовали гидро-
фобный и гидрофильный ха-
рактер поверхности кремне-
зема при адсорбции на ней
ионов цетилтриметиламмо-
ния. Когда кремнезем или
очищенная стеклянная по-
верхность вступают в кон-
такт с 10~7М водным рас-
твором бромида цетилтри-
метиламмония, то поверх-
ность кремнезема начинает
адсорбировать органический
агент и адсорбция идет до тех пор, пока не образуется монослон
этого вещества. При этом поверхность оказывается гидрофоб-
ной. Однако, если в растворе присутствует дополнительное коли-
чество этого агента, поверхность полностью не высыхает, когда
ее извлекают из раствора, и в действительности остается влаж-
ной при условии, что концентрация агента составляет более
чем 1 (У 4 моль/л. Критическая концентрация мицелл в системе
равна ~ 10~3 моль/л, поэтому очевидно, что при очень низких
концентрациях на кремнеземе будет адсорбироваться один мо-
нослой этого агента с гидрофобными группами, направленными
наружу. Это придает поверхности гидрофобные свойства, если
Рис. 4.22. Схема формирования агрега-
тов в виде листочков из частиц кремне-
зема.
Частицы имеют возможность приближаться
к краям листочка, где энергетический барьер
за счет сил отталкивания оказывается мень-
шим. чем на поверхности. Внизу показано по-
перечное сечение листочка из слипшихся между
собой частиц кремнезема.
Коллоидный кремнезем — концентрированные золи 533
в растворе отсутствует избыток агента. Но если при концентра-
ции мицелл 10~3 моль/л агента содержится больше, то тогда
формируется двойной адсорбированный слой и катионы распо-
лагаются снаружи, снова превращая поверхность кремнезема
в гидрофильную, которая при этом оказывается способной
к смачиванию.
Коллоидный кремнезем, подвергшийся флокуляции благодаря
образованию «гидрофобных связей» между гидрофобными уча-
стками на поверхности кремнеземных частиц, имеет следующую
характерную особенность: при добавлении смешиваемого с во-
дой спирта, например пропилового, наблюдается повторное дис-
пергирование флокулята за счет «смачивания» поверхности.
Гидрофобные группы пропилового спирта ориентируются по на-
правлению к гидрофобной поверхности частиц кремнезема так,
что спиртовые гидроксильные группы, направленные наружу,
превращают поверхность в гидрофильную. При повторной реге-
нерации и высушивании кремнезема спирт испаряется, и по-
верхность снова становится полностью гидрофобной.
Как отмечали Матиевич и Оттевилл [297], для адсорбции
катионного ПАВ на поверхности коллоидных частиц необ-
ходимо, чтобы частицы по крайней мере имели определен-
ный наименьший размер. Когда ПАВ в системе присутствует
только в необходимом для флокуляции количестве, поверхность
коллоидных частиц становится гидрофобной и происходит коа-
гуляция. Но когда вводится избыточное количество ПАВ, про-
исходит формирование второго слоя вследствие вандерваальсо-
вого притяжения между углеводородными цепочками. Тогда
ионизированные группы второго наружного слоя ориентируются
по направлению к раствору, и частицы начинают разделяться и
пептизировать с переменой знака заряда на поверхности.
Природа «гидрофобной связи» в водных растворах между
гидрофобными участками молекулярных размеров была выяв-
лена Клоцем [298]; затем этот вопрос обсудил Шерага [299]
в связи с исследованиями белков; дальнейшее тщательное изу-
чение было выполнено Немети и Шерага [300]. Козман [301]
определил «гидрофобную связь» как стремление неполярных
групп сцепляться друг с другом в водных средах. Это похоже
на образование мицелл с внутримолекулярной связью, подобно
тем, что существуют в водных растворах моющих веществ. Коз-
ман [302] представил общее обсуждение вопроса о «гидрофоб-
ных связях».
Исследования показывают, что ионы четвертичных аммоние-
вых оснований с длинными цепями, таких, как цетилпиридин,
адсорбируются на поверхности кремнезема примерно при крити-
ческой концентрации мицелл [303]. Ион алкиламмония занимает
на поверхности кремнезема площадь, примерно равную 33—
534
Глава 4
45 А2 [304]. Фактически эти ионы адсорбируются на кремнезем-
ной поверхности в виде димеров [305].
В результате флокуляции под воздействием катионного ПАВ,
такого, как бромид цетилтриметиламмония, золь кремнезема
может стать высоко тиксотропным, что полезно при его приме-
нении в качестве связующего вещества [306].
Для изучения флокуляции кремнезема, вызванной действием
поверхностно-активных веществ, оказывается интересным то, что
избыточное количество катионного ПАВ может быть оттитро-
вано анионным ПАВ с применением в качестве индикатора
бромфенолового синего [307].
Флокуляция под действием органических полимеров
Ламер и Хили [220] показали в своем обзоре широкие воз-
можности процессов адсорбции макромолекул на поверхности
раздела твердое тело—жидкость и флокуляции. Для каждой си-
стемы полимер—коллоид в пределах узкой области концентра-
ций флокулянта наблюдается максимальная флокуляция.
На начальной стадии, когда в системе присутствует слиш-
ком мало полимера, он не способен вызывать флокуляцию,
включающую адсорбцию полимера на поверхности частиц. При
этом оказывается, что число полимерных молекул недостаточно,
чтобы образовать мостики между всеми частицами кремнезема.
Такая ситуация может быть просто временной, так как при
дальнейшем перемешивании, по крайней мере для ряда систем,
полимер адсорбируется предпочтительно в точках контакта
между частицами, и, таким образом, некоторая доля кремне-
земных частиц начинает флокулировать, а основная масса на-
ходится в диспергированном состоянии. Когда в системе при-
сутствует избыточное содержание полимера, флокуляция может
происходить посредством временного образования мостиковых
связей между частицами. Но при последующем перемешивании
такие мостики могут разрываться, и поверхность частиц пол-
ностью покрывается адсорбированным полимером, что приводит
к повторному диспергированию. Ионная сила раствора также
является важной характеристикой, так как она оказывает за-
метное воздействие на свертывание линейных ионных молекул
в клубок или же их распрямление, влияя тем самым на поведе-
ние молекул при формировании мостиков.
Когда длина цепочки флокулирующего полимера увеличи-
вается, например, в пять раз, концентрация полимера, необ-
ходимая для благоприятного протекания флокуляции коллоид-
ных частиц, может быть снижена в сто раз, особенно в обла-
сти, где молекулярная масса полимера меньше 1000. Это свя-
Коллоидный кремнезем — концентрированные золи
535
зано с тем, что при низкой молекулярной массе каждая поли-
мерная молекула не может образовывать мостик между более
чем двумя частицами. При большей длине цепочки действие мо-
лекулярной массы становится гораздо менее важным. Отсюда
вытекает, что для флокуляции тонкодисперсных сусйензий крем-
незема флокулирующий полимер должен иметь цепочку доста-
точной длины, чтобы образовать мостик между частицами; та-
кой фактор оказывается заметным только тогда, когда катион-
ный полимер присутствует в системе в достаточном количестве,
чтобы нейтрализовать заряд на частицах кремнезема. Поли-
меры с очень высокой молекулярной массой вызывают повтор-
ное диспергирование флокулята [308].
В' процессе флокуляции, согласно данным Ламера [309],
преобладающим фактором, приводящим к образованию мости-
ков между частицами, является адсорбция полиэлектролита,
но не электростатическое взаимодействие. Автор представил
математический анализ кинетики флокуляции и дефлокуляции
при динамическом равновесии системы. В соответствии с этими
соображениями, флокуляция не может быть непосредственно
объяснена в рамках теории ДЛФО. Электростатическое взаи-
модействие, с которым главным образом имеет дело теория
ДЛФО, не является определяющим при рассмотрении флоку-
ляции. Основными факторами представляются специфические
химические взаимодействия между коллоидными частицами и
вызывающими флокуляцию агентами. Теория ДЛФО по суще-
ству не учитывает роль адсорбции различных веществ из рас-
твора на коллоидных частицах, но рассматривает добавляемые
к раствору вещества только с точки зрения их влияния, оказы-
ваемого на свойства водной среды, а также электростатические
и молекулярные силы между коллоидными частицами. Общий
обзор физических аспектов флокуляции коллоидов при воздей-
ствии полимеров был представлен в работе [310].
Геллер [311] в своем обширном обзоре показал возможные
случаи воздействий, оказываемых макромолекулярными соеди-
нениями в дисперсных системах. Он обсудил адсорбционные
характеристики полимерных молекул, которые, с одной стороны,
могут адсорбироваться на поверхности только своей концевой
частью, причем оставшаяся часть полимерной цепочки будет
направлена радиально от поверхности, и, с другой стороны, мо-
гут адсорбироваться плашмя, когда все части молекулы присое-
диняются к поверхности. Конечно, могут иметь место обе ситуа-
ции в зависимости от природы самого полимера и от того, на-
сколько велик избыток полимера по сравнению с количеством,
требуемым для формирования плоского мономолекулярного
слоя.
536
Глава 4
Ботем и Тейс [312] исследовали и описали различные воз-
можные ситуации, имеющие отношение к взаимодействию поли-
мера с частицами кремнезема. Они отметили, что, согласно мо-
стиковой модели, полимеры с высокой молекулярной массой вы-
зывают флокуляцию коллоидных дисперсий благодаря фор-
мированию мостика между смежными полимерными сегментами,
если число таких сегментов и их длина достаточны и если на
кремнеземных частицах имеется свободная поверхность для
присоединения участков мостика. С другой стороны, система,
подвергаемая флокуляции, становится дисперсной, если поверх-
ность частиц настолько покрывается полимером, что на поверх-
ности частиц не остается свободных мест для образования мо-
стиковой связи. Первоначально между частицами могут сфор-
мироваться мостики, но вполне вероятно, что локализованные
на поверхности избыточные концентрации полимера, которые
на начальной стадии образовывали мостики, будут перераспре-
деляться равномерно по всей поверхности частицы кремнезема.
Кроме того, если поверхность частицы оказывается открытой,
то полимерные цепочки стремятся скрутиться и адсорбируются
на поверхности только одной частицы, а не образуют мостики
между двумя частицами.
Взаимодействие кремнезема и белков с возникновением во-
дородных связей было рассмотрено Айлером в первой моногра-
фии [8] и позже исследовалось Крагом и Лэнгстоном [313] и
Бергманом и Нельсоном [314]. Образование мостиков показано
на основании результатов, полученных Крагом и Лэнгстоном,
которые наблюдали, что флокуляция происходит в такой си-
стеме, когда лишь одна треть желатина от его максимально воз-
можного содержания адсорбируется на поверхности кремне-
зема. Когда же адсорбируется весь желатин, то толщина ад-
сорбированного слоя превышает монослойное покрытие, и часть
цепочек распространяется в раствор. Они также продемонстри-
ровали, что подобный полимерный агент флокуляции не адсор-
бируется в монослоях. Прежде всего формируется разупорядо-
ченная структура с молекулярными цепочками, выступающими
от поверхности частиц, что способствует образованию полимер-
ных мостиков при столкновении частиц. Однако, когда при пе-
ремешивании такие связи разрываются, происходит медленная
перестройка поверхностного слоя, сопровождаемая адсорбцией
свободных цепочек, так что поверхность частиц полностью по-
крывается слоем желатина, после чего между частицами не мо-
гут уже образоваться мостики. Авторы подчеркивают, что pH
как существенный фактор процесса флокуляции влияет главным
образом на конфигурацию адсорбированного слоя желатина,
а не на общий заряд кремнеземных частиц. Цепочки желатина
могут иметь длину 12 000 А, поэтому флокуляция частиц посред-
Коллоидный кремнезем — концентрированные золи
537
ством образования мостиков из этих цепочек может происходить,
даже если частицы находятся на расстоянии 500 А друг от
друга. При таких условиях флокуляция происходит, несмотря
на то что на частицах имеется относительно высокий ионный
заряд.
Бергман и Нельсон сообщили о необычном наблюдении,
когда глобулин проявлял значительное сродство по отношению
к кремнезему, гораздо большее, чем альбумин или желатин. Ав-
торы предположили, что подобное сродство имеет отношение
к физиологическому воздействию кремнеземных частиц на био-
логические ткани.
Катионные полимеры. Взаимосвязь между электрокинети-
ческим потенциалом и флокуляцией под действием полимера
была изучена Рисом [315], который отметил, что, как только
коллоидная частица покрывается слоем полимера, на ней появ-
ляется тот же самый заряд, что и на полимере, и начинает наб-
людаться повторное диспергирование. Аналогичные исследова-
ния были выполнены Рисом и Мейерсом [316] с использованием
микрофореза и электронно-микроскопических наблюдений мо-
дельных коллоидных частиц и полимерных флокулянтов. Агенты
флокуляции полиаминного типа, по-видимому, распростра-
няются от поверхности частицы на расстояние 20—300 А. Фло-
куляция происходит посредством двух одновременных процес-
сов: нейтрализации заряда и образования мостиковой связи
из полимерных цепочек между частицами. После этого избыточ-
ный полимер меняет потенциал на обратный, и происходит пов-
торное диспергирование. Шилук [317] изучал адсорбцию
поли [(1,2-диметилвинилпиридин) метилсульфата] на кремнеземе.
Он пришел к заключению, что полимерные цепочки лежат
плоско на поверхности в том случае, когда в системе отсутст-
вует избыточное содержание полимера.
Механизм адсорбции полимеров, содержащих ионы четвер- 4.
тичных аммониевых оснований, на кремнеземе при низких зна-
чениях pH совершенно не ясен. Точка нулевого заряда крем-
незема находится при рН~2, однако Деревянко и др. [318]
сообщают, что максимальная адсорбция поли(2-метил-5-винил-
пиридинхлорида) наблюдалась при pH 2,9 и что монослой та-
кого полимера адсорбировался на кремнеземе при pH 0,5
или 2,9.
Согласно Зильбербергу [319], при равновесной адсорбции
макромолекулу с гибкими длинными цепями из разбавленного
раствора на твердой поверхности только некоторая доля цепо-
чек располагается на твердой поверхности раздела, тогда как
другие находятся далеко от нее в растворе. Автор обсуждает
возможность того, что концентрированный слой полимера на
поверхности может притягивать на большем пространстве по-
538
Глава 4
верхностную фазу, особенно если концентрация слоя близка
к критической концентрации для разделения фаз, т. е. коацер-
вации.
Теория структуры флокулирующих частиц была представ-
лена Сёзерлендом [289]. Плотность флокулированных осадков
может широко изменяться; она понижается по мере того, как
частицы стремятся агрегировать в цепочки. Цепочки из частиц
и объемные флокулированные осадки образуют, вероятно, неза-
висимо от природы флокулированную систему, на которую не
влияет поляризация частиц.
Грегори [320] на основании своих исследований о коагуля-
ции латексных частиц пришел к заключению, что флокуляция
и повторная стабилизация системы могут быть просто объяс-
нены с точки зрения нейтрализации и перемены знака зарядов.
Можно только сделать вывод, что взаимодействие латексных
частиц отличается от взаимодействия кремнеземных частиц.
Айлер [321] исследовал влияние размеров частиц коллоид-
ного кремнезема на количество требуемого для флокуляции и
обращения зарядов катионного полимера. Выбранным для экспе-
римента полимером был поли [ (R-метиламмоний) метилсульфат]
(где R— метил, акрилоил, оксиэтил, диэтил) [322]. Для частиц
диаметром менее 500 А количество флокулянта, имеющего длину
молекулярных цепочек 1500 А, требуемого для осаждения одной
единицы массы кремнезема, обратно пропорционально диаметру
частиц, тогда как для частиц больших размеров это количество
обратно пропорционально квадрату диаметра частицы. По-ви-
димому, при использовании флокулирующего агента данного
типа для формирования мостика между частицами размером
более 400 А в диаметре в точках контакта необходимы одна
или несколько полимерных цепочек, тогда как для частиц мень-
шего размера на каждую точку с мостиковой связью требуется
только всего несколько сегментов такой цепочки. Единичная
цепочка может простираться от одной точки контакта до дру-
гой, связывая таким образом несколько частиц кремнезема
вместе.
В данном исследовании использовался простой капельный
анализ проб для обнаружения либо избыточного содержания
кремнезема, либо избытка полимера во всплывающем слое
жидкости после завершения флокуляции. Метод основан на ад-
сорбции монослоя находящегося в жидкости избыточного ве-
щества на полированной черной стеклянной пластинке. Его при-
сутствие или отсутствие визуально обнаруживается посредством
нанесения монослоя кремнеземных частиц диаметром 150 нм,
что приводит к появлению в отраженном свете ярко окрашен-
ной интерференционной картины. Айлер обнаружил, что катион-
ный полимер можно титровать 0,02 н. раствором тетрафенил-
Коллоидный кремнезем — концентрированные золи
539
бората натрия в смеси бензилового спирта воды с соотношением
20:80 при рН~4 с использованием индикатора бромфеноло-
вого синего. Вместо тетрафенилбората натрия можно применять
додецилсульфат натрия. Однако невозможно выполнять такое
титрование в присутствии кремнезема, поскольку отмеченные
реактивы удаляют катионный полимер с его поверхности.
Полиэтиленимин (ПЭИ) сильно адсорбируется на поверх-
ности кремнезема. Линдквист и Стреттон [323] изучали флоку-
ляцию, используя полимеры в области молекулярных масс от
1760 до 18400. Для исследования был выбран кремнезем марки
людокс-АМ—золь кремнезема, модифицированный алюминатом
и способный сохранять отрицательный заряд в пределах pH 3—
10. Почти во всей области указанных значений pH ПЭИ пол-
ностью и необратимо адсорбируется на частицах кремнезема.
Критическая концентрация флокуляции (к.к.ф.) выше pH 9 и
в отсутствие каких-либо солей зависела от молекулярной массы
полимера. В точке к.к.ф. частицы кремнезема еще несут на по-
верхности отрицательный заряд. По-видимому, коагуляция выше
pH 9 обусловлена мостиковыми связями между частицами, об-
разуемыми полимером, который при таком значении pH имеет
низкий заряд. Между величинами логарифмов (к.к.ф.) и ка-
тионного заряда, находящегося на ПЭИ, наблюдается линейное
соотношение с отрицательным наклоном прямой.
Полимеры, образующие водородные связи. Только при ней-
тральном или низком значении pH, когда поверхность кремне-
зема обладает слабым отрицательным зарядом или же такой
заряд вовсе отсутствует, частицы кремнезема могут флокули-
ровать под действием ряда полимеров, которые в этой области
pH имеют либо неионный, либо слабокатионный характер.
Айлер [324] обсудил это явление и показал, что когда поверх-
ность кремнезема заряжается путем повышения pH или моди-
фицирования поверхности алюмосиликат-ионами с тем, чтобы
сохранить поверхность заряженной при низком значении pH,
взаимодействие кремнезема с указанными типами полимеров
значительно понижается. По-видимому, рассматриваемое взаи-
модействие с кремнеземом обусловлено водородными связями,
образующимися между электронно-донорными атомами в поли-
мере и нейтральными силанольными группами на поверхности
кремнезема.
Кремнезем взаимодействует с ламинарином — линейным по-
лисахаридом, состоящим из 20 единиц p-D-глюкопиранозы, свя-
занных между собой через атомы С-1 и С-3, при значении pH
5—6, как отметили в своей работе Холт и Вент [325]. Ниже
в связи с рассмотрением коацервации будет упомянуто, что
кремнезем также связывается с поливиниловым спиртом при
низком значении pH. Полиэфиры адсорбируются на кремнеземе
540
Глава 4
из воды, и их молекулы лежат плоско вдоль кремнеземной по-
верхности [326]. Исследование взаимодействия полиэфиров
с кремнеземом в четыреххлористом углероде, выполненное с при-
менением ИК-спектроскопии, показало образование водородных
связей между кислородными атомами эфира и атомами водорода
поверхностных силанольных групп [327]. Работа Рубио и Кит-
ченера [328] представляет особый интерес. Была изучена фло-
куляция водных суспензий образцов кремнезема, имевших раз-
личные типы поверхностей, под действием полиэтиленоксида
(ПЭО) с высокой молекулярной массой. Этот полимер связы-
вается с силанольными группами через кислородные атомы
эфира посредством водородных связей. Кроме того, на тех уча-
стках, где поверхность кремнезема оказывается гидрофобной,
по-видимому, появляется «гидрофобная связь» с углеводородной
цепочкой —СН2СН2—. Так же как и Айлер [324], Рубио и Кит-
ченер пришли к заключению, что ионизация групп SiOH ослаб-
ляет сродство неионных соединений. Такое явление истолковы-
вается как эффект «высаливания». Согласно другой интерпрета-
ции, предложенной Айлером [324], это явление — следствие про-
странственных помех по отношению к противоиону—катиону
в растворе вблизи заряженного участка.
Полиакриламид (ПАА) находит широкое применение как
флокулянт, и его взаимодействию с кремнеземом уделяется осо-
бое внимание. Кузькин и др. [329] сообщили, что ПАА вызы-
вает флокуляцию отрицательно заряженных минеральных ча-
стиц ниже значения pH 8 и его эффективность повышается
с увеличением молекулярной массы. Этот полимер при pH 1,2
имеет слабо выраженные катионные свойства. Грайот и Китче-
нер [330] показали, что водородная связь оказалась видом
взаимодействия при флокуляции под действием этого полимера.
Эффект, оказываемый полиакриламидом в нейтральном раст-
воре, был наиболее заметен при использовании пирогенного
кремнезема, на поверхности которого имеется только ограничен-
ное число силанольных групп. Полимер образует водородные
связи с этими группами, как показывает тот факт, что коагуля-
ция совершенно меняется при воздействии конкурирующих аген-
тов с низкими молекулярными массами, способными образовы-
вать водородные связи. Удивительно то, что, когда поверхность
кремнеземных частиц становится полностью гидратированной,
полиакриламид не вызывает никакой флокуляции. Но если
кремнезем снова дегидратируется при 300°С и повторно диспер-
гируется, то опять появляется возможность флокуляции. Это
различие оказывается настолько поразительным, что процесс
регидратации поверхности кремнезема при действии таких ка-
тализаторов, как HF или ионы ОН , можно контролировать по
степени коагуляции. Вполне очевидно, что водородная связь
Коллоидный кремнезем — концентрированные золи
541
образуется только с отдельной, изолированной силанольной груп-
пой на поверхности, но возможно также, что определенную роль
играет «гидрофобная связь» между сегментами углеводородных
цепочек и гидрофобными участками силоксано-вой поверхности.
С другой стороны, флокулирующее действие этого полимера на
отрицательно заряженные алюмосиликаты осуществляется пос-
редством некоторого ионного механизма, на который не влияют
агенты, образующие водородные связи. Сополимер акриламида и
•его катионный мономер оказываются более эффективными коа-
гулянтами для кремнезема при pH 6—7,2. В этой области pH
может происходить образование как ионных, так и водородных
связей [331].
Известь и жирные кислоты. При высоких значениях pH
{свыше 11) кремнезем адсорбирует . ионы кальция, которые
в свою очередь адсорбируют ионы стеарата, превращая таким
образом поверхность кремнезема в гидрофобную, что может
быть использовано для удаления кремнезема из железной руды
путем флотации. Крахмал при таком значении pH, очевидно,
адсорбируется на оксиде железа, препятствуя его взаимодейст-
вию со стеаратом, и, таким образом, железо остается в суспен-
зии [332].
Когда присутствует кальций, кремнеземный «шлам» может
•флокулировать под действием гуаровой смолы. Хотя гуаровая
смола, вероятно, носит неионный характер, она связывается не-
которым образом (может быть, путем координации через гид-
роксильные группы) с частицами кремнезема, а при высоких
значениях pH на смоле адсорбируются ионы кальция [333].
Взаимодействие полиоксисоединений с атомами кальция или
стронция на поверхности кремнезема при высоких pH имело бы
смысл исследовать ввиду известного образования сахарата
стронция, в котором участвует координационная связь кислород-
ных групп сахара с ионами металла.
Флокуляция в органических жидкостях. Росс и Шеффер
[334] описали поведение различных тонкодиспергированных ча-
стиц в органических жидкостях, включая полярные твердые ве-
щества и жидкости. Чем более полярна поверхность частицы,
тем более полно она способна смачиваться какой-либо поляр-
ной жидкостью; при этом в большей степени протекает дефло-
куляция и становится более плотным осажденный продукт —
уменьшается его седиментационный объем. Существование на
поверхности гидрофобных частиц небольших гидрофильных об-
ластей или участков заметно влияет на поведение частиц в жид-
ких средах. Так, гидрофобное твердое диспергированное веще-
ство в неполярной жидкости, например в жидком углеводороде,
в отсутствие гидрофильных участков на поверхности твердого
вещества приводит к образованию плотно осажденного флоку-
542
Глава 4
лята. Но в том случае, когда на поверхности твердых частиц
имеется некоторая доля отдельных гидрофильных участков, что
вызывает формирование цепочек из таких частиц, флокулят ока-
зывается гораздо более объемистым. Если добавляется поверх-
ностно-активное вещество, то оно способно покрывать гидро-
фильные участки, превращая их в гидрофобные, и в таком слу-
чае седиментационный объем снова уменьшается.
Добавление воды к несмешивающимся неполярным жидко-
стям увеличивает седиментационный объем твердых частиц, со-
держащих полярные участки, поскольку вода способствует фор-
мированию мостиков. С другой стороны, если частично гидро-
фобные частицы диспергируются в воде, то седиментационный
объем увеличивается из-за связывания, возникающего между
гидрофобными участками на частицах. Этот объем понижается,
когда добавляется полярный растворитель, такой, как диоксан,
который сообщает поверхности способность смачиваться водой
за счет адсорбции полярного органического вещества на гидро-
фобных участках. Эти факторы важно рассматривать в тех
случаях, когда кремнезем используется в качестве загустителя.
В неполярных жидкостях или маслах, загущенных крем-
неземом, некоторые присадки вызывают пластикацию гелей или
консистентных смазок. Уайтман и Чессик [335] исследовали
действие полярных соединений и следов воды в такой системе.
Коацервация
Термин «коацервация» означает образование «жидкого
осадка» в результате взаимной коагуляции коллоидных систем.
Коацервация фактически включает коагуляцию, однако необ-
ходимо учитывать, что мостики между частицами или коллоид-
ными единичными образованиями оказываются до такой сте-
пени неустойчивыми, что система может существовать в дина-
мическом равновесии, т. е. связи формируются, распадаются
и вновь восстанавливаются. В этом случае сетка коагулята
может стягиваться до максимально возможной степени. Таким
образом, основная жидкая фаза исключается из сетки прокоагу-
лировавших частиц до тех пор, пока коагулят не становится вы-
сококонцентрированным, но все еще остается жидким, поскольку
мостики или связи между коллоидными частицами нестабильны
и могут непрерывно разрываться и заново формироваться. Клас-
сическим примером служит жидкая фаза, которая отделяется
при смешивании концентрированных растворов желатина и
аравийской камеди.
Айлер [8] описал большой ряд коацерватов, имеющих водо-
родные связи. Формирование таких связей происходит при дей-
ствии на подкисленные коллоидные растворы кремнезема или
Коллоидный кремнезем — концентрированные золи
543
поликремневых кислот некоторых полярных органических соеди-
нений, которые способны смешиваться с водой, но, однако, мо-
гут высаливаться из раствора, захватывая с собой кремнезем.
Одним из наиболее ярких примеров коацервата может слу-
жить связанный водородными связями комплекс, образуемый
простым диэтиловым эфиром диэтиленгликоля с поликремневой
кислотой [8]. В том случае, когда к этой смеси добавляется
соль в кислом растворе, от рассола отделяются два содержащих \
органические вещества слоя. Более тяжелая фаза содержит ;
около 40 % кремнезема, тогда как в верхнем слое находится J
только ~1,5 % SiO2 (см. рис. 4.20).
Коацервация, очевидно, происходит только тогда, когда по-
верхность частиц кремнезема покрывается адсорбированным
слоем органических молекул, в котором полярные группы, ориен-
тированные по направлению к силанольным группам поверхно-
сти и связанные с ними водородными связями, еще достаточно
подвижны и могут перегруппировываться до тех пор, пока раз-
мещающиеся снаружи части молекул не будут состоять в основ-
ном из гидрофобных углеводородных сегментов. В результате
частицы становятся в некоторой степени гидрофобными и отде-
ляются в виде подобной маслу коацерватной фазы. Айлер [336]
показал, что в случае поливинилового спирта (ПВС) и коллоид-
ного кремнезема коацерват образуется при pH 3—5, только
если в системе присутствует достаточное количество этого поли-
мера, чтобы как раз покрыть поверхность кремнезема. Этот слу-
чай подробно рассматривался в гл. 3. Ввиду описанного Флиром
критического действия электролита, оказываемого на флоку-
ляцию золя иодида серебра, когда в качестве флокулирующего
агента добавляется ПВС, подобные исследования, которые мо-
жно было бы провести с кремнеземом, представили бы значи-
тельный интерес. Флир и Ликлема [338] сообщили, что ПВС
сильно адсорбируется на кремнеземе, причем наиболее сильно
при pH 3, а при pH 8 происходит частичная десорбция. Это по-
казывает, что при адсорбции имеет место образование водород-
ных связей. Точно такие же наблюдения были выполнены
Тедросом [339], который распространил свои исследования на
смеси ПВС с катионным ПАВ—бромидом цетилтриметиламмо-
ния (БЦТМА). Когда последний адсорбировался на кремнеземе
при pH 9,1, на поверхности кремнезема формировался двойной
слой и адсорбция ПВС возрастала. Аналогично когда ПВС ад-
сорбировался при низком значении pH, то возрастала адсорб-
ция ионов ЦТА+. Вероятно, гидрофобные цетиловые группы и
углеводородный каркас полимера соединялись посредством во-
дородных связей. Если это так, то определенные соотношения
ПВС и ионов ЦТА+ должны приводить к образованию коацер-
вата в пределах широкой области pH. Аналогично адсорбция
544
Глава 4
пвс может оказаться более высокой на пирогенном кремнеземе,
как это наблюдалось .в случае полиэтиленоксида в исследова-
нии Рубио и Китченера [328].
Формирование сферического кремнезема
под действием коацервации
Если один из компонентов в системе кремнезем—органиче-
ский коацерват подвергается полимеризации как раз в тот мо-
мент, когда жидкие капельки коацервата выделяются из водной
фазы, то такие капельки затвердевают в виде сферических ча-
стиц небольшого размера. После выжигания органического ве-
щества получаются сферические частицы пористого кремнезема,
которые при соблюдении некоторых условий могут оказаться
очень однородными по размеру. Например, когда при pH 2 6 %-
ный раствор поликремневой кислоты, приготовленный добав-
лением раствора силиката натрия с отношением SiO2: Na2O
3,3 к избыточному количеству энергично перемешиваемой
кислоты, подвергается старению до оптимального состояния и
затем смешивается в соответствующих пропорциях с полимером
простого эфира (например, нонаэтиленгликоля) или хлоргидра-
том М,М-диэтиланилина, то вначале получается гомогенный ра-
створ. Затем в ходе дальнейшего процесса полимеризации крем-
незема при обычной температуре наблюдается самопроизвольное
осаждение сферических частиц органического комплекса с крем-
неземом [340—342].
Коацервация с участием коллоидного кремнезема может
также происходить, если первоначально растворенный поляр-
ный органический полимер подвергается дальнейшей полимери-
зации в растворе при низком значении pH, когда может наблю-
даться формирование водородных связей с кремнеземом. Айлер
и Мак-Квестон [343] отмечали образование коацервата в виде
жидких капелек микронного размера в том случае, когда моче-
вина и формальдегид полимеризовались в кислом золе крем-
незема. Органические олигомеры, очевидно, формируют водо-
родно-связанный коацерват, содержащий кремнезем или дру-
гие гидрофильные коллоидные частицы. При правильно подоб-
ранных условиях капельки твердеют сразу же, как только вы-
деляются из раствора. Это позволяет получать однородные по
размеру сферические частицы, которые могут иметь диаметр
в интервале 0,5—20 мкм. После выжигания органического по-
лимера остается порошок кремнезема в виде пористых одно-
родных по величине частиц сферической формы, пригодных для
использования в хроматографии.
Коллоидный кремнезем — концентрированные золи
545
Агрегация упорядоченных структур.
«Благородный опал»
В том случае, когда коллоидные частицы чрезвычайно одно-
родны по размеру и форме, они могут соединяться, образуя
очень однородный регулярно упакованный агломерат, точно так
же как молекулы располагаются в кристаллической решетке.
В действительности получается как бы «кристалл», составлен-
ный из коллоидных частиц. Однако из-за того, что размеры та-
кой «решетки» оказываются слишком большими, интерферен-
ционные законы Брэгга не могут быть приложимы к рент-
геновским лучам, но остаются справедливыми по отношению
к видимой области света, имеющей длины волн во много раз
больше, чем рентгеновское излучение. В результате получается,
что при рассмотрении в лучах отраженного света под опреде-
ленными углами упорядоченные агрегаты предстают заметно
окрашенными.
Структуры такого типа, образованные из очень однородных
частиц коллоидного кремнезема диаметром 100—500 нм, обна-
руживаются в природных условиях и называются «благород-
ным опалом». Из-за необычной природы этого явления следует
рассмотреть его несколько более подробно, а также аналогич-
ные явления, имеющие место для других веществ с однород-
ными частицами.
Структура опала
Термин «опал» в широком смысле охватывает многие типы
гидратированных аморфных кремнеземов, обнаруживаемых
в природе: от отложений внутри бамбука, называемых «таба-
шир» [344], микроскопических кремнеземных образований
внутри живых биологических тканей и до массивных минераль-
ных осадков вблизи горячих источников. Такой кремнезем амор-
фен в том смысле, что он не дает резкой картины дифракции
рентгеновских лучей, хотя для некоторых разновидностей было
показано, что кремнезем состоит из субмикроскопических кри-
сталлитов кристобалита с некоторым содержанием воды между
кристаллами. Однако «благородный опал», показывая блестя-
щие переливающиеся цвета, почти полностью аморфен и иден-
тифицируется как «опал А» [345]. Такой опал дает размытую
дифракционную полосу, соответствующую межплоскостному
расстоянию в решетке 4,1 А, и не проявляет эндотермического
эффекта на кривой дифференциального термического анализа
при 150°С, что характерно для некоторых других опаловых
кремнеземов [346]. «Благородный опал» добывается в несколь-
9 Заказ № 250
546
Глава 4
ких районах мира; раньше центром его добычи была Венгрия,
а в настоящее время — главным образом Австралия, хотя не-
большие его запасы обнаруживаются и в других местах.
Структура «благородного опала» впервые была описана Джо-
унсом, Сандерсом и Сегнитом [347] и Сандерсом [348], которые
наглядно показали, что он состоит из частиц аморфного крем-
незема сферической формы диаметром 150—350 нм, характери-
зующихся только одной размытой широкой полосой на рентге-
нограмме и отсутствием каких-либо определенных четких линий
или пиков. На электронно-микроскопическом снимке, получен-
ном Сандерсом [348], представлена удивительная структура
одинаковым образом упакованных агломератов, состоящих из
кремнеземных сферических частиц, причем в промежутках
между сферами находилось вещество с несколько более низ-
ким значением показателя преломления, что и приводило к воз-
растанию интенсивности интерференционных цветовых оттенков
в отраженном свете. Автор показал, что в том случае, когда
размер отдельных сфер отличался от единого размера и, сле-
довательно, нарушалась регулярность упаковки, получающиеся
при этом свободные места рассеивали свет, давая молочное
окрашивание, характерное для опалов более низкого качества.
Когда не видно вообще никакой окраски, опал называется «от-
беленным». Сегнит, Стивенс и Джоунс [349] исследовали роль
воды в опале, применяя методы дифференциального термиче-
ского анализа, ИК-спектроскопии и ЯМР. Большинство опалов
содержит 4—9 % воды, но в «благородном опале» при понижен-
ной влажности теряется менее 1 % воды. От 20 до 70 % подоб-
ной воды удерживается «химически», т. е. представляет собой
гидроксильные группы, связанные с расположенными на внут-
ренней поверхности опала атомами кремния, а оставшаяся часть
воды находится в настолько малых по размеру или закрытых
порах, что даже при повышенных температурах она удаляется
очень медленно.
В генезисе «благородного опала» участвуют три различных
процесса: формирование однородных частиц, отложение их ре-
гулярным образом и связывание таких частиц вместе. Как бу-
дет показано ниже, агрегация, приводящая к регулярным, упо-
рядоченным множествам, может являться естественным следст-
вием чрезвычайной однородности самих сферических частиц.
Механизм формирования в природных условиях однородных
сферических образований размером 0,1 —1,0 мкм пока неизве-
стен. Джоунс с соавторами [350, 351] показали, что некоторые
минералы, такие, например, как биотит, разрушаются под дей-
ствием кислоты, и образовавшийся растворимый кремнезем
претерпевает непрерывное переосаждение с последующим фор-
мированием коллоидных частиц, которые агрегируют всфериче-
Коллоидный кремнезем — концентрированные золи
547
ские частицы большего размера. Последние могут транспортиро-
ваться в виде частиц золя к месту их отложения. Отмечалось,
что в среде, состоящей из отложений опала, обнаруживаются
большие содержания сульфатов. Сферические частицы могут
также формироваться при термическом растворении и осажде-
нии кремнезема.
Даррах и др. [352] указывают на три важных фактора, не-
обходимых для образования «благородного опала» по крайней
мере в случае австралийских месторождений: а) обильный за-
пас легко растворимого кремнезема; б) засушливый климат и
зона, ограниченная приповерхностными грунтовыми водами
с четко выраженными задерживающими слоями, например та-
кими, как бентонитовые пласты, предохраняющие подобное фор-
мирование от высыхания благодаря сохранению в этой зоне ти-
пичного раствора, содержащего вплоть до 3 % растворимых
сульфатов и хлоридов и 0,008 масс.% растворимого кремнезема;
в) присутствие полостей, сформированных различными путями,
в которых частицы кремнезема способны собираться и упоря-
дочивать свое расположение. Авторы утверждают, что золь крем-
незема, состоящий из частиц размером 10 нм, будет образовы-
вать частицы диаметром 150—200 нм в процессе его продолжи-
тельного старения, но необходимые для этого условия не были
определены. Условия для подобного процесса агрегации должны
быть очень специфическими, поскольку золи с частицами разме-
ром 10 нм могут подвергаться старению и гелеобразованию,
а также концентрированию и флокуляции при изменяющихся
в широкой области условиях, без появления каких-либо боль-
ших сферических образований. Существуют некоторые данные,
свидетельствующие о том, что сферы могут осаждаться в слабо-
вязкой среде, подобной 0,1 %-ному золю кремнезема, состоящему
из небольших частиц, которые способны флокулировать при
действии солей.
Вначале полученная структура опала оказывалась пластич-
ной, но в дальнейшем другие порции кремнезема проникали
в поры. Чтобы окрашивание было наибольшим, промежутки
между сферическими частицами не должны быть полностью за-
полнены, в них должна сохраняться только прозрачная масса.
Вместо связывающего, цементирующего действия в данном слу-
чае, вероятно, происходит вовлечение отложений из более рас-
творимого кремнезема в форме геля, состоящего из чрезвы-
чайно небольших частичек SiO2, которые способны удерживать
воду внутри пор.
Коул и Монроу [353] исследовали ленточную структуру
опала, называемую двойниковой. Они обнаружили, что имеются
полосы с гексагональной упаковкой частиц, которые распола-
гаются между полосами с тетрагональной упаковкой, причем
9*
548
Глава 4
подобные полосы, по данным электронной микроскопии, имеют
ширину всего в несколько сферических частиц.
Повышенное содержание воды при низкой пористости, как
показывают данные, полученные по адсорбции азота, наводит
на мысль, что сферические частицы размером 200 нм в свою
очередь составляются из плотно упакованных меньших по раз-
меру частиц диаметром 10—20 нм, поверхность которых по-
крыта слоем групп SiOH. Еще меньшие промежутки между та-
кими малыми частицами должны составлять только 10—20 А
в диаметре, так что вода внутри промежутков должна удержи-
ваться прочно. Подобная структура, предложенная Джоунсом
и Сегнитом [354], была подтверждена электронно-микроскопи-
ческими снимками, кривыми дегидратации образцов и исследо-
ваниями методом ИК-спектроскопии. Грир [355] пришел к за-
ключению, что сферические частицы присоединяются к струк-
туре опала посредством механизма винтовой дислокации в про-
цессе роста кристалла.
Дифракция света на «благородном опале» приводила к по-
явлению дифракционных картин, которые интерпретировались
по аналогии с теорией дифракции рентгеновских лучей. Сан-
дерс [356] обнаружил, что сферические частицы кремнезема
были упакованы в слоях гексагонально, а слои обычно распола-
гались произвольным образом. Имеются некоторые параллель-
ные области упорядоченных, обычно гранецентрированных, ку-
бических упаковок.
Опал, в котором сверкающие цвета появляются на темном
фоне, представляется наиболее ценным и, по-видимому, наиме-
нее изученным. Эффект вызывается не только тем, что темной
является подложка, но, кроме того, может иметь место феномен
световой ловушки, подобно пучку иголочек, просматриваемому
с определенных позиций. Без сомнения, также существуют и
другие эффекты, как, например, рассеяние света темным веще-
ством через матрицу. В качестве примера могут служить чер-
ные опалы [357]. Обнаруженное в порах темное органическое
вещество напоминало какой-то углеводород. После прокалива-
ния образца плотность обесцвеченного опала повышалась до
2,0—2,2 г/см3, так как органические вещества и вода были уда-
лены. Сандерс и Даррах [358] в дальнейшем подробно описали
микроструктуру опала. На представленном электронно-микро-
скопическом снимке (рис. 4.23) видны одинаковые сферы в об-
ластях однородных множеств [359]. На этом снимке австралий-
ского опала можно различить необычную структуру сфериче-
ских образований, составленных из еще меньших по размеру
частиц в виде располагающихся вокруг центрального ядра
слоев. В других опалах в частицах различается большое число
сглаженных, концентрически расположенных колец. По суще-
Коллоидный кремнезем — концентрированные золи
549
Рис. 4.23. Электронно-микроскопический снимок структуры опала, слегка
протравленного кислотой HF.
Ископаемый образец получен из месторождения Купер Педи, Австралия (Х19 500). Печа-
тается по разрешению доктора Дж. В. Сандерса [359].
ству все такие кольца вырастают вместе в одну относительно
плотную сферу и обнаруживаются при осторожном травлении
образца. Удивительно, что наблюдаемые изломы часто проходят
через центры слоев таких сферических образований, а не через
зону контакта между частицами. Для многих образцов разлом
на поверхности частицы кажется гладким, и просматривается
только серия пятнышек, объясняемых присутствием полостей
между первоначальными сферами, которые росли почти одно-
временно.
Сферические частицы упакованы в виде однородных мно-
жеств в различных меняющихся по размерам зонах, между ко-
550
Глава 4
торыми произвольно образуются разрывы. Такие разрывы могут
быть продольными, выделяя зоны в виде длинных ленточек, так
что при рассмотрении образца под оптическим микроскопом
видны тончайшие параллельные спрямленные линии с характер-
ной однородной окраской. Привлекательность опала зависит от
размеров и разнообразия цветовой гаммы областей, определяе-
мых в свою очередь размерами и распределением образовав-
шихся разрывов. Чистота цвета и отсутствие бликов от рассеян-
ного белого света зависят от наличия наиболее благоприятных
небольших различий между средними значениями показателей
преломления плоскостей, одна из которых проходит через
центры сферических частиц, а другая — через полости между
сферами. Если такое различие слишком мало, то опал оказы-
вается почти прозрачным с минимальным интерференционным
окрашиванием; если же различие слишком велико, то будет
рассеиваться только белый свет. Как было описано Айлером
[360], подобные эффекты наиболее легко различимы, когда
пористая белая масса синтетического опала пропитываемся
жидкостями с различными показателями преломления.
Сандерс отметил, что размер сферических частиц можно
быстро оценивать визуально. При рассматривании опала под
различными углами наблюдается цвет с наибольшей длиной
волны (Zmax) (при изменении цвета от фиолетового до красного
длина волны возрастает от 300 до 700 нм). Тогда диаметр сфе-
рической частицы получается делением значения длины волны
на 2,5. Следовательно, размеры частиц в благородном опале
могут изменяться приблизительно от 100 до 300 нм. Для одного
какого-либо выбранного образца такой размер частиц, как пра-
вило, однороден. Все цвета видимого спектра наблюдаются
только в том случае, когда размер сферических частиц состав-
ляет около 300 нм. При еще большем диаметре сферических об-
разований можно различать окраску «второго порядка», напри-
мер светло-вишневый цвет, получаемый смешением фиолетового
и красного цветов.
Структура оболочки сферических частиц, по-видимому, ха-
рактерна для некоторых типов опалов. Так, размер частицы, на-
ходящейся внутри концентрических слоев—оболочек, которых
может быть до пяти, в некоторых опалах составляет 50 нм. В та-
ких случаях одиночные частицы диаметром 50 нм часто обнару-
живаются и в полостях, расположенных между большими сфе-
рами. Для других опалов поперечное сечение сферических об-
разований имеет вид концентрических колец, подобно кольцам
в сечении ствола дерева, образованных, как кажется, из боль-
шого числа тончайших частичек. Сандерс и Даррах установили,
что в лабораторных условиях из разбавленных золей с части-
цами диаметром 50 нм в процессе медленной агрегации фор-
Коллоидный кремнезем — концентрированные золи
551
мируются частицы размером 150 нм. Вероятно, много меньшие
по размеру частицы могут также осаждаться вокруг зародышей.
Это, очевидно, происходит в пустотах грунтов или горных пород,
часто образующихся в результате разложения органического
вещества, а затем при осаждении или при концентрации крем-
незема в процессе испарения воды получаются частицы SiOz.
Таким образом, как уже обсуждалось, в подобных случаях дол-
жны, несомненно, вступать в игру явления разделения или упо-
рядочивания фаз. В лабораторных условиях рост таких «кри-
сталлов» в концентрированных золях идет в радиальном
направлении по отношению к наружной поверхности плотного,
состоящего из осажденных частиц слоя.
Результаты исследований, выполненных Балакиревым и др.
[361], в основном оказались в соответствии с выводами, достиг-
нутыми другими учеными.
Общие сведения о благородном опале имеются в нескольких
монографиях [362—364].
Другие агрегаты с упорядоченной структурой
Самопроизвольное размещение однородных коллоидных ча-
стиц в плотно упакованные, в высокой степени упорядоченные
агломераты представляет собой по существу вид кристаллиза-
ции, в которой кристаллы составляются не из молекул, а прямо
из частиц. Олфри и др. [365] описали кристаллы, состоящие из
латексных частиц. Частицы в таких различавшихся между со-
бой кристаллах имели размеры в области 100—1000 нм. Однако
в одном каком-либо выбранном кристалле частицы были очень
однородны по размеру. Подобные упорядоченные расположения
частиц были отмечены в ряде других систем, особенно имеющих
частицы размером 100—300 нм, поскольку в отраженном свете
становилось особенно наглядным интерференционное окрашива-
ние. Только в последние годы было признано, что структура
«благородного опала» имеет ту же природу. Поскольку способ
формирования больших частиц коллоидного кремнезема в при-
родных условиях и их отложения упорядоченным образом еще
неполностью выяснен, то представляет определенный интерес
суммировать известные данные, касающиеся образования одно-
родных субмикронных частиц, сил, вызывающих построение упо-
рядоченных агрегатов, а также некоторые примеры получения
подобных структур в лабораторных и в природных условиях.
Общие вопросы «кристаллизации» или высокоупорядоченной
агрегации супермолекулярных структурных единиц были рас-
смотрены Лаком, Клиром и Весслау [366], которые описали
наблюдения по вирусным кристаллам и плотно упакованным
552
Глава 4
латексным частицам, а также развили теорию интерференцион-
ного окрашивания таких систем в отраженном свете. Дефонтане,
Джексон и Миллер [367] связали оптическую дифракцию с упа-
ковкой наблюдаемых под микроскопом сферических частиц раз-
мером 2,68 мкм при использовании лазерного излучения с дли-
ной волны 632,8 нм. Теория упаковки сферических частиц
в упорядоченные множества была рассмотрена в работах
[368—371].
Вслед за Олфри и др. [365] возникновение интерференцион-
ных цветовых оттенков в полистироловом латексе изучалось ря-
дом ученых. Некоторые авторы [366—372] объясняли образо-
вание цветовых оттенков отражением видимого света по Брэггу.
Образование упорядоченных множеств в присутствии разупо-
рядоченных областей изучалось Кригером, Хилтнером и Папи-
ром [373]. Авторы обнаружили, что дальнодействующие силы
отталкивания, обусловленные имеющимися на поверхности ла-
тексных частиц анионными зарядами, удерживали частицы по-
рознь до тех пор, пока не добавлялось некоторое количество
электролита, позволявшее частицам сближаться, вследствие чего
происходил переход порядок—беспорядок.
Дальнодействующее взаимодействие диспергированных одно-
родных по размеру частиц может приводить к образованию пе-
риодической коллоидной структуры (ПКС), в которой частицы
располагаются достаточно далеко и не касаются друг друга.
К тому же избыток частиц в системе может приводить к такому
их расположению, когда наблюдается некоторое отталкивание
частиц [374]. Теория сдвиговых суммарных напряжений в неко-
торых избыточных системах согласуется с экспериментальными
данными для полиакрилонитриловой дисперсной системы.
В серии статей [375—379] был представлен обзор литера-
туры и результатов детальных исследований по данному во-
просу. Частицы могут образовывать упорядоченные множества,
в которых фактически отсутствует контакт частиц между собой.
В таких множествах все частицы имеют один и тот же размер,
а все другие размеры исключаются. Это явление предусматри-
вает фазовый переход, заключающийся в том, что концентра-
ция одной из фаз превышает некоторую объемную долю, обычно
равную 0,5±0,1, благодаря чему формируется вторая, более
концентрированная фаза, внутри которой частицы расположены
упорядоченно. Такое явление известно как переход Кирквуда—
Алдера [380—382] и рассматривается как чисто статистический
эффект, не требующий для объяснения введения потенциала при-
тяжения. Переход оказывается запрещенным, когда ионные
силы отталкивания превышают некоторый низкий уровень. Та-
кой переход может иметь место для суспензий в водных и не-
водных жидких средах. В водных системах переход будет за-
Коллоидный кремнезем — концентрированные золи
553
прещен даже при наличии очень однородных по размеру частиц,
если система имеет высокое содержание электролитов.
Большое разнообразие переливающихся радужных окрасок
в биологических материалах и в неорганических кристаллах счи-
талось следствием подобных дифракционных, интерференцион-
ных явлений в единообразно упорядоченных структурах. Авторы
работы [383] упоминают такие системы, как опал, некоторые
полевые шпаты, кристаллы хлорида калия, раковины моллюсков,
раковины ископаемых плеченогих, кристаллизованные вирусы,
насекомые и, по их собственным данным, кожа мешотчатого
полоза. Гринвальт [384] показал, что раскраска крыльев ко-
либри является следствием упорядоченного множества пузырь-
ков. Уильямс и Смит [385] изучили вирус, встречающийся
в виде ярко переливающихся окрашенных кристаллов, содер-
жащих 25 % личинок долгоножки. Этот вирус был впервые опи-
сан Ксеросом [386] и, согласно данным Боудена и Смита [387],
выглядит в точности, как «благородный опал». Переливаю-
щийся красками опал обнаруживается в нижних частях зубных
пластинок морских брюхоногих моллюсков (блюдечек), описан-
ных Ловенштамом [388а]. Сферические гранулы диаметром
10—25 мкм найдены в коже голотурии (Molpadia intermedia,
Holothuroidea), причем каждая гранула составлена из сфериче-
ских частиц кремнезема диаметром 100—190 нм, смешанных
с частицами такого же размера, но состоящих из «ферритина»,
т. е. кальциймагнийжелезо(Ш)фосфата. Это соответствует крем-
неземным частицам, имеющим тот же самый размер и обнару-
женным в «благородном опале» [3886].
Образование однородных по размерам
неорганических частиц
Хотя однородные частицы определенных типов латексов и
вирусов были известны еще до 1950 г., примеров неорга-
нических систем с однородными частицами в области разме-
ров 100—500 нм, необходимой, чтобы продемонстрировать ин-
терференционное окрашивание, было мало, если они вообще
имелись. Изодисперсные золи приготовляли из золота, серебра,
серы, хлорида серебра и сульфата бария, но не из кремнезема
[389]. В более ранних исследованиях попытки приготовить ча-
стицы кремнезема в области указанных размеров были безус-
пешны, поскольку не было известно, каким образом повысить
размер частиц. Фрейндлих [390] пробовал получить стабильные
золи с концентрацией свыше 10 % БЮг, однако был введен в за-
блуждение тем, что добавление щелочи, которая, как он знал,
должна была стабилизировать отрицательно заряженные ча-
стицы, приводило лишь к гелеобразованию.
554
Глава 4
Частицы кремнезема, достаточно большие и однородные по
размеру, способные образовывать единообразные множества,
проявляющие ярко сверкающие интерференционные цветовые
оттенки, были впервые приготовлены Сирсом. Он использовал
процесс «наращивания», предложенный Бечтольдом и Снайде-
ром, и наблюдал окрашивание в золе, который осаждался перед
этим в течение двух лет [360]. Позже Штобер, Финк и Бон [68]
разработали процесс, в котором для приготовления очень одно-
родных сферических частиц диаметром 0,05—2 мкм использо-
вался этилсиликат. Частицы такого типа могут быть осаждены
тем же самым способом для получения окрашенных слоев.
Матиевич и др. [391] описали приготовление однородных ча-
стиц из других материалов, но в той же самой области разме-
ров для того, чтобы также получать интерференционное окра-
шивание при осаждении. Авторы разработали методы для при-
готовления однородных по размеру сферических частиц водных
оксидов Сг3+, Al3+, Zr4+ и Сп2+. К тому же Кейтон и Матиевич
[392] приготовили однородные сферические частицы гидрати-
рованного оксида алюминия в той же области размеров. Подоб-
ные частицы основного сульфата хрома были описаны Беллом
и Матиевичем [393]. Уотсон, Карделл и Геллер [394] под-
робно исследовали известные в течение многих лет «слои Шил-
лера» тектоидов p-FeOOH, показывающие блестящее расцве-
чивание. Айлер приготовил сферические частицы СгРО4-хН2О
сине-зеленого цвета, которые осаждались с образованием слоя
однородных частиц, проявлявших красный цвет в отраженных
лучах света. Нет никакого сомнения, что будут обнаружены и
другие типы неорганических частиц.
Синтез опала
Формирование структур, подобных ярко расцвеченному, свер-
кающему красками опалу, путем осаждения золей наблюдалось
в 1953 г. Сирсом и Айлером, последующие исследования были
описаны в 1965 г. [360]. Твердые массы, напоминающие по
внешнему виду «благородный опал», были получены в процессе
медленного высушивания исходного водного осажденного слоя
и пропитывания пористой массы органическим веществом, имев-
шим показатель преломления, близкий к показателю для аморф-
ного кремнезема. Был получен патент [395] на синтетический
опалоподобный материал, стабилизированный пропиткой высу-
шенного пористого твердого вещества золем кремнезема с очень
маленькими по размерам частицами или же способным к поли-
меризации органическим веществом, таким, чтобы показатель
преломления полученного сцементированного вещества откло-
Коллоидный кремнезем. — концентрированные золи
555
нялся от показателя преломления кремнезема в пределах
±0,01—0,05.
О синтезе опала было заявлено в 1967 г. [396]. С этого
времени синтетические опалы стали доступными благодаря
Гилему из Швейцарии [397]. Они имели внешний вид и многие
свойства природного опала, например степень гидратирования,
преломления света, аморфный характер. Плотность их была
равна 2,113 г/см3, показатель преломления 1,45, твердость 6,15
по шкале Мооса [398]. Сообщалось, что синтетический черный
опал неотличим от соответствующего природного опала по ви-
зуальной оценке, некоторые различия наблюдались только
в флуоресценции. Метод приготовления синтетического опала
является секретом производящей этот материал фирмы «Гилем».
Джон Слокум [399, 400] совсем недавно разработал другой
материал, известный под названием «Slocum Stone». Были при-
готовлены образцы с яркой интерференционной окраской все-
возможных оттенков, имеющих более широкую область фоновой
окраски, чем любой природный опал. Могут быть воспроизве-
дены все типы опалов. Однако, несмотря на все сходство, полу-
ченный материал не может называться «синтетическим опа-
лом», поскольку имеет уникальный, характерный только для
него внешний вид, и его состав отличается от состава опала.
Кроме того, он негидратирован, непорист, более тверд, имеет
более высокую плотность (2,4—2,5 г/см3) и более высокий пока-
затель преломления (1,51). Цвета, по-видимому, возникают от
интерференционных пленок, диспергированных по всей матрице.
Метод производства этого материала не был раскрыт.
Уилкокс [401] запатентовал состав, который, как утвер-
ждается, имеет внешний вид опала, но представляет собой
в основном органическое вещество. Автор проводил гомоген-
ное заполнение всей массы бесцветной прозрачной органической
полимерной матрицы диспергированными частицами TiO2 диа-
метром 0,05—0,1 мкм. Объемная доля TiO2 составляла от
10-4 до 10%.
Адсорбция частиц кремнезема на поверхностях
Коллоидные частицы способны притягиваться и удержи-
ваться на плоских поверхностях под действием тех же самых
-сил, которые вызывают притяжение или отталкивание между
двумя коллоидными частицами. Некоторые основные положения
такой адсорбции были рассмотрены в обзоре Джиргенсонса и
Страуманиса (см. [53], с. 101). Айлер [402] показал возмож-
ность адсорбции коллоидного кремнезема на оксиде алюминия)
и коллоидного оксида алюминия на кремнеземе примерно npi^
pH 4. Как только поверхность, несущая ионные заряды, покры-)
556
Глава 4
вается монослоем противоположно заряженных коллоидных
частиц, знак ее заряда меняется, и так как заполненная поверх-
ность уже несет на себе заряды, принадлежащие адсорбиро-
ванным частицам, то никакой дальнейшей адсорбции не будет
происходить.
Можно наносить на поверхность много отдельных слоев кол-
лоидных частиц, один слой за другим. Когда очищенная гидро-
фильная кремнеземная поверхность, такая, например, как по-
верхность кварцевого стекла, несущая отрицательные заряды,
приводится в контакт с золем коллоидного оксида алюминия,
то на- поверхности стекла адсорбируется один слой частиц
оксида алюминия. При этом результирующий заряд на поверх-
ности оказывается противоположным исходному, и, следова-
тельно, оксид алюминия адсорбироваться далее не будет. Когда
избыточное количество оксида алюминия вымывается, на по-
верхности остается только один адсорбированный монослой
частиц оксида алюминия. Слой частиц кремнезема после этого
может быть отложен на поверхности, покрытой оксидом алю-
миния, если использовать раствор, содержащий коллоидный
кремнезем. Таким образом, адсорбируется один слой частиц
кремнезема, а избыточный кремнезем можно удалить вымы-
ванием.
Повторяя этот процесс, можно нанести на поверхность одно-
родный полислой любой желаемой толщины [403]. Адсорби-
руемый оксид алюминия может находиться в растворе основ-
ного хлорида алюминия, в котором частицы настолько малы,
что их размер трудно определить. Другим вариантом может
быть адсорбция катионного полимера с целью образования
положительно заряженного слоя на поверхности кремнезема,
на котором уже затем осаждается слой кремнеземных частиц.
Этим способом можно формировать пленки толщиной, равной
одной четверти длины волны видимого света, т. е. ~150 нм.
Поскольку пленки оказываются пористыми, то их показатель
преломления приблизительно равен средней величине между
показателем преломления воздуха и показателем преломления
подложки из кварцевого стекла, что обеспечивает превосход-
ные условия для развития интерференционного окрашивания.
Подробное описание этого способа и его прикладное значение
приводятся в патенте [404].
Такие адсорбированные пленки оказываются очень пластич-
ными и легко стирающимися, однако путем обработки гидро-
лизованными этилсиликатом или тетраизопропилтитанатом
удается сделать их твердыми. Такие пленки можно также
упрочнить путем спекания при повышенной температуре.
Для практического их использования особенно интересны
низкая отражательная способность подобных покрытий, обра-
Коллоидный кремнезем — концентрированные золи
557
зование изолирующих слоев на проводящих поверхностях и
проводящих слоев на непроводящих подложках, формирование
оксидных пленок на металлах, отложение коллоидного графита
с целью улучшения электропроводности поверхности или отло-
жение на поверхности твердых смазок. Чередующиеся прово-
дящие и непроводящие электрический ток слои могли бы быть
нанесены на подложку для их использования в электронных
печатных схемах.
Указанный способ оказывается полезным для качественного
обнаружения небольших количеств коллоидных веществ в ра-
створе. Основным моментом в этом способе является использо-
вание индикатора — золя, имеющего частицы с диаметром, рав-
ным примерно одной четверти длины световой волны, что дает
возможность легко их наблюдать при адсорбции монослоя этих
частиц на черной стеклянной подложке. Так, например, орга-
нические вещества катионного типа при низких концентрациях
можно обнаруживать посредством обработки поверхности чер-
ного стекла испытуемыми растворами. Поверхность промы-
вается или высушивается, затем на нее осаждается золь крем-
незема с размером кремнеземных частиц 100—200 нм, исполь-
зуемый в качестве индикатора; избыток золя вымывается,
и, наконец, пластинка повторно высушивается. Частицы крем-
незема прилипают во всех тех местах стеклянной подложки,
где было адсорбировано катионное вещество из испытуемого
раствора. Полученная кремнеземная пленка может в таком
случае легко просматриваться по интерференционному окраши-
ванию.
Как показано на рис. 4.24, этим способом можно также
детектировать небольшие по размеру частицы коллоидного
кремнезема в разбавленных растворах. Вначале на стеклянной
поверхности из раствора осаждается положительно заряженный
слой, например коллоидных частиц оксида алюминия или ка-
тионного полимера, затем избыток вещества смывается с по-
верхности и, наконец, испытуемый раствор, содержащий отри-
цательно заряженные коллоидные частицы, например кремне-
зема, приводится в контакт с частью стеклянной поверхности,
после чего стеклянная подложка высушивается. Присутствие
малых частиц кремнезема на поверхности стекла можно обна-
ружить посредством последующей обработки всей стеклянной
поверхности частицами кремнезема размером 100—200 нм,
которые способны прилипнуть только к положительно заряжен-
ной поверхности, если на ней не были адсорбированы какие-
либо отрицательно заряженные коллоидные частицы. Подобные
тестовые методики можно разработать для детектирования от-
рицательно или положительно заряженных частиц при содер-
жании коллоидных веществ в растворе менее чем 0,01 масс. %.
558
Глава 4
При этом оказывается, что чем меньше по размеру частицы,
тем более чувствительная методика. Однако в испытуемом
Рис. 4.24. Схемы процессов адсорбции и диффузии, лежащих в основе методов
обнаружения небольших коллоидных частиц кремнезема.
Слева-, большие по размеру частицы кремнезема А адсорбируются на покрытой оксидом
алюминия поверхности с образованием видимой пленки В, но не адсорбируются на участ-
ках. предварительно подвергавшихся действию небольших частиц кремнезема С, являю-
щихся невидимыми. Справа-, в золе, состоящем из смеси больших и малых частиц кол-
лоидного кремнезема, частицы меньшего размера диффундируют более быстро по на-
правлению к покрытой оксидом алюминия поверхности О и не допускают медленнее Дви-
гающиеся большие частицы Е в область D. (По данным Айлера [403].)
растворе, содержащем большие по размеру частицы, не должны
находиться маленькие частицы, что также изображено на
рис. 4.24.
О приблизительном размере частиц в коллоидной системе
можно судить по различиям в окраске, возникающим благодаря
Рис. 4.25. Схемы слоев кремнезема и оксида алюминия, адсорбированных на
поверхности черного стекла (G).
Слой, состоящий из небольших частиц кремнезема, и второй слой оксида алюминия, по-
казанный на рисунке в виде коротких черных черточек, в системе А повышают суммар-
ную толщину пленки до h по сравнению сив контрольной системе В. (По данным Ай-
лсра [403].)
возрастанию суммарной толщины полислоя после того, как
частицы изучаемого образца были осаждены на стеклянной
поверхности. Подробности этого метода (рис. 4.25) были опи-
саны Айлером [403].
При проведении такого рода работ из системы должны быть
исключены следы адсорбированных жиров или же других гид-
Коллоидный кремнезем — концентрированные золи 559
рофобных молекул. Вейл и Марбо [405] отметили влияние
адсорбированных катионов на адсорбцию следов жирных
кислот. Айлер наблюдал, что стеклянная подложка, обрабаты-
ваемая кислотой с целью удаления катионов, остается гидро-
фильной в атмосфере лабораторного помещения гораздо более
продолжительное время, чем после ее выдерживания в кон-
такте с многозарядными катионами или с положительно заря-
женными коллоидными частицами, которые способны адсорби-
ровать жирные кислоты из воздушной среды в течение несколь-
ких часов. Для получения более толстых отложений коллоидного
кремнезема может быть использовано наблюдение, что на
стеклянной подложке могут быть адсорбированы состоящие из
частиц агрегаты, правда ограниченного размера (диаметром
менее 1 мкм), если они скоагулированы и частично редиспер-
гированы под действием незначительного избытка катионного
ПАВ или катионного полимера. В таком случае агрегаты несут
положительный заряд и способны адсорбироваться вплоть до
формирования одного слоя, состоящего из этих агрегатов.
Адгезия монослоя высушенных кремнеземных частиц и
стеклянной поверхности может быть достигнута посредством
полирования поверхности пудрой, состоящей из дискретных,
однородных по размеру частиц гидрофильного коллоидного
кремнезема диаметром 100—300 нм [406]. Очевидно, что
частицы связываются с ровной поверхностью в большей сте-
пени, чем между собой, и, таким образом, к поверхности при-
липает один слой частиц. В некоторых случаях при тщательно
выполненном шлифовании поверхности и контролируемых усло-
вий может сформироваться второй слой частиц.
Измерение адгезии частиц к поверхностям было описано
Виссером [407], который использовал известную силу сдвига,
возникающую в воде вследствие перемещения частиц относи-
тельно поверхности. Теория адгезии или отделения частиц на
поверхности от потока золя была развита Гутовски [408].
Дэвис и Ренейд [409] непосредственно измеряли адгезию
частиц на различных поверхностях методом центрифугирования.
Золи кремнезема, содержащие частицы
с модифицированными поверхностями
Сразу же как только частицы кремнезема сформировались,
их поверхность может оказаться модифицированной в резуль-
тате присоединения к ней различных атомов или групп, спо-
собных изменить физическое и химическое поведение частиц.
Так, например, если поверхность частицы кремнезема пол-
ностью покрывается одним слоем оксида алюминия, даже тол-
щиной в одну или две молекулы, то частица ведет себя так,
560
Глава 4
как если бы вся она представляла собой твердый оксид алюми-
ния, т. е. несет положительный заряд и является устойчивой
при низких значениях pH.
Хемосорбция монослоя углеводородных групп обусловли-
вает поведение частицы кремнезема как одной большой угле-
водородной молекулы.
Отрицательно заряженные поверхности
Алюмосиликатные ионы
Одну из важных модификаций представляет собой поверх-
ность кремнезема, получаемая при его взаимодействии с алю-
минат-ионами. Существует очень сильное специфическое взаи-
модействие между оксидами алюминия и кремния, что доказы-
вается чрезвычайно низкой растворимостью алюмосиликатных
минералов, таких, например, как глиноземы. Особая взаимо-
связь между алюминием и кремнием объясняется, вероятно,
тем, что для обоих атомов координационное число по отноше-
нию к атому кислорода может при подходящих обстоятельствах
иметь значение 4 или 6, а также потому, что и А1, и Si имеют
приблизительно один и тот же атомный диаметр. Поскольку
алюминат-ион А1(ОН)Г* геометрически подобен Si(OH)4, то
он может быть введен на поверхность SiO2 или может вступать
на ней в обмен, образуя, таким образом, алюмосиликатные
участки, имеющие фиксированные отрицательные заряды.
Анионный характер алюминия в кристаллических минера-
лах, когда он замещает кремний в четырехкоординированном
состоянии, был давно признан Полингом [410] и вскоре после
этого в алюмосиликатных катализаторах крекинга [411]. Обра-
зование алюмосиликатного аниона посредством реакции алю-
минат-иона с поверхностью кремнезема представляется следую-
щей схемой [412] :
Милликен, Миле и Облад [413] показали, что такой анион
устойчив только в присутствии катиона, но не иона водорода,
Коллоидный кремнезем — концентрированные золи 561
так как свободная кислота нестабильна, и что должен присут-
ствовать избыток кремнезема. Значение pH также является
важным фактором. Так, только около 15 % оксида алюминия
в кремнеземе было стабилизировано в адсорбированной форме
при pH 6, когда в качестве стабилизирующего катиона брали
ион аммония. Ниже pH 3, когда существует водородная форма,
атом алюминия возвращается в шестикоординированное со-
стояние.
Такое поведение алюминия используется при модифициро-
вании поверхности коллоидного кремнезема, поэтому частицы
кремнезема будут оставаться отрицательно заряженными
вплоть до pH 3 в противоположность очень чистому кремнезему,
-который отрицательно заряжен в результате адсорбции гидро-
ксил-ионов выше pH 7, но теряет заряд в кислом растворе.
Александер и Айлер [414], таким образом, получили модифи-
цированный золь кремнезема, который оказался устойчивым
в нейтральной области pH, т. е. в тех условиях, когда немоди-
фицированный золь кремнезема быстро превращается в гель.
Для модифицирования этим способом золя кремнезема, содер-
жащего частицы размером 15 нм, по реакции с алюминатом
натрия требовалось всего только 0,66 масс. % А12О3, нанесен-
ного на кремнезем. Это соответствует только одному алюмо-
силикатному центру на каждые 20 силанольных групп на
поверхности, но поскольку такие центры, вероятно, распреде-
лены равномерно, то они располагаются друг от друга на рас-
стоянии всего 15 А. Различие между этим типом модифициро-
ванного золя кремнезема и немодифицированным золем, содер-
жащим только небольшие количества алюминия, обычно
присутствующие в коммерческих продуктах, было изучено
Алленом и Матиевичем [250, 251, 415]. Повышенная устойчи-
вость в пределах более широкой области pH расширяет
масштаб практического применения подобных систем [416,
417].
Коллоидные частицы кремнезема могут также покрываться
пленками толщиной 3—25 нм, в которых, согласно данным
Александера [418], атомное отношение кремния к алюминию
может меняться в интервале от 1:1 до 10:1. Можно также
приготовлять золи с частицами, которые целиком состоят из
алюмосиликата [419], но подобные системы выходят за рамки
настоящего обсуждения.
Влияние на свойства. Воздействие, оказываемое вводимыми
на кремнеземную поверхность алюмосиликатными ионами, про-
является при сравнении свойств выше рассмотренного модифи-
цированного алюминатом золя людокс-АМ (зарегистрирован-
ная марка фирмы «Дюпон де Немур», г. Уилмингтон, штат
Делавэр, США) и немодифицированного золя кремнезема
10 Заказ № 250
562
Глава 4
людокс-HS, из которогой первый был приготовлен. Матиевич
и соавторы показали, что в области pH 4—6, где частицы
кремнезема несут на поверхности очень небольшой заряд,
модифицированные частицы, по данным электрофореза, сохра-
няли значительный по величине заряд. К тому же многозаряд-
ный катион La3+ вызывает коагу-
ляцию только модифицированного
золя.
Айлер [324] приготовил серию
золей, модифицированных введе-
нием различных количеств алюми-
ната. Одна из серий приготовля-
лась из немодифицированного золя
с размером частиц 22 нм (людокс-
ТМ), а другая—из золя с разме-
ром частиц 14 нм (людокс-HS).
Степень покрытия алюминат-иона-
ми составляла 1,8—25 % от общего
числа силанольных групп на поверх-
ности (с учетом поверхностной кон-
центрации таких групп, равной 8
SiOH/нм2). Для стабилизации алю-
минат-ионами требуется, чтобы на
поверхности частицы каждый атом
алюминия был окружен тремя ато-
мами кислорода, связанными в свою
очередь с атомами кремния. Это
означает, что не более чем 25 %
поверхностных атомов кремния мо-
жет быть замещено атомами алюминия. При этом предполага-
ется, конечно, что слой, непосредственнно примыкающий снизу
к поверхности кремнезема, напорист и недоступен для реакции
с ионами А1 (ОН)Г.
Такое покрытие может быть оттитровано катионным ПАВ,
которое адсорбируется тем сильнее, чем большее число анион-
ных центров присутствует на поверхности при pH 3,5
(рис. 4.26).
Используя выше приведенные серии модифицированных
золей, Айлер продемонстрировал, что с увеличением заряда на
поверхности частицы менее активно вступают в реакции с веще-
ствами, способными образовывать водородные связи. Так, про-
теины, поливиниловый спирт и полиэтиленгликоли вызывают
коагуляцию немодифицированных золей кремнезема при pH
3—5, однако коагуляция тормозится, если поверхность крем-
незема оказывается заряженной в большей степени в резуль-
тате предварительной обработки алюминатом.
Рис. 4.26. Зависимость концен-
трации коагулянта от степени
покрытия поверхности кремне-
зема алюмосиликат-ионами.
Для коагуляции 50 мл 1 %-ного
золя с частицами кремнезема раз-
мером 14 нм при указанной на гра-
фике доле покрытия поверхности
частиц алюмосиликат-ионами тре-
буется определенный объем 1 %-ного
раствора бромида цетилтриметил-
аммония (БЦТМА).
Коллоидный кремнезем — концентрированные золи
563
Модифицированный оксидом алюминия золь значительно
более устойчив в отношении гелеобразования в области pH
4—6, в которой немодифицированный золь наиболее быстро
Рис. 4.27. Зависимость времени гелеобразования от нормальности NaCl.
Показана стабильность модифицированного алюмосиликатом золя кремнезема лю-
цокс-АМ (сплошная линия) по сравнению с немодифицированным золем людокс-HS
(пунктир) при 25 °C. Цифрами на рисунке показаны значения pH.
превращается в гель (рис. 4.27), а также наименее чувствите-
лен к присутствию соли.
Другие анионы
Другие элементы, по-видимому, ведут себя иначе, чем алю-
миний в отношении образования анионных центров на поверх-
ности кремнезема. Шафер и Рой [420] отметили, что ион Сг3+
не может образовывать чтырехкоординированную ассоциацию
с атомами кислорода в отличие от алюминия. Однако Вагнер
[421] утверждал, что стабилизированные золи могут полу-
чаться в случае пирогенного кремнезема, приготовленного
сжиганием SiCl4 вместе с А1С13 или TiCU. Казалось бы, что
отрицательный заряд на поверхности должен был возникать
в присутствии А13+, но не Ti4+. Возможно, что частицы просто
покрываются оксидами А12О3 или TiO2 и при этом заряжаются
положительно при низких значениях pH.
Железо, по-видимому, было бы подходящим для формиро-
вания ферросиликат-аниона, но атом Fe может оказаться
слишком большим по размеру, как и Сг. Тем не менее некоторое
специфическое действие должно проявляться, поскольку при-
сутствие ионов Fe3+ в золях кремнезема не вызывает окраши-
10*
564
Глава 4
вания в коричневый цвет в той степени, как это имеет месте
в случае Fe(OH)3. Хацель, Гордон и Шок [422] сообщили,
что при смешивании золя кремнезема при pH 2,2 с раствором
соли железа(III) такое окрашивание обесцвечивалось даже
после того, как повышалось значение pH.
Чтобы сохранить отрицательный заряд на частице кремне-
зема при низких значениях pH, т. е. в тех условиях, когда на
поверхности чистого кремнезема имеется совсем незначитель-
ный заряд, требуется, чтобы замещающий атом не только имел
валентность менее 4, но также обладал сильным сродством
к кремнезему при низких pH и оставался бы четырехкоорди-
нированным по отношению к кислороду. Всем этим требованиям
отвечает только алюминий. Действие бора, очевидно, не было
исследовано, но связь Si—О—В гидролитически нестабильна.
Был описан [423] другой тип отрицательно заряженного
покрытия золя кремнезема. Фосфаты Се, Hf, Sn, Ti или Zr
в коллоидной форме способны вступать в реакцию с коллоид-
ным кремнеземом, после чего такие системы регулируются до
pH 9 добавлением какого-либо летучего основания. Многоза-
рядные ионы металлов в комплексных соединениях, вероятно,
присоединяются к кремнеземной поверхности, и указанные фос-
фаты создают в коллоидной системе отрицательный заряд,
распространяющийся на все частицы.
Положительно заряженные частицы
Покрытия оксидами поливалентных металлов
Отрицательный заряд на поверхности кремнезема может
измениться на противоположный в результате адсорбции на
поверхности избыточного количества вещества, обладающего
положительным зарядом. Обращение зарядов в коллоидных си-
стемах давно известно, но концентрированный кремнеземный
золь такого типа с частицами, обладающими положительным
зарядом на поверхности, впервые был выделен Александером
и Болтом [424]. Авторы определили, что покрытия оксидами
поливалентных металлов могут быть нанесены на всю поверх-
ность частиц и обеспечить максимальную устойчивость золя.
Сюда относятся оксиды трех- и четырехвалентных металлов,
например алюминия, хрома, галлия, титана и циркония. Пред-
почтение следует отдать золю с содержанием 26 % кремнезема
и 4 % А12О3, в котором положительно заряженные частицы
сопровождаются противоионами — хлорид-ионами. Для изго-
товления такого продукта подкисленный золь кремнезема сме-
шивали с основной солью металла, содержавшей чрезвычайно
небольшие коллоидные частицы оксида металла, которые
Коллоидный кремнезем — концентрированные золи
565
и адсорбировались на поверхности кремнезема. Миндик и Ревен
[425] приготовляли основные соли металлов в присутствии золя
кремнезема смешиванием золя с солью металла и последующим
удалением большей части анионов соли ионным обменом.
Подобным образом золь с покрытием частиц оксидом титана
приготовляли посредством гидролиза титансодержащего орга-
нического соединения в стабилизированном кислотой золе
кремнезема при pH <2 и последующего нагревания смеси для
того, чтобы вызвать осаждение оксида титана на поверхности
кремнеземных частиц [426, 427]. Коварик [428] получил патент
на частичное покрытие оксидом металла в виде А12О3. Покры-
тие получалось смешиванием А12(ОН)5С1 с золем кремнезема
и удалением ионов С1~ с помощью анионита в бикарбонатной
форме. Для устранения хлорид-ионов можно использовать
основной ацетат алюминия, стабилизированный борной кисло-
той [429].
Одной из характерных особенностей таких золей является
то, что их можно высушить и повторно пептизировать.
Поскольку в растворе покрытые оксидом алюминия золи крем-
незема не образуют растворимого кремнезема и на поверхности
частиц отсутствуют силанольные группы, то при высушивании
золя до состояния порошка силоксановые связи между отдель-
ными частицами не будут формироваться. Хлоридные противо-
ионы остаются на поверхности, и порошок может быть повторно
диспергирован при погружении его в воду при pH 3—5 [430].
Устойчивость таких положительно заряженных золей была
исследована Катсанисом и Матиевичем [431]. При низких
значениях pH некоторые анионы способны оказывать дестаби-
лизирующее действие, как это и следует ожидать от полива-
лентных анионов. Дестабилизация при очень низких значениях
pH наблюдалась в результате выщелачивания с поверхности
ионов А13+.
Проведены многочисленные исследования по взаимодейст- 1
вию многозарядных ионов металлов с поверхностью кремне-
зема. Никольский и др. [432] изучили образование мостиков
Zr—О—Si и пришли к заключению, что реакционная способ-
ность основных ионов Zr понижается в процессе их полимери-
зации. Хили, Купер и Джеймс [433] нашли, что ионы Fe3+
и Сг3+ адсорбируется на частицах кремнезема, сообщая им
положительный заряд. Химизм гидролиза катионов Al, Zr, Th
и Сг был обобщен Баесом и Месмером [434]. В работе [435]
было показано, что именно полимерные гидролизованные раз-
новидности в растворе нитрита алюминия, а не одиночные
ионы А13+ могут вызывать появление обратного по знаку за-
ряда в золях галогенида серебра. То же самое справедливо
и для обращения заряда в случае золей кремнезема.
566
Глава 4
Многозарядные органические катионы
Хотя многозарядные органические катионы, в том числе
катионные полимеры, вызывают флокуляцию кремнезема при
их добавлении к отрицательно заряженному золю, однако в том
случае, если они присутствуют в избыточном количестве, про-
исходит обращение заряда. Редиспергирование наблюдается
в особенности тогда, когда частицы кремнезема оказываются
слишком большими по сравнению с длиной полимерной цепочки,
так что каждая частица может покрываться одной или более
полимерными молекулами. Если же частица кремнезема отно-
сительно невелика, то полимерные цепочки с большей вероят-
ностью образуют мостиковые связи между частицами, и тем
самым редиспергирование предотвращается [321].
Повторное диспергирование может также происходить,
когда к золю добавляется такое количество катионного ПАВ,
которого достаточно, чтобы образовать двойной электрический
слой вокруг каждой частицы, и такой слой будет окружать
каждую сферическую мицеллу. Однако подобные положительно
заряженные золи не нашли никаких практических приложений,
по всей вероятности, из-за относительно большого (по сравне-
нию с массой частицы) количества органического вещества,
расходуемого на каждый слой вокруг кремнеземных частиц.
Золи с модифицированными поверхностями —
органозоли
Силанольная поверхность частиц кремнезема ограничивает
их диспергирование в органических жидкостях — низших спир-
тах, амидах и кетонах. С другой стороны, адсорбция способных
хемосорбироваться молекул с расположенными на внешней
стороне углеводородными группами приводит к тому, что ча-
стицы кремнезема становятся полностью дисперсными, образуя
органозоли в широком ряду органических жидкостей, включая
углеводороды.
Имеются разнообразные способы связывания органических
групп с поверхностью кремнезема: а) присоединение органиче-
ских ионов; б) образование связей Si—О—С и кремневых слож-
ных эфиров; в) образование связей кремний—углерод в крем-
нийорганических группах.
Органические ионы
Золи, стабилизированные щелочью, не могут редиспергиро-
вать в воде сразу же после их высушивания, но если стабили- 1
зирующие систему противоионы натрия вначале замещаются
Коллоидный кремнезем — концентрированные золи 567
на ионы тетраметиламмония, то подобный золь в таком случае
может быть высушен до состояния порошка, который легко
повторно растворяется в низших спиртах с образованием золя.
Монослой органических катионов на поверхности частиц явно
предотвращает контактирование кремнеземных поверхностей
после высушивания частиц [205]. Используя ион четвертичного И
аммониевого основания, содержащего углеводородную группу * \
с длинной цепью, Айлер [436] обнаружил, что после обработки
кремнезем образовывал золь в органических растворителях.
Был предложен ряд патентов для изготовления органофиль-.
ного кремнезема посредством добавления органических аминов-
или четвертичных аммониевых соединений в присутствии не-
смешивающейся с водой органической жидкости, чтобы вызы-
вать перемещение кремнезема из воды в органическую фазу.
Хэтевей [437] использовал в качестве агента переноса такие
соединения, как RCONH (CH2)6NH2+7lH, где R —алкильная
группа с длинной цепью и п 1—4. Воссос [438] использовал
для этой цели четвертичные аммониевые ионы с длинной цепью.
Коварик и Воссос [439] получили патент на высушенные по-
рошки такого типа.
Органические анионы могут присоединяться к поверхности
кремнезема посредством катионов металлов. Например, золь
кремнезема с частицами, покрытыми одним из оксидов трехва-
лентных металлов Al, Fe или Сг, модифицируется путем про-
ведения хемосорбции с 2-этилгексилфосфатом и образует ста-
бильный органозоль [440].
Этерификация
Поверхность частиц кремнезема можно сделать устойчивой
в течение длительного срока посредством превращения неко-
торой доли гидроксильных групп SiOH в группы SiOR (алкокси-
группы) в результате реакции со спиртами, приводящей к об-
разованию сложного эфира на поверхности (этерификация).
В некоторых случаях, когда диспергированный кремнезем вво-
дился в органические жидкости, имеющие в составе гидроксиль-
ные группы, не было полной ясности, в достаточной ли мере
система безводна, чтобы протекала этерификация кремнезем-
ной поверхности:
=SiOH + ROH -> =SiOR + Н2О
Когда порошок пирогенного кремнезема с дегидратирован-
ной поверхностью подвергался диспергированию в спирте,
в конце концов достигалась по крайней мере частичная этери-
фикация.
568
Глава 4
Этерификация коллоидного кремнезема первоначально изу-
чалась на очень небольших по размеру частицах поликремневой
кислоты; этот вопрос уже рассматривался во второй и третьей
главах [441]. Поскольку такая поликремневая кислота имеет
низкую молекулярную массу, относительно большая часть буто-
ксигрупп включается в образуемый продукт.
После того как стали доступны однородные коллоидные
частицы кремнезема определенного размера, появилась воз-
можность при этерификации поверхности показать, что полу-
ченный продукт действительно содержит мономолекулярный
слой ориентированных бутоксигрупп, заместивших некоторую
долю гидроксильных групп на поверхности кремнезема, так что
внешняя поверхность частицы по существу представляет слой
углеводородных групп. Эти группы могут быть алифатическими,
если применяется такой спирт, как бутиловый, или же арома-
тическими, когда используется бензиловый спирт. Природа
углеводородной поверхности оказывает влияние на раствори-
мость частиц и на их способность диспергировать. Например,
когда частицы кремнезема диаметром 17 нм подвергаются
этерификации под действием бензилового спирта, конечный
продукт — высушенный порошок — может растворяться в бен-
золе с образованием прозрачного раствора, но не растворим
в алифатическом растворителе, таком, например, как керосин.
С другой стороны, когда тот же самый кремнезем этерифици-
руется алифатическим октадециловым спиртом с разветвленной
цепью, то полученный порошок легко растворяется в керо-
сине [442].
Кроме того, имеется возможность этерифицировать поверх-
ность частиц гликолем, при этом каждая молекула гликоля
присоединяется одним концом к поверхности кремнезема по-
средством связи —SiOC—эфир, а другой ее конец оказывается
на внешней поверхности частицы в виде полярной гидроксиль-
ной группы. Такая коллоидная система способна образовывать
устойчивые золи в полярных органических растворителях, но
не в углеводородах [443]. С другой стороны, кремнеземная
поверхность может покрываться слоем углеводородных групп,
если какой-либо гликоль, например гексаметиленгликоль,
вступает в реакцию этерификации с кремнеземной поверхностью
таким способом, что обе гидроксильные группы направлены
вовнутрь, а поверхность оказывается этерифицированной.
На агрегированные порошки кремнезема, этерифицированные
таким способом, был получен патент [444], но этот принцип
применим также и к золям кремнезема. Избытка гликоля сле-
дует избегать; используется такое количество гликоля, которого
как раз достаточно, чтобы при покрытии кремнеземной поверх-
ности оба конца гибкой молекулы присоединялись к поверх-
Коллоидный кремнезем — концентрированные золи
569
ности. Для удаления воды посредством азеотропной дистилля-
ции используется углеводородный растворитель. Поверхность,
таким образом, превращается в органофильную и гидрофобную
с минимальной затратой органического вещества, так как
углеводородные цепочки имеют тенденцию располагаться
плоско вдоль поверхности, поэтому органофильное покрытие
имеет толщину всего в несколько ангстрем.
Эти примеры показывают, что свойства коллоидного крем-
незема могут быть коренным образом изменены за счет нане-
сения мономолекулярного слоя какого-либо другого вещества,
связанного с поверхностью, так как поведение коллоидной
системы после модификации определяется главным образом
свойствами такого поверхностного слоя.
Наиболее полно реакция гидроксильных групп на поверх-
ности кремнезема осуществляется с метиловым спиртом. Бродж
[445] обнаружил, что до ~ 5,5 метоксильных групп может
присоединиться к 1 нм2 поверхности. Авторы работы [446]
подтвердили, что реакция с метанолом охватывает вплоть до
95 % всех первоначально имеющихся на поверхности водного
золя силанольных групп, поверхностную концентрацию которых
они приняли равной 5—6 ОН-групп/нм2.
Превращение водного золя в органозоль наиболее просто
осуществляется путем добавления смеси высококипящего сме-
шивающегося с водой многоатомного спирта с кипящей водой.
Если золь вначале был деионизирован, то кремнезем остается
диспергированным, и при конечной высокой температуре кипе-
ния органической жидкости процесс этерификации протекает
быстро по мере того, как вода удаляется [447—449].
Айлер [450] сообщил, что в присутствии избытка гликоля
только одна из двух его гидроксильных групп вступала в реак-
цию этерификации с поверхностью кремнезема, так что наруж-
ная поверхность модифицированного образца состояла из
алкильных гидроксильных групп, и, таким образом, получен-
ный золь мог смешиваться как с водой, так и со спиртами.
Однако новые наблюдения показали, что при добавлении воды
в таком количестве, чтобы на поверхности частиц оказался
только ее монослой (около 8 молекул Н2О/нм2), золь проявлял
характерную неустойчивость и превращался в гель. Если воды
добавлялось большее количество, то золь оказывался более
устойчивым.
Аналогично могут приготовляться золи в эфирах гликолей,
которые достаточно полярны, чтобы смешиваться с водным
золем кремнезема в процессе испарения воды, но обычно орга-
нозоль формируется в несколько более летучем растворителе.
Стабильные золи, содержащие вплоть до 60 % SiO2, могут быть
приготовлены в смешивающемся с водой алкоксиэтаноле
570
Глава 4
ROC2H4OH, где R — низшая алкильная группа [451]. Однако,
как утверждалось, золи, получаемые в простом моноэтиловом
эфире этиленгликоля, из которых вода удалялась в вакууме
при невысокой температуре, не содержали этерифицированного
кремнезема [452]. Если это так, то устойчивость золя должна
быть следствием «стерической стабилизации», вызванной обра-
зованием водородных связей между силанольными группами
поверхности кремнезема и атомами кислорода эфирных и гид-
роксильных групп органического вещества. В более раннем
патенте описано, что такой тип золя подвергался нагреванию
с целью его стабилизации, причем подобное воздействие, ве-
роятно, оказывала этерификация [453].
Порошок кремнезема, способный редиспергировать с обра-
зованием органозоля, может быть получен этерификацией всей
поверхности коллоидных частиц перед высушиванием золя
[442]. Исходный золь должен быть освобожден от ионных
примесей и прежде всего перемещен из воды в способную
смешиваться с водой полярную органическую жидкость, как,
например, «-пропиловый спирт. Затем золь этерифицируют на-
греванием под давлением или же переносят в другой спирт,
после чего нагревают до полной этерификации поверхности
и высушивают. Природа образовавшегося органического покры-
тия определяет растворимость коллоидных частиц в данном
органическом растворителе. На подобный способ приготовления
способного диспергировать порошка с органическим покрытием
был позднее получен патент Миндиком и Куртисом [454].
Удаление соли из водного золя при низких значениях pH
можно осуществить разбавлением золя способной смешиваться
с водой органической жидкостью, например диметилформами-
дом или этиленкарбонатом, которая осаждает соль. Затем
смесь дистиллируется под вакуумом для получения безводного
органозоля. В случае сложных карбонатных эфиров необходи-
мые для этерификации гидроксильные группы получаются,
вероятно, в результате гидролиза [455]. Для достижения ста-
билизации системы не требуется полная этерификация поверх-
ности частиц, если дисперсионной средой является вещество,
образующее прочные водородные связи. Золи, у которых под
действием способного смешиваться с водой спирта этерифици-
ровано немного менее половины поверхности частиц, оказы-
ваются устойчивыми в избытке спирта [456].
Для проведения этерификации реакционной смеси и промо-
тирования самой реакции были предложены различные методы
дегидратации. Обычно используют азеотропную дистилляцию.
Как только смесь становится обезвоженной, ее многократно
нагревают под давлением выше атмосферного с целью ускоре-
ния реакции этерификации. Вода может быть удалена из си-
Коллоидный кремнезем — концентрированные золи 571
стемы с помощью молекулярных сит [457]. Кремнезем может
находиться в эмульгированной смеси, состоящей из воды и
несмешивающегося с ней растворителя, такого, например, как
смесь из минерального масла, эфира цетилолеилгликоля и
эмульгирующего агента. Эту смесь дегидратируют в выпарном
аппарате, работающем по принципу непрерывной подачи тон-
кого слоя раствора [458]. Согласно данным Рицнара [459],
воду можно удалять реакцией с кеталем [459].
Образование силильного покрытия
Еще более устойчивая органофильная поверхность на части-
цах кремнезема получается при взаимодействии с алкилхлор-
силанами, которые присоединяются к поверхности с помощью
органосилильных групп. Например, в работе Айлера [460]
было показано, что поверхность коллоидного кремнезема по-
крывается триметилсилильными группами. Автор перемещал
кремнезем из водной среды в триэтилфосфат, высушивал золь,
добавлял триметилхлорсилан, затем проводил нагревание и
удалял избыточное содержание реактива и растворителя испа-
рением под вакуумом. Получаемый твердый продукт проявлял
способность диспергировать с образованием золей в бензоле,
простом эфире и хлороформе, но не в воде.
Авторы работы [461] исследовали дисперсные системы,
состоящие из частиц кремнезема, полностью гидрофобизиро-
ванных метилсилильными группами, способными покрывать по-
верхность в системе пропанол—вода. Наблюдалось, что поверх-
ность частиц и значения поверхностного натяжения подобны
соответствующим значениям для парафина. Смачивание по-
рошка происходило в водно-спиртовых смесях, содержащих
более 15 % пропанола, 32 % этанола и 50 % метанола. По-ви-
димому, диспергирование обусловливается не электростати-
ческим отталкиванием, а скорее свободной энергией поверхно-
сти раздела, которая имеет непосредственное отношение к про-
цессу смачивания.
Согласно данным Айлера [462], частицы кремнезема в золе
могут превращаться в органофильные в результате реакции
в среде вода—трет-бут иловый спирт с добавлением моно- или
диметилхлорсиланов. Такие частицы с модифицированной по-
верхностью переводятся в какую-либо органическую фазу и
дегидратируются под действием дистилляции.
Гидрозоль кремнезема, состоящий из частиц диаметром
3—5 нм, эмульгированный в среде минерального масла, ока-
зывался обезвоженным и вступал в реакцию с полидиметил-
силоксановым маслом с образованием гидрофобной антипенной
присадки [463].
572
Глава 4
Коммерческие типы коллоидного кремнезема
Вследствие постоянно происходящих изменений в развитии
и потребностях производства наблюдается обновление ассорти-
мента коммерческих кремнеземных золей: появляются новые
виды, а некоторые исчезают из продажи; поэтому невозможно
предсказать, какие виды золей останутся в будущем. Приме-
нявшиеся в прошлом и используемые в настоящее время типы
золей представлены в табл. 4.3. В таблицу включены и неко-
торые виды продукции, которых нет в продаже сейчас, но
о них упоминалось в более ранних технических публикациях.
Большая часть золей содержит частицы диаметром 5—50 нм
при содержании 30—50 масс. % SiC^. Большинство таких золей
стабилизировано либо гидроксидом натрия, либо аммиаком
при pH 8—10. Несколько типов золей стабилизировано при
низких значениях pH посредством проведения специальной
очистки, замещением воды каким-либо полярным органическим
растворителем или же за счет образования на частицах поло-
жительного заряда нанесением оксида алюминия. Коммерче-
ские золи состоят из дискретных частиц с очень небольшой
степенью агрегации, так как при высоких концентрациях крем-
незема любая, сколько-нибудь значительная степень агрегации
должна весьма сильно вызывать повышение вязкости.
Дисперсные кремнеземные системы могут быть приготовлены
из некоторых разновидностей пирогенных кремнеземных по-
рошков; о них будет упоминаться в гл. 5. В порошках, имею-
щих объемные плотности менее чем ~0,08 г/см3, индивидуаль-
ные частицы оказываются настолько слабо связанными между
собой, что их можно диспергировать, по крайней мере до
агрегатов коллоидных размеров, в водной среде при pH 9 путем
энергичного механического перемешивания. Порошки, имеющие
наиболее низкие значения объемной плотности и удельной по-
верхности или же наибольшие размеры первичных частиц,
могут быть диспергированы до состояния золей наиболее полно.
Поскольку во многих библиографических ссылках, касающихся
вопросов использования золей, не сообщается об источниках
получения таких золей, то подобные вопросы применения золей
всех видов будут рассмотрены в следующем разделе.
Применение коллоидного кремнезема
В этом обзоре невозможно показать относительную прак-
тическую ценность или важность различных специфических
приложений таких типов кремнезема. Поэтому данный раздел
в основном явится путеводителем по имеющимся в литературе
Золь, фирма-изготовитель Марка, сорт SiO2, % Стабилизатор Отношение SiO2 : Na2O pH Диаметр частиц, нм Удельная поверхность, м2/г Промышленный бюллетень
тип содержа- ние, %
изо 30 Na2O 0,65 46 10,0 8 375 К-1030 (1976)
1030 30 Na2O 0,40 75 10,2 11-13 190-270
1140 40 Na2O 0,40 100 9,7 15 200 К-1140 (1972)
1050 50 Na2O 0,35 143 9,0 17-25 120-176 К-1050 (1976)
1034А ж 34 — 3,1 16-20 135-190 К-1034 (1972)
1129 3 30 — 3,5 16-25 150 Спецификация (1974)
Е127
2325 м 35 — 5 20 150 К-2325 (1975)
43J25
2327 к 40 NH3 0,10 — 9,3 20 150 К-2327 (1975)
41D01
2600 л 57 Na2O 0,10 — — 20 150 Спецификация (1973)
1060 50 Na2O 0,25 — 8,5 60 50 К-1060 (1975)
40D04 ы 50 — — — 4,0 16-22 —
D2I49» 30 NH3 0,03 — 9,5 12-15 210
D2361° 30 A12O3 1,8 — 3,8 20 150
Нанкол (Nyacol, Inc., Ашленд, США) 215 830 15 30 Na2O Na2O 0,75 0,45 11 10,7 3-4 8 — Данные представ лены в 1976 г.
1430 30 Na2O 0,35 10,4 14
1440 40 Na2O 0,48 10,4 14 —
2050 50 Na2O 0,48 10 20 —
2034А п 34 — — 3,5 20 —
2046 ЕС 46 Нет данных 9,8 20 —
1430NH3 30 NH3 0,2 9,4 13-15 —
Сноутекс (Nissan Chem. Industries, Lid., Токио, Япония; Нью-Йорк, США) 20 30 20 30 Na2O Na2O 0,35 0,6 — 9,5-10 9,5 — 10,5 10-20 10-20 — Данные получены в 1977 г.
С 20 Na2O 0,2 — 8,5-9,0 10-20 —
№ 20 Na2O 0,04 — 9,0-10,0 10-20 —
ОР 20 — 3-4 10-20 —
Ситон (Monsanto, Ltd., Лондон, Англия) W15 W30 15 30 Нет данных — 9,8-10,6 9,8-10,6 36 36 75 75 № 53—3(E) М-Е-2 (1975)
Коллоидный кремнезем — концентрированные золи
577
вопросам, относящимся к применениям в исследовательских
целях. Только небольшая доля опубликованных патентов свя-
зана с такого рода использованием коллоидных кремнеземов,
поэтому может быть упомянута в данном разделе. Обширный
перечень данных по коллоидным коммерческим системам был
собран в Monsanto Technical Bulletin IC/SCS-237 вплоть при-
мерно до 1967 г. (см. табл. 4.3).
Никакого постоянного применения коллоидный кремнезем
не находил, пока не стали доступными концентрированные,
стандартизированные, устойчивые коммерческие золи. Шверин
[464] в 1915 г. получил разбавленный золь в результате про-
цесса электролиза раствора силиката натрия. В 1933 г. Грпсс-
бах [465] составил библиографию, относящуюся к приготовле-
нию и использованию коллоидного кремнезема. 10%-ный золь,
стабилизированный аммиаком, начала производить фирма
I. G. Farbenindustries. Многие из казавшихся тогда возмож-
ными применений в‘ настоящее время осуществлены: использо-
вание в бумажном и текстильном производстве, в качестве
наполнителя резиновых изделий, в керамических и огнеупорных
материалах. В 1945 г. Уайт [5] от фирмы Monsanto Chemical
запатентовал способ превращения посредством автоклавной
обработки щелочного геля кремнезема в относительно стабиль-
ный 20 %-ный золь с неоднородными по размеру частицами.
В 1941 г. Бёрд [4] из National Aluminate Со., а в 1951 г.
Бечтольд и Снайдер [6] от фирмы Е. I. du Pont de Nemours Co.
запатентовали способы удаления натрия из раствора силиката
натрия посредством ионного обмена и выращивания частиц до
желаемого размера в процессе концентрирования золя выпари-
ванием. Благодаря этим работам были созданы стабильные
прозрачные золи, содержащие 30 % кремнезема в виде одно-
родных частиц диаметром 10—15 нм. С того времени масштабы
практического применения коллоидного кремнезема выросли,
и ниже перечисляются виды его использования в соответствии
с теми задачами, для которых нужен такой кремнезем.
1. Приготовление силикагелей с необходимыми характери-
стиками: удельной поверхностью, размером пор, механической
прочностью, определяемыми размером частиц коллоидного
кремнезема; например для использования таких силикагелей
в качестве основных катализаторов и адсорбентов.
2. Получение загущенных и связанных волокнистых и гра-
нулированных материалов посредством введения золя и после-
дующего высушивания системы до образования жесткой струк-
туры геля. Это находит применение при изготовлении, напри-
мер, точных литейных форм, отформованных огнеупорных
изделий и высокотемпературных изоляционных материалов.
II Заказ № 250
578
Глава 4
3. Увеличение коэффициента трения между трущимися по-
верхностями («невидимый песок»), что используется, например,
на железнодорожных рельсовых путя-х, натертых вощеных
полах и для текстильных волокон.
4. Приготовление средств для уменьшения склеивания и
слипания органических пленок, а также для снятия статических
зарядов с таких пленок.
5. Проведение обработок поверхностей для устранения воз-
можности их загрязнения. Это достигается путем заполнения
золем микропор пористых материалов и получения ультраполи-
рованной олеофобной их поверхности, чтобы исключить накоп-
ление на них загрязняющих частиц. Такой обработке могут
подвергаться, например, ткани, бумага и окрашенные по-
верхности.
6. Придание различным поверхностям гидрофильных, олео-
фобных свойств благодаря присутствию на поверхности крем-
неземных частиц высокополярных групп SiOH. Примером
могут служить печатные формы, используемые для офсетной
печати.
7. Повышение или понижение адгезии между поверхностями
в зависимости от применяемой подложки и способа нанесения
коллоидного кремнезема. Это находит применение при обра-
ботке поверхностей, например органических пленок, стекол и
металлов.
8. Использование в качестве компонента: а) для создания
тонких тугоплавких, непроводящих пленок на электропроводных
поверхностях, например при обработке металлических пласти-
нок, используемых в сердечниках трансформаторов; б) для
создания электропроводных пленок на непроводящих материа-
лах, например при получении графитированных покрытий на
бумаге. '
9. Использование в качестве агентов, вызывающих форми-
рование поперечных связей, загустителей и наполнителей в ор-
ганических полимерных материалах, что обеспечивается посред-
ством связывания полимерных цепочек с равномерно распре-
деленными коллоидными частицами кремнезема. Это находит
применение при получении, например, искусственных кож, изде-
лий из вспененного латекса, эластомеров.
10. Употребление в качестве агента для полирования крем-
невых пластин.
11. Придание системам определенных поверхностно-актив-
ных свойств: флокулирующих, коагулирующих, диспергирую-
щих, стабилизирующих, эмульгирующих, суспендирующих; при-
дание антипенных свойств.
12. Регулирование вязкости систем: загущение, застудне-
вание.
Коллоидный кремнезем — концентрированные золи 579
13. Образование адсорбционных пленок на поверхностях;
получение оптических эффектов.
14. Использование в фотографии в качестве компонента
в многослойных пленочных системах.
15. Применение в биологических исследованиях; получение
питательных сред или сред при исследованиях методом центри-
фугирования.
16. Получение реакционноспособного кремнезема.
Приготовление катализаторов, силикагелей,
адсорбентов
Процесс приготовления кремнеземсодержащих материалов из
коллоидного кремнезема, а не из силиката натрия имеет очевид-
ные преимущества, связанные с легкостью присоединения кол-
лоидного кремнезема к другим компонентам катализатора.,
минимальной процедурой промывания для удаления нежелатель-
ных солей и возможностью получения более широкопористой
однородной структуры за счет формирования геля из относи-
тельно больших однородных коллоидных частиц. Преимущест-
вом приготовления основного катализатора из сферических
частиц коллоидного кремнезема, размещенных в плотно упако-
ванную систему, оказывается то, что при повышенной темпе-
ратуре масса катализатора не может легко сжиматься и
разрушаться или спекаться, и, таким образом, однородные
поры, образуемые между однородными частицами с единооб-
разной упаковкой, обеспечивают постоянное значение удельной
поверхности и высокую степень каталитической актив-
ности [467].
Маатман и Пратер [466] обсудили получение из коллоид-
ного кремнезема катализатора с однородным распределением
необходимых для катализа компонентов, созданного на основе
силикагеля. Трудно достичь однородного распределения пу-
тем пропитки предварительно таблетированного геля раство-
ром катализатора. Золь кремнезема, содержащий соли метал-
лов, может быть высушен распылением или вымораживанием
с образованием небольших сферических частиц геля, которые
затем могут уплотняться до желаемой степени [467]. Золь
можно превратить в тонкодисперсный порошок путем диспер-
гирования в органическом растворителе, частично смешиваемом
с водой, гелеобразования кремнезема и отгонки жидкой
фазы [468].
После того как катализатор был осажден на частицах крем-
незема, для повышения прочности можно осадить и высушить
кремневую кислоту [469]. Авторы работы [470] показали
некоторые типичные примеры использования коллоидного крем-
11*
580
Глава 4
Рис. 4.28. Электронно-микроскопические снимки наполнителя для хромато-
графической колонки.
<1 — стеклянная микробусинка, покрытая пористым слоем кремнезема, — диаметр мик-
робусинки, dp/30 — толщина покрытия; б — покрытие пористым кремнеземом.
незема при приготовлении основного катализатора. По-види-
мому, каталитической активностью частиц коллоидного крем-
незема, добавляемых к дизельному горючему, объясняется воз-
можность предупреждения образования загрязнений [471].
Формирование пористых микросфер для использования
в хроматографических колонках уже ранее описывалось в связи
с коацервацией [343]. Наполнение хроматографических коло-
нок другого типа проводилось с применением многослойного
способа. С целью достижения наибольшей эффективности хро-
матографических разделений необходимо так заполнять ко-
лонки, чтобы имелась наибольшая поверхность и наибольший
объем пор адсорбента при минимальной толщине последнего.
Это удалось достичь Киркленду, который осаждал несколько
слоев кремнеземных коллоидных частиц на поверхности стек-
лянных шариков и тем самым получил однородную пористую
пленку. Процессы диффузии в такой пленке протекают быстро.
Отсутствие пористости в самих шариках устраняет возмож-
ность появления медленной диффузии при хроматографических
измерениях [472]. Покрытие, нанесенное на стеклянный шарик
рассматриваемого типа, показано на рис. 4.28. Структура и
применение набивок для хроматографических колонок было
описано в ряде статей [473, 474].
Коллоидный кремнезем — концентрированные золи
581
Неорганические связующие и загущающие вещества
Одним из сравнительно давно предложенных применений
коллоидного кремнезема, полученного из силиката натрия
ионным обменом, было его использование для повышения проч-
ности керамических цементов [475]. При подобного рода при-
менениях коллоидный кремнезем действует при обычной тем-
пературе как связующее или загущающее вещество, поскольку
превращается в твердый гель. Эффективность цементов нахо-
дится в прямой зависимости от прочности геля, и теория этого
вопроса уже обсуждалась выше в данной главе.
В некоторых случаях коллоидный кремнезем использовался
благодаря своей высокой химической активности. Так, когда
стеклянный порошок покрывается коллоидным кремнеземом,
то его можно формовать. При нагревании кремнезем расплав-
ляется в стекле и получается твердое спекшееся изделие [476].
Прочность при добавлении коллоидного кремнезема в качестве
связующего, очевидно, усиливается при погружении системы
в спиртовую среду и образовании смеси с этилсиликатом
[477, 478]. В том случае, когда необходимо использовать кол-
лоидный кремнезем как связующее для кремнеземного порошка,
то более прочные связи будут образовываться ниже темпера-
туры расстекловывания, что достигается добавлением к золю
кремнезема борной кислоты (1—5%) с целью понижения
температуры спекания [479]. Сочетание коллоидного кремне-
зема и кислых фосфатов поливалентных металлов приводит при
нагреве к образованию прочных связей, вероятно, вследствие
того, что появляется некоторое количество соединений кремне-
зема с фосфат-ионами. Однако при высокой температуре
фосфаты, как правило, вызывают понижение прочности.
Повышенная прочность связей в керамических изделиях
достигается за счет использования смесей, состоящих из пер-
вичного кислого фосфата алюминия и коллоидного кремнезема,
при их добавлении к тонкодисперсным тугоплавким порошкам
циркония, оксида циркония или оксида алюминия [480]. В ре-
зультате реакции Р2О5 с кремнеземом при относительно низкой
температуре образуется высокая прочность связей в тугоплав-
ких композициях. Так, золь кремнезема может смешиваться
с фосфатом аммония или с другими первичными фосфатами при
относительно низком значении pH, и такой золь при минималь-
ных его количествах находит применение в качестве связую-
щего вещества для тугоплавких порошков [481, 482]. Согласно
данным Ли [483], связующие свойства коллоидного кремнезема
улучшаются за счет добавления растворимого четвертичного
силиката аммония, такого, например, как силикат тетраметил-
582
Глава 4
аммония, который оказывается в достаточной степени щелоч-
ным, чтобы взаимодействовать с коллоидным кремнеземом и
вызывать его коалесценцию в цементируемой структуре.
Рейтер [484] показал возможность типичного применения
коллоидного кремнезема в качестве связующего для тугоплав-
ких частиц с целью получения огнеупорной керамики. Автор
описал способную отливаться в форму смесь коллоидного крем-
незема и измельченных в порошок тугоплавких зерен вместе
с агентом желатинизации, чтобы вызвать схватывание смеси.
Мазиляускус [485] представил обзор работ, имевшихся
в 1958 г., об использовании коллоидного кремнезема в керами-
ческих изделиях. Прочные связи в подобных керамических
веществах образуются при относительно низкой температуре.
Шоуп приготовил гели кремнезема, обладающие очень вы-
сокой прочностью, из смесей силиката калия и коллоидного
кремнезема, о чем уже упоминалось при рассмотрении вопро-
сов о полисиликате калия в гл. 2 (см. лит. к гл. 2 [97, 96]).
Такие композиции могут иметь высокую прочность связи в1 тех
случаях, когда допускается присутствие небольших количеств
соли калия.
Формующиеся тугоплавкие изделия
Тугоплавкие изделия различных типов приготовлялись или
из одного чистого кремнезема, или при использовании кремне-
зема в качестве связующего для керамических частиц. Бергна
[486] описывает изготовление прозрачных брусков, подобных
брускам из «плавленого кварца», путем горячего прессования
(при 1200°С и давления 140 кг/см2 в течение 5 мин) чистого
кремнеземного порошка с добавлением полученного распыли-
тельной сушкой коллоидного кремнезема.
Бергна и Симко [487] за счет уплотнения и спекания пред-
варительно очищенного и высушенного распылением коллоид-
ного кремнезема получали непрозрачные изделия, похожие на
изделия из «плавленого кварца», но обладавшие в два раза
большей прочностью на поперечный разрыв. Это достигалось
вследствие образования в изделиях цилиндрических или тонне-
леподобных пор, которые действовали как «ограничители тре-
щин». Такая новая, ранее не существовавшая структура
получается в процессе уплотнения сферических гранул геля
кремнезема диаметром 1 мкм. При прессовании, перед тем как
вся масса подвергается обжигу, остаются макропоры между
гранулами. Внутри сферических гранул первичные частицы
коллоидного кремнезема полностью спекаются, образуя непо-
ристый кремнезем, тогда как между сферами остаются макро-
поры, формирующие связанную трехмерную сетку каналов,
Коллоидный кремнезем — концентрированные золи
583
которые не сокращаются полностью в процессе спекания всей
массы. Такое изделие имеет плотность, равную 91—99 % рас-
считанной. Если же температура повышается до такой точки,
когда все макропоры полностью сокращаются, и при этом
достигается 100 %-ная рассчитанная теоретически плотность
материала, то прочность изделия при этом падает до прочности
обычного плавленого кварца.
Формующиеся изделия из чистого кремнезема могут при-
готовляться в результате спекания прочного, плотного геля
кремнезема, приготовленного из смеси силиката калия и сили-
ката натрия, как это описывал Шоуп (см. лит. к гл. 2, [96]).
Тугоплавкий материал приготовлялся смешиванием колло-
идного кремнезема и основного хлорида алюминия, взятых
в таких соотношениях, чтобы образовать муллит. Используя
коллоидный кремнезем в качестве связующего для муллитового
дорошка, формуют муллитовое огнеупорное изделие при 1300°С
[489]. Подобным же образом огнеупорные изделия из силлима-
нита получались в результате связывания силлиманитного по-
рошка смесью коллоидного кремнезема и основного хлорида
алюминия, действующей на порошок как связующее. Для по-
лучения изделия вся масса подвергалась обжигу при 1300—
1400°С [488].
Не относящиеся, строго говоря, к керамике формующиеся
при обычной температуре металлические изделия, обладающие
повышенной теплопроводностью, могут приготовляться из ме-
таллических порошков с добавлением коллоидного кремнезема
и латекса в качестве связующей смеси [490]. Когда коллоид-
ный кремнезем смешивается с частицами тугоплавкого порошка
и такая смесь отливается в форме, то при этом керамические
частицы могут оседать и сегрегировать. К тому же по мере
высушивания всей массы коллоидный кремнезем будет мигри-
ровать к поверхности раздела с водой, и поэтому внутренняя
часть оказывается обедненной связующим веществом. По ука-
занной причине, как -правило, необходимо принимать меры,
чтобы гель кремнезема оставался внутри массы, или по край-
ней мере надо создать условия для развития тиксотропии,
предотвращающей миграцию коллоидного кремнезема. Это
достигается либо путем регулирования pH до области значе-
ний, где происходит застудневание в течение известного пе-
риода времени, либо добавлением агента, способного затормо-
зить процесс гелеобразования. Таким веществом может
являться соль кремнефтористоводородной кислоты, медленно
выделяющая HF. Другим подходом может оказаться добавле-
ние порошкообразного силикатного стекла щелочных металлов,
которое будет медленно растворяться и вызывать гелеобразо-
вание [491].
584
Глава 4
Связующие для волокон
Неорганическое связующее для неорганических волокон
приготовляется посредством диспергирования глины в коллоид-
ном кремнеземе с последующим подкислением до pH 3,5 и до-
бавлением соли алюминия, например формиата. Блок, изготов-
ленный из изоляционных стеклянных волокон, связанных
с указанным составом, остается превосходным материалом при
повышенной температуре [492]. Смесь коллоидного кремнезема
и поливинилового спирта запатентована как связующее при
получении изоляционного материала из стеклянных волокон,
причем такой материал остается связанным даже после того,
как поливиниловый спирт выжигается [493]. Коллоидный крем-
незем может притягиваться к неорганическому волокнистому
материалу в результате введения положительно заряженного
крахмала в коллоидный кремнезем. Этим улучшается связы-
вание смесей, содержащих асбестовые и алюмосиликатные
волокна [494]. Жаростойкий отражающий изоляционный мате-
риал приготовляется за счет связывания волокнистого титаната
калия со смесью из латекса и коллоидного кремнезема [495].
Коллоидный кремнезем находит применение в качестве свя-
зующего при получении материалов из высокожаростойких алю-
мосиликатных волокон [496]. Для того чтобы поддерживать
равномерное распределение связующего, используется такой
загуститель, как акриловый полимер [497]. Моор [498] сме-
шивал коллоидный кремнезем с различными видами латекса,
коагулировавшего после того, как связующее взаимодейство-
вало с волокнами. Придавать прочность и жесткость органиче-
ским листовым волокнистым материалам, а также листам
бумаги можно добавлением коллоидного кремнезема [499—502].
При изготовлении форзацной бумаги, используемой для рифле-
ния, ее жесткость улучшается за счет пропитки коллоидным
кремнеземом [503]. Добавление от 1 до 5 % коллоидного крем-
незема в определенного вида бумажные массы придает бумаге
прочность, жесткость и т. п. [504]. Нежелательное свойство
полиамидных волокон расщепляться и расслаиваться в значи-
тельной мере устраняется путем пропитки коллоидным кремне-
земом. Кожа способна разбухать и уплотняться после погло-
щения коллоидного кремнезема [505].
Огнеупорные покрытия
Основным применением коллоидного кремнезема, как это
показано в ряде соответствующих патентов, являются защит-
ные покрытия, наносимые для оказания противодействия вы-
Коллоидный кремнезем — концентрированные золи 585
зывающим эрозию эффектам, наблюдаемым при действии
расплавленной стали на «поддон» или на основание изложницы,
в которую сталь отливается. Такое покрытие продлевает жизнь
основанию изложницы и способствует извлечению стального
слитка [506]. В составы покрытий, наносимых на поддон,
входят тонко измельченное аморфное кварцевое стекло и кол-
лоидный кремнезем. Однако в связи с выделением газообраз-
ного водорода из примесей, содержащихся в расплавленном
металле, проблема защиты осложняется. Поэтому в состав
покрытий добавляется также определенное содержание водо-
растворимых красителей. Подобные ингибиторы, кроме того,
препятствуют выделению водорода из суспензий тонкодисперс-
ных металлов, таких, как цинк, и проникновению водорода
в связующее вещество — коллоидный кремнезем. Покрытия
представляют собой концентрированные шламы, состоящие из
тщательно отобранного по размеру тугоплавкого порошка, пере-
мешанного с концентрированным золем кремнезема, имеющим
обычно размер коллоидных частиц 10—50 нм в диаметре. Этот
состав должен быть таким, чтобы при распылении на горячий
поддон он мгновенно бы затвердевал и дегидратировал и со-
здавалось бы плотное, хорошо связанное покрытие [508]. Для
большей прочности Снайдер [509] предложил использовать
кремневую кислоту с низкой молекулярной массой (менее чем
90 000). Согласно Полларду [510], стабилизатор дисперсии,
добавляемый с целью предотвращения оседания тугоплавких
зерен, находящихся в связующем веществе — золе, оказывает
благоприятное действие. Автор запатентовал «Xanthomonas» —
гидрофильный коллоидный кремнезем, поступивший в продажу
наряду с загустителем под названием «Kelzan». Другие составы
были запатентованы Керфоттом и Баером [511]. Рушер [512]
описывает еще более усложненные составы, которые он исполь-
зовал в качестве загустителей, агентов против выделения газов
адгезионных промоторов, смачивающих реагентов и других
типичных усовершенствованных добавок, применяемых для та-
кого весьма специализированного вида продукции.
Изложницы для литья металлов
Коллоидный кремнезем уже давно применяется в литейном
производстве. Одним из важных его использований является
употребление в качестве связующего при изготовлении огне-
упорных изложниц для точного промышленного литья. Приме-
нение началось в 1945 г., когда впервые стал доступен коллоид-
ный кремнезем. Коллинз [513] предложил пластичную смесь
Для изготовления огнеупорных изложниц, которая включала
в себя огнеупорный наполнитель и водный коллоидный кремне-
586
Глава 4
зем вместе с небольшим количеством агента, промотирующего
гелеобразование. Клохерти и Эмблем [514] обсудили исполь-
зование золей кремнезема в этой области. Рейтер [515] запа-
тентовал основную идею об использовании коллоидного крем-
незема в качестве связующего в подобных составах, количество
электролита в которых регулируется, чтобы катализировать
процесс гелеобразования.
Эмблем, Маунтфорд и Морели [516] разработали сложный
состав, включающий коллоидный кремнезем и органическое
полимерное связующее вещество, который используется при
изготовлении внутренней оболочки изложниц, предназначенных
для точного промышленного литья.
Технические статьи по вопросу об использовании золей
кремнезема при изготовлении изложниц были опубликованы
Эмблемой [517, 518].
Были опубликованы различные патенты, охватывающие во-
просы нанесения восковых матриц для литья, смешиваемых
с золем кремнезема и кислотой [519]; нанесения покрытий
концентрированным золем в смеси подкисленный спирт—вода
[520]; использования красителя в слое из шлама, который
окрашивается после высушивания покрытия [521]; применения
оксида алюминия, покрывающего золь кремнезема [522].
Быстрое наращивание керамической изложницы вокруг
восковой литейной формы осуществлено Бейером, Моором и
Рушером [523] способом, который не требует высушивания
системы после каждого нанесения слоя. Приготовлялись две
суспензии, содержащие такой керамический порошок, как цир-
кон, причем в одной суспензии порошок находился в концент-
рированном стабилизированном щелочью золе коллоидного
кремнезема, а в другой — в положительно заряженном, покры-
том оксидом алюминия коллоидном кремнеземе. Литейная
форма попеременно окуналась в указанные смеси или обрызги-
валась последними, причем каждая смесь вызывала коагуляцию
и гелеобразование в другой смеси [523]. Положительно заря-
женный шлам может стабилизироваться таким комплексообра-
зующим реагентом, как лимонная кислота [524]. Другие
методы включают попеременное окунание формы в керамиче-
ские шламы коллоидного кремнезема или силиката натрия,
а основная соль алюминия используется или как коагулянт
[525], или как поликатионный агент, вызывающий отстаивание
[526]. Формовочные стержни для литейных изложниц могут
приготовляться с добавлением коллоидного кремнезема в ка-
честве связующего для формовочной смеси и кремнеземного
порошка наряду с добавлением агентов застудневания и сма-
чивания [527].
Коллоидный кремнезем — концентрированные золи
587
Добавки в различные материалы
Некоторые виды отделочных материалов, например гипс,
могут составляться с добавлением содержащего воду коллоид-
ного кремнезема, что позволяет получать гипсовые слепки
превосходной прочности [528, 529]. Однако экспериментальные
исследования показывают, что коллоидный кремнезем не может
добавляться к содержащим кальций цементным смесям, кото-
рые оказываются слишком щелочными, поскольку под воздей-
ствием кальция при высоких значениях pH кремнезем начинает
застудневать. В портландцементе это вызывает вспучивание и
растрескивание [530], но песок или волокна можно покрывать
коллоидным кремнеземом, чтобы предотвратить миграцию вред-
ных примесей или растворенных веществ в такой цемент [531].
Огнестойкий термоизоляционный материал низкой плот-
ности может связываться с коллоидным кремнеземом. Одним
из видов такого материала является формующаяся пена, со-
стоящая из растворимого силиката и коллоидного кремнезема,
причем вспенивание достигается за счет добавления катионного
ПАВ. В пену можно включать полиалкилсилоксан для улучше-
ния водостойкости. Другим распространенным материалом
является перлит, связанный с растворимым силикатом и кол-
лоидным кремнеземом. Этот материал особенно пригоден как
заменитель асбестовой изоляции на стальных трубопроводах
[532—534]. При добавлении аммиака к 45 %-ному золю крем-
незема получается пластичный гель, который перемешивается
с перлитом, после чего материал высушивается. Такой способ
дает возможность получать прочные легкие изделия [535].
С целью формирования абразивных частиц алмазный поро-
шок связывается в гранулы большего размера добавлением
коллоидного кремнезема; алмазный порошок, суспендированный
в 30 %-ном золе кремнезема, вводится в перемешиваемый
2-этил-1-гексанол; затем порошок погружается в бутанол.
Благодаря этому способу получаются гранулы кремнезема
с внедренными в них алмазными частицами. Для придания
твердости такие гранулы спекаются при 500°С [536].
Эффекты трения
Высокий коэффициент трения, такой, который наблюдается
между поверхностями наждачной бумаги, однако, без видимых
шероховатостей, может быть получен, если использовать раз-
нообразные поверхности с нанесенным на них коллоидным
кремнеземом. Одним из первых приложений коллоидного крем-
незема, уже давно используемых в большом объеме, оказалось
588
Глава 4
его добавление в содержащую воск мастику для натирки полов,
с тем, чтобы такие полы были менее скользкими [537, 538].
Пауерс и Харрисон [539] применили коллоидный кремнезем
для обработки неплотно прилегающих волокон перед процес-
сом их прядения, чтобы понизить эффект проскальзывания и
повысить предел прочности на растяжение.
Волокна
Были опубликованы многочисленные патенты, касающиеся
специфических составов или условий применения коллоидного
кремнезема к волоконным тканям для улучшения их фрикцион-
ных свойств [539—551]. Достаточно высокий коэффициент тре-
ния получается, когда коллоидный кремнезем с нанесенным на
него ПАВ типа катиона четвертичного аммония применяется
для обработки текстиля [552]. Вследствие подобных эффектов
коллоидный кремнезем нашел также применение при обработке
шерсти [553, 554].
Другое применение — нанесение кремнеземного покрытия на
органическое волокно, когда нить должна подвергаться пиро-
лизу с целью формирования новой химической структуры, но
при этом в процессе температурного воздействия в течение
определенного периода такое волокно необходимо поддержи-
вать механически, по мере того как оно проходит через пла-
стичное состояние. Бернетт и Загер [555] покрывали полиакри-
лонитриловые волокна коллоидным кремнеземом, чтобы обес-
печивать их механическое усиление до тех пор, пока в процессе
нагревания волокно приобретет новое состояние—структуру
с поперечными связями, способную самостоятельно поддержи-
вать необходимую механическую прочность. Благодаря улучшен-
ным фрикционным свойствам волокон ткани получаются более
прочными к истиранию [556]. Для применения к волоконным
тканям пирогенный кремнезем предварительно диспергируется
в воде с добавлением ПАВ [557]. Благодаря нанесению окра-
шенных оксидов металла с добавлением коллоидного кремнезема
и с последующим нагреванием для придания такому покрытию
прочного связывания с подложкой предотвращается эффект
проскальзывания стеклянных волокон и одновременно при-
обретается стойкое окрашивание поверхности волокна [558].
Чтобы не допускать проскальзывания нитей в узелках при
изготовлении рыболовных сетей из найлона, на такие узлы
наносится смесь, состоящая из коллоидного кремнезема с до-
бавлением CH3[H2N(CH2)4]Si(OEt)2 и воды [559].
Коллоидный кремнезем — концентрированные золи
589
Бумага
Необходимость увеличения трения между наполненными
бумажными мешками, сложенными в кучу в кузове грузового
автомобиля, совершенно очевидна для водителя, в практике
которого встречаются случаи, когда его груз перемещается
к одному борту. Уилсон [560] повысил фрикционные свойства
посредством обработки поверхности бумажных контейнеров
коллоидным кремнеземом, после чего наполненные бумажные
мешки не скользили при движении автомобиля [560]. Когда
стали применять коллоидный кремнезем при обработке бу-
маги, идущей на изготовление бумажных контейнеров, то воз-
никла проблема контроля за нанесением кремнезема на бумагу.
Тэрнер [561] использовал бесцветный лактоновый состав,
который распылял на бумагу; при этом в присутствии коллоид-
ного кремнезема бумага приобретала голубой цвет. Фирма
National Cash Register Со. [562], используя тот же самый тип
реакции, которая делает возможным применение бумаги, «не
реагирующей на углерод», разработала систему копирования.
Бесцветный лактоновый краситель не дает , синего окрашива-
ния с целлюлозой, но при контакте с бумагой, на поверхность
которой нанесены частицы коллоидного кремнезема или глины,
несущие водородные ионы, образуется контрастное синее окра-
шивание. Бесцветный лактоновый краситель может также
найти применение для разработки способа тайного печатания
на поверхности бумаги с нанесенным на нее коллоидным крем-
неземом. Добавление к золю кремнезема некоторого количества
тонкодисперсной глины, а также антипенной присадки и сма-
чивающего реагента повышает фрикционные свойства крафт-
бумаги при обработке ее такой смесью [563]. Воссос [564]
запатентовал использование больших по размеру (вплоть до
60 нм) частиц коллоидного кремнезема. Были составлены
рецептуры, которые при смешивании коллоидного кремнезема
с полиэтиленгликольдиолеатом, глицерином и глутаральдегидом
придавали бумаге превосходные свойства, предотвращающие
ее скольжение [565]. Для того чтобы коллоидный кремнезем
мог использоваться в качестве загустителя бумаги при одно-
временном сохранении большего количества кремнезема на ее
поверхности, водный коллоидный кремнезем загущается путем
добавления к нему диспергирующего агента, представляющего
собой кремнеземный порошок с низкой плотностью [566].
Флетчер [567] в своем обзоре показал возможности примене-
ния и характеристики подобных покрытий, применяемых для
устранения эффектов скольжения бумаги.
590
Глава 4
Стальные рельсы
Коллоидный кремнезем -улучшает тяговое усилие локомо-
тивов, особенно когда на рельсах присутствуют масляные
пленки. Рецептуры смесей, наносимых на рельсы, включают
коллоидный кремнезем, вводимый в смешивающийся с водой
органический растворитель, и смачивающие реагенты [568].
Другие поверхности
Скользящие детали клавиатуры музыкальных инструментов
могут обрабатываться коллоидным кремнеземом для того,
чтобы повысить «площадь соприкосновения» и уменьшить про-
скальзывание пальцев [572]. Качество поверхности чертежной
пленки из полиэтилентерефталата улучшается в отношении
способности воспринимать карандаш и чернила при примене-
нии коллоидного кремнезема [573]. Точно так же полиолефи-
новая пленка или бумага, покрытая полиолефиновой пленкой,
хорошо воспринимают чернила в результате применения кол-
лоидного кремнезема наряду с добавлением растворимого
в кислоте пленкообразующего вещества [574].
Незагрязняющиеся поверхности
Пленка коллоидного кремнезема, нанесенная на поверх-
ность волокон, например на волокна ковровых изделий, значи-
тельно снижает захват загрязняющих частиц и придает более
чистый внешний вид ковру после обработки пылесосом. Требо-
валось, чтобы коллоидный кремнезем создавал гладкую, прочно
удерживаемую на волокне пленку, к которой не должны при-
липать частицы грязи, особенно в связи с необходимостью
заполнения зазоров и щелей на поверхности волокна, которые
в противном случае заполнялись бы темными частицами за-
грязнений. Схожие эффекты наблюдались на раскрашенных
красками поверхностях, на синтетических пластмассовых тка-
нях, оконных шторах и обоях [575]. Такого рода использова-
ние коллоидного кремнезема было запатентовано Когованом
и Фредеричи [576]. Флорио и Рейнард [577] сообщили о преи-
муществе применения двух водных коллоидных оксидов метал-
лов одновременно. Для повышения адгезии коллоидного крем-
незема к волокнам предлагалось добавлять в препарат раство-
римый фосфат алюминия [578].
Рашер и Иетс [579] описали теорию заполнения коллоид-
ным кремнеземом субмикроскопических поверхностных неров-
ностей, которые имеются, например, на раскрашенных поверх-
Коллоидный кремнезем — концентрированные золи 591
ностях. Чтобы понизить степень загрязнения гидрофильной
поверхности, такой, как поверхность обивочной ткани, придать
ей гидрофобные свойства и устойчивость против загрязнений,
полидиметилсилоксановое масло смешивают с коллоидным
кремнеземом, используя при этом в качестве эмульгатора ме-
таллическое мыло. Такой смесью обрабатывают ткань и затем
высушивают [580]. Золь кремнезема с улучшенными свойст-
вами для получения покрытий, предохраняющих ткани от
загрязнений, получают посредством подкисления коллоидного
кремнезема и добавления небольшого количества алюминие-
вой соли муравьиной кислоты, чтобы придать кремнезему
прочное связывание с волокнами [581]. Чтобы понизить
жесткость, а также улучшить стойкость против загрязнений,
можно использовать комбинированный состав из коллоидного
кремнезема и коллоидных частиц глины, такой, например, как
монтмориллонит [582].
Даже металлические поверхности можно обрабатывать
путем адгезии углеродной сажи, что повышает их устойчивость
против загрязнений. Металлические полоски от подъемных
жалюзи покрывают коллоидным кремнеземом, нагревают до
120°С и затем полируют тальком [583]. Препарат для быстрой
очистки ковров составляют из коллоидного кремнезема и диа-
томовой земли [584]. Для получения водо- и грязеотталки-
вающих покрытий для тканей предусматривается их обработка
коллоидным кремнеземом и метакрил'атхромхлоридом марки
Volan (торговая марка фирмы Е. I. du Pont de Nemours
and Co.) вместе c 1,1-дегидроперфтороктаноловой смолой,
полиэтиленгликолем, глиоксалем и ацетатным буферным рас-
твором в смешанных спиртах. После высушивания при 150°С
ткань имеет высокую устойчивость против загрязнений [585].
В том случае, когда обрабатывают такой материал, как найлон,
с целью придания ему грязеотталкивающих свойств и пред-
отвращения накапливания статических электрических зарядов,
предпочтение отдается положительно заряженным частицам
оксида алюминия, покрытых слоем кремнезема [586]. Ферч
[587] обсудил основные положения использования коллоид-
ного кремнезема в качестве препарата для удаления статиче-
ских электрических зарядов.
Получение гидрофильных поверхностей
Кремнезем — одно из немногих веществ в природе, способ-
ных полностью смачиваться водой. Он становится гидрофоб-
ным только тогда, когда покрывается мылообразующими
многозарядными ионами металлов или органическими основа-
592
Глава 4
ниями. Наблюдаемая повсюду смачиваемость песков морских
побережий подтверждает, что поверхность кремнезема имеет
гидрофильный характер.
Способность поверхности смачиваться водой важна и при
печати литографским способом, когда печатающие поверх-
ности, содержащие масляные чернила, должны быть гидрофоб-
ными и смачиваться чернилами, но непечатающие поверхности,
способные смачиваться водой, должны быть постоянно гидро-
фильными и очень устойчивыми от постепенного накопления
на них масляных чернил. В этой области опубликовано мно-
жество патентов. Перенос масла, используемого при печати,
предотвращается разбрызгиванием коллоидного кремнезема
[588]. Гидрофильная природа печатных форм, используемых
при «плоской печати» для размножения газет, поддерживается
или восстанавливается за счет использования коллоидного
кремнезема [589, 590]. Новый тип литографской печатной формы
запатентован Марроном [591]: гидрофильная пленка коллоид-
ного кремнезема удерживает дисперсное, поглощающее чернила
вещество, которое выделяется на поверхности под давлением
или при нагревании для образования печатающих зон. Анало-
гичное действие предусматривается при офсетной плоской
печати [592].
Кенни [593] показал ценность водных оксидных коллоид-
ных систем для смачивания гидрофобных поверхностей водой.
Он заявил, что некоторые такие системы могут смачивать
любую из известных гидрофобных поверхностей без каких-либо
химических реакций. Однако Айлер нашел, что смачивание с ис-
пользованием золя кремнезема происходит на некоторых гидро-
фобных поверхностях только при низких значениях pH и при
оптимальном размере частиц кремнезема. После того как
изучаемую поверхность смачивали золем, промывали и высу-
шивали, эта поверхность смачивалась повторно только потому,
что на ней оставался адсорбированный монослой кремнеземных
частиц, связанных с поверхностью. Вид связи зависит от раз-
новидности гидрофобной поверхности. Например, металличе-
ская поверхность, являющаяся гидрофобной вследствие адсор-
бированной пленки, состоящей из жирных кислот (смазки),
становится гидрофильной в результате того, что кремнезем
замещает некоторую долю жирной кислоты и оказывается
связанным с оксидной поверхностной пленкой металла. Поверх-
ность будет оставаться гидрофильной до тех пор, пока на ней
в достаточной степени размещены частицы кремнезема. Такое
явление имеет место главным образом при нейтральном или
низких значениях pH и ускоряется в присутствии способных
смешиваться с водой органических растворителей (например,
спирта), которые помогают удалять жирную кислоту.
Коллоидный кремнезем — концентрированные золи 593
С другой стороны, органические полимерные пленки, такие,
как полиэфир или полиамид, оказываются гидрофобными, по-
скольку полярные группы обычно направлены внутрь, что не
позволяет образоваться взаимным водородным связям. Когда
же вводится золь кремнезема при pH 2—4, то амидные связи
или концевые аминогруппы, если они имеются, поворачиваются
наружу и образуют водородные связи с кремнеземом. В случае
полиэфира коллоидный оксид алюминия оказывается наилуч-
шим смачивающим реагентом, так как он вступает в- реакцию
с карбоксильными концевыми группами. На поверхности поли-
тетрафторэтилена в реакции участвуют карбоксильные конце-
вые группы фторкарбонового ПАВ, обычно применяемого
в эмульсионной полимеризации, которые и обеспечивают сма-
чивание коллоидными оксидами. Грот [594] заявил, что для
того, чтобы получить пористую гидрофильную мембрану из
политетрафторэтилена, коллоидный кремнезем следует вводить
на поверхность полимера с тем, чтобы обеспечить проводимость
ионов через поры.
Модифицирование адгезионных поверхностей
Коллоидный кремнезем можно использовать на разных по-
верхностях либо с целью повышения их адгезии по отношению
к другим веществам (фактически путем огрубления поверх-
ности, если вводимый кремнезем оказывается сцепленным
с поверхностью), либо с цельй понижения адгезии в других
случаях посредством удерживания на расстоянии способные
к «слипанию» поверхности.
Усиление адгезии
Если частицы кремнезема прочно сцеплены с поверхностью
или включены в поверхность, то субмикроскопические неров-
ности и полярность такой поверхности возрастают и адгезия
второго вещества обычно улучшается. С другой стороны, если
кремнезем присутствует в виде рыхлого, несвязанного покры-
тия или же кремнезем применяется в смеси с силоксаном или
с фторуглеродным полимером, то адгезия второго вещества
понижается.
Частицы кремнезема включают в поверхность полиэтилено-
вой пленки, чтобы улучшить адгезию покрытий термопластиче-
ских полимеров [595]. Поверхность фторуглеродного полимера
становится сцементированной в результате нанесения покры-
тия, состоящего из смеси диспергированного политетрафтор-
этилена и коллоидного кремнезема, и последующего нагрева-
ния такой поверхности при температуре свыше 500°С в течение
12 Заказ № 250
594
Глава 4
нескольких минут [596]. На пергаментной бумаге, используе-
мой для разделения невулканизированных листов резины, адге-
зия, как это требуется по условиям, должна была происходить
лишь на одной стороне бумаги, но не на другой. Поэтому одну
сторону бумаги обрабатывают коллоидным кремнеземом с кон-
центрацией 0,2—2,9 г/м2 поверхности и этой стороной накла-
дывают бумагу на листы резины. Бумага прилипает к резине,
и такую резину можно без опасений сворачивать в рулоны при
ее хранении [597].
Пленка полиэтилена прочно прилипает к бумаге при на-
греве, если бумага была покрыта слоем коллоидного кремне-
зема [598].
Покрытие, состоящее из политетрафторэтилена с добавле-
нием силиката щелочного металла и коллоидного кремнезема,
обладает улучшенными адгезионными и электроизоляционными
свойствами, особенно при нанесении на металлические поверх-
ности [599].
Адгезия окрашенных покрытий на стеклянных волокйах
улучшается путем первоначальной обработки волокон коллоид-
ным кремнеземом и их нагреванием при температуре немного
ниже температуры плавления [600]. Коллоидный кремнезем
также добавляется к клеям [601], а спрессованный со смолой,
используемой в качестве связующего, может применяться для
получения слоистых целлюлозных тканей [602].
Коллоидный кремнезем вместе с щавелевой кислотой нано-
сился и «сжигался» на поверхности железа и стали перед фор-
мированием химического покрытия из оксалата или фосфата.
Этот исходный продукт, состоящий из оксалата и реакционно-
способного кремнезема, вероятно, подвергался пиролизу с об-
разованием участков прочно связанного кремнезема и силиката
железа, которые затем закрепляли химическое покрытие [603].
С целью обеспечения повышенной адгезии к органическим
полимерам кремнеземное покрытие на металлической подложке
готовится путем смешивания коллоидного кремнезема с раз-
бавленным растаором аммониевой соли карбоксильного поли-
мера при pH 6,5 и последующим наложением смеси в виде
пленки на поверхность черного металла. После высушивания
благодаря органическому полимеру сводится к минимуму обра-
зование сетки волосных трещин на таком кремнеземном покры-
тии. Может быть применена альгиновая кислота [604].
Хромовая кислота используется для формирования ингиби-
рующей противокоррозионной пленки на поверхности цинка
или оцинкованного железа. Пленка с улучшенными свойствами
для более надежной защиты от коррозии с хорошей адгезией
красочных покрытий получается при смешивании коллоидного
кремнезема с раствором Н2СгО4 [605]. По-видимому, хромовая
Коллоидный кремнезем — концентрированные золи
595
кислота образует смесь гидратов Ре2О3 и Сг2О3, в которой
кремнезем способен коагулировать и отлагаться на поверх-
ности. Адгезия политетрафторэтилена к металлической поверх-
ности кухонной посуды улучшается посредством включения
коллоидного кремнезема в исходное покрытие [606]. В поли-
эфирном стекловолокне послойная адгезия между смолой и
стекловолокном может быть повышена в результате смешива-
ния коллоидного кремнезема с обычным ненасыщенным алкил-
аминосиланом, который представляет собой вещество для от-
делочных покрытий. При этом поверхность покрывается
частицами коллоидного кремнезема, на которых имеются реа-
гирующие с полимером поверхностные группы [607]. Элмес
[608] утверждает, что введение в стекловолокнистый шинный
корд сложного состава из коллоидного кремнезема в смеси
резорцинол—формальдегид с добавлением полибутадиенсти-
рол-2-винилпиридина, дает возможность получить более проч-
ную адгезию корда к смесям из натурального и синтетического
каучука [608]. Саката и Танака [609] сообщили, что получено
четырехкратное повышение адгезии полиэфирного шинного
корда к смесям из натурального и синтетического каучука,
когда такой корд предварительно обрабатывался хлорфенолом,
а кремнезем в диспергированном виде добавлялся в каучук
перед его вулканизацией.
Введение 2,6 % коллоидного кремнезема в полиолефиновую
смолу перед полимеризацией с. последующим травлением
с целью выделения кремнезема на поверхности формируемого
изделия позволяет покрывать такой полимер металлом с по-
мощью электролиза [610].
Понижение адгезии
Может показаться неожиданным, что кремнезем может не
только повышать адгезию к поверхностям, но и понижать
адгезию между подобными поверхностями. Хорошо известно,
что добавление небольшого количества сухого кремнеземного
порошка с низкой плотностью предотвращает спекание грану-
лированных материалов; это рассматривается в качестве при-
мера в гл. 5. В этом разделе будут показаны примеры, в кото-
рых золи, добавляемые к различным материалам, вызывают
понижение адгезии.
Маленькие стеклянные шарики становятся непылящими
в потоке благодаря обработке поверхности шариков смесью,
состоящей из водного коллоидного кремнезема, длинноцепочеч-
ной четвертичной аммониевой соли, содержащей алкильные
группы, и изопропилового спирта [611].
12*
595
Глава 4
Коллоидный кремнезем введен в Венгрии в перечень средств
для приготовления фармацевтических препаратов как офи-
циально утвержденный ингредиент [612]. Благодаря ему стало
возможным приготовлять разнообразные лекарства в дисперги-
рованной твердой форме.
При восстановлении железной руды в псевдоожиженном
кипящем слое частицы руды имеют тенденцию к слипанию
вместе еще до того, как закончится процесс восстановления.
Нанесение кремнеземной пленки на частицы решает эту про-
блему [613].
Небольшие частицы кремнезема на поверхности полиэфир-
ной пленки действуют как прокладки, предохраняющие пленку
от склеивания и слипания. В этом случае кремнезем может
осаждаться из паров при протекании реакции между SiCK
и Н2О [614]. Слипание листового целлофана не происходит,
если его поверхность обрабатывается диспергированным пиро-
генным кремнеземом в водном растворе полиэтиленимина или
кремнеземом, смешанным с меламинформальдегидной смолой
на стадии ее растворения [615]. Возможно, что наблюдаемый
эффект противослипания материалов объясняет, почему кол-
лоидный кремнезем, как отмечал Бретон [616], в охлаждаю-
щих агентах, применяемых при резке металлов, предотвращает
возможную сварку стружки и улучшает качество обрабатывае-
мой поверхности.
Композиционные покрытия
Органические составы, используемые для различных по-
крытий, с улучшенными адгезионными, прочностными, электри-
ческими свойствами и стойкие в течение длительного времени
получаются в результате Добавления коллоидного кремнезема
к органическим полимерным дисперсным системам [617—623].
В неорганических покрытиях кремнезем может использо-
ваться как основной компонент или как связующее вещество
в смесях. Для покрытий, состоящих главным образом из кол-
лоидного кремнезема, проблема заключается в том, каким
образом предотвратить усадку и образование сетки волосных
трещин. Этот вопрос обсуждался в связи с рассмотрением
прочности гелей. Почти невозможно себе представить, что
можно получить толстое кремнеземное покрытие, которое не
являлось бы пористым. Если исходить из измельченного в по-
рошок «плавленого кварцевого» стекла с отсортированными по
размеру частицами для получения плотной упаковки и суспен-
дировать такой порошок в минимально возможном количестве
концентрированного золя коллоидного кремнезема, также
имеющего несколько размеров частиц для плотной упаковки,
Коллоидный кремнезем — концентрированные золи 597
то можно получить высушенное изделие, имеющее максималь-
ную прочность и минимальную пористость, которое проявляет
минимальную усадку в процессе последующего спекания при
1000°С или даже выше [219].
Свойство коллоидного кремнезема образовывать пленки
улучшается путем введения диспергированных микроволокни-
стых материалов в золь, чтобы добиться минимального обра-
зования сетки волосных т-рещин. Барбарас [624] утверждает,
что целесообразно использование хризотил-асбеста, который
в форме коротких волокон может диспергировать до коллоид-
ного состояния при введении его в стабилизированные щелочью
золи кремнезема. В этом случае образование волосных трещин
также сводится к минимуму, и прочность сетки улучшается за
счет добавления некоторых водорастворимых полимеров, хотя
многие полимеры оказывают вредное воздействие. Было за-
явлено, что желатин [625], способные полимеризоваться цик-
лические сульфониевые амфотерные ионы [626] и не вызываю-
щий гелеобразования агент флокуляции, такой, например, как
линейный углеводородный полимер с четвертичными имидазо-
лами в боковых цепях [627], оказывают благотворное влияние.
Образование сетки волосных трещин также сводится к ми-
нимуму, если покрытие медленно образуется'посредством раз-
брызгивания золя на слегка нагретую поверхность [628].
Неорганическое красочное покрытие для асбестовых пане-
лей может изготовляться с добавлением коллоидного кремне-
зема в качестве связующего. Прочное водостойкое красочное
покрытие приготовляется при комбинировании коллоидного
кремнезема, гидроксида лития и силиката калия или фосфата
щелочного металла, глины и пигмента, и вся эта смесь спе-
кается в присутствии водяного пара [629]. Коллоидный крем-
незем, стабилизированный силикатом тетраэтаноламмония,
применяется как связующее для оксида железа и глинистых
пигментов [630]. Термостойкое покрытие, необходимое при
работе с асбестом или металлами, образованное из коллоид-
ного кремнезема и кислого фосфата магния, затвердевает при
200°С [631].
Стальные рабочие поверхности, находящиеся в горячих
условиях, покрывают тонкодисперсным порошком металличе-
ского хрома, связанного с коллоидным кремнеземом, чтобы
предотвратить образование окалины, если металл обрабаты-
вается горячей прокаткой при 1090°С [632]. Когда такие
металлы, как сталь, цинк или алюминий погружаются в кислый
золь кремнезема, содержащий сахар, то на металле осаждается
невидимая пленка кремнезема, и после соответствующей теп-
ловой обработки стойкость металла в отношении химических
воздействий повышается [633]. Коллоидный кремнезем играет
598
Глава 4
роль защитного покрытия при гальванической обработке
стали [634].
В том случае, когда горячий металл подвергается закалке
в воде, скорость его охлаждения высока, но если используется
золь кремнезема, то скорость закаливания металла заметно
снижается. В соответствии с этим при последующем отпуске
стали скорость охлаждения можно контролировать, если
использовать разные концентрации кремнезема. Этот эффект,
без сомнения, связан с образованием изолирующего кремне-
земного покрытия на горячей металлической поверхности [635].
Покрытие, предохраняющее сталь от коррозии, получается
из смеси, состоящей из коллоидного кремнезема, соединений
трехвалентного или шестивалентного хрома и катионов щелоч-
ного металла, взятых в определенных соотношениях с после-
дующим отвердением слоя [636].
Очень тонкий изоляционный слой может размещаться
между металлическими проводящими слоями в слоистых много-
канальных проводниках. Золь кремнезема наносится на са^ый
нижний проводящий слой, после чего получается пленка тол-
щиной 10 нм. Операция повторяется несколько раз с тем,
чтобы все поры были затянуты, и верхний проводящий слой
накладывается на такой непроводящий, лишенный пор слой.
В кремнеземной пленке могут быть оставлены окошечки,
если требуется наличие проводника между непроводящими
слоями [637].
Изоляционные покрытия требуется также наносить на пла-
стины из магнитной стали, применяемые в пластинчатых
сердечниках трансформаторов. Робинсон [638, 639] изобрел
составы для покрытий, в которых коллоидный кремнезем яв-
ляется связующим для огнеупорных материалов, таких, напри-
мер, как оксид магния. В другом типе покрытия, предназначен-
ного для тех же целей, используются коллоидный кремнезем
и фосфат аммония, причем к последнему Добавляется Р2О5 для
проведения реакции с кремнеземом и связывания его со сталью
[640]. Чистое кремнеземное покрытие наносится на сталь
посредством добавления к коллоидному кремнезему желатина
в качестве временного связующего в пропорции 1,5—3 ч. жела-
тина на 8 ч. кремнезема. Этот состав, накладываемый в виде
пленки, может затем нагреваться до распада желатина, и на
поверхности стекла или металлов остается чистое кремнеземное
покрытие. Улучшенное сцепление покрытия с металлами полу-
чается в том случае, когда поверхность металла сначала
покрывается пленкой коллоидного кремнезема. При последую-
щем нагревании металла на воздухе пленка улучшается и
образуется непроницаемое, хорошо сцепленное конечное покры-
тие, способное снизить дальнейший процесс окисления металла
Коллоидный кремнезем — концентрированные золи
599
при высокой температуре и улучшить устойчивость против
коррозии [641]. Декоративное покрытие получается на метал-
лических изделиях следующим способом: сначала проводят
анодирование поверхности в щелочном растворе, содержащем
коллоидный кремнезем, при этом формируется очень тонкая,
невидимая изоляционная пленка на определенных участках
поверхности, образуя неразличимый узор; затем вводят сверху
этого слоя блестящий никелевый электрод, благодаря чему
удается проявить невидимые ранее узоры на обрабатываемой
поверхности [642].
Устойчивые против коррозии покрытия могут получаться,
если предусматривается последующее нагревание накладывае-
мого на поверхность полимера для его расплавления или же
превращения в непроницаемую пленку. По первому способу на
металл наносится смесь из фенольной смолы и коллоидного
кремнезема с добавлением полиолефинов, затем смесь расплав-
ляется на поверхности [643]. Четвертичный аммониевый поли-
силикат (Quram 200), смешанный с акрилэтиленовым сополи-
мером и этиламином, прогревается на стальной подложке при
300°С, а затем прокаливается при 800°С в восстановительной
атмосфере и образует черное, прочно сцепленное блестящее
покрытие с хорошими изоляционными свойствами [644].
Фторуглеродное покрытие может быть нанесено на металл
или на керамику совместно с метилцеллюлозой и коллоидным
кремнеземом для получения хорошей адгезии [645]. В подоб-
ных покрытиях для кухонной посуды, как правило, предусмат-
ривается использование коллоидного кремнезема или полиси-
ликата [646—648].
Для проводящих электрический ток покрытий, которые
требуется использовать при повышенной температуре, в основ-
ном применяют графит, связанный с коллоидным кремнеземом.
Было предложено [649] использовать смесь кремнезема и гра-
фита, взятых примерно в равных частях, для получения
проводящей пленки на листах бумаги или асбеста, через кото-
рые пропускается электрический ток, выделяющий тепло до
3 Вт на площади 1 см2. Электрическое сопротивление поверх-
ности составляет 1,5—150 Ом/см2. Автор обнаружил впослед-
ствии, что при нагревании такого покрытия при 150°С на него
может наноситься клей из силиката натрия для склеивания
с асбестовым покрытием без изменения исходного сопротив-
ления [650].
В тканях из стекловолокна, используемых для нанесения
типографских красок и чернил, требуется покрытие коллоид-
ным кремнеземом либо в виде грунтового слоя [651], либо
путем включения кремнезема в связующее вещество из органо-
силана для нанесения пигмента [652].
600
Глава 4
При многослойных покрытиях на бумаге слой коллоидного
кремнезема может действовать подобно резервному слою,
сохраняющему вступающие в реакции соединения. Так, в чув-
ствительных к давлению покрытиях основной кремнеземный
слой удерживает соли меди и никеля, которые вступают в ре-
акцию и образуют окрашивание, когда в выше расположенном
слое под давлением разрываются оболочки микрокапсул, со-
держащих дитиоамидоксим [653].
Покрытия на кораблях, танкерах
Специальный тип покрытий используется для стали, сопри-
касающейся с сырой нефтью и морской водой, как это имеет
место в морских танкерах, транспортирующих нефть. Такое
покрытие включает неорганическую краску, содержащую цин-
ковый порошок, который обеспечивает катодную защиту от
коррозии на локализованном участке стали. Этот вопрос уже
рассматривался в гл. 2 в связи с применением силикатов лития.
Некоторые дополнительные положения заключаются в сле-
дующем.
Как указывалось в некоторых ранее опубликованных па-
тентах, в красках с большим содержанием цинка важное зна-
чение имеет кремнеземный компонент. Коллоидный кремнезем
вступает в реакцию с тонкодисперсным цинком с образованием
силиката цинка в коллоидной форме, в котором суспендирован
избыточный металлический цинк [654]. Водонерастворимое
связующее для содержащих цинк покрытий приготовляется
смешиванием стабилизированного щелочью коллоидного крем-
незема и гидроксида лития, взятых в соответствующих про-
порциях [655]. Для избежания выделения газа из смесей кол-
лоидного кремнезема с цинковым порошком в них вводятся
красители — индигоидные соединения [656]. По другому ре-
цепту предусматривается введение полисиликатного раствора
четвертичного аммония, размолотого вместе с оксидом свинца,
играющего роль связующего для цинка [657]. Некоторые про-
блемы возникают от различных примесей, содержащихся
в цинковом порошке, способных промотировать реакцию с окру-
жающей систему средой [658]. Адгезия красителей такого типа
по отношению к стальной поверхности улучшена за счет добав-
ления стиролакриловой смолы вплоть до 2 % в диспергиро-
ванном виде [659].
Необрастающая в морской воде водорослями поверхность,
окрашенная краской для кораблей, содержащей оксид трибу-
тилолова, связывается с коллоидными кремнеземом и дисперги-
рованной смолой, причем поверхность кремнезема превра-
Коллоидный кремнезем — концентрированные золи 601
щается в органофильную посредством добавления катионного
ПАВ [660].
Армирование органических полимеров
Упрочняющее или армирующее влияние, которое оказывает
коллоидный кремнезем в органических полимерах, пленках и
волокнах, изменяется в таких широких пределах, что подобные
воздействия не классифицированы. Коллоидный кремнезем
включался в полиолефины [661], в термопластические органи-
ческие полимеры [662], полиамиды [663] и в другие типы
полимеров [664]. Армирование полисилоксанов коллоидным
кремнеземом в разнообразных формах осуществляется весьма
специализированными технологическими способами, которые
выходят за пределы настоящей монографии. Благодаря сопо-
лимеризации коллоидного кремнезема и растворимого поли-
эфирного силиката образуется прочная водонепроницаемая
масса [665]. Упрочнение резины посредством введения кремне-
земных порошков представлено в гл. 5. Водные золи кремне-
зема используются в резиновой промышленности в основном
для загущения резины с открытыми ячейками, находящейся во
вспененном состоянии. Такой кремнезем, осажденный на стен-
ках пор, очевидно, оказывает фрикционное действие, делая
пену менее легко сжимаемой и, таким образом, повышая допу-
стимую несущую нагрузку [666—668]. Введение всего лишь
3 % SiO2 повышает сопротивление на сжатие примерно на 90 %
[669]. Наилучшие результаты были получены с золями кремне-
зема, не содержащими примеси металлов и с размером частиц
золя всего 1—3 нм в диаметре по сравнению с частицами
диаметром 8 нм [670].
Пленки из поливинилового спирта могут загущаться и пере-
водиться в нерастворимую форму в воде после введения кол-
лоидного кремнезема [671—673]. Твердый, способный формо-
ваться материал на основе ПВС приготовляется из смеси,
содержащей раствор полимера и коллоидный кремнезем, с до-
бавлением глицерина как пластификатора. Прочные, жесткие,
с высоким значением сопротивления удару листы из такого
материала получались горячим прессованием [674].
Полировальный реагент для кремниевых
пластин
В электронной промышленности пластинчатые заготовки
для полупроводников, отрезанные от монокристаллов кремния,
полируются до чрезвычайно высокой степени чистовой обра-
602
Глава 4
ботки с применением в качестве полирующей среды- коллоид-
ного кремнезема при высоких значениях pH. Даже самые
тончайшие стандартные абразивные порошки оказываются
слишком грубыми для таких целей. Уолш и Герцог [675] под-
робно описывают эту проблему. Улучшенный состав золя крем-
незема заявлен Сирсом, которому удалось стабилизировать
частицы кремнезема против их растворения в щелочной среде
путем нанесения на такие частицы покрытия, содержащего
алюмосиликатные анионные группы. Это дало возможность
работать при значениях pH 11 или даже выше с целью увели-
чения скорости полирования [676]. ,
Частицы кремнезема очень высокой чистоты и диаметром
10—20 нм формировались в результате гидролиза (СгНдОДЭ!
в системе бутанол—вода—аммоний. Осажденный кремнезем
высушивали и диспергировали при pH 11 в присутствии
(CH3)4NCH для получения полировального реагента (основа-
ние, вероятно, способствует подавлению процесса растворения
кремнезема при высоком значении pH) [677]. Частицы кремне-
зема размером менее 100 нм, находящиеся в шламе при pH 10,
как указывают авторы работы [678], загущались водораство-
римым производным целлюлозы. Суспензия, состоящая из тон-
чайших частиц осажденного кремнезема в растворе четвертич-
ного аммониевого силиката при pH 10, также может исполь-
зоваться для полирования [679].
Воздействие ПАВ
Окура, Гото и Мураи [680] представили обзор, в котором
основательно разобрали химические вопросы, связанные с фло-
кулирующим действием кремнезема. Проблемы коагуляции при
обработке кремнезема в водной среде в дальнейшем были
обсуждены в работе [681]. Несмотря на то что коммерческий
коллоидный кремнезем оказывает флокулирующее действие,
кремневая кислота, полимеризованная из смеси силиката нат-
рия и кислоты, оказывается гораздо дешевле и, вероятно,
и более действенной, если только она приготовляется при
контролируемых условиях. Использование подобных активи-
рованных золей кремнезема при флокуляции рассматривалось
в гл. 3.
Эффекты диспергирования
Эффекты диспергирования и суспендирования, в частности
при производстве эмульсионных полимеров, продолжают вызы-
вать интерес. Коллоидный кремнезем используется в качестве
диспергирующего агента при производстве определенного вида
Коллоидный кремнезем — концентрированные золи
603
органических сополимеров, что дает возможность, следова-
тельно, избегать применения органических ПАВ или веществ,
которые вызывают понижение поверхностного натяжения при
диспергировании. Высокое значение поверхностного натяжения
является определенным преимуществом при нанесении покры-
тий методами погружения или в потоке [682]. Эмульсионная
полимеризация на основе стирола или акриловых мономеров
с использованием коллоидного кремнезема в качестве агента
суспендирования позволяет избегать применения органических
ПАВ, которые уменьшают термическую устойчивость поли-
мера [683].
Когда коллоидный кремнезем частично покрывается адсор-
бированными катионными органическими молекулами, он ока-
зывается очень устойчивым эмульгатором для эмульсий масла
в воде. Вероятно, непокрытые участки частиц кремнезема
остаются в контакте с водной фазой, тогда как органофильные
участки (там, где адсорбированы мицеллы) находятся в кон-
такте с фазой масла. Широкий набор подобных катионных
веществ перечисляется в каталогах, и имеются описания их
применений при эмульсионной полимеризации органических
мономеров [684].
При микрокапсулировании стабилизирование эмульсии про-
исходит посредством образования прочных герметичных пленок
вокруг каждой капельки. Такие пленки можно получить, как
описывает Брокетт [685], за счет соединения коллоидного
кремнезема и желатина. По-видимому, в дальнейшем целе-
сообразно проводить исследование по взаимодействию коллоид-
ного кремнезема или с полимерами в катионной форме, или
с катионными ПАВ в масляной среде, или с полимерными
эмульсиями и другими дисперсными фазами в воде, в особен-
ности с точки зрения образования систем с высокой степенью
стабилизации поверхности раздела фаз.
Моющие средства могут быть улучшены путем добавления
кремнезема. При удалении масла с поверхности стали колло-
идный кремнезем, возможно, адсорбируется на оксидной
пленке, покрывающей металл, и помогает вытеснить жирные
кислоты. Таким образом, подкисленный золь кремнезема
в спирте или в других способных смешиваться с водой раство-
рителях обезжиривает поверхность металла [686]. Коллоидный
кремнезем может замещать фосфат в анионных моющих сред-
ствах, если он получается из силиката натрия и кислоты, обра-
зующей моющее средство [687].
Частицы коллоидных силикатов многовалентных металлов,
например частицы глины и асбеста, адсорбируют коллоидный
кремнезем, добавление которого приводит к улучшению таких
дисперсий. Вязкость системы, состоящей из диспергированных
601
Глава 4
частиц асбеста в воде, удается понизить без применения силь-
ной щелочи, подобной силикату натрия, только за счет введе-
ния кремнезема [688, 689].
Противопенное действие
Механизм, с помощью которого «неорганические наполни-
тели» оказывают противопенное действие в силоксановых жид-
костях, был изучен Повичем [690]. Он определил, что по мере
добавления кремнезема заметно повышается эффективность
этих жидкостей. Кремнезем не понижает давления в пене, но
значительное поглощение примесей или изменение вязкости
системы оказывается достаточным, чтобы вызвать наблюдаемый
эффект. Водный золь кремнезема можно использовать при его
эмульгировании в масле, при отгонке воды. Путем проведения
реакции кремнезема с полисилоксаном можно гидрофобизовать
кремнеземную поверхность [691]. Вместо силиконового масла
допустимо применение несмешиваемого с водой оксиэти^ен-
оксипропиленового полимера, к которому вместе с эмульгато-
ром добавляется коллоидный кремнезем [692].
Полагают, что противопенное действие имеет место, когда
большая гидрофобная частица попадает на стенки пузырька.
Вследствие низкой адгезии воды к поверхности такой частицы
последняя действует подобно «дырке», и поэтому пленка пу-
зырька разрывается. Глейм, Хентов и Башиков [693а] иссле-
довали кинетику разрушения пены. Кулкарни, Годдард и Кан-
нер [6936] изучили механизм антипенного действия кремнезема.
Модифицирование вязкости путем гелеобразования
Когда необходимо загустить жидкости, то обычно приме-
няются рыхлые, объемистые кремнеземные порошки, а не кон-
центрированные золи. Точно так же, как кремнезем, ставший
неполярным в результате его обработки гидрофобизующим
реагентом, агрегирует в воде с образованием густой массы, так
и гидрофильный кремнезем агрегирует в неполярных жидкостях
до состояния геля. Следовое содержание воды помогает «сце-
ментировать» гидрофильные частицы кремнезема вместе в трех-
мерную сетку в масляной среде. Уайтман и Чессик [694] иссле-
довали механизм разрушения геля путем введения определенных
добавок. Коллоидный кремнезем загущает сернокислотный
электролит в процессе хранения аккумуляторных батарей [695].
Порошок пирогенного кремнезема используется как загуститель
в полиэфирных смолах, маслах, эпоксидных клеящих вещест-
Коллоидный кремнезем — концентрированные золи
605
вах, полихлорвиниловых пастах и в материалах, применяемых
для уплотнения [696].
Необычный эффект наблюдается, когда частицы кремнезема
диспергированы с неионным ПАВ (типа полиэтиленоксида)
в нафтеновой жидкой фазе: когда жидкость между электро-
дами перемешивается, возникает электрический ток и, наоборот,
вязкость смеси повышается при пропускании тока [697].
Различные оптические эффекты, цветообразование,
фотография
Эффекты окрашивания «благородного опала» уже обсуж-
дались в связи с рассмотрением упорядоченной агрегации,
а появление интерференционных цветовых оттенков на одно-
родных слоях частиц кремнезема описывалось при рассмотре-
нии вопроса об адсорбции частиц на поверхностях.
Многочисленные патенты были опубликованы по покры-
тиям, подавляющим отраженный свет. В них кремнезем в раз-
ных формах осаждается в виде тонкого, хорошо сцепленного
с подложкой слоя, обычно на поверхности стеклянных или
пластмассовых пластинок или линз. Покрытие, полученное
с «низко гидратированным кремнеземом», было описано
в 1945 г. [698]. Пленки коллоидного кремнезема осаждаются
различными способами [699—704].
Пленка коллоидного кремнезема на стеклянной подложке
ослабляет яркий свет, когда правильно подобрана толщина
плёнки [705]. Как уже выше описывалось, Айлер разработал
метод наращивания полислоев, состоящих из однородных по
размеру коллоидных частиц, на различных поверхностях до
любой желаемой толщины [403, 404]. Было разработано про-
тивоотражающее покрытие, составленное из двух слоев, тол-
щины которых составляли одну восьмую длины световой
волны, причем один слой состоял из коллоидного кремнезема,
а другой — из фторированного полимера с показателем пре-
ломления 1,3—1,45 [706]. Различное содержание влаги вслед-
ствие ее конденсации в порах приводит к изменениям опти-
ческих свойств пленок, изготовленных из пористого кремнезема
[707]. Другие предложенные противоотражающие слои состоят
из Многослойных пленок, в состав которых входит ТЮ2 и SiO2
или SiO2, SnO2, ZrO2 [708, 709].
«Черное зеркало», прозрачное для инфракрасной области,
приготовляется путем, осаждения слоев никеля (частицы раз-
мером 10—15 нм) и SiO2 (80—90 нм); такое зеркало приме-
няется для исследований, связанных с поглощением солнечной
энергии [710]. С другой стороны, можно приготовлять пленки
на стеклянной подложке, которые обладают очень высокой
606
Глава 4
отражательной способностью; такие пленки получены осажде-
нием ТЮ2 и SiO2 [711]. Пленки толщиной более 1 мкм
склонны к растрескиванию и шелушению и отходят от стекла,
если только не содержат пористые слои и не предусмотрено
их нанесение на полированную подложку [712]. Как описали
Ведан и др. [713], методом эллипсометрии можно определять
как толщину тонких пленок, так и показатель преломления.
Чтобы получить интерференционные цвета разных оттенков,
пигмент из слюды покрывается TiO2 и SiO2 при правильном
подборе толщины слоя [714]. Покрытие внутри колб электри-
ческих ламп для получения рассеянного света без его погло-
щения формируется из коллоидного кремнезема [715, 716].
Слой кремнезема может также использоваться для связывания
'фосфора с внутренней поверхностью ламповых колб [717].
Покрытие для 'получения рассеянного света может быть при-
готовлено нанесением коллоидного кремнезема на стеклянную
подложку посредством распыления при 100—400°С [718]. Даже
кожу можно матировать. Косметический состав, содержащий
3—7 % коллоидного кремнезема, исключает необходимость
применения пудры [719].
Фотографическая и диазотипная бумаги покрываются кол-
лоидным кремнеземом [720—723]. В фотопленках типа поля-
роид коллоидный кремнезем играет существенную роль при
формировании слоя, в который переносится и затем разви-
вается позитивное изображение за счет почернения серебра.
Более подробно составы, включающие коллоидный крем.незем,
описаны в серии из восьми патентов Лэндом и др. [724].
Грэй [725] с целью восстановления серебра включил в слой,
в котором развивается изображение, кроме коллоидного крем-
незема силансодержащие соединения, в том числе одно соеди-
нение, содержащее связи кремний—водород.
В фотографии также используются поверхности на рентге-
новских пленках с нанесенными коллоидным кремнеземом и
отвержденным желатином [726], коллоидным кремнеземом
и модифицированными эмульсиями с галогенидом серебра для
повышения чувствительности [727], коллоидным кремнеземом
и поливиниловым спиртом, находящимся вместо желатина
в слоях, в которых развивается изображение [728].
я
Применение в биологических исследованиях,
измерение градиента плотности
Коллоидный кремнезем находит применение как плотная
среда для разделения биологических материалов методом
центрифугирования. Пертофт [729] сообщил об использовании
Коллоидный кремнезем — концентрированные золи
607
в своих исследованиях смеси коллоидного кремнезема лю-
докс-HS с полиэтиленгликолем, имевшим молекулярную массу
300—3700, в трис-буферном растворе при pH 7,5 по мере того,
как изменялись градиент плотности раствора в центрифуге
и степень выделения вируса. Этот общепринятый метод был
описан Пертофтом и Лаурентом [730]. Неповрежденные об-
разцы хлоропласта также разделялись в растворах людокса
Литлтоном [731]. Моргентхалер и др. [732, 733] использовали
людокс-АМ —• модифицированный алюминатом золь кремне-
зема, который стабилен в нейтральной среде и имеет меньшую
тенденцию образовывать комплексы с водородными связями
с некоторыми видами белка. Полиэтиленгликоли оказывали
некоторое воздействие на систему, вероятно, из-за конкури-
рующих эффектов образования водородных связей с другими
группами.
Наблюдались некоторые структурные изменения при раз-
делении клеток крови в среде коллоидного кремнезема [734].
Добавление поливинилпирролидона (ПВП) в качестве конку-
рирующего с кремнеземом агента, образующего водородные
связи, очевидно, предотвращает взаимодействие кремнезема
с клетками крови [735].
В своем первоначальном исследовании, предназначенном
для определения градиента плотности среды в растворе, Ма-
тейко и Копак [736] нашли, что коллоидный кремнезем —
единственно подходящий материал. Почти через четверть века
еще раз было подтверждено, что кремнезем пригоден для
таких исследований [737а]. Однако коллоидный кремнезем
еще имел тот недостаток, что показывал относительно слабое
взаимодействие с биологическим материалом во всей области
значений pH и не был способен удержать освобождаемый орга-
нический полимер. Недостаток, очевидно, удалось устранить
путем разработки специально составленного для такой задачи
типа кремнезема (марка Percoll) [7376], который представлял
собой комбинацию коллоидного кремнезема людокс-HS (диа-
метр частиц 17 нм) и поливинилпирролидона. В этом образце
поверхность частиц кремнезема была покрыта монослоем
адсорбированных молекул полимера. Соответствие выбранной
молекулярной массы полимера, оптимальное соотношение
между размером молекулы полимера и площадью поверхности
кремнезема и значение pH, равное 8,8, явились, вероятно,
необходимыми факторами, позволившими достичь такой хоро-
шей стабильности полученного концентрированного золя, что
его удалось стерилизовать посредством автоклавной обра-
ботки [737в—737д].
Эксперименты, выполненные Волффом и Пертофтом [737е],
показали, что ПВП не вытесняется с поверхности кремнезема
608
Глава 4
при воздействии полиэтиленгликоля с молекулярной массой ।
4000, который, как известно, также способен прочно связы-
ваться с поверхностью кремнезема. Другим интересным свой-
ством выбранной системы является то, что этот состав гораздо
меньше подвержен гелеобразованию или коагуляции в присут- j
ствии солей, чем исходный немодифицированный кремнезем,
и образец несет на поверхности лишь малый по величине (
ионный заряд или же такой заряд оказывается экранирован-
ным [7376].
Использование силикагеля в качестве питательной среды
рассматривается в гл. 7, поскольку такие кремнеземы приго-
товляются иным способом. Там же будут представлены и кол- 1
лоидные кремнеземы, применяемые как питательная среда. Г
Источники получения химически реакционноспособного «
кремнезема |
Коллоидный кремнезем гораздо более реакционноспособен,
чем большинство тонкодисперсных порошкообразных песков,
не только из-за того, что удельная поверхность коллоидного
кремнезема в несколько сотен раз больше, но и также вслед-
ствие того, что растворимость аморфного кремнезема в воде
примерно в 10 раз превышает растворимость кристаллического
кварца.
Растворимые силикаты
Кремнезем быстро деполимеризуется в присутствии сильной
щелочи. Таким образом, коллоидный кремнезем можно пре-
вратить в раствор полисиликата натрия, содержащий 4,2—
6,0 моль кремнезема на один моль оксида натрия [738], или
в раствор полисиликата лития и стабилизированный литием
золь с соотношением 4—25 моль кремнезема на один моль
оксида лития [739]. Такие составы не могут быть получены
посредством растворения силикатных стекол натрия или лития,
так же как не могут приготовляться растворением песка
в щелочи, поскольку силикат лития не растворим в горячей
воде. При более низких силикатных отношениях приготовление
растворов из коллоидного кремнезема имеет преимуществом
лишь большую скорость реакции.
Изделия из кремнезема
Так как очень небольшие частицы кремнезема спекаются при
относительно низкой температуре, разнообразные виды изде-
лий из кремнезема наиболее легко могут быть изготовлены
Коллоидный кремнезем — концентрированные золи 609
из коллоидного кремнезема. Кремнеземные сферические ча-
стицы с развитой пористостью приготовляют смешиванием
порошкообразного плавленого кварца и коллоидного кремне-
зема, содержащего гидроксильные поверхностные группы, и
формированием гранул, которые пропускают через зону на-
грева при температуре, равной 2000°С. Выделяющаяся при
этом вода образует поры внутри гранул [740]. До некоторой
степени сходный процесс для изготовления пустотелых сфер
SiO2 с использованием только одного коллоидного кремнезема
был запатентован Уолшем [741]. Кварцевые волокна более
легко приготовляются путем их скручивания из палочек, кото-
рые в свою очередь получены из порошкообразного кварца,
связанного коллоидным кремнеземом [742].
Для приготовления синтетических кварцевых кристаллов
коллоидный кремнезем мог использоваться в качестве источ-
ника растворимого кремнезема, получаемого при автоклавной
обработке в условиях высокой температуры [743]. При изго-
товлении горячим прессованием прозрачных изделий из квар-
цевого стекла вода, выделяющаяся из частиц коллоидного
кремнезема, выступает в роли катализатора, позволяющего
формировать полностью спекшиеся изделия [744]. Самое
прозрачное кварцевое стекло имело наиболее высокое содер-
жание остаточных групп ОН.
Оккерс и де Бур [745] дали общую оценку реакционной
способности аморфного кремнезема. Для' понижения темпера-
туры спекания кремнеземных порошков, предназначенных для
горячего прессования, Айлер ввел оксид бора при равномер-
ном распределении его по всей массе аморфного кремнеземного
порошка [746]. Чтобы исключить воду из порошка кремнезема
перед получением изделия, порошок можно нагревать в атмо-
сфере газообразного хлора при 600—1000°С [747]. Пленка из
чистого кремнезема вокруг стеклянного волокна с более высо-
ким коэффициентом термического расширения повышает проч-
ность "на разрыв в результате появления сжимающих напря-
жений в стекле при его охлаждении [748].
Составы стекол
Однородные стекла могут приготовляться посредством до-
бавления других компонентов, тщательно перемешиваемых
с коллоидным кремнеземом [749]. Химическая реакционная
способность коллоидного кремнезема играет роль при нанесе-
нии на стекло или огнеупорные материалы окрашенных или
проводящих покрытий, в которых образуется остеклованный
связующий материал без нанесения повреждений самой под-
ложке [558, 750].
13 Заказ № 250
610
Глава 4
Формирование твердых силикатов — цементы
Самая древняя из известных реакций аморфного кремне-
зема — это реакция приготовления цемента в Древнем Риме,
по которой известь смешивалась с песком и с раздробленным
до коллоидных размеров кремнеземом вулканического про-
исхождения, добывавшимся вблизи Поццуоли (Италия) и на
греческом острове Тира (Санторин). Это явилось основой для
приготовления чрезвычайно непроницаемого цементного обли-
цовочного материала, использовавшегося .в строительстве во-
доемов по всему Средиземноморью и в строительстве зданий
по всей территории Римской империи.
Без этого материала никогда не смогли бы быть построены
некоторые древние громадные величественные здания [751].
Песок и известь не смогли бы образовать подобного цемента.
Реакция между Са(ОН)2 и SiO2 изучалась в течение многих
лет. Прочность специфического соединения скоутита—силика-
токарбоната кальция 6CaSiO3-СаСО3-хН2О [752] была про-
демонстрирована тем фактом, что Айлер нашел вначале: при-
нятые за кварц кристаллы длиной 5 мм, которые выросли на
стенках внутри бутыли, изготовленной из легкоплавкого стекла.
В этой бутыли в течение 20 лет хранился 30 %-ный золь крем-
незема с размером частиц 14 нм при pH -—9,5. Колпачок от
бутыли был потерян и, по-видимому, в бутыль попадал угле-
кислый газ из атмосферы. . В скоутите кремнезем существует
в виде циклогексасиликатных анионов. Реакция извести и
кремнезема изучалась Гринбергом [753, 754] и Ассарссоном
[755]. Образование и свойства силиката кальция в портланд-
цементе исследовались и продолжают постоянно изучаться
вследствие очевидного практического значения.
Другие реакции и их практическое применение
Муллит может быть изготовлен при температуре ниже точки
плавления из коллоидного кремнезема и основного хлорида
алюминия [756]. Гидратация трикальцийалюмината ускоряется
благодаря применению небольших количеств коллоидного
кремнезема [757].
Реакция коллоидного кремнезема при получении силиката
магния замедляется при добавлении небольших количеств
оксида алюминия и зависит от скорости растворения кремне-
зема [758]. Кроме того, гидроксид магния понижает раствори-
мость кремнезема и замедляет реакцию между коллоидным
кремнеземом и солями магния [759].
Образование глиноподобных минералов из песка идет чрез-
вычайно медленно, если вообще такая реакция возможна, но
Коллоидный кремнезем — концентрированные золи 611
оно будет происходить при благоприятных условиях из кол-
лоидного или аморфного кремнезема. Синтез монтмориллонита
в автоклаве при 300°С показан Гранквистом [760]. Гекторит —
коллоидная форма силиката магния в виде похожих на лен-
точки кристаллов молекулярной толщины — был синтезирован
из геля кремнезема (вероятно, лучшим исходным веществом
был бы золь кремнезема) Гранквистом и Поллаком [761].
В том случае, когда приготовляются изоляционные мате-
риалы, в которых в качестве связующего используется известь,
то добавление коллоидного кремнезема повышает прочность,
если основными компонентами берутся песок или диатомовая
земля [762, 763].
Высокая реакционная способность коллоидного кремнезема
проявляется в реакциях с фосфорной кислотой [764]. Однако
фосфорная кислота будет адсорбироваться на поверхности
кремнезема, затормаживая такую реакцию, если только не
добавлять более сильную кислоту, например H2SO4 [765].
Коллоидный кремнезем, прокаленный в пламени, вступает
в реакцию с СН3Л1С12 с образованием метилхлорсиланов [766].
Такие металлы, как платина и вольфрам, закаливаются за
счет введения коллоидного кремнезема в соответствующий
гидроксид металла перед восстановлением или же в результате
адсорбции коллоидного кремнезема на поверхности тонкодис-
персного металла перед его уплотнением и спеканием
[767, 768].
Коллоидные силикаты
Хотя коллоидные силикаты и были рассмотрены в более
ранней монографии Айлера [8], библиография в этой области
чрезвычайно возросла и поэтому здесь не будет рассматри-
ваться. Шварцен-Аллен и Матиевич представили обзор по по-
верхностным свойствам и по коллоидной химии глин, сопро-
водив его превосходным библиографическим материалом [769].
ЛИТЕРАТУРА
1. Vail J. G., Soc. Chem. Ind. London, 44, 214 (1925).
2. Treadwell W. D., Wieland W., Helv. Chim. Acta, 13, 842 (1930).
3. Griessbach R., Chem. Ztg., 57, 253. 274 (1933).
4. Bird P. G., пат. CHIA 2244325 (National Aluminate Co.), 1941.
5. White J. F., пат. США 2375738 (Monsanto Chemical Co.), 1945.
6. Bechtold M. F„ Snyder О. E., пат. США 2574902 (Du Pont), 1951.
7. Rule J. M., пат. США 2577484, 2577485 (Du Pont), 1951.
8. Айлер P., Коллоидная химия кремнезема и силикатов. Пер с англ.—
М.: Госстройиздат, 1959.
9. Alexander G. В., пат. США 2750345 (Du Pont), 1956.
10. Atkins R. С., пат. США 3O12973'(Du Pont), 1961.
Н. Sippel R., пат. США 3440176 (Du Pont), 1959.
13*
612
Глава 4
12а. Allen L. S., англ. пат. 673196 (Monsanto Chemical Co.), 1952.
126. Napper D. H., Hunter R. G., Med. Tech. Publ., Int. Rev. Sci., Phys.
Chem. Ser. One, 7, 241 (1971) (Butterworth, London) (Chem. Abstr.,
78, 151867z).
13. Fyre W. S., McKay D. S., Am. Mineral., 47, 83 (1962).
14. Oehler I. H., Nature Phys. Sci., 241, (107), 64 (1973).
15. Albrecht W. L., пат. CHIA 3440174 (Nalco Chemical Co.), 1969.
16. Франц, пат. 1517324 (Nalco Chemical Co.), 1968.
17. Mindick M., Vossos P., пат. CHIA 3538015 (Nalco Chemical Co.), 1976.
18. Her R. K-, пат. CHIA 2885366 (Du Pont), 1959.
19. Iler R. K-, Wolter F. J., пат. CHIA 2631134 (Du Pont), 1953.
20. Barrett J. W., англ. пат. 1006845, 1965.
21. Bartholin M., Guyot A., Coll. Polym. Sci., 253, 623 (1975).
22. Albrecht W. L„ пат. США 3673104 (Nalco Chem. Co.), 1972.
23. Klosak E. J., пат. США 3462374 (Nalco Chemical Co.), 1969.
24. Reven L. E., Blake D. G., пат. США 3128251 (Nalco Chemical Co.),
1964.
25. Irani F. А., пат. США 3789009 (Du Pont), 1974.
26. Reuter R., Tozyldo A. J., пат. США 2929790 (Nalco Chemical Co.),
1960.
27. Sippel R. I., пат. США 3440176 (Du Pont), 1969.
28. Weldes H. H., Boyle F. A., Bobb J. S., пат. США 3440175 (Philadelphia
Quartz Co.), 1969.
29. Cummings W., пат. США 3607774 (Monsanto Chemicals Co.), 1971.
30. Egbregt J., пат. США 3770650 (AKZO N. V.), 1973.
31. Linton H. R., пат. США 3639133 (Du Pont), 1972.
32. Iler R. K-, пат. США 2727008 (Du Pont), 1953.
33. Mindick M., пат. США 3502593 (Nalco Chemical Co.), 1970’
34, Alexander G. B„ McWhorter J. R„ пат. США 2833724 (Du Pont), 1958.
35. Weldes H. H., Derolf M. R., пат. США 3346334 (Philadelphia Quartz
Co.), 1967.
36. Berkheimer E. R„ пат. США 2978419 (Atlantic Refinery Co.), 1961.
37. McNally P. H., Rosenberg N. W., пат. США 3113112 (Diamond Alkali
Co.), 1963.
38. Vossos P. H., пат. США 3714064 (Nalco Chemical Co.,), 1973.
39. Reuter R., Reven L. E., пат. США 3029151 (Nalco Chemical Co.), 1958.
40. Marotta R., пат. США 3711419 (Monsanto Chemical Co.), 1973.
41. Yates P. X., “Thermodynamic Stabilization of Highly Dispersed Systems:
Prevention of Particle Growth of Colloidal Amorphous Silica by Hydro-
xyl Ion Adsorption” presented at the Divisional Colloid Chemistry Sym-
posium on Colloidal Silica and Silicates, 137th Meeting of the American
Chemical Society, Cleveland, Ohio, April 13, 1960.
42. Baruch-Weill M., Ann. Phys., 13 (4), 1159 (1959).
43. Walton A. G., The Formation and Properties of Precipitates, Wiley,
New York, 1967, p. 155.
44. Derjaguin B.V., Landau L.D., Acta Physicochim., USSR, 14, 633 (1941).
45. Verwey E. J. W., Overbeek I. Th. G., Theory of Stability of Lyophobic
Colloids, Elsevier, Amsterdam, 1948.
46a . Loeb A. L., Overbeek J. Th. G., Wiersema P. H., The Electrical Double
Layer Around a Spherical Particle, M. I. T., Press, Cambridge, Mass.,
1961.
466. Ottewill R. H., J. Colloid Interface Sci., 58, 357 (1977).
47. Napper D. FL, Ind. Eng. Chem. Prod Res. Div., 9 (4), 467 (1970).
48. Roweler G. С. I., Chem. Phys. Lett., 8 (3), 275, (1971).
49. Visser J., Adv. Colloid Interface Sci., 3 (4), 331 (1972).
50. Kitchener I. A., Faraday Disc., 52, 379 (1971).
51. Matijevic E., J. Colloid Interface Sci., 43, 217 (1973).
Коллоидный кремнезем — концентрированные золи
613
52. Webb J. Т., Bhatnagar Р. D., Williams D. G., J. Colloid Interface Sci.,
49, 346 (1974).
53. lirgensons B„ Straumanis M. E., Colloid Chemistry, Mcmillan, New
York, 1962, p. 132.
54. Atkins R. С., пат. США 3012973 (Du Pont), 1961. '
55. Vossos P. EL, Mindick M., пат. США 3582494 (Nalco Chemical Co.),
1971.
56. Marotta R., пат. США 3342748 (Monsanto Co.,), 1967.
57. Smitham J. B., Evans R., Napper D. H., J. Chem. Soc., Faraday Trans.
(1), 71 (2), 285 (1975).
58. Bagchi P., J. Colloid Interface Sci., 47 (1), 86 (1974).
59. Luvisi G. W., пат США 3108970 (Nalco Chemical Co.), 1963.
60. Gandon L. el al., англ. пат. 861378 (Bonzel-Maleta, Societe Industrielle
de Products Chemie), 1959.
61. Feldi C. A., Reven L. E„ пат. ФРГ 1244745 (Nalco Chemical Co.), 1967.
62. Yates P. С., пат. США 3630954 (Du Pont), 1971.
63. Wolter F. J., пат. США 2601352 (Du Pont), 1952.
64. Iler R. К., Dalton R. L., J. Phys. Chem., 60, 455 (1956).
65. Mindick M„ Reven L. E., пат. США 3342747 (Nalco Chemical Co.), 1967.
66. Alexander G. B„ Iler R. K-, J. Phys. Chem., 57, 932 (1953).
67. Radczewski О. E., Richter EL, Kolloid-Z., 96, 1 (1941).
68. Stdber W., Fink A., Bohn E„ J. Colloid Interface Sci., 26, 62 (1968).
69. Balthis J. H„ пат. США 2614994, 2614995 (Du Pont), 1952.
70. McMillan D., пат. США 3591518 (Du Pont), 1971.
71. Greer R. T., Scanning Electron Microsc., 4 (1), 153 (1971).
72. Brusset EL, LeRat B. et al., Ann. Chim. Paris, 9, 477 (1954).
73. Lederer L., Schurz J., Janzon K. EL, Monatsh. Chem., 103 (5), 1299
(1972).
74. Ogino K., Kuronuma L, J. Colloid Interface Sci., 56, 629 (1976).
75. Barrett W. T., Moises S. G., Vanik M. С., пат. США 3097175 (W. R. Grace
and Co.), 1963.
76. Fitch F. T., Sanchez M. G., Vanik M. С., пат. США 3091592 (W. R. Grace
and Co.), 1963.
77. Cho I. S., Wadsworth M. E., Trans. Soc. Min. Eng. AIME, 287 (1963).
78. Kitahara S., Oshima F., Nippon Kagaku Zasshi, 87 (4), 316 (1966) (Chem.,
Abstr., 65, 11387g).
79. Alexander G. B., Iler R. K-, Wolter F. J., пат. США 2601235 (Du Pont),
1952.
80. Alexander G. B., Iler R. К., пат. США 2605228 (Du Pont), 1952.
81. Armstrong J. E., Cummings W. P., пат. США 2956957 (W. R. Grace
and Co.), 1960.
82. Teicher EL, канал, пат. 609186, 609190 (Monsanto Chemical Co.), 1960.
83. White J. F., пат. США 2285477 (Monsanto Chemical Co.), 1942.
84. Marcheguet H. G. L., Gandon A. L., пат. США 3026267 (Nobel-Bozel,
Paris, France), 1962.
85. Weldes H. H„ Boyle F. A., Bobb I. S. S., пат. США 3440175 (Philadel-
phis Quartz Co.), 1969.
86. Power W. H., пат. США 3051657 (Monsanto Chemical Co.), 1962.
87. Sanchez M. G., канад. пат. 586261 (W. R. Grace and Co.), 1959.
88. Iler R. К, пат. США 3668088 (Du Pont), 1972.
89. Vaquero M. G., испан. пат. 263758 (Chem. Abstr., 56, 9885f).
90. Prajapati M. N., Talpade C. R., Indian. Chem. Manuf., 12 (1), 13 (1974)
(Chem. Abstr., 82, 103661p).
91. Tripp T. G., пат. США 3645865 (Monsanto Chemical Ltd.), 1972.
92. Boari G., Merli C., Passino R., Teravantl T., Desalination, 15(1), 3
(1974).
93. Iler R. K, Wolter F. L, пат. США 2631134 (Du Pont), 1953.
614
Глава 4
94. Mindick М., Reven L. E., пат. США 3468813 (Nalco Chemical Co.), 1969.
95. Dirnberger L. А., пат. США 2703314 (Du Pont), 1955, Dirnberger L. A.,
Nelson R. T., пат. США 2974109 (Du Pont), 1961.
96. Iler R. K-, пат. США 2727008 (Du Pont), 1959; Reuter R., Tozyldo A. J.,
пат. США 2929790 (Nalco Chemical Co.), 1969; Weldes H„ Boyle F. A.,
Bobb J. S. S., пат. США 3440175, 1969; Weldes H. H„ Blackgood M.R.D.,
пат. США 3346334 (Philadelphia Quartz Co.), 1967; Marotta R-,
пат. США 3374180 (Monsanto Chemical Co.), 1968.
97. Wolter F. G., пат. США 2650200 (Du Pont), 1953; 2671056 (1954).
98. Shannon R. F., пат. США 3083167 (Ownes Corning Fiberglas Corp.),
1963.
99. Hoffman G. W., пат. США 3649556 (N. L. Industries, Inc.), 1972.
100. Matejka Z., Chem., Prum., 22 (7), 356 (1972) (in Czech).
101. Iler R. К., пат. США 3756958 (Du Pont), 1973.
102. Praetorious M., Wolf К., герм. пат. 432418 (1922).
103. Neundlinger K-, пат. США 1834420 (1931).
104. Trail Н. S., пат. США 2572578 (Monsanto Chemical Со.), 1951.
105. Ahlberg J. Е., Simpson Е. А., пат. США 2900348 (W. R. Grace and Со.),
1959.
106. Legal С. С., пат. США 2724701 (W. R. Grace and Со.), 1955.
107. Mertz G., пат. ФРГ 2006021 (Badische Anilin and Soda—Fabrik A. G.),
1971.
108. Бубырева H. С., Виндас Б.П.— Коллоидн. ж., 1959, т. 21, вып. 4, с.’388.
109. Flachsbart Н., Stober W., J. Colloid Interface Sci., 30, 568 (1969).
110. Stober W„ пат. США 3634558 (U. S. Atomic Energy Commission), 1972.
111. Brinsmead К. H., Brown W. B., Jr., пат. США 3313737 (British Indus-
tries Corp.), 1967.
112. Schnurch R., Koster А., пат. США 3346335 (Philadelphia Quartz Co.),
1967.
113. Montenyohl V. I., Olson С. M., пат. США 2614993 (Du Pont), 1952.
114. Bobb J. S. S., пат. США 3650977 (Philadelphia Quartz Co.), 1972.
115. Chilton H. T. J., пат. США 3654105 (Monsanto Chemical Co.), 1972.
116. Lofman L. A., Thereault J. R., пат. США 2984629 (Cabot Corp.), 1961.
117. Kloepfer H., Meir-Ewert H., герм. пат. 877891 (Deut Gold u. Silber
Scheideanstalt), 1953.
118. Wagner E., пат. США 2951044 (Deutsche Gold und Silber Scheideanstalt,
Vormals Roessler), 1960.
119. Aerosil, Degussa, Inc., New York, 1970 (Technical bulletin).
120. Cab-O-Sil, CGEN-7, Cabot Corp., Boston, 1970.
121. Spencer W. B., Smith W. R., Cosman A. F., пат. США 3024089 (Cabot
Corp.), 1962.
122. Clapsdale L. G„ Syracuse M. G., пат. США 2630410 (Union Carbide and
Chemical Co.), 1953.
123. Deutsche Gold und Silber Anstalt, пат. ФРГ 1667460, 1971.
124. Deutsche Gold und Silber Anstalt, англ. пат. 1326574, 1973.
125. Deutsche Gold und Silber Anstalt, нидерланд. пат. Appl. 6512426, 1966.
126a. Schaefer D. P., Gamage L. P., пат. США 4054536 (Nalco Chemical Co.),
1977.
1266. Graham T., Ann. Phys. Leipzig, 190, 187 (1861); Phil. Trans. R. Soc.
London, 151, 204 (1861); J. Chem. Soc., 15, 216 (1862); J. Chem. Soc.,
17, 318 (1864).
127. Monet G. P„ пат. США 2773028 (Du Pont), 1956.
128 Kuhn A. T., Industrial Electrochemical Processes, Elsevier, Amsterdam,
1971.
129. Iler R. К., пат. США 3969266 (Du Pont), 1976.
130. Chilton H. T. J., пат. США 3560400 (Monsanto Chemical Co.), 1971.
131. Michaels A. S„ Chem. Eng. Progr., 64, 31 (1968).
Коллоидный кремнезем-—концентрированные золи
615
132. Porter М. С., Michaels A. S., Chem. Tech., 56 (Jan. 1971).
133. Literature References to the Uses of Amicon Ultrafiltration Systems,
Publication 428A, AMICON Corp., Lexington, Mass., 1973.
134. Porter M. C., Ind. Eng. Chem. Prod. Res. Develop., 11 (3), 234 (1972).
135. Overbeek J. Th. G., in H. R. Kruyt, Ed. Colloid Science, Vol. 1, Elsevier,
Amsterdam, 1952, p. 72.
136. McGahan J., пат. США 3148110 (Du Pont), 1964.
137. Nickerson R. F., пат. США 2823186 (Monsanto Chemical Co.), 1958.
138. Michalski R. J., Sapienza M. S., Wolfson L. I., пат. США 3377275 (felalco
Chemical Co.), 1968.
139. Michalski R. J., пат. США 3336236 (Nalco Chemical Co.), 1967.
140. Bolt G. H„ J. Phys. Chem., 61, 1166 (1957).
141. Kuhn W. E., Ultrafine Particles, Wiley, New York, 1963, p. 104.
142. Alexander G. B., Iler R. K., J. Phys. Chem., 57, 932 (1953).
143. Watson J. H. L„ Anal. Chem., 20, 576 (1948).
144. Greene R. S. B., Murphy P. J., Posner A. M., Quirk J. P., Clays, Clav
Miner, 22 (2), 185 (1974).
145. Pertoft H., Philipson L., Oxelf ell P., Hoglund S., Virology 33, 185,
(1967).
146. Bailey J. T., Beattie W. H., Booth C., J. Chem. Educ., 39, 196 (1962).
147. Underwood E. E., Quantitative Stereology, Addison-Wesley, Reading,
Mass., 1970.
148. Dezelic Gj., Wrischer M., Decide Z., Kratohvil J. P., Kolloid-Z., 171, 42
(1960).
149. Maron S. H., Lou R. L. FL, J. Polym. Sci., 15, 29 (1954).
150. Trap H. J. L„ Hermans I. J., Rec. Trav. Chim., 73, 167 (1954).
151. Goring D. A. R., Senez M., Melanson B., Huque M. M., J. Colloid Sci.,
12,412 (1957).
152. Dezelic Gj., Kratohvil J. P., J. Phys. Chem., 66, 1377 (1962).
153. Dezelic Gj., Kratohvil J. P., Kolloid-Z., 173, 38 (1960).
154. Greenberg S. A., Chang T. N., Jarnutowski R., J. Polym. Sci., 58, 147
(1962).
155. Jennings B. R., Jerrard H. G., J. Polym. Sci. (A), 2, 2025 (1964).
156. Bonnelycke E., Dandliker W. B., J. Colloid Sci., 14, 567 (1959).
157. Claesson S., Ohman J., Ark. Kern., 23 (1), 77 (1965).
158. Brill O. L„ Weyl C. G„ Schmidt P. W., J. Colloid Interface Sci., 27,
429 (1968).
159. Janosi A., Kratky 0., Sekora O., Monatsh, Chem., 100, 1973 (1969).
160. Dragsdore R. D., J. Appl. Phys., 27, 620 (1956).
161. Lowen W. K., Broge E. C., J. Phys. Chem., 65, 16 (1961).
162. Hermans J. J., Ryke A. M., J. Colloid Sci., 13, 508 (1958).
163. Burns G. P., Chilton H. T. J., англ. пат. 1190980 (Monsanto Chemical
Ltd.), 1970.
164. Coll H., Fague G. R., Robillard К A. (Eastman Kodak), 49th National
Colloid Symposium, Clarkson College of Technology, Potsdam, N. Y„
June 16—18, 1975.
165. Small H., J. Colloid Interface Sci., 48, 147 (1974).
166. Small H., пат. США 3865717 (Dow Chemical Co.), 1975.
167a. Stoisets R. F., Poehlein G. W„ Vanderhoff J. W., J. Colloid Interface
Sci., 57, 337 (1976).
1676. Giddings J. C., Yang J. F., Meyers M. .V., Anal. Chem., 46, 1917, 1924
(1974)1
167b. Yang F. J., Meyers M. N„ Giddings J. C., J. Colloid Interface Sci. (1978).
168. McCann G. D., Vanderhoff J. W., Strickler A., Sacks T. L, Sep. Purif.
Methods. 2, 153 (1973).
169. Iler R. K.., J. Colloid Interface Sci., 53, 476 (1975).
170. Goto K., Okura T., Jap. Anal., 4, 175 (1955).
616
Глава 4
171. Goto К, J. Chem. Soc. Jap. Pure Chem. Sect., 76, 729 (1955).
172. Baumann H., Z. Anal. Chem., 217, 241 (1966).
173. Emmett P. H., Symposium of New Methods for Particle Size Deter-
mination, American Society for Testing Materials, Philadelphia, March 4,
1941, p. 95.
174. Nelson F. M., Eggersten F. T., Anal. Chem., 30, 1387 (1958).
175. Eberly P. E„ Jr., J. Phys. Chem., 65, 1261 (1961).
176. Ettre L. S., Cieplinski E. W., in W. E. Kuhn, Ed., Ultra-Fine Particles,
.Wiley, New York, 1963, p. 393.
177. Lowen W. K, Broge E. C., J. Phys. Chem., 65, 16 (1961).
178. Shapiro I., Kolthoff I. M., J. Am. Chem. Soc., 72, 776 (1950).
179. Kautsky H., Michel R., Z. Naturforsch., 7 (6), 414 (1952).
180. Unger K, Vydra F., J. Inorg. Nucl. Chem., 30, 1075 (1968).
181. Stryker L. J., Matijevic E., Adv. Chem. Ser., 79, 44 (1968) (American
Chemical Society); Kolloid-Z„ Z. Polym., 233, 912 (1969); J. Colloid
Interface Sci, 31, 39 (1969); 42, 232 (1973).
182. Sears G. W„ Anal. Chem., 28, 1981 (1956).
183. Bryant К C., J. Chem. Soc., 1952, 3017.
184. Bolt G. H., J. Phys. Chem., 61, 1166 (1957).
185. Heston W. M„ Iler R. K, Sears G. W„ J. Phys. Chem., 64, 147 (1960).
186. Grahame D. C., Chem. Rev., 41, 441 (1947).
187. Perram J. W., J. Chem. Soc., Faraday Trans., 69 (7), 993, (1973).
188. Parks G. A., Chem. Rev., 65, 177 (1965).
189. Allen L. H., Matijevic E., Mestes L., J. Inorg. Nucl. Chem., 33, 1239
(1971).
190. Tadros Th. F., Lyklema J., J. Electroanal. Chem., 17, 267 (1968).
191. Yates D. E., Healy T. W, J. Colloid Interface Sci., 55, 9 (1976).
192. Vysotskii Z. Z., Strazhesko D. N., in D. N. Strazhesko, Ed., Adsorption
and Adsorbents, Vol. 1, Wiley, New York, 1973, p. 55.
193. Yates D. E., Levine S., Healy T. W., J. Chem. Soc., Faraday Trans., 70
(10), 1807 (1974).
194. Mysels К J'., Introduction to Colloid Chemistry, Interscience, New York,
1959, Chapter XY.
195. Graham David, Chem. Revs., 41, 441 (1947).
196. Lyklema J., Molecular Forces in Colloidal Stability (Pontif. Acad. Sci.
Scr. Var., 31), Laboratory for Physical and Colloid Chemistry of the
Agricultural University, Wageningen, Netherlands, 1967.
197. Napper D. H., Ind. Eng. Chem. Prod. Res. Develop., 4, 467 (1970).
198. Sheludko A., Colloid Chemistry, Elsevier, New York, 1966.
199. Kruyt H. R., Colloid Science, Elsevier, New York, 1952.
200. Long R. D., Ross S, J. Colloid Interface Sci., 26, 434 (1968).
201. Deraska J., Yaeger E., Hovorka F., Quantitative Measurements of Colloi-
dal Vibration Potentials, Technical Report No. 2, Office of Naval Rese-
arch, Contract NONR 1439 (04)-Project No. NR 384—305.
202. Younger P. R., Zimmerman G. O., Chase С. E., Drost-Hansen W., J. Chem.
Phys., 58, 2675 (1973).
203. Horning S. C„ Shay C. W., пат. США 2601291 (Du Pont), 1952.
204. Iler R. К, пат. США 2668149 (Du Pont), 1952.
205. Wolter F. J., пат. США 2601352 (Du Pont), 1952.
206. Lawrence M., Vivian H. E., Aust., J. Sci., 12, 96 (1961).
207. Kruyt H. R., Ed., Colloid Science, Vol. 2, Elsevier, New York, 1952, p. 26.
208. Mooney M., J. Colloid Sci., 6, 162 (1951).
209. Simha R., J., Appl. Phys., 23, 1020 (1952).
210. Vand V., J. Phys. Chem., 52, 277, 300 (1948).
211. Ford T. F„ J. Phys. Chem., 64, 1168 (1960).
212. Thomas D. G, J. Colloid. Sci., 20, 267 (1965).
213. Lewis T. B., Nielsen L. E., Trans. Soc. Rheol, 12, 421 (1968).
Коллоидный кремнезем — концентрированные золи
617
214. Fedors R. F., J. Colloid Interface Sci., 46, 545 (1974).
215. Manley R. St. J., Mason S. G., Can. J. Chem., 32, 763 (1954).
216. Happel J., J. Appl. Phys., 28, 1288 (1957).
217. Ливанский Ю. E.— Коллоидн. ж., 1973, т. 35, с. 286.
218. Greenberg S. A., Jarnutowski R., Chang T. M., J. Colloid Sci., 20, 20
(1965).
219. Лег R. К, пат. США 2956958 (Du Pont), 1960.
220. LaMer V. K-, Healy T. W., Rev. Pure Appl. Chem., 13, 112 (1963).
221. Gruber E„ Knell W. L., Colloid Polym. Sci., 253 (6), 462 (1975).
222. Visser J., “Adhesion of Colloidal Particles”, in E. Matijevic Ed., Surface
and Colloid Science, Vol. 8, Wiley, New York, 1976, p. 3.
223. Her R. K., J. Phys. Chem., 56, 680 (1952).
224. Matijevic E., Kratohvil S., Stickels J., J. Phys. Chem., 73, 564 (1969).
225. Baxter S., Bryant К. C., J. Chem. Soc., 1952, 3024.
226. Penner S. S., J. Polym. Sci., 1, 441 (1946).
227. Brode A. P., Brown A. G., Hoff R., J. Colloid Sci., 8, 264 (1953).
—228. Thomas I. L., McCorkle К. H., J. Colloid Interface Sci., 36, 110 (1971).
229. Reuter R., пат. США 3029151 (Nalco Chemical Co.), 1962.
230. Dexter A. R„ Tanner D. W., Nature Phys. Sci., 230, 177 (1971).
231. Yates P. С., канад. пат. 878444 (Du Pont), 1971.
232. Sippel R. D„ пат. США 3397153 (Du Pont), 1968.
233. Белоцерковский Г. M. и др.— ЖПХ, 1970, т. 43, с. 1380.
234. Liehtenbelt J. W. Th., Ras Н. 1. М. С., Wiersema Р. Hv J. Colloid Inter-
face Sci., 46, (3), 522 (1974).
235. Чернобережский И. M„ Голикова Е. В.— Коллоидн. ж., 1972, т. 34 (5),
с. 793.
236. Van Olphen Н., Mysels К. J., Phys. Chem. Enriching Topics from Col-
loid and Surface Science IUPAC Commission 1.6, Theorex Inc., La Jolla,
Calif., p. 12.
237. Harding R. D., J. Colloid Interface Sci., 35, 172 (1971).
238. Hamaker H. C., Rec. Trav. Chirn., 55, 1015 (1936); 56, 3 (1937).
239. Rouweler G. C., Overbeek J. Th. G., Trans. Faraday Soc., 67 (7), 2117
(1971).
240. Tabor D., Winterton R. H. S„ Nature, 219, 1120 (1968).
241. Dumont F., Watillon A., Faraday Soc. Disc., 52, 375 (1971).
242. Depasse J., Watillon A., J. Colloid Interface Sci., 33, 431 (1970).
243. Wiese G. R„ Healy T. W., J. Colloid Interface Sci., 51, 427 (1975).
244. Burton E. F., Colloid Symp. Monogr., 4, 132 (1926).
245. Deryagin B., Trans. Faraday Soc., 36, 203 (1940).
246. Verwey E. J. W., J. Phys. Chem., 51, 631 (1947); Verwey E. J. W., in
J. Alexander, Ed., Colloid Chemistry, Reinhold, New York, 1950, Vol. 7,
p. 47; Overbeek J. Th. G., in H. R. Kruyt, Ed., Colloid Science, Part I,
Elsevier, Amsterdam, 1952, p. 245.
247. Verwey E. I. W., Overbeek J. Th. G., Theory of the Stability of Lyopho-
bic Colloids, Elsevier, Amsterdam, 1948; Booth F., Progr. Biophys. Bio-
phys. Chem., 3, 131 (1953).
248. Symposium “Coagulation and Flocculation”, Disc. Faraday Soc., 18,
9 (1954).
249. Allen L. H., Matijevic E., J. Colloid Interface Sci., 31, 287 (1969)
(paper 1).
250. Allen L. H., Matijevic E., J. Colloid Interface Sci., 33, 420 (1970)
(paper 2).
251. Allen L. H., Matijevic E., J. Colloid Interface Sci., 35, 66 (1971)
(paper 3).
252. Watillon A., J. Colloid Interface Sci., 33, 430 (1970).
253. Depasse Warlus J. Colloid Interface Sci., 56, 618 (1976).
618
Глава 4
254. Garner W. E., 10th Symposium Colston Research Society, University of
Bristol, England, 1958 (discussion); Everett D. H., Stone F. S., Eds.,
Structure and Properties of Porous Materials, Academic Press, New York,
1958, p. 285.
255. Tadros E., ' Lyklema I., J. Electroanal. Chem. Interface, Electrochem., 22,
1 (1969).
256a . Healy T. W., James R. 0., Cooper R., Adv. Chem. Ser., 79, 62 (1968)
[American Chemical Society],
2566. McFadyen P., Matijevic E., Kolloid-Z. Z. Polym., 251, 665 (1973).
257. O’Melia C. R., Stumm W., J. Colloid Interface Sci., 23, 437 (1967).
258. Ruehrwein R. A., Ward D. W., Soil Sci., 73, 385 (1952).
259. Hahn H. H., Stumm W., J. Colloid Interface Sci., 28, 134 (1968).
260. Hahn H. H., Stumm W., Adv. Chem. Ser., 79, 91 (1968). [American -
Chemical Society],
261. Stumm-W., O’Melia C. R., J. Am. Water Works Assoc., 60, (5), 514
(1968).
262. Stumm W., Huang С. P., Jenkins S. R., Croat. Chem. Acta, 42, 223
(1970).
263. Burwell R. L„ Pearson R. G„ Haller G. L„ Tjok P. B„ Shock S. P„
Inorg. Chem., 4 (8), 1123 (1965).
264. Hohl H., Stumm W., J. Colloid Interface Sci., 55, 281 (1976).
265. Goto K-, Bull. Chem. Soc. Jap., 29, 740 (1956).
266a. James R. 0., Healy T. W., J. Colloid Interface Sci., 40 (1), 42,’ 53
(1972).
2666. Schindler P. W., Fiirst B., Dick R., Wolf P. V., J. Colloid Interface Sci.,
55, 469 (1976).
267. Matijevic E., Kratohvil S., Stickles I., J. Phys. Chem., 73, 564 (1969).
268. Porter R. A., Weber W. J., Jr., J. Inorg. Nucl. Chem., 33, 2443 (1971).
269. Cassel E. A., Matijevic E., A. I. Ch. E. J., 17, 1486 (1971).
270. Mangravite F. G., Cassel E. A., Matijevic E., J. Colloid Interface Sci.,
39, 357 (1972).
271. Healy T. W., Wiese G. R., Yates D. W., Kavanagh В. V., J. Colloid In-
terface Sci., 42, 647 (1973).
272. Iler R. K-, J. Colloid Interface Sci., 43, 399 (1973).
273. Harding R. D., J. Colloid Interface Sci., 10, 164 (1972).
274. Hogg R., Healy T. W., Fuerstenau D. W., Trans. Faradav Soc., 62, 1638
(1966).
275. Чернобережский И. M., Голикова Е. В., Гирфанова Т. Ф.— Сб. Поверх-
ностные силы в тонких пленках и устойчивость коллоидов.— М.: Наука,
1975, с. 256—261.
276. Madi Bolyos A., Kesztus J., Szabo K-, Acta Phys. Chim. Debrecina,
18, 112 (1973).
277. Buzagh A., Rohrsetzer S.. Acta Chim. Acad. Sci., Hung., 10, 427 (1957).
278. Buzagh A., Kolloid-Z., 146, 133 (1956).
279. Wiese G. R., Healy T. W., Trans. Faraday Soc., 66, 490 (1970).
280. Ottewill R. H., Shaw J. N., Faraday Soc. Disc., 42, 154 (1966).
281. Matijevic E., Faraday Soc. Disc., 42, 173 (1966).
282. Kotera A., Furusawa K-, Kudo K-, Kolloid-Z. Z. Polym., 240, 837 (1970).
283. Frens G., Kolloid-Z. Z. Polym., 250, 736 (1972); Colloid Polym. Sci.,
253 (6),->500 (1975).
284. Boehm H. P., Schneider M., Z. Anorg. Allgem. Chem., 301, 326 (1959).
285. Goodman J. F., Disc. Faraday Soc., 42, 64 (1966).
286. Herkenhoff E. С., пат. США 2423022 (American Cyanamid Co.), 1947.
287. Tschapek M., Torres Sanchez R. M., J. Colloid Interface Sci., 54, 460
(1976).
288. Rubio J., Goldfarb J., J. Colloid Interface Sci., 36 (2), 289 (1971).
289. Sutherland D. N., J. Colloid Interface Sci., 25, 373 (1967).
Коллоидный кремнезем — концентрированные золи
619
290. Smellie R. Н., LaMer V. К., J. Colloid Interface Sci., 23, 589 (1958).
291. Tanford C., The Hydrophobic Effect, Wiley, New York, 1973.
292. Smith E., Turnbull J. S., пат. США 2888354 (Metropolitan Vickers)
(1958).
293. Void R. D., Sivaramakrishnan N. H., J. Phys. Chem., 62, 984 (1958).
294. Fuerstenau D. W., J. Phys. Chem., 60, 981 (1956).
295. Alexander G. B., Iler R. K-, пат. США 2801902 (Du Pont), 1957.
296. O’Connor D. J., Sanders J. P., J. Colloid Sci., 11, 158 (1956).
297. Matijevic E., Ottewill R. H., J. Colloid Sci., 13, 242 (1958).
298. Klotz I. M„ Science, 128, 815 (1958).
299. Scheraga H. A., J. Phys. Chem., 65, 1071 -(1961).
300. Nemethy G., Scheraga H. A., J. Phys. Chem., 66, 17 (1962).
301. Kauzmann W., Advances in Protein Chemistry, Vol. 14, Academic Press,
New York, 1959, p. 38.
302. Kauzmann W., “Hydrophobic Bonding”, in С. B. Anfinsen et al., Eds.,
Advances in Protein Chemistry, Vol. 14, Academic, New York, 1959, p. 40.
303. Thoma K, Ullmann E., Wolferseder E., Arch. Pharm., 299 (12), 1020
(1966).
Y 304. Lagaly G„ Weiss A., Kolloid-Z. Z. Polym., 238, 485 (1970).
305. Ter-Minassian-Saraga L., J. Colloid Interface Sci., 51, 211 (1975).
306. Smith E., пат. США 2888354 (Metropolitan Vickers, Ltd.), 1959.
307. Barr T„ Oliver I., Stubbings W. V., J. Soc. Chem. Ind., 67, 45 (1948).
308. Dixon J. K, LaMer V. K, Li Cassian, Messinger S., Linford H. B.,
J. Colloid Interface Sci., 23, 465 (1967).
309. La Мег V. K, “Colloid Stability in Aqueous and Non-aqueous Media”,
Disc. Faraday Soc., 42, 248 (1966).
310. Linke W. F„ Booth R. E„ AIME Trans., 217, 364 (1960).
311. Heller W., Pure Appl. Chem., 12, 249, 265 (1966).
312. Botham R., Theis C., J. Colloid Interface Sci., 31, 1 (1969).
313. Kragh К. M„ Langston W. G., J. Colloid Sci., 17, 101 (1962).
314. Bergman L, Nelson E. S., J. Colloid Sci., 17, 823 (1962).
315. Ries H. E., Jr., Nature, 226, 72 (1970).
316. Ries H. E., Meyers B. L., J. Appl. Polym. Sci., 15, 2023 (1971).
317. Shyluk W. P., J. Polym. Sci [A-2], 6 (2), 2009 (1968).
318. Деревянко Л. А., Меркушев О. M, Николаев А. Ф., Лавров Л. С,—
ЖПХ, 1971, т. 44, с. 2276.
319. Silberberg A., J. Colloid Interface Sci., 38, 217 (1972).
320. Gregory I., J. Colloid Interface Sci., 42, 448 (1973); “Adsorption of
Polycations on Negative Particles”, 49th National Colloid Symposium,
Clarkson College of Technology, Potsdam, N. Y„ June 16—18, 1975.
321. Her R. K, J. Colloid Interface Sci., 37, 364 (1971).
322. Hayek M„ пат. США 2741568 (Du Pont), 1956.
323. Lindquist G. M., Stratton R. A., J. Colloid Interface Sci., 55, 45 (1976).
324. Iler R. K, J. Colloid Interface Sci., 55, 25 (1976).
325. Holt P. F„ Went C. W„ Trans. Faraday Soc., 55, 1435 (1959).
326. Howard G. I., McConnel P„ J. Phys. Chem., 71, 2974 (1967).
327. Rupprecht H„ Liebl L. H„ Kolloid-Z. Z. Polym., 239 (2), 685 (1970).
328. Rubio J., Kitchener J. A., J. Colloid Interface Sci., 57, 132 (1976).
329. Кузькин С. Ф., Небера В. П„ Якубович И. А., Золин С. Н.— Изв. высш,
учебы, зав. Цветы, металлы, 1963, т. 6, с. 36.
330. Griot О., Kitchener J. A., Nature, 200, 1004 (1963); Faraday Soc. Trans.,
61, 1026, 1032 (1965).
331. Джалилов A. T., Габриелян Н. А., Сухинина Л. А.— Синтез высокомол.
соед., 1972, т. 70, с. 7.
332. Sorensen R. Т., Frommer D. W., U. S. Bur. Mines Rept., Invest., 6719
(1966) (Chem. Abstr., 64, 13810).
333. Peterson L. E., Opie J. W., Ind. Eng. Chem., 50, 1013 (1958).
620
Глава 4
334. Ross S., Schaeffer H. M., J. Phys. Chem., 58, 65 (1954).
335. Wightman J. P„ Chessick J. /., J. Phys. Chem., 66, 1217 (1962).
336. Iler R. K„ J. Colloid Interface Sci., 51, 388 (1975), пат. США 3738957
(Du Pont), 1973.
337. Fleer G. J., Polymer Adsorption and Effect in Colloidal Stability, Veen-
man H. and Zonen N. V., Wageningen, 1971.
338. Fleer G. J., Lyklema J., J. Colloid Interface Sci., 46, 7 (1974).
339. Tadros Th. F., J. Colloid Interface Sci., 46, 528 (1974).
340. Iler R. K-, J. Phys. Chem., 56, 673 (1952).
341. Iler R. K-, J. Am. Chem. Soc., 74, 2929 (1952).
342. Kirk J. S, пат. США 2383653 (Du Pont), 1945 (Example 5).
343. Iler R. K-, McQuestion H. I., пат. США 3855172 (Du Pont), 1974;
4010242, 1977.
344. Jones L. H. P., Milne A. A., Sanders J. V., Science, 151, 464 (1966).
345. Jones J. B., Segnit E. R., J. Geol. Soc. Aust., 18 (1), 57 (1971).
346. Jones J. B„ Segnit E. R., Nature, 198 (4886), 1191 (1963).
347. Jones J. B., Sanders J. V., Segnit E. R., Nature, 204, 990 (1964).
348. Sanders J. V., Nature 204, 1151 (1964).
349. Segnit E. R., Slevens T. J., Jones J. B., J. Geol. Soc. Aust., 12 (2), 211
(1965).
350. Jones J. B„ Biddle J., Segnit E. R„ Nature (5043), 1353 (June 25, 1966).
351. Jones J. G., Segnit E. R., Aust. J. Sci., 29, 129 (1966). ,
352. Darragh P. J., Gaskin A. J., Terrell В. C., Sanders J. V., Nature (5018),
13 (Jan. 1, 1966).
353. Cole S. H„ Monroe E. A., J. Appl. Phys, 38 (4), 1872 (1967).
354. Jones J. B., Segnit E. R., Mineral. Mag, 37 (287), 357 (1969).
355. Greer R. T„ Nature, 224, 1199 (1969). ’
356. Sanders J. V., Acta Cryst, A24, 427 (1968).
357. Мицюк Б. M., Багмут H. H., Матяш И. В., Федотов Ю. В.— Минерал,
1974, т. 8, с. 105.
358. Sanders J. V., Darragh Р. J., Mineral Rec. 2 (6), (1971); Germs and
Gemol, 291 (Summer, 1965).
359. Electron micrograph supplied bv courtsy of Dr. Sanders.
360. Iler R. K, Nature 207, 472 (1965).
361. Балакирев В. Г., Бутузов В. ГЕ, Гусельник Ю. В., Цинобер Л. И.—
Пробл. кристаллогр, 1971, с. 220.
362. Leechman F., The Opal Book, Ure Smith, Sidney, London, 1961.
363. Eyles W. C, The Book of Opals, Chas E. Tuttle Co. Rutland, Vt, 1964.
364. Kalokerinos A., Australian Precious Opal. Arco. Publ. Co, New York,
1972.
365. Alfrey T„ Bradford E. R., Vanderhoff J. W., Oster G., Opt. Soc. Am,
44, 603 (1954).
366. Luck W„ Klier M„ Wesslau H., Naturwis. 14, 485 (1963).
367. De Fontaine D., Jackson K- A., Miler С. E., Am. J. Phys, 37 (8), 789
(1969).
368. Kubitschek H. E., in W. E. Kuhn, Ed, Utrafine Particles, Wiley, New
York, 1963, p. 438.
369. Bradford E. B„ Vanderhoff J. W., J. App. Phys, 26, 864 (1955).
370. Smith A. F., Nature Phys, Sci, 246, 10 (1973).
371. Ravey J. C., J. Colloid Interface Sci, 50, 545 (1975).
372. Luck W., Klier M., Wesslau H., Ber. Bunsenges Phys. Chem, 67, 75, 84
(1963).
373. Krieger I. M., Hiltner P. A., Papir Y. S, Polym. Prepr, 11 (2), 690
(1969).
374. Efremov I. F., Lukashenko G. M., Us’yarov O. G. in В. V. Derjagin, ed.
Research in Surface Forces, Vol. 4, Consultants Bureau, New York, 1975.
375. Hachisu S., Kobayashi Y., Kose A., Colloid Interface Sci, 42, 342 (1973).
Коллоидный кремнезем — концентрированные золи
621
376. Kose A., Hachisu S., Colloid Interface Sci., 44, 330 (1973).
377. Kose A., Hachisu S., J. Colloid Interface Sci., 46, 460 (1973).
378. Hachisu S., Kobayashi U., J. Colloid Interface Sci., 46, 470 (1974).
379. Hachisu S., Kose A., Kobayashi Y., Takano K-, J. Colloid Interface Sci.,
55, 499, (1976).
380. Kirkwood J. G„ J. Chem. Phys., 7, 919 (1939).
381. Alder B. J., Wainright T. E., Phys. Rev., 127, 359 (1962).
382. Alder B. J., Hoover W. G., Young D. A., J. Chem. Phys., 49, 3688 (1968).
383. Monroe E. A., Monroe S. E., Science, 159, 97 (1968).
384. Greenwait С. H., Humming Birds, The American Museium of Natural
History, Doubleday & Co., Garden Citv., N. Y, 1960, Natl. Geogr., 658
(Nov. ’I960); 100 (Jan. 1963).
385. Williams R. C„ Smith К- M., Nature, 179, 119 (1957).
386. Xerox N„ Nature, 174, 562 (1954).
387. Bowden F. C., Smith K-, Chem. Ind., 17, 517 (1957).
388a. Lowenstam H. H., Science, 171, 487 (1971).
3886. Lowenstam H. A., Rossman G. R., Chem. Geol., 15, 15 (1975).
389. Kruyt H. R. Colloid Science, Vol. 1, Elsevier, Amsterdam, 1952, p. 74,
390. Freundlich H., Colloid and Capillar Chemistry, Methuen, London, 1925,
pp. 626—627.
391. Matijevic E., Bell A., Brace R., McFadyen P., J. Electrochem, 120, 893
(1973).
392. Catone D. L., Matijevic E., J. Colloid. Interface Sci., 48, 291 (1974).
393. Bell A., Matijevic E„ J. Phys. Chem., 78, 2621 (1974).
394. Watson J. H. L„ Cardell R. R„ Heller W„ J. Phys, Chem., 66, 1757
(1962). \
395. Gaskin A. Darragh P. J., пат. США 3497367 (Commonwealth Scien-
tific and Industrial Research Organization Australia), 1970.
396. Anonymous, New Sci. Aust, 5, 21 (1967).
397. Gilem S. A., 42, Chemin des Coudriers, 1211, Geneva (28).
398. Liddicoat R. T., Gems Gemol, 309, 1974.
399. Slocum J., M. D. I. Corp, 3417 Rochester Rd, Royal Oak, Mich.
400. Growningshield R., Gems Gemol, 14, 362 (1975).
401. Willcox О, пат. США 3907581 (Du Pont), 1975.
402. Iler R. K., J. Am. Ceram. Soc, 47 (4) 194 (1964).
403. Iler R. K-, J. Colloid Interface Sci, 24, 569 (1966).
404. Iler R. К, пат. США 3485658 (Du Pont), 1969.
405. Weyl W. A., Marboe E. C. The Constitution of Glasses, Vol. 2, Wiley,
New York, 1967.
406. Iler R. K-, J. Colloid Interface. Sci, 38 (2), 496 (1972).
407. Visser J., Paper 01, 49th National Colloid Symposium, Clarkson Col-
lege of Technology Potsdam, N. Y, June 14—16, 1975.
408. Gutowski W„ Inz. Chem, 3 (3) 477 (1973) [Chem. Abstr. 80, 87777].
409. Davies R., Ranade M. B., 49th National Colloid Symposium, Clarkson
College of Technology Potsdam, N. Y, June 16—18 1975.
410. Pauling L., Nature of the Chemical Bond, Cornel University Press,
Ithaca, N. Y, 1948.
411. Thomas C. L., Ind. Eng. Chem, 41, 2564 (1949).
412. Tamele M. W, Disc. Faraday Soc. (8), Heterogeneous Catalysis 270
(1950).
413. Milliken T., Jr, Mills G., Oblad A., Disc. Faraday Soc. (8) “Hetero-
geneous Catalysis” 279 (1950).
414. Alexander G. ~B., Iler R. К., пат. США 2897797 (Du Pont) 1959.
415. Matijevic E., Mangravite F. I., Cassell E. A., J. Colloid Interface Sci,
35, 560 (1971).
416. ' News Item, Chem. Eng. News, 39, 69 (October 2, 1961).
417. Simko F. A., Soap Chem, Spec, 39 (1), 97, 99, 101, 111 (1963).
622
Глава 4
418. Alexander G. В., пат. США 2913419 (Du Pont), 1959.
419. Alexander G. В., пат. США 2974108 (Du Pont), 1961.
420. Shafer M. W., Roy R., J. Am. Chem. Soc., 78, 1087 (1965).
421. Wagner E., пат. США 2951044 (Deut. Gold u. Silber Scheidenstalt), 1960.
422. Hazel F„ Gordon M., Schock R. K, Jr., J. Am. Chem. Soc., 71, 2256
(1949).
423. Yates P. С. пат. США 3634286 (Du Pont), 1972.
424. Alexander G. B., Bolt G. H., пат. США 3007878 (Du Pont), 1961.
425. Mindick M., Reven L. E., пат. США 3139406 (Nalco Chemical Co.) 1964.
426. Mindick M., Thompson А. С., пат. США 3244639 (Nalco Chemical Co.),
1966.
427. Mindick M., Thompson А. С., пат. США 3252917 (Nalco Chemical Co.),
1966.
428. Kovarik I. F., пат. США 3864142 (Nalco Chemical Co.), 1975.
429. Moore E. P., пат. США 3620978 and 3745126 (Du Pont), 1971 and 1973.
430. Chavannes I. L. В., англ. пат. 1271251 (Chem. Fabr. Pfersee GMBH),
1974.
431. Katsanis E. P„ Matijevic E., 49th National Colloid Symposium, Clarkson
College of Technology Polsdam N. Y., June 14—16, 1975.
432. Никольский Б. П., Андреев В. И., Дюбцев Р. И., Поляков М. С.— Ра-
диохимия, 1974, т. 16, с. 92.
433. Healy Т. W., Cooper R., James R. О., Am. Chem. Soc. Div. Water. Air
Waste Chem.. 7 (2), 91 (1967).
434. Baes C. F., Mesmer R. E. The Hydrolysis of Cations, Wiley, New York,
1976.
435. Matijevic E., Janauer G. E., Kerker M., J. Colloid Sci., 19, 333 (1964).
436. Iler R. К. пат. США 2692863 (Du Pont), 1954.
437. Hathaway F. S., пат. США 3485592 (Du Pont), 1969.
438. Rossos P. H. пат. США 3629139, 3652329 (Nalco Chemical Co.), 1971
and 1972.
439. Kovarik J. F., Vossos P. H., пат. США 3660301 (Nalco Chemical Co.),
1971.
440. Schaefer D. P., пат. США 3625856 (Nalco Chemical Co.), 1971.
441. Iler R. K., Pinkney P. S., Ind. Eng. Chem., 39, 1379 (1947).
442. Iler R. К, пат. США 2801185 (Du Pont), 1957.
443. Alexander G. В., пат. США 2921913 (Du Pont) 1960.
444. Broge E. С., пат. США 2739078 (Du Pont), 1956.
445. Broge E. С., пат. США 2736668 (Du Pont), 1956.
446. Akabayashi H. Yoshida A., Otsubo У., Kogyo Kagaku Zasshi, 68 (3),
429 (1965).
447. Stossel E„ пат. США 3004921 (1961).
448. Slack G. H., Copeland К. В., пат. США 2810738 (Copeland and Slack
Inc.), 1957.
1972.
449. Plata L. J., Vossos P. H., пат. США 3699049 (Nalco Chemical Co),
450. Iler R. К., пат. США 2739076 (Du Pont), 1956.
451. Iler R. К, пат. США 2974105 (Du Pont), 1961.
452. Rossos P. H., канад. пат. 954765 (Nalco Chemical Co.), 1974.
453. Albrecht W. L., Mindick M., пат. США 3351561 (Nalco Chemical Co.),
1967.
454. Mindick M., Curtis T. С., пат. США 3336235 (Nalco Chemical Co.),
1967.
455. Winyall M. E., пат. США 3660302 (W. R. Grace & Co.), 1972.
456 Albrecht W. L., Mindick M., пат. США 3351561 (Nalco Chemical Co.),
1967.
457. Akabayashi H., Yoshida A., Shimada M., япон. пат. 67-14698 (Nissan
Chem. Ind.), 1967 [Chem. Abstr., 69, 61823s],
Коллоидный кремнезем — концентрированные золи
623
458. Preston W. J., Lumley G. I., англ. пат. 965123 (Monsanto Chemical
Ltd.), 1964.
459. Ryznar J. W., пат. США 3004920 (Nalco Chemical Co.), 1961.
460. Iler R. К, пат. США 2801185 (Du Pont), 1957.
461. Benitez R., Contreras S., Goldfarb J., J. Colloid Interface Sci., 36, 146
(1971).
462. Iler R. К, пат. США 2786042 (Du Pont), 1957.
463. Youngs R. W., пат. США 3634288 (Nalco Chemical Co.), 1972.
464. Schwerin В., пат. США 1132394 (1915).
465. Griessbach R., Chem. Ztg. 57, 253, 274 (1933).
466. Meatman R. W., Prater C. D., Ind. Eng. Chem., 49, 253 (1957).
467. Bergna H. E., Simko F., пат. США 3284369 (Du Pont), 1966.
468. Drexel R. E., пат. США 2757073 (Du Pont), 1956.
469. Gentles R. P., пат. США 3425958 (British Hydrocarbon Chemical Ltd.),
1966.
470. McClellan W. R„ пат. США 3415886 (1968); Stiles A. B., McClellan W. R.,
пат. США 3497461 (1970); Sowards D. M., Stiles А. В., пат. США
3518206 (1970); McClellan W. R„ Stiles А. В., пат. США 3678139 (1972)
(all Du Pont).
471. Perolin Co., нидерланд. пат. 6410045 (1965).
472. Kirkland I. I., пат. США 3505785 (Du Pont), 1970.
473. Kirkland I. I., Anal. Chem., 37, 1458 (1965); 41, 218 (1969); J. Chroma-
tog. Sci., 7, 7 (1969); 7, 361 (1969); 8, 72 (1970); 10, 593 (1972).
474. Unger K., Renge P., Schick-Kalb I., Straube B., Z. Anal. Chem., 264,
267 (1973). /
475. Carter W. К, пат. США 2329589 (National Aluminate Corp.), 1943. .
476. Helgesson С. I., Persson B. S. S., Milsson О. I. II., пат. США 3883337
(Arbman Stig. А. В.). 1975.
477. Shaw С., англ. пат. 898103 (Zirconal Ltd.), 1962.
478. Fujita T., Wake M., пат. США 3682668 (Kobota Iron and /Machinery
Works), 1967.
479. Takano К, япоп. пат. 73-89210 (Hitachi Ltd.), 1973.
480. Noble R. D„ Bradstreet S. W., Rechter H. L., пат. США 2995453 (Ar-
mour Research Foundation), 1961.
481. Iler R. К, пат. США 3041205 (Du Pont), 1962.
482. Yates P. С., пат. США 3650783 (Du Pont), 1972.
483. Lee W. M., пат. США 3024125 (Pennsalt Co.), 1962.
484. Reuter R., пат. США 2856302 (National Aluminate Corp.), 1958.
485. Maziliauskus S., Ceram. Age., 72, 32 (1958).
486. Bergna H. E„ Ind. Quim., 28 (3), 123 (1970).
487. Bergna H. E., Simko F. А., пат. США 3301635 (Du Pont), 1967.
488. Haywood F. W., MacDiarmid I. R., англ. пат. 1332735 (Refractory Mold-
ings and Castings Ltd.), 1973.
489. Albert R. E., пат. США 3615778 (Du Pont), 1971.
490. Self I. M., пат. США 3674526, 3668168 (Du Pont), 1972.
491. Mercer R. S4 пат. США 2914413 (Pennsalt Corp.), 1958.
492. Iler R. K-, lelinek A. G., пат. 2886466 (Du Pont), 1959.
493. Nickerson R. F., пат. США 3024145 (Monsanto Chemical Co.). 1962.
494. Brown R. L., Sears G. W., пат. США 3224927 (Du Pont), 1965.
495. Edlin F. E., пат. США 3311585 (Du Pont), 1967.
496. Campbell R. А., пат. США 3077413 (Carborundum Co.), 1961 [Chem.
Abstr., 55, 25196e],
497. Myles T. А., пат. ФРГ 2331137 (Carborundum Co.), 1972 [Chem. Abstr.
80, 99493z],
498. Moore E. P„ пат. США 3752679—3752681, 3752689 (Du Pont), 1974.
499. Bauer I., пат. США, 2570750 (Fred Whitaker Co.), 1951.
624
Глава 4
500. McCarthy J. J. et al., пат. США 2570830 (Monsanto Chemical Co.),
1951.
501. Dempcy D., Rule J. M., пат. США 2980558 (Du Pont), 1961.
502. Iler R. К., пат. США 2801938 (Du Pont), 1957.
503. Colom-Pastor I. F., Martinez-Martinez F. J., Invest. Tec. Pap., 6 (19),
27 (1969).
504. Sun G. C., Shell T„ пат. США 3616196 (Du Pont), 1971.
505. Haag I. L„ пат. США 2663614 (Du Pont), 1956.
506. O’Shea B. J., пат. США 3184813 (Nalco Chemical Co.), 1965.
507. Rusher R. L„ пат. США 3421907—3421909 (Du Pont), 1969.
508. Sortwell E. T., англ. пат. 1101248 (Alchem, Ltd.), 1968.
509. Snyder W. T„ пат. США 3357481 (Nalco Chemical Co.), 1967 [Chem.
Abtsr., 68, 3246lw],
510. Pollard L., канад. пат. 784599 (Alchem. Ltd.), 1968.
511. Kearfott J. B., Baer С. E., пат. США 3509936 (Nalco Chemical Co.),
1970.
512. Rusher R. L., пат. США 3455705 (Du Pont), 1969 [Chem. Abstr., 71,
63755m],
513. Collins P. F., пат. США 2380945 (Austenal Labs. Inc.), 1945.
514. Cloherty D. J., Emblem H. G., Ind. Chem., 31, 111 (1955).
515. Reuter R., пат. США 2856302 (National Aluminate Corp.), 1958.
516. Emblem H. G., Mountford N. D. G., Merely C. W., пат. США 3051669
(Rolls Royce Ltd), 1962.
517. Emblem H. G., Compt. Rend Congr. Int. Chim. Ind. Liege, 31 (1958)
[publ. as Ind. Chim Beige. Suppl., 1, 874 (1959)].
518. Emblem H. G., Ind. Chem., 31, III (1955).
519. Strachan M. N., пат. США 2701902 (Monsanto Chemical Ltd.), 1955.
520. Reuter R„ пат. США 3222737 (Nalco Chemical Co.), 1965.
521. Baer С. E., пат. США 3436235 (Eutectic Eng. Co.), 1969.
522. Preece R. E., пат. США 3445250 (Nalco Chemical Co.), 1969.
523. Beyer J. N., Moore E. P., Jr., Rusher R. L., пат. США 3748157 (Du Pont),
1973.
524. Moore E. P„ пат. США 3764355 (Du Pont), 1973.
525. Moore E. P., пат. США 3748157 (Du Pont), 1973.
526. Moore E. P„ пат. США 3754945 (Du Pont), 1973.
527. Reuter R„ пат. США 2949375 (Nalco Chemical Co.), 1960.
528. Boehm B., Schaupp K., -Schober P., Simons D., пат. ФРГ 2110058 (Far-
benfabrik Bayer A. G.), 1972.
529. Newell W. J., Madden J. E., пат. США 3042537 (1962).
530. Pelts C. R„ Am. J. Sci., 254, 32 (1956).
531. Dove L. D., пат. США 2623828 (Porolith Inc.), 1952.
532. Weidman V. W., Yates P. С., пат. США 3725095 (Du Pont), 1973.
533. Gerow S. А., пат. США 3661602 (Du Pont), 1972.
534. Gerow S. A., Weidman V. W„ пат. США 3658564 (Du Pont), 1972.
535. Shannon R. E., пат. США 3810773 (Owens-Corning Fiberglas Corp.).,
1974.
536. Howard R. N., Sowman H. G., пат. ФРГ 2414047 (Minnesota Mining
and Mfg. Co.), 1974.
537. Iler R. К., пат. США 2597871 (Du Pont), 1952.
538. Iler R. К., пат. США 2726961 (Du Pont), 1955.
539. Powers D. H., Harrlson'D. H., пат. США 2443512 (Monsanto Chemical
Co.), 1948.
540. DiMaio V., пат. США 2515949 (Monsanto Chemical Co.), 1950.
541. Marshall M. D., пат. США 2515960 (Monsanto Chemical Co.), 1950.
542. Marshall M. D., пат. США 2515961 (Monsanto Chemical Co.), 1950.
543. Powers D. H., Harrison J. H., пат. США 2526684 (Monsanto Chemical
Co.), 1950.
Коллоидный кремнезем — концентрированные золи
625
544. Powers D. Н., Harrison J. Н., пат. США- 2527329 (Monsanto Chemical
Со.), 1950.
545. Skalkeas В. G., пат. США 2590659 (Monsanto Chemical Со.), 1952.
546. Powers D. Н., пат. США 2635056 (Monsanto Chemical Со.), 1953.
547. Kingsford С. L., пат. США 2693427 (Monsanto Chemical Со.), 1954.
548. Rassin Е. Н., пат. США 2696444 (Monsanto Chemical Со.), 1954.
549. Nickerson R. F., пат. США 2701218 (Monsanto Chemical Co.), 1955.
550. Skalkeas B. G., пат. США 2724657 (U. S. Dept, of Agriculture), 1955.
551. Simko F. A. Jr., Thompson С. В., пат. США 2910374 (Du Pont), 1959.
552. Vossos P. H., пат. США 3652329 (Nalco Chemical Co.), 1972.
553. Reider M. J., пат. США 2541780 (G. W. Bollman & Co.), 1951.
554. Bauer J., пат. США 2570750 (Fred Whittaker Co.), 1951.
555. Barnett I., Zager S. E., пат. США 2799915 (Johns Manville Corp.), 1957.
556. Payne J., Smith E., Br. Rayon Silk J., 30 (353), 91 (1953).
557. Dithmar K., Text Prax, 10, 1134 (1955).
558. Caroselli R. F., пат. США 2754221—2754224 (Owens-Corning Fiberglas
Co.), 1956.
559. Crooks L. L., Harshfeld J. J., белы. пат. 641992 (Monsanto Co.), 1964.
560. Wilson 1. V., пат. США 2643048 (Monsanto Chemical Co.) 1953.
561. Turner L., пат. США 3032401 (Du Pont), 1962.
562. Brockett B. W., пат. США 3179600 (National Cash Register Co.), 1965.
563. Leptein E. E., пат. США 2872094 (St. Regis Paper Co.) 1959.
564. Vossos P. H., пат. США 3649348 (Nalco Chemical Co.), 1972.
565. Payne С. C., Vossos P. H., пат. США 3901987 (Nalco Chemical Co.),
1975.
566. Payne С. С., пат. США 3754984 (Nalco Chemical Co.), 1974.
567. Fletcher C. H„ Jr., Tappi, 56 (8), 67 (1973).
568. Fisher F. G„ Allen R. K, Luvisi G. W., Trans. ASME, 80, 1037 (1958).
569. Luvisi G. W., канад. пат. 567315 (National Aluminate Corp.), 1958.
570. Nohejl T. С., канад. пат. 567300 (National Aluminate Corp.). 1968.
571. Ryznar J. W., пат. США '2877716 (Nalco Chemical Co.), 1959.
572. Bott L. L., пат. США 3547663 (Nalco Chemical Co.), 1970.
572a. Jewett H. А., пат. США 2675730 (1954).
573. Moede J. A., Schumacher D. F., пат. США 3547664 (Du Pont), 1970.
574. Woodward A. I., Challis A. J., Way A. P., пат. США 3676189 (Wiggins
Teape Res. & Deve., Ltd.), 1972.
575. News item, Chem. Week, 76, 74 (1955).
576. Cogovan E. J., Frederici E. D., пат. США 2622307 (Mohawk Carpet
Mills, Inc.), 1952.
577. Florio P. A., Rainard L. W., пат. США 2734835 (Alexander Smith, Inc.),
1956.
578. Florio P. А., пат. США 2786787 (Mohasco, Inc.), 1957.
579. Rusher R. L„ Yates P. С., пат. США 2877142 (Du Pont), 1959.
580. Remers N. A., Aarons R., пат. США 2881146 (Du Pont), 1959.
581. Aarons R., Schlatter R., пат. США 2891874 (Du Pont), 1959.
582. Aarons R., Bugosh J., пат. США 3033699 (Du Pont), 1962.
583. Walsh J. S., Milton C. L., пат. США 2978349 (Eastern Products Cor.),
1961.
584. Shaeffer V. E., Gardner G., Falivene P. J., Dillarstone А., пат. США
3630919 (Colgate-Palmovile Co.), 1971.
585. Nagai Y., япон. пат. 70-41639, 1970 [Chem. Abstr., 76, 101147с].
586. Payne С. C., Bloemke R. E., Schaefer D. P., пат. США 3901992 (Nalco
Chemical Co.), 1975.
587. Ferch H., Seifen, Oele, Fette Wachse, 101 (2)^51 (1975).
588. Bainbridge J. P., Jr., пат. США 2673520 (Monsanto Chemical Co.), 1954.
589. Jewitt C. L., Case J. M., пат. США 2714066 (Minnesota Mining and
Mfg. Co.), 1955.
14 Заказ № 250
626
Глава 4
590. Nichols О. S., пат. США 2780168 (One-half to J. H. Schneider), 1957.
591. Marron T. U., пат. США 2800077 (A. В. Dick Co.), 1957.
592. Blake R. К, пат. США 3547641 (Du Pont), 1970.
593. Kenney J. T„ пат. США 3657003 (Western Electric Co.), 1972.
594. Grot F. G., пат. США 3702267 (Du Pont), 1972.
595. Wolinsky L. E„ пат. США 2909443 (Du Pont), 1959.
596. Doban R. С., пат. США 2906658 (Du Pont), 1959.
597. McIntyre J. W., Hechtman I. F., пат. США 2803560 (Kimberly-Clark
Cor.), 1957.
598. Rusher R. L., пат. США 3380876 (Du Pont), 1968.
599. Huntsberger J. R., пат. США 2825664 (Du Pont), 1958.
600. Caroselli R. F., пат. США 2754221, 2754223 (Owens-Corning Hiberglas
Corp.), 1956.
601. Iler R. К., пат. США 2833661 (Du Pont), 1958.
602. Powers D. H., пат. США 2555506, 1951.
603. Keller H., Brodt R., Lampatzer К., пат. ФРГ 1287888 (Metallgesellschafl
A. G.), 1969.
604. Cupery M. E„ Iler R. К, пат. США 3013897 (Du Pont), 1961.
605. Okada H., Tamura H., пат. США 3506499 (Yawata Iron and Steel Co.),
1970.
606. Strolle С. FL, пат. США 3655604 (Du Pont), 1972.
607. Bunger P., Levitsky M., пат. США 3689300 (Du Pont), 1972.
608. Elmes О. С., пат. США 3896253 (General Tire and Rubber Co.), 1975.
609. Sakata M., Tanaka M., япон. пат. 73-81930 (Mitsuboshi Belting Ltd.),
1973.
610. Hogan J. P., пат. США 3627649 (Phillips Petroleum Co.), 1971.
611. DeVries E. R., пат. США 3177083 (Prismo Safety Cor.), 1962.
612. Nikolics K-, Weltler I., Gyogyszereszet, 10 (2), 53 (1966) [Chem. Abstr.,
65, 207If].
613. Gransden I. F., Sheasby I. S., Can. Metall. Q., 13 (4), 649 (1974).
614, Anderson J. C., Heffelfinger С. L, пат. США 3808027 (Du Pont), 1974.
615. Debus E., пат. ФРГ 1470784 (Deut. Gold u. Silber-Scheideanstalt), 1971.
616. Breton E. I., пат. США 2968999 (Du Pont), 1961.
617. Seymour R. В., пат. США 2467339—2467342 (Monsanto Chemical Co.),
1949.
618. Ikeda С. K-, пат. США 2592147 (Du Pont), 1952.
619. Iler R. K-, пат. США 2597872 (Du Pont), 1952.
620. McBride R. T., пат. США 2921869 (Du Pont), 1960.
621. Bullitt О. H„ Storrow R. R„ пат. США 3069375 (Du Pont), 1962.
622. Cull N. L., пат. США 3314911 (Esso Research & Engineering Co.), 1967.
623. Abress M. S„ McDowell M. J., пат. США 3687885 (Du Pont), 1972.
624. Barbaras G. D., пат. США 3057744 (Du Pont), 1962.
625. Nickerson R. F., пат. США 2799658 (Monsanto Chemical Co.), 1957.
626. Payne С. C., Vossos P. H., пат. США 3711416 (Nalco Chemical Co.),
1972.
627. Schmidt D. L„ Smith H. B„ Waites E. Г., пат. США 3813363 (Dow Che-
mical Co.), 1974.
628. Dempcy D. F., пат. США 3013898 (Du Pont), 1961.
629. Shimano M., япон. пат. 74-44564 (Kubota Ltd.), 1974 [Chem. Abstr., 83,
117136d].
630. Unilever Ltd., англ. пат. 1059084, 1967 [Chem. Abstr., 66, 88368x],
631. Iwata Y., япон. пат. 73-34938 (Dainippon Printing Co., Ltd.), 1973.
632. lackson P. R., пат. ФРГ 2311627 (International Nickel Co.), 1973.
633. Pearlman M. В., пат. США 3796608, 1973.
634. Adams N. M., пат. США 3177085 (Nalco Chemical Co.), 1965.
635. Cox I. I., Jr., пат. США 2910395 (Du Pont), 1959.
Коллоидный кремнезем — концентрированные золи
627
636. Vessey С. A., Albon С., Holker К., пат. США 3706603 (Albright and
Wilson, Ltd.), 1972.
637. Koepp R. L., Havas J., пат. США 3663277 (National Cash Register),
1972.
638. Robinson J. С., пат. США 2641556 (General Electric Co.), 1953.
639. Robinson J. С., пат. США 2809137 (General Electric Co.), 1957.
640. Allen W. S„ Romig О. E„ пат. США 2811473 (U. S. Steel Corp.), 1957.
641. Cupery M. E., Jackson R. P., Wright W. H., пат. США 3133829 (DuPont),
1964.
642. Frey S. S., пат. США 2780591 (Oakite Products, Int.),- 1957.
643. Aabrust P., Berg H., Heil Rath E., франц, пат. 2030373 (Metallgesell-
schaft A. G.), 1970.
644. U. S. S. Engineers and Consultant, Inc., пат. США 3839256 (1975).
645. Gummerman О. пат. США 3684755 (Du Pont), 1972.
646. Engelhardt E. H„ пат. США 3514425 (Du Pont), 1970.
647. Vest R. D„ пат. США 3546318 (Du Pont), 1970.
648. Strolle С. H., пат. США 3655604 (Du Pont), 1972.
649. Smith-Johannsen R., пат. США 2803566 (S-J Chemical Co.), 1957.
650. Smith-Johannsen R., пат. США 3179544 (Chemelex, Inc.), 1965.
651. Gottfried W., Horst F„ пат. ГДР 48-563, 1966 [Chem. Abstr. 65, 1557411].
652. Mastrianni V., пат. США 3717500 (Owens-Corning Fiberglas Corp.),
1973 [Chem. Abstr., 78, 137864q],
653. Haas H. С., пат. США 3287154 (Polaroid Corp.), 1966.
654. Smith-Johannsen R., пат. США 2995529 (Chemelex, Inc.), 1961.
655. McMahon W. M., Abba C. G., пат. США 3130061 (American Pipe and
Construction Co.), 1964.
656. Rusher R. L., пат. США 3421907 (Du Pont), 1969.
657. W-eldes H. H„ Netting D. J., пат. США 3372038 (Philadelphia Quartz
Co.), 1964-.
658. Wright M. G. B., Paint. Varn. Prod., 61 (10), 65 (1971).
659. Suzuki K- et al., япон. пат. 74-15721 (Nissan Chemical Ind.), 1974 [Chem.
Abstr., 82, 87770г].
660. Plum H., Runggas F., Van Haaren M., Krase H., пат. ФРГ 2240487
(Schering A. G.), 1974.
661. Pruett R. D., пат. США 3156666 (Du Pont), 1964.
662. Pruett R. D., пат. США 3061577 (Du Pont), 1962.
663. Symons N. К, пат. США 2874139 (Du Pont), 1959.
664. Dithmar К, пат: США 2819173 (Deut. Gold u. Silber-Scheideanstalt),
1958.
665. Adams N. M., пат. США 3371712 (Nalco Chemical Co.), 1968.
666. Iler R. К., пат. США 2760941 (Du Pont), 1956.
667. Bennett B., Novy L. E., Simko F. A., Jr., пат. США 3057750 (Du Pont),
1962.
668. Farrow J. E., Vossos P. H., пат. США 3582502 (Nalco Chemical Co.),
1971.
669. Talalay A., Coffey W. D„ Talalay J. А., пат. США 2926390 (В. F. Good-
rich Co.), 1960.
670. Farrow J., Vossos P. H., англ. пат. 1314704 (Nalco Chemical Co.)
[Chem. Abstr., 75, 50253d],
671. Seymour R. В., пат. США 2467340 (Monsanto Chemical Co.), 1949.
672. Iler R. К., пат. США 2833661 (Du Pont), 1958.
673. Iler R. К., пат. США 3773776 (Du Pont), 1973 [Chem. Abstr., 80, 61193p],
674. Kogishi S., Ishihara T., япон. пат. 75-02049 (Kuraray Co.), 1975.
675. Walsh R. J., Herzog A. H., пат. США 3170273 (Monsanto Chemical Co.),
1965 [Chem. Abstr. 62, 11293a; 66, 12527с].
676. Sears G. W., пат. США 3922393 (Du Pont), 1975.
14*
628
Глава 4
677. Farkas М., Fikar Е., Rozsa Р., Zoltai G., венгер. пат. 6844 (1973) [Chem.
Abstr., 80, 64977И].
678. Tredinnick В. C., Gambale R. J., Dupree P. M., пат. США 3715842 (Tizon
Chemical Corp.), 1973.
679. Cromwell W. E., пат. США 3807979 (Philadelphia Quartz Co.), 1974.
680. Okura T., Goto R., Murat M., Mem. Fac. Eng. Hokkaido Univ., 11, 25
(1960).
681. Goto R., Yotsuyanagi T., Kagaku to Kogyo, 16, 1406 (1963).
682. Bennett B., Novy L. E., Simko F. A., Jr., пат. США 3057750, 1962; англ,
пат. 870945, 1961 [Chem. Abstr., 55, 26480] (both Du Pont).
683. Wibur В. С., пат. США 3536875 (Atlantic Richfield Co.), 1970.
684. Dow Chemical Co., англ. пат. 952351, 952352, 1964 [Chem. Abstr., 61,
1304d, 13493e],
685. Brockett B. W., пат. США 3179600 (National Cash Register Co.), 1965.
686. Ryznor J. W., пат. США 3037886 (Nalco Cehmical Co.), 1962.
687. McDonald L., Soap Chem. Spec., 47, 42 (June 1971).
688. McDonald L., пат. США 3899447, 1975.
689. Lipsett S. G., пат. США 3582460 (Asbestos Corp.), 1971.
690. Povich M. J., AIChE. J., 21 (5), 1016 (1975).
691. Youngs R. W„ пат. США 3634288 (Nalco Chemical Co.), 1972.
692. Hayano R., япон. пат. 75-01475 (Nippon Oils and Fats Co.), 1975.
693a. Глейм В. Г., Хейтов В. 10., Башиков М. М.— Коллоидн. ж., 1966, т.' 28,
с. 648; 1968, т. 30, с. 607.
6936. Rulkarni R. D., Goddard Е. D., Runner В., J. Colloid Interface Sci., 59,
468 (1977).
694. Wightman J. P., Chessick J. J., J. Phys. Chem., 66, 1217 (1962).
695. Rauter R., пат. США 3180760 (Marc. Inc.), 1965.
696. News item, Chem. Week, 95, 53 (1964).
697. Anonymous, Chem. Eng. News, 46 (June 1966).
698. Geffcken W. G., Berger E., пат. США 2366516 (Allen Property Custo-
dian), 1945.
699. Moulton H. R., пат. США 2432484 (American Optical Co.), 1947.
700. Thomsen S. M., Nicoll F. H., пат. США 2505629 (Radio Corp, of Ame-
rica), 1950.
701. Moulton H. R., пат. США 2601123 (American Optical Co.), 1952.
702. Wills J. H., Hazel J. F„ пат. США 2649388 (Philadelphia Quartz Co.),
1953.
703. McLean S., пат. США 2639999 (General Electric Co.), 1953.
704. Moulton H. R., пат. США 2531945 (American Optical Co.), 1950.
705. Heany J. А., пат США 2442976 (1948) [Chem. Abstr., 42, 6073f],
706. Land E. H., Bloom S. M., пат. США 3883368 (Polaroid Corp.), 1974.
707. Первеев А. Ф., Золотарев E. M., Егеров П. П., Муранова Г. A.— Оптика
и спектроскопия, 1972, т. 32, с. 607.
708. Крылова Т. Н., Соколова Р. С.— Оптика и спектроскопия, 1957, т. 2,
с. 254.
709. Первеев А. Ф., Муранова Г. А., Фролова Н. П.— Оптико-механ. пром.,
1972, т. 39, с. 11.
710. Еолтун М.— Ж. прикл. спектр., 1970, т. 12, с. 350.
711. Крылова Т. Н.— Оптика и спектроскопия, 1958, т. 4, с. 217.
712. Александров Г. А.— Оптико-механ. пром., 1967, т. 34, с. 35.
713. Vedan R., Lai R., Lukes F., Srinivasan R., J. Opt. Soc. Am., 58, 526
(1968) [Chem. Abstr., 68, 100073v],
714. Rlenke E. F„ Lewis G. L„ пат/ США 3650790 (Du Pont), 1972.
715. Anderson I. T., пат. США 2740726 (General Electric Co.), 1956.
716. Werner L. C., Lopenski S. A., Rainone N. J., пат. США 2806444 (Wes-
tinghouse Electric. Corp.), 1957.
Коллоидный кремнезем — концентрированные золи
629
717. Thorington L., Peterson R. Е., пат. США 2806968 (Westinghouse Elec-
tric Corp.), 1957.
718. Self J. M„ Yates P. С., пат. США 3552992 (Du Pont), 1971.
719. Lanzet M., 3266995 (Yardley of London, Inc.), 1966.
720, Jahoda E., пат. США 2433515 (H. P. Andrews Paper Co.), 1947.
721. Von Glahn W. FL, Stanely L. N., пат. США 2566709 (General Aniline and
Film Corp.), 1951.
722. Frederick J. E., пат. США 2784089 (General Aniline and Film Corp.),
1957.
723. Unkauf H. С., пат. США 2805158 (Eugene Dietzgen Co.), 1957.
724. Land E. H. et al., пат. США 2698236—2698238; 2698245; 2705676;
2759825; 2765240; 2774667 (Polaroid Corp.), 1954—1956.
725. Gray R. FL, пат. США 2878121 (Du Pont), 1959.
726. Pakunov J., франц, пат. 2005840 (Eastman Kodak Co.), 1969.
727. Saleck IF., Himmelmann IF., Huckstadt H., Meyer R., белы. пат. 766095
(Agfa-Gevaert), 1971.
728. Dick Co. А. В., нидерланд. пат. Appl. 6500536, 1965.
729. Pertoft H., Anal. Biochem., 38, 506 (1970).
730. Pertoft FL, Laurent T. C., (Gerritsen T., Ed.), Modern Separation Met-
hods of Macromolecules and Particles, Vol. 2, Wiley-Interscience, New
York, 1969, p. 71.
731. Lyttleion J. IF., Anal. Biochem., 38, 377 (1970).
732. Morgenthaler J. J., Price C. A., Robinson J. M., Gibbs M., Plant Physiol.,
54 (4), 532 (1974).
733. Morgenthaler J. J., Marsden M. P. F., Price C. A., Arch. Biochem. Bio-
phys., 168, (1), 289 (1975).
734. Lindquist L, Nillsson O., Ronquist G., Uppsala J. Med. Sci., 79 (1), 1
(1974).
735. Pertoft FL, Back O., Lindall-Kiesseling K., Exp. Cell. Res., 50 (2), 355
(1968).
736. Mateyko G. M„ Kopak J. J., Exp. Cell. Res., 17, 524 (1959).
737a. Wolff D. A., Pertoft FL, Biochem., Biophys. Acta., 286, 197 (1972).
7376. Percoll is the registered trademark of Parmacia Fine Chemicals AB,
Uppsala, Sweden.
737b. Pertoft FL, Laurent T. C., A New Density Gradient Material, “Percoll”,
Anal Biochem., (1977) [in press].
737r. Price C. A., Reardon E. M., Guillard R. R. L., “Centrifugation of Plank-
ton in Percoll”, J. Limnol. Oceanogr. (1977).
737д. Pertoft FL, Laurent T. C., (Catsimpoolas N., Ed.), Methods of Cell Sepa-
ration, Vol. 1, Plenum, New York, 1977.
737e. Wolff D. A., Pertoft FL, J. Cell. Biol., 55, 579 (1972).
Z38._ Iler R. К, пат. США 3492137 (Du Pont), 1970.
739. Iler R. К, пат. США 2668149 (Du Pont), 1952.
740. Fisher I. R., Rigterink M. D., пат. США 2883347 (Bell Telephone Labs,
Inc.), 1959.
741. Walsh R. J., пат. США 3161468 (Monsanto Co.), 1964.
742. Potter C., Lindenthal J. IF,, пат. США 3177057 (Engelhardt Inc.), 1965.
743. Broge E. C., Iler R. K-, пат. США 2680677 (Du Pont), 1954.
744. McCarthy G. G„ Roy R., McKay J. M., J. Am. Ceram. Soc. (Dis.), 54,
637 (1971).
745. Okkerse C.; DeBoer I. II., (De Boer J. H. Ed.), Reactivity of Solids,
Elsevier, New York, 1961.
746. Iler R. К, пат. США 3761936 (Du Pont), 1973.
747. Elmer T. FL, Nordberg M. E., пат. США 3459522 (Corning Glass Co.),
1969.
748. Krohn D. A., Cooper A. R., J. Am. Ceram. Soc., 52, (12), 661 (1969).
749. Roy R., J. A. Ceram. Soc., 52, 344 (1969),
630
Глава 4
750. Tarnopol М. S., пат. США 2694649 (Pittsburgh Plate Glass Co.), 1954.
751. “Cement”, Encyclopaedia Britannica, 14th ed., The Encyclopaedia Bri-
tannica Co., Chicago, Vol. 5, 110; also “Rome”, Vol. 19, p. 465.
752. Wieker W„ Hahn U., Z. Chem., 13 (5), 195 (1973).
753. Greenberg S. A., J. Phys, Chem., 61, 373 (1957).
754. Greenberg S. A., J. Phys. Chem., 65, 12 (1961).
755. Assarsson G. 0., J. Phys. Chem., 61, 473 (1957); 62, 223 (1958).
756. Albert R. E„ пат. США 3615778 (Du Pont), 1971.
757. DeJong J. G. M., Stein H. N., Stevels J. M., J. Appl. Chem., 19 (1), 25
(1969).
758. Смирнова H. Г., Душина А. Д., Алексовский В. Б.— Изв. АН СССР,
Неорган. материалы, 1966, т. 2, с. 284.
759. Hast N., Ark. Kern., 9, 343 (1956).
760. Granquist W. T., пат. США 3252757 (National Lead Co.), 1966.
761. Granquist W. T., Pollack S. S., Proc. Natl. Conf. Clays, Clay Miner., 8,
150 (1960).
762. Shea F. L. Jr., Hsu J. L, Steebels J. M., J. Appl. Chem., 119, 125 (1969).
763. Shea F. L., Jr. et al., пат. США 2698256 (Great Lakes Carbon Corp.),
1954.
764. Makart H., Helv. Chim. Acta, 50, 399 (1967),
765. Мицюк Б. M.— Ж- неорг. хим., 1972, т. 17, с. 903.
766. Anderson R. C., Sleddon G. J., Chem. Ind. London, 1960, 1335.
767. Streicher J. S., пат. США 2752665 (J. T. Baker & Co.), 1956.
768. MacCragh A. P., Patil A. S., Ashby G. E., пат. США 3713816 (W. R. Grace
Co.), 1973.
769. Swartzen-Allen S. L., Matijevic E., Chem. Rev., 74, 385 (1974).
Глава 5
СИЛИКАГЕЛИ И ПОРОШКИ
Определения
Рассматриваемые здесь формы кремнезема * представляют
собой твердые вещества, имеющие величины удельных поверх-
ностей более -~5 м2/г. По своему строению такие вещества
могут быть чрезвычайно разнообразными: от больших по
размеру твердых масс до субмикроскопических частиц, а по
степени гидратации — от почти безводного кремнезема до
пластичных, студенистых масс, содержащих 100 ч. воды на 1 ч.
SiO2. Общей чертой силикагелей и порошков является то, что
они состоят из первичных кремнеземных частиц коллоидного
размера, имеющих диаметр от 1 до 100 нм.
Структура геля кремнезема может быть представлена
в виде связанной, жесткой трехмерной сетки из соприкасаю-
щихся между собой частиц коллоидного кремнезема. Форми-
рование кремнеземного геля путем полимеризации кремневой
кислоты подробно рассматривалось в гл. 3, а его образование
посредством агрегации частиц коллоидного кремнезема —
в гл. 4.
Кремнеземный порошок может содержать небольшие гра-
нулы геля кремнезема или сцепленные агрегаты, состоящие
в свою очередь из частиц субмикронного размера, причем такие
агрегаты соединяются вместе в чрезвычайно непрочные сетки.
Теоретически порошок кремнезема может состоять из раздель-
ных, дискретных кремнеземных частиц, но в том случае, когда
диаметр частиц менее 100 нм, подобные частицы самопроиз-
вольно слипаются вместе в рыхлые, неплотные агрегаты.
И только когда дискретные частицы оказываются много боль-
шими по размеру, т. е. их диаметр находится в интервале
5—50 мкм (5000—50 000 нм), когезионные силы становятся
настолько слабыми, что частицы уже не притягиваются друг
к другу и оказываются «пылящими» — при малейшем колеба-
нии воздушной среды образуют «дым».
* В отечественной научно-технической литературе . твердый высушенный
гель носит название «силикагель», а невысушенный гель с большим содер-
жанием воды называется «гидрогелем». В ряде стран терминология отличается
от принятой в нашей стране.— Прим,, ред.
632
Глава 5
Имеются и такие типы кремнезема, которые можно пред-
ставить как пограничные между силикагелями и порошками.
В том случае, когда связи между первичными коллоидными
частицами очень слабы и способны легко разрываться при
Упаковка
Рис. 5.1. Схемы поперечных сечений различных структурных разновидностей
силикагелей и порошков кремнезема.
А—частицы малого размера, плотная упаковка, коалесценция слабая; В — частицы
малого размера, открытая упаковка, коалесценция слабая, С — частицы большого раз-
мера, плотная упаковка, коалесценция слабая, D — частицы большого размера; открытая
упаковка, коалесценция слабая; 5 —частицы большого размера, плотная упаковка,
коалесценция сильная; F — частицы большого размера, открытая упаковка; коалесцен-
ция сильная. Под коалесценцией понимается степень связывания двух частиц или их
совместного формирования.
механических воздействиях, такой кремнезем можно класси-
фицировать как порошок. С другой стороны, когда коллоидные
частицы формируются в виде прочно связанной структуры, то
кремнезем классифицируется как силикагель, несмотря на то,
что размер гранул этого кремнезема может доходить до не-
скольких микрон.
На рис. 5.1 схематически представлены такие меняющиеся
величины, как размер частиц, плотность их упаковки, порис-
тость, способность частиц к слипанию или же прочность агре-
гирования.
Типы гелей
Гели кремнезема формируются в жидкой среде, как правило
в водной. Термины алкогель (спиртовый гель) и аквагель
(водный гель, гидрогель) относятся к гелям, поры которых
Силикагели, и порошки
633
заполнены соответствующими жидкостями, т. е. спиртом или
водой. Ксерогель (силикагель) представляет собой гель, из
которого удалена жидкая среда, так что структура оказывается
сжатой, а величина пористости — до некоторой степени пони-
женной за счет сил поверхностного натяжения, действовавших
в процессе удаления жидкости. Аэрогелем называется спе-
циальный тип ксерогеля, жидкая фаза из которого была уда-
лена таким способом, который позволяет предотвращать
какое-либо сжатие или изменение структуры при удалении
жидкости. В частности, это выполняется нагреванием запол-
ненного жидкостью ‘ геля, обычно алкогеля, в автоклаве выше
критической точки используемой жидкой фазы до исчезновения
поверхности раздела жидкость—пар с последующим удалением
такого пара.
Пористое стекло представляет собой по существу особую
форму силикагеля.
Типы порошков
Кремнеземные порошки можно классифицировать в соответ-
ствии со способом приготовления, вызывающим появление
характерных особенностей порошка.
Порошкообразные силикагели приготовляются посредством
размалывания или очень тонкого измельчения ксерогелей.
Первичная структура геля при этом остается неизменной.
Силикагели шаровидной формы приготовляются путем раз-
деления первичной кремневой кислоты или коллоидного золя
на мелкие капельки перед процессом гелеобразования. Струк-
тура такого геля сходна со структурой массивных, сплошных
образцов гелей, сформированных при тех же самых условиях.
Такие капельки можно суспендировать в воздушной среде
и затем высушивать или же суспендировать в несмешиваю-
щейся жидкости, в которой капельки затвердевают. Подобные
капельки могут также формироваться в водной фазе посред-
ством коацервации коллоидного кремнезема при добавлении
органическоого агента; образующиеся' капельки затем затвер-
девают.
Осажденный кремнезем образуется в том случае, когда пер-
вичные частицы кремнезема коагулируют в водной среде в виде
рыхлых агрегатов, которые выделяют, промывают и высуши-
вают. Коагуляцию можно осуществлять посредством добавле-
ния соли с высокой концентрацией или добавления других
коагулянтов, таких, например, как аммиак, способных смеши-
ваться с водой растворителей или некоторых типов органиче-
ских веществ. Когда первичные частицы по размеру больше
чем 5—10 нм, то они могут лишь слабо связываться друг
634
Глава 5
с другом. Если затем создаются условия для образования
структуры с открытой упаковкой, то частицы могут легко отры-
ваться и диспергировать в таких средах, как масла или каучук.
Аэросилы, или пирогенные кремнеземы — порошки, приготов-
ленные конденсированием кремнезема из паровой фазы при
повышенной температуре. Пар кремнезема можно получать
следующими способами: а) прямым испарением SiO2; б) вос-
становлением SiO2 до летучего соединения SiO, которое затем
повторно окисляется; в) окислением, летучих соединений крем-
ния, таких, как хлориды или сложные эфиры; г) гидролизом
паровой фазы SiF4.
Филлокремнезем (листоподобный) и лепидоидальный (че-
шуйчатоподобный) кремнезем менее известны и плохо охарак-
теризованы, но в основном состоят из частиц слоистой или
пластинчатой формы. Как уже рассматривалось в гл. 1, су-
ществует несколько типов таких кремнеземов в зависимости
от способа их получения.
Органофильные кремнеземы приготовляются из всех выше-
приведенных типов порошков путем нанесения на поверхность
мономолекулярного хемосорбированного слоя, состоящего из
органических групп, присоединенных к поверхностным атомам
через связи Si—С, Si—О—С, Si—O~N+—С, Si—О—М—О—С
(где М — многовалентный катион металла).
Алюмосиликатные анионы могут внедряться в кремнезем-
ную поверхность, создавая на ней более сильный анионный
заряд с большей катионообменной способностью и большей
кислотной каталитической активностью.
Быстро растет поток научно-технических публикаций,
имеющих отношение к различным аспектам тонкодисперсных
кремнеземов, как правило связанным с какими-либо конкрет-
ными приложениями или областями технологии. В ряде обзо-
ров [1 — 10в] в большинстве случаев достаточно подробно
обобщены разнообразные аспекты силикагелей и порошков.
Несмотря на большой объем литературы, кажется, что раз-
личные проблемы этой темы еще не сведены вместе в один
удобный источник информации.
Физические характеристики
Большинство методов получения характеристик приложимо
в равной мере и к силикагелям, и к порошкам. Фон Вузах [Юг]
в 1937 г. четко установил переменные величины, которые
должны приниматься в расчет при определении агрегирован-
ных структур:
1. Размер и форма первичных частиц.
Силикагели и порошки
635
2. Пространственное распределение частиц, включая поря-
док и плотность упаковки (см. рис. 5.1).
3. Прочность связи между частицами (коалесценция).
Автор далее указал, что размер частиц влияет на вели-
чину удельной поверхности; кроме того, при данной плотности
упаковки размер частиц определяет и размер пор или «меж-
мицеллярных промежутков», 'полностью пронизывающих агре-
гат или гель.
Размер первичных частиц
Методы определения подробно рассматривались в гл. 4
в связи с описанием коллоидных частиц в кремнеземных золях,
причем такие частицы во многих случаях перед получением их
характеристик высушивались до силикагеля или порошка.
Поэтому те же самые методы приложимы и в настоящем
случае.
Электронно-микроскопические снимки
Даже в компактных силикагелях, когда первичные частицы
упакованы плотно вместе, представляется возможным разли-
чать первичные частицы в тонком срезе кусочка геля. Приме-
няя методики, разработанные Коханом и Уотсоном [11] для
частиц газовой сажи, можно измерять видимый диаметр частиц
в тех случаях, когда частицы располагаются таким образом,
что их силуэты на поперечном срезе охватывают более поло-
вины круга. После измерения размеров нескольких . сотен
частиц можно подсчитать средние величины их диаметров,
выражаемые как среднечисленный диаметр dn или как средне-
поверхностный диаметр ds. Последняя величина представляет
собой диаметр частицы, имеющей удельную поверхность, рав-
ную средней величине удельной поверхности, подсчитанной для
всех измеренных частиц:
где п; — число частиц в г-м интервале размеров диаметров,
для которого среднее значение диаметра равно dp k — число
таких интервалов.
Величина удельной поверхности Sc может быть подсчитана
из среднеповерхностного диаметра ds при допущении, что
частицы представляют собой плотный аморфный кремнезем
636
Глава 5
с удельной массой 2,2 г/см3. Таким образом, Sc равна 2720/ds
(Sc имеет размерность в м2/г, a ds выражен в миллимик-
ронах) .
Медалиа и Хекман [12, 13] описали метод определения раз-
меров первичных частиц и агрегатов из подобных частиц в га-
зовых сажах, который должен быть применим в равной мере
и к кремнеземным порошкам. Данный метод позволяет опре-
делять число частиц, составляющих агрегат, измерять объем,
соответствующий значению пористости, а также размер первич-
ных частиц. Аналогичное исследование пористости для случая
мезопористого силикагеля было представлено Хавардом и
Уилсоном [14]. Этот силикагель рассматривался в качестве
стандартного образца с известной удельной поверхностью
(Gasil I), полученной Эвереттом и др. [15] и равной
286,1 + 3,5 м2/г. Хавард и Уилсон получили электронно-микро-
скопические снимки при достаточно высоком разрешении, из
которых был подсчитан средний диаметр частицы, составивший
8,8 нм. Но при этом получилось некоторое расхождение.
Соответствующее значение удельной поверхности таких неболь-
ших частиц должно было бы составлять приблизительно 2750/d
(где d — диаметр частицы в миллимикронах); если допустить,
что плотность кремнеземных частиц равна 2,2 г/см3, то удель-
ная поверхность должна составлять примерно 2750/8,8, или
312 м2/г. На самом же деле эта величина по измерениям ока-
залась равной 286 м2/г. Такое различие объясняется тем, что
вокруг каждой точки контакта между соседними сферическими
частицами существует пространство или кольцеобразная щель,
настолько узкая по размеру, что в нее не могут проникать
молекулы азота, используемые при измерении удельной по-
верхности (диаметр молекулы азота, как известно, равен
0,4 нм). В случае же больших по размеру кремнеземных частиц
значение удельной поверхности, подсчитанное по определяемым
из электронно-микроскопических снимков диаметрам частиц,
находится в лучшем согласии со значением удельной поверх-
ности, измеренным адсорбционными методами (см. лит.
к гл. 4 [142]).
Определение удельной поверхности
Величина удельной поверхности наиболее надежно изме-
ряется широко распространенным методом адсорбции азота
или других'газов и паров. Однако для методов, предусматри-
вающих адсорбцию каких-либо специфических ионов или мо-
лекул из раствора (водного либо органического), требуется
более простое оборудование, и, как подтверждается, они ока-
зываются более удобными, в особенности для текущей работы.
Силикагели и порошки
637
Во всех случаях использования адсорбционных методов
ключевым представляется следующий вопрос: разделены ли
первичные кремнеземные частицы между собой настолько,
чтобы поры между ними оказывались доступными для адсор-
бированных ионов или молекул и чтобы поверхность кремне-
зема могла покрываться по крайней мере единичным слоем
адсорбированного вещества? Наблюдались многочисленные
примеры, когда в микропористых силикагелях молекулы воды
или ионы ОН- оказывались способными проникать внутрь пор,
а молекулы азота или какие-либо другие большие по размеру
молекулы не могли в них входить. Принимается, что в твердых
адсорбентах, для которых измеряется удельная поверхность,
поры имеют такой размер, при котором определяемая порами
величина поверхности оказывается в основном доступной для
данного адсорбируемого вещества.
Другие свойства твердого тела, являющиеся функцией
удельной поверхности, также могут быть измерены, но, однако,
соответствующие методы не нашли широкого применения.
В обзоре Молла [16], включающем сведения, взятые прибли-
зительно из 150 литературных ссылок, были рассмотрены раз-
личные способы измерения удельной поверхности, разработан-
ные до 1954 г., для всех типов твердых адсорбентов. Джой [17]
в своем обзоре перечислил методы с применением адсорбции
азота, которые были развиты к 1953 г. В 1969 г. проходил сим-
позиум [18] по общим проблемам определения удельных
поверхностей, на котором особенное внимание уделялось аспек-
там адсорбционного-метода БЭТ.
Адсорбция газов и паров. Этой теме посвящено большое
число обзорных работ, кроме того, она являлась основным
вопросом, который рассматривался на многочисленных симпо-
зиумах [1—8, 18, 19].
Были выбраны стандартные вещества для измерения удель-
ной поверхности. Одним из таких адсорбентов является гидро-
ксилированный кремнеземный порошок, не имеющий пор,
«Fransil EF» [20] с удельной поверхностью 38,7 м2/г. Впослед-
ствии в качестве подходящих стандартов для проведения
исследовательских работ такого рода в лабораториях всего мира
было решено использовать два типа углеродной газовой сажи
и два типа кремнеземных порошков. Два последних имеют
обозначения ТК-800 (удельная поверхность 165,8 + 2,1 м2/г)
и Gasil I (286,2 ±3,5 м2/г) [21].
Уже давно появилась необходимость установить величину
площадки, занимаемой одной молекулой азота при адсорбци-
онных измерениях на непористой поверхности адсорбента
с точно геометрически определенной площадью. Были исполь-
зованы однородные образцы кристаллов и стеклянных шариков,
638
Глава 5
измеренные под микроскопом. Типичным примером может
служить работа Дица и Тернера [22а], которые применили
однородные по размеру стеклянные волокна марки Е с точно
измеренным диаметром. Были получены следующие значения
площадок, занимаемых отдельной адсорбированной молекулой
при температуре 77,4 К: для азота 16,2 А2, для аргона 13,8 А2,
для криптона 20,2 А2. Аналогичные значения были установ-
лены еще раньше другими методами. Макклеллан и Харнсбер-
гер [226] рекомендовали следующие значения: для азота (при
— 195°С) 16,2 А2, для аргона (при —195 и —183°С) 13,8 А2,
для криптона (при —195°С) 20,2 А2, для бензола (при 20°С)
43,0 А2 и для н-бутана (при 0°С) 44,4 А2.
Азот— наиболее широко распространенный адсорбат, при-
меняемый для определения удельной поверхности по низко-
температурной адсорбции при —196°С. Аргон и криптон при
наличии аналогичных измерительных установок также могут
успешно использоваться, поскольку известны соответствующие
значения площадок для этих атомов [23]. Как было показано
в работе Безина и Берубе [24], преимуществом криптона ока-
зывается его способность проникать в меньшие по размеру поры.
Брокхофф [25] представил общий обзор по адсорбции азота
на твердых адсорбентах и включил в него различные способы
объяснения получаемых данных на примерах широкого набора
пористых твердых тел.
В методе БЭТ Брунауэра, Эмметта, Теллера [26—28] для
расчета значения удельной поверхности на основании получен-
ной изотермы адсорбции используется главным образом азот
в качестве адсорбата при температуре измерения —196°С.
Иннес [29] разработал быстрый автоматический способ для
получения и измерения изотерм адсорбции. Липпенс и Хермане
[30, 31] описали соответствующую аппаратуру более подробно.
Кроме того, было разработано коммерческое оборудование,
основанное на использовании статического равновесного ме-
тода. Такое оборудование не нуждается в предварительной
градуировке и автоматически выдает значения удельной по-
верхности в виде цифровых данных (например, прибор фирмы
Micromeritics, Inc.).
Несмотря на то что было предложено большое число урав-
нений, описывающих изотерму адсорбции, уравнение БЭТ
сохраняет свою практическую пользу уже в течение многих лет.
Разработанные впоследствии способы оказываются полезными
для некоторых определенных ситуаций, но тем не менее урав-
нение БЭТ все еще широко используется. Уравнение БЭТ запи-
сывается в виде
Р __ 1________। (с — 1) р /! \
Va (Ро — Р) Vmc “Г vmcp0 v f
Силикагели и порошки
639
va — количество адсорбированного пара при равновесном
давлении р; моль адсорбата на 1 г адсорбента; vm — емкость
монослоя на поверхности, т. е. число молей пара в расчете на
1 г адсорбента, требуемое для покрытия всей поверхности
плотным монослоем адсорбированных молекул;
ро — давление насыщенного пара при выбранной темпе-
ратуре;
с — константа.
Если построить график зависимости величины р/и(р0— р),
откладываемой по оси ординат, от относительного давления
р/ро, нанесенного на ось абсцисс, то получается прямая линия,
имеющая наклон (с—l)/vmc и отсекающая отрезок на оси
ординат, равный l/vmc. Константа с является функцией теп-
лоты адсорбции и зависит от химической природы поверх-
ности.
Зная величину vm, рассчитываемую из известных экспери-.
ментальных данных при использовании уравнения (1), можно
определить удельную поверхность как:
5БЭТ = адЛ.1О-20
где Sest — удельная поверхность, определяемая методом БЭТ,
м2/г; ат — площадка, занимаемая отдельной адсорбированной
молекулой на поверхности, A2; N — число Авогадро, равное
6-Ю23.
Значение ат для азота может несколько меняться в зави-
симости от вида поверхности, но обычно оно принимается
равным ~ 16,3 А2 для окисленных поверхностей.
Для того чтобы иметь возможность сравнивать различные
твердые кремнеземные адсорбенты, удобно использовать так
называемое приведенное значение vs, которое определяется как
vs = v/v0,4, где Уод — объем адсорбированного пара при р/р0 =
= 0,4. Каррутерс и др. [20] провели сравнение адсорбции
азота на четырех типах кремнеземных порошков; графическая
зависимость v/vm как функция от vs (где vm — значение
емкости монослоя, определяемое из уравнения БЭТ) является
прямо пропорциональной.
Метод БЭТ дает возможность получать одни и те же ре-
зультаты независимо от того, гидроксилирована или дегидро-
ксилирована поверхность кремнезема. Так, в работе Красиль-
никова и др. [32] при измерении удельной поверхности на
19 образцах кремнезема, различающихся в значительной
степени по стадиям дегидратации поверхности, оказалось, что
эффект дегидратации не влиял практически на измеряемую
величину удельной поверхности и не превышал 5%. Ловен и
Броудж [33] показали, что, по мере того как исходный крем-
неземный порошок с полностью гидроксилированной поверх-
640
Глава 5
ностью подвергался дегидратации, то наряду с некоторым
уменьшением величины поверхности вследствие спекания об-
разца наблюдалось заметное понижение константы с в уравне-
нии БЭТ, что соответствовало более низкой энергии адсорбции:
Температура Удельная Количество Константа с
дегидратации, поверхность, SiOH-rpynn
СС м2/г на 1 нм2
120 182 10 104
620 170 3 53
810 141 2 45
При повторной гидратации поверхности значение с восста-
навливалось примерно до 100, тогда как удельная поверхность
оставалась без изменений. Коберштейн и Волл [34] также
исследовали воздействие степени гидроксилирования поверх-
ности на адсорбцию азота при различных температурах.
Можно было предположить, что микропористые мате-
риалы — неподходящие объекты для определения удельной
поверхности методом БЭТ. Однако Брунауэр [35] отметил, что
так как малые по размеру поры заполняются при более низком
парциальном давлении, чем поры большего размера, то наблю-
дается некоторая компенсация при изучении такого рода образ-
цов, которая ведет к совпадению значения, получаемого мето-
дом БЭТ, с истинным значением удельной поверхности при
условии, что отсутствуют поры с радиусами, меньшими при-
близительно 12 А.
Абсолютный метод Гаркинса и Юра [36], используемый
для подсчета удельной поверхности из адсорбционных данных,
очевидно, дает более согласующиеся результаты, чем метод
БЭТ, когда изучается адсорбция различных веществ на разного
рода твердых адсорбентах. Метод основан на применении
эмпирического уравнения
lgp/Po = -B~ А/и«
где А и В — константы; va — объем адсорбируемого пара;
р/ро — парциальное давление адсорбата.
Изображая графически зависимость lgp/ро от Va2> полу-
чают прямую линию с величиной наклона s.
Из этих данных рассчитывается удельная поверхность по
соотношению
S = k/s2
где S — удельная поверхность, м2/г; k — константа для дан-
ного адсорбата, определяемая «абсолютным» методом [36].
Некоторые значения константы k приведены ниже:
Пары Температура, °C k
Азота -195,8 4,04
Бутана 0 13,6
Воды 25 3,83
Силикагели и порошки
641
Многие исследователи впоследствии проверяли соответствие
между методом Гаркинса и Юра и методом БЭТ на основании
рассчитанных значений удельной поверхности из адсорбционных
данных. Согласно Древингу и др. [37], уравнение БЭТ выпол-
няется на силикагелях при значениях относительных давлений
р/ро в интервале 0,035—0,33, а уравнение Гаркинса и Юра —
в интервале 0,075—0,58.
Шулл [38] предложил метод t-диаграммы, согласно кото-
рому количество адсорбированного азота вычерчивается как
функция толщины адсорбированной пленки азота на гладкой,
непористой поверхности. Кранстон и Инкли [39] собрали имею-
щиеся доступные данные и опубликовали метод составной /-диа-
граммы, которым они и другие исследователи широко пользо-
вались для получения характеристик пористых твердых тел.
Де Бур и др. [4] изучали возможное приложение этого ме-
тода к большому числу твердых образцов адсорбентов, в кото-
рых отсутствовали микропоры, используя уравнение
t = 3,54va/vm
где / — толщина адсорбированной пленки азота, A; va — объем
адсорбированного азота; vm—объем адсорбированного азота
при образовании мономолекулярного слоя.
Удельная поверхность St подсчитывается из уравнения
St= 15,47va/t (для N2)
Величина / выбирается из таблицы или из уравнения, свя-
зывающего / и р/р0 и основанного на большом объеме экспери-
ментальных данных. Удельная поверхность 5г имеет размерность
м2/г, va — объем газообразного азота, адсорбированного при
нормальных значениях температуры и давления, см3/г.
Более подробное описание и примеры использования этого
метода изложены в серии статей де Бура и др. [40—42] и обоб-
щены Броекхоффом и Линсеном (см. ссылку [4]).
С целью вычисления / из известного отношения р/ро Долли-
мор и Хил [43] изучили 36 образцов кремнезема и оксида алю-
миния и пришли к заключению, что пригодна формула, пред-
ставленная де Буром:
где / — выражается в ангстремах.
Различные исследователи делали попытки улучшить перво-
начальную /-кривую, предложенную Кренстоном и Инкли, глав-
ным образом считая, что отсутствуют какие-либо поры в рас-
15 Заказ А’э 250
642
Глава 5
сматриваемом адсорбенте. Гиргис [44] предложил несколько
иной тип /-кривой, но для получения данных необходимо исполь-
зовать специальное уравнение
t = 6,15 — 2,25 In (In ро/р) + Ю,5 (р/р0)4
/-Диаграмма для непористого, но очень тонко дисперсного
объемистого кремнезема показывает отклонение от /-кривой
стандартного вида. Этот факт, как полагают де Бур и др., обус-
ловлен адсорбцией вещества вблизи точек контакта очень не-
больших частиц кремнезема. Авторы предложили уравнение
адсорбции с учетом геометрии упаковки частиц.
Данный метод оказывается полезным для адсорбентов,
в которых отсутствует капиллярная конденсация ниже р/ро
0,7—0,8. Уравнение можно применять вплоть до значения р!ръ =
= 0,3. Тернан [46] предложил теоретическое уравнение для
/-кривой, не требующее каких-либо констант для корректирова-
ния с экспериментальными данными. Основанное на положениях
термодинамики с учетом дисперсионных сил по Леннарду-
Джонсу, это уравнение устанавливает связь между относитель-
ным давлением и толщиной пленки в хорошем согласии с экспе-
риментальными данными.
Трудность метода /-кривой заключается в том, что необхо-
димы данные о количестве адсорбированного вещества в моно-
слое, т. е. надо знать удельную поверхность образца. Таким об-
разом, применение этого метода следует ограничивать изотер-
мами при условии, что метод БЭТ дает однозначное положение
точки В или емкости монослоя. Однако Брунауэр [47] отметил,
что удельная поверхность микропористых адсорбентов в обыч-
ных случаях может быть надежно измерена методом БЭТ. Сле-
довательно, наличие микропор не исключает использование
/-метода.
В работе [48] опубликованы данные для нескольких /-кри-
вых, относящихся к случаю сорбции воды на таких поверхностях
твердых веществ, для которых были известны дифференциаль-
ные теплоты адсорбции. Чтобы метод был пригоден для данной
поверхности, /-кривая должна соответствовать теплоте адсорб-
ции воды на этой поверхности.
В «cts-методе» Синга [49] вычерчивается зависимость сц от
х, где as = x!xs, х— количество адсорбированного газа и xs —
количество адсорбированного газа при выбранном значении от-
носительного давления (стандартное состояние). Как правило,
считается, что as равно 1,0 при р/ро=О,4. Такое относительное
давление выбрано потому, что ниже этой точки образуется лишь
монослойное покрытие поверхности и происходит заполнение
микропор, тогда как гистерезисные петли, возникающие вслед-
Силикагели и порошки
643
ствие заполнения пор, в которых имеет место явление капилляр-
ной конденсации, наблюдаются при более высоком относитель-
ном давлении.
Удельная поверхность высчитывается из наклона линейного
участка графика после того, как уже определен «стандартный
наклон» для непористого стандартного порошка с известным
значением удельной поверхности. Бамбани и др. [50] провели
сравнение этого метода с методом БЭТ для множества пористых
и непористых кремнеземов. В том случае, когда у образцов от-
сутствовали микропоры, наблюдалось превосходное согласие
в значениях удельных поверхностей, определяемых этими двумя
методами.
Метод Киселева основан на термодинамическом рассмотре-
нии полной адсорбционно-десорбционной петли гистерезиса
с привлечением явления поверхностного натяжения жидкого ад-
сорбата [6, 51, 52]. Однако этот метод полезен только для ад-
сорбентов, обладающих мезопорами, а также вынуждает про-
водить измерение целиком всей изотермы.
В методе Каганера [53] используется уравнение, которое
наиболее применимо в области низких относительных давлений
р/ро от 10-5 до 3-10-2:
lg иа = 1g vm — D (lg -^-)2
где D = 2,3 kR2T2. Удельная поверхность подсчитывается из ве-
личины емкости монослоя vm, которая получается из графиче-
ской зависимости адсорбционных данных с использованием от-
меченного уравнения при экстраполировании зависимости к зна-
чению lgPo/p = O.
Метод Френкеля—Хелси—Хилла [54—56] может приме-
няться при высоких значениях относительных давлений, когда
толщина адсорбционной пленки составляет несколько молеку-
лярных слоев. Изотерма в этом случае может описываться сле-
дующим уравнением:
Ро (vlvm)s
где показатель степени s определяется по понижению поверх-
ностных сил с увеличением расстояния от поверхности. Пирс
[57] показал, что для адсорбции азота данное уравнение ока-
зывается справедливым и за пределами мономолекулярного
слоя и что для р/р0> 0,3 изотерма записывается как
/ v \2,75_ 1,305
\ Vm ) ~ igPo/P
15*
644
Глава 5
где vm — объем адсорбированного азота при монослойном по-
крытии; v — адсорбированный объем при давлении р; ро — дав-
ление насыщенного пара.
Поскольку в этом случае молекулы азота адсорбируются на
лежащем ниже слое азота, то такая адсорбция не зависит от
самой подложки.
Метод непрерывного потока * для измерения удельной по-
верхности в настоящее время нашел широкое применение. Ме-
тод основан на адсорбции азота или какого-либо другого ве-
щества из потока инертного газа. Нельсен и Эггертсен [58]
дали описание метода, а Ли и Стросс [59] подтвердили его
возможности. Были введены многочисленные усовершенствова-
ния, и в настоящее время стали доступными компактные ком-
мерческие приборы (например, модель прибора фирмы Quan-
tachrome Corp.). Дальнейшие подробности метода описаны в ра-
боте [60]. Смесь известного состава, например, состоящая из
20 % N2 и 80 % Не, пропускается через предварительно обезга-
женный образец адсорбента при комнатной температуре и да-
лее через детектор теплопроводности (катарометр). При быстром
охлаждении образца в жидком азоте адсорбент поглощает неко-
торое количество азота, но затем быстро достигается насыщение.
Это вызывает временное понижение содержания азота в потоке,
что регистрируется в виде пика, направленного вниз от нулевой
линии на непрерывно движущейся ленте самописца. Когда же
образец быстро разогревается до комнатной температуры, то
азот десорбируется; при этом на самописце регистрируется пик,
направленный вверх от нулевой линии. Получаемые на эталон-
ных образцах адсорбентов с известной удельной поверхностью
(которая определяется обычным методом БЭТ) площади пиков
и/или их высоты служат для проведения калибровочных изме-
рений. Обычно расчет ведут по десорбционным пикам. Выпускае-
мые коммерческие приборы такого рода позволяют замерять ин-
тегральное значение площади пика и непосредственно давать
в цифровом виде величину удельной поверхности образца.
Газиев, Яновский и Бражников [61] отметили, что этот ме-
тод не учитывает имеющие место изменения теплоты адсорбции,
которые соответствуют изменениям константы с в уравнении БЭТ.
Обзоры, посвященные данному методу, появились в 1972 г.
[62, 63]. Поммиер, Джуллет и Тейчнер [64] продемонстриро-
вали, что этим методом можно определять очень небольшие зна-
чения удельных поверхностей. Ловелл [65], используя вместо
азота криптон, проводил измерения удельных поверхностей
вплоть до 0,019 м2/г. Пайн, Синг и Турк [66] сравнивали этим
* Этот метод также носит название динамического метода тепловой де-
сорбции.— Прим. ред.
Силикагели и порошки
645
методом адсорбцию аргона и азота на гидроксилированных по-
верхностях образцов кремнеземов и нашли величину площадки,
занимаемой одной молекулой аргона, равной 18,2 А2. Азот как
адсорбат более предпочтителен при измерении удельных поверх-
ностей микропористых твердых тел.
Могут использоваться и другие газы и пары, особенно в тех
случаях, когда некоторые затруднения вызывает применение ап-
паратуры охлаждения для создания температуры жидкого воз-
духа. Так, Киселев и Камакин [67] для измерения удельной по-
верхности и пористых свойств адсорбентов использовали мета-
нол при комнатной температуре. При относительном давлении
р/ро=О,1 удельная поверхность оказалась равной 145а м2/г,
где а — количество адсорбированного метанола, ммоль/г, или
приблизительно 4 молекулы СН3ОН на 1 нм2. Фуран при 23°С
и бутан и изобутан при 0°С образовывали монослойные покры-
тия, для них были вычислены площадки, приходящиеся на одну
молекулу в монослое: 42, 54 и 53 А2 соответственно [68].
Аммиак при температуре кипения дает монослойные покрытия,
изменяющиеся в зависимости от природы поверхности кремне-
зема [69]. Моноксид азота (NO) адсорбировался в температур-
ном интервале 181—293 К, что определялось измерением маг-
нитной восприимчивости [70]. При р?Ро = О,214 адсорбированный
бензол образовывал монослой на поверхности кремнезема; из
этих данных можно было вычислить удельную поверхность ад-
сорбента [71]. Исходя из основных положений, Киселев [72]
провел вычисления изотерм адсорбции, измеренных на силика-
гелях, которые различались по величине удельной поверхности,
размерами пор и степени гидроксилирования поверхности.
Пары воды можно использовать как адсорбат, если предус-
матривается, что поверхность кремнезема полностью гидрокси-
лирована. Для тех образцов кремнезема, поверхность которых
после соответствующей химической обработки или дегидратации
при термообработке кремнезема выше 300°С становилась ча-
стично гидрофобной, изучение адсорбции воды оказывается по-
лезным для определения только оставшихся гидрофильными
участков поверхности. Однако для силикагелей и порошков, сфор-
мированных в водной среде и высушенных при умеренных тем-
пературах, изотерма адсорбции воды дает заслуживающие до-
верия определения удельной поверхности кремнезема *. Адсорб-
* Удельная поверхность силикагелей и кремнеземных порошков может
определяться по адсорбции пара воды при условии предельно гидроксилиро-
ванного состояния поверхности (т. е. состояния, когда с поверхности уда-
лена физически адсорбированная вода, но еще практически остаются неза-
тронутыми поверхностные силанольные группы) и отсутствия у образцов
кремезема микропор, соизмеримых по диаметру с размером молекулы воды.—
Прим, перев.
646
Глава 5
ция паров воды изучалась, например, Гансом, Бруксом и Бой-
дом [73] и Кантро, Брунауэром и Вайсом [74], которые наш|ли,
что удельную поверхность кремнеземов можно вычислять, ис-
пользуя уравнение БЭТ. Адсорбция измерялась гравиметриче-
ским методом по приращению массы образца, находящегося
в эксикаторе в вакуумных условиях, по мере того, как он ад-
сорбировал воду в течение 7 сут при 25°С. Источником паров
воды с заданным равновесным давлением являлся водный раст-
вор соли, помещенный в отдельную колбочку, находящуюся
также в эксикаторе. Относительные давления пара воды изме-
нялись от 0,07 до 0,33 за счет использования различных насы-
щенных солевых растворов. При проведении измерений этим
методом затрачивается много времени, но для подобных экспе-
риментов требуется только простая лабораторная стеклянная
посуда. Белякова, Джигит и Киселев [75] исследовали адсорб-
цию воды на силикагелях, структурные характеристики которых
менялись в широких пределах. Они сообщили, что одна моле-
кула воды при монослойном покрытии занимает площадку *,
равную 25 А2.
Адсорбция тетрахлорида углерода измерялась при 28°С
посредством микровзвешивания, причем получался линейный
участок на графике, построенном в координатах БЭТ. Опре-
деленное по графику значение удельной поверхности образца
согласовывается со значением, найденным по адсорбции азота.
При этом не наблюдалось никакого химического взаимодейст-
вия [76]. Для диоксида углерода, изученного при температуре
—78°С на том же самом оборудовании, которое использовалось
при проведении адсорбционных изменений с азотом при —196°С,
были получены те же самые результаты в пределах 10 % [77].
Применение метода потока при пропускании смеси бутана
с диоксидом углерода позволило выполнить исследования,
в которых десорбированный бутан извлекали из смеси, а его
объем измеряли после поглощения диоксида углерода в щелоч-
ном растворе [78].
Как уже упоминалось в гл. 4, адсорбция из растворов вза-
имосвязана с величиной удельной поверхности коллоидного
кремнезема. Эти и подобные методы могут использоваться
также и для кремнеземных порошков и силикагелей. В каче-
стве адсорбатов берутся органические соединения из органиче-
ских и водных растворов, а также применяются органические
и неорганические катионы и ионы ОН- из воды. Полярные сое-
динения, находящиеся в неполярных (углеводородных) раство-
* При условии предельно гидроксилированного состояния поверхности
кремнезема.— Прим. ред.
Силикагели и порошки
647
рителях, способны адсорбироваться на полярных поверхностных
группах SiOH. Адсорбция на дегидроксилированной силокса-
новой поверхности еще недостаточно хорошо понята. Однако
имеются доказательства того, что в воде силоксановая поверх-
ность кремнезема проявляет гидрофобные свойства. Она адсор-
бирует только гидрофобные вещества или группы.
Удельные поверхности образцов кремнеземов с гидрофиль-
ной (гидроксилированной) поверхностью измерялись посред-
ством адсорбции нитрофенола из воды или бензола [79] и
фенола из декана или тетрахлорида углерода [80, 81]. Подоб-
ная система не должна содержать воду, поэтому органические
растворители следует обезвоживать с помощью молекулярных
сит. Фенол можно определять титрованием бромом или интерфе-
рометрией. Стеариновая кислота адсорбируется на кремнеземе
из безводного метанола и определяется по разности в показа-
ниях, определяемых потенциометрическим титрованием ще-
лочью. Полученные результаты показывают, что каждая моле-
кула стеариновой кислоты занимает площадку, равную только
20,6 А2 [82].
Как уже упоминалось в гл. 4, ионы Zn(en)2+ адсорбируются
на силикагелях и порошках и, согласно данным Унгера и
Выдры [83], могут использоваться для определения удельных
поверхностей порошков кремнезема, не имеющих микропор.
Уменьшение содержания цинка в растворе при pH 6—9, опре-
деляемое титрованием, дает возможность измерять удельную по-
верхность кремнезема примерно в пределах точности ±3 %.
При монослойном покрытии на 1 нм2 приходится около 1,26 мо-
лекул, т. е. площадка, занимаемая одной адсорбированной мо-
лекулой, равна 79 А2.
Адсорбция красителей на кремнеземных порошках, проводи-
мая с целью оценки значений удельных поверхностей, известна
уже давно, поскольку для ее изучения достаточно простого ко-
лориметрического метода. Ряд исследователей изучали адсорб-
цию метиленового голубого, а также факторы, влияющие на
такую адсорбцию [84, 85]. Авторы работ [86, 87] выполнили
сравнение адсорбции из растворов ряда катионных красителей,
в том числе метиленового голубого и кристаллического фиоле-
тового. Исследования адсорбции проводились на различных
кремнеземных порошках из растворов красителей в «-нитрофе-
ноле или в воде, и полученные результаты были скорректиро-
ваны со значениями удельных поверхностей, найденных мето-
дом БЭТ по адсорбции азота и криптона, а также методом элек-
тронной микроскопии.
Адсорбция катионных поверхностно-активных веществ (кати-
онных мыл) на кремнеземе изучалась Тер-Минасян-Шарага [88]
и Бийстербошем [89], которые показали, что на поверхности
648
Глава 5
может формироваться либо монослой, либо двойной слой ПАВ.
Подобные сложные образования на поверхности дают и неко-
торые красители, способные образовывать мицеллы. Авторам
пришлось разработать специальные условия эксперимента для
каждого адсорбата с тем, чтобы устранить возможное появле-
ние некорректных результатов. Кроме того, на адсорбцию таких
ионных разновидностей оказывает влияние величина pH и со-
держание примесей алюмосиликат-ионов на кремнеземной по-
верхности. Метод применения катионных красителей и ПАВ
имеет свои ограничения: он зависит от скорости, с которой мо-
гут сравниваться значения удельных поверхностей при проведе-
нии экспериментов на серии кремнеземных порошков одного и
того же типа.
Неионные красители, такие, как метиловый красный, при-
годны для определения удельной поверхности образцов кремне-
зема, имеющих гидроксилированную поверхность, и в особен-
ности для кремнеземов с частично дегидроксилированной или
оказавшейся частично органофильной поверхностью. Ловен и
Броудж [33] использовали метод Шапиро и Кольтгофа [90],
который включал физическую адсорбцию метилового красного
из бензола, взятого в качестве растворителя, на полярной по-
верхности кремнезема. В параллельно проводимых эксперимен-
тах осуществлялась реакция между триметилхлорсиланом
(CH3)3SiCl и силанольными поверхностными группами после
дегидратации кремнезема при повышенных температурах, так что
на поверхности образовывалось определенное количество хемо-
сорбированных групп (CH3)3SiO в зависимости от оставшегося
количества групп SiOH. Ясно, что для образцов кремнезема
с полностью гидроксилированной поверхностью, не обладаю-
щих микропорами, значение удельной поверхности может
измеряться с использованием любого реактива (см. рис. 5.2).
Каждая молекула метилового красного занимает площадку
116 А2, а каждая группа (CH3)3Si — 45 А2.
Страйкер и Матиевич [91] предложили использовать моно-
молекулярную адсорбцию Hf(OH)4 на кремнеземе из нейтраль-
ного раствора с целью определения удельной поверхности, при-
меняя в качестве меченого атома радиоактивный гафний. Ав-
торы работы [92] сообщили, что попытки повторить данный
способ оказались неудачными. Последовавший затем обмен
краткими сообщениями между двумя группами авторов не внес
ясности в этот вопрос.
Ф.енилтрихлорсилан C6H5SiCl3 вступает в реакцию с предва-
рительно высушенными силикагелями количественно; на осно-
вании необратимо прореагировавшего с силанольными поверх-
ностными группами количества реагента можно оценить удель-
Силикагели и порошки
649
ную поверхность. Отмечается, что на 1 нм2 приходится 2,16 фе-
нильных группы (или 3,6 мкмоль/м2) [94].
Титрование кремнезема щелочью в солевом растворе в ин-
тервале pH 4,5—9,0 представляет собой очень продуктивный,
быстрый метод для определения удельной поверхности гидро-
юоо/к
Рис. 5.2. Зависимость между поверхностной концентрацией силанольных групп,
температурой дегидратации и адсорбцией.
X— дЛЯ тримстилсилильных групп; В — для молекул метилового красного.
ксилированного кремнезема. Ионы ОН- достаточно малы по
своему размеру, поэтому они проникают в такие небольшие
поры, которые оказываются недоступными даже молекулам
азота. Подробности этого метода титрования Сирса рассмат-
ривались в главах 3 и 4. В том случае, когда процесс ведется
с силикагелями или порошками, равновесие в конечной точке
титрования при pH 9 достигается довольно медленно. Мефферт
и Лангенфельд [95] исследовали процесс титрования кремне-
земных порошков, применив автоматический титратор для до-
стижения конечной точки титрования. Абендрот [96] показал
необходимость использования высококонцентрированных раст-
воров соли для того, чтобы оказалось возможным титрование
поверхности внутри тонких пор кремнезема.
Были разработаны хроматографические методы, в которых
использовалась адсорбция веществ из жидкой фазы. Если име-
ется достаточно большое количество образца кремнезема, то им
можно набить хроматографическую колонку диаметром 2 мм.
Измерением длины окрашенной зоны в хроматографической ко-
650
Глава 5
лонке при пропускании через нее метилового красного в бензоле
можно определить удельную поверхность кремнезема [97].
Хоффман и др. [98] измерили адсорбцию спиртов Ci—С4 при
различных концентрациях их растворов в бензоле. Из получен-
ных данных авторы рассчитали площадки, занимаемые на по-
верхности кремнезема физически адсорбированными молеку-
лами следующих спиртов:
Спирт
Метанол
Этанол
1-Пропанол
2-Пропанол
1-Бутанол
Молекулярная площадка, А2
19,9
25,5
30,2
30,8
34,4
Позже Хоффман и др., использовав метод, разработанный
Френкелем и др. [99], исследовали жидкостную хроматографи-
ческую систему СН^ОН—Н2О — кремнезем и определили удель-
ную поверхность взятого кремнезема, которая оказалась в хо-
рошем согласии со значением удельной поверхности, найденной
методом БЭТ по адсорбции азота. Авторы пришли к заключе-
нию, что использование систем низшие спирты—бензол дает
исследователям простой метод измерения удельной поверхно-
сти кремнезема, для которого требуется минимум оборудования
и не нужно высокое профессиональное умение.
В другом методе измерения удельной поверхности образцов
кремнезема с гидроксилированной поверхностью применялся
в качестве адсорбата октадециловый спирт, определение кото-
рого проводились с помощью газовой хроматографии. Серпинет
и др. [100] использовали этот метод, чтобы проконтролировать
точность метода потока, в котором в качестве адсорбируемого
вещества был взят азот.
Диэлектрические измерения. Этот метод использовался для
исследования адсорбции жидкостей и паров на кремнеземе. По
мере того как бензол проникает в микропоры, его взаимодейст-
вие с поверхностью изменяет ход диэлектрической изотермы
кремнезема. После нагревания образца кремнезема также наб-
людаются изменения в ходе изотерм [101]. В более ранних ис-
следованиях [102] было отмечено отсутствие закономерности
в наклоне экспериментальной кривой, выражающей зависимость
электрической емкости кремнезема от количества адсорбирован-
ной воды. Куросаки [103] нашел, что для относительной влаж-
ности ниже 40 % адсорбция воды подчиняется уравнению БЭТ,
но на кривых емкости и диэлектрических потерь появляются
разрывы, что объясняется изменением свободы вращательного
Силикагели и порошки
651
движения адсорбированных молекул на кремнеземной поверх-
ности.
Диэлектрическая константа для данного типа кремнеземного
порошка связывается с наличием поверхностных групп SiOH
•и, следовательно, с величиной удельной поверхности. Для пиро-
генного кремнезема, у которого понижена концентрация сила-
нольных групп, наблюдается и более низкое значение диэлек-
трической константы [104].
Термические эффекты. Результаты многочисленных измере-
ний теплот адсорбции на образцах кремнезема с известными
значениями удельных поверхностей были описаны в целом ряде
работ. Однако надо отметить, что на полученные результаты
могли влиять многие факторы, кроме того, оборудование для
проведения подобных исследований достаточно сложно. Гаркинс
и Юра [105] разработали абсолютный метод, в котором обра-
зец порошка изотермически приводится в равновесие с паром
воды вплоть до того момента, когда поверхность кремнезема
покроется пленкой воды, после чего образец погружается вводу,
находящуюся в высокочувствительном калориметре. По количе-
ству выделяющейся при этом теплоты, выраженной в эргах на
грамм SiO2, деленной на полную поверхностную энергию воды
(118 эрг/см2), можно определить удельную поверхность кремне-
зема. Данный метод применим только для ограниченного числа
типов кремнезема. Теплоты смачивания различных порошков,
в том числе порошков кремнезема, также были связаны с удель-
ной поверхностью [106].
Одним из осложняющих факторов является более высокое
значение теплоты адсорбции в очень тонких микропорах [107],
правда, его можно использовать для получения характеристики
пористости кремнеземов с известными удельными поверхностями.
Другой важной переменной величиной является степень гидрата-
ции поверхности кремнезема, что может приводить к трехкрат-
ному различию в показаниях для теплот смачивания в бензоле
или в циклогексане [108].
Теплота смачивания в воде, равная 160±3 эрг/см2, была оп-
ределена для кремнеземов с гидроксилированной поверхностью
в области удельных поверхностей 8—150 м2/г в отсутствие мик-
ропор. Тэйлор и Хокки [109] сообщили, что это значение оста-
ется постоянным в указанной области, доказывая тем самым по-
добие поверхностных свойств изученных образцов кремнезема.
Во всех случаях поверхность кремнезема дегидратировалась
при повышенной температуре по мере спекания при 700—1040°С
в течение различных временных интервалов с целью пониже-
ния удельной поверхности. Затем поверхности образцов кремне-
земов регидроксилировались до получения поверхностной кон-
652
Глава 5
центрации силанольных групп, равной 4,71 ОН-групп/нм2. Та-
ким образом, по крайней мере теоретически, удельную поверх-
ность кремнезема можно определять после соответствующей
подготовки поверхности кремнезема путем измерения теплоты
смачивания в воде.
Метод вдавливания ртути. Давление р, необходимое для
того, чтобы ртуть входила в пору, связано с диаметром поры
обратно пропорциональной зависимостью. Следовательно, объем
ртути v, попавшей в пору при данном давлении, позволяет из-
измерить объем поры, а значит, и «длину» этой поры. Из таких
данных может быть вычислена внутренняя поверхность поры.
Однако на практике поры в образце сильно различаются по
своим размерам, и поэтому удельная поверхность кремнезема
определяется интегралом
“max
S =------J—g- ( р dv
a cos и J
где о и 0 — поверхностное натяжение и краевой угол, образуе-
мый ртутью со стенкой поры, соответственно; и0 и Umax —
объемы, занимаемые ртутью при ее вдавливании в поры под
давлением, равным 1 атм, и под максимальным давлением соот-
ветственно. Данный метод, в общем, может быть использован,
только когда удельная поверхность 5 составляет менее чем
100 м2/г [110].
Проницаемость газа. Сопротивление уплотненного порошка
по отношению к газовому потоку при определенных условиях
может использоваться для измерения размеров частиц или ве-
личины удельной поверхности. Методы подобного рода описаны
Арнеллем [111] и Дерягиным, Фридляндом и Крыловой [112].
Такие методы, вероятно, в настоящее время редко используются,
поскольку стал доступным быстрый метод потока, включающий
адсорбцию азота на кремнеземе.
Концентрация гидроксильных групп. Измерение количества
«связанной воды» на поверхности кремнезема после того, как
поверхность оказывается полностью гидратированной, а обра-
зец высушен в вакуумных условиях при температуре ниже
100°С, прямо дает величину удельной поверхности. Для образ-
цов кремнеземов, в которых отсутствуют микропоры, удельная
поверхность, определяемая таким методом, находится в согласии
со значением удельной поверхности, найденным методом БЭТ
по адсорбции азота. Согласно данным Журавлева и Киселева
[113], в том случае, когда поверхностная концентрация ОН-
групп измерялась методом дейтерообмена на более чем 40 раз-
Силикагели и порошки
653
личных типах аморфного кремнезема *, она соответствовала
значению 5,0 ОН-групп/нм2. В случае микропористых или так
называемых лепидоидальных и гидратированных кремневых
кислот содержание ОН-групп определяет «поверхность» в том
смысле, что оно позволяет учитывать все атомы кремния, кото-
рые не полностью связаны через атомы кислорода с окружаю-
щими атомами кремния.
Изучая различные коллоидные и осажденные разновидности
кремнезема, Тивари и др. [114] обнаружили, что пики диффе-
ренциального термического анализа (ДТА), площади которых
пропорциональны числу групп SiOH, подвергшихся дегидрации,
могут быть прямо скоррелированы с величиной удельной поверх-
ности, определяемой методом БЭТ. Содержание ОН-групп в рас-
чете на единицу поверхности, измеренной методом БЭТ, не за-
висит от размера частиц или пор, если разновидности кремнезе-
мов не содержат микропоры**.
Малоугловое рассеяние рентгеновских лучей
Использование этого метода для определения размеров пер-
вичных частиц кремнезема было рассмотрено в гл. 4. В выпол-
ненных сравнительно давно исследованиях Шулла, Элкина и
Росса [115] были составлены данные по дифракции с другими
структурными характеристиками силикагеля. Кроме того, Ба-
стик и Файвр [116] нашли, что размеры «мицеллярной поверх-
ности» силикагелей, подсчитанные из данных по дифракции,
коррелировали со значениями удельных поверхностей, измерен-
* Для предельно гидроксилированных аморфных кремнеземов методом
изотопного обмена, проводимого между поверхностными силанольными груп-
пами SiOH и введенной порцией тяжелой воды D2O, можно оценивать вели-
чины удельных поверхностей по соотношению з=аонЮ3/аон = «он103/8,0,
где аон—8-0 мкМОН/м2 (или 5,0 ОН-групп/нм2)—физико-химическая кон-
станта для типов аморфного кремнезема с предельно гидроксилированной
поверхностью, полученная на основании экспериментальных данных методом
дейтерообмена и методом БЭТ по адрсорбции криптона для большого числа
различных аморфных кремнеземов (см. табл. 6.1); «он (ммольОН/г)—коли-
чество силанольных групп на поверхности кремнезема, отнесенное к единице
массы SiO2, определяемое методом дейтерообмена (или каким-либо
другим независимым методом); s (м2/г) — удельная поверхность образца
(см. Л. Т. Журавлев, Сб. «Основные проблемы теории физической адсорб-
ции»/Под ред. М. М. Дубинина и В. В. Серпинского.— М.: «Наука», 1970,
стр. 309).
Масс-спектрометрические варианты метода описаны Л. Т. Журавлевым
в гл. 17, Р. Л. Гореликом, Л. Т. Журавлевым в гл. 18 в книге «Экспери-
ментальные методы в адсорбции и молекулярной хроматографии»/Под ред.
А. В. Киселева и В. П. Древинга.— М.: изд-во МГУ, 1973.— Прим. ред.
** См. Л. Т. Журавлев, А. В. Киселев.— Ж. физ. хим., 1965, т. 39,
с. 453, н лит. к гл. 5 [113].— Прим. ред.
654
Глава 5
ных по адсорбции азота. Шулл обнаружил, что средние диа-
метры частиц в различных гелях (2,9—8,8 нм) соответствовали
измеренным удельным поверхностям при допущении, что плот-
ные сферические частицы кремнезема связывались вместе
в трехмерную сетку геля с незначительной потерей площади
вблизи точек контакта между соседними частицами. Какудо
и др. [И7] ввели поправки к кривой рассеяния для кремнезема,
а также представили результаты измерения диаметров первич-
ных частиц силикагелей. Этим методом был определен диаметр
частиц коллоидного кремнезема людокс-HS; он оказался рав-
ным 15,5 нм [118]. Брадачек и др. [119] получили хорошо сог-
ласующиеся величины для пирогенных кремнеземов, измерив
размеры частиц методом рассеяния (8,2 нм) и методом элек-
тронной микроскопии (8,9 нм).
Как будет обсуждено ниже, структура геля кремнезема мо-
жет также быть выявлена этим методом.
Размеры агрегатов — частиц порошка, гранул геля
В порошках и гелях первичные частицы в конечном счете
всегда собираются в образования, называемые по-разному:
«вторичные частицы», «кластеры» или «агрегированные ча-
стицы». В настоящей книге они будут упоминаться просто как
«агрегаты». Под «первичными частицами» будут в основном
приниматься непористые частицы, хотя в нескольких особых слу-
чаях частицы могут содержать микропоры размером менее 5 нм,
как это указывалось в гл. 4. Первичные частицы, связанные
вместе в агрегатах посредством силоксановых связей, дисперги-
руются с механическим разрывом.
Кремнеземные порошки, состоящие из дискретных непори-
стых частиц или гранул, размер которых превышает полми-
крона, например порошки, приготовленные помолом массив-
ного куска кремнезема, выходят за пределы рассмотрения на-
стоящей книги. В гл. 4 были рассмотрены дискретные частицы
кремнезема, поверхность которых покрыта большими органи-
ческими катионами, оксидом алюминия или хлорид-анионами.
Такие частицы могут быть высушены до порошкообразного со-
стояния, а порошки способны самопроизвольно диспергировать
в воде. В гл. 4 также рассматривались дискретные кремнезем-
ные частицы, поверхность которых покрыта хемосорбирован-
ными алкокси- или органосилильными группами, так что полу-
ченные из них высушцванием коллоидные порошки могут само-
произвольно редиспергировать в органических растворителях.
Наглядное представление о таких чрезвычайно малых по
размеру первичных частицах дает следующее сравнение. Если
Силикагели и порошки
655
бы небольшую гранулу типичного силикагеля диаметром 1 мм
можно было расширить или увеличить до 50 м, то тогда размер
первичных кремнеземных частиц, образующих гранулу, состав-
лял бы примерно 1 мм в диаметре!
Размер агрегата может измеряться рядом стандартных ме-
тодов, включая просеивание. В настоящее время имеются сита
с размером отверстий вплоть до нескольких микрон. В некото-
рых типах пирогенного кремнезема, иногда называемых «дымо-
вой кремнезем» или «белая сажа», связи между первичными
частицами чрезвычайно слабы, и таким образом, агрегаты
оказываются настолько непрочными, что их размеры, измеряе-
мые методом просеивания, меняются по мере того, как порошок
подвергается механическому воздействию. С другой стороны,
агрегаты, полученные из осажденных кремнеземов, и в особен-
ности из высушенных кремнеземных гелей, оказываются доста-
точно прочно связанными, и поэтому при просеивании полу-
чаются воспроизводимые измерения их размеров.
Так называемые методы смачивания могут использоваться
в тех случаях, когда порошки суспендированы в жидкой фазе,
обычно в воде в присутствии ПАВ для устранения возможной
флокуляции агрегатов. Тогда размеры определяются путем от-
бора агрегатов из суспензии и измерением их под оптическим
микроскопом.
Пригодны разнообразные методы седиментации и отмучи-
вания, но они оказываются ценными лишь только после стан-
дартизации метода на определенный, заданный тип порошка или
геля при постоянной плотности изучаемых агрегатов. То же са-
мое относится и к измерениям проницаемости потока воздуха
или жидкости. Наружная поверхность пористых агрегатов, яв-
ляющаяся косвенной мерой величины агрегата, может быть оп-
ределена посредством измерения «проницаемости окружающего
газа под давлением». Газ под давлением пропускают через слой
исследуемого порошка и замеряют скорость газового потока и
градиент давления. В данном случае может быть приспособлена
та же аппаратура, что и для измерения удельной поверхности
по адсорбции азота методом потока (например, прибор Quanta-
sorb, поставляемый фирмой Quantochrome Corporation).
Полная библиография по вышеприведенным методам вплоть
до 1950 г. собрана в справочном руководстве [120]. Для иссле-
довательских целей простейшим доступным методом обычно яв-
ляется влажное или сухое просеивание. Однако Ван ден Хул и
Ликлема [121] рассмотрели важный вопрос о взаимозависимо-
сти размеров первичных частиц и размеров агрегатов, а также
исследовали различные методы, особенно «отрицательную» ад-
сорбцию. Этот метод может оказаться практичным для крем-
неземных гелей, имеющих определенные размеры пор.
656
Глава 5
«Отрицательная» адсорбция — довольно неопределенное вы-
ражение, относящееся к такому явлению, когда в суспензии, об-
разуемой частицами адсорбента в растворе, один из компонен-
тов раствора оказывается сконцентрированным в большей сте-
пени, а второй компонент, наоборот, в меньшей степени вблизи
поверхности адсорбента на расстоянии одного-двух молекуляр-
ных диаметров по сравнению с концентрациями компонентов
в объеме жидкости. Так, например, в суспензии кремнезема,
имеющего на поверхности отрицательный заряд, в водной среде
анионы будут испытывать отталкивание в непосредственной бли-
зости от поверхности, и особенно вблизи узких пор. Ликлема и
Ван ден Хул [122] включили метод «отрицательной» адсорбции
в шесть методов определения удельной поверхности кремнезема
и других тонкодисперсных твердых веществ. Так, для кремне-
земного порошка с удельной поверхностью 56 м2/г, измеренной
методом БЭТ, значение удельной поверхности по методу «отри-
цательной» адсорбции оказалось равным 34,5 м2/г.
Характеристики пор
Измерение и определение пористости возможно только для
таких агрегатов, которые механически достаточно прочны и
на которые не будут оказывать воздействие методы исследова-
ния. Например, получение характеристик пористости посредст-
вом измерения методом вдавливания ртути возможно для обыч-
ных силикагелей, используемых в качестве катализаторов, од-
нако структура аэрогелей или осажденных кремнеземов должна
при этом методе разрушиться, и полученные результаты оказы-
ваются бессмысленными. С другой стороны, измерение размеров
пор путем заполнения их жидким азотом — значительно менее
разрушающий способ, а анализ методом малоуглового рассея-
ния рентгеновских лучей, очевидно, совершенно не разрушает
структуру.
Структура агрегатов и гелей рассматривалась в гл. 3, неко-
торые вопросы относительно формирования гелей из дискрет-
ных коллоидных частиц обсуждались в гл. 4. Объем литературы
по данной теме огромен, особенно по вопросам, касающимся
использования силикагелей в качестве основных катализаторов,
а также по возникшей недавно проблеме использования кремне-
зема для набивки хроматографических колонок. Опубликован
ряд исчерпывающих обзоров, касающихся вопросов пористости
кремнеземов [1—6]. Унгер [6] представил особенно четкое и
конкретное описание природы пор и их характеристики приме-
нительно к кремнезему.
Имеется монография [123] по пористым структурам, в ко-
торой рассмотрены также методы измерений.
Силикагели и порошки
657
Следующие структурные меняющиеся величины характери-
зуют пористость:
1. Удельная поверхность (размерность м2/г). В эту величину
входит понятие суммарной поверхности твердой фазы. За
исключением чрезвычайно небольших агрегатов субмикронного
размера, наружная поверхность незначительна по сравнению
с площадью поверхности стенок пор. Методы измерения удель-
ной поверхности обсуждались в предыдущем разделе.
2. Удельный объем пор vp, измеряемый в миллилитрах на
грамм, представляет собой суммарный объем пор, отнесенный
к грамму твердого вещества. Эта величина обычно опреде-
ляется посредством измерения объема жидкости, необходимой
для заполнения собственно пор, а не промежутков между агре-
гатами.
3. Средний диаметр пор dp (измеряется в ангстремах).
4. Распределение пор по размерам, выражаемое в виде
функции распределения
kopl^dp = f(dp)
5. Степень, до которой вход в поры большего размера огра-
ничивается наличием пор меньшего диаметра (поры, имеющие
форму «бутылки с узкими открытыми концами» или форму «пу-
зырька для чернил»).
Подобные параметры обычно получаются с помощью следую-
щих экспериментальных методов:
1. Изотермы адсорбции газов и паров, посредством кото-
рых количество адсорбата ха измеряется как можно более
точно в пределах всей области относительных давлений
р/ро 0—1,0.
2. Метод ртутной порометрии — измерение объема ртути,
которая вдавливается в поры, в зависимости от давления. Поры
должны быть совершенно несмачивающимися по отношению
.к проникающей в них жидкости, поэтому ртуть является един-
ственно подходящей жидкостью, доступной при обычной темпе-
ратуре.
3. Малоугловое рассеяние рентгеновских лучей представ-
ляет сведения о расстоянии между поверхностями раздела
твердое тело—газ, т. е. дает значение диаметра пор (или диа-
метра частиц).
4. Электронная микроскопия оказывается полезным мето-
дом для подтверждения математических моделей, выдвигае-
мых при интерпретации адсорбционных данных и данных по
вдавливанию ртути.
5. Проницаемость газов в некоторых случаях оказывается
измеримой функцией от среднего размера пор.
16 Заказ № 250
658
Глава 5
6. Недоступность вхождения в поры ионов и молекул из-
вестного размера может использоваться при соответствующих
условиях для измерения размеров пор *.
7. Объем поглощаемой жидкости дает возможность изме-
рить объем пор, если такие измерения проводятся при подхо-
дящих контролируемых условиях.
Поскольку адсорбционные характеристики твердых тел за-
висят от размеров пор, то удобно классифицировать поры как
субмикро-, микро-, мезо- (или переходные) и макропоры. Эта
терминология была предложена, а затем несколько изменена
Дубининым [10, 124], который дал следующее распределение:
Тип пор Диаметр, А
Микро- От 10—12 до 26—38
Мезо- От 30—32 до 2000—4000
Макро- Свыше 2000—4000
Размер частиц и упаковка
В течение многих лет структура силикагелей оставалась
предметом дискуссий. Структура геля зачастую представлялась
в виде сетки с поперечными связями, составленной из молеку-
лярных «цепочек» поликремневой кислоты, подобно структуре
органических гелей. Однако в 1926 г. Френдлих предложил,
а в 1940 г. Кармен обосновал предложение рассматривать
структуру силикагеля как состоящую из первичных сфериче-
ских частиц.
До того как была открыта возможность приготовлять од-
нородные частицы кремнезема с диаметром больше 5—10 нм,
силикагели получались из типов кремнезема, сформирован-
ных в результате подкисления силиката натрия или гидролиза
сложных кремневых эфиров. Даже с помощью электронного
микроскопа было трудно различить структуру таких гелей. Од-
нако из рассмотрений Айлера [2], Киселева [3], Высоцкого
(в работе [5]) и Унгера [6], несомненно, следует, что силика*
гели и кремнеземные порошки первоначально образуются из
«корпускулярных» или «глобулярных» дискретных частиц. Шу-
гар и Губа [125] разработали способ приготовления тонких
срезов из очень тонкодисперсных структур геля, что позволило
получить снимки с 105-кратным увеличением. Благодаря этому
удалось различить, что структура в действительности представ-
ляла собой подобные волокнам нити, состоящие из цепочек гло-
* Так называемый молекулярноснтовый эффект, т. е. недоступность ми-
кропор для молекул адсорбата.— Прим. ред.
Силикагели и порошки
659
бул. Были измерены размеры пор и диаметров частиц и подсчи-
таны значения удельной поверхности и среднего размера пор,
которые удовлетворительно согласовывались со значениями,
полученными по адсорбции азота. Леонтьев и Лукьянович [126а]
при проведении электронно-микроскопических исследований раз-
личных кремнеземов использовали метод углеродных реплик,
получаемых со структур силикагелей.
Образование цепочек из частиц кремнезема сферической
формы в структуре геля было подробно рассмотрено в гл. 3.
В данном разделе описывается влияние, оказываемое на пори-
стость размерами частиц и их упаковкой.
Модель типичной поры в компактном силикагеле с кубиче-
ской упаковкой сферических частиц показана на рис. 5.3а.
Объем пор составляет 48 % от общего объема. Поскольку
52 % приходится на объем самого кремнезема, который имеет
плотность 2,2 г/см3, то удельная пористость оказывается равной
0,42 см3/г.
Еще в 1954 г. Киселев [1266] подсчитал значения удельной
поверхности, объемов пор, размеров пор, а также координацион-
ное число средней частицы в зависимости от изменения плот-
ности упаковки таких сферических частиц. Как видно из
рис. 5.36, если геометрия упаковки остается без изменений, то
диаметр пор убывает с уменьшением диаметра частиц. С дру-
гой стороны, при изменении упаковки частиц постоянного раз-
мера диаметр пор может изменяться. К тому же оба фактора —
упаковка и размер частиц—могут изменяться одновременно
таким образом, что диаметр пор сохраняется постоянным.
На основании экспериментальных данных считается, что все
гели и осажденные кремнеземы имеют тенденцию сжиматься под
действием сил поверхностного натяжения воды, определяющих
усадку по мере того, как вода удаляется при высушивании из
пор. Если не принимать специальных мер предосторожности,
чтобы укрепить структуру и понизить влияние сил поверхност-
ного натяжения, то образовавшийся влажный осадок, или гель,
оказывается сильно уплотненным до приблизительно одного и
того же координационного числа, равного 5—6. В этом случае
диаметр пор пропорционален размеру частиц и изменяется об-
ратно пропорционально величине удельной поверхности. Унгер
[6] дает следующую взаимосвязь диаметра пор по данным Ду-
бинина и удельной поверхности:
Тип пор Диаметр пор, о А Удельная поверхность м2/г
Микро- <20 >500
Мезо- 20-2000 500-10
Макро- >2000 <10
16*
660 Глава 5
Рис. 5.3а. Модель «элементарной поры», образующейся в случае регулярной
упаковки сфер при координационном числе 6.
Координационное число Объем % Объем пор, смэ/г
1 2
12 » ® 74.5 25,5 0,155
СП № Ж 52 48 0,42
3 5 95 8,6
3-2-3 1,3 98,7 35
$-2-2-3 0,83 99,17 . 54
Рис. 5.36. Схемы структур с различной пористостью.
Размеры первичных частиц и координационное число (число частиц, касающихся каждой
единичной частицы) определяют объем пор и их средний диаметр. В действительности
структуры трехмерны. 1 — объем твердого вещества; 2 — объем пор.
Силикагели и порошки
661
Однако такая связь не всегда существует; так, в некоторых
рыхлых аэрогелях, имеющих очень низкую плотность, могут
быть и поры большого диаметра, и высокие значения удельной
поверхности.
Взаимосвязь размеров частиц и их упаковки с характеристи-
ками пор рассматривалась на примерах нескольких моделей.
Масон [127] рассмотрел произвольную упаковку сфер, поскольку
в силикагелях упаковка, конечно, не является регулярной (за
исключением опала, который обсуждался в гл. 4). Он рассмат-
ривал пору в виде тетраэдрического узла, в котором центры
соседних сфер соединены, хотя при этом сферы не обязательно
касаются друг друга. Поры, следовательно, определяются коор-
динатами центров сфер. Свойства таких пор оценивались на
ЭВМ по составленной программе. Согласно Масону, произволь-
ная упаковка сфер дает объемную плотность, равную 0,63, что
оказывается примерно средним значением между значением
0,52, характерным для открытой (кубической) упаковки, и
значением 0,72 для закрытой (гексагональной) упаковки.
Упаковка частиц изучалась экспериментально, начиная
с очень тонкодисперсных кремнеземных порошков, состоящих
из однородных по размеру частиц и находящихся в рыхлом
состоянии с низким значением объемной плотности, когда каж-
дая единичная частица касается только лишь двух, трех или
четырех соседних частиц. Когда же такая система механически
спрессовывается, то число контактов между частицами стано-
вится все больше и больше.
Были подсчитаны [128] адсорбционные характеристики и
геометрия упакованных сферических частиц. Используя коор-
динационные числа 4, 6 и 8, авторы этой работы вывели урав-
нение для подсчета количества вещества, адсорбированного при
различных значениях относительных давлений.
Эвери и Рамзай [129] также подсчитали характеристики
различных способов упаковки однородных по размеру сфер,
представленные в табл. 5.1. Хотя в действительности в гелях, ве-
роятно, отсутствует подобная регулярность упаковки, тем не
менее из данных по пористости геля можно оценить значение
координационного числа п. Если средний диаметр частиц изве-
стен, то по плотности геля можно приблизительно подсчитать
диаметр пор. При этом заранее предполагается, что все пер-
вичные частицы в геле однородны по размеру, и это в общем
оказывается справедливым, если только не принимаются особые
условия, позволяющие иметь в системе частицы двух размеров.
Мейсснер, Михаэле и Кайзер [130] рассмотрели другие рас-
положения упаковок при значениях доли ф, приходящейся на
твердую фазу ([1—ф]—доля, относящаяся к пористости),
подсчитанной для разных координационных чисел. Авторы ссы-
662
Глава 5
лались на ранее выполненные работы Хиша и Лавеса [131] и
Манегольда, Хоффмана и Соффа [132] с приблизительно та-
Объемная пористость,
см3/см3 Радиус пор
Рис. 5.4. Зависимость объемной пористости и радиуса пор от координацион-
ного числа сферических частиц, сформированных в трехмерную сетку, подоб-
ную сетке геля.
А — объемная пористость (т. е. объем пор, отнесенный к единице объема твердого ве-
щества, см3/см3); В — радиус пор в горловинах; С — радиус пор в полостях в зависимо-
сти от изменения координационного числа сферических частиц, Я—радиус частицы.
Штриховая линия проведена на основании уравнения Мейсснера, Михаэлса и Кай-
зера [130].
ким же общим подходом. Мейсснер предложил общее урав-
нение
п = 2 ехр (2,4</>)
для которого были подсчитаны некоторые величины, представ-
ленные на рис. 5.4. При этом доля объемной пористости при-
нимается равной 1 —ф.
Чтобы более точно смоделировать действительные структуры
гелей, целесообразно провести исследования плотно, но произ-
вольно упакованных сфер. Скотт [133] сообщает, что при рых-
лой произвольной упаковке однородных сфер значение объем-
ной плотности равно 0,60, тогда как при плотной произвольной
Силикагели и порошки
663
Таблица 5.1
Характеристики геометрических тел, получаемых из упакованных сфер
радиусом 7? по данным [129]
Тип упаковки Координа- ционное число п Пористость, см3 пор на см3 тела Кажущаяся плотность тела а, г/см3 Радиус вписанной в полость сферы Радиус вписанной в горловину окружности
Гексагональная 12 0,260 1,63 0,225 Р 0,155 Р
(плотная) Тетрагональная 10 0,302 1,54 0,414 Р 0,291 Р 0,265 Р
(центрирован- ная) Гексагональная 8 0,395 1,33 0,527 Р 0,155 Р 0,414 Р
(простая) Кубическая (про- стая) Тетрагональная 6 0,476 1,15 0,732 Р 0,155 Р 0,414 Р
4 0,666 0,73 1,00 Р 0,732 Р
а Принимается, что истинная плотность самих кремнеземных сфер равна 2,2 г/см3.
упаковке получается значение 0,64. Эти результаты обсужда-
лись Берналом, Масоном и Найтом [134, 135]. Действие, ока-
зываемое произвольной упаковкой сфер на капиллярные свой-
ства системы, было описано Масоном [127].
Потери площади поверхности частиц вследствие
их упаковки
Эвери и Рамзай [129] методом конденсации паров приго-
товили частицы SiO2 (а также ZrO2) диаметром ~4 нм в виде
рыхлого порошка и затем прессовали порошок под давлением
вплоть до 15 500 кг/см2. В исходном распушонном порошке не
представлялось возможным выявить какие-либо определенные
«поры», но когда получалась связанная масса, то уже обнару-
живалось приблизительное соотношение между давлением и
координационным числом частиц кремнезема, которое можно
представить в следующем виде:
Давление, кг/см2 Удельная поверхность м2/г
0 636
1550 522
7 750 373
15 500 219
Координацион- Пористость.
ное число п см3 пор иа 1 см3 твердого вещества
3 —
5,6 0,51
9,8 0,33
— 0,204
664
Глава 5
В работе отмечается, что частицы легко спрессовываются
до состояния, приблизительно отвечающего кубической упа-
ковке, когда около 50 % объема твердого вещества составляют
поры. Однако при давлении 15500 кг/см2 пористость падает
до значения 0,204, что оказывается даже меньше, чем в случае
совершенных, плотно упакованных сфер, когда пористость равна
0,255. При таком большом давлении некоторые из частиц могут
сплющиваться, поэтому расчетные формулы оказываются не
применимы.
Авторы приняли, что удельная поверхность прессованного
порошка оценивается приблизительным уравнением
S = S0(l-„^)
где S— образующаяся после прессования удельная поверхность
из плотных сфер радиусом R (в нанометрах), м2/г; п — число
контактов единичной сферы с другими сферами; d — принятый
диаметр молекулы азота, равный 0,375 нм; Sg — исходная удель-
ная поверхность, получаемая из обособленных сфер. Для диск-
ретных сфер Sg = 2750/2R (м2/г), где R— радиус в нанометрах.
Подстановка Sg и d в выше приведенное уравнение дает
SR2 — 1375/?+ 128,9ц = 0
Таким образом, представляется возможным подсчитать ра-
диус первичной сферы R из известных данных по удельной по-
верхности и удельному объему пор, а также можно оценить
значение координационного числа. При подстановке данных
для S и п из выше представленной таблицы по указанным урав-
нениям определены следующие значения R и SG:
S (измеренное), п /? (рассчитан- Sq (рассчитан-
м2/г ное), нм ное), м2/г
552 5,6 1,91 719
373 9,8 1,95 705
Однако приведенное уравнение может стать неприменимым,
когда частицы малы, поскольку оно предполагает, что размер
молекулы азота незначителен по сравнению с размерами крем-
неземных частиц.
Более строгое выражение с учетом потери участка поверх-
ности при контактировании двух сфер радиусом R дается в под-
писи к рис. 5.5.
В таком случае для массы однородно агрегированных сфер
результирующая удельная поверхность S будет составлять
только долю от значения удельной поверхности Sg, имевшей
место до того, как такие сферы соединялись вместе:
е <? 71 „ а (2R + а) А
S Sq п 4Л>2 )
Силикагели и порошки
665
В том случае, когда известно изменение S в зависимости от п,
значение R можно подсчитать из последнего уравнения путем
подстановки
2750
2R
где а и R даются в нанометрах. Согласно Брокхоффу и Лин-
сену [4], диаметр молекулы азота должен быть взят равным
Рис. 5.5. Схематическое изображение неиспользуемой площади вблизи точки
контакта между двумя сферическими частицами при адсорбции азота.
R— радиус сферической частицы, нм; а — радиус молекулы азота, равный 0,177 нм; b —
радиус недоступной для адсорбции круговой площади:
Приблизительная величина неиспользуемой площади в расчете на один контакт одной
частицы составляет ЛЬ2. Доля неиспользуемой площади поверхности сферических частиц
составляет в расчете на один контакт a(‘2R + a)/4R2; немного более точное значение равно
0,5а/?/(/? + #)2, но оно менее удобно при решении уравнения относительного радиуса R.
3,54 А, а следовательно, а=0,177 нм. Тогда уравнение запишется
в виде
S/?3—137,5/?2+121,7/г^—10,76/г = 0 (1)
Объем пор pv, выражаемый в сантиметрах кубических на
грамм SiO2, можно перевести в величину пористости р/ по урав-
нению
п __ Ру
/7^ + 0,455
где р/ выражается в кубических сантиметрах пространнства пор,
отнесенных к 1 см3 кремнезема. Далее можно определить сред-
нее значение координационного числа, используя кривую А, по-
казанную на рис. 5.4. Тогда по данному измеренному значению
удельной поверхности S и полученному координационному числу
666
Глава 5
п можно подсчитать из уравнения (1) значение радиуса ча-
стицы /?. После этою вычисляется значение 5g из соотношения
5G = 2750/2/?. Ниже представлены данные Эвери и Рамзая:
Давление, Средняя Координацион- Подсчитанное Подсчитанное
КГ / СМ 2 удельная ное число п значение R, значение Sq
поверхность, м2 /г нм М2,' г
0 636 . — —
1550 522 5,6 2,02 680
7750 373 9,8 2,10 655
Рассчитанные значения Sg хорошо согласуются со значе-
нием 5 = 636 м2/г, измеренным для исходного рыхлого, неспрес-
сованного порошка, который должен иметь очень небольшое
координационное число. Это обстоятельство указывает на то,
что многие частицы в исходном состоянии могут удерживаться
в сетке, имея лишь одну точку контакта.
Удобной для применения формой уравнения является
Lf = п • 0,0885/7? — 0,0075/Т?2
где Lf — доля суммарной поверхности сферических частиц, те-
ряемая вследствие того, что каждая сфера с радиусом R со-
прикасается с п окружающими ее сферами.
На основании дальнейших исследований де Бур и др. [45]
получили несколько более точное выражение
Lf = 0,0885л. 177)2
В этом случае для подсчета радиуса частиц R при известных
измеренных значениях 5 и п используется следующее уравнение:
о (Z? + 0,177)2 - 0,885/1/? (/?+0,177)2
R (/? + 0,177)2
Барби [8] провел аналогичные расчеты.
Такие подсчеты не применимы, когда сферические частицы
упаковываются настолько плотно, что поры оказываются слиш-
ком малыми, а единичная молекула азота — слишком большой,
чтобы войти в пору и заполнить ее по ширине. Следовательно,
при более плотно спрессованных образцах кремнезема удель-
ная поверхность, измеренная по адсорбции азота, оказывается
слишком заниженной по сравнению с истинной. Этот случай
обсуждался авторами работы [136], которые показали, что
истинное значение удельной поверхности в таком случае может
оказаться в 3,63 раза большим по сравнению с кажущимся зна-
чением, а также то, что по методу адсорбции азота нельзя уста-
новить наличие пор с диаметрами менее чем 1,4 нм. Даже поры,
Силикагели и порошки
667
диаметр которых равен при измерении по адсорбции азота
2,0 нм, на самом деле имеют меньший диаметр, более близкий
к 1,0 нм. Этим фактом, вероятно, объясняется то, что диаметры
пор для прессованного кремнезема с диаметром частиц 4 нм
в два раза превышают диаметры пор, рассчитанные из гео-
метрической упаковки частиц.
Потеря доли удельной поверхности при вступлении в кон-
такт сферических частиц наблюдалась также при прессовании
аэрогеля с одновременным измерением изотерм адсорбции, из-
меняющихся в зависимости от давления прессования. Такие из-
мерения нельзя выполнять для силикагелей, из которых удалена
вода при высушивании, так как подобные структуры уже сжа-
ты в результате сил поверхностного натяжения, действовавших
в процессе высушивания. С другой стороны, высокопористые
аэрогели приготовляются без сжатия структуры, так что можно
получить небольшие по размеру частицы с высоким значением
удельной поверхности, с широкими порами и очень большим
объемом пор.
Николаон и Тейчнер (137] выполнили измерения изотерм
адсорбции азота на исходном аэрогеле, а также на образцах
аэрогеля после механического сжатия и уплотнения, как это
представлено в табл. 5.2 и на рис. 5.6. Исходный аэрогель пред-
ставлял собой чрезвычайно легкое твердое вещество, объем ко-
торого был равен 100 см3, а масса всего 5 г (иР = 8 см3/г). Од-
нако после заполнения этого вещества жидким азотом и после-
дующего испарения азота под действием поверхностного
натяжения такой жидкости происходила усадка структуры и ве-
личина vp понижалась до 2,38 см3/г. После того как этот поро-
шок кремнезема механически прессовался (в вакуумных усло-
виях), получались уже другие кривые изотерм, показанные на
рис. 5.6.
Айлер использовал вышеприведенное кубическое уравнение
(1), чтобы подсчитать радиус первичной частицы аэрогеля R.
Результаты этого подсчета показаны в табл. 5.3. Может выз-
Таблица 5.2
Характеристики прессованного аэрогеля
(по данным [137])
Обозначения на рис. 5. 6 Давление при прессовании, кг/см2 Объем пор, см3/г Удельная поверхность, м2/ г
А 0 2,38 906
В 2 000 1,28 858
С 10 000 0,40 660
668
Глава 5
Рис. 5.6. Изотермы адсорбции и десорбции азота на аэрогеле.
4 — образец аэрогеля рыхлый, неуплотненный; В — образец уплотнен под давлением
2000 кг/см2; С — образец уплотнен под давлением 10 000 кг/см2. По данным [137].
вать некоторое удивление тот факт, что подсчитанное значение
удельной поверхности для случаев разделения неконтактирую-
щих частиц оказывается значительно большим по сравнению
с измеренным по адсорбции азота для образца, который лишь
Таблица 5.3
Расчет размеров первичных частиц прессованного аэрогеля
Объем пор, см3/г «Пористость, см3/ см3 аэрогеля Координацион- ное число п Удельная поверхность, м2/г (эксперимент) R, нм (расчет) 8 , м2/г (J (расчет)
2,38 0,84 “3,35 906 1,06 1297
1,28 0,74 3,7 858 1,08 1273
0,4 0,47 6,2 660 1,00 1375
Среднее значение 1315
Силикагели и порошки
669
слегка спрессовывался за счет удаления жидкого азота. Срав-
нительно согласующиеся между собой значения для R находи-
лись в интервале 1,00—1,08 нм, а соответствующее среднее зна-
чение удельной поверхности составило 1315 м2/г.
Такие исследования являются наглядным доказательством
того, что свойства гелей кремнезема можно объяснить на основе
трехмерных агрегатов непористых аморфных сферических ча-
стиц кремнезема, различающихся по размерам и по значениям
плотности упаковки, даже для случая частиц, имеющих размер
только 20 А.
Характеристики пор по изотермам адсорбции
Синг [8] представил исключительно четкое и сжатое описа-
ние изотерм адсорбции и способов их интерпретации на основе
распределения пор по размерам. Как показано на рис. 5.7,
Рис. 5.7. Зависимость объема адсорбированного пара и заполнения пор
жидкостью от парциального давления. (По данным Айлера [2], с разрешения
Cornell University Press.)
пары таких веществ, как азот или вода, адсорбируются в пори-
стом силикагеле или в порошке во всевозрастающих количест-
вах по мере увеличения относительного давления пара р/рр,
где р0 — давление насыщенного пара выбранной в качестве ад-
сорбата жидкости.
Начинаясь с давления, равного нулю (точка А), типичная
изотерма адсорбции содержит несколько ступеней. Прежде
всего наблюдается, что с ростом давления возрастает доля по-
верхности, покрываемая адсорбируемыми молекулами (точка В).
В определенной точке на кривой (вблизи С) поверхность пол-
670
Глава 5
ностью покрывается одним слоем молекул. На этой стадии бу-
дут также заполняться адсорбатом поры, которые имеют диа-
метр только в два—три раза больше, чем диаметр молекулы
адсорбируемого вещества. При более высоком значении отно-
сительного давления (точка D) начнут заполняться поры боль-
шего размера. Когда давление близко к давлению насыщения
Ро (точка £), то жидкость заполнит все поры, что дает возмож-
ность измерить объем пор. Вначале предполагали, что диаметр
пор мог быть подсчитан из уравнения Кельвина [138], получен-
ного на основании теории, согласно которой жидкость остается
в порах даже в том случае, когда давление пара оказывается
ниже, чем давление жидкой фазы, так как давление пара жид-
кости в таких порах понижается из-за эффектов поверхностного
натяжения в тонких капиллярах. Уравнение Кельвина [138]
связывает давление пара р, при котором пар будет конденсиро-
ваться в цилиндрическом капилляре радиусом гк:
—2oV cos 0
Гк~ RT • 2,303 lgp/po
o' — поверхностное натяжение жидкого азота при его темпера-
туре кипения (—195,8°С), равное 8,85 эрг/см2; 9 —угол смачи-
вания, принимаемый равным нулю, так что cos 0=1; V — моль-
ный объем жидкого азота, равный 34,7 см3; 7? — газовая постоян-
ная, равная 8,314-107 эрг/(град-моль); Т — абсолютная темпе-
ратура, равная 77 К.
В случае азота уравнение Кельвина упрощается:
. _ 4,146
Гк ~ IgPo/P
где гк—-радиус капилляра, который заполняется при парциаль-
ном давлении р/ро. Однако это оказывается справедливым в ос-
новном только для капилляров сравнительно большого размера.
Когда же диаметр капилляра приближается к молекулярным
размерам, то необходимо принимать во внимание следующее:
когда р/ръ превышает 0,2—0,3, то даже плоская поверхность ста-
новится покрытой мономолекулярным слоем адсорбата. Напри-
мер, при р/ро=О,93 образуется слой, равный по толщине четы-
рем молекулам (14 А). Уравнение Кельвина указывает, таким
образом, на дополнительное количество адсорбата, добавляе-
мое к количеству, которое сконденсировалось на плоской поверх-
ности при данном значении р/р0- Следовательно, если толщина
адсорбированного слоя на плоской поверхности составляет t, то
тогда радиус поры гР, которая заполняется при р/ро, будет ра-
вен rP = t+rk.
Как показано на рис. 5.8, А, если после полного заполнения
снова понижать давление, то часто ход изотермы не совпадает
Силикагели и порошки
671
с начальным вследствие того, что в этом случае требуется не-
сколько более низкое давление, чтобы испарить жидкость. По-
добная «гистерезисная петля» появляется потому, что в боль-
шинстве структур силикагелей в порах имеются полости, диа-
метр которых превышает размер прохода, ведущего к ним.
Когда из такой поры удаляется жидкость, то давление будет
определяться меньшим диаметром канала, ведущего к полости.
Этот канал удерживает мениск жидкости, по мере того как
опорожняется внутренняя полость большего размера.
Рис. 5.8. Типичный вид изотерм адсорбции азота,
о
А — мезопористый силикагель с радиусом пор 36 А;; В — микропористый силикагель
О
с радиусом пор 10 А. (По данным Айлера [2], с разрешения Cornell University Press.)
Совсем другая ситуация имеет место, если все поры оказы-
ваются очень небольшими по размеру, так что их диаметр пре-
вышает диаметр молекулы адсорбата всего лишь в несколько
раз. Например, могут существовать поры диаметром 20 А, когда
адсорбатом является азот, диаметр молекулы которого состав-
ляет 3,54 А. Как показано на рис. 5.8, В, в этом случае поры
способны заполниться даже при низком давлении, и поэтому
трудно определить точку на кривой, соответствующей заполне-
нию монослоя.
Макбайн [139] ввел термин «персорбция», имея в виду ад-
сорбцию в порах, диаметр которых равен лишь одному или двум
диаметрам молекулы адсорбата. Не так давно поры, имеющие,
как правило,, диаметр менее 20 А, стали называться «микропо-
рами». В таких порах теплота адсорбции заметно выше, чем на
плоских поверхностях, и эти поры могут оказаться заполненными
при более низком давлении, чем то, которое соответствует обра-
зованию мономолекул яркого слоя на плоской поверхности. Неко-
торые силикагели, несмотря на то что в них имеются поры
большого размера, обладают еще и некоторым объемом, опреде-
ляемым присутствием в образце очень малых пор [140]. Как по-
стулировал Кармен [141], такие поры могут возникать в виде
672
Глава 5
трещин между коллоидными частицами, составляющими гель.
Шерешевский и Рассел [142] исследовали адсорбцию паров
спирта на произвольно упакованных стеклянных шариках и наб-
людали эффект капиллярной конденсации в щелевых зазорах
около точек контакта шариков. Такая система по существу
представляет собой крупномасштабную модель вероятной струк-
туры некоторых разновидностей силикагелей, в частности тех,
которые приготовляются высушиванием золей, содержащих од-
нородные коллоидные частицы кремнезема. С другой стороны,
подобные малые поры могут также иметь место в большом
числе случаев, когда гели приготовляются из очень небольших,
плотно упакованных частиц. Таким образом, плотно упако-
ванные частицы диаметром 1-—2 нм неизбежно должны форми-
ровать гели, пронизанные порами почти молекулярных размеров.
Согласно данным Коулинха [143] и Брунауэра [144], можно
подразделять силикагели на четыре группы в зависимости от
радиуса пор:
1. Силикагели с порами молекулярных размеров (менее
чем 2 нм в диаметре), проявляющие явление персорбций, но
без капиллярной конденсации.
2. Силикагели с порами около 2 нм в диаметре, показы-
вающие гистерезис для воды, но не для больших по размеру
молекул и сильно выраженную капиллярную адсорбцию в ин-
тервале р/р0 0,5—0,8.
3. Силикагели, обладающие порами диаметром 3—10 нм
(микропоры) и проявляющие гистерезис для относительно боль-
ших по размеру молекул, а также капиллярную адсорбцию для
них, но только при более высоких значениях р/р0.
4. Силикагели с порами, большими чем 10 нм в диаметре,
которые показывают капиллярную адсорбцию только тогда,
когда значение р/р0 приближается к единице (аэрогели и пи-
рогенные кремнеземы).
Беринг, Дубинин и Серпинский [145] классифицировали
поры диаметром менее чем 3 нм как «микропоры», а поры диа-
метрами 3—200 нм как «переходные поры».
Если бы было возможным просверлить параллельные ци-
линдрические каналы в твердом кремнеземе, то тогда можно
было бы приготовить серию образцов с каналами, отличаю-
щимися по размеру, но с постоянным значением объема пор
в расчете на 1 г кремнезема. В таком случае изотермы адсорб-
ции должны были бы проявляться в форме кривых, представ-
ленных на рис. 5.9. Поскольку для отдельного образца все
поры имеют один и тот же размер, то все они должны запол-
ниться одновременно, когда достигается некоторое характерное
для данного образца давление пара. Кроме того, поскольку
все поры однородны по размеру, то никакого гистерезиса не
Силикагели и порошки
673
Рис. 5.9. Гипотетический вид
изотерм адсорбции азота.
Три пористых адсорбента имеют
одинаковый удельный объем пор,
причем однородные поры цилиндри-
ческой формы различаются по диа-
метрам, указанным в ангстремах.
Р/Ро
Рис. 5.10. Изотермы адсорбции воды на различных силикагелях и соответст-
вующие кривые распределения пор по размерам (по данным Киселева (146]).
17 Заказ № 250
674
Глава 5
должно наблюдаться. При наличии очень больших пор, факти-
чески бесконечно большого размера по сравнению с размером
молекулы воды, поры заполняются только при значении
р/ро=1,О.
Подобная идеализированная ситуация, конечно, фактиче-
ски не имеет места в реальных силикагелях, поры в которых
представляют собой неправильной формы пространства между
беспорядочно расположенными первичными сферическими ча-
стицами кремнезема. В качестве примеров, связанных с при-
менением на практике таких систем, на рис. 5.10 показаны изо-
термы адсорбции пара воды и соответствующие кривые распре-
деления пор по размерам, рассматриваемые Киселевым [146]
для серии силикагелей, каждый из которых имеет относительно
однородные по размерам поры. Объемы пор оказались по су-
ществу одинаковыми для силикагелей 2 и 3 и для силикагелей
4 и 5, что подтверждается примерно одинаковым количеством
воды, требуемой для заполнения пор. Таким образом, для сили-
кагелей 2 и 3 это количество составляет 50 ммоль Н2О в расчете
на 1 г кремнезема, что равно 0,9 см3 Н2О/г SiO2. Площади под
кривыми распределения пор по размерам для тех же двух образ-
' цов 2 и <3 примерно равны и также выражают значение, равное
0,9 см3/г. Эти изотермы характерны для типов силикагелей, при-
готовляемых стандартными способами.
Объем пор
Объем пор определяется по объему жидкого азота, необхо-
димого-для заполнения пор при приближении относительного
давления к значению р/ро=1*. Объем жидкости равен 1,55Х
Х10 3 Vg, где Vg — объем газа при нормальных температуре
и давлении. Как показано на рис. 5.10, такой объем четко вы-
ражается в конце соответствующей кривой изотермы. Это
остается справедливым до тех пор, пока образец кремнезема
имеет определенную прочность и сохраняет заданную форму,
т. е. занимает определенный объем и имеет измеримую плот-
ность. Однако в случае очень непрочных аэрогелей или рых-
лых, легко поддающихся прессованию порошков, изотермы тя-
нутся вверх без измеримого излома, по мере того как жидкий
азот в объеме образца начинает конденсироваться.
Чтобы определить суммарный объем пор, Лард и Браун
[147] предложили использовать метод потока, посредством ко-
торого смесь азота с гелием, содержащая 96,7 % азота, при-
* Обычно эта величина в отечественной научно-технической литературе
носит название предельного объема сорбционного пространства и выража-
ется в сантиметрах кубических на грамм.— Прим. ред.
Силикагели и порошки
675
водится в равновесие с образцом при температуре жидкого
азота. Затем образец нагревают до 25°С и измеряют выделяю-
щийся объем газообразного азота. Для этого метода доста-
точно использовать простую аппаратуру, предназначенную для
измерения объемов. По-видимому, для очень широкопористых
адсорбентов должна использоваться смесь, содержащая 99 %
или даже более азота, чтобы обеспечить хорошее заполнение
пор.
Анализ по поглощению масла, продолжительное время ис-
пользовавшийся для оценки газовой сажи и пигментов, может
дать значение объема пор для кремнеземов. В этом случае
используется нелетучая жидкость, способная легко проникать
в поры кремнезема. Если подразумевать под термином «масло»,
например, гликоль, то количество этого «масла», адсорбирован-
ного кремнеземным порошком, является косвенным показате-
лем пористости. При смешивании масла с кремнеземом вся
масса остается порошкообразной до тех пор, пока поры не за-
полнятся. В точке заполнения порошкообразный, сыпучий ха-
рактер образца исчеза'ет и может быть отформована общая
масса, удерживаемая посредством поверхностного натяжения,
возникающего в результате образования тонкой пленки жидко-
сти на внешней поверхности пористых агрегатов [148]. Однако
для случая тонкодисперсных агрегатов кремнезема пространство
между такими небольшими агрегатами также заполняется мас-
лом, и объем масла, необходимого, чтобы приготовить связан-
ную массу, приблизительно будет равен объему, необходимому
для связывания агрегатов кремнезема, с учетом объема пор и
при допущении, что указанные агрегаты однородны по раз-
мерам.
Размер пор и распределение пор по размерам .
Такие характеристики могут оцениваться несколькими спо-
собами из изотерм десорбции. Брокхофф и Линеен [149] пред-
ставили по этому вопросу достаточно подробный обзор. В до-
полнение к трудоемкому приему точного измерения изотерм
адсорбции в большинстве методов предусматривается проведе-
ние отдельно расчетов для большого числа интервалов рас-
сматриваемой изотермы. Однако при значительно усовершен-
ствованном способе измерения и выдачи получаемых резуль-
татов, возможности для обработки получаемых данных и со-
ставления программ для обсчета размеров пор на ЭВМ подоб-
ная работа значительно упрощается.
В настоящее время доступны два типа коммерческих прибо-
ров для проведения подобного рода измерений. В одном ис-
пользуется вакуумная система, как и в оригинальном методе
17*
676
Глава 5
БЭТ (прибор Micromeritics), а в другом — система потока газа
(прибор Quantachrome). Изотерма с 10—15 равновесными точ-
ками может быть измерена в течение нескольких часов, и зна-
чения удельной поверхности и распределение пор по размерам
могут быть получены довольно быстро [150, 151].
За прошедший век были разработаны различные матема-
тические приближения для расчета распределения пор по раз-
мерам.
В большинстве методов предусматривается построение
кривой, поскольку необходимо учитывать то, что на относи-
тельно гладкой поверхности в отсутствие пор происходит ад-
сорбция и адсорбционная пленка оказывается толщиной
в несколько молекулярных слоев, прежде чем давление пара до-
стигает значения р/ро=1,О, соответствующего образованию
жидкости. Очевидно, в такой толстой пленке, состоящей из не-
скольких слоев, свойства азота не будут теми же самыми, что
и для нормальной жидкости. Как уже отмечалось, определение
пор по размерам требует не только использования уравнения
Кельвина для подсчета размеров пор, которые заполняются
жидким азотом, имеющим свойства нормальной жидкости, но
также и знания толщины адсорбционной пленки на внутренней
поверхности пор, еще не заполненных азотом.
Чтобы получить экспериментальные данные, учитывающие
толщину пленки, исследуемый кремнезем не должен содержать
микропор. Харрис и Синг [152] изучили ряд таких образцов
кремнезема (с удельной поверхностью менее 12 м2/г) и пока-
зали возможность проведения усредненной по рассмотренным
ими образцам изотермы в виде зависимости величины v/vm от
р/р0. Однако с тех пор были проведены многочисленные иссле-
дования на соответствующих непористых кремнеземах с целью
точного определения ^-значений. Бебрис, Киселев и Никитин
[153] приготовили очень однородный широкопористый кремне-
зем, не содержащий микропор, посредством термообработки пи-
рогенного кремнезема (аэросила) в парах воды при 750°С, по-
лучив указанный кремнезем с удельной поверхностью около
70—80 м2/г и порами диаметром около 400 А. Обычно прини-
маемые значения толщины пленки t для различных значений
р/р0 при использовании азота основаны на данных Липпенса,
Линсена и де Бура [154] и де Бура, Линсена и Осинда [155].
В табл. 5.4 представлены типичные ^-значения в зависимо-
сти от р/ро. Приводимое ниже уравнение дает возможность под-
считывать толщину пленки с учетом большей части опублико-
ванных данных по усредненным значениям t при относительном
давлении р/р0 выше 0,3:
,_______4,58
(~lgp/A))I/3
Силикагели и порошки
677
Таблица 5.4
Парциальное давление азота и толщина пленки
азота, адсорбированного на непористой поверхности
при температуре —195°С (по данным [154])
Р! Ра с А р.Ра о t, А Р/Р» Л А
0,3 5,6 0,66 8,0 0,90 12,7
0,4 6,2 0,70 8,5 0,93 14,4
0,5 6,8 0,76 9,2 0,96 17,3
0,56 7,2 0,80 9,9 0,98 22,1
0,6 7,5 0,86 11,2 0,99 27,8
Как описано Брокхоффом и Линсеном [156], многие иссле-
дователи сделали вклад в развитие методов расчета распреде-
ления пор по размерам из изотерм адсорбции. Первоначальный
подход и общее уравнение, разработанные Барретом, Джойне-
ром и Халендой [157], были доведены до конца Пирсом [158]
и позднее Кранстоном и Инкли. Последующее развитие этой
проблемы было подробно описано Грегом и Сингом [7].
Метод Кранстона и Инкли. Кранстон и Инкли (39), исполь-
зуя известную толщину пленки t адсорбированного азота на
внутренних стенках пор наряду с заполнением пор азотом
по механизму, описываемому уравнением Кельвина, разрабо-
тали метод расчета объема и размеров пор из десорбционной
или адсорбционной ветвей изотермы. Расчет ведется на участке
изотермы выше р/р0>0,3, где уже имеется адсорбированный
по крайней мере мономолекулярный слой азота.
Метод представляет собой ступенчатую процедуру подсчета,
которая, хотя и проста, но предусматривает такие подсчеты
на каждом очередном этапе. Изотерма десорбции состоит из
серии экспериментальных точек, в каждой из которых имеются
данные по измеренному объему адсорбированного газа при оп-
ределенном давлении. Начиная от точки р/ро=1,0 при пол-
ностью заполненных порах, ступенчато понижают давление
и при этом на каждом этапе замеряют адсорбированный объем
(это относится к изотерме десорбции, но процедура расчетов
будет той же самой и при рассмотрении изотермы адсорбции).
По мере того как давление понижается от значения pi/p0 до
Pilpo-, справедливыми оказываются следующие положения:
1. Объем жидкого азота Wnq испаряется из пор, образуя
при этом газ объемом AVg, который обычно выражается в куби-
ческих сантиметрах при нормальных условиях в расчете на 1 г
адсорбента.
678
Глава 5
2. Объем AV«g жидкого азота, который был удален из пор
в интервале размеров их радиусов между ц и г2, оставляет на
стенках этих пор пленку азота толщиной t2.
3. В порах, опорожненных на предыдущих этапах, толщина
пленки азота на стенках понижается от В до t2.
Читателю, незнакомому с этим вопросом, может помочь схе-
матическое изображение процесса, представленное на рис. 5.11.
На рисунке показано поперечное сечение образца с идеали-
зированными порами цилиндрической формы, которые разли-
чаются по своим диаметрам. Видно, что когда давление в си-
стеме понижается от щ (положение А) до р2 (положение В),
толщина пленки азота на стенках опорожненных капилляров
уменьшается от t\ до t2, количество жидкого азота уменьшается
в результате десорбции и при этом увеличивается число пустых
пор.
В положении А (рис. 5.11) имеется одна частично запол-
ненная пора диаметром 2гь в которой жидкий азот находится
в данный момент в равновесии с паром при давлении pi.
Аналогично имеем в положении В одну пору диаметром 2г2,
в которой содержится жидкий азот, находящийся в равновесии
при давлении р2. В указанных порах радиус определяется как
rp=t+rk, где rh— радиус, подсчитываемый из уравнения Кель-
вина при данном давлении. Расчеты основаны на использова-
нии следующих уравнений. Пусть L — длина, равная суммар-
ной длине всех опорожненных пор с радиусами в интервале от
Г\ до r2, а гр — среднее значение радиуса. Тогда суммарный
объем испаренного жидкого азота Vuq на данном этапе равен
Vliq = 3,14 (гр - /2)2 L + (t2 - h) £ А
где А — поверхность адсорбционной пленки, оставшейся в ука-
занных опорожненных порах.
Средний объем пор радиусом гр составляет
АКР — nfpZ
Исключив величину L, получим
АКР = [-7-^7; J" \Уид - (t2 - tx) £ А]
Так как гР— ^ = г?г, где rh находится из уравнения Кельвина, то
Г fp 12
Объем выделившегося газа, измеряемого при давлении р
и температуре ТК, соответствует объему жидкости
Силикагели и порошки
679
В-Р2/Ро
Рис. 5.11. Схема воображаемого адсорбента с набором цилиндрических пор,
показанных в разрезе, когда азот адсорбируется при двух давлениях pi и Рг-
А—давление pi. Все поры, имеющие радиус менее заполнены жидким адсорбирован-
ным веществом. Адсорбционная пленка имеет толщину /ь а радиус Кельвина в поре,
заполненной под воздействием поверхностного натяжения, равен .
5 — давление Рг (Pa<Pi). Большее число пор не заполнено, поры с радиусами менее г2
остаются заполненными. Адсорбционная пленка имеет толщину /2, меньшую, чем ti. Ра-
также становится меиьшим, чем г. . L
диус Кельвина г.
«2
— суммарная длина пор, опо-
рожненных по мере того, как давление понизилось от pt до рг (см. текст).
Площадь А внутренней поверхности рассматриваемых пор
при допущении, что они являются цилиндрическими, оказы-
вается равной
Л = 2 (1/рДр) 104
где Vp выражается в сантиметрах кубических, а радиус гр —
в ангстремах.
680
Глава 5
Используя данные по десорбции, расчеты начинают при
р/ро вблизи 1,0, когда поры фактически заполнены жидким
азотом. Крэнстон и Инкли описали поэтапные подсчеты объема
пор и поверхностей опорожненных пор. Тем не менее детали-
зация такого рассмотрения будет полезна.
Расчеты выполняются на каждом этапе при фиксирован-
ном давлении, начиная с заполненных пор и относительного
давления р/р0, близкого к 1,0. Для каждого этапа подсчиты-
ваются следующие значения:
1. Среднее значение гн из двух кельвиновских радиусов rht
и Гкг при соответствующих давлениях pi и р2, выражаемое
в ангстремах. Каждое значение подсчитывается из уравнения
Кельвина
4,146
гк = п---1—
lgPo/Л-
2. Толщины пленок t\ и i2 при давлениях рх и р2, выражае-
мых в ангстремах. Каждая толщина t берется из таблиц или
определяется из уравнения
t = 4,583/(lg р0/р),/3
3. Средний радиус пор гр в данном интервале:
fp~ 0,5 (г*, -|- Л~|- Гк2 + t2)
4. Значение t = t\ — t2, выраженное в ангстремах.
5. Объем десорбированного жидкого азота AVnq в расчете
на единицу массы адсорбента, АУид = 1,55-10~3 AVg, см3/г, где
AVg — объем выделившегося газообразного азота, приведенный
к нормальным условиям, см3.
6. Объем жидкого азота, теряемый на данном этапе за счет
утоньшения пленок на стенках пор и равный (А/) • (у А), где
У А — поверхность стенок всех пор, опорожненных в процессе
десорбции на всех предыдущих этапах (или ДА для первого
этапа). Указанный объем равен (А/) • (2 А) 10~4 и имеет раз-
мерность см3, поскольку At выражено в ангстремах, а у А —
в квадратных метрах.
7. AA = 2(AVlig)-rP-W.
8. Значение У А находится посредством суммирования всех
значений ДА из предыдущих этапов.
Указанный процесс расчета является необходимым на каж-
дом этапе такого ступенчатого метода. Серия расчетов выпол-
няется для каждого этапа поочередно по мере понижения дав-
лений, и полученные результаты сводятся в таблицы.
Совокупный объем пор Vc, начиная от р//?о = 0,3 и вплоть до
наибольшего значения р/ро, представляет собой просто сумму
Силикагели и порошки
681
значений &Vnq, получаемых на каждом этапе. Как правило,
вычерчивается графическая зависимость Ус от lg гр.
Совокупная поверхность Ас — это общая сумма значений АЛ,
получаемых на каждом этапе. Если микропоры отсутствуют, то
Ас обычно составляет значения, достигающие 85—100 % вели-
чины поверхности, определяемой методом БЭТ. Так как пос-
ледняя получается при измерениях в области более низких
значений р/р0 от 0 до 0,3, то такое согласие указывает на от-
сутствие микропор в образце.
Крэнстон и Инкли пришли к заключению, что для многих
силикагелей рассмотренный метод целесообразно использовать
и в обратном направлении, начиная от значения /?/ро = О,3 и
проводя измерения и расчеты на последующих этапах по мере
получения изотермы адсорбции.
Хоуген [159] представил дальнейшее обсуждение метода
Кранстона и Инкли и дал некоторые полезные номограммы.
Однако оказалось не так просто перевести систему уравнений
в способ практических расчетов, именно поэтому обсчет рас-
смотренных выше этапов был показан столь детально.
Распределение пор по размерам может оцениваться из
i-диаграммы в соответствии с данными Брокхоффа и де
Бура [160].
Микропоры. Особого рода проблемы возникают при изме-
рении и описании пор чрезвычайно малых размеров. В данной
книге невозможно дать обзор всего имеющегося огромного ли-
тературного материала, появившегося за последнее десятиле-
тие, но будет предпринята попытка дать описание некоторых ас-
пектов по этой проблеме, сопроводив их примерами.
Согласно Брунауэру, в основном принято считать, что «ме-
ханизм адсорбции молекул в микропорах не достаточно хо-
рошо понят» [35]. Синг [161] в 1976 г. констатировал, что «не
разработано ни одного заслуживающего доверия метода для
определения распределения микропор по размерам». Однако
совершенно ясно, что адсорбция в микропорах отличается ко-
ренным образом от адсорбции на поверхности стенок широких
пор и на открытых поверхностях и что молекулы в таких тон-
ких порах подвергаются действию притяжения со стороны ок-
ружающего твердого тела и находятся в состоянии сильного
сжатия. Дубинин [124] обсудил теорию адсорбции в подоб-
ных условиях, которая включает понятие «объема микропор»,
более точно описывающее процесс, чем понятие поверхности
таких пор.
Согласно Оккерсу [162], удельная поверхность в микропо-
ристых материалах не может быть определена, если радиус
микропоры оказывается менее 12 А. Этот автор использовал
термин «субмикропора», подразумевая под этим понятием
682
Глава 5
то же самое, что и другие исследователи, в том числе Айлер,
который применял термин «микропора». Оккерс обобщил воз-
можное применение ряда уравнений, которые были предложены
для пор наименьших размеров.
Рис. 5.12. Типичные /-диаграммы
для двух кремнеземных порошков,
имеющих примерно одинаковое
значение удельной поверхности.
А — микропористый образец; В — мак-
ропористый образец. VL — объем ад-
сорбированного жидкого азота, см3/г;
t — толщина адсорбционной пленки
азота.
Рис. 5.13. Типичные с^-диаграммы.
А — широкопористый силикагель без
наложения ограничений на мономоле-
кулярную и полимолекулярную адсорб-
цию, но в отсутствие адсорбции в ка-
пиллярах; В — конденсация в мезопо-
рах,, вследствие чего данная кривая
отклоняется от прямой Л; С — адсорб-
ция в микропорах; заполненный объем
микропор составляет Vp, as=x/xs, где
х — количество адсорбированного ве-
щества при р/ро=О,4О и х — количество
адсорбированного вещества при дру-
гих относительных давлениях.
Как четко было продемонстрировано Брокхоффом и Линсе-
ном [163], микропоры могут быть обнаружены путем изучения
изотерм адсорбции, изображенных в виде /-кривых. Если на
графике линия, изображающая зависимость Va от t, отклоняется
вниз в сторону /-оси, то это является указанием на присутст-
вие в образце микропор. Аналогичные графики, полученные
Михаилом [164], представлены на рис. 5.12 для двух силика-
гелей. Поскольку значения удельных поверхностей образцов
близки, то линии на /-диаграммах имеют примерно одинаковый
наклон. Для силикагеля А, который является микропористым и
плотным, /-кривая начинает отклоняться вниз в сторону /-оси
при относительном давлении р/ро=О,1. Для мезопористого си-
ликагеля В, имеющего низкую плотность, /-кривая отклоняется
вверх примерно при р/ро = О,5, т. е. когда начинают заполняться
широкие поры. В подобных гелях, имеющих однородные по раз-
меру поры, легко показать наличие микропор. Однако для мно-
Силикагели и порошки
683
гих силикагелей большая доля поверхности принадлежит мезо-
порам и только небольшая часть — микропорам. В таком слу-
чае отклонение от линейности на ^-кривой трудно определять.
Миевилл [165а] исследовал твердые материалы смешанной
структуры, обладавшие мезопорами и микропорами. Он при-
менил метод ^-диаграммы и показал, что в таком образце со
смешанной структурой 10 % составляют микропоры.
Используя as-диаграмму, Синг [8] показал наличие мезо-
пор по отклонению от линейности по отношению к <%®-оси при
более высоких значениях а®. Наличие микропор доказывается
отклонением кривой в сторону а®-оси при более низких значе-
ниях а®. Экстраполяция линейного участка к оси х позволяет
определить объем микропор (рис. 5.13). Авторы работы [50]
провели дальнейшие исследования в этом направлении с боль-
шим набором кремнеземов и дали объяснение отклонениям на
основе представлений о микропорах и мезопорах.
Рамзай и Эвери [1656] получили данные по адсорбции азо-
та в плотных спрессованных микропористых кремнеземах. Они
строили по своим данным графики, используя уравнение*
[165в]
In -L_= _ D (RT In
Xm k P0 )
Пирогенный кремнеземный порошок с размером частиц 3—4 нм
прессовался до получения значений объемов пор 0,22—0,11 см3/г
(плотности упаковки кремнезема составляли 67—80 %), что
соответствовало образованию пор диаметром 22—12 А. На гра-
фиках, представленных в координатах указанного уравнения,
видно уменьшение наклонов линий для серии образцов, что сви-
детельствует о происходящих в них изменениях в области от
полного заполнения объема пор до монослойного покрытия
(когда монослой адсорбата заполняет и наиболее тонкие поры).
В этой работе константа С на графике, построенном в коорди-
натах БЭТ, имела значение 73 для исходного, непрессованного
порошка и увеличивалась от 184 до более чем 1000 по мере
.того, как диаметр пор уменьшался от 22 до 12 А.
Метод «модельных пор» (МП). Брунауэр, Михаил и Бо-
дор [166] разработали метод для определения характеристиче-
ского распределения пор по размерам, включая даже часть об-
ласти, занимаемой микропорами.
По методу Крэнстона—Инкли, включающему также Ккри-
вую и уравнение Кельвина, кривые, характеризующие пористую
структуру образца, могут рассчитываться для пор радиусами
от 10 до 150 А. Однако полученные результаты' зависят от при-
* Уравнение Дубинина—Радушкевича—Каганера,—Прим. ред.
684
Глава 5
нятого допущения о цилиндрической форме пор. Так как на
самом деле поры не являются цилиндрическими, то расчет рас-
пределения пор по размерам не отражает реального положения
дел, особенно при наличии небольших по размеру пор.
В методе «модельных пор» вводится понятие гидравлический
радиус» а, определяемый как rh = V/S, где V — объем по-
ристой системы и S — поверхность стенок пор. Соотношение
применимо к порам любой формы. Значения V и S подсчиты-
ваются из изотерм адсорбции или десорбции. Когда происходит
десорбция и какая-то группа пор оказывается опорожненной»
то монослой молекул азота остается на их стенках при давле-
нии р. Опорожненное пространство поры называется «сердцеви-
ной». Эта величина представляет собой десорбированный объем
по мере того, как давление понизилось от р0 до р.
Данный метод отличается от метода Кранстона и Инкли
еще и тем, что в нем вместо уравнения Кельвина используется
уравнение Киселева
у ds == Ар da '
где у — поверхностное натяжение; ds — поверхность, которая
исчезает по мере заполнения поры; Ар — изменение химиче-
ского потенциала, da — число молекул жидкости, находящихся
в поре. (Уравнение Кельвина представляет собой
случай приведенного выше уравнения Киселева, если
риваются поры цилиндрической формы.) Изменение
ского потенциала рассчитывается по уравнению
= — 7?7’1п(р/ро).
Интегрирование дает
частный
рассмат-
химиче-
—Ар =
as
s = — ( - RT In da
V J Ро
ah
где аи — число адсорбированных молекул в начале гистере-
зисной петли и as — число адсорбированных молекул при
насыщении.
Последнее уравнение интегрируется графически по этапам:
1. При десорбции tti молей вещества относительное давле-
ние р/ро понижается от 1,0 до 0,95.
2. Образующийся объем всех сердцевин будет равен про-
изведению а\ на мольный объем адсорбата; для случая азота
он составляет 34,6 ai см3.
3. Si—площадь поверхности образующихся сердцевин
определяется по уравнению
as
— ( — RTRi-^-da
V I Ро
1
Силикагели и порошки
685
Интегрирование проводится графически.
4. rh — гидравлический радиус, равный образующемуся
объему сердцевин (этап 2), деленному на площадь поверхности
таких сердцевин (этап 3).
Затем на n-м этапе, когда десорбируется ап молей, наблю-
дается следующее:
1. Понижение относительного давления р/ро от рп/ро до
pn-1/po-
2. Образующийся объем сердцевин равен 34,6 ап см3. Однако
когда вещество десорбируется, то добавляется некоторый объем
адсорбата va со стенок пор, образовавшихся на предыдущих
этапах. Этот объем vn подсчитывается на основании построе-
ния /-кривой, что позволяет определить значение А/, т. е.
уменьшение толщины жидкой пленки по всей суммарной
поверхности сердцевин, образовашихся до этого момента.
Объем, таким образом, равен произведению А/ на суммарную
поверхность сердцевин. Введение такой поправки является
ключевым моментом расчета.
3. Разность ап — уп дает значение объема вновь образуе-
мых сердцевин на n-м этапе.
4. Площадь поверхности новых сердцевин Sn определяется
графическим интегрированием, как и гна предыдущих этапах.
Указанного объяснения достаточно, чтобы показать разли-
чие между таким «скорректированным методом модельных
пор» и методом Крэнстона—Инкли. Для более подробного
ознакомления с описанием метода и примерами расчетов необ-
ходимо обратиться к первоисточнику [166].
В большинстве случаев метод' «модельных пор» дает мень-
шее значение радиуса пор в максимуме кривой распределения,
чем то, которое получается по методу Крэнстона и Инкли.
Например, для образцов с радиусами пор в интервале 5—10 А
при использовании изотермы десорбции по данному методу
получено значение радиуса в максимуме кривой распределения
около 6 А, а по методу Крэнстона—Инкли 10 А. Ханна и др.
[167] для широкого набора различных силикагелей получили
хорошее согласие в значениях размеров пор, используя азот
или кислород в качестве адсорбата при двух различных темпе-
ратурах эксперимента. В некоторых случаях, отмеченных в дан-
ной работе, образцы кремнезема содержали как микро-, так
и мезопоры.
Стандарт для определения размеров пор. Гавард и Уилсон
[168] описали применение метода «модельных пор» на образце
мезопористого кремнезема Gasil(I), состоящего из сфер со
средним радиусом 4,38 нм, упакованных с координационным
числом 4. Такой кремнезем является одним из стандартов
686
Глава 5
SCI/IUPAC/NPL для определения удельной поверхности и мо-
жет также использоваться в качестве стандарта для определе-
ний размеров пор и для калибрования аппаратуры, работаю-
щей на принципе метода БЭТ во всей области давлений.
МП-метод был продемонстрирован Михаилом, Брунауэром
и Бодо [164]. Они показали применимость этого метода к изу-
чению микропор, а «скорректированного метода модельных
пор» — к исследованию пор большого размера. При примене-
нии этого метода к силикагелю, имеющему как микро-, так
и мезопоры, МП-метод дает совокупное значение поверхности
пор, согласующееся со значением, найденным методом БЭТ.
Этот факт указывает, что, несмотря на возражения, выдвигае-
мые против применения метода БЭТ для изучения микропо-
ристых образцов, данный метод, как можно надеяться, способен
давать надежные данные по удельным поверхностям даже
и в этих случаях.
Подробное рассмотрение структуры пор для пяти силикаге-
лей, выполненное Хагемасси и Брунауэром [169], может
считаться типичным для работ подобного рода, в которых
оценивалась структура пор с применением МП-метода. В этой
статье проводилось сравнение паров воды и азота в качестве
адсорбатов, и полученные данные находились в достаточно
хорошем согласии, давав значения диаметров пор в максиму-
мах кривых распределения, равные 4,1 и 4,6 А соответственно.
Однако для адсорбентов, имеющих какие-либо гидрофобные
участки поверхности, должен использоваться азот.
Супермикропоры. Этот термин был предложен Дубининым
[170] для пор с диаметрами 1,3—30,0 нм. Таким образом,
классификация пор по размерам будет следующей:
Тип пор Радиус', А
.Микро- <6—7
Супермикро- От 6—7 до 15—16
Мезо- >15—16
Основанием для подобной предлагаемой классификации
является то, что детальному исследованию могут быть под-
вергнуты супермикропоры и мезопоры, но не микропоры.
МП-метод подвергался критике [171], после чего последо-
вало опровержение критических замечаний [47].
Ультрамикропоры или субмикропоры. Такие поры имеют
радиус менее 3 А. Механизм, благодаря которому подобные
поры заполняются, оставался главной темой обсуждений.
Очевидно, что если наименьшая по размеру из известных мо-
лекула газа (гелия) не способна проникать в пору, то значит
поры просто не существует, поскольку это подтверждается
Силикагели и порошки 687
экспериментом. Таким образом, нижний предел размеров пор,
при котором эти поры могут обнаруживаться, зависит от раз-
мера используемой молекулы адсорбата.
Основным вопросом является рассмотрение ситуации, когда
молекула входит в пору, диаметр которой менее чем в два
раза превышает размер молекулы. В этом случае вандервааль-
сово взаимодействие очень сильно, и теплота адсорбции за-
метно выше, чем на плоской поверхности. Следовательно,
подобная ситуация отличается от той, когда может происхо-
дить образование единичного полимолекулярного ?лоя или
капиллярное заполнение пор.
Согласно Доллиморе и Хилу [136], поры, диаметр которых,
вероятно, равен 7—10 А, если они определяются на основании
изотерм адсорбции азота, на самом деле имеют диаметр только
лишь 4—5 А. Субмикропоры в силикагеле, приготовленном из
частиц золя размером всего ~10 А, оказываются настолько
малыми, что даже в них не могут войти молекулы криптона.
Известно, что монокремневая кислота быстро полимеризуется
при низких значениях pH с образованием частиц примерно
такого же размера. Доллиморе и Хил [172] приготовили
такой гель по способу высушивания вымораживанием 1 %-ного
раствора монокремневой кислоты при температуре ниже 0°С.
Поскольку при испарении и вымораживании удалялось боль-
шое количество' воды, то значение pH системы составляло
в процессе гелеобразования 1—2, т. е. именно то значение,
когда наблюдается наиболее медленный рост частиц. Такой
кремнезем можно было бы назвать «пористым», так как в та-
кие «поры» проникали молекулы гелия (и только лишь эти
молекулы). Отметим, что молекулы гелия также проникают
в плавленый кварц. Так что при общепринятом подходе такой
кремнезем считается непористым.
Изостерическая теплота адсорбции. Значение теплоты ад-
сорбции в микропорах оказывается аномально высоким. Синг
и Рамакришна [173] обнаружили, что посредством тщатель-
ного выбора адсорбатов и применения с^-метода исследования
можно различать капиллярную адсорбцию и адсорбцию на
высокоэнергетических поверхностных центрах. Показано, что
в интервале р/р0 0,01—0,2 изостерическая теплота адсорбции
азота на силикагеле, не содержащем мезопоры, остается по
существу постоянной на уровне 2,0 ккал/моль. На силикагеле,
содержащем мезопоры, наблюдается падение теплоты от 2,3 до
2,0 ккал/моль, а на микропористом силикагеле изостерическая
теплота падает от 2,7 до 2,0. Изостерическая теплота qst под-
считывается из изотерм адсорбции по уравнению Клаузиуса—
Клайперона.
688
Глава 5
Микропористость может просто характеризоваться построе-
нием графика зависимости изостерической теплоты от р/ро,
получаемого по изотермам адсорбции азота.
Были проведены [174] калориметрические исследования
микропористости, в которых измерили теплоту, выделяемую
в процессе адсорбции бензола на силикагеле. Они подтвердили,
что энергия адсорбции оказывается наивысшей в микропорах,
и измерили поверхность, которая была еще доступна для ад-
сорбции молекул азота при разных стадиях адсорбции бензола.
Дубицрн [10] охарактеризовал микропористость, используя
уравнение
а=[-ехр вт" 0g
где а — количество адсорбированного вещества; Т—абсолют-
ная температура; Ж— предельный объем микропор; v* — моль-
ный объем адсорбата; В — параметр, являющийся характери-
стикой размеров микропор.
В том случае, когда в образце присутствуют поры двух
размеров, то а выражается как сумма двух подобных членов,
различающихся величинами Wo и В.
При постоянной температуре уравнение принимает вид
lga = C- £>(lg-^)2
где С и D могут быть рассчитаны из изотерм адсорбции и
превращены в величины Wo и В. Дубинин использовал этот
метод для получения характеристик образца силикагеля, содер-
жащего микропоры с диаметрами в интервале 20—40 А.
Указанный метод еще дорабатывается [7].
Адсорбаты, различающиеся по размеру молекул. Такие
адсорбаты можно использовать при исследовании путем по-
строения /-кривых для того, чтобы получить распределение
микропор по размерам. Михаил и Шебл [175] использовали
такие вещества, как вода, метанол, пропанол, бензол, гексан
и тетрахлорид углерода. Различия в получаемых данных свя-
зывались с размером пор образца кремнезема, а также со
степенью гидроксилирования его поверхности. Молекулы боль-
шей части перечисленных адсорбатов не подходят для
измерения поверхностей кремнеземов, содержащих тонкие
поры [176].
Бартелл и Бауэр [177] еще раньше выполнили исследова-
ния с этими парами при температурах 25, 40 и 45°С. Фу и
Бартелл [178], применяя метод свободной поверхностной энер-
гии, определяли величину поверхности с использованием в ка-
честве адсорбатов различных паров. Они нашли, что значения
Силикагели и порошки
689
поверхностей в этом случае в общем согласовывались со зна-
чениями, определенными по адсорбции азота.
Для измерения поверхности в твердых материалах, содер-
жащих микропоры такого размера, который затруднял про-
никновение в них относительно больших молекул азота, может
быть использована вода. МП-метод, или «скорректированный
метод модельных пор», был применен авторами работы [179]
при изучении гидратированного силиката кальция.
Суммарный адсорбционный объем для различающихся по
размеру молекул.
Другим способом определения микропористых характери-
стик является проведение измерений при относительных давле-
ниях вблизи насыщения [180]. Различия в адсорбционных
объемах показывают, что такой объем и размер пор не позво-
ляют большим выбранным молекулам адсорбата проникать
в них, в то время как наименьшие используемые молекулы,
такие, например, как молекулы воды, обнаруживают «полное»
проникновение в эти поры, определяемое адсорбционным
объемом.
Когда микропоры оказываются слишком малыми, чтобы
в них могли войти молекулы метанола или бензола, тогда они
еще в состоянии поглощать воду. Высоцкий и Поляков [181]
описали такой тип силикагеля, который приготовлялся из
кремневой кислоты и дегидратировался при низкой темпера-
туре.
Грег и Лангфорд [182] разработали новый подход, так
называемый метод предадсорбции, для определения микропор
в углях в присутствии мезопор. Сначала проводили -адсорбцию
нонана, который проникал в микропоры при 77 К, затем отка-
чивали его при обычной температуре, однако микропоры оста-
вались заполненными. После этого методом БЭТ по азоту
обычным образом измеряли поверхность образца, причем ре-
зультаты такого определения согласовывались с геометрически
измеренной поверхностью, которую находили методом элек-
тронной микроскопии. Подобный метод предадсорбции для
исследования микропор, без сомнения, может быть использован
применительно к кремнезему, но в таком случае, вероятно,
должен использоваться значительно более полярный адсорбат
с целью блокирования микропор, такой, например, как деканол.
Рентгеновское рассеяние под малыми углами. Риттер и
Эрих [183] использовали данный метод и сравнили получен-
ные результаты с данными адсорбционных измерений. Лонгмен
и др. [184] сравнили метод рассеяния с методом вдавливания
ртути. Еще раньше возможности этого метода были описаны
Порай-Кошицем и др. [185], Породой [186а] и Имеликом,
Тейчнером и Картеретом [1866].
18 Заказ № 250
690
Глава 5
Метод вдавливания ртути. Ртуть не смачивает поверхность
кремнезема, и требуется приложить высокое давление, чтобы
заставить жидкую ртуть войти в поры небольших размеров.
Вашбурн [187] вывел уравнение
2о cos 0
где р — равновесное давление; а — поверхностное натяжение
ртути (480 дин/см); 0 — краевой угол смачивания между
ртутью и стенкой поры (140°); гр — радиус поры.
Из этого уравнения следует, что произведение ргп = 70 000
в случае, если р выражено в атмосферах, а гР — в ангстремах.
В поры радиусом 100 А ртуть способна проникать при давле-
нии выше 700 атм. Следовательно, для проникновения ртути
в микропоры необходимо применять очень высокие давления.
Одна из проблем заключается в том, что если силикагель
не очень прочен, то структура образца разрушается под воз-
действием внешнего давления ртути еще до того, как ртуть
сможет проникнуть в тонкие поры. Именно по этой причине
для исследовательских целей метод измерения адсорбционных
изотерм азота предпочтителен. Тем не менее для прочных
твердых тел, подобных промышленным кремнеземным катали-
заторам, метод ртутной порометрии является гораздо более
быстрым не только с точки зрения проведения самого экспери-
мента, но и для обработки данных с целью построения кривых
распределения пор по размерам.
Коммерческие ртутные порозиметры широко доступны,
а усовершенствованные варианты данного метода описаны
в работах [188, 189]. Де Уит и Шолтен [190] сравнили резуль-
таты, полученные методом ртутной порометрии, с результатами
методов, основанных на адсорбции азота. Они пришли к за-
ключению, что метод вдавливания ртути вряд ли может
использоваться при исследовании пор, диаметр которых меньше
10 нм (т. е. радиус меньше 50 А). В случае прессованного
порошка аэросила радиус пор, определенный по вдавливанию
ртути, в максимуме кривой распределения оказался равным
около 70 А, тогда как метод адсорбции азота давал значения
75 и 90 А при расчете кривой распределения разными методами.
Расхождение может быть обусловлено искривленным мениском
ртути радиусом около 40 А, имеющим более низкое (почти на
50%) поверхностное натяжение, чем в случае контакта ртути
с плоской поверхностью. Согласно Цвейтерингу [191], наблю-
дается превосходное согласие между указанными методами,
когда диаметр пор имеет величину около 30 нм. Подробное
описание работы на коммерческом ртутном порозиметре (или
пенетрометре), введение необходимых поправок и собственно
Силикагели и порошки.
691
метод расчета размеров пор представили Фревел и Крессли
[192]. Авторы дали также теоретические порозиметрические
кривые для случаев разных упаковок однородных по размеру
сфер.
Различные эффекты в микропорах
Диэлектрические изотермы (25°С, Гмгц). В работе [193]
при построении диэлектрических изотерм, когда бензол адсор-
бировался на силикагеле, было обнаружено, что изотерма
искривлена при низких значениях р/ро, когда адсорбция про-
исходит в микропорах; при более высоких относительных дав-
лениях наблюдался линейный ход изотермы.
Проникающая гель-хроматография. Этот метод довольно
трудоемок при измерении размеров пор, если кремнезем при-
меняется в качестве материала для набивки хроматографиче-
ской колонки. Метод основан на определении глубины проник-
новения в кремнезем молекул или полимеров различных разме-
ров, которое происходит до тех пор, пока не начнут препят-
ствовать такому проникновению эффекты поглощения по-
рами [184].
Ионная эксклюзия. Поры силикагелей, погруженных в рас-
творы электролитов, стремятся не допустить ионы внутрь. Этот
эффект использовался для сравнения размеров ионов с исполь-
зованием определенного силикагеля с известным диаметром
пор. Маатман и др. [194] опубликовали большой материал по
этому вопросу, но здесь цитируются только наиболее типичные
статьи. По-видимому, можно было бы определять неизвестный
размер пор силикагелей, если применять данный метод, исполь-
зуя ионы известного размера. (См. также раздел «Отрицатель-
ная адсорбция».)
Мессбауэровский эффект. Мессбауэровские спектры ионов,
находящихся в небольших порах, могут использоваться для
регистрации различия в размерах пор. Гольданский и др. [195]
сообщили, что температурная, зависимость таких спектров
заметно меняется при варьировании размеров пор. Так,
в спектрах ионов S2+n наблюдалось, что в узких порах коле-
бания ионов были ограничены.
Природа поверхности кремнезема
Химия поверхности кремнезема будет рассматриваться
в гл. 6. Однако для полноты характеристик силикагелей и по-
рошков следует указать методы для определения по крайней
мере физических свойств кремнеземных поверхностей в порах.
Типы поверхностей. Поверхности могут быть классифициро-
ваны следующим образом:
18*
692
Глава 5
1. Полностью гидроксилированная поверхность, когда по-
верхностная структура обрывается силанольными группами
SiOH. Такая поверхность легко смачивается водой и водо-
растворимыми органическими молекулами. Все типы кремне-
земов, у которых удалена вода путем их высушивания при
температуре менее чем 150°С, относятся к данному типу.
2. Силоксановая поверхность состоит большей частью из
атомов кислорода, причем каждый атом кислорода связывается
с соседними атомами кремния. Обычно на такой поверхности
также присутствует незначительная доля изолированных или
парных групп SiOH. К данному типу относятся пирогенные
кремнеземы, сконденсированные из парообразного состояния.
Кроме того, для гидроксилированных образцов кремнезема,
которые подвергаются дегидратации при ~1000°С, силоксано-
вая поверхность формируется за счет удаления молекул воды,
образуемых из смежных силанольных групп.
3. Органическая поверхность формируется за счет хими-
чески или физически присоединенных органических моледул
или радикалов. Образовавшаяся поверхность может проявлять
следующие свойства:
а) гидрофобное и сильно органофильное поведение, когда
расположенные снаружи группы являются углеводородными;
б) гидрофильное и олеофобное поведение, когда присоединен-
ные органические группы содержат расположенные снаружи
группы С—ОН или другие сильно полярные группы; в) и гид-
рофобное и олеофобное поведение, когда поверхностными яв-
ляются фторуглеводородные группы.
Поверхность кремнезема является объектом быстро расши-
ряющейся области исследований в связи с применением в хро-
матографических колонках кремнеземных набивок, используе-
мых как носители и адсорбенты. Здесь можно упомянуть
только о нескольких характерных методах, разработанных
в прошлом для изучения поверхности кремнезема.
Гидроксилированная поверхность может оцениваться рядом
способов, как это обсуждается с некоторыми подробностями
в работе Барби [196].
Теплота адсорбции. Ход изотермы адсорбции азота ме-
няется в зависимости от степени гидроксилирования поверх-
ности кремнезема, что проявляется в изменении константы С
уравнения БЭТ. Это обстоятельство можно использовать в ка-
честве приблизительной оценки относительного соотношения
между силоксановыми и силанольными группами на частично
гидратированной поверхности. На основании данных Ловена
и Броуджа [33] было предложено соотношение
С = 30 + 7,4й
где h — число силанольных групп на 1 нм2 поверхности.
Силикагели и порошки
693
Значение константы С понижалось также почти до 22, когда
поверхность кремнезема покрывалась 1-бутоксигруппами по-
средством этерификации некоторой части находящихся на
поверхности силанольных групп. Остальные силанольные
группы покрывались большими по размеру бутильными груп-
пами и не подвергались действию азота. Аналогичным образом
значение константы С заметно изменяется, когда силанольная
поверхность кремнезема покрывается триметилсилильными
оксигруппами в результате реакции с триметилхлорсиланом.
Адсорбция красителя метилового красного. Измерение ад-
сорбции некоторых красителей из бензола является индика-
цией степени гидроксилирования поверхности кремнезема при
условии, что структура кремнезема имеет достаточные по
размеру поры, чтобы они были полностью доступными для
молекул красителя. Метод применяется для порошков и сили-
кагелей с относительно большими порами, но не используется
для плотных силикагелей. Такой метод был описан Ловеном
и Броуджем [33].
Инфракрасное поглощение. Этот метод обычно применяется
и широко используется для изучения поверхностных силаноль-
ных групп. Метод более эффективен, когда он используется
в комбинации с реакцией дейтеро-водородного обмена
НгО—D2O *. Метод ИК-поглощения более полно обсуждается
в гл. 6; кроме того, сведения о нем обобщены в работе Барби
[196]. Этот метод дает информацию о природе связей, возни-
кающих на гидроксилированной поверхности кремнезема.
К тому же часто можно определить природу связи органиче-
ских молекул с такой поверхностью. Однако методом ИК-погло-
щения в большинстве случаев можно получить только полу-
количественные данные.
Химический анализ, проведенный после удаления летучих
или растворимых соединений, не обязательно показывает, что
оставшееся на поверхности органическое соединение находится
в виде связанного однородного мономолекулярного слоя.
Однако отсутствие силанольных групп указывает на наличие
характерного покрытия на поверхности. Полезные сведения
дает измерение изотерм адсорбции до и после удаления органи-
ческого вещества с поверхности при таких условиях, когда не
изменяется структура самого кремнезема. Если на поверхности
находится известное процентное содержание связанного орга-
нического вещества в виде слоя молекулярной толщины, то
знание удельной поверхности, а также объема и диаметра пор
кремнезема позволяет подсчитать толщину такого слоя.
См. примечание на стр. 868.— Прим. ред.
694
Глава 5
Органическое вещество обычно может быть удалено посред-
ством погружения образца в 70 %-ный раствор HNO3 с после-
дующим медленным нагреванием до 400°С на воздухе, чтобы
полностью завершить окисление. При такой обработке боль-
шинство образцов кремнезема не изменяется.
Прочность агрегатов — связь между частицами
кремнезема
Возникновение связей между индивидуальными частицами
в кремнеземных агрегатах и гелях рассматривалось в гл. 3
(см. рис. 3.19). «Перешейки» между частицами могут форми-
Рис. 5.14. Схема формирования «пере-
шейков» между частицами.
Вверху: процессы растворения и повтор-
ного осаждения перестраивают кремне-
зем; внизу: процесс осаждения продол-
жается за счет дополнительно введенного
кремнезема. Штриховые линии: исходные
сферические частицы кремнезема перед
упрочнением структуры.
Рис. 5.15. Схема определения коэф-
фициента коалесценции посредством
частичного растворения: гр и гр —
различающиеся степени укрепления
структуры; гп — радиус перешейка
между частицами; (гР — гп)—ра-
диус диспергированных частиц.
роваться по двум процессам, схематически показанным на
рис. 5.14 и 5.15. Согласно первому механизму процесс идет
путем растворения и повторного осаждения (он подробно рас-
смотрен в гл. 3). По второму механизму процесс идет посред-
ством дополнительного осаждения кремнезема, поступающего
из пересыщенного раствора. Последний процесс требует для
своего осуществления тщательного контроля, и о нем упоми-
нается лишь в нескольких работах. В любом из этих двух
случаев частицы связываются вместе до такой степени, кото-
Силикагели и порошки
695
рая определяется как «коэффициент коалесценции». Способы,
благодаря которым это достигается, будут рассмотрены ниже;
здесь же перечисляются только методы измерения такой сте-
пени коалесценции.
Электронно-микроскопические снимки
Этим методом можно показать появление связей между
частицами, особенно если диаметр частиц превышает 15—20 нм.
Обычно это имеет место в случае некоторых кремнеземных
порошков, но, вообще говоря, для общепринятых целей такое
появление связей наблюдать затруднительно.
Если можно измерить диаметр частиц d, то теоретически
рассчитанная удельная поверхность 8С для сферических частиц
может подсчитываться из сотношения Scd = 2750, где Sc имеет
размерность м2/г, a d выражено в нанометрах. Когда частицы
прочно коалесцированы, то величина 'потерянной площади
поверхности в местах контактов между частицами такова, что
удельная поверхность, измеренная по адсорбции азота Sjv,
оказывается меньше, чем величина 8С. Отношение Sc: 8к может
служить в качестве независимой характеристики степени коа-
лесценции, если координационное число (или упаковка) струк-
туры остается без изменения. Оно устанавливается уже после
того, как агрегаты сформированы.
Метод неполного растворения
Метод приложим к кремнеземным порошкам низкой плот-
ности со значениями удельных поверхностей в области 60—
400 м2/г. Александер, Айлер и Уолтер [197] подробно показали
способ, посредством которого 1 г кремнеземных частиц суспен-
дируют в 100 мл воды при 50°С при поддержании pH в интер-
вале 11,0—11,5 и непрерывном добавлении 1 н. раствора NaOH.
Мутность суспензии измеряют достаточно часто путем про-
пускания пучка света с длиной волны 400 нм через образец
толщиной 1 см. Одновременно измеряют количество раство-
ренного мономерного кремнезема, отбирая порции образцов
объемом 0,1 мл и исследуя их кремнеземно-молибдатным мето-
дом. Суммарный кремнезем измеряется в суспензии посред-
ством добавления щелочи NaOH, содержание которой доводят
до 0,5 н., после чего смесь нагревают в течение 2 ч в сосуде
(из нержавеющей стали), устойчивом к щелочи. Анализ вы-
полняется тем же методом.
Если процентное содержание растворенного кремнезема
вычерчивают как функцию процента светопропускания, то на
графике наблюдается точка перегиба. Она показывает то
696
Глава 5
среднее количество кремнезема, которое должно раствориться,
чтобы первичные частицы, составляющие агрегат, стали дис-
персными. Как можно видеть из рис. 5.15, две сросшиеся сферы
радиусом гр не будут распадаться при равномерном растворе-
нии кремнезема с поверхности до тех пор, пока толщина рас-
творенного слоя не станет равной радиусу поперечного сечения
шейки гп. Коэффициент коалесценции С в таком случае удобно
определять как ту долю кремнезема от его суммарного коли-
чества, которая растворилась к моменту, когда скорость дис-
пергирования агрегатов частиц достигает максимума:
, (Гр —Г„)3
“ ДД3
Гп = Гр [ 1 - (1 - С)1/3]
Например, когда С = 0,5, то гп составляет 20 % величины
радиуса частицы; при С = 0,75 гп равно 37 % радиуса частрцы.
Данный метод был использован применительно к осадку,
состоящему из кремнеземных частиц .радиусом около 12 нм.
Такой осадок имел коэффициент коалесценции С = 0,47, из
которого было вычислено гп1гР — 0,19 и гп = 2,3 нм. Добавляе-
мый кремнезем осаждался на исходной кремнеземной суспен-
зии до тех пор, пока отношение добавляемого кремнезема
к исходному не составило 2:1. При этом радиус исходной
частицы должен был возрасти в 31/э раза, или до значения
17,5 нм (размер частиц, определяемый из величины удельной
поверхности, был равен 16,5 нм). Образец имел измеримую
величину коэффициента коалесценции С = 0,69, г„/гр = 0,32
и гп — 5,3 нм. Добавление кремнезема продолжали вплоть до
получения соотношения, равного 4 ч. осажденного кремнезема
на 1 ч. исходного. Вновь определялись радиус частицы и сте-
пень коалесценции. Полученные данные суммировании и при-
ведены ниже:
Радиус частиц, нм Радиус перешейков, нм
Гр Дгр Гп Дг„ ^Гп11\Гр
12 — 2,3 — —-
16,5 4,5 5,3 3,0 0,67
20,5 7,5 7,8 5,5 0,73
Можно видеть, что приращение радиуса частицы оказы-
вается примерно равным приращению радиуса перешейка.
Однако последняя величина должна была бы нарастать более
интенсивно по сравнению с нарастанием на частице. Возможно,
что осаждение кремнезема идет более быстро на открытой
поверхности частиц, чем в расщелинах около шейки.
Силикагели и порошки
697
Данный метод также представляет возможность прослежи-
вать за спеканием относительно больших коллоидных сфери-
ческих частиц кремнезема радиусом 50 нм при нагревании их
в течение часа на воздухе при повышенной температуре. Были
получены следующие данные:
Температура, Т, °C Коэффициент коалесценции, С гп, нм Константа 0,025 (Т—150)/гЛ
200 0,06 2 0,65
300 0,11 3,8 1,00
400 0,17 6,0 1,05
600 0,28 10,4 1,09
800 0,43 17,1 0,95
1000 0,53 22,2 0,96
Как отмечалось выше, радиус шеек по необъяснимым при-
чинам возрастает пропорционально разности (Т—150).
Механическая прочность агрегатов
Другую возможность для сравнения коалесценции частиц
в структурах силикагелей, имеющих одинаковый размер частиц
и их упаковку, дает измерение механической прочности.
Мейснер, Михаэле и Кайзер [130] вывели уравнения для
определения прочности агрегатов сферических частиц с дан-
ным координационным числом га и с заданной силой сцепления
между сферами. На основании общего уравнения, связываю-
щего координационное число с объемной долей твердого
вещества в образце, ими было выведено следующее уравнение
для расчёта прочности на раздавливание:
РD~2 = Кф енр (7,2ф)
где Р — раздавливающая нагрузка; D — диаметр агломерата;
ф — доля от общего объема, занимаемая сферами; К — кон-
станта, являющаяся функцией диаметра сферических частиц
и прочности сцепления между двумя частицами. Обращалось
внимание на очень быстрое понижение прочности в ряду образ-
цов со все более открытыми упаковками при условии, что все
другие факторы останутся неизменными.
Пористость, СМ’/СМ’
силикагеля
0,26 а
0,30
0,5б
0,6
Относительная прочность
100
71
12
5
а Плотная регулярная упаковка.
6 Открытая упаковка.
698
Глава 5
Уравнение, адекватное написанному, характеризует проч-
ность агломератов, состоящих из тонкодисперсных частиц
оксида цинка при значении объемной пористости 0,5—0,7.
Силикагели
Этот раздел ограничивается рассмотрением силикагелей,
в него не включены алюмосиликатные гели и катализаторы. Не-
которые вопросы, связанные с поверхностью алюмосиликатов,
обсуждаются в гл. 6.
Теория формирования и структуры силикагелей, образую-
щихся в результате полимеризации кремневых кислот, была
приведена в гл. 3, а аналогичные вопросы, связанные с приго-
товлением гелей из золей, содержащих дискретные коллоидные
частицы, рассматривались в гл. 4. Характеристики силикагелей
были даны в предшествующем разделе. Здесь же будут опи-
саны методы приготовления силикагелей и изменяющиеся тех-
нологические режимы, которые оказывают воздействие , на
свойства получаемых силикагелей; а также определяют их
использование.
Происхождение и история силикагелей освещены в ряде
обзоров, опубликованных в течение прошедшего полувека,
начиная с работы Волфа и Преториуса, вышедшей в 1928 г.,
[198]. Вейл обобщил вопросы истории, производства и исполь-
зования коммерческих силикагелей в 1952 г. [199]. Теория
структуры геля, образуемой из частиц, и поведение таких
систем описаны Айлером в 1955 г. [2]. Обзор Неймарка охва-
тывал главным образом работы советских авторов, вышедшие
до 1953 г. [200]. Примерно в те же годы был опубликован
обзор Ромоса и Вега [201].
Два десятилетия спустя появилась новая серия обзоров.
Киселев рассмотрел структуру и поведение силикагелей в про-
цессе высушивания [202]. Кондо [203] в 1972 г. представил
обзорную работу по химическим и физическим свойствам сили-
кагелей. Обзор Неймарка и Шейнфайн [204], в котором
затрагиваются аналогичные вопросы, появился в СССР в 1973 г.
К тому же в это время увидел свет один из обзоров Барби
[8], который наиболее полно охватывал данную тему.
Начиная с 1970 г. активность в проведении исследователь-
ских работ и рост публикаций стимулировались все возрастаю-
щей необходимостью получения гелей с особой структурой для
применения в хроматографических колонках, весьма необхо-
димых при проведении химических анализов и контролировании
технологических процессов.
Механизм образования геля кремнезема из кремнеземных
частиц описан в гл. 4. Гели, получаемые из коллоидного крем-
Силикагели и порошки
699
незема, наряду с гелями, формируемыми из силиката натрия
и других веществ, рассматриваются ниже совместно с вопро-
сами, относящимися к факторам, регулирующим характери-
стики гелей.
Исходные материалы для получения
силикагелей
Получение из растворимых силикатов
и минералов
Силикат натрия был и, вероятно, останется в будущем наи-
более дешевым источником получения относительно чистой
кремневой кислоты, из которой приготовляется силикагель.
Однако некоторые природные коллоидные алюмосиликаты,
включая определенные разновидности глин, могут под дейст-
вием кислоты образовывать в виде конечного продукта пори-
стый, гидратированный кремнезем, способный в некоторых
случаях формироваться в гель (205]. Подобный исходный ма-
териал может стать наиболее важным, если такие глины будут
одновременно служить и источником получения алюминия.
Кроме того, определенные разновидности ортосиликатных ми-
нералов, легко поддающиеся обработке кислотой, могут ока-
заться выгодными при получении силикагелей. Например, Фле-
ниген и Гроус [206] нашли, что высокопористые силикагели
с удельной поверхностью 600—900 м2/г и очень тонкими порами
могут быть приготовлены из волокнистого силиката кальция —
волластонита путем растворения минерала в кислоте с после-
дующим гелеобразованием в кислом растворе.
Мюрата и Шлехт [207] обобщили действие кислот на раз-
личные силикатные минералы в виде следующих положений.
Те силикаты, которые содержат силикатные радикалы с низкой
молекулярной массой или же имеют непрерывные кремнекисло-
родные каркасы, в которые включено достаточное число атомов
железа или алюминия (удаляемых кислотами), образуют сту-
денистые кремнеземы. С другой стороны, минералы, имеющие
силоксановые структуры большой протяженности, такие, на-
пример, как цепочки SiO3, двойные цепочки Si-iOn и листочки
Si2O5, оказываются не способными застудневать, но дают
нерастворимые кремнеземные псевдоморфозы. Согласно дан-
ным Брунауэра [208] для глаукосила, т. е. кремнезема, выде-'
ленного из глауконита и имевшего структуру, подобную листоч-
кам слюды, значение удельной поверхности было равно 82 м2/г.
Если в структурах алюмосиликатов содержится более чем дПа
атома алюминия на каждые три атома кремния, то, по данным
Мюрата и Шлехта [207], при действии кислоты каркасная
700
Глава 5
сетка разрушается во многих местах с образованием «остров-
ков», состоящих из силоксановых фрагментов, из которых
получаются студенистые кремнеземы.
Патрик и Мак-Гавак [209] исследовали силикагели с точки
зрения их важного практического применения в качестве ад-
сорбентов. Прочно связанные силикагели, которые можно было
нагревать до красного каления без разрушения или потери
адсорбционной способности, производились посредством смеши-
вания довольно крепких растворов, содержащих силикат натрия
с отношением SiO2:Na2O 3,3:1 и избыточное количество
соляной кислоты, что позволяло формировать гель, который
затем промывали и медленно высушивали. За период с 1920
по 1950 г., как указывал в своей монографии Вайл [199], было
разработано большое число способов подкисления и гелеобра-
зования растворов, получаемых из растворимых силикатов,
повышения механической прочности силикагелей, снижения
усадки и увеличения их пористости. Процесс медленного высу-
шивания является существенным для предотвращения раз-
дробления кусочков геля, возникающего из-за более сильной
усадки наружных слоев в таком материале. Высокая концент-
рация кремнезема (вплоть до 15 г на 100 мл) в застудневаю-
щих растворах дает возможность получать плотные и механи-
чески прочные силикагели. Волф и Бейер [210] в своем обзоре
рассмотрели взаимосвязь между условиями приготовления
силикагеля из кислоты и силиката и свойствами конечного
продукта. Основное положение заключается в том, что при
промывании горячей водой увеличивается размер первичных
частиц и понижается удельная поверхность. Выдерживание
при pH >7 приводит к аналогичному эффекту. Если вода
в гидрогеле замещается органической жидкостью, имеющей
более низкое поверхностное натяжение, то формируемый сили-
кагель будет давать меньшую усадку при высушивании, сохра-
няя большие по размеру поры.
Обычно при промышленном производстве наиболее часто
используются кислоты H2SO4, НС1 и СО2 в указанной после-
довательности с точки зрения масштабов их применения. Для
того чтобы получить наиболее твердые и прочные силикагели,
гель, как правило, формируют при низких значениях pH и при
комнатной или даже более низкой температуре. Это позволяет
сохранить очень малый размер первичных частиц, что способ-
ствует увеличению пористости и распределения частиц по раз-
мерам. В результате последующих обработок силикагель
приобретает прочность.
* Чтобы изготовить силикагель максимальной прочности, си-
ликат на начальной стадии разбавляют до возможно меньшей
степени при смешивании с кислотой; в этом случае гелеобра-
Силикагели и порошки
701
зование протекает очень быстро. Например, золь кремнезема,
содержащий 13—17 % SiO2, приготовлялся путем смешивания
потоков раствора силиката натрия и кислоты при таких соот-
ношениях, когда достигалось pH 6—10,5. При смешивании
продукт разбрызгивался, и капельки застудневали менее чем
за секунду, находясь в виде суспензии в воздушной среде,
а затем падали в воду для дальнейшего процесса старе-
ния [211].
Непрерывно разрабатываются новые рецепты для частных
разновидностей силикагелей. Например, Виньял и Аккер [215]
получили широкопористый силикагель с удельной поверхностью
менее чем 320 м2/г и объемом пор 1 см3/г путем приготовления
гидрогеля из кислоты и силиката, смешанных в такой пропор-
ции, чтобы получить значение pH 9,8—10,4. Такой гель под-
вергали старению в течение 50 мин, затем подкисляли до pH
2—3, выдерживали до 5 ч, после чего опять подвергали старению
продолжительностью до 4 ч при pH 8, промывали и высуши-
вали. Высокое значение pH позволяло первичным частицам
расти и коалесцировать, формируя желаемую структуру, после
чего была необходима кислотная обработка с целью удаления,
окклюдированных ионов натрия. Вероятно, при удалении таких
ионов в образце остаются микропоры, которые закрываются
в процессе старения при pH 8. Множество аналогичных спосо-
бов приготовления силикагелей, кажущихся довольно произ-
вольными и сложными, можно объяснить на основе теперь уже
известной химии такой системы.
При другом подходе следует использовать соединение, ко-
торое медленно выделяет кислоту. Долго применяли такое
соединение, как Na2SiF6, по определенным рецептам. Однако
способные диспергировать в воде альдегиды, амиды и сложные
эфиры также нашли применение для специальных целей.
Концентрированный 30 %-ный раствор глиоксаля (6,5 ч.) сме-
шивали с концентрированным (36°В.) раствором силиката
натрия (100 ч.), при этом протекала реакция, и после выдер-
живания в течение суток вещество образовывало чрезвычайно
твердый гель, который промывали 4 н. раствором H2SO4 и
водой и высушивали. Реактив, который оказывал замедляю-
щее нейтрализующее действие, равномерно смешивался с си-
ликатом с тем, чтобы кремнезем полимеризовался при очень
высокой концентрации и окончательно получался очень проч-
ный гель [212]. Аналогично когда необходимо было сформи-
ровать очень прочный силикагель, например, в порах бетона
для придания ему водонепроницаемых свойств, то сложные
эфиры, такие, как глицерин или гликолевые ацетаты, смеши-
вали с концентрированным силикатным раствором непосред-
ственно перед использованием, с тем чтобы такой раствор в те-
702
Глава 5
чение двух часов превратился в гель непосредственно в том
месте, куда он был помещен [213]. Смесь Н2О2 и парафор-
мальдегида, добавляемая к раствору силиката натрия, превра-
щает его в пену, после отвердения образующую легкую, обла-
дающую изоляционными свойствами структуру силика-
геля [214].
Скорость гелеобразования. Вот уже в течение многих лет
изучается продолжительность времени, требующегося для пре-
вращения в гель определенного типа золя кремневой кислоты,
который приготовлялся из кислоты и силиката. Принимается,
что время гелеобразования обратно пропорционально скорости
полимеризации или скорости формирования геля. В серии ста-
тей Хард и соавторы [216а] сообщили фактически обо всех
факторах, оказывающих влияние на время гелеобразования
золей кремневой кислоты, приготовляемых из кислоты и сили-
ката натрия с отношением SiO2:Na2O 3.3:1. Необходимо
также сослаться на исследование Сена и Гоша [2166], кото-
рые, применяя разные кислоты, вывели уравнения, связываю-
щие время гелеобразования с pH, концентрацией и темпера-
турой. Как сообщалось в гл. 3, время гелеобразования дости-
гает максимума при значении pH ~2. В области pH 3—5
время гелеобразования изменяется в прямой зависимости от
концентрации ионов Н+ (или в обратной зависимости от кон-
центрации ионов ОН-). В интервале pH 4—9, как показано
на рис. 5.16, минимум продолжительности времени гелеобра-
зования наблюдается вблизи области нейтральности. Эти дан-
ные основаны главным образом на результатах Меррилла и
Спенсера [217]. Ниже будет отмечено, что концентрация ионов
натрия, пропорциональная концентрации кремнезема, обеспе-
чивает наиболее быструю скорость процесса гелеобразования
при pH 7—8, тогда как в отсутствие солей коллоидный крем-
незем превращается в гель наиболее быстро при pH 5—6
(см. гл. 4). Различные кислоты вызывают наибольшую ско-
рость гелеобразования при немного различающихся значе-
ниях pH.
Хилд, Коатес и Эдвардс [218] нашли, что дальнейшее
добавление солей ускоряет гелеобразование при pH 7—8, при-
чем очень эффективными являются ионы кальция.
При определенной концентрации кремнезема золи кремне-
вой кислоты, приготовленные из силикатов натрия с более вы-
сокими отношениями SiO2:Na2O, испытывают более медлен-
ное гелеобразование, вероятно, вследствие более низкой кон-
центрации ионов натрия.
Авторы работы [219] представили обзор, в котором рас-
смотрели возможные воздействия одновалентных электролитов
на время гелеобразования. Они сообщили результаты иссле-
Силикагели и порошки
703
Рис. 5.16. Зависимость времени гелеобразования для золей, приготовленных
из силиката натрия с отношением 3,25 SiO2 : Na2O и H2SO4 при 25 °C, от pH.
Концентрации кремнезема: 1 — 1 %; 2—1.5%; 3 — 2,5%; 4 — 3,5 %; 5 — 6,0 %. (Данные
для кривых 1—4 взяты из работы Меррила и Спенсера [217]; кривая 5— неопубликованные
результаты Айлера.)
дования золей кремневой кислоты при pH 2,7, когда действие
соли менее заметно, тогда как большая часть предыдущих
работ была проведена при более высоких значениях pH.
Способность катионов ускорять гелеобразование повышается
в ряду Li+, Na+ и К+, а анионов —-в ряду NOT, Cl-, Вг-
и I-. По необъяснимой причине ионы I- действуют в пять раз
эффективнее, чем ионы NOT. Подобные воздействия ионов
прямо пропорциональны концентрации соли, и не имелось дока-
зательств о возможной «критической концентрации коагу-
ляции».
Айлер исследовал ранние стадии полимеризации-и форми-
рования геля при pH 1,7 и отношении SiO2:Na2O 3,3:1.
Он показал, что скорость полимеризации и время гелеобразо-
вания были обратно пропорциональны квадрату концентрации
кремнезема. Высоцкий и Стражеско [221] провели аналогич-
ные тщательные измерения для такого типа золей кремневой
кислоты при концентрациях SiO2 1—2 % в пределах значений
pH 0—4 и точно так же нашли острый максимум времени
гелеобразования при pH 1,7, что также точно соответствовало
обратной зависимости от квадрата концентрации кремнезема.
Иногда оказывается удобным приготовлять золь, время геле-
704
Глава 5
образования которого было бы воспроизводимым. Время геле-
образования оказывается менее всего чувствительным к кис-
лотности среды при данном значении pH, когда время геле-
образования минимально (см. ниже). Значения pH, отвечающие
минимуму времени гелеобразования, изменяются в зависимости
от концентрации кремнезема. Ниже приведенные данные отно-
сятся к золям, приготовленным при 25°С посредством добав-
ления одного объема силиката натрия с отношением 3,25
к одному объему интенсивно перемешиваемой разбавленной
кислоты H2SO4 такой крепости, чтобы pH изменилось до 2.
Таким образом, для получения 2 %-ного золя SiO2 силикатный
раствор должен содержать 4 % SiO2. Затем pH повышалось до
конечного значения путем очень осторожного добавления 1 н.
раствора NaOH при интенсивном перемешивании. Были полу-
чены следующие результаты:
% SiO2 pH Минимальное время гелеобразования, мин
0,5 6,9 260
1,0 7,3 53
1,5 7,7 10,5
2,0 8,05 з,з
3,0 8,35 0,5
4 0 8,7 0,15
Такая точка гелеобразования не указывает на завершен-
ность формирования структуры геля. Относительно данной
точки можно сказать лишь то, что это точка, где происходит
образование растущих областей твердых микрогелей, которые
суспендированы в золе, достигая 50 % от всего объема, вслед-
ствие чего вязкость резко повышается (см. обсуждение меха-
низма гелеобразования в гл. 3). Оставшиеся области золя еще
продолжают затвердевать, и это имеет место по крайней мере
в течение такого времени, которое соответствует периоду геле-
образования. В этот период гель начинает сжиматься по мере
того, как сетка дает усадку и выделяет воду. Продолжитель-
ность синерезиса в несколько раз превышает время гелеобра-
зования. Высоцкий и Стражеско [221] показали, что такой же
процесс агрегации частиц продолжается во время синерезиса,
как и в процессе гелеобразования. Это доказывает, что pH
влияет на скорость синерезиса точно так же, как и на скорость
гелеобразования, и такое воздействие минимально при pH 1,7.
Получение из коллоидного кремнезема
Формирование теля из коллоидного кремнезема обсужда-
лось в гл. 4. Коллоидный кремнезем в настоящее время досту-
пен в виде концентрированных золей, различающихся размерами
Силикагели и порошки
705
содержащихся в них частиц. Такой кремнезем, без сомнения,
будет играть все возрастающую роль в промышленном про-
изводстве силикагелей.
Проведение реакции между растворимым силикатом и кис-
лотой в течение длительного времени являлось общепринятым
способом промышленного производства силикагелей. Однако
существуют определенные ограничения, которые всеми при-
знаются. Например, невозможно вырастить частицы кремнезема
большего размера до того, как начнется процесс формирова-
ния геля. При низких значениях pH, когда кремневая кислота
устойчива достаточно долго, так что становится возможным
отливать ее в желаемую форму, первичные частицы сохраняют
размер менее 2—3 нм в диаметре. При pH 4.—10, за исключе-
нием низких концентраций кремнезема, соль натрия вызывает
очень быстрое осаждение или гелеобразование, так что рост
дискретных частиц до начала формирования геля оказывается
невозможным. Диаметр частиц геля на конечной стадии состав-
ляет 3—7 нм [222]. Таким образом, когда силикат натрия
используется при изготовлении геля, то какое-либо дальнейшее
уменьшение удельной поверхности или увеличение диаметра
пор должно проводиться путем последующей обработки сили-
кагеля, например нагреванием образца в воде или при высокой
температуре в парах воды. Вымывание соли представляет собой
медленный процесс, и в результате получается большое коли-
чество отработанной сточной воды. Чтобы избежать этого,
раствор силиката натрия можно вначале пропустить через
ионит в водородной форме, и если процесс выполняется при
низкой температуре, то можно обеспечить выход 12 %-ного золя
кремневой кислоты [223]. Если только не идет дальнейшего
превращения в коллоидные частицы большего размера, то по-
лучается точно такой же силикагель с частицами малого раз-
мера и тонкими порами, как и в случае применения способа
кислотной обработки силиката.
Как было показано в гл. 4, способы горячего ионного обмена
или электродиализа с использованием более концентрирован-
ного раствора силиката натрия дают возможность получать
коллоидный кремнезем с заданными большими размерами
частиц при концентрации по крайней мере 12—15 °/о SiO2-
Остаточные небольшие количества натрия могут быть удалены
путем проведения дальнейшего ионного обмена. Изменяя кон-
центрацию и размер кремнеземных частиц в золе, который
затем подвергается гелеобразованию, представляется возмож-
ным приготовить силикагель с заранее заданным размером
пор, объемом пор и удельной поверхностью.
Существует одно различие между силикагелями, приготов-
ляемыми из стабилизированного коллоидного кремнезема, и
19 Заказ № 250
706
Глава 5
силикагелями из кремневой кислоты, которое заключается
в том, что коммерческий коллоидный кремнезем готовится
в горячих, слабощелочных условиях, так что отпадает необхо-
димость промывания водой, и частицы кремнезема освобож-
даются от микропор. С другой стороны, частицы, приготовлен-
ные полимеризацией кремневой кислоты обычно при pH <7
и при температуре менее 80°С, а также часто в присутствии
солей натрия, могут оказаться микропористыми. Это положе-
ние обсуждалось Высоцким и др. [221], которые подтвердили,
что поры могут быть доступными для молекул воды, но не для
больших молекул.
Получение гидролизом соединений,
содержащих кремний
Приготовление силикагелей из SiCl4 обработкой сложных
кремневых эфиров оказывается, как правило, слишком до-
рогим процессом, за исключением случая, когда требуется
получить чрезвычайно чистый кремнезем. Согласно данным
Ленера [224], очень плотные силикагели приготовлялись путем
гидролиза SiCl4 и сжатия отмытого геля под давлением
42 270 кг/см2. Содержание воды в образовавшемся белом
порошке было 12,3—12,9%, что соответствовало отношению
SiO2: Н2О 2:1. Первичные частицы в таком геле имели,
вероятно, размер менее чем 1 нм и при таком большом давле-
нии должны были быть полностью плотно упакованными и
коалесцированными. Шварц и Рихтер [225] приготовили сили-
кагель путем гидролиза SiCl4 при 0°С с последующей дегидра-
тацией образца жидким аммиаком с целью получения про-
дукта, соответствующего отношению SiO2: Н2О. Авторы работы
[226] приготовили силикагель путем гидролиза паров SiCl4
в воде. Гель высушивали постепенным нагреванием до 75°С,
затем выдерживали при 280°С в течение 4 ч, вновь промывали
с последующим повторным нагревом до 280°С, повторяя эту
процедуру четыре раза. Силикагель имел удельную поверхность
455 м2/г (что соответствует диаметру первичных частиц 6 нм).
Средний диаметр капиллярных пор составлял 8,2 нм.
Формирование силикагеля наблюдалось в исследованиях,
проводимых с помощью электронного микроскопа Радчевским
и Рихтером [227]. Золи кремнезема приготовляли гидролизом
тетрахлорида кремния с последующей очисткой электродиали-
зом. Такой золь содержал частицы менее чем 10 нм в диаметре.
При выдерживании образца при pH 6,8 наблюдались агре-
гаты, подобные губке, образовавшиеся из индивидуальных
частиц. Полученные таким способом полимеры кремнезема
были тщательно исследованы методом рентгеновской дифрак-
Силикагели и порошки
707
ции, а также методом электронной дифракции. Никаких при-
знаков кристалличности не было обнаружено, поскольку такой
кремнезем показывал широкие интерференционные полосы,
характерные для кварцевого стекла. Вполне вероятно, что
силикагели, имеющие наиболее тонкую микропористую струк-
туру, могут легко приготовляться при низкой температуре и при
низком значении pH путем гидролиза соединения с одним
атомом кремния, как, например, SiCU (см. ниже раздел, по-
священный субмикропористым гелям).
Силикагель с широкими порами приготовлялся путем гид-
ролиза этилсиликата до образования полиэтоксисилана (ПЭС).
Гидролиз проводили в спирте с ограниченным содержанием
воды и кислотного катализатора, затем добавляли углеводо-
род, например гексан, и на конечном этапе прибавляли аммиак
при быстром перемешивании, чтобы осуществить эмульсионную
полимеризацию. Унгер и Шарф [228] по этому методу при-
готовляли пористый силикагель в форме микрошариков для
использования в хроматографических колонках. Диаметр пор
был 116—160 А. Такой гель мог быть получен при постоянном
значении удельной поверхности 420 м2/г просто путем исполь-
зования ПЭС при постепенной полимеризации, когда молеку-
лярная масса менялась в области 75—2000. Объем пор мог
изменяться от 0,5 до 2,0 см3/г. Добавлением гексана перед
гелеобразованием можно было увеличить объем пор до
>3,0 см3/г, но при этом возникали дополнительные макропоры
вплоть до 1000 или даже до 3000 А в диаметре.
Отличительной особенностью этого типа силикагеля яв-
ляется то, что поры не сплющиваются в процессе высушива-
ния, как это имеет место для силикагелей, полученных из
силиката натрия и кислоты. Возможно, что в серии образцов,
приготовленных из ПЭС с различной молекулярной массой,
самые широкие поры дает ПЭС с наименьшей массой, так как
при постоянной концентрации кремнезема более прочная струк-
тура образуется из меньших по размеру первичных частиц.
В этом случае усадка получается меньшей в результате дей-
ствия сил поверхностного натяжения при высушивании.
Удельные поверхности всех таких силикагелей близки между
собой, т. е. размер первичных частиц, формирующих сетку
геля, остается одним и тем же, но меняется лишь плотность
упаковки. Если бы на стадии гидрогелей такие образцы про-
мывали спиртом и простым эфиром вплоть до полного обезво-
живания и превращали бы в аэрогели, то объемы пор и диа-
метры пор должны были бы быть, вероятно, близкими для
всех образцов.
Кажется возможным, что чистые силикагели почти любого
известного структурного типа могут приготовляться с исполь-
19*
708
Глава 5
зованием SiC14(C2H5O)4Si или других летучих способных гид-
ролизоваться соединений кремния. При проведении научных
исследований с целью изучения каких-либо явлений, включая
химию поверхности или коллоидную химию кремнезема, на
силикагелях, приготовленных иным способом, следует соответ-
ствующим образом готовить и образцы для сравнения, лучше
одним из приведенных выше способов получения чистых сили-
кагелей, чтобы быть уверенным, что никакие следовые загряз-
нения, подобные примесям натрия или алюминия, не оказывают
воздействия на получаемые результаты.
Факторы, определяющие характеристики
силикагелей
Конечные получаемые характеристики полностью сформи-
рованных высушенных силикагелей определяются теми физи-
ческими и химическими условиями, которые существовали на
каждом этапе или же процессе приготовления. Основные фак-
торы следующие:
1. Размер первичных кремнеземных частиц к моменту на-
чала их агрегирования в сетку геля.
2. Концентрация первичных частиц в растворе и, следова-
тельно, плотность образуемой сетки геля.
3. Значение pH, концентрация, температура и продолжи-
тельность обработки, которые имели место в процессе, когда
гидрогель подвергался старению или другим процедурам в то
время, как он находился во влажном состоянии (сюда вклю-
чается возможный этап замещения воды другой, способной
смешиваться с водой жидкостью, например спиртом).
4. Величина механического давления или силы сдвига,
прикладываемые к гелю до или в процессе высушивания.
5. Такие условия, как температура, давление, pH и содер-
жание соли, а также поверхностное натяжение жидкой среды,
во время испарения жидкости из пор геля.
6. Температура, продолжительность и тип окружающей ат-
мосферы, в которой гель нагревается уже после его высуши-
вания.
Корпускулярная теория формирования геля была описана
Айлером в 1955 г. [2]. Киселев и Лыгин [229] подтвердили
с помощью электронно-микроскопических исследований, что
по мере полимеризации кремневой кислоты частицы размером
вплоть до 5—10 нм могут образовываться на стадии золя,
и этот размер сохраняется в структуре геля.
Силикагели и порошки
709
Влияние величины pH на размер первичных частиц
В том случае, когда диаметр коллоидных частиц в золе
оказывается больше чем ~5 нм, как это имеет место для
коммерческих коллоидных кремнеземов, имеющих в основном
частицы размером 5—60 нм, силикагели с удельными поверх-
ностями от 550 до 50 м2/г соответственно могут непосредственно
приготовляться из таких коллоидных кремнеземов. Величина
pH оказывает слабое действие на получающееся значение
удельной поверхности силикагеля. Однако, когда силикагель
приготовляется из кремневой кислоты, образуемой подкисле-
нием силиката натрия или гидролизом галогенида или слож-
ного эфира кремния, размер первоначально образуемых частиц
составляет только 1—2 нм, поскольку их формирование про-
исходит обычно при низких значениях pH. Размер, до которого
частицы вырастают по мере формирования геля, его промы-
вания и высушивания, возрастает с увеличением значения pH
выше 6. В данном случае величина pH является единственным,
наиболее важным фактором, определяющим конечную величину
удельной поверхности силикагеля.
Как обсуждалось в гл. 3, формируются прежде всего поли-
кремневые кислоты с низкой молекулярной массой. Они, оче-
видно, могут быть пропущены через ультрафильтр. Сен и Гош
[230] продемонстрировали такую возможность, проводя иссле-
дования на целлофановом ультрафильтре, который после
определенного момента не должен был больше пропускать
кремнезем. Так, при pH 2,3 для свежеприготовленного 3 %-ного
золя SiO2, полученного из кислоты и силиката, они обнару-
жили, что 94 % всего кремнезема проходило через ультра-
фильтр в течение 216 ч, тогда как полный временной период,
необходимый для гелеобразования, составлял 264 ч, или, следо-
вательно, кремнезем проходил через фильтр за период времени,
составлявший 82 % от времени гелеобразования. Как только
начиналась агрегация, то такой ультрафильтр быстро заби-
вался. Усиление помутнения состава указывает на рост агре-
гатов в такой кислой системе, когда первичные частицы
оказываются слишком малыми, чтобы рассеивать много света.
В том случае, когда исходной является кремневая кислота,
повергаемая гелеобразованию при pH 2—6, процесс при более
высоких значениях pH приводит к формированию больших по
размеру частиц еще до того, как начинает образовываться
гель. После высушивания геля при 110°С получается силика-
гель с более низкой удельной поверхностью. Так, в 1947 г.
Плнк и Дрейк [231] при изучении силикагелей, приготовлен-
ных при исходной концентрации SiO2, равной 4 %, и при раз-
710
Глава 5
личных значениях pH, определили следующие значения удель-
ных поверхностей:
pH Удельная поверхность,
м2/г
2,7 656
5,3 413
8,7 404
11,0 354
По мере увеличения значений pH подсчитанные диаметры
частиц увеличивались от 4,2 до 7,7 нм, тогда как диаметры пор
возрастали от 6,4 до 13,8 нм. Интересным было и то, что
кремнезем подвергался гелеобразованию в более концентриро-
ванном растворе (более высокая плотность упаковки), но
плотность конечного высушенного силикагеля оказывалась
меньшей, по-видимому, из-за того, что более прочные струк-
туры при высушивании подвержены сжатию в меньшей
степени.
С тех пор было выполнено много аналогичных исследований.
Для подобных серий силикагелей не только наблюдается наи-
большее значение удельной поверхности, когда гель приготов-
ляется при низких значениях pH, но и содержание так назы-
ваемой «связанной воды» также соответственно оказывается
высоким в связи с тем, что поверхность покрывается группами
SiOH. Типичным примером исследования является работа
Гуха и Гупта [232], которые получили силикагели с различаю-
щимися плотностями путем их приготовления при различных
значениях pH. Еще до этого Синг и Мэдели [233] изготовили
силикагели при различных значениях pH и привели следующие
данные по величинам удельных поверхностей и содержанию
воды:
pH Удельная поверхность, м2/г (эксперимент) Содержание нао,% Удельная поверхность, м2 (г)-1 (расчет)
3,7 592 6,5 785
4,0 578 5,0 605
4,63 505 4,2 508
5,76 335 3,2 387
Видно, что содержание воды примерно пропорционально
удельной поверхности. Рассчитанные значения удельной поверх-
ности, указанные в данной таблице, получены при условии, что
вся вода присутствует в виде групп SiOH на поверхности,
и допущении, что концентрация силанольных групп на
полностью гидроксилированной поверхности составляет
Силикагели и порошки
711
5ОН-групп/м2 *. Следует упомянуть несколько подобных иссле-
дований. Авторы работы [234] измерили размер частиц в зо-
лях, полученных из силиката натрия с отношением SiO2: Na2O3
3,22: 1 в процессе гелеобразования при pH 3,25; 3,53 и 4,05.
Начальный размер частиц, измеренный методом рассеяния
света, составлял 10—20 А; по мере того, как шел наблюдаемый
визуально процесс гелеобразования, эти частицы вырастали
до 40—50 А, а затем формировались агрегаты размером вплоть
до 1200 А. Гартнер и Грис-
бах [235] приготовили ана-
логичные золи при pH 3—-6
и показали, что средний ра-
диус пор получаемых сили-
кагелей менялся постепенно
от 10 до 70 А с постепен-
ным понижением удельной
поверхности. Это снова под-
тверждает, что чем выше
значение pH, тем большим
получается размер первич-
ных частиц.
Оккерс и де Бур [236]
выполнили более тщатель-
ное исследование на основе
их прежних, давно получен-
ных результатов. Они под-
твердили, что значение pH
влияет главным образом на
Рис. 5.17. Влияние pH на характеристики
геля для случая, когда силикат натрия и
кислота вступают в реакцию с образова-
нием кремневой кислоты (5 % SiO2).
I — время гелеобразования, мин; 2 — удельная
поверхность, м2/г; 5 — объем пор, см3/г. (По
данным Оккерса и де Бура [236].)
размер частиц, который устанавливается еще до того, как
начинает формироваться гель. Концентрация кремнезема —
лишь вторичный фактор, поскольку после того, как гель испы-
тывает усадку в процессе высушивания, удельная поверхность
и размер пор силикагеля определяются размером первичных
частиц, плотность упаковки которых приблизительно одна
и та же независимо от концентрации. Согласно их наблюде-
ниям (см. рис. 5.17), размер частиц быстро возрастал в области
pH 4—8, что подтверждается заметным спадом величины
удельной поверхности, начиная от 800 м2/г (при диаметре
частиц 3—4 нм) и до 200 м2/г (при диаметре 14 нм). Когда
размер частиц составляет меньше —5 нм, то силы, вызываю-
щие усадку и действующие в процессе высушивания в порах
очень малого диаметра, могут спрессовывать такие частицы
в произвольную, плотную упаковку (SiO2 занимает 60 % от
объема, т. е. пористость равна 0,3 см3/г).
См. стр. 653.— Прим. ред.
712
Глава 5
При pH 5—6 частицы с несколько большим размером,
например 6 нм, образуют большие поры, и благодаря этому
силы сжатия ослабевают настолько, что уже не могут уплот-
нить структуру в такой же степени. Таким образом, в этом
случае получается силикагель с более открытой структурой
и значением объема пор 0,8 см3/г, что соответствует плотности
упаковки сферических частиц кремнезема только на 36 % от
объема.
При еще более высоком значении pH и большем размере
частиц прочность силикагеля понижается, поскольку умень-
шается степень коалесценции, причем силикагели ослабляются
настолько, что образцы сжимаются еще больше, а объем пор
понижается.
Необходимо подчеркнуть, что отмеченные взаимозависимости
имели место тогда, когда гели промывались и высушивались,
но не подвергались дальнейшему старению или термообработке
во влажном состоянии после того, как силикагель был сфор-
мирован.
Шейнфайн и Неймарк [237] рассмотрели теорию, в настоя-
щее время являющуюся общепринятой, согласно которой крем-
незем вначале образует частицы небольших размеров. Затем
частицы соединяются вместе и формируют сетку геля. Особый
интерес вызывает представленный этими авторами широкий
набор силикагелей, у которых удельные поверхности и объемы
пор изменялись независимо. Так, были приготовлены силика-
гели с удельной поверхностью в интервале 100—800 м2/г, при-
чем любой из этих образцов можно было приготовить
с объемом пор в интервале от 0,45 до ~1,7 см2/г. В случае
силикагеля с удельной поверхностью 650 м2/г объем пор мог
быть получен в интервале 0,35—2,37 см3/г. Чтобы осуществить
это, требуется не только выполнять надлежащие условия про-
цесса формирования геля, но и проводить промывание при
регулируемых значениях pH, а также высушивание при опреде-
ленных условиях.
Прочность влажных гелей
Прочность гелей, приготовляемых из коллоидного кремне-
зема, иная, чем гелей, полученных из кремневой кислоты, что
рассматривалось в гл. 4. Здесь будет описано самопроизвольное
упрочение связей между частицами в том случае, когда они
связываются вместе. Однако это относится к гидрогелю, достиг-
нувшему точки гелеобразования и затем подвергнутому про-
цессу старения в течение времени, по крайней мере равного
длительности гелеобразования, но не подвергавшегося на
каком-либо этапе специальной термической обработке. Проч-
Силикагели и порошки
713
ность увлажненного геля может не иметь прямого отношения
к свойствам высушенного силикагеля, так как при этом
в структуре происходит много изменений, например, может
иметь место усадка образца.
Хорошо известно, что золи поликремневой кислоты, в ко-
торых частицы кремнезема или полимерные единичные обра-
зования, составляющие только 20—30 А в диаметре, могут
формироваться в гели при концентрациях кремнезема ниже
0,5—1,0 масс. %, но все же практичнее оказываются коммер-
ческие золи кремнезема с частицами 50—300 А в диаметре,
которые не образуют гелей при таких низких концентрациях.
Одна из причин заключается в том, что скорость формирова-
ния геля оказывается до такой степени низкой, что подобные
гели наблюдаются редко. Другая причина состоит в том, что,
когда такие разбавленные золи формируют гель, структура
оказывается настолько хрупкой и слабой, что стремится
сжаться, образуя разбухший осадок.
Очевидно, и в случае частиц меньшего размера при данной
концентрации кремнезема в единице объема оказывается
гораздо больше частиц, поэтому формируется значительно
более тонкая сетка. Но еще важнее, что суммарное число
силоксановых связей,' скрепляющих такую сетку, становится
много больше. Рассмотрим, например, формирование геля из
частиц диаметром d (в нанометрах) при концентрации Cd
(в граммах SiO2 на 1л). Объемная концентрация, выражаемая
в кубических сантиметрах SiO2 на 1 л, составляет Cd/2,2, где
2,2 г/см3 — удельная масса коллоидного кремнезема.
Так как объем единичной частицы (в сантиметрах куби-
ческих) равен (л/6)<Д-10-21, то число частиц Ав 1 л будет
равно 0,87 • 1021 • Cri_!cC.
Чтобы оценить число силоксановых связей, которые могут
образоваться между частицами данного диаметра d в нейтраль-
ном или кислом растворе, необходимо обратиться к геометрии
двух частиц, находящихся в контакте. В точке контакта не
только происходит конденсация поверхностных силанольных
групп с образованием силоксановых связей, но также кольце-
вой зазор вокруг точки контакта заполняется до такой степени,
пока не образуется отрицательный радиус кривизны г, равный
5—10 А (см. рис. 5.18). В растворах монокремневой кислоты,
приготовленных способом деионизации или очень быстрым
подкислением разбавленного раствора силиката натрия до
pH 2, происходит относительно быстрое образование частиц,
имеющих такой радиус кривизны, что значение равновесной
растворимости падает ниже 0,02 %. Это пересыщение вызывает
осаждение кремнезема или влечет за собой локальное пере-
распределение кремнезема, создавая перешеек диаметром М
714
Глава 5
между частицами, как только они присоединяются друг
к другу всего несколькими силоксановыми связями.
Прочность связи между частицами будет пропорциональна
площади контакта, т. е. величине /И2. Прочность геля, напри-
мер, на растяжение пропорциональна сумме площадей по-
перечных сечений всех перешейков между частицами геля,,
которая в свою очередь зависит от числа перешейков, прихо-
дящихся на единицу площади поперечного сечения; последняя
величина зависит от концентрации кремнезема.
Рис. 5.18. Схема коалесценции частиц, вызывающей формирование прочных
связей между частицами.
Подсчет значения М (в нанометрах) для частиц радиусом R
и перешейка с отрицательным радиусом кривизны г, как можно
видеть из геометрического построения (рис. 5.18), дает соот-
ношения
(г + 7?)2 = (4- + г)2 + /?2
М = -2г ±2(г2 + 2г7?)1/2
Пусть величина Т пропорциональна прочности геля на рас-
тяжение, которая в свою очередь пропорциональна числу
составленных из частиц цепочек, пересекающих заданное по-
перечное сечение геля, и площади контакта между частицами
в цепочке. Число цепочек, состоящих из частиц, будет пропор-
ционально №/3.
Тогда Т = K.N2/3M2, где К — константа.
Так как d = 2/?, то
Г2/3
Т = К,-^-[-г+ (г2 + 2г/?)1/2]2
7_к'сГ[-^ + ^--^(гг+г«)''г]
Т = К'С:Л7(<<1
Силикагели и порошки
715
где К' и К" — новые константы, a f —функция, заключенная
в скобках.
Если допустить, что г — I нм, то
т = кст [4г + 4- -дН1 + ад)1/2]
т = KCTf! (/?)
где fi (R) — новая функция, заключенная в скобках.
Если принять, что г — 3 нм, что может иметь место при
длительном старении геля или при более высоком значении pH,
то
3
R
4-(9 + 67?)1/2]
Т ~ R.'cdl3f3(R)
где fs(R)— новая функция.
Значения fi(R) для г — 1,0 нм и для г = 3,0 нм даны
в табл. 5.5.
Для золей, приготовленных при низких значениях pH и
обычной температуре, г составляет около 1 нм. Если золи
приготовляются в нейтральной или щелочной области pH, то
Таблица 5.5
Значения функции f(R) для расчета относительной
прочности силикагеля при заданной концентрации
кремнезема
R г=1а г=Зб
1 0,268
2 0,191 0,31
3 0,151 0,27 (см. рис. 5.1)
5 0,107 0,211
7,5 0,080 0,168
15 0,046 0,108
30 0,025 0,064
50 0,016 0,043
а Может быть принято для силикагелей, получаемых
при pH 1—3 и 25°С.
б Может быть принято для силикагелей, прошед-
ших продолжительное старение при 25°С или более высоких
аиачеииях pH.
716
Глава 5
происходит дальнейшее развитие коалесценции и радиус г мо-
жет оказаться равным 3 нм. При данных условиях образова-
ния геля (pH, температура) вышенаписанное уравнение должно1
предсказывать возможные комбинации концентрации и раз-
мера частиц, которые будут соответствовать гелям равной
прочности. Например, если частицы 2 %-ного золя радиусом
1 нм образуют гель определенной прочности, то можно под-
считать, какую концентрацию частиц необходимо иметь при
радиусе частиц г = 7,5 нм, чтобы образовать гель равной проч-
ности.
Принимая г=1 нм и определяя значение функции f(R)
при соответствующем значении R из табл. 5.5, имеем
СТ (0,08) = 22/3 (0,27)
С15= 12,5%
Если гелеобразование проводят при комнатной температуре
и pH ~3, то расчеты показывают следующие значения ^ля
гелей примерно равной прочности:
Радиус частиц, нм Требуемая концентра-
ция SiO2> %
1 2
3 4,8
Айлер подтвердил эти данные, приготовив золи указанных
концентраций и размеров частиц. При этом выбиралось pH 3,
раствор Na2SO4 имел постоянную концентрацию 0,20 н., золи
помещались в сосуды равного объема. Золи застудневали
через 2—3 сут. После 5 сут такие золи «прозванивались»
с одинаковой, четко прослушиваемой частотой, когда ударяли
по сосудам с содержимым. Это показывало, что все образцы
имели примерно одни и те же модули упругости. Одинаковая
прочность подтверждалась и тем, что стеклянная палочка диа-
метром 5 мм проходила внутрь каждого из исследуемых гелей
на глубину 1 см примерно при равных усилиях, прикладывае-
мых к палочке и составляющих 275 ± 50 г.
Согласно данным Высоцкого и Стражеско [238а], проч-
ность гидрогеля, полученного из кремневой кислоты при фикси-
рованной концентрации кремнезема, имела острый максимум,,
когда он формировался при pH 1,7. Этого следовало ожидать,
так как рост частиц проявляет минимум около этой точки,,
поэтому частицы наименьшего размера формируют наиболее
прочные гели.
Минимальная концентрация при формировании геля.
Наблюдалось, что золь, приготовленный из свежеподкислен-
ного силиката натрия, в котором первичные частицы имеют
Силикагели и порошки
717
только около 1—2 нм в радиусе, будут образовывать слабый
гель, когда устанавливается pH 5,5 при концентрации кремне-
зема до 0,5 масс. % или около 0,25 объемн. %. Основываясь на
вышеприведенной теории равной прочности гелей, можно
ожидать следующих минимальных концентраций кремнезема,
при которых будет формироваться гель из силиката натрия
с отношением SiO2: Na2O 3,25 : I при pH 5—6:
Радиус частиц, Концентрация,
НМ объемн. % масс. %
1 0,25 0,5
3 0,6 1,3
5 0,9 2,0
7,5 2,2 4,8
Айлер приготовил три вида золей, не содержащих солей,
с размерами коллоидных частиц 3, 5 и 7,5 нм в радиусе, имев-
ших одинаковую концентрацию 0,6 об. °/о SiO2 при pH 5,5.
Один из образцов с радиусом частиц 3 нм застудневал в тече-
ние 5 сут, тогда как образец с наибольшим радиусом частиц
не превращался в гель даже в течение 10 сут.
Размер частиц и плотность упаковки
в высушенных гелях
Конечный размер частиц и способ, посредством которого
частицы связываются вместе, очевидно, обусловливают физи-
ческие характеристики геля, в частности его адсорбционные
свойства. Такие изменяющиеся величины рассматривались
в гл. 3, а также в настоящей главе в разделе, посвященном
рассмотрению характеристик геля.
Концентрация кремнезема во время формирования геля не
является важным фактором, определяющим свойства конеч-
ного продукта при условии, что не происходило дальнейшего
усиления структуры гидрогеля перед процессом высушивания.
Оккерс [162] четко показал, что силикагели, получаемые
из кремневой кислоты, могут приготовляться с различными
размерами частиц (т. е. с разными значениями удельных
поверхностей) путем варьирования величины pH. Однако
когда гели готовились из золей с частицами заданного оди-
накового размера, то они высушивались до одних и тех же
конечных значений удельной поверхности и объема пор неза-
висимо от концентрации кремнезема в золях и в гидрогеле
(1,2-5,0 % SiO2).
Объяснение, по-видимому, заключается в том, что при
заданном размере частиц усадка гелей происходит примерно
до одной и той же плотности упаковки при высушивании неза-
718
Глава 5
висимо от различий в концентрациях кремнезема на стадии
гидрогеля после его образования.
Однако гели, образованные при различных pH и затем
хорошо промытые и высушенные, имеют специфическое соот-
ношение между pH и объемом пор. Эти соотношения, рассчи-
танные на основании интерполированных данных Оккерса,
показаны на рис. 5.19.
Рис. 5.19. Соотношение между объемом пор и удельной поверхностью (изме-
ренной методом БЭТ) промытых и высушенных силикагелей, приготовленных
из силиката натрия и кислоты при различных значениях pH.
Штриховая линия, вероятно, истинные значения удельных поверхностей (по данным
Оккерса [162]).
Понижение объема пор с возрастанием размера частиц
(или с уменьшением удельной поверхности) при изменении pH
от 10 до 6 может быть объяснено на основе того, что коэффи-
циент коалесценции, или ширина шеек между частицами, ста-
новится больше для более мелких частиц, так что такой полу-
чаемый силикагель оказывается механически прочнее и более
устойчив по отношению к усадке. (Например, при частицах
большого размера самопроизвольно происходящая коалесцен-
ция оказывается настолько слабой, что частицы сближаются
лишь с образованием упаковки, равной 50 об. %, а получаемый
объем пор составляет 0,45 см3/г, как это показано на рис. 5.19
для образца с очень низкой удельной поверхностью.) Однако
силикагели с меньшим размером частиц имеют поры с мень-
шими радиусами. При удельной поверхности 600 м2/г и объеме
пор 0,8 см3/г частицы имеют размер только около 3,5 нм в диа-
Силикагели и порошки
719
метре, а радиус пор оказывается не более чем ~20 А. В не-
которой точке этой области силы сжатия, возникающие от
поверхностного натяжения воды в процессе ее удаления из пор
при высушивании, становятся огромными. Частицы в этом
случае произвольным образом плотно упаковываются, а по-
ристость достигает 40 об.%, иначе говоря, объем пор стано-
вится равным 0,30 см3/г. Как раз это значение и наблюдается
фактически, так как несколько более тонкие поры, образован-
ные ниже pH ~5,5, сжимаются при высушивании.
Добрускин и др. [2386] отметили появление более плотной
упаковки в ксерогелях, образованных из первичных частиц
большего размера. Большие по размеру частицы связываются
друг с другом менее сильно и поэтому могут перемещаться и
перестраиваться в более плотную структуру при высушивании.
Остается еще объяснить повышение объема пор, которое
происходит при еще более низких значениях pH. Почти нет
сомнений, что гели, образуемые при pH <5, фактически со-
держат еще меньшие по размеру частицы. Однако это не
выявляется при измерениях удельной поверхности методом
БЭТ, которая остается примерно равной 800—850 м2/г для
силикагелей, образованных при более низких pH. Это может
быть объяснено тем, что при измерениях методом БЭТ про-
исходит потеря площади вокруг точек контакта постепенно
уменьшающихся частиц (см. выше раздел по характеристикам
силикагелей). Таким образом, истинная поверхность силикагеля
может непрерывно увеличиваться, но она не может быть пол-
ностью измерена по адсорбции азота. Однако всевозрастающий
объем пор силикагелей, приготовленных при более низких pH,
оказывается реальным явлением, хотя и не легко объяснимым.
Вероятно, что, по мере того как диаметр частиц еще пони-
жается (от 3,5 до 1—2 нм), все более усиливается коалесцен-
ция, и увеличение прочности на сжатие оказывается большим,
чем те смещения, которые возникают за счет возрастающих
сил поверхностного натяжения, когда поры становятся более
мелкими. Как раз при pH 3,4 Оккерс измерил радиус пор,
оказавшийся равным только 10 А. Вода в подобных порах
фактически не имеет более своих нормальных свойств, поэтому
поверхностное натяжение может оказаться меньшим, чем пред-
полагается, из-за сильного молекулярного притяжения со сто-
роны стенок пор.
Во всяком случае, интересно знать, какова причина того
факта, что в микропористом силикагеле, полученном при очень
низких pH из 10 н. раствора НС1, содержится наиболее высо-
кий объем пор, равный 0,5 см3/г.
Частицы смешанных размеров. Из смеси частиц разных
размеров образуется силикагель с более широкими порами,
720
Глава 5
например, с более высоким значением пористости и с более
низкой объемной кажущейся плотностью. Сиппел [238в] обна-
ружил, что смесь, содержащая 60 % частиц размером 12 нм
и 40 % частиц размером 25 нм, давала силикагель с величиной
кажущейся плотности, меньшей, чем получается при выборе
лишь одного типа частиц любого из указанных размеров.
По-видимому, логично предположить, что частицы одного раз-
мера должны упаковываться более равномерно с более высо-
кой плотностью по сравнению со смесью частиц разных разме-
ров, когда один тип частиц препятствует другому. Однако
следует иметь в виду, что если частицы двух размеров разли-
чаются между собой по диаметрам более чем в восемь или
десять раз, то частицы меньшего размера могут входить в за-
зоры между большими частицами, и в таком случае общая
плотность упаковки окажется выше, а пористость такой смеси
окажется меньше, чем в случае одного типа частиц.
Повышенное значение пористости
при удалении наполнителей
Имеется большой объем литературы, в особенности патент-
ной, в которой рассмотрены вопросы введения инертных
материалов до начала процесса гелеобразования, причем такие
материалы впоследствии могут быть удалены с целью получе-
ния большей пористости. В перечень подобных веществ вклю-
чены водорастворимые соли, растворимые в кислых средах
порошки, органические вещества, которые можно выжигать,
и металлосодержащие соединения, удаляемые кислотой или
аммиаком. Некоторые из подобных систем сыграли важную
роль при изготовлении катализаторов и были описаны
Вейлем [199].
Можно было бы предположить, что водорастворимые орга-
нические полимеры должны быть особенно пригодными для
введения с целью повышения пористости или по крайней мере
создания сетки тонких каналов, способствующих увеличению
скорости диффузии реагирующих веществ при их проникнове-
нии внутрь катализатора. Однако поведение таких полимерных
веществ оказывается не столь простым. Большинство полиме-
ров не образует молекулярные однородные растворы ни с рас-
творимыми силикатами, ни с коллоидным кремнеземом.
По-видимому, разделение фаз имеет место на микроуровне,
особенно в процессе гелеобразования. Обычным явлением ока-
зывается то, что после удаления полимера из высушенного
геля его остатки либо сохраняются в более или менее изоли-
рованных полостях, либо оказываются способными прерывать
Силикагели и порошки
721
структуру геля и, следовательно, в заметной степени ослаблять
структуру кремнезема.
Однако имеются примеры того, что присутствие некоторых
полиэлектролитов улучшает прочностные свойства силикагеля
в высушенном состоянии перед тем, как электролит выжи-
гается. Кроме того, в определенных случаях, по-видимому,
достигается получение силикагеля с бипористой структурой.
В работе [239] патентуется силикагель, представляющий собой
неорганические агрегаты в виде гранул, связанных между собой
спекшимся неорганическим связующим, причем между грану-
лами имеются микропоры. Такие микропоры остаются после
выжигания формирующего пористость органического вещества.
Согласно Монтгомери и Парсонсу [240], повышенное зна-
чение объема пор с радиусом пор в интервале 20—10 000 А
образовывалось, когда 30 % или более водорастворимого
полимера, такого, как поливиниловый спирт или полиэтилен-
гликоль, соответствующим образом вводилось в силикагель,
а впоследствии удалялось окислением. В таком случае вначале
формировался промытый гидрогель кремнезема, который сразу
же смешивался с водным раствором полимера. Затем гель
высушивали и медленно нагревали на воздухе вплоть до вы-
жигания органического вещества.
Формование гранул силикагеля
Для многих применений необходимо иметь зерненую, или
гранулированную, форму силикагеля. Вместо обычного про-
цесса гранулирования или непрерывного формования измель-
ченного в порошок геля вместе со связующим для придания
кермнезему формы сферических гранул в процессе гелеобра-
зования можно применить следующие способы:
а) формирование небольших капелек золя в процессе
сушки распылением;
б) впрыскивание капелек в несмешивающуюся жидкость и
последующее их застудневание путем химического воздействия
или нагревания. В другом способе для приготовления сфери-
ческих гранул размером всего лишь несколько микрон пред-
усматривается коацервация, посредством котороой частично
полимеризованный или коллоидный кремнезем связывается
с водорастворимым органическим соединением с образованием
жидкого смешанного вещества, выпадающего из раствора
в виде капелек с последующим их затвердеванием. Чешуйки
геля могут формироваться посредством замораживания вод-
ного золя или гидрогеля. Когда образуются кристаллы льда,
между ними концентрируется и спрессовывается кремнезем.
Получающаяся при этом форма гранул силикагеля зависит от
20 Заказ № 250
722
Глава 5
размера и формы кристаллов льда. При высушивании тонких
слоев силикагель может получаться в виде ленточек. Многие
из первоначально предложенных способов формования были
описаны Вейлем [199], но в последнее время были разрабо-
таны новые способы.
Метод сушки при распылении раствора кремневой кислоты,,
который приготовлялся из силиката натрия ионообменным
способом в водородной форме, был запатентован Бейли [241].
По другому методу [242] золь приготовляется посредством
частичной нейтрализации раствора силиката натрия при pH
9,6—10,9 с последующим подкислением золя перед его сушкой
распылением. Такой способ, вероятно, впервые позволил полу-
чить коллоидные частицы размером около 3—4 нм при высо-
ком значении pH, причем выделяемая затем дополнительно
кремневая кислота с низкой молекулярной массой при низком
значении pH воздействует в качестве связующего для прида-
ния прочности структуре силикагеля и сохранения высоких
значений удельной поверхности (965 м2/г) и объема pop
(0,75 см3/г). По этому способу сохраняется относительно
открытая упаковка гранул, так что кремнезем занимает всего
лишь 38 °/о всего объема силикагеля.
По патенту Бергна и Симко [243] деионизованный золь
коллоидного кремнезема сушится распылением, что дает воз-
можность получать сферические гранулы силикагеля микрон-
ного размера. Удельная поверхность такого кремнезема опре-
деляется размером частиц исходного золя.
Эмульсионная полимеризация, или диспергирование капелек
золя в несмешивающейся жидкости перед процессом формиро-
вания геля, представляет направление, которое, как это было
рассмотрено в обзоре Вейла [244], было закреплено многочис-
ленными патентами. В типичном способе капельки подкислен-
ного раствора силиката натрия, формируемые механически, по
мере того как они затвердевают, пропускаются через масло,
откуда попадают в нижерасположенный слой воды с целью
вымывания солей. Ле Пейж, Бью и Дюшен [245] сохраняли
небольшое количество щелочи в сформированных по этому
способу сферических гранулах кремнезема, поскольку при вы-
сушивании щелочь оказывает промотирующее воздействие на
рост и коалесценцию первичных частиц. После прокаливания
такого кремнезема в течение 1 ч при 600°С сохранялся объем
пор, равный 0,9 см3/г при значении удельной поверхности
310 м2/г.
Силикагели с различающимися по размерам сферическими
гранулами могут быть получены путем изменения размеров
капелек золя кремневой кислоты, попадающих в несмешиваю-
щуюся жидкость перед их застудневанием. Для получения
Силикагели и порошки
723
небольших сферических гранул золь кремневой кислоты, при-
готовленный из кислоты и силиката натрия, может быть отре-
гулирован по величине pH с тем, чтобы он превращался в гель
в течение заранее выбранного временного интервала. Золь
эмульгируют в масле с добавлением эмульгатора до тех пор,
пока капельки не затвердеют. При другом варианте поликрем-
невая кислота при низком значении pH с добавлением соли
может быть экстрагирована в органическую жидкость, способ-
ную образовывать водородные связи, как это описывалось
в гл. 4. Такое жидкое сложное соединение может затем быть
эмульгировано обратно в водную фазу до тех пор, пока ка-
пельки не начнут застудневать. Этот процесс может быть
ускорен повышением значения pH до 5.
Дрексель [246] эмульгировал раствор кремневой кислоты,
приготовленный ионным обменом, в «-бутиловом спирте, пред-
варительно насыщенном водой. При этом добавлялся аммиак,
чтобы повысить pH до значения, достаточного для превраще-
ния в гель капелек водного золя. Такую суспензию затем на-
гревали, чтобы упрочнить полученный гель.
Гринг [247] впрыскивал раствор силиката натрия в масло,
насыщенное сернистым ангидридом, который застудневал
в виде капелек. Риттер [248] приготовлял эмульсию из сили-
ката натрия в тетрахлорэтилене и добавлял H2SO4, чтобы под-
нять значение pH до 6 и сформировать гель в виде мелких
шариков. Шварц [249] смешивал растворы силиката натрия
и ацетата аммония в носике емкости, по которому капельки
застудневающей смеси направлялись в масло, с целью их
отвердения. Моул [250а] получал раствор кремневой кислоты,
из которого затем капельки вводились в горячее масло с ам-
миаком, добавленным, чтобы вызвать процесс гелеобразования.
В несколько отличающейся системе, разработанной Кум-
мерле [2506], используются «-бутиловый спирт и хлорид нат-
рия, чтобы экстрагировать кремневую кислоту из подкислен-
ного раствора силиката натрия в жидкое сложное вещество,
способное образовывать водородные связи при низком значе-
нии pH. Такое вещество диспергируется на капельки и поли-
меризуется с формированием «шариков» геля при введении
в качестве катализатора ионов F~.
Сферические гранулы могут приготовляться из сложных
кремневых эфиров. По методу Кольшуттера и Мима [251]
следует частично гидролизовать этил- или метилсиликат
в спирте с количеством воды, немного меньшим, чем рассчи-
танное теоретически значение, и с использованием соляной
кислоты в качестве катализатора. Полиэтоксисилоксан пред-
ставляет собой масло, образующее эмульсию в водно-спирто-
вой смеси и затвердевающее с получением шариков геля
20*
724
Глава 5
диаметром 0,2—0,5 мм и диаметром пор 20 А. Унгер и Шарф
[252] исследовали влияние .варьирования молекулярных масс
полиэтоксисилоксана от 700 до 2000 с использованием основ-
ного катализатора для завершения гидролиза и застудневания
эмульсионных капелек. В последующем добавляли жидкий,
углеводород, чтобы разбавить капельки и достичь более низкой
плотности геля [253]. По этому методу получаются сфериче-
ские гранулы размером от 1 мкм до 1 мм с объемом пор
в интервале 0,3—4,2 см3/г и диаметром пор 20—800 А.
В несколько похожем процессе этилсиликат эмульгировали
в воде с добавлением сильной кислоты в качестве катализа-
тора, а затем нагревали до 90°С. После этого добавляли соль,,
вследствие чего происходило формирование сферических гранул
[254а]. В другом варианте подобного способа Томас [2546]
приготовлял частично гидролизованный этилсиликат, дисперги-
ровал его в воде и гексане и добавлял триэтиламин, чтобы
вызвать застудневание капелек. Шарики диаметром 50—
500 мкм имели удельную поверхность 500—700 м2/г.
Образование сферических частиц геля посредством коацер-
вации в присутствии соединений, способных образовывать
водородные связи, было рассмотрено в гл. 4. В зависимости
от размеров частиц кремнезема, которые меняются в области
10—20 А для частиц поликремневой кислоты и вплоть до
200 нм для частиц коллоидного кремнезема, могут быть при-
готовлены силикагели почти со всеми возможными сочета-
ниями величин удельной поверхности, объема пор и диаметра
пор. В методе Куммерле [255] предусматривается добавление
к перемешиваемому разбавленному (1,8 °/о SiO2) раствору
силиката натрия примерно 10 % н-гексилового эфира диэтилен-
гликоля с последующим подкислением H2SO4, чтобы понизить
pH до 6,7. После 20 мин перемешивания получались сфериче-
ские гранулы плотного силикагеля диаметром 10 мкм. Такой
полиэфир является реагентом, способным образовывать водо-
родные связи, поэтому он сильно адсорбируется кремнеземом
при низких значениях pH.
В методе замораживания золи концентрируются до тех пор,
пока частицы кремнезема не начнут вступать в контакт. Как
показано Хацелем и др. [256], формирование геля происходит
быстро, если только значение pH не оказывается равным 2—3,
когда наблюдается более медленное застудневание. Авторы
изучили это явление достаточно подробно. Вследствие того
что гранулы геля при некоторых условиях замораживания
получаются в форме листочков, такой силикагель называется
«лепидоидальным кремнеземом». Этот тип силикагеля широко
изучался [257, 258]. Хинц, Руттлофф и Тойфель [259] изучали
удельные поверхности и пористость силикагелей, получаемых
Силикагели и порошки
725
замораживанием, а структура подобных силикагелей была
описана Колосенцевым, Белоцерковским и Плаченовым [260].
Доллимор и Шинглес [261] приготовляли разбавленную крем-
невую кислоту из силиката натрия ионным обменом, а затем
раствор замораживали и получали микропористый силикагель.
Вулф [262] запатентовал условия, при которых более пред-
почтительно получается хлопьевидный силикагель. В работе
[263] описано воздействие температуры замораживания на
получаемый объем пор. Бутчер и Симпсон [264] запатентовали
способ высушивания замораживанием кремневой кислоты,
получаемой ионным обменом, после ее «стабилизирования»
гидроксидом аммония. Более высокое значение pH, вероятно,
способствовало росту частиц перед процессом гелеобразования,
так что удельная поверхность частиц силикагеля диаметром
около 60 А составляла 450 м2/г, а диаметр пор 70 А.
В работе [265] показано, что можно получать конечный
продукт с меньшим содержанием воды, если осажденный из
воды гель после образования уплотняют посредством замора-
живания до формирования гранул, воду выжимают и кремне-
зем отфильтровывают. Путем измерения изотерм адсорбции
были изучены изменения свойств гидрогелей после их замора-
живания [266]. Согласно данным Хальберштадта и др. [267],
электронно-микроскопические исследования показывают, что
структура геля после замораживания совершенно отлична от
структуры, которая была до замораживания. Волькин, Поно-
марев и Золтавин [268] рассмотрели общие вопросы замора-
живания и оттаивания коллоидных систем.
Определенные условия высушивания способствуют получе-
нию гранул силикагеля заданной формы. По мере того как
высушивается тонкий слой геля, происходит усадка кремне-
зема, приводящая обычно к растрескиванию слоя на чешуйки
или на ленточки. Частицы силикагеля в форме волокон шири-
ной 5—25 мкм и длиной 5—10 см получаются путем высуши-
вания пленки из концентрированного золя кремнезема, нане-
сенной на инертную поверхность. Такой силикагель разрывается
на параллельные ленточки, особенно когда высушивание про-
водят вдоль поверхности в одном направлении [269]. «Микро-
шарики», или пустотелые дискретные сферические частицы,
формируются в том случае, когда водные растворы высуши-
ваются распылением с добавлением небольшого количества
вещества, образующего газ, например карбоната аммония,
способного раздувать образующиеся капельки [270].
726
Глава 5
Обработка гидрогелей
Как только структура геля сформирована, она может быть
в дальнейшем модифицирована во влажном состоянии путем
обработок, приводящих к упрочению структуры кремнезема
без значительного воздействия на пористость, причем иногда
такая обработка называется «армированием силикагеля» (см.
в гл. 4 рассмотрение вопроса о прочности гелей), или к увели-
чению размеров пор и понижению удельной поверхности
посредством растворения и переосаждения кремнезема, огруб-
ляющему текстуру геля.
Такие процессы были описаны Айлером [2]. Недавно вы-
полненные исследования подтвердили, что кремнезем имеет
корпускулярную или «глобулярную» структуру в тот момент,
когда он превращается в гель. Такая структура подвергается
дальнейшим превращениям и перегруппировке, когда образец
кремнезема испытывает старение, нагревание в присутствии
влаги, высушивание и прокаливание. Эти этапы были обобщены
Высоцким, Голинской, Колычевым, Стрелко, Стражеско, Шейн-
файн и Неймарком в превосходно написанных главах, охваты-
вающих различные аспекты формирования и процесса старения
гелей [5].
Хотя нагревание геля в воде в автоклавных условиях и
упоминается здесь, этот вопрос более подробно будет рассмот-
рен в разделе, посвященном гидротермальным обработкам.
Упрочнение геля
Упрочнение может быть выполнено тремя обычно применяе-
мыми способами: 1) осаждением дополнительного кремнезема
на имеющейся структуре силикагеля; 2) добавлением «актив-
ного кремнезема» или поликремневой кислоты к золю (частицы
золя по размеру превышают ~5 нм) в процессе гелеобразова-
ния; 3) путем термического старения гидрогеля до определен-
ной ограниченной степени для увеличения коалесценции частиц.
Осаждение дополнительного кремнезема. Если после обра-
зования частицы геля SiO2 диспергируются в суспензии, то
такой кремнезем может быть упрочнен путем осаждения до-
полнительного количества кремнезема из пересыщенного
раствора. Александер, Айлер и Уолтер [271] описали способ,
посредством которого «активный кремнезем» или кремневая
кислота с низкой молекулярной массой могут быть выделены
в суспензию при поддержании pH 9—10,5 при 95°С с такой
скоростью, что добавляемый кремнезем будет осаждаться
равномерно по всей структуре силикагеля. Вполне очевидно,
что способ не применим к макроскопическим гелям. Такой
Силикагели и порошки
727
способ будет рассмотрен ниже в связи с вопросами получения
осажденных порошков кремнезема.
Добавление активного кремнезема перед процессом геле-
образования. В некоторых случаях упрочнение может дости-
гаться за счет присутствия активного кремнезема во время
процесса формирования геля. Силикагели, получаемые в ре-
зультате гелеобразования из золей коллоидного кремнезема
с частицами 5—100 нм в диаметре, становятся все более
слабыми по мере возрастания размеров частиц. Для исполь-
зования кремнезема в качестве связующего или как катали-
заторов и адсорбентов прочность можно заметно повысить за
счет присутствия некоторого количества кремневой кислоты
в процессе формирования геля. В самом деле, кремневая
кислота образует прочные мостики из геля кремнезема между
коллоидными частицами. Таким образом, коллоидные частицы
формируются в жесткую, внутренне связанную пористую трех-
мерную сетку при условии, что кремневая кислота с низкой
молекулярной массой полимеризуется на таких коллоидных
частицах в виде прочного плотного цемента, состоящего из
геля кремнезема.
Иетс [272] запатентовал способ приготовления силикагелей
такого типа, которые содержат большие, очень однородные
поры, но в то же время достаточно прочны и жестки. Пори-
стость таких силикагелей составляет около 50 % по объему,
показывая тем самым, что в высушенном силикагеле частицы
приблизительно размещены в кубической упаковке. Типичный
силикагель приготовлялся смешиванием 55,7 % золя коллоид-
ного кремнезема, имеющего частицы размером 112 нм, с 90 г
раствора полисиликата натрия, содержащего 50 г SiO2 и
11,1 г Na2O, и подкислением 50 %-ным водным раствором
уксусной кислоты примерно до pH 7. Такая смесь застудневала
в течение приблизительно 2 мин, затем ее подвергали старе-
нию в течение 10 ч, промывали и высушивали в вакуумных
условиях. После нагревания при 600°С удельная поверхность
составляла 26 м2/г, средний диаметр пор был равен 60 нм,
а предел прочности на поперечный излом 140 кг/см2.
Когда же такой силикагель прокаливали при 1100°С в те-
чение 1 ч, то он давал усадку вплоть до плотности, достигаю-
щей 98 % теоретического значения плотности, а предел проч-
ности на поперечный излом повышался до 350 кг/см2.
Вместо более широко применяемого силиката с отношением
SiO2: Na2O 3,25:1 использовался полисиликат натрия, по-
скольку первый оказывается несовместимым с коллоидным
кремнеземом, вызывая его застудневание даже перед подкис-
лением. Шоуп описал изготовление изделий с использованием
подобного процесса гелеобразования (см. лит. к гл. 2 [96]).
728
Глава 5
Рис. 5.20а. Стадии старения геля.
а — гель кремнезема, который после формирования подвергается высушиванию. Усадка
геля при высушивании ведет к образованию небольшого объема пор с малыми диамет-
рами; б — влажный гель подвергается термическому старению, при этом наблюдаются
повышенная коалесценция и небольшая усадка при высушивании. Диаметры пор больше,
чем в высушенном геле а; в — дальнейшее старение при повышенной температуре или
обработке в автоклаве. Структура становится более грубой, наблюдается малое значе-
ние удельной поверхности и еще большие по размеру поры, но объем пор сохраняется
таким же, как и в случае б; г — гель распадается на части, имеющие вид закругленных
частиц неправильной формы.
Он обнаружил, что силикат калия с любым силикатным соот-
ношением оказывается совместимым с концентрированным
коллоидным кремнеземом.
Старение гидрогеля. Общепринятым способом упрочнения,
не производящим каких-либо сильных изменений в структуре
геля, является термическое старение гидрогеля до оптималь-
ного предела. В том случае, когда такой процесс проводится
и дальше, это ведет к огрублению структуры кремнезема.
В зависимости от характера исходного геля, а также от тем-
пературы, продолжительности и pH процесса старения струк-
тура геля может проходить через стадии, показанные на
рис. 5.20а. Механизм старения представляет собой простое
растворение кремнезема на участках структуры геля с наи-
меньшими радиусами кривизны и осаждение кремнезема на
Силикагели и порошки
729
Рис. 5.206. Электронно-микроскопический снимок силикагеля, соответствую-
щего стадии б рис. 5.20а.
участках с наибольшими радиусами кривизны или в областях
наибольшей толщины. Этот механизм обсуждался в гл. 3 и был
подробно описан в 1956 г. Александером, Броуджем и Айле-
ром [273].
Шейнфайн и Неймарк [5] рассмотрели процесс старения
при различных условиях, в частности описали влияние вели-
чины pH. Скорость происходящих в процессе старения измене-
ний возрастает с повышением pH, и сам процесс зависит от
факторов, влияющих на скорость осаждения и скорость раство-
рения кремнезема, как это было рассмотрено в гл. 1.
Должно быть понятным, что такая простая стадия, как
вымывание из геля солей, также является стадией «старения»,
а значение pH промывной воды оказывается решающим фак-
тором в том случае, когда гели приготовляются из кислоты
и силиката. Кроме того, конечные свойства подобных гелей
зависят от значения pH, как того, при котором гель формиро-
вался, так и того, при котором его промывали водой (т. е.
подвергали старению) перед высушиванием. Неймарк и Сли-
някова [274] опубликовали превосходную серию данных, пока-
зывающих, как указанные значения pH влияют на величины
удельной поверхности и объема пор силикагелей, во всех
остальных отношениях приготовляемых одинаковым образом.
730
Глава 5
Наибольшее значение удельной поверхности этой серии сили-
кагелей было 725 м2/г при объеме пор 0,32 см3/г, а наибольшее
значение объема пор было 0,84 см3/г при удельной поверх-
ности 478 м2/г.
Шейнфайн и др. [5] первыми выделили две отчетливо раз-
личимые стадии процесса термического старения:
1. При наибольшей степени влажности процесс старения
геля с последующим высушиванием ведет к наименьшей
усадке кремнезема; при этом объем пор и диаметр пор стано-
вятся большими, тогда как удельная поверхность испытывает
очень небольшое изменение.
2. В процессе дальнейшего старения величина удельной
поверхности начинает понижаться, тогда как размер пор про-
должает возрастать. Однако при этом наблюдается лишь
незначительное дальнейшее изменение объема пор. Разъясне-
ние отмеченных стадий понятно из рис. 5.20а. На первой стадии
происходит только возрастание коалесценции или степени свя-
зывания между первичными частицами. Это ведет к такому
упрочнению геля, что он уже получает меньшую усадку при
высушивании.
Упрочнение гидрогеля для получения более слабого по струк-
туре высушенного силикагеля. Как описали Александер,
Броудж и Айлер [273], при термическом старении силикагеля
в присутствии влаги получается объемистая, быстро диспер-
гируемая форма кремнезема, которую можно легко размалы-
вать для дальнейшего использования в эластомерах в качестве
армирующего реагента или в маслах для приготовления кон-
систентных смазок. Были описаны условия обработки для упроч-
нения слабой структуры гидрогеля посредством нагревания
образца в воде до тех пор, пока его удельная поверхность не
понижалась на 10—50 %, после чего влага удалялась спиртом.
Существенно, что подобный гель формируется при достаточно
низкой концентрации в воде из кремнеземных частиц такого
размера, когда их поверхность в единице объема составляет
20—75 м2/см3. Так, для частиц диаметром 7 нм при величине
удельной .поверхности около 400 м2/г должна быть создана
подходящая концентрация кремнезема 5—20 % перед тем, как
начнется гелеобразование. Гель затем нагревается во влаж-
ном состоянии до тех пор, пока коэффициент коалесценции не
достигнет значения около 0,5.
Без такого упрочнения, когда используется только терми-
ческое старение, при высушивании силикагеля получаются
твердые плотные гранулы, но если приготовление идет по спо-
собу, представленному выше, то получаются пластичные гра-
нулы, легко диспергируемые до частиц коллоидных размеров.
Получаемый этим способом типичный конечный продукт погло-
Силикагели и порошки
731
щал масло в количестве 4,54 см3/г, что соответствует объемной
пористости 0,91 см3/см3, или среднему координационному
числу ~3. Диаметр пор составлял примерно 27 нм, а диа-
метр первичных частиц был около 9 нм. На основании извест-
ной степени коалесценции был рассчитан диаметр шейки
между соседними частицами ~2 нм.
Как было обсуждено в связи с измерением коэффициента
коалесценции, кажется аномальным, что очень низкая меха-
ническая прочность высушенного продукта объясняется упроч-
нением гидрогеля, проводимым с целью понижения усадки при
последующем высушивании.
Дополнительные факторы при термическом старении.
Нагревание гидрогеля в воде или в растворах солей при
80—100°С в основном вызывает упрочнение всей структуры, но
не изменяет структуру пор. Оккерс и де Бур [275] нагревали
серию гелей SiO2 в течение 1—4 сут при 80 °C в воде, кисло-
тах и в растворах хлорида калия и обнаружили, что если
силикагель имел удельную поверхность более 200 м2/г, то для
него наблюдалось понижение поверхности при незначительном
изменении объема пор. Очевидно, что в этом случае поры
увеличивались в размере. При pH 2 эффект был незначителен,
но в нейтральном или в щелочном растворе, в особенности
в присутствии соли, текстура силикагеля заметно огрублялась.
Например, удельная поверхность понижалась от 752 до
452 м2/г, тогда как радиус пор возрастал от 13 до 22 А, но при
этом объем пор оставался на уровне 0,50 см3/г.
Интересно, что при 80°С не наблюдалось вовсе никакого
эффекта, если только силикагель не имел удельной поверхности
свыше 200 м2/г. Поведение золя кремнезема, нагреваемого при
80°С, когда не происходит роста частиц, совершенно иное, если
только удельная поверхность золя не превышает 200—300 м2/г.
Шейнфайн, Стас и Неймарк [276] наблюдали продолжи-
тельное старение силикагеля с начальным значением удельной
поверхности 920 м2/г в воде при pH 6,8 и комнатной темпера-
туре. Удельная поверхность падала от 725 до 420 м2/г, тогда
как радиус пор возрастал от 9 до 43 А. Однако пористость при
этом также возрастала от 0,31 до 0,90 см3/г. Таким образом,
структура становилась прочнее, и усадка при высушивании
образца уменьшалась. Однако армирование структуры стано-
вилось намного больше, чем усадка, когда образцы вначале
промывали уксусной кислотой, а затем высушивали. Поскольку
поверхностное натяжение уксусной кислоты составляет только
одну треть поверхностного натяжения воды, то армированный
силикагель имел больший объем пор, равный 2,36 см3/г, что со-
ответствует силикагелю с очень низкой кажущейся плотностью.
732
Глава 5
Погружение геля SiO2 в разбавленные растворы гидро-
ксида аммония при 50—85°С приводит к сильному огрублению
текстуры силикагеля. Гиргис [277] сообщил, что даже смачи-
вание силикагеля при pH 10—11 в течение 1 сут при 20°С вы-
зывало падение удельной поверхности от 650 до 467 м2/г при
соответствующем повышении радиуса пор.
Дженкинс и Шварц [278] представили данные по влиянию
концентрации аммиака на степень понижения удельной поверх-
ности (от 430 до 126 м2/г) и на степень увеличения диаметра
пор (от 85 до 397 А). Подобные эффекты воздействия значений
pH, присутствия солей, кислот и щелочей на процесс старения
были описаны рядом исследователей [279—284].
Необходимо отметить, что гидрогели, содержащие ионы по-
ливалентных металлов в частности алюминия (т. е. в случае
алюмосиликатных гелей), оказываются менее чувствительными
к изменениям при старении во влажных условиях, так как
оксиды металлов понижают растворимость кремнезема. Так,
Аккер [285] при добавлении 2—6 % солей металла получил
тонкопористый силикагель с высокой адсорбционной способ-
ностью по отношению к воде при низком значении влажности.
Аналогично Престон, Вельтман и Хуббард [286] увеличили
способность силикагеля к адсорбции воды в результате смачи-
вания его раствором алюминиевой соли муравьиной кислоты
перед высушиванием.
Можно было бы ожидать, что, поскольку силикагель не рас-
творяется при низком значении pH, кислоты не будут оказывать
такого влияния на процесс старения во влажных условиях, как
это наблюдается для случая регулируемого значения pH.
Однако, Шейнфайн и др. [287] обнаружили, что обработка
гидрогеля сильными кислотами (HCl, HNOs или конц. H^SCK)
перед высушиванием ведет к повышению объема пор в высу-
шенном силикагеле без понижения величины удельной поверх-
ности. Это может быть вызвано тем, что кислота промотирует
коалесценцию между частицами без какого-либо влияния на
рост частиц или огрубление текстуры. С другой стороны, обра-
ботка 8 н. H2SO4 вызывала падение удельной поверхности от
700 до 300 м2/г, и в то же время происходило возрастание
объема пор. Такое различие может проявиться в том, что
гель SiO2 фактически оказывается более растворимым в 8 н.
H2SO4, чем в какой-либо другой кислоте, поэтому оно связано
только со степенью внесенных изменений в структуру силика-
геля.
В любом случае сильные кислоты (рН<2) промотируют
термическое старение силикагеля. Однако маловероятно, чтобы
щелочь, выступающая в роли промотора, не могла растворять
кремнезем, если она добавляется в избытке.
Силикагели и порошки
733
Из всего вышесказанного следует, что процессы старения и
термообработки действуют в одном и том же направлении:
понижается величина удельной поверхности и возрастает раз-
мер пор. Последний может также увеличиваться за счет раство-
рения некоторого количества кремнезема. Неймарк и Слиня-
кова [279] сообщили, что обработка силикагеля 0,5 н. раство-
ром КОН или разбавленной кислотой HF может вызвать уве-
личение пор от 7 до 37 или от 37 до 47 А соответственно. Если
бы кремнезем растворялся равномерно со всей поверхности,
должно было бы наблюдаться возрастание удельной поверх-
ности. Однако, вероятно, что области с меньшими радиусами
кривизны будут растворяться более быстро, так что факти-
чески значение удельной поверхности может понижаться.
Термическое старение в воде выше 100°С под давлением
в автоклавных условиях приводит к заметно большим струк-
турным изменениям, чем этого можно достичь при 100°С. Если
система является нейтральной или щелочной, то гель БЮг
сохраняет свою форму и пористость, но в то же время удель-
ная поверхность понижается, а поры становятся очень боль-
шими. В щелочной среде гель может диспергировать до золя
(см. лит. к гл. 4, [102—108]). Воздействие жидкой воды,
а также водяного пара на силикагели будет подробно рассмат-
риваться в настоящей главе.
Усадка при высушивании, получение
ксерогелей
Еще в 1911 г. Зигмонди [288] обнаружил, что силикагель
состоит из очень тонких гранул, разделенных между собой фак-
тически очень тонкими капиллярными каналами. Он показал,
как с этих позиций объясняются явления, наблюдаемые при де-
гидратации и регидратации силикагелей. Например, понижение
поверхностного натяжения паров воды есть прямое следствие
очень малых размеров таких капилляров.
Так как частицы геля SiO2 образуют трехмерную сетку или
короткие цепочки, состоящие из частиц, жестко связанных
вместе, и поскольку частицы кремнезема по существу довольно
жесткие, то очевидно, что по мере высушивания гидрогеля и
его усадки вследствие поверхностного натяжения воды в порах
такая сетка будет закручиваться или сморщиваться [289, 290].
Причина сжатия агрегатов кремнезема в процессе высушивания
подробно рассматривалась Бикерманом [291]. Окончательная
структура силикагеля будет зависеть от структуры исходного
геля, сформированного в растворе, но она оказывается лишь
сжатой и искаженной модификацией последней.
734
Глава 5
Однако наиболее важным моментом в связи с рассмотрением
силикагеля оказывается следующее. По мере того как струк-
тура испытывает усадку, определенное число связей между крем-
неземными частицами, пронизывающих всю структуру, должно
испытывать разрыв, поскольку кремнеземные частицы по су-
ществу являются жесткими. Если структура силикагеля оказы-
вается довольно уплотненной, а его масса хрупкой, то такой
силикагель, вероятно, будет растрескиваться. Если плотность
упаковки низка и гель остается пластичным и студенистым, так
как цепочки, состоящие из очень небольших по размеру частиц,
сохраняют гибкость (даже стеклянные волокна оказываются
гибкими), то такой гель дает усадку в основном без растрески-
вания, поскольку всегда будет оставаться достаточный участок
сетки, способный в любой момент сцементировать массу об-
разца. И все же усадка силикагеля оказывается необратимой.
Вероятно, после того как некоторые связи между частицами раз-
рываются, отдельные части сетки освобождаются и могут всту-
пать в контакт с другими частями, поэтому формируются но-
вые контакты и новые связи. Таким образом, плотность упа-
ковки возрастает, а диаметр пор уменьшается.
Усадка в процессе высушивания происходит до тех пор,
пока механические напряжения, возникающие в силикагеле, не-
способны противостоять давлению, воздействующему на струк-
туру благодаря поверхностному натяжению жидкости, находя-
щейся на границе раздела фаз в силикагеле. Как показал Бар-
кас [290], силы сжатия, действующие на силикагель, возра-
стают с уменьшением диаметров капилляров. Такое сжатие
сходно с силами, способствующими сближению стеклянных пла-
стинок, помещенных вертикально в жидкость [291]. Силы,
действующие на пластинки, обратно пропорциональны расстоя-
нию между пластинками. Когда любая влажная масса измель-
ченного в порошок материала высушивается, то возникающие
капиллярные силы сдавливают гранулы порошка, при этом по-
верхность твердого материала, смоченного жидкостью, имеет
по существу нулевой краевой угол (рис. 5.21).
Александер и Джонсон [292] также отметили, что жидкость,
помещенная в капилляр, имеет пониженное давление пара и, на-
ходясь под натяжением, передает его стенкам капилляра. На-
пример, Фостер [293] обнаружил, что в капиллярах диаметром
~1,5 нм спирты адсорбируются при относительном давлении
около 0,5. Это соответствует подъему спирта по капилляру до
такой высоты, когда давление пара на конце столбика равно
половине давления у его основания (рис. 5.22). Такая высота,
подсчитанная из формулы для подъема жидкости в капилляре
(с использованием величины поверхностного натяжения спирта),
оказывается равной 8 км, что соответствует гидростатическому
Силикагели и порошки
735
давлению 700 кг/см2. Это означает, что если поставить верти-
кально очень высокую трубку, заполненную сухим, твердым си-
ликагелем, нижний конец трубки погрузить в эту жидкость
и поместить подобный прибор в замкнутую систему, заполнен-
ную только парами спирта, то жидкий спирт должен был бы
подняться на высоту 8 км. (На этой высоте давление паров
Рис. 5.21. Схемы действия сил, вызывающих усадку силикагеля в процессе его
высушивания, сходных с силами, заставляющими смоченные стеклянные пла-
стинки стягиваться вместе. (По данным Айлера [2], с разрешения Cornell
University Press.)
спирта составляло бы примерно половину давления у основа-
ния трубки по той же самой причине, которая вызывает пони-
жение атмосферного давления с высотой.) Для того чтобы из-
влечь жидкий спирт из куска подобного силикагеля, необходимо
было бы приложить «отсасывающее разрежение», равное
700 кг/см2. Таким образом, можно представить себе, что если
жидкий спирт испаряется из куска силикагеля, имеющего поры
диаметром 1,5 нм, то этот кусок будет подвергаться очень силь-
ному сжатию (такого же порядка величины). Однако для сили-
кагелей, имеющих большие по размеру капилляры, например
диаметром 15 нм, будут развиваться силы сжатия всего лишь
порядка 140 кг/см2.
Именно по этой причине в случае крупнозернистых силика-
гелей или осажденных типов кремнезема, содержащих частицы
размером более 20—30 нм, когда промежутки внутри или поры
имеют такой же порядок величины, высушенный материал
сильно не сжимается, а оказывается легким и без труда превра-
щается в порошок.
736
Глава 5
Были подробно исследованы [294] изменения, происходящие
в силикагелях при их высушивании. Образующиеся внутрен-
ние напряжения были связаны с так называемым «загрязнен-
ным растрескиванием», имеющим место при высушивании. Та-
кой силикагель сжимается силами поверхностного натяжения,
причем первичные кремнеземные частицы сферической формы
Колонна с
силикагелем
Давление паров
спирта Р0/2
Спиртовая атмосфера
Высота
подъема жидкости в
силикагеле 8 нм
Рис. 5.22. Высота, которую могла бы достичь жидкость (спирт), поднимаясь
по капиллярам силикагеля, указывет на существование сильного сжатия, воз-
действующего на силикагель, когда жидкость испаряется из пор (см. текст).
(По данным Дилера [2], с разрешения Cornell University Press.)
упаковываются более плотно вместе, а их координационное
число повышается. Соотношения между объемом пор, плот-
ностью упаковки сферических частиц и координационным чис-
лом были рассчитаны Киселевым [1266], который также связал
эти величины с диаметром пор. Если допустить, что частицы
имеют заданный небольшой размер (порядка 3 нм в диаметре),
а координационное число возрастает при сжатии кремнезема
от 3,2 до 7, то можно вычислить, что удельная поверхность, спо-
собная адсорбировать азот, понижается примерно от 900
до 300 м2/г.
Обычно не придается значение тому факту, что чем больше
размер куска геля кремнезема, тем сильнее его сжатие и
усадка. Имелик [295] указал на эксперимент, выполненный
Фостером и Торпом [296], в котором кусок геля разделяли на
две части. Первую часть разрезали на кубики с размером ребра
Силикагели и порошки
737
3—5 см, а вторую часть дробили на небольшие кусочки. После
этого обе части высушивали одинаковым способом. Объем пор
в первой части оказался равным 0,39 см3/г, тогда как во второй
части 0,50 см3/г, что указывает на меньшую усадку.
Силикагели с низкой кажущейся плотностью получаются
только тогда, когда процесс усадки при удалении жидкой фазы
устраняется. Этого можно достигнуть следующими способами:
1) упрочнением' силикагеля посредством его армирования,
в результате чего повышается прочность связей между части-
цами кремнезема, что противодействует силам, вызывающим
усадку;
2) повышением сил поверхностного натяжения путем рас-
ширения диаметра пор при старении или гидротермальной об-
работке;
3) замещением воды какой-либо другой полярной жидкостью
с более низкой величиной поверхностного натяжения, например
спиртом;
4) нагреванием заполненного жидкостью геля под давле-
нием выше критической точки, когда отсутствует граница раз-
дела фаз жидкость—пар и пар удаляется (так называемый
«процесс получения аэрогеля»);
5) образованием гидрофобной поверхности кремнезема.
Способы 1 и 2 были обсуждены в связи с рассмотрением
процесса термического старения.
Высушивание с использованием жидкостей
с низкой величиной поверхностного натяжения
Вместо воды для высушивания гелей SiO2 иногда приме-
няют другие жидкости с более низкой величиной поверхност-
ного натяжения. Берман [297] дегидратировал силикагель из
аммиака. Тетер [298] промывал алюмосиликатный гель мета-
нолом и высушивал его в автоклаве. Керби [299] получал по-
рошковидный, тонкодисперсный гель с низкой плотностью по-
средством обработки осажденного геля кремнезема смешивае-
мыми с водой жидкостями, такими, как спирты или кетоны,
с последующим высушиванием образцов при атмосферном дав-
лении. Арчибальд [300] предложил аналогичный процесс с тем
отличием, что гель, смоченный органической жидкостью, нагре-
вался под давлением в области от 7 кг/см2 до критической
точки, после чего жидкость испарялась. Пирс и Кимберлин
[301] приготовляли адсорбенты, смешивая гидрогель кремне-
зема с н-бутанолом и дистиллированной водой до тех пор, пока
в азеотропной смеси частицы геля больше не слипались. Ча-
стицы геля приготовляли путем суспендирования капелек золя
21 Заказ № 250
738
Глава 5
в бутаноле, который извлекал воду и промотировал процесс
гелеобразования.
Был предложен осушенный растворитель, в частности для
дегидратации сферических частиц силикагеля или гранул. Си
и Бейли [302] получали катализатор в виде прочных шариков
посредством застудневания капелек золя в несмешивающейся
жидкой среде с последующим промыванием геля органическими
жидкостями, (спиртами), чтобы извлечь воду перед высушива-
нием. Неймарк и Шейнфайн [303] показали, что использование
уксусной кислоты и спиртов играет важную роль для пониже-
ния поверхностного натяжения и смачивания поверхности пор.
Простое удаление 95 % воды из геля путем ее замещения ор-
ганическим растворителем сохраняет поры, заполненные такой
жидкостью, имеющей гораздо более низкое поверхностное на-
тяжение, чем вода, поэтому при высушивании структура геля
сжимается в меньшей степени. Это позволяет в качестве загу-
стителя в маслах и в смолах использовать силикагель с более
слабой структурой, который легко диспергируется [304а]'.
Этерификация или другие методы, позволяющие гидрофоби-
зировать поверхность силикагеля обработкой в органической
жидкости, дают возможность дополнительно понизить адгезию
жидкости на поверхности раздела с твердым веществом и тем
самым еще больше уменьшить усадку силикагеля при его вы-
сушивании.
Важным моментом является выбор полярной жидкости, спо-
собной смачивать поверхность кремнезема. Например, если воду
в порах поочередно замещать ацетоном, а затем гексаном, то
после дегидратации силанольной поверхности при повышенной
температуре в тонких порах еще будет оставаться вода, тогда
как снаружи будут находиться углеводородные группы. Такая
система еще будет способствовать усадке материала. Однако
если вначале поверхность гидрофобизуется так, что поры пол-
ностью заполняются органическим соединением, то такой крем-
незем может высушиваться с меньшей усадкой. Сирианни и Ла-
дингтон [3046] добавляли олеат аммония и высококипящее
легко окисляемое масло, чтобы покрыть этой смесью поверх-
ность силикагеля. При нагревании для удаления воды в порах
оставалось до 60 % по массе органического вещества, которое
затем удалялось при последующем нагревании до 500°С. В ре-
зультате получались гранулы, имеющие кажущуюся плотность
всего лишь 0,04 г/см3, которые легко подвергались диспергиро-
ванию с образованием распушенного порошка, подобного аэро-
гелю.
Силикагели и порошки
739
Высушивание без усадки, получение
аэрогелей
Согласно данным Кистлера, аэрогель представляет собой
гель, в котором жидкая фаза замещена газообразной. Это поз-
воляет избежать усадки, которая происходит, если гель высу-
шивается непосредственно из жидкости. Таким образом, по-
видимому, аэрогель можно было бы идентифицировать как
гель, дающий усадку и теряющий объем пор при смачивании
жидкостью и последующем высушивании. Для его получения
необходима жидкость, такая, как «-пропиловый спирт, которая
смачивает поверхность пор, имеет очень низкое поверхностное
натяжение и испаряется при комнатной температуре.
В соответствии с таким определением разработаны способы
приготовления аэрогелей, исключающие процесс, предложенный
Кистлером. Некоторые из подобных аэрогелей получили назва-
ние «эстерсилы».
Кистлер- [305] приготовлял кремнеземные аэрогели посред-
ством замещения большей части воды в геле спиртом, нагре-
вания такого геля в автоклаве выше критической температуры
спирта с тем, чтобы отсутствовал мениск между жидкостью и
газовой фазой, и последующего удаления паров. В таком спо-
собе при удалении жидкой фазы структура геля не подверга-
лась воздействию сил сжатия, которые возникают на границе
-раздела жидкость—газ благодаря поверхностному натяжению.
Тот факт, что структура аэрогеля близка к структуре исход-
ного влажного геля, был продемонстрирован Маршаллом [306].
Автор проводил гелеобразование кремневой кислоты в водно-
спиртовых растворах при различных концентрациях кремнезема
и затем превращал такие гели в аэрогели. В табл. 5.6 представ-
лены данные, показывающие что кажущаяся плотность аэро-
геля прямо пропорциональна концентрации S1O2 в спирте, в ко-
тором кремнезем застудневал. Плотность упаковки частиц
кремнезема в аэрогеле, таким образом, близка к плотности упа-
ковки первоначально получаемого геля. Приготовленный Кист-
лером аэрогель, содержащий до 0,02 г/см3 кремнезема, представ-
лял собой очень легкое, прозрачное, слегка опалесцирующее
твердое вещество.
Способ производства аэрогеля в промышленном масштабе,
включая также рассмотрение проблемы нагревания и охлаж-
дения получаемого продукта, являющегося хорошим термоизо-
лятором, был описан Уайтом [307] и другими [308]. Харви
[309] изучил адсорбцию азота, бутана и воды на кремнезем-
ном аэрогеле, имевшем удельную, поверхность 740 м2/г, диа-
метр частиц (первичных) 2,5 нм и широкую область распреде-
21*
740
Глава 5
Таблица 5.6
Сравнение плотностей спиртового геля и аэрогеля
(по данным [306])
Содержание SiO^ в спиртовом золе Кажущаяся плотность аэрогеля
масс. % г /100 мл (а) г ' см3 г /100 мл (Ь) а b
3,89 3,1 0,035 3,5 0,88
5,66 4,5 0,050 5,0 0,90
5,93 4,75 0,051 5,1 0,93
6,20 ' 5,0 0,056 5,6 0,89
7,25 5,8 0,071 7,1 0,82
ления пор по размерам, причем наибольший объем пор наб-
людался в капиллярах, имевших диаметр 40 нм. Адсорбция
воды вызывала коалесценцию первичных частиц в таком геле
и усадку геля с потерей объема пор. Было сделано заключение,
что по поверхностным свойствам аэрогель подобен другим си-
ликагелям, но отличается от них по объему и размеру пор.
Кроме того, было обнаружено [310], что крупнопористый аэро-
гель SiO2 давал усадку при обработке водой, а после высуши-
вания образовывал структуру мелкопористого ксерогеля с не-
значительным изменением в величине удельной поверхности.
Значения удельных поверхностей до и после усадки соответ-
ственно составляли 796 и 813 м2/г, тогда как объем пор пони-
жался от 3,90 до 0,66 см3/г, а диаметр пор уменьшался от 20
до 3,2 нм.
Ван Нордстранд, Крегер и Рис [311] отметили эффекты спе-
кания и измельчения аэрогеля при сопоставлении с ксерогелем.
Опп использовали измерение изотерм низкотемпературной ад-
сорбции азота, чтобы проследить за изменением структуры та-
кого геля. Спекание в вакуумных условиях вызывало пропор-
циональное понижение адсорбции при всех значениях давле-
ний. Аэрогель прежде всего терял свои наиболее крупные поры.
Измельчение аэрогеля, очевидно, ведет к достаточному диспер-
гированию структуры, что способствует появлению больших
микропор или пустот между отдельными фрагментами геля
в результате помола. Когда р/р0 приближается к 1,0, количе-
ство адсорбированного азота бесконечно возрастает, так что
не представляется возможным найти определенное значение
объема пор.
Силикагели и порошки
741
Уайт и Уилсон [312] запатентовали аэрогели с высокой проз-
рачностью, пригодные для термоизоляции. Такой аэрогель дол-
жен быть исключительно чистым (с содержанием менее чем
0,01 % примесей металлов), чтобы не происходил рост частиц
при получении геля.
В других модификациях способа получения аэрогеля к кис-
лому золю в смеси этанол—вода добавляют мочевину, чтобы
вызвать разложение с образованием аммиака и получить нейт-
ральный продукт [313]. Вместо спирта при изготовлении аэро-
геля под давлением можно использовать гидрогель в смеси
•с крезолом, при нагревании которого вначале выпаривается вся
вода, а затем крезол. Очевидно, что при такой повышенной тем-
пературе поверхностное натяжение на поверхности раздела фаз
•оказывается настолько низким, что гель испытывает только
очень небольшую усадку. Остаточный крезол, вероятно в хе-
мосорбированном состоянии, удаляется нагреванием на воздухе
до 800°С [314].
Таулли [315] запатентовал органофильный аэрогель с улуч-
шенной способностью к диспергированию в органической среде.
Автор нагревал полученный аэрогель под давлением в присутст-
вии паров спирта, которые могли покрывать поверхность геля
этоксигруппами, хотя природа органической добавки в продукте
не была ясна. Прозрачные кремнеземные аэрогели с очень низ-
кими значениями кажущейся плотности в области 0,18—
0,35 г/см3, согласно данным Тейшнера и др. [316], оказались
подходящими при изучении эффекта Черенкова для частиц
с высокими энергиями, получаемых на протонном ускорителе.
Аэрогели с такими низкими плотностями получали гидролизом
этилсиликата в спирте с минимальным содержанием воды с уда-
лением паровой фазы при температуре выше критической. Не-
которые разновидности полученных прозрачных аэрогелей имели
удельную поверхность 1000 м2/г (что соответствует диаметру
частиц кремнезема всего лишь 20—30 А), объем пор 18 см3/г
и кажущуюся плотность 0,05 г/см3. Смесь, состоящую из метил-
ортосиликата Si(OCH3)4 в метаноле (10 % по объему), уксус-
ной кислоты с концентрацией 0,175 н. и воды (4 моль воды на
1 моль сложного метилового эфира), нагревали в автоклаве
до 250°С (критическая температура СН3ОН равна 242°С). Пары
удаляли в вакуумных условиях и охлаждали аэрогель в атмо-
сфере азота. На использование низших спиртов от метилового
'.до бутилового в таком способе был получен патент [317].
В серии статей Николаон и Тейшнер [318] описали методы
приготовления и превращения гидрогелей в спиртовые гели,
механическую и термическую стабильность и химическую струк-
туру аэрогелей. Аэрогель, вероятно, является наиболее легким
742
Глава 5
(с наименьшей кажущейся плотностью) связанным твердым ве-
ществом, которое вообще можно приготовить.
Гидротермальная обработка
В этом разделе рассматриваются влажные кремнеземные
гели, которые не подвергались высушиванию, а также высу-
шенные гели, которые в дальнейшем были обработаны водой.
Строго говоря, любой процесс, в который вовлечены гель
кремнезема и вода в любых формах (старение или нагревание),
мог бы называться «гидротермальной» обработкой. Однако
этот термин в основном означает то, что гель подвергается дей-
ствию воды или водяного пара при температуре выше 100°С.
Может также предусматриваться нагревание геля в потоке влаж-
ного воздуха, но обычно имеется в виду автоклавная обработка,
когда процесс проводят в воде или в водяном паре при тем-
пературе выше 100°С и давлении, превышающем атмосферное.
В 1952 г. Рис [319] описал заметное различие в свойствах
силикагеля, нагреваемого в парах воды вместо нагревания на
воздухе или в вакууме. Спекание кремнезема протекает двумя
различными путями в зависимости от того, присутствуют или
же нет пары воды.
В водяных парах спекание или уменьшение поверхности,
может происходить в результате перемещения аморфного крем-
незема с поверхности более широких капилляров и заполнения
более тонких капилляров или пор, что приводит к увеличению
среднего размера пор. Водяной пар представляет собой один
из основных факторов, ответственных за ухудшение качества
катализаторов крекинга, в особенности при работе под давле-
нием. В водяных парах наблюдается минимальное изменение
объема пор; внешняя форма гранул силикагеля также сильно
не меняется, но при этом имеет место огрубление структуры,
сопровождаемое повышением размеров пор и понижением удель-
ной поверхности. Следовательно, в присутствии паров воды
кремнезем становится более подвижным и перемещается вдоль
поверхности, заполняя поры наименьших размеров. Это приво-
дит к дальнейшему расширению больших по размеру пор без
какой-либо усадки скелета силикагеля. При сравнении силика-
гелевых катализаторов, модифицированных оксидом алюминия
или оксидом магния, оказывается, что силикагели с добавкой
оксида магния устойчивее по отношению к спеканию при воз-
действии паров воды и, таким образом, находят большее приме-
нение в последние годы.
Спекание другого рода происходит при воздействии высокой
температуры в воздушной атмосфере пли в вакууме; этот воп-
рос рассматривается ниже. В результате такого процесса струк-
Силикагели и порошки
743
тура силикагеля становится совершенно иной по сравнению
•со структурой, получающейся при спекании в присутствии па-
ров воды. В вакуумных условиях силикагель подвергается
усадке, поэтому поверхность кремнезема понижается, хотя при
этом размер пор остается без изменений.
Вейл [320] следующим образом представил первый тип
спекания в присутствии паров воды:
«При той температуре, когда твердое вещество становится подвижным,
система понижает свою поверхностную энергию за счет уменьшения общей
площади поверхности. Неровности каждого отдельного зерна, которые вы-
даются в пространство пор, уносятся, так что каждое зерно становится более
округлым. Удаляемое вещество используется для заполнения присутствующих
зазоров. Следовательно, рассматриваемый процесс ведет к уменьшению
удельной поверхности и сглаживает как индивидуальные частицы, так и
поры в них».
Отмечается, что такой процесс может повышать механиче-
скую прочность из-за увеличения массы в областях контакта
между составными частями материала, но нет никаких очевид-
ных доводов, почему данный процесс должен вызывать усадку
массы вещества. Согласно представленной схеме, как только
начинают заполняться капилляры, заполнение ускоряется по
мере того, как диаметр пор уменьшается, а другие капилляры
больших размеров соответственно растут.
Механизм перестройки структуры силикагеля в том случае,
когда силикагель нагревается в воде в автоклаве, был обобщен
Киселевым и др. [321] по следующим направлениям.
Растворимость мелких частиц (глобул), образующих скелет
силикагеля, выше растворимости больших по размеру частиц.
Таким образом, кремнезем, полученный при растворении малых
частиц, осаждается вначале около мест контактов частиц, там
где имеется отрицательный радиус кривизны. Ниже 200°С такой
процесс оказывается основным наряду с осаждением кремне-
зема на поверхностях больших по размеру частиц. При этом
наблюдается только умеренное уменьшение удельной поверх-
ности кремнезема. При 200—300°С, когда кремнезем более ра-
створим, указанная реакция протекает уже гораздо быстрее и
может происходить десятикратное увеличение размеров частиц
с соответствующим понижением удельной поверхности, но Тем
не менее структура скелета кремнезема остается корпускуляр-
ной. Сетка становится грубее, а составляющие скелет частицы
возрастают в диаметре по мере того, как исчезают более тон-
кие ответвления в исходной кремнеземной сетке. При более
высокой температуре, предположительно выше критической,
наблюдается уже неравномерный рост частиц и начинают воз-
никать большие участки непористой массы внутри огрублен-
ного в сильной степени скелета кремнезема.
744
Глава 5
Жидкая или паровая фаза
Гидротермальная обработка приводит к различным резуль-
татам в зависимости от того, находится ли кремнезем в паро-
вой или в жидкой фазе. Согласно данным Чертова, Джамбае-
вой и Неймарка [322], если гель обрабатывается в паровой
фазе, то после его высушивания объем пор становится боль-
шим, чем в кремнеземе, не прошедшем такой обработки. Если
же образец полностью погружается в жидкую воду, то после
обработки объем пор понижается. Никакого объяснения авторы
не дали, но можно предположить, что образец кремнезема, по-
груженный в воду,-должен испытывать некоторую усадку во
время последующего высушивания, тогда как образец крем-
незема, обработанный в водяном паре, усадке не подвергался,
поскольку его поры не были заполнены водой.
Термопаровая обработка. Те же авторы измерили скорость
изменения удельной поверхности в том случае, когда силика-
гель нагревался в паровой фазе (в парах воды). Было выве-
дено выражение —dSjdt = kSn, где S — удельная поверхность,
м2/г; t — время, ч. Значения показателя степени п определяются
по уравнению
/2 = 9(1 - 0,00222 Т)
где Т — температура, °C и k= 10!0’2ехр(_33220/7’). Энергия акти-
вации такого процесса оказалась равной 66 ккал/моль.
Бебрис, Киселев и Никитин [323] показали, что кремнезем-
ные порошки могут быть превращены в гели. Так, порошок
аэросила растирался в водной среде до получения густой па-
сты, которая после высушивания и последующих обработок
имела следующие характеристики:
Обработка Сушка при 140°С Удельная поверхность, м2, г 192 Объем пор, см3/г 1,32 Средний диаметр пор, о А 300
Нагревание на воздухе при 940°С в течение 4 ч 128 1,22 375
Нагревание в парах во- ды при 800°С в тече- ние 6 ч 89 1,17 530
Поры оказались неоднородными, но после нагревания в па-
рах воды они становились однородными. Робинсон и Росс [324]
исследовали изменение размеров пор в процессе нагревания
при 500°С в перегретом водяном паре. Они наблюдали, что
удельная поверхность силикагеля после нагревания на воздухе
Силикагели и порошки
745
при 500°С уменьшалась от 660 до 600 м2/г, но после термопа-
ровой обработки при этой же температуре в течение 15 ч она
становилась равной 335 м2/г. Объем пор не изменяется при
термопаровой обработке, а диаметр пор увеличивается от 25
до 45 А. Однако существует доказательство того, что при об-
работке в парах воды может появляться некоторое количество
пор размером менее 10 А (см. ниже обсуждение вопроса о мик-
ропористости) .
Силикагелевые катализаторы, которые были обработаны
в парах воды под давлением в области температур 200—450°С
и затем нагревались на воздухе до 500°С, имели прочную
структуру, способную противостоять внутренним напряжениям,
возникающим при смачивании и высушивании, что в противном
случае приводило бы к растрескиванию силикагеля при его ис-
пользовании. В 50-е годы было предложено большое число
патентов на применение подобных силикагелей [325, 326а].
Простое высушивание в перегретом паре воды, по-видимому,
приводит к упрочнению структуры, так что получается сили-
кагель с низкой кажущейся плотностью. Дженкинс и Шварц
[3266] приготовили гидрогель, содержащий 7 % S1O2, который
образовывался в течение 25 с в 1,1 н. растворе соли при pH
10,8. Такой гель подвергался старению в течение 1 ч при ком-
натной температуре, а затем разрезался на куски. Оставшееся
в образце некоторое количество NaOH замещали на сульфат
аммония, гель промывали и высушивали в парах воды в тече-
ние 4 ч при температуре 120°С, после чего образец нагревали
на воздухе при 205°С. Полученный силикагель имел кажущуюся
плотностью всего только 0,16 г/см3.
Другие дополнительные примеры не оставляют никакого сом-
нения в том, что при нагревании силикагелей в водяных парах
при температуре менее 700сС форма и размер частиц силика-
геля, или гранул, остаются неизменными, тогда как внутренняя
структура подвергается сильному огрублению, сопровождае-
мому значительным понижением удельной поверхности и рас-
ширением пор, хотя объем пор при этом не изменяется.
Была исследована [327] кинетика структурных изменений,
происходящих по мере того, как силикагели нагревались в па-
рах воды при атмосферном давлении и температурах 478—
863°С. В случае алюмосиликатного геля скорость потери удель-
ной поверхности S в зависимости от времени t была описана
уравнением
где k и п — константы при заданных условиях старения. Од-
нако для силикагелей после того, как удельная поверхность
743
Глава 5
понижалась примерно наполовину от исходной величины, даль-
нейшее ее уменьшение происходило более быстро, чем это пред-
сказывается данным уравнением. В работе представлены дан-
ные по изменению значений удельных поверхностей и объемов,
пор для различных типов силикагелей. Как правило, удельная
поверхность не падает более чем на 10—20 % при продолжи-
тельности обработки 10 ч, если только температура термопаро-
вой обработки не превышает 600°С.
В том случае, когда исходный силикагель микропористый и
в нем отсутствуют какие-либо загрязнения, термопаровая обра-
ботка может перекрыть поры и привести к образованию непро-
ницаемого кварцевого стекла. Например, когда пленку ацетата
кремния (тетраацетоксисилан) наносят из ацетона на кремние-
вую пластинку, то пленка гидролизуется на воздухе с образо-
ванием тонкого слоя чрезвычайно тонкопористого силикагеля.
При нагревании такого образца вплоть до 800°С в парах воды
формируется непроницаемое покрытие SiOs [328].
Киселев, Никитин и др. [329] подтвердили, что гидротер-
мальная обработка при 250°С и давлении 50 бар не изменяет
объем пор (1,1- см3/г), несмотря на' то, что удельная поверх-
ность понижается от 300 до 2 м2/г, а диаметр пор повышается
от 100 до 16 000 А. Было также сообщено [330], что никакого
изменения объема пор после гидротермального «старения» крем-
незема не отмечалось. Такая обработка дала возможность
изготовить кремнеземный носитель для катализатора, устой-
чивый к воздействию высоких температур при проведении
разных реакций, в том числе реакции с водяным паром. Бул-
манн [331] проводил гидротермальную обработку трех сили-
кагелей с небольшими, средними и большими размерами пор
в водяных парах вплоть до 176 гС и отмечал во всех слу-
чаях, что объем пор оставался постоянным, т. е. не наблюда-
лось никакой усадки.
Чертов, Джамбаева и Неймарк [332] проводили авто-
клавную обработку гидрогеля кремневой кислоты при низ-
ком значении pH и 90—215°С в течение 2—400 ч. В результате
были получены силикагели, которые испытывали меньшую
усадку при высушивании. Силикагели имели удельную поверх-
ность в области 421—48 м2/г и объем пор 0,76—1,96 см3/г.
После обработки отмечалось увеличение размеров первичных
частиц, образующих структуру силикагеля, а также увеличе-
ние диаметров пор.
Выбрав за основу коммерческие коллоидные кремнеземы,
Хилл [333] проводил гелеобразование относительно концент-
рированных золей кремнезема с частицами диаметром 7 и
15 нм, с последующей автоклавной обработкой этих гелей
в парах воды в течение одних суток или около этого при 180—
Силикагели и порошки
747
190°С. Полученный силикагель с наинизшей кажущейся плот-
ностью имел удельную поверхность 119 м2/г (частицы диамет-
ром 23 нм) и объем пор 1,14 см3/г, что соответствовало пори-
стости силикагеля 0,71 см3/см3, или координационному числу
~4. В некоторых случаях для промотирования процесса застуд-
невания добавляли небольшое количество фторсиликата ам-
мония, но последний способствует также понижению величины
удельной поверхности.
Нагревание в жидкой фазе. Де Бур и Влеескенс [334]
очень четко показали эффект нагревания силикагеля при его
погружении в жидкую воду. Очищенный силикагель, приготов-
ленный из SiCl4 и, следовательно, не содержащий щелочи, вы-
сушивали и нагревали на воздухе. Другой образец выдержи-
вали в воде в автоклавных условиях в течение 6 ч. Были полу-
чены следующие результаты:
Температура, СС На воздухе В воде
удельная поверхность ,м2/г ооъем пор, , см3/г удельная поверхность, м2,. г ооъем пор . см3/г
После 530 0,965 530 0,965
высуши-
вания
230 — — 80 0,92
459 498 0,89 —. —•
650 477 0,85 — —.
890 439 0,705 — —
На воздухе при температуре выше 600°С силикагель быстро
начинает давать усадку и объем пор понижается. При непре-
рывном нагревании при 450 или 650°С вначале (после 6 ч об-
работки) наблюдается небольшое изменение значений удель-
ной поверхности и пористости, но затем эти величины практи-
чески не изменяются вплоть до 96 ч. Однако при 890°С такие
изменения протекают непрерывно по мере того, как происходит
объемная диффузия; фактически силикагель медленно и одно-
временно по всей массе «течет». При нахождении в воде мень-
шие по размеру частицы растворяются и наблюдается даль-
нейшее увеличение размеров больших частиц, хотя размеры
самого силикагеля изменяются мало. При этом объем пор
остается почта неизменным.
Развитие микропор. Полностью гидроксилированная поверх-
ность широких пор была получена при автоклавной обработке
силикагеля в воде, высушивания и прокаливании образца до
900—1000°С в течение 6—10 ч, с последующим регидроксили-
рованием поверхности посредством погружения образца в воду
и его высушиванием ниже 150сС [335]. Однако при нагрева-
нии выше 1000°С в образце развивались микропоры. (Это могло
748
Глава 5
происходить вследствие образования микрокристаллов кристо-
балита, которые подвергались объемным изменениям, вызываю-
щим при охлаждении появление микротрещин.)
С другой стороны, Горелик, Журавлев, Киселев и др. [336]
обнаружили, что максимальная микропористость развивается
в том случае, когда исходный силикагель с удельной поверх-
ностью 210 м2/г, не содержащий микропор, подвергался гидро-
термальной обработке при 130—150°С. Дальнейшие экспери-
менты, выполненные этими авторами [337], показали, что ульт-
рапоры, возникающие в образце аэросила, оказались более
тонкими (в них проникала вода, но не входили молекулы N2
или СНзОН), чем подобные поры на ксерогеле. Оба образца
имели приблизительно одинаковое значение удельной поверх-
ности. Такие микропоры, очевидно, образуются внутри тонкого
слоя поликремневой кислоты, осажденной на плотной поверх-
ности кремнезема. Подобный слой, вероятно, появлялся из ра-
створа при охлаждении и осаждался при температуре ниже
100°С. В противном случае такой слой не должен был быть
микропористым. К тому же, хотя значение pH и не было ука-
зано, похоже, что pH суспензии ксерогеля был немного выше,
что и привело к несколько большим размерам пор, возникаю-
щих в осажденном слое.
Воздействие присадок, загрязнений. Присутствие значитель-
ной добавки оксида алюминия, как это имеет место в алюмо-
силикатных катализаторах, уменьшает те физические измене-
ния, которые происходят при нагревании в области 478—863°С
в парах воды при атмосферном давлении [337]. В случае чи-
стого кремнезема .это происходит вследствие диффузии кремне-
зема в присутствии водяного пара, благодаря чему микроскопи-
ческие кластеры, состоящие из небольших кремнеземных частиц,
превращаются в сплошную массу, как если бы они подверга-
лись локализованному расплавлению. Это ведет к понижению
объема пор, что и происходит при температурах выше 700°С.
В присутствии оксида алюминия подвижность кремнезема пони-
жается, отчасти по той же причине, что и в случае, когда оксид
алюминия понижает растворимость кремнезема. Были прове-
дены обширные исследования структурных изменений, проис-
ходящих в алюмосиликатных гелях под воздействием водяного
пара при температурах выше 700°С [338, 339]. Локализованная
природа уплотнения отдельных корпускулярных кластеров
с образованием участков сплошной массы наблюдалась под оп-
тическим микроскопом посредством окрашивания нагретого
алюмосиликатного катализатора красителем метиловым крас-
ным из бензола. Исходный катализатор окрашивался равно-
мерно в розовый цвет, но после указанного процесса термиче-
ского старения на катализаторе появлялись бесцветные лока-
Силикагели и порошки
749
лизованные области, где имело место уплотнение вещества
с образованием неокрашиваемых участков.
Находящиеся в кремнеземе щелочные примеси (Na+)
остаются в силикагеле и значительно способствуют структур-
ным изменениям, происходящим при гидротермальной обра-
ботке. Акшинская и др. [340] сообщили, что хорошо промытый
силикагель подвержен меньшим изменениям при температуре
275°С. Этими же авторами изучено воздействие автоклавной
гидротермальной обработки силикагеля в течение 4 ч при
температурах вплоть до 360°С. Диаметр пор очень чистого
силикагеля, подвергавшегося автоклавной обработке при 270°С,
возрастал от 9 до 150 нм, в то время как объем пор пони-
жался от 0,94 до 0,87 см3/г. При 360°С диаметр пор увеличи-
вался до 1250 нм, а объем пор понижался до 0,63 см3/г. Нагре-
вание силикагеля на воздухе в течение 6 ч при 900°С приводило
приблизительно к тому же самому результату, что и обработка
в воде в автоклаве в течение 4 ч при 295°С, т. е. к формирова-
нию пор диаметром 180 нм.
Силикагели иногда подвергаются автоклавной обработке
в воде при средних щелочных условиях (за счет добавления
аммиака) для того, чтобы обеспечить высокую прочность струк-
туры и сформировать широкие поры диаметром 100—1000 А
[341]. Интересно, что при гидротермальных обработках измене-
ния в очень чистых силикагелях происходят более быстро, чем
в некоторых коммерческих силикагелях. Без сомнения, это яв-
ляется следствием присутствия в коммерческом силикате нат-
рия оксида алюминия А12О3 в количестве до 0,1 %,, который за-
тем остается и в силикагеле [321].
Нагревание на воздухе и в вакууме
По мере того как силикагель нагревается до все более вы-
соких температур, образец теряет воду за счет ее удаления из
пор и, кроме того, посредством дегидратации поверхности при
удалении силанольных групп. Процессы гидратации и дегидра-
тации рассматриваются в гл. 6. Здесь же описываются только
происходящие при этом изменения физической структуры крем-
незема.
На первом этапе, вплоть до 300°С, главным образом выяв-
ляются потери вещества^ (воды) из пор; часто этот этап назы-
вают «активацией», поскольку после него силикагель имеет
более высокую адсорбционную способность по отношению
к парам. Важным фактором на данном этапе оказывается влия-
ние воды, удерживаемой на поверхности (предположительно
в виде силанольных групп). Если образец силикагеля быстро
750
Глава 5
нагревают до высокой температуры, то выделяющаяся вода
оказывает заметный каталитический эффект, так что поверх-
ность образца значительно уменьшается. Однако постепенное
нагревание позволяет более полно освободиться от воды еще
до того, как будет достигнута температура спекания кремне-
зема, причем эффекты спекания и потери удельной поверхности
проявляются в меньшей степени. Авторы работы [342] пока-
зали, что главным эффектом в вакуумных условиях оказывается
быстрое удаление выделяющихся паров воды, которые вслед-
ствие этого меньше влияют на структуру кремнезема.
В области температур 150—600°С происходит небольшое
(если вообще наблюдается) изменение удельной поверхности или
структуры пор кремнезема. Однако в этой области образуются
более прочные связи в местах контактов, или в «перешейках»,
между первичными частицами, т. е. наблюдается возрастание
коэффициента коалесценции.
При еще более высоких температурах удельная поверхность
понижается, силикагель подвергается усадке и объем пор также
неизбежно уменьшается. Однако размер остающихся пор обычно
сохраняется приблизительно неизменным, что вообще говоря,
трудно себе представить.
Киселев [3, 343а] утверждает, что в вакуумных условиях
удельная поверхность и объем пор понижаются пропорцио-
нально друг другу и что первичные сферические частицы прояв-
ляют текучесть и расходуются одновременно, т. е. наблюдается
объемная диффузия. Патрик, Фройер и Руш [3436] показали,
что капилляры в силикагеле начинают затягиваться, когда
кремнезем нагревается до 700°С. Миллиган и Хешфорд [344]
провели тщательное исследование воздействий повышенных
температур на силикагель, имеющий удельную поверхность
380 м2/г. Образцы нагревали в течение 2 ч при 200—1000сС.
Изучение образцов методом дифракции рентгеновских лучей ни
в одном случае не выявило какой-либо кристаллизации. Нагре-
вание вплоть до 500°С не сказывалось на величине удельной
поверхности или на структуре пор. Слабое спекание наблюда-
лось при 650°С, и более быстрые потери удельной поверхности
имели место при более высоких температурах, но при этом ди-
аметр пор оставался постоянным и равным ~5,6 нм. Было по-
казано, что в коммерческом силикагеле с диаметром пор около
2 нм и в алюмосиликатном ксерогеле, имевшем диаметр пор
около 4 нм, не наблюдалось никаких изменений среднего раз-
мера пор по мере развития процесса спекания [345, 346]. По-
видимому, поры разрушаются каким-то способом по мере того,
как кремнезем переходит из пористого состояния в непори-
стое, хотя размер остающихся пор не претерпевает каких-либо
изменений.
Силикагели и порошки
751
Даже при спекании рыхлого, объемистого аэрогеля, когда
величина усадки по объему составляла 83 %, Тейшнер и Нико-
лаев [316] обнаружили, что средний диаметр пор dp, который
равен отношению удельного объема пор к удельной поверхно-
сти, изменялся лишь на. небольшую величину:
Температура, °C S, м2/г V , см3/г V
300 1004 1,48 14,8
500 800 1,26 15,8
700 477 0,9 18,9
900 162 0,3 18,5
Согласно данным Кольшуттера и Кампфа [347], выполнив-
ших аналогичные исследования, нагревание силикагеля от 100
до 1000°С не вызвало никаких изменений в значениях диамет-
ров пор, которые оставались равными 22 А, тогда как удельная
поверхность понизилась от 618 до 341 м2/г. Истинная плотность
кремнезема, измеренная по гелию, изменилась от 2,18 до 2,22
г/см3, а структура, определяемая по дифракции рентгеновских
лучей, оставалась аморфной. При этом наблюдалось падение
объема пор от 0,57 до 0,33 см3/г, так что кажущаяся плот-
ность силикагеля возрастала от 0,79 до 0,98 г/см3, т. е. объем-
ная усадка составляла 19 %, а линейная усадка 6,8 Не-
значительная часть представленных в литературе данных
по спеканию является невоспроизводимой, поскольку, как те-
перь известно, многие из изученных силикагелей содержали не-
известные, но, без сомнения, существенные количества приме-
сей. Одно из объяснений вышеприведенного явления, благодаря
которому некоторые поры исчезают, но другие еще остаются
с неизменным размером, заключалось в том, что в отдельных
небольших областях кремнезема могли быть локализованы
примеси, такие, например, как ионы натрия, которые промоти-
ровали спекание вблизи своего местонахождения.
Однако в настоящее время это кажется маловероятным.
Так, Гудмен и Грегг [348] приготовили очищенный от ионов
силикагель, который они использовали в своих более тщательно
выполненных исследованиях по спеканию кремнезема, не содер-
жавшего примесей. Силикагель был приготовлен гидролизом
тетраэтилсиликата (тетраэтоксисилана). Диаметр микропор
оставался постоянным, равным 6,0±0,3 А при изменении тем-
пературы от 200 до 800°С, тогда как удельная поверхность и
объем пор понижались таким образом, что их отношение оста-
валось постоянным. Удельная поверхность падала от 700 до
450 м2/г, в то время как происходила объемная усадка сили-
кагеля.
Следовательно, весьма значительным фактом является то,
что при спекании силикагель дает усадку: объем пор пони-
752
Глава 5
жается пропорционально уменьшению удельной поверхности,
тогда как размер пор остается неизменным. Это представляется
так, как если бы некоторая часть силикагеля полностью под-
вергалась усадке до образования плотного твердого кремнезема,
тогда как оставшаяся часть кремнезема не претерпевала бы
никаких изменений.
Другой характерной особенностью является следующее.
В том случае, когда температура нарастает ступенчато, каж-
дый раз последующая доля силикагеля дает усадку до плотного
состояния, а удельная поверхность быстро падает до более низ-
кого значения, которое остается постоянным на следующей тем-
пературной ступени. Де Бур [349] рекомендовал обратить вни-
мание на этот факт, указывая, что для каждой более высокой
ступени температуры вплоть до ~700°С при выдерживании
кремнезема в течение 6 ч удельная поверхность падала до оп-
ределенного значения, после чего ее изменение происходило
очень медленно. Иначе говоря, наблюдалась быстрая потеря
величины удельной поверхности до некоторого определенного
значения при заданной температуре, и дальнейшие изменения
замедлялись. Возможно, такое поведение влечет за собой усадку
структуры силикагеля в определенных локализованных микро-
скопических областях кремнезема до подавления всякой пори-
стости, т. е. до образования в этих областях сплошного кремне-
земного стекла SiC>2. Если подобные области распределены
в виде сетки по кремнезему, то весь объем силикагеля должен
сокращаться, а поверхность и объем пор должны уменьшаться,
тогда как оставшиеся области должны оставаться неизменными
при сохранении исходного диаметра пор.
Объяснение этого механизма основано на допущении о том,
что склонность соседних частиц кремнезема сливаться вместе
или спекаться противостоит стремлению внутренних напряже-
ний сохранить частицы дискретными. Такие напряжения соз-
даются тогда, когда твердая сплошная сетка кремнезема начи-
нает давать усадку по трем координатным направлениям одно-
временно. Но если в локальной области, состоящей из двух ча-
стиц, начинается процесс их слияния и такая микрообласть
способна отделиться от других соседних областей, то спекание
двух отмеченных частиц будет протекать более быстро. Это
приводит к нарушению баланса сил в локальной области, так
что во всей такой области имеет место локализованный про-
цесс спекания до образования сплошной массы кремнезема. При
каждой более высокой температуре образуется новый ряд лока-
лизованных областей, поэтому происходит дальнейшее спекание.
Силикагели и порошки 753
Спекание однородных структур
Если все частицы в силикагеле оказываются очень однород-
ными по размеру и упаковываются однородным способом, то
спекание будет происходить внезапно, как только достигается
определенная температура, и все поры будут подавляться одно-
временно. Айлер наблюдал подобное явление в случае плотно
упакованных однородных кремнеземных сферических частиц
диаметром 200 нм (см. раздел о синтетическом опале в гл. 4).
Такой белый непрозрачный кремнезем проявляет быструю
усадку, когда достигается определенная температура (1000—
1200°С в зависимости от содержания ионов Na+), и он превра-
щается в прозрачное непористое кварцевое стекло SiO2 гораздо
ниже нормального значения температуры размягчения.
Вероятно также, что при такой однородной структуре сили-
кагеля температура спекания становится более низкой из-за
более мелких пор. Таким образом, как только при определен-
ной температуре группа частиц начинает коалесцировать, то
поры между ними становятся меньше и скорость, с которой
поры затягиваются, все возрастает до тех пор, пока поры не пе-
рекроются полностью.
Сравнение обычных плотных, тонкопористых силикагелей
с аэрогелем, для которого характерна открытая пористая струк-
тура, показывает, что при данной температуре в вакуумных
условиях спекания более устойчивой оказывается широкопори-
стая структура. Рис [319] подтвердил, что в подобном случае
твердые поверхности таких широких «пор» оказываются на-
столько далеко разделенными, что их сближение и «слияние»
в процессе нагревания происходят медленно. Волф и Бейер
[350] также показали, что для силикагеля с более низкой плот-
ностью, имеющего относительно меньшее число контактов ча-
стиц и более низкое координационное число, при высокой тем-
пературе наблюдаются наименьшие степень усадки и потеря
удельной поверхности. Авторы получили высушиванием две
порции.одного и того же силикагеля, причем первая порция (I)
формировалась из жидкой фазы с низким поверхностным натя-
жением (уксусная кислота), а вторая порция (II) — из воды:
Удельная поверхность, м2/г
исходный после
силикагель нагревания
при 900 СС
708 300
504 50
Силикагели, имеющие только однородные микропоры, спо-
собны спекаться полностью до получения теоретической плотно-
22 Заказ № 250
Силикагель Диаметр пор,
А
I 69
29
754
Глава 5
сти кремнезема при относительно низкой температуре. Малан-
чук и Стюарт [351] сообщили, что силикагель с порами диа-
метром 10 А спекался при необычно низком значении темпе-
ратуры:
Температура, °C Удельная поверхность,
м2у г
25 700
800 500
900-1000 0
Производство отлитых в форме изделий из чистого прозрач-
ного кварцевого стекла влечет за собой решение двух проблем:
а) синтезирование такого типа силикагеля, который не растре-
скивался бы при высушивании, и б) затвердевание изделия из
промытого и высушенного силикагеля без возможного про-
цесса расстекловывания. Шоуп и др. (см. лит. к гл. 2 [96, 97])
решили эту проблему посредством использования совместимого
раствора, состоящего из растворимого силиката (калия) и кол-
лоидного кремнезема с высоким содержанием SiC>2. При фор-
мовании с добавлением однородно распределенного подкисляю-
щего реагента (формамида) силикагель становится чрезвычайно
прочным вследствие высокого содержания кремнезема и очень
тонкопористой структуры. После промывания кислотой с целью
удаления щелочного металла отформованное изделие из си-
ликагеля высушивалось и спекалось при 1350°С. Затвердевание
протекало быстро, сопровождаясь однородной объемной усад-
кой силикагеля, так что получалось конечное, сохранившее
форму изделие из прозрачного плавленого кварцевого стекла.
Щелочные металлы, загрязнения и кристаллизация
При некоторой температуре выше 1000°С может начаться
процесс расстекловывания в результате образования центров
кристаллизации, состоящих из кристаллов кристобалита. Этот
процесс в сильной степени катализируется присутствием сле-
довых количеств загрязнений, особенно щелочных металлов
(натрия). С другой стороны, если силикагель берется очень
чистым, то он не расстекловывается, согласно данным Гудмена
и Грегга [348], даже в течение 5 ч при 1400°С. Фактически
наблюдается изменение межатомных связей внутри S1O2 уже
при умеренных температурах, хотя силикагель остается аморф-
ным в том смысле, что методом дифракции рентгеновских лучей
не может быть обнаружена какая-либо кристаллическая форма
кремнезема. Фрейссард и Имелик [352] пришли к заключению
на основании данных, полученных методом ПК-поглощения,
что существуют небольшие упорядоченные области со струк-
Силикагели и порошки
755
турами, напоминающими либо кварц, либо тридимит, либо
кристобалит. Аэрогели показывали структуру кварца, а ксеро-
гели— структуру тридимита, но оба вида аморфного кремне-
зема при температуре выше 600—800°С показывали изменения
с образованием связей, характерных для кристобалита. На тем-
пературу, при которой удельная поверхность быстро падала
до нуля, оказывали заметное влияние даже следы щелочных
примесей. Рис [319] обнаружил, что для аэрогеля, приготов-
ленного посредством конденсации из паровой фазы и поэтому
содержавшего очень низкие концентрации натрия и других ще-
лочных примесей, удельная поверхность приближалась к нулю
только при температуре выше 1100°С. Удельная поверхность
коммерческого силикагеля падала до нуля при температуре
около 1040°С, а для аэрогеля, для которого было известно, что
он приготовлялся по способу, предусматривающему включение
сульфата натрия, удельная поверхность оказывалась равной
нулю при 900°С.
Допустимые концентрации натрия в силикагелевых катали-
заторах настолько низки, что химические методы анализа могут
оказаться недостаточно чувствительными. Метод активацион-
ного анализа с использованием нейтронного потока, позволяю-
щего превращать атомы натрия в радиоактивный изотоп, ока-
зывается настолько чувствительным, что можно определять
содержание натрия до 0,0003 % со средним квадратичным от-
клонением 0,00002 % [353].
Структура силикагеля разрушается, а аморфный кремнезем
кристаллизуется при достаточно низкой температуре, когда
в нем присутствует 5 мол. % оксидов щелочных металлов. Ли-
тий промотирует процесс кристаллизации до образования
кварца, а натрий—до образования кристобалита всего лишь
при 700°С. Калий действует почти также быстро при 700°С
[354]. Удельная поверхность подобных силикагелей, содержа-
щих оксиды щелочных металлов, понижается почти до нуля
при 650—700°С [355]. Когда такой силикагель отбирают на
промежуточном этапе при его усадке, затем подвергают обра-
ботке кислотой, промывают и высушивают, то полученный крем-
незем содержит очень широкие поры диаметром 300—1200 А.
Кроме того, остаются микропоры, вероятно, после того, как
натрий извлекается из тонкого стекловидного слоя на поверх-
ности образца. Может быть получено любое значение удель-
ной поверхности от 70 до 6 м2/г путем охлаждения образца при
правильно выбранной температуре.
На скорость превращения аморфного кремнезема в кристал-
лический сильно влияет присутствие различных веществ, кото-
рые могут служить центрами кристаллизации, а также веществ,
которые вызывают лабильное'состояние связей Si—О—Si, что
22»
756
Глава 5
приводит к большей подвижности структуры кремнезема. Напри-
мер, как это было отмечено Милликеном, Миллсом и Обладом
[356], добавление 2 % AI2O3 к силикагелю при нагревании до
1160°С дает более прочную кристаллическую решетку кристо-
балита, чем в случае чистого силикагеля, не содержавшего ок-
сида алюминия. С другой стороны, при еще большем содержа-
нии оксида алюминия кристаллическая решетка кристобалита
начинает ослабевать. Наиболее важной концентрационной точ-
кой оказывается такая, когда незначительное количество оксида
алюминия в наибольшей степени упрочняет кристаллическую ре-
шетку кристобалита в кремнеземе после нагревания силикагеля
до высокой температуры. Эти авторы констатировали, что по-
скольку кристобалит имеет ту же самую структуру, что и алю-
минат калия, но в отсутствие ионов калия, то влияние кремне-
зема на склонность оксида алюминия к кристалличности, ве-
роятно, оказывается очень большим. Аналогично структура
алюмината влияет на ориентацию кремнекислородных тетраэд-
ров, примыкающих к алюминатной структуре, так что на гра-
нице раздела, вероятно, существуют отчетливые кристобалит-
ные кольца. Такое влияние кристаллизации, или «кристобалит-
ных центров кристаллизации», по-видимому, простирается
только на глубину в несколько молекул внутрь частицы крем-
незема. Эти кристобалитные центры кристаллизации могут дей-
ствовать как затравочные центры для роста кристаллов, как
это наблюдалось, когда силикагелевые катализаторы с низким
содержанием оксида алюминия нагревались до 1150°С.
Наблюдалось, что при адсорбции щелочи на силикагеле
имеет место значительное различие в зависимости от того, бу-
дет ли силикагель затем нагреваться в вакууме или на воз-
духе [357, 358]. При адсорбции 1,5-10~3 моль Na+ в расчете на
1 г кремнезема нагревание в вакууме всего до 150°С понижает
удельную поверхность от 695 до 380 м2/г, но на воздухе удель-
ная поверхность падала до 175 м2/г. Кажется вероятным, что
в вакуумных условиях щелочь удерживает меньшее количество
воды, и поэтому эффект, связанный с понижением поверхно-
сти, оказывается не столь большим. При указанном адсорби-
рованном количестве либо ионов Na+, либо ионов К+ большая
часть поверхности утрачивается, и кристаллизация до кристо-
балита происходит только при 800—850°С.
Было описано [359] необычное, бурно развивающееся явле-
ние при довольно быстром нагревании силикагеля до 800—1000°Ср
причем кремнезем содержал ~ 0,05 % предварительно адсор-
бированных ионов натрия. Силикагель вначале давал усадку
так, что поры закрывались, но затем внезапно поры прорыва-
лись или расширялись, и силикагель сильно напоминал лопаю-
щиеся зерна кукурузы. Причина подобного действия в обоих
Силикагели и порошки
757
случаях одна и та же. Исходный силикагель имел удельную
поверхность, измеренную методом БЭТ, 700 м2/г, объем пор
0,40 см3/г и содержание связанной воды в виде поверхностных
групп SiOH около 6 %. При соответствующем необходимом
количестве адсорбированных на поверхности кремнезема ионов,
натрия гранулы силикагеля блокировались при температуре
около 800°С еще до того, как вся вода успевала выделиться, и
такая масса вела себя подобно чрезвычайно вязкой жидкости,
которая затем вспенивалась за счет достижения определенного
внутреннего давления прорвавшегося водяного пара. Если со-
держание натрия слишком низко, то вязкость системы оказы-
вается слишком высокой и непроницаемая оболочка не может
сформироваться. Когда же содержание натрия, излишне вы-
соко, то вязкость системы становится слишком низкой, и в этом
случае водяной пар выделяется еще до того, как давление пара
дойдет до критической величины.
Оксид бора понижает температуру спекания кремнезема и,
несомненно, будет ускорять понижение удельной поверхности
силикагелей при высокой температуре. Однако оксид бора ока-
зывает задерживающее воздействие на проявление расстекло-
вывания, поскольку процесс кристаллизации до состояния кри-
стобалита происходит менее легко в борсодержащем стекле
SiO2—В20з [360]. Согласно де Буру [361] литий представляет
собой сильный «минерализатор», так как промотирует процесс
кристаллизации аморфного кремнезема. В большом количестве
(20 %) LiCl превращает аморфный кремнеземный порошок
в кварц в течение 2 ч при 700°С [362]. Воздействие ионов нат-
рия заключается в том, что они способны промотировать обра-
зование и разрыв связей Si—О—Si, повышая подвижность
структурных элементов. Очень низкая концентрация натрия уже
оказывается эффективной, но его присутствие в подобных,
очень малых количествах показывает, что воздействие будет
проявляться только в окрестности вокруг каждого атома нат-
рия. Следовательно, спекание и структурные перестройки мо-
гут иметь место только в локализованных зонах, что приводит
к образованию гетерогенной структуры [363а].
Спекание и расстекловывание силикагеля, по-видимому,
можно понижать теми же самыми способами, благодаря кото-
рым подобные процессы могут замедляться в стеклах с высо-
ким содержанием кремнезема. Элмер и Нордберг [3636] пока-
зали, что азотирование пористого стекла при 500—1000°С, про-
водимое в атмосфере аммиака, понижает расстекловывание.
Элмер и Мейсснер [ЗбЗв] ввели углерод в пористое стекло
в виде фурфурола, который полимеризуется и подвергается пи-
ролизу до образования углерода в отсутствие воздуха и при вы-
сокой температуре. Это ведет к дегидроксилированию поверх-
758
Глава 5
ности кремнезема, и, таким образом, удаление воды способст-
вует размягчению кремнезема при высокой-температуре.
Силикагели с нестандартными структурами
Субмикропористые силикагели — непроницаемый
кремнезем
В отличие от ранее известных способов приготовления об-
разцов кремнезема с как можно большими порами представляет
некоторый интерес синтез силикагелей с такими малыми по-
рами, насколько это только возможно (менее 3 А), В такой
поре каждая молекула азота или какая-либо другая молекула,
настолько малая, что может входить в пору, по существу окру-
жена стенками поры и очень прочно удерживается - силами
Ван-дер-Ваальса [364, 365]. Отсюда и появился термин «пер-
сорбция». В наименьшие по размеру поры входят молекулы
воды, но не входят молекулы азота, а еще меньшие поры впу-
скают только молекулы гелия. Могут иметь место неоднородно-
сти и прерывности твердой структуры с еще меньшими разме-
рами, но их нельзя называть порами, поскольку ни один из ато-
мов, какой бы малой величины он ни был, не может проник-
нуть в них.
Субмикропористые типы кремнезема получают двумя спосо-
бами: 1) из твердого силикатного стекла или кристалла, как
правило до некоторой степени гидратированного, посредством
удаления катионов без распада или растворения исходной
структуры; 2) из дискретных, чрезвычайно небольших по раз-
меру (диаметром 10—20 А) кремнеземных частиц посредством
их спрессовывания в плотно упакованную структуру. Спрессо-
вывание частиц происходит либо благодаря силам поверхност-
ного натяжения, возникающим в процессе высушивания кремне-
зема или удаления жидкости, либо путем сжатия кристаллами
льда в процессе замораживания частиц, либо просто за счет
приложения высокого механического давления.
Ряд примеров по «гидратированным аморфным типам крем-
незема» был описан в гл. 1. В действительности такие веще-
ства представляли собой силикагели, в которых поры были на-
столько малы, что содержавшаяся в них вода имела очень низ-
кое давление пара. Подобные силикагели образуются в том
случае, когда раствор поликремневой кислоты с низкой моле-
кулярной массой, например раствор кубического октамера, рас-
смотренный в гл. 3, испаряется при низком значении pH. Если
это значение после концентрирования до стадии образования
геля составляет ~2, то частицы остаются при этом неагреги-
рованными. После высушивания образуется плотная масса,
Силикагели и порошки
759
которая очень сильно спрессована за счет сил поверхностного
натяжения, возникающих в процессе высушивания, когда вода
удаляется из чрезвычайно тонких пор (см. лит. к гл. 1 [НО,
111]). Доллиморе и Хил [172] получали субмикропористый
кремнезем только тогда, когда указанное оптимальное количе-
ство остаточной кислоты присутствовало в системе.
Субмикропористый силикагель получали также в том слу-
чае, когда не допускалась агрегация первоначально сформиро-
ванных частиц кремневой кислоты, и их рост останавливали на
стадии образования коротких цепочек перед тем, как частицы
концентрировали и высушивали до твердого состояния. Если
агрегация частиц происходит в растворе, то поры в этих агре-
гатах сохраняются и в высушенном силикагеле. Однако если
силикагель подвергали чрезвычайно высокому давлению с тем,
чтобы выдавить воду, как об этом сообщал Ленер [366], то
тогда частицы переходили в плотно упакованное состояние.
Силикагель, о котором сообщал Ленер, приготовляли из крем-
невой кислоты, образуемой при гидролизе SiCl4; вероятно, он
состоял из частиц диаметром всего 10—15 А. Такие частицы
могут соответствовать приблизительно кубическому октамеру
(см. гл. 3), и при плотной упаковке частиц должны возникать
небольшие поры, из которых вода не может легко выходить.
В любом случае состав высушенного кремнезема соответст-
вовал соотношению (HSiOi.s)п после того, как кремнезем уплот-
нялся при давлении, превышающем 105 кг/см2.
Рамзай и Эвери [367] приготовили образцы уплотненного
кремнезема с порами молекулярного размера прессованием
кремнеземных порошков. Авторы сообщили о том, что при мак-
симальном сжатии образовывались только микропоры, хотя суб-
микропоры, вероятно, также присутствовали.
Субмикропористые образцы кремнезема содержат поры,
которые имеют тот же размер, что и поры в цеолитах, однако
неоднородны по размеру. Если исходить из частиц кремневой
кислоты, однородных по размеру, то из них можно получать
кремнезем, содержащий более однородные поры, чем это до-
стигалось ранее.
Непроницаемый силикагель может формироваться в том
случае, когда диаметр частиц составляет менее 10—15 А [172].
Если такие частицы плотно упаковываются, то будут присут-
ствовать лишь субмикропоры, причем остающиеся поры запол-
няются прочно удерживаемыми молекулами воды. Попытки
упрочить подобные пленки посредством включения небольших
количеств водорастворимых органических соединений (полиме-
ров) обычно ведут лишь к тому, что появляется фактор, ме-
шающий образованию плотной упаковки частиц и способствую-
щий ослаблению получаемой пленки. С практической точки
760
Глава 5
зрения проблема заключается в том, что золи кремневой кис-
лоты с такой низкой молекулярной массой или таким неболь-
шим размером частиц оказываются нестабильными, даже когда
они концентрируются при pH 2, т. е. когда агрегация частиц
будет протекать наиболее замедленно. Непроницаемый кремне-
зем образуется только в том случае, когда свежеприготовлен-
ный раствор быстро высушивают до образования тонкой пленки,
или из сконцентрированного методом замораживания твердого
кремнезема, который получается в виде псевдоморфной формы и
занимает пространство между кристаллами льда.
Субмикропористые типы кремнезема часто называют по-
разному в зависимости от способа их получения, и при этом
можно не распознать, что они принадлежат к одному и тому
же классу. Поры, образуемые в них, могут составлять вплоть
до 50 % от объема всей массы кремнезема и тем не менее иметь
диаметры только такого размера, что в них могут разместиться
небольшие молекулы. Например, посредством проведения реак-
ции между твердым метасиликатом щелочного металла, высу-
шенным до содержания воды менее 6 моль, и конц. H2SO4 Бло-
унт [368] обнаружил, что он называл термином «силикодиби-
сульфат» соединение SiO(HSO4)2 и сходные с ним составы.
При смешивании с водой соединение .образует «силикомуравьи-
ную кислоту», которую также можно классифицировать как
субмикро- или микропористый силикагель. Автор, кроме того,
нашел ряд аналогичных «соединений». Эти образования могут
случайно иметь простой стехиометрический состав и из-за силь-
ной ассоциации компонентов в некотором смысле быть класси-
фицированы как «соединения».
Некоторые «лепидоидальные» типы кремнезема, описанные
в гл. 1, получившие такое название благодаря физической форме
частиц порошка, принадлежат данному классу. Фактиче-
ски многие формы так называемых «гидратированных кремне-
земов» получаются путем замораживания растворов кремневой
кислоты с низкой молекулярной массой.
Согласно данным Акшинской и др. [369], силикагели, содер-
жащие как ультрапоры, так и мезопоры, теряют ультрапори-
стость при их нагревании на воздухе при 800°С или в парах
воды при 750°С.
Пористые стекла
Высокопористая форма почти чистого кремнезема может
быть получена с величиной пор в тех же областях размеров, как
это имеет место для многих силикагелей. В настоящее время
коммерческим является лишь один тип пористого стекла:
Vycor-7930. Прежде всего при высокой температуре приготов-
Силикагели и порошки
761
ляется гомогенное боросиликатное стекло. Затем такое стекло
выдерживается при более низкой температуре, при которой на-
чинают разделяться две несмешивающиеся жидкие фазы. При
тщательном контролировании состава может быть достигнуто
соответствующее соотношение этих двух фаз, так что фазы на-
чинают выделяться в виде взаимопроникающих сеток. По мере
того как со временем структуры обеих фаз становятся грубее,
на любой стадии можно вызвать затвердевание системы, чтобы
остановить процесс ликвации. Фаза стекла, обогащенная бором,
способна растворяться в сильной кислоте, так что остается бога-
тое кремнеземом высокопористое стекло [371]. Обычно такой
кремнезем не.называется «силикагелем», поскольку последний
термин относится, как правило, к пористому кремнезему, который
формируется посредством гелеобразования золя кремнезема и,
таким образом, состоит из сетки соприкасающихся частиц.
Vycor-7930 состоит из 96,3 % SiO2, 2,95 % В20з, 0,04 %
—R2O3 и RO2. Плотность этого пористого стекла составляет
1,5 г/см3, объем пустот, выраженный в процентах от общего
объема стекла, 28 %, средний диаметр пор 4 нм, удельная по-
верхность 200 м2/г и модуль разрушения 420 кг/см2. Структура,
выявленная на электронно-микроскопических снимках, пред-
ставлена в обобщающей статье Элмера, Нордберга и др. [372].
В статье дается также библиография.
Специфические адсорбенты
Приготовление «специфических адсорбентов», имеющих
сродство по отношению к определенным молекулам, было вна-
чале описано Дикки [373], а также обсуждалось Полингом
[374]. Такими веществами были силикагели, которые избира-
тельно адсорбировали отдельный краситель в ряду метил-,
этил-, «-пропил- и «-бутилоранж
R2N—C6H4N=N—C6H4SO3H,
'где R— одна из упомянутых алкильных групп. Бернард [375]
подтвердил наблюдения Дикки. Такой силикагель вначале при-
готовляется из силиката натрия и кислоты в присутствии инди-
видуального красителя, который должен адсорбироваться. Крем-
незем затем высушивают на воздухе, измельчают в порошок и
экстрагируют метанолом в течение нескольких дней, чтобы уда-
лить весь способный извлекаться краситель. Силикагель, при-
готовленный в присутствии данного красителя, имел большую
адсорбционную способность по отношению к выбранному кра-
сителю, чем образец силикагеля, приготовленный для сравне-
ния в отсутствие красителя. Адсорбция такого красителя на
силикагеле, очевидно, зависит от имеющихся в молекуле кра-
762
Глава 5
сителя катионных центров, таких, например, как группа диал-
киламмония, и было показано, что специфичность подобных си-
ликагелей зависела от строения диалкиламиногруппы.
При определенных условиях гелеобразования и высушива-
ния полимеризация кремнезема, очевидно, ведет к образованию
поверхности силикагеля с конфигурацией, которая формируется
вокруг каждой первоначально присутствующей молекулы кра-
сителя, так что такая поверхность впоследствии проявляет спе-
цифическую адсорбционную способность по отношению к моле-
кулам данного типа. Это означает, что кремнеземные частицы,
составляющие силикагель с рассматриваемым типом структуры,
должны быть молекулярных размеров.
Согласно данным Курти и Коломбо [376], разделение опти-
ческих. изомеров камфаросульфокислоты и миндальной кислоты
достигалось с помощью силикагеля, приготовленного специаль-
ным способом. 30 мл раствора силиката натрия плотностью
1,4 г/см3 с отношением Na2O:SiO2 3,34:1 и содержащего 2 г
D-камфаросульфокислоты, разбавляли 250 мл и доводили
pH до 4. Кислоту затем экстрагировали метанолом и смесьd-
и ь -камфаросульфокислот подвергали хроматографическому
разделению с использованием этого силикагеля. Элюат прояв-
лял левовращающую активность, следовательно, силикагель ад-
сорбировал D-камфаросульфокислоту. Утверждалось, что ана-
логичным методом достигалось некоторое разделение D- и
ь-изомеров миндальной кислоты.
Экспериментальная трудность при работе с силикагелями
этого типа состоит в том, что в водной суспензии продолжает
происходить перегруппировка структуры с уменьшением вели-
чины удельной поверхности. Кроме того, если силикагель не был
тщательно проанализирован на присутствие органического сое-
динения, которое могло попасть в силикагель в процессе его
формирования, то остается некоторое сомнение в том, пол-
ностью ли такой реагент был экстрагирован до проведения ис-
пытаний на селективность. Однако в этом случае следует быть
уверенным, что оптическая активность элюата не вызвана при-
сутствием какого-либо оставшегося на силикагеле левовращаю-
щего органического вещества, поскольку элюат проявлял лево-
вращающую активность, тогда как при приготовлении силика-
геля первоначально использовалось правовращающее соеди-
нение.
Дикки [377] провел дальнейшие исследования силикагелей,
приготовленных для изучения специфической адсорбции метил-
оранжа. При формировании силикагеля в присутствии такого
красителя существенным было требование понижения значений
pH, температуры и содержания соли. Некоторые из красителей
не могли быть экстрагированы из высушенного силикагеля.
Силикагели и порошки
763
В связи с этим возник вопрос, будет ли позже наблюдаться спе-
цифическая адсорбция, вызванная присутствием оставшихся
молекул красителя. Холдеман и Эмметт [378] исследовали спе-
цифическую адсорбцию на образцах кремнезема после их ста-
рения в течение года при комнатной температуре и обнаружили,
что подобная специфичность медленно понижалась.
Беккетт и Андерсон [379, 380], используя в своих работах
хинин, по-видимому, подтвердили такое явление специфической
адсорбции. Начиная с 1957 г. в Европе и в Советском Союзе
публиковались исследования по этой теме. В работе, выполнен-
ной в ПНР [381], были получены положительные результаты
на типах осажденного кремнезема при изучении адсорбции пи-
колинов, пиридина, хинолина и акридина. Однако Высоцкий,
Дивнич и Поляков [382] поставили вопрос о том, что молекулы
красителя, которые остаются неэкстрагированными, могут на-
ходиться не вблизи поверхности, а внутри кремнезема и по-
этому не будут давать вклад в специфическую адсорбцию. Было
сделано заключение, что «специфичность» представляет собой
«отпечаток» в структуре на поверхности первичных частиц крем-
незема и проявляется по мере того, как гель высушивается
в контакте с «формующим агентом», который затем уда-
ляется [383].
Необычное утверждение было высказано Патрикеевым [384],
заключающееся .в том, что силикагель, подвергаемый поверх-
ностной ориентации в присутствии определенных микроорганиз-
мов, впоследствии ускорял рост этих и некоторых других мик-
роорганизмов. (Вероятно, трудно доказать, что при этом не были
адсорбированы некоторые органические вещества, благотворно
влияющие на микроорганизмы.)
Продолжали появляться успешные доказательства того, что
силикагели могут специфически адаптироваться к адсорбции
различных типов молекул: гормонов [385], большого ряда раз-
новидностей органических оснований и их изомеров [386, 387]
и даже ферментов [388].
Улучшенный способ приготовления специфических адсорбен-
тов был описан Канаболоцким и др. [389]. Не только золи и
гели, но также и аэрогели могут быть использованы в качестве
источника кремнезема при подобных исследованиях. Кроме
того, такие силикагели с «отпечатками» сохраняют свою специ-
фичность даже после нагревания кремнезема до 250°С [390].
Адсорбция пропилоранжа на силикагеле, приготовленном пред-
варительно с этим красителем, после чего краситель удалялся,
оказалась в 20—30 раз выше по сравнению с адсорбцией на
контрольном силикагеле без красителя. Известны обзоры по
этой теме [391—394]. Природа «отпечатков» или «следов», ос-
тавляемых на кремнеземной поверхности молекулами, которые
764
Глава 5
впоследствии должны специфически адсорбироваться на дан-
ном кремнеземе, была исследована Бартелсом [395]. Автор
пришел к заключению на основании исследований, выполненных
методом дифракции рентгеновских лучей, что кремнезем, сфор-
мированный путем полимеризации кремневой кислоты в при-
сутствии 1,10-фенантролина (который затем удалялся), имел
«геометрически ограниченную двумерную структуру», которая
способна специфически адсорбировать оставившую отпечаток мо-
лекулу. Однако двумерная структура формируется, когда крем-
незем полимеризуется в присутствии многих органических осно-
ваний, т. е. когда образуются частицы наподобие чешуек или
хлопьев. Специфичность, таким образом, не связана с формой,
а скорее с природой поверхности. Кроме того, Канаболоцкий
и др. [396] пришли к заключению, что в некоторых случаях ад-
сорбент, обеспечивавший высокую специфичность, фактически
не отличался по своей текстуре от обычных силикагелей.
Бартелс [387] заключил при исследовании ряда органиче-
ских оснований, что «отпечаток» состоял из «двумерного распо-
ложения зарядов».
При сравнении адсорбции различных молекул, похожих на
молекулу, оставившую «отпечаток», можно прийти к заключе-
нию, что при специфической адсорбции определяющими яв-
ляются стерические факторы [397]. Имеется определенная воз-
можность того, что оставившие отпечаток молекулы, которые
не могут быть удалены растворителями, не оказываются внутри
кремнезема, как предположили Высоцкий, Дивнич и Поляков
[382], но связываются посредством образования стабильных
связей Si—С. Стрелко, Канаболоцкий и Высоцкий [398] позже
обнаружили, что, когда силикагели высушиваются в присутст-
вии органического вещества, процесс усадки силикагеля ведет
к разрыву связей Si—О—Si, образуя свободные радикалы, спо-
собные в свою очередь создавать связи Si—С. Это может по-
казаться удивительным, если не вспомнить о таком факте, что
кремнезем покрывается группами Si—R, где R — углеводород-
ная группа, причем такое покрытие создается просто разма-
лыванием кремнезема в углеводородной среде. Однако даже
если такой «отпечаток» и состоит из химически связанных мо-
лекул, то, по-видимому, маловероятно, что эти молекулы могут
проявлять подобное специфическое притяжение только лишь
для молекул своего собственного вида.
Подводя итоги, необходимо отметить, что явление специфи-
ческой адсорбции, проявляемое на силикагелях, приготовлен-
ных в присутствии вещества, которое затем должно адсорби-
роваться на кремнеземе, действительно существует, но в на-
стоящее время мало что известно относительно оставленных на
поверхности «отпечатков».
Силикагели и порошки
765
Осажденные кремнеземные порошки
Рассмотрение, которое здесь приводится, относится только
к кремнеземным порошкам, представляющим собой совокуп-
ность агрегатов, состоящих из первичных частиц коллоидных
размеров, причем агрегаты не имеют общих точек срастания
в отличие от макроскопических гелей в процессе их приготов-
ления. Это произвольное различие, но оно позволяет разграни-
чивать силикагели, представляющие собой массивные, грану-
лированные или измельченные в порошок формы, от всех дру-
гих типов тонкодисперсных кремнеземных порошков, приготов-
ляемых осаждением из газовой фазы или из раствора.
Вообще говоря, полученные осаждением кремнеземные по-
рошки отличаются от силикагелей, которые были превращены
в порошок, более открытой структурой, т. е. более высоким зна-
чением удельного объема пор. Оба вида порошков состоят из
агрегатов, образованных в свою очередь из частиц коллоид-
ного размера; но обычно структура силикагеля оказывается
спрессованной по мере того, как гель высушивается. Аэрогели
представляют собой исключение, и когда они измельчаются
в порошок, то трудно отличить- такой порошок от некоторых
разновидностей осажденных кремнеземов, но тем не менее аэро-
гели включены в разряд силикагелей.
Терминология оказывается ненадежной в том случае, когда
частицы кремнезема имеют размер более 50 нм. Такие частицы,
после того как они сконцентрированы и высушены из водной
фазы при рН<7, образуют относительно рыхлый осадок, кото-
рый трудно классифицировать как силикагель или порошок.
Александер [399] наблюдал в этом случае, что в отсутствие
соли или частиц меньшего размера коэффициент коалесценции
составлял менее 5 % •
Кремнезем, осажденный из раствора
силиката натрия
Наблюдается близкая взаимосвязь между формированием
силикагеля и образованием осадка. При осаждении концент-
рация кремнезема оказывается более низкой, а частицы объе-
диняются вместе в агрегаты под действием сил коагуляции.
В отсутствие агента коагуляции кремнезем не осаждается из
раствора ни при каком значении pH.
Как было показано в гл. 3 и 4, в кислом растворе кремне-
вая кислота полимеризуется до чрезвычайно небольших по раз-
меру частиц, которые собираются в виде цепочек и образуют
766
Глава 5
сетку геля, пронизывающую водную фазу по всему объему. При
низких концентрациях частицы могут собираться в неустойчи-
вые студенистые массы, но не будут заполнять всего объема.
В щелочном растворе кремнезем полимеризуется до образова-
ния дискретных коллоидных частиц, которые вырастают до 4—
5 нм в диаметре и более и остаются затем в виде стабильного
золя.
Если раствор кремневой кислоты, не содержащий солей,
очень быстро превращают в щелочной с pH 7—8 добавлением
аммиака или разбавленного раствора гидроксида натрия, то
вначале может образоваться разжиженный гель или раствор
может загустевать, но вскоре снова разжижается по мере того,
как тонкая сетка геля распадается и образуются большие по
размеру коллоидные частицы. Если концентрация превышает
некоторое критическое значение, то гель может сохраняться
и становиться более грубым по своей структуре, но при этом
никакого осаждения не происходит.
Берби [400] определяет «осажденный кремнезем» как ^вы-
сушенный кремнезем, для которого характерным является от-
сутствие как длинных, так и коротких элементов структуры».
Это определение основано на том факте, что в геле каждая
частица связана с окружающими в определенном усредненном
геометрическом соотношении, т. е. все участки геля схожи по
своей структуре. Это, вероятно, не относится к осажден-
ному кремнезему. Однако в настоящее время, как полагают,
типы кремнезема следует классицифировать на основании спо-
соба их приготовления без каких-либо попыток определять их
структуру, за исключением случаев, когда структура однозначно
определяется по известным свойствам кремнезема. Согласно
Вейзеру [401], единственным различием между осажденным
кремнеземом и гелем кремнезема является то, что осадок вклю-
чает в себя только часть той жидкости, в которой происходит
его образование.
Коагуляция кремнезема в присутствии
ионов натрия
В том случае, когда частицы кремнезема присутствуют в го-
рячей суспензии при pH 9—10, они начинают коагулировать,
как только концентрация ионов натрия превосходит примерно
0,3 н.
В растворе силиката натрия с обычным соотношением
SiO2: Na2O 3,3: 1 оказывается, что нормальность ионов натрия
составляет 0,1 С, где С — концентрация кремнезема, выражае-
мая в г/100 мл.
Силикагели и порошки
767
Эти простые факты лежат в основе многих процессов оса-
ждения кремнезема. Во всех них предусматривается нейтрали-
зация раствора силиката натрия кислотой с тем, чтобы колло-
идные частицы кремнезема росли в слабощелочном растворе и
могли флокулировать под воздействием ионов натрия из раство-
римых натриевых солей. Кроме того, должен быть принят также
во внимание фактор, вызывающий усиление или упрочнение
структуры агрегатов.
Когда кремнезем необходимо диспергировать в органическом
полимере, чтобы вызвать упрочнение полимера, или диспергиро-
вать в какой-либо жидкости, чтобы вызвать ее загущение, об-
разцы кремнезема, полученного осаждением, должны приготов-
ляться так, чтобы их легко можно было измельчить или диспер-
гировать до единичных первичных частиц или до очень неболь-
ших кластеров коллоидных размеров. Осажденные агрегаты
должны быть относительно большими по размеру, чтобы крем-
незем легко было промывать и высушивать. Однако структура
внутри самих агрегатов должна оставаться открытой и иметь
большие поры. Когда такие агрегаты будут подвергаться дейст-
вию сил сжатия или поперечных сил сдвига, например, если
кремнезем в измельченном состоянии вносится в резину, струк-
тура таких агрегатов может быть легко раздроблена и разру-
шена до коллоидных размеров. Если первичные частицы крем-
незема оказываются плотно упакованными, как в аэрогелях, то
прилагаемые обычные механические усилия не способны раз-
дробить их на отдельные части, по крайней мере нельзя полу-
чать раздробленные кусочки, заметно меньшие по размеру
500 нм.
Силы, вызывающие усадку при высушивании, рассматрива-
лись в предыдущем разделе, посвященном силикагелям. Те же
самые силы действуют и при высушивании осажденного кремне-
зема. Для того чтобы получить механическим помолом тонко-
дисперсный кремнезем, образующие осадок агрегаты не должны
быть чрезмерно сжаты силами поверхностного натяжения даже
в микроскопическом масштабе на локальных участках. В таком
случае требования, предъявляемые к процессу высушивания
осадка из водной фазы, будут следующими:
1. Диаметр первичных частиц должен быть больше ~15 нм,
обычно он составляет 20—30 нм.
. 2. Коалесценция или упрочнение частиц в агрегатах должны
достигать такой величины, чтобы структура агрегатов не давала
усадки при высушивании.
Более низкий предел, налагаемый на размер первичных ча-
стиц, необходим просто потому, что в малых частицах поры
мельче. Агрегаты с более мелкими порами оказываются более
прочными или имеют большую степень коалесценции, поэтому
768
Глава 5
способны противостоять силам, вызывающим усадку. Однако
при избыточном усилении структура агрегатов становится слиш-
ком прочной, и такие агрегаты трудно диспергировать путем
измельчения. Когда размер частиц равен 10—15 нм и высуши-
вается осадок, находившийся в жидкости, отличающейся от
воды и имеющей более низкое поверхностное натяжение, нет не-
обходимости добиваться слишком высокого упрочнения агрега-
тов, и получаемый конечный продукт остается в дисперсном со-
стоянии. Даже в том случае, когда такой кремнезем промы-
вается пропанолом и высушивается, осажденный кремнезем
с величиной удельной поверхности >300 м2/г (т. е. с размером
частиц меньше 10 нм в диаметре) все же еще чрезмерно дает
усадку, и такие агрегаты нелегко диспергировать с целью ис-
пользования подобного кремнезема в качестве пригодных на-
полнителей.
В большинстве процессов осаждения предусматривается про-
сто частичная нейтрализация раствора силиката натрия в при-
сутствии растворимой соли для того, чтобы вызвать коагуляцию.
Чисто эмпирические процедуры описаны в большом числе1 па-
тентов, однако основные этапы при формировании пригодных
для указанных целей продуктов теоретически не были осмыс-
лены. Эти этапы оценивались и раскрывались прежде всего
в патентах, но не были освещены в научной литературе.
Таких этапов три. Как показано на рис. 5.23, они аналогичны
этапам, предусматриваемым при формировании гелей кремне-
зема, но протекают в иных условиях. Сюда относятся:
1. Образование и рост коллоидных частиц.
2. Коагулирование частиц с образованием агрегатов, что
приводит к получению суспендированного осажденного кремне-
зема.
3. Упрочнение агрегатов.
Замечательно то, что осажденные кремнеземы, широко раз-
личающиеся по структуре и по своим свойствам, могут быть
приготовлены просто смешиванием кислоты и силиката натрия
различными способами при соответствующих временных интер-
валах и температурах. При этом необходимо знание условий
реакции в горячем растворе с тем, чтобы управлять следующими
процессами:
1) образованием зародышей частиц;
2) ростом частиц до желаемого размера;
3) коагуляцией частиц с целью образования агрегатов при
контролируемых значениях pH и концентрации ионов Na+;
4) упрочнением структуры агрегатов до желаемой степени
без дальнейшего процесса образования зародышей.
Чтобы получить конечный продукт с возможно большими по
размеру первичными частицами до того, как такие частицы нач-
Силикагели и порошки
769
Золи кремнезема
Кислота
Силикат
натрия
▼
Коагулят
(с пониженным зарядом)
Шлам коагулированного
кремнезема
Шлам упроченного
кремнезема
Рис. 5.23. Схема процесса упрочнения кремнеземных агрегатов. В виде сфер
показаны частицы кремнезема. (По данным Айлера [2], с разрешения Cornell
University Press.)
Срастание частиц
при добавлении
„активного“кремнезема
нут агрегировать, может оказаться целесообразным повысить их
размер выше предела, достигаемого при самопроизвольном про-
цессе вызревания по Оствальду (около 5 нм при 25°С и 10 нм
при 80—110°С). Рост дискретных частиц осуществляется спосо-
бами, рассмотренными в гл. 4. Известна методика приготовле-
ния кремнезема из кислоты и силиката натрия [402]. Такую
или аналогичную процедуру необходимо использовать в каче-
стве промежуточного этапа до того, как произойдет агрегация
частиц и упрочнение структуры, если необходимо, чтобы конеч-
ный продукт имел размеры частиц значительно больше 12—
15 нм.
Коагуляция может также быть вызвана добавлением неболь-
ших количеств солей поливалентных металлов, например, каль-
ция, или путем временного понижения значений pH до точки
минимальной устойчивости золя, например, до pH 5—8. Однако,
агрегацию в области pH 8—И обычно проводят в присутствии
соли натрия, полученной из силиката.
Как только агрегаты, состоящие из кремнеземных частиц же-
лаемого размера, получены (обычно в форме хлопьевидного
осадка), к суспензии добавляется «активный» кремнезем, чтобы
вызвать упрочнение агрегатов. По определению Рула [403],
«активным» кремнеземом является любой кремнезем, находя-
щийся в молекулярной или в коллоидной форме в водном рас-
творе, способный деполимеризоваться до мономера за интервал
23 Заказ № 250
770
Глава 5
времени не более чем 10 мин, когда раствор разбавляется гид-
роксидом натрия до pH ~ 12, а концентрация SiO2 составляет
около 0,2% по массе при температуре около 30°С, и в отсутствие
других катионов, кроме ионов натрия. Количество сформировав-
шегося мономера в любой данный момент времени определяется
по количеству образовавшейся кремнемолибденовой кислоты,
измеряемой колориметрическим методом.
«Активный» кремнезем приготовляется добавлением разбав-
ленных растворов силиката натрия и кислоты непосредственно
к суспензии, содержащей кремнеземные агрегаты, в таких про-
порциях, чтобы иметь возможность выделять «активный» крем-
незем, в то время как в суспензии поддерживаются щелочные
условия при температуре выше 60°С. В присутствии гидроксил-
ионов, используемых в качестве катализатора, «активным» крем-
неземом оказывается такой, который при добавлении к системе
срастается с поверхностью агрегатов кремнезема. Значение pH
суспензии во время такого процесса поддерживается в пределах
8—11.
Количество кремнезема, необходимого для «упрочнения»
агрегатов, характеризуется так называемым «отношением на-
ращивания», которое определяется как частное от деления ко-
нечной массы кремнезема в системе на массу агрегированного
кремнезема в суспензии перед началом реакции. Таким образом,
при отношении наращивания, равном 3: 1, агрегаты, состоящие
из частиц размером 5 нм, будут превращаться в агрегаты с «уп-
рочненной» или сетчатой структурой, состоящей из частиц диа-
метром около 7 нм. Отношение наращивания менее чем 4 : 1 бо-
лее предпочтительно, поскольку обычно нежелательно цементи-
ровать вместе первичные кремнеземные частицы, входящие
в агрегаты, до такой степени, чтобы агрегаты в дальнейшем не-
возможно было раздробить на составляющие частицы, когда ко-
нечный продукт измельчается, например для использования
в резине.
На этапе упрочнения кислота и силикат должны добавляться
с такой контролируемой скоростью, чтобы освобождаемый «ак-
тивный» кремнезем, или кремнезем с низкой молекулярной мас-
сой, полимеризовался бы на суспендированном кремнеземе, но
не образовывал новых частиц. Это определяется по следующим
измерениям удельной поверхности кремнезема в системе, при-
чем ее возрастание показывает, что «активный» кремнезем до-
бавляется слишком быстро и что в таком случае образуются
новые зародыши. Типичная скорость добавления составляет 10 г
«активного» кремнезема в час в расчете на 1000 м2 поверхности
кремнезема в суспензии.
Образование регулируемых по размеру частиц и упрочне-
ние агрегатов. Александер, Айлер и Уолтер [271] впервые опи-
Силикагели и порошки
771
сали способ приготовления такого типа кремнезема. Поскольку
этот способ был описан только в патентах, то здесь он рассмат-
ривается более подробно.
Во-первых, золь, состоящий из частиц размером 5—7 нм,
приготовлялся следующим образом: 425 ч. раствора силиката
натрия, содержащего 2,39 г SiO2/100 мл и имевшего молярное
отношение SiO2:Na2O 3,25, нагревали до 35°С и интенсивно пе-
ремешивали при добавлении 162 ч. 2,4%-ного раствора H2SO4
с постоянной скоростью в течение 30 мин. При этом нейтрали-
зовалось примерно 80% щелочи и значение pH понижалось до
9,7±0,2, измеряемого при 25°С. Такой прозрачный золь нагре-
вали до 95°С в течение 15 мин, чтобы частицы выросли до 5—•
7 нм в диаметре. Щелочь, освобождаемая в результате умень-
шения поверхности кремнезема, вызывала повышение pH золя
до 10,1.
Затем 85,4 ч. раствора силиката натрия (13,22 г SiO2/100 см3)
и примерно 95 частей раствора кислоты (4,65% H2SO4) одно-
временно медленно (в течение 2 ч при 95°С) добавляли к бы-
стро перемешиваемому раствору. Выбранные пропорции позво-
ляли -поддерживать pH 10,3±0,2. Концентрация ионов натрия
повышалась приблизительно до 0,27 н., и щелочь нейтрализова-
лась примерно на 80%. При этой процедуре кремнезем перехо-
дил в осадок, на котором осаждался остаток кремнезема. Затем
добавляли около 32 ч. раствора кислоты в течение 20 мин для
того, чтобы понизить pH до 5.
После старения раствора при 90°С в течение 4 ч образцы
кремнезема отфильтровывали и высушивали. Удельная поверх-
ность этого кремнезема была равна 335 м2/г, и коэффициент
коалесценции составлял 45 % • Контрольные опыты показали, что
агрегаты состояли из первичных непористых частиц диаметром
10 нм.
Основной продукт отфильтровывали после добавления 0,2 %
бромида цетилтриметиламмония, нанесенного на SiO2 для улуч-
шения флокуляции, затем продукт промывали ацетоном и высу-
шивали при комнатной температуре. Полученный порошок
с удельной поверхностью 335 м2/г был легким и мягким, погло-
щал льняное масло в количестве 476 мл/100 г SiO2, имел кажу-
щуюся плотность 0,12 г/см3 и легко подвергался диспергирова-
нию в масле. Из контрольных опытов по поглощению масла
можно было заключить, что удельный объем пор составлял по
крайней мере 2 см3/г и в этом отношении превосходил некото-
рые коммерческие аэрогели.
В другом патенте Александера и др. [271] подробно описан
нашедший большее практическое применение осажденный крем-
незем с более низким значением удельной поверхности, который
может быть выделен из водной фазы высушиванием, однако
23*
772
Глава 5
сохраняет открытую структуру и легко поддается диспергирова-
нию. Основное отличие от вышеприведенного кремнезема заклю-
чается в том, что исходный золь приготовлялся в более концен-
трированном растворе: 123 ч. раствора силиката натрия, содер-
жавшего 3,71 г SiO2/100 см3, нагревали до 90°С и затем к нему
добавляли 10,1 ч. 13%-ного раствора H2SO4 в течение 30 мин.
После этого одновременно добавляли в течение 2 ч 119 ч. рас-
твора силиката натрия, содержащего 8 г SiO2/100 см3 и 111 ч.
3,11 %-ного раствора H2SO4. В конце реакции прибавляли до-
полнительное количество кислоты, чтобы понизить pH до 7.
К полученному шламу приливали 3 ч. 1 %-ного раствора бро-
мида цетилтриметиламмония для улучшения фильтрации. Влаж-
ный кремнезем промывали для удаления соли, после чего дово-
дили pH до 8 и высушивали кремнезем при 110°С. Этот кремне-
зем имел удельную поверхность 132 м2/'г, поглощал масло
в количестве 207 см3/100 г SiO2, его кажущаяся плотность со-
ставляла 0,22 г/см3, диаметр первичных частиц был ~20 нм,
а коэффициент коалесценции 60%. Подобный кремнезем ока-
зался исключительно эффективным агентом, используемым для
упрочнения резины. По поглощению масла было показано, что
удельный объем пор составляет около 1,0 см3/г.
Конечно, можно использовать другие источники получения
первичных кремнеземных частиц, но они в общем оказываются
более дорогостоящими, чем упомянутые способы приготовления
из силиката натрия, применяемого в начальной стадии процесса.
Однако, когда необходимо избегать примесей натрия в готовом
продукте, коллоидный кремнезем с частицами соответствующего
размера может быть получен из других веществ путем флокуля-
ции и осаждения, например, регулированием нитрата аммония
до значения pH 9—10 за счет добавления гидроксида аммония
и упрочнением не содержащей натрия кремневой кислоты, при-
готовленной из силиката ионным обменом.
Для каждого типа осажденного кремнезема имеется опти-
мальная степень упрочнения, способствующая достижению мак-
симальной диспергируемости. Очевидно, такое упрочнение не
следует проводить в большей мере, чем это необходимо для пре-
дотвращения усадки структуры агрегатов в процессе их высу-
шивания.
В другом видоизмененном способе в качестве кислоты исполь-
зовали СО2. В таком варианте СО2 подавали в раствор, имев-
ший силикатное отношение SiO2 : Na2O 3,25 и содержавший
3,5% SiO2 (нормальность 0,35 н. по натрию), при 95°С в тече-
ние 40 мин до тех пор, пока отношение СО2 : Na2O становилось
равным 1,2 и достигалось pH 10. Интересно отметить, что при
подобных условиях получались частицы размером 16 нм в виде
золя. Требуемая концентрация ионов натрия для того, чтобы
Силикагели и порошки
773
вызвать коагуляцию, не оставалась неизменной, но несколько
возрастала с увеличением размеров частиц. Коагуляция и упроч-
нение достигались затем путем непрерывного добавления СО2
совместно с раствором силиката натрия с целью получения по-
вышенного отношения, равного 4:1. Конечный продукт, состоя-
щий из коалесцированных в высокой степени частиц диаметром
около 26 нм, исследовали под электронным микроскопом при
коэффициенте коалесценции 70% и удельной поверхности
60 м2/г, определенной по методу БЭТ. Такой кремнезем легко
измельчался в каучуке и давал превосходные результаты при
получении резины с высокими показателями прочности на растя-
жение и разрыв и высоким значением модуля упругости.
Метод выделения упрочненных кремнеземных агрегатов ока-
зывается важным. Когда удельная поверхность конечного про-
дукта составляет менее 200 м2/г, кремнезем может быть извле-
чен посредством фильтрации и удаления воды высушиванием
продукта, выделенного из слежавшегося осадка на фильтре.
С другой стороны, в том случае, когда удельная поверхность
продукта оказывается больше ~200 м2/г, предпочитают заме-
щать воду какой-либо другой жидкостью, например спиртом или
кетоном, чтобы получаемая структура не давала сильной усадки
или не уплотнялась при высушивании. Александер, Броудж и
Айлер [273] описали способ приготовления диспергируемого
кремнезема с высокой удельной поверхностью.
Несколько похожий способ для приготовления объемистого,
рыхлого силикагеля, используемого в качестве адсорбента, был
запатентован Маротта [404]. Этот автор первым сформировал
микрогель из кислоты и силиката при рН<6 и температуре
ниже 50°С с последующим упрочнением такого кремнезема пу-
тем добавления дополнительного количества силиката и кис-
лоты при 70—95°С. Высокое значение удельной поверхности, со-
ставлявшее 300—600 м2/г, поддерживалось наряду с высокой по-
ристостью и низкой кажущейся плотностью (0,05—0,10 г/см3,
или 3—7 фунтов на 1 куб. фут). Дальнейшие модификации и
усовершенствования этого общепринятого способа были описаны
в ряде патентов и публикаций [405—412].
Имеются некоторые указания того, что отчасти похожие ре-
зультаты могут быть получены не в горячем растворе, а при
комнатной температуре посредством выдерживания системы по-
сле частичной нейтрализации раствора силиката натрия в тече-
ние времени, необходимого для формирования, агрегирования и
коалесценции частиц. Так, Уосон и Ван-дер-Хеем [413] пред-
ложили кремнезем, имеющий удельную поверхность 100 м2/г и
величину поглощения масла 170 см3 в расчете на 100 см3. Крем-
незем получали подкислением раствора силиката натрия при
комнатной температуре на начальном этапе осаждения, а затем
774
Глава 5
проводили старение образца в течение 15 мин перед заверше-
нием нейтрализации и достижением значения pH 5,8. Похожий
способ был описан в работе [414], за исключением того, что
добавление кислоты прекращалось, когда отношение SiO2: Na2O
находилось в области 5,5—7,0, после чего процесс протекал со
скоростью в соответствии с выведенным уравнением. Осажден-
ный кремнезем получался с удельной поверхностью 100—
600 м2/г, не изменяющейся при высушивании.
Авторы работы [415] привели еще один пример получения
осажденного кремнезема с низкой кажущейся плотностью, по
свойствам напоминавшего аэрогель и оказавшегося очень устой-
чивым по отношению к спеканию вплоть до 1000°С. Раствор си-
ликата натрия 1,08 н. по щелочи, содержавший 10,8% SiO2, ох-
лаждали до 5°С и 40% щелочи медленно нейтрализовали в те-
чение 1 ч добавлением 12,75% H2SO4. Силикатное отношение
SiO2 : Na2O составляло 3,25; таким образом, введенного количе-
ства кислоты было достаточно на данном этапе, чтобы повысить
отношение до 5,4. Следовательно, большая доля кремнезема
должна была превратиться в коллоидное состояние. После этого
в течение 45 мин завершали нейтрализацию и получали кремне-
зем в виде осадка. В данном способе исключалось образование
плотного геля. Частицы кремнезема, выросшие и объединив-
шиеся между собой при нагревании при 95°С в течение 3 ч, про-
мывали последовательно водой, концентрированным раствором
NH4NO3 для удаления оставшегося натрия посредством ионного
обмена, ацетоном для удаления избыточной влаги до содержа-
ния воды менее 1% и, наконец, ацетон удаляли дистилляцией.
Полученный кремнеземный порошок имел удельную поверхность
268 м2/г, объем пор 2,52 см3/г и средний диаметр пор 365 А, при-
чем он оставался неизменным после прогрева в течение 3 ч при
983°С.
Этот способ подробно приводится здесь, так как является,
по-видимому, методом приготовления широкопористого кремне-
зема с низкой кажущейся плотностью, т. е. иным вариантом по
сравнению со способом нейтрализации в горячем растворе, и мо-
жет проводиться при высокой концентрации кремнезема.
Проведение только стадии роста частиц и их коагуляции без
этапа упрочнения дает в результате тонкодисперсный легкий по-
рошок при условии, когда первичные частицы оказываются на-
столько большими, что поры в них достаточно велики и силы,
вызывающие усадку при высушивании, слишком слабы, чтобы
вызвать уплотнение всей массы кремнеземного порошка. Так,
согласно раннему патенту, предложенному Александером, Айде-
ром и Уолтером [416], коллоидные частицы выращивали до-
37 нм в диаметре проведением реакции между силикатом и кис-
лотой в горячем растворе при поддержании концентрации ионов
Силикагели и порошки
775
натрия 0,2—0,27 н. и pH 9,5—10,3. После этого частицы подвер-
гали коагуляции посредством добавления в достаточной степени
разбавленного раствора хлорида кальция, затем их отфильтро-
вывали, промывали и высушивали. Этот легкий порошок под-
вергался диспергированию в каучуке, но не был хорошим уп-
рочняющим реагентом по сравнению с порошком, получаемым
по способу, когда частицы вначале формировались в упрочнен-
ные агрегаты перед процессом высушивания.
Коагуляция солями натрия или аммония
Существует много патентов, в которых предлагается сочета-
ние (кислот с добавлением солей натрия или солей аммония, ко-
гда воздействие оказывают как кислоты, так и соли. Часто при
этом в системе имеется добавочный свободный аммиак. В том
случае, когда реакции протекают при комнатной температуре,
тот факт, что осадки будут состоять из микроскопических частиц
микропористых силикагелей, по-видимому, не вызывает сомне-
ний. Такие силикагели находят незначительное применение в ка-
честве дисперсных наполнителей,, но широко используются для
других целей. Когда же реакции проводятся в горячих раство-
рах, происходит формирование совсем других продуктов и, по-
видимому, получаются некоторые разновидности легкодисперги-
руемых материалов.
Арсем и Райт [417] получили тонкодисперсный кремнезем
путем проведения реакции силиката натрия с хлоридом аммо-
ния, кипячением смеси до образования «кремневой кислоты»
(в результате чего выделялся аммиак), фильтрованием и высу-
шиванием получаемого конечного продукта. После высушивания
при 100°С последний содержал 15% связанной воды, следова-
тельно, должен был представлять собой тонкодисперсный мик-
ропористый силикагель. Хейзер [418] приготовил осажденный
силикагель, имевший большие поры и способный поглощать до
100% по массе и даже более воды из воздуха, насыщенного
влагой. Продукт получался посредством реакции между сили-
катом натрия и солью аммония при определенных условиях и
комнатной температуре. Частицы такого силикагеля оказы-
ваются сферическими по форме и имеют размеры в области от
0,5—5 мкм при значении объемной плотности в интервале 0,16—
0,24 г/см3.
Кремнезем может быть также получен в виде осадка при ре-
акции между силикатом натрия и солью натрия, например суль-
фатом натрия, с последующим подкислением получаемого геля
и промыванием. Приготовленный этим способом [419] гель пред-
ставлял собой скопление большого количества гранул, каждая
из которых была подобна губке, содержащей ультрамикроско-
776
Глава 5
пические поры. Аналогичный способ приготовления в горячем
растворе был разработан Уосоном [420]. Крейг и Киркхем [421]
получили патент на использование для подробной цели бикарбо-
ната натрия.
В другой серии способов основу составляет одновременное
проведение нейтрализации силиката натрия и осажденного
кремнезема солью аммония. Эккер и Санчез [422] приготовили
5%-ный золь кремнезема в аммиаке пропусканием раствора си-
ликата натрия через ионообменную смолу в аммониевой форме
с последующей коагуляцией кремнезема за счет добавления кон-
центрированного раствора соли аммония при pH 8—9,5 и тер-
мического старения в течение 4 ч. Такой способ обеспечивал
получение осажденного кремнезема высокой чистоты с удельной
поверхностью 414 м2/г, объемом пор 1,46 см3/г и диаметром пор
141 А.
Деринг и др. [423] исследовали воздействие NH4CI и NaCI
на процесс коагуляции кремневой кислоты и влияние упрочне-
ния кремнезема на свойства наполнителя для каучука. Эккер
и Виньял [424] приготовили нейтрализацией концентрирован-
ного раствора силиката натрия огрубленный слипшийся крем-
незем с частицами размером 100 мкм, объемом пор 1,75 см3/г
и удельной поверхностью 43 м2/г. По этому способу авторы
в процессе синтеза последовательно добавляли довольно кон-
центрированный раствор аммиака, уксусную кислоту и ацетат
аммония в определенных количествах; полученную смесь нагре-
вали до 90°С в течение 1 ч и затем промывали раствором карбо-
ната аммония для удаления адсорбированного натрия.
При другом варианте способа [425] концентрированный ам-
миак добавляли в качестве агента для коацервации силиката
натрия, после чего силикат частично нейтрализовали до pH
10,6—11,2, осаждали, отфильтровывали и промывали сначала
кислотой, затем водой, высушивали и измельчали в порошок.
В этом случае не проводили старения или нагревания, чем, ве-
роятно, и объясняется высокое значение удельной поверхности
(449 м2/г). Полученные частицы (скорее всего частицы пори-
стого микрогеля) имели размер 50—75 нм и объем пор 2,4 см3/г,
что поясняет низкую величину объемной плотности кремнезема,
равную всего лишь 0,06 г/см3.
Приведенные примеры показывают возможность формирова-
ния частиц пористого микрогеля, которые оказываются коллоид-
ного размера, поэтому усадка при высушивании не происходит.
В некоторых случаях частицы микрогеля все же сохраняют ми-
кропористость в процессе высушивания, образуя таким образом
бипористую систему.
При определенных условиях, однако, подобные микропоры
способны закрываться или уменьшаться в размере, так что из*
Силикагели и порошки
777
мерения, выполненные методом БЭТ по адсорбции азота, реги-
стрируют только внешнюю поверхность частиц микрогеля. Та-
ким образом, подобный продукт может иметь низкое значение
удельной поверхности, в особенности когда процесс проводится
в горячем растворе, а полученный осадок подвергается старению
при высоком значении pH [424].
Коагуляция под действием ионов кальция
Этот процесс лежит в основе получения важной группы раз-
новидностей осажденного кремнезема. Босс [426] описал обра-
зование двух кремнеземистых продуктов, которые оказались
пригодными в качестве армирующих наполнителей в эластоме-
рах и пластмассах. Первым был применен силен, представляю-
щий собой очень тонкодисперсный силикат кальция, приготов-
ленный путем осаждения-силиката натрия, имевшего силикатное
отношение 3,3, совместно с хлоридом кальция при тщательно
контролируемых условиях. Босс отметил, что имевшие до неко-
торой степени щелочный характер первичные частицы диамет-
ром 35 нм образовывали агрегаты при высушивании, что затруд-
няло диспергирование такого продукта при его использовании
в каучуке. Покрытие каждой индивидуальной частицы с целью
предупреждения агрегации представлялось сомнительным, по-
скольку в значительной мере возрастала стоимость при исполь-
зовании органического вещества, необходимого для покрытия
поверхности частиц хотя бы мономолекулярным слоем. Однако
такая проблема была решена путем применения некоторой при-
садки. На этой основе затем был разработан влажный способ
получения слегка гидратированного, тонкодисперсного кремне-
зема при относительно низкой щелочности и с превосходными
характеристиками по отношению к диспергированию. Этот про-
дукт, называемый Hi—Sil, имеет размер частиц 25 нм. С тех
лор еще ни разу не удавалось получить улучшенные разновид-
ности такого продукта с меньшими по размеру частицами. Со-
гласно данным Аллена [427], наилучший кремнеземный напол-
нитель приготовляется посредством осаждения силиката каль-
ция, обработки осадка с помощью NH4C1 (для образования
СаС12) и пропаривания аммиаком. Струи раствора силиката
натрия с силикатным отношением 3,3_ и раствора СаС12—NaCI
смешивали с помощью центробежного насоса, затем добавляли
NH4C1 и кипятили смесь в течение 4 ч для удаления NH3, после
чего ее обрабатывали кислотой НС1 при 30°С в течение 16 ч.
Продукт отфильтровывали, промывали и высушивали. Струк-
тура окончательно получаемого кремнезема в этом случае опре-
деляется стадией кипячения в щелочных условиях.
778
Глава 5
Коагуляция под действием органических веществ
Коагуляцию в растворимой системе силикат натрия—кис-
лота можно проводить различными способами. Добавляемыми
веществами могут быть:
1. Смешиваемые с водой растворители, такие, как ацетон.
При высоком значении pH они вызывают коагуляцию силиката
натрия, и при этом образуется осажденный либо твердый, либо
полужидкий коацерват, способный вступать в реакцию с кис-
лотой.
2. Органические катионные соединения, которые представ-
ляют собой полимерные или поверхностно-активные вещества,
поскольку имеют присоединенные большие углеводородные
группы, способные вызывать коагуляцию силикатных ионов и
частиц кремнезема в пределах широкой области pH.
3. Полярные органические соединения, являющиеся поли-
мерными, например полиэтиленоксид, взаимодействуют с крем-
неземом при низких значениях pH посредством образования
водородных связей, что приводит к формированию осадков.
4. Водорастворимые полярные соединения с низкой молеку-
лярной массой, например пропилтриэтилфосфат или диметил-
формамид, взаимодействующие с кремнеземом при низких зна-
чениях pH. Осажденный продукт получается при добавлении
растворимых солей.
Во всех случаях, если значение pH оказывается менее 11,
кремнезем подвергается полимеризации с образованием тонко-
дисперсного продукта, который может быть отделен от коагу-
лянта.
Коагуляция кремнезема. В кислом растворе коллоидные ча-
стицы кремнезема подвергаются флокуляции при воздействии
полимерных веществ, способных образовывать водородные связи.
Был перепробован большой ряд полимеров (в том числе поли-
виниловый спирт), которые смешивались в условиях водной
среды до или после процесса осаждения с целью получения тон-
кодисперсного кремнезема из раствора при низких значениях pH
[428]. Алифатические амины с длинной цепью вызывают коагу-
ляцию коллоидного кремнезема и приводят к образованию лег-
кого, рыхлого порошка [429]. Катионные ПАВ адсорбируются
на поверхности кремнезема и промотируют процесс коагуляции.
Такой способ, использованный авторами работы [430], дает воз-
можность получать тонкодисперсный чистый кремнезем из вод-
ной среды путем гидролиза этилсиликата аммиаком. Объемная
плотность получаемого кремнезема составляла около 0,1 г/см3.
Осаждение кремнезема в кислом растворе в виде коацервата,
состоящего из маленьких жидких капелек, было описано в гл. 4.
г
Силикагели и порошки
779
Кремнезем, используемый в хроматографических колонках, мо-
жет приготовляться подобным способом [431, 432].
Осажденный кремнезем, имеющий форму листочков и полу-
чаемый флокуляцией коллоидного кремнезема четвертичным ам-
мониевым основанием, с длинной цепью, уже рассматривался
выше [433].
Коагуляция силиката натрия перед его нейтрализацией. При
применении типичного способа, предложенного Мойером [434],
раствор силиката натрия вливался в метанол, в результате чего
образовывалась суспензия, состоящая из тонкодисперсных сфе-
рических частиц силиката натрия. Затем частицы нейтрализо-
вались проведением реакции с кислотой. По этому способу по-
лучались глобулярные частицы силикагеля диаметром около
1—2 мкм с закругленными сглаженными краями. Канхофер
[435] для приготовления небольших по размеру частиц силика-
геля смешивал алкиленполиамин с силикатом натрия перед тем,
как проводить реакцию с кислотой. Получались силикагелевые
частицы размером 5—20 мкм с необычайно низкой кажущейся
плотностью. Вероятно, полиамин воздействует в качестве агента
флокуляции и сводит к минимуму усадку геля.
При другом, отчасти похожем способе агентом выступает та-
кое вещество, которое вызывает коагуляцию раствора силиката
натрия, например спирт, NaCl или NH3. Это вещество добав-
ляется в количестве, достаточном для того, чтобы происходило
помутнение смеси посредством «кластерного эффекта», но не
образовывался осадок. Согласно данным Бейкера и Аустина
[436], при быстром подкислении такой смеси происходит оса-
ждение кремнезема в виде частиц размером 11—22 нм с удель-
ной поверхностью 69—301 м2/г. Продукт подобного типа,- как
известно, представляет собой армирующий наполнитель для
каучука, обладающий уникальными свойствами.
Осаждение кремнезема из фторидного раствора
Коагуляция при воздействии фторид-иона при низких значе-
ниях pH представляется уникальным явлением, очевидно не свя-
занным с процессом коагуляции под действием катионов. Теоре-
тически это объясняется тем, что группы SiOH на поверхности
по крайней мере частично замещаются группами SiF, в резуль-
тате чего поверхность становится гидрофобной. Подобные крем-
неземы частицы затем подвергаются флокуляции под воздейст-
вием гидрофобных связей.
Предложен ряд способов, в которых предусмотрено осажде-
ние кремнезема в присутствии фторид-ионов. Например, Гевекке
[437] впускал пары тетрафторида кремния в горячий раствор
карбоната натрия для того, чтобы получить тонкодисперсный
780
Глава 5
кремнеземный осадок. Алсфельд [438] приготовил в высокой
степени объемистый, рыхлый, тонкодисперсный гидратирован-
ный кремнезем из силиката натрия по реакции с фторидом ам-
мония в растворе гидроксида аммония. Свендсен [439] предло-
жил способ для приготовления тонкодисперсного кремнезема из
кремнеземистых осадков посредством нагревания кремнезема
или силикатов в присутствии фторида аммония до образования
паров, предположительно содержащих диаминотетрафторид
кремния, которые затем гидролизовались в воде. Продукт, со-
держащий только 2% воды и имеющий кажущуюся плотность
0,16—0,22 г/см3, был пластичным и образовывал связанную
массу, которая затвердевала при прокаливании. Продукт про-
зрачен при погружении в масло, но не прозрачен, когда поме-
щается в воду. Он характеризуется как «а-кремнезем». С дру-
гой стороны, при гидролизе таких паров в условиях относи-
тельно низкой температуры в аммиачном растворе получается
вещество, гидратированное до более высокой степени. Оно со-
держит 7% воды при 110°С и 5% Н2О при 250°С. В таком слу-
чае высушенный на воздухе продукт имеет кажущуюся плот-
ность 0,48—0,64 г/см3 и показатель преломления более низкий,
чем для аморфного кремнезема; это связано, по-видимому, с тем,
что продукт гидратирован.
Подобные вещества, вероятно, схожи с продуктами, впервые
описанными Шварцем и Лиде [440]. Авторы сообщили, что пу-
тем гидролиза тетрафторида кремния в кипящей воде обра-
зуются непрозрачные, незастудневающие чешуйки, которые по-
сле восьмисуточного диализа содержали 95% воды и немного
отличались от продукта, полученного гидролизом SiF4 при 0°С,
по внешнему виду или скорости дегидратации. Однако химиче-
ская реакционная способность этих двух продуктов совершенно
различна. Первый продукт, называемый «p-кремнеземом» и при-
готовляемый в кипящей воде, состоит из больших первичных ча-
стиц и в растворе образует полисиликат, полимеризованный до
большей степени. Гидролиз тетрахлорида кремния при 100°С
приводит к образованию продукта, проявляющего все свойства
«а-кремнезема», получаемого гидролизом тетрафторида крем-
ния при 0°С. Джекобсон [441] описал похожий, родственный
продукт, называемый «распушенным кремнеземом». Такой мате-
риал, получаемый гидролизом паров тетрафторида кремния
в воде, оказывается в чрезвычайной степени распушенным,
имеющим кажущуюся объемную плотность всего лишь
0,0248 г/см3, и он настолько легок, что способен течь подобно
воде. Его показатель преломления, равный 1,45, близок к пока-
зателю преломления аморфного кремнезема. Частицы такого
кремнезема состоят из микроскопических чешуек и содержат
92,86% SiO2 и 7,14% воды.
Силикагели и порошки
781
Никерсон и Баркерт [442] гидролизовали 15—25%-ный рас-
твор H2SiF6 аммиаком при pH 6—8 с тем, чтобы непрерывным
способом получать тонкодисперсный кремнезем. Согласно ана-
логичному патенту Борсоса [443], кремнезем образуется в виде
гидратированных частиц размером 30 мкм.
Осаждение кремнезема из органических жидкостей
Кремнезем с низкой объемной плотностью (всего 0,016 г/см3)
и удельной поверхностью 300 м2/г, пригодный в качестве на-
полнителя для каучука, приготовляется посредством осаждения
из спирта кремнезема, полученного из этилсиликата [444]. Так,
по этому способу смесь 6,8 мл (C2HsO)4Si и 20 мл конц. NH4OH
добавляли в 280 мл спирта, где проходила реакция между ними.
Способ, близко связанный с указанным, был разработан Што-
бером [445] для приготовления золей.
Осаждение кремнезема из коллоидных золей
Во всех вышеприведенных способах кремневая кислота под-
вергалась полимеризации почти в одной и той же системе, в ко-
торой затем кремнезем осаждался. Возможно, конечно, начи-
нать процесс, исходя из ранее приготовленного золя кремнезема
с частицами любого коллоидного размера, и вызывать осажде-
ние золя путем флокуляции или коагуляции с тем, чтобы полу-
чить высушенный продукт. Однако, если только не приняты спе-
циальные меры предосторожности и не используются специаль-
ные методы приготовления, получаемый осадок при высушива-
нии дает усадку с образованием прочного силикагеля, особенно
если первичные частицы имеют размер менее 25—50 нм. Для
того чтобы получить легкий, диспергируемый порошок, необхо-
димо либо упрочнять образуемый гель, либо изготовлять крем-
незем, по крайней мере с частично гидрофобной поверхностью,
чтобы свести к минимуму усадку. Способы флокуляции или коа-
гуляции с использованием катионов органических оснований,
имеющих длинные цепи, уже обсуждались ранее. Такие про-
цессы ведут к образованию объемистых, подобных чешуйкам
агрегатов, и по этой причине они были рассмотрены в гл. 4 (см.
рис. 4.21).
Осаждение кремнезема из паровой фазы.
Пирогенный кремнезем
Конденсация безводного пара SiO2, вероятно, имеет иной
механизм и другую кинетику по сравнению с конденсацией
Si(OH)4 в том случае, когда последний образуется при гидро-
782
Глава 5
лизе SiCU или Si(OC2H5)4 в пламени в присутствии паров воды.
Амелин [446] отметил, что при температуре пламени 1270 К
давление пара SiO2 составляет всего лишь 10 8 мм рт. ст., так
что наблюдаемая при этом очень высокая степень пересыщения
ведет к образованию большого числа небольших зародышей
процесса конденсации размером 1—10 нм.
Ульрих [447] в дальнейшем рассмотрел факторы, регулирую-
щие размер частиц в процессе конденсации из пара, и пришел
к заключению, что образованные в самом начале очень неболь-
шие по размеру частицы приобретают поступательные скорости
больших молекул газа, и конечный размер частиц определяется
такими факторами, как столкновение и коалесценция частиц.
Логарифм величины конечного размера частицы пропорциона-
лен логарифму времени роста частицы. Этим объясняется на-
блюдаемое увеличение размера частицы с возрастанием концен-
трации кремнезема; на том же основании можно предположить,
что окончательно сформированная частица будет, вероятно,
иметь микропористую структуру. Однако так как первичные ча-
стицы имеют диаметр только 10—20 А и плотно упакованы, то
внутренние поры конечных частиц обычно оказываются непро-
ницаемыми по отношению к адсорбции молекул азота и обна-
ружить их можно только путем адсорбции воды.
Берби [8] представил электронно-микроскопические снимки
образцов кремнезема, полученных конденсацией из различных
паров, и сопоставил их свойства. Плотность самих частиц ока-
залась 2,2 г/см3, т. е. равной плотности плавленого кварца или
аморфного кремнезема. Несмотря на то что частицы конденси-
руются при температурах, когда кристаллические формы оказы-
ваются стабильными, тшетно строить предположения об их
«кристаллической структуре», поскольку такие частицы, без вся-
кого сомнения, имеют ту же самую структуру, что и кремнезем-
ное стекло, получаемое при резком охлаждении. Дифракция
рентгеновских лучей указывает на стекловидное или аморфное
состояние кремнезема.
Испарение SiO2
Кремнезем может испаряться непосредственно при чрезвы-
чайно высокой температуре в плазменной струе [448—450]. Ко-
гда конденсация происходит в отсутствие водорода, а дисперги-
рование— в масляной среде, такой, например, как дибутилфта-
лат, то кремнеземный порошок оказывается нетиксотропным.
Было признано, что группы SiOH на поверхности частиц необ-
ходимы для удерживания кремнеземных частиц в виде тиксо-
тропной сетки посредством образования водородных связей в том
случае, когда такой кремнезем находится в относительно непо-
Силикагели и порошки
783
лярной жидкой среде. Как было отмечено в гл. 4, дискретные
кремнеземные частицы с полностью этерифицированной поверх-
ностью и не имеющие на ней неприкрытых групп SiOH превос-
ходно диспергируются даже в углеводородных растворителях
при высокой концентрации кремнезема, давая незначительное
повышение вязкости. Полностью обезвоженная поверхность
S1O2 органофильна и гидрофобна, и если такой кремнезем дис-
пергировать в воде при значении pH 2, чтобы задержать про-
цесс гидратации поверхности, то, без сомнения, будет разви-
ваться некоторая степень тиксотропии благодаря образованию
гидрофобных связей, как это отмечалось на других частично
гидрофобных частицах кремнеземного порошка. Хаусман [451]
запатентовал установку с использованием азота в качестве га-
зоразрядной плазмы для испарения кремнезема.
Как описал Берби [8], введение в струю газоразрядной
плазмы какого-либо вещества, приводящего к образованию
групп SiOH на поверхности кремнезема, способствует тому, что
наблюдается тиксотропия кремнезема в маслах. Авторы работы
[452] отметили, что когда такая плазма содержала водород или
аммиак, то шло некоторое восстановление кремнезема, вероятно,
до более летучего SiO; когда последний повторно окислялся,
шло образование конечного продукта с очень высоким значе-
нием удельной поверхности.
Окисление пара SiO
Хотя при очень высокой температуре (около 2000°С) крем-
незем испаряется и его пары конденсируются в виде тонкодис-
персного вещества, процесс испарения можно осуществлять при
гораздо более низкой температуре путем введения восстанови-
теля для образования SiO, который имеет температуру субли-
мации ~ 1700°С. При окислении моноксида кремния повторно
образуется диоксид кремния, который конденсируется в виде
«дыма». Поттер [456] приготовил тонкодисперсный кремнезем
посредством продувания паров моноксида кремния через обо-
греваемую камеру вместе с газом-окислителем. Фон Строх [457]
получил патент на применение полученного таким способом SiO2
в качестве наполнителя для резины. Кроме того, Рейк и Бирнс
[458] (фирма Permanente Metal Corp.) запатентовали способ
приготовления тонкодисперсного кремнезема из моноксида крем-
ния при температуре в области 1250—1450°С.
При использовании углеводородов в качестве «стабилизи-
рующей жидкости» в плазменной горелке, согласно данным Кю-
глера, Сннса и Зильбергера [453], даже более грубые частицы
кремнезема при их подаче в струю восстанавливались до пара
SiO. Эти авторы разработали также конструкцию горелки.
784
Глава 5
Кармен и др. [454] описали электрод с «жидкостной конвек-
цией». Так называемые «кремнеземы дугового разряда» просто
получаются испарением и, по крайней мере частично, превра-
щаются в SiO благодаря присутствию паров углерода, возни-
кающего в дуговом разряде, после чего SiO повторно окис-
ляется. Твердые кремнеземные продукты, получившие наимено-
вание Arc Silicas, представляют собой кремнеземные кластеры
размером 2—3 мкм, состоящие в свою очередь из частиц диа-
метром 15 нм [455].
Был запатентован [459] процесс установления равновесия
пара SiOg, выделившегося из дуговой печи, в которой в резуль-
тате реакции между песком и коксом и окисления пара SiO
до SiOg получаются частицы размером 8—28 нм. Согласно дру-
гому способу [460], песок и кокс испарялись из подобной печи
при 1500—2000°С, газовую смесь СО и SiO смешивали с водя-
ным паром и воздухом с целью охлаждения смеси до 350—400°С
и сконденсированный кремнезем отделяли центрифугированием.
Промежуточное соединение кремнезема затем подвергалось тер-
мической обработке при 200— 700°С в псевдоожиженном слое.
Конечный продукт состоял из кремнеземных частиц размером
5—150 нм и имел удельную поверхность в области 50—300 м2/г
при объемной плотности всего 15—30 г/л (0,015—0,03 г/см3).
Наиболее своеобразная форма тонкодисперсного кремнезема
была описана Неметчеком и Хофманом [461]. SiO приготов-
лялся посредством реакции между Si и SiOg в вакуумных усло-
виях в трубчатой печи при 1200°С. Между реакционной смесью
и SiO, который конденсировался вблизи от нее, по прошествии
48 ч образовывалось множество полых трубочек или волокон,
представляющих собой аморфный кремнезем с диаметром нитей
30—70 нм и толщиной стенок 6—15 нм. Такое вещество в конце
концов закупоривало реакционную трубку.
Окисление и гидролиз паров SiCli
Пары тетрахлорида кремния при высокой температуре могут
либо подвергаться окислению, либо гидролизоваться. SiCl4
можно сжигать в атмосфере кислорода и получать SiO2 и С12
[462]. По-видимому, С12 мог бы быть включен в повторный цикл
с целью получения дополнительного количества SiCl4 из отходов
кремния, сжигаемого в газообразном хлоре. Такое- сгорание
кремния протекает столь же легко, как и сгорание угля в печи.
Однако можно использовать более простое оборудование в том
случае, когда SiCl4 смешивается с СН4 и сжигается с образова-
нием тонкодисперсного SiO2 и НС1. По-видимому, НС1 можно
было бы окислять до С12 для применения в повторном цикличе-
ском процессе, если экономически это целесообразно.
Силикагели и порошки
785
Способ высокотемпературного пламенного гидролиза SiCl4
был запатентован в 1942 г. Клопфнером [463], а соответствую-
щие реакции были изучены Вагнером и Брюннером [464] и
Боде, Фечем и Фрацшером [465]. Некоторые свойства такого
кремнеземного порошка были рассмотрены в обзоре Лофтмана
[466], который дал описание промышленного технологического
процесса. Уайт и Даффи [467] констатировали, что в США
SiCl4 сжигается в смеси с Н2 и О2 по технологии фирмы Godfrey
L. Cabot Corporation, использующей способ, который еще
в 1940 г. был предложен в Германии фирмой Deutsche Gold-und-
Silber Scheideanstalt Vormals Roessler (Degussa). SiCl4 при-
готовляется посредством реакции хлорирования SiC.
Описана [468] аппаратура, в которой можно приготовлять
целый ряд тонкодисперсных оксидов металлов из обезвоженных
хлоридов (Ti, Al, Si, Fe, Zr). Такие оксиды могут быть получены
в виде однородных частиц, диаметр которых лежит в интервале
от 10 нм до 60 мкм. Николина [469] исследовала возможность
применения СН4 вместо Н2 в качестве топлива. Она обнару-
жила, что при температуре пламени 793°С кремнезем имел
удельную поверхность 112 м2/г. Биттнер и др. [470] запатенто-
вали некоторые определенные значения концентраций вступаю-
щих в реакцию паров и скоростей потоков реагентов, посту-
пающих к горелке, необходимых для получения высокой кон-
центрации НС1 при низком содержании С12 с целью достижения
возможности регенерации кислоты. Для получения кремнезем-
ного порошка с высокой удельной поверхностью использовалась
также охлаждаемая горелка с регулируемой заглушкой, что
обеспечивало хорошее предварительное смешивание газообраз-
ных реагентов и понижение температуры пламени до 1000—
1200°С [471]. С другой стороны, в [472] было сообщено, что
продукт с наивысшей удельной поверхностью, равной 160 м2/г,
был получен при температуре всего лишь 690°С и что в течение
последующей обработки с целью удаления НС1 кремнезем на-
чинал «кристаллизоваться» при ~500°С.
Типы кремнезема, приготовляемого по этому способу, обще-
известны как коммерческие продукты под названиями «кабосил»
в США [473] и «аэросил» в Европе [474] *. Структура таких
кремнеземов в достаточной мере была обсуждена в работе
Берби [8], в частности с указанием величины удельной поверх-
ности, которая теряется в местах контактов между первичными
сферическими частицами. Было сделано заключение, что перво-
начально сконденсированные частицы имеют диаметр около
* В Советском Союзе по указанной технологии выпускается несколько
марок аэросилов, а также аэросилов с химически модифицированной поверх-
ностью (на Калушском химико-металлургическом комбинате, УССР).— Прим,
ред.
24 Заказ № 250
786
Глава 5
1 нм. Они настолько плотно упаковываются (высокое значение
координационного числа), что лишь небольшое количество мо-
лекул азота способно проникать в микропоры между подоб-
ными частицами. Таким образом, «вторичные частицы» пред-
ставляют собой частицы, определяющие значение удельной
поверхности и обычно идентифицируемые на электронно-микро-
скопических снимках. Такие частицы можно рассматривать как
«первичные» в объемистой, состоящей из агрегатов рыхлой
структуре, имеющей низкое координационное число, равное -~3.
Такое заключение Берби было основано на исследованиях, про-
веденным Генсом и Кейлом и обсужденных Брукхоффом и Лин-
сеном [475].
Спенсер, Смит и Косман [476] получили чрезвычайно пори-
стые сферические частицы кремнезема, состоящие из агрегиро-
ванных небольших частиц диаметром 25 А, причем удельная по-
верхность такого кремнезема составляла 980 м2/г. Такой мате-
риал был получен путем адсорбции влаги иа поверхности сфери-
ческих гранул, состоящих из газовой сажи, выдерживания э^их
гранул в парах (CH)2SiCI2 при 25°С и последующего выжигания
углерода при 500°С.
Окисление и гидролиз паров сложных
эфиров кремния
Время от времени в литературе появляются сведения о тон-
кодисперсном кремнеземе, аналогичном по характеристикам
кремнезему/ получаемому из SiCl4, который был приготовлен из
сложных эфиров кремния. Такой способ представляет особен-
ный интерес, когда необходим кремнезем очень высокой чи-
стоты, поскольку сложный эфир легче получить в чрезвычайно
чистом состоянии [477]. Был изучен [477] гидролиз паров бу-
тил- и метилсиликатов в присутствии влажного азота при 950°С
и получен кремнезем с частицами диаметром 100 нм. Возможно,
конечно, получать частицы с широким набором размеров, варьи-
руя условия сжигания.
Гидролиз паров SiF4
Вероятно, одной из наиболее важных разработок представ-
ляется производство тонкодисперсного кремнеземного порошка
из пара SiF4, пригодного для применения в качестве наполните-
лей. Эта система оказалась более сложной по сравнению с гид-
ролизом SiCl4, поскольку HF в отличие от НС1 может воздей-
ствовать на кремнезем, изменяя ход реакции на обратный и
образуя дополнительно H2SiF6 и SiF4. Потенциально представ-
ляется возможным получать в большом объеме побочный про-
дукт SiF4 при промышленном производстве фосфатных удобре-
Силикагели и порошки
787
ний, причем такое вещество можно использовать для приготов-
ления кремнезема. Другим практическим вопросом оказывается
то, что при таком способе использование силиката натрия вызы-
вает расходование кислоты и щелочи и ведет к образованию
побочного продукта Na2SO4, вследствие чего возникает проб-
лема его удаления. С другой стороны, при гидролизе SiF4 кис-
лота HF, как это теоретически представляется, могла бы быть
повторно включена в цикл для обработки песка, при которой по-
лучался бы не слишком большой объем побочных продуктов.
К сожалению, подобный вопрос не настолько прост, так как для
регенерации и концентрирования HF, вероятно, всегда будут тре-
боваться дополнительный расход химических реактивов, дорого-
стоящее оборудование и специальные средства техники безопас-
ности на производстве.
Были опубликованы многочисленные патенты, имеющие отно-
шение к этому способу получения кремнезема, в которых пре-
дусматривается много подробностей в отношении регенерации
и повторных циклических этапов гидролиза. Поэтому затрудни-
тельно определить, какой же из предлагаемых вариантов, если
таковой вообще имеется, пригоден для практического использо-
вания [478—480]. Геллер, Уайт и Ганн [481] предложили спо-
соб проведения реакции HF с кристаллическим кремнеземом
(песком) при 120—180°С. Приготовленный SiF4 смешивали
с кислородом и метаном, сжигали полученную смесь в реакторе
с охлаждаемыми стенками и регенерировали SiO2. Согласно
данным Томита [482], распушенный кремнезем в форме волокон
получается в том случае, когда водяной пар и SiF4 вступают
в реакцию на стенках реактора при 500—800°С. Типичные во-
локна имели длину ~5 мм и диаметр 0,5 мкм. Масса получае-
мого продукта имела кажущуюся плотность 8 г/л или 0,008 г/см3.
Согласно заявленному Флеммертом патенту [483], тонкоди-
сперсный SiO2 и кислота HF получаются из смеси SiF4, Н2О и
Н2, сжигаемой при специальных условиях. Дрисколл [484] утвер-
ждает, что кремнезем можно производить прямым вводом SiF4
в пламя в камере, сгорания, конструкцию которой он описы-
вает. Удельная поверхность получаемого продукта находится
в прямой зависимости от концентрации SiF4 в реакционной га-
зовой среде.
В Швеции производится тонкодисперсный кремнезем флуо-
сил [485]. Реакция между SiF4, Н2 и О2 проводится при 1600—
2200°С по способу, защищенному несколькими патентами.
Согласно термодинамическим расчетам Смирновой и др.
[486], в присутствии избыточного содержания Н2О гидролиз
SiF4 с образованием SiO2 и HF может протекать на 99% при
температуре ниже 900°С. Получаемый кремнезем необходимо
удалять из горячего газа при высокой температуре реакции, ис-
24*
788
Глава 5
пользуя керамические фильтры, а кислоту HF из этого кремне-
зема— путем продувания воздухом и водяным паром через
SiO2 при 600°С. Такаги и Сува [487] сообщили, что рассматри-
ваемая реакция должна протекать в области 500—900°С при
величине отношений Н2О : S1F4, равной 4—10. Энергия актива-
ции этого процесса составляла 13 ккал/моль, а удельная поверх-
ность кремнезема менялась в области от 250 до 50 м2/г при
возрастании отношения Н2О : SiF4.
Полное регенерирование K2SiF6 и его превращение в SiF4
при избыточном содержании H2SO4 было описано Зайцевым
и др. [488]. Кроме того, в этой работе были рассмотрены спо-
собы превращения SiF4 в чистые вещества SiO2, SiC и S13N4.
Кремнеземные порошки в природных условиях
Рассмотрение этих веществ в общем выходит за пределы на-
стоящей книги, хотя в некоторых случаях кремнеземные по-
рошки имеют достаточно высокое значение удельной поверхно-
сти и по крайней мере некоторые их фракции могут иметь раз-
меры коллоидных частиц. Ниже упоминается несколько при-
меров.
Кварцевое стекло, получаемое плавлением высококачествен-
ного кварцевого песка, размалывается на шаровой мельнице до
образования тонкодисперсного порошка марки GP-31, в котором
размер 99% всех частиц менее 30 мкм, а 21% из них менее
1 мкм. Из наиболее тонких фракций такого порошка, очевидно,
можно было бы приготовить золь коллоидного аморфного крем-
незема [489]. Подобный порошок, используемый как наполни-
тель эпоксидной смолы, вызывает заметное уменьшение ее тер-
мического расширения.
Частицы кварца коллоидных размеров могут быть получены
из южноиллинойсского (США) трепела, представляющего собой
тонкозернистую осадочную кремнистую породу [490]. Порода
представляется «аморфной» в том смысле, что частицы не имеют
характерных для кристаллов угловых граненых форм,- Микро-
кристаллические частицы имеют средние размеры 1—10 мкм и,
очевидно, ведут свое происхождение от скелетов морских орга-
низмов. Благодаря низкой стоимости такой кремнезем широко
используется как наполнитель [491].
Аморфный, тонкодисперсный кремнезем, напоминающий
аэрогель по некоторым характеристикам, встречается в Японии.
Он сформировался под воздействием природного раствора
H2SO4 на силикаты вулканического происхождения. Аморфный
характер этого кремнезема подтверждается отсутствием типич-
ного рисунка при исследовании методом дифракции рентгенов-
ских лучей, высоким значением удельной поверхности и, нако-
Силикагели и порошки
789
нец, характерной растворимостью в 2 н. растворе NaOH при
100°С [492].
Диатомовая земля, или диатомит, представляет собой встре-
чающуюся в природе форму тонкодисперсного кремнезема, кото-
рый имеет большое практическое значение в промышленности
благодаря удельной поверхности и размеру частиц, приближаю-
щимся к соответствующим величинам для кремнеземов в кол-
лоидном состоянии. Коммерческие продукты из диатомита со-
держат частицы диаметром в несколько микрон, но они обла-
дают характеристической тонкопористой структурой скелета,
которая позволяет получить удельную поверхность около 20 м2/г
[493]. Содержание кремнезема в таком материале доходит до
94%. Кремнезем состоит из скелетных «раковин» — оболочек
большого множества микроскопических водяных одноклеточных
водорослей, по форме напоминающих диск или овал. Эти водо-
росли, известные как диатомеи, обычно состоят из одиночных
клеток, окруженных двумя створками, каждая из которых напо-
ловину закрывает клетку и состоит из прозрачного кремнезема
толщиной примерно в одну десятитысячную миллиметра. Такие
створки имеют тонкую структуру из полостей и разделяющих их
перегородок, напоминающих кружева, которые отличаются чрез-
вычайной красотой и сложностью .узора. В живых организмах
и в недавно образовавшихся отложениях кремнезем оказывается
аморфным, но поскольку возраст большей части природных от-
ложений диатомовой земли составляет миллионы лет, то крем-
незем становится в ней микрокристаллическим (см. рис. 7.2).
Очень тонкодисперсный аморфный кремнезем, содержащий
небольшое количество углерода, может быть получен прокали-
ванием рисовой шелухи при контролируемых условиях. Сооб-
щается, что такой кремнезем оказывается хорошим армирую-
щим наполнителем для резины, конкурентоспособным с терми-
ческими углеродными черными сажами [494].
Имеется высококремнеземистый, подвергшийся изменениям и
состоящий из вулканического пепла продукт, который оказы-
вается настолько реакционноспособным по отношению к извести,
что образует основной компонент цемента, используемого еще
со времен Римской империи. Айлер провел исследование об-
разца такого продукта, взятого из подлинного вулканического
отложения в Поццуоли, близ Неаполя (Италия), и обнаружил,
что продукт представляет собой очень тонкодисперсный, аморф-
ный кремнеземный порошок с величиной удельной поверхности
более 100 м2/г. Этот природный кремнезем обсуждался в гл. 4
в связи с рассмотрением вопроса об использовании коллоидного
кремнезема в цементах.
По-видимому, большинство аморфных форм кремнезема, об-
разующихся в природных условиях, в течение миллиона лет
790
Глава 5
кристаллизуются до микрокристаллического кварца и, таким
образом, становятся гораздо менее растворимыми и менее реак-
ционноспособными.
Микрокристаллические гидратированные
типы кремнезема
Порошки,такого типа неизменно приготовлялись из кристал-
логидратов солей металлов, большей частью из солей щелочных
металлов или комплексных солей, посредством удаления катио-
нов кислотой. Подобные кристаллические полисиликаты рас-
сматривались в гл. 2, а кристаллические гидратированные крем-
неземы— в гл. 1. До сих пор такие типы кремнезема редко ис-
следовались и не представляют практического интереса.
После дегидратации подобные кристаллы становятся субми-
кропористыми, так что в них проникает вода и небольшие по
размеру ионы. Они имеют слоистую структуру, которая в неко-
торых типах кремнезема способна расширяться и сжиматься
в процессе ионного обмена или в процессе гидратации и деги-
дратации, что напоминает аналогичные эффекты, наблюдаемые
в глинистых слоистых минералах. Таким образом, слоистые
структуры отличаются от цеолитоподобных структур алюмоси-
ликатов, имеющих жесткую, трехмерную решетку. На рис. 5.24
представлен микроснимок необычных чрезвычайно тонких ли-
сточков кремнезема, полученных из синтетического полисили-
ката натрия [495].
Гидрофобные, органофильные кремнеземные
порошки
Рассмотрение данного вопроса ограничивается порошками
кремнезема, которые подвергаются поверхностному модифици-
рованию в результате образования хемосорбированных молекул,
глубоко изменяющему адсорбционные характеристики или
способность к диспергированию кремнеземных частиц в жидких
системах. Подробное рассмотрение химии поверхности представ-
лено в гл. 6.
Интерес, проявляемый к гидрофобным, органофильным дис-
персным кремнеземам примерно с середины настоящего столе-
тия, был стимулирован несколькими очевидными практическими
потребностями: 1) необходимостью использования армирующего
наполнителя не черного цвета для каучука, идущего на изготов-
ление резиновых покрышек с боковой поверхностью белого
цвета; 2) разработкой загустителей масла для приготовления
консистентных смазок, превосходящих по своим свойствам такие
Силикагели и порошки
791
Рис. 5.24. Микрофотография кристаллического гидратированного кремнезема
(из работы Айлера [495]).
широко используемые присадки, как кальциевые мыла; 3) созда-
нием эффективных армирующих наполнителей для силиконового
каучука, который в то время разрабатывался. Более того, целый
ряд используемых в настоящее время способов приготовления
тонкодисперсных образцов кремнезема в тот период находились
на стадии разработки и одновременно изучались вопросы моди-
фицирования поверхности кремнезема.
По различным причинам выпуск некоторых из таких гидро-
фобных типов кремнезема в настоящее время прекращен, дру-
гие еще производятся, а также созданы новые типы. Однако, по
мере того как стоимость углеводородов и получаемых углерод-
ных черных саж возрастает, в конечном счете может возобно-
виться интерес к гидрофобному кремнезему, способному ча-
стично, если не полностью, заменить в крупнотоннажном произ-
водстве черную газовую сажу, применяемую в настоящее время
как существенный компонент в резиновых покрышках. Прово-
792
Глава 5
димые в прошлом исследования ограничивались лишь модифи-
цированием поверхности кремнезема органическими соедине-
ниями, но не затрагивали проблем размеров частиц, степени
формирования сетки, составленной из этих частиц, и оптималь-
ного значения коалесценции, необходимого для упрочнения си-
стемы при использовании подобного кремнезема. Кроме того,
вполне вероятно, что для каждого определенного вида эласто-
мера требуется индивидуальная степень покрытия поверхности
и оптимальный тип поверхностных органических групп.
Наибольшее распространение получили пять типов поверхно-
стей кремнезема, модифицированных следующими способами:
1) адсорбированными органическими катионами или полика-
тионами;
2) адсорбированными многозарядными ионами металлов
с присоединенными органическими кислотными группами (ме-
таллические мыла);
3) этерифицированными спиртами, образующими с поверх-
ностными силанольными группами покрытие, имеющее груп’пы
SiOR;
4) полимерными органическими покрытиями различных
типов;
5) поверхностными группами Si—О—SiRs или подобными,
многократно связанными группами (SiCOaSiRa, которые обра-
зуются при реакции поверхностных силанольных групп с крем-
нийорганическими промежуточными соединениями.
Только последний тип поверхностной модификации, как ока-
залось, нашел наиболее широкое применение, вероятно, вслед-
ствие простоты использования и стабильности образующейся
модифицированной поверхности. Однако имеет смысл рассмот-
реть все типы таких модифицированных поверхностей.
Поверхности с адсорбированными органическими
катионами
Адсорбция на поверхности кремнезема органических катио-
нов, таких, как замещенные аммониевые ионы с длинными це-
пями, применяется при отделении кремнезема методом флотации
от руд [496]. Например, в случае когда хлоргидрат додецила-
мина добавляется к суспензии тонко размолотого известняка,
содержащего кремнезем, додециламмониевые ионы образуют ад-
сорбированный слой на кремнеземе. Благодаря этому кремнезем
становится гидрофобным и флотируется при барботировании
воздуха через смесь.
Органические катионы с длинными цепями удовлетворяют
всем требованиям, необходимым для возникновения сильной ад-
Силикагели и порошки
793
сорбции. Полярная концевая группа молекулы заряжена поло-
жительно, поэтому в нейтральном или в щелочном растворе она
притягивается к отрицательно заряженной поверхности кремне-
зема и одновременно гидрофобные углеводородные цепи моле-
кулы стремятся освободиться от воды и присоединиться друг
к другу, формируя адсорбированный монослой. На основании
этого общего принципа были запатентованы многочисленные
практические способы. Так, полианионы полиэтилена, смешан-
ные с органическими длинноцепочечными кислотами, образуют
соединения, в которых реализуется отмеченный принцип [497].
Айлер [489] получил патент на тонкодисперсный кремнезем, по-
крытый азотсодержащими органическими соединениями, напри-
мер длинноцепочечными аминами и четвертичными аммоние-
выми соединениями.
При применении тонкодисперсного кремнезема в качестве
загустителя масла при приготовлении консистентной смазки до-
бавление длинноцепочечного амина [499] или четвертичных ам-
мониевых катионов [500, 501] улучшает стабильность системы
в присутствии воды.
Катион соли четвертичного аммониевого основания с длин-
ной цепью, которая растворима в масле, может использоваться
для сохранения органофильности и гидрофобности кремнезема
при его измельчении в масляной среде в процессе приготовления
консистентной смазки. Было сообщено [502] об использовании
соли карбаминовой кислоты
У°
R'R'NC-N+R1R2R3R4
\о
где R с разными индексами — атом водорода и углеводородные
группы, имеющие от 1 до 16 атомов углерода, причем по край-
ней мере одна из таких групп R, присоединенная к атому азота,
имеет длинную цепь. Нет сомнения, что существует множество
комбинаций ионосодержащих аммониевых солей с длинными це-
пями, которые могли бы быть использованы для образования
гидрофобных покрытий на поверхности кремнезема. Однако по-
добные системы имеют два недостатка. Прежде всего молеку-
лярная масса соединения такого типа оказывается слишком
большой в случае тонкодисперсных образцов кремнезема с ве-
личиной удельной поверхности 200—300 м2/г. Поэтому модифи-
цирующий поверхность кремнезема продукт имеет очень
высокое содержание относительно дорогостоящего органического
вещества. Последнее, кроме того, термически не всегда устой-
чиво. Применение подобного способа для превращения поверх-
ности кремнеземов в гидрофобное состояние, по-видимому, оста-
нется ограниченным.
794
Глава 5
Поверхности с адсорбированными многозарядными
катионами металлов и органическими анионами
Стабилизация консистентной смазки, загущенной кремнезе-
мом, в присутствии основного стеарата алюминия, вероятно,
предусматривает адсорбцию алюминия на поверхности кремне-
зема с присоединенными анионами жирной кислоты [503]. Крем-
неземные наполнители для эластомеров улучшают свои свой-
ства, если поверхность кремнезема превращается в органофиль-
ную с помощью основных комплексов хрома типа
где RCOO-— метакрилат-ион или олеат-ион [504]. Вполне ве-
роятно, что могли бы быть предложены и другие органофильные
покрытия типа металл — карбоксилатный ион.
Этерификация поверхности
В классе гидрофобных силикагелей и кремнеземных порош-
ков, заявленном в патенте Айлера [505], предусматривается хи-
мическое присоединение мономолекулярного слоя из первичных
или вторичных алкоксигрупп, содержащих от 2 до 18 атомов
углерода, к поверхности кремнезема. Поскольку такая поверх-
ность оказывается покрытой группами Si—OR, аналогичными по
своим связям сложным кремневым эфирам, то получаемые про-
дукты получили название «эстерсилы». Большинство завершен-
ных покрытий наносится путем нагревания кремнезема вместе
со спиртом в отсутствие избыточной влаги в течение 1 ч при
190°С для первичных спиртов или же при температуре вплоть
до 275°С для вторичных спиртов. Для частичных покрытий крем-
незема оказываются вполне эффективными гораздо более про-
стые способы обработки. Структура самого кремнезема остается
неизменной при этерификации поверхности, но конечный продукт
становится органофильным и гидрофобным. Когда образец эс-
терсила нагревается в атмосфере кислорода при 500°С с целью
удаления покрытия, затем повторно увлажняется путем погру-
жения в воду и вновь высушивается, то получающийся в таком
случае кремнезем невозможно отличить от исходного материала.
Силикагели и порошки
795
Для спиртов с нормальной неразветвленной цепью, таких, на-
пример, как «-бутиловый спирт, требуется примерно три эфир-
ных группы, чтобы покрыть площадь в 1 нм2. Это соответствует
площади 30—35 А2, занимаемой каждой бутильной группой.
Группы OR химически связаны и не могут десорбироваться в ус-
ловиях высокого вакуума при 150°С. Полное завершение покры-
тия поверхности подтверждается тем, что получаемый продукт
не адсорбирует краситель метиловый красный из раствора бен-
зола, тогда как исходный гидрофильный кремнезем с силаноль-
ной поверхностью адсорбирует мономолекулярный слой такого
красителя. Эстерсилы приготовлялись из различных кремнезе-
мистых материалов, включая аэрогели, тонкодисперсные по-
рошки и тонкодисперсные или волокнистые силикатные мине-
ралы после их обработки кислотой.
Особый вид эстерсила, наиболее перспективный для практи-
ческого использования, приготовлялся из золя кремнезема с ча-
стицами размером ~8 нм путем последующего застудневания
золя при pH 5,5, упрочнения структуры полученного геля нагре-
ванием при pH 5,5 и повторного упрочнения структуры геля
либо дополнительным нагреванием, либо осаждением некоторого
количества кремнезема. После этого кремнезем переводят
в среду «-бутилового спирта посредством азеотропной дистилля-
ции до тех пор, пока вся вода не будет удалена. Последующее
нагревание проводится в автоклаве для завершения процесса
этерификации поверхности, чтобы поверхность кремнезема ока-
залась полностью покрытой бутоксигруппами и снаружи не ос-
тавалось ни одной группы SiOH. Такой высушенный продукт
представлял собой чрезвычайно легкий, с низкой кажущейся
плотностью порошок, подобный аэрогелю, за исключением того,
что был полностью гидрофобным и органофильным, имел вели-
чину удельной поверхности в интервале 275—325 м2/г и обладал
способностью полностью диспергироваться в кремнийорганиче-
ских эластомерах, маслах, красках и т. д. Беллард и др. [506]
описали этот способ модифицирования с некоторыми подробно-
стями. Усадка такого геля предотвращалась посредством нагре-
вания смеси воды и н-бутилового спирта выше критической тем-
пературы растворения и последующим выпариванием, когда
происходит фракционная отгонка воды [507а].
.Частично гидрофобный и органофильный кремнезем можно
приготовить очень просто путем использования метода, запатен-
тованного Гобелем [5076]. Тонкодисперсный кремнеземный по-
рошок дегидратировался, а его поверхность дегидроксилирова
лась до оптимальной степени путем нагревания кремнезема в те-
чение 1,5 ч на воздухе при 470—530°С. Удельная поверхность,
измеренная методом БЭТ, составила 297 м2/г, но окончательно
величина удельной гидроксилированной поверхности этого же
796
Глава 5
образца оказалась равной только 184 м2/г. Когда такой порошок
охлаждали, а затем повторно разжижали при 118°С в бутиловом
спирте при атмосферном давлении или даже просто подвергали
его действию паров бутанола в закрытом объеме в течение не-
дели, то происходила этерификация порошка, и его поверхность
примерно наполовину покрывалась присоединенными бутокси-
группами.
На полностью этерифицированной поверхности «-бутокси-
группы оказываются достаточно устойчивыми. Например, об-
разцы подобного порошка, которые закапывались в землю и на-
ходились там в течение года, оставались после этого еще сухими
и пылили, даже если находились, под дождем. В качестве загу-
стителя консистентной смазки, используемой в горнодобываю-
щем оборудовании, такой продукт проявил очень высокую стой-
кость против деструкции при воздействии воды. Однако при
продолжительном выдерживании в воде при высокой темпера-
туре эстерсильное покрытие не может конкурировать с покры-
тием из метилсилильных групп, поскольку последнее не подвер-
жено гидролизу.
Был изучен целый ряд групп сложных эфиров. Некоторые из
них обсуждались в гл. 4 в связи с рассмотрением вопроса об
органозолях. Имеется много групп общего типа SiOR, где R мо-
жет а) представлять собой нитро-, хлор- или алкоксиалкил
[508]; б) содержать замещенные кислоты, такие, как меркапто-,
феноло-, фосфо- или карбаминовые кислоты [509]; в) содер-
жать фторуглеродные группы, например, ОСН2(Ср2)иХ, где X —
атом Н или Рип 1 —17 [510]; г) представлять собой группы
ORN+ и ORS+, где N и S входят в состав «ониевых» ионов;
д) входить в состав группы OROH, причем в данном случае R —
это двухатомная углеводородная группа, такая например как
гликоль [512], ненасыщенная группа [513] или амино-
группа [514].
Броудж обнаружил, что больше всего на поверхности может
располагаться метоксигрупп по сравнению с какими-либо дру-
гими типами групп (вплоть до 5,8 метоксигруппа/нм2) в про-
цессе проведения модифицирования при достаточно высоких
температуре и давлении и в безводных условиях с целью полу-
чения гидрофобной поверхности [515]. Впоследствии эстерсилы
с метоксигруппами привлекли внимание ряда исследователей.
Так, Чертов и др. [516] в качестве этерифицирующего реагента
использовали диазометан. Баверез и Бастик [517] обнаружили,
что в том случае, когда поверхность кремнезема была вначале
частично дегидроксилирована при высокой температуре, метило-
вый спирт СН3ОН вступал в реакцию как с оставшимися на по-
верхности группами SiOH, так и, по-видимому, раскрывал по-
верхностные группы SiOSi [517].
Силикагели и порошки
797
Китахара и Асано [518] исследовали систему кремнезем—
метиловый спирт в широкой области вплоть до высоких темпе-
ратур и давлений и обнаружили, что на этерифицированной по-
верхности содержится до 5 метоксигрупп/нм2. Они также выпол-
нили аналогичные исследования с высшими спиртами.
Этерифицированная поверхность в случае пирогенного крем-,
незема может получаться путем его обработки в парах
(C4H9O)4Si при температуре вплоть до 300°С, но без контакта
с воздухом с добавлением некоторого количества сухого ам-
миака в качестве катализатора [519].
Следует также обратиться к главам 4 и 6, в которых пред-
ставлена библиография по этерификации поверхностей коллоид-
ного кремнезема, силикагелей и кремнеземных порошков.
Кремнийорганические покрытия
В ранее применявшемся для гидрофобизации поверхности
кремнезема методе нужно было смешивать метилсиликонат нат-
рия с силикатом натрия перед проведением подкисления [520].
Слинякова и Куренная [521] приготовили силикагели с различ-
ными относительными количествами поверхностных групп
СН2=СН—Si и изучили адсорбционные свойства таких образ-
цов. Силиконовое масло может адсорбироваться на поверхности
кремнезема и вступать с ней в реакцию с выделением тепла
[522, 523]. Монослойное покрытие, состоящее из метилсилиль-
ных групп, наносилось на тонкодисперсный кремнезем посредст-
вом реакции с паром (CHs^SiCh [524] или с аминосиланами
[525]. Аэрогели с покрытием из кремнийорганических промежу-
' точных соединений оказываются гидрофобными и не дают
усадку при погружении образцов в воду и последующем высу-
шивании, но усадка появляется, когда образцы погружаются
в спирт, и последний смачивает поры [526]. Покрытие из крем-
нийорганического полимера может наноситься на кремнезем
[527, 528]. Гидрофобное покрытие может быть нанесено на
кремнезем в водной суспензии посредством проведения реакции
с CH3Si(OH)3, получаемом из сложного этилового эфира или из
соли натрия [529]. Силиконовая консистентная смазка приготов-
лялась замещением воды в силикагеле на способный смеши-
ваться с водой растворитель, а затем замещением растворителя
на диметилсилоксановую разновидность силиконового масла
[530].
Описываются многочисленные разновидности способов, ис-
пользуемых для гидрофобизации пирогенных кремнеземов по-
средством их обработки парами кремнийорганических соедине-
ний. Как довольно давно уже показал Кистлер [531] на
примере обработки аэрогелей, алкилхлорсиланы в смеси с ам-
798
Глава 5
миаком вступают в реакцию с поверхностными группами SiOH.
Однако при проведении такой реакции, по-видимому, в продукте
сохраняются остаточные количества хлоридов [531]. В предло-
женных более позже патентах используются методы нанесения
гидрофобных покрытий, в которых не содержатся нежелатель-
ные остаточные вещества. Пирогенный кремнезем можно гидро-
фобизировать путем его обработки силазаном, таким, на-
пример, как R3SiNH(SiR2NH)nSiR3, где R — алкильная
группа с числом углеродных атомов от Ci до С6 и п 0—3.
Фактически, как правило, используется гексаметилдисилазан
(CH3)3SiNHSi(CH3)3 [532, 533]. При его реакции с группами
SiOH на поверхности образуются группы SiOSi(CH3)3 и аммиак
NH3. Такая реакция протекает полностью, т. е. вплоть до со-
стояния, пока вся поверхность кремнезема не покроется ука-
занными группами в течение нескольких минут при комнатной
температуре [534].
При другом подходе проводится реакция между поверх-
ностью кремнезема и HSiCl3, а затем реакция образовавшихся
поверхностных групп SiH с этиленом или бутадиеном, которые
присоединяются своими алкильными группами к поверхности
[535].
Буеч и Оливер [536] выдерживали в контакте с парами си-
лана, вызывающими органофилизацию кремнеземной поверхно-
сти, свежеобразованный кремнезем в области температур 150—
500°С по мере того, как такой образец подвергался охлаждению
[536]. Обработка кремнезема парами (СН3)3SiO (СН3) и родст-
венными соединениями с добавлением 1% катализатора амин-
ного типа, такого, как бутиламин, может проводиться в замкну-
том контейнере в течение двух недель при комнатной темпера-
туре, для того чтобы получить поверхностное покрытие хорошего
качества. Получаемый модифицированный кремнезем находит
применение в полисилоксановых эластомерах в качестве арми-
рующего наполнителя [537].
Унгер, Берг и Галлей [538] изучали термическую стабиль-
ность монослойного покрытия CgHsSiEs на кремнеземе. При ре-
акции с СбН3—SiCl3 на поверхности образуются группы
CeH5Si(Cl)=, а затем С1 удаляется в результате гидролиза.
Размер пор кремнезема может оцениваться посредством изме-
рения степени, до которой поры меньшего размера закупори-
ваются таким реагентом. Большое число подобных реакций мо-
дифицирования кремнеземных поверхностей было выполнено
в связи с необходимостью получения набивочного материала для
хроматографических колонок, играющего роль адсорбентов и
носителей. Эти вопросы рассматриваются ниже..
Кремнеземы, несущие на поверхности органические группы,
имеют свойства, совершенно отличающиеся от свойств немоди-
Силикагели и порошки
799
фицированных кремнеземов. Бурвелл [539] рассмотрел в своем
обзоре исследования, в которых получались покрытия поверх-
ности кремнезема различными типами органических групп, на-
пример аминогруппами, способными в свою очередь адсорбиро-
вать атомы меди за счет образования координационной связи,
а также, адсорбировать БЮг и СОг, как это можно было пред-
видеть для случая поверхности, покрытой аминогруппами и
имеющей основной характер. Разнообразен набор других типов
органических групп, способных присоединяться к атомам крем-
ния на поверхности через углеродные цепи. Группы в соедине-
ниях, способные вызывать каталитическое воздействие в случае
однородного раствора, сохраняют свой каталитический эффект,
когда эти группы удерживаются на кремнеземной поверхности
и располагаются на ней так, что они направлены кнаружи от по-
верхности:
«Поверхность кремнезема представляет собой подобие удобной доски,
утыканной гвоздиками, к которым может быть присоединен целый ряд раз-
личных групп, что оказывается очень полезным при превращении гомоген-
ных катализаторов в гетерогенные и при приготовлении новых типов катали-
заторов» (Бурвелл).
Органические полимерные покрытия
Паддингтон и Сирианни разработали органические полимер-
ные покрытия. Кремнеземы приготовлялись органофильными,
если полимерные покрытия представляли собой полистирол,
льняное масло и алкидную смолу [540, 541]. Уэлде [542] зая-
вил о получении полимерного покрытия как продукта реакции
полиизоцианата и многоатомного спирта (глицерина) на поверх-
ности кремнезема. Согласно данным Эккера, Санчеза и Крамма
[543], гидрофобный полимер валерианового альдегида форми-
руется на поверхности кремнезема, когда исходный кремнезем
нагревается вместе с альдегидом при температуре 200°С, при из-
быточном давлении 2,5 кг/см2 в течение 4 ч. Реакция поверхно-
сти кремнезема с карбодиимидом, таким, например, как цикло-
гексанкарбодиимид СбН11К=С=1ЧСбН11, ведет к образованию
органофильной поверхности [544], которую, вероятно, можно
представить как изомочевину. Согласно данным Эккера [545],
нагревание кремнезема с фурфуролом под давлением при тем-
пературе 250°С в течение 4 ч приводит к отложению на кремне-
земе органофильной поверхностной пленки.
Силикагели с ионообменными поверхностями
Чистая поверхность кремнезема представляется настолько
слабокислотной, что в нейтральном растворе наблюдается
только лишь слабая степень обмена однозарядных катионов на
800
Глава 5
ионы водорода поверхностных групп SiOH. К тому же ионный
обмен, затрагивающий многозарядные ионы металлов, услож-
няется вследствие образования ковалентных связей между ато-
мами металла и атомами кислорода поверхностных групп SiOH,
так что обратимый обмен затрудняется или даже оказывается
невозможным, в том случае, когда адсорбируются такие ионы,
как Сп2+ или Со2+.
Поверхность кремнезема переходит в гораздо более сильное
кислотное состояние с возрастанием катионообменной способ-
ности, составляющей примерно один катионный заряд в рас-
чете на 1 нм2, посредством обработки силикагеля или кремне-
земного порошка в растворе NaA102 при значении pH 9—10.
В этом случае происходит формирование алюмосиликатных цен-
тров, в которых атом алюминия координирован с четырьмя ато-
мами кислорода, как и атом кремния, с образованием на поверх-
ности отрицательного заряда:
I < NaAIO, I 0 „
—О—Si—ОН --------> —О—А1—ОН Na®
Ионообменные и каталитические свойства алюмосиликатных
центров в Н+-форме рассматриваются ниже в гл. 6. Так назы-
ваемые «синтетические цеолиты», т. е. гели, приготовляемые пу-
тем реакции между силикатом и алюминатом натрия, сохраняю-
щие натриевую форму, были использованы для воды, но оказа-
лись нестабильными при низких значениях pH и, в общем, не
пригодными для катионного обмена.
Более стабильные ионообменники, образуемые на кремнезем-
ном носителе, подразделяются на два класса: модификации с не-
органической поверхностью и кремнийорганические модифика-
ции, когда поверхность покрывается органическими ионообмен-
ными группами, удерживаемыми на поверхности благодаря воз-
никновению связей углерод—кремний.
Неорганические ионообменные центры
Когда на поверхности кремнезема адсорбируется оксид тория
или оксид циркония, вероятно, в виде многоосновных катионов
или чрезвычайно небольших по размеру, положительно заря-
женных коллоидных окисных частиц, тогда поверхность стано-
вится положительно заряженной и может вести себя как анионо-
обменник. Такая поверхность в свою очередь необратимо адсор-
бирует фосфатные ионы; подобные поверхностные анионы
Силикагели и порошки
801
в таком случае могут вести себя как обменные центры для ка-
тионов:
s'-°x /Л О
/ \ Na®
о (У ^03
На самом деле структура поверхности, конечно, оказывается
более сложной, чем это представлено на схеме. Согласно дан-
ным Каутского и Весслау [546], катионообменник подобного
типа, полученный на основе кремнезема, можно было бы исполь-
зовать для выделения таких ионов металлов, как Ag+, Cu2+, Fe2+
и Са2+. Йетс [547] описал ионообменные поверхности этого типа.
Он приготовлял коллоидные фосфаты Ti3+, Zr4+, Sn4+, Hf4+ и Ce3+,
которые адсорбировались на поверхности кремнезема. Преиму-
ществом приготовления ионообменника на основе кремнезема
по сравнению с ионообменником, полностью состоящим из фос-
фата поливалентного металла, является то, что кремнезем гора-
здо легче сформировать со структурой, обладающей широкими
порами и высоким значением удельной поверхности, что необ-
ходимо для получения эффективных эксплуатационных качеств.
Кремнезем берется за основу для аммониевого молибдено-
фосфатного обменника, который может использоваться для ре-
генерирования ионов цезия из кислого раствора [548, 549]. Ка-
летка и Конечны [550] также использовали силикагель в каче-
стве подложки, на которую наносили нерастворимые соединения
гексацианоферратов никеля, меди, кадмия или цинка, способные
действовать как катионообменники. Они могут удалять следовые
количества ионов цезия даже из раствора сильной азотной кис-
лоты. Авторы измерили обменную способность таких различных
соединений.
Органические связанные ионообменные центры
В настоящее время представляется, что любой тип ионной
или хелатной органической группы может быть присоединен
к поверхности силикагеля с открытыми широкими порами или
к поверхности кремнеземного порошка для того, чтобы получить
пригодные ионообменные свойства. Унгер [6] обобщил различ-
ные способы, посредством которых на поверхности пористого
кремнезема можно образовать связи Si—С. Этот вопрос будет
обсужден ниже в гл. 7. Здесь же приводится рассмотрение
только специфических ионообменников данного типа.
Егоров и др. [551] прививали стирол, акриловую кислоту и
винилфосфиновую кислоту к поверхности кремнезема посредст-
25 Заказ № 250
802
Глава 5
вом облучения газовой фазы рентгеновскими лучами и быст-
рыми электронами; и затем ионные группы вводили на поверх-
ность в результате сульфирования, хлорметилирования и амини-
рования. Обменная емкость таких ионообменников составляла
1—2 мг-экв/г.
Унгер и Берг [552] обработали силикагель CeHsSiCh и затем
сульфировали фенильные группы, чтобы получить катионооб-
менник с сульфогруппами на поверхности, емкость которого
была равна 0,46 мг-экв/г. Унгер, Берг и Наймах [553, 554а]
в дальнейшем описали свойства и возможное использование
таких ионообменных порошков, полученных на основе кремне-
зема, в хроматографии. Чтобы приготовить анионообменник, по-
ристый кремнезем с удельной поверхностью 600 м2/г орошали
раствором (3-хлорпропил) трихлорсилана в ксилоле и высуши-
вали, после чего 3-хлоргруппы, образовавшиеся на поверхности
кремнезема, вступали в реакцию с раствором дибутиламина
в толуоле при 140°С. Такой продукт имел обменную емкость
0,5 мг-экв/г [5546]. Силикагель, содержащий аминогруппы, по-
глощал сухой газ СО2 из воздуха, но для возможности адсорб-
ции H2S и SO2 в воздухе должна содержаться влага [554в].
Аминопропильные группы также можно прививать к поверхно-
сти кремнезема посредством реакции с (3-аминопропил)триэто-
ксисиланом [555а]. При использовании продукта в качестве ад-
сорбента хелатного типа с целью улавливания ионов меди по-
верхностные аминогруппы вводят в реакцию с /г-нитробензоил-
хлоридом, затем образовавшиеся группы NO2 восстанавливают
дитионатом до групп NH2. После этого /г-аминобензиловые
группы подвергают диазотированию и реакции с 8-гидрооксихи-
нолином, представляющим собой вещество, способное образовы-
вать хелатные соединения. Полученный обменник имеет темно-
красный цвет и способен поглощать катионы тяжелых металлов
вплоть до чрезвычайно низких концентраций.
Упоминается об обменнике, называемом «кремнелактат», ко-
торый фактически может быть получен посредством этерифици-
рования поверхности молочной кислотой с образованием групп
8ЮСН(СНз)СООН. Силикагель или кремнеземный порошок
вначале нагревают до 230°С с целью дегидратации, а затем под-
вергают воздействию молочной кислотой в автоклаве при тем-
пературе 150°С. Катионная обменная емкость полученного про-
дукта составляет 0,6 мг-экв/г [556].
Другой способ создания ионообменных центров, предложен-
ный Павлик [5556], заключается в проведении реакции поверх-
ности кремнезема с Cl2Si (СН3) CH2CH2CN с образованием по-
крытия, состоящего из групп SiOSi (Cl)СНзСН2СН2СМ, способ-
ных гидролизоваться до групп СООН. В работе встречается
утверждение, что гидроксильные группы лимонной или молоч-
Силикагели и порошки
803
ной кислоты, находящейся в парообразном состоянии, способны
этерифицировать поверхностные группы SiOH с образованием
присоединенных групп СООН. Однако стабильность подобных
покрытий вызывает сомнения, если только они не используются
в ионообменных процессах в неводных хроматографических си-
стемах.
Коммерческие силикагели и кремнеземные порошки
Любой перечень коммерческих продуктов быстро становится
устаревшим, но дает по крайней мере некоторые представления
о том, как много разновидностей кремнезема, обсуждаемых
с теоретических позиций, оказываются практически важными.
Такие коммерческие материалы приведены в табл. 5.7 наряду
с их основными свойствами. Не представляется возможным об-
судить область применения каждого продукта или класса крем-
незема, поскольку во многих случаях практического использо-
вания кремнеземные продукты, относящиеся к разным катего-
риям, оказываются конкурентоспособными между собой. Хотя
такие продукты сгруппированы отчасти в соответствии со спо-
собом их изготовления, нельзя четко провести различие, напри-
мер, между осажденными кремнеземами и тонкодисперсными
силикагелями, поскольку их свойства могут оказаться подоб-
ными. Вообще говоря, если данный кремнезем рекомендуется
в качестве армирующего наполнителя для эластомеров, для ко-
торых является существенным, чтобы кремнезем был способен
диспергировать до частиц, близких по размерам к первичным
частицам, то, вероятно, выбранный кремнезем не следует клас-
сифицировать как силикагель. Некоторые кремнеземные мате-
риалы не являются в настоящее время доступными, но они
включены в таблицу, так как упоминались прежде в литературе.
Применение силикагелей и кремнеземных порошков
Давно установившиеся, традиционные применения силикаге-
лей и порошков были рассмотрены в обзоре Вейла [557]
в 1952 г. Наиболее распространено применение силикагелей
в гранулированной или шариковой форме, по-видимому, в каче-
стве основных катализаторов и как адсорбентов или осуши-
телей.
На основании опубликованной научной, патентной литера-
туры, а также литературы производственных фирм использова-
ние силикагелей и кремнеземных порошков можно сгруппиро-
вать в соответствии с их следующими назначениями:
Упрочнение, загущение и отверждение органических твердых
веществ.
25*
Таблица 5.7
Коммерческие силикагели и кремнеземные порошки
Фирма- изготовитель Продукт Удельная поверхность, измеренная по методу БЭТ, м2/ г Поглощение масла, г/г Объемная а плотность, г/л Примечание
Типы кремнезема, полученные осаждением
из раствора силиката натрия
PPG Industries Ловел 143 1,6 —
Ltd. (США) Хисил ЕР 60 1,8 —
Хисил 422 45 1,0 280
Хисил 233 150 1,5 160
Хисил (Orig) 100 — 112 Содержал 3 % СаО
Bayer A. G. Силкосил С 80 1,6 192
(ФРГ) Силкосил S 150-200 2,0 176
Degussa (ФРГ) Дуросил 55 1,7 128
VN2 120 2,5 160
VN3 200 2,4 160
K320DS 230 2,1 50
FK300DS 350 2,5 85
FK700 700 1,6 280
Hoesch (ФРГ) KS160 60 2,3 160
KS300 125 2,4 176
KS404 140 — 176
Ketjen NV (Гол- Кетьенсил 195 3,0 176
ландия)
Pechiney (St. Go- Левелит 380 2,0 —
bain) (Франция) MIS-412 -
Si-France (Фран- Тиксосил 38 90 2,8 —
ция)
Philadelphia
Quartz Со.
(США)
J. М. Huber
(США)
Кусо
Н-40
G-32
G-30
F-22
F-20
WR-50
WR-82
Цеосил
100
110SD
Бюллетень QUSO 18-1/5/75
200 1,8 160 Гидрофильный
300 2,5 64 и
300 2,5 160
325 2,6 64
325 2,6 160
130 — 160 »»
120 — 160
125-150 1,9 100-120
125-150 1,9 100-120
Ксерогели (гидрогели), аэрогели
Davison Chem. Гранулированные Промышленный бюллетень фирмы
Div. (W. R. Grace силикагели «Дэвисон», 1977 г. 12 марок, раз-
Corp.) (США) личающихся по размерам частиц
Марки Объем пор, см3/г
Номера от 01 до 44 720—760 700 0,43
81 600 590 0,70
407—408 720-760 750 —
923—950 700 735 0,40
Силоид
Марки Размеры частиц, мкм
60, AL-1 675 0,6 460 9
72 340 2,2 176 4
74 320 2,0 255 8
80 340 2,6 112 5,6
161, 162 175 1,55 240 7-9
166 220 2,3 112 4
Серия 200 270—320 3,0-3,1 80-112 2-4
Фирма-изготовитель Продукт Удельная поверхность, измеренная по методу БЭТ, м2/г Поглощение масла, г/г Объемная а плотность, г/л Примечание
Серия 300 250 2,5 160190 3,7-7
Glidden Pigments 600 Силкрон - 95 1,8 335 20 Размер агрегированных частиц, мкм
(США) G-100r 275 2,7 112 3
G-130® 300 2,2 96 3
G-300 = 300 1,9 160 4,2
G-310B 300 1,9 160 5
G-500 «•в 200 1,75 192 5
G-5106’B 225 1,8 208 6
G-520 в 200 1,75 192 5
G-530 в 225 1,8 208 6
G-550 в' в 200 1,75 192 5
G-600г 325 2,1 176 4,7
G-601г 325 2,1 176 6,5
G-602 г 320 2,05 176 7,1
G-603г 320 2,05 176 10,1
G-604г 320 2,05 176 12,0
G-900 г 625 0,8 400 8,5
G-910r 700 0,9 400 9,0
Типы осажденного кремнезема, полученные из раствора силиката натрия
Monsanto Chem. Сантосел Спецификация Monsanto Dev. Dec.
Со. (США) 21, 1950
С 260 280
54 270 280
FR-C 350 280
62 350 280
68 350 280
Degussa (ФРГ) Аэросил Бюллетень фирмы «Дегуеса»
R-972 120 + 30 32-48 № 301-0366 Гидрофобный (Бюлл. 3002)
МОХ-170 я 170 + 30 40—56
МОХ-80 я 80 + 15 40-80
МОХ-60 « 60+ 15 56-100
СОК-84 150 ± 25 56-100
ОХ-50 ТТ-600 2491-380 50+ Ю 200 + 50 380 ± 40 40-56 40-56 40-56 14 + 2 °/о AI2O3
2491 300 + 30 40-56
Аэросил С е 200 + 25 125
Аэросил UC е 200 ± 25 40—56
Godfrey L. Cabot Corp. (США) Кабосил М-5 200 + 25 37
MS-7 200 + 25 60—80
MS-75 255 + 15 60-80
HS-5 325 + 15 37
ЕН-5 325 + 40 3-7
S-17 400 + 20 60-80
Nynas-Petrolenm (Швеция) Флуосил 200U ж 220 + 25 40+ 10
300U ж 300 + 25 40 + 10
200U ж 100 + 20 100 + 20
ЗООС3 300 + 30 100 + 20
Н 190 + 25 100 + 20 Гидрофобный; 90 % SiOa
250С 260 + 25 100 + 20
250CNG 240 + 25 80+ 15 Сорт, не образующий глянца
а 160 г/л=10 фунт/фут3.
б Примерно 10—20 % потери массы при прокаливании.
В Поверхность подвергалась обработке.
г Поверхность не подвергалась обработке.
Д МОХ — смешанный оксид, содержащий 0,3—1,3 % АЬОз-
е С — прессованный, UC — непрессовапный.
ж и — непрессованный.
з С — прессованный.
808
Глава 5
Понижение адгезии между поверхностями твердых веществ.
Повышение адгезии клеев.
Повышение вязкости и тиксотропии в жидкостях.
Создание разнообразных оптических эффектов.
Получение эффектов, связанных с изменением поверхност-
ного состояния.
Создание гидрофобных эффектов.
Применение в качестве поглотителей.
Основные катализаторы.
Источник получения реакционноспособного кремнезема.
Образование ядер конденсации в облаках.
Наполнение хроматографических колонок.
Кресс [558] обобщил основные области применения, а Тей-
чер [559] перечислил характерные кремнеземные аэрогели и
ксерогели и их использование. В данной книге не делается ника-
ких попыток представить обзор по всей литературе, имеющей
отношение к кремнеземным катализаторам или адсорбентам,
или же достаточно полно рассмотреть какую-либо одну область
применения, упоминавшуюся выше. Взамен этого обсуждаются
некоторые из действующих механизмов и отмечаются вопросы,
представляющие технический интерес.
Упрочняющие и другие воздействия в органических
твердых веществах
Резина
В качестве армирующего наполнителя в наибольшем мас-
штабе применяется углеродная черная сажа. Но сейчас уже на-
блюдается тенденция частичной или полной замены углеродной
сажи кремнеземом, о чем свидетельствует развитие производ-
ства тонкодисперсного кремнезема, способного придавать такие
же свойства вулканизированной резине. Одним из примеров
подобного наполнителя является кремнезем, имеющий удельную
поверхность 60 м2/г, величину поглощения масла 1,8 см3/г и, сле-
довательно, обладающий очень открытой сетчатой структурой,
что дает возможность легко диспергировать такой кремнезем
путем его измельчения на мельнице. Это обеспечивает возмож-
ность изготовления упругих прочных резиновых изделий, имею-
щих цвета, отличающиеся от черного [560]. Типичный анализ
такого продукта выявляет присутствие в нем 1,7% NaCl и 0,8%
СаО, показывая тем самым, что он, вероятно, является осажден-
ным кремнеземом, содержащим некоторое количество адсорби-
рованных ионов кальция. Размер частиц, равный 40 нм, оказы-
вается большим, чем диаметр частиц 22 нм кремнезема, исполь-
зуемого ранее в качестве армирующего наполнителя [561]. Это
можно объяснить наличием улучшенных динамических свойств
Силикагели и порошки
809
такой резины и более быстрым процессом вулканизации вслед-
ствие пониженной адсорбции ускорителя этого процесса.
Из-за возрастания важности применения кремнеземов в ка-
честве армирующих наполнителей ниже будут рассмотрены бо-
лее подробно, в частности, некоторые сведения из опубликован-
ной литературы, так как большая доля информации оказывается
в основном недоступной, за исключением патентной литературы.
Примерно в 1959 г. в статьях Вагнера и Селлерса [562] и
Бахмана и др. [563] были исчерпывающим образом рассмот-
рены упрочняющие эффекты, вызываемые кремнеземами различ-
ных типов. Свойства были перечислены для осажденных и пиро-
генных кремнеземов и для аэрогелей, бывших доступными в то
время, наряду с рассмотрением обширной библиографии. Было
показано, что количество «связанного каучука», отнесенное
к единице массы наполнителя, характеризует армирующее дей-
ствие последнего. Такой наполнитель вводится в измельченном
виде в каучук при отсутствии каких-либо других присадок, и
смесь нагревается при различных температурах. Растворимый
компонент каучука после этого экстрагируется растворителем.
Связанный каучук формируется по механизму образования сво-
бодных радикалов.
Гесслер и Ренер [564] и Де Франческо и др. [565] исследо-
вали действие дегидратации поверхности кремнезема и нанесен-
ных на кремнезем органических покрытий на упрочнение полу-
чаемой резины.
Обзор по механизмам упрочнения и воздействиям, оказывае-
мым широким набором наполнителей на физические свойства
эластомеров, дополненный 47 библиографическими ссылками,
был опубликован Смитом [566а]. Салвадор [5666] исследовал
эффекты замещения некоторой доли углеродной сажи на крем-
незем в природном каучуке. Полное замещение дает более низ-
кие свойства, но при соблюдении соотношения 15SiC>2: 35С на-
блюдалось усиление величин относительного разрывного удли-
нения и раздира, а также термического старения, однако при
этом понизились модуль и упругость материала.
Эстерсилы, кремнеземы с этерифицированной поверхностью,
особенно подробно были изучены как наполнители и запатенто-
ваны Айлером [567]. Такие кремнеземы могли подвергаться дис-
пергированию в разнообразных эластомерах, даже если частицы
кремнезема достигали в диаметре всего лишь 5—7 нм. Приго-
товлялись кремнеземы с частицами разных размеров, которые
имели различные степени сетчатого строения или плотности упа-
ковки первичных частиц, составляющих агрегаты. Сравнивались
упрочняющие свойства кремнеземов при наличии гидрофобного
и органофильного покрытия поверхности, состоящего из буто-
ксигрупп, или же в отсутствие подобного покрытия.
810
Глава 5
В табл. 5.8 суммированы данные, взятые из указанного па-
тента. Испытания проводились с природным каучуком при со-
ставлении смеси, равной 60 ч. по массе наполнителя на 100 ч.
каучука, с добавлением 3 % серы и оптимальных количеств
бензотиазил- и тиурамдисульфида М, взятых в качестве ускори-
телей вулканизации. При этом наблюдалось оптимальное время
вулканизации с достижением, как правило, наивысшей прочно-
сти резины на раздир.
Для каждого данного типа кремнезема сравнение неэтерифи-
цированной гидрофильной поверхности кремнезема с соответст-
вующей этерифицированной поверхностью (н-бутильные группы)
показывает, что в случае А—С этерификация повышала полу-
чающиеся прочности на растяжение и на раздир, причем наи-
меньшие по размеру частицы давали наибольший эффект. Слу-
чаи А и В схожи по структуре получаемого материала и по
армирующему действию кремнеземов. Этерифицированный
кремнезем С дает более высокие значения прочностей на растя-
жение и на раздир и более высокую твердость, но, однако,-при-
водит к более низкому модулю, чем это имеет место для вулка-
низированного каучука с наполнителем из черной сажи марки
EPC Black. D представляет собой легкодиспергируемый напол-
нитель со структурой низкой плотности или низкой степенью
сетчатого строения, что доказывается по пониженному значению
объемной плотности, когда материал спрессовывался при давле-
нии в интервале 0,2—105 кг/см2. Диспергирование ведет к очень
низкому значению модуля, подобного тому, который получается
в отсутствие сетчатого строения из дискретных, неагрегирован-
ных этерифицированных частиц (см. табл. 5.9). Образец, упроч-
ненный углеродной сажей марки ЕРС, включен в табл. 5.8 для
сравнения.
В случае Е кремнеземный аэрогель придает заметно лучшие
свойства резине при этерифицировании поверхности н-бутило-
вым спиртом. В случае F демонстрируется, что этерификация
первичными спиртами со все более длинными цепями не ведет
к заметному изменению упрочняющих свойств, которые остаются
все же лучшими по сравнению с использованием неэтерифици-
рованного кремнезема.
В случаях G и Н частицами являются коллоидные волокна
с этерифицированными кремнеземными поверхностями. Наибо-
лее заметным оказывается эффект, проявляемый вследствие
анизометрии формы частицы: в сильной степени вытянутые ча-
стицы приводят к появлению очень высокого значения модуля.
Фактически все зарегистрированные свойства, получаемые для
этерифицированного, обработанного кислотой глинистого мине-
рала— аттапульгита, оказываются сходными со свойствами ма-
териала при наполнении его углеродной сажей марки ЕРС.
Силикагели и порошки
811
В параллельно выполненных дополнительных исследованиях и
наблюдениях были подтверждены приведенные выше резуль-
таты. На природном каучуке были испытаны некоторый интер-
вал размеров кремнеземных частиц, сетчатое строение или сте-
пень формирования цепочечной структуры, а также степень
покрытия силанольной поверхности кремнезема группами «-бу-
тилового эфира при оптимальном содержании ускорителей вул-
канизации и времени вулканизации. Этерификация поверхности
ведет к снижению требуемого количества таких ускорителей.
Действие степени этерификации или обратной ей величины —
доли поверхности, которая остается гидрофильной и полярной,
на упрочняющие свойства определялось следующим образом:
отбирался такой кремнеземный порошок, который имел струк-
туру, позволявшую полностью диспергировать этот порошок
в случае, когда он вводился в каучук. Такой кремнеземный по-
рошок, осушенный ацетоном, был полностью гидрофильным. При
орошении порошка «-бутиловым спиртом этерифицировалась
только часть его поверхности. После измельчения порошка по-
лярные участки поверхности кремнезема состояли из неэтери-
фицированных силанольных групп, а также из «голых» пятен,
образованных при отрыве друг от друга кремнеземных частиц,
связанных в сетчатую структуру. В случае другого образца по-
верхность кремнезема, имевшего сетчатое строение, полностью
этерифицировалась путем автоклавной обработки в среде бута-
нола. После введения измельченного кремнезема в каучук по-
лярными участками поверхности были только «голые» пятна,
образовавшиеся в местах разрыва частиц. Наконец, в случае
четвертого образца некоторое количество ранее этерифицирован-
ного кремнезема, имевшего сетчатое строение, подвергали по-
молу на шаровой мельнице в среде бутилового спирта и затем
повторно обрабатывали в автоклаве. Образовавшиеся в резуль-
тате частицы имели очень небольшую долю полярной поверх-
ности от всей поверхности кремнезема даже после диспергиро-
вания образца в каучуке.
Как показано в табл. 5.9, полярные или гидрофильные уча-
стки поверхности на кремнеземных частицах (соответствующие
окисленным участкам поверхности на частицах углеродной
сажи) способствуют загущению материала. Такое действие пол-
ностью аналогично загущению масла при получении консистент-
ной смазки с кремнеземом такого типа. При хорошей способно-
сти к диспергированию и при фиксированном размере кремне-
земных частиц этерификация поверхности кремнезема не
приводит к какому-либо воздействию на прочность при растяже-
нии или на прочность при раздире. Однако поскольку твердость
и значение модуля оказались меньше для полностью этерифи-
цированных частиц, то разрывное удлинение было большим, так
Таблица 5.8
Характеристики эстерсилов как армирующих наполнителей и армированных резин (по данным Дилера [569])
Эстерсил иа £ О <Я 5- = * Ч 0> (П . 5о« >>Е S Объемная плотность при сетчатой структуре, г/см3 Тип поверхности, степень этерификации Удельная поверхность красителя, м2/г Прочность, кг/см2 Разрывное удлинение, % Модуль, кг/см2 (300 %) Серповидный раздир, кг/см Твердость по Шору
под нагрузкой
0,2 кг/см2 | 110 кг/см2
A SiO2 осажденный а 97 0,160 0,492 Гидрофильная 97 180 460 105 18 73
S1O2 этерифицирован- ный 97 0,160 0,492 95 % 6 5 235 540 105 113 70
В Хисил 100 0,175 0,441 Гидрофильная 99 115 540 50 21 55
Хисил этерифициро- ванный 100 0,175 0,441 90 % 6 9 230 540 115 104 72
С SiO2 основной 335 0,13 — Гидрофильная 208 180 520 100 30 87
SiOa этерифицирован- ный 277 0,13 — 100 % 6 0 320 700 75 160 82
D SiO2 осажденный 144 0,10 0,64 Гидрофильная 144 300 660 80 102 70
SiO2 этерифицирован- ный 144 0,10 0,64 100 % 6 0 295 725 50 151 65
ЕРС черный — — — — — 280 480 165 86 72
Эстерсил я о ® * Ч о и С4 И Ч О >> Е S Объемная плотность при сетчатой структуре, г/см3 Тип поверхности, степень этерификации Удельная поверхность красителя, м2/г Прочность, кг / см2 Разрывное удлинение, % Модуль, кг/см2 (300 %) Серповидный раздир, кг/ см Твердость по Шору
под нагрузкой
0,2 кг/см2 | НО кг/см2
Е Аэрогель «Сантосел С» 157 — — Гидрофильная 114 220 580 85 27 86
Этерифицированный 157 — — 80 % 6 25 320 620 105 140 80
F SiO2 осажденный 160 — — Гидрофильная 160 190 410 130 35 84
SiO2 этерифицирован- ный 138 0,18 — 86 % 6 19 285 520 155 144 77
138 0,18 __ 67 % в 45 275 540 135 95 76
138 0,18 — 75 % г 34 290 540 155 143 78
G Хризотил-асбест эте- рифицированный 125 — — 100 о/о 6 0 240 330 225 76 77
Н Аттапульгит этерифи- цированный 157 57 % 6 67 280 440 200 100 71
а Из патента США 2657149.
б Этерифицирована м-бутильными группами.
в Этерифицирована изопропильными группами.
Этерифицирована изоамильными группами.
814
Глава 5
, Таблица 5.9
Характеристики резин, армированных тонкодисперсным кремнеземом
с группами SiOBu на поверхности
Кремнеземный наполнитель
Неэтерифицированный 200 490 1,0 115 89 34
Частично этерифициро- ванный перед диспер- гированием 315 670 2,1 95 87 125
Полностью этерифициро- ваиный перед диспер- гированием 345 730 2,5 75 78,5 125
Полностью ванный, и: на шаровс Полностью рованный вие сетчат туры Диаметр частиц, нм >терифициро- шельченный й мельнице »терифици- в отсутст- ой струк- Удельная поверхность, м2/г 345 760 2,6 60 71,5 125
25 110 280 740 2,1 38 56 116
17 160 310 815 2,5 32 65 155
10 275 350 800 2,8 40 72 155
что истинное значение прочности на растяжение, отнесенное
к единице поперечного сечения при разрыве (пропорционально
Т-Е), оказывается наивысшим для полностью этерифицирован-
ных частиц.
Для того чтобы совершенно исключить влияние полярных по-
верхностных групп и сетчатого строения частиц, золи, имевшие
однородные кремнеземные частицы, превращали в органозоли
посредством их этерификации еще в состоянии золя, описанной
в гл. 4. Как показано в табл. 5.9, понижение размеров частиц
ведет к возрастанию прочности на растяжение и разрывного
удлинения наряду с низким значением модуля, но еще при высо-
кой прочности на раздир. Для большинства видов наполнителей
(которые, как правило, содержат долю полярной поверхности)
подобные небольшие по размеру частицы обычно способствуют
получению очень твердых образцов вулканизированной резины
и высокого значения модуля благодаря связыванию частиц на-
полнителя через полярные участки их поверхностей.
Силикагели и порошки
815
Получающееся в итоге представление об упрочнении резины
состоит в том, что прочности на растяжение и на раздир и об-
щая величина жесткости заметно повышаются в том случае, ко-
гда очень небольшие частицы наполнителя (диаметром 5—
10 нм) полностью диспергированы и находятся в виде разде-
ленных, дискретных частиц внутри матрицы. Для получения
хорошего диспергирования подобные небольшие по размеру ча-
стицы должны быть, вероятно, гидрофобными. Когда же на ча-
стицах наполнителя имеются полярные и гидрофильные участки
поверхности, то такие частицы стремятся собраться вместе в виде
цепочки благодаря связыванию подобных участков, которые не
могут смачиваться углеводородным веществом матрицы эласто-
мера. Это придает жесткость структуре резины, т. е. ведет к по-
вышению значений модуля и твердости.
Сможет или нет кремнеземный наполнитель в конечном счете
заменить углеродную сажу в протекторах покрышек, где расхо-
дуется основное количество углеродной сажи, будет зависеть от
разработки необходимой адекватной износостойкости покрышек.
Будет ли необходим в этом случае гидрофобный кремнезем, за-
висит от того, насколько низкой будет стоимость получения эте-
рифицированной поверхности кремнезема в сравнении с покры-
тием кремнезема органосилильного типа, которое оказывается
достаточно устойчивым при подобном использовании. Замена
углеродной сажи на кремнезем представляется относительно не-
сложной проблемой, так как, согласно обширному обзору Ваг-
нера и др. [563], кремнезем и углеродная сажа, имеющие близ-
кие по размеру частицы, обеспечивают одинаковую степень
упрочнения, когда с кремнеземом используются подходящие,
способные с ними соединяться реагенты. В крупномасштабном
деле по изготовлению покрышек замена некоторой доли угле-
родной сажи на кремнезем привела бы к улучшению профили-
рования протекторов и к понижению отходов производства. В по-
крышках, используемых для легковых автомобилей, вероятно,
при оптимальном подборе нужных композиций можно достичь
повышения их износостойкости.
Силиконовые эластомеры
Тонкодисперсный кремнезем несомненно является необходи-
мым компонентом для силиконовых эластомеров. Вероятно, что
подобный требуемый тип кремнезема стал высокоспециализиро-
ванным, но в настоящее время никакой информации не имеется.
В более раннем патенте Буече [568] кремнезем с этерифициро-
ванной поверхностью использовался в качестве реагента, осу-
ществляющего поперечную связь между цепями в органополи-
S16
Глава 5
силоксановом эластомере. Согласно данным Кидвелла [569],
материал Estersil марки Varlon [570] оказался значительно
лучшим по сравнению с пирогенным кремнеземом, который
имел близкие по размеру частицы, для целей упрочнения метил-
фенилвинилполисилоксанового эластомера, образуя в результате
вулканизат с величиной прочности на растяжение вплоть до
91 кг/см2 и разрывным удлинением, равным 880%.
Как сообщили Зеликин и др. [571], гидрофобные пирогенные
кремнеземы были применены в качестве армирующих наполни-
телей в силиконовом каучуке, давая значения прочности на ра-
стяжение 71 кг/см2 и разрывного удлинения 460%. Однако уп-
рочнение этого типа эластомера, вероятно, в настоящее время
значительно улучшено благодаря новым способам введения ча-
стиц кремнезема с оптимальными характеристиками.
Разнообразные органические полимеры
Покрытый кремнийорганическим соединением кремнезем дает
превосходное упрочнение в случае трифторхлорэтиленвинило-
вого сополимера [572]. Олефины сополимеризовались с кремне-
земом, покрытым непредельными алифатическими углеводород-
ными группами [573]. Введением эстерсила в полиэтилен проч-
ность на разрыв в поперечном направлении по отношению
к усилию, прикладываемому в машине для испытания на раз-
рыв, повышают на 50% без потери других свойств [574]. Этери-
фицированный кремнезем с метильными поверхностными груп-
пами подвергался измельчению в полиэтиленовом прессовоч-
ном порошке, давая тонкодиспергированные частицы при
10%-ном наполнении смеси. Подобная дисперсная система была
получена в полиметилметакрилате [575]. С модифицированной
(этерифицированной) диэтиленгликолем поверхностью кремне-
зем оказывался более хорошо смешиваемым и совместимым
с полиэфирными смолами, а также мог применяться в качестве
наполнителя мебельного лака, используемого при нанесении по-
следнего верхнего слоя [576].
Волокнистый SiO2, приготовленный окислением SiO для уп-
рочнения полиуретановой смолы с образованием поперечных
связей между цепями за счет введения 1,5-нафталиндиизоциа-
ната, образует материал, обладающий прочностью на растяже-
ние 490 кг/см2, модулем, равным 135 кг/см2, и разрывным удли-
нением 700 % [577].
За последние десять лет, по-видимому, уменьшилось ежегод-
ное число литературных ссылок, имеющих отношение к упроч-
нению полимеров с использованием кремнезема. Трудно сказать,
вызвано ли это тем, что данная тема уже широко исследована
с неутешительными результатами, или же тем, что пригодные
Силикагели и порошки
817
новые типы кремнеземов оказались недоступными для проведе-
ния испытаний в различных полимерах. Для дальнейшего про-
движения вперед могут потребоваться совместные усилия по
экспериментальному подбору типов кремнезема с различными
размерами частиц, структурами и характеристиками поверхно-
стей, которые могут быть использованы в наиболее важных по-
лимерах.
Очень разнообразно использование кремнеземов при приго-
товлении микропористых полимеров. Посредством включения
кремнезема в мономер, применяемый для получения ионообмен-
ных смол, дополнительную пористость получают последующим
растворением и удалением кремнезема путем воздействия раз-
бавленной кислотой HF [578].
Кремнезем вводился в виде этилсиликата в мономер, взятый
в качестве предварительной исходной смеси, необходимой для
получения полиэфирных смол [579]. Вода, освобождаемая
в процессе конденсационной полимеризации, например, при при-
готовлении полиэтиленсукцината гидролизует сложный эфир
с выделением кремнезема в основном в виде сополимера. Если
такой кремнезем удаляется, то это должно привести к образо-
ванию чрезвычайно тонких пор.
Стана и Калдервуд [580] при изучении мембран из ацетата
целлюлозы, применявшихся для обратного осмоса, обнаружили,
что мембраны, содержавшие примерно 0,6% кремнезема, состо-
явшего из частиц размером 7 нм, нормально функционировали
без каких-либо изменений в течение 56 сут, тогда как мембраны,
не содержавшие кремнезема, понижали свою пропускную спо-
собность на 50% через 17 сут. При содержании кремнезема,
равном 50 объемн. %, получается в пять раз более высокая
пропускная способность, чем при использовании мембраны из
ацетата целлюлозы с низкой проницаемостью без кремнезема
[581].
Резина, покрытая тканью, которая оказывается в высокой
степени стойкой против прохождения жидкой воды, но в то же
время легко пропускает водяные пары, может быть изготовлена
посредством покрытия ткани латексной пленкой, смешанной
с гидрофобным кремнеземным аэрогелем, для обеспечения пори-
стости пленки [582].
Лед способен упрочняться за счет введения гидрофильного
кремнезема. Так, Нейар, Ленель и Анселл [583] обнаружили,
что когда во льду присутствует 0,5—1 объемн. % однородно дис-
пергированных кремнеземных частиц, то скорость процесса пла-
стической деформации такого льда под давлением оказывается
в 10—30 раз медленнее, чем в случае чистого льда.
Полиэтиленоксидгликоли превращаются из воскообразного
. состояния в прочное твердое вещество посредством включения
26 Заказ № 250
818
Глава 5
тонкопористого кремнезема, который способен связываться че-
рез водородные связи с эфирными группами и, следовательно,
осуществлять поперечную связь между цепями полимера. Этот
эффект используется при формировании стенок капсул, в кото-
рые помещаются различные материалы при капсулирова-
нии [584].
Пониженная адгезия
Меры против слипания листового материала или для предот-
вращения адгезии между слоями липких материалов в течение
длительного времени включали применение порошков талька
или крахмала. Но некоторые кремнеземные порошки обладают
чрезвычайно небольшими по размеру частицами и низким зна-
чением показателя преломления, что делает их для подобных
целей более эффективными и менее заметными. Слеживание по-
рошков или гранул, перемещающихся или рекристаллизующихся
при хранении, также предотвращается добавлением кремнезема,
который является нетоксичным и в основном инертным. Клей-
кость, или липкость, поверхности является проблемой того же
типа, что и склонность свеженанесенных красок или чернил да-
вать отпечатки. Очевидно, небольшие кремнеземные агрегаты
способны прилипать к поверхности и поглощать следовые коли-
чества жидкой фазы или осадка на такой пластичной поверхно-
сти, повышая ее твердость или вязкость и создавая «осушаю-
щий» эффект. Кроме того, при измельчении порошков небольшое
количество тонкодисперсного кремнезема во взятой порции об-
разует тонкую пленку на свежеобразованных поверхностях ча-
стиц измельченного порошка, удерживая частицы от контакта,
позволяя им повторно объединяться, если частицы снова подвер-
гаются уплотнению.
Покрытие, получаемое в результате погружения кремнезем-
ного аэрогеля в силиконовое масло, дает возможность дисперги-
ровать такой кремнезем в воде с образованием на поверхности
частиц органосилановых групп. Это позволяет предотвратить
адгезию при подогревании пищевых продуктов, помещенных
в бумажные пакеты, выдаваемые на авиалайнерах, когда такие
пакеты подвергаются нагреву [585].
Эффект, предупреждающий слипание полимерных пленок,
очевидно, может быть достигнут добавлением тонкодисперсного
кремнезема к перемешиваемым мономерам перед их полимери-
зацией. Хоппе и Бен [586] заявили, что приблизительно 0,5%
кремнезема, нанесенного на полимер, способствует понижению
адгезии на 50% • Природа такой пленки, содержащей кремнезем,
исследовалась посредством сканирующего электронного микро-
скопа, и было обнаружено, что полимер, окружающий выпук-
Силикагели и порошки
819
лость, которую создает частица кремнезема, оказывается менее
растворимым вблизи частицы. Такое искажение полимерной
пленки наблюдалось посредством колец Ньютона в отраженном
свете [587].
Существует множество применений кремнезема, в которых
предусматривается использование эффекта антислеживания по-
рошков. Типичным примером является применение пирогенного
кремнезема с целью предупреждения слеживания порошков,
предназначенных против потения, при их распылении пульвери-
затором в виде аэрозолей из емкостей [588], а также исполь-
зование лекарственных порошков — дустов, которые распы-
ляются над стаей птиц [589].
Повышенная адгезия
Имеются примеры, когда адгезия не понижается, а наоборот,
возрастает, т. е. когда частицы кремнезема находятся в системе
не в виде слабо связанных агрегатов, но в виде диспергирован-
ных частиц в среде клеящего вещества — адгезива, которое от-
верждается при контакте с твердой поверхностью. Такие крем-
неземные частицы оказывают положительное воздействие, если
они сильно адсорбируются на границе раздела твердое тело —
жидкая среда. В таком случае в действительности частицы уве-
личивают площадь твердой поверхности на границе раздела,
с которой затем связывается клей. Например, добавление 10%
пирогенного кремнезема в жидкую среду — бутилцианакрилат
вызывает повышение прочности и адгезии системы, применяемой
в хирургии, когда подобный адгезив полимеризуется и связы-
вается с кожей пациента [590]. Такой тип кремнезема исполь-
зуется как загуститель в компонентах эпоксидных клеев, когда
кремнезем также улучшает адгезию клея к некоторым твердым
поверхностям.
Повышенная вязкость, тиксотропия
Иногда становится непонятным, почему в дисперсионной среде,
содержащей значительные пропорции дискретных, полностью
дезагрегированных частиц в жидкости, не наблюдается замет-
ного возрастания вязкости и не развивается тиксотропия. Так,
органозоли, содержащие 20—30% SiO2, оказываются совсем
жидкими. И лишь только когда кремнеземные частицы коллоид-
ного размера формируют сетку, распространяющуюся по всему
объему жидкости, будут наблюдаться подобные эффекты за-
гущения.
Янг и Чессик [591] описали такие эффекты, вызываемые гид-
рофильными и гидрофобными частицами кремнезема в загу-
26*
820
Глава 5
щаемых полярных и неполярных жидкостях, но эти авторы не
развили полного представления о подобной системе. Витман и
Чессик [592] исследовали воздействие полярных (гидрофиль-
ных) кремнеземов при загущении неполярных (углеводородных)
жидкостей после добавления небольших количеств воды и н-
гептиловых производных, включая кислоту, хлорид, альдегид,
амин и спирт. Анализ седиментационных объемов для гидро-
фильного тонкодисперсного кремнезема, находящихся в декане
в присутствии перечисленных присадок, показал, что спирт и
амин препятствовали образованию мостиковых связей между
частицами и, таким образом, осадок стремился занимать наи-
меньший объем.
Механизм действия
Нельзя понять общего поведения кремнезема в таких си-
стемах до тех пор пока поочередно не будут рассмотрены сле-
дующие переменные величины:
1. Размер первичных частиц.
2. Степень диспергирования частиц, т. е. в какой мере ча-
стицы разделены или агрегированы.
3. Размер агрегатов.
4. Относительная энергия связи мест контактов частица—
частица в зависимости от поверхности раздела частица—жидкая
фаза.
5. Адсорбция присадок на частицах.
На рис. 5.25 представлены различного типа поверхности при
разных комбинациях. Кремнезем в исходном состоянии пред-
ставляется в виде двух классов частиц: одиночных дискретных
частиц и микроагрегатов. На каждый из этих случаев накла-
дываются три условия:
1. Немодифицированная силанольная поверхность, гидро-
фильная.
2. Частично органофильная поверхность, например, частично
этерифицированная.
3. Полностью органофильная поверхность, например, поверх-
ность, покрытая углеводородными группами.
Третий класс частиц образуется, когда микроагрегаты раз-
рываются на отдельные кусочки при их измельчении, так что
при этом создаются в местах разрыва на агрегатах «голые» ги-
дрофильные пятна, но уже меньшего размера.
Поведение всех этих типов частиц в таких жидких средах,
как вода, спирт (бутанол) и углеводороды (гексан), иллюстри-
руется на рис. 5.26, и может быть кратко описано следующим
образом. Если вся поверхность целиком для каждой частицы не
смачивается соответствующей жидкостью, то несмоченные по-
Силикагели и порошки
821
Поверхность
Частицы Полярная полярная-непопярная Неполярная
Гидрофильная Гидроорганофильная Органофильная
Рис. 5.25. Схематическое изображение поверхностей одиночных частиц, агрега-
тов больших размеров и агрегатов небольших размеров.
Каждая разновидность показана в виде гидрофильных поверхностей, частично органо-
фильных и полностью органофильных (перед измельчением). Гидрофильная поверхность
обозначена тонкими линиями; органофильная поверхность — толстыми линиями; обнажен-
ные гидрофильные участки после размалывания — волнистыми линиями.
Жидкости
Тип крем незема Полярная (вода) Полярная- неполяр ная (спирт) Неполярная (гексан)
Полярный Дискрет- ные час- тицы Небольшие агрегаты °°СО о ° °о ° ООО О°О°О°
8 %
Частично полярный Дискретные частицы Небольшие аг о с s аты О С Q о°о° о° гж
Неполяр- ный Дискретные частицы небольшие агрегаты О о оо о ° ои о о о о о О о ° О О
cP
Рис. 5.26. Эффекты вязкости и тиксотропии, наблюдаемые для некоторых типов
кремнезема, находящихся в различных жидкостях.
Дискретные частицы: гидрофильные, частично гидрофобные и полностью гидрофобные.
Агрегаты небольших размеров после помола: первоначально гидрофильные, частично
гидрофильные и полностью гидрофобные; в дальнейшем с дополнительными гидрофиль-
ными участками.
822
Глава 5
верхности двух различных частиц способствуют сцеплению этих
частиц между собой.
Таким образом, в воде частицы с гидрофобными неполяр-
ными поверхностями прилипают друг к другу благодаря обра-
зованию гидрофобных связей.
В масле частицы с гидрофильными полярными поверхно-
стями сцепляются между собой вследствие возникновения водо-
родных связей.
В спирте, когда происходит смачивание обоих видов поверх-
ностей кремнезема, частицы не прилипают друг к другу. Гид-
рофильные поверхности частиц смачиваются благодаря адсорб-
ции молекул спирта, повернутых своими концевыми Труппами
ОН в сторону кремнеземной поверхности. Гидрофобные поверх-
ности кремнеземных частиц смачиваются благодаря наличию
углеводородных групп в молекулах спирта (или таких групп
у других молекул, имеющих полярные и неполярные сегменты).
Как показано на рис. 5.26, в воде формируется сетка из ча-
стиц, если эти частицы имеют полностью гидрофобные или ча-
стично гидрофобные поверхности. Сетка из кремнеземных частиц
будет образовываться в масле, если частицы имеют полностью
гидрофильные или частично гидрофильные поверхности. Сетка
из кремнеземных частиц оказывается наиболее объемистой в том
случае, когда частицы кремнезема имеют как гидрофильные, так
и гидрофобные участки поверхности. Но никакой сетки не будет
формироваться в спирте, и вязкость системы зависит только от
объемной доли, занимаемой агрегатами в жидкости. Поскольку,
никакой сетки не образуется в этом случае, то наблюдается
очень слабая тиксотропия.
Это, конечно, слишком упрощенная картина, поскольку сте-
пень смачивания и гидрофильных, и гидрофобных поверхностей
будет зависеть как от самого спирта, так и от присутствия дру-
гих полярных и неполярных веществ.
Термин «смачивание» означает просто низкое значение меж-
фазной граничной энергии между жидкостью и поверхностью
твердого тела.
В том случае когда целиком вся поверхность частицы не сма-
чивается жидкостью, то частицы слипаются вместе в плотные
комочки. С другой стороны, когда лишь только определенные
участки в виде пятен на каждой из частиц неспособны смачи-
ваться, то такие частицы сцепляются вместе только по таким
пятнам, и при этом образуются более открытые сетки.
Формирование сетки максимальной величины происходит
в масляной среде (гексане), когда имеются только несмоченные
участки гидрофобных частиц кремнезема, представляющие собой
«голые» пятна, которые образуются путем разрыва на кусочки
(при измельчении) высокопористых агрегатов с открытой струк-
Силикагели и порошки
823
турой, что ведет к превращению их в меньшие по размеру аг-
регаты.
Формирование максимальной сетки и явление тиксотропии
имеют место, когда небольшие трехмерные кремнеземные агре-
гаты связываются вместе в еще большие трехмерные агрегаты,
которые способны распространиться по всему объему жидкой
Рис. 5.27. Схема загущения геля.
Наибольший эффект загущения и тик-
сотропия вызываются благодаря тому,
что агрегаты небольших размеров свя-
зываются вместе через имеющиеся не-
смоченные участки в обширную трех-
мерную сетку геля, которая’ распро-
страняется на всю жидкую среду. За-
черченные площадки в местах контак-
тов между небольшими по размеру аг-
регатами указывают на образование
полярных или водородных связей,
когда помол гидрофобных агрегатов
производится в масляной среде, или на
появление гидрофобных участков, когда
агрегаты, которые уже были частично
гидрофобными, подвергались механи-
ческому диспергированию в воде.
среды (рис. 5.27). Агрегаты связываются вместе через полярные
участки поверхности на кремнеземных частицах в случае масля-
ной среды или же через гидрофобные участки поверхности в слу-
чае водной среды. Такие связи, включающие образование соот-
ветственно водородных связей или гидрофобных связей, легко
разрываются под действием сил сдвига и легко восстанавли-
ваются, когда вся кремнеземная масса остается в покое и не
подвергается каким-либо воздействиям. Таким образом уста-
навливается высокая степень тиксотропии. Различные аспекты
использования кремнеземов в качестве загустителей в разнооб-
разных жидких средах при одновременном воздействии вторич-
ных присадок обсуждаются в типичных для такого рода работ
статьях [593—596]. В частности, характеристики пирогенного
кремнезема (аэросила) были получены в исследованиях Рупп-
рехта и Либла [597] и Каспера и др. [598]. В некоторых слу-
чаях были добавлены ПАВ.
Консистентная смазка
Ниже рассматривается несколько патентов, срок действия
которых уже истек, связанных с использованием гидрофобного
тонкодисперсного кремнезема в качестве загустителя среды
с целью превращения смазочного масла в консистентную смазку,
824
Глава 5
поскольку информация по данному вопросу не появлялась в тех-
нической литературе.
Гидрогели кремнезема подвергали измельчению в масляной
среде с добавлением олеофильного ПАВ, например четвертич-
ного аммониевого соединения с длинной цепью — хлорида диме-
тилдиоктадециламмония или металлических мыл. Использова-
лись и глины; такой способ применялся, кроме того, к тонкодис-
персному кремнезему [599]. Это лишь один из многих патентов,
имевших отношение к загустителям смазок, приготовляемых из
бентонитовых глин [600].
Упрочненный, с низкой плотностью и этерифицированной по-
верхностью кремнезем (эстерсил) был запатентован как загу-
ститель смазок, приготовляемых из нефтяных [601] и силиконо-
вых масел [602]. Кремнезем, поверхность которого покрыта
как эфирными бутоксигруппами, так и силиконовым маслом,
обеспечивал стабильность консистентной смазки в течение более
продолжительного времени [603]. Брендли [604] обнаружил, что
добавление некоторых полярных, образующих водородные связи
соединений, как, например, трикрезилфосфат, заметно повышало
адгезию консистентной смазки, приготовленной с введением эс-
терсила, к стальной поверхности при продолжительной эксплуа-
тации во влажных условиях, что предохраняло сталь от закали-
вания и останавливало процесс коррозии.
Мейер и Брендли [605] описали свойства эстерсиловых сма-
зок и сообщили, что такие смазки не подвержены абразивному
износу и в меньшей степени поддаются изменениям при повы-
шенных температурах по сравнению со смазками, приготовляе-
мыми с добавлением металлических мыл.
Согласно более недавним работам Фукса и др. [606], эстер-
сил с бутоксигруппами на .поверхности приводил к улучшению
устойчивости смазки к окислению.
В процессе развития работ, связанных с такого рода загу-
стителями смазок, было достигнуто лучшее понимание тиксо-
тропных эффектов, имевших место в системах, содержавших
тонкодисперсные частицы и агрегаты частиц в жидких средах.
Для приготовления органофильного кремнезема, используе-
мого в консистентной смазке, Фронзак [607] запатентовал в ка-
честве присадки карбонат алкилена С2—Cis- Это соединение, ве-
роятно, адсорбировалось на кремнеземе и могло даже вступать
в реакцию с поверхностными группами SiOH с образованием
покрытия эфирными группами на поверхности кремнезема. ‘
В работе [608] кремнезем покрывался фенолформальдегид-
ной смолой из раствора ацетона и подвергался термообработке;
было обнаружено, что получалась хорошая устойчивость си-
стемы при воздействии сил сдвига, когда такой кремнезем ис-
пользовался в качестве загустителя консистентной смазки.
Силикагели и порошки
825
Согласно Блаттенбергеру [609], загущенные кремнеземом
смазки предохраняют от застудневания при старении путем до-
бавления диолов, таких, например, как 2-метил-2,4-пентандиол
или 2-этил-1,3-гександиол. Такие диолы, несомненно, сильно
адсорбируются на поверхности кремнезема посредством обра-
зования двойных водородных связей.
Экономическая целесообразность применения загущенных
консистентных смазок изменилась в 50-е годы, поскольку обна-
ружилась среди других факторов относительно большая пригод-
ность стеарата лития в качестве водостойкого загустителя сма-
зок, что снизило интерес к такого рода использованию гидро-
фобных типов кремнезема.
Краски, грунтовки, чернила
Загущение и тиксотропические эффекты, достигаемые в этих
классах продуктов в результате применения кремнезема, оказа-
лись по литературным данным слишком многочисленными,
чтобы их можно было здесь цитировать. Обычно достигается не-
сколько эффектов при введении кремнезема, а именно матиро-
вание или понижение блеска, предотвращение образования
осадка из пигмента при хранении продукта, стабилизация эмуль-
сии, а также возможность нанесения красящих веществ без об-
разования капель. Однако нет какой-либо доступной информа-
ции относительно использования кремнеземов в таких продуктах
по сравнению с другими загустителями. По-видимому, матиро-
вание является тем эффектом, которое находит применение
в красках и в- чистовых покрытиях, о чем наиболее часто упо-
минается в бюллетенях фирм. Для чернил характерным при
использовании кремнезема обычно является регулирование их
вязкости.
Фармацевтические и косметические препараты
Общий обзор по использованию пирогенного кремнезема
в фармацевтических и косметических препаратах был опубли-
кован Ферчем [610]. Прозрачность высокопористого с высоким
значением удельной поверхности кремнезема позволила разра-
ботать прозрачные зубные пасты, содержащие кремнезем и ги-
дрофильный органический полимерный загуститель, вместе
с глицерином, маслом, очищающим средством и ароматизирую-
щим веществом [611, 612]. Разнообразные комбинации зубных
паст и реагентов, препятствующих образованию зубного камня,
включены в другие составы [613, 614].
Другие в отличие от загущающих эффекты, как, например,
использование кремнезема в косметических препаратах с целью
826
Глава 5
удаления жира из кожи, упоминаются в разделе об использо-
вании кремнезема в качестве адсорбента.
Разнообразные композиции
Электростатическое сцепление предусматривает проявление
электровязкостного эффекта, когда диспергированный кремне-
зем находится в масле. Например, тонкопористый кремнезем и
моноолеат глицерина, как было описано Мартинеком и Классом
[615], находились в диспергированном состоянии в масле, за-
полняющем узкий зазор между двумя сцепляющимися пласти-
нами. Такие пластины способны свободно вращаться относи-
тельно друг друга, но происходит их сцепление, когда к пласти-
нам прикладывается трехфазный переменный ток напряжением
2000 В. Это ведет к тому, что кремнезем загущает масло до со-
стояния геля, который уже передает вращающий момент от од-
ной пластины к другой. Аналогичная электровязкостная жид-
кость подробно описывается в другом патенте [616]. Класс
[617] исследовал параметры подобной системы. Такое явление
объясняется возникновением взаимодействий между поляризо-
ванными двойными слоями и между этими слоями и кремнезем-
ными частицами.
Действие температуры на электрореологию исследовалось
Шульманом и Мацепуро [618]. Были описаны присадочные па-
тентованные составы [619, 620].
Кристаллы больших размеров, которые не могут быть выра-
щены в воде, выращиваются в среде геля кремнезема. Струк-
тура геля предотвращает конвекцию и позволяет равномерно
протекать процессу диффузии компонентов. Такой рост моно-
кристаллов тетрагидрата тартрата кальция с присадкой неодима
был описан Рубиным [621], а монокристаллов оксида однова-
лентной меди — Бреннером [622]. Эти и другие кристаллы вы-
ращивались Хенишем, Деннисом и Ханок [623], которые также
исследовали действие облучения на скорость образования цент-
ров кристаллизации. Достаточно подробно рост кристаллов
HgC12-2H2O изучался Курцем [624], оксалата кальция — Биса-
иллоном и Таваши [625]. Армингтон и О’Коннор [626] описали
аппаратуру, в которой имеется возможность подводить к гелю
кремнезема с двух сторон реагирующие вещества.
Каши [627] представил обзор по росту кристаллов в кремне-
земных гелях.
Алкилнитраты, гидразин и другие виды ракетного топлива
загущаются до состояния геля или до состояния смазки за счет
введения рыхлого объемистого силикагеля [628].
Для того чтобы ввести кислоту в желаемый участок резер-
вуара с нефтью, ее предварительно загущают кремнеземом. При
Силикагели и порошки
827
разбавлении водой кислота сразу же в том месте, где она вво-
дится и смешивается, способна разжижаться и становиться ак-
тивной [629]. Кремнезем также может быть использован для
загущения кислоты в свинцовых аккумуляторных батареях.
Добавление кремнезема в качестве загустителя пен усили-
вает их противопожарные свойства [630].
Оптические эффекты, матирование
Красители часто дают настолько сильное окрашивание, что
их необходимо разбавлять в какой-либо прозрачной среде для
возможности получения наибольших цветовых эффектов. Это
в особенности справедливо для некоторых флуоресцентных кра-
сок. Родамин адсорбируется на измельченном в порошок сили-
кагеле, так что добавление нескольких процентов этого краси-
теля дает интенсивное флуоресцентное окрашивание [631].
Другим путем улучшения свойств сильно нерастворимых ор-
ганических пигментов оказывается возможность образования
центров кристаллизации для получения кристаллов таких ве-
ществ в значительно более тонкодисперсной форме без их из-
мельчения. Так, фталоцианин синтезируют в присутствии очень
тонкодисперсного кремнезема, и последний покрывается пигмен-
том. Получаемый продукт примерно на две трети состоит из
кремнезема, и это позволяет извлечь богатые цветовые оттенки
из подобного органического соединения, которое таким образом
распространяется по окрашиваемой поверхности [632].
Матирование и удаление эффектов блеска в красках, пласт-
массах, а также в печатных красках, вероятно, представляет со-
бой наиболее широкое использование кремнеземных порошков,
как можно об этом судить на основании сведений, опубликован-
ных в литературе фирм-изготовителей. Многочисленные сорта
ксерогелевых порошков, представленные в табл. 5.7, по-види-
мому, обеспечивают получение различной степени матирования
при самых разнообразных типах отделочной покраски.
Эффекты, связанные с изменением состояния
поверхности
Стабилизация эмульсий
Кремнеземные частицы как с гидрофильной, так и с органо-
фильной поверхностями будут, очевидно, собираться на границе
раздела фаз масло—вода. Это обеспечивает стабилизацию гра-
ницы раздела фаз даже в большей степени, чем применение
лишь одного только ПАВ. Хотя, в частности, подобное исполь-
зование кремнезема не является обычным, обработка поверхно-
828
Глава 5
сти раздела кремнеземом, выступающим в роли эмульгатора, ве-
дет, как правило, к стабилизации эмульсионных систем, таких,
например, как краски, где кремнезем может также выполнять
и другие функции.
Гидрофильная поверхность
Полиамидные волокна можно сделать гидрофильными путем
их обработки в суспензии, содержащей 1% высокопористого,
тонкодисперсного силикагеля с удельной поверхностью 400 м2/г
и диаметром пор 10—20 нм. Такой кремнезем образует на по-
верхности прилипшую пленку геля, которая ведет к улучшению
промывания и очистки волокна [633].
Антипенная присадка
Механизм, по которому пузырьки в пене прорываются из-за
присутствия, по крайней мере частично, гидрофобных частиц
кремнезема, обсуждался в гл. 4. Тонкодисперсные кремнеземные
агрегаты с гидрофобной в достаточной мере поверхностью, спо-
собные суспендироваться в углеводородном или в силиконовом
масле, по всей вероятности, должны содержать некоторые гидро-
фильные участки, благодаря которым такие агрегаты удержи-
ваются на поверхности пузырьков. Кремнезем обрабатывается
силиконовым маслом и нагревается до 245°С, после чего подоб-
ные кремнеземные агрегаты вступают в реакцию с поверхностью
пузырьков, суспендированных в масляной среде [634]. К маслу
могут также добавляться ПАВ [635].
Гидрофобные эффекты
Гидрофобные или водоотталкивающие эффекты могут полу-
чаться при введении в систему тонкодисперсных гидро-
фобных кремнеземов, частицы которых при адсорбции способны
образовывать очень тонкий адсорбционный слой на обычно на-
ходящихся в гидрофильном состоянии поверхностях. Такой эф-
фект, безусловно, оказывает воздействие против слеживания
гидрофобных частиц кремнезема, входящих как составная часть
во влажные порошки.
В наиболее существенном патенте [636] в этой области при-
менения эстерсилов предусматривается практическое использо-
вание многих видов материалов, в том числе бумаги и тканей,
которые приобретают высокие водоотталкивающие свойства
вследствие наложения невидимой адсорбционной пленки, состоя-
щей из гидрофобных коллоидных частиц кремнезема.
Силикагели и порошки
829
Применяемые в условиях морской воды красящие маркирую-
щие порошки состоят из частиц гидрофобного кремнезема, со-
держащих краситель, которые, следовательно, растворяются
в значительно большей степени вблизи поверхности воды
по сравнению с растворимостью после погружения в морскую
воду [637].
«Сухая» вода
Чрезвычайно интересный пример гидрофобизации представ-
лен в патенте, в котором дается способ получения сухой порош-
кообразной воды, приготовляемой путем покрытия полученных
помолом тонкодисперсных частиц льда гидрофобным кремнезе-
мом [638]. Когда такой порошок льда нагревается до комнат-
ной температуры, то система остается в виде «сухого» свободно
растекающегося белого порошка, который при рассмотрении под
микроскопом состоит из индивидуальных сферических капелек
прозрачной чистой воды, причем каждая капелька покрыта
почти невидимой пленкой кремнезема. При механическом пере-
мешивании такой порошок оседает с образованием жидкой
воды, но если система не подвергается воздействию и остается
в спокойном состоянии в закрытом сосуде для предотвращения
испарения, то она оказывается устойчивой в течение нескольких
недель. Таким способом могут приготовляться в порошкообраз-
ной форме и водные растворы, если в них не содержатся смачи-
вающие реагенты.
Концентрированный пероксид водорода (20—70% Н2О2) мо-
жет аналогичным образом превращаться в устойчивый порошок
путем вибрирующего перемешивания жидкости с кремнеземным
порошком [639].
Поглотитель
Наиболее широко кремнеземный поглотитель, вероятно, ис-
пользуется в качестве осушителя для изделий в упаковке, кото-
рые могут подвергаться коррозии или порче под воздействием
влаги. Ниже приводятся следующие различные наблюдения.
Инсектицидная активность тонкопористого кремнезема обсу-
ждается в гл. 7. Маслянистые или воскообразные компоненты
препятствуют потере влаги в процессе того, как инсектицидные
кутикулы адсорбируются кремнеземом; в результате этого насе-
комое умирает от дегидратации. Механизм действия кремнезема
описан Эбелингом [640]. Для подобных целей особенно эффек-
тивными оказываются гидрофобные эстерсилы [641].
Средства для удаления пятен состоят из очищающего рас-
створителя, в котором суспендирован кремнезем. Кремнеземные
830
Глава 5
частицы удерживаются на поверхности ткани, и когда раствори-
тель испаряется с такой поверхности, то загрязняющее вещество
переносится с ткани на кремнезем, который затем счищается.
Поглотители для удаления разлива нефти также находят
применение, причем в них определенную роль могут играть син-
тетические гидрофобные типы кремнезема. Однако для погло-
щения плавающей нефти используется гидрофобный, широко
распространенный и имеющий низкую стоимость перлит. Перлит
становится гидрофобным благодаря его обработке раствором
метилсиланолята натрия в воде. Указывается, что по некоторой
причине присутствие порошкового алюминия улучшает эффек-
тивность реакции гидрофобизации перлита [642].
Концентрированный озон может быть получен путем погло-
щения этого газа на силикагеле в интервале температур от
— 117 до 20°С [643].
Полисахариды, согласно данным Кеннеди, Баркера и Уайта
[644], предпочтительно адсорбируются на поверхностях кремне-
зема, покрытых предварительно полиароматическими молёку-
лами. Кажется удивительным, что углеводородная поверхность
должна иметь какое-то особое сродство для подобных, в высо-
кой степени гидрофильных молекул. Кремнеземистые пористые
материалы, покрытые адсорбционным слоем 1,3-диаминобен-
зола, способны адсорбировать гликоген и разветвленные поли-
сахариды, но не могут адсорбировать нейтральные или заря-
женные моносахариды.
Ионы урана, циркония и ниобия или ионы оксония адсор-
бируются кремнеземом из раствора азотной кислоты вплоть до
pH 0, а в случае ниобия — даже для 10 М раствора кислоты.
Слабая щавелевая кислота удаляет адсорбированные ионы та-
ких металлов [645]. На силикагеле цирконий способен отде-
ляться от гафния [646].
Способы определения цвета можно сделать в несколько раз
более чувствительными посредством проведения испытаний на
поверхности измельченного в порошок силикагеля; например,
дитизон в случае Hg2+ и K4Fe(CN)g с НОг+ [647].
Глицерин может селективно адсорбироваться на силикагеле
из отходов ферментации спиртных напитков [648]. Поры такого
силикагеля были, вероятно, очень тонкими.
Антиокислители в виде полиоксиароматических соединений
способны адсорбироваться на тонкопористом силикагеле такого
типа, который используется в качестве наполнителя резин, и мо-
гут диспергироваться в полиэтилене. Оказывается, подобные ан-
тиокислители имеют столь же хорошие качества, как и углерод-
ная сажа, но получаемая полиэтиленовая пленка-в данном слу-
чае становится прозрачной [649].
Силикагели и порошки
831
Косметические препараты могут иметь в своем составе крем-
незем в качестве адсорбента для поглощения природных масел.
Сальный волосяной покров головы можно обрабатывать суспен-
зией кремнезема в воде с целью поглощения масла и создания
исцеляющих условий [650]. Косметические препараты, содержа-
щие пирогенный кремнезем, отличаются тем, что они совершенно
незаметны, предохраняют кожу лица от маслянистого блеска и
такие препараты можно смывать даже теплой водой [651].
Катализаторы
Ниже упоминается только несколько различных применений
кремнезема в качестве катализаторов.
Аэрогели
Тейчнер и др. [316] приготовили катализаторы из кремне-
зема с очень высоким значением удельной поверхности (около
600 м2/г). Подобный катализатор, представляющий собой смесь
оксидов NiO—AI2O3, нанесенную на кремнеземную подложку,
способен окислять изобутилен до ацетона и метилакролеина без
образования СО и СО2 даже при температуре 305°С.
Подложка для митохондрий
Ферменты адсорбировались на поверхности кремнезема, и
было обнаружено ‘сохранение их активности. Но тот факт, что
митохондрии (частицы, представляющие собой образования, вы-
деляемые из живых клеток, и состоящие из сложных фермент-
ных систем) можно подобным же образом иммобилизовать на
кремнеземе, дает возможность раскрыть целые новые области
исследований в биохимии [652а]. Другие содержащие мембраны
частицы, или органеллы, могут аналогичным образом фиксиро-
ваться на кремнеземе, например в виде хлоропластов и микро-
сом печени. Поверхность кремнезема должна быть прежде всего
превращена в органофильную посредством ее обработки с нане-
сением алкилсилильных групп. Затем подобные биологические
образования могут прилипать к поверхности, давая монослойное
покрытие при температуре около 27°С, но они способны десор-
бироваться при 5°С. Природа такого эффекта непонятна, но
можно сделать предположение, что поскольку водородные связи
становятся более прочными при 5°С, то вода тем или иным об-
разом вытесняет эти частицы с поверхности, которые должны
удерживаться на ней гидрофобными связями. Подобные гидро-
фобные связи имеют место, и они используются для закрепления
ферментов на кремнеземной поверхности [6526].
832
Глава 5
Мигрирование атомов водорода
Было доказано, что для катализатора гидрирования, пред-
ставляющего собой нанесенную на кремнеземную подложку
платину, при введении на поверхность кремнезема хемосорбиро-
ванных метилсилильных групп не обеспечивался сколько-нибудь
значительный эффект активности процесса гидрирования [653].
Известно, что молекулы водорода расщепляются на водородные
атомы на поверхности платины, и такие атомы водорода спо-
собны затем мигрировать или «растекаться» по всей окружаю-
щей оксидной поверхности. Тейчнер и др. [653] доказали, что
вдоль поверхности оксида алюминия мигрируют атомы водо-
рода, возникающие во вставленной в оксид алюминия сетке из
платинового катализатора. После того как такая сетка уда-
ляется, оставшиеся атомы водорода образуют активные центры
на поверхности оксида алюминия, так что может происходить
гидрирование этилена и бензола. Однако Кунг, Букс и Бэрвелл
[654] обнаружили, что никакой подобной активации не проис-
ходит на поверхности кремнезема. При нанесении на кремнезем-
ную поверхность хемосорбированных групп (СНз)зЗЮ наблю-
дается ослабление мигрирования атомов водорода, но каталити-
ческая активность остается неизменной и является присущей
исключительно лишь платине.
Реакционноспособный кремнезем
Высокие значения удельной поверхности и скорости раство-
рения аморфного кремнезема позволяют проводить необходимые
реакции при значительно более низких температурах, чем это
требуется для измельченного в порошок кристаллического крем-
незема. Повышенная химическая реакционная способность кол-
лоидных кремнеземов была рассмотрена в гл. 4. Прозрачное
плавленое кварцевое стекло образуется при давлении 140 кг/см2
и температуре 1200°С из кремнеземного порошка с размером
первичных частиц 15 нм, тогда как для получения такого мате-
риала в виде выдуваемых в форму изделий требуется темпера-
тура 2000°С [655].
Удивительным является то, что тонкопористый аморфный
кремнезем вступает в реакцию с аммиаком только при 550°С
с образованием 812^0 и при 1250°С с образованием SisN^ От-
сюда сразу возникает вопрос, представляется ли возможным
изготовлять горячим прессованием изделия из таких огнеупор-
ных материалов, как получена серия оксинитрида кремния.
Кварцевое стекло с дефицитом кислорода, которое лишь с тру-
дом подвергается расстекловыванию, приготовляется посреДст-
Силикагели и порошки
833
вом проведения реакции измельченного в порошок бора с аморф-
ным кремнеземом [657]. Редкоземельные оксиды можно форми-
ровать в. виде кремнеземного соединения с внутренне однородной
структурой еще до появления стеклообразного состояния: про-
водят гидролиз паров SiCl4 в водном растворе редкоземельной
соли, затем" высушивают и окончательно расплавляют материал.
В результате получается флуоресцентное стекло [658].
Аморфный тонкопористый кремнезем оказывается настолько
реакционноспособным, что его можно использовать для приго-
товления синтетических глинистых минералов. Гекторит, или со-
содержащий магний бентонит, оказывается пригодным для из-
готовления электроизоляционного материала после превращения
его в форму, содержащую серебро [659]. Каолин образуется
в гидротермальных условиях при 200—300°С с использованием
силикагеля в качестве источника получения кремнезема [660].
Карбид кремния высокой чистоты в виде светло-желтого по-
рошка приготовляется путем реакции гидролиза SiCl4 в рас-
творе сахара с образованием силикагеля и с последующим на-
гревом в инертной атмосфере при температуре 1800°С в тече-
ние 4 ч [661].
Добавление от 1 до 2 % пирогенного кремнезема к портланд-
цементу улучшает его свойства благодаря высокой реакцион-
ной способности кремнезема [662].
В водном растворе ионы цинка вступают в реакцию с широ-
копористым силикагелем с образованием силиката цинка вплоть
до тех пор, пока на поверхности кремнезема не будет сформи-
ровано примерно семь слоев силиката. Поры, имеющие неболь-
шой диаметр, ограничивают дальнейшее образование силикат-
ных слоев, и поэтому такая реакция протекает недостаточно
полно [663].
Образование зародышей в облаках
Цеттльмойер, Чессик и Техеурекджан [664] открыли, что для
образования центров кристаллизации льда, или первого этапа
формирования дождевых капель в облаке, оказываются актив-
ными' частицы кремнезема диаметром 30—100 нм при условии,
что поверхность таких частиц состоит из мозаично расположен-
ных на ней участков, представляющих собой гидрофильные
пятна на гидрофобной поверхности. Гидрофобные участки этой
поверхности должны составлять около 20—30 % Взаимодейст-
вие пара воды с поверхностями различных твердых оксидов
было объяснено благодаря проведенным исследованиям мето-
дами калориметрии с определением теплот смачивания, измере-
ния диэлектрических потерь и отражательной ИК-спектроско-
пии [665].
27 Заказ № 250
834
Глава 5
Боучер [666] обобщил вопросы теории и практических при-
ложений, относящихся к проблеме образования центров кри-
сталлизации льда в облаках. Очевидно, что подобный эффект
образования зародышей может достигаться в некоторой мере
частично дегидратированной поверхностью кремнезема при не
очень высокой температуре, но, вернее, за счет частичного по-
крытия гидроксилированной поверхности хемосорбированными
гидрофобными органическими группами или органосилильными
группами.
Подобное применение кремнезема в недалеком будущем ста-
нет практически важной проблемой, если только будет пол-
ностью достигнута экономическая выгода по сравнению с ис-
пользованием для этих целей иодида серебра.
Наполнение хроматографических колонок
При наполнении колонок кремнезем может быть использо-
ван в трех направлениях:
1. В качестве адсорбента с поверхностью, содержащей сила-
нольные группы или совершенно другие полярные, неполярные
или Ионообменные органические группы, присоединенные к по-
верхности.
2. Как пористое вещество, способное удерживать неподвиж-
ную жидкую фазу, которая и будет являться активным адсор-
бентом.
3. В качестве микропористой набивки, содержащей однород-
ные по размеру поры, внутри которых наименьшие молекулы
способны диффундировать с большой скоростью, немного боль-
шие по размеру молекулы — гораздо медленнее или же вовсе не
иметь возможности диффундировать. В таком случае размеры
пор позволяют использовать эксклюзивную хроматографию для
определения распределения полимеров по их молекулярным
массам.
Каждое из отмеченных направлений накладывает некоторые
требования в отношении применяемых кремнеземов, которые
должны обладать определенными структурными характеристи-
ками, но обычно такие требования сводятся к следующим.
Частицы кремнеземного порошка должны быть сферическими
по форме, чтобы можно было достигать однородной упаковки из
таких частиц; они должны быть однородными по размеру с тем,
чтобы обеспечивался равномерный поток через поперечное сече-
ние колонки в любом месте; сферические частицы должны иметь
размер в области' 5—50 мкм с целью получения максимальной
эффективности работы колонки в пределах практически наблю-
даемых падений давления по длине колонки; частицы должны
быть однородными по пористости и иметь оптимальные струк-
Силикагели и порошки
835
турные характеристики, определяемые в. зависимости от кон-
кретного типа использования.
При некоторых применениях наиболее четкие хроматографи-
ческие разделения получаются при использовании частиц, кото-
рые имеют твердую сплошную сердцевину, но обладают порами
в поверхностном слое. Такой пористый слой вмещает в себя не-
подвижную жидкую фазу поглотителя (адсорбента), на котором
подвергаемые разделению молекулы могут быстро достигать
равновесия без какого-либо замедления процесса за счет диффу-
зии таких молекул к центрам упакованных сферических частиц.
Литература по этой теме с 1960 г. выросла до таких огром-
ных масштабов, а типы специально разработанных кремнеземов
для наполнения хроматографических колонок размножились на-
столько, что не представляется возможным отразить все эти во-
просы в настоящем обзоре.
Унгер [6] обобщил данные по способным изменяться струк-
турным характеристикам кремнеземов, используемых для на-
полнения колонок, включая характеристики пор, удельную по-
верхность и различные разновидности модифицированных по-
верхностей. Он также описал способ приготовления пористых
микросферических частиц с контролируемым размером пор по-
средством гидролиза этилортосиликата в эмульсионной системе
[667].
Айлер и Мак-Квестон [668], используя процесс коацервации,
приготовили другой тип микросферических пористых частиц для
применения в хроматографии. В этом случае для получения од:
нородных пор желаемого размера применяли коллоидные ча-
стицы одинакового размера. Способ наполнения хроматографи-
ческих колонок такого типа был запатентован Кирклендом
[669]. Однородные по размеру глобулы диаметром 5—10 мкм
приготовлялись из однородных плотных, более мелких кремне-
земных частиц [670]. Описаны их хроматографические характе-
ристики [671, 672]. Киселев и др. [673, 674] изучили влияние
размеров пор на хроматографическое разделение. Микросферы
с поверхностной пористостью могут быть изготовлены путем оса-
ждения слоев, состоящих из частиц коллоидного кремнезема, на
поверхности стеклянных шариков, на которых наращивается од-
нородное пористое покрытие, способное удержать неподвижную
фазу, играющую роль адсорбента. Киркленд и соавторы [675—•
678] описали характеристик^ подобных систем. Микросфериче-
ские частицы с широкими порами используются в эксклюзивной
или гель-хроматографии. Приготовление таких кремнеземных
материалов и их использование для разделения растворимых по-
лимеров по молекулярным массам описано в ряде статей [679—
683]. Диаметры пор в таких частицах составляли 200—1500 А.
Соотношение, связывающее диаметр пор и удельную поверх-
27*
836
Глава 5
ность, обычно выражается как произведение DA, определяемое
интервалом значений 15 000—40 000, где D — диаметр, выражен-
ный в ангстремах, и А — удельная поверхность, м2/г [680].
Модифицирование поверхности кремнезема осуществляется
путем нанесения молекулярных хемосорбированных покрытий
различными способами с целью изменения характерных хрома-
тографических параметров. Некоторые разновидности модифи-
цированных поверхностей, обладающие ионообменными свойст-
вами, уже обсуждались. Этерификация спиртами исходной
силанольной поверхности, приводящая к замещению на ней си-
ланольных групп разнообразными органическими группами, ис-
пользовалась в многочисленных работах, начиная с Киркленда
[684], который оценил важность этерифицированного кремне-
зема с «-бутильными группами на поверхности. В данном слу-
чае на поверхности возникают связи Si—О—С. Этерификация
выполнялась также по реакциям с полиэтиленгликолем [685],
различными алифатическими спиртами [686] и 3-оксипропио-
нитрилом [687]. ,
Присоединение органических групп к поверхности кремне-
зема прямыми связями Si—С, как правило, приводит к образо-
ванию более устойчивого по отношению к гидролизу покрытия.
Различные полярные и ионообменные группы можно присоеди-
нять к углеводородным группам, связанным с атомом кремния,
в результате чего можно получить широкий набор модифициро-
ванных поверхностей (см. раздел данной главы, посвященный
кремнезему с ионообменными поверхностями). Унгер и др. сде-
лали обзор типичных работ в этом направлении. Модифицирую-
щие покрытия состоят из таких групп, как —CgHg, —СН3 [688];
-СН2ОН, —CH2CN, —СН(ОН)СН(ОН)—, —СН2СН=СН2,
— (СН2)3СбН4СЬЬС1 [689]; —CH2CeH4NO2, —CH2CeH4NH2,
—CH2C6H4SO4H [690].
Авторы работы [691] модифицировали поверхность органи-
ческими группами, связанными с функциональными группами
алифатических цепей, например —СООН, —SO3H, —NH2, —CN
или NO2, посредством связей Si—N—С. Поверхностные группы
SiOH исходного кремнезема вначале вступают в реакцию
с SOC12, в результате чего превращаются в группы SiCl, кото-
рые затем реагируют с аминами.
Другим подходом является адсорбция на исходной кремне-
земной поверхности ионов металлов, которые способны образо-
вывать координационные комплексы с разделяемыми органиче-
скими веществами. Описано [692], что адсорбция ионов кадмия
на поверхности кремнезема способствовала улучшению разделе-
ния ароматических аминов.
Вероятно, следовало бы также использовать возможность об-
разования относительно стабильных, неспособных к ионизации
Силикагели и порошки
837
связей между ионами Сг3+, адсорбированными на поверхности
кремнезема, и группами —СООН, особенно алифатических кис-
лот с длинной цепью. Для модификации гидратированной по-
верхности, покрытой СГ2О3, можно было бы применить концевые
полярные группы, изменяющие ее полярность и формирующие
гидрофобные связи между параллельными, близко расположен-
ными углеводородными цепями. Это дало бы возможность ста-
билизировать все покрытие.
Новый тип ионообменной поверхности стеклянных микроша-
риков был предложен Кирклендом [693]. На ней были пооче-
редно адсорбированы слои противоположно заряженных частиц.
Вначале на чистой стеклянной поверхности адсорбировался мо-
нослой, состоящий из частиц сильноосновной ионообменной
смолы размером 0,1—0,5 мкм. Затем на этом слое смолы адсор-
бировался второй слой из частиц коллоидного кремнезема раз-
мером 0,015 мкм. Процесс повторяли до тех пор, пока не нара-
щивалась пористая пленка, обладающая анионообменными
центрами. Такая пленка достаточно эффективна для хромато-
графических целей.
ЛИТЕРАТУРА
1. Kuhn W. Е., Lamprey И., Sheer С., Ultrafine Particles, Wiley, New York,
1963.
2. Дилер P. К. Коллоидная химия кремнезема и силикатов. Пер. с англ.—
М.: Госстройиздат., 1959.
3. Everett D. W., Stone F. S., Eds., The Structure and Properties of Po-
rous Materials (Proc. 10th Symp. Colston Res. Soc., Univ. Bristol, March
24—27, 1958), Academic, New York; Butterworths, London, 1958.
4. Linsen B. G., Ed., Physical and Chemical Aspects of Adsorbents and
Catalysts, Academic, New York, 1970. Имеется ' перевод: Строение и
свойства адсорбентов и катализаторов. Под. ред. Б. Г. Линсена. М.:
Мир, 1973.
5. Strazhesko D. Л'., Ed., Adsorption and Adsorbents, Vol. 1, Wiley, New
York, 1973.
6. Unger Klaus, “Structure of Porous Adsorbents”, Angew. Chem. Int. Ed.,
11 (4), 267 (1972).
7. Грег С., Синг К. Адсорбция, удельная поверхность, пористость. Пер.
с англ.— М.: Мир, 1970.
8. Barby D., “Silicas”, in G. D. Parfitt and K. S. W. Sing, Characterization
of Powder Surfaces, Academic, New York, 1976, p. 353.
9. Sing K- S. W., “Surface Characterization-Physical”, Ref. 8, p. 21.
10a. Dubinin M. M„ Adv. Colloid Interface Sci., 2, 217 (1968).
106. Hermans P. H., in H. R. Kruyt, Ed., Colloid Science, Vol. 2, Elsevier,
New York, 1949. pp. 483—651.
10b. Weiser H. B., Colloid Chemistry, 2nd ed., Wilev, New York, 1950,
pp. 307—326.
10г. Von Buzagh A., Colloid Systems, The Technical Press, London, 1937,
p. 149.
11. Cohan L. H., Watson J. H. L., Rubber Age, 68, 687 (1951).
12. Medalia A. I., Heckman F. A., Carbon, 7, 567 (1969).
838 Глава 5
13. Medalia A. I., J. Colloid Interface Sci., 24, 393 (1967).
14. Havard D. C., Wilson R., J. Colloid Interface Sci., 57, 276 (1976).
15. Everett D. W., Parfitt G. D„ Sing K. S. W., Wilson R., J. Appl. Chem.
Biotechnol., 24, 199 (1974).
16. Moll W. L. H., Kolloid-Z., 138, 114 (1954).
17. Joy A. S., Vacuum, 33, 355 (1953).
18. Everett D. H., Ed., Surface Area Determination (Proc. Int. Symp. 1969),
Butterworth, London, 1970.
19. “The Size and Shape Factor in Colloidal Systems”, Disc. Faraday Soc.,
11 (1951) (Aberdeen).
20. Carruthers J. D., Cutting P. A., Day R. E., Harris M. R., Mitchell S. A.,
Sing K. S. W., Chem. Ind. London, 1968, 1772.
21. Everett D. H„ Parfitt G. D„ Sing K. S. W„ Wilson R., “SCI/IUPAC/NPL
Project on Surface Area Standards”, J. Appl. Chem, Biotechnol., 24
(4/5), 199 (1974) [Chem. Abstr. 81, 176670e],
22a. Dietz V. R., Turner N. H., Ref. 18, p. 43.
226. McClellan A. I., Harnsberger H. F., J. Colloid Interface Sci., 23, 577
(1967).
23. Micale F. I., “Determination of Vm and Effective Cross-Sectional Areas
of Ar, N2, Kr for Determination of Specific Surface Areas”, 49th Col-
loid Symposium, Clarkson College of Technology, Potsdam, N. Y., June
14—16, 1975.
24. Vezina S., Berube У., J. Colloid Interface Sci., 42 (1), 79 (1973).
25. Broekhoff J. С. P., Ref. 4, p. 1.
26. Brunauer S., “The Adsorption of Gases and Vapors”, Physical Adsorption,
Vol. 1 (Princeton University Press, Princeton, N. J., 1945).
Имеется перевод: Брунауэр С. Адсорбция газов и паров. М.: ИЛ, 1948.
27. Brunauer S., Emmett Р. Н., J. Am. Chem. Soc., 57, 1754 (1935).
28. Brunauer S.,'Emmett P. H„ Teller E., J. Am. Chem. Soc., 60, 309 (1938).
29. Innes W. B„ Anal. Chem., 23, 759 (1951).
30. Lippens В. C., Hermans M. E. A., Powder Metallo,- 7, 66 (1961).
31. Lippens В. C., thesis, Delft, (1961).
32. Красильников К Г., Киселев В. Ф., Капитонова Н. В., Сысоев Е. А.—
Ж. физ. химии, 1957, т. 31, с. 1448.
33. Lowen W. К-, Broge Е. С., J. Phys. Chem., 65, 16 (1961).
34. Koberstein Е., Voll М., Z. Phys. Chem. (Frankfurt), 71 (4—6), 275
(1970).
35. Brunauer S., J. Colloid Interface Sci., 41, 613 (1972).
36. Harkins W. D., Jura G., J. Am. Chem. Soc., 66, 1366 (1944).
37. Древинг В. П., Киселев А. В., Лихачева О. А.— ЖФХ, 1951, т. 25, с. 710.
38. Shull С. G., J. Am. Chem. Soc., 70, 1405 (1948).
39. Cranston R. W., Inkley F. A., Adv. Catal., 9, 143 (1957).
40. De Boer J. H., Linsen B. G., Osinga Th. J.. J. Catal., 4, 643 (1965).
41. -De Boer J. H., Lippens В. C., Linsen B. G., J. Catal., 3, 32, 38, 44 (1964);
4, 319, 649 (1965).
42. De Boer J. H., Lippens В. C., Linsen B. G. et al., J. Colloid Interface
Sci., 21, 405 (1966).
43. Dolllmore D., Heal G. R., J. Colloid Interface Sci., 33, 508 (1970).
44. Girgis B. S., J. Appl. Chem. Biotechnol., 23 (1), 19 (1973).
45. De Boer J. H., Linsen B. G., Broekhoff J. С. P„ Osinga Th. G., J. Catal.,
11, 46 (1968).
46. Ternan M., J. Colloid Interface Sci., 45, 270 (1973).
47. Brunauer S., J. Colloid Interface Sci., 39, 435 (1972); 41, 613 (1972).
48. Hagymassy J., Jr., Brunauer S., Mikhail R. Sh., J. Colloid Interface Sci.,
29, 485 (1969).
49. Sing K- S. W., Ref. 8, p. 34.
Силикагели и порошки
839
50. Bhambhani М. R., Cutting Р. A., Sing К. S. W., Turk D. И., J. Colloid
Interface Sci., 38 (1), 109 (1972).
51. Киселев А. В.— Успехи химии, 1945, т. 14, с. 367.
52. Kiselev А. V., in Dubinin М. М., Methoden zur Structurutersuchung an
hochdispersen und pordsen Stoffen, Akademie Verlag, West Berlin, 1961,
p. 132.
53. Каганер M. Г,— ДАН СССР, 1957, т. 116, с. 251.
54. Frenkel J., Kinetic Theory of Liquids, Oxford University Press, 1946.
55. Halsey G. D., J. Chem. Phys., 16, 931 (1948).
56. Hill T. L., Adv. Catal., 4, (1952). J. Chem. Phys., 14, 263, 441 (1946);
15, 767 (1947); 17, 580, 668 (1949).
57. Pierce Conway, J. Phys. Chem., 64, 1184, (1960).
.58. Nelsen. F. M., Eggertsen F. T., Anal. Chem., 30, 1387 (1958).
59. Lee C. F., Stross F. H., Div. Anal. Chem., Am. Chem. Soc., 135th Meet.,
Boston, Mass., April 9, 1959.
60. Ettre L. S., Cieplinski E. W., Ref. 1, p. 396.
61. Газиев Г. А., Яновский M. И., Бражников В. В. Кинетика и катализ,
1960, т. 1, с. 548.
62. Hopfe V., Marx G„ Z. Chem., 12, 370 (1972).
63. Gosslain P-, Ind. Chim. Beige, 54, 27 (1972).
64. Pommier B., Juillet F., Teichner S. J., Bull. Soc. Chim. Fr., 1972, 1268.
65. Lowell S„ Anal. Chem., 45 (8), 1576 (1973).
66. Payne D. A., Sing K- S. W., Turk D. H., J. Colloid Interface Sci., 43,
287 (1973).
67. Киселев А. В., Камакин H. M.— ДАН СССР, 1951, т. 80, с. 393.
68. Fejes D., Kira’ly J-, Schay G., Z. Anorg. Allgem. Chem., 300, 72 (1959).
69. Basiik J., Ind. Chem. Beige. Suppl., 1, 859 (1959) [Chem. Abstr. 47,
9101g],
70. Solbctkken A., Reyerson L. H., J. Phys. Chem., 63, 1622 (1959).
71. Добычин Д. П., Челинская T. Ф.— ЖФХ, 1959, т. 33, с. 204.
72. Kiselev А. V., Proc. Symp. Colston Res. Soc.,. 10, 195, 236 (1958) [Chem.
Abstr., 53, 21031a],
73. Gans D. M., Brooks U. S., Boyd G. E., Ind. Eng. Chem. Anal. Ed., 14,
396—399 (1942).
74. Kantro D. L., Brunauer S., Wise С. H., Adv. Chem., 33, 199 (1961).
75. Белякова Л. Д., Джигит О. M., Киселев А. В.— ЖФХ, 1957, т. 31,
с. 1577.
76. Cutting Р. A., Sing К- S. W., Chem. Ind. London, 1969 (9), 268.
77. Ходаков Г. С. Тр. ВНИИ новых стройматериалов, 1960, вып. 3, с. 132.
78. Ruzicka Z., Chem. Prumo, 22, 352 (1972) [Chem. Abstr., 77, 79806].
79. Giles С. H., D’Silva A. P., Trans. Faraday Soc., 65, 1943 (1969).
80. Boehm H. P., Gromes W., Angew, Chem., 71, 53, 65 (1959).
81. Kunowski H., Hofman U., Angew. Chem., 67 (11), 289 (1955).
82. Yarnto D„ Metallurgica, 82 (492), 1959 (1970).
83. Unger K-, Vydra F., J. Inorg. Nucl. Chem., 30, 1075 (1968).
84. Пошкус Д., Казлаускас P.— Коллоидн. ж., 1965, т. 27, с. 738.
85. Зеликин М. В., Сытник Л. В., Каменская Н. П. Тр. НИИ осн. химии,
вып. 15, 1963, с. 97.
86. Giles С. Н., D’SUva А. Р., Trivedi A. S., Ref. 18, р. 317.'
87. Giles С. Н., Trivedi A. S., Chem. Ind. London, 40, 1426 (1969).
88. Ter-Minassian-Saraga L. et al-, Adv. Chem., Ser., 43, 232 (1964); J. Col-
loid Interface Sci., 22, 311 (1966); J. Chim. Phys., 63, 1279 (1966);
C. R. Acad. Sci. C„ 270, 1354 (1970); J. Colloid Interface Sci., 51, 211
(1975).
89. Bijsterbosch В. H., J. Colloid Interface Sci., 47, 186 (1974); 51, 212
(1975).
90. Shapiro L, Kolthoff I. M„ J. Am. Chem. Soc., 72, 776 (1950).
840
Глава 5
91. Stryker L. J., Matijevic E., J. Colloid Interface Sci., 31, 39 (1969).
92. Murphy P. J., Posner A. M., Quirk J. P., J. Colloid Interface Sci., 39,
427 (1972).
93. Matijevic E., Stryker L. J., J. Colloid Interface Sci., 42, 232 (1973).
94. Unger R., Berg R., Gallei E., Kolloid-Z. Z. Polym. (234) (2), 1108
(1969)7
95. Meffert A., Langenfeld A., Fresenius Z., Anal. Chem., 249 (4), 231
(1970).
96. Abendroih R. P„ J. Phys. Chem., 76 (18), 2547 (1972).
97. Benesi H. A., Anal. Chem., 32, 1410 (I960).
98. Hoffman R. L., McConnell D. G., List G. R., Evans C. D., Science, 157,
550 (1976).
99. Frenkel E. N., Evans C. D., Moser H. M., McConnell D. G., J. Am. Oil
Chem. Soc., 38, 130 (1961).
100. Serpinet J. et al., J. Chim. Phys. Physicochim. Biol., 71 (6), 949 (1974).
101. Bouton L. H., Clark B. R., Coleman M. F., Thorp J. M., Trans. Faraday
Soc., 62, 10 (2928), (1966).
102. McIntosh R., Rideal E. R., Snelgrove J. A., Proc. Rov. Soc. London,
A208, 292 (1951).
103. Rurosaki S., J. Chem. Soc. Jap. Pure Chem. Sect., 73, 606 (1952).
104. Frommer M. A., Ish-Shalom M., J. Colloid Interface Sci., 21, 170 (1966).
105. Harkins W. D., Jura G., J. Chem. Phys., 11, 430 (1943); J. Am. Chem.
Soc., 66, 1356 (1944).
106. Okuda S., Uei L, Yogyo Kyokai Shi, 67, 321 (1959) [Chem. Abstr., 55,
62с].
107. Cancela Dios et al., J. Chim. Phys. Physiochem. Biol., 67 (3), 609 (1970)
[Chem. Abstr., 73, 48, 783s],
108. Whalen J. W-, J. Phys. Chem., 66, 511 (1962).
109. Taylor J. A. G., Hockey J. A., J. Phys. Chem., 70, 2169 (1966).
110. Rootare H. M., Prenzlow C. J., J. Phys. Chem., 71, 2733 (1967).
ill. ArnellJ. C„ Can. J. Res., 25A, 191 (1947).
112. Дерягин Б. B„ Фридлянд P., R рылова В.— ДАН СССР, 1948, т. 61,
с. 653.
113. Журавлев Л. Т., Ниселев А. В., см. [18], с. 155.
114. Tiwari R. N„ Naidu S. R., Ganguli N. C., Sen S. P., Technology, 7 (3),
134 (1970).
115. Shull C. G., Elkin P. B., Roess L. C., J. Am. Chem. Soc., 70, 1410—1414
(1948).
116. Bastick J., Faivre R., C. R. Acad. Sci., 242, 1166 (1956).
117. Rakudo M., Rasai N., Rimura M., Ribota Y„ Watase T., Nippon Kagaku
Zasshi, 78, 821 (1957) [Chem. Abstr., 52,.5089e],
118. Tamagusuku S., Shuin T., Jap. J. Appl. Phys., 9 (4), 356 (1970).
119. Bradaczek H., Pleith R., Schueller R., Glastech. Ber., 42 (3), 96 (1969).
120. Perry J. H., Ed., Chemical Engineer’s Handbook, 3rd ed., McGraw-Hill,
New York, 1950, p. 1109.
121. Van den Hui G. G., Lyklema J., J. Am. Chem. Soc., 90, 3010 (1968).
122. Lyklema J., Van den Hui H. J., Ref. 18, p. 341.
123. Linsen B. G., Van den Heuvel A., in E. A. Flood, ed., The Solid-Gas
Interface, Vol. 2, Dekker, New York, 1967, p. 1025.
124. Dubinin M. M., J. Colloid Interface Sci., 23, 487 (1967).
125. Sugar L, Guba F., Acta Chim. Acad. Hung., 7, 233 (1955).
126a. Леонтьев E. А., Лукьянович В. M.— ЖФХ, 1958, т. 32, с. 1922. Сб. Ме-
тоды исследования структуры высокодисперсных и пористых тел. М.:
АН СССР, 1958, с. 19—44.
1266. Диселев А. В,— ДАН СССР, 1954, т. 98, с. 431.
127. Mason S. G., J. Colloid Interface Sci., 35, 279 (1971).
Силикагели и порошки
841
128. Dollimore D., Heal G. R., Pore Struct. Prop. Mater. Proc. Int. Symp.
Rilem/Iupac, Part 1, Academia, Prague, 1973, p. A73.
129. Avery R. G., Ramsay J. D. F., J. Colloid Interface Sci., 42, 597 (1973).
130. Meissner H. P., Michaels A. S., Raiser R., Ind. Eng. Chem., Process
Des. Div., 3, 202 (1964).
131. Heesch H., Laves F., Z. Krist., 85, 443 (1933).
132. Manegold E., Hoffman R„ Soff R., Kolloid-Z., 56, 142 (1931).
133. Scott G. D„ Nature, 188, 908 (I960); 194, 956 (1962).
134. Bernal J. D„ Mason J., Nature, 188, 910 (I960).
135. Bernal J. D., Mason J., Rnight R. R., Nature, 194, 957 (1962).
136. Dollimore D., Heal G. R., Nature, 208, 1092 (1965).
137. Nicolaon G. A., Teichner S. J., Bull. Soc. Chim. Fr., (1968) (11), 4343.
138. Thompson J. (Lord Kelvin), Phil. Mag., 47, 448 (1871).
139. McBain J. W., Colloid Symposium Monograph, Vol. 4, Williams and
Wilkins, Baltimore, 1926, p. 1.
140. Brunauer S., Ref. 26, p. 345.
141. Carman P. C., J. Phys. Chem., 57, 56 (1953).
142. Shereskefsky J. L., Russel E. R., J. Phys. Chem., 57, 55 (1953).
143. Coelingh M. B„ Kolloid-Z., 87, 251 (1939).
144. Brunauer S., Ref. 26, pp. 394—408.
145. Bering В. P., Dubinin M. M., Serpinsky V. V., J. Colloid Interface Sci.,
21,378 (1966).
146. Riselev A. V., Ref. 3, p. 195.
147. Lard E. W„ Brown S. M., J. Catal., 25 (3), 451 (1972).
148. Standard for 1952, Vol. 4, American Society of Testing Materials, Phila-
delphia, p. 195.
149. Broekhoff J. С. P., Linsen B. G., Ref. 4, p. 1.
150. Beaven С. H. J., Eadington P., A Computer Program for Surface Area
Calculations by a BET Method”, Chem. Ind., 1484—1487 (1966).
151. Roberts B. F., “A Procedure for Estimating Pore Volume and Area
Distributions from Sorption Isotherms”, J. Colloid Interface Sci., 23,
266—273 (1967).
152. Harris M. R., Sing R. S. W., Chem. Ind. (London), 487-488 (1959).
153. Бебрис H. R., Киселев А. В., Никитин Ю. C.— Коллоидн. ж., 1967, т. 29,
с. 326.
154. Lippens В. С., Linsen В. G., De Boer J. H., J. Catal., 3, 32 (1964).
155. De Boer J. H., Linsen B. G., Osind T. J., J. Catal., 4, 643 (1965).
156. Broekhoff J. С. P„ Linsen B. G., Ref. 4, p. 13.
157. Barrett E. P., Joyner L. G., Halenda P. P., J. Am. Chem. Soc., 73, 373
(1951).
158. Pierce C., J. Phys. Chem., 57, 149 (1953).
159. Hougen O. A., Ind. Eng. Chem., 53, 7, 509 (1961).
160. Broekhoff J. С. P„ De Boer J. H., J. Catal., 9, 8, 15 (1967).
161. Sing R. S. W., Ref. 8, p. 45.
162. Okkerse C., “Submicroporous and Macroporous Silica”, thesis, Delft
Uitgevers Maatschappij, N. V. Delft, Netherlands, 1961.
163. Broekhoff J. С. P., Linsen B. G., Ref. 4, p. 33.
164. Mikhail R. Sh., Brunauer S., Bodor E. E., J. Colloid Interface Sci., 26,
54 (1968).
165a. Mieville R. L., J. Colloid Interface Sci., 41, 371 (1972).
1656. Ramsay J. D. F., Avery R. G„ J. Colloid Interface Sci., 51, 205 (1975,).
165b. Raeanep M. Г.— ЖФХ, 1959, t. 33, c. 2202.
166. Brunauer S., Mikhail R. Sh., Bodor E. E., J. Colloid Interface Sci., 24,
451 (1967).
167. Hanna R. M., Odler I., Brunauer S., Hagymassey J., Jr., Bodor E. E.,
J. Colloid Interface Sci. ,45, 38 (1973).
168. Havard D. C., Wilson R., J. Colloid Interface Sci., 57, 276 (1976).
842
Глава 5
169. Hagymassey J., Jr., Brunauer S., J. Colloid Interface Sci., 33, 317 (1970).
170. Dubinin M. M., J. Colloid Interface Sci., 46, 351 (1974).
171. Marsh H., Rand B„ J. Colloid Interface Sci., 33, 478 (1970); 40 121
(1972).
172. Dollimore D„ Heal G. R„ Faraday Soc. Trans., 59, 2386 (1963).
173. Sing K. S. W., Ramakrishna V. R., Colloq. Int. CNRS, No 201, 435
(1972).
174. Dios-Cancela Gonzalo, Rouquerol F., Rouquerol J., J. Chim Phys Chem.
Biol., 67 (3), 609 (1970) [Chem. Abstr. 73, 48783s],
175. Mikhail R. Shr., Shebl F. A., J. Colloid Interface Sci., 38, 35 (1972).
176. Mikhail R. Shr., El-Akkad T., J. Colloid Interface Sci., 51, 260 (1975).
177. Bartell F. E., Bower J. E., J. Colloid Sci., 7, 80 (1952).
178. Fu У., Bartell F. E., J. Am. Chem. Soc., 73, 308 (1951).
179. Bodor E. E„ Skalny J., Brunauer S., J. Colloid Interface Sci, 34, 560
(1970).
180. Dollimore D., Shingles T., J. Chem. Soc., A, 1971 (7), 872.
181. Высоцкий 3. 3., Поляков M. В.— Коллоидн. ж., 1961, т. 23, с. 216.
182. Gregg S. J., Langford J. F., Trans. Faraday Soc., 65, 1394 (1969).
183. Ritter H. L„ Erich L. C., Anal. Chem., 20, 665 (1948).
184. Longman G. W., Wignall G. D. et al., Colloid Polym. Sci., 252 (4), 298
(1974).
185. Порай-Кошиц E. А. и dp.— ДАН СССР, 1952, т. 86, с. 985. >
186а. Porod G., Kolloid-Z., 124, 83 (1951).
1866. Imelik В., Teichner S., Carteret У., J. Chem. Phys., 48, 438 (1951).
187. Washburn E. W„ Phys. Rev., 17, 273 (1921).
188. Ritter H. L., Drake L. C., Ind. Eng. Chem. Anal. Ed., 17, 782 (1945).
189. Guyer A., Bohlen B., Guyer A., Helv. Chem. Acta, 42, 2103 (1959).
190. DeWit L. A., Scholten J. J. F., J. Catal., 36, 36 (1975).
191. Zweitering, Ref. 3, p. 287.
192. Frevel L. K-, Kressley L. J., Anal. Chem., 35, 1492 (1963).
193. Thorp J. M., Woolf J. B., Trans. Faraday Soc., 63, 2068 (1967).
194. Maatman Russel W., et al., J. Miss. Acad. Sci., 8, 193, 201 (1962); 9,
44 (1963). -
195. Гольданский В. И., Неймарк И. Е., Плачинда А. С., Суздалев И. Л.
Пористая структура катализаторов и процессы переноса в гетерогенном
катализе/Под ред. Г. К. Борескова, Новосибирск, Наука, СО АН СССР,
1970, с. 155—163.
196. Barby D., Ref. 8, р. 353.
197. Alexander G. В., Iler R. К-, Wolter F. J., пат. США 2731326 (Du Pont),
1956.
198. Wolf К., Praetorius М., Metallbourse, 18, 453, 789, 1293, 2245 (1928)
[Chem. Abstr., 23, 2787].
199. Vail J. G., Soluble Silicates (ACS Monograph Series), Reinhold, New
York, 1952, vol. 1, p. 158; vol. 2, p. 549.
200. Неймарк И. E.— Успехи химии, 1956, т. 25, с. 748.
201. Romos R. de C., Vega M. N., Rev. Cienc. Apl. Madrid, 8, 11, 132 (1954).
202. Kiselev A. V., Trans. Faraday Soc. Disc. (52), 14 (1971).
203. Kondo S., Hyomen, 10 (6), 321 (1972).
204. Неймарк И. E., Шейнфайн P. Ю. Силикагель, его получение, свойства
и применение. Киев, «Наукова думка», 1973.
205. Овчаренко Ф. Д., Тарасевич Ю. И. и др.— Коллоидн. ж., 1973, т. 35,
вып. 3, 467.
206. Flanigen Е. М., Grose R. W., пат. США 3494874 (Union Carbide Corp.),
1970.
207. Murata К. J-, Schlecht W. G., U. S. Geol. Surv. Bull., 950, 25—33, 35—
82 (1946).
208. Brunauer, Ref. 26, p. 298.
Силикагели и порошки
843
209. Patrick W. A., McGavack I., J. Am. Chem. Soc., 42, 946 (1920); пат.
США 1297724 (1919).
210. Wolf F„ Beyer H„ Kolloid-Z., 165, 151 (1959).
211. Takigawa K., Ohba У, япон. пат. 73— 13834 (Asahi Chem. Ind.), May,
1973 [Chem. Abstr., 80, 17009j],
212. Gandon L., Lehman R. L., Marcheguet H. G. L., Tarbouriech F. P. M.,
пат. США 3028340 (Soc. Nobel Bozel), 1962.
213. Plaisted A. C., Pearson L. L., пат. ФРГ 2258111, 1973 [Chem. Abstr., 79,
96289f],
214. Holmes S. T., пат. США 3095312 (1963).
215. Winyall M. E„ Acker E. G, пат. США 3501269 (W. R. Grace Co.), 1970;
also англ. пат. 1077908, 1967.
216a. Hurd С. B. et al., J. Phys. Chem, 36, 604, 2194 (1932); 37, 321 (1933);
40, 21 (1936); 41, 553 (1937); 44, 57, 847 (1940); 45, 588 (1941); J. Am.
Chem. Soc, 62, 2767 (1940); 66, 388 (1944).
2166. Sen К. C, Ghosh B. N„ J. Indian Chem, 31, 803, 807 (1954) [Chem.
Abstr, 49, 1292bd].
217. Merrill R. C., Spencer R. W., J. Phys. Chem, 54, 806 (1950).
218. Heald I. A., Coates К. B., Edwards J. E., J. Appl. Chem. London, 5,
425 (1955).
219. Mookerjee S. K., Niyogi S. K-, Cent. Glass and Ceram. Res. Inst. Bull,
22, 1 (1975).
220. Iler R. K, J. Phys. Chem, 57, 604 (1953).
221. Высоцкий 3. 3., Стражеско Д. H., см. [5], с. 58.
222. Канторович С. И., Лаврова К. А. и др.— Коллоидн. ж, 1973, т. 35,
вып. 5, 935.
223. Mindick М., Reven L. Е., пат. США 3468813 (Nalco Chemical Со.).
1969.
224. Lenher V, J. Am. Chem. Soc, 43, 391 (1921).
225. Schwarz V. R„ Richter H„ Вег, 62B, 31 (1929).
226. Bartell F. E., Donahue D. J., J. Phys. Chem, 56, 665 (1952).
227. Radczewski О. E., Richter H., Kolloid-Z, 96, 1 (1941).
228. Unger K-, Scharf B., J. Colloid Interface Sci, 55, 377 (1976).
229. Киселев А. В., Лыгин В. И.— Коллоидн. ж, 1958, т. 20, с. 47.
230. Sen К. С., Ghosh В. N., J. Indian Chem. Soc, 33, 404 (1956).
231. Plank С. J., Drake L. C., J. Colloid Sci, 2, 399 (1947).
232. Cuha D., Gupta K., J. Indian Chem. Soc, 50 (2), 86 (1973) [Chem.
Abstr, 79, 80881].
233. Sing K. W, Madeley I. D., J. Appl. Chem. London, 3, 549 (1953).
234. Outouma H., Ukihashi H., Bull. Chem. Soc. Jap, 38 (10), 1634 (1965).
235. Gartner K., Griessbach R., Kolloid-Z, 160 (1), 21 (1958).
236. Okkerse C., de Boer J. H., Mme Cyrot, J. Chim. Phys, 57, 534 (1960).
237. Шейнфайн P. Ю„ Неймарк И. E.— Кинетика и катализ, 1970, т. 8,
с. 433.
238а. Высоцкий 3. 3., Стражеско Д. Н., см. [5], с. 62.
2386. Добрускин В. X. и др.— Ж. прикл. химии, 1967, т. 40, вып. 11, с. 2443.
238в. Sippel R. I., пат. США 3397153 (Du Pont), 1968.
239. Hisamine Kobayashi et al., пат. США 3526602 (Kabushiki Kaisha Shiki-
shima MUL-B-11), 1970.
240. Montgomery D. S., Parsons В. I., пат. США 3417028, 1968.
241. Bailie J. С., пат. США 2457970 (Standard Oil of Indiana), 1949.
242. Winyall M. E., Ellicott, Acker E. G., пат. США 3607777, 1971; франц,
пат. 2011162 (W. R. Grace Co.), 1970.
243. Bergna H. E., Simko F. А. пат. США 3284369, 1966; пат. США 3301635,
1967 (both Du Pont).
244. Vail J. G, Ref. 199, Vol. 2, p. 560.
844
Глава 5
245. LePage М., Beau R., Duchene J., франц, пат. 1473240 (Prod. Chim.
Pechinev-Saint-Gobain), 1967 [Chem. Abstr., 68, 990441].
246. Drexel R. E., пат. США 2757073 (Du Pont), 1956.
247. Gring J. L., пат. США 2641583 (Sinclair Reforming Inc.), 1953.
248. Ritter H. S., пат. США 2921839 (Columbia Southern Chem. Corp.), 1960.
249. Schwartz А. В., пат. США 3165279 (Socony Mobil Oil Co.), 1962.
250a. Moehl R. W., пат. США 2759898 (Universal Oil Prods.), 1956.
2506. Kummerle H. F., пат. США 3489516 (Owens Illinois Co.), 1970.
251. Kohlschutter H. W., Mihm U., Rolloid Z. Z. Polym., 243, 148 (1971).
252. Unger K-, Scharf B., J. Colloid Interface Sci., 55, 377 (1976).
253. Unger K-, пат. ФРГ 2155281 (Merck G. m. b. Ы.), 1973 [Chem. Abstr.,
79, 94153h],
254a. Burzynski A. J., Martin R. E., пат. США 3321276 (Owens Illinois Inc.),
1967.
2546. Thomas I. M., пат. США 3709833 (Owens Illinois Inc.), 1973.
255. Kummerle H. F., пат. США 3656901 (Owens Illinois Inc.), 1972.
256. Hazel I. F. et al., J. Phys. Coll. Chem., 51, 415 (1947); пат. США
2561304 (1951); J. Colloid Sci., 5, 532 (1950); Science, 110, 161 (1949).
257. Lottermoser A., Langenscheid F., Kolloid-Z., 58, 336 (1932).
258. Kautsky H., Chapter 1, Refs. 92—94.
259. Hinz W., Ruttloff H., Teufel A., Silik. Tech., 13, 378 (1962) [Chem. Abstr.,
58, 10758].
260. Колосенцев С. Д., Белоцерковский Г. M., Плаченоё Т. Г.— Ж- прикл.
химии, 1975, т. 48, вып. 1, с. 252.
261. Dollimore D., Shingles Т., J. Colloid Interface Sci., 29, 601 (1969).
262. Wulf G., пат. ФРГ 1268122, 1968.
263. Шарыгин Л. М., Чуклачев В. Г.— Ж. физ. химии, 1968, т. 42, с. 2120.
264. Butcher И. J., Simpson Е. А., пат. США 3681017; франц, пат. 2098070;
пат. ФРГ 2132222, 1972 [Chem. Abstr., 76, 74371m],
265. Burke О. W., Jr., пат. США 3401017, 1968.
266. Шарыгин Л. М., Чуклачев В. Г.— Коллоидн. ж., 1969, т. 31, с. 459.
267. Halberstadt Е. S., Henisch Н. К-, Nickl I., White Е. W., J. Colloid Inter-
face Sci., 29, 467 (1969).
268. Волькин В. В., Пономарев Е. И., Золтавин В. Л.— Коллоидн. ж., 1973,
т. 35, вып. 1, с. 144.
269. Wainer Е., Mayer Е. F., пат. США 3096144 (Horizons, Inc.), 1963.
270. Veach F., Burhans R. W., пат. США 2797201 (Standard Oil Co. of Ohio),
1957 [Chem. Abstr., 52, 798e],
271. Alexander G. B., Iler R. K-, Wolter F. J., пат. США 2731326 (Du Pont),
1956.
272. Yates P. С., канад. пат. 878444 (Du Pont), 1971.
273. Alexander G. B., Broge E. C., Iler R. К. пат. США 2765242 (Du Pont),
1956.
274. Неймарк И. E., Слинякова И. В,—Коллоидн. ж., 1956, т. 18, с. 219.
275. Okkerse С., de Boer J. Н., in J. H. de Boer, Ed., Reactivity of Solids,
Elsevier, Amsterdam, 1960, p. 240; see also Silic. Ind., 27, 195 (1962).
276. Шейнфайн P. Ю., Стас О. П., Неймарк И. Е.— Коллоидн. ж., 1965,
т. 27, вып. 6, с. 916.
277. Girgis В. S„ Z. Phys. Chem. (Frankfurt), 83 (1—4), 75 (1973).
278. Jenkins E. E., Schwartz А. В., пат. ФРГ 1226741 (Mobil Oil Corp.).
1968.
279. Неймарк И. E., Слинякова И. В.—Доповщи АН УРСР, 1955, № 5,
с. 469.
280. Слинякова И. В., Неймарк И. Е.— Коллоидн. ж., 1958, т. 20, с. 84.
281. Dollimore D., Heal G. R., J. Appl. Chem. London, 12 (10), 445 (1962).
282. Шейнфайн P. Ю., Липкинд В. А., Стас О. П., Неймарк И. Е.— Кол-
лоидн. ж., 1964, т. 26, вып. 6, с. 734.
Силикагели и порошки
845
283, Стас О. П., Шейнфайн Р. Ю., Неймарк И. Е.— Коллоидн. ж., 1967,
т. 29, с. 256; 1970, т. 32, вып. 1, с. 104.
284. Naidu S. R., Tiwari R. N., Ganculi N. C., Sen S. P., Technology, 9
(2—3), 130 (1972) [Chem. Abstr., 78, 140684].
285. Acker E. G., пат. США 3313739, 1967.
286. Preston L., Veltman L., Hubbard S. S., пат. США 2698062 (W. R. Grace
Co.), 1954.
287. Шейнфайн P. Ю. и dp.— Коллоидн. ж., 1961, т. 23, с. 756; 1963, т. 25,
с. 732; Кинетика и катализ, 1970, т. 8, с. 433.
288. Zsigmondy R., Z. Anorg. Allgem. Chem., 71, 356 (1911).
289. Hermans P. H., Ref. 106, pp. 536, 571—573.
290. Barkas W. W„ Trans. Faraday Soc., 42, 147 (1946).
291. Bikerman J. J., Surface Chemistry for Industrial Research, Academic,
New York, 1948, p. 33.
292. Alexander A. E., Johnson P., Colloid Science, Vol. 2, Clarendon Press,
Oxford, 1946, p. 607.
293. Foster A. G., Proc. Roy. Soc. London A147, 128 (1934); A150, 77 (1935).
294. Manegold E., Huertel M., Kolloid Z., 116, 66 (1950).
295. Imelik B., Ref. 3, p. 237.
296. Foster A. G., Thorp J. M., Ref. 3, p. 227 [includes references to Foster’s
related works].
297. Behrman A. S., пат. США 2400907, 1946.
298. Teter J. W., пат. США 2330640 (Sinclair Refining Co.), 1943.
299. Kearby К. К., пат. США 2429319 (Standard Oil Development Co.), 1947.
300. Archibald R. C„ пат. США 2438379 (Shell Development Co.), 1948.
301. Pierce J. A., Kimberlin C. N., пат. США 2454941, 1948; пат. США
2474910, 1949 (both Standard Oil Development Co.).
302. See M. J., Bailie J. С., пат. США 2455445 (Standard Oil Development
Co.), 1948.
303. Неймарк И. E., Шейнфайн P. Ю.— Коллоидн. ж., 1953, т. 15, с. 145.
304а. Пат. ФРГ 22'47413, 1972 [англ. пат. 1365784 (W. R. Grace Со.)].
3046. Sirianni A. F., Puddington I. Е., Сап. J. Chem. Eng., 42, 42 (1964).
305. Kistler S. S., J. Phys. Chem., 36, 52—64 (1932); пат. США 2093454
(1937) and 2249767 (1941); Nature, 127, 711 (1931); Kistler S. S., Fi-
scher E. A., Freeman I. R., J. Am. Chem. Soc., 65, 1909 (1943).
306. Marshall M. D., пат. США 2285449 (Monsanto Chemical Co.), 1942.
307. White J. F., Trans. Am. Inst. Chem. Eng., 38, 435 (1942); Chem. Ind.,
51, 66 (1942).
308. Anonymous, Chem. Met. Eng., 50, 144 (1943).
309. Harvey E. N., Jr., “Absorption Studies on Silica”, thesis, Princeton Uni-
versity, 1941.
310. Johnson M. F. L., Ries H. E., J. Am. Chem. Soc., 72, 4289 (1950).
311. Van Nordstrand R. A., Kreger W. E., Ries H. E., Jr., J. Phys. Colloid
Chem., 55, 621 (1951).
312. White J. F., Wilson W. S., пат. США 2807588 (Monsanto Chemical Co.),
1957.
313. Nickerson R. F., пат. США 2927083 (Monsanto Chemical Co.), 1960.
314. Неймарк И. E., Шейнфайн P. IO. Авт. свид., СССР, 107276, 1957.
315. Taulli T. А., пат. США 3346507 (Monsanto Chemical Co.), 1967.
316. Teichner S. J., Nicolaon G. A., Vicarini M. A., Gardes G. E. E., Adv.,
Colloid Interface Sci., 5, 245 (1976).
317. Teichner S. J., Nicolaon G. А., пат. США 3672833 (State of France),
1972; see also франц, пат. 1544835, 1968 [Chem. Abstr., 71, 93165m].
318. Nicolaon G. A. Teichner S. J., Bull. Soc. Chim. Fr., 1968, 1900, 1906,
3107, 3555, 4343.
319. Ries H. E., Jr., in Advances in Catalysis and Related Subjects, Vol. 4,
Academic, New York, 1950, p. 87.
846
Глава 5
320. Weyl W. A., “Atomistic Interpretation of the Mechanism of Solid State
Reactions and of Sintering, Crystal Chemistry as Applied to Ceramics”,
Fifth Annual Symposium, School of Mineral Industry, Pennsylvania
State College, State College, Pa., June 26, 1952.
321. Киселев А. В. и dp.— Коллоидн. ж., 1966, т. 28, вып. 5, с. 662- 1969,
т. 31, с. 388.
322. Чертов В. М„ Джамбаева Д. Б., Неймарк И. Е.— Укр. хим. ж., 1965,
т. 31, вып. 11, с. 1149.
323. Бебрис Н. К., Киселев А. В., Никитин Ю. С.— Коллоидн. ж., 1967, т. 29,
вып. 3, с. 326.
324. Robinson Е., Ross R. A., Can. J. Chem., 48, 2210 (1970).
325. Schwartz А. В., пат. США 2914486 (Socony Mobil Oil Со.), 1952.
326а. Weise Р. В., пат. США 2746935 (Socony Mobil Oil Со.), 1956.
3266. Jenkins Е. Е., Schwartz А. В., пат. США 3079234 (1963); see also
2480628, 2940830 (Socony Vacuum Oil Co.) [Chem. Abstr., 58, 9658e].
327. Schlaffer W. G., Adams C. R., Wilson J. N„ J. Phys. Chem., 69, 1530
(1965).
328. Chakrabarti V. K-, Josh V., J. Indian Inst. Sci., 58 (1), 28 (1976).
329. Киселев А. В., Никитин Ю. С. и dp.— Коллоидн. ж., 1968, т. 30, вып. 6,
с. 842; 1969, т. 31, с. 525.
330. Bohlen В., Buehlmann М., Guyer A., Chimia (Aarau), 20, 32 (1966)
[Chem. Abstr., 65, 11381f],
331. Buehlmann M. R., Promotionsarbeiten No. 3540, 112 (1964) [Chem. Abstr.,
65, 144, 766].
332. Чертов В. M., Джамбаева Д. Б., Неймарк И. Е.— Коллоидн. ж., 1965,
т. 27, с. 279.
333. Hill Т„ пат. США 3647709 (BP Chemicals Ltd., London), 1972.
334. De Boer J. H., Vleeskens J. M., K. Ned. Akad. Wet., 60, 234 (1957)
[Chem. Abstr., 51, 17324f]_
335. Якобсон Д„ Киселев А. В., Кузнецов В. В., Никитин Ю. С.— Коллоиди.
ж., 1970, т. 32, с. 41.
336. Горелик Р. Л., Журавлев Л. Т., Киселев А. В. и др.— Коллоидн. ж.,
1971, т. 33, вып. 1, с. 51.
337. Горелик Р. Л., Давыдов В. Я-, Журавлев Л. Т. и др.— Коллоидн. ж.,
1973, т. 35, вып. 3, с. 456.
338. Schlaffer W. G., Morgan С. Z., Wilson J. N., J. Phys. Chem., 61, 714
(1957).
339. Adams C. R„ Voge H. H„ J. Phys. Chem., 61, 722 (1957).
340. Акшинская H. В., Безносова В. E„ Киселев A. В., Никитин Ю. C.—
Ж. физ. химии, 1962, т. 36, с. 2277; Коллоидн. ж., 1970, т. 32, с. 160,
341. Le Page М., пат. ФРГ 2127649; франц, пат. 2093176 (Prod. Chim. Pechi-
ney-Saint-Gobain), 1971 [Chem. Abstr., 76, 50497g].
342. Mme. Gallard-Hasid, Jones H., Imelik B., Bull. Soc. Fr., 1959, 608 (1959).
343a. Киселев А. В., Никитин Ю. С. и dp.— Коллоидн. ж., 1967, т. 29, с. 95.
3436. Patrick W. A., Froyer J. C. W., Rush R. J., J. Phys. Chem., 31, 1511
(1927).
344. Milligan W. O., Hachford H. H., Jr., J. Phvs. Colloid Chem., 51, 333
(1947).
345. Van Nordstrand R. A., Kreger W. E., Ries H. E., Jr.. J. Phys. Colloid
Chem., 55, 641 (1951).
346. Bastick J., Bull. Soc. Chim. Fr.. 20, 437 (1953).
347. Kohlschutter H. W., Kampf G,, Z. Anorg. Chem., 292, 298 (1957).
348. Goodman J. F., Gregg S. J., J. Chem. Soc., 1959, 694.
349. De Boer J. H., Ref. 3, p. 291.
350. Wolf F. H., Beyer H., Z. Anorg. Chem., 300, 33 (1959).
351. Malanchuk. M., Stuart E. B., Ind. Eng. Chem., 50, 1207 (1958).
352. Fraissard J., Imelik B., J. Chim. Phys., 59, 415 (1962).
Силикагели и порошки
847
353. Кеппа В. Т., Conrad F. J., Taianta, 15 (4), 418 (1968) [Chem. Abstr.,
69, 8202m], '
354. DeKeyser W. L„ Cypres R., Silic. Ind., 26, 237 (1961).
355. Акшинская H. В., Киселев А. В., Никитин Ю. C.— Коллоидн. ж., 1967,
т. 29, вып. 1, с. 11.
356. Milliken Т. Н., Jr., Mills G. A., Oblad A. G., “Heterogeneous Catalysis”,
Disc. Faraday Soc., 8, 279 (1950).
357. Mougey C., Rossetti J. F., Imelik B., Ref. 3, p. 266.
358. Godon-Renou J., Fraissard J., Rossetti J. F., Imelik B., Bull. Soc. Chim.
Fr., 1962, 1156.
359. Kondo S., Fujiwara F., Muroya M., J. Colloid Interface Sci., 55, 421
(1976).
360. Kumar S., Nag В. B., Trans. Indian Ceram. Soc., 27 (3), 103 (1968)
[Chem. Abstr., 70, 108748j],
361. De Boer J. H., Ref. 3, p. 290.
362. Bassett D. R., Boucher E. A., Zettlemoyer A. C., J. Mater. Sci., 7, 1379
(1972).
363a. Uytierhoeven J., Andre J. et al., Bull. Soc. Chim. Fr., 1965, 1804.
3636. Elmer T. H., Nordberg M. E., J. Am. Ceram. Soc., 50, 275 (1967).
363b. Elmer T. H., Meissner H. E., J. Am. Ceram. Soc., 58, 206 (1976).
364. Van der Waals J. D., Versl. K. Ned. Akad. Wet., 5, 150 (1897); 6, 160
(1898); 7, 350 (1899).
365. Broekhoff J. С. P., Van Dongen R. H., cm. [4], c. 79—87.
366. Lenher V., J. Am. Chem. Soc., 43, 391 (1921).
367. Ramsay J. D. F., Avery. R. G., in S. Modry, Ed., Pore Struct. Prop. Ma--
ter. Proc. Int. Symp. Rilem/Iupac, 1973, Vol. 3, Academia Press, Prague,
1974, pp. B37—B45.
368. Blount D. H., пат. США 3929972 (1975); see also 3937782, 3954941,
3956466, 3960747, 3962067, 3962111, 3979362, 3993737.
369. Акшинская H. В., Давыдов В. Я., Киселев А. В., Никитин Ю. С.—
Коллоидн. ж., 1966, т. 28, с. 3.
370. Trademark registered by Corning Glass Works.
371. Nordberg M.E., Hood H.P., пат. США 2215039 (1934); 2106744 (1938);
2286275 (1942); 2315329 (1943) (all Corning Glass Works).
372. Elmer T. H., Nordberg M. E., Mater. Des. Eng. (Dec. 1962); J. Am.
Ceram. Soc., 53, 171 (1970).
373. Dickey F. H., Proc. Natl. Acad. Sci. U. S., 35, 227 (1949).
374. Pauling L., Chem. Eng. News, 27, 913 (1949).
375. Bernhard S. A., J. Am. Chem. Soc., 74, 4946 (1952).
376. Curti R., Colombo U., J. Am. Chem. Soc., 74, 3961 (1952); Gazz. Chim.
Ital., 82, 491 (1952).
377. Dickey F. H„ J. Phys. Chem., 59, 695 (1955).
378. Haldeman R. G., Emmett P. H., J. Phys. Chem., 59, 1039 (1955).
379. Beckett A. H., Anderson P., Nature, 179, 1074 (1957); J. Pharm. Phar-
macol., 11, 258T (1959).
380. Beckett A. H., Anderson P., Nature, 179, 1075 (1957).
381. Waksmundzki A., Rosz. Chem., 32, 323 (1958).
382. Высоцкий 3. 3., Дивнич Л. Г., Поляков М. В.— Докл. АН СССР, 1961,
т. 139, № 6, с. 1400.
383. Высоцкий 3. 3., Дивнич Л. Г. и др.— Коллоидн. ж., 1961, т. 23, с. 211.
384. Патрикеев В. В,— ДАН СССР, 1962, т. 146, с. 707.
385. Патрикеев В. В.— Изв. АН СССР, сер. хим., 1963, с. 1031.
386. Bartels Н., Erlenmeyer Н., Helv. Chim. Acta., 48 (2), 285 (1965).
387. Bartels H., Brijs В., Helv. Chim. Acta, 49 (5), 1621 (1966).
388. Гребенщикова О. Г. и др.— Вопросы мед. химии, 1975, т. 21, вып. 4,
с. 396.
389. Канаболоцкий В. А. и др. см. [5], с. 180, 185.
848
Глава 5
390. Стрелке В. В. и др,— ЖФХ, 1967, т. 41, с. 872; 1968, т. 42, с. 1219.
391. Waksmundzki A. et al., Przem. Chem., 42 (4—5), 193 (1963).
392. Erlenmeyer H., Bartels H., Helv. Chim. Acta, 47, 1285 (1964),
393. Bartels H., Z. Anorg. Allgem. Chem., 350, 143 (1967).
394. Bartels H., Prijs B., Adv. Chromatogr., 11, 115 (1974).
395. .Bartels H., J. Chromatogr., 30 (1), 113 (1967).
396. Канаболоцкий В. А. и др.— Адсорбция и адсорбенты, Респ. межвед.
сб. 1972, вып. 1, с. 105, с. 107.
397. Галинская В. И., Стрелка В. В., Стражеско Д. Н.— ДАН СССР, 1973,
т. 210, вып. 2, с. 468.
398. Стрелка В. В., Канаболоцкий В. А., Высоцкий 3. 3.— ЖФХ, 1968, т. 41,
вып. 5, с. 1219.
399. Alexander G. В., пат. США 3041140 (Du Pont), 1962.
400, Barby D., Ref. 8, p. 402.
401. Weiser H. B., Colloid Chemistry, 2nd ed., Wiley, New York, 1953, p. 307.
402. Alexander G. B., Iler R. K-, Wolter F. J., пат. США 2601235 (Du Pont),
1952
403. Rule J. M., пат. США 2577489 (Du Pont), 1951.
404. Marotta R., пат. США 3428425 (Monsanto C.,), 1969.
405. Hoesch, англ. пат. 945523, 1964.
406. Degussa, англ. пат. 1043282, 1966.
407. Degussa, англ. пат. 1105618, 1132831, 1968.
408. Nauroth Р., Becker А., франц, пат. 1352354 (Degussa), 1964.
409. Burke О. W. et al., англ. пат. 1122588—1122589, 1968; пат. США
3325249, 1967.
410. Koninklijke Zwavelzuur-Fabrieken Voorheen Ketjen, Neth. Appl. 6502791,
1966 [Chem. Abstr., 66, 3061 le).
411. Unilever N. V., франц, пат. 2222313, 1974 [Chem. Abstr., 82, 172332n],
412. Funnell N. A., Sirianni A. F., Puddington I. E., Can. J. Chem. Eng.,
49 (4), 541 (1971).
413. Wason S. K., Van der Heern P., пат. США 3893840, 1974, пат. ФРГ
2344805 (Soc. Fr. Silic. Spec. Sifrance), 1974 [Chem. Abstr., 81, 27773p],
414. Donnet I. B., Baudru B., Coudurier M., англ. пат. '1382340; франц.,
пат. 2159580 (Soc. Fr. Silic. Spec. Sifrance), 1973 [Chem. Abstr., 80,
61626g]; англ. пат. 1382340.
415. Aboutboul H. A., Kreckler J. H., Kirch W., пат. США 3652215 (National
Distillers & Chem. Corp.), 1972; пат. ФРГ 1940093, англ. пат. 1284086,
1294689 [Chem. Abstr., 73, 123923t],
416, Alexander G. B., Iler R. K-, Wolter F. I., пат. США 2601235 (Du Pont),
1952
417. Arseni W. C., Wright I. G. E., пат. США 1270093 (General Electric Co.),
1918.
418. Heuser R. V., пат. США 2114123 (American Cyanamid Co.), 1938.
419. Англ. пат. 203158 (J. Crosfield and Sons, Ltd.), 1922.
420. Wason S. К., пат. ФРГ 2344316 (Soc. Fr. Silicat. Spec. Sifrance), 1974
[Chem. Abstr., 81, 6571 lx].
421, Craig T. I. I., Kirkham А., англ. пат. 299483 (Peter Spence & Sons,
Ltd.), 1927.
422, Acker E. G., Sanchez M. G., пат. США 3081154 (W. R. Grace Co.), 1963.
423. Деринг H. А. и dp.— Хим. технол., 1970, т. 16, с. 7.
424. Acker Е. G., Winyall M. E„ пат. США 3787561, 1973; пат. ФРГ 2221721,
1972 (W. R. Grace & Со.) [Chem. Abstr., 78, 60294u],
425, Acker E. G., Winyall M. E., англ. пат. 1340230 (франц, пат. 2107652)
(W. R. Grace & Co.), 1972.
426. Boss A. E., Chem. Eng. News, 27, 677 (1949).
427. Allen E. M., пат. США 2910375 (Columbia Southern Chem. Corp.), 1959.
428. Degussa, франц, пат. 1529058, 1968.
Силикагели и порошки
849
429. Iler R. К., пат. США 2663650 (Du Pont), 1953.
430. Chiola V., Ritsko J. E., Vanderpool C. D., пат. США 3556725 (Sylvania
Elect. Prods., Inc.), 1971.
431. Iler R. K-, McQueston H. J., пат. США 3855172 (Du Pont), 1974.
432. Kirkland J. J., J. Chromatogr., 83, 149 (1973).
433. Alexander G. B., Iler R. K., Ref. 295, Chapter 4.
434. Moyer E. F., пат. США 2386337, 1945.
435. Kanhofer E. R., пат. США 2348072 (Universal Oil Products Co.), 1944.
436. Baker C. L., Austin J. F., пат. США 3208823 (Philadelphia Quartz Co.),
1965.
437. Gewecke F., пат. ФРГ 731343 (Deutsche Solvay-Werke, A. G.), 1937.
438. Alsfeld Max, пат. ФРГ 486951, 1927.
439. Svendsen S. S., пат. США 1859998, 1932; 1959747—1959749 (Clay Re-
duction Co.), 1934.
440. Schwarz R., Liede 0., Chem. Вег., 53B, 1680 (1920).
441. Jacobson C. A., J. Phys. Chem., 40, 413 (1936).
442. Nickerson J. D., Burkert G. M., пат. США 3271107 (Internat. Minerals
and Chem. Corp.), 1966.
443. Borsos А., пат. ФРГ 2121152 (Ruder. Topion. Basen Bor, Indrust. Hemij.
Proiz. Prahovo), 1972.
444. Patil A. S., Wirth D. G., Jr., Yeager J. L., пат. ФРГ 2131941 (W. R. Grace
Co.), 1970.
445. Stober W., Refs. 68 and 110, Chapter 4.
446. Amelin A. G., Kolloidn. Zh, 29 (1), 16 (1967).
447. Ulrich G. D„ Combust. Sci. TechnoL, 4 (2), 47 (1971).
448, Barnes W. R., англ. пат. 1211702 (1966).
449. Barnes W. R„ Barby D., англ. пат. 1211702.
450. Barby D., Ref. 8, p. 397.
451. Houseman J., пат. США 3668108 (Hercules, Inc.), 1972 [Chem. Abstr.,
77, 77184k],
452. Everest D. A., Sayce I. G., Shelton B., Symp. Electrochem. Eng., 1971,
2, 108 (1973).
453. Kugler T., Sins, Silberger J., пат. США 3733387 (Lonza Ltd.), 1973. '
454. Korman S., Sheer C., Angier D. J., Shaw H., Fine Part. Int. Conf. Paper
2nd 1973, Electrochemical Society Inc., Princeton, N. J., 1974, p. 153.
455. Pittsburgh Plate Glass Co., Chem. Week, 97, 30 (1965).
456. Potter H. N., пат. США 886637 (George Westinghouse), 1908.
457. Von Stroh G., англ. пат. 589636 (Permanente Metal Corp.), 1945; пат.
США 2428252 1947.
458. Reik R., Byrns А. С., пат. США 2428178 (Permanente Metal Corp.), 1947.
459. Biegler H., Neugebauer W., Kempers H., пат. США 3311451, 1967; пат.
ФРГ 1180723 (Degussa), 1964 [Chem. Abstr., 62, 3687g].
460. Illigen A., Neugebauer W., пат. США 3674430 (Degussa), 1968 [Chem.
Abstr,, 74, 101258y],
461. Nemetschek Th., Hof man U., Z. Naturforsch., 9b (2), 166 (1954).
462. Tanako J., Kato A., Yato, Yogyo Kyokai Shi., 81 (5), 179 (1973).
463. Kloepfner H., нем. пат. 762723; 830786 (1942).
464. Wagner E., Brunner H., Angew. Chem., 72, 744 (1960).
465. Bode R., Feach H., Fratzscher H., Paint, Oil, Color J., 415 (1968).
466. Lof Iman K. A., Ref. 1, p. 196.
467. White L. J., Duffy G. J., Ind. Eng. Chem., 51, 232 (March 1959).
468. Cuer P., Long J., Teichner S. J., Bull. Soc. Fr. Ceram., 72, 13 (1966)
[Chem. Abstr., 66, 77797k],
469. Николина В. Ю.— Хим. пром., 1962, с. 48.
470. Bittner F. et al., англ. пат. 121279 (Degussa), 1968.
471. Moore G. E., пат. США 3772427 (General Electric Co.), 1973.
28 Заказ № 250
850
Глава 5
472. Гит С., Yun С. G., Jang Н. D., Hwahak Kwa Hwahak Kongop (North
Korea), 16 (6), 324 (1973) [Chem. Abstr., 81, 138147g].
473. Cabosil registered trademark of Godfrey L. Cabot Co.
474. Aerosil trademark of Degussa, Germany.
475. Broekhoff J. С. P., Linsen B. G., Ref. 4, p. 27.
476. Spencer W. B., Smith W. R., Cosman A. F., пат. ФРГ 1080982 (God-
frey L. Cabot Inc.), 1960 [Chem. Abstr., 55, 26310g].
477. Gass J. L., Juillet F., Teichner S. J., Bull. Soc. Chim. Fr., 2 (Part 1),
429 (1973) [Chem. Abstr., 78, 140717z],
478. Secord R. N., пат. США 2886414 (Godfrey L. Cabot Corp.), 1959.
479. Engelson G. E. et al., пат. США 2631083, 1953.
480. Broughton, пат. США 2535036, 1950.
481. Heller G., White J. W., Gunn E. F., Jr., белы. пат. 618184 (Columbian
Carbon Co.), 1962.
482. Tomita T., пат. США 3540844 (Konoshima Chem. Co.), 1970 [Chem.
Abstr., 74, 55009t],
483. Flemmert G. L., пат. ФРГ Appl. P-22-51-787.9, Oct. 20, 1972; Disclosure
Document 2251787.
484. Driscoll R. E., пат. ФРГ 2132429 (Cities Service Co.), 1972; also пат.
США 3660025, 1972.
485. Fluosil trademark of A. B. Nynas-Petroleum, Stockholm, Sweden.
486. Смирнова 3. Г., Никитина H. 3. и др.— Ж. прикл. химии, 1967, т. ,40,
вып. 8, с. 1667.
487. Takagi J., Suwa М., Asaigarasu Kenkyu Hokoku, 17 (2), 99 (1967)
[Chem. Abstr., 68, 116035y],
488. Зайцев В. А., Пушков Ю. Г. и др.— Хим. пром., 1970, т. 46, вып. 12,
с. 906.
489. Bulletin 1975, Glasrock Prods, Inc., Atlanta, Ga.
490. Thomas J. et al., Ill. State Geol. Surv. Ind. Min. Notes., 40 (1970).
491. “Imsil” Bulletin No. 201, Illinois Minerals Co., Cairo, Ill.
492. Yamaoka K-, Utsugi H., Ganseki Kobutsu Kosho Gakkaishi, 68 (11), 346
(1973) [Chem. Abstr., 81, 80372].
493. Johns-Mansville Corp., The Story of Diatomite (1953).
494. Matrecon Inc., Oakland, Calif., Chem. Eng. News, 15 (May 9, 1977).
495. Her R. K, J. Colloid Sci, 19 (7), 648 (1964).
496. Kirby J. E., Gillson J. L., пат. США 2221485 (Du Pont), 1941.
497. Christman L. J. et al., пат. США 2364272 (1944); Jayne D. W. et al.,
пат. США 2312414 (1943); Jayne D. W. et al., пат. США 2336015 (1943).
Maust and Hollingsworth, пат. США 2343221 (1944); Mead and Maust.
пат. США 2357419 (American Cyanamid Co.), 1944.
498. Her R. K-, пат. США 2663650 (Du Pont), 1953.
499. Англ. пат. 672650 (N. V. de Bataafsche Petroleum Maatschappoj) 1952.
500. Stress F. H., пат. США 2625508 (Shell Development Co.), 1953.
501. Peterson W. H., пат. США 2629691 (Shell Development Co.), 1953.
502. Remes N. R., Martinek T. W., Froncziak E. T., пат. США 2913409 (Pure
Oil Co.), 1959.
503. Нем. пат. 829198 (N. V. de Bataafsche Petroleum Maatschappij) 1952.
504. Bachman J. H., Sellers J. W., Wagner M. P., канад. пат. 594065 (Co-
lumbia Southern Chem. Corp.), 1960.
505. Her R. K-, пат. США 2657149 (Du Pont), 1953 [Chem. Abstr, 48, 2336с].
506. Ballard С. C, Broge E. C„ Iler R. K-, John D. S. St., McWhorter J. R„
J. Phys. Chem, 65, 20 (1961).
507a. McWhorter J. R., пат. США 2810207 (Du Pont), 1957.
5076. Goebel M. T., пат. США 2736669 (Du Pont), 1956.
508. Her R. K-, пат. США 2739074 (Du Pont), 1956.
509. Berry K-, Joyce R. M., Kirby J. E., пат. США 2757098 (Du Pont), 1956.
510. Her R. K-, пат. США 2739077 (Du Pont), 1956.
Силикагели и порошки
851
511. Iler R. К, пат. США 2789075 (Du Pont), 1956.
512. Iler R. К, пат. США 2789076 (Du Pont), 1956.
513. Iler R. К, пат. США 2728740 (Du Pont), 1955.
514. Iler R. К, пат. США 2739075 (Du Pont), 1956.
515. Broge E. С., пат. США 2736668 (Du Pont), 1956.
516. Чертов В. №... Шейнфайн Р. Ю„ Кругликова Н. С., Неймарк И. Е.—
Укр. хим. ж., 1961, .т. 27, с. 190.
517. Baverez М., Basfick J., Bull. Soc. Chim. Fr. 1965 (12), 3662 [Chem.
Abstr., 64, 9577g],
518. Kitahara S., Asano T. et al., Chapter 1, Refs. 205—209.
519. Laufer S., пат. ФРГ 2057730 (Degussa), 1972.
520. Krieble R. H., Elliott I. R., пат. США 2441422—2441423, 1938.
521. Slinyakova I. B., Kurennaya L. I., Ref. 5, p. 138.
522. Anonymous, Munitions Supply Labs. (Aust.) Inf. Circ., 8 (1947); J. Am.
Ceram. Soc., 32, 157 (1949).
523. Welsh T. H., Wolf R. F., пат. США 2940947 (Columbia Southern Chem.
Corp.), 1956.
524. Bidaud A. F., Societe des Usines Chimiques Rhone Poulene, пат. США
2567316, 1937; франц, пат. 943915, 1947.
525. Франц, пат. 949783, (Campagnie Francais, Thomson-Houston) 1949.
526. Vail I. G., Ref. 199, Vol. 1, p. 175.
527. Anonymous, Chem. Eng. News, 29, 3017 (1951); Sirianni A. F. and
I. E. Puddington, австр. пат. Appl. 8, 627 (National Research Council
of Canada), 1952.
528. Sirianni A. F., Puddington I. E., пат. США 2583603—2583606, 1952.
529. Alexander G. B„ Iler R. К., пат. США 2801186, 1957; 2886460, 1959
(Du Pont).
530. Barry A. I., пат. США 2645588 (Dow Chem. Co.), 1955.
531. Kistler S. S„ пат. США 2589705, 1952. ’
532. Wilcox N. D„ Klein I. M„ Hudon F. А., пат. США 3600326 (U. S. Dept.
Army), 1971.
533. Аристова В. Г. и др.— ДАН СССР, 1973, т. 211, вып. 1, с. 130.
534. Laufer S., Roy W., пат. ФРГ 2043629 (Degussa), 1972.
535. Wagner G. H., пат. США 2705222 (Union Carbide and Carbon Corp.),
1955.
536. Beuche A. M„ Oliver C. S., пат. США 2993809 (General Electric Co.),
1961.
537. Nitzshe S. et. al., пат. ФРГ 1951620 (Wacher Chemie G. m. b. H), 1971.
538. Unger K, Berg K, Gallei E., Kolloid-Z. Z. Polym., 234 (2), 1108 (1969).
539. Burwell R. L., Jr., Chemtech., 370 (June 1974).
540. Anonymous, Chem. Eng. News, 29, 3017 (1951).
541. Disclosed in Ref. 528.
542. Weldes H. H. W., пат. США 3208867 (Philadelphia Quartz Co.), 1965.
543. Acker E. G., Sanchez M. G., Kramm D. E., пат. США 3180754 (W. R. Grace
Co.), 1965.
544. Gebura S. E., пат. США 3556829 (Interpace Corp.), 1971.
545. Acker E. G., пат. США 3377139 (W. R. Grace Co.), 1968.
546. Kautsky H., Wesslau H., Z. Natyrforsch., 9B, 569 (1954).
547. Yates P. С., пат. США 3634286 (Du Pont), 1972.
548. Caletka R., Konecny C., Radiochem. Radioanal. Lett., 12 (6), 325 (1972)
[Chem. Abstr., 78, 128770d].
549. Dolezal J. et al., J. Radioanal. Chem., 21 (2), 381 (1974) [Chem. Abstr.,
82, 77453а].
550. Caletka R., Konecny C., Ustav Jad. Vyzk. Cesk. Akad. Ved. (Rep.),
2643-Ch (1971) [Chem. Abstr.. 78, 131649b]; J. Radioanal. Chem. 14(2);
255 (1973).
28*
852
Глава 5
551. Егоров Е. В., Морозов Ю. Л. и др.— Сб. Радиационная химия поли-
меров. М.: Наука, 1966, с. 168—170.
552. Unger К-, Berg К-, Z. Naturforsch. В, 24 (4), 454 (1969); пат. ФРГ
2225904 (Merk Pat. G.m. b. H), 1973.
553. Unger К., Nyamah D., Chromatographia, 7 (2), 63 (1974) [Chem. Abstr.,
80, 115789h],
554a. Unger K-, Berg K-, Nyamah D., Lathe Th., Colloid Polym. Sci., 252 (4),
317 (1974).
5546. Meiller F., пат. ФРГ 2433409 (Rhone-Progil), 1975.
554в. Бурушкина T. Н., Стрелка В. В. и др. Адсорбция и адсорбенты, Респ.
межвед. сб. 1972, вып. 1, с. 94—98.
555а. Hill J. М., J. Chromatog., 76 (2), 455 (1973).
5556. Павлик Г. Е., Чуйко А. А., Хабер Н. В., Храновский В. А. и др. Сб.
Спектроскопия атомов и молекул./Под ред. Ю. П. Егорова. Киев: «Нау-
кова думка», 1969, с. 401—404.
556. Чуйко А. А. и др., Авт. свид. СССР, 206580, 1968.
557. Vail J. G., Ref. 199, Vol. 2, Chapter 9.
558. Kress W. (Degussa), Double Liaison, 18 (186), 63 (1971) [Chem. Abstr.,
75, 7475г].
559. Teicher Harry, “Aerogels and Hydrogels”, in T. C. Patton, ,Ed., Pigment
Handbook, Vol. 1. Wiley, New York, 1973.
560. “Hi-Sil” EP, Bulletin 85B (1977); trademark of PPG Industries, Ltd.
561. “Hi-Sil” 233, Bulletin 85A (1977); Plastics Technol. 1, 112 (1955); trade-
mark of PPG Industries, Ltd.
562. Wagner M. P., Sellers J. W., Ind. Eng. Chem., 51, 961 (1959).
563. Bachman J. H., Sellers J. W., Wagner M. P., Wolf R. F., Rubber Chem.
Technol., 1286 (December 1959).
564. Gessler A. M., Rehner I., Jr., Rubber Age, 875 (Sept. 3, 1955).
565. De Francesco A. J., Alling R. D. et al., Rubber World, 134, 866 (1956).
566a. Smith D. A., Rubber J., 150 (9), 54 (1968).
5666. Salvador A., Ind. Gomma (Ital.), 18 (12), 54 (1974) [Chem. Abstr., 83,
11798f],
567. Iler R. К., пат. США 2727876 (Du Pont), 1955.
568. Beuche A. M., пат. США 2914502 (General Electric Co.), 1954.
569. Kidwell A. S., De Francesco A. J. et al., пат. США 2982755 (Connecticut
Hard Rubber Co.), 1961.
570. Valron was a trademark of E. I. du Pont de Nemours & Co. for an este-
rified silica with a butoxy surface of about 300 m2g~1.
571. Великин M., Стремовский P. А. и dp.— Tp. НИИ осн. хим., 1970, вып. 21,
с. 13.
572. Robb L. Е., Wolf D. R., пат. США 2954359 (Minnesota Mining Mgf.
Co.), 1960.
573. DeGrotenhuis T. А., пат. США 2742378 (General Tire and Rubber Co.),
1956.
574. Iler R. K-, пат. США 2857355 (Du Pont), 1958.
575. Braendle R. О., пат. США 2751366 (Du Pont), 1956.
576. Ватаманчук В. И. и др.— Хим. технол. (Киев), 1975, вып. 1, с. 58.
577. Nichols R. F„ Weisz R. О., пат. США 2814604 (1957).
578. Wagner Н. В., J. Polym. Sci., 25, 500 (1957).
579. Taylor L. J., пат. США 3891954, 1975. •
580. Stana R. R., Calderwood A. S., пат. США 2129014 (Westinghouse Elect.
Corp.), 1971.
581. Klunker F„ Harriott P„ AIChE Symp. Ser., 68 (124), 340 (1972).
582. Bartell F..E., пат. США 2893962, 1959.
583. Nayar H. S., Lenel F. V., Ansell G. S., J. Appl. Phys., 42 (10), 3786
(1971).
584. Fanger G. О., пат. ФРГ 2125652 (National Cash Register Co.), 1971.
Силикагели и порошки
853
585. Dumas I. R., пат. США 2774674 (Chim. Rhone-Doulenc), 1956.
586. Hoppe L„ Behn R„ пат. ФРГ 1944619 (Wolff Walsrode A. G.), 1971.
587. ’ Griffin G. J. L., Appl. Polym. Symp., 16, 67 (1971) [Chem. Abstr., 75,
130413w].
588. Gorum L. А., пат. США 3798317 (Du Pont), 1974.
589. Banford J. A., Reuter G. L., пат. США 3268404 (Richardson-Merrill,
Inc.), 1966.
590. Squibb E. R. and Sons, Inc., франц, пат. 2036384 (1970), 1261281.
591. Young G. J., Chessick J. J., J. Colloid Sci., 13, 358 (1958).
592. Wightman J. D., Chessick J. J., J. Phys. Chem., 66, 1217 (1962).
593. Sirianni A. F., Meadus F. W., Puddington I. E., Can. J. Chem., 42 (12),
2916 (1964) [Chem. Abstr., 62, 8419b],
594. Huettenrauch F., Suess W., Schmeiss U., Pharmazie, 24 (5), 293 (1969)
[Chem. Abstr.. 71, 64543с].
595. Заседина T. И. и др.— Коллоидн. ж. 1971, т. 33, вып. 3, с. 352.
596. Laugner R., Am. Perfum. Cosmet., 81 (10), 51 (1966).
597. Rupprecht H., Liebl H„ Kolloid-Z. Z. Polym., 250 (7), 719 (1972).
598. Kasper H. et al., Pharm. Acta Helv., 37, 48, 73, 123 (1962) [Chem. Abstr.,
57, 969g],
599. Нидерланд. пат. 74490, (N. V. de Bataafsche Petrol. Maatsch.) 1954.
600. National Lead Co., Chem. Eng., 59, 226 (1952); Chem. Eng. News, 27,
1595 (1949).
601. Iler R. K-, пат. США 2676148 (Du Pont), 1954.
602. Iler R. K-, пат. США 2705700 (Du Pont), 1955.
603. Alexander G. В., пат. США 2818385 (Du Pont), 1957.
604. Braendle R. О., пат. США 2746922 (Du Pont), 1956.
605. Meyer G. C., Braendle R. O., Inst. Spokesman (Natl. Lubr. Grease Inst.),
18, 8—10 (1954).
606. Фукс И. Г. и др.— Хим. технол. топл. масел., 1970, т. 15, вып. 11, с. 48.
607. Fronczak Е. Т., пат. США 2939840 (Pure Oil Со.), 1960.
608. Christian I. В., Inst. Spokesman (Natl. Lubr. Grease Inst.), 35 (6), 188
(1971).
609. Blattenberger I. W., пат. США 2752310, 1956.
610. Ferch H., Pharm. Ind., 32 (6), 478 (1970).
611. Kirchgassner H., пат. США 3705940 (Degussa), 1972.
612. Harth H., Sturm L., пат. ФРГ 2220662 (Blendax-Werke, R. Schneider
Co.), 1972.
613. Unilever N. V., Neth., Pat. Appl. 71-02423, 1972.
614. Pader M., Wiesner W., англ. пат. 1264292 (Unilever, Ltd.), 1972.
615. Martinek T. W„ Klass D. L„ пат. США 3250726 (Union Oil Co.), 1966.
616. Pure Oil Co., Нидерланд. пат. Appl. 6406114, 1965 [Chem. Abstr., 65,
3400e],
617. Klass D. L., J. Appl. Phys., 38 (1), 67 (1967).
618. Шульман 3. П., Мацепуро А. Д.— Вести АН БССР, сер. физ. энерг.,
1973, вып. 4, с. 125,
619. Pure Oil Со., франц, пат. 1403950 (1965).
620. Winslow W. М„ пат. США 2417860, 3047507 (WEFCO, Inc.), 1962.
621. Rubin В. F„ AIChE, J. 15 (2), 206 (1969) [Chem. Abstr., 70, 118882m],
622. Brenner W„ Nature, 222 (5188), 79 (1969).
623. Henisch H. K-, Dennis I., Hanoka I. I., J. Phys. Chem. Solids, 26 (3),
493 (1965).
624. Kurz P. F., Ohio J. Sci., 66 (3), 284 (1966) [Chem. Abstr., 65, 9781d).
625. Bisaillon S., Tawashi R., J. Pharm. Sci., 64 (3), 458 (1975) [Chem. Abstr.,
82, 13216m],
626. Armington A. F., O’Connor J. I., пат. США 3578414 (U. S. Air Force)
1971 [Chem. Abstr., 75, 41773w],
627. Kachi S., Nippon Kessho Gakkaishi, 16 (1), 115 (1974).
854
Глава. 5
628. Scanlon J. J., Fisch К., пат. США 3116187 (U. S. Army), 1963 [Chem.
Abstr., 60, 7864d],
629. Christopher С. А., пат. США 3876007 (Texaco Co.), 1975 [Chem. Abstr<
83, 824971].
630. Fink H„ Koerner G. et al., пат. США 3655554 (Goldschmidt TH. A. G),
1972 [Chem. Abstr., 77, 795211].
631. Vukasovich M. S., англ. пат. 1191483 (Sherwin-Wiliams Co.), 1970.
632. Pitrot А., пат. США 3307960 (National Lead Co.), 1967 [Chem. Abstr.,
66, 116792b],
633. Brzezinski S., Int. Koloristenkongr. (Budapest), 4, 166 (1962).
634. Miller J. R. et al., пат. США 3714068 (Philadelphia Quartz Co.), 1973
[Chem. Abstr., 78, 161, 589z],
635. Boylan F. J., пат. США 3076768 (Hercules Powder Co.), 1963; 3408306
(1968) [Chem. Abstr., 21, 358h],
636. Iler R. К, пат. США 2733160 (Du Pont), 1956.
637. Hochwalt С. А., пат. США 2935481 (Monsanto Chem. Co.), 1960.
638. Schutte DI, Schmitz F. T., Brunner H., пат. США 3393155 (Degussa),
1968.
639. • Dohr M., Berekoven В., пат. ФРГ 2013763 (Degussa), 1971.
640. Ebeling W., Hilgardia, 30 (18), 531 (1961) [Chem. Abstr., 55, 21, 452g],
641. Goddin A. H., Sharp S. S., пат. США 2818340 (Du Pont), 1957.
642. LeDorze D., франц, пат. 2120549, 1972. ,
643. Атьяшева Л. Ф. и др.— ЖФХ, 1972, т. 46, вып. 10, с. 2602.
644. Kennedy I. F., Barker S. A., White С. A., Carbohydr. Res., 38 (1), 13
(1974) [Chem. Abstr., 82, 58026x],
<645. Никольский Б. П. и др.— Радиохимия, 1959, т. 1, с. 283.
646. Griffen R. Н., Iowa State Coll. J. Sci., 31, 422 (1957).
647. Коренманн И. M. и др.— Труды химии и хим. технол., 1960, т. 3, с. 572.
648. Polak F., Przem. Chem., 37, 83 (1958) [Chem. Abstr., 52, 14, 030i].
649. Hawkins W. L. et al., J. Appl. Polym. Sci., 5, S-15 (1961); пат. США
3259603 (Bell Telephone Labs), 1966.
650. Roney В. H., англ. пат. 1125800, 1966.
651. Mende W. C., Mehaffey R. J., пат. ФРГ 2113453 (Colgate Palmolive Co.),
1971.
652a. Worthy W., Chem. Eng. News, 17 (Feb. 9, 1976).
6526. Anonymous, Chem. Eng. News, 23 (May 30, 1977).
653. Teichner S. I., Mazabrard A. R., et al., Int. Conf. Colloids Surfaces —
ACS, IUPAC, Puerto Rico, June 21—25, 1976.
654. Kung H. H., Brooks В. 1., Burwell R. L., Jr., J. Phys. Chem., 78, 875
(1974).
655. Bergna H. E„ Ind. Quim., 28 (3), 123 (1970).
656. Marchand R., Lang I., C. R. Acad. Sci., Paris, Ser. C, 264 (11), 969
(1967).
' 657. Wagstaff F. E„ пат. США 3472667, 1969.
658. Best W. V., Hughes R. L., пат. США 3459673 (Owens-Illinois. Inc.), 1969
[Chem. Abstr., 71, 84, 259t],
659. Daimon N., Izawa T., пат. ФРГ 2423197, 1974 [Chem. Abstr., 82, 87,
869e],
660. Rayner J. H„ Colloq. Int. Cent. Natl. Res. Sci. Paris, 105, 123 (1962)
[Chem. Abstr., 57, 12, 131d],
661. Prener J. S., пат. США 3085863 (General Electric Co.), 1963.
662. Newell W. J., Madden J. E., пат. США 3135617, 1964.
663. Алексеева И. П. и др.— Коллоидн. ж., 1973, т. 35, вып. 2, с. 233.
664. Zettlemoyer А. С., Chessick I. J., Teheurekdjian Д'., пат. США 3272434,
1966.
665. Zettlemoyer А. С., McCafferty Е., Croat. Chem. Acta, 45 (1), 173 (1973)
[Chem. Abstr., 79, 83793г].
Силикагели и порошки
855
666. Boucher Е. A., Zettlemoyer А. С., Ed., Nucleation, Dekker, New York,
1969, p. 527.
667. Unger K-, Schick-Kalb J., Straube B., Coll. Polym. Sci., 253 (8), 658
(1975).
668. Iler R. K-, McQueston H. J., Chapter 4, Ref. 343.
669. Kirkland I. I., пат. США 3782075 (Du Pont), 1974.
670. Kirkland J. J., J. Chromatogr. Sci., 10, 593 (1972).
671. Kirkland J. J., J. Chromatogr., 83, 149 (1973); in S. G. Perry, Ed., Gas
Chromatography, Applied Science Publishers Ltd., Sussex, England, 1973,
p. 39; Gas Chromatogr. Int. Symp. Eur., 9, 39 (1972).
672. Williams R. C. et al., Am. Lab., 45—46, 48, 50 (1973).
673. Kiselev A. V., Nikitin Y. S. et al., Anal. Chem., 36 (8), 1526 (1964).
674. Kiselev A. V., J. Chromatogr., 49, 84 (1970).
675. Kirkland J. J., см. лит. к гл. 4, [472—474]; рис. 4.28.
676. Kirkland J. J., пат. США 3505785 (Du Pont), 1970.
677. Kirkland I. J., J. Chromatogr. Sci., 8, 309 (1970); 9, 206 (1971); 10,
129 (1972).
678. Destefano J. I., J. Chromatogr. Sci., 12, 337 (1974).
679. De Vries A. J., LePage M., Beau R., Guillemin C. L„ Anal. Chem., 39,
935 (1967).
680. LePage M., Beau R., de Vries A. J., J. Polym. Sci. (C), No. 21, 119
(1968).
681. Kiselev A. V., Khokhlova T. D., Eltekov Y. A., in E. S. Kovats, Ed., Co-
lumn Chromatogr. Int. Symp. Sep. Methods, 5th (1969), Saverlaender A. G.
Aarau, Switzerland, 1974, p. 124.
682. Frolov I. I., Vorob’eva R. G. et al., J. Chromatogr., 80 (2), 167 (1973).
683. Рябинина T. И., Хохлова T. Д., Эльтеков Ю. А. Сб. Макромолекулы
на границе раздела фаз.— Киев: «Наукова думка», 1971, с. 133—138.
684. Kirkland J. J., 4th Int. Symp. Gas. Chromatogr., Academic, 1963, p. 77.
685. Uiklein M., Halasz I., J. Chromatogr., 80 (1), 1 (1973).
686. Waksmundzki A., Chem. Anal. (Warsaw.), 16 (1), 43 (1971); 17 (5/6).
1223 (1972); 18 (4), 727 (1973).
687. Halasz I., Sebestian L, Angew. Chem. Int. Ed., 8, 543 (1969).
688. Unger K-, Ringe P., J. Chromatogr. Sci., 9 (8), 463 (1971).
689. Novotny M., Bektesh L. S., Grohman K-, J. Chromatogr., 83, 25 (1973).
690. Unger K, Nyamah D., Chromatographia, 7 (2), 63 (1974).
691. Brust O.-E., Sebestian I., Halasz I., J. Chromatogr., 83, 15 (1973).
692. Kunzru D., Frei R. W.. J. Chromatogr., 12 (4), 191 (1974).
693. Kirkland J. J., пат. США 3488922 (Du Pont), 1970.
2
Глава 6
ХИМИЯ ПОВЕРХНОСТИ КРЕМНЕЗЕМА
Свойства различных типов аморфного кремнезема с высоким
значением удельной поверхности, начиная от мельчайших кол-
лоидных частиц и до макроскопических силикагелей, зависят
в большой степени от химии поверхности твердой фазы. Это
практически важно в технологии катализаторов крекинга, при
обработке минерального сырья, использовании керамических из-
делий и адсорбентов. Аморфный кремнезем также непосредст-
венно применяется в производстве кремнеземных наполнителей
и загустителей в органических системах, включая краски, чер-
нила, эластомеры и смазочные материалы. В течение последних
50 лет была получена достаточно ясная картина природы крем-
неземных поверхностей, и на основе химического модифициро-
вания таких поверхностей были получены новые продукты.
Габер [1] высказал точку зрения, что атом на поверхности
твердого вещества только частично насыщен по своим связям
со стороны поверхности и, следовательно, обладает «остаточ-
ными валентностями» с внешней стороны. Ленгмюр [2] развил
эту идею в том направлении, что такие «остаточные валентно-
сти» ответственны за адсорбцию посторонних атомов или моле-
кул на поверхности, и сформулировал следующее уравнение для
адсорбционного равновесия при допущении, что выполняется
закон действующих масс
рЦх/т) — \/ab + р/Ь,
где р — давление паров адсорбируемого вещества; х/т — коли-
чество адсорбированного вещества на 1 г адсорбента; а — кон-
станта; b — величина, пропорциональная максимальному числу
молекул адсорбируемого вещества, которое может быть разме-
щено на единице площади поверхности.
Это допущение означает, что когда поверхность покрывается
одним слоем адсорбированных молекул, то никакой второй слой
не адсорбируется.
Там, где сильно химическое взаимодействие между адсорби-
рованной молекулой и атомами, на поверхности образуется пол-
ный мономолекулярный слой, даже когда концентрация адсор-
бированного вещества в жидкой или газовой фазе, соприкасаю-
Химия поверхности кремнезема
857
щейся с поверхностью, относительно низка. Дальнейшая адсорб-
ция второго слоя может происходить только за счет взаимодейст-
вия более слабых вторичных сил, распространяющихся от поверх-
ности, вблизи адсорбционного слоя. В таких случаях образова-
ние первого слоя обозначается термином «хемосорбция» и счи-
тается, что она подчиняется вышеприведенному уравнению изо-
термы адсорбции Ленгмюра. Образование второго слоя, если
оно происходит, определяется как «физическая адсорбция». Од-
нако если между атомами поверхности и молекулами адсорби-
руемого вещества в первом слое существует только слабое взаи-
модействие, то второй слой может начинать образовываться
даже до того, как завершилось заполнение первого слоя. В та-
ком случае адсорбция полностью обозначается термином «фи-
зическая адсорбция».
Ключевой характеристикой силоксановой (SiOSi) поверхно-
сти SiO2 является то, что так называемые остаточные валент-
ности реагируют с водой, так что при обычной температуре по-
верхность становится покрытой силанольными (SiOH) группами.
В настоящей главе рассматривается хемосорбция молекул на
силоксановой или силанольной поверхностях кремнезема, когда
адсорбируется одиночный слой молекул с образованием кова-
лентных или ионных связей с атомами на поверхности. Кроме
того, образуются водородные связи между полярными атомами,
такими, например, как атом кислорода простого эфира и атом
водорода силанольной группы на поверхности. Во всех случаях
существует сила связи между специфическим атомом адсорби-
рованного вещества и атомом на поверхности, так что поверх-
ность кремнезема покрывается только одним слоем, тогда как
второй слой не адсорбируется.
Примечание. В тех случаях, когда не показано, что атом кремния Si
имеет валентность, равную четырем, как, например, при записи SiOH или
SisOH, имеется в виду, что кремний находится на поверхности кремнезема
и что он связан посредством трех силоксановых связей SiOSi с тремя со-
седними атомами кремния.
Обзоры и основные выводы
Начало публикаций обзоров по химии поверхности кремне-
зема относится к 1950 г. Айлер [3] в 1955 г. рассмотрел пове-
дение силанольных групп на поверхности, в особенности их спо-
собность к дегидратации и к химическому взаимодействию,
включая этерификацию и хемосорбцию органических молекул.
Еще в 1936 г. Киселев [4] предположил, что активная по-
верхность силикагеля покрыта ОН-группами, связанными с кар-
касом SiO2, а в 1940 г. Карман [5] обнаружил, что на по-
верхности безводного SiO2 при добавлении воды образуются
858
Глава 6
силанольные группы. В 1959 г. Белякова и др. [6] своим обзо-
ром за 10 лет внесли вклад в химию силанольных групп, пока-
зав, что поверхностные силанольные группы являются центрами,
на которых адсорбируются молекулы воды, и что многие орга-
нические молекулы, имеющие полярные группы, адсорбируются
и удерживаются на SiOH-группах посредством водородной
связи. Кроме того, они представили следующие данные. Если
при построении изотерм адсорбции концентрацию воды выразить
числом микромолей на 1 м2 SiO2, то получается, что сильно раз-
личающиеся виды аморфного кремнезема с гидратированными
поверхностями адсорбируют одно и то же количество воды на
единицу поверхности при одинаковом низком парциальном
давлении воды. Таким образом, природа твердой фазы крем-
незема влияет очень незначительно; все типы поверхности
кремнезема оказываются подобными при условии, что в кремне-
земе отсутствуют микропоры. Концентрация SiOH-групп на по-
верхности кремнезема, по-видимому, составляла 11 ±1 микро-
моль ОН/м2 или 6,6 ОН-групп/нм2. В обзор была включена об-
ширная библиография вплоть до 1959 г.
В 1966 г. Боэм [7, 8] опубликовал два детальных обзора по
химии поверхностных групп на различных твердых веществах,
включая кремнезем. Сравнивались данные по числу ОН-групп
на квадратный нанометр, получаемые различными методами,
были рассмотрены этерификация и другие реакции на поверхно-
сти. В 1967 г. Хайр [9] рассмотрел ряд вопросов химии поверх-
ностей кремнезема, включая гидратацию и реакции на поверх-
ности, в частности, путем изучения ИК-спектров. В 1968 г. Снай-
дер [10] обсудил структуру поверхности кремнезема, придавая
особое значение адсорбционным характеристикам и использо-
ванию типов пористого кремнезема в хроматографии. В 1970 г.
Оккерс [И] в расширенно^ обзоре, посвященном приготовле-
нию и свойствам пористого кремнезема, описал гидроксилиро-
ванную поверхность кремнезема и ее структуру, опираясь на тот
факт, что на поверхности, вероятно, только одна группа ОН при-
ходится на один атом кремния. Этот же автор суммировал дан-
ные по дегидратации. В 1972 г. Сноуинк и Вебер [12] рассмот-
рели те же самые вопросы, что и Боэм, расширив библиогра-
фию. В 1976 г. Барби [13] представил расширенный обзор по
таким типам кремнезема, как гели и порошки, с детальным
обсуждением природы поверхностных силанольных групп.
Общий обзор по структуре и химии оксидных поверхностей
в связи с вопросами адсорбции был опубликован Киселевым
в 1971 г. [14]. Различные аспекты химии поверхности кремне-
зема при ее контакте с водой были представлены Визе и др.
[15] в общей статье по взаимодействию в системе коллоид—
вода.
Химия поверхности кремнезема 859
При наличии этих многочисленных обзоров может возник-
нуть сомнение в целесообразности приводимого здесь еще од-
ного обзора. Однако очевидно, что ранее придавалось особое
значение в основном природе гидратации поверхности, и ни в од-
ном из указанных обзоров не приводились различные аспекты
химии поверхности, включая поведение поверхности в контакте
с водой и органическими соединениями. Поэтому полезно соб-
рать эти сведения в одном месте.
Природа поверхности кремнезема
В настоящее время для исследования состава и структуры
поверхностей твердых тел применяются разнообразные химиче-
ские и физические методы. Они были- описаны достаточно под-
робно Кейном и Ларраби [16]. Многие из этих методов более
пригодны для исследования макроскопических пойерхностей, чем
порошков и коллоидов, но среди них имеются и такие, которые
еще не были использованы при изучении кремнезема. В этой
главе особое внимание будет уделено таким методам, которые
еще предстоит опробовать в химии поверхности твердого тела,
поскольку применявшиеся ранее методы достаточно подробно
рассмотрены в цитируемой литературе.
Очень хороший обзор по природе поверхности кремнезема
представил, как известно, в 1965 г. Хокки [17]. Более поздний
обзор [18] касается вопросов присоединения органосилильных
групп к поверхности.
Структура подповерхностного слоя кремнезема
Хотя было найдено, что различные формы кремнезема, по-
видимому, одинаково адсорбируют воду [6], оказалось бы уди-
вительным, если бы SiOH-группы на чистых поверхностях все-
возможных кристаллических и аморфных форм вели себя в точ-
ности одинаково. Действительно, Штобер [19] показал, что
SiOH-группы на поверхности редкой, чрезвычайно плотной
формы кремнезема, такой, как стишовит, в котором каждый
атом кремния находится в координации с шестью атомами кис-
лорода, ведут себя так же, как ОН-группы на оксиде алюминия,
если их поведение рассматривать с точки зрения образования
водородной связи. Стишовит адсорбировал поливинилпиридин-
N-оксид очень слабо, тогда как поверхности всех других форм
кремнезема адсорбировали его сильно. Так как плотности, на-
пример, кварца и разновидностей аморфного кремнезема неоди-
наковы, следовало бы ожидать некоторых небольших различий,
которые отсюда вытекают. Однако в работе Мейера и Хаккер-
мана [20] показано, что размеры частиц или радиусы кривизны
860
Глава 6
поверхности могут быть, по-видимому, более важными по зна-
чимости, чем различия в плотности между аморфными и обыч-
ными кристаллическими состояниями кремнезема.
Определение величины поверхности
Термин «поверхность», как это будет пониматься, означает
границу непористой твердой фазы. По традиции «поверхность»
обычно означает границу, которая непроницаема по отношению
к азоту — наиболее широко используемому адсорбируемому ве-
ществу для измерения удельной поверхности. Однако могут су-
ществовать микропоры, в которые проникают молекулы воды,
но не азота. Так как поверхность микропор трудно определить,
то под термином «поверхность» будет в общем пониматься та
величина, которая измеряется обычным методом БЭТ по ад-
сорбции азота.
Даже небольшое количество загрязнений, находящихся на
поверхности, может-значительно изменить ее свойства. Пусть,
- например, в силикагеле при значении удельной поверхности
200 м2/г имеется 0,01 масс. % натрия в виде примеси. Если бы
весь натрий располагался на поверхности, то на площади
100 нм2 находился бы один атом натрия, т. е. эти атомы раз-
мещались бы друг от друга на расстоянии 10 А, если бы они
были распределены равномерно. Фоуке и Бургесс [21] пока-
зали, что поверхность кремнезема способна захватывать атомы
натрия. Даже чистейший кварц абсорбирует 1013 атомов на
1 см2 в пределах толщины поверхности 100 А. На поверхности
создается отрицательно заряженный оксидный ион. Если такую
поверхность протравить HF, то она становится незаряженной,
но после нескольких недель выдержки при комнатной темпера-
туре атомы натрия выходят на поверхность из внутреннего
объема вещества. Если объемная концентрация составляла
0,0002 % Na, то после прокаливания при 1000°С на поверхности
накапливалось 2 % натрия.
Так как кремнезем, содержащий 0,0002 % Na, считается чрез-
вычайно чистым, то можно считать, что в большинстве гелей и
осадков кремнезема натрий может играть скрытую, еще непоз-
нанную роль.
Гидроксилированная поверхность
Так как атомы кремния на поверхности аморфного кремне-
зема находятся по определению не точно в регулярном геомет-
рическом порядке, то очевидно, что гидроксильные группы, при-
соединенные к соответствующим атомам кремния, не будут
в точности на равном расстоянии друг от друга (рис. 6.1).' Сле-
Химия поверхности кремнезема
861
довательно, не все гидроксильные группы эквивалентны по
своему поведению в отношении адсорбционных явлений или хи-
мических реакций.
£
-°\
,н' н. н
о7 V
-S ^,-25-170’С
° с
® Нг°
н /н
". ? ? Поверхность
“—Si--Si;-----------
о
выше
н н н н
О '(/ с/ 0 х О
1111 /х
— Si—Si—Si- -Si Si—Si—
zi\
„ '---xf
© HO OH p
H Si H г1"5кГ)
i / x i u'O > O.w
0 0 0 O (j)
— Si--Si--Si — Si-Si-
WW0?0
175° C
©
Поверхность
Поверхность
© H@ © H ®
“ n4
0 0 0 04 X)
/ \ I / \ 1 \н/«
-Si — Si--Si--Si--Si--Si-- Si—
oz ixozIY/!хох !xoz 1 Y) zi Y)
u 0 0 0 0 0u 0 u 0 u
Поверхность
Рис. 6.1. Схемы расположения возможных типов гидроксильных групп на по-
верхности аморфного кремнезема.
а—-смежные, расположенные рядом (вицинальные), гидратированные; б — смежные
(вйцинальные), безводные; в — силоксановые группы, дегидратированные; г — гидрокси-
лированная поверхность; д — одиночная (свободная) гидроксильная группа; е — парные
и строенные гидроксильные группы; ж — смежные, взаимно связанные водородными
связями.
Примечание. Типы групп е и ж, вероятно, не существуют на высушенной поверх-
ности.
Кроме того, можно представить, что в водном растворе до-
полнительные молекулы монокремневой кислоты могут конден-
сироваться на поверхности, причем к атомам кремния могли бы
присоединиться две или даже три гидроксильные группы (как
это показано на рис. 6.1 в положениях е и ж). Хотя существо-
ванием таких групп, как было постулировано, объясняются не-
которые данные, вероятно, что они впоследствии конденсируются
и на высушенной поверхности остаются только SiOH-группы.
При высушивании кремнезема молекулы воды, связанные во-
дородной связью, как показано на рис. 6.1, удаляются при обыч-
ной температуре в вакууме или при 150°С на воздухе.
862
Глава 6
Концентрация силанольных групп, силанольное число, т. е.
число гидроксильных групп, находящихся на единице поверхно-
сти, было предметом многих исследований и дискуссий.
Белякова и др. [6] нашли, что концентрация силанольных
групп оказывалась примерно одинаковой для различных типов
Рис. 6.2. Факторы, влияющие на число гидроксильных (силанольных) групп,
находящихся внутри и на поверхности первичной частицы кремнезема и спо-
собных образовывать молекулы воды.
а — подповерхностная гидроксильная группа искривляет поверхность, что способствует
появлению на ней парных и строенных силанольных групп; б — гидроксильная группа,
расположенная вблизи поверхности, удерживается более прочно и поэтому вызывает
увеличение числа групп на единице поверхности; в — число гидроксильных групп на
частицах кремнезема очень небольшого размера (менее 100 нм) в расчете на единицу
поверхности понижено; г — гидроксильные группы на частице большего размера распо-
лагаются ближе друг к другу и способны образовывать более прочно удерживаемые пары
гидроксильных групп, связанных между собой водородными связями.
кремнезема. С другой стороны, довольно различающиеся резуль-
таты были сообщены другими исследователями. Как указали
Дубинин, Беринг и Серпинский [22] в 1964 г., на поверхности
образцов аморфного кремнезема, полученных различными пу-
тями, упаковка тетраэдров, состоящих из атомов кислорода и
одного атома кремния, не является регулярной и может изме-
няться. Когда частицы кремнезема образуются в воде, то на
число ОН-групп можно воздействовать несколькими путями,
как это показано на рис. 6.2:
1. Подповерхностные неконденсированные «захороненные»
SiOH-группы, расположенные ниже поверхности силоксановой
Химия поверхности кремнезема
863
сетки, увеличивают искривления на поверхности. Когда они на-
ходятся более глубоко в объеме, то могут быть удалены только
при высокой температуре.
2. Некоторые SiOH-группы могут быть расположены как раз
в пределах поверхности, поэтому они увеличивают среднюю
плотность упаковки ОН-групп на единице поверхности, как это
показано на схеме б.
3. На частицах с очень небольшим радиусом кривизны на-
личие искривлений с положительным радиусом приводит к тому,
что SiOH-группы удерживаются на большем расстоянии друг
от друга, так что могут образовывать более слабые водородные
связи между собой. Поэтому такие группы удаляются более
легко при повышенной температуре, как это имеет место в слу-
чае в по сравнению с г.
4. В зазорах — в точках контакта между частицами, где
имеется отрицательный радиус кривизны, например в микропо-
рах,— поверхностные ОН-группы, наоборот, расположены ближе
друг к другу, и поэтому дегидратация более затруднена.
5. В частицах, образованных при низкой температуре и низ-
ком значении pH из еще меньших по размеру частиц путем их
агрегации, имеются микропоры или субмикропоры, как это опи-
сано в гл. 4, с гидроксилированными поверхностями, в которых
сильно удерживается вода. Величина таких поверхностей не из-
меряется методом БЭТ из-за того, что молекулы азота не могут
проникать в микропоры, и поэтому концентрация ОН-групп на
квадратный нанометр кажется аномально высокой.
Вода на гидроксилированной поверхности
В детальном исследовании поведения воды на поверхности
кремнезема, дегидратированного при 700°С, Клир и Цеттльмойер
[23] показали, что молекула воды располагается на SiOH-rpyn-
пах так, что ее атом кислорода направлен к группе ОН, по край-
ней мере на начальных стадиях адсорбции. Тогда водородносвя-
занные кластеры, состоящие из молекул НгО, могут образовы-
ваться даже еще до того, как на всех SiOH-группах произошла
адсорбция молекул Н2О и образовались поверхностные
SisOH : ОН2-группы. Это означает, что на изолированных SiOH-
группах вода менее прочно адсорбируется, чем на уже ранее
адсорбированных молекулах воды:
Si.sOH 4- Н2О = SisOH : ОН2 6,0 ккал/моль
SisOH:ОН2 + хН2О = Si^OH: ОН2 (ОН2) 10,5 ккал/моль
где Sis представляет собой атом кремния на поверхности. Вода
стабильна в кластере, состоящем из пяти или шести молекул,
почти так же, как и в жидкости.
864
Глава 6
На поверхности, которая полностью гидроксилирована, моле-
кулы Н2О сначала покрывают все SiOH-группы вследствие об-
разования многократных водородных связей. Кроме того, на ос-
новании измерений вязкости, выполненных Далтоном и Айлером
[24], доказано, что монослой молекул воды оказывается более
или менее лишенным подвижности на незаряженных SiOH-rpyn-
пах поверхности за счет образования водородных связей, когда
коллоидные частицы находятся в воде и полностью гидроксили-
рованы.
Представление о том, что такая заторможенность распрост-
раняется на несколько молекулярных диаметров в водную фазу,
недостаточно обосновано. В противном случае вязкость была бы
больше, чем получается для адсорбированного монослоя. Кроме
того, Ликлема [25] на основании изучений дисперсной системы
Agl сделал заключение, что электрокинетический потенциал сов-
падает с внешним потенциалом слоя Гельмгольца и что «на гра-
нице раздела фаз не образуется заторможенного слоя воды до-
статочной толщины».
В экспериментах по измерению вязкости воды между сфери-
ческой и плоской поверхностями плавленого кварца зарегистри-
ровано увеличение вязкости, когда поверхности отстояли друг от
друга дальше, чем на 160 нм [26]. Однако измерения проводи-
лись не при pH 2, т. е. не в изоэлектрической точке кремнезема,
поэтому поверхность кремнезема даже в чистой воде должна
была нести отрицательный заряд и должен был образовываться
двойной ионный слой, что авторами не принималось в расчет.
В первых двух молекулярных слоях воды на поверхности
вязкость, несомненно, достаточно высока, так как первый слой
воды в основном находится в равновесии с SisOH-группами и
связан водородными связями с ними. Более того, на такую вяз-
кость влияет смещение частиц, поскольку временной фактор
играет определенную роль в образовании и разрыве водородных
связей.
Поверхность кристаллического кварца существенно отли-
чается от поверхности аморфного кремнезема. Это было пока-
зано на. примере такой характерной особенности, как адсорбция
монокремневой кислоты (гл. 1). Подобная необычная адсорбция
на поверхности кварца может быть обусловлена особой геомет-
рической подгонкой молекул адсорбируемого вещества по отно-
шению к адсорбенту, и нет необходимости связывать это только
с упорядочением самих молекул воды. Тем не менее, по-види-
мому, имеется веское доказательство того, что на некоторых ти-
пах поверхностей смежные с поверхностью молекулы воды могут
принимать некоторую степень упорядоченности. Если твердое
вещество имеет кристаллическую поверхность, на которой атомы
расположены упорядоченно* то можно представить и упорядочен-
Химия поверхности кремнезема
865
ность в расположении адсорбированных молекул воды (льдопо-
добная структура), которая в результате образования водород-
ных связей может распространяться на некоторое расстояние от
поверхности в глубь раствора. Дрост-Хансен [27] суммировал
доказательства существования подобного явления. Однако нет
убедительного подтверждения, что такое упорядоченное струк-
турирование воды может быть индуцировано поверхностью
аморфного кремнезема, на которой SiOH-группы не имеют регу-
лярного расположения на расстояниях, превышающих размеры
между двумя или тремя силанольными центрами.
Состояние адсорбированной воды в пленке толщиной только
в несколько молекул можно представить в виде различных си-
туаций. Плустер и Гитлин [28] измерили теплоемкость пленок
воды, адсорбированных на кремнеземе. В данном положении
надо учитывать также существование границы раздела фаз жид-
кость— пар с некоторым поверхностным натяжением, которое
отсутствует, когда поверхность кремнезема погружена в объем
жидкой воды. В случае тонких пленок толщиной только 50—75 А
наблюдается максимум теплоемкости при температуре ниже 0°С,
и фазовый переход лед—вода при этом подавляется. В таких
тонких пленках, лежащих между границами раздела фаз твер-
дое тело—жидкость и жидкость—пар, силанольная поверхность,
по-видимому, оказывает воздействие на структуру воды на рас-
стоянии в несколько молекулярных диаметров от поверхности
при температуре около 0°С, когда, как известно, происходит об-
разование сильных водородных связей.
Было бы интересно изучить термическое расширение воды
вблизи 0°С в 30—40 %-ном золе, состоящем из кремнеземных
частиц размером 100 А, в изоэлектрической точке при pH 2,
когда имеют место только водородные связи. В этом случае все
молекулы воды находятся на расстоянии до 100 А от силаноль-
ной поверхности. Если бы влияние силанольной поверхности
распространялось очень далеко, то максимальная плотность не
наблюдалась бы при 4°С. Сам кремнезем имеет по существу
нулевой коэффициент расширения, и им можно пренебречь. При
изучении свойств воды в адсорбированном состоянии на поверх-
ности кремнезема рассмотрены эффекты, связанные с использо-
ванием ЯМР [29, 30], измерены теплоемкость [31], энтропия
[32] и давление пара [33].
Электропроводность поверхности
В водной суспензии, свободной от растворимых электроли-
тов, поверхность кремнезема в определенной степени является
проводником, так как она заряжена, а противоионы способны
обеспечить перенос заряда. Очевидно, что поверхность с нане-
29 Заказ № 250
866
Глава 6
сенным алюмосиликатным слоем, имеющая в качестве противо-
ионов Н+, должна бы давать наивысшую проводимость, схожую
с проводимостью катионообменной смолы в Н+-форме. Однако
такая проводимость не обусловливается исключительно только
подвижностью ионов в диффузионном двойном слое, как это
было предложено Бикерманом [34]. Киттака [35] пришел к за-
ключению, что проводимость связана с переносом ионов не
только в диффузионном, но также в «фиксированном» двойном
слое. Однако так как величина pH не была измерена и так как
ион Н+ имеет гораздо большую подвижность, чем любой другой
ион, то вполне возможно, что истинная роль этого иона еще не
раскрыта. Так, например, в экспериментах Киттака наивысшая
электрическая проводимость была отмечена в растворах с наи-
большим числом ионов Н+, а именно в растворе А1С13 и осо-
бенно Th(NO3)4, где значение pH было более низким.
Интересные наблюдения электропроводности на сухой по-
верхности кремнезема были выполнены Муройя и Кондо [36].
Они приготовили из силикагеля прочную пластину толщиной
1 мм с площадью на одной стороне, равной 3 см2. Электрбпро-
водность измерялась в интервале 150—650°С с помощью золо-
тых электродов, помещенных с обеих сторон пластины. Удельная
поверхность силикагеля составляла 650 м2/г, а пористость
0,33 мл/г. Ясно, что электропроводность обеспечивалась прото-
нами SiOH-групп. Такая проводимость, определяемая площадью
поверхности внутри пор, изменялась в области 10-20—10-17 ом-1
при 25—160°С на сухой обезвоженной SiOH-поверхности. Обра-
зец при 600°С имел дегидроксилированную поверхность, его про-
водимость была 10 й ом ', а при охлаждении она уменьшалась
до гораздо более низкой степени. Экстраполированное значение
проводимости при 25°С, по-видимому, составляло 10~30—
10-40 ом-1. Силанольная поверхность представляет собой, оче-
видно, зону проводимости.
Отличие адсорбированной воды от силанольных групп
Для определения числа силанольных групп на поверхности
требуется различать SiOH-группы и физически связанную или
водородносвязанную воду. Мысленно можно представить, что
некоторые молекулы воды могут настолько сильно удержи-
ваться, что при температуре, требуемой для десорбции, часть
силанольных групп может также теряться в результате дегидра-
тации. Вероятно, это происходит в микропорах, где энергия ад-
сорбции воды очень высока, а силанольные группы более легко
дегидратируются.
Де Бур и Влеескенс [37] нашли, что кремнезем во время
высушивания на воздухе при 120°С теряет всю физически адсор-
Химия поверхности кремнезема
867
бированную воду, но при 110°С еще остается вода, если в окру-
жающей атмосфере есть влага. Это справедливо, конечно, когда
нет никаких микропор; в противном случае адсорбированная
вода может удерживаться в микропорах вплоть до 180°С, хотя
поверхность уже начинает терять гидроксильные группы [38].
Авторы обнаружили, что на поверхностях, которые нагревались
или обрабатывались в автоклаве или нагревались и повторно
неоднократно смачивались, число ОН-групп, приходящееся на
квадратный нанометр, имеет тенденцию приближаться к значе-
нию 4,5—5,0 после высушивания при 120°С. Однако силикагели
и гидратированные типы кремнезема, полученные из минералов,
которые никогда не нагревались, имеют, как было подсчитано,
гораздо большее содержание гидроксильных групп на единице
поверхности. Другие исследователи также наблюдали, что при
высушивании даже широкопористых гелей или коллоидного
кремнезема вплоть до 150°С, но не выше общее содержание
воды достигало 6—8 групп ОН/нм2 (табл. 6.1).
Нолл, Дамм и Фаусс [44] сообщили, что реактив Карла Фи-
шера реагирует с водой, но не с силанольными группами сили-
кагеля. Вычитая содержание воды, полученное таким способом,
из общего количества, выделившегося при прокаливании до
1100°С из силикагеля, они заключили, что поверхность содер-
жала 5,2 ОН-групп/нм2. Айлер наблюдал, что в образцах сили-
кагеля, высушенных при 170°С, общее количество воды равня-
лось 8 ОН-групп/нм2, из них около 2,3 ОН-групп/нм2 реагиро-
вало быстро с реактивом Карла Фишера; с оставшимися
в количестве 5,7 ОН-групп/нм2 группами шла продолжительная
медленная реакция. Келлум и др. [45] сообщили о том же са-
мом явлении и экстраполировали затем линейную часть кривой
к нулевому моменту времени, чтобы подсчитать эквивалентное
содержание воды. С целью определения содержания SiOH-групп
Келлум и соавторы предложили способ титрования, в котором
используется в качестве основания раствор литийалюминийди-и-
бутиламида в диметоксиэтане и применяется N-фенил-п-амино-
азобензол как индикатор, если титрование ведется в смеси те-
трагидрофурана и пиридина (1 : 1).
Реакция SiOH-групп с дибораном представляет собой другой
подход. Авторы работы [46] сравнили действие дибор'ана и ре-
актива Гриньяра и нашли, что последний реагирует много бы-
стрее. Фрипья и др. [47], используя реактив Гриньяра и изме-
ряя количество метана, выделяющегося в результате реакции
с SiOH-группами, нашли, что на 1 нм2 находится 4,6 группы ОН.
Это же было подтверждено Моримото и Наоно [48], которые
обнаружили хорошее согласие с потерей массы при прокалива-
нии, показывающее, что такой реактив реагирует со всеми ОН-
группами.
29*
<3
sr
а
<3
Поверхностная концентрация гидроксильных групп для различных типов SiO:
Литература О О —* « <3 СО ''Ф ''Ф "ф "ф
Количество групп ОН на 1 нм2 «и 2 . _ 2 га Й Г**» Ф g Л С;О О —со сч °Л? L? S о сс сс~ о о оо ю 'Ф 1 | 00 lo о s °. « S О § § “Э а +1 я Ш tf G> & 2-^ а а. и
Метод исследования Я В, К в. R Щ о & с ® с ® 2 g ” с с я ° ф 2 ф S я е 5 S я 3 g § ЛЯ я о я о я н 53 я аЙ о О Cfl CQ г , CQ Р-< со <тч s SU sU к SU я S ч «а сз е; о я о ф е- о Я 0< Ф Ct, га о «V (дЗ о с ЙО E S о t=( — 0^02: о у о и \о ф о ,й СО-" j - О О’ Л, ОИЛ S S н 2 s о о,— X о. S4 о г ,си ° tic Н с с с s«- S
Удельная поверхность, м2/г О о ю ю о сч о о о — о Г- О ОСС С СФС1 О 1 1 о? — — —• —• ю — 1 1 О Ю о
высушивания 1 время 16 ч 4 сут 4 сут 2 ч 17 ч 17 ч 10200 ч
о о X Л Е? (D X О. атмосфера К 5, 1 * R » 1 1 1 О сз m m
Условия предва температура, °C О о ЮО О ООО о о ю— СЧ СЧЮСЧ СЧ Ю 1 — _ Т— —< о о
Тип SiO2 lit 1 я S , , ччч ’§2 « я 2 й а « 2 gsgsgs £>> 3§ g&soSg- ‘SS’SS-SS ®g. лдлдлд я о S и m о 2 га о rt« я s я s я s s о Я О 5 ° Е я ф Л 3 a S s ° s щ сш °* и ® s з a s и га га et ®з ct, г, ™ ял *я ч я 3 я .§«§«§«< Й 1 |®-я идssаf ч g-’Я §-«§« g. §£> S о £ § g 2 .. " Я а о фЯлдЯ^ЯЗч> м л 1 Я я f Е cd Ф п । feaSa«sSa и о ° g ° я S Й •& g я g 5 s воозоВяогаЛ§зЯ*ч |ggg§g§§ go|«§§S-tt§Sg.agS 5§Ч§Ч§Чф §чаи£о«ояям:«'—и Ut( t[ tt g g а 2
а Включенные в русский перевод опубликованные (в том числе [43])
неопубликованные данные Журавлева и соавторов.— Прим. ред.
Химия поверхности кремнезема 869
Распространенной точкой зрения является то, что низкотем-
пературное высушивание кремнезема под вакуумом является
единственным путем для удаления адсорбированной воды, не за-
трагивающим гидроксильных групп. Бермудец [49] показал ме-
тодом ЯМР, что при 110°С в течение 6 ч удаляется не только
вода, но также некоторое количество силанольных групп с по-
верхности и некоторая часть внутренней воды.
Вирцинг [50] использовал полосу ИК-поглощения 5265 см-1,
чтобы идентифицировать воду и четко отличать ее от различных
типов гидроксильных групп. Такая полоса появляется, когда мо-
лекулы НгО связываются с SiOH-группами. Этот способ также
позволяет определять ОН-группы, когда допускается адсорбция
воды на кремнеземе. Баверец и Бастик [51] также отличали ад-
сорбированную воду от групп SiOH, используя ИК-поглощение.
Как будет обсуждено ниже, главной проблемой представ-
ляется не различие между молекулами воды и группами SiOH,
находящимися на поверхности, а скорее различие между этими
поверхностными группами и водой, удерживаемой внутри крем-
неземных частиц в виде групп SiOH, которую можно удалить
лишь при высокой температуре.
Ланге [52] утверждает, что на гидроксилированном кремне-
земе имеются два вида адсорбированной воды, один из которых
десорбируется при прогреве образца в интервале 25—105°С,
а другой — в интервале 105—180°С. Он называет первый тип
«физически адсорбированной водой», а второй тип «водой, свя-
занной водородными связями». Для удаления физически адсор-
бированной воды требуется энергия активации 6,6—8,2 ккал/моль,
тогда как для удаления второго типа воды требуется энергия
активации 10 ккал/моль. Повторная адсорбция водородносвя-
занной воды хорошо описывается изотермой Ленгмюра. Физи-
чески адсорбированная вода сорбируется в соответствии со вто-
рым типом изотерм адсорбции * с завершением монослойного
покрытия при р/ро = О,18.
Вероятно, что водородносвязанная вода представляет собой
воду, адсорбированную только на тех силанольных группах, ко-
торые не имеют сильной водородной связи с соседними группами
на поверхности.
Манк, Баран и Янковская [53а] использовали метод ЯМР,
чтобы различить лабильные протоны и группы ОН на поверхно-
сти кремнезема и группы ОН внутри частиц. Расстояние между
группами SiOH, оцененное в этой работе, составляло 5,0—5,4 А,
что должно примерно соответствовать поверхностной концентра-
* По классификации Бруиауэра, Деминга, Деминга и Теллера (БДДТ)
(см. Брунауэр С. Адсорбция газов и паров. Пер. с англ,—М.: ИЛ, 1948).—
Прим. ред.
870
Глава 6
ции 3,9 ОН-групп/нм2 при распределении на двумерной гексаго-
нальной решетке или 3,7 ОН/нм2 на тетрагональной решетке.
Квливидзе [536] в 1964 г. опубликовала перечень ранее выпол-
ненных методом ЯМР работ и привела свои данные, полученные
этим методом на широкопористом силикагеле, дегидратирован-
ном в вакууме при 200°С. Все измерения, в том числе адсорбции
воды, проводились при низкой температуре в интервале 83—•
273 К.
Гидроксильные группы и молекулы воды внутри
частиц кремнезема
По мере продолжения исследований стало ясно, что почти
все разновидности аморфного кремнезема содержат силаноль-
ные группы не только на поверхности, но и по всей структуре
частиц. Такие захваченные силанольные группы могут возникать
различными путями в зависимости от процесса, посредством
которого частицы кремнезема были образованы.
В случае пирогенного кремнезема, как это было рассмотрено
в гл. 5, частицы сферической формы диаметром 10—20 нм обра-
зуются посредством агрегации элементарных первичных частиц
размером всего лишь 1—2 нм, полученных как продукт конден-
сации из пламени. Такие первичные частицы содержат некото-
рое количество поверхностных групп SiOH, которые оказы-
ваются захваченными внутри большой конечной частицы.
Последняя спекается в пламени, достигая по плотности прибли-
зительно теоретического значения.
В силикагелях, приготовленных из силиката натрия, первич-
ные частицы поликремневой кислоты, обычно имеющие размер
2—3 нм, по мере агрегации и в процессе «старения» геля захва-
тывают некоторое количество групп SiOH. Было показано [54],
что поверхностные группы могут в действительности захваты-
ваться, когда силикагель сильно сжимается.
Когда кремнеземные частицы выращиваются в воде в про-
цессе автоклавной обработки при высокой температуре, можно
ожидать, что кремнеземная структурная сетка будет уплотняться
и освобождаться от внутренних групп SiOH. Однако Давыдов
и Киселев [55] показали, что на самом деле остается вода, за-
хваченная внутри структуры. Как подробно рассмотрено Кисе-
левым [14], при высушивании такого образца вода может уда-
ляться, оставляя после себя микропоры.
В том случае, когда коллоидные частицы формируются в рас-
творе способом «наращивания» при рН~9, как об этом упо-
миналось в гл. 5, некоторое незначительное количество ионов
натрия адсорбируется как раз в момент осаждения кремнезема
на растущих частицах. Такие ионы связываются с водой, и даже
Химия поверхности кремнезема 871
после их удаления путем продолжительной кислотной обработки
и ионным обменом, глубоко в структуре в небольшом количестве
остаются прочно удерживаемые молекулы воды или силаноль-
ные группы.
Еще одним источником появления внутренних групп SiOH
представляется возможная диффузия молекул Н2О внутрь твер-
дой структуры SiO2. Доремус [56] утверждает, что точно так
же, как Н2О диффундирует внутрь прозрачного кварцевого сте-
кла при высокой температуре, молекулы воды могут проникать
внутрь аморфного образца SiO2 на расстояние до 150 А, где не-
большое количество такой воды может оставаться в виде спа-
ренных групп SiOH.
Довольно неожиданным оказалось, что пирогенный кремне-
зем, который получался как продукт конденсации из пламени,
все еще содержал микропоры. Томпсон [57] показал методом
ИК-спектроскопии, что сохраняется небольшой по величине, но
все же значительный объем микропор, достигающий 0,01 см3/г,
причем в таких порах вода способна очень сильно удерживаться.
С наружной поверхности частиц кремнезема вода удалялась от-
сасыванием в условиях высокого вакуума при 26°С, но удалить
ее из тонких капилляров, в которых молекулы воды, по-види-
мому, прочно связаны водородными связями с окружающими со
всех сторон стенками микропор, в этих условиях не удалось.
Вода продолжает удаляться из пор при нагреве вплоть до 400°С,
но выше указанной температуры капилляры перекрываются
в результате образования силоксановых связей Si—О—Si; впо-
следствии при проведении повторной гидратации образца поры
не открываются.
По некоторым данным [58] пирогенный кремнезем аэросил
при 100°С содержит на поверхности только 35 % групп SiOH от
полного их содержания, а ксерогель — только 61 %. С другой
стороны, когда кремнеземные частицы выращиваются посредст-
вом постепенного осаждения растворимого кремнезема в горя-
чем щелочном растворе, можно было бы ожидать, что только
лишь отдельные гидроксильные группы в очень небольшом ко-
личестве удержатся в объеме частицы. Однако никаких фак-
тических доказательств, которые могли бы подтвердить правиль-
ность такого положения, не было представлено. Доказательст-
вом могло бы служить сравнение суммарного содержания
гидроксильных групп, выраженного в зависимости от удельной
поверхности, для кремнеземных частиц различных размеров при
условии, что все образцы приготовляются одним и тем же спо-
собом. Это количество следует экстраполировать к значению, от-
вечающему концентрации групп ОН, находящихся внутри очень
больших по размеру частиц, когда величина поверхности стано-
вится незначительной.
872
Глава 6
Когда кремнезем подвергается обработке в автоклаве
при высокой температуре, то в этом случае вода может диффун-
дировать внутри твердой фазы, стремясь достичь равновесной
концентрации. Согласно Чертову и др. [59], после подобной ги-
дротермальной обработки образцы кремнезема со значениями
удельных поверхностей в интервале 21—600 м2/г содержат
1 миллимоль эквивалентов воды внутри частиц кремнезема
в расчете на 1 г SiO2, что должно составлять около 0,9%.
В процессе автоклавной обработки величина удельной поверхно-
сти понижалась, но концентрация поверхностных гидроксильных
групп оставалась равной 5,6 группа ОН/нм2. Однако при этом
содержание воды в объеме сохранялось неизменным, равным
0,9% [60].
Из этих данных следует со всей очевидностью, что при рас-
чете концентрации и определении поведения силанольных групп
на поверхности, группы ОН внутри частиц кремнезема должны
быть исключены из рассмотрения. К сожалению, во многих ис-
следованиях этого не было сделано. Только позже было понято,
что внутренняя связанная вода может быть удалена из образца
кремнезема при высокой температуре, а образующиеся микро-
поры закрываются в этих условиях. На прошедших такую обра-
ботку образцах кремнезема можно проводить исследования де-
гидратации и регидратации с получением воспроизводимых
результатов. Но даже и в этих случаях для избежания неодно-
значности интерпретации необходимо измерять содержание ги-
дроксильных групп на поверхности таким методом, который
фиксировал бы только поверхностные группы SiOH.
Присутствие внутренней воды (или групп SiOH, которые, ве-
роятно, появляются в объеме кремнезема парами, по мере того
как идет гидролиз внутренних силоксановых связей Si—О—Si)
также вызывает наложение полос при проведении исследований
методом ИК-спектроскопии. Как внутренние, так и наружные
группы SiOH дают полосы поглощения. Допущение о существо-
вании парной гидроксильной группы [=Si(OH)2] на поверхно-
сти, которое привлекается для объяснения высокого содержания
жидкой воды на кремнеземе, предварительно не подвергав-
шемся прогреву, в настоящее время является предметом дискус-
сий. По-видимому, образование внутренних несконденсирован-
ных силанольных групп является более легко объяснимым.
Содержание гидроксильных групп на одном
квадратном нанометре
В настоящее время, по-видимому, уже установилась общая
точка зрения, что .на сглаженной, непористой, термически ста-
билизированной аморфной поверхности кремнезема, которая
Химия поверхности кремнезема
873
полностью гидроксилирована *, содержится 4—5 групп SiOH на
1 нм2** (или на площади 100 А2), причем такие группы сохра-
няются, когда образец кремнезема прогревается при 120—150°С.
Это значение поверхностной концентрации гидроксильных
групп цитируется в большинстве обзорных статей [4—13]. Та-
кая поверхность кремнезема получается после прогревания по-
рошка или силикагеля до повышенной температуры с целью
удаления удерживаемых внутри частиц молекул воды и групп
ОН, перекрывания микропор и последующей регидратации об-
разца кремнезема в воде.
Армистед и др. [61] показали, что на регидратированном
пирогенном кремнеземе на поверхности содержится 4,6 ОН-груп-
пы/нм2, причем из них 1,4 ±0,1 составляют свободные гидро-
ксильные группы, не связанные водородными связями с сосед-
ними группами, а 3,2±0,1 взаимно связаны попарно водород-
ными связями. Кроме того, существуют внутренние группы
SiOH в количестве 1,6 ОН-групп/нм2, которые необратимо уда-
лялись при прокаливании. Ниже представлены данные по ИК-
спектрам:
Тип групп
Идентифицируемая
полоса поглощения,
см-1
Одиночные ОН-группы на по-
верхности
Парные ОН-группы на поверх-
ности, связанные водород-
ными связями
Внутренние ОН-группы, свя-
занные водородными свя-
зями
Молекулярная вода
3750
3540
3650
3400, 1627
Атомы дейтерия тяжелой воды вступают в обмен с атомами
водорода только поверхностных гидроксильных групп, но не
групп ОН, находящихся внутри кремнезема. Журавлев и др.
[62] сравнили потери массы, получаемые в результате прокали-
вания образца кремнезема, с содержанием групп SiOH на по-
верхности, найденным методом дейтерообмена. Эксперимент
проводился на образце силикагеля с удельной поверхностью
340 м2/г и диаметром пор ПО А. Авторы нашли, что полное со-
* В отечественной научной литературе в подобном случае часто исполь-
зуется выражение «предельно гидроксилированное состояние поверхности».—
Прим. ред.
** Это значение поверхностной концентрации гидроксильных групп для
различных разновидностей аморфного кремнезема первыми независимо полу-
чили и обосновали голландские ученые Я. X. де Бур и И. М. Влеескенс и
советские ученые Л. Т. Журавлев и А. В. Киселев.— Прим. ред.
874
Глава 6
держание воды в образце было больше, чем связанной, пред-
ставляющей собой поверхностные гидроксильные Труппы, при-
чем поверхностная концентрация групп ОН оказалась равной
5,2 ОН-групп/нм2.
Рис. 6.3. Зависимость концентрации ОН-групп на поверхности кремнезема от
температуры прокаливания образцов SiO2.
Затененная площадь — область экспериментальных данных, полученных для различных
типов кремнезема Давыдовым, Киселевым н Журавлевым (43]. Дегидратация прокален-
ного (700 °C) и затем регидратированного кремнезема осуществлялась: 1 — на воздухе;
2— в вакууме (по данным Тейлора, Хоки и Петика [84]); 3 — данные для образцов крем-
незема, предварительно не прокаленных (на основании результатов, полученных Уиттер-
ховеном и др. [83], Фрипья и др. [47], Журавлевым и Киселевым [85], Тейлором, Хоки и
Петикой [84, 86]).
Результаты, полученные методом дейтерообмена, были обоб-
щены Давыдовым, Киселевым и Журавлевым [43] и сопостав-
лены с данными химических методов, в которых использовались
LiCH3 и MgCH3I; эти данные представлены на рис. 6.3 и
табл. 6.1. Позднее Агзамходжаев, Журавлев, Киселев и Шенге-
лия [63] применили этот метод на серии гидроксилированных
силикагелей, аэрогелей и других видах кремнезема, имевших
различные размеры пор, и обнаружили, что поверхностная кон-
центрация групп ОН менялась в интервале 4,2—5,7 ОН-групп/нм2.
Используя подобный метод, Мэдели и Ричмонд [64] установили,
что на четырех образцах кремнезема с удельной поверхностью
в интервале 374—701 м2/г концентрация групп ОН на поверхно-
сти составляла только 4,27—4,63 ОН-групп/нм2 без заметного
отклонения. Эти данные свидетельствуют; по-видимому, о воз-
можностях выбранного метода, так как дейтерообмен, несом-
ненно, ограничивается поверхностью. Удельную поверхность
Химия поверхности кремнезема
875
лучше всего определять по адсорбции криптона методом БЭТ,
так как природа поверхности в этом случае меньше сказывается
на результатах, чем при использовании азота в качестве адсор-
бата. Смесь Н2О—D2O, полученная после обмена, анализируется
с помощью масс-спектрометра *.
Вирцинг [50, 65] показал, что число групп ОН может быть
также определено по адсорбции на них воды с одновременным
измерением получающейся, единственно возможной при подоб-
ных исследованиях ИК-полосы поглощения 5265 см-1. Исследо-
вался хорошо спеченный образец кремнеземного порошка
фирмы «Мэллинкродт», который многократно нагревался до
800°С, охлаждался и каждый раз повторно регидратировался.
Его удельная поверхность составляла 166 м2/г; очевидно, обра-
зец не имел микропор. После высушивания при 120°С образец
содержал 5,2 ОН-групп/нм2, как показал метод прокаливания
до высоких температур, или 4,0 ОН-групп/нм2 по определению
методом ИК-спектроскопии. На образце аэрогеля было найдено
значение концентрации гидроксильных групп на поверхности,
равное 4,4 ОН-групп/нм2.
Согласно данным де Бура и Влеескенса [66], на исходном
кремнеземе, который не подвергался термической обработке, со-
держалось 6,2 ОН-групп/нм2, но после повторного увлажнения,
нагревания и высушивания поверхность образца выравнивалась
на молекулярном уровне, и достигалось значение 4,6±
±0,2 ОН-групп/нм2. На таком высушенном образце при пол-
ностью гидроксилированной поверхности примерно половина из
имеющихся силанольных групп присутствует в виде попарно
связанных водородными связями групп ОН. При беспорядочном,
произвольном распределении поверхностных групп представ-
ляется очевидным, что многие группы SiOH будут находиться
на поверхности в виде одиночных, неспаренных групп [67].
Согласно данным Андерсона и др. [68], на гидроксилирован-
ной поверхности в присутствии адсорбированной воды могут на-
ходиться два типа молекул воды и два типа групп SiOH. Моно-
мерная молекула Н2О может быть связана с одиночной груп-
пой SiOH. Кластер, состоящий из молекул Н2О, может образо-
вывать сетку, связанную водородными связями. Группы SiOH
могут быть несвязанные и связанные с молекулами Н2О.
Бермудец [69] использовал протонный ядерный магнитный
резонанс для определения числа поверхностных силанольных
групп даже при условии покрытия поверхности адсорбированной
водой толщиной вплоть до трех монослоев. Силикагель имел
удельную поверхность 800 м2/г, а найденная концентрация сила-
нольных групп составила 7-10 6 моль/м2 или 4,2 ОН-групп/нм2.
* См. прим. ред. на стр. 653.
876
Глава 6
Как будет обсуждено в связи с процессами дегидратации и
регидратации поверхности кремнезема, многие из упоминав-
шихся данных, показывавших более чем 4,6 ОН-групп/нм2, веро-
ятно, являются следствием присутствия в образце внутренних
групп SiOH, которые, как это принималось авторами, были от-
несены к поверхностным.
Согласно данным Боема [8], определение числа гидроксиль-
ных групп ОН на поверхности химическими методами дает про-
тиворечивые результаты. Он указал, что при проведении боль-
шинства химических реакций, таких, например, как этерифика-
ция или хлорирование, в подобную реакцию вступает только
примерно около половины всех присутствующих на поверхности
групп ОН от известного полного числа, равного 5 ОН-групп/нм2,
определенного эффективным методом водородного обмена или
по потере воды.
Возможно, что подобные замещающие группы слишком ве-
лики, чтобы размещаться на каждом исходном центре SiOH.
Когда замещающая группа (например, этоксильная группа, или
атом хлора) всего лишь ненамного больше группы ОН, то при-
мерно только каждая вторая группа ОН сможет заместиться,
поскольку положение лежащих ниже атомов кремния фиксиро-
вано. Однако метильная группа оказывается достаточно неболь-
шой и способна присоединиться к каждому центру SiOH. Как
сообщалось в другом разделе данной книги, к поверхности крем-
незема на площади 1 нм2 могут присоединяться от четырех до
пяти метоксигрупп. Именно поэтому использование таких реак-
тивов, как СНзЫ и CHaMgl, в работе Фрипья и др. [47] ока-
залось успешным. Другим веществом, содержащим метильные
группы, является составное соединение, в котором присутствуют
диметилцинк и тетрагидрофуран (ZnMe2-2 ТГФ). Это соедине-
ние использовал в своей работе Ханке [70], который измерял
концентрацию групп ОН на поверхности аэросила по опреде-
лению объема выделяющегося метана СН4.
Единственным другим эффективным заместителем достаточно
малого размера, позволяющим замещать полностью все группы
ОН на поверхности, представляется атом фтора, но оказалось
трудным создать поверхность типа SiF без того, чтобы не раз-
рушить структуру SiO2.
Лифландер и Штобер [71] использовали раствор триизобу-
тилалюминия в гептане с целью проведения реакции с поверх-
ностными группами SiOH. После промывания образца гептаном
для удаления избыточного количества реактива поверхность
кремнезема удерживала после гидролиза 5,7 мкмоль А1:
SivOH + AlRs—^Si.OAIRa-^-SisOAI (ОН)2 + 2RH
Химия поверхности кремнезема
877
Теоретическая величина концентрации
поверхностных гидроксильных групп
Расчет числа силанольных групп на поверхности может быть
осуществлен различными способами. Простейшим является спо-
соб, предложенный Айлером [3], полностью основанный на гео-
метрических рассмотрениях и учете плотности аморфного крем-
незема. Этим способом, описанным в гл. 1, определено, что
7,8 атома кремния должно содержаться на 1 нм2 на поверхности
или очень близко к поверхности. Автор первоначально допу-
скал, что на поверхности должно содержаться 7,8 SiOH-групп/нм2.
Однако Боем [8] указал, что поскольку не все атомы кремния
точно находятся на границе раздела фаз, а могут располагаться
выше или ниже этой границы, то только половина всех поверх-
ностных атомов кремния должна нести группы ОН, и поэтому
поверхностная концентрация таких групп должна быть равной
только 3,9 ОН-групп/нм2.
Так как плотность и показатель преломления аморфного
кремнезема близки к соответствующим величинам кристобалита
и тридимита, то Айлер предположил, что концентрацию поверх-
ностных гидроксильных групп можно было бы оценить по этим
кристаллическим структурам. Исследование кристаллической
грани {100} p-кристобалита, которая была выбрана автором,
привело к заключению, что на каждом участке площади поверх-
ности, равном 50,2 А2, имелось два более низко расположенных
уровня атомов кремния, не несущих гидроксильных групп, и два
выше расположенных уровня атомов кремния, в которых каж-
дый атом Si удерживал по две группы ОН. Это давало концен-
трацию 8 ОН-групп/нм2. Аналогичные расчеты для поверхности
тридимита дали концентрацию 4,6 ОН-групп/нм2. Пери и Хенсли
[67] продолжили рассмотрение кристобалита и отметили, что
если выбирается грань {100}, то действительно каждый атом
кремния должен удерживать две группы ОН, давая значение
концентрации ~8 ОН-групп/нм2. Если бы такие гидроксильные
группы могли попарно удаляться произвольным образом, то, со-
гласно расчету, выполненному по методу Монте-Карло, на по-
верхности должно было бы оставаться 4,56 ОН-групп/нм2 в виде
либо парных групп ОН (две группы на одном атоме Si), либо
смежных, вицинальных групп ОН (одна группа на одном атоме
Si, но две группы ОН расположены рядом).
Так как ранее полученные экспериментальные данные
(табл. 6.1) давали значение концентрации около 8 ОН-групп/нм2,
то принималось, что полученные в теоретических расчетах бо-
лее высокие значения концентраций групп ОН были более веро-
ятными. Такое положение получило некоторую поддержку
878
Глава 6
в связи с появлением работ Беляковой, Джигит и Киселева [72]
и Жданова и Киселева [73], которые определили, что концен-
трация ОН составляет до 12 мкмоль/м2, или до 7,2 ОН-групп/нм2.
Однако де Бур и Влеескенс [74, 75] позже указали, что по-
скольку p-кристобалит кристаллизуется в виде октаэдра, то кон-
центрация гидроксильных групп должна подсчитываться на ок-
таэдрической грани {111}. Такая поверхность показана на
рис. 6.4. Рассчитанное значение концентрации гидроксильных
Рис. 6.4. Расположение атомов на октаэдрической грани [Ш] кристобалита,
содержащей 4,55 SiOH-группа/нм2.
Большие кружки — атомы кислорода; маленькие кружки — атомы кремния на поверхно-
сти; штрих-кружки — положение гидроксильных групп на поверхности, присоединенных
к нижележащим атомам кремния. Масштаб размеров атомов не сохранен.
групп составляет 4,55 ОН-групп/нм2. Экспериментальные опре-
деления подтвердили этот результат.
Из данных, которые появились к настоящему времени, сле-
дует, что термически стабилизированная поверхность аморфного
кремнезема действительно очень напоминает октаэдрическую
грань и что значение концентрации 4,6 ОН-групп/нм2 стало
обычно наблюдаемым экспериментально значением для регидра-
тированных образцов кремнезема, которые были «сглажены» и
прокалены при высокой температуре, а затем полностью регид-
ратированы и высушены.
Дегидратация и регидратация
По мере повышения температуры образца кремнезема гидро-
ксильные группы на поверхности конденсируются с образова-
нием силоксановых связей и с выделением воды. Уменьшение
концентрации гидроксильных групп на поверхности с ростом
температуры широко изучалось различными исследователями,
Химия поверхности кремнезема
879
и полученные результаты были обобщены в обзорах Барби [13],
Оккерса [11] и Стрелке [76], которые представили 168 библио-
графических ссылок по этой теме.
Это явление можно изучать, начиная с термически устойчи-
вой, полностью гидроксилированной поверхности и прослежи-
вая удаление гидроксильных групп в виде выделяющейся воды
в потоке воздуха или азота или в вакууме. Поскольку пары
воды являются сильным катализатором, способным перегруппи-
ровать силоксановые связи, то не удивительно, что данные по
дегидратации будут оставаться трудно воспроизводимыми, за
исключением случаев, когда водяной пар удаляется в условиях
вакуума или же исследуемый образец кремнезема берется
в очень небольшом количестве и распределяется в виде тонкой
пленки для того, чтобы обеспечить быстрое удаление такого
пара.
При другом подходе следовало бы начинать с исходной не-
поврежденной поверхности кремнезема, образуемой в инертном
газе или в вакууме. Антонини и Хохштрассер [77], применяя
метод электронного парамагнитного резонанса (ЭПР), исследо-
вали разорванные связи на образце плавленого кварца, который
распылялся в вакууме. Наблюдалось образование на поверхно-
сти центров Si+, которые могли реагировать с СО2, формируя
комплекс Si+CO~. Такая поверхность теряет свою активность
по мере того, как захватывает воду или другие примесные за-
грязнения, поступающие из образца. Жданов [78] сообщил, что
гидратация подобной поверхности происходила во много раз
быстрее, чем поверхности, которая предварительно дегидрати-
ровалась при высокой температуре.
Каутскому и Мишелю [79], по-видимому, удалось посредст-
вом окисления силоксана §1бО3Н6 приготовить такую разновид-
ность кремнезема, поверхность которой не имела силанольных
групп. Совершенно не похожая на обычные гидратированные
кремнеземы, эта форма SiO2 не адсорбировала родаминовый
краситель и не образовывала флуоресцирующий продукт.
Вероятно, из-за трудности приготовления полностью лишен-
ного водорода тонкодисперсного кремнезема с достаточно одно-
родной поверхностью и высоким значением удельной поверхно-
сти было проведено лишь немного работ по изучению процесса
гидратации. Такой кремнезем мог бы быть получен путем испа-
рения и конденсации SiO2 в сухой атмосфере кислорода.
Диц и Тернер [80] использовали в своих работах нетрону-
тые обработкой кварцевые волокна, содержавшие 3 % В2О3,
хотя следует отметить, что присутствие бора на поверхности мо-
жет усложнить истолкование получаемых результатов. Наибо-
лее значительным в работе явилось наблюдение, что свежеобра-
зованная обезвоженная поверхность волокон быстро регид-
880
Глава 6
роксилируется в результате адсорбции на ней молекул воды.
К тому же когда поверхность дегидроксилируется при 800°С, то
регидратация протекает гораздо медленнее по сравнению со слу-
чаем, когда поверхность дегидроксилируется при более низкой
температуре. Авторы считают, что на поверхности существуют
«центры», на которых начинается регидратация, и число таких
центров уменьшается при повышении температуры.
Дегидратация
Вероятно, по дегидратации кремнезема были проведены
сотни исследований, но лишь результаты нескольких работ поз-
воляют точно оценить степень дегидроксилирования поверхности
кремнезема по мере повышения температуры. Получаемые дан-
ные не особенно согласуются, но этот факт не должен удивлять,
поскольку образцы кремнезема очень сильно различаются по
своим структурным особенностям, а также по содержанию сле-
довых примесей. Например, присутствие некоторого количества
ионов К+ на поверхности кремнезема может понижать темпе-
ратуру дегидроксилирования на 100—200°С [81].
Поскольку дегидратация представляется неравновесным про-
цессом, то скорость, с которой вода покидает поверхность на
любой стадии процесса, будет являться функцией температуры
и концентрации оставшихся силанольных групп. Таким образом,
чтобы получить представляющее достаточную ценность значение
концентрации гидроксильных групп на поверхности, выражаемое
числом ОН-групп на 1 нм2 при данной температуре, продолжи-
тельность нагрева или конечная минимальная скорость потери
воды с поверхности должны быть определенными. Однако на-
блюдается относительно быстрая потеря воды в течение первых
6—10 ч после ступенчатого подъема температуры до нового
уровня, а затем вода удаляется с поверхности гораздо медлен-
нее, и этот процесс может происходить в продолжении несколь-
ких суток. Обычно дегидратация проводится в течение опреде-
ленного фиксированного времени, после чего, как показывает
эксперимент, в ходе более продолжительной дегидратации мо-
гут иметь место лишь «незначительные» дальнейшие изменения.
Имелик и соавторы [82] отметили, что различные типы крем-
незема могут иметь различающуюся плотность расположения
групп SiOH на поверхности и разную термическую устойчивость
таких групп. Однако на рис. 6.3 была представлена температур-
ная зависимость поверхностной концентрации силанольных
групп (число групп ОН на 1 нм2) для широкого ряда различных
кремнеземов, измеренной способом, позволяющим определять
только группы ОН на поверхности, но не внутри частиц крем-
Химия поверхности кремнезема
881
незема, после того как образцы подвергались термической обра-
ботке при заданной повышенной температуре.
Измеренная Кертойзом и др. [87] в еще более высокой об-
ласти температур концентрация оставшихся на поверхности
групп SiOH представлена ниже:
Температура, °C Число ОН-групп/нм2
700 1,2
800 0,9
900 0,65
1000 0,4
Как правило, начальная концентрация на полностью гидро-
ксилированной поверхности составляет примерно 5 ОН-групп/нм2
при 150°С и только ~ 1 ОН-групп/нм2 при 800°С. Силанольные
группы начинают конденсироваться п удаляться в виде воды
в заметной степени выше ~ 170°С. При 400°С немного менее по-
ловины исходных гидроксильных групп оказываются удален-
ными от поверхности, но большая часть остальных групп сохра-
няет попарно смежное расположение по крайней мере это имеет
место для каждой второй группы ОН. При таком состоянии по-
верхности вода может адсорбироваться и, следовательно, по-
верхность легко регидратируется. Выше 400—450°С уже боль-
шое число гидроксильных групп удалено с поверхности, и появ-
ляются обширные силоксановые участки, регидратация которых
затруднена [88]. При ~750°С на поверхности присутствуют
только одиночные свободные, неспаренные группы SiOH с вели-
чиной поверхностной концентрации 1,3 ОН-групп/нм2, как это
показал в своих работах Торп [89], используя измерение ди-
электрических изотерм и другие методы.
Дзпсько, Вишневская и Чесалова [90] показали возможность
проведения термической дегидратации поверхности кремнезема
без уменьшения величины его удельной поверхности. Вероятно,
эти авторы первыми пришли к правильному заключению, что
физически адсорбированная вода удалялась при 115°С и что
большая доля оставшейся «связанной» воды присутствовала
в виде слоя гидроксильных групп на поверхности кремнезема,
так как относительное процентное содержание этой «связанной»
воды оказалось пропорциональным удельной поверхности для
серин образцов. Они также пришли к заключению, что в интер-
вале 115—600°С все большее число гидроксильных групп кон-
денсировалось с выделением воды при условии лишь минималь-
ного снижения величины удельной поверхности. По оценкам
этих авторов, каждая группа ОН занимала на поверхности пло-
щадь, равную 15 А2, так что поверхностная концентрация дол-
жна была составлять 6,7 ОН-групп/нм2.
30 Заказ № 250
882
Глава 6
Согласно данным Боема [8], расстояния между группами ОН
на гидроксилированной поверхности могут отличаться. Некото-
рые гидроксильные группы расположены ближе друг к другу,
поэтому между ними образуются более прочные водородные
связи. Такие группы проявляют полосу ИК-поглощения 3520 см-1.
Другие группы, расположенные дальше друг от друга, более
слабо связаны между собой водородными связями. Их обнару-
живают по полосе поглощения 3660 см-1. Когда происходит на-
грев образца, то группы ОН, которые не так сильно взаимно
связаны водородными связями, удаляются первыми, т. е. вна-
чале исчезает полоса 3660 см-1 [91—94].
Полосы поглощения были идентифицированы следующим об-
разом:
Тип группы ОН Полоса, см-’
Изолированные, одиночные группы ОН или «свободные»' гидроксильные группы 3745 -3750
Изолированные пары, состоящие из двух смежных, соседних (вицинальных) групп SiOH, взаимосвязанных водородными связями 3650-3660
Смежные парные группировки, состоящие из двух смежных групп SiOH; между группировками имеется водородная связь 3540-3550
Адсорбированные на гидроксильных груп- пах молекулы воды 3400-3500
Хейр [9] обобщил данные по изучению поверхности кремне-
зема методом ИК-спектроскопии и дал описание, каким образом
идентифицировать различные полосы поглощения.
Кондо и Муройя [95] наблюдали отдельные стадии процесса
дегидратации, используя комбинацию методов: дифференциаль-
ный термический анализ, термогравиметрический анализ и ИК-
спектроскопию. Представленные ниже стадии сопровождаются
исчезновением соответствующей полосы поглощения ИК-спектра:
Температура, °C Исчезающая полоса
поглощения, см-J
300 3230
400 3460
500 3620
Полоса при 3750 см !, отвечающая свободным, изолированным
группам SiOH, сохраняется вплоть до высокой температуры.
С точки зрения не вызывающего сомнения присутствия внут-
ренних гидроксильных групп не представляется возможным оп-
ределить, какие же изменения происходят на указанных ста-
диях. Было бы очень интересным повторить подобные иссле-
дования, используя в качестве образца аэрогель, не содержа-
Химия поверхности кремнезема 883
щий, натрия, который был бы подвергнут термической обра-
ботке с целью стабилизации при 1000°С, а затем регидратации
в кипящей воде в течение суток или около этого. Подобный
кремнезем с незначительной долей гидратации внутри частиц
должен иметь удельную поверхность 100—200 м2/г, которая
могла бы оказаться достаточной для указанных выше методов
исследования.
Кунавич, Джонс и Хокки [96] и Бермудец [49, 69] иденти-
фицировали различные типы силанольных групп по их реакции
с ВС1з. Другие реакции, посредством которых изучались сила-
нольные группы, обобщены в следующем разделе настоящей
главы.
Де Бур и Влеескенс [37] нашли, что значение числа групп
ОН на 1 нм2 оказывается несколько меньшим, когда кремнезем
нагревается до 650°С и охлаждается, а затем регидратируется
в жидкой воде при 90°С и снова высушивается при 120°С. Если
такой процесс повторяется, то концентрация достигает вели-
чины 4,6 ОН-групп/нм2. Если исходный образец кремнезема на-
гревают один раз и сразу до 890°С, после чего регидратируют
и высушивают, то значение снова становится равным
4,6 ОН-групп/нм2.
Таким способом было продемонстрировано, что сглаженная
прокаленная поверхность кремнезема, когда она полностью
гидратирована, имеет одну гидроксильную группу ОН, прихо-
дящуюся на один поверхностный атом Si, а концентрация таких
групп составляет 4,6 ОН-групп/нм2. Кроме того было показано,
что когда образец кремнезема прокаливается, то внутренняя
вода и (или) внутренние группы ОН также удаляются и струк-
тура кремнезема уплотняется до такой степени, что при повтор-
ной гидратации поверхности меньшее количество воды будет
входить внутрь объема образца.
После рассмотрения появившейся доступной библиографии
Оккерс [11] пришел к заключению, что не имеется реального
доказательства существования постулированных парных групп
=Si(OH)‘2 и тем более существования строенных групп
—Si(OH)3 на любой высушенной поверхности кремнезема. Если
кремнезем имеет кажущееся содержание гидроксильных групп
более чем 4,6 ОН-групп!нм2, то избыточное содержание воды по
сравнению с этой величиной должно представлять собой захва-
ченную внутрь образца воду в виде внутренних групп SiOH *.
Как было показано Чертовым и др. [60], внутренняя вода и
внутренние гидроксильные группы удаляются начиная с 200°С.
* К подобному заключению на основании выполненных ими работ при-
шли ранее советские исследователи Л. Т. Журавлев и А. В. Киселев с со-
авторами (см. лит. [43, 62, 63 и 85] к наст. гл. и [113] к гл. 5).— Прим. ред.
30*
884
Глава 6
Максимальная скорость удаления воды в расчете на 1 градус
подъема температуры, наблюдается примерно при 500°С для
всех исследованных образцов кремнезема. Однако для почти
полного удаления температура должна достигать примерно
1000°С. При этой температуре поверхность кремнезема в силь-
ной степени дегидратируется до силоксанового состояния по-
верхности.
Механизм дегидроксилирования был рассмотрен Хокки и Пе-
тика [97]. Они отметили, что более вероятна миграция прото-
нов, чем гидроксильных групп. Авторы предполагают, что про-
тоны мигрируют с образованием напряженных кислородных мо-
стиков. Конечно, интересно определить, будет ли процесс деги-
дратации происходить более быстро в присутствии паров воды
с низкой концентрацией по сравнению с вакуумными условиями.
Имеется некоторое доказательство, представленное Кондо,
Фудживара и Муройя [98], что при увеличении содержания
ионов натрия в силикагеле от 0,0005 до 0,0060 % процесс деги-
дратации несколько облегчается. Наиболее очевидным эффек-
том, объясняющим такое поведение кремнезема, представляется
понижение температуры спекания, при которой происходит бы-
строе понижение величины удельной поверхности. Но во всех
случаях авторы отмечали небольшое спекание ниже 700°С. Ко-
гда содержание ионов натрия в образце наиболее низко, то по-
верхностная концентрация гидроксильных групп в зависимости
от температуры прокаливания образцов кремнезема, выражае-
мая как число групп ОН на 1 нм2, вероятно, будет приближаться
к верхней границе затененной площади, представленной на
рис. 6.3.
Для полной дегидратации пористого кремнезема очень эф-
фективен нагрев образца в атмосфере сухого хлора при 600—
1000°С. Этот способ, по-видимому, пригоден для получения оп-
тического стекла, полностью освобожденного от силанольных
групп [99]. При другом способе следует проводить реакцию та-
кого кремнезема, как аэросил, с SiCl4 при 400°С, а затем си-
стему нагревать до 700°С. Образующаяся в результате поверх-
ность, по существу, не содержит групп ОН [100].
Дегидратация поверхности кристаллического кварца, как мо-
жно было ожидать, протекает более равномерно, так как все ее
центры должны быть в химическом отношении одинаковы (за
исключением ребер). Штобер [101] изучил это явление, исполь-
зуя кварцевые частицы различных размеров, и пришел к заклю-
чению, что дегидратация по крайней мере на поверхности
кварца происходит по стадиям, показанным на рис. 6.5. Штобер
обнаружил, что даже после тщательной термической обработки
образца при 100°С на поверхности кварца на каждых двух си-
ланольных группах удерживается одна молекула чрезвычайно
Химия поверхности кремнезема
885
прочно адсорбированной воды. По-видимому, поведение аморф-
ного кремнезема совсем иное. Очевидно, на поверхности кварца
существуют группы SiOH, способные образовывать сильные во-
дородные связи. Возможно, именно с этим связана специфиче-
ская сильная способность такой кристаллической поверхности
адсорбировать полислои монокремневой кислоты из раствора,
как было показано в работе Баумана и описано в гл. 1 [151].
©3 •'© нН Н :'© н'; Н Ж ;'© Н; Н
© щн/ © © \ н,/ © © - дз / ©
-Si-©" ©-&-©-&-© ~©-Si-© - Si-© ©-Si-©
Г ' Si I ! 'Si I I 'Si I
(6)
© + © + © +
-Si-©x ©-Si-©~Si~©^ ©-Si-©-Si-©^ ©-Si-©
I V । i V I 1 s[ ।
/ \ /X / X
(S)
\ /©„ © ©ч © Д ©
Si Siz xSi Siz 4 Si Siz
''©©' '©©' /©©'
'Si zSi ©
Рис. 6.5. Стадии дегидратации поверхности кристаллического кварца.
а — вода, необратимо адсорбированная на поверхности кварца; б — поверхность кварца
сразу же после термической дегидратации; в — безводная силоксановая поверхность
после термической дегидратации. (По данным Штобера [101].)
Другим указанием на большое различие между поверхно-
стями аморфного кремнезема и кварца являются данные, пред-
ставленные Янгом и Баршем [102а], которые обнаружили, что
теплота адсорбции воды на поверхности кварца, с которой пред-
варительно была удалена вода, оказалась примерно в два раза
большей по сравнению с теплотой адсорбции на поверхности
аморфного кремнезема, а именно 285 и 120—190 эрг/см2 соот-
ветственно.
Егоров и соавторы [1026] представили интересную схему
природы гидратации и дегидратации кремнеземной поверхности,
а также показали результаты, связывающие эти процессы
с удельной поверхностью кремнезема и его адсорбционными
свойствами.
Влияние размеров частиц и диаметров пор. Согласно дан-
ным Брунауэра и др. [103], подтвержденным в работе Уолена
[104], частицы небольшого размера, например диаметром 3,7 нм,
способны дегидратироваться при более низкой температуре, чем
886
Глава 6
частицы больших размеров (6,4 нм). Уолен обнаружил, что
кремнезем с удельной поверхностью 330 м2/г имел 7 ОН-групп/нм2
при 110°С, а образец с удельной поверхностью 650 м2/г содер-
жал 5,5 ОН-групп/нм2.
Дзисько, Вишневская и Чесалова [105] провели дегидрата-
цию образцов силикагелей с диаметрами пор 10, 20 и 27 А при
серии значений температур обработки и измеряли содержание
оставшейся воды. Силикагели с меньшими размерами пор удер-
живали наибольшее количество воды, как это показано
в табл. 6.2. Эти результаты можно объяснить на основании того,
что удаление поверхностных гидроксильных групп, связанных
между собой водородными связями, при повышенной темпера-
туре затруднено. Вероятно, что механизм дегидратации преду-
сматривает термическую диссоциацию протона, который мигри-
рует и присоединяется к гидроксильной группе, образуя моле-
кулу воды. Для подобной диссоциации необходима затрата
большей энергии, если бы атом водорода располагался между
смежными атомами кислорода.
Таблица 6.2
Характеристики силикагелей с различным диаметром
пор (по даииым [105])
Температура дегидратации, °C Удельная поверхность, м2/г Количество связанной воды, % Концентрация групп ОН на 1 НЦ2
Диаметр пор 10 А
115 400 6,5 10,8
300 480 4,4 6,1
600 375 3,4 6,0
700 280 1,5 3,6
Диаметр пор 20 А
115 540 6,1 7,5
300 500 4,3 5,7
600 . 400 2,9 4,8
700 340 1,7 3,3
Диаметр пор 27 А
115 450 3,8 5,6
300 500 4,0 5,3
600 420 2,3 3,6
700 210 1,1 3,5
Химия поверхности кремнезема
887
На рис. 6.6 схематически показаны эффекты, которые опре-
деляются размером частицы, когда ее радиус кривизны поло-
жителен, и диаметром поры, когда радиус кривизны отрицате-
лен. Кажется вполне логичным, что внутри небольшой по раз-
меру поры гидроксильные группы должны ближе располагаться
друг к другу, и, таким образом, их состояние стабилизируется
Рис. 6.6. Влияние кривизны по-
верхности кремнезема на де-
гидроксилирование.
а — небольшой положительный ра-
диус кривизны (частицы малого
размера) приводит к меньшему чис-
лу водородных связей между гид-
роксильными группами, поверхность
дегидроксилируется наиболее легко;
б — большой радиус кривизны (ча-
стицы большого размера, плоская
поверхность) определяет относи-
тельно большее число гидроксиль-
ных групп, связанных водородными
связями, поверхность дегидроксили-
руется менее легко; в — небольшой
отрицательный радиус кривизны (в
небольших порах) обусловливает
наибольшее число водородносвязан-
ных гидроксильных групп, поверх-
ность дегидроксилируется наиболее
трудно.
посредством образования водородных связей; при этом могут
проявляться и другие силы взаимодействия.
По мере того как силикагель дегидратируется, отношение
числа одиночных (свободных) гидроксильных групп к числу
смежных, соседних (реагирующих) гидроксильных групп, вза-
имно связанных водородными связями, становится все более
зависящим, как это признал Снайдер [10], от диаметра пор. Он
установил, что поверхность, образуемая стенками широкой
поры, оказывается более похожей на поверхность «кристалличе-
ского» кремнезема и, следовательно, будет содержать большее
число свободных гидроксильных групп. Важна, конечно, не сте-
пень кристаллизации, а геометрия поверхности. Кристалличе-
ские грани являются плоскими, и в этом отношении они по-
добны поверхности большой поры, когда последнюю можно рас-
сматривать как «плоскую» с позиций атомного масштаба. Но
в небольших порах, диаметром менее 100 А, отрицательная кри-
визна заставляет сближаться друг с другом силанольные
группы, так что в этом случае возможно образование большего
числа взаимных водородных связей между группами ОН, что
888
Глава 6
приводит к большей их устойчивости относительно удаления при
высокой температуре.
Снайдер и Уорд [106] представили данные, показывающие
зависимость от диаметра пор отношения Sr/St — доли гидро-
ксильных групп, способных заметно реагировать, т. е. образо-
вывать водородные связи, к общему числу гидроксильных групп.
Диаметр пор оказывает небольшое воздействие, пока он более
100 А, но затем, когда диаметр пор уменьшается от 100 до 50 А,
отношение Sr/Sf возрастает от 0,05 до 0,8. Поскольку при кон-
центрации гидроксильных групп на поверхности 4,6 ОИ-групп/нм2
атомы кремния должны находиться друг от друга на среднем
расстоянии около 4,7 А, то можно наглядно представить, что
группы ОН не должны вступать в заметный контакт друг с дру-
гом до тех пор, пока диаметр пор не станет менее 100 А (см.
рис. 6.6). При диаметре поры 50 А ее радиус только примерно
в пять раз превышает расстояние Si—Si, и группы ОН должны
находиться гораздо ближе друг к другу, чем на плоской по-
верхности.
После дегидратации при высокой температуре внутри таких
небольших пор имеется, следовательно, большее количество пар
гидроксильных групп, связанных водородными связями, чем
в порах большего диаметра.
Снайдер и Уорд предположили, что при реакции с таким реа-
гентом, как (CH3)3SiCl, подобная пара гидроксильных групп
значительно более реакционноспособна по сравнению с одиноч-
ной группой ОН. В действительности один из атомов кислорода
пары может начать координировать с приближающимся атомом
кремния молекулы триметилхлорсилана, тогда как атом С1 этой
молекулы взаимодействует с атомом водорода второй группы
ОН на поверхности:
сн3 снз
СН3—Si—С1
Т н
оно
Si Si
Такое согласованное взаимодействие невозможно в случае оди-
ночной изолированной группы SiOH, и поэтому для проведения
реакции с ней потребуется более высокая температура.
Бакырджиев [107] в противоположность вышесказанному
рассчитал эффект влияния кривизны поверхности на концен-
трацию гидроксильных групп для четырех силикагелей, дегидра-
Химия поверхности кремнезема
889
Рис. 6.7. Изменение кривизны
поверхности от положительно-
го к отрицательному значению
в процессе старения силикагеля.
а — несостаренный силикагель (боль-
шая часть поверхности в порах
имеет положительный радиус кри-
визны); б — состаренный силикагель
(большая часть поверхности крем*
незема в порах имеет отрицатель-
ный радиус кривизны). Слегка утол-
щенные линии показывают участки
поверхности с положительным ра-
диусом кривизны.
на цис. 6.7. если силика-
тированных при 300—600°С. Он со-
общил, что в более узких порах де-
гидратация происходит при более
низкой температуре. Это противо-
речит выше представленным экспе-
риментальным данным, полученным
Дзисько, Вишневской и Чесаловой.
Однако здесь имеется почва для
неопределенности. Силикагель с бо-
лее узкими порами может фор-
мироваться из меньших по размеру
частиц кремнезема. Как показано
гель не подвергался процессу старения, то поверхность внутри
пор должна состоять в основном из небольших сферических
частиц с положительным радиусом кривизны. В этом слу-
чае гидроксильные группы должны быть менее сильно связаны
водородными связями и, следовательно, их легче удалять в виде
молекул воды. Таким образом, возможно, что в одном и том же
исходном силикагеле процесс старения может превратить кри-
визну большей части внутренней поверхности кремнезема из по-
ложительной в отрицательную, как это показано на рис. 6.7,6.
В подобном состаренном геле, чем меньше диаметр пор, тем
более стабильными становятся группы ОН.
Регидратация
Исследования, проведенные Янгом и Баршем [92, 102а], от-
четливо показали, что молекулы воды адсорбируются только на
гидроксилированной поверхности кремнезема, но не на силокса-
новой, которая по существу оказывается гидрофобной. Однако
процесс регидратации должен включать в качестве первого этапа
адсорбцию воды, так что гидратация, вероятно, имеет место и
на силоксановых кислородных центрах после того, как она
890
Глава 6
прошла на силанольных центрах. Авторы нашли, что поверхно-
сти кремнезема, дегидратированные при нагреве образцов
вплоть до 400—425°С, легко повторно гидратировались, тогда
как при более высокой температуре предварительной обработки
образцов процесс регидратации постепенно становился все мед-
леннее. При температуре предварительной обработки 425°С на
поверхности остается около 2 ОН-групп/нм2, или 40—45% ис-
ходной концентрации (4,6 ОН-групп/нм2). Дегидратированные
силоксановые центры, вероятно, являются обособленными при
таком состоянии поверхности. На этом этапе теплота адсорбции
воды в расчете на единицу поверхности кремнезема оказывается
максимальной.
Регидратация частично дегидроксилированной поверхности
катализируется щелочью [108]. Однако высокое значение pH
также содействует уменьшению величины удельной поверхности
при продолжительном контакте с водой. Добавление небольшого
количества NH4OH обеспечивает сохранение области pH, в ко-
торой этот нежелательный эффект устраняется. Процесс реги-
дратации, разумеется, замедляется при рН<5.
Снайдер [10, 109] сообщает, что регидратация частично де-
гидратированной силоксановой поверхности даже в воде идет
чрезвычайно медленно при 25°С. При 95°С кремнезем должен
выдерживаться в воде в течение нескольких часов. Однако та-
кую процедуру можно проводить только с термически стабили-
зированными, отожженными образцами кремнезема с низкой
удельной поверхностью. Кремнеземные порошки с высокой
удельной поверхностью будут испытывать сильные изменения
в структуре, и величина удельной поверхности при этих усло-
виях у них снижается.
Регидратация высокодегидратированной поверхности, веро-
ятно, начинается вслед за гидратацией на участках, где имеются
свободные гидроксильные группы, поскольку, по данным ИК-
спектроскопии, характеристическая полоса поглощения таких
групп ослабляется [91]. Часто встречающиеся в литературе вы-
сказывания о том, что поверхность кремнезема, дегидратирован-
ная при 1000°С, не может быть снова полностью гидратирована,
оказываются неверными, как это показано в работе Агзамход-
жаева, Журавлева, Киселева и Шенгелия [ИО]. Авторы обна-
ружили, что чем более высока степень предварительной дегидра-
тации поверхности кремнезема, тем более продолжительное
время требуется для регидратации. Кремнезем аэрогель, кото-
рый прокаливался при 1100°С в течение 10 ч и имел удельную
поверхность 144 м2/г, содержал всего лишь 0,06 ОН-групп/нм2.
После кипячения такого образца в воде в течение 60 ч величина
удельной поверхности все еще оставалась равной 108 м2/г, и
в результате проведенной регидратации было полностью восста-
Химия поверхности кремнезема
891
новлено значение концентрации 4,5 ОН-групп/нм2. Для полной
регидратации дегидратированной поверхности с концентрацией
0,66 ОН-групп/нм2, полученной после предварительной термо-
обработки при 900°С в течение 10 ч, потребовалось выдержива-
ние образца кремнезема в воде при комнатной температуре в те-
чение нескольких лет.
Вероятно, что регидратация идет вначале на гидроксильных
группах и что образующиеся гидроксилированные участки по-
верхности распространяются в виде отдельных пятен по мере
того, как гидратация продолжается вдоль границы между гид-
роксилированными и силоксановыми участками на поверхности.
Данные, представленные Волковым, Киселевым и Лыгиным
[111], по-видимому, подтверждают это представление, так как
на сильно дегидратированной поверхности, полученной после
термообработки при 900—1100°С, вода вначале хемосорбирова-
лась за счет раскрытия напряженных силоксановых связей с об-
разованием силанольных групп и воды. После этого происхо-
дила адсорбция воды предпочтительнее на силанольных груп-
пах, чем на силоксановых участках поверхности. Дальнейший
разрыв силоксановых связей мог происходить только вдоль гра-
ниц вблизи таких пятен.
Поверхностная энергия
Для образцов кремнеземов, которые имели различающиеся
известные удельные поверхности при разных степенях дегидро-
ксилирования, Брунауэр и др. представили [ЮЗ] следующие
значения полной поверхностной энергии (энтальпии), определяе-
мой из данных по теплоте растворения в смесях HNO3—HF
(определения проводились при 23°С):
Энергия, эрг/см2
Чистая силоксановая поверх- 259 ± 3
ность
Чистая силанольная поверх- 129 ± 8
ность
Теплота гидратации 130 ± 7
Следовательно, теплота гидроксилирования силоксановой по-
верхности, необходимая для превращения ее в силанольную при
23°С. составляет 259± 13 кал/г воды, или 4660±230 кал/моль.
Киселев [И2] сообщил, что когда силикагели с различными
Удельными поверхностями нагревались предварительно при
300°С, то степень гидроксилирования поверхности понижалась
с возрастанием величины удельной поверхности. Одновременно
поверхностная энергия таких образцов кремнезема возрастала.
892
Глава 6
Между этими величинами наблюдалась следующая зависи-
мость:
Удельная
поверхность,
м2/г
100
700
Содержание групп
SiOH, мкмоль/м2
5,0
3,3
Поверхностная
энергия, эрг/см2
170
200
Как было предсказано Айлером [3] и отмечено Брунауэром
и др. [ЮЗ], полная энергия силанольной поверхности оказы-
вается лишь немного выше, чем полная поверхностная энергия
жидкой воды, равная 118,5 эрг/см2 при 25°С.
Гуд [ИЗ] в своем обзоре дал развитие и обоснование тео-
ретических вопросов, связанных с поверхностной свободной
энергией твердых веществ и межфазовых границ раздела. Тео-
рия рассматривает физическую адсорбцию, смачивание и разде-
ление фаз, но не касается вопросов, связанных с необратимыми
процессами, такими, как, например, хемосорбция. ' ,
Теплота смачивания поверхности кремнезема
Вполне очевидно, что если проводить исследования с микро-
пористыми типами кремнезема, имеющими поры разных раз-
меров, то о природе таких поверхностей на основании измерений
теплот смачивания сказать можно очень немногое. При прове-
дении подобных экспериментов необходимо рассматривать
только типы кремнезема, не содержащие микропор.
Основываясь на ранее полученных Патриком [114] результа-
тах по теплотам смачивания поверхностей различных кремне-
земных порошков с разной степенью гидратации поверхности,
Айлер [3] экстраполировал данные для изученных образцов
к нулевой и к 100%-ной степени покрытия поверхности гидро-
ксильными группами и получил следующие значения:
Теплота смачивания,
эрг/см2
Силанольная поверхность 190
Силоксановая поверхность 130
Разность теплот 60
Однако не было уверенности, что образцы не содержали некото-
рой доли микропор.
Тэйлор, Хокки и Петика [84] получили данные по теплотам
смачивания в воде поверхности образцов, которые предвари-
тельно прокаливали и затем регидратировали. Опыты проводи-
лись при различных степенях дегидроксилирования, достигае-
мых путем нагревания образца до таких температур, когда еще
Химия поверхности кремнезема 893
не наблюдалось ни спекания, ни каких-либо изменений удель-
ной поверхности. Для полностью гидроксилированного кремне-
зема теплота смачивания составляла 160±3 эрг/см2, и эта ве-
личина оказалась не зависящей от удельной поверхности образ-
цов кремнеземов в интервале 8—150 м2/г.
Теплота смачивания образцов кремнезема, дегидратирован-
ных до различных степеней путем нагревания образцов при
разных температурах, оказалась величиной, трудно поддаю-
щейся обобщению и оценке, если только образец кремнезема
предварительно не прокаливали при 700°С, чтобы исключить
микропоры и поверхностные группы ОН, и затем регидратиро-
вали. В противном случае, как показали Тэйлор, Хокки и Пе-
тика [84], теплота смачивания оказывалась неодинаковой при
разных температурах проведения измерений (27 и 45°С). Это
обусловлено, по крайней мере частично, различными располо-
жениями групп SiOH на поверхности, которые могут претерпе-
вать изменения при смачивании. С другой стороны, прокаленный
и затем регидратированный кремнезем имеет стабильную по-
верхность, у которой одни и те же значения теплот смачивания
при 27 и 45°С, и эти значения возрастают по мере увеличения
концентрации гидроксильных групп на поверхности:
Концентрация гидроксильных групп, число ОН-групп/нм2 Теплота смачивания (приблизительное значение), Эрг/см2
0 117 (экстраполи-
рованное
значение)
2 122
3 130
4,7 160
Наибольшего доверия заслуживает значение теплоты смачива-
ния 117 эрг/см2 для силоксановой поверхности, хотя оно и ока-
зывается несколько ниже значения, равного 130 эрг/см2, подсчи-
танного Айлером. Более высокие значения, полученные другими
исследователями, могут, вероятно, быть объяснены тем, что об-
разцы кремнезема содержали микропоры или же их поверхность
не была термически стабилизирована прокаливанием до высо-
ких температур. Тэйлор, Хокки и Петика показали, что теплота
смачивания поверхности предварительно непрокаленного крем-
незема при максимальной степени гидроксилирования 4,7 ОН-
групп/нм2 может приближаться к значению 200 эрг/см2.
Существуют различные, требующие исследования факторы,
которые оказывают влияние на теплоту смачивания непрокален-
ных, предварительно гидроксилированных типов кремнеземов.
Например, Тэйлор, Хокки и Петика нашли, что дегидратация
894
Глава 6
в вакууме приводит к более высокому значению теплоты сма-
чивания, чем это имеет место при термообработке на воздухе.
В последнем случае, вероятно, водяной пар, который удаляется
не сразу, способствует перегруппированию поверхностных групп
SiOH.
Тэйлор и другие [115] разработали дифференциальный ка-
лориметр для измерения теплот смачивания твердых веществ.
Для проведения экспериментов в воде и бензоле были исполь-
зованы типы пирогенного кремнезема (аэросилы) с надежно оп-
ределенными характеристиками.
Несмачивание, или гидрофобный характер, проявляемый си-
локсановой поверхностью во время ее контакта с жидкостью,
позволило Ласковски и Китченеру [116] прийти к заключению,
что работа адгезионных сил воды при ее контакте с твердой по-
верхностью складывается из трех составляющих: 1) дисперсион-
ных сил (сил Ван-дер-Ваальса); 2) гидратации неионных поляр-
ных центров, т. е. связывание молекул воды с группами SiOH;
3) диссоциации.
Второй фактор определяет способность поверхности кремне-
зема к смачиванию. Краевой угол и электрокинетический потен-
циал, образуемые между водой и прозрачным кварцевым сте-
клом, измерялись на поверхности, которая предварительно была
гидрофобизирована проведением реакции с (СНз)з$1С1. Через
некоторое время после начала контакта между водой и образ-
цом исходная гидрофобная поверхность приобретала способ-
ность к смачиванию (краевой угол становился равным нулю),
несмотря на то что метильные группы все еще сохранялись на
поверхности. После удаления физически адсорбированной воды
поверхность снова становилась гидрофобной. (Вполне вероятно,
что поверхность не покрывалась полностью метильными груп-
пами.) В том случае, когда поверхность покрывается близко
расположенными углеводородными группами, то она не прояв-
ляет такого обратимого поведения; это было доказано на при-
мере эстерсилов, поверхность которых покрыта плотно упако-
ванными бутильными группами, что позволяет сохранять гидро-
фобные свойства при нахождении образца в воде в течение
месяцев.
Теплота смачивания полярными жидкостями [117, 118] по-
нижается по мере дегидроксилирования поверхности кремне-
зема. Даже в случае метанола теплота смачивания на пол-
ностью дегидроксилированной поверхности составляет 75 эрг/см2
по сравнению со значением 50 эрг/см2 для воды. По-видимому,
теплота смачивания зависит от возникающих ассоциаций, в ко-
торых объединяются полярные группы органической молекулы
и полярные силанольные группы на поверхности, и ассоциаций,
Химия поверхности кремнезема 895
состоящих из углеводородных участков органической молекулы
и гидрофобной силоксановой поверхности. Полностью гидрокси-
лированная поверхность кремнезема при погружении в воду не
проявляет никаких гидрофобных характеристик.
Физическая адсорбция неионных соединений
с низкой молекулярной массой
Число публикаций по исследованию физической адсорбции
на кремнеземе за последние 50 лет составляет около 1000 или
даже больше. В этом разделе будут рассмотрены только типич-
ные примеры по физической адсорбции неионных соединений.
Адсорбция ионных веществ представлена ниже.
Методы и аппаратура для изучения адсорбционных явлений
(ИК-спектроскопия, ядерный магнитный резонанс, измерения
диэлектрических свойств) здесь не будут описаны, но с ними
можно ознакомиться по первоисточникам, на которые даются
ссылки.
Образование водородных связей между электроотрицатель-
ными атомами или л-электронами молекул адсорбируемого ве-
щества и атомами водорода силанольных групп на поверхности
кремнезема играет основную роль в процессе адсорбции моле-
кул из пара или неводного раствора. Образование водородных
связей оказывается также главным фактором при адсорбции не-
диссоциированных молекул из водного раствора. Роль водород-
ной связи была рассмотрена в обзорных работах Хайра [9],
Литтла [119], Киселева и Лыгина [120].
Адсорбция паров
Обратимая адсорбция означает, что в процесс вовлекаются
только физические силы межмолекулярного взаимодействия и
что десорбция имеет очень низкую энергию активации. Сюда
относятся адсорбаты, молекулы которых образуют водородные
связи с поверхностью. Однако возможны и такие случаи, когда
существует сразу несколько точек, в которых молекула присое-
диняется^через водородные связи к поверхности, как, например,
С2Н5ОС2Н4ОС2Н4ОС2Н5. Десорбция такого вещества может ока-
заться по существу необратимой, или по крайней мере изотерма
адсорбции имеет вид изотермы Ленгмюра. Некоторые' много-
кратно связанные водородными связями поверхностные комп-
лексы могут оказаться настолько стабильными, что их следует
рассматривать по существу хемосорбированными.
896
Глава 6
Исследование адсорбции газов и паров на силикагелях и
кремнеземных порошках проводилось главным образом для по-
лучения необходимых характеристик твердых веществ. Кроме
того, подобные данные представляются существенными для
оценки практических достоинств силикагелей, используемых
в качестве адсорбентов. Обширный обзор по физической адсорб-
ции газов и паров, который был дан в первой части гл. 5, можно
также использовать при рассмотрении настоящего раздела.
Здесь же будет представлено только несколько аспектов по дан-
ной теме, причем они ограничиваются в основном примерами ад-
сорбции на поверхностях кремнезема, не содержащих микропор.
Проведенные многочисленные исследования ясно показы-
вают, что адсорбция полярных молекул или ароматических со-
единений посредством л-связей происходит наиболее сильно на
поверхностных силанольных группах, не связанных водородными
связями с соседними группами ОН. Так, на кремнеземе, деги-
дратированном при 500°С, когда на поверхности остается около
2,7 ОН-групп/нм2, на каждой группе ОН при комнатной темпе-
ратуре адсорбируется одна молекула C2H5NH2 [121]. Согласно
данным Бастика [122], подобным образом на группах SiOH при
стехиометрическом соотношении адсорбируется аммиак.
В небольшой монографии Киселева [123] кратко изложены
вопросы, касающиеся явления адсорбции на различных поверх-
ностях, и особенно на кремнеземных поверхностях. Роль водо-
родной связи в адсорбции различных классов соединений была
раскрыта в исследованиях, проведенных методом ИК-спектро-
скопии. Например, этим методом изучалась адсорбция диэтило-
вого эфира [124], пиррола [125] и трет-бутилового спирта [126].
Прочность водородной связи определяется смещением полосы
ИК-поглощения, относящейся к валентным колебаниям изоли-
рованных групп ОН. Семпелс и Роуксет [127] сравнили таким
методом электронодонорные характеристики для ряда веществ,
образующих слабые водородные связи. Они показали, что водо-
родные связи оказываются одинаковыми как в растворе, так
и на поверхности адсорбента.
Эффект, возникающий в результате адсорбции молекул,
имеющих различные конфигурации, рассматривался на примере
молекулы С1С2Н4С1. Было показано, что отношение цис- и транс-
изомеров для такой молекулы возрастало от 1,0, когда вещество
находилось в растворе СНС13, до 1,9 при ее адсорбции на по-
верхности кремнезема. При адсорбции ацетилацетона наблюда-
лось, что в образование водородной связи включалась только
энольная форма. Данное наблюдение подтверждает, что наибо-
лее благоприятной молекулярной формой при адсорбции дан-
ного вещества на поверхности кремнезема оказывается та, ко-
Химия поверхности кремнезема
897
торая обеспечивает образование наибольшего числа водородных
связей:
Стерические ограничения могут помешать образованию во-
дородных связей и сильно ослабить адсорбцию на поверхности
кремнезема, как это имеет место в случае 2-хлорпиридина, ко-
гда большой атом хлора, очевидно, мешает молекуле близко по-
дойти к поверхности [129].
Активизированный алюмосиликатный гель А12О3—SiO2 не ад-
сорбирует пар пиридина в той же степени, как чистый кремне-
зем. Природу адсорбционных явлений на алюмосиликатах до-
статочно подробно рассматривал в своем обзоре Хайр [9]. Он
отмечал, что на активированном алюмосиликатном геле един-
ственными гидроксильными группами, на которых может про-
исходить адсорбция, оказываются группы SiOH. Но поскольку
на единице площади поверхности число таких групп ОН в слу-
чае алюмосиликата оказывается меньшим, чем для гидроксили-
рованного кремнезема, то, следовательно, в первом случае ад-
сорбция пиридина понижается.
Согласно данным Близнакова и Поликаровой [130], адсорб-
ция аммиака на поверхности кремнезема оказывается не столь
простым процессом. Повышенное количество аммиака, необра-
тимо связанного с поверхностью, сохраняется ниже 70°С. Бойль
и Гоу [131] показали, что адсорбция аммиака имеет частично
химическую и частично физическую природу. Пиридин также
дает сложную картину при адсорбции на кремнеземе [132], ко-
торая была несколько прояснена в результате применения ИК,-
спектроскопии при изучении адсорбции замещенных пириди-
нов [133].
Была хорошо исследована адсорбция паров аминов. Благо-
даря своей основности эти пары сильно адсорбируются на цент-
рах SiOH, причем на центрах, являющихся более слабыми осно-
ваниями, адсорбция происходит посредством образования водо-
родной связи (Si.sOH : NHs-nftn), а на центрах, представляю-
щих собой более сильные основания,— путем солеобразования
(SisO-+NH4_n/?n). Бартелл и Добай [134] обнаружили, что
диэтил- и моно- и дибутиламины образовывали монослои на по-
31 Заказ № 250
898
Глава 6
верхности кремнезема при р/р0, равной всего лишь 0,01, тогда
как последующее заполнение пор происходило при р/р0 0,5—0,8.
Каждая из указанных молекул амина занимала следующую пло-
щадь:
Амин
(С2Н.5)2 NH
(С4Н9)2 NH
C4H9NH2
Площадь, А2
43
57
33
Авторы работы [135] предположили, что при адсорбции бро-
мистого метила гидроксильные группы на поверхности пироген-
ного кремнезема распределены неравномерно и не распола-
гаются попарно, как это принято считать,
Чессик и Цеттльмойер [136] нашли, что при измерениях ин-
тегральных теплот образования монослоев воды и органических
паров на поверхности кремнезема величины АН возрастали ли-
нейно с увеличением —TAS. 1
В работе [137] описан самописец, который при использова-
нии электромагнитных весов автоматически регистрировал изо-
термы адсорбции. Прибор вычерчивает изотермы адсорбции на
диаграммной бумаге в интервалах давлений от 760 до 10~"7 мм
рт. ст. и температур от —196 до +500°С для любого адсорбата,
который не вызывает коррозию стали.
Влияние дегидроксилирования поверхности
на адсорбцию
Вода. Шапиро и Кольтхофф [138] обнаружили при иссле-
довании адсорбции красителя метилового красного из раствора
бензола, что силикагель частично терял адсорбционную способ-
ность по мере дегидратации даже при температурах предвари-
тельной обработки, когда еще не происходит понижение удель-
ной поверхности вследствие спекания образца. Следовательно,
краситель метиловый красный должен адсорбироваться на по-
верхностных группах SiOH, но не адсорбируется на дегидрати-
рованных силоксановых участках поверхности Si—О—Si. Они
также нашли, что некоторое количество воды адсорбируется па-
раллельно с адсорбцией красителя, когда предварительно нагре-
тый силикагель охлаждают и выдерживают при различных зна-
чениях влажности. В том случае, когда давление пара воды ста-
новилось ниже, чем давление пара жидкой фазы, вода
адсорбировалась не на дегидратированной поверхности кремне-
зема, а только на группах SiOH. Поскольку пары воды не ад-
Химия поверхности кремнезема
899
сорбировались на дегидратированных поверхностях, то процесс
регидратации шел медленно, если силикагель подвергался дейст-
вию только водяных паров.
Свежеобразованная, обезвоженная поверхность стекла также,
очевидно, регидратируется медленно. Например, Брунауэр [139]
отмечает, что различия в теплотах адсорбции воды на поверхно-
сти силикагеля и на свежеобразованной поверхности стекла мо-
жно объяснить тем, что на поверхности силикагеля имеется слой
хемосорбированной воды, который «притягиваем» воду, и по-
этому такая адсорбция происходит легко, тогда как свежеобра-
зованная поверхность стекла может быть «лишена водных заро-
дышей», которые могли бы порождать адсорбцию. В последнем
случае требовалось достижение более высокого давления пара
для того, чтобы образовать первый слой воды на поверхности.
Брунауэр также цитирует Троутона [140], который сообщил, что
стеклянная вата, высушиваемая в течение 70 ч при 160°С над
Р2О5, адсорбировала воду более трудно, но если вата высуши-
валась неполностью, то вода адсорбировалась легко. Все эти
данные означают, что дегидратированная поверхность фактиче-
ски гидрсфобна. Электроны, принадлежащие атомам кислорода
в напряженных силоксановых мостиках, не настолько свободны,
чтобы включиться в-образование водородной связи.
Как было показано Ждановым [141], дегидратация поверх-
ности кремнезема вызывает появление гистерезисной петли при
измерении изотерм адсорбция — десорбция паров воды, причем
такой гистерезис первый раз проявляется на дегидратированном
силикагеле. (Вероятно, Жданов обнаружил этот эффект, свя-
занный с дегидратацией кремнезема, еще до того, как выпол-
нили свою работу Дзисько, Вишневская и Чесалова [90].) Жда-
нов пришел к заключению, что Н2О адсорбируется главным
образом на группах ОН, но оксидные мостики, получаемые в ре-
зультате дегидратации, могут быть регидратированы до обра-
зования групп ОН за счет адсорбции воды. Доказательством
того, что Н2О адсорбируется на группах ОН, согласно Жданову,
является следующий факт. Когда силикагель нагревался при
500, а не при 300эС, то образец дополнительно терял 2,2 ммоль
групп ОН в расчете на 1 г SiO2. При сравнении изотерм адсорб-
ции, измеренных после предварительного нагревания силикагеля
при 300 и 500°С соответственно, и при. значении относительного
давления р/ро = 0,1, образец, который нагревался при 500°С,
адсорбировал на 1,95 ммоль НгО/г SiO2 меньше, чем образец,
который нагревали при 300°С. При р/р0 = 0,2 эта разница соста-
вила 2,7 ммоль НгО/г БЮг- Таким образом, при низком относи-
тельном давлении при потере каждой группы ОН с поверхности
воды адсорбировалось приблизительно на одну молекулу
меньше.
900
Глава 6
Следовательно, можно представить адсорбцию самой пер-
вой порции адсорбированной воды на частично дегидратирован-
ной поверхности кремнезема следующим образом:
нннннн нннн
ООО о о
Н Н Н О НН
ООО Z \ о о
—Si—О—Si—О—Si—О—Si—О—Si—о—Si—О—Si-
Изучение адсорбции воды только на изолированных парах
групп ОН, связанных внутри пары водородными связями [142],
привело к неожиданным результатам. На изотерме адсорбции
проявились определенно выраженные ступеньки при давлениях
примерно 9,5 и 16 мм рт. ст. Первая пара групп ОН, находя-
щаяся на поверхности, адсорбировала одну молекулу Н2О, тогда
как на второй ступеньке адсорбировалось две молекулы Н2О,
всего с учетом нечеткой третьей ступеньки, вероятно, ад-
сорбировалось шесть молекул Н2О. Все остальные группы ОН
на поверхности кремнезема, находящиеся вокруг каждой изоли-
рованной пары групп SiOH, исключались посредством проведе-
ния реакции с триметилхлорсиланом (СНз)зВ1С1, который не
вступал в реакцию с парными гидроксильными группами. Таким
образом, гидрофильные участки адсорбировали Н2О, формируя
кластер по следующей схеме:
О
О—Si—о—н
/
/
о
О—Si—о—н
О
н\ /н
Ъ—н - о
ZH \ н
X х
н н н
/О—Н
Н н
Пригожин и Фрипья [143] в последующем подробно рас-
смотрели механизм взаимодействия воды с поверхностью крем-
незема. Выяснилось, что молекулы Н2О, адсорбированные на
изолированных группах SiOH, вступают в реакцию с соседними
силоксановыми участками поверхности, образуя две новые
группы SiOH. Смежные, вицинальные группы SiOH также ад-
сорбируют воду, которая затем гидратирует силоксановую по-
верхность. Постулируется, что молекулы воды, связанные силь-
ной водородной связью, диссоциированы в гораздо большей сте-
Химия поверхности кремнезема
901
пени и, таким образом, оказываются в состоянии разрывать
силоксановую связь и раскрывать мостик Si—О—Si.
Специфический характер адсорбции Н2О на группах SiOH
при низком давлении паров воды предлагалось использовать как
способ для получения харатеристики состояния гидроксилиро-
вания поверхности [144а]. Дубинин [1446] придавал важное
значение группе SiOH при адсорбции любого вещества, у кото-
рого имеется составляющая, способная образовывать донорно-
акцепторное взаимодействие, т. е. водородную связь.
Как показано Бэкером и Сингом [145], поведение воды на
гидроксилированных и дегидроксилированных образцах кремне-
зема, обладающих микропорами, оказывается сложным. Деги-
дроксилированная непористая поверхность кремнезема лишь ча-
стично покрывается адсорбированной водой на той стадии, кото-
рая обычно соответствовала бы монослойному покрытию при
выполнении определения методом БЭТ. Но если имеются микро-
поры, то даже при предварительной дегидратации кремнезема
при такой же высокой температуре молекулы воды адсорбиро-
ваны более сильно и образуют приближающееся к монослою
покрытие, определяемое методом БЭТ. Существует вероятность
того, что в микропорах дегидратация протекает менее полно, и,
по-видимому, результаты Бэкера и Синга подтверждают эту
мысль. Незначительное число гидроксильных групп, оставшихся
в подобных небольших порах, могло бы играть роль центров
адсорбции.
Инертные газы. Уже подчеркивалось, что теплота адсорбции
азота на кремнеземе уменьшается по мере того, как поверхность
дегидратируется [40]. Аристов, Киселев и др. [146] нашли, что
'аргон не реагировал на степень гидроксилирования поверх-
ности и, таким образом, лучше пригоден для поверхностей, отли-
чающихся химическим составом. Это подтвердили авторы ра-
боты [147], которые использовали для молекулы аргона пло-
щадь, равную 16,6 А2.
Органические и другие молекулы. Для полярных молекул ад-
сорбата группы SiOH представляют собой центры, на которых
происходит адсорбция. Следовательно, для достижения макси-
мальной адсорбции поверхность кремнезема не должна содер-
жать адсорбированную воду, но должна иметь наибольшую кон-
центрацию групп SiOH. Для этих целей оптимальным оказы-
вается предельно гидроксилированный кремнезем, который
подвергался термообработке при ~175°С. Кроме того, согласно
Снайдеру [10], для полиненасыщенных углеводородов или мо-
лекул, обладающих несколькими полярными группами, наилуч-
шим является предельно гидроксилированное состояние поверх-
ности, позволяющее обеспечить большее число абсорбционных
902
Глава 6
центров, которые могут совмещаться с молекулами адсорбата.
По данным Хайра и Хертла [148] и Кларк-Монкса и Эллиса
[149], наиболее сильная адсорбция органических соединений
происходит на изолированных, обладающих невозмущенными
колебаниями гидроксильных группах, расположенных на терми-
чески дегидратированной поверхности кремнезема. Молекулы
таких адсорбируемых веществ, как ароматические углеводороды,
взаимодействуют с подобными гидроксильными группами при
соотношении 1:1.
Когда на поверхности находятся смежные гидроксильные
группы, попарно взаимно связанные водородными связями, на-
блюдается лишь незначительная их склонность к образованию
водородных связей с рассматриваемыми молекулами адсорбата.
Исключение составляют адсорбируемые молекулы, атомы кото-
рых имеют сильную электронно-донорную способность, как это
имеет место для молекул воды или метанола. В действительно-
сти вода вступает во взаимодействие преимущественно на таких
центрах. Как только молекула воды адсорбируется, у каждого
центра адсорбции освобождается атом водорода, активный по
отношению к адсорбции воды благодаря возможности образова-
ния водородной связи: Это может быть представлено схематично
так:
Атомы кислорода простых эфиров также могут связываться
при адсорбции с такими парными группами ОН, разрушая их
взаимные водородные связи [124].
Кертойз, Давыдов, Киселев и др. [87] получали изотермы
адсорбции, а также использовали спектральные и калориметри-
ческие методы для изучения адсорбции разнообразных типов ор-
ганических молекул на предельно гидроксилированной и до вы-
сокой степени дегидроксилированной поверхностях кремнезема.
Авторы пришли к заключению, что прочность водородных связей
для адсорбированных молекул рассматриваемых типов на крем-
неземной поверхности оказывается той же самой, что и в рас-
творе. Используемые образцы кремнезема приготовлялись с об-
Химия поверхности кремнезема 903
разованием непористых, однородных поверхностей, и теплоты
адсорбции и спектры ИК-поглощения измерялись при различных
степенях покрытия как для гидроксилированной, так и для де-
гидроксилированной поверхностей кремнезема. Полученные од-
нозначные, не вызывающие сомнений данные гораздо легче под-
даются интерпретации по сравнению с результатами предыду-
щих исследователей, которые в своих экспериментах ' не
использовали таких, совершенно различных типов поверхностей.
Прочность водородной связи приблизительно оценивалась
как разность теплот адсорбции на гидроксилированной поверх-
ности при 50%-ной степени покрытия и на дегидроксилирован-
ной поверхности, когда степень покрытия гидроксильными груп-
пами была наименьшей. В работе было получено хорошее со-
гласие между рассчитанными энергиями связи, найденными из
данных по теплотам адсорбции и из спектральных данных, для
которых были выведены уравнения. Было показано, что теплота
адсорбции представляет собой сумму теплоты конденсации п
теплоты образования водородной связи.
Представленные данные показывают, что исследованные мо-
лекулы адсорбатов приблизительно подразделяются на три
класса с позиции силы их адсорбционного взаимодействия с ги-
дроксилированной поверхностью.
Слабое взаимодействие: Ar, N2, ССК, н-С6Н14, циклогексан.
Взаимодействие промежуточной силы: CH3NO2, С6Н6,
СН3СОО2Н5, CH3CN, тетрагидрофуран, диоксан, ацетон, (С2Н5)О.
Сильное взаимодействие: пиридин, (C2H5)3N.
Когда на кремнеземе адсорбируется трет-бутиловый спирт,
то легко обнаруживается формирование водородных связей. Да-
выдов, Киселев и Лыгин [150] нашли, что спиртовая гидроксиль-
ная группа связывается водородной связью только со свобод-
ными группами SiOH на кремнеземной поверхности, но не с си-
ланольными группами, которые оказываются достаточно близко
расположенными друг к другу и связываются водородными свя-
зями. Третичный спирт, кроме того, не взаимодействует с обра-
зованием связанного с поверхностью сложного эфира в той сте-
пени, как это имеет место..для первичного и вторичного спиртов.
Джигит, Киселев и др. [151] обнаружили, что теплота адсорбции
составляла 18 ккал/моль, но она резко понижалась, если поверх-
ность кремнезема дегидратировалась вследствие уменьшения
числа образующихся водородных связей. По мере того как по-
верхность кремнезема становилась все более дегидратирован-
ной, адсорбция метанола не понижалась столь сильно, как это
наблюдалось при адсорбции воды [152]. Вероятно, метильная
группа спирта имела возможность при адсорбции связываться
с гидрофобным силоксановым участком поверхности, примыкаю-
щим к силанольной группе.
904
Глава 6
В случае адсорбции паров амина остается невыясненной та
доля в образуемой связи, которая определяется либо водород-
ной связью, либо ионным притяжением:
SiOH 4- NR3 = SiOH Ч- : NR3
SiOH + NR3 = SiCT + HNR^
Очевидно, такая связь включает оба вида взаимодействия, при-
чем доля каждого зависит от реакционной основности атома
азота и кислотности атома водорода силанольной группы. Од-
нако для случая четвертичного аммониевого иона, например для
иона (CH3)4N+, необходимо допускать существование только
ионного притяжения.
Бауэр и Штобер [153] при изучении адсорбции моно-, ди-
или триэтиламина на образцах кремнезема, имевшего как гидро-
ксилированную, так и дегидроксилированную поверхность, вна-
чале наблюдали образование монослоя, а затем измерили теп-
лоты адсорбции при двухслойном заполнении, когда адсорбция
оказывается обратимой.
Хертл и Хайр [154] измерили теплоты адсорбции 23 соедине-
ний при полосе ИК-поглощения 3750 смД относящейся к по-
верхностным гидроксильным группам. Изостерическая теплота
адсорбции обычно представляется усредненной величиной по
всей суммарной поверхности образца. Такая поверхность может
быть и неоднородной, как, например, в случае частично дегидра-
тированной поверхности кремнезема. Хайр [155] разработал
спектроскопический метод для определения теплот адсорбции,
когда адсорбция происходит на центрах специфического типа.
Полидиметилсилоксановое масло адсорбируется на поверх-
ности кремнезема независимо от того, является ли такая поверх-
ность гидроксилированной или дегидроксилированной. При мо-
лекулярной массе этого масла 350000 одна молекула занимает
площадь 6-104 А2 при толщине слоя 10 А. Согласно данным Ки-
селева, Новиковой и Эльтекова [156], молекула располагается
на поверхности настолько плоско, насколько это позволяет на-
пряженное состояние цепи лежащей молекулы.
Диоксид серы адсорбируется на силикагеле при начальной
теплоте адсорбции 25—30 ккал/моль для первой адсорбируемой
порции, равной 0,01 мкмоль/м2, с последующим падением теп-
лоты до 6—7 ккал/моль для случая дегидроксилированной по-
верхности и 12 ккал/моль для гидроксилированной поверхности
кремнезема. Изотермы определялись на разного типа силикаге-
лях Глассом и Россом [157]. Была также изучена адсорбция
CH3NH2.
Химия поверхности кремнезема
905
Адсорбция из растворов в неионных системах
Неводные растворы
Физическая адсорбция из раствора определяется здесь до-
вольно произвольно и не затрагивает адсорбцию ионов, которая
рассматривается в следующем разделе. По-видимому, в данном
случае не существует какого-либо общего правила, основанного
на структуре или физических свойствах органических молекул,
для предсказания их относительного сродства к силанольной по-
верхности определенных бинарных или более сложных смесей.
Поскольку величина рКа силанольной поверхности сходна с ве-
личиной рК.а для воды, а энергия межфазной границы раздела
оказывается очень небольшой, то, вероятно, взаимодействия ор-
ганических молекул с водой будут соответствовать межмолеку-
лярным взаимодействиям с силанольной поверхностью.
В растворах или в жидких смесях силы, которые приводят
к концентрированию каких-либо молекул данного вида на гра-
нице раздела жидкость—силанольная поверхность, вероятно, оп-
ределяются следующими факторами:
а) склонностью к разделению системы на фазы, что находит
отражение в избыточной концентрации молекул на границе раз-
дела жидкость—пар;
б) энергией взаимодействия гидроксильной группы SiOH как
донора или акцептора электронов с соответствующей молекулой.
Роберт [158] Обнаружил, что молекула этилового спирта при
адсорбции из гептана на силикагеле (удельная поверхность Sn~
= 400 м2/г) занимала площадь 18 А2. Автор пришел к заключе-
нию, что адсорбированные молекулы спирта располагались пер-
пендикулярно поверхности, причем спиртовые гидроксильные
группы были связаны водородной связью с группами ОН сила-
нольной поверхности кремнезема. Молекула бензола занимала
площадь 55 А2 и, следовательно, лежала плашмя. Молекулы ук-
сусной кислоты, по-видимому, образовывали двойной слой, так
как площадь, занимаемая одной молекулой, составляла только
9,1 А2. Джоунс и Аутридж [159] исследовали адсорбцию на си-
ликагеле смесей, состоящих из бутанола и бензола, как для слу-
чая жидкой, так и для паровой фазы. Метанол и бензол конку-
рировали между собой по отношению к кремнеземной поверхно-
сти, но бензол замещался бутанолом по мере того, как
концентрация бутанола в жидкой фазе возрастала до 100%.
Об аналогичных исследованиях систем, содержащих этанол, ци-
клогексан и бензол, сообщалось в работе [160].
Методом ИК-спектроскопии Гриффитс, Маршалл и Рочестер
[161] провели обширные исследования роли водородной связи
90S
Глава 6
в подобных системах. Они показали, что атомы кислорода таких
молекул, как диэтиловый эфир или ацетон в процессе адсорбции
на кремнеземе из СС14, способны образовывать водородную-
связь с группами SiOH. Они также отметили, что сила замещен-
ных пиридиновых оснований изменяется, и это вызывает различ-
ную степень расширения полосы поглощения валентного колеба-
ния групп ОН и различные смещения по длинам волн полос ИК-
поглощения
C6H5N:H-OSi
Для измерения ИК-спектров молекул на границе поверхно-
сти раздела жидкость—твердое тело была разработана аппара-
тура, в которой для образцов использовались , подогнанные кю-
веты, расположенные в герметично закрытой циркуляционной
системе, в которой может быть создан вакуум и которая может
быть присоединена к системам очистки и хранения жидкостей
[162].
Ганиченко, Киселев и Красильников [163] провели измере-
ния адсорбции спиртов из четыреххлористого углерода на по-
верхности образцов кремнезема, которые были дегидроксилиро-
ваны до разных степеней. Гидроксильные группы молекул
спирта ориентировались по. направлению к .силанольным груп-
пам SiOH на поверхности. С увеличением длины цепи молекул
взаимодействие спиртов с гидратированными до разных степеней
поверхностями кремнезема становилось все более одинаковым,
и степень покрытия поверхности спиртами приближалась к ве-
личине 3 мкмоль/м2 для «-гексилового спирта. Длинноцепочеч-
ные спирты адсорбировались с образованием монослойного по-
крытия даже на частично дегидроксилировацной поверхности.
Бонецкая и Красильников [164] исследовали для ряда спиртов
влияние размеров пор кремнезема на адсорбцию из четыреххло-
ристого углерода.
Как показано в работе [165] при изучении адсорбции холе-
стерина из бензола, молекулы холестерина лежат плоско на
гидроксилированной поверхности, причем каждая молекула за-
нимает площадь 94 А2.
Адсорбция карбоновых кислот из неполярных растворителей
лучше всего объясняется тем, что карбоксильные группы обра-
зуют водородные связи с поверхностью кремнезема в соответ-
ствии с данными Эдлера и Шпрингера [166]. Сорбция кислот
из неполярных растворителей понижается с увеличением числа
атомов, углерода в молекуле в следующем порядке: уксусная,
пропионовая, кротоновая, бензойная и пальмитиновая кислоты.
Кроме того, адсорбция понижается при замене растворителей
в таком порядке: тетрахлорид углерода, толуол, нитробензол,
диоксан и вода. Важную роль в таких системах играет степень
Химия поверхности кремнезема
907
связывания посредством водородных связей между растворен-
ным веществом, растворителем и кремнеземом.
Проводилось сравнение адсорбции нормальных жирных кис-
лот из бензола и гексана. Бензол взаимодействует с группами
SiOH посредством л-связей, тогда как гексан такими связями
не обладает. Согласно данным Армистеда, Тилера и Хокки
[167], кислоты Св-и при адсорбции из бензола образуют только
ограниченное по величине покрытие, составляющее 0,5 мол./нм2,
тогда как в случае гексана величина покрытия оказалась в ин-
тервале 1,85—1,4 мол./нм2 для жирных кислот Cg-ie. Причиной
низкой степени покрытия в бензоле'является то, что в нем мо-
лекулы кислот С6—16 подвержены димеризации за счет образо-
вания водородных связей, причем димеры находятся в равнове-
сии с мономерными молекулами кислот, концентрация которых
в этом растворителе низка, а адсорбируются только лишь моно-
мерные молекулы.
На кремнеземе, часть поверхности которого этерифицирована
в результате реакции со спиртом н-BuOH, в системе лауриновая
кислота—гептан молекулы- кислоты адсорбируются только на
неэтерофицированных участках поверхности [165]. Рахлевская
и др. [168] сравнивали адсорбцию кислот С]7 и Ci8 и олеиновой
кислоты из смесей декан—уксусная кислота.
Как показано в работе [169], кислота Ci8 адсорбировалась
на пирогенном кремнеземе в гораздо меньшей степени, чем спирт,
Ci8 или амин С18, хотя некоторое количество ненасыщенных це-
пей у кислоты, очевидно, было разорвано.
По данным Маршалла и Рочестера [170],, олеиновая и лино-
левая кислоты адсорбируются из тетрахлорида углерода на гид-
роксилированной поверхности силикагеля. Спектры ИК-погло-
щения показывают, что водородные связи образуются со смеж-
ными силанольными группами, вероятно, следующим образом:
Si—0-Н--О,ч
н CR
Si.,-о о
Н'
В том случае, когда кремнезем с адсорбированной на нем лино-
левой кислотой высушивали и затем нагревали, то на поверхно-
сти сохранялись хемосорбированные линолеатные группы, ко-
торые оставались устойчивыми до 280°С. Указывалось, что име-
лись некоторые доказательства взаимодействия силанольных
поверхностных групп кремнезема с двойными связями в цепи.
Адсорбции ароматических углеводородов из смесей таких
растворителей, как предельные углеводороды, уделялось значи-
тельное внимание, без сомнения, в связи с разработкой практи-
908
Глава 6
ческих методов разделения подобных систем. Данные адсорбции
показывают, что ароматические молекулы предпочтительно ад-
сорбируются на силанольных группах, и наилучшее разделение
может быть достигнуто на мелкопористых силикагелях, в кото-
рых имеются поры достаточного размера, чтобы ароматическая
молекула могла входить внутрь такой поры (например, диамет-
ром около 20 А для бензола или 45—70 А для триметил- или
алкилбензолов) [171—173]. Винклер и др. [174], используя метод
ЯМР для изучения протонной релаксации, исследовали взаимо-
действие бензола с группами SiOH на поверхности кремнезема
и среднее значение длины перемещений адсорбированных мо-
лекул. Наблюдалась тенденция углеводородных молекул к ад-
сорбции на поверхностных группах SiOH, которую можно
сравнивать с поверхностным натяжением между углеводородами
и водой, возникающим на границе раздела фаз [175].
Фенолы предпочтительно адсорбируются на силанольной по-
верхности из неполярных растворителей, что может быть исполь-
зовано для определения взаимодействия между растворителем
и.поверхностью кремнезема. Такая система изучалась ДэвиСом,
Дешаром и Иббитсоном [176]. Авторы отметили относительную
степень, до которой растворитель еще был способен адсорбиро-
ваться совместно с фенолом. Взаимодействие растворителей
с группами SiOH на поверхности возрастает в следующем по-
рядке: гексан, циклогексан, СС14, СНзСвНз, С6Нб.
Нитроалканы в ряду от метана до бутана и нитробензол
адсорбируются на кремнеземе из бензола, ацетона, а также из
водных смесей. Джоунс и Миллс [177] вывели необходимые
уравнения, чтобы скорректировать полученные результаты. Ад-
сорбция лекарственного сырья на кремнеземе также, возможно,
представляет определенную важность при составлении рецептур
для фармацевтических препаратов и для проведения анализа.
Руппрехт и Киндл [178] показали, что алкалоиды адсорбируются
посредством образования водородных связей между SiOH и ос-
новными атомами азота.
Неорганические соли, растворимые в органических раствори-
телях, адсорбируются на поверхности кремнезема, особенно зна-
чительно в тех случаях, когда в системе присутствует неболь-
шое, оптимальное количество воды [179]. Такое явление может
быть использовано для удаления примесных солевых загрязне-
ний из органических растворителей, получаемых в каких-либо
технологических процессах в потоке. Большинство неорганиче-
ских солей слаборастворимо в таких растворителях, как напри-
мер, ацетон, но соли L1NO3, Nal и L1C1 до некоторой степени
растворимы, когда в системе присутствует небольшое содержа-
ние воды. Поскольку силикагели способны адсорбировать как
анионы, так и катионы, то ионный обмен в подобных системах
Химия поверхности кремнезема 909
не имеет места. Возможно, что вода, когда ее содержание мало,
концентрируется на силанольной поверхности, и соль удержи-
вается в этом тонком слое воды. Если в систему добавляются
высшие спирты, которые, как известно, концентрируются на си-
ланольной поверхности за счет образования водородных связей,
то никакой адсорбции солей не происходит. Избыточное содер-
жание воды повышает растворимость соли в растворителе, и по-
этому никакой адсорбции солей -на кремнеземе не наблюдается.
Диэлектрические эффекты. Циклогексан и бензол адсорби-
руются на микропористом силикагеле с одновременным измере-
нием диэлектрической изотермы, п-электроны молекул бензола
в процессе его адсорбции способны взаимодействовать с сила-
нольными группами, расположенными на противоположных
стенках пор. Это приводит к изменению наклона изотермы по
сравнению с первоначальным ее ходом. В гексане такой эффект
не проявляется. Подобные диэлектрические эффекты можно ис-
пользовать для измерения объема микропор, когда эти поры со-
держат гидроксильные группы на внутренних стенках [180, 181].
Водные растворы, неионные системы,
водородная связь
Как обсуждалось в гл. 3 в связи с образованием раствори-
мых, а в некоторых случаях и нерастворимых комплексов, свя-
занных водородными связями с поликремневой кислотой, может
иметь место взаимодействие нейтральных полярных органиче-
ских молекул, в состав которых входят атомы кислорода или
азота, обладающие свободными электронными парами. Точно та-
кие же типы связей участвуют в адсорбции из воды на границе
раздела фаз вода—силанольная поверхность.
Как детально рассматривалось в гл. 4 (см. лит. к гл. 4 [162—
191]), при значении pH выше нейтрального на поверхности крем-
незема создается возрастающая концентрация отрицательных
зарядов и образование водородной связи около таких заряжен-
ных центров предотвращается. При этом нет никакого разли-
чия, является ли таким заряженным центром поверхностный ион
SiO- или же это будет алюмосиликатный анион AlSiO~. Про-
тивоион, обычно катион Na(H2O)+, остается вблизи такого за-
ряженного центра и мешает приблизиться к поверхности моле-
кулы адсорбируемого вещества (рис. 6.8).
С другой стороны, при низком значении pH, когда на поверх-
ности имеется лишь очень незначительный заряд, силанольная
поверхность может покрываться монослоем молекул воды, свя-
занных с поверхностью водородными связями. Если в системе
присутствуют водорастворимые полярные органические моле-
910
Глава 6
н2о
Рис. 6.8. Схема уменьшения числа водо-
родных связей между молекулой адсор-
бируемого вещества и поверхностью
кремнезема около заряженных участков
поверхности.
Большой по размеру гидратированный ион нат-
рия при своем перемещении над заряженным
участком поверхности препятствует присоеди-
нению молекул адсорбата (например, полиэти-
леноксида и изопропилового спирта), способ-
ных образовывать водородные связи с поверх-
ностью.
молекулярных размеров. Отсюда
кулы, то они конкурируют
с водой за выход к сила-
нольной поверхности.
Донахью и Бартелл [182]
изучили адсорбцию воды из
водно-спиртовых смесей
(«-бутил, гексил и гептил)
на силикагеле с удельной
поверхностью 455 м2/г и
диаметром глобул 8—9 нм.
Авторы пришли к заключе-
нию, что полярные гидро-
ксильные группы на конце
молекулы спирта притягива-
ются к поверхности кремне-
зема, но эти молекулы вы-
тесняются водой, если кон-
центрация воды в спирте
увеличивается. По-видимо-
му, вода адсорбируется в
капиллярах, при этом обра-
зуется обогащенная водой
фаза. При исследовании ад-
сорбции «-бутанола было
определено, что площадь,
занимаемая одной молеку-
лой, составляет 39,6 А2 при
25°С и 33,8 А2 при 45°С. Ав-
торы сравнивают эти вели-
чины с площадью 21,6 А2,
подсчитанной на основании
следует, что молекулы спирта
в данном случае не столь плотно упакованы на поверхности
кремнезема, как это имеет место в жидких мономолекулярных
слоях высших спиртов на поверхности жидкой воды.
Для амидов остается невыясненным вопрос, будет ли водо-
родная связь с поверхностью образовываться через атом азота
или же в ее образовании участвует атом кислорода, как это, на-
пример, имеет место для диметилацетамида
сн3 О
I 8
SiyOH : N — С—СН3
' I
сн.,
N(CH3).
Si5OH:O = C
CH,
Нехтшейн [183] утверждает, что по крайней мере в присутст-
вии только изолированных, свободных групп SiOH на частично
Химия поверхности кремнезема
911
дегидратированной поверхности кремнезема с силанольной груп-
пой SiOH взаимодействует именно группа СО.
Степень вытеснения одной органической молекулы другой
с поверхности кремнезема может быть измерена. Эксперимен-
тально наблюдаемые факторы, которые упоминаются здесь, опи-
саны в гл. 3, а относительные «эффективности образования во-
дородных связей» различных классов соединений были, предста-
влены в табл. 3.11. По-видимому, основными являются три
фактора:
1. Прочность водородной связи. Как упоминалось в преды-
дущем разделе, измерения, выполненные Кертойзом и др. [87],
показывают, что более прочные водородные связи образуются
с более слабыми основаниями, такими, например, как пиридин,
через атом азота и более слабые — через атомы кислорода про-
стого эфира или кетогрупп, имеющих определенное сходство.
2. Соотношение числа углеводородных групп к числу поляр-
ных атомов в молекуле. Это.отношение непосредственно связано
с тенденцией углеводородных групп или цепочечных сегментов
к освобождению от воды и концентрированию на поверхности
раздела. Это еще один аспект поверхностной активности.
3. Число электронодонорных атомов, приходящихся на одну
молекулу адсорбата, т. е. число точек присоединения молекулы
к поверхности (см. гл. 3).
Известно значительное количество публикаций, касающихся
адсорбции азотных оснований из водных растворов, но, как пра-
вило, эти данные рассматриваются с точки зрения ионизирован-
ных аммониевых форм. Из-за наличия сильной конкуренции со
стороны воды лишь немногие полярные соединения с низкой
молекулярной массой способны сильно адсорбироваться на по-
верхности кремнезема (полимеры, образующие большое число
связей с поверхностью, будут рассмотрены в следующем раз-
деле).
Адсорбция из растворов сильных солей. Как было описано
в гл. 3, вещества с низкой молекулярной массой, образующие
водородные связи и обладающие лишь одним или двумя элек-
тронодонорными атомами в расчете на одну молекулу, взаимо-
действуют с группами SiOH только в том случае, когда добав-
ляется хорошо растворимая соль (NaCl пли Na2SO4). Вероятно,
она понижает концентрацию воды путем образования гидратных
ионов; так, например, в 30 %-ном растворе NaCl две трети всей
воды присутствует в системе в виде ионов Na(H2O)+.
По-видимому, необходимо провести некоторые дополнитель-
ные исследования адсорбции на кремнеземе водорастворимых
органических соединений с низкой молекулярной массой из от-
носительно концентрированных солевых растворов при pH 2—4,
912
Глава 6
когда может происходить образование водородных связей. Ис-
следования, выполненные до настоящего времени, затрагивали
только процессы осаждения или фазового разделения поликрем-
невой кислоты или коллоидного кремнезема для определения
полноты покрытия поверхности адсорбатом. Имеется некоторая
неясность относительно того, что разные неорганические соли
оказывают различное воздействие, возможно зависящее от сте-
пени их гидратации. Ничего не известно также относительно
действия, оказываемого различными анионами.
Вероятно, исследованиям в этой области уделялось недоста-
точное внимание из-за того, что не было очевидных практических
приложений, хотя, как это было впервые открыто Кирком (см.
лит. в гл. 3 [162]), комплексы кремневой кислоты с водородными
связями пригодны для дубления кож.
Ионизация и поверхностный заряд
Характеристики поверхности аморфного кремнезема большей
частью получены при изучении коллоидных порошков и силика-
гелей. Константы диссоциации моно- и поликремневых кислот
были рассмотрены в гл. 3. Природа поверхностного заряда на
кремнеземе обсуждалась достаточно подробно в гл. 4. Рассмот-
рение теории поверхностного заряда и двойного электрического
слоя дано в обзорной работе Визе и др. [15].
Гидроксилированная поверхность
Как обсуждалось в гл. 4, гидроксилированная поверхность
кремнезема имеет точку нулевого заряда (т. н. з.) при рН~2.
При более высоких значениях pH, вплоть до рН~6, концентра-
ция отрицательных зарядов- возрастает, но остается еще очень
низкой и повышается только тогда, когда pH возрастает до
~ 10,7. Между тем при pH 8—9 кремнезем растворяется и обра-
зует силикатные ионы HSiO~, так что в системе концентрация
этого электролита будет увеличиваться по мере возрастания pH,
особенно выше pH 9.
Концентрация зарядов на гидроксилированной поверхности
кремнезема при различных значениях pH и концентрациях элек-
тролита NaCl (данные Болта) была представлена на рис. 4.10.
Основным в адсорбции или обмене катионов является степень
диссоциации групп SiOH на поверхности. Величина рКа изме-
няется от 6,5 в отсутствие нейтрализации до ~9,2 при 50%
нейтрализации, как это было показано на рис. 3.3. Поскольку
рКа групп SiOH, которые первоначально ионизируются на по-
верхности аморфного кремнезема, составляет 6,5, такая поверх-
ность будет гораздо более кислой, чем монокремневая кислота,
у которой рЛ'а = 9,8. Как и для всех поликислот, кислотность,
Химия поверхности кремнезема
913
зависящая от остающихся на поверхности групп SiOH, будет
понижаться по мере того, как возрастает их степень диссоциа-
ции. Полная диссоциация достигается лишь тогда, когда значе-
ние pH становится немного ниже той точки, при которой насту-
пает растворение кремнезема, и то только в присутствии рас-
твора сильной соли (см. гл. 5).
Посредством кулонометрического титрования поверхности
силикагеля Шиндлер и Камбер [184а] измерили собственную
кислотность (Ktnt) поверхностных силанольных групп при по-
стоянной ионной силе в 0,1 М растворе NaC104 при 25°С:
IgKmz = -6,8 ± 0,2.
Паркс [1846, 185] провел широкие исследования изоэлектри-
ческой. точки (и. э. т.) и т. н. з. поверхностей твердых оксидов
и вывел уравнение для расчета заряда на поверхности. Он по-
лучил в изоэлектрической точке на поверхности дегидратиро-
ванного образца SiO2 pH 2,4, а для гидратированной поверхно-
сти значение pH, равное 1,8.
Помимо упоминавшихся работ (к гл. 3 [30—58] и к гл. 4
[184—199]), необходимо указать на следующие исследования.
Ликлема [186] дал разъяснение относительно выражений
«точка нулевого заряда» и «изоэлектрическая точка». Он от-
метил, что эти точки определялись различными способами.
Точка и. э. т. измеряется с помощью методов электрофореза,
и получаемое значение pH соответствует моменту, когда заряд
вблизи плоскости скольжения отсутствует. Точка т. н. з. опре-
деляется по кривым титрования, и, следовательно, фиксируется
значение pH, когда ни кислота, ни основание не расходуются
на образование заряда на поверхности.
В действительности добавлением соли к золю кремнезема
можно увеличить адсорбцию противоионов внутри слоя Штерна,
лежащего близко к поверхности, что будет способствовать даль-
нейшей адсорбции ионов ОН". Это приводит к увеличению сум-
марного числа заряженных центров, однако при электрофорезе
заряд снаружи плоскости скольжения будет понижаться. Таким
образом, точки т. н. з. и и. э. т. могут перемещаться в противо-
положных направлениях. Однако в разбавленных растворах обе
точки оказываются очень близкими.
Соотношение между т. н. з. для неорганических твердых ок-
сидов и их теплотой смачивания в воде оказывается линейным.
Эти наблюдения были выполнены Хили и Фурстенау [187], ко-
торые объяснили такое поведение систем напряженностью элек-
тростатического поля, определяющей адсорбцию и диссоциацию
воды на поверхности твердых оксидов. Точка нулевого заряда
была измерена в капилляре из прозрачного кварцевого стекла
и оказалась равной 2,5 ± 0,2 [188]. Это значение не обязательно
должно соответствовать т. н. з. для коллоидных кремнеземов
32 Заказ № 250
914
Глава 6
и гелей, и последние, по-видимому, чаще имеют значения,
т. н. з. ~2.
Хайр и Хертл [189] и позднее Маршалл и др. [190] провели
исследования по определению кислотности поверхности кремне-
зема. Методом ИК-спектроскопии они получили значение рКа,
равное 7,2, на основании измерений смещений полосы валент-
ных колебаний групп ОН, вызываемых присутствием органиче-
ских соединений (ацетон, фенолы) с различной степенью кислот-
ности.
Шиндлер и др. [191] определяли константу диссоциации по-
верхностных групп SiOH методом титрования основанием в 1 н.
растворе NaCIO4 в присутствии или в отсутствие солей,, двух-
или трехвалентных металлов. Константы химического равнове-
сия образовавшихся поверхностных комплексов также были
измерены. Суммарное число способных ионизироваться групп
SiOH на поверхности силикагеля или пирогенного кремнезема
оказалось в области 5,5—5,8 ОН-групп/нм2. Для титрования по-
верхности пирогенного кремнезема (аэросила-200) было выве-
дено эмпирическое уравнение
lg [Si5O_] = 5,2 lg (pH) — 5,35
где [SiO] — концентрация групп SiO-, моль/кг SiO2, при зна-
чении удельной поверхности 1,60-109 см2/кг. Суммарное число
центров SiOH Со составило 1,45 моль/кг:
Со = [SCOH] + [SCO-]
Выразив число зарядов ОН- в расчете на 1 нм2, получаем
1g (ОН/нм2) = 5,2 lg (pH) - 4,78
Ахмед [192] измерил плотность зарядов на поверхности
кварца в зависимости от pH. Такая поверхность была получена
обработкой НС1, а не щелочью NaOH с целью удаления воз-
можно образовавшейся пленки аморфного кремнезема. Плотно-
сти .зарядов оказались выше, чем это было сообщено Болтом
для аморфных образцов SiO2 (см. рис. 4.10). Значения, полу-
ченные в 1 М растворе KNO3 и интерполированные из графиче-
ских зависимостей, представлены ниже (1—данные Ахмеда,
2 — данные Болта, 3 — данные Шиндлера *):
Число зарядов ОН“/нм2
pH 1 2 3
10 2,52 1,81 2,6
9 1,45 1,06 1,52
8 0,76 0,61 0,82
Из приведенных данных видно, что результаты хорошо согла-
суются. Следует учесть также, что использованные указанными
авторами образцы кремнезема имели разные поверхности:
* Указанные значении, полученные в 1,0 М растворе, рассчитаны по
приведенному выше уравнению.
Химия поверхности кремнезема
915
180 м2/г (Болт), 0,043 м2/г (Ахмед), что на несколько порядков
меньше, и 160 м2/г (Шиндлер). Значения, представленные Шинд-
лером, оказались несколько иными, чем данные Болта, вероятно,
вследствие того, что аэросил был в незначительной степени мик-
ропористым.
Дегидроксилированная поверхность
Поверхность пирогенного кремнезема- или же поверхность,
которая подвергалась дегидратации при очень высокой темпе-
ратуре, не будет быстро регидратироваться; фактически для ре-
гидратации могут потребоваться недели, если образцы кремне-
зема находятся в виде нейтральной суспензии. Теперь ясно, что
кремнезем ведет себя в суспензиях совершенно иначе, чем крем-
незем с полностью гидратированной поверхностью. Следует
указать, что константы диссоциации изолированных. одиночных
и спаренных вицинальных силанольных групп на частично де-
гидратированной поверхности кремнезема не измерялись. В нейт-
ральном и кислом растворе скорость регидратации мала и по-
добные измерения оказались бы возможными. Вследствие того
что такие центры на дегидроксилированной поверхности разде-
лены, можно было ожидать, что поверхность окажется несколько
более ионизированной, т. е. группы SiOH окажутся более кис-
лыми, когда находятся поблизости от атомов кислорода сило-
ксановых мостиков. Доказательство этого факта было предста-
влено Кириченко и Высоцким [193], которые проводили иссле-
дования при низких значениях pH с тем, чтобы частично
дегидроксилированные поверхности не подвергались бы регид-
роксилированию в ходе экспериментов. Они измеряли ионизацию
поверхности кремнезема при pH от 1,5 до 6—7, измеряя одно-
временно адсорбцию ионов Rb+. Кремнезем, который не нагре-
вался выше 300°С, адсорбировал много меньше ионов Rb+, чем
тот же самый силикагель, прокаленный при температурах
вплоть до 1000°С. Данные этих авторов были представлены на
рис. 3.6.
Те же самые авторы предположили, что на частично дегидра-
тированной поверхности с имеющимися напряженными связями
Si—О—Si ион ОН~ более легко захватывается атомом Si, кото-
рый увеличивает при этом свое координационное число до 5.
В таком случае образуется анионный центр, который предста-
вляет собой сильную кислоту:
° _
Si—ОН + ОН Н ' Н;° > + нз°+
\ v AJrl
/°
32*
916
Глава 6
Абендрот [194] измерил плотность зарядов на пирогенном
кремнеземе в 0,1 н. растворах хлоридовСэ, К и Li и в 1,0; 0,1
и 0,01 н. растворах КС1 при pH 2—10. Значения оказались выше,,
чем для полностью гидроксилированной поверхности кремне-
зема, но они понижались при регидратации Поверхности. К со-
жалению, степень поверхностной гидратации не была известна.
Плотности поверхностных зарядов в случае солей Cs, К и Li раз-
личались между собой менее чем на 20%.
Дегидратированная поверхность гидрофобна. Это подтвер-
ждается тем фактом, что частицы кремнезема флокулируют
в воде при низких значениях pH, когда оставшиеся на поверх-
ности силанольные группы неионизированы. Было найдено [195],
что флокуляция пирогенного кремнезема в воде понижается по
мере того, как дисперсная система подвергается старению, но
после повторного нагревания при 300°С и редиспергирования
кремнезем снова флокулировал. Такое явление оказалось ти-
пичным для частиц кремнезема, на которых имеются участки, не
смачиваемые водой, и которые, следовательно, стягиваются под
воздействием поверхностного натяжения, как только такие уча-
стки на различных частицах приходят в соприкосновение, т. е.
образуется «гидрофобная связь». Марото и Грайот [196] иссле-
довали электрофоретическую подвижность частиц кремнезема,
которые предварительно были дегидратированы до различной
степени.
В дальнейшем было исследовано поведение дегидратирован-
ного кремнезема при флокуляции [197а]. Величина коагуляции
С определялась по концентрации NaCl, необходимой для того,
чтобы кремнезем флокулировал и чтобы через 24 ч оставалась
прозрачная суспензия. При сравнении свежеполученного диспер-
гированного в воде пирогенного кремнезема с коллоидным
кремнеземом, который был полностью гидроксилирован, наблю-
далось лишь незначительное различие. Это объяснялось тем,
что либо еще оставалось на поверхности образца аэросила-800
достаточное число остаточных групп SiOH, либо же его поверх-
ность быстро регидратировалась, и поэтому поведение пироген-
ного кремнезема при флокуляции было близко к поведению
полностью гидроксилированного коллоидного кремнезема. Од-
нако, после того как пирогенный кремнезем полностью дегидра-
тировался при 1100°С, он вел себя подобно дробленому кварцу.
Ниже pH 9—10 дегидратированные частицы ведут себя прямо
противоположно гидроксилированным частицам кремнезема.
Чем ниже значение pH, тем более чувствительной становится
суспензия по отношению к процессу флокуляции под воздейст-
вием электролита (рис. 6.9).
Возможным объяснением является то, что дегидроксилиро-
ванные частицы, если они полностью гидрофобны, будут флоку-
Химия поверхности кремнезема
917
лировать при отсутствии на них заряда. Однако при рН>2 наб-
людается увеличение поверхностного заряда на очень неболь-
шом числе оставшихся групп SiOH. Флокуляция будет иметь
место только в том случае, когда электрокинетический потен-
циал достаточно понижен за счет увеличения концентрации
соли. Такое поведение характерно для гидрофобных коллоидных
Рис. 6.9. Зависимость критической концентрации электролита NaCl(С), необ-
ходимой для флокуляции образцов кремнезема с гидроксилированной и де-
гидроксилированной поверхностью, от величины pH.
1 — гидроксилированная поверхность; 2 — дегидроксилированная поверхность. (По дан-
ным Чапека и Торрес-Санчеса [197а].)
частиц. С другой стороны, для образцов кремнезема с пол-
ностью гидроксилированной поверхностью флокуляция проис-
ходит только при высоких значениях pH и высокой концентра-
ции соли, и механизм совершенно другой. В таком случае ион
натрия ведет себя как связующее звено между заряженными
центрами, расположенными на двух соседних частицах. Чем
ниже становится величина поверхностных зарядов по мере по-
нижения значения pH, тем большая концентрация ионов натрия
требуется, чтобы вызвать процесс флокуляции.
При pH>10 дегидроксилированный кремнезем будет фло-
кулировать точно таким же образом, как и гидроксилированный.
Остается невыясненным, происходит ли это из-за того, что обе
разновидности достигли одинаковой концентрации заряженных
центров на поверхности, или же вследствие того, что суспензия
дегидратированного кремнезема достаточно долго подвергалась
старению при высоких значениях pH, в результате чего поверх-
ность регидроксилировалась. Регидроксилирование происходит
относительно быстро при pH >10.
918
Глава 6
Природа анионных заряженных центров
В гл. 4 при рассмотрении работ [184—206] обсуждались бо-
лее подробно практические стороны вопросов, связанных с на-
личием зарядов на кремнеземных частицах в водных суспен-
зиях. Данные, показывающие взаимозависимость между зарядом
на поверхности и'величиной pH, полученные Болтом и пред-
ставленные на рис. 4.10, вероятно, наиболее надежны.
Природа поверхностного заряда является основой для пони-
мания некоторых свойств системы, например ионного обмена.
Сначала напомним, что существуют так называемые клас-
сические модели адсорбции ионов на твердых •поверхностях (ве-
роятно, модель становится классической, когда ее используют
для расчетов достаточно долгое время). Такие модели хорошо
описывают реальные системы разбавленных растворов элек-
тролитов при соотношении компонентов 1 : 1, таких, например,
как соли щелочных металлов, когда определяющими являются
ионные силы. В случае же, когда картину определяют силы дру-
гого вида, вызывающие образование координационных или ко-
валентных связей между группами SiOH и ионом металла или
гидрофобных связей между адсорбированными органическими
катионами, то подобные теоретические подходы оказываются не-
применимыми.
Основу всех ионных теорий представляет уравнение Нернста
для расчета работы, совершаемой ионом при его перемещении
в растворе из бесконечности до точки на твердой поверхности.
Затем появилась теория диффузного двойного слоя Гуи—Чэп-
мэна, основанная на уравнениях .Пуассона—Больцмана. Со-
гласно этой теории, движение катионов вблизи поверхности
поддерживается тепловой энергией, причем катионы притяги-
ваются к поверхности соответствующими отрицательными заря-
дами. Этот же закон применим и для описания того, как моле-
кулы окружающей землю атмосферы удерживаются вблизи по-
верхности под действием сил земного притяжения. Затем было
понято, что катионы больших размеров не могли приближаться
к отрицательным зарядам на поверхности так же, как катионы
меньших размеров. Штерн ввел поправку^учитывающую размер
иона, и предложил рассматривать некоторый слой, который за-
тем стал называться «слоем Штерна». В этом слое вблизи отри-
цательно заряженной поверхности накапливается определенное
количество, катионов, которые в основном оказываются затор-
моженными. Таким образом, формируется «плотный двойной
электрический слой».
Затем было выяснено, что катионы могут, перемещаться еще
ближе к поверхности и образовывать.координационную или ко-
Химия поверхности кремнезема 919
валентную связь с группами SiOH. Несмотря на то что природа
этого дополнительного связывания с поверхностью в некоторых
случаях представлялась спорной, при математической обработке
подобной модели возникла необходимость во введении дополни-
тельного «специфического адсорбционного потенциала». Потен-
циал называется «специфическим», так как он различен для
каждого иона металла и для каждого вида поверхности.
- Проблема заключается в том, что, по-видимому, остаются
невыясненными те критерии, в силу которых катионы металлов
могут перемещаться из слоя Штерна в «специфический адсорб-
ционный» слой, который можно было бы называть «координаци-
онно связанным» слоем. Вначале привилегия такого перехода
признавалась за многозарядными ионами металлов, такими, как
ион кобальта, способными, как известно, образовывать коорди-
национные комплексы. Позже были включены в рассмотрение
двухзарядные ионы, например ионы кальция. Вероятно, затем
будут признаны также ионы щелочных металлов, чтобы пере-
нести основные положения специфической адсорбции на .кон-
центрированные растворы. Штамм и др. [1976, 197в] сыграли
основную роль в признании важности такой координации или
формирования комплексов между ионами металлов и поверх-
ностью окисных твердых .тел.
. В гл. 5 обсуждалась возможная роль координационной связи
в качестве мостикового механизма в процессе коагуляции частиц
кремнезема при воздействии катионов, в том числе при дейст-
вии ионов натрия.
Поведение двойного электрического слоя вокруг частиц крем-
незема, например в разбавленных растворах KNO3, может быть
объяснено теорией Гуи—Чэпмана—Штерна, за исключением
лишь того факта, что высокая плотность поверхностных зарядов
не согласуется с низким значением потенциала двойного слоя.
С целью объяснения этих данных, согласно Тедросу и Ликлема
[198], необходимо ввести в рассмотрение как отрицательно за-
ряженные группы в зоне поверхности, так и противоионы. На ос-
новании этого положения авторы выдвинули гипотезу о том, что
поверхность должна состоять из пленки геля кремнезема, содер-
жащей микропоры, т. е. величина поверхности должна быть
больше, чем измеряемая методом БЭТ. Перрам [199] также под-
держал эту идею. Однако, как будет показано ниже, полученные
впоследствии сведения показали, что во многих случаях нет не-
обходимости предполагать существование пленки геля.
Теория «связывания центров»
В ряде теорий принимается, что ион натрия не может приб-
лизиться к поверхности и нейтрализовать ее отрицательный за-
920 Глава 6
ряд за счет образования связи. Противоположное положение
было высказано авторами работ [200], а именно что адсорбиро-
ванный противоион образует ионную пару, расположенную не-
посредственно вблизи поверхности или прямо на поверхности.
В этом случае для объяснения имеющихся экспериментальных
данных.нет необходимости допускать существование какого-
либо слоя геля. Такая концепция в настоящее время поддер-
жана Смитом и др. [201], которые продемонстрировали в ряде
экспериментов, выполненных на палочках из кварцевого стекла,
что поверхностный заряд по величине оказался тем же самым,
как сообщалось Болтом [202]. Смит и др. [201] использовали
в качестве радиоактивных индикаторов Na24 и Вг82, что позво-
лило им измерить адсорбцию ионов на уплотненном участке
двойного электрического слоя при исследовании непористого
кремнезема. Эти результаты явились прямым доказательством,
опровергшим идею существования пленки геля на поверхности
и подтвердившим модель связывания центров.
Йетс, Левине и Хили [200] представили математическую
теорию и уравнения, выражающие величину поверхностного
заряда в зависимости от pH при различных концентрациях
электролита. Если выбираются определенные значения электри-
ческих емкостей для межфазной границы, то такая модель
воспроизводит характерные свойства системы кремнезем—вода,
а именно высокую плотность зарядов на поверхности и умерен-
ное значение электрокинетического потенциала. Выбор подоб-
ных электрических емкостей в качестве модели предполагает,
что диполи ионных пар расположены почти параллельно по-
верхности и что катионы находятся очень близко или входят
частично в пределы плоскости поверхности.
Отвергая гипотезу пленки геля на поверхности, Йетс и Хили
[203а] доказали, что свежеосажденный кремнезем марки BDH
имеет поверхность, которая оказывается проницаемой по отно-
шению к протонам и к противоионам, однако когда образец
кремнезема нагревается до температур, не превышающих 500°С,
то поверхность становится проницаемой только для протонов,
но не для противоионов. Наконец, когда кремнезем нагревается
до 800°С, поверхность делается непроницаемой и для протонов,
и для противоионов.
Одно из положений теории «связывания центров» заклю-
чается в том, что поверхностный потенциал такой оксидной по-
верхности относительно раствора не может задаваться уравне-
нием Нернста, в котором учитывается отрицательный «разма-
занный» по всей поверхности заряд. Б теории «связывания
центров» считается, что ионные центры представляют собой дву-
мерное множество, состоящее из дискретных положительно заря-
женных, нейтральных и отрицательно заряженных центров на
Химия поверхности кремнезема
921
поверхности раздела кремнезем—вода. Бизе и др. [15] предста-
вили расчеты, в которых принимается, что противоионы могут
располагаться только напротив специфических, отрицательно за-
ряженных центров на поверхности.
Представляется, что кремнезем уникален среди нераствори-
мых оксидных твердых веществ благодаря тому, что значение
рА на поверхности в точке нулевого заряда (рАт. н. з. = 2) от-
стоит исключительно далеко от значения рАа 6—7, соответст-
вующего начальной ионизации поверхностных групп SiOH. Та-
кая разность, равная пяти единицам, гораздо больше, чем для
других оксидных твердых веществ (три единицы pH или даже
меньше). Согласно Бизе и др. [15], представленная -модель
«связывания центров» объясняет поведение поверхностного по-
тенциала для всех оксидных твердых веществ, в том числе и для
SiOa. Характерная особенность кремнезема, очевидно, заклю-
чается в следующем: противоионы, т. е. ионы Na+, распола-
гаются почти в той же самой плоскости поверхности, где и опре-
деляющие поверхностный потенциал ионы SiO-, что совершенно
отличается от других оксидов. Вероятно, это обусловливается
относительно открытой сеткой ионов кислорода, расположенных
на поверхности, с достаточно большими расстояниями между
такими ионами, что позволяет, по крайней мере частично, акко-
модировать адсорбированные катионы.
Дэвис, Джеймс и Лекки [2036] поддержали теорию «связы-
вания центров», сделав дальнейЩий шаг в ее развитии. Они
предположили, что поверхностные комплексы образуются до-
полнительно к имеющейся простой ионизации групп ОН на по-
верхности даже в случае ионов натрия. Использовалась про-
грамма, составленная для ЭВМ, с целью нахождения равновес-
ного состояния в растворе, причем принималось во внимание
большое число переменных величин. Полученные результаты
показывают, что либо катион с заряженным центром на поверх-
ности образует «ионную пару», либо «поверхностный комплекс»
оказывается несуществующим. Важно то, что даже простые од-
нозарядные катионы металлов, по-видимому, связываются с по-
верхностью благодаря координации с атомом кислорода группы
SiOH. Такие поверхностные группы более кислые, чем группы
ОН монокремневой кислоты, которая имеет, вероятно, гораздо
меньшую тенденцию к образованию комплексов с ионами ме-
таллов (см. гл. 2).
Силы, обусловливающие адсорбцию ионов
Хотя поверхность кремнезема имеет слабую кислотность,
многие ионы довольно прочно и даже необратимо удерживаются
силами, которые возникают дополнительно к ионному притяже-
•922
Глава 6
нию. Несмотря на более чем полувековой период научных изы-
сканий, подобная аномальная ситуация понята еще не до конца.
Справедливо то, что в случае однозарядных катионов Na+,
К+, NH+, по-видимому, в процесс включается простой ионный
обмен. Однако и при этом наблюдаются различающиеся меха-
низмы в зависимости от значений pH. Например, ионы натрия
и калия воздействуют аналогичным образом в процессе фло-
куляции кремнезема в области pH 7—10, но при более высоких
значениях pH ионы калия не будут вызывать флокуляцию,
тогда как ионы натрия продолжают оказывать такое действие.
Обсуждение вопроса о природе ионных заряженных центров
показало, что существуют специфически связанные с поверх-
ностью ионы, которые удерживаются другими силами, отличаю-
щимися от сил ионного притяжения. Такие силы, вероятно, под-
разделяются на два класса. В первый включаются ковалентные
связи между катионом металла ЛМ+ и силанольной группой:
SisOH + (Н2О)хМу+ = Si50M(y-I) + (Н2О)Л-_ i + Н2О +н +
во второй класс — гидрофобные связи, например между длинно-
цепочечными гидрофобными катионами, такими, как
(Ci6H3i)NJ (CH3)3- В данном случае- играет роль поверхностное,
натяжение воды, которая не смачивает углеводородные группы.
Поверхностное натяжение воды удерживает вместе такие смеж-
ные адсорбированные группы, направляя их от поверхности. Од-
нако азотсодержащие катионы и сопутствующие им анионы ад-
сорбируются на поверхностных группах SiOH.
Только в присутствии «нейтрального электролита», напри-
мер NaCl, разбавленного в отношении 1:1, когда значения
рНта.з. и рНи.э.т очень близки, рассматриваемая система го-
раздо легче поддается исследованию с использованием матема-
тической модели.
Однозарядные катионы: ионы металлов
и низших аминов
Адсорбция ионов Na+, К+, Rb+ и др. неоднократно изуча-
лась, и здесь будет рассмотрено лишь несколько примеров. Мно-
гие более ранние экспериментальные данные носят только каче-
ственный характер вследствие несовершенных методов приготов-
ления образцов и неизвестных значений удельных поверхностей.
Кириченко и др. [204] разработали методику получения об-
разцов кремнезема для ионообменных исследований с воспро-
изводимыми значениями удельных поверхностей. Они пригото-
вили серию силикагелей гидротермальной обработкой с продол-
жительностью пребывания образцов в автоклаве 10—20 ч в ин-
Химия поверхности кремнезема
923
тервале температур 90—300°С. К сожалению, они затем не
удалили поверхностный слой обработкой разбавленной щелочью
с тем, чтобы убрать микропористый кремнезем, который осаж-
дался из пересыщенного раствора по мере охлаждепя системы.
Как и следовало ожидать, чем выше была температура обра-
ботки, тем более низким получалось значение удельной поверх-
ности. Однако при более высокой температуре предварительной
обработки при последующем охлаждении на небольших участках
поверхности осаждалось большее количество растворимого
кремнезема, образуя более толстый слой микропористого крем-
незема (см. гл. 5). Авторы приписали более высокую величину
адсорбции возросшей «гидратации» поверхности за счет образо-
вания групп Sis (ОН)2 и Sis (ОН)з, однако не доказали, что по-
добные группы существуют и не сомневались в правильности
своих выводов о том, что большее число групп ОН размещалось
на-едипице площади поверхности, измеренной методом БЭТ, хотя
с точки зрения недавно полученных данных относительно микро-
пористости поверхности кремнезема, подвергавшегося гидротер-
мальной обработке в автоклаве, дополнительные группы SiOH,
вероятно, находятся .внутри микропср, не доступных молекулам
азота. (Это является хорошим примером того, насколько сложно
получить однородную, сглаженную, непористую поверхность
кремнезема с высоким значением удельной поверхности для про-
ведения принципиальных исследований.)
Адсорбцию и обмен ионов К'-, Ва2+ и Mg2+ из растворов хло-
ридов на поверхности кремнезема исследовали методом ЯМР
[205]. Были зарегистрированы различающиеся по ширине линии
адсорбированной-воды НоО.
Число центров на единице поверхности, доступных для ад-
сорбции ионов NaT, было тем же самым, что и для ионов Кд;
,однако после предварительной адсорбции олсинамина число
центров осталось неизменным только для ионов К~, но понизи-
лось на 34 % лля ионов Na+. Таким образом, поверхность стала
селективной по отношению к нонам К [206]. Согласно данным
Абендрота [194], адсорбционное поведение ионов щелочных ме-
таллов от Li до Cs на кремнеземной поверхности различается
менее чем на 20%. Необходимо отметить, что для большинства
данных, полученных при изучении-однозарядных ионов, «адсорб-
ция» заключается главным образом в удерживании иона в на-
ружном слое Гельмгольца и увеличивается параллельно с возра-
станием поверхностного заряда (см. раздел о природе ионных
заряженных центров).
Низшие амины адсорбируются в виде замещенных ионов ам-
мония. Такие соединения заслуживают детального рассмотре-
ния, так как поверхность силикагеля, несущая подобные'адсор-
бированные ионы, представляет собой хороший адсорбент, осо-
924
Глава 6
бенно для веществ со слабокислыми свойствами. К тому же
силикагели, несущие на поверхности ионы аммиака или амина,
селективно проявляют сродство по отношению к ионам опреде-
ленных металлов, которые в этом случае прочно адсорбируются.
Регулируемая таким способом селективная адсорбция была про-
демонстрирована Неймарком и др. [207].
Катионы щелочноземельных металлов
и магния
Кэрролл и Фримен [208] измеряли относительную адсорб-
цию ионов металлов на поверхности кремнезема, определяя сте-
пень вытеснения ионами предварительно адсорбированных кра-
сителей. Наиболее сильно шла адсорбция ионов Са2+, затем Ва2+
и Sr2+.
В том случае, когда ион Са2+ адсорбируется на кремнезем-
ной поверхности, то он может замещать только один ион Н+:
Si.OH + Са2+ = Si5OCa+ + Н+
Об этом было сообщено Боемом и Шнайдером [209] и подтвер-
ждено Айлером [210]. Гринберг [211] показал, что ион кальция
адсорбировался выше pH 5; адсорбированное количество было
пропорционально удельной поверхности. Оказалось, что на 1 нм2
адсорбировалось примерно четыре иона Са2+. На основании ра-
нее сделанных Айлером допущений, что концентрация гидро-
ксильных группа равна 8 ОН-групп/нм2, можно считать, что
каждый ион Са2+ был связан с двумя группами SiOH. Однако
число групп на единице поверхности составляет примерно
4,6 ОН-групп/нм2; таким образом, каждый ион Са2+ связывается
с одной группой SiOH. К такому заключению пришли теперь
на основании другой информации о поверхностной концентрации
гидроксильных групп. Тедрос и Ликлема [212] сделали анало-
гичный вывод.
Многозарядные катионы металлов
Для получения дополнительной информации и примеров сле-
дует обратиться к библиографии, приведенной в предыдущих
главах. Так, в гл. 1 рассматривалось воздействие, оказываемое
адсорбированными ионами Ва2+ и АР+ на растворимость крем-
незема; в гл. 3 было описано взаимодействие кремневой кислоты
с некоторыми ионами металлов;в гл. 4 показано, что адсорбиро-
ванные на поверхности коллоидного кремнезема катионы уча-
ствуют в процессе коагуляции (см. лит. к гл. 4 [256—268]); ад-
сорбция ионов на силикагелях и порошках изложена в гл. 5.
Химия поверхности кремнезема
925
По-видимому, общее правило заключается в том, что кремне-
зем, суспендированный в растворах большинства солей полива-
лентных металлов, начинает адсорбировать ионы металла, когда
значение pH возрастает на 1—2 единицы выше, чем величина
pH, при которой осаждается гидроксид соответствующего ме-
талла. Это явление необычно, так как естественно было бы пред-
положить, что адсорбция должна происходить только тогда,
когда значение pH достаточно высоко, чтобы кремнеземная по-
верхность становилась ионизированной и на ней образовывался
отрицательный заряд. Число заряженных центров на поверхно-
сти кремнезема мало при pH 5 и равно нулю при pH 2 (см.
главы 3 и 4), но все же поверхность адсорбирует определенные
многозарядные катионы очень сильно и при низких значениях
pH, как это имеет место, например, для ионов UO22+, Fe3+, Al3+
и Сг3+ [213, 214]. Однако эти ионы не являются единственными
простыми ионами, которые могут адсорбироваться. При тех зна-
чениях pH, когда ионы сильно адсорбированы, они оказываются
полимеризованными до различных степеней и существуют уже
в виде чрезвычайно небольших по размеру положительно заря-
женных коллоидных оксидных частиц или многоатомных ка-
тионов.
Одиночные катионы, подобные таким, как UO2+, U4+, Pu4+,
•адсорбируются при удалении с поверхности одного иона Н+
в расчете на один ион металла. К тому же ионы Th4+, Fe3+ и А13+
адсорбируются аналогичным образом при низком значении pH,
когда в отсутствие таких ионов группы SiOH обычно не дол-
жны ионизироваться [215, 216].
Визе и др. [217] обсудили вид адсорбции ионов, при которой
их адсорбированные количества превышают эквивалентный по-
верхностный заряд. Когда ионы способны адсорбироваться даже
при значении т. н. з., такая адсорбция называется «сверхэкви-
валентной».
Колыпуттер и др. [218] в качестве типичного примера ис-
следовали адсорбцию на кремнеземе ионов алюминия А13+, гид-
ролизованных ионов А13+ или многоосновных катионов. Авторы
измеряли изменения величины pH в растворах А1С13, гидроли-
зованных до различных степеней, по мере того, как происходила
адсорбция разновидностей ионов алюминия.
В связи с изучением флокуляции коллоидного кремнезема
Матиевич, Мангравите и Кассел [219] определили то значение
pH, при котором ионы А13+ в нитратном растворе вызывали
флокуляцию. В этом примере был выбран коллоидный кремне-
зем людокс-АМ, на котором содержалось примерно 0,4 центра
SiA10~, предварительно нанесенных на поверхность кремне-
зема, чтобы вызвать появление на ней отрицательных зарядов
926
Глава 6
при pH, пониженном до 3. (Этот случай отличается от немоди-
фицированной поверхности кремнезема, когда поверхностный
заряд становится очень небольшим ниже pH 4,5). Адсорбция
ионов алюминия происходила и вызывала флокуляцию при pH 3,
т. е. когда отрицательный заряд на поверхности был частично
нейтрализован. Затем выше pH 3,3 поверхность постепенно при-
обретала положительный заряд по мере того, как все большее
количество ионов алюминия адсорбировалось, после чего ча-
стицы кремнезема начинали вести себя подобно частицам оксида
алюминия. Таким образом, флокуляция происходила при pH
6,0—6,5, иначе говоря, при том же самом значении pH, при ко-
тором гидроксид алюминия осаждался из нитратного рас-
твора.
Наблюдалось также хорошее доказательство того факта, что
ионы А13+, которые, очевидно, полимеризовались в течение пе-
риода флокуляции, составлявшего 24 ч,. адсорбировались затем
в виде поликатионов Als(OH)4+ или по крайней мере в виде не-
которой разновидности полиона, в котором на два атома Алю-
миния приходился один положительный! заряд. Как было пока-
зано [220], структуру основного поликатиона алюминия после
продолжительного старения или старения при повышенной тем-
пературе можно представить как А^ОДОН^ДН^С))^. Матие-
вич [221] отметил, что имеется пороговое значение pH, при ко-
тором катионы вызывают флокуляцию отрицательно заряжен-
ных частиц коллоидного кремнезема. При этом значении и выше
него катионы адсорбируются на поверхности кремнезема. В от-
сутствие алюмосиликатных анионов на кремнеземной поверхно-
сти адсорбция разновидностей катионов А13+ происходит при
pH 3,5—3,75.
Число хлорид-ионов, находящихся вблизи ионов А13+, кото-
рые адсорбированы на кремнеземе с дегидроксилированной по-
верхностью, зависит от степени гидроксилирования поверхности.
Согласно данным Кольцова и др. [222а], при предельно гидро-
ксилированном состоянии поверхности алюминий адсорбиро-
вался в виде (SisO)2AlCl, но на дегидратированных образцах
кремнезема только единичная поверхностная группа SiOH всту-
пает в реакцию с образованием SisOAlCl2.
Когда ион металла склонен к сильному комплексообразова-
нию и в действительности ведет себя подобно катиону сильного
основания, то, очевидно, такой ион перемещается ближе к по-
верхности и индуцирует появление на поверхности отрицатель-
ных зарядов (SiO-), причем число отрицательных зарядов на
поверхности будет равно числу зарядов катионов; при этом
высвобождается эквивалентное число ионов Н+. Выдра и Мар-
кова [2226] изучили это явление на этилендиаминовых ком-
Химия поверхности кремнезема
927
плексах ионов Ag~, Zn2+, Cn2i, Со2+ и Со3+. В случае ионов ко-
бальта в растворах NH<OH комплекс СЬ(?Шз)3+ оказался на-
столько стабильным, что адсорбировался на кремнеземе без
изменений. Однако комплекс Со2+(Н20)6 адсорбируется предпоч-
тительнее, чем комплексный аммиакат [223].
Маатман [224] обсудил обычно применяемые методы, ис-
пользуемые для обработки поверхности раздела кремнезем—
раствор. Ввиду близкого расположения групп SiOH на поверх-
ности он представил кремнеземную поверхность в виде поли-
дентатного лиганда.
Интересно отметить, что не только кремнезем обладает ок-
сидной поверхностью, которая склонна к образованию хелатов.
Хол и Стамм [225] нашли, что ион РЬ2+ прочно адсорбируется
на поверхности у-А12О3 при pH 7, т. е. когдц такая поверхность
еще несет некоторое число положительных зарядов. Вероятно,
.атом свинца координирует с атомом алюминия на поверхности
в виде А1
/°\
\)Z
Pb. Несмотря на то что при данном значении pH
кремнезем заряжен отрицательно, тогда как оксид алюминия
заряжен положительно, сродство иона РЬ2+ к поверхности оксида
алюминия оказывается на несколько порядков больше, чем к по-
верхности кремнезема. Таким образом, специфическая адсорб-
ция ионов зависит не от величины ионного заряда'и кислотности
поверхности, а от геометрии координационных связей, которые
могут образоваться.
Адсорбция ионов железа исследовалась менее подробно, чем
ионов алюминия. Потт и Мак-Николь [226] изучили координаци-
онную структуру и валентность ионов железа. Они наблюдали
за поведением ионов Fe3+, адсорбированных на кремнеземе,
после чего адсорбент нагревали до 400°С. Было показано, что
ионы Fe3+ выше 400°С принимали тетраэдрическую координа-
цию, а при 800°С кристаллизовались до cc-Fe2O3. Шенк и Вебер
[227] исследовали взаимодействие, железа с кремнеземом в кол-
лоидном растворе и нашли, что кремнезем катализировал окис-
ление двухвалентного железа до трехвалентного. Адсорбция
ионов Fe3+ и Сг3+ на кремнеземе исследовалась Хили, Купером
и Джеймсом [228] в зависимости от величины pH и ионной
силы. Полученные различия в адсорбции этих ионов были объ-
яснены на основе понятий координационной химии.
Боем [8] рассмотрел роль образования координационных
структур и отметил, что адсорбция многоатомных ионов по су-
ществу фактически необратима. Образование хелатных поверх-
ностных соединений, вероятно, требует участия групп SiOH на
928
Глава 6
поверхности, которые замещают некоторое число молекул воды
из иона металла:
SisOH
SisOH
н2о ,он2п+
+ н2о- м он,
Н2О он2
и Н,(ч-|)+
Sis—Q 9 рн2
/М:, + н*+ н2о
sis—с/ (5 он,
н.
Замещение групп, координированных на ионах металла, по-
верхностными группами SiOH было экспериментально исследо-
вано Кольтхоффом и Стенжером [229]. Они обнаружили, что
когда ион Cu(NHs)2+ адсорбировался на поверхности кремне-
зема, то отношение NHs: Си становилось менее чем 4: 1. В не-
которых случаях [230] в реакцию включались три силанольные
группы, как, например, это имело место для иона Zn2+:
3SisOH + ZnCI2 + 2NH4C1 = [(SisO)3 Zn2+Cl]2- + 2NH4+ + 3HC1
Хили, Джеймс и Купер [231] сообщили о сильной адсорбции
ионов металла на кремнеземе. При рН<6 кобальт адсорбиро-
вался из 10"5 М. раствора Со2+ неспецифически, как и при обыч-
ном ионном обмене. Выше pH 6,5, но ниже точки начала осаж-
дения Со(ОН)2 ионы кобальта перемещались в поверхностный
слой кремнезема, при этом одна из шести молекул воды, окру-
жающих ион кобальта, замещалась одной группой SiOH:
SisOH + Со (Н2О)|+ = SisO : -> Со (Н2О)§+
Н
Следовательно, знак заряда на кремнеземной поверхности из-
менялся на обратный, и поверхность становилась положительно
заряженной в соответствии с характером ионов гидроксида ко-
бальта.
Кикучи и др. [232] использовали измерение магнитной вос-
приимчивости, чтобы определить конфигурацию медных ком-
плексов с ацетилацетоном и дифенилпикрилгидразидом на си-
ликагеле. Очевидно, адсорбция тетрагональных плоских молекул
ацетилацетоната происходила в виде квадратных лоскутов, так
что молекулы адсорбата состыковывались по краям наподобие
плоского кристалла.
Бурвелл и др. [233а] также изучали поведение некоторых
координационных комплексов, адсорбированных на кремнеземе.
Авторы показали, что когда комплексные ионы адсорбируются
на поверхности кремнезема, то ион Si4O может проникать
внутрь и замещать лиганд из координационной сферы атома ме-
Химия поверхности кремнезема
929
талла, образуя, таким образом, ковалентную связь между этим
комплексом и поверхностью кремнезема:
[Cl2Co(en)2]* + OSi, [С12Со(еп)2] ’
OSi,
(зеленое окрашивание)
[C!2Co(en)2p OSi,
4- НС1
'О
st.
en = nh2c2h,nh2. (розовое окрашивание)
Аналогичные реакции имеют место с ионами цис-Со (еп)2Х
XNO2C1+ и (en)2PtBr+.
Полное покрытие всей поверхности кремнезема оксидом цир-
кония использовалось для обеспечения более прочного закреп-
ления на адсорбенте монослоев биохимических веществ, напри-
мер энзимов [2336]. Подобные пористые стекла, покрытые окси-
дом циркония, нашли применение; их преимущества, которые
заключаются в заметно большей инертности и нерастворимости,
описаны формой-изготовителем [233в].
Адсорбция из раствора сильной кислоты. Ряд металлов,
обладающих высокой валентностью, т. е. находящихся в наи-
большей степени окисления элемента, обычно способны сильно
адсорбироваться при pH 2 или ниже, а некоторые элементы мо-
гут адсорбироваться даже из раствора сильной кислоты. Напри-
мер, Fe3+ адсорбируется при pH 2. Сурьма, молибден, вероятно,
ванадий, тантал, ниобий, торий, цирконий, уран, плутоний и про-
тактиний адсорбируются при pH 0 или ниже.
По-видимому, еще мало известно относительно механизма,
благодаря которому ионы с такой различной основностью спо-
собны связываться с поверхностью кремнезема. Формирование
хелатных поверхностных соединений или совмещение кислород-
ных связей в подобных оксикатионах, как, например, в UO2+,:
с соответствующими атомами на поверхности кремнезема, веро-
ятно, способствует образованию множества ковалентных связей.
Обычно принято думать, что при промывании кремнезема
сильной кислотой удаляются все загрязнения. Однако несколько
элементов поразительно сильно адсорбируется и в этом случае.
Сурьма адсорбируется из раствора крепкой азотной кислоты
[234]. Хром в виде иона Сг6+, согласно данным Диббса [235],
остается на поверхности плавленого кварцевого стекла после
обработки адсорбента очищенным раствором H2CrO4 + H2SO4
и даже после промывания смесью HNO3 и H2SO4, причем на
поверхности остается 0,1 мкг/см2 хрома. Однако при обработке
адсорбента высушенным газообразным НС1 при температуре
180°С удается удалить все ионы Сг3+ и Сг6+ в основном в виде
СгО2С12 [236]. Ниобий и цирконий настолько сильно адсорби-
33 Заказ № 250
930
Глава 6
руются на кремнеземе, что могут избирательно извлекаться из
растворов солей урана [237].
Обращение знака заряда на поверхности кремнезема. Рас-
творимые гидролизованные ионы Th4+, Zr4+, Ве2+, Zn2+, Fe3+
и А13+ способны прочно адсорбироваться на кремнеземе, по-
этому когда они содержатся в избыточном количестве по сравне-
нию с тем содержанием, которое требуется для образования по-
крытия на поверхности кремнезема, то положительный поверхно-
стный заряд меняется на отрицательный. Гидролизованные
полимерные разновидности или основные соли металлов адсор-
бируются на кремнеземе при значительно меньшей величине pH,
чем это наблюдается для простых гидратированных ионов. Ме-
ханизм изменения знака заряда, как рассматривалось в гл. 4
в связи с обсуждением вопроса о коллоидных частицах кремне-
зема, в равной мере хорошо применим ко всем кремнеземным
поверхностям (см. лит. к гл. 4 [424—435]). Подробное рас-
смотрение примера, связанного с изменением знака заряда, ис-
следованного в работе [219], приводилось выше при описании
адсорбции ионов алюминия. Как отметили Джеймс, Визе и Хили
[276], в дисперсных системах, в которых наблюдается коагуля-
ция под воздействием гидролизованных ионов металла, нет ни-
какой очевидной корреляции между электрокинетическим потен-
циалом и устойчивостью коллоидной системы. Это показывает,
что теория ДЛФО, по-видимому, не может быть применена.
Авторы работы сравнивали адсорбционное поведение ионов
Со2+, La3+, Th4+ на одном и том же образце SiO2.
Прежде всего необходимо рассмотреть концентрацию катио-
нов в растворе в отличие от их общего количества в системе,
в которое входят также катионы, адсорбированные на твердой
поверхности. Так как обычно в системе известно лишь суммар-
ное количество катионов, то концентрация коллоидных частиц,
т. е. суммарная поверхность кремнезема в системе, должна со-
храняться настолько малой, чтобы большая часть ионов металла
все еще находилась в растворе.
По мере того как концентрация ионов металла возрастает
в кремнеземном золе, поверхность частиц кремнезема постепенно
покрывается адсорбированными поликатионами металла, кото-
рые в конце концов закрывают всю поверхность. При протека-
нии этого процесса величина отрицательного заряда исходного
электрокинетического потенциала поверхности кремнезема на-
чинает уменьшаться, проходит через нуль и окончательно при-
нимает положительное значение, соответствующее значению по-
тенциала оксида алюминия, когда поверхность кремнезем-а пол-
ностью покрывается поликатионами.
Авторы отмечают, что коагуляция происходит, когда электро-
кинетический потенциал заключен в интервале от —14 до
Химия поверхности кремнезема
931
+ 14 мВ. Представляет интерес вопрос о соотношении долей,
занимаемых на поверхности катионными и анионными участками
при этих крайних значениях электрокинетического потенциала.
Такие соотношения могут зависеть в некоторой степени от раз-
меров коллоидных частиц кремнезема и поликатионов.
Подобные воззрения приложимы только к системам, в кото-
рых поликатионы адсорбируются и способны покрывать поверх-
ность кремнезема, и, следовательно, при этом может происхо-
дить обращение знака заряда поверхности на обратный. Однако
они не применимы к коагулирующим системам, имеющим «плот-
ный двойной электрический слой» вокруг частиц в растворах
простых электролитов, разбавленных в отношении 1:1. Следует
отметить, что и в случае катионов больших размеров, как, на-
пример, К+ и (CH3)4N+, может также происходить обращение
знака заряда на поверхности, когда она становится полностью
покрытой этими большими катионами.
Краткие выводы и обзоры. Известны некоторые общие об-
зоры по ионообменным свойствам кремнеземной поверх-
ности и силикагелей, опубликованные за последние 25 лет, но
лишь в немногих рассматриваются все аспекты этой темы. Бен-
тон и Элтон [238] подсчитали энергию адсорбции ионов, нахо-
дящихся в слое Штерна. Душина и др. [239] показали взаимо-
связь между величиной pH и адсорбцией ионов металлов, ко-
торую они описали на основе растворимости , поверхностных
соединений [240].
Минимальная концентрация ионов металла при специфиче-
ской адсорбции (МКСА) была измерена Вакацуки, Фурукава
и Кавагучи [241] на кремнеземе при разных значениях pH для
15 катионов. Специфичность была увязана со способностью
атома металла образовывать ковалентные связи и понижалась
в ряду в следующем' порядке: Ga3+, Al3+, Fe3+, Cr3+, V3+, Со2+,
Cu2+, Zn2+, Ва2+, Sr2+, Са2+, Mg2+, Rb+, К+ и Na+. Константы
равновесия для адсорбции ионов металлов на поверхности крем-
незема были измерены Далангом и Стаммом [242].
Лигандные свойства поверхностных силанольных групп для
ионов металлов (Fe3+, Cu2+, Cd2+ и Pb2+) были измерены Шинд-
лером и др. [191], а также были определены константы адсорб-
ционного равновесия’. Величина адсорбции ионов металлов в за-
висимости от pH показана на рис. 6.10.
SlsOH + Мг+ = SisOM‘’“1 + н+
2SisOH + Мг+ = (SisO)2 Мг~ 1 + 2Н+ *BS2
Наблюдалось линейное соотношение между 1g и lg KiB2,
где К\В2— константа устойчивости гидрокомплексов ионов ме-
талла. Это показывает, что как лиганды SROH и НОН имеют
33*
932
Глава 6
сходство, и объясняет давно известный экспериментальный факт,
что комплекс силиката металла осаждается при таком значе-
нии pH, которое только незначительно ниже, чем pH начала
осаждения гидроксида металла в отсутствие кремнезема.
Арланд и др. [213] исследовали механизм адсорбции обычных
и гидролизованных (основных) ионов металла. Была предло-
жена термодинамическая модель системы.
I. рн
Рис. 6.10. Зависимость суммарного процентного содержания адсорбированных
ионов металлов от величины pH на гидроксилированной поверхности кремне-
зема. (По данным Шиндлера и др. [191]).
Джеймс и Хили [243] предположили, что при адсорбции гид-
ролизуемых ионов (поликатионов) на оксидной гидратирован-
ной поверхности энергетические изменения за счет кулоновских
сил и химических превращений, благоприятствующих адсорбции,
уравновешиваются энергией сольватации, которая противодей-
ствует этому процессу.
Библиография по адсорбции ионов металлов на кремне-
земе. Таблица 6.3 может служить в качестве руководства при
поиске литературы, хотя и не является исчерпывающей. В таб-
лицу включены литературные ссылки, относящиеся в основном
к гидроксилированным образцам кремнезема. Комплексы ионов
металлов включены в таблицу для тех случаев, когда лиганды
определены, в других случаях можно допустить, что в качестве
лигандов приняты ОН- или Н2О. Степень основности или сте-
пень полимеризации обычно не указываются.
Химия поверхности кремнезема
933
Таблица 6.3
Адсорбция ионов металлов на кремнеземе
Элемент Литература Примечание
Однозарядные ионы
Таллий 244 Комплекс с этилендиамином
Серебро 244 То же
245 Комплексы с аммиаком и этилендиамином
Двухзарядные ионы
Кальций 208 Найдена зависимость от размеров частиц
Стронций, барий Магний 211 212 241 239 208 241 246 239 SiO2 Взаимодействие с литием Адсорбционные данные Адсорбционные данные
Марганец 247 Координирован одним ионом SiO-
Никель 244 Комплекс с этилендиамином
Кобальт Медь 245 247 248 249 231 241 233а 244 245 247 250 276 191 Комплексы с аммиаком и этилендиамином Координирован двумя ионами SiO- Комплекс с аммиаком Возможная координация с полярными ли- гандами Комплексы с хлором и этилеидиамином Комплекс с этилендиамином Комплексы с аммиаком и этиленди амином Координирован одним ионом SiO- Изучено состояние Со на поверхности См. рис. 6.10
Цинк 229 244 251 248 ' 245 247 239 241 230 244 245 Комплекс с этилеидиамином Несколько лигандов Комплексы с аммиаком и этилендиамином Координирована одним ионом SiO- Адсорбционные данные Комплекс с этилеидиамином Комплекс с аммиаком
247 239 Координирован двумя ионами SiO- Адсорбционные данные
934
Глава 6
Элемент Литература Примечание
Хром (II) 252 Стабилизированные комплексы Сг2+—NH3
Кадмий 191 См. рис. 6.10
253 Взаимодействует с поверхностью в амми-
ачном растворе
239 Адсорбционные данные
Олово (II) 254 Использован эффект Мессбауэра
255 То же
Свинец 191 См. рис. 6.10
256 РЬ удаляется из расплавленного висмута
Палладий 257
239 Адсорбционные данные
Т рехзарядные ионы
Иттрий 241
Лантан Галлий Алюминий Хром (III) 246 276 241 215 216 213 218 219 241 221 258 213 214 241 231 259
258
Железо (III) 213 214 215 216 226 260 191 228 258
Сурьма 234
Адсорбируется выше pH 2,5
Адсорбируется ион (СгС12-4Н2О)+, но не
Сг3+
Адсорбируется при pH >4 в виде гидро-
ксида
См. рис. 6.10
Значение pH адсорбции зависит от анио-
нов
Адсорбируется из сильнокислых растворов
Четырехзарядные ионы
Цирконий
275 Адсорбируется из кислого раствора
261 Выделение из гафния
Химия поверхности кремнезема
935
Элемент Литература Примечание
Гафний Тантал Ниобий Торий Протактиний Плутоний Хром (VI) Дихромат (Сг2О72~) Уран (VI) (или UO22+) 262 263 264 263 262 261 264 265 275 258 216 266 ' 276 267 268 215 216 269 270 Шее 271 272 273 214 213 215 216 274 Адсорбируется только в виде продуктов ги- дролиза Адсорбируется на гидрофобной поверхности SiO2 ’ Адсорбируется коллоидный ZrO2; Zr4+ в 2 н. растворе HNO3 Адсорбируется на гидрофобной поверхно- сти SiO2 Адсорбируется только в виде продуктов гидролиза Выделение из циркония Адсорбируется даже из 10 н. кислоты; от- деляется от ниобия Адсорбируется в виде нейтральных ком- плексов; адсорбируется из растворов ки- слот вплоть до 6 н. Адсорбируется из кислых растворов Адсорбируется при pH >1,5 Вероятно, адсорбируется в виде полика- тионов Сильно адсорбируется, отделяется от Th, U, Zr, Hf, Nb Ионы Pu (ОН) И ” "> + адсорбируются при рН>1 Адсорбируется из 10~7 М раствора золя при нейтральном pH Адсорбируется при pH не выше 0,6—4,5; присутствует в виде частиц при pH 1,5— 8,5 тизарядные ионы Ион стабилен до 565°С; пригоден как ка- тализатор полимеризации SiOCr(O)2OCr(O)2OH Адсорбируется на SiO2, нагретом до раз- личных температур Ион UO22+ замещает один ион Н+
936
Глава 6
Неионные реакции на поверхности кремнезема
Ряд реакций уже рассматривался в связи с определением
групп SisOH. Многочисленные реакции перечислены и кратко
обсуждены Хайром [9]. Девел, Бартман и др. [277, 278] изу-
чили множество поверхностных реакций для получения покры-
тий SisOR и SisR в тот период, когда только стала признаваться
возможность образования хемосорбированных молекулярных
покрытий на кремнеземе. Например, было продемонстрировано
использование гексаметилдисилазина как особенно перспектив-
ный метод химического модифицирования.
Стрелко и Канаболоцкий [279] также классифицировали ме-
ханизмы ряда поверхностных реакций, представленных
в табл. 6.4. Реакции на поверхности выполняются в газовой фазе
или в растворе, как правило, в отсутствие воды или влаги. При-
водится некоторое количество библиографических ссылок, из ко-
торых может быть получена дальнейшая информация.
Гидрофильные покрытия на кремнеземе
Для некоторых применений желательно, чтобы поверхность
кремнезема или стекла смачивалась водой. Но в то же время
должны отсутствовать различные характерные ионные, гидро-
фобные или водородные связи, которые возникают при адсорб-
ции органических молекул. Известно, что поверхность кремне-
зема адсорбирует и денатурирует белки, после чего она
становится гидрофобной в результате образования покрытия угле-
водородными группами. Такая поверхность способна адсорби-
ровать органические молекулы путем образования гидрофобных
связей.
Смачиваемая поверхность может быть получена в результате
нанесения слоя, снаружи которого произвольно расположены
группы первичного спирта, такие, как SisOSi (СН)зСН2 (СН2)ПОН.
Таким способом можно образовать нейтральную гидрофильную
поверхность, причем, изменяя величину п, можно варьировать
степень полярности поверхности и ее смачиваемость.
Блекман и Хэрроп [323] описали приготовление очень устой-
чивой смачиваемой водой поверхности на стеклянной подложке
нанесением на стекло ненасыщенного силана, из которого затем,
вероятно, при окислении двойной связи образовывался диол.
Такая поверхность может иметь ряд преимуществ по сравне-
нию с поверхностью, несущей только одиночные концевые гидро-
ксильные группы. Для формирования устойчивых гидрофильных
поверхностей аллильные группы лучше подвергать окислению
перманганатом калия.
Химия поверхности кремнезема
937
Таблица 6.4
Механизмы реакций на поверхности кремнезема
Реакция Литература
Образование связей Si.—О—С
SisOH + ROH = SisOR + Н2О 9
SisOH + CH2N2 = S!sOCH3 + N2 (соблюдать меры 9, 280
предосторожности!)
Поверхность после дробления + ROH-i-SisOR-группы 281
SisOH + НСООН = SisOCH (ОН)2 (вероятно, образуется 282
хемосорбированное
соединение)
SisOH + линолевая кислота^хемосорбированная линолевая 170
кислота (нагревание после
адсорбции)
SisOH + RCOOH = SisOCOR + Н2О 9
SisOH + RCOC1 = SisOCOR + HC1 9
Образование связей Si—0—Si
При 230 °C
SisOH + SiH4 = SisOSiH3 + H2O 283, 284
SisOH + CISi (СН3)з = SisOSi (CH3)3 + HC1 9
SisOH + (C2HsO)3SiR = SisOSi (OC2H5)2R + C2H5OH
2SisOH + (EtO)3 SiCH2C6H4SO3H = (SisO)2 Si (OEt) X 285
X CH2C6H4SO3H + 2ЕЮН
Образование связей Si—галоген
S’sOH + NH4F = SisF + NH3 + H2O 9, 286
SisOH + HF = SisF + H2O 287
2SisOH + SOCI2 = 2SisCl + 2HC1 + SO2 9, 288
2SisOH + 2C12 = 2SisCl + 2HC1 + O2 9
2SisOH + CC14=* 2SisCl + CO2 + 2HC1 9
SisOH + SiCU = SisCl + оксихлориды кремния 289
SisOH + SiCl4 = SisOSiCl3 + HC1
2SisOH + SiCl4 = Sis-0 C1 + 2HC1 9
^Si^
Sis-o^ \:i
938
Глава 6
Реакция Литература
При > 350 °C SisOH + ССЦ = S1C1 + СОС12 + НС1 9, 290
Образование связей SI—С Si5OH -J- LiCgHg == Si5OLi -f- 291
SisOH + SOCh + n-BrMgC6H4OSi (CH3)3 в C5H5 + ТГФ 292
— Si^CfjH.iOH (после гидролиза) Поверхность S1O2 после дробления + стирол 281
-> SiC—полистирол SisCl + LiR = SisR + LiCl —
SisCl + RMgX = Si5R + MgXCl —
Реакции с участием водорода, воды, аммиака, циана
SisOH + Н2 -> SisOH • Н (высокочастотный разряд) 293
SiOSi4 + H2O =2SisOH —
хнх 9
2 SisOH + NH, = Si, ~?Sis '"'O SisOSi + NH3 -- SisOH + S isNH2 294
При 600 °C SisOH + NH3 = Sis-NH2 + H2O 295
При 800 °C Si4NH2 + O2 = Sb-0 + N2 + H2O 295
SisOH + (CN)2 = SisNCO + HCN 2SisOH + HCN-> SisOH + SisCN 296
Другие реакции на поверхности кремнезема Реакции полимеризации этилена,ацетилена в присутствии 9
следов натрия, HCN * SisC6H5 +HNO3-> SisC6HsNO3 восстановление диазотирование SisC5H5NO2 SisCeH5NH2 SisC6H5N2+Cl SisC6HaN2+Cl + С3Н5СН = СНг-> полимеризация прививкой, 297
полистирольное покрытие
Химия поверхности кремнезема
939
Реакция Литература
С бором
2Sis0H + Н3ВВН3 = 2SisOBH2 + 2Н2
SisOH + BCI3 = SbOBCh + НС1
2SUOH + BCI3 = (SisO)2BCl + 2HC1
SisOH + BF3 = SisOBF2 + HF
OSi,
SisOH + TiCI4 - B Br, - Si,O^ О В-OSis
Si„Cr 4) B-OSis
xosu
2SisOH 4- В (OH)3 = (SisO)2 BOH 4- 2H2O
(SisO)2 BOH + MeOH = (SisO)2 BOMe + H2O
С серой
Sis + SO3-> SisOS (O2) OH
С фосфором
Si40H 4- P2O5 (газ) -> фосфорсиликатная пленка
2SisOH 4- P2O5 4- H2O -> (SisO)2 PCOH
2SisOH 4- POC13 (газ) = (SisO)2 POC1 4- 2HC1
3Si5OH 4- POCI3 (газ) = (SisO)s PO 4- 3HC1
nSisOH 4- PC13 = (SisO)„ РС13-Л
С алюминием
SisOH 4- A1R3 = SisOAlRp 4- RH
SisOH 4- Al (изо-Вн)з = SisOAl (изо-Ви)2 4- C4H9
SisOH 4- AlBr3 = SisOAlBr2 4- HBr
SisOH 4- AICI3 = SisOAlCl2 4- HC1
2SisOH 4- A1C13 = (SisO)2 A1C1 4- 2HC1
С титаном
SisOH 4- TiCl4 = SisOTiCh 4- HC!
2SisOH 4- T1CI4 = (SisO)3 TICI2 4- 2HC1
С оловом
2Si5OH 4- SnCl4 = (SisO)2 SnCh 1- 2HC1
SisOH 4- SnCl4 = SisOSnCls 4- HC1
SisSnCl3 4- H2O SisOSn (OH)3
9, 291
298, 299
13, 69, 300
301, 302
61, 302, 303
304
305, 306
307
I 308
309
311
310
301, 304, 308
8
71
312
300, 301
67, 313
312, 314,
315, 301
316
317
317
940
Глава 6
Реакция
Литература
С молибденом
2Si,OH 4- M0CI5 = (Si,O)2 M0CI3 + 2НС1
(Si,0)2 M0CI3 + ЗН2О = (Si,0)2Мо (0Н)3 4- 3HC1
С ванадием.
Si,ОН 4- VOCI3 = Si,OVCl2 4- НС1
2Si,0H 4- VOC13 = (Si,0)2 V0C1 4- 2HCI
При 180 °C
3Si,0H 4- VOCI3 = (Si,0)з У0 4- 3HC1
С хромом
2Si,0H 4- CrOCh = (Si,0)2 Cr02 4- 2HC1
При>330 °C
(Si,O)2 CrO2-> (Si,0)2 CrO 4- O2
2 Si,OH 4- Н2СГО4 = (Si,0)2 СЮ2 4- 2НгО
318
318
319
320, 321
310, 320, 321
315
301
322
Гидрофобная поверхность кремнезема
В гл. 5 были рассмотрены различные типы мономолекуляр-
ных покрытий на кремнеземных порошках. В этой главе будут _
затронуты некоторые химические аспекты следующих хемосор-
бированных молекулярных слоев:
а) адсорбированные органические катионы (азотистые основа-
ния, четвертичные аммониевые ионы);
б) адсорбированные многозарядовые ионы металлов, к которым
присоединяются карбоновые кислоты с длинной цепью;
в) этерифицированные силанольные группы;
г) органосилильные покрытия;
д) адсорбированные линейные полимеры с образующими водо-
родные связи боковыми группами, дающие предельную гид-
рофобизацию, если вся поверхность кремнезема доступна
полимеру.
Блекман и Хэрроп [323] изучили устойчивость некоторых из
перечисленных гидрофобных покрытий под воздействием водя-
ного пара. Они нашли, что наиболее стабильными являются по-
лиметилсилоксановые группы и (СН3)з5Ю-группы.
Гидрофобная поверхность представляет интерес не только
из-за ее водоотталкивающих свойств; подобные поверхности
Химия поверхности кремнезема
941
имеют значение для достижения минимальной адгезии в техно-
логических процессах флотации при обогащении минерального
сырья.
Органические катионы и основания
Механизм адсорбции органических оснований на кремнезем-
ной поверхности изменяется в зависимости от силы основания
и значения pH раствора. Адсорбция аминов при нейтральном pH
может протекать посредством образования водородных связей
с поверхностью кремнезема:
SbOH4-NR3 = SbOH:NR3
Амины с длинными цепями могут адсорбироваться в виде
ионов аммония при условии достаточно высокого значения pH,
чтобы образовался отрицательный заряд на поверхности крем-
незема:
Si5OH + RNH2 = Si5OH3NR
Определяющим фактором для подобного рода покрытий яв-
ляется размер и тип углеводородной группы. Если R представ-
ляет собой длинноцепочечную алифатическую углеводородную
группу, то такие поверхностно-активные группы стабилизи-
руются за счет образования гидрофобных связей между ними на
поверхности. При очень низкой концентрации вещества в рас-
творе на поверхности адсорбируется только одиночный слой,
и поверхность становится гидрофобной. Однако при более вы-
сокой концентрации, даже ниже критической концентрации ми-
целлообразования, на поверхности формируется двойной слой
(рис. 6.11). На рис. 6.11 показана схема адсорбции длинноцепо-
чечных аминов на кремнеземе, например ацетата додециламина,
причем при pH 7 ниже 25°С, по-видимому, имеет место «физи-
ческая» адсорбция, скорее всего вследствие образования водо-
родных связей. Выше 25 °C, согласно данным Болла и Фурсте-
нау [324], идет процесс хемосорбции (вероятно, ионных форм).
Такая адсорбция вызывает появление больших энтропийных эф-
фектов, свидетельствующих об образовании гидрофобных связей
между адсорбированными додецилгруппами, что ведет к ста-
билизации всего адсорбированного слоя.
Элтон [325] описал изменение величины краевого угла при
смачивании водой поверхности кремнезема по мере возрастания
концентрации катионного ПАВ. Для случая БЦТМА (бромида
942
Глава 6
цетилтриметиламмония) наблюдения привели к следующим ре-
зультатам:
Концентрация БЦТМА, Краевой угол, град
моль/л
о о
10-7 84
10-6 до
10-“ 90
2 • 10-4 68
5 • 10"4 51
10-з о
Рис. 6.11. Схема адсорбции мицеллы четвертичного аммониевого катиона
с длинной цепью.
Мицелла, адсорбированная на поверхности, вырастает в размере за счет мицелл из рас-
твора. (По данным Айлера [3], с разрешения Cornell University Press.) '
Так как критическая концентрация мицеллообразования
(к. к. м.) для БЦТМА равна 8,5-10~4, то ясно, что, если концент-
рация БЦТМА заметно ниже к. к. м., на поверхности образуется
гидрофобный монослой с краевым углом 90°, но при концентра-
ции БЦТМА, равной и выше к. к. м., формируется уже бимоле-
кулярный слой и, следовательно, краевой угол снова падает до
нуля. Ниже к. к. м., по мере того как кремнеземная поверх-
ность превращается в гидрофобную, содержание БЦТМА в очень
разбавленном растворе истощается и раствор становится про-
зрачным. Происходящие в такой системе изменения на молеку-
лярном уровне были подробно описаны Тер-Минасян-Шарагой
[326], который наблюдал, как большая доля осаждаемого угле-
водородного покрытия образовывалась из вещества, которое
Химия поверхности кремнезема
943
концентрировалось на поверхности границы раздела фаз воз-
дух—вода,
Бреслер и соавторы [327] представили интересные сведения
о нанесении покрытий на пористое стекло при использовании
нескольких различных типов четвертичных аммониевых ионов.
Ниже представлены полученные ими данные, выраженные авто-
рами в микромолях в расчете на 1 г пористого стекла, имевшего
удельную поверхность 18 м2/г и диаметр пор 1120 А, пересчи-
танные на, число молекул адсорбированного вещества, приходя-
щееся на 1 нм2 (средние значения):
Катион Число молекул при
pH 3 pH 6 pH 9
h-C16H33N (СН3)3 0,8 1,3 1,9
CSH5N (С2Н5)з 0 0,15 0,5
CsH5N (СН3)2СН2СбН5 0 0,3 0,7
Адсорбция в области pH 3—6 длинноцепочечных катионов
показывает важное значение гидрофобных связей, которые за-
метно ослабляются при укорачивании длины цепи молекул.
В этой области pH добавление диоксана, который ведет себя
как ПАВ, «смачивающее» гидрофобные цепи и разрушающее
гидрофильные связи, понижает адсорбцию всех указанных ионов
до нуля. При pH 9 ионное притяжение является основной си-
лой, обусловливающей адсорбцию, и поэтому адсорбция ионов
еще происходит.
При повышении концентрации электролита происходит уве-
личение заряда на поверхности кремнезема, что в свою очередь
вызывает возрастание адсорбции алкильных четвертичных аммо-
ниевых ионов с длинными цепями, как, например, цетилпири-
дина [328]. Теплота адсорбции в подобных системах примерно
равна нулю, а энтропия адсорбции обусловливается ориентацией
длинных углеводородных цепей-, расположенных в хемосорбиро-
ванной пленке [329]. Площадь поверхности, занимаемая од-
ним длинноцепочечным четвертичным аммониевым ионом
С1бНззЙ(СНз)+, составляет 60—65 А.
Другие примеры адсорбции органических катионов на по-
верхности кремнезема описаны в связи с рассмотрением флоку-
ляции в гл. 4.
Бийстербош [330] выполнил детальное исследование адсорб-
ции RN+(CH3)3 (где R н-С12Н23 или «-С1вН3з) на различных ти-
пах кремнезема. Он подтвердил, что в воде идет образование
двойного слоя на поверхности кремнезема вблизи к. к. м. Бли-
жайшие к поверхности органические катионы нейтрализуются
группами SiO- на поверхности кремнезема, а катионы, нахо-
944
Глава 6
дящиеся дальше в глубине раствора, сопровождаются эквива-
лентным числом анионов Вг_, как это показано на рис. 6.12, а.
Значение гидрофобных связей или формирования мицелл
было раскрыто, когда вместо воды стали использовать водный
1 М раствор н-бутанола. При pH 10, когда поверхность крем-
незема сильно заряжена, необратимо адсорбировался монослой
ионов C16H33N (СНз)+ (т. е. ионов ЦТМА) даже при их наи-
(а)
S>02
Рис. 6.12. Схемы стадий адсорбции на поверхности кремнезема додецил- и це-
тилтриметиламмониевых ионов.
а — двойной слой; б — монослой в присутствии молекул спирта ВиОН; в — единичная мо-
лекула в присутствии молекул спирта ВиОН; г — спаренный (дуплекс) слой. М~ — ион
HSiOg . (По данным Бийстербоша [330].)
меньшей концентрации в растворе. При этом никакого второго
слоя не образовывалось, а также не адсорбировались бромидные
ионы. Поверхность стала покрываться при гораздо более низких
концентрациях ЦТМА в спиртовом растворе по сравнению
с водным раствором. Объяснение этого факта заключается
в том, что гидрофобный слой, на поверхности покрывается пере-
вернутым слоем спиртовых молекул н-BuOH, как это показано
на рис. 6.12, б. При более низком покрытии поверхности могут
быть адсорбированы и одиночные ионы ЦТМА, что изображено
на рис. 6.12, в, так как молекулы спирта окружают углеводо-
родную цепь. Это вызывает понижение энергии поверхности раз-
дела между углеводородом и водой.
Некоторые из полученных Бийстербошем кривых, показываю-
щих адсорбцию додецилтриметиламмониевых ионов (ДТМА)
на гидроксилированной поверхности кремнезема, представлены
на рис. 6.13, а. Он дает следующее объяснение двух уровней,
наблюдаемых при адсорбции на стадиях 2 и 3.
Стадия 1. Согласно Прусту и Тер-Минасян-Шараге [331], на
первом этапе адсорбции длинные цепи лежат почти параллельно
Химия поверхности кремнезема
945
Концентрация дтМА,ммоль/л
Рис. 6.13. Зависимость электрофоретической подвижности и адсорбции ионов
додецилтриметиламмония ДТМА+ и ионов Вг~ на полностью гидроксилирован-
ной поверхности частиц кремнезема BDH (полученного осаждением) (а) от
концентрации ДТМА в растворе при pH 10 и на частично дегидроксилирован-
ной поверхности кремнезема Cab-0-SiI (б).
I — электрофоретическая подвижность положительно заряженных частиц; 2 — поверх-
ностная концентрация ионов ДТМА+, адсорбированных из воды; 3 — поверхностная кон-
центрация ионов Вг~, адсорбированных из воды: 4 — поверхностная концентрация ионов
ДТМА+, адсорбированных из 1 М раствора н-бутилового спирта в воде; в последнем
случае ионы Вг- не адсорбировались. (По данным Бийстербоша [330].)
поверхности, несколько перекрывая друг друга, причем каждая
молекула занимает площадь НО А2, что соответствует поверхно-
стной концентрации 1,5 мкмоль/м2 при концентрации ДТМА
в растворе 1 ммоль/л. На этой стадии кривая, отражающая при-
сутствие заряда на частицах, не проявляется (рис. 6.13 а), что
свидетельствует о величине концентрации заряженных групп на
поверхности кремнезема, примерно равной 1,5 мкмоль/м2, или
0,9 5Ю~-групп/нм2. При pH 10 концентрация силикатных
34 Заказ № 250
946
Глава 6
ионов, находящихся в равновесии с мономером,.составляет около
0,004 н.; по данным Болта (см. рис. 4.10), плотность поверхност-
ных зарядов должна быть равной приблизительно 1,0 SiO -
групп/нм2, что примерно совпадает с величиной, найденной на
стадии 1.
Стадия 2. При переходе от стадии 1 к стадии 2 цепочечные
молекулы плотно упаковываются, как показано на рис. 6.13, а,
и затем, на стадии 2, выстраиваются вертикально к поверхности.
При этом поверхность все более положительно заряжается по
мере того, как все большее число дополнительных ионов ДТМА+
начинает удерживаться на поверхности посредством гидрофоб-
ных связей со смежными цепями. Согласно Прусту и Тер-Мина-
сян-Шараге [331], каждая молекула, когда она адсорбируется
вертикально к поверхности, занимает площадь около 60 А2, что
соответствует поверхностной концентрации, равной 2,8 мкмоль/м2.
Это, вероятно, объясняет ступеньку при величине поверхностной
концентрации около 3 мкмоль/м2, наблюдаемую на рис. 6.13, а.
Однако так как такие дополнительные катионы присоединяются
в виде двойного слоя к поверхности, то они не могут нейтрали-
зоваться дополнительными поверхностными зарядами. Они дол-
жны иметь противоионы — анионы в растворе. Однако анализ по-
казывает отсутствие каких-либо адсорбированных ионов Вг~.
Такими противоионами должны быть силикатные ионы НБЮз,
которые неизбежно образуются в растворе при pH 10. Добавлен-
ные ионы ДТМА должны находиться в плотно упакованном слое,
причем у половины всех имеющихся в слое четвертичных аммо-
ниевых ионов концы направлены от поверхности, как это пока-
зано на рис. 6.12, г. Действительно, даже с самого начала про-
цесса адсорбции некоторые ионы ДТМА+ должны иметь катион-
ную N+’ГОЛОВку, направленную наружу от поверхности.
Такой монослой с чередующейся ориентацией ионов носит
название «спаренного (дуплексного) монослоя». Подобный мо-
нослой является одним из типов, предложенных Тер-Минасян-
Шарагой [326] для описания адсорбции димеров.
Однако возникает вопрос, почему для данного типа монослоя
противоионами оказываются ионы HSiO“, а не Вг-. Действи-
тельно, анализ показывает отсутствие адсорбированных ионов
Вг на этой стадии. Поскольку концентрация ионов Вг в рас-
творе превышает концентрацию ионов HSiOy, то должна-быть
некоторая стерическая причина, почему ионы Вг~ не распола-
гаются вблизи двойного слоя. Ниже будет показано, что в спа-
ренном монослое углеводородные цепи способны плотно упако-
вываться вместе, так как только каждая вторая углеводородная
цепь может совмещаться с поверхностью своей более широ-
кой катионной группой (CH3)3N+R, как показано на рис. 6.12, г.
Химия поверхности кремнезема 947
Плотность упаковки, или поверхностная концентрация в таком
спаренном монослое, следовательно, составляет около
3 мкмоль/м2, что соответствует 1,8 молекул/нм2. Площадь, за-
нимаемая одной молекулой на поверхности, составляет 55 А2.
К более просторному размещению этих молекул в слое лучше
приспосабливаются силикатные ионы.
Стадия 3. Как видно из рис. 6.13, а, на этой стадии начи-
нают адсорбироваться ионы Вг и формируется обычный двой-
ной слой. Ионы ДТМА+, находящиеся в спаренном слое, таким
образом, полностью перестраиваются, как показано на рис. 6.12, а.
На единицу площади (1 нм2) в положении 6.12, а приходится
большее число ионов ДТМА+, чем в положении 6.12, г. При зна-
чении поверхностной концентрации 2,0 мкмоль/м2 каждый ион
ДТМ.А+ занимает площадь 83 А2 и каждой молекуле, присоеди-
ненной к поверхности, соответствует одна молекула, повернутая
катионом наружу, в раствор, причем ближайшим ионом в рас-
творе оказывается ион Вг~.
При переходе от стадии 2 к стадии 3 дополнительно адсор-
бируется 1 мкмоль ионов ДТМА+, что приводит к повышению по-
верхностной концентрации от 3 до 4 мкмоль/м2, и одновременно
адсорбируются 2 мкмоль ионов Вг- на 1 м2.
Если поверхность образца кремнезема марки Cab-0-Sil ча-
стично дегидроксилирована и ее большая часть оказывается си-
локсановой и гидрофобной, то, как показано на рис. 6.13, б,
уже при низких концентрациях ионов ДТМА+ начинает форми-
роваться двойной слой, что обнаруживается по адсорбции ионов
Bi~. Вероятно, на силоксановых участках поверхности адсор-
бируется некоторое количество ионов ДТМА+, причем гидрофоб-
ные концы ионов обращены к гидрофобной силоксановой по-
верхности, а катионы направлены в раствор.
В заключение Бийстербош продемонстрировал, что в присут-
ствии н-бутилового спирта на поверхности кремнезема с гидро-
ксилированной и с частично дегидроксилированной поверхно-
стью адсорбируется монослой с концентрацией примерно
2,0 мкмоль/м2. По-видимому, это соответствует максимальной
плотности упаковки ионов ДТМА+ на поверхности, когда кати-
онные (СНз)зЖ-группы обращены к поверхности (см.
рис. 6.12, б).
Совершенно ясно, что в этой системе главную роль играют
гидрофобные связи. Приведенные в табл. 6.5 численные данные,
полученные из графических зависимостей Бийстербоша, показы-
вают, что максимальные значения поверхностных концентраций
на кремнеземах достигались для ионов ЦТМА п ДТМА на обоих
указанных типах кремнеземных поверхностей как в случае вод-
ной фазы, так и 1 М водного раствора н-BuOH. Эти данные
также показывают, что в присутствии молекул спирта м-ВпОН
34*
948
Глава 6
Таблица 6.5
Максимальная поверхностная концентрация адсорбционных катионных ПАВ
при различных значениях pH (по данным [330])
рн Концентрация ДТМА, мкмоль/м2 Концентрация LIT.VLA, мкмоль/м2
в воде в к-ВиОН в воде в «~ВиОН
Кремнезем «кабосил»
8 1,4 0,2 2,8 0,45
9 3,2 0,7 4,5 1,2
10 4,5 2,0 4,5 2,5
BDH-кремнезем
8 3,9 1,7 5,3 1,3
9 4,0 2,0 ' 5,4 1,4
10 4,0 2,0 5,5 .2,0
основным фактором, вызывающим адсорбцию, оказывается при-
сутствие зарядов на поверхности кремнезема, притягивающих
катионы с длинными цепями. В воде проявляется дополнитель-
ный фактор — стремление гидрофобных катионов, если из них
не сформировались мицеллы посредством гидрофобных связей,
избавиться от воды и повернуться катионной головкой к поверх-
ности кремнезема. Это четко прослеживается при pH 8, когда
проявляется гораздо более сильная адсорбция из водной фазы,
чем адсорбция из смеси бутанол—вода, так как основным фак-
тором в последнем примере оказывается только слабый поверх-
ностный заряд (см. табл. 6.5). Длинноцепочечные ионы ЦТМА+
проявляют более сильную тенденцию к освобождению от воды
с образованием мицелл.
Примеры, когда суммарная адсорбция ионов из воды дости-
гала 4,5 и 5,5 мкмоль/м2, т. е. адсорбированное количество было
больше по сравнению с нормальным двойным слоем, могут быть
объяснены более высокой плотностью упаковки во внешней части
двойного слоя, достигаемой в результате ступенчатого располо-
жения расширенных катионных групп при вертикальном напра-
влении ионов к кремнеземной поверхности.
При адсорбции четвертичных аммониевых ионов ионное при-
тяжение оказывается вторичным фактором, что также было по-
казано Блекманом и Хэрропом [332]. Они наблюдали, что ионы
типа RN (СН3)* , где R — алифатическая цепь, могли легко смы-
ваться с поверхности кремнезема, если R-группа оказывалась
короче, чем Се. Подобным образом для цепей С12—Cis было за-
мечено понижение точки к. к. м. примерно в 15 раз, и, как по-
Химия поверхности кремнезема
949
казано Бийстербошем, концентрация, необходимая для форми-
рования монослоя на поверхности кремнезема, понижается по
крайней мере в 8 раз. Механизм адсорбции четвертичных аммо-
ниевых ионов на кремнеземе все еще остается спорным. Блек-
манн и Хэрроп пришли к заключению на основании исследова-
ний ИК-спектров, что ионный обмен в таких случаях не проис-
ходит; вместо него наблюдалось ослабление полосы поглощения
групп SiOH, находящихся на поверхности кремнезема, которое
показывает, что атом кислорода силанольной группы связывался
до некоторой степени с Ж-катионом. Последнее напоминает
своеобразный комплекс хлоргидрата диэтиланилина и кремневой
кислоты (см. лит. в гл. 3 [170]). Конечно, вполне возможно, что
при низком значении pH ионный обмен не включается в меха-
низм адсорбции; однако выше pH 8 ионообменный механизм
представляется убедительным.
Применение катионных ПАВ при отделении кремнезема от
руд посредством пенной флотации типично для технологических
процессов, как это показано Хедбергом [333] в приведенном им
сравнении методов обогащения различных типов руд.
Эффекты гидрофобизации
В исследовании О’Коннора и Сандерса [334] было обнару-
жено, что очень разбавленный раствор четвертичного аммоние-
вого ПАВ придавал поверхности кварцевого стекла гидрофоб-
ный характер при опускании образца в раствор и последующем
высушивании. После того как раствор стекал, поверхность сразу
же становилась сухой и водоотталкивающей. Но когда исполь-
зовался более концентрированный раствор, поверхность не ста-
новилась гидрофобной до тех пор, пока не смывался избыточный
раствор. Авторы обнаружили следующие соотношения между
концентрацией, выше которой поверхность оставалась гидро-
фильной, когда образец извлекался из раствора (это показы-
вало, что на поверхности адсорбировался двойной слой), и кон-
центрацией, выше которой происходило формирование мицелл
в растворе (точка к. к. м.):
Длина цепи Минимальная концентрация в растворе
при образовании при формировании
двойного слоя на мицелл в растворе
кварцевом стекле
Додецил С12 0,032 0,0016
Цетил Сю 0,0001 0,0010
Таким образом, в случае катионов с цепью Ci6 двойной слой,
который является просто адсорбированной мицеллой, форми-
950
Глава 6
руется при гораздо более низких концентрациях, чем значения
к. к. м. Для катионов с целью Ci2 положение меняется на про-
тивоположное.
Катионные красители
Катионные красители по своему поведению подобны катион-
ным ПАВ: образуют мицеллы в растворах и сильно адсорби-
руются на силанольной поверхности. Согласно данным- Эллин-
гема и др. [335], молекулы катионных красителей адсорби-
руются посредством ионообменного механизма вплоть до обра-
зования монослоя. Если концентрация превышает к. к. м., то
краситель дополнительно адсорбируется в виде мицелл, которые
могут образовать покрытие на поверхности кремнезема толщи-
ной вплоть до 50 монослоев. Если образец кремнезема промы-
вается, то на поверхности удерживается лишь монослой. Было
обнаружено, что для таких основных красителей, как метилено-
вая синь, кристаллический фиолетовый, малахитовая зелень
и некоторых других, адсорбционное равновесие достигалось
в течение 10 мин. Образование мицелл на поверхности кремне-
зема обнаруживалось по смещениям спектральных полос ИК-
поглощения.
Поскольку адсорбция носит главным образом ионный харак-
тер, значение pH должно быть достаточно высоким, чтобы вы-
звать заметную ионизацию силанольных групп и способствовать
адсорбции монослоя красителя. Саукель [336] обнаружил, что
такое значение pH составляло около 6. Уникальный органиче-
ский основной краситель, который действует в качестве индика-
тора на группы SiOH в присутствии других органических
веществ, содержащих гидроксильные группы, на поверхности бу-
маги, описывался в гл. 4 (см. лит. к гл. 4 [561, 562]). Лейкоос-
нование, рассматриваемое ниже, претерпевает структурные из-
менения в процессе адсорбции на поверхностных группах SiOH,
и его окраска изменяется из бесцветной в голубую. Этого не
происходит в присутствии воды или спиртов, которым оказы-
вается предпочтение при образовании водородных связей
с группами SiOH. Кроме того, такое вещество остается бесцвет-
ным в присутствии хлороформа, атом водорода которого играет
роль акцептора электронов. Когда краситель вводится в кон-
такт с кремнеземом в тетрахлориде углерода, то образуется го-
лубое окрашивание. Если же образец смачивается спиртом или
водой, то голубой цвет исчезает. Окрашивание появляется вто-
рично при испарении растворителя. Это соединение 3,3-бис (п-ди-
Химия поверхности кремнезема
951
этиламинофенил) -6-диметиламинофталид, или кристаллический
фиолетовый лактон, имеет структурную формулу
Алюмосиликатные поверхности
Необходимо упомянуть о заметном различии между алюмо-
силикатной поверхностью (многие неорганические силикаты,
глины) и поверхностью кремнезема. Даже очень небольшие ко-
личества алюминия на поверхности кремнезема приводят к по-
вышению отрицательного поверхностного заряда и существенно
видоизменяют адсорбцию на ней катионных веществ (см.
рис. 4.26 и лит. к гл. 4, [324, 410—419]). Именно по этой причине
в случае глин особенно эффективными оказываются длинноце-
почечные четвертичные аммониевые ионы при приготовлении
органофильных загустителей консистентных смазок и других ор-
ганических жидкостей.
Углеводородные группы, присоединяемые
посредством поливалентных металлов
Комплексные катионы металлов необратимо адсорбируются
на поверхности кремнезема. Так, карбоксилатный комплекс
хрома адсорбируется на кремнеземе с образованием монослоев:
4СГ
Если R — длинноцепочечная алкильная группа (например, сте-
аратогруппа), то поверхность кремнезема оказывается в сильной
степени гидрофобной [337, 338]. Каждая стеаратогруппа зани-
952
Глава 6
мает площадь 30 А2. Когда RCOO представляет собой метакри-
латогруппу, то подобное соединение, адсорбированное на стек-
ловолокне, оказывает антистатическое действие, т. е. снимает
статические электрические заряды, а также играет роль свя-
зующего между исходной смолой, взятой для изготовления слои-
стого пластика, и поверхностью стекловолокна [339].
Монослои карбоновых кислот также могут накладываться на
поверхность кварцевого стекла в два этапа: вначале проводится
адсорбция многозарядных ионов металла на поверхности
кремнезема, а затем обработка образца мыльным щелоком. Ис-
пользуются такие металлы, как кальций, барий или магний
[340]. Полученная таким путем поверхность кремнезема гидро-
фобна, поэтому на нее можно повторно наносить покрытия
в процессе флотации добавлением извести с последующим вве-
дением стеарата натрия [341]. Гаудин и Фурстенау [342] пока-
зали, что в процессе флотации кварца ионы бария, адсорбиро-
ванью в слое Штерна, затем адсорбировали лаурат-ионы, кото-
рые превращали поверхность кварца в гидрофобную; в этом
процессе барий получил название «активатора». Флотация крем-
незема из руд имеет важное промышленное значение. Ионы
кальция используются в качестве «активатора» для флотации
кремнезема с добавлением мыльного щелока. Интересно, что
стеарат-ионы должны также сообщать железной руде гидрофоб-
ный характер, и, таким образом, руда будет всплывать с пеной
в процессе флотации. Однако, если вначале добавляется крах-
мал, то он, адсорбируясь на оксидах железа, сохраняет их гид-
рофильность и, таким образом, может понижать флотируемость
руды. Вероятно, поликарбоксильные группы в крахмале (или
в окисленном крахмале), присоединенные к поверхности оксида
железа в большом числе точек, не могут замещаться стеарат-
ионами, которые гидрофобЛ и несут точно такой же по знаку
заряд, что и крахмал, поэтому не способны проникать сквозь
толстый гидрофильный анионный слой адсорбированного крах-
мала [343].
Вейл и Марбо [344] обсудили целый ряд интересных прояв-
лений гидрофильности кварцевого стекла. Они отмечают, что
термин «чистая поверхность» кварцевого стекла обычно озна-
чает, что такая поверхность полностью гидрофильна. Возникаю-
щая гидрофобность в значительной мере обусловливается при-
сутствием адсорбированных многозарядных ионов металла. Если
такие ионы находятся на поверхности, то жирные кислоты, следы
которых всегда присутствуют в виде паров в воздухе, способны
всего лишь за несколько часов адсорбироваться, превращая по-
верхность в гидрофобное состояние. Авторы рассмотрели также
роль органических кислых комплексов Сг3+ на модифицирован-
ных поверхностях кварцевого стекла.
Химия поверхности кремнезема
953
Образование эфиров на поверхности кремнезема
В гл. 5 кратко уже рассматривались силикагели и кремне-
земные порошки с этерифицированной поверхностью.
Реакция этерифицирования, если она проводится до конца,
превращает гидрофильную поверхность кремнезема в полностью
гидрофобную, как это было описано Айлером [345]. Условия
проведения таких реакций в дальнейшем были раскрыты Бел-
лардом и др. [346]. Краситель метиловый красный адсорби-
руется на группах SiOH, но адсорбция падает до нуля, когда
всего лишь некоторая часть групп SiOH была этерифицирована.
Ловен и Броудж [347] дали следующее объяснение этому факту.
Когда приблизительно три «-бутоксигруппы присоединялись
к поверхности, то их размер оказывался таким, что непрореаги-
ровавшие группы SiOH становились как бы отгороженными та-
кими группами и поэтому нереакционноспособными. Кроме того,
более низкая теплота адсорбции азота (т. е. низкое значение
константы С в уравнении БЭТ) показывает, что молекулы азота
не могут достичь групп SiOH, лежащих ниже м-бутоксигрупп,
несмотря на то, что присутствие групп SiOH подтверждается
анализом по методу сжигания, поскольку анализ показывает
присутствие избыточного водорода по сравнению с тем, которое
соответствует группам С4Н3.
Покрытие поверхности
Количество молекул спирта, которые должны вступать в ре-
акцию с поверхностью и образовывать завершенный монослой,
зависит от размера и формы алкильных групп. Так, например,
разветвленная «кустистая» группа будет охватывать большую
площадь, чем группа с нормальной прямой цепью, как это пока-
зано на рис. 6.14. Число эфирных групп, требуемое для покрытия
заданной площади, можно оценить из экспериментально уста-
новленного факта, согласно которому каждая молекула спирта
будет-покрывать площадь 0,14/г нм2, где п — число разветвле-
ний». Последнее определяется как максимальное число таких
атомов углерода, каждый из которых отделяется от атома кисло-
рода равным числом атомов углерода. Это соответствует рас-
стоянию между углеводородными группами в наиболее широком
месте, если бы такую молекулу удалось развернуть на плоскости
(рис. 6.15; наибольшее расстояние ограничено стрелками). От-
меченное соотношение оказывается справедливым для значений
п, превышающих 2. Когда имеется только одно ответвление, как,
например в «-бутаноле, каждая бутоксигруппа покрывает пло-
щадь около 32 А2. Если п = 5, как в случае 5,7,7-триметил-2-
954
Глава 6
Рис. 6.14. Схематическое изображение поверхности кремнезема, этерифициро-
ванной спиртом Си с сильно разветвленной цепью (вверху) и «-бутиловым
спиртом (внизу). (По данным Айлера [3], с разрешения Cornell University
Press.)
Химия поверхности кремнезема
955
(1,3,3-триметилбутил) -1 -октанола, то
каждая группа, как было определено, по-
крывает площадь около 70 А2.
Показано, что площадь, покрываемая
одной н-бутокси-группой, очень близка
к площади, занимаемой одной молеку-
лой н-бутиламина, адсорбированной из
газовой фазы (33 А2, [348]), но превы-
шает площадь, занятую одной молекулой
н-бутанола в монослое на поверхности
воды, которая оказалась равной 23,7 А2
[349].
Таким образом, безусловно, не каж-
дый атом кремния на поверхности спосо-
бен вступать в реакцию с алкоксигруп-
пой вследствие стерических ограничений.
Как отмечал Гаркинс [349], даже в слу-
чае физически адсорбированных поляр-
ных молекул, «присутствие поверхностной
с с с
хс/
— с с с с с
\ \ • /
с с
с с с
с с
\ /
с
с
Рис. 6.15. Углеродный
скелет разветвленной ал-
кильной Группы Cig,
имеющей «число развет-
влений», равное 5. (По
данным Айлера [3], с
разрешения Cornell Uni-
versity Press.)
полярной группы де-
лает местоположение молекулы весьма зависимым от разновид-
ности кристаллической решетки на поверхности, на которой про-
исходит адсорбция, ввиду чего должны происходить колебания
толщины адсорбционной пленки».
Штобер, Бауэр и Томас [350] измерили максимальное число
RO-групп, которое можно было этерифицировать на единице
площади (1 нм2), и получили следующие результаты: СН3О 4,7;
С2Н5О 3,7; «-С3Н7О 3,5; Н-С4Н9О 3,2; н-СвНпО 3,8 группы. Такая
степень этерификации достигалась в течение 6 ч при 200°С в ав-
токлаве при обработке кремнезема в безводном спирте. В связи
с измерениями растворимости аморфного кремнезема в спиртах
при высоких температурах и давлениях Китахара и др. [351,
352] сообщили о том, что поверхность полностью этерифициро-
валась этанолом и пропанолом при 200°С, в результате чего по-
верхностные концентрации становились равными 3,0 группы ЕЮ
и 3,0 группы РгО в расчете на 1 нм2. Ряд других исследований
по этерификации поверхности кремнезема различными спиртами
был проведен Фрипья и др. [353].
Степень покрытия поверхности алкоксигруппами может быть
определена по адсорбции паров воды, которые, как отмечалось,
адсорбировались только на чистой силанольной поверхности
[354]. Макеева, Николаева и Болотова [355] разработали метод
измерения гидрофобности с воспроизводимостью получаемых
значений до 1 %, который также заключался в определении сте-
пени этерификации [355].
Цуцуми и Такахаси [356] применили широкий набор методов
при исследовании поверхности, этерифицированной «-бутиловым
956
Глава 6
спиртом. Методом ИК-спектроскопии они подтвердили, что
группы SiOH заменялись на группы SiOBu. Кроме того, теплота
смачивания этерифицированной поверхности кремнезема в воде
понижалась на 50—70 % от значения теплоты смачивания гидро-
ксилированной поверхности.
Киселев и др. [357] сообщили об этерификации поверхности
резорцином и гликолем с образованием мозаичной структуры та-
кой поверхности. Резорциновая поверхность адсорбировала гек-
сан, азот и аргон более сильно, чем это наблюдалось для гли-
колевой поверхности, вероятно, из-за наличия в первом случае
ароматических колец.
Условия проведения реакций
Этерификация как пример необратимой адсорбции спиртов
исследовалась Штобером [358] и Беляковой и Киселевым [359].
Как первоначально было обнаружено Айлером [345], изучение
степени этерификации в процессе обработки разными спиртами
при различных условиях орошения и температуре пара 150°С,
а также в автоклаве при 250°С показало, что полная этерифика-
ция возможна только при условиях, создаваемых в автоклаве
или при высокой температуре орошения [360—362].
Действие, оказываемое различающимися по структуре изо-
мерами пентанола, определяет скорость и величину хемосорбции
(этерификации) кремнезема, дегидратированного предвари-
тельно до разной степени. Активность изомеров понижается
в следующем порядке: нормальный, изо-, вторичный, нео- и тре-
тичный изомеры; в работе [363] также обсуждается механизм
действия этих изомеров.
Воздействие воды на процессы хемосорбции или этерифика-
ции поверхности кремнезема было измерено Бауэром и Штобе-
ром [364] при работе с четырьмя низшими спиртами при 250°С.
Небольшие количества воды значительно снижали степень эте-
рификации.
Симпсон и Дойль [365] предложили упрощенную процедуру
приготовления гидрофобного силикагеля. Влажный стандарт-
ный силикагель измельчали в мельнице с гидроприводом до
размера частиц 1—20 мкм и погружали при 100—215°С в «-де-
циловый спирт. Проходила экзотермическая реакция, после чего
избыточный спирт удаляли испарением.
Реакция этерификации промотировалась слабыми основа-
ниями, такими, как амины; но если амины содержались в про-
дуктах гидролиза, то также наблюдалось ускорение реакции
этерификации [366]. Влияние полярности групп и непредельно-
сти алкильной цепи на скорость реакции оценивалось в ра-
боте [367].
Химия поверхности кремнезема
957
Спирт, используемый для получения гидрофобного силика-
геля посредством автоклавной обработки, не должен быть пол-
ностью безводным, если пары сбрасываются при высокой тем-
пературе. Броудж [368] предложил способ модифицирования,
по которому силикагель суспендировался в водно-спиртовой
смеси, имевшей массовое соотношение в интервале от 2,38 : 1 до
19 : 1 и нагревавшейся до 270—375°С перед тем, как давление
сбрасывалось, а система охлаждалась в контакте с воздухом.
Очевидно, хлористый водород также является катализатором
процесса этерификации и может иметь то преимущество, что не
адсорбируется на получаемом продукте и не гидролизует его.
Гросс [369] этерифицировал пирогенный кремнезем в потоке
газа, содержащего хлористый водород и алифатический спирт.
Ряд исследователей этерифицировали кремнезем в смеси из
спирта и углеводорода. Так, в работе [370] применяли цетило-
вый спирт и додекан в условиях орошения, причем вода непре-
рывно отделялась от конденсата [370]. Кремнезем этерифициро-
вали также в смеси спирта с гексаном [377]. Было установлено,
что третичные спирты оказываются не способными этерифициро-
вать кремнезем. Эксперименты, выполненные Айлером, пока-
зали, что грег-бутиловый спирт, например, быстро разлагается
до бутена и воды.
Росси, Мунари и Ценгари [372] сообщили о кинетических ис-
следованиях процесса этерификации поверхности выбранного
ими кремнезема различными спиртами. Спирты с неразветвлен-
ными цепями с числом атомов углерода от 3 до 16 испытывались
посредством автоклавной обработки кремнезема при избыточ-
ном количестве спирта и температуре 200°С в течение 0,25—4 ч.
Реакция SisOH + ROH= SisOR + H2O была первого порядка, кон-
станты скорости реакции менялись от 1,37-10~2, причем мини-
мальное значение константы проявлялось для цепи С8. Энергия
активации составляла 17—20 ккал/моль. Чуйко и др. [373] изу-
чали кинетику реакции при использовании н-бутилового спирта.
Они сообщили, что энергия активации подобной реакции соста-
вляла 13 ккал/моль для полностью гидроксилированного крем-
незема, но становилась меньше после дегидратации поверх-
ности.
Этерифицированный метоксигруппами кремнезем
Метильные группы оказываются единственными достаточно
небольшими по размеру группами, способными вступить в реак-
цию со всеми группами ОН па поверхности кремнезема при их
концентрации, равной 4,6 ОН-групп/нм2. Броудж [375] нашел,
что повторная автоклавная обработка с применением безводного
метанола приводила при 290°С к такой стадии этерификации
958
Глава 6
частиц кремнезема диаметром 17 нм, когда весь продукт стано-
вился гидрофобным и содержал 5,8 МеО-групп/нм2. Штобер^
Бауэр и Томас [350] сообщили о присоединении 4,7 МеО-
групп/нм2.
В том случае, когда поверхность только частично этерифи-
цирована, оставшиеся группы SiOH могут быть превращены
в группы SiCl по реакции с SOC12. Было сообщено [374], что
суммарное число групп SisOCH3 + SisCl остается постоянным.
Асано и Китахара [376] приводят данные о том, что, когда крем-
незем вначале дегидратировался при 500°С, а затем обрабаты-
вался метанолом в автоклаве, было обнаружено пять радикалов
МеО на 1 нм2 поверхности кремнезема.
Согласно данным Мертенса и Фрипья [377], реакция мета-
нола с аэрогелем, вероятно, проходит через образование прото-
нированного промежуточного соединения СН3ОН^ . Авторы изу-
чили кинетику такой реакции при 150—190°С и показали, что
происходит этерификация групп SiOH, а образующаяся вода
затем разрывает силоксановые связи.
В качестве реагента для метилирования исследовался диазо-
метан, но оказалось, что реакция этерификации завершается не-
полностью, причем имеется опасность взрыва [8].
Термический распад групп SisOCH3 наблюдается примерно
при 500°С, как это показано Абаяши и др. [378], обнаружив-
шими методом ДТА экзотермический эффект при этой темпера-
туре.
Реакция спиртов с дегидроксилированной
поверхностью
Гобель [379] высказал предположение, что частично дегидро-
ксилированная поверхность кремнезема действует подобно ан-
гидриду кислоты. Физически адсорбированные молекулы спирта
вступают в реакцию даже при 25°С с образованием поверхно-
стных групп эфира. Для того чтобы это произошло, необходимо
активировать поверхность, поддерживая оптимальную темпера-
туру дегидратации при ~550°С. При такой температуре на по-
верхности остается примерно 2 группы SiOH на 1 нм2, а полу-
чаемый по реакции конечный продукт содержит 2 «-ВиО-
группы/нм2, однако механизм реакции неизвестен. Мертенс [380]
методом ИК-спектроскопии получил данные, подтверждающие,
что в процессе такой реакции идет вскрытие силоксановых мо-
стиков.
Полностью безводная растрескавшаяся поверхность также
вступает в реакцию со спиртом, если последний присутствовал
во время образования трещин. При помоле массивного куска
Химия поверхности кремнезема
959
кремнезема в бутаноле происходит образование гидрофобного
порошка [381].
При реакции метанола с частично дегидратированной по-
верхностью кремнезема происходит этерификация оставшихся на
поверхности групп SiOH, а затем новые порции метанола будут
реагировать с силоксановыми участками поверхности [382].
На дегидратированной поверхности пористого стекла, которое
содержало 2 °/о В2О3, метанол будет связываться водородными
связями с группами SiOH и ВОН, и даже при 30°С происходит
реакция с напряженными силоксановыми кислородными мости-
ками. При 200°С идет формирование как групп BSOCH3, кото-
рые образуются в результате расщепления исходных групп, так
и групп SisOCHs [383]. Как было показано Кунатом [384], сило-
ксановые мостики, возникающие в результате дегидратации по-
верхности при 500°С, вступают в реакцию с CD3OH при 25°С.
Когда этерифицированный кремнезем нагревают, то при 450°С
образуется полиметилен
SisOCD3-> SisOD + (CD2)„
Реакция углеводородов с дегидроксилированной
поверхностью
По-видимому, недостаточно внимания было уделено откры-
тию, сделанному в 1955 г. Броуджем [385], показавшим, что не-
насыщенные по концевым группам алифатические углеводороды
вступают в реакцию с поверхностью кремнезема, которая дегид-
ратировалась при 400—700°С и имела примерно 2 ОН-группы/нм2.
Реакцию с октаном-1 проводят в течение нескольких часов по
способу орошения. При этом на 1 нм2 поверхности обнаружи-
вается приблизительно одна группа октила, которая присоеди-
няется к поверхности после того, как весь непрореагировавший
октан был удален. Получаемый в конечном итоге продукт
в основном оказывается органофильным и остается в «-бутаноль-
ном слое после встряхивания смеси с водой.
Происходят также реакции между термически активирован-
ной поверхностью и изобутиленом, диизобутиленом и триизобу-
тиленом. В последнем случае по крайней мере две двойных связи
должны вступать в реакцию, так что молекула оказывается рас-
положенной плоско на поверхности кремнезема, поскольку зна-
чение поверхностной концентрации составляет всего лишь
0,85 молекул/нм2, и конечный продукт получается высокогидро-
фобным. Возможно, что последующие исследования в указанных
направлениях приведут к получению устойчивого гидрофобного
кремнезема при минимальном содержании углеводородных
групп на поверхности. Некоторые предельные углеводороды
также можно использовать для получения гидрофобной поверх-
960
Глава 6
ности, если кремнезем, находящийся в контакте с углеводородом
в воздушной среде, нагревать ниже точки кипения органического
соединения. Бойлан [386] предложил способ, согласно которому
смесь, состоящая из 10 % кремнезема и белого парафинового
масла, нагревается на воздухе при перемешивании и темпера-
туре 250°С в течение 1 ч и затем охлаждается. После удаления
масла гексаном и высушивания образуется гидрофобный поро-
шок. Неизвестно, происходило ли на первом этапе процесса
окисление масла до спирта или же совместное дегидрирование
и окисление масла до алкена. Подобная смесь масла с кремне-
земом может с успехом применяться в качестве антипенной при-
садки. Как катализаторы могут использоваться микроколичества
нафтенатов Со, Мп или РЬ.
Этерификация в микропорах
Саукель [387] описал необычную форму этерифицировацного
кремнезема. Лепидоидальный кремнезем, о котором упомина-
лось в гл. 1 и 5, представляет собой микропористый силикагель,
имеющий форму тонких листочков, сформировавшихся путем за-
мораживания кремневой кислоты с низкой молекулярной массой.
Такой кремнезем удерживает примерно одну группу ОН в рас-
чете на один атом кремния, как этого и можно было ожидать
для первичных кремнеземных частиц, имеющих всего лишь не-
сколько нанометров в диаметре. Этерификация приводит к полу-
чению образца, для которого характерным является отношение
МеО : Si, равное 1 : 3, а при применении высших спиртов соот-
ношение RO : Si составляет 1:4.
Если принять, что группа РгО (или более высокая алкокси-
группа) покрывает площадь 0,33 нм2 и имеется 4 единицы SiO2
в расчете на 1 алкоксигруппу/то удельная поверхность такого
кремнезема составит 820 м2/г. Это соответствует размеру первич-
ных кремнеземных частиц около 3,4 нм в диаметре, что является
вполне подходящим для силикагелей, приготовленных из крем-
невой кислоты с низкой молекулярной массой при низких pH
температурах.
Замещенные спирты
В литературе упоминаются следующие замещенные спирты:
3-монохлорпропанол, п-крезол [388]; R/OR"OH, C1R"OH,
NO2R"OH, ROC(O)R"OH (где R' СиН2та+1 и R" (СН2)„ [389];
NH2C2H4OH [390, 391]; алициклические спирты CnH2n-iOH [392]
и галоидсодержащие спирты X(CF2)„CH2OH [393]. Очевидно,
Химия поверхности кремнезема
961
некоторые из этих соединений не способствуют образованию гид-
рофобных поверхностей.
Гидролиз эфирных групп
Устойчивость покрытий, содержащих эфирные группы, к гид-
ролизу заметно возрастает по мере того, как углеводородные
группы на поверхности достигают состояния плотной упаковки,
когда ни одна из силанольных групп, находящихся под эфир-
ными группами на поверхности, не будет способной поддержи-
вать адсорбцию воды. Такое покрытие оказывается очень ста-
бильным по отношению к процессу гидролиза до тех пор, пока
не образуются незащищенные («голые») места. Однако следует
отметить, что большинство силикагелей и порошков содержит
агрегаты, составленные из частиц сферической формы, которые
связываются в местах контакта. После этерификации поверхно-
сти частицы могут отделяться друг от друга при механическом
воздействии, при этом обнажаются пятна в местах разрыва, ко-
торые затем и подвергаются гидратации. В таких пятнах начи-
нается гидролиз углеводородного покрытия, который протекает
затем со всевозрастающей скоростью. Бауэр и Штобер [394]
также обнаружили «автокаталитический» ход гидролиза.
Этерифицированная поверхность оказывалась достаточно
стабильной, когда такие типы кремнезема использовались в ка-
честве консистентных смазок, но при применении этерифициро-
ванных образцов кремнезема для армирования эластомеров,
вероятно, требуется гораздо большая стабильность, рассчитан-
ная на долгосрочное действие. Возможно, что предварительное
покрытие поверхности группами (СНз)3БЮ (до 10—20%) перед
ее этерификацией бутиловым спиртом предотвратит распростра-
нение гидролиза и поможет создать более устойчивую гидро-
фобную поверхность с минимальными затратами средств. При
этом предполагается, конечно, что связи (СН3)зБ1—О—Sis более
\ устойчивы в отношении гидролиза, чем связи я-С^эОББ. Од-
нако необходимо вначале установить, насколько это окажется
справедливым. Айлер наблюдал, что при полностью этерифици-
рованной поверхности, на которой достигалась максимальная
плотность упаковки алкильных или других углеводородных
групп, гидролиз протекал очень медленно даже в кипящей воде.
Но в конце концов гидролиз начинается, что можно установить
по потере содержания углерода или по повышению адсорбции
красителя метилового красного. Скорость удаления алкокси-
групп увеличивается до тех пор, пока не будет удалена при-
мерно половина всех групп; когда групп остается намного
меньше, скорость понижается. Экспериментальные данные по-
казывают, что гидролиз происходит на границе между этерифи-
35 Заказ № 250
962
Глава 6
цированным и гидроксилированным участками, и как только
этот процесс начинается в отдельных местах в виде пятен, то
гидролиз ускоряется по мере того, как каждое гидрофильное
пятнышко вырастает по периферии.
Авторы работы [395] измерили устойчивость этерифицирован-
ной поверхности, полученной по реакции с «-октанолом, причем
эксперименты проводились при 2 % влажности и 190—250°С.
Гидролиз при этой температуре влечет за собой протекание ре-
акции SisOR + SisOH с образованием ROH и SisOSis. Согласно
Хаскину [396], гидролиз сложных эфиров кремневой кислоты
в воде протекает следующим образом:
SisOEt + Н18ОН = Si[8OH + EtOH
Это согласуется с результатами, описанными в работе [397],
в которой были использованы оптически активные вторичные
спирты для доказательства того, что реакция трансэтерифика-
ции происходит с расщеплением связи Si—О.
Термическая стабильность различных типов эфирных групп,
например разветвленных, ненасыщенных бензильных и диоль-
ных, определялась Уцуги и Хорикоси [398а], которые приме-
няли методы дифференциального термического анализа и термо-
гравиметрии. Поверхностные группы окислялись на воздухе при
температурах выше 200°С. Термический распад зависел от раз-
новидности спирта. Образовавшийся при 200—400°С углерод
окислялся при нагревании до 400—600°С.
Ланге [3986] наблюдал, что метоксигруппы на поверхности
распадались на воздухе при 235°С, а н-бутоксигруппы удалялись
при 215°С. Гидрофильная поверхность, этерифицированная эти-
ленгликолем или 1,2-пропиленгликолем, испытывала термора-
спад при 225 и 250°С соответственно. По-видимому, термическая
стабильность не столь значительно зависит от природы присо-
единенных к поверхности органических групп.
Органические группы, присоединенные
посредством связей С—Si
Перечень большинства поверхностных реакций с силаноль-
ными группами был представлен выше в данной главе. Однако
были отражены не все типы реакций, посредством которых на
поверхности кремнезема, а также на поверхности стекла могут
формироваться гидрофобные и органофильные покрытия. Ней-
марк [399] представил перечень сорока типов органосилильных
групп на поверхности кремнезема.
Химия поверхности кремнезема
963
Соединения типа R3SiX, R2SiX2 и RSiX3 (где X — галоид)
вступают в реакции с поверхностными группами SiOH, образуя
хемосорбированный слой, состоящий из органосилильных групп.
В частности, для проведения подходящей реакции оказывается
полезным соединение (CH3)3SiNHSi (СН3)3, поскольку оно обра-
зует с адсорбированной водой только NH3 и летучее соединение
(CH3)3SiOSi (СН3)3, а с группами SiOH формирует на поверхно-
сти группы SisOSi(CH3)3 и аммиак NH3. Хайр и Хертл [400] по-
казали, что вступают в реакцию именно те поверхностные
ОН-группы, которые не связаны попарно водородными связями.
Энергия активации реакции составляет 18,5 ккал/моль. Скорость
реакции равна А[ехр(—18 500/RT)] [ОН]2017, где А — константа,
[ОН] — доля групп ОН, присутствующих на поверхности в дан-
ный момент времени, и 9 — доля групп ОН, на которых к этому
же моменту времени размещены молекулы физически адсорби-
рованного реагента. Только лишь небольшая доля находящихся
на поверхности парных групп ОН, взаимно связанных водород-
ными связями, в конечном счете вступает в реакцию.
Боксаньи, Лиардон и Ковац [18] опубликовали обширный
обзор по реакциям н-алкилдиметилсиланолов и н-оксиалкилси-
ланолов с кремнеземной поверхностью. Этот обзор охватывает
большую часть вопросов, касающихся получения органосилиль-
ных покрытий, особенно на поверхности пирогенных типов крем-
незема, с целью формирования монослоя, состоящего из углево-
дородных цепей длиной вплоть до 20-атомов углерода.
Хайр [401] изучил кинетикуреакций ClnSi (СН3)4-И (п 1—4),
(CH3Si)2NH, GeCU и BCR с поверхностью пирогенного крем-
незема, который предварительно нагревался до 800°С для устра-
нения микропористости. После нагревания на поверхности оста-
вался только один вид свободных групп ОН, валентные
колебания которых идентифицировались по острому пику ИК-
поглощения при 3747 см-1. Предполагалось, что такие реагенты
будут вступать в реакцию второго порядка на поверхности крем-
незема, но на самом деле оказалось, что 40 % поверхностных
групп вступали в реакцию первого порядка
SisOH + CI2S1 (СН3)2= SBOSi (Cl) (СН3)2
Оставшиеся 60 % гидроксильных групп вступали во взаимодей-
ствие по реакции второго порядка
(SisOH)2 + CI2Si (СН3)2 = (SisO)2 Si (СН3)2
В том случае, когда с поверхностью кремнезема реагировал
ClSi(CH3)3, 80 % всех гидроксильных групп вступали в реакции,
отличавшиеся от реакции первого порядка. Даже если все ис-
35*
964
Глава 6
ходные группы SiOH энергетически равноценны, очевидно, их
распределение по поверхности различно, некоторые из них ока-
зываются близко расположенными, и при этом образуются пар-
ные группы ОН, на которых может протекать реакция второго
порядка.
Авторы работы [402] описали наблюдавшуюся ими необыч-
ную одностороннюю поверхностную реакцию. Она протекала при
взаимодействии хризотил-асбеста с Me3SiCI и HCI в среде про-
панола. Волокна асбеста, которые имеют диаметр только 20—
40 нм, как известно, представляют собой полые трубочки, обра-
зуемые в результате свертывания ленточки по ее длине. Обра-
ботка кислотой ведет к удалению магния из хризотил-асбеста,
и по мере того, как ленточка разворачивается, Me3SiCI вступает
в реакцию только лишь с той одной стороной этой силоксановой
ленточки, на которой размещаются группы SiOH. Эта сторона
становится частично покрытой группами SiOSi(CH3)3. В хло-
роформе такие ленточки набухают и раскручиваются, но после
того, как растворитель испаряется, ленточки скручивается
снова.
Силикагель, приготовленный в результате покрытия поверх-
ности группами SisOSi(C3He)NH3CI, представлял собой адсор-
бент, на котором наблюдалась обратимая адсорбция SO2, удер-
живаемый на поверхности в виде ионов SO2C1~ [403].
Большое внимание привлекли реакции кремнезема
с (CH3)3SiCl, (CH3)2SiCI2 и CH3SiCl3. Одно из первых исследо-
ваний реакций MesSiCI с кремнеземом было выполнено Штобе-
ром [404]. Тот же автор отметил воздействие, которое оказы-
вала на адсорбцию обработка поверхности аммиачной водой.
В работах [405—407] рассмотрено влияние предварительного
спекания кремнезема при температурах вплоть до 1000°С. Реак-
ция с водой проходила при 25СС, но реакция с группами SiOH
протекала только частично при высокой температуре. Армистед
и Хокки [88] обнаружили, что Me3SiCl и Me2SiCI2 реагировали
избирательно и полностью с изолированными группами SiOH,
тогда как MeSiCl3 и SiCI4, кроме того, вступали в реакцию
с некоторой долей парных групп SiOH, состоящих из смежных,
связанных водородной связью двух гидроксильных групп, рас-
положенных на частично дегидратированной поверхности крем-
незема. Из-за определенного размера группы (CH3)3Si только
лишь около 60 % всех свободных групп SiOH вступало в реак-
цию [408]. Поверхность, предварительно дегидратированная при
380°С, сохраняла достаточное число групп SiOH, обеспечивав-
ших присоединение монослоя из групп Me3SiO при такой темпе-
ратуре. Скорость реакции была наибольшей с Me3SiNHSiMe3,
понижаясь затем для Me3SiOH и далее для Me3SiCI [409].
Химия поверхности кремнезема
965
Берг и Унгер [410] провели соответствующее изучение ре-
акций с PhSiCl3, Ph2SiCl2 и Ph3SiCl (где Ph группа СеН5) и по-
казали, что идут следующие селективно протекающие реакции:
a zcl SiO^ zCl
SiOH + Si = = Si + 2HC1 (1 )
SiOH Cl Ph SiO Ph
Cl
SiOH + Ph2SiCI2 = SiOSi—Ph + HCI ' (2)
Ph
SiOH + Ph3SiCl - SiOSiPh3 + • HCI (3)
Реакция (1) происходит до тех пор, пока кремнезем не де-
гидратируется при температуре свыше 400°С; в противном слу-
чае идет реакция с участием только одного атома С1. Продукт
реакции можно прогидролизовать для того, чтобы удалить хлор.
Каждая молекула покрывает площадь 2,16 нм2.
Чуйко и др. [411] провели аналогичные эксперименты. Они
обнаружили, что Me2SiCl2 вступал в реакцию со свободными
группами SiOH при температуре выше 250°С с образованием
SisOSi(Cl) (Ме)2, но при 250°С получилась группа
Si„-O-Si(Me)2
О
Sis-O-Si(Me);
Герцберг и Эрвин [412] представили доказательство, что по-
верхностные реакции с участием метилхлорсиланов оказываются
не такими простыми, как было принято думать. В их экспери-
менты была включена тритиевая радиоактивная метка для ме-
тильных групп. Легко понять, что в большинстве указанных
примеров только один атом С1 дихлор- или трихлорсилана спо-
собен вступать в реакцию с одной группой SiOH, тогда как дру-
гие атомы С1 подвергаются гидролизу и связываются попереч-
ными связями между собой или «полимеризуются». Однако эти
авторы пришли к заключению, что в отсутствие «воды» на по-
верхности никакой реакции между хлорсиланами и стеклом не
происходило, но в ее присутствии шло образование метилполиси-
локсановых полимеров, которые покрывали стекло. Еще более
удивительным было то, что Me2SiCl при контактировании с по-
верхностью из нержавеющей стали или со стеклом претерпевал
разложение и диспропорционирование, а образовавшиеся при
этом побочные продукты гидролизовались и формировали гид-
966
Глава 6
рофобное покрытие. Однако, как только поверхность стекла пол-
ностью покрывалась, такое покрытие сохранялось на стекле, не
подвергаясь никакому дальнейшему распаду.
Для проведения реакций с кремнеземной поверхностью
обычно более пригодны соответствующие алкоксилорганосиланы,
чем хлорсиланы. Хертл [413] сообщил, что Me3SiOMe и
Me2Si(OMe)2 вступают в реакцию только с силанольными груп-
пами, не связанными водородными связями. На частично дегид-
ратированной поверхности большее число изолированных, не
связанных водородными связями групп ОН становится доступ-
ным для реакции, поэтому реакционная способность такой по-
верхности возрастает.
Коуллинг и Колб [414] методом ИК-спектроскопии предста-
вили доказательство того, что идет образование силоксановой
связи между (Me)3SiR и группами SisOH на поверхности стекла.
Когда свежеобразованная поверхность, полученная при раз-
ломе кремнезема, подвергается действию олефина, такого, как,
например, стирол, то образуются связи углерод—кремний. Бен-
сон и Кастль [281] изучили активность поверхности, полученной
размалыванием плавленого кварца в шаровой мельнице в атмо-
сфере гелия. Они обнаружили, что если кремнезем не подвер-
гался воздействию влаги, которая сразу же уничтожала актив-
ность, то активность поверхности постепенно снижалась в тече-
ние 10 сут. Реакция, вероятно, является следствием присутствия
напряженных связей Si—О—Si или поверхностных центров
SisO_ и Si+, которые вступают в следующую реакцию:
Si+ + 2СН,СН,С(.Н, = Si СН,Ш,С..Н, + стирол+
Активный стирол продолжает затем полимеризоваться на по-
верхности до тех пор, пока не разрушится свободный радикал:
Si (СН2СН)ПСН2СН+С6Н5
I
с6н5
Класс поверхностных групп на кремнеземе будет иметь практи-
ческую важность тогда, когда удастся осуществить сополимери-
зацию подобных групп с мономерами, обеспечивая таким обра-
зом прочное связывание кремнезема с образованием полимера.
Несомненно, наиболее выгодно таким способом обрабатывать
стекловолокно, с тем чтобы оно химически связывалось с поли-
эфирными или эпоксидными смолами в получаемых слоистых
материалах. Без таких связей вода будет проникать вдоль гра-
ницы раздела и прочность материала снизится, что может иметь
губительные последствия, например, для лодок, изготовленных
из стекловолокна.
Кроме хорошего связывания в конечном продукте должно
быть обеспечено свойство, характерное для обрабатываемого
Химия поверхности кремнезема
967
подобным образом стекловолокна: быстрое и полное смачивание
его жидкой смолой. Ли [415] исследовал поверхностные свой-
ства стекловолокна, обработанного следующими коммерческими
органосиланами:
a) RCH2CH2CH2Si (ОСН3)3 (где R-NH2CH2CH2NH_, СН2С (СН3) СОСГ,
СН2=СНСН2О_, С1_, NH2_; HS_); б) CH2=CHSi (ОСН3)3.
В настоящее время все еще продолжается дискуссия по во-
просу, является ли такая связь результатом фактически проис-
ходящего в процессе полимеризации химического связывания
смолы со стеклом через подобные реагенты на поверхности или
же результатом более полного смачивания полимером обрабо-
танной поверхности. Вероятно необходимыми оказываются оба
фактора.
Соединения типа СН=С (СН3) CONHCaH4Si (ОС2Н5)з могут
вступать в реакцию с поверхностью кремнеземных частиц, ис-
пользуемых в качестве армирующего наполнителя, причем такие
частицы диспергируются в полимеризационной системе, в кото-
рой образуется эластомер. Группы —Si(OC2H5)3 реактива гид-
ролизуются и вступают в реакцию с силанольными группами,
находящимися на поверхности кремнезема, что приводит к фор-
мированию невосприимчивого к воде, химически связанного
ненасыщенными связями покрытия, которое после полимериза-
ции становится частью полимерной структуры эластомера с по-
перечными связями [416].
Органические соединения, содержащие литий, очевидно,
представляют ценность для присоединения углеводородных
групп к кремнеземной поверхности после превращения групп
SiOH в группы SiCl. Унгер, Томас и Адриан [417] представили
интересные примеры таких систем. Они проводили реакции
между силанольными группами и SiCR или SOC12, используя си-
ликагели с величиной удельной поверхности 70—465 м2/г и со
средней поверхностной концентрацией гидроксильных групп 5,3
ОН-групп/нм2. Поверхностная концентрация получающихся
групп SiCl составляла 80 % концентрации ранее находившихся
на поверхности групп ОН, т. е. ее среднее значение было
4,2 атома С1 на 1 нм2. Такая поверхность затем взаимодейство-
вала с различными органическими соединениями лития LiR,
благодаря которым группы SiCl на поверхности замещались на
группы SiR. Число присоединенных к поверхности углеводо-
родных групп в расчете на 1 нм2 будет в зависимости от раз-
мера понижаться в следующей последовательности: 3,2 фениль-
ной; 2,0 нафтильной; 1,1 трифенилметильной группы. Сообща-
лось, что такие покрытия устойчивы в воде при pH 0—9.
968
Глава 6
Уайт [418] сообщил о термостабильности углеводородных
покрытий. В качестве поверхностных групп выбирались Et3Si—,
Me3Si—Me2Si=, McSi= и CH2=CHSi=. Все группы оказы-
ваются достаточно устойчивыми на воздухе вплоть до 200°С.
Винильные группы разрушаются при 300°С, метильные и этиль-
ные группы — при 500°С на воздухе. Фенильные группы немного
более устойчивы, чем метильные.
Адсорбция на гидрофобных поверхностях
Бабкин и Киселев [419] исследовали адсорбционные свойства
«химически модифицированного кремнезема», в частности образ-
цов с поверхностными группами Si(CH3)3. Такие триметилси-
лильные группы располагаются на поверхности, образуя покры-
тие почти с плотной упаковкой (93%). Были выполнены рас-
четы дисперсионных сил между н-гексаном или бензолом
и слоем метильных групп, а также лежащим под ним кремне-
земом SiO2. Адсорбция паров углеводородных соединений пони-
жается очень сильно после того, как кремнезем покрывается
слоем, состоящим из метильных групп; теплоты адсорбции ока-
зались равными—ЗЗООкал/моль для н-гексана и —4100 кал/моль
для бензола, т. е. гораздо ниже теплот конденсации.
На рис. 6.16 представлены типичные изотермы адсорбции,
полученные Киселевым [123] на одном из том же силикагеле.
Выявлены очень четкие различия, определяемые тем, является
ли поверхность гидроксилированной и гидрофильной или же она
покрыта триметилсилильными группами и гидрофобна. Вода, ме-
танол и бензол сильно адсорбируются в первом случае благо-
даря образованию водородных связей или л-связей с силаноль-
ными, а не с метилсилильными группами. Адсорбция азота про-
исходит главным образом за счет дисперсионных сил и поэтому
в гораздо меньшей степени зависит от природы поверхности.
Унгер и др. [410] изучали влияние типов поверхностных групп
на адсорбционные свойства. В их работе сравнивались число
гидроксильных групп, приходящееся на единицу площади по-
верхности кремнезема, и изостерические теплоты адсорбции
бензола и метанола:
Группа Число групп на 1 нм2 Теплота адсорбции, ккал/моль
бензола метанола
HOSi 8 6,9 12,1
Ph (ОН) Si 3,6 4,6 9,3
Ph2Si 2,6 4,4 7,2
Ph3S I 1,9 1,3 1,7
CH3Si 4,5 1,6 2,2
Химия поверхности кремнезема
969
На адсорбцию сильно влияет присутствие гидроксильных
групп. Так, в случае поверхностных групп Ph2Si некоторое коли-
чество групп ОН может еще находиться на поверхности в виде
Ph (ОН) Si, чем, вероятно, объясняется получаемое более высо-
кое значение теплоты адсорбции по сравнению с Ph3Si.
Рис. 6.16. Изотермы адсорбции (по данным Киселева [123]).
/ — гидроксилированная поверхность кремнезема; 2 — гидрофобно покрытые триметил-
силильными группами поверхности.
Существуют небольшие участки между триметилсилильными
группами, которые оказываются не полностью покрытыми; на
этих участках наблюдается более высокий адсорбционный потен-
циал для молекул, достаточно небольших по размеру и имею-
щих электронодонорный атом (N или О), способный образовы-
вать водородную связь с группами SiOH [421J.
Адсорбция, воды
Цеттльмойер и Хсинг [422] изучали адсорбцию воды на по-
верхности кремнезема, который предварительно обрабатывали
органосиланом и который имел поэтому наибольшее из возмож-
ных число органосилильных групп на поверхности, что сооб-
щило ему высокую степень гидрофобности. Тем не менее на по-
верхности оставалось еще достаточное число групп SiOH, кото-
рые могли вступать во взаимодействие с гораздо большими по
970
Глава 6
размеру группами (CH3)3Si. Под последними, как под зонти-
ками, еще оставалось некоторое число групп SiOH, не вступив-
ших в исходную реакцию модифицирования. Возникал вопрос,
будут ли при высокой влажности или при высоком парциальном
давлении паров воды под триметилсилильными группами наб-
людаться участки, где молекулы воды могли бы адсорбиро-
ваться.
На гидроксилированной поверхности кремнезема имеется
около 4,6 SiOH-групп/нм2, но из них только 2,45 группы
(CH3)3Si образуют на 1 нм2 полное триметилсилильное покры-
тие и, следовательно, на поверхности должно еще оставаться
2,1 ОН-групп/нм2*. Полностью гидроксилированный кремнезем
Hi-Sil модифицировали при взаимодействии с различными возра-
стающими количествами гексаметилдисилазана (CH3)3SiNHSiX
X (СНз)з (ГМДС) для того, чтобы получить различные степени
покрытия поверхности группами (CH3)3Si. После этого были из-
мерены изотермы адсорбции воды. Удивительным оказалось то,
что количество адсорбированной воды лишь слегка уменьшалось
с возрастанием степени покрытия вплоть до заполнения моно-
слоя. По-видимому, вода адсорбируется ниже групп (CH3)3Si,
а именно на расположенных под ними силанольных группах.
Действительно, понижение адсорбции с ростом степени модифи-
цирования наблюдалось только около тех точек, где группы
(CH3)3SiOSis были сцеплены с поверхностью:
Число групп ГМДС иа 1 ны2(Д) Покрытие поверхности, % Адсорбция, моль Н2О/и.м2(В) д + в
0 0 15,8 15,8
0,73 29,6 14,8 15,6
1,28 52,2 14,1 15,4
2,45 100 13,0 15,5
Однако при более высоком давлении пара воды, когда уже
должен был бы сформироваться полислой Н2О на немодифици-
рованном кремнеземе Hi-Sil, при наличии триметилсилильного
гидрофобного покрытия, тормозящего процесс, наращивание
слоя воды не происходит.
Аналогичные исследования были проведены с пирогенным
дегидроксилированным кремнеземом Cab-0-Sil, который имел на
* В работах Давыдова, Журавлева и Киселева (см. Ж. физ. хим., 1964,
т. 38, с. 2047, и ссылку [43] в данной главе) методом дейтерообмена было
показано, что оставшиеся под слоем триметилсилильных групп на поверхности
кремнезема силанольные группы SiOH вступали в реакцию изотопного об-
мена с тяжелой водой D2O при комнатной температуре. Концентрация таких
групп оказалась равной 2,15 ОН-групп/нм2, а концентрация триметнлсилиль-
ных групп была 2,45 группы (СН3) Si па 1 нм2, что совпадает с приводимыми
данными.— Прим. ред.
Химия поверхности кремнезема
971
1 нм2 поверхности всего 2,5 группы SiOH, тогда как остальные
участки представляли собой силоксановую поверхность. В дан-
ном случае на поверхности имелась как раз необходимая кон-
центрация групп ОН, чтобы присоединилось покрытие из групп
(CHshSi. Однако лежащие под триметилсилильными группами
силоксановые участки поверхности были гидрофобными, поэтому
наблюдалась очень слабая адсорбция воды.
Алзамора, Контрелас и Кортес [423] сообщили о заполнении
пор водой на частично гидрофобной поверхности. Влияние ча-
стично покрытой поверхности, оказываемое на дейтероводород-
ный обмен в группах SiOH, рассмотрено в работе [424].
Уцуги и Нисимура [425] исследовали поверхностные свойства
модифицированных силикагелей, этерифицированных 1-пентано-
лом. На каждом квадратном нанометре было 3 группы РеО
и 1,5 группы SiOH. На последних размещались пентильные
группы, которые образовывали плотно упакованный слой на по-
верхности. На образцах с различной степенью покрытия поверх-
ности изучалась адсорбция паров Аг, Н2О и СвНн. Поры в си-
ликагелях были настолько малы, что монослой пентильных
групп эффективно понижал диаметр и объем пор, а также
удельную поверхность.
Авторы работы [426] также этерифицировали силикагели
спиртами с числом углеродных атомов от 1 до 14 и измеряли
изотермы адсорбции Аг, Н2О и W-C7H15, а также определяли
теплоты смачивания в случае двух последних адсорбатов. Изо-
термы адсорбции воды на этерифицированных группами МеО
и ЕЮ силикагелях показали, что поры заполнялись водой, од-
нако при модифицировании высшими спиртами поры не запол-
нялись водой, так как их поверхность оказывалась гидрофобной.
На адсорбцию аргона природа поверхностных групп не оказы-
вала влияния. Наиболее интересным представляется тот факт,
что когда использовался метод БЭТ на частично покрытых моди-
фицированных поверхностях, то сумма величин поверхностей, из-
меренных по Н2О и по гептану, была равна величине поверхно-
сти, измеренной по аргону. Другими словами, вода адсорбиро-
валась как монослой только на оставшихся непокрытыми груп-
пах SiOH, а гептан адсорбировался в виде монослоя только на
гидрофобных (этерифицированных) участках поверхности.
Кроме того, наблюдалось, что на частично этерифицированных
поверхностях более длинные углеводородные цепи покрывали
большее число групп SiOH, так как подобные цепи не распола-
гались вертикально к поверхности. Однако, как отмечается
в других работах, когда поверхность полностью этерифицирова-
лась спиртами с неразветвленной углеродной цепью, то во всех
случаях покрывалась площадь примерно 33 А2 в расчете на одну
молекулу независимо от длины цепи спирта. Такие молекулы
972
Глава 6
должны плотно упаковываться, а их цепи должны располагаться
под прямым углом к поверхности кремнезема.
Было также найдено [426], что теплота смачивания частично
этерифицированного кремнезема в гептане возрастала с ростом
длины цепи алкоксигрупп (вероятно, при равной степени по-
крытия поверхности). К тому же теплота смачивания в воде
возрастала линейно с ростом площади поверхности, занимаемой
группами SiOH, измеренной по адсорбции воды. В другой ра-
боте [427] авторы показали, что свободная энергия смачивания
лучше отражала свойства подобных поверхностей, чем теплота
смачивания.
Интересно, что в очень небольших по диаметру порах сили-
кагеля с гидрофильной поверхностью теплота адсорбции воды
оказывается повышенной. Однако в случае использования ад-
сорбентов с менее полярными группами на поверхности теплота
адсорбции в таких небольших порах может стать отрицатель-
ной, вероятно, вследствие того, что молекулы, подобные молеку-
лам тетрахлорида углерода, не смачивают поры. Однако Бабкин
и Киселев [428] показали, что очень небольшие поры с органо-
фильными стенками могут образовываться посредством гидро-
фобизации пирогенного кремнезема группами (CHa^Si с после-
дующим прессованием такого порошка под давлением 10 т/см2.
Свойства полученного материала оказались совсем иными, так
как теплоты адсорбции бензола и тетрахлорида углерода за-
метно возросли. Это явление может быть положено в основу спо-
соба удерживания в порах обычных летучих органических жид-
костей в условиях относительной нелетучести, например для за-
медленного выделения таких жидкостей.
Образование центров кристаллизации льда. Интересное яв-
ление возникает при адсорбции паров воды на гидрофильных
группах, расположенных на гидрофобной поверхности. В дискус-
сии по проблеме адсорбции воды на дегидратированной поверх-
ности кремнезема Хертл и Хайр [142] сообщили, что молекулы
воды адсорбировались более упорядоченным образом на оди-
ночных группах SiOH или на изолированных спаренных группах
SiOH по сравнению с адсорбцией на полностью гидроксилиро-
ванной поверхности.
Авторы работы [147] при исследованиях адсорбции воды изу-
чили природу гидроксильных групп на частично дегидратиро-
ванной поверхности кремнезема и роль таких групп как центров
кристаллизации. Очевидно, эта роль заключается в индуциро-
вании определенной степени упорядоченности в отдельных кла-
стерах молекул воды, присоединяемых к небольшим полярным
центрам на гидрофобной поверхности, что в конечном счете ве-
дет к формированию зародышей кристаллизации льда. Такая
поверхность представляет собой основу для проявления эффекта
Химия поверхности кремнезема
973
рассеивания облаков, что было продемонстрировано Цеттльмойе-
ром, показавшим как некоторые гидрофобные кремнеземные
пылевидные порошки способны играть точно такую же роль, ка-
кую играют гидрофобные кристаллы иод ид а серебра.
Бассетт, Боучер и Цеттльмойер [429] обсудили вопросы
взаимодействия поверхности кремнезема с водой с точки зрения
явления рассеивания облаков и обобщили ранее полученные
экспериментальные данные, известные по литературе, касаю-
щиеся этой темы [430]. Цеттльмойер [431] отметил, что на гид-
рофобной поверхности вода адсорбируется только благодаря
проявлению сил Ван-дер-Ваальса или «дисперсионных сил», так
как не происходит образования водородных связей, которое воз-
можно только на очень небольших участках. После того как
первая молекула воды адсорбируется при относительно высоком
парциальном давлении, дальнейшая адсорбция происходит по-
средством наращивания кластера или микрокапельки воды, но
не путем распределения молекул воды по всей поверхности.
Адсорбция инертных газов
Как сообщили Ловен и Броудж [347], в случае, когда сила-
нольная поверхность кремнезема этерифицирована «-бутиловым
спиртом, значение константы С в уравнении БЭТ адсорбции
азота оказывается более низким. То же было получено Щерба-
ковой и Словенкой [432] для поверхности, этерифицированной
метильными группами, а также для поверхности кремнезема,
покрытой хлором, когда группы SiOH были замещены на
группы SiCl.
Адсорбция органических полимеров
на поверхности кремнезема
В большинстве экспериментальных работ, выполненных
в этой области, взаимодействие водорастворимых полимеров
с частицами кремнезема в суспензии изучалось, как правило,
в связи с практическими приложениями, касающимися про-
цесса флокуляции или очистки воды. Однако некоторое число
аналогичных исследований было проведено и в неводных рас-
творах.
Те же самые основные силы, которые определяют адсорбци-
онный процесс в случае более простых молекул, как это рас-
сматривалось в предшествующих разделах, действуют и при ад-
сорбции полимеров. Основное различие заключается в большем
числе точек присоединения какой-либо выбранной полимерной
молекулы с поверхностью. Это ведет к понижению до нулевого
974
Глава 6
значения вероятности одновременного разрыва всех связей с по-
верхностью, являющейся главным фактором энтропии.
По-видимому, почти повсеместно признается тот факт, что
полимеры в растворе формируют мономолекулярный слой на по-
верхности раздела фаз твердое тело—жидкость. Адсорбционные
силы могут быть различными: от ионного притяжения до гидро-
фобных или водородных связей.
Эйрих [433] подробно описал и классифицировал конформа-
ционные расположения полимерных молекул в адсорбирован-
ном монослое. Молекулы полимера могут иметь связывающие
центры вдоль всей цепи, но в любом случае всегда имеется связь
с поверхностью на концах цепи, где расположены полярные
атомы, такие, например, как азот, или кислород, или по двойной
связи С=С. Полимеры могут быть линейными, разветвленными
или с поперечными связями между цепями. Отмеченные фак-
торы определяют, как именно будут адсорбироваться полимер-
ные молекулы: а) в виде сферической губки, как это имеет ме-
сто в случае высоко разветвленной молекулы крахмала; б) цак
беспорядочно закрученная спираль, сегменты которой присоеди-
няются к поверхности; в) подобно червеобразной цепи, лежащей
плоско на поверхности; г) в виде цепи, присоединенной к поверх-
ности только одним концом и направленной вертикально к ней,
подобно волоску на парике.
Другая сторона вопроса, которая не рассматривалась Эйри-
хом, состоит в том, будут или нет точки присоединяться к по-
верхности фиксированно и относительно необратимо, удержи-
вая специфические связи между атомами полимерной молекулы
и твердой поверхностью или они будут находиться в подвижном
равновесии, когда полимерная молекула удерживается около по-
верхности под действием сил ионного притяжения.
В качестве примера, в котором рассматриваются специфи-
ческие связи и ионное притяжение, приводится [434J сопоставле-
ние полимерной молекулы (А), содержащей четвертичные ам-
мониевые катионы, с предшествующей ей молекулой полимера
(В), когда вторая молекула идентична первой, за исключением
того, что катионами являются третичные аминогруппы
(рис. 6.17). Если 0,5 %-ные растворы этих полимеров при
pH 4—5 наносят на два различных участка чистой поверхности
кварцевого стекла и затем промывают в течение нескольких
секунд, то каждый полимер на поверхности образует прилип-
ший слой. В случае полимера четвертичного аммониевого осно-
вания А, когда поверхность кремнезема еще сохраняется влаж-
ной, на предварительно обработанном очищенном участке обра-
зуется заметно более толстая пленка, чем на неочищенном.
После высушивания поверхность с адсорбированным полимером
А легко смачивается водой. В случае полимера В происходит
Химия поверхности кремнезема
975
образование водородных связей между третичными аминогруп-
пами и такими полимерами и группами SiOH на поверхности
кремнезема. После того как вода удаляется, поверхность сразу
же становится сухой. Вероятно, для полимера В все амино-
группы оказываются направленными к поверхности и присоеди-
ненными к ней, тогда как углеводородные цепи направлены
наружу от поверхности, что сообщает полученной пленке гидро-
фобность. Когда влажные полимерные пленки промываются
в струе воды в течение примерно одной минуты, то полимер А
Рис. 6.17. Схема адсорбции
полимеров: ионное взаимодей-
ствие (вверху) и образование
водородных связей (внизу).
Оба полимера идентичны за исклю-
чением того, что полимер А имеет
боковые группы C2H4N+(CH3)2, обо-
значенные Q+. а полимер В имеет
группы C2H4NH+(CH3)2, где группа
СНз обозначена буквой М. Поли-
мер представляет собой полимери-
зованный сложный метакрилатный
эфир .N.N-диэтилэтаноламина и его
метильное четвертичное производ-
ное. А — толстая гидрофильная по-
лимерная пленка; В — тонкая гид-
рофобная полимерная пленка.
Н Н Н Н Н Н Н
О ОООООО
Si 02
начинает смываться, поскольку отсутствуют какие-либо фикси-
рованные связи с поверхностью, а полимер В с поверхности не
удаляется. Вероятно, что подобные различия наблюдаются и для
полимеров, адсорбированных на поверхности кремнезема из
.. органических растворителей.
Адсорбция из водного раствора
Адсорбция полимеров на поверхности частиц золя кремне-
зема и происходящие при этом процессы флокуляции и коацер-
вации рассматривались более подробно в гл. 4 (см. лит. к гл. 4
[308—339]).
Выполнен значительный объем исследований по адсорбции
полимеров на силикагелях и кремнеземных порошках, но полу-
ченные результаты часто оказываются сомнительными. Кремне-
земная поверхность — это поверхность небольших по размеру
частиц, причем такие частицы собираются в малые кластеры или
даже в большие агрегаты, и поэтому молекула полимера, конфи-
гурация которой представляет, как правило, закрученную спи-
раль, не в состоянии достичь всех участков поверхности крем-
незема. Разновидности пирогенного кремнезема с отчасти откры-
той структурой агрегатов и осажденных кремнеземов с открытой
до некоторой степени структурой находят наибольшее при-
менение при изучении адсорбции полимеров.
976
Глава 6
Большую часть доступных водорастворимых полимерных мо-
лекул составляют линейные углеводородные цепи либо с поляр-
ными или ионными группами, присоединенными к каждому вто-
рому или к каждому третьему атому углерода (поливиниловые
спирты, ПВС), либо с эфирными или имидными звеньями, ре-
гулярно повторяющимися внутри цепи (полиэтиленоксид или
полиэтиленимин). Если отсутствуют какие-либо заместители,
вводимые для предотвращения свободного вращения относи-
тельно связей С—С, то такие полимерные молекулы могут ле-
жать плоско на поверхности кремнезема. Это было показано
Айлером для случая ПВС (см. рис. 3.63). Сигиура [435] также
пришел к заключению, что молекула ПВС лежит плоско, причем
ее гидроксильные группы связаны водородными связями с груп-
пами SiOH на поверхности кремнезема. Вывод Гринленда [436]
о том, что молекула ПВС не адсорбируется на гидроксилиро-
ванном (непрокаленном) кремнеземе, может быть обусловлен
недостаточно низким значением pH в его эксперименте, по-
скольку при pH 6—7 поверхность кремнезема имеет достаточный
ионный заряд, который способен предотвратить образование во-
дородных связей с ПВС, если только не добавляется некоторое
количество соли, чтобы вызвать уплотнение двойного слоя.
Полиэтиленоксид
Рубио и Китченер [437] исследовали с некоторыми подробно-
стями адсорбцию полиэтиленоксида (молекулярная масса
больше 106) с одновременным изучением воздействия, оказывае-
мого на адсорбцию разной степенью дегидроксилирования по-
верхности. На рис. 6.18 представлена зависимость концентра-
ции насыщения полиэтиленоксида при адсорбции на поверхности
кремнезема от значения pH. Концентрация ПЭО на поверхно-
сти кремнезема возрастает, оставаясь в равновесии с концент-
рацией ПЭО в растворе, но она приближается к значению на-
сыщения или к предельному значению, когда концентрация ПЭО
в растворе превышает ~0,008 %. При низком значении pH,
когда может образоваться наибольшее число водородных связей,
гидроксилированный (полученный осаждением) кремнезем спо-
собен адсорбировать на 1 нм2 площади максимальное число
сегментов цепи молекулы ПЭО, равное семи сегментам оксида
этилена — С2Н4О.
Поливиниловый спирт
Поведение ПВС на кремнеземе ранее уже описывалось с не-
которыми подробностями (см. рис. 4.20, лит. к гл. 4, [336, 339]
и лит. к гл. 3, [169]). Поливиниловый спирт адсорбируется на
Химия поверхности кремнезема
977
поверхностных группах SiOH посредством образования водород-
ных связей между спиртовыми гидроксильными группами в сег-
ментах — (СН2СНОН)П— полимерной молекулы и группами
SiOH на поверхности кремнезема. Это имеет место только в слу-
чае незаряженной поверхности кремнезема при низком значении
pH, а также когда гидрофобная углеводородная цепь покрывает
поверхность кремнезема таким образом, чтобы сделать частицы
Рис. 6.18. Зависимость концентрации насыщения полиэтиленоксида на поверх-
ности кремнезема от величины pH (по данным Рубио и Китченера [437]).
Равновесная концентрация полимера в растворе 0,008 масс. %. А — гидроксилированная
поверхность кремнезема, полученного методом осаждения; В —частично дегидроксили-
рованная поверхность кремнезема, полученного «пламенным» высокотемпературным
способом.
кремнезема гидрофобными и вызвать процесс коагуляции. Од-
нако Тедрос [438] показал, что адсорбция ПВС протекает даже
в нейтральном и основном растворе, если бромид цетилтриме-
тиламмония (БЦТМА) адсорбируется вначале, до адсорбции
ПВС. В таком случае образуются гидрофобные связи между
сегментами —СН2СН2— цепи ПВС и длинными углеводород-
ными цепями четвертичных аммониевых катионов, находящихся
на поверхности кремнезема.
Согласно данным Тедроса, ПВС образует с поверхностно-
активными веществами мицеллы, которые затем действуют как
зародыши полимеризации. Это происходит в результате возник-
новения гидрофобных связей между гидрофобной углеводород-
ной цепью ПАВ и гидрофобным каркасом полимера. Таким об-
разом, каждый компонент в подобной системе может самостоя-
тельно взаимодействовать с поверхностью кремнезема путем
образования водородных связей и адсорбции ионов, поскольку
35 Заказ № 250
978
Глава 6
Н20
SiOg
О ОН-группы
S Ион SiO ;
I
^Углеводород- j
мая цепь |
Ф Ион четвер-
тичного аммо-
ниевого осно-
вания
Рис. 6.19. Схемы возможных комбинаций, образуемых молекулами поверх-
ностно-активного вещества (ПАВ) н поливинилового спирта (ПВС) при гх
адсорбции на поверхности кремнезема (по данным Тедроса [438]).
1 — при низком значении pH; монослой ПВС иа поверхности кремнезема с образова-
нием иа ПВС гидрофобносвязанного монослоя ПАВ; 2 — при высоком значении pH; мо-
иослой ПАВ на поверхности кремнезема с образованием на ПАВ гндрофобносвязанного
монослоя ПВС; 3 — в нейтральном растворе; одиночные молекулы ПАВ, адсорбирован-
ные на поверхности кремнезема, покрыты гпдрофобносвязанной частью цепи ПВС, про-
стирающейся к другим частицам кремнезема.
ни спиртовые гидроксильные группы ПВС, ни группы (CH3)3N +
ПАВ не препятствуют формированию гидрофобных связей.
На поверхности кремнезема ПАВ может образовывать двойные
слои с различным возможным расположением (рис. 6.19).
Это подтверждается также и тем, что добавление к способ-
ному флокулировать кремнезему небольшого количества кати-
онного ПАВ в комбинации с ПВС с высокой молекулярной
массой может вызвать синергическое действие, так как ПВС дол-
жен в этом случае представлять собой мостик между кремне-
земными частицами и присоединяться к кремнезему через ПАВ.
Гидрофобные связи оказываются достаточно прочными, чтобы
параллельно протекала адсорбция аниона с длинной цепью, до-
децилбензосульфоната (ДБС), при условиях, когда такой анион
не должен.был бы адсорбироваться.
Тедрос [438] также показал, что адсорбция ПВС макси-
мальна при и. э. т. кремнезема и, кроме того, что идет образо-
Химия поверхности кремнезема
979
вание гидрофобных связей, если поверхность кремнезема пред-
варительно дегидроксплирована (оптимально при 700°С).
Катионные полимеры
Катионные полимеры прочно адсорбируются из воды на по-
верхности кремнезема во всей области значений pH в отсутст-
вие каких-либо полярных растворителей. Взаимодействие таких
полимеров с кремнеземом рассматривалось в гл. 4 (см. лит.
к гл. 4 [315—323]). В том случае, когда углеводородная цепь
полимера содержит четвертичные аммониевые ионы на корот-
ких боковых цепях, для такой полимерной молекулы появляется
возможность располагаться плоско вдоль поверхности. В случае
аммониевой соли поли(Н-метилдиэтилэтилметакрплата) каж-
дый сегмент катионного полимера покрывает площадь 1—2 нм.
Для покрытия больших по размеру частиц кремнезема с почти
плоскими по форме локальными участками поверхности тре-
буется меньшее количество полимера в расчете на единицу по-
верхности. Такой полимер испытывает незначительную конфи-
гурационную заторможенность на поверхности; это подтвер-
ждается тем фактом, что предшествующая ему форма, третичный
амин, адсорбируется с участием всех своих аминогрупп, обра-
щенных в сторону плоской поверхности, превращая ее в гидро-
фобную [434]. Однако когда после этого на такую поверхность
накладываются коллоидные частицы кремнезема, то некоторое
число аминогрупп разворачивается и адсорбируется уже на
этих небольших частицах, удерживая их тем самым на плоской
поверхности. Когда избыточный золь кремнезема смывается, на
поверхности еще сохраняется слой адсорбированных кремнезем-
ных частиц, и вся система остается гидрофильной.
Хостетлер и Сван [439] показали трудности измерения ад-
сорбции полимера на силикагеле или на сглаженных частицах
кремнеземного порошка. Адсорбция на пористом кремнеземе ре-
гулируется возможностью свободного проникновения выбран-
ного полимера в поры. Так, в случае полиэтиленимина (ПЭИ)
ниже pH 9 полимерная молекула вследствие ее ионизированного
состояния расширяется в растворе до такой степени, что оказы-
вается неспособной входить в поры. Только в узкой области
pH 10—12 иминогруппы не ионизированы, и гидрофобная поли-
мерная цепь будет плотно свертываться в спираль. Тогда поли-
мерная молекула оказывается настолько небольшой по размеру,
что становится способной проникать в поры силикагеля и адсор-
бироваться посредством небольшого числа ионов (СгН^ЬМИ-,,
еще остающихся при этом значении pH. Выше pH 12 таких
ионов оказывается слишком мало, чтобы вызвать адсорбцию.
36*
980
Глава 6
Положение полностью меняется, когда поверхность кремне-
зема, взятого в виде частиц золя, оказывается непористой и до-
ступной раствору полимера. На отрицательно заряженной по-
верхности кремнеземных частиц, модифицированной алюмина-
том, молекулы ПЭИ адсорбируются в области pH 3—10 [440].
Белки
Адсорбция белков и других биологических полимеров чрез-
вычайно сложна, поскольку в ней участвуют водородные связи
с группами ОН, NH или СО, ионные связи через четвертичные
аммониевые ионы, присутствующие в некоторых разновидностях
белков, и в особенности связи гидрофобной природы, ' возни-
кающие между сегментами протеиновых цепей и зависящие от
их конфигурации. Взаимодействие поверхности кремнезема
с желатином обсуждалось в гл. 3 (см. рис. 3.11, лит. к гл. 3
[856]), а с белками и с родственными веществами будет рас-
смотрено в гл. 7 (см. лит. к гл. 7 [249—273]). Данная тема, вы-
зывает постоянное внимание вот уже в течение более четверти
века. Еще в 1954 г. Холт и Боукотт [441а] измерили адсорбци-
онную способность на превращенном в порошок кварце с изве-
стной величиной удельной поверхности по отношению к ко-
ровьему альбумину. Из полученных данных можно подсчитать,
что при монослойном покрытии на 1 нм2 поверхности удержи-
валось около 4 амидных сегментов, принимая усредненное зна-
чение молекулярной массы амидного сегмента равным 100. По-
видимому, такая величина адсорбции является правдоподобной,
если рассматривать протеиновую цепь в форме спирали. Макси-
мальная адсорбция наблюдалась при pH 5—6. Те же авторы
[4416] исследовали поведение белков и аминов с длинными це-
лями, получаемых в виде мономолекулярных пленок на поверх-
ности раздела фаз воздух—вода, когда ниже этой поверхности
вводилась кремневая кислота. Белки более прочно связывались
при их изоэлектрической точке; такое связывание может проис-
ходить между органическими катионными группами молекулы
и заряженными участками на поверхности кремнезема и, кроме
того, путем образования водородных связей.
В работе, выполненной на стекле, Бреслер и др. [442] вы-
явили отчетливые граничные значения области pH, внутри кото-
рой наблюдалась адсорбция белков. При оптимальном значении
pH и низкой концентрации белков, находящихся в равновесной
системе, число амидных сегментов (при средней молекулярной
массе сегмента, равной 100), приходящееся на 1 нм2, состав-
ляет, вероятно, около девяти для альбумина и около семи для
у-глобулина. Оптимальные значения pH были 5 при адсорбции
альбумина и 6 при адсорбции у-глобулина, т. е. близки к изо-
Химия поверхности кремнезема
981
электрическим точкам. Оба вида белка полностью вымывались
из кремнезема при четко определенных значениях pH, равных
7,5 и 8,0 соответственно.
В специализированной области хроматографического разде-
ления биохимических веществ накопление получаемых данных,
без сомнения, будет постоянно продолжаться, что должно при-
вести к более детальной картине адсорбции на кремнеземе ве-
ществ с высокими молекулярными массами. Серьезную проб-
лему, однако, представляет собой денатурация белков, когда
скрученные в спирали и связанные водородными связями бел-
ковые конфигурации разрываются под действием сил, стремя-
щихся распрямить молекулы вдоль поверхности. С целью опре-
деления молекулярных размеров полимерных молекул методом
эксклюзивной хроматографии кремнезем следует сформировать
в виде совершенных структур с регулируемым размером пор
[443]. Однако при таком использовании адсорбции полимеров
на поверхности кремнезема необходимо избегать некоторых не-
желательных моментов, и, кроме того, еще нет полностью удов-
летворительного способа, пригодного для модифицирования по-
верхности, чтобы сделать ее инертной по отношению ко всем
адсорбционным силам.
Адсорбция полимеров на дегидроксилированных
кремнеземах
Значительное число работ по адсорбции полимеров на пиро-
генном кремнеземе в водной суспензии было выполнено еще до
того, как стало известно, что поверхность такого кремнезема
дегидроксилирована и на отдельных участках гидрофобна.
Крайот и Китченер [444] отметили, что некоторые неионные
водорастворимые органические полимеры вызывали процесс
флокуляции дисперсных систем пирогенного кремнезема, но не
оказывали такого действия в случае осажденного кремнезема.
Однако после старения в воде пирогенный кремнезем стано-
вился менее чувствительным к флокуляции. Эти авторы также
показали, что после того, как осажденный гидроксилированный
кремнезем дегидроксилировался при 730 °C, он вел себя уже по-
добно пирогенному кремнезему. Гринленд [436] еще ранее отме-
тил точно такие же эффекты при использовании поливинило-
вого спирта.
В настоящее время ясно, что флокуляция частично дегидро-
ксилированных частиц включает присоединение углеводородных
сегментов вызывающего флокуляцию полимера к гидрофобным
силоксановым участкам посредством образования гидрофобных
связей. Рубио и Китченер [437] продемонстрировали, что крем-
незем, гидрофобизированный в результате проведения поверх-
982
Глава 6
ностной реакции с метилхлорсиланом, адсорбировал полиэти-
леноксид (ПЭО) и подвергался флокуляции точно так же, как
и дегидроксилированный кремнезем.
Сходство дегидроксилированного кремнезема с гидрофобным
было доказано теми же авторами посредством контрольного
эксперимента, в котором порошки кремнезема встряхивались со
смесью воды и диэтилового эфира. Оба типа кремнеземного по-
рошка обнаруживались в эфирном слое, тогда как гидроксили-
рованный кремнезем не переходил в этот слой. Гидроксилиро-
ванный (полученный осаждением) кремнезем после дегидрокси-
лирования становился способным к адсорбции ПЭО. Кремнезем
оказался наиболее чувствительным к процессу флокуляции по-
сле нагревания в области температур 500—800°С. Рубио и Кит-
ченер [437] представили полезный литературный обзор по дан-
ной теме.
Из рис. 6.18 видно, что на дегидроксилированном (пироген-
ном) кремнеземе количество адсорбированного полимера пони-
жается с повышением pH до той же самой поверхностной кон-
центрации адсорбированного вещества, которая достигается на
гидроксилированном кремнеземе при низком значении pH. В по-
следнем случае адсорбированное количество понижается с уве-
личением pH. Отсюда можно сделать вывод о том, что при нейт-
ральном и низком значениях pH гидрофобные центры на дегид-
роксилированном кремнеземе' притягивают неплотные полимер-
ные спирали, так как число адсорбированных сегментов С2Н4О
полимерной цепи возрастает, значительно превышая монослой.
По мере того как pH увеличивается до 10, концентрация адсор-
бированного ПЭО на поверхности уменьшается до ~7 С2Н4О-
групп/нм2, т. е. до той же самой концентрации, которая дости-
гается на гидроксилированном кремнеземе при pH 2. Отсюда
следует, что гидрофобные участки при pH 10 имеют примерно
то же самое число центров для образования гидрофобных связей
с группами —С2Н4О— что и число групп SiOH на гидроксили-
рованном кремнеземе в отсутствие заряда на поверхности при
pH 2, обеспечивающее образование водородных связей с ато-
мами кислорода эфирных групп С—О—С.
Действие солей
Такие соли, как NaCl, обычно способствуют адсорбции по-
лимеров на кремнеземе (за исключением солей, имеющих ка-
тионы или анионы, которые взаимодействуют с определенными
полярными атомами полимерной цепи с образованием комплекс-
ных ионов). Когда концентрация соли низка, то повышение кон-
центрации увеличивает число заряженных центров на поверх-
ности кремнезема при данном значении pH, и за одинаковый
Химия поверхности кремнезема
983
интервал времени противоионы перемещаются ближе к заряжен-
ным центрам на поверхности, причем незаряженные силаноль-
ные участки на ней оказываются более доступными для проте-
кания адсорбции.
При высоких концентрациях добавление соли оказывает дру-
гое действие, например понижает активность воды при образо-
вании гидратированных ионов. В этом случае, напоминающем
эффект высаливания, большая часть полимеров может подвер-
гаться осаждению. Подобное действие, приводящее к осажде-
нию или разделению фаз, заставляет перемещаться молекулы
полимера по направлению к границе раздела кремнезем—вода
задолго до того, как начинает происходить какое-либо разделе-
ние в системе, в которой отсутствует соль.
Кажется аномальным тот факт, что добавление NaCl при
pH 5,9 вызывает повышение адсорбции ПЭО, хотя известно, что
добавление соли увеличивает концентрацию заряженных цент-
ров на поверхности кремнезема и, следовательно, должно пони-
жать возможность образования водородных связей. Однако до-
бавленный электролит также уплотняет толщину двойного слоя,
поэтому противоионы натрия перемещаются ближе к отрица-
тельно заряженным центрам (что доказывается понижением
электрокинетического потенциала). Следовательно, большая мо-
лекула ПЭО в виде произвольно закрученной спирали может
приблизиться более близко к поверхности и образовать водород-
ные связи с нейтральными группами SiOH. Но при рН~ 11 кон-
центрация противоионов — катионов Na+ — на поверхности или
вблизи нее становится настолько большой, что полностью пред-
отвращает адсорбцию ПЭО.
При оптимальных условиях при pH 2 имеется 7 сегментов
—С2Н4О— полимерной цепи, приходящихся на 1 нм2 поверхно-
сти. Это означает, что адсорбированный полимер лежит не
вполне плоско на поверхности, но образует петлю, находящуюся
в окружающей водной среде. К такому заключению пришли
также Говард и Мак-Коннелл [445].
Адсорбция из неводных растворителей
Степень, до которой линейные полимерные цепи способны
адсорбироваться плоско на силанольной поверхности кремне-
зема, зависит от силы водородной связи между группами SiOH
и полярными группами полимера, а также от возникающей кон-
куренции из-за воздействия растворителя, на полимер и поверх-
ностные группы SiOH. Таким образом, цепи ПЭО ложатся пло-
ско тогда, когда полимерные молекулы откладываются на сила-
нольной поверхности кремнезема, частично это происходит из-за
того, что гидрофобная углеводородная цепь отталкивается от
984
Глава 6
воды. Если растворитель хорошо совместим с углеводородами,
и особенно если он содержит активный атом водорода (напри-
мер, СНС13), способный образовывать водородные связи с эфир-
ными группами, то полимер ПЭО адсорбируется на кремнеземе
только по периодически расположенным эфирным группам. Сле-
довательно, на основании данных Говарда и Мак-Коннелла
[445], цепи образуют петли, направленные от поверхности в сто-
рону растворителя.
Авторы работы [446] нашли, что полиметилметакрилат ад-
сорбировался на кремнеземе из толуола, располагаясь плоско на
поверхности, так что атомы кислорода могли образовывать водо-
родные связи с группами SiOH. Однако когда использовались
растворители, например, содержащие атомы кислорода (в эфир-
ных, гидроксильных или кетогруппах), которые сами способны
формировать водородные связи с группами SiOH, то никакой
адсорбции не происходило, так как растворитель был способен
конкурировать с полимером на поверхности кремнезема.
Как показали Ботхэм и Тейс [447], в растворах, в которых
присутствовали смеси полимеров, ацетат этиленвинила, содер-
жащий группы СО, вытеснял полистирол с поверхности кремне-
зема в растворе трихлорэтилена. Однако совсем не было оче-
видным, что поливинилацетат должен вытеснять этилцеллюлозу,
которая в свою очередь замещала полиметилметакрилат. Поли-
мерная цепь, содержащая большее число точек присоединения
(атомы кислорода эфирных или карбонильных групп), как
и следовало ожидать, замещала полимерную цепь с меньшим
числом таких групп. В полимерных смесях тенденция к разде-
лению фаз находится во взаимосвязи со степенью более пред-
почтительной адсорбции по отношению к какому-либо поли-
меру. В полимерах, содержащих очень незначительное число
центров, способных связываться с поверхностью, большие поли-
мерные сегменты или петли могут удерживаться у поверхности
всего лишь немногими точками присоединения [448].
Ботхэм и Тейс [449] исследовали влияние пространственной
упорядоченности на адсорбцию полиизопропилатацетата из
растворителя СС12=СНС1. Во всех случаях основная доля сег-
ментов цепи оказывалась прочно связанной с кремнеземом, но
были отмечены некоторые различия между атактическим, син-
диотактическим и изотактическим полимерами. Основные вы-
воды связывались с флокуляцией; длинные полимерные цепи
образовывали мостики, связывающие каждую единичную частицу
кремнезема с соседними. Однако если первоначальная концент-
рация полимеров оказывалась слишком высокой, так что по-
верхность каждой частицы немедленно покрывалась сегментами
различных полимерных цепей, направленными в раствор вокруг
частицы, то совсем не оставалось участка на поверхности, спо-
Химия поверхности кремнезема 985
собного присоединить цепь, простирающуюся от соседней ча-
стицы кремнезема. С другой стороны, если концентрация поли-
меров была очень низкой, каждая цепь была в состоянии адсор-
бироваться и окутывать частицу до тех пор, пока вся поверх-
ность не покрывалась ею и на поверхности не оставалось сво-
бодных участков, способных присоединять полимерные цепи,
простирающиеся от другой частицы.
В работах [450, 451] сообщалось в дополнение к имевшимся
данным о проведении исследований адсорбции на кремнеземе
полиалкилакрилатов из органических растворителей. При ад-
сорбции из додекана полимерная цепь вытягивалась в достаточ-
ной мере плоско на поверхности кремнезема, причем 36 % по-
лимера связывалось с поверхностью, и толщина полимерной
пленки составляла только 20—40 А. На толщину пленки не ока-
зывала влияния молекулярная масса полимера. Однако никакой
адсорбции не происходило из растворителей, которые могли
сами связываться посредством водородных связей с поверхност-
ными группами SiOH. Удивительно, что из такого растворителя,
как MeCN, на кремнеземе адсорбировался изотактический поли-
мер, но не адсорбировался обычный полимер, имевший произ-
вольную конфигурацию. Адсорбированный полимер быстро уда-
лялся при воздействии метанола.
Полиэтиленгликоли, адсорбированные из растворителей на
кремнеземе, исследовались Киллменом и др. [452, 453]. Они
обнаружили, что приблизительно каждый второй атом кисло-
рода эфирных групп был связан водородными связями с поверх-
ностными группами SiOH. Авторы определили теплоты смачива-
ния. Атомы кислорода эфирных групп образовывали гораздо
более сильные водородные связи, чем вода или МеОН, так что
адсорбция полимеров наблюдалась из обоих растворителей. Ко-
роткие полимерные цепи (молекулярная масса 600) адсорбиро-
вались на одиночных частицах кремнезема, но для относительно
длинных по сравнению с размером кремнеземных частиц цепей
имело место образование мостиков между частицами и наблю-
дался процесс флокуляции.
Осаждение полислоев, образуемых из полиионов
и заряженных частиц
Поочередное осаждение взаимно реагирующих между собой
разновидностей на поверхности кремнезема было продемонстри-
ровано Айлером, который показал, что на заряженной поверх-
ности из раствора адсорбируется единичный слой противопо-
ложно заряженных по знаку полиионов или коллоидных частиц.
Затем раствор смывается и второй слой заряженных частиц
986
Глава 6
с противоположным по знаку зарядом по отношению к первому
слою накладывается на образец, а избыточный раствор опять
уделяется. Этот способ осаждения подробно рассматривался
в гл. 4 (см. лит. к гл. 4 [402—409]).
Аналогично представляется возможным осаждать чередую-
щиеся слои, состоящие из катионных и анионных органических
полимеров, что позволяет наращивать на кремнеземе проводя-
щие электричество покрытия. Кольцов, Алесковский и Волкова
[454] выполнили поочередные реакции на кремнеземе. Напри-
мер, хлорид, такой, как TiCl4, вступает в реакцию на поверхно-
сти с образованием монослоя и гидролизуется, после чего сле-
дующий монослой осаждается и гидролизуется и т. д. Одиноч-
ные молекулярные слои различных оксидов могут налагаться на
поверхность поочередно. Аналогично небольшие поры в силика-
геле могут постепенно перекрываться ступенчатым способом по-
средством проведения схожей серии реакций с SiCl4.
Алюмосиликатная поверхность
Некоторые характеристики и свойства алюмосиликатных
ионов на поверхности кремнезема рассматривались в гл. 3, где
была приведена константа химического равновесия реакции об-
разования алюмосиликата, и в гл. 4, в которой были описаны
способ образования и свойства золей кремнезема, модифициро-
ванных за счет введения алюмосиликата. Влияние оксида алю-
миния как примеси на растворимость кремнезема упоминалось
в гл. 1, а его влияние на токсичность кремнезема будет рас-
смотрено в гл. 7.
Необходимо по крайней мере упомянуть о химии поверхно-
сти алюмосиликатных катализаторов, которые имеют важное
значение с точки зрения технического и коммерческого исполь-
зования. Однако объем литературы по таким катализаторам на-
столько велик, что не представляется возможным дать здесь
соответствующий обзор.
Большая часть алюмосиликатных катализаторов представ-
ляют собой силикагели, в которые введена существенная доля
ионов АЮГ , являющихся изоморфными по отношению к SiO2.
Связанные алюмосиликатные ионы SiAlO~ сообщают поверхно-
сти сильно кислотное состояние. Хайр [9] представил обоснован-
ный обзор по поверхностным свойствам алюмосиликатных гелей,
природе кислотности и способам ее измерения.
На рис. 6.20 показаны различия между адсорбированным
ионом А13+ и алюмосиликатным анионным центром. Когда та-
кой центр в гидратированной форме, или так называемый брен-
стедовский кислотный центр, дегидратируется, то образуется не-
Химия поверхности кремнезема
987
Рис. 6.20. Схемы алюмосиликатной поверхности.
а — адсорбированный ион АР+ с координационным числом 6; б — алюмосиликатные ак-
тивные центры — бренстедовский кислотный центр; в — устойчивый бренстедовский кис-
лотный центр; а — дегидратированная поверхность — льюисовский кислотный центр.
ионный центр — льюисовский кислотный центр. Если оксид алю-
миния расположен непосредственно под поверхностью, то появ-
ляется бренстедовский кислотный центр, более устойчивый по
отношению к дегидратации [463].
Общие принципы образования и свойства алюмосиликатной
поверхности были изучены в 50-е годы [455—461]. Разработаны
структурные правила, в соответствии с которыми объединены
кремнезем и оксид алюминия [462]. Постепенно более подробно
была изучена структура поверхности алюмосиликата [463—466].
Кислотность такой поверхности измерялась многочисленными
методами [467—474].
988
Глава 6
Активные центры, свободные радикалы,
активный кислород, озон
Существуют данные, показывающие, что, по мере того как
при высокой температуре поверхность кварца лишается гидро-
ксильных групп, происходит выделение водорода. Этого не про-
исходит с аморфным кремнеземом, тем не менее его поверхность
проявляет определенные окислительные свойства. Бондаренко
и др. [475] предположили, что незавершенные тетраэдры SiO+
изменяют свою ориентацию так, что атомы кислорода появ-
ляются на поверхности,, причем такие атомы способны удаляться,
оставляя на поверхности объемный дефект. Красильников, Кисе-
лев и Сысоев [476] обнаружили, что на такой поверхности могла
быть измерена степень окисления путем йодометрического тит-
рования с использованием 0,01 н. раствора гипосульфита. По-ви-
димому, когда поверхность кремнезема дегидратируется в,ва-
кууме при высокой температуре, а затем подвергается действию
кислорода при 20°С, то кислород закрепляется на поверхности
и образуется нечто напоминающее поверхностный пероксид,
тогда как азот оказывается неактивным. Авторы также утвер-
ждают, что облучение силикагеля коротковолновым ультрафио-
летовым светом приводит к образованию поверхностных центров,
способных адсорбировать кислород. Подобные эффекты оказы-
ваются более заметными на силикагеле с удельной поверхно-
стью 400 м2/г, чем на образце с поверхностью 695 м2/г. Однако
эти эффекты оказываются слабыми, поскольку на тысячу исход-
ных групп SiOH на поверхности кремнезема приходится только
один такой центр, адсорбирующий кислород.
Согласно данным Чуанга и Тао [477], напряженные поверх-
ностные силоксановые группы SiOSi до некоторой степени ока-
зываются способными образовывать свободные радикалы. Та-
кие радикалы уничтожаются при воздействии воды или нитро-
метана. Радикалы могут обнаруживаться по аннигиляции
позитронов. Возможность наблюдения за аннигиляцией О-пози-
трония в силикагеле представляется уникальным способом ис-
следования размеров частиц, размеров пор и природы поверх-
ности кремнезема.
Поверхность кремнезема, способная вступать в реакции с ор-
ганическими веществами, образуется, когда плавленое кварцевое
стекло размалывается в шаровой мельнице [281, 478]. Хаузер
[479] сообщил, что атомарный кислород выделялся при получе-
нии свежеобразованной поверхности кремнезема. Шофилд,
Ральф и Грин [480] обобщили в своем обзоре эти и другие дан-
ные, показывающие, что когда кварц размалывался в присут-
Химия поверхности кремнезема
989
ствии монооксибензойной кислоты, то шло образование 3,4-ди-
оксибензойной кислоты. Относительно присутствия свободных
радикалов никакого доказательства не было получено. у-Излуче-
ние вызывало гидроксилирование бензойной кислоты, и этот про-
цесс промотировался в присутствии кремнезема.
Стрелко, Высоцкий и Ганюк [481] исследовали эффекты де-
гидратации кремнезема в присутствии веществ, способных поли-
меризоваться (стирол, винилацетат, тиофен), предполагая, что
образуемые свободные радикалы могли бы промотировать про-
цесс полимеризации. При этом наблюдалось много различных
эффектов, и в большинстве случаев поверхность кремнезема
способствовала образованию неэкстрагируемого органического
вещества. Эти факты наводят на мысль о том, что дальней-
шие исследования в подобном направлении могли бы стать
ценными.
В том случае, когда этерифицированный метиловым спиртом
пирогенный кремнезем подвергается воздействию высокой тем-
пературы в вакуумных условиях и модифицирующий слой рас-
падается, на поверхности остается очень незначительное число
активных центров, состоящих, вероятно, из смежных поверхно-
стных атомов кремния, связанных между собой очень активными
атомами кислорода [482, 483]. Если в группах RSR или ROSis
органический остаток R содержит более одного атома углерода,
то никакой активации не наблюдается, так что образование та-
ких своеобразных центров наблюдается только в присутствии
на поверхности метильных групп.
Обработка пирогенного кремнезема в парах метилхлорсила-
нов ведет к образованию радикалов СН3, которые, согласно экс-
периментальным данным, полученными методом ЭПР, остаются
устойчивыми на кремнеземном порошке даже при 94°С [484].
Метильные радикалы, стабилизированные в микропорах, обра-
зуются в том случае, когда Ме3А1 вступает в реакцию с гидро-
ксилированной поверхностью кремнезема, а затем образец под-
вергается воздействию осушенных воздуха или кислорода [485].
Пероксидные кремнеземные поверхности, согласно данным
Литковец и др. [486], приготовляются путем проведения реакции
между кремнеземом и кремнийорганическими хлорпероксидами
типа RmSi(O2R)nCl4-m-n.
При облучении ароматических соединений, адсорбированных
на силикагеле, на поверхности появляются ариловые радикалы,
которые остаются хемосорбированными. Шевец и др. [487] изу-
чили оптимальные условия термообработки такого силикагеля
с целью получения максимальной хемосорбции.
Боем [8] представил обзор по воцросу образования свобод-
ных радикалов, возникающих при облучении кремнезема УФ-
или у-излучением. у-Лучи обычно вызывают образование сво-
990
Глава 6
бодных радикалов, но не оказывают действия на структуру
кремнезема.
Быстрые нейтроны полностью разрушают структуру кремне-
зема: в более плотных формах кремнезема, подобных кварцу,
плотность при этом понижалась от 2,65 до 2,31 г/см3, тогда как
для прозрачного кварцевого стекла плотность наоборот возра-
стала от 2,22 до 2,30 г/см3. Состояние кремнезема с плотностью
2,30 г/см3 представляется наиболее интересным с точки зрения
изучения его микропористости. Если такой кремнезем нагре-
вается, то из возбужденного состояния кремнезем, вероятно,
возвращается обратно к исходному состоянию [488].
у-Излучение, получаемое от 6°Со, отщепляет группы ОН
и вызывает образование свободных валентностей, что ведет
к изменению адсорбционных характеристик кремнезема [489].
Подобная радиация, приводящая к усилению каталитической
активности кремнезема, вызывала также красное окрашивание
силикагеля, подобное окрашиванию фуксином, которое затем
обесцвечивалось при 25°С и взаимодействии с Н2, но не с, Ог;
более медленно кремнезем терял окрашивание при взаимодей-
ствии с Н2О. Такой цвет объясняется присутствием положи-
тельно заряженных электронных дырок на локальных участках,
где присутствует избыточный кислород [490].
Предпринималось много попыток изучения полимеризации
органических мономеров на поверхности силикагелей или алю-
мосиликатных гелей под воздействием радиации. Помимо того
что происходит процесс полимеризации, образуются свободные
радикалы, а полимеры распадаются с укорачиванием длины це-
пей, никаких других неожиданных результатов не было полу-
чено. Прививка полимера к поверхности частиц кремнезема,
по-видимому, мало отличается от тех случаев, когда исполь-
зуются катализаторы полимеризации [491]. Однако введение
диспергированного кремнезема, состоящего из чрезвычайно тон-
ких частиц, в мономеры еще до процесса полимеризации, веро-
ятно, могло бы привести к созданию новых разновидностей сме-
шанных неорганических и органических полимеров с полезными
свойствами.
В другом типичном направлении исследований радиацион-
ных эффектов были получены данные, подтверждающие, что на
силикагеле, обработанном в вакууме при температуре выше
400°С, при облучении возникают центры окрашивания, которые
при взаимодействии с водородом обесцвечиваются. Присутствие
таких добавок, как N2O или SFe, которые захватывают элек-
троны в процессе облучения кремнезема, способствуют образо-
ванию центров окрашивания. Эффективное значение сродства
для образования положительно заряженных электронных дырок
составляет 11 эВ [492]. у-Облучение кремнезема приводит к об-
Химия поверхности кремнезема
991
разованию электронов и положительно заряженных дырок, рас-
пространяющихся как по всему объему, так и на поверхности
[493]. Кроме того, если присутствует СО2, то он захватывает
электроны во время облучения с образованием СО~ [494]. До-
полнительно возникающая адсорбция кислорода и азота на си-
ликагеле после того, как образец подвергался у-облучению, ис-
следовалась методом ИК-спектроскопии [495]. Центрами ад-
сорбции протонов являлись положительно заряженные дырки,
стабилизированные на атомах кислорода, находящихся вблизи
атомов А! [496]. Комбинирование у-излучения и нейтронной
бомбардировки приводило к серо-желтому окрашиванию крем-
незема, а также вызывало частично протекающую необратимую
дегидратацию поверхности [497]. С другой стороны сообщалось,
что у-облучение вызывало дегидратацию кремнезема и погло-
щение кислорода [498]. Написан обзор [499] по группам на по-
верхности кремнезема, в том числе свободным радикалам.
Согласно данным Ерматова и др. [293], в том случае, когда
силикагель подвергается высокочастотному разряду в атмосфере
водорода, последний сильно адсорбируется. При этом атомарный
водород и ионизированные молекулы водорода вступают в реак-
цию с короткоживущими центрами, образующимися на поверх-
ности кремнезема. Вероятно, атомарный водород остается на по-
верхности.
Озон адсорбируется на силикагеле и может десорбироваться
без какой-либо реакции или распада. Изотермы адсорбции были
измерены в интервале от —117 до 20°С Атьякшевой и др. [500].
Согласно данным Реймшусселя и Маунтфорда [501], адсорбция
озона при комнатной температуре повышалась, если некоторое
количество воды уже присутствовало на поверхности кремне-
зема. Если кремнезем предварительно нагревался до 150°С, то
наблюдалось максимальное значение концентрации озона при
заполнении монослоя, когда измерения проводились при темпе-
ратуре —80°С. Это дало возможность предположить, что озон
наиболее прочно адсорбируется на силанольной поверхности.
Теплота адсорбции озона на силикагеле, у которого отсутствуют
микропоры, составляет около 2,6 ккал/моль [501]. Когда сили-
кагель вначале нагревался до 200—400°С, а затем после охла-
ждения подвергался облучению УФ-лучами в атмосфере кисло-
рода, то наблюдалась адсорбция кислорода [502]. Каждая пара
близко расположенных групп SiOH связывалась с одной моле-
кулой кислорода. Количество адсорбированного кислорода воз-
растало в высокочастотном разряде.
Относительно большинства из вышеприведенных реакций
можно сказать только то, что рассматриваемые соединения при-
сутствовали лишь в очень небольших концентрациях и количе-
992
Глава 6
ствах и не наблюдалось каких-либо макроскопических их вели-
чин, имеющих практическое значение. Ни одно из упомянутых
соединений не могло быть приготовлено каким-либо иным, более
легким способом. Тем не менее из типов кремнезема с высокими
значениями удельных поверхностей и очень рыхлой, открытой
структурой, например объемистых, прозрачных аэрогелей, при-
меняемых в качестве подложки для других элементов, могут
быть разработаны новые, селективные, светочувствительные,
активированные катализаторы.
ЛИТЕРАТУРА
1. Haber F., Z. Elektrochem., 20, 521 (1914).
2. Langmuir I., J. Amer. Chem. Soc., 40, 1361 (1918).
3. Айлер P. К. Коллоидная химия кремнезема и силикатов. Пер. с аигл.—
М.: Госстройиздат, 1959.
4. Киселев А. В.— Коллоидн. ж., 1936, т. 2, с. 17.
5. Carman Р. С., Trans. Faraday Soc., 36, 964 (1940). >
6. Белякова Л. Д., Джигит О. М., Киселев А. В., Муттик Г. Г., Щерба-
кова К. Д.— Ж. физ. химии., 1959, т. 33, с. 551.
7. Boehm Н. Р., Angew. Chem., 5, 533 (1966).
8. Boehm Н. Р., Adv. Catal., 16, 226 (1966) [361 refs.].
9. Hair M. L., Infrared Spectroscopy in Surface Chemistry, Dekker. New
York, 1967, p. 79.
10. Snyder L. R., Principles of Adsorption Chromatography, Dekker. New
York, 1968, p. 156.
11. Оккерс К. Сб. Строение и свойства адсорбентов и катализаторов. Пер.
с англ. Под ред. Б. Г. Линсена.— М.: Мир, 1973.
12. Snoeyink A., Weber W., “Surface Functional Groups on Carbon and
Silica”, in J. F. Danielli, M. D. Rosenberg, and D. A. Cadenhead, Eds.,
Progress in Surface and Membrane Science, Vol. 5, Academic, New York,
1972, p. 63.
13. Barby D., “Silicas”, Chapter 8, in G. D. Parfitt and K. S. W. Sing, Eds.,
Characterization of Powder Surfaces, Academic, New York, 1976, p. 353.
14. Kiselev A. V., Trans. Faraday Soc. Disc., 1971 (52), 14.
15. Wiese G. R., James R. 0., Yates D. E., Healy T. IF., “Electrochemistry
of the Colloid-Water Interface”, J. О. M. Bockris, Ed., Mtp. Int. Rev.
Sci., Phys. Chem., Vol. 6, Series 2, Butterworth, London, New York,
1976, p. 53.
16. Kane P. F., Larrabee G. B., Characterization of Solid Surfaces, Plenum,
New York, 1974.
17. Hockey J. A., Chem. Ind. London, 1965, 57 (January).
18. Bokasanyi L., Liardon O., Kovats E., Adv. Colloid Interface Sci.. 6, 95
(1976).
19. Stober W., Beitr. Silikose-Forsch., H 89, 87 (1966).
20. Meyer D. E., Hackerman N., J. Phys. Chem., 70, 2077 (1966).
21. Fowkes F. M., Burgess T. E., in G. Goldfinger, Ed., G. Goldfinger, Ex.,
Clean Surfaces (Symposium North Carolina State University, Dekker),
New York, 1970.
22. Dubinin M. M., Bering В. P., Serpinskii V. V., in J. F. Danielli,
K. G. A. Pankhurst, and A. C. Riddeford, Eds., Recent Progress in Sur-
face Science, Academic, New York, 1964, p. 42.
23. Klier K-, Zettlemoyer A. C., J. Colloid Interface Sci., 58, 216 (1977).
Химия поверхности кремнезема
993
24. Dalton ft. L., Iler ft. К., J. Phys. Chem., 60, 955 (1956).
25. Lyklema I., J. Colloid Interface Sci., 58, 242 (1977).
26. Peschel G., Aidfinger К. H., J. Colloid Interface Sci., 34, 505 (1970).
27. Drost-Hansen W., J. Colloid Interface Sci., 58, 251 (1977).
28. Plooster M. N„ Gitlin S. N„ J. Phys. Chem., 75, 3322 (1971).
29. Zimmerman J. ft., Holmes B. G., Lasater I. A., J. Phys. Chem., 60, 1157
(1956).
30. Morariu V. V., Mills ft., J. Colloid Interface Sci., 39, 406 (1972).
31. Gloates ft. I., Conrad V. H„ Soil Sci., 77, 313 (1954).
32. LeBot I., LeMontaguer S., J. Phys. Radium, 16, 163 (1955).
33. Lakhanpal M. L., Sud ft. K., Puri B. ft., J. Phys. Chem., 59, 160 (1955).
34. Bikerman I. I., Trans. Farday Soc., 36, 154 (1940); Kolloid.-Z., 72, 100
(1935).
35. Kittaka S., J. Colloid Interface Sci., 55, 431 (1976).
36. Muroya M„ Kondo S., Bull. Chem. Soc. Jap., 47 (6), 1533 (1974).
37. De Boer J. H., Vleeskens J. M., K. Ned Akad. Wet. Proc. Ser. B, 60,
23, 45, 54 (1957); 61, 2, 85 (1958).
38. Okkerse C. [11], p. 251.
39. Shapiro I., Weiss H. G., J. Phys. Chem., 57, 219 (1953).
40. Lowen W. K, Broge E. C., J. Phys. Chem., 65, 16 (1961).
41. Iler ft. K., personal observation.
42'. Erkelens J., Linsen B. G., J. Colloid Interface Sci., 29, 464 (1969).
43. Davydov V. Ya., Kiselev A. V., Zhuralev L. T., Trans. Faraday Soc., 60,
2254 (1964).
44. Noll W„ Damm K, Fauss ft., Kolloid-Z„ 169, 18 (1960).
45. Kellum G. E. et al., Anal. Chem., 39, 341, 1623 (1967); 40, 2058 (1968).
46. Van Tongelen M., Uytterhoeven J. B., Fripiat I. J., Bull. Soc. Chim. Fr.,
1965, 2318.
47. Fripiat J. J., Uytterhoeven I. et al., J. Phys. Chem., 66, 800 (1962); Bull.
Soc. Chim. Fr., 1965, 1800.
48. Morimoto T., Naono H., Bull. Chem. Soc. Jap., 46, 2000 (1973).
49. Bermudez V. M., J. Phys. Chem., 74, 4160 (1970).
50. Wirzing G., Naturwissenschaften, 50, 13, 466 (1963).
51. Baverez M., Bastick I., C. R. Acad. Sci., 260 (14), 3939 (1965).
52. Lange K. ft., J. Colloid Sci., 20, 231 (1965).
53a. Манк В. В., Баран А. А., Янковская Г. Ф.— Укр. хим. ж., 1972, т. 38.
с. 934.
536. Квливидзе В. Я,—ДАН СССР, 1964, т. 157, С. 158.
54. Cornier G., Baverez М., Bastick I., Bull. Soc. Chim. Fr., 1968 (7), 2707.
55. Давыдов В. Я-> Киселев А. В.— ЖФХ, 1963, т. 37, вып. 11, с. 1404.
56. Doremus ft. Н., J. Phys. Chem., 75, 3147 (1971).
57. Thompson W. К., Proc. Brit. Ceram. Soc., 5, 143 (1965).
58. Uytterhoeven I., Hellinckx E., Fripiat I. I., Silic. Ind., 28, 241 (1963)
[Chem. Abstr., 59, 5805d].
59. Чертов В. M„ Джамбаева Д. Б. и др.— ДАН СССР, 1965, т. 161, с. 345.
60. Чертов В. М., Джамбаева Д. Б. и др.— ЖФХ, 1966, т. 40, с. 520.
61. Armistead С. G., Tyler A. I., Hambleton F. Н., Mitchell S. A., Hockey I. А.,
J. Phys. Chem., 73, 3947 (1969).
62. Журавлев Л. Т., Киселев А. В.— Коллоидн. ж., 1962, т. 24, с. 23; Жу-
равлев Л. Т., Киселев А. В., Найдина В. П., Поляков А. Л.— ЖФХ,
1963, т. 37, с. 2258.
63. Агзамходжаев А. А., Журавлев Л. Т„ Киселев А. В., Шенгелия К. Я.—•
Изв. АН СССР, сер. хим., 1969, вып. 10, с. 2111.
64. Madeley I. D., Richmond ft. С., Z. Anorg. Allg. Chem., 389 (1), 92
(1972).
65. Wirzing G., Naturwissenschaften, 51, 211 (1964).
37 Заказ № 250
994
Глава 6
66. De Boer I. H., Vleeskens J. M., K. Ned. Akad. Wet. Proc. Ser. B, 61,
2 (1958); Angew Chem., 70, 383 (1958).
67. Peri J. B., Hensley A. L„ J. Phys. Chem., 72, 2936 (1968).
68. Anderson J. H., Wickersham K. A., Surf. Sci.. 2, 252 (1964); 3 (8), 290
(1965).
69. Bermudez V. M., J. Phys. Chem., 75, 3249 (1971).
70. Hanke W7., Z. Anorg. Allg. Chem., 395, 191 (1973).
71. Lieflander M., Stober W7., Z. Naturforsch., 15, 411 (1960).
72. Белякова Л. Д„ Джигит О. M., Киселев А. В.— ЖФХ, 1957 т 31,
с. 1577.
73. Жданов С. П., Киселев А. В.—ЖФХ, 1957, т. 31, с. 2115.
74. De Boer I. Н., Angew. Chem., 70 (13), 383 (1958).
75. De Boer I. H., Vleeskens I. M., K. Ned. Akad. Wet. Ser. B, 61, 2 (1958).
76. Стрелка В. В., Адсорбция и адсорбенты, Респ. межвед. сб., вып. 2,
1974, с. 65.
77. Antonini J. F., Hochstrasser G., Surf. Sci., 32, 665 (1972).
78. Жданов С. П,— ДАН СССР, 1957, т. 115, с. 557.
79. Kautsky Н., Michel R., Z. Naturforsch., 7В, 414 (1952).
80. Dietz V. R„ Turner N. H., J. Phys. Chem., 75, 2718 (1971).
81. Ливеровская H. В., Кольцов С. И.— ЖФХ, 1965, t. 39, c. 408.
82. Frissard Imelik B., Bull. Soc. Chim. Fr., 1963, 1712; Mathieu M. V.,
Imelik B., J. Chim. Phys., 59, 1189 (1962); Naccacche C., Imelik B.,
J. Chim. Phys., 59, 359 (1962); Bull. Soc. Chim. Fr., 1961 (3), 553’.
83. U ytterhoeven I., Sleex M. et al., Bull. Soc. Chim. Fr., 1965, 1800.
84. Taylor I. A. G., Hockey I. A., Pethica B. A., Proc. Brit. Ceram. Soc., 5,
133 (1965).
85. Журавлев Л. T., Киселев A. B.— ЖФХ, 1965, t. 39, c. 236.
86. Taylor I. A. G., Hockey I. A., J. Phys. Chem., 70, 2169 (1966).
87. Curthoys G., Davydov V. Y., Kiselev A. V. et al., J. Colloid Interface
Sci., 48, 58 (1974).
88. Armistead C. G., Hockey I. A., Trans. Faraday Soc., 63, 2549 (1967).
89. Thorp I. Al. et al., J. Phys. Chem., 66, 1086 (1962); 67, 2617 (1963);
Trans. Faraday Soc., 55, 442 (1959); 61, 962, 975 (1965); 62, 1928, 2287
(1966); 63, 2068 (1967); 64, 2190 (1968); 65, 869, 1741 (1969).
90. Дзисько В. А., Вишневская А. А., Чесалова В. С.— ЖФХ, 1950, т. 24,
с. 1416.
91. McDonald R. S., J. Am. Chem. Soc., 79, 850 (1957); J. Phvs. Chem., 62,
1168 (1958).
92. Young G. I., J. Colloid Sci., 13, 67 (1958).
93. Киселев А. В., Лыгин В. И.— Коллоидн. ж., 1959, т. 21, с. 581.
94. Friplat I. I., Gastuche Al. С., Brichard R., J. Phys. Chem., 66, 805 (1962).
95. Kondo S., Muroya Al., Bull. Chem. Soc. Jap., 43, 2657 (1970).
96. Kunawicz I., Tones P„ Hockey A., Trans. Faraday Soc., 67, 848 (1971).
97. Hockey I. A., Pethica B. A., Faraday Soc. Trans., 57, 2247 (1961).
98. Kondo S., Fujiwara F., Muroya M., J. Colloid Interface Sci., 55, 421
(1976).
99. Elmer T. H„ Nordberg Al. E., пат. США 3459522 (Corning Glass Works,
Inc.), 1969.
100. Pohle W„ Fink P.. Z. Chem., 12 (10), 394 (1972).
101. Stober W., Kolloid-Z., 145, 17 (1956).
102a. Young G. J., Bursh T. P., J. Colloid Sci., 15, 361 (1960).
1026. Egerov Al. Al., Kvlividze IF. I., Kiselev V. F., Krasilnikov G. K.. Koi.
loid-Z. Z. Polym., 212, 126 (1966).
103. Brunauer S., Kantro D. L., Weise С. H., Can. J. Chem.. 34. 1483 (1956).
104. Whalen I. IF., J. Phys. Chem., 71, 1557 (1967).
105 Дзисько В. А., Вишневская А. А., Чесалова В. С.— ЖФХ, 1950, т. 24,
с. 1416.
Химия поверхности кремнезема
995
106. Snyder L. R„ Ward I. W., J. Phys. Chem., 70 (12), 394 (1966).
107. Бакырджиев И.— ДАН СССР, 1966, т. 168, с. 618.
108. Mills G. A., Hinden S. G, J. Am. Chem. Soc., 72, 5549 (1950).
109. Snyder L. R., Sep. Sci., 1, 191 (1966).
ПО. Агзамходжаев А. А., Журавлев Л. T., Киселев А. В., Шенгелия К- Я.—•
Коллоидн. ж., 1974, т. 36, с. 1145.
111. Волков А. В., Киселев А. В., Лыгин В. И.— ЖФХ, 1974, т. 48, с. 1214.
112. Киселев В. Ф.— ЖФХ, 1960, т. 34, вып. 3, с. 332.
113. Good F. /., J. Colloid Interface Sci., 59, 398 (1977).
114. Patrick W. A., “The Properties of Highly Desiccated Silica Gel”, Sym-
posium on Catalysts and Reaction Mechanism, sponsored by the Cata-
lyst Club of Philadelphia, University of Pennsylvania, April 28, 1951.
115. Tyler A. Taylor J. A. G., Pethica B. A., Hockey I. A., Trans. Faraday
Soc., 67 (2), 483 (1971).
116. Laskowski L, Kitchener J. A., J. Colloid Surface Sci., 29, 670 (1969).
117. Ильин Б. В., Киселев В. Ф., Красильников К- Г.— Вестник Моск, уни-
вер., сер. мат., механ., астрои., физ., хим., 1958, т. 13, вып. 2, с. 223.
118. Егоров М. М., Красильников К. Г., Сысоев Е. А.— ДАН СССР, 1956,
т. 108, с. 103.
119. Литтл Л. Инфракрасные спектры адсорбированных молекул. Пер.
с англ,-—М.: Мир, 1969.
120. Киселев А. В., Лыгин В. И. Инфракрасные спектры поверхностных сое-
динений и адсорбированных веществ.— М.: Наука, 1972.
121. Ross R. A., Taylor А. Н., Proc. Brit. Ceram. Soc., 5, 167 (1965).
122. Bastick I., C. R. Acad. Sci., 247, 203 (1958).
123. Kiselev A. V., Adsorption (Exhibits of Soviet Science, Technology and
Culture Exhibition, New York, 1959), USSR Academy of Sciences, Mos-
cow, 1959.
124. Джигит О. M., Киселев А. В., Муттик Г. Г.— Коллоидн. ж., 1961, т. 23,
с. 504.
125. Elkington Р. К, Curthoys G„ J. Colloid Interface Sci., 28, 331 (1968).
126. Meye .W., Koezinger H., Acta. Cient. Venez. Supl., 24, 170 (1973).
127. Sempels R. E., Rouxhet P. G., Bull. Soc. Chim. Belg., 84, 361 (1975).
128. Yoshino T., J. Chem. Phvs., 23, 1564 (1955).
129. Kagel R. 0., J. Phys. Chem., 74, 4518 (1970).
130. Bliznakov G., Polikarova R., J. Catal., 5, 18 (1966).
131. Boyle T. W., Gaw W. J. et al., J. Chem. Soc., 1965, 240.
132. Kubelkova L. K-, Лги P., Collect. Czech. Chem. Commun., 37, 2853
(1972) [Chem. Abstr., 78, 128764e],
133. . Knoezinger H., Surf. Sci., 40, 339 (1974).
134. Bartell F. E., Dobay D. G., J. Am. Chem. Soc., 72, 4388 (1950).
135. Van Cauwelaert F. H., Van Assche I. B., U ytterhoeven J. B., J. Phys.
Chem., 74, 4329 (1970).
136. Chessick I. I., Zettlemoyer A. C., J. Phys. Chem., 65, 1672 (1961).
137. Gorski С. H., Stettler L. E., J. Colloid Interface Sci., 37, 918 (1971).
138. Shapiro I., Kolthoff I. M., J. Am. Chem. Soc., 72, 776 (1950).
139. Брунауэр С. Адсорбция газов и паров. Пер. с англ.— М.: ИЛ, 1948.
140. Trouton F. Т., Proc. Roy. Soc. London Ser. A, 79, 383 (1907).
141. Жданов С. П— ДАН СССР, 1949, т. 68, с. 99.
142. Hertl W„ Hair M. L„ Nature, 223, 1 151 (1969).
143. Prigogine M., Fripiat I. I., Bull. Soc. Roy. Sci. Liege, 7—10, 449 (1974).
144a . Mikhail R. S., Hashed S., Sing K. S., in S. Modry, Ed., Pore Struct.
Prop. .Mater. Proc. Int. Symp. Rilem-Iupac (1973), Vol. 4, Academia,
Prague, 1974, p. 157.
1446. Дубинин M. M.— Известия АН СССР, отд. хим. наук, 1960, с. 1620.
145. Baker F. S„ Sing К. S. IF., J. Colloid Interface Sci.,’55, 605 (1976).
146. Аристов Б. Г., Киселев А. В. и др.— ЖФХ, 1963, т. 37, с. 2520.
37*
996
Глава 6
147. Bassett D. R., Boucher E. A., Zettlemoyer A. C., J. Colloid Interface
Sci., 27, 649 (1968).
148. Hair M. L„ Hertl W„ J. Phys. Chem., 73, 4269 (1969).
149. Clark-Monks C., Ellis B., J. Colloid Interface Sci., 44, 37 (1973).
150. Давыдов В. Я., Киселев А. В., Лысин В. И,—ЖФХ, 1963, т. 37, с. 469.
151. Джигит О. М., Киселев А. В. и др.— Коллоидн. ж., 1962, т. 24, с. 241.
152. Егоров М. М. и др.— Вестник Моск, универ, сер. матем., механ., астрой.,
физ. хим., 1957, т. 12, с. 35; Ильин Б. В. и др., там же, 1958, т 13,
с. 203.
153. Bauer G„ Stober W., Kolloid-Z., 167, 27 (1959).
154. Hertl W„ Hair M. L., J. Phys. Chem., 72, 4676 (1968),
155. Hair M. L.. J. Colloid Interface Sci., 59, 532 (1977).
156. Киселев А. В., Новикова В. H., Эльтеков JO. А.— ДАН СССР 1963
т. 149, с. 131.
157. Glass R. W„ Ross R. A., Can. J. Chem., 50, 1241, 1666 (1972).
158. Robert L„ C. R. Acad. Sci., 234, 2066 (1952).
159. Jones D. C., Outridge L„ J. Chem. Soc., 1930, 1574.
160. Matayo D. R., Wightman J. P„ J. Colloid Interface Sci., 35, 354 (1971).
161. Griffiths D. M., Marshall K-, Rochester С. H., J. Chem. Soc. Faradav
Trans. I, 70 (2), 400 (1974).
162. Low M. J. D., Hasegawa M., J. Colloid Interface Sci., 26, 95 (1968).
163. Ганиченко Л. Г., Киселев В. Ф., Красильников К. Г.—ДАН СССР, 1959,
т. 125, с. 1277.
164. Бонецкая А. К., Красильников К. Г., ДАН СССР, 1957, т. 114, с. 421.
165. Николаев А. Л., Мигуров Е. А., Вовченко Г. Д.— ЖФХ, 1973 т 47
с. 1554.
166. Elder A. L., Springer R. A., J. Phys. Chem., 44, 943 (1940).
167. Armistead С. G., Tyler A. J., Hockey J. A., Trans. Faraday Soc., 67,
493, 500 (1971).
168. Рахлевская M. H. и dp.— ЖФХ, 1973, t. 47, c. 2354.
169. Low M. J. D., Lee P. L., J. Colloid Interface Sci., 45, 148 (1973).
170. Marshall K-, Rochester С. H., J. Chem. Soc. Faraday Trans. I, 71, 1754
(1975).
171. Eltekov Y. A., Khopina V. V., Kiselev A. V., J. Chem. Soc. Farad. Trans.
I, 68 (5), 889 (1972).
172. Morimoto T., Naono H., Bull. Chem. Soc. Jap., 45, 700 (1972).
173. Веремчук M. K-, Квитковский Л. H.— Нефтепереработка и нефтехимия.
Респ. межвед. сб., вып. 7, Киев: Наукова думка, 1972, с. 88—91.
174. Winkler Н., Nagel М., Michel D., Pfeifer Н., Z. Phys. Chem. Leipzig,
248, 17 (1971).
175. Robert L., Kessaissia Z., Tabak G., C. R. Acad. Sci. Ser. C, 276, 451
(1973).
176. Davis К- M. C., Deuchar J. A., Ibbitson D. A., J. Chem. Soc. Faraday
Trans. 1, 69 (6), 1117 (1973).
177. Jones D. C., Mills G. S., J. Chem. Soc., 1957, 213.
178. Rupprecht H., Kindi G., Arch. Pharm., 308, 46 (1975) [Chem. Abstr., 82,
. 144896л].
179. Maatman R., Geertsema A. et al., J. Phys. Chem., 72, 97 (1968).
180. Boulton L. H., Clark B. R., Coleman M. F., Thorp J. M., Trans. Faraday
Soc., 62, 2928 (1966).
181. Thorp J. M., Woolf J. B., Trans. Faraday Soc., 63, 2068 (1967).
182. Donahue D. J., Bartell F. E., J. Phys. Chem., 56, 665 (1952).
183. Nechtschein J., Bull. Soc. Chim. Fr., 3 (1), 913 (1973).
184a. Schindler P.t Kamber H. R., Helv. Chim. Acta, 51, 1781 (1968).
1846. Parks G. A., “Aqueous Surface Chemistry of Oxides and Complex Oxide
Materials”, in Equilibrium Concepts in Natural Water Systems. (Adv.
Chem. Ser. 67), American Chemical Society, Washington, D. C., 1967.
Химия поверхности кремнезема
997
185. Parks G. A., Chem. Rev., 65, 177 (1965).
186. Lyklema J., Faraday Soc. Disc., 52, 318 (1971).
187. Healy T. W., Furstenau D. W., J. Colloid Sci., 20, 376 (1965).
188. Wiese G. R., James R. O., Healy T. IF., Faraday Soc. Disc., 52, 302
(1971).
189. Hair M. L„ Hertl W„ J. Phys. Chem., 74, 91 (1970).
190. Marshall K., Ridgewell G. L., Rochester С. H., Simpson J., Chem. Ind.
London, 1974 (19), 775.
191. Schindler P. W., Furst B., Dick R., Wolf P., J. Colloid Interface Sci.,
55, 469 (1976).
192. Ahmed S. M„ Can. J. Chem., 44, 1663 (1966).
193. Кириченко Л. Ф., Высоцкий 3. 3.— ДАН СССР, 1967, т. 175, с. 635.
194. Abendroth R. Р., J. Colloid Interface Sci., 34, 591 (1970).
195. Griot О., Kitchener J. A., Nature, 200, 1004 (1963).
196. Maroto A. J. G., Griot O., An. Asoc. Quim. Argent., 58, 181 (1970).
197a. Tschapek M., Torres-Sanchez R. M., J. Colloid Interface Sci., 54, 460
(1976).
1976. Stumm W., Huang С. P., Jenkins S. R., Croat. Chem. Acta, 42, 223
(1970).
197b. Stumm W., Hohl H., Dalang F., Croat. Chem. Acta (1977) [in press].
198. Tadros T. F., Lyklema J., J. Electroanal. Chem. 17, 267 (1968).
199. Perram J. IF., J. Chem. Soc., Faraday Trans., 69, 993 (1973).
200. Yates D. E., Levine S„ Healy T. IF., J. Chem. Soc. Faraday Trans. I, 70,
1807 (1974).
201. Smit W„ Holten C. L. M. et al., J. Colloid Interface Sci., 63, 120 (1978).
202. Bolt G. H., J. Phys. Chem., 61, 1166, 1967.
203a. Yates D. E„ Healy T. W„ J. Colloid Interface Sci., 55, 9 (1976).
2036. Davis J. A., James R. O., Leckie J. O., J. Colloid Surface Sci., 63, 480
(1978).
204. Кириченко Л. Ф., Чертов В. M., Высоцкий 3. 3., Стражеско Д. Н.—
ДАН СССР, 1965, т. 164, с. 618.
205. Morariu V. V., Mills R., J. Phys. Chem. Frankfurt, 85 (1—4), 38 (1973).
206. Starer-Mendes H„ C. R. Acad. Sci. Ser. C, 273 (23), 1573 (1971).
207. Неймарк И. E., Чертов В. M., Шейнфайн Р. Ю., Кругликова Н. С.—
ДАН СССР, 1960, т. 132, с. 1356.
208. Carroll В., Freeman Е., J. Phys. Chem., 58, 335 (1954).
209. Boehm Н. Р., Schneider М., Z. Anorg. Chem., 301, 326 (1959).
210. Iler R. K-, J. Colloid Interface Sci., 53, 476 (1975).
211. Greenberg S. A., J. Phys. Chem., 60, 325 (1956).
212. Tadros E., Lyklema J., J. Electroanal. Chem. Interfacial Electrochem.,
22, 1 (1969).
213. Ahrland S., Grenthe I. et al., Acta Chem. Scand., 14, 1059 (1960).
214. Stanton J. H., Maatman R. W., J. Colloid Sci., 18, 132 (1963).
215. Stanton J. H., Maatman R. W. et al., J. Phys. Chem., 68, 757 (1964).
216. French С. M., Howard J. P., Chem. Ind. London, 1956, 572.
217. Wiese G. R. et al., Ref. 15, p. 61.
218. Kohlschutter H. W., Gerost H. et al., Z. Anorg. Allg. Chem., 308, 190
(1961).
219. Matijevic E., Mangravite F. I., Cassell E. A., J. Colloid Interface Sci.,
35, 560 (1971).
220. Johansson G. [71] of Chapter 3 and discussion.
221. Matijevic E., J. Colloid Interface Sci., 43. 217 (1973).
222a. Кольцов С. И. и dp.— Изв. высших учебных заведений, химия и химии,
технол., 1973, т. 16, вып. 10, с. 1475.
2226. Vydra F., Markova'V., J. Inorg. Nucl. Chem., 26, 1319 (1964).
223. Комиссаренков A. A. и dp.— Изв. АН СССР, Неорган. материалы, 1971,
т. 1, с, 2195,
998
Глава 6
224. Maatman R. W, U. S. At. Energy Comm. COO-1354-5 (1965).
225. Hohl H„ Stumm W„ J. Colloid Interface Sci., 55, 281 (1976).
226. Pott G. T., McNicol B. D., Disc. Faraday Soc., 52, 121 (1971).
227. Schenk f. E., Weber W. f., fr., J. Am. Water Works Assoc., 60, 199
(1968).
228. Healy T. W., Cooper R., fames R. O., Am. Chem. Soc. Sect. Div. Water
Air Waste Chem. Prepr, 7 (2), 91 (1967).
229. Kolthoff I. M„ Stenger V. A., J. Phys. Chem., 36, 2113 (1932).
230. Kozawa A., J. Inorg. Nucl. Chem., 21, 315 (1961).
231. Healy T. W., fames R. O., Cooper R., Adv. Chem. Ser., 79, 62 (1968).
232. Kikuchi K., Bernard H. W., et al., J. Phys. Chem., 69, 3654 (1965).
233a. Burwell R. L., fr., Pearson R. G., et al., Inorg. Chem., 4, 1123 (1965).
2336. Weetal H. H., Havewala N. B., Enzyme Engineering, Wilev, New York,
1972, p. 241.
233в. Biomedical Supports, Corning Biological Products Dept., Medfield, Mass.,
1973.
234. Eschrick H, Herrera-Huertas M., Tallberg K-, U. S. At. Energy Comm.
NP-18738 (1970) [Nucl. Sci. Abstr., 25 (14), 32793 (1971)].
235. Dibbs H. P., Can. J. Chem., 40, 565 (1962).
236. Малыгин А. А., Волкова A. H. и др.— Ж. общей химии, 1972, т. 42,
вып. И, с. 2373.
237. Maroto A. f. G. et al., Radiochem. Radioanal. Lett., 20, 121 (1975)
[Chem. Abstr., 82, 130715г].
238, Benton D. P., Elton G. A. H., Trans. Faraday Soc., 49, 1213 (1953).
239.' Dushina A. P., Kondrasheva A .L. et al., lonnyi Obmen, 132 (1970)
[Chem. Abstr., 74, 116290b],
240. Душина А. П„ Алесковский В. Б.— Ж. общей химии, 1968, т. 38, вып. 7,
с. 1419.
241. Wakatsuki Т., Furukawa Н., Kawaguchi К., Soil Sci. Plant. Nutr. Tokyo,
20, 353 (1974) [Chem. Abstr., 82, 160671г].
242. Dalang F., Stumm W., Int. Conf. Colloids Surf., San Juan, Puerto Rico,
June 21________25 1976
2. 43. fames R. O., Healy T. W., J. Colloid Interface Set., 40, 65 (1972).
244. Vydra F., Markova V., Collect. Czech. Chem. Commun., 30 (7), 2382
(1965) [Chem. Abstr., 63, 9048а].
245. Smith G. W., facobson H. W., J. Phys. Chem., 60, 1008 (1956).
246. Read A. D., Kitchener f. A., J. Colloid Interface Sci., 30, 391 (1969).
247. Taniguchi K. et al., Bull. Inst. Chem. Res. Kyoto Univ., 49, 212 (1971);
Nippon Kagaku Zasshi, 91 (6), 529 (1970) [Chem. Abstr, 73, 80980].
248. Душина А. П„ Алесковский В. Б. и др.— Ж. прикл. химии, 1970, т. 43,
вып. 11, с. 2443; 1972, т. 45, вып. 1, с. 33; 1973, т. 46, с. 1643.
249. Каверинский В. А., Бороков В. Ю. и др.— Кинетика и катализ, 1974,
т. 15, с. 819.
250. Бротиковский О. И., Швец В. А. и др. Кинетика и катализ, 1972, т. 13,
вып. 5, с. 1342.
251. Hathaway В. f., Lewis С. Е., J. Chem. Soc. А, 15, 2295 (1969).
252. Hierl С., Krauss Н. L., Z. Anorg. Allg. Chem, 401 (3). 263 (1973).
253. Калатзей E. А., Душина А. П. и др.— Ж. прикл. химии, 1974, т. 47,
вып. 3, с. 481.
254. Суздалев И. П., Гольданский В. И. и др.— Ж. эксп. теорет. физики,
1965, т. 49, вып. 5, с. 1424.
255. Goldanskii V. I., Neimark I. Е. et al., in G. K. Boreskov, Ed, Porous
Struct. Catal. Transp. Proc. Heterog. Catal. Symp, 1968, Akad. Kiado,
Budapest, 1972, p. 293.
256. Thompson K. D., Nucl. Sci. Abstr, 28 (9), 23969 (1973); Rep. IS-T-594,
Dept. NT[S (1973).
257. Sulcek Z., Vasak M., Dolezal f., Microchcm. J, 16 (2), 210 (1971).
Химия поверхности, кремнезема
999
258. Vydra F., Calba J., Collect. Czech. Chem. Commun., 32 (10), 3530 (1967)
[Chem. Abstr., 67, 103120].
259. Cornet D., Burwell R .L., Jr., J. Am. Chem. Soc., 90 (10), 2489 (1968).
260. Schenk J. E., Weber W. J., Jr., J. Am. Water Works Assoc., 60, 199
(1968).
261. Caletka R., J. Radioanal. Chem., 6 (1), 5 (1970).
262. Caletka R., Radiokhimia, 12 (3), 448 (1970). 0 ‘
263. Caletka R., Radiokhimia, 12 (4), 549 (1970). i
264. Caletka R., Kyrs M., Collect. Czech. Chem. Commun., 29, 1150 (1964).
265. Caletka R., Radiokhimia, 12 (4), 554, 558 (1970).
266. Kohlschuetter H. W., Katzenmayer W., Z. Anorg. Allg. Chem., 329 (1—2),
163 (1964).
267. Caletka R., Collect. Czech. Chem. Commun., 37 (5), 1684 (1972).
268. Davydov Y. P„ Radiokhimiya, 14 (2), 210 (1972).
269. Rozzell T. C., Andehnan J. B., Non-Equil. Syst. Natur. in Water Chem.
(Adv. Chem. Ser.), American Chemical Society, Washington, D. C., 1971,
p. 106.
270. Гребенщикова В. И., Давыдова Ю. П.— Радиохимия, 1965, т. 7, вып. 2,
с. 191.
271. Pullukat Т. J., Makromol. Chem., 176 (8), 2479 (1975).
272. Maatman R. W., Kramer A., J. Phys. Chem., 72, 104 (1968).
273. Zaki M. R., Abd-El-Moneim I., Z. Anorg. Allg. Chem., 360 (3—4), 208
(1968).
274. Кабышев Г. Я,—ДАН СССР, 1959, т. 127, вып. 2, с. 373.
275. Akatsu Е. et al., J. Nucl. Sci. Technol. Tokyo, 2 (4), 141 (1965)-.
276. James R. O., Wiese G. R., Healy T. U7., J. Colloid Interface Sci., 59,
381 (1977).
277. Devel H„ Wartman J. et al., Helv. Chim. Acta., 42, 1160, 1166 (1959).
278. Wartmann J., Devel H., Chimia Buenos Aires, 12, 82 (1958).
279. Стрелко В. В., Канаболоцкий В. А.— Коллоидн. ж., 1971, т. 33, вып. 5,
с. 750.
280. Tsutsumi К., et al., Nippon Kagaku Kaishi, 1973 (8), 1374.
281. Benson R. E., Castle J. E., J. Phys. Chem., 62, 840 (1958).
282. Fraissard J., Caillat R. et al., J. Chim. Phys., 60, 1017 (1963).
283. Ganyuk L. N. in D. N. Strazhesko, Ed.t Adsorption and Adsorbents,
Vol. 1, Wiley, New York, 1973.
284. Ганюк Л. H. Адсорбция и адсорбенты, Респ. межвед. сб., вып. 1, Киев:
Наукова думка, 1972, с. 98.
285, Wolf F., Beyer Н., Haedicke U., J. Prakt. Chem., 24 (3—4), 154 (1964).
286. Elmer T. H., Chapman I. D., Nordberg M. E., J. Phys. Chem., 67, 2219
(1963).
287. Чукин Г. Д.— Ж. прикл. спектроск., 1974, т. 21, с. 879.
288. Uytterhoeven J., Naveau Н., Bull. Soc. Chim. Fr., 1962, 27.
289. Hair M. L., Hertl W„ J. Phys. Chem., 77, 2070 (1973).
290. Тертых В. А. и др. Сб. Физико-химич. механика и лиофильность дис-
персных систем, вып. 4, Киев: Наукова думка, 1973, с. 37.
291. Boehm И. Р., Schneider М., Wistuba Н.. Angew Chem., 77, 622 (1965).
292. Ikeno S., японск. пат. 75-18424 (Matsushita Elect. Wks.), 1973.
293. Ерматов С. E. и др.— Изв. АН Каз. ССР, серия физ.-мат., 1972, т. 10,
вып. 4, с. 71.
294. Blomfield G. A., Little L. Н., Can. J. Chem., 51, 1771 (1973).
295. Fink Р., Marx G., Meyer К, Z. Chem.. 13, 314 (1973).
296. Ruttenberg F. E., Low M .J. D., J. Am. Ceram. Soc., 56, 241 (1973),
297. Fery N., Hoene R., Hamann K., Angew. Chem. Int. Ed., 11, 337 (1972).
298. Fripiat J. J., Van Tongelen M., J. Catal., 5, 158 (1966).
299. Shapiro I., пат. США 2829981, 1958.
300. Peglar R. J., Hambleton F. H., Hockey J. A., J. Catal., 20, 309 (1971).
1000
Глава 6
301. Fink Р., Pforr G., Rackow В., Z. Chem., 10, 424 (1970); 12, 451 (1972).
302. Rhee К. H., Basila M. R., J. CataL, 10, 243 (1968).
303. Morrow B. A., Devi A., J. Chem. Soc. Faraday Trans. I, 68 (3), 403
(1972).
304. Кольцов С. H., Ухова T. В., Волкова А. И.— Ж. прикл. хим., 1974,
т. 47, с. 20.
305. Heyer W„ Wolf E., Z. Anorg. Allg. Chem., 393, 50 (1972).
306. Hair M. L., Hertl W., J. Phys. Chem., 77, 1965 (1973).
307. Киселев А. В., Лыгин В. И., Щепалин К- Л.— Кинетика и катализ,
1971, т. 12, с. 185.
308. Кельцев Н. В. и др.— ЖФХ, 1970, т. 44, с. 1592, 2246.
, 309. Eldridge J. М., Balk Р., Trans. Met. Soc., AIME, 242, 539 (1968).
310. Волкова A. H. и др.— Ж. общей химии, 1973, т. 43, с. 724.
311. Low М. J. D., Ramamurthy Р., Chem. Commun., 1967 (12), 609.
312. Kochlschutter Н. W., Bogel U., Fortschrittsber. Kolloide Polym., 55,
29 (1971).
313. Кольцов С. H., и dp.— Изв. высших учебных заведений, химия и хи-
мии. технология, 1973, т. 16, с. 1475.
314. Шарыгин Л. М.— Кинетика и катализ, 1973, т. 14, с. 1600.
315. Волкова А. Н. и др.— Ж. общей химии, 1972, т. 42, с. 1431.
316. Раховский Р. Р., Кольцов С. И., Алексовский В. Б.— Ж. неорг. хим.,
1970, т. 15, с. 3158.
317. Шарыгин Л. М. и др.— ЖФХ, 1971, т. 45, с. 2684.
318. Волкова А. Н., Малыгин А. А. и др., авт. свидет. СССР 440338, 1974.
319. Малыгин А. А. и др.— Ж. общей химии, 1973, т. 43, с. 1436.
320. Малыгин А. А.— Изв. высших учебных заведений, химия и химии, тех-
нология, 1973, т. 16, с. 1471.
321. Кольцов С. Н. и др.— ЖФХ, 1973, т. 47, с. 988.
322. Hierl G., Krauss Н. L., Z. Anorg. Allg. Chem., 415, 57 (1975).
323. Blackman L. C. F., Harrop R., J. Appl. Chem., 18, 37 (1968).
324. Ball B., Furstenau D. W., Disc. Faraday Soc., No. 52, 361 (1971).
325. Elton G. A., 2nd. Int. Congr. Surf. Activ. III. Electrical Phenomena —
Solid Liquid Interface, Butterworth, London, 1957, p. 161.
326. Ter-Minassian-Saraga L., Adv. Chem. Ser., 43, 232 (1964).
327. Бреслер С. E. и dp.— Коллоидн. ж., 1974, т. 36, с. 592.
328. Rupprecht Н., Ullmann Е., Thoma К-, Fortschrittsber. Kolloide Polym.,
55, 45 (1971).
329. Ter-Minassian-Saraga L., C. R. Acad. Sci. Ser. C, 262, 1304 (1966); 252,
1596 (1961); Soc. Chem. Ind., 25, 272 (1967).
330. Bijsterbosch В. H., J. Colloid Interface Sci., 47, 186 (1974).
331. Proust J. E., Ter-Minassian-Saraga L., C. R. Acad. Sci. Ser C., 274 (12),
1105 (1972).
332. Blackman L. C. F., Harrop R., Nature, 208, 777 (1965).
333. Hedberg M. C., J. Am. Oil. Chem. Soc., 47, 177 (1970).
334. O'Connor D. J., Sanders J. V., J. Colloid Sci., 11, 158 (1956).
335. Allingham M. M., Cullen J. M. et al., J. Appl. Chem., 8, 106 (1958).
336. Saukel H„ Z. Naturforsch., 18 (4), 332 (1963).
337. Her R. К, пат. США 2273040 (Du Pont), 1942.
338. Iler R. K-, Ind. Eng. Chem., 46, 766 (1954).
339. Yates P. C., Trebilcock I. W., “The Chemistry of Chromium Complexes
used as Coupling Agents”, 16th Annu. Meet., Reinforced Plastics Div.,
Soc. Plastics Ind., 1961.
340. Badollet M. S., пат. США 2194683 (Johns-Manville Co.), 1940.
341. Herkenhoff E. С., пат. США 2423022 (American Cyanamid Corp.), 1947.
342 Gaudin A. M., Furstenau D. W., Min. Eng. 7 AIME Trans., 202, 66
(1955).
Химия поверхности кремнезема
1001
343. Sorensen R. Т., Frommer D. W., U. S. Bur. Mines Rep. Invest., 6719
(1966).
344. Weyl IF. A., Marboe E. C., The Constitution of Glasses, Vol. II, Part 2,
Wiley, New York, 1967, pp. 1033—1065.
345. Iler R. К., пат. США 2657149 (Du Pont), 1953 [Chem. Abstr., 50, 11564].
346. Ballard С. C., Broge E. C., Iler R. K-, St. John D. S., McWhorter J. R„
J. Phys. Chem., 65, 20 (1961).
347. Lowen IF. K., Broge E. C., J. Phys. Chem., 65, 16 (1961).
348. Bartell F. E., Dobay D. G., J. Am. Chem. Soc., 72, 4388 (1950).
349. Harkins W. D., The Physical Chemistry of Surface Films (Rheinhold,
New York, 1952, pp. 211, 245).
350. Stober W., Bauer G., Thomas K-, Ann. Chem., 604, 104 (1957); Kolloid-Z.,
149, 39 (1956).
351. Kitahara S., Motnii T., Yonekura A., Fukuoka Kyoiku Daigako Kiyo,
Dai-3-BU, Rika-Hen, 22, 121 (1972).
352. Kitahara S., Asano T., Hirowatari T., Nippon Kagaku Zasshi, 92, 377
(1971).
353. Fripiat J. J., Uytterhoeven J. et al., Helv. Chim. Acta, 43, 176 (1960).
354. Miyata K., J. Chem. Soc. Jap. Pure Chem. Sect., 87, 116 (1966).
355. Макеева E. Д., Николаева И. H., Болотова Л. В. Тр. ВНИИ по пере-
работке нефти, вып. 10, 1967, с. 333.
356. Tsutsumi К-, Takahashi Н., Nippon Kagaku Kaishi, 1972 (10), 1800.
357. Киселев А. В., Королев А. Я. и др.— Коллоидн. ж., 1963, т. 25, с. 140.
358. Stober W., Kolloid-Z., 145, 17 (1956).
359. Белякова Л. Д., Киселев А. В.— ЖФХ, 1959, т. 33, с. 1534.
360. Stober W., Lief lander М., Bohn Е., Beitr. Silikose-Forsch., Spec. Vol. 4,
111 (1960) [Chem. Abstr., 57, 3739h],
361. Stober W., Bauer G., Die Staublungenerkrankungen, 3, 31 (1957).
362. Bauer G., Beitr. Silikose-Forsch. Sonderb., 2, 321 (1956).
363. Lambert R., Singer N., J. Colloid Interface Sci., 45, 440 (1973).
364. Bauer G„ Stober W., Kolloid-Z., 160, 142 (1958).
365. Simpson E. A., Doyle C. F., пат. США 3720532 (W. R. Grace & Cd.),
1973.
366. Utsugi H., Matsuzawa T., Akoshima A., Shikizai Kyokaishi, 48 (6), 372
(1975).
367. Azrak R. B., Angell C. L., J. Phys. Chem., 77, (26) 3048 (1973).
368. Broge E. С., пат. США 2680696 (Du Pont), 1954.
369. Gross А., пат. США 2973282 (Degussa) (1956).
370. Utsugi H., Matsuzawa T., Zairo, 24 (262), 638 (1975).
371. Utsugi H., Nishimura S., Kano T., Zairo, 21, 225, 528 (1972).
372. Rossi C., Munari S., Cengarie L., Chim. Ind. Milan, 41, 1067 (1959).
373. Чуйко А. А. и dp.— Коллоидн. ж., 1973, т. 35, с. НО.
374. Gokcek С., thesis, University of Heidelberg, Germany; see Ref. 8.
375. Broge E. С., пат. США 2736668 (Du Pont), 1956.
376. Asano T., Kitahara S., J. Chem. Soc. Jap., Pure Chem. Sect., 91, 109
(1970).
377. Mertens G., Fripiat J. J., J. Colloid Interface, Sci., 42, 169 (1973).
378. Abayashi H., Yoshida A. et al., J. Chem. Soc. Jap. Ind. Chem. Sect., 70,
156 (1967).
379. Goebel M. T., пат. США 2736669 (Du Pont), 1956.
380. Mertens G., in J. M. Serratosa, Ed., Reunion Hispano-Belga Miner. Ar-
cilla (1970), J. M. Cons. Super. Invest. Cient. Madrid, 1971.
381. Duel H., Gentili R., Helv. Chim. Acta, 39, 1586 (1956).
382. Bavarez M., Bastik J., C. R. Acad. Sci., 258, 3683 (1964).
383. Low M. J. D., Harano У., J. Res. Inst. Catal. Hokkaido Univ., 16 (1),
271 (1968) [Chem. Abstr., 70, 81224w].
384. Kunath D„ Z. Chem., 11 (12), 471 (1971).
1002
Глава 6
385. Broge Е. С., пат. США 2866716 (Du Pont), 1958.
386. Boylan F. J., пат. США 3591519 (Hercules, Inc.), 1971.
387. Saukel H., Angew. Chem., 76, 603 (1964).
388. Utsugi H., Nishimura S., Horikoshi H., Zairo, 22 (238) 673 (1973).
389. Iler R. K-, пат. США 2739074 (Du Pont), 1956.
390. Чуйко А. А., Тертых В. А. и др.— ДАН СССР, 1969, т. 186, вып. 2,
с. 358.
391. Киселев А. В., Лыгин В. И., Соломонова И. Н.— ЖФХ, 1970, т. 44,
вып. 5, с. 1249.
392. Utsugi Н., Matsuzawa Т., Akoshima A., Zairo, 24 (262), 632 (1975).
393. Goebel М. Т„ пат. США 2739077 (Du Pont), 1956 [Chem. Abstr., 50,
11564].
394. Bauer G., Stober W., Kolloid-Z., 160, 142 (1958).
395. Ahmed A., Gallei E., Unger K., Ber. Bunsenges. Phys. Chem., 79, 66
(1975).
396. Хаскин И. Г —ДАН СССР, 1952, т. 85, с. 129.
397. Utsugi И., Horikoshi Н., Matsuzawa Т., J. Colloid Interface Sci., 50 (1),
154 (1975).
398а. Utsugi Н., Horikoshi Н., Nippon Kagaku Kaishi, 1972 (12), 2244.
3986. Lange K. R-, Chem. Ind. London, 1969, 1273.
399. Neimark I. Y., Neftekhimiya, 3, 149 (1963).
400. Hair M. L„ Hertl IT., J. Phys. Chem., 73, 2372 (1969); 75, 2181 (1971).
401. Hair M. L., J. Colloid Interface Sci., 60, 154 (1977).
402. Frazier S. E., Bedford J. A., Hower J., Kenney M. E., Inorg. Chem., 6,
1693 (1967).
403. Burwell R. L., Leal O., J. Chem. Soc. Chem. Communic., 1974, 342.
404. Stober IF., “Silyl Derivatives of SiO2”, Beitr. Silikose-Forsch. Sonderb.,
2, 333 (1956).
405. Kohlschutter H. H., Kampf G., Z. Anorg. Allg. Chem., 292, 298 (1957).
406. Kohlschutter H. H., Best P., Wirzing G., Z. Anorg. Chem., 285, 236
(1956).
407. Wirzing G., Kohlschutter H. W., Z. Anal. Chem., 198, 270 (1963).
408. Eakins W. J., Ind. Eng. Chem. Proc. Des. Dev., 7 (1), 39 (1968).
409. Тертых В. А. и др.— ЖФХ, 1973, т. 47, вып. 1, с. 158.
410. Berg К., Unger К., Kolloid-Z. Z. Polym., 234, 1108 (1969); 246, 682
(1971).
411. Чуйко А. А. и др. Физико-химич. механика и лиофильность дисперсных
систем, вып. 4, Киев: Наукова думка, 1972, с. 43.
412. Herzberg W. J, Erwin W. R., j. Colloid Interface Sci., 33, 172 (1970).
413. Hertl W„ J. Phys. Chem., 72, 3993 (1968).
414. Koelling I. G., Kolb К. E., Chem. Commun., 1965, 6.
415. Lee L. H., J. Colloid Interface Sci., 27, 751 (1968).
416. Wiggell J. В., пат. США 3649335 (Du Pont), 1972.
417. Unger K-, Thomas W., Adrian P., Koll. Z. Z. Polym., 245, 45 (1973).
418. White T. E., Proc. Anniv. Tech. Conf. (Soc. Plastics Ind.), Reinforced
Plastics Div. 20th, Chicago, 1965 [Chem. Abstr.. 63, 7675с].
419. Бабкин И. Ю., Киселев А. В.~ ДАН СССР, 1959, т. 129, с. 357.
420. Unger К-, Berg К-, Gallei Е., Erdei G., Fortschrittsber. Kolloide Polym.,
55, 34 (1971).
421. Бабкин И. Ю., Киселев А. В.— ЖФХ, 1962, т. 36, с. 2448.
422. Zettlemoyer А. С., Hsing Н. Н., J. Colloid Interface Sci., 58, 263 (1977);
Prog. Colloid Polym. Sci., 61, 54 (1976).
423. Alzamora L., Contrelas S., Carte’s J., J. Colloid Interface Sci., 50, 503
(1975).
424. Лыгин В. И., Киселев А. В.— Коллоидн. ж., 1961, т. 23, с. 250.
425. Utsugi Н., Nishimura S., Zairo, 22 (238), 680 (1973).
Химия поверхности кремнезема
1003
426. Utsugi Н., Nishimura S., Shimazaki М., Nippon Kagaku Kaishi, 1972
(И), 2007.
427. Utsugi H„ Nishimura S„ Zairo, 20 (213), 737 (1971); 21 (225), 534
(1972).
428. Бабкин И. Ю., Киселев А. В.— Коллоидн. ж., 1962, т. 24, с. 554.
429. Bassett D. R., Boucher Е. A., Zettlemoyer А. С., J. Colloid Interface Sci.,
27, 649 (1968); 34, 436 (1970).
430. Zettlemoyer A. C„ McCafferty E., Croat. Chem. Acta, 45, 173 (1973)
[Chem. Abstr., 79, 83793г].
431. Zettlemoyer A. C., Ed., Nucleation, Dekker, New York, 1969.
432. Щербакова К. Д., Словецкая К. И.— ДАН СССР, 1956, т. 111, с. 855.
433. Eirich F. R., J. Colloid Interface Sci., 58, 423 (1977).
434. Iler R. K., personal observation.
435. Sugiura M., Proc. Jap. Congr. Mater. Res., 15, 123 (1972) [Chem. Abstr.,
77, 153993г].
436. Greenland D. J., Meded. Fac. Landbouweten Sch. Rijksuniv. Gent, 37 (3),
915 (1972).
437. Rubio J., Kitchener J. A., J. Colloid Interface Sci., 57, 132 (1976).
438. Tadros Th. F., J. Colloid Interface Sci., 46, 528 (1974).
439. Hostetler R. E., Swan J. W.t J. Polym. Sci. Polym. Chem. Ed., 12, 29
(1974).
440. Lindquist G. M., Straiten R. A., J. Colloid Interface Sci., 55, 45 (1976).
441a . Holt P. F., Bowcott J. E. L., AMA Arch., Ind. Hyg. Occup. Med., 9,
503 (1954).
4416. Holt P. F., Bowcott J. E. L., Biochem. J., 57, 471 (1954).
442. Бреслер С. E. и dp.— Коллоидн. ж., 1973, т. 36, с. 748.
443. Snyder L. R., Kirkland J. I., Modern Liquid Chromatography, Wiley,
New York, 1974.
444. Griot O., Kitchener J. A., Trans. Faraday Soc., 61, 1026, 1032 (1965).
445. Howard G. I., McConnell P., J. Phys. Chem., 71, 2974 (1967).
446. Forsman W. C., H amort E., Hughes R. E., Macromolecules, 4 (2), 193
(1971), [Chem. Abstr., 75, 25817].
447. Botham R. A., Theis C„ J. Polym. Sci. C, (30), 369 (1970).
448. Липатов JO. С. и dp.—ДАН СССР, 1974, т. 218, с. 1144.
449. Botham R., Theis C., J. Colloid Interface Sci., 31, 1 (1969).
450. Fontana В. I., Thomas J. R., J. Phys. Chem., 65, 480 (1961).
451. Miyamoto T., Cantow H. Makromol. Chem., 162, 43 (1972).
452. Killmann E., Eckart R., Makromol. Chem., 144, 45 (1971).
453. Killmann E., Winter K-, Angew. Makromol. Chem., 43, 53 (1975).
454. Кольцов С. И., Алесковский В. Б., Волкова А. Н. Адсорбенты, их полу-
чение, свойства и применение. Труды III Всес. совещания по адсорбен-
там, 1969,/Под. ред. М. М. Дубинина.— Л.: Наука, 1971, с. 29.
455. Tamele М. W., Heterogeneous Catalysis (Disc. Faraday Soc., 8), 270—
274 (1950).
456 Milliken T. H., Jr., Mills G. A., Oblad A. G., Heterogeneous Catalysis
(Disc. Faraday Soc., 8), 279 (1950).
457. Basila M. R., J. Phys. Chem., 66, 2223 (1962).
458. Роев Л. M., Филимонов В. H., Теренин А. Н.— Оптика и спектроскопия,
1958, т. 4, с. 328.
459 Oblad A. G., Hinden S. G., Mills G. A., J. Am. Chem. Soc., 75, 4096
(1953).
460. Хаскин И. Г.—ДАН СССР, 1952, т. 85, с. 129.
461. Sempels R. Е„ Rouxhet Р. G., J. Colloid Interface Sci., 55, 263 (1976);
J. Chem. Soc. Faraday Trans. 1, 70, 2021 (1974).
462. Lowenstein W., Am. Mineral., 39. 92 (1954).
463. Bourne К- H., Cannings F. R., Pitkethly R. C., J. Phys. Chem., 74, 2197
(1970).
1004
Глава 6
464. Jepson W. P., Jeffs D. G., Ferris A. P., J. Colloid Interface Sci., 55,
454 (1976).
465. Healy T. W., Wiese G. R., Yates D. E., Kavanagh. В. V., J. Colloid Inter-
face Sci., 42, 647 (1973).
466. Певзнер И. 3. и др.— Ж. прикл. химии, 1974, т. 47, с. 2758.
467. Гейшин П. А., Демидова Н. А., Соломка К А.— Коллоидн. ж., 1974,
т. 36, вып. 4, с. 760.
468. Fomi L., Catal. Rev., 8 (1), 65 (1973).
468а. Benisi Н. A., J. Catal., 28, 176 (1973) [Chem. Abstr., 78, 48397х].
4696. Benisi H. A., J. Am. Chem. Soc., 78, 5490 (1956).
470. Hammet L. P., Deyrup A. J., J. Am. Chem. Soc., 54, 2721 (1932).
471. Benesi H. A., J. Phys. Chem., 61, 970 (1957).
472. Parry E. P„ J. Catal., 2, 371 (1963).
473. Leftin H. P., J. Phys. Chem., 64, 1714 (1960).
474. Damon J. P., Bonnier J. M., J. Colloid Interface Sci., 55, 381 (1976).
475. Бондаренко А. В. и dp.— ДАН СССР, 1961, т. 136, с. 157.
476. Красильников К. Г., Киселев В. Ф., Сысоев Е. А.— ДАН СССР, 1957,
т. 116, с. 990.
477. Chuang S. Y., Tao S. J., J. Chem. Phys., 54, 4902 (1951).
478. Brohn H., Paudert R., J. Prakt. Chem., 283, 63 (1960).
479. Hauser E. A., Silicic Science, Van Nostrand, Princeton, N. J., 1955.
480. Schofield P. J., Ralph B. J., Green J. H., J. Phys. Chem., 68, 472 (1964).
481. Стрелко В. В., Высоцкий 3. 3., Ганюк Л. Н. Высокомолек. соёдин.,
химич. свойства и модификация полимеров, Сб. статей, 1964, с. 19.
482. Morterra С., Low М. J. D., Chem. Commun., 1968 (23), 1491; J. Catal.,
28 (2), 265 (1973).
483. Low M. J. D., Shimizu M., McManus J. C., J. Chem. Soc. D, 1971 (11),
579.
484. Огенко В. M., Тертых В. А., Чуйко А. А. Физико-химич. механика и
лиофильность дисперсных систем, вып. 4, Киев: Наукова думка, 1973,
с. 200.
485. Peglar R. J., Murray J. et al., J. Chem. Soc. A, 1970 (13), 2170.
486. Литковец А. К. и др. Сб. Прикладные исследования в области химии
неорганических перекисных соединений. Рига, 1974, с. 206—210.
487. Шевец Д. И. и др., Адсорбция и адсорбенты. Респ. межвед. сб., вып. 1,
Киев: Наукова думка, с. 115.
488. Simon I., Phys. Rev., 103, 1587 (1956).
489. Стародубцев С. В. и др.— ДАН СССР 1959, т. 4, с. 1259.
490. KohnH. W„ Nature, 184, 630 (1959).
491. Ngoc Binh Dinh, Rabe J. R., Schnabel IF., Angew. Makromol. Chem.,
46, 23 (1975) [Chem. Abstr., 83, 179671].
492. Ogura H., Tachika J. et al., J. Nucl. Sci. Technol., 12, 167 (1975).
493. Любимова О. И., Котов А. Г.— Химия высоких энергий, 1970, т. 4,
вып. 1, с. 62.
494. Kinell Р. О. et al., Acta Chem. Scand., 24, 3265 (1970).
495. Абляев Ш. А., Алимова Л. Я., Сеттарова 3. С. Сб. Вторично-эмиссион-
ные и структурные свойства твердых тел. Ташкент: Фан, 1970, с. 184.
187.
496. Парийский Г. Б. и др.— Кинетика и катализ, 1965, т. 6, вып. 4, с. 625.
497. Громов В. В., Спицын В. И.— Атомн. энергия, 1963, т. 14, с. 491.
498. Тагиева М. М., Киселев В. Ф.— ЖФХ, 1961, т. 35, с. 680.
499. Donnet J. В., Bull. Soc. Chim. Fr. Spec. No. 3353 (1970).
500. Атьякшева Л. Ф. и др.— ЖФХ, 1972, т. 46, вып. 10, с. 2602; там же,
1973, т. 47, вып. 4, с. 996.
501. Reimschuessel Н. К-, Mountford G. Д., J. Colloid Interface Sci., 25, 558
(1967).
502. Ерматов С. Е., Тусеев С. Е.— Изв. АН Каз. ССР. физ. мат., 1972,
т. 10, вып. 6, с. 70, 73.
Глава 7
КРЕМНЕЗЕМ В БИОСФЕРЕ
Введение
С момента опубликования обзора [1] Айлера в 1955 г., в ко-
тором рассматривались вопросы о нахождении и роли кремне-
зема в живых организмах, был написан краткий обзор Гюнтером
и Абергом [2] о связи кремния с жизненными процессами.
Кроме того, появились небольшая монография Мона [3] и
книга Воронкова, Зелчана и Лукевица [4а]. Мон обобщил глав-
ным образом литературу, появившуюся за последнюю четверть
века, включив краткий обзор по химии кремнезема с точки зре-
ния проблем биологии, а также рассмотрел большой экспери-
ментальный материал, в котором отражено потребление кремне-
зема крысами при добавлении его в различных формах в пищу.
Воронков и соавторы представили обширный обзор по распро--
страненности кремнезема в природе, его возможной роли в про-
исхождении жизни, распределению во всех типах живых су-
ществ, токсичности кремнезема и применению в лечебных целях
недавно открытых органических производных кремнезема и
кремния, сопроводив свой обзор более чем 500 библиографи-
ческих ссылок.
Лео и Баргхорн [46] дали краткое изложение литературы
по вопросам биохимии кремнезема. Они обсудили циклические
формы переноса кремнезема, включая его круговорот в био-
сфере.
В настоящей главе кратко рассматриваются некоторые из
этих аспектов. Будет уделено внимание химии растворимых и
коллоидных форм кремнезема с точки зрения участия кремне-
зема в ряде биохимических процессов.
В описании многих биологических исследований чаще встре-
чается термин «кремний», чем «кремнезем». Поскольку имеется
мало доказательств того, что кремний встречается в какой-либо
биосистеме в форме, отличающейся от координации кремния
с кислородом, то автор считает возможным в настоящей главе
использовать термин «кремнезем» или SiO2.
Окремнение форм живых организмов уже было описано
в гл. 1, так как в этом вопросе не обнаружено сложных биохи-
мических проблем.
1006
Глава 7
Происхождение жизни
Несмотря на то- что кремний является одним из наиболее
распространенных элементов, он, как полагают, не имеет суще-
ственного значения для большинства живых организмов, тогда
как гораздо менее распространенный углерод является основ-
ным элементом жизни на Земле.
Однако было выдвинуто предположение, что первоначально
соединения кремния играли важную и, по всей вероятности, не-
обходимую роль в происхождении жизни. Гамов [5] отмечал,
что переход от неживой материи мог протекать очень посте-
пенно. Опарин [6] выдвинул постулат, согласно которому
жизнь возникла посредством ассоциации простых, встречаю-
щихся в природе углеродных соединений с неорганическими
веществами в коллоидной форме. Бернал [7] предположил, что
коллоидные силикаты, вероятно, играли каталитическую роль
в процессах формирования сложных органических молеку/i из
простых молекул. Он допускал также, что первоначальная атмо-
сфера Земли (до возникновения жизни) должна была состоять
из таких водородных соединений, как метан, аммиак, сероводо-
род и водяные пары. Как показал Миллер [8], аминокислоты
могут образовываться из метана, азота и водяного пара под
влиянием электрических разрядов, поэтому могли существовать
разнообразные органические соединения. Бернал высказал пред-
положение, что обогащение простых органических молекул
могло происходить при их адсорбции на коллоидных глинистых
минералах, имеющих очень большое значение удельной поверх-
ности и сродство по отношению к органическим веществам. Он
указал, что небольшие по размеру молекулы, присоединенные
к поверхности глины, способны удерживаться на ней не беспо-
рядочно, а в определенных положениях как по отношению
к поверхности глины, так и друг к другу. Таким образом, вслед-
ствие упорядоченного расположения эти молекулы могут взаи-
модействовать между собой с образованием более сложных сое-
динений, особенно в том случае, когда осуществляется подвод
энергии за счет падающего на поверхность света. Согласно Бер-
налу, вначале могло происходить формирование асимметрич-
ных молекул, которые характерны для живых организмов. Это
могло осуществляться путем более предпочтительной попарной
адсорбции асимметричных молекул на поверхности кварца, так
как кварц — единственный общеизвестный минерал, обладаю-
щий асимметричной структурой.
Необходимо иметь в виду, что хотя первоначально и могли
образоваться сложные органические молекулы благодаря подоб-
ным неорганическим катализаторам, однако такое превращение
Кремнезем в биосфере
1007
было всего лишь первым этапом из сотен или тысяч последую-
щих реакций, которые затем привели к возникновению первого,
действительно «живого» организма, т. е. организма, способного
размножаться. Поэтому говорить, что отмеченный первый этап
представлял собой стадию «происхождения жизни», это равно-
сильно утверждению, что первый извлеченный из недр земли
кусок железной руды послужил первопричиной появления авто-
мобиля.
Жанэ [9] обсудил в своем обзоре роль кремнезема в период
добиотической эволюции. В дальнейшем роль кремнезема в по-
степенном развитии жизни была рассмотрена Седлаком. [10] и
Высоцким, Даниловым и Стрелко [11]. Если коллоидные сили-
каты на первом этапе представляли собой молекулярную кри-
сталлическую решетку, играющую роль в происхождении жизни,
то необходимость в подобном строении отпадала, как только ус-
танавливалось высокоупорядоченное, активное, «живое» распо-
ложение органического вещества. Если гипотеза справедлива, то
структура живого вещества должна была бы унаследовать в не-
которой степени строение кристаллической решетки. Например,
должно было бы наблюдаться некоторое соответствие между
молекулярной структурой биологических веществ белкового
типа и межатомными расстояниями, характерными для поверх-
ностей таких коллоидных силикатов, как бентонит, палыгорскит
или каолинит.
Воронков, Зельчан и Лукевиц [4а] обобщили все подобные
предположения и гипотезы по этой проблеме, возникавшие за
последние 200 лет, включая возможности образования других
биологических форм во Вселенной на основе полимеров, имею-
щих связи Si—Si, Si—N или Si—С.
Наиболее ранние биологические формы
Несомненно, что наиболее древними ископаемыми остатками
живых организмов являются сине-зеленые водоросли, обнару-
женные в виде включений в шерте (микрокристаллическом
кремнеземе), открытые Баргхорном и Тайлером [12] и в дальней-
шем изученные многими исследователями [13—17]. Эти микро-
скопические биологические формы были, очевидно, тесно связаны
с растворимым и коллоидным кремнеземом, поскольку первона-
чально они укреплялись в силикагеле. Такие микроорганизмы
возникли 3,5 млрд, лет назад, вероятно, всего лишь через 1 млрд,
лет после образования Земли и задолго до формирования много-
клеточных биологических форм, быстро размножившихся
0,6 млрд, лет назад. Именно в течение этого интервала времени,
составившего 3 млрд, лет, сине-зеленые водоросли, как пола-
гают, превратили исходную восстановительную атмосферу
1008
Глава 7
Рис. 7.1. Микрофотография оболочки водоросли с адсорбированным коллоид-
ным кремнеземом.
Земли в атмосферу окислительную в результате выделения
кислорода при фотосинтезе. В настоящее время предполагают,
что процесс фотолиза воды в верхних слоях атмосферы с уда-
лением водорода в космическое пространство не смог бы обес-
печить образование большого количества кислорода в течение
докембрийского периода [18].
Еще нет доказательств того, что кремнезем — одно из необ-
ходимых веществ, принимающих участие в метаболизме сине-
зеленых водорослей, но по крайней мере один из типов таких
водорослей, по наблюдениям Айлера, неплохо растет в концент-
рированных 30 %-ных золях коллоидного кремнезема. Было об-
наружено, что на хорошо промытых клеточных оболочках име-
ется адсорбированный коллоидный кремнезем (рис. 7.1).
Олер и Шопф [19] провели исследования по фоссилизации
водорослей в силикагеле. Водоросли суспендировали в концент-
рированном коллоидном кремнеземе, переводили в гель кремне-
Кремнезем в биосфере
1009
зема и подвергали затем автоклавной обработке при достаточно
высокой температуре с целью превращения геля в микрокри-
сталлический кварц. В результате получалась похожая на шерт
масса, содержавшая внедренные в нее водоросли, подобные тем,
которые были найдены в древних окаменевших образцах.
Биологическое разрушение горных пород
Хотя большая часть вторичных минералов, таких, как глины,
может формироваться из первичных силикатных горных пород
посредством неорганических реакций в присутствии воды, од-
нако процесс эрозии может катализироваться органическими
реагентами. Джекс [20] представил обзор работ группы совет-
ских ученых, которые полагают, что во многих случаях в про-
цесс выветривания горных пород включается и биологическое
воздействие. Полынов [21] считал, что многие из нестойких
минералов, обнаруживаемых в настоящее время на поверхности
земли, давно бы исчезли, если бы они непрерывно не синтези-
ровались живыми организмами. Айдинян [22] сообщил, что на
поверхности горных пород, на которой растут лишайники, об-
наружены минералы в коллоидной форме, являющиеся продук-
том выветривания и имеющие такое же отношение SiO2 : R2O3
(оксиды железа или алюминия), что и в золе лишайника. Это
указывает на биологическое происхождение таких коллоидных
минералов. Глазовская [23] пришла к заключению, что водо-
росли и диатомеи представляют собой сильнодействующие
агенты эрозии, способные вырабатывать аморфный кремнезем и
синтезировать алюмосиликаты, подобные бейделлиту и монт-
мориллониту. Прилова [24] обнаружила, что лишайники выде-
ляют кислоты, которые разъедают твердые горные породы и
могут расщеплять кристаллы плагиоклаза на частицы меньшего
размера. Некоторые из таких глинистых минералов нонтронит-
бейделлитового типа, по-видимому, синтезируются в раститель-
ных тканях. Болышев [25] полагает, что сине-зеленые водоросли
вызывают разложение почвенных минералов и способствуют
переносу кремнезема и оксида алюминия в раствор и что крем-
незем, приготовленный подобным образом, пригоден для усвое-
ния некоторыми видами диатомей, которые обычно сопровож-
дают сине-зеленые водоросли. Александров и Зак [26] выделили
бациллу (В. siliceus), которая вызывала разложение нераство-
римых, содержащих поташ алюмосиликатов, причем получаемый
калий оказывался пригодным для усвоения растениями. Внесе-
ние посевного материала в почву, обогащенную азотом и фос-
фором, при небольшом содержании поташа в растворимой
форме вызывало повышение урожая зерновых (пшеницы, куку-
рузы) на 50—100 %. В проведенных в лабораторных условиях
38 Заказ № 250
1010
Глава 7
исследованиях Оберлис и Полманн [27] нашли, что на образцы
отполированного полевого шпата оказывали воздействие раз-
личные бактерии, как и в случае проведения соответствующих
опытов на стекле [28]. Кутузова [29] проводила обширные
исследования с множеством разнообразных микробов и минера-
лов. Она обнаружила, что значение pH в таких средах в некото-
рых случаях понижалось до 2, что вызывало распад алюмосили-
катов. Даже кварц выделял аморфный SiO2 под воздействием
бактерий рода Sarcina. Разнообразные типы бактерий, способ-
ных оказывать разрушение силикатов, были рассмотрены в об-
зоре Воронкова, Зелчана и Лукевица [4а].
Ассоциация с простейшими
Сведения о содержании кремнезема в микроорганизмах или
о наличии адсорбированного кремнезема на их внешней обо-
лочке, как это показано на рис. 7.1, трудно получить посред-
ством химического анализа. Отделение клеточных микроорга-
низмов от загрязняющих примесей, таких, как коллоидные
глины, также представляет собой проблему, которая делает
сомнительными сообщаемые в литературе данные о содержании
кремнезема в «золе», в особенности в исследованиях, выполнен-
ных еще до того, как стал применяться электронный микроскоп.
Вирусы
Сообщается, что даже в вирусах кремнезем является суще-
ственным компонентом. Фауст и Адамс [30] выделили кристал-
лический вирус, состоящий из полиэдрических частиц, из раз-
личных гусениц (Bombyx топ и др.) и обнаружили в нем со-
держание кремния, соответствующее концентрации 0,2—0,6 %
SiO2, что составляет существенную часть белковой матрицы.
Бактерии
Для некоторых определенных почвенных бактерий харак-
терно, что поглощение кремния в виде растворимого кремнезема
в культуральной среде сопровождается выделением фосфора.
Хейнен [31] изучил факторы, которые ускоряли и затормажи-
вали такой обмен. В отсутствие глюкозы кремний маскировался
избытком фосфата. Фракции в виде частиц, отделяемые от бак-
териальных оболочек, включались в метаболизм кремния [32].
За период с 1960 по 1967 г. Хейнен представил 14 статей,
в которых изложил детали этого вопроса.
Существенная роль кремнезема в метаболизме определенных
видов бактерий, а также взаимодействие бактерий с силикаге-
Кремнезем в биосфере
1011
лями и минералами исследовалась в широком масштабе, осо-
бенно в Советском Союзе. Эти работы обобщены в монографии
Воронкова, Зелчана и Лукевица [4а].
Грибы и лишайники
Тот простой факт, что грибы способны поглощать кремнезем,
когда к такой культуре добавляют растворимые силикаты, мо-
жет свидетельствовать только о том, что образующийся колло-
идный кремнезем адсорбируется на поверхности клеток. Однако
то обстоятельство, что поглощение кремния ускоряется в отсут-
ствие кислородных соединений трехвалентного фосфора, явля-
ется подтверждением определенной роли, которую кремний
способен играть в подобном метаболизме [4а].
Лишайники, представляющие собой симбиотическое соедине-
ние грибов и водорослей, вероятно, существовали с давних вре-
мен и, как уже ранее упоминалось, по-видимому, ответственны
за превращения горных пород в почву. Водоросли являются
фотосинтезирующими организмами, и они запасают энергию
в форме углеводов, тогда как грибы оказывают воздействие
на горную породу, запасаясь минеральными питательными ве-
ществами. Ввиду большой сложности структур и близкого
объединения с минеральными силикатами к результатам хими-
ческого анализа на содержание кремнезема необходимо отно-
ситься осторожно, но тем не менее сообщалось о том, что зола
содержит 10—20 % кремнезема. Данные по воздействию лишай-
ников на кварц не были подтверждены [4а].
Водоросли
Из тысяч видов водорослей известна одна группа, а именно
диатомовые водоросли, или диатомеи, образующие класс Diato-
шасеае или Bacillariophyceae, которые способны поглощать рас-
творимый кремнезем из воды при чрезвычайно низких его кон-
центрациях, причем такой кремнезем подвергается метаболизму
и осаждается в виде внешнего скелета. Согласно данным Кал-
верта [33], существует более чем 10 000 разновидностей диато-
мей; некоторые из них живут в пресной воде, а другие — в соле-
ной воде. Почти все разновидности схожи в том отношении, что
их наружные стенки наполнены кремнеземом. Эти растения
представляют собой одноклеточные организмы, состоящие из
двух частей, причем края одной части входят внутрь другой, что
напоминает соединение двух половинок коробочки от пилюль.
Помимо того что диатомовая водоросль упрятана в кремнезем-
ную оболочку, каждая ее клетка способна накапливать ка-
пельку нефти. Предполагается, что эта нефть наряду с другими
38*
1012
Глава 7,
углеводородами, образующимися в результате разложения
органического вещества диатомей, могла бы быть причиной фор-
мирования большой доли мировых запасов нефти. Отложения
диатомового ила на площади, достигающей 650 км в длину и
150 км в ширину, были обнаружены вдали от океанского побе-
режья в Африке. Органические глинистые сланцы, из которых,
как предполагают впоследствии получилась нефть, формирова-
лись из отмерших телец этих организмов, осаждавшихся на дне
древних океанов. Фотографии некоторых из этих великолепных
формирований, состоящих из микроскопических скелетов диато-
мей, представлены Калвертом.
Морская вода содержит только 0,0002—0,0014 % SiOa, что
гораздо ниже значения насыщения по отношению к раствори-
мости аморфного кремнезема [34]. Частично SiO2 находится
в виде суспендированных в воде небольших обломков от крем-
неземистых организмов [35]. Сивер [36] отметил, что основной
механизм получения кремнеземного осадка на поверхности
земли носит биохимический характер. В своем обзоре Воронкбв,
Зелчан и Лукевиц [4а] показали, что за осаждение кремнезема
ответственны разнообразные организмы. Скрытокристаллические
минералы кремнезема, например яшма и халцедон, вероятно,
представляют собой продукты преобразования очень древних
диатомитовых отложений. Примерно на две трети осажденный
кремнезем состоит из диатомей, а остальная его часть — из
радиолярий и губок.
На рост каждого вида диатомей существенное влияние ока-
зывает определенная минимальная концентрация кремнезема
в растворе. Увеличение содержания кремнезема от 0,00035 до
0,00083 % удваивает скорость роста одного из видов диатомей,
содержание кремнезема в которой составляет 4—22 % высушен-
ной массы организмов [37]. Однако некоторые разновидности,
содержащие всего лишь 0,4 % SiO2, могут получать достаточное
количество кремнезема для своего роста непосредственно из
обычной стеклянной посуды. Левин обнаружил, что коллоидный
кремнезем не будет поддерживать рост диатомей до тех пор,
пока он не деполимеризуется до состояния растворимого крем-
незема. Диатомовая водоросль Navicula pelliculosa нуждается
для достижения максимальной скорости роста в 0,0035 % рас-
творимого кремнезема [38]. Скорость роста в морской воде по-
нижается по мере того, как содержание растворимого кремне-
зема падает вследствие перенаселенности диатомей (цветение
диатомовых водорослей) [39]. Диатомеи способны снижать кон-
центрацию кремнезема в воде вплоть до значений, меньших чем
0,000008 %. В том случае, когда концентрация подобных клеток
становится высокой, начинает выделяться какой-либо ингибитор,
способный замедлять поглощение кремнезема [40]. Величина,
Кремнезем в биосфере
1013
соответствующая половине насыщения при поглощении кремне-
зема, изменяется для различных разновидностей диатомей в об-
ласти от 0,000005 до 0,00002 % SiO2 [41а]. Очевидно, что при
концентрации растворимого кремнезема ниже ~0,000006 %, он
уже оказывается недоступным для этих организмов [416].
Когда содержание кремния исчерпано, диатомовые клетки на-
Рис. 7.2. Электронно-микроскопический снимок кремнеземного скелета диато-
меи (из работы [1], с разрешения Cornell University Press). '
чинают покрываться студенистой оболочкой из полиуронида,
получаемого из остатков глюкуроновой кислоты [42].
В монографии [43а] обобщены сведения по данному вопросу
до 1962 г., а Воронков, Зелчан й Лукевиц [4а] представили
библиографию вплоть до 1975 г. В 1977 г. появилась моногра-
фия Вернера [436] по биохимии и физиологии диатомовых во-
дорослей.
Наружный скелет диатомей, состоящий из кремнезема, пред-
ставляет собой изумительно сложную конструкцию вплоть до
молекулярного уровня. Как видно из рис. 7.2, электронно-мик-
роскопические снимки показывают тончайшую структуру неко-
торых разновидностей диатомей. Ниже будет показано, что гео-
метрическая повторяемость структуры является характерным
свойством не только для больших по размеру частей скелета,
которые можно наблюдать в оптический микроскоп, но и про-
должается вплоть до мельчайших единичных образований, види-
5
1014
Глава 7
мых лишь под электронным микроскопом при X 100 000. Крем-
незем плотно окружен органическим веществом и не подвер-
гается прямому воздействию окружающей воды. Таким образом,
очевидно, что кремнезем может осаждаться внутри тканей
в четко определенных формах [44]. Дарли [45] представил
обзор, в котором рассматривается подобный процесс окремне-
ния. Образуемый скелет представляет собой микропористый
силикагель, который проявляет селективные свойства по отно-
шению к ионам. Следует отметить, что свойства кремнезема,
выделенного из недавно погибших клеток диатомей посредством
растворения органического вещества этих клеток в 70 %-ной
HNO3, сильно отличаются от свойств диатомовой земли, кото-
рая подвергалась уплотнению и даже кристаллизации в суб-
микроскопическом масштабе. Как наблюдал Айлер, свежевыде-
ленный кремнезем имел удельную поверхность, измеренную по
адсорбции азота, свыше 100 м2/г, и если его не высушивали при
повышенной температуре, то он обладал к тому же микропори-
стостью, чем и объясняются его селективные ионообменные
свойства. С другой стороны, удельная поверхность диатомовой
земли, имеющей возраст несколько тысячелетий, оказывается
по величине значительно меньшей, поскольку кремнезем в ней
стал микрокристаллическим [46а].
На представленных Панкрацем электронно-микроскопиче-
ских снимках при очень большом увеличении выявлено два вида
структур в кремнеземе, полученном из радиолярий и очищенном
кислотной обработкой. На рис. 7.3 показана тонкая кремнезем-
ная пластинка толщиной 500 А, выделенная на участке около
открытых каналов (образцы были приготовлены Хардом [466]).
Более темные участки кремнезема, по-видимому, состоят из
агрегированных первичных частиц диаметром ~200 А. Удель-
ная поверхность такого кремнезема должна иметь значение
около 140 м2/г. На рис. 7.4, очевидно, показана сплошная крем-
неземная матрица (более темное изображение), пронизанная
многочисленными порами или отверстиями диаметром 20—500 А.
Возникает вопрос, имеются ли в кремнеземной матрице жи-
вой клетки доступные участки, на которых находились бы
живые ткани, или же с ними соприкасается только водная фаза.
Пока неизвестен механизм, благодаря которому кремнезем оса-
ждается в предопределенной заранее форме. Наблюдение за
ранними стадиями формирования кремнеземного панциря диа-
томовых водорослей было описано Доусоном [46в].
Своеобразная темно-зеленая водоросль, как было установ-
лено Айлером, проявляла способность расти в концентрирован-
ном коллоидном кремнеземе при pH 9—10. Эта водоросль,
описанная Кингсбери [47], имела необычайно малые размеры:
ширину клетки ~1 мкм и длину ~2 мкм. По наблюдениям
Кремнезем в биосфере
1015
Рис. 7.3. Микрофотогра-
фия обработанного кис-
лотой образца биоген-
ного кремнезема.
Показан тонкий слой радио-
лярии, состоящий из первич-
ных частиц диаметром око-
ло 200 А . (Из работы [466]
с разрешения авторов.)
Рис. 7.4. Микрофотогра-
фия другого участка об-
разца биогенного крем-
незема, представленного
на рис. 7.3.
Видно, что кремнезем пред-
ставляет собой сплошную
матрицу, заполненную пора-
ми округлой формы (свет-
лые участки) диаметром 20-—
500 А. (Из работы [466] с
разрешения авторов.)
1016
Глава 7
Айлера, в водорослях этого типа клетки были ограничены обо-
лочкой или пленкой трубчатой формы, от которой во все сто-
роны распространялись тонкие волоконца на общее расстояние
около 3 мкм. Срез оболочки, на которой находится адсорбиро-
ванный коллоидный кремнезем, показан на снимке (рис. 7.1).
Добавление нитрата и фосфата приводило к заметному ускоре-
нию роста водоросли.
Был описан [48] удивительный случай появления кристаллов
кварца размером 100 нм на кремнеземной клеточной стенке
микроорганизма Chlorochytridion tuberculatum. Кажется чрез-
вычайно неправдоподобным, что кристаллы кварца таких не-
больших размеров могли бы присутствовать как загрязняющая
примесь; следовательно, можно только прийти к заключению,
что такие кристаллы сформировались in situ. Возникает вопрос,
почему кремнеземные скелеты разложившихся диатомей по-
вторно не растворяются в морской воде, которая в значительной
степени ненасыщена. Йоргенсон [49] обнаружил, что скелеты
недавно погибших диатомей растворялись в воде, но, как пока-
зано Левиным [50] и обсуждалось в гл. 1, скорость такого рас-
творения оказывается очень низкой в присутствии следовых
количеств алюминия и железа.
Метаболизм рассматриваемого кремнезема исследовался на
процессах роста диатомей путем измерения эффектов, возника-
ющих при недостаточном содержании кремнезема в растворе.
Левин обнаружил, что кремнезем не поглощался промываемыми
клетками диатомей до тех пор, пока в систему не добавлялось
соединение серы. Кадмий затормаживал поглощение кремне-
зема, возможно, потому, что связывал соединение серы. Погло-
щение кремнезема представляет собой также аэробный процесс
[51]. Соотношение между поглощениями изотопов 31SiO2 и 14СО2
показало, что при поглощении кремнезема большая часть угле-
рода входила в состав аминокислот, но когда поглощение SiO2
прекращалось, то углерод переходил в сахара [52]. В работе
[53] указывалось, что захваченный 31SiO2 вначале накапливался
в цитоплазме. Это почти без сомнения показывает, что кремне-
вая кислота присутствовала в химически связанном состоянии.
С внешней водной средой такая кислота не находилась в равно-
весии, но оказалась сконцентрированной в цитоплазме, где ее
содержание по сравнению с остальной частью было в 30 раз
больше. Поглощение тормозилось ингибиторами метаболизма,
такими, например, как 2,4-динитрофенол. Для поглощения и
осаждения кремнезема требовалась затрата энергии, что под-
тверждалось расходом нуклеозидтрифосфата [54].
О решающей роли кремнезема на ранних стадиях развития
диатомей, других водорослей и растений можно судить на осно-
вании наблюдаемых аномалий роста Cyclotella cryptica [55, 56].
Кремнезем в биосфере
1017
Кроме того, наблюдалось, что германиевая кислота оказывает
ингибирующее воздействие на кремневую кислоту в диатомеях.
В стенках диатомовых клеток было найдено такое соединение,
как 2,3-цис, тра«с-3,4-диоксипролин. Вопрос о том, как это со-
единение могло участвовать в механизме метаболизма и пере-
носе кремнезема, остается в сфере предположений [58].
Воронков, Зелчан и Лукевиц [4а] рассмотрели в своей книге
вопрос о возможности концентрирования кремнезема в водорос-
лях разных типов и представили обзор литературы по диато-
меям. Согласно данным Сулливана и Волкани [59], было обна-
ружено, что кремнезем стимулировал активность ТМП-киназы
и ДНК-полимеразы у водоросли С. fusiformis. Сулливан иссле-
довал подробно необходимые условия, налагаемые на взаимо-
действие кремневой кислоты с подобными ферментами [60].
Несомненно, что кремнезем играет очень существенную роль
< в метаболизме водорослей. В отсутствие кремнезема, по данным
Рейманна [61], вся клетка полностью становится дезорганизо-
ванной и не может продолжать деление. Возможно, что кремне-
зем играет определенную роль в ДНК водорослей, подобно тому
как он может оказывать воздействие на более развитые орга-
низмы.
Губки
Кремнезем губок также оказывается источником образования
некоторых кремнеземных минералов. Содержание кремнезема
в губках изменяется в широких пределах от 1 до 90 % [4а].
Сообщается, что твердые, жесткие губки имеют скелеты, состоя-
щие из спикул, или игл, или кристаллического «кубического
опала», или кремневой кислоты с кубической симметрией. Стек-
лянные губки очень богаты кремнеземом. Скелет губок состоит
из «кубического опала», сцементированного белковым вещест-
вом — спонгином. В четырехлучевых губках содержится «тетра-
эдрический опал». Предполагается, что прочный, плотный не-
органический природный материал — кремень, из которого
раньше изготовлялись наконечники стрел, получил свое происхо-
ждение от кремнеземистых спикул ископаемых губок [62].
Такой кремнезем может казаться «кристаллическим» только по
внешнему виду.
Губки способны поглощать так много кремнезема, что пони-
жают его содержание в воде внутренних морей. Вотинцев [63]
сообщает, что в воде озера Байкал содержится меньше SiO2
(2—4 мг/л), чем в воде его притоков (7—10 мг/л), благодаря
присутствию в озере кремнеземистых губок. Содержание крем-
незема в таких губках составляет примерно 30 % массы сухого
органического вещества, и мертвые остатки губок образуют
1018
Глава 7
типичный кремнистый осадок на дне озера. Кремнезем губок
в основном аморфен, несмотря на то что форма частиц застав-
ляет делать предположения о возможном существовании кри-
сталлов. В одном из видов губок Geodia gibberosa содержатся
небольшие шарики размером 55 мкм с твердым стекловидным
слоем на поверхности, покрытым небольшими выростами длиной
3,5 мкм; такие шарики были предложены для наполнения хро-
матографических колонок [64]. В некоторых разновидностях
губок аморфный кремнезем оказывается вмурованным в белко-
вую массу [65]. Длина спикул возрастала с понижением их
числа на поверхности отдельной губки по мере того, как увели-
чивалось силикатное содержание в среде, в которой происходил
рост губок, что было показано на примере роста пресноводных
губок [66].
Кремнистые валуны, залегающие на определенных уровнях
в меловых пластах в Англии, несомненно, сформировались
в результате постепенного синерезиса скелетов древних губок.
Каждый отдельный скелет постепенно подвергался усадке* и
превращался в валун округлой формы. Данный случай необычен
в том отношении, что понижение величины удельной поверхно-
сти происходило в течение периода, равного 80 млн. лет, и со-
провождалось очень небольшим уменьшением поверхностной
энергии на границе раздела фаз. Внутри валунов, как было от-
мечено Айлером, обнаруживаются замурованные белемниты,
устрицы и другие остатки.
Брюхоногие моллюски, голотурии
У моллюсков, называемых блюдечками (Р. vulgata), как
было показано в [67], зубцы терки состоят из богатых кремне-
земом волокон длиной 80 нм, вероятно связанных вместе по-
средством РегОз. У голотурий Molpadia (Holothurioidea) крем-
незем встречается в гранулах в форме сферических частиц
диаметром 100—190 нм, находящихся в наружном слое орга-
низма, смешанных со сферическими частицами такого же раз-
мера, но состоящими из «ферритина». Последний представляет
собой основной кальциймагнийжелезофосфат [68]. Биологиче-
ская роль кремнезема в голотуриях неизвестна, но поскольку
ферритин может служить в качестве источника железа для
такого организма, то, вероятно, кремнезем также может в нем
сохраняться как запасной источник питания. Это означает, что
в метаболизме подобного организма кремнезем способен играть
необычную роль [69].
Кремнезем в биосфере
1019
Растения
Вероятно, кремнезем является веществом, необходимым для
энергичного роста большинства растений, тем не менее, по-ви-
димому, он часто определяет вторичные эффекты. Например,
в некоторых растениях кремнезем участвует в построении опре-
деленных частей структуры, для других растений характерно
поглощение кремнезема из почвы, несмотря на то что кремне-
зем, казалось бы, и не выполняет какой-либо полезной функции.
Вопрос о значении кремнезема в питании растений остается
невыясненным вследствие того факта, что его присутствие
в некоторых растениях, по-видимому, усиливает их сопротивляе-
мость по отношению к грибковым заболеваниям, что способ-
ствует оздоровлению таких растений. Кроме того, на некоторых
почвах добавление растворимых силикатов ускоряет рост расте-
ний косвенным путем вследствие выделения фосфатных ионов,
адсорбированных на частицах почвы, и таким образом, повы-
шает суммарное количество усвояемого растением фосфата.
Необходимость в кремнии для растений не была доказана, за
исключением лишь нескольких отдельных примеров, поскольку
оказывается трудно удалить все следовые примеси кремния из
искусственных сред, создаваемых для ' выращивания растений
[70]. Шпрехер [71] считал, что кремнезем выполняет важную
биологическую функцию в стимулировании роста растений и,
вероятно, имеет некоторое значение при поддержании «физио-
логического равновесия» в питательных растворах в почве.
Обычно бывает трудно подтвердить, что кремний суще-
ственно влияет на рост растений. Однако, по крайней мере для
свеклы (Beta vulgaris), кремний, согласно данным Рэлея [72],
является важным элементом роста. Важность кремнезема в фи-
зиологии риса и ячменя была подчеркнута Окавой [73]. Напри-
мер, было показано, что кремнезем полезен для молодых расте-
ний как питательное вещество. Кремнезем также нужен для
того, чтобы побеги риса смогли быстрее распуститься, и в об-
щем случае, по-видимому, необходим для нормального роста
риса, особенно при формировании колосьев. По невыясненной
причине побеги молодого ячменя, вероятно, способны противо-
стоять похолоданиям, если в растворе, питающем эту культуру,
присутствует кремневая кислота в коллоидной форме. Липман
сообщил, что подсолнечник, по-видимому, также нуждается
в кремнеземе; в присутствии кремнезема повышался урожай его
семян. Дас и Мотирамани [75] в своих наблюдениях указали
на возможность того, что кремний мог поглощаться растениями
при замещении бора. Так, для маш-фасоли было обнаружено,
что при заболевании — пожелтении растения обогащались крем-
1020.
Глава 7
неземом, а содержание бора понижалось по сравнению со здо-
ровыми растениями.
Однако трудно определить, влияет ли на растение непосред-
ственно кремнезем или же он просто способствует изменению
окружающей микросреды, как это было показано в исследова-
ниях Онодера и Кагесима [76а] по изучению влияния коллоид-
ного кремнезема на рис. Добавление коллоидного кремнезема
к питательному раствору для подкормки побегов риса, по-ви-
димому, привело к тому, что растение стало переносить большие
содержания калия. Однако равным образом возможно и то, что
коллоидный кремнезем выступает в роли адсорбента ионов и,
следовательно, способен удерживать ионы калия на- своей по-
верхности, удаляя их из раствора и от растений.
Пыльца некоторых растений, таких, например, как Lychnis
alba, содержит 0,8 % кремния (или около 2% ЭЮг), сконцен-
трированного, очевидно, во внешних ее структурах, что повы-
шает сопротивляемость гниению или погодным воздействиям
[766].
Во многих случаях растворимый кремнезем, по-видимому,
попадает в 'растение с водой просто как инертный компонент,
а затем осаждается, где только возможно, по мере испарения
воды из листьев и повышения его концентрации. В других слу-
чаях осаждение кремнезема ограничивается некоторыми харак-
терными областями растений, но отсутствует в других областях.
Например, по мере того как кремнезем концентрируется и пре-
вращается в коллоидную форму, он оказывается неспособным
проходить через оболочки клеток и, следовательно, остается
там, где сконцентрировался. В конце концов в некоторых расте-
ниях кремнезем начинает вступать в обмен веществ данного
растения, поскольку он переносится и осаждается в очень точно
определенных формах, например в полых жгучих волосках
крапивы.
Природа кремнеземных отложений в растениях
Как правило, наблюдается, что кремнезем осаждается внутри
тканей растения в аморфной форме. Тем не менее было сообщено,
что в ряде случаев происходит осаждение и кристаллического
кремнезема, хотя, правда, не существует способа определения
того, не является ли такое осаждение следствием механических
пылевидных включений кристаллического кремнезема. Умемото
[77] утверждает, что при получении золы растений методом
низкотемпературной плазмы фактически удается избежать тер-
мических эффектов. (Это позволяет устранить возможную
опасность, появляющуюся при использовании сильных окисли-
телей.) Умемото сообщил, что, хотя вначале кремнезем был
Кремнезем в биосфере
1021
аморфным, после проведения исследований он оказался смешан-
ным с небольшим количеством а-кварца. Леннинг [78] и Стер-
линг [79] с полной уверенностью сообщили о присутствии
кварца в различных растениях.
Кремнеземные отложения в растениях встречаются обычно
в форме частиц характерной формы (фитолиты). Форма частиц
оказывается характерной для данного растения и сильно зави-
сит от разновидности растений [80]. В травах содержание крем-
незема может достигать 2 %, что вызывает смерть телят из-за
образования кремнеземных камней в мочевом тракте животного,
если к их рациону не добавляется соль, способствующая тому,
что животные начинают пить больше воды [81]. Такие фито-
литы проходят по кишечному тракту животных и накапливаются
в почве [82]. Фитолиты напоминают опал (гидратированный
аморфный кремнезем) и обнаруживаются в тканях различных
трав частично в виде трехмерного образования [83]. Это слу-
жит указанием на то, что кремнезем исключается из клеток
растений и осаждается фактически в виде силикагеля в проме-
жутках между клетками [84—86]. Кремнезем переносится в ра-
стении в виде Si (ОН) 4, затем концентрируется и застудневает
по мере испарения воды из листьев [87]. Неудивительно по-
этому, что края листьев сорго, пшеницы и ржи оказываются
окремненными в наибольшей степени, так как в этих участках
растений кремнезем концентрируется сильнее из-за наиболее
быстрой потери воды [88]. Было показано, что структура крем-
незема в целом ряде растений состоит из плотного силикагеля
с порами диаметром 1—10 нм, заполненными водой. Такой
кремнезем полностью аморфен [89]. Характер распространения
кремнезема в растении был изучен с помощью сподограмм, или
получения изображений листьев растений в виде золы [90], и
изменялся даже для различных разновидностей пшеницы. Лен-
нинг [91] провел сравнение показателя преломления и рентгено-
грамм подобного кремнезема и пришел к заключению, что он
представлял собой биогенный опал, поскольку образец оказался
аморфным и не очень пористым. Интересно, что выращенные на
одной и той же почве при идентичных условиях различные
растения накапливают сильно отличающиеся количества разных
элементов. Анализы высушенных веществ, выполненные Купе-
ром, Паденом и Митчелом [92], дали следующие результаты,
выраженные в мае. % элементов:
Элемент Хлопчатник Пшеница
Si 0,08 1,00
Са 1,21 0,13
А1 0,08 0,10
Р 0,41 0,11
N 2,42 0,53
1022
Глава 7
Различия в содержании кремнезема, конечно, нельзя объяс-
нить лишь разным количеством испаряемой воды. Было пока-
зано [93а], что на поглощение и осаждение кремнезема в пше-
нице могли оказывать влияние условия эксперимента.
Упрочнение частей растений
Хотя отложение кремнезема в виде фитолитов и не обяза-
тельно приносит пользу самому растению, тем не менее кремне-
зем, который распределяется определенным образом по струк-
туре растения, в особенности в стеблях, играет необходимую
роль по приданию прочности и жесткости растению; Такой эф-
фект обычен для многих тканей растений, включая стебли трав
и хлебных злаков, скорлупу или оболочки определенных разно-
видностей орехов, бамбука, определенных типов древесины,
а также колючек и жгучих волосков некоторых растений, напри-
мер крапивы. Затвердевшие за счет кремнезема кончики волос-
ков или колючек у некоторых растений обеспечивают им защиту
от травоядных животных [936].
Хвощи
Хвощи (Equisetum genus) содержат настолько много крем-
незема, что их применяли в домашнем хозяйстве в качестве
«мочалки» для чистки посуды. Они также использовались для
чистки зубов. Согласно данным Фризона [94], эти растения
веками применялись как абразивные материалы, причем один
тип употреблялся для шлифования дерева, а другой — для от-
делки домашних инструментов.
Кремнезем в хвоще Е. arvense осаждается в виде длинных
волокон в эпидермисе, а также выступает наружу в виде черве-
образных выростков до тех пор, пока вся поверхность не покры-
вается опаловым кремнеземом [95]. По данным Виховера и
Пруски [96], кремнезем, вероятно, встречается в эпидермисе
растений как органическое соединение с целлюлозным веще-
ством стенок клеток. Это заключение было сделано на основа-
нии того факта, что эпидермальная ткань, остающаяся после
растворения целлюлозы в медно-аммиачном растворе, состояла
из соединения кремнезема с органическим веществом. После
обработки кислотой HF она становилась мягкой и давала поло-
жительную реакцию на целлюлозу. Кроме того, ткань проявляла
значительную сопротивляемость микробам, способным разру-
шать целлюлозу. Кауфман и др. [97а—д] подробно описали
осаждение и распределение кремнезема, а также представили
обзор литературы [976] по кремнезему в хвощах (Equisetum).
Было обнаружено, что кремнезем осаждается внезапно на опре-
Кремнезем в биосфере
1023
деленной стадии клеточной дифференциации. Распределение
кремнезема в многолетнем хвоще (Equisetum hyemale var af-
fine) исследовалось с помощью сканирующего электронного
микроскопа с одновременным применением электронного микро-
зонда, что позволило определить концентрацию кремния в ка-
ждой точке структуры. Кремнезем осаждается только после
того, как стенка клетки полностью растянулась, но не в тех
участках стебля, которые еще продолжают расти. Это справед-
ливо также и для овса и риса.
Упоминавшийся выше микроволокнистый кремнезем в Equi-
setum arvense может формироваться тем же самым способом,
как и силикагель, выделяющийся внутри клеток с внутренней
поверхности оболочек в Avena sativa [95]. Как было показано
на электронно-микроскопических снимках Кауфманом и др.
[97в], кремнезем, по-видимому, в виде волокон диаметром
~ 120 А, находящихся на расстоянии 120 А друг от друга, вы-
тесняется со стороны внутренней поверхности стенок специаль-
ных «кремнеземных клеток». По мере того как параллельные
волокна вырастают и удаляются в сторону от поверхности обо-
лочки, вероятно, они испытывают синерезис, как и большинство
свежеобразованных гелей кремнезема. Однако благодаря нали-
чию ориентированной структуры усадка материала происходит
только по двум направлениям, и волокна вытягиваются вместе
в виде параллельных пучков или палочек диаметром примерно
600 А. На микроснимках видно, что такой высушенный кремне-
зем разрушается, но внутри живой клетки кремнезем образует
выпрямленные линии, по которым кремнеземные палочки на-
правлены от поверхности, подобно ворсу на ковре.
Такой кремнезем, сформированный посредством полимери-
зации при комнатной температуре, вероятно, состоит из плотно
упакованных первичных частиц SiO2 с поверхностными группами
SiOH, а также молекулами воды, прочно удерживаемыми
в микропорах между этими небольшими частицами, точно так
же, как это имеет место в микропорах силикагелей, полученных
в лабораторных условиях. Если провести высушивание при под-
ходящих условиях, то такой волокнистый силикагель должен
был бы иметь значение удельной поверхности более 400—600м2/г.
Волокна могут образоваться в результате биохимического
концентрирования и выделения Si (ОН) 4 на внешней стороне
оболочки клетки. Молекулы Si (ОН) 4 могут затем продиффун-
дировать через близко расположенные, как в сите, отверстия
диаметром 120 А в оболочке и подвергнуться полимеризации
вблизи каждого отверстия с внутренней стороны поверхности
оболочки клетки.
Кауфман [97г] предположил, что подобная полимеризация
кремнезема затормаживается в областях, где гормон гибберел-
1024
Глава 7
линовой кислоты вызывает понижение величины pH от 6,5 до
5,0 и даже ниже, как это отмечалось в вытянутых клетках [97д].
Такой эффект может оказаться настолько существенным, что
падение значения pH должно привести к стабилизированию
хелатных соединений кремния типа трополона и, таким образом,
замедлить выделение мономерного кремнезема (см. работу
Вейсса в [127]).
Бамбук
Твердость и жесткость бамбука частично могут быть припи-
саны присутствию кремнезема в его волокнистой структуре.
Однако избыток кремнезема сосредоточен в скоплениях силика-
геля, которые часто обнаруживаются в полостях бамбуковых
стеблей. Такой студенистый продукт, содержащий некоторое
количество органических веществ и известный под названием
табашир, использовался в восточных странах в качестве врачеб-
ного средства. Согласно данным Фризона [94], это вещество
было известно еще в древности в Китае и Индии, и о нем упо-
минал Одорико Порто, современник Марко Поло, живший
в XIV в. Интерес к этому необыкновенному веществу, по-види-
мому, периодически возрождается. Оно было исследовано евро-
пейскими учеными-химиками во второй половине девятнадца-
того столетия. После этого оно было фактически забыто вплоть
до 1926 г., когда Ракузин [98] опубликовал обзор сведений,
известных о табашире. Годом позже Уолтер [99] изучал специ-
фические физические свойства этого геля. Однако с тех пор ему
уделялось мало внимания, и он не был исследован современ-
ными методами. Согласно данным Ракузина, табашир был
также известен как бамбуковый сахар, так как он сладок на
вкус. Очевидно, что этот гель кремнезема к тому же содержит
сахара или какие-то другие органические вещества, получаемые
из соков бамбука. 99,9 % всей неорганической части табашира
составляет кремнезем. По-видимому, вследствие своей чистоты
табашир был запатентован ранее [100] для использования его
при приготовлении катализаторов крекинга.
Еще в 1791 г. табашир изучался Маци [101], который при-
готовлял из него силикат натрия. Через столетие физические
свойства табашира исследовались Коном [102] и Ван Бемеле-
ном [103]. Согласно данным Кона, такой гель имеет объем пор,
равный 0,75 мл/г. Прозрачные, однородные кусочки геля
аморфны, и под оптическим микроскопом нельзя различить
поры. Однако пористая масса вещества способна впитывать раз-
личные жидкости, образуя прозрачное твердое тело, похожее
на стекло. Через некоторое время после поглощения жидкости
масса становится флуоресцирующей. Окрашивая массу таба-
Кремнезем в биосфере
1025
шира различными веществами, Кон приготовлял синтетические
опалы и ониксы. Он утверждал: «Нигде в растительном или
в животном царстве не существует более странного вещества,
чем табашир». Уолтер [99] исследовал табашир в виде стекло-
видных кусочков массой 3—15 г. Отчасти такое вещество напо-
минает опал, получаемый очень медленным высушиванием геля
кремнезема. Показатель преломления табашира составляет
~1,18 и может быть изменен, если табашир помещать в раз-
личные жидкости. Прокаленный табашир способен поглощать
примерно 166 % воды от массы сухого вещества. Плотность
прокаленного табашира равна 0,54 г/см3, а кремнеземный скелет
составляет 25,7 % от общего объема. Он имеет приблизительно
такую же адсорбционную емкость по отношению к различным
жидкостям, что и коммерческий силикагель. Табашир, находя-
щийся в бамбуке, имеет необыкновенную пористость и способен
очень быстро поглощать раствор иода, метиленовую синь или
фенол. Джоунс, Милн и Сандерс [104] при исследовании
свойств табашира обнаружили, что он представляет собой опа-
ловидный гель кремнезема, состоящий из кластеров, составлен-
ных в свою очередь из кремнеземных частиц размером 10 нм.
Злаки
Многие виды злаков, тростников и соломы устойчивы к раз-
личным погодным условиям в сильной степени благодаря при-
сутствию в них кремнезема [94]. Рисовая шелуха очень бо-
гата кремнеземом. Блестящий эпидермис ротанговой пальмы,
нашедший применение при изготовлении мебели, пропитан
кремнеземом.
И солома, и зерна пшеницы содержат кремнезем. Содержа-
ние кремнезема в соломе колеблется в пределах 2—3 % и со-
ставляет примерно половину общего количества золы. В верх-
ней половине стебля пшеницы кремнезема в два раза больше,
чем в нижней части. Согласно данным Коппенета и др. [105],
зерна также содержат от 0,07 до 0,025 % SiO2. Чен [106] опре-
делял содержание кремнезема в пшенице на различных стадиях
ее роста.
Вследствие того что в зернах содержится кремнезем, полу-
чаемое из них пиво по существу оказывается насыщенным рас-
твором кремнезема. Согласно данным Стоуна и Грея [107, 108],
анализы 14 сортов пива показали присутствие в них 0,006—
0,01 % SiO2, который почти полностью попадал в пиво из солода,
изготовляемого с добавлением шелухи зерна. Такие злаки, как
овес и пшеница, имеют упрочненные стебли благодаря осажде-
нию кремнезема в эпидермисе клеток [109].
39 Заказ № 250
1026
Глава 7
Колючие растения
В некоторых частях растения содержится почти чистый крем-
незем, в частности в колючках или в шипах. Например, согласно
данным Ногера [110], на двух южноамериканских растениях,
Melinis minutiflora и Pappophorum silicosum, образуются легко
Рис. 7.5. Микроснимок волосков жгучей крапивы (из работы [1], с разреше-
шения Cornell University Press).
отделяемые колючки, содержащие 75—84 %' SiO2. Высушенные
цветы этих растений содержат 7,5 и 10 % SiO2 соответственно.
Как сообщалось, крапива содержит кремнезем в волосках.
В высушенной крапиве (стебли и листья) найдено 3,3 масс. %
SiO2 [111]. На рис. 7.5 показаны два таких волоска при неболь-
шом увеличении. В проходящем свете кончик волоска выглядит
как прозрачное стекло, а в свежем образце кончик заполняется
жидкостью, в которой просматривается несколько пузырьков.
По представленным Страсбургером и др. [112а] данным, такой
кончик представляет собой кремнеземистую трубочку, а основа-
ние волоска (в виде луковицы) содержит кальций. Жидкость,
находящаяся на кончике, способна выделяться, когда кончик
волоска проникает сквозь кожу человека и затем отламывается.
Эта жидкость в значительной степени ядовита и содержит бел-
ковый токсин. Некоторые разновидности тропической крапивы
не только причиняют боль, но и вызывают судороги.
Кремнезем в биосфере
1027
При легком прикосновении листьев крапивы к коже хрупкие
волоски проникают через нее, но в том случае, когда растение
захватывается быстро и сильно, волоски успевают отломиться
еще до проникновения в кожу. Эта мысль выражена в старин-
ном четверостишии* (предложенном Ф. К- Карлсоном, г. Уил-
мингтон, США):
Nettles '
Grasp it with a touch that’s gentle,
And it stings you for your pains,
Grasp it as a man of mettle,
And it soft as silk remains.
Anon.
В отсутствие кремнезема крапива не вызывает ожогов. Бар-
бер и Шон [1126] описали проведенные ими эксперименты,
когда крапива Urticaria dioica выращивалась в культуральном
растворе, практически свободном от кремнезема. В этом случае
листья растения проявляли лишь незначительную способность
вызывать ожог. После того как к культуральной среде добав-
лялся раствор кремнезема, жгучесть волосков крапивы через две
недели становилась более ощутимой, вероятно вследствие того,
что волоски приобретали жесткость из-за осаждения кремне-
зема.
В жестких волосках, покрывающих стебли некоторых типов
мака, кремнезем заполняет промежутки между плотно распо-
ложенными волокнами целлюлозы [ИЗ]. Согласно данным
Тинги и Пиллемера [114], колючие волоски защищают растения
благодаря тому, что они способны пронзать насекомых. Оче-
видно, подобные волоски, такие, как у крапивы и чертополоха,
защищают растения от поедания их животными. Такое свойство
кремнезема, находящегося в колючках растения, скорее всего
универсально, но фактически оно доказано только в нескольких
случаях.
Койке
Койке, или слезы Иовы (Coix lacryma L.), имеет твердые,
блестящие, с контрастными пятнышками семена, которые ис-
пользуются как бусы. Эпидермис настолько плотно пропитан
кремнеземом, что, по данным Фризона [94], этими семенами
можно царапать опал.
* Не следует к крапиве иежно прикасаться
Она ведь ядовита, боль резка.
Тут надо по-мужски внезапно, сильно взяться,
Тогда в руке останется, как шелк, она мягка.
Перевод Л. Т. Журавлева
39*
1028
Глава 7
Пальмы
Листья пальмы барассус пальмира, растущей в Индии, на
протяжении веков использовались как писчая бумага. Эти листья
содержат отчетливые кремнеземистые включения. Эндокарпий
костяной пальмы содержит слой вытянутых клеток, объединен-
ных вместе в столбчатое образование, причем каждая клетка
имеет воронкообразную полость, заполненную кремнеземом.
Фризон предложил способ-приготовления образцов для их ис-
следования оптическими методами и получил превосходно вы-
раженные иглы кремнезема, причем каждая из них была по-
крыта еще более тонкими иглами, что хорошо просматривалось
под микроскопом. Кремнеземистые включения имеются также
в эндокарпии кокосового ореха и в волокнах кокосовой пальмы,
липы, а также в манильской конопле.
Деревья
По данным Фризона [94], кремнеземистые включения в виде
плотных частиц кремнезема, находящихся внутри клеток, часто
обнаруживаются в тропических деревьях. Это приводит к за-
туплению пил и других инструментов. В некоторых породах
обнаруживалось вплоть до 3,18 мае. % SiO2, а для более чем
50 разновидностей тропических деревьев (например, тиковое
дерево) содержание кремнезема превышает 0,5 %. Однако ни-
каких следов кремнезема не было обнаружено в деревьях в зо-
нах с умеренным климатом. Древесина некоторых тропических
деревьев так прочна, что ее не могут повредить такие насеко-
мые, как, например, точильщик морской (Toredo). Было убе-
дительно показано, что подобная сопротивляемость обусловлена
присутствием кремнеземных частиц; деревья, содержащие более
чем 0,5 % SiO2, оказываются практически не поврежденными
насекомыми. Амос [115] представил перечень 400 кремнезем-
содержащих деревьев (с содержанием свыше 0,05 % SiO2), при-
надлежащих к 32 семействам, сопроводив его такими данными,
как содержание SiO2, сопротивляемость насекомым-точильщи-
кам и прикладные качества их древесины.
Амос и Дедсвелл [116] исследовали распространение крем-
незема в синкарпии лавролистной австралийской (Syncarpia
laurifoiia Теп.), которая получила всемирную известность бла-
годаря своей устойчивости против морского точильщика. Было
показано, что эта устойчивость прямо связана с содержанием
кремнезема (0,59 % SiO2), так как древесина того же вида де-
рева, но выращенного на Гавайских островах и содержащего
только 0,09 %- SiO2, таким свойством не обладала. Было выска-
Кремнезем в биосфере
1029
зано предположение, что частицы кремнезема, находящиеся
в дереве, разрушают грызущие ротовые органы этих насекомых,
но авторы также считают, что кремнезем может действовать и
как яд, поскольку он растворяется в очень слабом растворе
щелочи и, следовательно, должен переходить в растворимое
состояние в пищевом тракте этого насекомого.
Механизм поглощения, переноса
и осаждения кремнезема
Несмотря на заметную растворимость кремнезема в воде и
относительно большие объемы воды, движущиеся по растению,
а затем испаряющиеся из него, для всех растений наблюдается
невысокая степень окремнения. Фрей-Висслинг [117] считает,
что растения просто выделяют кремнезем как неусвояемое веще-
ство, находящееся в транспирационном потоке. С этих позиций
можно объяснить накопление кремнезема внутри полых стеблей,
как, например, в случае бамбука, но, однако, нельзя объяснить
формирование специфических, в высокой степени окремненных
элементов в отдельных частях растения, таких, например, как
жгучие волоски крапивы. Но как отмечает Фрей-Висслинг,
в большинстве растений кремнезем осаждается в перифериче-
ских участках тканей и вдоль проводящих сосудов. В этом от-
ношении кремнезем напоминает кальций, соли которого, слу-
чайно попадая в систему, осаждаются в некоторых растениях
почти так же, как и кремнезем.
Уиттенбергер [118] исследовал механизм, посредством кото-
рого кремнезем попадает в сок растения через корни ржи и
подсолнечника. Автор обнаружил, что при содержании 0,045 %
кремнезема в культуральном растворе растения накапливали
кремнезем прежде всего в корнях. Поскольку кремнезем рас-
творим только до ~0,01 %, становится понятным, что такое
накопление должно обусловливаться в основном отфильтровы-
ванием коллоидного кремнезема на корневых мембранах.
Однако при содержании кремнезема менее чем 0,015 % (что
приблизительно соответствует истинному раствору аморфного
кремнезема) кремнезем накапливался только в молодых побе-
гах и листьях, что указывает на перенос растворимого кремне-
зема вместе с транспирационным испаряющимся потоком. Было
показано, что при использовании глины в качестве источника
кремнезема корни выделяли вещество, способное переносить
кремнезем в раствор. Это было продемонстрировано путем от-
деления глины от корней с помощью коллодиевой мембраны.
При этих условиях не наблюдалось никакого поглощения крем-
незема. Было сделано заключение, что в естественных условиях
кремнезем, вероятно, поглощается растениями главным образом
1030
Глава 7
в виде растворимой кремневой кислоты и что растворимые си-
ликаты образуются благодаря распаду сложных силикатов.
В этом исследовании подчеркивается роль растений в процессах
эрозии горных пород и формирования почвы. Хольцапфель [119]
считает, что кремнезем может переходить в растворимое состоя-
ние под воздействием некоторых сахаров. Однако существуют
определенные соединения, подобные катехину, которые способ-
ствуют переходу кремнезема в растворимое состояние, и поэтому
их также, вероятно, следует включить в рассмотрение [120]
(см. гл. 1 и 3).
Амос и Дедсвелл [116] утверждают, что в способных погло-
щать кремнезем растениях протоплазменная поверхность корне-
вого волоска носит основной характер с преобладанием гидро-
ксильных групп, которые замещаются силикат-ионами. Те же
растения, корневые волоски которых имеют более кислые свой-
ства, вероятно, имеют активность ионов, обеспечивающую по-
явление благоприятной ситуации для адсорбции катионов;
Отмечается, что в других растениях могут существовать плаз-
матические мембраны, содержащие примерно равное число
кислотных и основных групп, которые, следовательно, должны
поглощать анионы и катионы примерно в равной мере.
Количество кремнезема в растворе понижается при добав-
лении солей металлов. Так, опрыскивание растений и почвы, на
которой выращивался рис, раствором сульфата меди понижало
количество кремнезема, отлагаемого в листьях растений [121].
Без сомнения, данный эффект является следствием образования
нерастворимого силиката меди и, таким образом, превращения
кремнезема в нерастворимое состояние.
Имеется очень мало сведений относительно того, что в расте-
ниях могут встречаться иные, чем свободный кремнезем, соеди-
нения кремния. Малфитано и Катуар [122] сообщили, что особо
чистый крахмал, полученный из картофеля и зерна, образовывал
золу, содержавшую SiOg. На этом основании предполагается,
что кремнезем мог присутствовать в форме химически связан-
ного соединения с крахмалом.
Энгель [123] изучил природу кремнезема в соломе ржи и
показал, что в ней присутствуют органические комплексы крем-
незема. При обработке соломы горячей водой или метанолом
после предварительной обработки смесью метанол—бензол из
нее могли образовываться неустойчивые органические соедине-
ния кремнезема. Такие соединения легко превращаются в не-
органические, нерастворимые полимеры SiO2. Также было полу-
чено небольшое количество кремнийорганического, способного
растворяться в простом эфире комплексного соединения, в кото-
ром, как нашел автор, присутствовала галактоза в соотношении
2 моль SiO2 на 1 моль сахара. Невозможно было определить,
Кремнезем в биосфере
1031
включал ли такой кремнеземный комплекс в эфирном экстракте
жирные кислоты и фосфорную кислоту наряду с небольшим
количеством пентозы, ассоциированной более прочными связями.
Эксперимент, проведенный на более позднем этапе роста расте-
ния, показал, что солома ржи содержала другой кремнеземный
комплекс, в котором отношение SiCh к галактозе было равно
1:1. По-видимому, кремневая кислота в подобной физиологиче-
ской структуре связывается как с компонентами сахара, так и
с другими компонентами. Примерно 18 % кремнезема в струк-
туре соломы ржи должно соединяться с целлюлозным каркасом,
поскольку именно такое количество кремнезема выделялось при
растворении целлюлозы в медно-аммиачном растворе.
Энгель [123] отметил, что поскольку осаждение кремнезема
в специфических частях растения, очевидно, достаточно хорошо
поддается контролю, то такое осаждение должно включаться
в определенные процессы метаболизма, в которых, следова-
тельно, должны участвовать органические соединения кремне-
зема.
В среде с низким содержанием кремнезема растения пше-
ницы могут в действительности терять кремнезем из тканей,
расположенных над уровнем земли. Это показывает, что крем-
незем может переноситься вниз, по направлению к корням,
благодаря процессу циркуляции внутри растения [124].
Для того чтобы понизить содержание кремнезема в траве
свинорой и сделать ее более вкусной для животных, листья
опрыскивали глифосфатом [125]. Определенное содержание
кремнезема понижает усвояемость кормовых растений; это было
подтверждено в экспериментах, в которых к растениям добав-
лялся растворимый силикат [126].
Вейсс и Герцог [127] впервые отделили и идентифицировали
чистое соединение кремния, относящееся к растениям. Они ис-
следовали хвойное дерево тую (Thuja plicata) и нашли хелатное
соединение кремния из туяплицина, которым оказался изопро-
пилтрополон (см. ниже раздел по метаболизму кремния).
Растворимый кремнезем и плодородие почв
Хотя кажется, что кремнезем не является важным для роста
большинства растений, тем не менее неоднократно было пока-
зано, что добавление растворимого силиката к почве или
к культуральным растворам давало благотворный результат
в том случае, когда имелся дефицит усвояемого фосфора. В на-
стоящее время выяснен механизм этого явления. Это происхо-
дит не потому, что растение способно утилизировать силикат
вместо фосфат-иона, как предполагалось вначале, а скорее
благодаря тому, что силикатный ион способен вытеснять фос-
1032
Глава 7
фат-ион с поверхности частиц почвы или коллоидного материала,
увеличивая таким образом содержание фосфора в рассматри-
ваемой системе.
Например, Сринивасан рассмотрел доступную информацию
о роли кремния в питании растений и пришел к заключению,
что силикат в' почве способствует поглощению фосфора. В дру-
гих исследованиях, выполненных этим же автором [128], было
показано, что растворимый кремнезем (или силикат-ион) ад-
сорбируется определенными компонентами почвы, в частности
глинами. Соотношение между концентрацией и степенью удер-
живания силикат-иона оказывается логарифмическим, что ука-
зывает на наличие адсорбции. Было продемонстрировано, что
гели оксида алюминия и оксида железа адсорбировали силикат-
ионы почти так же, как и почвы, образуя адсорбционный ком-
плекс, из которого силикат удаляется промыванием с большим
трудом. Далее было показано, что в том случае, когда почва
обрабатывается растворимым силикатом, фоСфат-ионы адсорби-
руются менее прочно. Силикагель не адсорбирует фосфат-ионы.
Следовательно, ясно, что добавление силиката может привести
к определенному эффекту в питании растения, поскольку сили-
кат вытесняет фосфат-ионы, находящиеся в адсорбированном
состоянии на поверхности почвы и, таким образом, делает фос-
фат более доступным для растения. Бастисс [129] также пока-
зал, что фосфат-ионы можно освободить из адсорбированного
состояния на некоторых почвах посредством добавления раство-
римого кремнезема. Этот прием особенно эффективен для лате-
ритных почв, на которых фосфат-ионы прочно адсорбируются.
Последние, становятся недоступными для растений из-за обра-
зования нерастворимых фосфатов железа и алюминия. В почвах
такого типа добавление силиката ведет к вытеснению адсорби-
рованных фосфат-ионов, так что в результате урожаи зерновых
удваиваются или утраиваются, если среда щелочная, видоизме-
ненная за счет добавления силиката, и возрастают вплоть до
пятикратного размера, если среда нейтральная. Отмечалось
также заметное увеличение в растении содержания SiO2, Р2О5
и железа. Вытеснение фосфат-ионов из некоторого вида почв
силикатом было также продемонстрировано путем измерения
изотерм адсорбции [130]. Обработка почв силикатами натрия
и калия вела к понижению их способности адсорбировать фос-
фат из раствора. Вероятно, силикат изолирует активные адсорб-
ционные центры коллоидной системы и сам удерживается более
сильно, чем фосфат-ионы. Это приводит к предотвращению
адсорбции фосфата.
При исследовании вытеснения анионов из почв путем добав-
ления растворимого силиката было показано [131], что фосфат-
ионы освобождались из адсорбированного состояния только
Кремнезем е биосфере
1033
в слабощелочной среде, так что такое вытеснение скорее осу-
ществлялось гидроксил-ионами или силикат-ионами, чем крем-
невой кислотой. При рН~7 растворимый кремнезем в основном
оказывается в неионизированной форме и поэтому лишь слабо
вытесняет фосфат-ионы. Определенное возрастание урожая
ячменя и суданской травы отмечалось в том случае, когда
к почве добавляли силикат кальция или магния, т. е. вещества,
которые, несомненно, в достаточной мере щелочные, чтобы
обеспечить образование некоторого количества силикатных
ионов. Отмечалось заметное поглощение кремнезема редькой,
ячменем и суданской травой, когда они выращивались на сили-
катных почвах.
Среди других наблюдений, относящихся к воздействию крем-
незема на питание растений, можно отметить следующие. В вод-
ной культуре ячменя растворимый силикат вызывал значитель-
ное повышение сухой массы растений, если в системе отмечался
недостаток фосфора [132]. Развитие листьев тормозилось при
недостатке фосфата и ускорялось при добавлении силиката.
В присутствии достаточного количества фосфора силикат ока-
зывал небольшое влияние. По данным Леммерманна и Висс-
мана [133], кремнезем дает повышение урожая определенных
видов культур, в частности бобовых и крестоцветных, только
в том случае, когда недостаточно содержание фосфорной кис-
лоты. Однако благотворное воздействие кремнезема может ока-
заться значительно слабее, когда в системе отмечается дефицит
поташа или азота. Указанные авторы [134] считают, что крем-
незем не изменяет функциональные возможности растения, но
способствует растворению фосфатных соединений.
Дюшон [135] пришел к заключению, что благоприятное дей-
ствие коллоидного кремнезема на урожаи культур в песчаной
среде при недостаточном содержании фосфорной кислоты, ис-
пользуемой в качестве удобрения, обусловлено главным образом
физическими свойствами коллоидной системы и заключается
в улучшении физического состояния песчаной почвы и исполь-
зования имеющегося фосфора. Хэмпл [136] пришел к аналогич-
ному заключению.
Количество доступного растениям растворимого кремнезема
в почвенной влаге в значительной мере определяется химиче-
ским составом. Свободные оксиды железа или алюминия погло-
щают и переводят в нерастворимое состояние кремнезем. Овес
поглощает кремнезем со скоростью, которая непосредственно
зависит от количества кремнезема в почвенных водах, и сум-
марное содержание кремнезема возрастает с увеличением коли-
чества испаряющейся воды. В этом случае кремнезем, таким
образом, играет лишь пассивную с биохимической точки зрения
роль [137].
1034
Глава 7
Органические кислоты, способные образовывать хелатные
соединения с Fe3+ или А13+, очевидно, приводят к выделению'
кремневой кислоты из соединения кремнезема с этими элемен-
тами в почве или, наоборот, удаляют указанные элементы с по-
верхности кремнезема, что позволяет ему растворяться. Погло-
щение кремнезема ускоряется при понижении значения pH
в почве [138]. Почву можно удобрять кремнеземом, который
переводится в растворимое состояние в виде комплекса с гума-
том аммония, аналогичного комплексу катехолата аммония
[139].
Фосфат-ионы, согласно данным Рейфенберга и Бучвальда
[140], способны вытесняться из почв и глинистых минералов,
таких, как монтмориллонит, причем этот эффект наименее вы-
ражен при pH 7,5. Однако силикат может вытеснять фосфат,
особенно при более высоких значениях pH.
Экстрагирование кремнезема растениями из почвы, целиком
состоящей из инертных горных пород, таких, как базальт, рио-
лит и кварцевый диорит, было исследовано Ловерингом и Эйфе-
лем [141], которые использовали хвощи, известные своей спо-
собностью аккумулировать кремнезем. В опытах применялась
циркулирующая деминерализованная вода. Подсчитано, что
количество кремнезема, поглощенного растениями, было эквива-
лентно количеству базальта в слое, площадь которого равна
0,4 га, а толщина составляет 0,3 м, образовавшегося за 5000 лет.
Было доказано, что по крайней мере часть поглощенного крем-
незема находилась в виде комплекса с ароматическим соеди-
нением.
По данным Эрхарта [142], растения играют некоторую роль
при формировании глинистых минералов; В почвах, бедных
кальцием и магнием, растения содержат А1 и Si в пропорциях,
соответствующих каолину, и вымершие ткани растений выде-
ляют прекаолинит, из которого впоследствии образуется глина.
По-видимому, это подтверждается данными Пейнеманна и Фер-
рейро [143], согласно которым в верхних почвенных слоях рас-
тениями производится образование фракций тонкодисперсного
аморфного кремнезема и глины путем формирования фитолитов
и прекаолина.
Усвояемость растениями кремнезема может оказаться отно-
сительно высокой в почвах, богатых гумусом. Сообщалось о том,
что в ФРГ имеется район, где почва содержит кремневую кис-
лоту в сочетании с подобными таннину компонентами гумуса,
стабильного при pH 9—10 [144]. Рис поглощает большее коли-
чество кремнезема из питательной среды, когда в систему до-
бавляется поли (2-винилпиридиноксид) [145]. Сообщалось, что-
найдено вещество, способное понижать токсичность коллоидного
кремнезема или поликремневой кислоты. Природа образуемого'
Кремнезем в биосфере
1035
комплекса, если он существует, еще не раскрыта (см. гл. 1),
изучено только воздействие pH.
Полезные и защитные воздействия кремнезема
Имеются убедительные доказательства того, что присутствие
кремнезема в определенных растениях ведет к повышению их
сопротивляемости в отношении грибковых заболеваний. Лунди
[146] пришел к заключению, что кремнезем не играет суще-
ственной роли в питании растений, но при осаждении в эпидер-
мисе он обеспечива’ет защиту растения от грибковых заболева-
ний, таких, например, как ржавчинный гриб.
Гермар [147] исследовал такое воздействие кремнезема на
хлебные злаки, в особенности на их сопротивляемость против
милдью. Рожь, ячмень и пшеницу выращивали в очищенном
кварцевом песке и снабжали коллоидным кремнеземом при раз-
личных скоростях его поступления, а также соответствующими
удобрениями. Кремнезем вызывал увеличение сухой массы рас-
тения в том случае, когда наблюдалось недостаточное содержа-
ние оксида калия. Кремнезем осаждается в эпидермисе листьев,
а также формируется в виде кремнеземистых образований, кото-
рые вместе составляют кремнеземный скелет растения. Дефицит
азота и избыток поташа способствуют накоплению кремнезема,
но подвод фосфора не оказывает влияния на аккумуляцию SiO2.
Гермар исследовал эффект присутствия кремнезема в листьях
и пришел к заключению, что SiO2 не влияет на механическую
прочность листа. Однако хлебные злаки, которые достаточно
хорошо обеспечивались кремнеземом, проявляли большую со-
противляемость по отношению к заражению милдью, очевидно,
вследствие осаждения SiO2 в эпидермисе, что делает последний
более устойчивым против воздействия фермента, выделяемого
грибковыми гифами. Сопротивляемость к грибкам, которые спо-
собны внедряться в растение через устьица, не повышалась
в присутствии кремнезема.
Вагнер [148] продемонстрировал важное значение кремне-
вой кислоты в повышении сопротивляемости растений против
порошковидных грибков милдью. В водных культурах недоста-
точное содержание кремнезема приводило к угнетению роста
таких растений, как рис, овес, ячмень, кукуруза, огурцы, табак,
кустовая фасоль и помидоры. Растения с дефицитом кремнезема,
несомненно, нуждаются в большем количестве воды при данной
скорости роста. Кремневая кислота действует благоприятно
в отношении накопления и лучшего использования кальция, фос-
фора, калия и магния в растениях. При изучении метаболизма
в растениях оказалось невозможным заместить фосфорную кис-
лоту на кремневую кислоту, поэтому кремнезем рассматривался
1036
Глава 7
как ценный компонент для этих растений. В экспериментах,
проводимых в лабораторных условиях, доля пораженных участ-
ков растений милдью оказалась обратно пропорциональной
содержанию кремнезема. Соммер [149] пришел к заключению,
что в случае проса, выращенного в культуральном растворе,
кремнезем повышал урожай семян и делал эти семена невос-
приимчивыми к заражению грибками.
Влияние кремневой кислоты на сопротивляемость риса про-
тив пирикуляриоза риса (грибковое заболевание) изучалось
Иоши [150]. Было определено, что она оказалась пропорцио-
нальной концентрации кремнезема в культуральной среде и
в самом растении риса. Однако эту сопротивляемость не связы-
вали с прочностью длинного узкого листа растения, измеренной
методом иглоукалывания. Такая прочность листа не сопостав-
лялась и с содержанием кремнезема, и фактически листья ока-
зались даже менее прочными в том случае, когда растение
выращивалось в присутствии кремнезема. Таким образом, было
установлено, что присутствие кремнезема защищало растение
от указанного заболевания, но механизм такой защиты остается
еще непонятым. Выполненное в дальнейшем исследование [151]
по воздействию кремнезема на сопротивляемость риса к забо-
леваниям показало, что магний усиливает благоприятное влия-
ние кремнезема, так как в отсутствие магния растение не погло-
щало кремнезем. При добавлении магния растение начинало
поглощать кремнезем, который затем накапливался в листьях
и повышал сопротивляемость растения против грибка Piricularia
oryzae.
Эффекты благоприятного воздействия кремнезема были рас-
смотрены в обзорах [152, 153], в том числе вопросы переноса
питательных веществ из почвы и повышения сопротивляемости
против насекомых и грибковых заболеваний. Никакой связи
между содержанием SiO2 в растениях пшеницы и сопротивляе-
мостью к гессенской мушке или устойчивостью против заболе-
ваний Лэннинг [154] не установил, но для некоторых сортов
сорго между этими факторами наблюдалась прямая корреляция
[155]. В лабораторных экспериментах по удобрению ячменя
силикагелем и опылению его силикагелевым порошком было
показано снижение заболеваемости милдью [156]. Многочислен-
ные исследователи сообщили о благотворном воздействии крем-
незема на рис. Листья растений становились твердыми и жест-
кими по мере того, как содержание кремнезема в них повыша-
лось от 0,3 до 13%; кроме того, получено определенное
доказательство контролируемости осаждения кремнезема мета-
болической активностью [157]. Сопротивляемость к личинкам
паразитических червей [158], а также к насекомым-точильщи-
кам в стеблях растений [159] возрастала. Другие благоприят-
Кремнезем в биосфере
1037
ные воздействия заключались в возрастании сопротивляемости
против коричневой пятнистости [123].
Энгель сообщил [123], что рожь, огурцы, помидоры, бобы
и табак при дефиците кремнезема становились более чувстви-
тельными к воздействию холода и к грибкам милдью. Некото-
рые исследования по воздействию кремнезема на рис наводят
на мысль, что кремнезем может играть не только благоприят-
ную, но, возможно, и существенную роль. Окамото [160] нашел,
что растворимый кремнезем в питательной среде вызывал повы-
шение скорости роста и конечную массу сухого растения, а также
увеличивал размеры частей растений и размер зерен.
В завершение обзора о важной роли кремнезема в растениях
необходимо упомянуть о том, что обширная библиография по
этим вопросам приведена в книге Воронкова, Зелчана и Луке-
вица [4а].
Насекомые
Содержание кремнезема в насекомых меняется в сильной
степени для разных видов. Например, в золе шпанской мушки
содержится вплоть до 15 % SiO2, а для определенных насеко-
мых, относящихся к роду Homoptera, в золе содержится до 88 %
S1O2- Однако в большинстве насекомых содержание кремнезема
составляет 1—2% [3]. Снова возникает вопрос, сколько крем-
незема могло присутствовать в этом случае в прилипших части-
цах почвы, а сколько — внутри насекомого. Тем не менее крем-
незем, несомненно, в действительности имеется внутри опреде-
ленных тканей насекомого, например придает твердость их
мандибулам [161].
Кремнезем взаимодействует с определенными насекомыми
физическим способом, не включаясь каким-либо образом в био-
химические процессы в насекомом. Это позволяет использовать
кремнезем как инсектицид. Любая курица избавляется от жи-
вущих на ней насекомых-пухоедов, купаясь в пыли, распушив
свои перья и стараясь, чтобы пыль проникла до самой кожи.
Вероятно, когда в XVII в. напудривали парики, то стремились
достичь той же цели!
Тонкодиспергированные твердые порошки с высокой удель-
ной поверхностью действуют как контактные инсектициды, по-
скольку они поглощают липиды (масла) из кутикулы насеко-
мого, организм которого при этом быстро дегидратируется.
Гидрофобный кремнезем действует в этом отношении сильнее,
чем гидрофильные порошки. Такие пылевидные препараты пли
дусты оказываются наиболее эффективными при низкой степени
влажности, но применение лишь одного осушителя, например
СаС12, который не является олеофильным, не оказывает инсек-
1038
Глава 7
тицидного действия [162]. Кремнеземный аэрогель, отчасти
органофильный благодаря способу его приготовления, при кон-
центрации 0,05 % тормозил активность долгоносика в зернах
[163]. При расходе 27 кг SiO2 в виде пирогенного кремнезема
(Cab—О—Sil) на 35 м3 было получено более продолжительное
защитное действие против точильщика зернового по сравнению
с химическими инсектицидами [164].
Тонкодисперсный порошок оксида алюминия также оказы-
вает инсектицидное действие. Когда гусеницы ползают по влаж-
ному дусту, то не наблюдается никакого эффекта из-за присут-
ствия влаги. Однако при этом кожный покров гусеницы теряет
свою маслянистую защиту вследствие адсорбции оксида алю-
миния, после чего насекомое обезвоживается и быстро погибает,
если его помещают в сухую атмосферу [165]. Смесь аэрогеля и
фторсиликата аммония оказалась особенно эффективной для
уменьшения активности клещей на содержавшихся в клетке
пресмыкающихся [166]. Силикагелевые инсектициды поражали
устойчивых к ДДТ комаров [167]; кремнеземный аэрогель, (мо-
дифицированный путем его обработки борной кислотой, ока-
зался эффективным против тараканов; такой препарат оказывал
смертельное действие, даже когда ротовые отверстия тараканов
были закрыты воском, чтобы этот инсектицид не попадал в их
пищеварительный тракт [168, 169].
Эбелинг [170] провел изучение необходимых характеристик
наиболее эффективных инсектицидных порошков. Он нашел,
что такой порошок должен иметь высокое значение удельной по-
верхности, достигаемое за счет наличия пор с диаметром более
2 нм, и высокую адсорбционную способность по отношению
к воску; при удалении последнего с кутикулы насекомое быстро
обезвоживается и погибает. Кремнеземная суспензия с добав-
лением клея представляет собой полезный препарат для обрыз-
гивания курятников с целью уничтожения пухоедов (Mallo-
phaga) и клещей [171].
Таршиш [172] представил обзор по применению инертных
пылевидных препаратов в качестве инсектицидов, в особенности
для уничтожения тараканов.'
Рыбы, земноводные, пресмыкающиеся, птицы
Отдельные доступные данные были подобраны Воронковым,
Зелчаном и Лукевицом [4а]. В некоторых случаях невозможно
было решить, какие из сообщаемых данных были надежными,
а какие превышали то количество кремнезема, которое теорети-
чески может находиться в виде растворимого SiOH4 в жидко-
стях рассматриваемого организма. Таким образом, если организм
содержал 50 % воды и 10 % золы и вода была насыщена аморф-
Кремнезем в биосфере
1039
ным кремнеземом, то кремнезема в золе должно было бы со-
держаться 0,05%. Обычно при анализе золы обнаруживается
не менее 0,1—0,2 % кремнезема. Кожа, перья, кишечник и т. д.,
т. е. те части тела, которые подвергаются контакту с веществами
из окружающей среды, имеют более высокое содержание крем-
незема. Например, в золе перьев может содержаться вплоть до
77 % кремнезема, что является достаточным доказательством
физической адсорбции коллоидного кремнезема или силикатов.
К тому же слишком высокое содержание кремнезема в более
ранних данных могло быть вызвано наложениями, возникаю-
щими благодаря присутствию фосфора при проведении кон-
трольных измерений кремнемолибдатным методом.
Тем не менее имеются некоторые примеры, когда при ана-
лизе крови, яиц и внутренних органов в золе обнаруживалось
вплоть до 1 % кремнезема, что не может быть объяснено физи-
ческой окклюзией или концентрированием растворимого кремне-
зема. Важная роль кремнезема в развитии цыплят будет рас-
смотрена в следующем разделе.
Млекопитающие, человек
Часто принималось, что для высокоорганизованных живых
существ, т. е. для животных, такой элемент, как кремний, не
играет существенной роли. Однако, Хольцапфель [173, 174] по-
казал, что кремнезем присутствует в следовых количествах
в большинстве животных.
Животные постоянно получают некоторое количество кремне-
зема из воды, но в отличие от растений они способны также
выделять кремнезем в растворе, так что не происходит никакого
накопления кремнезема в теле животного. Кремнезем, попадаю-
щий в организм через рот, конечно, почти безвреден. Кроликам
скармливали по 2 г силикагеля ежедневно в течение 30 недель,
и не было обнаружено никаких вредных последствий [175].
Силикат магния, который при действии кислоты легко выделяет
кремнезем, находит применение при терапевтическом лечении.
Оказывает ли силикагель при приеме внутрь благотворное
действие при некоторых обстоятельствах или же нет, этот вопрос
остается дискуссионным. Табашир, или силикагель, обнаружи-
ваемый внутри бамбука [94], по традиции считается ценным
веществом при лечении астмы и туберкулеза [98], и возможно,
что для подобного лечения есть некоторые практические осно-
вания, как это имело место с другими народными средствами.
Примерно в 1920 г. в Европе был проявлен некоторый инте-
рес к использованию кремневой кислоты в качестве терапевти-
ческого препарата против туберкулеза. Например, Гоннерманн
[176] отмечал, что лекарственные травы, применяемые сель-
1040
Глава 7
скими жителями средней Европы в качестве средства от тубер-
кулеза, были исключительно богаты кремнеземом. Был сделан
анализ золы, полученной от различных тканей животных, и об-
наружено, что, как обычно, в ней содержалось 2—10 % S1O2.
Это наводит на мысль, что кремнезем выполняет некоторые
важные физиологические функции. Однако необходимо учиты-
вать, что доля золы, полученной из тканей, очень невелика, так
что фактическое содержание кремнезема оказывается чрезвы-
чайно низким. Кале [177] допускал, что содержание кремнезема
в поджелудочной железе изменяется в зависимости от различ-
ных типов заболеваний. При туберкулезе поджелудочная железа
содержит меньшее количество кремневой кислоты, чем у здоро-
вого человека, тогда как в случаях раковых заболеваний в этой
железе обнаруживается большее содержание кремневой кислоты
по сравнению со здоровым органом при условии, что не прово-
дилось никакого хирургического вмешательства. В случае опе-
рации содержание кремнезема оказывалось меньшим, чем
у здорового человека. При беременности, как полагают, п^од
отбирает кремнезем из поджелудочной железы матери; таким
образом, объясняется повышение восприимчивости женщины
к туберкулезу в этот период. Утверждалось, что при проведении
опытов по исследованию туберкулеза на морских свинках раз-
витие заболевания тормозилось путем введения кремневой кис-
лоты. Одно время кремневая кислота приготовлялась.и прода-
валась для терапевтического лечения туберкулеза легких. Спо-
собы описания характеристик этого препарата, датируемые
1921 г., не дают возможности точно представить природу золей,
которые тогда использовались [178]. Однако, Кадиш [179] не
обнаружил каких-либо благотворных результатов при исполь-
зовании кремневой кислоты в 14 случаях лечения открытой
формы туберкулез», и, вероятно, после этого интерес к подоб-
ному методу лечения угас.
Данные по содержанию кремнезема в теле млекопитающих,
собранные Воронковым, Зелчаном и Лукевицом [4а], так же
сомнительны, как и упоминавшиеся в предшествующем разделе.
В золе тех частей тела животного, которые имеют непосред-
ственный контакт с веществами из окружающей среды (волосы,
желудок, кожа и кишечник), обнаруживают вплоть до 15 %
кремнезема. Однако во внутренних органах, крови, как правило,
содержится 0,1—-1,0 % Опять-таки невозможно судить о том,
насколько важны эти числа, если не известна доля такой золы
во всем образце. В тканях и жидкостях живых организмов
обычно содержится 0,0010—0,0100 % SiO2 от основного влаж-
ного компонента. Поскольку эти значения меньше растворимо-
сти аморфного кремнезема в физиологическом солевом растворе
при обычном для такого раствора значении pH, то нельзя судить
Кремнезем в биосфере
1041
о том, какая доля кремнезема, если она вообще имеется, связана
с биохимическими компонентами.
Огромный объем сведений о содержании кремнезема в теле
человека и млекопитающих определяется необходимостью изу-
чения проблемы силикоза. Были отмечены некоторые необычные
факты, например повышение содержания кремнезема в крови
по утрам по сравнению с вечером, а также повышенное его
содержание в пожилом возрасте. Очевидно, накопление крёмне-
земсодержащих пылей в легких увеличивается с возрастом.
С другой стороны, содержание кремнезема в артериальных
стенках сосудов оказывается более высоким (0,019 %) у мла-
денцев, а у взрослого человека средних лет оно почти в два
раза ниже [180].
В сыворотке крови человека концентрация кремнезема со-
ставляет 0,00079 % независимо от пола или возраста [181].
Аустин [127] сообщил, что в эритроцитах содержание SiO2
достигает 0,0092 %, из них 0,0042 % находятся в связанном со-
стоянии. Имеются данные [182], что во. всей крови в целом
находится 0,0016 % кремнезема, и его содержание достигает
0,0020 % в случаях заболевания силикозом. Однако, согласно
Хвапилу [127], количество кремнезема в кровяной сыворотке
зависит в большей мере от атмосферных условий, чем от сте-
пени повреждения организма силикозом. В других опытах про-
верка сотен людей показала, что в среднем во всей крови
содержание кремнезема составляет 0,00083 ±0,00024 % [183,
184], причем отсутствует какая-либо корреляция с полом, воз-
растом, родом занятий или с условиями, определяющими дея-
тельность легких. Однако этот уровень возрастал, когда соеди-
нения кремния вводились определенным образом. В таком слу-
чае могло бы показаться, что уровень содержания кремнезема
должен был бы отчасти изменяться для разных стран и геогра-
фических условий обитания. Сауэр и др. [185] подтвердили,
что содержание кремнезема в тканях подопытных животных не
зависело от концентрации кремнезема, добавляемого в рацион
животных. У .крыс содержание кремнезема в мышцах повыша-
лось по мере их роста, но затем понижалось в пожилом воз-
расте. Содержание кремнезема в мышцах увеличивалось при
добавлении кофеина [186]. Уровень кремнезема в крови изме-
нялся в обратной зависимости от содержания кремнезема
в мышцах.
Из этих противоречивых результатов становится очевидным,
что аналитические методы, применявшиеся в прошлом, давали
вводившие в заблуждение завышенные результаты. В работе
Бауманна [187] показано, что концентрация кремнезема в крови
заметно ниже, чем принималось раньше, и что кремнезем при-
сутствует почти полностью в виде свободной растворимой моно-
40 Заказ № 250
1042
Глава 7
кремневой кислоты. Бауманн обнаружил, что концентрация
кремнезема в красных кровяных тельцах животных была точно
такой же, что и в плазме крови. В обоих случаях кремнезем
присутствовал в виде мономера, поскольку обладал способно-
стью проходить через ультрафильтр. Во всей крови крупного
рогатого скота нормальная концентрация Кремнезема составляет
0,00019±0,00005 %. В крови человека его содержится 0,00004—
0,00005 %. Однако когда с питьевой водой вводится 50 мг рас-
творимого кремнезема, то весь кремнезем выделяется с мочой
в течение 10 ч. Максимальная концентрация кремнезема в моче
при этом изменяется от 0,02 и до 0,06 % в зависимости от
объема выделенной мочи, и кремнезем еще остается полностью;
мономерным. Оказалось, что кремневая кислота полимеризуется
с одной и той же скоростью и в моче, и в воде. Скорость удале-
ния кремнезема, выраженная в микрограммах в минуту, явля-
ется постоянной величиной независимо от объема выделяемой
мочи. Она пропорциональна количеству оставшегося в организме
кремнезема. Концентрация кремнезема в крови при этом дости-
гает 0,0002—0,0003 %. Если поглощается 300 мг растворимого
кремнезема, то его концентрация в крови достигает 0,0006 %.
Отношение концентрации выделяемого с мочой кремнезема
к его концентрации в плазме крови, составляющее около 100 : 1,
определяется тем, что почки выделяют кремнезем вместе со
всеми другими растворенными веществами, имеющими молеку-
лярную массу менее 70 000 при ультрафильтрации через мем-
браны почечных клубочков с образованием «первичной мочи».
Затем на следующем этапе в почечных канальцах биологически
активными мембранами поглощается около 99 % всей воды по
неизученному «активному» процессу. В результате в конечной
концентрированной, вторичной, моче остаются все вещества,
которые повторно не всасываются в кровь. Таким образом, кон-
центрация кремнезема, выделяемая с мочой, оказывается го-
раздо большей, чем его концентрация в плазме крови [187].
Японские исследователи обнаружили, что высокое содержа-
ние кремнезема в питьевой воде прямо коррелирует со смерт-
ностью, вызванной повреждением сосудов в центральной нерв-
ной системе. Содержание кремнезема в некоторых частях тела
повышалось в случаях возрастного атеросклероза, в особенности
в аорте [188]. С другой стороны, Лопер и Полет [189] пришли
к заключению, что, хотя в артериях крупного рогатого скота
и человека обнаружен кремнезем, его содержание выше в нор-
мальных артериях, чем в перерожденных. Кроме того, при каль-
цинировании артерий содержание кремнезема в крови оказы-
валось низким.
Кворнинг [190] нашел, что во влажной ткани аорты чело-
века содержится 0,012—0,015 % кремнезема [19'0]. С другой
Кремнезем в биосфере
1043
стороны, в сильно обызвествленной кальцием аорте кремнезема
оказалось только 0,005 %, хотя это, возможно, было обусловлено
высокой концентрацией солей кальция. Количество кремнезема
в органической части аорты могло оставаться достаточно вы-
соким.
Более низкое содержание кремнезема в случае вырожденных
артерий, вызываемое пищей с большим количеством холесте-
рина, позволило в ходе экспериментов изучить вопрос о том,
способствует ли введение соединений растворимого кремнезема
предотвращению подобного заболевания у крыс. Лопер и Лопер
[191] нашли, что введение внутривенно растворимого силиката
или введение через рот лизинсиликата приводило к уменьшению
количества новообразований (атером) от 100 до 40%, а моно-
метилтрисиланолсалицилат, вводимый внутривенно, понижал их
развитие до 11 %.
Согласно данным Чернота и Переса [192], метаболизм крем-
ния в организме регулируется стероидными гормонами и гормо-
нами щитовидной железы, поэтому изменение содержания крем-
ния с возрастом может быть обусловлено понижением гормо-
нальной активности. Продолжавшееся в течение 9 лет изучение
поведения кремния в теле животного позволило сделать сле-
дующий вывод: старение организма сопровождается пониже-
нием содержания кремния в мягких тканях у самок и повыше-
нием кремния в кальцинированных тканях у обоих полов [127].
Кремний играет определенную роль в гормональном балансе
системы. Комплекс, состоящий из NaOSiCH3(OH)2 и ортоокси-
бензоата натрия, понижает аномальные минеральные отложения
у крыс с удаленными яичниками и ведет к улучшению нарушен-
ного кальций-магниевого обмена, вызванного гормональными
изменениями вследствие овариотомии или старения животного.
В глазных нервах человека, как отмечал Семенов, содержа-
ние кремнезема повышалось с возрастом, особенно после 60 лет
[193а].
Кремнезем также участвовал в процессах возрастных склеро-
тических изменений идентичного типа вырождения, происходя-
щего в головном мозге молодых людей при болезни Алцгеймера.
Аустин [127] описал характеристические микроскопические
тельца, или тромбоциты, а также другие тельца (corpora ату-
lacea), которые появляются в среде определенных вырождаю-
щихся клеток головного мозга. Такие тельца содержат довольно
высокую концентрацию кремния: в некоторых областях мозга
вплоть до 1—2 масс. %.
Сообщалось также [1936], что при подобных заболеваниях
головного мозга в ядрах нейронов содержание алюминия повы-
шалось в 4 раза, но, по-видимому, еще более значительным было
увеличение количества кремния. Алюминий мог просто адсор-
40*
1044
Глава 7
бироваться на кремнеземе ввиду большого сродства между
кремнеземом и оксидом алюминия. Присутствие в высокой сте-
пени локализованного кремния в тромбоцитах может оказаться
только результатом полимеризации кремневой кислоты, выде-
ляемой из остатков окружающих поврежденных тканей, в кото-
рых при нормальном состоянии головного мозга обнаруживается
очень низкая концентрация кремния (см. следующий раздел).
Роль кремнезема в жизнедеятельности
млекопитающих
Как давно известно, кремний обнаруживается в определен-
ных видах низших организмов и растений и, вероятно, играет
для них важную роль, однако не было установлено, насколько
существенными для млекопитающих были следовые количества
кремнезема. Так как следы кремнезема присутствуют во всех
видах природной пищи, в воде и в переносимой по воздуху
пыли, то трудно было создать среду, полностью лишенную
кремнезема. Проблема была решена тогда, когда научились
приготовлять искусственную пищу, отфильтрованный воздух,
очищенную воду, полностью пластмассовую посуду и соответ-
ствующее лабораторное оборудование.
Количества кремнезема, найденные ранее в тканях живых
организмов, оказались в сильной степени завышенными по срав-
нению с результатами, полученными в настоящее время с аппа-
ратурой, совершенно лишенной стекла, при использовании хими-
ческих реактивов, в которых отсутствовал кремнезем. Наивыс-
ший уровень содержания кремнезема для большинства тканей
млекопитающих определяется теперь равным 0,0001—0,0002 %.
Однако его содержание выше (около 0,01 %) в соединительных
тканях и в областях активного кальцинирования, достигая 0,5 %
SiO2 при развитии костей в молодом организме.
Фауре [194] сообщил, что наиболее плотные и прочные кости
развивались у кроликов, которым добавляли в рацион 3 г SiO2
ежедневно в течение 3 мес. Содержание SiO2 в костях оказалось
в 10 раз выше по сравнению с его концентрацией в контроль-
ных особях. Отношение Са : Р оставалось постоянным, а крем-
незем, по-видимому, включался в процесс развития костей на
ранних этапах. Однако продолжительное введение кремнезема
вызывало воспалительные процессы в костях. Шварц с сотр.
[195—197] разработали новый способ определения требуемых
следовых элементов в животных при полном исключении загряз-
нений и обнаружили, что кремний играет важную роль. Почти
одновременно было обнаружено, что то же самое справедливо
для цыплят [198, 199].
Кремнезем, в биосфере
1045
Шварц [200] и Карлисл [201] нашли, что развивающиеся
костные структуры у зародышей оказывались деформирован-
ными в отсутствие кремния. Все это подтверждает, что кремне-
зем так или иначе важен для скелета и соединительных тканей.
Карлисл [127] сообщил, что кремний необходим в качестве
затравочного агента, образующего зародыши, вероятно, в форме
силиката кальция, для последующего осаждения на них фтор-
фосфата кальция. Шварц отмечает, что кремний играет суще-
ственную роль в соединительных тканях, где связывается в му-
кополисахаридах связями типа эфирных, хотя эта точка зрения
остается спорной. Пектин содержит 0,5 % кремнезема в связан-
ной форме. Некоторые сульфаты гиалуроновой и хондроитино-
вой кислот и белки в соединительных тканях содержат.связан-
ный кремнезем. В соединительных тканях животных и растений
кремнезем используется как агент для образования поперечных
связей [202].
Содержание кремния в питьевой воде (в виде растворимой
формы Si(OH)4 коррелирует до ограниченного предела с опре-
деленной статистически пониженной смертностью в различных
сообществах. Обычно высокие концентрации растворенных
твердых веществ понижают вероятность сердечно-сосудистых и
почечных заболеваний [203]. С другой стороны, аналогичное
исследование, проведенное в Англии, по-видимому, показало,
что высокое содержание кальция в питьевой воде (чае?) ока-
зывало действие, противоположное кремнию, причем последний
положительно коррелировал с фактором смертности [204а].
Взаимосвязь содержания кремнезема в теле с возрастом и здо-
ровьем организма остается неизвестной ввиду противоречивых
данных.
Данные Аустина, обсуждавшиеся выше, показывают, что
кремний, находящийся в следовых количествах в головном
мозге, может играть существенную роль. Обнаружено, что, хотя
и не наблюдается общего повышения содержания кремния с воз-
растом, он, по-видимому, концентрируется в микроскопических
тельцах в головном мозге людей, страдавших слабоумием и
умерших в старческом возрасте, или молодых людей, умерших
от болезни Алцгеймера. В обоих случаях имеет место схожее
повреждение головного мозга. Это наводит на мысль, что крем-
ний концентрировался в определенных участках головного
мозга в виде побочных продуктов его отравления. Аустин [127]
обобщил также свои данные, ранее описанные в серии статей
[2046, в]. Не было получено доказательств, что кремний при-
сутствовал в данных случаях в необычных количествах или же
играл какую-либо роль в процессах дегенерации.
Гранулы, содержащие кремний, по данным Мехарда и Вол-
кани [204г], также обнаруживались в митохондриях клеток
1046
Глава 7
печени, почек и селезенки нормальных крыс. В гранулах диа-
метром 25—75 нм под электронным микроскопом наблюдались
более мелкие, размером 5—10 нм, непрозрачные гранулы, кото-
рые имели высокое содержание кремния и фосфора. Функция
кремния неизвестна, но вполне вероятно, что он является ком-
понентом ферментов.
Токсичность кремнезема
Ввиду повсеместного присутствия по крайней мере некото-
рых форм растворимого кремнезема в природных водах не уди-
вительно, что он считается безвредным в продуктах питания и
потребляемых жидкостях. Мономерный кремнезем Si(OH)4 про-
никает во все жидкости и ткани тела в концентрациях, мень-
ших, чем его растворимость (0,01 %), и легко выделяется [187а,
205]. В крови человека, как в ее форменных элементах, так и
в плазме, концентрация кремнезема составляет 0,0001 %,
а в крови крупного рогатого скота 0,00022 %.
Прием внутрь небольших количеств аморфного кремнезема
в виде порошка, по-видимому, оказывается полностью безвред-
ным, так как он растворяется с образованием только мономера.
Такой кремнезем представляет собой безопасную присадку
к продуктам питания, и это позволяет использовать его в каче-
стве добавки против подгорания [206] в количествах вплоть
до 2 %.
В тканевых культурах селезенки и легких кроликов не было
отмечено никаких изменений, когда кремневую кислоту вводили
в концентрации 0,0065 % в течение 6 мес. Очевидно, кремнезем
добавляли в виде олигомера, но, вероятно, в таких условиях он
быстро деполимеризовался до мономера [207, 208]. В ранее
проведенных аналогичных экспериментах при содержании
0,0009—0,0021 % SiO2 также не было обнаружено никаких ток-
сических воздействий. Остается неясным, достигала ли моно-
кремневая кислота равновесной концентрации внутри клеток
подобных тканей.
Шил, Флейшер и Клемперер [209] продемонстрировали не-
токсичность Si (ОН) 4 и возможность его удаления. Насыщенные
растворы монокремневой кислоты при 38 °C, как было показано,
содержат 0,017 % SiO2 в равновесии с коллоидным аморфным
кремнеземом. Кремнезем, введенный в организм крыс в виде
силикагеля или золя, удалялся с мочой в концентрации 0,0184 %.
Это указывает на то, что кремнезем выделялся в виде моно-
мера и вызывал поглощение воды, при этом объем выделяемой
мочи удваивался или утраивался. Указанный мочегонный эф-
фект являлся единственным симптомом токсичности кремнезема,
Кремнезем в биосфере
1047
выявленного на крысах. (Не представляет ли собой насыщенный
раствор SiOH4 безвредное мочегонное средство?)
Чрезмерное поглощение растворимого кремнезема крупным
рогатым скотом при недостатке воды для удаления кремнезема
из системы может приводить к образованию камней в мочеис-
пускательном канале [210, 211], в особенности если концентра-
ция кремнезема в моче превышает 0,007—0,008 % [212]. Подоб-
ным образом у собак, в рацион кормления которых входили
в качестве значительной составной части силикатные веще-
ства, образовывались кремнеземные камни в почках, мочевом
пузыре и мочеиспускательном канале [213].
Кремнезем, вводимый в рацион питания крыс в больших
количествах в течение различных периодов времени, в конечном
счете весь выделялся в основном с мочой [214]. На основании
анализа выделяемых количеств кремнезема Минова [215] при-
шла к заключению, что кремнезем имеет в 29 раз большую
растворимость в организме, чем в физиологическом растворе. Зна-
чительная часть такого кремнезема должна так или иначе свя-
зываться в тканях. У крыс, имевших большую нагрузку в пита-
нии в виде силикагеля, в кишечном тракте формировались узе-
лочки [216]. Образование камней (при мочекаменной болезни)
может быть понижено, если вводить в рацион животных больше
соли, чтобы они больше пили воды и, таким образом, кремнезем
вымывался из организма [217]. Контрольное кормление овец
дало возможность получить соотношение между концентрацией
кремнезема и объемом мочи [218]. Высокое содержание кремне-
зема в крови и в моче крупного рогатого скота оказывается
симптоматическим признаком хронической гематурии мочевого
пузыря. Кремнезем сам по себе не является причиной этой бо-
лезни, которая, несомненно, возникает в результате присутствия
в сене какого-либо токсического органического соединения, воз-
можно таких веществ, как сапонины [219]. Керстен и Стаудин-
гер [220] в 1956 г. представили обзор по биохимическому дей-
ствию монокремневой кислоты.
Когда кремнезем вводится в тело не через рот, а каким-либо
иным путем, то почти неизменно получается некоторый токсич-
ный эффект, и кремнезем осаждается в различных частях тела.
Ниже приведены подтверждающие это примеры.
Кремнезем, силикат кальция и силикат натрия в тонкодис-
персной форме вводили собакам в виде аэрозолей, которые жи-
вотные вдыхали. Такой кремнезем полностью задерживался
в легких. Силикаты выделяли растворимый кремнезем, который
распространялся по телу и обнаруживался главным образом
в печени и в моче, а для удаления силиката кальция требова-
лось более продолжительное время [221]. Вдыхание кремнезема
в конце концов приводило к заболеванию силикозом.
1048
Глава 7
Введение внутривенно беременным крысам суспензии, состав-
ленной из пирогенного кремнезема (аэросила), во всех случаях
приводило к выкидышам [222],
Не удивительно, что непрерывное введение внутривенно кол-
лоидного кремнезема кроликам приводило к высоким уровням
концентрации кремнезема в печени, селезенке, сердце, легких и
почках и в конечном счете к гибели животных [223]. В том
случае, когда органофильные частицы кремнезема приготовля-
лись путем их предварительного этерифиЦирования спиртами
или алкилсиланолами, то сразу же после введения в организм
никакой реакции не наблюдалось в отличие от немодифициро-
ванного кремнезема, но по прошествии нескольких суток наблю-
далось токсическое действие [224].
Из тысяч проведенных в течение ряда лет экспериментов
на крысах и кроликах, когда кремнезем вводили во всех мысли-
мых его формах в систему кровотока, в результате чего живот-
ные погибали, было получено одно очевидное заключение: вве-
денный подобным способом кремнезем осаждается по всему
пути кровеносной системы и повреждает печень, легкие, селе-
зенку и почки. По-видимому, никаких кардинальных выводов
из такого, совершенно неестественного способа введения кремне-
зема в тело животного не было сделано, за исключением того,
что кролик способен переносить без каких-либо осложнений
введение ему кремнезема в виде золя [225] вплоть до 5 мг
в неделю и что такой кремнезем растворяется и выделяется
[225]. Когда кремнезем вводился в почечную артерию, это вы-
зывало образование фиброз, и у животного развивалась гипер-
тония [226].
Инъекция подкожно или внутрибрюшинно и вдыхание — бо-
лее естественные способы введения кремнезема с целью изуче-
ния его взаимодействия с различными видами тканей.
Первобытным племенам было хорошо известно, что растира-
нием определенных видов почв в надрезах, сделанных на коже,
можно получать прочные декоративные глубокие следы. Шелли
и Харли [227] рассмотрели этот вопрос. Когда какому-либо
человеку нанесены телесные повреждения, при которых кремне-
зем внедряется в ткани, образующиеся узелочки кремнеземной
гранулемы развиваются на протяжении до 10 лет. Этот эффект
появления твердых образований напоминает фиброз в тканях
легких. Впоследствии может также развиться саркома [228].
Сообщалось о многочисленных опытах, в которых исследовалось
введение кремнезема внутрибрюшинно. Вдыхание, конечно,
представляется наиболее общим путем, посредством которого
кремнезем оказывает опасное токсическое воздействие. Было
проведено множество контрольных исследований по воздействию
кремнезема при вдыхании в связи с проблемой силикоза.
Кремнезем в биосфере
1049
Микроволокнистый кремнезем, вероятно, получается в орга-
низме, когда хризотил-асбест вступает в реакцию с желудоч-
ными кислотами. Попадание асбеста в пищу оказалось причаст-
ным к возникновению рака желудка. Статистическое обследо-
вание, выполненное Мерлиссом [229], показало, что рис, который
подвергался обработке тальком и содержал поэтому неко-
торую долю асбеста, служит причиной высокой распространен-
ности рака желудка в Японии, где подобный рис — общераспро-
страненный продукт питания. Тальк удерживается на поверх-
ности рисовых зерен глюкозой, и это помогает сохранить
приятный вкус риса. Такая технология обработки риса находит
применение только в Японии. По-видимому, чистый тальк без-
вреден.
Необычный тип токсичности кремнезема проявляется при
вдыхании органофильных гидрофобных частиц кремнезема при
их высокой концентрации. Такой материал по некоторым свой-
ствам напоминает присадки, препятствующие вспениванию, и,
вероятно, способен вызывать депрессию оболочек легких прибли-
зительно по тому же механизму. В результате происходит воз-
никновение отека легких, ведущего к летальному исходу. Но
когда такой порошок вводится внутрибрюшинно, он оказывается
по существу инертным. В то же время идентичный кремнезем-
ный порошок, но без гидрофобного монослойного покрытия из
углеводородных групп, введенный по последнему из указанных
способов, вызывает перитонит. С другой стороны, когда точно
такой же не покрытый модифицирующим слоем порошок вды-
хался, то наблюдалось повреждение легких, но не столь быстро,
как в случае отека легких [230].
При инъекции внутрибрюшинно гидратированного силика-
геля подопытным крысам отмечалось его диспергирование с об-
разованием поликремневой кислоты с размером частиц ~1 нм.
Эти частицы попадали в кровь и проходили через капилляры
почечных клубочков, но на этом участке поглощенный кремне-
зем полимеризовался и вызывал повреждение почек [231].
Цитотоксичность
Основным различием между кремнеземными пылями и тон-
кодисперсными порошками, полученными из других материалов,
подобных глинистым минералам, углям и карбиду кремния,
оказывается то, что, когда последние достигают легкие, их ча-
стицы поглощаются макрофагами и переносятся через лимфа-
тическую систему в лимфатические узлы легких, где они и на-
капливаются. С другой стороны, кремнеземные частицы, погло-
щаемые макрофагами, убивают клетки макрофага, и поэтому
кремнезем не может быть удален. Доказательства подобных
1050
Глава 7
процессов были получены в многочисленных исследованиях, как
in vitro, так и in vivo, хотя механизм токсического воздействия
вот уже долгое время остается дискуссионным вопросом.
Измерения токсичности тонкодисперсных порошков, подоб-
ных пылям, in vitro включают применение макрофагов в тка-
невых культурах с указанием возможного воздействия пылевид-
ных частиц на поглощение кислорода, потребление глюкозы
и т. д. [232]. Маркс и Нагельшмидт [233] использовали этот
метод с целью сравнения токсичности целого ряда частиц пыле-
видных препаратов. Кремнезем оказался гораздо более токсич-
ным по сравнению с другими порошками.
В культурах макрофагов кристаллическая форма кремнезема
(тридимит) вызывала понижение поглощения кислорода и по-
давляла 'образование молочной кислоты, тогда как аморфный
кремнезем такого действия не оказывал. Если частица триди-
мита оказывалась покрытой слоем аморфного кремнезема или
А120з, то она становилась неактивной. С другой стороны, триди-
мит не оказывал никакого действия на полиморфноядерные
лейкоциты и на другие клеточные субстанции [234]. Коммоли
и Перин [235] высказали предположение, что кремнезем обна-
руживает свое токсическое действие на местах синтеза белка.
Для культур in vitro было показано, что механизм, благо-
даря которому кремнезем стимулировал развитие силикоза, за-
ключался в следующем: частицы кварца инактивировали макро-
фаги, способствуя, таким образом, более быстрому росту бакте-
рий [236]. Было показано, что из-за токсического действия
кремнезема на макрофаги подавляется реакция инородного тела
при трансплантациях кожи, тогда как при обычных условиях
кожа, как правило, отторгается вследствие воздействия макро-
фагов [237]. Ясно, что один из непосредственных эффектов,
вызываемых присутствием частиц кварца размером 0,1—1,0 мкм,
состоит в том, что эти частицы поглощаются клетками макро-
фага. В клетках частицы вступают в реакции с мембранами
лизосом, что приводит к выделению лизосомных ферментов.
Последние не только убивают макрофаги, но и распространя-
ются в окружающие ткани, что может вызвать фиброз. Действие
кремнезема на мембрану лизосомы заключается в том, что
такая частица, вероятно, разрывает мембрану, делая ее прони-
цаемой. При этом мембрана адсорбируется вокруг кремнеземной
частицы, так что могут выделяться ферменты [236, 238]. Зис-
ман, Хирш и Аллисон [239] дали другое указание на то, что
кремнезем вмешивается в окружающую систему по механизму
иммунизации животных. Мышей заражали вирусом herpes..sim-
plex, а затем им вводили впрыскиванием либо антимакрофаго-
вую сыворотку, либо суспензию, содержавшую частицы кварца
размером ~2 мкм. Оба вводимых препарата приводили к уве-
Кремнезем в биосфере
1051
личению смертности мышей. На основании этих фактов было
сделано заключение, что кремнезем инактивировал макро-
фаги.
Частицы кристаллического кварца с поверхностью, обрабо-
танной таким образом, чтобы удалить поверхностный слой
аморфного кремнезема, отличались по поведению от частиц не-
обработанного кварца, что обнаруживалось по их реакции
с макрофагами. Так, кварц с очищенной поверхностью взаимо-
действовал с макрофагами немедленно, и при этом повышалась
активность кислой фосфатазы, что не наблюдалось в случае
необработанного кварца [240а].
Штальдер и Штобер [2406] провели контрольные опыты на
всех формах кремнезема с частицами ~ 1 мкм для выяснения
их гемолитической активности. Было показано, что все формы
кремнезема, за исключением стишовита, оказались в достаточ-
ной мере активными. Активность убывала в ряду тридимит,
кварц, кристобалит, прозрачное (аморфное) кварцевое стекло и
коэсит. Кварц и тридимит были во многом схожи между собой
и более активны, другие формы оказались менее эффективными.
Это, по-видимому, позволяет установить прямую связь между
гемолитической активностью и активностью поверхностей ука-
занных типов кремнезема по отношению к возникновению сили-
коза. Штальдер [240в] продолжил исследования в этом напра-
влении и показал, что все силикагенные порошки проявляют
специфическое цитотоксическое воздействие вследствие повреж-
дения биологических мембран. Сильное притягивание таких мем-
бран к кремнеземной поверхности вызывает разрушение лизо-
сом с выделением кислых гидролаз и разрывом макрофагов.
Кремнеземная поверхность, по-видимому, обладает способностью
специфически притягивать мембрану, содержащую липиды, при
этом изменяется форма липидных мицелл и нарушается селек-
тивность ионов, так что происходит осмотическое набухание
системы, сопровождаемое распадом клеток.
Более точным было бы предположение о существовании
сильного притяжения подобной мембраны к поверхности крем-
незема, которое, вероятно, обусловливается присутствием фос-
фатидилхолина. Этот тип соединения представляет собой компо-
нент мембран в большинстве высших организмов и характеризу-
ется наличием группы, содержащей четвертичный аммониевый
ион. Недавно выполненные исследования показали сильное при-
тяжение к кремнеземной поверхности таких сильноосновных
О
II
катионов как (диглицерид) О—Р—О—СН2—СН2—N+(CH3)3.
О"
1052
Глава 7
Депассе [240г] поддержал идею о том, что литическое дей-
ствие частиц кремнезема на мембраны обусловливается силь-
ным взаимодействием с ионами триметиламмония, содержащи-
мися в мембранах.
Кремнезем в биохимических соединениях
Известно мало путей, по которым кремний может входить
в соединения с органическими молекулами. Как будет обсуждено
ниже, существует лишь косвенное доказательство того, что крем-
нийорганические соединения с прямыми связями углерод—крем-
ний могут синтезироваться в живых клетках. Не возникает
также и никаких связей азот—кремний. Даже образование свя-
зей кремний—кислород—углерод требует очень специфического
пространственного расположения атомов кислорода и углерода
в подобной органической молекуле, как, например, в молекулах
катехина или трополона, такого, чтобы могли сформироваться
хелатные кольца. Подобные соединения обсуждались в глдвах
1 и 3 со ссылками на работы Вейсса и Бауманна.
Конечно, могут участвовать и водородные связи, если идет,
хотя бы в некоторой степени, полимеризация кремневой кислоты.
Однако о возможном мономерном или полимерном состоянии
кремнезема в большинстве живых тканей, где атом кремния
участвует в процессах метаболизма или переноса, ничего пока
не известно.
Имея в виду распространенность кремнезема во многих ви-
дах биологических тканей, можно предположить, что существуют
и другие типы связей, которые следует распознать.
Первые попытки выделить соединения кремния из живых
тканей были предприняты Хольцапфелем [241]. Автор иссле-
довал молекулярную ассоциацию высших спиртов с кремневой
кислотой, обнаруженной в крови быка в концентрации 0,053 %,
а также подытожил результаты этерификации кремнезема холе-
стерином в ткани легких. Ткани анализировали методом влаж-
ного озоления в смеси с конц. H2O2 и конц. HNO3 при их соот-
ношении,! :5 и температуре 100°С. При этом имевшийся крем-
незем переводили в нерастворимую форму путем повторного
смачивания образца конц. НС1 и испарения досуха с последую-
щим вымыванием примеси Н3РО4 через фильтр. Хольцапфель
[242, 243] пришел к заключению, что в исследованных им тка-
нях человека и животных кремневая кислота находилась в сое-
динении с липоидными веществами и холестерином. Галактоза
присутствовала только как углеводный компонент в соединении
с кремнием.
Клейн и Нейенбург [244] показали, что кремнезем может
существовать в форме соединения с определенными жирами.
Кремнезем в биосфере
1053
С а-моностерином получались сложные ортоэфиры кремневой
кислоты и окончательно ортосиликат C^H^OigSi. Значения тем-
ператур плавления и растворимости таких кремнеглицеридов
ортокремневых эфиров близко напоминали соответствующие
величины подлинных глицеридов. Такие соединения, по-види-
мому, были достаточно устойчивыми при гидролизе и вследствие
их липоидной природы способны легко адсорбироваться биоло-
гическими тканями. Кауфманн [245] описал другое соединение
кремния с растворимыми липоидами, которое являлось сложным
этиловым эфиром силицилрецинолеиновой кислоты. Это соеди-
нение представляет собой прозрачную маслянистую жидкость,
нерастворимую в воде, устойчивую в отношении гидролиза и
хорошо растворимую в органических растворителях.
Соединения с полисахаридами
Кремнезем химически связывается в тканях с гликозамино-
гликанами и полиуронидами [246]. Было обнаружено, что при-
мерно 0,08 % SiO2 находилось в связанном состоянии с очищен-
ной гиалуроновой кислотой, хондроитин-4-сульфатом и гепарин-
сульфатом. При этом только одна молекула SiO2 приходится
на 200 повторяющихся полимерных молекул. Сообщалось также,
что кремнезем находится в связанном состоянии в пектине и
в альгиновой кислоте. Такой кремнезем не подвергается диализу
или не вступает в реакцию с молибденовой кислотой, оказы-
вается устойчивым при автоклавной обработке с 8 М раствором
мочевины и при взаимодействии с кислотами и основаниями.
Его можно выделить только крепким раствором щелочи. Веро-
ятно, кремнезем образует эфироподобные связи Si—О—С, но,
несомненно, подобная стабильность атома кремния должна
определяться некой хорошо защищенной пространственной кон-
фигурацией, при которой кремний окружен атомами кислорода,
как в хелатах. Неизвестно, находится ли такой кремнезем
в мономерной или же в низкомолекулярной полимерной форме.
Холт и Вент [247] сообщили, что соединение поликремневой
кислоты с пленкой ламинара (поли-1,3-глюкопиранозы) при
pH 6 становится жестким. Можно было бы предположить, что
такой полиоксиполиэфир образует водородные связи с кремне-
вой кислотой ниже pH 6, но, поскольку это соединение уже
существует при pH 6, некоторые самопроизвольные процессы
формирования хелатных соединений или этерификации, веро-
ятно, имеют место.
Взаимодействие поликремневых кислот с полисахаридами,
по-видимому, подлежит дальнейшему детальному исследованию.
Когда будут представлены надежные доказательства того, что
в сыворотке больных силикозом повышено содержание муко-
1054
Глава 7
протеинов и связанных с белками гексоз, а также более высок
общий уровень содержания кремнезема, может появиться неко-
торый общий фактор диагностики подобных заболеваний [248].
Возможно, существует некоторая форма ассоциации полисаха-
ридов с кремнеземом, поскольку начало жизни на Земле свя-
зано с такими простейшими организмами, как диатомеи, для
которых характерен тот факт, что при дефиците кремнезема
в клетках они покрываются полиуронидом, образуемым из
остатков глюкуроновой кислоты [42]. Нет никакого сомнения,
что наружный микропористый кремнеземный скелет диатомеи
сплошь пронизан этим полимером.
Соединения с белками
Уже давно известно, что кожу для изделий можно дубить
поликремневой кислотой, а альбумин способен коагулировать
с коллоидными кремнеземом. Подобные взаимодействия вклю-
чают образование водородных связей, как это обсуждалось
в главах 3 и 4. Взаимодействие кремнезема с монослоями белка,
находящимися на поверхности воды' дает дальнейшее объясне-
ние природы такой ассоциации.
Кларк, Холт и Вент [249] выполнили весьма важные иссле-
дования действия кремневой и поликремневой кислот в водном
растворе на мономолекулярные слои альбумина и найлона, на-
ходящиеся на поверхности раствора. Наблюдалось, что воздей-
ствие монркремневой или поликремневой кислот проявляется
по мере того, как кислота адсорбируется снизу на мономолеку-
лярном органическом слое. Пленки при этом становятся более
прочными и жесткими, так что сжимаемость мономолекуляр-
ного белка или слоя органического полимера, понижается. Наи-
более важным в этих наблюдениях оказалось то, что монокрем-
невая кислота совсем не адсорбировалась. Кремнезем должен
был сначала полимеризоваться, а затем уже мог адсорбиро-
ваться на монослое белка. В рассматриваемом случае полимер-
ные цепи белка становятся менее сжимаемыми, и это хорошо
подтверждает такие факты, основанные на влиянии величины
pH в процессе старения, что адсорбированная поликремневая
кислота под поверхностью пленки белка полимеризуется, фор-
мируя слой силикагеля. Этот кремнеземный слой толщиной всего
лишь, вероятно, в несколько ангстрем придает белковой пленке
жесткость, что предотвращает ее от последующего сжатия.
В случае найлона легко формировались мономолекулярные
слои, которые взаимодействовали с поликремневой кислотой
при pH 2—9 и становились особенно жесткими, или дублеными,
в интервале pH 4,5—6,5, т. е. именно тогда, когда, как известно,
Кремнезем в биосфере
1055
наблюдается максимальная скорость полимеризации кремнезема
при формировании геля.
Очевидно, молекулы поликремневой кислоты присоединяются
к белковой пленке сразу во многих точках. Если молекула
белка, находясь в растворе, свертывается в спираль и не мо-
жет полностью распрямиться и плоско расположиться на по-
верхности еще до добавления в систему кремнезема, то в та-
ком случае кремнезем образует поперечные связи в молекуле
белка и тем самым препятствует дальнейшему развертыванию
спирали белковой молекулы. Когда монослои желатина на по-
верхности раствора поликремневой кислоты оказываются сжа-
тыми, то СН2-группы пролиновых колец будут отталкиваться от
поверхности. Это влечет за собой сближение пептидных групп,
облегчая тем самым их связывание поперечными связями, обра-
зуемыми поликремневой кислотой. В результате такого про-
цесса пленка становится жесткой. Монослои, состоящие из син-
тетических полиамидов (найлона), также испытывали подоб-
ное «дубление» [250, 251].
Бычий альбумин хорошо адсорбировался на порошках
кварца и аморфного кремнезема при рН~6, причем альбумин
в растворе находился в равновесии с молекулами адсорбирован-
ного альбумина [252]. Айлер установил [253], что это как раз
то значение pH, ниже которого при адсорбции достаточного ко-
личества альбумина наблюдается флокуляция коллоидного
кремнезема, зависящая от концентрации соли в растворе. В от-
сутствие соли NaCl флокуляция наблюдалась при pH 4,4,
а в 0,1 н. растворе соли процесс флокуляции шел при pH 6,2.
Было отмечено также, что когда лишь 5 % поверхности кремне-
зема было покрыто алюмосиликатными анионами, то никакой
коагуляции в 0,1 н. растворе NaCl при рН>5,5 не происходило.
Таким образом, коллоидные алюмосиликаты (глины), обладаю-
щие большим анионным зарядом по сравнению с чистым крем-
неземом в нейтральном растворе, оказываются менее реакци-
онноспособными при взаимодействии с некоторыми белками.
Хотя поликремневая кислота и соединяется с монослоем
трипсина при pH 3,5—7, такая пленка не поддается дублению
[247]. Взаимодействие и взаимная коагуляция желатина с по-
ликремневыми кислотами подробно были рассмотрены в гл. 3.
Впоследствии Бергман и Нельсон [254] сравнили поведение
золей поликремневой кислоты при pH 2,5 и 7,5, не учитывая
того, что золь при низком значении pH состоял из частиц диа-
метром 2—3 нм, составляющих коллоидные агрегаты, тогда как
при pH 7,5 кремнеземные частицы были, вероятно, в два раза
большими по размеру и не объединялись в агрегаты, а находи-
лись в виде дискретных частиц. Больший размер частиц при
меньшем их числе при pH 7,5 объясняет тот факт, что в этих
1056
Глава 7
условиях меньшее количество желатина коагулирует при задан-
ном количестве кремнезема. Как показал Айлер [253], сущест-
вуют два пути адсорбции белков на кремнеземе. Те молекулы
белков, которые обладают сильноосновными катионными груп-
пами, способны адсорбироваться в нейтральном растворе на от-
рицательно заряженных центрах поверхности кремнезема при
pH 6—7. Однако белки, подобные желатину, связываются
с кремнеземом главным образом через водородные связи, и это
происходит только на неионизированной поверхности SiOH.
Следовательно, водородная связь оказывается наиболее прочной
в области pH 2—5, затем она ослабляется с повышением pH
по мере того, как заряд на кремнеземной поверхности увеличи-
вается. Алюмосиликатные анионные центры не способны обра-
зовывать водородных связей, но они могут прочно связывать
органические сильноосновные катионные группы.
Липопротеины можно удалять из человеческой сыворотки,
применяя коллоидный кремнезем. Другие типы белков при
этом остаются в сыворотке, и сыворотки становятся более ус-
тойчивыми в процессе хранения [255].
Денатурация белков, коагуляция крови
Когда белки определенного типа адсорбируются на поверх-
ности стекла или кремнеземе, они денатурируют, но затем, ко-
гда снова десорбируются, оказывается, что их структура под-
верглась необратимым изменениям. Полагают, что реакция бел-
ков с кремнеземом, который вводится в брюшину подопытных
животных, обусловливается подобной денатурацией. Рюттнер и
Ислер [256] отметили, что глобулины особенно сильно адсорби-
руются на поверхности кремнезема, и после десорбции обнару-
живается их денатурация. Шил, Флелшер и Клемперер [257]
на основании изучения взаимодействия кремнезема с белками
пришли к заключению, что возникающий при этом токсический
эффект является следствием денатурации белков.
Следовало бы ожидать, что меньшие по размеру частицы и,
таким образом, образцы кремнезема с большей удельной поверх-
ностью будут давать больший эффект денатурации. Однако
Лундгрен и Свенссон [258] обнаружили, что при заданном ко-
личестве кремнезема такой эффект увеличивался с увеличением
размера частиц, когда сравнивались частицы с диаметрами 10,
14 и 60 нм. Кроме того, оказалось, что токсичность пересыщен-
ного раствора монокремневой кислоты повышалась по мере
полимеризации кремнезема [259], Таким образом, ниже опре-
деленного низкого значения молекулярной массы или опреде-
ленного размера частиц поликремневая кислота вообще не вы-
зывает денатурацию белка, тогда как с увеличением размера
Кремнезем в биосфере
1057
частиц выше этого предельного значения денатурация возра-
стает. Однако выше указанного предела заметно снижается
величина удельной поверхности кремнезема, что вызывает по-
нижение его активности. Следовательно, когда частицы разме-
ром более 10 нм сравниваются в отношении их токсичности
при инъекции внутривенно кремнезема, следует иметь в виду,
что токсичность может уменьшаться с возрастанием размеров
частиц просто из-за понижения их удельной поверхности [260].
Было найдено, что гемолиз красных кровяных телец (эритро-
цитов) не происходил в присутствии монокремневой кислоты,
но начинался, как только кремнезем в процессе полимеризации
формировался в сравнительно большие по размеру частицы.
Так, уже через 20 мин при pH 7,4 0,2 %-ный раствор мономер-
ного кремнезема полимеризовался настолько, что начинал про-
являть активность [261]. При исходной концентрации моно-
мера 0,125 % гемолиз начинался только через 5 ч. Харли и
Марголис [262] установили, что гемолиз не происходил в при-
сутствии частиц кремнезема размером 4 или 3 нм, но наблю-
дался для частиц размером 5 нм и в дальнейшем с возраста-
нием размеров частиц (5,5; 6 и 30 нм) эффективность про-
цесса повышалась.
Как объясняет Марголис [263], действие кремнезема обус-
ловливается адсорбцией и денатурацией глобулярного белка—•
фактора Хагемана. Было обнаружено, что степень денатурации
возрастала с увеличением размеров частиц коллоидного кремне-
зема, который добавлялся в систему. Предложенный механизм
заключался в том, что на достаточно больших по размеру ча-
стицах или же на плоских поверхностях кремнезема при фор-
мировании монослоев молекула белка растягивается под дей-
ствием адсорбционных сил. Но в том случае, когда размер ча-
стиц кремнезема очень мал, молекулярные сегменты белка, не
раскрываясь, присоединяются сразу к различным кремнеземным
частицам. Для пояснения рассматриваемых эффектов приве-
ден рис. 7.6, аналогичный рисунку в работе Марголиса. В том
случае, когда молекула белка адсорбируется на большей по
размеру частице кремнезема или на образованном из малень-
ших частиц большом агрегате, цепь молекулы белка растяги-
вается, при этом некоторое число внутренних водородных свя-
зей, удерживавших молекулу белка в какой-либо специфической
конформации, оказываются разорванными. На одиночных ча-
стицах небольшого размера подобного растяжения молекулы не
происходит [264—266].
Активность процесса денатурации в расчете на 1 г кремне-
зема достигает максимума при кремнеземных частицах диамет-
ром 20—30 нм. Однако такая активность, рассчитанная на еди-
ницу площади поверхности кремнезема, быстро возрастает по
41 Заказ № 250
1058
Глава 7
мере того, как диаметр частиц увеличивается от 3 до 10 нм,
а затем возрастает более медленно с увеличением размеров ча-
стиц вплоть до 100 нм. В случае силикагеля компактный агре-
(6) (д)
(в) (е)
Рис. 7.6. Схема механизма денатурации белков под действием кремнезема,
предложенного Марголисом [263].
а — частицы кремневой кислоты диаметром 2—3 нм оказываются слишком малыми для
того, чтобы разделить витки молекулы белка и вызвать деструкцию ее структуры; б —
одиночная небольшая коллоидная частица кремнезема диаметром 5—10 нм вызывает
разрыв молекулы белка на ограниченном участке; в — частица кремнезема диаметром
более 20—30 нм вызывает глубокую деструкцию; а — небольшие частицы кремнезема,
которые подверглись агрегированию или превратились в гель и образовали цепочки дли-
ной более 10 нм, могут исказить конфигурацию и разорвать молекулу белка, когда вдоль
такой цепочки происходит образование водородных связей; д — агрегирование или геле-
образование частиц кремнезема размером 5—10 нм вызывает меиее разрушительную де-
струкцию, поскольку поверхность частиц имеет меньшую протяженность, чем в случае в;
е — спиральная молекула белка с расположенными на ией благодаря адгезии небольшими
частицами кремневой кислоты, как это имеет место при дублении.
гат, состоящий из небольших кремнеземных частиц диаметром
3 нм, очевидно, обеспечивает формирование достаточно разви-
той поверхности для активной денатурации белка. Однако в си-
ликагеле, состоящем из частиц диаметром 30 нм, уже каждая
отдельная частица действует в качестве активного участка де-
натурации. В любом случае активность, отнесенная к 1 г SiC>2,
кажется низкой из-за того, что только наружная поверхность
гранулы силикагеля участвует в таком процессе. Интересно от-
метить, что когда на эритроциты действовали кремнеземными
частицами размером 3 нм, то они, очевидно, покрывались не-
большими частицами в такой степени, что молекулы белка уже
не испытывали денатурацию при последующем воздействии
кремнеземных частиц большего размера.
Мицеллы, состоящие из свободных жирных кислот и по-
добно кремнезему проявляющие слабокислотный характер, спо-
Кремнезём в биосфере
1059
собны также, согласно данным Марголиса [267], вызывать
в токе крови денатурацию в соответствии с фактором Хагемана
и свертывание крови.
В зависимости от испытуемого животного материала должны
существовать некоторые основные различия либо в энергиях
адсорбции кремнезема на эритроцитах, либо в проявляемых
механических воздействиях. Ариенцо и Бресчано [268] сооб-
щили, что количество данного типа SiO2, необходимого, чтобы
вызывать гемолиз 106 клеток, составляло (в микрограммах)
0,104 для крысы, 0,173 для человека, 0,33 для кролика, 0,63 для
курицы, 1,17 для быка, 4,8 для лошади и до 80,0 для лягушки
с промежуточными в указанном интервале значениями для дру-
гих животных.
Наблюдения Холта и Вента [269] наводят на мысль о том,
что кремнеземные частицы размером меньше 5 нм и, следова-
тельно, безвредные могут стать токсичными в биологической си-
стеме за счет какого-либо типа полимеризации, стимулируемой
внутри данной системы. Авторы наблюдали, что полимеризация
кремнеземных частиц может иметь место, когда монослой белка
(инсулина), находящийся на поверхности жидкости — очень
разбавленного золя кремневой кислоты,— поглощает кремнезем
из раствора, и при последующем сжатии пленки частицы крем-
незема тесно сближаются. Такая полимеризация протекает наи-
более быстро в области pH 5,4—6,1, т. е. как раз при тех зна-
чениях pH, когда идет наиболее быстро процесс гелеобразова-
ния кремнеземных золей. Это означает, что, хотя поликремневая
кислота, состоящая из частиц размером меньше 5 нм, мо-
жет быть безвредной в биологической системе, частицы тем не
менее способны объединиться вместе в большие агрегаты бла-
годаря отмеченному выше механизму, особенно в том случае,
когда биологические мембраны могут сжиматься. Образовав-
шиеся большие агрегаты могут затем становиться активными
по отношению к денатурации белка.
Когда эритроциты, помещенные в суспендированную среду —
коллоидный кремнезем (с частицами диаметром 14 нм),— цент-
рифугировали, состоящая из клеток поверхность сморщивалась
с образованием зернистой структуры, которая, вне всякого сом-
нения, появлялась благодаря деформации, возникшей при ад-
сорбции коллоидных частиц [270] . Браун [271] наблюдал, что
липиды поверхности эритроцитов адсорбировались кремнезе-
мом. Депассе и Варлус [272] предположили, что сильное свя-
зывание частиц кремнезема с поверхностью эритроцитов обус-
ловливается присутствием в мембране четвертичных аммоние-
вых ионов.
Не все белки, адсорбированные кремнеземом, испытывают
денатурацию. Так по крайней мере у-глобулины, которые ад-
1060
Глава 7
сорбировались на измельченном в порошок кварце, сохраняли
свои антигенные специфические свойства. На основании подоб-
ных фактов авторы работы [273] пришли к заключению, что
иммунологические реакции, имеющие отношение к возникнове-
нию силикоза, могли и не быть следствием образования анти-
тел, действующих против белковых молекул сыворотки, изме-
нившихся под воздействием частиц кварца. Однако другие типы
белков, безусловно, испытывают денатурацию и могут ока-
заться причиной силикоза.
Соединения со специфическими веществами
и ферментами
Поверхность кварца катализирует окисление на воздухе
^/-пролина до получения оксипролина, что указывает на воз-
можное участие в подобном процессе некоторых типов активи-
рованной, ориентированной и специфической адсорбции [274].
Такой процесс заслуживает внимания потому, что оксипролин
выделяется в моче горнорабочих, у которых наблюдается за-
болевание силикозом на той стадии, когда содержание оксипро-
лина на 31 % выше, чем у здоровых людей. Суммарное содер-
жание оксипролина в крови и моче служит показателем актив-
ности фиброзообразования [275].
Сахара различаются по своей склонности соединяться с крем-
неземом. Хольцапфель [173, 276] обнаружил, что галактоза
адсорбировалась на кварце и вступала в реакцию с фенилгид-
разоном с формированием озазонов, тогда как арабиноза не
образовывалась. Галактозы адсорбировало в два раза больше,
чем лактозы, и в четыре раза больше, чем глюкозы.
Ферменты могут адсорбироваться на кремнеземе и при этом
сохранять свою активность. Бычий трипсин был адсорбирован
на частицах коллоидного кремнезема размером 14 нм, затем
с помощью глутаральдегида переведен в нерастворимую форму,
так что сохранялось 80 % его активности по отношению к эсте-
разе и 17 % его протеолитической активности. Покрытые ад-
сорбированным веществом кремнеземные частицы можно от-
делить цетрифугированием и затем повторно диспергировать
[277]. Фермент адсорбировали из 0,1 М боратного буферного
раствора при pH 8,5; при этом вокруг кремнеземных частиц
сферической формы диаметром ~ 14 нм формировался мономо-
лекулярный слой из молекул фермента, имевших диаметр 4 нм.
Таким частицы можно было наблюдать под электронным мик-
роскопом. Как только такое покрытие формировалось, оно очень
прочно связывалось с глутаральдегидом поперечными связями.
Фермент может также присоединяться к поверхности рых-
лого пирогенного кремнеземного порошка по реакции с ами-
Кремнезем в биосфере
1061
нопропилсилильными группами, которые были предварительно
хемосорбированы на поверхности исходного кремнезема. Можно
также одновременно использовать активатор. Рейнольдс и Мил-
лер [278] запатентовали получение трипсина такого типа с со-
хранением его свойств.
Ферментное ингибирование в присутствии олигомеров крем-
невой кислоты может быть следствием либо направленности
активных центров фермента в сторону кремнеземной поверх-
ности, либо денатурации молекулы фермента по механизму
Марголиса. Глюкоза-6-фосфатаза из микросом клеток печени
крыс ингибируется олигокремневыми кислотами при их содер-
жании 0,0.1—0,025 % S1O2 в отличие от некоторых других фос-
фатаз, например дегидрогеназы и оксидазы [279]. Кинг и др.
[280] сообщили, что подобным образом ингибируются ацетпл-
холинэстераза, сукцинатдегидрогеназа, эстераза и гиалурони-
даза; не подвержены ингибированию амилаза, панкреатическая
липаза, пепсин, щелочные и кислые фосфатазы, трипсин и
уреаза.
Согласно данным Роуселла и Леонарда [281], монокремне-
вая кислота не оказывает никакого действия на ферменты. Мо-
номер кремнезема при проникновении в клетки печени не взаи-
модействовал с ферментами и не блокировал клеточные мем-
браны, которые сохраняли нормальную проницаемость по отно-
шению к ионам и к небольшим молекулам, например молеку-
лам ацетата или мочевины.
Ферменты способны соединяться с поверхностью очень широ-
копористого силикагеля с образованием относительно устойчи-
вого закрепленного на поверхности ферментного катализатора
[282]. Широкопористый кремнезем с порами диаметром 51 нм
использовался как носитель, к поверхности которого присоеди-
нялись молекулы фермента протеазы бактерии Bacillus subtitle.
Вначале происходила реакция бифункциональной соли диазония
с поверхностью кремнезема, а затем реакция фермента с проти-
воположной от поверхности группой диазония. Фермент на крем-
неземном носителе был способен гидролизовать казеин; его
время полупревращения (время, за которое концентрация фер-
мента уменьшалась вдвое) превышало 7 мес, тогда как точно
такой же фермент, просто адсорбированный на исходном крем-
неземе, имел время полупревращения всего 2,5 мес.
Витамин С (аскорбиновая кислота) стабилизировался после
адсорбции на силикагеле при pH 6,5 [283]. По-видимому, мо-
лекулы витамина адсорбировались не за счет образования во-
дородных связей ввиду относительно высокого значения pH,
а, вероятно, удерживались благодаря хелатной реакции с си-
ланольными поверхностными группами кремнезема, так как мо-
лекула витамина обладает типичной парой гидроксильных
1062
Глава 7
групп, которые расположены на смежных атомах углерода, свя-
занных между собой двойной связью. Подобные гидроксиль-
ные группы характерны для катехина — хелатообразующего ре-
агента по отношению к атому кремния. Если бы в действитель-
ности обе гидроксильные группы молекулы витамина конденси-
ровались с одним поверхностным атомом кремния, то должно
было бы стабилизироваться поверхностное соединение с двой-
ной связью, устойчивое к окислению. Очевидно, в случае вита-
мина С не исследовалась возможность образования водораст-
воримого анионного хелатного соединения кремния.
Ориентация кристаллов низкомолекулярных пептидов и ами-
нокислот, когда они выращивались на кварцевых поверхностях,
указывает на эпитаксиальную согласованность, игравшую, воз-
можно, некоторую роль в добиотической эволюции [284].
Кремнезем в соединениях фосфора.
Нуклеиновые кислоты, ДНК, РНК ,
Ввиду устойчивости ДНК трудно представить возможность
обмена между Si(OH)4 и фосфатом в рибонуклеиновых кисло-
тах. Однако Шварц и Баронецкий [285] сообщили, что реакции
гидролиза, сопровождающиеся выделением свободной фосфор-
ной кислоты, ускорялись в присутствии Si (ОН) 4. Авторы пока-
зали, что наблюдался обмен кремневой кислоты с фосфатом
в рибонуклеиновой кислоте. Керстен и Штаудингер [286] ука-
зали на взаимодействие кремневой кислоты с никотинамидаде-
ниндинуклеотидом (NAD) и даже с аденозинтрифосфатом
(АТР), Позже возникли сомнения относительно этих наблюде-
ний, поскольку подобные данные могли быть вызваны случай-
ным присутствием бактерий.
В экспериментах Кикута и Шлипкотера [287] также была
показана возможность взаимодействия кремнезема с рибонук-
леиновой кислотой (РНК). Авторы нашли, что золь кремнезема,
вводимый крысам через трахею, вызывал в органической форме
узелковый фибриз. Но в том случае, когда кремнезем вводился
совместно с РНК, последствия оказывались гораздо более тяже-
лыми— образовывалась сильно развитая сеть коллагеновых
волокон [287]. Трипсин также усиливал действие кремне-
зема [288].
Согласно Хаугеру, Криту и Стаудингеру [289], мономерный
кремнезем оказывается более токсичным по отношению к выде-
ленным митохондриям по сравнению с поликремневой кисло-
той. Мономерный кремнезем вызывал набухание митохондрии
в печени крыс и подавлял окислительный процесс фосфорилиро-
вания. Поскольку в этих экспериментах использовалась только
Кремнезем в биосфере
1063
монокремневая кислота, то, возможно, происходило ее взаи-
модействие с нуклеиновой кислотой.
Как будет рассмотрено ниже в связи с проблемой силикоза,
присутствие частиц кварца в легких или брюшине приводит
к образованию необычного слоя с решетчатой структурой из
кристаллического железо(П)железо(П1) фосфосиликата. До на-
стоящего времени подобное вещество мало изучено, и также
остается неизвестным специфическое соединение, связанное
с токсичностью или с указанным заболеванием [127, 290].
Присутствие растворимого кремнезема оказывает воздейст-
вие на метаболизм фосфата. При концентрации 0,01 % олиго-
кремневые кислоты понижали ассимиляционный коэффициент
Р: О митохондрий клеток печени крыс, задерживая усвоение
фосфата на 30 % [291]. В том случае, когда в течение 3 мес
в рацион крыс вводили большие количества кремнезема, в по-
чечных канальцах формировались включения, в которых крем-
незем как-то соединялся с пуриновыми или пиримидиновыми ос-
нованиями [292]. Воронков и др_. предполагают, что кремневая
кислота в значительной мере замещает фосфорную в ДНК и
РНК, так что отношение Si : Р может сохраняться в интервале
от 1 : 20 до 1 :45. Ортокремневая кислота может образовывать
эфирные связи с группами ОН углеводных компонентов [293].
По данным Шварца и др. [294, 295], 0,0001 % Si(OH)4 ката-
лизирует гидролиз рибонуклеиновой кислоты, и кремневая кис-
лота обменивается с остатком Н3РО4. ^Монокремневая кислота
повреждает дыхание клеток посредством взаимодействия с NAD
(гомогенат печени), а также вступает в реакцию с АТР
[285, 286].
Мутации, атеросклероз, раковые опухоли
Поскольку Si (ОН) 4 может в значительной степени обмени-
ваться с фосфатом в нуклеиновых кислотах, то отсюда следует,
что в определенных точках вдоль цепи ДНК должны сущест-
вовать связи, которые по стабильности отличаются от остальных.
Можно представить два типа таких связей:
I
дезоксирибоза
О
О=Р-СГ
I
о
I
дезоксирибоза
I
дезоксирибоза
О
HO-Si-OH
6
I
дезоксирибоза
1064
Глава 7
Фосфатные или силикатные группы располагаются снаружи
двойной спирали ДНК, так что по крайней мере пространствен-
ное расположение способствует обмену. Однако, когда такие
группы оказываются введенными в цепь ДНК, ее стабильность
должна измениться, так как силикатная связь отличается от
фосфатной по стабильности, в особенности потому, что группа
SiOH должна, быть ионизирована, вероятно, в значительно
меньшей степени. Невозможно предсказать, будет ли введение
кремнезема способствовать повышению или понижению ста-
бильности ДНК-
Очевидно, существует некоторая аналогия между развитием
фиброза в тканях легких, пораженных силикозом, и развитием
атеросклеротических бляшек в артериях. В обоих случаях на-
блюдаются потеря эластичности и аномальное быстрое разра-
стание тканей. Кроме того, происходит частичное омертвение
ткани. Как отмечал Бендитт [296], клетки атеросклеротических
бляшек, по прежним представлениям, считались фибробластами,
но в настоящее время их идентифицируют как клетки, подоб-
ные клеткам гладкой мышечной ткани. Рост бляшки, по-види-
мому, включает мутацию, которая ведет к быстрому размно-
жению гладких мышечных клеток, что в свою очередь вызывает
наращивание стенок артерий и приводит к омертвению ткани
внутри массы самой атеросклеротической бляшки. Бендитт пе-
речисляет возможные факторы, способствующие появлению му-
тации, такие, как химические мутагены, вирусы и ионизирую-
щее излучение.
Вероятно, можно было бы включить и еще один фактор: со-
держание кремния в крови. Незначительные количества крем-
ния, как было показано, несомненно, представляются реальным
компонентом в нуклеиновых кислотах (ДНК, РНК)- Если бы
оказалось, что кремнезем влияет на возникновение мутации,
то тогда была бы установлена взаимосвязь между уровнями
содержания кремнезема и развитием атеросклероза.
Было представлено [297] доказательство того, что кремне-
зем может способствовать появлению атеросклероза. Было
найдено, что при данном заболевании наблюдается повышение
количества кальция в стенке артерии при значительном пони-
жении содержания кремнезема в ней. Де Францискис и Оли-
виеро [298] исследовали воздействие растворимого кремнезема
в питьевой воде (0,0061 %) на крыс с нормальным и повышен-
ным уровнями содержания холестерина в крови. При потребле-
нии подопытными крысами питьевой воды с высокой концент-
рацией кремнезема наблюдалось понижение высокого уровня
холестерина, а также снижение инфильтрации жиров, посту-
пающих в печень, по сравнению с контрольными особями.
Имеются данные, что присутствие силиката натрия в питьевой
Кремнезем в биосфере
1065
воде оказывало умеренное воздействие на аккумуляцию кальция
и других элементов в атеромах [299]. Как будет обсуждено при
рассмотрении кремнийорганических соединений, некоторые из
подобных соединений, по-видимому, подвергаются преобразо-
ваниям при метаболизме и могут поставлять кремнезем, кото-
рый предотвращает появление атеросклероза у кроликов при
диете с высоким содержанием холестерина. [300] .
Подобную роль кремнезем, возможно, играет и в раковых
опухолях. Так, изменение уровня содержания кремнезема в по-
жилом возрасте может влиять на вероятность появления мута-
ции. Воронков, Зелчан и Лукевиц [4а] обобщили немногочис-
ленные исследования по содержанию кремнезема в тканях и
биологических жидкостях у больных раком. В нейросаркомах
содержание кремнезема оказывается пониженным по сравне-
нию с другими областями организма. В тканевой жидкости под-
желудочной железы содержание кремнезема повышено в случае
заболеваний раком, но оно понижается до нормального уровня
после хирургической операции. У больных раком содержание
кремнезема в моче оказывается в 2,5—17 раз меньше, чем у здо-
ровых пациентов. Упоминается, что подобные наблюдения дают
возможность ранней диагностики рака. При раковой опухоли
мочевого пузыря, наоборот, возрастает содержание кремнезема
в моче. Кровь больных раком также содержит более высокие
концентрации кремнезема.
Защитное действие кремнезема в случаях раковых заболе-
ваний обосновывается тем, что рак редко начинается в обла-
стях организма с богатым содержанием кремнезема (или маг-
ния), но, однако, чаще возникает в участках, богатых кальцием,
так как кальций выступает в роли «враждебного кремнию эле-
мента». Воронков, Зелчан и Лукевиц [4а] даже постулируют,
что раковые заболевания чаще возникают у высших организ-
мов, поскольку они содержат меньше кремнезема в результате
эволюционных процессов. Однако существует много других фак-
торов, затрагивающих области распространения рака в орга-
низме, и кажется сомнительным, что рак мог бы быть непосред-
ственно связан с количеством кремнезема, потребляемого с пи-
щей или питьевой водой в отдельных географических районах.
Кроме того, некоторая часть вышеприведенной информации
имеет пятидесятилетнюю давность, поэтому результаты анализа
на содержание кремнезема можно подвергнуть некоторым сом-
нениям.
Поскольку имеются сведения о присутствии кремнезема
в ДНК и РНК и возможных изменениях в уровнях содержания
кремнезема у больных раком, вероятно, целесообразно проведе-
ние дальнейших исследований кремнезема как возможного фак-
тора, влияющего на заболевание раком. Исследования по раз-
1066
Глава 7
витию раковых заболеваний у подопытных животных, потреб-
ляющих аномально низкое или аномально высокое количество
монокремневой кислоты, могли бы привести к дальнейшему по-
ниманию этой проблемы.
Силикоз, пневмокониоз, фиброгенез
Наиболее распространенный путь попадания опасных коли-
честв кремнезема в организм человека — это вдыхание, хотя
имеются некоторые предположения, что вред может причинить
и коллоидный кремнезем, попадающий в открытые раны. Од-
нако возможность длительного воздействия последнего малове-
роятна, в то время как силикоз легких — обычное явление. Еще
в начале прошлого века в Англии была высока смертность про-
мышленных рабочих, которые на фабриках приготовляли тонко-
размолотый кремень, или кремнезем, для керамических изделий.
Поэтому женщины были вынуждены выходить замуж три-че-
тыре раза. Проблема силикоза в промышленности привела
к появлению строгих правительственных мер по снижению опас-
ности, которой подвергались рабочие. В большинстве стран рас-
пространенность этого заболевания в настоящее время мала.
Постоянно возрастающий объем научных исследований и
публикаций по силикозу и относящимся к нему вопросам химии
затрудняет рассмотрение этой проблемы в данной книге. Од-
нако здесь будут даны некоторые химические аспекты и обоб-
щены основные теоретические положения, связанные с силико-
зом. По данным Воронкова, Зелчана и Лукевица [4а], насчиты-
вается более 50 теорий, по-разному объясняющих механизм
действия кремнезема; эти авторы цитируют сотни работ.
Имеются краткие обзоры по проблеме силикоза [301—306].
Монография Холта (1955 г.), в которой насчитывается 657 биб-
лиографических ссылок, а также публикации и монография
Хеппльстона (1973 г.) будут рассмотрены более подробно.
Механизм возникновения силикоза
Воздействие, благодаря которому тонкодиспергированный
пылевидный кварц, попадающий в легкие, вызывает заболева-
ние силикозом, в течение долгого времени было дискуссионным
вопросом. Существуют фактические данные, показывающие, что
частицы кристаллического кварца размером ~ 1 мкм, отличаю-
щиеся от многих разновидностей пыли тем, что состоят из наи-
более твердого материала, подобного карборунду SiС [307],
вызывают структурные изменения в легких, включая фиброз,
отвердение ткани, снижение объема легких, и значительно по-
Кремнезем в биосфере
1067
вышают вероятность заболевания туберкулезом. Имеются две
группы теорий:
1. Более ранние теории, в которых предполагалось, что не-
которую роль играет растворимый кремнезем. Они базировались
на том, что монокремневая кислота способна вступать во взаи-
модействие с ДНК или с РНК и вызывать изменение в фер-
ментативных системах. Согласно наиболее распространенной
теории, кварц растворяется с образованием растворимого моно-
мерного кремнезема. Этот процесс сам по себе безвреден, однако
мономерный кремнезем полимеризуется затем до поликремневой
кислоты, которая, как известно, денатурирует белок и разру-
шает клеточные мембраны, т. е. в результате мономерный крем-
незем оказывается цитотоксичным.
2. Современные теории, по которым единственной причиной,
вызывающей силикоз, считается чистая поверхность частиц
кристаллического кремнезема, обычно кварца.
В течение долгого времени существовала путаница вслед-
ствие ошибочного допущения, что денатурация белка и цито-
токсическое действие кремнезема приводят к развитию фиброза
в затронутых силикозом легких. Наоборот, сильная цитоток-
сическая активность кремнезема, по-видимому, обратна его
фиброзной активности в том смысле, что наиболее активные
цитотоксические формы кремнезема, такие, как поликремневая
кислота, аморфный и коллоидный кремнезем, в наименьшей
степени активны в отношении образования волокнистой ткани
или фиброза. В настоящее время преобладает мнение, что
именно кварц в виде частиц размером 0,05—5 мкм при вдыха-
нии вызывает типичные заболевания силикозом или пневмоко-
ниозом, для которых характерно развитие тяжелой формы фиб-
роза легких.
Причина такого явления заключается в том, что макрофаги
или фагоциты, выступающие как защитные силы против ино-
родных включений, поглощают кварцевые частицы. В этом со-
стоянии поверхность кварца действует как специфический ге-
терогенный катализатор процесса возникновения некоего «фак-
тора», вероятно какого-то модифицированного фермента, кото-
рый, выделяясь затем из омертвевшего макрофага, способен
вызывать раздражение фибробластов, что приводит к получе-
нию избытка оксипролина. Это в свою очередь ведет к форми-
рованию аномально большого количества фиброзной ткани.
Для того чтобы дать высокую оценку тем огромным усилиям,
которые предпринимались в прошлом по этой сложнейшей про-
блеме, рассмотрим различные, ранее выдвинутые теории и свя-
занные с ними наблюдения. Это также даст возможность опи-
сать многие случайные, но тем не менее интересные наблюде-
ния, которые были зарегистрированы.
1068
Глава 7
Объем литературы по этой теме настолько огромен, что
здесь приведены библиографические ссылки лишь на недавно
опубликованные обзоры. Некоторое обобщение относящихся
к силикозу теоретических представлений, которые появились на-
чиная с 1955 г., состоит в следующем.
Шеперс [308] представил достаточно полный обзор по исто-
рии исследований силикоза с обсуждением 50 основных теорий,
которые были им классифицированы на химические, физиоло-
гические и биологические. Автор особо выделил тот факт, что
после начального взаимодействия кварцевой частицы с прото-
плазмой и разрушения фагоцитов дальнейшие изменения про-
исходят уже в присутствии некротической, отмирающей ткани.
Большая часть кремнезема может быть удалена, но биологиче-
ские изменения продолжаются, приводя к образованию фиброз-
ных тканей и больших количеств легочных липидов, так что
масса легких может возрасти в 30 раз (в расчете на сухую
массу), причем в ней на долю кремнезема может приходиться
менее 5 %.
Автор пришел к заключению, что теория растворимости здесь
не применима. Вместо нее рассматривались химические свойства
поверхности кремнеземных частиц, непосредственно участвую-
щих в реакциях с тканями. О физических теориях в общем су-
дили как о недостаточно пригодных для описания изучаемой
проблемы. Хотя и можно было установить некоторую корреля-
цию между величиной удельной поверхности или адсорбцион-
ной способностью альбумина и патогенностью кремнезема, но
были найдены и исключения. Объяснение, каким образом ча-
стицы кремнезема убивают фагоцит, весьма далеко от реаль-
ных биохимических взаимодействий, имеющих место в разви-
тии силикоза.
Штобер представил обзор по истории вопроса о силикозе
и три основные теории, в которых рассматривались а) раство-
римость кремнезема, б) поверхностные взаимодействия кремне-
зема и в) иммунные реакции, происходящие в присутствии
кремнезема. Затем автор обобщил обширные исследования,
выполненные с частицами всех известных форм кремнезема,
в которых поверхностные свойства и характеристики раствори-
мости сопоставлялись с биологическими реакциями [309]. Што-
бер указывал, что простая теория растворимости не может
быть корректной. Был отмечен следующий факт: если располо-
жить различные формы кремнезема в порядке возрастания ра-
створимости (кристаллические разновидности кремнезема, опал,
тонкодисперсный кремнезем с малыми по размеру частицами,
коллоидный кремнезем), то фиброгенная активность в этом
ряду понижается, тогда как токсичность по отношению к тка-
ням повышается.
Кремнезем в биосфере
1069
Что касается «поверхностной» теории, то с ее позиций
-растворимый кремнезем представляет собой несущественную,
случайную форму, тогда как именно поверхность, или граница,
раздела фаз кварцевой частицы способна вызывать изменения
в органических молекулах. Основная идея заключается в том,
что регулярное расположение атомов на поверхности кристал-
лической решетки действует как матрица, по которой совме-
щаются полярные группы белковых молекул. Последние таким
образом могут адсорбироваться и затем модифицироваться.
В теории иммунной реакции, которая не вступает в противо-
речие с другими теориями, делается попытка объяснить после-
дующие этапы при развитии силикозных'повреждений.
Отмечалось, что высокая скорость растворения стишовита
наряду с полным отсутствием его биологической активности,
по-видимому, позволила окончательно исключить из рассмот-
рения теорию растворимости. Стишовит представляет собой
форму кремнезема, подобную а-оксиду алюминия, в котором
атом кремния координирован шестью атомами кислорода. Ос-
новное отличие поверхностных свойств заключается в том, что
в стишовите в два раза более высока концентрация поверхност-
ных групп SiOH в расчете на единицу площади и что его по-
верхность не адсорбирует поли(винилпиридин-М-оксид), кото-
рый сильно адсорбируется другими кристаллическими модифи-
кациями кремнезема. Однако довольно высокая силикозная ак-
тивность кварца по сравнению с другими модификациями не
была объяснена.
Хеппльстон [310, 311] в своем обзоре рассмотрел основные
теории по фиброгенному действию кремнезема, классифициро-
ванные аналогичным образом с позиций: а) растворимости,
б) поверхностных свойств и в) иммунологических эффектов.
Иммунологическая теория имеет второстепенную важность, по-
скольку не объясняет первоначального действия кремнезема
в стимулировании быстрого разрастания соединительной ткани.
Теория растворимости была признана непригодной, так как не
давала необходимых объяснений. Эта теория не отвечала тре-
бованиям еще и потому, что ни известные поверхностные струк-
туры различных модификаций кремнезема, ни их адсорбцион-
ные характеристики по отношению к альбумину или у-глобу-
лпну не могли быть скоррелированы с биологической актив-
ностью. Однако Штобер [312] показал, что все кристаллические
формы кремнезема, за исключением стишовита, относительно
активны.
Хеппльстон отметил, что цитотоксические эффекты кремне-
земных частиц в отношении фагоцитов не были в нужной мере
скоррелированы с фиброгенезом и развитием силикоза. В дан-
ном случае он затронул ключевой вопрос, который в конце кон-
1070
Глава 7
цов был разрешен. Хеппльстон сослался [127, 310] на доказа-
тельство того, что кварц, взаимодействуя с компонентами кле-
ток макрофага, способствует возникновению некоего возбуди-
теля, который в свою очередь стимулирует образование колла-
гена, т. е. ведет к фиброзу. Вещество, играющее роль посред-
ника при развитии фиброза после повреждения клеток, является
не липидом, а, возможно, галактаном. Во всяком случае, как
полагают, высвобождается какой-то Н-фактор (оксипролиновый
фактор), который стимулирует образование коллагена в фибро-
бластах. Так как силикозные ткани содержат избыток липидов,
то вполне вероятно, что, как только начинается процесс, при-
сутствие одних лишь этих липидов может стимулировать раз-
витие фиброгенеза.
Хеппльстон [313] сообщил об удивительном наблюдении,
которое заключалось в том, что у крыс, не содержащих пато-
генных микроорганизмов (т. е. в отсутствуе фагоцитов), не
образовывались типичные силикозные узелки, которые, однако,
появлялись у нормальных крыс даже в том случае, когда ,им
вводили кремнезем респираторным путем. Этот факт поддер-
живает представление о том, что фагоциты участвуют в разви-
тии фиброза. Веллер и др. [314] утверждали, что у крыс, не со-
держащих патогенных микроорганизмов, кремнезем вызывал
появление патологических эффектов. Но кремнезем при этом
вводился внутрибрюшинно, поэтому полученные этими авторами
результаты не могут сравниваться с эффектами, возникающими
в легких при типичном развитии фиброза.
Хеппльстон исследовал стадии развития макрофагов в при-
сутствии кварцевых частиц. После разрушения макрофагов и
отделения их от частиц кварца и от остатков клеток методом
центрифугирования был получен раствор, содержавший отмечен-
ный выше фактор, оказавшийся способным вызывать фиброз.
Когда раствор вводили в выделенную культуру фибробластов,
то наблюдалось заметное стимулирование образования оксипро-
лина, который является характерным компонентом фиброзной
соединительной ткани. Автор показал, что такое условие было
необходимо для выращивания макрофага в присутствии квар-
цевых частиц. Дополнительное введение кварца для разруше-
ния макрофага не приводило к появлению указанного фактора.
Избыток кварца вызывал только лишь умерщвление макрофагов,
(цитотоксическое действие), но фиброз не развивался [127].
Хеппльстон и др. продемонстрировали также другой тип
воздействия, когда эпителиальные клетки в легких, реагировав-
шие на избыточное содержание кварцевой пыли тем, что произ-
водили огромные количества фосфолипидов (в частности, дп-
пальмитоилового типа), поглощающих кварцевые частицы п
Кремнезем в биосфере
1071 •
препятствующие их воздействию на макрофаги. В этом случае
фиброз не развивался [127, 315].
Эллисон [127] показал, что внутри макрофага лизосомные
мебраны, содержащие фосфолипиды, связаны водородными свя-
зями с равномерно расположенными группами SiOH на поверх-
ности кварца через атомы кислорода фосфатных групп эфира.
При таких условиях фиброгенный фактор, вероятно представ-
ляющий собой аномальный или видоизменный фермент, синте-
зируется и накапливается в клетке до тех пор, пока последняя
не погибает. Айлер [127] обобщил данные, касающиеся рас-
смотрения водородных связей между полярными органическими
соединениями и поверхностью аморфного кремнезема. Причина
остается неизвестной. Однако можно представить, что регуляр-
ное расположение групп SiOH на кристаллической поверхно-
сти кварца, возможно, способствует более совершенному форми-
рованию водородных связей с мембраной, чем в случае аморф-
ного кремнезема с достаточно хаотично расположенными по-
верхностными группами SiOH. Характеристики поверхности
кварца, безусловно, отличаются от характеристик аморфного
кремнезема. Например, на кварцевой поверхности адсорби-
руется мономерный кремнезем из раствора, тогда как на по-
верхности аморфного кремнезема такая адсорбция не проис-
ходит (см. гл. 1).
Саммертон и др. [316] предположили, что именно белковый
компонент, а не фосфолипид, адсорбировался в первую очередь,
так как только такой полимер, как белок, образует большое
число кооперативных связей и поэтому сильно адсорбируется.
Однако авторы не учли сильного гидрофобного связывания
между длинными алкильными группами смежных адсорбиро-
ванных фосфолипидных молекул, которое может также стаби-
лизировать адсорбированный слой.
Последующие этапы процесса, посредством которого ча-
стицы кварца вызывают фиброз, были предложены Эллисоном
[317—319] и обобщены Саммертоном и др. [316] с некоторыми
небольшими изменениями.
1. Кварцевая частица в альвеоле покрывается органиче-
ским полимерным веществом, образующим с кремнеземной по-
верхностью водородные связи, как это имеет место, например,
в случае белков.
2. Покрытая таким образом частица кварца может подвер-
гаться фагоцитозу, т. е. ее поглощает макрофаг (род лейкоци-
тов) .
3. Внутри макрофага лизосома и ее ферменты удаляют по-
крытие с кварцевой частицы.
4. Раскрытая кварцевая частица связывается водородными
связями с определенными компонентами внутренней поверх-
1072
Глава 7
ности лизосомной мембраны, которая в этом случае выделяет
гибель и растворение макрофага.
Однако позднее было сделано важное наблюдение, что та-
кие превращения не приводили к гибели макрофага [320].
Вместо этого повреждение макрофага происходит тогда, когда
он еще сохраняется живым с находящимися внутри клеток квар-
цевыми частицами. Кварцевая поверхность каким-то образом
повреждает лизосомы, так что при этом производится новый
фактор, вероятно фермент. Когда такой фермент выделяется из
омертвевшего макрофага, он побуждает фибробласты произво-
дить дополнительное количество оксипролина, который затем
связывается с образованием фиброзных тканей. Вероятно, по-
добное развитие процесса подтверждает наблюдения Марасаса
и Харингтона [3206] относительно того, что поверхность кварца
катализирует окисление на воздухе dZ-пролина до оксипролина.
Сопоставление аморфного и кристаллического кремнезема.
Влияние размеров частиц
- В реальных условиях производства силикоз, включая фиб-
роз легких, вызывается не состоящей из частиц аморфного
кремнезема пылью, но скорее всего кварцевой пылью. Однако
в многочисленных исследованиях брались частицы разных раз-
меров, но не учитывалось их кристаллическое или аморфное
состояние.
Тщательное сравнение порошков кварца, кристобалита, три-
димита и плавленого кварцевого стекла, имевших во всех слу-
чаях одни и те же размеры, частиц (в основном 1—4 мкм) и
одинаковые удельные поверхности, было проведено Кингом
и др. [321]. Пылевидные препараты вводились в виде суспен-
зии в легкие крыс, после чего наблюдалось развитие силикоза.
Все формы кремнезема вызывали легочный фиброз. Однако
воздействия, оказываемые аморфным (плавленым) кремнезе-
мом, оказались наименее тяжелыми. Кварц и кристобалит при-
мерно в равной степени вызывали типичные симптомы силикоза.
Такой же порядок фиброгенной активности был установлен
Ингльбрехтом и Нагельшмидтом [322]. Штобер [323а] сделал
заключение, что существует не очень большое различие между
образцами аморфного кварцевого стекла и кварца в возникно-
вении силикоза при некоторых определенных экспериментальных
условиях, если брались частицы равного размера. Эта точка
зрения была поддержана исследованиями, проведенными in
vitro Бауманом и Раше [3236], которые продемонстрировали
значительное различие в биологической активности кристалли-
ческого кварца с чистой поверхностью и точно такого же квар-
цевого порошка, на котором был осажден слой аморфного крем-
Кремнезем в биосфере
1073
незема. Кристаллическая поверхность оказалась токсичной по
отношению к культуре клеток альвеолярных макрофагов собаки,
тогда как аморфная, кремнеземная поверхность оставалась по
существу инертной. Были использованы частицы трех размеров:
0,6; 1,2 и 2,0 мкм, а толщина аморфного слоя, осажденного на
кристаллической поверхности, составляла всего три или четыре
молекулярных слоя.
С практической точки зрения существует очень немного ус-
ловий, когда аморфный кремнезем (кварцевое стекло) мог бы
содержать опасные для человека частицы размером 1—4 мкм.
Почти все аморфные кремнеземные пылевидные образцы со-
стоят из частиц, величиной по крайней мере на два порядка
меньше. С другой стороны, почти все кремнеземные пыли с ча-
стицами размером 1—4 мкм, встречающиеся в промышленных
условиях, являются кристаллическими, как правило кварцевыми.
Таким образом, можно констатировать, что на практике
аморфные кремнеземы не вызывают силикоза. Кроме того, даже
при введении поправки на различие в размерах частиц необхо-
димо учитывать, что кварцевая поверхность (и особенно триди-
митовая) более фиброгенна, чем поверхность аморфного крем-
незема.
Как было показано Шеперсом [324] и Клостеркёттером
[325], пирогенный порошок, состоящий из аморфных кремне-
земных частиц субмикронного размера, обычно представляю-
щих собой агрегаты, составленные из первичных частиц коллоид-
ного размера, не вызывал силикоза. Контрольные исследования
по введению кремнезема респираторным путем крысам и кро-
ликам показали, что имели место раздражения и бронхиальные
осложнения, но не развивался фиброз, и все животные верну-
лись в нормальное состояние после окончания испытаний. Осаж-
денный аморфный кремнезем также не был фиброгенным, [326].
Действительно опасными являются частицы твердых образ-
цов кристаллического кремнезема в любой форме (за исклю-
чением стишовита) с размерами 5,0—0,5 мкм. Еще меньшие
частицы кристаллического кремнезема (0,05—0,5 мкм) могут
оказаться даже более опасными, но эксперименты с ними, по-
видимому, не проводились. Возможно, что описанные в литера-
туре случаи значительно более высокой силикозной активно-
сти коэсита (размер частиц менее 0,5 мкм) по сравнению с ак-
тивностью частиц кварца (размером 1—3 мкм) были обуслов-
лены меньшим размером частиц коэсита [327а].
Даже наиболее мелкие частицы порошков аморфного крем-
незема могут временно вызывать узелковые формы пневмоко-
ниоза; например, для пирогенного аэросила максимальный допу-
стимый уровень концентрации кремнеземной пыли в атмосфере
рабочих помещений составляет 1 мг/м3 [3276].
42 Заказ № 250
1074
Глава 7
Восприимчивость к силикозу
Было обнаружено, что восприимчивость к силикозу в значи-
тельной степени зависит от курения и потребления алкоголя:
продолжительность жизни пьющих и курящих в условиях запы-
ленности сокращается на 6—7 лет, тогда как непьющий человек
при идентичных условиях живет более чем на 12 лет
дольше [328]. Увеличение концентрации кремнеземной пыли
на 50 % сокращает в среднем вдвое продолжительность жизни.
Индивидуальная восприимчивость к силикозу, без сомнения, ши-
роко варьирует, но подобный фактор трудно выделить. Рак лег-
ких не является следствием силикоза, и кремнезем не канце-
рогенное вещество. Однако силикоз повышает восприимчивость
к раковым заболеваниям курящих, поскольку в этом случае
ухудшается устранение канцерогенных соединений табачного
дыма из легких [329]. . .
-Необычное соединение в пораженных силикозом тканях
Вейсс [290] обнаружил необычное кристаллическое неорга-
ническое соединение, о котором уже говорилось выше, обра-
зующееся тогда, когда кварцевые частицы остаются в тканях
животных. При отборе проб легочной ткани людей, умерших
от силикоза, Вейсс [127] в 76 случаях обнаружил кристалличе-
ское вещество — гидратфосфатосиликата железа. Это вещество
не было обнаружено ни в одной из 11 проб, взятых из тканей
легких, не пораженных силикозом. Подобное соединение неиз-
вестно в природе, но синтезировано в легких или брюшной поло-
сти у крыс при осаждении тонкомолотого кварца. Вещество
синтезируется в условиях in vivo в течение 6—18 нёд. Его со-
четав может-быть выражен формулой
Мв55 (H2O)A.[Fe®+Feft (ОН) Si3,36Р0>64О,,]
Катионы М+ можно менять; кроме того, вода может замещаться
небольшими полярными органическими молекулами. Вещество,
вероятно, родственно основному фосфату кальциймагнийжелеза,
который, как было найдено, соединяется с кремнеземом -в орга-
низме морского животного — голотурии [69].
Неизвестно, играет ли какую-нибудь роль это соединение
в биологическом воздействии кварца. Оно может существовать
только в виде побочного продукта, образуемого в живой си-
стеме, поскольку все его компоненты, как оказалось, содер-
жатся в определенных концентрациях в растворе, с которыми
кристаллическая фаза данного вещества находится в равно-
весии.
Кремнезем в биосфере
1075
Теория растворимости
В настоящее время считают, что теория растворимости не
способна объяснять развитие типичного силикоза, который
включает и фиброз. Однако поликремневые кислоты и коллоид-
ные кремнеземы, конечно, оказывают и другие токсические воз-
действия на ткани и живые клетки. В большинстве работ, пер-
воначально направленных на поиски причин заболеваний сили-
козом и основанных на теории растворимости, не объяснялся
механизм образования фиброза и силикоза, но тем не менее
это направление представило большую часть сведений относи-
тельно других воздействий кремнезема.
В своей монографии по пневмокониозу Холт [330] собрал
много важных факторов: 1) наблюдения относительно того,
что поликремневые кислоты или коллоидный кремнезем денату-
рируют белки и проявляют цитотоксичность; 2) полученные
в экспериментах на моно молекулярных пленках доказательства
того, что белковые монослои сильно адсорбируют поликремне-
вую кислоту и тем самым постоянно изменяются и связываются
поперечными связями; 3) сведения о способности белковых
монослоев на поверхности воды концентрировать кремневую
кислоту путем ее адсорбции, а также стимулировать полимери-
зацию такой кислоты, когда общая концентрация кремнезема
настолько низка, что в иных условиях полимеризация кремне-
зема была бы невозможна.
В теории растворимости принимается, что повреждение тка-
ней вызывается присутствием в них поликремневых кислот или
коллоидных частиц кремнезема. Растворимый или мономерный
кремнезем Si(OH)4 оказывается инертным при низкой темпе-
ратуре. Такие полимерные формы кремнезема не образуются
до тех пор, пока концентрация растворимого кремнезема не
превысит растворимость аморфного кремнезема, которая оказы-
вается значительно большей чем 0,01 % SiOz. Однако кварц
растворяется только до ~ 0,001 % ВЮг. Одно из объяснений об-
разования поликремневой кислоты и коллоидных частиц из от-
носительно нерастворимого кварца заключается в том, что раст-
воримый кремнезем первоначально концентрируется с помощью
каких-то биологических процессов. В вышеприведенном пере-
числении установленных закономерностей (п. 3) Холт приводил
доводы, что кремнезем мог концентрироваться на мембранах,
и там полимеризоваться. Другой возможный путь самопроиз-
вольного концентрирования кремнезема состоит в следующем.
В экспериментах, выполненных Айлером, раствор монокремне-
вой кислоты, не насыщенный по отношению к аморфному крем-
незему, подвергался старению в нейтральном растворе, содер-
42*
1076
Глава 7
жащем небольшое количество бромида цетилтриметиламмония.
Кремнезем осаждался вместе с ПАВ. Судя по всему, мицело-
образующий катион способен притягивать и концентрировать
мономерный кремнезем так, что последний может полимеризо-
ваться вплоть до образования частиц, растворению которых пре-
пятствует окружающий их слой катионов, т. е. частица будет
находиться внутри мицеллы. Таким образом, можно предста-
вить, что внутри живых клеток катионные группы могут соби-
рать кремнезем и вызывать затем его полимеризацию. Процесс
идет до тех пор, пока полимер окажется настолько большим, что
станет способным денатурировать белок.
Еще одно объяснение образования поликремневой кислоты
и коллоидного кремнезема из кварцевых частиц заключается
в том, что свежеприготовленные кварцевые частицы содержат
какую-то фракцию, высокорастворимого кремнезема. Таким
образом, Холт предложил «расширенную теорию растворимо-
сти», в которой постулировалось, что пылевидные частицы
кремнезема могут вначале образовать пересыщенный раствор
кремнезема, который затем полимеризуется.. Как Холт [331],
так и Соффге [306] придавали значение растворению кремне-
зема, за которым следует полимеризация. Частицы кремнезема
собираются и концентрируются фагоцитами, и создаются высо-
кие локальные концентрации кремнезема. Коллаген адсорби-
руется на кремнеземе и затем связывается поперечными свя-
зями за счет кремневой кислоты способом, сильно напоминаю-
щим кремнеземное дубление.
Этой теории уже давно'придерживался Деттль [332], кото-
рый поставил следующий опыт. Кремнеземный порошок в кол-
лодиевой оболочке, через которую мог проходить только раст-
воримый кремнезем, был имплантирован в боку у кролика.
В этом случае у животного начиналась воспалительная реак-
ция. Однако никакой подобной реакции не происходило, когда
в коллодиевом мешочке кремнезема не содержалось.
Фабер [333] был одним из первых, кто предположил, что
растворимый кремнезем полимеризовался с образованием поли-
кремневой кислоты, которая представляла собой активный
агент. Другое направление экспериментальных наблюдений,
базирующихся на теории растворимости, заключалось в том, что
когда кварцевые частицы размером меньше 2 мкм вводились
в организм животного (например, в уши кролика), то кремнезем
осаждался в печени, легких, селезенке и костном мозге. Однако
частицы субмикронного размера могли быть внесены и в кро-
воток животного. Эти эксперименты, конечно, наводят на мысль,
что растворимый кремнезем присутствовал во всей живой си-
стеме.
Кремнезем в биосфере
1077
Возражение против «расширенной теории растворимости»
Холта состояло в том, что теория не давала никакого объяс-
нения, почему частицы кварца субмикронного размера оказы-
вались более токсичными, чем частицы таких же размеров более
растворимых типов кремнезема. Однако Ягер отметил необык-
новенно высокую растворимость нарушенного слоя на поверх-
ности кварца и предположил, что это обеспечивает получение
мономерного кремнезема, который затем диффундирует по орга-
низму, полимеризуется и образует токсичные поликремневые
кислоты [34]. Свежеизмельченный в порошок кварц имеет
высокое значение поверхностной энергии по сравнению с дру-
гими типами кремнезема, и, таким образом, очень небольшие
по размеру частицы кварца или острые углы и края более
крупных частиц могли служить источником мономерного крем-
незема, создавая его относительно высокую концентрацию
в течение более продолжительного периода времени, чем дру-
гие более растворимые модификации кремнезема. В последних
подобные области повышенной растворимости должны были бы
быстро исчезнуть. Как уже рассматривалось в гл. 1, поверх-
ностная энергия на границе раздела фаз кварц—вода состав-
ляет, вероятно, около 416 эрг/см2 в отличие от 50 эрг/см2 для
системы аморфный кремнезем—вода. В соответствии с эффек-
том Томпсона — Гиббса, кварцевые частицы с очень малыми ра-
диусами кривизны (т. е. либо частицы очень небольшого диа-
метра, либо острые углы и края) должны быть более раство-
римы по сравнению с частицами аморфного кремнезема с та-
кими же радиусами кривизны, Айлер [1] подсчитал, что при
радиусе кривизны 1,5 нм растворимость поверхности кварца
должна быть порядка 0,1 %.
Штобер и Арнольд [335] установили, что тонкоизмельчен-
ный кварц вследствие отмеченного выше эффекта может раст-
воряться в воде с образованием мономерного кремнезема с кон-
центрацией 0,008 %, даже если концентрация кварцевого по-
рошка в суспензии ограниченна. Вблизи поверхности частицы,
внедренной в ткань, концентрация растворенного мономера мо-
жет становиться еще более высокой, и, следовательно, возможна
его полимеризация с образованием вредной для организма поли-
кремневой кислоты. Мономер даже мог сначала продиффундиро-
вать через примыкающие мембраны, а затем образовать по-
лимер.
Было показано [336], что свежеизмельченный сухой кварц
оказывается более активным, чем порошок, полученный при
влажном помоле, при одинаковых размерах частиц в обоих
случаях. Это можно было бы объяснить получением более раст-
воримого кремнезема за счет острых углов и краев. При влаж-
ном помоле такие выступы с высокой поверхностной энергией
1078
Глава 7
должны подвергаться закруглению благодаря процессу раст-
ворение-осаждение. Об активности кремнезема судили по воз-
растанию содержания оксипролина в легких, который накапли-
вается одновременно с развитием фиброза. Другое объяснение
заключается в том, что в процессе влажного помола кварцевая
поверхность покрывается адсорбированным слоем аморфного
кремнезема и таким образом становится менее активной.
Имеются и другие факты в пользу применимости теории ра-
створимости для объяснения силикоза. Наименее растворимые
типы кремнезема оказываются и наименее вредными. Так, осаж-
дение ионов растворимого алюминия на поверхности кремне-
зема понижает растворимость, а также токсичность последнего
[337—340]. К тому же алюмосиликатные минералы, такие, как
глины, которые еще менее растворимы, чем кварц, не вызывают
силикоза. Сообщение о том, что пораженные силикозом -ткани
легких содержат сложные эфиры кремневой кислоты, например
холестерин [341], по-видимому, также поддерживает идею об
участии в подобных системах растворимого кремнезема. ,
Теория растворимости не была «заблуждением», поскольку
была основана на многих убедительных наблюдениях и объяс-
няла многие цитотоксические и другие вредные воздействия, ко-
торые могли происходить в организмах животных, когда крем-
незем вводился каким-либо неестественным способом. Однако,
хотя она и давала ошибочные данные, приверженцы этой тео-
рии упорно продолжали верить, что эти эффекты должны при-
водить к фиброзу, который характерен для силикоза.
Растворимый кремнезем не вызывает силикоза. Стишовит —
кристаллическая форма кремнезема — способен растворяться
в воде; при этом концентрация получаемого растворимого крем-
незема оказывается намного большей по сравнению с растворе-
нием кварца в подобных условиях [309]. Если растворимый
кремнезем представляет собой активный агент, способствующий
возникновению силикоза, то в таком случае стишовит должен
был бы проявить токсичность даже большую, чем кварц. Но на
самом деле стишовит безвреден.
Аморфный тонкодиспергированный кремнезем не вызывает
силикоза. У животных вдыхание кремнезема вызывало брон-
хит или интерстициальную пневмонию, но не силикоз. Однако
это как раз тот тип кремнезема, из которого легко получается
насыщенный раствор мономерного кремнезема в воде. Справед-
ливо, конечно, что введение очень большого количества дан-
ного типа кремнезема в легкие, под кожу или в полость тела
вызывало развитие плотной фиброзной ткани, что, по-видимому,
не имеет ничего общего с силикозом, возникающим под дей-
ствием кварца.
Кремнезем в биосфере
1079
Если растворимый кремнезем получался бы из кварца, то он
должен был бы обнаруживаться в легких. Уитли [342] не уда-
лось найти какой-либо формы аморфного кремнезема в легких
в тех случаях, когда вводился кварц. Кроме того, он не нашел
и никаких разновидностей поликремневых кислот в водных экс-
трактах кварца.
Идея о том, что силикоз порождается кремнеземом в раст-
воримой форме, получающимся при растворении свежеобразо-
ванных кварцевых частиц, оказалась ошибочной. Это было по-
казано Кингом и др. [343], которые перед проведением исследо-
ваний на легочных тканях подопытных животных удаляли вы-
сокорастворимый кремнезем из кварцевых частиц. Слой такого
растворимого кремнезема устранялся обработкой щелочью, так
что получаемый кварцевый порошок имел истинную раствори-
мость кварца (около 0,001 %). Однако этот порошок оказался
фиброгенноактивным. (Порошки кварца, предварительно обра-
ботанные HF, проявляли даже еще большую токсичность, воз-
можно, из-за того, что адсорбированный на кварце фтор нельзя
удалить промыванием водой; он может замещаться на ОН”
только при промывании основанием.)
Авторы работы [344] при изучении культуры эмбриональ-
ных миокардиальных клеток цыплят пришли к заключению, что
добавление как очень тонкодисперсного кварцевого порошка,
так и аморфного кремнезема не вызывает никаких воздействий.
Авторы полагали, что введенные частицы должны были нахо-
диться полностью внутри клеток, так что токсичность могла
быть обусловлена контактом поверхности частиц с цитоплаз-
мой, как и в случае введения кремнезема внутрь фагоцитов.
Таким образом, растворимый кремнезем, несомненно, не уча-
ствует в рассматриваемых воздействиях. Теорию растворимости
подкрепляли также наблюдения относительно токсичности поли-
кремневых кислот, проведенные не на легких, а на других ор-
ганах. Применяя внутрибрюшинные инъекции, Петерсон и
Уитли [345] решительно утверждали, что возникающий при этом
так называемый силикоз вызывался раствором кремневой кис-
лоты, полученным в результате растворения поверхности кварца,
и что токсичность проявляли исключительно мономерная или
олигомерные кремневые кислоты (поддающиеся контролю мо-
лпбдатным методом) при инъекциях внутрибрюшинно это спра-
ведливо, и нет никаких сомнений в том, что поликремневые
кислоты могут вызвать денатурацию белка п как следствие
воспалительные реакции. Однако при развитии истинного сили-
коза, т. е. при фиброзе легких, вдыхаемые кремнеземные ча-
стицы, по-видимому, вызывают некий добавочный эффект, имею-
щий другой механизм.
1080
Глава 7
Именно поверхность кварцевых частиц вызывает силикоз
при условии, что частицы имеют определенные размеры. Што-
бер [346] писал: «Кажется маловероятным, что выделяемая
с поверхности кремнеземной частицы кремневая кислота как-
либо воздействует на тот химический механизм, который вызы-
вает развитие силикоза».
Вещества, тормозящие развитие силикоза
Поскольку именно на твердой поверхности кремнезема про-
исходят процессы, связанные с притяжением клеточных мем-
бран и их разрушением, что вызывает силикоз, то, следова-
тельно, любое покрытие или химическое модифицирование крем-
неземной поверхности, снижающее адсорбцию компонентов кле-
точных мембран, особенно фосфатидилхолина, будет
предотвращать инициирование силикоза. Этого можно достичь
тремя основными способами:
1. Добавлением водорастворимого биологически инертного
органического полимера, такого, например, как поли (винилпи-
ридин-М-оксид) (ПВПО), который адсорбируется на кремнезем-
ной поверхности более сильно, чем компоненты клеточных мем-
бран.
2. Добавлением какого-либо источника получения раствори-'
мого оксида алюминия,.благодаря чему оксид алюминия будет
адсорбироваться на поверхности кремйезема и изменять ее хи-
мические свойства, уменьшая ее фиброгенность.
3. Нанесением на кремнеземную поверхность хемосорбиро-
ванного слоя какого-либо вещества, не обладающего фиброген-
ностью или же способного превращать поверхность в инертную,
например монослоя из алкильных групп.
Поли(винилпиридин-М-оксид). В 1958 г. были найдены сое-
динения, способные компенсировать развитие силикоза в тка-
нях' животных, подвергшихся воздействию сйликозогенных
форм кремнезема. Соединение под шифром 48/80 неизвестного
состава, представлявшее собой какое-то органическое основа-
ние, оказалось эффективным, как это было показано Марксом
и др. [347—349], но слишком токсичным для человека. Соеди-
нение 48/80 способно просто покрывать поверхность кремнезема.
Те же авторы сообщили, что определенные феназины и вызы-
вающие выделение гистамина реагенты снижали токсичность
кремнезема. Некоторые защитные свойства наблюдались при
применении полимиксина В и гексиламина.
Позже Шлипкётер и др. [350—352], а также Бек, Врач и
Брокхауз [353] сообщили, что поли (винилпиридин-М-оксид) ока-
зался эффективным веществом, обладающим свойствами пре-
пятствовать развитию силикоза. Оно, несомненно, было эффек-
Кремнезем в биосфере
1081
тивным при подкожном введении даже через 30 сут после
инъекции кремнезема в трахеи.
Может быть простым совпадением то, что мономерное сое-
динение 1-оксипиридин-М-оксид является уникальным при обра-
зовании хелатного соединения с кремнием (см. гл. 1). Хелат-
ная связь не может образовываться с ПВПО, за исключением
тех случаев, когда в процессе биологического окисления этот
полимер переходит в 1-оксипроизводное.
Этот полимер по существу является единственным вещест-
вом, выступающим в роли антагониста по отношению к кремне-
зему, которое интенсивно исследуется на протяжении почти
20 лет. Вероятно, это вещество уникально благодаря комбина-
ции двух сильноэлектроотрицательных атомов — азота и кисло-
рода. Это объясняет его способность к сильному взаимодейст-
вию с группами SiOH на кремнеземной поверхности, которое
может быть представлено следующей схемой:
-1
СН2 СН—сн
сн—с n=o = н о; si
сн2 СН=СН и Г) [ с; СН—сн
СН—С( N=o •. Н О [ si Хсн=сн НО! Si
Другим полимером, с которым также проводили исследова-
ния, был поливинилпирролидон (ПВП), используемый в каче-
стве заменителя плазмы крови. Он также адсорбируется на по-
верхности кремнезема, но в общем оказывается менее эффек-
тивным, вероятно, из-за его менее сильной адсорбции по
сравнению с ПВПО.
Эффект, получаемый от применения ПВПО оказывается,,
очевидно, системным. Так, Клостеркёттер [354а] обнаружил,
что при подкожных инъекциях он оказывал защитное действие
в печени и других органах от повреждений, вызываемых инъек-
циями внутрибрюшинно кремнезема. Марчизо и Комолли [3546]
продемонстрировали, как ПВПО защищал макрофаги in vitro от
повреждений, вызываемых кремнеземом.
Мон [354в] сообщил, что коллоидный кремнезем, вводимый
внутривенно крысам, вызывал повреждение печени, которое
предотвращалось введением ПВПО, имевшего молекулярную
массу около 13 500. В аналогичных экспериментах на крысах
было обнаружено,' что поливинилпирролидон также компенси-
1082
Глава 7
ровал вредное влияние кремнезема, но оказался значительно
менее эффективным. Кроме того, приходилось вводить в 10 раз.
большую дозу по сравнению с ПВПО, которого требовалось
всего лишь 10 мг на одну особь. ПВП также усиливал повреж-
дение почек.
Используя электромикрографию в своих исследованиях, в ко-
торых ПВПО метили тритием и наносили в определенные ме-
ста вблизи проявляющихся частиц серебра, авторы работы
[354г] показали, что в образовавшихся грануломах ПВПО ока-
зывался связанным с находящимися в организме частицами
тридимита.
В культурах фагосом ПВПО вызывал задержку или предот-
вращал выделение токсичных лизосомных ферментов, которые
выделялись при добавлении кремнеземного пылевидного препа-
рата [355]. Очевидно, ПВПО проникал в клетку и здесь соеди-
нялся с кремнеземом (по всей вероятности, образовывал покры-
тие на кремнеземной частице) и тем самым препятствовал
адсорбции и последующей денатурации белков. Таким образом,
лизосомные мембраны оставались неповрежденными, и не вы-
делялись ферменты, способные умертвить макрофаг изнутри.
Имеется доказательство того, что ПВПО способствовал вос-
становлению состояния здоровья организма, подвергнувшегося
вредному воздействию кремнезема. Это вещество понижало со-
держание гранулой в печени крыс после их образования вслед-
ствие инъекции кремнезема в селезенку [356]. Были продемон-
стрированы также и обратные эффекты, когда кремнезем ис-
пользовался преднамеренно, чтобы разрушить макрофаги и тем
самым подавить отторжение трансплантата. Трансплантаты
кожи на хвостах крыс отторгались под действием макрофагов,
но становились более устойчивыми, когда внутрибрюшинно
вводили кремнезем с целью разрушения макрофагов. Инъек-
ция ПВПО полностью предотвращала этот иммунодепрессивный
эффект кремнезема [357]. Подобным образом исследовались
трансплантаты костного мозга, которые в обычном случае от-
торгались. Однако предшествующая инъекция внутренне крем-
неземных частиц диаметром 5 мкм приводила к разрушению
макрофагов и снижала отторжение. И в этом случае, если перед
инъекцией кремнезема вводили ПВПО, то кремнезем не оказы-
вал никакого действия [358].
Токсические эффекты от вдыхания кремнезема понижались
посредством введения внутрь трахеи. ПВПО (молекулярная
масса 40 000—80 000), но последний, однако, вызывал крово-
течение и бронхит [359, ЗбО]. К тому же ПВПО стимулировал
удаление кремнеземной пыли, а также защищал макрофаги
легких от- разрушения их кремнеземом. Причем все вызванные
введением кремнезема повреждения исчезали после обработки,
Кремнезем в биосфере
1083
проводимой в течение года. Вещество с молекулярной массой,
равной 50 000—150 000, оказывалось более эффективным, чем
с массой 2000 [361, 362]. Удаление из легких Т1О2, являющегося
несиликозогенным порошком, не стимулировалось введением
ПВПО [363]. Введение ПВПО крысам, однако, снижало число
заболеваний силикозом, который был вызван введением крем-
незема в легкие и трахеи. Оно уменьшалось примерно на 2/з
даже через 140 дней после того, как был осажден кремнезем
[364]. Противотуберкулезное средство — изониазид, вводимое
совместно с ПВПО, оказалось очень эффективным для живот-
ных, зараженных туберкулезом и подвергнутых воздействию
кремнезема [365].
Замедленное поражающее воздействие хризотил-асбеста
в легких или in vitro, не подавлялось введением ПВПО [366].
Вероятно, в этом случае чрезвычайно острые волокна (диамет-
ром—250 А) проникали механически через мембраны незави-
симо от того, была ли покрыта кремнеземная поверхность ПВПО
или нет. Терапевтическое воздействие ПВПО было рассмотрено
в обзоре Бархада, Ротару и Лазареску [367], включающем 45
библиографических ссылок (обсуждение асбестоза см. ниже).
Механизм действия ПВПО. Имеется некоторое доказатель-
ство того, что ПВПО играет двойную роль благодаря его ад-
сорбции как на поверхности кремнезема, так и на клеточных
мембранах, причем последние при этом упрочняются [368].
Посредством ПВПО достигается защитное действие также от
других токсических возбудителей [369]. В отличие от этого
-поливинилпирролидон не способен защищать клеточные мем-
браны вследствие того, что адсорбируется не на них, а только
на кремнеземе, так что он, вероятно, менее эффективен в пре-
дупреждении силикоза.
Именно благодаря присутствию в ПВПО N-оксидной группы
достигается благоприятное действие, поскольку некоторые по-
лимеры, обладающие такими группами, оказались активными
в подавлении лизиса макрофагов, вызываемого кремнеземом
[370]. Оба вещества, как ПВПО, так и ПВП, понижали гемо-
литическую активность кремнезема, причем эффективность ока-
залась обратно пропорциональной молекулярной массе веще-
ства [371]. Вполне вероятно, что адсорбция этих полимеров на
кремнеземе определяется образованием водородных связей, по-
скольку точно такая же обратная взаимосвязь между образо-
ванием водородных связей и молекулярной массой обычно на-
блюдается при формировании водородных связей между поли-
эфирами и другими полярными полимерами и кремнеземом.
Холт и соавторы в серии экспериментов, проведенных
в 1966—1969 гг., исследовали механизм взаимодействия ПВПО
1084
Глава 7
с кремнеземом. Изучение методом УФ-поглощения показало, что
между N-оксидом и группами SiOH образуются прочные водо-
родные связи [372]. При сравнении действий изомеров таких
веществ оказалось, что поли(2-винилпиридиноксид), а не 4-ви-
нил-изомер является антагонистом кремнезему в культурах
макрофагов [373]. Отмечается, что в случае 2-винил-изомера
поливиниловая углеводородная цепь располагается -ближе, чем
4-винил-полимер, к кремнеземной поверхности, к которой как
раз и присоединяются N-оксидные группы. Замещение алкиль-
ных групп на пиридиновое кольцо снижало активность подоб-
ных веществ, вероятно, из-за стерических препятствий.
Не обнаружено никакой связи между эффективностью изо-
мера в его противодействии кремнезему и его способностью оса-
ждать кремнезем. Наблюдалось, что 4-винил-изомер, но не 2-ви-
нил-изомер, по-видимому, связывался поперечными связями при
его выдерживании с монокремневой кислотой. Однако в на-
стоящее время очевидно, что подобное образование попереч-
ных связей является следствием не присутствия монокремневой
кислоты как таковой, а медленного формирования олигомера,
так как такое поперечное связывание происходило только при
концентрации ЗЮг 0,06 %, при которой мономер полимери-
зуется, но не при 0,03 %, когда процесс полимеризации очень
замедлен [373, 374].
Доказательство того, что ПВПО связывается с кремнеземом
в биологических тканях, было представлено Фромме, Штобером
и Штрекером [375]. Они применили ПВПО с радиоактивной
меткой и получали электронно-микроскопические снимки, чтобы
показать, что это вещество концентрируется вокруг частиц крем-
незема (ПВПО может также находиться на клеточных мембра-
нах, но в таком случае он слишком сильно «размазан» по мем-
бране и не может быть обнаружен).
Один из факторов, рассматриваемых при терапевтическом
использовании ПВПО, состоит в том, что он не выделяется,
а осаждается в тканях, т. е. образуется ретикулоэндотелиаль-
ная система (фагоциты) [376].
В 1963 г. Шлипкётер [377] указал в обзоре все известные
вещества, которые действуют в качестве ингибиторов силикоза,
процитировав 91 библиографическую ссылку. Воронков, Зелчан
и Лукевиц [4а] в своей книге привели 39 источников, относя-
щихся к исследованиям ПВПО.
p-Аминопропионитрил. В работах [378, 379] показано, что
это соединение служило ингибитором процесса образования си-
ликозного фиброза. Однако в дальнейшем никаких работ не
было представлено.
Неионное ПАВ. Габор и др. [380] утверждали, что соеди-
нение С8Н17С6Н4О(СН2СН2О)8СН2СН2ОН действовало подобно
Кремнезем в биосфере
1085
ингибитору при силикозе, когда оно вводилось крысам респира-
торным путем. Поскольку экспериментально показано, что по-
лиэфиры такого типа сильно адсорбируются на кремнеземной
поверхности, то действие такого вещества может быть подобно
действию ПВПО.
Стероидные гормоны. Имеется Сообщение Казанцевой и Аре-
тинского [381] о том, что гидрокортизон тормозил развитие
силикоза у крыс. Это наводит на мысль, что изучение адсорбции
стероидов из раствора Рингера на поверхности кремнезема мо-
гло бы подтвердить данное положение.
Воздействие оксида алюминия на силикоз. Как уже упоми-
налось в связи с рассмотрением теории растворимости, Хал-
дане [337] еще в начале нашего века признавал, что оксид алю-
миния проявлял антагонистический кремнезему эффект, т. е.
предотвращал его действие при развитии силикоза. В то время,
когда теория растворимости имела широкое хождение, это по-
ведение оксида алюминия казалось логичным, поскольку было
известно, что при смешении оксида алюминия с кремнеземом го-
раздо меньшее количество растворимого кремнезема появля-
лось в растворе. Теперь такая «поверхностная» теория требует
дальнейшего пояснения.
В течение многих лет был очевиден тот факт, что глинистые
минералы в виде пылей, а также другие алюмосиликаты не вы-
зывают силикоза. Следовательно, когда оксид алюминия вво-
дится совместно с кремнеземом, то на поверхностях существует
некоторое соединение из этих оксидов, делающее поверхности
безвредными. Такой тип обмена рассматривался в предыдущих
главах. Как показано в гл. 4 на основании работы Айлера, соз-
дание алюмосиликатных центров на поверхности кремнезема
в основном понижает стремление белков адсорбироваться на
поверхности из нейтрального раствора.
Однако пока еще не ясно, почему отрицательно заряженная
алюмосиликатная поверхность не проявляет даже еще более
сильного притяжения по отношению к катионным группам хо-
лина в клеточных мембранах по сравнению с действием по-
верхности чистого кремнезема. Можно только предположить,
что для подобного взаимодействия необходимы как ионное при-
тяжение, так и образование водородных связей. Как показано
Айлером [253], алюмосиликатные ионы препятствуют образо-
ванию водородных связей. На этом основании следует принять,
что относительно низкая концентрация оксида алюминия дол-
жна понижать губительное для клеточных мембран притяже-
ние к кремнеземной поверхности.
Вдыхание-, аэрозолей, получаемых из.суспензий гидроксида
алюминия или других содержащих алюминий растворов, пред-
отвращает силикоз при последующей ингаляции силикозоген-
1086
Глава 7
ного кремнезема [382]. - Кроме того, могут быть использованы
пылевидные препараты из металлического алюминия, или из
аморфного гидратированного оксида алюминия, или из колло-
идного оксида алюминия [383, 384]. Шеперс [385а] показал,
что растворимый кремнезем не оказывал вредного действия,
а растворимый кремнезем, который остается в растворе
(0,0002 %), в смеси, состоящей из суспендированного кремне-
зема и оксида алюминия, оказывался физиологически инерт-
ным. Эти формы оксида алюминия типичны для многих работ,
в которых оксид алюминия рассматривался как антисиликоз-
ное вещество. Добавление цинка в рацион подопытных живот-
ных приводило к торможению развития силикоза. Согласно
данным Чвапила [127, 3856], цинк ингибирует различные функ-
ции макрофагов и, таким образом, может понижать образова-
ние веществ, стимулирующих фиброз.
Оксид железа не проявлял антисиликозного эффекта [386],
оказывая лишь временное действие.
Кремнеземы с нанесенными покрытиями. Такие кремнеземы
не имеют никакой практической важности в предупреждении
силикоза, так как покрытие должно наноситься перед ингаля-
цией. Как отмечалось в других разделах (см. например, гл. 4),
поверхность тонкодисперсного кремнезема может вступать
в реакции со спиртами или алкилхлорсиланами, в результате
чего она покрывается молекулярным слоем органического ве-
щества. В теле животного такие покрытые кремнеземные ча-
стицы временно предотвращали силикозные реакции, но в ко-
нечном счете метаболические процессы, по-видимому, приводят
к удалению таких покрытий, после чего кремнеземные частицы
начинают действовать обычным образом [387, 288а].
Асбестоз, или заболевания, вызванные микроиглами
Эффект, возникающий при вдыхании микроскопических игло-
подобных частиц длиной обычно меньше 5 мкм, отличается от
эффекта, вызываемого кремнеземом с обычной формой частиц.
Теперь признается, что рак легких может возникать (главным
образом у курящих в возрасте 25—30 лет) под влиянием вды-
хания асбестовой пыли. Так как асбестовые минералы являются
силикатами, то можно было бы предположить, что вынос из
них катионов металлов, приводящий к образованию иглоподоб-
ных псевдоморфоз кремнезема, представляет собой причину
возникновения заболеваний. Однако, согласно данным Сели-
коффа [127], причина не в этом, так как иглоподобные, но не
содержащие кремнезема частицы оксида алюминия или стекла
вызывали аналогичные воздействия, по крайней мере, у подо-
пытных животных, у которых такой тип рака развивается болеб
Кремнезем в биосфере
1087
быстро. К тому же эффект, вызываемый асбестом, не ингиби-
руется введением ПВПО, который оказывался ингибитором дей-
ствия кремнезема [366].
Поскольку такое заболевание, по-видимому, обусловливается
механическим повреждением тканей и не связано с химической
природой микроигольчатых частиц пылевидного препарата, то
думается, что следовало бы применять более широкий и выра-
зительный, чем «асбестоз», термин, например «микроацикулез»,
или заболевания, вызванные микроиглами.
Благотворные воздействия кремнезема
Присутствие следовых количеств кремнезема необходимо для
жизнедеятельности животных; кроме того, кремнезем, по-види-
мому, оказывает и некоторые полезные воздействия. В част-
ности, полезной является его способность подавлять процесс
отторжения трансплантата, о чем уже упоминалось. Оборот-
ная сторона этого эффекта — снижение иммунитета к болезням.
Вайл [3886] сообщил, что «гидратированный порошок с от-
ношением Na2O : 3SiO2 .добавляли в течение многих лет в корм
крупного рогатого скота, чтобы компенсировать естественную
нехватку некоторых компонентов, которая вредно влияет на
рост поголовья скота, а также чтобы сделать- более гибкими
суставы постаревших животных».
В опытах на кроликах при прикладывании к ранам порошка
аморфного кремнезема с частицами размером 0,1 — 1 мкм ока-
залось, что кремнезем стимулировал заживление ран, не вызы-
вая образования гранулой инородных тел [389]. Согласно дан-
ным Грундманна [390а], коллоидный кремнезем помогает за-
живлению ран, поглощая токсические вещества и стимулируя
развитие соединительной ткани.
Присутствие кремнезема, ио-видимому, снижает уровень со-
держания холестерина в крови. Аномально низкий уровень со-
держания кремнезема в артериях, пораженных атеросклерозом,
как уже обсуждалось, показывает, что кремнезем может играть
некоторую благотворную роль в системе кровообращения.
Метаболизм кремния
Мало что известно о способах, которыми кремний погло-
щается и утилизируется в живых организмах. Кремний в виде
Si (ОН) 4 в небольших количествах должен попадать в большин-
ство водных областей живых организмов. Без сомнения, крем-
ний способен диффундировать в любой орган, куда может про-
никать вода или глицерин. Вероятно, кремний не способен про-
никать через липидные слои мембран.
1088
Глава 7
Однако можно предположить, что для переноса, концентри-
рования и осаждения кремния в организме, по-видимому, дол-
жно происходить образование соединений хелатного типа. Вейсс
и Герцог [127] нашли, что в растениях при pH 4,0—6,8 обра-
зуются катионные кремниевые комплексы производных тропо-
лона. Подобный комплекс мог бы быть перенесен и сконцен-
трирован, а затем, если значение pH повышалось до 7, выде-
лялась и осаждалась кремневая кислота в свободном состоянии.
Энергия, необходимая для переноса, концентрирования и выде-
ления кислоты, может оказаться совсем небольшой. Она дол-
жна главным образом расходоваться на реакции, требуемые для
незначительного изменения величины pH в локализованном
районе. Как только локальная концентрация SiO2 оказывается
выше 0,01—0,02 %, особенно в присутствии четвертичных аммо-
ниевых ионов (например, фосфолипидов), происходит поли-
меризация с образованием твердого кремнезема.
Вполне возможно, что в животном организме в метаболизме
кремния может участвовать и другой тип хелатного соединения
(например, анионные комплексы типа катехина). В таком слу-
чае, по Бауманну [3906], хелатное соединение оказывается ста-
бильным только при pH>7, а выделение кремневой кислоты
в свободном состоянии идет ниже этого значения pH. Никаких
хелатных соединений кремния не было выделено из. тканей жи-
вотных. Однако присутствие целого ряда молекул со структу-
рой, напоминающей катехин, как, например, катехоламины, не
исключает возможности образования такого типа соединений
в организме животных. Подобные хелатные соединения обсуж-
дались в гл. 1 и 2.
Способ, благодаря которому кремнезем собирается около
зародышей полимеризации в специфических центрах организма,
остается вопросом предположений и гипотез. Для того чтобы
образовались твердые частицы кремнезема в суспензии в вод-
ной среде, необходимо сильное пересыщение раствора моно-
мера. Однако если какое-либо органическое вещество оказы-
вается способным адсорбировать монослой кремневой кислоты,
которая затем полимеризуется до образования пленки кремне-
зема, то кремнезем будет продолжать осаждаться на таком уча-
стке из раствора при условии небольшого пересыщения в этой
области. Так, на основании изучения состава стенок клеток
нескольких разновидностей диатомей Хекки и др. [390в] пред-
положили, что кремневая кислота способна связываться на
богатой гидроксильными группами клеточной поверхности
белка, на которой содержатся большие количества серина и
тропеошша. Такой белковый слой удерживается на полисаха-
ридах клеточной стенки. Гидроксильные группы аминокислот-
ных сегментов полисахаридов формируют поверхность, на ко-
Кремнезем в биосфере
1089
торой близко расположенные между собой группы ОН в свою
очередь создают матрицу с особым сродством к кремневой
кислоте. Смежные молекулы кремневой кислоты могли бы затем
полимеризоваться с образованием гидратированной кремнезем-
ной поверхности, на которой уже в последующем происходило
бы осаждение кремнезема из окружающего раствора.
Силикагель как культуральная среда
Использование золей кремнезема в качестве среды для по-
лучения градиента плотности при разделении клеток и других
биологических компонентов методом центрифугирования рас-
сматривалось в гл. 4. Кроме того, при проведении биологиче-
ских исследований кремнезем может применяться в качестве но-
сителя или подложки культуральной среды. Такую кремнезем-
ную подложку можно стерилизовать предварительной автоклав-
ной обработкой, а при необходимости ее можно изготовлять
в очень чистой форме, свободной от других неорганических или
органических загрязнений.
Мюллер и Холм [391] описали подготовку силикагелевых
пластинок для выращивания определенных типов бактерий. Золь
кремневой кислоты получали добавлением раствора силиката
натрия к буферному раствору соляной кислоты при pH ~ 4,5.
Силикагель помещали в чашки Петри, промывали, пропитывали
питательным раствором и затем обрабатывали в автоклаве.
Для получения особо чистой среды промежуточный золь можно
было подвергнуть диализу перед тем, как он превращался
в гель [392]. Улучшенный способ введения питательных веществ
непосредственно в кремневую кислоту, из которой затем в сте-
рильных условиях обазуется гель, что позволяет исключить ста-
дию диализа, был разработан Стержесом [393]. Более простой
способ [394] состоит в следующем. Смешивают 700 мл 0,5 н. ра-
створа НС1 с 300 мл неорганического питательного раствора.
К 1 ч. объема 0,5 н. раствора силиката натрия добавляют 1 ч.
объема известковой воды и к этой смеси приливают 1 объемную
часть кислого питательного вещества. Полученную смесь раз-
ливают в чашки Петри, подвергают старению в течение 2 ч
и выдерживают в сушильной печи при 100°С в течение 1 ч.
Ингельман и соавторы [395, 396] предложили использовать
сложный тетраметиловый эфир ортокремневой кислоты. Для
этой цели чистые золи кремнезема готовили ионным обменом,
затем их стерилизовали и хранили для последующего примене-
ния. Добавление питательного раствора вызывало застуднева-
ние [397]. Синг [398] и Темпль [399] также использовали си-
ликагель в качестве культуральной среды. Применение доступ-
ных коммерческих растворов коллоидного кремнезема заметно
43 Заказ № 250
1090
Глава 7
упрощает приготовление силикагелевых сред. Воспроизводимый
способ изготовления силикагелевой подложки из коммерческого
золя для культуральных сред, содержащих различные типы пи-
тательных растворов, был детально описан Кингсбери и Барг-
хорном [400]. Прамер [401а] подробно изложил условия полу-
чения гелей, очищенных от солей пропусканием раствора чи-
стого кристаллического силиката натрия через ионообменник
для удаления натрия и превращения продукта в силикагеле-
вую культуральную среду. Бухагьяр [4106] описал способ очи-
стки коммерческого коллоидного кремнезема, применяемого для
приготовления определенных сред для выращивания микробио-
логических культур, а также условия гелеобразования.
Кремнийорганические соединения
До настоящего времени ни в животных, ни в растительных,
ни в неорганических объектах природы не обнаружено каких-
либо кремнийорганических соединений, в которых органический
радикал соединялся бы с атомом кремния связью углерод—
кремний. Исключением, вероятно, является лишь упоминаемый
Хейненом [402] микроорганизм Proteus mirabilis, который мо-
жет синтезировать целый ряд сложных органических эфирных
и амидных производных кремневой кислоты, включающих
связи Si—О—С и Si—N—С, а также некоторое число связей
Si—С. Хотя трудно представить такую биохимическую реакцию,
как —SiOH + H—С =—SiC—; + Н2О, она должна представляться
не менее замечательной, чем обычные биохимические превра-
щения при фотосинтезе, в результате которых происходит реак-
ция СО2 + 4Н2 = СН4 + 2Н2О и образуются связи С—С.
В прошлом веке тысячи соединений со связями Si—С были
синтезированы и частично испытаны на возможные биохими-
ческие эффекты. Согласно данным Воронкова [403, 127], осо-
бенно широко исследовались соединения такого типа в Рос-
сии. Замечательным примером может служить группа 1-арил-
силатранов, которые проявляли высокую токсичность. Как от-
мечает Воронков, в руках преступников такие вещества могли
бы стать едва уловимым ядом, поскольку они не имеют цвета,
запаха, вкуса и оказываются настолько сильнодействующими,
что их следовые количества, приводящие к гибели, трудно обна-
ружить в теле жертвы. Основную структуру силатранов состав-
ляет кремневая кислота, этерифицированная по трем группам
ОН путем реакции с N(C2H4OH)3, а четвертая связь кремния
идет на органическую группу, такую, как арильная, при этом
возникает связь Si—С.
н-Хлорфенпльное производное используется как родентицид
(для уничтожения грызунов), поскольку это соединение быстро
Кремнезем в биосфере
1091
инактивируется в организме погибшего грызуна и труп оказы-
вается нетоксичным для других животных. В случае использо-
вания «-СНзСбН481 (OCaH^sN в этих целях смертельная доза
для мышей составляет приблизительно 0,00002 % или 0,2 мг/г.
Согласно Воронкову, многочисленные органические произ-
водные кремния находят применение в медицине; например,
утверждают, что «мивал» и «мигуген» — соединения с нераскры-
той структурой — относятся к эффективным противораковым
препаратам, а соединения класса XYZSi(CH2)sNRR'— к анти-
биотикам. Другие кремнийорганические вещества стимулируют
развитие соединительной ткани, промотируют рост волос и пре-
дотвращают их выпадение, а также улучшают метаболизм
жиров. Однако все эти вещества пока находятся на ранней ста-
дии их разработки и применения. Согласно Воронкову, Грида-
линовичу и Зелчану [404а], некоторые соединения кремния,
введенные крысам, вызывали дистрофические изменения в клет-
ках карциномы Уокера и образование коллагена.
Фессенден и др. [4046] показали, что кремнийорганические
соединения настолько подвержены биологическому окислению
в организме, что даже происходит разрыв связи Si—С:
-SiH- SiOH-> -SiOSi-
-SiCH3-> - S1CH2OH
-SiC6H5 -> -SiCGH6OH
-Si(OH2),z CH.3 -> -SiOSi-
Следовательно, трудно утверждать, что какой-либо специ-
фический физиологический эффект обусловливается частной
структурой, вводимой в организм. Как уже упоминалось ранее
в связи с рассмотрением атеросклероза, такое соединение, как
монометилтрисиланол-О-оксибензоат натрия, по-видимому, пре-
дупреждает подобное заболевание у крыс при рационе с высо-
ким содержанием холестерина [300].
Большое число соединений кремния в настоящее время на-
ходится в стадии исследования, и нет сомнения, что будет полу-
чено много новых фармацевтических средств. Удивительное от-
крытие [127], что 2,6-ц«с-дифенилгексаметилциклотетрасилок-
сан оказывал точно такой же гормональный эффект, как и
эстроген, несмотря на отсутствие данных относительно их моле-
кулярного подобия, говорит о том, что могут иметь место и дру-
гие сюрпризы. Силатраны Воронкова [127] и силиконазотные
«силофармовые» материалы Вааннагатва [127], в которых на-
считывается свыше 10 000 индивидуальных веществ, представ-
ляются лишь началом исследований в этой области.
43*
1092
Глава 7
Аналитические проблемы
В добавление к аналитическим методам, упоминавшимся
в гл. 1, следует перечислить существующие в настоящее время
проблемы, которые связаны с определением следовых содержа-
ний кремнезема в биологических образцах.
Бертранд [405] отметил, что при молибдатном методе опре-
деления кремния в присутствии фосфора мог образовываться
фосфомолибдат, приводящий к завышению результатов. Воз-
можен и обратный эффект, когда при обычном колориметриче-
ском определении фосфора получающаяся фосформолибденовая
кислота может также включать и соединения кремния, если не
приняты соответствующие меры предосторожности. По этой при-
чине, вероятно, в прошлом при анализе не отличали кремний от
фосфора. Исаакс [406] разработал метод определения кремния
в присутствии фосфора, который был затем проверен Фулге-
ром [407]. Было показано, что при точном выполнении мето-
дики анализа действительно можно определять кремний в при-
сутствии фосфатов, хотя значения, по-видимому, оказываются
несколько заниженными, если содержание фосфата достаточно
велико. Причиной этого может служить замедление фосфатами
восстановления кремнемолибдата. Однако в пределах концент-
раций фосфора, обнаруживаемых в золе тканей животных, его
мешающее действие может-быть устранено путем небольшого
повышения активности системы перед добавлением восстано-
вителя.
Гор и Шолл [408] констатировали, что определение микро-
количеств кремнезема в биологических тканях представляет
собой одну из наиболее трудных проблем аналитической химии.
Гравиметрический метод определения по потере массы SiF4,
когда зола биологического образца обрабатывается смесью HF
и H2SO4, дает завышенные результаты. Сообщалось, что обыч-
ный колориметрический метод определения кремния дает неточ-
ные результаты в присутствии фосфора и железа, а в биоло-
гических объектах как раз присутствуют и фосфор, и железо.
Гор и Шолл описали улучшенный метод отделения фосфорной
кислоты от кремнезема и последующего определения кремне-
зема молибдатным методом после восстановления до молибде-
новой сини. По рекомендуемой ими процедуре можно опреде-
лять вплоть до 2 мкг кремнезема из навески образца, рав-
ной 2 г.
Существуют модификации вышеописанного способа, в кото-
рых не требуется полное отделение фосфора от кремнезема, но
они зависят от основного этапа восстановления кремнемолиб-
деновой кислоты до молибденовой сини в присутствии фосфоро-
Кремнезем в биосфере
1093
молибденовой кислоты. Обычно подчеркивается, что заслужи-
вающие доверия результаты получаются при исключении за-
грязнений образца кремнеземом из стеклянной посуды и дру-
гих предметов, устранении влияния фосфора и достижении мак-
симальной чувствительности при проведении колориметрических
измерений [409—411]. Бауманн [3906] рекомендовал методику
анализа кремнезема в крови и в других биологических мате-
риалах (см. лит. к гл. 1 [320]).
Мак-Гавак, Лесли и Као [412], а также Кинг [413] разра-
ботали специальные приемы для устранения наложений, возни-
кающих из-за присутствия фосфора в биологических образцах
при проведении анализа молибдатным методом.
Чтобы определить, присутствует ли кремний в виде органи-
ческого комплекса, Холт и Йетс [414] добавляли к культураль-
ной ткани растворимый кремнезем, меченный изотопом 31Si,
а также вводили этот изотоп в брюшинную полость крысы с по-
следующим выделением растворимых в спирте соединений, со-
держащих 31Si. Однако они отметили, что извлекаемый мате-
риал мог состоять из мицелл полимеров кремневой кислоты,
покрытой органическими веществами. Большая часть 31Si в тка-
нях оставалась в нерастворимом виде.
Заключение
Количество элементов, которые считаются существенными
для жизненных процессов, по-видимому, постепенно возрастает
по мере того, как экспериментальные методы анализа совер-
шенствуются. Нельзя отрицать возможность того, что все эле-
менты могут оказаться необходимыми для обмена веществ
живых существ. Жизнь и разум — это высшие формы сущест-
вования материи. Жизнь, возможно, одна из форм проявления
бесконечности в ее химическом разнообразии и сложности
электронной структуры.
ЛИТЕРАТУРА
1. Айлер Р. К. Коллоидная химия кремнезема и силикатов. Пер. с англ.—
И.: Госстройиздат, 1959.
2. Hunter М. J., Aberg В., Acta Pharmacol. Toxicol Suppl., 36 (3), 9 (1975).
3. Mohn G., Uber das Verhalten der Kieselsaure in der belebten Natur.,
Ann. Univ. Sarav. Med. 21 (2), 63—136 (1974) [Universitat des Saar-
landes-Saarbrucken],
4a . Воронков M. Г., Зелчан Г. И., Лукевиц Э. Я. Кремний и жизнь. 2-е изд.—
Рига: Зинатне, 1978.
46. Leo R. F., Barghoorn Е. S., Acta Cient. Venez., 27, 231 (1976).
5. Gamow G., Biography of the Earth, New American Library of World
Literature, New York, 1941.
6. Опарин А. И. Происхождение жизни.— M.: Госполитиздат, 1945.
1094
Глава 7
7. Bernal J. D., Physical Basis of Life, Routledge and Kegan Paul. Lon-
don, 1951.
8. Miller Stanley L., Science, 117, 3045 (1935).
9. Janet J., Р.-V., Seances Soc. Sci., Phys. Nat. B., 1970, 69 (1972).
10. Sedlak W., Role of Silicon in the Biochemical Evolution of Life, PWN,
Warsaw, 1967, 84 pp. in Polish.
11. Высоцкий 3. 3., Данилов В. И., Стрелка В. В.— Усп. соврем, биологии,.
1967, т. 63, вып. 3, с. 362.
12. Barghoorn Е. S., Tyler S. A., Science, 147, 463 (1965).
13. Cloud Р. Е„ Jr., Science, 148, 27 (1965).
14. Oro J., Nooner D. W., Zlatkis A., Wikstrdm S. A., Barghoorn E. S.,
Science, 148, 77 (1965).
15. Schopf J. W., Barghoorn E. S., Maser M. D., Gordon R. O., Science. 149-
1365 (1965).
16. Barghoorn E. S., Schopf J. IE., Science, 150, 337 (1965).
17. Barghoorn E. S., Schopf J. IE., Science, 152, 758 (1966).
18. Margolis L., Walker J. C. G., Rambler M., Nature, 264, 620 (1976).
19. Oehler J. H„ Schopf J. IE., Science, 174, 1229 (1971).
20. Jacks G. V., Sci. Prog., 41, 301 (1953).
21. Полынов Б. Б.— Почвоведение, 1945, с. 325; 1948, с. 594.
22. Айдинян П. К.—ДАН СССР, 1949, т. 67, с. 729.
23. Глазовская М. А.— Тр. Почвенного ин-та им. В. В. Докучаева. 1950,
т. 34, с. 28.
24. Ярилова Е. А.— Тр. Почвенного ин-та им. В. В. Докучаева, 1950, т. 34,
с. ПО.
25. Болышев Н. И.— Почвоведение, 1952, с. 403.
26. Александров В. Г., Зак Г. А.— Микробиология, 1950, т. 19, с. 99.
27. Oberlies F., Pohlmann G., Naturwissenschaften, 45, 513 (1958).
28. Adams P. B., New Sci., 1969, 25 (Jan. 2).
29. Кутузова P. C.— Микробиология, 1969, т. 38, вып. 4, с. 714.
30. Faust R. M., Adams J. R., J. Invertebr. Pathol., 8 (4), 526 (1966).
31. Heinen IE., Arch. Microbiol., 45 (2), 145 (1963).
32. Heinen IE., Arch. Biochem. Biophys., 120 (1), 86, 93, 101 (1967) [Chem.
Abstr., 66, 122126].
33. Calvert R., Diatomaceous Earth, Chemical Catalog Co., New York, 1930.
34. Mellors IE., Inorganic and Theoretical Chemistry, Vol. 6, Longmans,
Green, London, 1925, 143; E. Rabin, Wiss. Meeresunters. Kiel, 11. 311
(1910).
35. Лисицын А. П., Богданов Ю. А. Океанологические исследования: Сб:
статей, вып. 18, 1968, с. 5—45.
36. Siever R., Am. Mineral.. 42, 821 (1957).
37. Lewin J. C., Can. J. Microbiol., 3, 427 (1957); J. Gen. Microbiol., 18,
418 (1958).
38. Lewin J. C., Plant Physiol., 30, 129 (1955).
39. Paasche E„ Mar Biol., 19 (2), 117 (1973).
40. Jorgensen E. G., Physiol. Plant., 6, 301 (1953; 8, 840 (1955).
41a. KUham S. S„ J. Phycoh, 11 (4), 396 (1975).
416. Paasche E., Mar. Biol., 19 (3), 262 (1973).
42. Lewin J. C., J. Gen. Microbiol., 13, 162 (1955).
43a. Lewin J. C., Lewin R., Biochemistry and Physiology and Algae, Acade-
mic, New York, 1962.
436. Werner D., Ed., The Biology of Diatoms, Botanical Monograph, Vol. 13,
Blackwell, Oxford, 1977.
44. Reimann В. E. F., Lewin J. C., Volcani В. E., J. Cell. Biol., 24 (1), 39
(1965).
45. Darley W. M., Bot. Monogr., 10, 655 (1974).
46a. Helmcke J. G., Naturwissenschaften, 41, 254 (1954).
Кремнезем в биосфере
1095
466. Hurd D. С., Pankratz Н. S., personal communication of research in pro-
gress.
46b. Dawson P. A., J. Phyco!., 9, 353 (1973).
47. Kingsbury J. M., Biol. Bull., 110, 310 (1956).
48. Brandenberger E., Frey-Wyssling A., Experientia, 3, 492 (1947).
49. Jorgenson E. G., Physiol. Plant., 8, 846 (1955).
50. Lewin J. C., Geochim. Cosmochim. Acta, 21, 182 (1961).
51. Lewin J. C., J. Gen. Physiol., 37, 589 (1954); 39, 1 (1955).
52. Coombs J-, Volcani В. E„ Planta, 82, 280 (1968).
53. Azam F., Hemmingsen В. B., Volcani В. E., Arch. Microbiol., 97 (2), 103
(1974).
54. Coombs J., Halicki P. J-, Hansen О. H., Volcani В. E., Exp. Cell Res.,
47, 302, 315 (1967), Chem. Abstr., 68, 1116, 1117.
55. Werner D., Ber. Dtsch. Bot. Ges., 81 (9), 425 (1968) [Chem. Abstr., 71
54021].
56. Werner D., Z. Naturforsch., 23b, 268 (1968).
57. Karie I. L., Daly J. W., Witkop B., Science, 164, 1401 (1969).
58. Nakajima T., Volcani В. E., Science, 164, 1400 (1969).
59. Sullivan C. W., Volcani B. E., Biochim. Biophys. Acta, 308 (2), 212
(1973).
60. Sullivan C. II7., Univ. Calif. San Diego, Diss. Abstr. Int. B, 32 (9), 5295
(1972) [Chem. Abstr., 77, 16435w],
61. Reimann В. E., personal communication to the author, 1965.
62. Encyclopaedia Britannica, 14th ed., Vol. 21, p. 255.
63. Вотинцев К- К— ДАН СССР, 1948, т. 62, с. 661.
64. Webb J. L., Smith V. Е„ Wells Н. №., J. Gas Chromatogr., 3(11), 384
(1965).
65. Нис A., DeVos L., С. R. Acad. Sci. Ser. D, 275 (13), 1399 (1972).
66. Ре J., Arch. Biol., 84 (1), 147 (1973).
67. Runham N. W. et al., Z. Zellforsch. Mikrosk. Anat., 99 (4), 608 (1969).
68. Lowenstam H. A., Science, 171, 487 (1971).
69. Lowenstam H. A., Rossman G. R., Chem. Geol., 15, 15 (1975).
70. Hutchinson G. E., Soil Sci., 60, 29—40 (1945).
71. Sprecher A., Bull. Soc. Bot. Geneve, ser., 23 (4), 155—192 (1911).
72. Raleigh G. J., Plant Physiol., 14 (4). 823—828 (1939).
73. Okawa Kinsaku, J. Sci. Soil Manure Jap. 10, 95—110, 216—243, 414—
428 (1936); 11, 23—26 (1937); 14, 703—718 (1940).
74. Lipman С. B., Soil Sci., 45, 189—198 (1938).
75. Das S. C., Motiramanl D. P., Curr. Sci. India, 18, 46—47 (1949).
76a. Onodera L, Kageshima J., J. Sci. Soil Manure Jap., 10, 318—333 (1936).
766. Crang R. E., May G., Can. J. Bot., 52, 2171 (1974).
77. Umemoto K-, Zasshi Yakugaku, 94 (3), 380 (1974) [Chem. Abstr., 81,
132747].
78. Lanning F. C., Am. Soc. ortic. Sci., 76, 349 (1960).
79. Sterling C.. Am. J. Bot., 54 (7), 840 (1967).
80. Parry №. D., Smithson F., Ann. Bot. 28, 169 (1964); Nature, 179. 975
(1957).
81. Simon E. M., Hwang S„ Science, 155, 696 (1967).
82. Smithson F., Nature, 178, 107 (1956).
83. Parry D. W„ Smithson F., Ann. Bot. London, 22, 543 (1958).
84. Peinemann K., Ferreiro E. A., An. Edafol. Agrobiol., Madrid, 31, 1011
(1972) [Chem. Abstr., 79, 114363g],
85. Christian G, Bull. Mens. Soc. Linn. Lyon, 33 (7), 282 (1964) [Chem.
Abstr., 62, 4318h],
86 Lanning F. R., Ponnaiya B. W. X., Crumpton C. F.. Plant Physiol., 33,
339 (1958).
1096
Глава 7
87. Peinemann N„ Tschapek M., Grassi R., Z. Pflanzenernaehr. Dueng. Bo-
denkd., 127, 126 (1970) [Chem. Abstr., 74, 86953а].
88. Handreck K. A., Jones L. H. P., Plant Soil, 29 (3), 449 (1968).
89. Schwab G. M., Wahl B., Naturwissenschaften, 43, 513 (1956).
90. Umemoto K., Chem. Pharm. Bull. Jap. 21 (6), 1391 (1973) [Chem. Abstr.,
79, 123927w],
91. Lanning F. C., J. Agric. Food. Chem., 11, 435 (1963).
92. Cooper H. P., Paden W. R., Mitchell J. H., Trans. Int. Congr. Soil Sci.
Amsterdam, 1, 236 (1950).
93a. Holzapfel L., Engel W., Arch. Biochem. Biophys., 83, 268 (1959).
936. Kaufman P. B. et al., Am. J. Bot., 63 (5), 635 (1976).
94. Prison E., Microsc. Entomol. Mon. 7, 38 (1948) [Chem. Abstr., 42, 7842].
95. Laroche J., Rev. Gen. Bot., 76 (905—906), 483 (1969) [Chem. Abstr., 73,
11376k],
96. Viehoever A., Prusky S. C., Am. J. Pharm., 110, 99 (1938).
97a . Kaufman P. B., LaCroix J. D., Rosen J. J., Bigelow W. C., Dev. Biol.,
31 (1), 124 (1973) [Chem. Abstr., 78, 145252d],
976. Kaufman P. B., Bigelow W. C., Schmidt R., Ghosheh N. S., Am. J. Bot,
58 (4), 309 (1971).
97b. Kaufman P. B., Petering L. B., Soni S. L., Phytomorphology, 20 (3),
281 (1970), issued 1971.
97r. Kaufman P. B., personal communication. ,
97д. Kaufman P. B. et. al., Plant Physiol., 58, 670 (1976).
98. Rakusin M. A., Chem. Zig., 75, 568 (1926).
99. Wolter Helmuth, Z. Angew. Chem., 40, 1113 (1927).
100. N. V. de Bataafsche Petroleum Maatschappij, Neth. Pat. 53471, 1942.
101. Made J. L., Phil. Trans., 81, 368 (1791).
102. Cohn F. J., Beitr. Biol. Pflanz., 4, 365 (1887).
103. Van Bemmelen J. M., Z. Anorg. Allg. Chem., 13, 292 (1897).
104. Jones L. H. P., Milne A. A., Sanders J. V., Science, 151, 464 (1966).
105. Coppenet M., Ducet G., Guerillot J., Kahane E., Ann. Agron., 17, 564
(1947).
106. Chene Marcel, Papier, 46, 209 (1943).
107. Stone I., Gray P. P., Wallerstein Labs. Commun., 11, 301—318 (1948).
108. Stone I., Gray P. P., Am. Soc. Brew. Chem. Proc., 1948, 76—94.
109. Kaufman P. B., Bigelow W. C., Petering L. B., Drogosz F. B., Science,
166, 1015 (1969).
110. Noguera Gomez E., Rev. Brasil, chim. Sao Paulo, 10, 396 (1940).
111. Iler R. K-, unpublished investigation.
112a. Strasburger E., Jost L., Schench H., Karsten G., Lehrbuck der Botanik,
10th ed. Gustav Fischer, Jena, 1910, p. 100.
1126. Barber D. A., Stone M. G. T., Exp. Bot., 17, 569 (1966).
113. Iler R. K., personal observation.
114. Tingey W. M„ Pillemer E. A., Sci. News (August 21, 1967).
115. Amos G. L., Aust. Commonw. Sci. Ind. Res. Org. Bull., 267, 55 (1952).
116. Amos G. L., Dadswell H. E., J. Council Sci. Ind. Res., 21, 190 (1948).
117. . Frey-Wyssling A., Ber. Dtsch. Bot. Ges., 48, 179, 184 (1930).
118. Whittenberger R. T., Am. J. Bot., 32, 539 (1945).
119. Holzapfel L., Z. Anorg. Allg. Chem., 273, 186 (1953).
120. Rosenheim A., Raibmann B., Schendel G., Z. Anorg. Allg. Chem., 196,
160 (1931).
121. Akai Shigeyasu, Takahashi Minoru, Morimoto Taiji, Nakazato Hirosuke,
Igaku To Seibutsugaku, 17, 295 (1950).
122. Malfitano G., Catoire M., C. R. Acad. Sci., 174, 1128 (1922).
123. Engel W„ Angew. Chem., 64, 601 (1952); Plant., 41, 358 (1953).
124. Holzapfel L., Engel IF., Arch. Biochem. Biophys., 83, 268 (1959).
Кремнезем в биосфере
1097
125. Coupland D., Caseley J. С., J. Exp. Bot., 26 (90), 138 (1975) [Chem.
Abstr., 83, 2133Н].
126. Smith G. S., Urquhart N. S., J. Anim. Sci., 41 (3), 882 (1975) [Chem.
Abstr., 83, 177074s],
127. “Biochemistry of Silicon and Related Problems”, Nobel Symp. No. 40,
Stockholm Aug. 23—26, 1977, Plenum, London.
128. Sreenivasan A., Curr. Sci. India, 3, 193 (1934); Proc. Indian Acad. Sci.,
IB, 607 (1935); 2, 201 (1935); 3, 258 (1936); 3B, 283 (1926); Soil Sci.,
54, 27 (1942).
129. Bastisse E. M., Ann. Agron., 16, 463 (1946).
130. Laws W. D„ Soil Sci., 15, 89 (1951).
131. Toth S. J., Soil Sci., 47 (2), 123 (1939).
132. Brenchley W. E., Maskell E. J., Warington K, Ann. Appl. Biol., 14 (1),
45 (1927).
133. Lemmermann O., Wiessman H., Z. Pflanzenernaehr. Dueng. Bodenkd.
(A), 1 (4), 185 (1922).
134. Lemmermann O., Wiessman H., Sammet K, Z. Pflanzenernaehr. Dueng.
Bodenkd., 4 (5), 265 (1925).
135. Duchon E., Z. Pfanzenernaehr. Dueng. Bodenkd., 4 (5), 316 (1925).
136. Hampl Jan, Chem. Listy, 38, 125 (1944).
137. Jones L. H. P., Handreck К A., Plant Soil, 23 (1), 79 (1965) [Chem.
Abstr., 67, 81544p],
138. Grib J., Lanning F. C., Trans. Kans. Acad. Sci., 73 (3), 399 (1970)
[Chem. Abstr., 75, 139805h],
139. McCormack R. W., пат. США 3552943 (Scientism Laboratories, Inc.),
1971.
140. Reifenberg A., Buchwald S. J., J. Soil Sci., 5, 106 (1954).
141. Lovering T. S., Engel C., U. S. Geol. Surv. Prof. Pap., 594-B (1967).
142. Erhart H„ C. R. Acad. Sci. Ser. D, 262, 602 (1966).
143. Peinemann N., Ferreira E. A., Z. Pflanzenernaehr. Dueng. Bodenkd.,
132, 1 (1972).
144. Dr. Eitel Wilhelm, personal communication, 1948.
145. Wynn P. D., Ann. Bot. London, 39 (162), 815 (1975).
146. Lundie M., S. Afr. J. Sci., 9 (10), 263 (1913).
147. Germar B., Z. Pflanzenernaehr. Dueng. Bodenkd. (A), 35, 102 (1934).
148. Wagner Fritz, Phytopath. Z., 12, 427 (1940).
149. Sommer A. L., Univ. Calif. (Berkeley) Publ. Agric. Sci., 5, 57 (1926)
[Chem. Abstr., 21, 2917].
150. Yosii Hazime, Bull. Sci. Fak. Terkult. Kyusyu Imp. Univ., 9, 277, 292,
297 (1941).
151. Ishizuka Yoshiaki, Hayakawa Yasuo, J. Sci. Soil Manure Jap., 21, 253
(1951).
152. Sticher H., Bach R., Schweiz Landwirtsch. Forsch., 4, 352 (1965); J. Sci.
Food Agric., 17 (9), 111 (1966).
153. Ponnaiya B. W. X., Madras Agric. J., 56 (2), 49 (1969).
154. Lanning F. C., J. Agric. Food Chem., 14, 350 (1966).
155. Lanning F. C., Linka У., J. Agric. Food Chem., 9, 463 (1961).
156. Grosse-Breuckmann E., Z. Pflanzenkr. Pflanzenschutz, 65, 689 (1958).
157. Tanaka A., Park У. D., Soil Sci. Plant Nutr. Tokyo, 12 (5), 1966 [Chem.
Abstr., 67, 18602г].
158. Shigeyasu, Ann. Phytopathol. Soc. Jap., 17, 109 (12), 1953.
159. Sasamoto Kaoru, Uyo-Kontyn, 9, 108 (1953) [Chem. Abstr., 48, 3578с].
160. Okamoto Y., Nippon Sakamotsu Gakkai Kiji, 39 (2), 139, 151, 242 (1970)
[Chem. Abstr., 73, 130262e],
161. Viehoever A., Prusky S. C., Am. J. Pharm., 110, 99 (1938).
162. Krishnakumari M. L., Majumder S. K, Nature, 193, 1310 (1962).
1098
Глава 7
163. King D. R., Morrison E. O., Sandman J. A., J. Econ. Entomol., 55, 506
(1962).
164. LaHeu D. W., U. S. Dept. Agric. Mark. Res., 860 (1970).
165. Hurst H., Dis. Faraday Soc., 3, 206 (1948).
166. Tarshish I. B., J. Econ. Entomol., 53 (5), 903 (1960).
167. Micks D. W., J. Econ. Entomol., 53 (5), 915 (1960).
168. Ebeling W„ Reierson D. A., Pence R. J., Viray M. S., Pestic. Biochem.
Physiol., 5 (1), 81 (1975).
169. Moore R. C„ Conn. Agric. Exp. Stn., New Haven Bull., 740 (1973).
170. Ebeling E., Hilgardia, 30 (18) 531 (1961) [Chem. Abstr., 55. 21452g],
171. Lamina J., Kruner N., Dtsch. Tieraeztl. Wochenschr., 73 (6), 124 (1966)
[Chem. Abstr., 65, 4575а].
172. Tarshish I. B„ Pest Control, 27 (6), 14—28 (1959) [Chem. Abstr., 53,
20675g],
173. Holzapfel L., Z. Elektrochem., 55, 577 (1951).
174. Holzapfel L., Angew. Chem., 63, 286 (1951).
175. Gye IF. E., Purdy W. I., Br. J. Exp. Pathol., a, 238 (1924).
176. Gonnermann M., Z. Physiol. Chem., 99, 255 (1917); J. Chem. Soc., 112
(1); 494 (1917).
177. Kahle H„ Beitr. Klin. Tuberk., 47, 296 (1921).
178. Anonymous, Chem. Ztg., 45, 1249 (1921).
179. Kadisch E., Beitr. Klin. Tuberk., 53, 111—117 (1922).
180. Kvorning S. A., J. Gerontol., 5, 23 (1950) [Chem. Abstr., 45. 7214b],
181. Kvorning S. A., Kirk E., J. Gerontol., 4, 16 (1949).
182. Gohr H., Boenniger W., Z. Gesamte Inn. Med., 7, 455 (1952).
183. Worth G., Campen G., Hoppe-Seylers Z. Physiol. Chem., 288, 155 (1951).
184. Worth G., Klin. Wochenschr., 30, 82 (1952).
185. Sauer F. et al., Can. J. Biochem. Physiol., 37, 1173 (1959).
186. Kukhtina A. P., Mikroelem. Med., 1972 (3), 123.
187a. Baumann H., Z. Physiol. Chem., 319, 38 (1960); 320, 11 (1960).
1876. Baumann H., Bochum, Germany, personal communication.
188. Akiya S., Misawa T„ Motohashi N. et al., Bull. Tokyo Med. Dent. Univ.,
6, 383 (1959) [Chem. Abstr., 55, 5737h],
189. Loeper L., Polet С., C. R. Soc. Biol., 151, 263 (1957).
190. Iler R. K-, unpublished investigation.
191. Loeper L, Loeper J., C. R. Soc. Biol.. 161 (3), 589 (1967).
192. Charnot K, Peres G., Ann. Endocrinol., 32 (3), 397 (1971) [Chem. Abstr.,
75, 138641g],
193a. Семенов Г. И. Сб. Микроэлементы и их применение в сельском хозяй-
стве и медицине Сибири и Дальнего Востока/Под. ред. О. В. Макеева
и др., 1967, с. 557. Сб. Микроэлементы нерв. сист. Материалы Всес.
сими. Баку, 1963, с. 84.
1936. Arehart-Treichel J., Sci. News, 112, 218 (1977).
194. Faure Ch., Maroc Med., 53 (572), 572 (1973).
195. Schwarz K-, Milne D. B., Nature, 239, 333 (1972).
196. Smith J., Schwarz K„ J. Nutr., 93, 182 (1967).
197. Schwarz K., in W. G. Hoekstra, Ed., Trace Element Metab. Anim. Proc.
Int. Symp., 2nd, 1973, University Park Press, Baltimore, 1974, p. 355.
198. Carlisle E. M., Science, 167, 279 (1970); 178, 619 (1972).
199. Carlisle E. M., Fed. Proc. Fed. Am. Soc. Exp. Biol., 31, 700 (1972).
200. Schwarz K., in A. Chavey, Ed., Nutr. Proc. Int. Congr., 9th, 1972, 1,
96 (1975).
201. Carlisle E. M„ Ref. 197, p. 407.
202 Schwarz K-, Fed. Proc. Fed. Am. Soc. Exp. Biol., 33 (6), 1748 (1974);
Proc. Nat. Acad. Sci., 70, 1608 (1973).
203 Saver H. I., Proc Water Qual. Conf., 16, 39 (1974) [Chem. Abstr., 82,
102960 у].
Кремнезем в биосфере
1099
204а. Elwood Р. С., Abernathy AL, Martin М., Lancet, 2 (7895), 1470 (1974)
[Chem. Abstr., 82, 107143u],
2046. Austin J. H. et al., Prog. Brain Res., 40, 485 (1973).
204b. Austin I. H. et al., Arch. Neurol., 25, 198 (1971); 27, 549 (1972).
204r. Mehard C. W., Volcani В. E., Cell Tissue Res., 174, 315 (1976).
205. Baumann H., Beitr. Silikose-Forsch., Sonderb., 4, 19 (1960).
206. Anonvmous, Fed. Regist., 32, 12606 (Aug. 31, 1967) [Chem. Abstr., 60,
4689g; 65, 9612с].
207. Knake E., Peter H., Beitr. Silikose-Forsch., 68, 37 (1960) [Chem. Abstr.,
53, 9466с].
208. Knake E., Peter H., Beitr. Silikose-Forsch., 63, 35 (1959).
209. Scheel L. D., Fleischer E., Klemperer F. W., AMA Arch. Ind. Hyg. Occup.
Med.. 8, 564 (1953).
210. Bailey С. B„ Science, 155, 696 (1967).
211. Keeler R. F., Ann. N. Y. Acad. Sci., 104, 592 (1963).
212. Forman S. A., Whiting F., Conell R., Can. J. Comp. Med. Vet. Sci., 23,
157 (1959).
213. Ehrhart L. A., McCullagh K. G., Proc. Soc. Exp. Biol. Med., 143 (1),
131 (1973).
214. Mohn G., Beitr. Silikose-Forsch., 94, 25 (1968) [Chem. Abstr., 68,
112802g],
215. Минова К. Ф.— Гигиена труда и проф. заболев., 1969, т. 13, вып. 8,
с. 50.
216. Desai С. Н., Burkman A., Salisbury R., J. Am. Pliarm. Assoc., 47, 589
(1958).
217. Whiting F., Connell R., Forman S. A., Can. J. Comp. Med. Vet. Sci., 22,
332 (1958) [Chem. Abstr., 53, 23901].
218. Nottle M. C., Aust. J. Agric. Res., 17 (2), 175 (1966).
219. Georgiev R., Wien Tieraerztl. Monatsschr., 44, 78, 200 (1957) [Chem.
Abstr., 51, 1072511].
220. Kersten W., Staudinger J. I., Beitr. Silikose-Forsch. Sonderb., 2, 353
(1956).
221. Michon R., C. R. Acad. Sci., 243, 2194 (1956).
222. Arienzo R., Malabo M., Rass. Med. Sper., 16 (5), 218 (1969) [Chem.
Abstr., 74, 96842с].
223. Ammon R., Mohn G., Beitr. Silikose-Forsch. Sonderb., 3, 355 (1958)
[Chem. Abstr., 56, 15773g],
224. Strecker F. J., Beitr. Silikose-Forsch. Sonderb., 4, 121 (1960) [Chem.
Abstr., 57, 4975cd],
225. Gye W. E„ Purdy W. J., Br. J. Pathol.. 3. 75 (1922).
226. Campbell M., Proc. Soc. Exp. Biol. Med., 85, 590 (1954).
227. Shelly W. B., Hurley H. J., J. Invertebr. Dermatol., 34 (2), 107 (1960).
228. Druckrey H., Schmahl D., Naturwissenschaften, 41, 534 (1954).
229. Merliss R. R., Science, 173, 1141 (1972).
230. Schepers G. W. H., Toxicol. Appl. Pharmacol., 1, 487 (1959).
231. Policard A., Collet A. et al., J. Urol. Med. Chir., 65/585 (1960) [Chem.
Abstr., 55, 8638e]; J. Biophys. Biochem. Cvtol., 9, 236 (1961) [Chem.
Abstr., 55, 17859а].
232. Marks J., James D. M., J. Pathol. Bacteriol.. 77, 401 (1959) [Chem. Abstr..
53. 151801].
233. Marks J., Nagelschmidt G., AMA Arch. Ind. Health, 20, 383 (1959)
[Chem. Abstr., 54, 3794а].
234. Kessel R. W. I., Monaco L., Marchisio M. A., Br. J. Exp. Pathol., 44,
351 (1963).
235. Comolli R., Perin A.. Proc. Soc. Exp. Biol. Med., 113, 289 (1963).
236. Allison A. C„ D’A Hart P„ Br. J. Exp. Path., 49, 465 (1968).
237. Pearsall A'. A'., Weiser R. S., J. Reticuloendothelial Soc., 5. 107 (1968).
238. Allison A. C., Harington J. S., Birbeck M., J. Exp. Med., 124, 141 (1966).
1100
Глава 7
239. Zisman В., Hirsch М. S., Allison A. C., J. Immunol., 101 (5), 1155 (1970).
240a. Koshi K, Sabake H., Ind. Health Jap., 3 (3—4), 140 (1965) [Chem. Abstr.,
65, 14322b],
2406. Stalder K, Stober IF., Nature, 207 (4999), 874 (1965).
240b. Stalder K-, Beitr. Silikose-Forsch. (Pneumokoniose), 22 (5), 237 (1970).
240r. Depasse J., J. Colloid Interface Sci., 60, 414 (1977).
241. Holzapfel L„ Z. Elektrochem., 47, 327 (1941); Naturwissenschaften, 30,
33—44 (1943); Staub, 21, 141 (1944).
242. Holzapfel L., Arbeitsmedizin, 21, 79 (1942).
243. Holzapfel L„ Kolloid-Z„ 115, 137 (1949).
244. Klein G., Nienburg H., Chem. Вег., 69B, 2066 (1936).
245. Kaufmann H. P., Klin. Wochenschr., 14, 1420 (1935).
246. Schwarz K, Proc. Nat. Acad. Sci. U. S., 70 (5), 1608 (1973).
247. Holt P. F., Went G. W., Trans. Faraday Soc., 55, 1435 (1959).
248. Кляцкина H. K.— Гигиена труда и проф. заболев., 1969, т. 13, вып. 8,
с. 56.
249. Clark S. G., Holt Р. F., Went С. W., Faraday Soc. Trans., 53, 1500
(1957).
250. Holt P. F., Went C. W., Vortr. Originalfassung Intern. Grenzflaechen-
aktive Stoffe, 3rd Cologne, 2, 49 (1960) [Chem. Abstr., 57, 126751].
251. Holt P. F., Clark S. G„ Biochem. J., 60 (1955).
252. Holt P. F., Bowcott I. E. L.,.MAK Arch. Ind. Hyg. Occup. Med., 9,
503 (1954); Biochem. J., 57, 471 (1954).
253. Iler R. K, J. Colloid Interface Sci., 55, 25 (1976).
254. Bergman I., Nelson E. S., J. Colloid Sci., 17, 823 (1962) [Chem. Abstr.,
58, 7400а].
255. Stephan W., Roka L., Z. Klin Chem. Klin. Biochem., 6 (3), 186 (1968)
[Chem. Abstr., 69, 17412h].
256. Rilttner J. R., Isler К. M., Schweiz. Med. Wochenschr., 86, 63 (1956)
[Chem. Abstr., 50, 65286].
257. Scheel L. D., Fleischer E., Klemperer F. W., AMA Arch. Ind. Hyg. Occup.
Med., 8, 564 (1953) [Chem. Abstr., 48, 13448h].
258. Lundgren, Swensson A., Acta. Med. Scand., 145, 84 (1953).
259. Glomme I., Holmquist С. E. et al.. MA A Arch. Ind. Health, 17, 204
(1958).
260. Swensson A., Glomme I., Bloom G., AMA Arch. Ind. Health, 14. 482
(1956).
261. Charache P., MacLeod C., White P., J. Gen. Physiol., 45, 1117 (1962).
262. Harley J. D., Margolis J., Nature, 189 (4769), 1010 (1961).
263. Margolis J., Aust. J. Exp. Biol. Med. Sci., 39, 249 (1961).
264. Margolis J., Ann. N. Y. Acad. Sci., 104 (1), 133 (1963).
265. Margolis I., Proc. 8th Int. Congr. Hematol., Tokyo, Pan. Рас. Press,
Tokyo, 1960, pp. 4—10.
266. Harley I. D., Margolis /., Nature, 189, 1010 (1961).
267. Margolis I., Aust. J. Exp. Biol. Med. Sci., 40, 505 (1962).
268. Arienzo R., Bresciano A., Rass. Med. Spec, 16 (3), 135 (1969).
269. Holt P. F., Went C. W., Biochem. J., 65, 13p (1957).
270. Lindquist I., Nilsson O., Ronquist G., Upsala J. Med. Sci., 79 (1), 1
(1974).
271. Brown E. A., J. Cell. Comp. Physiol., 50, 49 (1957).
272. Depasse I., Warlus I., J. Colloid Interface Sci., 56, 618 (1976).
273. Pernis B., Clerici E., Ghezzi I., Med. Lav., 50, 405 (1959) [Chem. Abstr.,
54, 21225g],
274. Marasas L. W., Harington I. S., Nature, 188, 1173 (1960).
275 Федькина Л. Г., Аретинский Б. В.— Тр. Центр, н.-и. и проект.-кон-
структ. ин-та профилакт. пневмокониозов и техн, безопасности, 1971.
вып. 5, с. 136.
Кремнезем в биосфере
1101
276. Engel W., Holzapfel L., Kolloid-Z., 119, 60 (1950).
277. Haynes R., Walsh K. A., Biochem. Biophys. Res. Commun., 36 (2), 235
(1969); пат. США 3796634 (USHEW), 1974.
278. Reynolds J. H., Miller R. E., пат. ФРГ 2106214 (Mosanto Co.), 1971
[Chem. Abstr., 75, 108529s],
279. Krisch K-, Hoppe-Seylers Z. Physiol. Chem., 314 , 211 (1959).
280. King E. J., Schmidt E. et al., Enzymologia, 17, 341 (1956) [Chem. Abstr.,
51, 14850а].
281. Rowsell E. V., Leonard R. A., Biochem. J.. 66, 3P (1956).
282. Messing R. A., Stinson J. R., Mol. Cell. Biochem., 4 (3), 217 (1974)
[Chem. Abstr., 82, 53464d].
283. Leclat P.. C. R. Acad. Sci., 224, 845 (1947).
284. Seifert H„ Naturwissenschaften, 46, 261 (1959).
285. Schwarz R., Baronetsky E., Naturwissenschaften, 43, 68 (1956).
286. Kersten W„ Staudinger H., Naturwissenschaften. 43. 68 (1956). 44. 600
(1957).
287. Kikuth W., Schlipkoter H. W., Naturwissenschaften, 43, 68 (1956).
288. Schnaidman I. M., Raiklin I. M., Byul. Eksp. Biol. Med.. 75 (6)’. 27 (1973)
[Chem. Abstr., 79, 74543].
289. Hauger R., Krisch K, Staudinger H„ Beitr. Silikose-Forsch. Sonderb., 5,
69 (1963) [Chem. Abstr., 61, 3602e],
290. Weiss A., Angew. Chem. Int. Ed., 1, 165 (1962); Beitr. Silikose-Forsch.,
4, 51 (1960); Hoppe-Seylers Z. Phys. Chem., 324, 153 (1961), Z. Physiol.
Chem., 324, 153 (1961).
291. Kersten W. et al., Hoppe-Seylers Z. Physiol. Chem., 313, 109 (1958).
292. Pecchiai L., Med. Sper., 24, 262 (1953).
293. Воронков M. Г., Скоробогатова В. И., Бугмейстер Е. К, Макар-
ский В. В.— ДАН СССР, 1975, т. 220, вып. 3, с. 723.
294. Schwarz R„ Wiss. Forschungsber. Naturwiss. Reihe, 66, 113 (1958).
295. Schwarz R., Baronetsky E., Angew. Chem., 68, 573 (1956).
296. Benditt E. P., “The Origin of Atherosclerosis”, Sci. Am., 236 (6), 74
(1977).
297. Loeper J., Loeper J.. C. R. Soc. Biol., 152, 563 (1958). [Chem. Abstr.,
53, 3445i],
298. De Franciscis P., Oliviero G„ Rass. Med. Sper., 17 (1—2), 24 (1970)
299. Charnot Y., Desmaris-Delecraz D., Peres G., C. R., Soc. Biol., 166 (8—
• 9), 996 (1972) [Chem. Abstr., 78, 145982s],
300. Gendre P., Cambar R„ Bull. Soc. Pharm. B., Ill (1), 3 (1972) [Chem.
Abstr., 82, 93025t],
301. Butenandt A., Beitr. Silikose-Forsch. Sonderb, 5 (5), v—vi (1963); Ein-
brodt H. J. et al., vii—ix [Chem. Abstr., 60, 15106с].
302. Brown H. V.. Am. Ind. Hyg. Assoc. J., 26 (3), 212 (1965).
303. Garcia M., Acta Cient. Compostelana, 7 (3—4), 225 (1970).
304a. Thomas K, Stober IT., Naturwissenschaften, 1, 22 (1968).
3046. Thomas K, Embrodt H. J., Scoedel W., Beitr. Silikose—Forsch Sonderb.,
6, 3—18 (1963; publ. 1965) [Chem. Abstr., 63, 17021].
305. Hofmann U., Ber. Disch. Keram. Ges., 39, 272 (1962).
306. Soffge К. H., Neues Jahrb. Mineral. Monatsh., 1957, 12.
307. Lundgren K. D., Swenson A., Acta Med. Scand., 145, 84 (1953).
308. Schepers G. W. H., Ind. Med. Surgery. 29 (7). 326, (8) 359. (9) 434
(1960).
309. Stober W„ Beitr. Silikose-Forsch. H, 89. S. 1/113 (1966); Arch. Environ.
Health, 16, 706 (1968).
310. Heppleston A. G„ Br. Med. Bull., 25 (3). 282 (1969); Nature, 214, (5087)
521 (1967).
311. Heppleston A. G., “Biological Response to Silica”, in E. Kulonen, Ed.,
1102
Глава 7
Biol. Fibroblast, Sigrid Juselius Found. Symp. 4th., 1972, Academic,
New York, 1973, pp. 529—537.
312. Stober W„ Beitr. Silikose-Forsch. Sonderb., 1, 15 (1955).
313. Heppleston A. G„ Nature, 213, 199 (1967).
314. Weller W., Haacks H., Guzy J. K., Heine W., Beitr. Silikose-Forsch.
(Pneumokoniose), 26 (3), 263 (1974) [Chem. Abstr., 82, 107166d],
315. Heppleston A. G., Fletcher K., Wyatt I., Experientia, 28, 938 (1972);
Br. J. Exp. Pathol.. 55. 384 (1974).
316. Summerton J., Hoenig S., Butler С. II, Chvapil M., Exp. Mol. Path., 26,
113 (1977).
317. Allison A. C., Arch. Int. Med., 128, 131 (1971).
318. Allison A. C., Harrison J. S„ Birbeck Af., J. Exp. Med., 124, 151 (1966).
319. Allison A. C., Harrison I. S., Birbeck Al., Nash T., in C. N. Davies, Ed.,
Inhaled Particles and Vapors, Vol. 2, Pergamon, Oxford, 1967, p. 121.
320a. Heppleston A. G., Styles I. A., Nature, 214 (5087), 521 (1967); Fortsch.
Staublungen Forsch., 1967 (2), 123.
3206. Marasas L. W., Harington I. S., Nature, 188, 1173 (1960).
321. King E. I., Mohanty G. P., Harrison С. V., Nagelschmidt G., Br. J.
Ind. Med., 10, 9—17 (1953).
322. Ingelbrecht I. M., Nagelschmidt G., AMA Arch. Ind. Health, 17, 287
(1958).
323a. Stober W., Beitr. Silikose-Forsch. Sonderb., 6, 35 (1964).
3236. Bauman H., Rasche B., Ber. Silikose-Forsch. Bergbau-Berufsgenossen-
schaft, 1 (1971).
324. Schepers G. W. H., “The Biological Action of Flame Process Submicron
Amorphous Silica Dust”, AMA Arch. Ind. Health, 16, 125, 203, 363
(1957).
325. Klosterkotter W„ Int. Congr. Occup. Health, Madrad, 1963, (2), 517
(1964) [Chem. Abstr., 64, 7263b],
326. Beyers P. D., Gage I. C., U. Ind. Med., 18, 295 (1961).
327a. Breiger H., Gross P., Beitr. Silikose-Forsch., 81, 43 (1964).
3276. Klosterkotter W., Arch. Hyg. Bakteriol., 150 (6), 542 (1966) [Chem.
Abstr., 66, 1142161].
328. Suciu I., Prodan L., Clujul. Med., 45 (2—3), 303 (1972).
329. Мокроусова К. А., Шабунина H. К. и др.— Вопросы онкол., 1973. т. 19,
вып. 5, с. 3.
330. Holt Р. F., Pneumoconiosis, Edward Arnold, London, 1957.
331. Holt В. F., Sc. Prog., 44 (175), 442 (1956).
332. Kettle E. H„ J. Ind. Hyg., 8, 491 (1926).
333. Faber O. M„ Staub.. 3, 372 (1936).
334. lager R., Kolloid-Z., 119, 165 (1950).
335. Stober W„ Arnold M„ Kolloid-Z., 174, 20 (1961).
336. Gothe С. I., Lidstrom L., Swensson A., Med. Lav., 63 (8—9), 357 (1971).
337. Haldane I. S., Seventh Rep. Expl. in Mines Comm., London, 1914 [see
also Ref. 308].
338. Germer L. H., Storks К. H., Ind. Eng. Chem. Anal. Ed., 11, 583 (1939).
339. Crombie D. W. et al., Can. Med. Assoc. J., 50, 318 (1944).
340. lacobs A. W., Eng. Min. J., 147, 70; 148, 84 (1946).
341. Holzapfel L., Naturwissenschaften, 30, 185 (1942); Arbeitsmedizin, 21,
79 (1942); Z. Ver. Dtsch. Ing., 87, 605 (1943); Naturwissenschaften, 31,
386 (1943); Staub.. 21, 141 (1944); Z. Physiol. Chem., 281, 203 (1944).
342. Wheatley K., Research (London), 10, 76 (1957).
343. King E. J., Mohanty G. P., Harrison С. V., Nagelschmidt G., AMA Arch.
Indust. Hyg. Occup. Med., 455 June (1953).
344. Policard A., Collet A., Rey L., C. R. Acad. Sci.. 249, 16 (1959) [Chem.
Abstr., 53, 22460g],
Кремнезем в биосфере
1103
345. Paterson М. S., Wheatley К.-H., Minist. Fuel Power (G. В.) Safety
Mines. Res. Establ., Rep., 124 (1955).
346. Stober IV'., Arch. Environ. Health. 16, 706 (1968).
347. Marks J., James D. M., Morris T. G., Br. J. Ind. Med., 15, 1 (1958).
348. Marks J., Br. J. Ind. Med., 16, 166 (1959).
349. Marks J., Br. J. Ind. Med., 14, 81 (1957).
350. Schlipkoter H. W„ Brockhaus A., Klin. Wochenschr., 39. 1182 (1961)
[Chem. Abstr., 56, 7955b],
351. Schlipkoter H. W., Dolgner R., Brockhaus A., Ger. Med. Mon., 8, 509
(1963); Dtsch. Med. Wochenschr., 88 (39), 1895 (1963) [Chem. Abstr.,
60, 22481].
352. Schlipkoter H. W„ Beck E. G„ Med. Lav., 56 (6-7). 485 (1965) [Chem.
Abstr., 64, 4151e],
353. Beck E. G., Bruch J., Brockhaus A., Z. Zellforsch. Mikrosk. Anat, 59,
568 (1963).
354a. Klosterkotter W., Beitr. Silikose-Forsch. Sonderb., 6, 291 (1963, publ.
1965).
3546. Marchisio M. A., Cotnolli R., Med. Lav., 55 (6—7). 401 (1964).
354b. Mohn G„ Beitr. Silikose-Forsch., 88, 1 (1965).
354r . Fromme H. G., Stober W., Strecker F. J., Hoppe-Sevlers Z. Physiol. Chem.,
346,74 (1966).
355. Allison A. C., Harington J. S., Birbeck M., J. Exp. Med., 124, 141 (1966).
356. Heeren E„ Eger W., Einbrodt J. H., Beitr. Silikose-Forsch (Pneumoko-
niose), 23 (3), 137 (1971) [Chem. Abstr., 76, 121663w],
357. Rios A., Simmons R. L., Transplantation, 13 (3). 341 (1972) [Chem.
Abstr., 77, 28885f],
358. Lotzova E., Cudkowic G., J. Immunol.. 113 (3), 798 (1974) [Chem. Abstr.,
81, 118420П].
359. Безродных А. А., Каспаров А. А. и dp.— Гигиена труда и проф. забо-
лев., 1973, т. 12, с. 20.
360. Шнайдман И. М, Райхлин Н. Т.— Бюллетень эксперим. биологии и ме-
дицины. 1973, т. 75, вып. 6, с. 27.
361. Богдановская Н. И., Толстая М. С.— Гигиена и санитария, 1973, т. 38,
вып. 4, с. 102.
362. Богдановская Н. И.— Гигиена труда и проф. заболев., 1973, т. 12, вып. 5,
с. 22.
363. Klosterkotter W., Gono F., in W. H. Walton, Ed., Inhaled Particles,
Vol. 3, Proc. Int. Symp. 3rd, 1970 (Unwin Bros., 1971) [Chem. Abstr.,
77, 57245с].
364. Chung C. W., Kalullik Taehak Uihakpu Nonmunjip (Korea), 27, 79
(1974) [Chem. Abstr., 83, 383851].
365. Фролова H. H., Озерова Л. В., Мордовский Г. Г.— Проблемы туберкул.,
1975, вып. 3, с. 67.
366. Davis J. М. G.. J. Exp. Pathol., 53 (6), 652 (1972) [Chem. Abstr., 78,
67070b],
367. Barhad В., Rotaru G., Lazarescu I., Igiena, 20 (8), 451 (1971) [Chem.
Abstr., 76, R68004d],
368. Шнайдман И. M.— Гигиена труда и проф. заболев., 1974, т. 13, вып. 1,
с. 19.
369. Antweiler Н., Beitr. Silikose-Forsch. (Pneumokoniose), 22 (3), 93 (1970)
[Chem. Abstr., 74, 85829с].
370. Ferruti P., Marchisio M. A., Med. Lav., 57 (8—9), 481 (1966) [Chem.
Abstr., 69, 17723k],
371. Sanchez de Alaiz M. J., Jimenez-Torres N. V., Med. Segur. Trab., 21
(82), 12 (1973) [Chem. Abstr., 83, 54060b],
372. Holt P. F„ Nasrallah E. T„ Nature, 211, 878 (1966), J. Chem. Soc. B.
1968 (3), 233 [Chem. Abstr., 68, 87717q].
1104
Глава 7
373. Holt Р. F., Lindsay H., Nasrallah E. T., Nature, 216 (5115), 611 (1967)
[Chem. Abstr., 68, 87717q].
374. Holt P. F., Lindsay H., J. Chem. Soc. B, 1969 (1), 54 [Chem. Abstr.,
70, 47939y].
375. Fromme H. G., Stober W., Strecker F. J., Hoppe-Seylers Z. Physiol.
Chem., 346, 74 (1966).
376. Gruenspan M., Antweiler H., Marcic L Beitr. Silikose-Forsch. (Pneumo-
koniose), 23 (3), 101 (1971) [Chem. Abstr., 76, 121898b],
377. Schlipkoter H. W., Med. Lav., 54 (5), 405 (1963).
378. Stalder K-, Strecker F. J., Beitr. Silikose-Forsch. Sonderb., 6, 161 (1965).
379. Levene C. L, Bye L, Saffioti U., Br. J. Exp. Pathol., 49, 152 (1968).
380. Gabor S., Zugravu E. et al., Igiena, 21 (6), 329 (1972) [Chem. Abstr.,
79, 14141q],
381. Казанцева С. В., Аретинский Б. В.— Tp. Центр, н.-и. и проект.-кон-
структ. ин-та профилакт. пневмокониозов и техн, безопасности. 1971,
вып. 5, с. 155.
382. Daniel Н., Martin J. С., LeBouffant L., in W. Н. Walton, Ed., Inhaled
Particles, Vol. 3, Proc. Int. Symp. 3rd, 1970, Unwin Bros., 1971.
383. Sander O. A., in F. A. Patty, Ed., Industrial Hygiene and Toxicology,
Vol. I, 2nd rev. ed., Interscience, New York, 1958.
384. Schepers G. UL H., 9th Conf. McIntyre Res. Found. Toronto, 1958.
385a. Schepers G. IF. H., Toxicol. Appl. Pharmacol., 3, 188 (1961). ,
3856. Chvapil M., in S. D. Lee, Ed., Biochemical Effects of Environmental
Pollutants, Ann. Arbor Science Publishers, Ann. Arbor. Mich.. 1977,
Chapter 16 Environ. Health Perspect., 9, 283 (1974).
386. Кочеткова T. А. Борьба с силикозом. Сб. статей, АН СССР, вып, 7,
1967, с. 272—278.
387. Klosterkotter W„ Arch. Hyg. Bakteriol., 152.(1), 7 (1968) [Chem. Abstr.,
69, 21753m].
388a. Островская И. С., Тимченко А. Н. Борьба с силикозом. Сб. статей
АН СССР, вып. 9, 1974, с. 206.
3886. Vail J. G., see Chapter 2, Ref. 1, Vol. 2, p. 597; Jersey Bull Dairy World,
55, 1642, 1652 (1936), Rural New Yorker, 96, 284 (1937).
389. Jotten K. W., Klosterkotter IF.’, Die Medizinsche, 1076, 1093 , (1956)
[Chem. Abstr., 50, 17103h],
390a. Grundmann G., Zentralbl. Chir., 75, 158 (1950); Chem. Zentralbl. I,
2375 (1950) [Chem. Abstr., 48, 824d; 11444а].
3906. Baumann H., Beitr. Silikose-Forsch. Sonderk., 4, 43 (1960).
390b. Hecky R. E., Mopper K-, Kilham P., Degens E. T., Mar. Biol., 19, 323
(1973).
391. Muller D., Holm F., Zentralbl. Bakteriol. Parasitenkd., Part II, 105. 131
(1942).
392. Olszewski \V., Kohler H., Bas-u. Wasserfach, 85, 483 (1942).
393. Sterges A. J., J. Bacteriol., 43, 317 (1942).
394. Sterges A. J., J. Bacteriol., 44, 138 (1942).
395. Ingleman B., Jutlander I., Nature, 156, 572 (1945).
396. Ingleman B., Laurell H., J. Bacteriol., 53, 364 (1947).
397. Taylor A. S., J. Gen. Microbiol., 4, 345—237 (1950).
398. Singh B. N., Nature, 157, 302 (1946).
399. Temple K- L., J. Bacteriol., 57, 383 (1949).
400. Kingsbury J. M., Barghoorn E. S., Appl. Microbiol., 2 (1), (1954).
401a. Pramer D., Appl. Microbiol., 5, 392 (1957).
4016. Buhagiar R. W. M., J. Gen. Microbiol., 1 (1977).
402. Heinen W., Arch. Microbiol., 37, 199 (1960); 41, 229 (1962); 45, 145,
162, 172 (1963); 52, 49, 69 (1965); Arch. Biochem. Biophys.. 110, 137
(1966); 111, 236 (1966); 120, 86, 93, 101 (1967).
403. Voronkov M. G., Chem. Br., 9, 411 (1973).
Кремнезем, в биосфере
1105
404а. Воронков М. Г., Гридалинович Г. А., Зелчан Г. И.— ДАН СССР, 1971,
т. 200, вып. 4, с. 967.
4046. Fessenden R. I. et al., J. Med. Chem., 7, 561, 695 (1964); 8, 604 (1965);
9, 262 (1966); 10, 810 (1967); 11, 1070 (1968); Adv. Drug Res.. 4, 95
(1967).
405. Bertrand G., Bull. Soc. Chim. Biol., 6, 656 (1924).
406. Isaacs L., Bull. Soc. Chim. Biol., 6, 157 (1924).
407. Foulger J. H„ J. Am. Chem. Soc., 49, 429 (1927).
408. Gohr H., Scholl Otto, Beitr. Klin. Tuberk., B102, 29 (1949).
409. Ohlmeyer P., Olpp V., Z. Physiol. Chem., 281, 203—207 (1944). .
410. Kahler H. L. et al., Ind. Eng. Chem. Anal. Ed., 13, 536 (1941).
411. Gajatto S., Studi Sassaresi, 22, 259 (1944).
412. McGavack T. H., Leslie I. G., Kao Y-K T., Proc. Soc. Exp. Biol. Med.,
110,215 (1962).
413. King E. J., Stacy B. D. et al., Analyst, 80, 441 (1955).
414. Holt P. F„ Yates D. M„ Biochem. J., 54, 300 (1953).
44 Заказ № 250
ПРЕДМЕТНЫЙ. УКАЗАТЕЛЬ
Агрегаты частиц см. Порошки крем-
неземные
Агрегация частиц 75, 495—507. См.
также Коагуляция, Флокуляция
влияние размера частиц 445—446
вязкости при 349
механизм 303—308
образование геля 301—321, 428—
502 •
предотвращение 436—437, 441—
443
скорость 366—368
стадии 445—446, 497—498
тепловые эффекты 19—20
терминология 495—497
упорядочивание структуры при
545—555
Адгезия 164—165, 497
монослоя 559
понижение 595—596, 818—819
усиление 593—595, 819
Адсорбаты ‘
для исследования микропор 688—
689
для определения удельной по-
верхности см. Поверхность удель-
ная
классификация 903
Адсорбенты
приготовление из золей 579—580
специфические 761—764
Адсорбция
изотермы, применение
----для измерения удельной по-
верхности 479—480, 636—650
----для характеристики пор
669—691
метод
— Боунауэра—Эммерта—Т еллера
(БЭТ) 638-641
— /-диаграм.мы 641—644, 682
— Каганера 634
— Киселева 634
— a-метод Синга 632—634, 683
— непрерывного потока 644—645
— Френкеля—Хелси—Хилли 634
теплота 651—652, 692—693
— изостерическая 687—688
Адсорбция кремнезема 555—559
из растворов на
------гидроксидах металлов 112
------оксиде алюминия 555—
557
па кремнеземе 54—62
на эритроцитах 1059
Адсорбция на кремнеземе 895—985
аминов 897, 904, 923—924, 941 —
942
аммиака 897
белков 980—981, 1056—1060, 1071
витамина С 1061 —1062
влияние дегидроксилирования
898—891
водорода 991
воды 889, 899—901, 969—973
газов инертных 901, 973
газов и паров 637—650, 895—898
глицерина 830
диэлектрические эффекты 909
жирных кислот 907
из воды 909—912, 975—983
— кислот 929—930
— неводных растворов 905—909,
983—985
— растворов сильных электроли-
тов 911—912
ионов металлов
— двухзарядных 924, 927—929,
933—934
— многозарядных 830. 924—929,
934—935
— однозарядных 922—924, 933
катионов комплексных 951—952
красителей 693, 761—764, 950—
951
молекул органических 901—904,
909_911
— полярных 896, 901
на гидрофобных поверхностях
968—973
неионных веществ 895—912
обзоры по 931—935
образование водородных связей
при 902—903, 909—912
обращение знака заряда 930—931
озона 830, 991
Предметный указатель
1107
осаждение полислоев 556, 985—
986
оснований органических 941—942
----влияние концентрации элек-
тролитов 943
-------pH 943, 948
-----стадии 944—947
полиалкилакрилатов 985
поливинилового спирта 976—979
полимеров катионных 979—980
полимеров органических 973—985
----- влияние солей 982—983
----из водных растворов 975—
983
полиметилметакрилата 984
полиэтиленоксида 976
спиртов 906, 956. См. также Эте-
рификация кремнезема
углеводородов ароматических
907—909
ферментов 831, 1060—1062
холестерина 906
Азот как адсорбат 479
для измерения объема пор 674—
675 '
для определения удельной по-
верхности 638—645
Аквагель 632
Алкогель 632
Альбумин 394—395, 1054, 1068
Алюминий, адсорбция ионов на крем-
неземе 925—927, 930, 931, 934
Алюминия оксид
адсорбция кремнезема 555—557
влияние на растворимость SiOg
82—84, 113
как присадка в силикагелях 748—
749
свойства инсектицидные 1038
соосаждение с SiO2 125
Алюмосиликат-ионы
влияние на свойства золей 561—
563
в гелях 897
в кремнеземных порошках 634
модифицирование поверхности
кремнезема 560—561, 951, 986—
988
Алюмосиликаты
взаимодействие с белками 1055
разрушение биологическое 1009
Аммиак
адсорбция на кремнеземе 897
как адсорбат для определения
удельной поверхности 645
р-Аминопропионптрил, роль
в борьбе с силикозом 1084
44*
Амины, адсорбция на кремнеземе 897
длинноцепочечных 941—942
низших 904, 923—924
Аморфный кремнезем см. Кремнезем
аморфный
Анализ атомно-абсорбционный 134
Анализ химический 134—143, 1092—
1093
биологических материалов 1092—
1093
золей 467—468
Капельный 538—539
концентрирование для 141 —142
обнаружение коллоидов 556—557
отделение фосфора 1092—1093
растворы стандартные 143
силикагелей 693—694
титрование 144—147
— йодометрическое 988
Антиокислители, адсорбция на крем-
неземе 830
Арилсилатраны 1090
Асбестоз 1086—1087
Атеросклероз 1042—1044, 1063—1066,
1087
Аэрогели 755, 831
получение 739—742
этерификация 958. См. также Эте-
рификация кремнезема
Аэросил 457. 634, 785, 871, 914, 1048.
См. также Кремнезем пирогенный
Бактерии 1061. См. также Вирусы,
Микроорганизмы
обмен кремния и фосфора 1010
роль кремнезема в метаболизме
1010—1011
Бамбук 1024—1025, 1039. См. также
Растения
Барий, адсорбция ионов на кремне-
земе 931, 933
Белки
адсорбция на кремнеземе 980—981,
1056—1060, 1071
взаимодействие с
----алюмосиликатами 1055
----кремнеземом 397, 399—400,
536—537, 1054—1056
Бериллий, адсорбция ионов на крем-
неземе 930
Вирусы 1010. 1050. См. также Ми-
кроорганизмы
Витамин С, адсорбция на кремнезе-
ме 1061 — 1062
1108
Предметный указатель
Вода
адсорбция 889, 899—901, 969—973
— на гидрофобной поверхности
962—972
---- дегидратированной поверх-
ности 889—890
----пирогенном кремнеземе 970—
971
----этерифицированном кремне-
земе 971—972
взаимодействие с поверхностью
кремнезема 898—901
внутри частиц кремнезема 870—
872
как адсорбат для определения
удельной поверхности 645—646
морская 25, 26, 82, 111, 1012
обработка гелей 742—749, 872
обработка золями 401—404
отличие от силанольных групп
866—870
поведение на гидроксилирован-
ной поверхности 863—865
смачивание поверхности 892—895
содержание в кварцевом стекле
16
«сухая» 829
хемосорбированная 899
Водород, адсорбция на силикагеле
991
Водородные связи см. Связи водо-
родные
Водоросли 26, 1007—1009, 1011—1017.
См. также Диатомеи
выращивание в коллоидном крем-
неземе 1012
нахождение в шерте 125
— сине-зеленые, ранние формы
жизни 1007—1009
Воды геотермальные 26—27, 119, 127,
132—133, 380
Вязкость 819—827
в золях 327—332, 441, 491—495
----до начала агрегации 327—
328
при гелеобразовании 317—320,
344
Газы инертные, адсорбция на крем-
неземе 901, 973
Газы и пары, адсорбция на кремне-
земе 637—650, 895—898
Галлий, адсорбция ионов на кремне-
земе 931, 934
Гафний, адсорбция ионов на кремне-
земе 935
Гелеобразование
влияние коагулянтов 315—316,
702—704
— pH 241—242, 498—500, 702—
712
вязкость при 317—320, 344
замораживанием 724—725
из золей 503—506
путем агрегации 301—321, 428—
502
-------влияние размеров частиц
499—500
-------температуры 501—502
------- электролитов 500—501
-------pH 498—499
скорость
— влияние концентрации кремне-
зема 499—500, 703
-------органических жидкостей
500—501
-------размера частиц 499—500
-------температуры 501—502
-------электролитов 500. 702—703
-------pH 702—704
формирование микрогеля 313—
315
Гели. См также Адсорбция на крем-
неземе, Гидрогель, Силикагели ij
диспергирование в золь 455—458
коалесценция частиц 714, 730, 767
--------неполная 309—313
коммерческие 803—807
нагревание
— в вакууме 749—753
— в воде 747
— в парах 742—743, 744—747
— на воздухе 744, 747, 749—753
микрогель
— влияние на ультрафильтрацию
золей 465
— формирование 313—315
обработка гидротермальная 742—
749, 872
осаждение кремнезема 109—125
пептизация 453—454
пленки 556—557
плотность 316
получение
— гидролизом этилсиликата 44—
45
— из гидролизуемых соединений
кремния 706—708
— из золей 503—506
— из коллоидного кремнезема
704—706
— из растворимых силикатов 164
прочность, теория 502—506
Предметный указатель
1109
размер частиц
----влияние на агрегацию 445—
446
---------гелеобразование 499—
500
структура 316—317, 631
типы 632—633
усадка 503—505
Гидрогель 726—733
старение 728—730
— термическое 731—733
упрочнение 726—728
Гидролиз
кварца 57
коэсита 57
кристобалита 57
монокремневой кислоты 180, 285
мономерных соединений кремния
244—246
паров
— сложных эфиров 786
— SiCl4 784—786
— SiF6 786—788
силикатов щелочных металлов
124, 182—183
соединений кремния 706—708
стишовита 57
тридимита 57
этилсиликата 44—45
эфирных групп на поверхности
961—962
Гидрофобные поверхности см. По-
верхность кремнезема
Гиллиспит 35
Глицерин, адсорбция на кремнеземе
830
Голотурии 1018. См. также Кремне-
зем в биосфере
Гормоны стероидные, роль в .борьбе
с силикозом 1085
Грибы 1011. См. также Растения
Группы силанольные
определение 10
отличие от адсорбированной воды
866—870
Губки 1017. См. также Кремнезем
в биосфере
Дегидратация см. Поверхность крем-
незема
Дезоксирибонуклеиновая кислота
(ДНК) см. Кислоты нуклеиновые
Дерягина—Ландау—Фервея—Овербе-
ка теория 438, 519, 522, 535
/-Диаграмма см. Адсорбция
Диатомеи 1011—1017. См. также Во-
доросли
Диатомит 789
Диоксид серы, адсорбция на силика-
геле 904
Диоксид углерода как адсорбат при
определении удельной поверхности
646
Дисиликат-ионы 169—170
Дисиликат натрия 35
Диэлектрические измерения 650—651
Диэлектрические эффекты при ад-
сорбции 909
Дубление 399—400, 1055
Желатин, реакции с кремнеземом
394—395
Желатинирование 497
Железо
адсорбция ионов на кремнеземе
925, 929, 930, 931, 934
модифицирование поверхности
кремнезема 563—564
Жирные кислоты
адсорбция на кремнеземе 907
взаимодействие с кремнеземом
1053
как агенты флокуляции 541
Жиры, взаимодействие с кремнеземом
1053
Заболевания растений грибковые
1035—1037. См. также Растения
Заряды на поверхности 912, 919—921
обращение знака 930—931
частиц золей 482—491
--------природа 483—487
--------противоионы 487—491
Земноводные 1038—1039. См. также
Кремнезем в биосфере
Злаки 1025, 1030—1031, 1035. См.
также Растения
Золи. См. также Кислота монокрем-
невая, Кислоты поликремневые, Кол-
лоиды
«активированные» 401—404
анализ химический 467—468
влияние алюмосиликат-ионов на
свойства 561—563
вязкость 327—332, 441, 491—495
— до начала агрегации 327—328
гелеобразование 503—506
действие анитипенное 604,
замораживание 521
изодисперсные 553—554
1110
Предметный указатель
концентрация максимальная 438—
441
концентрированные 421—443
— размер частиц 421—430, 439
молекулярная масса, определение
471—472
обогащение 459—466
— выпариванием воды 459—466
— ультрафильтрацией 461—465
----влияние микрогеля 465
----ограничения метода 463—
465
— центрифугированием 460
— электродекантацией 465—466
оптические эффекты 605—606
органозоли 566—571
очистка 458—459
— диализом и электродиализом
458—459
— ионным обменом 458
поглощение света 472—474
плотность упаковки частиц 440
получение
— гидролизом соединений крем-
ния 454—455
— диспергированием пирогенного
кремнезема 455—458, 525
— из растворимых силикатов
164, 448. 449
— ионным обменом 426,430,451 —
453
— пептизацией гелей 453—454
— растворением элементарного
кремния 455
— способом «наращивания» 425,
427, 430
— электродиализом 449—451
применение 572, 577—608
— в качестве добавок 587
— в печатных формах 592
— в полировальных материалах
601—602
— в производстве адсорбентов
579—580
------загустителей 581—582,
604—605
------изложниц для литья 585—
587
------катализаторов 579—580
------моющих средств 603
------неорганических связую-
щих 581—582
------силикагелей 577
------формующихся изделий
582—584
------эмульсионных полимеров
602—604
— в фотографии 605—606
— для защиты от загрязнений
590—591
— для изменения адгезии 593—
596
— для композиционных покрытий
596—601
----модифицирования поверхно-
стей 593—596
----огнеупорных покрытий 584—
585, 597—598
----разделения биологических
материалов 606—608
----усиления трения 587—590
промывание 459
стабилизация 429—430, 431—443
— адсорбционные эффекты при
431—432
— влияние концентрации ионов
натрия 432—433, 440
----pH 432
— дисперсий 466
— механизм 436—437
— органическими ионами 438—
441
— путем образования ионных за-
рядов 567—571
—стерическая 441—442
— термодинамика 431—436
ультрацентрифугирование 475
характеристики, способы установ-
ления
— анализ химический 467
— измерение pH 468
— определение концентрации
электролита 468
— рассеяние рентгеновских лх’чей
474
частицы
— агрегация 495—498. См. также
Агрегация частиц
— заряд на поверхности 482—
491
---------природа 483—487
---------противоионы 487—491
— микропористость 446
— модифицированные поверхно-
сти 559—566
------отрицательно заряженные
560—564
------положительно заряжен-
ные 564—566
— обнаружение 556—557
— определение размера 469—478
— поверхность удельная 478—
482. См. также Поверхность
удельная
П редметный указатель
1111
— пористые 443—446, 492
— с некремнеземной сердцевиной
447
— распределение по размерам
475—476
— скорость растворения 481—482
— удлиненные 446
— характеристики 22, 469—495
этерификация 567—571. См. так-
же Этерификация кремнезема
Изложницы для литья металлов 585—
586
Иммунологическая теория см. Реак-
ции иммунные
Инсектицидное действие
оксида алюминия 1038
порошков кремнезема 829, 1037—
1038
Ионы металлов
адсорбция на кремнеземе 830,
922—935
влияние на коагуляцию кремне-
зема 766—775
----кристаллизацию силикаге-
лей 754—757
----растворимость SiO2 83—85,
114—115
высаливающее действие 184—186
комплексообразование с силикат-
ионами 213—215
модифицирование поверхности
кремнезема 563—564
реакции с монокремневой кисло-
той 259—265
----поликремневыми кислотами
404—405
Иттрий, адсорбция ионов на кремне-
земе 934
Иетса теория стабилизации системы
431—436
Кабосил (Cab—О—Sil) 457, 785, 947,
970
Каганера метод см. Адсорбция
Кадмий, адсорбция ионов на кремне-
земе 934
Калий, адсорбция ионов на кремне-
земе 931
Калия силикат
высаливание 184—186, 223—227
гидролиз 182—183
коллоидообразование 183
получение 157—160
растворы
— коммерческие 160—162
— полимеризация 183
— применение 163—166
— природа 167—176
— равновесия в. 170—176, 179—
180
Кальций, адсорбция ионов на крем-
неземе 931, 933
Каолин 25, 833
Капельный анализ см. Анализ хими-
ческий
Катехин 86
как ингибитор растворения SiO2
106
комплексы с кремнием 215—216
Кварц 29, 788
адсорбция белков 980
биогенное действие 1050—1051,
1067, 1068
гидролиз 57
образование зародышей НО—111
растворимость 49—52, 58—62
растворы водные, термодинамиче-
ские константы 17
поверхность удельная 58. См.
также Поверхность удельная
токсичность 1075—1080
Кварцевое стекло см. Стекла
Киселева метод см. Адсорбция
Кислота кремнемолибденовая
использование в анализе 135—141
получение 265—266
состав 273—275
структура 266—267
Кислота монокремневая 21—28, 243—
265
биогенное действие 1062
гидролиз 180, 285
ионизация 23, 73
классификация разновидностей
267
конденсация 43, 340, 861
константы диссоциации 248—252
летучесть в водяных парах 23
осаждение из воды 117—125
------скорость 120—125
пересыщенные растворы 14—15.
26, 117—119
полимеризация 13, 21, 340—341,
См. также Полимеризация
— влияние оксида алюминия 25
получение
— гидролизом мономерных со-
единений 244—246
1112
Предметный указатель
— из силикатов 187—188, 243—
244, 246—247
растворимость 21. См. также Рас-
творение кремнезема
реакции 23, 257—265
— с ионами металлов 259—265
----кислотами 258—259
---- молибденовой кислотой
188—190
триметилсилильные производные
192—194
устойчивость 257
характеристики 247—257
эфиры 191. См. также Этерифи-
кация кремнезема
Кислота фтороводородная
как катализатор растворения 91
-------полимеризации 287—289, 343
Кислоты нуклеиновые 1062—1066
Кислоты поликремневые 22
денатурация белков 1075
деполимеризация 268, 275
— в кислом растворе 300, 343
коагуляция 280—285. См. также
Коагуляция
комплексы с органическими со-
единениями 382—394
конденсация 295
— механизм 285—287
модели 293
определение размеров частиц
278—280
полимеризация, стадии 291—292
получение из силиката натрия
381—382
разновидности 267
— разделение 275—277
реакции с
-------альбумином 394—395, 1054
--------белками 397, 399—400, 1055
--------желатином 394—395
--------катионами металлов 404—
405
--------молибденовой кислотой 267—
273
--------целлюлозой 398—399
сополимеризация с органически-
ми соединениями 394—397
токсичность 1075—1080
трпметилсилильные производные
355
эфиры 191, 394, 400—401. См.
также Этерификация кремнезема
Китит 29
Клеи 163, 164—165, 819
Коагулянты 509—521
катионы двухзарядные 515—516
— многозарядные 516—521
— однозарядные 511—515
электролиты 510—511
Коагуляция 496, 497, 507—525, 765—
779
влияние ионов металлов 766—775
— концентрации кремнезема 521—
522
— размера частиц 522—525
крови 1056—1060
механизм 507—510
в присутствии
---ионов кальция 777
------ натрия 766—775
------натрия и аммония 775—
777
---органических веществ 778—
779
Коалесценция частиц см. Гели
Коацерваты 388—392, 542—544
с водородными связями 542—543
поливинилового спирта 543 ,
Коацервация 320—321, 497, 542—544,
835
формирование сферических ча-
стиц 544
Кобальт, адсорбция ионов на крем-
неземе 928—929, 930, 931, 933
Койке 1027. См. также Растения
Коллоидообразование, моделирование
303—308
Коллоиды. См. также Золи
биогенное действие 1048
гидрофильные 507
гидрофобные 507
деполимеризация 142—143, 477,
608
коагуляция 507—525
обнаружение на поверхностях
144, 556—557
осаждение из воды 118
------скорость 130—133
полимеризация 183—184
применение 577—608
— для закаливания металлов 611
--- приготовление синтетическо-
го кварца 609
------стекол 609
------цемента 610
реакционная способность 609—611
типы коммерческие 572—576
-токсичность 1075—1080
Колориметрические методы анализа
см. Анализ химический
Комплексы, адсорбция на кремнезе-
ме 951—952
Концентрация ионов водорода pH
Предметный указатель
1 113
' влияние на гелеобразование 241 —
242, 498—500, 702—712
--------полимеризацию 289—290,
298—300, 321—327, 337—381 ,
--------------------------размеры частиц кремнезема
709—712
-------------------------- флокуляцию 526
измерение 468
Концентрирование для анализа 141 —
142
Коэсит 29
биогенное действие 1051
гидролиз 57
поверхность удельная 58. См. так-
же Поверхность удельная
растворение, скорость 93
Крапива 1026—1027. См. также Ра-
стения
Красители
адсорбция на кремнеземе 479—
480, 693, 761—764, 950—951
как адсорбаты для определения
удельной поверхности 646
Кремень 1017
Кремневая кислота см. Кислота мо-
нокремневая, Кислоты поликремне-
вые
Кремнезем «активный» 421—428
Кремнезем аморфный 38—48
биогенное действие 1072—1074.
См. также Кремнезем в биосфере
биогенные формы 47—48, 788
гидратированный 39, 44—47
микроформы 38—39, 40, 113
осаждение из воды 118
пирогенный 42. См. также Крем-
незем пирогенный
растворимость в воде 13—14,62 —
65, 69—71
---------- влияние электролитов 68
----------примесей 82—88, 113
----------размера частиц 74—82
----------в гидротермальных усло-
виях 68—69
— в морской воде 26, 82
— в перхлорате натрия 66—68
— в расплавленных солях 90
— в спиртах 89—90
— в щелочных средах 71—73
растворы водные, термодинами-
ческие функции 17
токсичность 1072—1073, 1078—
1079
частицы
— зависимость состава от разме-
ра 17
— содержание ОН-групп 18
Кремнезем в биосфере
биогенное действие 1046—1052
роль в жизнедеятельности
--------микроорганизмов 1090
--------млекопитающих 1039—
1089
-------- птиц 1044
--------пресмыкающихся 1038—
1039
--------растений 1028—1031
--------рыб 1038—1039
--------человека Ю39—1089
— в метаболизме фосфата 1062—
1063
содержание в крови 1041—1044,
1052, 1064, 1065, 1068, 1075—1080'
--------моче 1042, 1047
-------- почвах 1031—1035
--------растениях 1020—1031
--------тканях животных 1041, 1044,
1048, 1052
соединения биохимические
--------выделение из тканей 1052
--------с белками 1054—1056
--------жирами 1053
--------полисахаридами 1053—
1054
--------сахарами 1060
--------ферментами 1060—1062
токсичность 1046—1049, 1068
цитотоксичность 1049—1052, 1070
Кремнезем кристаллический
адсорбция Si (ОН) 4 54—62
биогенное действие 1072—1073.
См. также Кремнезем в биосфере
модификации 28—38
— гидратированные 34—38
растворимость в воде 14, 53—54
----------влияние примесей 82—88
----------размера частиц 74—79
Кремнезем лепидоидальный 37, 760
Кремнезем пирогенный 683, 970, 989
адсорбция полимеров 981—982
гидрофобизация 797—799
дегидроксилирование поверхности
915—921
диспергирование 455—458
микропоры в частицах 871
поверхность частиц 873
получение 781
— гидролизом паров SiF6 786—
788
— испарением SiO2 782—783
— окислением пара SiO 783—784
— окислением и гидролизом па-
ров SiCl4 784—786
---------------сложных эфиров 786
1114
П редметный указатель
реакции с
----органическими группами
963—964
----ферментами 1060—1061
титрование поверхности 914—915
Кремнезем М 38
Кремнезем О 32
Кремнезем W 30—31
Кремнезем X 32
Кремний в растениях 1019—1020,
1035—1037
упрочнение стеблей 1022—1029
Кремнийорганические соединения
1090—1092
Кристобалит 29, 754
гидролиз 57
поверхность удельная 58. См.
также Поверхность удельная
растворение, скорость 93
растворимость 53
токсичность 1072—1073
цитотоксичность 1051
Кровь
гемолиз 1056—1060
содержание кремнезема 1041 —
1044, 1052, 1064, 1065, 1068,
1075—1080
Ксерогель 633, 733—738, 755. См.
также Гель
Лантан, адсорбция ионов на кремне-
земе 930, 934
Лепидоидальный кремнезем см. Крем-
незем лепидоидальный
Литийорганические соединения 967
Лития силикат 133, 200—201, 205—
206
Лишайники 1009, 1011. См также Ра-
стения
Лю доке-AM 539, 561, 607
Людокс-HS 561, 607, 654
Магадиит 36—37, 219—220
Магний, адсорбция ионов па кремне-
земе 931, 933
Магния оксид 115
Магния силикат, получение 610—611
Макрофаги 1049—1050, 1067, 1070—
1072, 1076
Марганец, адсорбция ионов на крем-
неземе 933
Медь, адсорбция ионов на кремне-
земе 931, 933
Меланофлогит 31
Метод БЭТ см. Адсорбция
Метод непрерывного потока см. Ад-
сорбция
Микроорганизмы 1090
ископаемые 1007—1008
содержание кремнезема 1010,
1016
Минералы кремнеземные 27, 48, 126,
192, 219—220, 699, 1018
разрушение биологическое 1009—
1010
Модифицирование поверхности крем-
незема
алюминат-ионами 560—561, 951,
986—988
анионами 563—564
золями 559—566, 593—596
Моллюски 1018. См. также Кремне-
зем в биосфере
Мономер см. Кислота монокремневая
Морская вода 82, 111
содержание кремнезема 25, 1012
Насекомые 1037—1038. См. также
Кремнезе.м в биосфере
Натрий, адсорбция ионов на кремне-
земе 931
Натрия силикат 156—196
высаливание 184—186, 223—227
гидролиз 124, 182—183
коагуляция 779
коллоидообразование 183
кристаллический 35
получение осажденного кремне-
зема из 765—766
— триметилсилильных производ-
ных 192—194
приготовление 157—160
растворение в расплаве Na2SO4X
ХЮН2О 181
растворы
— коммерческие 160—162
— полимеризация 183
— применение 163—166
— природа 167—176
— равновесия в 170—176, 179—
182
— свойства 162—163, 176—182
Никель, адсорбция ионов на кремне-
земе 933
Ниобий, адсорбция ионов на кремне-
земе 830, 935
Нуклеации теория 295—298
Обогащение золей см. Золи
Обработка
Предметный указатель
1115
воды 401—404
гидротермальная 68—69, 742—
749, 872
Образование центров кристаллизации
льда 972—973 ‘
Озон, адсорбция на силикагеле 830,
991
N-Оксиды 87
Олигомеры 290—295, 362—368, 1061.
См. также Поликремневые кислоты
Олово, адсорбция ионов на кремне-
земе 934
Опал 545—555
генезис 546—547
дифракция света 548—550, 553
размер частиц 550
синтез ‘554—555
структура 545—551
фиброгенное действие 1068
Органические соединения см. также
Поверхностно-активные вещества
адсорбция на кремнеземе 941 —
948
влияние на растворимость крем-
незема 85—88
водородносвязанные 382—394
комплексообразование с кремни-
ем 215—218
Осаждение кремнезема 109—125
биогенное 118
из воды 109—111. 117—125
гидроксидами 112—115 '
.механизм ПО, 117—120, 768—770
этапы 768
Очистка золей см. Золи
Палладий, адсорбция ионов на крем-
неземе 934
Пептизация см. Гели
Пероксисиликаты 228—229
Персорбция 671—672
«Пирит блокированный» 77
Плавиковая кислота см. Кислота фто-
роводородная
Плутоний, адсорбция ионов на крем-
неземе 925, 935
Пневмокониоз 1066—1086
узелковые формы 1073
Поверхностно-активные вещества
(ПАВ) 1076, 1084—1085
адсорбция на кремнеземе 941
гидрофобпзация поверхности
кре.мнезема 949
как агенты флокуляции 529—534
как адсорбаты для определения
удельной поверхности 647
отделение кре.мнезема из руд 949
роль в борьбе с силикозом 1084—
1085
Поверхность алюмосиликатная 986—
988
анионные центры 986
Поверхность кремнеземная
адсорбция ионов металлов на
924—926, 933—935
гидроксилированная 860—878
— концентрация ОН-групп 868,
870—878
------зависимость от темпера-
туры прокаливания 874
------теоретическая величина
877—878
— плотность зарядов 914—917
— поведение воды на 863—865,
866—870, 901
— силанольные группы 866—870
------реакции 867
— точка изоэлектрическая 913
— точка нулевого заряда 912
— электропроводность 865—866
гидрофильная, получение 591—
593
гидрофобная 940—973
— адсорбция органических осно-
ваний 941—953
— дегидроксилирование 915—918
-- механизм 884
дегидратация 878—889, 899
— влияние размеров частиц и
диаметров пор 885—889
— кристаллического кварца 884—
885
— скорость 880
— число ОН-rpvnn на 1 нм2 при
880—881
дегидроксилированная
— влияние на адсорбцию 898—
891
— реакция со спиртами 958—960.
См. также Этерификация кремне-
зема
------углеводородами 959—960
заряды на 919—921
— обращение знака 930—931
ионообменная 799—803, 836—837
модифицированная 836—837
— алюмосиликат-ионами 560—561,
951, 986—988
— анионами 563—564
— многозарядными катионами 794
— органическими катионами и
анионами 792—794
природа 859—878
радикалы свободные на 988—992
1116
Предметный указатель
реакции
— механизмы 937—940
— неионные 936
— с органическими группами
962—968
регидратация 878—880, 889—891,
899
смачивание 892—895
— теплота 892—893, 972
структура подповерхностного
слоя 859—860
теплота гидроксилирования 891
центры активные 988—992
— анионные 915, 918—921
энергетические характеристики
20, 891—895
этерифицированная 794—797. См.
также Этерификация кремнезема
— адсорбция полимеров 981—982
— устойчивость 961—962
Поверхность удельная, определение
адсорбаты
— азот 638—645
— аммиак 645
— бутан и изобутан 645
— вода, пары 645—646
— диоксид углерода 646
— катионные ПАВ 647
— красители 647, 648
— моноксид азота 645
— стеариновая кислота 647
— тетрахлорид углерода 646
— фенол 647
— фуран 645
методы
— адсорбции азота 479
— адсорбции красителей 479—480
— диэлектрических измерений
650—651
— измерений числа ОН-групп
652—653
---проницаемости газа 652
— расчета по величине частиц
470—471
— ртутной порозиметрии 652
— Синга а-метод 478—482
— Сирса 480—481
— титрования щелочью 649
— хроматографические 649—650
стандартные вещества 637—650
Подложки 1089—1090
для митохондрий 831
Покрытия
гидрофильные 591—592, 936
кремнеземные
— композиционные 596—597
— для контакта с морской водой
600—601
— незагрязняющиеся 590—591
— на оксиде тория 124
— противокоррозионные 205—206
— термостойкие 597—598
— электропроводящие 599
кремнийорганические 797—799
полимерные 799
частиц кремнезема
----алюмосиликатными ионами
560—563
----анионами 563—564
----катионами органическими
566
----оксидами поливалентных ме-
таллов 564—566
----силильные 571
Поливиниловый спирт, адсорбция на
кремнеземе 543, 976—979
Поли(винилпиридин-М-оксид), роль
в борьбе с силикозом 1080—1084
Поливинилпирролидон, роль в борьбе
с силикозом 1081
Полимеризация 14, 15
влияние концентрации 337, 358—
361, 367, 368, 374, 375, 379—380
— температуры 380—381
— фторид-ионов 289, 343
— электролитов 357—358
— pH 362—368, 373—374, 379-
380
механизм 348, 350—352, 361—362,
364—368, 369—372
мономеров органических на по-
верхности кремнезема 990
при pH 2—7 289—290, 298—300,
337—357
при pH 7 357—372
при рН>7 321—327, 372—381
размер частиц при 339—340, 344
растворов силикатов 182 — 184
----влияние солей 184—186
рост частиц при 320—327, 358,
372—373
скорость 338, 341—343. 352—357,
375—379
стадии 238—241
теория 237—241
тепловые эффекты 332—335
в тканях организмов 1088—1089
эмульсионная 397—398, 722—724
Полимеры органические, адсорбция
на кремнеземе 973—985
Полиметилметакрилат, адсорбция на
кремнеземе 984
Полисахариды, взаимодействие с крем-
неземом 1053—1054
Предметный указатель
1117
Полисиликаты 196—206
кристаллические 218—223
в растворах силикатов 172—176,
197—206
Полиэтиленоксид, адсорбция на крем-
неземе 976
Порозиметрия ртутная см. Поры в ча-
стицах силикагелей
Порошки кремнеземные 633—637
агрегаты частиц
— определение размеров 654—656
— прочность 694—698
-----методы исследования 695—
697
--- механическая 697—698
— характеристики пор 656—663
-----по изотермам адсорбции
669—691
гидрофобные 790—799
— токсичность 1049
микрокристаллические 790
органофильные 634
осажденные 633—634, 765—788
— в присутствии солей Са2+ 777
---------Na+ 766—775
---------Na+ и NH4+ 775—777
---------органических веществ
778—779
— из золей 781
— из органических жидкостей 781
— из паровой фазы 781—788
— из раствора силиката натрия
765—766
— из фторидных растворов 779—
781
— упрочнение агрегатов 770—775
применения 803, 808—831
— в антипенных присадках 828
косметических препаратах
825—826, 831
консистентных смазках 823—
825
---- красках 825
----- органических полимерах
816—818
----- силиконовых эластомерах
815—816
----синтезах 832—833
-----фармацевтических препара-
тах 825—826
— для армирования и упрочнения
резины 808—815, 817
----дождеобразования 833—834
----изменения адгезии 818—819
--- матирования 827
---наполнения хроматографи-
ческих колонок 834—837
----повышения вязкости 819—
827
----создания гидрофобных эф-
фектов 828—831
----стабилизации эмульсий 827—
828
---- удаления пятен 829—830
— разнообразные 826—827, 829—
831
в природных условиях 788—790
размер первичных частиц 635
свойства
— влияние алюмосиликатных ио-
нов 634
— инсектицидные 829, 1037—1038
— каталитические 831—832
типы коммерческие 803—807
тонкодисперсные
— биогенное действие 1066
— цитотоксичность 1049—1052
частицы
— определение размеров 635—636,
653—654
----удельной поверхности 636—
654. См. также Поверхность удель-
ная
Поры в частицах силикагелей
измерение объема 670, 674—675
-------по объему азота 674—675
-------по поглощению масла 675
мезопоры 683
метод «модельных пор» 683—685
микропоры 681—683, 747—748
— эффекты в них 691
определение размеров 685—686
поверхность пор
-------типы 691—692
----методы исследования 692—
694
порозиметрия ртутная 690—691
распределение по размерам 669,
672, 675—681
субмикропоры 681, 686—687,
758—760
супермикропоры 686
ультрамикропоры 686—687
характеристика по изотермам ад-
сорбции 669—691
Почвы 1031 —1035. См. также Крем-
незем в биосфере
Происхождение жизни 1006—1007
Протактиний, адсорбция ионов на
кремнеземе 935
Птицы 1038—1039. См. также Крем-
незем в биосфере
1118
Предметный указатель
Рак 1063—1066
желудка 1049
Рассеивание облаков 972—973
Растворение кремнезема
влияние размера частиц 95—104
— солей 104—105
— pH 94—95
ингибиторы 105—106
кинетика 90—108
нестабильность пересыщенных
растворов 15
Растворимость кремнезема
аморфного 13—14, 62—65, 69—71
— в морской воде 26, 82
влияние оксида алюминия 82—84,
ИЗ
— ионов металлов 83—85, 114—
115
—фтороводородной кислоты 91
кварца 49—52, 58—62
кварцевого стекла 15
кристаллического кремнезема 14,
53—54
кристобалита 53, 93
монокремневой кислоты 21
стишовита 93
тридимита 52—53
Растения
метаболизм, роль кремнезема
в 1019—1020
поглощение и перенос кремнезе-
ма 1028—1031
отложения кремнеземные, приро-
да 1020—1022
содержание кремнезема 1020—
1031
сопротивляемость грибковым за-
болеваниям 1035—1037
упрочнение стеблей и листьев
1022—1029
Реакции иммунные 1069
Резина, армирование и упрочнение
кремнеземом 808—815, 817
Рибонуклеиновая кислота (РНК) см.
Кислоты нуклеиновые
Родентициды 1090—1091
Рубидий, адсорбция ионов на кремне-
земе 931
Рыбы 1038—1039. См. также Крем-
незем в биосфере
Сахара, взаимодействие с кремнезе-
мом 1060
Свинец, адсорбция ионов на кремне-
земе 934
Связи водородные
в коацерватах 542—543
образование при адсорбции 896—
897, 902—903, 909—912
Серебро, адсорбция ионов на крем-
неземе 933
Силатраны 1090—1091
Силикагели
агрегаты частиц см. Порошки
кремнеземные
адсорбция
— антиокислителей 830
— водорода 991
— глицерина 830
— ионов ниобия 830
урана 830
циркония 830
— красителей 761—764
— озона 830, 991
— ферментов 831, 1060—1062
анализ химический 693—694
высушивание
— без усадки 739—742
— усадка при 733—737
— условия 725
гранулы, формование 721—726
дегидратация 886—887
ионообменные поверхности в 799—
803
как культуральная среда 1089—
1090
кристаллизация 754—758
мезопористые 636
порошкообразные 633
— с шаровидными частицами 633
приготовление
— гидролизом кремнесодержащих
соединений 706—708
— из золей 577, 579—580
----коллоидного кремнезема
704—706
----минералов 699—704
----растворимых силикатов 699—
704
применение 803, 805. См. также
Порошки кремнеземные
прочность 712—717
размер первичных частиц 635
расстекловывание 754—758
свойства адсорбционные 964
спекание 753—754. 757
субмикропористые 758—760
типы коммерческие 803—807
характеристики физические 634—
635
частицы
—плотность упаковки 717—719
— пористость повышенная 720—
721
Предметный указатель
1119
— потери площади поверхности
663—669
— размеры 719—720
----влияние pH на 709—712
----определение 635—636, 653—
654
—удельная поверхность 636—654
этерпфицированные 971. См. так-
же Этерификация кремнезема
Силикалит 33
Силикат .листоподобный 36
Силикаты растворимые см. Калия си-
ликат, Натрия силикат
Силикоз 1047, 1051, 1060, 1064, 1066—
1086
влияние типа кремнезема на
1072—1073
восприимчивость к 1074
вещества, тормозящие развитие
1080—1086
механизм возникновения 1066—
1072
роль оксида алюминия 1085—
1086
Синга a-метод см. Адсорбция
Сирса метод см. Поверхность удель-
ная
Система кремнезе.м—вода 12—21
взаимосвязь размера и состава
частиц 17—19
зависимость энергии от размера
и состава частиц 19—21
модели 12—13
природа 14
термодинамические характеристи-
ки 16
Спирты
адсорбция на кремнеземе 903, 906,
956
в реакциях этерификации 809—
811, 814—815, 953—962
Средства моющие 163, 603
Стекла
кварцевые 29, 609, 788
— гидролиз 57
— гидрофильность 952
— гидрофобизация 949
— растворимость 15
— реакции с органическими ве-
ществами 988—989
— содержание воды в 16
— токсичность 1072—1073
— цитотоксичность 1051
пористые 633, 760—761
растворимые 227—228
Стишовит 21, 29
биогенное действие 1069
гидролиз 57
растворение, скорость 93
токсичность 1072—1073
удельная поверхность 58
цитотоксичность 1051
Стронций, адсорбция иснсв на крем-
неземе 931, 933
Структура гелей 316—317, 631
спала 515—555
Сурьма, адсорбция ионов на кремне-
земе 934
Табашир 1024—1025, 1039
Таллий, адсорбция ионов на кремне-
земе 933
Тантал, адсорбция ионов на кремне-
земе 935
Таумасит 21
Терминология 10, 495—497
Термодинамические функции 17, 19—
20, 332—335, 431—436,’ 651—652, 692—
693, 891—895, 972
Тетрасиликат, получение 218, 222. См.
также Полисиликаты
Титрование поверхности см. Поверх-
ность удельная
Ткани растений и животных 26—27,
88
анализ 140—141
окремнение 125—130
содержание кремнезема 1020—
1031
Торий, адсорбция ионов на кремне-
земе 925, 930, 935
Тридимит 12, 29
гидролиз 57
растворение, скорость 93
растворимость 52—53
токсичность 1072—1073
—цитотоксичность 1051
Туберкулез 1039—1040, 1066—1067
Углеводороды ароматические, адсорб-
ция на кремнеземе 907—909
Удаление кремнезема из воды 109,
115—117
Ультрафильтрация см. Золи
Уран, адсорбция ионов на кремнезе-
ме 830, 925, 929, 935
Фагоциты см. .Макрофаги
Ферменты, адсорбция и взаимодейст-
вие с кремнеземом 831, 1060—1062
Фиброгенез 1066
1120
П редметный указатель
Фиброз 1070—1073, 1078—1079
Филлокремнезем 634
Флокуляция 75, 132, 236, 496, 525—
542
агенты 526
— жирные кислоты 541
— известь 541
— катионные ПАВ 529—534
— полиакриламид 540
— полимеры 534—537, 537—541
влияние pH на 526
механизм 526—529, 917
в органических жидкостях 541
скорость 521
Флотация 952
Фосфор, обмен с кремнием 1010,
1062—1066
Фторид-ион
влияние на
-------осаждение кремнезема 779—
781
----растворимость SiO2 85, 91—
93, 107—108
как катализатор полимеризации
275, 289, 343
при приготовлении кремнеземных
порошков 779—781
Френкеля—Хелей—Хилли метод см.
Адсорбция
Хвощи 1022—1024. См. также Расте-
ния
Холестерин, адсорбция на кремнезе-
ме 906
Хризотил-асбест 36, 954, 1049, 1083
Хром, адсорбция ионов на кремнезе-
ме 929, 931, 934, 935
Хроматографические методы
ионный обмен 115—117, 426, 430,
451, 453, 458
определения удельной поверхно-
сти 649—650, 834—837
Целлюлоза, реакции с поликремневы-
ми кислотами 398—399
Центры на поверхности 800—803, 915,
918—921, 986—992
Цинк, адсорбция ионов на кремнезе-
ме 928, 930, 931, 933
Цирконий, адсорбция ионов на крем-
неземе 830, 930, 934
Шерт 125, 1007—1009
Эластомеры силиконовые 815—816
Эмульсии, стабилизация 827—828
Эстерсилы 809
Этерификация кремнезема 809—811,
814—815, 953—962
в биохимических соединениях
1053
гидролиз эфирных групп 961—962
дегидроксилированной поверхно-
сти 958—960
кинетика 957
метоксигруппами 957—958
в микропорах 960
силикагелей 971
степень покрытия поверхности
953—956
токсичность эфиров 1078
условия проведения 956—957
Этилсиликат, гидролиз для получе-
ния гелей 44—45
СОДЕРЖАНИЕ
Глава 4. Коллоидный кремнезем — концентрированные золи.......... 421
Определение коллоидного кремнезема. Историческая справка . . . 421
Рост и стабилизация дискретных частиц......................... 422
Увеличение размера частиц при добавлении «активного» крем-
незема ..................................................... 423
Способы приготовления частиц размером менее 10 нм.......... 428
Стабилизация системы для предотвращения роста частиц (тео-
рия Пауля Йетса)........................................... 431
Стабилизация для предотвращения агрегации................... 436
Стабилизация путем образования ионных зарядов............. 438
Добавление соли для понижения вязкости.................... 441
Стерическая стабилизация.................................. 441
Пористые частицы............................................ 443
Удлиненные частицы.......................................... 446
Частицы с некремнеземной сердцевиной........................ 447
Методы приготовления золей.................................... 447
Нейтрализация растворимых силикатов кислотами............. 448
Электродиализ ............................................ 449
Ионный обмен.............................................. 451
Пептизация гелей.......................................... 453
Гидролиз соединений кремния............................... 454
Растворение элементарного кремния......................... 455
Диспергирование пирогенного кремнезема...................• 455
Очистка, обогащение, стабилизация дисперсий................... 458
Ионный обмен................................................ 458
Диализ и электродиализ ..................................... 458
Промывание.................................................. 459
Обогащение.................................................. 459
Выпаривание воды.......................................... 459
Центрифугирование......................................... 460
Ультрафильтрация.......................................... 460
Электродекантация ........................................ 465
Стабилизация дисперсий ..................................... 466
Характеристики золей.......................................... 466
Химический анализ.......................................... 467
Измерение pH.............................................. 468
Концентрация электролита.................................. 468
Характеристики частиц ...................................... 469
Размер частиц............................................. 469
Удельная поверхность...................................... 478
Ионный заряд на поверхности частиц.......................... 482
Природа ионного заряда . ................................. 483
Противоионы и двойной электрический слой.................. 487
Вязкость.................................................... 491
Агрегация частиц.............................................. 495
Определения................................................. 495
Гелеобразование ............................................ 498
45 Заказ № 250
1122
Содержание
Влияние величины pH.....................................
Влияние размеров частиц и концентрации SiOz ............
Электролиты и органические жидкости.....................
Температура ............................................
Теория прочности гелей .................................
Коагуляция...............................................
Механизм коагуляции....................................
Коагуляция под действием электролитов .................
Однозарядные катионы как агенты, формирующие мостиковую
связь ..................................................
Коагуляция под действием двухзарядных катионов металлов
Коагуляция под действием многозарядных катионов основных
солей металла ..........................................
Влияние концентрации кремнезема и других факторов . . . .
Влияние размера частиц..................................
Частично дегидратированная поверхность............ , . .
Флокуляция...............................................
Флокуляция под действием катионных поверхностно-активных
веществ.................................................
Флокуляция под действием органических полимеров........
Коацервация ..............................................
Формирование сферического кремнезема под действием коа-
цервации ...............................................
Агрегация упорядоченных структур. «Благородный опал» . , . .
Структура опала ........................................
Другие агрегаты с упорядоченной структурой..............
Образование однородных по размерам неорганических частиц
Синтез опала ...........................................
Адсорбция частиц кремнезема на поверхностях .............
Золи кремнезема, содержащие частицы с модифицированными по-
верхностями ...............................................
Отрицательно заряженные поверхности......................
Алюмосиликатные ионы....................................
Другие анионы...........................................
Положительно заряженные частицы..........................
Покрытия оксидами поливалентных металлов................
Многозарядные органические катионы......................
Золи с модифицированными поверхностями — органозоли ....
Органические ионы.........................................
Этерификация............................................
Образование силильного покрытия.........................
Коммерческие типы коллоидного кремнезема...................
Применение коллоидного кремнезема..........................
Приготовление катализаторов, силикагелей, адсорбентов . . . .
Неорганические связующие и загущающие вещества...........
Формующиеся тугоплавкие изделия.........................
Связующие для волокон...................................
Огнеупорные покрытия....................................
Изложницы для литья металлов............................
Добавки в различные материалы...........................
Эффекты трения...........................................
Волокна ................................................
Бумага..................................................
Стальные рельсы.........................................
Другие поверхности......................................
Незагрязняющиеся поверхности.............................
Получение гидрофильных поверхностей......................
498
499
500
501
502
507
507
510
511
515
516
521
522
525
525
529
534
542
544
545
545
551
553
554
555
559
560
560
563
564
564
566
566
566
567
571
572
572
579
581
582
584
584
585
587
587
588
589
590
590
590
591
Содержание 1123
Модифицирование адгезионных поверхностей..................... 593
Усиление адгезии........................................... 593
Понижение адгезии.......................................... 595
Композиционные покрытия...................................... 596
Покрытия на кораблях, танкерах............................. 600
Армирование органических полимеров........................... 601
Полировальный реагент для кремниевых пластин................. 601
Воздействие ПАВ.............................................. 602
Эффекты диспергирования.................................... 602
Антипенное действие ....................................... 604
Модифицирование вязкости путем гелеобразования............... 604
Различные оптические эффекты, цветообразование, фотография 605
Применение в биологических исследованиях, измерение гради-
ента плотности............................................... 606
Источники получения химически реакционноспособного кремне-
зема ........................................................ 608
Растворимые силикаты....................................... 608
Изделия из кремнезема...................................... 608
Составы стекол............................................. 609
Формирование твердых силикатов — цементы................... 610
Другие реакции и их практическое применение................ 610
Коллоидные силикаты .......................................... 611
Литература . . . •............................................ 611
Глава 5. Силикагели и порошки..................................... 631
Определения.................;................................ 631
Типы гелей................................................... 632
Типы порошков................................................ 633
Физические характеристики............................, . . . . 634
Размер первичных частиц..................................... 635
Электронно-микроскопические снимки........................ 635
Определение удельной поверхности ......................... 636
Малоугловое рассеяние рентгеновских лучей................. 653
Размеры агрегатов — частиц порошка, гранул геля............. 654
Характеристики пор .......................................... 656
Размер частиц и упаковка.................................. 658
Потери площади поверхности частиц вследствие их упаковки 663
Характеристики пор по изотермам адсорбции................... 669
Объем пор................................................. 674
Размер пор и распределение пор по размерам............... 675
Различные эффекты в микропорах............................ 691
Природа поверхности кремнезема.............................. 691
Прочность агрегатов — связь между частицами кремнезема . . 694
Электронно-микроскопические снимки........................ 695
Метод неполного растворения............................... 695
.Механическая прочность агрегатов......................... 697
Силикагели.................................................... 698
Исходные материалы для получения силикагелей................ 699
Получение из растворимых силикатов и минералов............ 699
Получение из коллоидного кремнезема....................... 704
Получение гидролизом соединений, содержащих кремний . . . 706
Факторы, определяющие характеристики силикагелей............ 708
Влияние величины pH на размер первичных частиц............ 709
Прочность влажных гелей................................... 712
Размер частиц и плотность упаковки в высушенных гелях . . 717
Повышенное значение пористости при удалении наполнителей 720
Формование гранул силикагеля................................ 721
45*
1124
Содержание
Обработка гидрогелей........................................ 726
Упрочнение геля............................................. 726
Усадка при высушивании, получение ксерогелей................ 733
Высушивание с использованием жидкостей с низкой величи-
ной поверхностного натяжения.............................. 737
Высушивание без усадки, получение аэрогелей................. 739
Гидротермальная обработка................................... 742
Жидкая или паровая фаза................................... 744
Нагревание на воздухе и в вакууме........................... 749
Спекание однородных структур.............................. 753
Щелочные металлы, загрязнения и кристаллизация............ 754
Силикагели с нестандартными структурами..................... 758
Субмикропористые силикагели — непроницаемый кремнезем . . 758
Пористые стекла........................................... 760
Специфические адсорбенты.................................. 761
Осажденные кремнеземные порошки............................... 765
Кремнезем, осажденный из раствора силиката иатрия ..... 765
Коагуляция кремнезема в присутствии ионов натрия.......... 766
Коагуляция солями натрия или аммония...................... 775
Коагуляция под действием ионов кальция.................... 777
Коагуляция под действием органических веществ............. 778
Осаждение кремнезема из фторидного раствора................. 779
Осаждение кремнезема из органических жидкостей.............. 781
Осаждение кремнезема из коллоидных золей.................... 781
Осаждение кремнезема из паровой фазы. Пирогенный кремне-
зем ........................................................ 781
Испарение SiOa............................................ 782
Окисление пара SiO........................................ 783
Окисление и гидролиз паров SiCU........................... 784
Окисление и гидролиз паров сложных эфиров кремния .... 786
Гидролиз паров SiF«....................................... 786
Кремнеземные порошки в природных условиях..................... 788
Микрокристаллические гидратированные типы кремнезема .... 790
Гидрофобные, органофильные кремнеземные порошки............... 790
Поверхности с адсорбированными органическими катионами . . 792
Поверхности с адсорбированными многозарядными катионами
металлов и органическими анионами........................... 794
Этерификация поверхности.................................... 794
Кремнийорганические покрытия................................ 797
Органические полимерные покрытия............................ 799
Силикагели с ионообменными' поверхностями................... 799
Неорганические ионообменные центры.......................... 800
Органические связанные ионообменные центры.................. 801
Коммерческие силикагели и кремнеземные порошки................ 803
Применение силикагелей и кремнеземных порошков............... 803
Упрочняющие и другие воздействия в органических твердых ве-
ществах .................................................... 808
Резина.................................................... 808
Силиконовые эластомеры.................................... 815
Разнообразные органические полимеры....................... 816
Пониженная адгезия.......................................... 818
Повышенная адгезия.......................................... 819
Повышенная вязкость, тиксотропия............................ 819
Механизм действия......................................... 820
Консистентная смазка...................................... 823
Краски, грунтовки, чернила................................ 825
Фармацевтические и косметические препараты................ 825
Содержание 1125
Разнообразные композиции................................ 826
Оптические эффекты, матирование........................... 827
Эффекты, связанные с изменением состояния поверхности . *. . 827 .
Стабилизация эмульсии.................,................. 827«1
Гидрофильная поверхность................................... 828
Антипенная присадка ....................................... 828
Гидрофобные эффекты.......................................... 828
«Сухая» вода............................................... 829
Поглотитель ............................................... 829
Катализаторы................................................. 831
Аэрогели................................................... 831
Подложка для митохондрий................................... 831
Мигрирование атомов водорода............................... 832
Реакционноспособный кремнезем................................ 832
Образование зародышей в облаках.............................. 833
Наполнение хроматографических колонок........................ 834
Литература..................................................... 837
Глава 6. Химия поверхности кремнезема............................. 856
Обзоры и основные выводы....................................... 857
Природа поверхности кремнезема................................. 859
Структура подповерхностного слоя кремнезема.................. 859
Определение величины поверхности............................. 860
Гидроксилированная поверхность .............................. 860
Вода на гидроксилированной поверхности..................... 863
Электропроводность поверхности ............................ 865
Отличие адсорбированной воды от силанольных групп .... 866
Гидроксильные группы и молекулы воды внутри частиц крем-
незема ................................................... 870
Содержание гидроксильных групп на одном квадратном нано-
метре ..........................,....................... 872
Теоретическая величина концентрации поверхностных гидро-
ксильных групп............................................ 877
Дегидратация и регидратация.................................. 878
Дегидратация............................................... 880
Регидратация .............................................. 889
Поверхностная энергия........................................ 891
Теплота смачивания поверхности кремнезема.................... 892
Физическая адсорбция неионных соединений с низкой молекуляр-
ной массой.................................................... 895
Адсорбция паров ............................................. 895
Влияние дегидроксилирования поверхности на адсорбцию . . 898
Адсорбция из растворов в неионных системах................... 905
Неводные растворы.......................................... 905
Водные растворы, неионные системы, водородная связь . . . 909
Ионизация и поверхностный заряд................................ 912
Гидроксилированная поверхность............................... 912
Дегидроксилированная поверхность............................. 915
Природа анионных заряженных центров.......................... 918
Теория «связывания центров»................................ 919
Силы, обусловливающие адсорбцию ионов........................ 921
Однозарядные катионы: ионы металлов и низших аминов . . . 922
Катионы щелочноземельных металлов и магния................. 924
Многозарядные катионы металлов............................. 924
Непонные реакции на поверхности кремнезема..................... 936
Гидрофильные покрытия на кремнеземе............................ 936
Гидрофобная поверхность кремнезема............................. 940
1126
Содержание
Органические катионы и основания....................... 941
Эффекты гидрофобизации.................................... 949
Катионные красители................................... 950
Алюмосиликатные поверхности............................... 951
Углеводородные группы, присоединяемые посредством полива-
лентных металлов ........................................... 951
Образование эфиров на поверхности кремнезема............ 953
Покрытие поверхности.................................. 953
Условия проведения реакций................................ 956
Этерифицированный метоксигруппами кремнезем........... 957
Реакция спиртов с дегидроксилированной поверхностью . . . 958
Реакция углеводородов с дегидроксилированной поверхностью 959
Этерификация в микропорах............................. 960
Замещенные спирты..................................... 960
Гидролиз эфирных групп................................ 961
Органические группы, присоединенные посредством связей С—Si 962
Адсорбция на гидрофобных поверхностях................... 968
Адсорбция воды .... ....................................... 969
Адсорбция инертных газов.............................. 973
Адсорбция органических полимеров на поверхности кремнезема 973
Адсорбция из водного раствора........................... 975
Полиэтиленоксид....................................... 976
Поливиниловый спирт......................................' 976
Катионные полимеры..............-.......................... 979
Белки...................................................... 980
Адсорбция полимеров на дегидроксилированных кремнеземах 981
Действие солей ............................................ 982
Адсорбция из неводных растворителей........................ 983
Осаждение полислоев, образуемых из полиионов и заряженных
частиц........................................................ 985
Алюмосиликатная поверхность................................... 986
Активные центры, свободные радикалы, активный кислород, озон 988
Литература.................................................... 992
Глава 7. Кремнезем в биосфере.................................... 1005
Введение..................................................... 1005
Происхождение жизни........................................ 1006
Наиболее ранние биологические формы.......................... 1007
Биологическое разрушение горных пород........................ 1009
Ассоциация с простейшими..................................... 1010
Вирусы...................................................... 1010
Бактерии.................................................... 1010
Грибы и лишайники............................................ 1011
Водоросли .................................................. 1011
Губки....................................................... 1017
Брюхоногие моллюски, голотурии.............................. 1018
Растения..................................................... 1019
Природа кремнеземных отложений в растениях......... 1020
Упрочнение частей растений................................. 1022
Хвощи..................................................... 1022
Бамбук..........•......................................... 1024
Злаки..................................................... 1025
Колючие растения.......................................... 1026
Койке..................................................... 1027
Пальмы.................................................... 1028
Деревья................................................... 1028
Механизм поглощения, переноса и осаждения кремнезема . . . 1029
Содержание • 1127
Растворимый кремнезем и плодородие почв.................... 1031
Полезные и защитные воздействия кремнезема................. 1035
Насекомые.................................................... 1037
Рыбы, земноводные, пресмыкающиеся, птицы...................... 1038
Млекопитающие, человек........................................ 1039
Роль кремнезема в жизнедеятельности млекопитающих.......... 1044
Токсичность кремнезема..................................... 1046
Цитотоксичность .......................................... 1049
Кремнезем в биохимических соединениях...................... 1052
Соединения с полисахаридами ........................'. . . 1053
Соединения с белками...................................... 1054
Денатурация белков, коагуляция крови...................... 1056
Соединения со специфическими веществами и ферментами . . 1060
Кремнезем в соединениях фосфора. Нуклеиновые кислоты,
ДНК, РНК.................................................. 1062
Мутации, атеросклероз, раковые опухоли.................... 1063
Силикоз, пневмокониоз, фиброгенез.......................... 1066
Механизм возникновения силикоза............;.............. 1066
Сопоставление аморфного и кристаллического кремнезема.
Влияние размеров частиц................................... 1072
Восприимчивость к силикозу................................ 1074
Необычное соединение в пораженных силикозом тканях . . . 1074
Теория растворимости...................................... 1075
Вещества, тормозящие развитие силикоза ................... 1080
Асбестоз, или заболевания, вызванные микроиглами.......... 1086
Благотворные воздействия кремнезема........................ 1087
Метаболизм кремния............................................ 1087
Силикагель как культуральная среда ........................ 1089
Кремнийорганические соединения................................ 1090
Аналитические проблемы........................................ 1092
Заключение.................................................... 1093
Литература.................................................... 1093
Предметный указатель......................................... 1106
УВАЖАЕМЫЙ ЧИТАТЕЛЬ!
Ваши замечания о содержании книги, ее оформ-
лении, качестве перевода и другие просим присылать
по адресу: 129820, Москва, И-110, ГСП, 1-й Риж-
ский пер., д. 2, издательство «Мир».
Ральф К. Айлер
ХИМИЯ КРЕМНЕЗЕМА
в 2-х частях, ч. 2
Научный редактор Н. А. Васина
Мл. научный ред. И. И, Землячева
Художник В. Завадовская
Художественный редактор М. Кузьмина
Технический редактор Л. Бирюкова
Коррректор В. С. Соколов
ИБ № 2795
Сдано в набор 26.02.82. Подписано к печати
01.11.82. Формат 60Х90‘У1б. Бумага типографская
№ 2. Объем 22,25 бум. л. Гарнитура литературная.
Печать высокая. Усл. печ. л. 44,5. Усл. кр.-отт.
44,50. Уч.-нзд. л. 48,17. Изд. № 3/1594. Тираж
2700 экз. Зак. 250. Цена 5 р. 80 к.
ИЗДАТЕЛЬСТВО «МИР»
129820, Москва, И-110, ГСП. 1-й Рижский пер., 2.
Ленинградская типография № 8 ордена Трудового
Красного Знамени Ленинградского объединения
«Техническая книга» им. Евгении Соколовой
Союзполиграфпрома при Государственном коми-
тете СССР по делам издательств, полиграфии
и книжной торговли.
190000, г. Ленинград, Прачечный переулок, 6.