

















































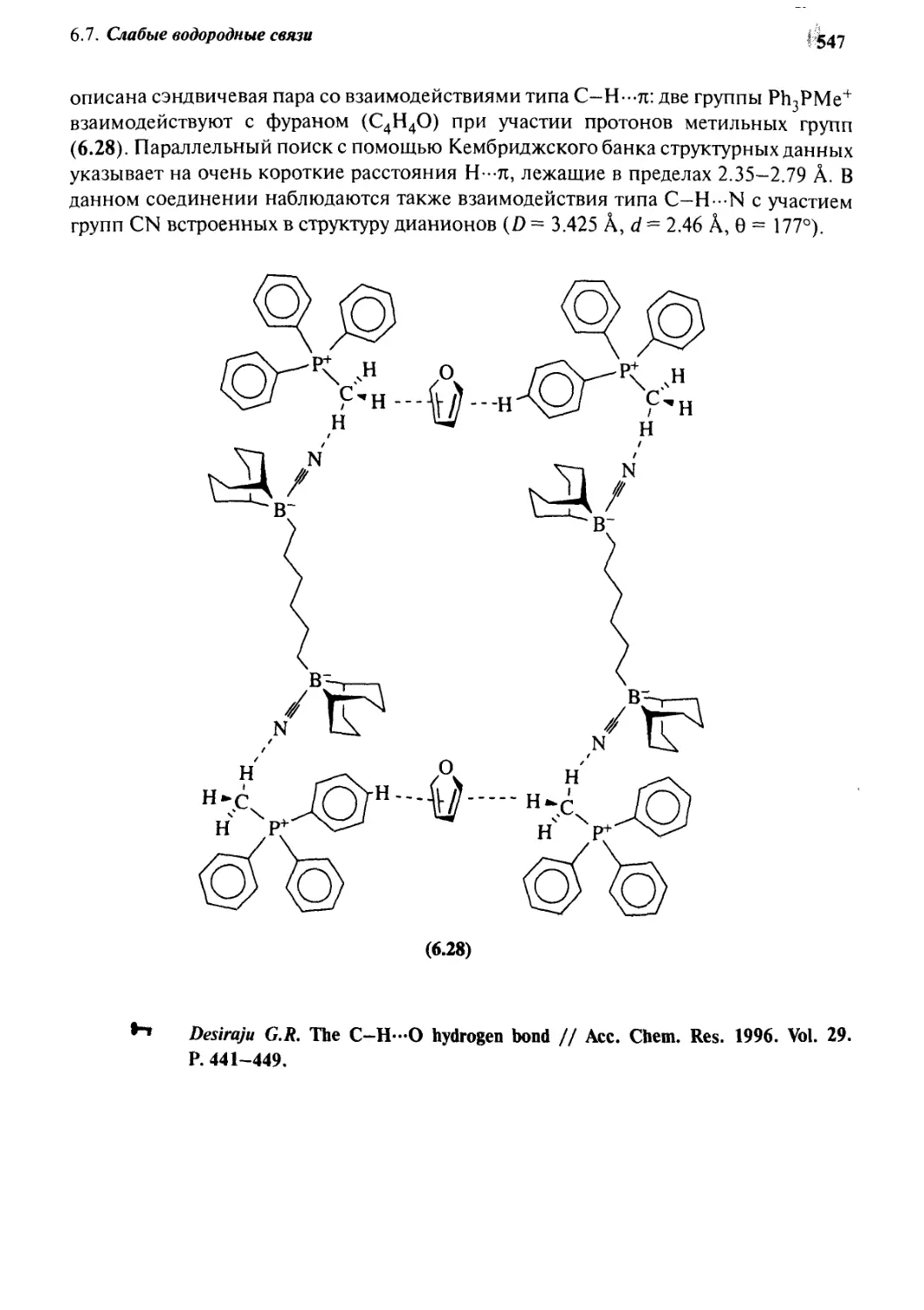








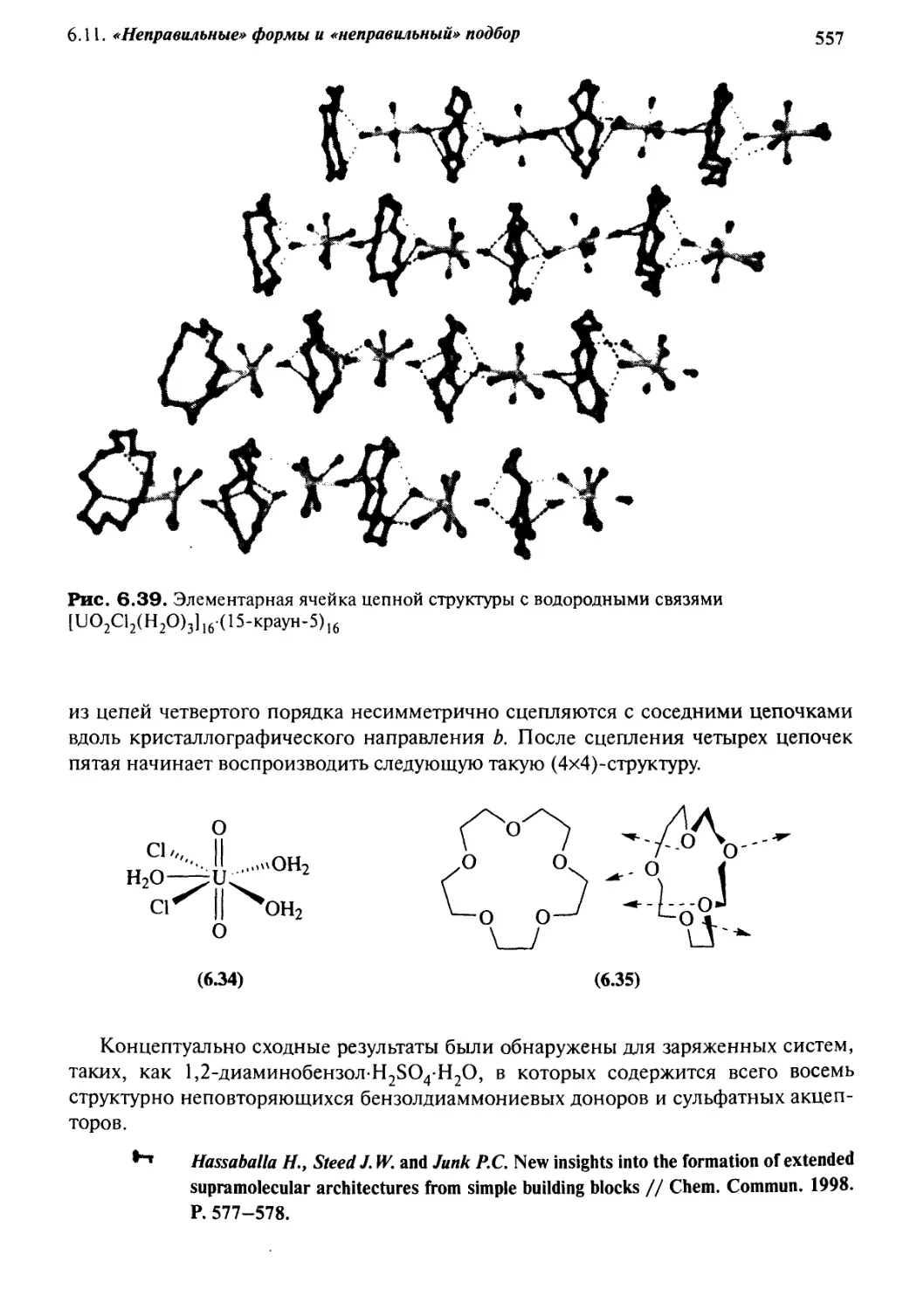






















































































































































































































Автор: Стид Дж. В. Этвуд Дж. Л.
Теги: органическая химия химия молекулярная биология молекулярная физика
ISBN: 978-5-94628-303-8
Год: 2007




Текст
химия
СУПРАМОЛЕКУЛЯРНАЯ
Jonathan W. Steed
King's College, London
Jerry L. Atwood
University of Missouri, Columbia
John Wiley & Sons, Ltd
Chichester • New York • Weinheim • Brisbane • Singapore -Toronto
ДЖ.В.СТИД, Дж.Л. ЭТВУД
химия
В двух томах
Том 2
Перевод с английского
кандидата химических наук И. Г. Варшавской,
кандидата химических наук Б. И. Харисова,
кандидата химических наук О. В. Белуженко,
кандидата химических наук И. С. Васильченко,
доктора химических наук Ю. А. Алексеева
Под редакцией
академика РАН, профессора А. Ю. Цивадзе,
доктора химических наук, профессора В. В. Арсланова,
доктора химических наук, профессора А. Д. Гарновского
МОСКВА
ИКЦ «АКАДЕМКНИГА»
2007
УДК 547
ББК 24.2
С80
Стид Дж. В., Этвуд Дж. Л.
Супрамолекулярная химия. Пер. с англ.: в 2 т. / Джонатан В. Стид, Джерри Л.
Этвуд. - М. : ИКЦ «Академкнига», 2007. - ISBN 978-5-94628-303-8.
Т. 2. - 2007. - 416 с. : ил. - ISBN 978-5-94628-307-6.
Книга является всеобъемлющим изданием в области супрамолекулярной химии и по
разностороннему освещению ее фундаментальных и прикладных аспектов, и по ясному и
четкому изложению ее основных концепций, понятий, определений. Большое внимание
уделено состоянию и перспективам развития основных разделов супрамолекулярной
химии.
Рассмотрены невалентные взаимодействия, создание супрамолекулярных ансамблей,
вопросы современной химии краун-соединений и антикраунов, фуллеренов, геликатов,
дендримеров, супрамолекулярных фото- и биоактивных структур, твердых и жидких
кристаллов, химических сенсоров, нелинейно-оптических материалов и молекулярных
устройств, а также вопросы химии жизненно важных процессов. Предлагаемая книга —
ценное руководство для становления и развития супрамолекулярной химии в нашей стране.
Эта монография может служить учебником для молодых специалистов.
Для специалистов различных областей, студентов, аспирантов и преподавателей
высших учебных заведений.
ISBN 0-471-98831-6 (англ.)
ISBN 0-471-98791-3
ISBN 978-5-94628-303-8 (рус. общий)
ISBN 978-5-94628-307-6 (т. 2)
© John Wiley & Sons, Ltd, 2000
© ИКЦ «Академкнига», 2007
Оглавление
Том 2
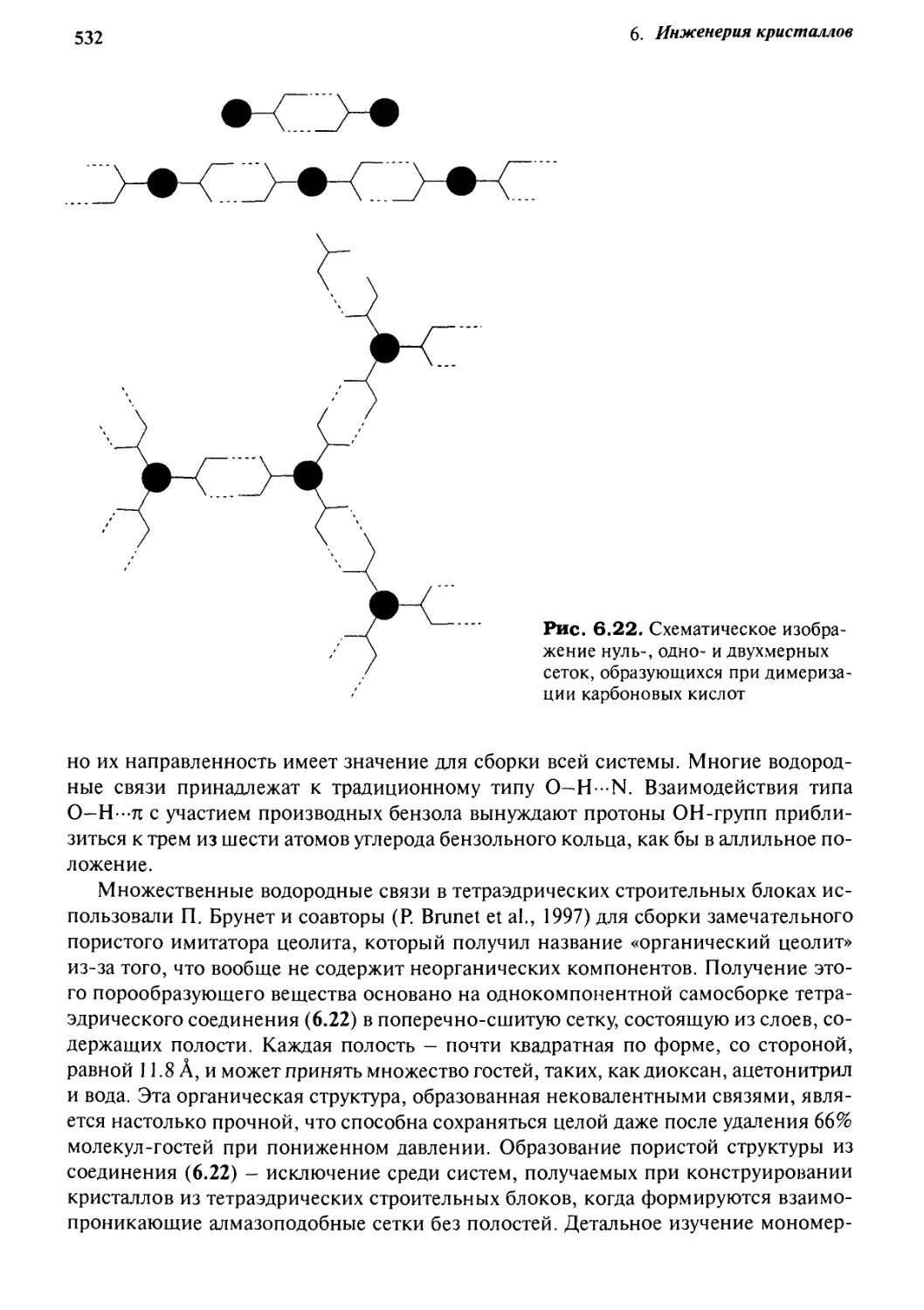
О Инженерия кристаллов 491
6 л
6.2
6*3
Общие вопросы
6.1.1
6.1.2
6.1.3
6.1.4
6.1.5
6.1.6
6.1.7
Введение
Межмолекулярные взаимодействия
Особая роль водородных связей
Анализ набора графов
Рост кристаллов
Разрушение кристаллов
Стратегии дизайна при инженерии кристаллов
Предсказание структуры кристаллов
6.2.1
6.2.2
Расчетные методы
Поавила Эттео
491
491
494
494
501
504
514
515
517
517
521
Кембриджский банк структурных данных 525
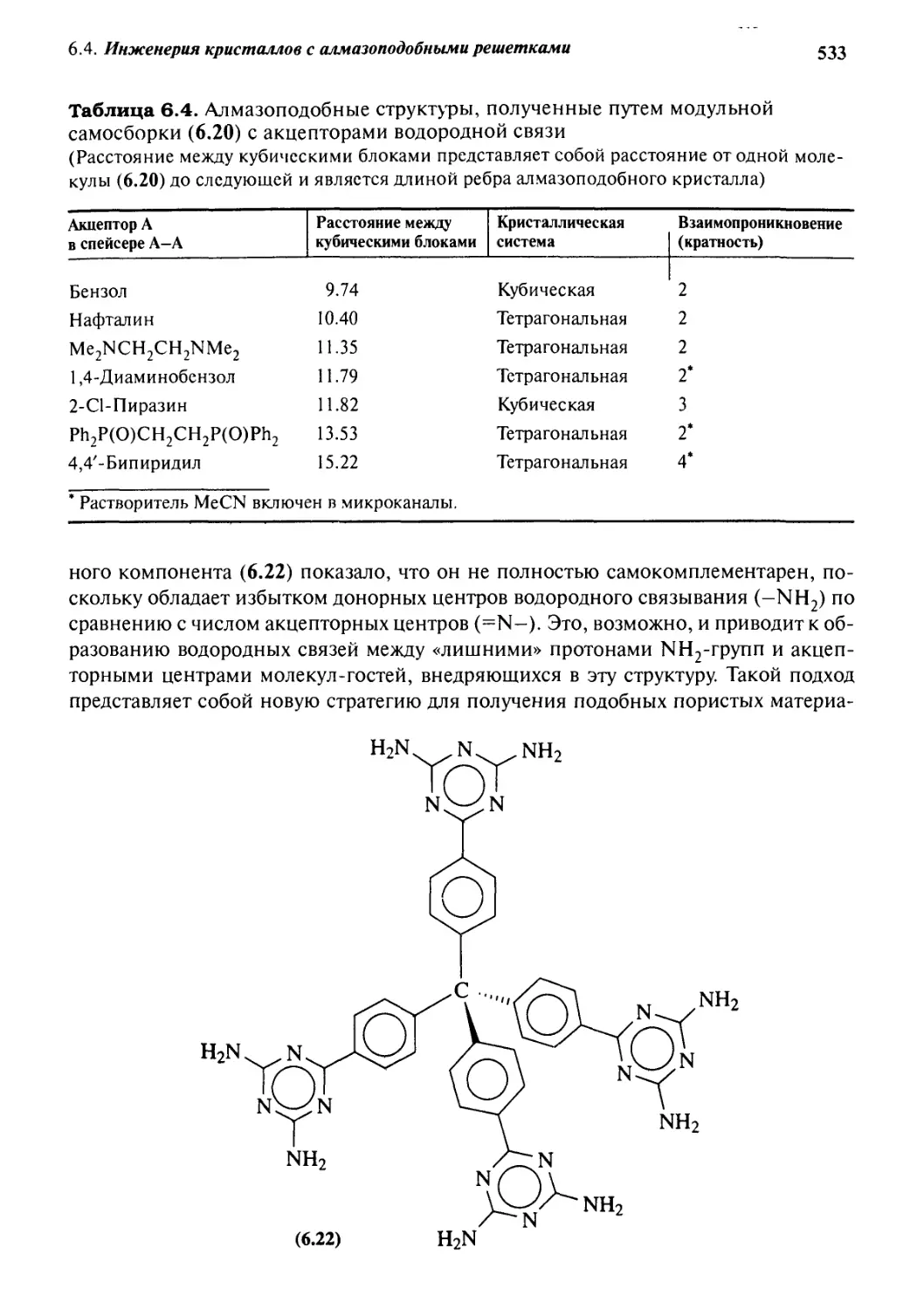
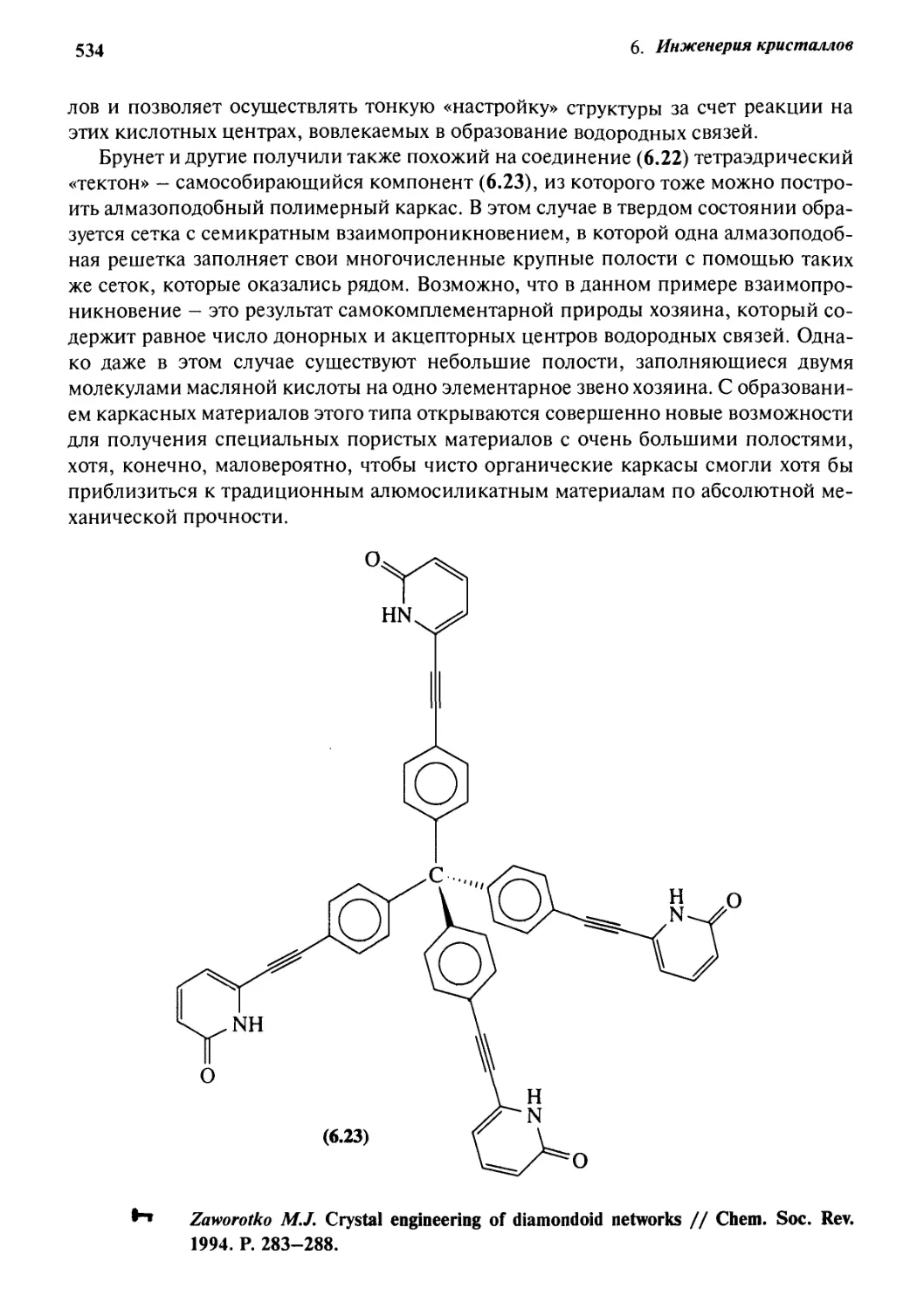
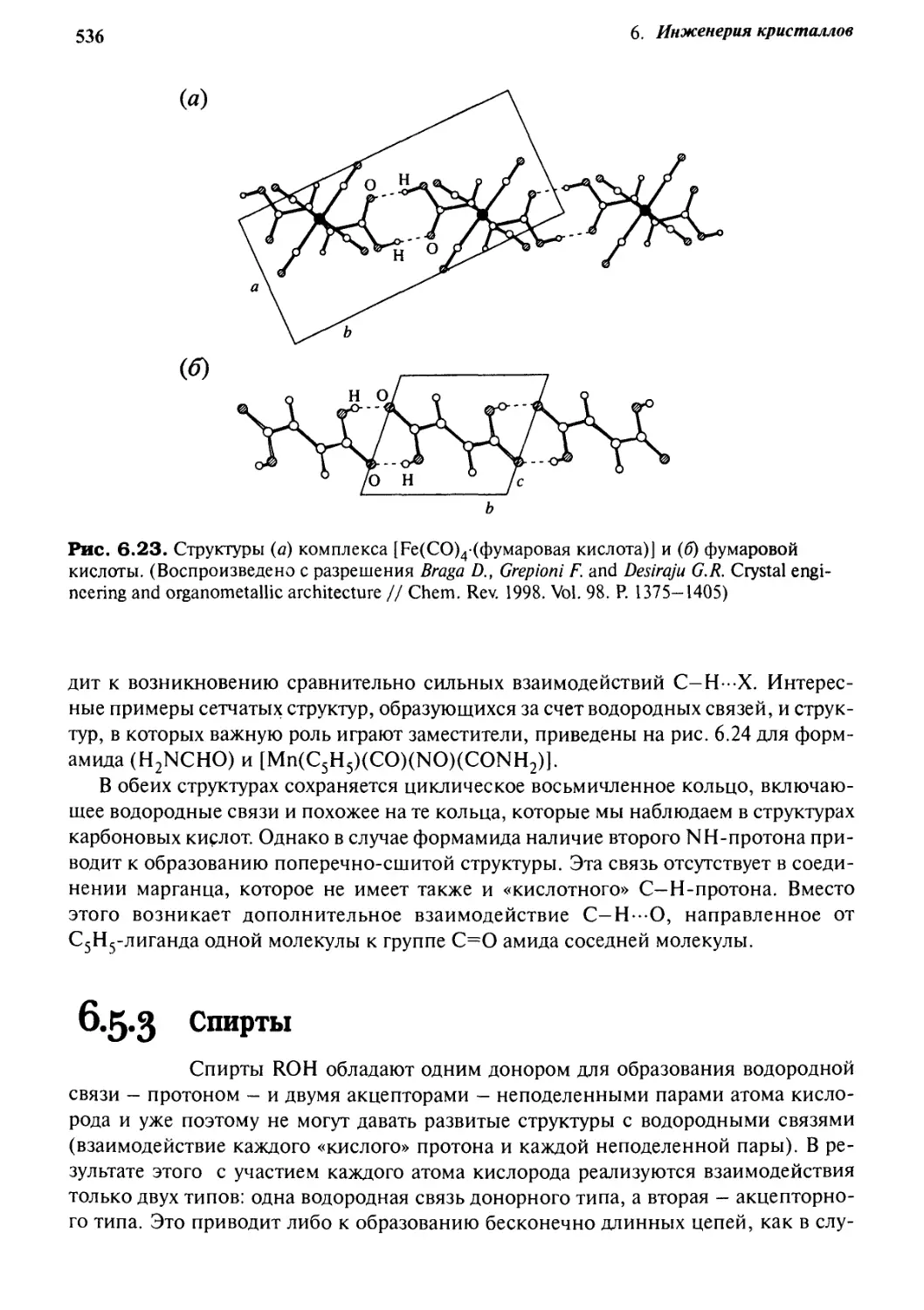
6.4 Инженерия кристаллов с алмазоподобными
решетками 529
6.5 Инженерия кристаллов с водородными связями 535
6.5.1 Димеры карбоновых кислот 535
6.5.2 Амиды 535
6.5.3 Спирты 536
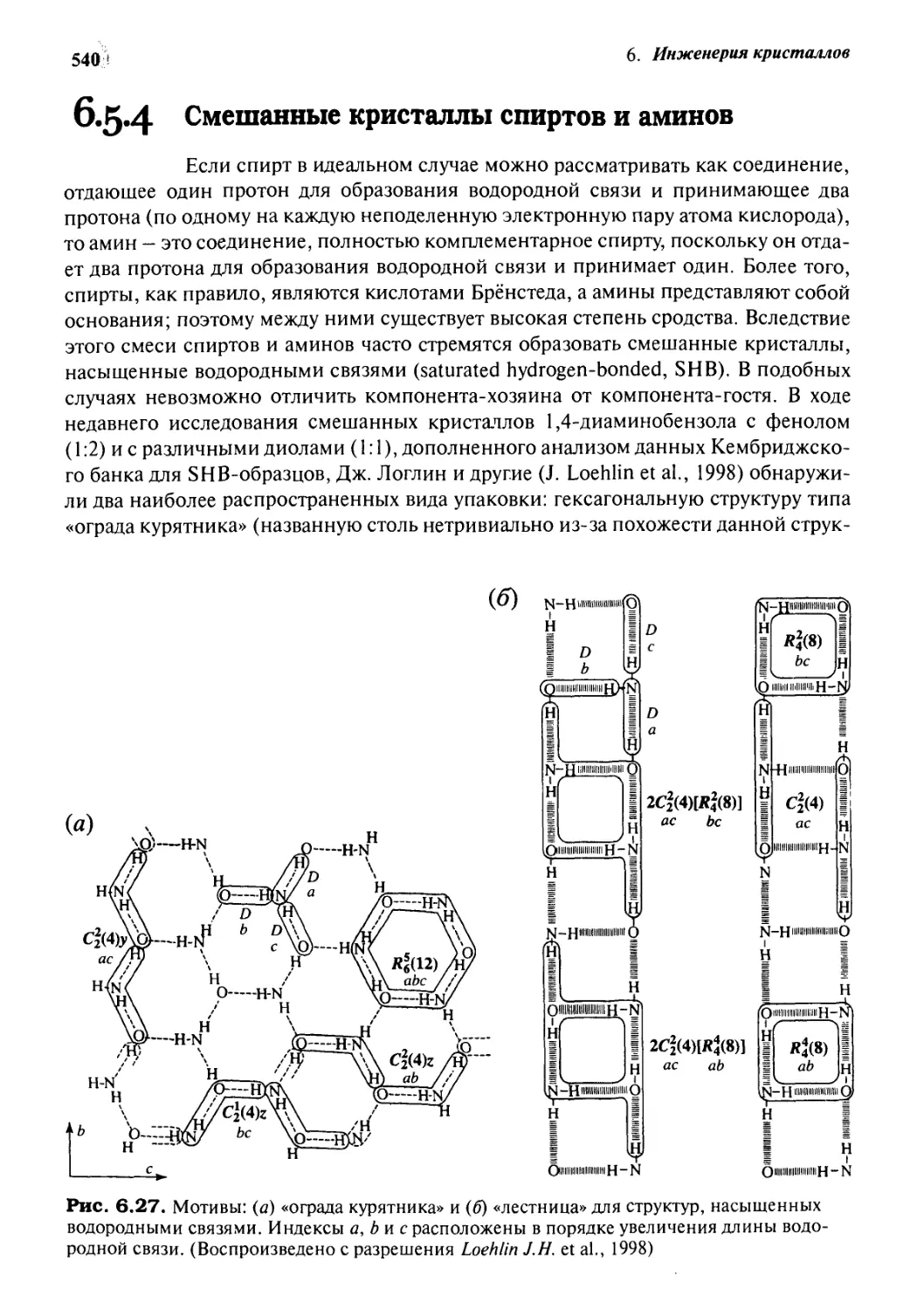
6.5.4 Смешанные кристаллы спиртов и аминов 540
6.5.5 Смешанные кристаллы аминов с аминами 541
6.6 Водородные связи монооксида углерода 542
6 Слабые водородные связи 544
g) Оглавление
6.8 Водородные связи с металлами и гидридами металлов 548
6.8.1 Прямое взаимодействие с металлами 548
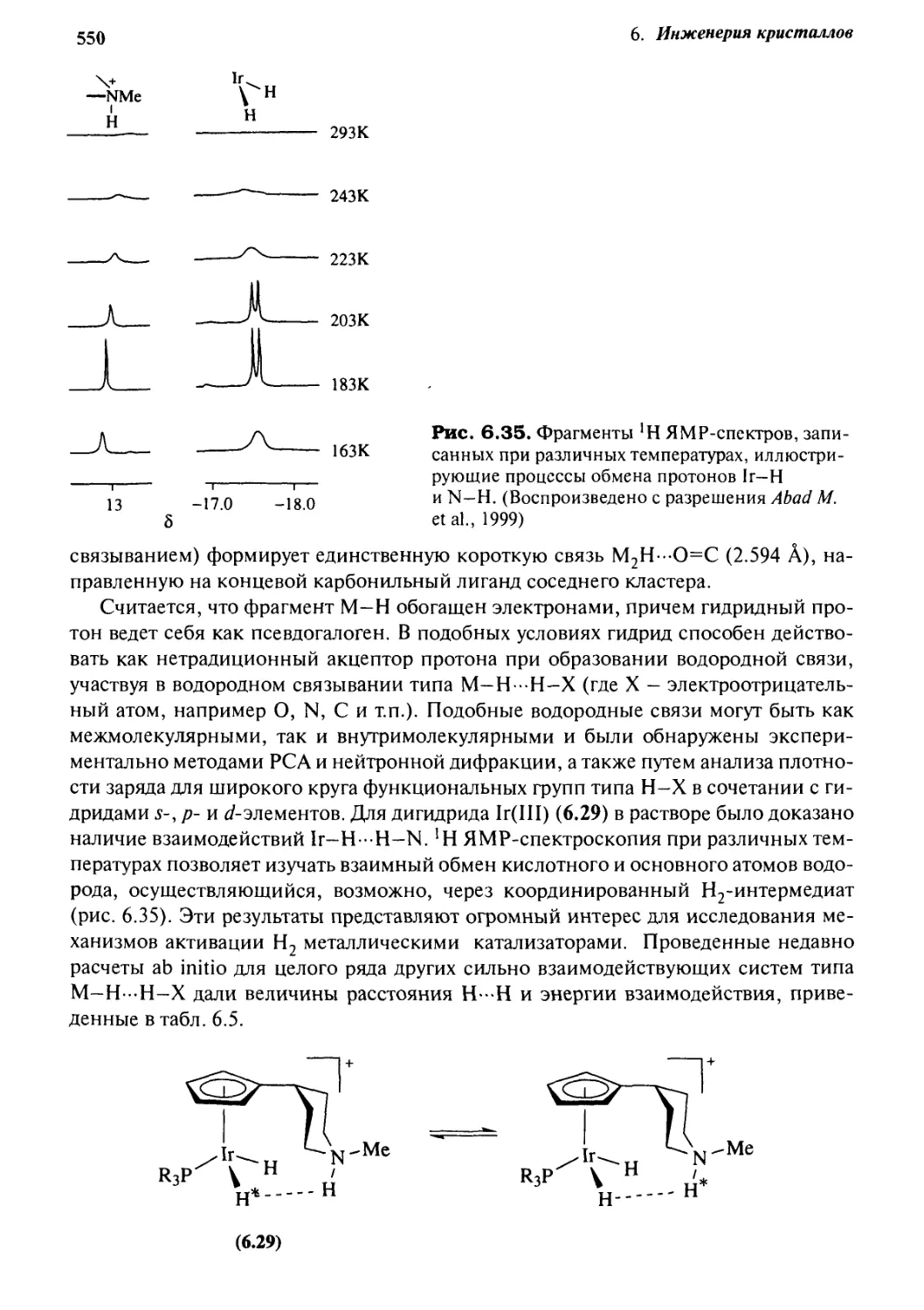
6.8.2 Взаимодействие с гидридами металлов 549
6.Q 71-71-Стэкинг-взаимодействия 551
«к/
Прочие взаимодействия 554
6.11 «Неправильные» формы и «неправильный» подбор 555
6.11.1 Хозяева типа «колесо-ось» 555
6.11.2 «Неправильно» подобранные партнеры 556
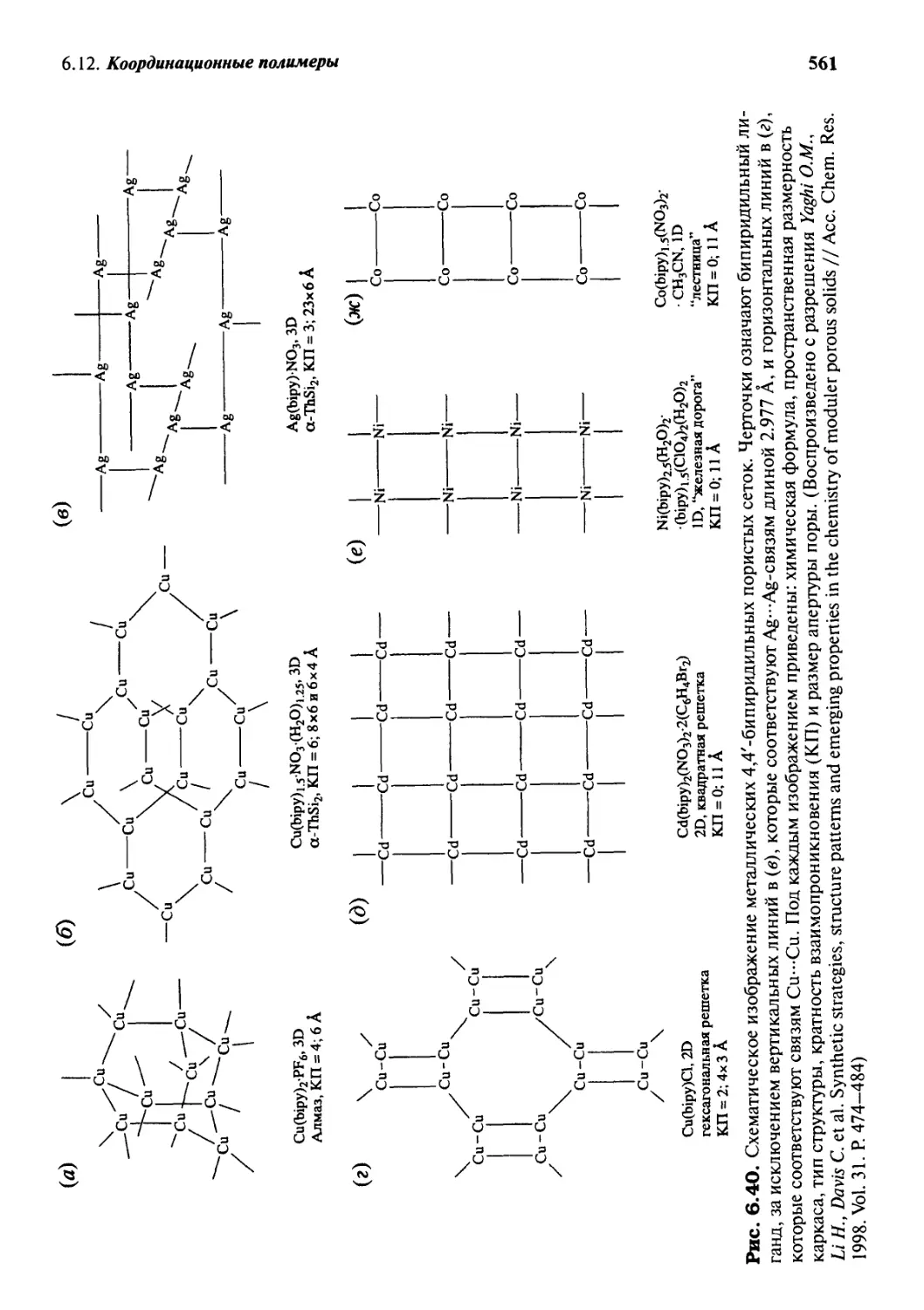
6.12 Координационные полимеры 558
6 Л. 3 Биомиметические (биоподражательные) структуры 562
Смешанные кристаллы: включения типа
«песочные часы» 567
Учебные задания 572
Мысленный эксперимент 574
Литература 574
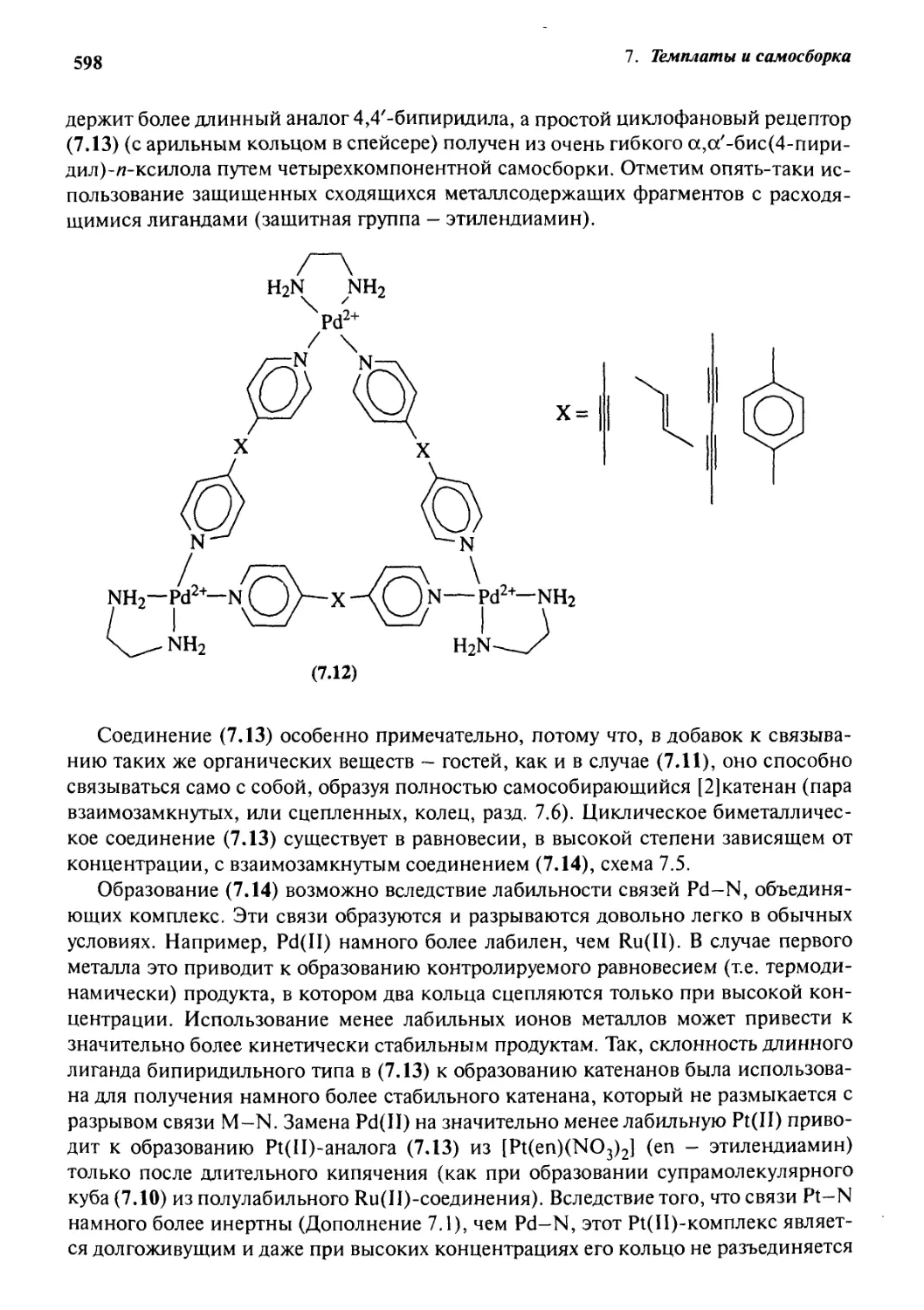
у Темплаты и самосборка
576
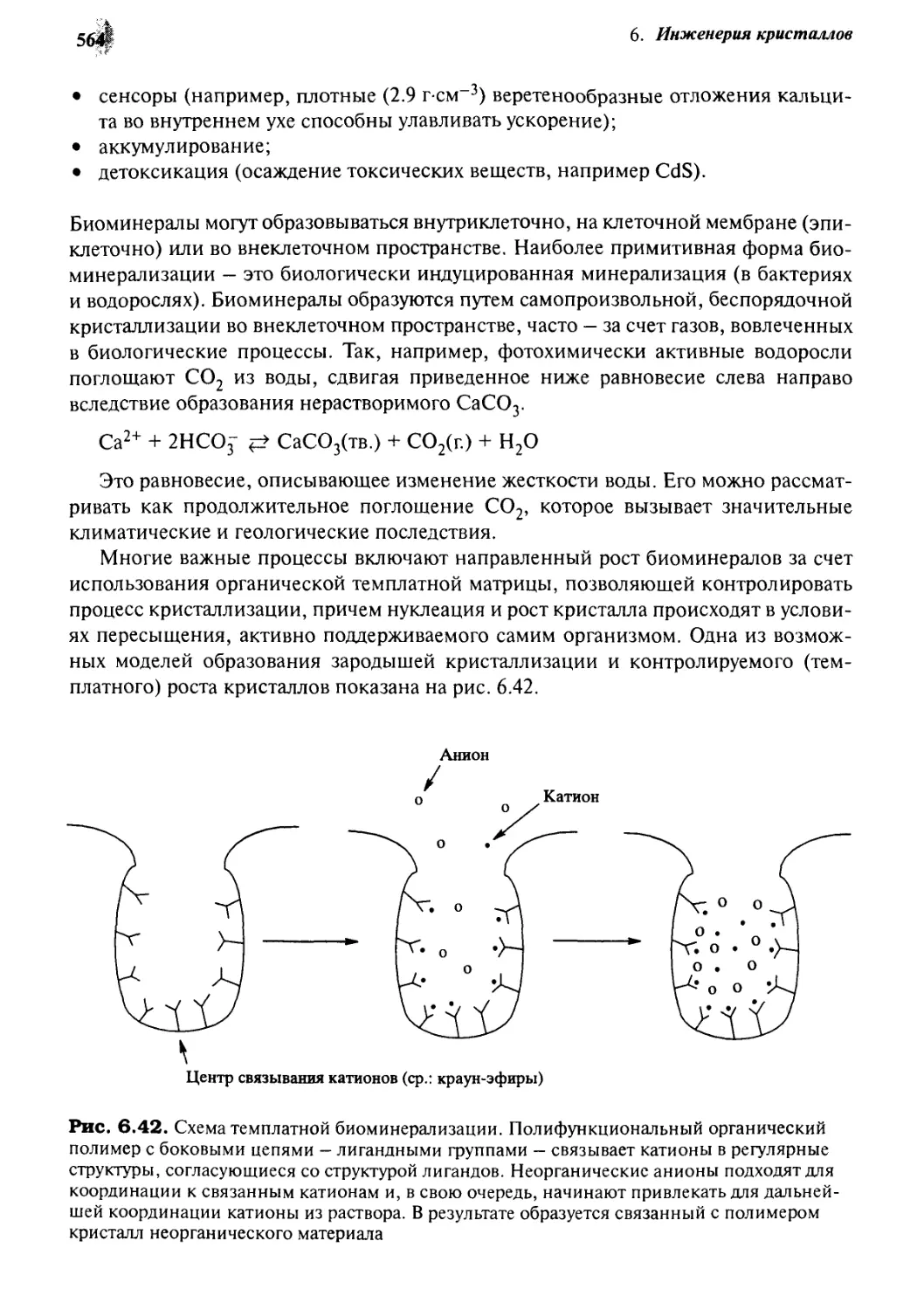
Введение 576
7.1.1 Цели и задачи 576
7.1.2 Терминология 578
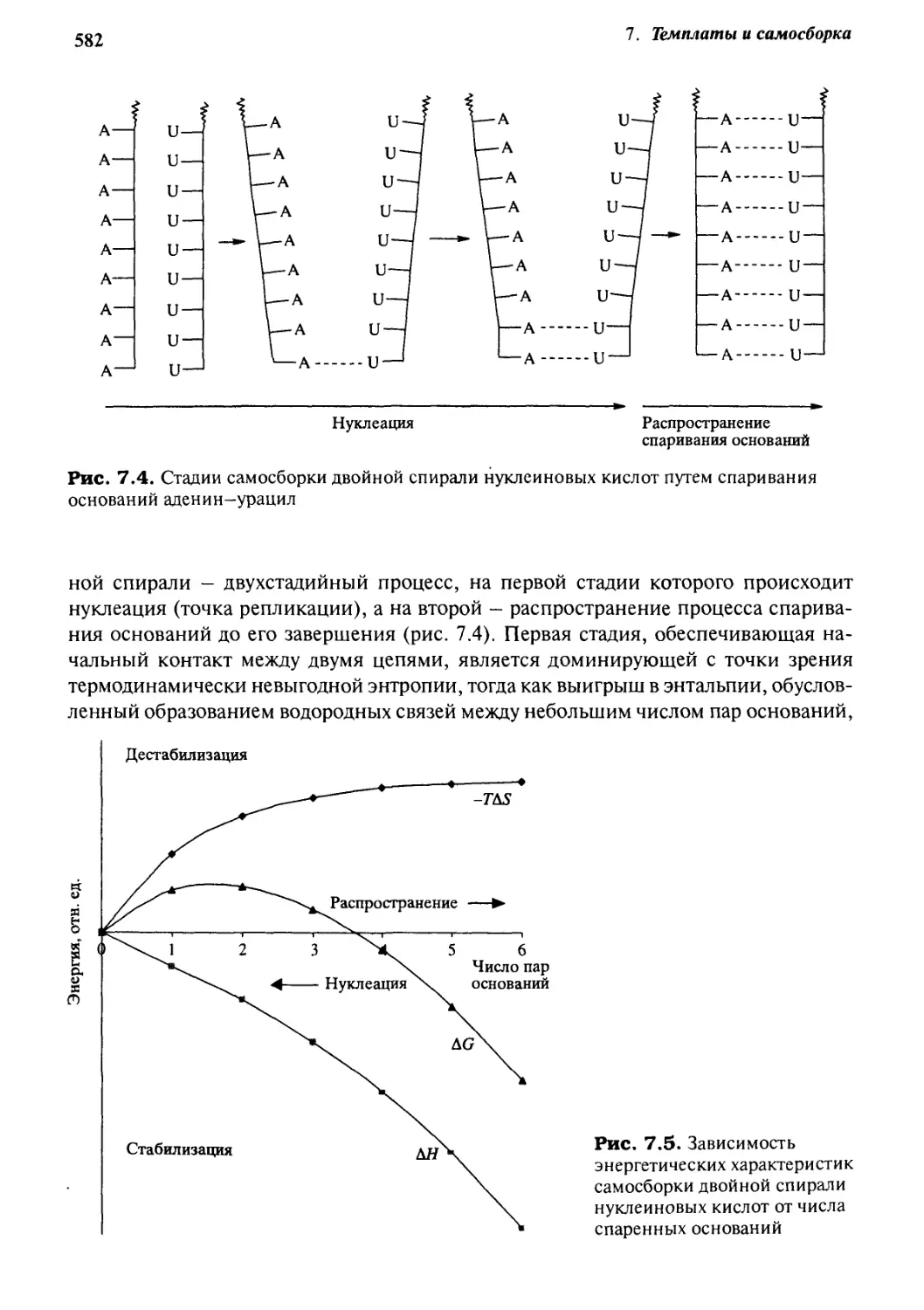
Биохимическая самосборка 580
7.2.1 Вирус табачной мозаики 580
7.2.2 Строгая самосборка 581
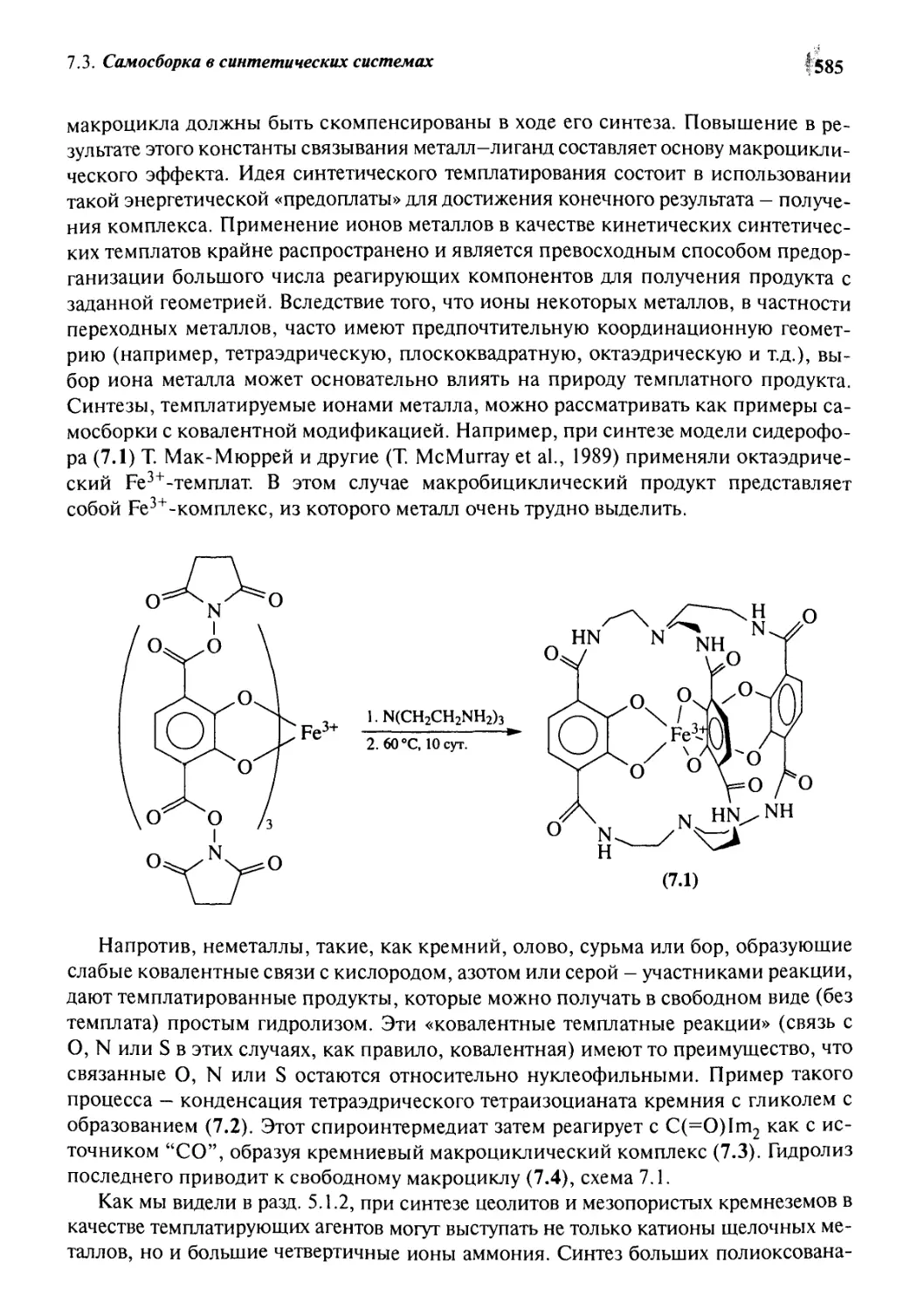
7.2.3 Самосборка с ковалентной модификацией 583
Самосборка в синтетических системах:
кинетический и термодинамический подходы 584
7.3.1 Темплатные эффекты в синтезе 584
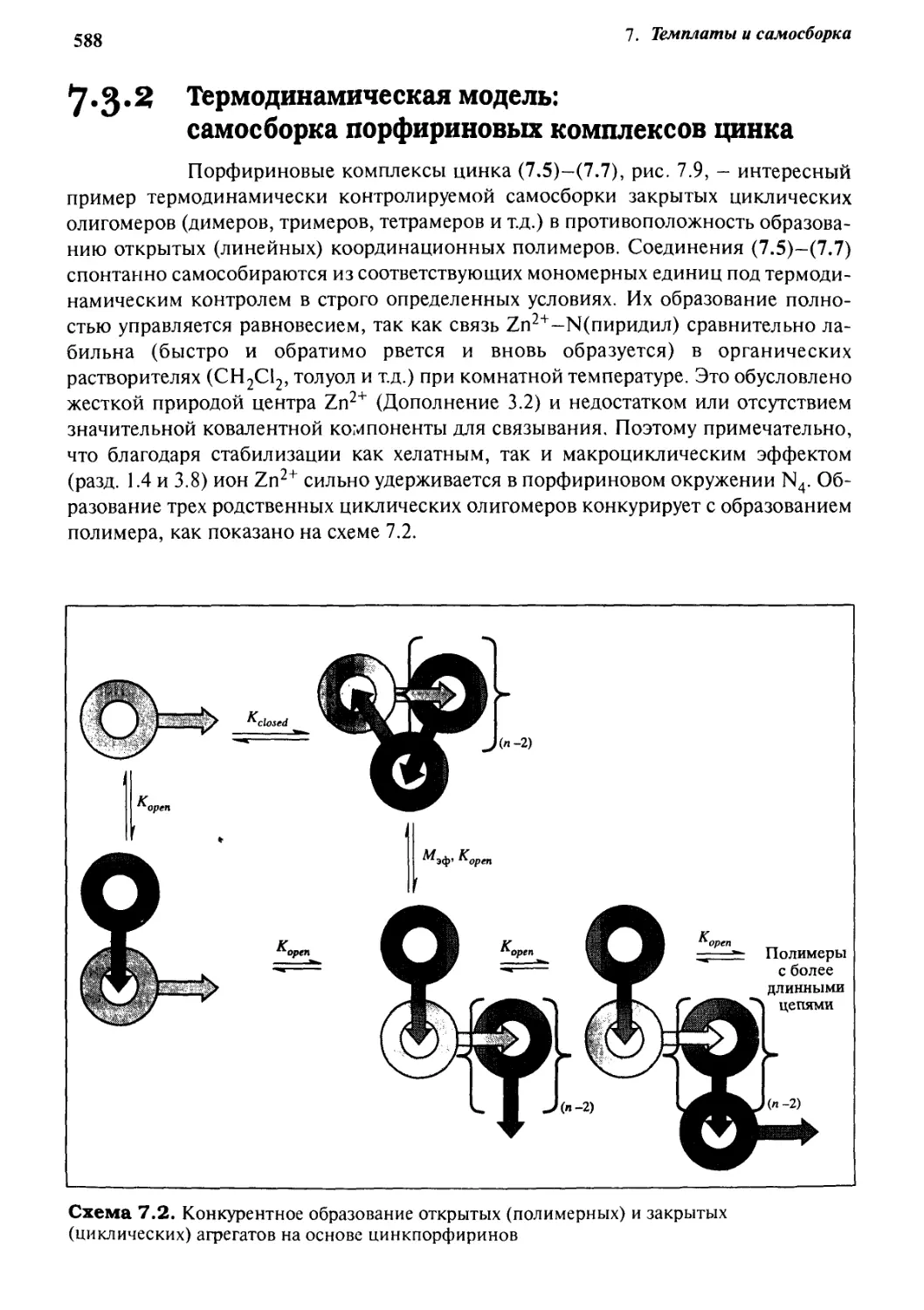
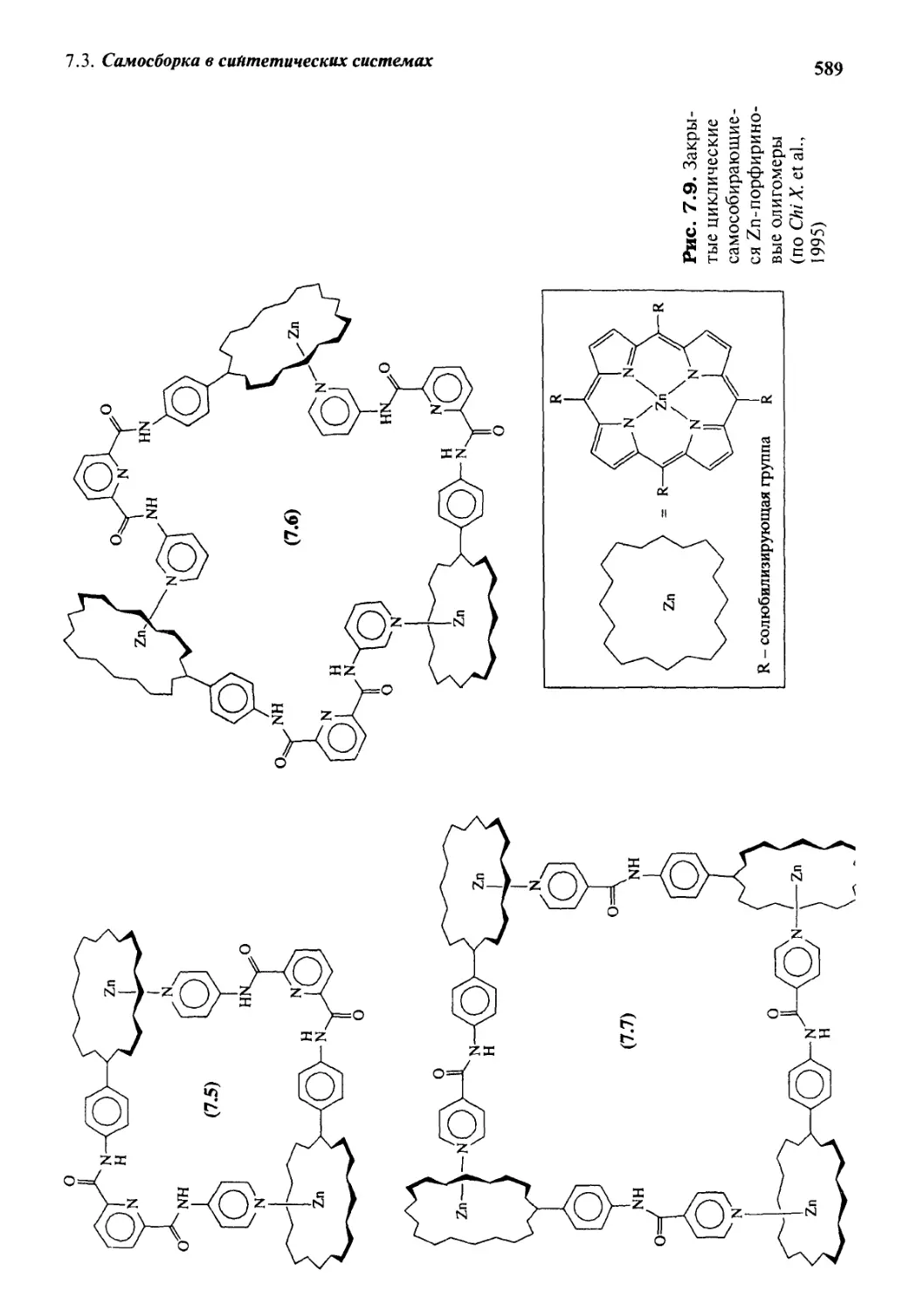
7.3.2 Термодинамическая модель:
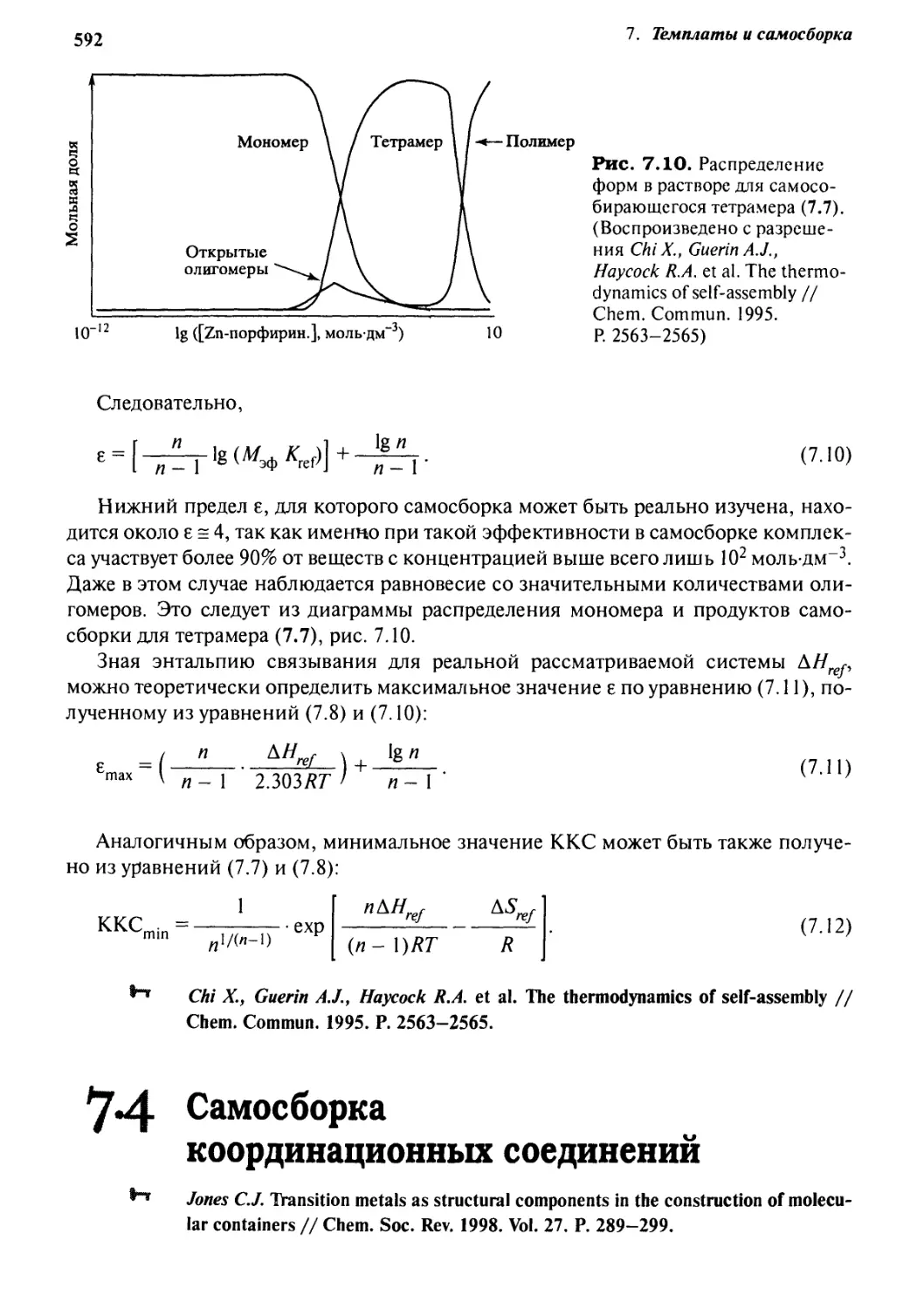
самосборка порфириновых комплексов цинка 588
Оглавление
487^
7-4 Самосборка координационных соединений 592
7.4.1 Принципы дизайна 593
7.4.2 Супрамолекулярный куб 593
7.4.3 «Молекулярные квадраты» и «молекулярные коробки» 597
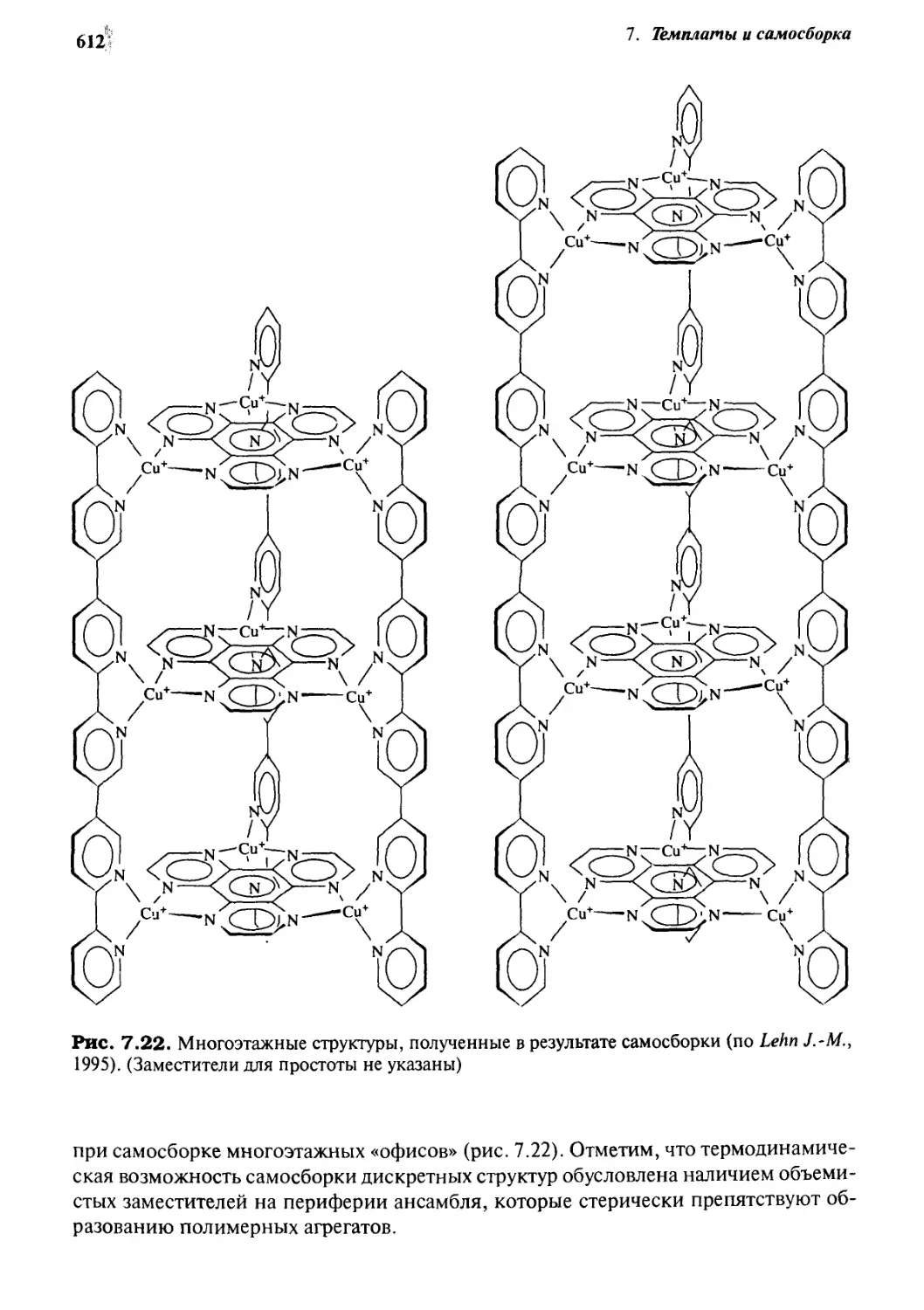
7.4.4 Металлические ансамбли 609
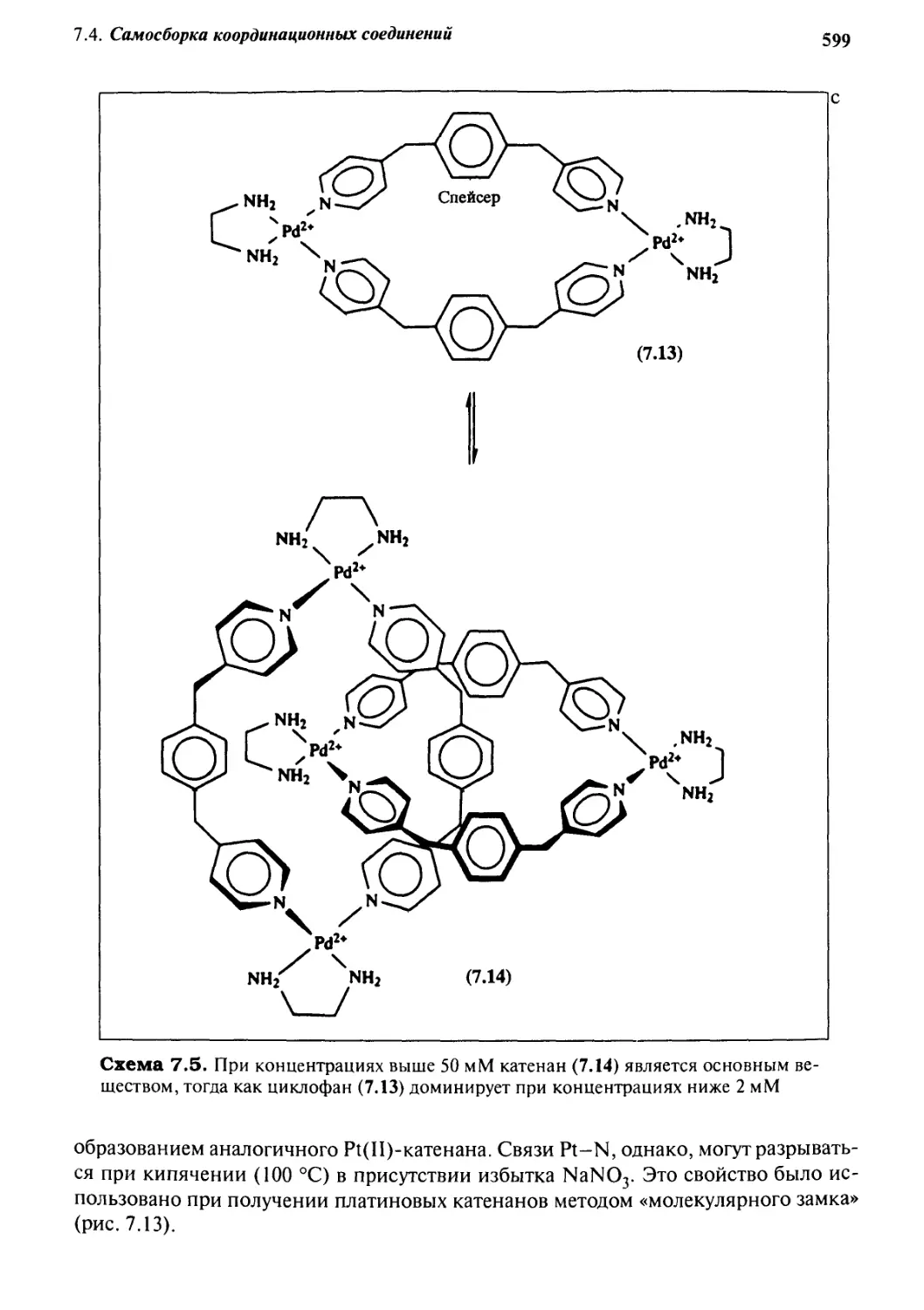
J Jj Самосборка закрытых комплексов
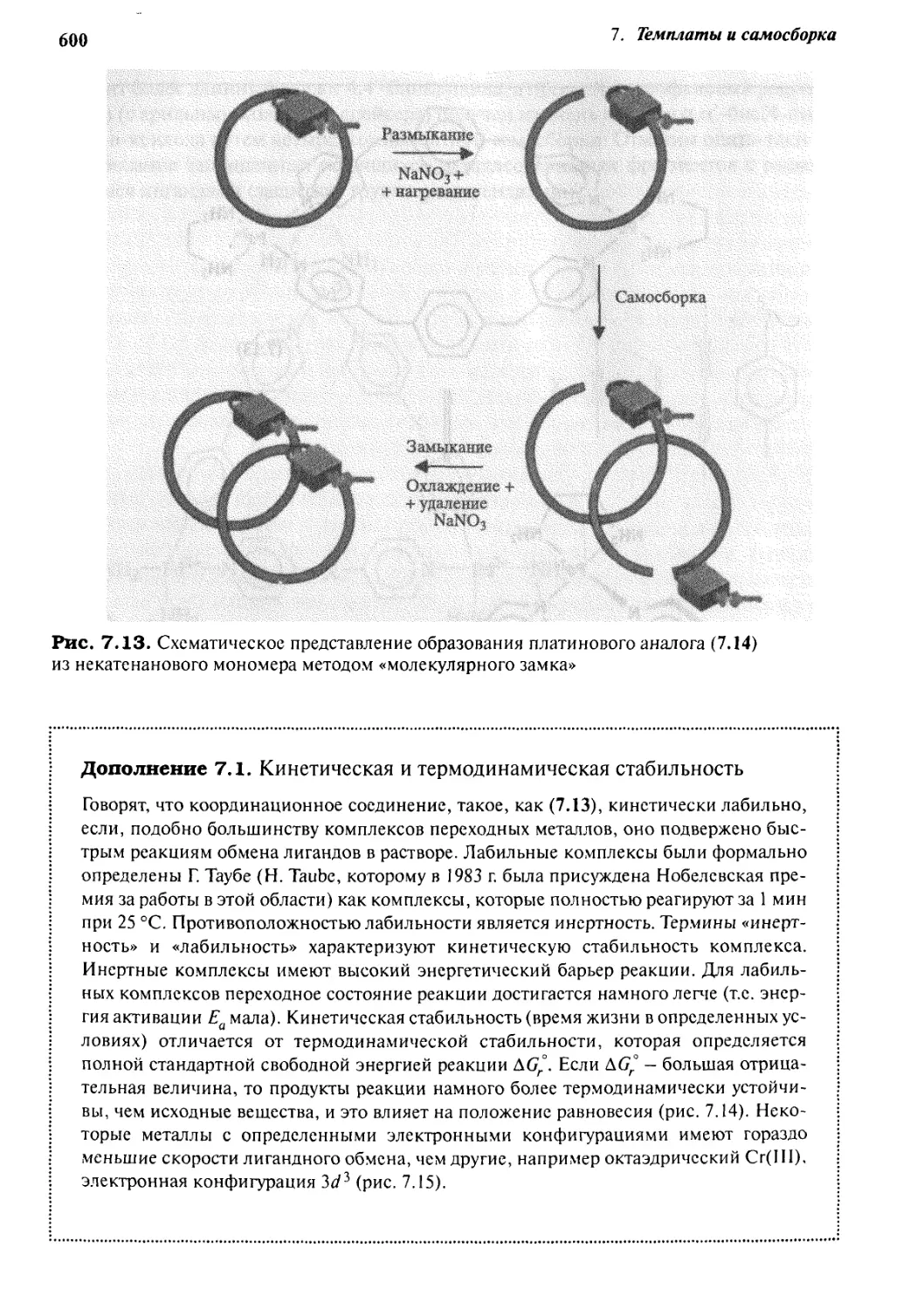
с помощью водородных связей 61 з
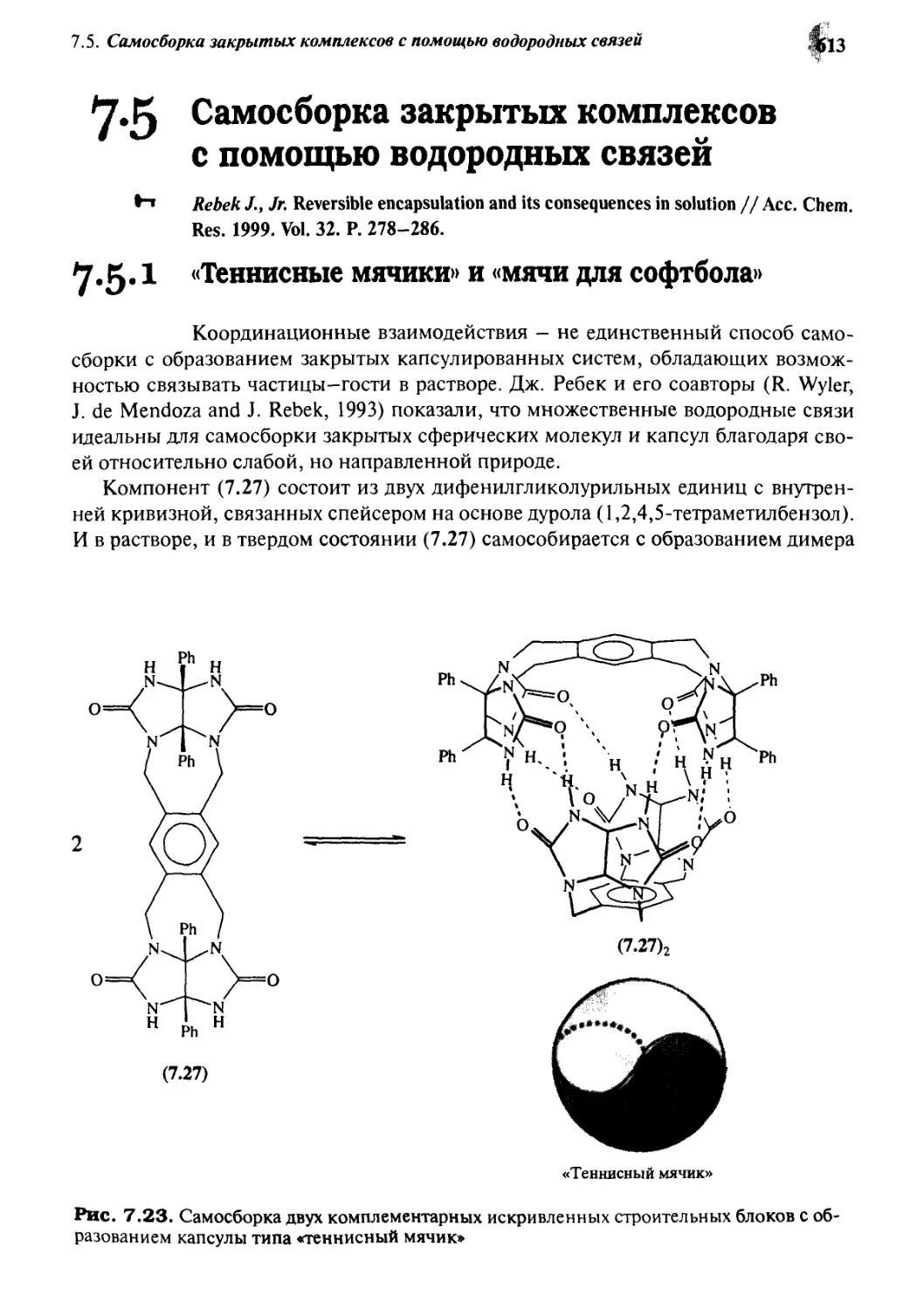
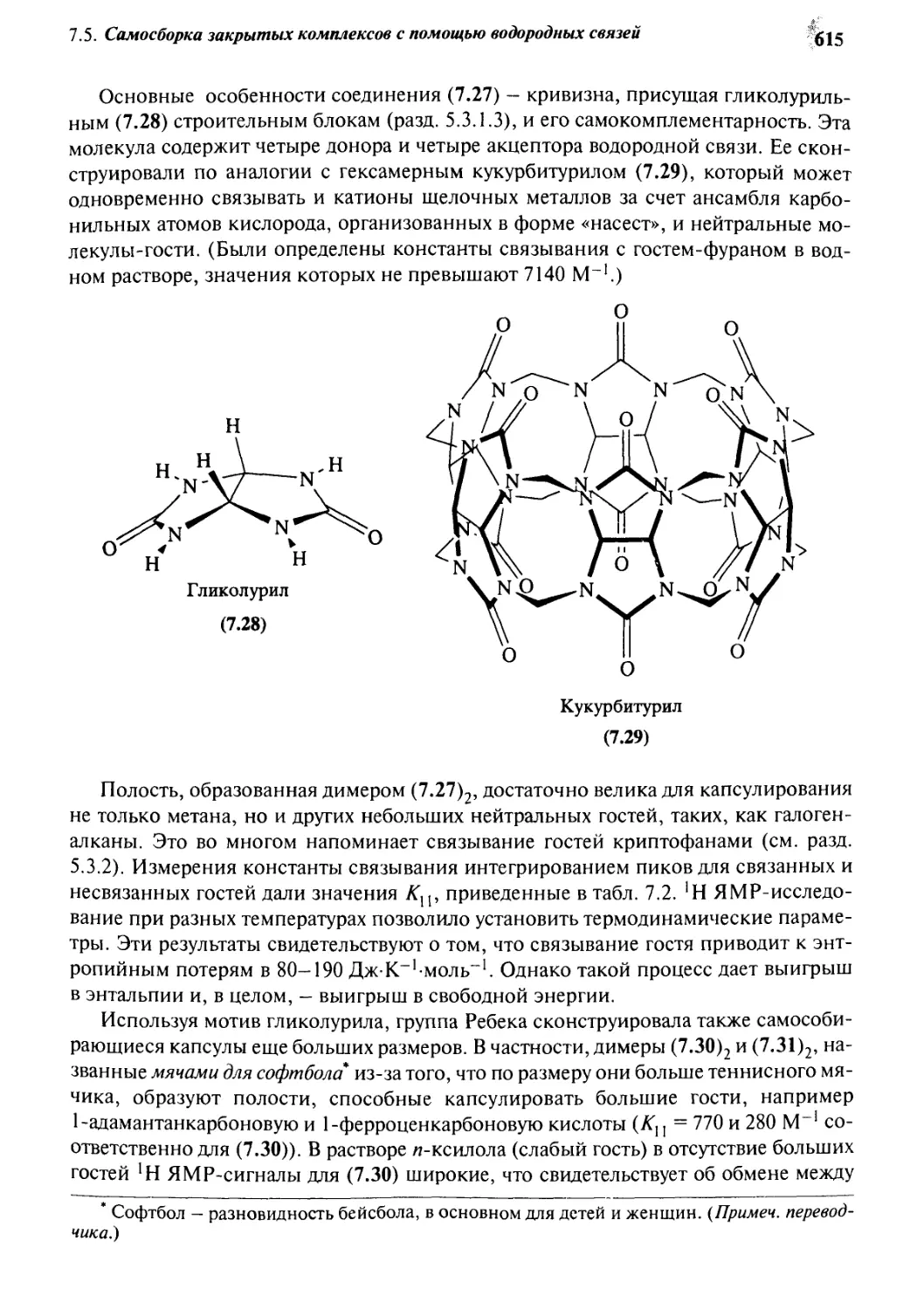
7.5.1 «Теннисные мячики» и «мячи для софтбола» 613
7.5.2 Гигантские самособирающиеся капсулы 619
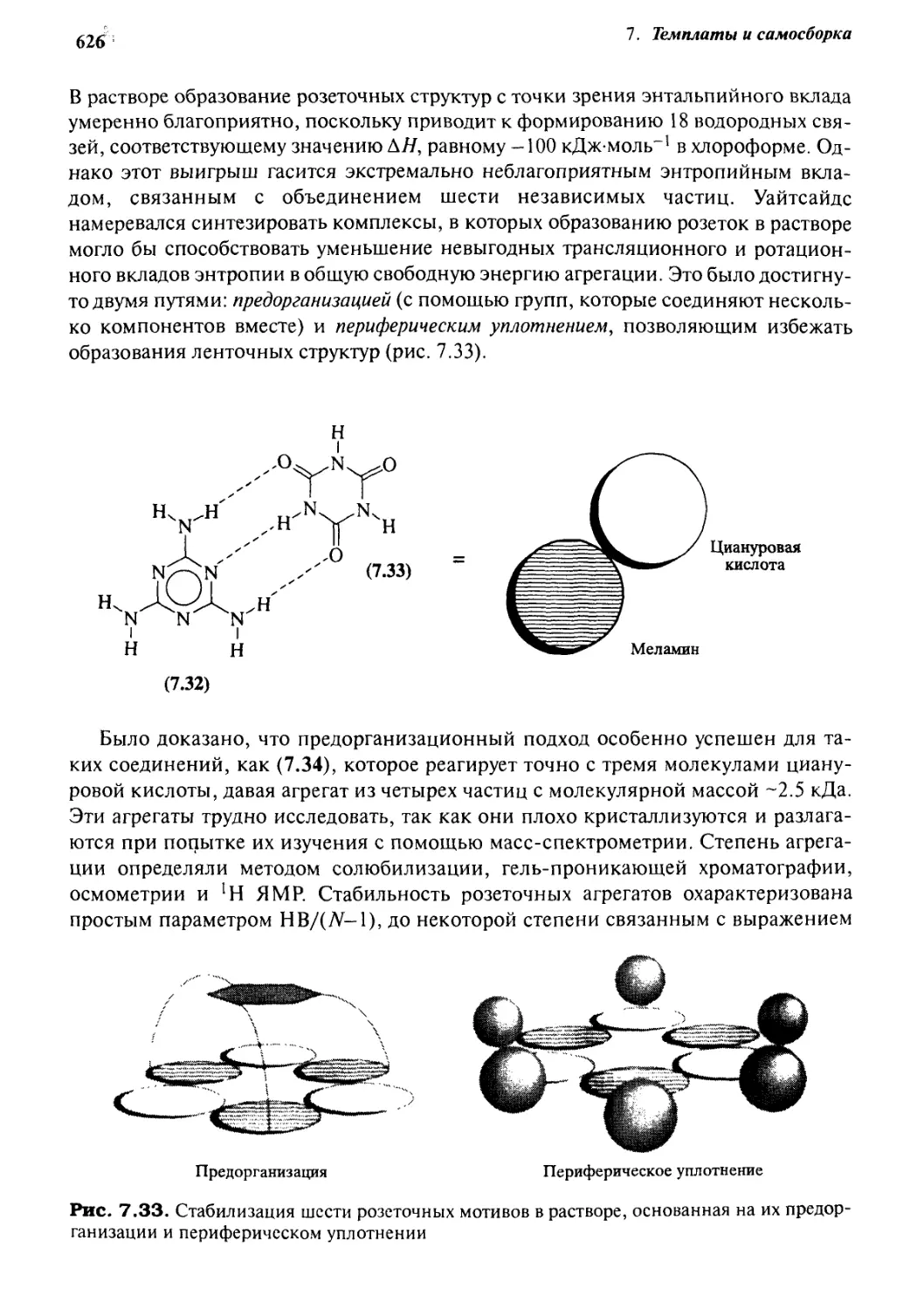
7.5.3 Розетки 625
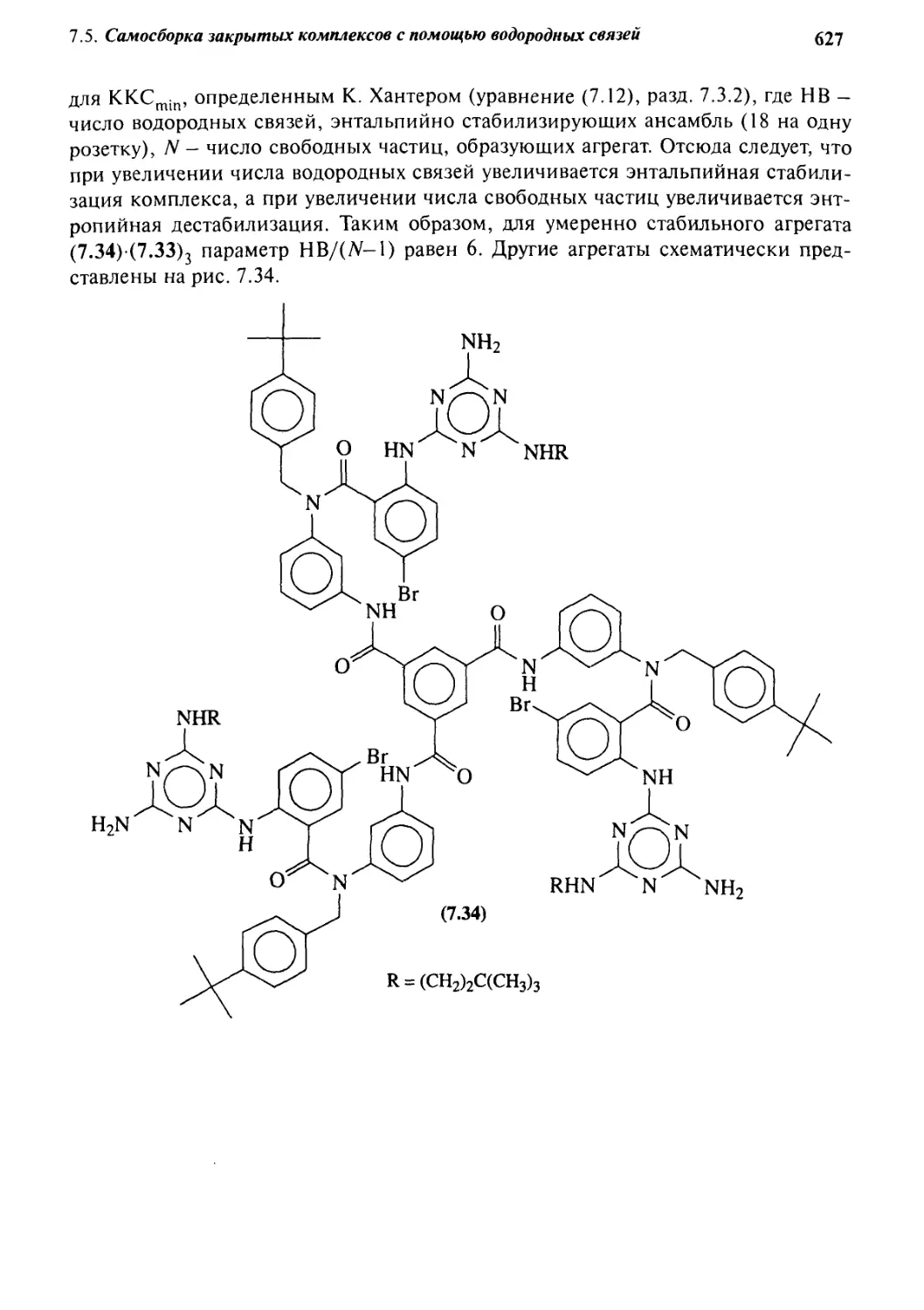
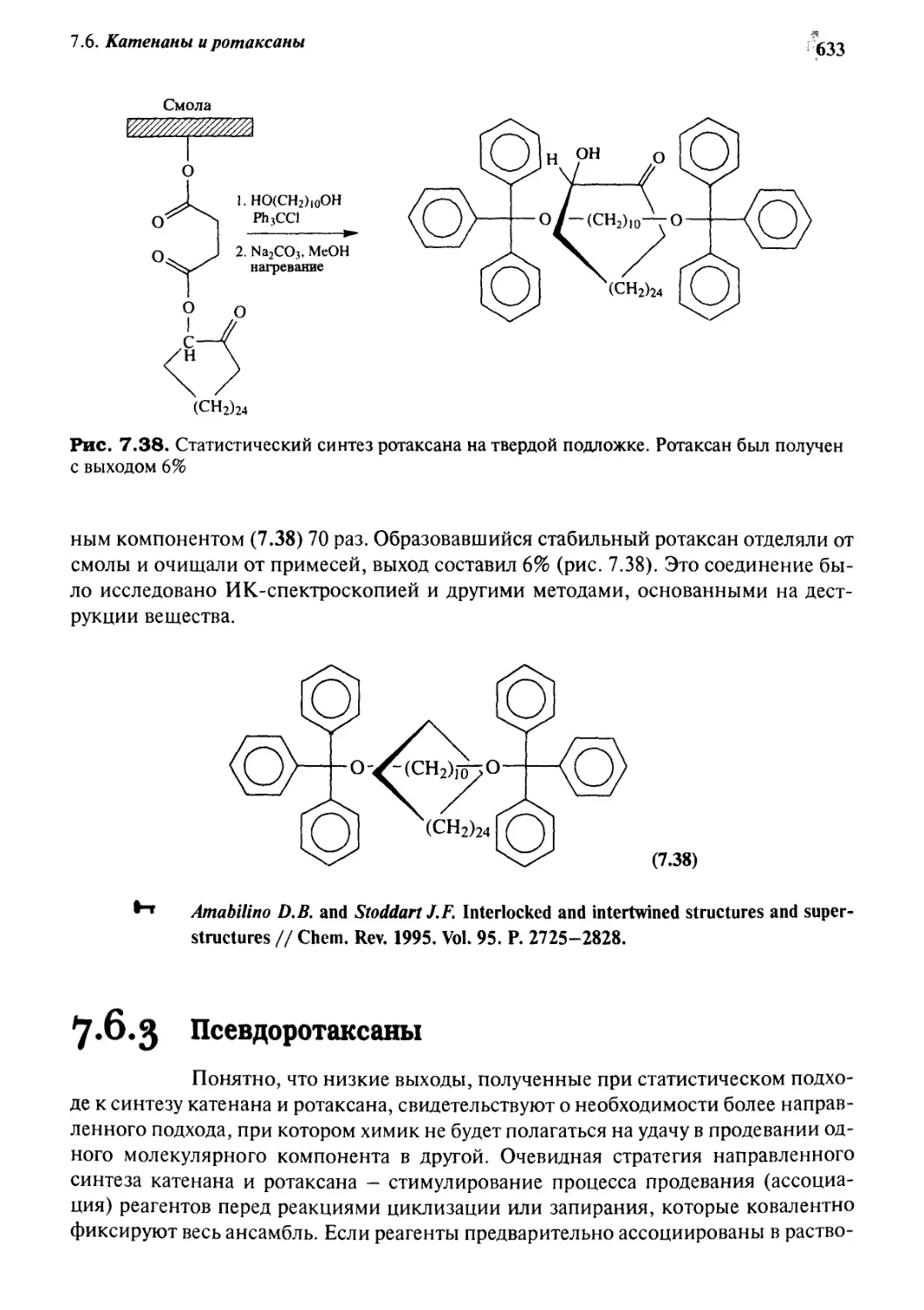
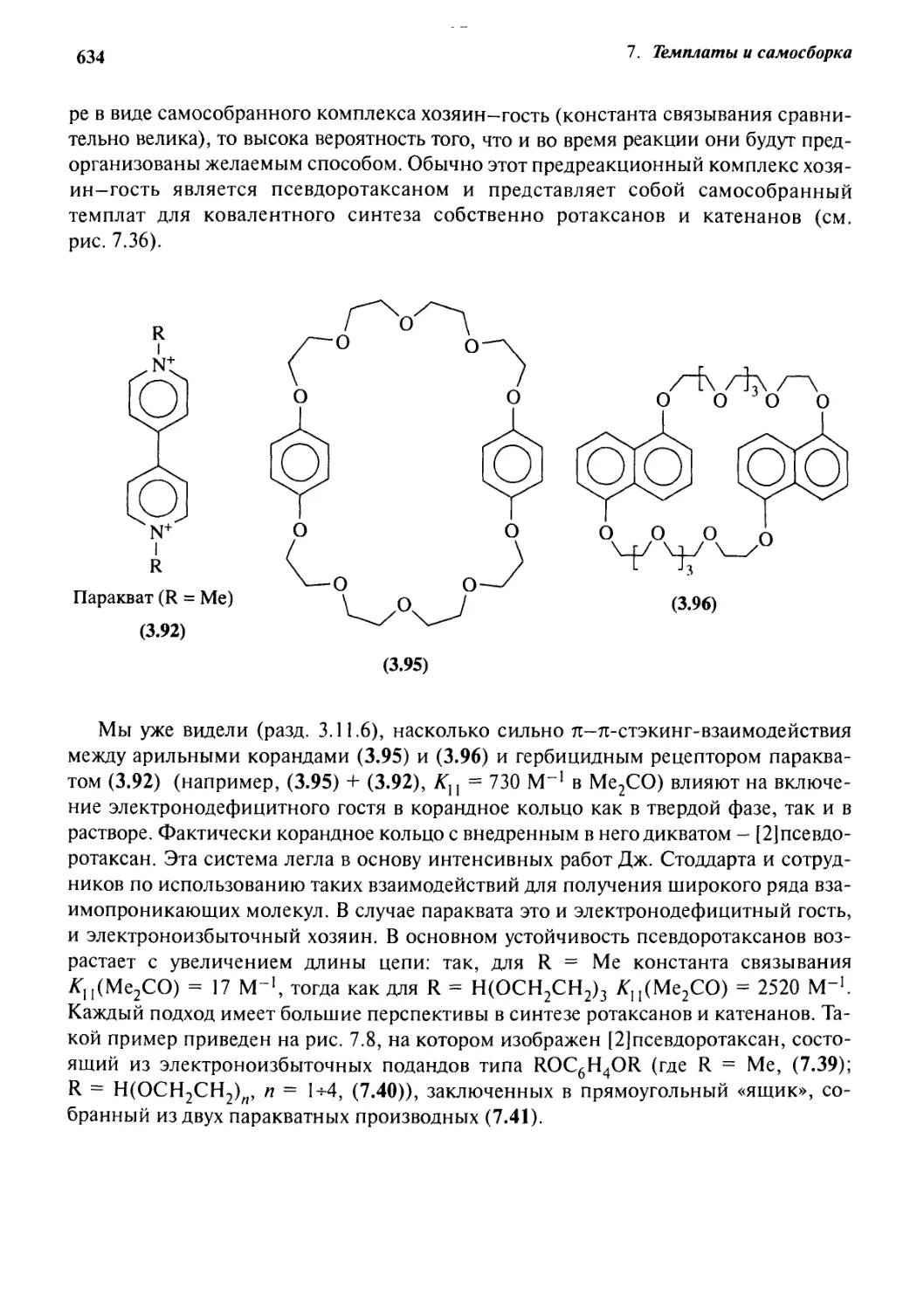
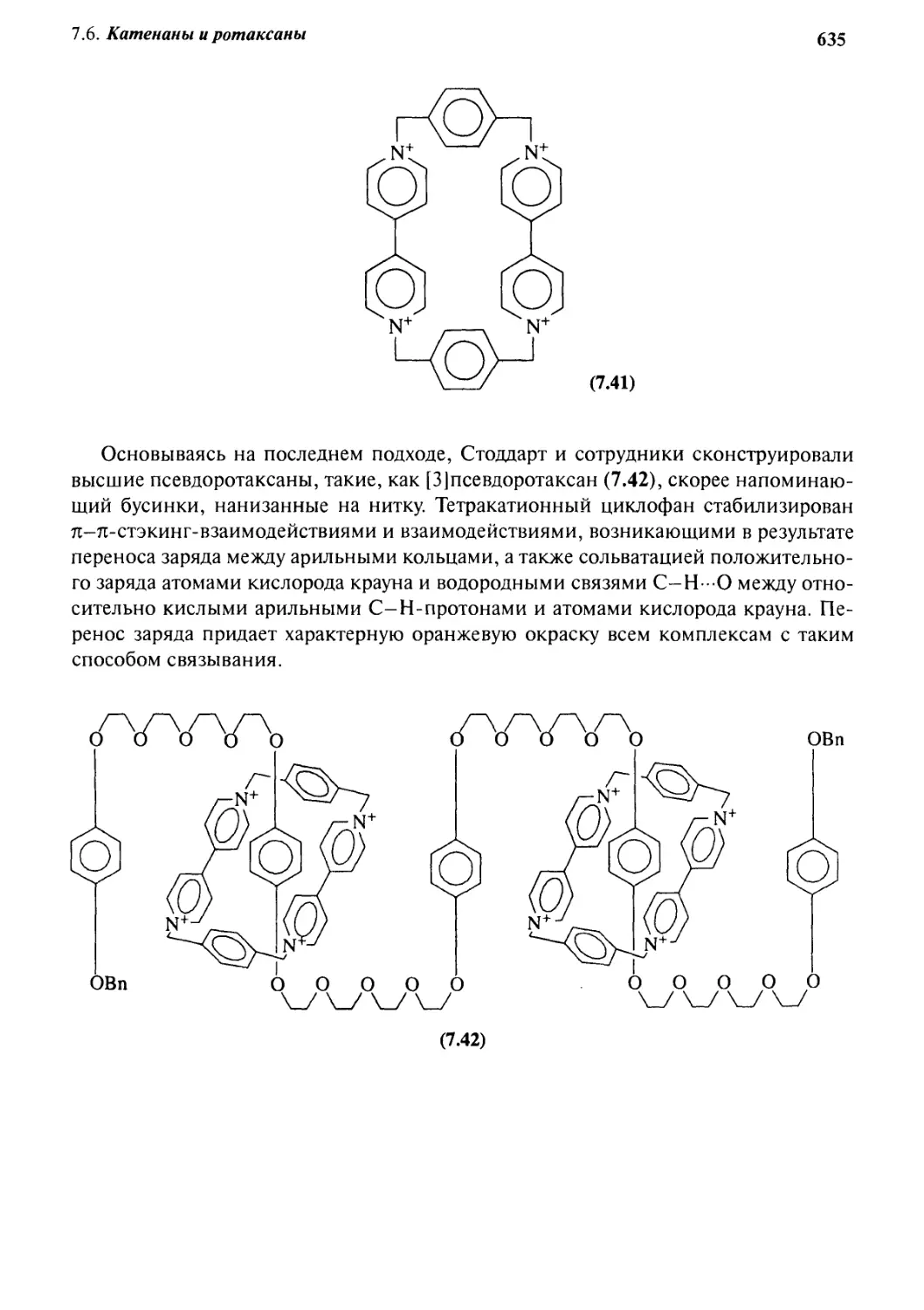
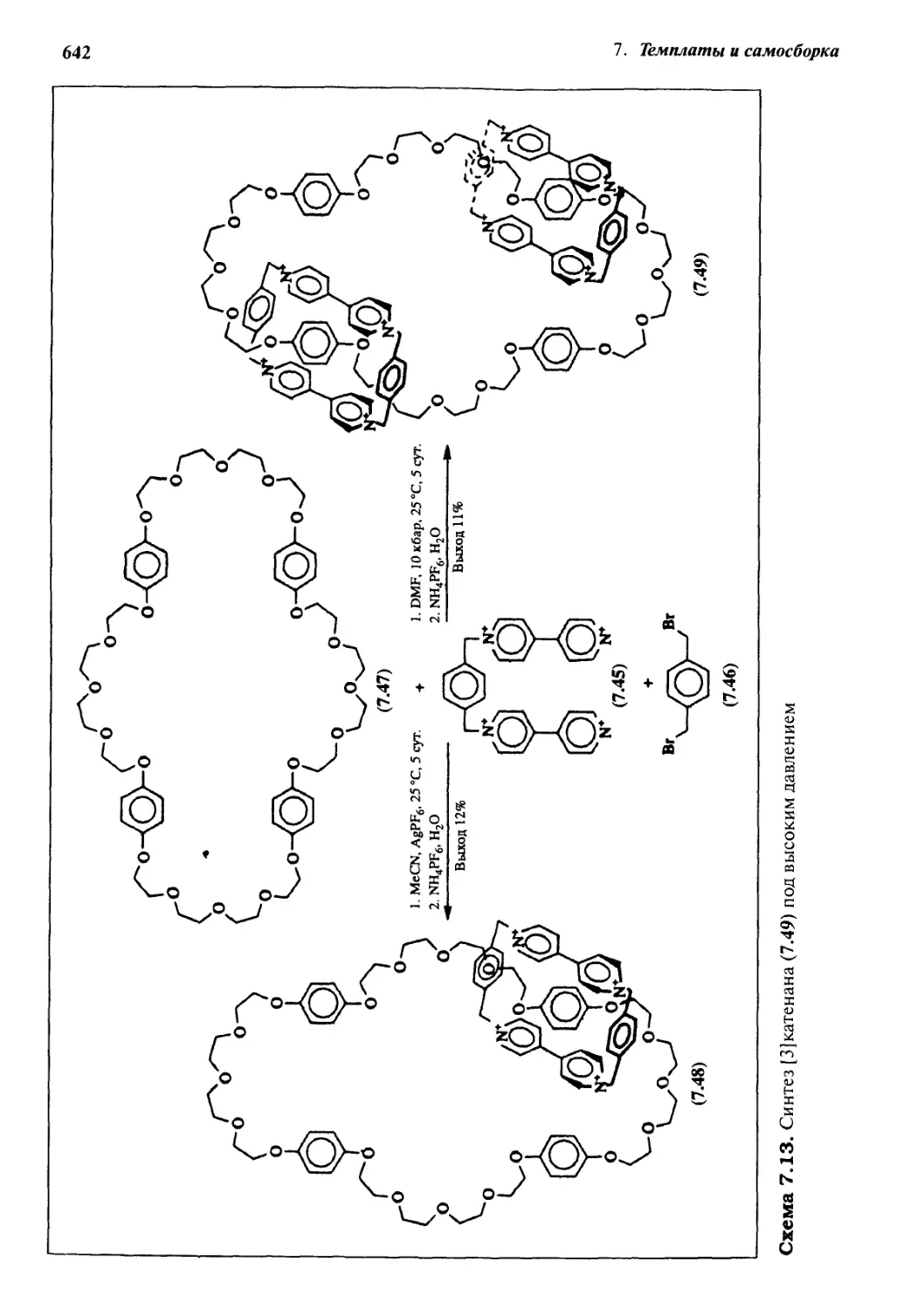
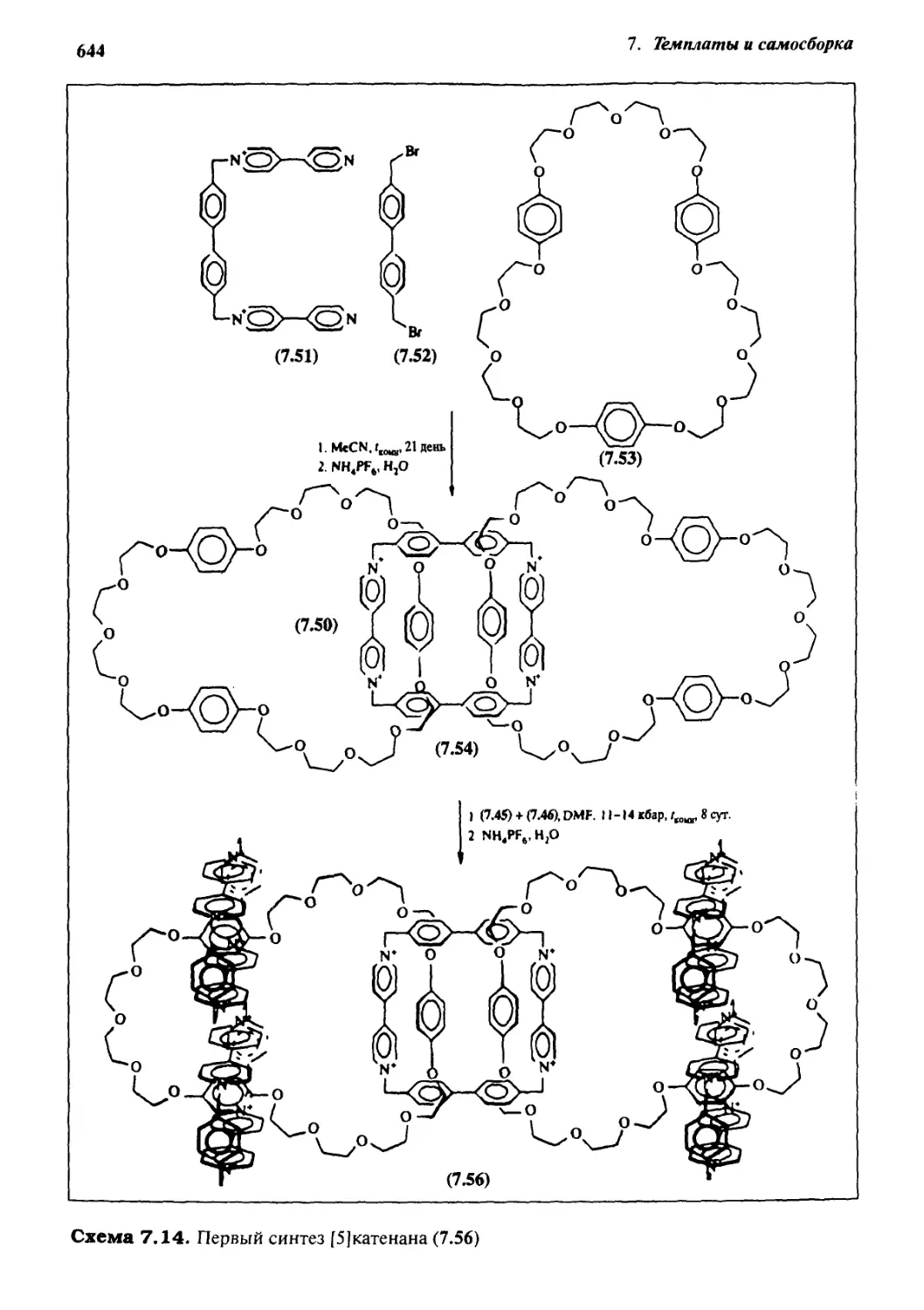
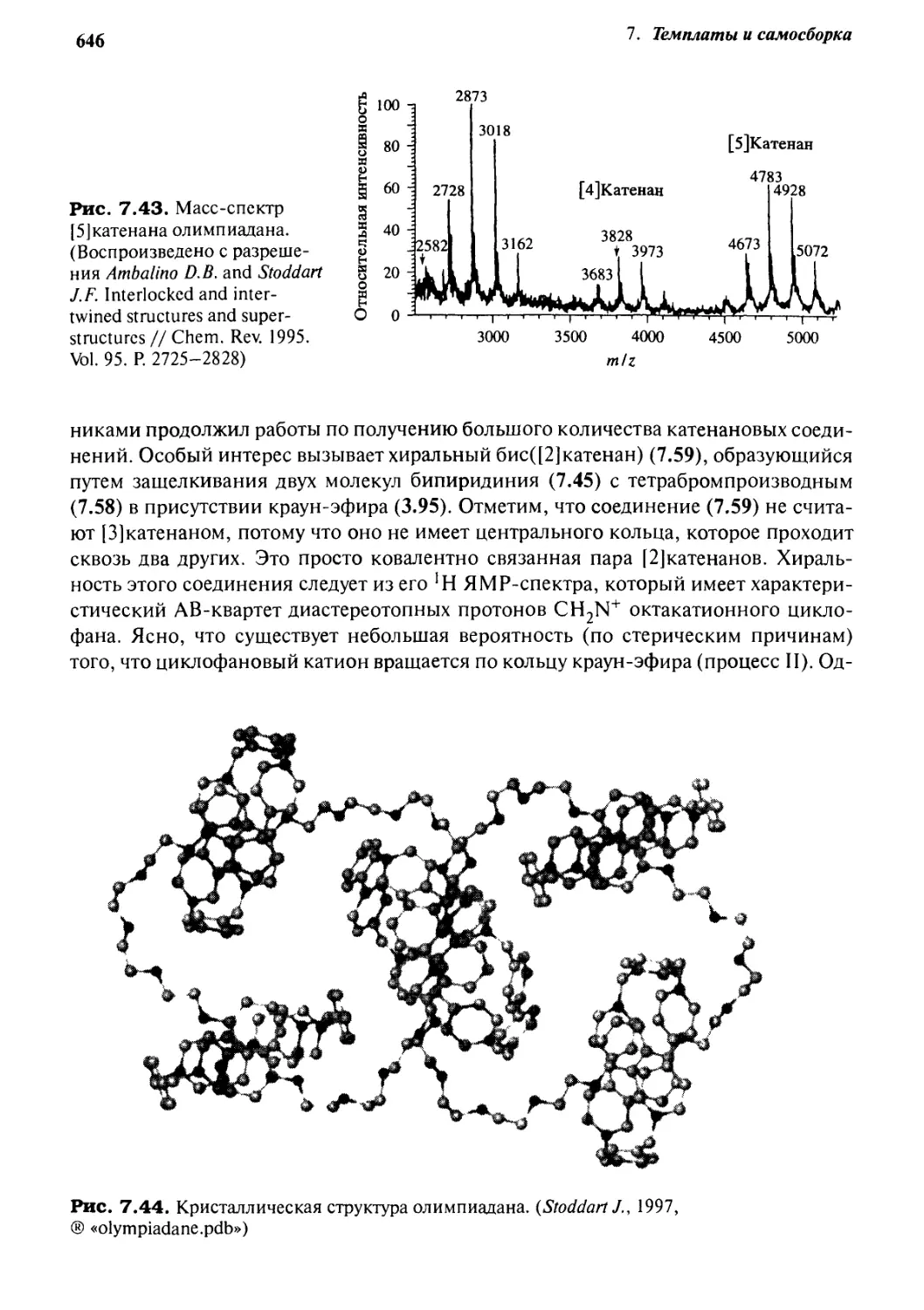
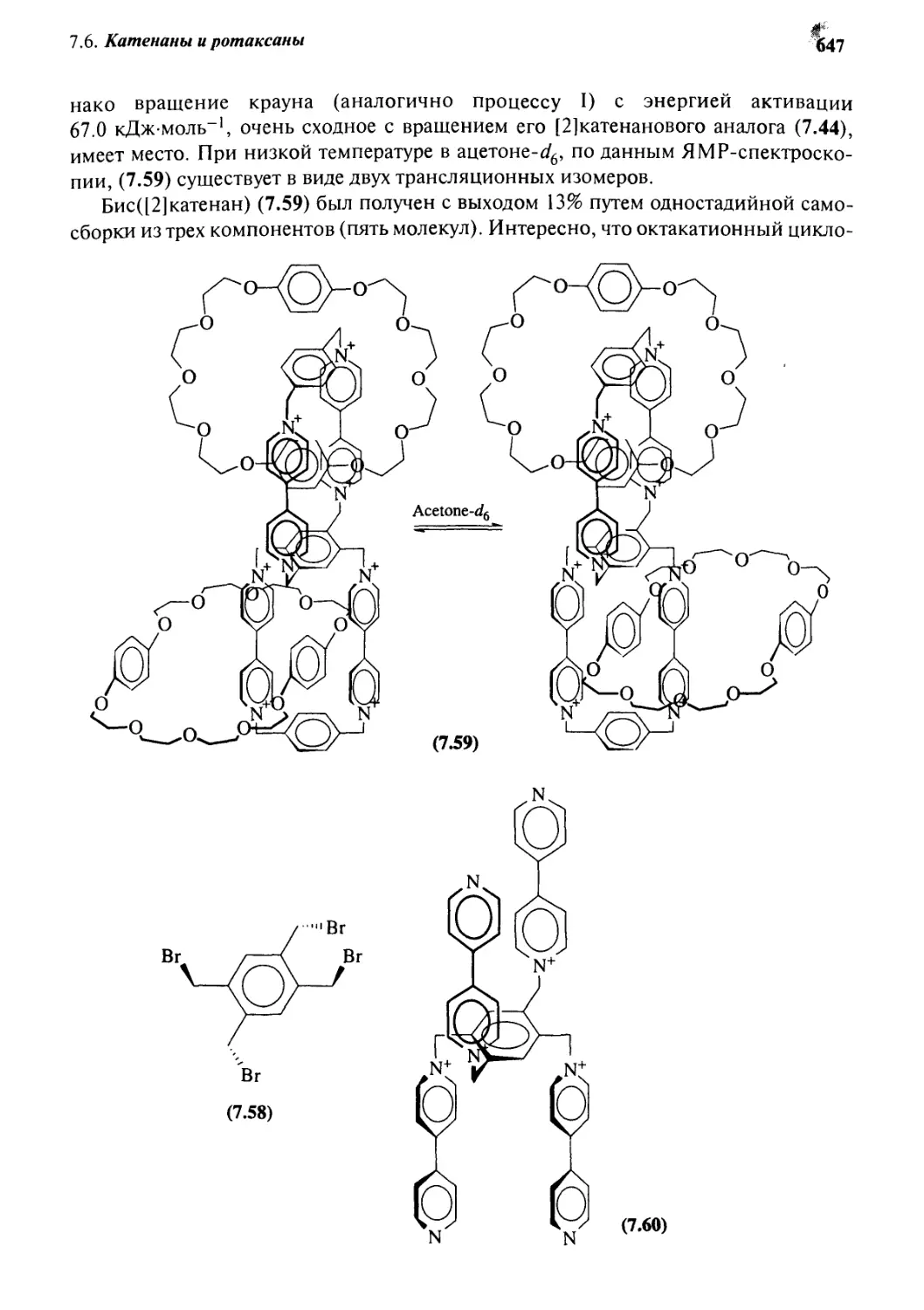
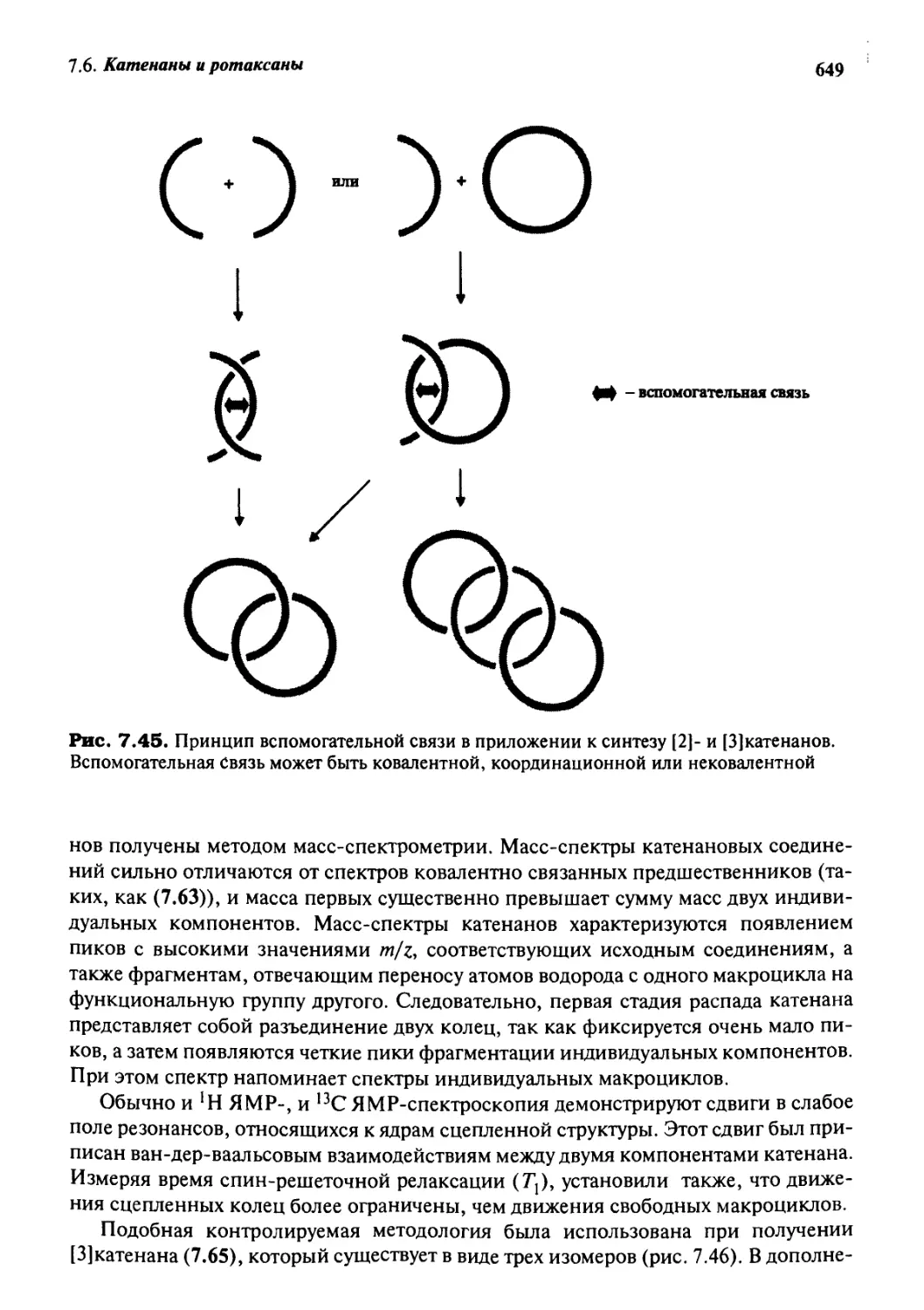
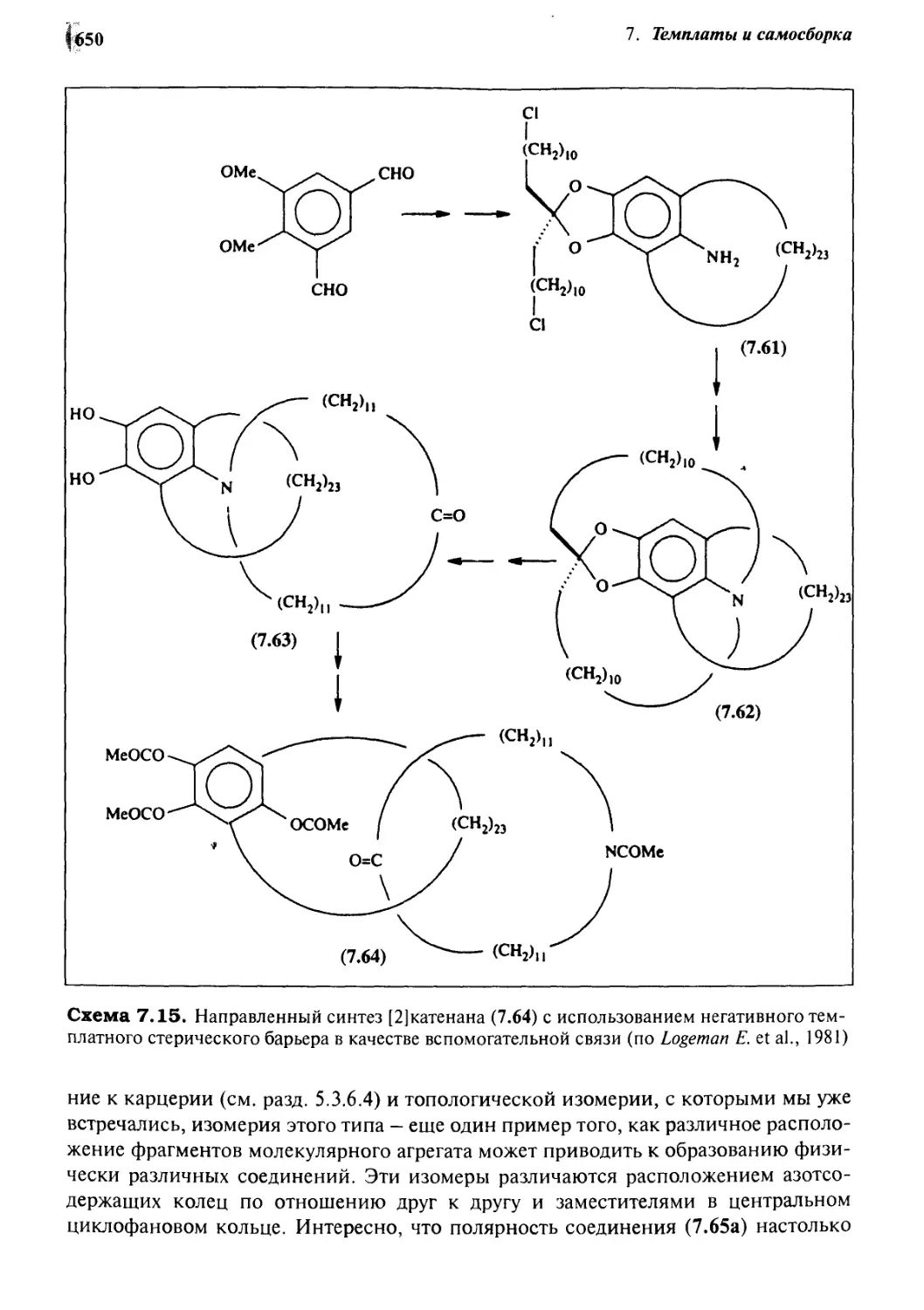
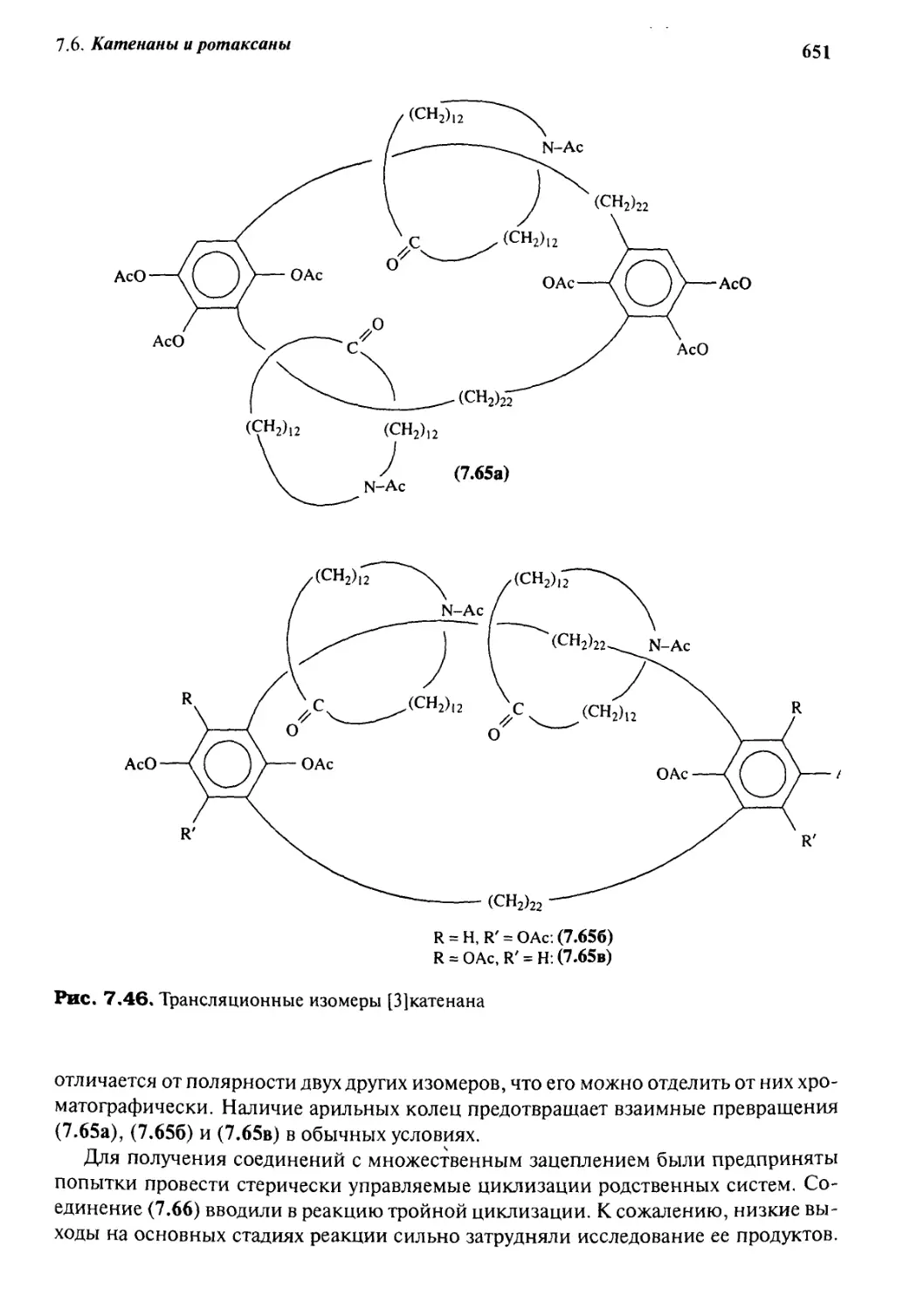
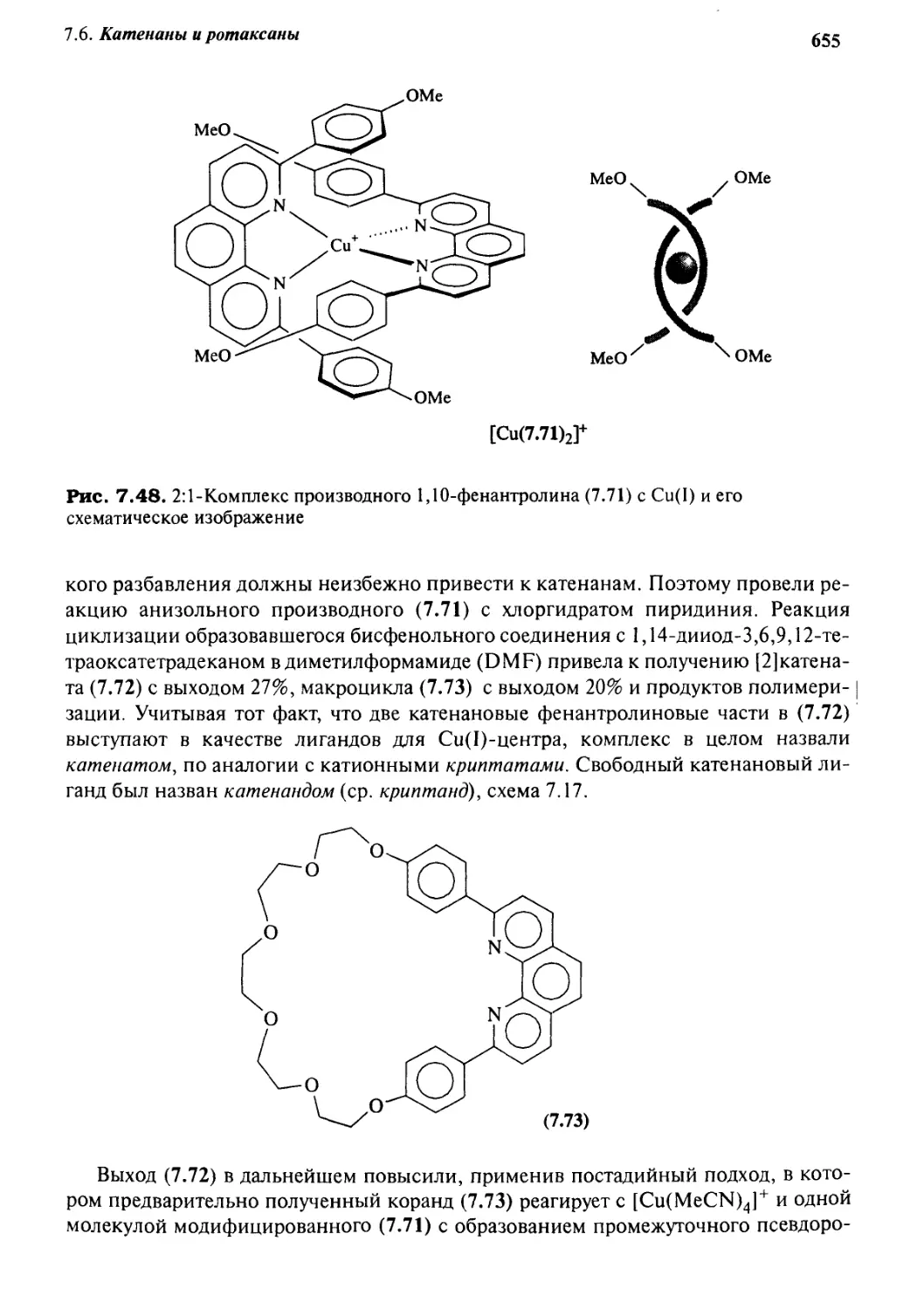
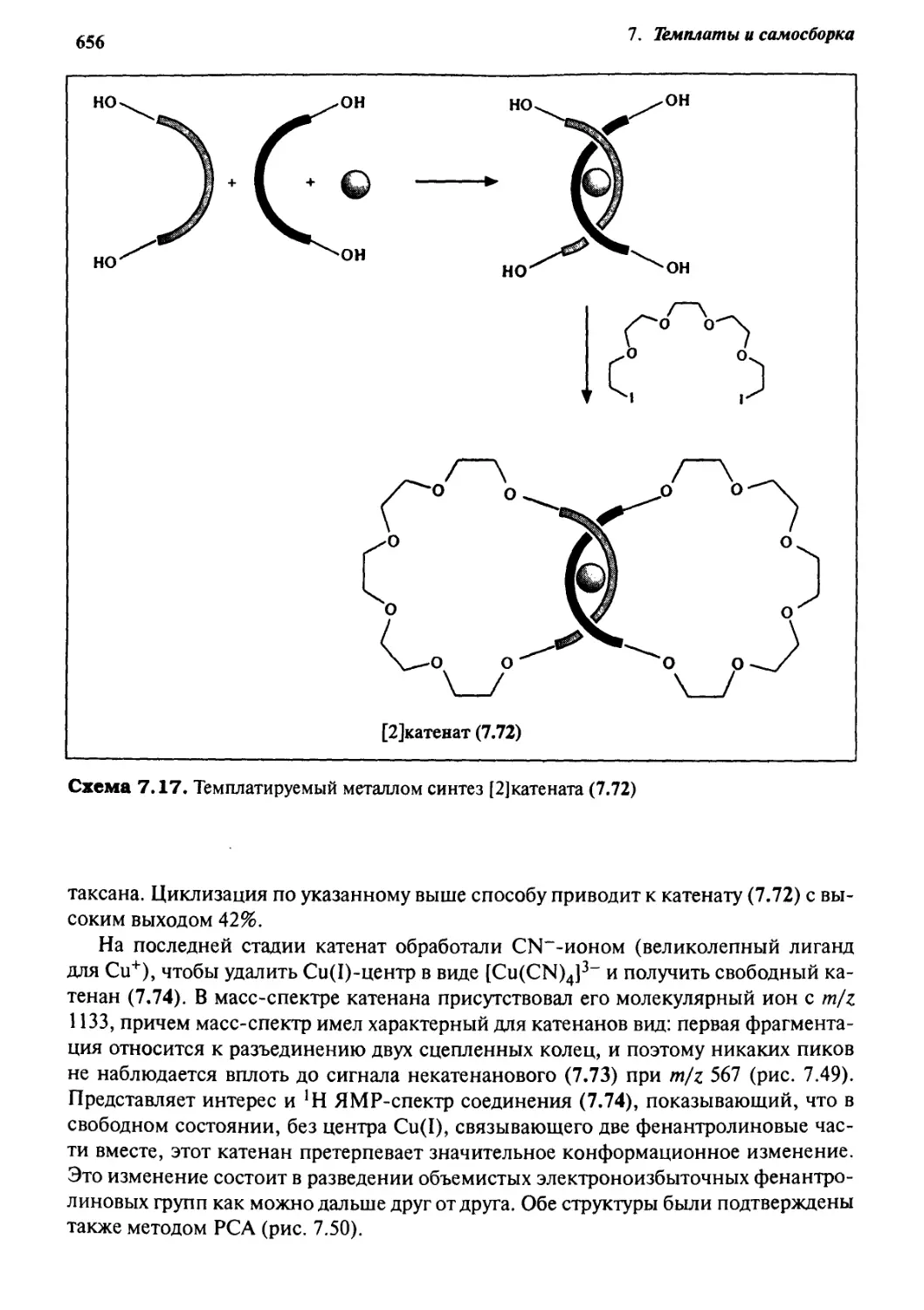
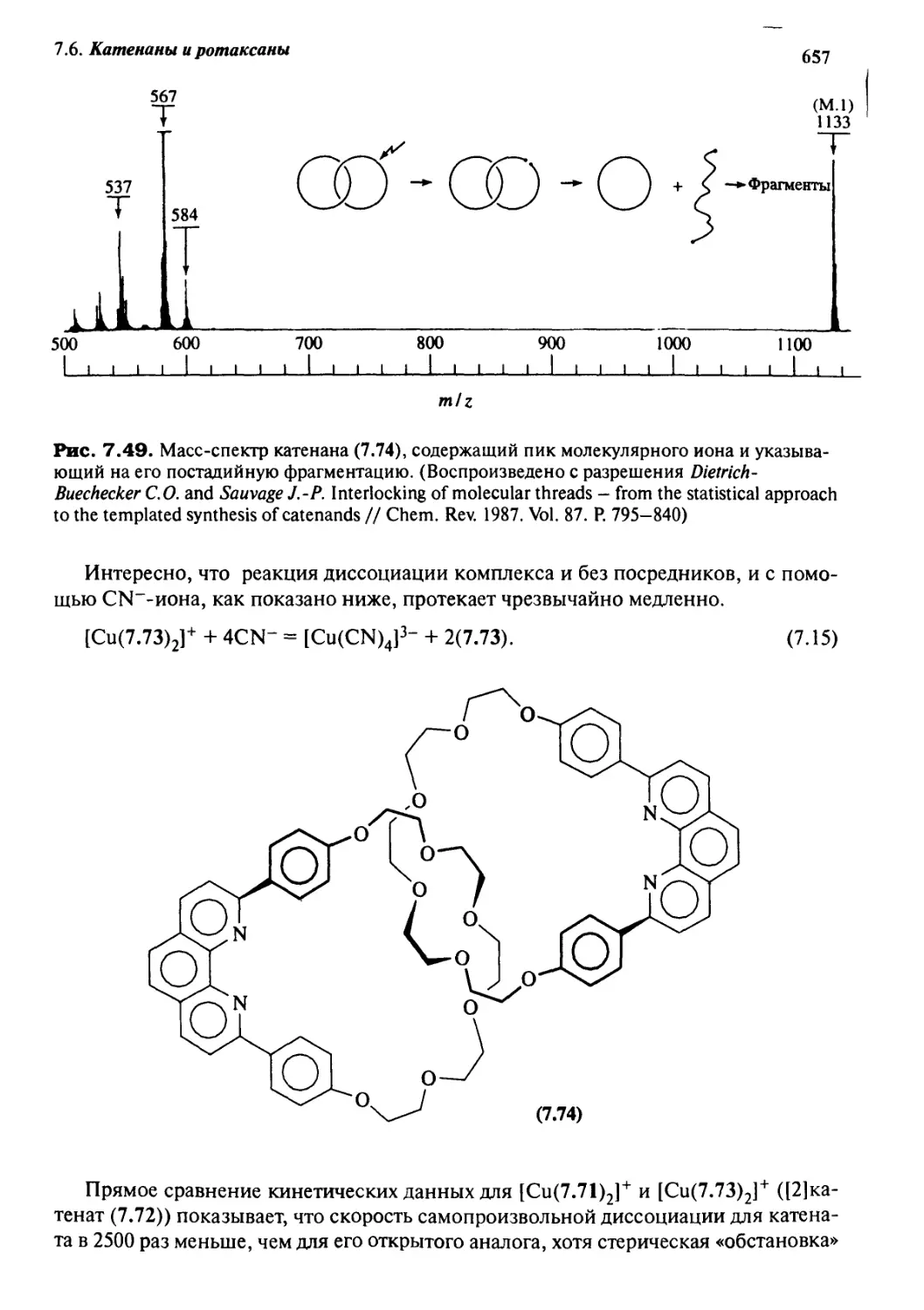
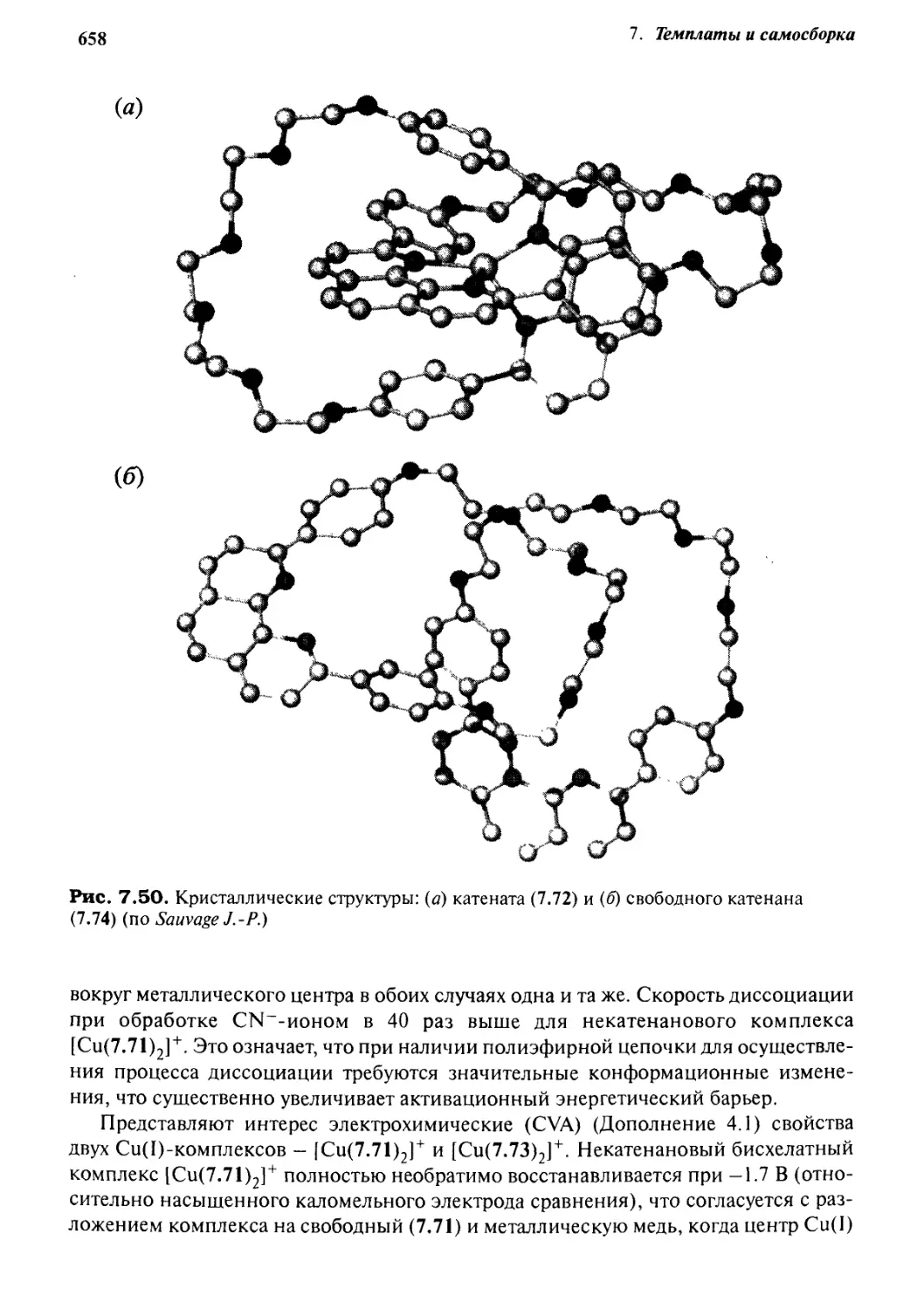
Катенаны и ротаксаны 628
7.6.1 Введение 628
7.6.2 Статистический подход
к синтезу катенанов и ротаксанов 632
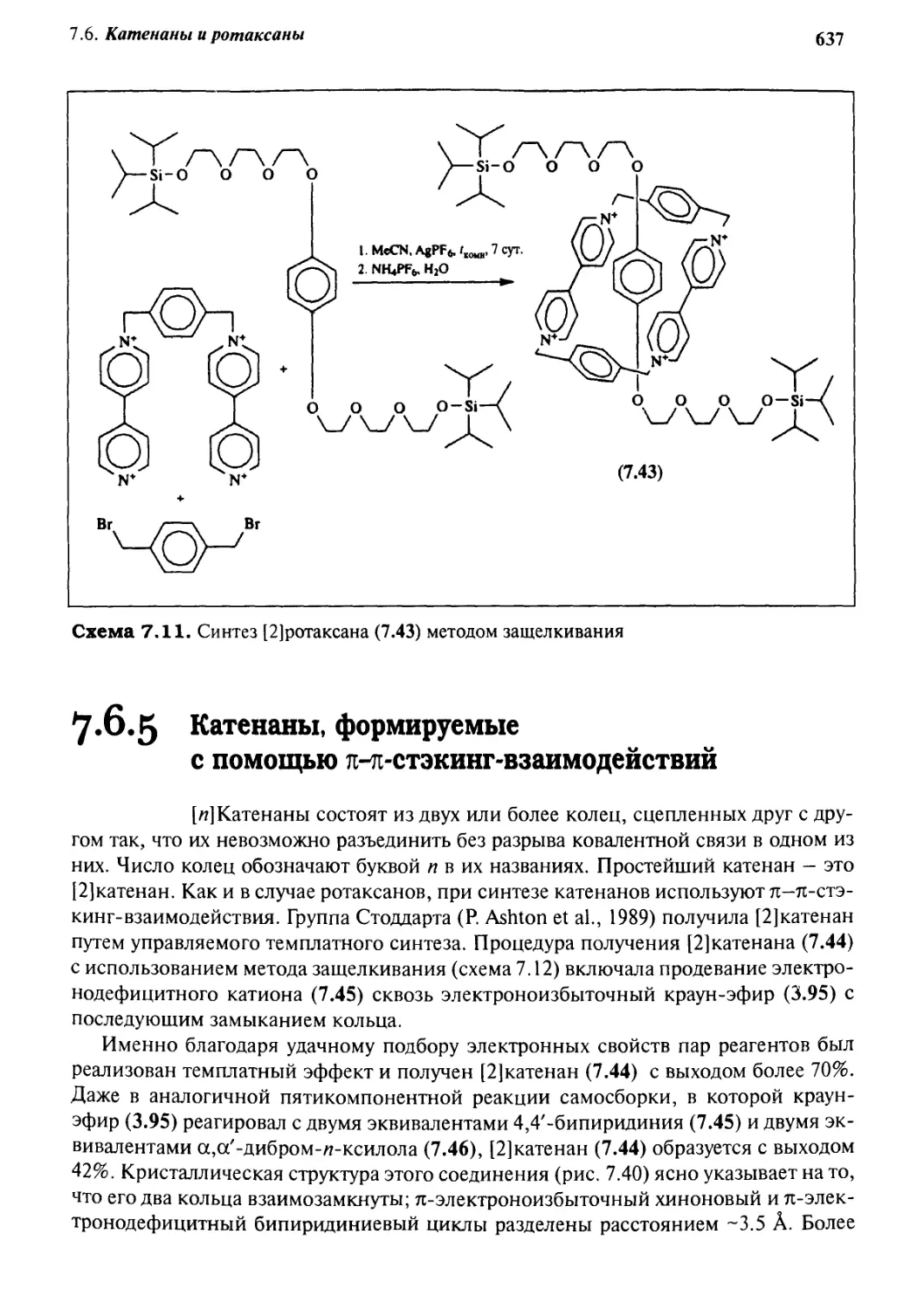
7.6.3 Псевдоротаксаны 633
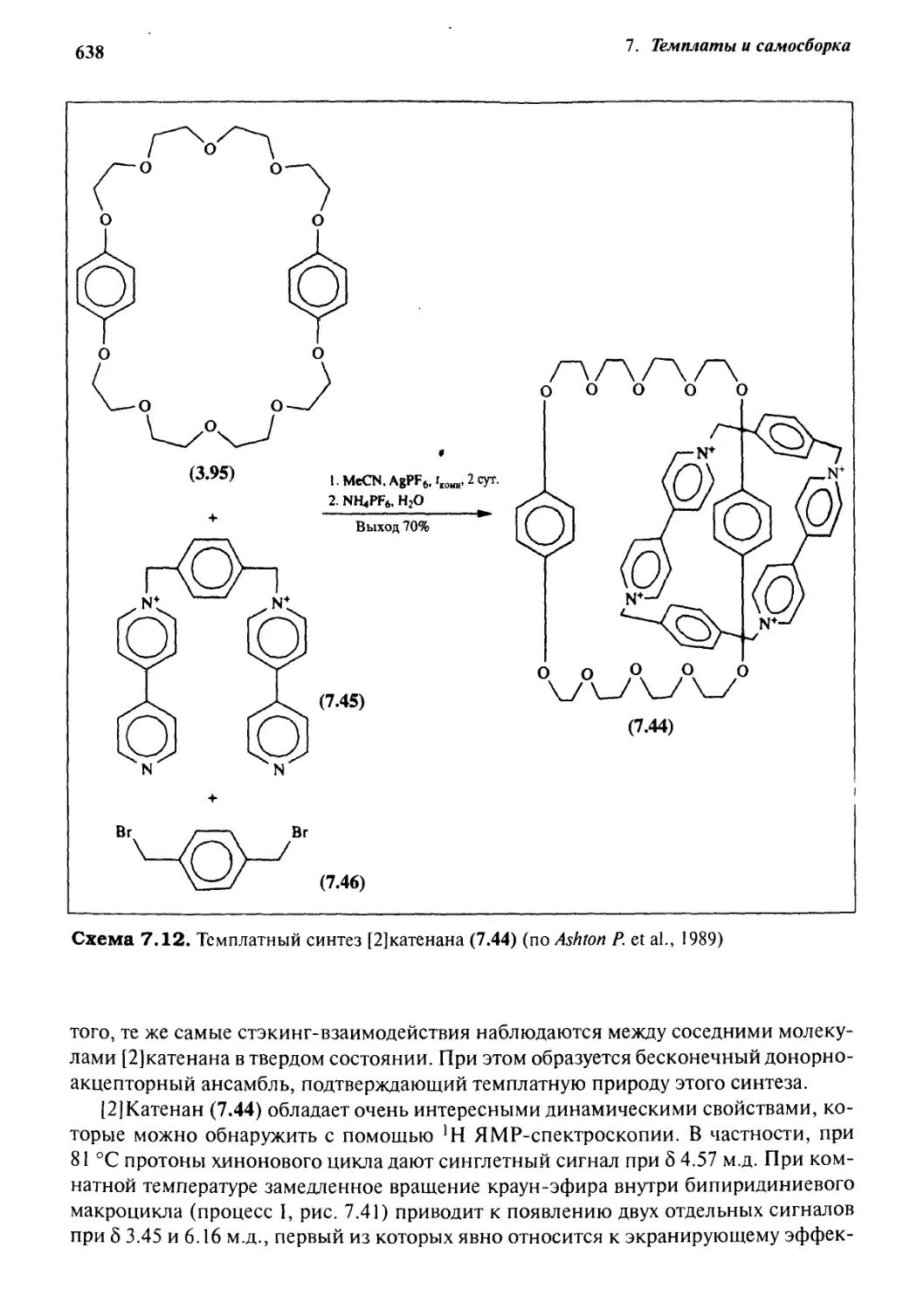
7.6.4 Ротаксаны 636
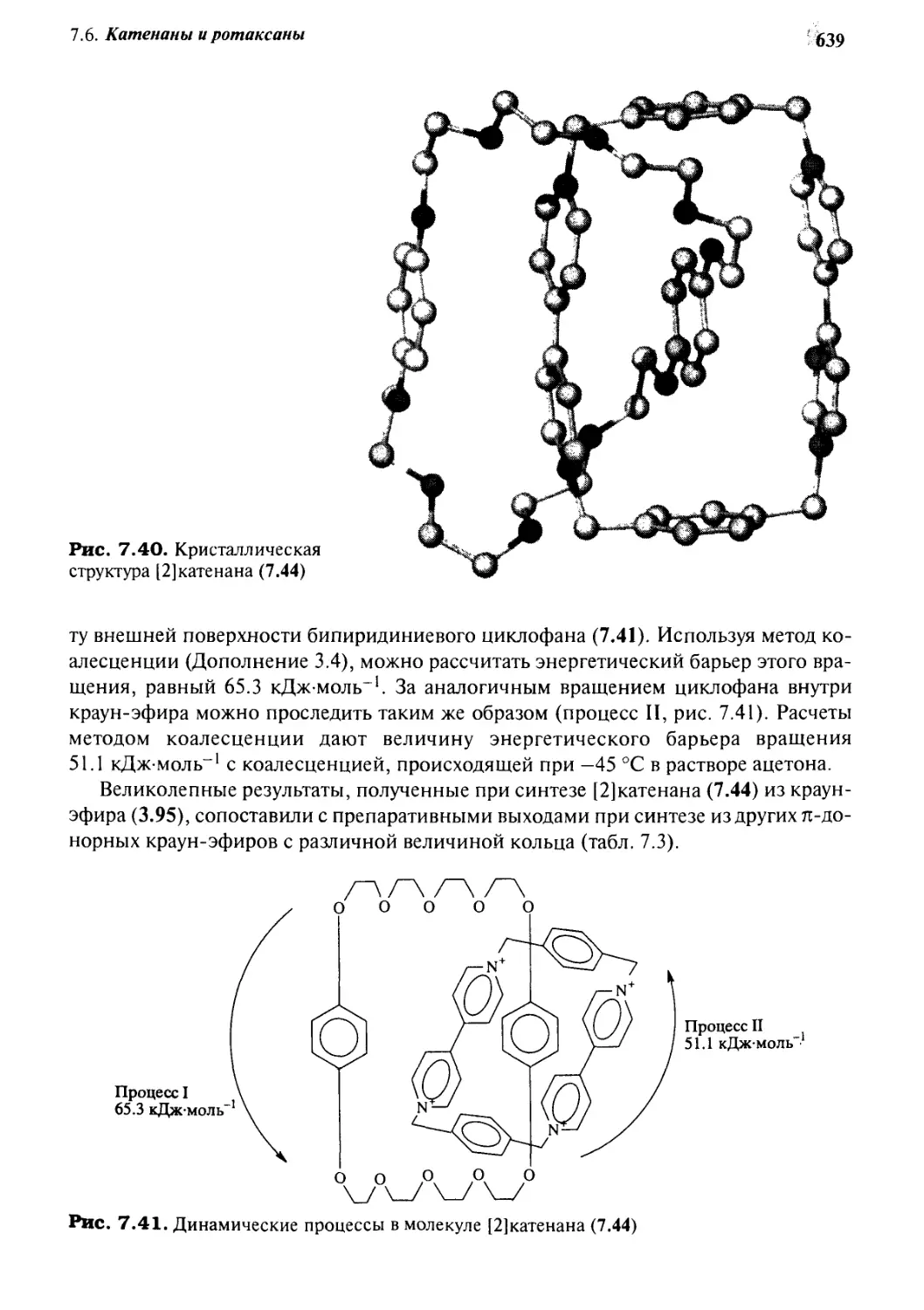
7.6.5 Катенаны, формируемые с помощью
л—71-стэкинг-взаимодействий 637
7.6.6 Принцип «вспомогательной связи» в синтезе катенанов 648
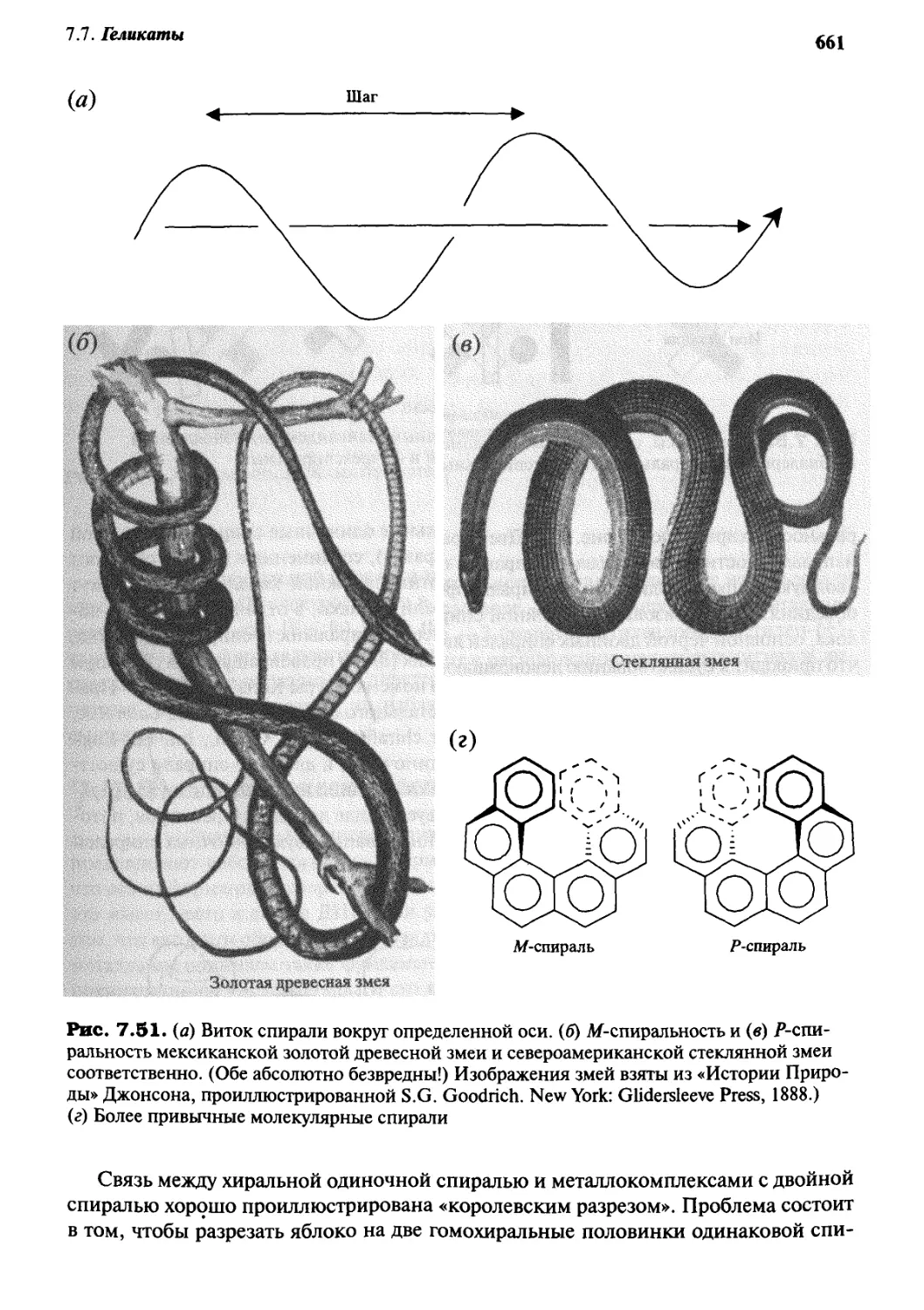
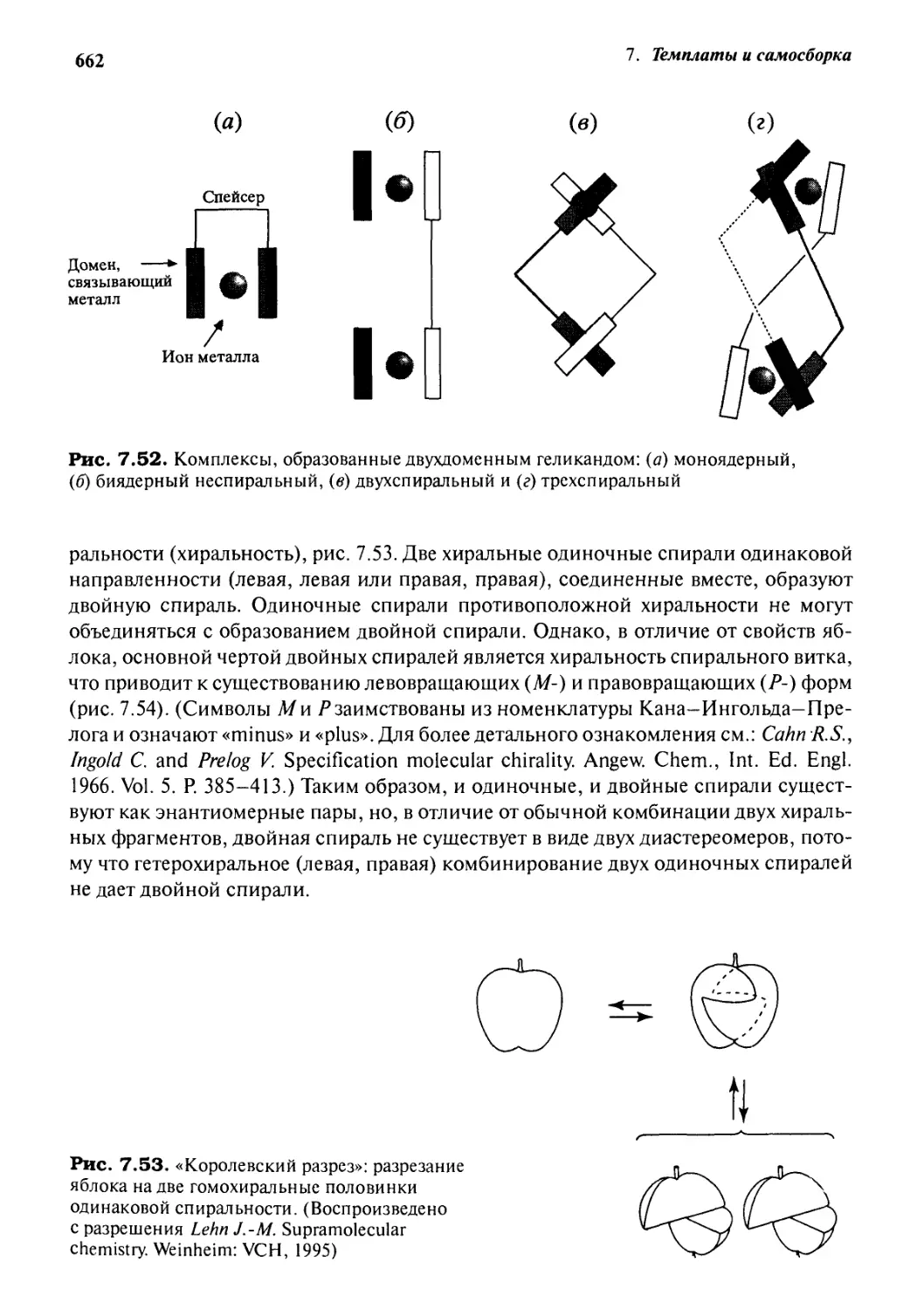
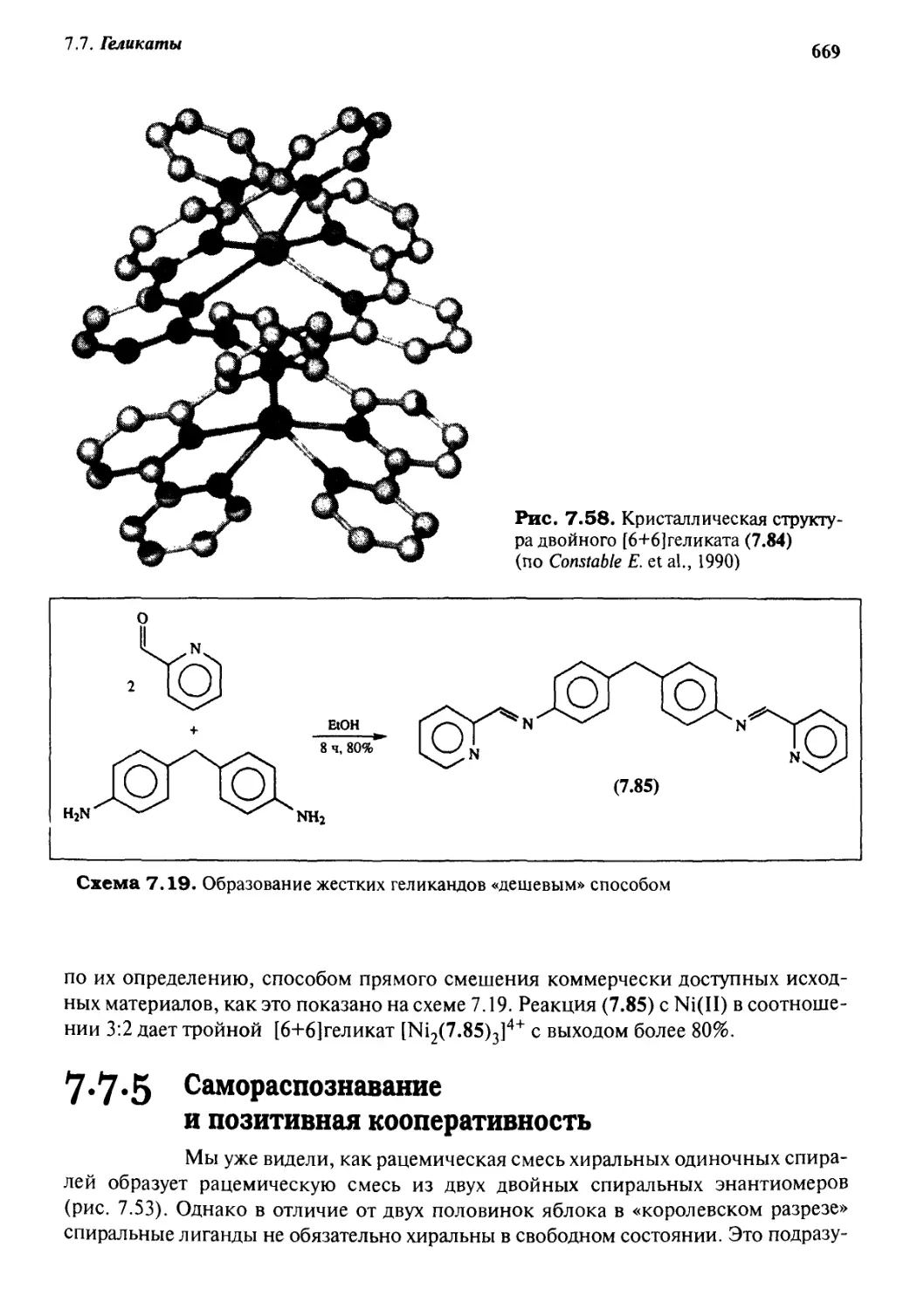
Геликаты 660
7.7.1 Введение 660
7.7.2 Синтетические аспекты 663
7.7.3 ]4+4] Геликаты 665
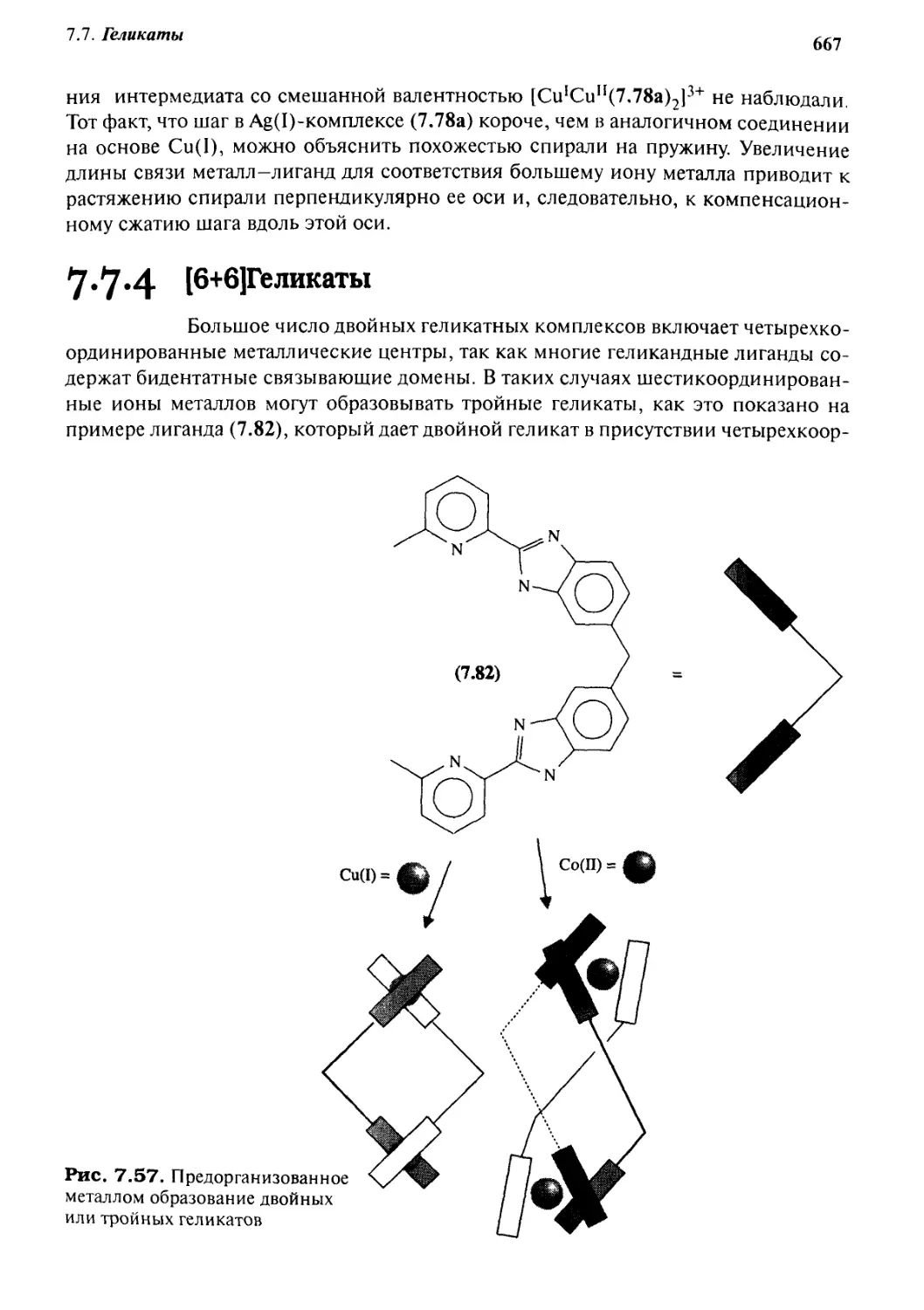
7.7.4 [6+6] Геликаты 667
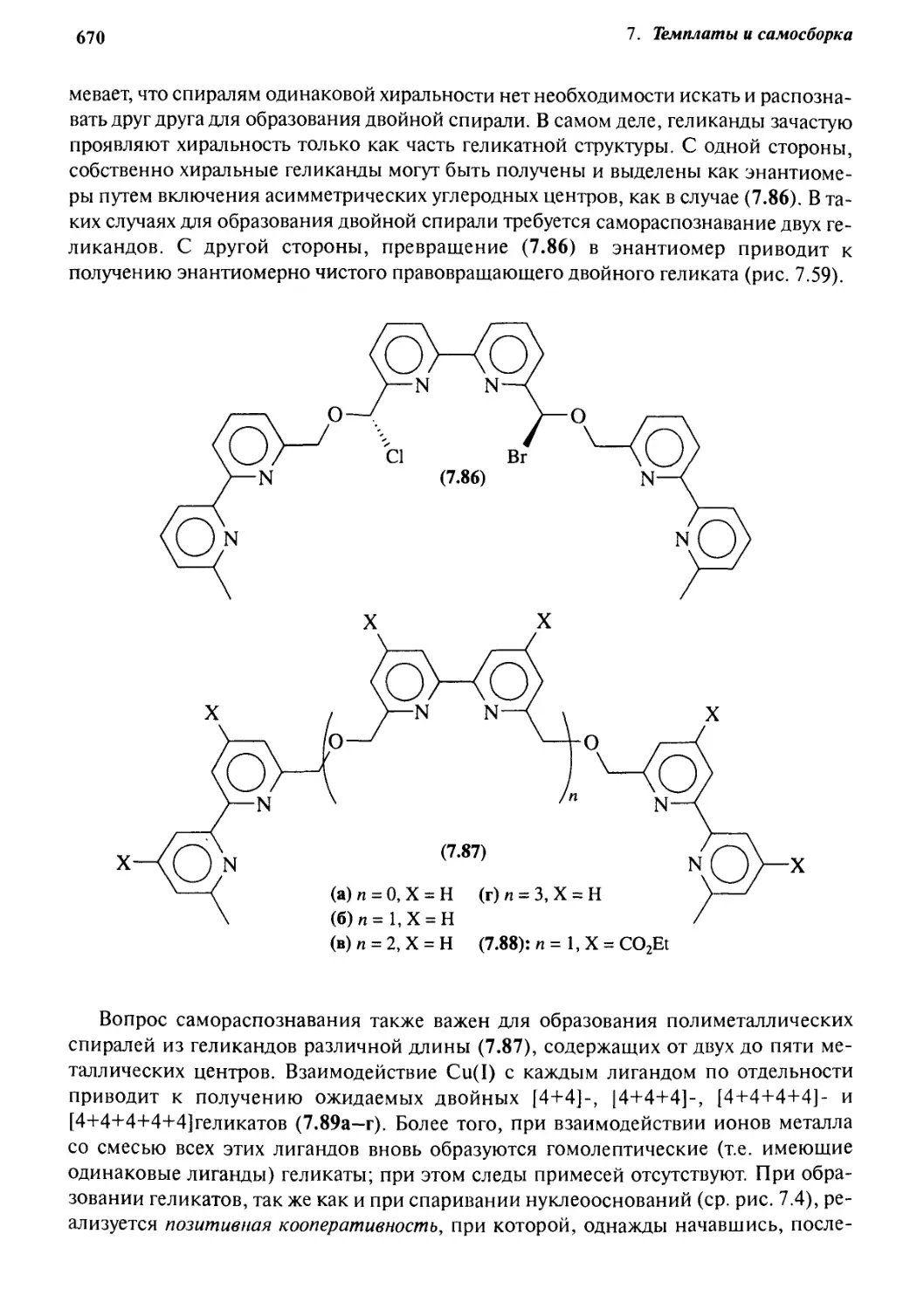
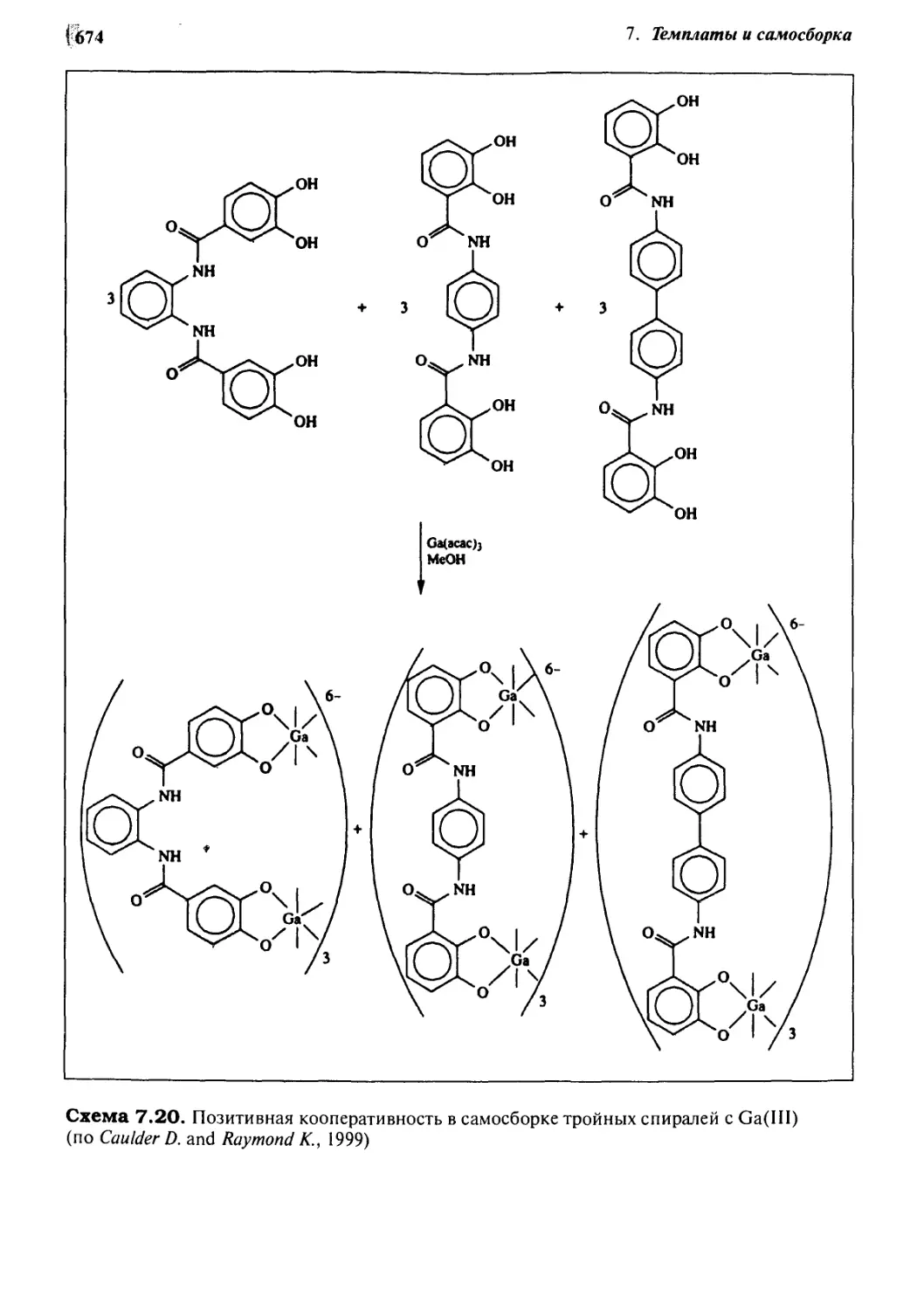
7.7.5 Самораспознавание и позитивная кооперативность 669
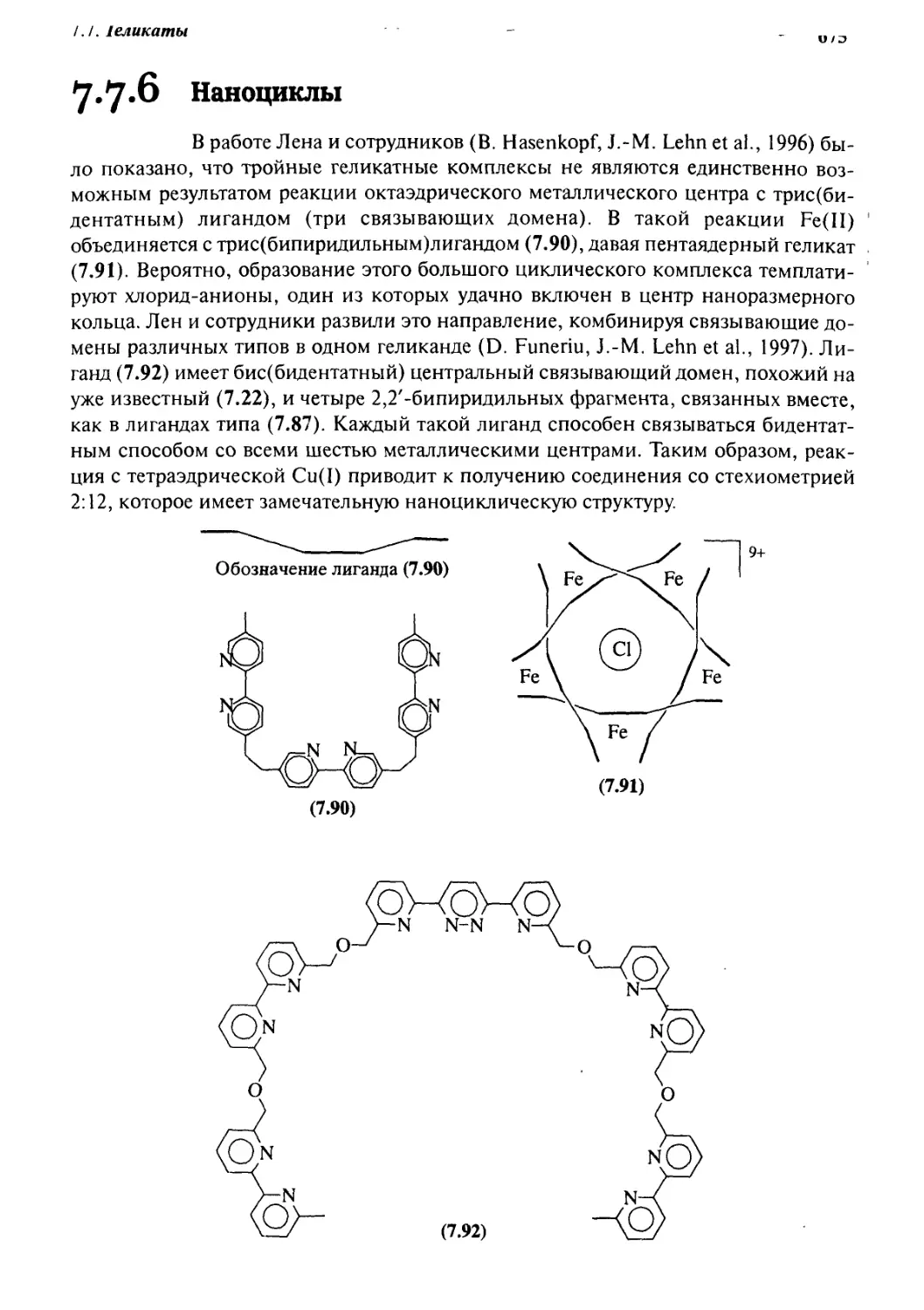
7.7.6 Наноциклы 675
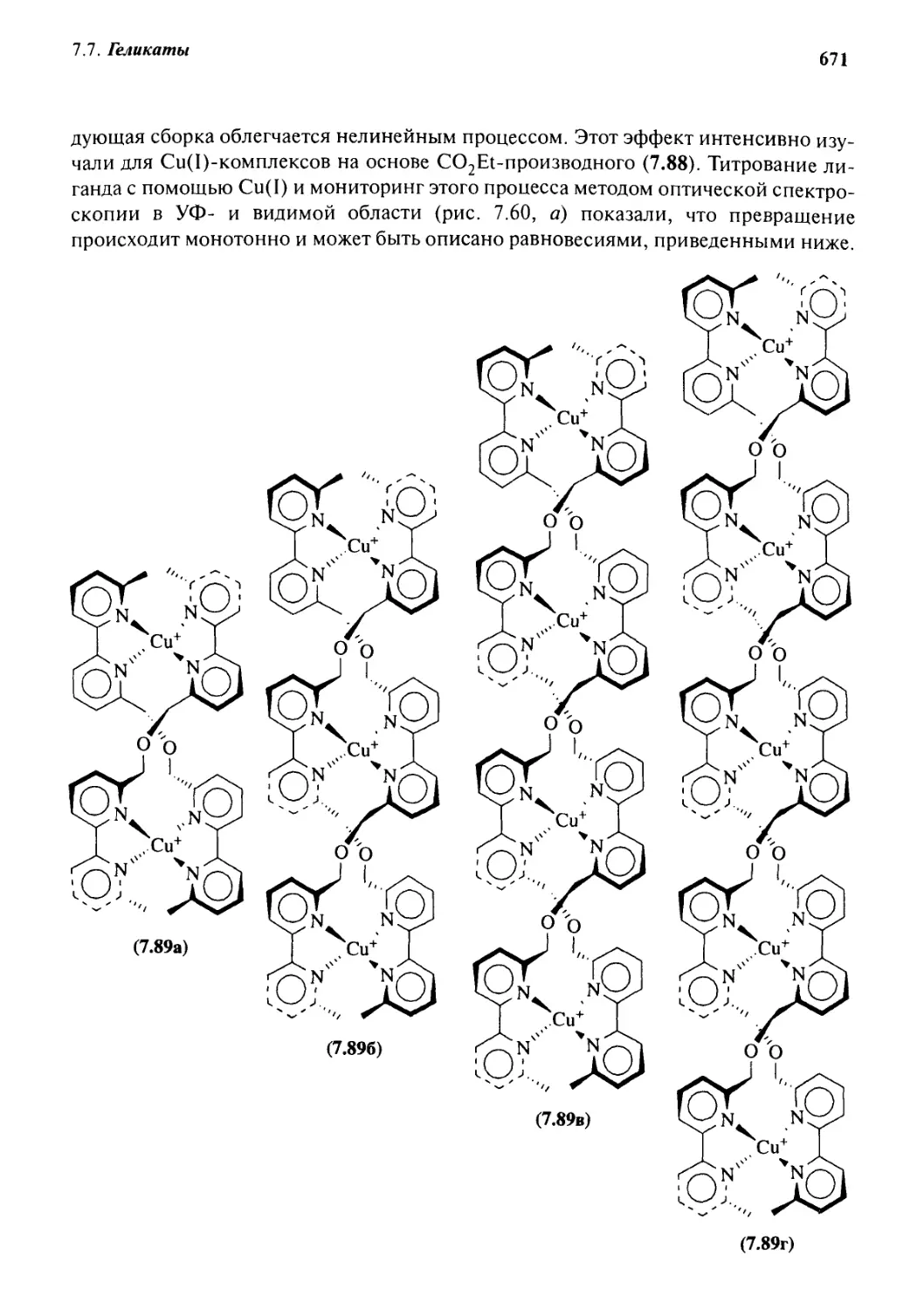
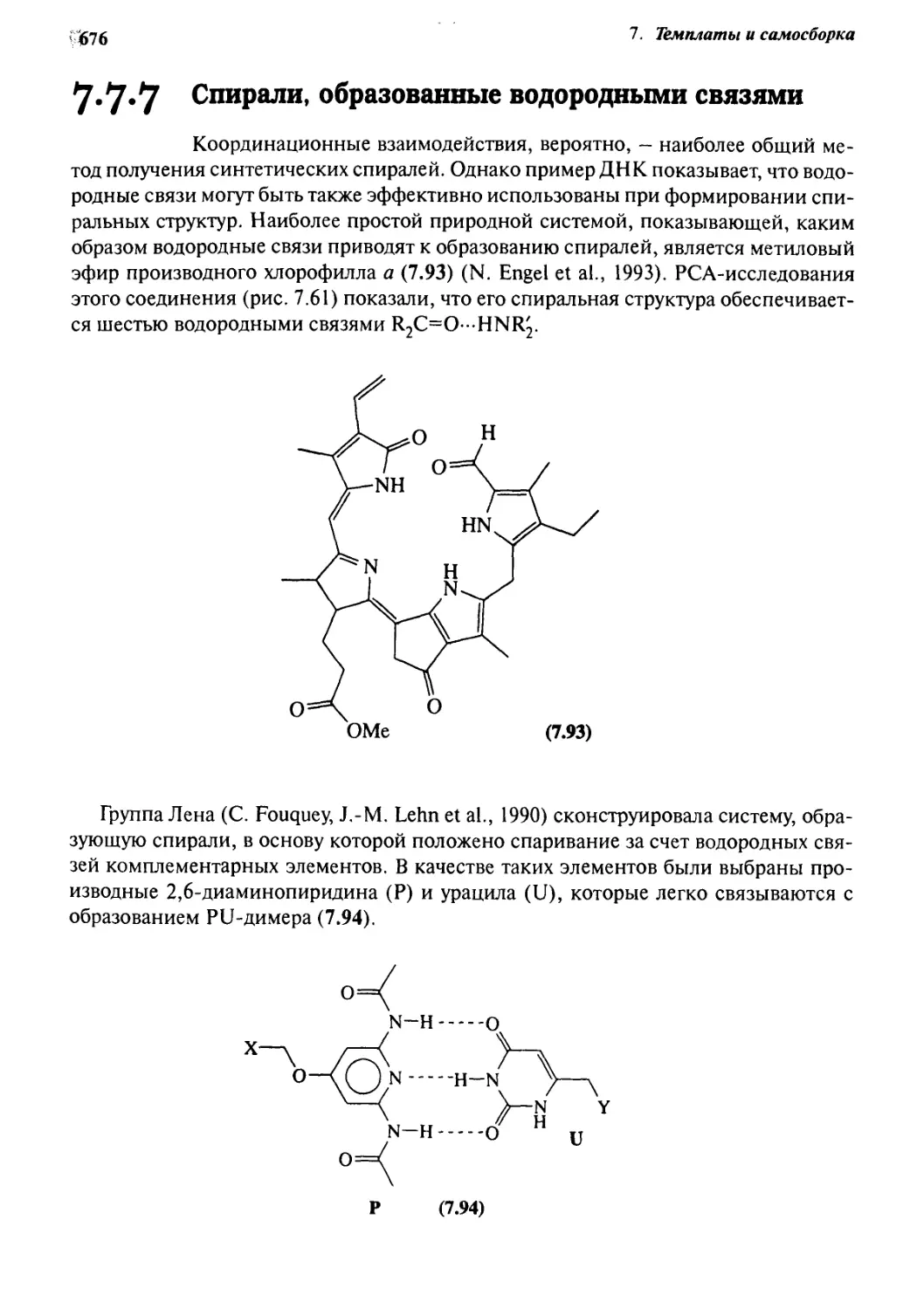
7.7.7 Спирали, образованные водородными связями 676
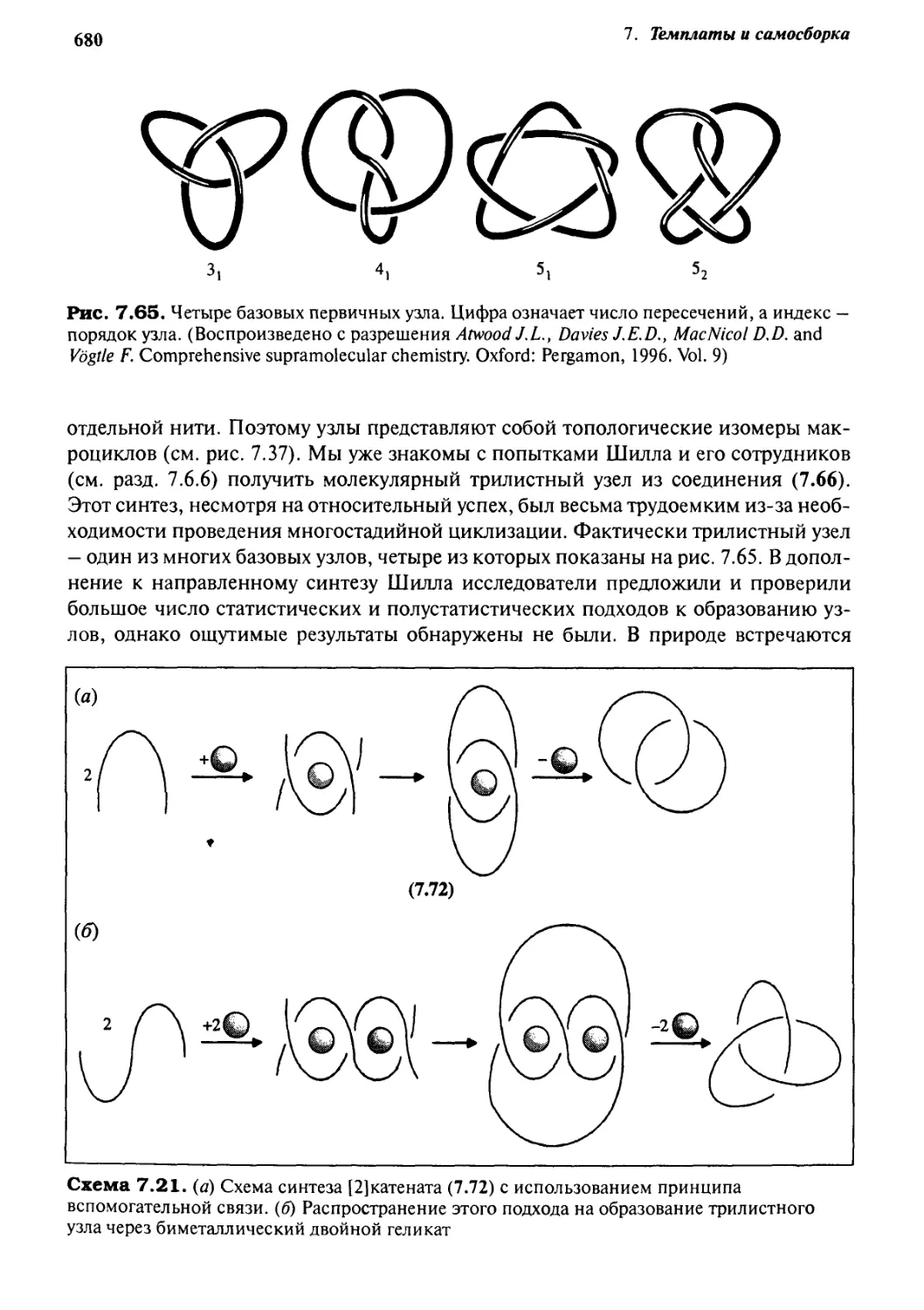
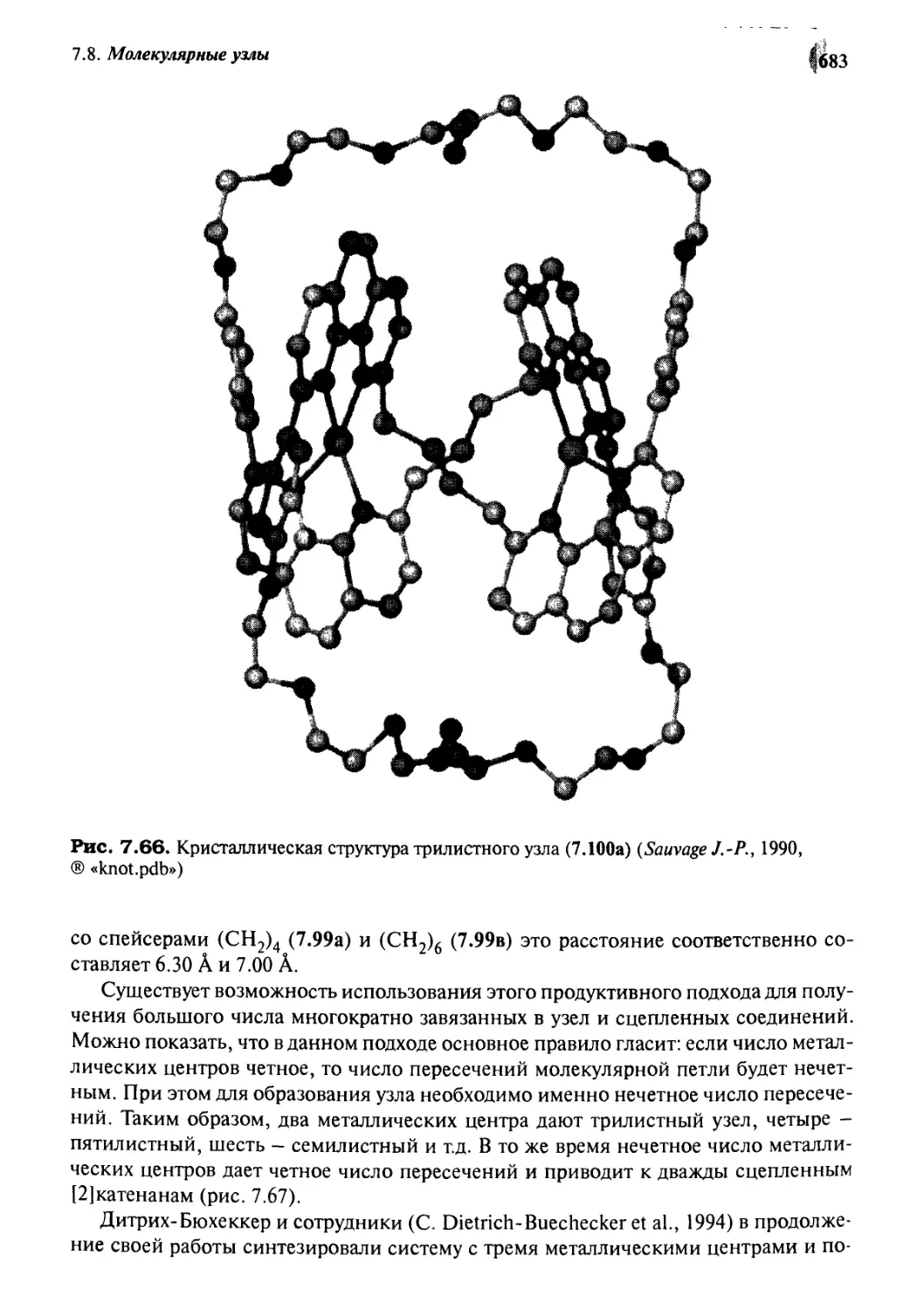
7.8 Молекулярные узлы
679
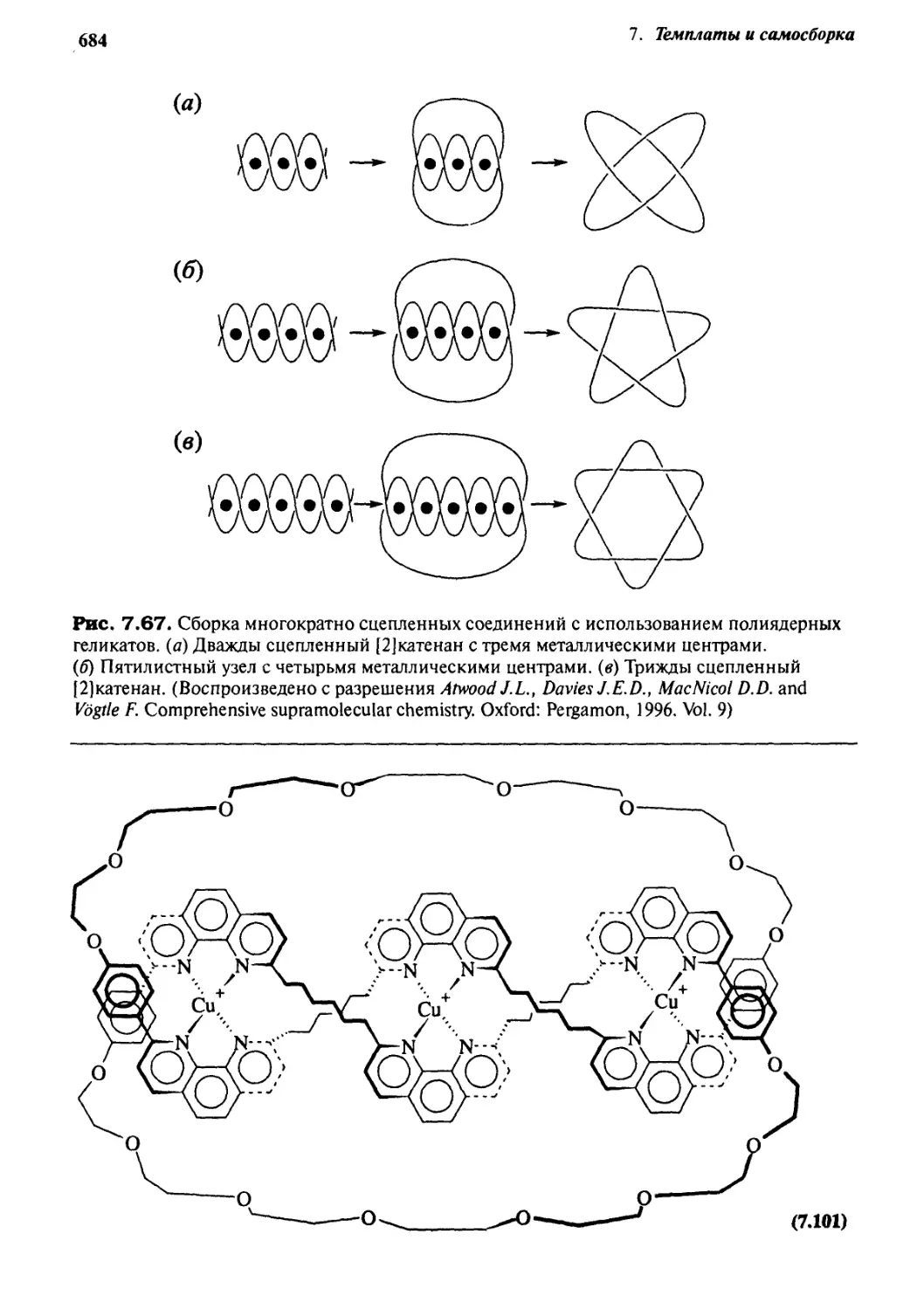
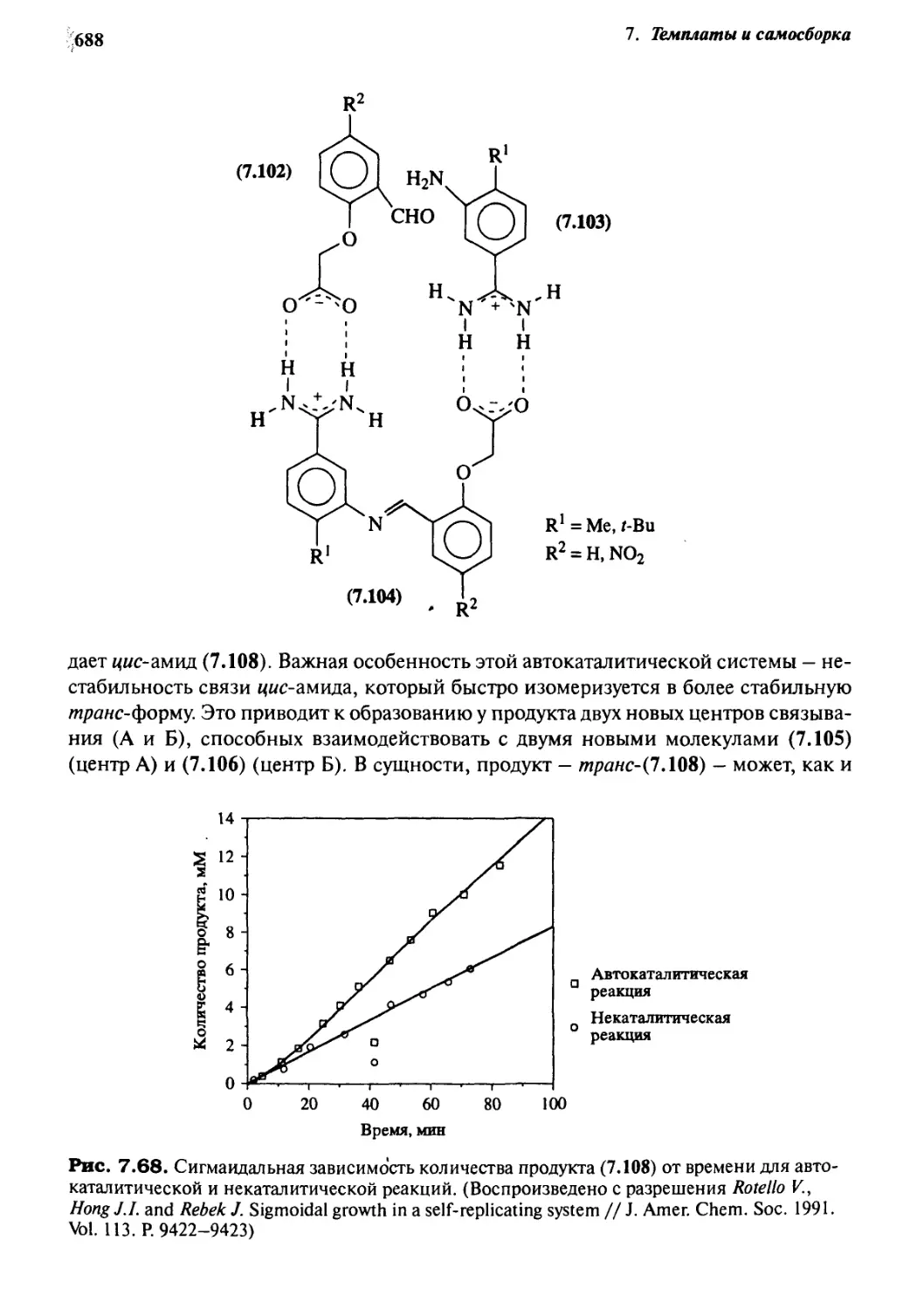
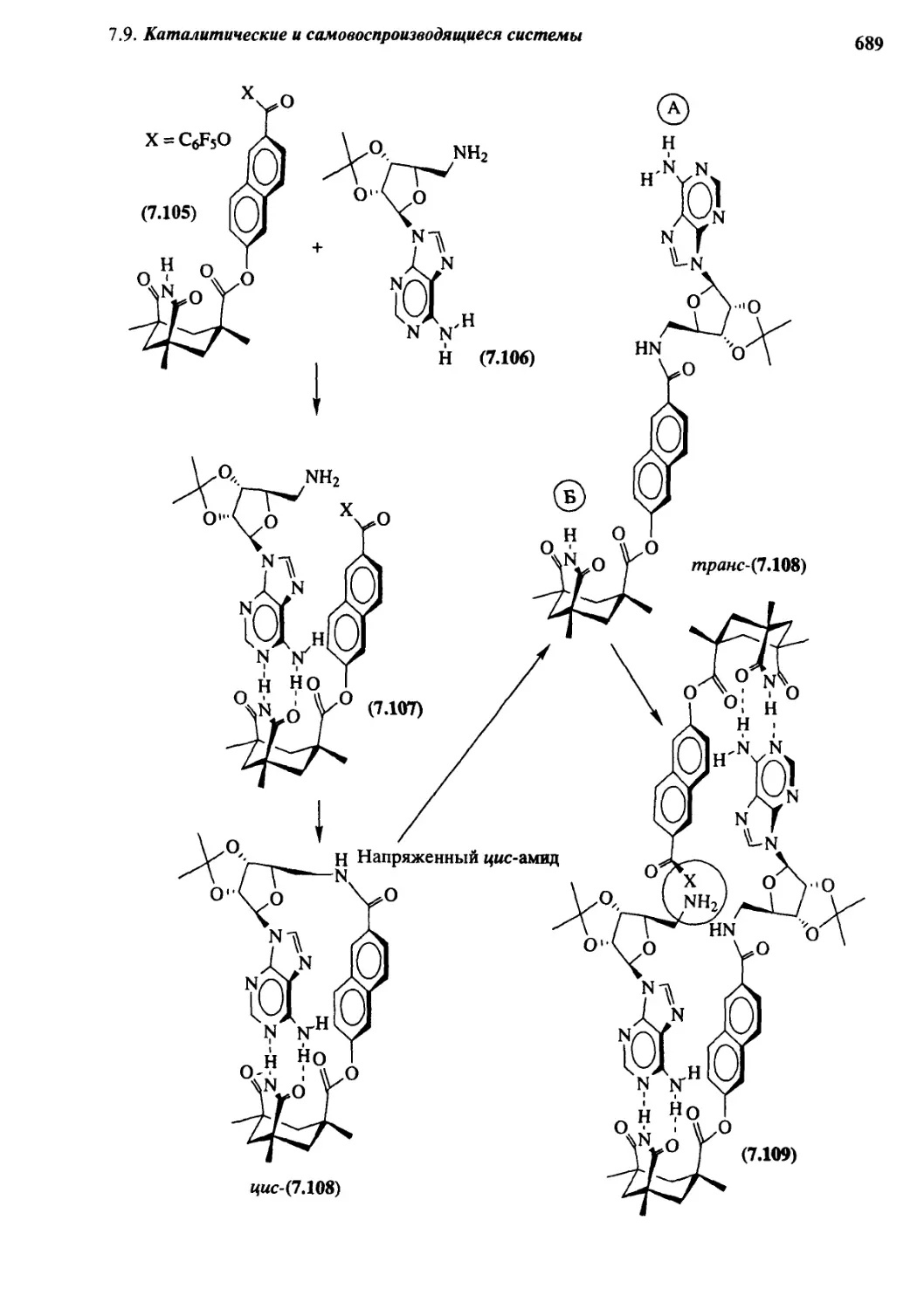
,С| Каталитические и самовоспроизводящиеся системы 685
Учебные задания 690
Мысленный эксперимент 691
Литература 692
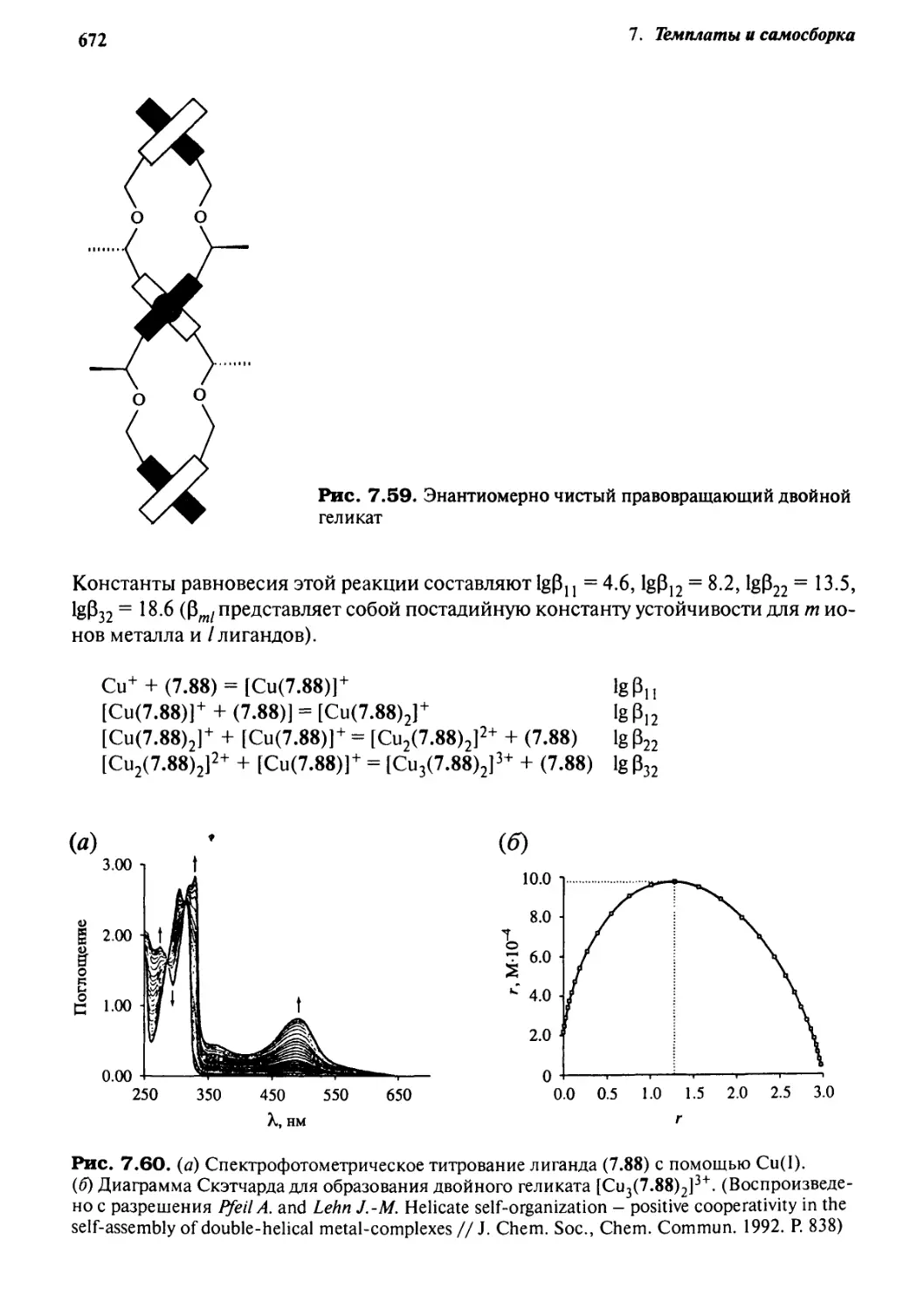
Оглавление
8
Молекулярные устройства 695
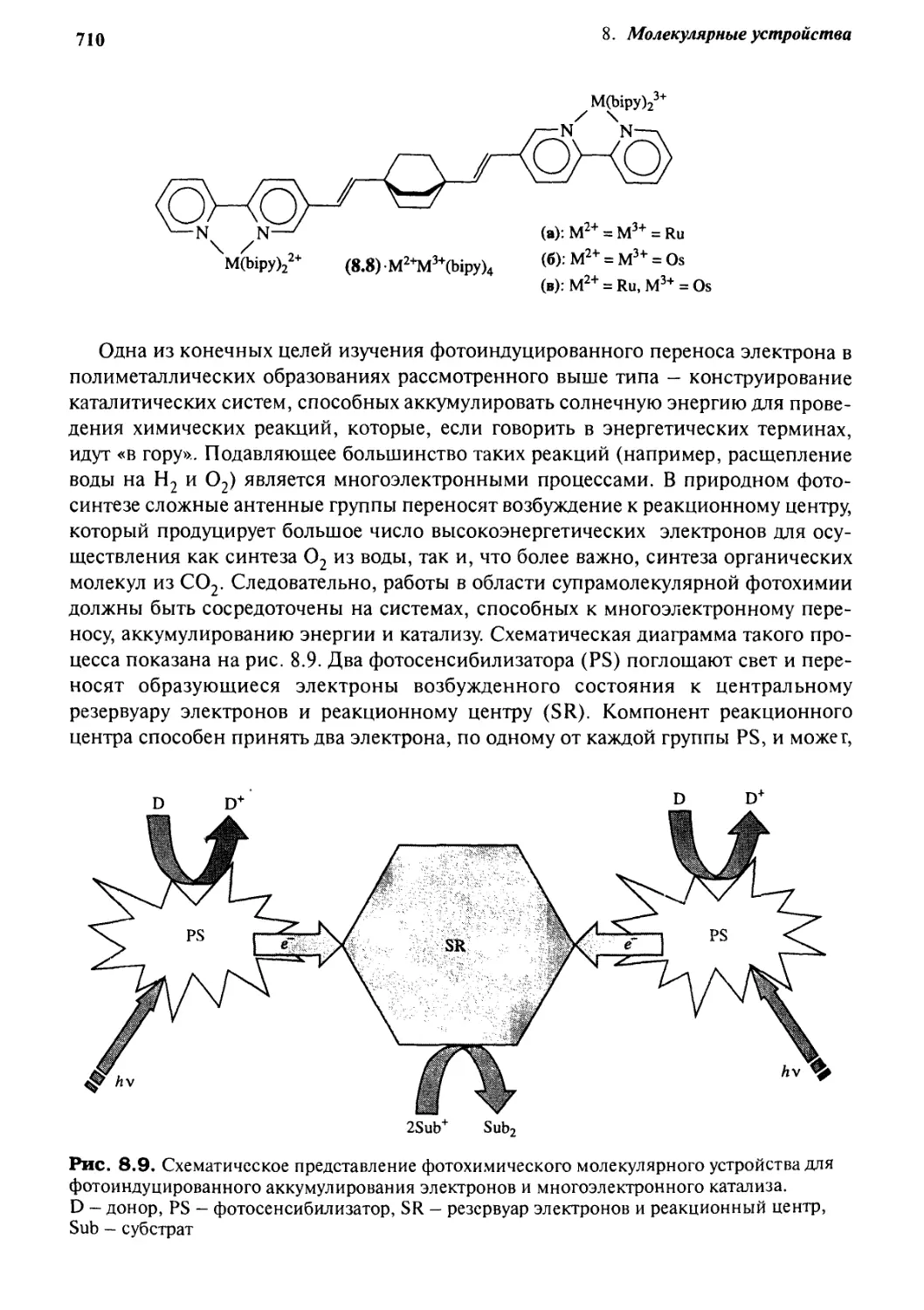
8.1 Введение 695
8.1.1 Философия молекулярных устройств 695
8.1.2 Что такое супрамолекулярное устройство? 696
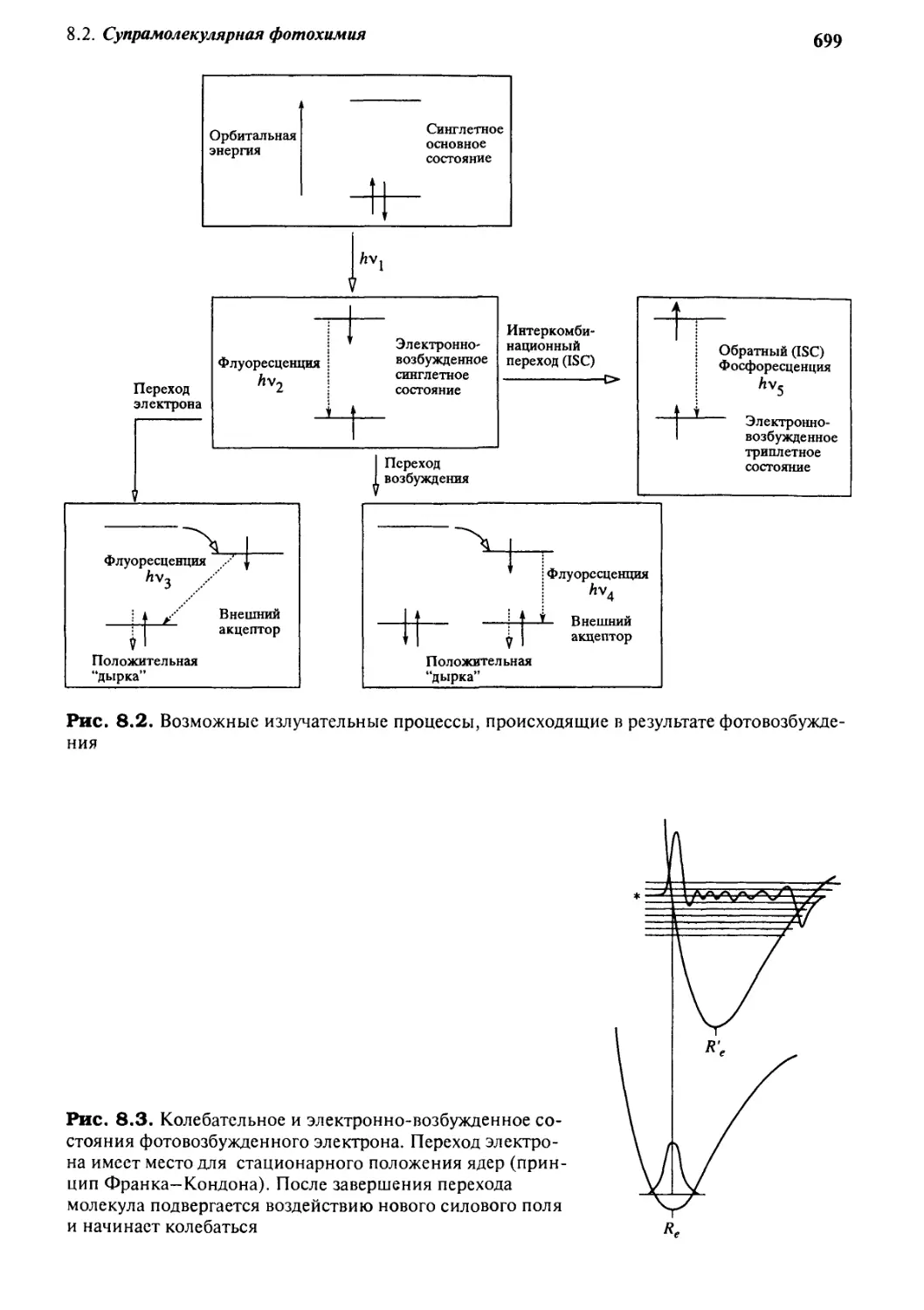
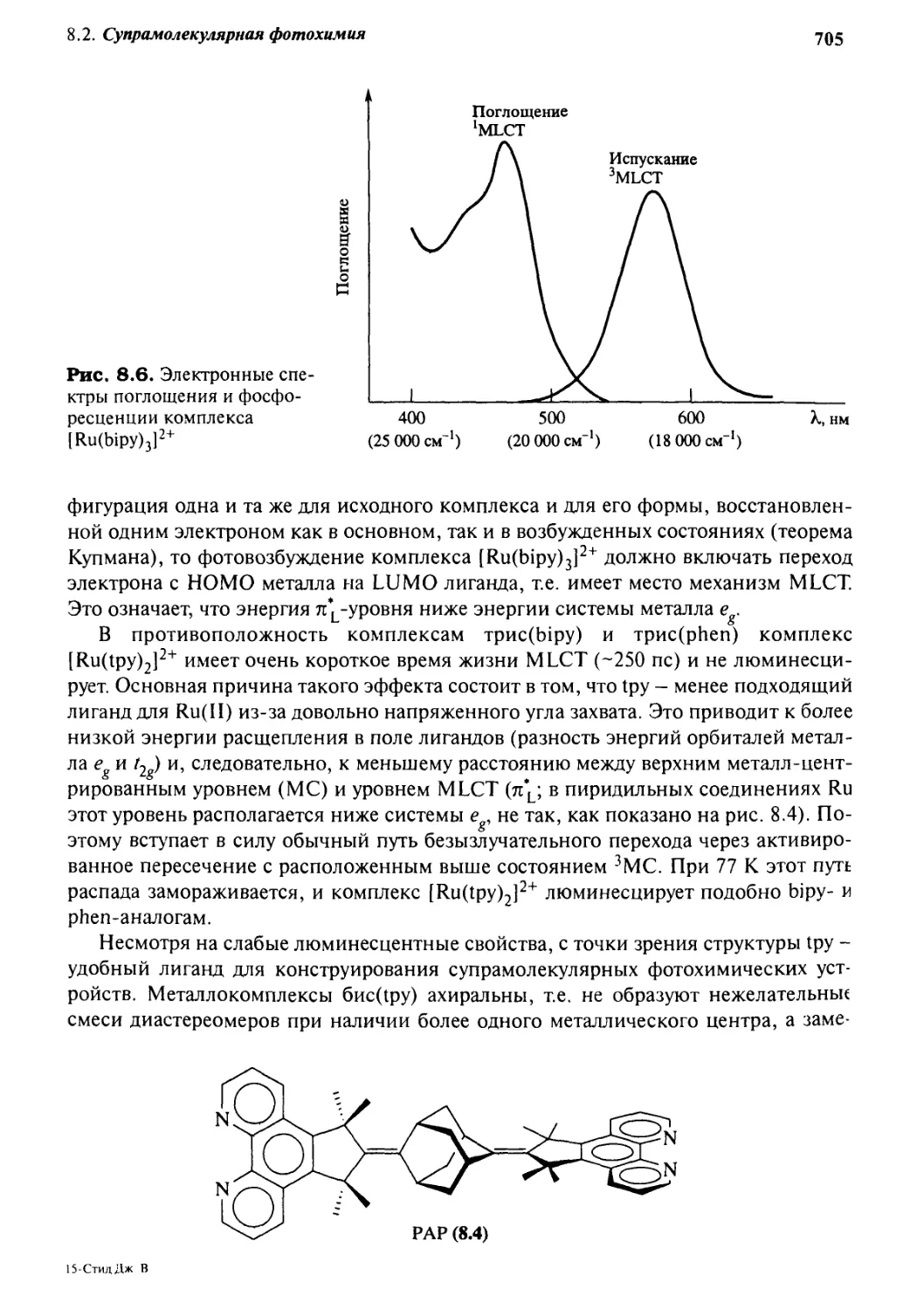
8.2 Супрамолекулярная фотохимия 698
8.2.1 Основы фотохимии 698
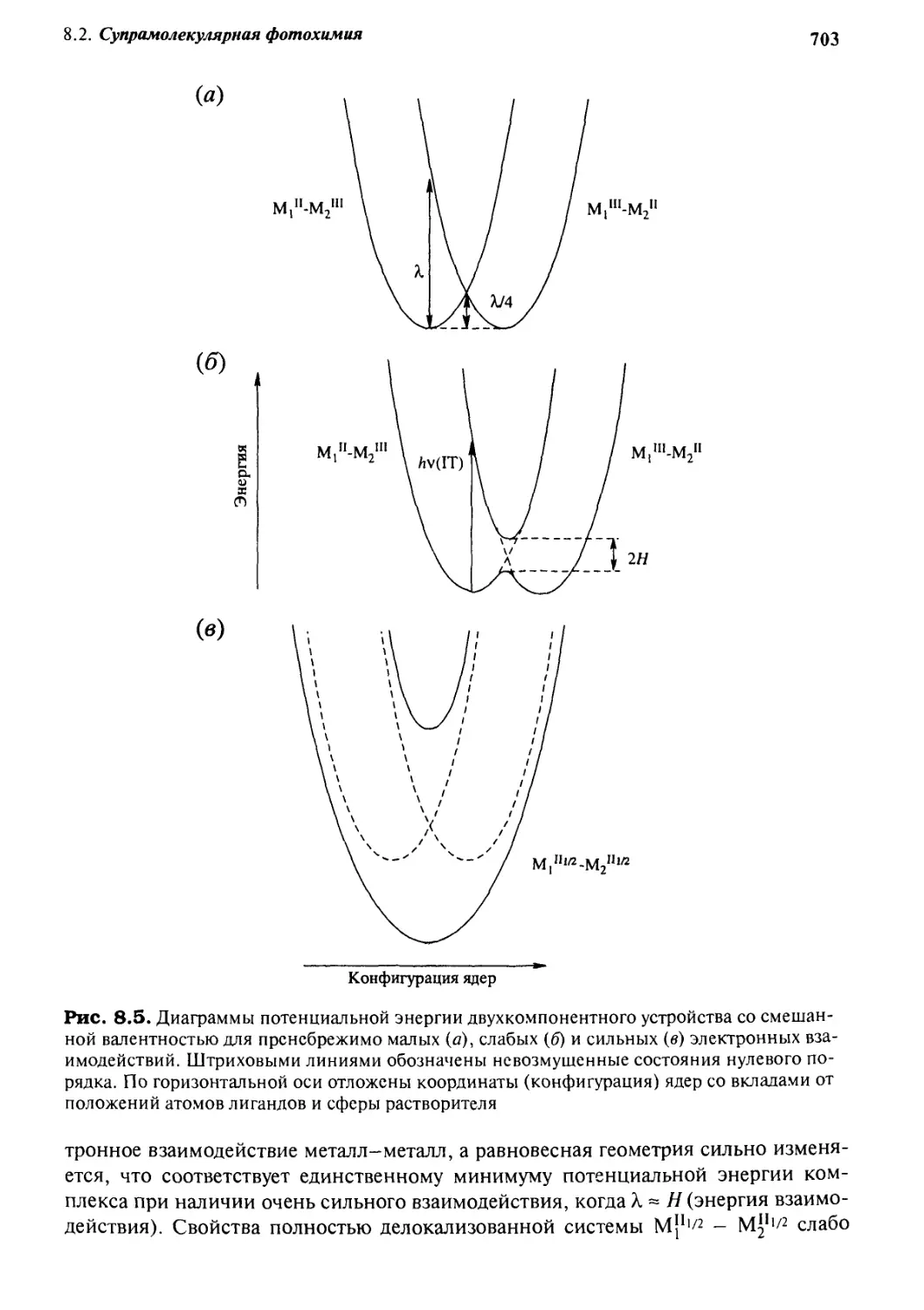
8.2.2 Биметаллические системы
и системы со смешанной валентностью 702
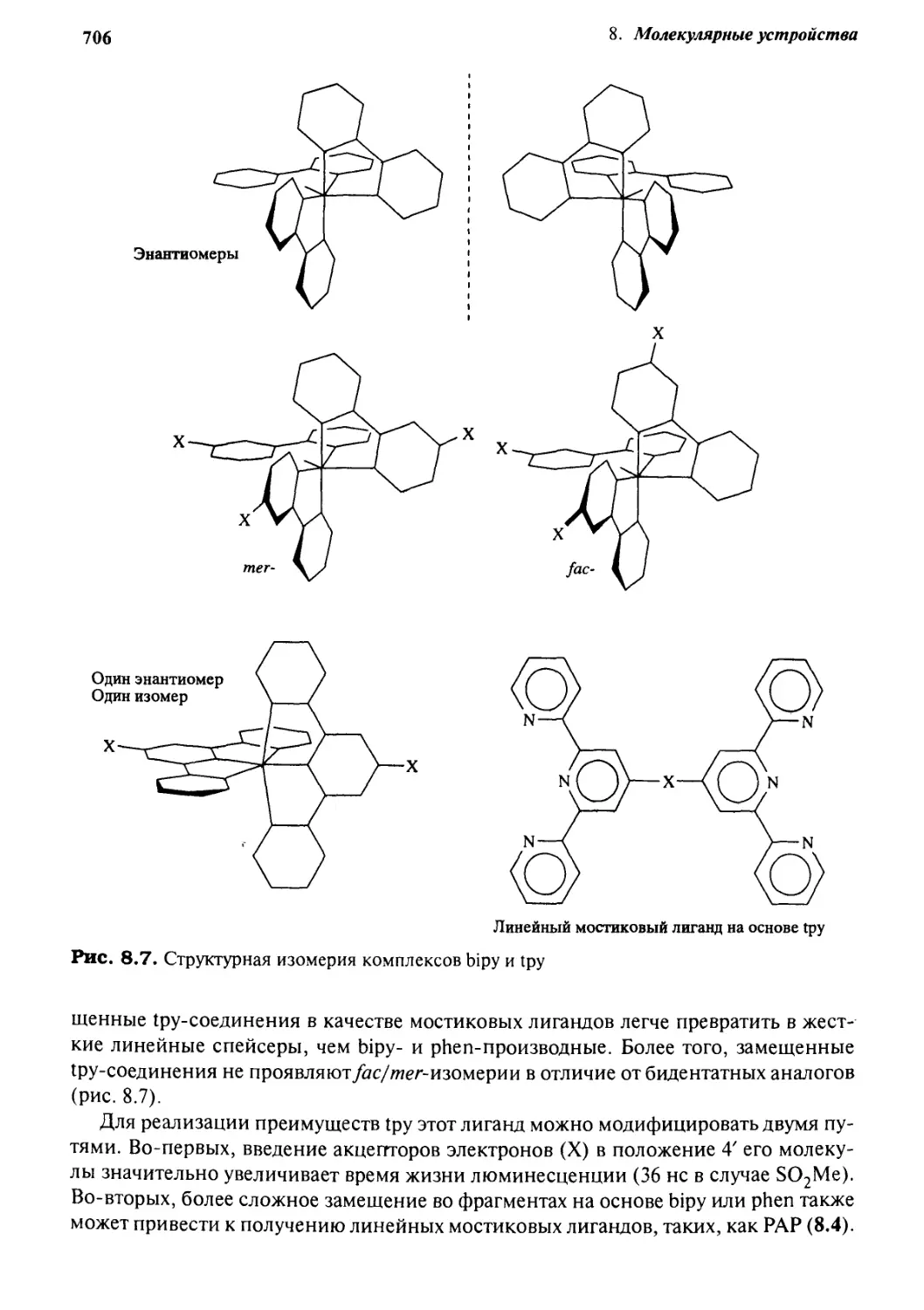
8.2.3 Бипиридил и его друзья 704
8.2.4 Фото- и электрохимические устройства
на основе бипиридила 707
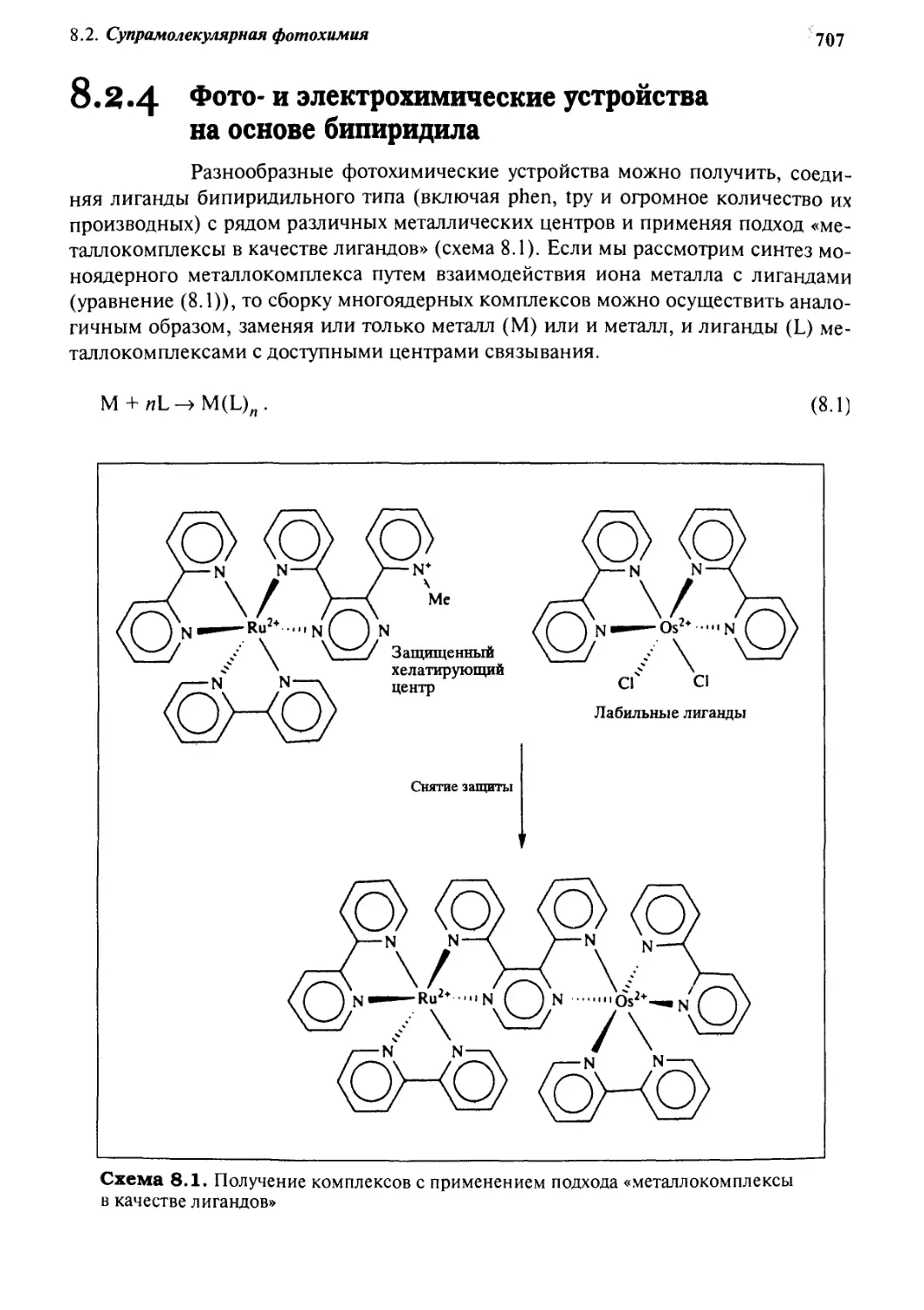
8.2.5 Устройства для преобразования света 711
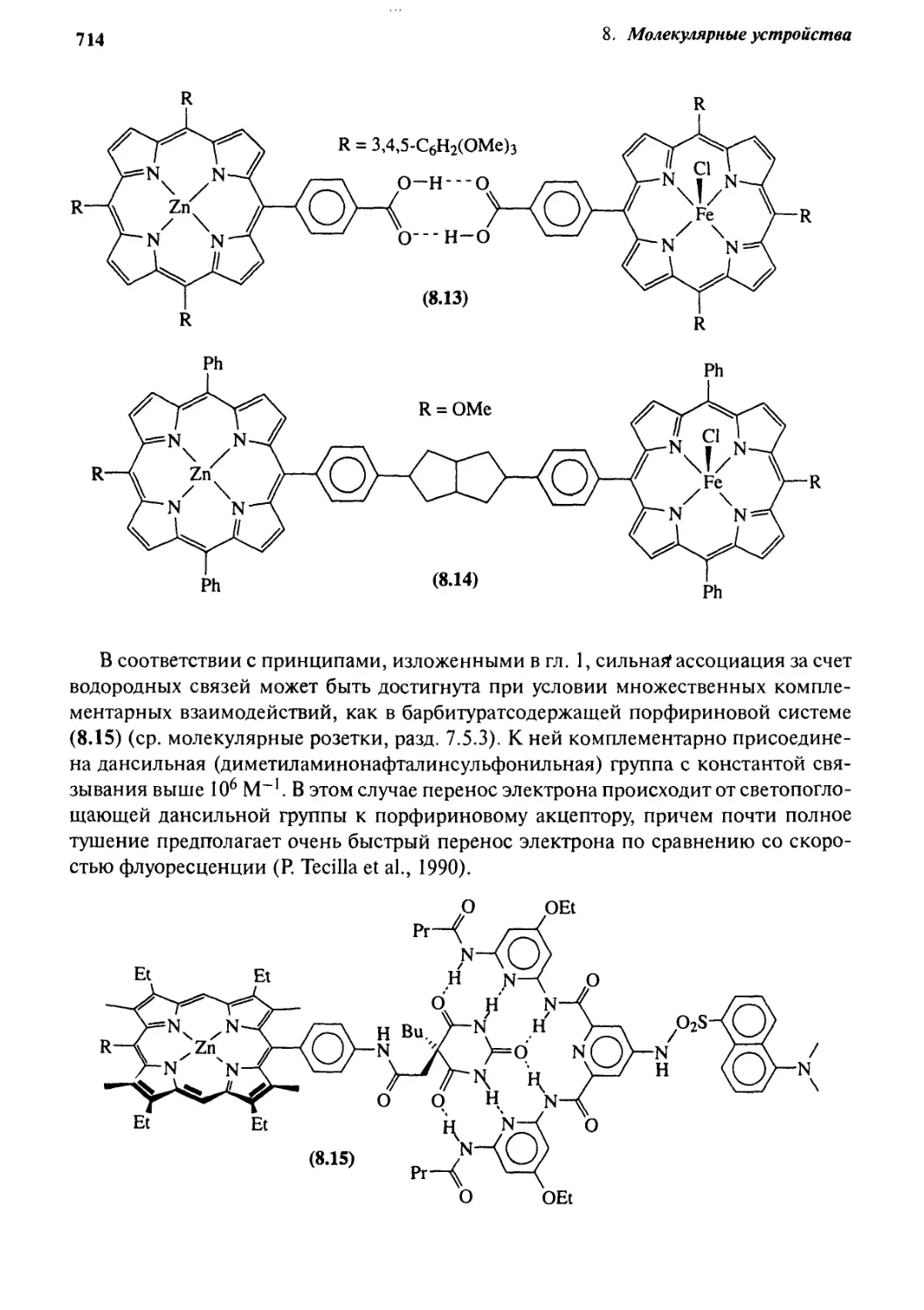
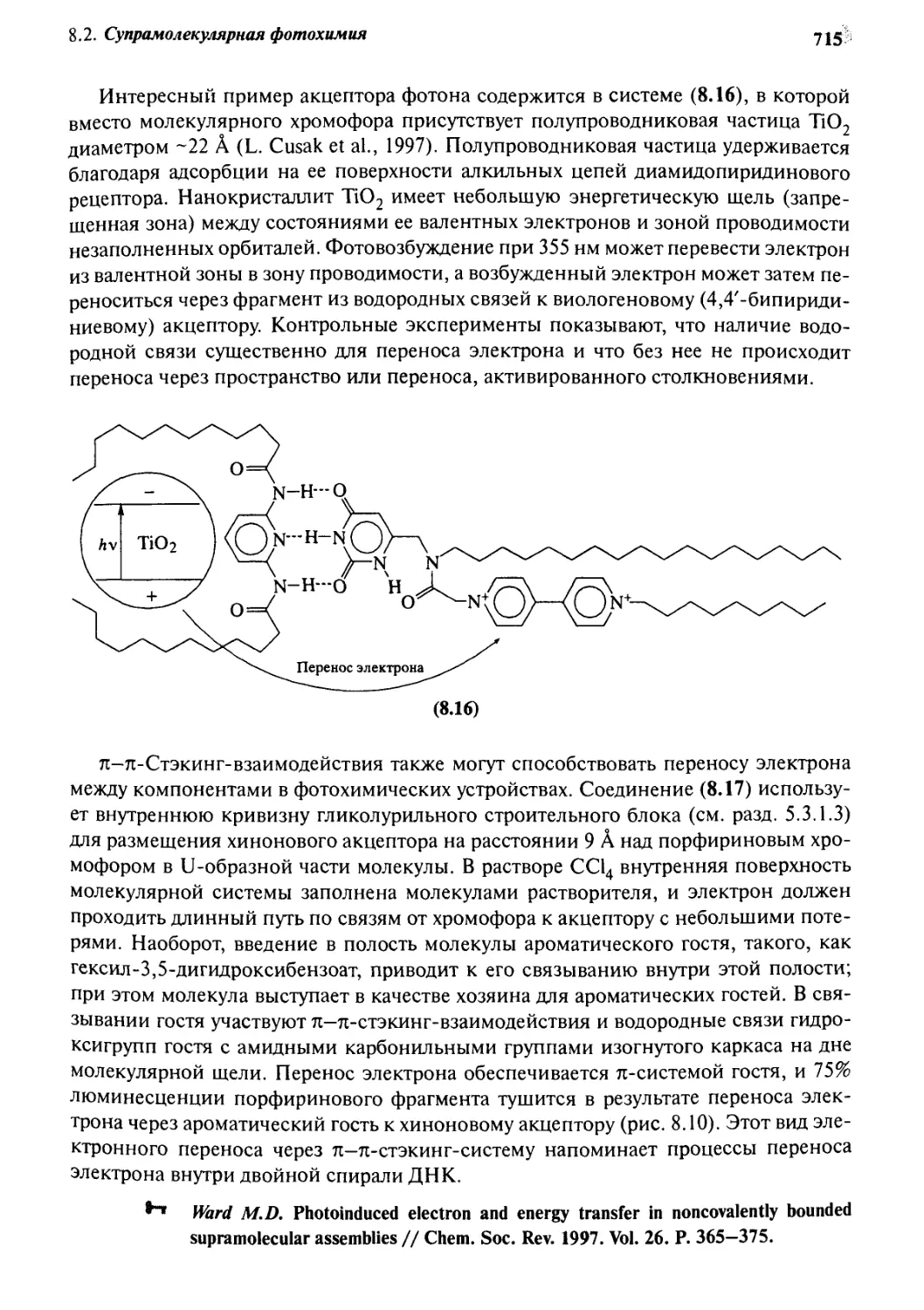
8.2.6 Нековалентно связанные системы 712
8. g Информация и сигналы: семиохимия 716
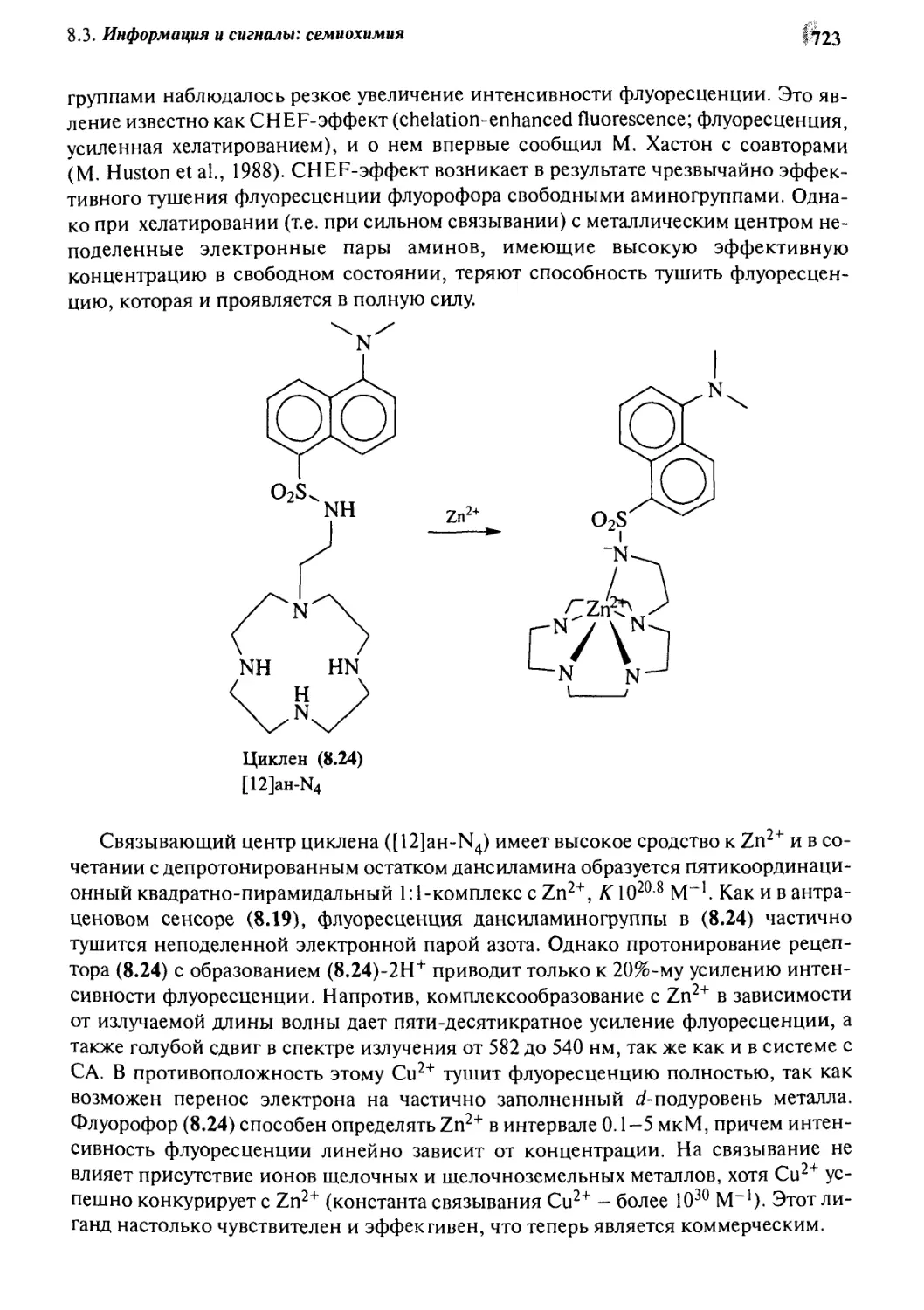
8.3.1 Фотохимические сенсоры 717
8.3.2 Флуорофоры 721
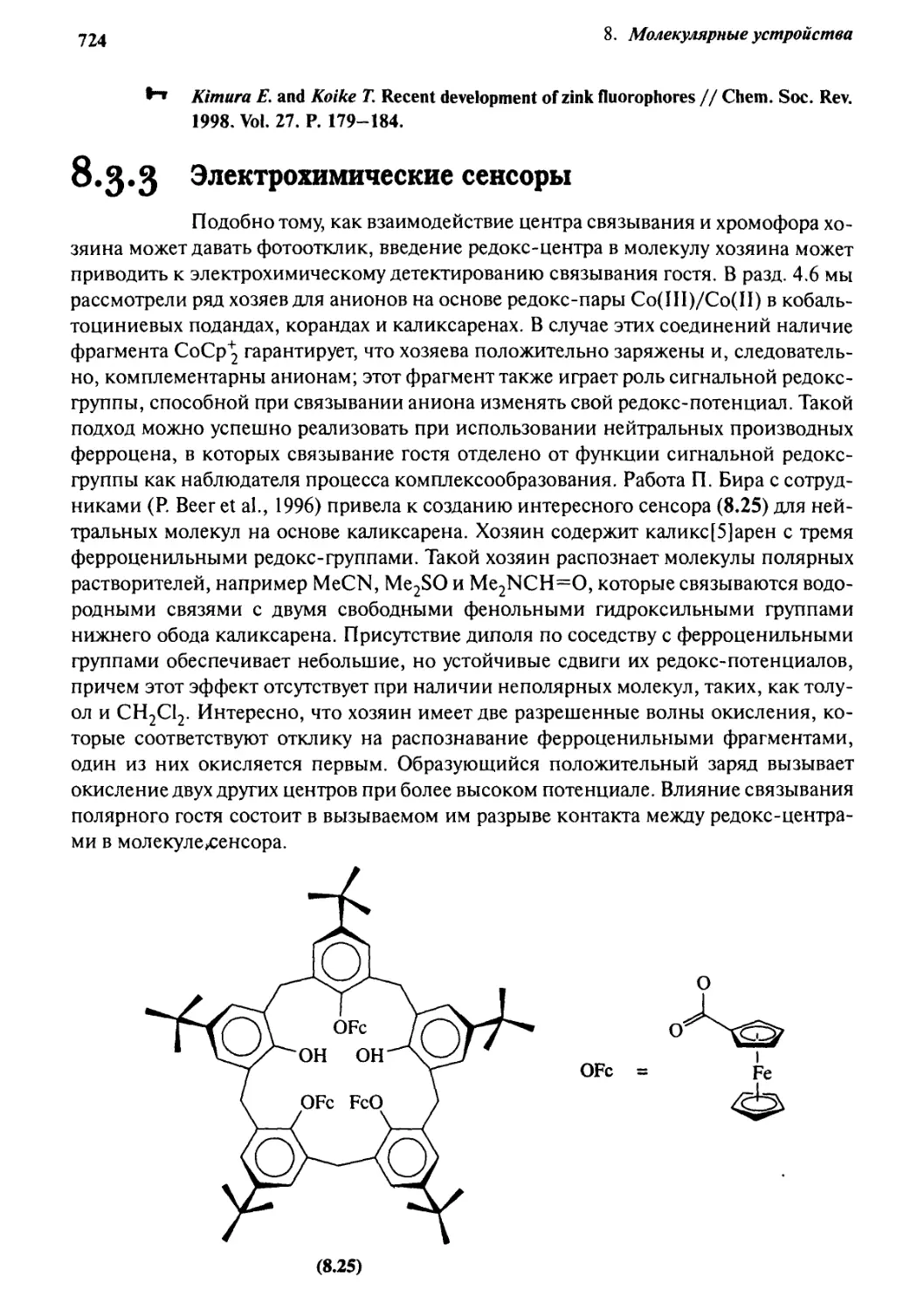
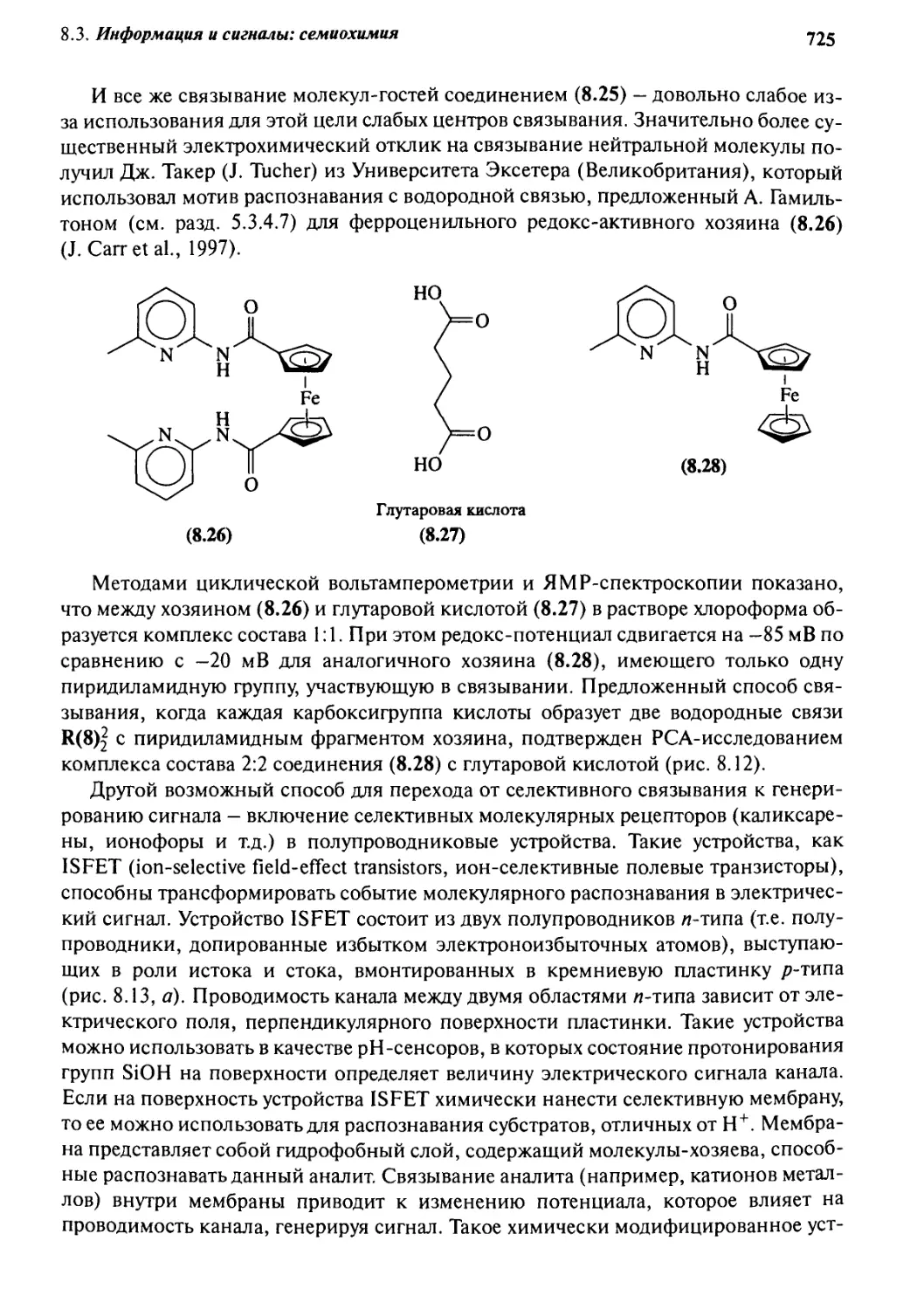
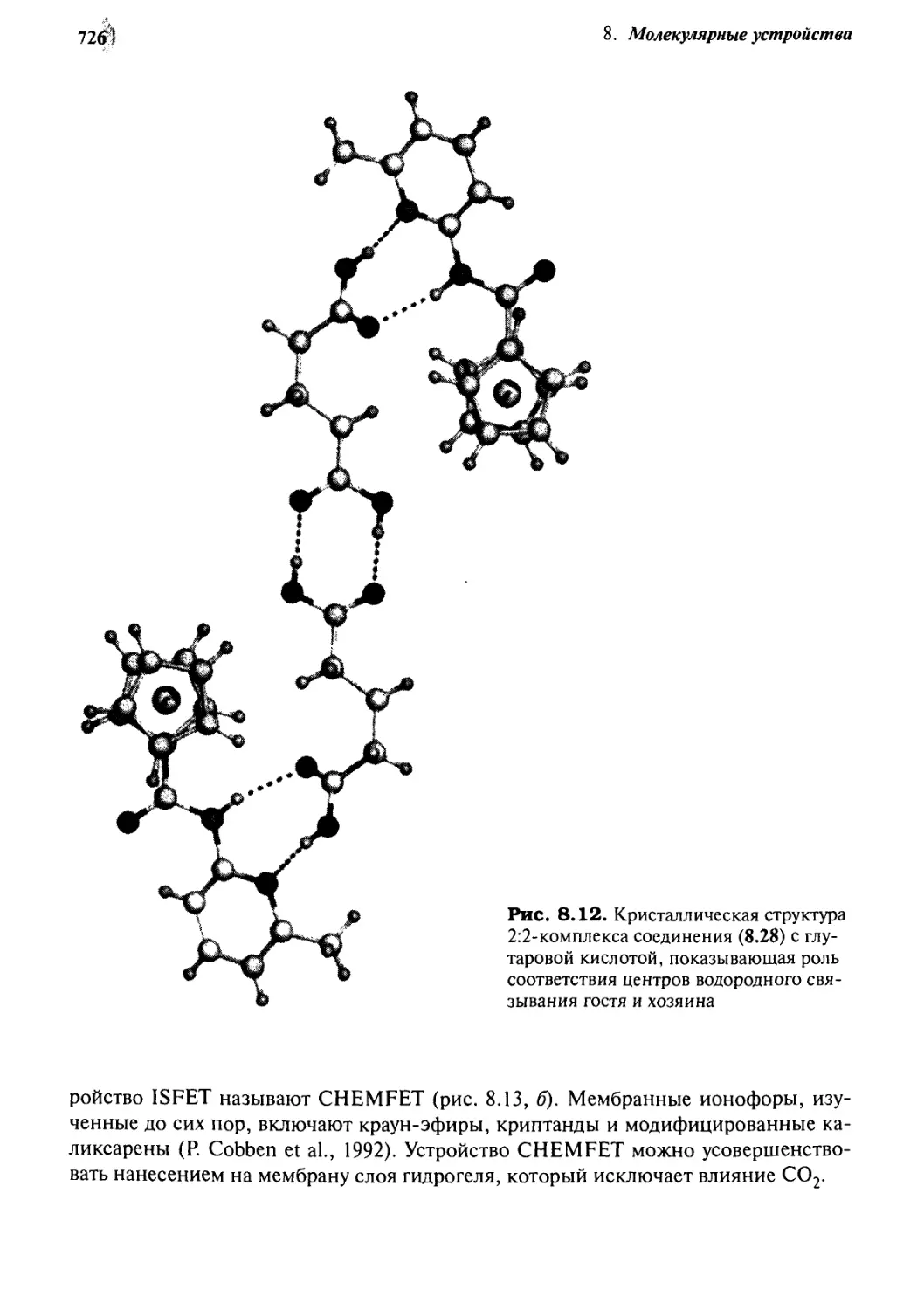
8.3.3 Электрохимические сенсоры 724
8.4 Молекулярные электронные устройства:
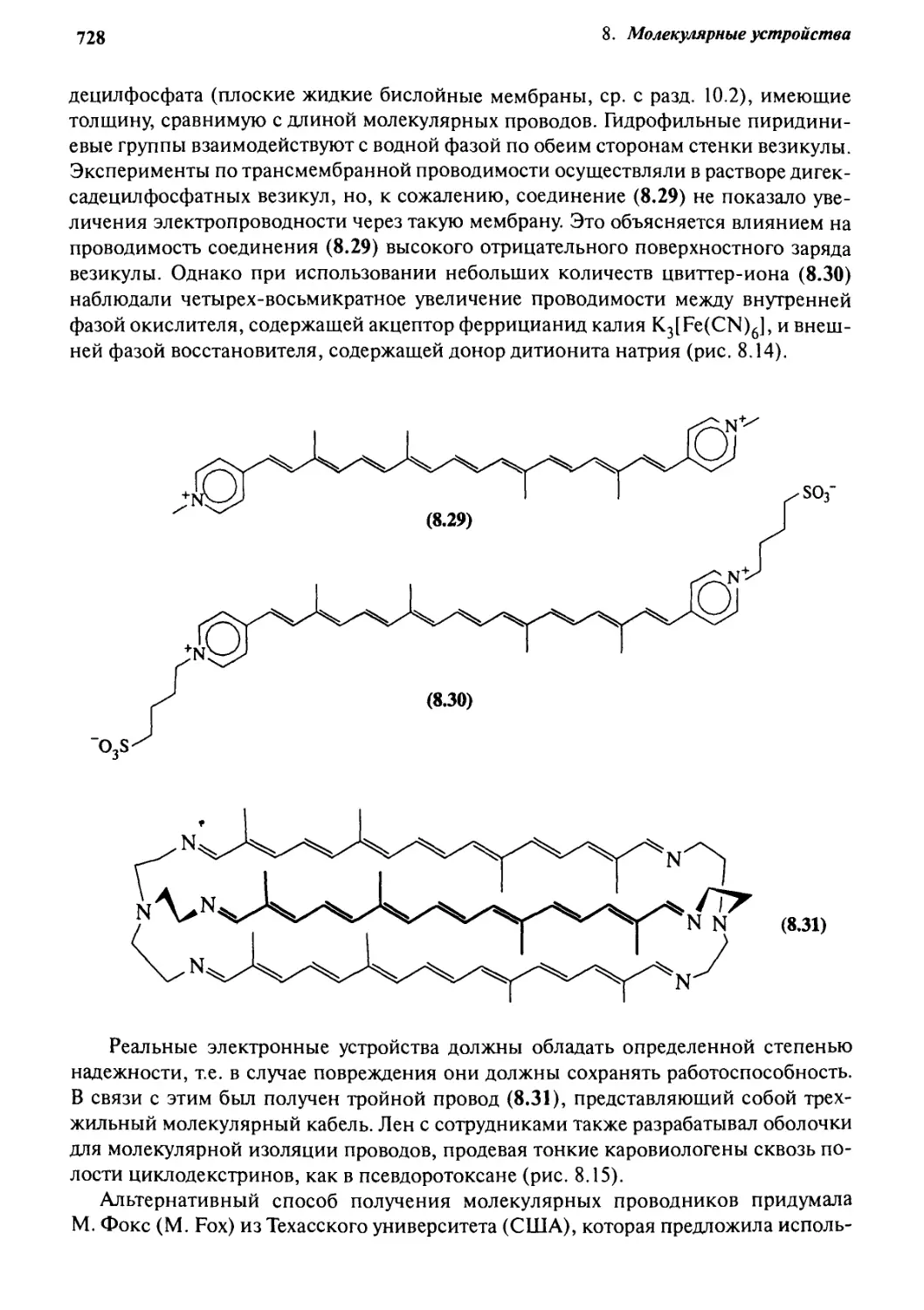
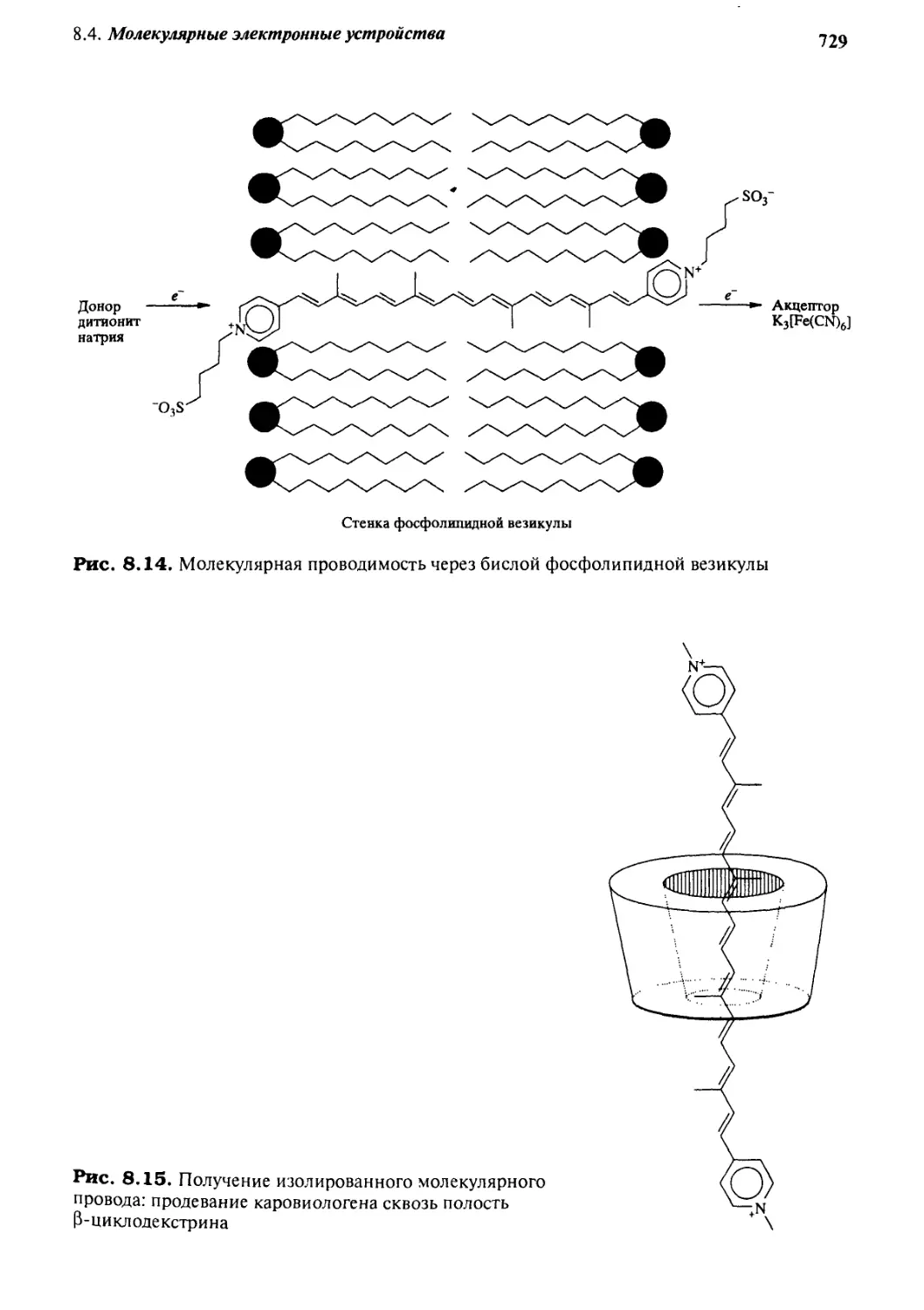
переключатели, провода и выпрямители 727
8.4.1 Молекулярные провода 727
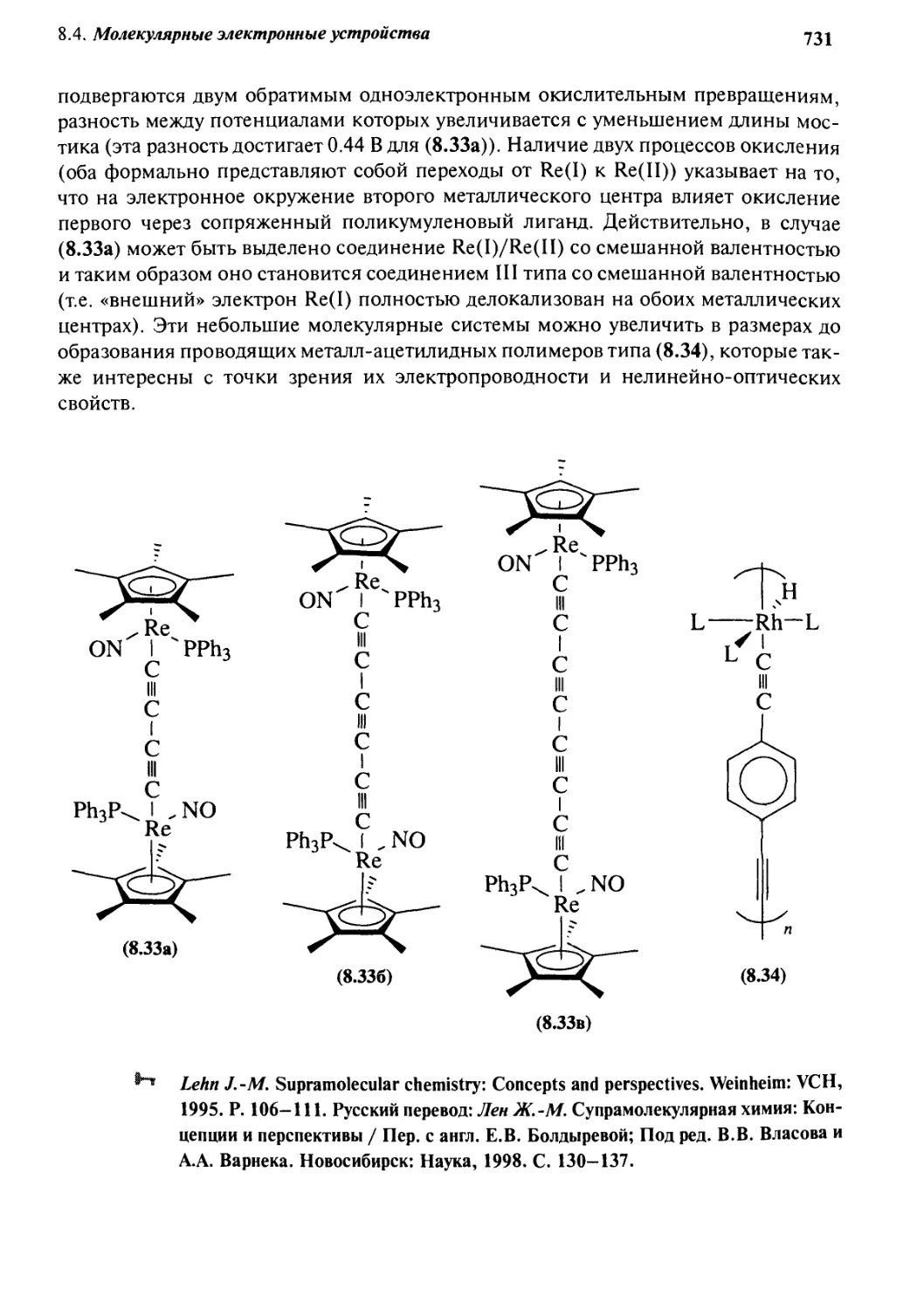
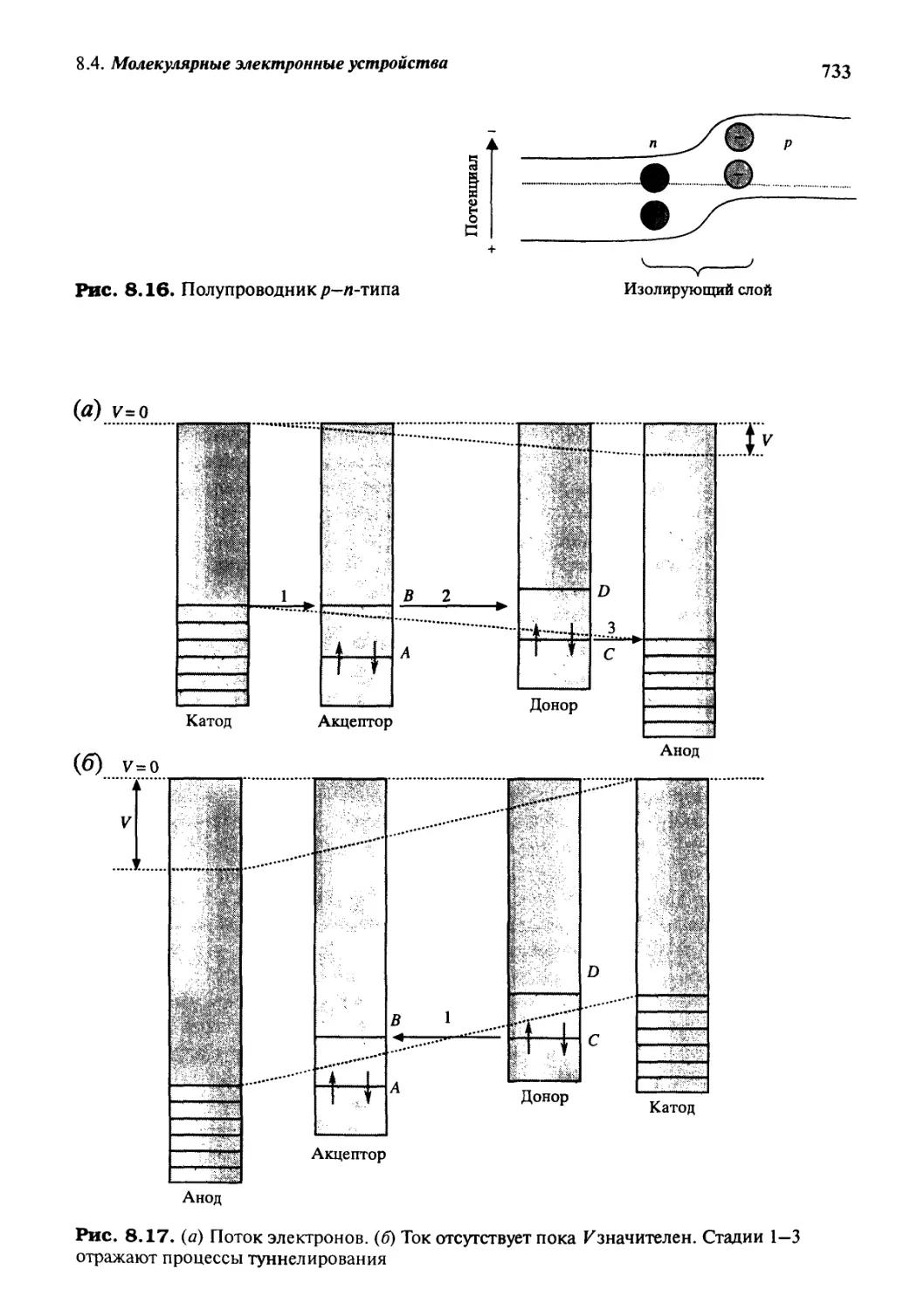
8.4.2 Молекулярные выпрямители 732
8.4.3 Система 1,2-дитиенилэтена как переключатель 734
8.4.4 Электропереключаемая люминесценция 736
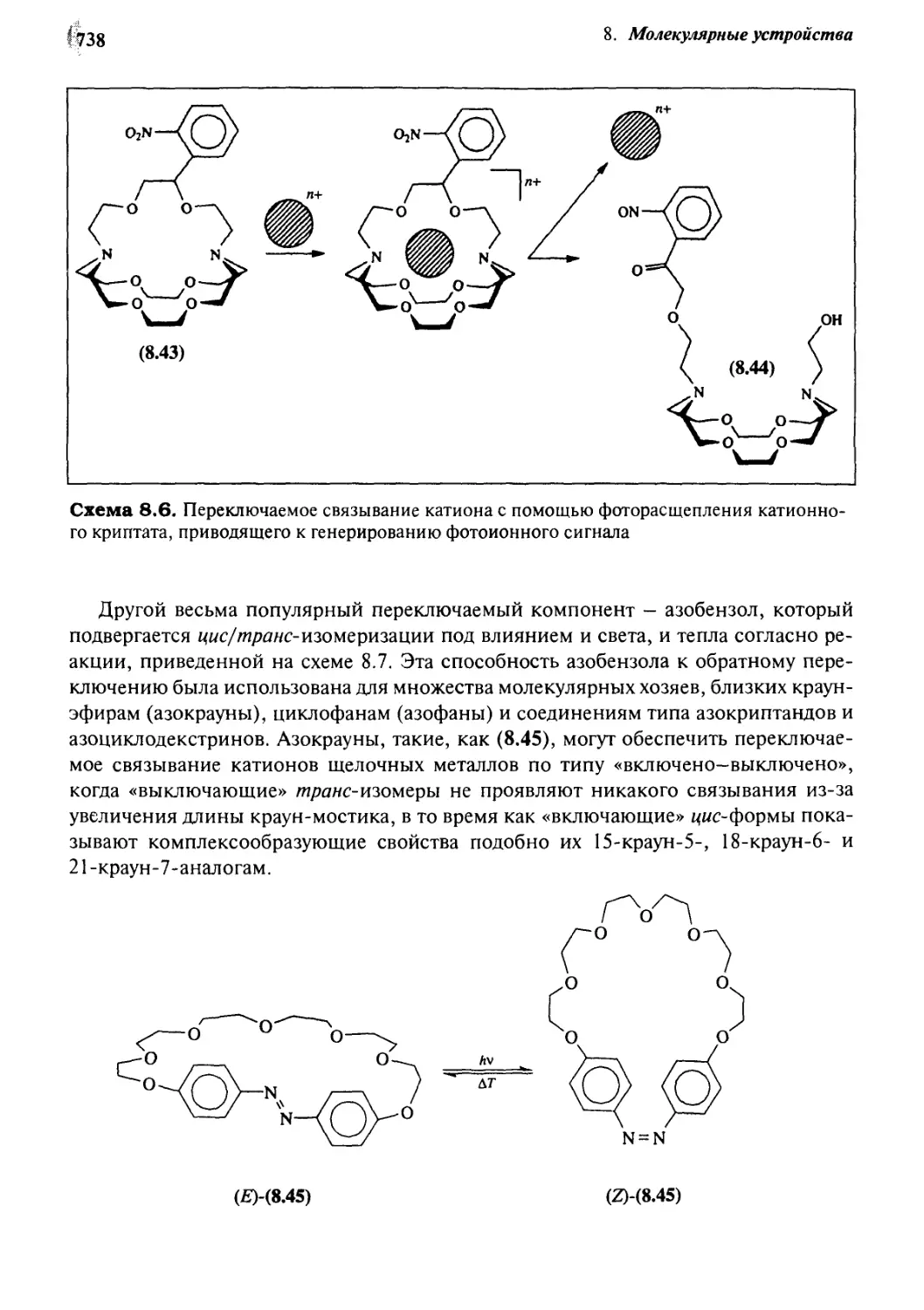
8.4.5 Переключаемое связывание 737
8.4.6 Аллостерические переключатели 739
8*5 Молекулярные машины
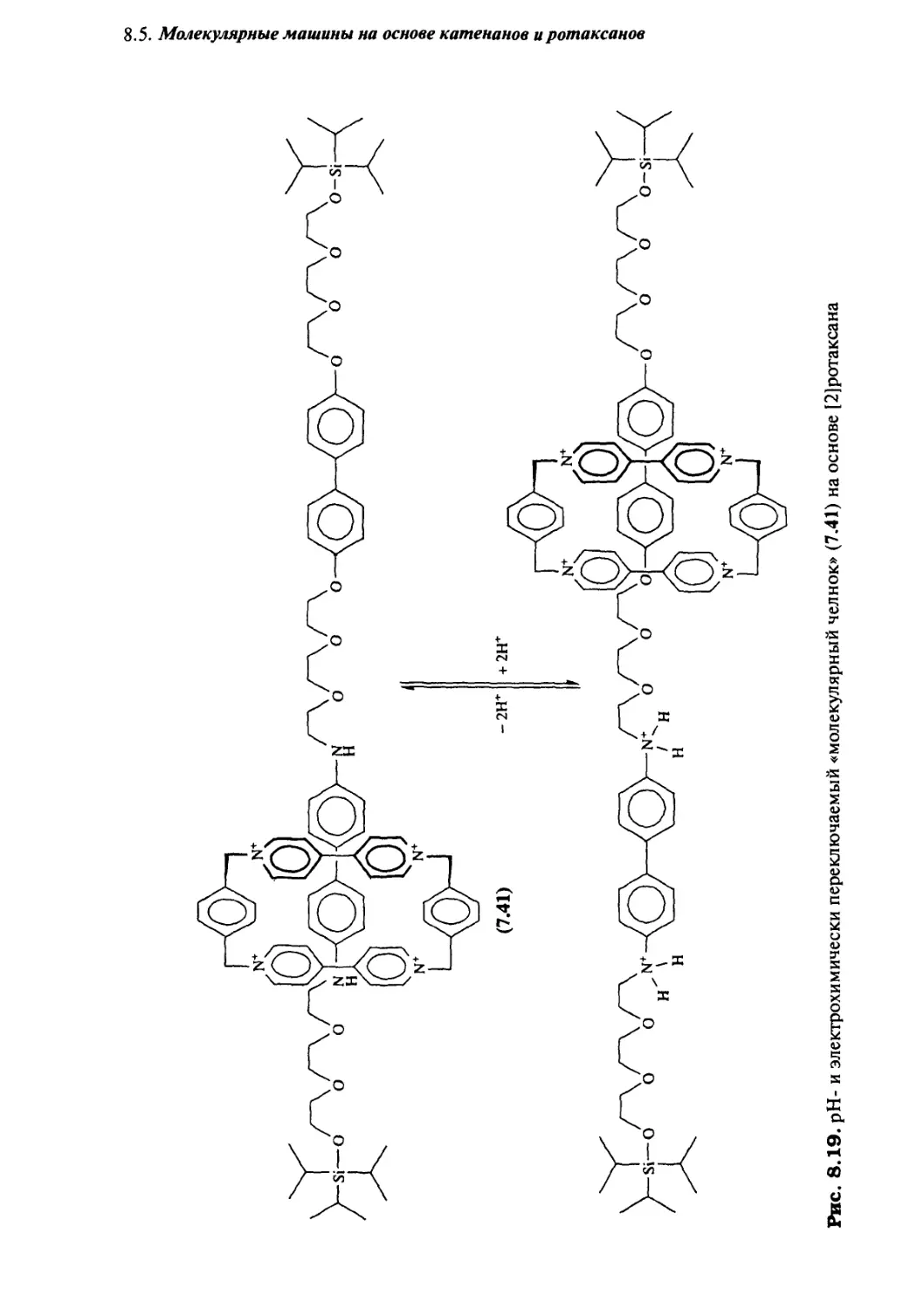
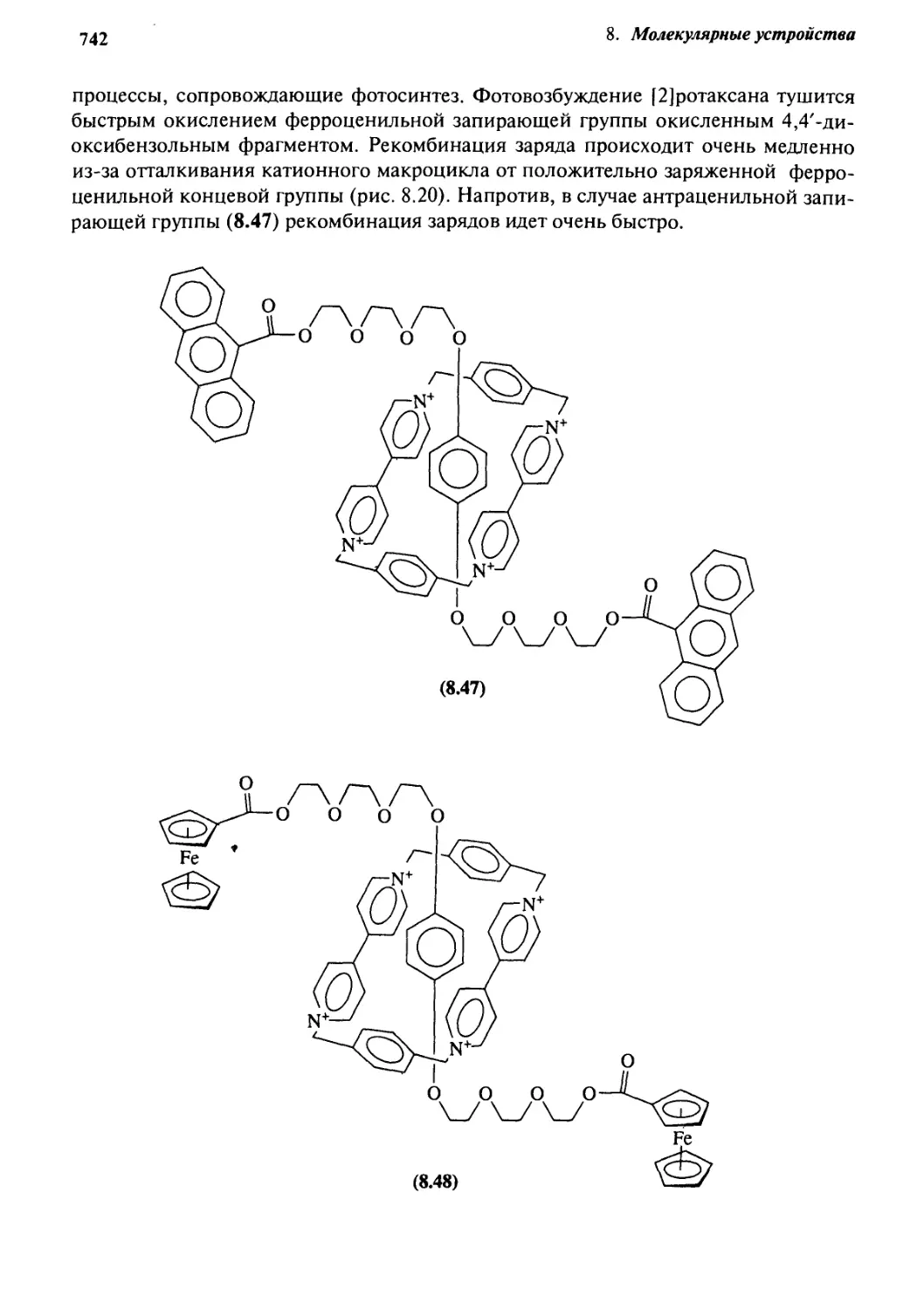
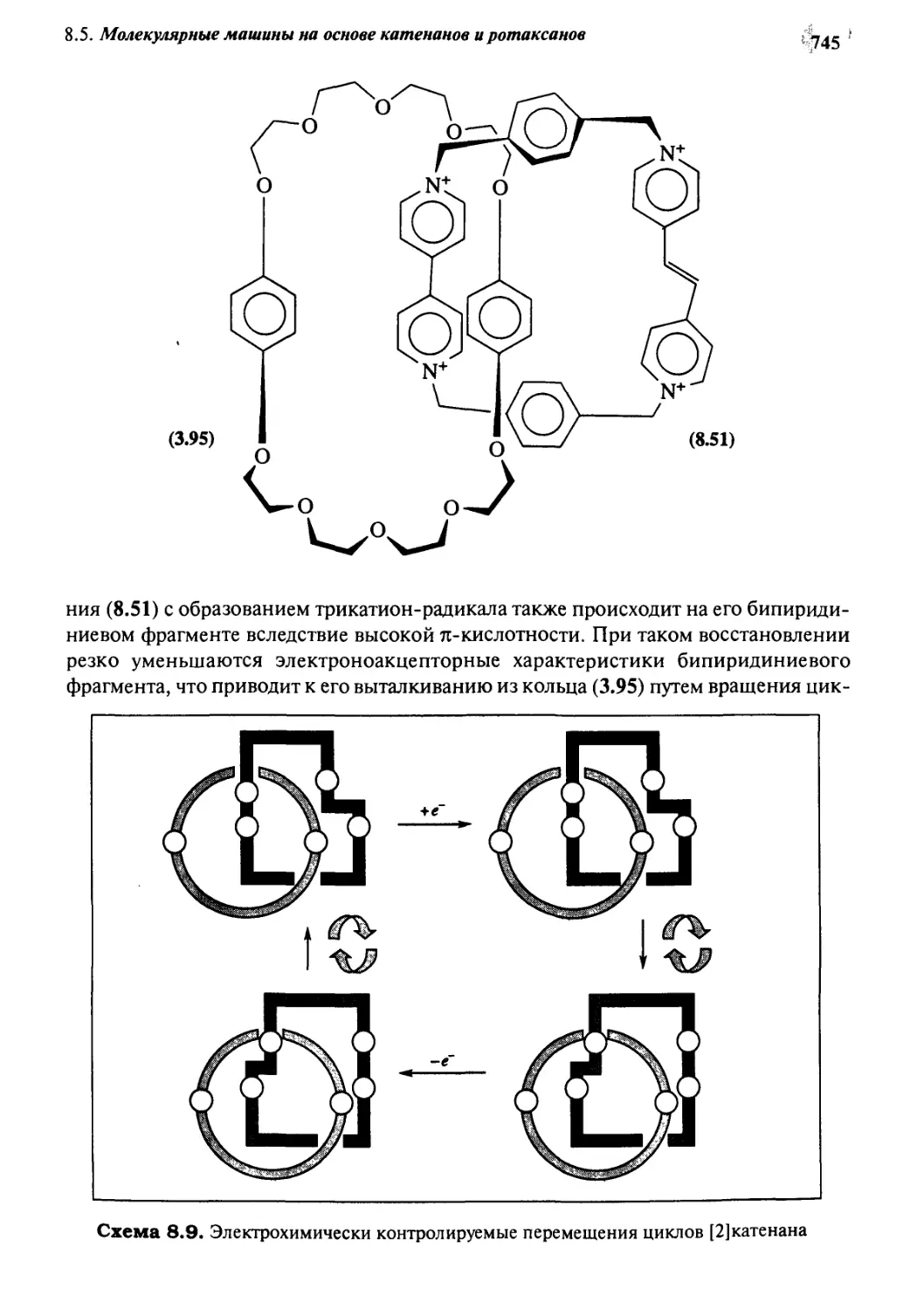
на основе катенанов и ротаксанов 740
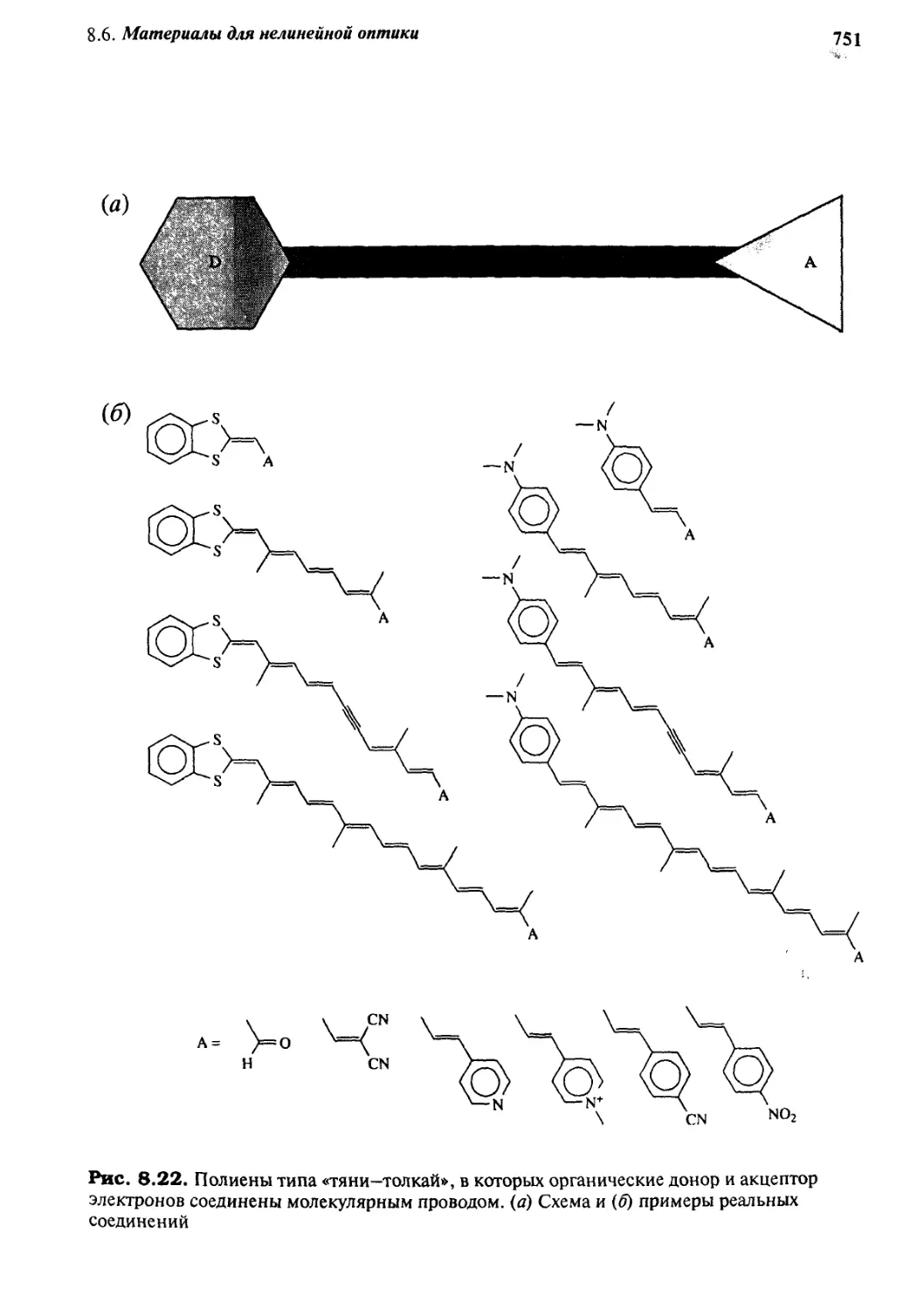
8.6 Материалы для нелинейной оптики 746
8.6.1 Основы нелинейной оптики 746
8.6.2 Материалы для нелинейной оптики второго порядка 749
8.6.3 Материалы для нелинейной оптики третьего порядка 753
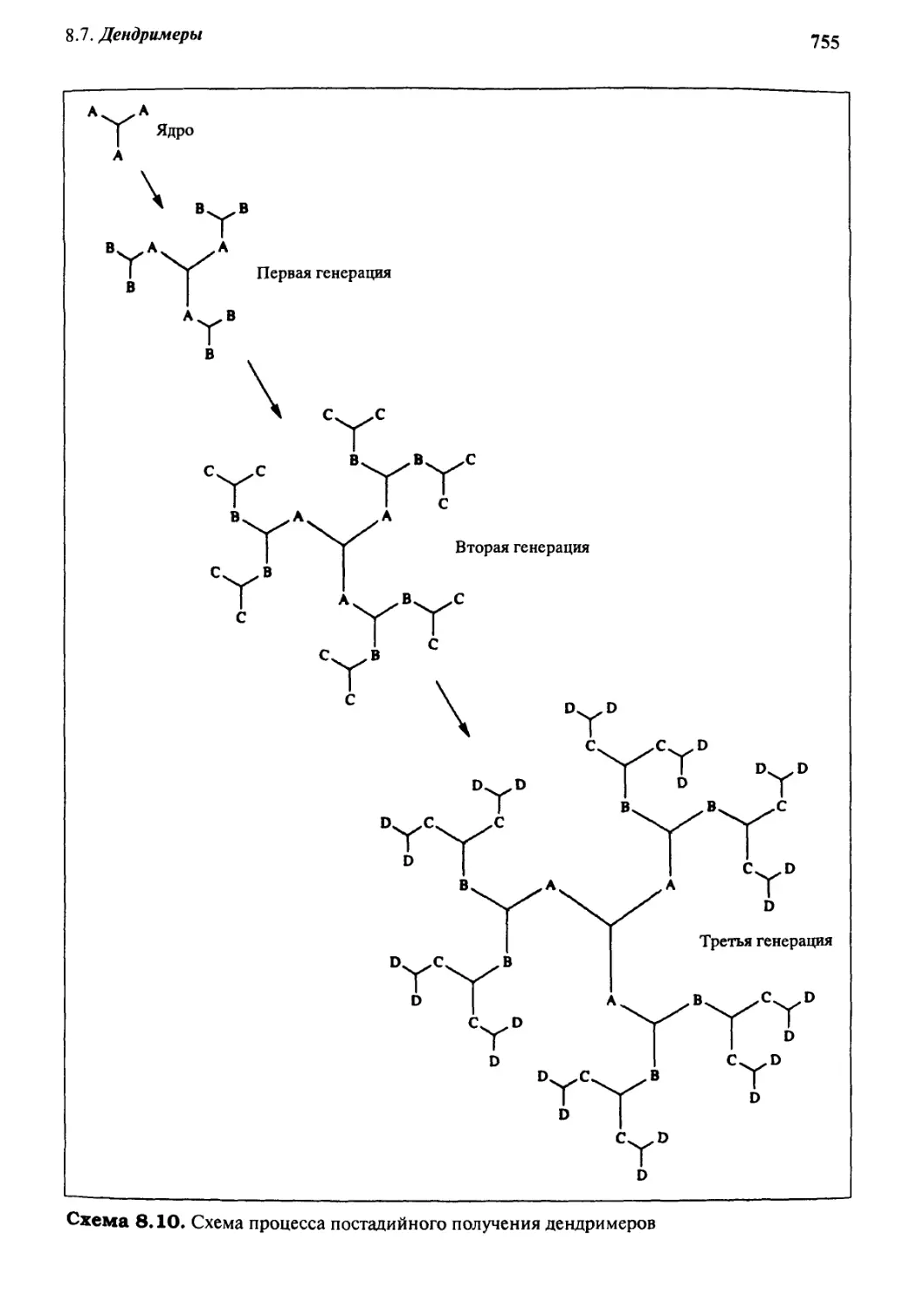
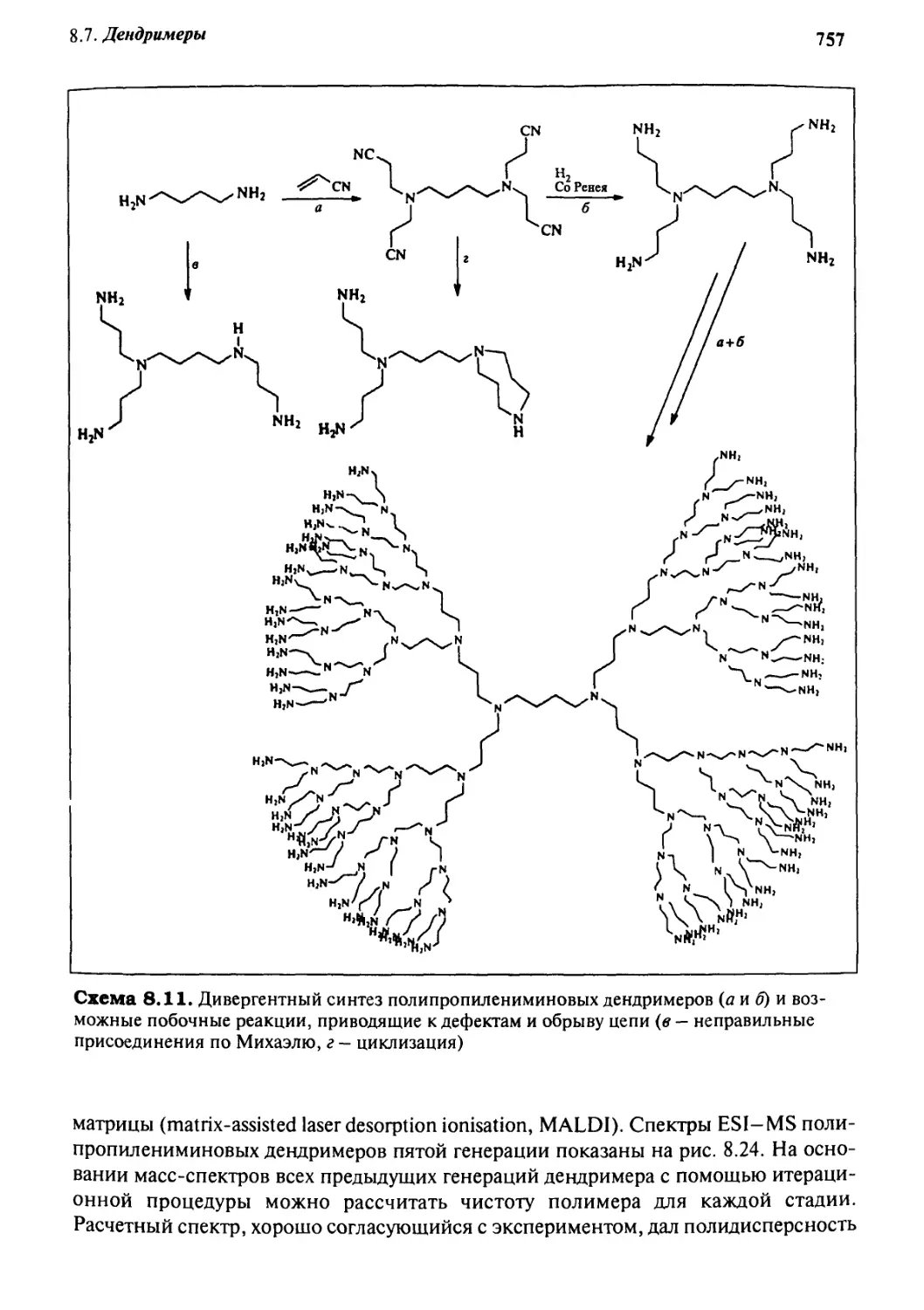
8 Дендримеры 754
8.7.1 Получение и свойства 756
Оглавление
9
489»
8.7.2 Химия хозяин-гость для дендримеров 759
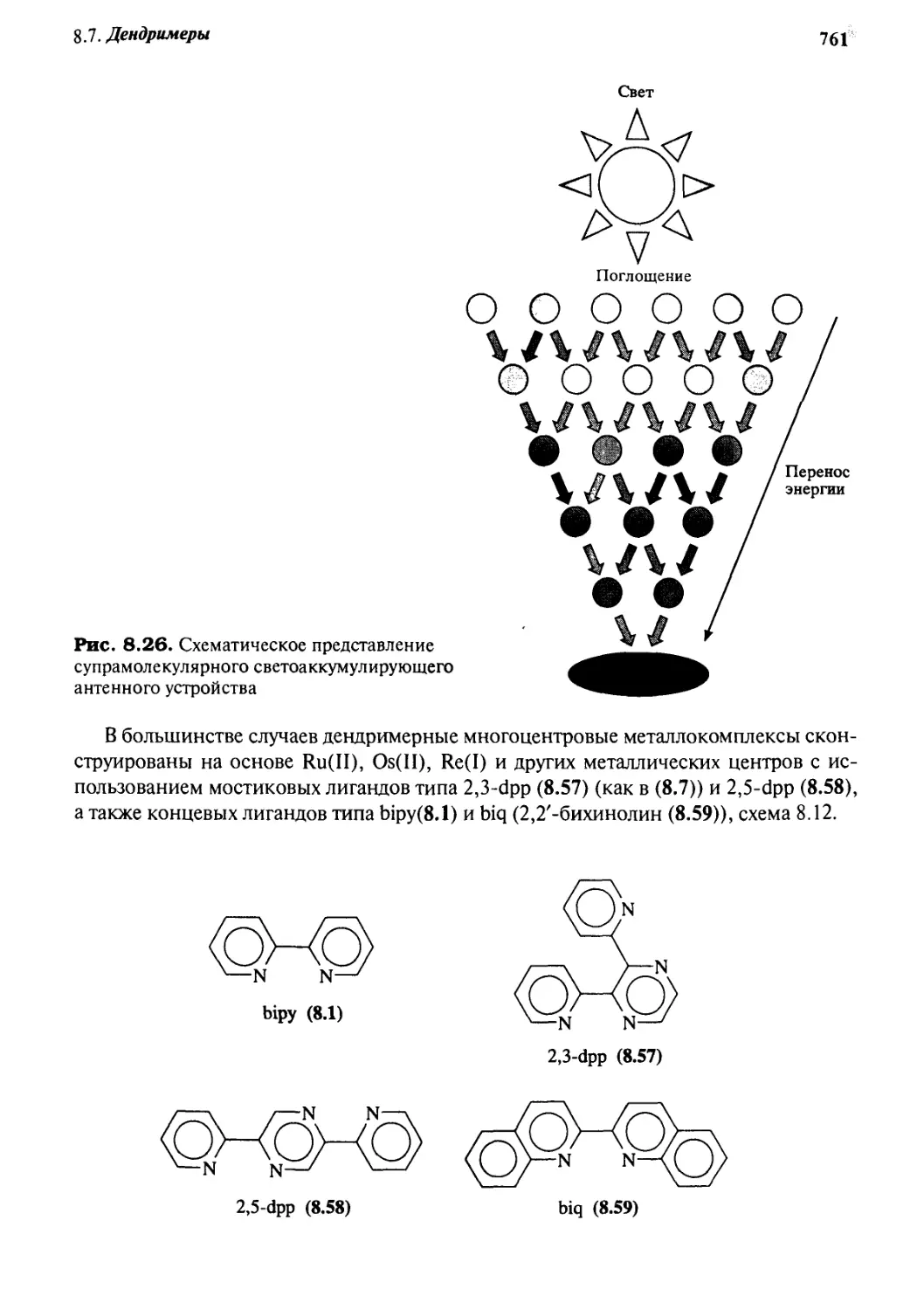
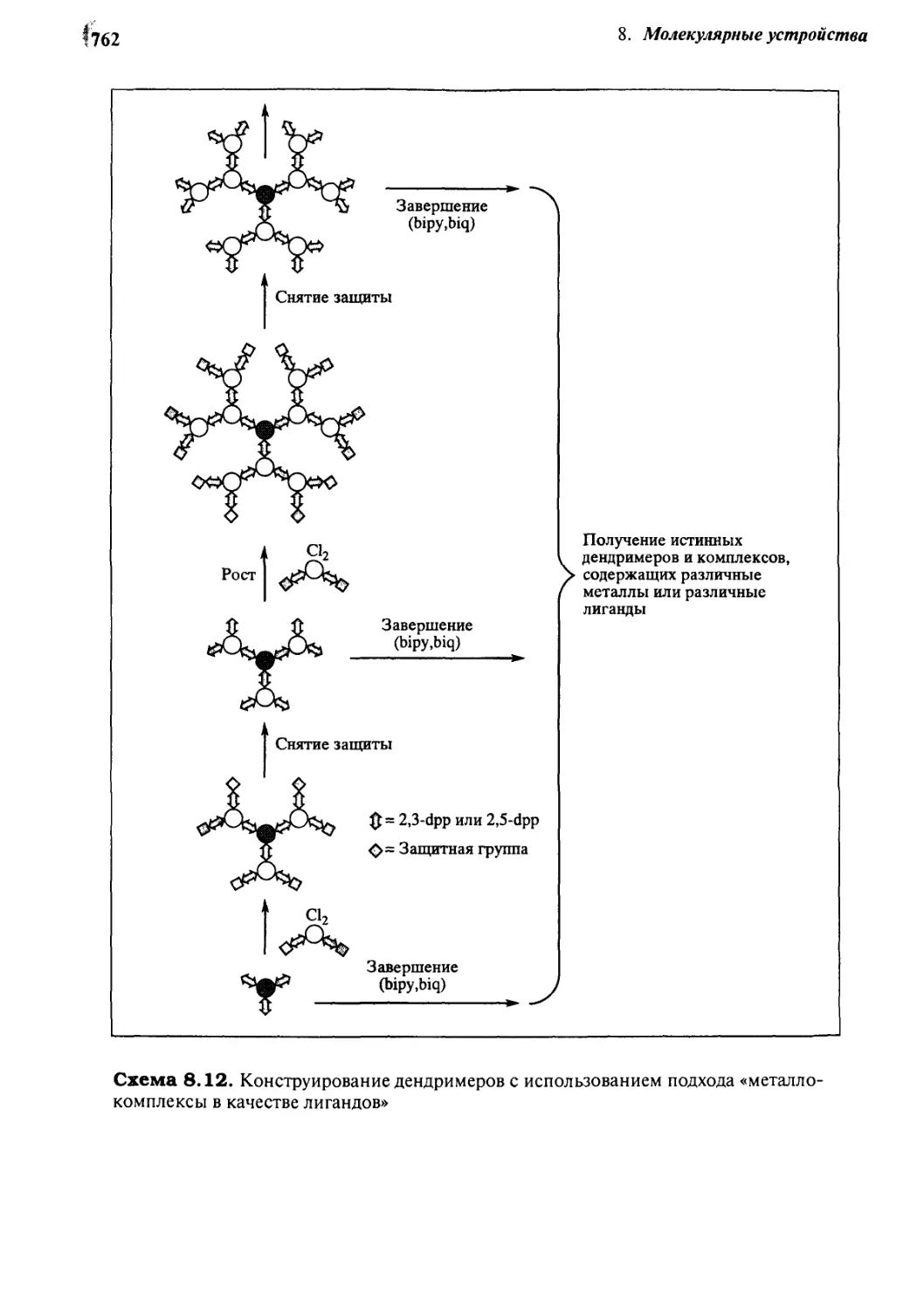
8.7.3 Фотохимические устройства на основе дендримеров 760
Учебные задания 764
Литература 765
Биомиметика 767
Q. 1 Введение 767
9.1.1 Супрамолекулярная биохимия 767
9.1.2 Характеристики биологических моделей 768
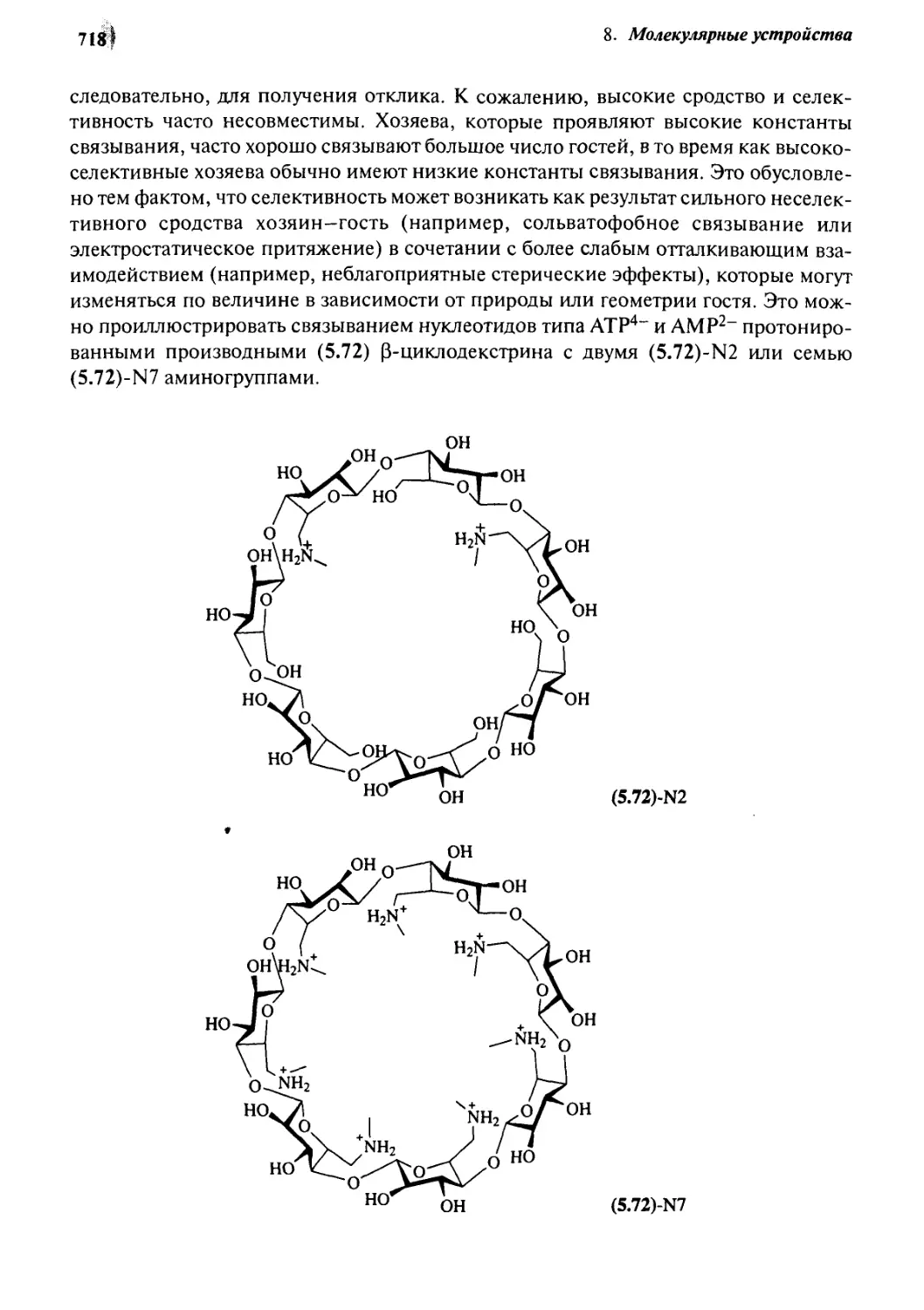
0.2 Характеристики ферментов 770
9.2.1 Определение и структура 770
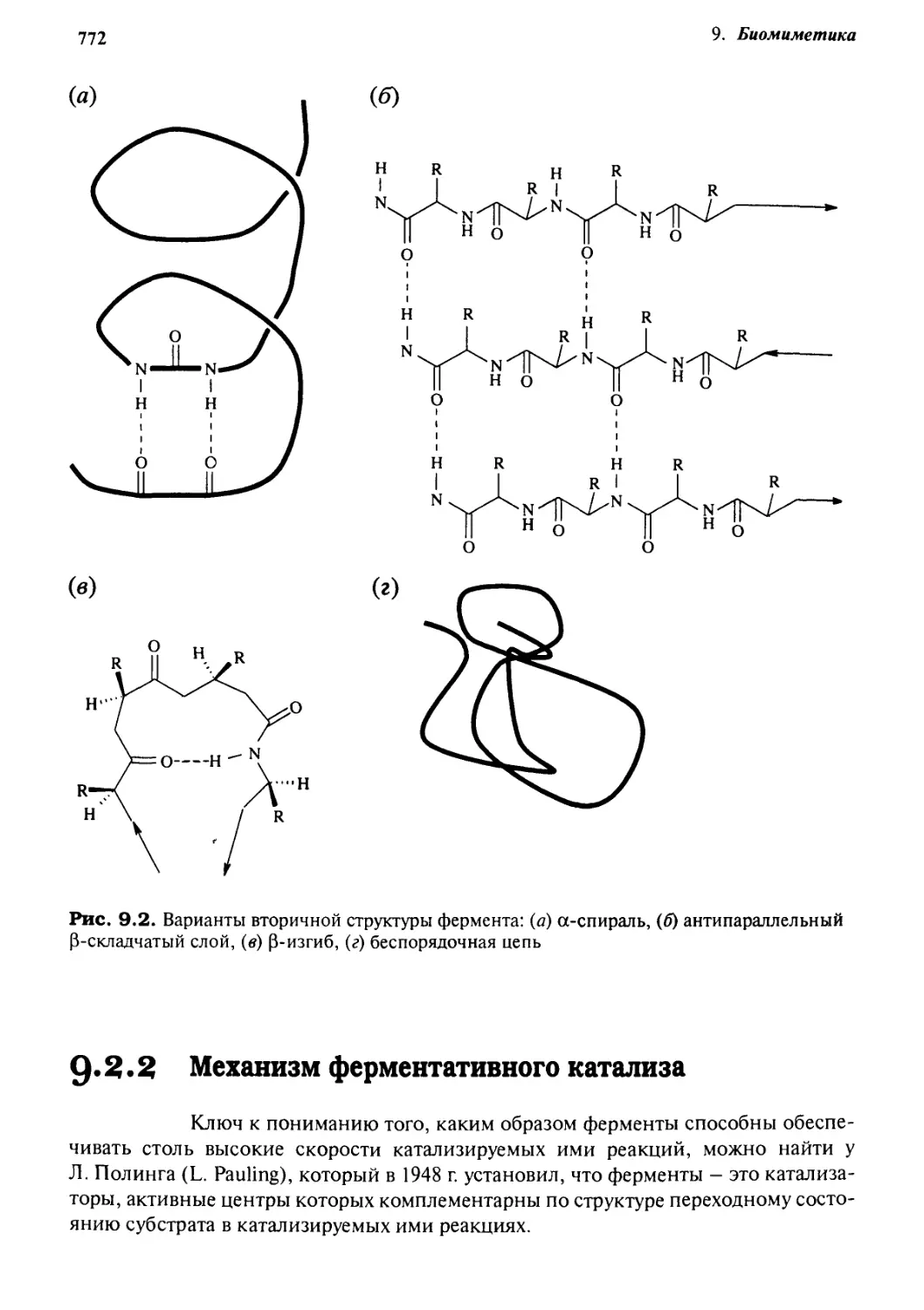
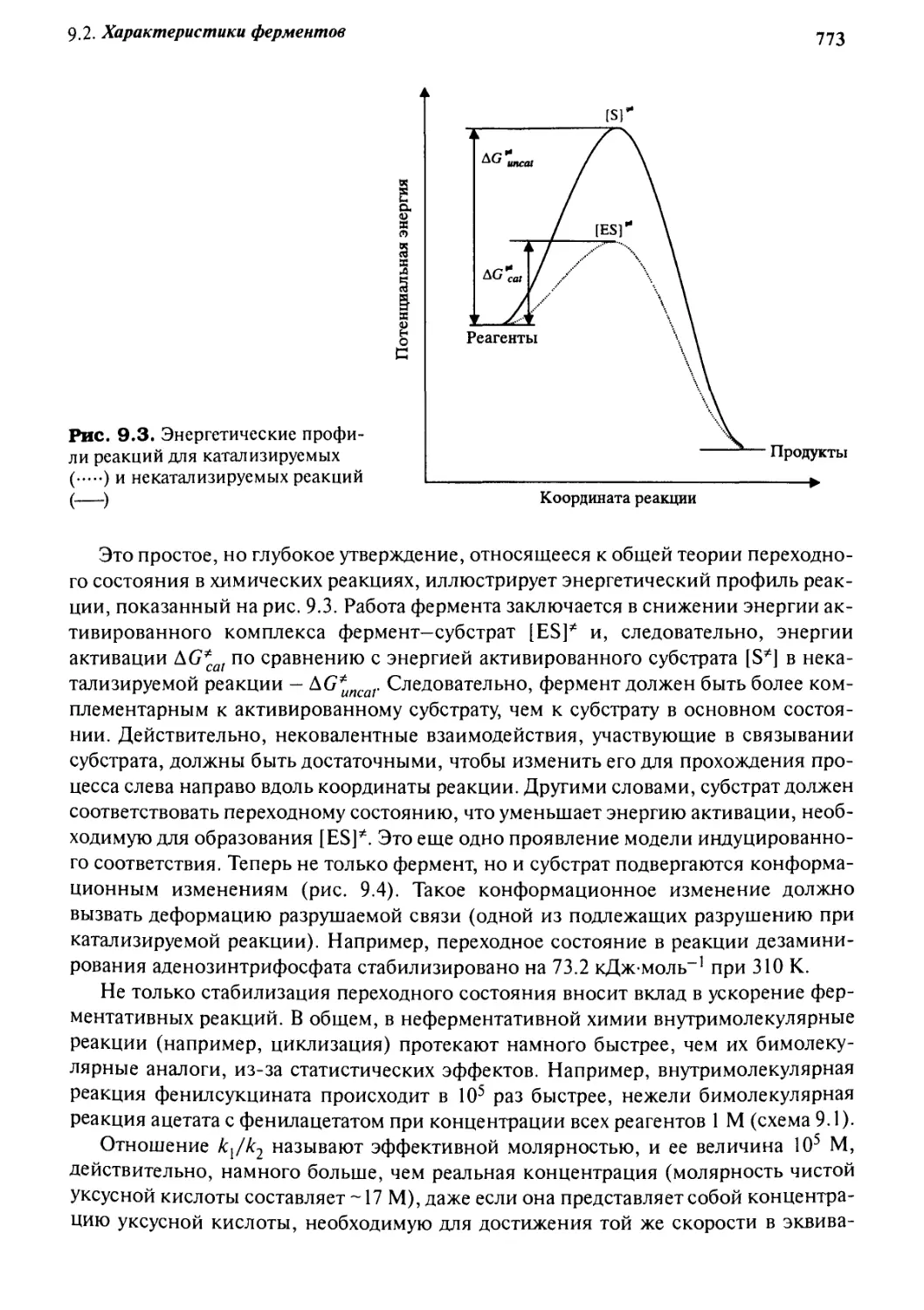
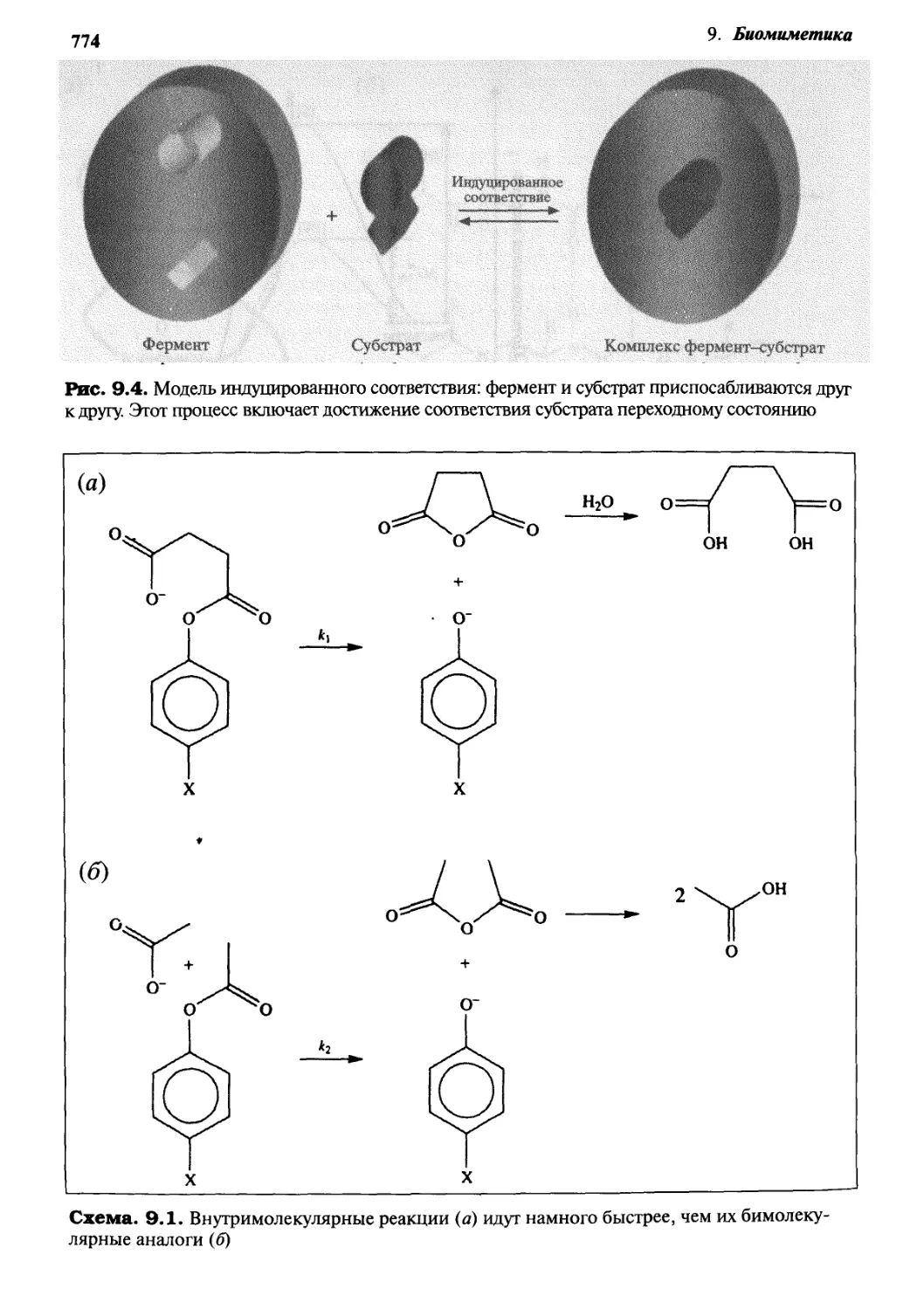
9.2.2 Механизм ферментативного катализа 772
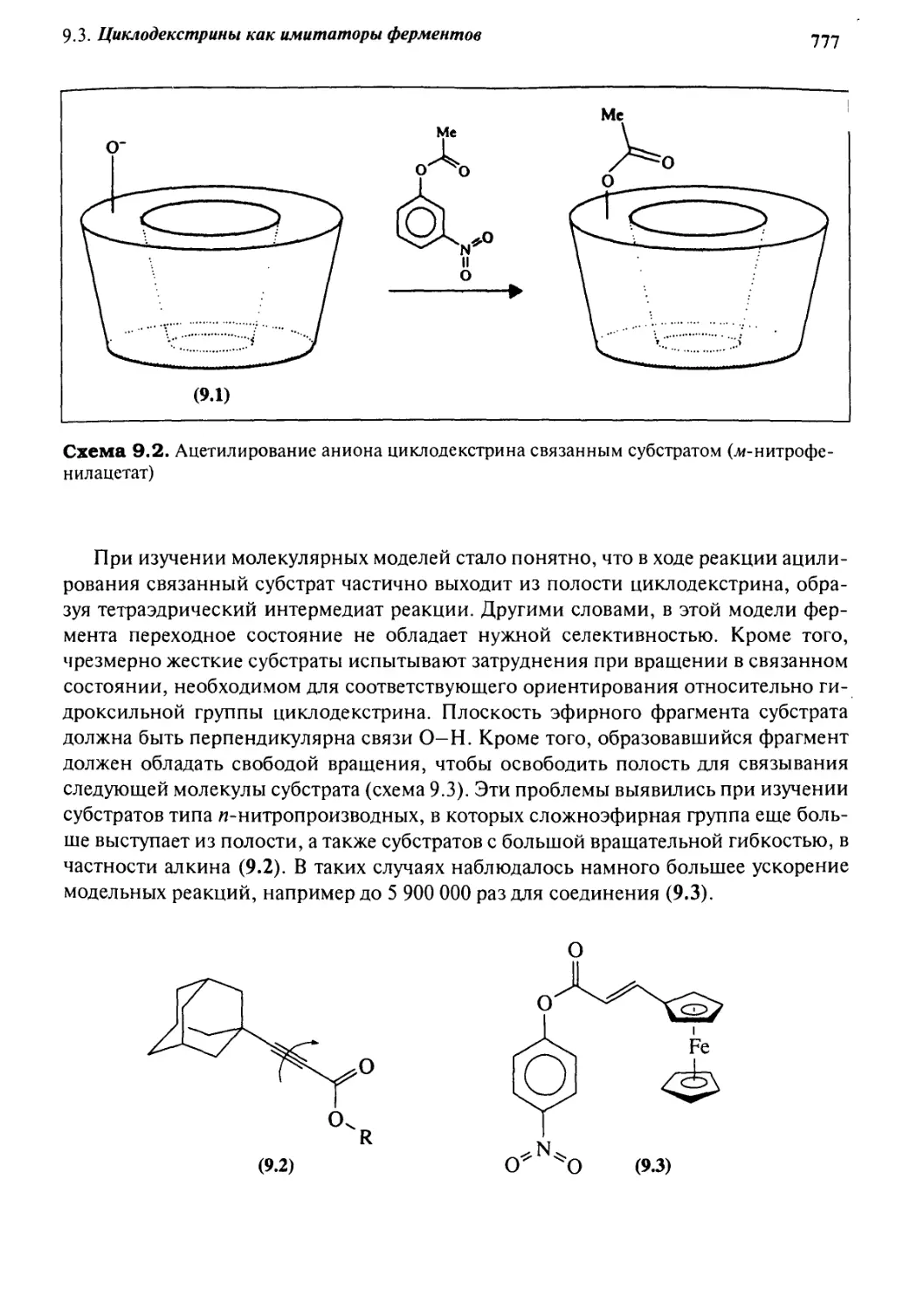
Q.g Циклодекстрины как имитаторы ферментов 775
9.3.1 Моделирование ферментов
с использованием циклодекстринов как хозяев 776
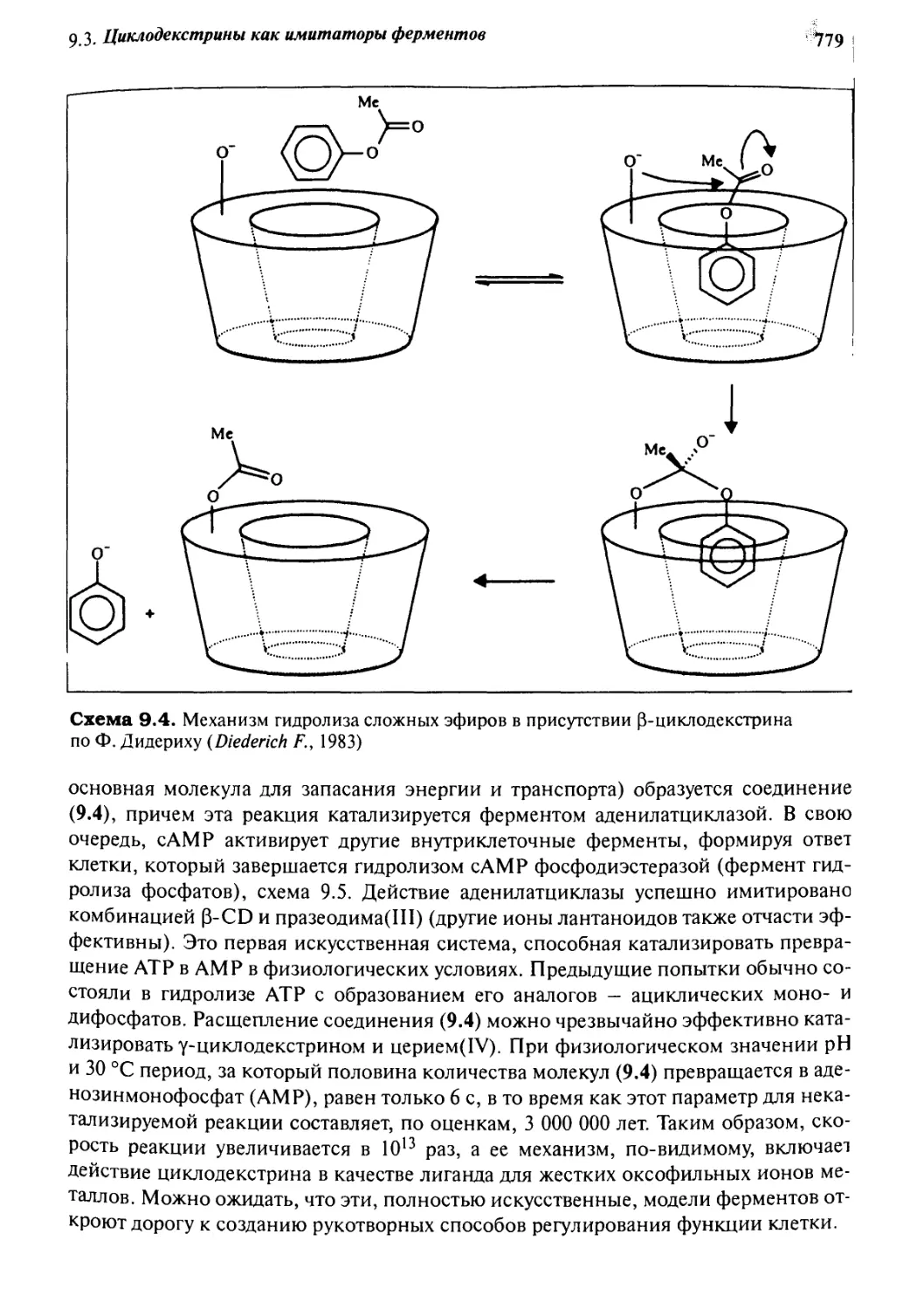
9.3.2 Циклодекстрины как имитаторы эстераз 778
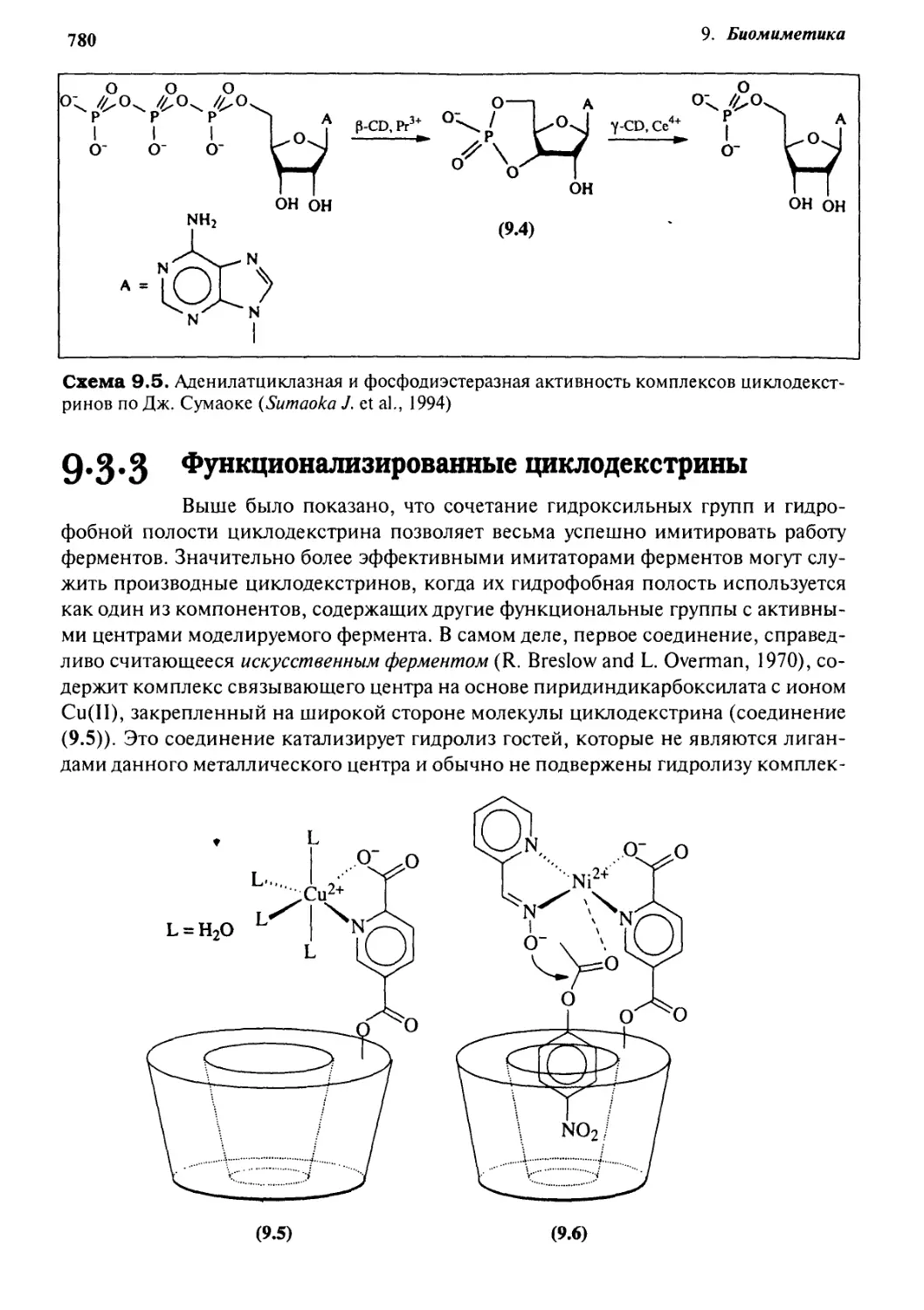
9.3.3 Функционализированные циклодекстрины 780
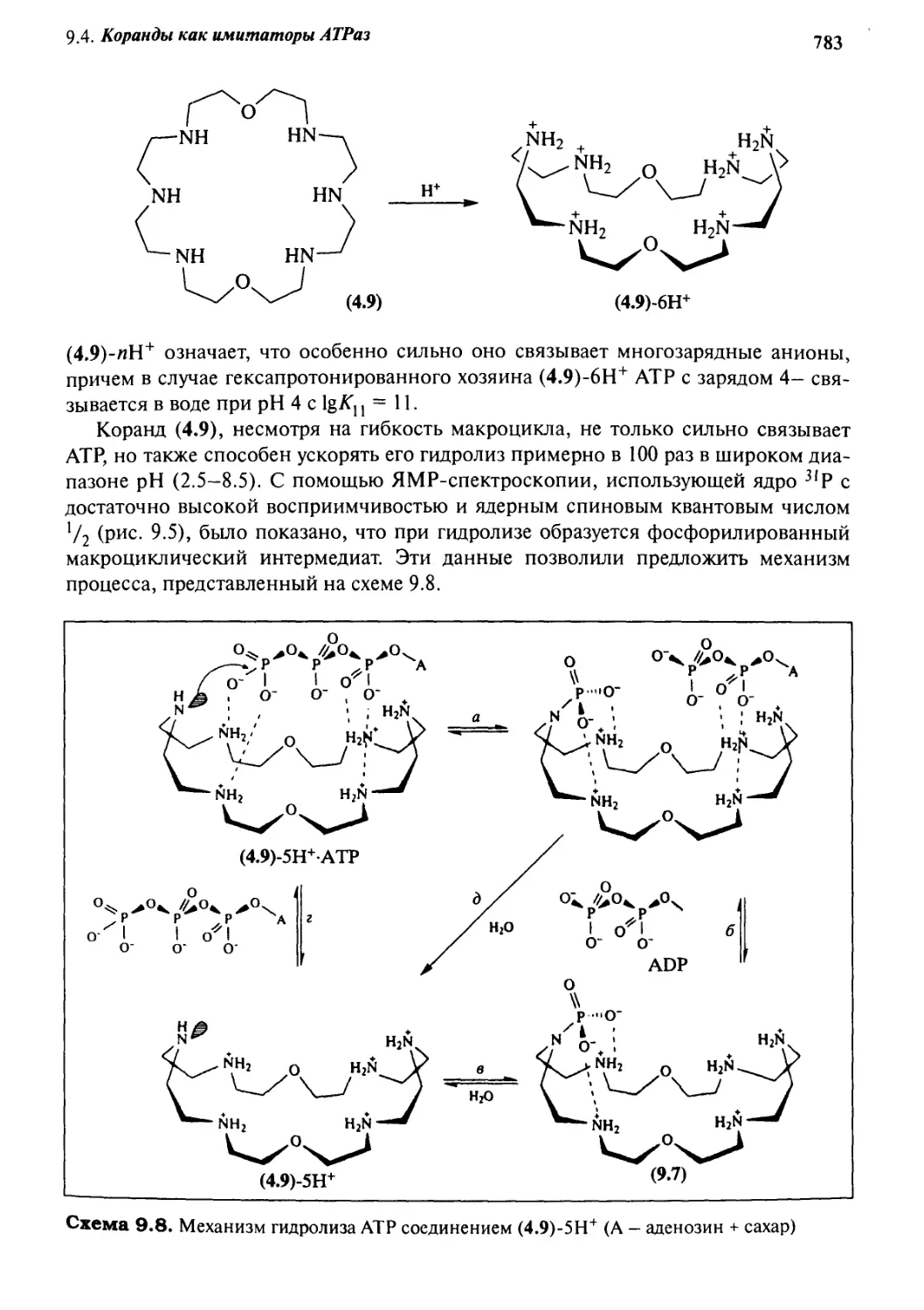
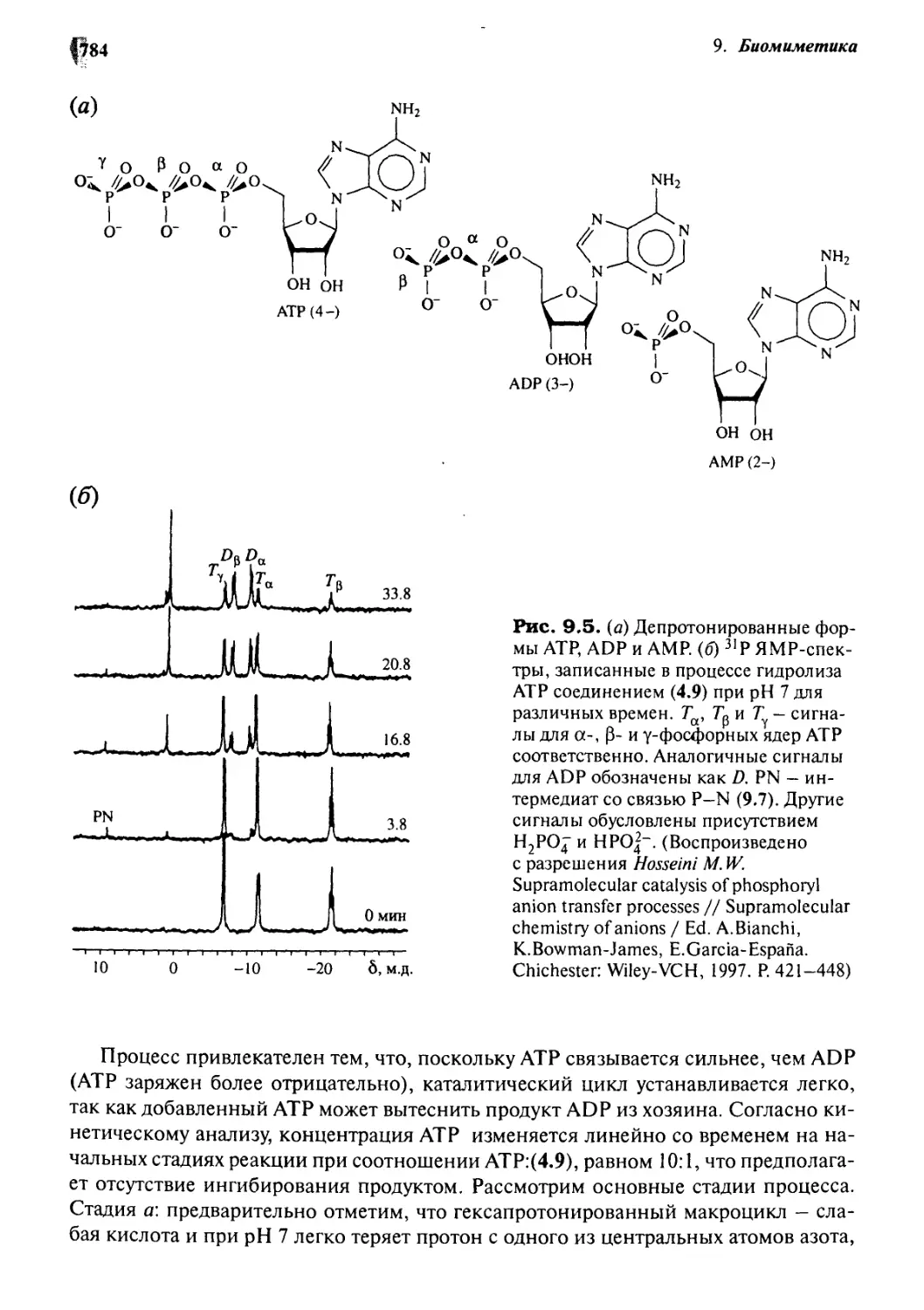
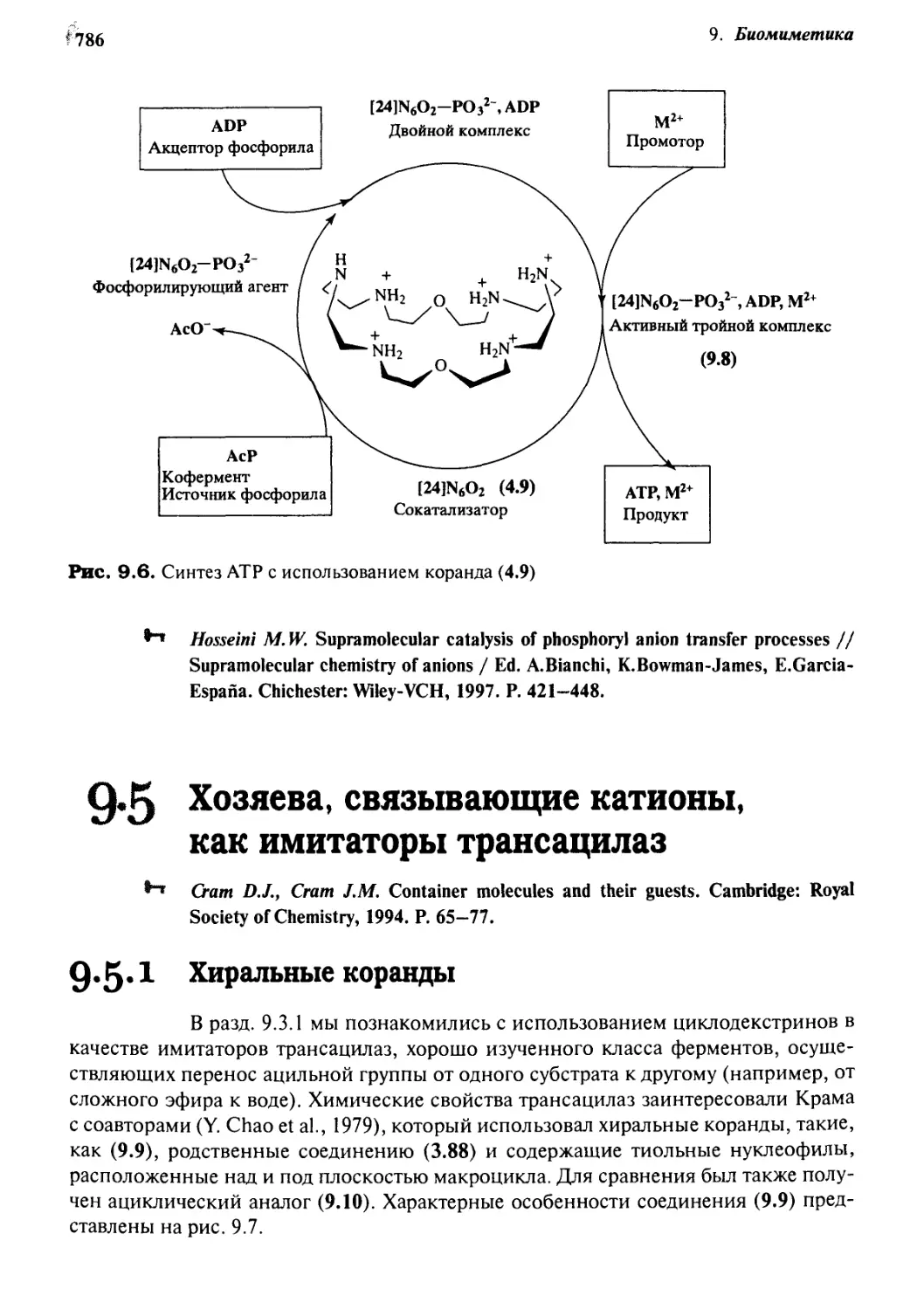
9*4 Коранды как имитаторы АТРаз 782
9*5 Хозяева, связывающие катионы,
как имитаторы трансацилаз 786
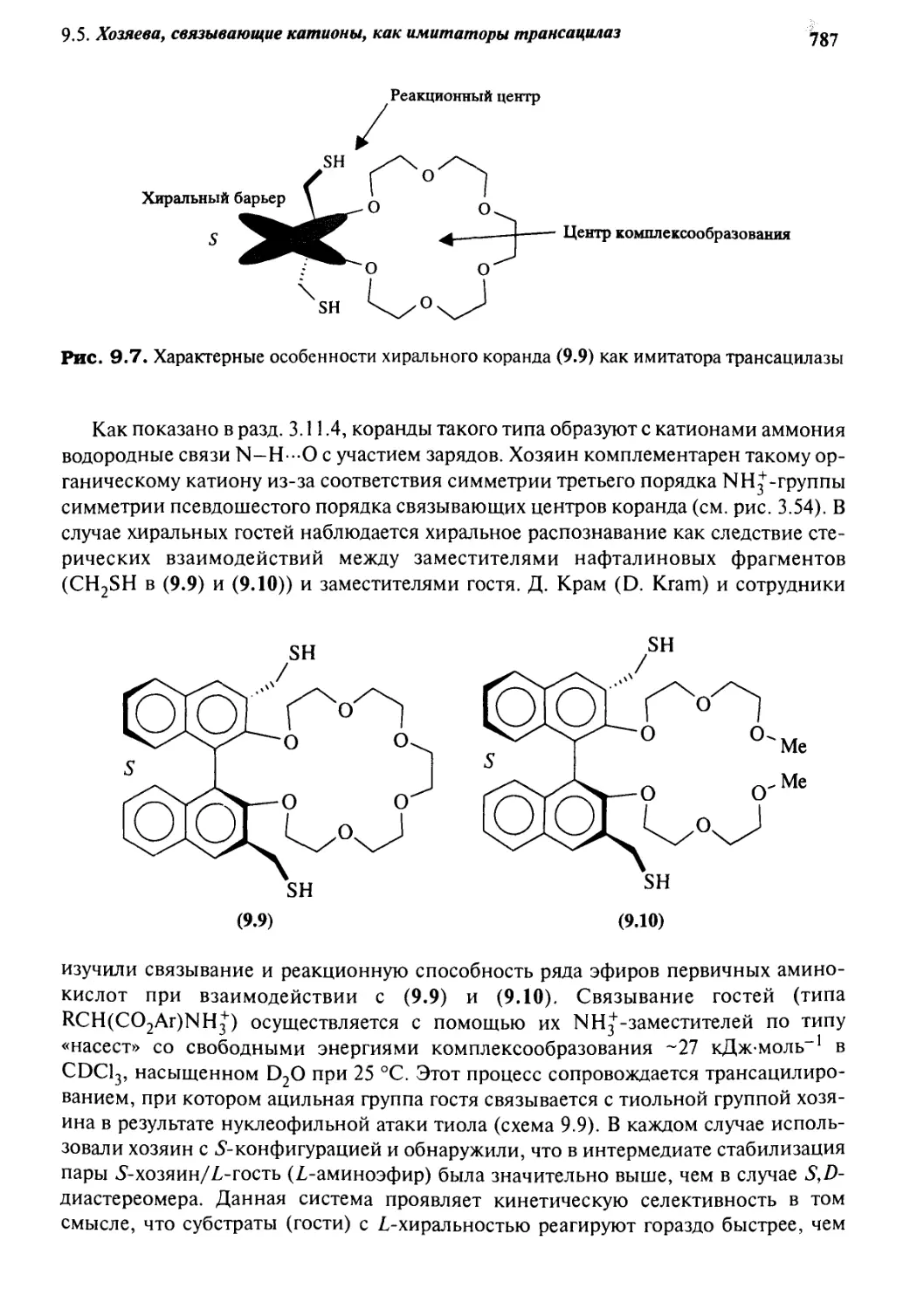
9.5.1 Хиральные коранды 786
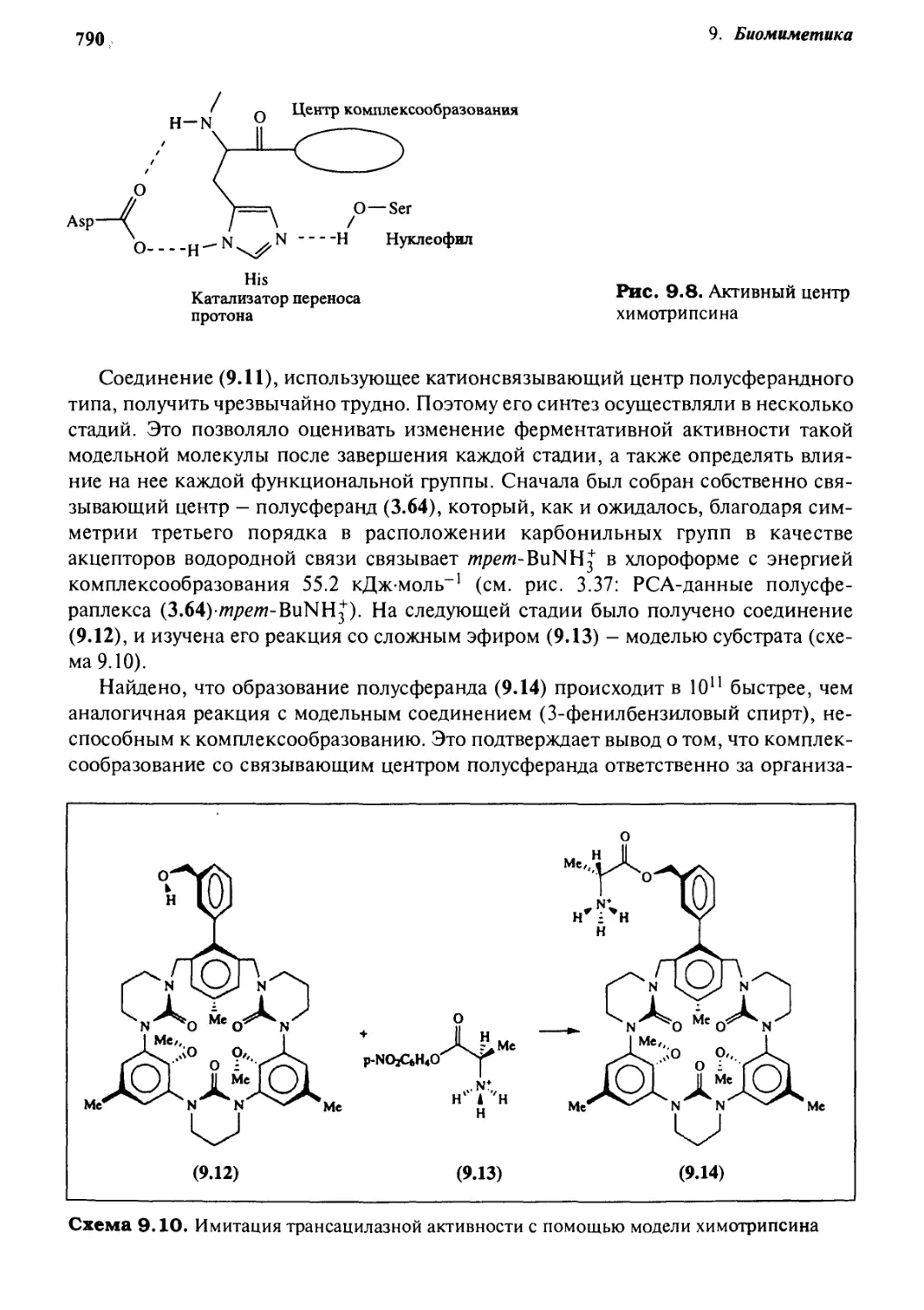
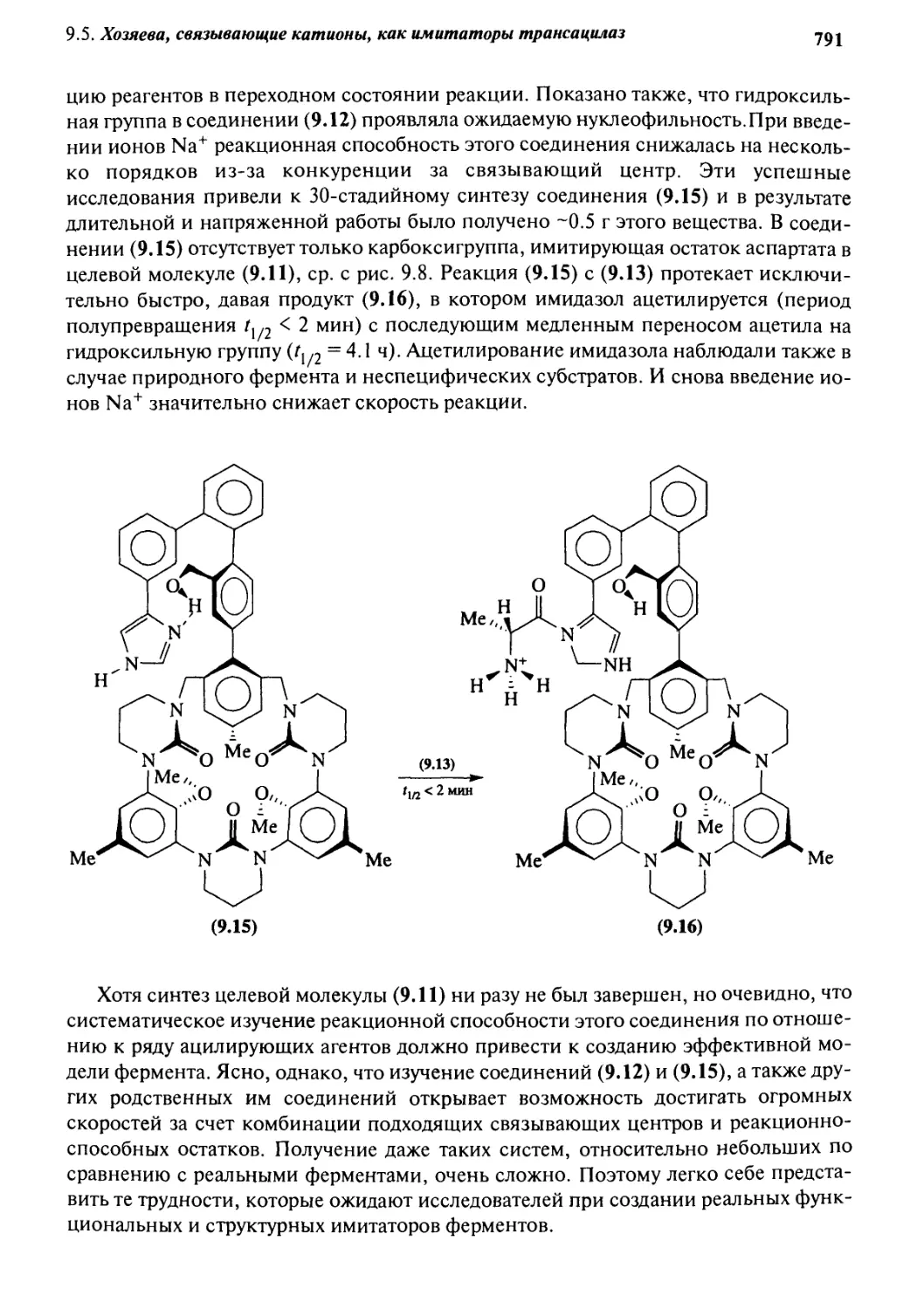
9.5.2 Имитация структуры и функции 789
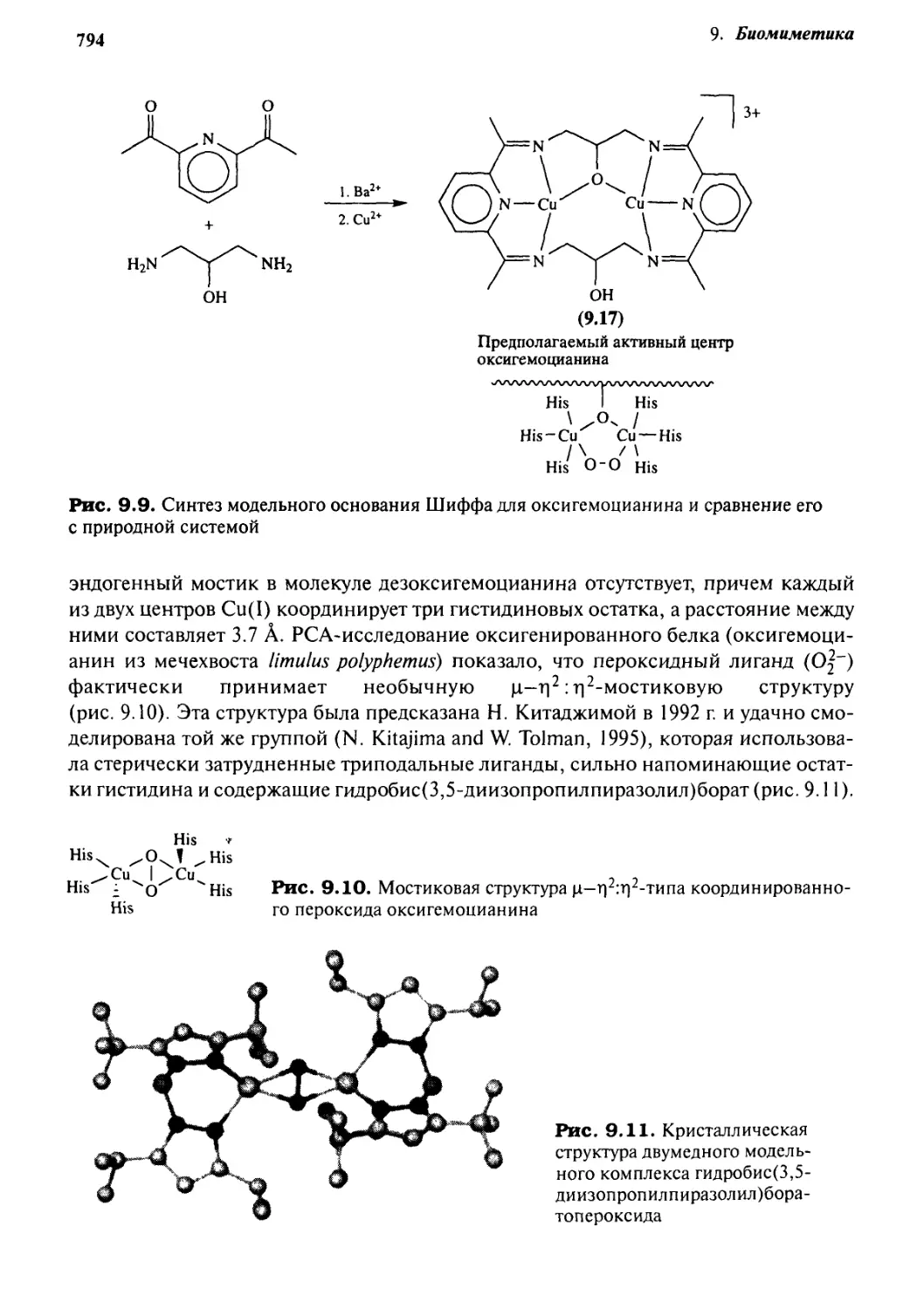
Q.6 Металлобиоцентры 792
9.6.1 Модели гемоцианина 793
9.6.2 Цинксодержащие ферменты 795
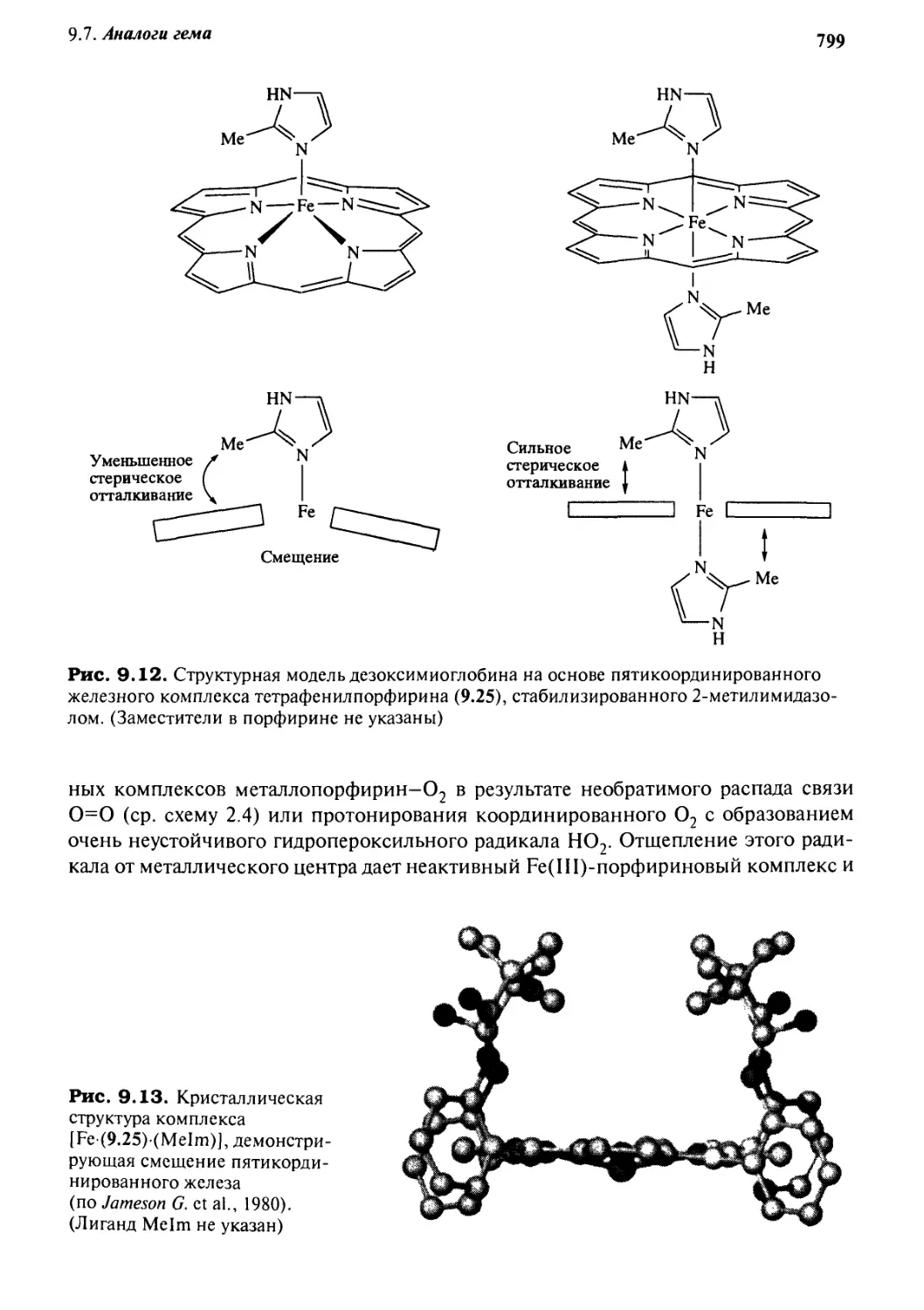
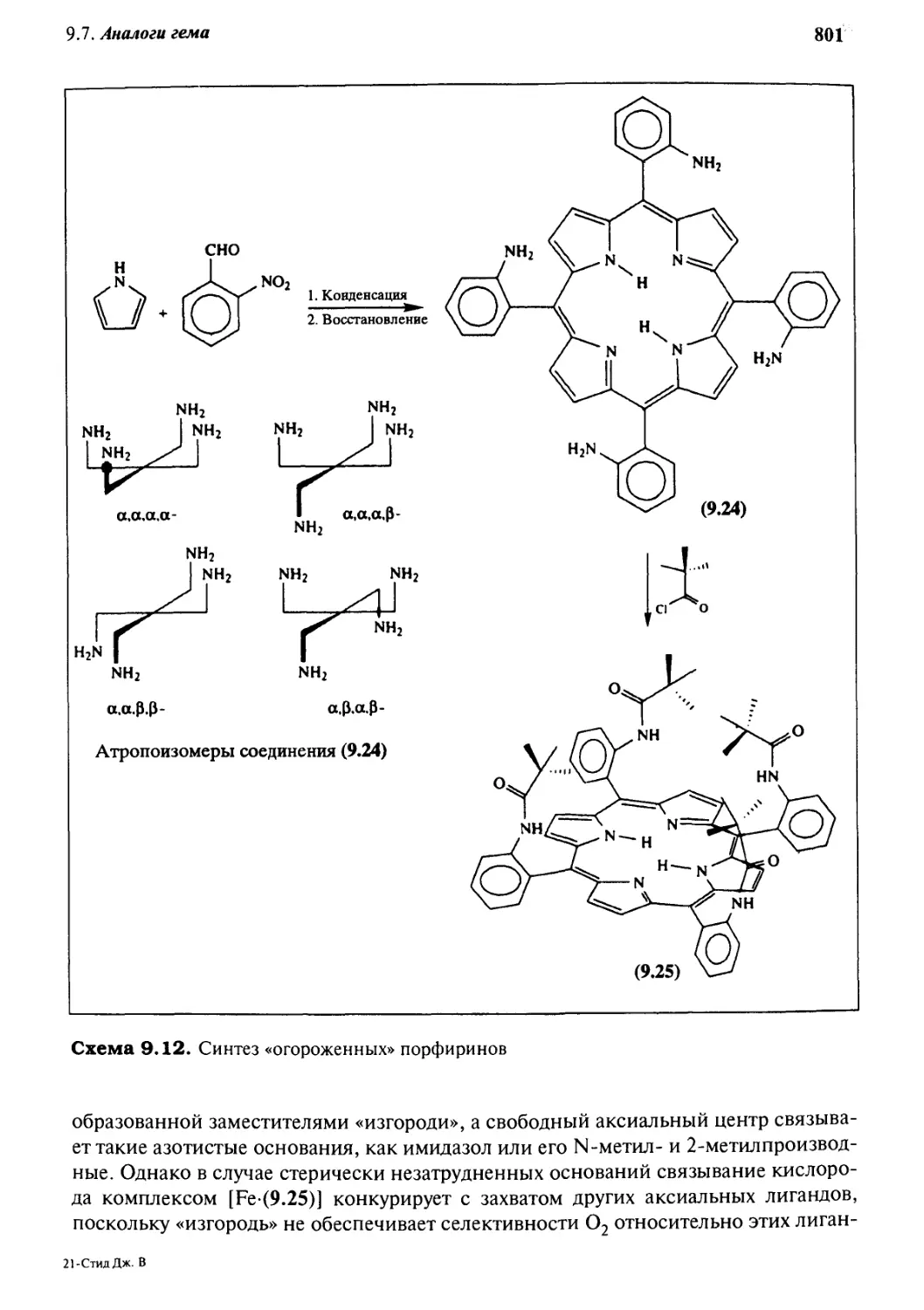
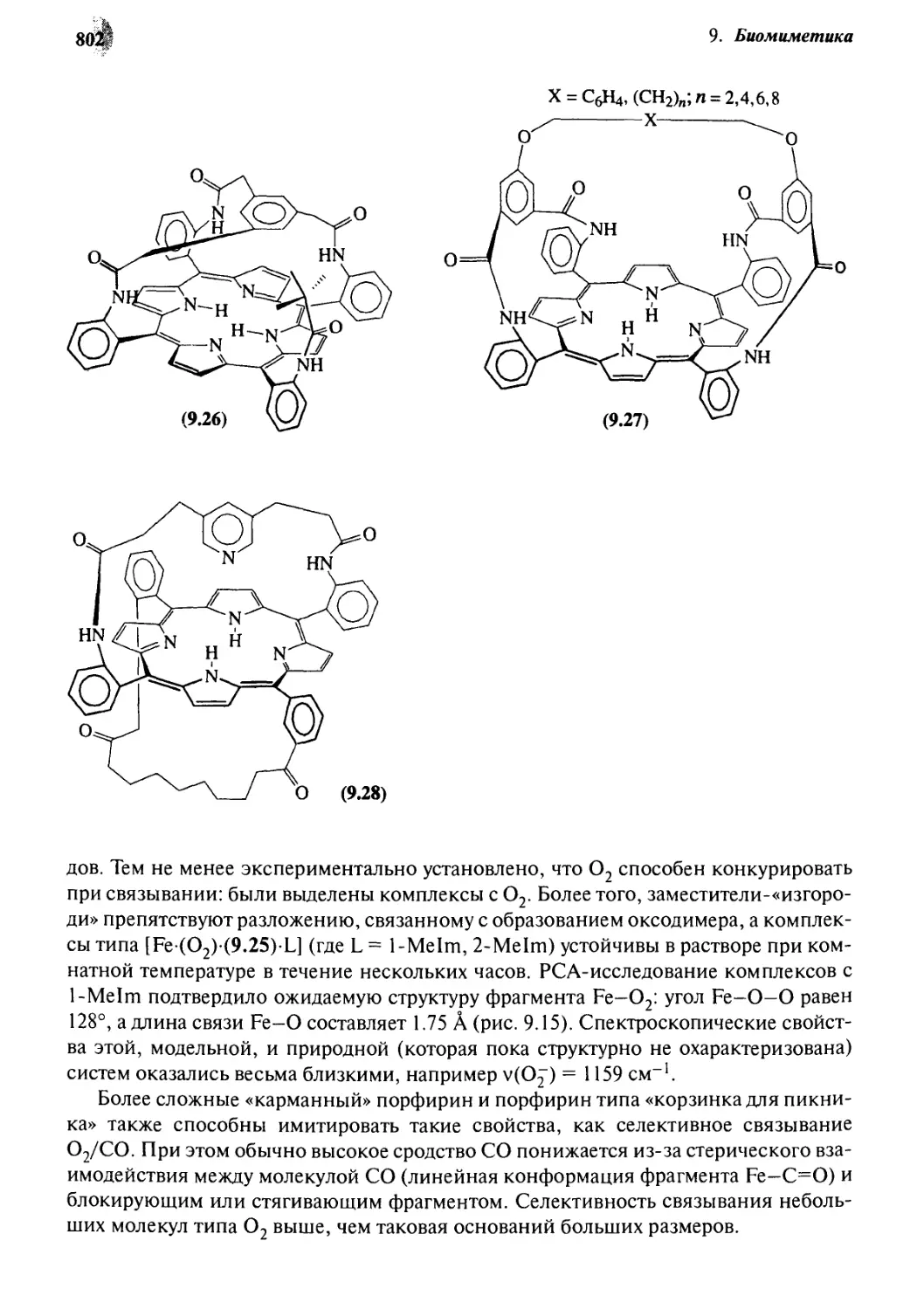
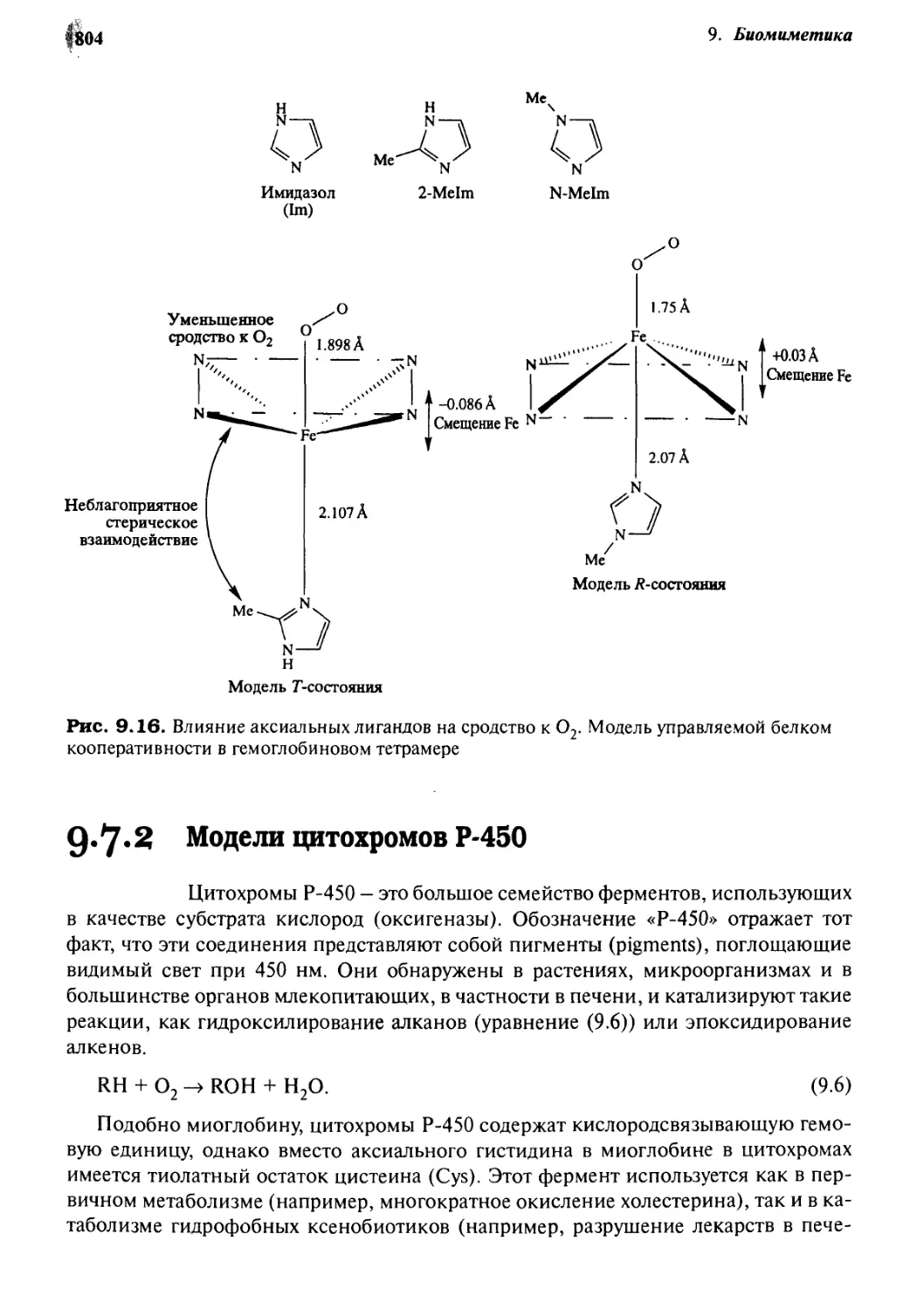
Q. *J Аналоги гема 797
9.7.1 Модели связывания и транспорта кислорода 797
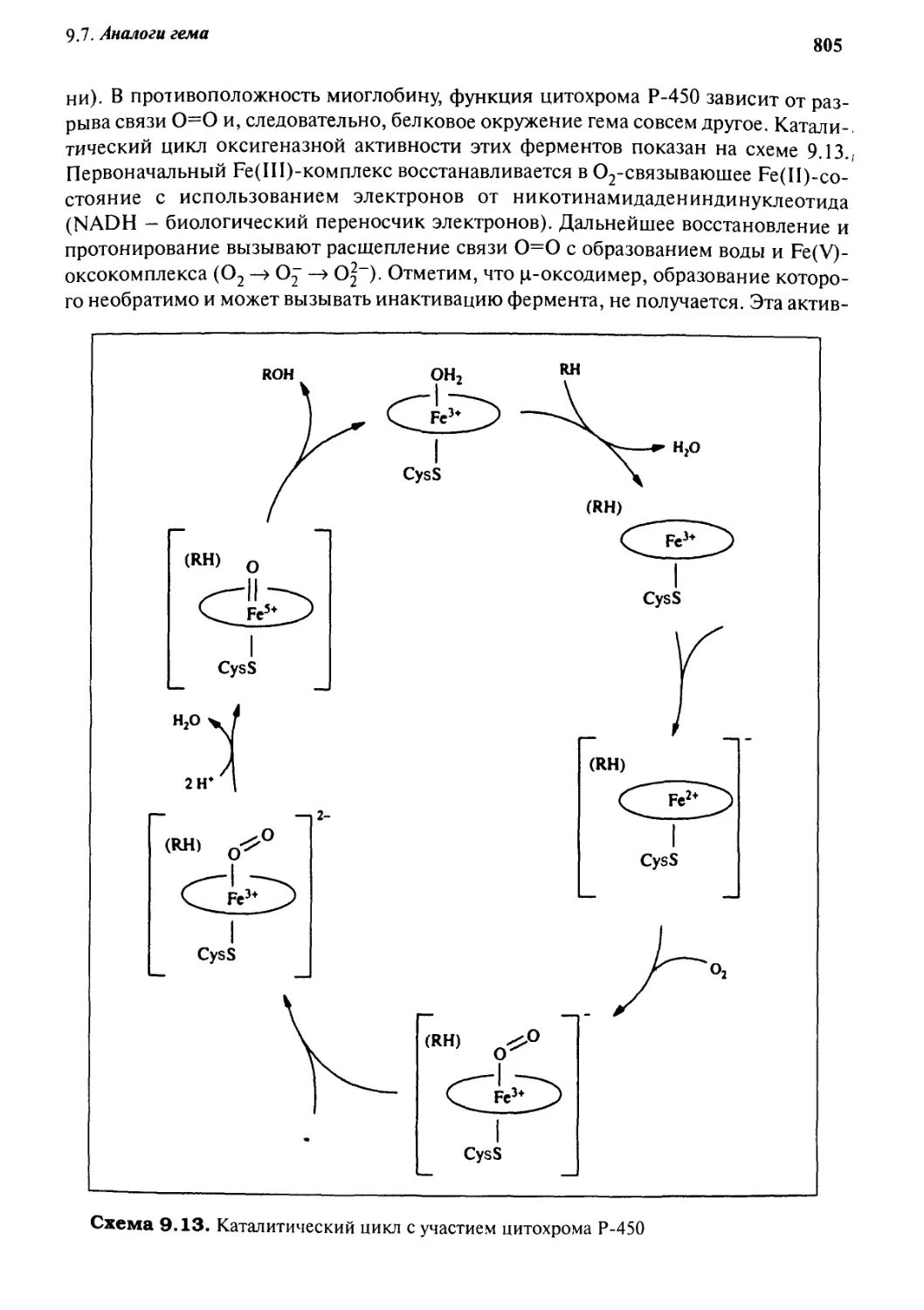
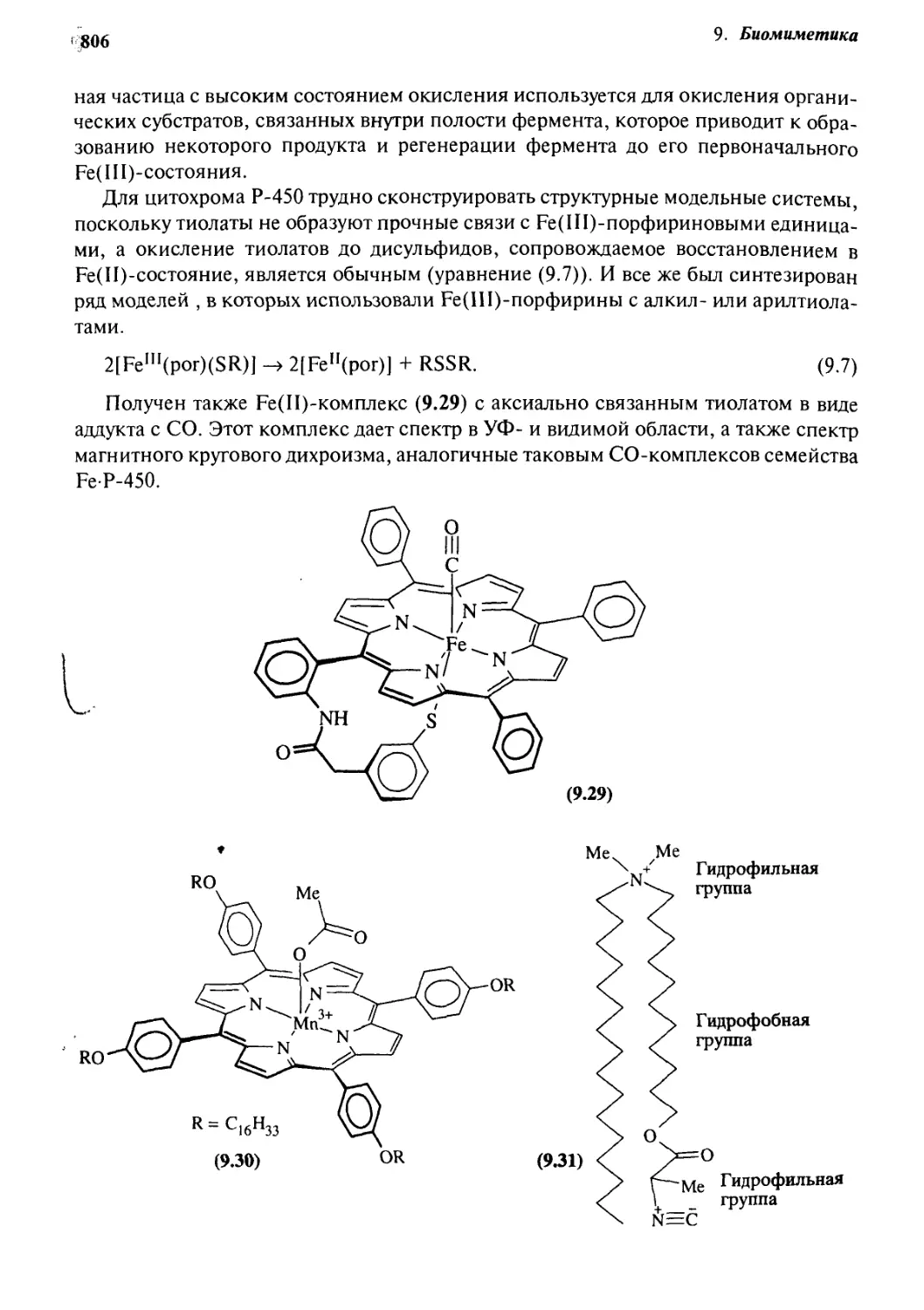
9.7.2 Модели цитохромов Р-450 804
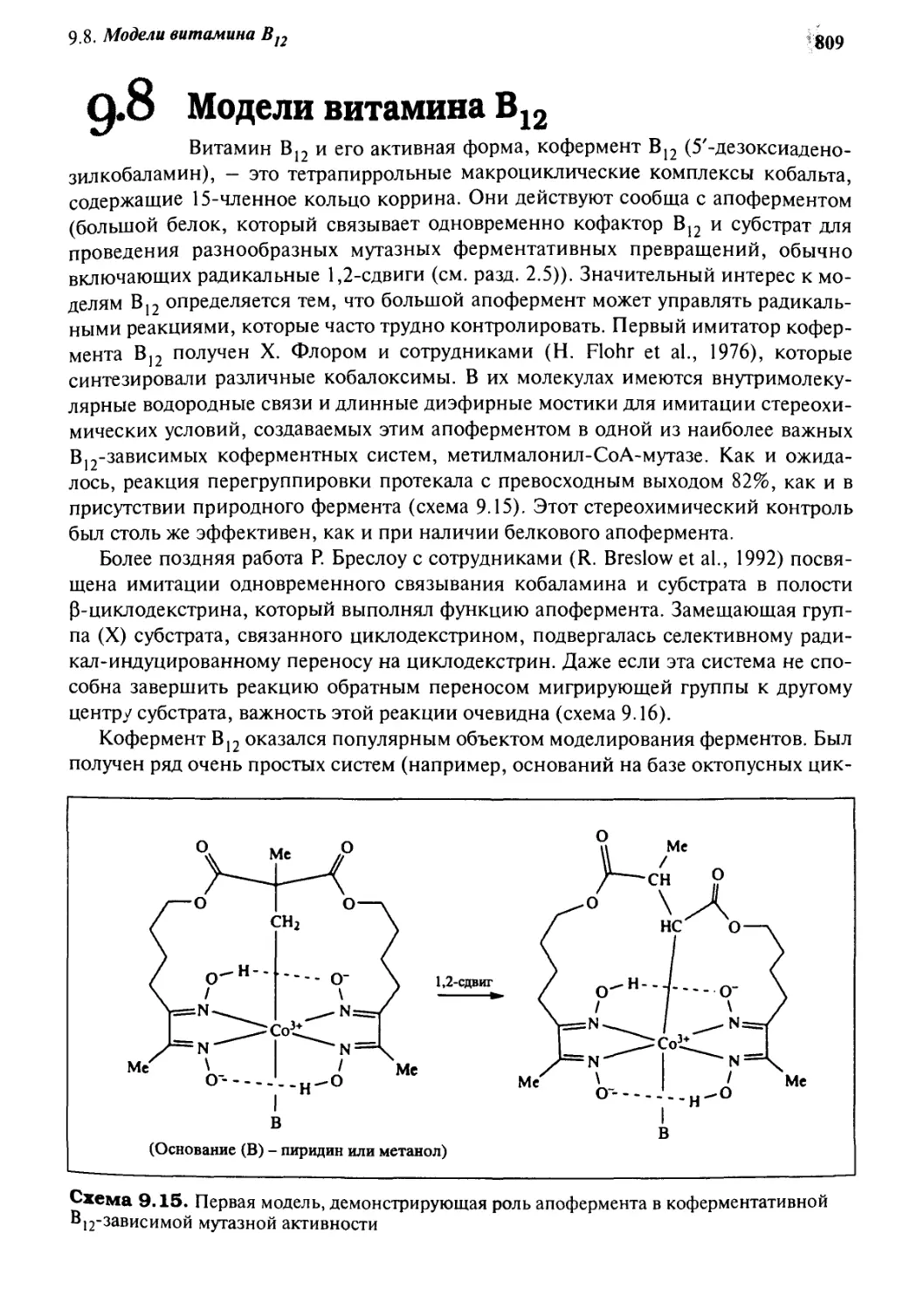
9*8 Модели витамина В12 809
Учебные задания 810
Мысленный эксперимент 811
Литература 811
490 Оглавление
1О Жидкие поверхности раздела,
жидкие кристаллы
и жидкие клатраты т
10.1 Порядок в жидкостях 813
10.2 Поверхностно-активные вещества
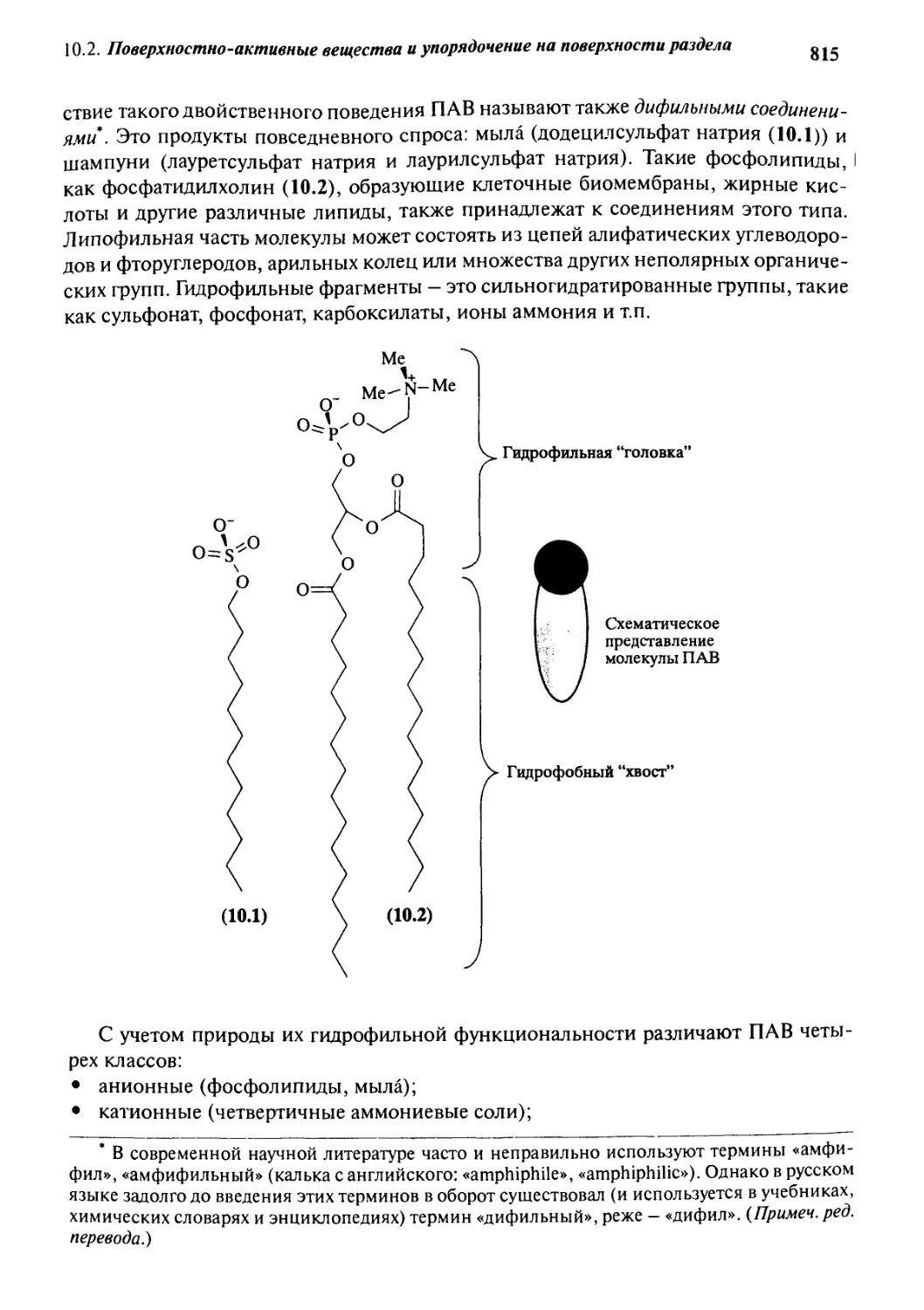
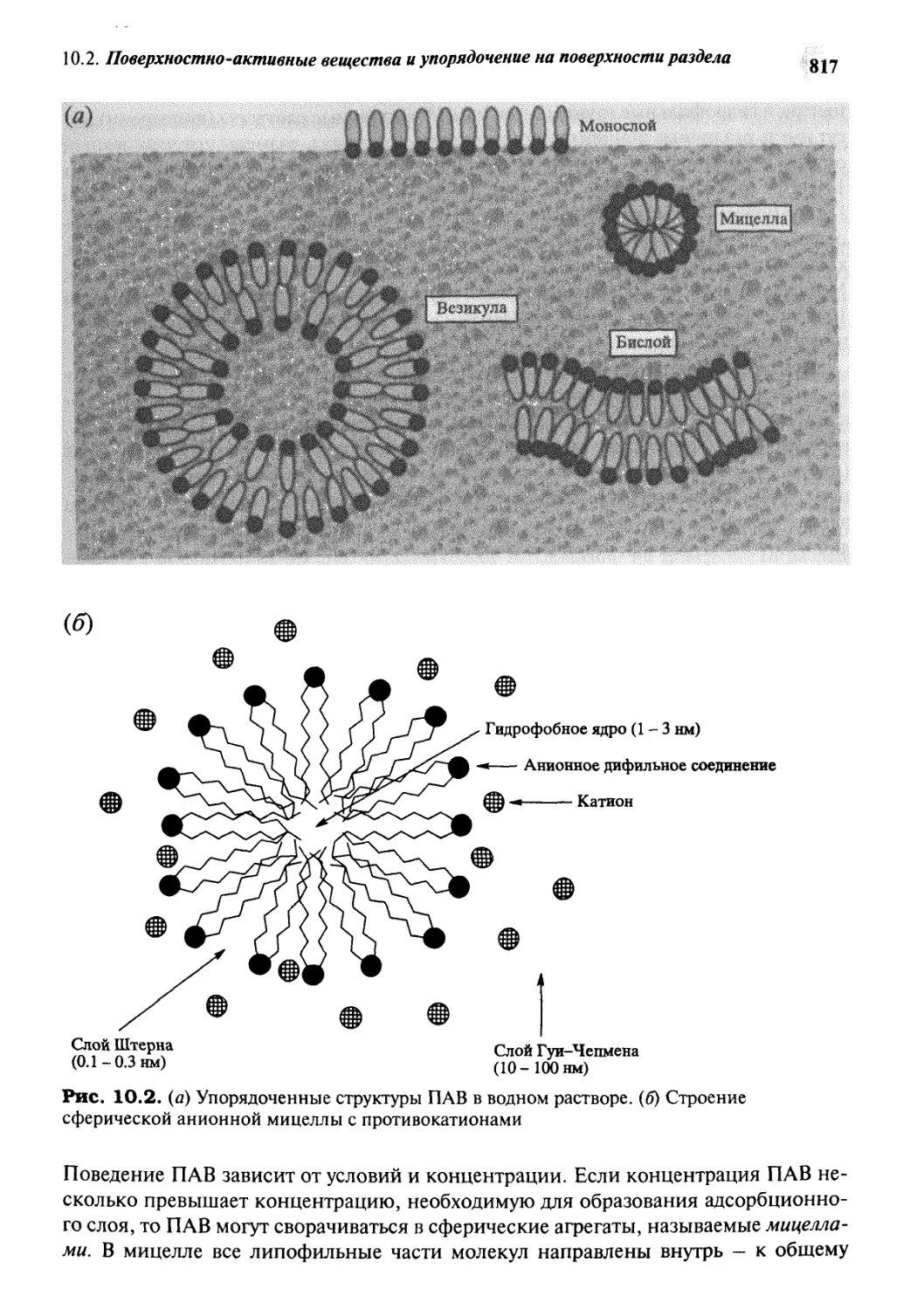
и упорядочение на поверхности раздела 814
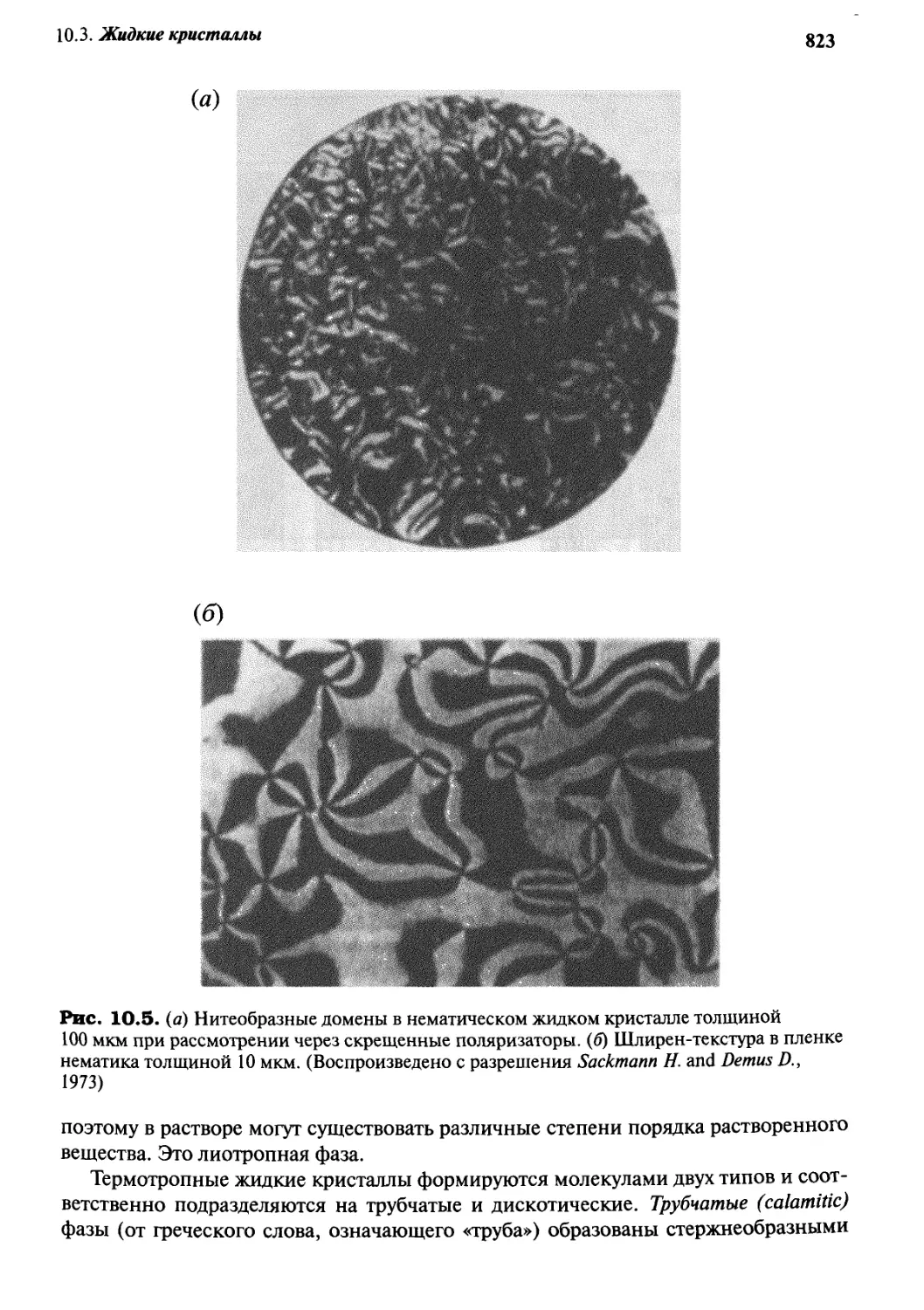
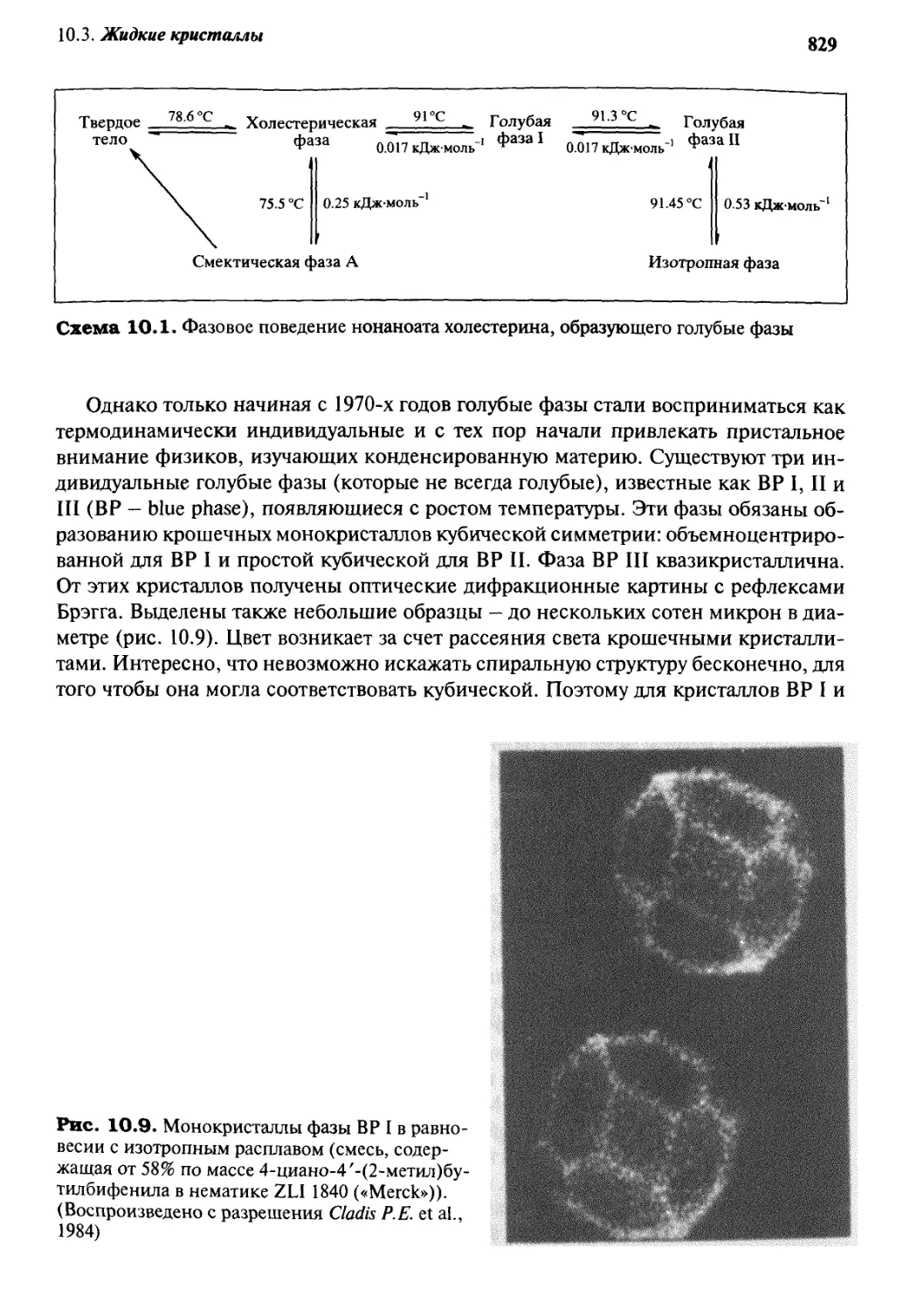
10.3 Жидкие кристаллы 820
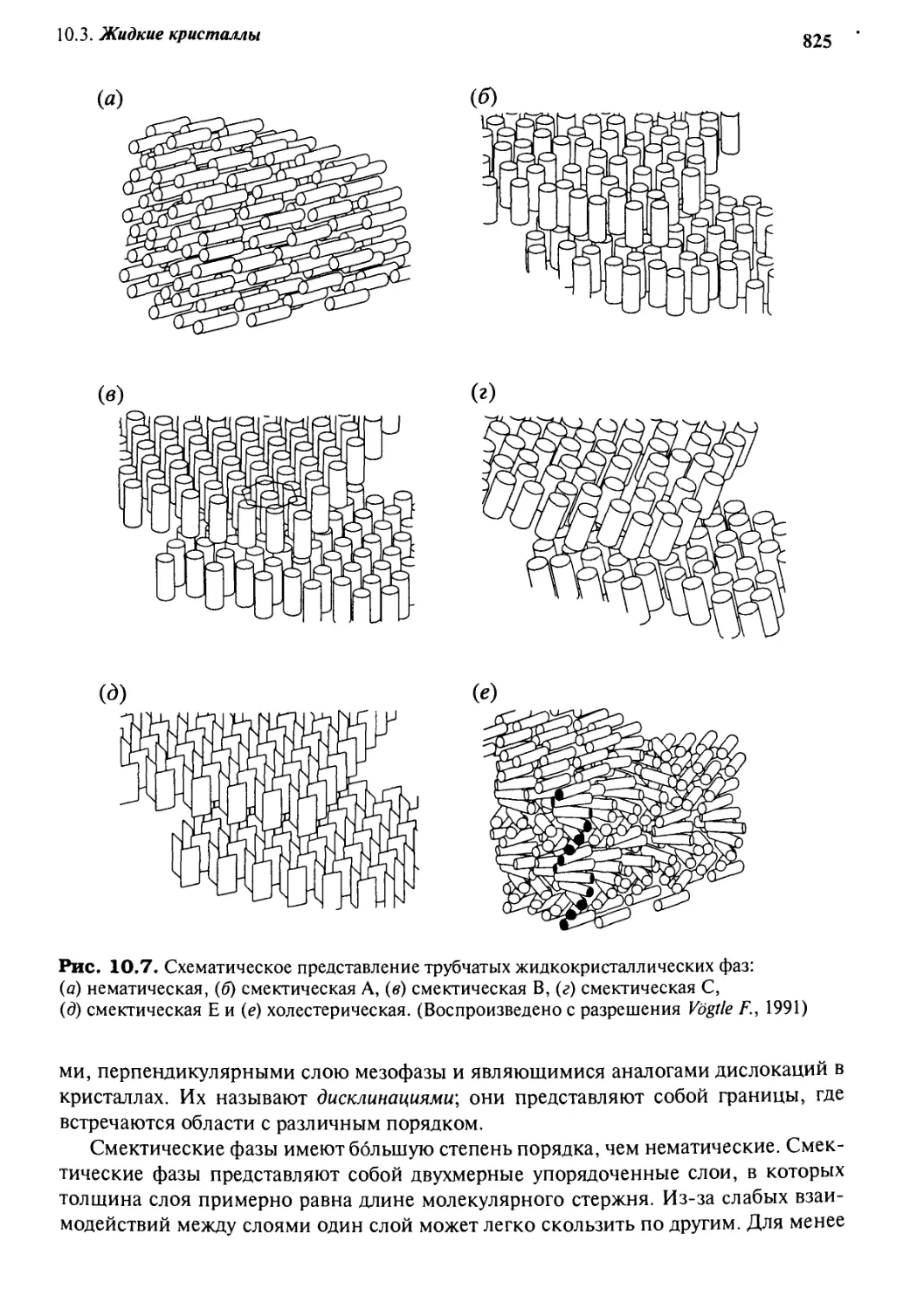
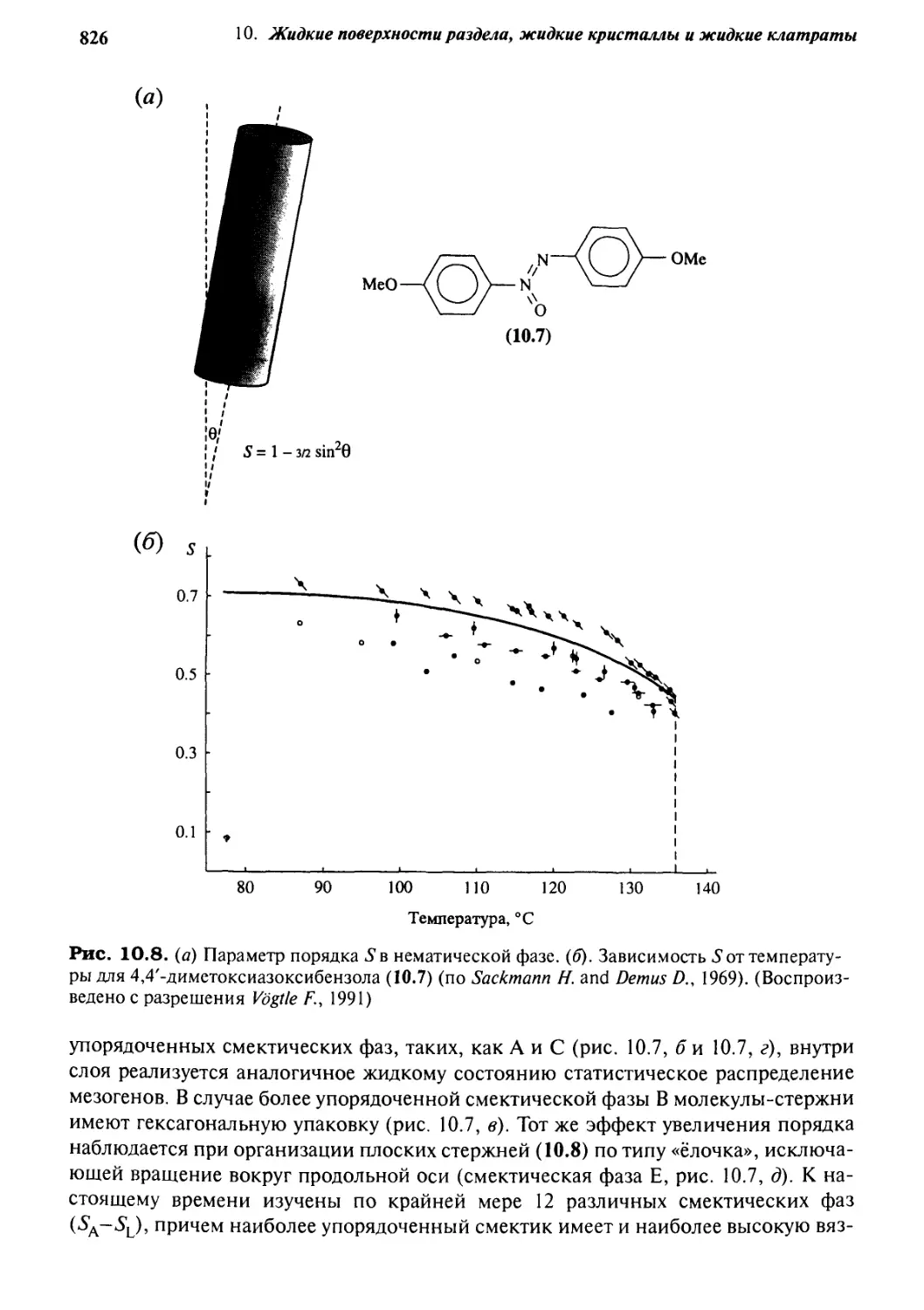
10.3.1 Природа и структура 820
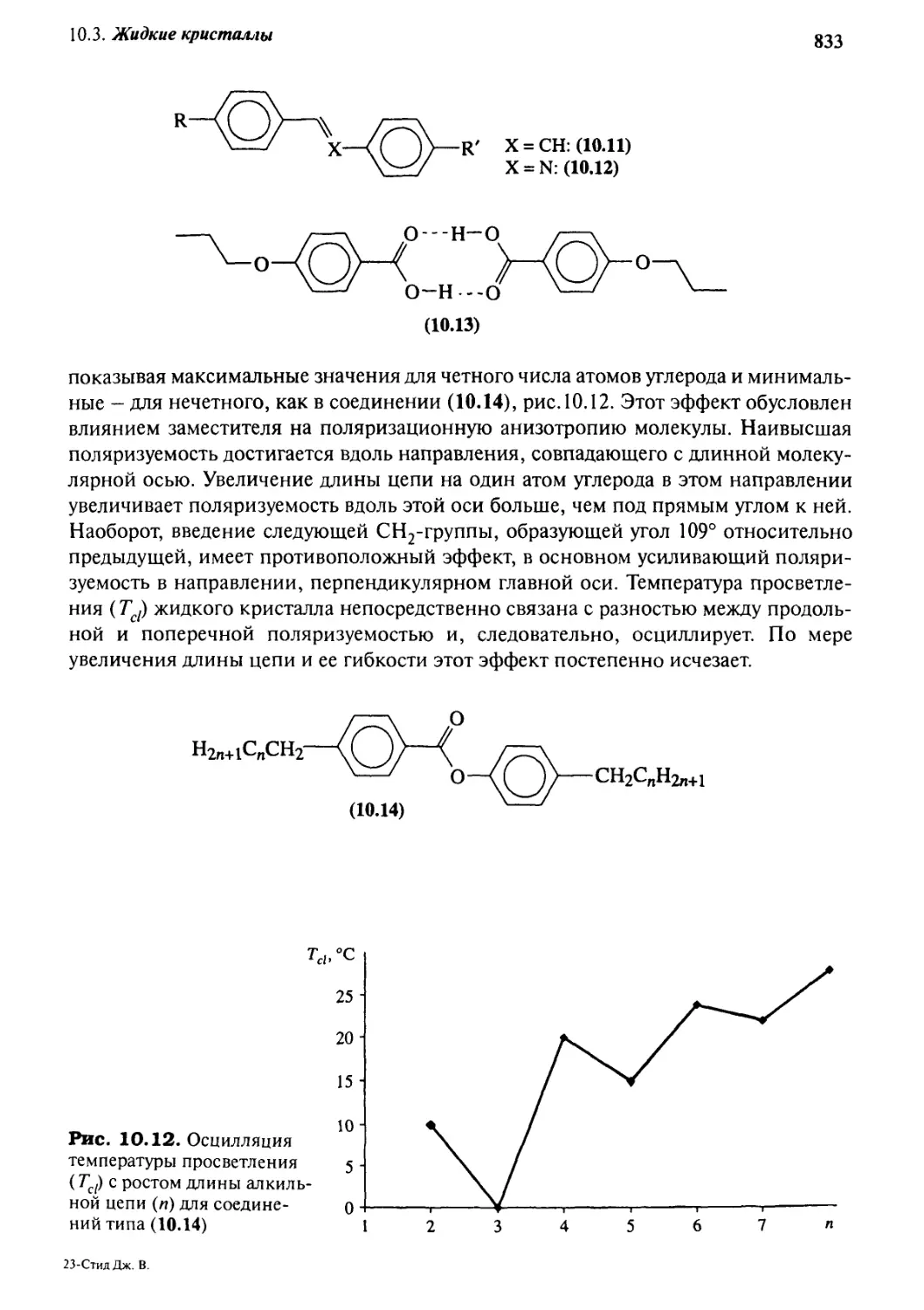
10.3.2 Дизайн жидкокристаллических материалов 831
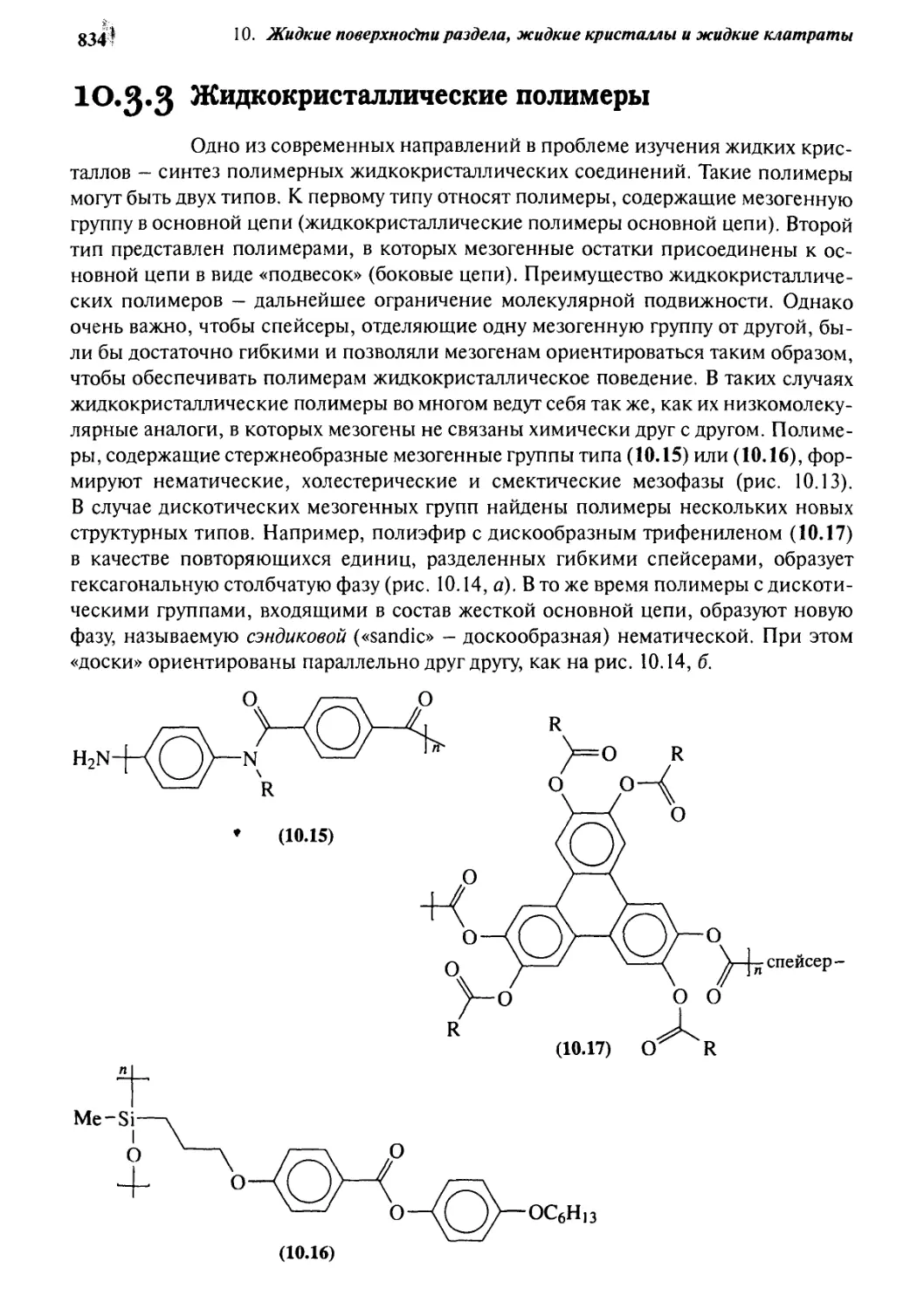
10.3.3 Жидкокристаллические полимеры 834
10.3.4 Жидкокристаллические дисплеи 835
10.4 Жидкие клатраты 837
10.4.1 Открытие и свойства 837
10.4.2 Ион оксония в химии жидких клатратов 842
Учебные задания 845
Литература 846
Предметный указатель 847
Дополнительная литература 883
Q Инженерия кристаллов
Нет более ответственной задачи в химии,
чем установление геометрической структуры молекулы.
Грамотное решение этой задачи делает бессмысленными
любые дальнейшие рассуждения о ее структуре и
обеспечивает надежную основу для понимания физических,
химических и биологических свойств данного соединения.
Р. Хофман
6.1 Общие вопросы
*"* Desiraju G.R. Crystal engineering: The design of organic solids. Amsterdam: Elsevier,
1989.
6.1.1 Введение
В молекулярной химии основной строительный блок (или тектон) —
это атом. Атомы взаимодействуют друг с другом, образуя молекулы с ковалентны-
ми связями. В супрамолекулярной химии, говоря очень упрощенно, процесс
самосборки кристаллов эквивалентен процессу образования молекулы из атомов в
молекулярной химии. В этом случае строительными блоками - тектонами - являются
уже молекулы (а также ионы), причем при взаимодействии друг с другом молекулы
образуют связи нековалентной природы (см. разд. 1.7) и дают в растворах
кристаллы или агрегаты всевозможных форм (рис.6.1).
Процесс кристаллизации, по определению, — это процесс самосборки в том
смысле, что молекулы компонента (кристаллические или супрамолекулярные тек-
тоны) должны сами найти и узнать друг друга в растворе, а также принять
оптимальную взаимную ориентацию в рамках отведенного времени. Образовавшийся
агрегат, будучи уже ансамблем, должен продолжать поиск других аналогичных тек-
тонов. В конце концов, этот процесс приведет к образованию упорядоченного
зародыша кристаллизации, обладающего некоторой стабильностью. Мы, однако,
обязаны провести четкое различие между самосборкой кристалла и самосборкой в
растворе (которая будет подробно рассмотрена в гл. 7).
• Самосборка кристалла — неравновесный процесс, в ходе которого конечная
структура материала определяется как кинетическими, так и
термодинамическими факторами. Эта структура в большинстве случаев зависит от условий
кристаллизации; при этом, как правило, образуются полиморфные структуры
(кристаллы, составленные из одинаковых тектонов, но упакованных различным
492
6. Инженерия кристаллов
antagona
Vt поп potelf fieri angulus solidus absque tribus planis sic поп
poteft fieri corpus physicum sine в $ дг (<• -i О
*
Sal efculentum
Ommia in mansura, et numero, et pondere difpofuifti, fap ij
Ameihy flus
Рис. 6.1. Примеры правильных
конфигураций в кристаллических
веществах и природных формах.
Иллюстрация заимствована из
работы шотландского ученого
У. Дэвидсона (Дэвиссона), который
один из первых упомянул
кристаллографию как «новую
дисциплину» (Davissone W. Les
elements de la philosophie de l'art du feu,
ou Chemie. Paris, 1657)
образом). Структуры, которые образуются быстро, могут доминировать над
другими структурами, даже если последние более стабильны.
• Самосборка в растворе - термодинамически контролируемая равновесная реак-.'
ция, в ходе которой компоненты способны проходить через целый ряд возмож-'1
ных структур до тех пор, пока не сформируется структура, обладающая макси-.
мальной устойчивостью. Реакция протекает в течение бесконечно большого
промежутка времени, поэтому возможные «ошибки» в составлении «ансамблям
могут быть «исправлены» в процессе распада комплекса, а затем — повторного
комплексообразования.
Синтез молекул и синтез кристаллов отличаются друг от друга как масштабами, так
и многообразием. Что касается масштаба, то атомы и молекулы представляют
собой микроскопические частицы, поведение которых подчиняется квантово-меха-
ническим законам. Кристаллы же являются макроскопическими объектами,
обладающими дальним порядком и объемными свойствами (электрическая
проводимость, теплоемкость и т.п.). Что же касается многообразия, то ковалентная
связь представляет собой сложное, достаточно хорошо изученное взаимодействие,
характеризующееся высокой прочностью на небольших расстояниях, направлен-
6.1. Общие вопросы 493
ностью и синтетической гибкостью. Синтез кристаллов представляет собой
значительно более сложный для управления процесс, поскольку включает в себя
манипулирование синергетической системой, в которой имеют место взаимодействия как
дальнего, так и ближнего порядка. Некоторые из них быстро ослабевают с
увеличением расстояния (например, энергия индукционных и дисперсионных
взаимодействий убывает обратно пропорционально шестой степени расстояния между
взаимодействующими партнерами), тогда как другие, например водородные, связи
могут эффективно действовать на очень больших расстояниях. Поэтому не
удивительно, что в то время как структурный синтез сложных молекул (таких, как
природные соединения, карцеранды и т.п.) стал реально осуществимым, все еще в
значительной степени не реализованы возможности предсказания и, следовательно,
контроля за структурообразованием практически всех (за исключением самых
простых) кристаллов.
Область инженерии (конструирование) кристаллов обрела свою
индивидуальность благодаря А. фон Хиппелю (A. von Hippel), который детально описал ее
основы в 1962 г., используя термин «молекулярное конструирование». Современная
инженерия кристаллов восходит к началу 1960-х годов, когда она изначально
развивалась как метод изучения региоселективности и распределения продуктов в
молекулярных реакциях, протекающих в твердом состоянии; причем изучали
соединения, относящиеся к а,3-ненасыщенным карбоновым кислотам и л-хинонам,
а сама область получила название топохимия (G. Schmidt, 1964). Эта область
быстро развивалась, особенно после появления современных кристаллографических
методик (в частности, использование четырехкружных дифрактометров в начале
1970-х годов, а позднее — использование детекторов CCD для кристаллографии
малых молекул, см. Дополнение 2.1). В настоящее время она охватывает большой круг
вопросов, таких, как твердофазные межмолекулярные взаимодействия,
направленное предсказание, получение и интерпретация различных структур, а также синтез
новых молекулярных строительных блоков и кристаллических материалов. Более
того, данная область включает и синтез твердых материалов, например цеолитов,
используемых в нефтедобывающей и нефтехимической промышленности (см.
разд. 5.1.2). Инженерия кристаллов состоит из двух основных разделов: анализа и
синтеза. Такие ведущие специалисты в области инженерии кристаллов,как Д.
Брага (D. Braga) и Ф. Грэпиони (F. Grepioni) из Университета Болоньи (Италия), а
также Г. Дезирайя (G. Desiraju) из Университета Хайдарабада (Индия) определяют
взаимодействие между этими двумя областями следующим образом:
«Разум и воображение чудесным образом переплетаются при поиске новых функциона-
лизированных твердых веществ, тогда как эксперимент и расчеты одинаково важны
для предсказания и дизайна кристаллических структур». (Braga D., Grepioni F. and
Desiraju G.R.//Chem. Rev. 1998. Vol. 98. P. 1375.)
Следовательно, инженерия кристаллов представляет собой конструирование и
получение кристаллического материала на основе знаний или, по крайней мере,
представлений о стерических и топологических особенностях имеющихся
строительных блоков, а также об их способности^ межмолекулярным взаимодействиям.
494 6. Инженерия кристаллов
Такая способность была названа межмолекулярной валентностью по аналогии с
общепринятой валентностью атома элемента. Наряду с этим будут необходимы
сведения по кинетике и термодинамике процессов образования центров
кристаллизации, знание условий получения кристаллов различных типов плотной упаковки и
понимание важности их взаимодействий, которые, действуя сообща, «монтируют»
кристаллический объект.
*"» Desiraju C.R. Review of general principles // Comprehensive supramolecular
chemistry / Ed. J.L. Atwood, J.E.D. Davies, D.D. MacNicol and F. Vogtle. Oxford:
Pergamon, 1996. Vol. 6. P. 1-22.
6.1.2 Межмолекулярные взаимодействия
В области инженерии кристаллов, более, чем в любой другой области
супрамолекулярной химии, следует особенно внимательно отнестись к синергети-
ческой взаимосвязи межмолекулярных нековалентных взаимодействий. В
отсутствие какого-либо одного доминирующего взаимодействия (что, как правило, и
имеет место) анализ и предсказание структуры формирующегося кристалла являются
очень сложными задачами. В разд. 1.7 были обсуждены нековалентные
взаимодействия различных типов. Обычно большинство из этих сил имеют
электростатическую природу. Для инженерии кристаллов наиболее важны зависимость этих сил от
расстояния и их направленность. Силы, действующие на средних расстояниях, в
частности взаимодействия Ван-дер-Ваальса, определяют форму молекулы и вносят
наибольший вклад в общую устойчивость кристалла, а также являются движущей
силой процесса образования плотной упаковки кристалла. Силы, действующие на
больших расстояниях, например водородные связи, отличаются значительно
большей направленностью и даже могут приобретать характер ковалентных связей.
Ионные силы, действующие на очень больших расстояниях, например
взаимодействия между катионами металлов и гетероатомами, оказывают весьма
специфическое влияние на регулирование процесса образования кристаллической структуры.
6.1*3 Особая роль водородных связей
Водородные связи рассматривают как ключевые взаимодействия в
супрамолекулярной химии и это особенно справедливо по отношению к инженерии
кристаллов. Водородная связь характеризуется как высокой прочностью (до
120 кДжмоль), так и отчетливой направленностью. Дж. Джеффри (G. Jeffrey) из
Университета Питтсбурга (США) изучил время жизни различных водородных
связей и разбил их на три основные категории - сильные, средние и слабые — в
соответствии с энергиями, выделяющимися при данных взаимодействиях. Основные
характеристики водородных связей этих трех классов приведены в табл. 1.4. Такая
классификация в достаточной степени условна, поскольку, как мы увидим в
последующих разделах, на самом деле имеется некий «континуум водородных связей», в
рамках которых наиболее слабая «сильная» связь оказывается сравнимой по
энергии с наиболее сильной «средней» связью и т.д. Кроме того, более слабые и более
сильные взаимодействия часто действуют согласованно, и поэтому бывает довольно
6.1. Общие вопросы
495
трудно понять при исследовании общей структуры кристалла, какое именно
взаимодействие определяет ее. Это видно, в частности, при анализе структуры «6:1 -цик-
ламера» циклогександион-бензол, F.1N-С6Н6 (М. Etter et al., 1986), которая была
выбрана в качестве логотипа для нового журнала «Crystal Engineering
Communications» (Royal Society of Chemistry, Великобритания, 1999). Красивейшая
циклическая структура удерживается за счет очень коротких, «средних» по силе
водородных связей О-Н-"О=С с расстоянием R -О, равным 2,579 А, которые, по
всей видимости, и определяют структуру комплекса. Однако этот «цикламер»
(а конкретно, цикла-6-мер, поскольку он включает в себя шесть остатков)
образуется только в присутствии бензола. Молекула бензола принимает участие в
образовании шести протяженных водородных связей С—Н -О с длиной 3.00 А, при этом
С-Н-связи направлены непосредственно на сш/-неподеленную пару атома
кислорода кетона. При кристаллизации из тиофена, пиридина, спиртов или хлороформа
возникает линейная структура, которая не включает молекулы гостя. Это
указывает на то, что у кетона F.1) вероятность образования циклической или линейной
структуры практически одинакова. Тем не менее «слабые» взаимодействия с
молекулой бензола являются достаточно сильными, для того чтобы способствовать
образованию циклической структуры.
О"
F.1NС6Н6
Более ранние исследования структур, включающих водородные связи типа
X—Н--Х, при помощи РСА (см. Дополнение 2.1) показали, что положение
указанного атома водорода невозможно установить, так как этот метод исследования
определяет электронную плотность, которая на атомах водорода, естественно, очень
мала. Для установления положений атомов водорода используют метод дифракции
нейтронов на монокристалле, который крайне эффективен именно при решении
этой задачи (Дополнение 6.1). К сожалению, этот метод дорог и требует много вре-
6. Инженерия кристаллов
мени, поскольку единственным известным источником нейтронов служат ядерные
реакторы. Кроме того, метод детектирования нейтронов не очень чувствителен, что
является следствием отсутствия заряда у зондирующих частиц. Результат всего
перечисленного - почти полное отсутствие информации о положениях атомов
водорода в кристаллах. Это заставило кристаллографов ввести критерий оценки
расположения водородной связи, учитывающий расстояние XX между несвязанными
атомами. В соответствии с этим критерием расстояние между такими атомами
(которое может быть совершенно точно определено при помощи методик РСА), как
правило, должно быть меньше суммы ван-дер-ваальсовых радиусов двух атомов,
предположительно участвующих в образовании водородной связи. Ван-дер-вааль-
совы радиусы для элементов, представляющих интерес в контексте данной книги,
составляют l.lOA для водорода, 1.40А для кислорода, 1,50А для азота, 1.80А для
хлорид-иона, 1.95А для бромид-иона и 2.15А для иодид-иона. Вот почему следует
ожидать, что длины водородных связей типа N-H---X будут меньше, чем 2.50, 2.60,
2.90, 3.05 и 3.25 А для X = О, N, C1, Вг и I соответственно. В настоящее время после
появления современных детекторов рентгеновских лучей и криокристаллографии
(изучение дифракции при низких температурах, как правило, составляющих
100-250 К) стало возможным устанавливать локализацию атомов водорода и
определять их позиции с точностью до ± 0.02 А. Полученные таким образом
координаты атома водорода сильно зависят от деформационных колебаний Х-Н-связи
(рис. 6.2), при этом длина связи искусственно укорачивается за счет смещения
электронной плотности данного атома водорода по направлению к тому атому, с
которым он связан. Тем не менее если учесть все эти эффекты, то можно разработать
значительно более надежные критерии для характеризации водородных связей,
основанные на информации и о направленности вектора Х-Н, и о расстояниях Н---Х,
а также Х—Х. Результаты этих исследований показывают, что критерий,
основанный на оценке только расстояния XX, относительно нечувствителен к прочности
водородной связи и его нельзя использовать при анализе многоцентровых связей,
когда атом водорода взаимодействует с двумя или с большим числом акцепторов.
•""» Jeffrey G.A. An introduction to hydrogen bonding. Oxford: Oxford University Press,
1997.
Кажущееся усредненное
положение атома Н
Деформационное колебание связи Х-Н
Рис. 6.2. Деформаци-
Кажущаяся длина связи Х-Н QHHOe укорачивание
-► Истинная длина связи Х-Н ковалентных связей
6.1. Общие вопросы 497
Дополнение 6.1. Дифракция нейтронов от монокристалла
Рентгеновская дифракция и дифракция нейтронов от монокристаллов являются
эффективными методами изучения молекулярной и кристаллической структуры
твердых веществ; оба метода основаны на близких принципах. Действительно, для
обработки результатов экспериментов, полученных как с помощью нейтронов, так и
рентгеновских лучей, можно применить одинаковое программное обеспечение. В
отличие от экспериментов с рентгеновскими лучами, которые выполняют в
большинстве современных университетов и во многих заводских лабораториях, дифракцию
нейтронов используют в меньшей степени из-за ее дороговизны. Единственный
доступный источник нейтронов — ядерный реактор. Скорость быстрых нейтронов
замедляется таким образом, что их длина волны составляет ~1 А, что сопоставимо с
расстоянием между атомами в кристаллах. После коллимации исследователи получают
более или менее монохроматический нейтронный пучок, подходящий для
дифракционных экспериментов. К сожалению, интенсивность потока нейтронов очень низка
практически во всех источниках, за исключением лишь нескольких специальных
источников особого назначения, и поэтому для экспериментов по дифракции
нейтронов часто требуются довольно крупные кристаллические образцы A-5 мм; ср. с
0.02—0.5 мм для нужд современной CCD-рентгенографии) и длительное время сбора
информации. В связи с этими экспериментальными сложностями неудивительно, что
по дифракции нейтронов проводят лишь незначительное число экспериментов. Тем
не менее такая дифракция имеет одно очень важное преимущество перед
дифракцией рентгеновских лучей, поскольку рассеяние нейтронов происходит на атомных
ядрах и не зависит от электронной плотности, что имеет место при рентгеновских
исследованиях. Именно поэтому положения легких атомов в кристалле, в частности
атома водорода, можно легко установить (хотя в большинстве случаев дейтерий
заменяют на водород, для того чтобы уменьшить фоновое рассеяние — еще одно
экспериментальное усложнение). Таким образом, ясно, что при изучении водородного
связывания, когда точное расположение атомов водорода имеет первостепенное значение,
лишь эксперименты по дифракции нейтронов могут дать точную информацию о
положении каждого водородного атома. Более того, на координаты атомов,
определяемые в ходе эксперимента по дифракции нейтронов, не оказывает влияния участие
электронной плотности атома водорода в образовании ковалентных связей, что
сдвигает атомы водорода по направлению к тому атому, с которым данный атом водорода
связан, при этом укорачивается длина данной X—Н-связи. Как правило,
качественный эксперимент по дифракции нейтронов способен дать расстояния для связи X—Н
с точностью до ± 0.002 А, а углы — с точностью до ± 0.01°. Эти данные можно
сравнить с результатами, полученными в ходе низкотемпературных рентгеновских
измерений, обеспечивающих наиболее высокую точность. В последнем случае ошибки
определения этих параметров, по крайней мере, на порядок выше, чем при
использовании метода дифракции нейтронов, даже в отсутствие атомов, более
тяжелых, чем углерод или кислород.
498 6. Инженерия кристаллов
6.1.3.1 Сильные водородные связи
Сильные водородные связи для твердого состояния вещества, как
правило, реализуются в сильных кислотах, таких, как HF в ионе HF^, или с гидрдтиро-
ванным протоном (ион оксония), Н3О+, Н5О^, НуО^, или в общем виде Н+(Н2О)Л
(обычно п = 1 -s- 6, хотя в масс-спектрометрах в газовой фазе были обнаружены
кластеры с п > 20). Сильные водородные связи наблюдают также в комплексах
«протонной губки» F.2), в которых протон более или менее равномерно распределен
между двумя NMe2-rpynnaMH, находящимися в непосредственной близости друг от
друга. В этом случае присутствие протона фактически уменьшает невыгодные
взаимодействия неподеленных электронных пар двух атомов азота и прочность данной
водородной связи увеличивается за счет взаимодействий между неподеленной
парой атома азота и положительным зарядом «обнаженного» протона.
Me,, .-Н+^ .лМе
'N N^,
Me
Н+-комплекс "протонной губки"
ГУ
F.2)
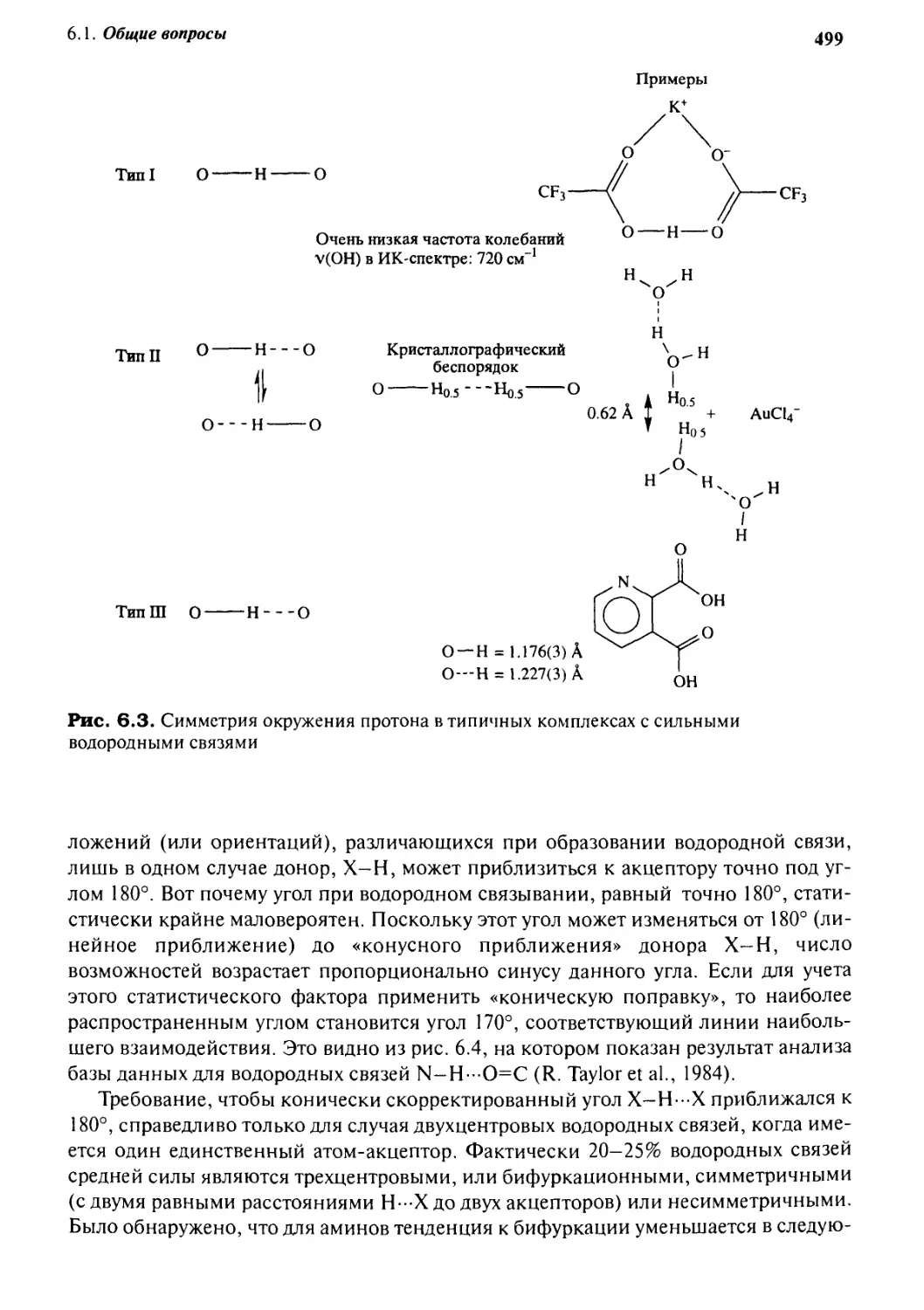
Сильная водородная связь характеризуется углом X - Н - X (где X = F, О, N),
близким к 180°, и коротким расстоянием Х-Х, что приводит к увеличению длины
ковалентной связи X — Н. В результате этого протон оказывается почти посередине
между двумя электроотрицательными атомами. Фактически известно лишь
несколько случаев, когда такой протон, действительно, равно удален от двух атомов-
акцепторов (рис. 6.3, тип I). Как правило, из-за кристаллографической
неупорядоченности вещества создается впечатление о равноудаленности протонов (тип II), а
в случае образцов с очень низкой симметрией положения протонов можно точно
определить (тип III). В целом, рассматриваемый протон находится на плоской
поверхности потенциальной энергии, причем на его точное расположение сильно
влияет общее поле кристалла.
6.1.3.2 Водородные связи средней силы
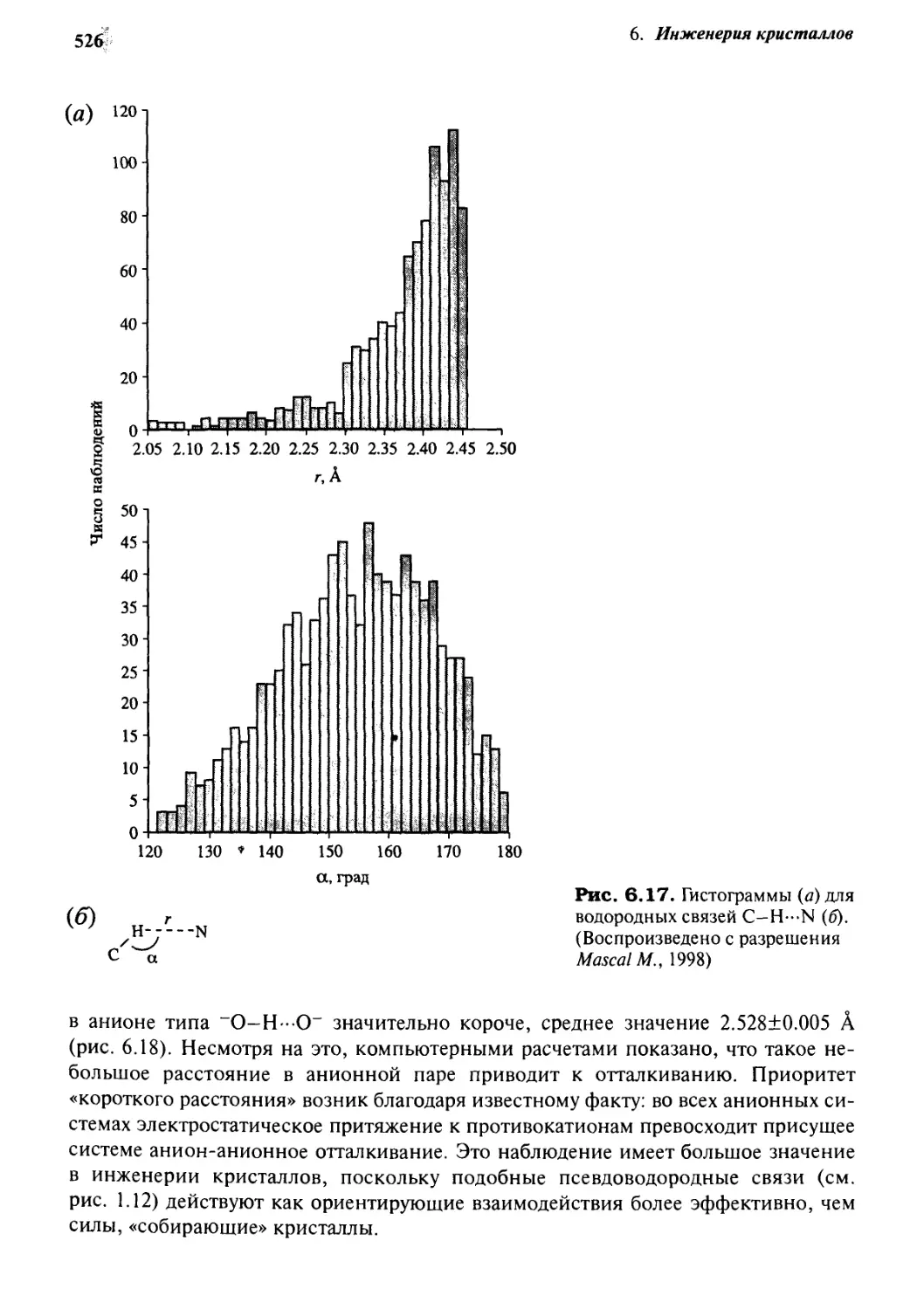
Водородные связи средней силы наиболее распространены и чаще
всего реализуются в случае атомов водорода, связанных с электроотрицательными
атомами (в частности, с атомами кислорода), причем в биологических системах они
встречаются повсеместно (например, стабилизируют третичную структуру белков).
Длины водородных связей могут различаться более чем на 0.5 А, а углы Х-Н--Х -
изменяться от 178 до 140°. Анализ данных по водородным связям, содержащихся в
Кембриджском банке структурных данных (разд. 6.3), показывает, что наиболее
распространенным является угол Х-Н-Х, равный -155°. Причина этого может
быть объяснена исходя из статистических выкладок. Так, из всех возможных распо-
6.1. Общие вопросы
499
Тип! О Н
О
Очень низкая частота колебаний
v(OH) в ИК-спектре: 720 см
О Н О
•j^r, тт О Н О Кристаллографический
беспорядок
О Но.5---Но5 О
О---Н О
ТипШ О Н О
0.62 А 1 °5 + АиСЦ-
Н(M
О —Н = 1.176C) А
О—Н = 1.227C) А
Рис. 6.3. Симметрия окружения протона в типичных комплексах с сильными
водородными связями
ложений (или ориентации), различающихся при образовании водородной связи,
лишь в одном случае донор, Х-Н, может приблизиться к акцептору точно под
углом 180°. Вот почему угол при водородном связывании, равный точно 180°,
статистически крайне маловероятен. Поскольку этот угол может изменяться от 180°
(линейное приближение) до «конусного приближения» донора X—Н, число
возможностей возрастает пропорционально синусу данного угла. Если для учета
этого статистического фактора применить «коническую поправку», то наиболее
распространенным углом становится угол 170°, соответствующий линии
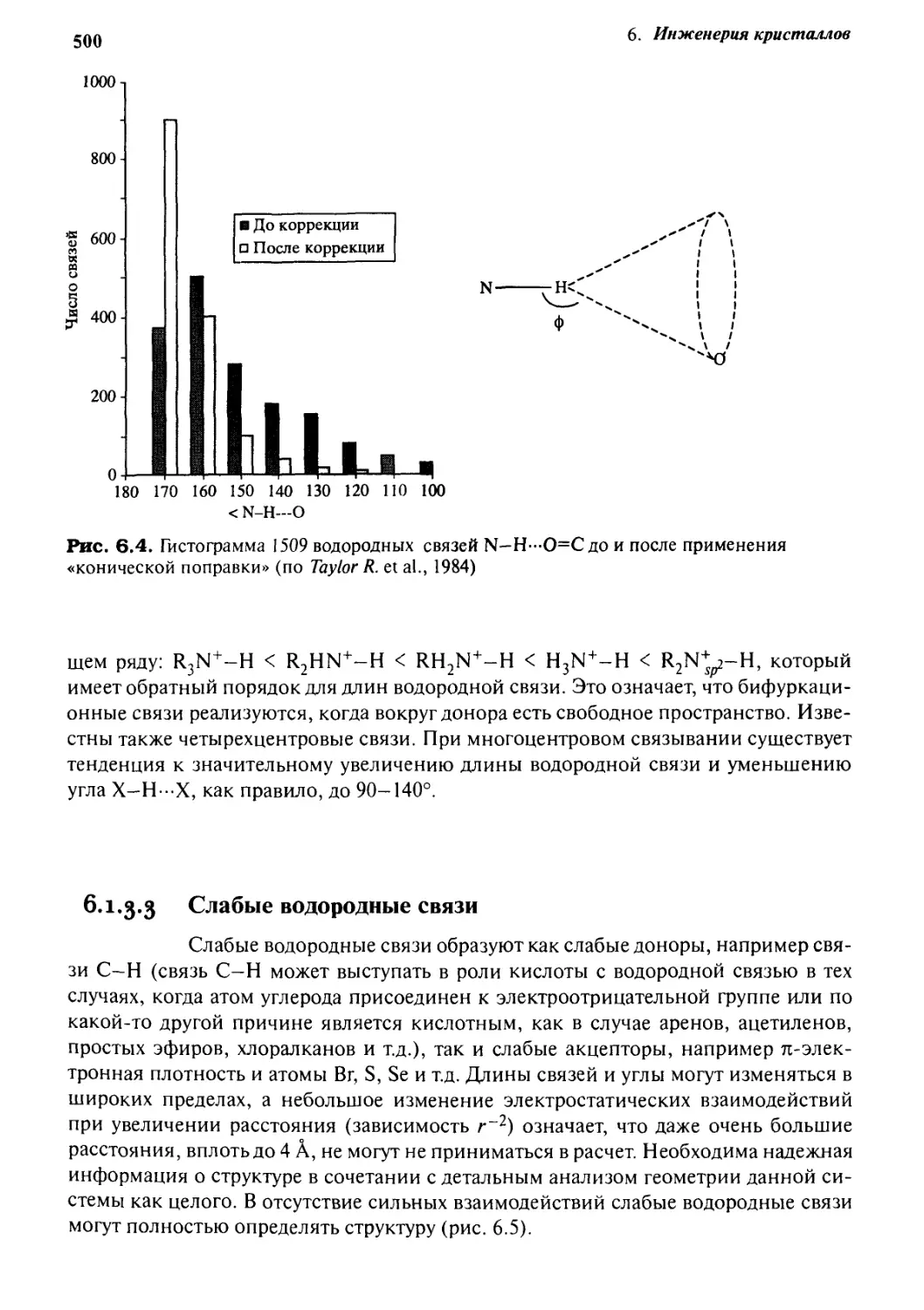
наибольшего взаимодействия. Это видно из рис. 6.4, на котором показан результат анализа
базы данных для водородных связей N-H-O=C (R. Taylor et al., 1984).
Требование, чтобы конически скорректированный угол X—Н- X приближался к
180°, справедливо только для случая двухцентровых водородных связей, когда
имеется один единственный атом-акцептор. Фактически 20-25% водородных связей
средней силы являются трехдентровыми, или бифуркационными, симметричными
(с двумя равными расстояниями Н-Х до двух акцепторов) или несимметричными.
Было обнаружено, что для аминов тенденция к бифуркации уменьшается в следую-
500
1000
800-
g 600-
6. Инженерия кристаллов
В До коррекции
Р После коррекции
N-
180 170 160 150 140 130 120 ПО 100
< N-H—О
Рис. 6.4. Гистограмма 1509 водородных связей N—Н—О=С до и после применения
«конической поправки» (по Taylor R. et al., 1984)
щем ряду: R3N+-H < R2HN+-H < RH2N+-H < H3N+-H < I^N^-H, который
имеет обратный порядок для длин водородной связи. Это означает, что
бифуркационные связи реализуются, когда вокруг донора есть свободное пространство.
Известны также четырехцентровые связи. При многоцентровом связывании существует
тенденция к значительному увеличению длины водородной связи и уменьшению
угла Х-Н-Х, как правило, до 90-140°.
6.1.3.3 Слабые водородные связи
Слабые водородные связи образуют как слабые доноры, например
связи С—Н (связь С—Н может выступать в роли кислоты с водородной связью в тех
случаях, когда атом углерода присоединен к электроотрицательной группе или по
какой-то другой причине является кислотным, как в случае аренов, ацетиленов,
простых эфиров, хлоралканов и т.д.), так и слабые акцепторы, например тг-элек-
тронная плотность и атомы Br, S, Se и т.д. Длины связей и углы могут изменяться в
широких пределах, а небольшое изменение электростатических взаимодействий
при увеличении расстояния (зависимость г~2) означает, что даже очень большие
расстояния, вплоть до 4 А, не могут не приниматься в расчет. Необходима надежная
информация о структуре в сочетании с детальным анализом геометрии данной
системы как целого. В отсутствие сильных взаимодействий слабые водородные связи
могут полностью определять структуру (рис. 6.5).
6.1. Общие вопросы
Рис. 6.5. Гексагональная сетчатая структура 1,3,5-трицианобензола (по Reddy D. et al.,
1993а)
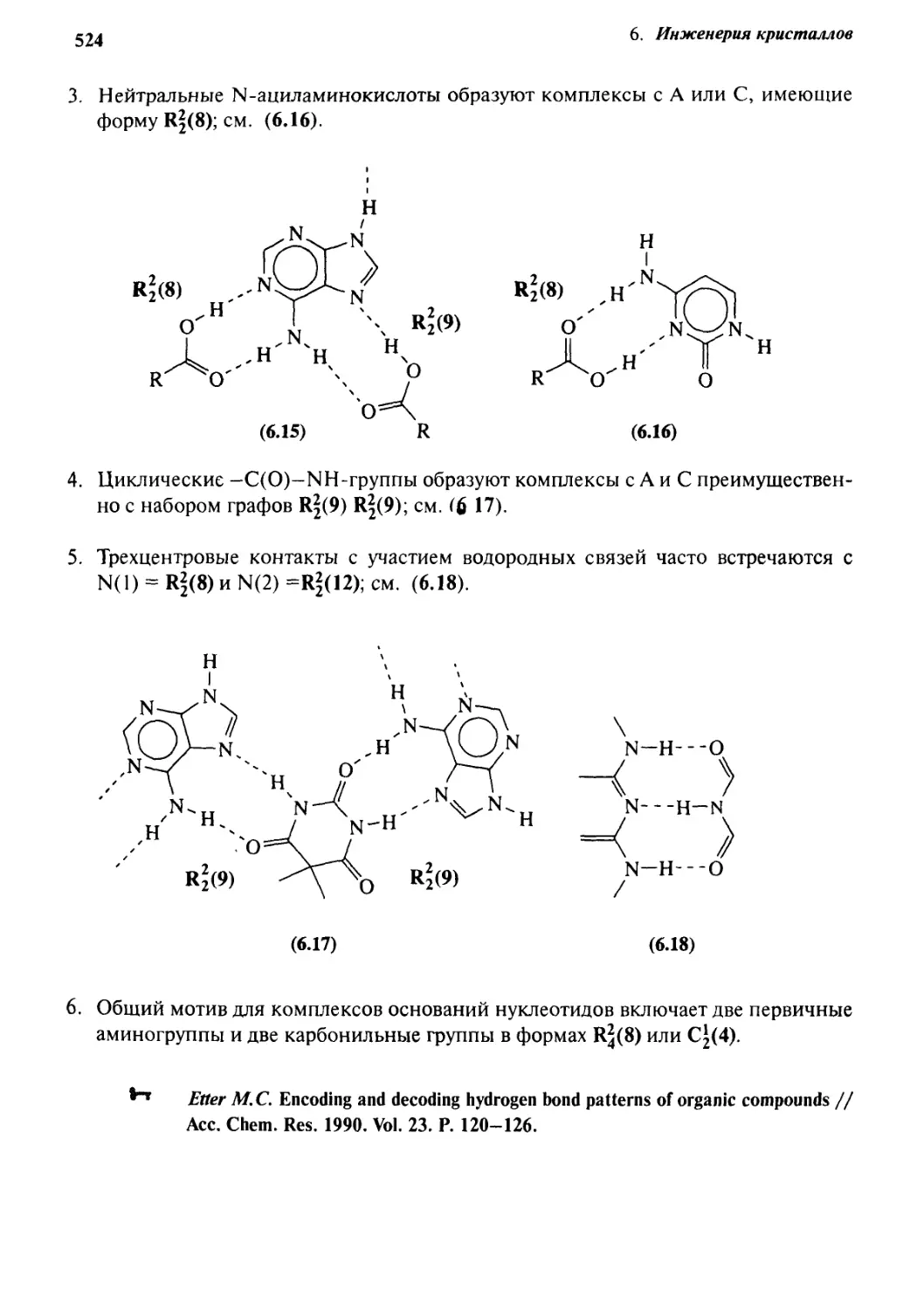
Анализ набора графов
Выше был рассмотрен вопрос о существовании индивидуальных
водородных связей и значении их взаимной ориентации, определяющей форму
молекулы в структуре твердого вещества. Поэтому представляется важной разработка
специального языка, при помощи которого мы могли бы описывать способ сборки
ансамблей из сеток водородных связей в кристаллы. В частности, этот язык может
быть полезен при идентификации кристаллических форм, топологий и способов
ассоциации родственных групп в весьма разнообразных, с точки зрения химии,
структурах.
Такая номенклатура — анализ набора графов — была применена к водородным
связям М. Эттер (М. Etter, Университет штата Миннесота, США) и развита
Дж. Бернстайном (J. Bernstein, Университет Бен-Гуриона, Израиль) и Р. Дэвисом
(R. Davis, Университет штата Техас, Остин, США).
При использовании анализа набора графов все кристаллические структуры,
содержащие водородные связи, можно свести к комбинациям четырех простых струк-
502
6. Инженерия кристаллов
тур, обозначенных следующим образом: С — цепи (от англ. «chains»), R - кольца
(от англ. «rings»), S — фрагменты с внутримолекулярными водородными связями
(от англ. «self»), D — другие фрагменты конечных размеров (от англ. «discrete»).
К этим основным обозначениям добавляют нижний индекс, указывающий на
число доноров водородной связи в данном фрагменте (d), а также верхний индекс,
указывающий на число акцепторов (а). Общее число атомов (я), включая Н, в
данном фрагменте называют степенью фрагмента и указывают в скобках после
обозначения фрагмента. Таким образом получают полное обозначение набора графов
(дескриптор) Gad(n), рис. 6.6.
Эту номенклатуру легко использовать в простых случаях, когда имеются
водородная связь лишь одного типа (единственный возможный по симметрии) и одна
разновидность фрагментов. Структуру, содержащую водородную связь только
одного типа, называют мотивом, и она будет иметь один или более дескрипторов
набора графов, с ней связанных. В более сложных случаях, когда в структуре
присутствует более одного мотива, можно приписывать дескрипторы набора графов
каждому мотиву по отдельности, считая, что все остальные мотивы отсутствуют.
Такие дескрипторы называют набором графов первого уровня и все вместе они
образуют единичный набор графов, обозначаемый символом Nj.
G - обозначение фрагмента
С - цепь
R - кольцо
S - сам (внутримолекулярно)
D - дискретный (межмолекулярный
фрагмент конечных размеров)
Н—N
СD)
а - число акцепторов водородной связи
п - число атомов во фрагменте
d - число доноров водородной связи
SF)
avid опускают, когда a = d-\
п опускают для тех случаев D, когда
D
имеется лишь одна водородная связь
Рис. 6.6. Примеры составления обозначений для наборов графов
6.1. Общие вопросы
503
Рис. 6.7. Отнесения набора графов для
бинарного уровня а-глицина. (Воспроизведено
с разрешения Bernstein J., Davis R.E., Shimoni L.
and Chang N.-L. Patterns in hydrogen bonding:
functionality and graph set analysis in crystals //
Angew. Chem., Int.Ed.Engl. 1995. Vol. 34.
P. 1555-1573)
Наиболее интересные случаи часто возникают при взаимодействии более чем
одного мотива. Рассмотрим, например, структуру аминокислоты а-глицина
(NH^CH2CO^~), у которой существуют две отдельные водородные связи, а н b
(рис. 6.7). В качестве добавления к двум наборам графов первого уровня,
описывающим а и b по отдельности, мы имеем также бинарный набор графов N2,
включающий как а, так и Ь. В случае глицина фактически существуют два варианта для
цепей, содержащих водородные связи а и />, а именно С2F), названный основным
бинарным набором графов, и С2A0), названный комплексным набором графов,
поскольку последний обладает более высоким значением п для тех же самых двух
водородных связей. Тем не менее ни тот, ни другой из этих двух наборов бинарных
графов не описывает наиболее очевидную характерную черту данной системы —
кольцо, которое обозначают как ^A6).
У системы, включающей три водородные связи {a, b и с), может существовать
три бинарных набора графов (наборы графов второго уровня), каждый из которых
содержит водородную связь более одного типа (последние могут даже представлять
связь одного и того же типа в двух разных по симметрии формах, при этом они
вовсе не должны быть химически различными): N2 (ab), N2(ac) и N2(bc). Мы будем
иметь также набор графов третьего уровня, N3 (a, b, с) и так далее.
Взаимосвязь между наборами графов первого и второго уровня может быть
проиллюстрирована примером, приведенным на рис. 6.8, на котором изображены две
совершенно различные водородные связи, образующие восьмичленный цикл.
Кажется весьма заманчивым немедленно записать дескриптор в виде R2(8) для этой
части структуры. Однако, если быть точными, он представляет собой набор графов
второго уровня, поскольку включает две различные водородные связи. Строго
говоря, нам следует вначале распознать два отдельных D-мотива (по одному для
водородной связи каждого типа), т.е. Nj = DD, а уже затем записывать N2 = ^(8).
Анализ наборов графов потенциально представляет собой мощный инструмент
инженерии кристаллов. Тем не менее его применение, вероятно, будет зависеть от
развития компьютерных программ, способных осуществлять автоматизированный
анализ и в более сложных случаях. Конечно, такое программное обеспечение
504
6. Инженерия кристаллов
Рис. 6.8. Набор графов первого
уровня для восьмичленного цикла — DD.
Дескриптор 1^(8) описывает набор графов
второго уровня
потребует от пользователей адекватного ввода информации и, в частности, ответа
на основной вопрос: о составляющих водородной связи.
*~* Bernstein /., Davis R.E., Shimoni L. and Chang N.-L. Patterns in hydrogen bonding:
Functionality and graph set analysis in crystals // Angew. Chem., Int. Ed. Engl. 1995.
Vol. 34. P. 1555-1573.
6.1.5 Рост кристаллов
Hulliger J. Chemistry and crystal growth // Angew. Chem., Int.Ed. Engl. 1994.
Vol. 33. P. 143-162.
6.1.5.1 Образование центров кристаллизации и рост кристаллов
При изучении структуры кристалла дифракционными методами
существует опасность увлечься анализом «межмолекулярных взаимодействий
притяжения» (диполь-дипольное взаимодействие, водородные связи, взаимодействия Ван-
дер-Ваальса и т.п., которые были подробно рассмотрены в разд. 6.1.2, 6.1.3) и
пренебречь фактическим механизмом формирования кристалла, т.е. тем
механизмом, в соответствии с которым упомянутые силы «собирают» кристалл. Детальное
рассмотрение явления роста кристалла выходит за рамки настоящей работы. Более
подробное обсуждение этого вопроса можно найти в ключевой ссылке к данному
разделу. Как известно, процесс осаждения кристаллов включает начальное кластер-
образование — слипание частиц растворенного вещества, находящихся в
пересыщенном растворе, в стабильные или неустойчивые агрегаты (образование центров
кристаллизации) с последующим ростом кристалла, как только данный ансамбль
достигнет критического размера. Схема этого процесса показана ниже.
1. Образование агрегатов из тектона А:
А +А = А2,
А2 + А = А3,
6.1. Общие вопросы :505
2. Образование или коалесценция кластеров:
3. Образование критического зародыша (центр кристаллизации) А* и его рост:
а;+а=ай+1,
4. Молекулярное наращивание на макроскопических поверхностях кристалла (Q.):
О. + А = &А.
До того как будет достигнут размер критического зародыша кристаллизации А*,
частицы докритического размера могут образовываться и вновь растворяться в
соответствии со статистическими флуктуациями тепловой энергии. Как только
критический размер сферического центра кристаллизации оказывается достигнут,
начинает происходить наращивание кристалла аналогично тому, как растет цепь в
радикальных цепных реакциях. Как показали эксперименты, основанные на
рассеянии света, растущие кристаллы окружены частично упорядоченным слоем
растворителя, толщина которого составляет несколько микрометров. Конечная структура
кристалла и его морфология (форма) зависят от термодинамики системы (включая
нековалентные межмолекулярные взаимодействия), ее кинетики, а также от
эффектов переноса массы и тепла. Свободная энергия, требующаяся для образования
критического зародыша кристаллизации, ДG*, в условиях того или иного процесса
кристаллизации может быть как положительной, так и отрицательной; при этом
она зачастую может быть понижена за счет соприкосновения кристаллизующихся
частиц со стенками реакционного сосуда или за счет присутствия примесей даже в
очень незначительных концентрациях. Небольшие количества примесей могут
также сдерживать рост кристаллов на поверхности одной или нескольких граней. На
этом явлении основан принцип действия кинетических ингибиторов роста,
используемых при добыче природного газа для предотвращения образования газовых
гидратов (см. Дополнение 5.1).
6.1.5*2 Рост кристаллов на поверхности раздела воздух—жидкость
В работе Лахава (Lahav) из Вейцмановского научного института
(Израиль) предпринята попытка исследовать в растворе структуру ансамблей, которые
могут выступать вроли зародышей для последующей кристаллизации. В частности,
была изучена кристаллизация ахиральной аминокислоты - а-глицина F.3).
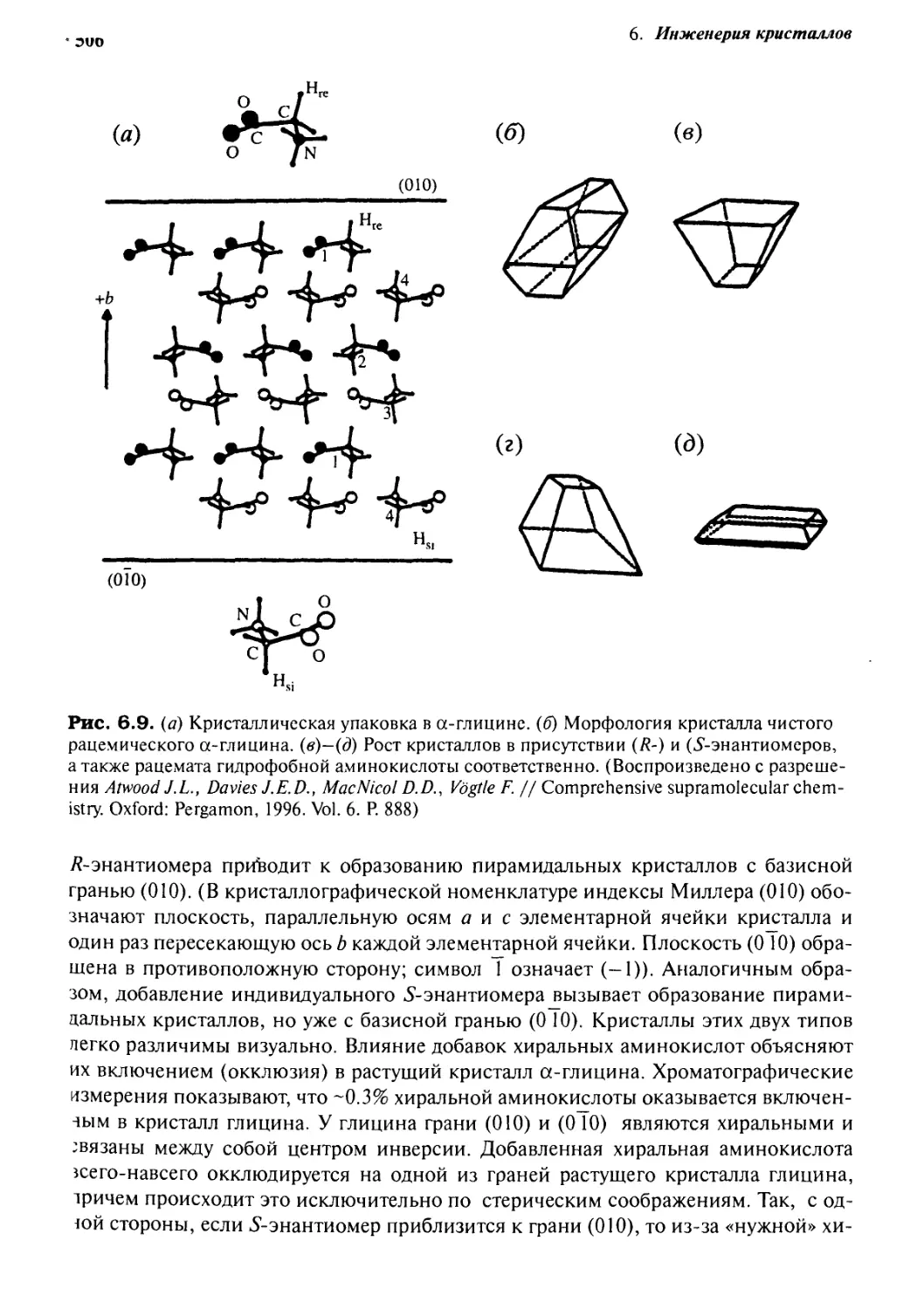
Структура чистого глицина, показанная на рис. 6.9, а, представляет собой центросимме-
тричные слои, обладающие центром инверсии (благодаря центрам инверсии
существует R- и S-хиральность хиральных образцов). Чистый а-глицин кристаллизуется
в бипирамидальной форме, показанной на рис. 6.9, б. Интересно, что добавление
разделенных хиральных аминокислот вызывает четкие изменения в морфологии
кристаллов (рис. 6.9, в - 6.9, д). Так, например, добавление индивидуального
6. Инженерия кристаллов
О
(а)
(б)
(в)
@10)
+b
(г)
(д)
@10)
Рис. 6.9. (а) Кристаллическая упаковка в а-глицине. (б) Морфология кристалла чистого
рацемического а-глицина. (в)—(д) Рост кристаллов в присутствии (R-) и (^-энантиомеров,
а также рацемата гидрофобной аминокислоты соответственно. (Воспроизведено с
разрешения Atwood J.L., Davies J.E.D., MacNicol D.D., Vdgtle F. // Comprehensive supramolecular
chemistry. Oxford: Pergamon, 1996. Vol. 6. P.
/?-энантиомера приводит к образованию пирамидальных кристаллов с базисной
гранью @10). (В кристаллографической номенклатуре индексы Миллера @10)
обозначают плоскость, параллельную осям awe элементарной ячейки кристалла и
один раз пересекающую ось Ь каждой элементарной ячейки. Плоскость @ 10)
обращена в противоположную сторону; символ 1 означает (-1)). Аналогичным
образом, добавление индивидуального S-энантиомера вызывает образование
пирамидальных кристаллов, но уже с базисной гранью @ 10). Кристаллы этих двух типов
пегко различимы визуально. Влияние добавок хиральных аминокислот объясняют
их включением (окклюзия) в растущий кристалл а-глицина. Хроматографические
измерения показывают, что -0.3% хиральной аминокислоты оказывается
включенным в кристалл глицина. У глицина грани @10) и @ 10) являются хиральными и
связаны между собой центром инверсии. Добавленная хиральная аминокислота
зеего-навсего окклюдируется на одной из граней растущего кристалла глицина,
тричем происходит это исключительно по стерическим соображениям. Так, с од-
юй стороны, если S-энантиомер приблизится к грани @10), то из-за «нужной» хи-
6.1. Общие вопросы 507
ральности этой аминокислоты ее боковая цепь с «неправильной» ориентацией
должна вызвать непреодолимое стерическое отталкивание от поверхности
кристалла глицина. С другой стороны, ^-энантиомер - желанный гость для грани @10). На
самом деле, окклюзия того или иного стереоизомера аминокислоты замедляет рост
кристалла на окклюдированной грани и, следовательно, влияет на его морфологию.
Если же кристаллы глицина выращивают в присутствии неразделенных смесей R-
и S-энантиомеров аминокислот, то замедляется рост кристалла на обеих
упомянутых выше гранях, следствием чего является образование пластинчатых кристаллов.
NH2 N
Глицин (Gly) Ч ' 2
v *
--<.-- Y^q H R - пальмитоил
F.3) МЕ-Пальмитоиллизин F.4)
Если же добавляемые хиральные аминокислоты содержат гидрофобный
заместитель (например, ГЧ8-пальмитоил -(/?,5)-лизин F.4)), то ориентация и морфология
кристаллов могут стать управляемыми. Гидрофобные цепи Ы8-пальмитоиллизина
«высовываются» из водной фазы, образуя монослой на поверхности раздела
воздух-вода. Кристаллы а-глицина легче воды и поэтому будут накапливаться и расти
именно на ее поверхности. В присутствии индивидуального Л-энантиомера
Ы8-пальмитоил-(/?)-лизина грань @10) растущих кристаллов а-глицина обращена
вверх, тогда как S-энантиомер вызывает рост кристаллов а-глицина с экспозицией
грани @ 10) вверх, к монослою. Образование монослоя из рацемической смеси
обоих энантиомеров «темплатной» аминокислоты приводит к получению случайной
смеси индивидуальных кристаллов а-глицина с преимущественной ориентацией
вверх, к монослою той или другой грани. Следует отметить, что кристаллы
а-глицина, выращенные в отсутствие темплатной матрицы, являются бипирами-
дальными. Исследование кристаллической упаковки твердых (/?,5)-лизина и
а-глицина показывает, что они очень похожи. Это позволяет предположить, что
рост кристаллов а-глицина обеспечивается зародышами кристаллизации -
фрагментами лизина. При этом хиральность лизина будет определять ориентацию
первого слоя в кристалле а-глицина. Обнаружение смешанных кристаллов в случае
добавления рацемата «темплатной» аминокислоты наводит на мысль о такой
организации монослоя Ме-пальмитоил-(/?,5)-лизина, при которой образуются
изолированные R- и S- домены (рис. 6.10). Более подробная информация о монослоях
будет представлена в разд. 10.2.
Интересная особенность этого явления состоит в том, что если одна из граней
растущего кристалла глицина в ходе процесса кристаллизации блокирована
(например, за счет флотации на поверхности раздела воздух-вода), то кристаллы
глицина будут окклюдировать только один энантиомер из рацематной смеси,
находящейся в растворе (именно тот энантиомер, который «соответствует»
неблокированной грани), тогда как второй энантиомер аминокислоты будет
оставаться в растворе, обогащая его. Фактически этот процесс представляет собой
508
6. Инженерия кристаллов
(Л)-монослой
С18 С,8 С18 С18 С18 С18 С|8
E)-монослой
С,8 С18 с18 С,8 С18 С18 С18 С18
(Л)-аминокислота
Рис. 6.10. Образование пирамидальных кристаллов а-глицина на монослоях
ЫЕ-пальмитоил-(/?,Л)-лизина. (Воспроизведено с разрешения Atwood J.L., DaviesJ.E.D.,
MacNicol D.D., Vogte F. // Comprehensive supramolecular chemistry. Oxford: Pergamon, 1996.
Vol. 6. P. 889)
частичное разделение двух хиральных аминокислот. Более того, этот эффект
усиливается, если хиральные аминокислоты, остающиеся в растворе, действуют, подобно
Ы8-пальмитоил-(/?,5)-лизину, как темплаты, управляющие ростом кристаллов
глицина с той или иной ориентацией граней. Если данный раствор обогащен,
например, управляющим /?-энантиомером, то последний будет инициировать рост
кристаллов по граням @10), обращенным вверх, а это будет сопровождаться окклюзией
более удаленного S-энантиомера на растущих гранях @10) в растворе. Благодаря
этому раствор станет еще более обогащенным именно /?-энантиомером. Этот
«механизм усиления» был использован для практического разделения хиральных
аминокислот. Можно также предположить, что именно этот механизм был реализован
для разделения хиральных аминокислот в предбиотических условиях и сыграл
важную роль на начальных стадиях возникновения жизни. Этот основополагающий
«эффект Адама» будет обсуждаться в следующем разделе.
*"* Wessbuch /., Addadi L, Berkovitch- Yellin Z. et al. Spontaneous generation and
amplification of optical activity in a-amino acids by enantioselective occlusion into cen-
trosymmetric crystals of glicine // Nature (London). 1985. Vol. 310. P. 161—164.
6.1.5.3 Индукция хиральности: эффект Адама
Явление нуклеации зародышей кристаллизации представляет
особенный интерес, поскольку, в отличие от большинства химических реакций, которые
являются результатом взаимодействия ~1020 индивидуальных молекул,
образование активных центров кристаллизации часто происходит в результате лишь не-
жольких актов взаимодействия, а иногда — лишь одного. Это означает, что, в отли-
^е от большинства разделов химии, данный раздел не подчиняется
:татистическим закономерностям, характерным для ансамблей большого размера.
Это обстоятельство особенно уместно иметь в виду применительно к концепции
6.1. Общие вопросы 509
хиральности в химии и в биохимии. Почему, например, подавляющее большинство
природных биологических молекул существует в виде только одного оптического
изомера? Организм человека легко поглощает лишь один из энантиомеров
глюкозы, однако состоятельные приверженцы низкокалорийных диет могут позволить
себе подслащивать пищу «инвертированными» сахарами противоположной
хиральности (по отношению к формам, встречающимся в природе), которые не
усваиваются организмом человека. Аналогично все аминокислоты, входящие в состав
белков высших организмов, относятся к L-ряду. Может ли это быть связано с
основополагающим «эффектом Адама», согласно которому вся земная жизнь
произошла благодаря единичному начальному хиральному акту взаимодействия или
благодаря единичной молекуле, которая явилась шаблоном для всей последующей
эволюции?
В предыдущем разделе мы уже рассматривали влияние хиральных темплатов на
рост рацемических кристаллов ос-глицина. В отсутствие таких темплатов
образование кристаллов рацемической смеси молекул, которые по отдельности являются
хиральными, как правило, приводит к центросимметричным структурам; в них
присутствуют оба энантиомера, связанных между собой центром инверсии (см.
Дополнение 6.2). Если хиральное соединение осторожно разделить на изомеры с
тем, чтобы присутствовал только один энантиомер, оно будет кристаллизоваться
именно в той хиральной кристаллической форме симметрии (нецентросимметрич-
ная пространственная группа), которая соответствует энантиомеру, находящемуся
в растворе. Такой кристалл не сможет содержать инверсионных элементов
симметрии, поскольку инверсионная симметрия автоматически обращает хиральность.
Имеются, однако, некоторые химические системы, из которых образуются хираль-
ные кристаллические структуры, даже несмотря на то, что образец в растворе не хи-
рален. Хиральность твердых тел - в большой степени результат способа, при
помощи которого молекулы были упакованы в кристалл, чем следствие присутствия в
молекулах индивидуальных хиральных центров. В подобных случаях хиральность
кристаллического зародыша будет определять хиральность макроскопического
кристалла. Поскольку образование зародышей кристалла не является
статистически предсказуемым, оно может приводить к неожиданным эффектам, например к
спонтанному разрешению, т.е. к образованию кристаллов единственной энантио-
мерной формы, несмотря на то, что образование либо D-, либо L-формы (право-
или левовращающей) равновероятно.
Это было с изяществом показано для кристаллов NaC103 Дж. Мак-Брайдом
(J. McBride) и Р. Картером (R. Carter) из Йельского университета (США). В
растворе NaC103 представляет собой ахиральное соединение, однако в твердом состоянии
он образует кристаллы, относящиеся к хиральной кубической пространственной
группе />2,3. Хиральность индивидуального кристалла может быть установлена по
его вращению плоскости поляризованного света (±3.6°-мм~! при длине волны
546 нм). В 1898 г. было установлено, что если подвергнуть кристаллизации
очищенный пересыщенный раствор NaC103, не оказывая на него никакого возмущающего
воздействия, то в образце, состоящем из 3137 кристаллов, полученных в 46
отдельных экспериментах по кристаллизации, 50.08% кристаллов обладали /)-хиральнос-
тью, а оставшиеся — I-хиральностью, т.е., как и ожидалось, ни одна из энантио-
ми
6. Инженерия кристаллов
Рис. 6.11. Видеоизображение кристаллизации, протекающей при перемешивании
раствора NaClO3 магнитной мешалкой, (а) Первый контакт между вращающейся палочкой
и растущим родительским кристаллом с А-хиральностью. {б) Дочерние кристаллы,
образовавшиеся в результате перемешивания в течение 1 мин (обратите внимание на то, что
верхушка родительского кристалла почти не пострадала после контакта с вращающейся
палочкой), (в) Увеличенные изображения дочерних призм. (Воспроизведено с разрешения
Me Bride J.M. and Carter R.L. Spontaneous resolution by stirred crystallisation // Angew. Chem.,
Int. Ed. Engl. 1991. Vol. 30. P. 293-295)
морфных форм не имеет предпочтения перед другой. Однако, отталкиваясь от
результатов Кондепуди (Kondepudi), Мак-Брайд и Картер осуществили тот же самый
эксперимент в перемешиваемом растворе. В такой системе образование центров
кристаллизации достаточно маловероятно, поэтому они смогли получить образец,
в котором монокристалл (обладающий А-хиральностью) был выращен по
направлению, заданному палочкой магнитной мешалки. Эти исследователи сделали
видеозапись данного процесса и «поймали на камеру» точный момент, в который
растущий кристалл в первый раз «наткнулся» на вращающуюся палочку магнитной
мешалки. В течение 30 с после этого столкновения в растворе образовалось более
5000 дочерних кристаллов. Все эти кристаллы обладали L-хиральностью. Подтекст
этого ясен: при каждом столкновении с вращающейся палочкой в пересыщенный
раствор направляется поток кристаллитов, обладающих L-хиральностью, которые
действуют как вторичные центры кристаллизации в ходе роста дочерних
кристаллов, имеющих ту же хиральностью, что и родительский кристалл. Никаких
кристаллов с /)-хиральностью обнаружено не было. Видеоизображение приведено на
рис. 6.П. Это означает, что одно единственное столкновение с микроскопическим
зародышем кристаллизации, которое определяет хиральность родительского
кристалла, далее приводит к той же самой хиральности для целого образца, несмотря на
то, что обе формы могут образовываться с одинаковой вероятностью.
McBride J.M. and Carter R.L. Spontaneous resolution by stirred crystallisation //
Angew. Chem., Int. Ed. Engl. 1991. Vol. 30. P. 293-295.
6.1. Общие вопросы
511
Дополнение 6.2. Симметрия молекул и кристаллов
В молекулах имеется пять элементов симметрии: тождество, зеркальные плоскости,
оси собственного вращения (простые поворотные), оси несобственного вращения
(зеркально-поворотные) и инверсия. Полные данные об этих элементах симметрии и
о соответствующих им операторах можно найти в любом учебнике по химии. Эти
сведения приведены на рис. 6.12.
Симметрия всех этих типов характерна и для молекул в кристаллах, а, кроме
этого, подобная молекулярная симметрия может в рамках одного кристалла связывать
одну молекулу с другой. Например, зеркальная плоскость или центр инверсии будет
связывать один энантиомер с другим в кристалле, содержащем рацемическую смесь
двух хиральных молекул. Так называемых операторов точечной симметрии вполне
достаточно, чтобы описать симметрию единичной молекулы для любого
спектроскопического приложения. В совокупности они дают 36 точечных групп молекулярной
симметрии (в частности, вода принадлежит к точечной группе C2v).
В кристаллографии нам придется иметь дело с трансляционной симметрией, так
же как и с симметрией точечных групп, а это означает, что мы должны внести два
дополнительных оператора для трансляционной симметрии (рис. 6.13):
• винтовая ось (вращение + трансляция),
• плоскость скольжения (отражение + трансляция).
В конце концов было решено использовать более удобную для кристаллографии
специальную систему символов симметрии — систему Германа—Могена, отличную от
системы обозначений Шёнфлиса (табл. 6.1).
Зеркально-поворотная ось
второго порядка (S2) - вращение
с последующим зеркальным
отражением
Тождество (Е) - действия
fP отсутствуют, фигура остается
J; -** без изменений
Простая поворотная ось
второго порядка (С2)
Зеркальное отражение (о)
Инверсия (О
Рис. 6.12. Операции молекулярной симметрии
512
Ь. Инженерия кристаллов
Трансляция
Отражение перпендикулярно
направлению трансляции
Плоскость скольжения Винтовая ось второго порядка
Рис. 6.13. Операции трансляционной симметрии в кристаллах
Таблица 6.1. Обозначения симметрии по системам Шёнфлиса
и Германа—More на, используемым в спектроскопии и кристаллографии
соответственно
Операция
или элемент симметрии
Собственное вращение
Несобственное вращение
Отражение
Инвеосия
Спектроскопия
q
*4
*3
а
/
Кристаллография
2 (число раз до совпадения)
4
6
3
4
6
/и
Г
Винтовая ось
Плоскость скольжения
пт (вращение на 180° с последующей
трансляцией на т/п длины элементарной
ячейки параллельно оси вращения;
например, 2, — это поворот
на 180° + перенос на половину длины
ячейки)
о, Ь, с, п, d (отражение + трансляция на
половину длины ячейки в указанном
направлении; например, а — это
трансляция в направлении х)
о. i. \juiujuK ьиприсы
Сочетание операторов указанных выше пяти точечных групп симметрии с двумя
операторами трансляционной симметрии дает 230 различных возможных
комбинаций, называемых кристаллографическими пространственными группами. Каждый
кристалл принадлежит к одной из таких пространственных групп, так же как каждая
молекула принадлежит к точечной группе. Наиболее простая (и в наименьшей
степени симметричная) точечная группа (обладающая только оператором тождества) — это
группа Су Кристаллографической пространственной группой с наинизшей
симметрией является примитивная триклинная пространственная группа Р\. Большинство
элементарных ячеек кристаллов (основные строительные блоки любого кристалла)
содержит не одну, а несколько молекул, связанных между собой симметрией
пространственных групп. Уникальную, несимметричную, часть элементарной ячейки
называют асимметрической ячейкой. Применение симметрии пространственных групп к
асимметрической ячейке приводит к полной элементарной ячейке кристалла.
Повторение элементарной ячейки бесконечное число раз во всех трех направлениях (при
помощи трансляции) дает полную структуру кристалла.
Инженерия кристаллов была бы крайне сложной дисциплиной, если бы все 230
пространственных групп были одинаково распространены. Было бы очень трудно
предсказать, какая симметрия кристалла будет предпочтительной для данной
молекулы. К счастью, на практике всего две пространственные группы, Р2]/с и Р1, отвечают
за более чем 60% из 200 000 кристаллических структур, имеющихся в Кембриджском
банке структурных данных к моменту написания этой книги. Наряду с несколькими
другими группами они охватывают огромное число известных структур (табл. 6.2).
Следует отметить широкую распространенность винтовых осей второго порядка (ось
2j соответствует «ёлочному» способу упаковки, наблюдающейся, в частности, для
кристаллического бензола, см. рис. 1.17). Это энергетически весьма выгодный способ
упаковки молекул. Симметрия инверсии также широко распространена, причем
больше всего — среди металлоорганических соединений. И наконец, за исключением
вращения второго порядка (С^ или 2) в С2/с, другие элементы симметрии молекул,
как правило, отсутствуют. Это ясно указывает на то, что такие формы, связывающие
одну молекулу с другой, не приводят к эффективной кристаллической упаковке.
Таблица 6.2. Наиболее часто встречающиеся пространственные группы
Пространственная
группа
Операция
или элемент симметрии
(в добавление
к тождественности)
Относительная распространенность,
органические
соединения
металлоорганияеские
соединения
j
Р\
/■2,2,2,
Р2Х
С 2/с
РЬса
Рпа2]
РЬсп
Р\
Винт, скольжение 100
Инверсия 44.6
Винт 45.7
Винт 29.2
Вращение второго порядка, 16.0
скольжение
Инверсия, скольжение 12.2
Винт, скольжение 4.7
Инверсия, скольжение 2.2
Отсутствует 4.0
100
59.1
12.0
6.8
22.1
9.9
4.1
2.7
1.7
З-СтидДж. В.
514
6. Инженерия кристаллов
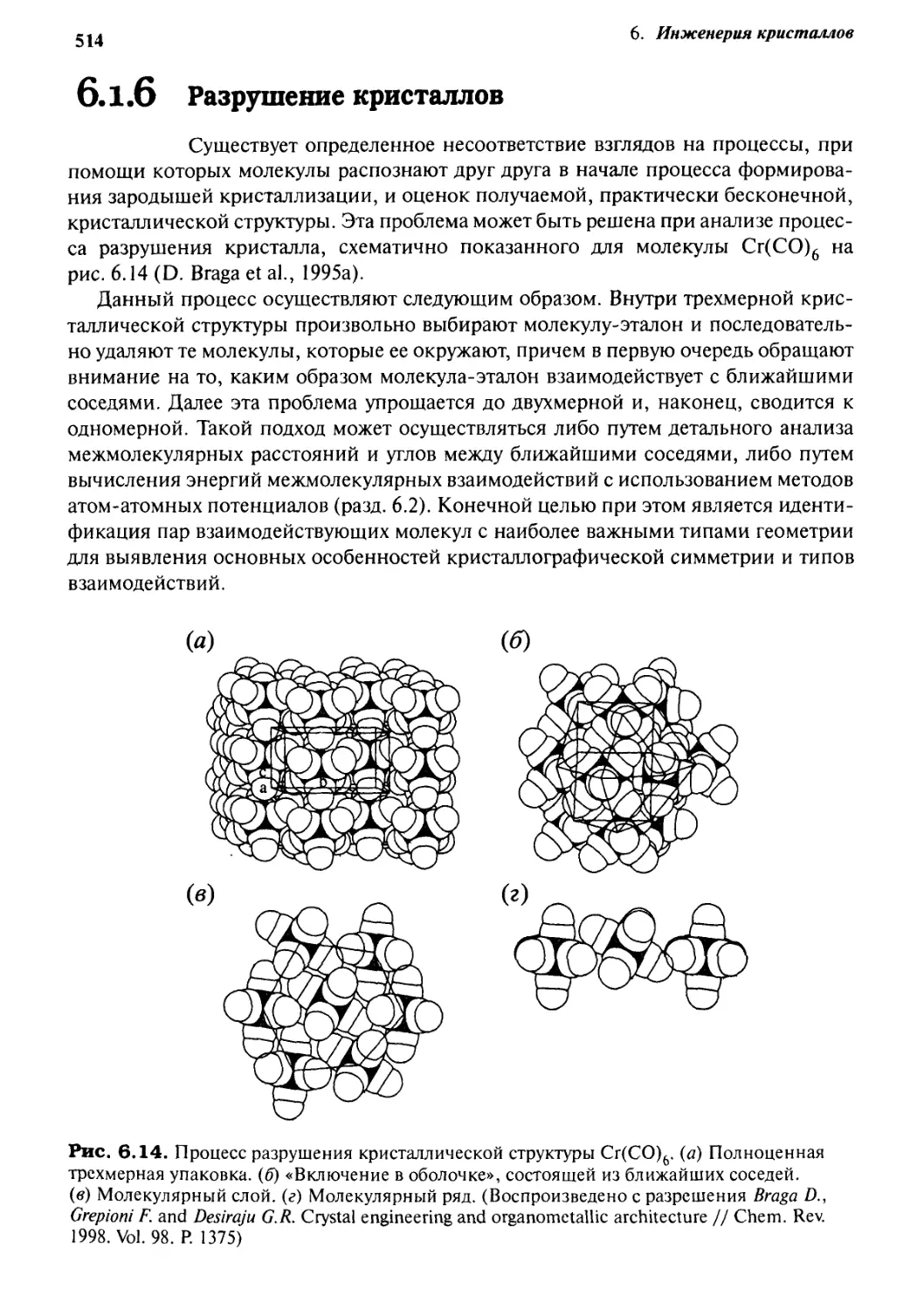
6.1.6 Разрушение кристаллов
Существует определенное несоответствие взглядов на процессы, при
помощи которых молекулы распознают друг друга в начале процесса
формирования зародышей кристаллизации, и оценок получаемой, практически бесконечной,
кристаллической структуры. Эта проблема может быть решена при анализе
процесса разрушения кристалла, схематично показанного для молекулы Сг(СОN на
рис. 6.14 (D. Braga et al., 1995a).
Данный процесс осуществляют следующим образом. Внутри трехмерной
кристаллической структуры произвольно выбирают молекулу-эталон и
последовательно удаляют те молекулы, которые ее окружают, причем в первую очередь обращают
внимание на то, каким образом молекула-эталон взаимодействует с ближайшими
соседями. Далее эта проблема упрощается до двухмерной и, наконец, сводится к
одномерной. Такой подход может осуществляться либо путем детального анализа
межмолекулярных расстояний и углов между ближайшими соседями, либо путем
вычисления энергий межмолекулярных взаимодействий с использованием методов
атом-атомных потенциалов (разд. 6.2). Конечной целью при этом является
идентификация пар взаимодействующих молекул с наиболее важными типами геометрии
для выявления основных особенностей кристаллографической симметрии и типов
взаимодействий.
Рис. 6.14. Процесс разрушения кристаллической структуры Сг(СОN. (а) Полноценная
трехмерная упаковка, (б) «Включение в оболочке», состоящей из ближайших соседей.
(в) Молекулярный слой, (г) Молекулярный ряд. (Воспроизведено с разрешения Braga D.,
Grepioni F. and Desiraju G.R. Crystal engineering and organometallic architecture // Chem. Rev.
1998. Vol. 98. P. 1375)
6.1. Общие вопросы Ц§15
•""* Braga D., Grepioni F., Desiraju G.R. Crystal engineering and organometallic
architecture // Chem. Rev. 1998. Vol. 98. P. 1375.
6.I.7 Стратегии дизайна при инженерии кристаллов
Дизайн при инженерии кристаллов в значительной степени основан
на предвидении. Он включает информацию об оптимальных межмолекулярных
расстояниях в системе, наряду с молекулярными формой, топологией и
электронными свойствами, для того чтобы выбрать и подготовить один или более
компонентов, способных в твердом состоянии спонтанно образовать желаемую
кристаллическую структуру. Далее эти компоненты смешивают в нужном соотношении в
подходящем растворителе и подвергают кристаллизации с помощью целого ряда
способов, начиная от медленного испарения или медленного охлаждения и
заканчивая более сложными способами, такими, как конвекция, диффузия жидкости в
жидкости или диффузия пара. Участие растворенной фазы крайне необходимо,
поскольку хотя смеси твердых компонентов в отсутствие растворителя все же могут
спонтанно сформировать желаемую фазу в результате «притирки», кинетически эти
процессы протекают чрезвычайно медленно и полученные продукты, как правило,
непригодны для дифракционного анализа монокристалла.
Дизайн кристаллов такого типа чаще приводит к поражению, чем к успеху, а
сами предсказания структуры кристаллов находятся пока на ранних стадиях своего
развития (разд. 6.2). В целом подавляющее большинство известных к настоящему
времени структур хозяев было открыто случайно, а не в результате дизайнерских
изысканий. Тем не менее, поскольку наши познания относительно многих важных
факторов, управляющих рассматриваемыми процессами, все же растут, то можно
надеяться, что со временем, опираясь на более мощные методы расчетов и
моделирования кристаллов, мы придем к более значительным успехам.
Рассмотрим кристалл, составленный из двух отдельных молекулярных
компонентов (супрамолекулярный тектон). На одном конце спектра взаимодействий его
можно рассматривать как комплекс хозяин—гость, или клатрат, в котором
кристаллическая сетка, образованная одним компонентом, служит хозяином по
отношению к другому компоненту. Для такого комплекса необходимо, чтобы хозяин
образовывал практически инвариантную сетку, не зависящую от природы гостя. Это
означает, что хозяин должен уметь «приспосабливаться» к самым разнообразным
гостям. Целый ряд веществ, удовлетворяющих этому требованию, был рассмотрен
в разд. 5.2 (например, тримезиновая кислота, мочевина, спиральные тубуланды,
CTV и т.п.). Дизайн подобных веществ значительно облегчается, если
молекулярная структура хозяина может быть изменена без нарушения строения основного
мотива кристаллической упаковки, что делает возможным «настройку» структуры.
Подобная манипуляция не осуществима, например, для мочевины (см. разд. 5.2.1),
тогда как структуры алициклических диолов семейства спиральных тубуландов (см.
разд. 5.2.2) могут быть модифицированы. Простые стратегии дизайна, которые
использовали для сеток хозяев, включают выбор частично несимметричных, или
изогнутых, молекул, обладающих «неправильными» формами, которые не могут
обеспечить плотную упаковку и не способны включать в себя молекулы второго
gj£ 6. Инженерия кристаллов
компонента — гостя, даже если последние подходят по размеру (разд. 6.11.1).
Однако во многих подобных системах не просматривается эта очевидная связь между
молекулярной структурой и структурой кристалла.
На другом конце этого спектра двухкомпонентных взаимодействий находятся
тектоны, обладающие комплементарными формами или функциональностью
(например, образующие водородные связи кислота и основание). Такие тектоны,
будучи смешаны, кристаллизуются совместно, а не образуют отдельные кристаллы
каждого компонента. В подобных случаях различие между хозяином и гостем
становится весьма условным, а сама терминология хозяин—гость теряет смысл.
Стратегии дизайна кристаллов могут основываться на электронной природе
заместителей (донор и акцептор, кислота и основание) или на их пространственном
расположении в пределах молекулы.
F.5)
Идея «неправильной» формы молекулы хозяина и сильно взаимодействующих
функциональных групп — в качестве инструмента для дизайна кристаллов — была
развита Э. Вебером и сотрудниками (Е. Weber et al., 1988) в «координационно-кла-
тратном» подходе, согласно которому хозяин не только имеет «неправильную»
форму и, следовательно, «плохо» упаковывается, но при этом у него еще есть отдельный
центр для связывания гостя (например, водородными связями), рис. 6.15.
Характерный пример - 9,9'-спирофлуорен-2,2'-дикарбоновая кислота F.5), которая
взаимодействует с частицами-гостями за счет образования водородных связей с кар-
боксигруппами СООН, но не может быть закристаллизована в качестве
индивидуального вещества из-за своей «неправильной», скрученной формы.
«Неправильная» форма хозяина
Рис. 6.15. «Координационно-клатратный»
Центр связывания гостя подход Вебера
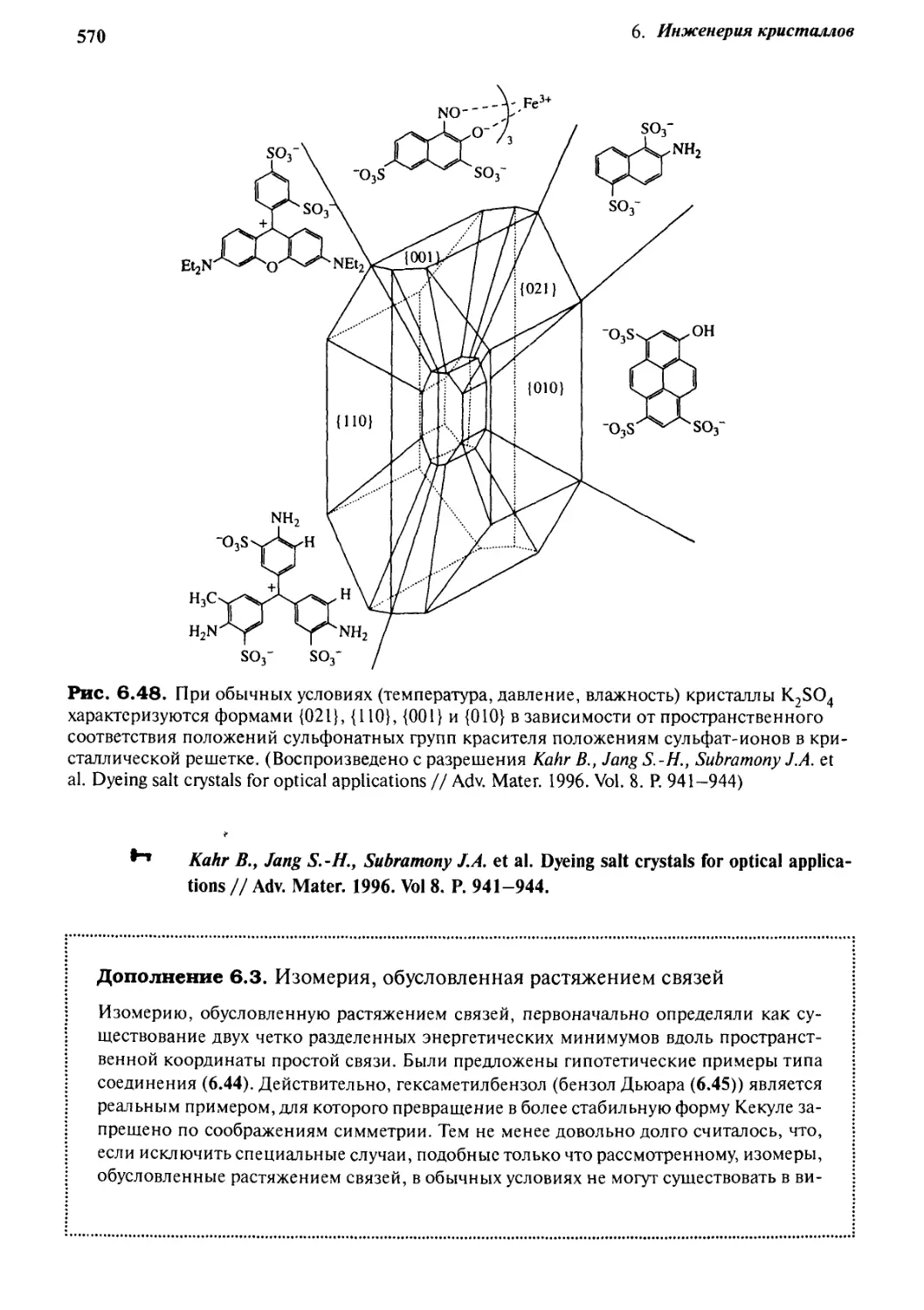
6.2. Предсказание структуры кристаллов
sit
На основании геометрии кристаллов клатратов фенола и гидрохинона (ось
шестого порядка) Д. Мак-Никол (MacNicol) предложил структуры шестичленных
хозяев. Упаковка кристалла гидрохинона сводится к образованию структуры с
расположением ароматических колец вверх-вниз (см. разд. 5.2.3); при этом остается
полость, достаточно большая, для того чтобы принять гостей, даже таких, как фул-
лерен С60. Путем замены шестичленного кольца с водородными связями F.6) на
подобное же кольцо, но с ковалентными связями, был получен целый ряд хозяев,
например F.7), молекулы которых в твердом состоянии объединяются таким
образом, что по своей способности к включению гостей напоминают своего
предшественника — гидрохинона.
F.7)
Другая очевидная стратегия дизайна заключается в подборе геометрически
совместимых доноров и акцепторов водородных связей. Эти пары могут быть либо
взаимно совместимыми, как, например, карбоновые кислоты, либо донорно-акцеп-
торными, как спирты и алкиламины. Особый интерес представляет дизайн
нецентросимметричных кристаллов для использования их как при хиральном
разделении (ср. с три-0-тимотидом, разд. 5.2.4), так и в нелинейной оптике (удвоение
или утроение частоты лазерного луча при прохождении его через кристаллы,
генерирующие вторую гармонику, разд. 8.5). Эта задача разрешается путем введения
асимметрического центра в молекулу хозяина. Остальные методы — это темплати-
рование хиральных решеток хиральными кристаллическими добавками или
внесение выделенных кристаллов одного из энантиомеров в качестве затравки.
6.2 Предсказание структуры кристаллов
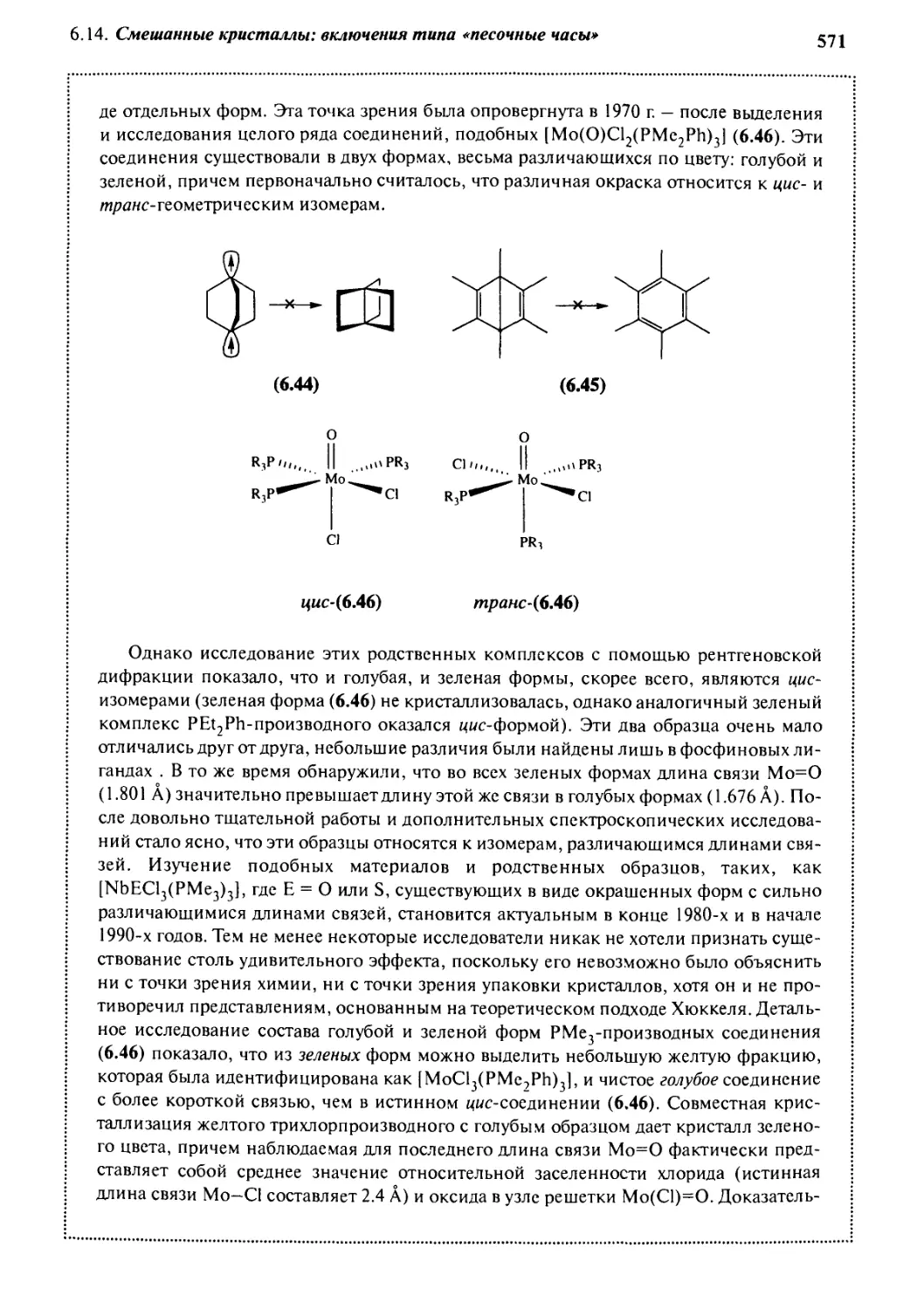
6.2.1 Расчетные методы
В ключевой ссылке к данному разделу А. Гавеццотти (A. Gavezzotti,
1994) из Миланского университета (Италия) на свой собственный вопрос: «Можно
ли предсказывать кристаллические структуры!» честно отвечает: «Нет». Эта задача
gjg 6. Инженерия кристаллов
еще более усложняется из-за распространенности полиморфизма - способности
твердых веществ существовать в более чем одной стабильной (или, по крайней
мере, продолжительно метастабильной) форме, часто с очень различающимися
физическими свойствами. Проблемой становится предсказание не только данной
кристаллической структуры, но и всех кристаллических структур, которые могут быть
стабильными в рассматриваемых условиях, с учетом как кинетических, так и
термодинамических эффектов. Кристаллические структуры, которые можно предсказать
и, следовательно, воспроизвести, пользуются большим спросом. Современный
подход к предсказанию кристаллической структуры включает сбор информации и
ее анализ, сопровождающийся все возрастающим числом организационных
мероприятий. В частности, Кембриджский банк структурных данных представляет
собой бесценный источник для систематизации и статистического анализа данных об
экспериментальных структурах, а также для сравнения их с предсказанными и
вычисленными.
Несмотря на тот факт, что предсказание кристаллических структур - сложная
задача, был предпринят целый ряд попыток, все более и более успешных, чтобы
«подобраться» к данной проблеме с расчетной стороны. Вычисление
кристаллической структуры часто оказывается полезным, в особенности в сочетании с
дополнительными данными, полученными из других (как правило, экспериментальных)
источников. В табл. 6.3 показаны дополняющие друг друга экспериментальные и
теоретические методы, которые можно использовать для предсказания
кристаллической структуры.
Одним из наиболее общих теоретических подходов является метчэд
атом-атомных потенциалов, изначально базирующийся на вычислении и минимизации
полной энергии решетки - параметра, который можно экспериментально проверить,
измеряя теплоту сублимации данного кристалла. Величины потенциалов
взаимодействия между парами атомов (например, Н---Н, С---Н, С—С, С---С1 и т.д.)
как для структур с водородными связями, так и без таких связей были получены
эмпирически. Эти расчетные данные хорошо согласуются с экспериментальными
теплотами сублимации. Такие параметры применяют при расчетах поверхностей
потенциальной энергии кристалла, для того чтобы найти термодинамический
минимум (энергия решетки). Отправной точкой в этих расчетах является структура с
беспорядочно ориентированными молекулами. Считают, что различия между
структурами, обусловленные энтропийными эффектами, пренебрежимо малы. В
ходе данного процесса получают и новые сведения о симметрии кристалла
(пространственная группа). Самый удобный подход — использование наиболее общих
элементов симметрии кристаллов (инверсия, винтовые оси, плоскости скольжения
и трансляция, см. Дополнение 6.2) для создания вероятных структур димеров или
небольших кластеров молекул (метод кластеров). Далее вычисляют энергии
взаимодействия этих кластеров друг с другом, суммируя потенциалы взаимодействия
каждой пары атомов, в принципе — до бесконечности, а на практике — до
определенного предела. Построение стабильных кластеров или агрегатов упрощается,
когда имеют место сильные доминирующие взаимодействия. Этот метод,
использованный Гавеццотти в его программе PROMET, применим и для органических
кристаллов. Метод был распространен на металлоорганические структуры
6.2. Предсказание структуры кристаллов
519
Таблица 6.3. Экспериментальные и теоретические методы предсказания
кристаллической структуры
(по GavezzottiA. Are crystal structures predictable? //Ace. Chem. Res. 1994. Vol. 27. P. 309-314)
Экспериментальные методы
Теоретические методы
Изучение зародышеобразования
Изучение морфологии
Систематический поиск полиморфных
структур
Стандартизованное изучение растворения
Рентгеноструктурный анализ
монокристалл (см. Дополнение 2.1)
порошок (метод Ритвелда)
Термохимия
DSC (см. Дополнение 5.2)
TGA (см. Дополнение 5.2)
измерение давления пара
Твердофазный ЯМР
Ультрамикроскопия
сканирующая электронная микроскопия
(SEM)
просвечивающая электронная
микроскопия (ТЕМ)
микроскопия атомных сил (AFM)
Использование данных Кембриджского
банка структурных данных
Расчет систем образцов
Эмпирические методы
Методы, основанные на определении
потенциалов кристалла
Методы, основанные на динамике решетки
Поиск потенциалов гиперповерхностей
Метод Монте-Карло
Метод молекулярной динамики
М. Шмидтом и Ю. Энглертом (М. Schmidt and U. Englert, 1996), подход которых в
виде схемы показан на рис. 6.16. Отталкиваясь от исходной молекулярной
структуры (которая может быть установлена по рентгеновским данным, расчетным или
спектроскопическим методом), а также от предположения о геометрии кристалла,
они применили понижающийся алгоритм, для того чтобы минимизировать обшую
энергию упаковки. Этот метод корректно воспроизводит структуру таких веществ,
как Ni(COL.
Методы подобного рода, основанные на атом-атомных потенциалах и
осуществляющие «просеивание» большого числа возможных структур, — это примеры
«статических» методов. Как правило, они довольно успешны, особенно если
подкрепляются дополнительными экспериментальными данными, полученными при
помощи методов, описанных в табл. 6.3. В альтернативном «динамическом»
подходе применяют метод Монте-Карло или метод молекулярной динамики. В этих
методах используют большое число беспорядочно ориентированных молекул, т.е.
статистический анализ проводят для достаточно крупного образца. Межмолекулярные
взаимодействия рассчитывают с применением эмпирических потенциалов
(аналогично подходу, использующему атом-атомные потенциалы). Всему ансамблю
молекул отводят время на его эволюцию — это предусматривает решение классических
уравнений движения для каждой молекулы — или выводят среднюю величину в
520
6. Инженерия кристаллов
Молекулярная
структура
Пространственная
группа
Беспорядочная
упаковка
Минимизация
энергии решетки
Повторная оценка
с повышенной точностью
Более высокая
симметрия
Рис. 6.16. Схема для
предсказания кристаллической структуры
металлоорганических соединений
с использованием метода атом-
атомных потенциалов
рамках большого конфигурационного пространства. Окончательный результат
такого подхода — предсказываемое упорядоченное равновесное состояние, которое
должно соответствовать данной кристаллической структуре. Динамические
методы лимитированы временем, в течение которого идут расчеты, и поэтому с ростом
числа молекул в образце ситуация с расчетным временем ухудшается
экспоненциально. В принципе, динамические методы значительно более информативны, чем
статические подходы, так как их можно использовать для исследования всего
фазового пространства системы как функции температуры, давления и т.п. Тем не
менее в настоящее время успех подобных исследований в органической химии
ограничивается бензолом и некоторыми другими простыми органическими
молекулами.
Gavezzotti A. Are crystal structures predictable? // Ace. Chem. Res. 1994. Vol. 27.
P. 309-314.
6.2. Предсказание структуры кристаллов $$1\.
6.2.2 Правила Эттер
В свете трудностей предсказания даже простых кристаллических
структур удивительно, что некоторые исследователи, работающие в области
инженерии кристаллов, отваживаются делать прогнозы относительно поведения
веществ в твердом состоянии. Однако в случае систем с водородными связями
достаточной силы, когда имеется доминирующее взаимодействие, которое определяет
кристаллическую упаковку небольших молекул правильной формы, вполне можно
попытаться предсказать ожидаемую кристаллическую структуру. Для этой цели
М. Эттер (М. Etter) из Университета штата Миннесота (США) предложила набор
правил, обеспечивающих предсказание структуры органических соединений с
водородными связями:
1. Все эффективные доноры и акцепторы протонов участвуют в водородном
связывании.
2. Внутримолекулярные водородные связи шестичленных циклов имеют
преимущество перед межмолекулярными водородными связями.
3. Наиболее активные доноры и акцепторы протонов, оставшиеся после
образования внутримолекулярных водородных связей, дают межмолекулярные
водородные связи друг с другом.
F.8)
Что касается правила 1, к эффективным донорам протонов относят протоны
карбоновых кислот, амидов, мочевин, анилинов, имидов, фенолов и т.д. Правило 2
базируется на тех же идеях, что и хелатный эффект в неорганической химии (см.
разд. 1.4), т.е. на выгодном энтропийном вкладе. Пяти- и семичленные циклы,
образованные внутримолекулярными водородными связями, встречаются достаточно
часто, однако не установлено, каким образом эти, потенциально более
напряженные, системы реагируют на образование межмолекулярных водородных связей.
Правило 3 можно проиллюстрировать на примере изучения продуктов совместной
кристаллизации 2-аминопиримидина и карбоновых кислот. В частности, в формуле
F.8) видно, как наиболее активные доноры протонов (кислотные ОН-группы)
попарно связываются с наиболее активными акцепторами протонов (атомы азота цикла).
В дополнение к этим основным правилам в рамках некоторых групп
органических соединений могут быть также сделаны обобщения, которые приводятся ниже.
522 6. Инженерия кристаллов
6.2.2.1 Нитроанилины
1. Протоны аминогрупп образуют водородные связи с нитрогруппами.
2. Возможно образование одной или более межмолекулярных водородных связей
типа «амино—нитро».
3. Агрегаты, возникшие благодаря межмолекулярным водородным связям с
участием мета- и ля/?я-заместителей, не будут иметь центр симметрии.
4. Взаимодействие «амино—нитро» обычно приводит к трехцентровой водородной
связи, R^D) F.9).
5. opwo-Замещенные первичные нитроанилины образуют, скорее, двухцентровые
межмолекулярные водородные связи с набором графов A^F) F.10), чем трех-
центров ые .
О / // \
—N Н—N —N N—
о х о---н
F.9) F.10)
6.2.2.2 Диарилмочевины
1. Атомы водорода NH-групп предпочитают занимать я«яш-положение по
отношению к карбонильной группе и образовывать трехцентровые связи с
С=О-группой мочевины, СD) [1^F)] F.11).
2. Продукты совместной кристаллизации образуются в виде [^(б)] F.12), если в
лшиа-положении ароматических колец имеются электронодефицитные
заместители (в формуле F.12) не показаны), подобные группам NO2, а также если у
молекул гостей имеются акцепторные группы, более сильные, чем участвующий
в межмолекулярном водородном связывании атом кислорода С=О-группы
мочевины.
F.12)
6.2. Предсказание структуры кристаллов 523
3. При образовании продуктов совместной кристаллизации протоны NH-rpynn
дают трехцентровые водородные связи с группами-акцепторами,
4. Нитрогруппы л^/яя-нитрозамещенных диарилмочевин обычно не используют в
качестве акцепторов при образовании водородных связей с атомами водорода
NH-групп мочевины, если при этом присутствуют молекулы-гости,
являющиеся эффективными акцепторами протонов.
6.2.2.3 Продукты совместной кристаллизации
карбоновых кислот с 2-аминопиримидином
1. В образовании водородных связей участвуют как протоны NH-rpynn, так и
атомы азота цикла.
2. Молекулы 2-аминопиримидина предпочитают образовывать водородные связи
с кислотами, а не между с собой.
3. 2-Аминопиримидин образует циклические формы 1^(8) как с кислотами, так и
сам с собой.
4. Два протона NH-групп не обязательно образуют водородные связи с
идентичными группами. То же самое справедливо и для обоих атомов азота цикла.
5. Для данных структур протоноакцепторная способность уменьшается в
следующем ряду: NA) > NC) > кислотный С=О > NB) или ND); см. F.13) и F.14).
Мономер™* LWJ LWJ °бдРоарГнГЙсвя„
F.13) F.14)
6. Способность выступать в качестве доноров протонов уменьшается в следующем
ряду: кислотный ОН > NHA) > NHC) > NHB).
6.2.2.4 Продукты совместной кристаллизации
оснований нуклеотидов
1. Аденин (А) и цитозин (С), в противоположность тимину и урацилу, образуют
продукты совместной кристаллизации со многими органическими
соединениями, имеющими кислотный характер.
2. Продукты совместной кристаллизации А и С с карбоновыми кислотами
существуют в формах с наборами графов 1^(8) или R^(9); см. F.15).
524
6. Инженерия кристаллов
3. Нейтральные N-ациламижжислоты образуют комплексы с А или С, имеющие
форму R|(8); см. F.16).
F.15)
О NV^
А ж' Y
R О О
Н
F.16)
4. Циклические -С(О)-ЫН-группы образуют комплексы с А и С
преимущественно с набором графов R^(9) R^(9); см. F 17).
5. Трехцентровые контакты с участием водородных связей часто встречаются с
NA) = R^(8) и NB) =R|A2); см. F.18).
4.N
\
N-H--0
N---H-N
//
N-H--0
F.17)
F.18)
6. Общий мотив для комплексов оснований нуклеотидов включает две первичные
аминогруппы и две карбонильные группы в формах R^(8) или
Etter М.С. Encoding and decoding hydrogen bond patterns of organic compounds //
Ace. Chem. Res. 1990. Vol. 23. P. 120-126.
6.3. Кембриджский банк структурных данных 525
Кембриджский банк
структурных данных
Кембриджский банк структурных данных (Cambridge Structural
Database, CSD) представляет собой источник для проведения сравнительного
анализа всех данных (рентгеноструктурных и нейтронографических) по структуре
«малых молекул» (т.е. не являющихся биологическими макромолекулами),
содержащих «органические атомы углерода» (органические, металлоорганические,
координационные соединения и т.п.). Эта база данных может быть дополнена за
счет Брукхейвенского банка данных по белкам (Brookhaven Protein Data Bank,
BPDB), Базы данных по структурам неорганических кристаллов (Inorganic Crystal
Structure Database, ICSD) и Картотеки кристаллографических данных по металлам
(Metals Crystallographic Data File, MDF), и вполне вероятно, что в скором будущем
эти источники станут доступны для широких кругов научной общественности.
Кембриджский банк — бесценный источник данных для тех исследователей,
которые берутся конструировать кристаллы. К 1999 г. эта база данных уже содержала
свыше 200 тысяч структур, а, по оценкам специалистов, к 2010 г. она будет включать
более 0,5 млн структур. Помимо легкости нахождения индивидуальных структур в
Кембриджском банке по сравнению с традиционным подходом к изучению
научной литературы, огромный размер выборки в этой базе данных означает, что может
быть выполнен статистический анализ межмолекулярных взаимодействий и
структурных мотивов. Осуществляя поиск и анализ данных, имеющихся в этой базе,
можно обнаружить новые научные факты. Эти факты могут сильно отличаться от
ожиданий исследователей, которыми они руководствовались, начиная актуальное
для них исследование структуры. Это вполне вероятно, поскольку подобный
кристаллографический эксперимент всегда дает в цифровом виде полную информацию
о молекулярной структуре, с которой можно дальше работать и которую можно
анализировать за рамками прежней интерпретации. Например, анализ
распределения положений атомов водорода относительно несвязывающих гетероатомов
позволяет выявить оптимальную геометрию водородных связей и определить влияние
заместителей. Анализ базы данных предпринял М. Маскал (М. Mascal, 1998),
чтобы выяснить тривиальный факт наличия водородной связи С—Н • N.
Статистический анализ расстояния H---N (г) иуглаС-H-N (а), рис. 6.17, выявил изобилие
взаимодействий такого рода с г < 2.45 А (сумма ван-дер-ваальсовых радиусов Н и N) и
120° < а < 180°. Без учета конической поправки (рис. 6.4) распределение угла а, как
и ожидалось, имело максимум -155-160°. Наблюдалось также большое число
низких значений г, вплоть до 2.05 А (ср. рис. 1.14).
Данные Кембриджского банка структурных данных были использованы и
Д. Брагой с соавторами (D. Braga et al., 1998), наряду с ab initio расчетами, для тоге
чтобы проверить справедливость взаимосвязи прочность—длина, согласно которой
короткие межмолекулярные контакты было принято считать сильными
межмолекулярными взаимодействиями. Анализ расстояний 0-0 в нейтральных образцах,
подобных карбоновым кислотам, с водородной связью О—Н---0 дает распределение
с максимумом ~2.652+0.001 А. В противоположность этому расстояния ~О-О"
5263
6. Инженерия кристаллов
2.05 2.10 2.15 2.20 2.25 2.30 2.35 2.40 2.45 2.50
г.А
эи ■
45-
40-
35-
зо-
25"
20-
15-
ю-
5-
о-
п
п
п г
ш
If
1
_
f
' Г
1
120 130 * 140 150 160 170 180
а,град
Рис. 6.17. Гистограммы (о) для
водородных связей С—H---N (б).
(Воспроизведено с разрешения
MascalM., 1998)
в анионе типа ~О-Н--О~ значительно короче, среднее значение 2.528+0.005 А
(рис. 6.18). Несмотря на это, компьютерными расчетами показано, что такое
небольшое расстояние в анионной паре приводит к отталкиванию. Приоритет
«короткого расстояния» возник благодаря известному факту: во всех анионных
системах электростатическое притяжение к противокатионам превосходит присущее
системе анион-анионное отталкивание. Это наблюдение имеет большое значение
в инженерии кристаллов, поскольку подобные псевдоводородные связи (см.
рис. 1.12) действуют как ориентирующие взаимодействия более эффективно, чем
силы, «собирающие» кристаллы.
6.3. Кембриджский банк структурных данных
527
Рис. 6.18. Гистограммы расстояний 0-0
в производных карбоновых кислот для
взаимодействий типа О-Н-0 и -О-Н--О-.
(Воспроизведено с разрешения Braga D. et al., 1998)
150 -
100 -
50 -
0 -
■
_ r^ljl
1
1
2.4
10 -
5 -
2.5
2.6 2.7 2.8
O-H-O, A
2.4 2.5 2.6 2.7 2.8
-o-h-o-, A
Еще на заре функционирования Кембриджского банка структурных данных
С. Найбург и К. Файерман ( S. Nyburg and С. Faerman, 1985) предостерегали других
исследователей по поводу межмолекулярных взаимодействий на небольших
расстояниях. Анализ несвязанных, в том числе и водородными связями, контактов
Х-Х (X = N, О, F, Cl, Br, I, S, Se) показал, что в то время как взаимодействия с
относительно неполяризуемыми атомами N, О и F давали умеренно полусферическое
распределение по расстояниям Х---Х (г) в зависимости от угла ц, для случаев, когда
\х{ = ц2, более крупные атомы - Cl, Br, I, S и Se - давали распределение более
эллиптической формы. Это явление известно под названием «полярное уплощение»
(рис. 6.19). Оно было интерпретировано с помощью несферических ван-дер-вааль-
совых радиусов, когда такой радиус рассматриваемого атома в направлении связи
С-Х ( т.е. при \х ~ 90°) является более коротким, чем радиус, который
перпендикулярен этой связи (т.е. при |Л = 0°).
•"» Allen F.H., Kennard О., Watson D.G. Crystallographic databases: Search and
retrieval of information from the Cambridge Structural Database // Structure
correlation / Ed. H.-B. Biirgi and J.D.Dunitz. Weinheim: VCH, 1994. Vol. 1. P. 71-109.
(а)
18 (б) /
4.00 3.00 2.00 1.00 0 4.00 3.00 2.00 1.00 0
X
\
с
5.00 4.00 3.00 2.00 1.00 0
г-.
5.00 4.00 3.00 2.00 1.00 0
18
4.00 3.00 2.00 1.00 0 4.00 3.00 2.00 1.00 О
Рис. 6.19. (а) Полярная диаграмма распределения расстояний Х---Х (г) для различных углов \хх при несвязанных контактах
(X = Н, О, Hal), (б) На осях отложено расстояние Х-Х (г), угол цх отсчитывается по часовой стрелке от вертикали.
(Воспроизведено с разрешения Nyburg S. and Faerman С, 1985)
I
•8
6.4. Инженерия кристаллов с алмазоподобными решетками
529
6.4 Инженерия кристаллов
с алмазоподобными решетками
Кристаллическая структура алмаза — метастабильной аллотропной
формы углерода — включает 5р3-гибридизованные атомы углерода,
последовательно связанные между собой, с образованием бесконечного «молекулярного
кристалла», в котором каждый атом связан с четырьмя другими атомами с помощью
единичных ковалентных связей. Структурная единица кристалла состоит из
строительных блоков (тектоны), напоминающих органическую молекулу адаманта-
на (рис. 6.20). Алмаз сам по себе — не супрамолекулярное соединение, поскольку
связи у него — ковалентные. Тем не менее если анализировать структуру алмаза с
позиций инженерии кристаллов, то атом углерода нужно описывать как
«самокомплементарный тектон с четырьмя тетраэдрически расположенными центрами
связывания». По-видимому, алмазоподобную кристаллическую структуру можно
получить и из любого другого — супрамолекулярного — тектона, который будет
удовлетворять данному критерию. Такими тектонами являются молекулы воды и
дигидрофосфата калия КН2РО4 (potassium dihydrogenphosphate, PDP). Молекула
воды имеет два донорных центра для водородной связи и два акцепторных, которые
вместе дают алмазоподобные (кубические) полиморфные структуры обычного
гексагонального льда (\h, см. разд. 5.1.1). Аналогично, Н2РО^ — анион PDP — образует
алмазоподобную сетку еще большего размера. Несмотря на то, что алмаз, вода и
Структуры углерода, воды и дигидрофосфата калия (PDP),
указывающие на их тетраэдрическую природу
7-
О
-p..
У
о
н
Четыре донорно- Два донорных и два акцепторных
акцепторных центра центра в каждом соединении
связывания
Структура алмаза с выделенным
адамантаноидным фрагментом
Кубический
лед
PDP
Пустое пространство,
заполненное за счет
взаимопроникновения
алмазоподобных сеток
Рис. 6.20. Алмазоподобные сетки и их строительные блоки (тектоны). Лёд, находящийся
при нормальных условиях (lh), имеет гексагональную структуру, однако известны две его
алмазоподобные полиморфные модификации
530
6. Инженерия кристаллов
PDP сходны топологически, относительные размеры образуемых ими сеток сильно
различаются. Алмаз имеет плотноупакованную сетку и предствляет собой одно из
наиболее твердых и высокоплавких веществ. Неэффективная упаковка
кубического льда обуславливает аномально низкую плотность льда по сравнению с
плотностью жидкой воды. Наконец, сетка PDP так велика, что более 50% пространства в ее
кристалле было бы пустым, если бы одна алмазоподобная сетка не проникала в
другую (т.е. одна сетка PDP заполняет пустоты в другой, обладающей такой же
структурой). Образующуюся сетку называют алмазоподобной структурой с двукратным
взаимопроникновением.
Алмазоподобные кристаллы представляют значительный интерес из-за наличия
в их структурах пустого пространства, что приводит либо к взаимопроникновению
алмазоподобных сеток, либо к включению гостей. Поэтому были предприняты
значительные усилия, чтобы предотвратить взаимопроникновение алмазоподобных
структур, мешающее использованию полостей. Интересным представляется и тот
факт, что такие структуры, построенные из строительных блоков, обладающих
почти тетраэдрической симметрией, испытывают недостаток центров инверсии и
поэтому кристаллизуются в хиральных пространственных группах. Эти, довольно
необычные, структуры (табл. 6.2) могут быть полезными при создании устройств, в
которых используются сегнетоэлектрические, пьезоэлектрические,
пироэлектрические и нелинейно-оптические эффекты. Действительно, PDP в основном
используют как нелинейный оптический материал в лазерах с перестраиваемой
частотой (разд. 8.5). Эти особенности в сочетании с прочностью алмазоподобного
каркаса привели к широкомасштабным исследованиям таких сеток.
При конструировании большинства алмазоподобных сеток очень важно
сохранять симметрию строительных блоков, близкую к Td. Это может быть достигнуто с
помощью единственного самокомплементарного строительного блока, подобного
молекуле адамантан-1,3,5,7-тетракарбоновой кислоты F.19), обладающего четырьмя
взаимно комплементарными карбоксильными группами, или при помощи
модульного подхода, при котором применяют комбинирование узловых точек Td (или, по
крайней мере, S4). Конкретным примером такого подхода служит использование
кубического кластера [Мп(СОK(ц3~ОН)]4 F.20) и линейного спейсера А—А (рис. 6.21).
со2н
(СОKМп
НО2С/„
"СО2Н
н
Мп(СОK
Mn(COK'
-Mn(
\
F.20)
:оK
н
\
со2н
НО2С/„
"СО2Н
СО2Н
F.21)
6.4. Инженерия кристаллов с алмазоподобными решетками
531
х-х
X
х
?
X
X - самокомплементарная
донорно-акцепторная функциональная
группа
А - акцептор
D - донор
Рис. 6.21. Структура алмазоподобных сеток, полученная путем самосборки
самокомплементарных строительных блоков S4 или модульной самосборки взаимно комплементарных
узлов S4 с линейными спейсерами
Самокомплементарную структуру F.19) можно рассматривать как результат
последовательной трехмерной эволюции топологических нуль-, одно- и двухмерных
точек, цепей и слоев, образованных бензойной, терефталевой и тримезиновой
кислотами (см. разд. 5.2.2). Это схематически показано на рис. 6.22. Адамантан-1,3,5,7-
тетракарбоновая кислота F.19) образует алмазоподобную сетку со столь большими
полостями, что упаковка может стать плотной только за счет
взаимопроникновения (сцепление) пяти полностью независимых алмазоподобных сеток
(пятикратное взаимопроникновение); при этом совсем не остается свободного места для
включения гостей. Степень взаимопроникновения может быть уменьшена путем
модифицирования алмазоподобного каркаса. В случае производного диона F.21)
имеет место лишь трехкратное взаимопроникновение, в результате чего
обеспечивается пространство для карбонильных групп, которые выступают наружу и
заполняют полости. Так создается пространство, достаточное для включения молекул-
гостей уксусной кислоты.
Поистине громадные алмазоподобные структуры получил М. Заворотко
(М. Zaworotko, 1994) из Университета Виннипега (Канада), использовавший
стабильный кубический кластер кубен F.20). Соединение F.20) - наглядный пример
супрамолекулярного тектона, который является жестким донором водородной
связи с симметрией S4, но, вместе с тем, может выступать и в качестве слабого
акцептора водородной связи (атомы кислорода лигандов СО - очень слабые основания).
В результате этоттектон взаимодействует с огромным числом акцепторов
водородной связи (даже с такими слабыми акцепторами, как бензол) с образованием
алмазоподобных сеток с дву-, трех- и четырехкратным взаимным проникновением
(табл. 6.4). Несмотря на то, что некоторые из этих водородных связей слабые, имен-
532
6. Инженерия кристаллов
Рис. 6.22. Схематическое
изображение нуль-, одно- и двухмерных
сеток, образующихся при димериза-
ции карбоновых кислот
но их направленность имеет значение для сборки всей системы. Многие
водородные связи принадлежат к традиционному типу O—H--N. Взаимодействия типа
О—Н--71 с участием производных бензола вынуждают протоны ОН-групп
приблизиться к трем из шести атомов углерода бензольного кольца, как бы в аллильное
положение.
Множественные водородные связи в тетраэдрических строительных блоках
использовали П. Брунет и соавторы (P. Brunet et al., 1997) для сборки замечательного
пористого имитатора цеолита, который получил название «органический цеолит»
из-за того, что вообще не содержит неорганических компонентов. Получение
этого порообразующего вещества основано на однокомпонентной самосборке тетра-
эдрического соединения F.22) в поперечно-сшитую сетку, состоящую из слоев,
содержащих полости. Каждая полость - почти квадратная по форме, со стороной,
равной 11.8 А, и может принять множество гостей, таких, как диоксан, ацетонитрил
и вода. Эта органическая структура, образованная нековалентными связями,
является настолько прочной, что способна сохраняться целой даже после удаления 66%
молекул-гостей при пониженном давлении. Образование пористой структуры из
соединения F.22) - исключение среди систем, получаемых при конструировании
кристаллов из тетраэдрических строительных блоков, когда формируются
взаимопроникающие алмазоподобные сетки без полостей. Детальное изучение мономер-
6.4. Инженерия кристаллов с алмазоподобными решетками
533
Таблица 6.4. Алмазоподобные структуры, полученные путем модульной
самосборки F.20) с акцепторами водородной связи
(Расстояние между кубическими блоками представляет собой расстояние от одной
молекулы F.20) до следующей и является длиной ребра алмазоподобного кристалла)
Акцептор А
в спейсере А-А
Бензол
Нафталин
Me2NCH2CH2NMe2
1,4-Диаминобензол
2-С1-Пиразин
Ph2P(O)CH2CH2P(O)Ph2
4,4'-Бипиридил
Расстояние между
кубическими блоками
9.74
10.40
11.35
11.79
11.82
13.53
15.22
* Растворитель MeCN включен в микроканалы.
Кристаллическая
система
Кубическая
Тетрагональная
Тетрагонал ьная
Тетрагональная
Кубическая
Тетрагональная
Тетрагональная
Взаимопроникновение
(кратность)
2
2
2
2*
3
2*
4*
ного компонента F.22) показало, что он не полностью самокомплементарен,
поскольку обладает избытком донорных центров водородного связывания (—NH2) по
сравнению с числом акцепторных центров (=N—). Это, возможно, и приводит к
образованию водородных связей между «лишними» протонами NH2-rpynn и
акцепторными центрами молекул-гостей, внедряющихся в эту структуру. Такой подход
представляет собой новую стратегию для получения подобных пористых материа-
N^/NH2
NH2
F.22)
H2N
534
6. Инженерия кристаллов
лов и позволяет осуществлять тонкую «настройку» структуры за счет реакции на
этих кислотных центрах, вовлекаемых в образование водородных связей.
Брунет и другие получили также похожий на соединение F.22) тетраэдрический
«тектон» — самособирающийся компонент F.23), из которого тоже можно
построить алмазоподобный полимерный каркас. В этом случае в твердом состоянии
образуется сетка с семикратным взаимопроникновением, в которой одна алмазоподоб-
ная решетка заполняет свои многочисленные крупные полости с помощью таких
же сеток, которые оказались рядом. Возможно, что в данном примере
взаимопроникновение — это результат самокомплементарной природы хозяина, который
содержит равное число донорных и акцепторных центров водородных связей.
Однако даже в этом случае существуют небольшие полости, заполняющиеся двумя
молекулами масляной кислоты на одно элементарное звено хозяина. С
образованием каркасных материалов этого типа открываются совершенно новые возможности
для получения специальных пористых материалов с очень большими полостями,
хотя, конечно, маловероятно, чтобы чисто органические каркасы смогли хотя бы
приблизиться к традиционным алюмосиликатным материалам по абсолютной
механической прочности.
О
F.23)
Zaworotko M.J. Crystal engineering of diamondoid networks // Chem. Soc. Rev.
1994. P. 283-288.
6.5. Инженерия кристаллов с водородными связями 535
6.Р> Инженерия кристаллов
с водородными связями
В соответствии с репутацией водородных связей как «универсальных
взаимодействий» в супрамолекулярной химии работы по конструированию
кристаллов в основном затрагивали системы с водородными связями, нежели системы
со взаимодействиями любых других типов. В такой многопрофильной работе,
какой является настоящая книга, невозможно рассмотреть все множество систем с
водородными связями. Поэтому в последующих разделах содержится лишь
несколько характерных примеров использования сильных водородных связей и
водородных связей средней силы при инженерии кристаллов. Многие их этих примеров
относятся к металлоорганическим системам, исследование которых является
одним из направлений научной деятельности авторов. В целом, понятия о
межмолекулярных взаимодействиях и межмолекулярной валентности для органических и
металлоорганических систем очень близки, поскольку атом металла расположен не
на периферии супрамолекулчрного тектона и, следовательно, почти не принимает
участия в сборке кристалла. Однако именно в случае систем, содержащих металл,
мы сталкиваемся с большим разнообразием форм и топологий. Детали содержатся
в ссылках к данному разделу.
*"» Braga D., Grepioni F. and Desiraju G. Crystal engineering and organometallic
architecture // Chem. Rev. 1998. Vol. 98. P. 1375-1405.
6.5-1 Димеры карбоновых кислот
Супрамолекулярные тектоны, содержащие остатки карбоновых
кислот RCO2H, в твердом состоянии способны образовывать циклические димеры
типа 1^(8); в разд. 6.4 мы уже наблюдали, какую важную роль играет эта их
особенность при образовании взаимопроникающих алмазоподобных сеток. Тем не менее
такое взаимопроникновение редко происходит, если в твердом образце отсутствуют
большие полости, и поэтому дикарбоновые кислоты склонны образовывать
простые ленточные структуры, возникновение которых почти не зависит от наличия
других функциональных групп. На рис. 6.23 показаны топологически
эквивалентные «ленты» с наборами графов первого уровня R^(8) и СG) для фумаровой
кислоты и ее комплекса [Ре(СОL(фумаровая кислота)] (D. Braga et al., 1994).
} Амиды
С точки зрения инженерии кристаллов амидная группа представляет
чрезвычайный интерес. Этот интерес обеспечивается не только наличием
кислотных NH-групп (способных выступать в роли доноров протонов при образовании
водородных связей) и группы С=О (акцептор протонов), но и наличием протона
при атоме углерода амидной группы, который по сравнению с другими С—Н-про-
тонами обладает выраженным «кислотным» характером благодаря индуктивным
эффектам со стороны ближайших атомов кислорода и азота. Это зачастую приво-
536
6. Инженерия кристаллов
(а)
(б)
Рис. 6.23. Структуры (а) комплекса [Ре(СОL(фумаровая кислота)] и (б) фумаровой
кислоты. (Воспроизведено с разрешения Braga D., Grepioni F. and Desiraju G.R. Crystal
engineering and organometallic architecture // Chem. Rev. 1998. Vol. 98. P. 1375-1405)
дит к возникновению сравнительно сильных взаимодействий С—Н-X.
Интересные примеры сетчатых структур, образующихся за счет водородных связей, и
структур, в которых важную роль играют заместители, приведены на рис. 6.24 для
формамида (H2NCHO) и [Mn(C5H5)(CO)(NO)(CONH2)J.
В обеих структурах сохраняется циклическое восьмичленное кольцо,
включающее водородные связи и похожее на те кольца, которые мы наблюдаем в структурах
карбоновых кислот. Однако в случае формамида наличие второго NH-протона
приводит к образованию поперечно-сшитой структуры. Эта связь отсутствует в
соединении марганца, которое не имеет также и «кислотного» С—Н-протона. Вместо
этого возникает дополнительное взаимодействие С—Н---О, направленное от
С5Н5-лиганда одной молекулы к группе С=О амида соседней молекулы.
6.5-3 СПИРТЬ1
Спирты ROH обладают одним донором для образования водородной
связи — протоном - и двумя акцепторами — неподеленными парами атома
кислорода и уже поэтому не могут давать развитые структуры с водородными связями
(взаимодействие каждого «кислого» протона и каждой неподеленной пары). В
результате этого с участием каждого атома кислорода реализуются взаимодействия
только двух типов: одна водородная связь донорного типа, а вторая —
акцепторного типа. Это приводит либо к образованию бесконечно длинных цепей, как в слу-
6.5. Инженерия кристаллов с водородными связями
537
(а)
Рис. §.24. Структуры
(а) формамида H2NCHO
и (б) [Mn(C5H5)(CO)(NO)(CONH2)]
538
6. Инженерия кристаллов
R
1
1
чн
Цепь
ч
|
1
R
.Н''
R
1
чн
1
1
R
н ,-'
I .''
1-°'
Четырехчленный цикл
R
Шестичленный цикл
Рис. 6.25. Геометрические формы, образуемые молекулами спиртов за счет водородных
связей
чае спиральных тубуландов (разд. 5.2.2), либо к образованию циклов. Циклические
структуры обычно включают или четыре, или шесть ОН-групп (рис. 6.25).
В случае хинонов и фенолов, подобных соединению Дианина (разд. 5.2.3), чаще
образуется шестичленный цикл, который обладает высокой способностью давать
клатраты. Эта структура настолько прочна, а ее способность к образованию
соединений включения настолько хорошо известна, что ее использовали в качестве
модели для целого ряда ковалентных хозяев при реализации стратегии под условным
названием «стратегия шестичленного хозяина», например соединения F.5), в
котором водородные связи, образующие цикл, заменены на ковалентные связи. В целом
ряде объемистых комплексов эти циклические структуры воспроизводятся с
размером цикла, который почти не зависит от стерических особенностей остальной
части молекулы спирта. Так, [Mo(COJ(r|5-C5H5)(r|3-C7HI0OH)] дает четырехчленный
цикл, в котором все ОН-группы направлены внутрь молекулы, тогда как
объемистые металлоорганические группы участвуют в слабых взаимодействиях на
периферии молекулы. В то же время Ре2-димер [Fe2(COL(C5H4C2H4OHJ] образует
шестичленный цикл с водородными связями (рис. 6.26).
6.5. Инженерия кристаллов с водородными связями
539
(а)
Рис. 6.26. Структуры с четырех- и шести-
членными циклами, образуемые
молекулами (a) [Mo(COJ(ti5-C5H5)(ti3-C7H10OH)J
и (б) [Fe2(COL(C5H4C2H4OHJ]
540
6. Инженерия кристаллов
6.5-4 Смешанные кристаллы спиртов и аминов
Если спирт в идеальном случае можно рассматривать как соединение,
отдающее один протон для образования водородной связи и принимающее два
протона (по одному на каждую неподеленную электронную пару атома кислорода),
то амин — это соединение, полностью комплементарное спирту, поскольку он
отдает два протона для образования водородной связи и принимает один. Более того,
спирты, как правило, являются кислотами Брёнстеда, а амины представляют собой
основания; поэтому между ними существует высокая степень сродства. Вследствие
этого смеси спиртов и аминов часто стремятся образовать смешанные кристаллы,
насыщенные водородными связями (saturated hydrogen-bonded, SHB). В подобных
случаях невозможно отличить компонента-хозяина от компонента-гостя. В ходе
недавнего исследования смешанных кристаллов 1,4-диаминобензола с фенолом
A:2) и с различными диолами A:1), дополненного анализом данных
Кембриджского банка для SHB-образцов, Дж. Логлин и другие (J. Loehlin et al., 1998)
обнаружили два наиболее распространенных вида упаковки: гексагональную структуру типа
«ограда курятника» (названную столь нетривиально из-за похожести данной струк-
\п)—H-N
<*№-»# » е%_
H-N
S ц
GiiiiiiiiiiiiiiiiniiiiH-N
Рис. 6.27. Мотивы: (а) «ограда курятника» и (б) «лестница» для структур, насыщенных
водородными связями. Индексы a, b и с расположены в порядке увеличения длины
водородной связи. (Воспроизведено с разрешения Loehlin J.H. et al., 1998)
6.5. Инженерия кристаллов с водородными связями
541
туры на тонкую проволочную сетку, которой принято огораживать вольеры с
мелкой птицей) и структуру типа «лестница», составленную только из мотивов,
обладающих набором графов /)-типа (первого уровня), рис. 6.27. Наборы графов второго
уровня для обеих форм служат иллюстрацией сложности анализа при помощи
набора графов даже для этих, химически относительно простых, систем.
6.5*5 Смешанные кристаллы аминов с аминами
Комплементарность между числом доноров - протонов — при
образовании водородных связей и числом акцепторов — неподеленных электронных пар -
может быть также проиллюстрирована на примере кристаллической структуры
комплекса тетрафторбората аммония с уротропином (F.24), N4(CH2N). Донор
протонов NH4 совершенно комплементарен четырем тетраэдрически расположенным
третичным аминогруппам соединения-акцептора F.24). Тем не менее, несмотря на
тетраэдрическую природу строительного блока уротропина, алмазоподобная
структура не образуется (ср. с разд. 6.4), а получающийся комплекс обладает структурой
супервюрцита (ZnS), в которой ионы Zn2+ заменены на ионы NH4, а ионы S2~ — на
фрагменты F.24). Гексагональные «клетки» ионов NH^, напоминающие структуру
кристаллов льда, выступают в роли хозяев по отношению к тетраэдрическим
анионам BF^~ (рис. 6.28). Элегантную структуру этого комплекса предложили О. Эрмер
и А. Элинг в 1994 г. (О. Ermer and A. Eling), хотя впервые его получили в 1978 г.
Однако тогда возможность образования сетки водородных связей не была критически
осмыслена, даже несмотря на то, что расстояния для водородных связей N-N,
равные 2.999 и 3.025 А, оказались сравнительно короткими для взаимодействий
N—H-N (результат влияния заряда и эффекта сжатия аниона BF4 ). Представляется
Рис. 6.28. Два представления структуры супервюрцита [NH4F.24)]BF4, показывающие
включенный анион BF^
542 6. Инженерия кристаллов
интересным соотнести включение аниона BF^ в данную структуру с тетраэдричес-
ким соответствием для анионного хозяина D.63), см. разд. 4.10.
N| N
F.24)
6.6 Водородные связи
монооксида углерода
Монооксид углерода(П) представляет собой удобный лиганд для
металлов с низкими степенями окисления, поскольку способен вести себя подобно
тт-кислоте. Однако делокализация неподеленных электронных пар атомов
кислорода делает СО сравнительно плохим акцептором протона при образовании
водородной связи. Поэтому в случае таких соединений, как кубен Мп4 F.20), даже бензол
более предпочтителен в качестве акцептора, нежели СО, хотя в этом случае
начинают действовать и структурные, и электронные факторы. Тем не менее в отсутствие
лучших акцепторов водородное связывание с участием СО может быть заметным
фактором при образовании кристалла. Основность водородной связи
координированного СО возрастает при переходе от концевого способа связывания к мостико-
вому и далее — к тройному мостиковому (рис. 6.29); при этом СО становится более
похожим на кетон (двойная связь), чем на соединение с тройной связью. Это может
даже повлиять на молекулярную структуру образцов карбонилов металлов, если
имеются две или более структуры с мостиковыми связями СО различных типов,
близкими по энергии. В этих случаях водородное связывание с СО в твердом
состоянии осуществляется по мостиковому принципу.
При наличии «жестких» (сильных) доноров протонов для образования
водородной связи, таких, как спирты или карбоновые кислоты, СО редко принимает
участие во взаимодействиях такого рода. При этом функциональные группы — доноры
протонов для водородных связей — связываются, как правило, с другими частями
молекул, обеспечивающими более сильные взаимодействия, что было подробно
рассмотрено в разд. 6.2.2. В одном случае, а именно для структуры
[Сг(СОK{г|6-С6Н4(С5Н4МеОН)}], рис. 6.30, - реализуется синергетический харак-
о+ о66" о6"
III j| S
I / \ /Ci\
м м м м—1-м
М Рис. 6.29. Способы связывания СО
Концевое Мостиковое Тройное мостиковое и формальные заряды
6.6. Водородные связи монооксида углерода
543
Рис. 6.30. Кристаллическая структура [Сг(СОK{т16-С6Н4(С5Н4МеОН)}]. Показаны
водородные связи О-Н-О=С и С-Н-0 (по Braga D. et al., 1994)
тер взаимодействий; гидроксигруппа образует бифуркационную водородную связь
с двумя атомами кислорода от двух молекул СО. Длина этой связи сравнима с
длиной межмолекулярных водородных связей С-Н-О, направленных от
координированной арильнои группы к неподеленнои электронной паре гидроксильнои группы
B.29, 2.44 и 2.27 А соответственно).
Совершенно иная картина наблюдается при образовании водородных связей в
случае «мягких» доноров протонов, например С—Н-связей. Для этих доноров
водородные связи типа С—Н-О=С наиболее распространены в металлоорганических
системах. В результате исследования таких водородных связей (D. Braga et al.,
1995b) с помощью данных Кембриджского банка получили распределение
межмолекулярных расстояний С---0 от 3.2 до 3.9 А для взаимодействий С—Н-протона с
Рис. 6.31. Гистограмма
расстояний С-О, найденных с
использованием данных Кембриджского
банка структурных данных для
комплексов с водородной связью
типа С-Н- •0=С. Связи с более
основными мостиковыми СО-ли-
гандами имеют меньшую длину,
чем с концевыми лигандами.
(Воспроизведено с разрешения Braga D.
et al., 1995b)
30 i
25 ■■
20-
1
I
и
CD Концевое Fe-CO-связывание
ЕЯ Мостиковое Fe-CO-связывание
3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 4.0
Расстояние С—О
6. Инженерия кристаллов
мостиковым СО; расстояния в среднем оказались гораздо более короткими, чем
расстояния, возникающие между этим же протоном и концевым СО (рис. 6.31).
•""* Braga D. and Grepioni F. Hydrogen bonding interactions with the CO ligand in the
solid state // Ace. Chem. Res. 1997. Vol. 30. P. 81-87.
Слабые водородные связи
Концепция С—Н-связи, которая может участвовать в водородном
связывании кислоты, способной вступать в слабые взаимодействия с основными
акцепторами, в частности с кислородом, была впервые предложена Сутором (Sutor) в
1962 г. В то время эта идея встретила серьезное сопротивление, однако в свете
множества сообщений об исследованиях с помощью Кембриджского банка
структурных данных, а также на фоне открытия большого числа соединений с водородными
связями типа С—Н-0 наличие взаимодействий такого рода больше ни у кого не
вызывает сомнений, а расстояния С-О (D) между 3.0 и 4.0 А расцениваются как
значительные F.25). В случае, касавшемся взаимодействий С— Н-протонов с
координированным СО (см. разд. 6.6), мы видели, что расстояние D зависит от
основности акцептора — атома кислорода. Кроме этого, имеются еще два фактора, которые
определяют D: кислотность С—Н-связи и стерическое окружение протона связи
С-Н. Кажется очевидным, что из перечисленных факторов именно стерические
эффекты играют ведущую роль, ограничивая величину D узким диапазоном
значений 3.38—3.52 А вследствие простой компенсации невыгодных стерических
взаимодействий силами притяжения Ван-дер-Ваальса. В стерически незатрудненных
системах кислотность связи С—Н становится намного более важным фактором, и
практически единственным, поскольку она как бы «размазана» по достаточно
большому объему образца. На рис. 6.31 показано, как это отражается на основности
атома кислорода. Действительно, между значениями D и рКа СН-кислоты в DMSO
(Me2SO) существует заметная корреляция (рис. 6.32). Как правило, расстояния
С--О линейно возрастают с ростом рКа; при этом наиболее короткие расстояния в
диапазоне 3.1—3.2 А и были обнаружены в стерически незатрудненных системах с
водородными связями, образовавшимися за счет сильнокислых протонов связей
С-Н, как, например, в СНС13 и (CNJ=CHR.
D
F.25)
Один из наиболее элегантных комплексов со слабыми водородными связями,
комплекс 2-этиниладамантанола-2 F.26), методом дифракции нейтронов в 1996 г.
изучила Дж. Хоуард (J. Howard) с сотрудниками из Университета Дарема (Велико-
6.7. Слабые водородные связи
50
5451
40
30
20
10
о
3.0
*+-\ Н2С =CHR
RCHO
RC=CH
RRC(OH)CHCH
R(CO)C=CH>—•—i
PhCHRCN • •
•—*—' CHC13
(CNJCHR* (CNJCHRfri
RRCHNO2 A) R(SOJCHRCN
' R(CO)CHRCOR B) (RSO2JCHR
R(CO)CH2COR C)R(CO)CHRCO2R
R H R(CO)CHRCN
RH
D,
V
RR
I 1
3.1 3.2 3.3 3.4 3.5 3.6
Расстояние C-H-O(D), A
3.7
3.8
3.9
4.0
Рис. 6.32. Корреляция между кислотностью С—Н-кислот (рКа в DMSO) и длиной
водородной связи С--0 (D). В точках • стерические препятствия отсутствуют, тогда как в точках
▲ сближение групп С—Н и О затруднено. Для соединения (CNJCHR неклатратная
структура обозначена символом «я», тогда как клатратная — символом «6». (Воспроизведено
с разрешения Desiraju G.R. The С—Н-0 hydrogen bond // Асе. Chem. Res. 1996. Vol. 29.
P. 441-449)
британия); (F. Allen et al., 1996). Установленная ими структура показала, что
происходит взаимное усиление двух «сильных» водородных связей О—Н-0 и двух «слабых»
взаимодействий С—Н-0 за счет кислых алкиновых протонов, в результате чего
получается циклический тетрамер. Это исследование позволило установить, что
расстояние Н-0 (d) составляет 2.070 А и является одним из кратчайших расстояний,
Н
C^/
Н
/
Н
о-
н-
F.26L
5-СтидДж. В.
$46
6. Инженерия кристаллов
известных на сегодняшний день (ср. расстояние С---0 (D) составляет 3.135 А). Те
группы ОН, которые не участвуют в образовании водородных связей типа О—Н-О,
вступают в слабые взаимодействия иного сорта — с ацетиленовой л-электронной
плотностью. Межмолекулярные взаимодействия всех типов в данной системе
показаны на рис. 6.33.
Вызывает удивление тот факт, что даже очень слабые кислоты, такие, как ме-
тильные группы, могут участвовать в образовании достаточно прочных
межмолекулярных водородных связей. Например, при совместной кристаллизации (в
соотношении 1:1) 4-нитробензойной и 4-(Ы,Н'-диметиламино)бензойной кислот
(смешанный кристалл F.27)) образуется ленточная структура, содержащая
чередующиеся циклы издимеров карбоновых кислот (ср. разд. 6.5.1), а также восьмичлен-
ный цикл, который получается за счет водородных связей между двумя атомами
кислорода нитрогруппы и двумя протонами диметиламинового фрагмента.
н
о ---
F.27)
Тем не менее наиболее широко распространенные водородные связи типа
C-H--0 — не единственный пример слабых водородных связей, потенциально
пригодных для инженерии кристаллов. Взаимодействия такого рода были
обнаружены для систем: С—Н•••N, С—Н---С1 и С— Н -тт наряду со взаимодействиями, в
которых участвуют слабые акцепторы протонов, такие, как О—Н--тт и N—Н--тт. В
сообщении Р. Хантера (R. Hunter et al., 1994) из Университета Кейптауна (ЮАР)
Рис. 6.33. Кристаллическая
структура F.26) с водородными
связями типа О—Н-О, С—Н-0
и О-Н-я (по Allen F. et al., 1996)
6.7'. Слабые водородные связи !%47
описана сэндвичевая пара со взаимодействиями типа С-Н--тт: две группы Ph3PMe+
взаимодействуют с фураном (С4Н4О) при участии протонов метильных групп
F.28). Параллельный поиск с помощью Кембриджского банка структурных данных
указывает на очень короткие расстояния Н-тт, лежащие в пределах 2.35—2.79 А. В
данном соединении наблюдаются также взаимодействия типа C-H-N с участием
групп CN встроенных в структуру дианионов (£> = 3.425 A, d = 2.46 А, 0 = 177°).
F.28)
*"■* Desimju G.R. The C-H-0 hydrogen bond // Асе. Chem. Res. 1996. Vol. 29.
P. 441-449.
548
6. Инженерия кристаллов
6.8 Водородные связи с металлами
и гидридами металлов
6.8.1 Прямое взаимодействие с металлами
Атомы или ионы металлов обладают выраженным амфотерным
характером и поэтому могут выступать и как основания Льюиса, и как кислоты Льюиса в
зависимости от природы металла, его степени окисления, характера
вспомогательных лигандов и т.д. В металлоорганической химии взаимодействие а-электронной
плотности одинарной ковалентной связи, в том числе связи С—Н, с электроноде-
фицитным центром — атомом металла — явление хорошо известное, называемое
агостическим взаимодействием (связывание), рис. 6.34, а. Сильное агостическое
взаимодействие можно рассматривать как трехцентровую двухэлектронную связь; в
результате возникновения последней порядок связи С—Н уменьшается. Подобные
взаимодействия могут быть определены по следующим признакам:
• по уменьшению константы спин-спинового взаимодействия 'Усн в 13С ЯМР-
спектре (данная константа определяется а-электронной плотностью связи
С-Н);
• по высокопольному химическому сдвигу в 'Н ЯМР-спектре, возникающему в
результате экранирования электроположительным металлическим центром
(~0—10 м.д., как и в гидридах металлов);
• по уменьшению частоты валентных колебаний v(CH) в ИК- или рамановских
спектрах вследствие понижения колебательной силовой константы;
• по короткому времени релаксации спиновой решетки Т{ в ЯМР-спектре,
поскольку спиновая поляризация может быть легко перемещена к
металлическому центру.
(а)
(б)
(в) Ч
н V
I .
I
/
R""^O-
Агостическое взаимодействие
[Co(C5Me5)(PR3)(C2H5)]+
Ч
с ^о
/»
о
Межмолекулярное
псевдоагостическое (IPA)
взаимодействие
[R3NH] [Co(COL]
L'
Cu
L'
°TR
о
\
о-
Межмолекулярное
многоцентровое
гетероакцепторное (IMH)
взаимодействие
Рис. 6.34. Взаимодействия типа М-Н
6.8. Водородные связи с металлами и гидридами металлов 549
Как правило, агостические связи являются ковалентными, однако тот факт, что
они имеют очень небольшой диапазон изменения углов и расстояний, позволяет
классифицировать их как водородные связи типа М•••Н—С. Подобные же
взаимодействия, но со связями X—Н, принято называть межмолекулярными псевдоагости-
ческими (intermolecular pseudo-agostic, IPA); при этом в базе данных
Кембриджского банка имеется множество примеров, подтверждающих их существование
(D. Braga et al., 1997). Одно из наиболее характеристических 1РА-взаимодействий
реализуется в простой соли [R3NH+][Co(CO)^], рис. 6.34, б. Стерически
незатрудненная природа тетраэдрического комплекса Со(-1) допускает сближение с N-H-
группой и последующее образование усиленной зарядом IPA водородной связи
N+—Н-Со". Изучение методом дифракции нейтронов для случая R = Et
позволило установить расстояние N-Co, 3.666 А, и расстояние Н-Со, 2.611 А, причем все
три атома находятся почти на одной линии. Родственны 1РА-взаимодействиям
межмолекулярные многоцентровые гетероакцепторные (intermolecular multicentre
heteroacceptor, IMH) водородные связи, которые напоминают взаимодействия
обычных доноров типа X—Н (где X = О, N, С и т.п.) с М—X' (где М и X' обогащены
электронами). По существу, подобное взаимодействие является разновидностью
бифуркационной водородной связи, при образовании которой сам металл и
координированный гетероатом (X') действуют как два акцептора. Взаимодействие
типа IMH имеет место в соединении двухвалентной меди [CuL2L'2]-2H2O (L —
сопряженный анион N-ацетил-а-аланина, L' — N-метилимидазол), в котором
О—Н-группа воды взаимодействует одновременно с центром Cu(II) и с
координированным атомом кислорода (расстояние О—Н-0 составляет 2.027 А, расстояние
О-Н-Си - 2.568 А), рис. 6.34, в.
6.8.2 Взаимодействие с гидридами металлов
К настоящему времени собрано достаточно доказательств того, что в
дополнение к металлу, являющемуся акцептором водородной связи, фрагменты
М-Н могут выступать в роли как доноров протона при образовании водородной
связи, так и акцепторов — за счет атома водорода гидрида, несущего формальный
отрицательный заряд. В качестве нетрадиционного донора водородной связи атом
водорода фрагмента М—Н должен быть стерически доступным и иметь дефицит
электронов. Поскольку такой гидридный лиганд представляет собой
исключительно а-донор, его электронная природа будет зависеть от плотности электронов на
металлическом центре. Последняя в значительной мере определяется степенью
окисления металла, причем катионы металлов оттягивают на себя максимум
электронной плотности. Этот эффект особенно резко выражен, когда гидрид участвует
в двойной или даже тройной мостиковой координации более чем с одним
металлическим центром в зависимости от их электронной плотности. В подобных
примерах фрагмент М—Н представляет собой «мягкий» донор протона для водородной
связи и часто взаимодействует с «мягкими» акцепторами типа СО, причем
последний является широко распространенным солигандом в соединениях, образуемых
металлическими гидридами. Комплекс [Os3(COI0(pi-H)(pi-NCHCF3)] -
превосходный пример соединения, в котором гидридный лиганд (с двойным мостиковым
550
6. Инженерия кристаллов
V
—NMe
i
Н
293К
243К
223К
203К
183К
163К
13
-17.0
-18.0
Рис. 6.35. Фрагменты 'Н ЯМР-спектров,
записанных при различных температурах,
иллюстрирующие процессы обмена протонов Ir— H
и N—Н. (Воспроизведено с разрешения Abad M.
etal., 1999)
связыванием) формирует единственную короткую связь М2Н-О=С B.594 А),
направленную на концевой карбонильный лиганд соседнего кластера.
Считается, что фрагмент М—Н обогащен электронами, причем гидридный
протон ведет себя как псевдогалоген. В подобных условиях гидрид способен
действовать как нетрадиционный акцептор протона при образовании водородной связи,
участвуя в водородном связывании типа М—Н-Н—X (где X —
электроотрицательный атом, например О, N, С и т.п.). Подобные водородные связи могут быть как
межмолекулярными, так и внутримолекулярными и были обнаружены
экспериментально методами РСА и нейтронной дифракции, а также путем анализа
плотности заряда для широкого круга функциональных групп типа Н-Х в сочетании с
гидридами s-, р- и d-элементов. Для дигидрида Ir(III) F.29) в растворе было доказано
наличие взаимодействий Ir-H-H-N. !H ЯМР-спектроскопия при различных
температурах позволяет изучать взаимный обмен кислотного и основного атомов
водорода, осуществляющийся, возможно, через координированный Н2-интермедиат
(рис. 6.35). Эти результаты представляют огромный интерес для исследования
механизмов активации Н2 металлическими катализаторами. Проведенные недавно
расчеты ab initio для целого ряда других сильно взаимодействующих систем типа
М-Н-Н-Х дали величины расстояния Н-Н и энергии взаимодействия,
приведенные в табл. 6.5.
н---
6.9. п—п-Стэкинг-взаимодействия 551
Таблица 6.5. Вычисленные параметры для различных систем М—Н---Н—X
(по Orlova G. etal., 1999)
Комплекс
Расстояние Н... Н, А
Энергия взаимодействия,
кДжмоль
-1
Mo(COJ(PH3J(NO)(H)-H-F
Mo(COJ(NH3J(NO)(H)-H-F
Mo(COJ(NH3J(NO)(H)-H-OH
Re(C5H5)(CO)(NO)(H)--H-OH
Re(C5H5)(CO)(NO)(H)-H-OCF3
1.378
1.300
1.647
1.770
1.458
46.4
64.4
54.8
45.6
41.0
6.9 я-л-Стэкинг-взаимодействия
Природа тс—тс-стэкинг-взаимодействий, которые осуществляются
между ароматическими кольцами либо по типу «торец—плоскость» (С—Н-я), либо
по типу «плоскость—плоскость» (С---С), обсуждалась в разд. 1.7.6. Такие
взаимодействия важны для стабилизации молекулярных комплексов хозяин—гость в
растворе, когда молекулы гостей ароматические (см. гл. 5). С точки зрения инженерии
кристаллов как сами взаимодействия по типу тс—тс-стэкинга, так и их
направленность являются относительно слабыми. Поэтому их трудно предсказывать и
контролировать, в особенности в присутствии других, более сильных, взаимодействий.
Межмолекулярные тс—я-стэкинг-взаимодействия важны для ароматических
молекул, подобных бензолу, ибо благодаря им возникает характерный «елочный» мотив
упаковки (рис. 1.17). тс—тг-Стэкинг-взаимодействия типа «плоскость—плоскость»
ответственны за «пишущие» свойства графита; при этом один слой
шестиугольников типа «ограда курятника» скользит относительно другого. В случае крупных
полициклических ароматических углеводородов, структуры которых представляют
собой нечто среднее между структурами бензола и графита, важность стэкинг-вза-
имодействий возрастает с увеличением числа ароматических колец. Дж. Дезирайя
и А. Гавеццотти (G. Desiraju and A. Gavezzotti, 1989) разработали модель для
предсказания структуры ароматических углеводородов с конденсированными кольцами
(fused-ring aromatic hydrocarbons, FAH), которая основана на таких простых
понятиях, как «укладывание стопкой» и «скольжение» атомов углерода и водорода в
молекуле с учетом их топологической связности. Как правило, все структуры FAH-mo-
лекул подразделяются на четыре типа, зависящих от длины их кратчайшей
кристаллографической оси. Тенденция к принятию кристаллами формы,
характерной для того или иного типа, обусловлена относительным вкладом атомов углерода
и водорода в поверхностную площадь молекулы. Эти четыре типа перечислены
ниже и проиллюстрированы соответствующими примерами: нафталином, пиреном,
короненом и пентаценом (рис. 6.36).
1. Простая «ёлочка»; ближайшие соседи не параллельны; 5.4 А < кратчайшая ось <
<8.0А.
552
6. Инженерия кристаллов
Пирен
Коронен
Пентацен
Рис. 6.36. Четыре базовых типа
упаковки ароматических соединений;
короткая ось в каждом случае вертикальна.
(Воспроизведено с разрешения
Desiraju G.R. and Gavezzotti A., 1989)
2. Сэндвичеобразная «ёлочка»; «ёлочный» мотив состоит из диад, напоминающих
сэндвич; кратчайшая ось > 8.0-А.
3. уТип; взаимодействия между параллельно смещенными молекулами; 4.6 А <
< кратчайшая ось < 5.4 А.
4. Р-Тип; графитовые плоскости; кратчайшая ось < 4.2 А.
Какая кристаллическая структура будет при этом реализована, зависит от
оптимизации С-"С-взаимодействий, которые максимальны между параллельными
молекулами, уложенными со смещением в стопку по принципу «плоскость—плоскость»
на расстоянии радиусов Ван-дер-Ваальса (взаимодействия скольжения), а также от
оптимизации С-Н-взаимодействий, которые являются максимальными в
ориентированных под углом друг к другу молекулах вследствие своей электростатической
(г)
Переход.
163.9 К
Трихлинная форма
148, 123, 101 К
промежуточная
конформация
Моноклинная форма
ферроцена
293, 173 К
(среднее значение)
заторможенная
конформация
Переход,
242 К
Орторомбическая форма
98 К
заслоненная конформация
Рис. 6.37. Объемное представление трех полиморфных форм ферроцена: (а) триклинная,
(б) моноклинная, (в) орторомбическая. (Воспроизведено с разрешения Braga D., Grepioni F.,
Desiraju G.R. Crystal engineering and organometallic architecture // Chem. Rev. 1998. Vol. 98.
P. 1379). (г) Фазовые превращения ферроцена
6.9. к—п-Стэкинг-взаимодействия
553
природы (стэкинг-взаимодействия). у- и р-Структуры участвуют в образовании
кристаллов в тех случаях, когда важно максимизировать С-С-взаимодействия,
тогда как «ёлочный» мотив обеспечивает сильные С---Н-взаимодействия. В случае
сэндвичеобразных «ёлочек» важны взаимодействия обоих упомянутых типов.
Стэкинг-взаимодействия с участием плоскостей и торцов кристаллов важны
также в структурах, образованных ароматическими металлоорганическими
соединениями. Первая из синтезированных ароматических металлоорганических
молекул — ферроцен Fe(C5H5J — существует в виде трех различных кристаллических
форм, каждая из которых имеет «ёлочный» мотив из-за высокого содержания
атомов водорода на поверхности (рис. 6.37). Различия между этими тремя формами
(моноклинной, которая неупорядочена при комнатной температуре, а также двумя
низкотемпературными — триклинной и орторомбической) определяются
относительной ориентацией двух колец С5Н5. Это обстоятельство, в свою очередь, влияет
на направленность взаимодействий С—Н-С.
Подобно ферроцену, структура твердого бис(бензол)хрома Сг(С6Н6J похожа на
структуру «ёлочного» типа. Интересно отметить, что, если обогащенный
электронами Сг(С6Н6J подвергнуть совместной кристаллизации с электронодефицитным
C6F6, то образуется комплекс с переносом заряда Cr(C6H6JC6F6 красного цвета, в
котором осуществляется точное перекрывание типа «плоскость—плоскость» межд>
шестичленными кольцами двух упомянутых компонентов, отстоящих друг от друга
на 3.48 А (С. Aspley et al., 1999), рис. 6.38.
= Cr O = F O=C
Рис. 6.38. Донорно-акцепторное л—я-стэкинг-взаимодействие в Cr(C6H6JC6F6.
(Воспроизведено с разрешения Aspley C.J. et al., 1999)
554
6. Инженерия кристаллов
ЗЛО Прочие взаимодействия
В дополнение к классическим и неклассическим водородным связям и
тс-стэкинг-взаимодействию имеется достаточно сведений о существовании иных
атом-атомных взаимодействий, которые могут оказаться полезными при
конструировании кристаллов. Такие взаимодействия часто слабы, несмотря на то, что они
реализуются на расстояниях, меньших радиусов Ван-дер-Ваальса, и обычно
бывают направленными. Наиболее распространенные «короткие» взаимодействия в
кристаллах — это взаимодействия с участием поляризующихся атомов: Cl, Br, I, S,
Se и т.п.; можно сделать вывод о том, что любые силы притяжения между ними
могут быть обусловлены взаимодействиями индуцированных диполей. Спиральные
трубчатые хозяева (тубуланды), см. разд. 5.2.2.2, включают в себя молекулы,
подобные СС14, с расстояниями С1---С1, равными 3.54 А, и молекулу 12, который
взаимодействует с атомами кислорода включенных молекул этанола. Д. Редди и другие
(D. Reddy et al., 1993b) использовали взаимодействия CN-C1 для получения
молекулярных лент типа F.30) (R1 — R2 = Me), находящихся в твердом кристаллическом
состоянии. Связи N-C1 здесь необыкновенно короткие C.099 А) и довольно
направленные, причем угол C=N-C1 составляет 137°. Отметим отсутствие в этой
структуре любых других сильных межмолекулярных взаимодействий или
потенциально взаимодействующих групп. Замена одной из метоксигрупп на ОН-группу
(R1 = Н, R2 — я-октил) не приводит к разрушению ленточной структуры, хотя
расстояния N---C1 заметно удлиняются (до 3.4 А) из-за необходимости обеспечить
образование более сильных водородных связей О—Н---С1.
N---.._
-----С1
Атом-атомные взаимодействия совсем другого типа были обнаружены в
комплексах с двойной оболочкой типа [МпХ4{М1A8-краун-6)}4] [Т1Х4]2, где М1 = Rb, T1;
Мп = Zn, Си, Мп, Со; X = О, Вг, I. Эти интересные блоки образуют алмазоподобное
множество (см. разд. 6.4) тетраэдров TIX^, которые удерживаются вместе за счет
коротких связей Х-Х, сравнимых с таковыми в чистых галогенах. Алмазоподобная
полость окружена четырьмя молекулами 18-краун-6, расположенными тетраэдрически
относительно центральной внутренней полости, в которую включен анион МПХ|~~
или другие молекулы-гости. В случае ТП4 расстояния I-I составляют 3.69 А, что
вполне сопоставимо с короткими расстояниями I—I—I—I у 12, равными 3.50 А.
6.11. «Неправильные» формы и «неправильный» подбор
1555
Необходимо, однако, отметить, что множество коротких связей в твердом
состоянии может быть обусловлено «полярным уплощением» (см. разд. 6.3; рис. 6.19),
которое позволяет атомам подходить друг к другу на расстояния, более близкие, чем
предполагает простое сложение радиусов Ван-дер-Ваальса. При этом исключается
необходимость какого-либо специального межатомного притяжения.
6.11 «Неправильные» формы
и «неправильный» подбор
6Л1Л Хозяева типа «колесо-ось»
Правила плотной упаковки кристалла гласят, что даже в отсутствие
особых, сильных, взаимодействий притяжения дисперсионные силы и силы Ван-
дер-Ваальса все равно заставят молекулы упаковаться так плотно, как только это
возможно. Это неизбежно приводит к тому, что плотность органических кристаллов
является довольно постоянной и составляет примерно 1.4— 1.6 гсм~3. Органические
молекулы часто несимметричны и имеют неправильную форму, однако, несмотря
на это, они способны упаковываться сравнительно правильно; при этом
большинство из них демонстрируют достаточно высокую кристалличность без включения
молекул растворителя. Это происходит благодаря конформационной гибкости
таких молекул и «мягкой» природе взаимодействий Ван-дер-Ваальса в сочетании с,
как правило, выпуклой формой молекул, которая, в первом приближении,
является сферической. Наблюдаемое при образовании многих хозяев-клатратов (см. гл. 5)
включение растворителя очень часто — это следствие того факта, что такие клатрат-
образующие компоненты имеют неправильную (часто вогнутую) форму (например,
каликсарены, три-о-тимотид). Поскольку они не в состоянии упаковаться
эффективно, то должны включать так называемые молекулы-наполнители, чтобы
заполнить такие зазоры. Это простое соображение часто используют при инженерии
кристаллов, когда специально синтезируют молекулы, обладающие
«неправильной» формой. В частности, молекулы типа «колесо—ось», подобные молекулам
F.31) и F.32), имеющие узкий линейный ацетиленовый спейсер и объемистые
концевые группы, способны в твердом состоянии выполнять роль хозяев для
целого ряда гостей, поскольку необходимо заполнить пустой зазор между двумя
объемистыми «колесами».
6. Инженерия кристаллов
Металлоорганические соединения типа [{Ки(т13:т13-С8Н10Ме2)}2-(|ы-пиразин)]
F.33) ведут себя аналогично, причем в данном случае в зазор между двумя
объемистыми концевыми бис(аллильными) группами включены молекулы хлороформа.
F.33)
*"* Hart Н, Lin L.-T.W. and Ward D.L. Molecular design for hosts in crystalline
host-guest complexes // J. Amer. Chem. Soc. 1984. Vol. 106. P. 4043-4045.
6.11.2 «Неправильно» подобранные партнеры
Совершенно иной подход к проблеме «неправильных» молекул был
выбран Дж. Стидом (J. Steed) при инженерии кристаллов путем объединения
водородными связями множества донорно-акцепторных единиц. Взаимодействие аква-
комплексов металлов, используемых в качестве доноров, образующих
направленные водородные связи и обладающих неправильной формой, со стерически
«неподходящими» акцепторами протонов, в частности с краун-эфирами, приводит
к получению целого ряда высоконесимметричных структур. Эти соединения
характеризуются присутствием в кристаллофафической несимметричной ячейке
множества молекул, неэквивалентных по симметрии. Такая ситуация возникает из-за
необходимости химически идентичных субъячеек принимать различные формы
для их объединения в кристаллы, чтобы максимизировать межмолекулярные
взаимодействия, реализуемые с помощью водородных связей. Подобная структура,
образовавшаяся при взаимодействии гидратированного уранилхлорида иО2С12(Н2О)з
F.34) в качестве донора с 15-краун-5 F.35) в качестве акцептора, замечательна тем,
что она содержит 16 неповторяющихся комплексов уранилхлорида и 16
неповторяющихся молекул краун-эфиров, расположенных в виде решетки 4x4 в
асимметричной ячейке, представляющей собой неповторяющуюся часть элементарной ячейки
кристалла. Структура состоит из бесконечных цепей, в которых каждая пара ура-
нилхлорид—краун-эфир воспроизводит себя в каждом четвертом комплексе
(рис. 6.39). Такая структура комплекса - прямое следствие противоречия между
требованиями симметрии нелинейных векторов H2O-U-OH2 (~ 140 и 152°) и кон-
формацией 15-краун-5, в которой каждый макроцикл предоставляет две свои
поверхности: одну с тремя доступными атомами кислорода, а вторую — с двумя.
Результатом этого является нерегулярная цепь четвертого порядка, состоящая из четырех
пар комплексов уранилхлорид—краун-эфир, в которой ориентация предыдущей
пары определяет направление подхода следующей. Макроциклы краун-эфиров одной
6.11. «Неправильные» формы и «неправильный» подбор
557
Рис. 6.39. Элементарная ячейка цепной структуры с водородными связями
[иО2С12(Н2ОK]16A5-краун-5I6
из цепей четвертого порядка несимметрично сцепляются с соседними цепочками
вдоль кристаллографического направления Ь. После сцепления четырех цепочек
пятая начинает воспроизводить следующую такую Dх4)-структуру.
О
н,о—^ur1"*
О
о
С1
он2
о
о
F.34)
о о
^ -о
Ч
F.35)
Концептуально сходные результаты были обнаружены для заряженных систем,
таких, как 1,2-диаминобензолН25О4Н2О, в которых содержится всего восемь
структурно неповторяющихся бензолдиаммониевых доноров и сульфатных
акцепторов.
•""* Hassaballa #., Steed J. W. and Junk P.C. New insights into the formation of extended
supramolecular architectures from simple building blocks // Chem. Commun. 1998.
P. 577-578.
6. Инженерия кристаллов
6 12 Координационные полимеры
Большинство исследований по инженерии кристаллов,
рассмотренных в настоящей главе, посвящено действию «классических», сравнительно слабых
межмолекулярных сил, которые «собирают» тот или иной кристалл или же
участвуют в его «самосборке» при помощи ван-дер-ваальсовых сил, обеспечивающих
получение плотной упаковки и дополненных «направляющим» влиянием водородных
связей, тс-стэкинг-взаимодействий и т.п., как описано в разд. 6.1. Недавние
исследования в области конструирования неорганических кристаллов положили начало
рассмотрению значительно более сильных и направленных координационных
взаимодействий металл—лиганд в качестве средства для создания прочных
кристаллических конструкций. Идея образования подобных полимерных молекул,
удерживаемых вместе за счет бесконечных цепей с координационными взаимодействиями,
восходит к химии цеолитов (см. разд. 5.1.2) и клатратов хофмановского типа. Эти
соединения — одни из первых представителей координационных полимеров, для
которых также характерны взаимодействия типа «хозяин—гость» (см. разд. 5.1.4).
Конструирование молекулярных или супрамолекулярных материалов,
содержащих микроскопические поры или обладающих канальной структурой,
представляет собой сложную, но необыкновенно плодотворную область исследований.
Подобные материалы могут найти применение в нефтехимической промышленности
(ср. с получением такого простого сырья, каким, например, является метанол, с
помощью цеолитов), в экологии — для очистки окружающей среды, например для
разрушения летучих органических соединений. Возможность осуществления
направленных химических реакций на твердой матрице уже была показана для
соединений включения мочевины. В то же время привлекательный энантиоспецифичес-
кий синтез на хиральных каркасах еще не реализован. К настоящему времени в
перечисленных выше областях широко используют кремнеземы и алюмосиликаты,
обладающие микропористой структурой, подобной структуре цеолитов, из-за их
термической устойчивости (даже в отсутствие молекул—гостей), а также из-за
возможности изменять размеры каркасных молекул при темплатном синтетическом
подходе. К сожалению, этот метод не позволяет создавать более универсальные
хозяева, включая хиральные хозяева, вследствие слишком простой природы
строительных блоков (тетраэдрические фрагменты, подобные SiO^"). Приведенные
исследования показали, что более разнообразные каркасные структуры хозяев можно
получить при использовании неорганических координационных полимеров.
Конструирование жестких лигандов с расходящимися, точно позиционированными
центрами связывания предоставляет огромные возможности для синтеза
специальных пористых материалов. Ранее этот принцип был реализован группой О. Яги
(О. Yaghi) из Государственного университета штата Аризона (США). Эти
исследователи получили F.36), слоистый координационный полимер на базе
бензол-1,3,5-трикарбоновой кислоты F.37), (Yaghi et al., 1995). Спейсерами (S)
служили два пиридиновых лиганда, а весь ансамбль представлял собой двухмерный
координационный полимер слоистой структуры. Каждый слой взаимодействовал с
такими же слоями выше и ниже себя за счет слабых тс—я-стэкинг-взаимодействий
пиридиновых ароматических колец. Термогравиметрический анализ этого соеди-
6.12. Координационные полимеры
559
нения показал, что при 190 °С оно способно обратимо высвобождать
ароматические молекулы-гости из своих пор-каналов, тогда как общая кристаллическая
структура хозяина сохраняется вплоть до 350 °С. Это было показано рентгеновской
дифракцией порошкообразного образца и при изучении кристаллов с помощью
оптического микроскопа. Молекулы-гости могут быть вновь внедрены в
освободившиеся каналы хозяина. Такой способ включения гостей напоминает клатраты,
описанные Р. Хофманом (см. разд. 5.1.4). Однако обнаруженные термическая
устойчивость и обратимость связывания позволяют надеяться на то, что эти системы
могут оказаться весьма востребованными с точки зрения их практического
применения.
м-::"
Спейсер
о
F.36)
F.37)
В принципе, конструирование пористых координационных полимеров является
столь же простым делом, что и выбор комбинации лиганда с более чем одним
центром связывания, неспособного к хелатированию (расходящиеся центры
связывания), и металла, имеющего комплементарные расходящиеся центры связывания.
Результатом подобного сочетания и будет координационный полимер. Этот подход
получил развитие в ходе своего широкомасштабного применения для синтеза
самособирающихся дискретных молекулярных структур (разд. 7.4.1). Если лиганд
достаточно большой, то между соседними металлическими центрами будут оставаться
полости. Однако на самом деле ситуация не столь проста. Взаимодействия
металл—лиганд зачастую являются сильными и, следовательно, нелабильными. В
результате этого продукты могут быстро осаждаться в виде аморфных порошков; при
этом образуются в основном кинетические продукты, обладающие нерегулярной
структурой. Проблемы такого рода могут быть частично преодолены за счет
применения методик выращивания кристаллов, в которых используются процессы
диффузии, нагревания раствора и гелевой проницаемости. Более того, геометрию
взаимодействий металл—лиганд не всегда легко предсказать; при этом даже если
пористая структура образована, то нет никакой гарантии, что эти поры окажутся
доступными для молекул-гостей, находящихся вне кристалла. Тем не менее это
необходимое условие для использования подобных конструкционных материалов при
ионном обмене или в реакциях включения. Несмотря на указанные трудности, в
инженерии пористых твердых веществ был достигнут значительный прогресс. В
дополнение к F.37) лиганды, подобные 4,4'-бипиридилу (bipy, F.38)), и их
увеличенные аналоги типа F.39), наряду с тетраэдрическим Ge4Sj^ F.40), были применены
6. Инженерия кристаллов
в качестве прототипа строительных блоков, полученных с использованием
расходящихся лигандов.
F.38)
S"/,,,
/
' <3е
Ge
F.40)
В частности, лиганд F.38) образует огромное количество пористых сеток,
геометрия которых зависит от координационных требований данного металлического
центра. При этом в случае простых сеток взаимопроникновение не происходит (ср.
алмазоподобные сетки, разд. 6.4). Однако по мере возрастания сложности
начинают образовываться структуры с дву-, трех-, четырех- и шестикратным взаимным
проникновением, хотя многие из них все еще содержат поры и каналы с
включенными противоанионами (рис. 6.40). Например, четырехкратная
взаимопроникающая алмазоподобная структура [Cu(bipyJ][PF6] в порах шириной 6 А содержит
анионы PF^~, которые не могут быть заменены на другие анионы. В отличие от этой
структуры, более открытая структура (Cu(bipy), 5][NO3](H2O)l 25
(демонстрирующая все еще высокую степень взаимопроникновения) по мере удаления
кристаллизационной воды при нагревании начинает обменивать анионы. Так, небольшой по
размеру анион N0^* может быть заменен на SO^~ и BF^ .
Как лиганд F.38), так и увеличенный бипиридильный лиганд F.39) К. Пауэр и
другие (К. Power et al., 1998) использовали для получения прямоугольных сетчатых
соединений, аналогичных соединениям включения, описанным Хофманом. По-
видимому, дополнительный спейсер С2 в F.39) в большинстве случаев оказывает
влияние только на размеры ячейки, увеличивая ее. Единственное исключение —
реакция с Cu(II), в ходе которой образуется причудливая сетка [CuF.39J(NO3JL,'
рис. 6.41. Cu(II) представляет собой ион металла с конфигурацией cf9 и проявляет
сильный эффект Яна-Теллера, искажающий геометрию основного состояния. В
результате этого октаэдрический металлический центр имеет значительно более
длинные связи с аксиальными лигандами (ионы NOj), которые, в отличие от би-
пиридильных, проникают в ячейку сетки. Не совсем понятно, почему Cu(II) в
сочетании с этим лигандом принимает структуру NbO, в которой экваториальная
плоскость иона металла повернута на 90° по отношению к двум из его четырех
ближайших соседей, однако, возможно, что это вызвано трудностью заполнения более
вместительных полостей.
Взаимодействие [Мп(О2ССН3J(Н2ОL] с лигандом F.40) дает элегантную алма-
зоподобную структуру [NMe4]2[MnGe4S10], в которой противоионы NMe4 включе-
(а)
\
Of/
tr
"Си
Си
;си
С
Cu(bipyJPF6, 3D
Алмаз, КП = 4; 6 А
(г) \ /
Си-Си
\
.Си-Си
Си-Си
Ч /
Си-Си
Си-Си /Си-Си
\ /
Си-Си
Си-Си
/ \
Cu(bipy)Cl, 2D
гексагональная решетка
КП = 2;4хЗА
\
F)
Си Си
\ /
Си Си
\
.Си
Си Си
Си Си / \-
\ /U
Си Си
/ \
Cu(bipyI5NO3(H2OI 2S, 3D
a-ThSi2, КП = 6; 8x6 и 6x4 А
-Cd-
-Cd-
-Cd Cd-
Cd Cd Cd Cd
Cd Cd Cd Cd
-Cd Cd Cd Cd -
Cd(bipyJ(NO3J-2(C6H4Br2)
2D, квадратная решетка
КП = 0;11 А
(в)
-Ni-
Ni-
Ag-
Ag.
-Ag-
-Ag-
Ag-
Ag-
Ag-
-Ag-
'Ag.
-Ag-
Ag(bipy)NO3, 3D
a-ThSi2, КП = 3;23хбА
(Ж) j
-Ni-
Co-
Ni-
Ni-
Ni-
"Ag
Ag_
Ag-
-Co
Co Co
Ni(bipyJS(H2OJ-
(bipy),.5(ClO4J(H2OJ
ID, "железная дорога"
КП = 0; 11 A
Co Co
Co Co
Co(bipy), S(NO3J-
• CH3CN, ID
"лестница"
КП = 0; 11 A
to
>4
Рис. 6.40. Схематическое изображение металлических 4,4'-бипиридильных пористых сеток. Черточки означают бипиридильный ли-
ганд, за исключением вертикальных линий в (в), которые соответствуют Ag'-Ag-связям длиной 2.977 А, и горизонтальных линий в (г),
которые соответствуют связям Cu---Cu. Под каждым изображением приведены: химическая формула, пространственная размерность
каркаса, тип структуры, кратность взаимопроникновения (КП) и размер апертуры поры. (Воспроизведено с разрешения Yaghi O.M.,
Li H., Davis С. et al. Synthetic strategies, structure patterns and emerging properties in the chemistry of moduler porous solids // Ace. Chem. Res.
1998. Vol. 31. P. 474-484)
552 6. Инженерия кристаллов
I I I
.„'_L.'_L.'_L
Й II / II # II Рис. 6.41. Каркас типа NbO для
«. а — 11— • — 11— • — • L [CuF.39J(N03J]00. (Воспроизведено с раз-
/II * || * || решения Power K.N., Hennigar T.L. and
II / II / II Zaworotko M.J. X-Ray crystal structure of
- щ _ i шшт» _ j — ф шштфш, {Cu[ j ,2-bisD-pyridil)ethane]2(NO3J}/): The
'ill first example of a coordination polymer that
ф ф ф exhibits the NbO 3D network architecture//
III Chem. Commun. 1998. Vol. 5. P. 595)
ны в качестве гостей в крупные поры алмазоподобной решетки. Нагревание этого
соединения до 500 °С (аналогично получению цеолитов при кальцинации)
приводит к деструкции алкиламмониевого катиона с образованием свободного от гостей
пористого материала, который предельно стабилен благодаря своей, крайне
прочной неорганической структуре. Катионы NMe^" можно заменить целым рядом
двухвалентных металлов.
*""* Yaghi O.M., Li H., Davis С. et al. Synthetic strategies, structure patterns and
emerging properties in the chemistry of modular porous solids // Ace. Chem. Res. 1998.
Vol. 31. P. 474-484
Биомиметические
(биоподражательные) структуры
Результаты, изложенные в разд. 6.12, ясно указывают на колоссальный
прогресс, достигнутый исследователями в получении органических, металлоорга-
нических и неорганических кристаллических конструкций с помощью новых
функциональных материалов. В природе были также обнаружены неорганические
материалы, называемые биоминералами, которые образуются путем осаждения
кристаллических неорганических соединений на органических темплатах, например на
коллагеновых волокнах. Из биоминералов состоят, в частности, кости и зубы,
осуществляющие такие важные функции, как опорная и защитная (например, скелет,
яичная скорлупа и т.д.), основанные на твердости неорганического компонента. В
табл. 6.6 приведен перечень наиболее важных биоминералов. Фактически подобные
материалы «сделаны» как из органического, так и из неорганического «сырья».
Сравнительно быстро образующиеся хитиновые (полисахаридные) структуры
беспозвоночных и скелеты акул состоят преимущественно из органического материала.
Однако кости позвоночных «построены» из гидроксиапатита Са]0(ОНJ(РО4N; при
этом в состав органической матрицы входят коллагеновые волокна, гликопротеины
и мукополисахариды. Неорганический компонент придает этим материалам
твердость и сопротивление давлению, тогда как присутствие органической матрицы
обеспечивает эластичность, а также прочность на разрыв, на изгиб и сопротивление
разрушению. Органический компонент часто выполняет также темплатную функ-
6.13. Биомиметические (биоподражательные) структуры
563
Таблица 6.6. Некоторые важные биоминералы
Состав
Минеральная форма
(фаза)
рК (показатель
растворимости) при
рН 7 (произведение
растворимости К , М)*1
Местонахождение,
применение
Карбонат кальция
СаСО3
Фосфат кальция
Са10(ОНJ(РО4N
Оксалат кальция
СаС2О4-лН2О
Сульфаты металлов
CaSO42H2O
SrSO4
BaSO4
Аморфный кремнезем
SiO27jH2O
(=SiO/)(OHL_2/))
Оксиды железа
Fe3O4
a,y-Fe(O)OH
5Fe2O3-9H2O
Кальцит
Арагонит
Фатерит
Аморфная
Гидроксиапатит
Уэвеллит
Уэдделлит
Гипс
Целестин
Барит
Аморфная
Магнетит
Гетит,
лепидокрокит
Ферригидрит
8.42
8.22
7.6
7.4
13.0
8.6
-
4.2
6,5
10.0
Растворимость
< ЮОмг-дм
—
-
-
Внешние скелеты
(например, яичная
скорлупа, кораллы,
раковины
моллюсков), весовые
датчики
Внутренние скелеты
(кости и зубы
позвоночных)
Камни в почках,
аккумулирование
кальция
Весовые датчики
Внешние скелеты
Весовые датчики
Створки диатомей
и радиолярий;
защитные функции
в растениях
Магнитные датчики,
зубы хитонов*2
Зубы хитонов
Аккумулирование
железа
*' Ksp- [катион] [анион], рА" = -\gK (действителен для гетерогенных равновесий с
незатрудненным обменом между твердой и растворенной фазами).
*2 Разновидность моллюсков.
цию, вынуждая неорганический твердый компонент принимать подходящую
структуру в соответствии с генетической предрасположенностью.
Для биоминерала существенными являются низкая растворимость при
физиологических условиях и хорошая биологическая доступность, поскольку требуются
его значительные количества. Интересный пример - наличие целестина SrSO4 во
внешних скелетах некоторых морских организмов. Морская вода не насыщена
этим минералом и поэтому его накопление возможно только с помощью
специальных приемов (ср. ионные насосы, гл. 2). После смерти морского организма его
внешний скелет быстро растворяется. Биоминералы выполняют несколько важных
функций:
• механическая опора и защита;
• инструменты и оружие (например, зубы);
6. Инженерия кристаллов
• сенсоры (например, плотные B.9 гсм~3) веретенообразные отложения
кальцита во внутреннем ухе способны улавливать ускорение);
• аккумулирование;
• детоксикация (осаждение токсических веществ, например CdS).
Биоминералы могут образовываться внутриклеточно, на клеточной мембране (эпи-
клеточно) или во внеклеточном пространстве. Наиболее примитивная форма
биоминерализации - это биологически индуцированная минерализация (в бактериях
и водорослях). Биоминералы образуются путем самопроизвольной, беспорядочной
кристаллизации во внеклеточном пространстве, часто — за счет газов, вовлеченных
в биологические процессы. Так, например, фотохимически активные водоросли
поглощают СО2 из воды, сдвигая приведенное ниже равновесие слева направо
вследствие образования нерастворимого СаСО3.
Са2+ + 2НСО3- & СаСО3(тв.) + СО2(г.)
Н2О
3 & СаСО3(тв.) + СО2(
Это равновесие, описывающее изменение жесткости воды. Его можно
рассматривать как продолжительное поглощение СО2, которое вызывает значительные
климатические и геологические последствия.
Многие важные процессы включают направленный рост биоминералов за счет
использования органической темплатной матрицы, позволяющей контролировать
процесс кристаллизации, причем нуклеация и рост кристалла происходят в
условиях пересыщения, активно поддерживаемого самим организмом. Одна из
возможных моделей образования зародышей кристаллизации и контролируемого (тем-
платного) роста кристаллов показана на рис. 6.42.
Анион
Катион
Центр связывания катионов (ср.: краун-эфиры)
Рис. 6.42. Схема темплатной биоминерализации. Полифункциональный органический
полимер с боковыми цепями — лигандными группами — связывает катионы в регулярные
структуры, согласующиеся со структурой лигандов. Неорганические анионы подходят для
координации к связанным катионам и, в свою очередь, начинают привлекать для
дальнейшей координации катионы из раствора. В результате образуется связанный с полимером
кристалл неорганического материала
6.13. Биомиметические (биоподражательные) структуры
165
В соответствии с этим механизмом происходит формирование удлиненных
костей у позвоночных. Кости, подобные человеческой бедренной кости, почти на 30%
состоят из фибриллярного белка (коллаген) и неорганических компонентов,
внедренных в цементирующие гликопротеины. Минеральные фазы включают в
основном гидроксиапатит, а также небольшие количества карбоната кальция,
кремнезема, карбоната магния, ионов других металлов и цитрата. Нити коллагена
переплетаются друг с другом и образуют волокна размером 1.3x300 нм. С помощью
31Р ЯМР-спектроскопии было показано, что кислые функциональные группы
фосфатов служат для «привязки» этого белка к неорганическому компоненту, в
результате чего образуется правильная минералоподобная цепочка ионов Са2+ (рис. 6.43).
Получение искусственных органических—неорганических темплатных
материалов по технологии, описанной выше, представляет собой интересную, но сложную
задачу. Конструирование нано- и микрометровых неорганических частиц,
имеющих отчетливо выраженную структуру, может найти широкое применение при
создании миниатюрных деталей и механизмов, а также привести к новым открытиям
в области биосинтеза природных «композиционных материалов». За эту задачу
взялся Дж. Озин (G. Ozin) из Университета Торонто (Канада). Он решил воссоздать
красивейшие природные кремниисодержащие микроскелеты морских организмов,
таких, как диатомеи и радиолярии (рис. 6.44, а). Эту область химии называют
морфосинтезом, что означает получение веществ заданной конфигурации или
формы. Используя дифильные молекулы, например дигидрофосфат дециламмония
CjqHjjNH^HjPO^ F.41), состоящие из длинного липофильного «хвоста» и
гидрофильной аммониевой «головки», Озин и сотрудники наблюдали в растворе
образование сферических везикул. При этом происходила агломерация молекул дифилов
таким образом, что их липофильные участки оказывались направленными внутрь,
к центру каждой везикулы, а группы NH^, наоборот, были направлены наружу, для
того чтобы взаимодействовать с водной средой (дифильные молекулы и везикулы
будут детально рассмотрены в разд. 10.2). Если эту реакцию проводят в смешанном
растворителе вода-гликоль в присутствии соединения алюминия (А1РО4), то неор-
Коллагеновые
волокна
Рис. 6.43. Темплатное
биоосаждение
кристаллического гидроксиапатита
Минеральная фаза
Органический материал
566
6. Инженерия кристаллов
(а)
Рис. 6.44. (а) Микроскелет дискообразной диатомеи. (Воспроизведено с разрешения
Simpson T.L. and Volcani BE. Silicon and siliceous structures in biological systems. New York:
Springer-Verlag, 1981). (б) Искусственная биомиметическая форма, напоминающая диато-
мею. (Воспроизведено с разрешения Oliver S. and Ozin G. Skeletons in the beaker — synthetic
hierarchical inorganic materials//Adv. Mater. (Weinhem Ger). 1995. Vol. 7. P. 943)
ганический алюминийсодержащий компонент будет осаждаться на поверхности
каждой везикулы, образуя биомиметические формы изумительной красоты. Озину
посчастливилось на базе фосфата алюминия впервые получить твердые
образования миллиметровых размеров (сфероиды и полые раковины), имеющие
микрометровые поверхностные узоры или фигуры: поры, чашевидные углубления,
криволинейные мозаики, звездчатые додекаэдры и т.д. (рис. 6.44, б).
но-
о
/ он
F.41)
Интересно, что эти структуры, образовавшиеся на везикулярных темплатах,
отлично воспроизводятся (всегда образуются сфероиды или раковины), однако они
не обнаруживают дальнего порядка, что вполне согласуется с динамической
природой образующихся в растворе везикул. Озин объяснил эти результаты двумя
причинами. Во-первых, многофункциональной ролью гликоля-сорастворителя, а
во-вторых, влиянием процессов, напоминающих динамику клеток, когда везикулы
слипаются друг с другом, распадаются на более мелкие, меняют форму и, наконец,
разрушаются. Озин полагает, что все эти события, происходящие с везикулами,
могут так или иначе влиять на поверхностные узоры образующихся
макроскопических форм. Дополнительное свидетельство в пользу такой везикулярно-темплатной
модели было получено в ходе РСА-исследования кристаллической структуры ди-
6.14. Смешанные кристаллы: включения типа «песочные часы»
1567
фила F.41). Его структура состоит из бислоев, в которых NH^-группы и
Н2РС>4 -анионы образуют твердый гидрофильный субслой, напоминающий глину.
Гидрофильные субслои чередуются с гидрофобными областями, образованными
«частоколом» параллельно расположенных децильных цепей.
*"* Ozin G.A. Morphogenesis of biomineral and morphosynthesis of biomimetic forms //
Ace. Chem. Res. 1997. Vol. 30. P. 17-27.
6.14 Смешанные кристаллы:
включения типа «песочные часы»
В настоящей главе мы имели дело с образованием в основном чистых
кристаллов, хотя иногда они содержали два или более компонентов. Тем не менее в
разд. 6.1.5.2 мы отмечали, что случайный выбор между кристаллическими
решетками двух компонентов может привести к нестехиометрическим фазам, в которых
одна кристаллическая решетка является темплатной для образования другой. В тех
случаях, когда два указанных компонента не разделены на четко видимые домены,
можно прийти к ошибочному выводу о полном беспорядке в системе, поскольку
исследования строения кристаллов дают структуры, усредненные по всем
элементарным ячейкам данного кристалла. Поведение подобного рода — причина такого
явления, как изомерия, обусловленная растяжением связей, которое было
обнаружено в конце 1980-х годов (Дополнение 6.3). Однако, когда это явление становится
более отчетливо выраженным, оно приводит к образованию смешанных
кристаллов с очень интересными свойствами, в особенности в случае неорганических
полупроводниковых систем. Одним из наиболее наглядных примеров смешанных
кристаллов являются соединения включения типа «песочные часы» (HI).
Соединения включения типа «песочные часы» образуются за счет вкрапления
небольших количеств растворимых в воде молекул органических красителей типа
арилсульфонатов F.42) и F.43), в кристаллическую решетку таких неорганических
веществ, как K2SO4 или КН2РО4. Включение подобного красителя, которое
является нестехиометрическим и обычно имеет место в одной из тысячи элементарных
R
561
6. Инженерия кристаллов
Рис. 6.45. Схематическое изображение
окрашенных и неокрашенных участков, характерное
для соединения включения типа «песочные часы»
ячеек, легко обнаружить по окраске определенных областей неорганического
кристалла, которая придает этому кристаллу форму «песочные часы» (рис. 6.45).
Данные о включении дихроичного фуксина F.42) в кристаллическую решетку
сульфата калия были впервые опубликованы X. Бакли (Н. Buckley) в 1934 г., хотя
соединения включения типа «песочные часы» известны с 1854 г. Появление
современных методов исследования структуры позволило провести тщательный анализ
известных соединений включения этого типа, а также получить новые. Краситель
F.42) встраивается в растущую грань {110} кристалла K2SO4 благодаря
соответствию расположения групп SO^~ красителя положениям сульфатных групп в
кристаллической решетке K2SO4. Эта жесткая неорганическая решетка вынуждает
молекулы красителя осаждаться и удерживаться в четко определенной организации лишь
на комплементарных гранях. Измерение поляризационных спектров поглощения
для кристаллов с соединениями включения типа «песочные часы»,
ориентированных известным образом, позволило Б. Кару (В. Kahr) и его сотрудникам из
Университета Вашингтона (США) сопоставить экспериментальные и рассчитанные
спектры поглощения и на этом основании подтвердить модель «соответствия центров»,
описывающую процесс включения данного красителя в данную неорганическую
матрицу. Вооружившись сведениями о размерах и геометрии центра, который
включает молекулу красителя, Кар продолжил работу по подбору следующего
красителя - F.43), который, как оказалось, еще лучше подходил к данной
неорганической решетке (рис. 6.46). Так удалось получить соединение включения с молекулой
этого красителя, и это был первый синтез соединения включения типа «песочные
часы». Более поздние исследования были посвящены получению соединений
включения этого типа с целым рядом красителей (рис. 6.47), которые поглощались
различными гранями растущих кристаллов сульфата калия в зависимости от того,
соответствуют ли положения сульфонатных групп того или иного красителя
положениям сульфат-ионов в кристаллической решетке K2SO4. На рис. 6.48 показаны
различные варианты такого соответствия.
= 5.8 А, у = 7.5 А
г = 9.5А
х = 5.4 А, у = 7.7 А
г = 9.зА
Рис. 6.46. Соответствие решеток K2SO4
и красителя F.43). (Воспроизведено с
разрешения Kelly M.P. et al., 1994)
'O3S,
,SGR
N M N M = Co
V
O3S NH2 H3CO
Рис. 6.47. Соединения включения типа «песочные часы» на основе K2SO4 и ряда цветных красителей. (Воспроизведено с разрешения
Kahr В. and Subramony J.A.)
570
6. Инженерия кристаллов
NO
_._-V.Fe3+
SO,
SO
EtjN
SO,
sor
NH,
Рис. 6.48. При обычных условиях (температура, давление, влажность) кристаллы K2SO4
характеризуются формами {021}, {ПО}, {001} и {010} в зависимости от пространственного
соответствия положений сульфонатных групп красителя положениям сульфат-ионов в
кристаллической решетке. (Воспроизведено с разрешения Kahr В., Jang S.-H., Subramony J.A. et
al. Dyeing salt crystals for optical applications // Adv. Mater. 1996. Vol. 8. P. 941-944)
•""» Kahr В., Jang S.-H., Subramony J.A. et al. Dyeing salt crystals for optical
applications// Adv. Mater. 1996. Vol 8. P. 941-944.
Дополнение 6.З. Изомерия, обусловленная растяжением связей
Изомерию, обусловленную растяжением связей, первоначально определяли как
существование двух четко разделенных энергетических минимумов вдоль
пространственной координаты простой связи. Были предложены гипотетические примеры типа
соединения F.44). Действительно, гексаметилбензол (бензол Дьюара F.45)) является
реальным примером, для которого превращение в более стабильную форму Кекуле
запрещено по соображениям симметрии. Тем не менее довольно долго считалось, что,
если исключить специальные случаи, подобные только что рассмотренному, изомеры,
обусловленные растяжением связей, в обычных условиях не могут существовать в ви-
6.14. Смешанные кристаллы: включения типа «песочные часы»
571
де отдельных форм. Эта точка зрения была опровергнута в 1970 г. — после выделения
и исследования целого ряда соединений, подобных [Mo(O)Cl2(PMe2PhK] F.46). Эти
соединения существовали в двух формах, весьма различающихся по цвету: голубой и
зеленой, причем первоначально считалось, что различная окраска относится к цис- и
/и/гянс-геометрическим изомерам.
F.44)
R3P"<.,,.
Mo.
С)
•ci
F.45)
..,>>» PR3
цмс-F.46)
транс-FА6)
Однако исследование этих родственных комплексов с помощью рентгеновской
дифракции показало, что и голубая, и зеленая формы, скорее всего, являются цис-
изомерами (зеленая форма F.46) не кристаллизовалась, однако аналогичный зеленый
комплекс РЕ12Рп-производного оказался ^wc-формой). Эти два образца очень мало
отличались друг от друга, небольшие различия были найдены лишь в фосфиновых ли-
гандах . В то же время обнаружили, что во всех зеленых формах длина связи Мо=О
A.801 А) значительно превышает дли ну этой же связи в голубых формах A.676 А).
После довольно тщательной работы и дополнительных спектроскопических
исследований стало ясно, что эти образцы относятся к изомерам, различающимся длинами
связей. Изучение подобных материалов и родственных образцов, таких, как
[NbECl3(PMe3K], где Е = О или S, существующих в виде окрашенных форм с сильно
различающимися длинами связей, становится актуальным в конце 1980-х и в начале
1990-х годов. Тем не менее некоторые исследователи никак не хотели признать
существование столь удивительного эффекта, поскольку его невозможно было объяснить
ни с точки зрения химии, ни с точки зрения упаковки кристаллов, хотя он и не
противоречил представлениям, основанным на теоретическом подходе Хюккеля.
Детальное исследование состава голубой и зеленой форм РМе3-производных соединения
F.46) показало, что из зеленых форм можно выделить небольшую желтую фракцию,
которая была идентифицирована как [MoG3(PMe7PhK], и чистое голубое соединение
с более короткой связью, чем в истинном г<ш>соединении F.46). Совместная
кристаллизация желтого трихлорпроизводного с голубым образцом дает кристалл
зеленого цвета, причем наблюдаемая для последнего длина связи Мо=О фактически
представляет собой среднее значение относительной заселенности хлорида (истинная
длина связи Mo—C1 составляет 2.4 А) и оксида в узле решетки Мо(С1)=О. Доказатель-
572
6. Инженерия кристаллов
ство наличия такой примеси — относительное удлинение анизолропного
температурного эллипсоида для «зафязненного» оксидного центра. При этом фафик в
координатах «наблюдаемая длина связи Мо=О — фракционная заселенность хлорида» может
быть построен на основании данных, полученных для специально «зафязненных»
образцов (рис. 6.49). Было установлено, что подобная совместная кристаллизация
является причиной изомерии, обусловленной растяжением связей, также и для системы
[NbECl3(PMe3K], которая, как оказалось, содержит следы [NbCl4(PMe3K]. В
настоящее время изомерия такого рода — общепризнанный артефакт, характерный для
смешанных кристаллов. В то же время полемика по поводу этого явления помогла
установить важную истину: гомогенные кристаллы не обязательно представляют собой
химически чистое соединение.
2.4
1.6
0.0
0.2 0.4 0.6 0.8
Доля оксокомплекса
Рис. 6.49. Расстояние Мо(С1)=О как функция заселенности кислородного узла
кристаллической решетки в соединениях типа F.46). (Воспроизведено с
разрешения Gibson V.C. and McPartlin M. Bond stretch isomerism: Fact or artefact? J. Chem.
Soc, Dalton Trans. 1992. P. 947-956)
*""* Gibson V.C. and McPartlin M. Bond stretch isomerism: Fact and artefact? // J. Chem.
Soc, Dalton Trans. 1992. P. 947-956.
Учебные задания
6.1. Предложите структуры твердых соединений, которые могут соответствовать
следующим наборам графов и молекулам:
a) R|(8) (уксусная кислота);
бЖ66A2)(фенол);
в) R|F) ( нитробензол + анилин, 1:1);
г) R^D) (нитробензол + фенол, 1:1);
Учебные задания 573
д) S|E) ([4]резорцинарен);
е) СD) B-нитроацетанилид);
ж) SF) B-нитроацетанилид).
О
2-Нитроацетанилид
О
6.2. Установите единичный и бинарный наборы графов для пар оснований нуклео-
тидов, изображенных на рис. 2.30 и 2.31. Будет ли полезен анализ наборов
графов для того, чтобы провести различие между парами оснований Уотсо-
на-Крика и парами оснований Хугстена?
6.3. Укажите основные особенности, характерные для сильных, средних и слабых
водородных связей. Как Вы думаете, какие водородные связи будут наиболее
полезны при конструировании кристаллов?
6.4. Объясните тот факт, что бензол кристаллизуется в «ёлочной» форме
(рис. 1.17), тогда как высшие ароматические углеводороды предпочитают
структуры у-типа или типа графита.
6.5. Методом рентгеновской дифракции установлено, что при 298 К ОН-связь
имеет длину 0.85 А. При 100 К такая связь в структуре этого же соединения
имеет длину 0.92 А. Объясните это, а также, используя элементарную
тригонометрию, оцените угол, на который отклоняется ОН-связь при маятниковых
колебаниях и 298 К. (Колебаниями при 100 К можно пренебречь.) Как можно
точно измерить длину этой связи?
6.6. Расположите следующие кислоты, способные образовывать водородные
связи, в ряд в порядке увеличения силы их водородных связей: МеОН,
Ме2Р(О)ОН, RCO2H, CF3OH, MeNH2, Me2NH, PhOH, PhNH2, CH2C12,
CHC13, MeSH, C6H6, Me(CH2LMe, MeOMe, Me2NH+, Н3О+.
6.7. Предложите способы, при помощи которых можно экспериментально
установить различие между связями Х-Н в алканах, спиртах и аминах, которые:
а) являются свободными; б) участвуют в агостическом связывании; а)
участвуют в межмолекулярном псевдоагостическом взаимодействии.
6.8. Предскажите общий вид 'Н ЯМР-спектра для протонов Н1-Н4 следующего
соединения: а) в статической форме; б) при быстром вращении вокруг
вектора 1г-С5; в) при быстром обмене протонов Н'иН2.
574
6. Инженерия кристаллов
Мысленный эксперимент
Почему водородные связи в супрамолекулярной химии рассматривают как
«универсальные взаимодействия»? Составьте перечень характеристик, при помощи
которых можно отличить водородные связи от других нековалентных взаимодействий.
Литература
Abad, 1999 Abac/ M.M., Atheaux I., Maisonnat A., Chaudret B. Control of proton transfer by
hydrogen bonding in the protonated forms of the binucleophilic complex
[{n5-C5H4CH(CH2LNMe}Ir(PPh3)H2].//Chem. Commun. 1999. P. 381.
Allen, 1996 Allen F.H., Howard J.A.K., Hoy V.J. et al. First neutron diffraction analysis of an
O—H- л hydrogen bond: 2-Ethynyladamantan-2-ol. // J. Amer. Chem. Soc.
1996. Vol. 18. P. 4081.
Aspley, 1999 Aspley C.J., Boxwell C, Bull M.L. et al. A new combination of donor and
acceptor: Bis(rN-benzene)chromium and hexafluorobenzene form a charge-transfer
stacked crystal. // Chem. Commun. 1999. P. 1027.
Braga, 1994 Braga D., Grepioni F., Sabatino P., Desiraju G.R. Hydrogen-bonding in
organometallic crystals. 1. From carboxylic acid and alcohols to carbonyl-com-
plcxes. // Organometallics. 1994. Vol. 13. P. 3532.
Braga, 1995a Braga D., Grepioni F., Tedesco E., OrpenA.G. Crystal construction and molecular
recognition for [Cr(CON]. //J. Chem. Soc, Dalton Trans. 1995a. P. 1215.
Braga, 1995b Braga D., Grepioni F., Biradha K. ct al. Hydrogen-bonding in organometallic
crystals. 2. С—Н---0 hydrogen-bonds in bridged and terminal first-row metal-car-
bonyls.//J. Amer. Chem. Soc. 1995b. Vol. 117. P. 3156.
Braga, 1997 Braga D., Grepioni F., Tedesco E. et al. Hydrogen bonding in organometallic
crystals. 6. X— H-M hydrogen bonds and M-(H—X) pseudo-agostic bonds. //
Organometallics. 1997. Vol. 16. P. 1846.
*
Braga, 1998 Braga D., Grepioni F., Novoa J.J. Interanion О—Н---О" hydrogen bond like
interactions: The breakdown of the strength-length analogy. // Chem. Commun. 1998.
P. 1959.
Brunet, 1997 Brunei P., Simard M., Wuest J.D. Molecular tectonics. Porous hydrogen-bonded
networks with unprecedented structural integrity. // J. Amer. Chem. Soc. 1997.
Vol. 117. P. 2737.
Desiraju, 1989 Desiraju G.R., Gavezzotti A. From molecular to crystal-structure — polynuclear
aromatic hydrocarbons. //J. Chem. Soc, Chem. Commun. 1989. P. 621.
Ermer, 1994 Ermer O., Eling A. Molecular recognition among alcohols and amines — super-
tetrahedral crystal architectures of linear diphenol-diamine complexes and
aminophenols// J. Chem. Soc, Perkin Trans. 1994. P. 925.
Etter, 1986 Etter M.C., Urbanczyk-Lipkowska Z, Jahn D.A., Frye J.S. Solid-state structural
characterization of 1.3-cyclohexanedione and of a 6-1 cyclohexanedione-benzene
cyclamer, a novel host guest species. // J. Amer. Chem. Soc. 1986. Vol. 108.
P. 5871.
Литература
575
Hippel von, 1962 Hippel A.R. von. Molecular designing of materials. // Science. 1962. Vol. 138.
P. 91.
Hunter, 1994 Hunter R., Haueisen R.H., Irving A. The first water-dependent liquid clathrate —
X-ray evidence in the solid for а С—Н---Л (heteroarene) interaction. // Angew.
Chem., Int. Ed. Engl. 1994. Vol. 33. P. 566.
Kelley, 1994 Kelley M.P., Janssens В., KahrB., Vetter W.M. Recognition of dyes by K2SO4
crystals — choosing organic guests for simple salts. // J. Amer. Chem. Soc. 1994.
Vol. 116. P. 5519.
Loehlin, 1998 Loehlin J.H., Franz K.J., Gist L, Moore R.H. Supramolecular alcohole-amine
crystals and their hydrogen-bond patterns. // Acta Crystallogr. B. 1998. Vol. 54.
P. 695.
Mascal, 1998 MascalM. Statistical analysis of C---N hydrogen bonds in the solid state: There are
real precedents. // Chem. Commun. 1998. P. 303.
Nyburg, 1985 Nyburg S.C., Faerman C.H. A revision of van der Waals atomic radii for
molecular-crystals — N, O, F, S, Cl, Se, Br and I bonded to carbon. // Acta Crystallogr.
B. 1985. Vol. 4. P. 274.
Orlova, 1999 Orlova G, Scheiner S., Kar T. Activation and cleavage of H—R bonds through
intermolecular H—H bonding upon reaction of proton donors HR with 18-elec-
tron transition metal hydrides. // J. Phys. Chem. A. 1999. Vol. 103. P. 514.
Power, 1998 Power K.N., Hennigar T.L., Zaworotko M.J. X-Ray crystal structure of
{Cu[l,2-bisD-pyridyl)ethane]2(NO3J}w: The first example of a coordination
polymer that exhibits the NbO 3D network architecture. // Chem. Commun.
1998. P. 595.
Reddy, 1993a Reddy D.S., Goud B.S., Panneerselvam K. et al. C-H-N mediated hexagonal
network in the crystal-structure of the 1/1 molecular-complex 1,3,5-tricyanobenzene
hexamethylbenzene. // J. Chem. Soc, Chem. Commun. 1993a. P. 663.
Reddy, 1993b Reddy D.S., Panneerselvam K., Pilati Т., Desiraju G. Molecular tapes based on
CN-C1 interactions. //J. Chem. Soc, Chem. Commun. 1993b. P. 661.
Schmidt, 1964 Schmidt GM.J. Topochemistry. Part III. The crystal chemistry of some trans-cyn-
namic acids. //J. Chem. Soc. 1964. P. 2014.
Schmidt, 1996 Schmidt M. U., Englert U. Prediction of crystal structures. // J. Chem. Soc, Dalton
Trans. 1996. P. 2077.
Taylor, 1984 Taylor R., Kennard O., Versichel W. The geometry of the N-H-O=C hydrogen-
bond. 3. Hydrogen-bond distances and angles. // Acta Crystallogr. B. 1984.
Vol. 40. P. 280.
Weber, 1988 Weber E., Csoregh I., Ahrendt J. et al. Design of roof-shaped clathrate hosts -
inclusion properties and X-ray crystal-structures of a free host and of inclusion-
compounds with 1-BuOH and DMF. // J. Org. Chem. 1988. Vol. 53. P. 5831.
Yaghi, 1995 Yaghi O.M., Li G., Li H. Selective binding and removal of guests in a microporous
metal-organic framework. // Nature, 1995. Vol. 378. P. 703.
7
Темплаты и самосборка
Из комнаты в комнату я блуждаю,
И мой хозяин меня не замечает,
И по сию пору я не знаю:
Толь пленник тут я, то ли гость.
У. Уотсон A858-1936). Странности мира
Введение
*~" Philip D. and Stoddart J.F. Self-assembly in natural and unnatural systems // Angew.
Chem., Int. Ed. Engl. 1996. Vol. 35. P. 1155-1196.
• 1.1 Цели и задачи
Синтез даже низкомолекулярных веществ методами традиционной
органической или координационной химии зачастую может казаться
утомительным и однообразным процессом. С каждой стадией синтеза все больше и больше
продукта теряется, даже на относительно высокопроизводительных этапах, а цена -
и материалов, и труда специалиста, осуществляющего синтез, — резко возрастает.
В то же время основная цель родственных химии дисциплин, например
химической технологии, — максимальное упрощение химических синтезов. Однако
такой подход сегодня использовать невозможно, так как для решения современных
задач необходимы специалисты высокого класса, способные создавать
уникальные химические соединения. В полной мере это относится к получению интерме-
диатов и больших молекул, молекулярных систем и наноразмерных машин.
Создание таких больших агрегатов, чему многие специалисты в области
супрамолекулярной химии придают большое значение, проложит путь к
конструированию новых ультраминиатюрных компонентов для компьютерных,
электронных и оптических устройств (гл. 8). К настоящему времени уже проделана
огромная работа по созданию «запрограммированнных систем», в которых небольшие,
легко получаемые молекулярные компоненты автоматически собираются вместе,
образуя намного более крупные и более сложные агрегаты. Под термином
«запрограммированная система» мы понимаем такую химическую систему, в природе
молекулярных строительных блоков которой (в смысле размера, формы, симметрии
и электронных свойств их активных центров) содержится вся информация,
необходимая для получения желаемой суперструктуры. Такой супрамолекулярный
ансамбль собирается самостоятельно, т.е. без участия (использования) каких-либо
внешних воздействий (факторов).
7.1. Введение 577
Этот подход имеет принципиальное значение для понимания соотношения
между размерами структур и компонентов, которые, с одной стороны, принадлежат
к миру молекул, а с другой - используются в современной литографической
технике (в частности, в электронной промышленности). Производство электронных
молекулярных устройств должно вызвать небывалый скачок в скорости и
вычислительных возможностях компьютеров, а также расширить области применения
таких устройств из-за их небольших размеров. В самом деле, компьютерный
жесткий диск уже имеет считывающую головку, располагающуюся на высоте всего 25 нм
(или 200 атомов) над его поверхностью. Для функционирования молекулярные би-
стабильные устройства (переключатели) должны быть снабжены инфраструктурой
вход/выход, необходимой для их соединения с внешним миром. Более того, они
должны быть полностью управляемыми, обратимыми и удобными в обращении в
течение миллионов циклов «вкл. — выкл.». С одной стороны, современная
литографическая гравировальная техника позволяет производить компоненты кремниевых
плат размером примерно до 0,1 мкм (т.е. 1000 А, или 100 нм). И даже на этом уровне
мы сталкиваемся с проблемами туннелирования электронов и трудностями отвода
тепла, а также с более тонкими эффектами, такими, как квантово-механические
ограничения по носителям заряда, т.е. с влиянием размера компонента на
энергетическую диаграмму. С другой стороны, наибольшая на сегодняшний день
синтезированная и охарактеризованная супрамолекула имеет размеры ~10 нм и обладает
некоторыми полезными свойствами. Однако их недостаточно для выполнения тех
условий, которые требуются при производстве реальных приборов. Это
предполагает, что точка пересечения между миром электронной технологии и миром
химического синтеза находится в области размерности 10—1000 нм, в которой
функциональные супрамолекулы с четко определенными структурой и связями могут стать
основой для электронного конструирования нового типа. Прецедент уже имеется в
мире молекулярной биологии (гл. 2), где функциональные молекулярные
устройства размерами 1-10000 нм регулируют всю химию жизни. В самом деле, не
существует фундаментальной причины, почему искусственные молекулярные устройства
не могут действовать подобным образом. Подавляющее большинство таких
биохимических систем образуется путем самосборки, что обеспечивает экономное
потребление генетической информации, необходимой для их синтеза (разд. 7.2).
Возвращаясь к проблеме соотношения между современной электронной и химической
технологиями, следует выделить два различных подхода: «конструирование вниз»*
и «синтез вверх»**. Мы уже видели, что для реализации первого подхода —
«конструирование вниз» — возможностей очень мало вследствие современных
технологических (и не только) ограничений. Таким образом, остается второй подход —
разработка технологии «синтеза вверх» в требуемом диапазоне размеров и
функциональности с помощью супрамолекулярной химии.
С концепцией самосборки перекликается идея самовоспроизводства
(репликация): возможности молекулы или организма создавать собственную копию. Анало-
* Уменьшение размеров компонентов электронных устройств. {Примеч. ред. перевода.)
Синтез больших функциональных молекул или молекулярных ассоциатов, способных
выполнять заданные операции. {Примеч. ред. перевода.)
7-СтадДж В.
|i|78 7• Темплаты и самосборка
гия с биологическим (неполовым) воспроизводством очевидна, и возможность
самовоспроизводства была предложена в качестве основного критерия жизни.
Огромное количество примеров в природе демонстрирует, что самовоспроизводство —
это реальное и на самом деле ничем не примечательное событие на молекулярном
уровне. Деление биологической клетки включает расплетение двойной спирали
ДНК и темплатный синтез дочерней цепи путем спаривания оснований за счет
межцепных водородных связей. В результате образуется точная копия каждой
отдельной цепи, а следовательно, и две новые двойные спирали. Единичная цепь
ДНК содержит всю необходимую информацию для образования своей точной
копии без ошибок и вариаций. Для сравнения: обычный синтез даже небольшого
полинуклеотида постадийной конденсацией фосфатных остатков представляет
собой фундаментальную задачу. В самом деле, решение этой задачи при синтезе
олигонуклеотида путем полимеразной цепной реакции (polymerase chain reaction,
PCR) — процесса, при котором небольшие количества олигонуклеотидов темплати-
руют образование большого числа собственных копий, — послужило основанием
для присуждения в 1993 г. Нобелевской премии по химии К. Мюллису (К. Mullis) за
его работу в этой области (см. разд. 2.7.3). Мы уже ознакомились с действием тем-
платного эффекта (см. разд. 3.8), когда катион металла используется в качестве
простой сферической матрицы для сборки макроциклических органических лигандов.
В этой главе мы рассмотрим намного более общие области применения темплатно-
го эффекта и его предтечи - самосборки, включая синтез молекулярных узлов, ка-
тенанов и ротаксанов, а также попытки получения искусственных абиотических
(небиологических) самовоспроизводящихся систем.
.1.2 Терминология
Перед тем, как мы познакомимся с природными и синтетическими
темплатно-собирающимися и самособирающимися системами, важно
договориться об определениях, которыми мы будем пользоваться. Большая часть
терминологии супрамолекулярной химии все еще не устоялась, и многие термины еще не
имеют четких определений. Ж.-М. Лен (J.-M. Lehn) предложил иерархический порядок
терминов «темплатирование» (супрамолекулярное содействие синтезу, шаблониро-
вание), «самосборка» и «самоорганизация». Вместе эти термины охватывают
процессы, предоставляющие возможность запрограммированным молекулярным
компонентам, или тектонам (от греч. «tekton» — строитель), объединяться
самопроизвольно, строго определенным путем, образуя ансамбли, организация
которых происходит в одном, двух или трех измерениях и, возможно, (хотя и не
обязательно) во времени.
Мы уже убедились, что темплатный эффект (см. разд. 3.8) полезен в синтезе
благодаря временному или постоянному привлечению «вспомогательных» частиц
(например, катиона щелочного металла). Самосборка не обязательно должна включать
стадию темплатирования с участием, например, катиона металла. Поэтому
собственно темплатный эффект еще не является самосборкой, но, по определению
Лена, ее можно рассматривать как «отдельную стадию самосборки, которая содержит
несколько стадий, протекающих спонтанно в рамках одной операции». Среди про-
7.1. Введение
579 i
цессов самосборки мы должны различать молекулярную самосборку и супрамоле-
кулярную самосборку. Молекулярная самосборка включает образование ковалент-
ных связей как часть определенной химической процедуры, контролируемой сте-
реохимическими параметрами реакции и конформационными характеристиками
интермедиатов, например при получении макроциклов в результате конденсации
аминов и альдегидов (см. схему 3.20) или криптандов (см. гл. 3). Супрамолекулярная
самосборка подразумевает основанную на узнавании обратимую спонтанную
ассоциацию ограниченного числа тектонов (молекулярные компоненты),
протекающую под контролем сравнительно лабильных межмолекулярных нековалентных
взаимодействий, таких, как координационные взаимодействия, водородные связи
и диполь-дипольные взаимодействия. Обратимость супрамолекулярной
самосборки — важное свойство конечных систем выбирать имеющиеся в распоряжении
компоненты для образования термодинамически наиболее выгодной структуры. Все
это обеспечивает возможность «залечивания» и самоисправления дефектов, как в
биологических системах.
Интересен граничный случай между молекулярной (ковалентной) и
супрамолекулярной самосборками при образовании фуллеренов, в частности С60 и С70, и
родственных веществ, таких, например, как протяженные углеродные нанотрубки
(рис. 7.1), протекающем в парах утлерода при высоких температурах. Хотя, строго
говоря, это пример необратимого образования ковалентных связей, однако в таких
экстремальных условиях возможно обратимое образование даже сильных
ковалентных связей, что в некоторой степени роднит их с более слабыми супрамолекуляр-
ными взаимодействиями, реализующимися в обычных условиях.
Рис. 7.1. Ковалентная самосборка
фуллеренов и углеродных нанотрубок в экстремальных
условиях
ggQ 7. Темплаты и самосборка
Наконец, термин самоорганизация включает и взаимодействие между
составными частями самособирающихся объектов, и объединение этих взаимодействий,
приводящее к явлениям коллективного характера, как, например, при фазовых
переходах.
*~* Lindsey J.S. Self-assembly in synthetic routes to molecular devices. Biological
principles and chemical perspectives: A review // New J. Chem. 1991. Vol. 15. P. 153-180.
Биохимическая самосборка
. 2.1 Вирус табачной мозаики
Мы уже рассмотрели некоторые основные аспекты биохимической
самосборки в разд. 2.8. Однако в биохимии можно найти примеры некоторых
объектов, свойства которых легко интерпретировать в понятиях супрамолекулярной
химии. Один из таких объектов — вирус табачной мозаики. Эта система состоит из
спиральных частиц вируса размером ~300 х 18 нм (рис. 7.2). Центральная цепь РНК
заключена в оболочку из 2130 идентичных белковых субъединиц, каждая из которых
включает 158 аминокислот. По сравнению с чисто химическим объектом этот вирус
замечателен тем, что если его разобрать на составные части и эти фрагменты
смешать в физиологических условиях, то частица вируса в точности вновь
самособерется и вновь обретет присущие ей свойства. Механизм этого впечатляющего примера
самосборки хорошо известен и состоит в образовании дисковидного модуля путем
соединения двух слоев из оболочных белковых субъединиц. Этот белковый диск
затем преобразуется в два витка спирали при продевании петли РНК в центральное
отверстие диска. Процесс повторяется путем нанизывания следующих дисков до тех
пор, пока вирусная частица не будет полностью сформирована (рис. 7.3).
(а)
ioooA
Рис. 7.2. (а) Электронная микрофотография и (б) схематическое представление вируса
табачной мозаики. (Воспроизведено с разрешения Philip D. and Stoddart J.F. Self-assembly
in natural and unnatural systems//Angew. Chem., Int. Ed. Engl. 1996. Vol. 35. P. 1155-1196)
1.2. Биохимическая самосборка
Рис. 7.3. (а)—(г) Поэтапная самосборка вируса табачной мозаики. (Воспроизведено с
разрешения Philip D. and Stoddart J.F. Self-assembly in natural and unnatural systems // Angew.
Chem., Int. Ed. Engl. 1996. Vol. 35. P. 1155-1196)
Преимуществом такого модульного подхода к сборке вируса, при которой идеи
тичные субъединицы собираются в конечный объект, является то, что для програм
мирования сборки ансамбля требуется намного меньше генетической информа
ции, чем для кодирования локализации каждого остатка индивидуально
Формирование вируса табачной мозаики служит иллюстрацией той роли, которун
в природе играют нековалентные взаимодействия. Весь процесс самосборки управ
ляется взаимодействиями, не более сильными, чем водородные связи, электроста
тические и сольватофобные эффекты или другие очень слабые силы, действующи!
согласованно. Конечная структура представляет собой термодинамически выгод
ный продукт, а сам процесс имеет высокую константу равновесия (константа обра
зования). Слабая природа таких взаимодействий подразумевает, что если в процес
се сборки будет совершена ошибка, то она будет исправлена автоматическ!
благодаря тому, что процесс обратим, а «ошибочный» продукт просто не столь ста
билен, как результат правильной сборки.
.2,.2, Строгая самосборка
Из-за использования исключительно нековалентных взаимодействий
самосборку вируса табачной мозаики считают примером строгой самосборки, в
противоположность самосборке с ковалентной модификацией. В процессе строгой
самосборки конечный продукт образуется исключительно самопроизвольно, если
компоненты смешаны в правильных отношениях при определенном наборе условий
температура, рН, концентрация и т.д. Образование продукта должно быть
полностью обратимым и термодинамически выгодным. В сущности, вся информация
необходимая для протекания сборки, закодирована в ее составных частях.
Самый знаменитый пример процесса строгой самосборки - образование
двойной спирали ДНК (см. рис. 2.25) путем спонтанной ассоциации (за счет
водородных связей) комплементарных пар нуклеооснований, например, таких, как гуаниь
и цитозин (см. рис. 2.29). Организация структуры двойной спирали типа «застежка-
молния» очень интересна, так как с термодинамической точки зрения ассоциации
двух цепей спирали приводит к термодинамически невыгодному понижению
энтропии, означающему, что этот процесс требует преодоления значительного
термодинамического барьера. Исследование сравнительно небольших модельных
систем, например комплементарной пары нуклеиновых кислот на основе аденина и
урацила, (АI7 и (UI7, показывает, что сплетение двух цепей с образованием двои-
582
7. Темплаты и самосборка
А—
А—
А-
А—
А—
А
А—
А—
А—
U—
U—
и—•
и—
и
и—
и—
и—
и—
Нуклеация
—А И—
А и
А U—
— А U —
А U
А U —
|—А U —
\—А U —
L_A JJ
Распространение
спаривания оснований
Рис. 7.4. Стадии самосборки двойной спирали нуклеиновых кислот путем спаривания
оснований аденин—урацил
ной спирали — двухстадийный процесс, на первой стадии которого происходит
нуклеация (точка репликации), а на второй — распространение процесса
спаривания оснований до его завершения (рис. 7.4). Первая стадия, обеспечивающая
начальный контакт между двумя цепями, является доминирующей с точки зрения
термодинамически невыгодной энтропии, тогда как выигрыш в энтальпии,
обусловленный образованием водородных связей между небольшим числом пар оснований,
Дестабилизация
5 6
Число пар
оснований
Рис. 7.5. Зависимость
энергетических характеристик
самосборки двойной спирали
нуклеиновых кислот от числа
спаренных оснований
7.2. Биохимическая самосборка 583
сравнительно незначителен. После завершения первой стадии и образования
контакта между цепями за счет объединения нескольких пар оснований происходит
ассоциация каждой новой пары оснований, что приводит к дальнейшему увеличению
выигрыша энтальпии с незначительным изменением энтропии. Так формируется
двойная спираль (рис. 7.5).
У. 2.3 Самосборка с ковалентной модификацией
Термин «самосборка с ковалентной модификацией» в основном
применяют к системам, в которых образование ковалентных связей является
необратимым. Поэтому, в отличие от точной самосборки, конечный продукт не обязательно
должен быть системой с термодинамическим минимумом. На рис. 7.6 изображено
схематическое представление концепции самосборки этого типа, включающей
модификацию предшественника. Два самособирающихся компонента не могут про-
взаимодействовать, прежде чем не произойдет ковалентное химическое
превращение одного из них. Это может приводить к изменению его размера, формы или
ориентации, а также к удалению блокирующего элемента. После этого две
субъединицы готовы к самосборке. Самосборка с ковалентной модификацией может также
включать послесборочную обработку. В этом случае для сборки прекурсорного
комплекса, фиксируемого затем в желаемом состоянии ковалентной модификацией,
используются нековалентные силы. Такую процедуру можно рассматривать как
разновидность темплатного эффекта, когда части структуры, необходимые для темпла-
тирования исходной самосборки, можно впоследствии отщепить. Превосходным
биологическим примером этого является биосинтез гормона млекопитающих —
инсулина. Инсулин состоит из двух полипептидных цепочек (А и В), связанных парой
дисульфидных (-S-S-) мостиков. Восстановление этих мостиков ведет к распаду
молекулы инсулина. Однако, в противоположность вирусу табачной мозаики,
индивидуальные полипептидные цепочки не содержат информации, необходимой для
Ковалентная модификация
\
Самосборка
Рис. 7.6. Комбинация ковалентной модификации и самосборки
7. Темплаты и самосборка
А
-S-S-
Препроинсулин Проинсулин Инсулин
Рис. 7.7. Биосинтез инсулина самосборкой с последующей ковалентной модификацией
самосборки, и поэтому окисление продуктов восстановления не приводит к
образованию активного инсулина. Фактически инсулин синтезируют из полипептида
намного большего размера — препроинсулина — в две стадии с посттрансляционной
обработкой. Препроинсулин способен к самосборке с использованием нековалент-
ных сил в такой конформации, в которой фрагменты А и В синтезируемого
инсулина размещены в нужной относительно друг друга ориентации. Затем две цепи
необратимо сшиваются за счет образования двух дисульфидных связей, при этом
образуется проинсулин. Далее после надежного соединения цепей избыточную
часть полипептида можно удалить с образованием конечного продукта (рис. 7.7).
7.3 Самосборка в синтетических
системах: кинетический
и термодинамический подходы
1 Темплатные эффекты в синтезе
Различия между кинетическим и термодинамическим темплатными
эффектами в синтезе макроциклов были рассмотрены в разд. 3.8. Под действием
кинетического темплатного эффекта распределение продуктов реакции
циклизации претерпевает изменение из-за связывания реагирующих веществ в
относительно жесткий каркас около темплатного иона металла. В таких условиях реакция не
должна быть обратимой вследствие того, что темплатированное вещество не
обязательно будет термодинамически наиболее устойчивым продуктом реакции.
Действительно, в разд. 3.9 мы видели, что затраты на невыгодные взаимодействия внутри
7.3. Самосборка в синтетических системах
макроцикла должны быть скомпенсированы в ходе его синтеза. Повышение в
результате этого константы связывания металл-лиганд составляет основу макроцикли-
ческого эффекта. Идея синтетического темплатирования состоит в использовании
такой энергетической «предоплаты» для достижения конечного результата —
получения комплекса. Применение ионов металлов в качестве кинетических
синтетических темплатов крайне распространено и является превосходным способом предор-
ганизации большого числа реагирующих компонентов для получения продукта с
заданной геометрией. Вследствие того, что ионы некоторых металлов, в частности
переходных металлов, часто имеют предпочтительную координационную
геометрию (например, тетраэдрическую, плоскоквадратную, октаэдрическую и т.д.),
выбор иона металла может основательно влиять на природу темплатного продукта.
Синтезы, темплатируемые ионами металла, можно рассматривать как примеры
самосборки с ковалентной модификацией. Например, при синтезе модели сидерофо-
ра G.1) Т. Мак-Мюррей и другие (Т. McMurray et al., 1989) применяли октаэдриче-
ский Ре3+-темплат. В этом случае макробициклический продукт представляет
собой Ре3+-комплекс, из которого металл очень трудно выделить.
l.N(CH2CH2NH2K
2. 60 °С, Юсут.
G.1)
Напротив, неметаллы, такие, как кремний, олово, сурьма или бор, образующие
слабые ковалентные связи с кислородом, азотом или серой - участниками реакции,
дают темплатированные продукты, которые можно получать в свободном виде (без
темплата) простым гидролизом. Эти «ковалентные темплатные реакции» (связь с
О, N или S в этих случаях, как правило, ковалентная) имеют то преимущество, что
связанные О, N или S остаются относительно нуклеофильными. Пример такого
процесса - конденсация тетраэдрического тетраизоцианата кремния с гликолем с
образованием G.2). Этот спироинтермедиат затем реагирует с C(=0)Im2 как с
источником "СО", образуя кремниевый макроциклический комплекс G.3). Гидролиз
последнего приводит к свободному макроциклу G.4), схема 7.1.
Как мы видели в разд. 5.1.2, при синтезе цеолитов и мезопористых кремнеземов в
качестве темплатирующих агентов могут выступать не только катионы щелочных
металлов, но и большие четвертичные ионы аммония. Синтез больших полиоксована-
586 7. Темплаты и самосборка
но
OCNn NCO
X +2
OCN NCO l
VyV
o о о.
o о o-^
H2O
H H
G.4) G.3)
Схема 7.1. Ковалентный темплатный синтез 18-членного макроцикла G.4)
датных клеточных структур темплатируется ионами С1~ или комбинацией NH| и С1~.
Термодинамический темплатный эффект в синтезе макроциклов состоит в том,
что присутствие темплата — иона металла — стабилизирует систему
термодинамически или способствует удалению (например, осаждением) отдельного (обычно
циклического) продукта из равновесной смеси, сдвигая равновесие к этому
термодинамическому минимуму. Отсюда следует, что любое воздействие, приводящее к
термодинамической стабилизации, может сдвигать равновесие смеси компонентов
в сторону образования отдельного продукта согласно принципу Ле Шателье (если
на равновесную смесь воздействовать извне, то она будет реагировать таким
образом, чтобы уменьшить влияние внешнего воздействия).
На границе между кинетическим и термодинамическим синтетическими тем-
платными эффектами находятся процессы, включающие поэтапную самосборку с
последующей ковалентной модификацией. В роли темплатирующего агента можно
использовать электростатические силы, например, при синтезе циклических
молекул довольно необычных видов, в частности взаимопроникающих катенанов.
Электростатические л-л-стэкинг-взаимодействия между плоскостями электроноизбы-
точных и электронодефицитных арильных колец обеспечивают проникновение
такого кольца (гость) в полость макроцикла (хозяин), рис. 7.8. С одной стороны,
если образующееся соединение включения можно замкнуть в цикл, то в результате
получается взаимопроникающий катенан, в котором одно макроциклическое
кольцо необратимо продето сквозь другое. С другой стороны, присоединение к гостю
7.3. Самосборка в синтетических системах
587
Электроноизбыточный л-алкокси-
дизамещенный предшественник (гость)
Электронодефицитный бис(пиридиниевый)
хозяин
Рис. 7.8. Проникновение электроноизбыточного предшественника катенана или
ротаксана внутрь электронодефицитного макроцикла
объемистых концевых групп приводит к ротаксану, в котором гость, подобно
нитке, необратимо «вставлен в ушко» циклического хозяина. Начальное сближение
хозяина и гостя за счет п-тт-стэкинг-взаимодействий — пример термодинамической
самосборки, тогда как последующая ковалентная циклизация (катенан) или
введение «запирающих» концевых групп (ротаксан), которые фиксируют компоненты
относительно друг друга, контролируется кинетически. Вторую стадию в синтезе
катенанов выполняют в условиях высокого разбавления. Как только эта,
кинетически контролируемая, стадия завершается, пропадает необходимость в темплатном
взаимодействии, но она может и сохраняться. Химия катенанов и
ротаксанов подробно обсуждается в разд. 7.6.
Как мы уже видели в биологических системах, водородные связи также
являются мошной темплатирующей силой. Хотя индивидуальная водородная связь может
представлять собой слабое взаимодействие, совокупность комплементарных
водородных связей может стабилизировать большие агрегаты. Водородные связи были
использованы при создании первых самовоспроизводящихся систем (разд. 7.9). В
таких автокаталитических молекулах самокомплементарное амидное производное
под термодинамическим контролем способно организовывать тройной (трехком-
понентный) комплекс еще с двумя соединениями-предшественниками. Эти
соединения в тройном агрегате сближаются в строго определенной ориентации и
реагируют между собой, образуя копию темплатирующей молекулы. В таких система*
опять-таки используется темплатный эффект, в котором термодинамическая
самосборка сопровождается кинетической ковалентной модификацией.
*"» Busch D.H., Vance A.L. and Kokhinski A.G. Molecular template effect: Historica
view, principles and perspectives // Comprehensive supramolecular chemistry / Ed
J.L. Atwood, J.E.D. Davies, D.D. MacNicol, F. Vogtle. New York: Pergamon, 1996
Vol. 9. P. 1-42.
588
7. Темплаты и самосборка
7 • 3 • ^ Термодинамическая модель:
самосборка порфириновых комплексов цинка
Порфириновые комплексы цинка G.5)—G.7), рис. 7.9, — интересный
пример термодинамически контролируемой самосборки закрытых циклических
олигомеров (димеров, тримеров, тетрамеров и т.д.) в противоположность
образованию открытых (линейных) координационных полимеров. Соединения G.5)—G.7)
спонтанно самособираются из соответствующих мономерных единиц под
термодинамическим контролем в строго определенных условиях. Их образование
полностью управляется равновесием, так как связь Zn2+—Ы(пиридил) сравнительно
лабильна (быстро и обратимо рвется и вновь образуется) в органических
растворителях (СН2С12, толуол и т.д.) при комнатной температуре. Это обусловлено
жесткой природой центра Zn2+ (Дополнение 3.2) и недостатком или отсутствием
значительной ковалентной компоненты для связывания. Поэтому примечательно,
что благодаря стабилизации как хелатным, так и макроциклическим эффектом
(разд. 1.4 и 3.8) ион Zn2+ сильно удерживается в порфириновом окружении N4.
Образование трех родственных циклических олигомеров конкурирует с образованием
полимера, как показано на схеме 7.2.
Полимеры
с более
длинными
цепями
Схема 7.2. Конкурентное образование открытых (полимерных) и закрытых
(циклических) агрегатов на основе цинкпорфиринов
R - солюбилизирующая группа
Рис. 7.9.
Закрытые циклические
самособирающиеся Zn-порфирино-
вые олигомеры
(по ChiX. etal.,
1995)
590 7. Темплаты и самосборка
Систему в целом можно количественно описать как:
[Цикл]
L Closed' [Мономер]" * GЛ)
Это константа равновесия для самосборки закрытого циклического олигомера,
содержащего п мономерных единиц.
[(Олигомер)„]
" °Реп [(Олигомер)„_,][Мономер]' v ;
Это константа равновесия для открытой нециклической ассоциации мономера
с растущим линейным олигомером, содержащим п—\ мономерных единиц. Ее
трудно измерить, так как, строго говоря, для каждого шага открытой ассоциации она
должна иметь свое значение. Кроме того, измерение такого параметра даже для
одной стадии исключительно сложно из-за конкурентного образования закрытой
структуры. Однако К' можно оценить, если измерить константу равновесия для
ассоциации фрагмента Zn-порфирин с пиридильными лигандами, неспособными
формировать закрытую структуру. Константа ассоциации для этого случая (
вещество сравнения) обозначается К/, Кг~ К .
К К
-> r>i л. гt closed closed ,- -,ч
3. Эффективная молярность Л/эф = = . G.3)
К "pen Knref
Эффективная молярность — очень полезный параметр, определяющий
концентрацию, при которой образование полимера начинает конкурировать с
самосборкой закрытых олигомеров, т.е. это верхний предел, при котором циклическая
структура стабильна.
4. Критическая концентрация самосборки ККС = [Мономер],
«[Цикл]
где ^=1 G.4)
[Мономер]"
и
[Цикл] = А^Мономер]". G.5)
ККС — это минимальная концентрация мономера, при которой начинается
самосборка закрытой структуры. Ее определяют как концентрацию, при которой
комплекс собран наполовину, т.е. мольная доля мономерных единиц,
представленных в виде полностью собранного комплекса, равна 0.5. Зная величину Л/эф,
значение ККС можно рассчитать из уравнений G.4) и G.5) следующим образом:
7.3. Самосборка в синтетических системах
Таблица 7.1. Параметры самосборки для циклов G.5)—G.7)
(комнатная температура, СН2С12)
Комплекс
G.5)
G.6)
G.7)
л
2
3
4
Кпр дм3 моль
5.6-Ю-3
3.910-3
1.910-3
ККС, мольдм
з-ю-9
2-Ю-7
зз-ю-5
Af^, мольдм
6
100
0.6
«1
£
9.3
8.7
4.5
Подставляя величину Kclosed в уравнение G.3), получаем:
1
ККС -г—. г: —. : ~ . G.7)
>/(я-1)Д/^(я-1)^(я-1)
Данные для макроциклических соединений цинка G.5)-G.7) приведены
в табл. 7.1. Они ясно показывают, что макроциклы, состоящие из меньшего числа
мономерных единиц («), более стабильны, чем макроциклы с более высокими п.
Этому можно дать разумное объяснение на основании энтропийного фактора:
уменьшение числа степеней свободы с ростом числа агрегированных мономерных
единиц. Это существенно ограничивает число мономерных единиц, которые могут
соединяться с образованием закрытой структуры. Отсюда следует, что число
мономерных единиц в закрытом цикле может быть увеличено благодаря
преднамеренному уменьшению числа степеней конформационной свободы и предорганизации
этих единиц для циклизации, например путем стерических ограничений или
многоцентровых взаимодействий.
Экспериментальное значение А/Эф достаточно хорошо согласуется с
теоретическими максимальными значениями, которые можно получить из уравнения G.8)
(Н. Anderson etal., 1995).
Мэф,тах=ехР|—j^" • G.8)
Для указанных выше пиридиновых комплексов цинка было найдено, что
значения AS в толуоле равны ~50 Дж-К^-моль, и, следовательно, верхний предел для
А/Эф находится в области 400 моль-дм~3. Для тримера G.6) экспериментальное
значение (в СН2С12) 100 моль-дм близко к теоретическому максимуму; это
подразумевает, что система близка к геометрически оптимальной (т.е. мономерные
единицы высококомплементарны). Напротив, такие факторы, как напряжение кольца,
могут вносить вклад в понижение этого значения для димера G.5).
Величины МЭф и ККС образуют «концентрационное окно», внутри которого
закрытая самособранная структура термодинамически стабильна. Чем шире это
окно, тем более стабилен комплекс. Таким образом, общая эффективность процессе
самосборки (е) может быть выражена в значениях ККС и Л/эф.
е = 1ё(Мэф/ККС). G.9;
592
7. Темплаты и самосборка
Мономер
Открытые
олигомеры
- Полимер
lg ([Zn-порфирин.], мольдм )
10
Рис. 7.10. Распределение
форм в растворе для
самособирающегося тетрамера G.7).
(Воспроизведено с
разрешения Chi X., Guerin A.J.,
Haycock R.A. et al. The
thermodynamics of self-assembly //
Chem. Commun. 1995.
P. 2563-2565)
Следовательно,
G.10)
Нижний предел е, для которого самосборка может быть реально изучена,
находится около е = 4, так как именно при такой эффективности в самосборке
комплекса участвует более 90% от веществ с концентрацией выше всего лишь 102 мольдм.
Даже в этом случае наблюдается равновесие со значительными количествами оли-
гомеров. Это следует из диаграммы распределения мономера и продуктов
самосборки для тетрамера G.7), рис. 7.10.
Зная энтальпию связывания для реальной рассматриваемой системы АЯ-
можно теоретически определить максимальное значение е по уравнению G.11),
полученному из уравнений G.8) и G.10):
2303RT
+ .
п- 1
G.П)
Аналогичным образом, минимальное значение ККС может быть также
получено из уравнений G.7) и G.8):
1
KKCmin== „1Д/.-0
• ехр
яАЯ,
ref
AS.
ref
(п- \)RT
R
G.12)
Chi X., Guerin A.J., Haycock R.A. et al. The thermodynamics of self-assembly //
Chem. Commun. 1995. P. 2563-2565.
Самосборка
координационных соединений
Jones C.J. TYansition metals as structural components in the construction of
molecular containers // Chem. Soc. Rev. 1998. Vol. 27. P. 289-299.
7.4. Самосборка координационных соединений д^З
•4« 1 Принципы дизайна
Обобщая результаты для 2п2+-порфириновых комплексов, мы
обнаружили, что дизайн самособирающихся супрамолекулярных ансамблей, основу
которого составляют направленные взаимодействия любого вида (ковалентные,
координационные или водородные связи; ион-дипольные или диполь-дипольные
взаимодействия и т.д.), можно условно разделить на две категории:
взаимодействия, приводящие к образованию полимерных веществ или олигомерных агрегатов.
Представляют интерес соединения двух классов: полимеры при моделировании
канальных цеолитов, циклические олигомеры как молекулярные реакторы полукар-
церандного типа, а соединения обоих классов - как наноразмерные устройства.
М. Фуджита (М. Fujita) из Института молекулярных исследований (Япония)
показал, что тип агрегатов, получаемых на основе координационных соединений
переходных металлов (подразумевая, что они включают и направленное связывание
других видов), предсказуем и контролируется природой строительных блоков.
Строго говоря, мы заинтересованы и в «расходящихся», и в «сходящихся»
связывающих центрах. Вспомним определение хозяина как компонента, состоящего из
сходящихся связывающих центров. И наоборот, гость обладает расходящимися
связывающими центрами (см. разд. 1.1). Согласно Фуджите, это означает, что
образующийся комплекс хозяин-гость является дискретным (неполимерным)
веществом, поскольку сходящийся хозяин окружает расходящегося гостя. В соответствии
с этим подходом можно заключить, что если сходящийся лиганд не может окружить
гостя, то он образует дискретный агрегат большего размера, в который гость будет
включен (схема 7.3, я), так как в конечном итоге образование дискретных
соединений является термодинамически выгодным в соответствующем концентрационном
диапазоне. Если используется расходящийся лиганд, то дискретный комплекс
может образоваться только в том случае, когда металлоцентр трансформирован в
сходящийся, например, путем его блокировки лигандом (схема 7.3, б). Таким образом,
дискретные вещества образуются любой (комплементарной)
сходящейся/расходящейся парой. Наконец, если в двухкомпонентной системе оба вещества
расходящиеся, то должен сформироваться полимер (схема 7.3, в).
2 Супрамолекулярный куб
Дж. Томас с сотрудниками (S. Roche, С. Haslam, H. Adams, S.L. Heath,
J.A. Thomas, 1998) из Университета Шеффилда (Великобритания) попытался
методом самосборки изготовить супрамолекулярный куб, комбинируя линейные спей-
серы и пирамидальные структуры, которые должны были выступать
соответственно в качестве ребер куба и его углов. Глядя на кубическую фигуру, легко понять, что
для этого необходимо 8 пирамидальных структур и 12 спейсеров (рис. 7.11).
В качестве ребра был выбран расходящийся мостиковый лиганд 4,4'-бипи-
ридил F.38), тогда как углы были получены из Ru(II)-KOMmieKca
[Ru([9]aH-S3)Cl2(DMSO)]2+ G.8). На схеме 7.3, б представлен пример стратегии, в
соответствии с которой хлоридные и диметилсульфоксидный лиганды в G.8) легко
594
7. Темплаты и самосборка
(а)
Обнаженный металл
(расходящийся)
Сходящийся
лиганд
(б)
Защищенный металл
(сходящийся)
Расходящийся
лиганд
F)
Обнаженный металл
(расходящийся)
Расходящийся
лиганд
Схема 7.3. Конструирование капсулярных и полимерных самособирающихся структур
удаляются и остаются три сходящихся центра связывания. [9]aH-S3 действует в
качестве группы, блокирующей остальные три/ас-позиции октаэдрического
металлического центра. Реакция G.8) с тремя молекулами F.38) должна приводить к
замещению лабильных лигандов С1 и DMSO тремя 4,4'-бипиридилами и образованию
[Ru([9]aH-S3)D,4'-bipyK]2+ G.9) вследствие того, что бипиридильные лиганды
обладают сильными донорными и тьакцепторными свойствами, которые
стабилизируют Ru(II). Более сложная задача состоит в том, чтобы «убедить» четыре катиона
G.9) прореагировать еще с четырьмя молекулами G.8) для образования закрытой
кубической структуры. Это всего лишь один из возможных путей реакции. Вообще
может образовываться огромное количество разных олигомеров и полимеров, и в
7.4. Самосборка координационных соединений
$95
Спейсер
Угол
Рис. 7.11. Компоненты супрамолекулярного куба
отсутствие темплата (например, большого кубического аниона) не ясно, почему
должен собраться именно желаемый куб [{Ru([9]aH-S3)}8(^-4,4/-bipy),2]16+ G.10).
После завершения реакции (схема 7.4) сначала образуется сложная смесь
продуктов. Однако после кипячения в течение месяца состав реакционной смеси
продуктов упрощается, и, в конце концов, в 'Н ЯМР-спектре проявляется единичный
набор сигналов бипиридильных лигандов: кубическая структура самособралась
(рис. 7.12).
Это явно антиэнтропийное получение упорядоченной симметричной молекулы
можно объяснить с позиций процесса химического «естественного отбора».
Одновременная сборка 20 компонентов (8 углов и 12 ребер) - статистически крайне
маловероятна, особенно в отсутствие кинетического темплата. Однако вследствие
достаточно высокой лабильности иона Ru(II) его связи с лигандами будут постоянно
(хотя и медленно) разрушаться и восстанавливаться по мере приближения системы
Jf*\ SOM"
У Cl
G.8)
О
N
F.38)
NO2Me
!6NaPF6
HRu([9)aH-S3)}8D.4'-bipy)l2)(PF6I6
G.Ю)
Схема 7.4. Получение кубического соединения [{Ru([9]aH-S3)}8(n-4,4'-bipyI2]16+ G.10)
596
(а)
7. Темплаты и самосборка
(б)
(в)
(г)
(д)
9.6 9.2
8.8 8.4 8.0
б, м.д.
7.6 7.2
Рис. 7.12. 'Н ЯМР-спектры
(область бипиридила) продуктов
и исходных веществ:
(a)[Ru(l9]aH-S3)D,4'-bipyK]2+,
(б) после трех дней, (в) после
одной недели, (г) после четырех
недель, (д) выделенный продукт.
(Воспроизведено с разрешения
Roche S., Haslam С, Adams H. et al.
Self-assembly of a supramolecular
cube // Chem. Commun. 1998.
P. 1681-1682)
к равновесию. Фрагменты куба будут сосуществовать с мономерами и полимерами
и будут случайно образовываться мельчайшие количества кубического продукта
G.10).
Однако, в отличие от открытых продуктов, закрытая кубическая молекула
значительно более термодинамически стабильна и ее образование происходит почти
необратимо, так как разрушение куба требует искажения всей закрытой структуры,
а не простого разрыва единичной связи Ru—N. В результате, в течение крайне
длительного периода времени кубическая структура накапливается как конечный
продукт реакции. Весь этот процесс возможен лишь потому, что образование связи
Ru-N в трис(бипиридильных) комплексах обратимо. Томас и сотрудники
постулировали, что это результат неблагоприятных стерических взаимодействий между
орто-С— Н-протонами бипиридильных лигандов. Без этих, слегка
дестабилизирующих, взаимодействий Ru2+ должен быть слишком инертен для самосборки
кубической структуры за разумное время.
Следует отметить, что этот способ кубической самосборки является довольно
общим. Аналогичные кубические структуры таких соединений, как [{Со(триаза-
циклононан)}8(ц.-СЫ)J]12+ и [{Со(триазациклононан)}4{Сг(триазациклоно-
HaH)}4(|a-CN)]2]12+, также были получены. При этом использовали мостиковые
CN~-HOHbi мотива берлинской лазури. В таких случаях промежуточные квадратные
структуры должны быть защищены молекулами DMSO, блокирующими
неиспользуемые координационные центры.
7.4. Самосборка координационных соединений
4*3 «Молекулярные квадраты»
и «молекулярные коробки»
Концепция особой стабильности закрытых трехмерных жестких фигур
не ограничивается кубами. Обширные исследования были предприняты в области
конструирования объектов, получаемых самосборкой двух или более фрагментов,
которые могут распознавать друг друга. Цель этих работ состояла в получении
содержащих полость молекул и ансамблей, стабильных в растворе. Поэтому
исследование по комплексообразованию в растворах было направлено на выявление
способности карцерандов, полукарцерандов и близких им соединений защищать
чувствительные объекты-гости от влияния внешней среды (см. разд. 5.3.3). Как и в
случае наноразмерных компонентов, подход самосборки дает возможность
конструировать предельно большие полости, способные участвовать в более сложных
химических процессах и связывать или защищать значительно большие по размеру
(и, возможно, более функциональные) частицы-гости.
Еще до работы Томаса и сотрудников были получены близкие по составу
«молекулярные квадраты» и «треугольники». Жесткие спейсеры (ребра), подобные 4,4'-
бипиридилу, не допускают образования треугольников, а формирование ими
комплексных ансамблей ограничено необходимостью, чтобы углы связей при
металлоцентрах минимально отклонялись от 90° (октаэдрическая и
плоскоквадратная координационная геометрия). Так, М. Фуджита и сотрудники (М. Fujita et al.,
1991) получили «молекулярный квадрат» G.11), который в растворе может
функционировать в качестве хозяина для ароматических гостей, например для нафталина.
Константа связывания нафталина Кп равна 1800 М~'.
NH2—Pd2+— N ГУ) < О N—Pd2+— NH2
| V^7 V^7 | \
G.11)
Для синтеза аналогичных закрытых координационных макроциклов с менее чем
восемью компонентами, необходимыми для образования соединения G.11),
нужны более гибкие спейсеры-ребра. Например, ряд хозяев-«треугольников» G.12) со-
598
7. Темплаты и самосборка
держит более длинный аналог 4,4'-бипиридила, а простой циклофановый рецептор
G.13) (с арильным кольцом в спейсере) получен из очень гибкого а,а'-бисD-пири-
дил)-я-ксилола путем четырехкомпонентной самосборки. Отметим опять-таки
использование защищенных сходящихся металлсодержащих фрагментов с
расходящимися лигандами (защитная группа — этилендиамин).
G.12)
Соединение G.13) особенно примечательно, потому что, в добавок к
связыванию таких же органических веществ — гостей, как и в случае G.11), оно способно
связываться само с собой, образуя полностью самособирающийся [2]катенан (пара
взаимозамкнутых, или сцепленных, колец, разд. 7.6). Циклическое
биметаллическое соединение G.13) существует в равновесии, в высокой степени зависящем от
концентрации, с взаимозамкнутым соединением G.14), схема 7.5.
Образование G.14) возможно вследствие лабильности связей Pd—N,
объединяющих комплекс. Эти связи образуются и разрываются довольно легко в обычных
условиях. Например, Pd(II) намного более лабилен, чем Ru(II). В случае первого
металла это приводит к образованию контролируемого равновесием (т.е.
термодинамически) продукта, в котором два кольца сцепляются только при высокой
концентрации. Использование менее лабильных ионов металлов может привести к
значительно более кинетически стабильным продуктам. Так, склонность длинного
лиганда бипиридильного типа в G.13) к образованию катенанов была
использована для получения намного более стабильного катенана, который не размыкается с
разрывом связи M-N. Замена Pd(II) на значительно менее лабильную Pt(II)
приводит к образованию Р1(П)-аналога G.13) из [Pt(en)(NO3J] (en - этилендиамин)
только после длительного кипячения (как при образовании супрамолекулярного
куба G.10) из полулабильного Ки(П)-соединения). Вследствие того, что связи Pt-N
намного более инертны (Дополнение 7.1), чем Pd—N, этот Р1(П)-комплекс
является долгоживущим и даже при высоких концентрациях его кольцо не разъединяется
7.4. Самосборка координационных соединений
599
Схема 7.5. При концентрациях выше 50 мМ катенан G.14) является основным
веществом, тогда как циклофан G.13) доминирует при концентрациях ниже 2 мМ
образованием аналогичного Р1(П)-катенана. Связи Pt-N, однако, могут
разрываться при кипячении A00 °С) в присутствии избытка NaNO3. Это свойство было
использовано при получении платиновых катенанов методом «молекулярного замка»
(рис. 7.13).
600
7. Темплаты и самосборка
Размыкание
——♦>
NaNO3 +
+ нагревание
Самосборка
Замыкание
Охлаждение +
+ удаление
NaNO3
Рис. 7.13. Схематическое представление образования платинового аналога G.14)
из некатенанового мономера методом «молекулярного замка»
Дополнение 7.1. Кинетическая и термодинамическая стабильность
Говорят, что координационное соединение, такое, как G.13), кинетически лабильно,
если, подобно большинству комплексов переходных металлов, оно подвержено
быстрым реакциям обмена лигандов в растворе. Лабильные комплексы были формально
определены Г. Таубе (Н. Taube, которому в 1983 г. была присуждена Нобелевская
премия за работы в этой области) как комплексы, которые полностью реагируют за 1 мин
при 25 °С. Противоположностью лабильности является инертность. Термины
«инертность» и «лабильность» характеризуют кинетическую стабильность комплекса.
Инертные комплексы имеют высокий энергетический барьер реакции. Для
лабильных комплексов переходное состояние реакции достигается намного легче (т.е.
энергия активации Еа мала). Кинетическая стабильность (время жизни в определенных
условиях) отличается от термодинамической стабильности, которая определяется
полной стандартной свободной энергией реакции AG°. Если &G° — большая
отрицательная величина, то продукты реакции намного более термодинамически
устойчивы, чем исходные вещества, и это влияет на положение равновесия (рис. 7.14).
Некоторые металлы с определенными электронными конфигурациями имеют гораздо
меньшие скорости лигандного обмена, чем другие, например октаэдрический Сг(Ш),
электронная конфигурация Зс1ъ (рис. 7.15).
7.4. Самосборка координационных соединений
601
Переходное состояние
Стадия!
Реагенты
Стадия П
Энергия активации Еа
Свободная энергия реакции AG°
Продукты
Координата реакции
Рис. 7.14. Изменение свободной энергии в ходе экзотермической химической
реакции
Рассмотрим реакцию диссоциации октаэдрического комплекса, которую можно
разбить на две стадии (см. рис. 7.14):
1. Потеря лиганда с образованием квадратно-пирамидального пятикоординирован-
ного интермедиата:
М(Н2О)£+-» M(H2O)f + Н2О. G.13)
2. Реакция с другим лигандом (L) с образованием октаэдрического продукта:
G.14)
М(Н2О){?+ + L-> M(H2OML"+.
Рис. 7.15. Диаграмма
кристаллического поля для
октаэдрического иона */3-металла
dxi-y\ dz2
Сферический
ион
3/5д
dxy, dxz, dyz
Октаэдрический
602
7. Темплаты и самосборка
Энергия активации — энергия, необходимая для завершения стадии 1, —
компенсируется несколькими факторами, особенно энергией связи М—ОН2 и энергией
любой реорганизации при создании новой структуры. Последняя будет включать
изменение энергии стабилизации поля лигандов (ligand field stabilisation energy, LFSE). Это
выигрыш энергии при нахождении (/-электронов иона металла в несферическом
окружении лигандов (в нашем случае — октаэдрическом). Например, для Сг(Ш)
электронная конфигурация Зс/3 дает LFSE, соответствующую трем электронам,
стабилизированным двумя пятыми параметра расщепления поля лигандов (А).
Квадратно-пирамидальное переходное состояние реакции уже не является октаэдри-
ческим и поэтому не дает такого же расщепления валентных (/-орбиталей, что
приводит к потерям LFSE, которые должны быть скомпенсированы высокой энергией
активации реакции. Чем больше значение А, тем больше будут эти потери, хотя они и
регулируются LFSE в переходном состоянии (квадратная пирамида в нашем случае).
Разницу между значениями LFSE в основном и переходном состояниях называют
энергией активации поля лигандов (ligand field activation energy, LFAE); это параметр,
определяющий скорость всей реакции.
В соединениях, подобных G.13), и Pd(II), и Pt(II) имеют плоскоквадратную, а не
октаэдрическую геометрию; следовательно, их валентные электроны располагаются
на (/-орбиталях иным образом (рис. 7.16). Схему расщепления для плоскоквадратных
комплексов можно представить как образующуюся путем удаления двух аксиальных
лигандов из октаэдра (бесконечное тетрагональное искажение). Для электронной
конфигурации Js, как у Pd(II) и Pt(II), это приводит к образованию одной
незаполненной высокоэнергетической орбитали dx2 — у2. Такая конфигурация также имеет
высокую LFSE и, следовательно, является относительно инертной в зависимости от
точного значения энергии расщепления кристаллического поля А. Фактически
параметр А значительно возрастает при уменьшении номера группы в Периодической
таблице. Таким образом, Ni(II) и Pd(II) относительно лабильны при обмене лигандов,
тогда как значение А для Pt(II) достаточно велико при резком снижении скоростей
этих реакций в обычных условиях.
Плоскоквадратный
ион
dx1-y1,dzl
dj-y2
dxy, dxz, dyz
dxz. dyz
Тетрагональное
искажение
Рис. 7.16. Влияние перехода от октаэдрического к плоскоквадратному полю
лигандов за счет удаления аксиальных лигандов (тетрагональное искажение) на
относительные энергии орбиталей металла
7.4. Самосборка координационных соединений 603
При кипячении моноциклического РцЧО-аналога G.13) с NaNO3 кольцо
размыкается за счет повышения лабильности связей Pt—N. В этих условиях система
находится под термодинамическим контролем и при высоких концентрациях
самособирается в [2]катенан. На самом деле, этот процесс, вероятно, включает в себя
разрыв связи Pt—N, продевание образующегося линейного комплекса через
отверстие второго макроцикла за счет гидрофобных факторов и замыкание разорванной
координационной связи. При охлаждении до комнатной температуры и удалении
избытка соли из-за снижения лабильности связей Pt—N образуется катенан с двумя
продетыми друг в друга и надежно замкнутыми кольцами. Обработка «соль/тепло»
представляет собой эффективный ключ к замку. Образование катенанов с
металлическим каркасом рассмотрено в обзоре Н. Такеды и соавторов (N. Takeda et al.,
1999), а химия других видов катенанов будет обсуждаться в разд. 7.6.
Хотя соединения G.11)—G.13) и очень интересны, они, по существу, — всего
лишь двухмерные хозяева (корандного типа), которые, как мы уже видели (гл. 3),
более слабо связывают гостей, чем их трехмерные криптандные аналоги, и, что
более важно с точки зрения внутриполостной химии, не защищают гостей от внешней
среды над и под полостью. Концепция гибкого спейсера, рассмотренная на
примере соединения G.13), была распространена на систему с триподальным лигандом -
а,а',а"-трисD-пиридил)мезитиленом, способным принимать участие в пятиком-
понентной самосборке с образованием соединения G.15) - криптандного аналога
соединения G.13) (М. Fujita et al., 1995a). Этот трехмерный хозяин был получен в
воде с выходом более 90% только в присутствии подходящего большого гостя,
такого, как 2-фенилпропионовая кислота или 1-адамантанкарбоновая кислота. Менее
полярные гости меньшего размера, например и-ксилол, обеспечивают намного
более низкий выход комплекса, тогда как катионные гости вовсе не темплатируют его
образование. Такой вид самосборки, в которой информацию несет молекула гостя,
называют вынужденным соответствием; в нем образование хозяина определяется
размерами гостя. В принципе, разные гости в зависимости от их размера и свойств
могут управлять формированием различных олигомерных ансамблей (ср. темпла-
тируемое анионами образование пента- и гексаядерных систем G.90) и G.92), разд.
7.7.6). Этот эффект аналогичен кинетическому темплатному эффекту (гл. 3),
потому что образующиеся продукты довольно прочны и не обязательно обеспечивают
термодинамический минимум для системы. Более того, они не требуют
длительного присутствия темплатирующей молекулы. Аналогичным образом получены даже
большие трехмерные хозяева [{Pd(en)}12G.16)8]12+ с диаметром до 4.6 нм (М. Fujita
et al., 1995b), а их существование в растворе подтверждено экспериментами по
светорассеянию (в которых видимый свет рассеивается большими частицами с
размерами, соизмеримыми с его длиной волны). Супрамолекулярный
[{Pd(en)}12G.16)8]12+ может связывать одновременно четыре молекулы адамантан-
карбоновой кислоты внутри своей полости, хотя его структура стабильна даже в
отсутствие этого гостя. Примечательно, что процесс связывания обладает четким
аллостерическим эффектом: титрование сольватированного хозяина адамантан-
карбоновой кислотой в качестве гостя показывает немедленное комплексообразо-
вание 1:4 без образования промежуточных комплексов 1:1, 1:2 или 1:3. Таким
образом, смесь хозяина и гостя 1:1 дает сигналы только от свободного хозяина G5%) и
604
7. Темплаты и самосборка
комплекса 1:4 B5%). Это объясняется возрастающей способностью комплекса
связывать молекулы гостя, так как включение первых гостей делает полость все
больше и больше гидрофобной.
N.
G.15)
он
Темплатирующие гости
С развитием еупрамолекулярной химии концепция трехмерных
самособирающихся капсул все больше распространялась на крупные системы с целью
образования полостей, достаточно больших для капсулирования гостей большого размера
или более чем одного гостя. Как часть исследования систем, моделирующих
биологические макромолекулы с полостью, Л. Барбу и сотрудники (L. Barbour et al., 1998)
получил э/сзо-бидентатный лиганд G.17), обладающий следующими свойствами:
• растворимость в воде;
• наличие амидных групп и арильных колец, обычных для биологических систем;
• наличие активных расходящихся центров.
Любопытно, что даже без концевого лиганда типа этилендиамина этот лиганд легко
самособирается в присутствии Cu(NO3J, образуя закрытый, содержащий полость
ансамбль [Cu2G.17L(H2OL](NO3L-8H2O. Тот факт, что при этом не получается
полимерное соединение, — вероятно, результат темплатного эффекта другого вида,
состоящего в образовании водородных связей между аксиальными молекулами воды и
четырьмя нитрат-анионами, включенными в гигантскую полость ансамбля.
7.4. Самосборка координационных соединений
605
G.17)
2 Cu(NO3J
Н,0
о
О
Р> AQ
О
он2
[Cu2G.17L(H2OL](NO3L-8H2O
О
Самым неожиданным было то, что структура [Cu2G.17L(H2OLKNO3L-8H2O в
твердом состоянии содержит очень интересную часть (хотя и не предусмотренную
авторами, делавшими упор на биомиметический аспект) - соединенный
водородными связями кластер из 10 молекул воды. С точки зрения конструирования
кристалла молекулы воды, занимающие аксиальные места при ионах Си2+ и
направленные от полости, представляют собой доноры водородной связи, нуждающиеся в
акцепторах для стабилизации кристаллической решетки. Карбонильные атомы
кислорода на внешней стороне сфероидной структуры подходят для этой цели,
однако большой размер ансамбля препятствует сближению достаточного количества
групп С=О для взаимодействия с двумя О—Н-донорами. Вместо этого две внеполо-
стные молекулы воды двух различных ансамблей объединяются с помощью
водородных связей еще с восемью некоординированными молекулами воды, образуя
поразительный декамер из воды со структурой, очень напоминающей кубическую
606
7. Темплаты и самосборка
Рис. 7.17. Сравнение внеполостного декамера из молекул воды для
[Cu2G.17L(H2OL](NO3L-8H2O с элементарной ячейкой кубической фазы льда 1^. Три
кристаллографически различающиеся молекулы воды помечены буквами а, бив. Молекула а
скоординирована с центром Cu(II). (Воспроизведено с разрешения BarbourL. et al., 1998)
фазу льда Ic, рис. 7.17 (ср. с более часто встречающимся гексагональным льдом lh,
рис. 5.1).
Наконец, в 1999 г. группы М. Фуджиты (М. Fujita) и П. Станга (P. Stang)
независимо друг от друга смогли осуществить самосборку предельно больших капсул.
Применяя те же принципы, что и при сборке более мелких плоскоквадратных
систем, Фуджита провел реакцию [Pd(en)(NO3J] с треугольным гексадентатным ли-
гандом G.18) и получил гексаэдр, содержащий шесть треугольных лигандов,
связанных 18 ионами Pd(II). Заметим, что количество 6-6 = 36 донорных атомов
согласуется с 218 = 36 вакантными координационными центрами (без учета
лабильных ионов N0^). Внутренний объем капсулы достигает 900 А3 и содержит
определенное число нитрат-ионов. Самосборка этой капсулы высветила некоторые
проблемы, связанные с рентгеновской дифракцией супрамолекулярных
соединений: из общего числа 36 нитрат-ионов, присутствие которых ожидалось,
экспериментально смогли определить положения только 14 ионов, причем только пять из
них находились в полости. Кристаллическая структура этого ансамбля показана на
рис. 7.18.
Рис. 7.18. Шестигранный ансамбль из 18 единиц
Pd(enJ+ и шести лигандов G.18) (по Takeda N. et al.,
1999)
7.4. Самосборка координационных соединений
607
G.18)
Сразу после статьи Фуджиты в журнале «Nature» была опубликована работа
Станга о получении двух ансамблей G.19) и G.20), собранных из треугольных
строительных блоков. Станг и сотрудники использовали также плоскоквадратный ион
Pt2+ с лабильным лигандом (трифторметансульфонат - трифлат, Tf) и двумя
нелабильными лигандами (PPh3), находящимися в транс-геометрии, для получения
расходящегося металлического центра. Сходимость была достигнута за счет
второго компонента (пиридилоподобный лиганд или производное бензофенона),
имевшего V-образную форму. В результате реакции был собран кубооктаэдр G.19) или
G.20) - одно из полуправильных архимедовых тел (разд. 7.5.2), содержащее 24
центра Pt(II), схема 7.6. Структура ансамбля, рассчитанная методом ESFF (extended
systematic force field, расширенное систематическое силовое поле) представлена на
рис. 7.19.
*"» Fujita M. Metal directed self-assembly of two- and three-dimensional synthetic
receptors // Chem. Soc. Rev. 1998. Vol. 27. P. 417.
Рис. 7.19. Пространственная модель
ансамбля G.20). 12 Р1(П)-содержащих
кубооктаэдров рассчитаны методом
ESFF. (Воспроизведено с разрешения
Olenyuk В. et al., 1999)
608
7. Темплаты и самосборка
OTf
PhjP—Pt-PPh3
■X
Схема 7.6. Самосборка наноразмерных кубооктаэдрических структур G.19) и G.20).
Условия: СНХ1-,, комнатная температура, 10 мин, выход более 98 % (по Olenyuk В. et al.
1999)
7.4. Самосборка координационных соединений
609
Металлические ансамбли
Кроме закрытых капсул путем самосборки (рис. 7.20) было получено
большое количество реечных, лестничных и решеточных структур с
использованием полидентатных мостиковых лигандов типа «жесткий стержень». В качестве
примера можно отметить образование «молекулярной лестницы» G.21). И вновь
термодинамические силы приводят к образованию дискретных олигомерных
объектов, а не полимеров. В каждом случае информация для сборки всей
структуры — жесткость лигандов, количество и расположение донорных центров, а также
координационная геометрия металла - закодирована в индивидуальных
компонентах. Так, лиганд G.22) в присутствии 1.5 эквивалентов AgCF-jSO-j самособирается в
15-компонентной реакции с образованием молекулярной ЗхЗ-решетки
[Ag9G.22N]9+. Анион CF-jSOj использован потому, что в качестве лиганда для
мягкого Ag+ он не конкурирует с более активными N-донорами. В согласии с
решеточной структурой спектр 109Ag ЯМР содержит сигналы от трех различных групп,
окружающих ионы серебра, в соотношении 4:4:1. Это четыре иона металла в каждом
углу, четыре — в середине каждой стороны и один - центральный Ag+ A09Ag — ядро
со спином ]/2, как и 'Н, и высоким содержанием в природе, что делает
спектроскопию 109Ag ЯМР очень полезным инструментом для исследования ансамблей этого
металла.)
Ж.-М. Лен и сотрудники (P. Baxter, J.-M. Lehn, A. DeCian, 1993) также изучали
системы, состоящие из двух различных лигандов: один стержнеобразный, как
G.22), а другой дискообразный. Им удалось осуществить самосборку
цилиндрической «коробки», напоминающей офис с двумя этажами, состоящими из двух
трис(бидентатных) «полов» G.23) и трех 6,6///-диметил-2,2':5',3":6",2"'-тетрапири-
диновых G.24) «стен». Эти лигандные компоненты, соединенные шестью тетраэд-
рическими ионами Cu(I), образуют ансамбль G.25). Структура этого
замечательного комплекса, определенная с помощью РСА, представлена на рис. 7.21. «Этажи»
расположены слишком близко друг к другу, чтобы допустить включение гостя.
(а)
Рис. 7.20. (а) Реечная, (б) лестничная и (в) решеточная структуры, полученные
из лигандов типа «жесткий стержень» и ионов металлов
9-СтидДж В.
610
7. Темплаты и самосборка
Рис. 7.21. Кристаллическая
структура «офиса» G.25)
7.4. Самосборка координационных соединений
611
G.25)
Заместители для простоты не указаны
Лиганд G.24) - пример дитопного лиганда в том смысле, что он имеет два
2,2'-бипиридильных фрагмента, каждый из которых может хелатировать различные
металлические центры. Увеличение числа связывающих фрагментов, как в случае
G.22), приводит к тому, что большее число ионов металла связывается в линию.
Если координационные сферы этих ионов заняты концевыми лигандами типа самого
2,2'-бипиридила, то образуется «молекулярная рейка». Замена концевых лигандов
на мостиковые бипиримидилы типа G.26) приводит к лестничным структурам, как
в случае G.21). Развивая этот подход, Бакстер и соавторы получили линейные оли-
гопиридильные лиганды с восемью пиридиновыми кольцами и использовали их
612'
7. Темплаты и самосборка
Рис. 7.22. Многоэтажные структуры, полученные в результате самосборки (по Lehn J.-M.,
1995). (Заместители для простоты не указаны)
при самосборке многоэтажных «офисов» (рис. 7.22). Отметим, что
термодинамическая возможность самосборки дискретных структур обусловлена наличием
объемистых заместителей на периферии ансамбля, которые стерически препятствуют
образованию полимерных агрегатов.
7.5. Самосборка закрытых комплексов с помощью водородных связей Ш1Ъ
Самосборка закрытых комплексов
с помощью водородных связей
•"ч Rebek /., Jr. Reversible encapsulation and its consequences in solution // Ace. Chem.
Res. 1999. Vol. 32. P. 278-286.
.5.1 «Теннисные мячики» и «мячи для софтбола»
Координационные взаимодействия - не единственный способ
самосборки с образованием закрытых капсулированных систем, обладающих
возможностью связывать частицы—гости в растворе. Дж. Ребек и его соавторы (R. Wyler,
J. de Mendoza and J. Rebek, 1993) показали, что множественные водородные связи
идеальны для самосборки закрытых сферических молекул и капсул благодаря
своей относительно слабой, но направленной природе.
Компонент G.27) состоит из двух дифенилгликолурильных единиц с
внутренней кривизной, связанных спейсером на основе дурола A,2,4,5-тетраметилбензол).
И в растворе, и в твердом состоянии G.27) самособирается с образованием димера
G.27)
G.27J
«Теннисный мячик»
Рис. 7.23. Самосборка двух комплементарных искривленных строительных блоков с
образованием капсулы типа «теннисный мячик»
f^j4 7. Темплаты и самосборка
в форме теннисного мячика G.27J, рис. 7.23. Образование димера было
подтверждено:
• !Н ЯМР-спектроскогтией в растворе CDC13 и C6D6: NH-протоны значительно
сдвинуты в слабое поле по сравнению с модельными соединениями, которые не
образуют димерных капсул из-за наличия в их молекулах электроноакцепторных
карбоксильных атомов кислорода, связанных водородной связью с NH-протонами;
• масс-спектрометрией: пик молекулярного иона для димера наблюдается в
различных условиях ионизации;
• осмометрией: измеренная молекулярная масса соответствует димеру.
• Рентгеновской дифракцией: длина всех восьми водородных связей N—Н-0
равна 2.78-2.89 А (рис. 7.24).
Даже несмотря на все эти доказательства, образование димера G.27J было
настолько неожиданным, что Ребек захотел получить точное подтверждение факта
существования димера и в растворе, и твердом состоянии. Из-за того, что две половины
димера идентичны, трудно было быть абсолютно уверенным в том, что эффекты,
отражающиеся в ЯМР-спектре раствора, действительно, определяются
образованием димера. У Ребека не было возможности провести прямое сравнение с
индивидуальным мономером G.27) в тех же условиях. Окончательное доказательство
образования этих замечательных ансамблей было получено благодаря их способности
капсулировать метан. Добавление метана к раствору G.27J приводит к сдвигу
резонанса СН4 от 0.23 м.д. (свободный метан в CDC13) до —0.91 м.д., что согласуется с
расположением сигнала метана-гостя в области магнитного экранирования ариль-
ных колец хозяина. Подобные сдвиги наблюдали для широкого круга небольших
молекул-гостей. Этот значительный сдвиг в сильное поле является следствием
магнитной анизотропии арильных колец хозяина - производного дурола. Капсулиро-
ванный метан находится в области экранирования кольцевого тока (ср.
Дополнение 3.4).
Рис. 7.24. Кристаллическая структура
димера G.27Jтипа «теннисный мячик»
7.5. Самосборка закрытых комплексов с помощью водородных связей %15
Основные особенности соединения G.27) - кривизна, присущая гликолуриль-
ным G.28) строительным блокам (разд. 5.3.1.3), и его самокомплементарность. Эта
молекула содержит четыре донора и четыре акцептора водородной связи. Ее
сконструировали по аналогии с гексамерным кукурбитурилом G.29), который может
одновременно связывать и катионы щелочных металлов за счет ансамбля
карбонильных атомов кислорода, организованных в форме «насест», и нейтральные
молекулы-гости. (Были определены константы связывания с гостем-фураном в
водном растворе, значения которых не превышают 7140 М.)
Н
н
0
Гликолурил
G.28,
Кукурбитурил
G.29)
Полость, образованная димером G.27J, достаточно велика для капсулирования
не только метана, но и других небольших нейтральных гостей, таких, как галоген-
алканы. Это во многом напоминает связывание гостей криптофанами (см. разд.
5.3.2). Измерения константы связывания интегрированием пиков для связанных и
несвязанных гостей дали значения Кп, приведенные в табл. 7.2. *Н ЯМР-исследо-
вание при разных температурах позволило установить термодинамические
параметры. Эти результаты свидетельствуют о том, что связывание гостя приводит к
энтропийным потерям в 80-190 Дж-К^'-моль. Однако такой процесс дает выигрыш
в энтальпии и, в целом, — выигрыш в свободной энергии.
Используя мотив гликолурила, группа Ребека сконструировала также
самособирающиеся капсулы еще больших размеров. В частности, димеры G.30J и G.31 J,
названные мячами для софтбола* из-за того, что по размеру они больше теннисного
мячика, образуют полости, способные капсулировать большие гости, например
1-адамантанкарбоновую и 1-ферроценкарбоновую кислоты (Ки = 770 и 280 1VH
соответственно для G.30)). В растворе «-ксилола (слабый гость) в отсутствие больших
гостей 'Н ЯМР-сигналы для G.30) широкие, что свидетельствует об обмене между
* Софтбол — разновидность бейсбола, в основном для детей и женщин. (Примеч.
переводчика.)
616
7. Темплаты и самосборка
Таблица 7.2. Константы связывания
гостя, капсулируемого димером G.27J
(О °С, CDC13)
Гость
СНС13
CF4
СН2С12
с2н4
сн4
0.04
2.81
4.00
278
303
мономером G.30) и его димером и, следовательно, о том, что образование димера,
действительно, темплатируется гостем. Прибавление гостя приводит к резким
сигналам. Введение фенольной гидроксильной группы в молекулу хозяина, как в G.31),
делает образование димера еще более благоприятным процессом: добавляются
восемь водородных связей (типа О-Н-О=С), удерживающих два компонента вместе.
Ребек назвал это соединение мячом для софтбола с застежкой типа «молния».
Термодинамический анализ таких больших «софтбольных» соединений, содержащих
полость, показал, что, в отличие от небольшого G.27J, капсулирование гостя ими
определяется выигрышем энтропии: большие частицы-гости замещают
капсулированные молекулы растворителя (схема 7.7). В воде это должно было быть
эквивалентно гидрофобному эффекту. Однако большая часть химии «мячей для
софтбола» относится к ароматическим растворителям типа бензола и ксилолов.
О
X = Н: G.30)
X = ОН: G.31)
R = CO2-i-C5HH
Темплатирующие гости
Можно ожидать, что такой управляемый энтропией механизм капсулирования
гостя должен приводить к увеличению количества свободных частиц в конце этого
процесса. Отсюда следует, что внутри «мяча для софтбола» перед связыванием
гостя должна размещаться более чем одна молекула растворителя. Это было установ-
7.5. Самосборка закрытых комплексов с помощью водородных связей
617
Схема 7.7. Капсулирование гостя хозяином — «мячом для софтбола» — управляется
энтропийными (сольватофобными) эффектами
лено растворением «мяча для софтбола» в смеси двух разных, но родственных
растворителей - C6D6 и C6D5F - и с помощью ЯМР-спектроскопии (отсюда и
необходимость в дейтерированных растворителях). В случае одного растворителя
наблюдается только один «софтбольный» комплекс. Использование смеси растворителей
приводит к трем различным комплексам хозяин—гость, представляющим собой
«мячи для софтбола», которые содержат либо два бензольных гостя, либо две фтор-
бензольные молекулы, либо по одной каждого вида. Следовательно, «мяч для
софтбола» может капсулировать две молекулы растворителя.
Структуры «теннисный мячик» и «мяч для софтбола» были сконструированы в
виде капсул, которые могут выступать в качестве «реакционных сосудов» по
аналогии с полукарцерандами Крама и сотрудников (разд. 5.3.6). Преимущество «мячей
для софтбола» состоит в том, что их получить легче, потому что основная стадия ди-
меризации с образованием полости осуществляется путем самосборки. В синтезе
полукарцерандов это стадия кинетически контролируемого образования ковалент-
ной связи. Большие «мячи для софтбола» были использованы в качестве хозяев в
реакциях Дильса-Альдера. Полость хозяев, таких, как G.31J, достаточно большая
для одновременного капсулирования л-хинона и циклогексадиена-1,3. По
истечении одного дня наблюдалось образование капсулированного продукта
присоединения (схема 7.8). Контрольные эксперименты с хозяевами, неспособными к димери-
зации, или с гостями-диенофилами, слишком большими для размещения в
полости, показали, что реакция не происходит. К сожалению, эту систему невоз-
Схема 7.8. Катализируемое хозяином — «мячом для софтбола» — присоединение
по Дильсу—Альдеру
фил 7. Темплаты и самосборка
р
можно использовать в катализе, так как образование продукта присоединения
замедляется самоингибированием: продукт присоединения эффективно блокирует
полость, не допуская дальнейшего катализа реакции хозяином.
Интересно рассмотреть природу увеличения (стехиометрической) скорости этих
реакций внутри полости «мяча для софтбола». В обычных каталитических
реакциях, идущих, например, на центрах переходных металлов, значительную роль играет
статистический эффект. Реагенты сближаются друг с другом при одновременной
координации с металлом. Это чрезвычайно повышает их эффективную
концентрацию и, следовательно, скорость любой бимолекулярной реакции между ними.
Подобный эффект также наблюдается при образовании тройных комплексов в
органическом катализе (разд. 7.8). Можно легко рассчитать эффективную
концентрацию веществ-гостей в полости хозяев, таких, как G.31J, зная объем
полости, который составляет -300 А3. Пример расчета для двух молекул дейтериро-
ванного бензола приведен ниже.
Количество молекул C6D6 на полость: 2 (из 'Н ЯМР-эксперимента),
2 2
количество молей C6D6 на полость: ——- = 23 = 3.32-10~24,
iV л xj.yJJi' i\)
_ моли 3.3210"4 моль ,
концентрация СЛ), в полости: = 5 *— =11.1 мольдм~°.
0 ° объем 300-A0~yflM)J
Этот простой расчет свидетельствует о том, что внутриполостная концентрация
бензола превышает 10 М! Для сравнения, концентрация чистого жидкого бензола
равна 11.2 М (это значение легко вычисляется из плотности бензола 0.874 г-см~3 и
его молярной массы 78.11 гмоль). Внутриполостная область — это практически
чистый бензол. Если мы примем значение 81 А3 для молекулярного объема
бензола, полученное при моделировании молекулярной упаковки, то мы можем
показать, что в жидком бензоле и в полости хозяев типа «мячей для софтбола»,
содержащих две молекулы бензола, ~55% общего объема заполнено и 45% не заполнено.
Это наблюдение существенно для конструирования таких систем. Сравнительные
эксперименты показывают, что в основном гости или комбинации гостей, которые
занимают -55% объема полости, связываются хозяевами наиболее эффективно.
Доля объема, занятого гостем, может увеличиваться при специфических, энталь-
пийно выгодных, взаимодействиях, таких, например, как водородные связи между
гостями или между гостем и хозяином. Этот вывод особенно важен в случае крип-
тофанов, для которых наивысшее сродство при связывании проявляют гости,
имеющие размер, лишь немного меньший размера полости хозяина.
*"» Conn M.M. and Rebek /., Jr. Self-assembling capsules // Chem. Rev. 1997. Vol. 97.
P. 1647-1668.
7.5. Самосборка закрытых комплексов с помощью водородных связей § ^
7 • 5 • ^ Гигантские самособирающиеся капсулы
Поиск систем с капсулами еще больших размеров, особенно
устойчивых в растворе, в еще большей степени опирался на метод самосборки ансамблей,
способных капсулировать объемистые гости. Интересно, что последние результаты
по самособирающимся системам получены для каликс[41резорцинареновых
строительных блоков E.43), которые при ковалентном связывании образуют карцеранды
и полукарцеранды.
ОН
Каликс[4]резорцинарен
E.43)
R = СН3, CH2CH2Ph, ундецил и т.п.
Реакция E.43) с малоперспективным темплатным бакминстерфуллереном (С60)
в пропаноле-2 приводит к выделению в твердом виде самособранного полукарце-
ранда, в котором две резорцинареновые полусферы связаны экваториально
восемью молекулами пропанола-2 (одна на каждую гидроксильную группу верхнего
обода), рис. 7.25. Эти молекулы растворителя представляют собой эффективный
«клей», который связывает две половины капсулы вместе. Темплат С60 также
является составной частью структуры, отделяющей каждую капсулу от остальных. По-
видимому, роль С60 состоит во взаимодействии с арильными кольцами резорцин-
ареновых 2-фенилэтильных «ног» путем тг—тг-стэкинга. Объем полости составляет
~230 А3, что вполне достаточно для размещения в ней нескольких молекул о-ди-
хлорбензольных и пропанольных гостей. К сожалению, в твердом состоянии эти
гости сильно разупорядочены, а в растворе агрегаты не образуются.
В работе Дж. Этвуда и Л. Мак-Джилливрея (J. Atwood and L. MacGillivray, 1997)
были развиты основные принципы конструирования гигантских полукарцеранд-
ных хозяев методом самосборки, которые основаны на фундаментальных взглядах
древнегреческих математиков и философов в области геометрии. Этвуд и Мак-
Джилливрей показали, что основные усилия при таком синтезе требуются для
придания изначальной кривизны капсульным компонентам (см. разд. 5.3.1). Если в
создании трехмерной полости использовать неискривленные компоненты, то для
этого нужно взять много простых строительных блоков, как в синтезе супрамолеку-
лярного куба G.10), сборка которого зависит только от легкости изготовления
«ребер» и «углов». Этот подход контрастирует с намного более трудоемким
конструированием систем при использовании строительного блока G.27), которое требует
меньшего количества компонентов для капсулирования конечного объекта, потому
что такой блок уже содержит искривленные гликолурильные единицы. Фактически
трехмерная полость может быть образована только «плоскими» строительными
620
7. Темплаты и самосборка
Рис. 7.25. Кристаллическая структура кап-
сулярной бис(каликс[4]резорцинареновой)
части сокристалла на основе соединения
E.43) (R = CH2CH2Ph) с С60, связанной про-
панолом-2
блоками, если они комплементарны и могут быть соединены с помощью ковалент-
ных связей или нековалентных взаимодействий в трехмерные мозаики. Если
используют только один такой строительный блок, то образующаяся супрамолекула
будет представлять собой одно из пяти Платоновых тел (рис. 7.26). Тогда понятно,
что «плоскими» строительными блоками могут быть только треугольник, квадрат
или пятиугольник. Не существует других плоских правильных фигур, способных
при объединении полностью оградить трехмерное пространство. Это
автоматически означает, что невозможно оградить трехмерное пространство менее чем
четырьмя компонентами (минимум, необходимый для образования тетраэдра), если эти
компоненты изначально не искривлены, как в соединении G.27).
Если имеются два различных плоских компонента, то возможности
существенно расширяются. Комбинирование правильных двухмерных фигур двух типов для
Додекаэдр
Икосаэдр
Рис. 7.26. Пять Платоновых тел. Только эти
правильные трехмерные тела могут быть
собраны из двухмерных фигур одного типа
7.5. Самосборка закрытых комплексов с помощью водородных связей
(а)
Рис. 7.27. 13 Архимедовых полуправильных тел, собранных путем комбинирования двух
или более различных двухмерных фигур. Отметим, что в дополнение к треугольникам,
квадратам и пятиугольникам, допустимым в Платоновых телах, появляются также
шестиугольники, восьмиугольники и десятиугольники, но всегда в сочетании с другими фигурами.
В частности, структура (к) соответствует бакминстерфуллерену С60, которому для полного
ограждения пространства требуются пятиугольники, хотя в основном структура состоит из
шестиугольников
аналогичного полного ограждения трехмерного пространства дает 13 архимедовы
тел (рис. 7.27) и множество изоэдрических тел (призмы и антипризмы), полный пе
речень которых был приведен Каталаном (Catalan) в 1865 г.
Следует отметить, что гексаэдр, полученный Фуджитой на основе G.18), при
надлежит к каталановым телам, в то время как кубооктаэдры G.19) и G.20) соответ
ствуют структуре (б) на рис. 7.27. Структура (к), усеченный икосаэдр, хорошо изве
стна химическому сообществу как бакминстерфуллерен С60, составленный и
шести- и пятиугольников. Было точно установлено, и химически, и геометричесга-
что пятиугольники в этой структуре необходимы для осуществления полного тре>
мерного ограждения, так как без них формируются только открытые двухмерны
графитовые слои. Даже популярные сейчас нанотрубки (см. рис. 7.1), которые ее
стоят из шестиугольников, открыты с обоих концов, однако при добавлении к ни
пятиугольников образуются закрытые фуллерены (рис. 7.28).
Рис. 7.28. Фуллерен С60 - усеченный икосаэдр
I ^22 7 • Темплаты и самосборка
С учетом этих принципов Этвуд и сотрудники разработали синтез аномальной
молекулярной капсулы, геометрия которой соответствует геометрии усеченного
куба, структура (ё) на рис. 7.27. Реакция шести молекул каликс[4]резорцинарена
E.43), где R = Me или ундецил, с восемью молекулами воды приводит к
самосборке с образованием хирального сферического гексамера, объединенного 60
водородными связями (рис. 7.29), топология которого несколько напоминает строение
сферических вирусов типа гепатита В или человеческого римовируса. В случае ун-
децильного производного образующаяся структура также существует в растворе, а
ее форма напоминает небольшую обратную мицеллу (разд. 10.2). Усеченный куб
имеет полость диаметром 17.7 А и объемом 1375 А3 (ср. 300 А3 у самого большого
«мяча для софтбола»). Хиральность возникает из-за поворота резорцинареновых
(квадратных) граней примерно на 23° по отношению к кубу, сформированному
молекулами воды. В терминах инженерии кристаллов существование четырех
внутримолекулярных водородных связей у каждого резорцинарена означает, что каждый
из них является донором квадрупольной водородной связи. Вследствие
сферической природы капсулы и локализации молекул воды на центрах симметрии третьего
порядка каждая молекула воды способна участвовать только в трех водородных
связях и, следовательно, создавать грань треугольной формы. В результате, из 64
кислых протонов в ансамбле A6 от воды и 6-8 = 48 от резорцинаренов) только 60
участвуют в водородных связях внутри капсулы, а оставшиеся четыре, расположенные
тетраэдрически, образуют связи с соседними капсулами.
Если гексамер резорцинарена только напоминает обратную мицеллу, то л-суль-
фокаликс[4]арен E.50а) на самом деле формирует структуру наподобие везикулы с
внутренней и внешней водной фазой (G. Orret al., 1999). Направленные наружу
гидрофильные сульфогруппы верхнего обода стабилизируют везикулу,
взаимодействуя с катионами металла. Этот ансамбль, образующий капсулу с внутренним
объемом 1700 А3, представляет собой трехкомпонентную систему, состоящую из
пентанатриевой соли E.506), нитрата лантаноида, например La(NO3K-6H2O, и пи-
ридин-М-оксида, который добавляют в качестве солиганда для оксофильного
катиона лантаноида. Капсулообразующие компоненты берут в соотношении 2:1:2.
Однако если соотношение изменить на 2:1:8, то получается бесконечный спиральный
трубчатый ансамбль с тем же диаметром (ср. с вирусом табачной мозаики). Эти ре-
Рис. 7.29. Пространственная модель структуры
усеченного куба (е), рис. 7.27, образовавшегося
из шести молекул каликс[4]резорцинарена и
восьми молекул воды
7.5. Самосборка закрытых комплексов с помощью водородных связей
623
(а)
(б)
(в)
| 2.8 нм
Схема 7.9. Схематическое изображение организации сферических (а—в) и трубчатых
(г-е) ансамблей л-судьфокаликсD]арена E.50а). (ж~и) Образование агрегатов более
высокого порядка путем ассоциации сфер и трубок. (Воспроизведено с разрешения Orr G. et al.,
1999)
акции приведены на схеме 7.9, а структуры сферического и трубчатого ансамблей -
на рис. 7.30 и 7.31. По аналогии с архимедовыми телами изумительный по красоте
металлокаликсареновый ансамбль, показанный на рис. 7.30, имеет сходство с
большим ромбокубооктаэдром (ромбоусеченный кубооктаэдр), хотя и содержит
отклонения от правильной геометрии последнего.
7. Темплаты и самосборка
Рис. 7.30. Структура сферического лантаноидного л-сульфокаликс[4]аренового ансамбля.
(а) Пространственное изображение молекулы вдоль оси псевдопятого порядка. Пиридин-
N-оксид и один каликсарен изображены в виде палочек. Группы S03 выстраиваются на
поверхности сферы, арильные кольца формируют гидрофобную оболочку, полярное ядро
содержит 30 молекул воды и два иона Na+. (б) Поперечное сечение кластера [Na(H2ON]2
в ядре. (Воспроизведено с разрешения Atwood J.L., 1999)
Рис. 7.31. Структура
трубчатого лантаноидного л-сульфо-
каликс[4]аренового ансамбля.
(а) Пространственное
изображение, параллельное оси трубки,
показывает шесть витков
спирали. л-Сульфокаликс[4]ареновые
анионы, изображенные в виде
палочек, относятся к двум
виткам спирали. Витки спирали
образуются за счет и—л-стэкинг-
взаимодействий между
каликсаренами и пиридин-
N-оксидными гостями (также
показаны палочками).
(б) Поперечное сечение гидра-
тированных ионов Na+ и La3+
внутри цилиндрического
канала. (Воспроизведено с
разрешения Atwood J. L, 1999)
7.5. Самосборка закрытых комплексов с помощью водородных связей
*""* MacGillivray L.R. and AtwoodJ.L. Structural classification and general principles for
the design of spherical molecular hosts // Angew. Chem., Int. Ed. Engl. 1999. Vol. 38.
P. 1018-1033.
7*5*3 Розетки
Другой подход к пониманию пользы слабых взаимодействий при
конструировании больших структур и агрегатов выбрала группа Дж. Уайтсайдса
(G. Whitesides) из Гарвардского университета (США). Эти исследователи
установили, что комплементарные пары дискообразных молекул меламина G.32) и циану-
ровой кислоты G.33) могут образовывать за счет водородных связей кольца
(«розетки») и цепочки («ленты») в твердом состоянии, как показано на рис. 7.32.
Розетка
Лента
H
1
^N Nx
н y
ч
0
1
H
N
1'''
^N"
H
N
1
H
o
N
H
1
v
II
^
\
1
H
У
N
1
H
H
1
Рис. 7.32. Розеточный и ленточный мотивы в структуре твердого комплекса
меламинциануровая кислота
626
7. Темплаты и самосборка
В растворе образование розеточных структур с точки зрения энтальпииного вклада
умеренно благоприятно, поскольку приводит к формированию 18 водородных
связей, соответствующему значению АН, равному —100 кДжмоль в хлороформе.
Однако этот выигрыш гасится экстремально неблагоприятным энтропийным
вкладом, связанным с объединением шести независимых частиц. Уайтсайдс
намеревался синтезировать комплексы, в которых образованию розеток в растворе
могло бы способствовать уменьшение невыгодных трансляционного и
ротационного вкладов энтропии в общую свободную энергию агрегации. Это было
достигнуто двумя путями: предорганизацией (с помощью групп, которые соединяют
несколько компонентов вместе) и периферическим уплотнением, позволяющим избежать
образования ленточных структур (рис. 7.33).
Н
Циануровая
кислота
G.33)
н
Меламин
G.32)
Было доказано, что предорганизационный подход особенно успешен для
таких соединений, как G.34), которое реагирует точно с тремя молекулами циану-
ровой кислоты, давая агрегат из четырех частиц с молекулярной массой -2.5 кДа.
Эти агрегаты трудно исследовать, так как они плохо кристаллизуются и
разлагаются при попытке их изучения с помощью масс-спектрометрии. Степень
агрегации определяли методом солюбилизации, гель-проникающей хроматографии,
осмометрии и [И ЯМ Р. Стабильность розеточных агрегатов охарактеризована
простым параметром HB/GV-1), до некоторой степени связанным с выражением
Предорганизация
Периферическое уплотнение
Рис. 7.33. Стабилизация шести розеточных мотивов в растворе, основанная на их предор-
ганизации и периферическом уплотнении
7.5. Самосборка закрытых комплексов с помощью водородных связей
627
для KKCmin, определенным К. Хантером (уравнение G.12), разд. 7.3.2), где НВ -
число водородных связей, энтальпийно стабилизирующих ансамбль A8 на одну
розетку), N — число свободных частиц, образующих агрегат. Отсюда следует, что
при увеличении числа водородных связей увеличивается энтальпииная
стабилизация комплекса, а при увеличении числа свободных частиц увеличивается
энтропийная дестабилизация. Таким образом, для умеренно стабильного агрегата
G.34)G.33K параметр НВДЛМ) равен 6. Другие агрегаты схематически
представлены на рис. 7.34.
NH2
О HN N NHR
= (СН2JС(СН3)з
628
7. Темплаты и самосборка
4) to
«,
Число водородных
связей (НВ)
Число ч
6
9
4
4
7
10
5
3.8
4.5
6
6
6
6
9
Мол. ма
1.2
4.5
2.5
2.7
4.7
6.4
5.5
4 12
2 18
5.7
4.7
4.1
18
36
54
Рис. 7.34. Увеличение
относительной стабильности
производных комплекса
меламинциануровая кислота
с ростом параметра
HB/(jV—1). (Воспроизведено
с разрешения Whitesides G.M.,
Simarek E.E., MathiasJ.P. et al.
Noncovalent synthesis — using
physical-organic chemistry to
make aggregates // Ace. Chem.
Res. 1995. Vol. 28. P. 37-44)
Whitesides G.M., Simarek E.E., Mathias J.P. et al. Noncovalent synthesis — using
physical-organic chemistry to make aggregates // Ace. Chem. Res. 1995. Vol. 28.
P. 37-44.
Катенаны и ротаксаны
Raymo KM. and Stoddart J.F. Interlocked macromolecules // Chem. Rev. 1999.
Vol. 99. P. 1643-1666.
У .6.1 Введение
Катенаны - это соединения, молекулы которых состоят из двух или
более колец, механически сцепленных друг с другом без какого-либо химического
взаимодействия между ними. Их кольца невозможно разъединить без разрыва
химической связи. Катенаны называют по количеству сцепленных колец; например,
[2]катенан состоит из двух таких колец (рис. 7.35). Окончание «-«и» придано их
названию по аналогии с названием «алкш/ы»; обычно катенан входит в состав
органического фрагмента, хотя изредка он состоит только из углеводородных частей.
Термины «[я]катеня«д» и <<[я]катенат» также используют по аналогии с терминами
«криптянд» и «криптшя» в случаях, когда система сцепленных колец может
выступать в качестве лиганда для металлического центра. Катенанд — свободный лиганд,
7.6. Катенаны и ротаксаны
629
[2]Катенан ([2]катенанд)
[2]Катенат
[3]Катенан ([3]катенанд)
■мнима
CD CD
[2]Псевдоротаксан [2]Ротаксан [з]Ротаксан
Рис. 7.35. Номенклатура катенанов, ротаксанов и псевдоротаксанов
который образует катенатный комплекс в присутствии такого центра. По логике его
можно было бы называть катаплексом, но этот термин в литературе практически не
используется!
Ротаксаны — это соединения, молекулы которых состоят из макроцикла и
длинной открытой цепи, продетой сквозь этот цикл, подобно нитке сквозь ушко
иголки. Истинные ротаксаны опять-таки не могут распадаться на отдельные цикл и
цепь без разрыва химических связей, и поэтому линейная, цепочечная часть их
молекул содержит концевые, часто объемистые группировки, которые слишком
велики для прохождения сквозь циклический фрагмент. Ротаксаны без таких барьеров,
в которых «нитка» может выскальзывать из «иголки», называют псевдоротаксанами.
Псевдоротаксаны зачастую являются исходными соединениями для получения и
ротаксанов, и катенанов. Типичные синтетические процедуры включают темплат-
ную самосборку псевдоротаксана при участии иона металла, электростатических
сил или водородных связей и последующее образование цикла (катенаны) или
прививку к одному или обоим концам объемистых концевых групп (ротаксаны),
рис. 7.36. Образование этих веществ представляет не только академический
интерес. Их применение при конструировании сложных молекулярных приборов типа
переключателей, создании материалов с новыми физическими свойствами и
поверхностной иммобилизации каталитически фото- или редокс-активных веществ
1630
7. Темплаты и самосборка
Гость
[2]Катенан
[2]Псевдоротаксан
Хозяин
[2]Ротаксан
Рис. 7.36. Схема синтеза катенанов и ротаксанов в терминах подхода хозяин—гость
без необходимости изменения их свойств химической модификацией имеет
большие перспективы.
Химический состав катенана или ротаксана идентичен составу двух (или более)
отдельных компонентов, но продевание одного компонента сквозь другой имеет
важные последствия для физических и химических свойств образующегося
агрегата. Прежде всего, у таких сцепленных или продетых молекул существует особая
форма изомерии (отличная от традиционной химической изомерии, такой, как
цис-/транс- nfac-/mer-).
Катенаны являются топологическими изомерами их отдельных циклических
компонент, а форма такой изомерии катенанов родственна карцерии (разд. 5.3.6.4).
В этом контексте топология агрегата определяется количеством и типом точек
пересечения, если структура нарисована на плоскости (например, на куске бумаги).
Мы не можем нарисовать [2]катенан без, по крайней мере, двух точек пересечения,
тогда как два отдельных макроцикла не имеют ни одной точки пересечения. Эти два
топологических изомера являются, таким образом, принципиально различными.
Формально они гомеоморфны, т.е. не изотопны.
Интересно, что, следуя этому строгому определению, ротаксаны не являются
топологическими изомерами своих индивидуальных компонентов, так как, по
меньшей мере концептуально, цикл ротаксана всегда может соскользнуть с конца
7.6. Катенаны иротаксаны
631
Топологические изомеры
Топологические
диастереомеры
Топологические энантиомеры
Рис. 7.37. Топологическая изомерия, диастереомерия и хиральность [2]катенана,
тройного узла и дважды сцепленного [2]катенана
линейной части; при этом образуются отдельные компоненты. На самом деле это
справедливо только для псевдоротаксанов. В случае же истинных ротаксанов
разделение компонентов произойти не может из-за того, что запирающие (концевые)
группы слишком велики. Различие между реальной молекулой и ее
топологическим описанием состоит в том, что в топологии все компоненты могут
неограниченно растягиваться до тех пор, пока не порвутся. Существование топологических
изомеров подразумевает также существование топологических энантиомеров.
Например, хиральный катенан (или другая молекулярно взаимозамкнутая система)
хирален именно благодаря сцеплению, а не хиральности любого из компонентов
(рис. 7.37).
Ь-ч
Breault G.A., Hunter C.A. and Mayers P.C. Supramolecular topology // Tetrahedron.
1999. Vol. 55. P. 5262-5293.
632 ~l • Темплаты и самосборка
.6.2 Статистический подход
к синтезу катенанов и ротаксанов
Существует два различных подхода к синтезу катенанов:
статистический подход и подход, основанный на самосборке - «управляемый синтез».
Статистический подход учитывает небольшую вероятность того, что макроциклизация
может произойти в тот промежуток времени, когда линейный компонент проходит
сквозь макроциклический компонент. Поскольку вероятность этого события
весьма мала, то статистический путь приводит к низким выходам и в основном
представляет исторический интерес. Статистический подход впервые был использован
при синтезе [2]катенана G.35), схема 7.10, циклизацией длинноцепочечного ди-
эфира G.36), продетого сквозь цикл дейтерированного циклоалкана С34 G.37)
(Е. Wasserman, 1960). Хотя общий выход реакции менее 1%, существование катена-
на было четко доказано. Сравнительно полярное соединение — [2]катенан —
вместе с другими полярными продуктами макроциклизации и исходными веществами
отделили от свободного циклоалкана. Затем разорвали цикл ацилоина (гидрокси-
кетон) и собрали неполярную фракцию. Понятно, что дейтерированный
макроцикл в этой фракции мог возникнуть только из катенанового продукта.
Статистический подход использовали также и в синтезе ротаксанов. Ротаксаны
и псевдоротаксаны получили кипячением при 120 °С большого числа циклических
углеводородов, содержащих 11-39 групп -СН2—, с линейным углеводородом,
содержащим концевые трифенилметильные запирающие группы. Обнаружили, что
при такой (повышенной) температуре циклы больших размеров случайным
образом «проскальзывали» через концевые трифенилметильные группы. При
охлаждении в этой смеси образуется небольшое количество (менее 2%) ротаксана G.38).
Было найдено, что при комнатной температуре ротаксаны, содержащие
макроциклы менее С29, стабильны и не распадаются на компоненты, тогда как макроциклы
С33 и выше были предельно лабильны.
Чтобы увеличить статистические выходы, проводили повторяющиеся реакции с
циклическим компонентом, иммобилизованным на твердой подложке. Так,
макроцикл С28 был ковалентно закреплен на поверхности смолы и обработан гантелевид-
Y
OEt
/\
1. Na, ксилол, 140 °С
2. HOAc H0
C3IH57D,
OEt 3I 5
G.36) G.37) G.35)
Схема 7.10. Первый синтез [2] катенана G.35), основанный на статистическом подходе
7.6. Катенаны иротаксаны
633
Смола
НО(СН2)юОН
Ph3CCl
».
2. Na2CO3, MeOH
нагревание
О/-(СН2I0—О
(СН2J4
Рис. 7.38. Статистический синтез ротаксана на твердой подложке. Ротаксан был получен
с выходом 6%
ным компонентом G.38) 70 раз. Образовавшийся стабильный ротаксан отделяли от
смолы и очищали от примесей, выход составил 6% (рис. 7.38). Это соединение
было исследовано ИК-спектроскопией и другими методами, основанными на
деструкции вещества.
G.38)
*""* Amabilino D.B. and Stoddart J.F. Interlocked and intertwined structures and
superstructures // Chem. Rev. 1995. Vol. 95. P. 2725-2828.
7 »6 • 3 Псевдоротаксаны
Понятно, что низкие выходы, полученные при статистическом
подходе к синтезу катенана и ротаксана, свидетельствуют о необходимости более
направленного подхода, при котором химик не будет полагаться на удачу в продевании
одного молекулярного компонента в другой. Очевидная стратегия направленного
синтеза катенана и ротаксана - стимулирование процесса продевания
(ассоциация) реагентов перед реакциями циклизации или запирания, которые ковалентно
фиксируют весь ансамбль. Если реагенты предварительно ассоциированы в раство-
634
7. Темплаты и самосборка
ре в виде самособранного комплекса хозяин—гость (константа связывания
сравнительно велика), то высока вероятность того, что и во время реакции они будут пред-
организованы желаемым способом. Обычно этот предреакционный комплекс
хозяин—гость является псевдоротаксаном и представляет собой самособранный
темплат для ковалентного синтеза собственно ротаксанов и катенанов (см.
рис. 7.36).
оТо] юга
C.96)
C.95)
Мы уже видели (разд. 3.11.6), насколько сильно п—я-стэкинг-взаимодействия
между арильными корандами C.95) и C.96) и гербицидным рецептором параква-
том C.92) (например, C.95) + C.92), Ки = 730 М~' в Ме2СО) влияют на
включение электронодефицитного гостя в корандное кольцо как в твердой фазе, так и в
растворе. Фактически корандное кольцо с внедренным в него дикватом - [2]псевдо-
ротаксан. Эта система легла в основу интенсивных работ Дж. Стоддарта и
сотрудников по использованию таких взаимодействий для получения широкого ряда
взаимопроникающих молекул. В случае параквата это и электронодефицитный гость,
и электроноизбыточный хозяин. В основном устойчивость псевдоротаксанов
возрастает с увеличением длины цепи: так, для R = Me константа связывания
Ки(Ме2СО) = 17 М-1, тогда как для R = Н(ОСН2СН2K АГ,,(Ме2СО) = 2520 М.
Каждый подход имеет большие перспективы в синтезе ротаксанов и катенанов.
Такой пример приведен на рис. 7.8, на котором изображен [2]псевдоротаксан,
состоящий из электроноизбыточных подандов типа ROC6H4OR (где R = Me, G.39);
R = H(OCH2CH2)rt, n = l-f-4, G.40)), заключенных в прямоугольный «ящик»,
собранный из двух паракватных производных G.41).
7.6. Катенаны иротаксаны
635
G.41)
Основываясь на последнем подходе, Стоддарт и сотрудники сконструировали
высшие псевдоротаксаны, такие, как [3]псевдоротаксан G.42), скорее
напоминающий бусинки, нанизанные на нитку. Тетракатионный циклофан стабилизирован
я-я-стэкинг-взаимодействиями и взаимодействиями, возникающими в результате
переноса заряда между арильными кольцами, а также сольватацией
положительного заряда атомами кислорода крауна и водородными связями С— НО между
относительно кислыми арильными С-Н-протонами и атомами кислорода крауна.
Перенос заряда придает характерную оранжевую окраску всем комплексам с таким
способом связывания.
сГЛААГъ
ОВп
0 0 0 0 0
W \_У W W
ОВп
0 0 0 0 0
V7WWW
G.42)
636 7. Темплаты и самосборка
7*6.4 Ротаксаны
Наиболее очевидный путь получения ротаксана из псевдоротаксана -
прикрепление объемистого заместителя к одному или обоим концам продетой
молекулы. Этот прием называют методом «продевания» (рис. 7.39). Например,
реакция [2]псевдоротаксанов, образованных диэфиром G.39), диолом G.40) и бипири-
диниевым рецептором G.41), с три(изопропил)силилтрифторметансульфонатом в
присутствии лутидина (диметилпиридин) приводит к соответствующим триизо-
пропилсилилированным [2]ротаксанам G.43) с выходом -21%. Это же соединение
с выходом 14% было получено методом «защелкивания» (см. рис. 7.39), в котором
использована триизопропилсилилированная нить, по реакции, показанной на
схеме 7.11. И наконец, еще один подход к синтезу ротаксанов — метод
«проскальзывания», который включает использование концевой группы, которая достаточно
мала, чтобы пройти сквозь макроцикл при контролируемом нагревании
(кипячение), но не обладает энергией, необходимой для преодоления конформационного
барьера при низких температурах, как это наблюдалось в статистическом подходе к
синтезу ротаксана G.38).
CI
Защелкивание
п
— Продевание ■
D
Проскальзывание
Рис. 7.39. Способы синтеза ротаксанов путем самосборки электроноизбыточных
и электронодефицитных арильных фрагментов. (Воспроизведено с разрешения AtwoodJ.L.
DaviesJ.E.D., MacNicol D.D. and Vdgtle F. Comprehensive supramolecular chemistry. Oxford:
Pergamon, 1996. Vol. 9. P. 93)
7.6. Катенаны и ротаксаны
637
ч . Г\Г\Г\
)—si-o о о о
Вг
Г\ГЛГ\
Si-O О О О
l.McCN,A8PF4.fI[OMBOcyT.
2.
О О О O-Si
WWW
О О О O-Si—(
www Г\
G.43)
Схема 7.11. Синтез [2]ротаксана G.43) методом защелкивания
Катенаны, формируемые
с помощью я-я-стэкинг-взаимодействий
[п] Катенаны состоят из двух или более колец, сцепленных друг с
другом так, что их невозможно разъединить без разрыва ковалентной связи в одном из
них. Число колец обозначают буквой «вих названиях. Простейший катенан - это
[2]катенан. Как и в случае ротаксанов, при синтезе катенанов используют п—я-стэ-
кинг-взаимодействия. Группа Стоддарта (P. Ashton et al., 1989) получила [2]катенан
путем управляемого темплатного синтеза. Процедура получения [2]катенана G.44)
с использованием метода защелкивания (схема 7.12) включала продевание электро-
нодефицитного катиона G.45) сквозь электроноизбыточный краун-эфир C.95) с
последующим замыканием кольца.
Именно благодаря удачному подбору электронных свойств пар реагентов был
реализован темплатный эффект и получен [2]катенан G.44) с выходом более 70%.
Даже в аналогичной пятикомпонентной реакции самосборки, в которой краун-
эфир C.95) реагировал с двумя эквивалентами 4,4'-бипиридиния G.45) и двумя
эквивалентами а,а'-дибром-я-ксилола G.46), [2]катенан G.44) образуется с выходом
42%. Кристаллическая структура этого соединения (рис. 7.40) ясно указывает на то,
что его два кольца взаимозамкнуты; я-электроноизбыточный хиноновый и я-элек-
тронодефицитный бипиридиниевый циклы разделены расстоянием ~3.5 А. Более
638
7. Темплаты и самосборка
C.95)
1. MeCN. AgPF6, tI0MH, 2 сут.
2. NH^PFft, H;O
Выход 70%
о о о о о
G.45)
о о ° °
G.44)
G.46)
Схема 7.12. Темплатный синтез [2]катенана G.44) (по Ashton P. et al., 1989)
того, те же самые стэкинг-взаимодействия наблюдаются между соседними
молекулами [2]катенана в твердом состоянии. При этом образуется бесконечный донорно-
акцепторный ансамбль, подтверждающий темплатную природу этого синтеза.
[2]Катенан G.44) обладает очень интересными динамическими свойствами,
которые можно обнаружить с помощью 'Н ЯМР-спектроскопии. В частности, при
81 °С протоны хинонового цикла дают синглетный сигнал при 5 4.57 м.д. При
комнатной температуре замедленное вращение краун-эфира внутри бипиридиниевого
макроцикла (процесс I, рис. 7.41) приводит к появлению двух отдельных сигналов
при 5 3.45 и 6.16 м.д., первый из которых явно относится к экранирующему эффек-
7.6. Катенаны иротаксаны
1539
Рис. 7.40. Кристаллическая
структура [2]катенана G.44)
ту внешней поверхности бипиридиниевого циклофана G.41). Используя метод ко-
алесценции (Дополнение 3.4), можно рассчитать энергетический барьер этого
вращения, равный 65.3 кДж-моль. За аналогичным вращением циклофана внутри
краун-эфира можно проследить таким же образом (процесс II, рис. 7.41). Расчеты
методом коалесценции дают величину энергетического барьера вращения
51.1 кДж-моль~' с коалесценцией, происходящей при —45 °С в растворе ацетона.
Великолепные результаты, полученные при синтезе [2]катенана G.44) из краун-
эфира C.95), сопоставили с препаративными выходами при синтезе из других я-до-
норных краун-эфиров с различной величиной кольца (табл. 7.3).
Процесс I
65.3 кДжмоль
Процесс II
51.1 кДжмоль"
О О О О С
V7 \-У V-У W
Рис. 7.41. Динамические процессы в молекуле [2]катенана G.44)
7. Темплаты и самосборка
Таблица 7.3. Зависимость выходов [2]катенанов,
полученных согласно схеме 7.12, от размера
кольца краун-эфира
Краун-эфир
31-Краун-9
34-Краун-Ю
37-Краун-И
40-Краун-12
43-Краун-13
46-Краун-14
л
0
1
2
2
3
3
т
1
1
1
2
2
3
Выход, %
10
70
55
54
40
49
Очевидно, что 34-членное кольцо оптимально. В случае небольших колец
выходы очень быстро понижаются из-за малого соответствия друг другу исходных
соединений. Низкие выходы наблюдались также для более крупных краунов из-за
увеличения конформационной свободы их молекул. Большая гибкость таких колец
отражается и на энергии активации вращения кольца (процесс II), которая
примерно на 10.5 кДжмоль меньше для более крупных краун-эфиров.
Комплекс G.44) был также исследован методом циклической вольтамперомет-
рии (см. Дополнение 4.1), которая показала, что это соединение дает большое число
редокс-пиков по сравнению с числом пиков, соответствующих двум его
компонентам. Это прямой результат п—тг-стэкинг-взаимодействий. Электрохимическое
восстановление бипиридиниевого циклофана G.41) в катенане протекает в три стадии:
1. Первое одноэлектронное восстановление более доступной, менее стабильной
бипиридиниевой части вне полости краун-эфира (эта часть действует как
акцептор только для стабилизации гидрохинонового фрагмента).
2. Второе одноэлектронное восстановление бипиридиниевой части внутри
полости.
3. Наконец, двухэлектронное восстановление, соответствующее нейтрализации
обоих бипиридиниевых колец.
7.6. Катенаны иротаксаны
641?
В отличие от комплекса G.44) соединение G.41) претерпевает лишь два двухэле-
ктронных восстановления. Для G.44) не наблюдаются четыре одноэлектронных
процесса. Причина состоит в следующем: после первых двух одноэлектронных
процессов восстановления вращение кольца (процесс II, рис. 7.41) становится
настолько быстрым на шкале времен вольтамперометрии, что третий и четвертый
процессы одноэлектронного восстановления дают один пик двухэлектронного
восстановления. Циклические вольтамперограммы этих соединений представлены
на рис. 7.42.
Принцип самосборки, взятой на вооружение Стоддартом с сотрудниками,
оказался весьма плодотворным для синтеза катенанов. Начиная с синтеза [2]катенана
G.44) в 1989 г., эти ученые, используя описанную выше методологию, смогли
сконструировать [3]-, [4]- и даже [5]катенаны. Синтез [л]катенанов со значениями п > 2
должен основываться на соединениях с кольцом большого размера, которые могли
бы вместить части двух сцепленных колец в свои полости. Первые исследования в
этой области были сфокусированы на димере соединения C.95) — тетракис(л-фе-
нилен)-68-краун-20 G.47). В реакции с G.45) и G.46) в сравнительно мягких
условиях, сходных с показанными на схеме 7.12, образовался только [2]катенан G.48) с
низким выходом, что согласуется с большим размером кольца краун-эфира G.47)
(ср. табл. 7.3). Однако под давлением 10 кбар был получен аналогичный [3]катенан
G.49) с еще меньшим выходом (схема 7.13). Низкие выходы можно объяснить
высокой гибкостью 68-членного кольца.
Соединение G.48), как и его более мелкие [2]катенановые аналоги, проявляет
очень интересные динамические свойства, при которых небольшое катионное
кольцо циклофана ведет себя как молекулярный «поезд», который проезжает
четыре гидрохиноновые «станции» кольца крауна со скоростью 300 раз в секунду, что со-
-1.2 -0.6 0.0
Е, В (отн. НКЭ)
Рис. 7.42. Циклические вольтамперограммы циклофана G.41)
(а) и [2]катенана G.44) (б). (Воспроизведено с разрешения
Ambalino D.B. and Stoddart J.F. Interlocked and intertwined
structures and superstructures // Chem. Rev. 1995. Vol. 95. P. 2725-2828)
11-СтидДж. В.
r
о
;6
G.48)
1. DMF, 10 кбар, 25 "С, 5 сут.
2. NH4PF6, H2OО
3 1. MeCN, AgPF6, 25 "С, 5 сут.
2. NH4PF6> H2O
О% -^
Выход 12%
G.49)
G.46)
Схема 7.13. Синтез [3|катенана G.49) под высоким давлением
I
7.6. Катенаны и ротаксаны
643
ответствует энергии активации 59.0 кДж-моль. Если [2]катенан G.48) — «молеку-j
лярный поезд», то [3]катенан G.49) можно представить в виде «молекулярной
карусели». В растворе симметрия кольца крауна сохраняется. Это указывает на то, что
оба циклофана G.41) путешествуют по кольцу крауна (энергия активации
57.0 кДжмоль) согласованно и всегда остаются на диаметрально
противоположных гидрохиноновых «станциях».
Вслед за этой успешной работой Стоддарт и сотрудники показали, что, для тоге
чтобы конструировать даже несколько большие катенаны, циклофан G.41) нужне
заменить на акцепторную частицу больших размеров, способную вмещать два
гидрохиноновых фрагмента от различных молекул краун-эфира. Таким образом,
комбинирование большого кольца донора электронов — краун-эфира — с большими
циклофановыми катионами может привести к получению катенановых полимеров
Был синтезирован более крупный акцептор электронов - тетракатионный
циклофан G.50), в котором л-ксилольная часть бипиридиниевого фрагмента G.41)
замещена на л-бифенильный фрагмент.
G.50)
Ожидалось, что двухстадийный процесс, включающий защелкивание предшест
венников G.51) и G.52) вокруг двух молекул краун-эфира G.47), даст [3]катенан
содержащий в центре молекулу циклофана G.50), а последующее защелкивани
двух молекул G.45) и G.46) вокруг двух молекул краун-эфира в [3]катенане приведе
к желаемому [5]катенану. К сожалению, осуществление этой схемы закончилось не
удачей, которую Стоддарт и сотрудники приписали конформационной подвижное
ти краун-эфира G.47). Тогда они попытались применить ту же стратегию к краун
эфиру среднего размера - к G.53). Промежуточный [3]катенан G.54) получили npi
атмосферном давлении с выходом 3.5% и ввели его во вторую стадию. Реакция G.54
с G.45) и G.46) при сверхвысоком давлении привела к получению [4]катенана G.55
с 22%-м выходом и желаемого [5]катенана G.56) лишь в виде следов (схема 7.14;
Хотя выход ожидаемого продукта был удручающе низким, исследователи не отка
запись от этой стратегии и продолжили работу по синтезу макроскопических коли
644
7. Темплаты и самосборка
с
о
о
G.51) G.52) О
Г
О
I.MeCN.fKII8,21fleHb
2. NH4PF6, H,O
/ о
о
Q
G.53)
t
W
О N'
J
О
G-54)
J G.45) + G.46), DMF. М-14 кбар, Г,оиг 8 сут.
2 NH4PFVH,O
G.56)
Схема 7.14. Первый синтез [5]катенана G.56)
7.6. Катенаны и ротаксаны
645
честв [5]катенана. Они, в конце концов, достигли своей цели, еще раз изменив
структуру краун-эфира: на этот раз 1,4-дигидроксибензольные кольца в G.53)
заменили на 1,5-дигидроксинафталиновые G.57). Стратегия, представленная на схеме
7.14, привела к получению ожидаемого [5]катенана с удовлетворительным выходом
5% без использования высокого давления. Образовавшемуся соединению было
присвоено название «олимпиадан» из-за схожести его молекулы с символом
всемирных Олимпийских игр. Был также получен соответствующий [4]катенан с
выходом 31%. Рассказ о синтезе этой удивительной молекулы полностью приведен в
ключевой ссылке к этому разделу.
G.57)
Олимпиадан был охарактеризован 'Н ЯМР-спектроскопией, которая показала,
что эта молекула при 60 °С высокосимметрична вследствие быстрого вращения всех
фрагментов. Ее спектр при комнатной температуре значительно уширен, тогда как
при 0 °С резонансные линии, отнесенные к небольшим циклофанам G.41),
расщеплялись на два сигнала, соответствующих замораживанию вращательных
движений этих компонентов. Расчет энергии их вращения дал величину
61.5 кДжмоль. В масс-спектре [5]катенана четко фиксировались сигналы,
соответствующие олимпиадану в соединении с семью, восемью, девятью и десятью (из
возможных двенадцати) анионами PF^~, и аналогичные пики для [4]катенана
(рис. 7.43). Кристаллическую структуру олимпиадана определили участники
группы Д. Уильямса из Королевского колледжа (Лондон, Великобритания), которые
обнаружили его очень компактную упаковку (рис. 7.44). Этот результат — одно из
наиболее замечательных достижений супрамолекулярной кристаллографии.
Таким образом, подход, основанный на использовании
я-тг-стэкинг-взаимодействий в синтезе, подтвердил свою эффективность, и поэтому Стоддарт с сотруд-
646
7. Темплаты и самосборка
Рис. 7.43. Масс-спектр
[5]катенана олимпиадана.
(Воспроизведено с
разрешения Ambalino D.B. and Stoddart
J.F. Interlocked and
intertwined structures and
superstructures//Chem. Rev. 1995.
Vol. 95. P. 2725-2828)
100 -g
80 -=
60 -.
2873
[5]Катенан
4783
4928
3000
5000
никами продолжил работы по получению большого количества катенановых
соединений. Особый интерес вызывает хиральный бис([2]катенан) G.59), образующийся
путем защелкивания двух молекул бипиридиния G.45) с тетрабромпроизводным
G.58) в присутствии краун-эфира C.95). Отметим, что соединение G.59) не
считают [3]катенаном, потому что оно не имеет центрального кольца, которое проходит
сквозь два других. Это просто ковалентно связанная пара [2]катенанов. Хираль-
ность этого соединения следует из его 'Н ЯМР-спектра, который имеет
характеристический АВ-квартет диастереотопных протонов CH2N+ октакатионного цикло-
фана. Ясно, что существует небольшая вероятность (по стерическим причинам)
того, что циклофановый катион вращается по кольцу краун-эфира (процесс II). Од-
Рис. 7.44. Кристаллическая структура олимпиадана. (Stoddart /., 1997,
® «olympiadane.pdb»)
7.6. Катенаны иротаксаны
647
нако вращение крауна (аналогично процессу I) с энергией активации
67.0 кДж-моль, очень сходное с вращением его [2]катенанового аналога G.44),
имеет место. При низкой температуре в ацетоне-^6, по данным ЯМР-спектроско-
пии, G.59) существует в виде двух трансляционных изомеров.
Бис([2]катенан) G.59) был получен с выходом 13% путем одностадийной
самосборки из трех компонентов (пять молекул). Интересно, что октакатионный цикло-
Вг
G.58)
G.60)
54g 7. Темплаты и самосборка
фан до сих пор не выделен. Это означает, что электроноизбыточные краун-эфиры
темплатируют эту реакцию. Соединение G.59) не образуется при попытке
двойного защелкивания тетракатиона G.60) с а,а'-дибром-л-ксилолом G.46). Было
обнаружено, что выход бис([2]катенана) значительно возрастает при увеличении длины
ковалентного спейсера между двумя кольцами циклофанового катиона.
•""» Amabilino D.B., Ashton P.R., Reder A.S. et al. Olympiadane // Angew. Chem., Int.
Ed. Engl. 1994. Vol. 33. P. 1286-1290.
7.6.6 Принцип «вспомогательной связи»
в синтезе катенанов
Привлекательность методологии синтеза катенана, пионерами которой
были Дж. Стоддарт и сотрудники, — в ее относительной простоте, высоких выходах
и многосторонности. Благодаря возможности конструирования самособирающихся
систем хозяин—гость синтез катенанов стал программируемым. При этом
зацепление, осуществляемое методами самосборки, фиксируется ковалентной
модификацией. С учетом синтеза катенанов концепцию хозяин—гость можно обобщить в
виде принципа «вспомогательной связи», согласно которому хозяин и гость в
начальном комплексе, полученном с помощью самосборки, не различимы. В соответствии
с этим принципом также не имеет значения, какие силы удерживают их вместе. Они
могут быть электростатическими силами, ковалентными, координационными или
водородными связями и т.д. Вспомогательная связь — это инструмент настройки, с
помощью которого реагенты удерживаются в заданной позиции, обеспечивающей
получение желаемого катенанового продукта, рис. 7.45 (ср. с синтезом инсулина
методом самосборки с ковалентной модификацией, разд. 7.2.3).
Впервые принцип вспомогательной связи был использован в направленном
синтезе углеводородных катенанов Дж. Шиллом и А. Лютрингхаузом (G. Schill and
A. Liittringhaus), в качестве вспомогательной связи выступал стерический барьер.
Один из самых элегантных примеров такого подхода показан на схеме 7.15
(Е. Logeman et al., 1981). Эта процедура включает конструирование корандного
кольца, содержащего в качестве заместителя первичную аминогруппу G.61).
Эффект вспомогательной связи в этом предшественнике обеспечивается негативным
темплатным стерическим барьером, который разводит две хлоралкановые «руки» в
противоположных направлениях. Эти «руки» атакуют аминогруппу с
противоположных направлений, что приводит к образованию двойного анса-соединения
G.62) с высоким выходом. После завершения основной стадии циклизации,
G.61) —> G.62), необходимость во вспомогательной связи отпадает, и кольцо легко
отделяется от темплата при гидролизе НВг в уксусной кислоте с образованием ке-
тона G.63). И наконец, благодаря разрыву связи между атомом азота и
ароматическим кольцом высвобождается катенан G.64).
Существование катенановых соединений типа соединения G.64) установлено
различными спектральными методами. Очень важные данные о поведении катена-
7.6. Катенаны и ротаксаны
649
- вспомогательная связь
Рис. 7.45. Принцип вспомогательной связи в приложении к синтезу [2]- и [3]катенанов.
Вспомогательная Связь может быть ковалентной, координационной или нековалентной
нов получены методом масс-спектрометрии. Масс-спектры катенановых
соединений сильно отличаются от спектров ковалентно связанных предшественников
(таких, как G.63)), и масса первых существенно превышает сумму масс двух
индивидуальных компонентов. Масс-спектры катенанов характеризуются появлением
пиков с высокими значениями m/z, соответствующих исходным соединениям, а
также фрагментам, отвечающим переносу атомов водорода с одного макроцикла на
функциональную группу другого. Следовательно, первая стадия распада катенана
представляет собой разъединение двух колец, так как фиксируется очень мало
пиков, а затем появляются четкие пики фрагментации индивидуальных компонентов.
При этом спектр напоминает спектры индивидуальных макроциклов.
Обычно и [И ЯМР-, и 13СЯМР-спектроскопия демонстрируют сдвиги в слабое
поле резонансов, относящихся к ядрам сцепленной структуры. Этот сдвиг был
приписан ван-дер-ваальсовым взаимодействиям между двумя компонентами катенана.
Измеряя время спин-решеточной релаксации (Tj), установили также, что
движения сцепленных колец более ограничены, чем движения свободных макроциклов.
Подобная контролируемая методология была использована при получении
[3]катенана G.65), который существует в виде трех изомеров (рис. 7.46). В дополне-
№50
7. Темплаты и самосборка
С\
(СН2)
г'хо
ОМе.
ОМе'
О
.сно
N (СН2J3
СНО
(СН,).
2'23
G.62)
(СН2)„
(СН2JЭ
G.64)
(СН2)„
NCOMe
Схема 7.15. Направленный синтез [2]катенана G.64) с использованием негативного тем-
платного стерического барьера в качестве вспомогательной связи (по Logeman E. et al., 1981)
ние к карцерии (см. разд. 5.3.6.4) и топологической изомерии, с которыми мы уже
встречались, изомерия этого типа - еще один пример того, как различное
расположение фрагментов молекулярного агрегата может приводить к образованию
физически различных соединений. Эти изомеры различаются расположением
азотсодержащих колец по отношению друг к другу и заместителями в центральном
циклофановом кольце. Интересно, что полярность соединения G.65а) настолько
7.6. Катенины и ротаксаны
651
АсО
АсО
АсО
АсО
G.65а)
АсО
R'
R'
(СН2J2
R = Н, R' = ОАс. G.656)
R = ОАс, R' = Н: G.65в)
Рис. 7.46. Трансляционные изомеры [3]катенана
отличается от полярности двух других изомеров, что его можно отделить от них хро-
матографически. Наличие арильных колец предотвращает взаимные превращения
G.65а), G.656) и G.65в) в обычных условиях.
Для получения соединений с множественным зацеплением были предприняты
попытки провести стерически управляемые циклизации родственных систем.
Соединение G.66) вводили в реакцию тройной циклизации. К сожалению, низкие
выходы на основных стадиях реакции сильно затрудняли исследование ее продуктов.
||J52 ?• Темплаты и самосборка
Соединение G.66) получили в девять стадий; и его последующая многократная
циклизация привела всего лишь к 1.7% продукта, который представляет собой
смесь трех различных сцепленных соединений (изомеры).
G.66)
В общем, вследствие того, что эта молекула обладает симметрией второго
порядка, существует только два способа связывания хлорзаместителей 1—4 с
аминогруппами 5 и 6: связывание 1и2с5(пЗи4с6) или связывание 1иЗс5(а2и4с6).
Однако разнообразные возможности топологической изомерии и диастереоизомерии
приводят к четырем продуктам (без учета энантиомеров). При первом способе
связывания, в конце концов, образуется [2]катенан, тогда как при втором может
получаться макроциклическое соединение или сложная структура, названная трилист-
ным узлом, которая и была первоначальной целью этого синтеза (ср. разд. 7.8),
рис. 7.47. Были синтезированы все три продукта, и ЯМР-спектроскопические
результаты показали, что в качестве одного из них образовался предшественник для
трилистного узла. Конечные стадии получения нековалентно сцепленных структур
не были описаны.
Во всех до сих пор рассмотренных случаях предшественники и образующиеся
катенаны содержат большое количество функциональных групп, необходимых для
управления синтезом. Однако с начала синтеза катенанов остается задача, хотя бы
ради академического интереса, получить чисто углеводородный катенан,
состоящий из двух сцепленных циклоалканов. С одной стороны, очевидно, что из-за
ограниченной силы ван-дер-ваальсовых взаимодействий между алкановыми
предшественниками чисто статистический подход малопривлекателен для получения
ощутимых выходов. С другой стороны, направленный подход связан с введением
большого числа функциональных групп, от которых будет трудно избавиться. На
этом этапе арсенал достижений супрамолекулярной химии был дополнен
комбинацией статистического и последующего направленного подходов (схема 7.16).
Вначале был получен статистическими методами проскальзывания трансляционный
изомерный [2]ротаксан G.67). Объемистые трифенилметильные (тритильные)
запирающие группы предотвращали выдергивание нити ротаксана из цикла при
комнатной температуре. Метиленовые группы в а-положении к сульфонильным
заместителям обладали достаточной кислотностью для их протонирования в
присутствии алкиллитиевых реагентов. Полученную литиевую соль использовали
для синтеза ротаксанов с алкинильными заместителями, способными к
циклизации по Глазеру с образованием либо [2]ротаксана G.68), либо [2]катенана G.69).
7.6. Катенаны и ротаксаны
Ш53
Макроцикл
G.66)
Макроцикл
[2]Kai
Рис. 7.47. Возможные трижды циклизованные интермедиаты на основе соединения
G.66). (Воспроизведено с разрешения Chem. Rev. 1995. Vol. 95. P. 2733)
Восстановление образующегося алкинового макроцикла и удаление лишних
блокирующих групп дают или некатенановые макроциклы, или полностью
углеводородный катенан G.70).
Направленный подход Шилла — пример принципа вспомогательной связи,
который хотя весьма элегантен, но требует огромных усилий при синтезе и в
конечном счете приводит к низким выходам из-за необходимости тратить энергию на ко-
валентное связывание. При этом ковалентные связи вынуждают реагенты
принимать конформацию, подходящую для образования продукта со сцеплением.
Более очевидный и доступный пример вспомогательной связи — это координация с
катионом металла, используемая в кинетическом темплатном синтезе для простых
реакций макроциклизации, рассмотренных в гл. 3. Этот подход с огромным
успехом применил Ж.-П. Соваж (J.-P. Sauvage), один из первых студентов нобелевского
лауреата Ж.-М. Лена. Подход Соважа (С. Dietrich-Buechecker, P. Marnot,
J.-P. Sauvage et al., 1984) основан на наблюдении (твердо установленном благодаря
детальному ЯМР-исследованию), что ионы переходных металлов, например Cu(I),
в основном принимают тетраэдрическую координационную геометрию и в
присутствии бидентатных лигандов, таких, как производное 1,10-фенантролина G.71),
способны располагать эти лиганды взаимно ортогонально (рис. 7.48).
Сразу же стало понятным, что функционализация метоксильных заместителей
и макроциклизация с длинной полиэтиленгликольной цепочкой в условиях высо-
654
7. Темплаты и самосборка
(СИ,)
♦ R— SO2 (СН2J5 SO2 R
R — SOj—С (CH2),—С SO
(CH,).
\ H
\ R —SO,— С (CH^^-C SOj
H / Hi
R — S02— C-\~ (CHZ),—С —f SO2-R
G.69)
1 (CH2);
G.68)
G.70)
Схема 7.16. Статистический и направленный подходы к синтезу первого
углеводородного катенана G.70)
7.6. Катенаны и ротаксаны
655
ОМе
МеО
МеО
МеО
МеО
ОМе
ОМе
ОМе
[CuG.71J]+
Рис. 7.48. 2:1-Комплекс производного 1,10-фенантролина G.71) с Cu(I) и его
схематическое изображение
кого разбавления должны неизбежно привести к катенанам. Поэтому провели
реакцию анизольного производного G.71) с хлоргидратом пиридиния. Реакция
циклизации образовавшегося бисфенольного соединения с 1,14-дииод-3,6,9,12-те-
траоксатетрадеканом в диметилформамиде (DMF) привела к получению [2]катена-
та G.72) с выходом 27%, макроцикла G.73) с выходом 20% и продуктов
полимеризации. Учитывая тот факт, что две катенановые фенантролиновые части в G.72)
выступают в качестве лигандов для СиA)-центра, комплекс в целом назвали
катенатом, по аналогии с катионными криптатами. Свободный катенановый ли-
ганд был назван катенандом (ср. криптанд), схема 7.17.
G.73)
Выход G.72) в дальнейшем повысили, применив постадийный подход, в
котором предварительно полученный коранд G.73) реагирует с [Cu(MeCNL]+ и одной
молекулой модифицированного G.71) с образованием промежуточного псевдоро-
656
7. Темплаты и самосборка
[2]катенат G.72)
Схема 7.17. Темплатируемый металлом синтез [2]катената G.72)
таксана. Циклизация по указанному выше способу приводит к катенату G.72) с
высоким выходом 42%.
На последней стадии катенат обработали CN~-hohom (великолепный лиганд
для Си+), чтобы удалить СиA)-центр в виде [Cu(CNL]3~ и получить свободный ка-
тенан G.74). В масс-спектре катенана присутствовал его молекулярный ион с m/z
1133, причем масс-спектр имел характерный для катенанов вид: первая
фрагментация относится к разъединению двух сцепленных колец, и поэтому никаких пиков
не наблюдается вплоть до сигнала некатенанового G.73) при m/z 567 (рис. 7.49).
Представляет интерес и 'Н ЯМР-спектр соединения G.74), показывающий, что в
свободном состоянии, без центра Cu(I), связывающего две фенантролиновые
части вместе, этот катенан претерпевает значительное конформационное изменение.
Это изменение состоит в разведении объемистых электроноизбыточных фенантро-
линовых групп как можно дальше друг от друга. Обе структуры были подтверждены
также методом РСА (рис. 7.50).
7.6. Катенаны и ротаксаны
657
567
(МЛ)
1133
584
"Фрагменты
500 600 700 800 900 1000
I j i i i I i i i i I i i i i I i i i i I i i i i 1
1100
I I I I I I
mlz
Рис. 7.49. Масс-спектр катенана G.74), содержащий пик молекулярного иона и
указывающий на его постадийную фрагментацию. (Воспроизведено с разрешения Dietrich-
Buechecker CO. and Sauvage J.-P. Interlocking of molecular threads - from the statistical approach
to the templated synthesis of catenands // Chem. Rev. 1987. Vol. 87. P. 795-840)
Интересно, что реакция диссоциации комплекса и без посредников, и с
помощью CN~-HOHa, как показано ниже, протекает чрезвычайно медленно.
[CuG.73J]+ + 4CN- = [Cu(CNL]3" + 2G.73). G.15)
G.74)
Прямое сравнение кинетических данных для [СиG.71J]+ и [СиG.73J]+ ([2]ка-
тенат G.72)) показывает, что скорость самопроизвольной диссоциации для катена-
та в 2500 раз меньше, чем для его открытого аналога, хотя стерическая «обстановка»
658
7. Темплаты и самосборка
(а)
(б)
Рис. 7.50. Кристаллические структуры: (а) катената G.72) и (б) свободного катенана
G.74) (по SauvageJ.-Р.)
вокруг металлического центра в обоих случаях одна и та же. Скорость диссоциации
при обработке CN~-hohom в 40 раз выше для некатенанового комплекса
[СиG.71J]+. Это означает, что при наличии полиэфирной цепочки для
осуществления процесса диссоциации требуются значительные конформационные
изменения, что существенно увеличивает активационный энергетический барьер.
Представляют интерес электрохимические (CVA) (Дополнение 4.1) свойства
двух СиA)-комплексов - [СиG.71J]+ и [СиG.73J]+. Некатенановый бисхелатный
комплекс 1СиG.71J]+ полностью необратимо восстанавливается при —1.7 В
(относительно насыщенного каломельного электрода сравнения), что согласуется с
разложением комплекса на свободный G.71) и металлическую медь, когда центр Cu(I)
7.6. Катенаны и ротаксаны
659
восстанавливается до Cu@). Для катената [СиG.73J]+, наоборот, наблюдается
волна полностью обратимого восстановления при том же потенциале. Это
свидетельствует о том, что комплекс катенановый (два фенантролиновых фрагмента не могут
отодвигаться друг от друга) и поэтому способен стабилизировать Си@)-комплекс.
Действительно, синие растворы [СиG.73J]° устойчивы в атмосфере аргона в
течение нескольких дней. Вероятно, это соединение только формально является
комплексом Cu@); на самом же деле, дополнительный электрон находится на одном из
фенантролиновых фрагментов, образуя органический анион-радикал.
Соваж и сотрудники продолжили работы по получению [3]- и [4]катенанов,
таких, как G.75), аналогичным методом, используя бис- и трисфенантролиновые
производные значительно больших размеров, которые выступали в роли
центральных макроциклов.
G.75)
Dietrich-Buechecker CO., Sauvage J.-P. Interlocking of molecular threads - from
the statistical approach to the templated synthesis of catenands // Chem. Rev. 1987.
Vol. 87. P. 795-810.
660
7. Темплаты и самосборка
Геликаты
Piguet С, Bernardinelli С, Hopfgartner G. Helicates as versatile supramolecular
complexes // Chem. Rev. 1997. Vol. 97. P. 2005-2062.
7«7#1 Введение
Самособирающаяся структура двойной спирали ДНК явилась толчком
к появлению еще одной области супрамолекулярной самосборки, а именно - к
использованию ионов металлов для темплатирования сборки органических «нитей» в
спирали. Спиральные координационные комплексы, замечательные по своей сути,
были использованы в качестве отправной точки для получения широкого ряда
молекулярных узлов, например трилистных. Фундаментальным свойством
спирального соединения является хиральность, обусловленная направлением закручивания
вокруг его оси. Степень этого закручивания определяется шагом спирали:
расстоянием между двумя ее витками (рис. 7.51). Спиральные комплексы практически
всегда состоят из «молекулярных нитей», представляющих собой удлиненные полиден-
татные мостиковые лиганды с более чем одним «связывающим доменом», т.е. с
более чем одним набором донорных атомов, способных хелатировать различные
металлические центры. Связывающим доменом может служить 2,2'-бипиридиновая
G.76) или 1,10-фенантролиновая G.77) единица, так что нить будет формироваться
из 2,2':6/,2//:6",2/"-кватерпиридина G.78а), состоящего из двух бипиридильных
связывающих доменов. Спиральные металлосоединения и их лиганды имеют
номенклатуру, аналогичную номенклатуре криптандов и криптатов, катенанов и кате-
натов. Так, молекулярные нити (лиганды) типа G.78а), которые могут образовывать
спиральные комплексы, называют геликандами, а их металлокомплексы — геликата-
ми. В принципе, существуют три способа, при помощи которых лиганды, подобные
G.78а), могут связывать четырехкоординированный металлический центр. Эти
лиганды могут давать моноядерный плоскоквадратный комплекс 1:1, биядерный
неспиральный комплекс 2:2 или комплекс 2:2 в виде двойной спирали, обычно с тет-
раэдрической координационной геометрией металла. В присутствии
шестикоординированного металла могут также образовываться комплексы с
тройной спиралью, если три геликанда смогут уместиться около металлических центров
(рис. 7.52). Двойные спирали также могут быть получены, если оставшиеся
координационные центры металла занимают дополнительные лиганды.
G.76)
(a)R = H
(б) R = Me
7.7. Геликаты
661
(а)
Шаг
Л/-спираль
Р-спираль
Рис. 7.51. (о) Виток спирали вокруг определенной оси. (б) М-спиральность и (в) Р-ст-
ральность мексиканской золотой древесной змеи и североамериканской стеклянной змеи
соответственно. (Обе абсолютно безвредны!) Изображения змей взяты из «Истории
Природы» Джонсона, проиллюстрированной S.G. Goodrich. New York: Glidersleeve Press, 1888.)
(г) Более привычные молекулярные спирали
Связь между хиральной одиночной спиралью и металлокомплексами с двойной
спиралью хорошо проиллюстрирована «королевским разрезом». Проблема состоит
в том, чтобы разрезать яблоко на две гомохиральные половинки одинаковой спи-
662
7. Темплаты и самосборка
(а)
Домен, ►
связывающий
Спейсер
/
Ион металла
I
I
(б)
(в)
Рис. 7.52. Комплексы, образованные двухдомснным геликандом: (а) моноядерный,
(б) биядерный неспиральный, (в) двухспиральный и (г) трсхспиральный
ральности (хиральность), рис. 7.53. Две хиральные одиночные спирали одинаковой
направленности (левая, левая или правая, правая), соединенные вместе, образуют
двойную спираль. Одиночные спирали противоположной хиральности не могут
объединяться с образованием двойной спирали. Однако, в отличие от свойств
яблока, основной чертой двойных спиралей является хиральность спирального витка,
что приводит к существованию левовращающих (А/-) и правовращающих (Р-) форм
(рис. 7.54). (Символы Ми Рзаимствованы из номенклатуры Кана-Ингольда—Пре-
лога и означают «minus» и «plus». Для более детального ознакомления см.: Cahn R.S.,
Ingold С. and Prelog V. Specification molecular chirality. Angew. Chem., Int. Ed. Engl.
1966. Vol. 5. P. 385-413.) Таким образом, и одиночные, и двойные спирали
существуют как энантиомерные пары, но, в отличие от обычной комбинации двух хираль-
ных фрагментов, двойная спираль не существует в виде двух диастереомеров,
потому что гетерохиральное (левая, правая) комбинирование двух одиночных спиралей
не дает двойной спирали.
Рис. 7.53. «Королевский разрез»: разрезание
яблока на две гомохиральные половинки
одинаковой спиральности. (Воспроизведено
с разрешения Lehn J.-M. Supramolecular
chemistry. Weinheim: VCH, 1995)
7.7. Геликаты
663
Правовращающий
геликат
Левовращающий
геликат
Рис. 7.54. Хиральность двойных геликатов
Для экспериментальной проверки хиральности металлогеликатов использовали
методы РСА и ЯМР-спектроскопии в растворе. Введение хиральных сдвигающих
реагентов, таких, как [Eu(tfcK] (где tfc — 3-(трифторметилгидроксиметилен-(+)-
камфорат), в раствор геликатов приводит к расщеплению некоторых сигналов
лиганда в результате образования диастереомерных комплексов со сдвигающим
реагентом. Такое расщепление не наблюдается для свободных лигандов, которые
ахиральны.
7 • 2 Синтетические аспекты
Значительное число геликатов получено самосборкой, и поэтому они
представляют собой структуры с термодинамическим минимумом. Это означает,
что вся информация о сборке спирали должна быть заложена в самой системе, как
это имеет место в случае ДНК. Для рассматриваемой системы можно говорить о
том, что алгоритм сборки диктуется или металлом, или лигандом. Под «диктатурой»
металла мы понимаем такие фундаментальные свойства, как предпочтительное
координационное число металла и его координационная геометрия, а также другие
характеристики, например расстояния металл-лиганд. С одной стороны, металлы
с плоскоквадратной координационной геометрией, такие, как Pd(II) и Pt(II),
имеют склонность к образованию моноядерных неспиральных соединений, в которых
все донорные атомы геликандов типа G.78) расположены в одной плоскости, что
препятствует спирализации. С другой стороны, металлы с тетраэдрической
координационной геометрией, например Cu(I), образуют двойные геликаты, потому что
спейсер между связывающими доменами лиганда редко бывает достаточно гибким
для того, чтобы два домена смогли расположиться строго ортогонально
относительно друг друга.
(>64
7. Темплаты и самосборка
Для придания лиганду спиральности он должен быть предорганизован (обычно
стерически). Макроциклический лиганд G.79) уже обладает спиральностью
вследствие фиксированных ориентации бинафтильных заместителей и образует
моноядерный катион с двойной спиралью [СиG.79)]+. Аналогично лиганд G.80) уже
готов к образованию спирали благодаря жесткости своего каркаса, состоящего из
конденсированных шестичленных колец. В этой структуре два концевых кольца
сильно сближены, однако во избежание крайне невыгодных стерических
взаимодействий плоскость лиганда претерпевает спиральные искажения. Действительно,
связывание с ионами натрия приводит к образованию различных спиральных
структур (Т. Bell and H. Jousselin, 1994). В частности, мононатриевый комплекс
содержит однотяжную спираль, которая медленно рацемизуется при комнатной
температуре. Замещенный тетрапиридиновый лиганд G.786) в некоторой степени
предрасположен к спиральности из-за невыгодных стерических взаимодействий
между метальными заместителями. Это приводит к тому, что два центральных
пиридиновых кольца ориентируются некопланарно, нарушая плоскоквадратную
координацию, хотя этот лиганд может принимать и псевдопланарную геометрию в
случае Си(Н)-комплексов. Хиральный одиночный геликат может образоваться
даже из незамещенных полипиридиновых лигандов, таких, как 2,2/:6/,2":6",2"/:6'",2""-
пентапиридин G.81), в результате невыгодных стерических взаимодействий между
его концами. Это подтверждает кристаллическая структура его Ag+-KOMruieKca
[AgG.81)]+, рис. 7.55.
G.79)
G.80)
G.81)
7.7. Геликаты
665
Рис. 7.55. Кристаллическая структура одиночного геликата [AgG.81)]+
7*7*3 [4+4]Геликаты
При взаимодействии геликанда G.786), спиральная предорганизация
которого обеспечивается стерическими отталкиваниями между метильными
группами, с Cu(I) (в основном в форме относительно лабильных предшественников
типа [Cu(MeCNL]+) быстро образуется биядерный геликат [Си2G.78бJ]2+,
кристаллическая структура которого представлена на рис. 7.56 (J.-M. Lehn et al., 1983).
Биядерный комплекс [Cu2G.786J]2+ — пример [4+4]геликата, в котором [п + т]
представляют собой координационные числа двух металлических центров (для
каждого центра — четыре). В данном случае и тетраэдрические металлические
центры, и стерически предорганизованный лиганд предрасположены к образованию
геликата. Поэтому комплекс можно рассматривать как комплементарную систему
для образования геликата. Напротив, Cu(II) в основном имеет искаженную
эффектом Яна—Теллера октаэдрическую или квадратно-пирамидальную геометрию,
менее подходящую для синтеза геликата. Этот факт был использован и получили
интересные результаты. Одноэлектронное окисление СиA)-центров в [Си2G.78бJ]2+
до Cu(II) приводит к геликату Cu(I)—Cu(II) со смешанной валентностью. При
дальнейшем окислении геликат разлагается с образованием моноядерного Cu(II)-co-
единения (схема 7.18).
Использование геликанда G.78а), имеющего меньшие стерические
ограничения (и поэтому менее предорганизованного), также приводит к двойному
спиральному СиA)-комплексу, причем тетраэдрической координационной геометрии иона
металла вполне достаточно для темплатирования образования геликата.
Интересно, что расстояние Си--Си (которое соответствует шагу спирали) в неметилирован-
ном комплексе равно 3.17 А в противоположность 3.90 А в [Си2G.78бJ]2+. В анало-
666}
7. Темплаты и самосборка
МеЧО>
Рис. 7.56. (а) Кристаллическая структура двойного геликата [Си2G.78бJ]
(б) Его схематическое представление
гичном А§A)-комплексе [Ag2G.78a)9]2+ это расстояние составляет 3.10 А.
Следовательно, шаг спирали в большей степени определяется метильными группами, чем
предорганизацией лиганда. Ясно также, что наличие метальных групп
благоприятствует образованию двойной спирали, а для лиганда без метальных групп образова-
2[СипG.78б)]2+
Моноядерный
2[СигG.78б)]+
Моноядерный
Быстро
Медленно
[Сип2G.78бJГ
Двойная спираль
[Си'2G.78бJ]2+
Двойная спираль
[Cu'CunG.786J]3+
Двойная спираль
Схема 7.18. Влияние степени окисления меди на образование спирали
7.7. Геликаты
667
ния интермедиата со смешанной валентностью [CuICuIIG.78aJ]3+ не наблюдали.
Тот факт, что шаг в А§A)-комплексе G.78а) короче, чем в аналогичном соединении
на основе Cu(I), можно объяснить похожестью спирали на пружину. Увеличение
длины связи металл-лиганд для соответствия большему иону металла приводит к
растяжению спирали перпендикулярно ее оси и, следовательно, к
компенсационному сжатию шага вдоль этой оси.
У. 7 • 4 [6+6]Геликаты
Большое число двойных геликатных комплексов включает четырехко-
ординированные металлические центры, так как многие геликандные лиганды
содержат бидентатные связывающие домены. В таких случаях шестикоординирован-
ные ионы металлов могут образовывать тройные геликаты, как это показано на
примере лиганда G.82), который дает двойной геликат в присутствии четырехкоор-
Рис. 7.57. Предорганизованное
металлом образование двойных
или тройных геликатов
668
7. Темплаты и самосборка
динированной Cu(I) и тройной геликат с шестикоординированным Со(П). Это
можно рассматривать как прямой результат различной предорганизации двух
металлических центров (рис. 7.57).
Однако, возможно образование двойных [6+6]геликатов, если используются
тридентатные домены, связывающие металл. Шестикоординированный ион
металла может, таким образом, связываться с одним доменом от каждого лиганда-гели-
канда. Простейший пример такого соединения — лиганд с игривым названием
«2,Т:Ь',2":6",2'"\6'",2"":6"",2'""-сексипиридин» G.83). В присутствии большого
числа ионов металлов, например Cd2+, Fe2+, Co2+, Ni2+ и Cu2+, образуются
двойные спиральные 2:2-комплексы, схематически изображенные в виде структуры
G.84). Кристаллическая структура кадмиевого соединения показана на рис. 7.58.
G.83)
-Я?*ЧО> G-м>
Лиганд G.83) практически универсален, так как он может рассматриваться как
обладающий двумя тридентатными или тремя бидентатными связывающими
доменами, приводящими к образованию двойных [6+6]- или [4+4+4]геликатов. Можно
ожидать, что Си(Н)-аналог соединения G.84) будет подвергаться
электрохимическому восстановлению с образованием (через сложную серию интермедиатов) три-
металлического двойного [4+4+4]геликата. Однако использование шестикоорди-
нированных металлических центров в сочетании с лигандами более сложной
структуры, содержащими два разных связывающих домена, приводит к тройным
[6+6]геликатам. Наглядный пример такого подхода был описан сотрудниками
группы М. Хэннона (М. Harmon et al., 1997) из Университета Уорвика
(Великобритания), которые получили бис(бидентатный) иминный лиганд G.85) «дешевым»,
7.7. Геликаты
669
Рис. 7.58. Кристаллическая
структура двойного [6+6]геликата G.84)
(по Constable E. et al., 1990)
Схема 7.19. Образование жестких геликандов «дешевым» способом
по их определению, способом прямого смешения коммерчески доступных
исходных материалов, как это показано на схеме 7.19. Реакция G.85) с Ni(II) в
соотношении 3:2 дает тройной [6+6]геликат [Ni2G.85K]4+ с выходом более 80%.
7*7*5 Самораспознавание
и позитивная кооперативность
Мы уже видели, как рацемическая смесь хиральных одиночных
спиралей образует рацемическую смесь из двух двойных спиральных энантиомеров
(рис. 7.53). Однако в отличие от двух половинок яблока в «королевском разрезе»
спиральные лиганды не обязательно хиральны в свободном состоянии. Это подразу-
670
7. Темплаты и самосборка
мевает, что спиралям одинаковой хиральности нет необходимости искать и
распознавать друг друга для образования двойной спирали. В самом деле, геликанды зачастую
проявляют хиральность только как часть геликатной структуры. С одной стороны,
собственно хиральные геликанды могут быть получены и выделены как энантиоме-
ры путем включения асимметрических углеродных центров, как в случае G.86). В
таких случаях для образования двойной спирали требуется самораспознавание двух ге-
ликандов. С другой стороны, превращение G.86) в энантиомер приводит к
получению энантиомерно чистого правовращающего двойного геликата (рис. 7.59).
(а) п = О, X = Н
(б)и=1,Х =
(в) п = 2, X = Н G.88): п =
Вопрос самораспознавания также важен для образования полиметаллических
спиралей из геликандов различной длины G.87), содержащих от двух до пяти
металлических центров. Взаимодействие Cu(I) с каждым лигандом по отдельности
приводит к получению ожидаемых двойных [4+4]-, [4+4+4]-, [4+4+4+4]- и
[4+4+4+4+4]геликатов G.89а-г). Более того, при взаимодействии ионов металла
со смесью всех этих лигандов вновь образуются гомолептические (т.е. имеющие
одинаковые лиганды) геликаты; при этом следы примесей отсутствуют. При
образовании геликатов, так же как и при спаривании нуклеооснований (ср. рис. 7.4),
реализуется позитивная кооперативность, при которой, однажды начавшись, после-
7.7. Геликаты
671
дующая сборка облегчается нелинейным процессом. Этот эффект интенсивно
изучали для СиA)-комплексов на основе СО2Е1-производного G.88). Титрование ли-
ганда с помощью Cu(I) и мониторинг этого процесса методом оптической
спектроскопии в УФ- и видимой области (рис. 7.60, а) показали, что превращение
происходит монотонно и может быть описано равновесиями, приведенными ниже.
G.89a)
G.896)
G.89г)
672
7. Темплаты и самосборка
Рис. 7.59. Энантиомерно чистый правовращающий двойной
гели кат
Константы равновесия этой реакции составляют Ig3n = 4.6, Ig312 = 8.2, Ig322 = 13.5,
Ig332 = 18.6 фт1 представляет собой постадийную константу устойчивости для т
ионов металла и /лигандов).
Си+ + G.88) = [CuG.88)]+
[CuG.88)]+ + G.88)] = [CuG.88J]+
[CuG.88J]+ + [CuG.88)]+ = [Cu2G.88J]2+ +
[Cu2G.88J]2+ + [CuG.88)]+ = [Cu3G.88J]3+
lg C,,
lg p,2
G.88) lg C22
+ G.88) lg 332
(a)
3.00 -,
2.00
Д 100 К
0.00
250
650
(б)
0.0
Рис. 7.60. (а) Спектрофотометрическое титрование лиганда G.88) с помощью Cu(I).
(б) Диафамма Скэтчарда для образования двойного геликата [Си3G.88J]3+.
(Воспроизведено с разрешения Pfeil A. and Lehn J.-M. Helicate self-organization - positive cooperativity in the
self-assembly of double-helical metal-complexes//J. Chem. Soc, Chem. Commun. 1992. P. 838)
7.7. Геликаты 673fi
Наличие в спектрах четко выраженных изобестических точек (точки
пересечения спектров) указывает на присутствие только двух веществ и, следовательно, на
образование индивидуального триметаллического геликата. Позитивная коопера-
тивность - это вполне определенная характеристика, которая устанавливается
большим числом тестов. Система с многими равновесиями будет проявлять
позитивную кооперативность, если соотношение Кт+уКт больше, чем значение,
рассчитанное из уравнения G.16). Некооперативная (статистическая) система имеет
значение, равное рассчитанному по уравнению G.16), тогда как более низкая
величина означает негативную кооперативность.
Km+i = m(t-m)
(« + l)(r-m + l)' GЛ6)
К
где т - число занятых центров в соединении МШЦ, t - общее число центров.
Величины К — константы равновесия при образовании соответствующего вещества.
Позитивная кооперативность может быть описана диаграммой Скэтчарда,
представляющей собой выпуклую нисходящую кривую, как в случае [Си3G.88J]3+
(рис. 7.60, б). Параметр заселенности г— это среднее число занятых центров,
которое для системы [Си3G.88J]3+ определяется уравнением G.17). Некооперативные
реакции дают прямую линию, тогда как негативным кооперативным системам
соответствует вогнутая восходящая кривая.
G17)
Позитивная кооперативность — основа для разработки молекулярных
усилителей. Это свойство можно использовать для фильтрации ошибок, так как только
корректный ввод приведет к генерации каскада кооперативных структур.
Некоторые интересные примеры позитивной кооперативности были ранее
опубликованы К. Раймондом (К. Raymond) из Университета Калифорнии (Беркли,
США). В его системах как основной лигандный мотив используется дианион де-
протонированного 1,2-дигидроксибензола. Выбрав близкие химически, но
имеющие разную длину спейсеры между двумя связывающими доменами, Раймонд
осуществил селективную самосборку большого числа тройных спиралей и
координационных кластеров. В одном из примеров реакция смеси таких лигандов
трех типов с соответствующим числом катионов Ga3+ привела к селективному
образованию трех индивидуальных гомолептических тройных спиралей, схема 7.20
(D. Caulderand К. Raymond, 1999).
*"* Perlmutter-Hayman В. Cooperative binding to macromolecules. A formal approach //
Ace. Chem. Res. 1986. Vol. 19. P. 90-96.
13-СтидДж. В.
7. Темплаты и самосборка
О
NH
NH
О
.он
чон
,он
о
о nh
♦ > О
о
он
Ga(acacK
С
б-
NH
NH
О
■°\|
ч/ I4/
О I /3
Схема 7.20. Позитивная кооперативность в самосборке тройных спиралей с Ga(III)
(по Caulder D. and Raymond К., 1999)
./. 1еликаты
Наноциклы
В работе Лена и сотрудников (В. Hasenkopf, J.-M. Lehn et al., 1996)
было показано, что тройные геликатные комплексы не являются единственно
возможным результатом реакции октаэдрического металлического центра с трис(би-
дентатным) лигандом (три связывающих домена). В такой реакции Fe(II)
объединяется с трис(бипиридильным)лигандом G.90), давая пентаядерный геликат
G.91). Вероятно, образование этого большого циклического комплекса темплати-
руют хлорид-анионы, один из которых удачно включен в центр наноразмерного
кольца. Лен и сотрудники развили это направление, комбинируя связывающие
домены различных типов в одном геликанде (D. Funeriu, J.-M. Lehn et al., 1997). Ли-
ганд G.92) имеет бис(бидентатный) центральный связывающий домен, похожий на
уже известный G.22), и четыре 2,2'-бипиридильных фрагмента, связанных вместе,
как в лигандах типа G.87). Каждый такой лиганд способен связываться бидентат-
ным способом со всеми шестью металлическими центрами. Таким образом,
реакция с тетраэдрической Cu(I) приводит к получению соединения со стехиометрией
2:12, которое имеет замечательную наноциклическую структуру.
Обозначение лиганда G.90)
G.90)
G.92)
v^676 ^' Темплаты и самосборка
7-7*7 Спирали, образованные водородными связями
Координационные взаимодействия, вероятно, — наиболее общий
метод получения синтетических спиралей. Однако пример ДНК показывает, что
водородные связи могут быть также эффективно использованы при формировании
спиральных структур. Наиболее простой природной системой, показывающей, каким
образом водородные связи приводят к образованию спиралей, является метиловый
эфир производного хлорофилла а G.93) (N. Engel et al., 1993). РСА-исследования
этого соединения (рис. 7.61) показали, что его спиральная структура
обеспечивается шестью водородными связями R2C=O-HNR2.
Н
о
ОМе G.93)
Группа Лена (С. Fouquey, J.-M. Lehn et al., 1990) сконструировала систему,
образующую спирали, в основу которой положено спаривание за счет водородных
связей комплементарных элементов. В качестве таких элементов были выбраны
производные 2,6-диаминопиридина (Р) и урацила (U), которые легко связываются с
образованием PU-димера G.94).
G.94)
7.7. Гели каты
677
Рис. 7.61. Структура биомолекулы G.93), выделенной из культуры клеток и
организованной с помощью водородных связей R2C=O-HNR2
В тех случаях, когда заместителями X и Y являлись алкилэфирные или амидные
группы с длинной цепочкой, эти соединения приобретали жидкокристаллические
свойства (разд. 10.3). Хотя индивидуальные компоненты не проявляют мезомор-
физма, агрегат формирует метастабильную гексагональную колончатую структуру.
Если PU-димер «снабдить» алкильной оболочкой, чтобы индуцировать
жидкокристаллическое поведение, то сначала можно синтезировать дитопные рецепторы
типа РТР или UTU (Т — хиральный спейсер), а затем организовать самосборку
полимерной нити. Введение длинных алкильных групп в каркас дитопного элемента
(UTU или РТР) может привести к получению различных супрамолекулярных
полимеров, обладающих жидкокристаллическими свойствами. Экспериментально этот
подход был реализован путем конденсации комплементарных групп Р и U с длин-
ноцепочечными производными L-, D- или л/езо(М)-винной кислоты, которая
выступает в качестве спеисера с образованием различных изомеров дитопных
комплементарных рецепторов G.95) (РТР) и G.96) (UTU). Такие рецепторы
самособираются в полимерные нити (РТР, UTU)^. В этом примере хиральность L-
или D-изомеров придает хиральность индивидуальным полимерным нитям. В
зависимости от хиральности группы Т наблюдали различное поведение полученных
соединений. Образующиеся структуры были исследованы методами рентгеновской
дифракции и электронной микроскопии. Самым замечательным оказалось то, что
образовавшийся в результате самосборки /,-тартратный полимер (PLP, ULU),,
G.97) и его D-аналог самоорганизуются путем переплетения трех нитей с
образованием большой тройной спирали, закрытой оболочкой из алкильных заместителей
тартратного производного (рис. 7.62). Напротив, (РМР, ими)я-форма полимера
дает три нити (полосы) в конформации «зигзаг». Установлено, что оба вещества
представляют собой жидкие кристаллы, и их мезофаза имеет гексагональную
колончатую структуру. L-,D-CMQCb спонтанно разделяется на право- и левоврашаю-
щие спирали, что согласуется с данными по самораспознаванию (см. разд. 7.7.5).
678
7. Темплаты и самосборка
Рис. 7.62. Схематическое изображение модели
тройной спиральной организации трех
самособранных гомохиральных полимерных нитей
(РТР, UTU),, (Т = L или D, но не М). PU-пары,
объединенные водородными связями и
относящиеся к одной нити, представлены одинаковыми
кружками. Алкильные цепи торчат по бокам
цилиндра. (Воспроизведено с разрешения Lehn J.-M.
Supramolecular chemistry. Weinheim: VCH, 1995)
При высоких концентрациях нити собираются в большой полимерный агрегат в
виде волокна, затем они ветвятся подобно дереву. На рис. 7.63 показаны
электронные микрофотографии различных смесей. Следует особо отметить противополож-
(а)
(б)
(в)
Рис. 7.63.
Электронные
микрофотографии текстур,
полученных из смесей:
(а) PLP + ULU
(правовращающая
спираль),
(б) PDP + UDU
(левовращающая)
H(ff)PMP+UMU
(неспиральный
полимер).
(Воспроизведено с разрешения
LehnJ.-M. Supra-
molecular chemistry.
Weinheim: VCH, 1995)
1.%. Молекулярные узлы 679
H-N прп __, 1~Л ORO P
HN ORO ^ H-N VOv^^Vn-h
Vo
ORO /—ч H
" ' " ~ X/^v' "^ " v_N ~ " Y * Y U ^ IX-
>- О OR n-h О Н О OR NHq
Hf^-0 G.95) = FTP °Л i G.96) =UTU
R = C12H25
-H-N ° ORO r-V H"W ORO O—H-N ° ORO
OR N-H- -O H О OR ЙЛ)- HN ° °R
G.97)
T - производное L-, D- или мезо(М)-винной кислоты
ную закрученность тройных L- и D-спиралей (право- и левовращающие спирали
соответственно).
Молекулярные узлы
Временно отрешившись от химических проблем, рассмотрим узлы и
переплетенные фигуры, которые известны уже давно. Зачастую они представляют
собой причудливые конструкции, как, например, многократно переплетенные
сложные структуры, взятые из «Книги кельтов», датируемой VIII веком. Узел — это
символ неразрывных связей, и в этом качестве его элемент - три взаимно
сцепленных кольца (типа катенана) - использовала могущественная итальянская семья
Борроме. В работах голландского художника М.К.Эшера показаны прекрасные
образцы переплетений, многие из которых можно воспроизвести методами
современного синтеза (рис. 7.64).
Применение принципа вспомогательной связи к синтезу катенатов типа G.72)
(см. разд. 7.6.6) в сочетании с расширением представлений о родственных им гели-
катах (см. разд. 7.7), в конце концов, позволило синтезировать с высокими
выходами большое количество многократно сцепленных комплексов, названных
молекулярными узлами. В отличие от кольца катенанов, первичный элемент узлов — петля
Рис. 7.64. (а) и (б) Образцы кельтской вязи из «Книги кельтов», датируемой VIII веком.
(в) Символ семьи Борроме. (г) Трилистный узел в представлении голландского художника
М.К. Эшера. (Воспроизведено с разрешения Atwood J.L., Davies J.E.D., MacNicol D.D. and
Vogue F. Comprehensive supramolecular chemistry. Oxford: Pergamon, 1996. Vol. 9. P. 44)
680
7. Темплаты и самосборка
Рис. 7.65. Четыре базовых первичных узла. Цифра означает число пересечений, а индекс
порядок узла. (Воспроизведено с разрешения AtwoodJ.L., Davies J.E.D., MacNicol D.D. and
Vogtle F. Comprehensive supramolecular chemistry. Oxford: Pergamon, 1996. Vol. 9)
отдельной нити. Поэтому узлы представляют собой топологические изомеры
макроциклов (см. рис. 7.37). Мы уже знакомы с попытками Шилла и его сотрудников
(см. разд. 7.6.6) получить молекулярный трилистный узел из соединения G.66).
Этот синтез, несмотря на относительный успех, был весьма трудоемким из-за
необходимости проведения многостадийной циклизации. Фактически трилистный узел
— один из многих базовых узлов, четыре из которых показаны на рис. 7.65. В
дополнение к направленному синтезу Шилла исследователи предложили и проверили
большое число статистических и полустатистических подходов к образованию
узлов, однако ощутимые результаты обнаружены не были. В природе встречаются
Схема 7.21. (а) Схема синтеза [2]катената G.72) с использованием принципа
вспомогательной связи, (б) Распространение этого подхода на образование трилистного
узла через биметаллический двойной гели кат
7.8. Молекулярные узлы
681
ОН
НО
ОН
G.98)
(а)Х = (СН2L
(б)Х = (СН2J
(в)Х = (СН2N
(г)Х =
G.100)
многочисленные завязанные и переплетенные формы нитей ДНК, изображение
которых было получено при помощи электронной микроскопии. Механическая
прочность современных синтетических полимеров также обусловлена
переплетением их цепей. Исследования Э. Вассермана (Е. Wasserman, 1960) из компании
«Du Pont» по молекулярному моделированию привели к предположению, что
напряжения, возникающие на пересечениях полимерных нитей в узлах, могут вызы-
682 7. Темплаты и самосборка
вать механическое ослабление этих узлов. Очевидно, что если можно получить
достаточно большие макроциклы, то нет препятствий для сборки химически
стабильных узлов, подобно катенанам.
Вслед за синтезом [2]катената G.72) (схема 7.21, а) К. Дитрих-Бюхеккер и
Ж.-П. Соваж (С. Dietrich-Buechecker and J.-P. Sauvage, 1989) предположили, что
замена одиночного ядра Cu(I) в [СиG.71J]+ на биметаллический двойной геликат
приведет к предшественнику, который при циклизации немедленно даст трилист-
ный узел (принцип двойной вспомогательной связи, схема 7.21, б).
В соответствии с этим они получили дифенольный геликанд G.98а) с гибким
спейсером —(СН2L— между двумя связывающими доменами. Лиганд
синтезировали из 1,10-фенантролина и Li(CH2LLi в несколько стадий и ввели его в реакцию с
[Cu(MeCNL]+, чтобы получить геликат G.99а). Следуя своему методу синтеза кате-
нана, Дитрих-Бюхеккер и Соваж затем осуществили двойную циклизацию в
условиях высокого разбавления и после длительной хроматографической очистки
впервые выделили трилистный узел G.100а) с выходом 3%. Этот узел был
охарактеризован методами масс-спектрометрии и ЯМР-спектроскопии. Однако
замыкание такого большого кольца (86-членного) — статистически неблагоприятный
процесс. Кроме того, параллельно образовывалось большое число побочных
продуктов, содержавших хиральную двойную спираль. Был использован еще один
способ доказательства синтеза трилистного узла, состоявший в удалении
металлических центров с помощью KCN, для того чтобы получить свободные лиганды. В этих
условиях только завязанное узлом 86-членное кольцо обладает безусловной хираль-
ностью, которая и была продемонстрирована. Другие продукты — 84- и 43-членные
макроциклы — ахиральны в отсутствие металлов, темплатирующих спирали.
Проведенный позже РСА кристаллической структуры подтвердил образование
трилистного узла G.100а), рис. 7.66.
Низкий выход G.100а), при всей изобретательности синтетиков, заставил их
усовершенствовать этот метод. Особые проблемы создавали конкурентное
образование негеликатных 2:2-комплексов типа «плоскость—плоскость» на стадии
синтеза геликата и пространственное расположение четырех химически активных
концевых групп G.99). Поэтому с целью повысить выход геликата были получены
геликандные лиганды с различными спейсерами X G.986—г). Длину цепочки
полиэтил енгликоля, использованного на стадии циклизации, также варьировали. Все
испытанные соединения давали узлы типа узла G.100). Для геликандов G.986) и
G.98в) выходы также оказались низкими, лучший из них — 8% — был получен для
84-членного завязанного узлом макроцикла с X = (СН2N и пентаэтиленгликольной
цепочкой. Напротив, выходы до 24% были получены для не завязанных узлом мак-
роциклических топологических изомеров. Более жесткий лиганд G.98г),
подававший большие надежды, дал трилистный узел G.100г) с выходом 29% после
хроматографии. Такой высокий выход позволил исследовать свойства некоторых узловых
соединений и охарактеризовать их методами ЯМР и FAB-масс-спектрометрии (fast
atom bombardment - сравнительно мягкий метод ионизации). Причина такого
значительного повышения выхода не вполне понятна, хотя она может быть связана с
компактной геометрией соединений. В кристаллической структуре
предшественника G.99г) расстояние Си--Си равно 4.76 А, в то время как для предшественников
7.8. Молекулярные узлы
Р83
Рис. 7.66. Кристаллическая структура трилистного узла G.100а) (Sauvage J.-P., 1990,
® «knot.pdb»)
со спейсерами (СН2L G.99а) и (СН2N G.99в) это расстояние соответственно
составляет 6.30 А и 7.00 А.
Существует возможность использования этого продуктивного подхода для
получения большого числа многократно завязанных в узел и сцепленных соединений.
Можно показать, что в данном подходе основное правило гласит: если число
металлических центров четное, то число пересечений молекулярной петли будет
нечетным. При этом для образования узла необходимо именно нечетное число
пересечений. Таким образом, два металлических центра дают трилистный узел, четыре -
пятилистный, шесть - семилистный и т.д. В то же время нечетное число
металлических центров дает четное число пересечений и приводит к дважды сцепленным
[2]катенанам (рис. 7.67).
Дитрих-Бюхеккер и сотрудники (С. Dietrich-Buechecker et al., 1994) в
продолжение своей работы синтезировали систему с тремя металлическими центрами и по-
684
7. Темплаты и самосборка
(в)
У V
Рис. 7.67. Сборка многократно сцепленных соединений с использованием полиядерных
геликатов. (а) Дважды сцепленный [2]катенан с тремя металлическими центрами.
(б) Пятилистный узел с четырьмя металлическими центрами, (в) Трижды сцепленный
[2]катенан. (Воспроизведено с разрешения AtwoodJ.L., DaviesJ.E.D., MacNicol D.D. and
Vogtle F. Comprehensive supramolecular chemistry. Oxford: Pergamon, 1996. Vol. 9)
G.101)
7.9. Каталитические и самовоспроизводящиеся системы 585
лучили (бесспорно хиральный) дважды сцепленный [2]катенат G.101).
Синтетический метод оказался сходным с тем, который использовали при получении трили-
стных узлов G.100) и однократно сцепленного [2]катенатаG.72). Он включал
двойное замыкание кольца двух 60-членных макроциклов по реакции, катализируемой
Cs2CO3, двух дифенольных нитей предшественника с 1СН2(СН2ОСН2NСН21 в
DMF при большом разбавлении и 60 °С. Нужно отдать должное высокой
квалификации исследователей, выделивших после интенсивной хроматографической
очистки с выходом 2% соединение, которое затем было охарактеризовано методами
ЯМР и масс-спектрометрии.
•"* Chambron J.-C, Dietrich-Buechecker С. and Sauvage J.-P. TVansition metals as
assembling and templating species: synthesis of catenanes and knots //
Comprehensive supramolecular chemistry / Ed. J.L. Atwood, J.E.D. Davies, D.D.
MacNicol and F. Vogtle. Oxford: Pergamon, 1996. Vol. 9. P. 43-85.
У.Q Каталитические
и самовоспроизводящиеся системы
Одна из основных целей исследования самосборки — получение
систем, имеющих каталитическую активность природных ферментов, и
конструирование новых искусственных ферментов, способных выполнять такие операции,
которые не свойственны их природным аналогам. Простой пример того, как это может
быть достигнуто (с системами много меньших размеров, чем природные
ферменты), приведен на схеме 7.22 (Т. Kelly et al., 1989). Симметричный катализатор
(называемый бисубстратным реакционным темплатом) связывает с помощью
множественных водородных связей одновременно два реагента с образованием тройного
комплекса. В тройном комплексе два реагента сильно сближаются, что приводит к
шестикратному ускорению нуклеофильной атаки первичной аминогруппы на
бромалкановый заместитель. Эта система при всей своей простоте и
оригинальности страдает множеством недостатков, что нужно учитывать при разработке
катализатора. Во-первых, конечный двойной комплекс, безусловно, более стабилен, чем
исходный тройной комплекс, и поэтому он будет сразу отравлять катализатор или
ингибировать сам процесс. Улучшить ситуацию можно осаждением продукта в
виде соли гидробромида. Кроме того, идентичность природы двух связывающих
центров реагентов означает, что может образоваться много тройных комплексов,
которые не приводят к желаемой (внутримолекулярной) реакции, например, между
катализатором и двумя молекулами одного и того же субстрата. Эта проблема была
решена с помощью синтезированной несимметричной системы. Благодаря
точному распознаванию пары реагентов удалось повысить скорость в 12 раз.
Если катализатор сам является продуктом реакции, то система становится
самовоспроизводящейся, т.е. синтетическим аналогом ДНК. Получение синтетических
самовоспроизводящихся систем может приблизить химиков к пониманию
происхождения жизни.
686
7. Темплаты и самосборка
Реагенты
Каталитический тройной комплекс
I
Продукт, удаленный в виде соли с НВг
Схема 7.22. Каталитическая самосборка тройного комплекса, в которой используется
бисубстратный реакционный темплат (по Kelly T. et al., 1989)
Простейшая модель самовоспроизведения показана на схеме 7.23. Репликатор
(R) должен быть способен распознать и связать в тройной комплекс по крайней
мере два различных исходных компонента (С1 и С2), а также ускорить их химическую
реакцию друг с другом с образованием продукта, являющегося копией исходного R.
7.9. Каталитические и самовоспроизводящиеся системы
687
Схема 7.23. Простейшая модель самовоспроизведения. Синтез репликатора R ускоряется
связыванием компонентов С1 и С2 с образованием тройного комплекса [C1C2R]
Такая простая система будет постоянно конкурировать с некатализируемой
бинарной реакцией между С1 и С2.
Простейшие самовоспроизводящиеся системы, способ получения которых
представлен на схеме 7.23, были синтезированы по реакции Шиффа —
конденсацией альдегида G.102) с первичным амином G.103). Реакция катализируется ее
собственным продуктом G.104) через тройной комплекс [G.102)G.103)G.104)J.
Наиболее примечательная черта этой системы — то, что, по кинетическим данным,
тройной комплекс более стабилен, чем двойной [G.104)-G.104)]. Это значит, что по
завершении одиночного акта катализа новый продукт спонтанно вытесняется из
его темплатируюшего партнера двумя новыми молекулами реагента. Поэтому в
данной системе возможен экспоненциальный автокатализ. С точки зрения
энтропийного вклада этот процесс не выгоден и должна быть энтальпийная
компенсация, источником которой может быть сравнительно высокая прочность солевых
мостиков.
Более сложная (и, к тому же, противоречивая) самовоспроизводящаяся система
была сконструирована Дж. Ребеком (М. Famulok, J.S. Nowick and J. Rebek et ah,
1992). Автокаталитическая сборка основывалась на трикарбоксипроизводном Кем-
па G.105) с аксиальными заместителями. Взаимодействие G.105) с амином G.106)
приводит к самособирающемуся бинарному комплексу G.107), в котором неподе-
ленная пара электронов амина идеально расположена для атаки пентафторфениль-
ного эфирного заместителя в G.105). Компоненты комплекса G.107)
удерживаются вместе благодаря оригинальной комбинации комплементарной пары
водородных связей и высокоорганизующим п—я-стэкинг-взаимодействиям типа
«плоскость—плоскость» между электроноизбыточным нафталиновым
производным G.105) и электронодефицитным гетероциклическим фрагментом системы
G.106). Нуклеофильное замещение группы C6F5O в бинарном комплексе G.107)
688
7. Темплаты и самосборка
G.102) [О] H2N
чсно
R
G.104)
G.103)
R1 = Me, t-Bu
R2 = H, NO2
дает цис-шид G.108). Важная особенность этой автокаталитической системы -
нестабильность связи цис-амида, который быстро изомеризуется в более стабильную
транс-форму. Это приводит к образованию у продукта двух новых центров
связывания (А и Б), способных взаимодействовать с двумя новыми молекулами G.105)
(центр А) и G.106) (центр Б). В сущности, продукт - транс-G.108) - может, как и
14 -
12 -
10-
Q -
О
6"
4 -
2 -
п -
/
Автокаталитическая
реакция
Некаталитическая
реакция
20
40 60
Время, мин
80
100
Рис. 7.68. Сигмаидальная зависимость количества продукта G.108) от времени для
автокаталитической и некаталитической реакций. (Воспроизведено с разрешения Rotello V.,
HongJ.I. and Rebek J. Sigmoidal growth in a self-replicating system // J. Amer. Chem. Soc. 1991.
Vol. 113. P. 9422-9423)
7.9. Каталитические и самовоспроизводящиеся системы
689
Н Напряженный цмс-амид
G.109)
цас-G.108)
69Q 7. Темплаты и самосборка
в предыдущих каталитических и автокаталитических системах, образовывать
тройной комплекс G.109), в котором осуществляется организация двух
предшественников. Реакция между иммобилизованными G.105) и G.106) дает следующую
молекулу транс-{1.108), что приводит к самовоспроизведению. К сожалению,
образующийся димер (транс-7.108J самокомплементарен (константа ассоциации
80 000 М), так что дальнейший катализ ингибируется из-за необходимости
разрушения бинарного комплекса, с тем чтобы каждая из двух образующихся молекул
могла бы сформировать два новых тройных комплекса. Это означает, что при 25%-й
завершенности процесса равновесная концентрация G.109) составит всего лишь
3%. Несмотря на это, присутствие комплекса G.109) увеличивает скорость
образования продукта в 2 раза. Более того, при катализе имеет место сигмаидальная
зависимость скорости от времени, характерная для самовоспроизводящихся систем
(рис. 7.68). Эти кинетические измерения показывают, что хотя автокатализ не
эффектен, он является доминирующим направлением.
*""* Philip D. and Stoddart J.F. Self-assembly in natural and unnatural systems // Angew.
Chem., Int. Ed. Engl. 1996. Vol. 35. P. 1154-1196.
Учебные задания
7.1. Предскажите основную форму спектра 109Ag для решеточного комплекса,
состоящего из 16 ионов Ag(I), связанных восемью октадентатными лигандами,
изображенными ниже. Сколькими связывающими доменами обладает этот
лиганд? Ожидаете ли Вы, что он будет образовывать геликаты? Как будут
выглядеть 109Ag ЯМР-спектры аналогичного (Ag+ вместо Си+) лестничного
соединения G.21) и соединений, изображенных на рис. 7.22?
7.2. Рассмотрим систему, содержащую всего четыре индивидуальных катиона
Pd(enJ+ и четыре индивидуальные молекулы 4,4'-бипиридила. Допустим, что
каждый фрагмент является жестким и имеет углы N-Pd—N, близкие к 90°.
Пренебрегая ротамерами, рассчитайте количество возможных восьмикомпо-
нентных молекул, которые могут самособраться, если образование каждой
молекулы равновероятно. Рассчитайте также статистическую вероятность
образования молекулярного квадрата. Как изменится эта вероятность, если
возможно также образование: (а) четырехкомпонентных ансамблей и (б) двух-,
четырех- и шестикомпонентных ансамблей? Какой эффект может проявиться
при наличии восьми компонентов каждого типа? Сколько 20-компонентных
ансамблей кроме супрамолекулярного куба G.10) Вы можете предложить, ис-
Учебные задания 1б91
ходя из восьми индивидуальных катионов Ru([9]aH-S3J+ и 12
индивидуальных молекул 4,4'-бипиридила?
7.3. Константа связывания нити Т макроциклом С равна 100 М. Нить Т
претерпевает циклизацию со 100%-м выходом, образуя копию макроцикла С.
Рассчитайте относительные выходы копии С и [2]катенана СС, считая С и СС
одинаково стабильными, а реакцию Т с образованием С - быстрым одномо-
лекулярным процессом, на который не оказывает воздействия связывание с С.
Нить Т и макроцикл С представлены в мольном соотношении 1:1.
7.4. Полукарцеранд имеет внутреннюю полость объемом 600 А3 и действует как
катализатор реакции Дильса-Альдера циклогексадиена-1,3 (С6Н8) с л-хино-
ном (С6Н4О2), см. схему 7.8. Если уравнение скорости этой реакции имеет вид:
v = А:[С6Н8][С6Н4О2], то определите увеличение скорости при переходе от
раствора, содержащего 0.1 мольдм каждого реагента в отсутствие катализатора,
к катализируемой системе, используя эффективные концентрации внутри
полости полукарцеранда. Как Вы думаете, почему такое увеличение скорости не
достижимо на практике?
7.5. Используя кусочек картона и шесть отрезков веревки, постройте
топологическую модель соединения G.66). Следуя структуре, показанной на рис. 7.47,
скрепите кусочки веревки попарно так, чтобы у Вас получился один из
множества ковалентно сцепленных продуктов. Где необходимо разрезать веревку,
чтобы получить катенат или сцепленный продукт?
Мысленный эксперимент
Для приведенных ниже реагентов (L — лабильный лиганд) предложите возможные
закрытые олигомеры, которые могут образоваться: (а) если Си+ - жесткий тетраэд-
рический ион или (б) если центр Си+ обладает значительной гибкостью в
отношении координационной геометрии, а анионы различного размера используются для
темплатирования реакции. Какая система более подходит для образования
полимеров? Некоторые идеи можно почерпнуть в работе: Keller S.W. and Lopez S. A two-
dimensional geomimetic coordination polymer containing pentagonal cavities. J. Amer.
Chem. Soc. 1999. Vol. 121. P. 6306-6307.
N( )) (( )N
"'L \W/ \W/
Соотношение 1:1
692
7. Темплаты и самосборка
Литература
Amabilino, 1997 АтаЫНпо D.B., Ashton P.R., Boyd S.E. et al. The five-stage self-assembly of a
branched heptacatenane // Angew. Chem., Int. Ed. Engl. 1997. Vol.36. P. 2070.
Anderson, 1995 Anderson H.L., Anderson S., Sanders J.K.M. Ligand-binding by butadiyne-linked
porphyria dimers, trimers and tetramers // J. Chem. Soc, Perkin Trans. 1995.
P.2231.
Ashton, 1989 Ashton P.R., Goodnow T.T., Kaifer A.E. et al. A [2]catenane made to order //
Angew. Chem., Int. Ed. Engl. 1989. Vol.28. P. 1396.
Atwood, 1997 AtwoodJ.L., MacGillivray L.R. A chiral spherical molecular assembly held
together by 60 hydrogen bonds // Nature. 1997. Vol.389. P.469.
Barbour, 1998 Barbour L.J., Orr G.W., Atwood J.L. An intermolecular (H2O)A0) cluster in a
solid-state supramolecular complex // Nature. 1998. Vol. 393. P.671.
Baxter, 1993 Baxter P., Lehn J.-M., DeCian A. Multicomponent self-assembly - spontaneous
formation of a cylindrical complex from 5 ligands and 6 metal-ions // Angew.
Chem., Int. Ed. Engl. 1993. Vol.32. P.69.
Bell, 1994 Bell T.W., Jousselin H. Self-assembly of a double-helical complex of sodium //
Nature. 1994. Vol.367. P.441.
Caulder, 1999 Caulder D.L., Raymond K.N. Supermolecules by design // Ace. Chem. Res. 1999.
Vol. 32. P.975.
Chi, 1995 Chi X., Guerin A.J., Haycock R.A. et al. The thermodynamics of self-assembly //
Chem. Commun. 1995. P.2563.
Constable, 1990 Constable E.C., Ward M.D. Spontaneous assembly of a double-helical binuclear
complex of 2,2'-6',2"-6",2'"-6"/,2""-6"",2/""-sexipyridine // J. Amer. Chem.
Soc. 1990. Vol.112. P. 1256.
Dietrich- Dietrich-Buechecker CO., Marnot PA., Sauvage J.-P. et al. Copper(I) or ruthe-
Buechecker, 1984 nium(II) with built-in coordinates — synthesis and spectroscopic properties //
Nouv. J. Chim. 1984. Vol.8. P.573.
Dietrich- Dietrich-Buechecker CO., Sauvage J.-P. A synthetic molecular trefoil knot //
Buechecker, 1989 Angew. Chem., Int. Ed. Engl. 1989. Vol.28. P.189.
Dietrich- Dietrich-Buechecker CO., Guilhem J., Pascard C, Sauvage J.-P. Structure of a
Buechecker, 1990 synthetic trefoil knot coordinated to 2 copper(I) centers // Angew. Chem., Int.
Ed. Engl. 1990. Vol.29. P.I 154.
Dietrich- Dietrich-Buechecker С О., Lieze E., Nierengarten J.F. et al. Singly and doubly
Buechecker, 1994 interlocked [2]-catenanes - influence of the degree of entanglement on chemi-
Литература
693
cal-stability as estimated by fast-atom-bombardment (FAB) and electrospray-
ionization (ESI) mass spectrometries (MS) // J. Chem. Soc, Chem. Commun.
1994. P.2257.
Engel, 1993 Engel N., Gossauer A., Gruber K., Kratky С X-ray molecular-structure of a red
bilin derivative from chlorella-protothecoides. 4. Chlorophyll catabolism // Helv.
Chim. Acta. 1993. Vol.76. P.2236.
Famulok, 1992 Famulok M., Nowick J.S. , Rebek J., Jr. Self-replicating systems // Acta Chem.
Scand. 1992. Vol.46. P.315.
Fouquey, 1990 Fouquey C, Lehn J.-M., LevelutA.-M. Electron-microscopic study of supramole-
cular liquid-crystalline polymers formed by molecular-recognition-directed self-
assembly from complementary chiral components // Adv. Mater. 1990. Vol.2.
P.254.
Fujita, 1991 Fujita M., Yazaki J., Ogura K. Macrocyclic polynuclear complexes
[(en)MD,4'-bpy)]4(NO3)8 (M = Pd or Pt) as inorganic cyclophane - their
ability for molecular recognition // Tetrahedron Lett. 1991. Vol.32. P.5589.
Fujita, 1995a Fujita M., Nagco S., Ogura K. Guest-induced organization of a 3-dimensional
palladium^ I) cage-like complex — a prototype for induced-fit molecular recognition //
J. Amer. Chem. Soc. 1995a. Vol.117. P. 1649.
Fujita, 1995b Fujita M., Ogura D., Miyazawa M. et al. Self-assembly of 10 molecules into
nanometer-sized organic host frameworks // Nature. 1995b. Vol.378. P.469.
Funeriu, 1997 Funeriu D.P., Lehn J.-M., Baum G., Denske D. Double subroutine self-assembly;
spontaneous generation of a nanocyclic dodecanuclear Cu-I inorganic
architecture // Chem. Eur. J. 1997. Vol.3. P.99.
Hannon, 1997 Hannon M.J., Painting C.L., Jackson A. et al. An inexpensive approach to
supramolecular architecture // Chem. Commun. 1997. P.1807.
Hasenkopf, 1996 Hasenkopf В., Lehn J.-M., Kneisel B.O. et al. Self-assembly of a circular double
helicate//Angew. Chem., Int. Ed. Engl. 1996. Vol.35. P.1838.
Kelly, 1989 Kelly T.R., Zhao C, Bridger G.J. A bisubstrate reaction template // J. Amer. Chem.
Soc. 1989. Vol.111. P.3744.
Lehn, 1983 Lehn J.-M., Sauvage J.-P., Simon J. et al. Synthesis and metal-complexes of a
conformationally restricted quaterpyridine — crystal-structure of its dimeric
dinuclear Cu(I) complex, [Cu2(pqpJ]2+ // Nouv. J. Chem. 1983. Vol.7. P.413.
Lehn, 1995 Lehn J.-M. Supramolecular chemistry: Concepts and perspectives. Weinheim:
VCH, 1995.
Logeman, 1981 Logeman E., Rissler K., Schill G, Fritz H. Synthesis and spectra of 2 novel cate-
nanes// Ber. Dtsch. Chem. Ges. 1981. Vol.114. P.2245.
McMurray, 1989 McMurray T.J., Raymond K.N., Smith PH. Molecular recognition and metal-ion
template synthesis // Science. 1989. Vol.244. P.938.
Il94
7. Темплаты и самосборка
Olenyuk, 1999 Olenyuk В., WhitefordJ.A., FechtenkotterA., Stang P.J. Self-assembly of nanoscale
cuboctahedra by coordination chemistry// Nature. 1999. Vol.398. P.796.
Orr, 1999 Orr G.W., Barbour L.J., Atwood J.L. Controlling molecular self-organization:
Formation of nanometer-scale spheres and tubules // Science. 1999. Vol.285.
P. 1050.
Roche, 1998 Roche S., Haslam C, Adams H. et al. Self-assembly of a supramolecular cube //
Chem. Commun. 1998. P. 1681.
Takeda, 1999 Takeda N., Umemoto K., Yamaguchi K., Fujita M. A nanometer-sired hexahedral
coordination capsule assembled from 24 components // Nature. 1999. Vol.398.
P.794.
Wasserman, 1960 Wasserman E. The preparation of interlocking rings: A catenane // J. Amer.
Chem. Soc. 1960. Vol.82. P.4433.
Wyler, 1993 Wyler R., de Mendoza J., Rebek J., Jr. A synthetic cavity assembles through self-
complementary hydrogen-bonds // Angew. Chem., Int. Ed. Engl. 1993. Vol.32.
P. 1699.
Q Молекулярные устройства
Целое больше, чем его часть.
И.Тодхантер. Элементы Евклидовой геометрии
(Лондон, Макмиллан, 1880 (перевод с греч.))
8Л Введение
*~* Lehn J.-M. Supramolecular chemistry: Concepts and perspectives. Weinheim: VCH,
1995. P. 89-135.
8.1.1 Философия молекулярных устройств
Традиционная задача химии состоит в разработке подходов к синтезу
молекул. Если Вам удалось создать новые молекулы, то Вы можете получать и новые
вещества, и материалы или увеличивать количества уже известных. Химические ве-
щесТЖ^Пфасители и краски, металлы и пластмассы, стекло и керамика,
фармацевтические препараты, синтетические ткани и т.д. — это окружающие нас полезные
материалы. Вы можете перерабатывать эти материалы, носить изделия из них,
изменять их эстетические и противокоррозионные свойства, принимать их внутрь,
делать из них кухонную утварь и инструменты, а также множество других орудий
цивилизации. В каждом случае используется только одно главное свойство
химического соединения. Такими свойствами могут быть цвет (поглощение или
испускание света), твердость и механическая прочность, адгезия "и эластичность, эле-
ктро-_и_теплопррводность, химическая стойкость и т. д. Традиционно этим
занимались различные инженерные дисциплины, а позднее к ним добавилась электронная
инженерия. Все они имеют дело с объемными свойствами различных химических
веществ, из которых изготавливают компоненты для производства машин и
механизмов. Машина — это функциональное устройство, состоящее из ряда
взаимодействующих компонентов, выполняющих определенную задачу для достижения
полезного результата. Главное в машине или механизме, в отличие от химического
соединения, - что она делает, а не то, из чего она состоит. Поскольку детали
машины должны быть точно подогнаны друг к другу, чтобы взаимодействовать
определенным образом, они должны иметь макроскопические размеры. Тогда человек
может собрать их, используя острое зрение и сноровку. В последние годы электронная
промышленность выпускает микроскопические «машины» (устройства с размерами
компонентов порядка микрометров)*. В производстве этих машин участвуют маши-
Размеры элементов современных устройств, получаемых по литографической
технологии, достигают 100 нм. (Примеч. ред. перевода.)
696 8. Молекулярные устройства
ны нового поколения, обладающие существенно более высокой «сноровкой» и
«остротой зрения». «Микромашины» занимают меньше места, меньше подвержены
влиянию внешних воздействий и, что важнее всего, являются более
быстродействующими, а также способны выполнить более сложные задачи по сравнению со
своими громоздкими «предками». На очереди производство наноразмерных машин —
машин, размер которых сопоставим с размерами индивидуальных молекул. Эти
устройства обладают еще большим быстродействием и еще более сложны.
В разд. 7.1 и во всей главе 7 мы рассмотрели методы, с помощью которых
можно собрать большие молекулы или наноразмерные молекулярные ансамбли либо
индуцировать эти процессы самосборки. Самосборка относится к
фундаментальной проблеме синтетических наноструктур, в которых компоненты располагаются
определенным образом. В этой главе мы продолжим тему наноинженерии («нано»
означает 10~9 м), причем особое внимание уделим свойствам индивидуальных
компонентов и способам обмена информацией между ними, которые используют для
конструирования молекулярных и супрамолекулярных наноаналогов механизмов
реального мира.
8.1.2 Что такое супрамолекулярное устройство?
До сих пор мы пользовались определением супрамолекулярного
соединения (которое предложил Лен) как соединения, включающего нековалентные
взаимодействия (разд. 1.1). Это определение полностью соответствует химии
хозяин—гость и самособирающимся системам с термодинамическим контролем,
которые обсуждались в гл. 7. Однако для наноустройств такое определение уже нельзя
использовать, поскольку при конструировании супрамолекулярных устройств упор
делается на функциональные взаимодействия между компонентами, а не на
химическую природу их связывания. Это означает, что супрамолекулярное устройство
может быть полностью ковалентной молекулой, если оно обладает
характеристиками супрамолекулярной системы. Так, супрамолекулярное устройство можно
определить как комплексную систему, сконструированную из молекулярных
компонентов, обладающих определенными индивидуальными свойствами. Эти свойства
присущи молекулярному компоненту вне зависимости от того, является ли он
частью устройства или нет. Иными словами, энергия взаимодействия между
компонентами супрамолекулярного устройства должна быть малой в сравнении с
другими энергетическими характеристиками этой системы. Следовательно, не имеет
значения, как компоненты связаны в этом устройстве (ковалентно, водородными
связями, координационными взаимодействиями и т.д.); существенно только то,
что каждый компонент должен вносить в эту систему нечто уникальное и присущее
только ему. Если это не так и функционирование системы обусловлено всей
молекулой в противоположность отдельным компонентам, тогда лучше считать этот
комплекс «большой молекулой», а не супрамолекулярной системой.
Такое определение супрамолекулярного устройства не исключает традиционных
понятий «хозяин—гость» или «рецептор—субстрат». Акт молекулярного
распознавания между хозяином и гостем может быть одной из операций супрамолекулярного
устройства, которая обеспечивает связывание и сигнализирует о присутствии гостя.
8.1. Введение
697
Обычные компоненты супрамолекулярных устройств, изучаемых в настоящее
время, — фотохимические и редокс-активные молекулы, т.е. молекулы, способные
поглощать и/или излучать свет, и молекулы, способные отдавать или принимать
электрон. Определение супрамолекулярного устройства, составленного из этих
компонентов, иллюстрирует рис. 8.1. Если облучение молекулы (♦<->•) светом
сопровождается образованием возбужденных состояний, которые существенно
локализованы на одном из двух компонентов (♦ или •), или вызывает перенос
электрона от одного компонента к другому, тогда говорят, что это соединение супрамоле-
кулярно. Если возбужденное состояние существенно делокализовано по обоим
компонентам, тогда лучше считать этот комплекс просто большой молекулой.
Аналогичные соображения справедливы и по отношению к редокс-процессам. Следует
отдавать себе отчет в том, что характер взаимодействий между двумя компонента-
Супрамолекулярные
соединения
Частица
Большие молекулы
+е
+/- Возбужденное состояние с разделенными зарядами
Щ Электронно-возбужденное состояние
•*—► Спейсер любого типа или связь, удерживающие вместе два компонента
Рис. 8.1. Фотохимические и электрохимические критерии, используемые для отнесения
химически сложных частиц к супрамолекулярным соединениям или большим молекулам
6<jg 8. Молекулярные устройства
ми критическим образом зависит от природы связи между ними (<-»), будь то мос-
тиковый лиганд, жесткий или подвижный ковалентный спейсер либо нековалент-
ная связь.
8*2 Супрамолекулярная фотохимия
Использование фотохимически активных компонентов при
конструировании супрамолекулярного устройства представляется сегодня наиболее
универсальным. Процессы, индуцированные светом, имеют фундаментальное значение в
биохимических устройствах, таких, как фотосинтетические мембраны растений
(разд. 2.3). Светопоглощающие компоненты (хромофоры) легко доступны и
способны к разнообразным синтетическим превращениям, а свет легко вводится в
систему, находящуюся в различных физических состояниях (например, твердое тело,
жидкость или газ), или в среду (растворы в различных растворителях). Свет можно
использовать для индуцирования, разделения зарядов, инициирования катализа и
опроса сенсорных систем, а также для изменения состояний бистабильных
устройств (переключение). При включении фотохимически активных компонентов в
супрамолекулярный комплекс следует ожидать возмущения (или модуляции)
фотохимических свойств хромофоров, приводящего к ряду интересных и потенциально
полезных эффектов, таких, как миграция энергии, фотоиндуцированное
разделение зарядов, возмущение оптических переходов и поляризуемости, изменение ре-
докс-потенциалов основного и возбужденного состояний, управляемое светом,
селективное связывание и т.д.
*"* Balzani V. and Scandola F. Supramolecular photochemistry. Chichester: Ellis
Horwood, 1991.
8.2.1 Основы фотохимии
Когда на молекулярный хромофор воздействуют электромагнитным
излучением с длиной волны, соответствующей энергии перехода электрона в
электронно-возбужденное состояние, энергия излучения поглощается, вызывая его
переход с молекулярной орбитали основного состояния в состояние с более высокой
энергией. Такой процесс известен как первичное разделение зарядов: электрона,
имеющего высокую энергию, и положительно заряженной «дырки». Энергия
электрона в возбужденном состоянии либо рассеивается в виде тепла при релаксации
в растворителе (безызлучательный переход), либо превращается в излучение
(люминесценция), либо используется для проведения химической реакции (рис. 8.2).
Люминесценцию, включающую прямой излучательный переход, при котором
электрон из возбужденного синглетного состояния немедленно возвращается в
основное состояние, называют флуоресценцией. Флуоресцентное излучение обычно
имеет более низкую энергию, чем поглощенная, поскольку электрон, переведенный в
колебательно-возбужденное состояние, сначала релаксирует безызлучательно, а
затем, при переходе в основное состояние, излучает свет (рис. 8.3). Вот почему мно-
.2. Супрамолекулярная фотохимия
699
Орбитальная
энергия
Синглетное
основное
состояние
-н-
*v.
Переход
электрона
Флуоресценция
Av,
Электронно-
возбужденное
синглетное
состояние
Интеркомбинационный
переход (ISC)
Переход
возбуждения
Обратный (ISC)
Фосфоресценция
Электронно-
возбужденное
триплетное
состояние
Флуоресценция .■■
Av, /
Внешний
акцептор
Положительная
"дырка"
Флуоресценция
hyA
Положительная
"дырка"
Рис. 8.2. Возможные излучательные процессы, происходящие в результате
фотовозбуждения
Рис. 8.3. Колебательное и электронно-возбужденное
состояния фотовозбужденного электрона. Переход
электрона имеет место для стационарного положения ядер
(принцип Франка—Кондона). После завершения перехода
молекула подвергается воздействию нового силового поля
и начинает колебаться
700 8. Молекулярные устройства
гие флуоресцентные красители способны поглощать высокоэнергетический УФ-
свет, а флуоресцировать в видимой области.
Если изменяется спиновое состояние электрона (интеркомбинационный
переход), тогда он переходит в триплетное состояние. Формально это превращение
запрещено, но правило отбора AS= О (отражение того факта, что не существует
компонента электромагнитного излучения, который взаимодействует со спином
электрона) может быть смягчено вследствие спин-орбитального взаимодействия. В
результате этого формируется долгоживущее триплетное возбужденное состояние.
Колебательная релаксация вызывает переход этого состояния в состояние с более
низкой энергией, которое затем медленно релаксирует путем обратного
интеркомбинационного перехода (конверсия). Этот переход сопровождается
фосфоресценцией (разновидность люминесценции), частота излучения которой ниже частоты
поглощенного света. В присутствии внешнего акцептора электрона
(низкоэнергетическая незаполненная орбиталь соседней молекулы или компонента) электрон в
возбужденном состоянии может восстановить этот акцептор химически с
образованием долгоживущего состояния пространственно разделенных зарядов (вторичное
разделение зарядов). Окончательная рекомбинация сопровождается излучением
света другой частоты или выделением тепла. Наконец, энергия возбужденного
состояния может быть перенесена на внешний акцептор без переноса электрона. Этот
процесс называют переносом энергии (energy transfer, ET). Релаксация
образовавшегося вторичного возбужденного состояния сопровождается люминесценцией с
более низкой частотой по сравнению с частотой первоначально поглощенного света.
Такой процесс запускает каскад переносов энергии, как в фотосинтезе.
Эффекты, вызываемые фотовозбуждением, можно подразделить на три
категории:
1. Излучение поглощенной энергии в виде света (флуоресценция или
фосфоресценция).
2. Химическая реакция возбужденного состояния (вторичное разделение зарядов,
изомеризация, диссоциация и т.д.).
3. Безызлучательное колебательное тушение возбуждения растворителем.
В контексте супрамолекулярных устройств представляет интерес повторное
излучение в виде люминесценции для сенсорных и сигнальных систем, в то время как
химические реакции полезны в молекулярных переключателях и при фотокатализе.
Распространенными хромофорами являются комплексы переходных металлов,
которые можно использовать либо как светопоглощающую (воспринимающую)
часть супрамолекулярного устройства, либо как сигнальный и сенсорный
компоненты. В случае моноядерного комплекса переходного металла можно получить его
простое молекулярно-орбитальное описание путем линейной комбинации орбита-
лей металла и лигандов. Это удобное приближение, которое обычно используют
для описания электронных конфигураций как в основном, так и в
электронно-возбужденном состоянии, носит название «приближение локализованных
молекулярных орбиталей». Индивидуальные молекулярные орбитали (МО) можно
обозначить как М или L, если они принадлежат соответственно металлу или лиганду.
.2. Супрамолекулярная фотохимия
701
MLCT LC
м /
МС LMCT
Орбитали
металла
Молекулярные
орбитали
Орбитали
лиганда
Рис. 8.4. Диафамма энергетических уровней моноядерного октаэдрического комплекса
переходного металла, показывающая электронные переходы различных видов.
МС — переход, центрированный на металле, LC — центрированный на лиганде,
MLCT — перенос заряда от металла к лиганду, LMCT — перенос заряда от лиганда к металлу
Диаграмма энергетических уровней моноядерного комплекса металла (М) с лиган-
дами (L) показана на рис. 8.4.
В основном состоянии комплекса орбитали лигандов aL и nL обычно
заполнены. Орбитали лм заполнены по крайней мере частично (в зависимости от
состояния окисления металла), а другие орбитали всегда пусты. Поглощение света
изменяет эту заселенность. Переходы, центрированные на металле (metal-centred, МС),
иногда называемые d—d-переходами, или переходами в поле лигандов, обычно
имеют низкую энергию, часто соответствующую длинам волн видимого света и
включают переходы </-электронов металла из состояния /2 в состояние е .
Переходы, центрированные на лиганде (ligand-centred, LC), обладают значительно более
высокой энергией. Наиболее распространенным процессом является перенос
заряда (charge transfer, CT), который включает изменение орбитального квантового
числа (</-электроны становятся р-электронами или наоборот). Перенос заряда от
металла к лиганду (metal-to-ligand charge transfer, MLCT) особенно распространен, так
как включает переход электронов с высшей заполненной молекулярной орбитали
(highest occupied МО, HOMO) на низшую незаполненную молекулярную орбиталь
(lowest unoccupied МО, LUMO). Точный порядок расположения орбиталей в
соответствии с их стабильностью существенно зависит от природы металла и лигандов,
и часто уровень n*L располагается ниже уровня металла е (ст^), например, в
702 8. Молекулярные устройства
[Ru(bipyK]2+ (bipy — 2,2'-бипиридил). В некоторых случаях окисление или
восстановление приводит к нежелательной деструкции или разрыву связи металл-ли-
ганд, тогда как важнейшими условиями введения хромофора в супрамолекулярное
устройство являются его устойчивость и обратимость его
окислительно-восстановительных и фотохимических превращений.
8.2.2 Биметаллические системы
и системы со смешанной валентностью
По определению, супрамолекулярное устройство состоит из более чем
одного компонента, и поэтому может включать два фотохимически активных или ре-
докс-центра. Характер взаимодействия между этими двумя центрами будет сильно
зависеть от природы мостика между ними. В случае двух взаимодействующих
металлических центров, таких, как рассмотренные выше, можно различить три типа
поведения, обозначаемых как 1, II и III. Если взаимодействуют два электронных изомера
с локализованными валентностями, в которых металлические центры находятся в
состоянии окисления +2 или +3 (например, М^-М^11 и М^'-М^1), то существует
определенная равновесная геометрия (в терминах расстояний М—L в первой
координационной сфере и взаимодействий с сольватной оболочкой) для каждого «изомера». В
системе I типа два металлических центра, по существу, полностью изолированы и
ведут себя подобно изолированным моноядерным комплексам. Эта ситуация
представлена на рис. 8.5, а. Кривую потенциальной энергии каждого изомера можно
рассматривать как отвечающую возбужденному состоянию второго изомера, имеющего
энергию реорганизации X. В более широком смысле X представляет энергию,
необходимую для передвижения атомов лиганда и растворителя в их новые положения
равновесия в ответ на изменение состояния окисления металла. В точке пересечения оба
электронных изомера имеют одинаковую геометрию и одинаковую энергию. Обмен
электронами между ними маловероятен, даже если они приобретают энергию,
необходимую для достижения точки пересечения (в данном случае Х/4).
Однако в большинстве случаев между этими двумя металлическими центрами
существует некоторое электронное взаимодействие (рис. 8.5, б). При равновесной
геометрии взаимодействие металл—металл мало влияет на форму кривой
потенциальной энергии, но вблизи точки пересечения происходит смешивание состояний
нулевого порядка (случай изолированных состояний представлен на рис. 8.5, а).
Это «исключенное пересечение» — свойство частиц со смешанной валентностью II
типа. Такие системы все еще валентно локализованы и, следовательно, все еще су-
прамолекулярны в указанном выше смысле, но у них уже можно наблюдать новые
свойства, возникающие вследствие интервалентного взаимодействия
металл-металл, например оптический интервалентный переход (IT), в результате которого два
изомера взаимопревращаются. Необходимая для этого энергия может
предоставляться фотохимически.
Свойства систем III типа (их представитель — знаменитый Ru(II)-Ru(III)-hoh
Крейтца-Таубе [(NH3MRu(|i-nHpa3HH)Ru(NH3M]5+) находятся вне сферы супрамо-
лекулярной химии и соответствуют ситуации, при которой не наблюдается никакой
валентной локализации (рис. 8.5, в). В этом случае существует значительное элек-
.2. Супрамолекулярная фотохимия
703
(а)
м,"-м2
HI \л И
(б)
(в)
Ш/2
Конфигурация ядер
Рис. 8.5. Диафаммы потенциальной энергии двухкомпонентного устройства со
смешанной валентностью для пренебрежимо малых (а), слабых (б) и сильных (в) электронных
взаимодействий. Штриховыми линиями обозначены невозмушенные состояния нулевого
порядка. По горизонтальной оси отложены координаты (конфигурация) ядер со вкладами от
положений атомов лигандов и сферы растворителя
тронное взаимодействие металл-металл, а равновесная геометрия сильно
изменяется, что соответствует единственному минимуму потенциальной энергии
комплекса при наличии очень сильного взаимодействия, когда X ~ Н (энергия
взаимодействия). Свойства полностью делокализованной системы М]1'/2 -
слабо
#04
I. Молекулярные устройства
напоминают свойства моноядерных предшественников. Системы III типа
классифицируют как «большие молекулы».
8.2.3 Бипиридил и ег<> друзья
Одним из наиболее распространенных лигандов в супрамолекулярной
фотохимии является бидентатный хелат 2,2'-бипиридила (bipy) (8.1). Это
соединение представляет собой первый член гомологического ряда полипиридилов,
который включает 2,2':6/,2//-трипиридил (tpy) (8.3), сексипиридил, образующий гелика-
ты (разд. 7.7.4), и т.д.
Имея протяженные тг-системы, молекулы bipy, tpy и 1,10-фенантролин (phen)
(8.2) способны поглощать свет с последующим п~п*-переходом. Таким образом,
они способны сенсибилизировать координированные ионы металлов, выступая в
качестве световых антенн. Напротив, если фотовозбуждается сам ион металла, то
релаксация может происходить по механизму MLCT, при котором возбужденное
состояние металла восстанавливает лиганд путем переноса заряда на я^-орбиталь,
центрированную на лиганде.
На практике комплексы бипиридил а и фенантролина с металлом, такие, как
[Ru(bipyK]2+ и [Ru(phenK]2+, могут находиться в долгоживущих фосфоресцентных
возбужденных состояниях, возникающих в зонах лигандно-центрированного трип-
летного переноса заряда CMLCT). Времена их жизни - ~102-103 не в растворе при
комнатной температуре. Спектр фосфоресценции комплекса [Ru(bipyK]2+ показан
на рис. 8.6. Многое о природе фотовозбужденных состояний можно узнать при
изучении электрохимически восстановленных или окисленных частиц.
Электрохимическое окисление комплекса [Ru(bipyK]2+ включает удаление электрона металла
примерно при -1-1.25 В с образованием частицы с Ru(III) (потенциал относительно
насыщенного каломельного электрода (НКЭ) сравнения в MeCN). Замена одного
бипиридила на два хлоридных лиганда снижает потенциал до +0.32 В, а два я-ак-
цепторных лиганда СО повышают его до +1.9 В. Восстановление обратимо и
происходит на 71*ь-обитали, локализованной на лиганде при сохранении неизменной
электронной t^-конфигурации металла. При -54 °С в DMF можно наблюдать до
шести волн восстановления лиганда между -1.33 и -2.85 В, что соответствует
последовательному прибавлению одного, а затем двух электронов к каждому лиганду.
Эти электрохимические данные свидетельствуют о том, что если орбитальная кон-
.2. Супрамолекулярная фотохимия
705
Рис. 8.6. Электронные
спектры поглощения и
фосфоресценции комплекса
ы
[Ru(bipyV2+
В
о
о
Поглощение
1MLCT
Испускание
3MLCT
400
B5 000 см)
500
B0 000 см)
600
A8 000 см)
А., нм
фигурация одна и та же для исходного комплекса и для его формы,
восстановленной одним электроном как в основном, так и в возбужденных состояниях (теорема
Купмана), то фотовозбуждение комплекса [Ru(bipyK]2+ должно включать переход
электрона с HOMO металла на LUMO лиганда, т.е. имеет место механизм MLCT.
Это означает, что энергия я*ь-уровня ниже энергии системы металла е .
В противоположность комплексам трис(Ыру) и трис(рпеп) комплекс
[Ru(tpyJ]2+ имеет очень короткое время жизни MLCT (-250 пс) и не люминесци-
рует. Основная причина такого эффекта состоит в том, что tpy - менее подходящий
лиганд для Ru(II) из-за довольно напряженного угла захвата. Это приводит к более
низкой энергии расщепления в поле лигандов (разность энергий орбиталей
металла е и t2g) и, следовательно, к меньшему расстоянию между верхним металл-цент-
рированным уровнем (МС) и уровнем MLCT (n*L; в пиридильных соединениях Ru
этот уровень располагается ниже системы е , не так, как показано на рис. 8.4).
Поэтому вступает в силу обычный путь безызлучательного перехода через
активированное пересечение с расположенным выше состоянием 3МС. При 77 К этот путь
распада замораживается, и комплекс [Ru(tpyJ]2+ люминесцирует подобно bipy- и
phen-аналогам.
Несмотря на слабые люминесцентные свойства, с точки зрения структуры tpy -
удобный лиганд для конструирования супрамолекулярных фотохимических
устройств. Металлокомплексы бисеру) ахиральны, т.е. не образуют нежелательные
смеси диастереомеров при наличии более одного металлического центра, а заме-
РАР (8.4)
15-СтидДж В
706
I. Молекулярные устройства
Энантиомеры
Один энантиомер
Один изомер
Линейный мостиковый лиганд на основе tpy
Рис. 8.7. Структурная изомерия комплексов bipy и tpy
щенные tpy-соединения в качестве мостиковых лигандов легче превратить в
жесткие линейные спейсеры, чем bipy- и phen-производные. Более того, замещенные
tpy-соединения не проявляютуяс/тел-изомерии в отличие отбидентатных аналогов
(рис. 8.7).
Для реализации преимуществ tpy этот лиганд можно модифицировать двумя
путями. Во-первых, введение акцепторов электронов (X) в положение 4' его
молекулы значительно увеличивает время жизни люминесценции C6 не в случае SO2Me).
Во-вторых, более сложное замещение во фрагментах на основе bipy или phen также
может привести к получению линейных мостиковых лигандов, таких, как РАР (8.4).
1.2. Супрамолекулярная фотохимия
707
8.2-4 Ф°то~и электрохимические устройства
на основе бипиридила
Разнообразные фотохимические устройства можно получить,
соединяя лиганды бипиридильного типа (включая phen, tpy и огромное количество их
производных) с рядом различных металлических центров и применяя подход «ме-
таллокомплексы в качестве лигандов» (схема 8.1). Если мы рассмотрим синтез
моноядерного металлокомплекса путем взаимодействия иона металла с лигандами
(уравнение (8.1)), то сборку многоядерных комплексов можно осуществить
аналогичным образом, заменяя или только металл (М) или и металл, и лиганды (L) ме-
таллокомплексами с доступными центрами связывания.
wL->M(L),,.
(8.1)
Защищенный
хелатирующий
центр
Снятие защиты
Лабильные лиганды
Схема 8.1. Получение комплексов с применением подхода «металлокомплексы
в качестве лигандов»
708
8. Молекулярные устройства
Металл можно заменить моно- или олигоядерными комплексами с легко
обменивающимися лигандами, например типа [Ru(bipyJCl2] (8.5), а лиганды - моно-
или олигоядерными комплексами с некоординированными неподеленными
электронными парами, как в (8.6). В общем, это наиболее прямой способ получения
таких ненасыщенных строительных блоков путем простого контроля за
стехиометрией реакции, поскольку такие комплексы относительно инертны. При
необходимости можно защитить один из атомов азота N-метилированием.
Лабильные лиганды
(bipyJRu2t (bipyhRu2^
CI
(8.5)
Вакантный координационный центр
(8.6)
Таким образом были собраны удивительные металлокомплексы, например
22-ядерный [Ru{(^-2,3-dpp)Ru[(^-2,3-dpp)Ru{(^-2,3-dpp)Ru(bipyJ}2]2}3]44+ (8.7)
(где dpp — дипиридинопиразил), или металлодендримеры (разд. 8.7). Соединение
(8.7) - блестящий пример того, что для взаимодействия металлокомплексы
используют один и тот же мостиковый лиганд. Поэтому все 12 периферических Ru2+-ueH-
тров, не соединенных друг с другом регулярным образом, окисляются при одном и
том же потенциале в едином 12-электронном процессе.
(8.7)
ф = Ru
2+
Типичные соединения II типа со смешанной валентностью на основе жесткого
бис(бипиридильного) мостикового лиганда (8.8) синтезировал В. Бальзани с
8.2. Супрамолекулярная фотохимия
7091
сотрудниками из Университета Болоньи (Италия) (L. DeCola, V. Balzani et al., 1993).
Эти соединения можно получить как производные Ru11—Ru111, Os11—Os111 и
Ru11—Osm постадийным способом, рассмотренным выше, начиная с менее
лабильных комплексов Os с другим металлом. В каждом случае облучение приводит к
фотохимическому возбуждению Ми-центра, который действует как донор
электронов. Центр М11 переходит в состояние 3MLCT, обладающее высокой энергией, с
которого электрон передается на Мш-центр с образованием электронного изомера
исходного соединения (рис. 8.8). В случае комплексов Ru2 и Os2 типа (8.8а) и (8.86)
продукты неотличимы от исходных соединений, в то время как продукт со
смешанными металлами (8.8в) относится к типу Rum—Os11, который представляет собой
возбужденное состояние основного состояния Ru11—Os111. Этот комплекс
возвращается в основное состояние при термически индуцированном обратном переносе
электрона. В каждом случае перегруппировка ядер пренебрежимо мала, поскольку
не изменяется заселенность антисвязывающих орбиталей eg, которая сильно
влияет на расстояния М—L. Константа скорости релаксации системы со смешанными
металлами заметно ниже константы скорости любого другого процесса, что
согласуется с пониженной движущей силой этой реакции. Как супрамолекулярные
устройства такие соединения могут использоваться для разделения зарядов и
электронного переключения.
2.0-
ю
Б. 1-0 —|
u
03
*Run-L-OsnI
*Ru"-L-Ru'
тт
I \
I 1
I \
I \
I \
I l
I \
I \
I \
I ±
RuIn-L-(
/
I \
I 1
I \
I \
I \
I \
I I
I \
*0sn-L-0sni
Ruu-L-OsUI
Run-L-Ruin RuIn-L-Run
Osni-L-Osn
Рис. 8.8. Диаграммы энергетических уровней фотоиндуцированного переноса электрона
для соединения (8.8), показывающие возбуждение ( ), люминесценцию (—)
и безызлучательный переход ( )
710
I. Молекулярные устройства
M(bipyJ3
N N-
M(bipyJ2+
(8.8)M2+M3+(bipyL
(a): M2+ = M3+ = Ru
F): M2+ = M3+ = Os
(в): M2+ = Ru, M3+ = Os
Одна из конечных целей изучения фотоиндуцированного переноса электрона в
полиметаллических образованиях рассмотренного выше типа — конструирование
каталитических систем, способных аккумулировать солнечную энергию для
проведения химических реакций, которые, если говорить в энергетических терминах,
идут «в гору». Подавляющее большинство таких реакций (например, расщепление
воды на Н2 и О2) является многоэлектронными процессами. В природном
фотосинтезе сложные антенные группы переносят возбуждение к реакционному центру,
который продуцирует большое число высокоэнергетических электронов для
осуществления как синтеза О2 из воды, так и, что более важно, синтеза органических
молекул из СО2. Следовательно, работы в области супрамолекулярной фотохимии
должны быть сосредоточены на системах, способных к многоэлектронному
переносу, аккумулированию энергии и катализу. Схематическая диаграмма такого
процесса показана на рис. 8.9. Два фотосенсибилизатора (PS) поглощают свет и
переносят образующиеся электроны возбужденного состояния к центральному
резервуару электронов и реакционному центру (SR). Компонент реакционного
центра способен принять два электрона, по одному от каждой группы PS, и можег,
Рис. 8.9. Схематическое представление фотохимического молекулярного устройства для
фотоиндуцированного аккумулирования электронов и многоэлектронного катализа.
D — донор, PS — фотосенсибилизатор, SR — резервуар электронов и реакционный центр,
Sub — субстрат
5.2. Супрамолекулярная фотохимия
711
следовательно, осуществить двухэлектронное восстановление субстрата с
образованием ковалентно связанного продукта BSub+ —■► Sub2). Образующиеся
положительно заряженные «дырки» в PS-группах заполняются за счет внешних доноров
электронов, которые сами по себе не могут эффективно восстановить Sub+.
Ранее сообщалось (S. Molnar et al., 1994) о фотохимическом устройстве,
способном реализовать химические превращения такого вида. В этом соединении (8.9)
PS-группами являются центры Ru(II), a SR - это соединения Ir(III). В целом этот
комплекс представляет собой соединение II типа со смешанной валентностью.
Фотопоглощение для соединения (8.9) не очень эффективно, но оно может
катализировать электрохимическое восстановление СО2 до муравьиной кислоты путем
фотовозбуждения фрагментов Ru(II) и переноса двух электронов к 1гAП)-центру,
быстро восстанавливая его до Ir(I). Внешними донорами служат амины, например
диметиланилин. Центр Ir(I) восстанавливает затем СО2 до НСО2Н в присутствии
аре
N N- ч
(8.9)
Развивая этот подход, можно конструировать большие ансамбли антенных
компонентов, которые собирают энергию и переносят возбужденные электроны на
большие расстояния посредством множества «скачков», чтобы сконцентрировать
их в определенном реакционном центре. К таким системам относят фотопоглоща-
ющие полимеры с привитыми хромофорами и порфириновые ансамбли. Системы
на основе дендримеров, полученные из металлополипиридилов, также
представляют значительный интерес (разд. 8.7).
*"» Balzani К, Juris A., Venturi M. et al. Luminescent and redox-active polynuclear
transition metal complexes // Chem. Rev. 1996. Vol. 96. P. 759-833.
8.2*5 ^тройства для преобразования света
Явление люминесценции можно использовать для создания
фотохимических устройств, способных поглощать свет одной длины волны и излучать свет
другой длины волны. Эта простая задача решается с помощью любого
люминесцентного соединения. С точки зрения настройки длины волны излучения и
аккумулирования максимального количества падающего света супрамолекулярное
712
8. Молекулярные устройства
устройство обладает большой эффективностью. Если мы разложим процесс
преобразования света на стадии поглощения (absorbance, А), переноса энергии (energy
transfer, ET) и излучения (emittance, E), то функциональность такого устройства
можно обозначить как А—ЕТ—Е. Оптимальным является такое устройство, в
котором центр, собирающий свет, или антенна, и центр, излучающий свет, могут
настраиваться раздельно. Комбинация взаимодействующих центров дает устройство,
преобразующее свет с максимальной эффективностью.
Рассмотренный выше bipy-лиганд может играть роль антенны. Он легко
настраивается, и компоненты антенны можно получить из ряда биарильных и гетероби-
арильных лигандов bipy-типа. При этом люминесценцируют ионы металлов, таких,
как лантаноиды Eu(III) и ТЬ(Ш), с большим набором состояний/-электронов. В
этом случае взаимодействие между компонентами А и Е относится к
взаимодействию металл—лиганд. Эта концепция была реализована при конструировании
лантаноидных криптатов типа (8.10) и (8.11).
(8.10)
(8.11)
Обычно в водном растворе Еи3+ не люминесцирует, так как возбуждение
тушится молекулами этого растворителя без излучения. Однако комплексобразование с
трис(бипиридил)криптандным лигандом защищает этот ион металла от
растворителя и одновременно предоставляет светоаккумулирующие bipy-фрагменты.
Высокий заряд катиона Еи3+ и его макробициклическая стабилизация лигандом
означают, что эти комплексы очень устойчивы. Они ярко люминесцируют, причем (8.11)
проявляет особенно высокую эффективность преобразования энергии,
достигающую ~60% падающего света. Наличие N-оксидных заместителей стабилизирует
этот комплекс, чему способствует жесткость лиганда, благоприятная для иона
лантаноида, и увеличивает ЕТ-взаимодействие.
8.2.6 Нековалентно связанные системы
В примерах, рассмотренных нами до сих пор, процессы
фотовозбуждения и переноса электрона (еТ) или энергии (ЕТ) осуществляются в
сильносвязанных системах, удерживаемых вместе ковалентными связями или
координационными взаимодействиями со значительной степенью ковалентности. В этих случаях
имеется четкое пространственное соответствие между светоаккумулируюшей груп-
5.2. Супрамолекулярная фотохимия
713;
пой и внешним донором или акцептором (их называют тушителями, поскольку
они тушат люминесцентное излучение поглощенного света, предоставляя
альтернативный путь дезактивации возбуждения). Блестящий пример таких модульных
ковалентных систем - (8.12), синтетическое устройство, объединяющее два
основных компонента природной фотосинтетической системы: металлопорфириновый
хромофор и хиноновый тушитель в качестве внешнего акцептора электронов. Эти
два компонента связаны ковалентно длинным Tt-сопряженным мостиком. Перенос
электрона вдоль этого мостика заканчивается восстановлением хинона до семихи-
нона и, наконец, до гидрохинона.
H,iC5
Перенос электрона
н,,с5 \
Хромофор
(8.12)
Интересно создать аналогичные модульные (и, следовательно, супрамолекуляр-
ные) системы, в которых компоненты связаны исключительно нековалентными
взаимодействиями, такими, как водородная связь. Объединив концепции
самосборки и конструирования молекулярных устройств, можно получить гораздо более
сложные ансамбли. При этом важным моментом является конструирование
функциональных компонентов, содержащих комплементарные и взаимодействующие
центры связывания. Такой подход эффективен только тогда, когда благодаря неко-
валентным взаимодействиям может быть создан эффективный канал для еТ- или
ЕТ-процесса. С этой целью были собраны две порфириновые системы — (8.13) и
(8.14). В обеих системах происходит фотовозбуждение 7п(П)-порфиринового
компонента с последующим переносом электрона на фрагмент с Fe(III) и его
восстановление в Fe(II). Перенос электрона должен происходить через водородные связи
в (8.13) или пенталан с ковалентными а-связями в (8.14). Удивительно, что не
только обе системы эффективны при осуществлении этого процесса, но и константы
скорости переноса фотоэлектрона 8.1-109 и 4.3-109 с для (8.13) и (8.14)
соответственно показывают, что две обычные водородные связи в карбоновых кислотах
более эффективны при переносе возбужденного электрона, чем а-каркас в (8.14).
714
8. Молекулярные устройства
R = 3,4,5-С6Н2(ОМеK
О-Н---О
Ph
В соответствии с принципами, изложенными в гл. 1, сильная1 ассоциация за счет
водородных связей может быть достигнута при условии множественных
комплементарных взаимодействий, как в барбитуратсодержащей порфириновой системе
(8.15) (ср. молекулярные розетки, разд. 7.5.3). К ней комплементарно
присоединена дансильная (диметиламинонафталинсульфонильная) группа с константой
связывания выше 106 М~'. В этом случае перенос электрона происходит от светопогло-
щающей дансильной группы к порфириновому акцептору, причем почти полное
тушение предполагает очень быстрый перенос электрона по сравнению со
скоростью флуоресценции (P. Tecilla et al., 1990).
OEt
О Fl N
Н Bu >-N H
N X /=O N
OEt
$.2. Супрамолекулярная фотохимия
715^
Интересный пример акцептора фотона содержится в системе (8.16), в которой
вместо молекулярного хромофора присутствует полупроводниковая частица ТЮ2
диаметром ~22 A (L. Cusak et al., 1997). Полупроводниковая частица удерживается
благодаря адсорбции на ее поверхности алкильных цепей диамидопиридинового
рецептора. Нанокристаллит ТЮ2 имеет небольшую энергетическую щель
(запрещенная зона) между состояниями ее валентных электронов и зоной проводимости
незаполненных орбиталей. Фотовозбуждение при 355 нм может перевести электрон
из валентной зоны в зону проводимости, а возбужденный электрон может затем
переноситься через фрагмент из водородных связей к виологеновому D,4'-бипириди-
ниевому) акцептору. Контрольные эксперименты показывают, что наличие
водородной связи существенно для переноса электрона и что без нее не происходит
переноса через пространство или переноса, активированного столкновениями.
(8.16)
л-л-Стэкинг-взаимодействия также могут способствовать переносу электрона
между компонентами в фотохимических устройствах. Соединение (8.17)
использует внутреннюю кривизну гликолурильного строительного блока (см. разд. 5.3.1.3)
для размещения хинонового акцептора на расстоянии 9 А над порфириновым
хромофором в U-образной части молекулы. В растворе СС14 внутренняя поверхность
молекулярной системы заполнена молекулами растворителя, и электрон должен
проходить длинный путь по связям от хромофора к акцептору с небольшими
потерями. Наоборот, введение в полость молекулы ароматического гостя, такого, как
гексил-3,5-дигидроксибензоат, приводит к его связыванию внутри этой полости;
при этом молекула выступает в качестве хозяина для ароматических гостей. В
связывании гостя участвуют л—л-стэкинг-взаимодействия и водородные связи гидро-
ксигрупп гостя с амидными карбонильными группами изогнутого каркаса на дне
молекулярной щели. Перенос электрона обеспечивается л-системой гостя, и 75%
люминесценции порфиринового фрагмента тушится в результате переноса
электрона через ароматический гость к хиноновому акцептору (рис. 8.10). Этот вид
электронного переноса через л—л-стэкинг-систему напоминает процессы переноса
электрона внутри двойной спирали ДНК.
•""* Ward M.D. Photoinduced electron and energy transfer in noncovalently bounded
supramolecular assemblies // Chem. Soc. Rev. 1997. Vol. 26. P. 365-375.
716
8. Молекулярные устройства
О
О
О
Акцептор Изогнутый
каркас
(8.17)
Хромофор
изогнутый
каркас
Акцептор
Ароматический гость
Плоскость порфирина
Рис. 8.10. Фотохимический перенос электрона за счет я—я-стэкинг-взаимодействий
с ароматическим гостем
8*3 Информация и сигналы: семиохимия
Семиохимия — область супрамолекулярной химии, в самом широком
смысле связанная с сигнальными устройствами. Термин происходит от слова
«семиотика», означающего изучение знаков и символов, а также их использование
и интерпретацию. Так, в биохимии семиохимическими являются, например, такие
вещества, как феромоны, передающие сигнал от одного организма к другому.
Наиболее очевидная сфера применения семиохимии — производство молекулярных
сенсорных устройств, т.е. соединений, выполняющих двойную задачу - по
молекулярному распознаванию и по выдаче сигнала. Семиохимия, однако, гораздо шире,
чем просто сенсорика. Она также включает вопросы генерирования сигнала, его
обработку, передачу, усиление, превращение и детектирование. Очевидно, все эти
понятия ассоциируются с современными компьютерами и электронными
устройствами. Конструирование молекулярных электронных устройств подробно
обсуждается в разд. 8.4. А пока рассмотрим генерирование сигнала в химических
сенсорах.
I Fabbrizzi L. and Poggi A. Sensors and switches from supramolecular chemistry //
Chem. Soc. Rev. 1995. Vol. 24. P. 197-202.
8.3. Информация и сигналы: семиохимия
8.3 • 1 Фотохимические сенсоры
Основная концепция молекулярного сенсора представлена на
рис. 8.11. Субстрат (гость, аналит) должен приблизиться к рецепторной части
сенсора. Это акт молекулярного распознавания, причем рецепторами могут служить
любые системы, такие, как краун-эфиры, криптанды, кавитанды и т.д.,
рассмотренные в гл. 3—5. Связывание должно быть селективным для данного субстрата в
присутствии ряда других потенциальных гостей, и это зависит от системы и ее
окружения. Простого акта связывания, однако, недостаточно. Рецептор также должен
быть связан с сигнальным элементом, который реагирует на связывание гостя,
генерируя сигнал в виде электромагнитного излучения (фотохимический отклик),
тока (электрохимический отклик) или изменения другого параметра, который можно
измерить (например, цвета и рН). При этом автоматически подразумевается, что
спейсер, соединяющий сигнальный и рецепторный элементы, должен
обеспечивать сообщение между ними и что событие связывания должно запускать механизм
изменения свойств образовавшегося комплекса, которые отличаются от свойств
свободного гостя или хозяина, и соответственно генерировать сигнал. Можно
сформулировать следующие критерии конструирования сенсоров:
• стабильность,
• селективность по отношению к гостю (аналит),
• сродство гостя к хозяину,
• эффективное преобразование сигнала,
• излучение УФ-света, интенсивность которого доступна для регистрации, или
другого сигнала с измеримой интенсивностью,
• кинетически быстрое детектирование,
• простота передачи измеренного сигнала на записывающее устройство,
• доступность.
Одна из областей, для которых создают такого рода хозяева, — аналитическая
химия, где хозяина используют для связывания и распознавания небольших
количеств аналитов. Если концентрация аналита особенно низка, то требуется его
высокое сродство для получения измеримой концентрации комплекса и,
Сигнализирующий Спейсер Рецептор Субстрат
элемент
Рис. 8.11. Схематическое изображение устройства химического сенсора
TIJ
8. Молекулярные устройства
следовательно, для получения отклика. К сожалению, высокие сродство и
селективность часто несовместимы. Хозяева, которые проявляют высокие константы
связывания, часто хорошо связывают большое число гостей, в то время как
высокоселективные хозяева обычно имеют низкие константы связывания. Это
обусловлено тем фактом, что селективность может возникать как результат сильного
неселективного сродства хозяин—гость (например, сольватофобное связывание или
электростатическое притяжение) в сочетании с более слабым отталкивающим
взаимодействием (например, неблагоприятные стерические эффекты), которые могут
изменяться по величине в зависимости от природы или геометрии гостя. Это
можно проиллюстрировать связыванием нуклеотидов типа АТР4~ и АМР2~
протежированными производными E.72) 3-циклодекстрина с двумя E.72)-N2 или семью
E.72)-N7 аминогруппами.
НО
он
E.72)-N2
НО
E.72)-N7
8.3. Информация и сигналы: семиохимия Ш19
Высокопротонированное соединение E.72)-N7 сильно связывает АТР4~,
наряду с широким набором других нуклеотидных анионов (lgA'j, = 6), вследствие
притяжения между хозяином с семью положительными зарядами и отрицательно
заряженными гостями. Наоборот, благодаря более слабому связыванию соединение
E.72)-N2 обладает значительно более высокой селективностью и способно
отличить АТР от большого круга аденозин-сахар-фосфатных производных, включая ри-
бозу и дезоксирибозу.
Элегантный пример модульной флуоресцентной сенсорной системы — краун-
антраценовая система, полученная А.П. де Сильва (А.Р. de Silva). Она состоит из ла-
риат-эфира на основе антрацена (8.18), см. разд. 3.2.2. Фрагмент аза-18-краун-6
выступает в роли рецептора, селективного к ионам К+. Сигнальным элементом
является фрагмент антрацена. Сильная флуоресценция свободного антрацена
обусловлена п—л*-переходом. Однако в соединении (8.18) флуоресценция полностью
тушится в результате переноса электрона от неподеленнои электронной пары
азота. Короткий спейсер -СН2- вполне пригоден для осуществления эффективного
переноса электрона (схема 8.2). Однако в присутствии К+ эта неподеленная пара
включается во взаимодействие с металлическим центром, являющееся частью
взаимодействий макроцикла. В результате тушение флуоресценции антрацена
становится невозможным по чисто термодинамическим причинам (т.е. потенциал
окисления значительно снижается). Следовательно, при связывании К+ «включается»
флуоресценция антрацена, интенсивность которой можно легко измерить.
Сенсоры этого вида называют РЕТ-сенсорами (photon-induced electron transfer, фотоинду-
цированный перенос электрона).
Предложен аналогичный дитопный сенсор на основе антрацена, способный,
как и соединение C.86), разд. 3.11.3, распознавать диаммониевые катионы.
Использование антраценовой группы в качестве спейсера, как в соединении (8.19),
приводит к флуоресцентному распознаванию гостей типа Н3Л (CH2)/1NH3, которое
зависит от длины спейсера (F. Fages et al., 1993).
Перенос электрона \ О
Схема 8.2. Катионный сенсор на основе антрацензамещенного азакоранда (8.18)
720
8. Молекулярные устройства
О О
(8.19)
Флуоресцентные свойства фрагмента [Ru(bipyK]2+ также использованы в ряде
сенсорных устройств, особенно при определении анионов, когда можно
реализовать эффект от наличия положительного заряда в этом компоненте. Так, на основе
о
о
(8.21) (a):M = Fe
(б): М = Co
8.3. Информация и сигналы: семиохимия 721
бипиридила был получен (P. Beer and F. Szemes, 1995) сенсор на хлорид-ион.
Хозяева содержали связывающий азакорандный центр, который может
взаимодействовать с хлорид-ионом за счет четырех водородных связей N—H---C1 при
посредничестве связей С-Н-С1 и положительного заряда двух катионов [Ru(bipyK]2+.
1Н ЯМР-титрование показало, что соединение (8.20) связывает хлорид-ион с
константой -4-104 М в DMSO, а анион Н2РО^ не связывает вовсе, что обусловлено
жесткостью небольшого макроцикла. Наличие двух люминесцентных центров
усложняет флуоресцентное определение аналитов с помощью соединения (8.20).
Поэтому был синтезирован смешанный электрофотосенсор (8.21а, б). Эти соединения
связывают хлорид-ион с таким же сродством, как и (8.20), причем в присутствии
одного мольного эквивалента хлорида наблюдается значительное увеличение
(примерно в 3 раза) интенсивности флуоресценции соединения (8.216).
8*3* 2 Флуорофоры
Разработка флуорофоров (флуоресцентные и связывающие агенты) —
соединений для измерения распределения и концентрации биологически важных
ионов металлов, таких, как Zn2+, Ca2+ и Mg2+, — представляет особый интерес для
сенсорной технологии. Распределение кальция и магния в клеточной мембране
имеет большое значение для запуска и регуляции различных процессов в живой
клетке. Цинк — необходимый компонент ряда важных ферментов, например карбо-
ангидразы, и белков с мотивом «цинковые пальцы»; он также влияет на синтез
ДНК, апоптоз (запрограммированный клеточный суицид, например, при
образовании конечностей и пальцев в эмбрионах) и на экспрессию генов. Особое значение
имеет возможность определять один из этих катионов в присутствии других в
образцах тканей. Первым лигандом, способным выполнять такую функцию, был TSQ
(8.22), действующий как селективный флуоресцентный краситель при определении
Zn2+ в присутствии Са2+ и Mg2+. К сожалению, интенсивность флуоресценции
зависит от характера среды, и, поскольку стехиометрия соединений оставалась
совсем не ясной, эта система была далеко не идеальной. Улучшенный флуорофор —
цинкин (8.23), который, будучи нейтральным, способен диффундировать через ли-
пофильные клеточные мембраны. Проникнув в клетку, он гидролизуется эстераза-
ми и связывает Zn2+ как анионный хелатный лиганд. Образуются комплексы
состава 1:1 и 2:1 с константами связывания ~106-107 М при физиологическом рН.
Такое сродство достаточно для определения Zn2+ в живых клетках, но оно не
позволяет цинкину взаимодействовать с прочно связанным цинком в металлофермен-
тах и белках с мотивом «цинковые пальцы».
Конструирование намного более эффективных сенсоров на цинк основано на
данных о том, что в присутствии цинксодержащей карбоангидразы (СА)
интенсивность флуоресценции флуорофора дансиламида* резко возрастает, а его спектр
смещается в голубую область (схема 8.3). Этот эффект объясняют тем, что при
связывании лигандом закрепленного на ферменте Zn2+ лиганд находится под защитой
гидрофобного кармана фермента. При этом депротонирование дансиламида (с
образованием группы SO2NH~) также способствует связыванию катиона цинка.
* Более точное название — дансиламин. {Примеч. ред. перевода.)
Л 22
8. Молекулярные устройства
МеО
TSQ (8.22)
Цинкин (8.23)
Основываясь на этих данных, Э. Кимура (Е. Kimura et al.) с сотрудниками из
Университета Хиросимы (Япония) получил чрезвычайно эффективный цинковый
флуорофор — дансиламиноэтилциклен (8.24) (Т. Koike et al., 1996), депротониро-
ванная дансиламинофуппа которого обладает высоким сродством к катиону Zn2+.
При хелатировании металлического центра хромофором с несколькими амино-
NH2
Даней л амид
Максимум эмиссии 580 нм
Квантовый выход 5,5%
Карбоангидраза
рН7.4
Гидрофобный
карман
Гистидиновые
остатки
Максимум эмиссии 468 нм
Квантовый выход 84%
Схема 8.3. Усиление флуоресценции и сдвиг в голубую область эмиссионного спектра
дансиламида при связывании с карбоангидразой
1.3. Информация и сигналы: семиохимия
Р723
группами наблюдалось резкое увеличение интенсивности флуоресценции. Это
явление известно как CHEF-эффект (chelation-enhanced fluorescence; флуоресценция,
усиленная хелатированием), и о нем впервые сообщил М. Хастон с соавторами
(М. Huston et al., 1988). CHEF-эффект возникает в результате чрезвычайно
эффективного тушения флуоресценции флуорофора свободными аминогруппами.
Однако при хелатировании (т.е. при сильном связывании) с металлическим центром не-
поделенные электронные пары аминов, имеющие высокую эффективную
концентрацию в свободном состоянии, теряют способность тушить
флуоресценцию, которая и проявляется в полную силу.
Zn
2+
Циклен (8.24)
[12]aH-N4
Связывающий центр циклена ([12]aH-N4) имеет высокое сродство к Zn2+ и в
сочетании с депротонированным остатком дансиламина образуется пятикоординаци-
онный квадратно-пирамидальный 1:1-комплекс с Zn2+, К1020-8 М~'. Как и в
антраценовом сенсоре (8.19), флуоресценция дансиламиногруппы в (8.24) частично
тушится неподеленной электронной парой азота. Однако протонирование
рецептора (8.24) с образованием (8.24)-2Н+ приводит только к 20%-му усилению
интенсивности флуоресценции. Напротив, комплексообразование с Zn2+ в зависимости
от излучаемой длины волны дает пяти-десятикратное усиление флуоресценции, а
также голубой сдвиг в спектре излучения от 582 до 540 нм, так же как и в системе с
СА. В противоположность этому Си2+ тушит флуоресценцию полностью, так как
возможен перенос электрона на частично заполненный flf-подуровень металла.
Флуорофор (8.24) способен определять Zn2+ в интервале 0.1-5 мкМ, причем
интенсивность флуоресценции линейно зависит от концентрации. На связывание не
влияет присутствие ионов щелочных и щелочноземельных металлов, хотя Си2+
успешно конкурирует с Zn2+ (константа связывания Си2+ - более 1030 М). Этот ли-
ганд настолько чувствителен и эффективен, что теперь является коммерческим.
у 24 8. Молекулярные устройства
•""* Kimura E. and Koike Т. Recent development of zink fluorophores // Chem. Soc. Rev.
1998. Vol. 27. P. 179-184.
8*3*3 Электрохимические сенсоры
Подобно тому, как взаимодействие центра связывания и хромофора
хозяина может давать фотоотклик, введение редокс-центра в молекулу хозяина может
приводить к электрохимическому детектированию связывания гостя. В разд. 4.6 мы
рассмотрели ряд хозяев для анионов на основе редокс-пары Со(Ш)/Со(Н) в кобаль-
тоциниевых подандах, корандах и каликсаренах. В случае этих соединений наличие
фрагмента СоСр^ гарантирует, что хозяева положительно заряжены и,
следовательно, комплементарны анионам; этот фрагмент также играет роль сигнальной редокс-
группы, способной при связывании аниона изменять свой редокс-потенциал. Такой
подход можно успешно реализовать при использовании нейтральных производных
ферроцена, в которых связывание гостя отделено от функции сигнальной редокс-
группы как наблюдателя процесса комплексообразования. Работа П. Вира с
сотрудниками (P. Beer et al., 1996) привела к созданию интересного сенсора (8.25) для
нейтральных молекул на основе каликсарена. Хозяин содержит каликс[5]арен с тремя
ферроценильными редокс-группами. Такой хозяин распознает молекулы полярных
растворителей, например MeCN, Me2SO и Me2NCH=O, которые связываются
водородными связями с двумя свободными фенольными гидроксильными группами
нижнего обода каликсарена. Присутствие диполя по соседству с ферроценильными
группами обеспечивает небольшие, но устойчивые сдвиги их редокс-потенциалов,
причем этот эффект отсутствует при наличии неполярных молекул, таких, как
толуол и СН2С12. Интересно, что хозяин имеет две разрешенные волны окисления,
которые соответствуют отклику на распознавание ферроценильными фрагментами,
один из них окисляется первым. Образующийся положительный заряд вызывает
окисление двух других центров при более высоком потенциале. Влияние связывания
полярного гостя состоит в вызываемом им разрыве контакта между редокс-центра-
ми в молекуле»сенсора.
OFc =
(8.25)
8.3. Информация и сигналы: семиохимия
725
И все же связывание молекул-гостей соединением (8.25) — довольно слабое из-
за использования для этой цели слабых центров связывания. Значительно более
существенный электрохимический отклик на связывание нейтральной молекулы
получил Дж. Такер (J. Tucher) из Университета Эксетера (Великобритания), который
использовал мотив распознавания с водородной связью, предложенный А.
Гамильтоном (см. разд. 5.3.4.7) для ферроцен ильного редокс-активного хозяина (8.26)
(J. Carretal., 1997).
О
(8.28)
(8.26)
Глутаровая кислота
(8.27)
Методами циклической вольтамперометрии и ЯМР-спектроскопии показано,
что между хозяином (8.26) и глутаровой кислотой (8.27) в растворе хлороформа
образуется комплекс состава 1:1. При этом редокс-потенциал сдвигается на —85 мВ по
сравнению с —20 мВ для аналогичного хозяина (8.28), имеющего только одну
пиридиламидную группу, участвующую в связывании. Предложенный способ
связывания, когда каждая карбоксигруппа кислоты образует две водородные связи
R(8>2 с пиридиламидным фрагментом хозяина, подтвержден РСА-исследованием
комплекса состава 2:2 соединения (8.28) с глутаровой кислотой (рис. 8.12).
Другой возможный способ для перехода от селективного связывания к
генерированию сигнала — включение селективных молекулярных рецепторов (каликсаре-
ны, ионофоры и т.д.) в полупроводниковые устройства. Такие устройства, как
ISFET (ion-selective field-effect transistors, ион-селективные полевые транзисторы),
способны трансформировать событие молекулярного распознавания в
электрический сигнал. Устройство ISFET состоит из двух полупроводников «-типа (т.е.
полупроводники, допированные избытком электроноизбыточных атомов),
выступающих в роли истока и стока, вмонтированных в кремниевую пластинку /?-типа
(рис. 8.13, а). Проводимость канала между двумя областями «-типа зависит от
электрического поля, перпендикулярного поверхности пластинки. Такие устройства
можно использовать в качестве рН-сенсоров, в которых состояние протонирования
групп SiOH на поверхности определяет величину электрического сигнала канала.
Если на поверхность устройства ISFET химически нанести селективную мембрану,
то ее можно использовать для распознавания субстратов, отличных от Н+.
Мембрана представляет собой гидрофобный слой, содержащий молекулы-хозяева,
способные распознавать данный аналит. Связывание аналита (например, катионов
металлов) внутри мембраны приводит к изменению потенциала, которое влияет на
проводимость канала, генерируя сигнал. Такое химически модифицированное уст-
I. Молекулярные устройства
Рис. 8.12. Кристаллическая структура
2:2-комплекса соединения (8.28) с глу-
таровой кислотой, показывающая роль
соответствия центров водородного
связывания гостя и хозяина
ройство ISFET называют CHEMFET (рис. 8.13, б). Мембранные ионофоры,
изученные до сих пор, включают краун-эфиры, криптанды и модифицированные ка-
ликсарены (P. Cobben et al., 1992). Устройство CHEMFET можно
усовершенствовать нанесением на мембрану слоя гидрогеля, который исключает влияние СО2.
8.4. Молекулярные электронные устройства
727
Рис. 8.13. Схема устройств ISFET (а) и CHEMFET (б).
/ - гидрофобная мембрана, 2- гидрогель, 3- слой оксида (затвор), 4- канал, 5- изолирующая
смола, 6- электрод сравнения. S- исток, /)-сток, В- подложка
Молекулярные электронные
устройства: переключатели,
провода и выпрямители
К молекулярным электронным устройствам можно отнести такие
устройства, которые, по крайней мере концептуально, напоминают компоненты
электронных приборов и компьютеров. Если компоненты электронных устройств, в
частности провода, переключатели, выпрямители и т.д., можно собрать вместе, то
существует принципиальная основа для получения электронных устройств
молекулярных размеров (наноразмеров).
8 *4* 1 Молекулярные провода
Основные свойства молекулярного провода — способности соединять
два компонента (обычно донор и акцептор электрона) и проводить электрический
сигнал или импульс между ними. На молекулярном уровне большое значение
имеет перенос одного электрона. Электрический сигнал может передаваться на
значительное расстояние, например, через биологическую мембрану (~5 нм). Пример из
биохимии - трансмембранный ток, необходимый для передачи нервного импульса
(см. разд. 2.1). В биологических клетках эта проблема решается или переносом
ионов по каналам, или с помощью посредников-ионофоров. С точки зрения скорости
потока зарядов концепция молекулярных проводов ближе к механизму переноса по
каналам. Однако в идеале электронное устройство должно работать на потоке
электронов, а не на потоке катионов щелочных металлов.
Для решения этой проблемы группа Лена (Т. Arrhenius et al., 1986) синтезировала
ряд соединений (8.29)—(8.31) каротиноидного типа, называемых каровиологенами
из-за наличия в их молекулах виологеноподобных бипиридиниевых фрагментов.
Используя метод линейного дихроизма, Лен и сотрудники показали, что эти каро-
виологены способны пронизывать ламеллярные модельные мембраны из дигекса-
728
8. Молекулярные устройства
децилфосфата (плоские жидкие бислойные мембраны, ср. с разд. 10.2), имеющие
толщину, сравнимую с длиной молекулярных проводов. Гидрофильные пиридини-
евые группы взаимодействуют с водной фазой по обеим сторонам стенки везикулы.
Эксперименты по трансмембранной проводимости осуществляли в растворе дигек-
садецилфосфатных везикул, но, к сожалению, соединение (8.29) не показало
увеличения электропроводности через такую мембрану. Это объясняется влиянием на
проводимость соединения (8.29) высокого отрицательного поверхностного заряда
везикулы. Однако при использовании небольших количеств цвиттер-иона (8.30)
наблюдали четырех-восьмикратное увеличение проводимости между внутренней
фазой окислителя, содержащей акцептор феррицианид калия K3[Fe(CNN], и
внешней фазой восстановителя, содержащей донор дитионита натрия (рис. 8.14).
(8.30)
(8.31)
N'
Реальные электронные устройства должны обладать определенной степенью
надежности, т.е. в случае повреждения они должны сохранять работоспособность.
В связи с этим был получен тройной провод (8.31), представляющий собой трех-
жильный молекулярный кабель. Лен с сотрудниками также разрабатывал оболочки
для молекулярной изоляции проводов, продевая тонкие каровиологены сквозь
полости циклодекстринов, как в псевдоротоксане (рис. 8.15).
Альтернативный способ получения молекулярных проводников придумала
М. Фокс (М. Fox) из Техасского университета (США), которая предложила исполь-
I А. Молекулярные электронные устройства
729
Донор
дитионит
натрия
Акцептор
K3[Fe(CNN]
~O,S
Стенка фосфолипидной везикулы
Рис. 8.14. Молекулярная проводимость через бислой фосфолипидной везикулы
Рис. 8.15. Получение изолированного молекулярного
провода: продевание каровиологена сквозь полость
Р-циклодекстрина
730
]. Молекулярные устройства
зовать блоксополимеры, содержащие донорные и акцепторные элементы,
разделенные проводящим реле, как в соединении (8.32). Эти блоксополимеры
синтезировали методом метатезисной полимеризации с раскрытием кольца (ring opening
metathesis polymerization, ROMP), которая позволяет получить монодисперсные (с
узким молекулярно-массовым распределением) материалы в мягких условиях.
Полимеризация катализируется металлами и является примером «живой»
полимеризации, когда после ее завершения при дополнительном введении мономера (даже
структурно отличного) реакция запускается вновь. Таким образом можно строго
контролировать не только общую длину полимера, но и число мономерных единиц,
составляющих каждый полимерный блок. Эти полимеры - достаточно жесткие
системы (т.е. сохраняют форму на шкале времен переноса электрона) и,
следовательно, имеют стабильные электрические характеристики и обеспечивают
векториальный перенос электрона от донора к акцептору.
D (донор) =
I
Me
R (реле) =
Интересно, что скорости переноса электрона в подобных полипептидных
соединениях зависят от ориентации диполя пептидной спирали. Отсюда следует, что
перенос электрона в таких системах можно контролировать, задавая внешний
электрический потенциал. Этот подход можно использовать для управления
дальнодействием и переносом электрона, что необходимо для функционирования нано-
размерных оптоэлектронных устройств (М. Fox, 1999).
Образование молекулярных проводов не ограничивается органическими
проводниками. Благодаря введению в их молекулы ионов металлов с их изменяемыми
в широких пределах редокс-состояниями и фотохимической активностью, не
говоря уже о легко регулируемых электронных и геометрических характеристиках,
открываются большие возможности для синтеза новых металлсодержащих
молекулярных проводников. Например, жесткие стержнеобразные олигомеры (8.33а—в)
8.4. Молекулярные электронные устройства
731
подвергаются двум обратимым одноэлектронным окислительным превращениям,
разность между потенциалами которых увеличивается с уменьшением длины
мостика (эта разность достигает 0.44 В для (8.33а)). Наличие двух процессов окисления
(оба формально представляют собой переходы от Re(I) к Re(II)) указывает на то,
что на электронное окружение второго металлического центра влияет окисление
первого через сопряженный поликумуленовый лиганд. Действительно, в случае
(8.33а) может быть выделено соединение Re(I)/Re(II) со смешанной валентностью
и таким образом оно становится соединением III типа со смешанной валентностью
(т.е. «внешний» электрон Re(I) полностью делокализован на обоих металлических
центрах). Эти небольшие молекулярные системы можно увеличить в размерах до
образования проводящих металл-ацетилидных полимеров типа (8.34), которые
также интересны с точки зрения их электропроводности и нелинейно-оптических
свойств.
Ph3P\ I . NO
Re
, Re.
ON I PPh3
С
III
С
I
С
III
с
I
с
III
с
NO
PPh3
(8.33a)
(8.336)
(8.34)
(8.33b)
*""* Lehn J.-M. Supramolecular chemistry: Concepts and perspectives. Weinheim: VCH,
1995. P. 106-111. Русский перевод: Лен Ж.-М. Супрамолекулярная химия:
Концепции и перспективы / Пер. с англ. Е.В. Болдыревой; Под ред. В.В. Власова и
А.А. Варнека. Новосибирск: Наука, 1998. С. 130-137.
7321
8. Молекулярные устройства
8 «4* 2 Молекулярные выпрямители
Выпрямитель — это устройство, пропускающее поток электронов
только в одном направлении. В больших электронных устройствах его используют для
превращения переменного тока в постоянный. В обычных выпрямителях имеются
контакты между полупроводниками /ьтипа (электронодефицитный) и л-типа (эле-
ктроноизбыточный). Действие выпрямителя основано на появлении
изолирующего слоя в контактной области (рис. 8.16). Когда между полупроводниками/?- и л-ти-
па образуется соединение, электроны переходят от донора к акцептору вплоть до
нейтрализации заряда внутри контактной области и прекращения потока
электронов. Если к выпрямителю приложить внешнее напряжение, то в зависимости от
полярности такого напряжения возникает одна из двух возможных ситуаций. Если
знак приложенного потенциала таков, что катод (отрицательно заряженный
электрод) контактирует со стороной полупроводника л-типа, а анод — со стороной
полупроводника /?-типа, то изолирующий слой будет уменьшаться, а поток электронов
(ток) будет увеличиваться. Наоборот, при обратной полярности изолирующий слой
будет увеличиваться, препятствуя потоку электронов. При приложении
переменного тока электрический ток возникает только в случае надлежащей полярности, т.е.
переменный ток превращается в постоянный.
На молекулярном уровне точно такие же характеристики должны иметь супра-
молекулярные компоненты и, следовательно, молекулярный выпрямитель должен
состоять из донора и акцептора, разделенных изолирующим спейсером.
Соединения (8.35) и (8.36) содержат а-изолирующий спейсер, а донорно-акцепторные ка-
ротиноидные производные типа (8.37) разделены л-системой с более высокой
проводимостью.
Донор
ОМе Акцептор
ОМе
О (8.36)
(8.37)
8.4. Молекулярные электронные устройства
733
Рис. 8.16. Полупроводник р-л-типа
Изолирующий слой
(a) v=o
Jfc.
Катод
(б) V=0
Анод
■и-
В 2
Акцептор
Акцептор
....3.
Донор
Анод
Донор
Катод
Рис. 8.17. (а) Поток электронов, (б) Ток отсутствует пока ^значителен. Стадии 1—3
отражают процессы туннелирования
734 8. Молекулярные устройства
Действие молекулярного выпрямителя можно объяснить с помощью рис. 8.17 на
примере молекулы типа (8.35), присоединенной своими донорной и акцепторной
частями к аноду и катоду при различных сочетаниях полярности. Схема цепи,
показанная на рис. 8.17, я, обеспечивает протекание тока, поскольку движущий
потенциал V, требуемый для преодоления изолирующего слоя, относительно мал, т.е.
этот слой тонок. Для цепи, приведенной на рис. 8.17, б, движущий потенциал
должен быть значительно выше; поэтому движение потока электронов в таком случае
затруднено. Величина V определяется относительными энергиями орбиталей
донора и акцептора. В случае цепи, показанной на рис. 8.17, а, электроны с катода
переносятся на орбиталь В акцептора, как только потенциал становится достаточно
большим, чтобы вызвать перекрывание орбиталей донора и акцептора.
Аналогичным образом электроны переносятся с орбитали Сдонора к аноду. Из-за малой
разности энергий орбиталей В и Свозможно туннелирование электронов через
изолирующий спейсер. Электрон с орбитали В туннелируется на орбитали донора в
возбужденном состоянии Франка-Кондона, а затем безызлучательно релаксирует в
основное состояние; при этом электрическая цепь замыкается. Этот процесс
необратим, пока энергия В больше энергии С.
В обратной ситуации (рис. 8.17, б) непрерывный поток электронов достигается
только тогда, когда уровень D находится ниже уровня Ферми катода, что позволяет
электронам переходить от катода к D. Аналогично уровень Ферми анода должен
быть ниже уровня А. Для этого необходим потенциал, значительно больший, чем в
первом случае.
&~* Vogtle F. И Supramolecular chemistry. Chichester: Wiley, 1991. P. 313-319.
8.4* 3 Система 1,2-дитиенилэтена как переключатель
Превосходный компонент молекулярных устройств — система
1,2-дитиенилэтена, которую изобретательно использовали Лен и соавторы (S.Kawai,
S. Gilat, J.-M. Lehn, 1994) в ряде переключающих устройств. Основным
переключающим актом является переход дитиенилэтена между двумя стабильными формами
в зависимости от длины волны падающего излучения (схема 8.4).
Обратимость переключения обеспечивается путем чередования облучения
системы УФ- C65 нм) и видимым светом (более 600 нм). Кроме того, две формы —
открытая и закрытая — сильно различаются электронными свойствами. Закрытая
форма содержит полностью сопряженную л-систему вдоль мостика, что
обеспечивает надежную электронную связь между заместителями R1 и R2 через эту делока-
лизованную систему. В открытой форме R1 от R2 изолированы друг от друга.
Группа Лена нашла различные области применения этой функциональной
переключающей системе, опираясь на ее «компонентную» природу, которая, по
определению, может быть легко перестроена. В соединении (8.38) переключатель
связывает одинаковые заместители. Найдено, что в закрытой форме соединение (8.38)
легко восстанавливается электрохимически; это не характерно для открытой
формы. Таким образом, мы имеем прототип переключаемого молекулярного провода.
В случае соединения (8.39) R1 - электронодонорная, a R2 - электроноакцепторная
группа. Соответствующая молекула в закрытой форме — это электронно-коммун и-
.4. Молекулярные электронные устройства
R1
R1
R1
Открытая форма
•'■ "Ok
365 нм
>600hm
-R2
(8.38)
R<
CN
A
J
CN
F F
Закрытая форм;
(8.39)
R2
i
Схема 8.4. Формы бистабильного переключающего 1,2-дитиенилэтена
F %F
\ /C .^F
p^\ /*^p №hm _
У Л. >600нм
Открытая форма
BIS-OH (8.40)
i
Электрохимически
[ инертная форма
(в интервале -1 т +1 В)
>6QQhm
т ' X
Фотохимически
инертная форма
\ / F
\^он
Закрытая форма
BIS-OH
i
Е1/2 = -180мВ
(относительно НКЭ)
/Л
ЛоХл
Закрытая форма
BIS-Oq
Схема 8.5. Фото- и электропереключаемая система (8.40)
736
8. Молекулярные устройства
кационная система типа «тяни—толкай» («push—pull»), обладающая заметными
нелинейно-оптическими (nonlinear optical, НЛО) свойствами, что следует из большой
величины ее гиперполяризуемости (коэффициент Р). При облучении видимым
светом раскрывается кольцо переключателя и нарушается сообщение между двумя
концами молекулы, при этом гиперполяризуемость заметно уменьшается.
Нелинейно-оптические свойства будут рассмотрены в разд. 8.6.
Возможно, что из систем, основанных на этой концепции, наиболее интересна
система (8.40), представляющая собой фото- и электропереключаемое устройство
(схема 8.5). Как и в случае (8.38), открытая форма соединения (8.40) BIS-OH
электрохимически инертна. Фотопереключение в закрытую форму позволяет молекуле
легко окисляться в бис(хинон) BIS-Oq. Хиноновая форма фотохимически инертна
и не может перейти в открытую форму без восстановления. Систему (8.40) можно
использовать для надежного хранения информации, поскольку она является
химическим аналогом такого способа хранения информации, как EDRAW (erase direct
read after write, стирание прямого считывания после записи), - процесс, когда
информация записывается оптически при облучении УФ-светом и затем
закрепляется путем окисления. Информация может позднее считываться много раз, и, в
конце концов, ее можно стереть после восстановительной разблокировки (ср.
WORM-процесс (write once read many; однократная запись, многократное
считывание), когда информация записывается на носителе, таком, как CD-ROM, и может
затем считываться много раз по мере надобности).
8 «4*4 Электропереключаемая люминесценция
Другой пример комбинированного электрооптического
молекулярного устройства — пара (8.41) и (8.42). Переходы между этими компонентами легко
осуществляются электрохимически, поскольку окисление гидрохинонового фраг-
(8.41)
(8.42)
8.4. Молекулярные электронные устройства 737
Перенос энергии
или электрона
Излучение Излучение
отсутствует
Рис. 8.18. Электрофотопереключение
мента происходит перед окислением центра Ru(II). В восстановленной гидрохино-
новой форме остаток Яи(Ыру)з+ сильно люминесцирует из состояния MLCT, как
описано в разд. 8.2. Однако электрохимическое окисление в хиноновую форму
приводит к полному «выключению» люминесценции из-за способности хинона тушить
состояние MLCT путем чрезвычайно быстрого переноса электрона от металла к ли-
ганду. Хинон — эффективный акцептор электронов. Действительно, хиноны
играют значительную роль при разделении зарядов в фотосинтезе благодаря своим
низкоэнергетическим акцепторным л*-орбиталям (см. разд. 2.3). Следовательно, это
соединение действует как бистабильный электрофотопереключатель (рис. 8.18). И
окисленную, и восстановленную форму можно выделить, причем эта редокс-пара
полностью обратима и устойчива. Люминесценция тушится полностью (V. Goulle
etal., 1993).
8.4*5 Переключаемое связывание
Фотохимически индуцированные реакции можно использовать для
переключения светопоглощающего материала из одного состояния в другое с
последующим изменением его свойств. Эти свойства могут быть связаны с
молекулярной структурой, изменение которой приводит к важным последствиям для
химии такой системы. Например, фотохимическое переключение между одним
хозяином (который может прочно связывать катионы щелочных металлов) и
другим хозяином (с намного более слабым связыванием) можно использовать для
управляемого световым импульсом высвобождения катионов — фотоионного сигнала.
Такое поведение наблюдается у нитрофенильного производного [2.2.2]криптанда
(8.43). Криптанды прочно связывают катионы типа К+ даже в воде (см. разд. 3.3),
однако облучение при -330 нм вызывает раскрытие кольца и образование краун-
эфирного лиганда (BiBLE) (8.44) с боковой цепью. Это соединение имеет
значительно меньшее сродство к катионам металла (примерно в 104 раз), и,
следовательно, любой связанный ион металла немедленно высвобождается (схема 8.6).
17-СтидДж. В.
8. Молекулярные устройства
Схема 8.6. Переключаемое связывание катиона с помощью фоторасщепления катионно-
го криптата, приводящего к генерированию фотоионного сигнала
Другой весьма популярный переключаемый компонент — азобензол, который
подвергается ц«с/тря//с-изомеризации под влиянием и света, и тепла согласно
реакции, приведенной на схеме 8.7. Эта способность азобензола к обратному
переключению была использована для множества молекулярных хозяев, близких краун-
эфирам (азокрауны), циклофанам (азофаны) и соединениям типа азокриптандов и
азоциклодекстринов. Азокрауны, такие, как (8.45), могут обеспечить
переключаемое связывание катионов щелочных металлов по типу «включено—выключено»,
когда «выключающие» траяс-изомеры не проявляют никакого связывания из-за
увеличения длины краун-мостика, в то время как «включающие» цис-формы
показывают комплексообразующие свойства подобно их 15-краун-5-, 18-краун-6- и
21 -краун-7-аналогам.
Av
дг
(ЯМ8.45)
(ZM8.45)
8.4. Молекулярные электронные устройства
739
Схема 8.7. Переключение азобензола
Относительная стабильность цис- и ш/?анс-изомеров во многих устройствах,
содержащих производные азобензола, зависит от природы гостей, которая влияет
также на скорости как термической, так и фотохимической изомеризации.
8*4*6 Аллостерические переключатели
Аллостерическая система — это система, которая включает
конформационные взаимодействия между двумя и более связывающими центрами (ср.
гемоглобин). Так, связывание на одном центре вызывает конформационные изменения,
влияющие (по типу «включено—выключено») на связывающую способность
другого центра. Одним из первых примеров такой системы был макроцикл (8.46) на
основе бипиридила (J. Rebek, 1984). В свободном состоянии эта молекула не предор-
ганизована для связывания с любым из ее центров. Комплексообразование
бипиридильного центра с переходным металлом в низком состоянии окисления,
например с Ru(II), вызывает конформационное изменение, включающее вращение
вокруг оси пиридил—пиридил, которое переводит краун-эфирную группу в кон-
740 8. Молекулярные устройства
формацию, более предорганизованную для связывания. Точно так же связывание
щелочного металла краун-эфирным фрагментом свободного лиганда должным
образом организует фрагмент бипиридила для связывания гостя. Системы, подобные
этой, где два связывающих центра разделены только несколькими связями,
находятся на одном конце длинного ряда; другой конец занимают макромолекулы, в
которых аллостерические связывающие центры могут быть разделены большими
расстояниями.
Молекулярные машины
на основе катенанов и ротаксанов
Синтез и свойства механически сцепленных соединений, называемых
катенанами и ротаксанами, суммированы в разд. 7.6. Механическая природа связи
между компонентами ротаксана и катенана сильно напоминает механическую
связь в макроскопических машинах, что инспирировало множество работ по
фото-, редокс- и рН-переключаемым системам.
Вначале Дж. Стоддарт и сотрудники в рамках работ по синтезу [3]ротаксанов
попытались получить длинные, гантелеподобные нити, содержащие два и более элек-
троноизбыточных фрагментов арила и биарила. Однако в большинстве случаев
были получены только [2]ротаксаны. Структура [2]ротаксана, содержащего два
электроноизбыточных связывающих центра, позволяет «качать» макроцикл G.41)
вперед и назад между двумя крайними положениями подобно поезду на
железнодорожном пути с двумя станциями. В результате методом продевания был получен
[2]ротаксан, показанный на рис. 8.19; его выход составил 19%. Поскольку две
«станции» отличаются друг от друга, существуют значительные энергетические
предпосылки для задержки «поезда» у более обогащенного электронами бензиди-
нового фрагмента («азотная станция»). 'Н ЯМР-спектроскопия дает соотношение
двух трансляционных изомеров 84:16 в растворе CD3CN при -44 °С. Однако прото-
нирование трифторуксусной кислотой резко уменьшает электронную плотность на
основном бензидиновом фрагменте, заставляя макроцикл полностью переходить к
4,4'-диоксибензольному фрагменту. Подобный результат получен при
электрохимическом окислении «бензидиновой станции», дающем аналогичный
катион-радикал. Полная обратимость как рН-, так и электрохимически переключаемого
процесса приводит к получению ротаксанового молекулярного переключателя.
Большое число работ посвящено получению редокс- и фотопереключаемых
ротаксанов. В частности, найдено, что использование в молекуле нити (см. рис. 8.19)
вместо группы Si(/-PrK фотоактивных концевых групп, содержащих антрацениль-
ные заместители, или редокс-активных ферроценильных групп позволяет
контролировать скорость рекомбинации заряда при фотовозбуждении в образовавшихся
ротаксанах (8.47) и (8.48). Полагают, что фотовозбуждение лазерным импульсом
при 437 нм дает ион-радикальную пару за счет переноса электрона от электроноиз-
быточной нити к макроциклу. В случае ферроценильного производного (8.48)
наблюдали долгоживущие состояния с разделенными зарядами, что напоминает
v ИКОНО
МОМОН ° °
1
!
Рис. 8.19. рН- и электрохимически переключаемый «молекулярный челнок» G.41) на основе [2]ротаксана
742
8. Молекулярные устройства
процессы, сопровождающие фотосинтез. Фотовозбуждение [2]ротаксана тушится
быстрым окислением ферроценильной запирающей группы окисленным 4,4'-ди-
оксибензольным фрагментом. Рекомбинация заряда происходит очень медленно
из-за отталкивания катионного макроцикла от положительно заряженной
ферроценильной концевой группы (рис. 8.20). Напротив, в случае антраценильной
запирающей группы (8.47) рекомбинация зарядов идет очень быстро.
О
ОООО
WWW
(8.47)
г\г\г\
оооо
оооо
WWW
(8.48)
8.5. Молекулярные машины на основе катенанов и ротаксанов
743
Рис. 8.20. Большие времена жизни состояния с разделенными зарядами
для соединения (8.48)
Работа по псевдоротаксанам привела к получению системы с переключением
между двумя различными цветами (хромофорный переключатель), которое являет-:
ся результатом связывания двух разных нитей. Цвет комплекса, изменяющийся от
красного до пурпурного, зависит от того, какая нить, (8.49) или (8.50), связывается.
В исходном состоянии переключателя предпочтительно связывается более элек-
троноизбыточная 1,5-диоксинафталиновая группа соединения (8.49), придавая
пурпурную окраску комплексу в растворах MeCN. Нить (8.49) дитопна и может
выступать в роли хозяина для катионов щелочных металлов, особенно для К+, за счет
имеющегося в ее составе кольца 18-краун-6. Однако в комплексе, образовавшемся
при связывании ионов К+ краун-эфирной частью соединения (8.49), немедленно
возникает сильное электростатическое отталкивание между нитью и катионным
циклофановым паракватным производным G.41). Поскольку этот комплекс
является лишь псевдоротаксаном (запирающие группы отсутствуют), такое
отталкивание вызывает немедленное вытягивание нити из циклофана G.41) и связывание
последнего с другой нитью, (8.50), что придает раствору красное окрашивание.
Таким образом, присутствие бесцветных солей К+ можно определять просто при
визуальном контроле этого раствора (схема 8.8).
Несимметричные катенаны, подобно ротаксановому «челноку», также могут
подвергаться трансляционной изомерии (рис. 8.19). Был сконструирован [2]кате-
нан на основе асимметричного тетракатионного циклофана (8.51), содержащего
два различных центра распознавания, или две «станции»: бипиридиниевый и
т/?я//с-бипиридинийэтиленовый фрагменты. Этот тетракатионный циклофан
можно сцепить с бис(и-фенилен)-34-краун-10 C.95) тем же способом, что и соединение
G.48), разд. 7.6. ]И ЯМР-спектр полученного катенана в ацетоне показал, что
меньший бипиридиниевый фрагмент соединения (8.51) находится внутри электроноиз-
быточного краун-компонента C.95). Электрохимическое восстановление соедине-
8. Молекулярные устройства
У-\г\г\
-* О О (
О О
\ /
о о о
АЛ Г\
но о о
Красный
НО О О
&
(8.50) I (8.49)
О
о о
У—\г\г\
0й О О О
Пурпурный
G.41)
Электростатическое
отталкивание
+ (8.50)
Схема 8.8. Химически управляемый хромофорный молекулярный переключатель
на основе псевдоротаксана
8.5. Молекулярные машины на основе катенанов иротаксанов
4745
C.95)
(8.51)
ния (8.51) с образованием трикатион-радикала также происходит на его бипириди-
ниевом фрагменте вследствие высокой п-кислотности. При таком восстановлении
резко уменьшаются электроноакцепторные характеристики бипиридиниевого
фрагмента, что приводит к его выталкиванию из кольца C.95) путем вращения цик-
Схема 8.9. Электрохимически контролируемые перемещения циклов [2]катенана
74E 8. Молекулярные устройства
лофана (8.51). Теперь его место внутри полости краун-эфира займет более л-акцеп-
торный бипиридинийэтиленовый фрагмент. При окислении процесс идет в
обратном направлении (схема 8.9).
•""* Balzani К, Gomez-Lopez M, Stoddart J.F. Molecular machines // Асе. Chem. Res.
1998. Vol. 31. P. 405.
8*6 Материалы для нелинейной оптики
Возможность управлять частотой, фазой, поляризацией или
траекторией света имеет большое технологическое значение в таких областях, как
производство настраиваемых лазеров и аккумулирование оптических данных. В
настоящее время множество исследований посвящено как органическим, так и
неорганическим материалам, проявляющим нелинейно-оптические (НЛО)
эффекты, в частности генерацию второй гармоники (ГВГ), которая, попросту говоря,
вызывает удвоение частоты падающего света. Обычно их используют, например, для
получения синего цвета (длина волны —300 нм) из зеленого (длина волны 600 нм).
НЛО-материалы, такие, как дигидрофосфат калия (PDP), также применяют для
генерации суммарной частоты, когда смешивается свет от двух лазеров различной
частоты с образованием суммарной волны (частоты) на выходе. Инженерия
материалов с заданными НЛО-свойствами - важная научная задача, и, поскольку это
предмет модульного молекулярного дизайна, она адресована специалистам в
области супрамолекулярной химии. Прежде чем начать обсуждение НЛО-материалов и
их свойств, стоит коротко обсудить происхождение этих эффектов. Подробные
данные можно найти в ключевой ссылке.
*~* Marder S.R. Metal containing materials for nonlinear optics // Inorganic materials.
2nd ed. / Ed. D.W.Bruce, D.O'Hare. Chichester: Wiley, 1996. P. 121-169.
8.6.1 Основы нелинейной оптики
Когда электромагнитное излучение взаимодействует с материей, оно
вызывает колебания электронной плотности в материале с той же частотой, что и
частота падающего света. Поскольку в разрешенном во времени процессе
взаимодействия света и материи участвует много фотонов, неудивительно, что колебание,
вызванное одним фотоном, будет влиять на свойства этого материала, а это
«увидит» следующий фотон. Присутствие осциллирующего заряда приводит к
некоторому разделению зарядов и образованию осциллирующего индуцированного
диполя с моментом \i, который является функцией частоты падающего света (ш) и
пропорционален величине Еего электрического поля (рис. 8.21, а). Величина
индуцированного диполя при данном Ев основном определяется тем, насколько легко
оно деформирует электронную плотность в данном атоме или молекуле. Легкость
деформации электронной плотности - это ее поляризуемость. Поляризуемость —
8.6. Материалы для нелинейной оптики
747
(а)
О
(б)
(в)
А
i
1 1
t Т
Линейная
шая х
Нелинейная
Приложенное поле
Рис. 8.21. Индуцированная поляризация как функция времени, (а) Линейная
и (б) нелинейная поляризация, (в) Графическое представление поляризации в виде
зависимости от величины приложенного поля
тензорная величина, так как она может быть различной в различных направлениях.
Так, общая молекулярная поляризация (F) дается уравнением:
(8.2)
Здесь аДш) - тензор линейной поляризуемости при частоте ш, который
описывает линейную вариацию индуцированного диполя с электрическим полем.
Осцилляция поляризации (осциллирующий диполь) является, по существу, движущимся
зарядом, и, следовательно, должна вызывать излучение той же частоты, что и
осцилляция. Это приводит к таким оптическим эффектам, как двойное
лучепреломление и рефракция.
Уравнение (8.2) описывает полную картину, если только атомный или
молекулярный потенциал является классическим гармоническим потенциалом. Тем не
менее, когда на молекулу действуют очень интенсивные поля, например свет
лазера, материал становится настолько поляризованным, что его поляризуемость может
измениться и тогда индуцированная поляризация становится нелинейной
функцией силы поля (рис. 8.21, в). Для примера рассмотрим полярную связь С=О в
ацетоне. Электроотрицательность кислорода означает, что электронная плотность будет
поляризоваться легче по направлению к кислороду, чем к углероду. Поэтому
величина нелинейной поляризации будет зависеть от направления поля (рис. 8.21, б).
Напомним, что это тензорная величина, поскольку падающий свет может
распространяться в любом направлении (х, у или z) и индуцировать поляризационный
отклик в направлениях х, у или z. Такой асимметрический поляризационный отклик
можно разложить в ряд Фурье, содержащий компонент непосредственного
взаимодействия (direct coupling, DC) ц0, а также компоненты основной частоты и частоты
748 8- Молекулярные устройства
второй гармоники. Обычно используют аппроксимацию разложением в ряд
Тейлора. Строго говоря, этот ряд имеет бесконечное число членов и постепенно
становится более точным по мере увеличения их числа. При высокой силе поля
становятся важными второй и третий члены ряда.
ц = ^0 + (atpE+ */2$iJk)E-E+ */6(yiJk)EEE + - (8.3)
Основной компонент (ос£) линеен по En отражает линейно-оптические
свойства, обсуждавшиеся выше. Вторая (]/2ф)Е-Е), третья A/6(у)Е-ЕЕ) и последующие
гармоники нелинейны по Ей отвечают за НЛО-эффекты. Величины Р и у
называют коэффициентами квадратичной и кубической гиперполяризуемости
соответственно. Второй гармонический член описывает ГВГ, третий — эффекты утроения
частоты и т.д. Существенно, что, так как только усредненная по времени
асимметрично индуцированная поляризация приводит к НЛО-эффектам второго
порядка, молекула или кристалл должны быть нецентросимметричными, иначе эти
эффекты будут гасить друг друга. Тем не менее эффекты третьего порядка можно
наблюдать как в центросимметричных, так и в нецентросимметричных структурах.
Итак, каким образом взаимодействие света с материей, вызывающее уже
знакомые нам эффекты, сказывается на влиянии материи на свет (удвоение частоты и
т.д.)? В случае линейного вклада осциллирующий заряд излучает свет с частотой
колебаний, равной частоте падающего света. Для НЛО-вкладов осцилляция
поляризации ангармонична и содержит вклад ЕЕ в случае второго члена.
Электрическое поле в данный момент времени зависит от частоты излучения и
амплитуды волны Ео следующим образом:
£■=£■„ cos(o)/). (8.4)
Уравнение (8.3) можно переписать так:
V2((V)£o cos2(w/) + l/6(yiJk)El cos3(oH + - (8.5)
где cos2(w/) = V2 + l/2cosBo)t) и, таким образом, осциллирующая поляризация
второго порядка, индуцированная электромагнитным излучением, содержит
компонент, осциллирующий с частотой 2@, т.е. частота удваивается. Этот заряд,
осциллирующий с двойной частотой, будет, следовательно, давать излучение двойной
частоты в соответствии с величиной второго гармонического члена. По существу,
два фотона одной частоты, объединяясь, дают один фотон удвоенной частоты.
Аналогичным образом вклады с утроенной частотой описываются третьим
гармоническим членом ряда и т.д.
На самом деле это особый случай суммирования двух (или более) фотонов с
одинаковыми энергиями. Если свет от двух различных лазерных источников
суммируется, то мы имеем два электрических поля (ZTj и Е2) и две частоты (tOj и ш2). ГВГ-эф-
фект теперь заключается в сложении этих двух частот с образованием излучающего
компонента новой частоты, ш3 = Wj + @2. Таким образом настраиваемые лазеры с
переменной частотой ш3, использующие такие НЛО-материалы, как PDP, стали те-
8.6. Материалы для нелинейной оптики 749
перь реальностью. Однако одна из проблем ГВГ состоит в том, что доля излучения
с удвоенной частотой составляет очень небольшую часть интенсивности
падающего света. Понятно, что этот вклад зависит от величины члена 1/2Ф)ЕЕ.
Интенсивность света с удвоенной частотой можно увеличить путем увеличения Е (т.е.
мощности лазера), но необходимо также получать новые материалы с большой
гиперполяризуемостью (величина Р), что позволит увеличить эффективность этого
процесса.
Итак, каким образом можно создавать материалы с большим коэффициентом
гиперполяризуемости? Как химики мы немного представляем себе линейную
поляризуемость молекулы. Органические молекулы типа ацетилена более поляризуемы,
чем этан, из-за их я-электронных облаков. Неорганические материалы, такие, как
полупроводники с небольшой щелью HOMO—LUMO, более поляризуемы, чем
изоляторы. В общем, можно сказать, что поляризуемость связана со степенью дело-
кализации электронов, которая, в свою очередь, зависит от числа орбиталей в
рассматриваемой электронной системе, их гибридизации и степени межорбитального
взаимодействия. В случае гиперполяризуемости эта проблема менее очевидна.
Ангармоническая поляризация показывает наибольшее отклонение от линейного
вклада при высоких степенях искажения электронного облака. Это означает, что
мы не можем наблюдать НЛО-эффекты в случае неполяризуемых молекул, где это
искажение всегда мало. К тому же, нам нужна большая собственная
ангармоничность (т.е. отклонение потенциала от линейности, и, следовательно, большая
величина 3). В терминах молекулярного дизайна это означает, что мы нуждаемся в
молекуле с электронодонорной и электроноакцепторной частями. Всегда легче
поляризовать молекулярную электронную плотность по направлению к акцептору,
чем по направлению к донору, и, следовательно, донорно-акцепторные молекулы,
особенно органические, имеют одни из наибольших величин р. Сами по себе
большие величины Р, однако, недостаточны для того, чтобы ГВГ-материал обладал
необходимой эффективностью. Должны выполняться критерий нецентросимметрич-
ности (т.е. кристаллический материал должен кристаллизоваться в полярной
пространственной группе), а также другие критерии. Материал должен быть
бесцветным или только слегка окрашенным, чтобы не поглощать свет лазера с
последующим перегревом или плавлением. Он также не должен повреждаться при
действии света высокой интенсивности. Эти свойства более присущи неорганическим
соединениям, которые часто имеют более низкие величины Р, и, следовательно,
проблема получения органических и неорганических НЛО-материалов с
улучшенными характеристиками еще ждет своего решения.
8.6.2 Материалы для нелинейной оптики
второго порядка
Основные электронные критерии, применяемые при
конструировании ГВГ-устройств с большими значениями Р:
• большие изменения дипольного момента при возбуждении,
• большие дипольные моменты переходов,
• малая энергетическая щель между возбужденным и основным состояниями.
|%50 8. Молекулярные устройства
Мы уже встречались с некоторыми органическими донорно-акцепторными
молекулами типа молекулярных выпрямителей (8.35)—(8.37) и асимметричного
переключателя (8.39). Благодаря большому расстоянию между органическими донором
и акцептором электронов в этих системах и наличию между двумя частями
молекулы электронного сообщения (молекулярный провод) эти системы легко
поляризуются и имеют высокую гиперполяризуемость, удовлетворяющую указанным
критериям. Поэтому такие системы проявляют НЛО-свойства. Используя концепцию
молекулярных проводов, Лен предложил ряд полиенов типа «тяни-толкай» с
различной длиной провода на основе бензодитиа- или диметиламинофенильных до-
норных групп (D) и разнообразных акцепторных групп (А), рис. 8.22. Материалы с
более длинной цепью имеют большие величины 3, однако остается проблема
организовать так эти молекулы в регулярные нецентросимметричные ансамбли, чтобы
могли проявляться их НЛО-свойства. Величины 3 можно определить в
экспериментах по генерированию второй гармоники, индуцированной электрическим
полем (electric-field-induced second harmonic, EFISH). Этот метод позволяет провести
векториальное вычленение тензора гиперполяризуемости вдоль направления
молекулярного диполя и, следовательно, дает прямую информацию о собственной
оптической нелинейности молекулы (L. Cheng et al., 1991). Такая ориентация
достигалась путем включения соединения с длинной цепью (полиен) в однослойную
пленку Ленгмюра—Блоджетт из жирной кислоты. Жирные кислоты — примеры ди-
фильных соединений (разд. 10.2), они содержат гидрофильные «головки» (СО2Н) и
длинные гидрофобные «хвосты». На поверхности воды они образуют монослои, в
которых все «головки» направлены в водную фазу, а углеводородные цепи
(«хвосты») выступают над поверхностью жидкости. Упорядоченные системы, ведущие
себя таким образом, описаны в гл. 10. Если выбрана жирная кислота, длина молекулы
которой примерно равна длине молекулы донорно-акцепторного соединения с
гидрофобным молекулярным проводом между D- и А-фрагментами, то
НЛО-материал внедряется в этот монослой, причем все D- и А-группы имеют согласованную
нецентросимметричную ориентацию. Монослои, полученные таким образом,
проявляют заметные НЛО-свойства (рис. 8.23).
Аналогичные подходы применимы и к металлоорганическим материалам, где
присутствие металлоорганических остатков, таких, как карбонилы металлов и ме-
таллоцены, предоставляет широкие возможности для синтетического и
электронного конструирования. Изучены производные металлоценов типа (8.52) и показа-
X = Н, Me
л=1,2
Y = CN, СНО, NO2
М = Fe, Ru
(8.52)
8.6. Материалы для нелинейной оптики
751
(б)
А =
у v UN \
Рис. 8.22. Полиены типа «тяни—толкай», в которых органические донор и акцептор
электронов соединены молекулярным проводом, (а) Схема и (б) примеры реальных
соединений
752
8. Молекулярные устройства
\
\
\
но2с но2с но2с
\
\
но2с но2с но
Рис. 8.23. Смешанный монослой Ленгмюра-Блоджетт, состоящий из полнена типа
«тяни—толкай» и жирной кислоты
но, что они проявляют НЛО-свойства (табл. 8.1). Эти соединения имеют величины
Р, сравнимые с величиной Р органических соединений того же типа, но обладают
некоторыми особенностями из-за наличия собственных электронных состояний
металла.
Из табл. 8.1 ясно видно, что НЛО-свойства усиливаются даже при небольшом
увеличении длины цепи. Наличие метальных заместителей в циклопентадиениль-
ном кольце также значительно увеличивает как дипольный момент, так и
нелинейность (Р) за счет обогащения электронами донорного компонента. /яраяс-Соедине-
ния более нелинейны, чем их цис-аналоги, что согласуется с большим расстоянием
между двумя заместителями и усилением взаимодействий между ними.
Соединения Ru менее нелинейны, чем их Fe-аналоги, т.е. рутений — более слабый донор.
Это подтверждается более высоким потенциалом окисления рутенийсодержащих
металлоциклов. К сожалению, такие соединения имеют поглощение для состояния
с переносом заряда в видимой области спектра, и это может ограничить их исполь-
8.6. Материалы для нелинейной оптики
753
Таблица 8.1. НЛО-свойства соединений (8.52)*
м
Изомер
, нм
ц, 10-18СГСЭ
Ю-30 СГСЭ
Fe
Fe
Fe
Fe
Ru
Fe
Н
Н
Me
Н
Me
Me
1
1
1
1
1
2
Е-
Z-
Е-
Е-
Е-
Е,Е-
* Измерения проводили при 1.907 мкм
NO2
NO2
NO2
CN
NO2
NO2
356/496
325/480
366/533
324/466
370/424
382/500
4.5
4.0
4.4
4.6
5.1
4.5
31.0
13.0
40.0
10.0
24.0
66.0
зование для генерации второй гармоники в УФ- или видимой области, но их
применение в области телекоммуникаций, где используется излучение с длиной волны
0.8-1.5 мкм, имеет хорошие перспективы.
В интересной работе Б. Коу (В. Сое, 1999) из Университета Манчестера
(Великобритания) описал получение первого переключаемого НЛО-устройства. Коу с
сотрудниками удалось показать, что 11иA1)-комплекс (8.53) имеет очень большой
коэффициент Р, равный 1112-Ю0 СГСЭ, обусловленный интенсивным
низкоэнергетическим MLCT-возбуждением. Более того, величины Р можно настраивать,
манипулируя заместителями в бипиридиниевой части молекулы и заменяя амин-
ные лиганды, например, на пиридилы. Высокая способность этих комплексов к
переключению заключается в том, что НЛО-характеристики этой системы
снижаются примерно в 10—20 раз при окислении металлического центра до Ru(III) водным
подкисленным раствором пероксида водорода. Восстановление гидразином
приводит к регенерации системы.
(8.53)
8.6.3 Материалы для нелинейной оптики
третьего порядка
Факторы, влияющие на получение ГВГ-материалов, понятны
довольно хорошо. В то же время существует мнение, что использование стратегии
сопряженных полиенов для получения материалов, генерирующих третью гармонику,
с достаточно большими коэффициентами у при значительно более низкой цене
невозможно. Один из успешных подходов состоял в использовании сопряженных
754 8. Молекулярные устройства
полиалкинов типа (8.54), где L = Р(«-ВпK, с протяженной делокализацией вдоль
полимерной цепи (S. Guha et al., 1989). Действительный и мнимый компоненты
гиперполяризуемости третьего порядка, у' и у", составили соответственно 1552
и344-10-36СГСЭ.
L
Pt ^= •=.
L
(8.54)
8.7 Дендримеры
Одним из наиболее удивительных классов макромолекулярных
соединений, которай стал недавно актуальным с синтетической, структурной и
функциональной точек зрения, являются дендримеры (от греч. «dendron» — дерево; в
физиологии дендрит, или дендрон, — древоподобная разветвленная нервная клетка).
В идеале дендримеры, или каскадные молекулы, — это монодисперсные
макромолекулы с высокоразветвленной трехмерной структурой. Дендример можно
рассматривать как многокомпонентное соединение, вырастающее из центрального ядра,
подобно дереву. Таким образом, структурно, а часто и синтетически, дендример
берет начало от многофункционального «корня», или ядра. Из каждой
функциональной группы ядра вырастает другая многофункциональная повторяющаяся единица
(зачастую такое же химическое соединение, как само ядро) с образованием олиго-
мера, имеющего центральное ядро и несколько растущих из него функционализи-
рованных «ветвей». Это дендример первой генерации (первого поколения). Если на
функциональных группах первой генерации как на ядрах проводится операция по
выращиванию «ветвей», аналогичная предыдущей, то образуется намного
больший, более сильно разветвленный дендример второй генерации. В принципе этот
итерационный процесс можно повторять бесконечно, что, в конце концов,
приводит к четко определенному трехмерному полимеру, каждая молекула которого по
молекулярной массе идентична всем другим молекулам, если каждая стадия
реакции проходила без ошибок. Этот процесс показан на схеме 8.10.
Дендримеры интересны с различных точек зрения, например для химии
хозяин-гость и катализа, когда область ядра дендримера, которая часто
высокопориста, может выступать в роли хозяина, а его плотноупакованный внешний слой
экранирует внутреннюю область от внешней среды. Благодаря такой структуре
дендримера можно моделировать гидрофобные карманы фрагментов. Внешняя
поверхность дендримера, состоящая из множества идентичных центров связывания
субстратов, потенциально важна с точки зрения фармакологии. Дендримеры могут
также оказаться полезными при создании функциональных материалов и констру-
8.7. Дендримеры
755
Т Ядро
А
\
1
А
Первая генерация
YB
в
\
V
Y
C>VB
с
D
/BYC
с
Вторая генерация
\
\ D D
\ Y
D^v-D I Y
TV VY
YvB
D | /
Третья генерация
C. .D ^y" I '
Y T 1 D
D 1 CVD
DYc>vv^ d
CY°
D
Схема 8.10. Схема процесса постадийного получения дендримеров
756 8. Молекулярные устройства
ировании структурированных молекулярных устройств, используемых, например,
в технологии антенн. К тому же, четко определенная структурная связь между
генерациями, наряду с узким молекулярно-массовым распределением
(монодисперсность), облегчает анализ их свойств по сравнению с обычными полимерными
материалами, даже если дендримеры достигают очень высоких молекулярных масс и
проявляют свойства блочного материала. Детальный обзор химии дендримеров не
входит в рамки этой книги, но интересующийся читатель может обратиться к
ключевым ссылкам этого раздела. Мы же ограничимся кратким рассмотрением
синтеза дендримеров и некоторых интересных областей применения в контексте
молекулярных устройств.
*"* Bosman A. W.f Janssen H.M., Meijer E. W. About dendrimers: Structure, physical
properties and applications // Chem Rev. 1999. Vol. 99. P. 1665-1688.
•"* Fischer M. and Vogue F. Dendrimers: FVom design to application // Angew. Chem.,
Int. Ed. Engl. 1999. Vol. 38. P. 884-905.
8.7.1 Получение и свойства
Основная проблема химии дендримеров - чистота полимеров высоких
генераций, которая во многом определяется способом получения дендримера.
Существуют два главных пути синтеза дендримеров: дивергентный подход (см. схему
8.10), когда генерации наращиваются последовательно, и конвергентный подход,
когда дендример строится от внешней поверхности внутрь, но с последовательным
взаимодействием небольших фрагментов. При дивергентном подходе необходимо
провести множество реакций в одной и той же молекуле. В результате каждая
реакция должна протекать с очень высоким выходом и селективно, если мы хотим
избежать ошибок или дефектов. Например, в синтезе полипропилениминовых
дендримеров (схема 8.11) для получения пятой генерации требуется провести 248
реакций с 64 концевыми аминогруппами. Если каждая реакция селективна на
99.5%, то содержание бездефектного дендримера в смеси продуктов составит
0.995248 = 29%. При падении селективности до 99% мы получим только 8.3%
чистого дендримерного продукта. Возможные побочные реакции, приводящие к обрыву
цепи, показаны на схеме 8.11. В некоторых случаях чистота конечного дендримера
контролируется статистически, поскольку практически невозможно очистить
дендример на каждой стадии его роста. Однако это зависит от характера системы, и
многие исследователи используют традиционные методы очистки, по крайней
мере, для дендримеров начальных генераций.
Исследования дендримеров, особенно определение их чистоты (содержание
бездефектного дендримера), достаточно трудоемки из-за их больших размеров и
высокой симметрии. Для этих целей используют ЯМР, элементный анализ и
хроматографию (например, высокоэффективную жидкостную хроматографию, HPLC),
но этими методами невозможно надежно обнаружить малые количества примесей
в дендримерах высоких генераций. Наилучшим методом, как правило, является
масс-спектрометрия с использованием современных методов ионизации:
ионизации электрораспылением (electrospray ionisation, ESI) и лазерной десорбции из
8.7. Дендримеры
757
NHi
Схема 8.11. Дивергентный синтез полипропилениминовых дендримеров (а и б) и
возможные побочные реакции, приводящие к дефектам и обрыву цепи (в — неправильные
присоединения по Михаэлю, г — циклизация)
матрицы (matrix-assisted laser desorption ionisation, MALDI). Спектры ESI-MS
полипропилениминовых дендримеров пятой генерации показаны на рис. 8.24. На
основании масс-спектров всех предыдущих генераций дендримера с помощью
итерационной процедуры можно рассчитать чистоту полимера для каждой стадии.
Расчетный спектр, хорошо согласующийся с экспериментом, дал полидисперсность
8. Молекулярные устройства
Экспериментальные
Расчетные
5000
5500
6000
6500
7000
7500
mlz
Рис. 8.24. Экспериментальный и расчетный ESI—MS-спектры полипропилениминовых
дендримеров. (Воспроизведено с разрешения Bosman A. W., Jamsen H.M., Meijer E. W. About
dendrimers: Structure, physical properties and applications // Chem. Rev. 1999. Vol. 99. P. 1665-1668)
Mw/Mn = 1.002 (отношение экспериментально найденной молекулярной массы к
теоретической) и чистоту дендримера 23%.
При конвергентном подходе к синтезу дендримеров фрагменты строятся
раздельно и связываются с общим ядром на конечной стадии, минимизируя, таким
образом, число индивидуальных стадий для одной молекулы. При этом многие
трудности, встречающиеся при дивергентной стратегии, преодолеваются. На каждой
стадии реакции используется постоянное и небольшое число реакционных
центров, что минимизирует возможность возникновения дефектов и облегчает очистку,
по крайней мере, полимеров невысоких генераций.
Физические свойства дендримеров чрезвычайно разнообразны и зависят от
природы групп компонентов и взаимодействий между ними. Многие дендримеры
могут частично сворачиваться, что вызывает уменьшение их поверхностной
плотности и числа доступных поверхностных функциональных групп. Однако при
образовании открытой протяженной и пористой структуры существует
необходимость энтропийной компенсации. При высоких молекулярных массах поведение
дендримеров в растворах значительно отличается от поведения линейных
макромолекул. Объем, занимаемый дендримером, растет как функция третьей степени с
каждой генерацией, а его молекулярная масса увеличивается экспоненциально.
Это означает, что, например, вязкость дендримера не увеличивается с ростом
молекулярной массы, а достигает максимума при определенном номере генерации.
Такой параметр, как размер, также сильно зависит от природы растворителя, причем
дендримеры принимают более открытую структуру в растворителях, к которым они
имеют высокое сродство.
8.7. Дендримеры
759
8.7е 2 Химия хозяин-гость для дендримеров
Плотная внешняя оболочка дендримера любого типа приводит к двум
интересным видам поведения хозяин-гость: изоляция центра и включение гостя.
Изоляция центра происходит при дивергентном росте дендримера от
функционального ядра, которое с увеличением числа генераций все в большей степени
становится экранированным от внешней среды. Благодаря этому можно изучать
поведение ядра, не осложненное влиянием сольватации и самоассоциации. Например,
конструирование дендримера, содержащего полиэтиленгликольные «ветви», от
Ре(Ш)-порфиринового ядра дает изолированный от внешней среды
водорастворимый аналог гема (8.55), рис. 8.25 (P. Dandliker et al., 1995). Сравнение
электрохимических свойств дендримеров первой и второй генераций показало, что в случае
более толстого экрана (вторая генерация) для восстановления Ре(Ш)-центра
требуется на 420 мВ более отрицательный потенциал. Аналогично включение ядра
Fe4S4 в тетраэдрическую структуру дендримеров на основе полиароматических
простых эфиров, связанных с атомами Fe тиолатными лигандами (8.56), мешает как
кинетике, так и термодинамике восстановления этого кластера. Кластер
показывает более отрицательный потенциал восстановления и увеличение числа волн на
циклической вольтамперограмме для каждой последующей генерации дендримера.
Рис. 8.25. Экранирование железопорфиринового ядра дендримером второй генерации
4%6О ^- Молекулярные устройства
Den
Fe
Fe-
S S F4
(8.56)
В дополнение к химии ковалентного включения ядра низкая плотность вблизи
него, характерная для дендримеров, может приводить к включению в эту область
молекулы гостя с последующей его изоляцией от внешней среды. Это явление
называют топологическим захватом. Капсулирование гостя необратимо, если
внешний слой дендримера функционализирован объемистыми группами, что делает
этот слой жестким и непроницаемым. Превосходный пример такого подхода -
реакция полипропилениминового дендримера четвертой генерации
DAB-Den-(NH2N4 (где DAB - 1,4-диаминобутил) с активированным сложным
эфиром N—/-BOC—L-Phe (хиральная аминокислота L-фенилаланин, защищенная
mpem-бутильной сложноэфирной группой) в присутствии молекул-гостей, таких,
как флуоресцентный краситель бенгальский розовый, имеющий некоторое
сродство к третичным аминогруппам внутри дендримера. Это приводит к получению
дендримера DAB—Den—(NH—/-BOC—L-Phe)M с жесткой оболочкой, содержащего от
одной до четырех молекул бенгальского розового. Капсулированный краситель
показывает сильную флуоресценцию при 600 нм, в то время как для свободных
молекул она тушится при этой длине волны. Примечательно, что такой дендример
также индуцирует хиральность в ахиральном красителе. В случае одной захваченной
молекулы спектр кругового дихроизма (CD) напоминает спектр в УФ- и видимой
области. При захвате четырех молекул спектр CD соответствует четырем близко
расположенным друг к другу хромофорам с определенной ориентацией. Ясно, что,
в то время как дендримерные капсулы сами по себе ахиральны, полости вблизи
ядра обладают хиральностью (J. Jansen et al., 1995).
8.7.3 Фотохимические устройства
на основе дендримеров
Используя подход «металлокомплексы в качестве лигандов» (разд.
8.2.4), можно достаточно легко собрать полиметаллические катионные дендриме-
ры. Действительно, 22-ядерный комплекс (8.7) является примером дендримера
третьей генерации этого типа. Такие соединения могут служить превосходными
прототипами супрамолекулярных антенных устройств, способных аккумулировать свет
по механизму переноса энергии, аналогично биохимическим фотосинтетическим
мембранам (рис. 8.26).
8.7. Дендримеры
761
Свет
Поглощение
О О О О О О
о о о о о
Y/V/W
Перенос
энергии
Рис. 8.26. Схематическое представление
супрамолекулярногосветоаккумулирующего
антенного устройства
В большинстве случаев дендримерные многоцентровые металлокомплексы
сконструированы на основе Ru(II), Os(II), Re(I) и других металлических центров с
использованием мостиковых лигандов типа 2,3-dpp (8.57) (как в (8.7)) и 2,5-dpp (8.58),
а также концевых лигандов типа Ыру(8.1) и biq B,2'-бихинолин (8.59)), схема 8.12.
N
2,3-dpp (8.57)
2,5-dpp (8.58)
biq (8.59)
1762
8. Молекулярные устройства
Снятие защиты
Завершение
(bipy.biq)
Рост
С12
Завершение
(bipy.biq)
«д»
Снятие защиты
$= 2,3-dpp или 2,5-dpp
ф= Защитная группа
Завершение
ФЛ£* (bipy.biq) I
Получение истинных
дендримеров и комплексов,
содержащих различные
металлы или различные
лиганды
Схема 8.12. Конструирование дендримеров с использованием подхода «металло-
комплексы в качестве лигандов»
.7. Дендримеры
763
Ru(bipyJ2+
Os(bipyJ2+
Ru(biqJ2+
Рис. 8.27. Процессы переноса энергии в тетраядерных (первая генерация) комплексах
Энергию низшего MLCT-состояния в каждом металлическом фрагменте
(компонент) можно легко предсказать, причем она систематически изменяется в
соответствии с характеристиками металла и лигандов. Поэтому можно сконструировать
каскадные дендримеры с переносом электрона или энергии, систематически
варьируя энергию поглощения компонентов от одной генерации к следующей.
Компоненты с наиболее низкой энергией MLCT (например, Os(|i-2,5-dppK+) образуют
ядро дендримера, а компоненты с наиболее высокой энергией поглощения
ограничивают поверхность дендримера (например, Ru(biqJ((i-2,3-dppJ+), обуславливая
контролируемый поток энергии. Точно так же соответствующая организация
фрагментов обеспечивает почти любую комбинацию направлений переноса энергии.
Для тетраядерных комплексов реализованы все способы переноса энергии,
показанные на рис. 8.27. Аналогичные свойства имеют системы с шестью и десятью
компонентами.
При распространении этих идей на очень большие дендримерные системы,
чтобы получить плавно изменяющийся каскад с переносом энергии, вероятно,
необходим более широкий набор ионов металлов и лигандов. Можно использовать
такие ионы металлов, как Rh(III), Ir(III), Pd(II) Pt(II) и Pt(IV), хотя лиганды
пиридильного типа в этих случаях менее пригодны из-за неподходящей
конфигурации их энергетических уровней. В таких случаях более полезными могут оказаться
комбинации пиридильных и Ы-С~-карбанионных лигандов, подобных депротони-
рованному 2-фенилпиридину.
764
]. Молекулярные устройства
Учебные задания
8.1. Циклодекстриновый хозяин (HI, концентрация 10~2 мольдм) связывает два
гостя (А и В; 10~4 моль-дм~3) с небольшими значениями свободной энергии
связывания (AGA = 10 кДжмоль, ДGВ = 12 кДж-моль). Вычислите
процентное содержание гостей А и В, образовавших комплекс с этим хозяином при
равновесии. Выразите Ваш результат в виде фактора селективности
(связанные [В]/[А]). Второй хозяин (Н2) в тех же условиях дает значительно более
высокие значения свободной энергии связывания (Дбд = 30 кДж-моль,
АСВ = 32 кДжмоль). Вычислите концентрации связанных гостей и фактор
селективности. Полагая, что связывание можно рассматривать как часть
аналитического определения концентраций А и В, выясните, какой хозяин лучше
всего различает эти два аналита. Какие недостатки имеет этот хозяин при
таком аналитическом применении?
8.2. Дендример получен из тетрафункционального ядра, которое можно
рассматривать как сферу с радиусом 5 А. С каждой последующей генерацией
дендримера каждая функциональная группа добавляет еще 5 А к радиусу сферы и
предоставляет три новых связывающих центра для последующего роста.
Предполагая, что весь дендример имеет сферическую форму с максимальной
протяженностью, вычислите объем пустого пространства внутри дендримера для
каждой генерации и предскажите, сколько генераций можно добавить,
прежде чем дендример свернется. Как изменится эта ситуация, если радиус
строительного блока дендримера составит 4 А? (Объем сферы равен 4/з nf3-)
8.3. Тот же дендример образуется с выходом 98.5% для каждой отдельной стадии
(прибавление каждой сферической генерации). Каков будет выход чистого
дендримера каждой генерации? Можно ли таким способом сконструировать
дендример с числом генераций, достаточным для полной изоляции ядра
дендримера от внешней среды?
8.4. Используя строительные блоки, показанные ниже, а также подход «металло-
комплексы в качестве лигандов», разработайте стратегию получения (а) тетра-
ядерного Ru-комплекса и (б) комплекса, содержащего шесть ионов Os2+ и
четыре иона Ru2+. Если каждый металлический центр существует в двух
энантиомерных формах, сколько диастереомеров будет иметь каждое
соединение?
[RuCl2(bipyJ]
[OsCl2(bipyJ]
[RuCl2(MeCNL]
Литература
765
8.5. Светоаккумулирующее устройство поглощает УФ-излучение при 200 нм с
коэффициентом поглощения (е) 15 000 дм3-моль~'-см~'. Образец с
концентрацией 210~~5 моль-дм в оптической ячейке длиной 1 см облучали 15 мин
излучением с длиной волны 200 нм интенсивностью /0 = 4-Ю14 кванте. После
этого устройство излучает свет в видимой области при 600 нм. Рассчитайте
количество квантов, поглощенных при 200 нм в ходе эксперимента, и
количество квантов, излученных при 600 нм, в предположении, что эффективность
люминесценции составляет 100%. (Указание: следует использовать закон
Ламберта-Бера и определение спектральной поглощательной способности
(оптическая плотность).)
Литература
Arrhenius, 1986 Arrhenius T.S., Blanchard-Desce M., Dvolaitzky M. et al. Molecular devices —
caroviologens as an approach to molecular wires synthesis and incorporation into
vesicle membranes // Proc. Natl. Acad. Sci. USA. 1986. Vol. 83. P. 5355.
Beer, 1995 BeerP.D.,SzemesF. Remarkable chloride over dihydrogen phosphate anion
selectivity exhibited by novel macrocyclic bis[ruthenium(Il) bipyridyl] and rutheni-
um(II) bipyridyl-metallocene receptors // J. Chem. Soc, Chem. Commun. 1995.
P. 2245.
Beer, 1996 Beer P.D., Gale P.A., Chen Z. New bis- and гш-ferrocenoyl and rr/s-benzoyl lower-
rim substituted calix[5]arene esters: Synthesis, electrochemistry and X-ray crystal
structures // Supramol. Chem. 1996. Vol. 7. P. 241.
Carr, 1997 CarrJ.D., Lambert L., Hibbs D.E. et al. Novel electrochemical sensors for neutral
molecules// Chem. Commun., 1997. P. 1649.
Cheng, 1991 Cheng L.-T., Tarn W., Stevenson S.H. et al. Experimental investigations of
organic molecular nonlinear optical polarizabilities. 1. Methods and results on benzene
and stilbene derivatives // J. Phys. Chem. 1991. Vol. 95. P. 10631.
Cobben, 1992 Cobben P.L.H.M., Egberink R.J.M., Bomer J.G. et al. Transduction of selective
recognition of heavy-metal ions by chemically modified field-effect transistors
(chemfets) // J. Amer. Chem. Soc. 1992. Vol. 114. P. 10573.
Сое, 1999 Сое B.J. Molecular materials possessing switchable quadratic nonlinear optical
properties // Chem. Eur. J. 1999. Vol. 5. P. 2464.
Cusak, 1997 Cusak L., Nagaraja Rao S., Fitzmaurice D. Heterosupramolecular chemistry: Self-
assembly of an electron donor (TiO2 nanocrystallite)-acceptor (Viologen)
complex // Chem. Eur. J. 1997. Vol. 3. P. 202.
Dandliker, 1995 Dandliker P.J., Diederich F., Gisselbrecht J.-P. et al. Water-soluble dendritic iron
porphyrins: Synthetic models of globular heme proteins // Angew. Chem., Int.
Ed. Engl. 1995. Vol. 34. P. 2725.
DeCola, 1993 DeCola L, Balzani V., Barigelletti F. et al. Photoinduced energy and electron-
transfer processes in supramolecular species — tris(bipyridine) complexes of
Ru(II)/Os(II), Ru(II)/Ru(III), Os(II)/Os(IlI), and Ru(II)/Os(III) separated by
a rigid spacer // Inorg. Chem. 1993. Vol. 32. P. 5228.
76(j 8. Молекулярные устройства
Fages, 1993 Fages F., Desvergne J.-R, Kampke K. et al. Linear molecular recognition — spec-
troscopic, photophysical, and complexation studies on alpha, omega-alkanediyl-
diammonium ions binding to a bisanthracenyl macrotricyclic receptor // J. Amer.
Chem. Soc. 1993. Vol. 115. P. 3658.
Fages, 1999 Fox M.A. Fundamentals in the design of molecular electronic devices: Long-range
charge carrier transport and electronic coupling // Ace. Chem. Res. 1999. Vol. 32.
P. 201.
Goulle, 1993 Goulle V., Harriman A., Lehn J.-M. An electro-photoswitch - redox switching of
the luminescence of a bipyridine metal-complex // J. Chem. Soc, Chem.
Commun. 1993. P. 1034.
Guha, 1989 Guha S., Frazier C.C., Kang K., Finberg S.E. Measurement of the 3rd-order hyper-
polarizability of platinum polyynes // Optics Lett. 1989. Vol. 14. P. 952.
Huston, 1988 Huston M.H., Haider K.W., Czarnik A.W. Chelation-enhanced fluorescence in
9,10-6w(tmeda)anthracene // J. Amer. Chem. Soc. 1988. Vol. 110. P. 4460.
Jansen, 1995 Jansen J.F.G.A., de Brabander-van den Berg E.M.M., Meijer E. W. Induced chiral-
ity of guest molecules encapsulated into a dendritic box // Rec. Trav. chim. Pays-
Bas. 1995. Vol. 114. P. 225.
Kawai, 1994 Kawai S.H., Gilat S.L., Lehn J.-M. Dural-mode optical-electrical molecular
switching device//J. Chem. Soc, Chem. Commun. 1994. P. 1011.
Koike, 1996 Koike Т., Watanabe Т., Aoki S. et al. A novel biomimetic zinc(II)-fluorophore,
dansylamidoethyl-pendant macrocyclic tetraamine 1,4,7,10-tetraazacyclodode-
cane (cyclen) // J. Amer. Chem. Soc 1996. Vol. 118. P. 12696.
Molnar, 1994 Molnar S.M., Nallas G., Bridgewater J.S., Brewer K.J. Photoinitiated electron
collection in a mixed-metal trimetalHc complex of the form
([(bpyJRu(bpb)]2IrCl2)(PF6M (bpy = 2,2'-bipyridine and bpb = 2,3-bisB-
pyridyl)benzoquinoxaline) //J. Amer. Chem. Soc. 1994. Vol. 116. P. 5206.
Rebek, 1984 Rebek J., Jr. Binding forces, equilibria, and rates — new models for enzymic
catalysis // Ace. Chem. Res. 1984. Vol. 17. P. 258.
Tecilla, 1990 Tecilla P., Dixon R. P., Slobodkin G. et al. Hydrogen-bonding self-assembly of mul-
tichromophore structures // J. Amer. Chem. Soc. 1990. Vol. 112. P. 9408.
9
Биомиметика
Правила и модели разрушают гения и мастерство.
В. Хазлит A778-1830). Круглый стол. I. О вкусе
Введение
•""» Murakami К, Kikuchi J.-L, Hisaeda К, Hayashida О. Artificial enzymes // Chem.
Rev. 1996. Vol. 96. P. 721-758.
9.I.I Супрамолекулярная биохимия
Биологические системы сыграли огромную роль в развитии супрамо-
лекулярной химии. Множество синтетических супрамолекулярных систем было
создано в рамках биомиметического подхода, т.е. путем подражания (mimic)
структуре или функции более сложных биологических объектов*. Такие искусственные
абиотические (небиологические) молекулы или реакции будем называть моделями.
Под этим мы подразумеваем, что небольшие искусственные системы могут
обладать (частично или во всем объеме) свойствами реальных биологических систем.
Концепция биологической модели прекрасно изложена Д. Крамом в его
нобелевской лекции:
«Мало найдется ученых, знакомых с химией биологических систем на молекулярном
уровне, которых не вдохновил бы этот подход. Эволюция создавала химические
соединения, исключительная организация которых обеспечивала выполнение наиболее
сложных и тонких задач. Многие химики-органики, рассматривая кристаллические
структуры ферментативных систем или нуклеиновых кислот и проникая в чудеса
специфичности иммунной системы, не могут не мечтать о разработке и синтезе более
простых органических объектов, имитирующих работу этих природных соединений».
Cram D.J. The design of molecular hosts, guests, and their complexes (Nobel Lecture) //
Angew. Chem., Int. Ed. Engl. 1988. Vol. 27. P. 1009-1020.
Началом супрамолекулярной химии можно считать модель Фишера «ключ—замок»,
используемую в ферментативном катализе (см. разд. 1.3), которая увидела свет
задолго до появления сложных хозяев, таких, как карцеранды, криптанды или
самособирающиеся устройства и системы. Эта концепция соответствия формы субстра-
* Биомиметика (бионика, биогенез) — современное научное направление по
заимствованию у природы ценных идей и реализации их в виде оригинальных материалов, процессов и
технологий, имитирующих природные аналоги. (Примеч. ред. перевода.)
768
Биомиметика
та (гость) в сочетании с работами П. Эрлиха по антителам дала нам идею
рецептора (хозяин). Связывание рецептор-субстрат, часто чрезвычайно селективное,
имеет в биохимии большое значение. Классический пример — это связывание бомби-
кола рецепторным центром самца тутового шелкопряда Bombyx mori. Бомбикол —
половой аттрактант, выделяемый самкой шелкопряда. Самец возбуждается, если в
воздухе содержится 4.2-10~18 М бомбикола. Следовые феромоны также «работают»
в поразительно малых количествах. Одного миллиграмма следового феромона
муравья-листореза Atta texana достаточно для того, чтобы колонна муравьев 3 раза
обогнула земной шар.
Связывание рецептор-субстрат обратимо и контролируется термодинамически.
Как обсуждалось в разд. 1.6, селективность связывания количественно выражается
отношением констант связывания рецептора с конкурирующими субстратами.
Однако в живых системах, как ни в каких других объектах супрамолекулярной химии,
акт связывания — это лишь часть сложного процесса. Связывание биохимических
сигнальных соединений, например гормонов или нейромедиаторов, должно быть
прелюдией таких событий, как реакции дефосфорилирования, нервные импульсы
(деполяризация мембраны), сокращение мышцы и т.д. В отсутствие
функционирующей системы событие связывания, по существу, бесполезно. В значительной
степени связывание субстрата часто нужно для индуцирования конформационного
изменения рецептора, которое включает биохимический процесс. Фактически
энергия связывания субстрата используется рецептором для достижения
конформационного изменения, необходимого при запуске процесса, которое он один
осуществить не может. Такое поведение описывается моделью индуцированного
соответствия, а не моделью жесткого взаимодействия «замок—ключ» (рис. 9.1).
В процессах с участием ферментов, являющихся биологическими
катализаторами химических реакций, функционирующая система требует от фермента
кинетической селективности, когда адаптация фермента в системе обеспечивает ее
стабилизацию на каждой стадии вдоль реакционного пути. Достижение «живой
селективности» такого типа — более важная задача, чем просто влияние фермента
на равновесие, и, справедливости ради, следует сказать, что исследование реально
функционирующих моделей ферментов все еще находится в зачаточном состоянии.
Тем не менее о свойствах и функционировании биологических систем можно
многое узнать с помощью моделей, успешно имитирующих ту или иную часть всей
системы. Более того, некоторые абиотические функциональные системы,
действительно, существуют, например, среди рецепторов, подобных циклодекстринам,
которые способны осуществлять ферментативный катализ.
9*1.2 Характеристики биологических моделей
Биологические системы, в частности ферменты, обладают рядом
ценных свойств, которые необходимо воссоздать в модельной системе:
1. Ферменты обеспечивают высокие скорости реакций в мягких условиях.
2. Ферменты обладают высокой избирательностью структурного и хирального
распознавания субстратов.
769
Фсрмен!
t'yficrpar
Комплекс формет • cy
Рис. 9.1. Модели жесткого взаимодействия «замок—ключ» (а) и индуцированного
соответствия (б) при связывании субстрата ферментом
3. Каждая молекула фермента катализирует реакцию громадного числа молекул
субстрата и при этом не разрушается (большое число циклов).
4. Действие ферментов может быть блокировано соединениями-ингибиторами,
которые связываются с ферментом, но сами не участвуют в реакциях.
Желательно, чтобы модель помимо этих свойств обладала также и некоторыми
полезными свойствами, не характерными для биологических ферментов, такими, как
молекулярные массы, меньшие, чем 2000—3000 Да, небольшое число элементов хи-
ральности (чтобы минимизировать увеличение числа диастереомеров),
стабильность, позволяющая работать даже при высоких температурах, и растворимость в
большом числе растворителей. При попытках смоделировать биологическую
молекулу трудно ожидать, что полученная абиотическая система (по божественному
вдохновению или слепому провидению) в состоянии имитировать одновременно
все структурные, связывающие и каталитические особенности естественного
процесса. Природа затратила миллионы лет на разработку биологических процессов
19-СтидДж. В
9. Биомиметика
высокой сложности, в то время как технический прогресс в этой области и
понимание таких процессов пока находятся на не сравнимо более низком уровне. Поэтому
для достижения конечной цели нужно использовать несколько стратегий, среди
которых выделим две главные:
1. Система, эффективно связывающая субстрат или его упрощенный аналог, или,
что более правильно, аналог переходного состояния биологической реакции. Ее
можно назвать структурной моделью.
2. Система (обычно межмолекулярная), в которой для осуществления реакции не
требуется связывания субстрата. Ее можно назвать функциональной моделью.
Оба эти подхода дают простейшие модельные системы, которые, если они вообще
способны осуществлять желаемую биологическую реакцию, делают это
значительно медленнее и с меньшей селективностью, чем природный фермент и субстрат. По
мере усложнения такой модельной системы желательно обеспечить выполнение
ряда базисных требований, отмеченных Г. Дюга (Н. Dugas, 1989):
1. Модель должна иметь гидрофобный центр для связывания субстрата.
2. Модель должна обеспечивать центры водородного и/или электростатического
связывания, которые комплементарны таковым в субстрате.
3. Модель должна содержать подходящие каталитические группы.
4. Структура модели должна быть строго определенной и жесткой.
5. Модель должна быть водорастворимой и каталитически активной при
физиологических условиях.
*"» Kirby AJ. Enzyme mimics // Angew. Chem., Int. Ed. Engl. 1994. Vol. 33.
P. 551-553.
Характеристики ферментов
Kim D.H. Supramolecular aspects of enzymes // Comprehensive supramolecular
chemistry / Ed. J.L.Atwood, J.E.D.Davies, D.D.MacNikol, F.Vogtle. Oxford:
Pergamon, 1996. Vol. 4. P. 503-526.
9*2Л Определение и структура
Ферменты — это макромолекулы с молекулярной массой более 10 000 Да.
Они состоят из полипептидных цепей, которые, в свою очередь, являются
полимерами аминокислот белка (см. Дополнение 2.2). Полипептидные цепи
сворачиваются с образованием глобулярной структуры, содержащей щели и карманы на своей
поверхности. Связывание субстрата при ферментативном катализе происходит в
одном из этих карманов, называемом активным центром. Первоначальное
связывание — термодинамически контролируемый равновесный процесс, который
высокоселективен и обычно протекает по механизму индуцированного соответствия. Свя-
9.2. Характеристики ферментов j|7 71
зывание происходит посредством трехмерного контакта между ферментом и
субстратом и включает гидрофобные взаимодействия, водородные связи, солевые
мостики (ион—ион) и другие формы межмолекулярного взаимодействия. Связанные
субстраты претерпевают химические превращения иногда при скоростях,
приближающихся к скоростям диффузии, ~108 с"''М"' (реакция протекает настолько
быстро, насколько субстрат достигает фермента в соответствии с градиентом
локальной концентрации и термической флуктуацией). Суммарный процесс можно
представить уравнением (9.1), где Е, S и Р - фермент, субстрат и продукт
соответственно. Отметим, что фермент регенерируется после завершения реакции, как это
происходит в любом каталитическом процессе.
(9.1)
(9.2)
Специфичность и селективность данного фермента для конкурирующих
субстратов зависят и от констант скорости к{ и к \ для каждого субстрата (т.е. от
константы равновесия), и от ксаГ Обычно эта связь выражается через константу
специфичности кса/Ки. Таким образом, наиболее специфичные ферменты осуществляют
катализ быстро и не нуждаются в особенно сильном связывании.
Структуру фермента можно подразделить на первичную, вторичную и
третичную. Первичная структура — это последовательность аминокислотных остатков в
полипептидной цепи, которая задается при его синтезе. Вторичная структура
определяется упорядочением цепей в дискретные единицы или сегменты, такие, как
а-спирали и C-слои (рис. 9.2), а третичная структура — это глобулярный белок,
состоящий из вторичных структур. В формировании третичной структуры участвуют
водородные связи, стэкинг- и гидрофобные взаимодействия (ср. мицеллы,
разд. 10.2), а также часто — молекулы воды, которые глубоко погружены в фермент,
где они заполняют небольшие полости и действуют как «клей», связывающий
вторичные структуры. Именно третичная структура фермента ответственна за
организацию центра связывания: это тоже наиболее гибкий компонент структуры
фермента, позволяющий центру связывания деформироваться в ответ на связывание
гостя. Тем не менее третичная структура фермента строго определена, и ферменты
упорядочены столь высоко, что они могут кристаллизоваться.
Способ, в соответствии с которым происходит кодирование третичной
структуры для данной первичной аминокислотной последовательности, очень сложен и до
сих пор представляет неразгаданную тайну. Это один из вызовов природы,
адресованных специалистам по супрамолекулярной химии. Гидрофобные эффекты,
обуславливающие глобулярную форму белка, очень важны наряду со стерическими
эффектами, которые, возможно, в основном ответственны за такую организацию.
Тогда специфические направленные водородные связи определяют, какая из
небольшого числа глобулярных конформаций наиболее предпочтительна.
9. Биомиме тика
(в)
{б)
н R
Рис. 9.2. Варианты вторичной структуры фермента: (а) а-спираль, (б) антипараллельный
Р-складчатый слой, (в) Р-изгиб, (г) беспорядочная цепь
9« 2.2 Механизм ферментативного катализа
Ключ к пониманию того, каким образом ферменты способны
обеспечивать столь высокие скорости катализируемых ими реакций, можно найти у
Л. Полинга (L. Pauling), который в 1948 г. установил, что ферменты - это
катализаторы, активные центры которых комплементарны по структуре переходному
состоянию субстрата в катализируемых ими реакциях.
9.2. Характеристики ферментов
773
Рис. 9.3. Энергетические
профили реакций для катализируемых
( ) и некатализируемых реакций
Координата реакции
Это простое, но глубокое утверждение, относящееся к общей теории
переходного состояния в химических реакциях, иллюстрирует энергетический профиль
реакции, показанный на рис. 9.3. Работа фермента заключается в снижении энергии
активированного комплекса фермент—субстрат [ES]* и, следовательно, энергии
активации &G*cat по сравнению с энергией активированного субстрата [S*] в нека-
тализируемой реакции — kG*uncar Следовательно, фермент должен быть более
комплементарным к активированному субстрату, чем к субстрату в основном
состоянии. Действительно, нековалентные взаимодействия, участвующие в связывании
субстрата, должны быть достаточными, чтобы изменить его для прохождения
процесса слева направо вдоль координаты реакции. Другими словами, субстрат должен
соответствовать переходному состоянию, что уменьшает энергию активации,
необходимую для образования [ES]*. Это еще одно проявление модели
индуцированного соответствия. Теперь не только фермент, но и субстрат подвергаются конформа-
ционным изменениям (рис. 9.4). Такое конформационное изменение должно
вызвать деформацию разрушаемой связи (одной из подлежащих разрушению при
катализируемой реакции). Например, переходное состояние в реакции дезамини-
рования аденозинтрифосфата стабилизировано на 73.2 кДжмоль при 310 К.
Не только стабилизация переходного состояния вносит вклад в ускорение
ферментативных реакций. В общем, в неферментативной химии внутримолекулярные
реакции (например, циклизация) протекают намного быстрее, чем их
бимолекулярные аналоги, из-за статистических эффектов. Например, внутримолекулярная
реакция фенилсукцината происходит в 105 раз быстрее, нежели бимолекулярная
реакция ацетата с фенилацетатом при концентрации всех реагентов 1 М (схема 9.1).
Отношение kl/k2 называют эффективной молярностью, и ее величина 105 М,
действительно, намного больше, чем реальная концентрация (молярность чистой
уксусной кислоты составляет ~17 М), даже если она представляет собой
концентрацию уксусной кислоты, необходимую для достижения той же скорости в эквива-
774
9. Биомиметика
11пдуамровамвс
соотве гстииг
Фермойг
Субстрат
ко мнлскс фс рмс е г-с\ Cvtj >л г
Рис. 9.4. Модель индуцированного соответствия: фермент и субстрат приспосабливаются друг
к другу. Этот процесс включает достижение соответствия субстрата переходному состоянию
Схема. 9.1. Внутримолекулярные реакции (а) идут намного быстрее, чем их
бимолекулярные аналоги (б)
9.3. Циклодекстрины как имитаторы ферментов %75
лентной внутримолекулярной реакции. В сравнении с бимолекулярными
мономолекулярные процессы заканчиваются только потерей энтропии внутреннего
вращения и, таким образом, более благоприятны с точки зрения энтропийного вклада
на 54-59 кДжмоль. В ферментативных реакциях, если связь субстрата с
ферментом обладает достаточной прочностью, то комплекс ES действует как единая
молекула, и поэтому наблюдается увеличение скорости такого внутримолекулярного
процесса в 109. Фактически затраты энтропии на сближение двух реагентов
окупаются связыванием субстрата, а не реакцией.
Наконец важным фактором ускорения ферментативной реакции является также
десольватация связанного субстрата, что опять-таки окупается энергетическим
выигрышем при комплексообразовании. Поверхностные взаимодействия между
функциональными группами фермента и субстрата эффективно вытесняют
молекулы воды из сольватного окружения субстрата.
Несомненно, ферменты эволюционировали, приспосабливаясь к
высокоселективному решению этих специфических задач, и каждое семейство, несмотря на
общие аминокислотные строительные блоки, сильно отличается от других, так как
выполняет свои конкретные задачи. Общие черты всех ферментов - большие
размеры и сложность молекул. Уже эти факторы сильно затрудняют возможность
досконального понимания функции ферментов на модельных системах, поскольку
низкомолекулярные модели просто не могут приблизиться к сложности реальных
объектов. Итак, большой размер молекул, действительно, необходим или это
эволюционная случайность? По-видимому, такой размер ферменту требуется для того,
чтобы его свернутая структура могла обеспечить необходимую геометрию
связывающего центра и ориентацию в нем функциональных групп. Более того,
связывающий центр должен обладать некоторой контролируемой и четко определенной
гибкостью, чтобы обеспечить свободу движений субстрата в процессе его химического
превращения. Это одна из причин, объясняющих необходимость наличия у
фермента податливой третичной структуры белка. Возможно, также имеет значение
большое число функциональных групп на поверхности фермента, удаленных от
связывающего центра. Они могут служить для улавливания потенциальных
субстратов и увеличивать скорость их диффузии по направлению к активному центру.
Часто самые большие ферменты являются и самыми специфичными по
отношению к другим полипептидам в качестве их субстратов. Подсчитано, что для таких
взаимодействий площадь поверхности контакта должна составлять, по крайней
мере, 10 нм2. Видимо, небольшие белки с молекулярными массами меньше 10 000 Да
и общей площадью поверхности меньше 15 нм2 просто не в состоянии обеспечить
подходящую площадь контакта.
9*3 Циклодекстрины
как имитаторы ферментов
*""* Breslow R. and Dong S.D. Biomimetic reactions catalysed by cyclodextrins and their
derivatives // Chem.Rev. 1999. Vol. 98. P. 1997-2011.
776 9. Биомиметика
9* 3 • * Моделирование ферментов
с использованием циклодекстринов как хозяев
Из всех соединений, содержащих полости, возможно, именно цикло-
декстрины лучше всего подходят для конструирования небольших имитаторов
молекул ферментов. Читатели, незнакомые с химией циклодекстринов, могут
обратиться к разд. 5.3.2, в котором обсуждаются синтез и строение этих важных хозяев.
Общие критерии эффективности систем, способных имитировать структуру и/или
функцию биологических ферментов, приведены в разд. 9.1.2. В широком смысле
эти критерии состоят в том, чтобы система связывала свой субстрат (гость) быстро,
селективно и обратимо, обычно нековалентно, и катализировала химические
реакции этого субстрата. Цели такого химического подхода — лучшее понимание того,
как «работают» природные ферменты, развитие области промышленных
катализаторов, способных осуществлять неизвестные до сих пор превращения с высокой
эффективностью и селективностью, а также искусственное регулирование
биохимических реакций. В рамках указанных соображений весьма доступные циклодек-
стрины обладают рядом особенностей, которые делают их привлекательными в
качестве потенциальных имитаторов и моделей ферментов. Эти особенности, в
частности:
• растворимость в воде при физиологических значениях рН. Вода - это среда, в
которой действует большинство ферментов;
• Быстрое обратимое нековалентное связывание гостей и их медленное отделение
от реакционного центра, как в ферментах;
• четко определенные структуры и способы связывания гостей;
• широкий набор доступных производных;
• наличие в молекуле гидроксильных групп, которые могут принимать активное
участие в катализе, а также гидрофобной полости для связывания гостей;
• хиральность молекулярной полости, обеспечивающая энантиоселективность
связывания и катализа;
• реальная каталитическая активность.
Большой интерес среди нативных нефункционализированных циклодекстринов
вызывает анион (9.1), в котором одна из ОН-групп циклодекстрина депротонирова-
на, т.е. в этом циклодекстрине нуклеофильная функциональная группа находится в
непосредственной близости к гидрофобной связывающей полости. Было показано,
что сложные эфиры при связывании внутри циклодекстриновой полости могут аце-
тилировать анионный C-циклодекстрин (схема 9.2). Оказалось, что скорость этой
реакции примерно в 100 раз превосходит скорость аналогичного гидролиза ти-нитро-
фенилацетата в отсутствие циклодекстрина. С одной стороны, разумеется, реакция
ацетилирования — не каталитическая, а увеличение ее скорости — очень скромное
по стандартам ферментативных реакций. С другой стороны, молекулярная масса
циклодекстрина — меньше 2000 Да, т.е. его молекула очень «маленькая», и стадии
катализа можно было бы легко изучить. Более того, эта реакция близко соотносится с
действием серинпротеазных ферментов, когда ацильная группа переносится к сери-
новому гидроксилу на первой стадии ферментативной реакции.
9.3. Циклодекстрины как имитаторы ферментов
111
0"
1
с
—■
с
(9.1)
Me
I и
о
Me
п
Схема 9.2. Ацетилирование аниона циклодекстрина связанным субстратом (л*-нитрофе-
нилацетат)
При изучении молекулярных моделей стало понятно, что в ходе реакции ацили-
рования связанный субстрат частично выходит из полости циклодекстрина,
образуя тетраэдрический интермедиат реакции. Другими словами, в этой модели
фермента переходное состояние не обладает нужной селективностью. Кроме того,
чрезмерно жесткие субстраты испытывают затруднения при вращении в связанном
состоянии, необходимом для соответствующего ориентирования относительно ги-
дроксильной группы циклодекстрина. Плоскость эфирного фрагмента субстрата
должна быть перпендикулярна связи О—Н. Кроме того, образовавшийся фрагмент
должен обладать свободой вращения, чтобы освободить полость для связывания
следующей молекулы субстрата (схема 9.3). Эти проблемы выявились при изучении
субстратов типа я-нитропроизводных, в которых сложноэфирная группа еще
больше выступает из полости, а также субстратов с большой вращательной гибкостью, в
частности алкина (9.2). В таких случаях наблюдалось намного большее ускорение
модельных реакций, например до 5 900 000 раз для соединения (9.3).
(9.2)
(9.3)
778
9. Биомиметика
Схема 9.3. Тетраэдрический интермедиат и его вращение
2 Циклодекстрины как имитаторы эстераз
Уже давно известно, что циклодекстрины и их производные способны
катализировать огромное число биохимических и небиохимических превращений.
Основой катализа нативными (немодифицированными) циклодекстринами
является расположение реакционноспособных вторичных гидроксильных групп
непосредственно у входа в молекулярную полость. Одна из наиболее эффективных
реакций, катализируемых циклодекстринами, - гидролиз арильных сложных эфиров
и фосфатов (эстеразная активность). Например, в присутствии C-циклодекстрина
скорость гидролиза я-нитрофенильных сложных эфиров увеличивается до 750 000
раз. Механизм катализа с помощью этого циклодекстрина показан на схеме 9.4.
Поразительное ускорение отмечено также в реакциях образования и гидролиза
органических фосфатов, катализируемых циклодекстринами в сочетании с ионами
лантаноидов. Циклический 3',5'-аденозинмонофосфат (сАМР (9.4)) — важный
вторичный мессенджер межклеточных контактов в биологических системах. Когда
клетка получает стимул (первичный мессенджер*), из аденозинтрифосфата (АТР,
* Первичный мессенджер, как правило, — это гормон, связывание которого с рецептором
клетки запускает в ней химические процессы, активируя вторичный мессенджер. (Прим. ред.
перевода.)
9.3. Циклодекстрины как имитаторы ферментов
Схема 9.4. Механизм гидролиза сложных эфиров в присутствии Р-циклодекстрина
по Ф. Дидериху (Diederich F., 1983)
основная молекула для запасания энергии и транспорта) образуется соединение
(9.4), причем эта реакция катализируется ферментом аденилатциклазой. В свою
очередь, сАМР активирует другие внутриклеточные ферменты, формируя ответ
клетки, который завершается гидролизом сАМР фосфодиэстеразой (фермент
гидролиза фосфатов), схема 9.5. Действие аденилатциклазы успешно имитировано
комбинацией C-CD и празеодима(Ш) (другие ионы лантаноидов также отчасти
эффективны). Это первая искусственная система, способная катализировать
превращение АТР в AMP в физиологических условиях. Предыдущие попытки обычно
состояли в гидролизе АТР с образованием его аналогов — ациклических моно- и
дифосфатов. Расщепление соединения (9.4) можно чрезвычайно эффективно
катализировать у-циклодекстрином и цериемAУ). При физиологическом значении рН
и 30 °С период, за который половина количества молекул (9.4) превращается в аде-
нозинмонофосфат (AMP), равен только 6 с, в то время как этот параметр для нека-
тализируемой реакции составляет, по оценкам, 3 000 000 лет. Таким образом,
скорость реакции увеличивается в 1013 раз, а ее механизм, по-видимому, включает
действие циклодекстрина в качестве лиганда для жестких оксофильных ионов
металлов. Можно ожидать, что эти, полностью искусственные, модели ферментов
откроют дорогу к созданию рукотворных способов регулирования функции клетки.
780
9. Биомиме тика
о- /Яо.
он
он он
(9.4)
А =
Схема 9.5. Аденилатциклазная и фосфодиэстеразная активность комплексов циклодекст-
ринов по Дж. Сумаоке (Sumaoka J. et al., 1994)
9*3*3 Функционализированные циклодекстрины
Выше было показано, что сочетание гидроксильных групп и
гидрофобной полости циклодекстрина позволяет весьма успешно имитировать работу
ферментов. Значительно более эффективными имитаторами ферментов могут
служить производные циклодекстринов, когда их гидрофобная полость используется
как один из компонентов, содержащих другие функциональные группы с
активными центрами моделируемого фермента. В самом деле, первое соединение,
справедливо считающееся искусственным ферментом (R. Breslow and L. Overman, 1970),
содержит комплекс связывающего центра на основе пиридиндикарбоксилата с ионом
Cu(II), закрепленный на широкой стороне молекулы циклодекстрина (соединение
(9.5)). Это соединение катализирует гидролиз гостей, которые не являются лиган-
дами данного металлического центра и обычно не подвержены гидролизу комплек-
(9.5)
(9.6)
9.3. Циклодекстрины как имитаторы ферментов
781
сами Cu(II). Соединение (9.6) также оказалось очень эффективным катализатором
гидролиза. л-Нитроарильный субстрат связывается внутри полости
циклодекстрина и вовлекается во взаимодействия с ГчЩП)-центром и гидрокс ильным и группами
циклодекстрина. В результате нуклеофильной атаки оксимным лигандом
образуется продукт гидролиза. Катализатор регенерируется при гидролизе этого продукта.
Циклодекстрины, модифицированные по узкой стороне, также используют в
качестве моделей ферментов, в частности как имитаторы глиоксалаз. Глиоксалазы
катализируют изомеризацию а-оксоальдегидов в гидроксикислоты —
превращение, осуществляемое с участием тиолатных фрагментов. Этот процесс
смоделирован с помощью Р-циклодекстрина, содержащегося аминоэтановую тиольную
группу на узкой стороне. Субстратом служит 2-нафталинглиоксаль, который в
присутствии циклодекстрина перегруппировывается в соответствующую гидрокси-
кислоту с умеренной селективностью, превышающей, однако, селективность в
аналогичных реакциях без циклодекстрина. Такие реакции полезны для сравнения
двух систем (схема 9.6).
Если аминокислотные боковые цепи ферментов неэффективны для проведения
желаемой реакции, то фермент часто использует коферменты. Кофермент
связывается ферментом вместе с субстратом, и этот фермент катализирует бимолекулярную
реакцию между коферментом и субстратом (см. разд. 2.5). Предложена простая
модель синтеза а-аминокислоты путем трансаминирования пиридоксамином,
который в качестве заместителя привит на узкую сторону р-циклодекстрина. Пиридок-
самин способен превратить а-оксокислоты в а-аминокислоты даже без
циклодекстрина. Однако циклодекстриновая модель фермента оказалась более
селективной и трансаминировала субстраты с арильными кольцами, прочно
связанными с циклодекстрином, намного быстрее, чем субстраты, имеющие малое срод-
Нафталинглиоксаль
Схема 9.6. Имитация глиоксалаз путем модифицирования узкой стороны молекулы
циклодекстрина
7821
9. Биомиметика
СО2Н
Фенилпиро-
виноградная
кислота
H,N
Пиридоксамин
NH,
СО,Н
Фенилаланин
Схема 9.7. Трансаминирование фенилпировиноградной кислоты циклодекстрином
и пиридоксаминовым коферментом
ство к циклодекстрину. Так, (л-трет-бутилфенил)пировиноградная и фенилпиро-
виноградная кислоты трансаминировались соответственно в 15 000 и в 100 раз
быстрее, чем сама пировиноградная кислота, образуя соответственно (n-mpem-byivu\-
фенил)аланин и фенилаланин (схема 9.7).
9*4 Коранды как имитаторы АТРаз
Нуклеотидные полифосфаты, в частности аденозинтри-, ди- и
монофосфаты (ATP, ADP и AMP), — неотъемлемые участники энергетического цикла
широкого круга биологических процессов, таких, как фотосинтетическое и
окислительное фосфорилирование, сокращение мышцы и активный транспорт. Роль
АТР в снабжении энергией, необходимой для поддержания трансмембранного
электрического потенциала, рассмотрена в разд. 2.1. Эта энергия обеспечивается
ферментами, относящимися к АТРазам (Na+/K+-ATPa3a, в частности, для
потенциалов клеточных мембран). Функция АТРаз заключается в гидролизе концевого
фосфатного остатка трифосфатного «хвоста» АТР с образованием ADP и
неорганического фосфата (Н2РО^, Pj). При этом процессе высвобождается 35 кДж-моль
энергии и временно образуется фосфорилированный фермент.
Удивительно эффективная модель активности АТРаз была разработана Леном
на основе большого коранда [24]N6O2 D.9). Мы уже видели, что в присутствии
кислоты соединение D.9) протонируется и проявляет значительное сродство к
многозарядным анионам, причем связывание аниона осуществляется посредством
электростатических сил и водородных связей N+-H-X~ (разд. 4.4.3).
РСА-данные гексахлорида протонированного макроциклического соединения
D.9) показывают, что оно принимает конформацию, напоминающую карман; при
этом модельные исследования подтверждают существование такой конформации в
растворах. Электростатическая природа связывания анионов соединением
9.4. Коранды как имитаторы АТРаз
783
О
NH
HN
NH
HN
NH HN
О
D.9)
D.9)-6Н+
D.9)-«Н+ означает, что особенно сильно оно связывает многозарядные анионы,
причем в случае гексапротонированного хозяина D.9)-6Н+ АТР с зарядом 4—
связывается в воде при рН 4 с \gKu = 11.
Коранд D.9), несмотря на гибкость макроцикла, не только сильно связывает
АТР, но также способен ускорять его гидролиз примерно в 100 раз в широком
диапазоне рН B.5—8.5). С помощью ЯМР-спектроскопии, использующей ядро 31Р с
достаточно высокой восприимчивостью и ядерным спиновым квантовым числом
У2 (рис. 9.5), было показано, что при гидролизе образуется фосфорилированный
макроциклический интермедиат. Эти данные позволили предложить механизм
процесса, представленный на схеме 9.8.
D.9)-5Н+
(9.7)
Схема 9.8. Механизм гидролиза АТР соединением D.9)-5Н+ (А - аденозин + сахар)
NH7
NH2
9. Биомиметика
он он
АТР D-)
F)
I -. -- Л
33.8
ЛЛ L
PN
L
20.8
16.8
3.8
I! j[ 0 мин
о„ / ТО:1
онон
ADP C-)
1
О"
Uo^.1
И
1 1
он он
AMP B-)
10 0 -10 -20 6, м.д.
Рис. 9.5. (а) Депротонированные
формы ATP, ADP и АМР (б) 31Р ЯМР-спек-
тры, записанные в процессе гидролиза
АТР соединением D.9) при рН 7 для
различных времен. Та, Тп и Т —
сигналы для а-, Р- и у-фосфорных ядер АТР
соответственно. Аналогичные сигналы
для ADP обозначены как D. PN — ин-
термедиат со связью Р—N (9.7). Другие
сигналы обусловлены присутствием
Н2РО^" и НРО|~. (Воспроизведено
с разрешения Hosseini M. W.
Supramolecular catalysis of phosphoryl
anion transfer processes // Supramolecular
chemistry of anions / Ed. A.Bianchi,
K.Bowman-James, E.Garcia-Espana.
Chichester: Wiley-VCH, 1997. P. 421-448)
Процесс привлекателен тем, что, поскольку АТР связывается сильнее, чем ADP
(АТР заряжен более отрицательно), каталитический цикл устанавливается легко,
так как добавленный АТР может вытеснить продукт ADP из хозяина. Согласно
кинетическому анализу, концентрация АТР изменяется линейно со временем на
начальных стадиях реакции при соотношении АТР:D.9), равном 10:1, что
предполагает отсутствие ингибирования продуктом. Рассмотрим основные стадии процесса.
Стадия а: предварительно отметим, что гексапротонированный макроцикл -
слабая кислота и при рН 7 легко теряет протон с одного из центральных атомов азота,
9.4. Коранды как имитаторы АТРаз Щ&5
давая пентапротонированный хозяин с одной доступной неподеленной
электронной парой (см. схему 9.8). Нуклеофильная атака на концевой (у-) атом фосфора
сначала приводит к образованию пятикоординированного фосфорного интермеди-
ата, а затем — к фосфорилированию макроцикла и образованию связанного ADP.
Эта реакция идет с высокой скоростью, поскольку благодаря карманоподобной
конформации макроцикла аминная неподеленная электронная пара располагается
вблизи концевой фосфатной группы. Катализатор может быть регенерирован путем
диссоциации комплекса ADP (стадия б) с последующим гидролизом выделенного
интермедиата (9.7), (стадия в). Можно также гидролизовать связь P-N в
комплексе ADP водой (стадия д) и вновь ввести коранд [D.9)-5Н] в реакцию с АТР (стадия
г).
Общая скорость реакции составляет 0.064 мин. Такое низкое значение по
сравнению со скоростью реакции, катализируемой природными АТРазами
(примерно 103—104 мин), обусловлено существенно более простой структурой
комплекса. Однако сродство между компонентами и механизм процесса сходны для
обеих реакций. В модельной системе, как и в случае природных систем, большое
значение имеет введение ионов металлов; например, катионы Са2+ и La2+
значительно увеличивают скорость реакции. Природные АТРазы в своей «работе»
используют катионы Mg2+ и щелочных металлов. Влияние ионов металлов, скорее
всего, обусловлено образованием тройного комплекса D.9)М"+АТР, в котором
металл, скоординированный с несвязанными атомами кислорода АТР, выступает в
роли кислоты Льюиса и тем самым увеличивает нуклеофильность
координированной воды.
Интересным моментом является обратимость стадий реакции, показанных на
схеме 9.8. Подразумевается, что в определенных обстоятельствах система может
стать моделью не АТРазы, а АТРсинтазы. Это было достигнуто при использовании
фосфорилирующего кофермента ацетилфосфата (АсР, МеСО2РО2~) и катиона
двухвалентного металла (Mg2+ или Са2+) в качестве промотора. В этих условиях в
присутствии 10-кратного избытка АсР ~35% ADP были снова превращены в АТР.
Полагают, что этот синтез проходит через соединение (9.7) и тройной комплекс
(9.8), согласно циклу, представленному на рис. 9.6.
(9.8)
f786
9. Биомиметика
ADP
Акцептор фосфорила
[24]N6O2-PO32-, ADP
Двойной комплекс
[24]N6O2-PO32-
Фосфорилирующий агент
АсО
АсР
Кофермент
Источник фосфорила
[24]N6O2 D.9)
Сокатализатор
М2+
Промотор
[24]N6O2-r^3-
Активный тройной комплекс
(9.8)
Рис. 9.6. Синтез АТР с использованием коранда D.9)
*"* Hosseini M. W. Supramolecular catalysis of phosphory) anion transfer processes //
Supramolecular chemistry of anions / Ed. A.Bianchi, K.Bowman-James, E.Garcia-
Espana. Chichester: Wiley-VCH, 1997. P. 421-448.
Хозяева, связывающие катионы,
как имитаторы трансацилаз
*""» Cram D.J., Cram J.M. Container molecules and their guests. Cambridge: Royal
Society of Chemistry, 1994. P. 65-77.
9 • 5 • * Хиральные коранды
В разд. 9.3.1 мы познакомились с использованием циклодекстринов в
качестве имитаторов трансацилаз, хорошо изученного класса ферментов,
осуществляющих перенос ацильной группы от одного субстрата к другому (например, от
сложного эфира к воде). Химические свойства трансацилаз заинтересовали Крама
с соавторами (Y. Chao et al., 1979), который использовал хиральные коранды, такие,
как (9.9), родственные соединению C.88) и содержащие тиольные нуклеофилы,
расположенные над и под плоскостью макроцикла. Для сравнения был также
получен ациклический аналог (9.10). Характерные особенности соединения (9.9)
представлены на рис. 9.7.
9.5. Хозяева, связывающие катионы, как имитаторы трансацилаз
/
Реакционный центр
SH
Хиральный барьер
5
Центр комплексообразования
SH
Ряс. 9.7. Характерные особенности хирального коранда (9.9) как имитатора трансацилазы
Как показано в разд. 3.11.4, коранды такого типа образуют с катионами аммония
водородные связи N—Н-0 с участием зарядов. Хозяин комплементарен такому
органическому катиону из-за соответствия симметрии третьего порядка NH^-группы
симметрии псевдошестого порядка связывающих центров коранда (см. рис. 3.54). В
случае хиральных гостей наблюдается хиральное распознавание как следствие сте-
рических взаимодействий между заместителями нафталиновых фрагментов
(CH2SH в (9.9) и (9.10)) и заместителями гостя. Д. Крам (D. Kram) и сотрудники
(9.9)
изучили связывание и реакционную способность ряда эфиров первичных
аминокислот при взаимодействии с (9.9) и (9.10). Связывание гостей (типа
RCH(CO2Ar)NH^") осуществляется с помощью их NH^-заместителей по типу
«насест» со свободными энергиями комплексообразования ~27 кДжмоль в
CDC13, насыщенном D2O при 25 °С. Этот процесс сопровождается трансацилиро-
ванием, при котором ацильная группа гостя связывается с тиольной группой
хозяина в результате нуклеофильной атаки тиола (схема 9.9). В каждом случае
использовали хозяин с ^-конфигурацией и обнаружили, что в интермедиате стабилизация
пары 5"-хозяин//,-гость (£-аминоэфир) была значительно выше, чем в случае S,D-
диастереомера. Данная система проявляет кинетическую селективность в том
смысле, что субстраты (гости) с L-хиральностью реагируют гораздо быстрее, чем
788
9. Биомиметика
Устойчивый 5,1^интермедиат
SH
Ar
О ..Т .. о-
О
н н+к 0^
SH
АгСГ
Менее устойчивая 5,О-конфигурация
Схема 9.9. Стадия трансацилирования, осуществляемого соединением (9.9)
(Ar = p-C6H4NO2)
субстраты с £)-хиральностью, поскольку переходное состояние реакции более
устойчиво для S/L-пары. Эта кинетическая селективность сильно зависит от
природы производного аминокислоты, которая определяется свойствами его группы R.
Для небольших аминоэфиров, таких, как производные аланина (R = Me),
соотношение ks JkR j составляло единицу, т.е. селективность не наблюдалась. Однако
селективность быстро увеличилась с ростом объема R. Факторы селективности
скорости составили: 6.4 для R = Ме2СНСН2, 8.2 для R = С6Н5СН2 и 9.4 для R = Ме3СН.
Представляет также интерес различие в поведении макроцикла (9.9) и поданда
(9.10), табл. 9.1.
В случае всех первичных аминоэфиров реакционная способность соединения
(9.9), которое предорганизовано для связывания субстрата, в 102-103 раз больше,
чем таковая непредорганизованного поданда (9.10), который способен только к
неспецифическому связыванию. Величины Д(ДG*) отражают уменьшение энергии за
счет образования переходного состояния как стадии, лимитирующей скорость про-
Таблица 9.1. Факторы ускорения и разности свободных энергий реакций
трансацилирования /)-аминоэфиров соединениями S-(9.9) и 5-(9.10)
(тиол взят в 50-кратном избытке, EtOH:CH2Cl2 = 20:80 (по объему))
Аминоэфир (бромид)
Me2CHCH2CH(NH^)CO2Ar
C6H5CH2CH(NH3+)CO2Ar
Me2CHCH(NH^)CO2Ar
MeCH(NH+)CO2Ar
CH2(NH+)CO2Ar
C4H9N+CO2Ar*
* Соль эфира пролина.
^9.9)/*f9.10)
>1170
490
160
>130
>130
0.8
Д(ДС), кДж моль
>17.6
15.5
12.5
>12.1
>12.1
-0.4
9.5. Хозяева, связывающие катионы, как имитаторы трансацилаз
789
цесса относительно равновесного основного состояния. Только в случае
вторичного аминоэфира, производного пролина, не наблюдается ускорения. Этот субстрат
не способен к триподальному связыванию. Очевидно, что триподальный способ
связывания по типу «насест» снижает энергию активации реакции примерно на
10—20 кДжмоль. Такая модель ускорения при связывании подтверждается также
тем фактом, что введение ионов К+, конкурирующих с центром связывания
субстрата, дает 500-кратное ингибирование реакции переноса ацила.
К сожалению, скорость гидролиза ацилированного хозяина оказалась очень
низкой (в 10~4 раз ниже скорости трансацилирования), и, следовательно, эта
система не способна к регенерации катализатора. Поэтому лучше всего ее называть
частичным имитатором фермента.
8 Имитация структуры и функции
В то время как корандная система, описанная в предыдущем разделе,
способна имитировать функцию (реакционная способность) ферментов трансаци-
лазного класса, их центры связывания и реакционноспособные остатки мало
похожи на природные системы. Соединение (9.9), таким образом, не является
структурным имитатором. Структура типичной трансацилазы — химотрипсина —
показана на рис. 9.8. Помимо гидрофобного центра связывания этот фермент
содержит нуклеофильную гидроксильную группу серина, имидазольный остаток гис-
тидина и карбоксильную группу аспартата, причем эта система предорганизована
за счет водородных связей и других взаимодействий. С учетом этих особенностей
активного центра фермента Крам с помощью молекулярных СРК-моделей, как и в
случае сферандов (разд. 3.4), сконструировал структурный и функциональный
имитатор химотрипсина — соединение (9.11).
Нуклеофил
Катализатор
переноса протона
О" N' "О МеО
|Ме/,
о- >rWv";o о ?
Me
911
N N
Центр
комплексообразования
м
(9.11)
790
9. Биомиметика
H-N
Центр комплексообразования
О
О—Ser
N Н Нуклеофил
His
Катализатор переноса
протона
Рис. 9.8. Активный центр
химотрипсина
Соединение (9.11), использующее катионсвязывающий центр полусферандного
типа, получить чрезвычайно трудно. Поэтому его синтез осуществляли в несколько
стадий. Это позволяло оценивать изменение ферментативной активности такой
модельной молекулы после завершения каждой стадии, а также определять
влияние на нее каждой функциональной группы. Сначала был собран собственно
связывающий центр — полусферанд C.64), который, как и ожидалось, благодаря
симметрии третьего порядка в расположении карбонильных групп в качестве
акцепторов водородной связи связывает mpem-BuNH^ в хлороформе с энергией
комплексообразования 55.2 кДж-моль" (см. рис. 3.37: РСА-данные полусфе-
раплекса C.64)-т/?ет-ВиЫНз~). На следующей стадии было получено соединение
(9.12), и изучена его реакция со сложным эфиром (9.13) - моделью субстрата
(схема 9.10).
Найдено, что образование полусферанда (9.14) происходит вШ" быстрее, чем
аналогичная реакция с модельным соединением C-фенилбензиловый спирт),
неспособным к комплексообразованию. Это подтверждает вывод о том, что комплек-
сообразование со связывающим центром полусферанда ответственно за организа-
н .,
г. Me
(9.12)
Hv к H
н
(9.13)
Схема 9.10. Имитация трансацилазной активности с помощью модели химотрипсина
9.5. Хозяева, связывающие катионы, как имитаторы трансацилаз
791
цию реагентов в переходном состоянии реакции. Показано также, что гидроксиль-
ная группа в соединении (9.12) проявляла ожидаемую нуклеофильность.При
введении ионов Na+ реакционная способность этого соединения снижалась на
несколько порядков из-за конкуренции за связывающий центр. Эти успешные
исследования привели к 30-стадийному синтезу соединения (9.15) и в результате
длительной и напряженной работы было получено ~0.5 г этого вещества. В
соединении (9.15) отсутствует только карбоксигруппа, имитирующая остаток аспартата в
целевой молекуле (9.11), ср. с рис. 9.8. Реакция (9.15) с (9.13) протекает
исключительно быстро, давая продукт (9.16), в котором имидазол ацетилируется (период
полупревращения /, ,2 < 2 мин) с последующим медленным переносом ацетила на
гидроксильную группу (f| ,2 = 4.1 ч). Ацетилирование имидазола наблюдали также в
случае природного фермента и неспецифических субстратов. И снова введение
ионов Na+ значительно снижает скорость реакции.
(9.15)
(9.16)
Хотя синтез целевой молекулы (9.11) ни разу не был завершен, но очевидно, что
систематическое изучение реакционной способности этого соединения по
отношению к ряду ацилирующих агентов должно привести к созданию эффективной
модели фермента. Ясно, однако, что изучение соединений (9.12) и (9.15), а также
других родственных им соединений открывает возможность достигать огромных
скоростей за счет комбинации подходящих связывающих центров и реакционно-
способных остатков. Получение даже таких систем, относительно небольших по
сравнению с реальными ферментами, очень сложно. Поэтому легко себе
представить те трудности, которые ожидают исследователей при создании реальных
функциональных и структурных имитаторов ферментов.
9. Биомиметика
Q.U Металлобиоцентры
Многие белки и ферменты являются металлопротеинами, т.е. в ядрах
их активных центров содержатся ионы металлов (например, Zn2+ и Fe2+). Ион
металла может играть роль кислоты Льюиса или редокс-агента либо действовать как
функция, стабилизирующая структуру (как темплат и регулятор). Фактически ме-
таллофермент можно рассматривать как одно очень большое координационное
соединение, в котором белковая (органическая) часть молекулы представляет собой
лиганд для металлического центра. Отсюда следует, что модели ферментов можно
разрабатывать, воспроизводя ближайшую координационную сферу иона металла,
точно так же, как в соединениях типа (9.11), которые имитируют органический
активный центр. Обычно координационные соединения, воспроизводящие
металлсодержащие центры в биологических моделях (металлобиоцентры), являются
структурными моделями, т.е. имитируют структуру активного центра фермента, но
не его функцию или каталитическую активность. Существуют различные пути, с
помощью которых можно установить связь между информацией, получаемой при
изучении таких простых моделей, и более косвенными данными о поведении
реальных природных систем. Структурные модели дают информацию о таких
структурных параметрах, как конформации, длины связей и углы между ними, которую
можно сопоставить с данными исследований методами электронной
спектроскопии (в УФ- и видимой области), ЭПР и EXAFS. Например, первым ферментом,
который удалось закристаллизовать, была уреаза (из бобов), расщепляющая
мочевину на аммиак и СО2. Это открытие Дж. Самнера (J. Sumner) было отмечено
Нобелевской премией по химии 1946 г. В 1975 г. методами электронной
спектроскопии в УФ- и видимой области было показано, что этот фермент содержит никель.
Сопоставив эти данные с аналогичными данными для модельных
координационных соединений, сделали вывод о том, что ион Ni(II) в ферменте имеет октаэдри-
ческую конфигурацию и находится в окружении азотсодержащих донорных лиган-
дов. Это было подтверждено в 1995 г. РСА-исследованием родственной уреазы из
Klebsiella aerogenes. Модели, которые используют для имитации
спектроскопических или магнитных свойств фермента неизвестной структуры, называют
спекулятивными (speculative) моделями. Напротив, подтверждающие (corroborative) модели
применяют для непосредственной имитации известных структурных особенностей
координационного металлического центра в природном материале. Эту
информацию обычно дает кристаллическая структура фермента.
Для получения функциональной модели металлобиоцентра можно
использовать тот же концептуальный подход, который обычно применяют к органическим
системам, но в более усложненном виде. Успех в создании такой модели зависит от
той степени, в которой активность фермента связана с природой первой
координационной сферы металла. В общем, трудности в управлении окружением
металлобиоцентра возрастают почти экспоненциально по мере удаления от иона металла и
его первой координационной сферы.
Ионы металлов часто имеют несколько координационных центров, и в случае
удачной модели характеристики донорных групп на каждом центре должны быть
четко определены вне зависимости от того, участвует ли этот центр непосредствен-
9.6. Металлобиоцентры уд?
но в катализе или выполняет другую биологическую функцию. В моделях металло-
биоцентров часто используют макроциклические и хелатирующие полидентатные
системы, поскольку благодаря своей относительной жесткости они могут
имитировать предорганизацию активного центра фермента. Иминные макроциклы
(основания Шиффа), подобные открытым Куртисом, Бушем и Егером (Curtis, Busch and
Jager), рис. 3.45, оказались особенно полезными из-за своей способности прочно
связывать ионы переходных металлов. Использовали также и насыщенные азако-
ранды. Макроциклы представляют особый интерес в металлобиоцентрах,
содержащих два иона металла, которые должны располагаться на определенном расстоянии
друг от друга. Возможность изменения размера макроцикла с двумя металлокоор-
динированными доменами (например, в уреазах, имеющих два никелевых центра
на расстоянии ~3.7 А друг от друга) удобно использовать для регулирования
расстояния металл—металл. Несколько примеров моделей ферментов на основе
макроцикл ических и хелатных лигандов обсуждаются ниже.
*""* Fenton D.E. Metallobiosites and their synthetic analogues — a belief in synergism //
Chem. Soc. Rev. 1999. Vol. 28. P. 159-168.
Q.6.1 Модели гемоцианина
Гемоцианин — это металлопротеин, участвующий в транспорте О2 у
членистоногих и моллюсков. Даже система транспорта О2 в организме гигантского
кальмара, который может весить до 150 кг, основана на гемоцианине, а не на
гемоглобине (разд. 2.4). Структура активного центра гемоцианина в начале 1980-х годов
еще не была известна, но на основании спектроскопических данных уже тогда
были определены его некоторые характеристики:
• два Cu-центра, каждый из которых связан с тремя единицами гистидина;
• эндогенный (расположенный внутри молекулы) мостиковый донорный атом
(возможно, депротонированный кислород из остатка тирозина);
• сильное антиферромагнитное взаимодействие между ионами металлов с
образованием диамагнитного комплекса;
• тетрагональное окружение Си (^тах 345 нм, 8 = 20 000, по данным УФ- и
видимой спектроскопии);
• расстояние Cu--Cu 3.64 А, по данным EXAFS;
• резонансная рамановская частота v(O—О) 750 см~' (видимо, лиганд О^").
Исходя из этих данных, Д. Фентон с сотрудниками (N. Bailey, D. Fenton et al., 1987)
из Университета Шеффилда (Великобритания) получил Си(И)-биметаллическое
основание Шиффа (9.17). Этот комплекс, являющийся спекулятивной моделью,
воспроизводит многие свойства природного фермента, например расстояние
Cu-Cu, равное 3.64 А, эндогенный алкоксо-мостик и незначительное
антиферромагнитное взаимодействие (рис. 9.9).
Несмотря на использование, казалось бы, весьма разумного плана построения
этой модели, было установлено, что она не согласуется с данными РСА дезоксиге-
моцианина, выделенного из колючего омара (Panulirus interruptus). По этим данным,
794
9. Биомиметика
l.Ba2+
2. Cu2+
H2N
NH2
OH
OH
(9.17)
Предполагаемый активный центр
оксигемоцианина
/ /
His-Cu Cu —His
/\ / \
His 0-0 His
Рис. 9.9. Синтез модельного основания Шиффа для оксигемоцианина и сравнение его
с природной системой
эндогенный мостик в молекуле дезоксигемоцианина отсутствует, причем каждый
из двух центров Cu(I) координирует три гистидиновых остатка, а расстояние между
ними составляет 3.7 А. РСА-исследование оксигенированного белка (оксигемоци-
анин из мечехвоста limulus polyphemus) показало, что пероксидный лиганд @\~)
фактически принимает необычную \i—г\2 :г|2-мостиковую структуру
(рис. 9.10). Эта структура была предсказана Н. Китаджимой в 1992 г. и удачно
смоделирована той же группой (N. Kitajima and W. Tolman, 1995), которая
использовала стерически затрудненные триподальные лиганды, сильно напоминающие
остатки гистидина и содержащие гидробисC,5-диизопропилпиразолил)борат (рис. 9.11).
His,
His'
His
His
.
His Рис. 9.10. Мостиковая структура ц-г]2:г}2-типа
координированного пероксида оксигемоцианина
Рис. 9.11. Кристаллическая
структура двумедного
модельного комплекса гидробисC,5-
диизопропил пиразол ил )бора-
топероксида
9.6. Металлобиоцентры уо-
Работа по созданию более адекватных функциональных моделей, способных
обратимо связывать О2, продолжается.
Q.6.2 Цинксодержащие ферменты
Цинк, будучи очень важным элементом в биохимии, входит в состав
активных центров более 100 ферментов, из которых наиболее распространены
гидролазы (например, щелочная фосфатаза и карбоксипептидаза А), трансферазы
(например, ДНК- и РНК-полимераза), оксидоредуктазы (например, алкогольдегидро-
геназа и супероксиддисмутаза) и лизазы (карбоангидраза). Кроме того,
цинксодержащие фингерпротеины представляют собой сильные регуляторы. Во
многих таких системах ион Zn2+, не обладающий редокс-активностью, является
кислотой Льюиса, вокруг которой субстраты координируются, поляризуются и,
следовательно, активируются. Цинк действует и как темплат, а также как
структурирующий и регулирующий агент.
Карбоангидраза (СА) стала популярным ферментом для разработки ее
структурных моделей. Роль этого фермента состоит в катализе простой, но очень важной
реакции фиксации СО2 (уравнение (9.3)).
СО2 + Н2О = НСО3- + Н+. (9.3)
Активный центр в различных формах природного фермента изучен методом
РСА. Он содержит тетраэдрический центр Zn2+, координирующий три гистидино-
вых остатка и, в зависимости от рН, либо молекулу воды, либо гидроксид-ион.
Каталитическая активность этого фермента основана на льюисовской кислотности
центра Zn2+, который уменьшает рКа координированной воды с 14 до ~7. Такое
большое A07-кратное) увеличение кислотности — результат координации ионом
Zn2+ нейтральных лигандов (они приобретают положительный заряд),
взаимодействия с другими кислотными группами и наличия гидрофобной полости фермента
(ограничивает сольватацию). Механизм катализа включает стадии, показанные на
схеме 9.11. Основная стадия состоит в нуклеофильной атаке гидроксидного
комплекса цинка на СО2. Это дает пятикоординированный интермедиат, содержащий
бидентатный анион НСО^. Легко понять, что депротонирование воды с
образованием более нуклеофильного гидроксида — решающая стадия всего процесса.
Основание (В) ВН*
(HisKZn—ОН2 -^—*^-*~ (His)jZn—ОН
Схема 9.11. Основные стадии реакции, катализируемой карбоангидразой
796
9. Биомиметика
Для этого процесса была разработана удачная структурная модель, которая
содержит триподальный трис(пиразолил)борат-анион. При этом образуется
искаженная тетраэдрическая структура (9.18). Известен также аналог гидрокарбонатного
каталитического интермедиата с пятикоординированным 2п2+-центром в виде би-
дентатного нитрата [Zn{-n3-HBC-PhpzK}('n2-NO3)].
Н—
Bu-f
(9.18)
(9.19)
(9.20)
В своей работе Э. Кимура с сотрудниками (Е. Kimura et al., 1992) сосредоточил
усилия на макроцикле [12]aH-N3, который также образует тетраэдрический
цинковый аквакомплекс (9.19). Преимущество этого комплекса, как и комплекса в
природном ферменте, состоит в его дикатионном характере, что значительно
увеличивает кислотность координированной воды. Введение аниона SCN~ приводит к
образованию пятикоординированного комплекса (9.20) и, в конце концов, —
бис(тиоцианата). Образование таких пятикоординированных интермедиатов,
содержащих координированную воду или гидроксид и анионы, имеет большое
значение для катализа с участием СА. Однако в отличие от реальной системы ни (9.18),
ни (9.19) не содержат гидрофобной полости, которая играет важную роль в
реализации высокой активности и нуклеофильности этого фермента. Эту задачу решил
П. Волтон (P. Walton) из Йоркского университета (Великобритания), который для
имитации гидрофобных полостей фермента использовал основания Шиффа на
базе триподального цис-1,3,5-триаминоциклогексана (9.21). Конденсация этого
строительного блока с альдегидами и последующая реакция с Zn2+ дали ряд триподаль-
ных лигандов, в которых гидратированный ион цинка расположен на дне глубокого
гидрофобного колодца (9.22).
Ш
NH2
(9.21)
(9.22)
9.7. Аналоги гема 797
Аналоги гема
п.у. 1 Модели связывания и транспорта кислорода
Уникальный кооперативный механизм, в соответствии с которым
кислород связывается и переносится гемоглобином, содержащим железопротопорфи-
рин IX, подробно описан в разд. 2.4. Поразительные свойства этой системы
стимулировали разработку низкомолекулярных моделей, которые помогли бы
приблизиться к пониманию ее природы и предложить новые способы
осуществления химических превращений, транспорта и катализа. Система гемоглобина
особенно привлекательна для специалистов в области супрамолекулярной химии,
потому что концептуально она относительно проста (например, по сравнению с
ферментами). Связывание О2, по существу, — это термодинамически
контролируемое связывание нейтральных частиц-гостей, которые при этом не подвергаются
никаким химическим превращениям. Тогда, казалось бы, нет необходимости
усложнять модельные системы, вводя в них функции, связанные, например, с
кинетической селективностью вдоль пути реакции. Этот простой взгляд на связывание
О2 противоречит, однако, тому факту, что О2 — далеко не «безобидный» гость, а
химически весьма реакционноспособный. Поэтому гемоглобин выполняет
существенно более сложные задачи, такие, как предотвращение образования оксида или
ц-оксодимера, связывание и выделение кислорода в физиологических условиях
(определенные парциальные давления О2 в легких и мышечных тканях), а также
контроль за редокс-потенциалом Fe(II), необходимым для восстановления О2 в
супероксид. Все это делает гемоглобин интересным объектом для моделирования.
Очевидно, что при создании искусственных моделей, имитирующих связывание
кислорода, вполне логично использовать окисляемые комплексы переходных
металлов (особенно железа) с порфириновыми (рог) лигандами типа (9.23), как и в
природной системе. В таких случаях сродство О2, по существу, управляется
потенциалом восстановления металлического центра (£,0) в реакции М(л+1)+ + е~ = М"+.
В этом отношении особенно предпочтительны Fe(II) и Со(П). Эти два катиона
металла полезны также еще и потому, что при связывании порфиринового лиганда
они имеют вакантные центры аксиальной координации, необходимые для
связывания О2. Это обусловлено их предпочтительной октаэдрической координационной
геометрией. К сожалению, для точной имитации свойств природной системы
требуется только один аксиальный координационный центр, чтобы связать кислород.
Следовательно, для заполнения координационной сферы металла необходим еще
один аксиальный лиганд. В природной системе эту функцию выполняет
ближайший гистидиновый остаток. В случае катиона Fe(II), который является ионом
г/6-металла, имеются веские энергетические причинь. для связывания второго
аксиального лиганда, поскольку образовавшееся октаэдрическое поле лиганда
обладает достаточной силой, чтобы вызвать переход из высокоспинового состояния
(общее спиновое квантовое число S = 2) в низкоспиновое (S = 0),
сопровождающийся приростом энергии стабилизации поля лигандов (ср. Дополнение 7.1). В
этом случае соотношение констант равновесия Ки/Кп — 15 (уравнения (9.4) и
(9.5)). В природной системе жесткость, заданная третичной структурой белка, пре-
798
9. Биомиметика
пятствует приближению периферического остатка гистидина к вакантному
аксиальному координационному центру. Это исключает конкуренцию со стороны
белка за центр связывания О2. В случае Со(П) с ^-конфигурацией конкуренция со
стороны других аксиальных лигандов за центр связывания О2 не имеет места из-за
отсутствия спинового перехода.
[Fe(por)]2+ + L = [Fe(por)L]2+, Ku
[Fe(por)L]2+ + L = [Fe-(por)-L2]2+. Kn
(9.4)
(9.5)
R
В модельных системах на основе Fe(II) третичная структура белка отсутствует, и
поэтому проблема генерирования пятикоординированного металлического центра
должна решаться иначе. Некоторые из удачных подходов рассмотрены ниже.
1. Использование 2-метилимидазола (Melm) в качестве аксиального лиганда. Сте-
рические препятствия, обусловленные метильной группой, стабилизируют пя-
тикоординиррванные Ре(Н)-порфириновые комплексы, в которых смешение
железа относительно плоскости порфирина уменьшает стерические
взаимодействия с метильной группой (рис. 9.12). Образующийся 2-метилимидазольный
комплекс с производным тетрафенилпорфирина (9.25), рис. 9.13, — хорошая
структурная модель дезоксимиоглобина. Имидазол и его производные
оказались хорошими имитаторами гистидинового остатка белка.
2. Ковалентное связывание единственного аксиального лиганда (L) с каркасом
порфирина. Недостаток такого подхода — димеризация порфириновой системы
при повышенных концентрациях с образованием одного четырехкоординиро-
ванного и одного шестикоординированного металлических центров (рис. 9.14).
3. Наиболее элегантный, хотя синтетически менее доступный способ состоит в
совмещении одной стороны порфириновой системы с молекулярной клеткой,
так что связывание имидазол ьных аксиальных лигандов происходит только с
доступной плоскостью порфирина.
Для эффективного использования этих систем необходимо решить проблему их
низкой стабильности, обусловленную разложением низкомолекулярных модель-
9.7. Аналоги гема
799
Уменьшенное
стерическое
отталкивание
Сильное
стерическое
отталкивание
Рис. 9.12. Структурная модель дезоксимиоглобина на основе пятикоординированного
железного комплекса тетрафенилпорфирина (9.25), стабилизированного 2-метилимидазо-
лом. (Заместители в порфирине не указаны)
ных комплексов металлопорфирин-О2 в результате необратимого распада связи
0=0 (ср. схему 2.4) или протонирования координированного 02 с образованием
очень неустойчивого гидропероксильного радикала Н02. Отщепление этого
радикала от металлического центра дает неактивный Ре(Ш)-порфириновый комплекс и
Рис. 9.13. Кристаллическая
структура комплекса
[Fe(9.25)(MeIm)],
демонстрирующая смещение пятикорди-
нированного железа
(по Jameson G. ct al., 1980).
(Лиганд Melm не указан)
8QQ 9. Биомиметика
Рис. 9.14. Ковалснтное связывание единственного
аксиального лиганда
свободный НО2, который быстро диспропорционирует на О2 и Н2О2. В самом деле,
если природная система мутирует с образованием остатков аминокислот, таких, как
тирозин, на периферическом центре (кислородсвязывающий карман), то гем
остается в неактивном Ре(Ш)-состоянии. В модельных системах аналогичный эффект
дает связывание воды. Все эти нежелательные пути разложения в природном мио-
глобине исключаются благодаря третичной структуре белка, которая вводит
кислородсвязывающий центр в гидрофобный карман, предотвращая димеризацию и
доступ к кислотным группам. Одними из первых удачных моделей гема, в которых
учитывалась эта особенность, были «огороженные» порфирины (J. Collman and
С. Reed, 1973). Их получили в результате введения в порфириновое ядро
объемистых гидрофобных заместителей, образующих вертикальную «изгородь» вокруг же-
лезосвязывающего центра (схема 9.12). Синтез этих стерически защищенных
соединений (9.24) включает конденсацию нитрозамещенного бензальдегида с
пирролом и последующее восстановление NO2-rpynnbi до NH2-rpynnbi. Это дало
порфирин, существующий в виде ряда изомеров (называемых атропоизомерами),
различающихся относительной ориентацией 1МН2-заместителей над и под
плоскостью порфирина. Они не взаимопревращаются ниже 80 °С и могут быть разделены
хроматографически. Только а,а,а,а-изомер можно использовать для получения
«огороженного» порфирина (9.25). Нагревание выше 80 °С и последующее хрома-
тографическое разделение смеси изомеров может дать больше продукта (9.25).
Аналогично ряд «карманных» порфиринов (9.26) можно синтезировать, блокируя три
из четырех аминозаместителей, а порфирины типа «корзинка для пикника»
(например, (9.27)) образуются при блокировке всех аминогрупп. Подобным образом
реакции с аДа,3-изомером могут дать соединение с блокировкой заместителей на
обеих сторонах порфирина. Введение химически различающихся петель по обе
стороны плоскости макроцикла (9.28) способствует связыванию с центральным
атомом металла различных аксиальных заместителей.
Fe(II)-KOMruieKC соединения (9.25) при взаимодействии с О2 оказался
великолепной моделью оксигенированного миоглобина. Молекула О2 связана в полости,
9.7. Аналоги гема
801
СНО
1. Конденсация
►
2. Восстановление
NH2 NH2
NH2 I NH2 NH2 I NH2
| NH2 ^У | I У |
V
o.o.a,a-
Г
NH2
NH2
I NH2 NH2
NH2
H2N
NH2
a.a.p.p-
NH2
NH2
o.p.a.p-
Атропоизомеры соединения (9.24)
(9.24)
(9.25)
Схема 9.12. Синтез «огороженных» порфиринов
образованной заместителями «изгороди», а свободный аксиальный центр
связывает такие азотистые основания, как имидазол или его N-метил- и 2-метилпроизвод-
ные. Однако в случае стерически незатрудненных оснований связывание
кислорода комплексом [Fe(9.25)] конкурирует с захватом других аксиальных лигандов,
поскольку «изгородь» не обеспечивает селективности О2 относительно этих лиган-
21-СтидДж. В
8<Й
9. Биомиметика
и;п = 2,4,6,8
а
а
(9.27)
О.
О
О (9.28)
дов. Тем не менее экспериментально установлено, что О2 способен конкурировать
при связывании: были выделены комплексы с О2. Более того, заместители-«изгоро-
ди» препятствуют разложению, связанному с образованием оксодимера, а
комплексы типа [Fe(O2)(9.25)L] (где L = l-Melm, 2-MeIm) устойчивы в растворе при
комнатной температуре в течение нескольких часов. РСА-исследование комплексов с
l-Melm подтвердило ожидаемую структуру фрагмента Fe—О2: угол Fe-O—О равен
128°, а длина связи Fe-О составляет 1.75 А (рис. 9.15). Спектроскопические
свойства этой, модельной, и природной (которая пока структурно не охарактеризована)
систем оказались весьма близкими, например v^^) =1159 см.
Более сложные «карманный» порфирин и порфирин типа «корзинка для
пикника» также способны имитировать такие свойства, как селективное связывание
О2/СО. При этом обычно высокое сродство СО понижается из-за стерического
взаимодействия между молекулой СО (линейная конформация фрагмента Fe-C=O) и
блокирующим или стягивающим фрагментом. Селективность связывания
небольших молекул типа О2 выше, чем таковая оснований больших размеров.
9.7. Аналоги гема |ВОЗ
Рис. 9.15. Кристаллическая
структура комплекса [Fe(O2)(9.25)(MeIm)].
(Лиганд Melm не указан)
Были также предприняты попытки смоделировать наблюдаемую в природной
системе кооперативность* между четырьмя миоглобиновыми единицами, из
которых построен белок гемоглобина. Кооперативность жизненно важна для
функционирования этого транспортного белка в точно заданном интервале концентраций и
выражается в синергетическом увеличении сродства к О2 несвязанных единиц гема
при взаимодействии любой из них с О2 (см. разд. 2.4). Так, связывание О2 первой ге-
мовой единицей вызывает опосредованные белком конформационные изменения в
остальных трех единицах из Г-состояния с низким сродством в /^-состояние с
повышенным сродством. Различия между R- и Г-состояния ми легко понять,
используя «огороженные» модельные соединения. Найдено, что сродство комплекса
[Fe-(9.25)L] к О2 (и СО) значительно ниже (в 10-100 раз), когда L = 2-MeIm, в
отличие от стерически более благоприятного случая, когда L = N-Melm или L = Im.
Итак, в дезоксигенированном состоянии комплекс [Fe(9.25)B-Melm)] является
превосходной моделью дезоксигенированного Г-состояния гемовой единицы
гемоглобина с низким сродством. Пониженное сродство к кислороду — результат
неблагоприятных стерических взаимодействий между 2-MeIm и порфирином,
которые усиливаются при связывании О2, потому что переход от пяти- к
шестикоординированному железному центру сопровождается значительным
уменьшением смещения атома железа из плоскости порфиринового кольца
(рис. 9.16).
•""» Collman J.P. and Zhang X. Functional analogues of the oxygen binding and activating
haem proteins // Comprehensive supramolecular chemistry / Ed. J.L.Atwood,
J.E.D.Davies, D.D.MacNicoI, F.Vogtle. Oxford: Pergamon, 1996. P. 1-32.
Имеется в виду гомотропный положительный кооперативный эффект, состоящий в том,
что первоначальное связывание кислорода с одной субъединицей ускоряет связывание
кислорода с остальными субъединицами {Эллиот В., Эллиот Д. Биохимия и молекулярная
биология. М.: МАИ К «Наука/Интерпериодика», 2002. С. 373). См. разд. 2.4 - аллостерический
эффект. {Примеч. ред. перевода?)
804
9. Биомиметика
Ме'
Имидазол
(Im)
N
2-MeIm
Me
О
N
N-Melm
Уменьшенное
СрОДСТВО К О2
Неблагоприятное
стерическое
взаимодействие
4 -0.086 А
I Смещение Fe N~ "
t +0.03 А
Смещение Fe
2.07 А
О
Me
Модель Л-состояния
Модель Г-состояния
Рис. 9.16. Влияние аксиальных лигандов на сродство к О2. Модель управляемой белком
кооперативности в гемоглобиновом тетрамере
Модели цитохромов Р-450
Цитохромы Р-450 — это большое семейство ферментов, использующих
в качестве субстрата кислород (оксигеназы). Обозначение «Р-450» отражает тот
факт, что эти соединения представляют собой пигменты (pigments), поглощающие
видимый свет при 450 нм. Они обнаружены в растениях, микроорганизмах и в
большинстве органов млекопитающих, в частности в печени, и катализируют такие
реакции, как гидроксилирование алканов (уравнение (9.6)) или эпоксидирование
алкенов.
RH + О2 -> ROH
Н2О.
(9.6)
Подобно миоглобину, цитохромы Р-450 содержат кислородсвязывающую гемо-
вую единицу, однако вместо аксиального гистидина в миоглобине в цитохромах
имеется тиолатный остаток цистеина (Cys). Этот фермент используется как в
первичном метаболизме (например, многократное окисление холестерина), так и в
катаболизме гидрофобных ксенобиотиков (например, разрушение лекарств в пече-
9.7. Аналоги гема
805
ни). В противоположность миоглобину, функция цитохрома Р-450 зависит от
разрыва связи 0=0 и, следовательно, белковое окружение гема совсем другое. Катали-,
тический цикл оксигеназной активности этих ферментов показан на схеме 9.13
Первоначальный Ре(Ш)-комплекс восстанавливается в О2-связываюшее Fe(II)-co-
стояние с использованием электронов от никотинамидадениндинуклеотида
(NADH - биологический переносчик электронов). Дальнейшее восстановление и
протонирование вызывают расщепление связи 0=0 с образованием воды и Fe(V)-
оксокомплекса @2 -> О^ —> 0\~)- Отметим, что ц-оксодимер, образование
которого необратимо и может вызывать инактивацию фермента, не получается. Эта актив-
Схема 9.13. Каталитический цикл с участием цитохрома Р-450
806
9. Биомиметика
ная частица с высоким состоянием окисления используется для окисления
органических субстратов, связанных внутри полости фермента, которое приводит к
образованию некоторого продукта и регенерации фермента до его первоначального
Ре(Ш)-состояния.
Для цитохрома Р-450 трудно сконструировать структурные модельные системы,
поскольку тиолаты не образуют прочные связи с Ре(Ш)-порфириновыми
единицами, а окисление тиолатов до дисульфидов, сопровождаемое восстановлением в
Ре(П)-состояние, является обычным (уравнение (9.7)). И все же был синтезирован
ряд моделей , в которых использовали Ре(Ш)-порфирины с алкил- или арилтиола-
тами.
2[Fem(por)(SR)] -> 2[Fen(por)] + RSSR.
(9.7)
Получен также Ре(П)-комплекс (9.29) с аксиально связанным тиолатом в виде
аддукта с СО. Этот комплекс дает спектр в УФ- и видимой области, а также спектр
магнитного кругового дихроизма, аналогичные таковым СО-комплексов семейства
FeP-450.
О
(9.29)
Me. Me
\ + Гидрофильная
^ группа
R=C16H33
(9.30) OR
(9.31)
Гидрофобная
группа
Гидрофильная
группа
9.7. Аналоги гема
807
Значительный успех был достигнут и при конструировании функциональных
модельных систем, проявляющих оксидазную активность, в том числе систем, не
содержащих железа. В работе А. Шённинга (A. Schenning et al., 1994) были
получены системы, обладающие некоторыми особенностями природного фермента:
1. Металлопорфирин, связанный с мембраной.
2. Аксиальный лиганд.
3. Донор электронов (ср. NADH).
4. Переносчик электронов.
5. Мембранная система, которая удерживает компоненты внутри всего бислоя или
внутри водной прослойки.
Соединение ацетата марганца (9.30) представляет собой металлопорфирин с
длинными липофильными «щупальцами» для его закрепления на гидрофобной
мембране подобно тому, как белковая часть природной системы удерживает
порфириновый комплекс внутри липофильной части биологической фосфолипид-
ной клеточной мембраны. Обсуждаемая система включает искусственную мембрану,
состоящую из полимеризованного изоцианатного ПАВ (9.31), которое в водном
Полимерная стенка везикулы
Стенка
мембраны
везикулы
Pt
Комплекс
Мп-порфирин
"Т
MB,
Mnl
Mn
O2.2H*.
у \
Схема 9.14. Заполненный платиной везикулярный микрореактор, моделирующий работу
цитохрома Р-450. (MBox/red- переносчик электронов метиленовый голубой, окисленная
и восстановленная формы соответственно)
808
9. Биомиметика
растворе образует бислойные везикулы (разд. 10.2). Внутренняя водная область
этих везикул отделена от остального водного раствора бислойной мембраной из
ПАВ. В работе Шённинга с сотрудниками внутренняя область везикулы заполнена
коллоидальной платиной, полученной путем восстановления водного раствора
соли Pt(II) молекулярным водородом. Наночастицы платины выступают в роли
источника электронов и постепенно окисляются в Pt(III). Найдено, что эта система
катализирует эпоксидирование алкенов (которые в воде нерастворимы и,
следовательно, включены в мембрану везикулы), например стирола, а также
водорастворимых соединений типа 2,5-дигидрофурана. В качестве переносчика электронов
применяли редокс-активный краситель метиленовый голубой (П. Эрлих
первоначально его использовал для окрашивания клеток), схема 9.14. 2,5-Дигидро-
фуран дает 3,4-эпокситетрагидрофуран, а также продукт, получающийся при
раскрытии кольца. В случае стирола найдены только продукты раскрытия кольца.
Число каталитических циклов оказалось весьма небольшим (8 и 1.3 соответственно для
этих двух субстратов), однако система дает наглядное представление о
возможности использования принципов биологических процессов в абиотических
молекулярных устройствах.
Циклодекстрины также использовали как имитаторы цитохрома Р-450: железо-
порфириновый комплекс ковалентно закреплялся между двумя молекулами цикло-
декстрина, как в соединении (9.32). Связывание гидрофобных субстратов типа
циклогексена в полости циклодекстрина приводит к их селективному эпоксидиро-
ванию, причем источником кислорода служит иодозобензол. При наличии железо-
порфириновых комплексов, но отсутствии циклодекстриновой полости эта
реакция протекает с трудом.
(9.32)
9.8. Модели витамина В12
809
Q.O Модели витамина В12
Витамин В12 и его активная форма, кофермент В12 E'-дезоксиадено-
зилкобаламин), - это тетрапиррольные макроциклические комплексы кобальта,
содержащие 15-членное кольцо коррина. Они действуют сообща с апоферментом
(большой белок, который связывает одновременно кофактор В12 и субстрат для
проведения разнообразных мутазных ферментативных превращений, обычно
включающих радикальные 1,2-сдвиги (см. разд. 2.5)). Значительный интерес к
моделям В12 определяется тем, что большой апофермент может управлять
радикальными реакциями, которые часто трудно контролировать. Первый имитатор кофер-
мента В]2 получен X. Флором и сотрудниками (Н. Flohr et al., 1976), которые
синтезировали различные кобалоксимы. В их молекулах имеются
внутримолекулярные водородные связи и длинные диэфирные мостики для имитации стереохи-
мических условий, создаваемых этим апоферментом в одной из наиболее важных
В,2-зависимых коферментных систем, метилмалонил-СоА-мутазе. Как и
ожидалось, реакция перегруппировки протекала с превосходным выходом 82%, как и в
присутствии природного фермента (схема 9.15). Этот стереохимический контроль
был столь же эффективен, как и при наличии белкового апофермента.
Более поздняя работа Р. Бреслоу с сотрудниками (R. Breslow et al., 1992)
посвящена имитации одновременного связывания кобаламина и субстрата в полости
C-циклодекстрина, который выполнял функцию апофермента. Замещающая
группа (X) субстрата, связанного циклодекстрином, подвергалась селективному ради-
кал-индуцированному переносу на циклодекстрин. Даже если эта система не
способна завершить реакцию обратным переносом мигрирующей группы к другому
центру субстрата, важность этой реакции очевидна (схема 9.16).
Кофермент В12 оказался популярным объектом моделирования ферментов. Был
получен ряд очень простых систем (например, оснований на базе октопусных цик-
У-М
1,2-сдвиг
(Основание (В) - пиридин или метанол)
Схема 9.15. Первая модель, демонстрирующая роль апофермента в коферментативной
В,2-зависимой мутазной активности
9. Биомиметика
*СН, X
Схема 9.16. Одновременное связывание кобаламинового кофермента и субстрата
Р-циклодекстрином
лофанов), включая везикулярные модельные мембраны. Более подробные
сведения можно найти в ключевой ссылке этого раздела.
*"» Murakami Y., Kikuchi J.-L, Hlsaeda К, Hayashida О. Artificial enzymes // Chem.
Rev. 1996. Vol. 96. P. 721-758.
Учебные задания
9.1. На основе материала гл. 2 и 9 предскажите свойства, которыми могли бы
обладать (а) структурная и (б) функциональная модели гемоглобина.
9.2. Укажите разницу между первичной, вторичной и третичной структурами
белков. Что обычно отвечает за геометрию и свойства субстратсвязывающего
центра? Какими свойствами должен обладать связывающий центр в согласии
с «моделью индуцированного соответствия»?
Литература
811
9.3. Обсудите смысл утверждения Л. Полинга (L. Pauling, 1948), что «ферменты -
это молекулы, комплементарные по структуре переходным состояниям в
реакциях, которые они катализируют». Включите в Ваш ответ представления о
селективности ферментативного катализа.
9.4. Как Вы думаете, почему коранд D.9) столь эффективен в качестве имитатора
АТРазы? Для ответа используйте сведения об относительно низком сродстве
гостей типа катионов щелочных металлов и галогенид-ионов, приведенные в
гл. 3 и 4. Почему полусферанд, а не сферанд выбран в имитаторе фермента
(9.11) в качестве центрального ядра, связывающего катион?
9.5. Обсудите причины и следствия смещения в порфириновом и полипирроль-
ном макроциклах. Почему этот фактор лишь ограниченно применим к более
гибким хозяевам типа корандов?
Мысленный эксперимент
Укажите причины особой популярности циклодекстринов при получении
искусственных имитаторов ферментов. Попытайтесь применить другие «ядерные»
системы-хозяева в качестве имитаторов ферментов для реакций, катализируемых
производными циклодекстринов?
Литература
Bailey, 1987 Bailey N.A., Fenton D.E., Moody R. et al. Complexes of ligands providing
endogenous bridges. 4. Copper(II) complexes of macrocyclic schiff-bases derived from
2,6-diacetylpyridine and l,rt-diamino-rt'-hydroxyalkanes (n,n' = 3,2-4,2- and 5,3) —
synthesis, properties, and structures // J. Chem. Soc, Dalton Trans. 1987.
P. 2519.
Breslow, 1970 Breslow R., Overman L.E. An «artificial enzyme» combining a metal catalytic
group and a hydrophobic binding cavity // J. Amer. Chem. Soc. 1970. Vol. 92.
P. 1075.
Breslow, 1992 Breslow R., Duggan P.J., Light J.P. Cyclodextrin-B,2, a potential enzyme coen-
zyme mimic//J. Amer. Chem. Soc. 1992. Vol. 114. P. 3982.
Collman, 1973 Collman J.P., Reed C.A. Synthesis of ferrous porphyrin complexes. A hypothetical
model for deoxymyoglobin // J. Amer. Chem. Soc. 1973. Vol. 95. P. 2048.
Chao, 1979 Chao Y., Weisman G.R., Sogah G.D.Y., Cram D.J. Host guest complexation. 21.
Catalysis and chiral recognition through designed complexation of transition
states in transacylations of amino ester salts // J. Amer. Chem. Soc. 1979. Vol. 101.
P. 4948.
Diederich, 1983 Diederich F. On the way to enzyme models // Chem. Unserer Zeit. 1983. Vol. 17.
P. 105.
Dugas, 1989 Dugas H. Bioorganic chemistry // New York: Springer-Verlag, 1989.
812
9. Биомиметика
Flohr, 1976a Flohr #., Pannhorst W., Retey J. Synthesis and structure determination of an
intramolecularly alleviated bridged cobaloxime // Angew. Chem., Int. Ed. Engl.
1976. Vol. 15. P. 427.
Flohr, 1976b Flohr #., Pannhorst W., Retey J. Synthesis, structure determination and
rearrangement of a model for the active site of methylmalonyl CoA mutase with
incorporated substrate //Angew. Chem., Int. Ed. Engl. 1976. Vol. 15. P. 561.
Jameson, 1980 Jameson G.B., Molinaro K, Ibers J.A. et al. Models for the active site of oxygen-
binding hemoproteins. Dioxygen binding properties and the structures of
B-methylimidazole)-meso-tetra(a,a,a,a-o-pivalamidophenyl)porphyrinato
iron(II) - ethanol and its dioxygen adduct // J. Amer. Chem. Soc. 1980. Vol. 102.
P. 3224.
Kimura, 1992 Kimura E., Koike Т., Shionoya M., Shim M. A versatile macrocyclic [12]aneN3 for
interconversion of tetrahedral and trigonal bipyramidal zinc(II) complexes —
relevance to 4-coordinate <-» 00005-coordinate geometries of zinc(II) in carbonic-
anhydrase // Chem. Lett. 1992. P. 787.
Kitajima, 1995 Kitajima N., Tolman W.B. Coordination chemistry with sterically hindered
hydrotris(pyrazolyl)borate ligands — organometallic and bioinorganic perspectives
// Progr. Inorg. Chem. 1995. Vol. 43. P. 419.
Schenning, 1994 SchemingA.P.H.J., Hubert D.H.W., Esch J.H. et al. Novel bimetallic model
system for cytochrome-P-450 — effect of membrane environment on the catalytic-
oxidation // Angew. Chem., Int. Ed. Engl. 1994. Vol. 33. P. 2468.
Sumaoka, 1994 Sumaoka J., Miyama S., Yashiro M., Komiyama M. Enormous acceleration by
cerium(IV) for the hydrolysis of nucleoside 3',5'-cyclic monophosphates at pH 7
// J. Chem. Soc, Chem. Commun. 1994. P. 1755.
10
Жидкие поверхности раздела,
жидкие кристаллы
и жидкие клатраты
Вряд ли кто-нибудь будет доволен
нашествием воды и грязи.
И конечно же, только благоговейный, глаз
Увидит смысл в жидком состоянии.
Р. Брук A887-1915). Небеса
ЮЛ Порядок в жидкостях
До сих пор мы имели дело в основном с дальним порядком в твердых
телах (например, в кристаллах) и лишь немного касались объектов с ближним
порядком, реализуемым в жидких фазах, подобных стехиометрическим соединениям
включения типа «хозяин—гость». Действительно, считается, что жидкая фаза в целом
состоит из хаотически распределенных молекул, находящихся в постоянном
движении. Хотя и в жидких, и в твердых фазах должны осуществляться одни и те же «супра-
молекулярные» взаимодействия, сила этих взаимодействий во всех случаях, кроме
специально сконструированных соединений типа «хозяин—гость», мала по
сравнению с тепловой энергией молекул в изотропных жидкостях, особенно при высоких
температурах и/или при наличии слабовзаимодействующих компонентов. Однако
при сравнении жидкой и газообразной фаз становится ясным, что межмолекулярные
взаимодействия в жидкостях должны быть значительными для поддержания
конденсированного состояния. Так, комплементарные водородные связи в Н20 сохраняют
ее жидкой при атмосферном давлении и температурах вплоть до 100 °С C73 К).
Значительно более слабые водородные связи, характерные для H2S, обуславливают
кипение этого соединения при —61 °С B12 К), несмотря hi более высокую молекулярную
массу. Подтверждение наличия в чистой воде молекулярного порядка можно
получить из следующего эксперимента. Если образец твердой воды (лед), имеющей
кристаллическую структуру, очень медленно нагревать выше ее точки плавления,
постоянно изучая картину дифракции рентгеновских лучей, то можно наблюдать
образование переходной фазы, в которой индивидуальные (кристаллические)
рефлексы Брэгга исчезают. Это указывает на плавление. Вместе с тем, остаются
диффузные концентрические круги, как в случае рентгенограмм порошков, что
предполагает существование в жидкости остаточного порядка, который исчезает при
дальнейшем нагревании и достижении истинно изотропной фазы. Растворение
катионов щелочных металлов типа Cs+ обеспечивает ближний порядок в воде вследствие
образования вокруг такого катиона первичной сольватной оболочки примерно из
восьми молекул Н2О. Катион Li+, обладающий значительно большей поляризующей
способностью, в состоянии организовать вокруг себя как первую, так и вторую
координационные сферы. Эти сферы состоят из 12 молекул воды, поэтому гидратирован-
ный катион лития имеет больший радиус и, следовательно, меньшую подвижность.
о j^ 10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
Рассмотренные примеры показывают, что при соответствующих условиях в
определенных зонах жидкой фазы может наблюдаться молекулярный порядок. В
таких зонах, имеющих существенно большие размеры, чем одиночный комплекс
хозяин-гость, молекулы принимают строго определенную пространственную
ориентацию по отношению друг к другу. Фактически упорядоченные жидкости,
возможно, даже более важны, чем твердые тела, по крайней мере с биохимической
точки зрения. Однако их значительно труднее изучать, чем твердые тела. Один из
наиболее важных примеров упорядочения жидкости — образование биологических
клеточных мембран. Биологические мембраны формируют объемистые фосфоли-
пиды, которые вынуждены упорядочиваться в воде в результате комбинированного
действия гидрофобных эффектов и взаимодействий между растворителем и
гидрофильными «головками» фосфолипидов. При этом образуются капсулоподобные
везикулы (клетки), важные для биологических процессов. Клеточная мембрана
должна существовать в жидкой фазе для обеспечения быстрого биохимического
транспорта и химических реакций. В то же время прочная структура, подобная
твердотельной, характерная для биологических мембран, необходима для
сохранения мембранного потенциала, управления клеточной сигнальной системой и
защиты чувствительных внутриклеточных ферментов. Клеточная мембрана также
должна быть достаточно организованной для закрепления трансмембранных белков и
АТРаз. Она жизненно важна для векторного переноса возбуждения в
фотосинтетических мембранах. Упорядочение жидкости имеет чрезвычайно большое значение
в современных технологических устройствах, таких, как жидкокристаллические
дисплеи (liquid crystal displays, LCDs). Сам термин «жидкий кристалл»
подразумевает не только текучесть, свойственную жидкостям, но и некоторую степень кристал-
лоподобной упорядоченности.
В этой, заключительной, главе мы попытаемся кратко просуммировать роль су-
прамолекулярной химии и супрамолекулярных взаимодействий в образовании и
использовании структурированных жидких фаз. Объекты, относящиеся к
жидкокристаллическим материалам, весьма многочисленны; здесь, однако, мы
рассмотрим лишь их малую часть. Вместе с тем, по этому вопросу имеется обширная
литература, которой может воспользоваться заинтересованный читатель.
Fuhrhop J. -Н. and Koning J, Membranes and molecular assemblies, tbe synkinetic
approach. Cambridge: Royal Society of Chemistry, 1994.
1O.2 Поверхностно-активные вещества
и упорядочение на поверхности раздела
Поверхностно-активное вещество (ПАВ) — это соединение, молекула
которого состоит из двух частей с сильноразличающейся «растворимостью».
Гидрофильная часть этой молекулы имеет высокое сродство к воде, а липофильная часть
высокогидрофобна и, следовательно, «растворима» в органических средах. Вслед-
10.2. Поверхностно-активные вещества и упорядочение на поверхности раздела
815
ствие такого двойственного поведения ПАВ называют также дифилъными
соединениями . Это продукты повседневного спроса: мыла (додецилсульфат натрия A0.1)) и
шампуни (лауретсульфат натрия и лаурилсульфат натрия). Такие фосфолипиды,
как фосфатидилхолин A0.2), образующие клеточные биомембраны, жирные
кислоты и другие различные липиды, также принадлежат к соединениям этого типа.
Липофильная часть молекулы может состоять из цепей алифатических
углеводородов и фторуглеродов, арильных колец или множества других неполярных
органических групп. Гидрофильные фрагменты - это сильногидратированные группы, такие
как сульфонат, фосфонат, карбоксилаты, ионы аммония и т.п.
О"
о=У-°
о
A0.1)
V. Гидрофильная "головка"
Схематическое
представление
молекулы ПАВ
Гидрофобный "хвост"
J
С учетом природы их гидрофильной функциональности различают ПАВ
четырех классов:
• анионные (фосфолипиды, мыла);
• катионные (четвертичные аммониевые соли);
* В современной научной литературе часто и неправильно используют термины «амфи-
фил», «амфифильный» (калька с английского: «amphiphile», «amphiphilic»). Однако в русском
языке задолго до введения этих терминов в оборот существовал (и используется в учебниках,
химических словарях и энциклопедиях) термин «дифильный», реже — «дифил». {Примеч. ред.
перевода)
gj£ 10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
• амфотерные (цвиттер-ионные бетаины);
• неионные (жирные кислоты)*.
Гидрофобные эффекты, за которые ответственна липофильная часть дифильной
молекулы, способствуют агрегации этих молекул на поверхностях раздела, таких,
как воздух-вода. При этом гидрофильные «головки» обращены в водный слой, а
липофильные «хвосты» — в газовую фазу. Дифильные молекулы образуют монослои
(т.е. слои толщиной в одну молекулу), которые обычно изучают с помощью
поверхностных весов Ленгмюра, состоящих из прямоугольной ванны** с подвижным
барьером для изменения площади поверхности. Небольшое количество ПАВ в
органическом растворителе наносят на поверхность водной субфазы. Затем
органическому растворителю дают испариться, а барьер сжимает монослой. В
процессе сжатия степень организации молекул в монослое возрастает, и, наконец,
формируется конденсированный монослой. В таком упорядоченном монослое все
гидрофобные «хвосты» молекул ПАВ располагаются почти перпендикулярно водной
поверхности (рис. 10.1). Монослои, приготовленные таким образом, можно затем
перенести на различные твердые подложки (кремний, металлы, оксиды металлов и
т.д.) для дальнейшего изучения (A. Ulman, 1996). Силу, приложенную к барьеру,
можно измерить и соотнести с площадью поверхности монослоя***.
Образование монослоя на поверхности раздела между водной и липофильнои
фазами приводит к ряду поверхностных явлений, например к снижению
межфазного натяжения. К таким поверхностным явлениям относятся также растекание,
смачивание, образование дисперсий и эмульсий, солюбилизация и
ценообразование. Они лежат в основе механизма действия мыл, которые способствуют
растворению органических масел в воде.
Образование монослоя — не единственный тип поведения ПАВ. Могут также
образоваться и другие структуры ПАВ: мицеллы, бислои и везикулы (рис. 10.2).
Видная суофаза
Рис. 10.1. Монослои
ПА В на поверхности
водной субфазы в ванне
Ленгмюра
* Жирные кислоты относятся к анионным ПАВ (рКа ~5.0). Неионогенные ПАВ - это
эфиры жирных кислот и алифатические спирты. (Примеч. ред. перевода.)
** Часто используют круглую ванну. (Примеч. ред. перевода.)
*** С помощью поверхностных весов Ленгмюра измеряют зависимость поверхностного
давления (уменьшение поверхностного натяжения воды, вызываемое монослоем) от
площади в расчете на одну молекулу в монослое. Эта зависимость отражает фазовое поведение
монослоя, ориентацию и взаимодействия молекул в нем. (Примеч. ред. перевода.)
10.2. Поверхностно-активные вещества и упорядочение на поверхности раздела
817
Гидрофобное ядро A-3 нм)
• Анионное дифильное соединение
Катион
Слой Штерна
@.1-0.3 нм)
Слой Гуи-Чепмена
A0-IOOhm)
Рис. 10.2. (а) Упорядоченные структуры ПАВ в водном растворе, (б) Строение
сферической анионной мицеллы с противокатионами
Поведение ПАВ зависит от условий и концентрации. Если концентрация ПАВ
несколько превышает концентрацию, необходимую для образования
адсорбционного слоя, то ПАВ могут сворачиваться в сферические афегаты, называемые
мицеллами. В мицелле все липофильные части молекул направлены внутрь - к общему
gjg 10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
центру, а гидрофильные группы - наружу. Мицеллярные растворы (дисперсии)
могут иметь различные концентрации и состоять из мицелл, размеры которых зависят
от длины органической группы. Обычно диаметр мицеллы изменяется в пределах
1-3 нм*. Следует отметить, что ядро мицеллы заполнено органическими цепями
ПАВ и поэтому не может содержать молекул-гостей. Кроме гидрофобного ядра
мицеллы из анионных ПАВ имеют гидрофильный слой Штерна толщиной 0.3—0.6 нм,
который содержит анионные «головки» и некоторое количество противокатионов.
Мицелла окружена оболочкой из катионов и молекул растворителя толщиной
10-100 нм, называемой слоем Гуи-Чепмена. В целом мицелла напоминает
крошечную каплю масла в воде.
Концентрацию, при которой начинается образование мицелл, называют
критической концентрацией мицеллообразования (ККМ) и ее концептуально относят
к критической концентрации самосборки, рассмотренной в разд. 7.3.2. ККМ
можно измерить с определенной точностью, поскольку свойства смеси вода—ПАВ
(плотность, электропроводность, поверхностное натяжение и осмотическое
давление) резко изменяются при мицеллообразовании. На эту концентрацию сильно
влияет природа органической группы. Разветвленные органические радикалы
увеличивают ККМ, так как стерически им сложно плотно упаковаться в ядре
мицеллы, в то время как более длинные алкильные или арильные цепи уменьшают ККМ
из-за усиления гидрофобных взаимодействий. Для соединения A0.1) ККМ в воде
при 20 °С достаточно высока (8.1 мольдм), а мицеллы в среднем содержат ~62
мономерные единицы. Напротив, соединения с длинной цепью, такие, как
Me(CH2)n(OCH2CH2NOH, имеют ККМ ~0.1 моль-дм ** . Для образования
замкнутой внешней поверхности мицелле, обращенной в водную среду, требуется до
400 мономерных единиц. Мицеллообразование особенно благоприятно для таких
неионных ПАВ из-за отсутствия электростатического отталкивания заряженных
головных групп в слое Штерна. В некотором отношении мицеллы напоминают
дендримерные структуры (разд. 8.7); однако они не разветвлены и их плотность,
видимо, не возрастает при удалении от ядра.
По мере увеличения концентрации раствора могут быть реализованы структуры
мицелл нескольких типов, причем каждая — при своем значении ККМ. При
концентрации, значительно превышающей первую ККМ, наблюдается переход от
сферической к удлиненной цилиндрической форме мицелл, а выше третьей ККМ эти
структуры начинают агрегировать с образованием в растворе упорядоченных
структур большого размера (который можно определить дифракцией видимого света). В
результате получается лиотропный жидкий кристалл (разд. 10.3), обычно нематиче-
ского (стержнеобразного) типа.
В лиофильных органических растворителях с большим сродством к
гидрофобным частям ПАВ могут образоваться обратные мицеллы. В этих частицах полярные
группы направлены внутрь — к капсулированным молекулам воды, имеющимся в
следовых количествах, которые необходимы для образования обратных мицелл, так
* Диаметр мицеллы олеата натрия составляет 5 нм.
** Для большинства мицеллообразующих ПАВ величина ККМ располагается в области
небольших концентраций A0~5—10~2 моль-дм). (Примеч. ред. перевода.)
10.2. Поверхностно-активные вещества и упорядочение на поверхности раздела g jg
как сольватируют полярные головные группы. Органические «хвосты» направлены
наружу - в сторону растворителя. Эти образования напоминают крошечные капли
воды в масле. Увеличение содержания воды приводит к разрушению обратных
мицелл и образованию эмульсии, а в конце концов — к обычным мицеллам.
Возможно, наиболее интересным в химии ПАВ является образование бислоев и
везикул. Бислой можно рассматривать как протяженную слоистую мицеллу
(рис. 10.2). При механическом перемешивании, воздействии ультразвуком и т.д. эти
образования могут превращаться в везикулы, в которых бислой отделяет
внутреннюю водную фазу от внешней фазы. Эта структура сильно напоминает
биологические клетки, мембраны которых образованы фосфолипидными бислоями. Очень
интенсивное воздействие может даже привести к многократному складыванию
бислоев и образованию многослойных оболочек везикул, напоминающих
«рубашки» луковицы. Соблазнительно рассматривать эти системы как прообраз первых
биологических клеток. Действительно, растворение простых газов в океане должно
было сопровождаться химическими реакциями, протеканию которых
способствовали тепловое и УФ-излучение, а также космическая радиация. Это могло бы
приводить к образованию простых дифильных соединений и их растеканию в
монослои на поверхности океана. Механическое воздействие на поверхность раздела
вода-воздух должно было превратить эти монослои в бислой и везикулы с
включением в них других химических веществ, в частности аминокислот.
Такую структуру, в отличие от живой клетки, можно назвать протоклеткои'.
Процесс, посредством которого эта клетка эволюционировала в растущий
репродуцирующий организм и «научилась» кооперироваться с другими такими же
клетками с образованием многоклеточных организмов, является предметом обширной
дискуссии!
Следует отметить, что размеры биологических клеток значительно больше
размеров везикул. Вдобавок к внешней бислойной мембране клетка содержит
множество трансмембранных и связанных с мембранами белков и ферментов. Внутри
клетки также существуют мембраны, ограничивающие клеточные органеллы.
Простая модель везикулы не в состоянии объяснить механизм диффузионной
проницаемости живой клетки. Уже более 30 лет общепризнанной моделью, наилучшим
образом описывающей клетку, является жидкостно-мозаичная модель, которая
представляет клеточную мембрану как жидкую структуру (это, прежде всего,
жидкость). Клетка подвергается беспорядочным флуктуациям, приводящим к
появлению пор, а ее свойства — результат усредненных по времени равновесий и
механизмов накачки. Белки закреплены на клеточной мембране или пронзают ее, что
зависит от длины и природы якорных групп липофильного пептида, т.е. их можно
рассматривать как «острова» в липидном «море» (рис. 10.3). Эта модель
подтверждена электронно-микроскопическим методом замороженных сколов, а также
методом флуоресценции.
*""* Hoffmann H. and Ebert G. Surfactants, micelles and fascinating phenomena //
Angew. Chem., Int. Ed. Engl. 1988. Vol. 27. P. 902-912.
Вообще говоря, это не клетка, а модель фрагмента биомембраны. (Примеч. ред.
перевода.)
Ю- Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
Рис. 10.3. Жидкостно-мозаичная модель биологических клеточных мембран
Жидкие кристаллы
Chandrasekhar S. Liquid crystals. 2nd ed. Cambridge: Cambridge University Press,
1992.
IO.3.I Природа и структура
Жидкости, в которых существует истинный дальний порядок,
называют мезоморфными, или жидкокристаллическими, соединениями. Термин «жидкий
кристалл» подразумевает состояние вещества, сочетающее упорядоченность
кристаллического состояния твердого тела с легкой деформируемостью и динамикой,
характерными для жидкой фазы. Фактически жидкие кристаллы представляют
собой жидкую фазу (или несколько жидких фаз), промежуточную между изотропной
жидкой и твердой фазами. Такое анизотропное поведение — это в основном
следствие молекулярной формы. Молекулы, образующие жидкие кристаллы, называемые
мезогенами, в общем либо длинные и цилиндрические (стержнеобразные), как
4'-«-пентил-4-цианобифенил A0.3) и холестерилнонаноат A0.4), либо плоские и
круглые (дискообразные), как гексакис(D-октилфенил)этинил)бензол A0.5) и
бисC,4-динонилоксибензоил)метанатомедь(Н) A0.6). Мезогенами могут быть
полимеры или агрегаты, такие, как удлиненные мицеллы (разд. 10.2), или комплекс,
образованный водородными связями, на основе алкилзамещенных 2,6-диаминопи-
ридина и урацила, предложенный Леном (G.94), см. разд. 7.7.7). Сильная
анизотропия мезогенов заставляет их выстраиваться друг за другом с образованием
различных упорядоченных структур. Это отчасти результат межмолекулярных
взаимодействий и объемистости молекул, а также относительно высокой вязкости
жидкой фазы. Однако ничтожные возмущения, например частицы пыли или
неоднородности поверхности, могут привести к различной ориентации одной области
мезогенов относительно ее соседей. В результате этот жидкий материал с
макроскопическими доменами однородно ориентированных молекул становится видимым
невооруженным глазом: рассеянный свет дает яркую радужную картину. Порядок в такой
жидкости четко определяют с помощью поляризационного микроскопа (рис. 10.4):
10.3. Жидкие кристаллы
821
Рис. 10.4. Поляризационный микроскоп с нагревательным столиком
A0.5)
822
10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
Н19С9О ОС9Н19 H19C9O ОС9Н19 A0.6)
каждый домен вращает плоскополяризованныи свет, как твердый кристалл, давая
светлые и темные участки в зависимости от того, способен или нет
поляризованный свет проходить через анализатор (рис. 10.5).
Поскольку мезогены — это жидкости, а домены могут очень легко
деформироваться, то поведение жидкого кристалла должно сильно зависеть от температуры, и
это действительно так. Жидкокристаллическое поведение проявляется выше точки
плавления чистого твердого материала при температурах, еще низких, но уже
достаточных для медленного молекулярного движения, и энергиях межмолекулярных
взаимодействий, еще значительно превышающих тепловые энергии. По мере
увеличения температуры увеличивается и интенсивность молекулярного движения.
При этом жидкий кристалл может подвергаться дальнейшим фазовым изменениям,
часто проходя через состояния с менее упорядоченными мезофазами, прежде чем
окончательно расплавиться с образованием изотропной жидкости. Последний
процесс реализуется как только тепловая энергия становится достаточной, чтобы
индуцировать быстрое движение одной молекулы относительно соседних. Такое
термическое поведение — первая фундаментальная отличительная особенность жидких
кристаллов. Вещества, образующие мезофазы при нагревании твердых кристаллов,
называют термотропными, в то время как те соединения, которые образуют
мезофазы под влиянием растворителя или среды (например, мицеллы), называют лио-
тропными (рис. 10.6). Термотропные фазы образуются при медленном плавлении
кристаллического вещества. По мере роста температуры увеличиваются
молекулярные колебания до тех пор, пока межмолекулярные (супрамолекулярные) связи не
разрываются и не образуется жидкость. Примерно в 5% органических молекул не
все межмолекулярные связи разрываются сразу, и возникает
жидкокристаллическая фаза, в которой твердотельный порядок уже не существует, но все еще
присутствуют различные степени кристаллического порядка. С дальнейшим ростом
температуры различные межмолекулярные связи разрушаются одна за другой с
образованием все менее и менее упорядоченных жидкокристаллических фаз вплоть
до достижения изотропного расплава. Кроме плавления твердотельную решетку
можно разрушить, растворяя вещество в растворителе. Как и при плавлении,
возможно, что в зависимости от концентрации не весь кристалл разрушается сразу, и
10.3. Жидкие кристаллы 323
(б)
Рис. 10.5. (а) Нитеобразные домены в нематическом жидком кристалле толщиной
100 мкм при рассмотрении через скрещенные поляризаторы, (б) Шлирен-текстура в пленке
нематика толщиной 10 мкм. (Воспроизведено с разрешения Sackmann H. and Demus D.,
1973)
поэтому в растворе могут существовать различные степени порядка растворенного
вещества. Это лиотропная фаза.
Термотропные жидкие кристаллы формируются молекулами двух типов и
соответственно подразделяются на трубчатые и дискотические. Трубчатые (calamitic)
фазы (от греческого слова, означающего «труба») образованы стержнеобразными
324 10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
•ело
зсгь
I емператур
плавления ККМ
Анизотрот
жидкий
расплав зотропный
jacTBop
Температур
просветлеш ККМ
Изотропны . . . ■ этропный
жидкий ■ ■ : ^
расплав
Рис. 10.6. Основные типы жидких кристаллов
мезогенами. Совсем недавно выделенные и изученные дискотические (discotic)
фазы сформированы дискообразными соединениями. Трубчатые фазы могут быть
нематическими (от греч. «нема» — «нить»), смектическими (от греч. «смегма» -
«мыло») или холестерическими (от названия производных холестерина типа A0.4),
образующих мезофазы).
Нематическая фаза (рис. 10.7, а) характеризуется высокой степенью дальнего
ориентационного порядка молекул, но не дальним трансляционным порядком.
Другими словами, все мезогены ориентированы в одном направлении, но разупо-
рядочены в отношении скольжения вдоль этого направления (ось стержня).
Именно это легкое скольжение придает нематической фазе высокую степень текучести.
Нематическая фаза — наименее упорядоченная из всех трубчатых фаз, и в системах
с более чем одной мезофазой нематическая составляющая обычно появляется
последней непосредственно перед изотропным плавлением. Степень порядка в
нематической фазе определяется параметром S (рис. 10.8), который представляет собой
степень вращательной свободы стержнеобразной молекулы в направлении,
перпендикулярном ее продольной оси. Параметр S, равный единице, соответствует
идеально организованной нематической фазе, а изотропная жидкость имеет S = 0.
Нематические фазы характеризуются ярко окрашенными, быстро движущимися
пятнами, видимыми невооруженным глазом или под микроскопом, и шлирен-тек-
стурой (см. рис. 10.5, б). Шлирен-текстура обусловлена линейными сингулярностя-
10.3. Жидкие кристаллы
825
Рис. 10.7. Схематическое представление трубчатых жидкокристаллических фаз:
(о) нематическая, (б) смектическая А, (в) смектическая В, (г) смектическая С,
(д) смектическая Е и (е) холестерическая. (Воспроизведено с разрешения Vdgtle F., 1991)
ми, перпендикулярными слою мезофазы и являющимися аналогами дислокаций в
кристаллах. Их называют дисклинациями; они представляют собой границы, где
встречаются области с различным порядком.
Смектические фазы имеют большую степень порядка, чем нематические. Смек-
тические фазы представляют собой двухмерные упорядоченные слои, в которых
толщина слоя примерно равна длине молекулярного стержня. Из-за слабых
взаимодействий между слоями один слой может легко скользить по другим. Для менее
826
10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
{а)
МеО
ОМе
A0.7)
/ 5=1-з/2 sin26
(б) s
0.7
0.5
0.3
0.1
т
80 90 100 ПО 120
Температура, °С
130
140
Рис. 10.8. (а) Параметр порядка ^в нематической фазе. (б). Зависимость S от
температуры для 4,4'-диметоксиазоксибензола A0.7) (по Sackmann H. and Demus Z)., 1969).
(Воспроизведено с разрешения Vdgtle F., 1991)
упорядоченных смектических фаз, таких, как А и С (рис. 10.7, б и 10.7, г), внутри
слоя реализуется аналогичное жидкому состоянию статистическое распределение
мезогенов. В случае более упорядоченной смектической фазы В молекулы-стержни
имеют гексагональную упаковку (рис. 10.7, в). Тот же эффект увеличения порядка
наблюдается при организации плоских стержней A0.8) по типу «ёлочка»,
исключающей вращение вокруг продольной оси (смектическая фаза Е, рис. 10.7, д). К
настоящему времени изучены по крайней мере 12 различных смектических фаз
(^д-^), причем наиболее упорядоченный смектик имеет и наиболее высокую вяз-
10.3. Жидкие криста/мы g27
кость. Смектическая фаза D обладает довольно необычной кубической структурой,
которая не удовлетворяет условию слоистости. В общем, при нагревании
наблюдается переход от более упорядоченных фаз к менее упорядоченным в
последовательности: SE/SG -> SB -> SF -> Sc -> SD -> SA. Например, л-октилпроизводное
соединения A0.3) подвергается в общем трем фазовым переходам. Сначала оно плавится
при 24 °С, давая смектическую фазу А (энтальпия плавления
25.5 кДж-моль). При 34 °С она переходит в нематическую фазу @.13 кДж-моль),
которая при 42.6 °С превращается в изотропный расплав @.97 кДжмоль).
Фазовое поведение таких систем удобно исследовать с помощью поляризационного
микроскопа с нагревательным столиком. В этом случае можно видеть характерные
текстуры мезофаз и соотнести их с температурой образца (Дополнение 10.1).
омоно
Дополнение 10.1. Методы исследования мезофаз
Существуют три основных метода исследования мезофаз: поляризационная (с
нагревательным столиком) микроскопия, дифференциальная сканирующая калориметрия
(ср. Дополнение 6.1) и малоугловое рассеяние рентгеновских лучей.
Поляризационная оптическая микроскопия с нагревательным столиком
Этот метод, схематически изображенный на рис. 10.4, был первым и наиболее
простым из методов, примененных для изучения свойств мезофаз. Образец (обычно
менее 1 мг) помещают между двумя прозрачными покровными стеклами для
микроскопии и кладут на нагревательный (с точным контролем температуры) столик. Текстуру
образца изучают под микроскопом в процессе его медленного и контролируемого
нагревания и охлаждения. Плоскополяризованный свет, падающий на образец,
создается поляризатором, находящимся между источником света и образцом. За образцом,
ближе к наблюдателю, располагается второй поляризатор, повернутый на 90°
относительно первого. В отсутствие образца или в случае его изотропности свет не проходит
и поле микроскопа затемнено. Однако вследствие двойного лучепреломления жидких
кристаллов плоскость поляризованного света, проходящего через образец,
изменяется (он становится эллиптически поляризованным), и свет проходит через второй
поляризатор к наблюдателю. Визуализованные таким образом текстуры характерны для
данной мезофазы, причем их лучше всего наблюдать в процессе охлаждения образца.
Дифференциальная сканирующая калориметрия
Метод дифференциальной сканирующей калориметрии (differential scanning calorime-
try, DSC) описан в Дополнении 6.1. Этот метод удачно сочетается с методом
оптической микроскопии при работе с жидкими кристаллами. DSC дает информацию об
изменении энтальпии в процессе фазового перехода. Обычно она велика при плавлении
10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
и заметно уменьшается при переходе между мезофазами. Результаты, полученные
DSC-методом, полезны, так как они гарантируют, что изменения, наблюдаемые в
микроскопическом эксперименте, соответствуют именно фазовому переходу. Этот метод
позволяет идентифицировать такие фазовые превращения, которые не
сопровождаются существенными изменениями оптических картин.
Малоугловое рассеяние рентгеновских лучей
Мезоморфные фазы упорядочены и имеют периодичность, необходимую для
дифракции. Дифракция сильно уменьшается с углом, потому что порядок зачастую не
распространяется очень далеко, и поэтому измерения проводят при малых углах
рассеяния. Дифракционная картина от жидкого кристалла может дать важную информацию
о параметрах данной мезофазы и является единственным способом ее четкой
идентификации. Например, для смектической фазы можно наблюдать дифракционные
линии, по которым определяют межслоевую и внутрислоевую периодичность.
Сопоставление периода повторяемости с расчетными длинами молекул может дать
информацию об углах наклона молекул в слоях. Если образец упорядочен в
магнитном поле, то этот метод дает дополнительную информацию об ориентации мезогенов.
Холестерические фазы - разновидность спиральной нематической фазы с
шагом спирали (длина одного оборота спирали; ср. геликаты, разд. 7.7),
изменяющимся в пределах 200—2000 нм. Вместо параллельного выстраивания, как в нематике,
стержнеобразные молекулы изменяют свою ориентацию по отношению друг к
другу регулярным образом (рис. 10.7, ё). Холестерические мезогены оптически
активны. (Действительно, холестерические фазы можно получить, вводя немезогенные
хиральные молекулы в обыкновенную нематическую фазу, поскольку энергия
скручивания составляет лишь небольшую часть общей энергии, связанной с
молекулярным порядком.) Холестерические фазы имеют очень интересные оптические
свойства, такие, как селективное отражение циркулярно поляризованного света и
круговой дихроизм, а также чрезвычайно высокие величины оптического
вращения, достигающего 18 000 град-мм, по сравнению с несколькими десятками
градусов для молекулярной хиральности или несколькими сотнями для хиральных
фуллеренов типа С82. Холестерические мезофазы имеют привлекательные
радужные цвета. На них впервые обратил внимание австрийский ботаник Ф. Рейнитцер
в 1888 г. при изучении ацетата и бензоата холестерина. Спиральные
холестерические фазы с шагом менее —500 нм могут давать голубые фазы (названные так из-за их
цвета), которые обычно существуют только в очень узком температурном
интервале (~1 °С) перед образованием изотропного расплава. Разнообразие фазовых
переходов для нонаноата холестерина A0.4) представлено на схеме 10.1.
Голубые фазы известны с момента обнаружения жидкокристаллических
соединений. Рейнитцер в письме немецкому физику О. Леману писал о них в 1888 г.:
«При охлаждении (жидкой фазы бензоата холестерина] возникает фиолетовая и
голубая окраска, которая затем быстро исчезает, а соединение становится мутным,
оставаясь все еще жидким».
10.3. Жидкие кристаллы
829
Твердое _
тело
\
78.6 °С __ Холестерическая _ 91_°с_ _
i
\ 75.5 °С
\
Смектическая (j
за 0.017 кДжмоль
0.25 кДжмоль
>аза А
Голубая
фаза!
913 °с _ Голубая
0.017 кДж-моль-1 фа3
91.45 °С
Изотроп
all
0.53 кДжмоль
ная фаза
Схема 10.1. Фазовое поведение нонаноата холестерина, образующего голубые фазы
Однако только начиная с 1970-х годов голубые фазы стали восприниматься как
термодинамически индивидуальные и с тех пор начали привлекать пристальное
внимание физиков, изучающих конденсированную материю. Существуют три
индивидуальные голубые фазы (которые не всегда голубые), известные как ВР I, II и
III (ВР — blue phase), появляющиеся с ростом температуры. Эти фазы обязаны
образованию крошечных монокристаллов кубической симметрии: объемноцентриро-
ванной для ВР I и простой кубической для ВР II. Фаза ВР III квазикристаллична.
От этих кристаллов получены оптические дифракционные картины с рефлексами
Брэгга. Выделены также небольшие образцы — до нескольких сотен микрон в
диаметре (рис. 10.9). Цвет возникает за счет рассеяния света крошечными
кристаллитами. Интересно, что невозможно искажать спиральную структуру бесконечно, для
того чтобы она могла соответствовать кубической. Поэтому для кристаллов ВР I и
Рис. 10.9. Монокристаллы фазы ВР I в
равновесии с изотропным расплавом (смесь,
содержащая от 58% по массе 4-циано-4'-B-метил)бу-
тилбифенила в нематике ZLI 1840 («Merck»)).
(Воспроизведено с разрешения Cladis P.E. et al.,
1984)
830
10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатращы
(б)
Рис. 10.10. (а) Столбчатая
и (б) нематическая дискоти-
ческие фазы
ВР II характерны дефектные линии, или дисклинации, содержащие изотропную
жидкость и проходящие через решетку.
Дискотические фазы начали изучать только с 1977 г., хотя их существование
было постулировано Д. Форлендером (D. Vorlander) еще в 1923 г. Дискотики
формируют мезофазы двух индивидуальных типов — нематическую фазу и столбчатую
(рис. 10.10). Столбчатая мезофаза состоит из дисков, сложенных в стопки
(колонны). Сложенные регулярным образом, колонны могут формировать двухмерные
решетки различных типов: гексагональную, прямоугольную, наклонную и т. д.
Столбчатые мезофазы дают также соединения, получаемые путем замены плоского
ядра дискотического мезогена коническим, как в случае гексаэфира циклотрипи-
рокатехина A0.9). Столбчатые мезофазы, полые в середине, формируются
производными корандов, такими, как гекса(я-я-додецилоксибензоил)гексациклен
A0.10). Они относятся к тубулярным мезофазам и представляют интерес в качестве
моделей белков с ионными каналами (ср. разд. 2.1). Нематическая фаза дискотиков
менее упорядочена, поскольку в ней реализуется ориентационный порядок без
какого-либо трансляционного.
Лиотропные жидкокристаллические фазы состоят из двух и более компонентов,
один из которых обычно — дифильная молекула с гидрофильной и гидрофобной
частями (разд. 10.2), а другой - вода или, реже, иной растворитель. Тип
образующейся мезофазы зависит от концентрации ПАВ, причем для того, чтобы
взаимодействия между агрегатами ПАВ привели к образованию мезофазы, его концентрация
должна быть довольно высокой. Такими жидкокристаллическими агрегатами могут
быть ламеллярные бислои, мицеллы или цилиндры. Ламелляриые, или чистые,
фазы ПАВ состоят из плоских бислоев. Между ними находится вода, контактирующая
с их гидрофильными группами (ср. рис. 10.2). Порядок возникает в результате
ориентации бислоев. Эти слои могут изгибаться и образовывать мицеллярные или
везикулярные (ср. рис. 10.2) кубические, а также вязкие изотропные фазы. В них
сферические агрегаты образуют объемноцентрированную кубическую структуру. Может
также образоваться гексагональная фаза, в которой стержнеобразные мицеллы
10.3. Жидкие кристаллы
831
jj п. о Or
r4°V°T Г к
\ 9 Я P ? >o
(Ю.9)
R = С9Н19,
расположены параллельно друг другу и дают структуру, скорее напоминающую
столбчатую дискотическую фазу (рис. 10.11).
.3.2 Дизайн жидкокристаллических материалов
Чтобы выявить молекулярные характеристики, ответственные за
жидкокристаллические состояния систем, были систематически изучены свойства
большого числа мезогенов. Основные выводы из этих исследований приведены
ниже.
(б)
Рис. 10.11. (а) Ламеллярная, или чистая, фаза ПАВ и (б) гексагональная, или средняя,
фаза
332 Ю- Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
• Молекулы должны иметь вытянутую или плоскую форму и жесткий
молекулярный каркас.
• Длина молекулы должна превышать 1.3 нм A3 А), что согласуется с наличием
длинных алкильных цепей у многих жидких кристаллов, существующих при
комнатной температуре.
• Структура не должна быть разветвленной.
• Для усиления межмолекулярных дисперсионных взаимодействий молекулы
должны обладать высокой анизотропией поляризуемости. Этому
благоприятствует наличие легкополяризуемых групп и постоянных диполей.
• Соединение должно плавиться при невысокой температуре, что затрудняет
образование метастабильных монотропных* жидкокристаллических фаз.
Низкотемпературное мезоморфное поведение обычно технологически более полезно.
Введение алкильных концевых цепей способствует получению таких соединений.
Действительно, удлиненные структурно жесткие молекулы наиболее подходят для
получения соединений, проявляющих жидкокристаллические свойства. Поэтому
многие жидкокристаллические соединения содержат замещенные (в основном
яяря-замещенные) бензольные кольца, пара- Группировка обуславливает
вытянутую протяженную форму, например, производных стильбена A0.11) и оснований
Шиффа A0.12). т/?а«с-Конформация центральной двойной связи в этих
соединениях более эффективно обеспечивает жидкокристаллический порядок, чем
аналогичные £<ис-изомеры, благодаря более вытянутой молекулярной форме. Вместе с
тем, использование оснований Шиффа может оказаться проблематичным
вследствие гидролитической неустойчивости азометинов. Необязательно, однако, чтобы
центральная группа содержала ароматическое кольцо. Соединения на основе
насыщенных циклогексильных производных также проявляют жидкокристаллические
свойства. Отсюда следует, что наиболее важна именно их структурная жесткость,
обусловленная наличием кольцевой системы, а не дисперсионные взаимодействия
с участием арильных колец. Для усиления межмолекулярных взаимодействий
полярные группы можно вводить в любые части молекулы. Традиционно они
располагаются в центре мезогена (как в A0.7)) в окружении алкильных цепей. Однако и
концевые заместители, такие, как нитрильная, сложноэфирная или карбоксильная
группа, также эффективны и часто содержатся в современных технологически
важных жидкокристаллических соединениях, например в системах типа A0.3).
Важность размера и формы видна на примере димерного мезогена карбоновой кислоты
A0.13), образовавшегося за счет водородных связей. Водородная связь
обеспечивает этой молекулярной паре необходимую длину, и она достаточно сильна для
образования димера, имеющего время жизни, необходимое для образования мезофазы.
Алкильные цепи снижают температуру просветления** (Tcj) при плавлении
мезогена и повышают его молекулярную поляризуемость. Наблюдается сложная
зависимость температуры просветления от длины алкильной цепи: Тс1 осциллирует,
* Монотропными называют системы, у которых жидкокристаллическое состояние
проявляется ниже температуры плавления истинных кристаллов. (Примеч. ред. перевода.)
** Температурой, или точкой, просветления называют температуру перехода из мезофазы
в изотропную жидкость. (Примеч. ред. перевода.)
10.3. Жидкие tcpucmcuLibi
833
X = СН: A0.11)
X = N: A0.12)
о- --н-о
о-н --о
A0.13)
показывая максимальные значения для четного числа атомов углерода и
минимальные — для нечетного, как в соединении A0.14), рис. 10.12. Этот эффект обусловлен
влиянием заместителя на поляризационную анизотропию молекулы. Наивысшая
поляризуемость достигается вдоль направления, совпадающего с длинной
молекулярной осью. Увеличение длины цепи на один атом углерода в этом направлении
увеличивает поляризуемость вдоль этой оси больше, чем под прямым углом к ней.
Наоборот, введение следующей СН2-группы, образующей угол 109° относительно
предыдущей, имеет противоположный эффект, в основном усиливающий
поляризуемость в направлении, перпендикулярном главной оси. Температура
просветления (Тс1) жидкого кристалла непосредственно связана с разностью между
продольной и поперечной поляризуемостью и, следовательно, осциллирует. По мере
увеличения длины цепи и ее гибкости этот эффект постепенно исчезает.
н2п+1спсн2
A0.14)
Рис. 10.12. Осцилляция
температуры просветления
(Гс/) с ростом длины алкиль-
ной цепи (п) для
соединений типа A0.14)
23-СтияДж. В.
8344
10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
*3*3 Жидкокристаллические полимеры
Одно из современных направлений в проблеме изучения жидких
кристаллов - синтез полимерных жидкокристаллических соединений. Такие полимеры
могут быть двух типов. К первому типу относят полимеры, содержащие мезогенную
группу в основной цепи (жидкокристаллические полимеры основной цепи). Второй
тип представлен полимерами, в которых мезогенные остатки присоединены к
основной цепи в виде «подвесок» (боковые цепи). Преимущество
жидкокристаллических полимеров — дальнейшее ограничение молекулярной подвижности. Однако
очень важно, чтобы спейсеры, отделяющие одну мезогенную группу от другой,
были бы достаточно гибкими и позволяли мезогенам ориентироваться таким образом,
чтобы обеспечивать полимерам жидкокристаллическое поведение. В таких случаях
жидкокристаллические полимеры во многом ведут себя так же, как их
низкомолекулярные аналоги, в которых мезогены не связаны химически друг с другом.
Полимеры, содержащие стержнеобразные мезогенные группы типа A0.15) или A0.16),
формируют нематические, холестерические и смектические мезофазы (рис. 10.13).
В случае дискотических мезогенных групп найдены полимеры нескольких новых
структурных типов. Например, полиэфир с дискообразным трифениленом A0.17)
в качестве повторяющихся единиц, разделенных гибкими спейсерами, образует
гексагональную столбчатую фазу (рис. 10.14, а). В то же время полимеры с дискоти-
ческими группами, входящими в состав жесткой основной цепи, образуют новую
фазу, называемую сэндиковой («sandic» — доскообразная) нематической. При этом
«доски» ориентированы параллельно друг другу, как на рис. 10.14, б.
спейсер-
R
A0.17) О R
Me-Si
о
ОСбН'3
A0.16)
10.3. Жидкие кристаллы
835
(О)
(б)
с £ I'
Рис. 10.13. Мезогенные полимеры. Мезоген-
ные группы могут (а) входить в состав основной
цепи или (б) присоединяться как боковые группы
(а)
(б)
Рис. 10.14. Фазы дискотических полимерных мезогенов: (а) гексагональная столбчатая
и (б) сэндиковая нематическая
Покрытия из холестерических жидкокристаллических полимеров используют
на практике в качестве отражательных пленок и поляризаторов. Такой жидкий
кристалл, охлажденный ниже температуры стеклования, образует твердый полимер,
который хотя и аморфен, но содержит большие области «замороженного»
жидкокристаллического порядка. Эти структуры обнаружены и в природе в виде
радужной, почти металлической окраски жуков и других насекомых, возникающей
благодаря наличию спиральных холестерических структур в наружном слое защитного
панциря.
Жидкокристаллические дисплеи
Жидкие кристаллы применяют в ряде важных областей. Их
используют при высокоточном измерении температуры, когда изменения
жидкокристаллической фазы могут дать разрешение температуры вплоть до 0.007 К в интервале 1 К.
С меньшей точностью жидкокристаллическими термометрами можно измерить
температуру между —20 и +250 °С. Эти термометры дешевы и токсикологически
безопасны. Они находят применение в медицине, их используют даже в качестве
настенных термометров. Жидкие кристаллы применяют в аналитической химии, а
836
10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
также при измерении свойств вещества, зависящих от ориентации его молекул.
Растворение исследуемого соединения в жидкокристаллической нематической
фазе обеспечивает строгую ориентацию молекул этого гостя, облегчая измерение его
анизотропных свойств, в частности кругового дихроизма, методами ИК-спектро-
скопии, а также спектроскопии в УФ- и видимой области. Жидкие кристаллы
используют и в качестве стационарных фаз при хроматографическом разделении
близких по свойствам изомеров (например, дигалогенаренов), зачастую имеющих
различную растворимость в жидкокристаллических фазах. Их также широко
применяют в ЯМР-спектроскопии.
До сих пор наиболее важным является использование мезоморфных
соединений в жидкокристаллических дисплеях (liquid crystal displays, LCD). Вначале они
появились в наручных часах и карманных калькуляторах в виде хорошо знакомого
семисегментного дисплея (напоминающего цифру 8 с квадратными краями).
Современные LCD — одна из важнейших частей цветных мониторов компьютеров (с
добавками красителей) — представляют собой закрученные нематические ячейки
(twisted nematic cells, TNC), которые впервые были предложены Шадтом (Schadt) и
Хельфрихом (Helfrich) в 1971 г. Принцип действия LCD основан на изменении
ориентации молекул под влиянием электрического поля и результирующем изменении
оптических свойств ячейки. В TNC тонкий слой A0—15 мкм) нематического
жидкого кристалла заключен между двумя прозрачными стеклянными пластинками.
Поверхность пластинок предварительно обработана таким образом, что молекулы
ориентируются параллельно поверхности в одном направлении, подобно тому, как
в жидкокристаллической фазе мезоген самопроизвольно ориентируется благодаря
взаимодействию со своими соседями. Если эти пластинки повернуть относительно
друг друга на 90°, то в нематической фазе появляется спираль с углом закручивания
90° от одной поверхности ячейки до другой (рис. 10.15). Плоскополяризованный
Плоскость поляризации
света поворачивается на 90°
и он проходит через
* второй поляризатор
Стеклянные
пластинки
с ориентантом
Электроды
Скрещенные
поляризаторы
Падающий свет поляризован
Гомеотропно
ориентированный
мезоген: свет не проходит
через второй поляризатор
Источник тока
Рис. 10.15. Работа закрученной нематической ячейки. В отсутствие электрического поля
закрученный нематик вращает плоскость поляризованного света от одной поверхности
ячейки до другой: свет свободно проходит через ячейку. Приложенный потенциал (V)
делает ее непрозрачной
10.4. Жидкие клатраты 037
свет, проходящий через этот слой жидкого кристалла, изменяет направление
поляризации на 90°. Ориентированный таким образом жидкокристаллический слой
помещают между двумя скрещенными поляризаторами. В отсутствие электрического
поля этот дисплей прозрачен для света. Приложение напряжения к электродам
опорных поверхностей вызывает изменение ориентации молекул нематической
фазы. При этом реализуется гомеотропная ориентация*: закручивание исчезает,
прекращая вращение плоскополяризованного света и, следовательно, свет через ячейку
не проходит. В результате дисплей становится темным. Поскольку напряжение
требуется только для затемнения дисплея, затрата мощности исключительно мала. Это
делает TNC идеальным элементом для использования в портативных устройствах
(часы, небольшие компьютеры и т.п.). Такие дисплеи плоские и компактные, их
четкость возрастает с увеличением освещенности помещения. Однако они не работают
в темноте, а время отклика (~50 мкс) существенно больше, чем у
электронно-лучевых трубок A0 мкс). Это ограничивает их применение, например в телевизионных
системах.
Вероятно, в будущем станут доступными жидкокристаллические дисплеи со
значительно более быстрым откликом, в частности дисплеи на основе ферроэлект-
рических жидких кристаллов, таких, как л-децилоксибензилиден-л'-амино-2-ме-
тилбутилциннамат, обеспечивающий быстрое переключение между двумя
стабильными состояниями при приложении внешнего поля. Такие устройства можно
использовать для хранения оптической информации.
1О«4 Жидкие клатраты
1О.ф1 Открытие и свойства
Термин «жидкий клатрат» ввел в 1969 г. Дж. Этвуд из Университета
Алабамы (США) для описания фазового поведения ряда растворов солей в
ароматических растворителях типа бензола и толуола. В этом кроется некоторое
противоречие, поскольку клатраты, по определению, являются твердыми веществами (см.
разд. 1.2). Тем не менее концепция захвата гостя матрицей хозяина как результат
геометрического и топологического (в противоположность химическому)
соответствия одинаково приложима как к твердым, так и к жидким фазам. Существует
близкая аналогия между тем, что более строго можно назвать нестехиометрическими
жидкими соединениями включения, и обычными твердыми клатратами. Многое в
химии жидких клатратов еще не выяснено. Однако уже сейчас можно отметить
несколько важных свойств жидких клатратов, которые приведены ниже.
• Такие клатраты образуются из соли некоординирующегося аниона.
• Существуют специальные геометрические требования как для катиона, так и
для аниона.
• Образование даже небольших агрегатов (например, димеров, тримеров и т.д.) не
допускается.
Гомеотропная ориентация соответствует расположению длинных осей молекул мезоге-
на перпендикулярно опорной поверхности. {Примеч. ред. перевода.)
338 10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
• Жидкие клатраты полезны для селективного разделения ряда органических
соединений.
Первые жидкие клатраты были получены в результате реакции KN3 с чрезвычайно
чувствительным к воздуху и влаге А1Ме3, приводящей к образованию твердого
K[Al2Me6N3] A0.18). Это твердое вещество случайно смешали с толуолом, в
котором, как считалось, оно нерастворимо. При этом наблюдали быстрое растворение
белого твердого вещества с образованием двух несмешивающихся жидких слоев,
нижний из которых состоял из KlAl2Me6N3]-3.8C6H5Me, а верхний — из чистого
толуола. Нашли, что отношение алкилалюминиевой соли к толуолу в нижнем слое —
величина постоянная, не зависит от количества добавленного толуола (пока оно
выше некоторой критической величины) и выражается через А/А (число молекул
ароматического растворителя, приходящихся на один анион). Аналогичным
образом A0.18) образует жидкий клатрат с шестью молекулами бензола. Однако ни
моноалюминиевый комплекс K[AlMe3N3], ни KN3 не дают жидких клатратов с
бензолом. Это различие в поведении солей объясняется объемистостью
алкилалюминиевого аниона [Al2Me6N3]~ в жидком клатрате, эффективно
блокирующего доступ к наиболее благоприятным центрам связывания катионов К+,
расположенным вблизи группировки X—Nj этого аниона (рис.10.16). Таким образом,
размер и форма аниона чрезвычайно важны для образования жидкого клатрата.
Кроме того, склонность анионов к образованию жидких клатратов находится в
обратной зависимости от их способности к множественным взаимодействиям с
катионами. Описано много жидких клатратов на основе алкилалюминиевых
комплексов и ароматических растворителей, число молекул которых находится в интервале
между 1.3 и 42.2. Вместе с тем, наиболее распространены системы, содержащие
более четырех молекул ароматического растворителя.
Модель жидких клатратов, которая наилучшим образом объясняет их
поведение, изображена на рис.10.17. В жидкости существуют большие упорядоченные об-
о
о
о
о
N"
II
II
N+
II
II
О
о
о
о
о
о
о
N"
N
II
II
о
1
Л
/
и
н н
"f-T
н н
N Н Н
О к*
A0.18)
/
Центр связывания катиона К+
10.16. Центры, благоприятные для связывания катиона К+ азид-анионами
i/-u2ivie6N3]~, [AlMe3N3]~ и Nj: показаны ограничения, нарастающие по мере приближения
катиона к аниону и обусловленные ростом объема последнего. Это приводит к созданию
областей, заполненных растворителем (клатрация) даже в жидкой фазе, которая составляет
основу для образования жидкого клатрата
Рис
,Ме
10.4. Жидкие клатраты
839
-Анион
- Ароматический растворитель
- Катион
Рис. 10.17. Двухмерная модель жидких клатратов
ласти, в которых реализуются сильные взаимодействия анионов и катионов друг с
другом. Эти эффекты, а также стерические особенности анионов и/или катионов
предотвращают как обычное растворение, так и образование агрегатов, например
димеров и тримеров, которые могли бы индуцировать формирование
кристаллических зародышей. Роль ароматического растворителя заключается в поддержании
структуры и стабилизации заряда катиона и аниона путем интенсивных п—тг-стэ-
кинг- и тг-катион-взаимодействий, включающих квадруполь ароматического
кольца (см. разд. 1.7.5 и 1.7.6). Ароматический растворитель в жидком клатрате также
выступает в качестве гостя и, как в твердом клатрате, заполняет пустоты структуры,
что подтверждает обоснованность введения термина «жидкий клатрат».
Взаимодействие тг—катион, наблюдающееся в растворе, имеет ту же природу, что и в твердой
структуре Cs[Al2Me6N3]-2(«-KCRJKm), в которой катион Cs+ располагается между
двумя ароматическими гостями (рис. 10.18). Следует, однако, соблюдать большую
осторожность при сравнении результатов для твердого и жидкого состояний,
особенно в случае нежестких систем, подобных этой.
Рис. 10.18. Кристаллическая структура
Cs[Al2Me6N3]-2(rt-Kcwicm),
демонстрирующая я— катион-взаимодействия
#840 '0- Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
При рассмотрении процесса образования жидкого клатрата можно написать
уравнение реакции A0.1), отражающее равновесие при растворении твердого
предшественника типа A0.18) в ароматических растворителях или фазовый переход,
обусловленный ожижением твердого вещества.
MX + «S = MX-S,,. A0.1)
Это равновесие сильно зависит от температуры. Например, в случае A0.18) при
20 °С равновесие полностью сдвигается влево, а при 30 °С — полностью вправо.
Предельное (максимальное) значение п соответствует величине А/А. Однако
жидкие клатраты могут образовываться даже в том случае, когда количество
присутствующего растворителя меньше, чем значение А/А. Например, для A0.18)
минимальное пороговое значение п при инициировании образования жидкого клатрата
составляет -2.5, если S — бензол. Получившееся соединение, названное голодным
жидким клатратом, представляет собой гомогенный раствор. Этот раствор
продолжает поглощать растворитель до достижения величины А/А = 6, выше которой
помимо жидкого клатрата образуется чистая фаза растворителя. Очевидно, что
величина А/А не зависит от температуры. Она управляется тремя основными
факторами:
• для данных аниона и ароматического растворителя чем больше катион, тем
больше А/А;
• для данных катиона и ароматического растворителя чем больше анион, тем
больше А/А;
• для данных катиона и аниона чем больше ароматическая молекула, тем меньше
А/А.
Найдено, что величины А/А хорошо коррелируют с ван-дер-ваальсовыми
объемами ароматических молекул. При этом предполагается, что модель с полостями в
жидкости, которые заполняет ароматика, является адекватной. Постоянство
величины А/А дает основания считать, что катион-анионные взаимодействия приводят
к образованию пустот в растворителе, объем которых не зависит от характера
растворителя.
Основной интерес жидкие клатраты представляют с точки зрения их
применения для разделения, например, очень близких по структуре ароматических
соединений, таких, как бензол и толуол или изомеры ксилола (ср. методы, основанные на
использовании цеолитов, разд. 5.1.2). Разделение происходит благодаря большей
способности одного органического растворителя по сравнению с другим
стабилизировать фазу жидкого клатрата. Поэтому относительное содержание двух
растворителей (S1 и S2) в жидком клатрате и в объемной фазе должно различаться.
Количественно это различие выражают с помощью параметра селективности а, который
определяется уравнением A0.2).
a. tS'WPfe , <10.2)
I ibulk/i.^ hulk
где [S1]^ и [S2]LC - концентрация растворителей S1 и S2 в фазе жидкого клатрата,
10.4. Жидкиеклатраты о>*
Таблица 10.1. Разделение смесей растворителей состава 1:1
с помощью жидких клатратов
Исходное соединение
[NMe4][Al2Me6Cl]
[NMe4][Al2Me6NO3]
[NMe4][Al2Et6NO3]
[NPr4][Al2Me6I]
[NMe4][Al2Me6Cl]
[NPr4][Al2Me6I]
[NMe4][Al2Me6NO3]
[NMe4][Al2Et6NO3]
Смесь растворителей
Бензол/гексан
Бензол/гексан
Бензол/гексан
Бензол/гексен-1
Бензол/цикл ооктадиен-1,5
Бензол/дициклопентадиен
Бензол/мезитилен
Бензол/мезитилен
а
24.0
12.0
5.0
8.8
4.0
2.8
1.8
1.6
a ^'Ьм/А; и №2]bulk - концентрация растворителей S1 и S2 в их исходной смеси
в объеме.
При разделении смесей жидкие клатраты имеют определенные преимущества
перед твердыми аналогами (например, при образовании соединений включения
хофмановского типа, разд. 5.1.4) вследствие чрезвычайно быстрой кинетики
смешения, отсутствия временных затрат на процесс кристаллизации и легкости
разделения двух жидких фаз. Оказалось также возможным проводить разделение с
помощью жидких клатратов в режиме непрерывной экстракции. В табл. 10.1 приведены
значения а для некоторых бинарных смесей растворителей, которые разделяли с
помощью жидких клатратов.
Видно, что наиболее высокая степень разделения достигается в случае смеси
ароматических и алифатических растворителей. Влияние размера молекул
разделяемых соединений на эффективность процесса становится ясным при сравнении
результатов для гексана и гексена, наличие двойной связи в гексене-1 не сильно
увеличивает его включение в фазу жидкого клатрата. Наблюдается и влияние размеров
аниона: небольшие компактные анионы дают лучшее разделение.
Еще одна область применения жидких клатратов — ожижение угля. Уголь
содержит много низкомолекулярных и более тяжелых ароматических углеводородов.
Поскольку толуол имеет крайне малую эффективность при экстракции этих
соединений, контакт угля с жидким клатратом, полученным на основе толуола, при 50 °С
обеспечивает быстрое распределение 23% угольной массы в фазе жидкого клатрата.
Между углем и раствором жидкого клатрата не происходит никаких химических
реакций и соль, образующую жидкий клатрат, можно выделить, понижая
температуру. Этот процесс запатентован, его можно широко использовать в производстве
химического сырья и топлива при существенных изменениях в соотношении цен на
нефть и уголь.
•"-1 Atwood J.L, Liquid clathrates // Inclusion compounds / Ed. J.L. Atwood, J.E.D.
Davies, D.D.MacNicol. London: Academic Press, 1984. Vol.1. P. 375-405.
342 10 Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
1О.ф2 Ион оксония в химии жидких клатратов
Состав жидких клатратов далеко не ограничивается алкилалюминие-
выми комплексами. Вслед за первым синтезом таких соединений в этой области су-
прамолекулярной химии были начаты интенсивные исследования. Одна из
наиболее интересных систем на основе жидких клатратов получена при взаимодействии
макроциклического 18-краун-6 с газофазным НС1 в толуоле в присутствии
незначительного количества влаги. При этом образуется слой жидкого клатрата состава
(Н3О)-[18-краун-6][НС12]-3.8С6Н5Ме, из которого можно выделить кристаллы
состава [Н3О18-краун-6][НС12] A0.19). Реакция этого жидкого клатрата с двумя
молекулами воды дает еще один кристаллический продукт - (Н5О2)[Н3О18-краун-6]С12
A0.20), который также осаждается из этого клатратного слоя. Образованию
жидкого клатрата способствует большой объем катиона [Н3О18-краун-6]+ и небольшие
размеры галогенид-анионов, которые обеспечивают благоприятные я-катион-вза-
имодействия с толуолом и низкую энергию решетки. Оба эти соединения содержат
катион оксония Н3О+, удерживаемый внутри полости краун-эфира водородными
связями с атомами кислорода краун-эфира (рис. 10.19, а). В соединении A0.20)
большой ион оксония Н5О2 помещается вне полости. При изучении широкого
ряда родственных систем установили высокую селективность 18-краун-6 по
отношению к Н3О+. Катион Н3О+- первый член семейства катионов оксония H+(H2O)w и
в соответствии со своим эффективным ионным радиусом располагается между Na+
и К+. По размеру и топологии он комплементарен 18-краун-6 (симметрия третьего
порядка), и его включение реализуется путем образования трех бифуркационных
водородных связей типа О—Н-О. Этот ион полностью находится в полости
макроцикла. Тем не менее рассмотрение ряда комплексов, содержащих катион
(Н3О)[18-краун-6]+, показало, что атом кислорода выступает на 0.007-0.87 А из
плоскости краун-эфира и, следовательно, как эти комплексы, так и сам макроцикл
обладают определенной лабильностью. Интересно, что взаимодействия в твердых
структурах 18-краун-6, содержащих (непротонированную) воду, имеют совершенно
другой характер вследствие ограничений иной симметрии гостя, (^v, а также
большей длины водородных связей, образующихся без участия зарядов. Структуры
[18-краун-6]4Н2О, [18-краун-6]-8 Н2О и [18-краун-6]12 Н2О содержат одинаковые
краун-эфиры с симметрией D3d, взаимодействующие с помощью водородных
связей с четырьмя молекулами воды, две из которых расположены над плоскостью
краун-эфира, а две - под ней, рис. 10.19, б (A. Albert and D. Mootz, 1997).
Совместимость Н3О+ с [18]краун-6 привела к заключению о том, что
кристаллизация комплексов краун-эфиров и криптандов из кислой среды жидкого клатрата
могла бы стать прямым методом систематического выделения различных форм
иона оксония согласно размеру краун-эфира и топологическому соответствию.
Образование жидкого клатрата наблюдали при пробулькивании газообразного НС1 через
толуольные растворы большого числа краун-эфиров в присутствии следовых
количеств влаги. Кристаллизацию в этих растворах можно было вызвать прибавлением
различных металлсодержащих соединений, которые при взаимодействии с
избытком хлорида водорода давали бы большие неорганические галогенид-анионы. В
частности, карбонилы переходных металле реагируют с образованием анионов типа
10.4. Жидкие нлатраты
(а)
843
Рис. 10.19. Кристаллические структуры: (а) катиона [Н3О18-краун-6]+ в A0.19)
и (б) [18-краун-6]4Н2О
[М(СОMС1]~, [М(СОLС13]-, [М(О)С14(Н2О)]" и т.п. (М = Mo, W). Таким образом
были выделены комплексы ионов оксония с разнообразными краун-эфирами. При
таком соответствии крауна и оксониевого катиона друг другу по размеру и
симметрии наблюдали некоторую селективность для отдельных ионов оксония, в
особенности у краун-эфиров больших размеров. Так, 21-краун-7 (рис. 10.20) и дибензо-24-
краун-8 капсулируют H5OJ, а дибензо-30-краун-10 включает два иона Н3О+.
Последнее соединение содержит также молекулу толуола, что указывает на сущест-
344 Ю- Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
Рис. 10.20. Образование комплекса (Н5О2)[21-краун-7]+ в среде жидкого клатрата
(по Junk P.C. and AtwoodJ.L, 1997)
вование взаимодействий л-катион. Краун-эфиры меньших размеров, такие, как
15-краун-5 и 12-краун-4, ведут себя неоднозначно в зависимости от природы
аниона. Ионы оксония, интеркалированные между молекулами краун-эфиров,
отделяют их друг от друга, поэтому тип оксониевого иона зависит от характера
кристаллической упаковки в системе. Таким путем были выделены частицы Н7О^ и г^О^.
Наконец, стоит отметить, что не только краун-эфиры образуют большие
катионы, которые могут генерировать фазы жидких клатратов. Глубоко окрашенный
синий жидкий клатрат образуется при взаимодействии [2.2.2]криптанда с
газообразным НС1 в толуоле в присутствии СоС12. При длительном хранении было выделено
кристаллическое соединение {[2.2.2]криптанд-2Н+[СоС14]2~}2С6Н5Ме. Этот
комплекс содержит линейные группировки типа анион-катион-арен--катион-анион,
в которых л—катион-стэкинг-взаимодействия обеспечивают изоляцию протони-
рованных криптандных катионов друг от друга (рис. 10.21). Справедливость вывода
о реализации такого рода взаимодействий вытекает из сжатой структуры этого
криптанда, в которой расстояние N---N между головными атомами составляет
6.36 А. Это расстояние значительно короче, чем в таком же комплексе с хлорид-
анионом F.87 А), который не содержит толуол.
*"» Atwood J.L, Bott S. G., Junk P. C, May M. T. Anionic coordination complexes for Mo
and W which crystallise from liquid clathrate media with oxonium ion-crown ether
cations // J.Coord.Chem. 1996. Vol. 37. P. 89-105.
Учебные задания
845
Рис. 10.21. Взаимодействия л-катион в {[2.2.2]криптанд-2Н+[СоС14]2~}2С6Н5Ме
(по MacGillivray L.R. and AtwoodJ.L, 1996)
Учебные задания
10.1. Исходя из Вашего понимания нековалентных взаимодействий,
рассмотренных в гл. 1, предложите механизм образования монослоев на поверхности
воды, мицелл и других упорядоченных структур в жидкости. Как это
соотносится с образованием гидрофобных молекулярных комплексов циклофанов типа
«хозяин—гость» (см. гл. 5)?
10.2. Дифильное соединение на основе иона аммония имеет площадь поперечного
сечения 5 А2 *. Вычислите площадь поверхности, занимаемой 210~5 моль
этого соединения. Сколько молей потребуется для образования плотного
монослоя в ванне Ленгмюра площадью 10 см2? Предположите, как будет вести
себя система, если количество этого ПАВ увеличить по сравнению с
рассчитанным (а) в 2 раза и (б) в 100 раз.
10.3. Объясните, почему можно наблюдать более чем одну жидкокристаллическую
фазу для лиотропной системы. Почему при нагревании термотропного
жидкого кристалла нематическая фаза обычно предшествует образованию
изотропного расплава?
Реальная площадь такого дифильного соединения не может быть меньше 17 А .
{Примеч. ред. перевода.)
$46
10. Жидкие поверхности раздела, жидкие кристаллы и жидкие клатраты
Литература
Albert, 1997 Albert A., Mootz D. Formation and crystal structures of the hydrates of 18-crown-6
// Z. Naturforsch. B. 1997. Vol. 52. P. 615.
Cladis, 1984 Cladis P.E., Pieranski P., Joanicot M. Elasticity of blue phase-I of cholesteric
liquid-crystals // Phys. Rev. Lett. 1984. Vol. 52. P. 542.
MacGillivray, 1996 MacGillivray L.R., AtwoodJ.L. Structural reorganization of the doubly protonated
[2.2.2]cryptand through cation—я and charge-charge interactions: Synthesis and
structure of its [CoCl4]0.5C6H5CH3 salt // Angew. Chem., Int. Ed. Engl. 1996.
Vol. 35. P. 1828.
Junk, 1997 Junk PC, AtwoodJ.L. Synthesis and crystal structures of oxonium ion complexes
of 21-crown-7 and dibenzo-30-crown-10 // J. Chem. Soc, Dalton Trans. 1997.
P. 4393.
Sackmann, 1969 Sackmann H., Demus D. Properties and structures of the thermotropic liquid
crystal state // Forstschr. Chem. Forsch. 1969. Vol. 12. P. 349.
Sackmann, 1973 Sackmann H., Demus D. Problems of polymorphism in liquid crystals // Mol.
Cryst. Liq. Cryst. 1973. Vol. 21. P. 239.
Ulman, 1996 Ulman A. Formation and structure of self-assembled monolayers // Chem. Rev.
1996. Vol. 96. P. 1533.
Vogtle, 1991 Vbgtle F. Supramolecular chemistry. Chichester: J. Wiley & Sons, 1991.
Предметный указатель
Автокатализ 687
Агонист 100
Агостическое взаимодействие 548
Аденозиндифосфат (ADP) 65, 66,
240, 252, 256, 782, 784, 785
Аденозинмонофосфат (AMP) 779,
782, 784
Аденозинтрифосфат (АТР) 65, 66,
240, 252, 256, 778, 779,
782-786
депротонированная форма
784
переходное состояние,
реакция дезаминирования 773
свойства 65, 66
синтез 84, 86, 785, 786
ЯМР-спектр 31Р
Азакоранды
аналоги 141
антрацензамещенный 719
макроциклы 252-260
Азакрауны 129-135
Азид-анион, криптаты бис(трена)
250
Азобензол, переключаемое
связывание 737-739
Активный центр 770
Алкалиды 206-209
Алкиламмония катионы
свободные энергии
связывания 194
связывание криптофанами
191,415
Алмазоподобные решетки
529-534
Аминные лиганды 204
Аминокислоты белков 104-106,
метаболизм, метилмалонил-
СоА-мутаза 97, 98
D-Аминоэфиры, трансацилиро-
вание 788
Аммония катионы 155—157,
191-194
связывание азакорандами,
геометрия типа «насест» 191-193
связывание корандами
191-193
связывание лариат-эфирами,
стерические ограничения 155
связывание трехмерными
хозяевами 194
Анализ набора графов,
водородные связи 501-504
Аналоги гема
биологические имитаторы
797-808
модели, не содержащие железа
807,808
модели связывания и
транспорта кислорода 797—803
модели цитохромов Р-450
804-808
Анаэробные организмы 89
8-Анилино-1 -нафталинсульфонат
(ANS) 47,260,396
Анион BF;j~, кристаллическая
структура комплекса 289
Анионы
нуклеофильность 146
свойства 238, 239
«48
Предметный указатель
Антенное устройство светоакку-
мулирующее 82, 83, 760, 761,
хлорофилл а 80—82
каскад переносов энергии 82
Антикрауны 246, 282-286
комплекс ртуть-12-краун-4
на основе карборана с С1~ 285
сэндвичный полимер
комплекса с Вг~ 283
Антрацен, система
краун—антрацен 719
Апоферменты 97, 809
Аргинин 242,
«солевой мостик», 243
Ароматические кольца
я-я-взаимодействия 55, 56
эффекты кольцевого тока 197
Ароматические системы, правило
Хюккеля 78, 79
Ароматические углеводороды
с конденсированными
кольцами (FAH) 551
Архимедовы тела 607, 621
Асимметрическая ячейка 513, 556
Атропоизомеры 800
Ацетилхолин (АСН) 100,101
Аэробные организмы 89
АТРазы 65, 75, 76
имитация корандами 782—786
свойства 65
транспортное действие 65—69
ферментативное действие 75
Na+/K+-ATPa3a 65-68, 75-77,
782
Бакелит 210
Бакминстерфуллерен 347,
430-438, 619-621, см. также
Фуллерены
редокс-чувствительное
устройство 435
темплат 619
Бактериальные анионтранспорт-
ные белки 241, 242
белки, связывающие сульфаты
241,242
белки, связывающие фосфаты
241
Бактериофеофетин 84
Бактериохлорофилл а 80, 81
Барбитураты 363, 364, 402-404
Бенгальский розовый,
ассоциация дендример—DAB 760
Бенз-1,2-ин 428,429
Бензол
4,4 '-диметоксиазоксибензол
826
квадрупольный момент,
катион—тс-взаимодействия 53-55
кристаллическая структура 56
разделение смесей с помощью
жидких клатратов 841
я—71-стэкинг-взаимодействия, ?
55,56 i
6:1-цикламер циклогександи-
онбензол 495
л-Бензохинон, связывание
404-406
Бибрахиальные лариат-эфиры
(BiBLE), 132, 141,224,
одновременное образование
четырех связей 132
Бидентатные лиганды 36, 135
Биметаллические системы,
фотохимия 702-704
Биологические модели,
структурная и функциональная
768-770
Предметный указатель
Биологические системы 64—120
гемоглобин, связывание
и транспорт кислорода 88—96
ДНК 101-115
катионы щелочных металлов
65-76
кофермент (витамин) В12
96-99, 809, 810, см. Цианоко-
баламин
нейротрансмиттеры и гормоны
99-101
порфириновые и тетрапир-
рольные макроциклы 78, 79
фотосинтез 79—88
Биомиметика 767—810
Биомиметические структуры
562-567
Биоминералы 562—564
Биохимическая самосборка
115-119,580-584
вирус табачной мозаики 580,
581
ковалентная модификация
583, 584
строгая самосборка 581—583
2,2'-Бипиридил (Ыру) 50, 112,
660,704-710,712,761,763
4,4'-Бипиридил (Ыру) 593
Бислои 816, 817, 819, см. также
Мембраны биологических
клеток
Бис(трен) 250-252
Благородные газы 57
Большие молекулы, см. также
Супрамолекулярные частицы
696, 697
Бомбикол 768
л-т/?ет-Бутилкаликс[4]арен 57, 58
катализаторы межфазного
переноса 217-219
комплекс тетраэтилового
эфира с Na+ 213
конформеры 212
растворимость 217
Валиномицин 70, 71
Взаимодействия Ван-дер-Ваальса
57,246,418
дисперсионные (силы
Лондона) 57
обменно-отталкивающие 57
Взаимодействия, типы
агостические 548
водородные связи 51-53,
494-501
с гидридами металлов
549-551
с лариат-эфирами, стеричес-
кие ограничения 157
с металлами 548, 549
диполь-дипольные 51,321
ион-дипольные 50
ион-ионные 48, 49
катион-я- 53-55
координационные 286—290
координационно-клатратные
516
межмолекулярные 494, см.
также Взаимодействия Ван-
дер-Ваальса
немолекулярные 48—60
я—71-стэкинг- 55-57,
катенаны 637-648
супрамолекулярные
взаимодействия 48—60
Виагра 117-119
Витамин В12 96-99, 809, 810, см.
Цианокобаламин
850
Предметный указатель
модели, биологические
имитаторы 809,810
производные 96
Вода
декамер 605, 606
кислотность 795
окисление до кислорода,
катализированное марганцем
ж со
оЭ—о о
равновесие, описывающее
изменение жесткости воды
564
Водородные связи 51,183,276,
277, 402-407, 495-504,
535-551,813,832
амиды 535, 536
анализ набора графов 501—594
бифуркационные 499
ВДНК51-53, 107
в молекулярных устройствах
711,712
включение гостя, циклофан
402-407
димеры карбоновых кислот
52, 535
жидкие кристаллы 832
закрытые комплексы,
самосборка 613-628
инженерия кристаллов
535-542
комплекс краун-эфира с нит-
рометаном 54
мотивы «ограда курятника» и
«лестница» 540, 541
мотивы распознавания 724,
725
нейтральные хозяева 276, 277
нетропсин 114
определение мотива 502
свойства 52
с металлами и гидридами
металлов 548-551
взаимодействие с гидридами
металлов 549—551
прямое взаимодействие
с металлами 548, 549
с монооксидом углерода
542-544
сильные 51—53,498
системы М—Н—Н—X,
параметры 549-551
слабые 51-53, 500,501,
544-547
смешанные кристаллы аминов
с аминами 541, 542
смешанные кристаллы
спиртов и аминов 540, 541
спирали 676—679
спаривание оснований по Уот-
сону и Крику 52, 106, 167
спаривание оснований по Хуг-
стену 107
спирты 540
средние 51-53,498-500
темплатные эффекты 586
ферроценильный редокс-ак-
тивный хозяин 724
фосфаты и сульфаты 241, 242
КНС2О4 53
C-H-N 525, 526
С-Н-0 495, 543
О-Н-0 495,525
Х-Н-Х 495,496
Вольтамперометрия циклическая
267-269
Вольфрамат, модель для тетраэд-
рических анионов 243
Ворота ионного канала 69, 220
Времена релаксации ЯМР
хозяина и гостя 196
Предметный указатель
851
Вспомогательная связь 648-659
Вторая гармоника,
индуцированная электрическим полем
(EFISH) 750
Галогенид-анионы
константы связывания 270
координационные числа 249,
250
Галогенуглеводороды, связывание
криптофанами 411—414
Гексафторид серы 333
Гексациклен 141,142,252,253
Геликанд двухдоменный,
потенциальные комплексы 662
Геликаты 289, 660-679,
[4 + 4]геликаты 665-667,
кристаллическая структура
666
[6 + 6]геликаты 667-669,
кристаллическая структура
669
наноциклы 675
номенклатура 660
полиядерные 683, 684
самораспознавание и
позитивная кооперативность 669—674
синтетические аспекты 663,
664
спирали, образованные
водородными связями 676-679
темплаты и самосборка
660-669
Гем 78, 89
комплексы 78
модели Полинга и Вайсса 91,
92
тетрапиррольные макроциклы
78,79
Гемоглобин 89
аллостерический эффект 95
кооперативность, управляемая
белком 804
миоглобин 95
насыщение кислородом 95
связывание и транспорт
кислорода 88-96
сродство к кислороду, влияние
аксиальных лигандов 804
структура 89
Гемоцианин, модели,
биологические имитаторы 793—795
Генерация второй гармоники
(ГВГ) 746
Генетически модифицированные
(GM) продукты 108
Генетический код 105,106,108
Генная инженерия 108, 109
Геометрия типа
«гнездо» 93, 177, 194,213,262
«капсула» 33, 177
«насест» 33, 177, 178, 191, 194,
262,264,283,358,615
Гербициды 204—206
Гетерогенное разбавление 392
Гетерокраун-эфиры 181-184
Гетерокриптанды 184, 185,
константы связывания катионов
Ag+, К+иТ1+ 184
Гибридные хозяева катионов 139
Гигантские самособирающиеся
капсулы 619-625
«Гидридная губка» 278, 279
другие льюисовские
кислотные хелаты 278—281
«протонная губка» 279
хелатное поведение аниона
279
Гидриды металлов 549—551
852
Предметный указатель
Гидроксиапатит, биоосаждение
565
Гидрофобные эффекты 58—60,
354,399,400,406,816
связывание органических
гостей в водном растворе 59
энтальпийные и энтропийные
59
Гидрохинон 332—336, 433
структура полости 334
фенол и соединение Дианина,
стратегия шести хозяев
332-336
Гиперполяризуемость 736
Гликолурил 367—369, 615—618
Глины 311-315,359
Глиоксалазы, их имитация 781
сс-Глицин 503,506-508
кристаллическая упаковка 506
наборы графов 503
пирамидальные кристаллы,
монослои №-пальмитоил-
(ад-лизина 508
Голоферменты 97
Гомохиральные стопки 329, 330
Гормоны 99, 100
Гости 320
динамика обмена гостями
198-200
связывание в полярных и
неполярных средах 403
стадия, определяемая гостем
(GDS) 422
темплатирование 603
Гость, определение 28, 29, 244
График Вант- Гоффа 412
График Джоба 46
Графит 312,314-317
Гуанидиния катион 193, 242,
262-265
рецепторы на основе
гуанидиния 262-265
комплекс метилгуанидиния
с дигидрофосфатом 262,
кристаллическая
структура 263
хиральный хозяин, синтез
263
центр связывания анионов
242
связывание 193
Гуанозинтрифосфат (GTP)
117-119
Дансиламида производные,
сенсоры на цинк, флуорофоры
721-723
Дендримеры 754—763
дивергентный синтез 756, 757
итерационное получение 754,
755
конвергентный синтез 756—758
фотохимические устройства
на основе дендримеров
760-763
химия хозяин—гость для
дендримеров 759, 760
Диаграмма Скэтчарда 672, 673
1,4-Диаминобутил (DAB) 760
Диаммония катионы,
распознавание 193, 195
Диарилмочевины 521,522
Диастереомерия 631, 652,
тройной узел 631
Диатомеи, микроскелет 565
1,2-Дигидроксибензол 673
Дигидрофосфат калия 529
ос,со-Дикарбоновые кислоты
гости 256
димеризация 532, 535
Предметный указатель
853
распознавание в соответствии
с длиной 257
Дикват, связывание 204, 205, 634
Димеры типа «мяч для софтбола»
615-618
капсулирование гостя 616, 617
присоединение по Дильсу—Аль-
деру, катализируемое
хозяином 617
самосборка 615—618
Димеры типа «теннисный мячик»
613-616
кристаллическая структура 614
самосборка 613—616
связывание гостя, константы
616
Диметилртуть 336
Диметилформамид (DMF) 423
механизм обмена метильных
групп 199
Динамика обмена гостями 198
Динамическое спаривание
хозяин—гость 196
Дисклинации 825
Дисперсионные взаимодействия
при анионном связывании
56, 244
Дистамицин А 114
Дитопные рецепторы 195,196,
256, 257, 287
линейное молекулярное
распознавание катионов
диаммония 196
Дифенилгликолурил 613-618
Дифенилметан 260, 396-401
Дифильные рецепторы 68, 203,
204, 815, 816, см.
Поверхностно-активные вещества
Дифрактометр, прибор
с зарядовой связью (CCD) 73
Дифракция нейтронов 497, 544
Дифференциальная
сканирующая калориметрия (DSC),
соединения включения
349-353
Диффузионное рентгеновское
рассеяние 320, 325
ДНК 101-115
водородные связи 51
двойная спираль 51, 102
интеркаляция 112—115
направленный мутагенез 108,
109
спаривание оснований 52, 106,
107, 578
самосборка 581—583
связывание 110—115
структура и функции 101—107
полимеразная цепная реакция
(PCR) 109, 110
чувствительность комплексов
114
Домены связывающие 660
Железа катионы Fe2+ и Fe3+
89-96, 228-232, см. также
Гем, Гемоглобин, Миоглобин,
Порфирины
гексацианоферрат(Н)-анион
Fe(CNN]4~, суперкомплексы
253, 254
железосодержащие лиганды,
см. Сидерофоры
оксиды как биоминералы 563
хелатные Ре3+-лиганды,
величины рЛ/ 231
Жесткие ионы металлов 179
Жидкие клатраты 837-845
ион оксония в химии жидких
клатратов 842—845
854
Предметный указатель
открытие и свойства 837—841
Жидкие кристаллы 677, 814,
820-837
дизайн 831-833
жидкокристаллические
дисплеи (LCD) 835-837,
закрученные нематические ячейки
(TNC) 836
жидкокристаллические
полимеры 834,835
классификация 823, 824
применение 835—837
природа и структура 820—831
Жидкие фазы 813—846,
поверхностно-активные вещества и
упорядочение на поверхности
раздела 814-820
Жидкокристаллические фазы
голубые 829
дискотические 824,831
лиотропные 824, 830
нематические 824, 826, 828,
830,834
смектические 824-827, 829,
834
столбчатая 830, 831
сэндиковая 834
трубчатые 823, 825
холестерические 824, 825, 828,
829, 834, 835
Жидкостно-мозаичная модель
биологической клетки 819,
820
Жуки,окраска 835
Закрученные нематические
ячейки (TNC) 836
Закрытые комплексы 613-628
гигантские самособирающиеся
капсулы 619-625
димеры типа «теннисный
мячик» и «мяч для софтбола»
613-618
розетки 625-628
самосборка с помощью
водородных связей 613-618
«Замок—ключ», модель Фишера,
аналогия 34, 35, 767, 768
Змеи, спиральность 661
Изменение жесткости воды 564
Изменение pKfl макроцикла 255
Изоляция центра при росте денд-
римера 759
Изомерия, обусловленная
растяжением связей 570-572
Изомеры электронные 702, 709
Имидазолия катион, комплексо-
образование 193
Имитаторы трансацилаз
786-789
Индукция хиральности в
кристаллах 508-510
Индуцированное соответствие
243, 603, 768, 773, 774, см.
«Замок—ключ» 768
Инертные комплексы 600—602
Инженерия (конструирование)
кристаллов 491—573
алмазоподобные решетки
529-534
биомиметические структуры
562-567
водородные связи 494—501,
с монооксидом углерода
542-544
дифракция нейтронов от
монокристалла 497
история 491-494
Предметный указатель
855
Кембриджский банк
структурных данных 525-528
координационные полимеры
558-561
межмолекулярные
взаимодействия 494
«неправильно» подобранные
партнеры 556, 557
определение 493
предсказание структуры
кристаллов 517—524
прочие взаимодействия 554,
555
разрушение кристалла 514,
515
самосборка 491,492
синтез 492,493
соединения включения типа
«песочные часы» 567—570
стратегии дизайна 515—517
п—я-стэкинг-взаимодействия
553, 554
хозяева типа «колесо—ось»
555, 556
Инсулин 583,584,648
Интеркаляторы 112, сродство
кДНКПЗ
Интеркаляты 311—317,
классическая модель и модель Дау-
маса-Геролда 313,314
Информация и сигналы 716-727
нервный импульс 67
семиохимия 716—727
флуорофоры 721—724
фотохимические сенсоры
717-721
электрохимические сенсоры
724-727
Иодид-анион, связывание тетра-
эдрическими рецепторами
248
Ион Ru(II)-Ru(III) Крейтца-
Таубе 702
Ионизация электрораспылением
(ESI) 756
Ионная сила 273, 274
Ионные каналы 67-69
Ионофоры 68, 69
трансмембранные 126, 127
транспортные свойства 132
хромоионофор 128
Ион-селективные полевые
транзисторы (ISFET) 725
Кавиплекс 354
Кавитанды 32, 354-386, 408
определение 32, 354
синтез на основе цикловера-
трилена (CTV) по Краму 408
строительные блоки 308,343,
354,355
Кавитаты 33,354
динамика обмена гостей
364-366
определение 32,354
Калид 207
Калия катион К+
биохимия 65—76
мембранный транспорт 69—76
связывание, константы
172-175
гибидными каликсаренами
219-223
лариат-эфирами 155
содержание в биологических
системах 66
темплатный эффект для мак-
роциклических соединений
161, 162, 164
флуоресцентный сенсор 719
856
Предметный указатель
Калия соли
реакция с бензилхлоридом
вацетонитриле 147,148
сульфат калия,
кристаллические формы 570
Каликсарены 209—223, 271,
354-364,431,432
п-трет-бутил каликсарены
57, 58, 212, 217, 218, конфор-
меры 212
имитаторы ферментов 212
история 210—212
каликс[4]арен, структура
234
каликс[5]арен 212, 724,
электрохимический сенсор 724
каликс[4]резорцинарен 619
кристаллические структуры
комплексов 432
я-метилкаликс[4]арен, синтез
по Хайесу и Хантеру 210
определение комплекса 33, 34
равновесие межфазного
переноса катионов 217—219
внутриполостное
связывание Cs+ 218, 219
связывание катионов
металлов 218
разделение смеси С60 и С70
430
связывание анионов 270, 272,
255, 256
связывание катионов 213—217
конформационные аспекты
включения 215
туннелирование Ag+ 216
Ag+-7i-комплекс 216
связывание катионов
гибридными каликсаренами
219-223
макротрициклическая
бис(каликсареновая
клетка) 219-221
«перекачка» металла
«молекулярным шприцем»
222
процессы обмена катионом
металла, внутри-
и межмолекулярные 221
связывание К+ 219-222
Калике[4]пиррол 276
Калориметрическое титрование
47
Кальция катион Са2+
биоминералы 563—565
переносчик Са2+, поданды
126
сенсоры, флуорофоры 721
Карбид молибдена 149
Карбоангидраза (СА) 721, 795
Карбоновые кислоты
димеризация 535, 536
водородные связи 536
нуль-, одно- и двухмерные
сетки 532
кристаллические структуры
531-533,535,536
продукты совместной
кристаллизации с 2-аминопириди-
ном, правила Эттер,
предсказание структуры 523
Карбоксипептидаза А, рецепторы
анионов 243, 244
Карбонилы, диполь-дипольное
взаимодействие 51
Карбоплатин 110,111
Каровиологены 727, 728
Карцеплексы 417,419,423-426,
полукарцеплексы 417,420,
421
Предметный указатель
857
Карцеранды 417-423
карцерия 425, 426
определение и синтез
417-421
полукарцеранды 417,421,
617,619
реакции включения 426—429
связывание 423, 424
темплатные эффекты в синтезе
карцерандов 421—423
Каскадные молекулы, см. Денд-
римеры
Каталановы тела 621
Катализ краун-эфирами 145, 146
Катализ супрамолекулярный
685-690
Катализируемые и некатализиру-
емые реакции 773
Каталитические и
самовоспроизводящиеся системы 685—690
Каталитический крекинг 311
Катапинанды 237-240, 244, 245,
286
определение 237
связывание анионов 245
Катапинаты 248, 249
Катенаны (катенанды) 163, 206,
404, 598, 628-659
бис([2]катенан), синтез 647,
648
динамические свойства 638,
639
[2]катенан 629-632,637,641,
643, 647
вспомогательная связь,
трилистный узел 648—650,
680
выход 640
кристаллические структуры
639, 658
направленный синтез
649-654
темплатный синтез 638
электрохимически
контролируемые
перемещения циклов 745
ЯМР-спектр 'Н656
[3]катенан 629,641-643
изомеры 649-651
синтез 648—651
синтез под высоким
давлением 642
[4]катенан 641,643,645
[5]катенан, первый синтез 641,
643-646
кинетические и
термодинамические эффекты 587
макроциклизация в условиях
высокого разбавления 653,
655
молекулярные машины
740-745
номенклатура 629
олимпиадан 645,646
определение 628, 629
принцип вспомогательной
связи в синтезе катенанов
648-659
самосборка 598
синтез 630,632,648-659
статистический подход 632,
633
стерически управляемая
циклизация 651
я—я-стэкинг-взаимодействия
637-639
топологическая изомерия 631
трансляционная изомерия
743
хиральность 631
858
Предметный указатель
Катенаты 628, 629, 655-659
кристаллическая структура
658
синтез 656—659
Катион бипиридилдиамминпла-
тины(П), кристаллическая
структура 205
Катион—л-взаимодействия
53-55, 100,214,218,224,839,
845, схематическое
изображение, квадрупольный момент
бензола 55
Катионы, свойства 239
Катионы металлов (кислоты
Льюиса), координационные
взаимодействия 286—290
Катионы щелочных металлов
65-76, 179
константы связывания 181
мембранные потенциалы
65-68
мембранный транспорт
68-76
родопсин 76, 77
размер и свойства 179
Кинетика
связывание катионов 130,177
связывание нейтрального
гостя 364-366, 422
Кинетическая селективность
41-48, 149-159
Кинетическая стабильность
комплекса 600
Кинетический темплатный
эффект 131, см. Темплатный
эффект
Кислород 88-96,793-795,
797-804
аэробные и анаэробные
организмы 88, 89
модели связывания и
транспорта, аналоги гема, порфи-
рины 797-804
связывание и транспорт
гемоглобином 88—96
Кислотность С—Н-кислот,
корреляция с длиной водородной
связи С-Н-0 545
я-Кислоты 186, 187, 223-227
Классификация соединений
хозяин—гость 32-34
кавитат и клатрат 32, 33
нейтральные хозяева 33
пространственное
соотношение между гостем и хозяином
33
Клатранды, определение 32
Клатратный хлоргидрат 31,295,
296
Клатраты 32, 34, 295,
применение 295
Клатраты жидкие 837—845
Клатраты мочевины 318—324
алканоны и алкандионы
321-323
гости 320
полимеризация при
включении 323
соразмерные и несоразмерные
структуры 318—3 24
структура 318, 319
Клатраты неорганические
твердофазные 296-318
история 296
клатратные гидраты 296-302
образование 296, 297,
проблемы в газовой
промышленности 301, 302
применение 301, 302
свойства 300, 301
Предметный указатель
859
сравнение размеров гостей
и полостей, занятых, как
в простых гидратах 299
структуры 297-299
клатраты Хофмана и Вернера
317,318
твердые слоистые материалы
и интеркаляты 311—317
графитовые интеркаляты
315-317
классическая модель
и модель Даумаса— Геролда
313,314
характеристики и классы
311,312
цеолиты 303—311
Клатраты твердых органических
хозяев 318-353
гидрохинон, фенол и
соединение Дианина 332-336
канальные клатраты 324-332
клатраты мочевины 318—324
пергидротрифенилен 329—332
спиральные тубуланды
327-329
тетрафенилен 348,349
тримезиновая кислота (ТМА)
324-327
гексагональное
расположение молекул 326
структура слоя моногидрата
324,325
три-о-тимотид 337—343
циклотривератрилен (CTV)
343-348
«Книга кельтов» 679
Кобаламины 78, 96, 97
тетрапиррольные макроциклы
78
цианокобаламин, см.
Витамин В12, модели,
биологические имитаторы 809,810
Кобальтоцений 266
Кобальтпорфириновый комплекс
797,798
Ковалентные связи
деформационное
укорачивание 496
самосборка с ковалентной
модификацией 583, 584
Коелентеранды, определение 225
Комплекс карбида молибдена
148, 149
Комплекс кислородвыделяющий
(ОЕС) 85-88
Комплекс «матрешка» 384
Комплекс переходного металла,
молекулярно-орбитальное
описание 700
Комплекс PF^, трехкатионный
хозяин подандного типа 272
Комплексы из лигандов типа
«жесткий стержень» 609
Комплексы меди 39
Комплексы типа «рейки»,
«лестницы» и «решетки» 609—612
Комплементарность 40,
169-176, 193
определение 170
предорганизация 169—176
связывание хозяин—гость
40-41
Комплементарные
(дополняющие) цвета 81
Конденсация аминов с
альдегидами 391
«Коническая поправка» 499, 500
Конкин 370-372
Константы ассоциации, см.
Константы связывания
860
Предметный указатель
Константы образования, см.
Константы связывания
Константы связывания 42—48
галогенид-анионы,
зависимость от длины спейсера 270
гетерокриптанды 184
двухмерные хозяева,
связывание анионов 256, 257
жесткие ионы металлов 181
измерение 42—48
катионы
Ag+ 184
Ва2+ 158, 181
К4" 181, 184
РЬ2+ 181
Т1+ 181, 184
краун-эфиры 129, 151
криптанды 158, 184
лариат-эфиры 129, 156, 157
мягкие ионы металлов 181
образование суперкомплексов
255
поданды 129
связывание 43
«Конструирование вниз» 577
Конструирование хозяина 60, 61
Конструирование хозяина для
анионов 244-247
антикрауны 282-286
биологические рецепторы
анионов 240-244
«гидридная губка» и другие
льюисовские кислотные хела-
ты 278-282
константы связывания,
длина спейсера
дикарбокси-гостя 256, 257
[Рс1С14]2~-комплекс
соединения H10[30]aH-N{jJ+
254
расширенный порфирино-
вый хозяин с капсулиро-
ванным фторид-ионом 258
суперкомплексы азакоран-
дов 255
изменение рН 247—261
двухмерные хозяева
252-260
селективность формы
249-252
тетраэдрические рецепторы
247—249, криптанд
«футбольный мяч» и его
криптат с хлорид-анионом
249
циклофановые хозяева
260,261
концепции 244-247
координационные
взаимодействия 286-290
металлоорганические
рецепторы 265-273
нейтральные рецепторы 273,
274
основность Льюиса анионов
246
рецепторы на основе гуаниди-
ния 262—265
Конформации каликсаренов
1,2- и 1,3-альтернаты
(чередующиеся) 211,212,214,216,
277
коническая (конус) 212, 214,
216
частично коническая
(частичный конус) 212, 214, 216
Концентрационный градиент
67,68
Концепция жесткой концевой
группы 128, 392
Предметный указатель
861
Кооперативность (модель),
влияние аксиальных лигандов
на сродство к О2 803, 804
Кооперативность
позитивная 669—674
негативная 673
Координационная геометрия
анионов 287
Координационная химия 34, 35
«Координационно-клатратныи»
подход Вебера 516
Координационное число 246, 247
Координационные
взаимодействия 286-290
комплекс с Н2РО^ 288
координационная геометрия
анионов 287
Координационные полимеры,
инженерия кристаллов
558-562
Координационные (дативные)
связи 50
Координационные соединения
металлические ансамбли
609-612
«молекулярные квадраты»
и «молекулярные коробки»
597-608
принципы дизайна 593
самосборка 592—612
супрамолекулярный куб
593-596
Коранды 40,140
донорные центры кислорода
203
как имитаторы АТРаз
782-786
макроциклический эффект 38
модель активности АТРаз 782
номенклатура 140-142
определение 140
структуры 41, 141
хиральные 786—789
хиральный барьер 200
хозяева, свободные энергии
связывания ионов аммония
192
хозяева, связывающие
катионы, как имитаторы трансаци-
лаз 786-791
Кораплекс 142
Кораты 142
«Королевский разрез» 661,662
Корреляция силы и длины
водородной связи 52
Корриновое кольцо, тетрапир-
рольные макроциклы 78, 79,
96, 809
Коферменты 96,97,781
В12 96,97
модели, биологические
имитаторы 809, 810
производные 96, 97
F450, тетрапиррольные
макроциклы 78, 79
Краун-эфиры 121—126
аза-18-краун-6, комплекс с ни-
трометаном 54
азотсодержащие аналоги 252
ациклические аналоги (подан-
ды) 126
бензо-15-краун-5 148, 149
бензо-18-краун-6, темплатный
синтез 163
в химии жидких клатратов
842-845
гетерокраун-эфиры 181—184
диаза-18-краун-6, синтез
методом высокого разбавления
157, 166
862
Предметный указатель
диаметры полостей, сравнение
с диаметрами катионов
металлов 152
дибензо-18-краун-6 161, синтез
122, 125
дибензо-24-краун-8 153, 154,
843, 844
дибензо-30-краун-10 123, 153,
154, паракват (метилвиоло-
ген) 204
дициклогексил-14-краун-4
152
дициклогексил-18-краун-6
123, изомеры 125, 126, 143
изменение константы
связывания
как функция радиуса
катиона металла 151
как функция размера
кольца краун-эфира 151
капсулирование катиона
металла макроциклом 154
12-краун-4 183
15-краун-5 123, 183
18-краун-6 122-126, 141, 143,
148, 150, 151, 153, 160, 162,
183
в органическом
растворителе и гидрофильной среде
144
ион оксония 842-844
комплекс с К+ 143, 160
комплекс с Ag+ 182
21-краун-7 123,843,844
30-краун-10 154
межфазный катализ 144, 145
модели Кори—Полинга-Кол-
туна (СРК-модели) 136,
137, 194
номенклатура 140—142
окисление органических
субстратов КМпО4 146, 147
определение 122,
классификация комплексов 33
открытие 121 — 124
полимерные продукты
161-163
применение 124
растворимость 143, 144
ртуть- 12-краун-4 на основе
карборана 285
связывание иона оксония
842-845
связывание катионов, плато
селективности 150,151
связывание нитрометана 54
селективность связывания
катионов 149-154
синтез 122, 124-126, 161, 162
система краун—антрацен 719
скорость реакции 148
структуры 123
темплатный эффект 161,162,
164
торсионные углы 144
триаза-18-краун-6 247
трибензо-21-краун-7 154
угол захвата л и ганда 152,153
Крахмал, получение циклодекст-
ринов 378,379
Криоскопия 210
Криптанды 132-136, 157-159,
173, 176,248,249
гетерокриптанды, константы
связывания Ag+, K+ и Т1+ 184
константы связывания
катионов
щелочноземельных металлов 158
катионов щелочных
металлов 158
Предметный указатель
863
криптофикс 132
макробициклический эффект
159
макротрициклические крип-
танды 247—249
макроциклический эффект
159
номенклатура 140—142
определение 140
переключаемое связывание
737
селективность связывания
катионов 157—159
сепулькраты и саркофагины
135, 136
синтез 132-136; [1 + 1]-,
[2+2]- и [3+3]-циклосокон-
денсация 167-169
спелеанды 203
структуры 134, 141
«футбольный мяч» 134,191,
248, 249
Криптасферанды 139, 194
Криптаты 142, 158, 160,738
бис(трена) 250,251
катионов щелочноземельных
металлов 158
смешанные, синергетический
эффект 185-187
спелеаты 203
«футбольного мяча» 248, 249
Криптофаны 407-417
комплексы с галогенуглеводо-
родами 411-414
комплексы с катионами
алкиламмония 415,416
комплексы с метаном 416,
417
конкуренция с растворителем
414,415
конструкция молекулярных
контейнеров из изогнутых
строительных блоков 407-411
синтез 409
Кристаллы, плотная упаковка
в твердом состоянии 58, еж.
также Жидкие кристаллы
Критическая концентрация
агрегирования (САС) 400
Критическая концентрация ми-
целлообразования (ККМ)
818
Критическая концентрация
самосборки (ККС) 590
п-Ксилол, связывание циклофа-
новым хозяином 59
Кубен 531
Кубооктаэдры 607, 608
Кукурбитурил 367,368
«Купольность» 79, 92-94, 798,
799
Лабильные комплексы 600—602
Лазерная десорбция из матрицы
(MALDI) 757
Лактаноидные и-сульфока-
ликс[4]ареновые ансамбли
623, 624
Лантаноидов катионы 126,163
Лантаноиды, светопреобразую-
щие устройства 712
Лариат-эфиры 129—131, 141,
155-157,223,224
бибрахиальные лариат-эфиры
(BiBLE) 132, 141, 155,224
водородное связывание NH^",
стерические ограничения
155
комплекс с катионом металла
130
864
Предметный указатель
комплексы с К+ и Ag+ 223,
224
константы связывания 129,
156, 157
селективность связывания
катионов 149-159
N- и С-прикрепленные,
синтез 131
Ленты 625-627
Лёд
гексагональный (\h) 296, 297,
300,301,606
кубический AС) 606
Лиганды моно- и бидентатные
36,37
Лиганды, связывающие железо,
см. Сидерофоры
Лиганды типа «жесткий
стержень» 609
Лиотропные жидкие кристаллы
818,822
Лиотропные фазы 823, 824, 830
Липофильность 61, 143
Лития катион Li+
арильные соединения 138
электрид 208
Локализованные валентности
702
Льюисовские кислотные хелаты
278-282
Люминесценция 698, 699. 736,
737
светопреобразующие
устройства 711,712
тушители 713
Магния катион Mg2+
в хлорофиллах 79-88
транспорт щелочных металлов
75,76
Макробициклический эффект
37-39, 159-161,250,251
Макротрициклическая бис(ка-
ликсареновая) клетка,
связывание катионов гибридными
каликсаренами 219-222
Макротрициклические криптан-
ды 247-249
Макроциклические азакоранды,
суперкомплексы 254, 255
Макроциклические лиганды 78,
79
ковалентный темплатный
синтез 586
межфазный катализ, эффект
«обнаженного» катиона 146
синтез методом высокого
разбавления 165-167
синтез с удалением серы 392
Макроциклический эффект
35-39,78, 159-161,282
Масс-спектрометрия 419, 437
дендримеры 756—758
катенаны 645, 646, 649, 656, 657
Материалы для нелинейной
оптики 746-754
второго порядка 749—753
генерация второй
гармоники (ГВГ) 745
переключаемое устройство
753
полиены типа «тяни—
толкай» 736,750,751
молекулярная поляризация
индуцированная 747
общая 747,748
основы нелинейной оптики
746-749
коэффициент
гиперполяризуемости 748, 749
Предметный указатель
865
поляризуемость 746,747,
749
третьего порядка 753, 754
Машина, определение 695
Межмолекулярные
взаимодействия, инженерия кристаллов
494
Межмолекулярные
многоцентровые гетероакцепторные
водородные связи 549
Межфазный перенос 144,178,
217-219,259
Мезогены 820-831,834,835
Мезоморфные
(жидкокристаллические) соединения 820
Мезопоры 307
Мезофазы 822, 830
Меламин 625, структура
комплекса меламинциануровая
кислота 625—628
Мембранные потенциалы
65-68
Мембранный транспорт ионов
68-76, 127,273,388
канальный механизм 69
канальный механизм с
воротами 69
механизм с участием
переносчиков 69
Мембраны биологических клеток
68-70,814,819
Металлобиоцентры 792, 793
биологические имитаторы
792-796
модели гемоцианина 793—795
модели для имитации,
подтверждающие и
спекулятивные 792
Металлоорганические рецепторы
265-273
комплекс бис(амида) с Вг~,
кристаллическая структура
265, 266
комплекс каликсарена с С1~
271
комплекс поданда с PF^" 272
циклическая вольтамперомет-
рия 267-269
Металлопротеины 792
Металлы,
см. также Щелочные металлы
биметаллические системы
702-704
водородные связи с металлами
и их гидридами 548-551
катионы, равновесие
межфазного переноса 217—219
комплекс с лариат-эфиром
130
комплексы катион
металла—макроцикл 144-149
конструирование хозяина
60-61
координационные центры
792
металлические ансамбли
609-612
«молекулярные квадраты»
и «молекулярные коробки»,
самосборка 597—608
октаэдрический ион, диаграмма
кристаллического поля 601
перенос заряда от металла
клиганду(МЬСТ) 701
переходы, центрированные на
металле (МС) 701
селективность связывания
катионов 149-159
синтез «атом металла — пар
лиганда» 227
25-СтидДж. В.
866
Предметный указатель
системы со смешанной
валентностью 702—704
суперкомплексы макроцикли-
ческих соединений 253
Метан 416,417
клатратный гидрат 299-302
связывание криптофанами 416
Метациклофаны 390, 391
Метилвиологен, см. Паракват
Метиленовый голубой, структура
35, 807, 808
Метилимидазол (Melm),
структурная модель гема 798, 799,
801-804
Метилмалонил-СоА-мутаза,
аминокислотный метаболизм 99
Метод атом-атомных
потенциалов 518-520
Метод высокого разбавления
165-167,392,409,418
аппарат 165-167,392,409,418
Метод коалесценции 199, 200
Метод «молекулярного замка»
600
Метод растянутой тонкой
структуры рентгеновского
поглощения (EXAFS) 86, 87, 792,
793
Метоксид-ион 278
Микобактин 228
Миоглобин 89, 94-96
в гемоглобине 95
насыщение кислородом 95
Мицеллы 307,400,817-819,830,
обратные 818
«Мобил-процесс» 310, 311
Модели Кори—Полинга—Колтуна
(СРК-модели) 136, 137, 178,
194,210,418,789
Модель Вайсса 92
Модель ионного насоса 67
Модель Полинга 92
Модель связывания Чатта 187
«Молекулярная карусель» 643
«Молекулярный поезд» 643
«Молекулярный челнок» 740,741
«Молекулярный шприц» 222, 223
Молекулярный конструктор
LEGO 371
Молекулярные выпрямители
732-734
«Молекулярные квадраты» и
«молекулярные коробки»
597-608
Молекулярные орбитали (МО)
701
«Молекулярные пинцеты»
386-389
Молекулярные провода 727—731
Молекулярные сита 306
Молекулярные узлы 652,
679-685
многократно сцепленные
соединения 683—685
пятилистный 683, 684
сборка 679-684
трилистный 652, 679, 680,
682, 683
Молекулярные устройства
695-763
дендримеры 754—763
информация и сигналы
(семиохимия) 716—726
материалы для нелинейной
оптики 746—754
молекулярные машины на
основе катенанов и ротаксанов
740-746
определение 695
философия 695
Предметный указатель
867
фотохимия 698—716
«Молекулярные щели» 386—389
Молекулярные электронные
переключатели 734—740
аллостерические
переключатели 739,740
переключаемое связывание
737-739
система 1,2-дитиенилэтенакак
переключатель 734—736
электропереключаемая
люминесценция 736, 737
Монодисперсность 756
Монооксид углерода
водородные связи 542—544
связывание в гемоглобине 94,
802
Монослой ПАВ 816,817
Морфосинтез 565
Мотив «ёлочка», см. Упаковка
молекул по типу «ёлочка»
Мочевина 113, 318—324, ферро-
упрутие свойства комплекса
323
Мыла 815,816,824
Мягкие ионы металлов 179
Мягкие лиганды для мягких
ионов металлов 179—190
гетерокраун-эфиры 181-184
гетерокриптанды 184, 185
катионы щелочных металлов
179
основания Шиффа 187-190
смешанные криптанды
185-187
теория жестких и мягких
кислот и оснований (теория
HSAB) 180
МЕСАМ (комплексообразователь
железа) 230, 231, структура 230
Наноразмерная инженерия, см.
Молекулярные устройства
Направленный мутагенез 108,
109
Насыщенные водородными
связями (SHB) структуры 540
Натрия катион Na+
биохимия 65—77
содержание в биологических
системах 67
содид 207
транспорт, биологическая
мембрана 68, 69, 75, 76
Нейротрансмиттеры 99—101
Нейтральные молекулы,
связывание 295-438
внутриполостные комплексы
353-429
неорганические твердофазные
клатратные соединения
296-318
твердые клатраты
органических хозяев 318—353
фуллерены 430-438
Нейтральные рецепторы
273-277
анионсвязывающие хозяева
273
хозяева, образующие
водородные связи 276, 277
цвиттер-ионы 274, 275
Нековалентная координационная
химия анионов 237—240
Нековалентные взаимодействия
48-60
Нелинейно-оптические (НЛО)
свойства 330, 736, 746-754
Нервный импульс 67, 100
Нервные клетки, передача
информации 67
868-
Предметный указатель
Несоразмерные структуры 320,
321,329,339
Нестехиометрические
соединения 319
Нетропсин, водородные связи
114
Никотин 100, 101
Никотинамидадениндинуклеотид
(NADH) 805,807
Никотин-ацетилхолиновый ре-
цепторный белок (nAChR)
100, 101
Нитроанилины 522
Нонактин 70, 114
Нуклеиновые кислоты,
самосборка 581-583
Нуклеозиды 103, определение 261
Нуклеооснования 103
Нуклеотидные полифосфаты
782-786
Нуклеотиды, определение 103,
261
Нуклеофил анионный 146
Нуклеофильное замещение 146,
391
Обобщенная координационная
химия 61
Образование центров
кристаллизации (нуклеация) 504, 505,
508-510
Однократная запись,
многократное считывание (WORM) 736
Оксид азота 117-119
Оксид кремния как биоминерал
563
Олимпиадан 645, 646
Оптическое вращение энантио-
мера 338
Органические гости,
гидрофобное связывание 59
Ортоциклофаны 391
Основание Трёгера 386, 399
Основания Шиффа 30,187-190
жидкие кристаллы 832
конденсация 30,187-189
макроциклические 136,188,793
оксигемоцианин 794
1:1-циклоконденсация 189
«Офис» 609-612
кристаллическая структура 610
«полы» и «стены» 609, 611
Паракват (метилвиологен) 204,
205, структура 634
Параметр ЕТ C0) Димрота—Рей-
харда 402
Параметр Тафта (константа
индуктивности) 224
Парациклофаны 226, 227, 260,
261,390,391,394,401,402
Пептидные звенья 106
Пергидротрифенилен (РНТР)
329-332
Переключаемое связывание
737—739, см. также
Молекулярные электронные
переключатели
Переключатель
нелинейно-оптический 753
хромофорный молекулярный
744
Переключение 734-740
Перенос заряда от металла к ли-
ганду(МЬСТO01
Перенос электрона (еТ) 709-711
нековалентно связанные
системы 712—716
энергетические уровни 709
Предметный указатель
869
Перенос энергии (ЕТ) 700, 712
фотосинтез 82-84
фотохимия 698—702
Переносчики катионов 178
Переносчики электронов 204
Переходные металлы
моноядерный комплекс,
диаграмма энергетических
уровней 701
реакции дикатионов 182, 183
редокс-активный металл, СО-
субстрат 185, 186
связывание 182, 225
Периферическое уплотнение 626
Пероксид водорода 242
Пигменты светособирающие 81, 82
Пикраты щелочных металлов
константы скоростей
связывания и диссоциации 177
свободные энергии
связывания 169-175
экстракция 47, 48
Пиразин 423,424
Пиримидины 103
Пирокатехин, свойства 231
Платины катион Pt2+
катион бипиридилдиаммин-
платины(Н) 204, 205
лекарства 110, 111
связывание ДНК 110-112
Плато селективности 150,175
Платоновы тела 620, 621
Пленка Ленгмюра—Блоджетт 750
Поверхностно-активные
вещества (ПАВ) 814-819
классы 815,816
упорядочение на поверхности
раздела 814-820
Поверхностные весы Ленгмюра
816
Поданды 37, 126-129
комплекс с Eu(III) 127
комплекс с К+ 160
константы связывания 129
концепция жесткой концевой
группы 128
макроциклический эффект 38
определение 140
переносчики Са2+ 126
структуры 141
термодинамические вклады
в стабильность 160
трансмембранные ионофоры
126
подандный тригидроксиэти-
ленгликоль 127
«укутывание» катиона Rb+
криптофиксом-5 176
хелатный эффект 37
Податы, определение 142
Полиены типа «тяни—толкай»
750,751
Полимеразная цепная реакция
(PCR) 109,110
Полимеризация при включении
323
Полимерные продукты 161,162,
166
Полиморфизм 518
Политопные рецепторы 195—200
Полости 32
Полукарцеранды 417,421, 617, 619
Полупроводник TiO2 715
Полусферанды 139, 790
Поляризационная оптическая
микроскопия с
нагревательным столиком 820,821,827
Поляризованный свет 822
Полярное и неполярное
связывание гостей 403
870
Предметный указатель
Полярность кристалла 330
Пористые координационные
полимеры 558—562,
металлические 4,4'-бипиридильные
пористые сетки 561
Поросилы 303
Порфирины 78, 79, 89-94,
256-259, 588-592, 797-804
аналоги гема, модели
связывания и транспорта кислорода
797-804
атропоизомеры 800
«карманные» 800
комплекс с капсулированным
F" 258
комплексы цинка, самосборка
588-592
«корзинка для пикника» 800
«купольность» 92—94
лиганды 92
«огороженные» 92, 800-802
перенос электронов 713
расширенные 256—259
семиохимия 716
экранирование железопорфири-
нового ядра дендримером 759
Потенциометрическое титрование
44, 252
Правила Эттер 521—524
диарилмочевины 522
нитроанилины 522
продукты кристаллизации кар-
боновых кислот с 2-аминопи-
римидином 523
продукты кристаллизации
оснований нуклеотидов 523
Правило Хюккеля,
ароматические системы 78, 79
Предорганизация 40, 158,
169-178,206,245,282,626
динамический эффект 177,
178
кинетический эффект 177,178
связывание хозяин—гость 40,
169-176
термодинамические эффекты
169-176
Предсказание структуры
кристаллов 517-524
расчетные методы 517—520
экспериментальные методы
519
П реобразование света 711,712
Преобразование сигналов 67, см.
также Информация
и сигналы
Призмы 621
Применение растворов 144—148
Принцип Франка-Кондона 699
Присоединение по Дильсу—Аль-
деру 617
синтез конкина 370
катализируемое «мячом для
софтбола» 618
Пространственные группы 513
Протеинтирозинфосфатаза
(РТР-аза) 242
Противораковые препараты
карбоплатин 110,111
цисплатин ПО, 111
Протоклетка, происхождение
жизни 819
«Протонная губка» 278, 279
Процессы, индуцированные
светом, см. Фотохимия
Псевдоводородные связи 53
Псевдоротаксаны 629—631,
633—635, 743, хромофорный
молекулярный
переключатель 744
Предметный указатель
871
Пурины 103
Пурпурный бензол
123
Разделение (рекомбинация)
зарядов 85,742,743
Разрушение кристаллов 514, 515
Распознавание ионов 200—203
латеральное 193
молекулярное 34, 149, 195,
696,716
сферическое 133
хиральное 200-203, 264, 341
центральное 193
Распространение спаривания
оснований 582
Рассеяние рентгеновских лучей
малоугловое 827
Растворители, взаимодействия
хозяин—гость 170
Расширенное систематическое
силовое поле (ESFF) 607
Реакции включения 426—429,
водородные связи 402-407
Реакции типа реакции Вюрца 391
Реакция Митсонобу 391, 392
Резорцинарены 354-364, 418, 622
каликс[4]резорцинарен 355,
усеченный куб 621,622
[4]резорцинаренкаликс[4]арен
425
Рентгеноструктурный анализ
(РСА) 55,71-73,206,321,
322, 606
Рецептор хинона, синтез 405
Рецепторы анионов биологические
240-244
аргинин как центр связывания
анионов 242, 243
белки, связывающие фосфаты
и сульфаты 241,242
карбоксипептидаза А 243,244
Рецепторы гербицидов 204-206
Рецепторы дитопные и политоп-
ные 195
Рецепторы, координация и
модель «замок—ключ» 34, 35,
767, 768
Рибонуклеаза , самосборка 115
Рибонуклеиновая кислота (РНК)
103, 105-107
Рибосома 105
Родопсин 76, 77
Розетки 625-628
Рост кристаллов 504—513
образование центров
кристаллизации (нуклеация) 504,
505, 508
операции трансляционной
симметрии 512
рост на поверхности раздела
воздух—жидкость 505—508
симметрия молекул и
кристаллов 511
системы Шёнфлиса и
Германа—Могена 511
хиральность, эффект Адама
508-510
Ротаксаны 628-630, 632, 633,
636, 637, 652
кинетический и
термодинамический темплатные эффекты
586
молекулярные машины
740-743
состояние с разделенными
зарядами 743
«молекулярный челнок»
740,741
номенклатура 628
определение 628
872
Предметный указатель
[2]ротаксан, синтез
статистическим методом
проскальзывания 652
синтез 630,632,636,637
подход на основе
самосборки (защелкивание,
продевание,
проскальзывание) 632, 636, 637
статистический подход
632, 636, 652
Рутения катионы Ru2+ и Ru3+
593-596, 702, 705-709
22-ядерный металлокомплекс
708
[Ru(bipyJCl2] 708
[Ru(bipyK]2+ 702, 705,
спектры поглощения и
фосфоресценции 705
Ru(H)-Ru(III), ион Крейт-
ца—Таубе 702
Ряд Хофмейстера 250, 252
Сальварсан 35
Самоингибирование 618
Самовоспроизводство
(репликация) 578,685-690
Самоорганизация 580, 677, 678
Самораспознавание 669—673,
677
Самосборка 115-117, 578-581
биохимическая 115—117,
580-584
в синтетических системах
584-592
геликаты 660-679
ДНК 105-107, 115,581-583
закрытые комплексы,
водородные связи 613—628
гигантские
самособирающиеся капсулы 619—625
розетки 625—628
«теннисные мячики»
и «мячи для софтбола»
613-618
капсулярные и полимерные
самособирающиеся
структуры 594
каталитические и
самовоспроизводящиеся системы
685-690
катенаны и ротаксаны
628-659
координационные соединения
592-612
металлические ансамбли
609-612
«молекулярные квадраты»
и «молекулярные
коробки» 597-608
принципы дизайна 593
супрамолекулярный
куб 593-596
кристалл 491,492
молекулярная 579
молекулярные узлы 679—685
определение 115
с ковалентной модификацией
583, 584
модификация
предшественника 583
обработка после сборки 583
синтетические системы
кинетический и
термодинамический темплатные
эффекты в синтезе
584-592
термодинамическая модель
587-592, 626, порфирино-
вые комплексы цинка
588-592
Предметный указатель
873
строгая 581—583
супрамолекулярная 579
терминология 578—580
цели и задачи 576—578
Самособирающиеся
многоэтажные структуры 609—612
Сапфирин 256
Саркофагины, Со(Ш) и Pt(IV)
135, 136, 142
Светопреобразующие устройства
711,712
Сворачивание дендримеров 758
Связывание анионов 237—292
биологические рецепторы
240-244
конструирование хозяина 60,
61
Связывание катионов 121—232
заряд катиона 152
факторы, определяющие
селективность хозяина 149,152
Связывание нейтральных
молекул 295-438
Связывание органических
катионов 190-206
дитопные рецепторы 195-200
эффекты кольцевого тока
ароматического кольца
197
ЯМР-спектроскопия 197
дифильные рецепторы 203,
204
рецепторы гербицидов
204-206
связывание катионов аммония
корандами 191-194
связывание катионов аммония
трехмерными хозяевами 194
хиральное распознавание
200-203
устройство для разделения
энантиомеров 202
хиральный барьер 200, 201
Селективность
кинетическая и
термодинамическая 41-48, 149-159, 240,
768,787
связывание анионов 244—252,
273
связывание катионов
149-154, 170
связывание циклодекстринами
380-384
переходного состояния 311
формы 249-252
Семиохимия 716—727, см.
Информация и сигналы
Сенсоры 60, 716-727
флуорофоры 721—724
фотохимические 717—721
химические 717
электрохимические 724—727
PET 719
Сепулькраты, Со(Ш) 135, 136, 142
Серебра катион Ag+
комплекс 18-краун-6 с Ag+ 181
комплекс углеводородного ли-
ганда с Ag+ 226
туннелирование Ag+,
связывание каликсаренами 216
ЯМР-спектроскопия 109Ag
609
Сигмаидальная зависимость
скорости реакции от времени
688, 690
Сидеранды 231,232
Сидерофоры 228—232
природные 228—230
синтетические системы
230-232
87#!
Предметный указатель
МЕСАМ 230
темплатный синтез энтеробак-
тина 229
Симметрия молекул и кристаллов
511-513
Симпорт (перенос) Н+ и С1~-ани-
онов 259
Синапс 100
«Синтез вверх» 577
Синергетическое связывание
185-187,223,494
Системы со смешанной
валентностью, фотохимия 702—704
Слой Гуи—Чепмена 817,818
Слой Штерна 817,818
Соединение Дианина 332—336
Соединения включения
дифференциальная
сканирующая калориметрия (DSC)
349-353
клатраты Хофмана и Вернера
317,318,559
конформационные аспекты,
включение катиона в калик-
сарен 215
кристаллическое состояние 33
суперкомплексы металлов 253
термогравиметрический
анализ (TGA) 349-353
типа «песочные часы»
(смешанные кристаллы) 567—569
три-о-тимотид 337—340
циклодекстрины 378, 384
цикл отри вератрилен (CTV)
346-348
циклофаны 402-407
Соединения (комплексы)
хозяин—гость
геометрия типа «гнездо»,
«капсула», «насест» 177
график Джоба 46
классификация 32-34
конструирование хозяина 60, 61
определение 28, 29
предорганизация и компле-
ментарность 40,41,169,170
связывание 170, константы
43-48
термодинамическая
устойчивость 37
Соль Цейзе 53
Сольватация гостей и хозяев
246,414,415
конкуренция гостя и
растворителя 414,415
свободная энергия 152, 238
Соответствие размеров полости
хозяина и гостя-катиона
149, 150, 152,394
Сочетание алкинов,
катализируемое медью 391, 393
Спаривание оснований в ДНК
52, 106, 107, 578
по Уотсону и Крику 106, 107
по Хугстену 107
Спейсер 27
Спелеанды 203
Спелеаты 203
«Спиновое ингибирование» 87
Спирали 319, 327-329, 337-340,
660-663, 676-679, 828
Спиральность М- и Р- 338, 661
Спиральные тубуланды 327—329
Спирты 536, 538, структура 538
Стабилизация, обусловленная хе-
латным эффектом 38
Стадия, определяемая гостем
(GDS) 422
Статистический синтез катенанов
и ротаксанов 633
Предметный указатель
875
Статическая стабильность
комплекса 150
Стерическое соответствие
хозяин—гость 34
Стехиометрия 45
Стирание прямого считывания
после записи (EDRAW) 736
Структуры из лигандов типа
«жесткий стержень» 609
Сульфаты
водородные связи 241, 242
как биоминералы 564
Супервюрцит, структура 541
Суперкомплексы азакорандов
с анионами 253
азакоранды с большими
кольцами 253
константы связывания 255
Супероксиддисмутаза 242
Супрамолекулярная фотохимия
698-716
Супрамолекулярная химия
история 30-35, 206, 239
классификация соединений
хозяин—гость 32—34
комплементарность 40
конструирование хозяина 60,
61
координация 34, 35
определение 27-29
предорганизация 40
природа взаимодействий
48-60
развитие представлений 30—33
рецепторы 34, 35
термодинамическая и
кинетическая селективность 41—48
хелатный и макроциклический
эффекты 35-39
химия хозяин—гость 28, 29
Супрамолекулярное устройство,
определение 696
Супрамолекулярные
взаимодействия, типы 48-60
Супрамолекулярные частицы 27
большие молекулы 696, 697
история 34, 35, 206, 237
конструирование хозяина 60,
61
определение 696
фотохимические и
электрохимические критерии 697
Супрамолекулярный куб 593-596
компоненты 595
самосборка 593—596
Сферанды 40, 136-140
как хозяева для
катионов-гостей 139
классификация соединений
хозяин—гость 33
комплекс с Li+ 138,175
модели Кори—Полинга-Кол-
туна (СРК-модели) 136,137,
418
синтез, циклизация 138
структура гибридных хозяев
41
Сфераплекс 142
Таллия катион Т1+ 181, 184
Твердофазные реакции 342
Тектоны (строительные блоки)
491,515,516,529-531,535,
определение 578, 579
Темплатирование 578, 585
Темплатированныи рост
кристаллов 505-508
Темплатирующие гости 604, 616
Темплатная биоминерализация
564
Предметный указатель
Темплатный эффект 131,133,
161-165, 189, 190,229,578,
616,656
в синтезе 584—587
бензо-18-краун-6 163, 164
[2]катената 656
карцерандов 421—423
тетраазамакроциклов 164
ионы металлов, влияние
на циклизацию 163
кинетический 164
синтез методом высокого
разбавления 165—167
термодинамический 164
удаление темплатного катиона
Ni2+ 190
циклические и ациклические
продукты 162
1:1-циклоконденсация 189
[2 + 2]-циклосоконденсация
167-169
Темплаты 576—690, см. также
Самосборка
в синтезе 584—592
геликаты 660—679
закрытые комплексы,
водородные связи 613-628
каталитические и
самовоспроизводящиеся системы
685-690
катенаны и ротаксаны
628-695
координационные соединения
592-613
молекулярные узлы 679—684
терминология 578—580
цели и задачи 576—578
Теория жестких и мягких кислот
и оснований (HSAB) 180,
244
Термогравиметрический анализ
(TGA), соединения
включения 349-353
Термодинамическая
селективность 41,240
Термодинамическая стабильность
комплекса 600-602
Термотропные жидкие кристаллы
822, 823, трубчатые и диско-
тические 823,824
Тетрагидрофуран (ТН F) 208,
274, 282, 283
Тетрапиррольные макроциклы
78,79
корриновое кольцо 78, 79, 98,
809
комплексы магния 79—85
Тетраол, синтез соединений
с ацетальными мостиками
420
Тетрафенилен 348, 349
я-Толуолсульфонат (Ts) 125
Топологические диастереомеры
и энантиомеры 631, 652
Топологический захват 760
Топохммия 295, 493
Торсионные углы в комплексах
краун-эфиров 143, 144
Трансаминирование циклодекст-
рином и пиридоксаминовым
коферментом 782
Трансацилазы 787
£>-аминоэфиры 788
хиральные коранды 786, 787
хозяева, связывающие
катионы 786-791
Трансляционная изомерия 647,
651,652
1,3,5-Тригидроксибензол (ТНВ,
флороглюцин) 406
Предметный указатель
877
Тригидроксиэтиленгликоль
подандный 127
Трикислота Кемпа 387
Трилистный узел 631, 652, 680,
682, 683
базовый 680
диастереомерия 631
Тримезиновая кислота (ТМА)
324-327, трианион 277
Трис(пиразолил)бораты 225
Три-о-тимотид (ТОТ) 337-343
применение 342, 343
производные и родственные
хозяева 340
синтез 340
соединения включения
337-340
спонтанное разделение кон-
формаций 338
Трифлат-анион 226
1,3,5-Трицианобензол 500, 501
Тройной комплекс 685, 686
Тубулярные мезофазы 830
Тушители 713,719
люминесценция 713
флуоресценция 719
Углеводородные хозяева 225—227
Углерод (углеводные
эквиваленты), фотосинтетическое
получение 79, 80
Углеродные донорные и я-кис-
лотные лиганды 223-227
смешанные С-гетероатомные
хозяева 223—225
углеводородные хозяева
225-227
комплекс с Ag+ 226
комплекс парациклофана
сСг+ 227
Угол захвата лиганда 152, 153,
280, 705
Упаковка молекул по типу
«ёлочка» 56, 382, 552, 553, 826
Уравнение Гиббса 160
Уравнение Эйринга 200
Уранилнитрат 128
Уранилхлорид,гидрат 556
Урацил 676-679
Уреаза из бобов 792
Фактор занятости 416,617
Фенол 332, 333
Ферментативный катализ 34
Ферменты 767—796, см. также
Коферменты
активные центры 770, 790
апофермент 97, 809
голофермент 97
имитаторы
каталитические и
самовоспроизводящиеся системы
685-690
структурный 789
циклодекстрины
(как хозяева) 775—782
частичный 789
индуцированное соответствие
768, 769, 773, 774
искусственный 780
история 792
металлобиоцентры 792—796
модели 767-770,792,793
определение 770-772
перенос протона 243
размер молекул 775
свойства 768, 769
серинпротеазные 776
структура 770—772
трансмембранные 65
878
Предметный указатель
ферментативный катализ 34,
763, 768
механизм 772—775
модель «замок—ключ» 34,
767-769
характеристики 770—775
энтатическое состояние 94
Феромоны 768
Ферритин, самосборка 116
Ферроцен 53, 430
комплекс с ос-циклодекстри-
hom(cc-CD) 383
полиморфные формы 553
Фетодигитогеник 142
Фильтр селективности 74, 75
Фильтрация ошибки 673
Флуоресцентное титрование 46
Флуоресценция
определение 698—700
система краун—антрацен
719-721
Флуоресценция, усиленная хела-
тированием (CHEF) 722, 723
Флуорофоры, семиохимия
721-724
Формамид 536, структура 537
Фосфаты, водородные связи
241,242
Фосфолипидная везикула,
молекулярная проводимость 728,
729
Фосфолипиды 815, мембрана
69,70
Фосфоресценция, определение
700
Фотовозбуждение, последующие
излучательные явления
698-702
Фото- и электропереключаемая
система 735, 736
Фотоионный сигнал 737,
переключаемое связывание
737-739
Фотосинтез 79, 378, 709
окисление воды до кислорода,
катализированное марганцем
85-88
перенос энергии (ЕТ) 700
реакционный центр 85
тетрапиррольные комплексы
магния 79-85
фотосинтетическая мембрана
Фотосистемы (PS) I и II 85, 86
Фотохимические устройства
на основе дендримеров
760-763
сенсоры, семиохимия 716—727
Фотохимия 698-704
биметаллические системы
и системы со смешанной
валентностью 702—704
бипиридил, трипиридил
и 1,10-фенантролин 704—707
критерии для отнесения
частиц 697
накопление энергии 204
основы 698-702
светопреобразующие
устройства 711,712
системы с нековалентными
связями 712—715
фотохимические устройства
на основе дендримеров
760-763
фото- и электрохимические
устройства на основе бипири-
дила 707-711
Фталоцианины, суперфталоциа-
нин 190
Предметный указатель
879
Фторхлорбромметан 413
Фуллерены 358, 430-438, 579
бакминстерфуллерен 347,
430-438,619-621
как гости 430—433
как сверхпроводящие
соединения включения 436
как хозяева 433—436
открытие 437, 438
редокс-чувствительное
сенсорное устройство на основе бак-
минстерфуллерена 435
самосборка 579
структура 621
Фумаровая кислота и ее комплекс
535, структуры 536
«Хвостовое остаточное сродство»
390
Хелатирование 280,281
Хелатирующие агенты 278—282
Хелатное кольцо 38, 149, 152, 153
размер (угол захвата лиганда)
152, 153,280
энергия напряжения 38
Хелатный эффект 35—39
анионы 270,279,280
стабилизация 38
Хелаты 280, кристаллические
структуры 280,281
Химически модифицированные
ион-селективные полевые
транзисторы (CHEMFET)
726, 727
Химия растворения щелочных
металлов, история 206
Химия хозяин—гость 28, 29
Химотрипсин 789, 790,
активный центр 790
Хиральная хроматографическая
колонка 202
Хиральность 631
двойные геликаты 663
двухдоменный геликанд,
комплексы 662
индукция 508-510
коранды 786—789
трилистный узел 682
умножение 343
Хиральные капсулы 363
Хиральный барьер 200, 201,
определение 200
Хиральный сдвигающий реагент
663
Хлорид-анион, связывание тетра-
эдрическими рецепторами 248
Хлорофиллы 78, 80-85
спектр поглощения 81
тетрапиррольные макроциклы
78
этилхлорофиллид,
кристаллическая структура 83
Хозяева кремнийсодержащие,
связывающие анион 281
Хозяева на основе гликолурила
367-369
Хозяева на основе конкинов
370-372
Хозяева, связывающие катионы
121-232
алкалиды и электриды
206-209
бибрахиальные лариат-эфиры
(BiBLE) 132, 141, 155,
224
гибридные 139, 171-174
имитаторы трансацилаз
786-791
каликсарены 209—223
880
Предметный указатель
комплексы с катионами
металлов 149-159
комплексы с органическими
катионами 190—206
дитопные рецепторы
195-200
коранды 191—193
трехмерные хозяева 194, 195
коранды 160, 175,178
краун-эфиры 121 — 126,
143-154, 159-164, 166, 167,
169
криптанды 132-136, 157-160,
167-169, 171, 173, 176
лариат-эфиры 129—131,
155-157,223,224
мягкие лиганды для мягких
ионов металлов 179—190
номенклатура 140—142
поданды 126-129, 159-161,
175, 178,272
сидерофоры 228—232
сферанды 136-140, 171, 172,
175
углеродные донорные и я-кис-
лотные лиганды 223—227
Хозяева типа «колесо—ось» 555,
556
Хозяин, определение 28, 29,
244
Хрома катион Сг+
парациклофаны, комплекс
сСг+ 227
Сг(СОN, разрушение
кристаллической структуры 514
Хромионофор 128
Хромофоры 27,698,715
Хромофорное молекулярное
переключение, псевдоротак-
саны 743, 744
Цвиттер-ионы, нейтральные
рецепторы 274, 275
Цезиевый эффект 163, 392
Целлюлоза, получение циклодек-
стринов 378
Центры связывания хозяина
и гостя, сходящиеся и
расходящиеся 29
Цеолиты 303-311
синтез 307-309
катионы-темплаты и типы
образующихся цеолитов
308
состав и структура 303—307
зигзагообразные каналы
bZSM-5 307
топология каркаса
структуры 304, 305
MFI-типа в нефтяной
промышленности 309-311
Цеосилы 303
Цианокобаламин, см.
Витамин В,2
Циануровая кислота 625
структура комплекса мела-
минциануровая кислота
625-628
Циклобутадиен, внутриполост-
ной синтез 427, 428
Циклам 190
Циклическая вольтамперометрия
254,267-269,640,641,658
Циклические диолы 327-329
Циклический 3',5'-аденозинмо-
нофосфат (сАМТ), гидролиз
фосфодиэстеразой 778, 779
Циклодекстрины 372—386, 431
ацетилирование 777
как имитаторы ферментов
775-782,809,810
Предметный указатель
881
как имитаторы глиоксалаз
781
как имитаторы эстераз
778-780
как катализаторы гидролиза
сложных эфиров 778, 779
как хозяева 776—778
функционализированные
780-782
определение класса комплекса
33
применение 384—386
продевание каровиологенов
сквозь полости циклодекст-
ринов 728, 729
свойства 372—377
синтез 378
соединения включения
378-384
структуры 234, 373
упаковка 382
Циклосоконденсация 167—169
комплекс криптанда с NaC104,
кристаллическая структура
169
криптанды 167
криптанды на основе бипири-
дила 168
Циклотетравератрилен (С 1 IV) 345
Циклотривератрилен (CTV) 33,
203,271,272,343-348,
407-417,430
определение 33
синтез 345, 346
соединения включения
346-348
фазы 346
Циклофаны 59, 209, 226, 227,
260,261,389-407,390,391,
см. также Каликсарены
включение гостя с помощью
водородных связей 402-407
дифенилметан 396—401
изменение рН 261
клетки в водном и неводных
растворителях 401, 402
концепция пинцета 394—396
номенклатура 390,391
определение 389
связывание л-ксилола 59, 60
синтез 391—394
супрамолекулярная
самосборка 598
циклические вольтамперо-
граммы 641
Цинкин 721,722
Цинковые сенсоры 721—723,
флуорофоры 721—724
Цинкпорфириновые комплексы
588-592, 713, 714, самосборка
588-592
Цинксодержащие ферменты
721,795,796
Цисплатин 110, 111
Цитохромы Р-450 804-808
катализ реакций 804, 808
каталитический цикл оксиге-
назной активности 805
микрореактор, моделирующий
работу цитохрома 807
модели 804-808
циклодекстрины как
имитаторы цитохрома 808
Цитратсинтаза 242
Число А/А в жидких клатратах
838
Частичный имитатор фермента
789
882
Предметный указатель
Шампуни 815
Шлирен-текстура
823, 824
Экзотермическая реакция,
изменение свободной энергии
601
Электриды 206-209, 436,
первый электридный комплекс
207
Электропереключающаяся
люминесценция 736, 737
1,6-Элиминирование 391
Энтатическое состояние 94
Энтеробактин 229, 230, тем-
платный синтез 229
Энтропийные и энтальпийные
вклады хелатного и макро-
циклического эффектов
35-39
Эстеразы 778-780
/я/?я«с-Этерификация 263
Эфир Вильямсона, синтез 125
Эффект Адама, см. Индукция хи-
ральности в кристаллах
Эффект «обнаженного» аниона
146,207 1
Эффективная концентрация
гостя в полости хозяина 618
Эффективная молярность 590,
773
Эффективность самосборки 591,
592
Эффекты кольцевого тока
ароматического кольца 197
Ядерный эффект Оверхаузера
(NOE) 198,214,215
ЯМР-спектроскопия 138,
197-200, 265, 343, 396, 397
аденозинтрифосфатC1Р) 784
дитопные рецепторы ('Н) 195
каликсарены AН) 211
катенаны^Ни 13С) 649
криптанды A9F) 138
металлические ансамбли
A09Ag) 609
ртутьсодержащий хозяин
A99Hg) 281
спин-спиновое взамодействие
198
тетракатионный хозяин A9F)
289
ЯМР-титрование 44—46
Дополнительная
литература
К главе 1
1. Lehn J.-M., Ball P. Supramolecular chemistry / Ed. N. Hall. The new chemistry.
CUP, 2000. P. 300.
2. Scheinder H.-J., Yatsimirsky Л.К. Principle and methods in supramolecular
chemistry. Chichester: J.Wiley and Sons, 2000. 349 p.
3. Fuhrhop J.-H., Endisch С Molecular and supramolecular chemistry of natural
products and their model compounds. New York: M. Dekker, 2000. 602 p.
4. Dudzuik H. Introduction to supramolecular chemistry. Netherlands: Kluver
Academic Publishers, 2001. 364 p.
5. Encyclopedia of supramolecular chemistry/ Ed. J.L.Atwood, J.W.Steed. New York:
M. Dekker, 2004. 1500 p.
6. From the molecule to the nanoscale: synthesis and structure. Comprehensive
coordination chemistry II / Ed. M.Fudjita, A.Powell. Amsterdam - Oxford - New York
-Sydney - Tokyo: Elsevier - Pergamon Press, 2003. Vol. 6. 853 p.
7. From the molecule to the nanoscale: properties. Comprehensive coordination
chemistry II / Ed. R.A. Crentz. Amsterdam - Oxford - New York -Sydney -
Tokyo: Elsevier - Pergamon Press, 2003. Vol. 7. 817 p.
8. Synthetic coordination and organometallic chemistry / Ed. A.D. Garnovskii,
B.I. Kharisov. New York-Basel: M. Dekker, 2003. 520 p.
9. Supramolecular polymer / Ed. A. Ciferri. New York: CRC Press, 2005. 1000 p.
10. Lehn J.M. Dynamers: dynamic molecular and supramolecular polymers // Progr.
Polymer Sci. 2005. Vol. 30. P. 814.
11. Pratt L.R., Pohorille A. Hydrophobic effects and modeling of biophysical aqueous
solution interfaces//Chem. Rev. 2002. Vol. 102. P. 2671.
12. Скопенко В.В., Цивадзе А.Ю., Савранский Л.И., Гарновский А.Д.
Координационная химия. М.: Академкнига, 2006 (в печати).
13. Peter J.A. Practical guide to supramolecular chemistry. Chichester: J.Wiley, 2005.
214 p.
14. Badjica J.D., Nelson A., Cantrill S.J. et al. Multivalency and cooperativity in
supramolecular chemistry//Ace. Chem. Res. 2005. Vol.38. P.723.
15. Арсланов В.В., Шейнина Л.С, Калинина М.А. Супрамолекулярная химия
высокоорганизованных планарных систем // Современные проблемы
физической химии. М: Граница, 2005. С. 94-118.
16. Mulder A., Huskens J., Reinhoudt D.N. Multivalency in supramolecular chemistry
and nanofabrication // Org. Biomol. Chem. 2004. Vol. 2. P. 3409.
17. Reinhoudt D.N. and Crego-Calama M. Synthesis beyond the molecule // Science.
2002. Vol. 295. P. 2403.
18. Badjica J.D., Cantrill S.J., Grubbs R.H. et al. The exclusivity of multivalency in
dynamic covalent processes // Angew. Chem., Int. Ed. 2004. Vol. 43. P. 3273.
gg4 Дополнительная литература
19. Сумм Б.Д., Иванова Н.И. Объекты и методы коллоидной химии в нанохимии //
Успехи химии. 2000. Т. 69. С. 995.
20. БучаченкоА.Л. Нанохимия - прямой путь к высоким технологиям нового
века // Успехи химии. 2003. Т. 72. С. 419.
К главе 2
1. Itoh Т., Yano К., Inada Y, Fukushima Y. Photostabilized chlorophyll a in meso-
porous silica: absorption properties and photoreduction activity of chlorophyll a //
J. Amer. Chem. Soc. 2002. Vol. 124. P. 13437.
2. Шарова Н.П., Абрамова Е.Б. Повреждение и починка ДНК, или «на всякую
прореху найдется заплата» // Природа. 2004. № 11. С. 3.
3. Sakurai К., Shinkai S. Molecular recognition of adenine, cytosine and uracil in a
single-stranded RNA by a natural polysaccharide: schizophyllan // J. Amer. Chem.
Soc. 2000. Vol. 122. P. 4520.
4. Reha D., KabaWu M., Ryi6cek F. et al. 1. Nature of stacking interactions between
intercalators (ethidium, daunomycin, ellipticine, and 4,6-diamidine-2-phenylin-
dole) and DNA base pairs. Ab initio quantum chemical density functional theory
and empirical potential study// J. Amer. Chem. Soc. 2002. Vol. 124. P. 3366.
5. Smith K.M. Porphyrines // Comprehensive Coordination Chemistry II. Ed.
A.B.P.Lever. Amsterdam - Oxford - New York - Sydney - Tokyo: Elsevier -
Pergamon Press, 2003. Vol. 1. P. 493-506.
6. Me Keown N.B. Phtalocyanines // Ibid. P. 507-514.
7. Purrello R., Gurrieri S., Lauceri R. Porphirin assemblies as chemical sensors //
Coord. Chem. Rev. 1999. Vol. 190-192. P. 683.
8. Baginski M., Cybulska В., Gruszecki A. Interaction of polyene macrolide antibiotics
with lipid model membranes // Advances in planar lipid bilyaers and liposomes.
2006. Vol. 3. P. 269.
9. TaubertA., NapoliA., Meier W. Self-assemble of reactive amphiphilic block copoly-
mers as mimetics for biological membranes // Current opinion in chemical biology.
2004. Vol. 8. P. 598.
10. Sun L., Akermarck В., Ott S. Iron hydrogenase active site mimics in supramolecular
systems aiming for light-driver hydrogen production // Inorg. Chem. Commun.
2005. Vol. 8. P. 406.
11. Rappolt M., Amenitsch H., Strancar J. Phospholipid mesophases at solid interfaces:
in-situ X-ray diffraction and spin label studies // Adv. Coll. Interface Sci. 2004.
Vol. 111. P. 63.
12. Piccolo A. The supramolecular structure of humic substances: a novel
understanding of humus chemistry and implication in soil science // Adv. Agronomy. 2002.
Vol. 75. P. 57.
13. Rosi N.L., Mirkin C.A. Nanostructures in biodiagnostics // Chem. Rev. 2005.
Vol. 105. P. 1547.
14. Vluek A., Jr. The life and times of excited states of organometallic and coordination
compounds// Coord. Chem. Rev. 2001. Vol. 200-202. P. 933.
15. Стойкое И. И., Антипин Н.С., Коновалов А. И. Искусственные ионные каналы //
Успехи химии. 2003. Т. 72. С. 1190.
Дополнительная литература ««с
16. Мамардашвили Г.М., Мамардашвили Н.Ж., Койфман О.Н. Супрамолекулярные
комплексы порфиринов // Успехи химии. 2005. Т. 74. С. 839.
17. Качалова А.В., Зубин Е.М., Орецкая Т.С. Методы синтеза олигонуклеотидов,
содержащих реакционноспособные электрофильные группировки // Успехи
химии. 2002. Т. 71. С. 1173.
18. Коновалова Н.В., Евстигнеева Р.П., Лузгина В.Н. Синтетические
молекулярные системы на основе порфиринов как модели для изучения переноса
энергии при фотосинтезе // Успехи химии. 2001. Т. 70. С. 1059.
К главе 3
1. WuJ., Poke M.J., Wesdemiotis С Unimolecular chemistry of Li+- and Na+-coordi-
nated polyglycol radicals: A new class of distonic radical cations // J. Amer. Chem.
Soc. 2000. Vol. 122. P. 12786.
2. ВыЫ М., WipffG. Hydronium ion complex of 18-crown-6: where are the protons?
A density functional study of static and dynamic properties // J. Amer. Chem. Soc.
2002. Vol. 124. P. 4473.
3. Громов СП., Дмитриева С.Н., Ведерников А.И., Чуракова М.В. Новая
методология синтеза бензоазакраунсоединений трансформацией макроцикла бен-
зокраунэфиров// Изв. РАН. Сер. хим. 2004. С. 1362-1372.
4. Levitskaya T.G., Bryan J.C., Sachleben R.A. et al. A surprising host-guest
relationship between 1,2-dichloroethane and the cesium complex of tetrabenzo-24-crown-
8 // J. Amer. Chem. Soc. 2000. Vol. 122. P. 554-562.
5. Бауэр И., Хабихер В.Д., Антипин И.С, Синяшин О.Г. Фосфорные макроциклы
и криптанды // Изв. РАН. Сер. хим. 2004. С. 1348.
6. Соловьев В.П., Варнек А.А. Использование молекулярных фрагментов для
моделирования зависимости между структурой молекул и их комплексообразу-
ющими свойствами по отношению к катионам металлов // Изв. РАН. Сер.
хим. 2004. С. 1380.
7. Бухрум С, Арно Ф., Асфари 3., Висан Ж. Распознавание ионов каликсаренами:
связывание ионов щелочных и тяжелых металлов с помощью тиака-
ликс[4]арен-бис-краун[я]эфиров // Изв. РАН. Сер. хим. 2004. С. 1489.
8. Brewster R.E., Shuker B.S. Molecular recognition in methanol: The first example of
hydrogen-bond-mediated self-association of calix[4]arene in polar, protic solvent //
J. Amer. Chem. Soc. 2002. Vol. 124. P. 7902.
9. Hernandes-Molina R., Mederos A. Acyclic and macrocyclic Schiff base ligands //
Comprehensive coordination chemistry II. Ed. A.B.P.Lever. Amsterdam — Oxford —
New York - Sydney - Tokyo: Elsevier - Pergamon Press, 2003. Vol. 1. P. 411-446.
10. Vigato A.P., Tamburini S. The challenge of cyclic and acyclic Schiff bases and
related derivatives // Coord. Chem. Rev. 2004. Vol. 248. P. 1717.
11. Гарновский А.Д., Васильченко И.С Рациональный дизайн координационных
соединений металлов с азометиновыми лигандами // Успехи химии. 2002.
Т. 71. С. 1064; Russ. Chem. Rev. 2001. Vol. 71. P. 943.
12. Radecka-Paryzek W., Patromak V., Lisowski J. Metal complexes of polyaza and
polyoxaaza Schiff base macrocycles // Coord. Chem. Rev. 2005. Vol. 249. P. 2156.
igg(, Дополнительная литература
13. Sunatsuki У., Motoda У., Matsumoto N. Cupper(II) complexes with multidentate
Schiff-base ligands containing imidazole groups: ligand complex of
self-complementary molecule // Coord. Chem. Rev. 2002. Vol. 226. P. 199.
14. Ziessel R. Schiff-based bipyridine ligands. Unusual coordination features and meso-
morphism // Coord. Chem. Rev. 2001. Vol. 216-217. P. 195.
15. Vincenti M., IricoA. Gas-phase interactions of calixarene- and resorcinarene-cavi-
tands with molecular guests studied by mass spectroscopy // Int. J. Mass
Spectrometry. 2002. Vol. 214. P. 23.
16. Гарновский Л.Д., Василъченко И.С, Гарновский Д.А. Современные аспекты
синтеза металлокомплексов. Основные лиганды и методы (концепция
ЖМКО). Ростов-на-Дону: РГУ, Лаборатория перспективного планирования,
2000. 354 с.
17. Synthetic coordination and organometallic chemistry / Ed. A.D.Garnovskii, B.I.
Kharisov. New York-Basel: M. Dekker, 2003. 520 p.
18. Chartres J.D., Lindoy L.F., Meehan G.V. Transition and post-transition metal
systems incorporating linked synthetic macrocycles as structural elements //
Coordination Chemistry Reviews. 2001. Vol. 216-217. P. 249.
19. Kalinina M.A., Arslanov V.V., Zheludeva S.I., Tereschenko Е.Уи. The inversion of
amphiphilic cyclen selectivity toward transition metal ions in Langmuir monolayers//
Thin Solid Films. 2005. Vol. 472, N. 1-2. P. 232.
20. Нестеров СВ. Краун-эфиры в радиохимии. Достижения и перспективы //
Успехи химии. 2000. Т. 69. С. 840.
21. Громов СП., Дмитриева С.Н., Чуракова М.В. Фенилаза- и бензоазакраун-со-
единения с атомом азота, сопряженным с бензольным кольцом // Успехи
химии. 2005. Т. 74. С. 503.
К главе 4
1. Beer P.D., Oale P.A. Anion recognition and sensing: the state of the art and future
perspectives // Angew. Chem., Int. Ed. 2001. Vol. 40. P. 486.
2. Kubik S., Kirchner R., Nolting D., Seidel J. A molecular oyster: a neutral anion
receptor containing two cyclopeptide subjunits with a remarkable sulfate affinity in
aqueous solution // J. Amer. Chem. Soc. 2002. Vol. 124. P. 12752.
3. Hof F., Craig S.L., Nuckolls C, Rebek J. Jr. Molekulare verkapselung // Angew.
Chem. 2002. Bd. 114. S. 1556-1578.
4. Wedge T.J., Hawthorne M.F. Multidentate carborane-containing Lewis acids and
their chemistry: mercurcarborands // Coord. Chem. Rev. 2003. Vol. 240. P. 111.
5. Шур В.Б., Тихонова И.А. Перфторированные полимеркурмакроциклы как ан-
тикрауны. Применение в катализе // Изв. РАН. Сер. хим. 2003. С. 2401.
6. Functional synthetic receptors / Ed. T.Schnader, A.D.Hamilton. New York: Wiley
- WHC, 2005. 440 p.
7. Llinares J.M., Powell D., Bowman-Yames K. Ammonium based anion receptors //
Coord. Chem. Rev. 2003. Vol. 240. P. 57.
8. Bondy R.C., Loeb S.J. Amide based receptors for anion // Coord. Chem. Rev. 2003.
Vol. 240. P. 77.
Дополнительная литература ggy
9. Chio К., Hamilton A.D. Macrocyclic anion receptors based on directed hydrogen
bonding interaction // Coord. Chem.. Rev. 2003. Vol. 240. P. 101.
10. Davis A.P., Joos J.-B. Steroids as organizing elements in anion receptors // Coord.
Chem. Rev. 2003. Vol. 240. P. 191.
11. Gale PA. Anion coordination and anion-directed assembly: highlights from 1997
and 1998 // Coord. Chem. Rev. 2000. Vol. 199. P. 181.
12. Sessler J.L., Camiolo S., Gale PA. Pyrrolic and polypyrrolic anion binding agents
// Coord. Chem.. Rev. 2003. Vol. 240. P. 17.
К главе 5
1. Кузнецов Ф.А., Истомин В.А., Родионова Г.В. Газовые гидраты: исторический
экскурс, современное состояние, перспективы исследований. // Рос. хим.
журн. им. Д.И.Менделеева. 2003. Т. 47. С. 5.
2. Garcia #., Roth D.H. Generations and reactions of organic radical cations in zeolites
// Chem. Rev. 2002. Vol. 102. P. 3947.
3. Lee G.S., Nakawaga Y., Hwang S.-L. et al. Organocations in zeolite synthesis: fused
bicycle[/,/w,o]cations and the discovery of zeolite SSZ-48 // J. Amer. Chem. Soc.
2002. Vol. 124. P. 7024.
4. Chong K.C.W., Sivaguru J., Shichi T. et al. Use of chirality modified zeolites and
crystals in photochemical asymmetric synthesis // J. Amer. Chem. Soc. 2002.
Vol. 124. P. 2858.
5. Голубь А. С, Зайковский В.И., Леленко Н.Д. и др. Синтез и исследование
микроструктуры интеркаляционных соединений дисульфида молибдена с
азотсодержащими органическими молекулами // Изв. РАН. Сер. хим. 2004.
С. 1837.
6. Стойкое И. И., ГафиуллинаЛ.И., Ибрагимова Д. III. и др. Синтетические
рецепторы на основе функционализированного по нижнему ободу калике [4]арена
в молекулярном распознавании дикарбоновых, ос-гидрокси- и
ос-аминокислот// Изв. РАН. Сер. хим. 2004. С. 1125.
7. Ballester P., Shivanyuk A., FarA.R., Rebek J. Jr. A synthetic receptor for choline and
carnitine // J. Amer. Chem. Soc. 2002. Vol. 124. P. 14014.
8. Герасько О.А., СамсоненкоД.Г., Федин В.П. Супрамолекулярная химия кукур-
битурилов // Успехи химии. 2002. Т. 71. С. 840.
9. NakamuraA., Inoue Y. Electrostatic manipulation of enantiodifferetiating photocy-
clodimerization of 2-anthracenecarboxylate within y-cyclodextrin cavity through
chemical modification. Inverted product distribution and enhanced enantioselectiv-
ity // J. Amer. Chem. Soc. 2002. Vol. 124. P. 14014.
10. Abou-Hamdan A., Bugnon P., Saudach C. et al. High-pressure studies as a novel
approach in determining inclusion mechanisms: thermodynamics and kinetics of
the host-guest interactions for a-cyclodextrine complexes // J. Amer. Chem. Soc.
2000. Vol. 122. P. 592.
11. Bartoli S., Roelens S. Binding of acetylholine and tetramethylamrnonium to a cyclo-
phane receptor: anion's contribution to the cation-7i interaction // J. Amer. Chem.
Soc. 2002. Vol. 124. P. 8307.
ggg Дополнительная литература
12. Brotin Т., Lesage A., Emsley L., Collet A. 129Xe NMR spectroscopy of deuterium-
labeled cryptophane-A xenon complexes: investigation of host-guest complexation
dynamics // J. Amer. Chem. Soc. 2000. Vol. 122. P. 1171.
13. Станкевич И.В., Соколов В.И. Достижения химии фуллеренов // Изв. РАН.
Сер. хим. 2004. С. 1749.
14. Sternfeld Т., Hoffman R.E., Sounders M. et al. Two helium atoms inside fullerens:
probing the internal magnetic field in C^q and C^q // J. Amer. Chem. Soc. 2002.
Vol. 124. P. 8786.
15. Sun D., Tham F.S., Reed C.A. et al. Supramolecular fullerene-porphyrin chemistry.
Fullerene complexation by metalated «jaws porphyrine» hosts // J. Amer. Chem.
Soc. 2002. Vol. 124. P. 6604.
16. Hirsh A., Brettreich M. Fullerenes. Chemistry and reactions. New York: Wiley —
VCH, 2004. 435p.
17. Balch A.L., Olmstead MM. Structural chemistry of supramolecular assemblies that
place flat molecular surface around the curved exterios of fullerens // Coord. Chem.
Rev. 1999. Vol. 185-186. P. 601.
18. Mateo-Alonso A., Sooambar C, Prato M. Fullerene photoactive dyads assembled by
axial coordination with metals // Comptes Rendue Chimie. 2006. Vol. 9. P. 944.
19. Lee M., Cho B.-K, Zin W.-C. Supramolecular structures from rod-coil block copoly-
mers// Chem. Rev. 2001. Vol. 101. P. 3869.
20. Cornelissen J., Rowan A., Nolle R., Sommerdijk N. Chiral architectures from makro-
molecular building blocks // Chem. Rev. 2001. Vol. 101. P. 4039.
21. BrunsveldL., FolmerB.J.B., MeijerE. W., Sijbesma R.P. Supramolecular polymers//
Chem. Rev. 2001. Vol. 101. P. 4071.
22. Hapiot F., Tilloy S., Monflier E. Cyclodextrins as supramolecular hosts for
organometallic complexes // Chem. Rev. 2006. Vol. 106. P. 767.
23. Wenz G., Han B.-H., MullerA. Cyclodextrin rotaxanes and polyrotaxanes // Chem.
Rev. 2006. Vol. 106. P. 782.
24. Tao Y., Kanoh H., Abrams L., Kaneko K. Mesopore-modified zeolites: preparation,
characterization and applications // Chem. Rev. 2006. Vol. 106. P. 896.
25. КонаревД.В., Любовская Р.Н. Донорно-акцепторные комплексы и
ион-радикальные соли на основе фуллеренов // Успехи химии. 1999. Т. 68. С. 23.
26. Караулова Е.Н., Багрий Е.И. Фуллерены: методы функционализации и
перспективы применения производных // Успехи химии. 1999. Т. 68. С. 979.
27. Болталина О.В., Галева Н.А. Прямое фторирование фуллеренов // Успехи
химии. 2000. Т. 69. С. 661.
28. Евтюгии Г.А., Будников Г.К., Никольская Е.Б. Биосенсоры для определения
ингибиторов ферментов в окружающей среде // Успехи химии. 1999. Т. 68. С. 1142.
29. Шпигун О.А., Ананьева И.А., Буданова Н.Ю., Шаповалова Е.Н. Использование
циклодекстринов для разделения энантиомеров // Успехи химии. 2003. Т. 72.
С. 1167.
К главе 6
1. Войтеховский Ю.Л. О кристаллах, полиэдрах, радиоляриях, вольвоксах, фул-
леренах и немного о природе вещей // Природа. 2004. № 8. С. 19.
Дополнительная литература 339
2. Burrows A.D. Crystal engineering using multiple hydrogen bonds // Structure and
bonding. 2004. Vol. 108. P. 55.
3. Звездин А. К. Квантовая механика плененных фотонов. Оптические
микрорезонаторы, волноводы, фотонные кристаллы // Природа. 2004. № 10. С. 12.
4. Blake A.J., Campness N.R., Hubberstey P. et al. Inorganic crystal engineering using
self-assembly of tailored building-blocks // Coord. Chem. Rev. 1999. Vol. 183.
P. 117.
5. Damman P., Coppee S., Geskin V.M., Lazzaroni R. What is the mechanism of
oriented crystal grows on rubber polymer substrates & topography vs epitaxy // J. Amer.
Chem. Soc. 2002. Vol. 124. P. 15166.
6. Илюшин Г.Д. Моделирование процессов самоорганизации в кристаллообра-
зующих системах. Структурный механизм самосборки цирконосиликата
Na2ZrSi07 из супраполиэдрических кластеров // Изв. РАН. Сер. хим. 2004.
С.1392.
7. Beatty A.M. Open-framework coordination complexes from hydrogen-bonding
networks: toward host-guest complexes // Coord. Chem. Rev. 2003. Vol. 246. P. 131.
8. Kheobustov A.N., Blake A.J., Champnes N.R. et al. Supramolecular design of one-
dimensional coordination polymers based on silver(I) complexes of aromatic
nitrogen-donor ligands // Coord. Chem. Rev. 2001. Vol. 222. P. 155.
9. Tadakoro M. Nakasuji K. Hydrogen bonded 2,2'-bisimidazolate transition metal
complexes as a tool of crystal engineering // Coord. Chem. Rev. 2000. Vol. 198.
P. 205.
10. Navarro J.A.R., Lippert B. Simple 1:1 and 1:2 complexes of metal ions with hetero-
cycles as building blocks of discrete molecular as well as polymeric assemblies //
Coord. Chem.Rev. 2001. Vol. 222. P. 219.
11. Evans O.R., Lin W. Crystal engineering of NLO materials based on metal-organic
coordination networks // Ace. Chem. Res. 2002. Vol. 35. P. 511.
12. Тарасов Б.П., Гольдшлегер Н.Ф., Моравский А.П. Водородсодержащие
углеродные наноструктуры: синтез и свойства // Успехи химии. 2001. Т. 70. С. 149.
К главе 7
1. Цивадзе А.Ю. Супрамолекулярные металлокомплексные системы на основе
краунзамещенных тетрапирролов // Успехи химии. 2004. Т. 73. С. 6.
2. Cronin L. Macrocyclic and supramolecular coordination chemistry // Ann. Rep.,
Progr. Chem., Sect. A. 2004. Vol. 100. P. 323.
3. Holiday B.J., Mirkin C.A. Koordinationschemische Synthesemethoden zum Aufbau
supramolekularer Verbindungen // Angew Chem. 2001. Bd. 113. S. 2077.
4. Leininger S., Oleynik В., Stang P. T.J. Self-assembly of discrete cyclic nanostructures
mediated by transition metals// Chem. Rev. 2000. Vol. 100. P. 853.
5. Albrecht M. From molecular diversity to template-directed self-assembly - new
trends in metallosupramolecular chemistry //J. Indus. Phenom. Macrocycl. Chem.
2000. Vol. 36. P. 127.
6. Vi/arR. Anion-templated synthesis//Angew. Chem., Int. Ed. 2003. Vol. 42. P. 1460.
7. Prins L.J., Reinhoudt D.N., Timmerman P. Nichtovalente Synthese mit
Wasserstoffbriiken//Angew. Chem., Int. Ed. 2001. Vol. 40. P. 2382.
g90 Дополнительная литература
8. Лекегбу СВ., Beatty А.Л/., Helfrich B.A. A high-yielding supramolecular reaction //
J. Amer. Chem. Soc. 2002. Vol. 124. P. 14425.
9. Paraschiv V., Crego-Calamo M., Ichi-e T. et al. Molecular «chaperones» guide the
spontaneous formation of a 15-component hydrogen-bonded assembly // J. Amer.
Chem. Soc. 2002. Vol. 124. P. 7638.
10. Shivanyuk A., Rebek J. Jr. Social isomers in encapsulation complexes // J. Amer.
Chem. Soc. 2002. Vol. 124. P. 12074.
11. Kimizuka N. Self-assembly in mesoscopic dimension and artificial supramolecular
membranes // Curr. Opinion Chem. Biology. 2003. Vol. 7. P. 702.
12. Swiegers G.F., Malefeste T. Y. Classification of coordination polygons and polyhedra
according to their mode of self-assembly. 2. Review of the literature // Coord.
Chem. Rev. 2002. Vol. 225. P. 91.
13. Vdachin K.A., Wilson L.D., RipmeesterJ.A. Solid polyrotaxanes of polyethylene gly-
col and cyclodextrins: the single crystal X-ray structure of PEG-P-cyclodextrine //
J. Amer. Chem. Soc. 2000. Vol. 122. P. 12375.
14. Jones J.W., Zakharov L.N., Rheingold A.L., Gibson H.W. Cooperative host/guest
interactions via counterion assisted chelation: pseudorotaxanes from
supramolecular cryptands // J. Amer. Chem. Soc. 2000. Vol. 122. P. 12375.
15. Anderson M., Linke M., Chambron J.-C. et al. Porphyrin-containing [2]-rotaxanes:
metal coordination enhanced superexchange electron transfer between noncova-
lently linked chromophores// J. Amer. Chem. Soc. 2000. Vol. 122. P. 3526.
16. Wisner J.A., Beer P.D., Drew M.B.G., Sambrook M. Anion-templated rotaxane
formation // J. Amer. Chem. Soc. 2002. Vol. 124. P. 12469.
17. Jung J.H., John G, Yoshida K., Schimuzu T. Self-assembly structure of long-chain
phenyl glycoside influenced by the introduction of double bonds // J. Amer. Chem.
Soc. 2002. Vol. 124. P. 10674.
18. Albrecht M. «Let's twist again» - double stranded, triple stranded and circular heli-
cates//Chem. Rev. 2001. Vol. 101. P. 3457.
19. Han L., Hong M. Recent advances in the design and construction of helical
coordination polymers // Inorg. Chem. Commun. 2005. Vol. 8. P. 406.
20. Andrews P.C., Hardie M.J., Raston C.L. Supramolecular assemblies of globular main
group cage species // Coord. Chem. Rev. 1999. Vol. 189. P. 169.
21. Applegarth L, Goeta A.E., Steed J.W. Influence of hydrogen bonding on
coordination polymer assembly // Chem. Commun. 2005. P. 2405.
22. Turner D.R., Spencer E.C., Howard J.A.K. et al. A modular, self-assembled,
separated ion-pair binding system // Chem. Commun. 2004. P. 1352.
23. Vriezema DM., Aragonus M.C., Elemans J.A.A. Self-assembled nanoreactors //
Chem. Rev. 2005. Vol. 105. P. 1445.
24. Hoeben F.J.M., Jonkheijm P., MeijerE.W., SchenningA.P.H.J. About
supramolecular assemblies of7i-conjugated systems// Chem. Rev. 2005. Vol. 105. P. 1491.
25. Swiegers G.F., Malefeste T. Y. New self-assembled structural motifs in coordination
chemistry // Chem. Rev. 2000. Vol. 100. P. 3483.
26. Tasis D., Tagmatarchis N., Bianko A., Prato M. Chemistry of carbon nanotubes //
Chem. Rev. 2006. Vol. 106. P. 1105.
27. Шапиро Б.И. Молекулярные ансамбли полиметиленовых красителей //
Успехи химии. 2006. Т. 75. С. 484.
Дополнительная литература 391
28. Briihwiler D., Calzaferri G. Molecular sieves materials for supramolecular
organization // Microporous and mesoporous material for supramolecular organization.
2004. Vol. 72. P. 1.
29. Панова И.Г., Топчиева И.Я. Ротоксаны и полиротаксаны. Синтез и супрамо-
лекулярные устройства на их основе // Успехи химии. 2001. Т. 70. С. 28.
30. Герасько О.А., СамсоненкоД.Г., Федин В.П. Супрамолекулярная химия кукур-
битурилов // Успехи химии. 2002. Т. 71. С. 840.
31. Collin J.-R, Dietrich-Buchecker С, Натапп С. et al. Supramolecular systems: tem-
plating // Comprehensive coordination chemistry II . Ed. J.A.McCleverty, T.J.
Meyer. Amsterdam - New York: Elsevier-Pergamon Press, 2003. Vol. 7. P. 303.
32. Yeh R.M., Davis A.D., Raymond K.N. Supramolecular systems: self-assembly //
Comprehensive Coordination Chemistry II . Ed. J.A.McCleverty, T.J. Meyer.
Amsterdam—New York: Elsevier—Pergamon Press, 2003. Vol. 7. P. 327.
33. Лрсланов В.В. Полимерные монослои и пленки Лэнгмюра—Блоджетт. Поли-
тиофены // Успехи химии. 2000. Т. 69. С. 839.
34. Huie J. С. Guided molecular self-assembly: a review of recent efforts // Smart Mater.
Struct. 2003. Vol. 12. P. 264.
35. Corbellini F., Mulder A., Sartori A. et al. Assembly of a supramolecular capsule on a
molecular printboard//J. Amer. Chem. Soc. 2004. Vol. 126. P. 17050.
36. Love J. C, EstroffL.A., KriebelJ.K. et al. Self-assembled monolayers of thiolates on
metals as a form of nanotechnology// Chem. Rev. 2005. Vol. 10. P. 1103.
К главе 8
1. Алфимов М.В. Фотоника супрамолекулярных наноразмерных структур //
Изв. РАН. Сер. хим. 2004. С. 1303.
2. Брень В.А. Флуоресцентные и фотохромные хемосенсоры // Успехи химии.
2001. Т. 70. С. 1152.
3. McQuade D. Т., Pullen A.E., Swager T.M. Conjugated polymer-based chemical
sensors // Chem. Rev. 2000. Vol. 100. P. 2537.
4. Bargossi C, Fiorici M.C., Montalti M. et al. Recent development in transition metal
ion detection by luminescent chemosensors // Coord. Chem. Rev. 2000. Vol. 208.
P. 17.
5. Ashkenasy G., Ivanisevic A., Cohen R. et al. Assemblies of «hinged» ironporphyrins as
potential oxygen sensors // J. Amer. Chem. Soc. 2000. Vol. 122. P. 1116.
6. Irie M. Diarylethenes for memories and switches // Chem. Rev. 2000. Vol. 100.
P. 1685.
7. Yokoyama Y. Fulgides for memories and switches // Chem. Rev. 2000. Vol. 100.
P. 1717.
8. Vannikov A.V., Grishina A.D., Gorbunova Yu.G. et al. Photo refractive IR-spectrum
composites prepared from polyimide and ruthenium(II) tetra-15-crown-5-phthalo-
cyanine with axially coordinated triethylenediamine molecules // Rus. J. Phys.
Chem. 2006. Vol. 80, N 3. P. 453.
9. Bercovich G., Krongaus V., Weiss V. Spiropyrans and spirooxazines for memories and
switches// Chem. Rev. 2000. Vol. 100. P. 1741.
Ig92 Дополнительная литература
10. Feringa B.L., Delden R.A. van, Koumura N., Geerstema E.M. Chiroptical molecular
switches//Chem. Rev. 2000. Vol. 100. P. 1789.
11. Asakawa M., Brankato G., Fanti M. et al. Switching «on» and «off» the expression of
chirality in peptide rotaxanes // J. Amer. Chem. Soc. 2002. Vol. 124. P. 2939.
12. Kushmeric J.G., Holh D.B., Pollack S.K. et al. Effect of bond-length alternation in
molecular wires//J. Amer. Chem. Soc. 2002. Vol. 124. P. 10654.
13. Balzani V., CrediA., Raymo F.M., Stoddart J.F. Kiinstliche molekulare Maschines//
Angew. Chem. 2000. Bd. 112. S. 3484.
14. Ballardini R., Balzani V., Clemente-Leon M. et al. Photoinduced electron transfer in
a trade that can be assembled/disassembled by two different external inputs toward
molecular-level electrical extension//J. Amer. Chem. Soc. 2002. Vol. 124. P. 12786.
15. Balzani V., CrediA., Langford S.J. et al. Construction molecular machinery: a chem-
ically-switchable[2]-catenane// J. Amer. Chem. Soc. 2000. Vol. 122. P. 3542.
16. Koumura N., Geertsema E.M., Gelder M.B. van et al. Second generation light-driven
molecular motors. Undirectional rotation controlled by a single stereogenic center
with near-perfect photoequilibria and acceleration of the speed of rotation by
structural modification // J. Amer. Chem. Soc. 2002. Vol. 124. P. 5037.
17. Domingues Z, Dang H., Strouse M.J., Garcia-Garibay M.A. Molecular «compasses»
and «gyroscopes». I. Expedient synthesis and solid state dynamics of an open rotor
with a bis(triarylmethyl) frame//J. Amer. Chem. Soc. 2002. Vol. 124. P. 2398.
18. Le Bozec H., Le Bouder Т., Maury O. et al. Coordination chemistry for nonlinear
optics: a powerful tool for the design of octapolar molecules and supermolecules //
J. Optics A: Pure Appl. Optics. 2002. Vol. 4. P. 189.
19. Delaire J.A., Nakatani K. Linear and nonlinear optical properties of photochromic
molecules and materials // Chem. Rev. 2000. Vol. 100. P. 1817.
20. Grayson S.M., Frechet J.M.J. Convergent dendrons and dendrimers: from synthesis
to application//Chem. Rev. 2001. Vol. 101. P. 3819.
21. Белецкая И.П., Чучуркии А.В. Синтез и свойства функционально
замещенных дендримеров // Успехи химии. 2000. Т. 69. С. 699.
22. Freeman A.W., Коепе S.C., Malennfant P.K.L. et al. Dendrimers-containing light-
emitting diodes: toward site-isolation of chromophores // J. Amer. Chem. Soc.
2000. Vol. 122. P. 12385.
23. Andronov A., Gilat S.L., Frechet J.M.J. et al. Light harvesting and energy transfer in
light-dye-labeled poly(aryl ether)dendrimers // J. Amer. Chem. Soc. 2000. Vol. 122.
P. 1175.
24. Al-Jamal К. Т., Ramaswamy C, Florence P. Supramolecular structures from
dendrons and dendrimers // Advanced Drug Delivery Rev. 2005. Vol. 57. P. 2338.
25. G.A.Kinberger G.A., Cai W., Goodman M. Collagen mimetic dendrimers // J. Amer.
Chem. Soc. 2002. Vol. 124. P. 15162.
26. Fogtle F., Gestermann S., Hesse R. et al. Functional dendrimers // Progr. Polymer
Science. 2000. Vol. 25. P. 987.
27. Van Heerbeek R., Kamer P. C.J., Van Leeuwen P. W.N.M., Reek J. N. H. Dendrimers as
support for recoverable catalysts and reagent // Chem. Rev. 2002. Vol. 102.
P. 3717.
28. Crespo L, Sanclimens G., Pons M. et al. Peptide and amide bond-containing
dendrimers// Chem. Rev. 2005. Vol. 105. P. 1663.
Дополнительная литература 893§
29. Astruc D., Chardac F. Dendric catalysts and dendrimers in catalysis // Chem. Rev.
2001. Vol. 101. P. 2991.
30. Guldi D.M., SwartzA., Luo С et al. Rigid dendritic donor-acceptor ensembles:
control over energy and electron transduction // J. Amer. Chem. Soc. 2002. Vol. 124.
P. 10875.
31. Xu M.-H., LinJ., Ни Q.S., Pu L. Fluorescent sensors for the enantioselective
recognition of mandelic acid: signal amplification by dendritic branching // J. Amer.
Chem. Soc. 2002. Vol. 124. P. 14239.
32. Clark C, Pfenning В., Bocarsly Л.В. Photoinduced multielectron charge transfer
processes in group 8-platin cyanobridged supramolecular complexes // Coord.
Chem. Rev. 2000. Vol. 208. P. 33.
33. Schulz-Ekloff G., Wufrre D., Van Duffel В., Schoonheyd R.A. Chromophores in
porous silicas and minerals: preparation and optical properties // Microporus and
mesoporus Materials. 2002. Vol. 51. P. 91.
34. Yokoyama Ya., Kose M. Reversible control of properties of materials by thermally
irreversible photochromism // J. Photochem. Photobiol. A: Chemistry. 2004. Vol.
166. P. 9.
35. Smith D.K., Hirst A.R., Love C.S. et al. Self assembly using dendric building
blocks — towards controllable nanomaterials // Progr. Polym. Chem. 2005.
Vol. 30. P. 220.
36. Principles of physical chemistry - understanding molecules, molecular assemblies,
supramolecular machines / Ed. H.Kurr, H.-D.Forsterling. Chichester: Wiley, 2000.
970 p.
37. Richter M.M. Electrochemiluminescence (ECL) // Chem. Rev. 2004. Vol. 104.
P. 3003.
38. Burda C, Chen X., Narayanan R. et al. Chemistry and properties of nanocrystals of
different shapes// Chem. Rev. 2005. Vol. 105. P. 1025.
39. Gates B.D., Xu Q., Stewart M. et al. New approach to nanofabrication: molding,
printing and other techniques // Chem. Rev. 2005. Vol. 105. P. 1171.
40. Shimuzu Т., Masuda M., Minamikawa H. Supramolecular nanotube architectures
based on amphiphilic molecules// Chem. Rev. 2005. Vol. 105. P. 1401.
41. Kakkar A.K. Nano-organometallics: heterogenizing homogeneous catalyst via thin
film methodology// Chem. Rev. 2002. Vol. 102. P. 3579.
42. Ивановский A.JJ. Моделирование нанотубулярных форм веществ // Успехи
химии. 1999. Т. 68. С. 119.
43. Раков Э.Г. Методы получения углеродных нанотрубок // Успехи химии. 2000.
Т. 69. С. 41.
44. Раков Э.Г. Химия и применение углеродных нанотрубок // Успехи химии.
2001. Т. 70. С. 934.
45. Ивановский A.JI. Неуглеродные нанотрубки: синтез и моделирование //
Успехи химии. 2002. Т. 71. С. 203.
46. Valeur В., Leray I. Design principles of fluorescent molecular sensors for cation
recognition // Coord. Chem.. Rev. 2000. Vol. 205. P. 3.
47. Kalinina M.A., Golubev N.V., Rightman O.A. et al. A novel ultra-sensing composed
Langmuir-Blodgett membrane for selective calcium determination in aqueous
solutions // Sensors & Actuators, B, Chemical. 2006. Vol. 114. P. 19.
394 Дополнительная литература
48. Власов Ю.Г., Легин А.В., Рудницкая A.M. Мультисенсорные системы типа
электронный язык — новые возможности создания и применения химических
сенсоров // Успехи химии. 2006. Т. 75. С. 141.
К главе 9
1. Wullf G. Enzyme-like catalysts by molecularly imprinted polymers // Chem. Rev.
2002. Vol. 102. P. 1.
2. Haupt K., Mosbach K. Molecularly imprinted polymers and their use in biomimetic
sensors // Chem. Rev. 2000. Vol. 100. P. 2495.
3. Jelinek R., Kolusheva S. Carbohydrate biosensors // Chem. Rev. 2005. Vol. 105.
P. 5987.
4. Hill D.J., Mio M.J., Prince R.B. et al. A field guide to foldamers // Chem. Rev. 2001.
Vol. 101. P. 3893.
5. Но Т., Hioki Т., Yamaguchi T. et al. Development of a molecular recognition ion
gatting membrane and estimation of its pore size control // J. Amer. Chem. Soc.
2002. Vol. 124. P. 7840.
6. Ни Zh., Williams R.D., Tran D. et al. Re-engineering enzyme model active sites:
reversible binding of dioxygen at ambient conditions by a bioinspired copper
complex // J. Amer. Chem. Soc. 2000. Vol. 122. P. 3556.
7. Forman S.L., Fettinger J.C., Pieraccini S. et al. Toward artificial ion channels:
a lipophilic G-quadruplex // J. Amer. Chem. Soc. 2000. Vol. 122. P. 4060.
8. Kovbasyuk L., Krdmer R. Allosteric supramolecular receptors and catalysts // Chem.
Rev. 2004. Vol. 104. P. 3161.
9. Engeldinger E., Armspach D., Matt D. Capped cyclodextrins // Chem. Rev. 2003. Vol.
103. P. 4147.
10. Мисюров Д. Практическая нанотехнология // В мире науки. 2005. № 3. С. 20.
11. Ициксон Н.А., Зырянов Г.В., Чупахин О.Н., Матерн А.И. Гетеродитопные
рецепторы // Успехи химии. 2005. Т. 74. С. 820.
12. Gokel G. W., Leevy W.M., Weber M.E. Crown ethers: Sensors for ions and molecular
scaffolds for materials and biological models // Chem. Rev. 2004. Vol. 104. P. 2723.
К главе 10
1. Mueller A., O'Brien D.F. Supramolecular materials via polymerization of hydrated
amphiphiles// Chem. Rev. 2002. Vol. 102. P. 727.
2. Левашов А.В., Клячко Н.Л. Мицеллярная энзимология: методы и техника //
Изв. РАН. Сер. хим. 2001. С. 1638.
3. Elemans J.A., Rowan A.E., Notle R.J.M. Hierarchical self-assembly of amphiphilic
metallohosts to give discrete nanostructures // J. Amer. Chem. Soc. 2002. Vol. 124.
P. 1532.
4. Kondo J., Miyazwa H, Sakai H. et al. 1st anionic micelle with unusually long
lifetime: self-assembly of fluorocarbon-hydrocarbon hybrid surfactant // J. Amer.
Chem. Soc. 2002. Vol. 124. P. 6516.
5. Berihier D., Buffeteau Т., LegerJ.-M. et al. From chiral counterions to twisted
membranes//J. Amer. Chem. Soc. 2002. Vol. 124. P. 13486.
Дополнительная литература 895
6. Ishimura К. Photoalignment of liquid-crystal systems // Chem. Rev. 2000. Vol. 100.
P. 1847.
7. Shen D., Pegenau A., Diele S. et al. Molecular design of nonchiral bent core liquid
crystals with antiferroelectric properties // J. Amer. Chem. Soc. 2000. Vol. 100.
P. 1593.
8. Pietrasik U., Szydlowska J., Krywczynski A. et al. Re-entrant isotopic phase between
lammelar and column mesophases // J. Amer. Chem. Soc. 2002. Vol. 124. P. 8884.
9. Kolbel M., BeyersdorffT., Cheng X.H. et al. Design of liquid crystalline block
molecules with nonconventional mesophase morphologies: calamitic bolaamphiphiles
with lateral alkyl chains // J. Amer. Chem. Soc. 2001. Vol. 123. P.6809.
10. Zimerman #., Bader V., Poupko R. et al. Mesomorphism, isomerization and
dynamics in a new series of pyramidic liquid crystals // J. Amer. Chem. Soc. 2002. Vol. 124.
P. 15286.
11. Hartley C.S., Lazar C, Wand M.D., Lemieux R.P. Detection of chiral perturbation
in ferroelectric liquid crystals induced by an atropisomeric biphenyl dopants //
J. Amer. Chem. Soc. 2002. Vol. 124. P. 13513.
12. Dantlgraber G, Baumeister U., Diele S. et al. Evidence for a new ferroelectric
switching liquid crystalline phase formed by a carbosilane based dendrimer with banana-
shaped mesogenic units //J. Amer. Chem. Soc. 2002. Vol. 124. P. 14852.
13. Binnemans K. Ionic liquid crystals // Chem. Rev. 2005. Vol. 105. P. 4148.
14. Gimenez R., Lydon D.P., Serrako J.L. Metallomesogens: a promice or a fact //
Current Opinion in Solid State and Material Science. 2002. Vol. 6. P. 527.
15. Belanger S., Keefe M.H., Jennifer L. et al. Rapid derivatization of mesoporous thin-
film material based on Re(l)zinc-porphirin molecular squares: selective
modification of mesopore size and shape by binding of aromating nitrogen donor ligands //
Coord. Chem. Rev. 1999. Vol. 190-192. P. 29.
16. Serrano J.L., Sierra T. Helical supramolecular organization from metallorganic
liquid crystals // Coord. Chem. Rev. 2003. Vol. 242. P. 73.
17. Chang T.-M., Dang L.X, Recent advances in molecular simulations of ion solvation
at liquid interfaces // Chem. Rev. 2006. Vol. 106. P. 1305.
18. Пебалк Д.А., Барматов Е.Б., Шибаев В.П. Жидкокристаллические иономе-
ры — новый класс мезоморфных полимерных систем // Успехи химии. 2005.
Т. 74. С. 610.
Научное издание
Стид Джонатан В., Джерри Л. Этвуд
СУПРАМОЛЕКУЛЯРНАЯ ХИМИЯ
Том 2
Редактор М.Л. Франк
Художник И.А. Слюсарев
Дизайнер И. М. Кадченко
Компьютерный дизайн и верстка О.Д. Эшлиман
ИД №04284 от 15.03.2001.
Подписано в печать 01.11.2006. Формат 70x100/16. Гарнитура NewtonC.
Печать офсетная. Печ. л. 26. Тираж 1600 экз. Тип. зак. 2716.
Издательско-книготорговый центр «Академкнига»
117997, Москва, Профсоюзная ул., 90
По вопросам поставок обращаться
в отдел реализации ИКЦ «Академкнига»
Тел./факс: D95) 334-73-18
e-mail: bookreal@maik.ru, web-site: http://www.akademkniga.com
Отпечатано с готовых диапозитивов в
ОАО «Ивановская областная типография»
153008, г. Иваново, ул. Типографская, 6.
E-mail: 091-018@adminet.ivanovo.ru
Special thanks for
DNS-scan
Revenae ~ edit & encodina
химия
Jonathan W. Steed, King's College, London
and
Jerry L Atwood, University of Missouri, Columbia
Супрамолекулярная химия - это междисциплинарная
область, которая до сих пор активно сопротивляется
всем попыткам очертить ее рамки или дать
достаточно полное ее определение. Книга
«Супрамолекулярная химия» - это на сегодняшний день первый в
мировой литературе учебник, охватывающий
практически все разделы супрамолекулярной химии. В ней
рассказывается обо всем, что студент должен знать
для успешного начала своей научной деятельности в
этой области. Предполагая, что читатель уже
обладает определенным уровнем подготовки, авторы
включили в книгу и теоретические вопросы, относящиеся
к рассматриваемой области, и сведения о наиболее
важных экспериментальных методах исследования
супрамолекулярных систем. Значительное внимание
уделено истокам супрамолекулярной химии, ее
объектам, а также вопросам терминологии. Книга
содержит большое число примеров супрамолекулярных
соединений, вопросы и задачи для повторения
материала каждой главы.
Supramolecular Chemistry
Is the first integrated supramolecular book written specifically for students.
Is clearly structured and lucidly written with many examples,
applications and problems drawn from this diverse discipline.
Features a web site with additional information and the latest developments:
www.ch.kcl.ac.uk/supramol/textbook.htm
ISBN 978-5-94628-303-8
>BN 0-471-98831-6
ИКЦ «АКАДЕМКНИГА»
СУПРАМОЛЕКУЛЯРНАЯ